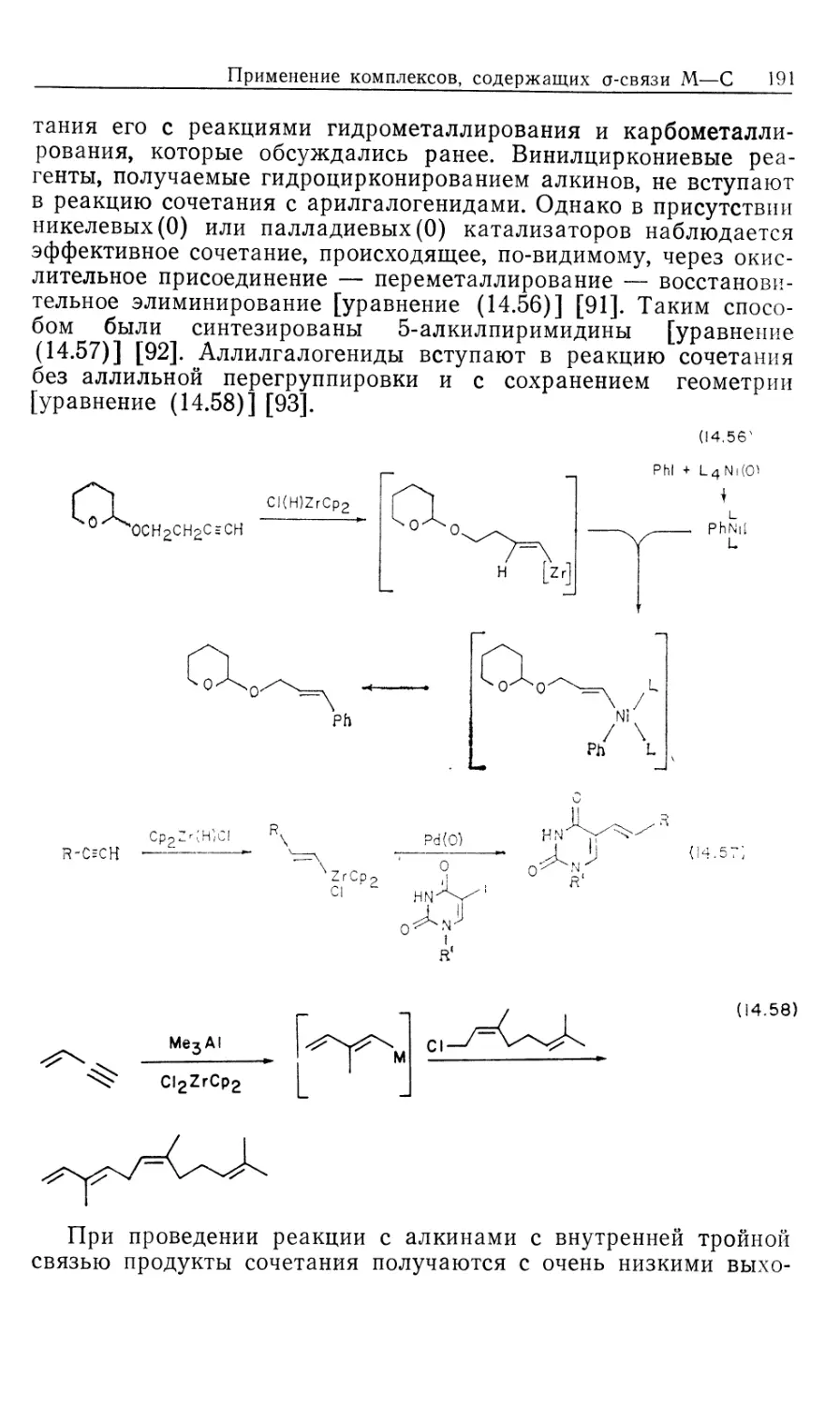
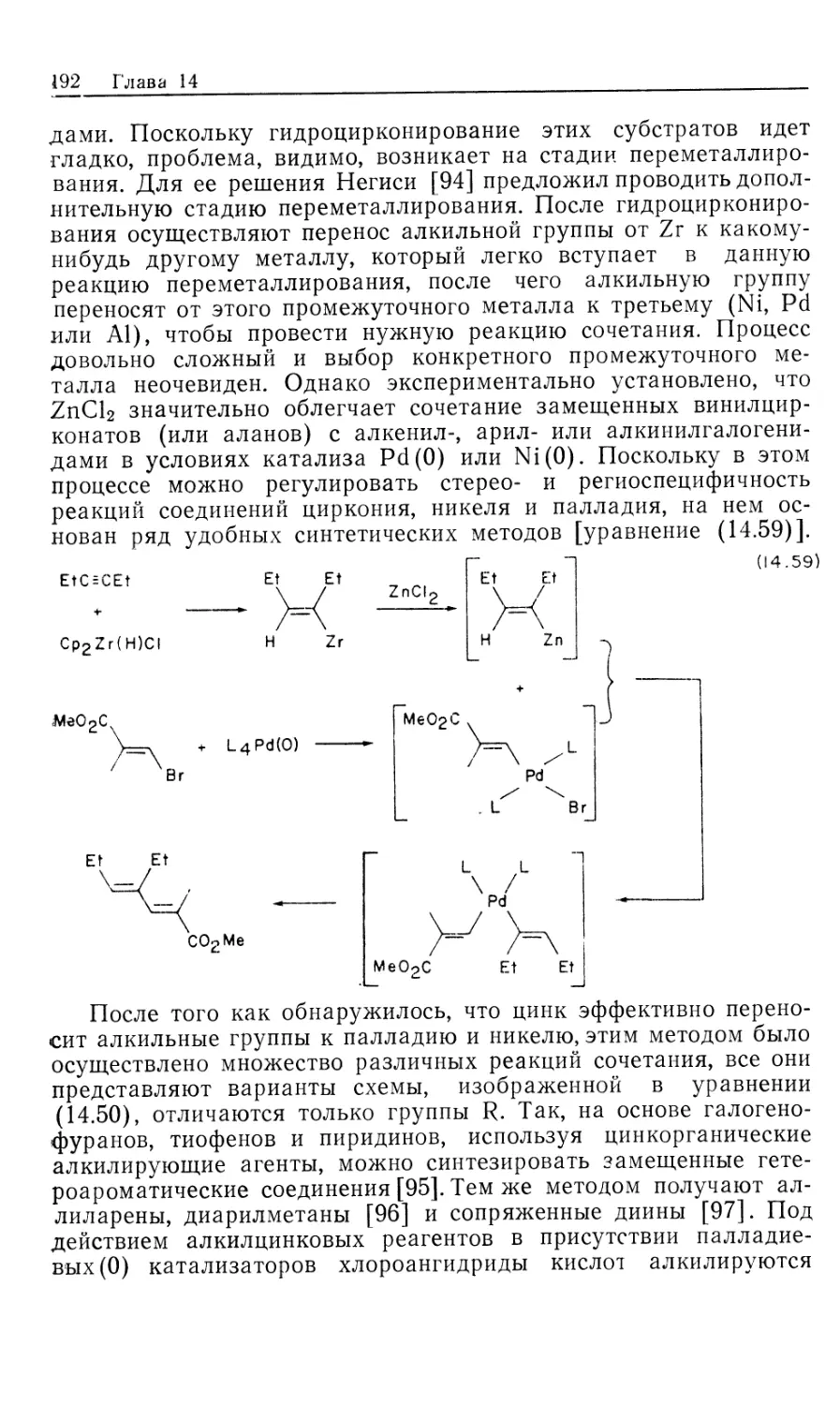
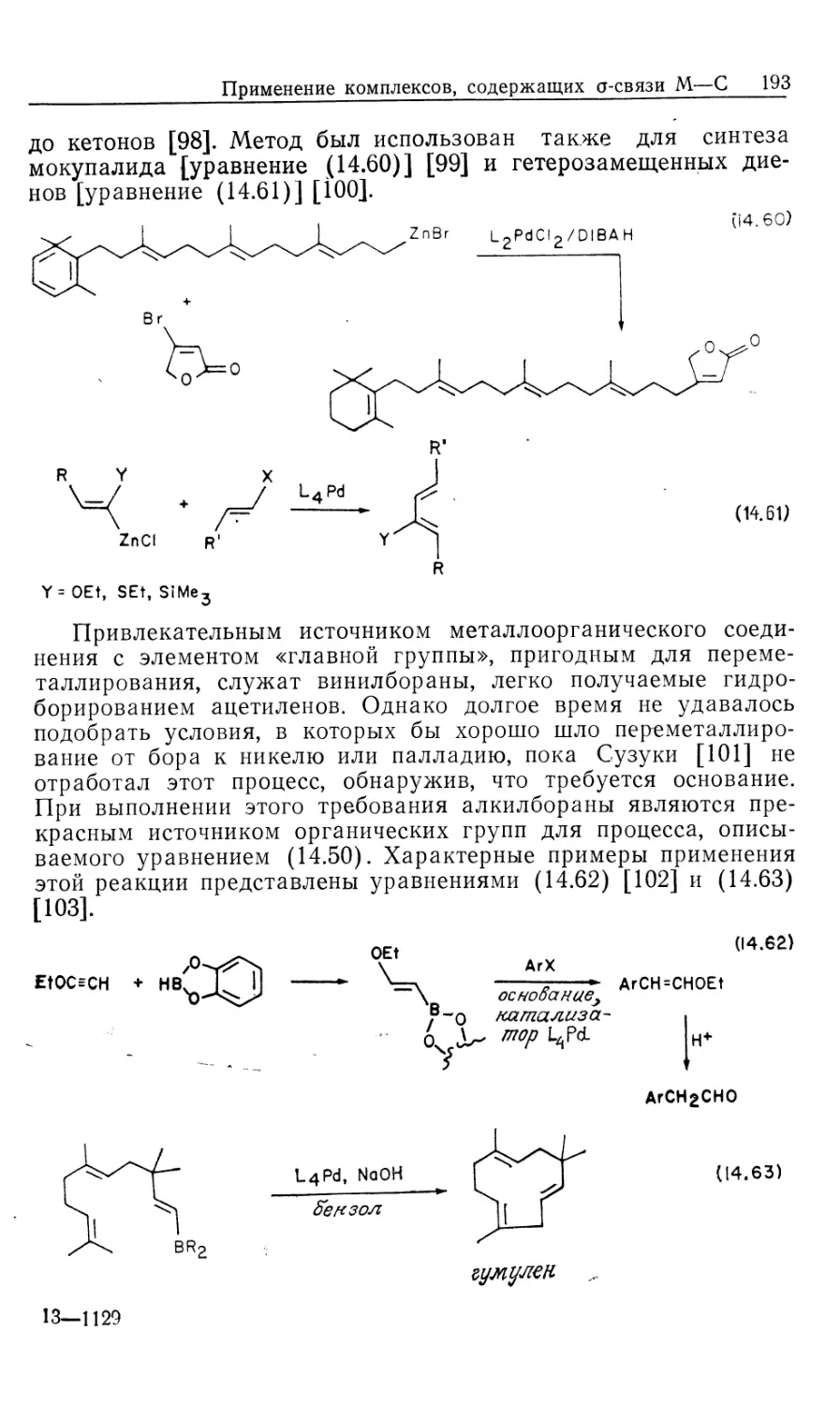
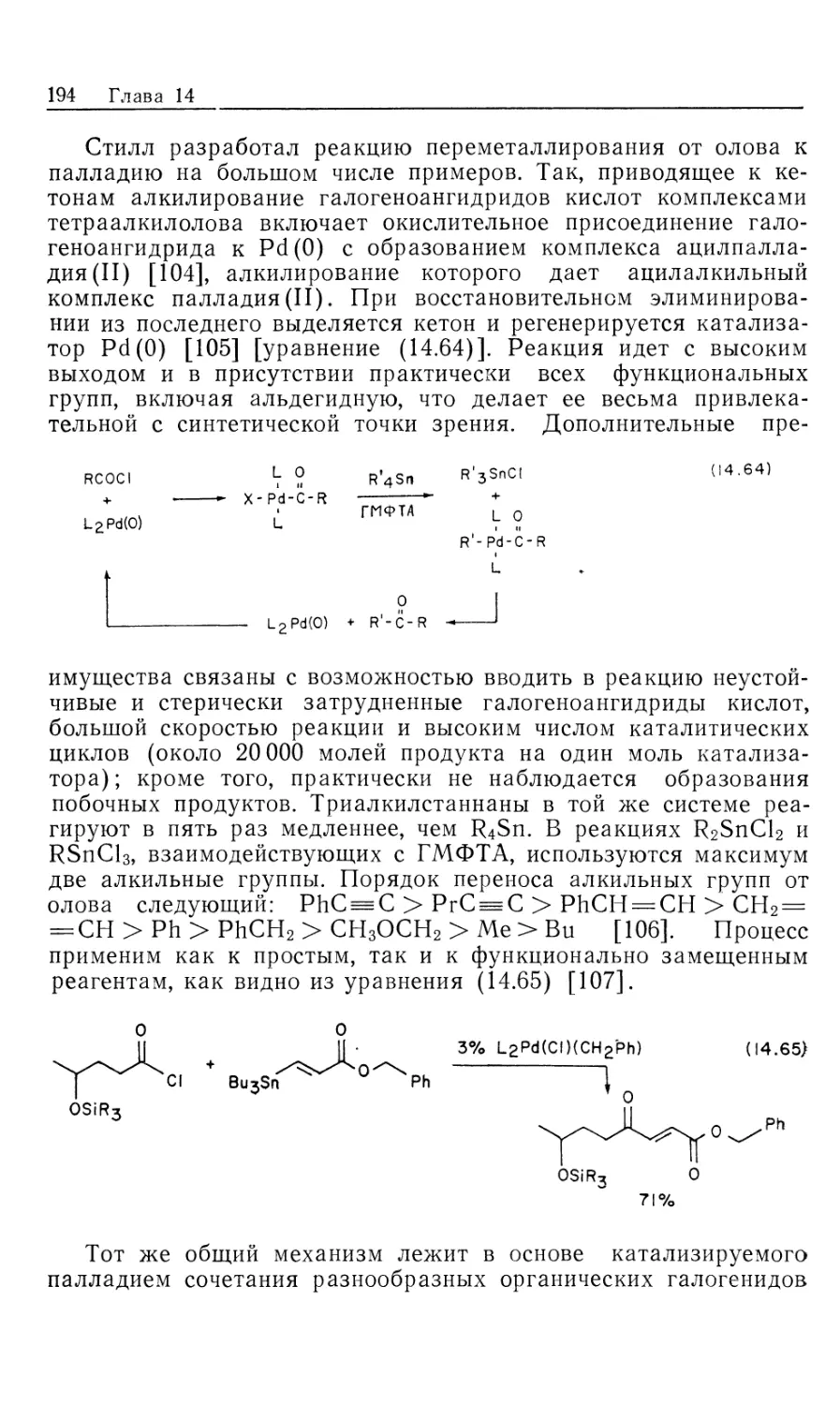
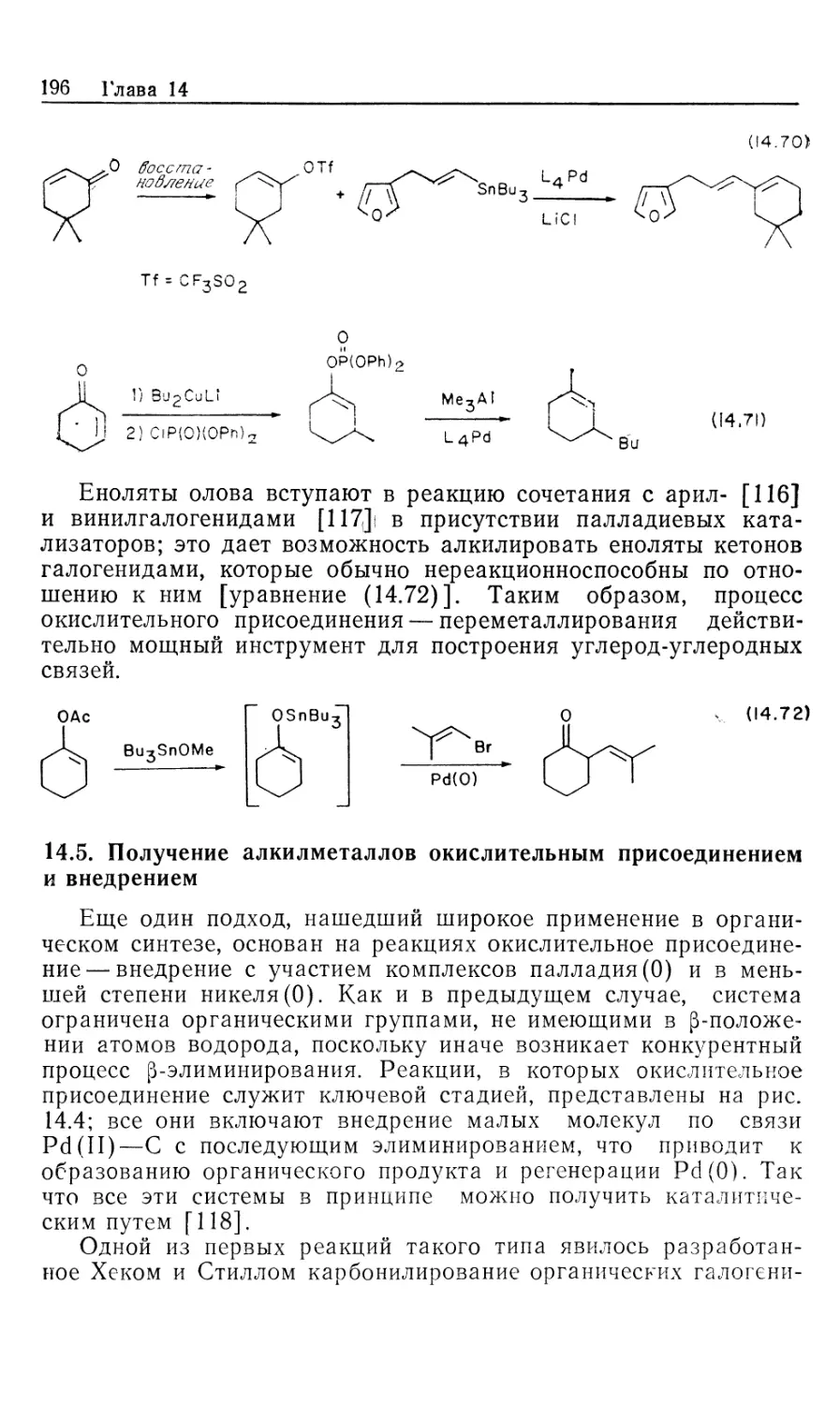
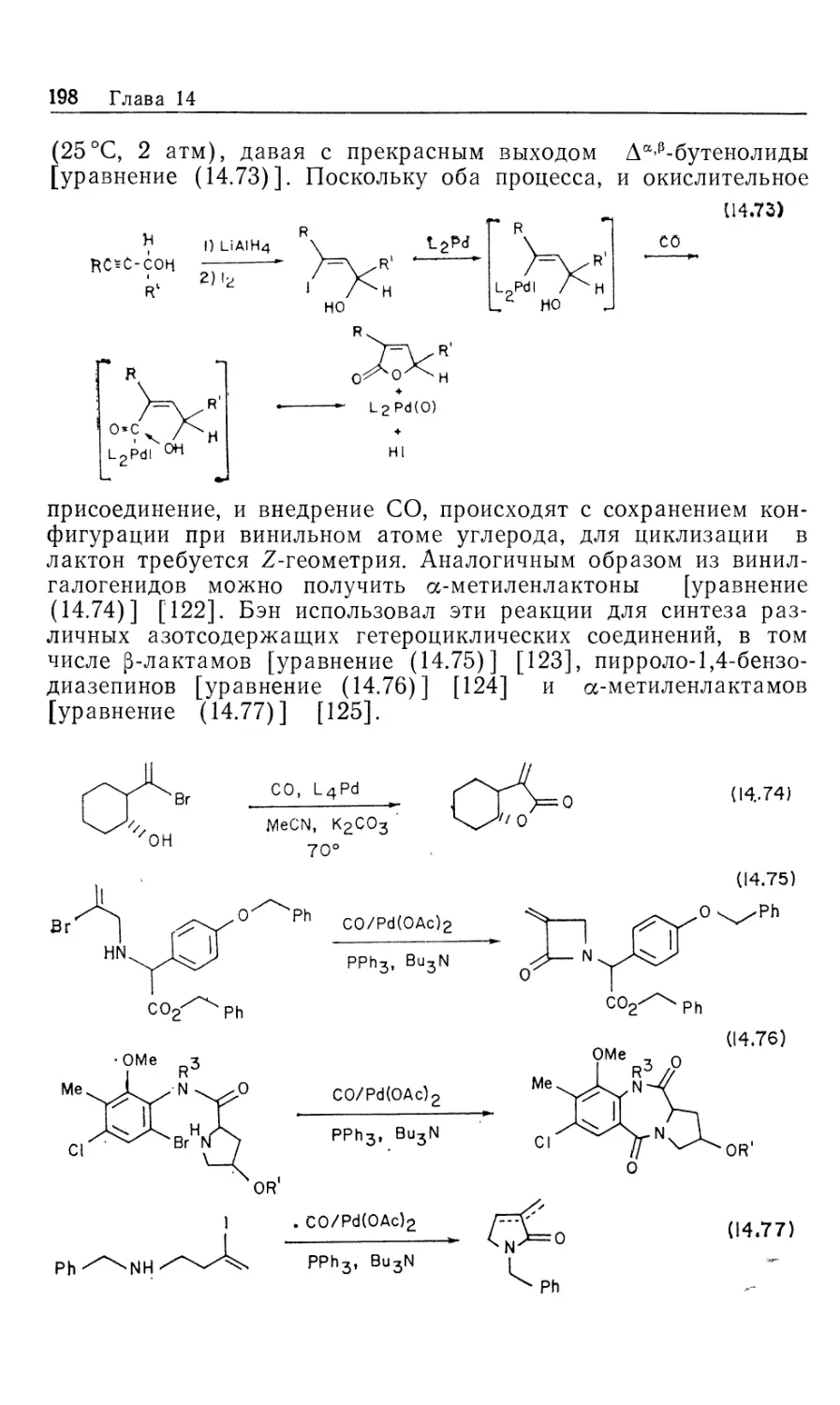
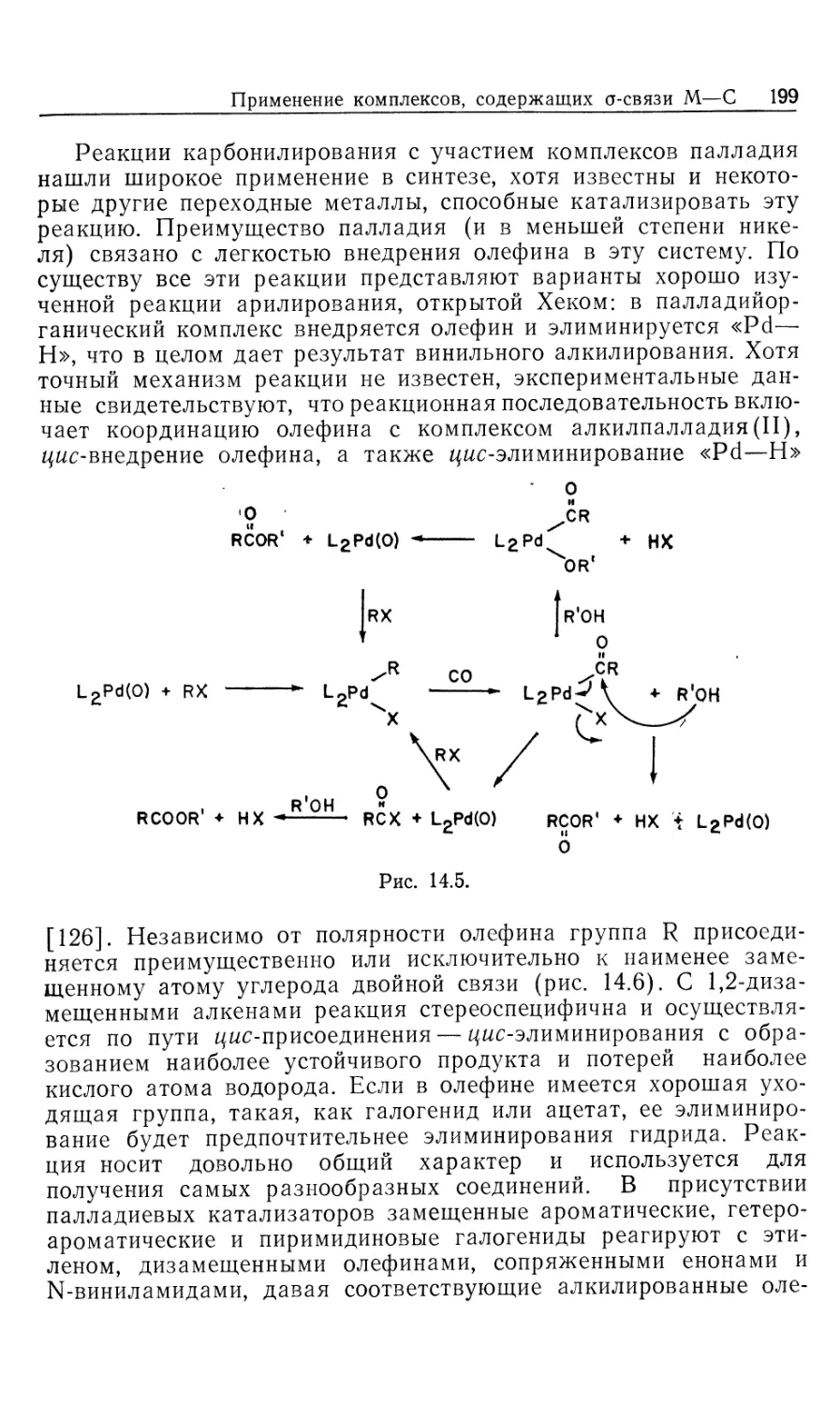
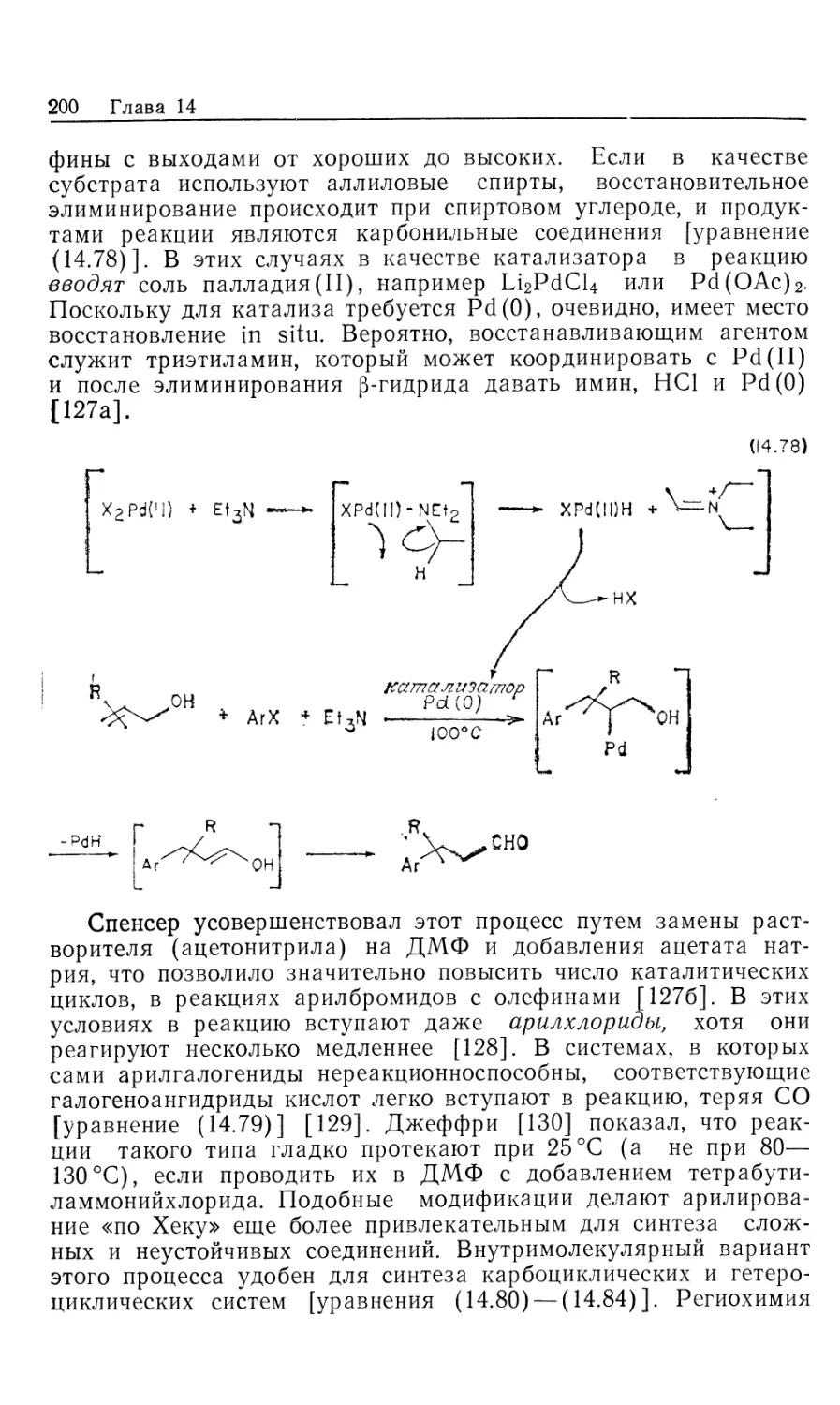
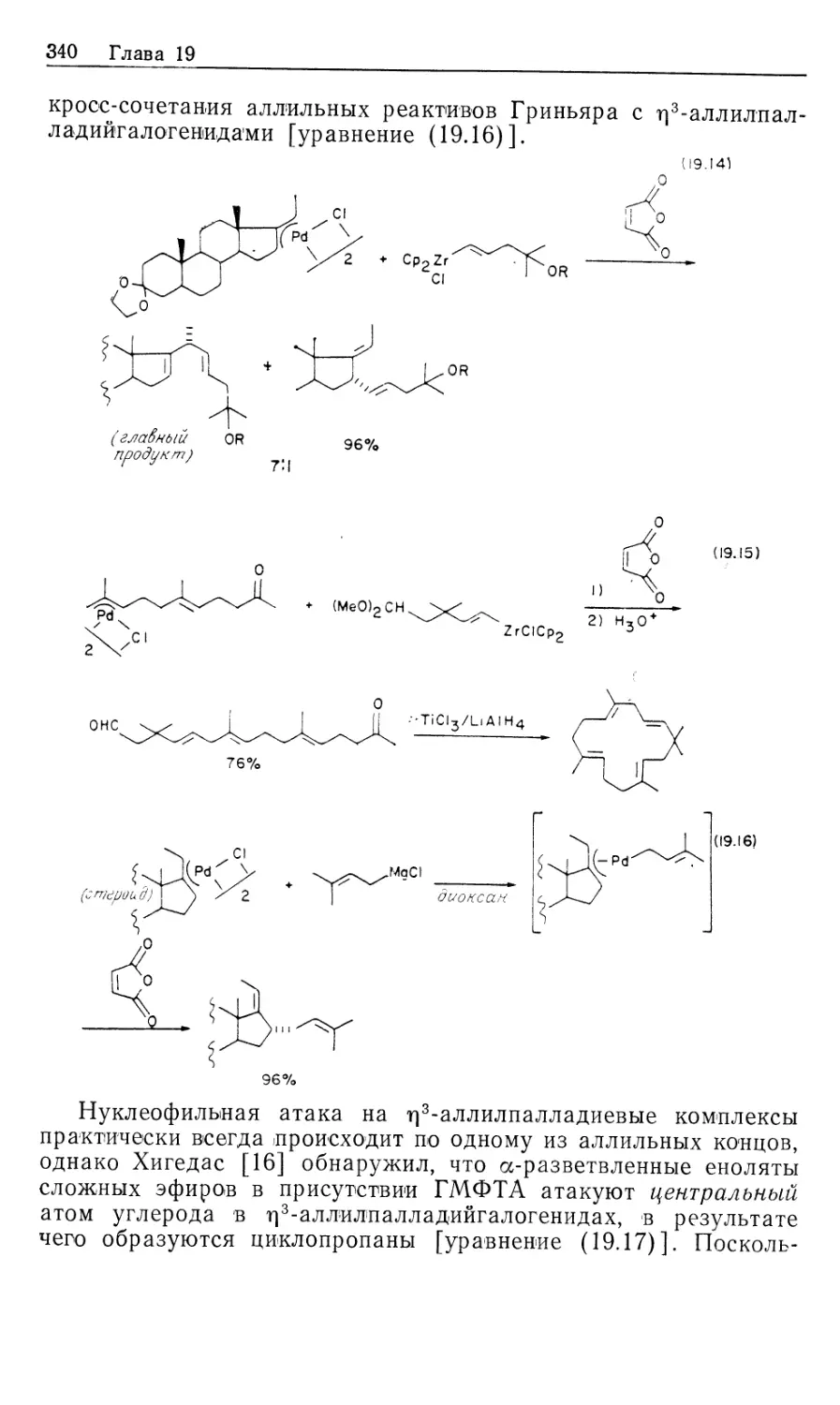
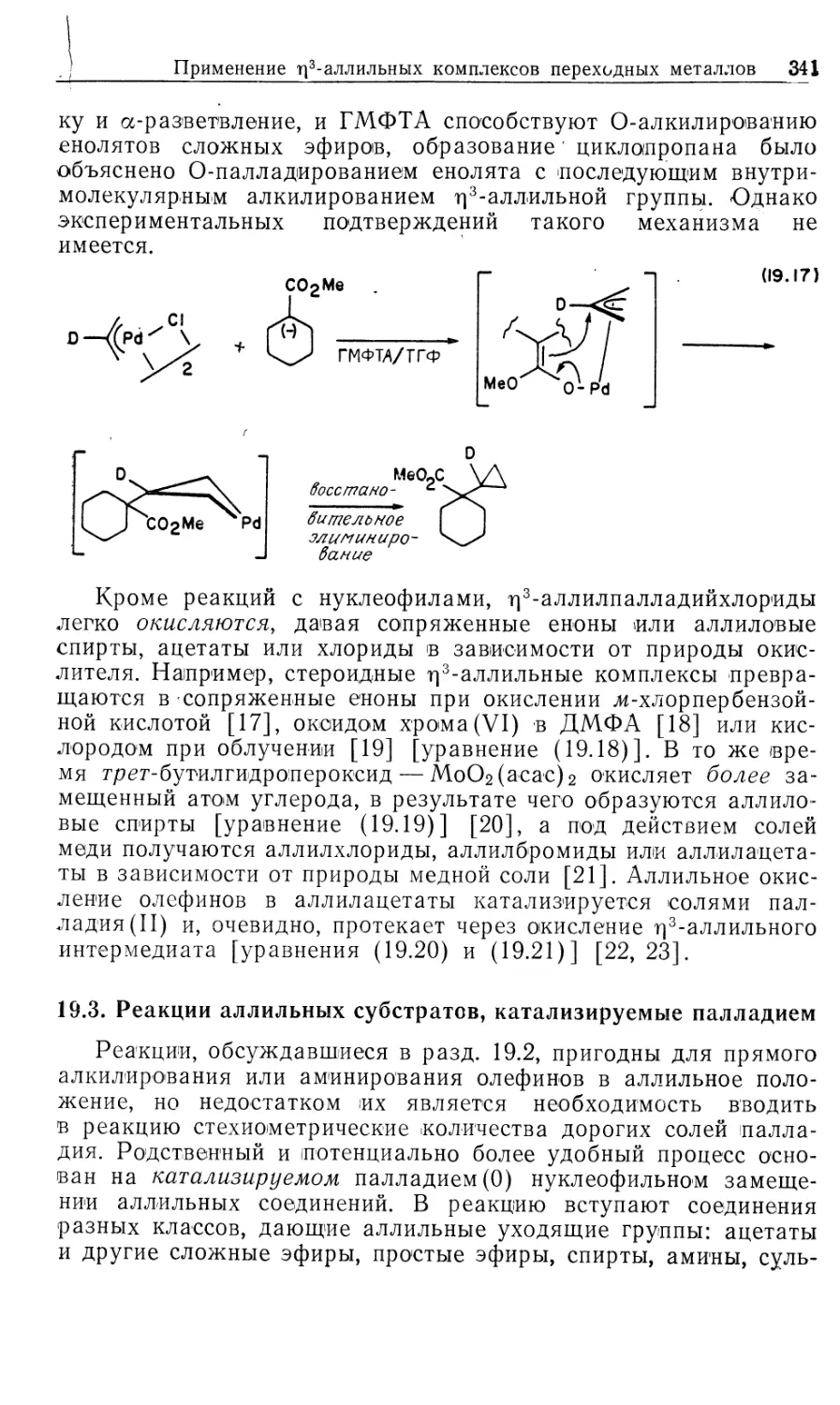
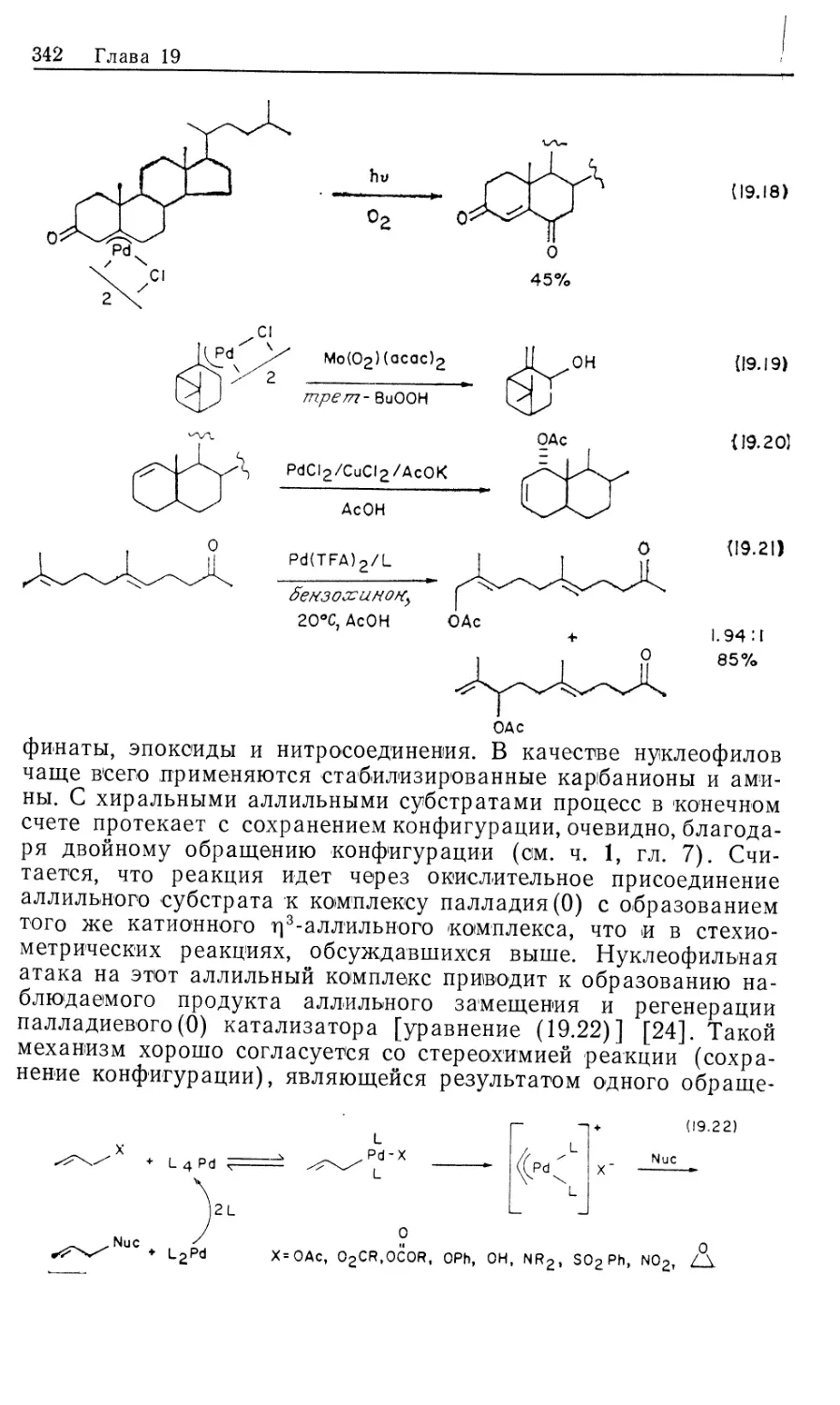
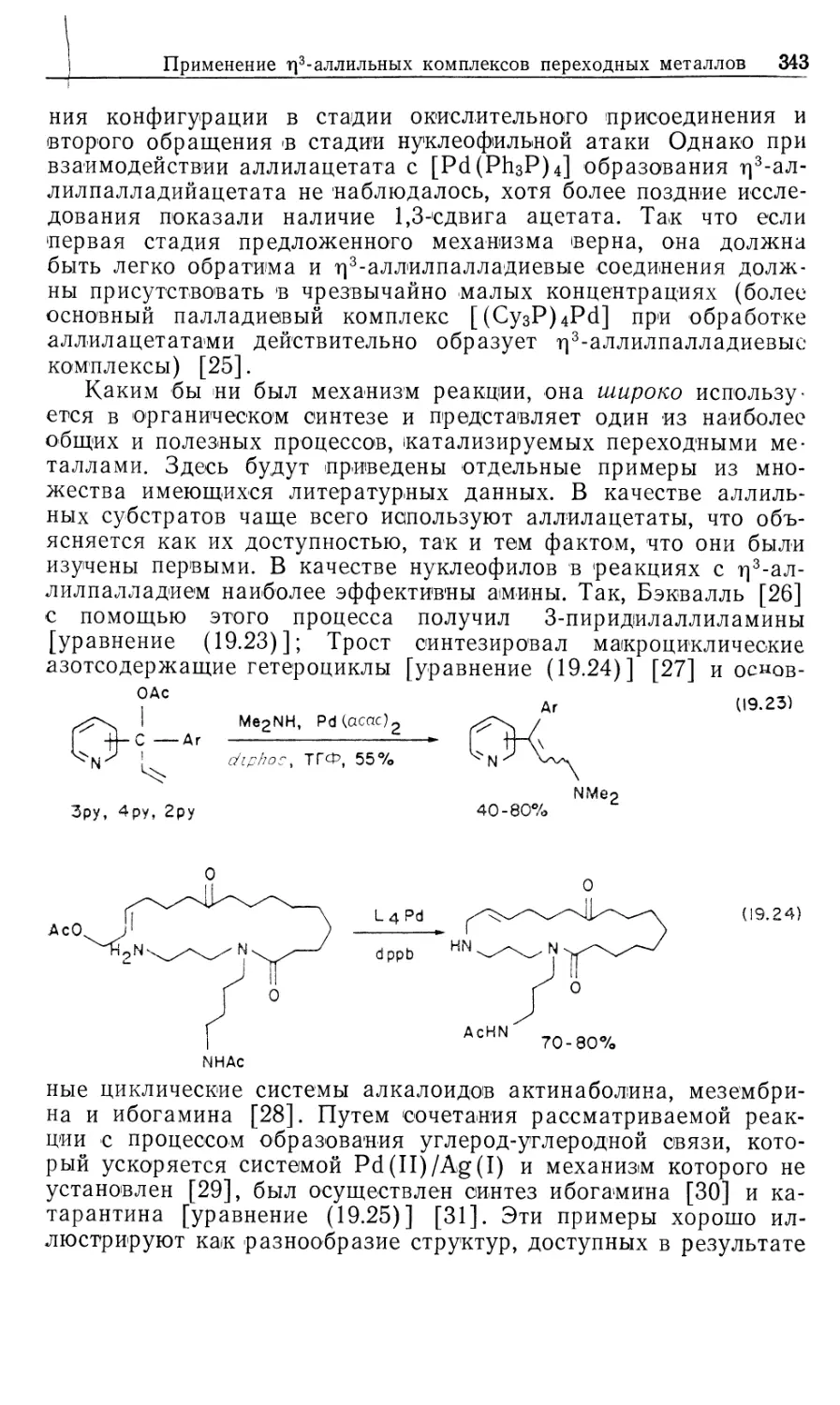
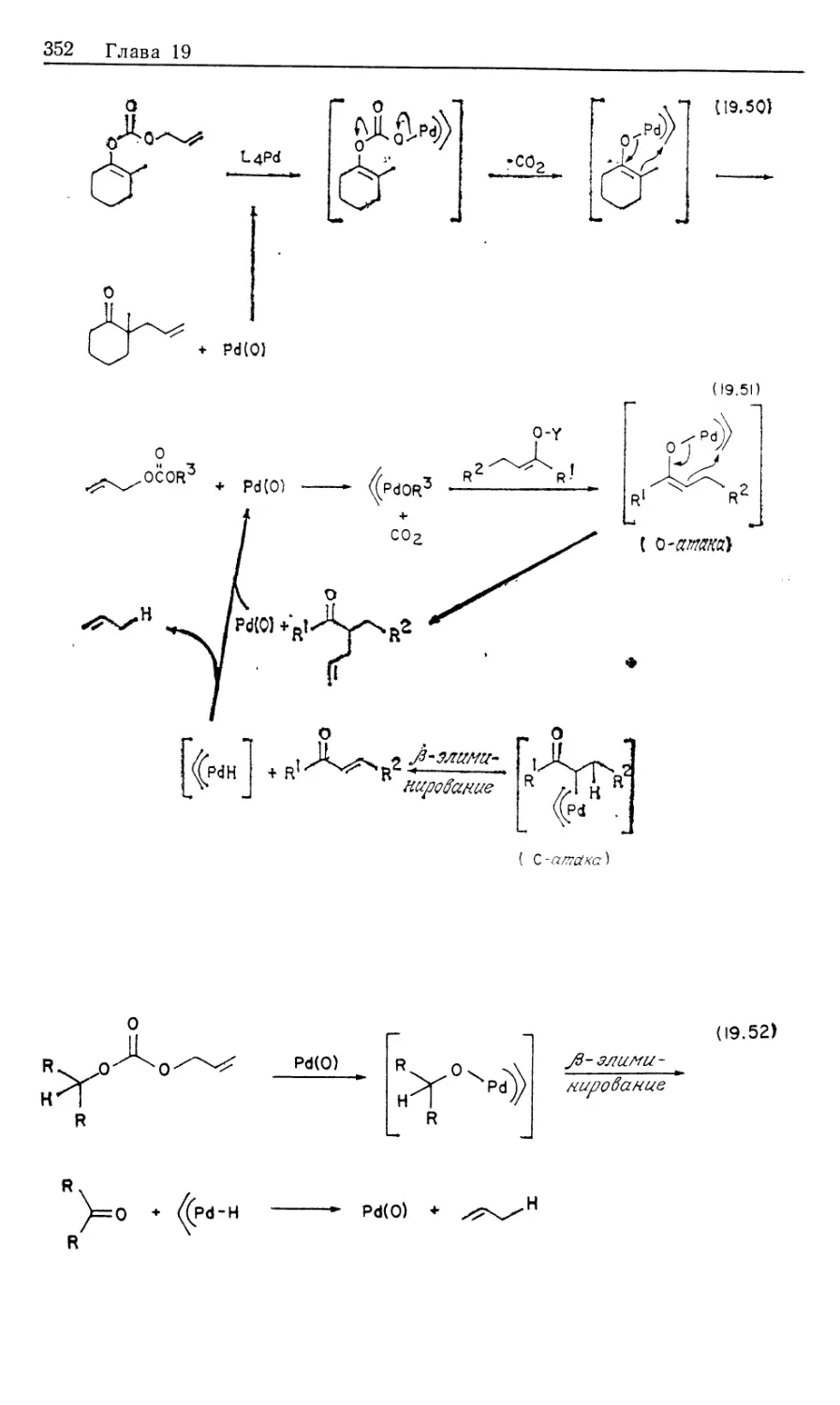
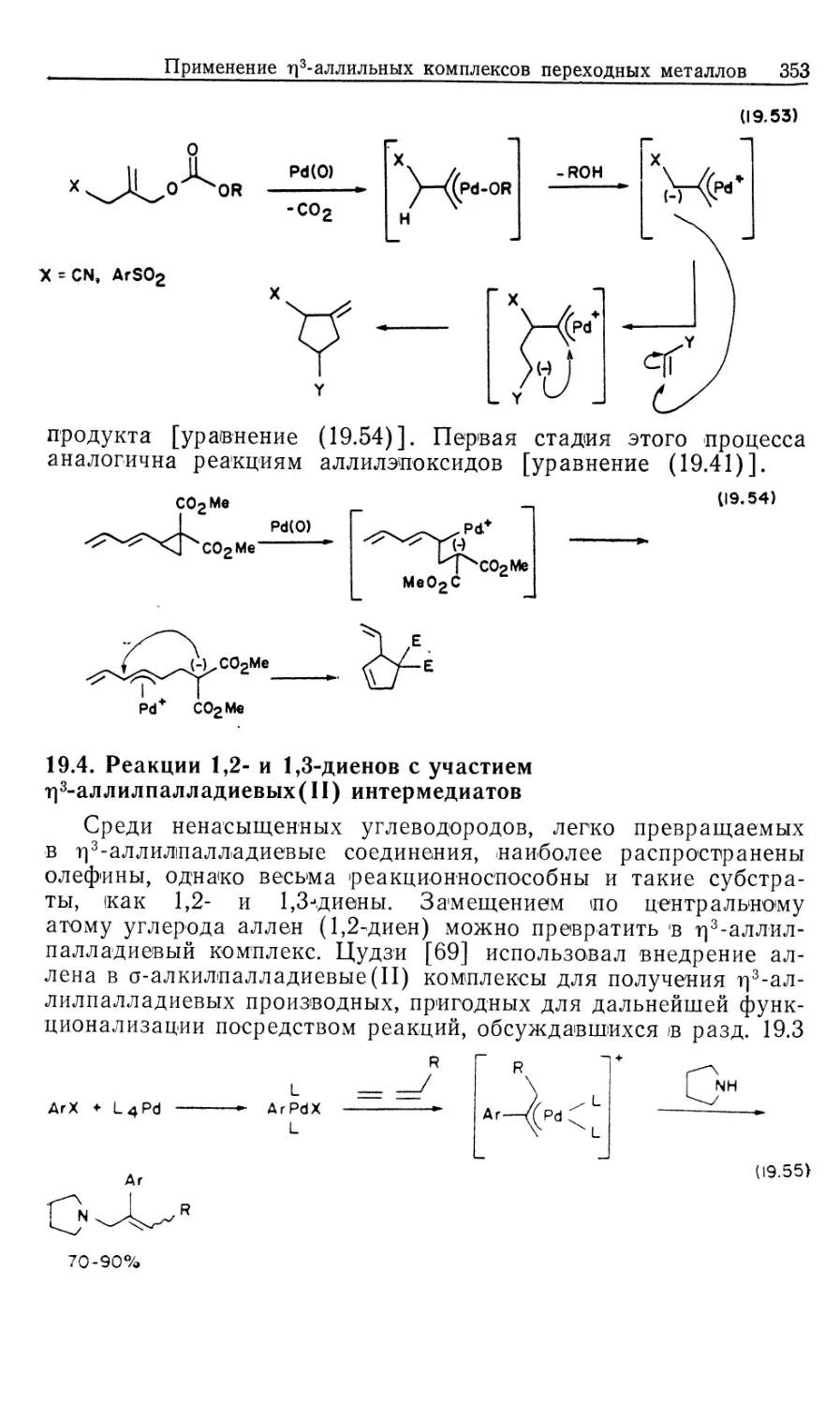
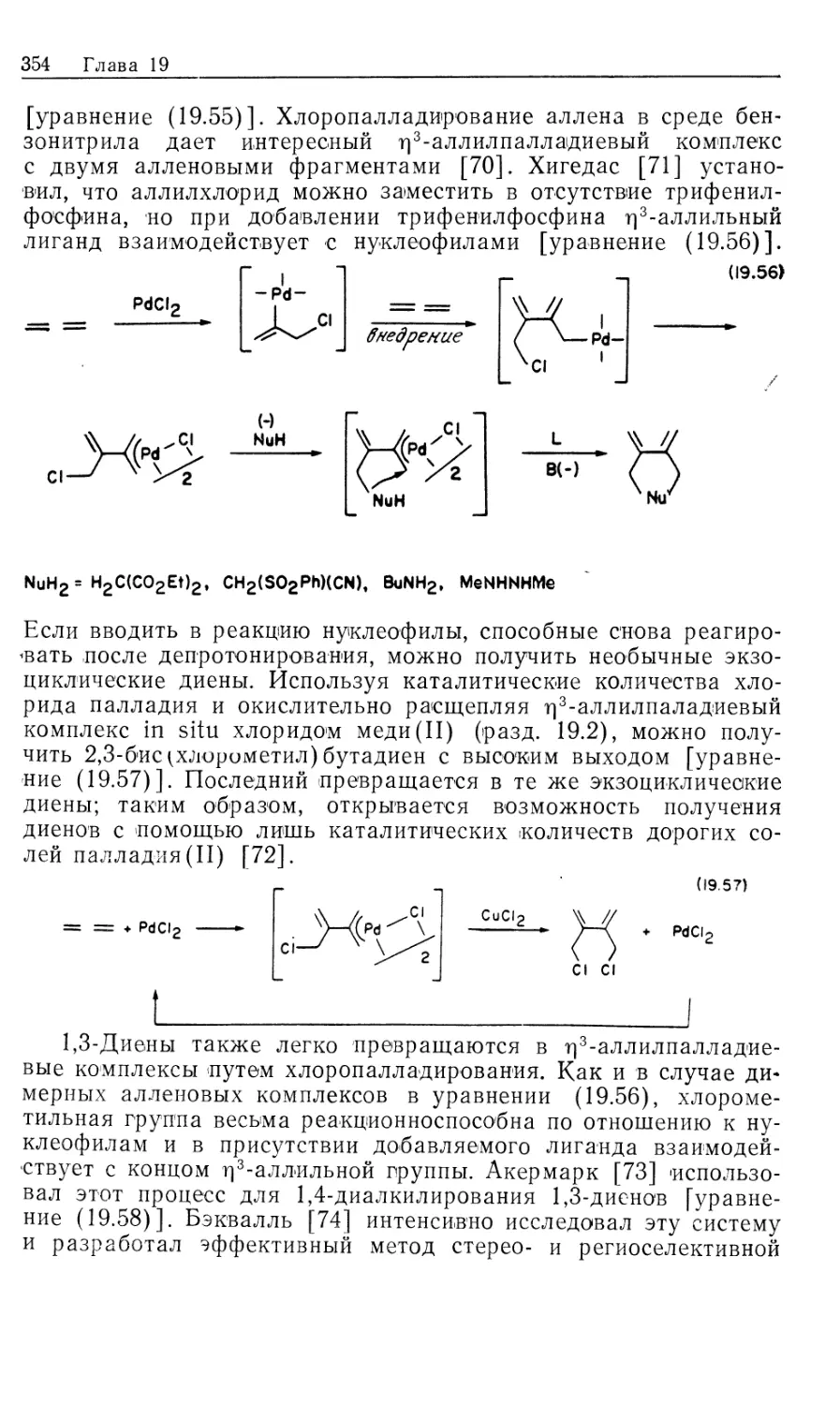
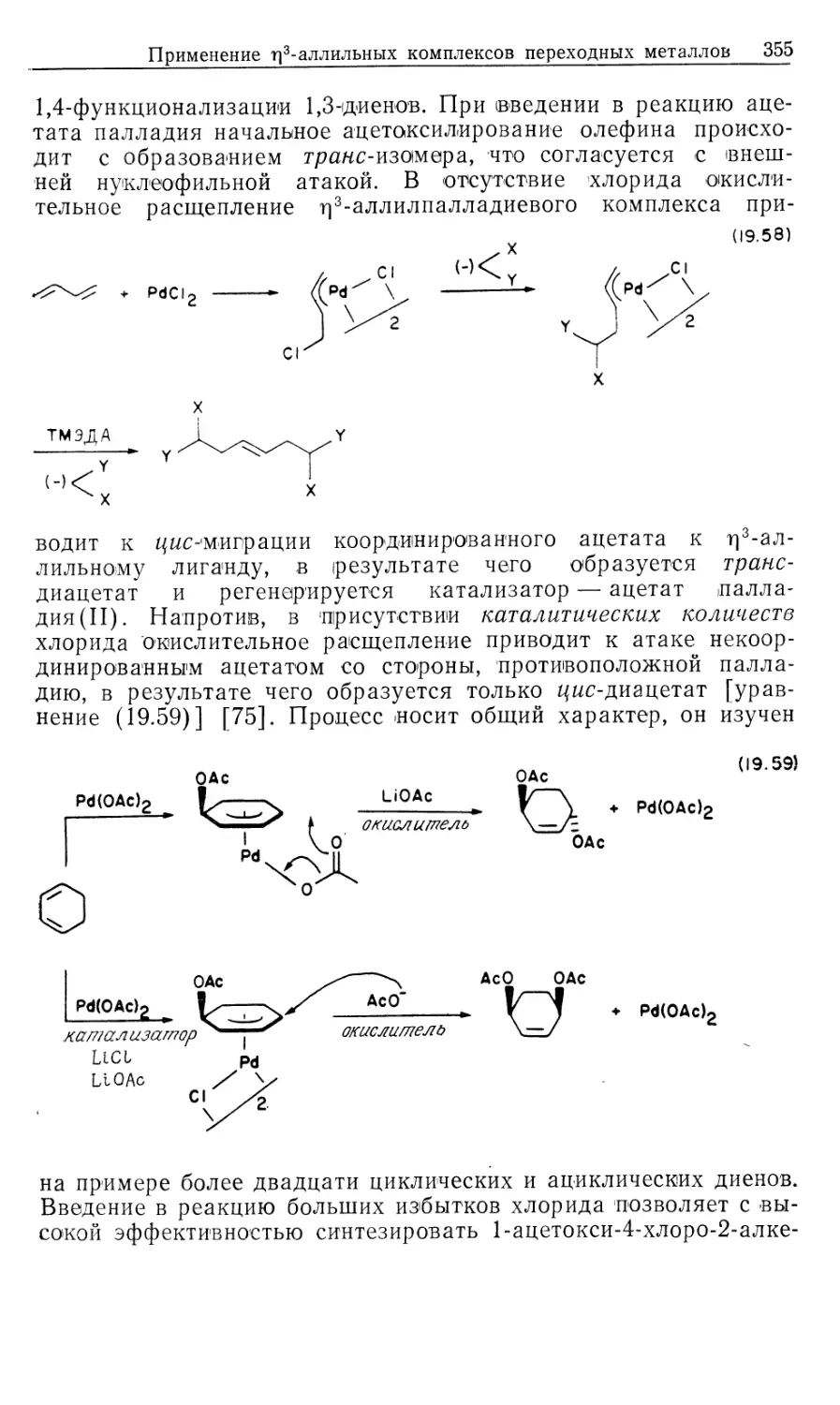
/




























































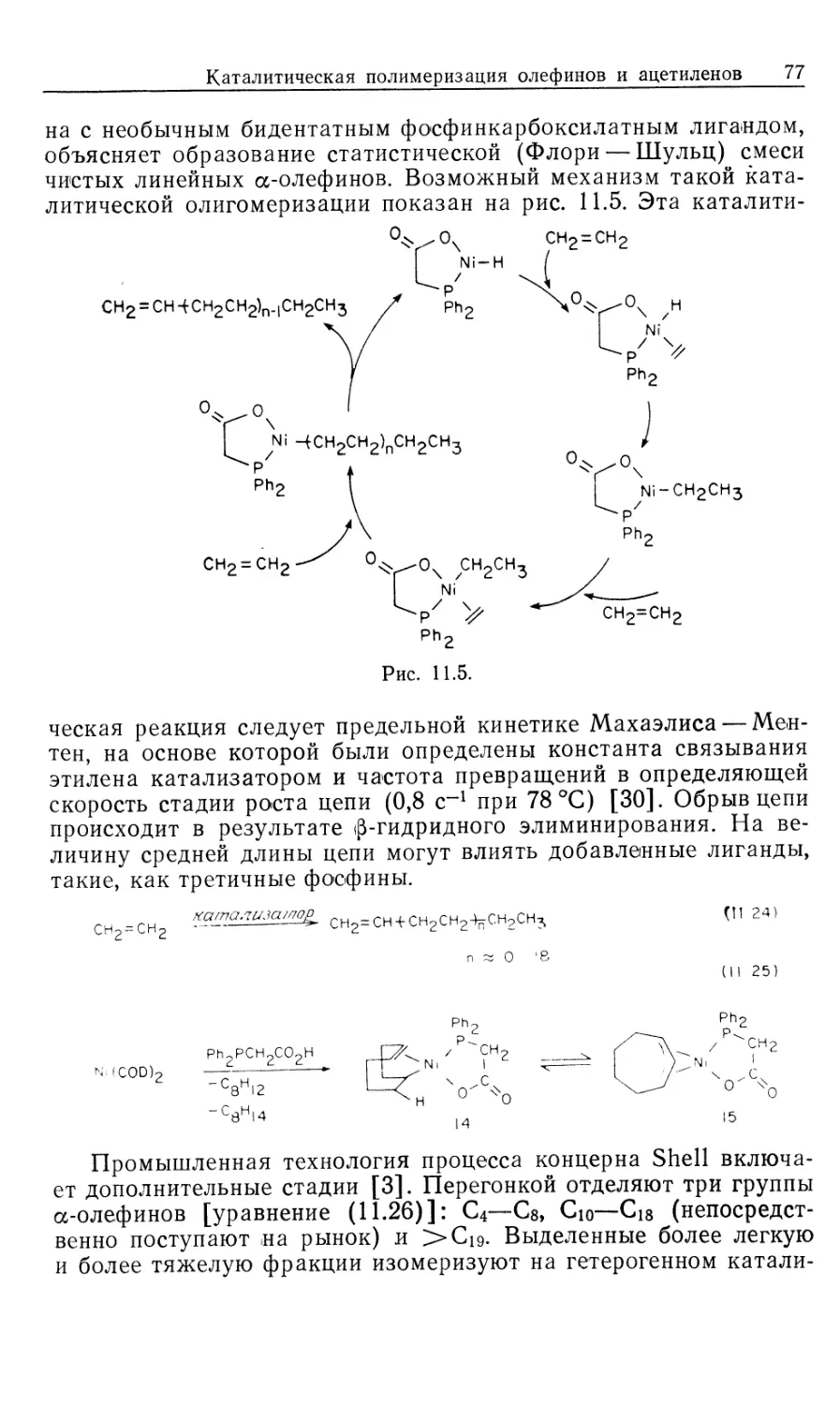










































































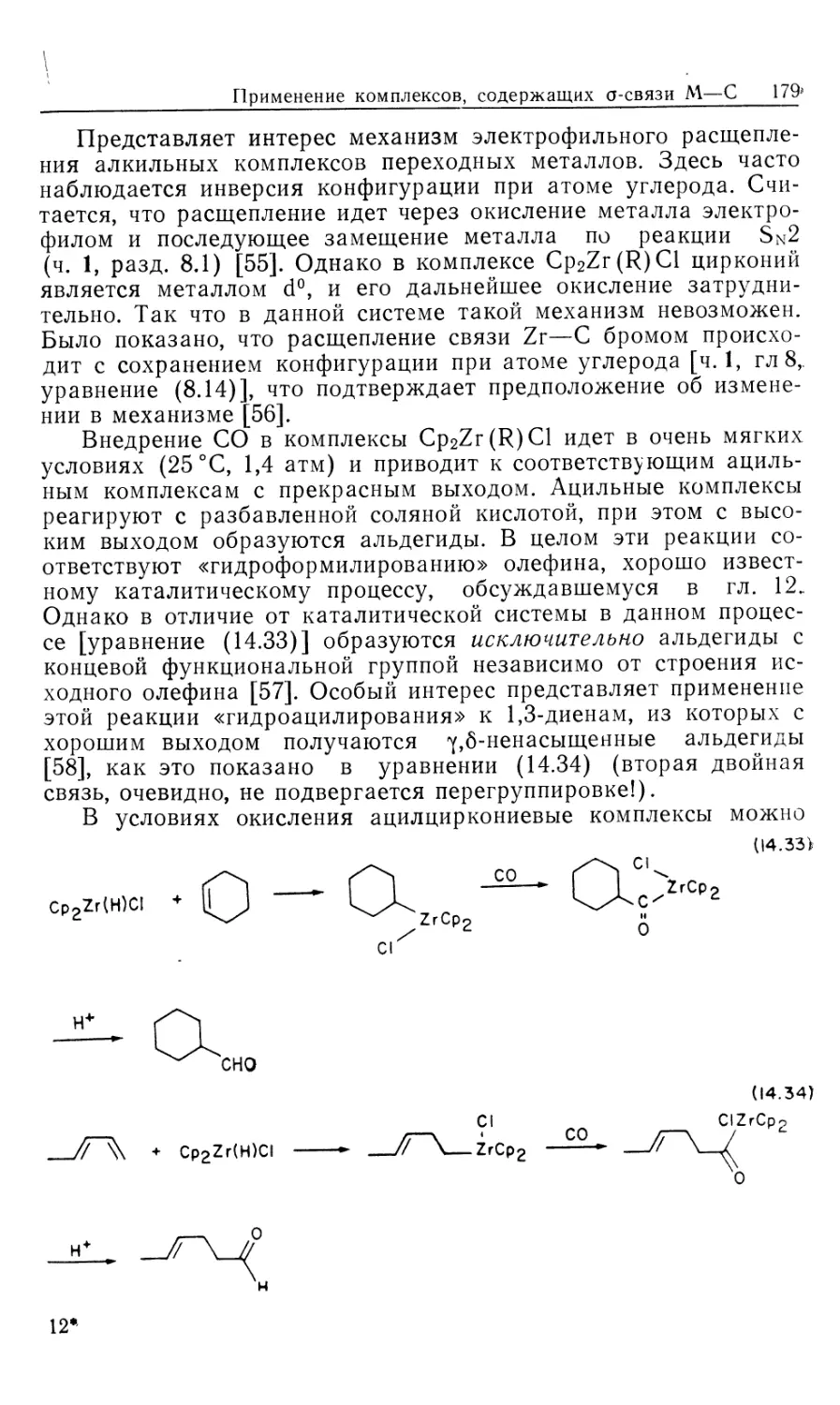
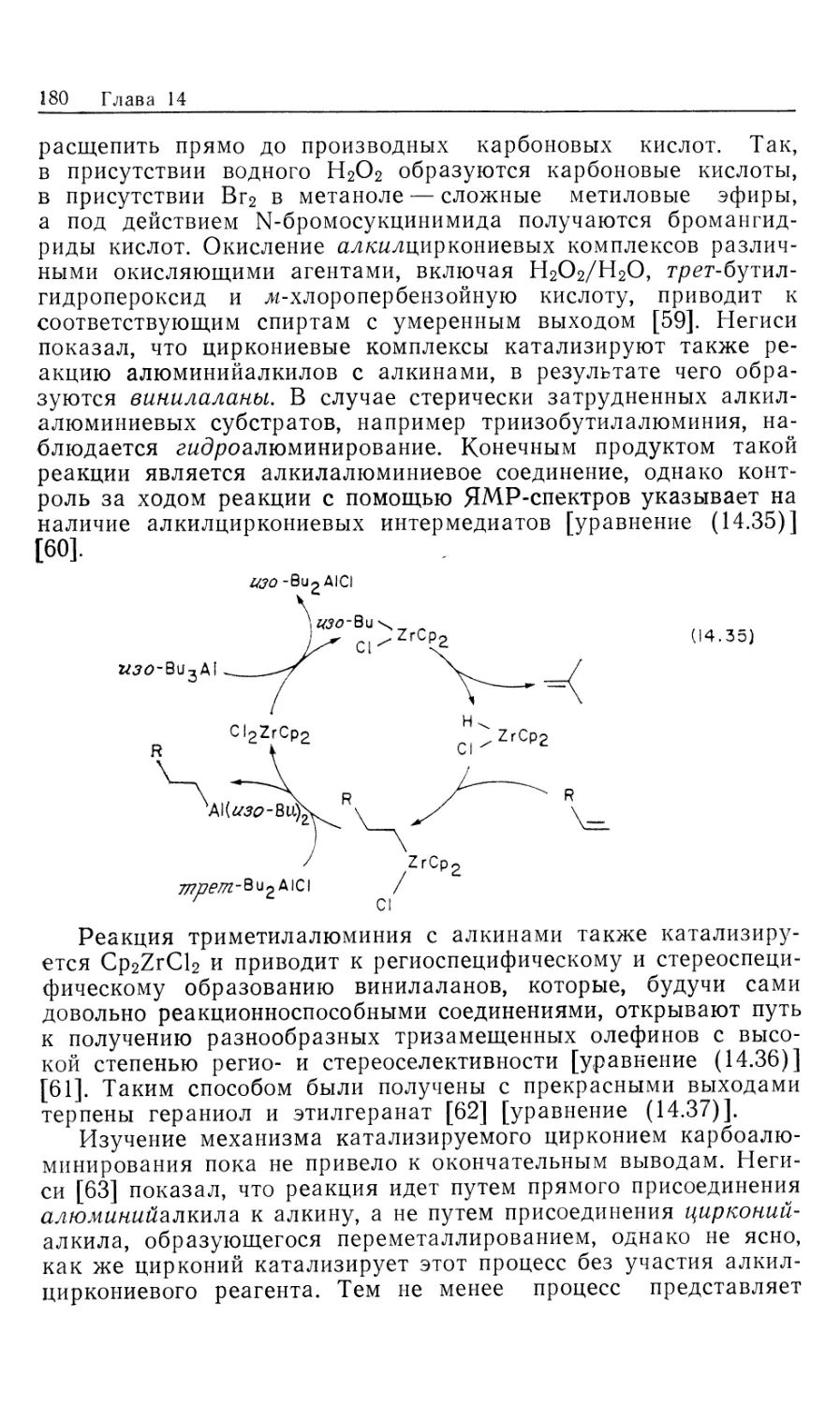
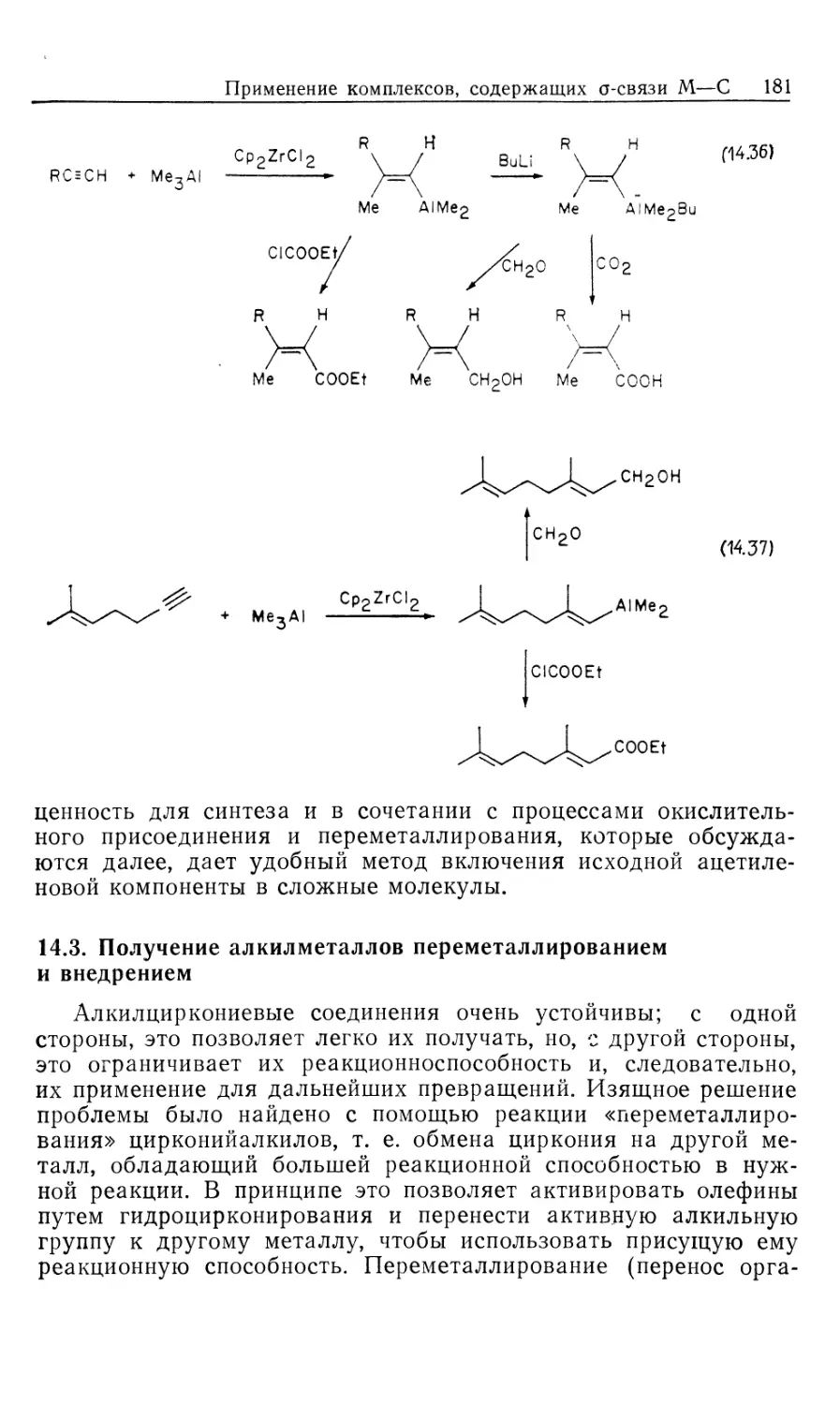

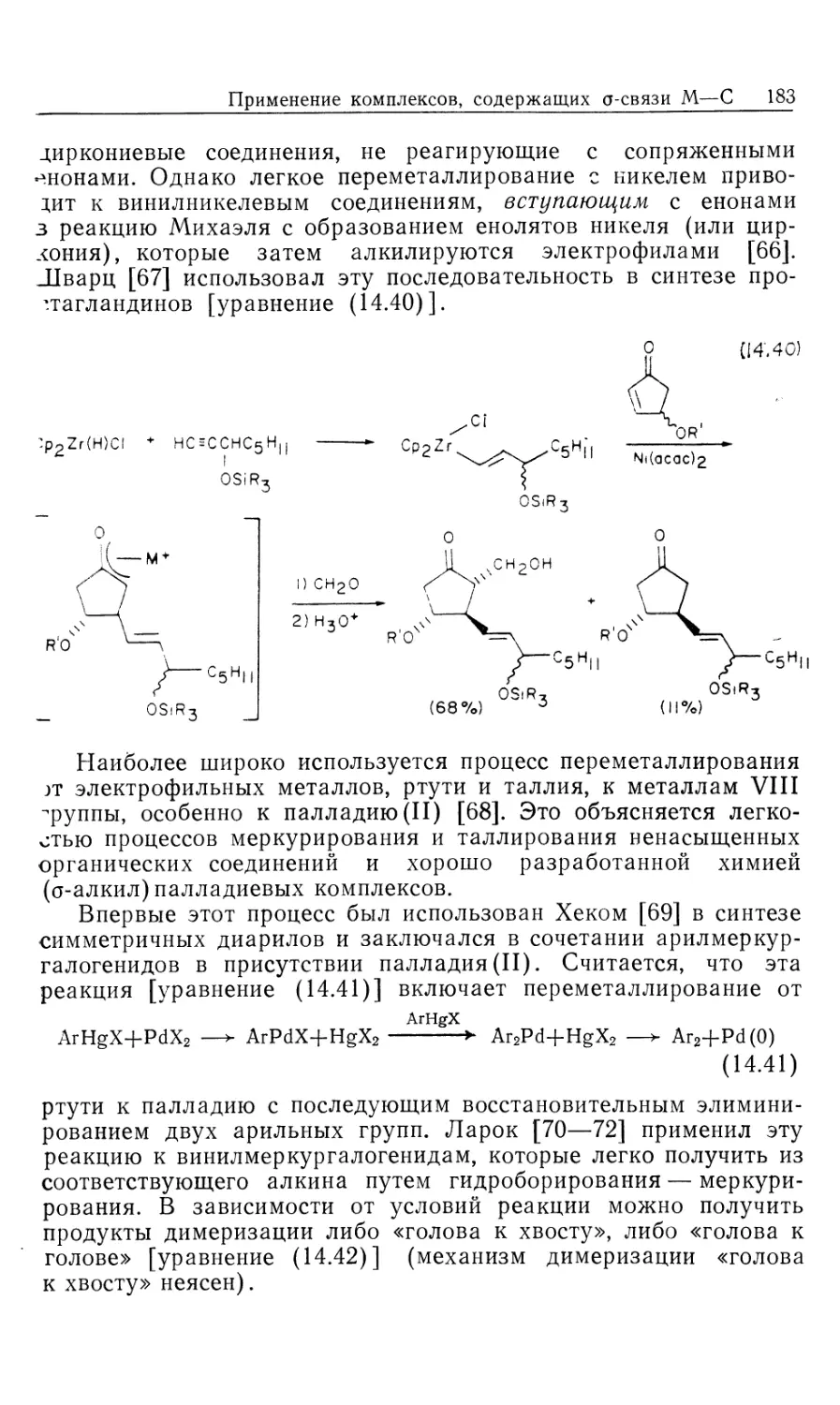
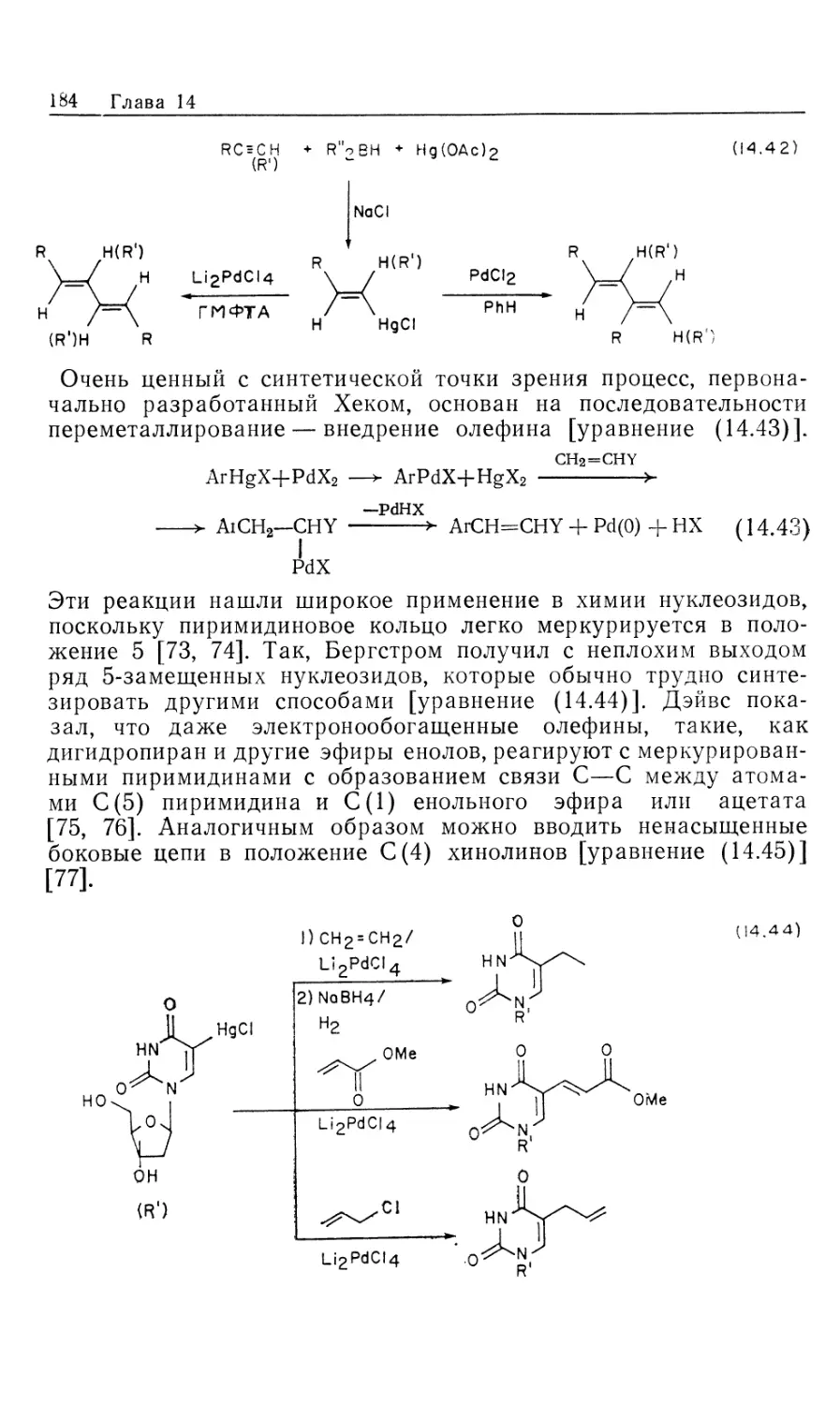
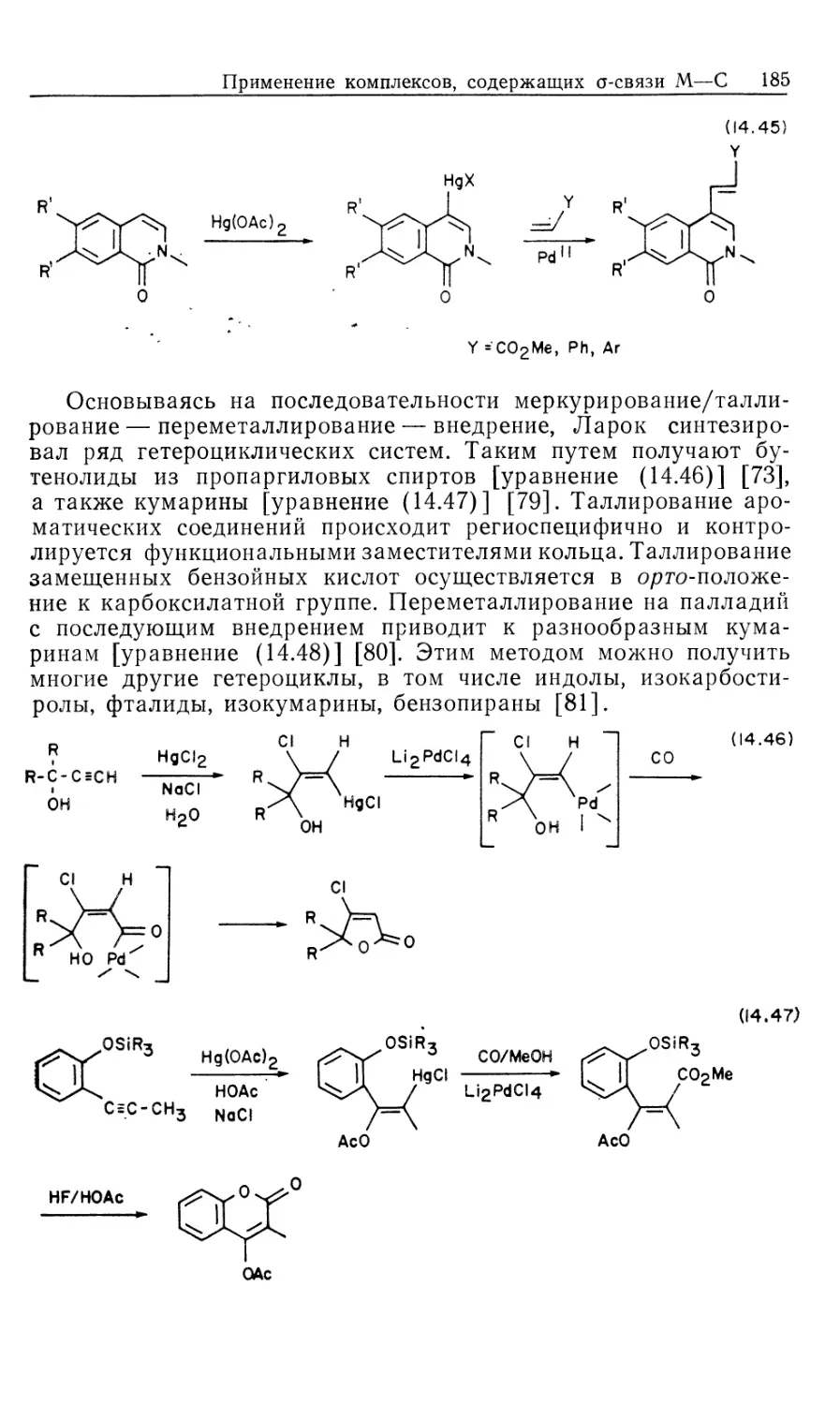
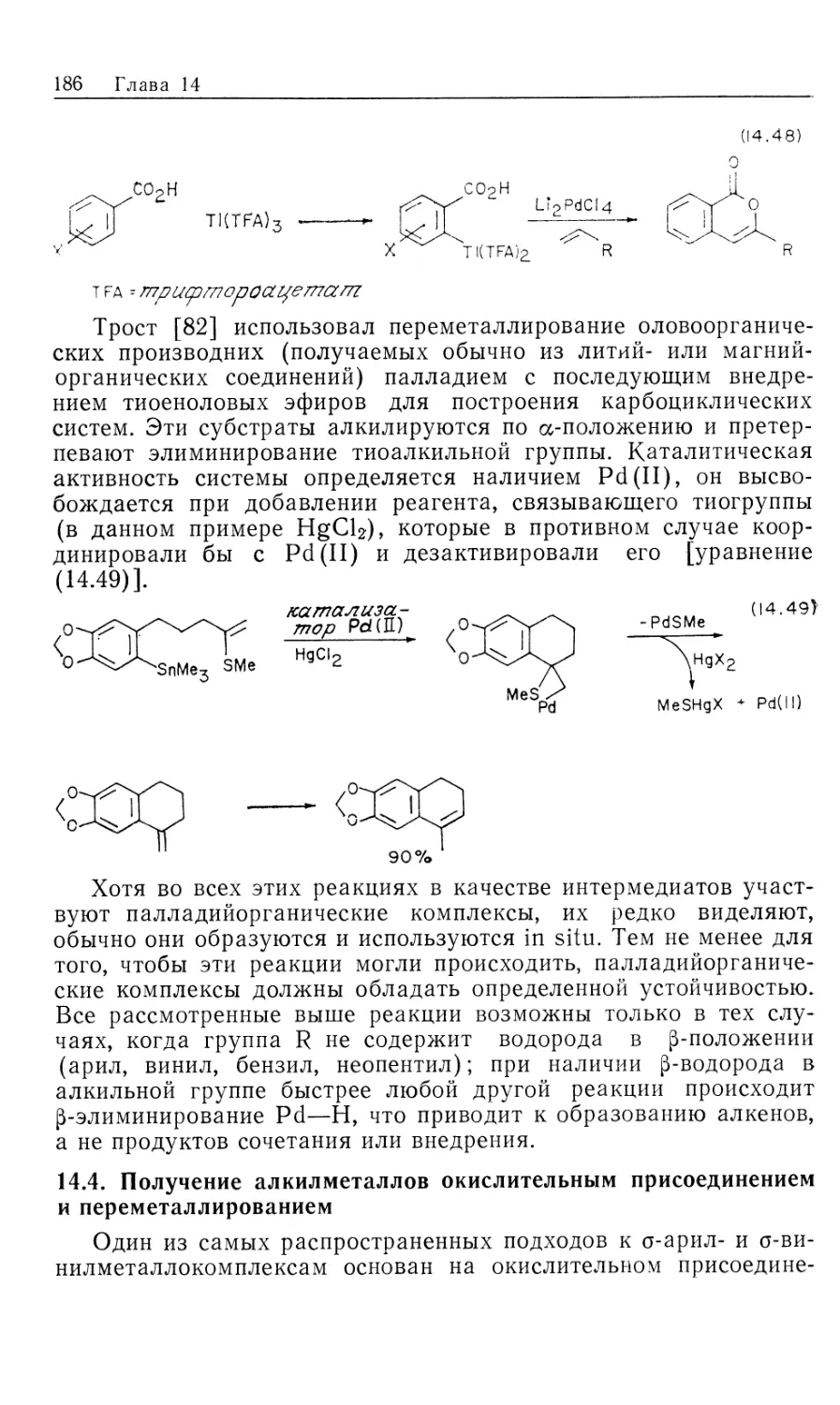



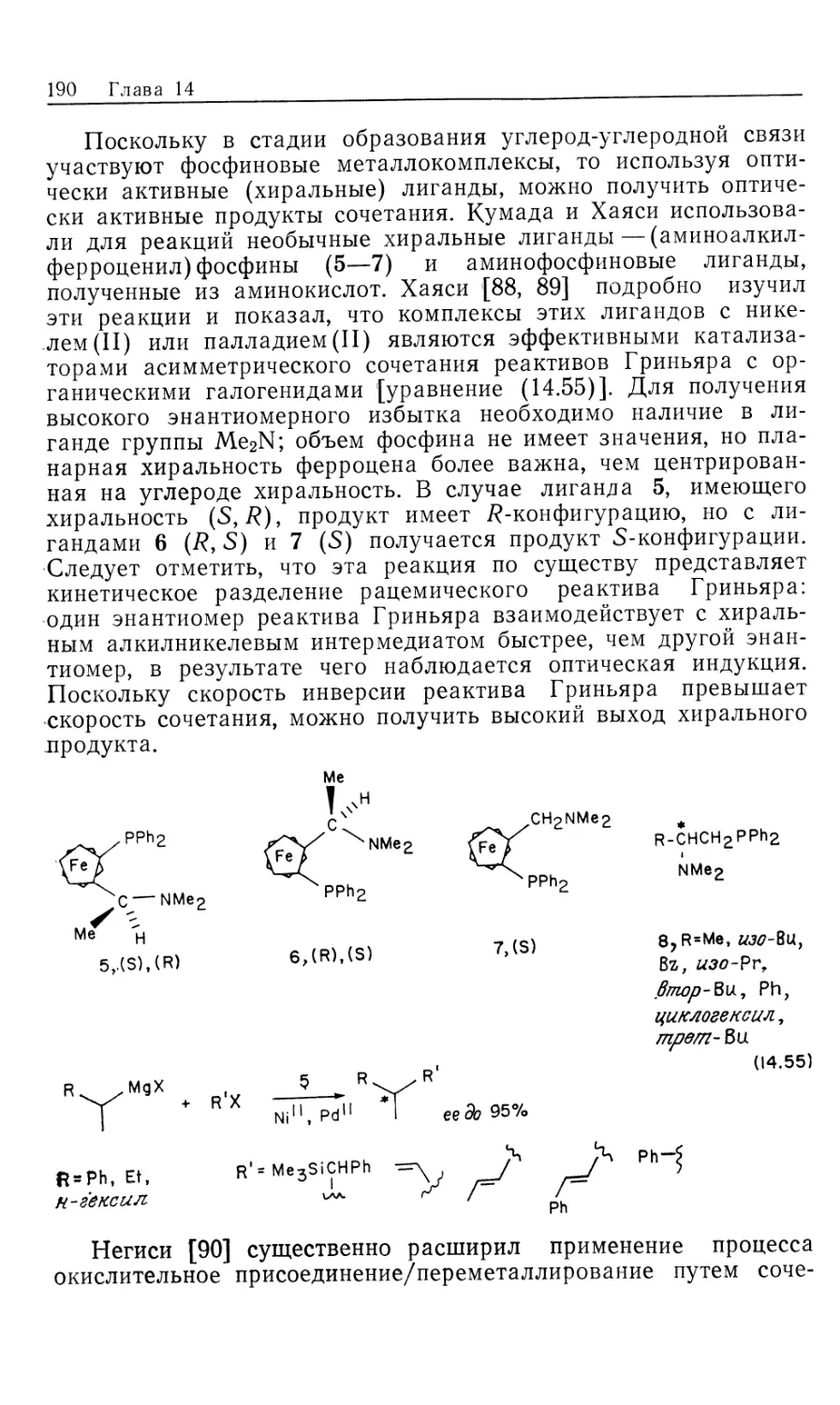










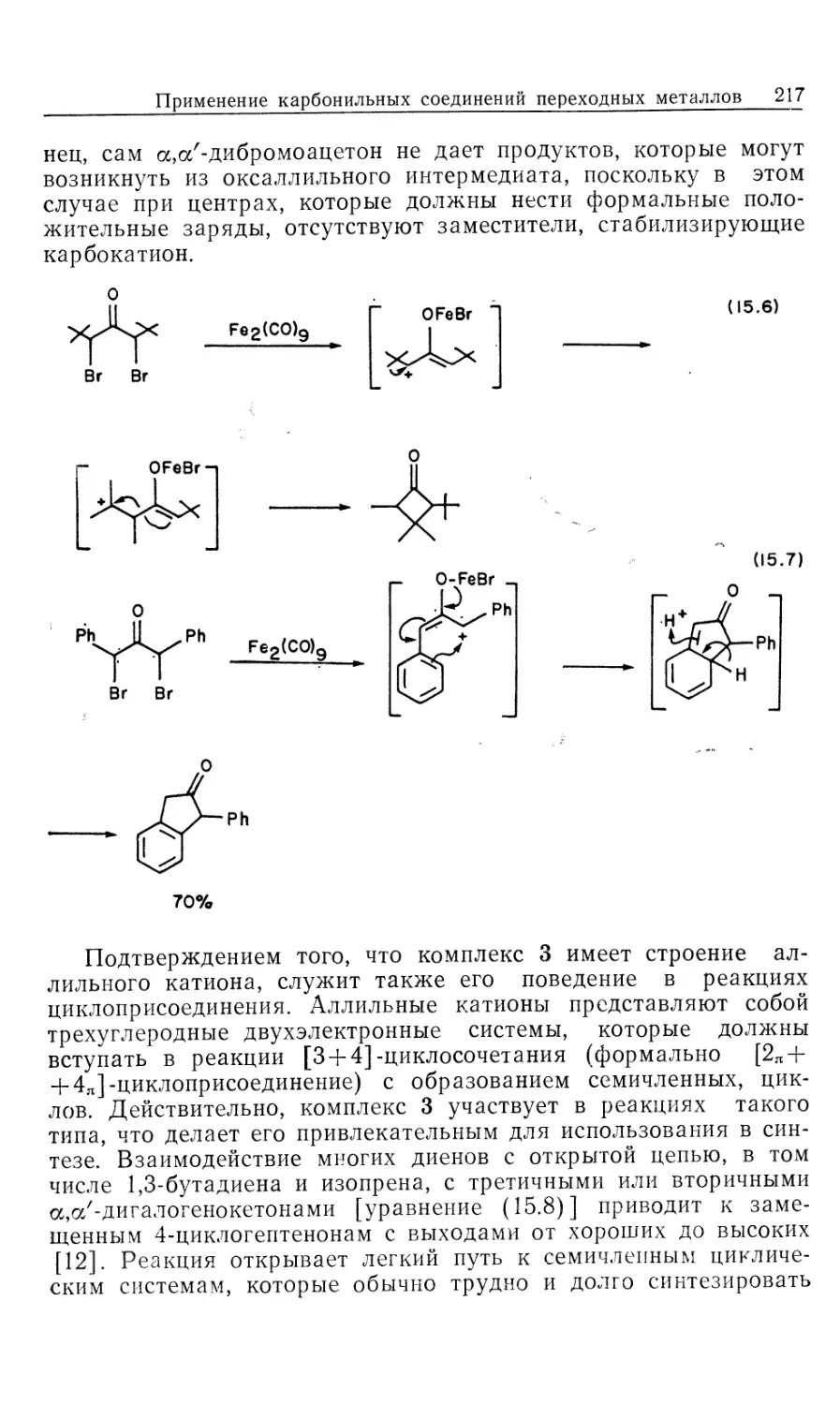
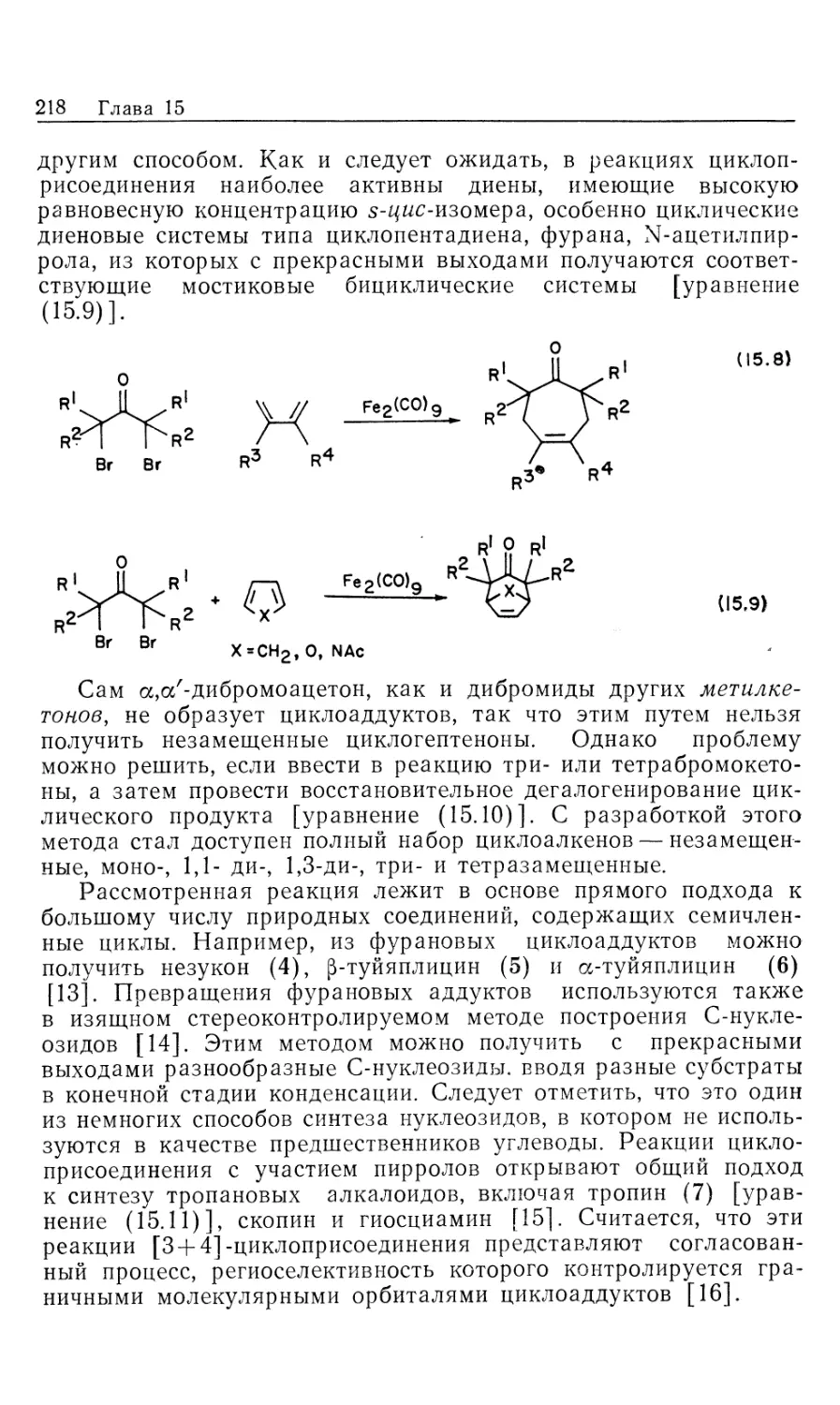
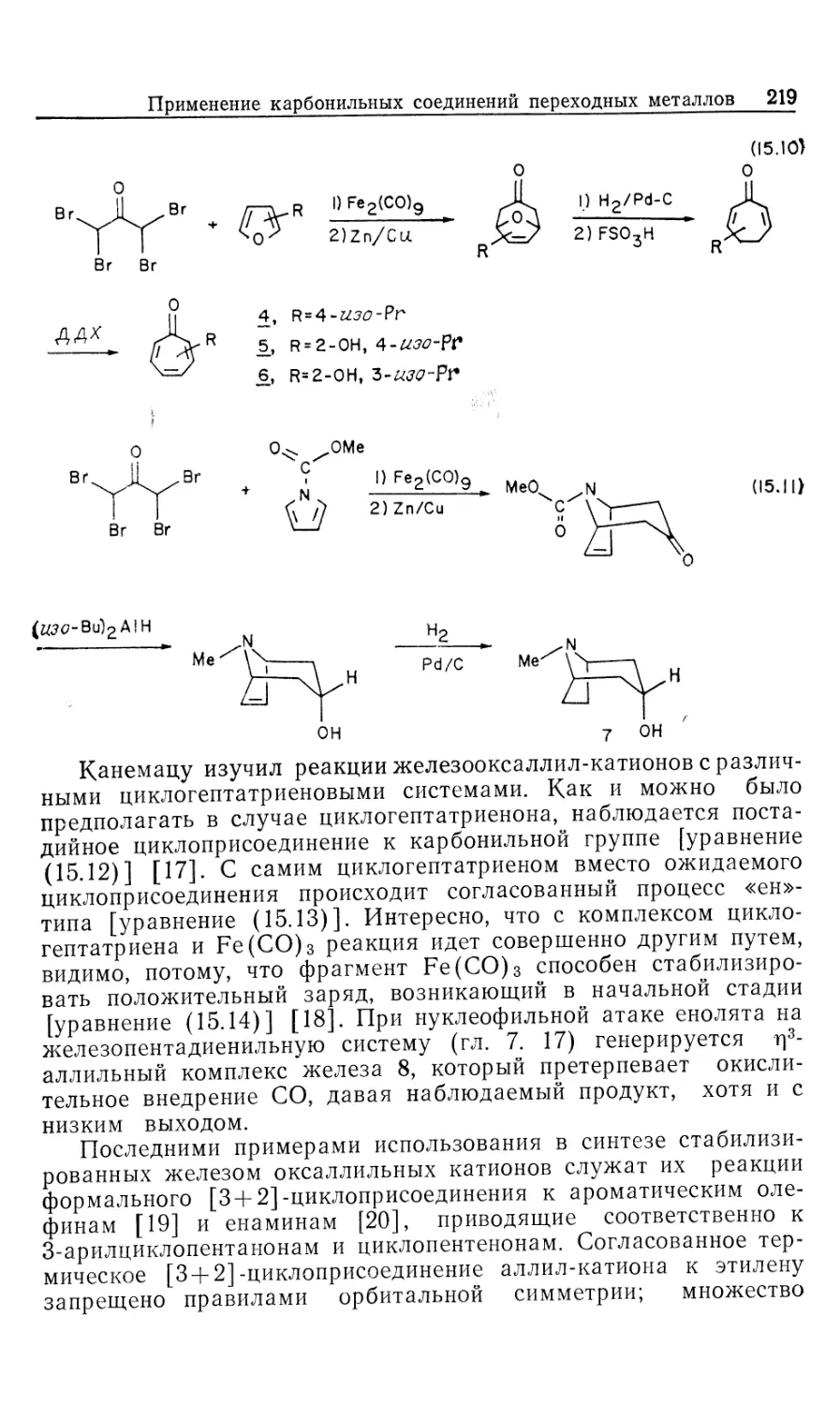
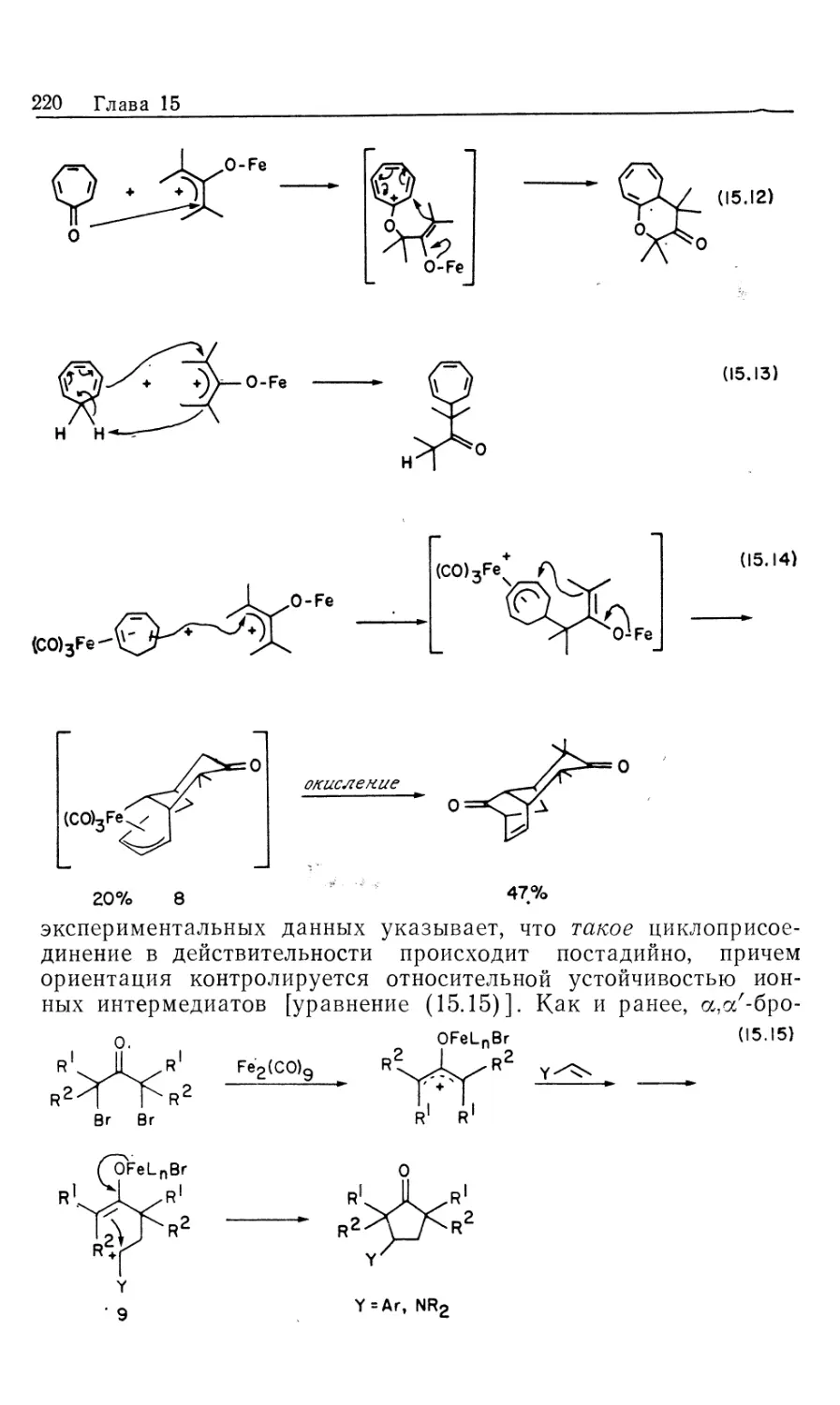
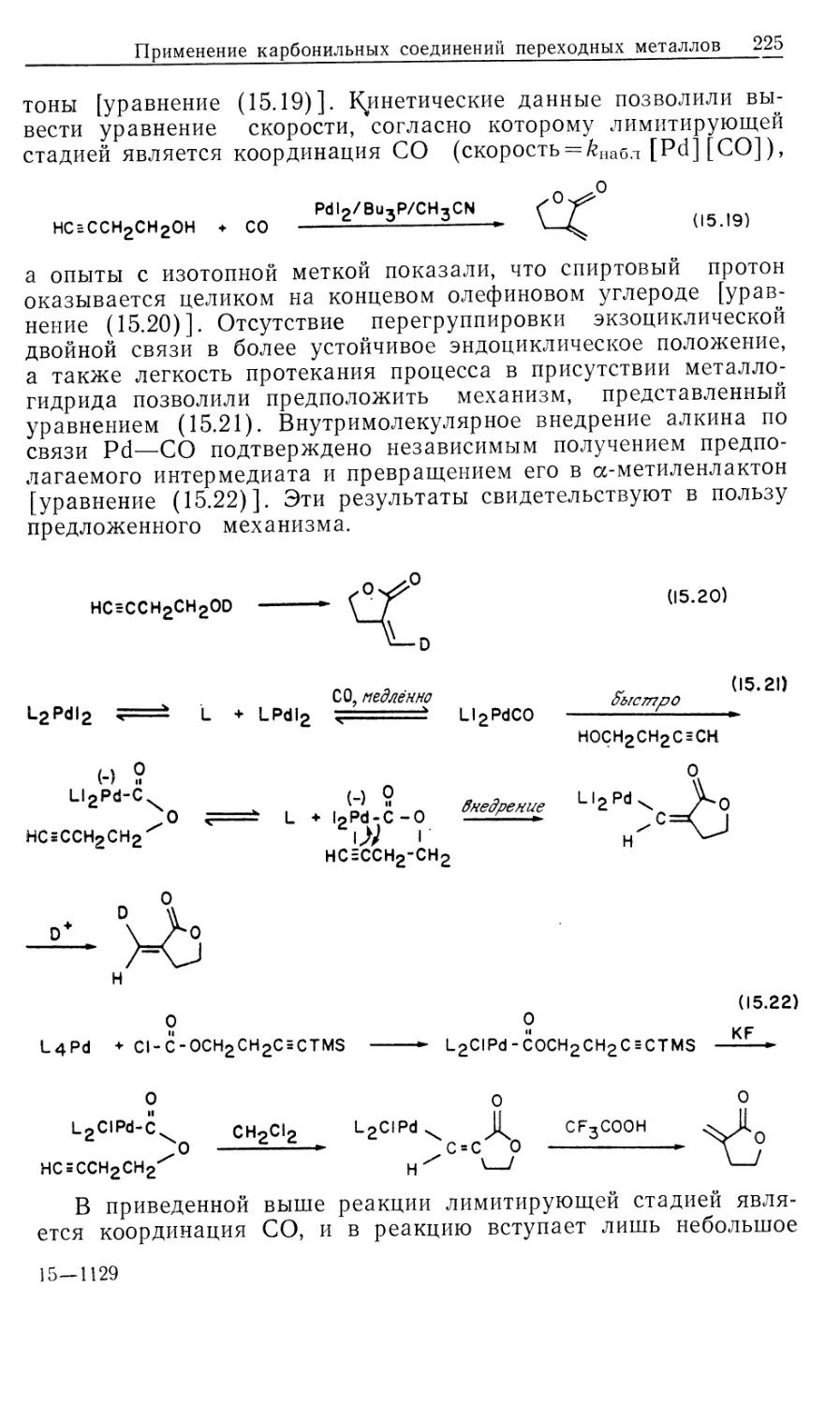


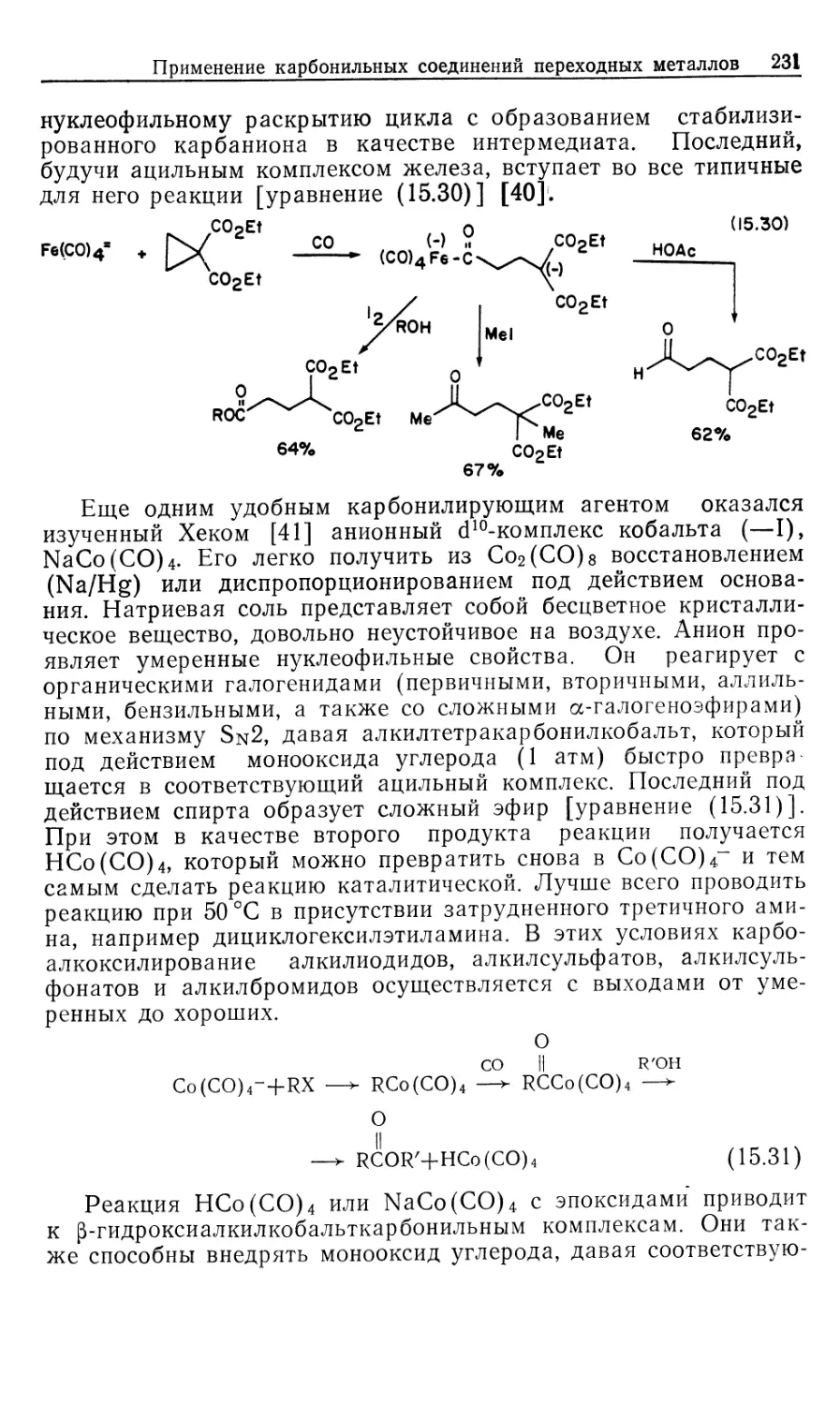













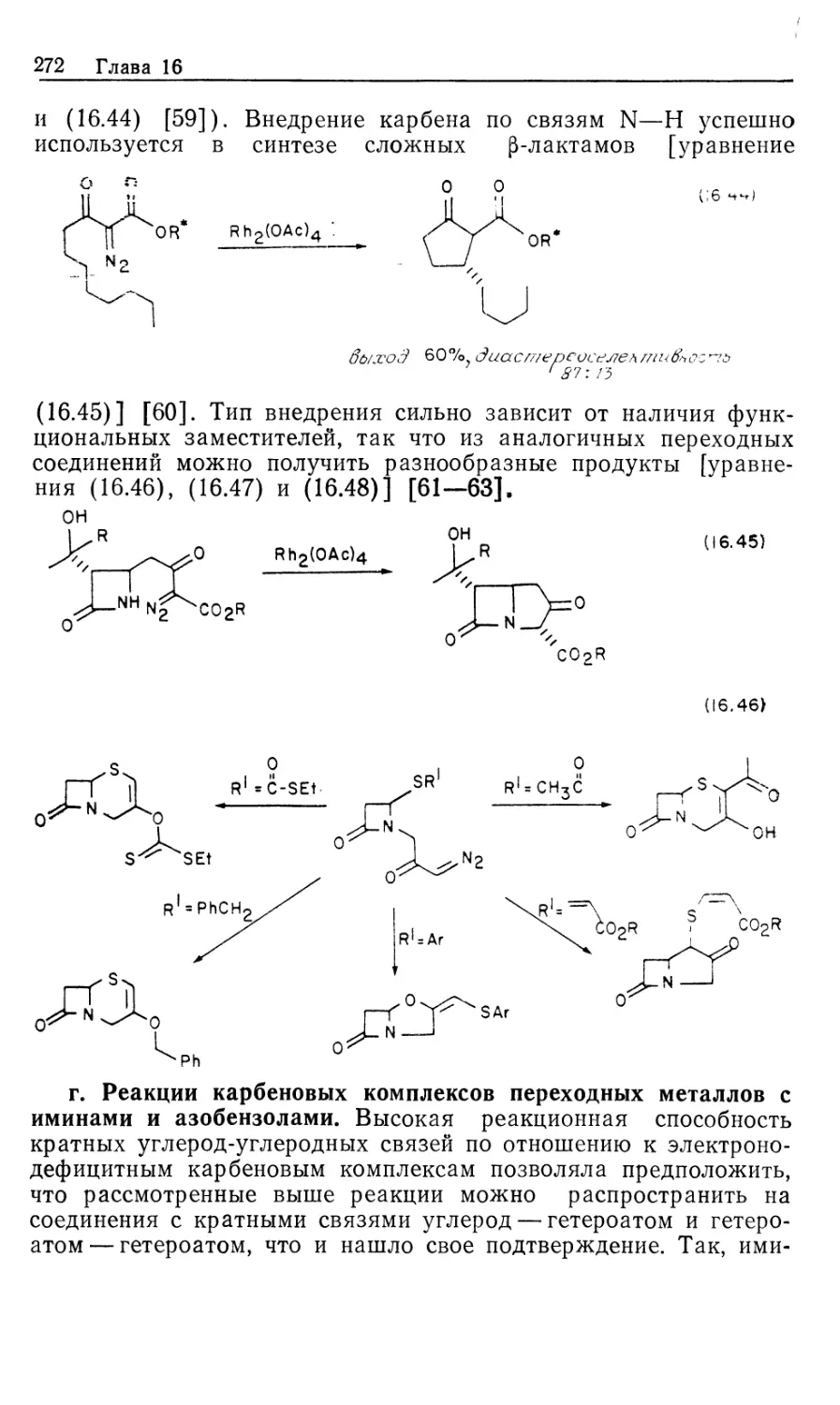
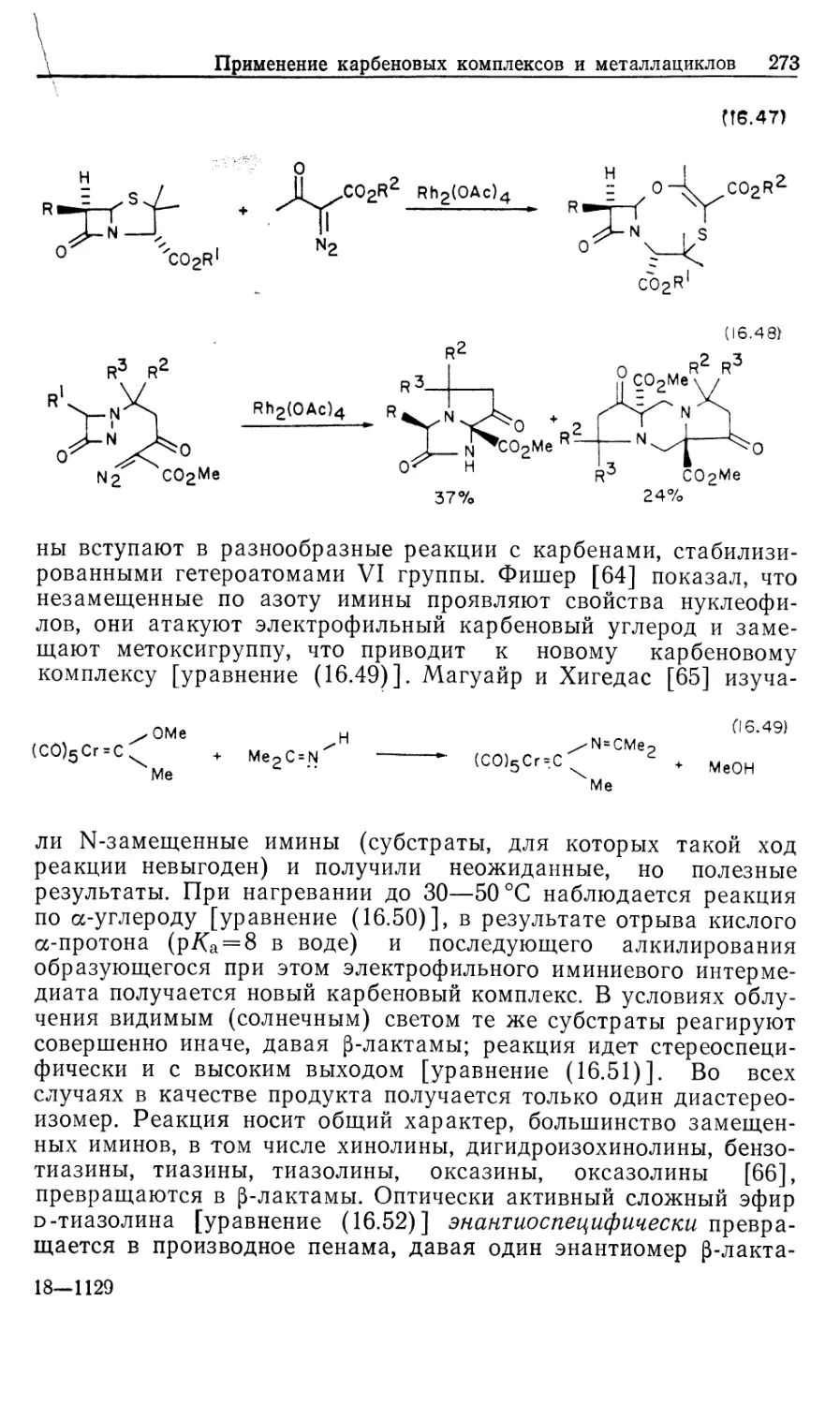
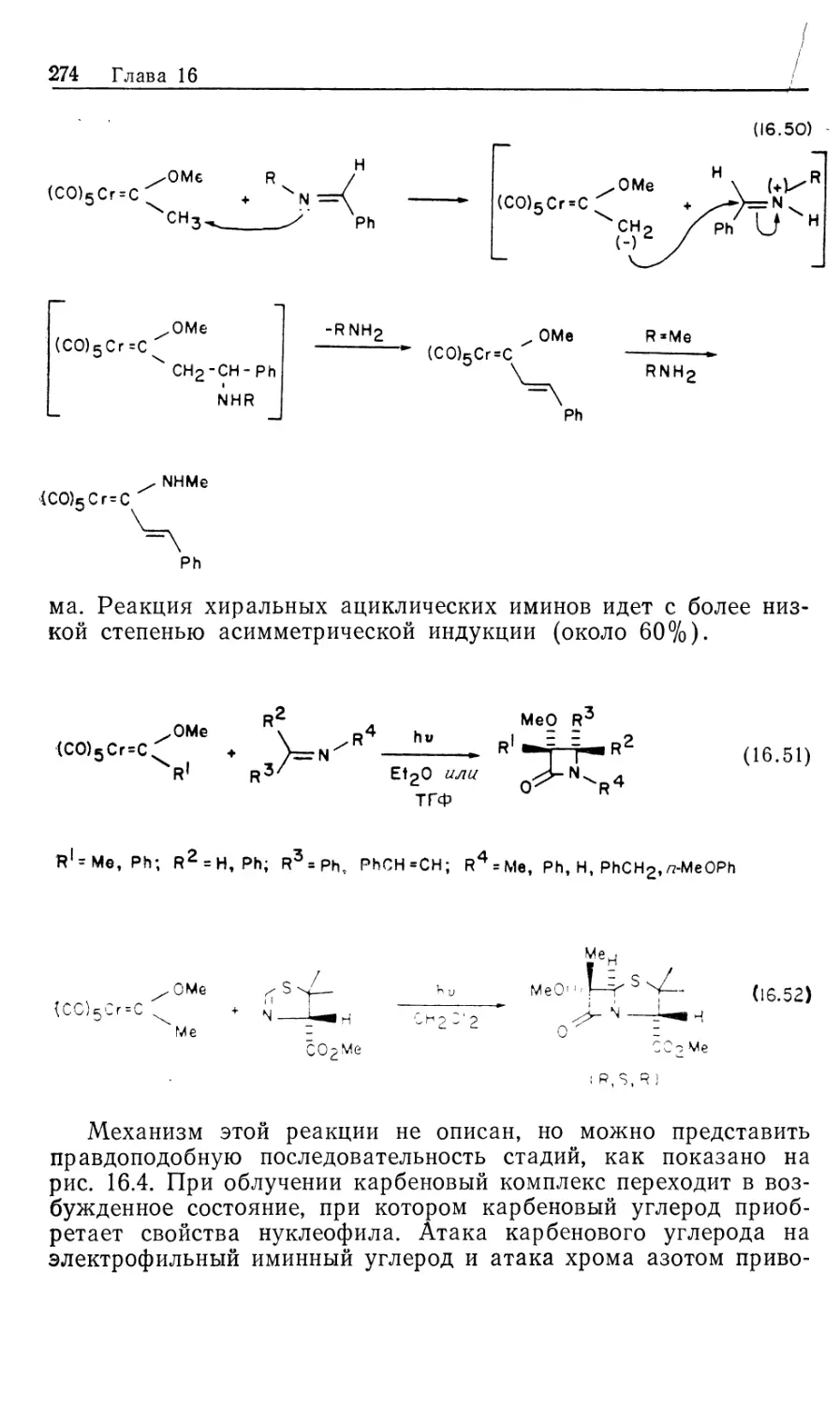
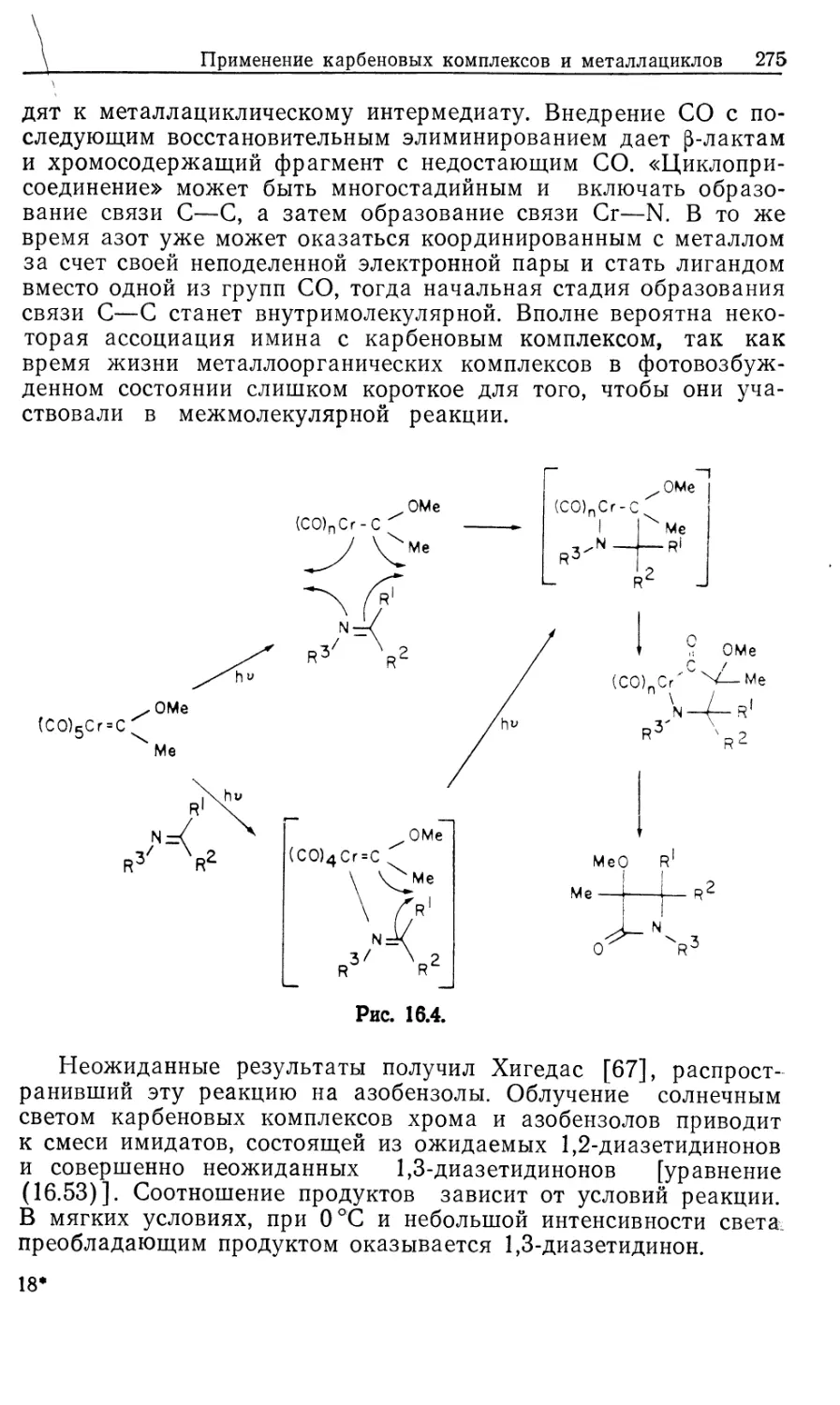












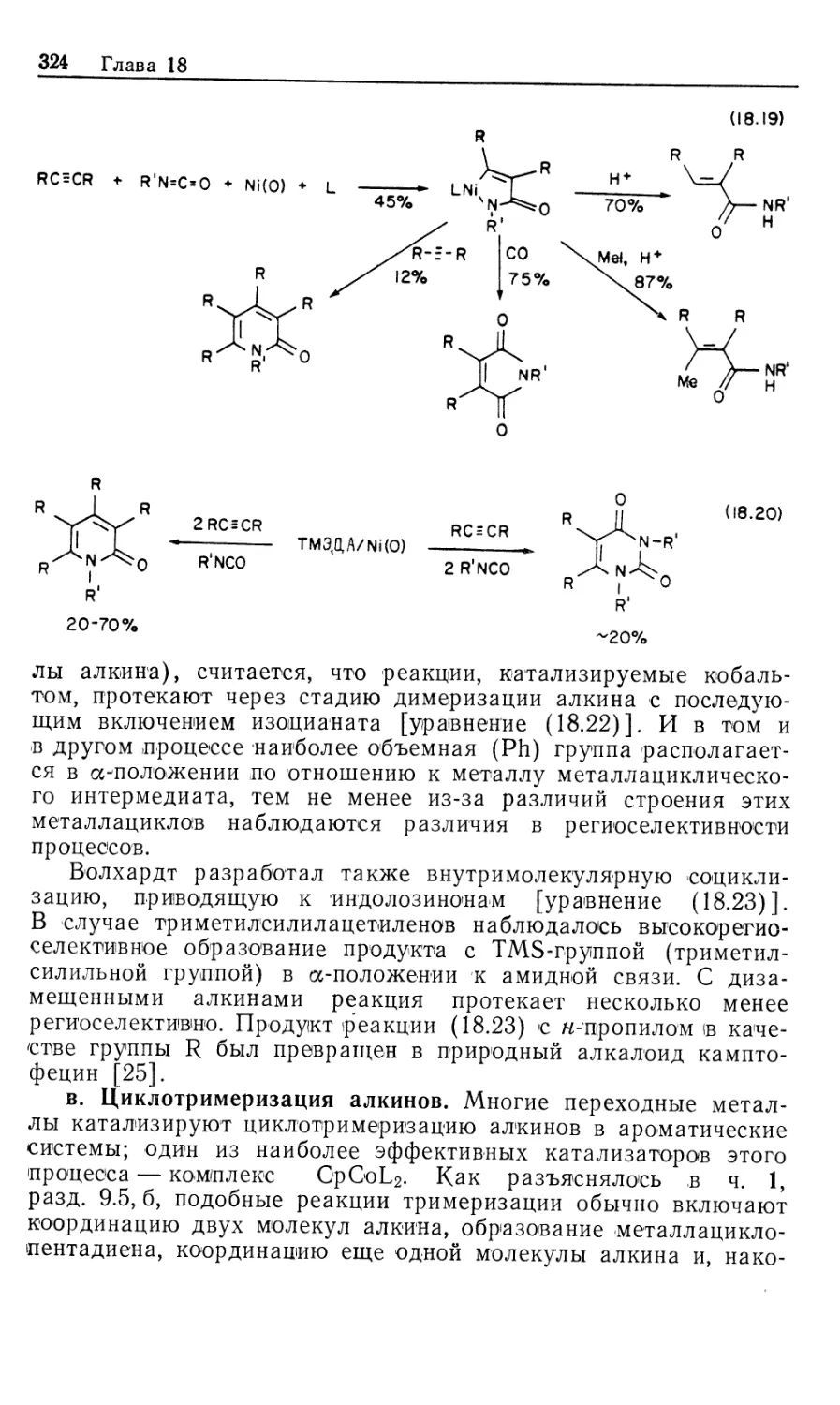
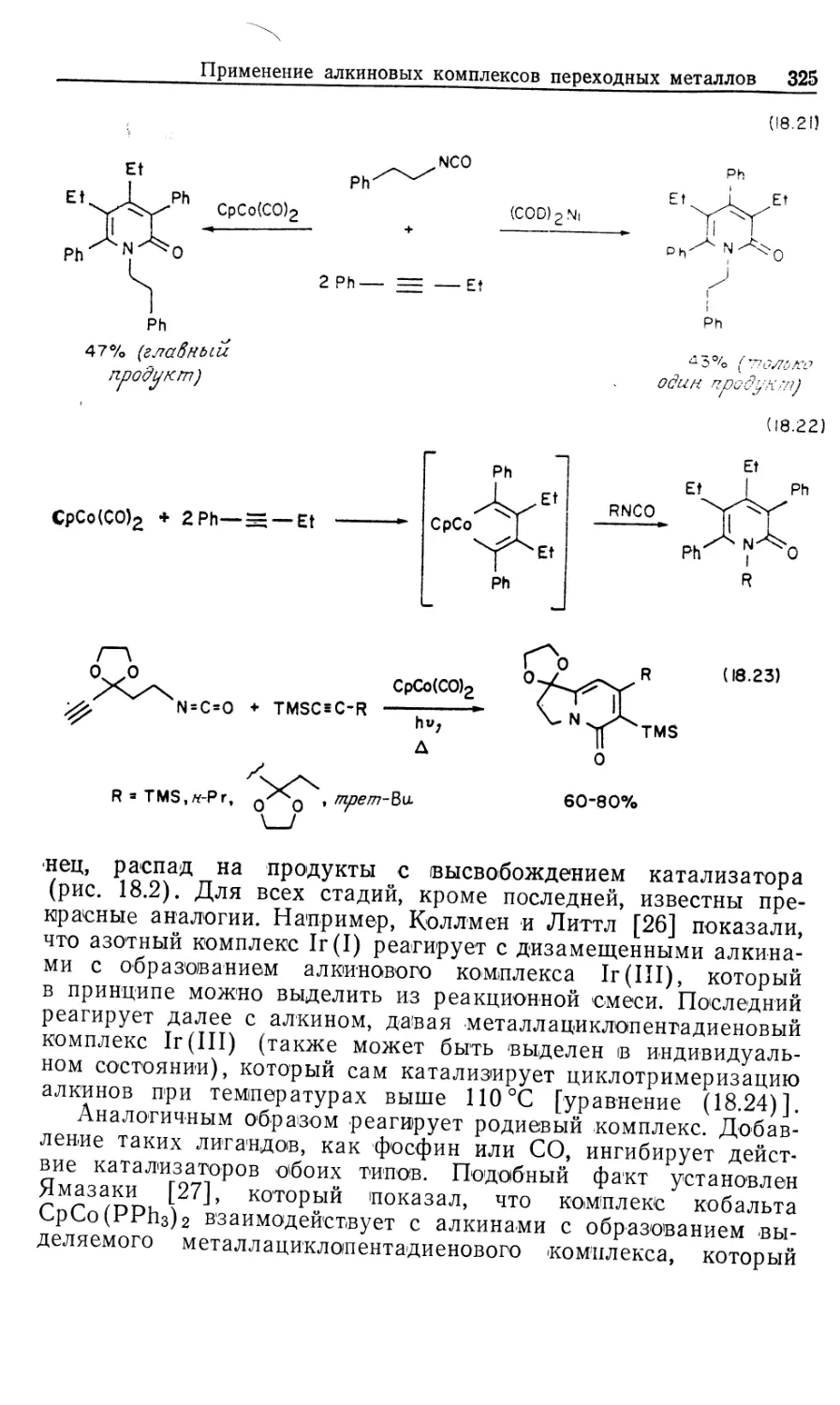
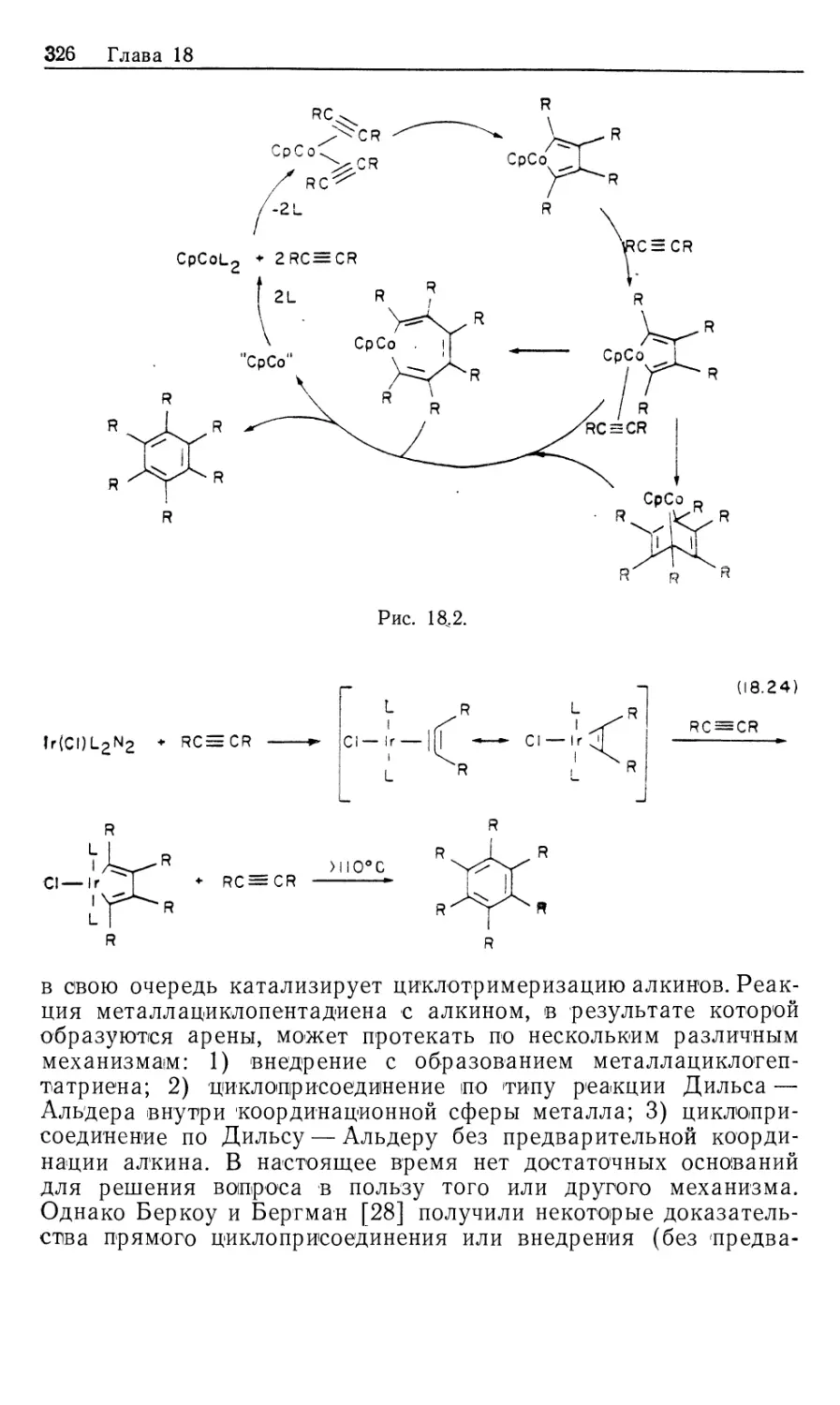

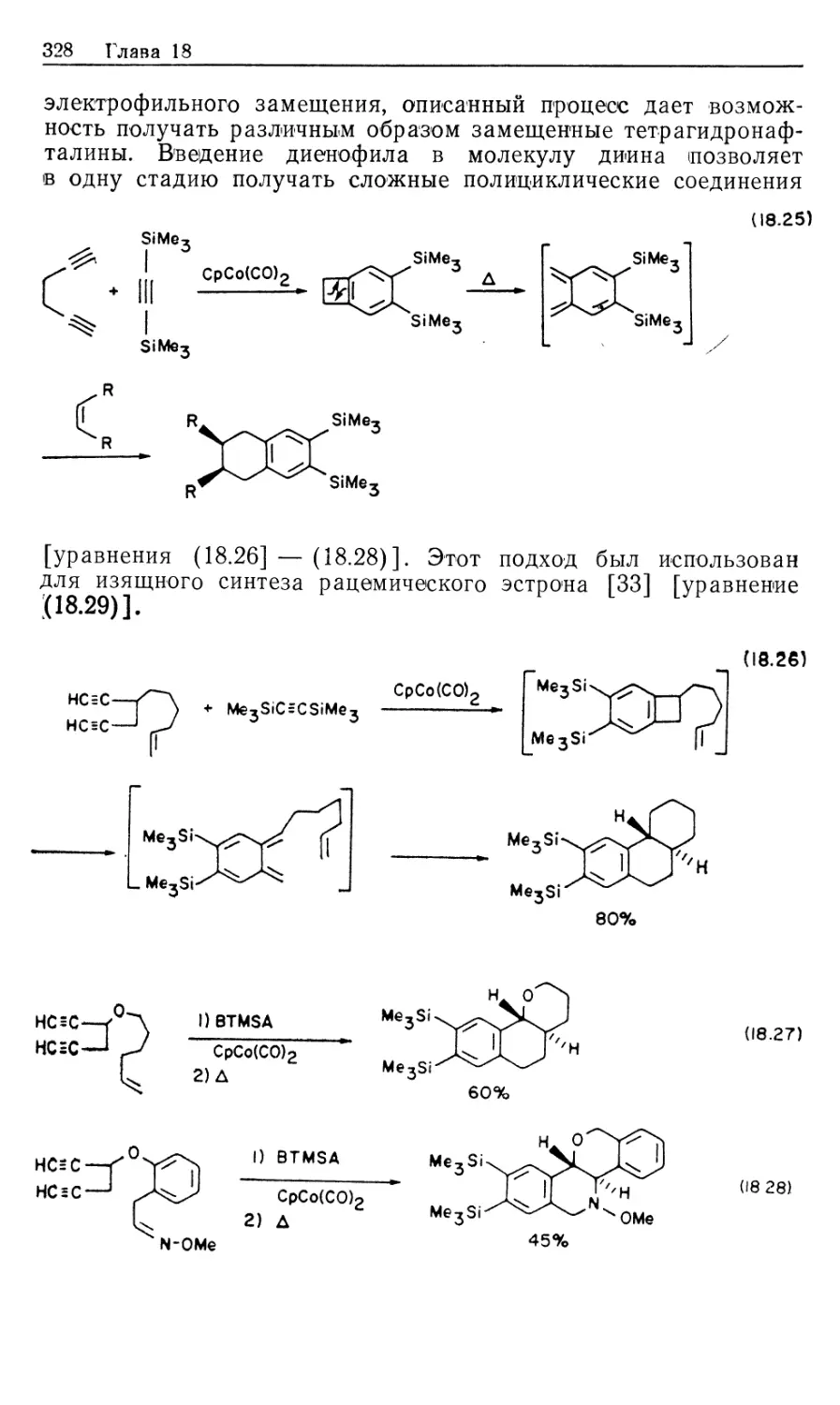



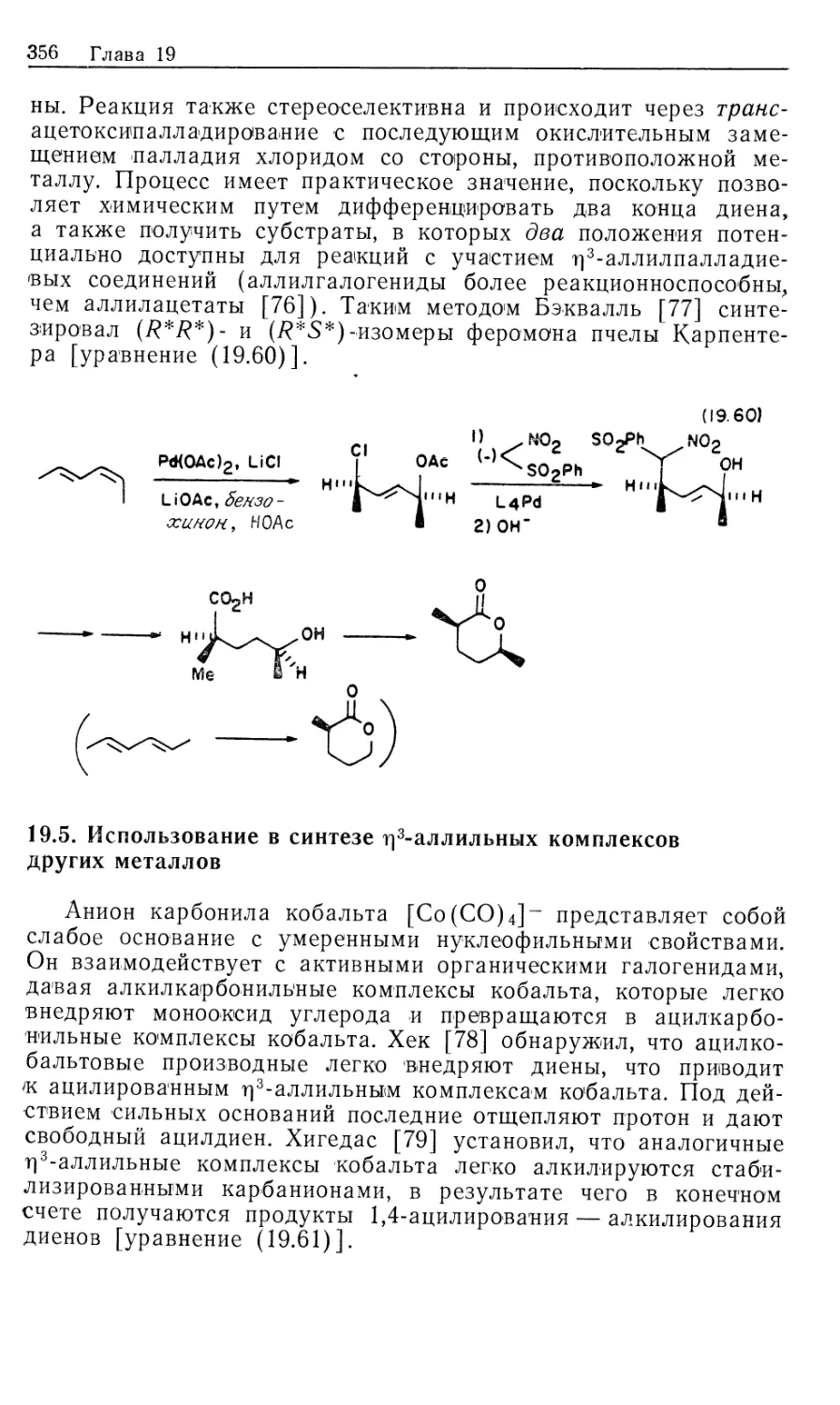







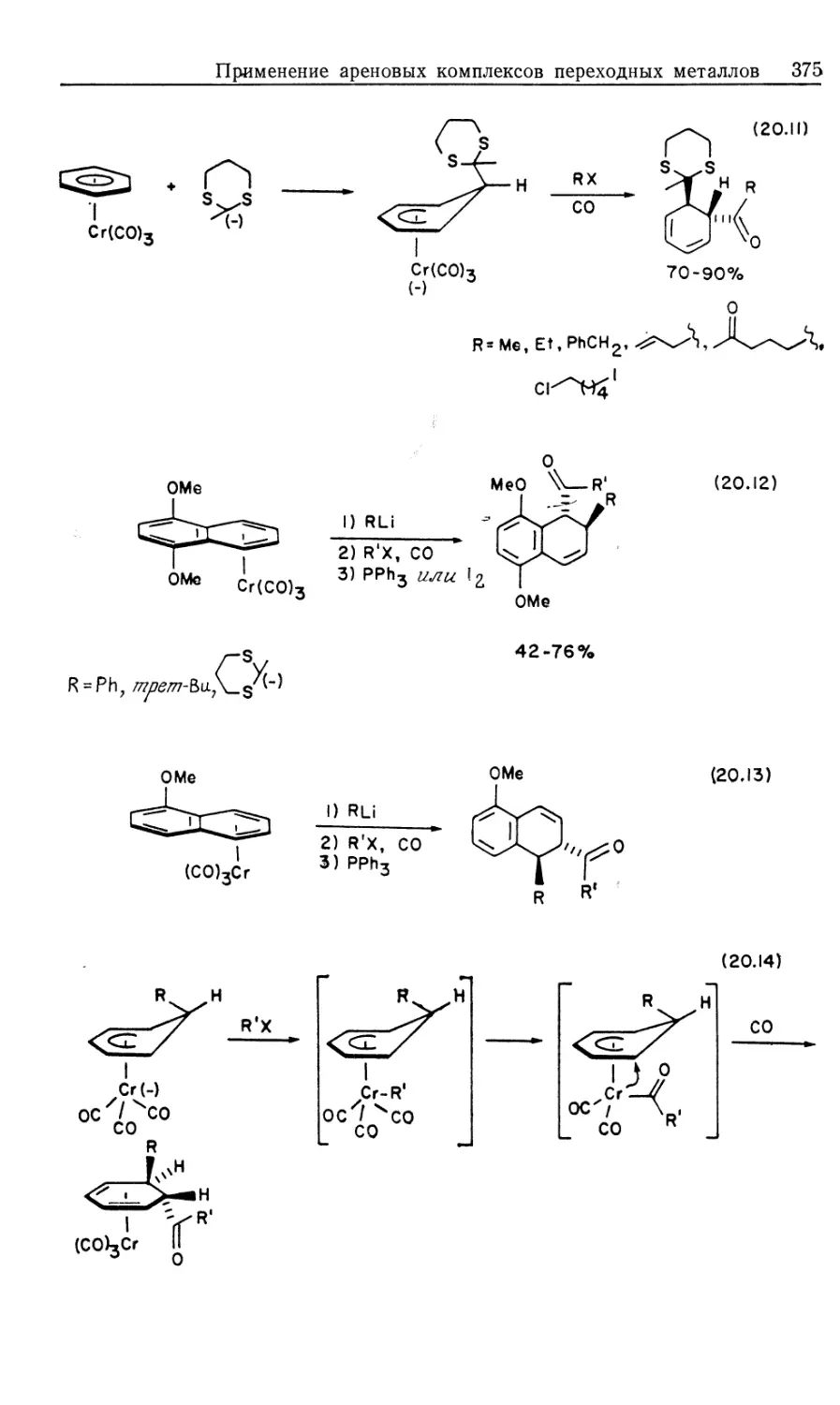
















Автор: Коллмен Дж. Хигедас Л. Нортон Дж. Финке Р.
Теги: органическая химия химия металлоорганическая химия
ISBN: 5-03-000277-4
Год: 1989




Текст
Principles and Applications
of Organotransition
Metal Chemistry
James P. Collman
Stanford University
Louis S. Hegedus
Colorado State University
Jack R. Norton
Colorado State University
Richard G. Finke
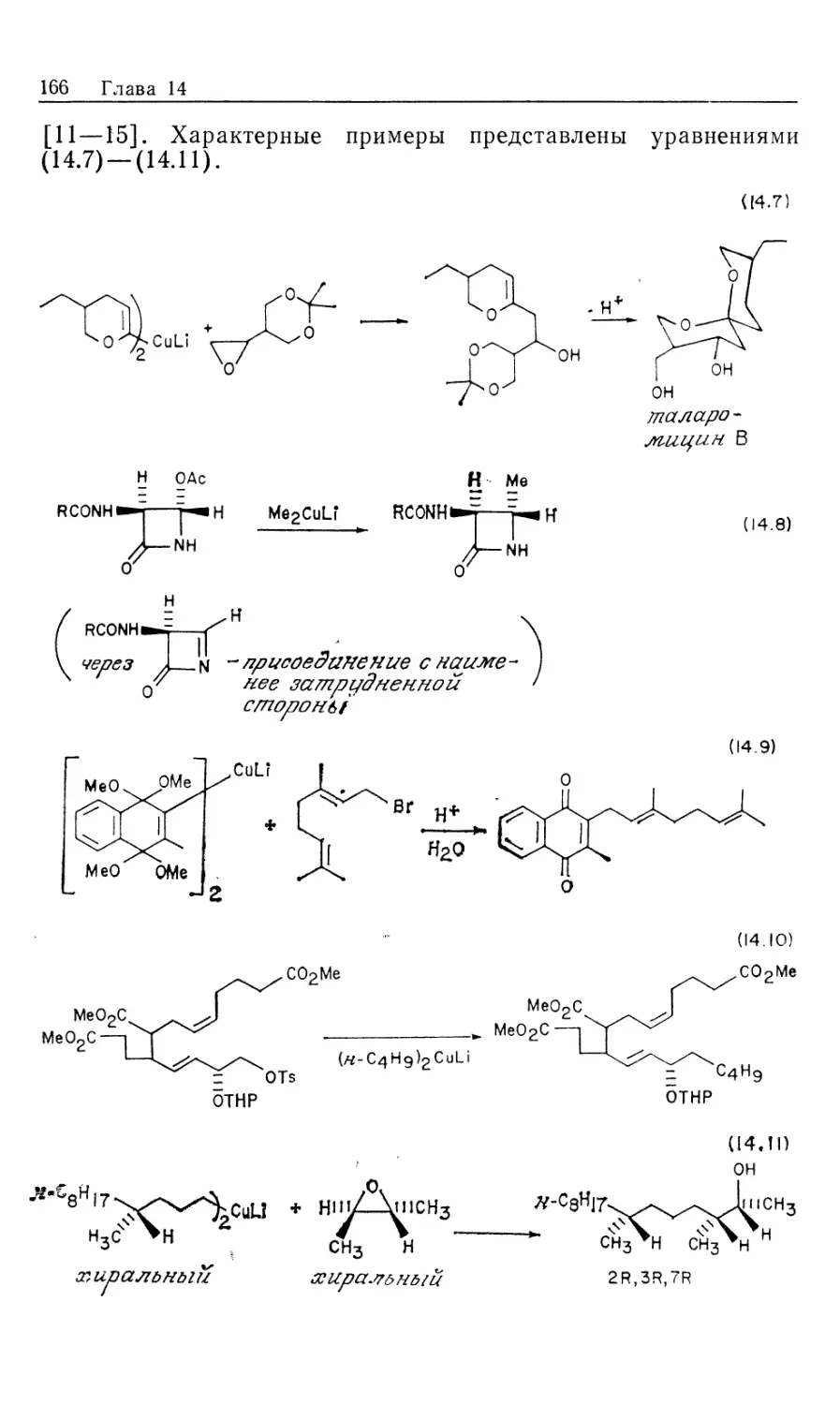
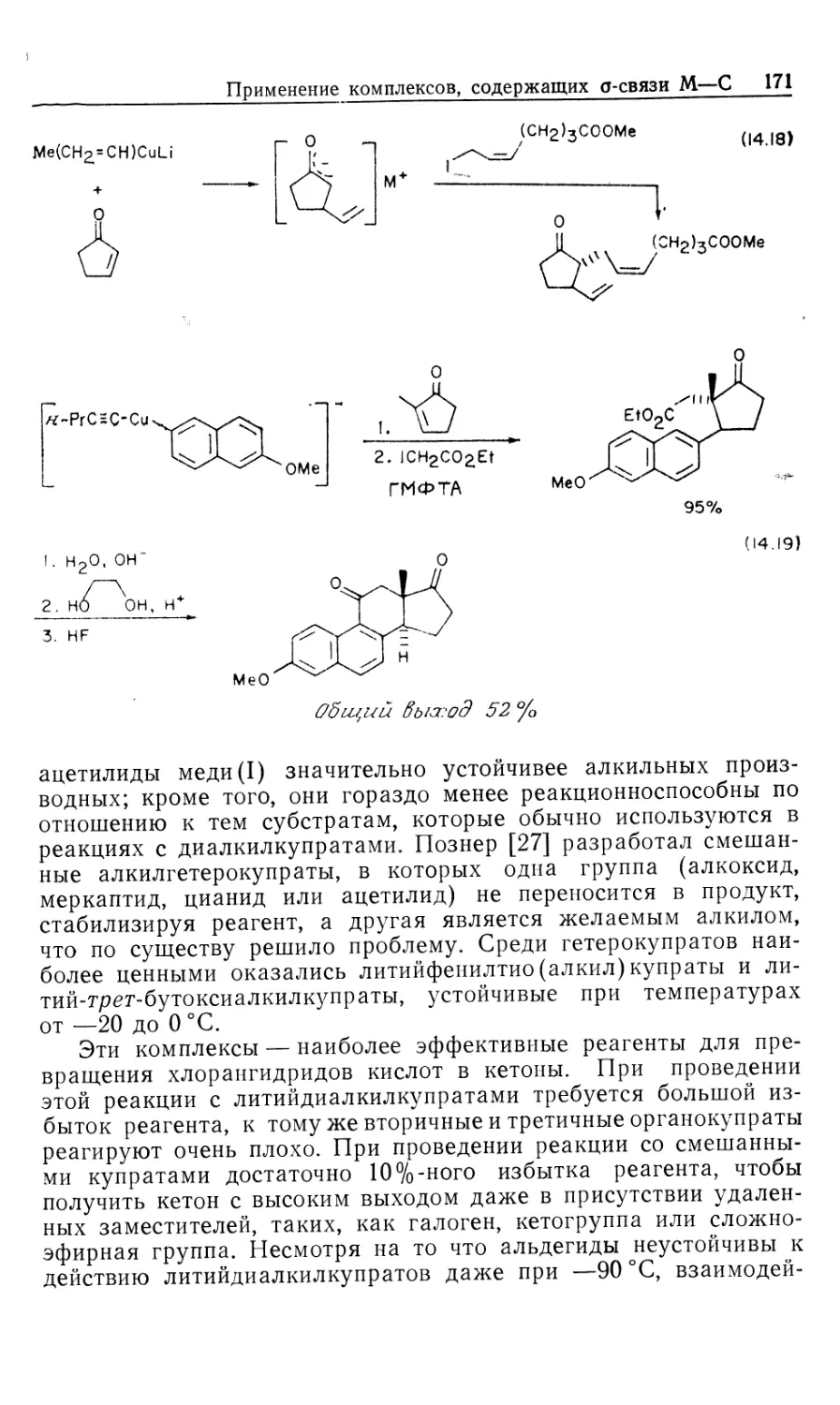
University of Oregon
University Science Books
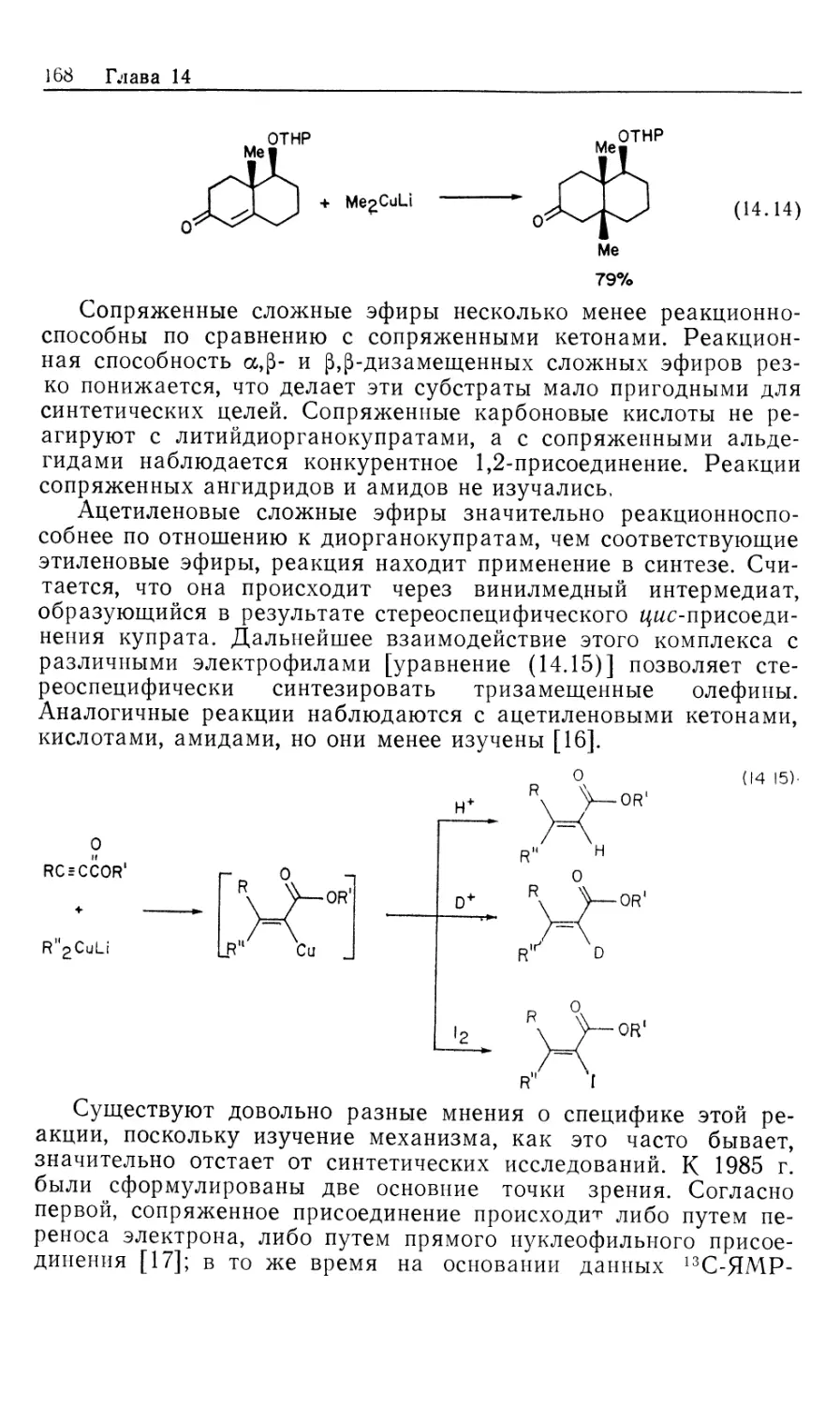
Mill Valley, California
Дж. Коллмен, Л. Хигедас,
Дж.Нортон, Р. Финке
МЕТАЛЛО-
ОРГАНИЧЕСКАЯ
ХИМИЯ
ПЕРЕХОДНЫХ
МЕТАЛЛОВ
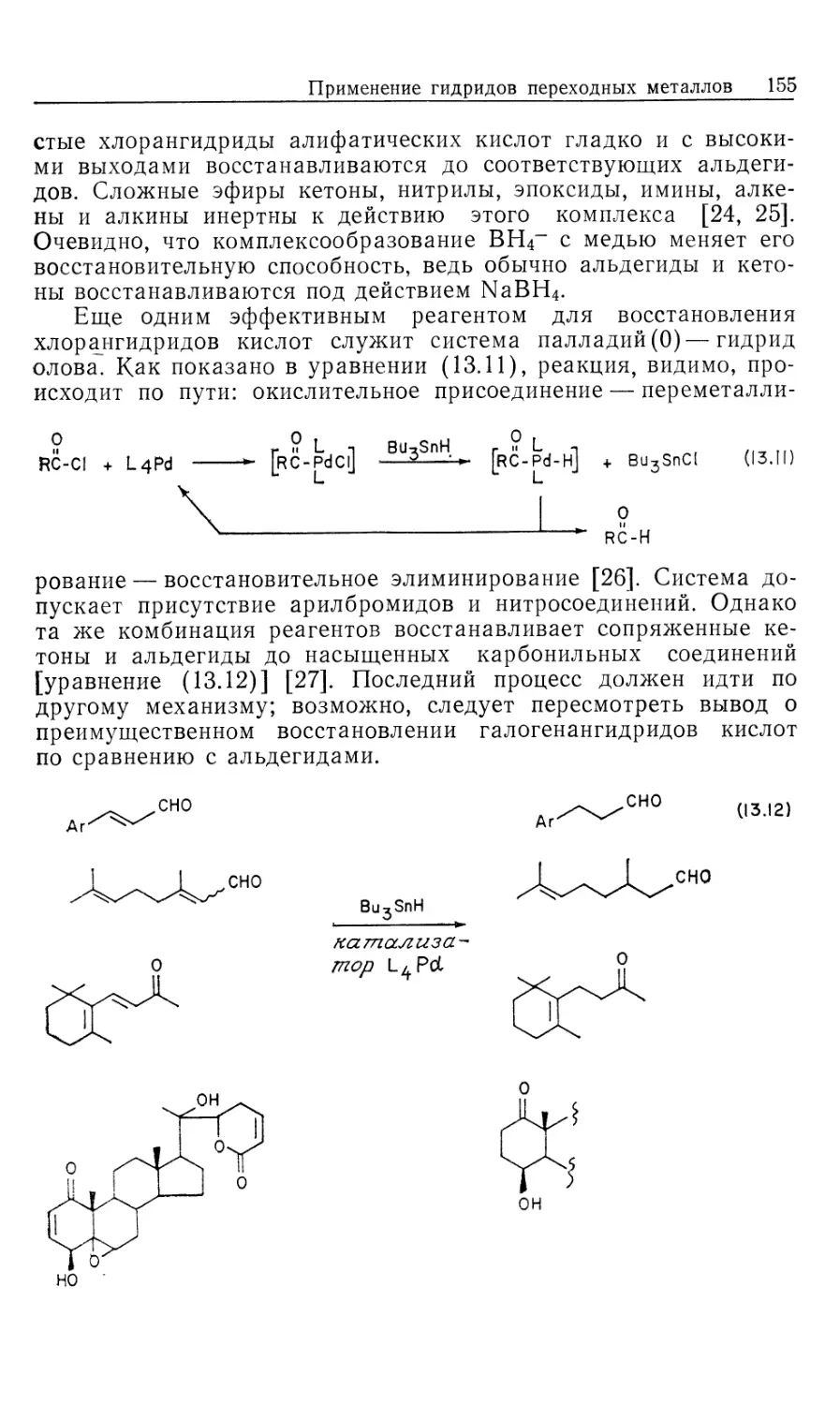
Основы и применения
В 2-х частях
2
Перевод с английского
канд. хим. наук 3. Е. САМОИЛОВОЙ
под редакцией
чл.-корр. АН СССР И. П. БЕЛЕЦКОЙ
Spliner
Москва «Мир» 1989
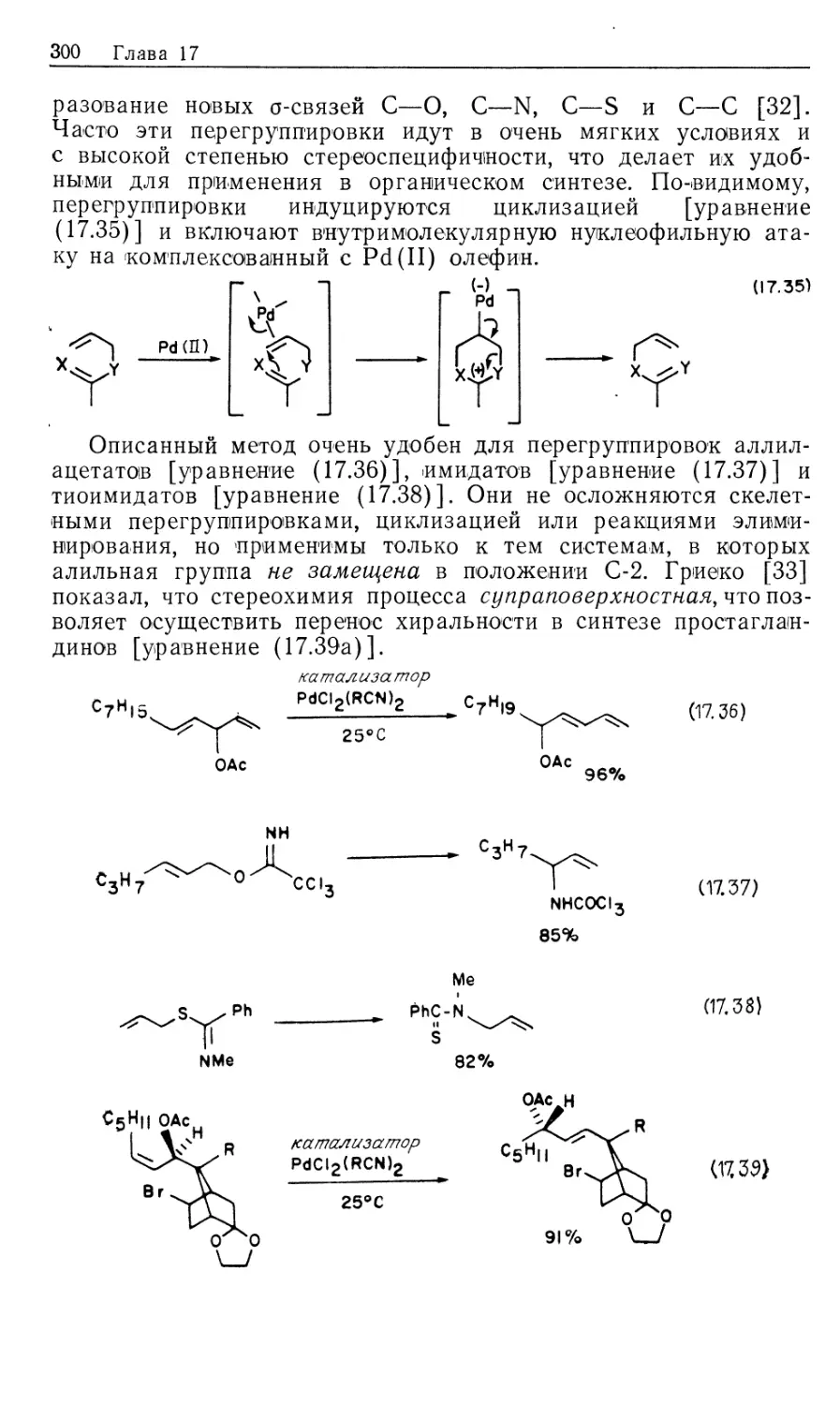
ББК 24.2
М54
УДК 547 + 546.3
Авторы: Коллмен Дж., Хигедас Л., Нортон Дж., Финке Р.
Металлоорганическая химия переходных металлов. Ос-
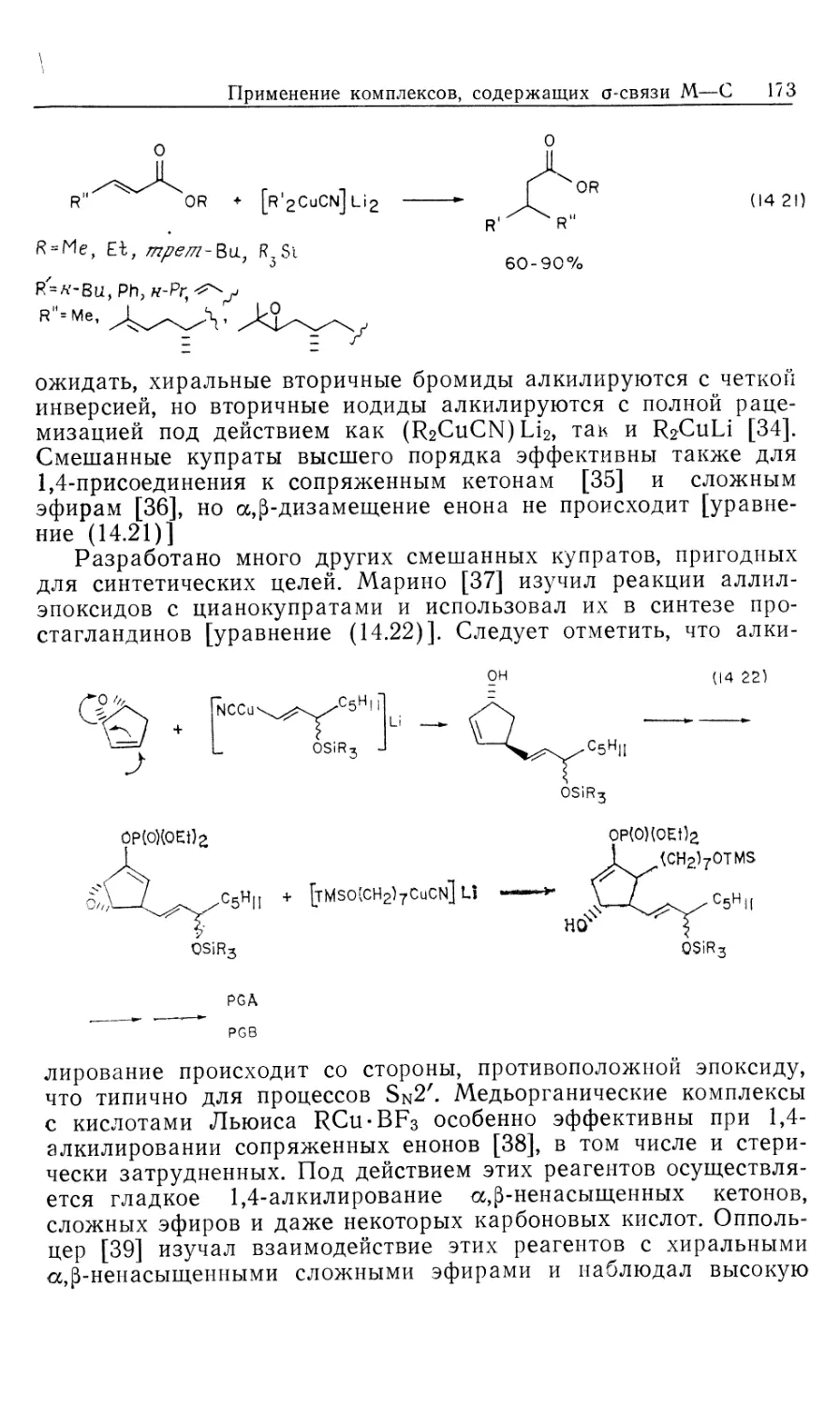
М54 новы и применения: В 2-х частях: ч. 2. Пер. с англ. — М.,
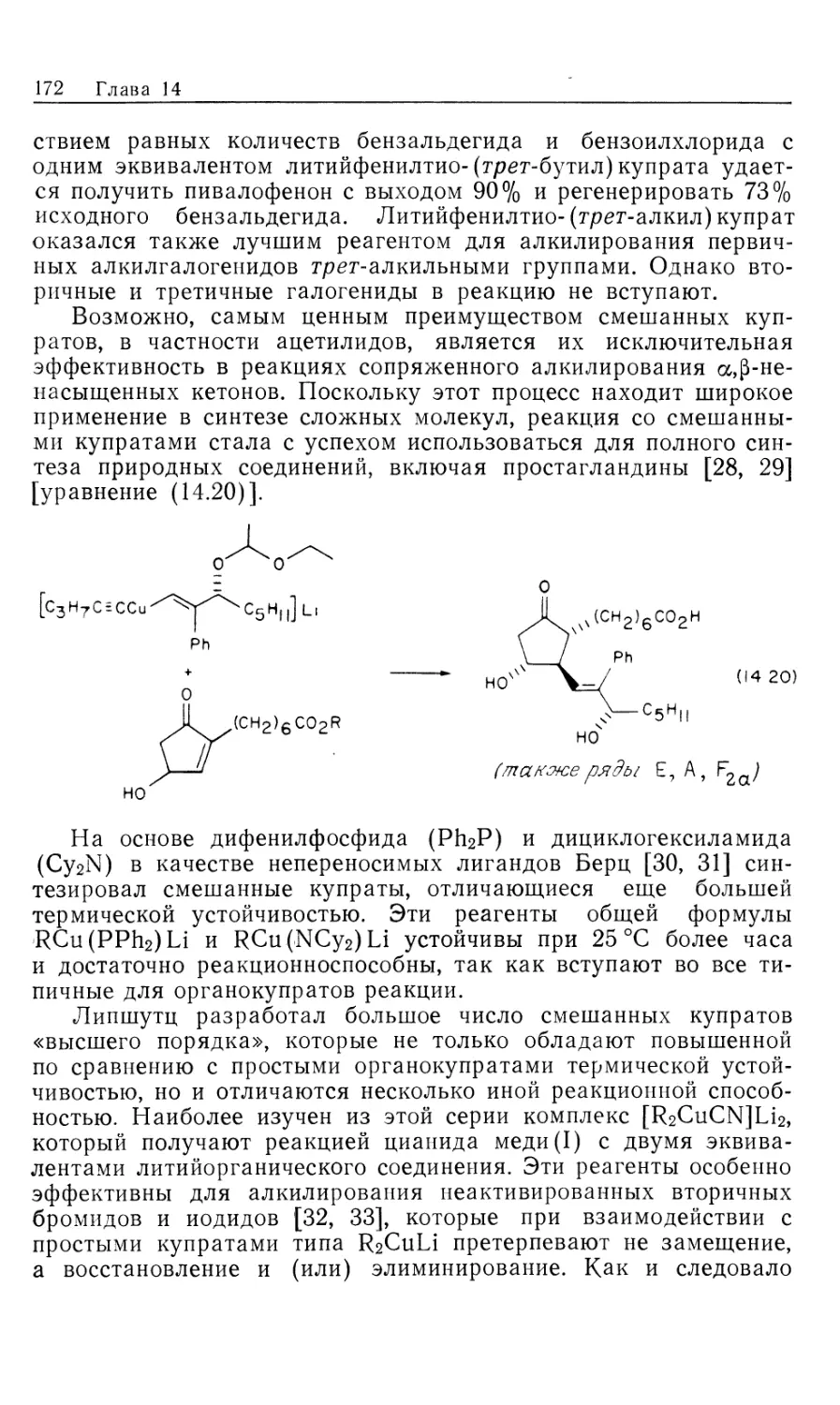
Мир, 1989.— 396 с, ил.
ISBN 5-03-000277-4
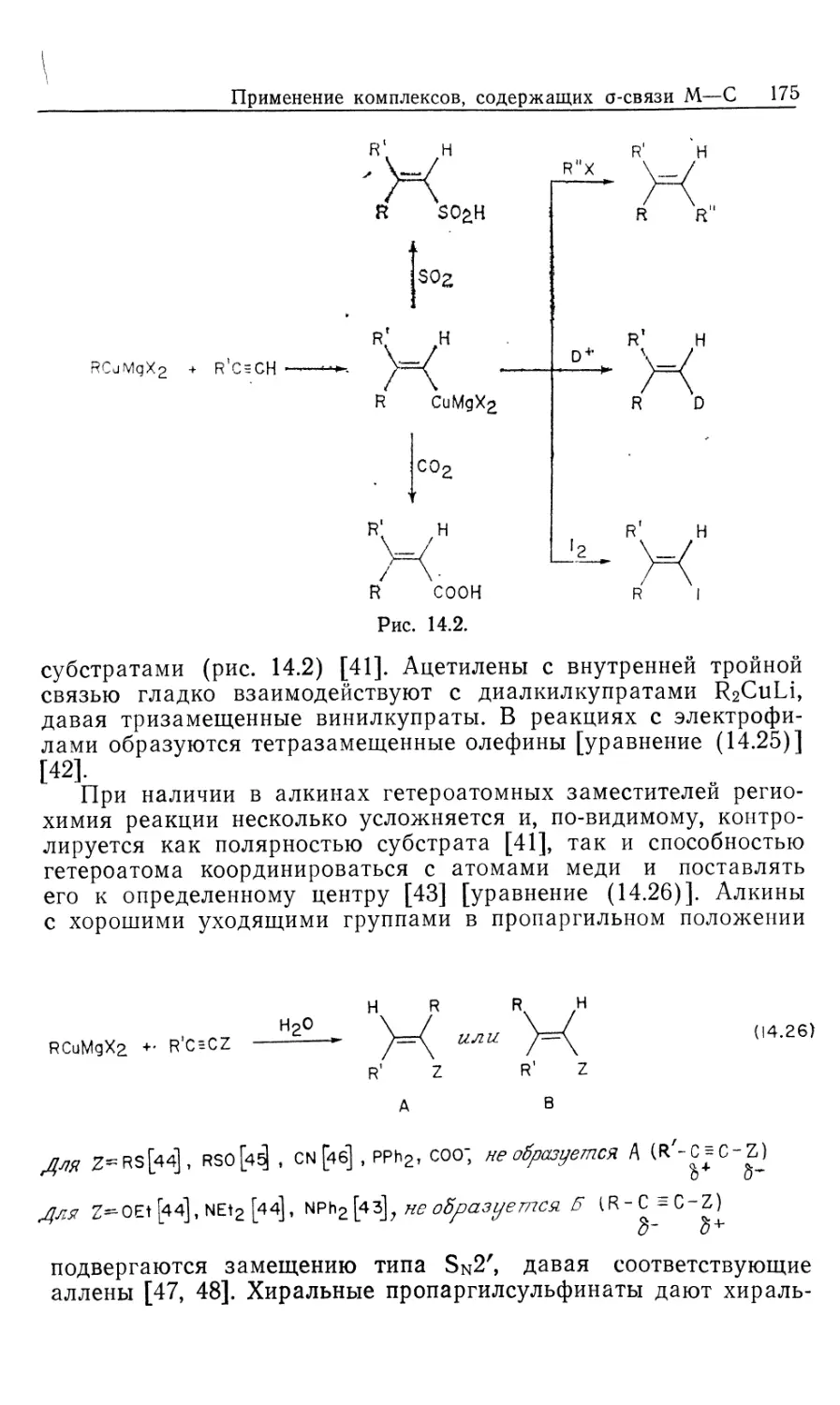
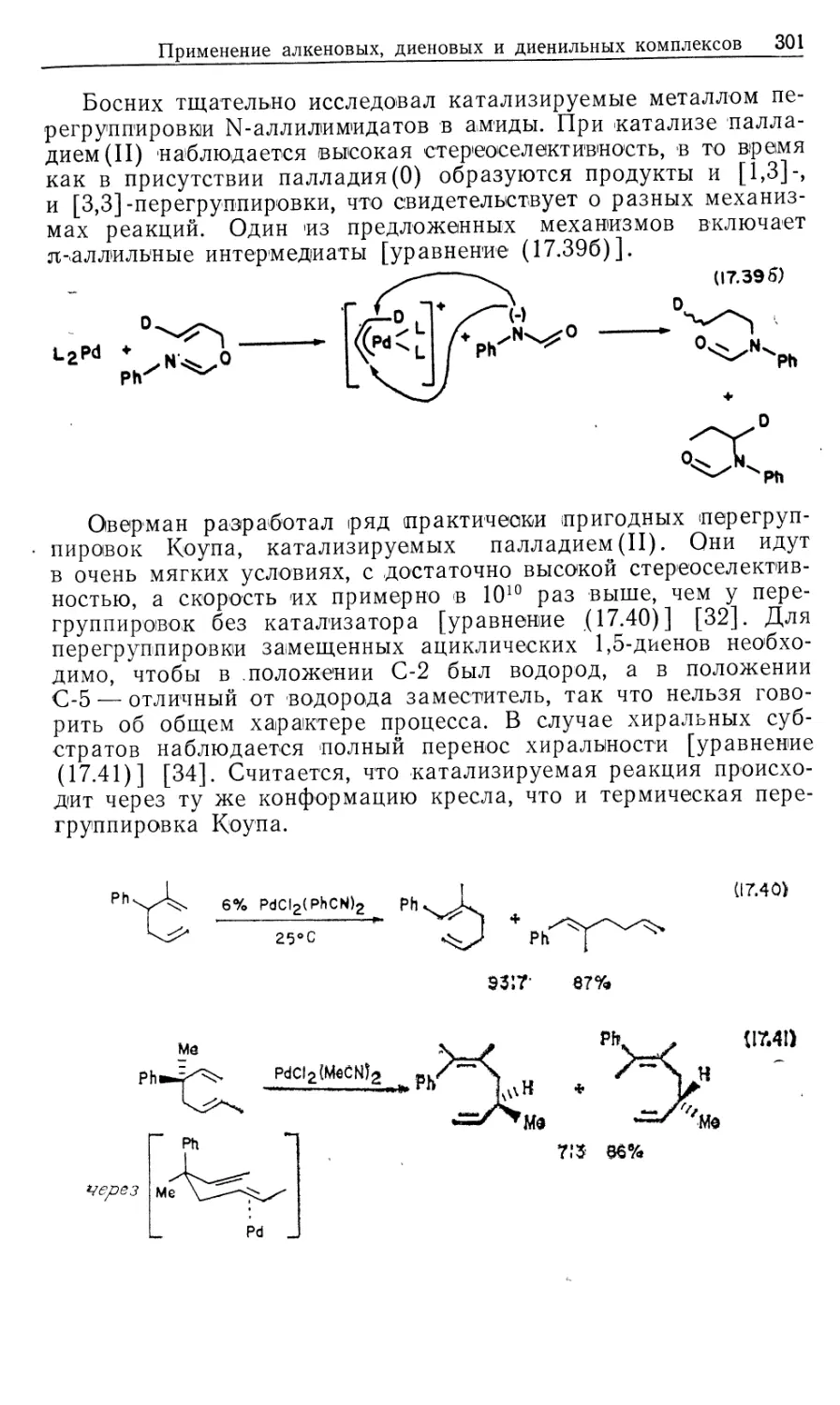
Учебное пособие, написанное коллективом авторов из США, посвящено ме-
таллоорганической химии, являющейся пограничной областью между органической
и неорганической химией. В книге удачно сочетаются как неорганические
аспекты химии переходных металлов, так и органические аспекты — основная часть ее
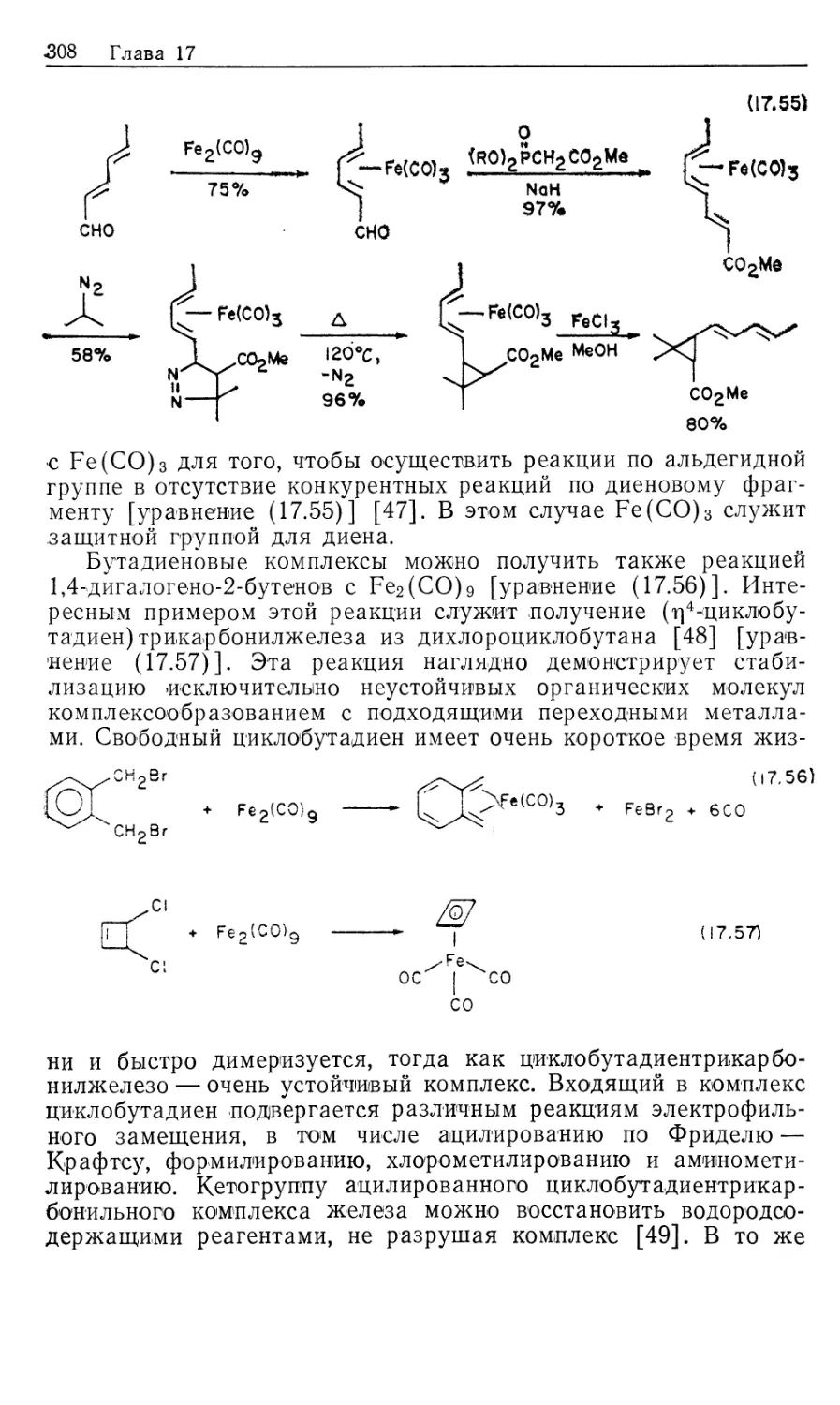
посвящена органическому синтезу с использованием комплексов переходных
металлов. Особое место занимают каталитические реакции.
На русском языке книга издается в 2-х частях. В ч. 2 рассматриваются
теоретические и практические основы металлокомплексного катализа.
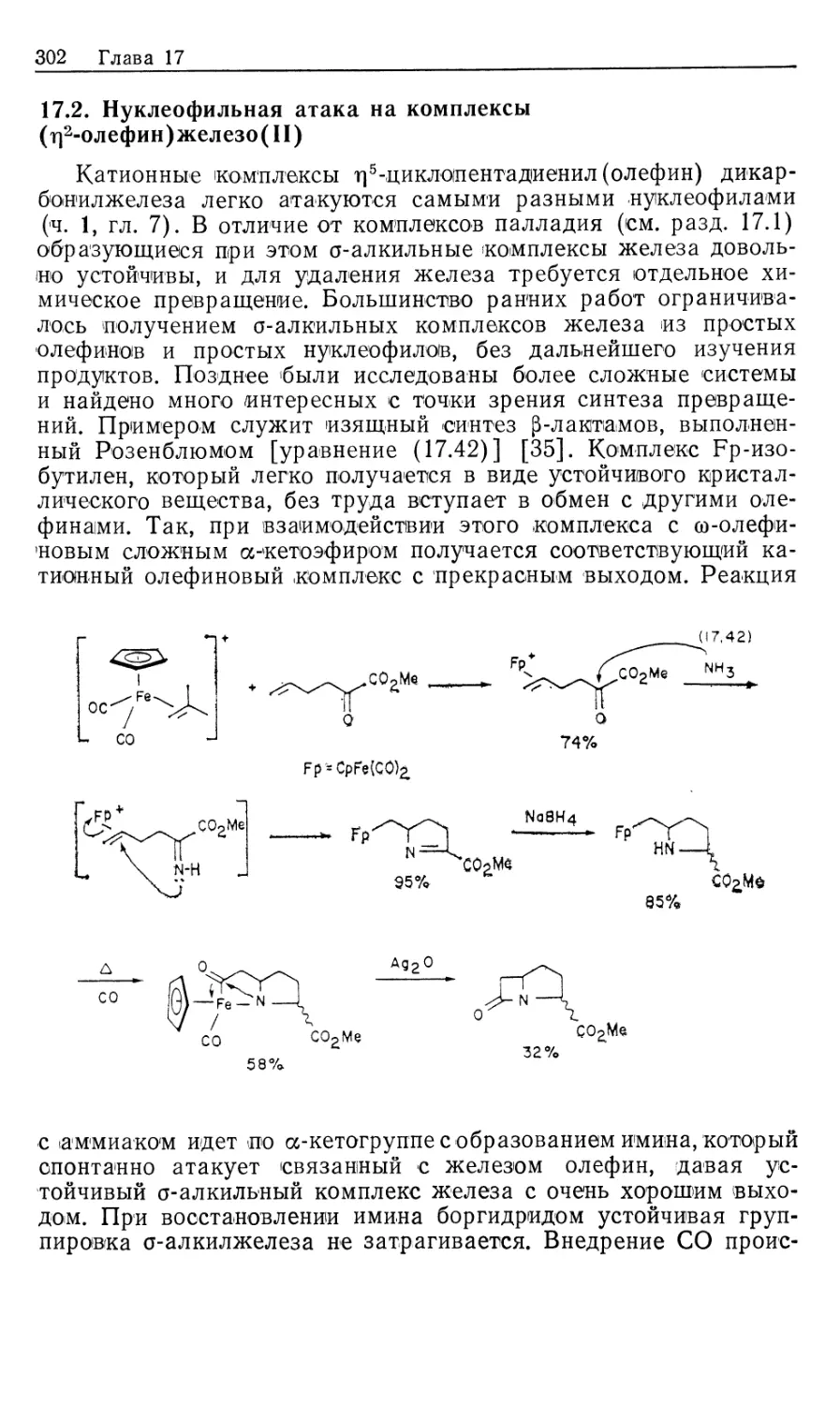
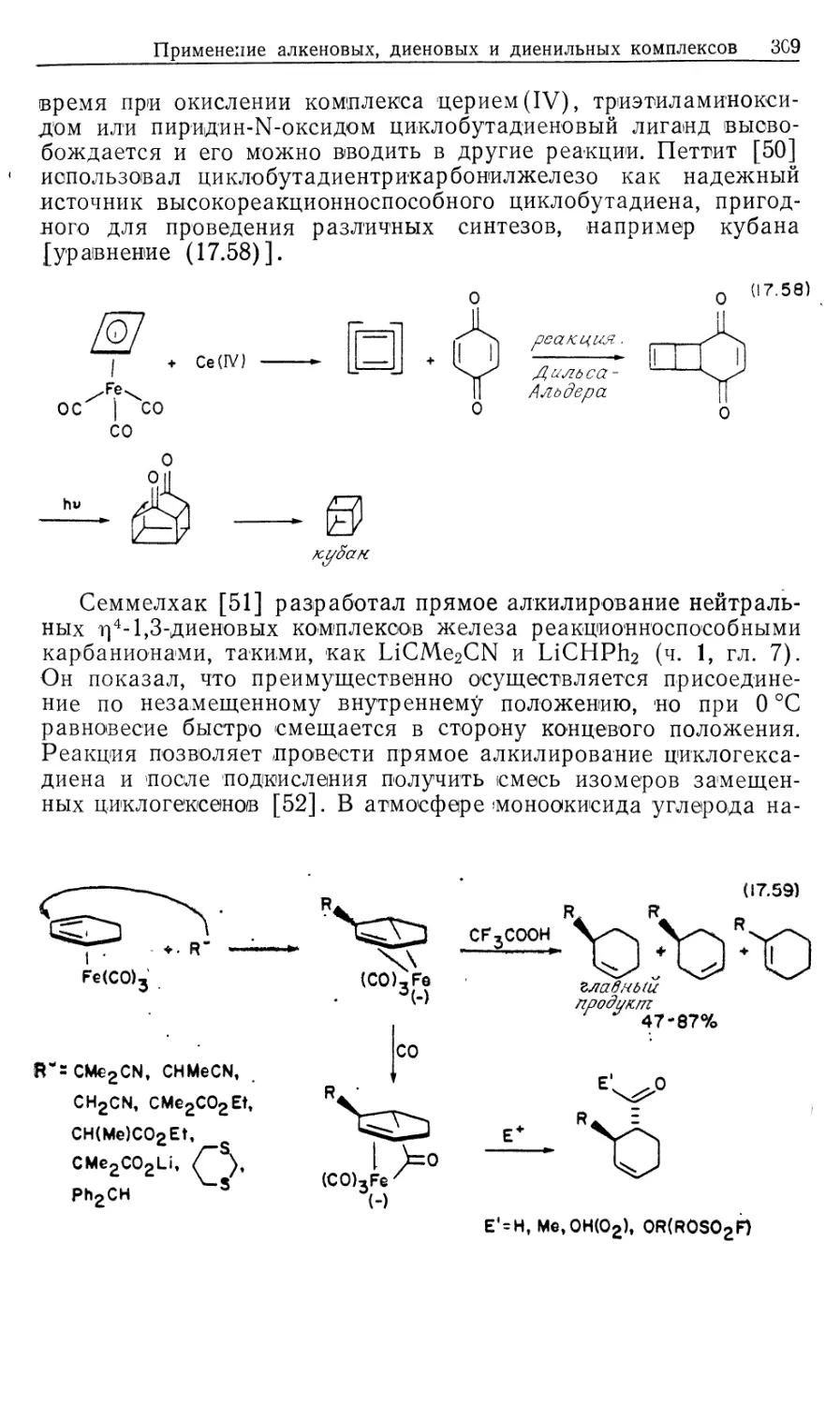
Для студентов, аспирантов, научных работников, инженеров-химиков,
работающих в области катализа.
1705000000-078 , я
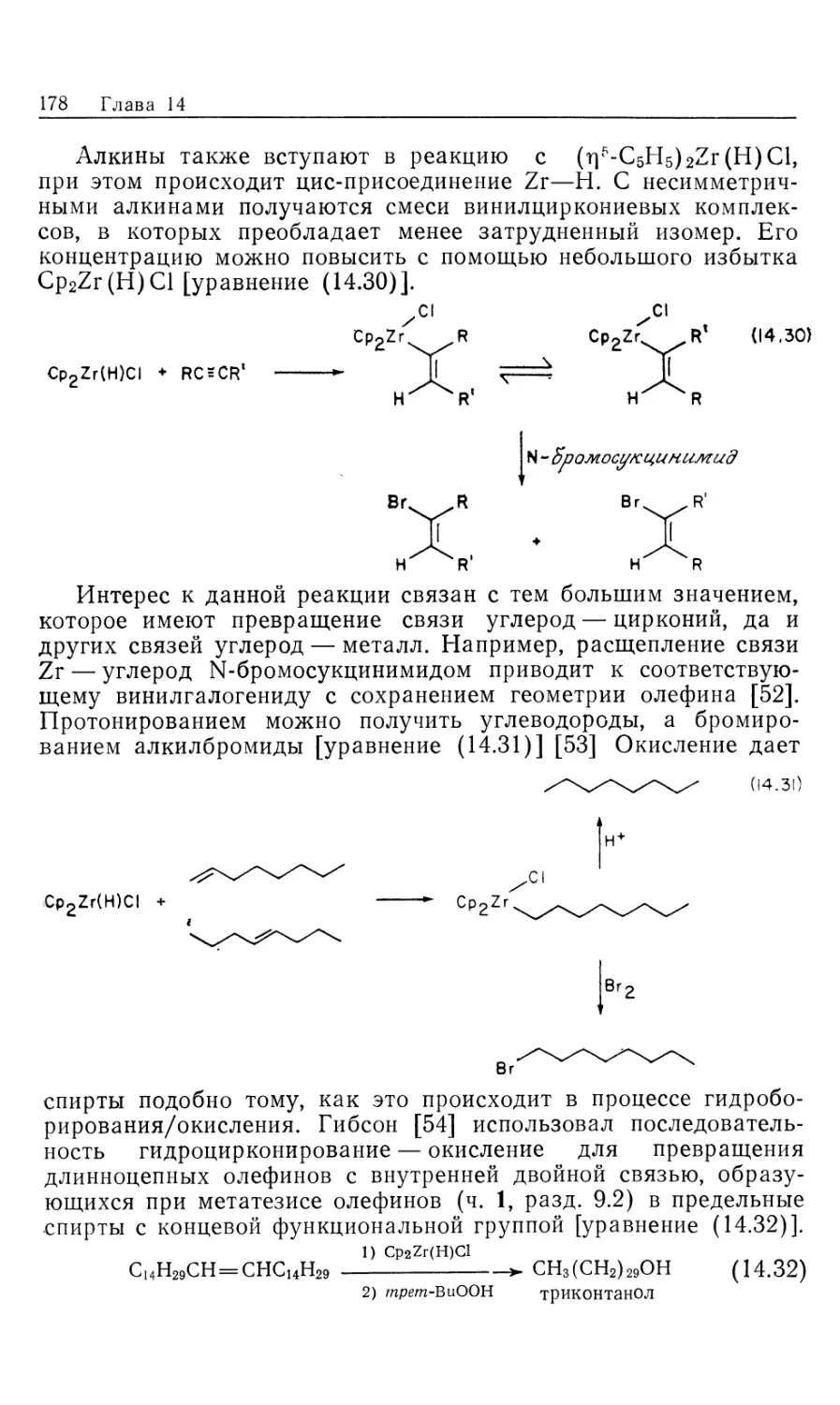
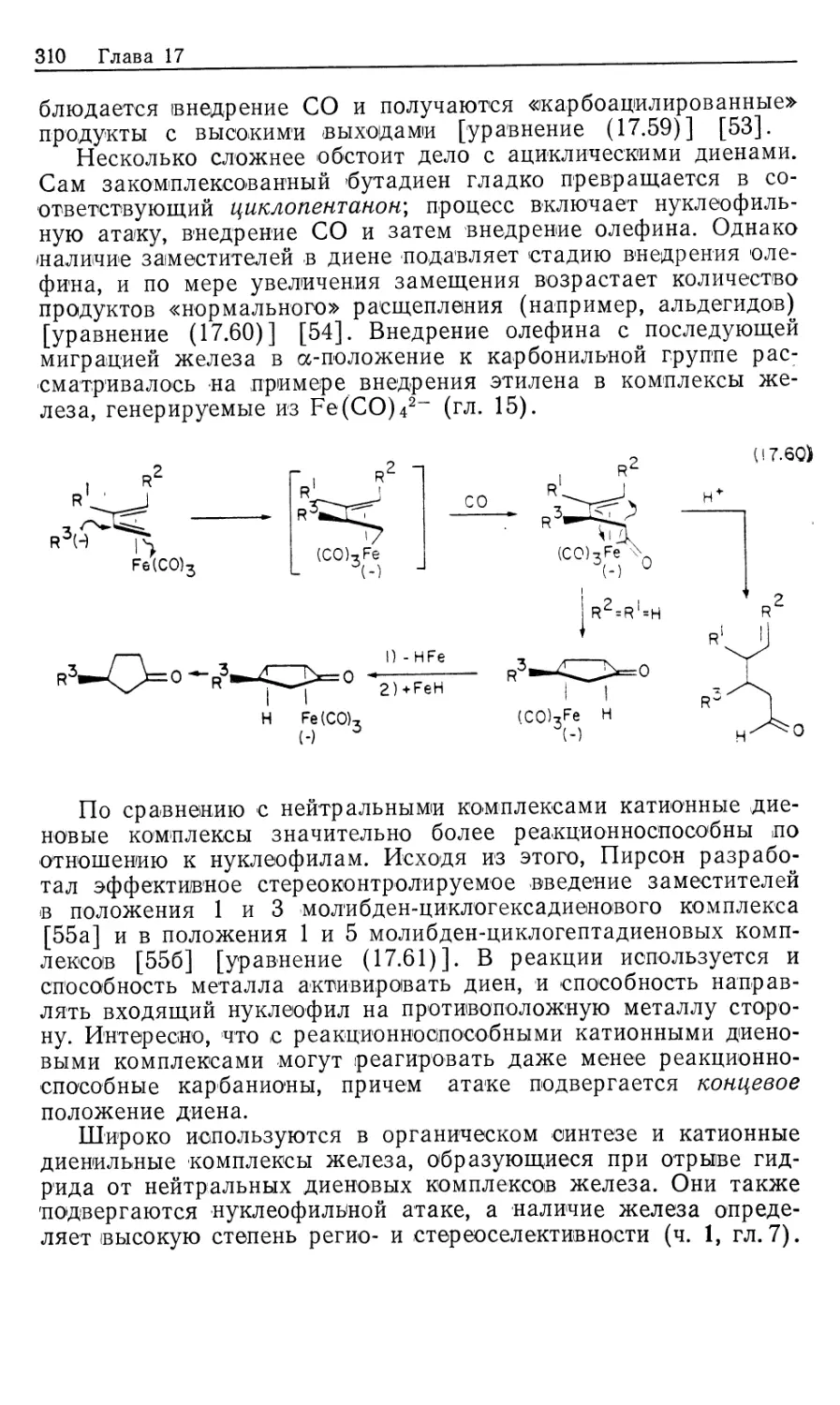
М 70-90 ББК 24.2
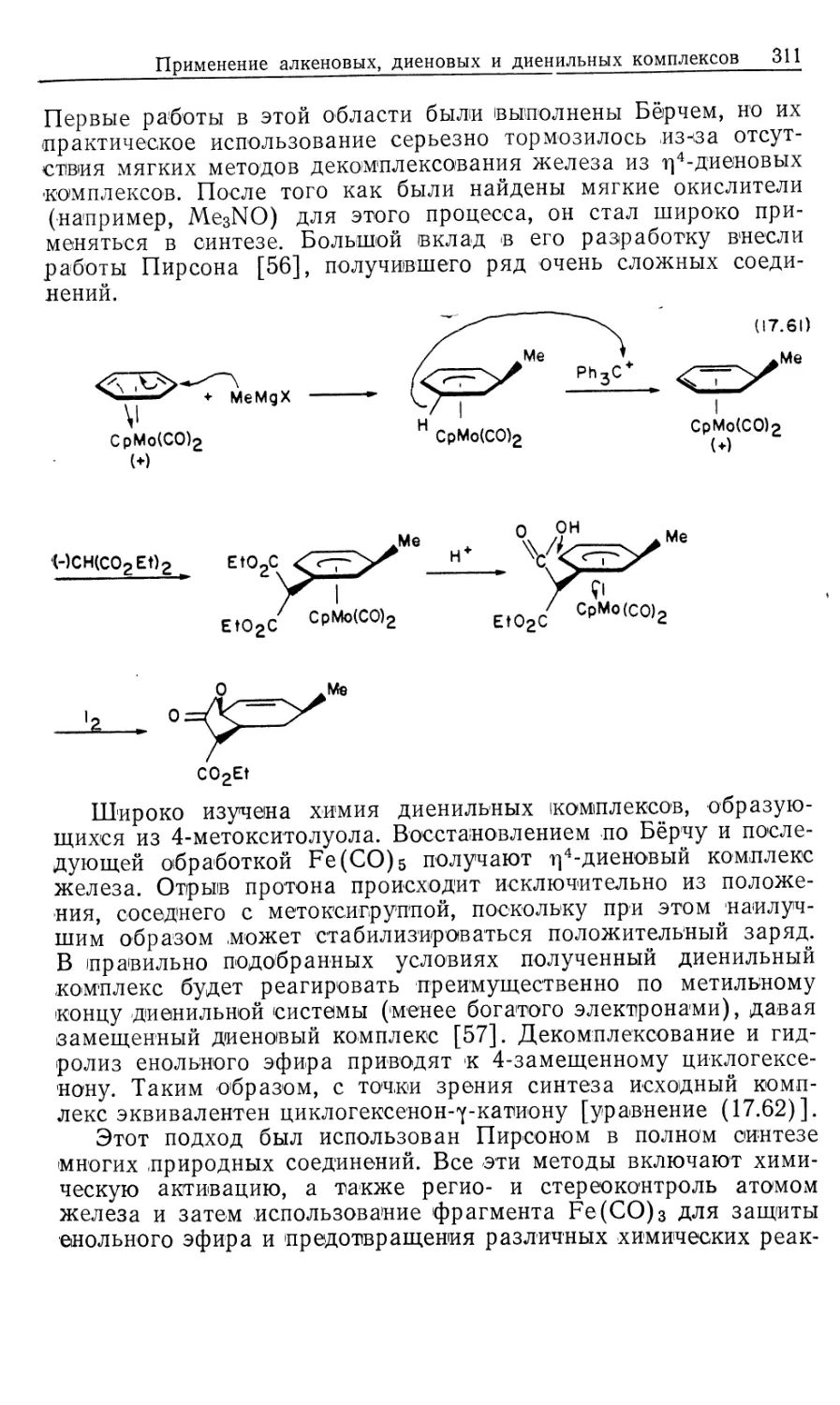
041(01)-89
Редакция литературы по химии
ISBN 5-03-000277-4 (русск.) © 1987 by University Science Books
ISBN 5-03-000278-2 © перевод на русский язык, «Мир», 1989
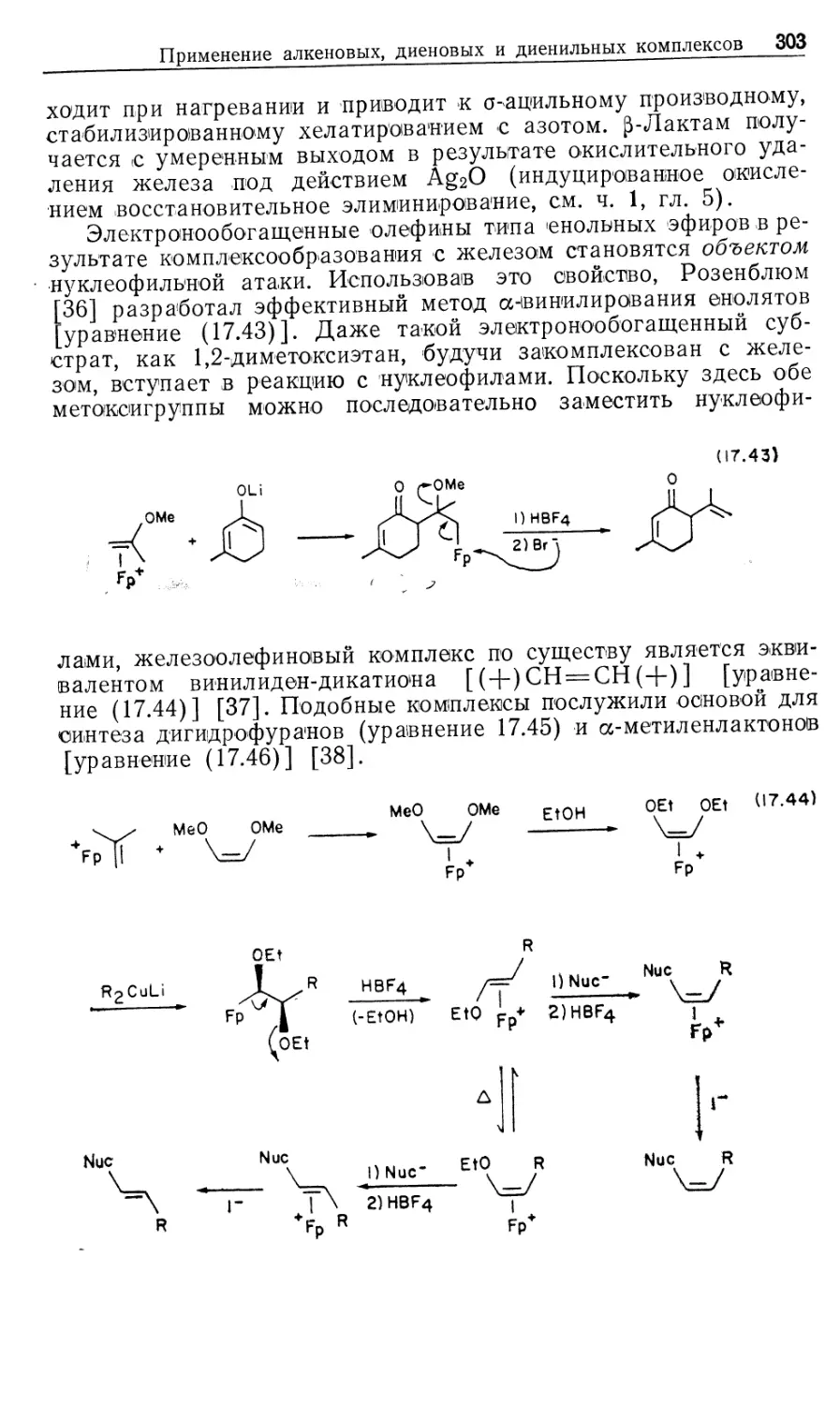
ISBN 0-935702-51-2 (англ.)
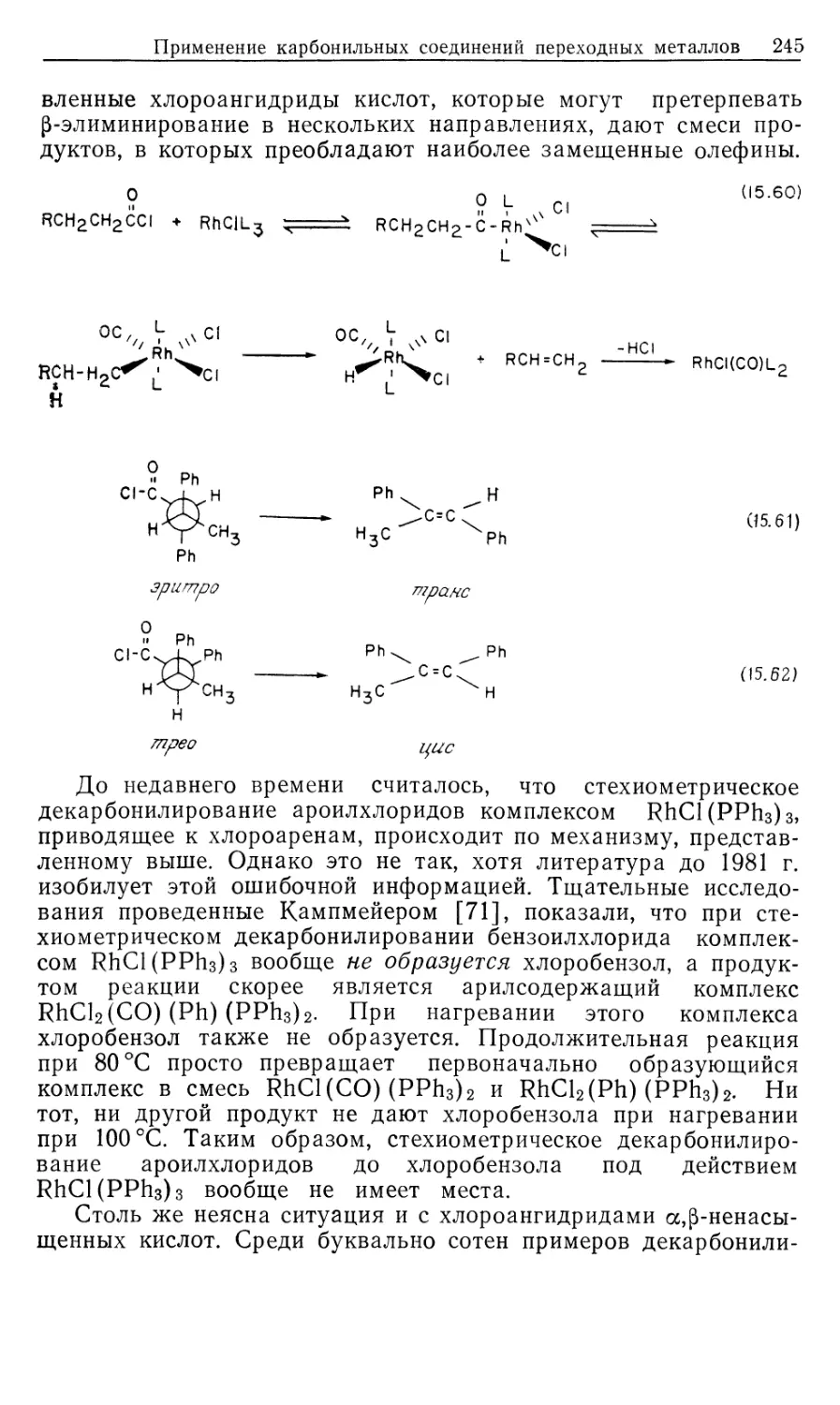
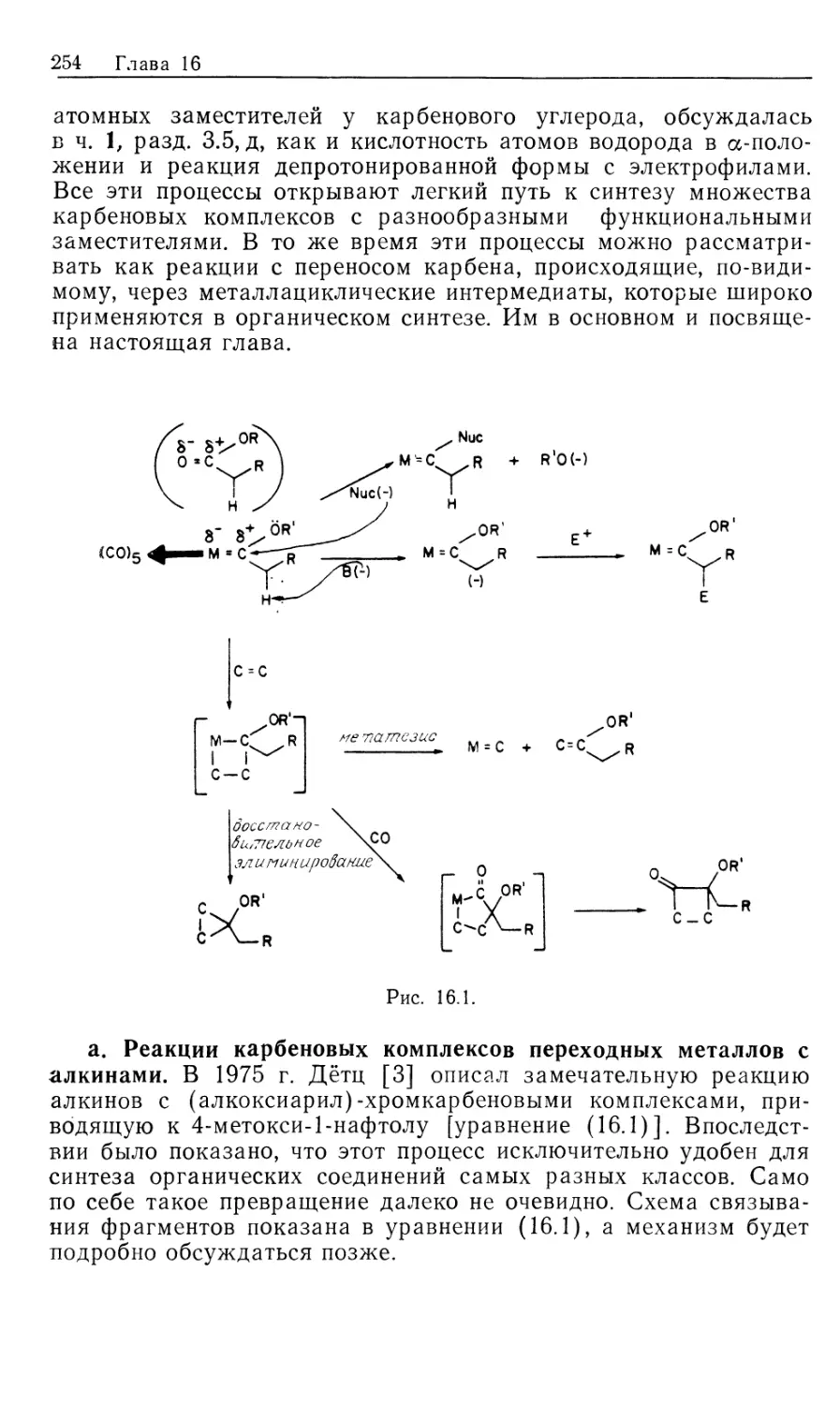
РАЗДЕЛ II
КАТАЛИТИЧЕСКИЕ ПРОЦЕССЫ
10
ГОМОГЕННЫЕ КАТАЛИТИЧЕСКИЕ ПРОЦЕССЫ:
ГИДРИРОВАНИЕ, ГИДРОСИЛИЛИРОВАНИЕ,
ГИДРОЦИАНИРОВАНИЕ
10.1. Введение
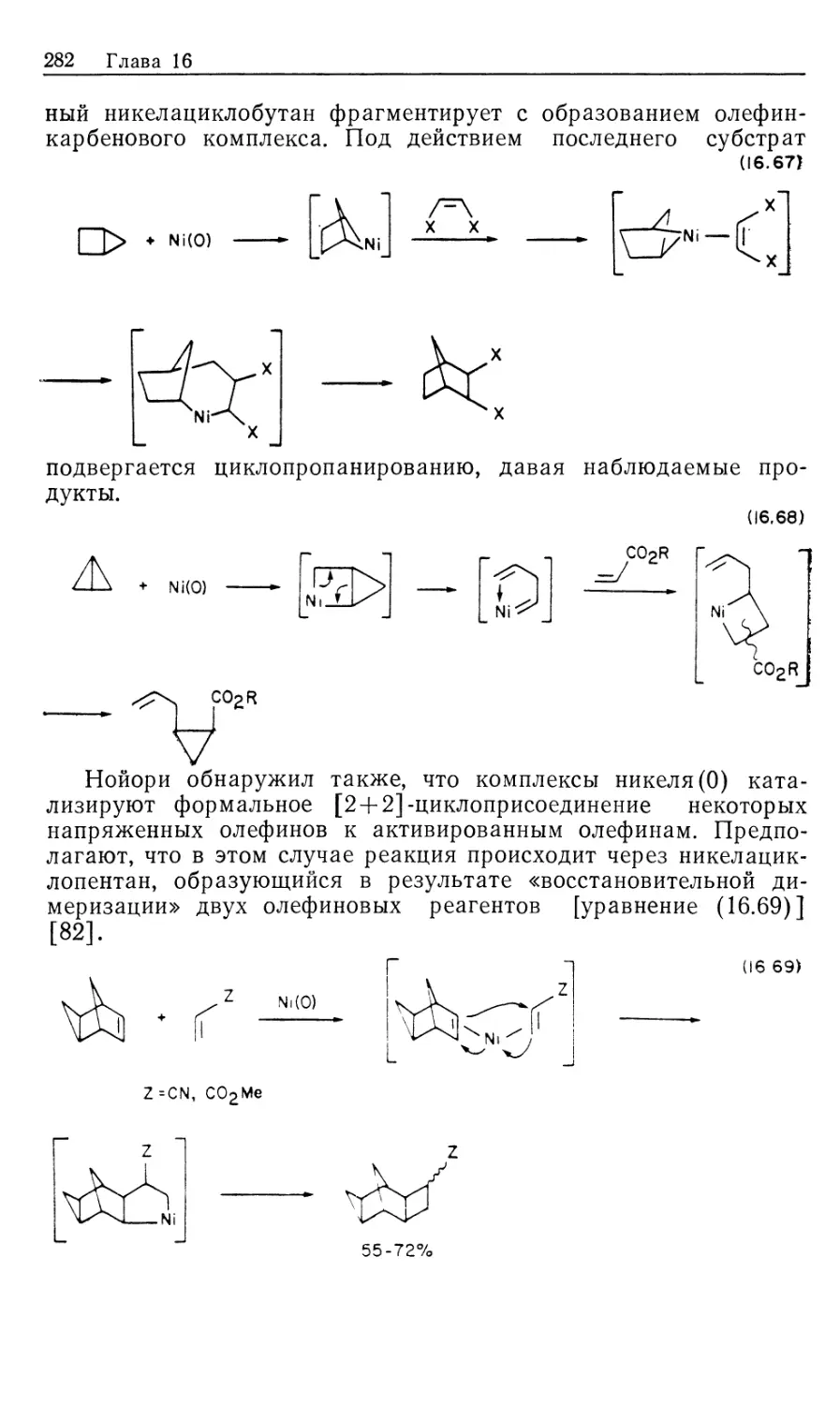
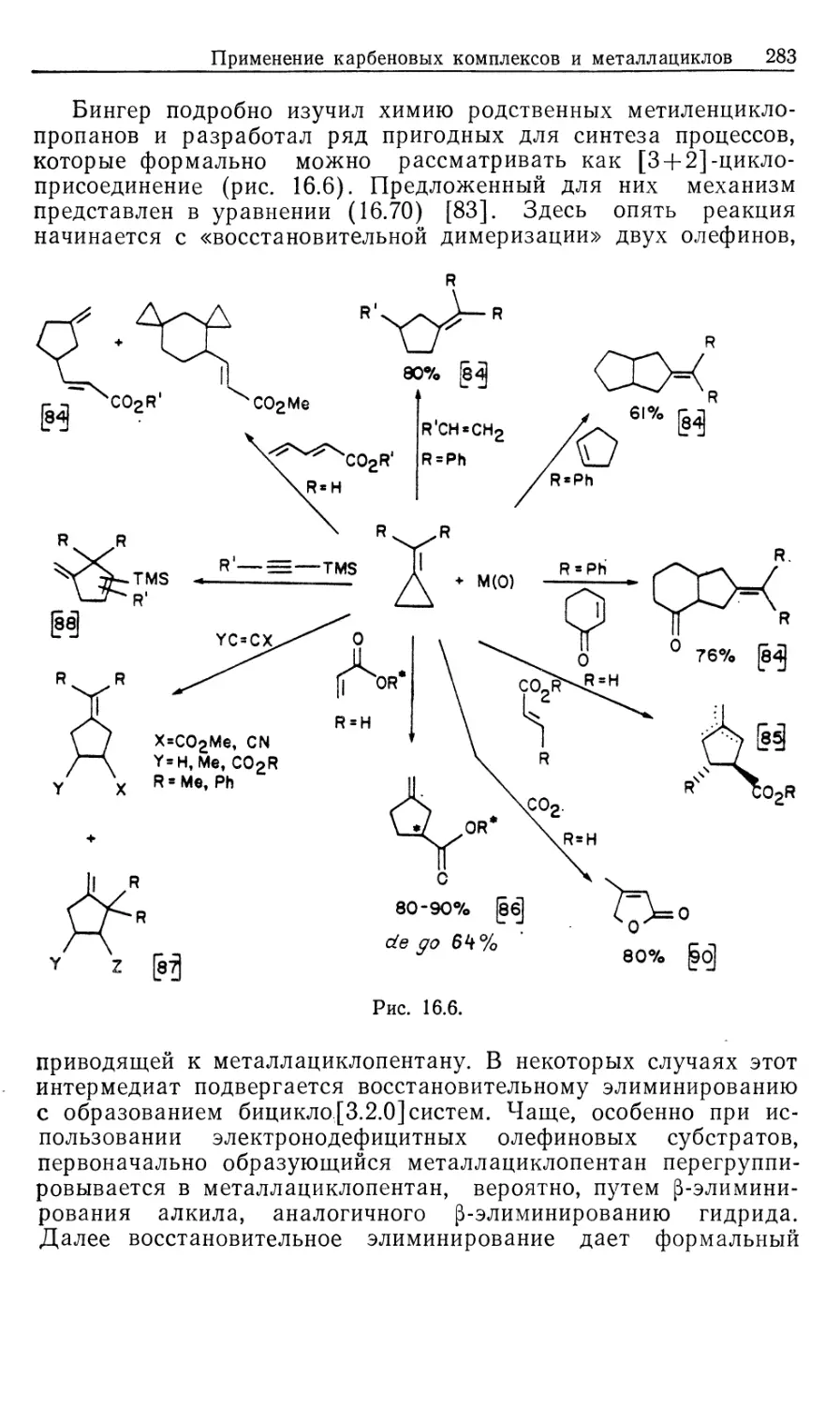
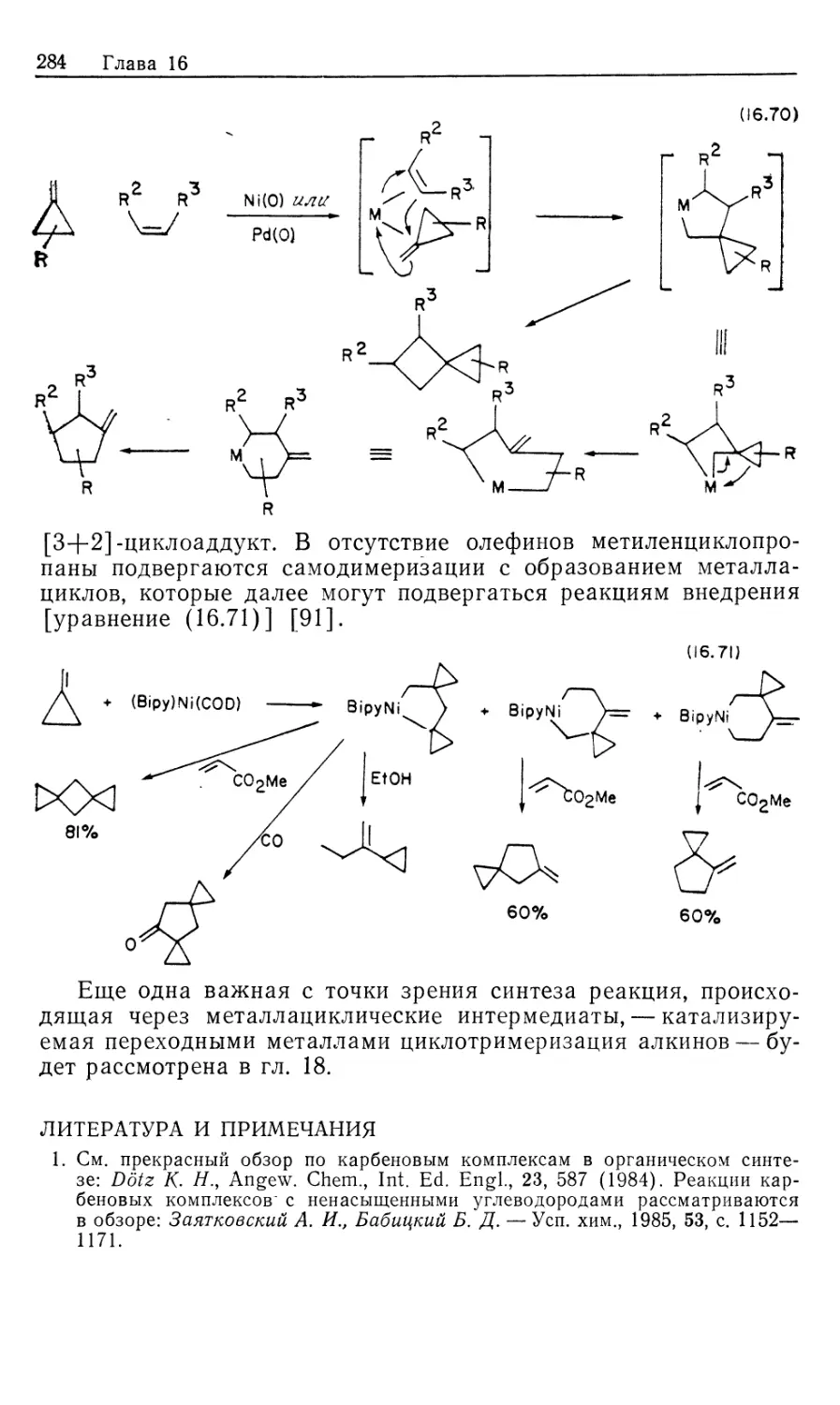
Важным 'классом металлоорганических реакций является
гидрирование органических соединений в условиях гомогенного
катализа. Наиболее изученными субстратами оказались олефи-
ны, хотя многие другие соединения, содержащие
функциональные группы, например, ацетилены, альдегиды, кетоны, нитросо-
единения, арены, также подвергаются гидрированию в
присутствии растворимых катализаторов. Известно множество
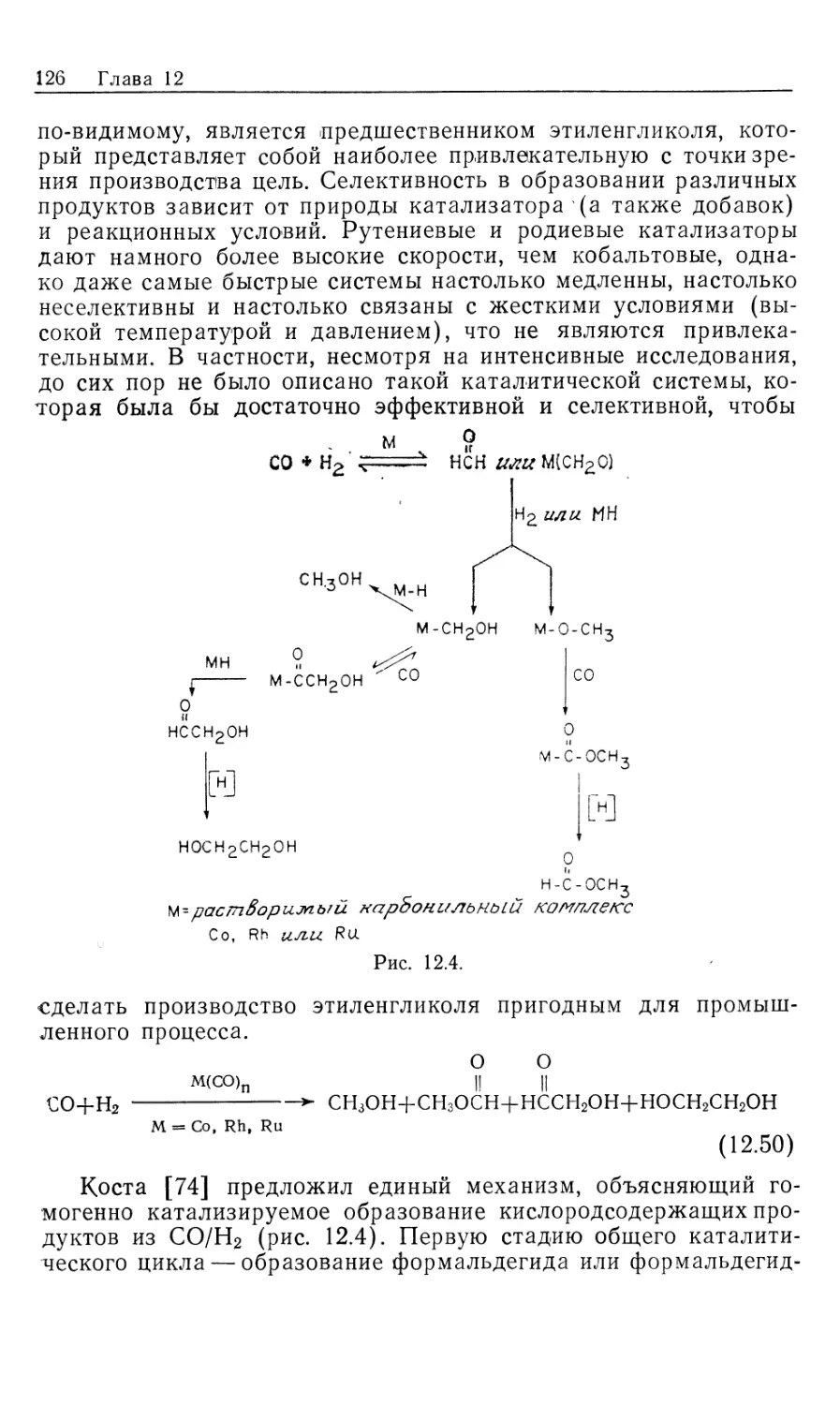
гомогенных катализаторов гидрирования [1], однако лишь немногие
нашли широкое .применение в органическом синтезе. Для
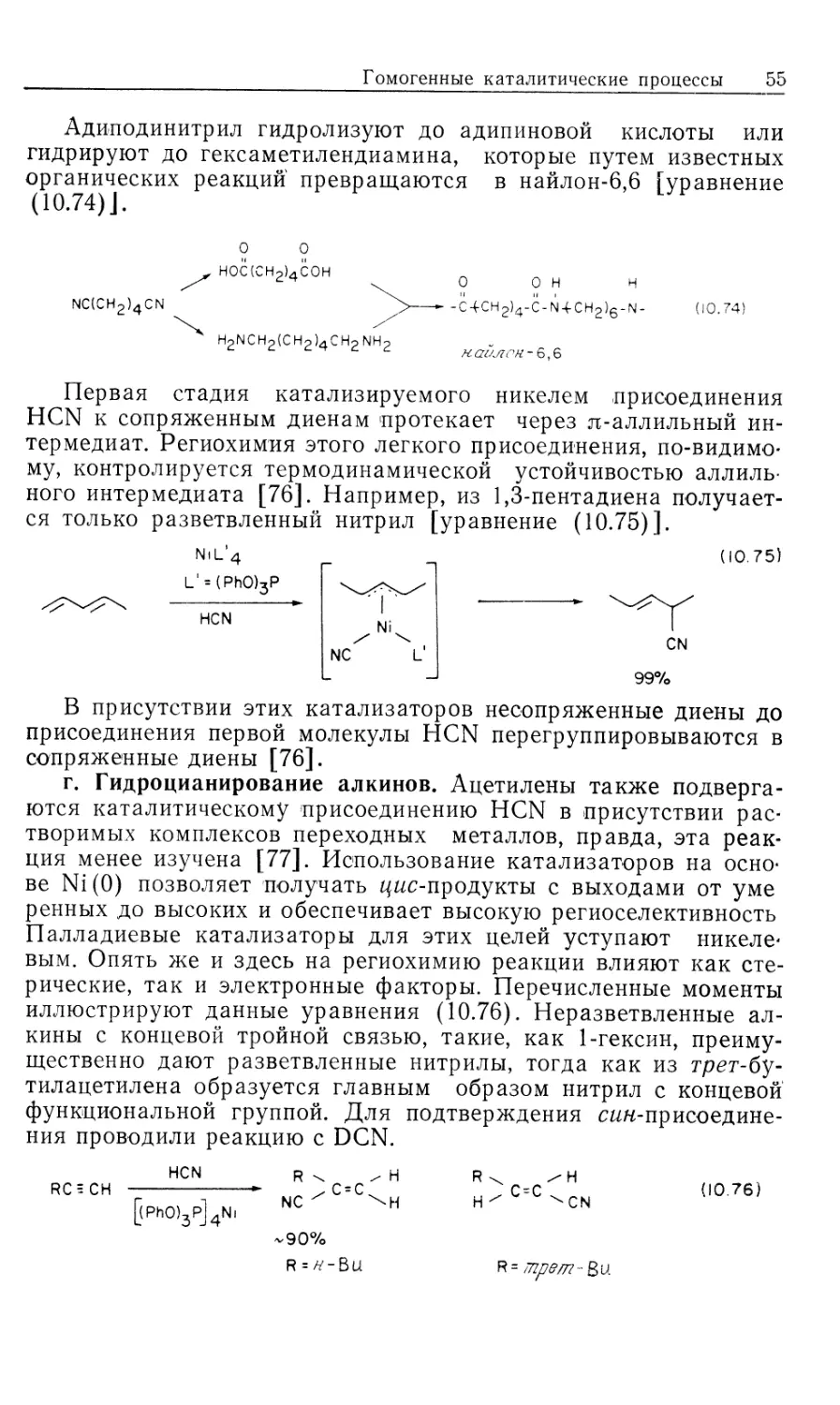
некоторых гомогенных катализаторов источником водорода
необязательно служит Н2. Такие катализаторы «переноса водорода»
образуют нужные гидриды металлов в качестве интермедиатов
при взаимодействии со спиртами, другими гидридами типа
NaBH4, муравьиной кислотой или при гидролизе карбонилсо-
держащих координационных соединений (в процессе конверсии
водяного газа, который обсуждался в ч. 1, гл. 6 и будет
рассмотрен в гл. 12). Поскольку такие катализаторы по своим
свойствам сходны с гомогенными катализаторами
гидрирования, они также рассматриваются в настоящей главе. Сюда же
включены процессы каталитического гидросилилирования и
гидроцианирования, концептуально подобные гидрированию.
10.2. Общее представление о гомогенном каталитическом
гидрировании олефинов
Многие растворимые комплексы переходных металлов
служат «предкатализаторами» в процессах гидрирования олефинов
и других ненасыщенных субстратов. Термин «предкатализатор»
или «предшественник катализатора» будет использоваться по
Глава 10
отношению к веществу, как правило, представляющему
известный комплекс, которое добавляют в колбу, чтобы вызвать
реакцию. «Предкатализатор» взаимодействует с водородом и (или)
субстратом, давая истинный катализатор, который можно
рассматривать как первый идентифицируемый комплекс,
непосредственно участвующий в каталитическом цикле. Например, один
или несколько лигандов могут диссоциировать или удаляться
из предкатализатора при гидрогенолизе. Лишь в немногих
случаях известна природа истинного катализатора.
Такая отличительная особенность гомогенных катализаторов
гидрирования, как селективность по отношению к субстрату,
определяет их преимущественное использование для многих
процессов, в частности для исключительно эффективного
асимметрического гидрирования прохиральных олефинов. К
основным недостаткам гомогенных катализаторов гидрирования
относятся чувствительность к примесям (особенно к следам
кислорода), способность вызывать перегруппировки олефинов,
трудность отделения продукта от катализатора (что составляет
главную проблему для всех без исключения гомогенных
катализаторов), а также то обстоятельство, что эти ценные
катализаторы редко удается регенерировать после завершения
реакции.
Большинство гомогенных катализаторов гидрирования
представляют собой координационно ненасыщенные соединения.
Вакантные координационные центры важны для комплексообра-
зования с субстратом и окислительного присоединения Н2.
Для синтеза соединений с вакантными координационными
центрами и, следовательно, для получения активных
катализаторов существует несколько стратегических путей. Так,
латентные координационные центры могут быть заняты слабоко-
ординированными лигандами, например в Сг (CO)3(CH3CN)3 и
[Rh(PR3)2(Me0H)2_|+. Фотолабильные лиганды можно
обработать в условиях фотохимического катализа, например, Сг(СО)б
и Fe(CO)5- Некоторые лиганды необратимо разрушаются при
гидрировании, например олефины или аллилы. Лиганды,
связанные с координационным центром не через один, а через
несколько атомов, например т]3-аллилы или орто-металлированные
группы, могут обнажать его в процессе обратимого раскрытия
хелатного кольца. Комплексы с пониженным координационным
числом склонны к ассоциации с образованием каталитически
неактивных форм. Присутствие немостиковых лабильных
лигандов, таких, как МеОН, или объемных лигандов типа Су3Р
создает защиту от подобной агрегации. В настоящей главе будут
приведены примеры упомянутых явлений.
Теперь перейдем к механизмам действия гомогенных
катализаторов в процессах гидрирования олефинов-. Эти многоста-
Гомогенные каталитические процессы
дийные процессы иллюстрируют применение основных классов
реакций, рассмотренных в первой части книги: замещение ли-
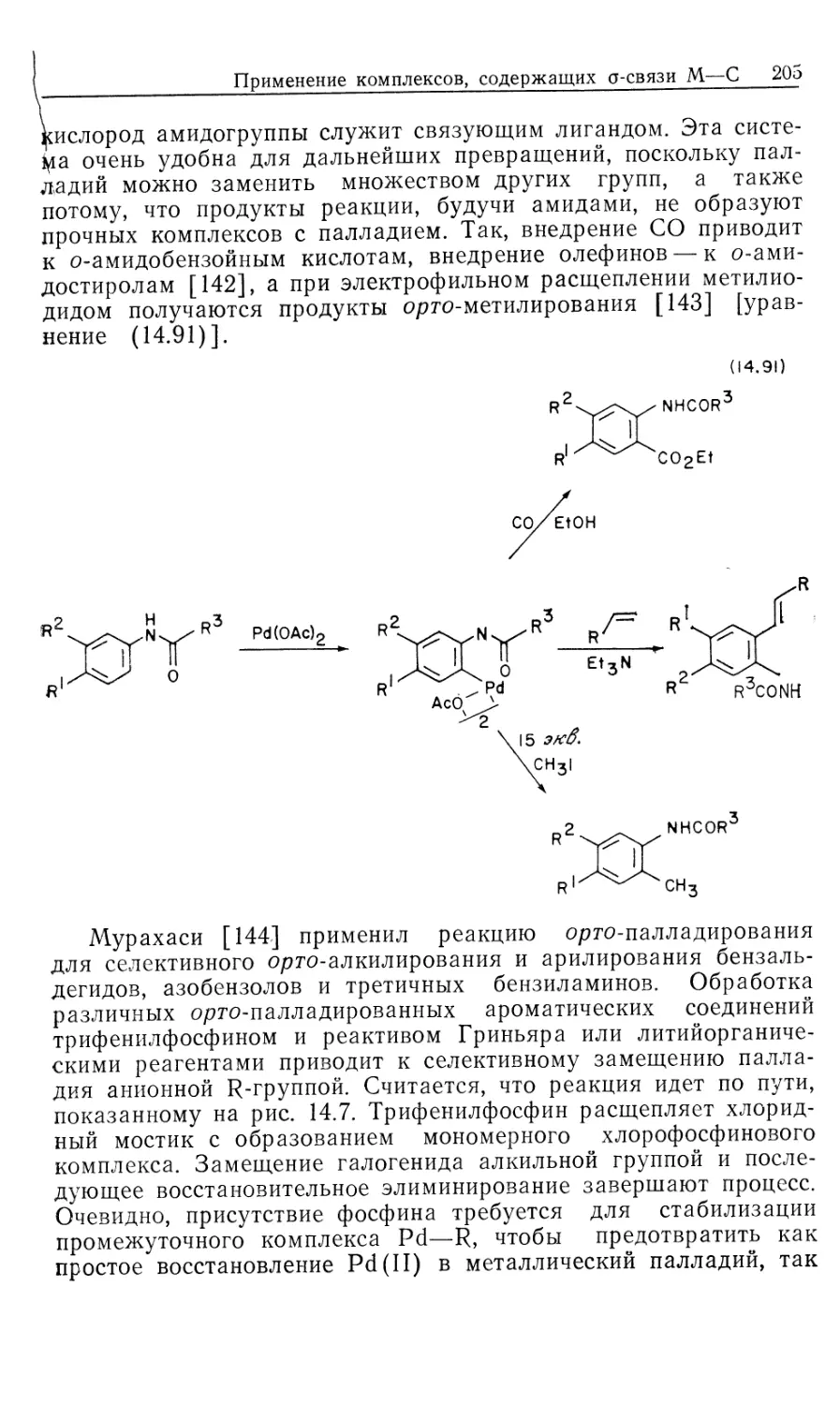
ганда, окислительное присоединение, восстановительное
элиминирование, миграционное внедрение.
10.3. Механизмы гидрирования олефинов в условиях
гомогенного катализа
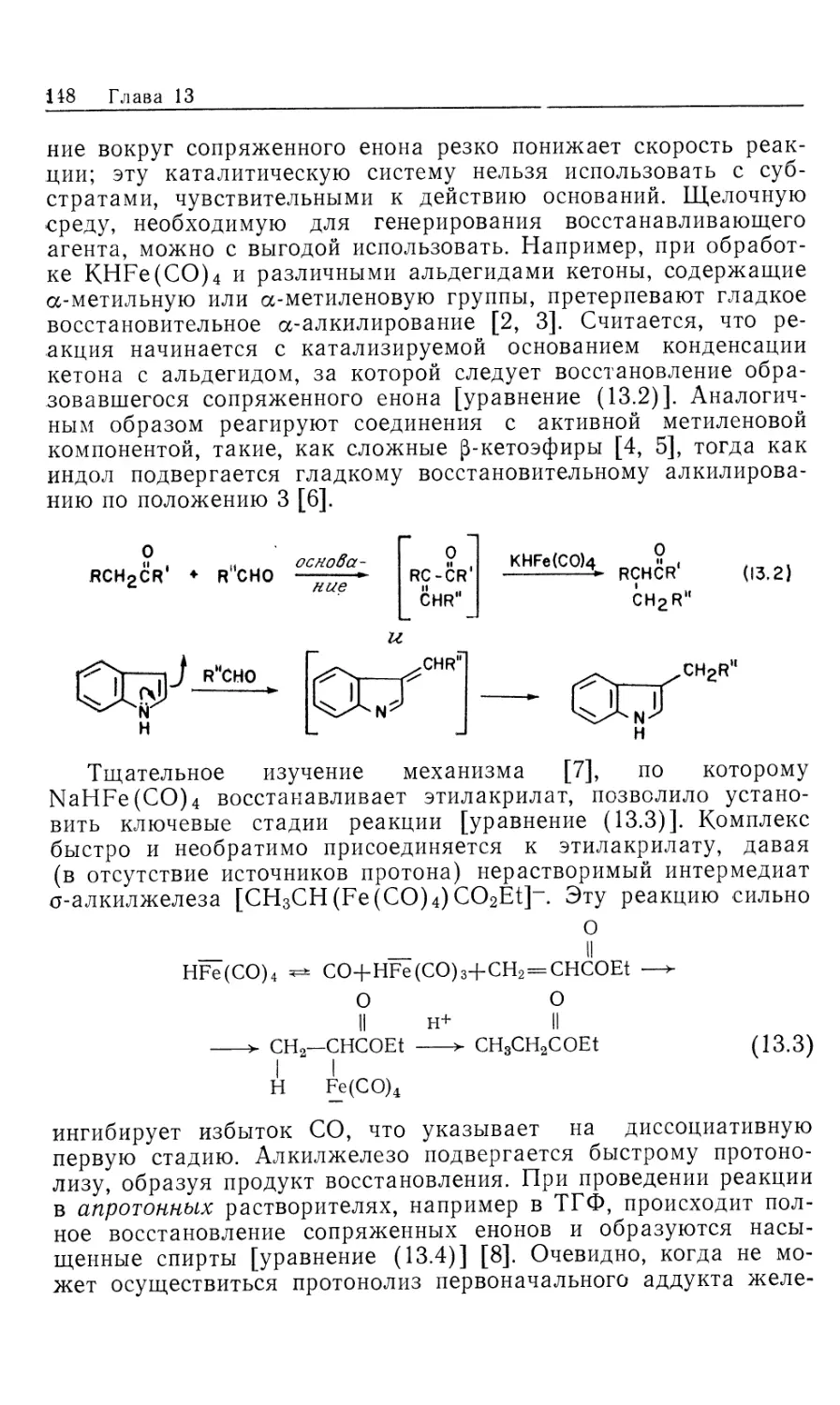
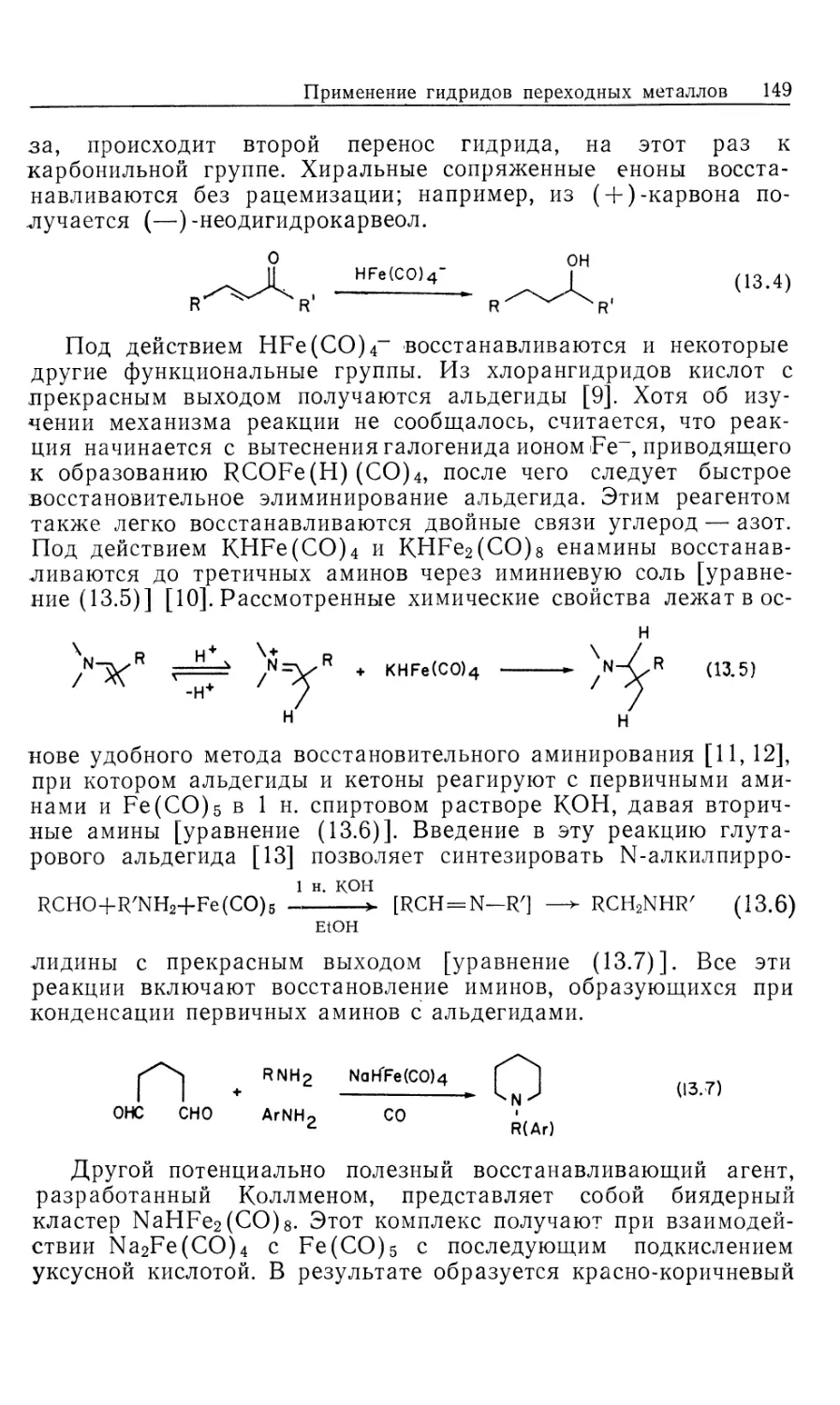
а. Основные представления. Установить механизм любой
реакции чрезвычайно сложно, особенно если эта реакция
представляет собой многостадийный каталитический процесс. Даже
если механизм считается установленным, это всего лишь
гипотеза, которая согласуется с фактами и, в частности, может
количественно предсказать наблюдаемое кинетическое поведение
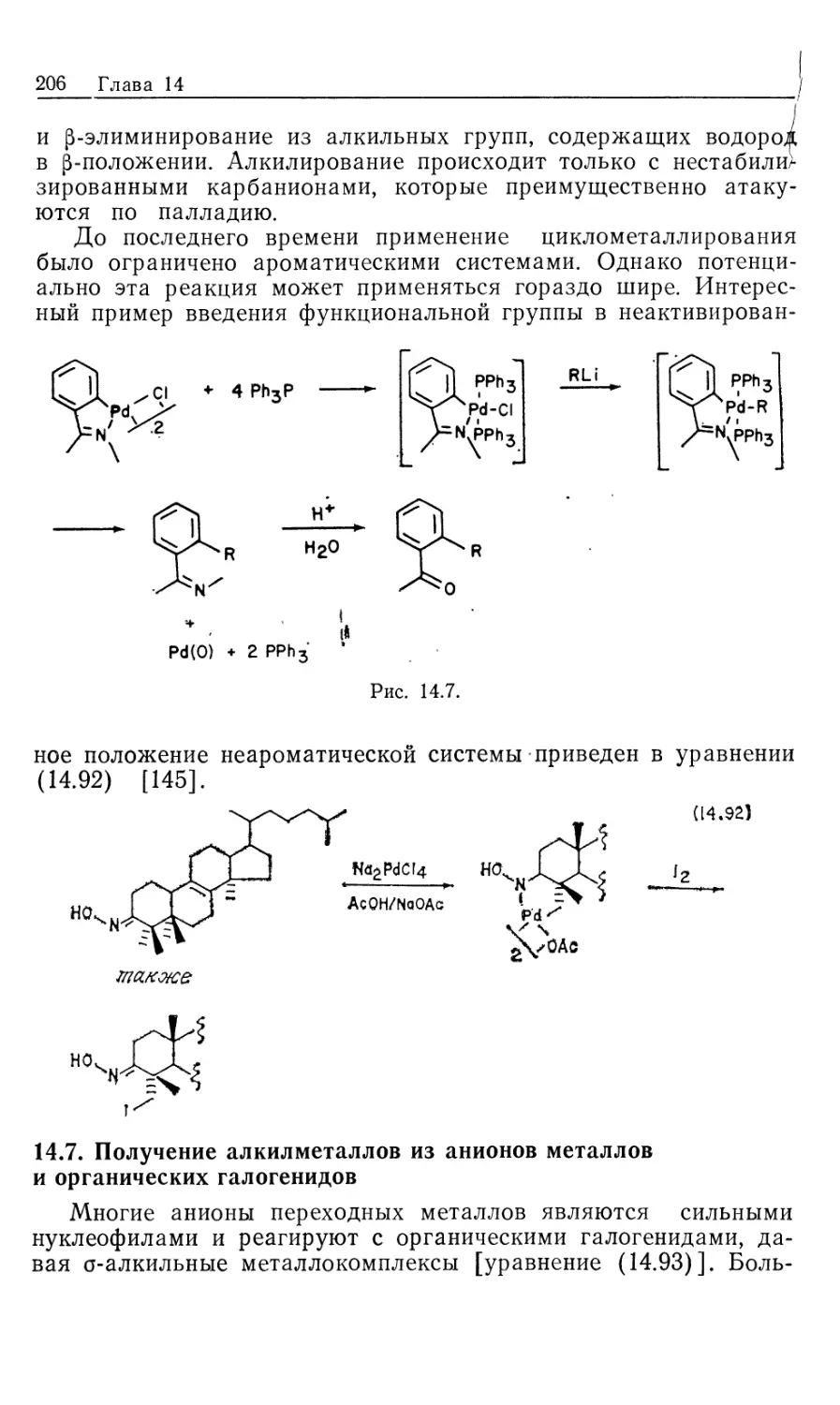
системы в определенной области переменных. Поэтому
неудивительно, что лишь для очень немногих случаев механизм
гомогенного каталитического гидрирования можно называть
«известным». В остальных случаях можно только предполагать
наличие вероятных стадий, согласующихся с определенными
экспериментальными данными.
Из проведенного Халперном [2] исчерпывающего
исследования механизма катализируемого родием гомогенного
гидрирования олефинов следуют два важных и полезных вывода. Во-
первых, те частицы, которые накапливаются в ходе
каталитической реакции в концентрации, достаточной для того, чтобы
идентифицировать их спектроскопически, зачастую не участву-
юг непосредственно в катализе. В действительности эти
непродуктивные комплексы тормозят каталитическую реакцию,
связывая часть катализатора и переводя его в неактивную форму.
Только установив связь между определенными комплексами и
кинетическими характеристиками каталитической реакции,
можно быть уверенными, что данный комплекс действительно
участвует в каталитическом процессе.
И второй вывод. Для того чтобы установить механизм
многостадийной каталитической реакции, необходимо независимо
вывести уравнения скорости индивидуальных стадий. Если
собраны параметры скорости и равновесия для отдельных стадий
и показано, что они количественно объясняют каталитический
процесс в целом, то можно говорить о том, что механизм
«известен». Недостаточно просто изучить влияние нескольких
переменных на общую кинетику .многостадийной каталитической
реакции, так как получаемые при этом зависимости могут ввести
в заблуждение. Внимательный читатель химической литературы
должен заметить, что приведенные выводы часто игнорируются.
Имеющиеся данные по механизмам гомогенного
каталитического гидрирования указывают, что перенос водорода к суб-
8 Глава 10 *
страту осуществляется промежуточно образующимися
гидридами металлов в дискретных стадиях, обычно в реакциях
внедрения и восстановительного элиминирования. Таким образом,
образование гидридов металлов из молекулярного водорода
является обязательной стадией в процессе гомогенного
каталитического гидрирования.
Известны три главных способа активации водорода:
окислительное присоединение [уравнение (10.1) J, гидрогенолиз
[уравнение (10.2)] и гетеролитическое расщепление водорода
[уравнение (10.3)] [За].
(10.2)
Все три реакции обсуждались в разд. 5.3, а [см. ч. 1,
уравнения (5.9) и (5.11)]. Для всех этих реакций требуется наличие
вакантного координационного центра («центра
ненасыщенности»), поэтому для всех трех реакций характерна
подверженность «отравлению» внешними лигандами типа СО. Чаще всего
встречается такой путь активации водорода, как окислительное
присоединение; напомним, что оно требует изменения
формальной степени окисления.
И гидрогенолиз [уравнение (10.2)], и гетеролитическое
расщепление водорода [уравнение (10.3)] могут включать в
качестве промежуточной стадии окислительное присоединение.
Например, окислительное присоединение с последующим
восстановительным элиминированием в целом приводит к гидрогеноли-
зу, но интермедиат 1 может оказаться недетектируемым
[уравнение (10.4)]
LnMX
X
LnM-H
- Н J
1
LnM-H + НХ (Ю.4)
Следует отметить, что X может даже быть атомом другого
металла, как это, по-видимому, имеет место в тех случаях,
когда катализатором гидрирования служит Со2(СО)8 [см. ч. 1,
уравнение (5.17) и обсуждение в разд. 5.3, в]. Аналогичным
образом, гетеролитическое расщепление водорода также может
включать предварительную стадию окислительного присоедине-
Гомогенные каталитические процессы
ния и идти через образование интермедиата 2, который
невозможно наблюдать [уравнение (10.5)J.
X
Н-В (10.5)
dn'2
Гетеролитическое расщепление может происходить в одну
стадию, без окисления металла. Это справедливо для тех
случаев, когда требующаяся в результате предварительного
окислительного присоединения более высокая степень окисления
недостижима. Так, лантаноорганические соединения общей
формулы (Ср2*МН)2, где Ср* = т15-(СНз)5С5, M = La, Nd, Sm, Lu,—
очень активные катализаторы гидрирования (частота
каталитических циклов до 30 с"1) [36]. Поскольку в этом случае
окислительное (присоединение энергетически невозможно, должно
иметь место гетеролитическое расщепление.
Упомянутые выше три способа активации водорода дают
логическую основу для деления гомогенных катализаторов
гидрирования на две главные категории:
1) гомогенные катализаторы, образующие моногидрид
(М—Н) в одной из стадий каталитического цикла;
2) гомогенные катализаторы, образующие дигидрид (МН2)
за счет присоединения Н2 в одной из стадий каталитического
цикла.
Эти два класса гомогенных катализаторов проявляют
некоторые различия в селективности и в механизмах реакции.
* б. Катализаторы гидрирования, образующие моногидриды.
Несмотря на то что детальный механизм действия моногидрид-
ных катализаторов ни в одном случае не установлен, очевидно,
что имеются две ключевые стадии: а) образование алкилметал-
ла 'в реакции моногидрида с олефиновым субстратом и б)
образование алкана в качестве продукта и регенерация
моногидрида. Рассмотрим возможные механизмы каждой из этих
ключевых стадий.
Образование алкилметалла в реакции гидрида с олефином
в принципе может происходить двумя путями: классическое
«миграционное внедрение» [уравнение (10.6)] и прямая
реакция, без предварительной координации олефина [уравнение
(10.7)]
\ / ||
М-Н N M-Vl ч ч М^ ^Н (10.6)
М-Н + С = С - м-С-С-Н (10.7)'
' \ / \
10 Глава 10
Миграционное внедрение [уравнение (10.6)] подробно
обсуждается в ч. 1, разд. 6.3, а. Напомним некоторые главные
особенности этой реакции, приобретающей особую важность для
понимания процесса гидрирования моногидридом. Прежде чем
произойдет внедрение, олефин должен образовать
координационную связь, а для этого необходимо наличие вакантного
координационного центра на гидриде металла. Конкурирующие
лиганды, блокируя этот центр, могут «отравить» катализатор.
Селективность по отношению к различным олефинам зависит
от их способности образовывать связь с металлом. Так, олефи-
ны с концевой двойной связью должны быть более реакцион-
носпособны, чем олефины с внутренней связью, а цис-олефины
более реакционноспособны, чем транс-изомеры.
Стадия миграционного внедрения также требует
^^-присоединения М—Н к олефину. Поскольку эта стадия обратима
[уравнение (10.6); часто встречающийся процесс, известный как
«^-элиминирование гидрида»], возможна перегруппировка оле-
фина, а также изотопный обмен между дейтерием в М—D и
атомами водорода в олефине. В особенности это характерно
для тех случаев, когда последующая стадия гидрогенолиза
относительно медленная. Действительно, реакция с участием мо
ногидридных катализаторов часто сопровождается
перегруппировкой олефинов и изотопным обменом.
Альтернативный путь, «прямое внедрение» [уравнение
(10.7)], не требует предварительной координации олефина,
поэтому конкурентное ингибирование лигандами здесь не имеет
места. Такого рода прямой процесс чаще встречается в случае
сопряженных олефинов, например 1,3-диенов, и стирола. Далее
в этой главе будет приведен пример прямого внедрения для
полициклических субстратов. Как правило, оно происходит по
радикальному механизму [см. уравнения (10.8) и (10.9)].
Повышенная реакционная способность сопряженных диенов
объясняется устойчивостью образующегося сопряженного
радикала [уравнение (10.8)]. Радикальное присоединение атома
водорода хорошо изучено на примере гидрирования а-метилсти-
рола под действием НМп(СО)б. Для установления радикального
механизма в этом случае Халперн [4] использовал химически
индуцированную динамическую поляризацию ядер (ХПЯ).
LnM—H+PhCH = CH2 —^ PhCH—сНз+LnM. (10.8)
PhCH—СНз+LnM. —+ PhCH—СН3 (10.9)
MLn
Вторая ключевая стадия каталитического гидрирования
олефина моногидридными комплексами — стадия образования
продукта — заключается в гидрогенолизе алкилметалла [уравне-
Гомогенные каталитические процессы 11
ние (10.10)]. Она может (происходить через биядерное
восстановительное элиминирование [уравнение (10.11)J,
обсуждавшееся в ч. 1, разд. 5.7.
M-R+H2 -ни м-H+R-H (10.10)
М—R+M'—Н -н- M--M'+R—H (10.11)
Механизм подобных реакций гидрогенолиза неясен. В
некоторых случаях действительно происходит окислительное
присоединение с последующим восстановительным элиминированием
[уравнение (10.12)]. При этом во избежание образования
20-электронного интермедиата необходимо наличие вакантного
координационного центра (легко генерируют вакантные
координационные центры, облегчающие протекание стадии
гидрогенолиза, г)3-аллильные комплексы). Альтернативная стадия
образования продукта — биядерное восстановительное
элиминирование [уравнение (10.11)]—также требует наличия вакантного
координационного центра на алкилметалле.
Н
| быстро
М—R+H2 ч=* Н—М—R ^Н—M+R—Н (10.12)
Рассмотрим для примера два моногидридных катализатора.
Уравнение (10.13) показывает селективное гидрирование
олефинов с концевой двойной связью, катализируемое
координационно насыщенным гидридом родия(I) 3 в мягких условиях.
В присутствии этого катализатора обнаруживается выраженная
селективность субстрата, что подтверждается отсутствием
реакции в случае попытки восстановления циклогексена [5].
25 °С, <1 атм, Н2 точ
RCH-=CH2 + Н2 ► RCH2CH3 (10.13)
HRh(CO) (РРЬз)з
3
Олефины с внутренней двойной связью не подвергаются
конкурентному восстановлению, а изомеризуются. Такие
функциональные группы, как RCHO, RCN, RC1, RCO2R/, не
восстанавливаются. Восстановление олефинов ингибируется при
добавлении PPh3. Этот факт указывает на то, что по крайней мере в
одной из стадий каталитического цикла необходим центр
ненасыщенности. В гл. 12 будет показано, что тот же комплекс 3
служит предкатализатором для «оксо»-реакции. Известно, что
оксо-катализаторы обычно относятся к катализаторам типа
моногидридов для гидрирования олефинов. Действительно,
гидрирование олефинов — это конкурирующая побочная реакция в
оксо-процессе, которая становится весьма значительной в
случае сопряженных диенов. Как будет показано далее,
оксо-катализаторы обычно промотируют гидрирование альдегидов до
12 Глава 10
спиртов, но для этого требуются повышенные давления и
температура, очевидно, из-за того, что альдегидная группа —
относительно неактивный субстрат.
Другой моногидридный катализатор гидрирования олефи-
нов, комплекс рутения 4, представляет собой редкий пример
ненасыщенного пятикоординационного с!6-комплекса.
По-видимому, три объемные фосфиновые группы затрудняют координацию
с другим крупным лигандом. Этот катализатор проявляет
высокую селективность по отношению к субстрату, восстанавливая
олефины с концевой двойной связью в 1000 раз быстрее, чем
олефины с внутренней связью [6] [уравнение (10.14)]. Скорость
восстано!вления различных олефинов в присутствии комплекса 4
соответствует их сродству к иону серебра; видимо, в уравнение
скорости входит координация олефина. Комплекс рутения 4
катализирует также обмен между D2 и протонами в орго-положе-
ниях трифенилфосфиновых лигандов. Последняя реакция
служит еще одним примером «ортометаллирования»,
рассматривавшегося в ч. 1, разд. 5.4, б. Эта побочная реакция может
осложнить введение изотопной метки.
RCH=CH2 + Н2 >- RCH2CH3 (10.14)
HRtCl(PPh3)3
4
в. Катализаторы гидрирования, образующие дигидриды.
Гомогенные катализаторы гидрирования этого класса наиболее
хорошо изучены и чаще всего используются. Сначала будут
обсуждены механизмы действия двух главных катализаторов этого
типа и проанализировано успешное асимметрическое
гидрирование прохиральных олефинов. Затем будут рассмотрены главные
особенности и области применения дигидридных катализаторов.
Основные характеристики катализатора Уилкинсона.
В 1964 г. Уилкинсон и Коффи независимо и почти
одновременно открыли замечательные свойства гомогенного катализатора
гидрирования олефинов RhCl(PPh3)3 (5), получившего
известность как «катализатор Уилкинсона». Этот катализатор широко
используется в органической химии благодаря своей
надежности, селективности, эффективности, высокой скорости и
большому числу каталитических циклов. ]Неханизм гидрирования
олефинов изучен Халперном [2а, 26], но сначала мы рассмотрим
синтез, селективность и условия реакции этого важного
катализатора.
Известно два метода получения катализатора Уилкинсона 5.
Первый метод весьма прост, но применим только к трифенил-
фосфину и некоторым другим триарилфосфинам [уравнение
(10.15)J. В результате восстановления родия(III) одним молем
трифенилфосфина наряду с целевым продуктом 5, представля-
Гомогенные каталитические процессы 13
ющим собой темно-красное кристаллическое вещество,
умеренно чувствительное к кислороду, образуется фосфиноксид. Хлор
в молекуле 5 можно заменить другим лигандом, бромом или
иодом по реакции типа SN2, характерной для четырехкоордини-
рованных с!8-комплексов (ч. 1, разд. 4,4а).
сн3сн2он
RhCl3(H2O)x + PPh3 >■ RhCl(PPh3)3+Ph3PO (10.15)
5
Другой метод получения комплекса 5 и родственных
производных более гибкий [уравнения (10.16), (10.17)] и состоит из
двух стадий. Сначала готовят умеренно растворимый бисэтиле-
новый комплекс 6 с помощью окислительно-восстановительной
реакции [уравнение (10.16)], аналогичной синтезу ацетальдеги-
да в присутствии палладиевого катализатора и воды в Вакер-
процессе (см. ч. 1, гл. 6 и 7, и гл. 17). Используя вместо
этилена циклооктен, можно получить более растворимый аналог 6.
Во второй стадии этилен или циклооктен замещают трифенил-
фосфином или другим третичным фосфином (L/) и получают
аналоги катализатора Уилкинсона. В этом методе соотношение
фосфина и родия может варьироваться; таким образом, можно
генерировать in situ катализаторы гидрирования, обладающие
различной степенью реакционной способности.
МеОН
RhCI3(H2O)x + СН2=СН2 I ^Rh ^ I + СН3СНО (10.16)
Несопряженные олефины и ацетилены в присутствии
катализатора Уилкинсона 5 подвергаются быстрому гидрированию при
комнатной температуре и обычном давлении водорода в
бензоле с полярным сорастворителем, например этанолом.
6 ?-+• IARhCl (L'=PPh3 5 или R'R2P) (10.17)
Полярные сорастворители могут облегчать миграционное
внедрение, являющееся лимитирующей стадией. Скорости
гидрирования моноеновых субстратов могут отличаться в 50 раз. Такую
разницу можно было бы объяснить стерическими эффектами,
влияющими на параметры связи в олефинах. Однако
некоторые олефины с сильной связью, например этилен и 1,3-бутади-
ен, при нормальных условиях восстанавливаются очень
медленно. Такие олефины действуют как мощные конкурирующие
ингибиторы простых моноенов. Как будет ясно из приведенного
ниже механизма, эти ингибиторы закрывают путь, начинаю-
14 Глава 10
щийся с присоединения водорода, и таким образом меняют
механизм реакции.
Другая особенность катализатора Уилкинсона, которую
необходимо учитывать для понимания механизма его действия,
состоит в отсутствии изотопного обмена между Н2 и D2 и
между протонами в растворителе или в олефиновом субстрате [1в].
В тщательно контролируемых условиях можно осуществить
чрезвычайно чистое ^^-присоединение водорода (или
дейтерия). Эта особенность становится понятной, если сравнить
относительные скорости ключевых стадий процесса.
Механизм гидрирования в присутствии катализатора
Уилкинсона. Проведенный Халперном [2а, 26] постадийный анализ
механизма действия катализатора Уилкинсона в процессе
гидрирования олефинов дает основу для понимания большинства
его характерных особенностей, упомянутых выше. Общая
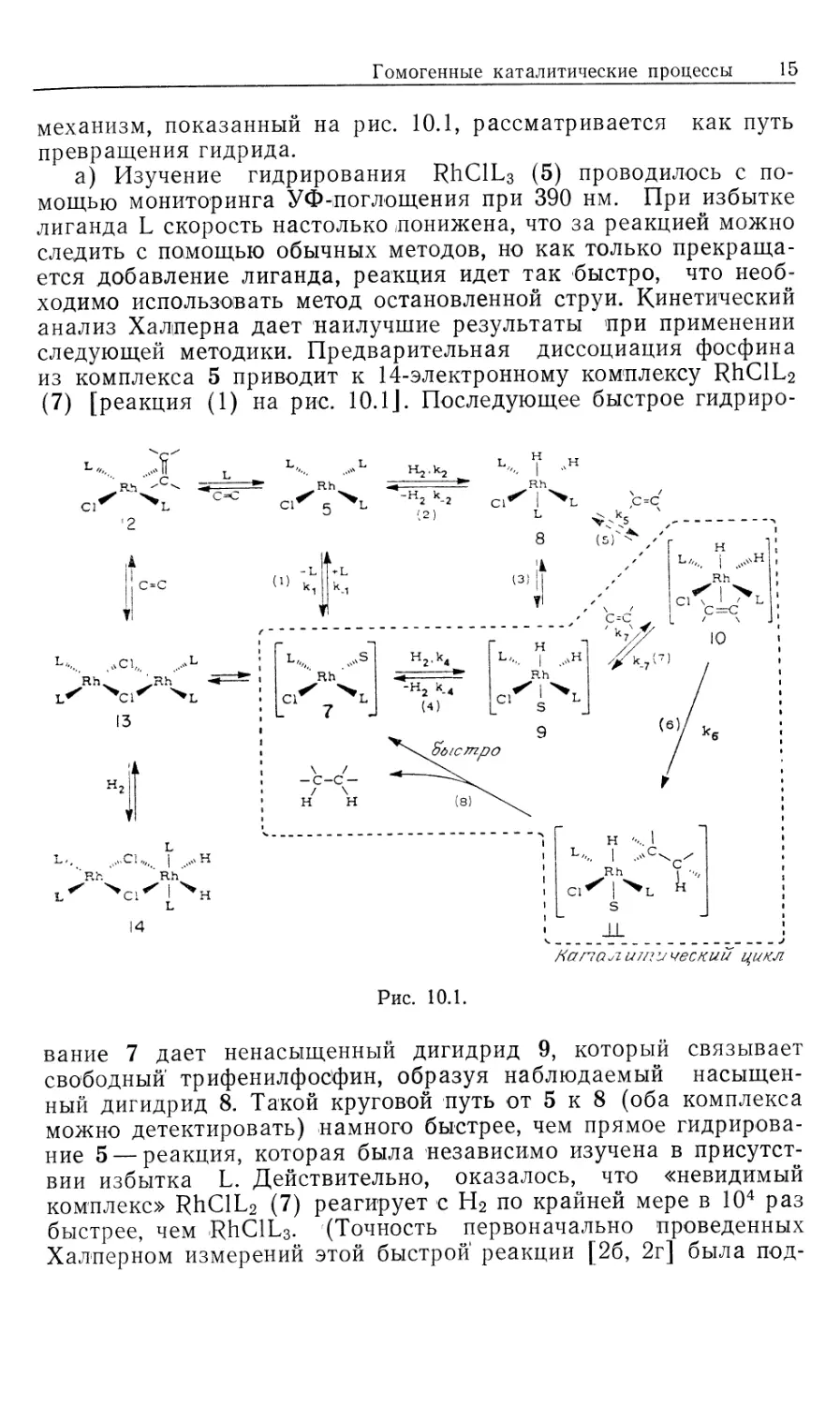
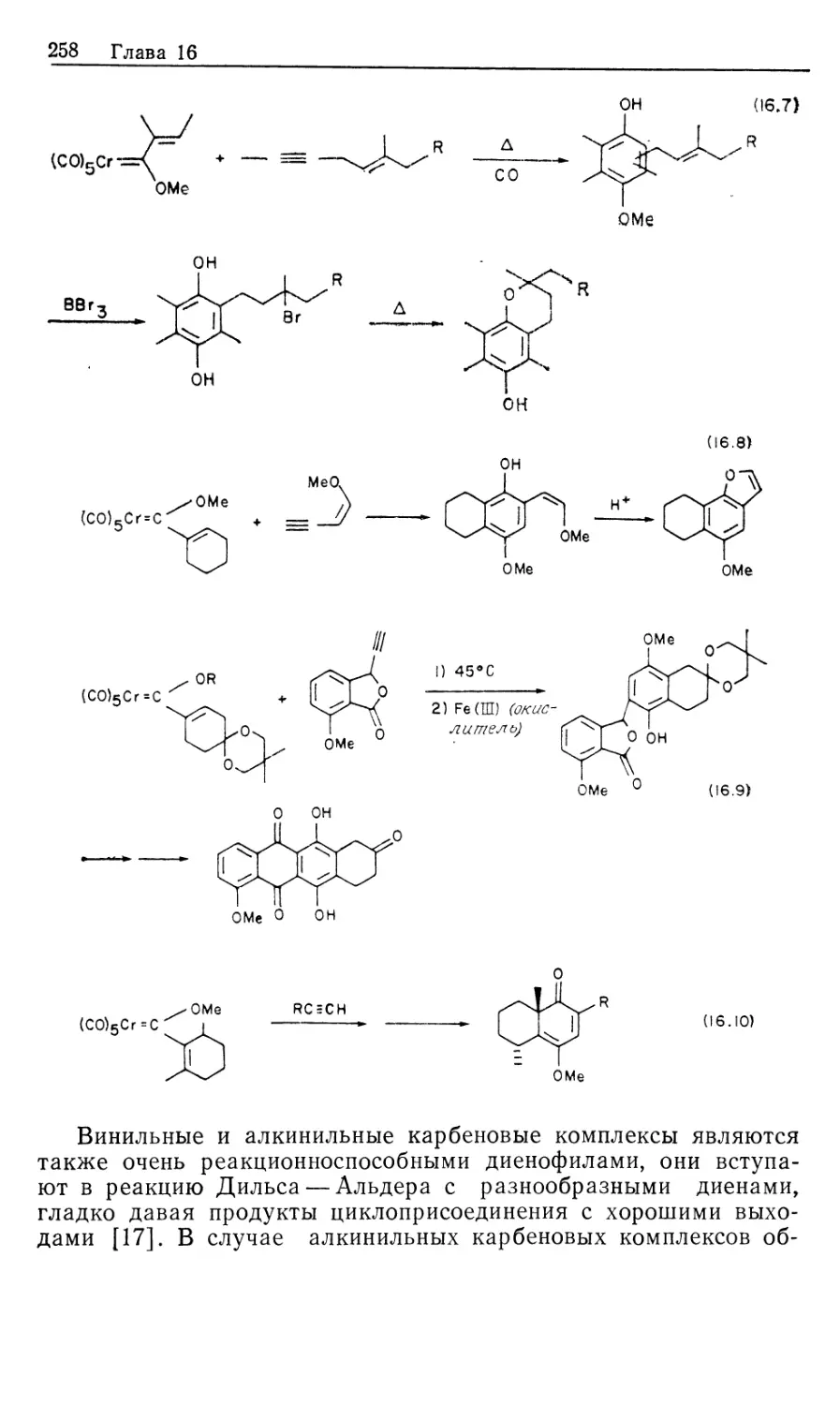
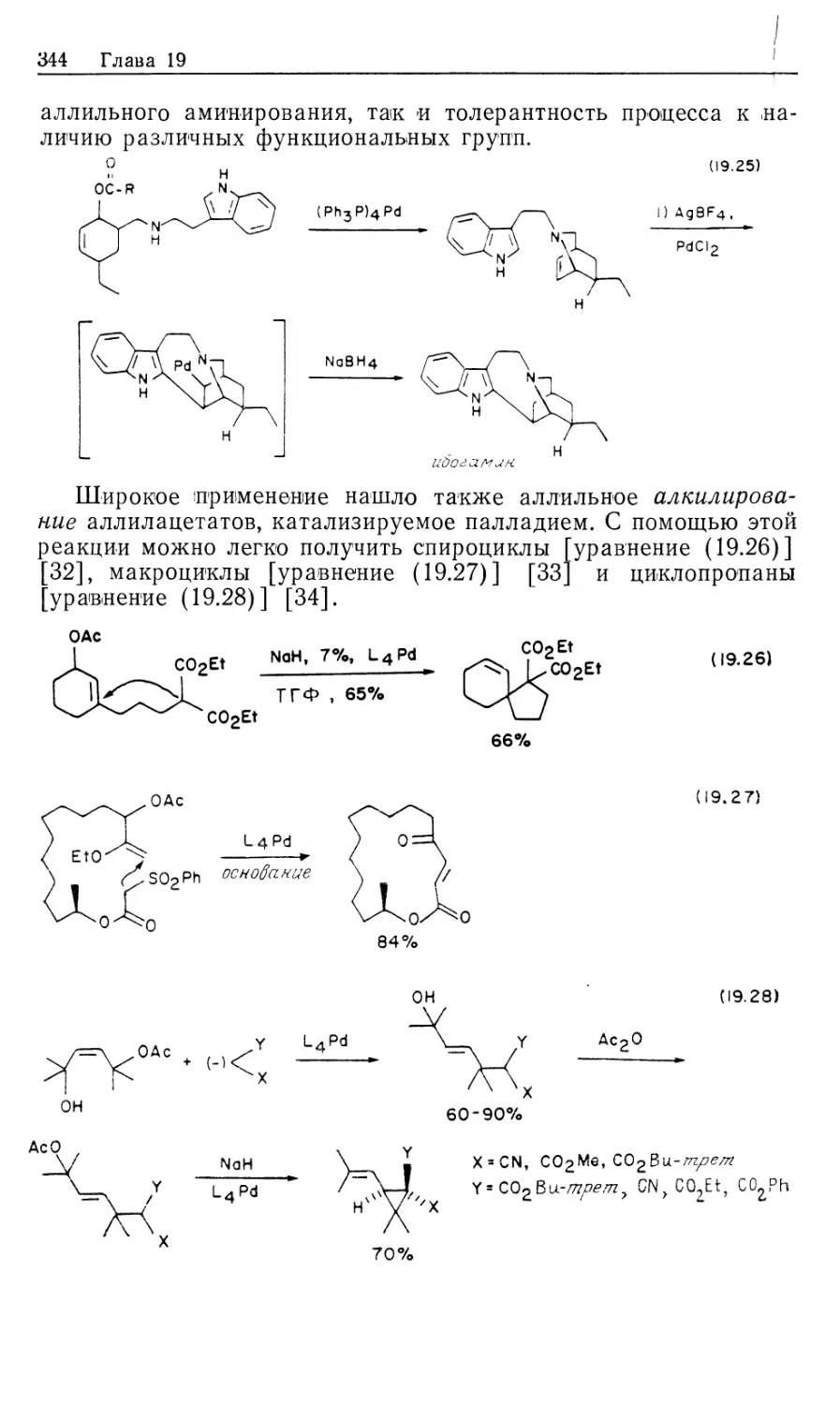
схема представлена на рис. 10.1.
Предлагаемый механизм был установлен в результате
отдельного изучения каждой стадии и вывода для нее уравнения
скорости. Сопоставление полученных результатов позволило
количественно объяснить кинетические закономерности общей
каталитической реакции в интервале определенных условий
эксперимента. На рис. 10.1 пунктиром обведены предполагаемые
промежуточные формы, они заключены в квадратные скобки
(7, 9, 10, 11); остальные соединения были либо выделены,
либо детектированы в растворе. Интересно, что из кинетических
данных следует, что именно соединения 7, 9, 10 и 11 являются
истинными комплексами, участвующими в каталитической
реакции.
Как отмечалось ранее, из этого результата следует важный и
практически полезный вывод: идентификация доминирующего
или детектируемого соединения в каталитической системе
может привести к неверной интерпретации механизма реакции.
Только в том случае, когда кинетическими и
термодинамическими измерениями определены роль комплексов и истинный путь
реакции, можно говорить о том, что механизм установлен.
Скорость каталитической реакции, представленной на рис. 10.1,
уменьшается с ростом числа идентифицируемых комплексов,
изображенных вне пунктирной линии.
Суммарную реакцию можно разделить на две части: а)
гидрирование 'RhClL3 и б) реакция RhH2ClL3 с олефином.
Поскольку реакция происходит в такой последовательности, общий
Гомогенные каталитические процессы
15
механизм, показанный на рис. 10.1, рассматривается как путь
превращения гидрида.
а) Изучение гидрирования RhClL3 (5) проводилось с
помощью мониторинга УФ-логлощения при 390 нм. При избытке
лиганда L скорость настолько лонижена, что за реакцией можно
следить с помощью обычных методов, но как только
прекращается добавление лиганда, реакция идет так быстро, что
необходимо использо(вать метод остановленной струи. Кинетический
анализ Хадперна дает наилучшие результаты при применении
следующей методики. Предварительная диссоциация фосфина
из комплекса 5 приводит к 14-электронному комплексу RhClL2
(7) [реакция (1) на рис. 10.1J. Последующее быстрое гидриро-
14
Каталитический цикл
Рис. 10.1.
вание 7 дает ненасыщенный дигидрид 9, который связывает
свободный трифенилфосфин, образуя наблюдаемый
насыщенный дигидрид 8. Такой круговой путь от 5 к 8 (оба комплекса
можно детектировать) намного быстрее, чем прямое
гидрирование 5 — реакция, которая была независимо изучена в
присутствии избытка L. Действительно, оказалось, что «невидимый
комплекс» RhClL2 (7) реагирует с Н2 по крайней мере в 104 раз
быстрее, чем RhClL3. (Точность первоначально проведенных
Халперном измерений этой быстрой реакции [26, 2г] была под-
16 Глава 10
тверждена прямым измерением скорости присоединения Н2 к
комплексу 7, генерируемому флеш-фотолизом [85].) Стадия
присоединения Н2 обратима; это следует из того факта, что в
ходе каталитического гидрирования олефина пара-обогащенный
Н2 оказывается в состоянии равновесия с орто —
пара-равновесной смесью. Это было показано с помощью рамановской
спектроскопии [7]. Как будет показано в следующем разделе, в
случае катионных катализаторов, когда окислительное
присоединение Н2 является лимитирующей стадией, за которой следуют
быстрые необратимые стадии, взаимопревращение орто — пара-
водорода отсутствует. Уилкинсон [8] первый предложил тот
же круговой путь гидрирования, катализируемый RhClL3
[реакции (1), (3) и (4) на рис. 10.1J, однако он исходил из
ошибочного допущения, что диссоциативное равновесие K = k\lk-\
сильно сдвинуто в сторону 14-электронного комплекса 7. Более
поздние измерения показали, что в действительности это
равновесие сдвинуто влево (/Ci = 1 -10~5 М). Такое расхождение
возникает из-за чувствительности RhClL3 к действию воздуха:
комплекс реагирует с кислородом, давая диссоциированный
Ph3PO и показывая заниженную величину молекулярного веса.
Для чувствительных к воздуху металлоорганических
соединений это серьезная проблема — измерения молекулярного веса
зачастую очень ненадежны.
«Невидимый» 14-электронный комплекс RhClL2 проявляет
выраженную тенденцию к димеризации (7—>\Ъ, рис. 10.1,
Дравн^Ю6 М"1). Гидрированный димер 14, охарактеризованный
Толменом и сотр. [9], не участвует прямо в каталитическом
гидрировании олефина. В присутствии Н2 равновесная реакция с
участием комплексов 13 и 14 незначительна, так как водород
реагирует с трехкоординированным комплексом 7,
предотвращая его димеризацию.
В отсутствие Н2 образуются олефиновые комплексы типа
12. В случае олефинов с умеренной способностью к связыванию,
например циклогексена, побочное образование комплексов типа
12 мало влияет на ход гидрирования олефина. Однако склонные
к жшплексообразованию олефины, например этилен, действуют
как конкурентные ингибиторы дигидридного пути реакции,
давая устойчивые комплексы типа 12.
i6) Кинетическое изучение отдельных реакций дигидрида 8
с циклогексеном, взятым в большом избытке (условия
псевдопервого порядка) позволило вывести уравнение скорости
[уравнение (10.18)]. Рассмотрение превращения 8 и 10 как предрав-
новесия (константа равновесия Къ) и последующего
превращения в 11 как лимитирующей стадии (константа скорости й6)
дает выражение для йнабл в уравнении (10.19) [для
последовательности реакций (3), (7), (6), (8)]. Величины &набл измерены
Гомогенные каталитические процессы 17
в широком интервале концентраций олефина и фосфина.
Уравнение, обратное уравнению (10.19), предсказывает, что
зависимости 1/^набл от отношения [L]/[C = C] должны быть
линейными, с наклоном прямой l/Kske и пересечением l/k6 [уравнение
(10.20)J. Такие линейные графики действительно были
получены, и из них рассчитаны величины Къ и k6. Скорость
миграционного внедрения (k6) самая низкая как в стадии б, так и в
цикле каталитического гидрирования в целом. Последующая
реакция [восстановительное элиминирование алкилгидрида,
реакция (8) на рис. 10.1] достаточно быстрая, так что процесс,
обратный реакции (6), кинетически незначителен. Этим
объясняется отсутствие изотопного обмена между D2 и олефиновым
субстратом.
—d [RhH2ClL3] /dt=kna6jl [RhH2ClL3] (10.18)
(10.20)
Интересный аспект механизма, представленного на рис. 10. Ц
заключается в том, что определяющая скорость стадия
миграционного внедрения [реакция (6)] не обнаруживает
зависимости скорости от концентрации свободного Ph3P. Поскольку
в k& включен разрыв связи Rh—Н, можно ожидать проявления
кинетического изотопного эффекта. Действительно, в реакции
циклогексена с дигидридом 8 наблюдался небольшой изотопный
эффект: /гНабл(Н)/&набл(О) = 1,15.
Центры ненасыщенности в соединениях, участвующих в
каталитическом цикле, могут занимать полярные сорастворители,
как это имеет место в интермедиатах, показанных на рис. 10.1.
Не считая этих случаев, когда возникает некоторая
неопределенность, стехиометрия «невидимых комплексов» основывается
на данных изучения кинетики и равновесия. Однако
относительное расположение объемных фосфиновых групп в комплексах
7, 9,, 10 и 11 неясно. Исходя из данных по спектрам ЯМР с
переносом спиновой плотности и по относительной реакционной
способности родственных комплексов, Браун [10] привел
доказательства в пользу того, что в активных формах 10 и 11 фос-
финогрушпы находятся в цис-положеяяи.
Комбинированные уравнения скорости из разделов (а) и (б)
количественно объясняют наблюдаемую скорость гидрирования
циклогексена. Поскольку реакция миграционного внедрения (k&)
является самой медленной стадией каталитического цикла, в
условиях постоянного давления Н2, высокой концентрации
олефина (~1 М) и без добавления фосфина скорость процесса дости-
2--П29
18 Глава 10
гает предельной величины, определяемой этой медленной
стадией. Суммарное уравнение скорости показывает, что некоторые
преимущества могут быть достигнуты модификацией
катализатора. Например, комплекс RhClL2 (7) можно иммобилизовать
и закрепить на жесткой подложке, что в принципе должно
предотвращать его димеризацию в 13.
Хотя показанный на рис. 10.1 механизм кажется
совершенно понятным, основа его может быть несколько иной в случае
небольших изменений реагентов. Например, если в качестве
лиганда используют стирол (который считается лучшим лиган-
дом, чем циклогексен), то уравнению скорости соответствует
другой, параллельный путь реакции, включающий координацию
двух молекул стирола. Ранние исследования Уилкинсона
показывают, что кинетика реакции существенно меняется при
использовании таких сильносвязывающих субстратов, как этилен
или 1,3-бутадиен; здесь более важным может стать путь,
включающий связывание олефина до присоединения Н2. Кроме
того, существенные изменения природы фосфина (например, при
использовании алкилзамещенных фосфинов) сильно
воздействуют на диссоциативные равновесия и тем самым меняют
механизм гидрирования.
Таким образом, рассматриваемая многостадийная реакция
очень сложна. Основной механизм реакции, подобно хамелеону,
приспосабливается к изменениям природы катализатора, лиган-
дов или субстрата. Тем не менее показанный на рис. 10.1
механизм не содержит каких-либо неожиданностей, а отражает
постадийную комбинацию родоначальных классов реакций,
обсуждавшихся в ч. 1, гл. 4—6.
Механизм гидрирования катионными родиевыми
катализаторами. Класс катионных родиевых дигидридных катализаторов
описывается общей формулой [RhL2(S)2]+, где S — молекула
полярного растворителя, например тетрагидрофурана (ТГФ)
или метанола; L2 — два третичных фосфиновых лиганда
(обычно хелатированных). Наиболее хорошо изученным
представителем этого семейства катализаторов является DIPHOS,
содержащий хелатированный бидентатный лиганд [11]. Этот
комплекс (15) заслуживает особого внимания, так как
использование такого типа катализаторов приводит к высокой энантиосе-
лективности при асимметрическом гидрировании амидокоричных
кислот в том случае, когда катализаторы содержат хиральные
фосфиновые лиганды.
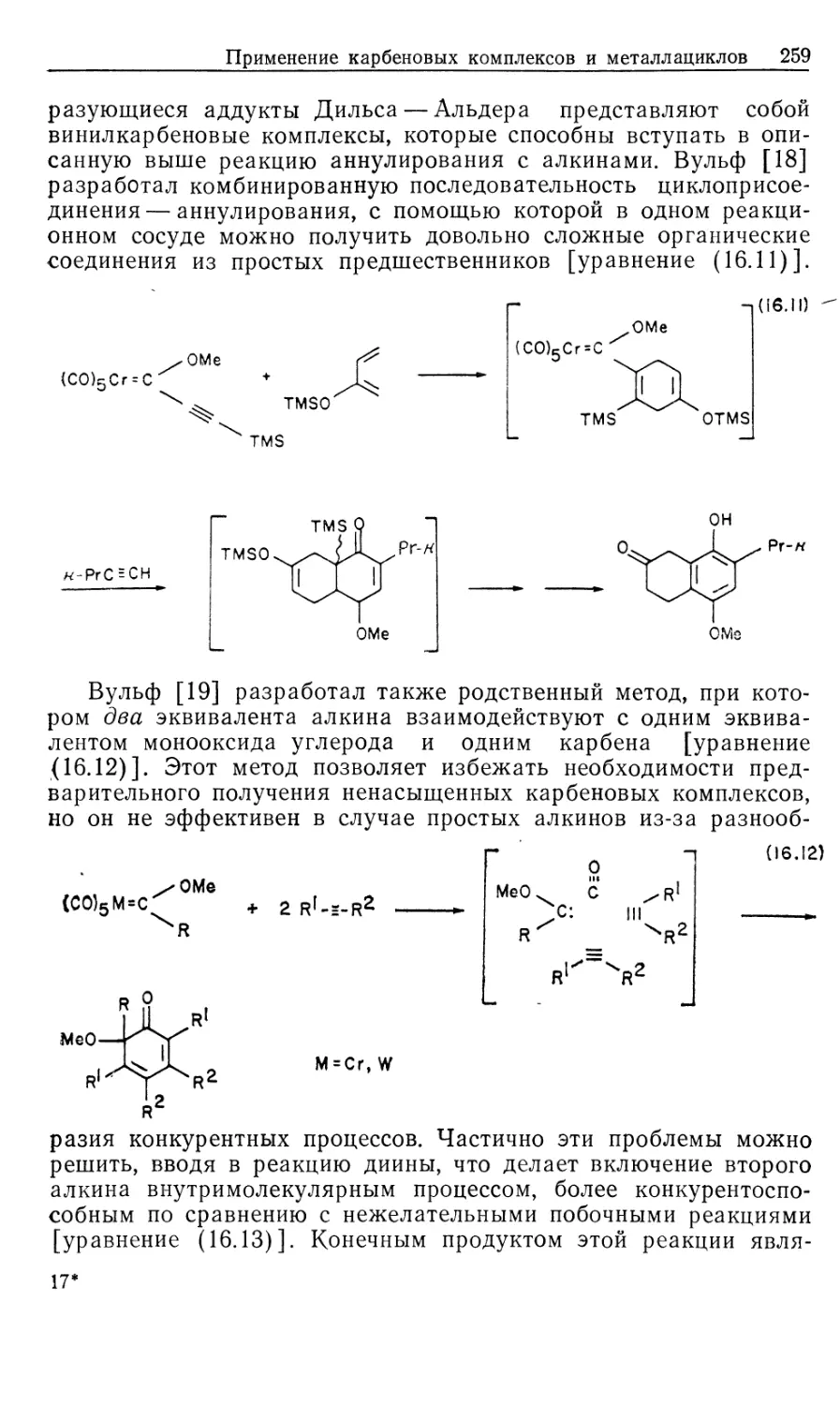
Механизм гидрирования олефинов в присутствии
катализатора DIPHOS подробно изучен Халперном [2в, 2д, 2е].
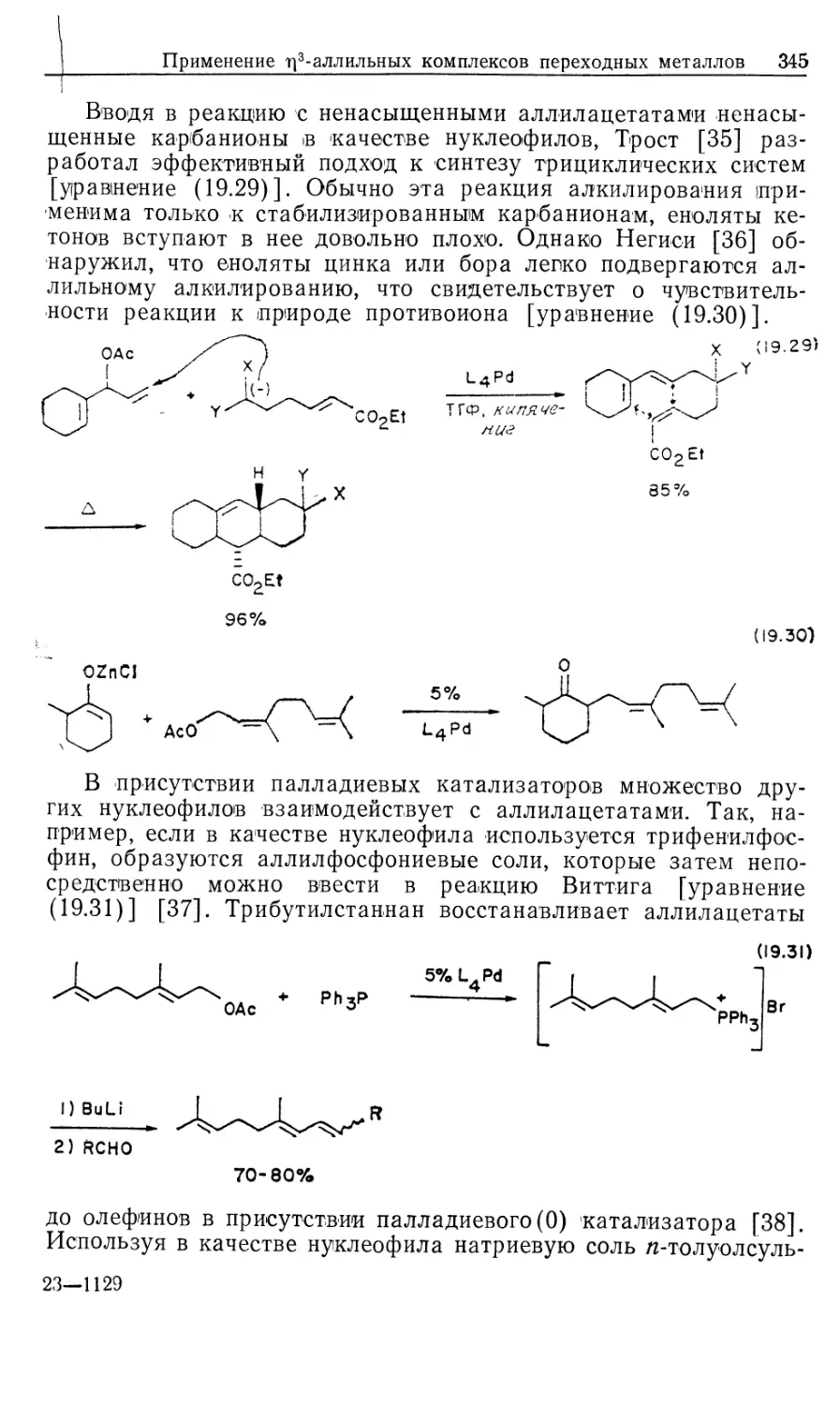
Установленный им механизм изображен на рис. 10.2. Он
значительно отличается от рассмотренного выше механизма
гидрирования с нейтральным катализатором Уилкинсона (рис. 10.1). Пер-
Гомогенные каталитические процессы
19
вое, что следует отметить,—это то, что катионный катализатор-
типа 15 генерируется in situ при гидрировании очень доступных,,
легко очищаемых диеновых комплексов [уравнение (10.21)].
Реакция служит удобным методом для создания центров
ненасыщенности, которые в данном случае заполняются
молекулами растворителя. Важно отметить и то обстоятельство, что
хотя комплекс 15 ненасыщенный, он не присоединяет другой моль
водорода. Поскольку две фосфиногруппы удерживаются в цис-
положении хелатным циклом, такой аддукт может иметь только
один гидрид, образующийся в результате цис-присоединения Н2>
вынужденного занимать транс-положение по отношению к фос-
финовому лиганду. Аналогичные комплексы с двумя монодентат-
Ph Ph
15
-PhCH2CH
NNHCOCH2
C-COOCH3
Rhni/ "nh
Ph ph S' CH3
18
Ph NHCOCH3
к ,
Рис. 10.2.
ными фосфиновыми лигандами способны присоединять второй
моль Н2.
\/
Ph2
(10.21)
Доминантный механизм гидрирования олефинов в
присутствии катализатора 15 включает путь через олефин, а не череч
гидрид. Кинетически важна следующая последовательность:
1) координация олефина, 2) окислительное присоединение Н2,
3) внедрение олефина в гидрид, 4) восстановительное
элиминирование (рис. 10.2). Три комплекса, 15, 16 и 18, кинетически
20 Глава 10
участвующие в каталитическом цикле, можно
спектроскопически детектировать и охарактеризовать; в этом состоит отличие
от механизма с участием нейтрального катализатора Уилкин-
сона.
Олефиновый комплекс 16 находится в состоянии равновесия
с катализатором 15. Сродство 15 к ненасыщенным молекулам,
выражаемое величиной /Сравш очень зависит от структуры оле-
фина, как это видно из уравнения (10.22). Даже ароматические
соединения, например бензол, координируются с 15; поскольку
эти арены не восстанавливаются, они оказываются
конкурентными ингибиторами гидрирования олефина, так как связывают
катализатор и переводят его в неактивную форму.
Происхождение большой константы ассоциации между 15 и
метил-(2)-а-ацетамидоциннаматом (MAC, 19) объясняется
кристаллической структурой аддукта 16, схематически
изображенного на рис. 10.2. Субстрат 19 образует хелат с атомом родия
через карбонильный кислород амидогруппы, а также через
нормальную (ц2)-координацию двойной связи. Аналогичной
оказалась структура комплекса 16, установленная по данным
спектров ЯМР на ядрах 31Р, 13С и !Н.
^Равя
[Rh(DIPHOS)]++HeHac. -<—>• [Rh(DIPHOS) (ненас.)] + (10.22)
15
О
1!
Нч /С—OR 19, R = CH3 (MAC)
20, R = C2H5 (EAC)
I!
О
Таким образом, между комплексами 15 и 16 (рис. 10.2)
быстро устанавливается равновесие, и даже при умеренных
концентрациях MAC оно сдвинуто в сторону олефинового
аддукта 16. При 25 °С лимитирующей стадией в общем цикле
гидрирования является окислительное присоединение Н2 к олефино-
вому аддукту 16 (k2), однако при пониженных температурах
(—40 °С) лимитирующей становится стадия восстановительного
элиминирования (&4, рис. 10.2). Это объясняется более низкой
энтальпией активации в стадии окислительного присоединения
(6 ккал/моль) по сравнению со стадией восстановительного
элиминирования (17 ккал/моль). Таким образом, при низких
температурах в растворе преобладает алкилгидридный комплекс
18, образующийся в результате миграционного внедрения
олефина в гидрид. Для характеристики комплекса 18 был
использован метод многоядерной ЯМР-спектроскопии, подтвердивший
ожидаемую региохимию реакции миграционного внедрения, ко-
Гомогенные каталитические процессы 21
торая происходит через пятичленный хелатный цикл,
включающий частично восстановленный субстрат. Так что в комплексе
18 гидрид оказывается связанным с ^-углеродом, а родий —
с а-углеродом олефина. Дигидридный олефиновый комплекс 17
(рис. 10.2) не был охарактеризован, из чего следует вывод, что
внедрение 17 в 18 происходит очень быстро. Напомним, что в
случае нейтрального катализатора Уилкинсона стадия
внедрения является лимитирующей.
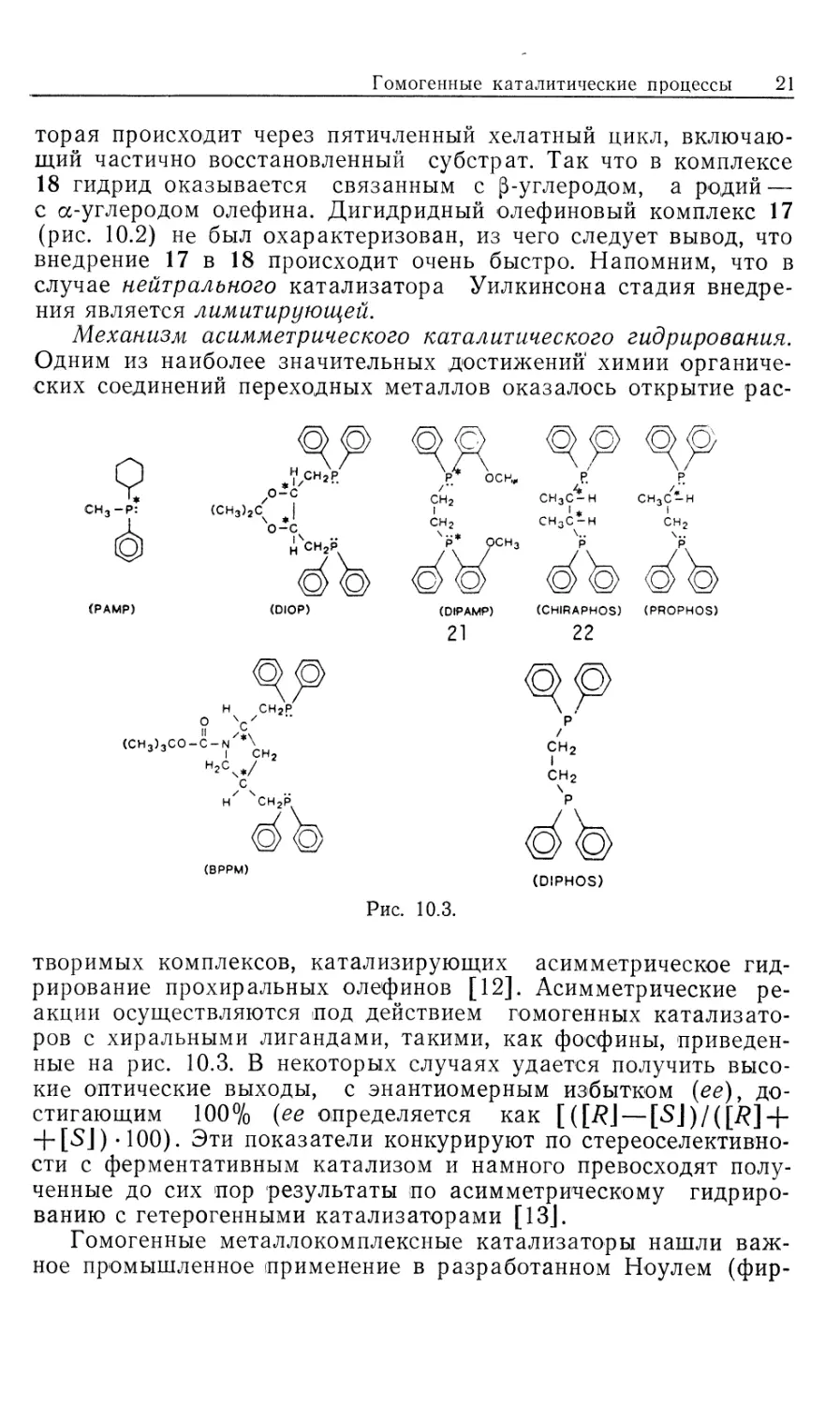
Механизм асимметрического каталитического гидрирования.
Одним из наиболее значительных достижении химии
органических соединений переходных металлов оказалось открытие рас-
СН3-Р:
СРАМР)
(DIPAMP) (CHIRAPHOS) (PROPHOS)
21 22
рр
н сн2р
(CH3)3CO-C-N *\
ч
с
нх чсн2р
Рис. 10.3.
творимых комплексов, катализирующих асимметрическое
гидрирование прохиральных олефинов [12]. Асимметрические
реакции осуществляются под действием гомогенных
катализаторов с хиральными лигандами, такими, как фосфины,
приведенные на рис. 10.3. В некоторых случаях удается получить
высокие оптические выходы, с энантиомерным избытком (ее),
достигающим 100% {ее определяется как [{[R\ — [S\)/([R] +
+ [SJ)-100). Эти показатели конкурируют по стереоселективно-
сти с ферментативным катализом и намного превосходят
полученные до сих пор результаты по асимметрическому
гидрированию с гетерогенными катализаторами [13J.
Гомогенные металлокомплексные катализаторы нашли
важное промышленное применение в разработанном Ноулем (фир-
22 Глава 10
ма Monsanto) процессе производства L-Dopa [уравнение
(10.23)] [14]. Этот препарат используется для лечения болезни
Паркинсона.
• I ,Dh/OD 1 с (10.23)
NHCOR П IDK'DD-1-'C' '
Н2, MeOH=S
2) Н3О+
L-Dopa
Реакция, представленная уравнением (10.23), типична для
большинства процессов селективного асимметрического
гидрирования [15]. Так, для гидрирования производных се-ацнлами-
докоричной кислоты в качестве катализатора используют кати-
онный родиевый комплекс с хиральным лигандом (чаще всего
хелатированными фосфинами). Центром хиральности может
быть атом фосфора или атом углерода в лиганде (рис. 10.3).
Прохиральные олефины других типов дают намного меньшие
величины ее (обычно менее 60%). Следует отметить, что
реакции с величиной ее ниже 90% не представляют практической
ценности. Чтобы объяснить столь узкие возможности
асимметрического каталитического гидрирования, необходимо понять
механизм реакции.
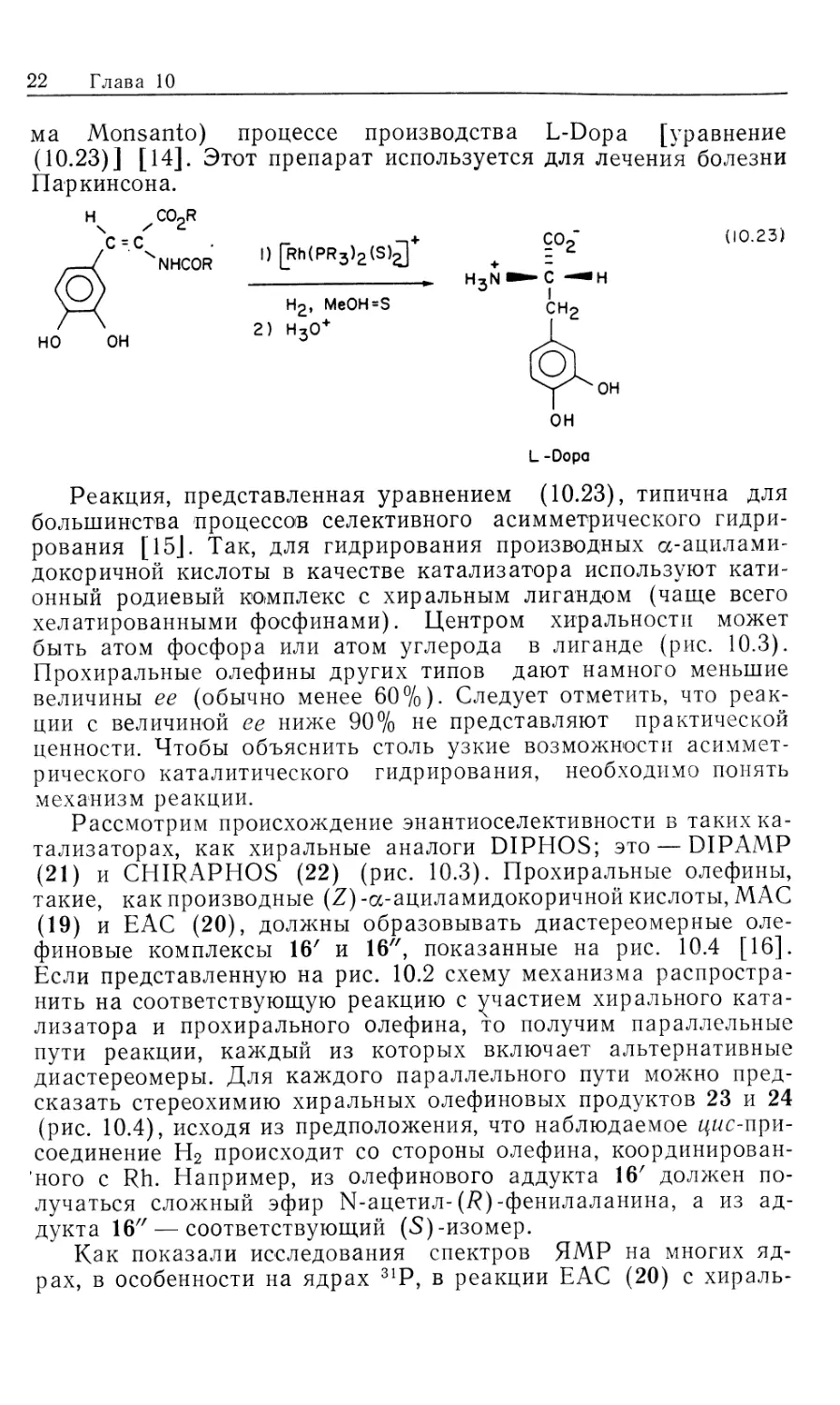
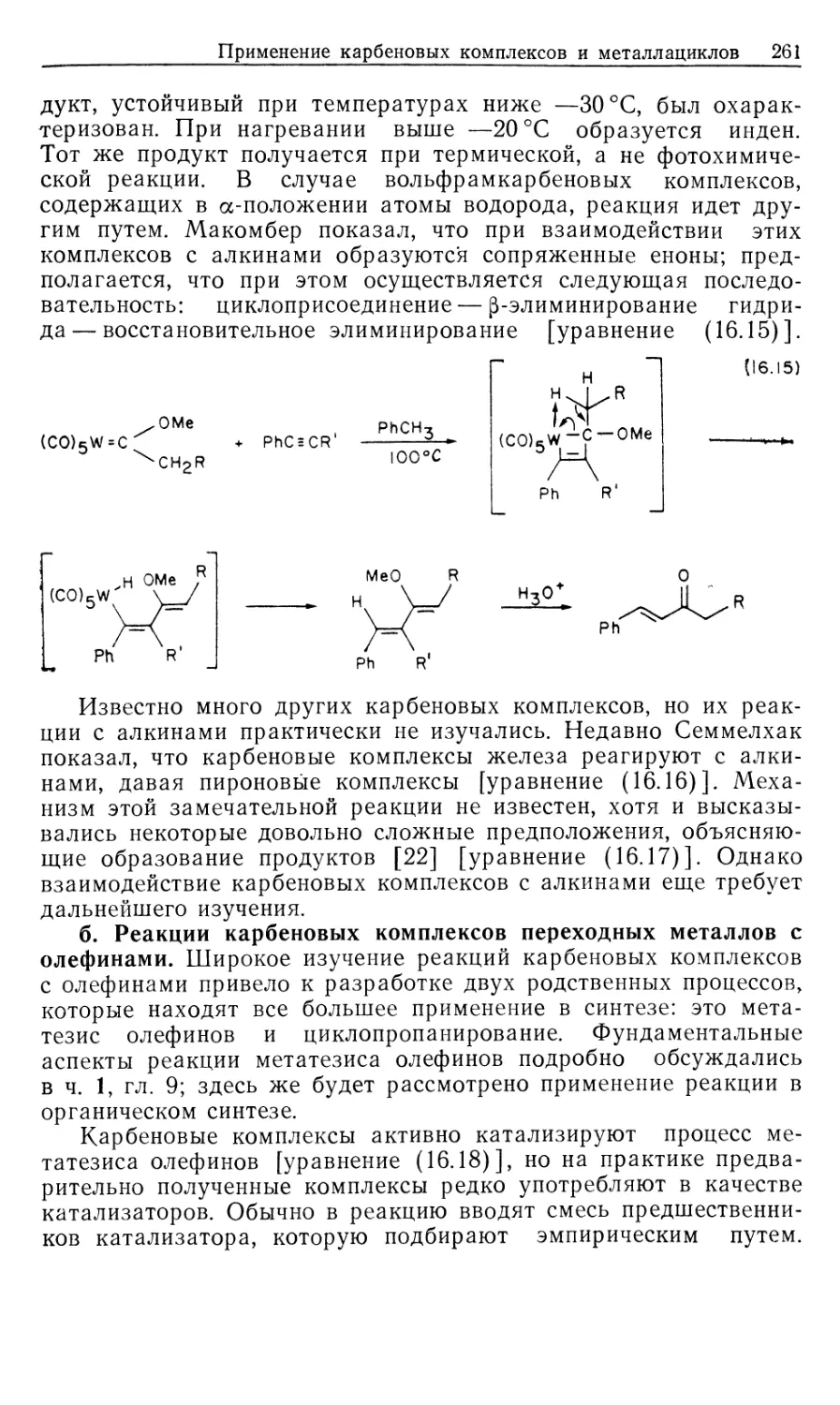
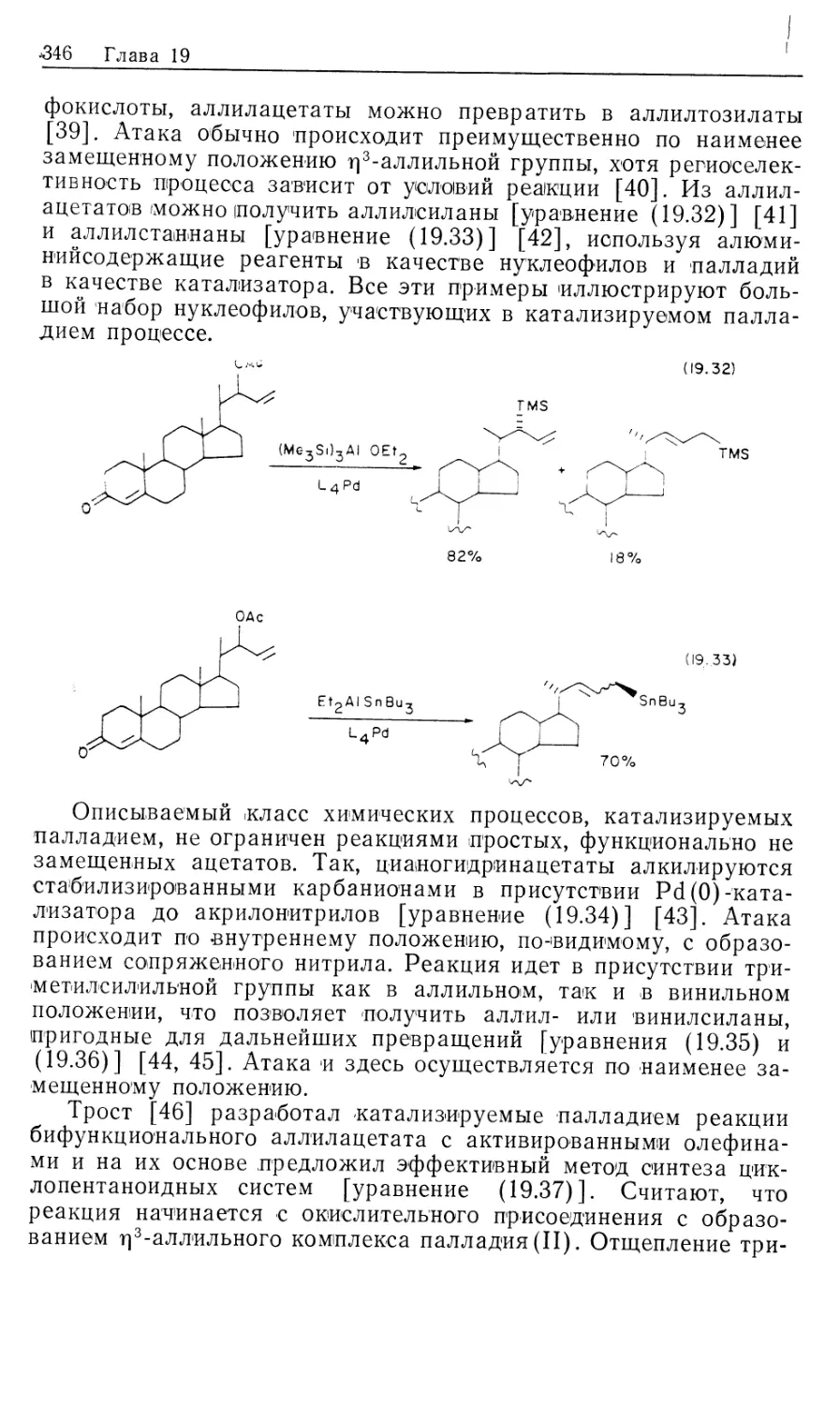
Рассмотрим происхождение энантиоселективности в таких
катализаторах, как хиральные аналоги DIPHOS; это — DIPAMP
(21) и CHIRAPHOS (22) (рис. 10.3). Прохиральные олефины,
такие, как производные (Z) -сс-ациламидокоричной кислоты, MAC
(19) и ЕАС (20), должны образовывать диастереомерные оле-
финовые комплексы 16' и 16", показанные на рис. 10.4 [16].
Если представленную на рис. 10.2 схему механизма
распространить на соответствующую реакцию с участием хирального
катализатора и прохирального олефина, то получим параллельные
пути реакции, каждый из которых включает альтернативные
диастереомеры. Для каждого параллельного пути можно
предсказать стереохимию хиральных олефиновых продуктов 23 и 24
(рис. 10.4), исходя из предположения, что наблюдаемое цис-при-
соединение Н2 происходит со стороны олефина,
координированного с Rh. Например, из олефинового аддукта 16/ должен
получаться сложный эфир М-ацетил-(7?)-фенилаланина, а из
аддукта 16"—соответствующий (5)-изомер.
Как показали исследования спектров ЯМР на многих
ядрах, в особенности на ядрах 31Р, в реакции ЕАС (20) с хираль-
Гомогенные каталитические процессы 23
ным лигандом (5,5)-CHIRAPHOS (22) образуется только один
диастереомер, 16' или 16". На первый взгляд может
показаться, что хиральность продукта определяется тем, какой вид
связывания субстрата оказывается предпочтительным. Это своего
рода разновидность механизма «ключ и замок», ведущего свое
H COOMe
Ph NNHCOMe
(* ^R
16
к 2 I [н2]
t
МеООС
17'
к з I S'
^CO2Me
18'
k4 |-[Rh(P* P)S2j +
0 V
гз <r)
/PhARh/P^
ме-4о^- \p;
L 16" J
[H2]
O2Me I
i 4
н
17"
Me
HN О
8"
-[Rh(P* P)S'2]*
1
Рис. 10.4.
происхождение от сформулированных ранее Фишером
представлений о стереоспецифичности ферментов. Однако такое
допущение некорректно', в действительности преобладающий энантио-
мер продукта образуется из минорного диастереомер а аддукта
катализатора с олефином (16)!
24
Глава 10
Ключевым экспериментом, позволившим установить верный
механизм, явилось определение структуры преобладающего
диастереомера иона [Rh(S,S-CHIRAPHOS) (EAC)]+ с помощью
рентгеноструктурного анализа монокристаллов его перхлората.
Согласно структуре этого иона, в реакции должен был
образоваться сложный этиловый эфир 1М-ацетил-(5)-фенилаланина,
в действительности же преобладающим продуктом оказался
7?-изомер.
Дополнительные подтверждения правильности сделанного
вывода получены при изучении гидрирования MAC (19)
родиевым комплексом (i?,/?)-DIPAMP (21). В этом случае с помощью
ЯМР можно определить наличие в растворе обоих диастерео-
меров, 16' и 16", но невозможно сделать отнесение их
абсолютной конфигурации [17]. При низких температурах, когда
отсутствует взаимопревращение диастереомеров, данные ЯМР
показывают, что минорный диастереомер намного быстрее реагирует
с Н2, образуя сначала гидридоалкил 18', а затем продукт.
ГмеООС -
LVR<
^ р \MUH°PZ,y ь
\рУ_ - — --/---
\ —
арный
\ \~—
\ Л
\\
\ \
\
\
\
\
L
1
■
МеООС
Р Н "~°
н^ "~~1 ( ^\ н
Ноордина/па реакции
Рис. 10.5.
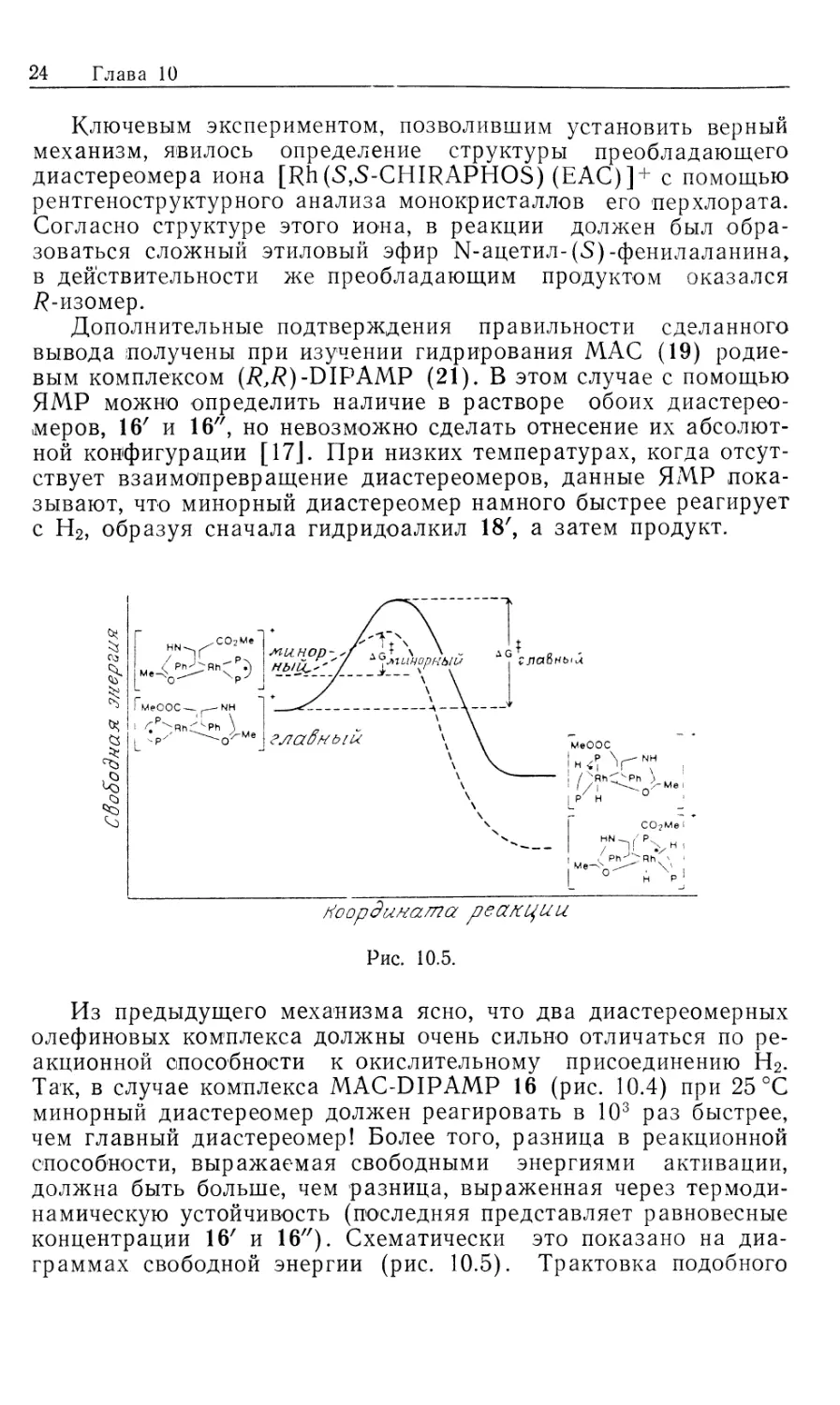
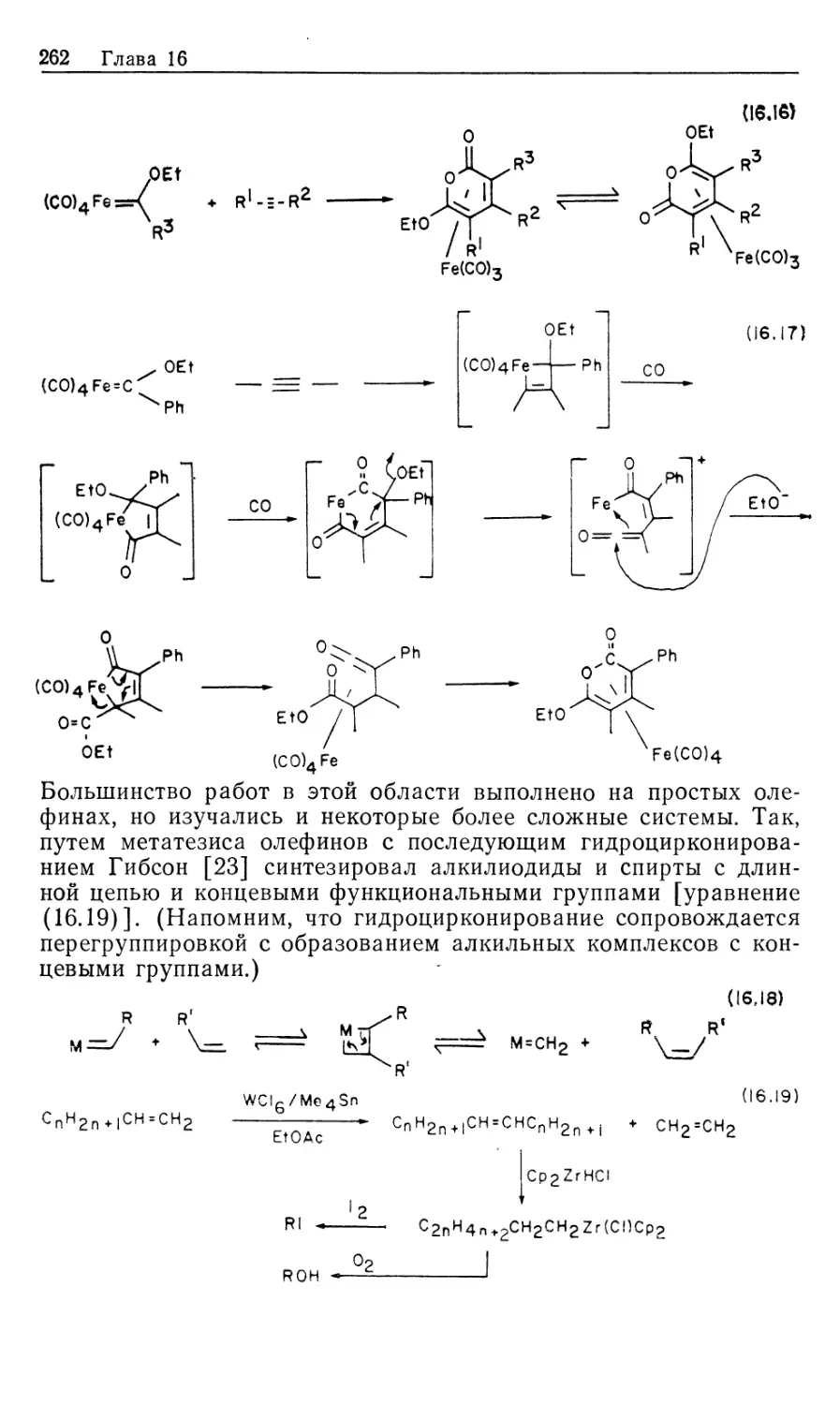
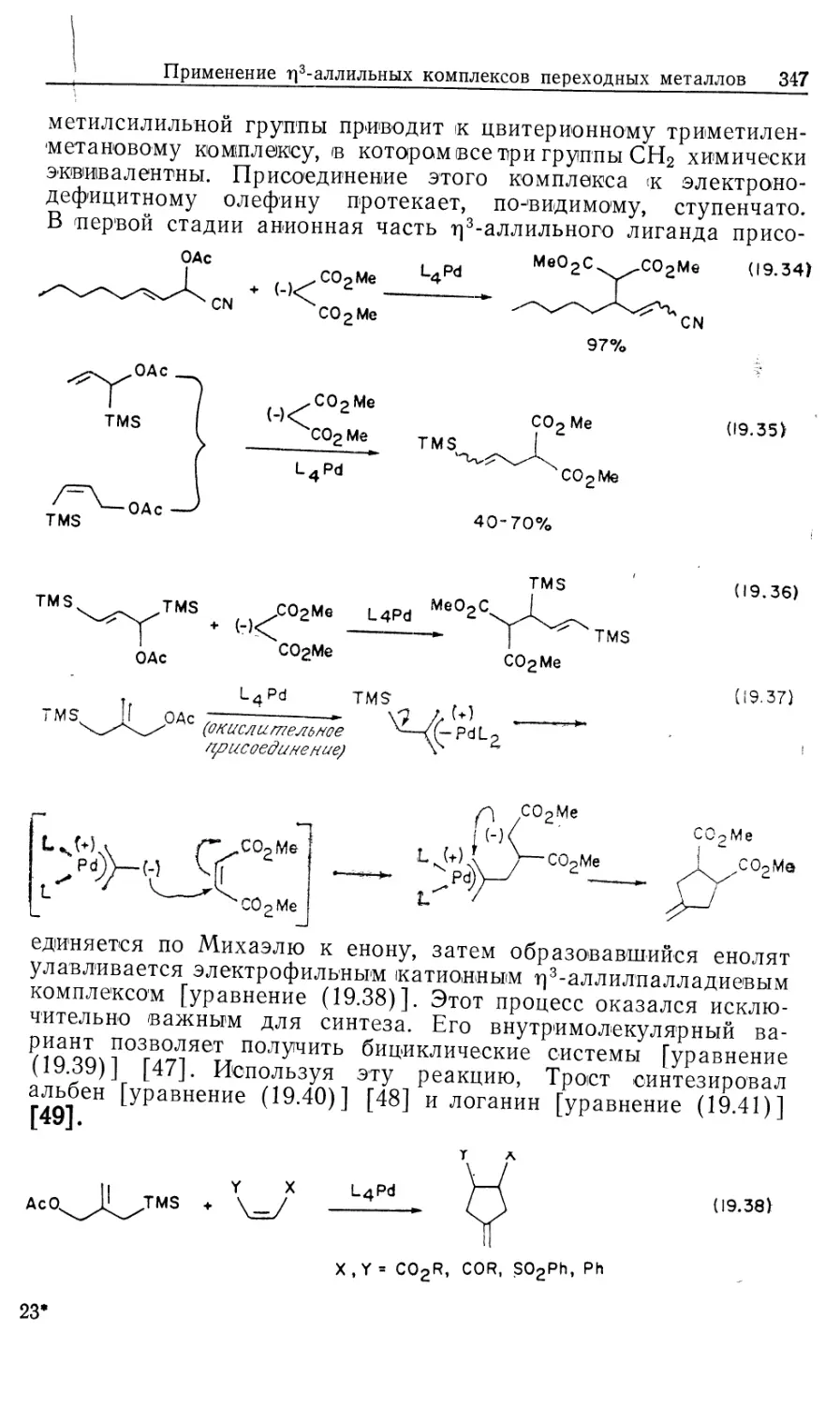
Из предыдущего механизма ясно, что два диастереомерных
олефиновых комплекса должны очень сильно отличаться по
реакционной способности к окислительному присоединению Н2.
Так, в случае комплекса MAC-DIPAMP 16 (рис. 10.4) при 25 °С
минорный диастереомер должен реагировать в 103 раз быстрее,
чем главный диастереомер! Более того, разница в реакционной
способности, выражаемая свободными энергиями активации,
должна быть больше, чем разница, выраженная через
термодинамическую устойчивость (последняя представляет равновесные
концентрации 167 и 16"). Схематически это показано на
диаграммах свободной энергии (рис. 10.5). Трактовка подобного
Гомогенные каталитические процессы 25
экспериментального результата непроста; к тому же
невозможно предсказать, какие именно компоненты должны проявлять
это необычное свойство — различные хиральные лиганды,
различные гомогенные катализаторы или различные ненасыщенные
субстраты.
Выводы из наблюдаемой каталитической энантиоселектив-
ности сводятся к следующему: 1) вероятно, лишь
ограниченный класс олефиновых субстратов, подобных MAC (19),
проявляет высокую стереоселективность и 2) систематическое
исследование других катализаторов асимметрического гидрирования
кажется безнадежным.
По-видимому, все остальные катализаторы
асимметрического гидрирования этих олефинов действуют по тому же
механизму. Тестом для такого рода явления служат два удобных
экспериментальных приема, позволяющие избежать сложного
кинетического и структурного анализа. Первый критерий связан с
обратной зависимостью между оптическим выходом и
давлением водорода. Согласно кинетическому анализу Халперна [2е],
при повышении давления водорода энантиоселективность
должна понижаться, а в некоторых случаях хиральность
преобладающего продукта должна быть обратной. Именно такая картина
наблюдалась в ряде примеров [18]. Высокая концентрация
водорода эффективно ускоряет процесс присоединения водорода
!по сравнению с взаимопревращением диастереомеров 16'и 16".
Второй, родственный критерий касается температурной
зависимости оптического выхода: последний уменьшается с
понижением температуры [18, 19]. Это легко объяснить, если учесть,
что взаимопревращение диастереомеров 16' и 16" (которое
является главным образом внутримолекулярным процессом,
происходящим путем диссоциации и рекоординации олефинового
конца хелатирующего енамида [17в]) замедляется при
пониженных температурах; в экстремальных случаях более
устойчивый диастереомер может оказаться «кинетически
вымороженным». И наоборот, известны случаи, когда величина ее
возрастает при повышении температуры реакции.
Во всех исследо'ванных примерах наблюдалась зависимость
ее либо от обоих факторов, давления водорода и температуры,
либо от одного из них. Вероятно, действие всех этих энантио-
селективных катализаторов осуществляется по механизму,
установленному Халперном и Брауном. Известны иридиевые
аналоги каталитических интермедиатов асимметрического
гидрирования; они охарактеризованы спектрами ЯМР [176].
26
Глава 10
10.4. Примеры применения дигидридных катализаторов
гидрирования олефинов
а. Катализатор Уилкинсона. Благодаря региоселективности,
стереоселективности, отсутствию изотопного перераспределения,
а также «терпимости» к наличию функциональных групп
катализатор Уилкинсона всегда превосходит более
распространенные гетерогенные катализаторы гидрирования олефинов. Ниже
приводятся некоторые примеры.
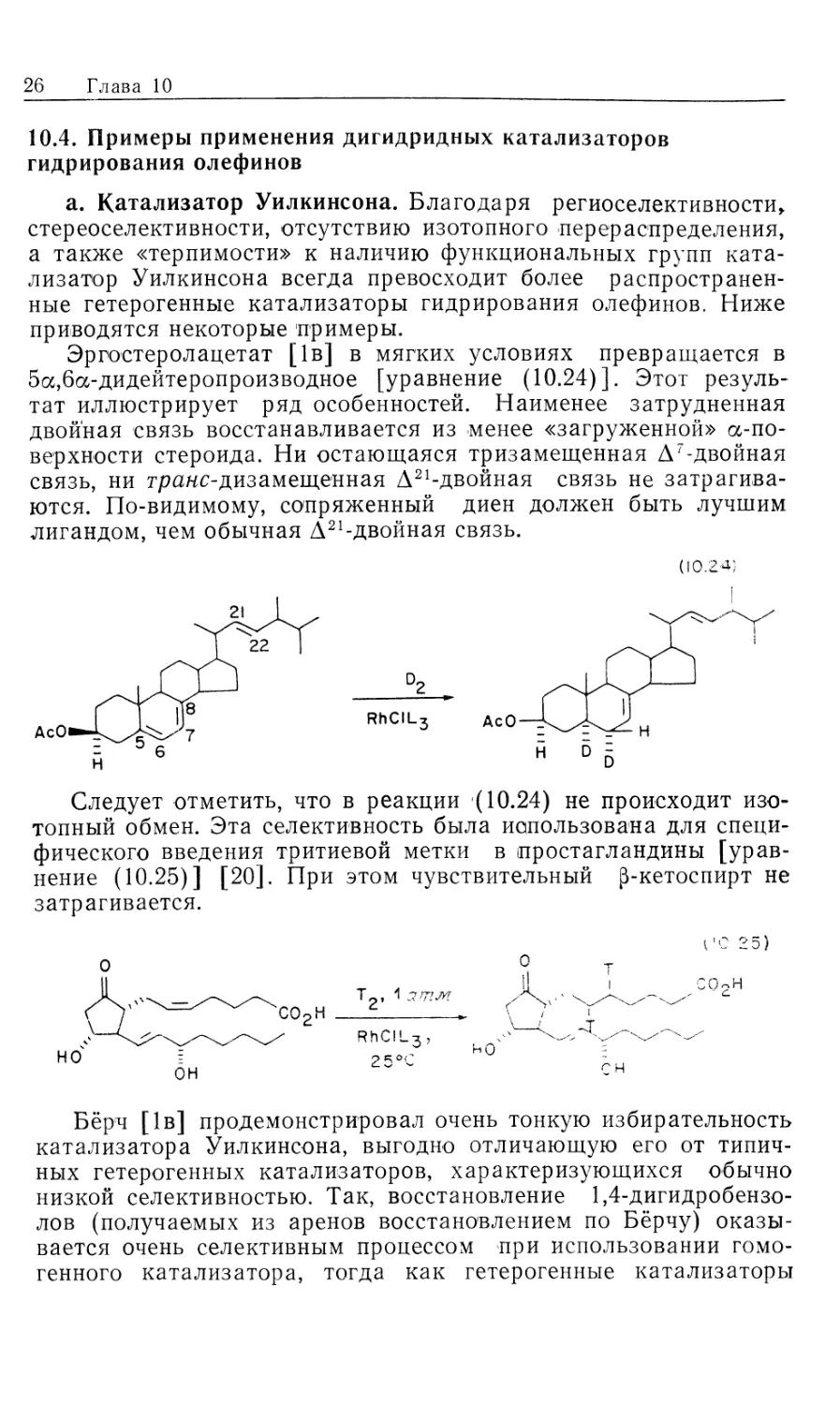
Эргостеролацетат [1в] в мягких условиях превращается в
5а,6а-дидейтеропроизводное [уравнение (10.24)]. Этот
результат иллюстрирует ряд особенностей. Наименее затрудненная
двойная связь восстанавливается из менее «загруженной» а-по-
верхности стероида. Ни остающаяся тризамещенная А7-двойная
связь, ни граяс-дизамещенная А21-двойная связь не
затрагиваются. По-видимому, сопряженный диен должен быть лучшим
лигандом, чем обычная А21-двойная связь.
(10.24}
АсО
Следует отметить, что в реакции (10.24) не происходит
изотопный обмен. Эта селективность была использована для
специфического введения тритиевой метки в простагландины
[уравнение (10.25)] [20]. При этом чувствительный р-кетоспирт не
затрагивается.
25°С
СН
1'С 25)
СО9Н
Бёрч [1в] продемонстрировал очень тонкую избирательность
катализатора Уилкинсона, выгодно отличающую его от
типичных гетерогенных катализаторов, характеризующихся обычно
низкой селективностью. Так, восстановление 1,4-дигидробензо-
лов (получаемых из аренов восстановлением по Бёрчу)
оказывается очень селективным процессом при использовании
гомогенного катализатора, тогда как гетерогенные катализаторы
Гомогенные каталитические процессы 27
эмотируют диспропорционирование и способствуют образо-
яю нежелательных побочных продуктов [уравнение (10.26) J.
1-3 r^V^l r^ir^l (Ю.26)
о
iOoMe
СО2Ме СО2Ме
96% 4%
49% 26%
з присутствии катализатора Уилкинсона можно селективно
гидрировать углерод-углеродную двойную связь в нитроолефи-
iax [уравнение (10.27)], в то время как гетерогенные
катализаторы обычно реагируют с нитрогруппой. Аналогичным образом
з винилацеталях под действием никеля Ренея отщепляется ал-
килгруппа, а в присутствии катализатора Уилкинсона происхо-
;ит только восстановление олефина [уравнение (10.28)].
oCHoNOo (Ю.27)
(10.28)
^ пл /
MeO
Большинство гетерогенных катализаторов подвержено от-
эавлению элементной ртутью, которая заполняет их поры и тем
:амым блокирует доступ субстрата к катализатору.
Катализатор же Уилкинсона можно с успехом использовать для гидроге-
толиза связей арил — ртуть [уравнение (10.29)].
/'7~\ RhCiL 5 г^\
( ( ))—НдОАс ч—+~ (/О) + Нд° + НОДс (10 29)
4 J 3 riniM Н о "" У
с.
Еще одну проблему предста&ляют арен-хромовые комплексы,
которые под действием гетерогенных катализаторов
разрушаются, но катализатор Уилкинсона их не затрагивает, и его мож-
то использовать для восстановления незакомплексованных оле-
dhhob [уравнение (10.30)].
28
Глава 10
(Ю 30)
Далее в настоящей глазе будет показано, что двойные связи
как в растворимых, так и в сетчатых полимерах можно
гидрировать в присутствии катализатора Уилкинсона, но по отношению
к гетерогенным катализаторам они инертны.
б. Иридиевый катализатор Крэбтри. Крэбтри [21]
разработал иридиевый предкатализатор 25 с высокой степенью
ненасыщенности, обладающий замечательными свойствами. Он
катализирует восстановление затрудненных олефинов, причем с
высокими скоростями, и (в противоположность большинству
гомогенных катализаторов) не подвергается действию ни
кислорода, ни метилиодида. Саггс применил катализатор Крэбтри для
гидрирования затрудненных двойных связей в стероидах
[уравнение (10.31)]. Реакция идет стереоселективно по ос-1поверхно-
сти.
С8Н17
(10.31)
СН2С!2,
SiO i
H H
Катализатор Крэбтри совершенно инертен к другихм
функциональным группам. Это иллюстрируется восстановлением
двойной связи в присутствии циклопропильной группы и геми-
нальной дибромгрушш [22]; и та и другая не инертны по
отношению к гетерогенным катализаторам [уравнение (10.32)]. Ли-
ганд COD в 25 подвергается гидрогенолизу при образовании
активного катализатора, чему, видимо, способствуют пиридин и
объемные лиганды Су3Р. Спирты и другие полярные
растворители дезактивируют этот катализатор.
25 , CHp
(10 32)
Вг Вг
Вг Вг
Гомогенные каталитические процессы 29
Таблица 10.1. Частота каталитических циклов при гомогенном
каталитическом гидрировании олефинов
Рредкатализатор
Ir(cod)(PCy3)(Py)] +
Ir(cod)(PMePh2)2] +
Ir(cod)(PMeP<h2)2] +
~'Rh(cod)(PPh3)2] +
"Ru(H)Cl(PPh3b]
RhCl(PPh3)3]
RhCl(PPh3)3]
Температура,
°C
0
0
0
25
25
25
0
Растворитель
CH2C12
CH2C12
CH3COCH3
CH2C12
C6H6
C6H6/Et0H
C6H6/Et0H
Частота каталитических
циклов, ч—1
1-гексен
6400
5100
10
4000
9000
650
60
циклогек-
сен
4500
3800
0
10
7
700
70
тетраметил-
этилен
4000
50
0
0
0
0
0
В табл. 10.1 сравниваются скорости гидрирования трех оле-
Ьинов, отличающихся степенью стерического затруднения,
з присутствии нескольких гомогенных катализаторов. Из при-
зеденных данных очевидно, что катализатор Крэбтри (25) го-
эаздо более реакционноспособен, чем нейтральные и более
затрудненные стерически родиевый и рутениевый катализаторы.
Сравнение данных второй и третьей строки таблицы
демонстрирует дезактивирующее влияние полярного растворителя на ка-
-ионный иридиевый катализатор; по-видимому, ацетон
выступает 'В роли конкурентного ингибитора. Данные четвертой
строки показывают, что катионный родиевый катализатор уступает
иридиевому аналогу в случае восстановления тетразамещенного
злефина, инертного в присутствии нейтральных катализаторов
ср. последние три строки).
Сообщалось, что внутреннее высвобождение водорода про-
чсходит с той стороны молекулы, которая несет спиртовую
группу. Катализатор Крэбтри особенно эффективен благодаря
высокому сродству к полярным лигандам [23]. Резко отличрое
"юведение гомогенного иридиевого катализатора в сравнении с
чалладием на угле иллюстрируется уравнением (10.33). Однако
^корость реакции в этом случае значительно (Понижается, при-
элизительно до 30 ч"1.
пагпали?атор
НО
ката/шзагпор Pd /С 20
катализатор = [i г (с О D) Р Су3р у] 9 9^ 9
(10.33)
30 Глава 10
в. Катализаторы VI группы, применяемые для селективного
восстановления диенов. В присутствии некоторых гомогенных
катализаторов диены селективно гидрируются до ц^омоноенов.
Наиболее изученные предкатализаторы этой реакции относятся
к типу 1/3Сг(СО)3, где L/з — арен (чаще всего нафталин) [24,
25], (CH3CN)3 или (СО)3 [26]. Общая особенность их
заключается в том, что ненасыщенный металлосодержащий фрагмент
(возможно, «Сг(СО)3») генерируется in situ путем термической
или фотохимической активации [26] координационно
насыщенного предкатализатора. Под действием этих катализаторов
осуществляется 1,4-присоединение водорода к сопряженным
диенам, приводящее к Z-моноену [уравнение (10.34)] [26, 27].
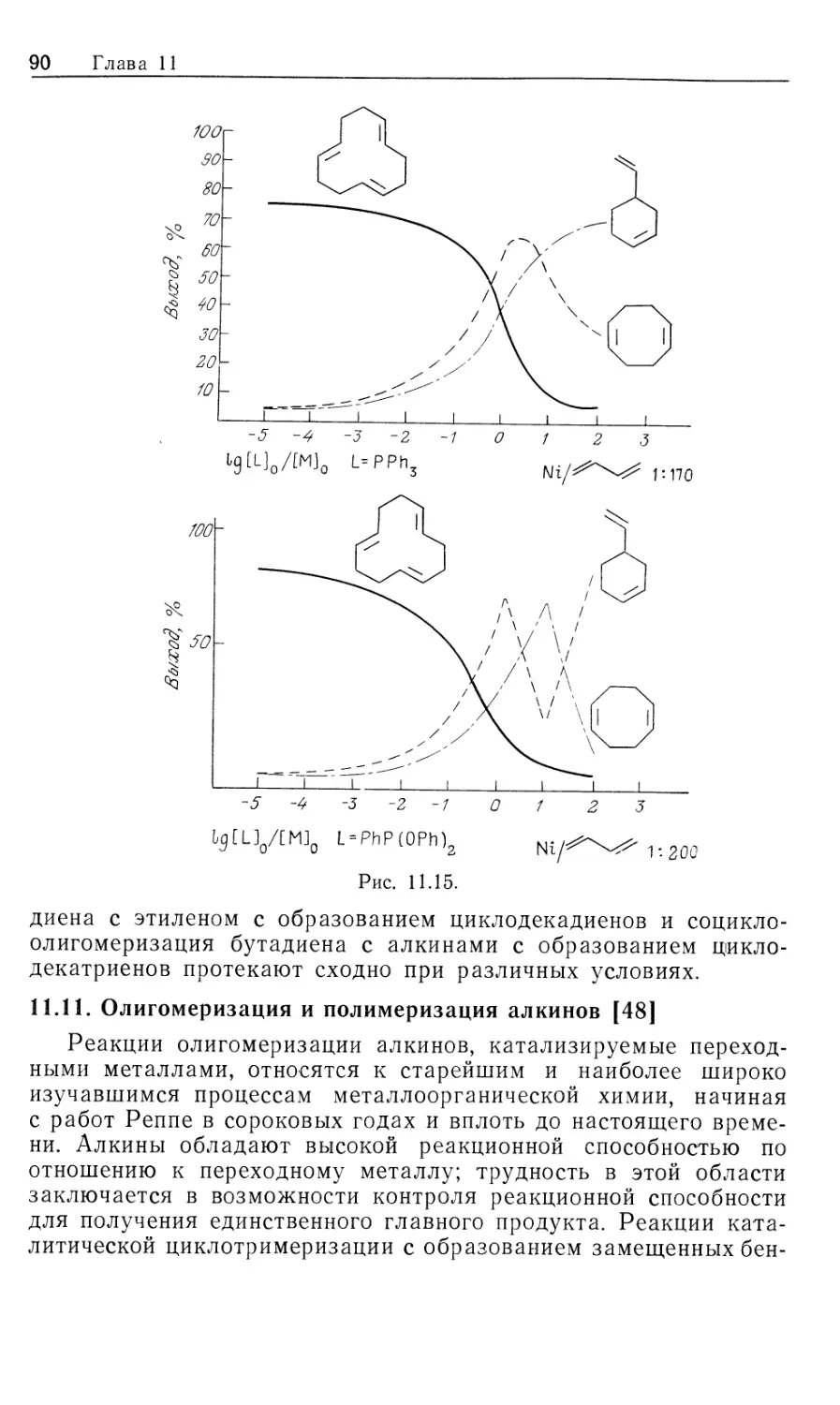
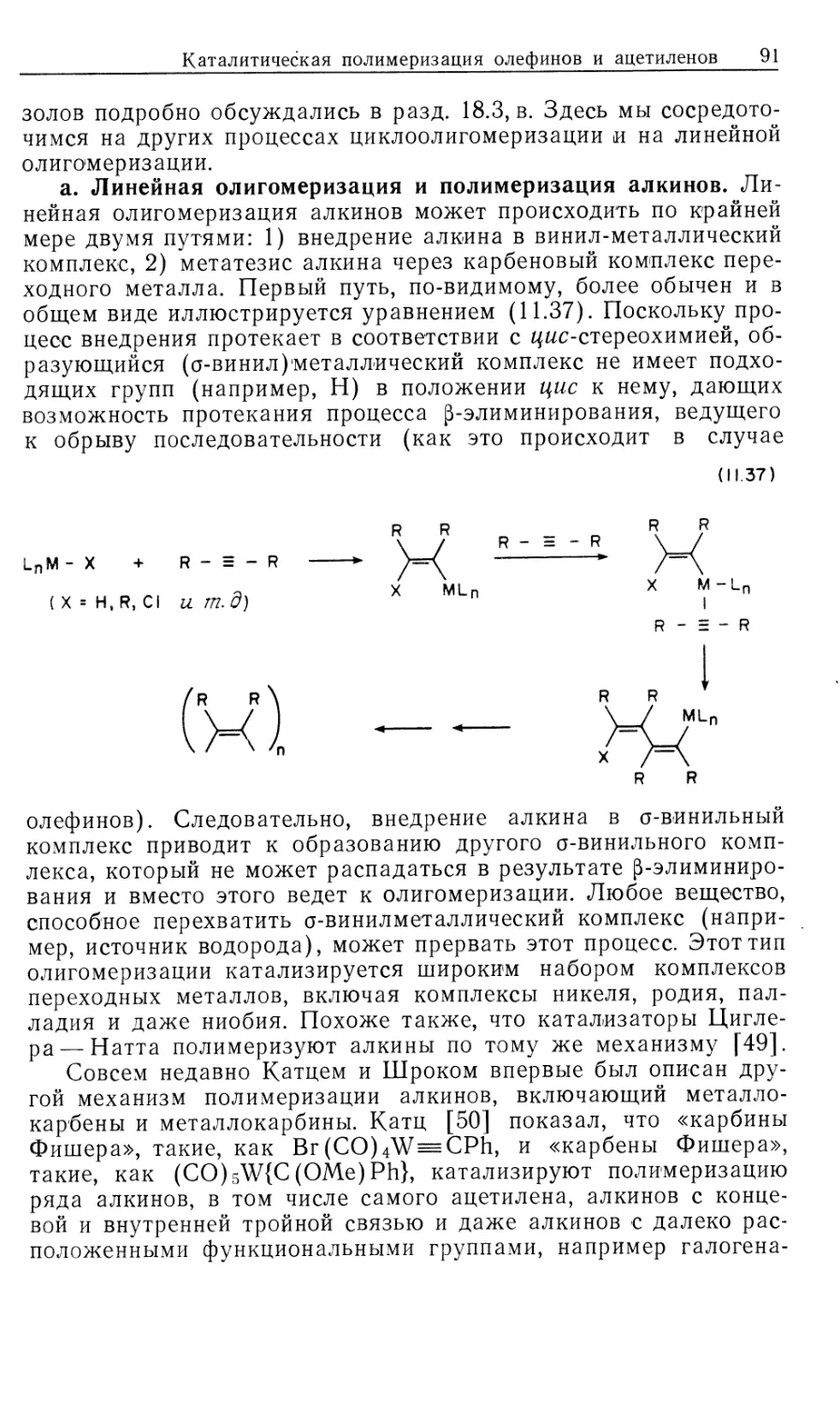
D D
■СО2Ме (10.34)
СО?Me D2, > \Ъатм N
!60°С
Наиболее эффективным и лучше всех изученным
катализатором данного класса является комплекс нафталина
(СюН8)Сг(СО)3; в его присутствии 1,4-диены гладко
гидрируются до Z-моноенов, миграция двойной связи при этом
минимальна, что служит дополнительным преимуществом
катализатора. Процесс представляет потенциальный интерес для
промышленности. Важной стадией обработки соевых бобов и семян
хлопка является каталитическое гидрирование содержащихся в
их маслах полиненасыщенных сложных эфиров. Однако в
присутствии гетерогенных катализаторов при гидрировании
образуются как £-олефины, так и смеси позиционных изомеров,
являющиеся нежелательными побочными продуктами.
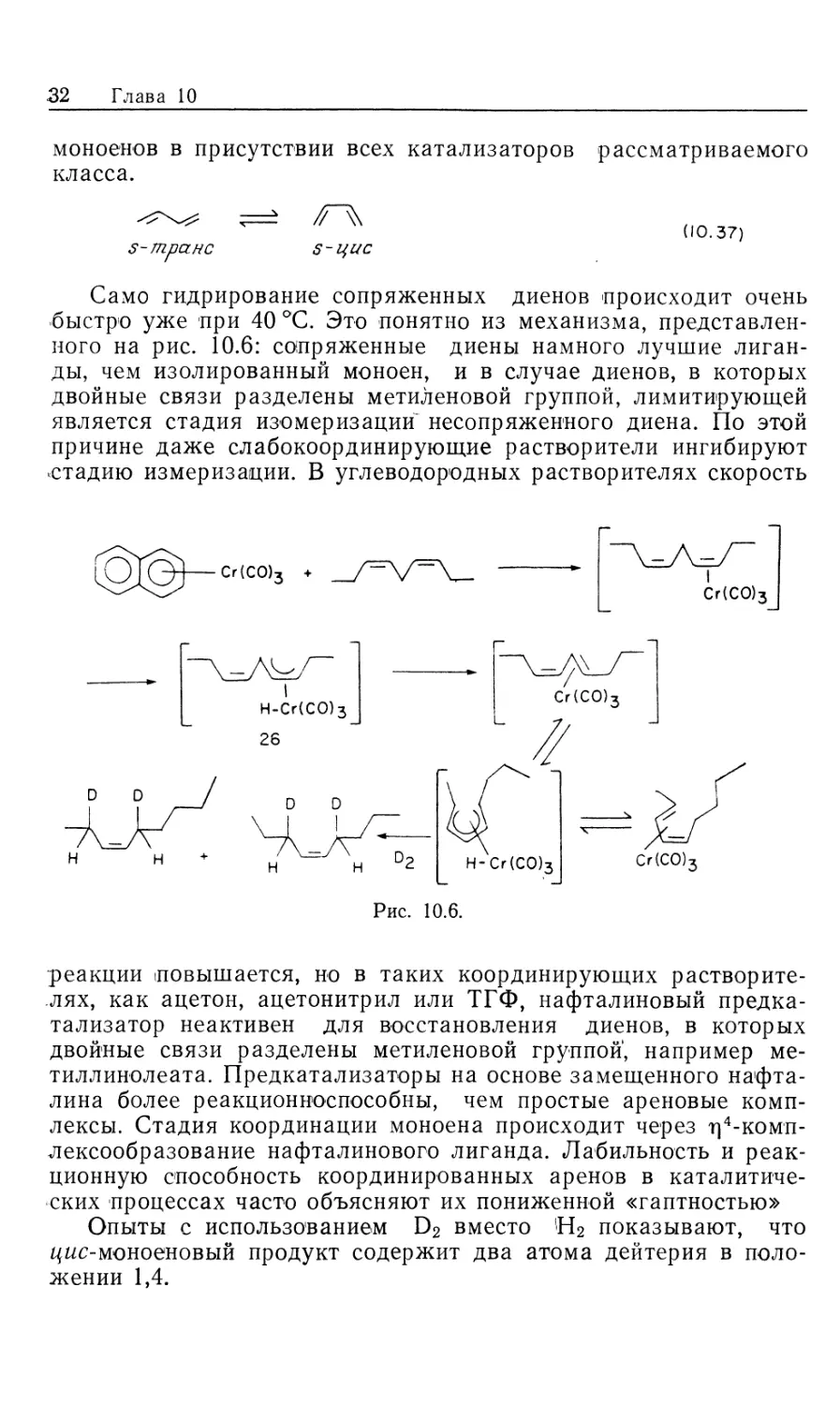
Такер и Райли [24] изучили этот процесс гидрирования,
сопровождающийся перегруппировками, и представили
убедительные доказательства вероятного механизма (рис. 10.6). Диен,
в котором две двойные связи разделены метиленовой
компонентой, сначала должен изомеризоваться в сопряженный цис,транс-
диен, который затем восстанавливается путем 1,4-присоедине-
ния Н2. Изомеризация происходит медленно и потому является
лимитирующей стадией, так что общая скорость реакции не
зависит от давления водорода. Показанный на рис. 10.6 процесс
начинается с координации ^мс-олефина; эта реакция ингибиру-
ется избытком нафталина и других конкурирующих лигандов.
Нафталин не подвергается гидрогенолизу и может быть
регенерирован интактным. Предполагается, что стадия изомеризации
происходит через я-аллилгидридный интермедиат 26, который
затем образует цис-транс-сопряжеяяыи диеновый комплекс.
Молекулярные модели показывают, что аллильная метиленовая
группа, связанная с цис-олефиноъои связью цис,транс-д#епового
Гомогенные каталитические процессы 31
комплекса так фиксирована, что должна подвергаться
внутримолекулярному С—Н-окислительному присоединению. Таким
образом, цис-транс-ляеяовыя комплекс может образовать дие-
нилгидрид и оказаться в равновесии с другим цис,транс-ляеяо~
вым комплексом. Каждый из этих комплексов после 1,4-гидри-
рования даст свой цис-мояоея. Так, метиллинолеат (метш-цис,
цг/с-9,12-октадекадиеноат), имеющий две разные двойные
связи и поэтому образующий два разных аллилгидридных
комплекса и два разных цис-транс-ляеяовых комплекса, дает в
результате гидрирования равные количества четырех различных
цис-мояоеяов (А9, А10, А11 и А12).
Предложенный механизм согласуется с результатами
независимых исследований, показавшими, что один сопряженный
диен, метил-^с,граяс-9,11-октадекадиеноат, дает два цис-яролук-
та: 9- и 10-октадеценоаты в эквимолярных количествах
[уравнение (10.35)J, а другой транс,транс-9,\ 1-октадекадиеноат
(который не может находиться в равновесии с другим диеновым
комплексом через диенилгидрид) дает только один изомер,
цис-10-мояоея [уравнение (10.36)J.
С—С
сн3(снг>7"' 9Х(сн2)7 ;о;)ме
С = С" 9 х(СН?)7СО,Ме
СН3(СН2>5
?)7СО,
(10.36)
Н (СН2)7СО2Ме
н^ >=< н2 н^ ^н
С = С ^ 9 Н — ** С ~- С
СН3^Н2)5/ II^H "Cr(CO)3" CH3(CH2)ff ю^(СН2)ьСО2Ме
Эти результаты показывают, что образование диенилгидрида
в случае транс,граяс-диена происходит медленно, а в случае
цис,транс-диена быстро (как это и следует из анализа
молекулярных моделей). ^с-Моноеновый продукт получается при
1,4-присоединении Н2 к 5-ц?^^-конформеру сопряженного диена
[уравнение (10.37)]. Только в S-^wc-конформации может
образоваться хелатированный диеновый комплекс. Лишь те диены,
которые способны принять 5-ц«с-конфигурацию, гидрируются до
32 Глава 10
моноенов в присутствии всех катализаторов рассматриваемого
класса.
s-транс
// \\
s-цис
(10.37)
Само гидрирование сопряженных диенов происходит очень
быстро уже при 40 °С. Это понятно из механизма,
представленного на рис. 10.6: сопряженные диены намного лучшие лиган-
ды, чем изолированный моноен, и в случае диенов, в которых
двойные связи разделены метиленовой группой, лимитирующей
является стадия изомеризации^ несопряженного диена. По этой
причине даже слабокоординирующие растворители ингибируют
стадию измеризации. В углеводородных растворителях скорость
-Сг(СО)3
I
Сг(СО)3
Сг(СО)3
Рис. 10.6.
реакции повышается, но в таких координирующих
растворителях, как ацетон, ацетонитрил или ТГФ, нафталиновый предка-
тализатор неактивен для восстановления диенов, в которых
двойные связи разделены метиленовой группой, например ме-
тиллинолеата. Предкатализаторы на основе замещенного
нафталина более реакционноспособны, чем простые ареновые
комплексы. Стадия координации моноена происходит через т]4-комп-
лексообразование нафталинового лиганда. Лабильность и
реакционную способность координированных аренов в
каталитических процессах часто объясняют их пониженной «гаптностью»
Опыты с использованием D2 вместо Н2 показывают, что
цис-мояоеяовыи продукт содержит два атома дейтерия в
положении 1,4.
Гомогенные каталитические процессы 33
10.5. Гомогенное каталитическое гидрирование аренов
Многие растворимые комплексы переходных металлов
служат катализаторами гидрирования ароматических циклов [29—
38J. Типичные примеры приведены в табл. 10.2. Эти
катализаторы можно разделить на две категории: а) системы, промоти-
рующие гидрирование всех, кроме одного цикла, в таких
полициклических ароматических субстратах, как нафталин или
антрацен; б) системы, промотирующие гидрирование простых
моноциклических аренов. Изучение механизма реакции
показывает, что к первой категории относятся гомогенные
катализаторы, однако во второй категории по крайней мере некоторые
активные катализаторы могут быть гетерогенными, представляя
собой суспендированные частицы металла («золи»). Рассмотрим
эти две категории катализаторов по порядку.
а. Гидрирование полициклических аренов. Наиболее
эффективный и лучше других изученный катализатор этой категории
получается из анионного гидридорутениевого шредкатализатора
27, открытого Грейем и Пецем [37а] и охарактеризованного
Халперном [376). В присутствии этого предкатализатора
антрацен гидрируется до 1,2,3,4-тетрагидроантрацена в относительно
мягких условиях, с частотой каталитических циклов более
50 ч"1 и с числом циклов более 100 [уравнение (10.38)].
ТГФ- 1001>с ^^гЯ^^^-Н (10.38)
10 атлм Нр 7
27 н н
Важно отметить, что и антрацен и нафталин частично
гидрируются в присутствии 27, но моноциклические арены этим
катализатором не восстанавливаются.
Координационная химия соединения 27 и его производных,
а также стехиометрическое гидрирование антрацена в его
присутствии были исследованы Халперном [376]. На основании его
выводов можно представить правдоподобный механизм
гидрирования полициклических аренов; однако о кинетическом
изучении полного каталитического дикла не сообщалось. Тем не
менее полученные Халперном результаты предоставляют
убедительные доказательства в пользу гомогенно катализируемого
реакционного цикла.
Мнимым катализатором является антраценовый комплекс
29, получающийся в результате двух реакций: необратимого
присоединения Н2 к 27 с образованием тригидрида 28
[уравнение (10.39)] и последующего взаимодействия 28 с антраценом,
приводящего к образованию красного антраценового компле-
3—1129
Таблица 10.2. Гомогенные катализаторы для гидрирования аренов
Предкатализатор
А. Е1зА1 + Щ11)-2-этилгексаноат
или Et3Al+(RCO2)2M
Б. т]3-СзН5Со[Р(ОМе)з]з
В. Комплекс N-фенилантранило-
вой кислоты с Rh(I)
Г. №С12(ВН4)(ДМФА)руа
Д. [(Ti5-C5Me5)RhCl2]2
Е. (Ti6-C6Me6)(r]4-C6Me6)Ru
Ж. (Ti6-C6Me6)Ru(H)Cl(PPh3)
3. [(Ti6-C6Me6)Ru]2|[(jx-H)2-
(jx-Cl)]Cl, генерируется in situ
из [RuCl2(ri6-C6Me6)]2H Na2CO3
И. [RuH2(PPh3)2(PPh2C6H4)]-
К. Со2(СО)8
Условия
7000 кПа,
150—210 °С
100 кПа, 25 °С
100 кПа, 20 °С
ДМФА
ДМФА
500 кПа, 50 °С,
15 экв. Et3N,
изопропанол
150—300 кПа,
90 °С
5000 кПа,
50 °С
5000 кПа,
50 °С
1000 кПа,
100 °С, ТГФ
20000 кПа,
200 °С
'Субстрат
о-Ксилол
C6D6
СбН6
CioHg
Пиридин
C6D6
СбНб, п-кси-
лол
СбНе
O6D6
Антрацен
Антрацен
Продукт
^ш>1,2-Диметил-
циклогексан
C6H6D6 (полно-
стью цис)
QHi2
Q0H18
Пиперидин
СбО6Нб (почти
полностью цис)
СбН12, цис-\,4-№-
метилциклогексан
СбН12
C6D6H6
>95%
1,2,3,4-Тетрагид-
роантрацен
9,10-Дигидроант-
рацен
Характерные особенности
Наиболее активен при
соотношении Al/Ni«3,5 : 1, черные рас-
i воры
Около 20 каталитических циклов
за 48 ч; неприменим в
присутствии групп NO2, F и CN
Антр ацен> нафталин> бензол
Хинолин восстанавливается, изо-
хинолин нет
225 каталитических циклов за
36 ч; бензол>толуол
Количественная регенерация
катализатора, с увеличением алкиль-
ного замещения скорость
уменьшается, значительное
образование циклогексена, интенсивный
обмен Н—D
Частота каталитических циклов
0,8 мин"1, циклогексены не
обнаружены
900Ю каталитических циклов за
36 ч, тиофен подавляет
активность, галогенобензолы
подвергаются гидрогенолизу
Производные бензола не
гидрируются
Т /Г
Имеются доказательства в поль
зу свободнорадикального
механизма
Литература
29
30
31
32
33
34
35
36
37
38
Гомогенные каталитические процессы 35
кса 29 [уравнение (10.40)]. В реакции (10.39) в процессе,
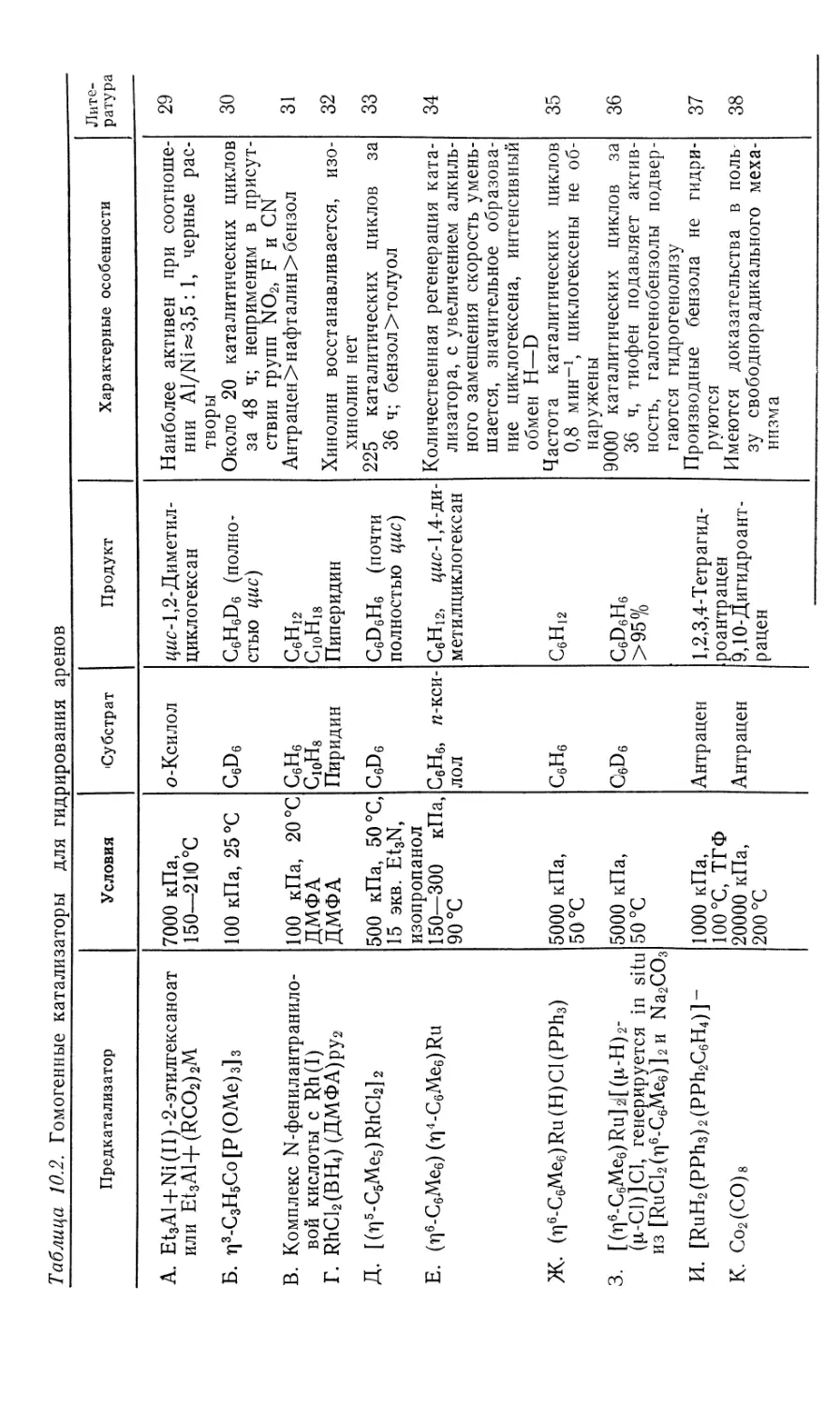
обратном ортометаллированию, генерируется координационный
центр, необходимый для окислительного присоединения Н2.
PPh3 I
3 C0 39)
24 v н< ,
н
28
(10.40)
L
H н H
28
I ' L
H |
H
30
\
L =
L' =
PPh5
PPhEt
Отнесение стереохимии, предложенное для нескольких
анионных рутениевых соединений этого ряда, было сделано на
основании близкого соответствия между их мультиядерными
спектрами ЯМР и спектрами аналогичных нейтральных
иридиевых комплексов установленного строения. Для примера
можно сравнить структуры 28 и 30 [уравнение (10.41)].
(10.41)
Весьма интересно превращение тригидрида 28 в
антраценовый комплекс 29 [уравнение (10.40)]. Эта реакция,
описываемая законом скорости для реакции нулевого порядка по
антрацену, видимо, происходит через предварительное
восстановительное элиминирование Н2 [уравнение (10.42)]. Моногидрид-
ный интермедиат 31, который невозможно детектировать
спектроскопически, отличается высокой степенью ненасыщенности и
потому очень подходит для образования т]4-антраценового
комплекса 29.
лимитирующая
стадия антрацен
фас-^иНзЬзГ >■ [RuHL3]- * 29 (10.42)
—Н2 быстро
28 31
36
Глава 10
Присоединение четырех молей Н2 к антраценовому
комплексу 29 приводит к тетрагидроантрацену и пентагидриду 32
[уравнение (10.43) J; реакция быстро протекает при 25 °С. Пен-
тагидрид реагирует с двумя молями антрацена, в результате
регенерируется комплекс 29 и образуется другой моль тетра-
гидроантрацена [уравнение (10.44)]. Эти две реакции, (10.43)
и (10.44), были изучены независимо, но вместе взятые они
составляют вероятный каталитический цикл. Вторая реакция
[уравнение (10.44)] более медленная и может быть
лимитирующей стадией в общем каталитическом процессе, показанном в
уравнении (10.38), однако неизвестно, является ли этот путь
доминирующим.
н
[RuН(L)2
29
4Нр
25°С
ТГФ
(10 43)
[RuH5(L)2]1"'
32
25°С
24 ч
[RuH(l) г
29
Н Н
Приведенный выше механизм иллюстрирует несколько
общих положений. В уравнениях (10.39) и (10.42) показаны два
различных процесса, при которых генерируются
координационные вакансии. ^-Координация антрацена в 29 объясняет ре-
гиохимию восстановления и тот факт, что эти катализаторы
неэффективны для восстановления моноциклических аренов —
бензола, толуола или тетрагидронафталина.
Полициклические ароматические соединения гидрируются
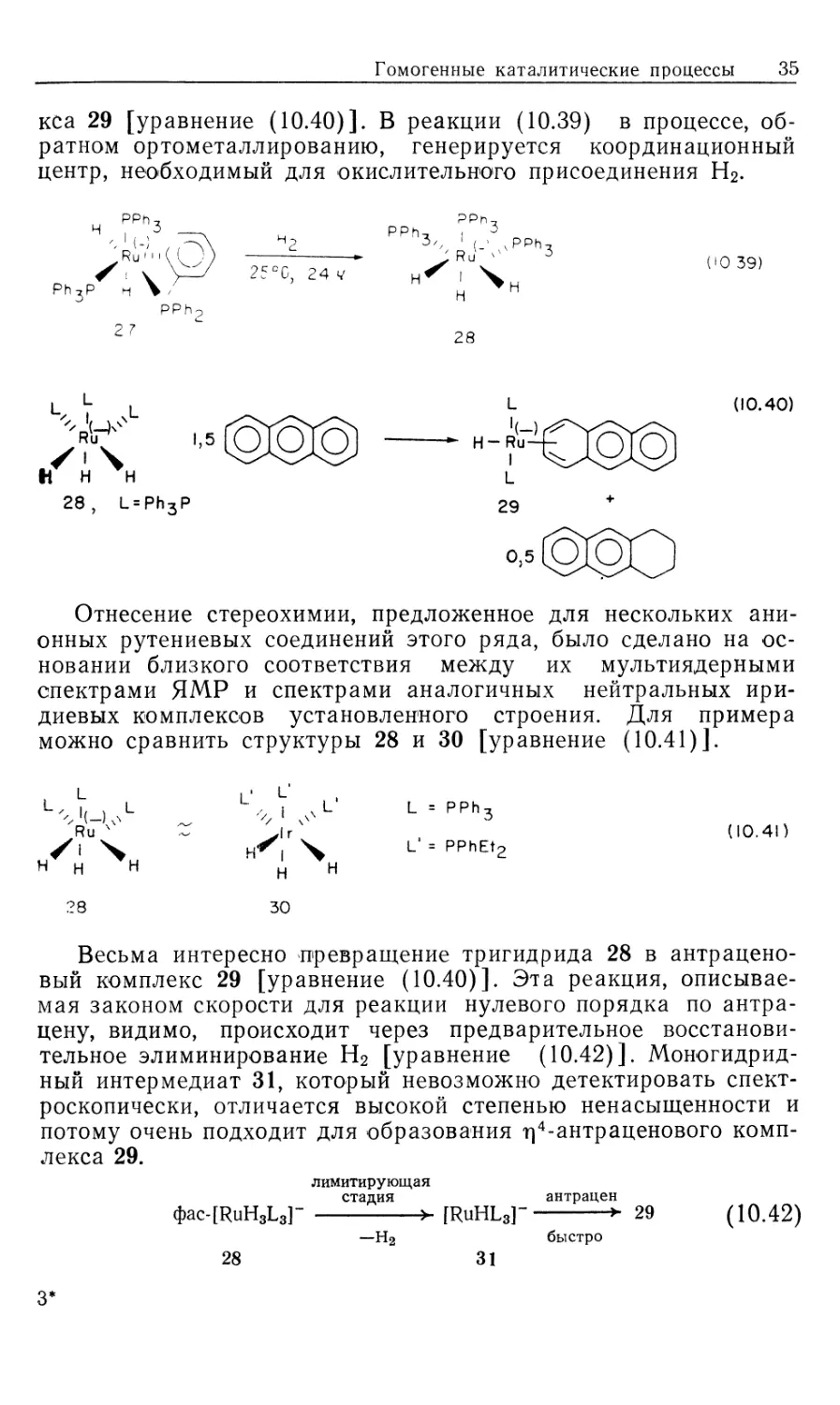
также в присутствии моногидридного катализатора НСо(СО)4
[уравнение (10.45)]. Реакция (10.45) очень отличается от
реакции, катализируемой комплексом рутения [уравнение (10.38)].
Так, ее региохимия совершенно иная — в качестве продукта
образуется только 9,10-дигидроантрацен в виде смеси цис- и транс-
стереоизомеров. 9,10-Диметилантрацен реагирует быстрее, чем
антрацен. Наблюдается обмен между дейтерием в положениях
9, 10 и Н2. Добавление монооксида углерода не понижает
скорость реакции и не приводит к образованию продуктов карбо-
нилирования. Реакция имеет первый порядок как по НСо(СО)4,
так и по ароматическому субстрату. Все эти факторы учитывает
Гомогенные каталитические процессы 37
НСо(СО)4
~ 200 атм Н
200°С
2 1
(10.45)
предложенный Халперном [38] свободнорадикальный механизм,
показанный на рис. 10.7. Лимитирующей стадией является
передача атома водорода от НСо(СО)4 к антрацену. Энтальпия
активации этой лимитирующей стадии зависит от сродства
антраценового субстрата к свободным радикалам. Рассмотрение
энергетики этой стадии для разных субстратов приводит к пред-
НСо(СО)4
•Со(СО)4
2«Со(СО)4
Со2(СО)8 +
Со2(СО)8
=± 2НСо(СО)4
НСо(СО)4
R Н
•Со(СО)4
Рис. 10.7. Предлагаемый свободнорадикальный механизм гидрирования
антрацена.
сказанному соотношению скоростей, которое уменьшается в
ряду антрацен : нафталин : бензол как 1000 : 10 : 1. Эти данные
хорошо объясняют тот факт, что радикальные пути моногидридно-
го каталитического гидрирования сильно зависят от природы
сопряженного субстрата. Например, простые производные
бензола нереакционноопособны, но стирол может реагировать по
подобного рода свободнорадикальному пути. Механизм такого
типа не требует предварительной координации субстрата, и
потому на него не влияет избыток СО. Диметилпроизводное имеет
более высокое сродство к радикалам, чем сам антрацен. Регио-
химию реакции можно объяснить в терминах наиболее
устойчивого радикального интермедиата. Все это свидетельствует о том,
что каталитическое гидрирование действительно осуществляется
радикальным путем.
38 Глава 10
Из представленного на рис. 10.7 механизма видно, что вторая
стадия заключается в быстром отрыве атома водорода от другой
молекулы НСо(СО)4. Димеризация радикала •Со\(СО)4 и гид-
рогенолиз Со2(СО)8 завершают каталитический цикл. Можно
ожидать, что любой гидрид 'переходного металла первого ряда
будет катализировать аналогичные реакции, при условии что
гидрид можно регенерировать гидрогенолизом.
б. Гидрирование моноциклических аренов. Как показано в
табл. 10.2, некоторые растворимые комплексы переходных
металлов служат предкатализаторами для гидрирования бензола
и его производных. Как правило, эти катализаторы либо
требуют высокого давления, либо реагируют очень медленно с очень
небольшим числом каталитических циклов за несколько часов.
Сообщалось о некоторых случаях высокой стереоселективности,
но экспериментальные детали очень разрозненны, к тому же
ничего не известно о конкретных проявлениях этой
стереоселективности, которые могли бы служить доказательством
гомогенности. Более того, для растворимых предкатализаторов этой
категории часто наблюдается индукционный период и наличие ме-
таллосодержащих осадков, указывающих на возможность
гетерогенного катализа. Предварительно уже говорилось о том,
насколько сложно определить истинную каталитически активную
частицу, даже для хорошо известных гомогенных
катализаторов гидрирования олефинов. Еще труднее это сделать в случае
катализаторов гидрирования моноциклических аренов. Здесь
главный вопрос заключается в том, являются ли активные
катализаторы металлическими «золями» размером от 10 до 40 А.
Далее мы будем часто возвращаться к этому моменту при
рассмотрении полимерных субстратов, используемых для
установления гомогенности катализатора.
С практической точки зрения не доказано, что «гомогеннные»
катализаторы гидрирования моиоциклических аренов
одновременно и селективны и достаточно реакционноспособны, чтобы
заменить хорошо разработанные гетерогенные катализаторы.
Однако последние страдают такими серьезными недостатками,
как тенденция катализировать изотопный обмен Н—D и
отсутствие стереоспецифичности. Этих недостатков должны быть
лишены гомогенные катализаторы гидрирования аренов, так что
проблема сводится к тому, чтобы их обнаружить и найти среди
них реакционноапошбные соединения.
Рассмотрим некоторые данные табл. 10.2, имея в виду как
практические моменты, так и соображения о механизме. Мало
изученные катализаторы, получаемые из растворимых карбок-
силатов .переходных металлов и алкилалюминия [29] (лункт А),
проявляют некоторые свойства, указывающие на гомогенность в
стереоспецифическом ^wc-восстановлении субстратов [уравне-
Гомогенные каталитические процессы 39
ние (10.46) J. Гомогенность этих катализаторов подтверждается
результатами гидрирования растворимого полистирола [29д],
но этот результат следует проверить еще на сетчатых
полимерах. Как явствует из экспериментов с рассеянием света,
темные растворы катализатора содержат очень мелкие частицы
[39J.
(10 46)
/—\ н?' 700 атм МеО2Сv/--^ ^c02Me
МеО2С—(О)—СО о Me - ,( У
100°С, Ni(acac)2 H '' Н
Аллилкобальтовый предкатализатор (пункт Б) настолько
неактивен, что практически непригоден [30]. Родиевый
предкатализатор (пункт В) [31], видимо, должен относиться к классу
полициклических аренов. Гидрирование пиридина боргидридсо-
держащим родиевым катализатором (пункт Г) [32]
представляет особый случай, который нельзя экстраполировать на простые
арены. Для родиевого катализатора (пункт Д) [33] не удалось
провести тест на гомогенность (см. ниже). Рутениевый
предкатализатор (лункт Е) [34] заслуживает дальнейшего изучения.
Утверждалось, что все предкатализаторы можно
регенерировать в неизмененном виде, но этот лиганд должен подвергаться
обмену, если в реакции участвует растворимый катализатор.
Рутениевый предкатализатор в пунктах Ж и 3 [35, 36]
содержит тот же лиганд, что и предыдущий предкатализатор. Все эти
катализаторы также заслуживают дальнейшего изучения
благодаря мягким условиям реакции и достаточно высокой частоте
каталитических циклов. Однако, как сообщалось [28],
родственное нафталинсодержащее производное рутения непригодно для
гидрирования аренов, если после индукционного периода не
выпадает твердый осадок (возможно, металлический рутений).
Соединения, представленные в пунктах И и К уже подробно
обсуждались ранее. Таким образом, очевидно, что только
соединения, представленные в пунктах Г и И, предлагают
практическую альтернативу гетерогенным катализатором.
10.6. Гомогенное гидрирование других функциональных групп
Известны, хотя и в меньшем количестве, растворимые
предкатализаторы, способствующие гидрированию таких
функциональных соединений, отличных от олефинов, как альдегиды, ке-
тоны, сложные эфиры, нитрилы, ангидриды, нитросоединения.
Однако все они гидрируются при использовании хорошо
известных гетерогенных катализаторов, поэтому не было стимулов
40 Глава 10
разрабатывать гомогенные катализаторы. Хиральные
гомогенные катализаторы представляются многообещающими для
асимметрического гидрирования несимметричных кетонов [15], но,
как было показано на примере прохиральных олефинов, лишь
для очень немногих субстратов был получен высокий энантио-
мерный избыток (ее) и не известно ни одного случая, когда бы
величина ее превышала 90%, а именно такая величина
требуется для практического асимметрического синтеза.
Важно отметить те субстраты, которые не
восстанавливаются в присутствии растворимых катализаторов. Так, не
известны какие-либо гомогенные катализаторы для гидрогенолиза бен-
зильных и бензгидрильных производных или циклопропанов;
поэтому использование этих полезных реакций ограничено пока
областью гетерогенного катализа.
Далее обобщены типичные примеры гомогенного
каталитического гидрирования субстратов, отличных от олефинов и аренов.
а. Альдегиды и кетоны. Комплексы металлов VIII группы
оказались наиболее эффективными катализаторами
гомогенного гидрирования кетонов до вторичных спиртов [40]. С этой
целью использовались такие фосфиновые комплексы, как
RhCl(PRsh(CO) [41], RuCl2(PPh3)3 [42], RuH2(CO) (PPh3)3
[42], Ru(CF3CO2)2(CO)(PPh3)2 [42] и [RhH2X2L2]+, где Х =
растворитель, L = PPh2Me или РМе3 [40а].
Большинство этих соединений являются также
катализаторами гидрирования олефинов. Как лиганды альдегиды и кетоны
обычно хуже олефинов, поэтому их восстановление протекает
медленнее и часто требует более жестких условий.
Гидрирование альдегидов относится к числу процессов, представляющих
интерес для промышленности, как стадия, обычно проводимая
во время или после гидроформилирования (разд. 12.3). Для
гидрирования кетонов чаще всего использовались катионные
родиевые катализаторы.
В последнее время для этой цели применяют комплексы
родия, содержащие координированные с азотом лиганды,
например 1,10-фенантролин, 2,2/-дипиридил, замещенные фенантроли-
ны [43]. Это первые случаи использования подобных лигандов
в гомогенном катализе. Развитие этого направления
представляется перспективным, если учесть склонность фосфинов к
деградации при окислительном присоединении по связи Р—С [44].
Широко изучалось асимметрическое гидрирование кетонов,
но даже лучшие результаты оказались разочаровывающими. Все
кетоны, восстанавливающиеся с достаточной величиной ее,
содержат вторую функциональную группу, способную к хелатиро-
ванию с металлом. Ситуация напоминает асимметрическое
гидрирование прохиральных олефинов и может возникать в
результате аналогичных факторов. Примером служит реакция (10.47),
Гомогенные каталитические процессы 41
где в качестве хирального фосфина (PRsb используется
1-[(5)-1/2-бис(дифенилфосфино)ферроценил]этанол, а
величина ее составляет 59%.
О
II н2
МеС - СО2Н >- МеСН(ОН)СО2- (10 47)
[Rh(PR3)2(S)2] +
б. Сложные эфиры карбоновых кислот. Сложные эфиры кар-
боновых кислот с трудом поддаются гидрогенолизу, даже при
использовании гетерогенных катализаторов требуется высокая
температура (200 °С) и давление более 70 атм. Большинство
гомогенных катализаторов не оказывает влияния на эту реакцию.
Однако открытый Греем и Пецем [37] анионный рутениевый
предкатализатор 27 промотирует гидрирование сложных эфиров
в относительно мягких условиях [уравнение (10.48) J. В этой
реакции важны стерические эффекты. Так, этилацетат и метил-
пропаноат реагируют медленнее, чем метилацетат, а метил-
бензоат вообще не восстанавливается. Большое значение имеет
и образование ионных л ар; так, краун-эфиры уменьшают
скорость реакции. Поскольку гетерогенные катализаторы довольно
инертны, есть все основания разрабатывать более совершенные
гомогенные катализаторы для гидрирования сложных эфиров.
О 10 атм Иг О
СН3СОСН3 "* СН3СН2ОН + СН3СОСН2СН3 + СН3ОН (10.48)
90°С, толуол
27 = [к (диглиМ^ [RuH2(PPh3)2(PPh2c6H4G "
в. Нитрилы. Нитрилы тоже с трудом поддаются
гидрированию. Их удается гидрировать с помощью анионного
рутениевого комплекса 27, но реакция идет медленно. Эффективным
гомогенным катализатором оказался родиевый комплекс 33, при
использовании которого реакция осуществляется в удивительно
мягких условиях [уравнение (10.49)] [45]. Реакция обратима,
в присутствии 33 первичные амины дегидрируются [уравнение
(10.50)].
R-CeeN ^™ТГФ R-CH2-NH* 45-loo°/o (Ю.49)
R = ft-Bu, изо-Pr, mpem-Bu, Bz, CH2=CHCH2, Ph
33
33 = RhH(PPr3-W3o)3
1 10 °С, 0,05 мм рт. ст.
PhCH2NH2 >• PhC=N + H2 (10.50)
33
г. Ангидриды карбоновых кислот. Ангидриды карбоновых
кислот мало изучались в качестве субстрата при гомогенном
42 Глава 10
каталитическом гидрировании. Интересный пример такой
реакции представляет гидрирование янтарного ангидрида в
присутствии рутениевого предкатализатора [уравнение (10.51)] [46,
47]. Продукт получается с выходом 50%; это объясняется
образованием воды, под действием которой молекула ангидрида
раскрывается, давая янтарную кислоту, инертную к
восстановлению в данных условиях.
■° со£н
0 н2
50% 50%
д. Нитросоединения. Нитрогруппа в ароматических нитросо-
единениях довольно легко восстанавливается под действием
многих реагентов, а также при гетерогенном каталитическом
гидрировании. Гомогенные комплексы часто оказываются
активными катализаторами гидрирования ароматических нитро-
групп, но редко используются в синтетических целях. Механизм
этих реакций недостаточно ясен. К тому же процессы'
восстановления иногда осложняются образованием азосоединений,
тогда как восстановлению алифатической нитр о группы мешает
образование оксимов через нитрозоинтермедиаты.
Разносторонними свойствами обладает открытый Мак-Куиллином комплекс
родия 34, содержащий в качестве одного из лигандов бор
гидрид [32]; он оказался эффективным катализатором для
гидрирования нитроарильных соединений. Реакция идет даже в том
случае, когда в качестве субстрата берут сетчатый полистирол
35 [уравнение (10.52)] [48]. Как будет показано далее, этот
результат подтверждает вывод о том, что активный катализатор
гомогенен.
24
Н2 (34)
10.7. Иммобилизация гомогенных катализаторов гидрирования
В 70-х годах обширные исследования были посвящены
гомогенным катализаторам на основе переходных металлов,
прикрепленным к нерастворимым носителям (подложкам); они
получили разные названия — иммобилизованные, нанесенные на
подложку, гибридные, закрепленные — и использовались во
многих реакциях: гидроформилировании, окислении этилена по
методу Вакера, гидрировании, гидросилировании, димеризации
Гомогенные каталитические процессы 43
олефинов, метатезисе олефинов, циклотримеризации ацетилена.
Этому предмету посвящен обзор [49]. Наиболее широко
изучены гибридные катализаторы гидрирования олефино©, которые
вкратце обсуждаются ниже.
В принципе гибридный катализатор мог бы сочетать
преимущества растворимых катализаторов (гомогенность, утилизация
всех атомов металла, большая скорость реакции и высокая
селективность) с такими достоинствами гетерогенных
катализаторов, как легкость отделения продукта от катализаторов,
регенерация и повторное использование дорогого катализатора.
В прочих отношениях гибридные катализаторы могут даже
превосходить растворимые аналоги. Например,
высоконенасыщенные центры в них могут стабилизироваться за счет
агрегации, их можно вводить в реакцию с такими растворителями,
в которых нерастворим гомогенный катализатор.
Однако практически гипотетические преимущества
гибридных катализаторов очень редко реализуются в лабораторном
эксперименте и никогда в промышленных процессах
(исключение составляют иммобилизованные ферменты, которые здесь
рассматриваться не будут). Неудивительно, что их
промышленная разработка приостановлена. Гибридные катализаторы
трудно приготовить и охарактеризовать; сложность работы с ними
усугубляется низкой скоростью реакции, невозможностью
выделить активный центр, и особенно их неустойчивостью к
вымыванию с подложки. Большинство этих катализаторов содержат ко-
валентно связанные фосфины, и главной причиной разрушения
катализатора является окислительное присоединение по связи
Р—С [44] (см. ч. 1, гл.5). Кроме того, литературные данные по
этим катализаторам, как правило, плохо документированы, не-
воспроизводимы и потому вводят в заблуждение, так что
экспериментатору не следует относиться к ним с доверием.
В качестве носителей используются главным образом
полистирол и диоксид кремния.
а. Катализаторы, связанные с органическими полимерами.
Сетчатый полистирол («гель») можно подвергнуть
функциональному замещению с помощью обычных органических
реакций при условии, что сетчатый полимер набухает в
растворителе, используемом для данной реакции. Катализатор, лиганды
которого связаны с полимером^ можно получить только в
растворителе, в котором этот полимер набухает. Однако в этих
условиях катализатор распределяется вдоль всей полимерной
цепи и даже в разбавленных растворах возникает агрегация,
так что практически невозможно выделить активный центр
[50]. Взаимодействие активных центров замедленно, но
происходит даже в макропористых смолах с минимальной
способностью к набуханию.
44 Глава 10
Помимо серьезных проблем с агрегацией и вымыванием,
связанные с полимерами катализаторы отличаются пониженной
скоростью реакции, что видимо связано с более медленной
диффузией субстрата к каталитическим центрам, хотя этот эффект
не самый важный. Например, активность нанесенного на
полимерную подложку аналога катализатора Уилкинсона
составляет около 7% активности растворимого комплекса [51].
Один из хорошо доказанных случаев повышенной активности
связанного с полимером катализатора описан Граббсом [49aJ.
Он приготовил титаноценовый предкатализатор 36,
прикрепленный к макропористому полистиролу со степенью сшивания 20%
[уравнение (10.53) J. В реакции гидрирования олефинов этот
полимерный катализатор проявлял активность в 60—120 раз
большую, чем соответствующий растворимый аналог, имеющий
выраженную тенденцию к агрегации.
3) MeLi
4) CpTiCI3
5)H-BuLl
Альтернативный подход к закрепленным катализаторам,
который хотя и мало изучался, но кажется многообещающим,
состоит в использовании ионообменных смол в сочетании с
ионными катализаторами. Например, на катионообменную смолу
наносят катионный катализатор, образующийся in situ из
Rh2Cl2(C8Hi4)2 и 2-аминопиридина в этаноле. В присутствии
такого катализатора гидрирование циклооктена происходит в
10 раз быстрее, чем с катализатором Уилкинсона [52].
б. Катализаторы, связанные с диоксидом кремния.
Гомогенные катализаторы, связанные с диоксидом кремния, должны
превосходить катализаторы, нанесенные на смолы.
Максимальная концентрация реакционноспособных свободных силаноль-
ных групп на обширной поверхности диоксида кремния (около
300 м2/г) составляет в грубом приближении единицу на 100 А2.
Самый простой метод закрепления функциональных групп на
SiO2 включает использование растворимых силилирующих
агентов, которые образуют «стеклянные связи» Si—О—Si с
поверхностью носителя. В гл. 12 будет рассмотрена реакция гидрофор-
милирования в присутствии гибридного катализатора, лиганды
которого связаны с диоксидом кремния. Потенциальные
преимущества жестких матриц SiO2 для иммобилизации
гомогенных катализаторов перекрываются трудностями их синтеза и
Гомогенные каталитические процессы 45
аналитического исследования, а также проблемами снятия с
носителя [54], поэтому диоксид кремния в качестве подложки
используется реже, чем смолы. Кроме того, высоконенасыщенные
комплексы на этих поверхностях могут образовывать частицы
металла с большой площадью поверхности. Даже малые
количества таких гетерогенных металлических катализаторов
могут быть настолько реакционноспособны, что подавят
каталитическую активность закрепленных металлокомплексов.
Указанием на наличие реакционноспособных частиц металла служит
каталитическое гидрирование бензола.
Таким образом, вопрос о применении гибридных
металлических катализаторов представляется противоречивым.
Практическое применение этих веществ ограниченно; более подробную
информацию можно найти в обзоре [49].
10.8. Тест на гомогенность катализаторов гидрирования
Многие гомогенные предкатализаторы проявляют признаки
гетерогенных катализаторов; к таким признакам относятся,
в частности, индукционные периоды и возможность
гидрирования бензола. Очень трудно установить, является ли активным
катализатором растворимый металлокомплекс или
суспендированные частицы металла [55]. Последние могут присутствовать
в следовых количествах в каталитических системах в условиях
восстановления, т. е. при высоком давлении водорода или в
присутствии сильных водородсодержащих восстанавливающих
агентов. Это особенно справедливо для растворимых предката-
лизаторов, которые, как известно, распадаются в ходе
каталитической реакции. Выделяемые после реакции осадки нельзя
считать датчиками истинного катализатора, поскольку у
больших частиц может быть такая малая площадь поверхности, что
они могут и не проявлять каталитическую активность в
измеримой степени. В случае промышленных гетерогенных
катализаторов главная функция таких носителей, как оксид алюминия или
углерод, заключается в стабилизации частиц металла, имеющих
малый размер, но большую площадь поверхности, и
предотвращении агрегации. Каталитическая активность при этом
возрастает, и один каталитический центр может участвовать в
образовании тысяч молекул продукта. Таким образом, очень малые
количества очень активного катализатора могут быть
«невидимы» в растворе, содержащем спектроскопически наблюдаемые
металлоорганические комплексы, сами по себе каталитически
неактивные.
Один из подходов к решению данной дилеммы был
предложен Уайтсайдсом [56]. Он состоит в том, что раствор
катализатора выдерживают в присутствии элементарной ртути, отрав-/
ляющей гетерогенные катализаторы путем блокирования пор
46 Глава 10
твердого вещества с большой площадью поверхности. Другой
подход заключается в использовании сетчатых полимеров в
качестве субстратов [48J. Другие подходы описаны в работе [43]
в гл. 5 (ч. 1).
Гетерогенные катализаторы, как нанесенные на подложку,
так и суспендированные («золи»), неактивны при гидрировании
субстратов в растворимых и особенно сетчатых полимерах.
Очевидно, даже малые частицы металла не могут эффективно
диффундировать через набухшую в растворителе смолу.
Наоборот, истинно гомогенные катализаторы эффективно промотируют
гидрирование субстратов, связанных с полимером, хотя и с
несколько меньшей скоростью. Связанный с полимером субстрат
должен набухать в растворителе, совместимым с гомогенным
катализатором. Применение этого теста на гомогенность
иллюстрируется уравнением (10.54) [48].
Некоторые катализаторы, гомогенность которых вызывала
сомнения в связи с условиями реакции или их селективностью
по субстрату, были проверены с помощью описанного критерия
и действительно оказались гомогенными. Среди них —
катализатор Мак-Куиллина [уравнение (10.52)] и катализатор Крэбтри
[уравнение (10.54)]. Достоинство теста состоит в том, что он
основан на самом каталитическом процессе, а не на
детектировании катализатора.
(10.54)
катализа- —N
Катализатор
RhCll_3
[Rh(NBD)L2]cl04
/СН3
V-сн
^сн3
Выход, %
100
90
Pd/C
[lr(COD)(P(iPr)3py)J PF6 100
Одним из вариантов теста является использование
полимерных каталитических ядов, таких, как политиолы. Найдено, что
они не оказывают влияния на гетерогенные катализаторы, но
тормозят действие гомогенных катализаторов [48].
Известен только один пример применения данного теста к
катализаторам гидрирования аренов [48]. Предкатализатор
[iRh(C5Me5)Cl2]2 неактивен при гидрировании аренов ни с
гранулированным сетчатым полистиролом, ни с гидратированным
бутадиен-стирольным сополимером [уравнение (10.55)], однако
олефиновые группы гидрируются в сетчатом полимере [уравне-
Гомогенные каталитические процессы 47
ние (10.56) J. По-видимому, эта система имеет гомогенную
компоненту, способную гидрировать олефин, но распадающуюся
при гидрировании арена.
[Rh(C5Me5)CI2]2, (l0.55)
шо-PrOH о
-СН2 CH-tt-CH2CH2CH-CH2-t- *" реакция не иает
ОН Н2, Et3N
CH2CI2, Et3N,
н2
Следовало бы применить описанные тесты для поиска
подходящих предкатализаторов, особенно для гидрирования
аренов.
10.9. Гомогенный катализ в реакциях с переносом водорода
В некоторых реакциях гидрирования вместо Н2 можно
использовать другие восстанавливающие агенты, налример
NH2NH2, NaBH4, спирты, НСО2Н, СО/Н2О, они описаны в
обзоре Бёрча [1в]. Показательным примером служит гидрирование
олефина муравьиной кислотой в присутствии рутениевого
катализатора [уравнение (10.57)J.
Ru(H)ClL3
RCH=CH2 + HCO2H >■ RCH?CH3 + CO2 (10.57)
ДМФА
Ряд гомогенных каталитических реакций с переносом
водорода изучил Хенбест [57]. При использовании комбинации из
соли иридийхлорида, влажного изопропилового спирта в
качестве донора водорода и такого лиганда, как триметилфосфит,
в кислой среде циклогексаноны восстанавливаются с
преимущественным образованием аксиальных спиртов. Вместо триметил-
фосфита можно использовать диметилсульфоксид (ДМСО) или
фосфористую кислоту. В некоторых случаях фосфит или ДМСО
оказываются первичными восстанавливающими агентами.
Пример представлен уравнением (10.58).
Н (!а58)
изо-РгОН, Н2О
97% 3%
Реагент чувствителен к стерическому затруднению в
субстрате и потому проявляет полезную регеоселективность. Так,
48 Глава 10
в стероидных субстратах восстанавливаются 2- и 3-кетогруппы,
тогда как кетогруппы в положении 11, 17 и 20 не
затрагиваются [уравнение (10.59)]. Следует обратить внимание на стерео-
селективное образование За (аксиального)-спирта.
о о
[II 1 Н21гС16, Р(ОМе)3
([Г изо-PrQHt НрО
^j ОНИ
94%
В этих условиях селективно восстанавливается олефиновая
группа сопряженных кетоолефинов [уравнение (10.60)].
i? _H_2_lrCl6 ?
Ph^ ^^ "'"^Ph ДМСО Ph^ ^ Ph
Для (проведения реакций, катализируемых различными
растворимыми комплексами переходных металлов, в качестве
восстанавливающей среды удобно использовать синтез-газ (СО+
+Н2). В некоторых случаях активным восстанавливающим
агентом оказывается такой интермедиат, как гидрид металла,
образующийся при гидролизе карбонильных соединений, а сам
водород в реакции не участвует. Таким образом Петтит [58]
восстанавливал ароматические нитросоединения [уравнение
(10.61)].
Z + Нг/СО ~^^^>^^NH2 (Ю6))
ТГФ, 43 атп
M=Fe{C0)5l Rb6(C0)|6 и т.д.
10.10. Гомогенное каталитическое силилирование
Гидросилилирование, т. е. присоединение связей Si—Н к оле-
финам, ацетиленам, альдегидам и кетонам, катализируется
многими растворимыми комплексами переходных металлов [59].
Известно также гидросилилирование с гетерогенными
катализаторами. Каталитическое гидросилилирование аналогично
гидрированию, но протекает более экзотермично (примерно на
5 ккал/моль). Реакция имеет большое значение для
производства некоторых производных кремния [60]. Известно также ка-
Гомогенные каталитические процессы 49
талитическое присоединение связей Ge—Н [61], но здесь оно
не будет рассматриваться.
Процесс каталитического гидросилилирования очень сложен.
Основываясь на известных стадиях реакции, можно
представить вероятные схемы, однако полный механизм процесса до
конца не понятен. Реакции осложняются рядом факторов:
индукционные периоды, невоспроизводимая кинетика, возможные
радикальноцепные процессы, перегруппировки и димеризация
олефинов, изотопный обмен у атома кремния и в олефине,
выраженная чувствительность продукта к природе гидросилили-
рующего агента. В некоторых случаях неясно, является ли
катализатор гидросилилирования гомогенным или гетерогенным.
Несмотря на эти осложнения, каталитическое гидросилили-
рование представляет потенциально удобный процесс, но делать
какие-либо обобщения было бы неразумно.
а. Механизм гидросилилирования олефинов. Некоторые
катализаторы гидрирования оказывают также каталитическое
действие при гидросилилировании. Исходя из анализа
продуктов и наблюдения индивидуальных стадий, можно представить
правдоподобный механизм каталитического
гидросилилирования (рис. 10.8). Предполагается, что обязательной стадией в
каталитическом гидросилилировании является окислительное
присоединение гидридов кремния [62]. Неясно, происходит ли
координирование олефинового субстрата до стадии
окислительного присоединения Si—Н, т. е. возникает тот же самый вопрос
об «олефиновом» или «гидридном» пути, который ставился при
рассмотрении каталитического гидрирования. В ряде случаев
вероятным кажется олефиновый путь, поскольку олефиновые
комплексы являются эффективными предкатализаторами, а не-
* R3siH
r3s;-c-c-h
Рис. 10.8.
4—1129
50 Глава 10
которые катализаторы гидросилилирования разлагаются в
присутствии гидридов кремния, когда олефин отсутствует.
Показанный на рис. 10.8 механизм включает две обратимые
стадии: окислительное цис-присоединение Si—Н (ki, k-i) и гид-
ридная миграция (k2, k-2), после чего следует необратимое
восстановительное элиминирование — стадия образования
продукта (&3). Обратимость первых двух стадий объясняет иногда
наблюдаемый изотопный обмен между олефином и связью Si—Н,
а также перегруппировку олефина, являющуюся обычной
побочной реакцией. Природа заместителей в силане может влиять
на региоспецифичность стадии внедрения в случае
несимметричных олефинов, а также на склонность олефинов к
перегруппировкам. При использовании меченных дейтерием силанов
можно наблюдать интенсивные перегруппировки между
гидридом кремния и олефиновым субстратом [63], но в других
случаях присоединение происходит без изомеризации олефина и
без изотопного обмена [64]. На примере нескольких
катализаторов показано, что в целом процесс гидросилилирования
протекает с сохранением конфигурации при хиральном атоме
кремния [65]; очевидно, конфигурация сохраняется в обеих
стадиях, и при окислительном присоединении, и при
восстановительном элиминировании.
б. Примеры гидросилилирования олефинов. Проводилось
сравнение относительных скоростей гидросилилирования этилена
в присутствии трех разных катализаторов, однако результаты
зависят еще и от природы силана и олефина [уравнение
(10.62)].
катализатор
PhMe2SiH + СНа=СН2 >- PhMe2S iCH2CH3 (10.62)
катализатор-Н2Р1С14>№С1Ьз>Со2(СО)8 (4000 : 200 : 1)
Изомеризация олефина и выраженная тенденция к
образованию я-алкилсиланов могут быть ответственны за образование
я-пентилсилана из цис-2-пеятеяа. в присутствии родиевого
катализатора [уравнение (10.63)]. Интересно, что более слабо
координирующий транс-2-иеятея в тех же условиях нереакционно-
способен [66].
Me
Me I
i —ч / RhCIL3 Si_Ph
PhSi-H + ^^ y^^^^^ \ (10.63)
! Me
Me
Гидросилилирование акрилонитрила катализируется
несколькими комплексами металлов VIII группы. В каждом
случае реакция высоко региоселективна и атом кремния
присоединяется к а-углероду [уравнение (10.64)].
Гомогенные каталитические процессы Ы
l
Rh(H)(CO)L*
— Me"f"Me (1064)
Me / \
Н3С CN
Гидросилилирование ацетилена протекает довольно сложно
и трудно предсказуемо. В случае ацетиленов с концевой
тройной связью (присоединение Si—Н нерегиоопецифично и
приводит к образованию двух моноаддуктов. Кроме того, может
иметь место как однократное, так и повторное присоединение,
иногда с выделением Н2 [67].
в. Гидросилилирование альдегидов и кетонов. По сравнению
с гидросилилированием олефиновых субстратов альдегиды и ке-
тоны легче вступают в реакцию благодаря тому сродству,
которое проявляет кремний по отношению к кислороду. Поэтому
неудивительно, что гидросилилирование карбонильных
соединений полностью региоспецифично, кремний всегда связывается
с кислородом, а гидрид — с углеродом. Последующий гидролиз
силиловых эфиров приводит к спиртам [68]. Таким образом,
процесс гидросилилирования с последующим гидролизом
служит альтернативой каталитическому гидрированию.
При использовании в качестве субстратов а,р-ненасыщен-
ных кетонов может восстанавливаться либо двойная связь,
либо карбонильная группа. Сообщалось, что моногидросиланы
реагируют исключительно путем 1,4-присоединения
(восстановление олефина), тогда как дигидросиланы дают
преимущественно продукты 1,2-орисоединения (восстановление кетона),
как это показано в уравнении (10.65).
R1 H
(10.65)
OSiR
R c.
н-с-с7
/ \
R* H
Jh3c
RCH-CHo
R1
3
0
-c-
R"
MS
R3
RhL2(S)2
HpSiRp
>
R
R1
I-
зо +
H
R"
^C .
H
2«
"^ R'
52 Глава 10
Наиболее изучено гидросилилирование применительно к
асимметрическому синтезу с использованием хиральных ли-
гандов, приведенных на рис. 10.3 [69J. Результаты напоминают
ситуацию с асимметрическим гидрированием прохиральных
олефинов. В случае простых кетонов и ^-ненасыщенных кето-
нов оптические выходы умеренные, и величина ее редко
превышает 50%. Более высокие оптические выходы достигнуты при
каталитическом гидросилилировании 1,4- и 1,2-дикарбонильных
соединений [уравнение (10.66). Вероятно, вторая
карбонильная группа действует как лиганд, превращая субстрат в хелат.
И степень энантиоселективности, и доминирующая
конфигурация продукта зависят от природы силана. На практике очень
трудно сделать выбор между возможными
экспериментальными вариантами, что должно быть хиральным — фосфин,
субстрат или силан. Более подробные данные можно найти в
обзоре [69J.
(10.66)
NpPhSiHg
lN(R) (PhCH2)(Ph)PMe (R), ее 85 %
10.11. Гомогенное каталитическое гидроцианирование
олефинов и ацетиленов
В отсутствие подходящих катализаторов HCN не
присоединяется к простым олефинам. Прочность связи Н—С
объясняется как слабой кислотностью HCN по Брёнстеду, так и его
неспособностью реагировать с олефинами ни по полярному, ни
по радикальному механизмам. Однако присоединение HCN к
олефинам и ацетиленам катализируется многими
растворимыми комплексами переходных металлов. Иногда вместо
ядовитого HCN удобнее использовать ацетонцианогидрин.
Катализируемое переходными металлами присоединение
HCN к олефинам изучается примерно с 1950 г. и отражено в
трех серьезных обзорах [70—72J. Главный стимул изучения
гидроцианирования связан с получением адиподинитрила,
которое было разработано в промышленном масштабе фирмой
DuPont в 1971 г. и явилось одним из успешных индустриальных
процессов, использующих гомогенные катализаторы на основе
переходных металлов [73].
а. Механизм. Концепция и механизм гомогенного
каталитического гидроцианирования тесно связаны с гидрированием и
гидросилилированием. Хотя детальный механизм
гидроцианирования до конца не ясен, можно записать последовательность
вероятных стадий, объясняющих некоторые из каталитических
Гомогенные каталитические процессы 53
циклов. Промежуточный гидрид металла образуется в
результате окислительного присоединения HCN к d8- или с110-комплек-
су [уравнение (10.67)]. За координацией олефина следует
внедрение [уравнение (10.68)]. Реакционный цикл завершается
восстановительным элиминированием алкилцианида [уравнение
(10.69)].
ч
М + HCN v s М-СЧ CO. 67)
.68)
(10.69)
Наличие этих стадий подтверждается различными наборами
данных в случае того или иного катализатора. Обратимое
внедрение гидрид — олефин объясняется изомеризацией
олефина и изотопным обменом, сопровождающими некоторые реакции.
Стадия внедрения [уравнение (10.68)] определяет региохимию
присоединения HCN; на нее влияет как природа олефинового
субстрата, так и катализатор. Для олефинов с концевой
двойной связью в присутствии объемных лигандов обычно
наблюдается присоединение против правила Марковникова. Возможно,
это связано со стерическими эффектами при внедрении
металл— гидрид. Мы еще встретимся с этим явлением при
рассмотрении каталитического гидроформилирования (разд. 12.3).
При использовании других катализаторов региохимия может
оказаться обратной [72].
Предполагается, что стадией, определяющей образование
шродукта, является восстановительное элиминирование
[уравнение (10.69) J, в некоторых случаях ее протекание облегчается
такими сокатализаторами, как кислоты Льюиса.
б. Гидроцианирование олефинов. Недавно стереохимические
исследования гидроцианирования в присутствии таких
катализаторов, как Ni(0) и Pd(O) привели к дополнительным
подтверждениям справедливости предложенного выше механизма. Так,
Джексон [74] и Бэквалль [75] независимо показали, что
каталитическое присоединение HCN стереоспецифично и приводит к
ci/я-продуктам, как, например, в случае дейтерированного алке-
на с концевой связью в уравнении (10.70).
\/ + HCN ^ Н|Л_^г) (10.70)
D D D CN
54 Глава 10
Интересно, что в реакциях присоединения HCN к алкенам
наиболее эффективны палладиевые катализаторы с дифосфино-
выми лигандами, способные образовывать семичленные хелат-
ные циклы [74J.
При использовании хиральных лигандов, например DIOP,
оптические выходы умеренны, величина ее не превышает 40%.
Но это не вызывает удивления, если учесть, что у субстрата нет
другого лиганда, чтобы образовать жесткий хелат с
катализатором. Как явствует из рассмотрения асимметрического
каталитического гидрирования, высокие значения ее получаются в
результате необычного явления. Реакции с DON в присутствии
тетракис(трифенилфосфит) никеля(0) и хлорида цинка идут с
интенсивным удалением дейтериевой метки как из продукта,
так и из регенерируемого олефина [75J. Это объясняет, почему
гидроцианирование норборнена приводит исключительно к эк-
зо-продукту [уравнение (10.71)]. Этот результат согласуется с
тем фактом, что связывание металла происходит по менее
затрудненной стороне алкена.
Pd(P(OPh) ) V /CN
HCN — - |
(10 7!)
в. Гидроцианирование диенов. Большой интерес представляет
каталитическое присоединение HCN к диенам. Как говорилось
ранее, двойное присоединение HCN к 1,3-бутадиену лежит в
основе промышленного синтеза адипиновой кислоты [73].
Считается, что реакция идет в три стадии и в результате получается
продукт с нужной региохимией [уравнения (10.72) и (10.73)]
[76]. Сообщалось, что для этой реакции в качестве
катализатора использовали фосфит никеля (0)—кислоту Льюиса.
Ni(O)
HCN CN
(10.72)
CN
64% 34%
^ Ni(O)
CN * ^ CN HCN
CN
В первой реакции происходит как 1,4-, так и 1,2-лрисоедине-
ние HCN [уравнение (10.72)]; последний продукт образуется в
меньшем количестве и затем перегруппировывается в основной
продукт [уравнение (10.73)]. Присоединение к более реакцион-
носпособному олефину с концевой двойной связью происходит
против правила Марковникова и приводит к адиподинитрилу
[уравнение (10.73)]. Точный механизм перегруппировки олефи-
на и региоспецифического присоединения HCN не известен.
Гомогенные каталитические процессы 55
Адиподинитрил гидролизуют до адипиновой кислоты или
гидрируют до гексаметилендиамина, которые путем известных
органических реакций превращаются в найлон-6,6 [уравнение
(10.74) J.
о о
Н0С(СН2)4С0Н ООН Н
NC{CH2)4CN > -C4CH2)4-C-N4CH2)6-N- (10.74)
HoNCHp(CHp)4CH?NHo
Первая стадия катализируемого никелем присоединения
HCN к сопряженным диенам протекает через л-аллильный ин-
термедиат. Региохимия этого легкого присоединения,
по-видимому, контролируется термодинамической устойчивостью аллиль-
ного интермедиата [76]. Например, из 1,3-пентадиена
получается только разветвленный нитрил [уравнение (10.75)].
NiL'4 ^ _, (10.75)
(PhO)3P
HCN
' "'^ , CN
NC L
99%
В присутствии этих катализаторов несопряженные диены до
присоединения первой молекулы HCN перегруппировываются в
сопряженные диены [76].
г. Гидроцианирование алкинов. Ацетилены также
подвергаются каталитическому присоединению HCN в присутствии
растворимых комплексов переходных металлов, правда, эта
реакция менее изучена [77]. Использование катализаторов на
основе Ni(O) позволяет получать цис-продукты с выходами от уме
ренных до высоких и обеспечивает высокую региоселективность
Палладиевые катализаторы для этих целей уступают никеле*
вым. Опять же и здесь на региохимию реакции влияют как сте-
рические, так и электронные факторы. Перечисленные моменты
иллюстрируют данные уравнения (10.76). Неразветвленные ал-
кины с концевой тройной связью, такие, как 1-гексин,
преимущественно дают разветвленные нитрилы, тогда как из трет-бу-
тилацетилена образуется главным образом нитрил с концевой
функциональной группой. Для подтверждения
гая-присоединения проводили реакцию с DCN.
HCN R^ n'H R\ ^H ,|П7в,
пр ; ли щ С - С Р~Р I U » О /
-90%
R = //-Bu R= шре/п- В и
56 Глава 10
д. Выводы. Продемонстрировано каталитическое
присоединение HCN к моноенам, диенам и ацетиленам. Для моноенов
лучше всего подходят палладиевые катализаторы с хелатирующими
фосфинами; отмечается высокая региоселективность и
умеренная стереоселективность (с хиральными лигандами).
Сопряженные диены реагируют с большей скоростью по механизму,
включающему я-аллильный интермедиат; для этих реакций
предпочтительнее никелевые катализаторы. Присоединение HCN к
ацетиленам катализируется комплексами никеля; для реакций
характерна сгш-стереохимия и хорошая региоселективность.
ЛИТЕРАТУРА И ПРИМЕЧАНИЯ
1. Гомогенному каталитическому гидрированию посвящены обзоры:
а) James В. R., in: Homogeneous Hydrogenation, Wiley, New York 1974;
б) James B. #., Adv. Organomet., Chem., 17, 319 (1979); в) Birch A. D.,
Williamson D. #., Org. React., 24, 1 (1976); r) Jardine F. #., Prog. Inorg. Chem.,
28, 63 (1981); д) Faller J. W., in: Homogeneous Catalysis with Metal Phos-
phine Complexes, Pignolet L. H. (Ed.), Plenum, New York, 1983, Chapter 2;
e) James B. R., in: Comprehensive Organometallic Chemistry, Wilkinson G.,
Stone F. G. A., Abel E. W. (Eds.), Pergamon, New York, 1982, Vol. 8, p. 285.
2. a) Damans F., Morel D., J. Mol. Catal., 3, 403 (1977—78); 6) Halpern J.,
Okamoto Т., Zakhariev Л., J. Mol. Catal., 2, 65 (1976); в) Chan A. S. C,
Halpern J., J. Am. Chem. Soc, 102, 838 (1980); r) Halpern J., Inorg. Chim.
Acta, 50, 11 (1981); д) Halpern J., Riley D. P., Chan A. S. C, Pluth J. J.,
J. Am. Chem. Soc, 99, 8055 (1977); e) Halpern /., Science, 217, 401 (1982).
3. а) Гетеролитическая активация водорода рассматривается в работе:
Brothers Р. /., Prog. Inorg. Chem., 28, 1 (1981); б) Jeske G., Lauke И.,
Mauermann H., Schumann H., Marks T. /., J. Am. Chem. Soc, 107, 8111
(1985).
4. Sweany R. L., Halpern /., J. Am. Chem. Soc, 99, 8335 (1977).
5. Sanchez-Delgado R. A., DeOchoa D. L., J. Mol. Catal., 6, 303 (1979).
6. Birch A. D., Williamson D. #., Org. React., 24, 1 (1976).
7. Brown J. M., Canning L. R., Downs A. J., Forster A. M., J. Organomet. Chem,
255, 103 (1983).
8. a) Osbom J. A., Jardine F. H., Young /. F., Wilkinson G., J. Chem. Soc (A),
1966, 1711; б) Этот 14-электронный интермедиат был генерирован и изучен
с помощью флеш-фотолиза. Wink D., Ford Р. С, J. Am. Chem. Soc, 107,
1794 (1985); 108, 4838 (1986).
9. Meakin P., Jesson J. P., Tolman C. A., J. Am. Chem. Soc, 94, 3240 (1972).
10. Brown J. M.t Canning L. R., Lucy A. R.f J. Chem. Soc, Chem. Commun.,
1984, 915.
11. Schrock R. R., Osbom J. A., J. Am. Chem. Soc, 98, 2134 (1976).
12. Эти реакции асимметрического гидрирования в условиях гомогенного
катализа рассматриваются в обзорах: a) Kagan Н. В., Frian J. С, «New
Approaches in Asymmetric Synthesis», Topics in Stereochemistry, 10, 175
(1977); 6) Morrison J. D., Masler W. F., Neuberg M. /(., Adv. in Catalysis,
25, 81 (1976); в) Kagan H. В., Pure Appl. Chem., 43, 401 (1975); r)
Morrison J. D., Masler W. F., Hathaway S., in Catalysis in Organic Synthesis,
Rylander P. N., Greenfield H. (Eds.), Academic, New York, 1976; д) Mar-
ko L, Ней В., Catal Rev., 8, 269 (1974); e) Valentine D., Scott J. W.,
Synthesis, 1978, 329; ж) Knowles W. S., Sabacky M. J., Vineyard B. D., Adv. Chem
Ser., 132, 274 (1974).
Гомогенные каталитические процессы 57
13. Harada /С, in: Asymmetric Synthesis, Morrison J. D. (Ed.), Academic, New
York, 1985, Vol. 5, Chapter 10.
14. Knowles W. S., Ace. Chem. Res., 16, 106 (1983) и цитированные в этой
статье работы.
15. Koenig /С. Е., in: Asymmetric Synthesis, Morrison J. D. (Ed.), Academic,
New York, 1985, Vol. 5, Chapter 3.
16. Рисунки 10.2, 10.4 и 10.5 взяты с разрешения авторов из работы [2е]
(рисунки 1, 2 и 4 соответственно).
17. a) Brown J. М., Chaloner P. A., J. Chem. Soc, Chem. Commun, 1980, 344;
6) Alcock N. W., Brown J. M., Derome A. E., Lucy A. R., J. Chem. Soc,
Chem. Commun., 1985, 575; в) Brown J. M., Chaloner P. A., Morris G. Л.,
J. Chem. Soc, Chem. Commun., 1983, 664.
18. Ojima I., Kogure Т., Yoda N., Chem. Lett., 1979, 495; Ojima I., Kogure Т.,
Yoda N., J. Org. Chem., 45, 4728 (1980).
19. Sinou D., Tetrahedron Lett., 22, 2987 (1981).
20. Koch G. K., Dalenberg J. W. J. Labelled Compounds, 6, 395 (1970).
21. Grabtree R., Ace Chem. Res., 12, 331 (1979) и цитированные в этой статье
работы.
22. Suggs J. W., Cox S. D., Crabtree R. H., Quick J. M., Tetrahedron Lett., 22,
303 (1981).
23. Crabtree R. H., Davis M. W.t Organometallics, 2, 681 (1983).
24. Tucker J. R., Riley D. P., J. Organomet. Chem., 279, 49 (1985).
25. Le Maux P., Jaouen G., Saillard J. У., J. Organomet. Chem., 212, 193 (1981);
Schroeder M. A., Wrighton M. S., J. Organomet. Chem., 74, C29 (1974).
26. Wrighton M. S., Schroeder M. A., J. Am. Chem. Soc, 95, 5764 (1973); Mir-
bach M. /., Tuyet N. P., Saus A., J. Organomet. Chem., 236, 309 (1982).
27. Frankel E. N., Selke E., Glass C. A., J. Am. Chem. Soc, 90, 2446 (1968).
28. Hull J. W., Jr., Gladfelter W. L, Organometallics, 3, 605 (1984).
29. a) Sloan M. F., Matlock A. S., Breslow D. S., J. Am. Chem. Soc, 85, 4014
(1903); 6) Kroll W. R., J. Catal, 15, 281 (1969); в) Lapporte S. /., Ann. N. Y.
Acad. Sci., 158, 510 (1969); r) Lapporte S. J., Schuett W. R., J. Org. Chem.,
28, 1947 (1963); д) KHnedinst K. A., Boudart M, J. Catal, 28, 322 (1973);
e) Lipovich V. C, Schmidt F. K-, Kalechits I. V., Chem. Abstr., 68, 59185w
(1968).
30. Stuhl L. S., Rakowski-DuBois M., Kirsekorn E. J., Bleeke J. R., Stevens A. E.,
Muetterties E. L., J. Am. Chem. Soc, 100, 2405 (1978).
31. Holy N. L., J. Org. Chem., 44, 239 (1979).
32. Love C. J., McQuillin F. /., J. Chem. Soc, Perkin Trans. 1, 1973, 2509.
33. Russell M. J., White C, Maitlis P. M., J. Chem. Soc, Chem. Commun., 1977,
427 и цитированные в этой статье работы.
34. a) Muetterties E. L., Bleeke J. R., Асе. Chem. Res., 12, 324 (1970); 6)
Johnson J. W., Muetterties E. L., J. Am. Chem. Soc, 99, 7395 (1977).
35. Bennett M. A., Huang T.-N., Smith A. K., Turney T. W., J. Chem. Soc, Chem.
Commun., 1978, 582.
36. Bennett M. A., Huang T.-N., Turney T. W., J. Chem. Soc, Chem. Commun ,
1979, 312.
37. a) Grey R. A., Pez G. P., Wallo A., J. Am. Chem. Soc, 102, 5948 (1980)
и цитированные в этой статье работы: б) Wilczynski R., Fordyce W. A., Hal-
pern /., J. Am. Chem. Soc, 105, 2066 (1983).
38. Feder H. M., Halpern /., J. Am. Chem. Soc, 97, 7186 (1975).
39. Falk J. C, in: Catalysis in Organic Synthesis, Rylander P. N , Greenfield H
(Eds.), Academic, New York, 1976, pp. 305—324.
40. a) Schrock R. R., Osborn J. Л., J. Chem. Soc, Chem. Commun., 1970, 567;
6) Bonvicini P., Levi A., Modena G., Scorrano G., J. Chem. Soc, Chem.
Commun., 1972, 1188; в) Levi A., Modena G., Scorrano G., J. Chem. Soc,
Chem. Commun., 1975, 6; r) Heil В., Toros S., Vastag S., Marko L J Orga-
58 Глава 10
nomet. Chem., 94, C47 (1975); д) Zassinovich G., Camus A., Mestroni G.,
Inorg. Nucl. Chem. Lett., 12, 865 (1976); e) Frediani P., Matteoli U., Bian-
chi M., Piacenti F., Menchi G., J. Organomet. Chem., 150, 273 (1978).
41. Vastag S., Ней В., Marko L., J. Mol. Cat., 5, 189 (1979).
42. Strohmeier W., Weigelt L., J. Organomet. Chem., 171, 121 (1979).
43. Pasternak H., Lancman E., Pruchnik F., J. Mol. Catal., 29, 13 (1985).
44. Разрыв связи Р—С в ходе реакций, катализируемых переходными
металлами, обсуждается в обзоре: Garrou P., Chem. Rev., 85, 171 (1985).
45. Yoshida Т., Okano Т., Otsuka S., J. Chem. Soc, Chem. Commun, 1979,
870.
46. Lyons J. E., J. Chem. Soc, Chem. Commun., 1975, 412.
47. Мог and P., Kayser M., J. Chem. Soc, Chem. Commun., 1976, 314.
48. Collman J. P., Kosydar K. M., Bressan M., Lamanna W., Garrett Т., J. Am.
Chem. Soc, 106, 2569 (1984).
49. Нанесенные на подложку гомогенные катализаторы описаны, в частности,
в обзорах: a) Grubbs R. #., Chemtech, 1977, 512; б) Hartley F. R., Ve-
zez P. N., Adv. Organomet. Chem., 15, 189 (1977); в) Hodge P., Chem. Brit.,
14, 237 (1978).
50. Вопрос о выделении активного центра в лигандах, связанных с
полимером, очень противоречив. Это явление обсуждается в следующих ключевых
статьях: a) Collman J. P., Hegedus L. S., Cooke M. P., Norton J. R., Dol-
cetti G., Marquardt D. N., J. Am. Chem. Soc, 94, 1789 (1972); 6) Crow-
ley J. L, Rapoport H., Ace Chem. Res., 9, 135 (1976); в) Grubbs R. H.,
Sweet E. M., J. Mol. Catal., 3, 259 (1977/78); r) Reed /., Eisenberger P.,
Teo B. K., Kincaid B. M., J. Am. Chem. Soc, 99, 5217 (1977); Reed J.,
Eisenberger P., Teo B. /t, Kincaid В. М, J. Am. Chem. Soc, 100, 2375 (1978);
д) Scott L. Т., Rebek J., Ovsyanko L., Sims C. L.., J. Am. Chem. Soc, 99,
625 (1977); e) Mazur S., Javalekshmy P. /., J. Am. Chem. Soc, 101, 677
(1979).
51. Grubbs R. H., Kroll L. C, Sweet E. M., J. Macromol. Sci. Chem., 7, 1047
(1973).
52. Zuber M., Banas В., Prunchnik F., J. Mol. Catal., 10, 143 (1981).
53. Murrell L. L. Advanced Materials in Catalysis, Academic, New York, 1977,
p. 235.
54. Neuberg M. K-, Ph. D. dissertation, Stanford University, 1978.
55. Hamlin J. E., Hirai K., Millan A., Maitlis P. M., J. Mol. Cat., 7, 543 (1980).
56. Whitesides G. M., Hackett M., Brainard R. L., Lavalleye J.-P., Sowinski A. F.f
Izumi A. N., Moore S. S., Brown D. W., Staudt E. M., Organometallics, 4,
1819 (1985).
57. См. обзор [1в].
58. Cole Т., Ramage R., Cann K, Pettit R., J. Am. Chem. Soc, 102, 6182
(1980).
59. Speier J. L., Adv. Organomet. Chem., 17, 407 (1978).
60. Noll W., Chemistry and Technology of Silicones, Academic Press, New York,
1968.
61. Green M., Howard J. A. N., Proud J., Spencer J. L., Stone F. G. A., Tsip-
si C. A., J. Chem. Soc, Chem. Commun., 1976, 671.
62. Chalk A. J., Harrod J. F., J. Am. Chem. Soc, 87, 16 (1965).
63. Ryan J. W., Speier J. L., J. Am. Chem. Soc, 86, 895 (1964).
64. Selin T. G., West R., J. Am. Chem. Soc, 84, 1863 (1962).
65. Sommer L. H., Lyons J. E., Fujimoto H., J. Am. Chem. Soc, 91, 7051 (1969).
66. Chalk A. /., J. Organomet. Chem., 21, 207 (1970).
67. Tamao K., Miyaki N., Kiso Y., Kumada M., J. Am. Chem. Soc, 97, 5603
(1975).
68. Ojima I., Yamamoto K., Kumada M., in: Aspects of Homogeneous Catalysis,
Reidel, Dordrecht, 1977, Vol. 3, p. 186.
Гомогенные каталитические процессы 59
69. Ojima /., Hirai К., in: Asymmetric Synthesis, Morrison J. D. (Ed.), Academic,
New York, 1985, Vol. 5, Chapter 4.
70. Brown E. S.> in: Aspects of Homogeneous Catalysis, Ugo R. (Ed.), Reidel,
Dordrecht, 1974, Vol. 2, p. 57.
71. Brown E. 5., in: Organic Synthesis by Metal Carbonyls, Wender I., Pino P.
(Eds.), Wiley, New York, 1977, Vol. II, p. 655 (есть русский перевод
первого издания книги: Органические синтезы через карбонилы металлов. Пер.
с англ./Под ред. Уэндера И., Пино П. — М.: Мир, 1970).
72. James В. R., in: Comprehensive Organometallic Chemistry, Wilkinson G,
Stone F. G. A., Abel E. W. (Eds.), Pergamon, New York, 1982, Vol. 8, p. 353.
73. a) Parshall G. W., Homogeneous Catalysis, Wiley-Interscience, New York,
1980, p. 70; 6) Parshall G. W., J. Mol. Catal., 4, 244 (1978); в) Tolman C. A.,
McKinney R. J., Siedel W. C, Druliner J. D., Stevens W. R., Adv. in
Catalysis, 33, 1 (1985).
74. Jackson W. R., Perdmutter P., Elmes P. S., hovel C. G., Thompson R. J.,
Haarburger D., Probert M. K- S., Smallridge A. J., Campi E. M., Fitzmauri-
ce N. J., Kerttesz M. A., in: Organic Synthesis, an Interdisciplinary
Challenge (5th IUPAC Symposium on Organic Synthesis), Blackwell, London
1986, p. 55.
75. Backvall J. E., Andell O. S., J. Chem. Soc, Chem. Commun., 1981, 1098.
76. Keim W., Behr A., Luhr H.-O., Weisser /., J. Catal., 78, 209 (1982).
77. Jackson W. R., Lovel C. G., Aust. J. Chem., 36, 1975 (1983).
11
КАТАЛИТИЧЕСКАЯ ПОЛИМЕРИЗАЦИЯ ОЛЕФИНОВ
И АЦЕТИЛЕНОВ
11.1. Введение
Данная глава посвящена вопросам полимеризации и олиго-
меризации олефинов и ацетиленов, в частности тем реакциям,
которые катализируются соединениями переходных металлов.
Важными являются как растворимые, так и нерастворимые
катализаторы, но мы будем уделять основное внимание
гомогенным катализаторам, так как гетерогенные менее изучены и не
являются основной темой данной главы.
Эти важные реакции образования углерод-углеродной связи
служат основой для создания многих известных промышленных
продуктов. Например, с помощью таких катализаторов
производят огромные количества (несколько миллионов тонн в год)
полиэтилена и полипропилена. Наиболее желательные
физические свойства этих полимеров связаны с их морфологией,
основой которой являются стереорегулярная природа и интервал
молекулярных весов этих соединений.
Химия этих процессов поистине необъятна, если судить по
количеству выполняемых учеными ежегодно исследований,
числу публикуемых книг, статей, патентов. При этом в ней много
спорного, особенно это относится к механизму (ам) стадии роста
полимерной цепи, идущей через образование связи углерод —
углерод. Подробности промышленных процессов являются
секретом производства и покрыты тайной. С учетом этих осложнений
будут приведены только несколько основных примеров, и
обсуждение механизмов реакций не будет исчерпывающим. За
дополнительными сведениями следует обратиться к обзорам
[i-6j.
Начало этим полимеризационным процессам было положено
пионерской работой Циглера и Натта, которые за свое
открытие были удостоены Нобелевской премией по химии 1963 г.
11.2. Механизмы полимеризации/олигомеризации моноенов
Полимеризация или олигомеризация простых моноенов
включает три основные стадии: (а) инициирование, (б) рост цепи и
(в) обрыв цепи [уравнение (11.1)]. Как правило, считают, что
каталитически активными центрами, на которых протекает по-
Каталитическая полимеризация олефинов и ацетиленов 61
лимеризация или олигомеризация, являются алкильные
комплексы переходных металлов.
(П.!)
R" или Н"
Стадия инициирования (а) обычно включает образование
ненасыщенного соединения переходного металла и алкила
LnM—R, часто по реакции между галогенопроизводным
переходного металла и электроположительным алкилом, например A1R3-
Другой возможный путь — получение вначале ненасыщенного
гидрида LnM—Н при действии подходящего
восстановительного агента и последующее внедрение алкена в этот гидрид, что
приводит к образованию каталитического центра LnM—R.
Реакция внедрения [(б) в уравнении (11.1)] протекает в
две стадии: координирование олефина и образование углерод-
углеродной связи. Внедрению олефинов обязательно должно
предшествовать его координирование, для этого требуется
вакантное место. Для эффективной полимеризации олефин
должен быть хорошим лигандом. Среди простых моноенов такие
реакции полимеризации обычно протекают только в случае
этилена и других незатрудненных олефинов с концевой двойной
связью (этилен>пропилен> 1-бутен). Олефины с внутренней
двойной связью обычно не полимеризуются при использовании
таких катализаторов. Более прочно связывающиеся олефины
являются конкурентными ингибиторами по отношению к более
слабо связывающимся. Известно, что многие катализаторы
полимеризации чувствительны к отравлению загрязнениями,
действующими как конкурирующие лиганды.
Вопрос о протекании второй стадии — процесса роста цепи—
является дискуссионным. Простейшей возможностью
протекания этой стадии образования углерод-углеродной связи
является миграционное внедрение координированного алкена в
координированном олефиновом субстрате («механизм Косее») [7].
Региохимия и стереохимия этого необратимого внедрения
определяют стереорегулярную природу растущей полимерной цепи.
Суммарная скорость стадии (б) является важной с точки
зрения контролирования молекулярного веса полимера; для
полимеров с высоким молекулярным весом эта стадия обычно очень
быстрая [2, 8], часто по скорости близкая к ферментативным
реакциям (до 10 000 превращений на один каталитический
центр в секунду). Такие очень высокие скорости усложняют
изучение кинетики роста полимерной цепи, так как приводят к
возникновению диффузионного контроля процесса подвода мо-
62 Глава 11
номера к активному центру, обусловленного экранированием
активного металлоалкильного центра полимерной цепью [2].
Существует несколько возможностей протекания стадии
обрыва цепи (в) [6]. Простейшей из них является обычное ^-гид-
ридное элиминирование с регенерацией исходного гидрида и
образованием шолимера или олигомера с концевой двойной
связью [уравнение (11.2)]. Другой путь обрыва полимерной
цепи — присоединение водорода [гидрогенолиз связи
металл-углерод; уравнение (11.3)]. Этот процесс, который иногда
используют для контроля области молекулярного веса, приводит к
образованию насыщенных полимеров [3].
LnM—CH2CH2—RP —*• LnM—H+CH2 = CH—RP (11.2)
LnM-CH2CH2-Rp -^l LnM-H+CH3CH2Rp (Ц.З)
Третий путь обрыва полимерной цепи, как и ^-гидридное
элиминирование, также является существенным. Он включает
перенос ^-гидрида между металлоалкилом и
координированным олефиновым мономером [уравнение (11.4)] [6]. В
некоторых случаях эта стадия становится особенно важной при
повышении температуры. Обрыв растущей полимерной цепи могут
вызвать и другие переносчики цепи, например алкилметаллы,
галогеноводороды, галогенопроизводные углеводородов.
? R
7СН2-СН- сн =СН (11.4)
м н * м
tfS^LJ сн2-сн3
Соотношение скоростей роста и обрыва цепи (kp и kt
соответственно) определяет молекулярный вес продукта.
Возможны три ситуации. В первом случае kp значительно больше kt,
при этом получается полимер с высоким молекулярным весом.
Во втором случае kp и kt близки, при этом образуются олиго-
меры с геометрическим молекулярно-весовым распределением
(типа «Шульца — Флори»). В третьем случае kt значительно
больше kp и это приводит к исключительному образованию ди-
меров.
Обычно полимеры с высоким молекулярным весом
образуются при использовании катализаторов, полученных из
переходных металлов, относящихся к начальным группам (IV, V и VI);
катализаторы, получаемые из металлов VIII группы,
преимущественно приводят к р-гидридному элиминированию и
образованию олигомеров или димеров [1]. Некоторых изменений
отношения kp/kt можно достигнуть, меняя природу лигандов. До-
Каталитическая полимеризация олефинов и ацетиленов 63-
норные лиганды, как известно, увеличивают, а акцепторные
уменьшают относительную скорость роста цепи. Есть данные,
что при увеличении степени окисленности входящего в
катализатор металла уменьшается скорость роста цепи [1].
Обычно считается, что гетерогенные и гомогенные
катализаторы полимеризации олефинов действуют по сходным
механизмам [3J; это предположение остается чисто умозрительным из-
за больших сложностей изучения гетерогенного катализа.
Характерным для гетерогенных катализаторов является
существование на их поверхности относительно небольшого числа
активных каталитических центров; тем не менее с помощью
гетерогенных катализаторов может быть достигнут больший контроль
стереорегулярности, особенно для изотактических
полимеров [2].
11.3. Механизм стадии роста цепи
Представления об образовании углерод-углеродной связи,
т. е. о стадии роста цепи при катализированной металлами
полимеризации олефинов, в течение длительного времени были
Рис. 11.1.
противоречивыми. Из ряда предположений следует выделить два
основных пути: четырехцентровое «миграционное внедрение»
(Косее [7]) и механизм вхождения карбена в металлацикл
(Грин [9J). На рис. 11.1 они изображены соответственно как
пути (а) и (б), общим для них является участие
ненасыщенного алкилметалла. В большинстве случаев в этой главе мы
используем при обсуждении традиционный механизм внедрения,
лучше других механизмов подтверждаемый различными
модельными реакциями. Однако необходимо учитывать, что в
зависимости от конкретного катализатора или определенных
условий реакции может превалировать любой из приведенных
механизмов (или даже какой-либо другой).
Если принять во внимание, что имеется много комплексных
соединений, в которых олефиновые и алкильные лиганды
находятся в £{ис-положении по отношению друг к другу и при этом
64
Глава 11
не претерпевают внедрения, то становится сомнительным
прямое миграционное внедрение олефина ло связи М — алкил,
предполагаемое в механизме Косее [11J. Ватсон [10] сообщила о
первой экспериментальной модели миграционного внедрения
олефина. Ее результаты суммированы в уравнении (11.5). Ряд
исследований с использованием ЯМР-спектроскопии меченных
изотопами веществ ясно показал, что имеет место региоспеци-
фичное внедрение олефина по связи М—алкил. Это внедрение
сопровождается двумя конкурирующими реакциями:
дальнейшей олигомеризацией олефина и ^-гидридным
элиминированием. Следует отметить, что механизм Грина, связанный с
образованием металлацикла [9J, требует соответствующего
окислительного присоединения к металлическому центру [10].
Комплекс лютеция(III) не может с легкостью участвовать в такой
двухэлектронной окислительно-восстановительной реакции.
сн,
(I I M
€р2 Lj-CH3
Cp2Lu-H
= С
СН2 =
сн,
Ch-
сн3
Cp2Lu-CH2-CH
сн.
сн2=снсн3
сн,
Ср
Ме5С5
Cp2Lu-CH2CH-CH2-CH
Флуд [11], используя хелатный комплекс (2,2-диметил-4-
пентен-1-ил) платину (II) с изотопной меткой, смог наблюдать
обратимое внедрение алкена по связи М—алкил [уравнение
(11.6)]. В среде CD^NO2 при 125 °С протекает обратимая
перегруппировка, описываемая кинетическим законом первого
порядка. Эта система искусно подобрана для предотвращения
конкурирующего ^-гидридного элиминирования. Промежуточное
производное циклобутилкарбинила 1 не наблюдалось, так как
оно термодинамически менее устойчиво.
(И.6)
<drnpe)
(dmpe) PI
(dmpe)Pt JL
D D
Существенным различием между двумя предложенными
путями является наличие в карбеновом механизме Грина миграции
Каталитическая полимеризация олефинов и ацетиленов 65
а-водорода. Ожидалось, что первичный кинетический изотопный
эффект (kH/kD) для стадии образования карбена должен быть
около 3. На этом основании Груббс [12] вел поиски кинетического
изотопного эффекта при сополимеризации по типу Циглера —
Натта этилена и пердейтероэтилена по уравнению (11.7).
Изотопный эффект для этой реакции измеряли с большой
точностью, определяя среднее количество дейтерированных звеньев в
полимерной цепи. Было установлено, что W&d равно 1,04+
+0,03. Как утверждает Груббс, «эти данные основательно
поддерживают механизм внедрения, не включающий миграцию
водорода на определяющей скорость стадии роста цепи» [12].
Однако этот нулевой результат фактически является результатом
«отрицательного эксперимента» и поэтому не вполне убедителен.
(М.7)
Р 1.EUAICI,
cd2=cd2 + сн2=сн2 + Cp2Ti -улна—* с2н5(сн2сн2)п (со2со2)пн
С!
Затем Груббс придумал искусный стереохимический
изотопный эксперимент, показанный на рис. 11.2. В этом эксперимен-
М О и М н
\ А \ A D
/С-Х IEt2AICI ( У I J
Cp2Ti D (CH2)n
M \ D Мч °- Н
сн9 V- CH- У^
2. bipy
2 Н^
м = cp2Tici ^- ^
Рис. 11.2.
те для исследования стереохимического изотопного эффекта при
внедрении олефина использовали титаноалкил 2 с
«привязанным» олефином. Как показано на рис. 11.2, внедрение а-олефи-
на в связь металл — углерод, монодейтерированную в а-поло-
жении, приводит к образованию диастереомеров. Если бы до
внедрения происходила активация а-водорода (механизм
Грина), нарушилась бы симметрия CHD и отношение диастереоизо-
меров должно было бы отражать отношение активаций С—Н к
С—D. При этом прямое миграционное внедрение показало бы
отсутствие изотопного эффекта. Изотопный эффект не был
обнаружен. Применяя 2Н-ЯМР нашли, что отношение цис/транс
5—П29
66 Глава 11
диастереоизомеров в металлоорганических и погашенных
кислотой органических продуктах равно l,00zh0,005 [13а].
Эта система поучительна еще и тем, что для индуцирования
внедрения (как и для полимеризации с добавленным этиленом)
необходимо введение сокатализатора EtAlCl2 — кислоты
Льюиса. Бипиридин гасит эти реакции, выводя из строя такой соката-
лизатор. Катионный комплекс Cp2Zr(CH3) (ТГФ)+ полимеризу-
ет этилен без кислоты Льюиса как сокатализатора [136].
Еще один экспериментальный результат противоречит ме-
таллациклобутановой гипотезе Грина. Методом 13С-ЯМР было
показано, что при использовании в качестве катализатора ме-
тильного комплекса, обогащенного 13С, ни одной метки 13С не
было обнаружено в метиленовом звене у конца полимера
[уравнение (11.8)] [14].
(И 8)
м-сн3
p-s полимер
М =
каталитический центр
■-сн,
Р-СНОСН
сн,
сн3
Р-СНо-СН
\
сн3
13^
Р-СИо-СН
d. \
СИ,
Имеется по крайней мере одна модельная система,
служащая доказательством того, что полимеризация олефинов может
идти по механизму Грина. Шрок [15] показал, что танталовый
комплекс 3 в присутствии этилена может приводить к
образованию «живого полимера». При этом комплекс 3 подвергается
обратимому внедрению своего карбенового лиганда, давая ал-
кильный комплекс и доказывая возможность образования ме-
таллациклобутана [уравнение (11.9)].
третгВм у
С —СН2
I I d
Та-СНо
\
Та =
Та(РМе3)212
Н
Та
пСН2=СН2
(11.9)
Брукхарт [16] предположил, что независимо от механизма
роста цепи существует параллелизм между «агостическими»
гидрид-олефиновыми комплексами [17] и аналогичными алкил-
олефиновыми комплексами, с легкостью участвующими в
реакции роста цепи. Это схематически изображено на рис. 11.3. Бо-
Каталитическая полимеризация олефинов и ацетиленов 67
лее вероятной, чем структура с концевым атомом водорода,
является агостическая структура с четырехцентровой двухэлек-
тронной связью М—Н—С (4 на рис. 11.3). Это — «задержанное»
внедрение. Предполагается, что аналоги таких систем, в которых
на месте водорода находится алкил, должны иметь низкие
энергии активации реакции внедрения олефина; это делает их
многообещающими кандидатами для каталитической полимеризации
олефинов. Брукхарт [16] проиллюстрировал это положение,
приготовив комплексы типа (QMes) (P)(OMe)3)CoR+, которые, как
было показано, при реакции с этиленом образуют живой
полимер.
LnM-H
L M-R
Рис. 11.3.
Наконец, следует упомянуть о еще трех хорошо
обоснованных для полимеризации или олигомеризации олефинов стадиях
образования связи углерод — углерод, катализируемых
металлами. К ним относятся: метатезис (циклических олефинов),
электрофильная активация и электроциклическое образование
металлациклопентановых интермедиатов. Каждая из них
рассмотрена в последующих разделах.
11.4. Полиэтилен
Из пластиков в США больше всего по объему производится
полиэтилена. Имеются два основных типа полиэтилена: высокой
плотности (линейный полимер с плотностью около 0,95,
температурой плавления вблизи 135°С) и низкой плотности
(разветвленный полимер с плотностью около 0,92 и широким
интервалом температур плавления) [3]. Полиэтилен высокой плотности
получают, используя катализаторы Циглера. Полиэтилен низкой
плотности вначале приготовляли, проводя свободнорадикальную
полимеризацию этилена при высоких давлениях (до 350 МПа
при 350 °С). Однако компания Union Carbide, применив в
качестве катализаторов координационные соединения, ввела очень
эффективный процесс, идущий при низких давлениях (0,7—
2,1 МПа при <100°С) [3].
68 Глава 11
Катализаторы Циглера обычно получают при взаимодействии
хлоридов титана с алюмоалкильным сокатализатором.
Предполагается, что активным каталитическим центром является тита-
ноалкил, входящий в комплекс, образованный алюминиевым
производным. В США в настоящее время для изготовления
полиэтилена высокой плотности преимущественно применяют
комплексы хрома, которые химически связаны с очень
плотными носителями, например с кремнеземом. Эти катализаторы
могут быть приготовлены различными способами. Представляет
интерес реакция между хромоценом и кремнеземом [уравнение
(11.10)]. Аналогичные катализаторы образуются при
восстановлении центров Cr(IV) водородом или монооксидом углерода.
Считают, что инициирующим центром в этих катализаторах
является гидрид хрома типа 6. Рост полимера происходит в
результате реакций, подобных приводимым ранее [уравнение
(пн)]
с
\
/
\
Si-OH
/ чо
\ /
.Si-OH
/
h
с2
э_н4
Ср2Сг
\
Сг СН2(
-^ 0
Si-
/
С2
-О
Сг
/
-0
6
н4
(II 11)
сг-сн2сн2(сн2сн2)гси2сн2сн3
Возможны два основных пути обрыва цепи: гидрогенолиз
связи металл—алкил или вызванное нагреванием ^-гидридное
элиминирование [3]. И в том и в другом случае образуется ме-
таллогидридный комплекс, способный инициировать новую
полимерную цепь [уравнение (11.12)]. Гидрогенолиз является
полезным методом получения полимеров с узким интервалом
молекулярных весов, а также для удаления полимера с
кремнеземного носителя; этот метод реализуется значительно легче с
титановыми катализаторами, чем с хромовыми. При fj-гидридном
элиминировании образуется олефин с концевой двойной связью,
который снова может реагировать с центрами роста
полиэтилена. В этом случае образуется разветвленный полиэтилен низкой
плотности.
Каталитическая полимеризация олефинов и ацетиленов
69
Все катализаторы, применяемые в промышленности для
получения полиэтилена (и полипропилена), являются
гетерогенными или коллоидными [2, 3]. Удаление катализатора из полимера
стоит дорого; кроме того, нежелательно оставлять
дорогостоящие компоненты катализатора, а также входящие в него
агрессивные ионы хлора. Обычно для решения этой проблемы
применяют крайне активные и эффективные катализаторы,
приводящие к образованию >104 кг полиэтилена (или полипропилена)
на 1 г катализатора [3, 6]. При этом катализатор остается в
полимере в виде следов. Следует отметить, что в отличие от
традиционных катализаторов Циглера кремнеземно-хромовый
катализатор не содержит галогениды.
11.5. Полипропилен
Физические свойства полипропилена, имеющие значение при
его технологических применениях, сильно зависят от стереорегу-
лярности и молекулярного веса этого полимера. Наиболее
пригодным является полимер с регулярной последовательностью
присоединения по типу «голова к хвосту» и высокой стереорегу-
лярностью (изотактический) [2, 3].
сн3 н
н сн, сн3 н
Рис. 11.4.
Известны три типа полипропилена [3—5]: изотактический
(7), синдиотактический (8) и атактический (9). Во всех этих
полимерах мономерные звенья присоединены по типу «голова
к хвосту». Однако они отличаются относительными ориентация-
ми чередующихся хиральных углеродов вдоль полимерной цепи
(рис. 11.4).
В идеале изотактический полимер должен иметь одинаковые
относительные конфигурации при всех центрах, образованных
хиральными углеродами метиновых групп (7 на рис. 11.4). Они
являются «псевдоасимметрическими центрами». На самом деле
некоторое количество центров имеет противоположную
зеркальную симметрию. Такая ситуация приводит к кристаллической
спиральной структуре, в которой каждая метильная группа
ориентирована вдоль внешней поверхности винтовой спирали.
Обычно образуется равное количество спиралей, закрученных
70 Глава 11
в правую или левую сторону. Эти гигантские молекулы
представляют собой зеркальноизомерные опирали. Изотактический
полимер (7) находит наибольшее применение в
промышленности; он менее растворим, более высокоплавок, имеет более
высокую прочность на растяжение [2].
Другая стереорегулярная «синдиотактическая» форма {8)
имеет конфигурацию с чередующимися соседними хиральными
центрами вдоль полимерной цепи; это вещество состоит из
гигантских мезомолекул. Синдиотактическая форма более
легкоплавка и меньше используется.
Атактический полипропилен (9) имеет хаотическую
ориентацию хиральных центров вдоль цепи; он аморфен, легкоплавок
и не находит спроса.
В настоящее время все промышленные катализаторы
получения полипропилена являются гетерогенными [3]. До сих пор
полипропилен с высоким содержанием изотактических структур
мог быть получен только при использовании гетерогенных
катализаторов Циглера — Натта. Было выполнено большое число
исследований, посвященных этой стереоспецифичес'кой
полимеризации. Их результаты соответствуют механизму, при котором
полимеризация происходит в результате внедрения комплекса
алкила с переходным металлом в ^^-координированный
пропилен по хиральному металлическому центру [4]. Так как
мономерный пропилен является прохиральным, его комплекс с хи-
ральным металлическим центром может привести к образованию
одного из двух диастереоизомеров, имеющих, естественно,
разные свободные энергии. Преимущественно образуется более
стабильный диастереоизомер. Предположительно этот процесс
протекает, как показано в уравнении (11.13), где
металлический центр является хиральным. Предполагают, что внедрение
олефина по механизму Косее происходит как стерео- (цис), так
и региоспецифично. Этот процесс приводит к образованию
единичного хирального центра (R или S) и полимера, построенного
по типу «голова к хвосту». Последующие внедрения в ходе роста
полимерной цепи дают центры с той же симметрией. Каждое
внедрение, как полагают, приводит к образованию первичной
связи металл — алкил, порождая структуру, построенную по
типу «голова к хвосту». Отметим, что эти катализаторы имеют
равное число R и S-центров, так что образующийся в
результате изотактический полимер представляет собой рацемическую
смесь право- и левозакрученных спиралей.
с-нъ сн3
^ ; ~**н *" Н ^»С \ *" изог[7акгг?ис^бс~ С' 13)
L м _ R I r и гл. д. кии полимер
LnM
_^ Каталитическая полимеризация олефинов и ацетиленов 71
Стереоспецифичность катализаторов получения изотактиче-
ского полипропилена обусловлена особенностями структуры их
поверхности [2]. Например, cc-TiCl3 приводит к образованию
полипропилена с высокой степенью изотактической стереорегуляр-
ности, а р-Т1С1з, который также является очень активным
катализатором полимеризации как пропилена, так и этилена, менее
склонен к образованию изотактического полипропилена.
Приведенный выше механизм требует, чтобы хиральность
центров изотактической полимерной цепи контролировалась
зеркальной симметрией активных центров, а не конфигурацией
углеродных атомов в растущей полимерной цепи [4, 6]. Имеется
ряд доказательств в пользу этой гипотезы. Например, если во
время роста полимера происходит «ошибка», процесс
полимеризации быстро возвращается к исходной стереорегулярности;
таким образом, полимеризационный центр является
«самокорректирующимся». Более того, на стереоспецифичность
полипропиленовых участков не влияет внедрение этиленовых звеньев
в растущую изотактическую полимерную цепь.
Детали образования синдиотактического полипропилена
менее очевидны [4, 18]. Предполагают, что внедрение идет цис-
стереоспецифично, однако его региоспецифичность
противоположна таковой в случае изотактического полипропилена;
возможно, рост цепи происходит с образованием вторичной
металл-углеродной связи [уравнение (11.14)]. Эта
региоспецифичность уменьшается с ростом температуры.
3с н сн, ,сн
2хХ u-Jv-CH2-R с = с л
чн ► н^"с —я—■*» синоиотак-
И I и™# тический
полимер
н3с н сн, ,снз
^ 7 ч
Предполагалось, что имеющий здесь место стерический
контроль при образовании каждого нового хирального центра
обусловлен хиральностью атома углерода в последнем пропилено-
вом звене. Это удалось доказать, используя растворимый
ванадиевый катализатор [18]. Синдиотактические полимеры не
являются в целом региоспецифичными [6].
Вплоть примерно до 1984 г. гомогенные катализаторы
приводили к образованию беспорядочного атактического и иногда
синдиотактического полипропилена, но не изотактического.
В настоящее время эта ситуация изменилась [19]. Катализатор,
получаемый из хирального, содержащего этиленовое звено
бис(инденил)цирконийдихлорида 10 в сочетании с циклическим
олигомером метилалюмоксаном ([А1(СН3)О]П) в качестве сока-
тализатора 11 позволяет получать полипропилен с высокой сте-
72
Глава 11
пенью изотактичности [8]. Для проведения полимеризации
использовали рацемат 10 [уравнение (11.15)], поэтому в
результате был получен рацемический изотактический полипропилен.
Активность этого гомогенного катализатора очень высока, почти
такая же, как у гетерогенных катализаторов. Были получены
высокий молекулярный вес и узкое молекулярно-весовое
распределение [8]. Высокая степень изотактичности (>95%) была
определена методом 13С-ЯМР высокого разрешения; этот метод
исключительно удобен для расшифровки стереохимической
последовательности в цепи полипропилена. Обнаружение стерео-
специфического гомогенного катализатора в конечном счете
может привести к детальному пониманию механизма стереоспе-
цифической полимеризации Циглера — Натта и, возможно,
к простому методу получения хиральных полиолефинов. Тот же
самый катализатор дает возможность получения изотактическо-
го полибутена. Мезоизомер титанового аналога соединения 10
приводит к образованию почти идеального атактического
полипропилена [19]. Этот результат совпадает с 'представлением
о том, что координирование прохирального пропиленавого
мономера с мезометаллическим центром не может приводить к
образованию диастереомеров; таким образом, ахиральный центр
роста цепи не может осуществлять стереохимический контроль
процесса полимеризации.
сн3сн=сн2
СН3С6Н5
катализа -
тор (10 + 11)
СН,
-(сн-сн2)- изотактический
(II.15)
= катализатор
11.6. Этилен-пропиленовые сополимеры
В отличие от гомополимеров полиэтилена и полипропилена,
являющихся пластиками, некоторые этилен-пропиленовые
сополимеры обладают эластичностью и могут быть использованы
Каталитическая полимеризация олефинов и ацетиленов 73
вместо каучука [3, 21]. Большинство промышленно значимых
этилен-пропиленовых эластомеров получают с помощью
катализаторов Циглера — производных растворимых соединений
ванадия (например, УОС13 + А12Е1зС1з в гептане). Обычно в
полимерную цепь включают небольшие количества диена, например ди-
циклопентадиена. Этот остающийся менее реакционноспособный
олефин впоследствии при вулканизации серой образует
поперечные сшивки. Получаемый в результате «вулканизованный»
эластомер не содержит двойных связей и поэтому по сравнению
с натуральным каучуком более устойчив к окислительной
деструкции, в частности под действием озона.
Активный каталитический центр, который, как полагают,
представляет собой V(III)—алкил, не очень селективен.
Внедрения пропилена происходят в соответствии с региохимией
обоих типов, в результате чего возникает неупорядоченное
чередование двух основных сомономеров. Такая беспорядочная
микроструктура является важной для получения желаемых эласто-
мерных свойств. В действительности эти мономеры проявляют
разную реакционную способность, которую можно предсказать
на основе их способности к равновесному связыванию (этилен>
>>пропилен>дициклапентадиен). С учетом этого -в идеале они
должны были бы являться конкурентными ингибиторами друг
для друга, конкурируя за встраивание в каталитический центр.
Их относительные соотношения в полимере таким образом
должны регулироваться изменением соотношения концентраций этих
мономеров в ходе полимеризации.
11.7. Полимеризация, включающая метатезис олефинов
с раскрытием цикла
Циклические олефины могут быть заполимеризованы с
помощью катализаторов метатезиса олефинов. Этот процесс
включает простое раскрытие цикла несущим цепь металл-карбе-
новым катализатором метатезиса по механизму,
обсуждавшемуся в ч. 1, разд. 9.2. Существует обзор, посвященный
полимеризации этого типа [22]. Эти процессы с метатезисом циклических
олефинов намного хуже изучены, чем традиционная
полимеризация Циглера — Натта. Полученные в последнее время
результаты свидетельствуют о богатых возможностях метатезисной
полимеризации.
В зависимости от конкретного используемого катализатора
могут быть достигнуты высокие степени селективности.
Существуют твердые правила, предсказывающие результат таких
реакций. Например, циклопентен может давать либо полностью
74 Глава 11
цис-, либо полностью транс-полкмер [уравнение (11.16)].
Последний является эластомером и имеет промышленное будущее.
-(СН.
i R3AI /
(11.16)
-(сн2)3
За исключением ненапряженных циклогексенов, большинство
незамещенных циклических олефинов могут полимеризоваться
под действием метатезисных катализаторов. Наиболее высока
реакционная способность напряженных олефинов (ч. 1,
разд. 9.1, г). Так, несмотря на то что тризамещенные олефины
обычно нереакционноспособны, 1-метилциклобутен
[уравнение (11.17)] вступает в реакцию [22, 23].
СН
3 катализатор
металл
(11.17)
Полимеризация норборнена в присутствии исходного титано-
карбена [уравнение (11.18)] была тщательно исследована Грубб-
сом [24], который доказал присутствие «живого полимера». Это
позволяет регулировать молекулярный вес. Отметим, что в
полимере сохраняется ^г/с-ориентация атомов углерода, находящихся
в голове моста, тем не менее получают смесь цис/транс-олефя-
новых изомеров.
сР2т
(II 181
Очень интересным приложением этого химизма является
полимеризация 3,4-диизопропилиденциклобутена (12) с
использованием в качестве катализаторов метиленовых
предшественников титаноцена [25]. Как показано в уравнении (11.19),
реагируют только напряженные циклические олефины, что приводит
к образованию первого из известных кросс-сопряженных
полимеров 13. Окислительный «допинг» этого полимера иодом дает
Каталитическая полимеризация олефинов и ацетиленов 75
молекулярный металл с необычайно высокой
электропроводностью. «Живой» полимер гасится метанолом.
12
19)
Следует заметить, что многие катализаторы метатезиса
олефинов ингибируются полярными функциональными группами.
Успешно была осуществлена полимеризация ненасыщенных
лактонов с раскрытием цикла [уравнение (11.20)] [26].
| 20)
4 (СН
11.8. Катализируемая металлом электрофильная полимеризация
Классический способ полимеризации олефинов включает
действие катионных инициаторов, например из источника протонов.
Катионная полимеризация олефинов и ацетиленов может быть
также вызвана очень электрофильным лалладиевым
комплексом [Pd(CH3CN)4] (BF4b [27]. Пример показан в уравнении
(11.21). Олафины и ацетилены, имеющие дефицит электронов, не
полимеризуются под действием этого катализатора. Как фосфи-
ны, так и ионы галогенов дезактивируют эту каталитическую
систему. В этом процессе используют преимущество, связанное
с электрофильной поляризацией координированного олефина
ионом Pd2+ (см. ч. 1, гл. 3 и 7).
Pd(CH3CN)4+
CH3NO2
(11.21)
Аналогичный катионный комплекс палладия катализирует со-
олигомеризацию СО и этилена, приводя к образованию поли-1,4-
дикетона [уравнение (11.22) ] [28]. Отметим, что присутствующие
в этом случае фосфиновые лиганды не дезактивируют
катализатор, однако добавление галогенид-ионов подавляет эту
реакцию. Возможный механизм реакции показан уравнением
(11.23). Предполагается, что алкилпалладиевый интермедиат
образуется в результате некоторой неконкретизируемой реакции.
Внедрение монооксида углерода приводит к образованию реак-
76
Глава 11
ционноспособного ацила, в который легко внедряется этилен, но
не СО (как отмечалось в ч. 1, разд. 6.1, ж, ацилы устойчивы по
отношению к миграционному внедрению СО). Полимерная цепь
может быть оборвана метанолом, который по реакции с палла-
дийацилом образует метиловый эфир (ч. 1, разд. 7.3). Отметим,
что внедрение этилена по связи Pd—ацил протекает значительно
легче, чем простая полимеризация этилена. Таким образом
получается сополимер 1:1.
Pd(CH3CN)nL4.n
CH2=CH2 + C0 СНС1Э, 25»С,50о^
\ со
Pd-R
,Pd-C-R
с2н4
о
\ II
Pd-CH2CH2C-R
(11.22)
11.9. Олигомеризация алкенов
Как уже отмечалось выше, реакции олигомеризации
олефинов протекают по механизмам, аналогичным полимеризацион-
,ным. Разница в случае катализаторов олигомеризации
заключается в том, что более быстрой является стадия обрыва цепи,
^-гидридное элиминирование. Могут использоваться как
катализаторы Циглера, так и растворимые соединения переходных
металлов последних групп (предшественники гидридов). Эти оли-
гомеризационные процессы широко обсуждались [1].
Соотношение реакционной способности олефинов соответствовало
относительному координационному сродству для каталитического
центра: этилен^>пропен>я-бутен>я-пентен. Только некоторые
разветвленные олефины или олефины с внутренней двойной
связью олигомеризуются легко. С точки зрения промышленности
наиболее интересным субстратом является этилен. Это
обсуждается в следующем разделе. Для несимметричных олефинов
продукты, как правило, являются разветвленными.
а. Процесс получения высших олефинов концерна Shell.
Первая стадия процесса получения высших олефинов концерна Shell
[29, 30] включает олигомеризацию этилена гомогенным
никелевым катализатором [уравнение (11.24)] [31]. Считают, что
активным катализатором является гидрид никеля, образующийся
in situ из исходных соединений типа 14 и 15 [уравнение (11.25)].
Этот катализатор селективен только для этилена в присутствии
других олефинов. Эта селективность, которая может быть связа-
Каталитическая полимеризация олефинов и ацетиленов 77
на с необычным бидентатным фосфинкарбоксилатным лигаадом,
объясняет образование статистической (Флори — Шульц) смеси
чистых линейных а-олефинов. Возможный механизм такой
каталитической олигомеризации показан на рис. 11.5. Эта каталити-
сн2=сн2
Ni-H /
сн2=снчсн2сн2)п.1сн2сн3
i
Ni 4CH2CH2)nCH2CH3
Ph2
сн2=сн2
Рис. 11.5.
ческая реакция следует предельной кинетике Махаэлиса — Меи-
тен, на основе которой были определены константа связывания
этилена катализатором и частота превращений в определяющей
скорость стадии роста цепи (0,8 с-1 при 78 °С) [30]. Обрыв цепи
происходит в результате ip-гидридного элиминирования. На
величину средней длины цепи могут влиять добавленные лиганды,
такие, как третичные фосфины.
н3 <П 24)
О '8
(II 25)
Ph^PCH2CO2H г~~"
;fCOD)9 ^
-с н ~ i v
О"
14
Промышленная технология процесса концерна Shell
включает дополнительные стадии [3]. Перегонкой отделяют три группы
а-олефинов [уравнение (11.26)]: С4—Се, Сю—Ci8
(непосредственно поступают на рынок) и >Ci9. Выделенные более легкую
и более тяжелую фракции изомеризуют на гетерогенном катали-
■о Глава 11
заторе в смесь олефинов с внутренней двойной связью. Более
легкую и более тяжелую фракции образующихся олефинов
с внутренней двойной связью затем подвергают рекомбинирова-
нию на гетерогенном катализаторе метатезиса (МоО3/А12Оз)
[уравнение (11.27)]. Образующийся в результате продукт
содержит линейные олефины Сю—Q8 с внутренней двойной
связью. Эти олефины затем гидроформилируют
[уравнение (11.28)], используя кобальтовый катализатор, который
одновременно изомеризует внутренние двойные связи (детали см.
в разд. 12.3, а). Полученные таким образом первичные спирты
Си—Cig используют как пластификаторы («жирные спирты»).
Таким образом, описанный процесс позволяет превращать
недорогой этилен в ценные а-олефины и первичные спирты.
Аналогичный процесс с использованием AlEt3 дает возможность оли-
гомеризации этилена в статистическую смесь а-олефинов [3].
Так как этот процесс протекает без участия переходных
металлов, он выходит за границы данного изложения.
«Ni-H»
СН2=СН2 >• СН2-СН(С2Н4)ПН С4-С8 41% (11.26)
С10-С18 40,5%
С20 и более 18,5%
(11.27)
R|CH=CHR2
R3CH=CHR4
C|0H|,CH=CHCH3
MoO3/AI2O3
HCo(CO)4
со/н2
RJCH
R3CH
Cl
+ 2II
R4Ch
и ll /^11
1 d.d. c.
\
0
II
2c-
R.CH
f- 'll
R4CH
-H
R?ch
+ HI
R3CH
(11.28!
H2 HC0(C0)4
cI3h27ch2oh
б. Димеризация алкенов путем внедрения в связи металл —
углерод. Для димеризации алкенов известны два основных
механизма: традиционное внедрение олефина в алкилметалл и
процесс с участием металлациклов. Первый механизм включает
тот же (самый каталитический цикл, что и полимеризация
олефинов, за исключением того, что скорость ^-гидридного обрыва
цепи намного выше, чем скорость роста цепи. Это обеспечивает
образование почти исключительно димеров. Этот вопрос также
был рассмотрен в обзоре [1].
Димеризация этилена в бутаны катализируется многими
соединениями переходных металлов, особенно металлов VIII
группы. Эти реакции с точки зрения промышленного производства
Каталитическая полимеризация олефинов и ацетиленов 79
интереса не представляют, поскольку бутены обычно дешевле
этилена [3J.
Димеризация пропилена обычно приводит к образованию
смеси продуктов состава С6, являющихся результатом различных
конкурирующих региоселективных актов внедрения. Четыре
скелетных изомера промежуточного алкилметалла могут получаться
в результате двух разных направлений внедрения по связи М—Н,
за которыми следуют два разных типа внедрения по связи М—R
[уравнение (11.29)]. Оба типа внедрений могут приводить к
образованию изомеров.
м-н
МСНМе2-
М-СН2СН2Ме
Me
1
М-СНСНгСНМе£-
М-СН2-СНСНМе2
Me
Me
i
М-СНСН2СН2СН2Ме
М-СН2СНСН2СН2Ме
Me
(11,29)
Прихмером такого процесса является димеризация пропилена
на никелевом катализаторе Циглера [уравнение (11.30)] [3].
Распределение изомерных димеров зависит от природы фосфина,
используемого с этим катализатором. Более объемные фосфины
приводят к образованию более разветвленных олефинов.
Заметим, что продукты образуются в виде смеси олефиновых
изомеров. Влияние лиганда в основном связано со стерическим
контролем второго внедрения пропилена, а не с первичным
внедрением гидрида. Эти процессы могут быть исключительно быстрыми.
Например, комплекс NiBr^-QHs) (РСу3) и EtAlCl2 в хлоро-
бензоле при 25 СС катализирует димеризацию пропилена со
скоростью 60 000 превращений в секунду [32].
снг=снсн3
.30)
СН2=СНСО2Ме
PdCI2(PhCN)2
МеО2С-СН=СН-СН2СН2СО2Ме
(И.31)
Наблюдалась также каталитическая димеризация
замещенных олефинов [1]. Реакции такого типа предлагают интересный
путь образования полимерных интермедиатов. Примером являет-
80 Глава 11
ся катализируемая палладием димеризация метилакрилата в
преимущественно линейный димер [уравнение (11.31)] [33].
Асимметрическую каталитическую содимеризацию двух
разных олефинов наблюдали в нескольких случаях [34]. Наиболее
заметный пример — обнаруженная Богданович и Вилке [35]
содимеризация этилена и норборнена, катализируемая (я-ал-
лил) никелевым катализатором Циглера [уравнение (11.32)].
В присутствии хиральных фосфинов (L*), в частности димен-
тил (изопропил)фосфина, при низких температурах удалось
получить до 80% ее. Реакции такого типа были изучены не очень
подробно, несмотря на то что они дают возможность получать
блоки хиральных конструкций, привлекательные для
фармацевтической промышленности.
сн2=сн2
катализатор I \ 1<>, (II 32}
в. Димеризация алкенов по металлациклическому
механизму. Некоторые комплексы переходных металлов первых групп,
имеющих низкую валентность, избирательно димеризуют
пропилен в 2,3-диметил-1-бутен. Такая селективная димеризация
свидетельствует о совершенно отличном механизме,
включающем металлациклический интермедиат [уравнение (11.33)]
[36, 37]. Высокая селективность образования структуры
«голова к голове» объясняется преимущественным образованием ме-
таллациклопентана с менее затрудненной первичной связью
металл— углерод. В то же время если заменить метильные группы
объемными неопентильными заместителями, наблюдается
димеризация «голова к хвосту», поскольку две большие по объему
алкильные группы не могут находиться у соседних атомов
углерода [38]. Эти металлациклические интермедиаты были
охарактеризованы спектроскопически. Как уже обсуждалось в ч. 1,
разд. 9.4, а, подобные металлациклопентаны являются
результатом электроциклизации двух олефинов на некотором реакцион-
носпособном металлическом центре в процессе типа
окислительного присоединения. Металлацикл, вероятно, распадается с
образованием олефина в две стадии: за ^-гидридным
элиминированием следует восстановительное элиминирование (ч. 1,
разд. 9.4,6). Подобный механизм димеризации олефинов через
образование металлацикла, по-видимому, является довольно
обычным для систем с катализаторами на основе переходных
металлов нециглеровского типа, не имеющих алюминиевых со-
катализаторов.
Каталитическая полимеризация олефинов и ацетиленов 81
/А~
СН
на тали-
|| 33),
катализатор ~- Zr(«-C4Hg)(dmpe) «-wTaCl2(Cp)(RCH = СН2)2
11.10. Олигомеризация и полимеризация
сопряженных диенов [39]
Олигомеризация и полимеризация сопряженных диенов,
катализируемые переходными металлами, представляют собой про-
мышленно значимые процессы, используемые для получения
материалов, начиная с синтетических каучуков (например,,
полностью ^/ic-полибутадиена и полиизопрена) и кончая
исходными веществами для синтеза найлона (например, циклододека-
триена). Для проведения этих процессов используют
катализаторы двух классов: катализаторы Циглера на основе кобальта
и титана (см. выше) и (я-аллил) никелевые и палладиевые
катализаторы. Хотя первые используются более широко (по крайней
мере для полимеризации), механизм действия последних
намного более понятен, поэтому именно они составят основу
изложения в этом разделе.
Независимо от катализатора практически все процессы
полимеризации и олигомеризации сопряженных диенов, согласно
существующим представлениям, протекают через образование
интермедиата— (т]3-аллил) металлического комплекса.
Возможны два пути образования этих т]3-аллильных комплексов из
сопряженных диенов. В случае предкатализаторов, включающих
гидриды металлов или алкилметаллы, т]3-аллильные комплексы
могут образовываться в результате внедрения диена по связи
металл — водород или металл — алкил [уравнение (11.34)].
Равновесие между т]3 и r^-формами сильно зависит от природы
других лигандов и является главным фактором, определяющим
дальнейшее направление реакции (см. ниже).
LnMR ^=^
R \ \ (11.34)
(Н) R >
(Н)
(vh
В то же время многие металлы низкой валентности, в
особенности Ni (0) и Pd(0), могут восстановительно димеризовать
бутадиен в соответствующий бис- (т]3-аллильный) комплекс
[уравнение (11.35)]. Этот процесс является важным для многих
6—1129
82
Глава 11
реакций циклоолигомеризации диенов (см. ниже). Дальнейшее
внедрение в эти т]3-аллильные интермедиаты диена или моноена
приводит к реакциям олигомеризации и полимеризации,
обсуждаемым в этом разделе.
(11.35)
а. Полимеризация 1,3-диенов. В промышленном
производстве полибутадиена и полиизопрена наиболее часто используют
катализаторы Циглера. Избирательность процесса сильно
зависит от конкретного катализатора на основе переходного металла
(табл. 11.1) [40]. Хотя эти различия в реакционной способности
Таблица 11.1. Стереоселективная полимеризация бутадиена
Катализатор
CoCl2/AlEt2Cl
MoO2(OR)2/AlEt3
Cr(acac)3/AlEt3
VCl3(TrO)3/AlEt2Cl
Структура полимера, %
транс-1,4
1
1
1—2
99
цис-1,4
98
3-6
0—3
0
1,2
1
92—96
(синдиотактический)
97—99
(изотактический)
1
не нашли объяснения, очевидно, что активными частицами
являются (т]3-аллильные) комплексы металлов и что структура
продукта определяется стереохимией стадии внедрения
бутадиена Общий механизм показан на рис. 11.6 [41]. Для лучшего
понимания ключевая стадия внедрения изображена
протекающей из т^-аллильного комплекса, хотя равновероятным является
прямое внедрение из т]3-аллильного комплекса.
Никелевые катализаторы [42] используются реже, чем
катализаторы Циглера, для полимеризации сопряженных диенов,
однако о них известно намного больше, благодаря обширным
исследованиям, особенно выполненным Вилке с соавторами
[426]. Предварительно образующиеся комплексы (т]3-аллил)
никеля и сами по себе являются активными катализаторами
этого процесса, но более часто активный катализатор образуется
in situ из соли никеля (II) и алкилалюминиевого реагента.
Вновь полимеризация протекает в результате внедрения
бутадиена в (т]3-аллил) никелевый комплекс, и стереохимия продукта
Каталитическая полимеризация олефинов и ацетиленов 8&
определяется геометрией (син или анти) активного т]3-аллильно-
го комплекса [уравнение (11.36)].
(II 36)
&эи
EtNiX *■ H-Ni-X + =
миниро-
ffavue
тра.чс-поли
бутадиен
Полимеризация других 1,3-диено>в и сополимеризация
различных 1,3-диенов происходят по аналогичным механизмам.
б. Олигомеризация и соолигомеризация сопряженных диенов.
Каталитическая димеризация и олигомеризация 1,3-диенов
могут приводить к очень большому числу линейных или
циклических продуктов, образующихся по аналогичным механизмам.
Образование основного продукта очень сильно зависит как от
металла, так и от вспомогательных лигандов, и о влиянии на
распределение продуктов известно многое (см. ниже). В этом
разделе будут обсуждены некоторые системы с хорошо изученным
механизмом. Для более детального ознакомления могут быть
рекомендованы соответствующие обзоры [39].
Линейная олигомеризация диенов. Хотя многие
каталитические системы приводят к линейной димеризации бутадиена,
одной из наиболее хорошо изученных и наиболее характерной
является система никель (0) —триэтилфосфит — морфолин,
предложенная Хаймбахом [43] (рис. 11.7). Восстановительная
димеризация бутадиена никелем (0) приводит к образованию бис-
(т]3-аллил) никелевого комплекса. Протонирование внутреннего
положения одной из т]3-аллильных групп морфолино'м приводит
к отщеплению протона в а-положении по отношению к другой
г|3-аллильной группе, в результате получается наблюдаемый ди-
мер бутадиена и регенерируется катализатор.
В случае несимметричных диенов типа изопрена возможно
образование четырех разных димеров («голова к голове»,
«голова к хвосту», «хвост к голове», «хвост к хвосту») и проблема
состоит в том, чтобы сделать максимальным выход желаемого
(обычно «голова к хвосту» для получения ациклических
терпенов). Такой процесс катализируют соответствующие палладие-
6*
84 Глава 11
Et3AI ♦ LnMX
инииииро
да пие
in-МГ)
троне -1,4- поли-
ffi/гпадивн
■ цис -1,4- полибутадиек
Рис. 11.6.
Ln— мУ>
\,г-полиЬута-
диен
ъые комплексы в присутствии муравьиной кислоты (источника
водорода) (рис. 11.8). В этой системе ключевым интермедиатом
снова является т]3-аллильный комплекс, хотя он образуется не
при восстановительной димеризации диена, а в результате
присоединения Pd—Н к диену. Отметим, что для получения
наблюдаемых продуктов требуется внедрение более замещенной
двойной связи изопрена по связи Pd—Н и менее замещенной
двойной связи по связи Pd—С. Причины специфичности этой
системы в настоящее время остаются неясными.
Каталитическая полимеризация олефинов и ацетиленов 85
Рис. 11.7.
нсоон
со.
+ R,P + НСООН
(внедрение)
оч
Рис. 11.8.
86
Глава 11
В отсутствие добавленных лигандов многие палладиевые
катализаторы превращают бутадиен в линейный тример додекатет-
раен (рис. 11.9). Великолепные доказательства показанного
механизма были получены Джолли [44], которому удалось
выделить и охарактеризовать комплекс 17 и diphos-аналог 18, а
также доказать их превращение в додекатетраены. В присутствии
добавленных лигандов образуются только димеры, возможно,
Pd(O)
(5и-падиен)
- я7
ипикирование
Рис. 11.9.
из-за того, что лиганды препятствуют координированию третьего
диена, необходимому для образования тримера.
Главной стадией всех этих олигомеризационных процессов
является внедрение одной из двойных связей диена по связи
аллил — металл. Аналогичным образом может происходить
внедрение олефина — главная стадия в полезном в общем
смысле процессе соолигомеризации 1,3-диенов с олефинами. Общая
схема процесса, приводящего к образованию содимера— 1,4-гек-
садиена, показана на рис. 11.10 [45]. В промышленных процес-
Каталитическая полимеризация олефинов и ацетиленов 87
сах в качестве катализаторов используют комплексы родия,
однако комплексы палладия, кобальта, никеля и железа
катализируют тот же общий процесс. Другие диены и другие олефины
также могут участвовать в этом процессе. В то же время
сополимеры 1,3-диенов и олефинов могут образовываться только
в том случае, если внедрение диена в а-алкильный интермедиат,
образовавшийся в результате внедрения олефина, протекает
с большей скоростью, чем ip-элиминирование и образование со-
LnM-H
)>
родание
м )
Рис. 11.10.
димера. Введение алкенов или алкинов в любой из
рассмотренных выше процессов может приводить к их встраиванию в
конечный продукт, делая доступным широкий набор соолигомеров.
Циклоолигомеризация 1,3-диенов. Комплексы никеля (0)
катализируют множество реакций циклоолигомеризации
бутадиена. Этот процесс, интенсивно изучавшийся Вилке [396, 426]
и сотрудниками, может приводить к образованию множества
продуктов в зависимости от реакционных условий (рис. 11.11).
Рис. 11.11.
Глава 11
Циклотримеризация бутадиена с образованием циклододекатри-
ена была одной из первых металлоорганических реакций,
механизм которой был тщательно изучен. Предполагаемая схема
реакции показана на рис. 11.12. Комплекс 21 был выделен и
н
>-40°С
20"
21
Рис. 11.12.
охарактеризован; было показано, что он входит в
каталитический цикл именно так, как предполагалось. Были выделены
также стабилизированные фосфинами аналоги 20 и доказана их
важная роль в реакциях циклодимеризации.
Комплекс 21, как показано на рис. 11.13, имеет собственную
к~с\гиге
21
+ Ni(PPh3)4
Каталитическая полимеризация олефинов и ацетиленов 89
богатую химию [46]. При проведении этого процесса в
присутствии хороших лигандов для комплексов никеля (0) [426],
таких, как фосфины или фосфиты, наблюдали циклодимеризацию
бутадиена (рис. 11.14). Специфичность получаемых димеров
определялась лигандом и отношением лиганд/металл.
В действительности реакция бутадиена с никелевыми (0)
катализаторами в присутствии различных лигандов является
исключительно сложной, в ходе этой реакции конкурентно
действует множество подвижных равновесий. Замечательно, что
стало возможным контролировать эту реакцию с целью получения
одного главного продукта. Недавно потенциально мощный
способ изучения и оптимизации сложных реакций,
устанавливающий связь между добавленными лигандами и изменением в рас-
И It
Q
П
Рис. 11.14.
пределении продуктов, был разработан Хаймбахом [47]. Он был
назван методом обратного титрования. На график наносили
отношение концентраций лигандов и комплекса металла
lg([L]o/[Ni]o), меняющееся в логарифмической шкале от —5 до
+ 2, й распределение продуктов в зависимости от этого
отношения, получая в результате контрольные карты «продукт —
концентрация лиганда». Эти карты могут служить источником
обширной информации, однако, по-видимому, более важной
является возможность с их помощью оптимизировать реакцию для
получения конкретного нужного продукта с минимальным
числом экспериментов.
На рис. 11.15 показаны две такие контрольные карты
«продукт— концентрация лигандов» для только что описанных
реакций. Они убедительно иллюстрируют чувствительность
распределения продукта к природе и концентрации добавленных
лигандов. Они также позволяют легко выбрать оптимальные условия
для получения нужного продукта. Социклоолигомеризация бута-
90
Глава 11
-3-2-7
L = PhP(0Ph)2
Рис. 11.15.
1:200
диена с этиленом с образованием циклодекадиенов и социкло-
олигомеризация бутадиена с алкинами с образованием цикло-
декатриенов протекают сходно при различных условиях.
11.11. Олигомеризация и полимеризация алкинов [48]
Реакции олигомеризации алкинов, катализируемые
переходными металлами, относятся к старейшим и наиболее широко
изучавшимся процессам металлоорганическои химии, начиная
с работ Реппе в сороковых годах и вплоть до настоящего
времени. Алкины обладают высокой реакционной способностью по
отношению к переходному металлу; трудность в этой области
заключается в возможности контроля реакционной способности
для получения единственного главного продукта. Реакции
каталитической циклотримеризации с образованием замещенных бен-
Каталитическая полимеризация олефинов и ацетиленов 91
золов подробно обсуждались в разд. 18.3, в. Здесь мы
сосредоточимся на других процессах циклоолигомеризации и на линейной
олиго'меризации.
а. Линейная олигомеризация и полимеризация алкинов.
Линейная олигомеризация алкинов может происходить по крайней
мере двумя путями: 1) внедрение алкина в винил-металлический
комплекс, 2) метатезис алкина через карбеновыи комплекс
переходного металла. Первый путь, по-видимому, более обычен и в
общем виде иллюстрируется уравнением (11.37). Поскольку
процесс внедрения протекает в соответствии с ^z/c-стереохимией,
образующийся (а-винил)'металлический комплекс не имеет
подходящих групп (например, Н) в положении цис к нему, дающих
возможность протекания процесса р-элиминирования, ведущего
к обрыву последовательности (как это происходит в случае
(11.37)
R - = - R
LnM -X + R - = - R
X = HsR,Ci и т.д) X MLn Л 7 ^П
R - = - R
R R \ R R
Hi У ML"
R R
ГН
олефинов). Следовательно, внедрение алкина в о-винильный
комплекс приводит к образованию другого а-винильного
комплекса, который не может распадаться в результате ^-элиминиро-
вания и вместо этого ведет к олигомеризации. Любое вещество,
способное перехватить а-винилметаллический комплекс
(например, источник водорода), может прервать этот процесс. Этот тип
олигомеризации катализируется широким набором комплексов
переходных металлов, включая комплексы никеля, родия,
палладия и даже ниобия. Похоже также, что катализаторы Цигле-
ра — Натта полимеризуют алкины по тому же механизму [49].
Совсем недавно Катцем и Шроком впервые был описан
другой механизм полимеризации алкинов, включающий металло-
карбены и металлокарбины. Катц [50] показал, что «карбины
Фишера», такие, как Br(CO)4W=CPh, и «карбены Фишера»,
такие, как (CO)5W{C(OMe)Ph}, катализируют полимеризацию
ряда алкинов, в том числе самого ацетилена, алкинов с
концевой и внутренней тройной связью и даже алкинов с далеко
расположенными функциональными группами, например галогена-
92
Глава 11
ми, сложноэфирными, нитрильными и другими группами. Ме-
таллокарбены вовлекаются в процесс в качестве интермедиатов,
и реакция, как полагают, протекает по механизму, схожему с
^метатезисом, показанному в уравнении (11.38).
(11.38)
= - R
и т. д.
Джоффрой [51] поддержал это предположение, получив кар-
бен-алкиновый комплекс 22 при низких температурах и показав,
что при нагревании он приводит к полимеризации алкина
[уравнение (11.39)]. Более того [52], предварительно образующиеся
карбеновые комплексы не требуются, так как активный
катализатор полимеризации алкинов может образовываться при
фотолизе IF (СО) 6 в присутствии алкинов с концевой тройной связью.
Считают, что исходный алкиновый комплекс 23 изомеризуется
[согласно уравнению (3.130) в ч. 1, разд. 3.6, в] в винилидено-
вый комплекс 24, который затем инициирует катализ
[уравнение (11.40)].
(11.39)
(COL
Ph
OMe
RCHCH
Ph
{C0)4W-
R
ОМе
(11.40)
W(CO)fi
RC=CH
hv
(CO)5W -
RC=CH
. полимер
R
I
III
I
H
23
(CO)5W=C=C
24
Каталитическая полимеризация олефинов и ацетиленов 93
Шрок [53] показал, что карбиновые (алкилидиновые)
комплексы Mo(VI) и W(VI) катализируют полимеризацию алкинов;
процесс, как полагают, включает циклоприсоединение алкинов
и к карбиновым, и к карбеновым интермедиатам |(рис. 11.16).
через
(RO)2 Mo
29
- (RO)-Mo^
26
+ =
32
Рис. 11.16.
Наблюдения показывают, что грет-бутилацетилен участвует
в реакции с алкилидиновым комплексом 25, давая алкилидено-
вый комплекс 26, который содержит три молекулы алкина.
Механизм, предлагаемый для образования 26, включает циклопри-
соединение ацетилена к 25 с образованием металлациклобута-
диена 27. Потеря ROH этим комплексом приводит к
образованию карбенового комплекса 28 с координированным алкином
[см. реакцию (9.59) в ч. 1, разд. 9.3]. Циклоприсоединение
алкина к карбену 28 с последующей фрагментацией и
образованием 30 приводят к росту цепи [в результате того же процесса,
что в уравнениях (11.38) и (11.39)], а кроме того, к регенерации
реакционноспособного карбенового центра для дальнейшей оли-
гомеризации. В случае комплекса 25 этот процесс обрывается
*94 Глава 11
внутримолекулярным захватом (32-^33) координированного ал-
кина после того, как произошло встраивание трех молекул
внешнего алкина. Однако в других системах процесс может
продолжаться, приводя к полимеризации алкина.
б. Циклоолигомеризация алкинов. Одной из первых работ,
посвященной катализируемой металлом олигомеризации
ацетилена, была работа Реппе [54], который показал, что сам
ацетилен может быть циклотетрамеризован в циклооктатетраен в
присутствии никелевых катализаторов при 80—120 °С и 10—25 атм
[уравнение (11.41)]. Монозамещенные алкины приводят к
образованию тетразамещенных циклооктатетраенов, а дизамещен-
ные алкины в реакцию не вступают. Вводя лиганды, например
фосфиновые, которые могут конкурировать с алкином за
координационный центр, реакцию можно направить в сторону цикло-
тримеризации с образованием смеси 1,2,4- и 1,3,5-тризамещен-
ных бензолов. С этой циклотримеризацией конкурирует
линейная олигомеризация, и в некоторых случаях линейные олигомеры
являются основными продуктами. Хотя дизамещенные
алкины не циклотримеризуются при действии никелевых
катализаторов, они часто могут быть соолигомеризованы с самим
ацетиленом [уравнения (11.42) и (11.43)].
4 СН = СН + NiX2(yravz?.) ► [I IJ 70% (11.41)
Ni(acac)2
3 HC = CH + RC = CR -►
L?N,(CO)p
2 ИС = СН + RC = CR ^— II! (1143)
Этой работе более тридцати лет, но механизм реакции до
сих пор остается неясным, хотя предлагались различные
варианты. Циклотримеризация алкинов кобальтовыми комплексами
была исследована наиболее широко и обсуждается в деталях
в разд. 18.3, в. Циклотеграмеризация типа реакции Реппе с
образованием циклооктатетраена может проходить аналогичным
образом, т. е. через образование металлациклопентадиена и ме-
таллациклогептатриена в качестве интермедиатов [уравнение
(11.44)]. (Возможность карбинового метатезиса, который
приводил бы к случайному набору алкиновых углеродов, была
исключена, благодаря тщательно выполненному исследованию
Фольгардта [55] с использованием меток.)
Каталитическая полимеризация олефинов и ацетиленов 95
(II 44}
С другой стороны, Вилке предложил биметаллический
механизм (рис. 11.17), основанный на наблюдении, что {Ni(COT)}2
сам по себе является катализатором этого процесса, и на
структурных исследованиях в близкой области химии комплексов
хрома [56]. В этом процессе две соседние частицы никелациклопен-
тадиена, стабилизированные координированием с циклооктатет-
Рис. 11.17.
раеном, взаимодействуют, образуя биникелевое соединение в
результате сочетания их соответствующих С4-звеньев. Цепь С8
в этих соединениях связана так же, как в только что
упоминавшихся соединениях хрома [Cr (ri-CsHsJJ^CsHs). Сочетание двух
концов Cs-цепей приводит к образованию {Ni(COT)}2, который
снова включается в цикл.
96 Глава 11
Хотя это обсуждение было ограничено рамками «само»- и
«гомо»-олигомеризационных процессов, алкины соолигомеризу-
ются с различными алкинами так же, как с 1,3-диенами и олефи-
нами. Число различных возможных продуктов поистине
потрясает, тем не менее процессы, по которым они образуются, скорее
всего представляют собой простые комбинации стадийных
процессов, рассмотренных в этой главе.
ЛИТЕРАТУРА И ПРИМЕЧАНИЯ
1. Keim W., Behr Л., Poper M., in: Comprehensive Organometallic Chemistry,
Wilkinson G., Stone F. G. A., Abel E. W. (Eds.), Pergamon, New York, 1982,
Chapter 52, p. 371.
2. Gates B. C., Katzer J. R., Schuit G. С. Л., Chemistry of Catalytic Processes,
McGraw-Hill, New York, 1979, p. 150.
3. Par shall G., Homogeneous Catalysis, Wiley-Interscience, New York, 1980,
p. 48.
4. Pino P., Mulhaupt R.y Angew. Chem., Int. Ed. Engl., 19, 857 (1980).
5. Boor /., Ziegler-Natta Catalysis and Polymerizations, Academic, New York,
1979.
6. Sinn H., Kaminsky W.t Adv. Organomet. Chem., 18, 99 (1980).
7. a) Cossee P., J. Catal., 3, 80 (1964); 6) Adman E. J., Cossee P., J. Catal., 3,
99 (1964).
8. Kaminsky W., Kulper K., Brintzinger H. H., Wild F. R. W. P., Angew. Chem.,
Int. Ed. Engl., 24, 507 (1985).
9. Ivin K. J., Rooney J. J., Stewart С D., Green M. L. H., Mahtab J. R.y J. Chem.
Soc, Chem. Commun., 1978, 604; Green M. L. #., Pure Appl. Chem., 100,
2079 (1978).
10. Watson P. L, J. Am. Chem. Soc, 104, 337 (1982).
11. Flood T. C, Bitler S. P., J. Am. Chem. Soc, 106, 6076 (1984).
12. Soto /., Steigerwald Af., Grubbs R. #., J. Am. Chem. Soc, 104, 4479 (1982).
13. a) Clawson L., Soto J., Buchwald S. J., Steigeerwald M. L., Grubbs R. #.,
J. Am. Chem. Soc, 107, 3377 (1985); 6) Jordan R. F., Bajgur C. S., Willett R.,
Scott В., J. Am. Chem. Soc, 108, 7410 (1986).
14. Zambelli A., Locatelli P., Sacchi M. C., Rigamonti £., Macromolecules, 13,
798 (1980).
15. Turner H. W., Schrock R. R., Fellmann J. D., Holmes S. J., J. Am. Chem. Soc,
105, 4942 (1983).
16. Schmidt G. F., Brookhart M, J. Am. Chem. Soc, 107, 1443 (1985).
17. Brookhart M., Green M. L #., J. Organomet. Chem., 250, 395 (1983).
18. Zambelli A., Locatelli P., Zannoni G., Bovey F. A., Macromolecules, 11, 923
(1978). „
19. Ewen J.'A., J. Am. Chem. Soc, 106, 6355 (1984).
20. Kaminsky W., Miri M., Sinn H., Woldt R., Makromol. Chem. Rapid Comm,
4, 417 (1983).
21. Cesca S., Macromol. Rev., 10, 1 (1975).
22. a) Grubbs R. #., in: Comprehensive Organometallic Chemistry, Wilkinson G.,
Stone F. G. A., Abel E. W. (Eds.), Pergamon, New York, 1982, Vol. 8,
Chapter 54, p. 502; 6) Dragutan V., Balaban А. Т., Dimonie M., Olefin
Metathesis and Ring Opening Polymerization of Cycloolefins, Wiley, New York, 1986;
в) Долгоплоск Б. А., Коршак Ю. В. —Yen. хим., 1984, 53, с. 65—86.
23. Katz Т. /., McGinnis J., Altus С, J. Am. Chem. Soc, 98, 606 (1976);
Wilson S. R., Schalk D. £., J. Org. Chem., 41, 3928 (1976).
24. Gilliom L. R., Grubbs R. #., J. Am. Chem. Soc, 108, 733 (1986).
25. Swager T. M., Grubbs R. #., J. Am. Chem. Soc, 109 (1987) в печати.
26. Ast W., Rheinwald G., Keber R., Makromol. Chem., 177, 1341 (1976).
27. Sen A., Lai T.-W.t Organometallics, 1, 415 (1982).
Каталитическая полимеризация олефинов и ацетиленов 97
28. Sen A., Lai T.-W., Organometallics, 3, 866 (1984).
29. Keim W., Kowaldt F. H., Goddard R., Kruger C, Angew. Chem., Int. Ed.
Engl., 17, 466 (1978).
30. Freitas E. R., Gum C. R., Chem. Engin. Progr., 75, 73 (1979).
31. Peuckert M., Keim W.t Organometallics, 2, 594 (1983).
32. Bogdanovic В., Spliethoff В., Wilke G., Angew. Chem., Int. Ed. Engl., 19,
622 (1980).
33. Pracejus H., Krause H. J., Oehme G., Z. Chem., 20, 24 (1980).
34. Kagan H. В., in: Comprehensive Organometallic Chemistry, Wilkinson G.,
Stone F. G. A., Abel E. W. (Eds.), Pergamon, New York, 1982, Vol. 8,
Chapter 53, p. 486.
35. Bogdanovic В., Непс В., Meister В., Pauling H., Wilke G., Angew. Chem.,
Int Ed. Engl., 11, 1023 (1972); Bogdanovic В., Angew. Chem., Int. Ed. Engl.,
12, 954 (1973).
36. Datta S., Fisher M. В., Wreford S. S., J. Organomet. Chem., 188, 353
(1980).
37. Schrock R. R., McLain S., Sancho /., Pure Appl. Chem., 52, 729 (1980).
38. McLain S. J., Sancho J., Schrock R. R.t J. Am. Chem. Soc, 101, 5451 (1979).
39. См. обзоры: a) Keim W., Behr A., Poper M., «Alkene and Alkyne Oligomeri-
zation, Cooligomerization and Telomerization Reactions», in: Comprehensive
Organometallic Chemistry, Wilkinson G., Stone F. G. A., Abel E. W. (Eds.),
Pergamon, New York, 1982, Vol. 8, pp. 371—462; 6) Jolly P. W.t «Nickel-
Catalyzed Oligomerization of Alkenes and Related Reactions», работа [39a],
Vol. 8, pp. 615—648.
40. Atlas S. M., Mark H. F., Catal Rev., 13, 1 (1976).
41. Nakamura A., Tsutsui M.y Principles and Applications of Homogeneous
Catalysis, Wiley, New York, 1980, p. 143.
42. a) Parshall G. W., Homogeneous Catalysis, Wiley, New York, 1980, pp. 55—
57; 6) Jolly P. W., Wilke G., The Organic Chemistry of Nickel, Wiley, New
York, 1975, Vol. 2, pp. 213—245.
43. Heimbach P., Angew. Chem., Int. Ed. Engl., 7, 882 (1968).
44. Benn R., Jolly P. W.f Mynott R., Schenker G., Organometallics, 4, 1136
(1985).
45. Работа [39a], p. 419.
46. Baker R., Chem. Rev., 73, 487 (1973).
47. Heimbach P., Schenkluhn #., Topicss Current Chem., 92, 45 (1980).
48. а) Работа [39a], pp. 410—430; б) работа [396], pp. 649—670; в)
Winter M. /., in: The Chemistry of the Metal-Carbon Bond, Patai S. (Ed.), Wiley,
New York, 1985, Vol. 3, p. 259.
49. Clake T. C, Yannoni C. S., Katz T. /., J. Am. Chem. Soc, 105, 7787 (1983).
50. a) Katz T. J., Ho T. H., Shih N-Y., Ying Y-C, Stuart V. I. W.y J. Am. Chem.
Soc, 106, 2659 (1984); 6) Katz T. J., Hacker S. M., Kendrick R. D.,
Yannoni С S.y J. Am. Chem. Soc, 107, 2182 (1985).
51. Foley #., Strubinger L. M., Targos T. S., Geoffroy G. L., J. Am. Chem. Soc,
105, 3064 (1983).
52. Landon S. /., Schulman P. M., Geoffroy G., J. Am. Chem. Soc, 107, 6739
(1985).
53. Strutz H., Dewan J. C., Schrock R. R.t J. Am. Chem. Soc, 107, 5999 (1985)
и цитированная в этой статье литература.
54. a) Hoogzand С, Hiibel W., «Cyclic Polymerization of Acetylenes by Metal
Carbonyl Compounds», in: Organic Synthesis via Metal Carbonyls, Wen-
der I., Pino P. (Eds.), Wiley-Interscience, New York, 1968, Vol. 1, pp. 343—
369 (есть русский перевод первого издания книги: Органические синтезы
через карбонилы металлов. Пер. с англ./Под ред. Уэндера И., Пино П. —
М.: Мир, 1973).
55. Colbom R. Е., Vollhardt К. Р. С, J. Am. Chem. Soc, 103, 6259 (1981).
56. Wilke G., Pure Appl. Chem., 50, 677 (1978).
7—1129
12
КАТАЛИТИЧЕСКИЕ РЕАКЦИИ С УЧАСТИЕМ
МОНООКСИДА УГЛЕРОДА
12.1. Введение
Каталитическое введение монооксида углерода в
органические соединения явилось предметом интенсивных исследований
с начала сороковых годов. На базе этих исследований были
созданы важные промышленные процессы [1—4]. Основным
содержанием этой главы является химизм этих промышленных
процессов, главным образом синтезы товарных химикатов.
Начиная примерно с 1973 г., когда ОПЕК ввела эмбарго на
нефть, резко возросли темпы как прикладных, так и
фундаментальных исследований в области химии монооксида углерода,
однако в настоящее время падение цен на нефть на мировом
рынке приводит к уменьшению значимости этих исследований.
Подъем и падение интереса к получению товарных химикатов
из «синтез-газа» (СО/Н2) являются следствием того факта, что
нефть представляет собой основное сырье для промышленности.
Синтез-газ может быть получен путем риформинга природного
газа (в^основном метана), нефтяного битума, сырой нефти и
особенно угля, наиболее широко распространенного и обильного
источника восстановленного углерода. В середине семидесятых
годов казалось, что уголь становится важным альтернативным
химическим сырьем, в восьмидесятых годах начинает казаться,
что таковым будет метан. В любом случае синтез-газ будет
первой ступенькой на пути к многочисленным полезным химикатам.
Синтез-газ уже используется для получения линейных
(нормальных) альдегидов, спиртов >и их производных в результате «оксо-
процесса», для производства метанола, а из него, применяя
процесс фирмы Monsanto, уксусной кислоты или уксусного
ангидрида в результате модификации этого процесса. Привлекательной
с точки зрения промышленности целью является получение эти-
ленгликоля из СО/Н2, но, несмотря на интенсивные исследования,
жизнеспособный процесс пока не создан. Гетерогенно
катализируемое превращение СО/Н2 в линейные углеводороды и олефины
(процесс Фишера — Тропша) и превращение полученного из
синтез-газа метанола в высокооктановый бензин с помощью
кислотных молекулярных сит (процесс фирмы Mobil),
осуществляются на практике сегодня; однако это нерентабельные
«политические» процессы, мощно поддерживаемые государственными
98
Каталитические реакции с участием монооксида углерода 99
субсидиями. Существуют и другие приложения каталитической
химии СО в тонкой химической технологии, но еще большее их
число ждет своего открытия или разработок.
В границах данного изложения механизмы каталитической
химии монооксида углерода являются предметом
первостепенной важности. Примеры, относящиеся к наиболее важным
классам реакций, обсуждавшихся в ч. 1, гл. 4—6, могут быть
найдены в этих главах. Будет интересно увидеть, как группа СО,
являющаяся сильно связывающим я-кислотным лигандом, может
быть превращена в множество продуктов, несмотря на свою
склонность к образованию насыщенных комплексов и снижению
d-электронной плотности реакционноспособного металлического
центра. Будет показан также характерный способ активации СО
путем диссоциативной хемосорбции, но этот путь представляется
возможным только для некоторых гетерогенных катализаторов.
12.2. Конверсия водяного газа и связанные с ней реакции
Синтез-газ (смесь СО и Н2) получают паровым риформин-
гом или неполным окислением углеводородов. Процесс
конверсии водяного газа (КВГ) [уравнение (12.1)] используют для
получения Н2 и побочного продукта СО2. Этот
равновесный процесс катализируется многими растворимыми
комплексами переходных металлов; при этом применяемые в
промышленности катализаторы являются типично гетерогенными: Сг2О3
при 350 °С или смесь оксидов меди и цинка при 200—300 °С [5].
СО+Н2О — СО2+Н2 (12.1)
Были изучены многие гомогенные катализаторы КВГ,
включая Ru3(CO)12, Pt(PR3)3, Rh(CO)2I2- и HFe(CO)4" [6—9].
На базе исследований этих гомогенных катализаторов КВГ для
соответствующих реакций может быть записан механизм,
включающий уже знакомые типы реакций [уравнения (12.2 и 12.3)].
О
+ -н+ II
М—СО+Н2О —>■ [М—С—ОН] —* М—Н+СОа (12.2)
1 2 3
+ + СО +
М—Н+Н —>- [М]+Н2 —>■ М—СО (12.3)
Нуклеофильная атака на электрофильный карбонил
металла 1 (ч. 1, разд. 7.1), как полагают, приводит к образованию
нестабильного карбогидрокси-комплекса 2, который декарбоксили-
руется с образованием гидрида металла 3. Механизм этого де-
карбоксилирования является сложным, может быть использован
любой из двух путей (ч. 1, разд. 3.4, а). В некоторых случаях
100 Глава 12
координационно ненасыщенная протонированная форма
(МСО2Н) теряет СО2 по реакции, аналогичной известному
р-гидридному элиминированию [10]. В других случаях анионная
форма (МСО2~) теряет СО2, образуя анион металла, который
затем, реагируя с протоном, образует гидрид 3 [11]. В
зависимости от катализатора и от условий может преобладать любой
путь. Протонолиз гидрида 3 приводит к образованию Н2 и в
присутствии СО регенерирует исходный катализатор 1,
завершая цикл. Подобные механизмы могут рассматриваться и для
гетерогенно катализируемых реакций КВГ. Важно заметить, что
гидрид металла 3 может быть перехвачен. В условиях КВГ
можно гидрировать такие субстраты, как кетоны, альдегиды, олефп-
ны и нитрогруппы.
В то же время в присутствии СО и олефинов может
проходить гидроформилирование, приводящее к образованию
альдегида и СО2 [уравнение (12.4)]. Этот процесс иногда называют
реакцией Реппе. Было показано, что стадия образования продукта
в этой реакции представляет собой биядерное восстановительное
элиминирование, включающее реакцию между гидридом железа
и ацилом железа [12]. Тот же катализатор восстанавливает
альдегид в соответствующий спирт. Другие металлы, такие, как
рутений и родий, также будут катализировать реакцию Реппе
карбонилирования алкенов.
Fe(CO)5
RCH=CH2 + CO + Н2О > RCH2CH2CHO + RCHCH3 + CO2 (12 4)
он- | v ;
СНО
12.3. Гидрокарбонилирование олефинов (оксо-реакция)
В присутствии некоторых гомогенных катализаторов —
производных переходных металлов — олефины реагируют с
синтез-газом (СО + Н2) с образованием альдегидов [уравнение (12.5)].
Этот важный промышленный процесс часто называют «гидр'о-
формилированием», так как формально происходит
присоединение элементов формальдегида по двойной связи. В
действительности формальдегид не является интермедиатом этой реакции,
которую также называют «оксо-процессом». В наиболее важных
промышленных оксо-процессах используют кобальтовый или
родиевый катализатор для превращения пропилена в смесь
масляного и изомасляного альдегидов.
катализатор Rh или Со
СН3СН=СН2 + СО + Н2 >- СН3СН2СН2СНО — CHgCHCHg
I
СНО
(12.5)
Каталитические реакции с участием монооксида углерода 101
В мире ежегодно производится несколько миллионов тонн
оксопродуктов [1]. Альдегиды с неразветвленной цепью
являются более предпочтительными, нежели с разветвленной.
На последующих стадиях масляный альдегид превращается
в я-бутанол, 2-этилгексанол или 2-этилгексановую кислоту
[уравнение (12.6)]. Основной промышленный продукт,
2-этилгексанол, превращают в эфиры фталевой кислоты, которые
используют как пластификаторы для поливинилхлоридных смол. Оксо-
процесс используется также для превращения линейных олефи-
нов, например 1-октена, в линейные спирты, которые применяют
в качестве детергентов [1]. Три промышленных катализатора
используются в оксо-процессе: Со2(СО)8, карбонил кобальта,
модифицированный фосфином, и модифицированная фосфином
родиевая система. Ниже мы обсудим и сравним кобальтовый
и родиевый катализаторы в контексте того, что известно об их
реакционных механизмах [13—15], а затем кратко опишем
некоторые другие оксо-катализаторы.
.сно
сн3сн2сн2сно
лизатор
-н2о
сн3(сн2)2сн =
ката-
лизатор\ н?
сн3(сн2)3сн
сн2сн3
сн3(сн2)3сн
(12.6)
сн2сн3
сн2он
чсн2сн3
2 -
а. Кобальтовые оксо-катализаторы. Первый оксо-катализа-
тор, Со2(СО)8, был открыт Реленом [4] в 1938 г., и до
настоящего времени это соединение является промышленно важным
катализатором. В интервале температур 120—170 °С и давлений
200—300 атм для смеси СО+Н2 образуется масляный альдегид
с выходом 70—80%.
Более высокие выходы желаемых линейных альдегидов
получают, используя модифицированный три-я-бутилфосфином
кобальтовый катализатор, предложенный фирмой Shell [16]. Хотя
этот модифицированный кобальтовый катализатор ведет процесс
более медленно и требует более высоких температур для
получения приемлемых с точки зрения экономики скоростей, он более
устойчив к разложению, чем Со2<(СО)8, и поэтому его можно
использовать при более низких давлениях (^100 атм). Фосфин-
замещенные кобальтовые катализаторы дают и дополнительные
преимущества: реализуются более высокие соотношения нор-
102 Глава 12
мальных и изо- (линейных и разветвленных) продуктов,
альдегид может быть отогнан от катализатора, упрощается технология
процесса. Использование модифицированных фосфинами оксо-
катализаторов позволяет превращать олефины с внутренней
двойной связью в нормальные спирты с концевой ОН-группой
через сочетание стадий изомеризации олефинов и региоселектив-
ного гидроформилирования. Однако модифицированные
фосфинами катализаторы ускоряют гидрирование некоторых олефинов,
являющееся нежелательной побочной реакцией. Эти
катализаторы превосходно работают в тех случаях, когда целевым
продуктом являются линейные спирты, в других случаях
предпочтительнее простой катализатор — карбонил кобальта, как,
например, в производстве 2-этилгексанола.
Несмотря на то что механизм катализируемых кобальтом
оксо-реакций был объектом многочисленных исследований и
обсуждений, он не является абсолютно понятным. Исследование
скоростей, анализ продуктов, инфракрасная спектроскопия в
условиях реакции, модельные реакции для разных стадий
реакционного цикла и изотопные метки — все это было использовано
в попытках разобраться в этом многостадийном процессе. Хотя
и существует возможность написания серии стадий, которые
в сумме описывают полный реакционный цикл, но
количественное понимание каталитического процесса на основе
кинетических параметров и параметров равновесия для каждой
индивидуальной стадии пока отсутствует.
Механизм, который обычно записывается для оксо-реакции,
был впервые предложен Хеком и Бреслоу [15] на основе
реакций .модельных органических карбонильных комплексов
кобальта. Рассмотрим последовательно каждую из этих стадий.
Предкатализатором, несомненно, является НСо(СО)4,
исключительно кислотный (ч. 1, разд. 3.4,6) «гидрид» переходного
металла. Он образуется в результате гидрирования Со2(СО)з
[уравнение (12.7)]—реакции, ингибируемой СО. В некоторых
случаях НСо(СО)4 приготовляют отдельно и вводят в оксо-ре-
акцию [17].
н2 —со
Со2(СО)8 — 2НСо(СО)4 ^=^ НСо(СО)3+СО (12.7)
+СО
Следующая стадия — «миграционное внедрение» олефина по
связи Со—Н, — очевидно, требует его предварительного
координирования, при этом одна группа СО должна быть потеряна
[уравнение (12.7)] перед внедрением олефина [уравнение
(12.8)]. Образование линейного или разветвленного альдегида
на стадии присоединения этого кобальтогидрида контролируется
региохимией процесса, тем не менее присоединение является
быстрым н обратимым.
Каталитические реакции с участием монооксида углерода 103
_с0 . RCH2CH2 (12.8)
+RCH = CH2 ^= 'S Со(СО)3
НСо(СО)4 ч Н-Со(С0)3
-RCH=CH2 ^ RCH-CH3
Со(СО)3
Обратимостью этой стадии объясняется то, что эти кобальто-
гидридные комплексы способны катализировать и изомеризацию
олефинов, и изотопный обмен между атомами дейтерия и
водорода в олефине. Из-за необходимости вакантного
координационного центра и изотопный эффект, и изомеризация олефина
тормозятся при более высоких парциальных давлениях СО.
Этот процесс изомеризации олефина/изотопного обмена
иллюстрируется реакцией о-тридейтерированных олефинов в
присутствии оксо-катализатора карбонила кобальта при
относительно низких давлениях СО/Н2 [18]. Принимая во внимание,
что небольшое количество дейтерия продуктом теряется, атомы
дейтерия распределены вдоль цепи почти статистически. Для
анализов альдегидный продукт превращали в сложный эфир
[уравнение (12.9)]. Полученные результаты соответствовали
представлениям о быстрой внутримолекулярной
перегруппировке кобальтоалкильного интермедиата.
1) Со2(СО)8, СО, Н2
CD3CH2CH2CH2CH=CH2 >• C6H10D3CO2Me (12.9)
2) окислитель, МеОН, Н+ v '
Следующей стадией каталитического цикла является еще
одно «миграционное внедрение» с образованием кобальтоацила.
Образующийся координационно ненасыщенный ацильный
комплекс кобальта 4 обратимо присоединяет молекулу монооксида
углерода [уравнение (12.10)]. При исследовании методом
ИК-спектроскопии стационарной реакции в стандартных
каталитических условиях [Со2(СО)з, 150 °С, 250 атм] в случае линейных
олефинов, в частности 1-октена, единственным обнаруживаемым
комплексом кобальта был ацильный RO(0)Co(CO)4 [19].
О О
II со II
RCo(CO)4 =<=* R_C-Co(CO)3 <=* R_C-Co(CO)4 (12.10)
4
Для стадии образования альдегида были предложены два
механизма: окислительное присоединение водорода к
ненасыщенному ацильному комплексу (с последующим
восстановительным элиминированием альдегида) или биядерное
восстановительное элиминирование с участием того же самого
ненасыщенного ацила и НСо(СО)4 [уравнение (12.11)]. С точки зрения
стехиометрии могут протекать обе реакции, но какая из них яв-
104 Глява 12
ляется важной в каталитических условиях, неочевидно.
Различные группы исследователей представляли доказательства в
пользу прямого окислительного присоединения Н2 [20а, б] и в
пользу биядерного варианта [20в—д]. Оба процесса должны ингиби-
роваться СО [61], поскольку для каждого необходим свободный
координационный центр на ацильном комплексе. Комплексы,
записанные в скобках, не могут наблюдаться в реакционных
условиях, поскольку это ненасыщенные комплексы, быстро
присоединяющие СО.
о
RCH «■ [Со2(СО)-^]
(12,11)
Реакции, описываемые уравнениями (12.7), (12.8), (12.10)
и (12.11), вместе составляют каталитический цикл. Соотношение
линейных и разветвленных альдегидов определяется обоими
уравнениями (12.8) и (12.10). Для линейных олефинов
определяющая скорость стадия включает ацильный комплекс
RC (О) Со (СО) 4. Модифицированный фосфином катализатор
фирмы Shell дает более высокое отношение линейных и
разветвленных продуктов, поскольку в этом случае сильнее региохими-
ческий контроль стадии присоединения кобальтогидрида к оле-
фину. При определении направления присоединения по или
против правила Марковникова, приводящего соответственно к
образованию разветвленного или линейного альдегида,
важными являются и стерические и электронные эффекты. Лиганд
РВи3 занимает большой объем, и соответствующий гидрид
НСо(СО)з(ИзР) представляет собой намного более слабую
кислоту Брёнстеда, чем НСо(СО)4 (ч. 1, разд. 3.4,6). Анализы,
выполненные методом ИК-спектроскопии, в условиях стационарной
оксо-реакции с модифицированным фосфином катализатором не
дали доказательств образования алкил- или ацил-кобальтовых
комплексов, наблюдались только димеры фосфинзамещенного
карбонила кобальта и гидридные комплексы [21]. По-видимому,
в случае этого модифицированного катализатора каталитический
цикл имеет другую определяющую скорость стадию.
б. Родиевые оксо-катализаторы. Простые карбонилпроизвод-
ные родия не представляют ценности как оксо-катализаторы.
При их использовании процесс осложняется конкурирующими
равновесиями, включающими термодинамически стабильные, но
Каталитические реакции с участием монооксида углерода 105
каталитически неактивные тетра- и гексамерные кластеры кар-
бонила родия. Тем не менее они способны образовывать
каталитически активный комплекс HRh(CO)4 [22] при высоких
давлениях СО/Н2 [уравнение (12.12)]. Несмотря на свою высокую
каталитическую активность, HRh(CO)4 приводит к
гидрированию и изомеризации олефинов и по сравнению с карбонилко-
бальтовыми катализаторами дает более низкое отношение
линейных продуктов к разветвленным.
Rh4(C0)|2 * * Rh6(C0)l6 (12.12)
HRh(C0)4
Добавление фосфиновых лигандов к исходным веществам для
получения карбонилродиевого катализатора дает возможность
получения активного катализатора с великолепной
селективностью образования желаемых линейных альдегидов. Эти ли-
ганды стабилизируют мономерные родиевые комплексы, ингиби-
руя образование кластеров. Такие системы приводят к
существенному увеличению отношения линейных и разветвленных
продуктов (вплоть до 30 : 1) и подавляют гидрирование и
изомеризацию олефинов, но тем не менее обеспечивают высокие
скорости, близкие к получаемым в случае их предшественников —
карбонилродиевых катализаторов.
Модифицированные фосфинами родиевые оксо-катализаторы
являются реакционноспособными даже при обычных давлениях
и температурах. Поэтому их можно использовать в
синтетических лабораториях, не прибегая к специальному оборудованию
для высоких давлений, редко доступному для академических
лабораторий. Существует ряд пригодных для оксо-реакций
родиевых предкатализаторов. Полезными являются металлический
родий на углероде, RhCl(CO) (PPh3)2 + Et3N, (acac)Rh(CO)2 и
HRh(CO) (РРЬз)з; последние два могут вводиться
непосредственно при обыкновенных давлении и температуре без
индукционного периода.
Такие модифицированные родиевые оксо-катализаторы были
введены в промышленное производство фирмой Union Carbide
в 1976 г. [23, 24] и впоследствии использовались несколькими
другими компаниями. Одновременно Уилкинсон с сотрудниками
[25, 26] исследовали эти реакции в обычных условиях и
предложили механизм процесса.
В присутствии избытка трифенилфосфина могут быть
достигнуты высокие выходы масляного альдегида с высокой
селективностью по линейному альдегиду (до 92% от общего выхода).
106 Глава 12
Промышленный процесс с участием пропилена можно проводить
в расплаве трифенилфосфина (ТПЛ = 79ОС) в качестве
растворителя при умеренных условиях (100 °С, до 50 атм). Лишь
небольшое количество пропилена гидрируется и превращается в
пропан, и в очень малой степени происходит гидрирование
альдегида. Таким образом, несмотря даже на то что родий намного
дороже кобальта (300: 1), благодаря умеренным условиям,
высокой селективности, большим выходам, улучшенной стабильности
катализатора, простоте отделения продукта и более высоким
скоростям родиевые каталитические системы способны
конкурировать с кобальтовыми в промышленном производстве. Тем не
менее фосфины в условиях реакции медленно разрушаются в
результате окислительного присоединения по связи
фосфор—углерод [27], поэтому потерю ценного фосфина необходимо
принимать во внимание в случае данного и, по-видимому,
большинства других модифицированных фосфинами промышленных
катализаторов.
0
RCH2CH2CH —^
н2 —^
RC
HRh(
*
{a) -CO
\ «.yquj
7
Рис.
:o)2l2
+ C0
ГЛ]| ^. ^
LUK.2 -^
5 ^^
CO)L2 ^~*^
12.1.
/ RCH=CH2
/
RCH2CH2Rh(CO)L2
CO
Механизм гидроформилирования с использованием
комплекса родия, модифицированного фоофинами, исследовали во
многих лабораториях, используя ИК-, ПМР- и 31Р-ЯМР-спектроско-
пию. Эти обширные исследования рассмотрены в обзоре [14].
Хотя детальный механизм не вполне понятен, можно записать
вероятный каталитический цикл (рис. 12.1). Отношение L/Rh
(где L — третичный фосфид) и стадия, определяющая скорость,
различны при разных условиях реакции. Для простоты на
рис. 12.1 изображено отношение L/Rh, равное 2: 1, но,
несомненно, другие комплексы также вносят вклад в общую реакцию.
На рисунке приведен только главный линейный алкильныи
комплекс.
Первая ступень (а) включает создание вакантного
координационного центра в 5, который необходим для последующих
координирования и внедрения олефина на стадии (б) с образова-
Каталитические реакции с участием монооксида углерода 107
нием 6. Уилкинсон [25] предложил дополнительную
параллельную схему, в которую включены 20-электронные интермедиа™,
однако это кажется маловероятным [14]. Региохимия стадии
присоединения гидрида контролирует селективность реакции.
Присоединение по правилу Марковникова (гидрид к концевому
атому углерода, который на рис. 12.1 не показан) привело бы
к образованию разветвленного продукта и могло бы нести
ответственность за изомеризацию олефина. Вопрос о том,
кинетически или термодинамически контролируется соотношение
линейных и разветвленных алкильных комплексов в условиях
стационарной каталитической реакции, неясен; несомненно, что это
соотношение зависит от условий реакции. При умеренных
давлениях ненасыщенный алкильный комплекс 6 необратимо
связывает СО, ингибируя изомеризацию олефина или превращение
в разветвленный алкильный комплекс.
Стереохимия стадии (б) на рис. 12.1 была исследована
[28]. В результате было продемонстрировано стереоспецифиче-
ское ц^с-присоединение D и DCO к 2-3-метил-2-пентену, как
показано в уравнении (12.13). £-Олефин дает такое же цис-ищсо-
единение. Эти результаты соответствуют ^^-присоединению
гидрида и родия с одной и той же стороны олефина, вслед за
которым происходит внедрение [стадия (в)] с сохранением
конфигурации. Это соответствует ожидаемой стереохимии таких
процессов внедрения (ч. 1, разд. 6.3).
М<4 J* ))DRh(C0)L3 Me^ чЧчН
■ 2) [о] ,
L=PPh3
Стадия (в) ведет к образованию ацильного комплекса 7,
который необратимо гидрируется в линейный альдегид на
стадии (г). Последняя реакция, как считают, является
определяющей скорость практически при любых условиях, тем не менее
детальный механизм стадии (г) неизвестен. Большинство
авторов предполагает простое окислительное присоединение Н2, за
которым следует восстановительное элиминирование альдегида
[уравнение (12.14)]. В другом случае стадия (г) может
включать биядерное восстановительное элиминирование с другим
гидридом родия [уравнение (12.15)]. Любой из процессов требует
одного вакантного координационного центра на ацильном
комплексе родия 7 и поэтому должен ингибироваться СО.
О О
II Н2 II быстро
R'—С—Rh(CO)L2 —>- R'— С—Rh(H)2(CO)L2
108 Глава 12
О
I!
R'-C-H+H-Rh(CO)L2 (12 14)
О
R'—С—Rh(CO)L2+HRh(CO)2L2 —►•
7
О
I! н2
—> R'C—H+Rh2(CO)3L4 —♦■ 2HRh(CO)L2 (12.15)
-со
Недавние исследования фосфин-замещенных комплексов
родия, изолированно расположенных на силикагеле, показали, что
только пары родиевых комплексов, расположенные достаточно
близко друг к другу, чтобы претерпеть биядерное
восстановительное элиминирование [как в уравнении (12.15)], могут быть
оксо-катализаторами. Полностью изолированные связанные сси-
ликагелем родиевые комплексы не проявляют никакой
каталитической активности [29]. Неясно, насколько этот результат может
быть экстраполирован на растворимые родиевые оксо-катали-
заторы.
Влияние различных реакционных переменных на оксо-про-
цесс, катализируемый родием, может быть систематизировано
с помощью схемы, показанной на рис. 12.1. Увеличение
концентрации фосфина замедляет скорость реакции, но увеличивает
отношение линейных и разветвленных продуктов и подавляет
как гидрирование, так и изомеризацию олефина. Это
соответствует росту региоселективности стадии присоединения
гидрида (б). Наличие пространственно-объемного фосфина или
нескольких фосфинов, по-видимому, способствует региохимизму
против .правила Марковникова, однако электронные эффекты
также могут играть значительную роль.
Увеличение давления СО сильно повышает тенденцию к
присоединению формильной группы к одному из атомов углерода
исходной двойной связи, снижая таким образом вероятность
образования изомеризованных продуктов. Это объясняется
ускорением стадии внедрения (в), приводящим к эффективному
подавлению обратимости стадии (б) (что может приводить к
изомеризации олефина). Таким образом, происходит захват комплекса 6
и принуждение его к движению вперед в соответствии с
каталитическим циклом, показанным на рис. 12.1.
Определяющая скорость стадия (г) требует свободного
координационного центра, поэтому избыток фосфина тормозит
суммарный процесс. Аналогичным образом может быть
объяснен и факт уменьшения скорости при более высоких давлениях
СО. С другой стороны, реакция имеет первый порядок по давле-
Каталитические реакции с участием монооксида углерода 109
нию водорода из-за того, что скорость определяется
стадией (г).
При достаточно высоких концентрациях олафина скорость
перестает быть чувствительной к концентрации олефина. В этом
состоянии стационарности главной формой катализатора
является ацильный комплекс типа 7; таким образом, можно сказать,
что катализатор «насыщается» олефиновым субстратом [30].
В то же время при низких концентрациях субстрата в
выражение для скорости концентрация олефина входит в первой
степени, и начальная стадия цикла становится определяющей
скорость [14].
Индивидуальную относительную реакционную способность
различных олефинов в условиях гидроформилирования грубо
можно считать аналогичной реакционной способности в
условиях каталитического гидрирования (ч. 1, гл. 10), однако это
зависит от того, какая стадия каталитического цикла является
лимитирующей при конкретном наборе условий. Например, если
скорость реакции имеет нулевой порядок по олефину,
катализатор уже насыщен олефином и относительные скорости для
индивидуальных олефинов будут зависеть не от стадии
координирования, а от некой другой стадии. При других обстоятельствах
относительные скорости будут действительно отражать
относительное сродство различных олефинов к комплексу металла.
Например, если в одной и той же колбе реагируют два олефина,
каждый из них является конкурентным ингибитором по
отношению к другому в ходе соперничества за доступный активный
катализатор. Поэтому их относительные скорости, вероятно,
будут отражать относительные способности этих двух олефинов
к равновесному связыванию даже в упоминавшихся выше
условиях насыщения.
Предшествующие объяснения являются в значительной
степени упрощенными; в случае такого большого числа сложных
переменных фактически разобраться в них невозможно.
Например, существует несколько конкурирующих реакционных циклов,
причем каждый включает отличное от другого отношение Rh/L
[14], а мы в нашем упрощенном обсуждении учитывали только
один из них.
Во многих исследованиях катализируемых родием оксо-реак-
ций объектом,рассмотрения являлись зависимости скорости и
селективности образования продукта от природы фосфина [14].
В случае простых олефиновых субстратов ни один из лигандов
не превосходит заметно простые триарилфосфины, большинство
дают худшую скорость или селективность, или и то и другое
вместе. Однако, как показано в уравнении (12.16), стерически
затрудненные фосфиты более действенны в случае затрудненных
олефинов [31].
110 Глава 12
Rh(CODMOAc),
н
L*= P(OR)3i R= -C(CF3)2,
Другой интересной перспективой является асимметрическое
гидроформилирование в присутствии катализаторов,
замещенных хиральными фосфинами. Если из прохиральных олефинов
удастся получить большие (выше 90%) энантиомерные
избытки (ее), возможно, эти реакции будут полезными при синтезе
природных продуктов и даже в фармацевтической
промышленности. К сожалению, самое высокое из полученных к
настоящему времени значение энантиомерного избытка составляет 73%,
а в большинстве случаев это величина, меньшая 40% [32а].
Следующая проблема заключается в том, что в случае олефинов
с концевой двойной связью желаемый продукт представляет
собой хиральный i/зо-альдегид, а не я-альдегид. Пример,
приведенный в уравнении (12.17), является лучшим из
опубликованных результатов [326].
PtCi?/SnCI2,
Н\ DIPHOL
С-СН? PhCH-СН* ♦ PhCHoCH2CHO + PhCH2CH3 Ul2 m
Ph^ H2/C° Ыо
56% (7 3% ее) 14% 20%
Реакция, описываемая уравнением (12.17), использующая
хиральный фосфин DIPHOL [33, 34], иллюстрирует образование
нежелательного я-альдегида и алкановых побочных продуктов,
а также использование другого металла — платины — в качестве
катализатора гидроформилирования. Перейдем теперь к
рассмотрению других оксо-катализаторов, помимо кобальта
и родия.
в. Другие переходные металлы как оксо-катализаторы.
Комплексы нескольких других переходных металлов использовались
в качестве оксо-катализаторов. Для повышения реакционной
способности и стабилизации катализатора в присутствии СО
и Н2 часто необходимы вспомогательные лиганды. Реакционная
способность различных карбонилов металлов как
оксо-катализаторов по отношению к кобальту выглядит следующим образом:
Rh>Co>Ru>Mn>Fe>Cr, Mo, W, Ni
103—104 1 Ю-2 1СГ4 10"6
Каталитические реакции с участием монооксида углерода 111
Мы уже упоминали платиновые оксо-катализаторы
[уравнение (12.17)] в связи с асимметрическими реакциями. Для этих
катализаторов нужен необычный вспомогательный лиганд
SnCl3~ и фосфин (обычно хелатообразующий). Используют
умеренные температуры (15—100°С) и давления (20—100 атм).
С помощью этих катализаторов были достигнуты высокие
отношения линейных и разветвленных продуктов (вплоть до 98%),
однако полезными представляются только алкены с концевой
двойной связью. Серьезной побочной реакцией может являться
гидрирование олефинов; кроме того, эти катализаторы не
обладают особенно высокой реакционной способностью и
стабильностью [35, 36].
Фосфин-замещенные иридиевые оксо-катализаторы
стабильны, но не очень реакционноспособны. Примечательным примером
является HIr (СО)зР(^зо-Рг)3, катализирующий гидроформили-
рование этилена при 14 атм и 50 °С. Анализ ИК-спектров этой
системы свидетельствует о последовательных гидридной, алкиль-
ной и ацильной стадиях в каталитическом цикле [37]. Неясно,
какая из стадий цикла определяет скорость, однако элемент
третьего ряда иридий должен претерпевать более медленные
реакции обмена лиганда (см. ч. 1, гл. 4) и более медленное
миграционное внедрение (ч. 1, гл. 6), чем элементы первого и второго
ряда, например кобальт или родий. Комплексы более тяжелых
металлов часто используют в модельных исследованиях
индивидуальных стадий реакций, быстро катализируемых более
легкими сородичами. Например, ацильныи комплекс иридия
гидрируется с образованием пропионового альдегида [уравнение
(12.18)]; эта реакция, по-видимому, требует свободного
координационного центра, поскольку ингибируется СО [25].
О О
II II
EtC—Ir(CO)3L2+H2 —+• Et-C—H+HIr(CO)2L2 (12.18)
Фосфиновый комплекс рутения(0) Ru(CO)3(PPh3)2 также
является оксо-катализатором. Этот предкатализатор, по-видимому,
действует, сначала отщепляя СО, а затем присоединяя Н2;
предполагают, что эти стадии являются лимитирующими в оксо-цик-
ле [38а]. При 100 °С и давлении водорода и монооксида
углерода 100 атм этот катализатор позволяет получить из олефинов
с концевой двойной связью линейные и разветвленные
продукты в соотношении только 3 : 1. Карбонилы рутения дают высокие
отношения линейных и разветвленных продуктов [386].
г. Различные аспекты оксо-реакции. Существует
возможность включения стадии биядерного восстановительного
элиминирования, по крайней мере, в некоторые оксо-процессы. Мы уже
112 Глава 12
обсуждали эффект блокирования этой стадии в случае
изолированно расположенных родиевых оксо-катализаторов [29]. Если
бы эта биядерная стадия была важной, оксо-катализаторы,
содержащие два разных металла, могли бы демонстрировать
весьма различное каталитическое поведение. Действительно, было
отмечено небольшое ускорение реакций гидроформилирования
при использовании гетерометаллических гомогенных
катализаторов [39, 40], однако механизм, приводящий к этому эффекту,
не был объяснен. Например, карбонильный комплекс кобальта
и рутения в соотношении 1 : 10 в 27 раз более активен, чем
обычный карбонил кобальта [39]. И выход продукта, и скорости
выше, чем сумма соответствующих величин для индивидуальных
катализаторов.
Превращение гомогенных оксо-катализаторов в гетерогенные
в результате связывания их посредством фоофиновых лигандов
с нерастворимым носителем (полимерным гелем [41] или сили-
кагелем [29, 34]) не приводит к получению лучших
катализаторов. В связи с этим подходом возникает ряд проблем.
Во-первых, оксо-реакции сильно экзотермичны (около 28 ккал/моль),
поэтому в случае быстродействующего гетерогенного оксо-ката-
лизатора возникает проблема диссипации тепла. Другая
проблема связана с «вымыванием» катализатора, это явление можно
подавить, только используя полидентатные лиганды,
обладающие высоким сродством к металлу, но эти лиганды часто
замедляют каталитическую реакцию! Последняя проблема
заключается в том, что иммобилизованные лиганды разрушаются в
результате окислительного присоединения по связи Р—С [27].
Такие реакции становятся дополнительным путем потери
катализатора.
Намного более перспективным подходом представляется
удерживание активного катализатора во второй жидкой фазе
[42]. Например, химики фирмы Rhone-Poulenc использовали
водорастворимый фосфин с родиевым оксо-катализатором в
реакторе с мешалкой, содержащем две несмешивающиеся фазы:
углеводород и воду. Такой «обратный межфазный катализ»
облегчает отделение продукта (в неводной фазе) от катализатора
(в водной фазе) фактически без потерь, обеспечивает высокие
скорости и прекрасный теплоперенос [42]. Однако проблема
разрушения фосфина, по-видимому, не может быть решена таким
способом. Основные принципы описанного метода показаны
в уравнении (12.19).
Было высказано предположение, что некоторые
катализируемые кобальтом оксо-реакции могут протекать по свободноради-
кальному механизму [43]. Эта возможность более
предпочтительна для таких субстратов, как стирол, который может
образовывать резонансно-стабилизированный бензильный радикал.
Каталитические реакции с участием монооксида углерода 113
неводная „сн сн сно (12.ig>
R-CH-CH.
5
перемешивание
HRh(CO)(TPPTSL
бодная
} дОатж
^2
н/изд ~ В4- %
небодная
(раза
сразсс
TPPTS= Р
()(
водный
рас/лвор
SO; Na
Оксо-реакции сопряженных олефинов приводят к сложной
смеси продуктов и до настоящего времени применения не нашли
[44, 45]. В частности, 1,3-бутадиен не дает чистого продукта
в случае кобальтовых катализаторов, а родиевые катализаторы
приводят к образованию смеси моно- и диальдегидов.
Образование нереакционноспособных я-аллильных комплексов является
проблемой в случае этого и других субстратов «(таких, как ал-
лены [46]).
В случае а,р-ненасыщенных эфиров главной конкурирующей
реакцией становится гидрирование олефина. Кроме того,
образуются смеси а- и р-формильных продуктов [уравнение (12.20)].
Соотношение этих продуктов, вероятно, определяется региосе-
лективным присоединением гидридного, комплекса родия к
сопряженному олефину, однако факторы, его контролирующие, не
понятны. Рост температуры способствует образованию
^-продукта, в то время как повышение давления или присутствие
третичного фосфина облегчает образование а-продукта.
Но/СО
СО2Ме
СОоМе
СНО
О
НС'
12.4. Каталитическое карбоалкоксилирование олефинов
Производные карбоновой кислоты могут быть получены па
каталитической реакции между монооксидом углерода и олефи-
нами в присутствии воды, спирта или амина. Эти реакции,
которые катализируются различными соединениями переходных ме~
8—П29
114 Глава 12
таллов [4], служат основой нескольких промышленных
процессов [1]. Один из предложенных механизмов процесса
карбоалкоксилирования олефинов включает сольволиз ацильно-
го интер|медиата [уравнение (12.22)]. Ацильный комплекс
типа 8 может образовываться в результате присоединения ме-
таллогидрида к олефину с последующим внедрением алкил —
карбонил [уравнение (12.21)]. Тем, что металлогидрид
превращается в ацильный комплекс 8, эти процессы напоминают гидро-
формилирование; разница заключается в том, что при карбоал-
коксилировании ацильный комплекс затем реагирует с нуклео-
филом, например водой, спиртом или амином [уравнение
(12.22)], в то время как в оксо-процессе ацильный комплекс
подвергается гидрогенолизу.
с=с о
Н-
II II со II I I
1_М >• м_с^С—Н >- М—С—С—С— Н (12.21)
8
о о
II I I HY I I II
М—С—С—С—Н * М—Н + Н—С—С—С—Y (12 22)
II II v ■ ;
8
Y=OH, OR, NR2
В промышленности процессы карбоалкоксилирования
используют для получения из линейных олефинов эфиров жирных
кислот. При карбоалкоксилировании, так же как в оксо-процессах,
важным моментом является селективность образования
линейных продуктов по отношению к разветвленным. Другая
проблема, связанная с этими реакциями, касается образования
водорода известным путем конверсии водяного газа
[уравнение (12.23)]; образующийся водород может приводить к
гидрированию и даже гидроформилированию олефина. Эти побочные
реакции могут стать опасными. Основным промышленным
катализатором карбоалкоксилирования олефинов является Со2 (СО) 8;
однако все большее внимание привлекают комплексы палладия,
благодаря их эффективности в мягких условиях.
катализатор М
со + н2о с > со2 + н2 (12.23)
Механизм Со—Н/олефин для катализируемого кобальтом
карбоалкоксилирования показан на рис. 12.2. Типичными
условиями являются 140—170 °С при 100—200 атм СО в метаноле.
Каталитические реакции с участием монооксида углерода 115
Присутствуют малые количества Н2. Получают около 70%
я-алкилового эфира наряду с 30% разветвленного эфира.
Главное различие между механизмом, показанным на рис. 12.2,
и гидроформилированием заключается в атаке ацильного
комплекса метанолом. При добавлении пиридина в эту реакцию
скорость возрастает и увеличивается региоселективность в
отношении линейных сложных эфиров [47а]. По-видимому, пиридин
атакует промежуточно образующееся ацильное производное
Со?(СО)с
н2
RCH=CH2
СО
(OC)4Co-CH2CH2-R
СО
Рис. 12.2.
и таким образом увеличивает скорость образования эфира
[уравнение (12.24)].
R~C-Co(CO)4
РУ
О
R-C.-py
Со(СО)4
МеОН
RC-OMe + НСо(С0)4 + РУ
Милштейн [476] опубликовал доказательства протекания
катализируемого Соя (СО) s процесса карбометоксилирования
бутадиена не через Со—Н, а через интермедиат СОСО2СН3 по
механизму, аналогичному записанному ниже в уравнении (12.26).
Этот процесс используют в производстве диметиладипината из
бутадиена [47в].
Так же как в случае оксо-процесса, катализируемое
кобальтом карбоалкоксилироеание можно проводить для смеси олефи-
нов с концевыми и внутренними двойными связями; в ходе
реакции происходит быстрая изомеризация олефина, приводя к ли-
116 Гла»а 12
нейным сложным эфирам в качестве главного продукта
[уравнение (12.25)].
(12.25)
Со2(СО)8
РУэ СО?
МеОН (н2)7
. 160°С? 150 атм
Катализ карбоалкоксилирования олефинов палладием идет
в более мягких условиях [48, 49]. Известны два класса
катализаторов, для каждого существует, по-видимому, свой механизм.
В присутствии кислоты (НС1) РсЮ^РРЬзЬ катализирует кар-
боалкоксилирование пропилена, давая преимущественно
разветвленный продукт. Было высказано предположение, что эта
реакция не включает внедрение олефина в гидридный комплекс
палладия. Вместо этого сначала может образовываться алкокси-
карбонильное производное 9, которое затем присоединяется
к олефину [уравнение (12.26)]. Предполагают, что региохимизм
этого внедрения должен приводить к образованию первичного
алкил-палладиевого интермедиата 10. Хлороводород, согласно
предположению, рвет связь Pd—С в 10, регенерируя
катализатор и образуя изобутират в качестве продукта
[уравнение (12.27)].
CO2R
L2PdCI2 11 l-2PdC0R —■•* L2Pd-CH2CHCH3 (12.26)
HCI (T°2R (12.27)
L2Pd-CH2CHCH3 L2PdCI2 + CH3CHCH3
При использовании палладиевого катализатора в условиях
нейтрального рН необходимо введение специальной добавки —
SnClV Хлорид олова1 (II) в виде лиганда SnCl3", как полагают,
приводит к образованию гидридного комплекса палладия 11
в присутствии [уравнение (12.28)] или в отсутствие водорода
[50]. Этот комплекс присоединяется к олефину с концевой
двойной связью преимущественно против правила Марковникова.
н2
Pd(SnCl3bL2 —+■ HPd(SnCl8)L2+H++SnCl3- (12.28)
И
Последующее внедрение СО в алкильные комплексы
палладия и алкоголиз ацильного производного завершают
каталитический цикл, аналогичный первому из механизмов,
предложенных для катализа кобальтом [уравнение (12.29)]. Лиганд
5пС13~ не только способствует образованию и стабилизации
Каталитические реакции с участием монооксида углерода 117
гидрида, но также оказывает очень сильное и благоприятное
влияние на региохимизм внедрения гидрида, приводящего к
образованию линейного сложного эфира [1, 50]. Отметим, что
в обоих механизмах интермедиа™, алкильные комплексы
палладия, имеют первичную структуру, эта преимущественная ре-
гиохимия, несомненно, является результатом влияния объемных
фосфинов.
со
HPd(SnCl3)L2+RCH = CH2 —> RCH2CH2Pd(SnCl3)L2 —■*■
О О
II R'OH II
—> RCH2CH2C—Pd(SnCl3)L2 —■*■ RCH2CH2C—OR'+HPdfSnCyLa
(12.29)
Предполагалось, что сольволиз этих ацилпроизводных
происходит по механизму присоединения — элиминирования, во
многом напоминающему простой гидролиз сложного эфира.
Модельная реакция, для которой этот механизм был предложен,
представляла собой показанную в уравнении (12.30) [51]
реакцию ацильного комплекса марганца. Подобно гидролизу эфира
этот процесс нуждается в кислотном или щелочном катализе.
О ОН О
II ROH | ||
СН3С—Мп(СО)5 <=± СН3—С—Мп(СО)5 > СН3С—OR+HMn(CO)5
OR
(12.30)
Многие другие комплексы переходных металлов
катализируют карбоалкоксилирование олефино'в, но менее эффективно, чем
катализаторы, рассмотренные в этом разделе.
12.5. Карбонилирование ацетиленов
Ацетилены, так же как олефшны, имеют высокое сродство
к переходным металлам. Поэтому не удивительно, что можно
карбонилировать ацетилен; известны процессы как моно-, так и
дикарбонилирования ацетилена. В Европе ацетилен
используется как промышленное сырье; его получают при крекинге метана.
Промышленный процесс с использованием карбонила никеля
превращает ацетилен в ценный мономер — акриловую кислоту
[уравнение (12.31)] [4].
Ni(C0)4 н \ ^ Н
HCSCH+CO*H2O — "^С = ССГ (12.31)
Дикарбонилирование ацетилена приводит к образованию
янтарной кислоты, катализатором служит карбонил кобальта
118 Глава 12
[уравнение ;(12.32)] [4]. Механизмы этих реакций, несомненно,
аналогичны механизмам карбоалкоксилирования олефинов [92].
Однако ацетиленовые С—Н-группы могут окислительно
присоединяться к низковалентным металлическим центрам.
Присоединения такого типа приводят к нежелательным побочным
реакциям. Еще одно осложнение связано с нестабильностью
ацетилена, в газовой фазе под давлением он может взрываться.
Со2(СО)8 / \
НС = СН ♦ 2СО ♦ 2Н?О НОоС СО2Н (12.32)
14О°С7 МОатм
Сложное, катализируемое металлом карбонилирование
ацетилена при более мягких условиях приводит к образованию
гидрохинона. Эта реакция [уравнение (12.33)], катализируемая
карбонилом рутения и солями родия, представляет
промышленный интерес. Механизм этой многостадийной реакции неясен.
Образование возможного интермедиата [52] иллюстрируется
стехиометрической реакцией, показанной в уравнении (12.34).
2НСнСН ♦ 2СО ♦ Н2 — [О] (2 33)
(fFe(C0)4 ♦ HC5CH [I -Н— Fe(C0)3 (I2.34)
12.6. Карбонилирование спиртов: процесс получения
уксусной кислоты фирмы Monsanto
Разработанный сотрудниками фирмы Monsanto процесс кар-
бонилирования метанола с образованием уксусной кислоты
[уравнение (12.35)] является из ряда вон выходящим,
технологически наиболее удачным примером гомогенного катализа [53,
54]. Эта чрезвычайно селективная реакция, идущая с высоким
выходом, кроме того, является очень быстрой, скорости ее
близки к скоростям ферментативных процессов. Более миллиона
тонн уксусной кислоты получают ежегодно с помощью этого
процесса. С экономической точки зрения технология
превосходит все другие способы производства уксусной кислоты.
Эффективный, идущий при низком давлении родий-иодидный процесс
аналогичен более раннему кобальт-иодидному процессу при вы-
Каталитические реакции с участием монооксида углерода 119
соких давлениях, разработанному фирмой BASF [4, 55], для
которого необходимы жесткие условия 210 °С и 700 атм,
обеспечивающие приемлемые скорости.
катализатор Rh, I~
СН3ОН + СО >- СН3СО2Н (12 35)
180 °С, 30—40 атм v ;
99%
Многое известно о механизме процесса фирмы Monsanto;
промежуточные реакционные стадии были исследованы
методами ИК- и ЯМР-спектроскопии с использованием хорошо
изученного предкатализатора Ph4As+[Rh(CO)2l2]~+MeI. Основные
черты предложенного механизма показаны на рис. 12.3.
О ^ Mel
[Rh(CO)2l2]~
[MeRh(CO)2l3]~
МеОН
9
МеС-1
?ть -
♦ HI
♦ н2о —
L J L-
Рис.
-*- Mel ♦ НоО
О
—*- МеС-ОН ♦ HI
|'[со]-[сн3он]-
12.3.
В качестве «предкатализатора» может быть введен
практически любой источник родия или иодида. В реакционных
условиях они превращаются в. [Rh(CO)2l2]~ и Mel. Определяющей
скорость стадией является окислительное присоединение мети-
лиодида к анионному комплексу родия (I) (рис. 12.3). Исходя из
обсуждения окислительного присоединения в ч. 1, разд. 5.5,6,
понятно, почему скорость этой реакции намного выше в случае
иодида, нежели бромида (который приводит к в 10 раз более
медленной реакции), и почему вспомогательные иодидные ли-
ганды у родия ускоряют процесс. Следует ожидать, что
кобальтовые катализаторы на этой стадии будут намного более
слабыми. Общее выражение для скорости показано на рис. 12.3, оно
согласуется с тем, что скорость определяется этой стадией
(заметим, что активной формой иодида является метилиодид).
Последующие стадии в каталитическом цикле представляют
собой миграционное внедрение и восстановительное элиминиро-
120 Глчва 12
вание. Эти знакомые реакции (ч. 1, разд. 6.1 и 5.6
соответственно) поотекают быстро и поэтому не являются лимитирующими
стадиями в каталитичеком процессе. Стадия восстановительного
элиминирования с образованием ацетилиодида наблюдалась
в модельной реакции. Однако в промышленных условиях может
также идти прямой гидролиз ацильного комплекса родия. В
зависимости от реакционных условий в реакции ацетилиодида
(или ацильного комплекса родия) с метанолом или с уксусной
кислотой могут образовываться метилацетат или ацетангид-
рид [56].
Механизм, показанный на рис. 12.3, зависит от
индивидуальных химических свойств иодида, который одновременно
является хорошим нуклеофилом, очень слабым протонным основанием,
хорошей уходящей группой и эффективным лигандом для родия.
Никакой другой элемент или группа, по-видимому, не способны
осуществлять все эти функции столь эффективно. Однако
коррозионная природа иодида представляет собой главную
техническую проблему в процессе фирмы Monsanto. Вместо иодида
можно использовать пентахлоротиофеноксид СбС158~~, но
скорость очень сильно уменьшается [57]. Отметим, что метил
является особой алкильной группой, так как претерпевает
наиболее быстрое окислительное присоединение (стадия,
лимитирующая процесс), а образующийся алкил не способен к ^-гидридно-
му элиминированию.
Кобальт менее реакционноспособен, чем родий, несомненно,
из-за более медленной стадии окислительного присоединения.
Более того, природа комплексов кобальта (I), по-видимому, иная,
чем комплексов родия (первые, вероятно, являются пятикоорди-
национными). Комплексы иридия активны, но менее селективны,
и механизм их действия более сложен, чем у аналогичных
родиевых систем. Форстер [58] показал, что существуют два главных
каталитических цикла для иридиевых систем в случае карбони-
лирования метанола, один из них включает нейтральные, а
другой— анионные иридиевые соединения. Конкурирующая
конверсия водяного газа также является проблемой при катализе
комплексами иридия.
Попытки иммобилизации катализатора образования уксусной
кислоты в процессе фирмы Monsanto оказались неудачными, что
не удивительно, если учесть изложенное выше. Во-первых, эта
реакция в достаточной степени экзотермична для возникновения
проблем, связанных с теплопереносом в газофазной
гетерогенной каталитической системе. Добавление таких лигандов, как
фосфины (которые будут необходимы для иммобилизации
катализатора), не только приведет к замедлению реакции, но и к
алкилированию некоординированных фосфинов метилиодидом.
Конечно, иодид-ионы не могут быть закреплены на носителе.
Каталитические реакции с участием монооксида углерода 121
Была предпринята изобретательная попытка иммобилизовать
катализатор типа Monsanto с помощью связанного с полимером
перхлоротиофенилата вместо иодида, но скорость и
стабильность катализатора оказались неудовлетворительными [59].
12.7. Родственные процессы карбонилирования,
промотируемые иодидом
Сообщалось о нескольких других каталитических реакциях
для производных спиртов с использованием иодидных
промоторов. Несмотря на то что большинство этих реакций не привело
к промышленным процессам, химия их поучительна и поэтому
заслуживает комментария.
Водный формальдегид, существующий в виде геминального
диола, может претерпевать карбонилирование в присутствии
соли родия и иодидного промотора [60] [уравнение (12.36)].
Главным нежелательным побочным продуктом является
уксусная кислота. Возможный механизм этой реакции аналогичен
механизму процесса фирмы Monsanto. Предполагаемый интерме-
диат НОСН21 карбонилируется в процессе, аналогичном карбо-
нилированию метилиодида. Уксусная кислота образуется при
последующем карбонилировании гликолевой кислоты в
малоновую, которая декарбоксилируется [уравнение (12.37)].
О
Rh(I) II
СН2(ОН)2+СО >■ НОСН2СОН+СН3СО2Н (12.36)
150°С, 1000 атм 86% 14%
HI Rh(I) -CO2
НОСН2СО2Н —■>- [1СН2СО2Н] —■>- НО2ССН2СО2Н —*■ СН3СО2Н
Го О2-37)
Если будет обнаружен достаточно сильный и селективный
катализатор, другая промотируемая иодидом реакция сможет
иметь промышленную значимость. По этой реакции два
недорогих исходных продукта, этиленоксид и этилен, превращаются
в более ценный продукт — тетрагидрофуран [60], однако ни
степень превращения, ни стабильность катализатора не пригодны
для внедрения в производство. По-видимому, этиленоксид
превращается в 2-иодоэтанол, который алкилируется в результате
внедрения этилена. Предполагают, что предпоследним продуктом
является 4-иодобутанол, как показано в уравнении (12.38).
(12.38)
Ru(CO)*L? С2Н4 г х п ^
н|32» [ho(ch2)2i] » [ho(ch2)4i]
L = Ph3P
122 Глава 12
Метанол может быть гомологизирован в более высокие
спирты с умеренной селективностью (40%) по отношению к этанолу.
Эта реакция катализируется Сог,(СО)8 в присутствии иодидного
промотора при давлении СО и Н2, равном 300 атм, и 160—
180 °С [1]. Ключом к этому процессу является расщепление
связей С—О эквивалентом HI. Важным фактором, по-видимому,
является сильно кислотный характер НСо(СО)4. Превращение
диметилового эфира в метилацетат, катализируемое солями
рутения в присутствии иодид-иона, представляет собой
родственный с точки зрения механизма процесс.
Последним промотируемым иодидом процессом,
осуществленным на практике компанией Tennessee Eastman в 1983 г.,
является карбонилирование метил ацетата в ацетангидрид [63а].
На начальной стадии из метилацетата и Lil или HI образуется
метилиодид [уравнение (12.39)].
О
СНзОАс
HI
СН31
+
НОАс
Rh(I)
Rh-СНз
СО
RhCCH3
(12.39)
Окислительное присоединение метилиодида к Rh (I) с
последующим карбонилированием дает ацильный комплекс родия.
Элиминирование Acl в присутствии LiOAc приводит к
образованию ацетангидрида [уравнение (12.40)]. В присутствии
водорода ацильный комплекс родия дает ацетальдегид, который,
реагируя с эквивалентом ангидрида, образует этил,и денди ацетат
[уравнение (12.41)] [636].
Ас
(12.40)
RhCU
хОАс
СН3ОАс ♦ СО + Н2
сн3сн
(12.41)
ОАс
пиколии ►
150° С, 140а/77Ж
В результате элиминирования уксусной кислоты из этилиден-
диацетата может быть получен винилацетат [уравнение (12.42)].
СН3СН(ОАс)2 —+ СН2==СН(ОАс)+НОАс (12.42)
12.8. Декарбонилирование
В принципе различные стадии циклов карбонилирования,
например процесса гидроформилирования, являются обратимыми.
В соответствии с этим альдегиды, хлороангидриды и другие ор-
Каталитические реакции с участием монооксида углерода 123
ганические карбонилсодержащие производные должны
претерпевать декарбонилирование, катализируемое переходными
металлами. На практике трудно добиться эффективного
каталитического декарбонилирования. Основной проблемой, связанной
с каталитическим декарбонилированием, является то, что
участвующий .в нем алкильный интермедиат может вступать в
реакцию ^-гидридного элиминирования, приводящую к олефиновым
и изомеризованным продуктам. Эти реакции были рассмотрены
в обзоре [64] и более детально будут обсуждены в разд. 15.3.
Стехиометрическое декарбонилирование альдегидов
«катализатором Уилкинсона» RhCli(PPh3)3 [уравнение (12.43)] протекает
в мягких условиях [65]. Образующийся карбонил родия (I) 12
прочно удерживает СО. Однако при достаточно высоких
температурах может происходить выброс СО, и в этих условиях само
соединение 12 действует как катализатор декарбонилирования
ароматических альдегидов [уравнение (12.44)]. Для таких
субстратов (5-гидридное элиминирование не является проблемой.
CH2CI2
RCHO+RhCl(PPh3)3 * RH+RhCl(CO)(PPh3)2+PPh3 (12.43)
25 °С v '
12
RhCl(CO) (PPh3)2
ArCHO *■ CO + ArH (12.44)
180°C ч
Подобное каталитическое декарбонилирование альдегида
можно проводить при более низких тем'пературах, используя
комплекс родия (I) с хелатообразующим фосфиновым лигандом
DIPHOS [66, 67]. В этом случае граяс-фосфин делает СО в его
промежуточно образующемся комплексе более лабильным.
Однако это не решает проблемы ^-гидрида. Кроме того,
гетерогенный катализатор — палладий на углероде — более подходит для
каталитического декарбонилирования ароматических
альдегидов [68].
Механизм этих реакций является микроскопически
обратимым соответствующему карбонилированию [69]. При декарбони-
лировании альдегидов лимитирующей скорость стадией может
быть либо окислительное присоединение альдегида к
низковалентному металлическому центру (ч. 1, гл. 5), либо (особенно
для каталитических реакций) потеря СО комплексом металла
в конце одного цикла. Стадия окислительного присоединения
протекает намного быстрее в случае галогеноангидридов, чем
альдегидов, но если используют алифатический хлорангидрид,
содержащий ^-водородные атомы, перегруппировка алкильного
производного металла, являющегося интермедиатом, остается
проблемой. Рассмотрим в качестве примера реакцию, описывае-
124 Глава 12
мую уравнением (12.45) [70]. Происходит изомеризация даже
хлороангидрида, что иллюстрирует обратимость всех стадий.
Pd(acac)2
СН3(СН2)14СОС1 >■ С
—со
+ + +
изомер изомер изомер
(12.45)
Исключительно эффективное декарбонилировани.е и
ароматических и алифатических альдегидов возможно при
использовании © качестве катализаторов порфириновых комплексов
рутения (II) [уравнение (12.46)] [71]. В течение 30 мин наблюдали
до 104 превращений. Реагируют как ароматические, так и
алифатические альдегиды, однако алифатические дают побочные
продукты. Необходимость присутствия кислорода позволяет
предположить радикально-цепной механизм.
Ru(TPP) (PPh3)2
СН3СН,СНСНО ^СН3СН2СН2СН2СН3 + СО
Р(и-Ви)3, СН2С12, MeCN, ток Аг, 50 °С
сн2
(12.46)
12.9. Восстановительное карбонилирование ароматических
нитросоединений
Ароматические нитросоединения реагируют с монооксидом
углерода с образованием СО2 и изоцианатов или в присутствии
спирта — уретанов [уравнение (12.47)]. Эти реакции катализи-
/ -ArNCO+2COQ (I2.47)
соли Ра / г-
ArNOo + ЗСО Ъ1Г<
г или Rh \^oy_ArNcOR t2COv
руются несколькими переходными металлами, но особенно
солями палладия и родия. Хотя такое катализируемое переходными
металлами восстановительное карбонилирование еще не
внедрено в промышленность, подобная каталитическая химия
вызывает значительный интерес; диизоцианаты, например толуиленди-
изоцианат (ТДИ) и дифенилметандиизоцианат (МДИ),являются
важными компонентами вспененных полиуретанов и
эластомеров [1]. Кроме того, используемая в настоящее время
технология включает фосген (С1СОС1), высокотоксичное летучее
вещество. Однако проведение реакции типа (12.47) в случае двух
Каталитические реакции с участием монооксида углерода 125
нитрогрупп, находящихся в одном кольце (как было бы нужно
для получения ТДИ), является сложным.
0СНи)-снз ocN—COh-CHo—(О)— nco
TDI MDI
Детальный механизм, в соответствии с которым происходит
восстановительное карбонилироеание, катализируемое
переходным металлом, неясен, но некоторые аспекты очевидны. В ходе
каталитической реакции, несомненно, реализуется окислительно-
восстановительный цикл с участием металла {например, Pd (II)
и Pd(0)]. Существуют две приемлемые возможности
образования изоцианата. Либо координированный СО атакуется
ароматическим амином [который образуется в предшествующем
катализируемом металлом восстановлении нитросоединения, как
в уравнении (12.48), либо происходит внедрение СО в нитрено-
вый комплекс [уравнение (12.49)].
Pd2+—CO+ArNH2 —* Pd°4-ArNCO+2H+ (12.48)
Pd = NAr+CO —* Pd(0)+ArNCO (12.49)
Путь, показанный уравнением (12.48), включает нуклеофиль-
ную атаку на карбонильный лиганд (тип реакции,
рассмотренной в ч. 1, разд. 7.1). Имеются также прецеденты для
альтернативного пути, описываемого уравнением (12.49). Нитреновые
комплексы, образующиеся при катализируемом металлом
разложении арилазидов, реагируют с СО с образованием изоциана-
тов [72]. Подобные нитреновые комплексы могут получаться
в реакции между восстановленным металлом и нитроареном
и действительно могут быть интермедиатами в катализируемом
металлом восстановлении нитробензола в анилин [73], которое
требуется в соответствии с уравнением (12.48). В отсутствие
детальных исследований, однако, эти альтернативные пути
являются только предположениями.
12.10. Гомогенно катализируемое гидрирование СО
Растворимые карбонильные соединения металлов (особенно
соединения родия, рутения и кобальта) катализируют
превращения синтез-газа (СО/Н2) в большое число разнообразных
кислородсодержащих продуктов [уравнение (12.50)] [74]. Для
этого необходимы высокие давления и температуры.
Первичными продуктами реакции являются метанол, метилформиат
и гликолевый альдегид; предполагают, что они образуются
параллельными конкурирующими путями. Гликолевый альдегид.
126 Глава 12
по-видимому, является предшественником этиленгликоля,
который представляет собой наиболее привлекательную с точки
зрения производства цель. Селективность в образовании различных
продуктов зависит от природы катализатора (а также добавок)
и реакционных условий. Рутениевые и родиевые катализаторы
дают намного более высокие скорости, чем кобальтовые,
однако даже самые быстрые системы настолько медленны, настолько
неселективны и настолько связаны с жесткими условиями
(высокой температурой и давлением), что не являются
привлекательными. В частности, несмотря на интенсивные исследования,
до сих пор не было описано такой каталитической системы,
которая была бы достаточно эффективной и селективной, чтобы
м
со * нг ч - s. нсн или м(снго)
Ие или МН
о
н-с-осн3
м=растворьипый нарБокильньсй комплекс
Со, Rh или Rut
Рис. 12.4.
сделать производство этиленгликоля пригодным для
промышленного процесса.
О О
М(СО)П || ||
ш+н2 >• сн3он+сн3осн+нссн2он+носн2сн2он
М = Со, Rh, Ru
(12.50)
Коста [74] предложил единый механизм, объясняющий
гомогенно катализируемое образование кислородсодержащих
продуктов из СО/Н2 (рис. 12.4). Первую стадию общего
каталитического цикла — образование формальдегида или формальдегид-
Каталитические реакции с участием монооксида углерода 127
ного комплекса — обычно рассматривают как определяющую
скорость. В действительности образование свободного
формальдегида из СО и Н2 термодинамически затруднено (изменение
свободной энергии составляет +6,5 ккал/моль при 25 °С
и +12,1 ккал/моль при 227 °С) [75]. Даже в случае
эффективных катализаторов, по-видимому, не достигается самая малая
равновесная концентрация формальдегида. Таким образом, все
процессы гидрирования монооксида углерода должны
«протискиваться» через это узкое кинетическое и термодинамическое
«окно».
Вслед за этой определяющей скорость стадией реакция
разветвляется: образуется любой из двух таутомерных комплексов
металла МСН2ОН или МОСН3. Металлометоксидный интерме-
диат приводит к образованию метилформиата. Как показано
на рис. 12.4, гидроксиметильный интермедиат может
превращаться либо в метанол (в результате восстановления), либо
в гликолевый альдегид НОСН2СНО в результате двухстадийно-
го процесса (внедрение СО с последующим восстановлением).
На второе направление после образования МСН2ОН могут
влиять многие факторы: растворитель, присутствие или
отсутствие различных промоторов и отношение парциальных
давлений СО и Н2. Эти факторы влияют на селективность
образования конечного продукта — метанола либо этиленгликоля.
После лимитирующей скорость стадии каждая стадия на
рис. 12.4 имеет аналог среди стехиометрических реакций,
наблюдавшихся в случае хорошо изученных комплексов. Эти
модельные реакции рассматривались в обзоре [76]. Некоторые из этих
характерных типов реакций обсуждались в предшествующих
главах, однако ряд важных аспектов такого Химизма следует
подчеркнуть в контексте схемы, показанной на рис. 12.4.
Лимитирующее скорость процесса образование
формальдегида обычно включает предварительное образование формильного
комплекса. Реакция внедрения карбонила по связи М—Н
[уравнение (12.51)], как правило, термодинамически невыгодна [за
исключением двух особых случаев Rh(OEP) [77а], и органоак-
тинида [776] (ч. 1, разд. 6.1, ж)]. Однако пример с октаэтил-
порфирином родия не является простой внутримолекулярной
реакцией. Формильные комплексы были также получены по
реакции между сильно восстанавливающими гидридами и электро-
фильными карбонильными лигандами [76].
н о
I I!
м—с=о *=* м—с—н (12.51)
На большую часть процессов межмолекулярного
восстановления координированного СО с образованием формильных:
128 Глава 12
комплексов влияют только гидриды электроположительных
металлов, таких, как цирконий, титан и алюминий. Столь сильные
восстановительные агенты не могут быть регенерированы
водородом. Поэтому такие системы — плохие модели
каталитического гидрирования СО гетерогенными или гомогенными
катализаторами. В связи с этим показанная в уравнении (12.52) реакция
между гидридным комплексом рутения и электрофильным
комплексом СО весьма интересна, так как теоретически гидридный
комплекс рутения мог бы регенерироваться водородом [78а].
Отметим, однако, что сообщалось о запутанной стехиометрии
этой реакции. Подобная межмолекулярная реакция является
привлекательной альтернативой простому
внутримолекулярному «внедрению» гидрида. Была продемонстрирована [786]
другая схема восстановления СО, включающая нуклеофильную
атаку на карбонил с образованием карбенового комплекса,
который впоследствии восстанавливается.
+ —со
3HRu(CO)4-+2CpRe(CO)2NO —>• HRu3(CO)n-+2CpRe(CO) (NO) (CHO)
(12.52)
Предполагают, что в ходе каталитического гидрирования СО
образуются очень малые, недетектируемые количества формиль-
ных комплексов, превращающиеся в результате последующей
реакции в формальдегид. Хорошо известным примером
моноядерного формальдегидного комплекса является т]2-осмиевое
соединение 13, которое не образуется при последующих переносах
гидрида к СО, а, более вероятно, получается в результате
прямой реакции формальдегида с нуль-валентным комплексом
осмия. Как показано в уравнении (12.53), этот формальдегидный
комплекс самопроизвольно превращается в формилгидридный
кохмплекс [79]. Эта реакция представляет собой
микроскопически обратимое образование формальдегида.
сн2о
Os(CO)2(PPh3)3 > Os(ri2-CH2O)(CO)2(PPh3h —+•
13
—>• Os(CHO)H(CO)2(PPh3)2 (12.53)
Стехиометрическое последовательное восстановление элек-
тронодефицитного координированного СО в гидроксиметильный
комплекс было проиллюстрировано Грэмом [80]. Эта
последовательность реакций [уравнение (12.54) ] является моделью
первых двух стадий предполагаемого каталитического процесса
(рис. 12.4).
NaBH4 NaBH4
CpRe(CO)2(NO)+ ►■ CpRe(CHO) (CO) (NO) >■
0 °C ТГФ/Н2О ТГФ/Н2О
CpRe(CH2OH)(CO)(NO) (12.54)
Каталитические реакции с участие^! мрнооксида углерода 129
В поисках стехиометрических примеров оставшихся стадий
превращения гидроксиметильного комплекса в гликолевый
альдегид, а затем в этиленгликоль следует обратиться к другим
комплексам. Показательным является превращение сс-ацилокси-
метильного комплекса марганца [уравнение \( 12.55)] [81а].
Сообщалось также о карбонилировании родственного а-гидрок-
сиалкильного комплекса [816].
ООО О
II н2 И || II
(OC)5MnCH2OCR —->- HCCH2OCR —->- HOCH2CH2OCR (12.55)
Некоторые стадии процесса, показанного на рис. 12.4,
по-видимому, облегчаются при образовании комплекса с кислотами
Льюиса. Известно, что эти кислоты промотируют и стадию
миграционного внедрения, и стадию восстановления [76] за счет
образования комплекса с кислородом ацила[82]. Роль кислот
Льюиса в промотировании образования ацильного комплекса
обсуждалась в ч. 1, разд. 6.1, ж.
Рассмотрев унифицированный механизм Коста (рис. 12.4)
и модельные для индивидуальных стадий реакции, возвратимся
к специфическим примерам гомогенно катализируемых реакций
между монооксидом углерода и водородом и
экспериментальным факторам, влияющим на селективность образования
продуктов и скорость. Самый слабый катализатор НСо(СО)4
требует жестких условий (1970 атм СО/Н2, 230 °С) для достижения
лишь умеренных скоростей, но анализ продуктов
свидетельствует в пользу общего механизма Коста, показанного на рис. 12.4.
Например, выражение для скорости [83, 84] имеет первый
порядок как по СО, так и по Н2, что соответствует лимитирующей
стадии образования формальдегида через металлоформил
[уравнение (12.56)]. Образованию этиленгликоля способствуют
высокие давления, высокие отношения Н2/СО и более низкие
температуры [85, 86]. Влияние реакционных переменных
свидетельствует об образовании метанола и этиленгликоля из
единственного интермедиата, скорее всего производного кобальта
НОСН2—Со, как показано на рис. 12.4. Этот кобальтовый
катализатор, вероятно, никогда не представит промышленного
интереса, так как хорошая избирательность образования
этиленгликоля требует более низких температур, но скорости
слишком малы даже при более высоких температурах.
НСо(СО)4 *=* (ОС)зСоСНО — >• (ОС)3Со(Н)(НСНО)
лимитирующая стадия
(12.56)
Обширные исследования, проведенные фирмой Union
Carbide, были направлены на реализацию процесса превращения син-
9—1129
130 Глава 12
тез-газа в этиленгликоль, катализируемого родием [87].
Несмотря на то что родиевые катализаторы по меньшей мере
в 10 раз сильнее, чем кобальтовые, скорости, достигаемые при
использовании родиевых комплексов, все-таки слишком малы,
чтобы быть пригодными для промышленных целей. Частота
превращений составляет лишь 0,1 с"1 на родии даже в жестких
условиях (280 °С, 1020 атм); кроме того, проблемой является
регенерация родия. При более высоких температурах
катализатор разрушается. Тем не менее эта система может давать
высокую селективность образования этиленгликоля в качестве
основного продукта; в наиболее благоприятных случаях можно
получить весовое отношение этиленгликоль/метанол, равное 90%.
Типичные условия для таких катализируемых родием
реакций приведены в уравнении (12.57). Роль промоторов, в частно-
О
Rh(CO)2 acac, ||
со+н2 —— >■ носн2сн2он+сн3он+сн3осн
' RCO2CS, тетраглим, 2-гидроксипи- ' '
1:1 ридин, 220—280 °С, 545—1020 атм
(12.57)
сти цеЗ'ИЙкарбоксилатов, может быть связана с образованием
и стабилизацией анионных кластеров карбонила родия,
например [Rh5(CO)i5]~, которые, как было показано, являются
основными соединениями родия, присутствующими в типичных
реакционных условиях. Эти кластеры были обнаружены и
идентифицированы методом инфракрасной спектроскопии с
использованием специальных инфракрасных кювет для высоких давлений
и температур [88]. Обычно предполагалось, что один или более
анионных родиевых кластеров ответственны за образование
этиленгликоля; -в условиях же, благоприятствующих образованию
метанола, основным соединением, присутствующим в системе,
является мономерный карбонил Rh(CO)4~. Это некое косвенное
доказательство, которое использовали в поддержку утверждения,
что истинным катализатором, участвующим в образовании
этиленгликоля, является кластер. Если это так, то это один из
весьма редких примеров, когда кластер переходного металла
фактически представляет собой гомогенный катализатор. Вероятные
аргументы в пользу кластерного катализатора касаются
возможностей образования карбонилов и гидридов металла на
различных центрах, возможностей биядерного восстановления СО и
стабилизации формила и других интермедиатов в результате
образования мостиков с соседними металлами. В то же время нет
оснований полагать, что гомогенный катализатор, приводящий
к образованию этиленгликоля, должен всегда быть кластером,
упоминавшийся выше кобальтовый катализатор является
мономером.
^__ Каталитические реакции с участием монооксида углерода 131
Фосфиноксиды вместе с солями цезия представляют собой
полезные промоторы, способствующие образованию этиленгли-
коля [89]. (Напомним, что в ч. 1, разд. 6.1, е было показано, что
R3PO может катализировать реакции внедрения СО.) Большое
количество кластеров карбонилов родия с заключенными в них
основными группами атомов, такие, как [Rh9(CO)2i]2~, было
получено и исследовано в качестве катализаторов синтеза эти-
ленгликоля [90], но не было найдено ни одного, существенно
превосходящего катализатор, образующийся in situ из
Rh(CO)2(acac).
Карбонил рутения Ru(CO)5 является селективным
катализатором для почти исключительного образования метанола и эти-
ленгликоля, как было показано методом ИК-спектроскопии при
высоких давлениях и кинетическими исследованиями (скорость
имеет первый порядок по рутению). Однако реакции с такими
непромотированными рутениевыми катализаторами требуют
высоких температур и давлений (230 °С и 2000 атм) и протекают
медленнее, чем катализируемые родием реакции [86, 91, 93].
Обнаружен ряд промоторов, включая полярные кислотные
растворители, сильно повышающих селективность рутениевых
катализаторов в образовании этиленгликоля. На основании
скоростей, селективности образования продуктов и результатов
инфракрасных исследований анионные кластеры карбонилов
рутения были привлечены в качестве действенных катализаторов
образования гликоля. В интересном примере, сообщенном Книфто-
ном [94], в качестве растворителя при катализируемом
рутением образовании этиленгликоля и его производных использовали
расплав соли — галогенида четвертичного фосфония [уравнение
(12.58)]; показано присутствие анионного кластера,
по-видимому, являющегося активным катализатором.
[HRu3(CO)u]-
СО+Н2 >- НОСН2СН2ОН+НОСН2СН2ОМе+МеОН
RP+C1-
(12.58)
Было также обнаружено, что уксусная кислота и ацетат
цезия улучшают селективность по отношению к гликолю и
повышают скорость катализируемой рутением реакции [95]. Иодид-
ион также способствует образованию гликоля и ускоряет
каталитическую реакцию [96]. Вновь предполагают, что важными
катализаторами служат анионные кластеры. Например, в
присутствии иодида предшественник катализатора Ru3i(CO)i2
превращается в смесь анионных кластеров [уравнение (12.59)],
которые сохраняются в условиях каталитической реакции.
7Ru3(CO)I2+9I-+3H2 _». 6[HRu3(CO)II]-+3[Ru(CO)3l3]-+9CO
(12.59)
132 Глава 12
Точная роль этих анионных кластеров в каталитической
реакции не очевидна, но существует ряд интригующих намеков.
Анионы гидридорутенийкарбонила (особенно кластеры) имеют
выраженный гидридный характер — являются активными
восстановительными агентами. Возможно, формильный интермеди-
ат образуется в результате бимолекулярной реакции между гид-
ридным комплексом рутения и отличным от него электрофиль-
\ным карбонильным комплексом рутения. Важными для контроля
реакционной способности гидрида являются эффекты
образования ионных пар [956]. Предполагают, что в процессе участвуют
два разных комплекса рутения. Это позволяет предположить
возможность получения более активного катализатора при
использовании двух различных металлов: одного для активации
СО по отношению к гидридной атаке, а второго — для
активации Н2 с целью образования очень реакционноспособного
гидрида. Такой эффект наблюдали в действительности. В патенте
фирмы ICI [97] сообщается о рутений-родиевом катализаторе,
более селективном, чем каждый из металлов в отдельности.
Умеренные селективности по отношению к этиленгликолю
обнаружены в реакциях, промотированных солями уксусной кислоты
при давлениях меньше 1500 атм и 240 °С [976].
12.11. Восстановительное карбонилирование аммиака
N-Метилформамиды могут быть получены непосредственно по
реакции между синтез-газом и аммиаком. Этот тип реакции
гомогенно катализируется рядом переходных металлов, однако
самыми лучшими катализаторами являются комплексы
рутения [98]. Типичный пример показан в уравнении (12.60).
Эффективны простые предшественники рутениевых катализаторов
H2+CO+NH3 —> (CH3)2NCHO+CH3NHCHO+H2NCHO
(12.60)
R11O2 или Ru3(CO)i2; добавки типа иодидов, кислот Льюиса или
фосфинов не могут быть эффективными промоторами процессов
такого типа. Другие металлы VIII группы (иридий, родий и
осмий) проявляют ограниченную каталитическую активность,
но она ниже, чем у рутениевых катализаторов. Комбинации
биметаллических катализаторов не являются эффективными.
Скорость этого процесса ниже даже скорости образования спирта
из синтез-газа. Эти низкие скорости ограничивают их
промышленный потенциал. Формальдегид чрезвычайно реакционноспо-
собен в этих условиях, давая те же самые продукты. Поэтому
представляется возможным, что в этом процессе интермедиатсм
Каталитические реакции с участием монооксида углерода 133
является формальдегид и что низкие скорости обусловлены
необходимостью прохождения через узкое термодинамическое
и кинетическое «формальдегидное окно», а также тем, что
аммиак представляет собой умеренный ингибитор активного
катализатора. Возможный механизм этого процесса показан на
рис. 12.5. Образующийся вначале формальдегид (возможно,
в виде комплекса металла) восстановительно аминируется в
метиламин, который затем карбонилируется. Другой
возможностью является образование формамидных интермедиатов и
последующее их гидрирование с образованием метиламинов.
Имеются некоторые примеры для таких реакций [99, 786].
лимитирующая стадия
СО+ Н2 *■ Н2СО
H2CO+NH3 -^ CH3NH2+H2O
CH3NH2+CO *=* (CH3)HNCHO
н2
CH3NH2+H2CO —+ (CH3)2NH+H2O
(CH3)2NH+CO *=± (CH3)2NCHO
Рис. 12.5.
Было показано, что метанол не может быть реальным интер-
медиатом в синтезах ДМФА и N-МФА из CO/H2/NH3, но
возможно его участие в параллельной реакции. Это было
продемонстрировано анализом продуктов в присутствии различных
количеств метанола. Однако метанол может включаться в
реакционные продукты при определенных условиях.
12.12. Окислительное сочетание монооксида углерода
Альтернативным по отношению к восстановительному
процессу прямого синтеза этиленгликоля из СО и Нг является
окислительное сочетание СО. Этот сложный многостадийный
процесс приводит к образованию эфиров щавелевой кислоты наряду
с диалкилкарбонатными побочными продуктами. Оксалаты
затем гидрируются в этиленгликоль с помощью гетерогенных
катализаторов.
Первая стадия окислительного сочетания гомогенно
катализируется смесью комплекса палладия и солями меди или железа
[уравнение (12.61)] [100]. В процессе образования оксалата
палладий (II) восстанавливается в палладий(0). Роль солей меди
и железа заключается в катализе обратного окисления Pd(0)
в Pd (II) с использованием в качестве окислителя кислорода; эта
134 Глава 12
схема напоминает катализируемый палладием Вакер-процесс
превращения этилена в ацетальдегид (ч. 1, разд. 7.4, а).
00
2CO+2ROH+1/2O2 ^^Рес^ ROCCOR+H2O (12.61)
Предполагают, что ключевая стадия в предшествующей
окислительной димеризации СО включает восстановительное
элиминирование бис (карбометокси) палладиевого (II) комплекса 14,
как показано в уравнении (12.62) [101]. Подобные реакции
исследовались фирмами Union Carbide Corporation и Ube
Industries, описавшими не содержавшую фосфинов систему,
катализатором окисления в которой служили оксиды азота [102].
О 0 0
со I! II ||
L2Pd(OAc)2 —> L2Pd(COMe)2 —> MeOC—COMe+PdL3CO
МеОН ПОАО\
L = PPh3 14 (IZ.bZ)
Подобное окислительное сочетание спиртов и этилена
предлагает потенциально привлекательный путь получения эфиров
акриловой и янтарной кислот [уравнение (12.64)] [103]. Эфиры
янтарной кислоты этим способом могут быть получены с
высокой селективностью (90%).
PdCl2
C2H4+CO+O2+ROH >■ H2C==CHCO2R+ROCH2CH2CO2R+
СО или FeCl3
+RO2CCH2CH2CO2R (12.63)
12.13. Гетерогенно катализируемые реакции водорода
с монооксидом углерода
а. Реакция Фишера — Тропша. Восстановительная
полимеризация СО в присутствии Н2 с образованием линейных
углеводородов, олефинов и спиртов [уравнение (12.64)]
представляет собой давно известный процесс, который в недавнее время
привлек очень большой интерес из-за возможности
использования угля в качестве источника получения топлива и жидкого
химического сырья. История процесса Фишера — Тропша (ФТ)
прослеживается вплоть до патентов Германии 1913—1914 гг.,
в которых описывалось образование углеводородов, алкенов и
кислородсодержащих соединений на гетерогенных кобальтовых
и железных катализаторах. Франц Фишер и Ганс Тропш, в
честь которых был назван процесс, в 1923—1925 гг. разработа-
Каталитические реакции с участием монооксида углерода 135
ли реакцию, катализируемую железом и щелочью. В 1936 г.
в Германии начало работать первое промышленное
предприятие, впоследствии в военные годы использовавшее
катализируемый кобальтом процесс ФТ для получения значительной доли
жидкого углеводородного топлива. С 1957 г. в ЮАР
функционирует крупный завод ФТ (SASOL), использующий в качестве
сырья каменный уголь, а в качестве катализатора железо [104].
С экономической точки зрения и производство жидких
углеводородных топлив, и получение химических полупродуктов из
синтез-газа в настоящее время невыгодно (исключение
составляет этиленгликоль), поэтому функционирование заводов ФТ —
это в значительной степени политическое решение. Из-за
эмбарго на нефть в начале семидесятых годов и как следствие
этого повышения цен на нефть во всем мире начали широко
проводить исследования, сфокусированные на этом и
родственных процессах. С 1926 г. этому вопросу было посвящено более
5000 статей и 5000 патентов. Процесс Фишера — Тропша и
близкие к нему химические реакции были рассмотрены в
обзорах [105, 106].
катализатор
СО+Н2 >- СН3(СН2)пСН2ОН+СНз(СН2)пСН = СН2+
А, Р
+ СН3 (СН2) пСН3+Н2О (12.64)
Общими чертами процесса ФТ являются следующие. Эта
гетерогенно катализируемая экзотермическая реакция приводит
к образованию изобилия продуктов, включающих линейные
а-олефины, алканы, спирты, альдегиды, карбоксикислоты,
сложные эфиры и даже арены. а-Олефины и спирты являются
первичными продуктами, в то время как алканы получаются при
последующем гидрировании алкенов. Селективность
образования продуктов зависит от множества факторов, таких, как
отношение СО/Н2, давление, температура, катализатор и способ
его получения, а также наличие промоторов, например
щелочных металлов в носителе катализатора. Полимеризационный
процесс ФТ, как правило, приводит к образованию
статистического (Флори — Шульца) молекулярно-весового распределения,
указывающего на то, что рост цепи подчиняется полимериза-
ционной кинетике. Отсутствие селективности при получении
продуктов и статистический молекулярно-весовой интервал
являются главными недостатками реакции ФТ; кроме того,
большое количество содержащегося в угле углерода необходимо
впустую превратить в СО2, чтобы получить необходимый для
реакции ФТ водород (по реакции конверсии водяного газа).
Великолепными катализаторами ФТ являются железо и
кобальт; при использовании железа необходимы более высокие
136 Глава 12
давления, и в случае обоих катализаторов требуется
присутствие щелочных промоторов. Рутений приводит к первичному
образованию линейных углеводородов с высокими молекулярными
весами. Никель первично образует метан и поэтому не является
истинным катализатором ФТ. Платина проявляет
незначительную активность, а родий приводит к образованию
кислородсодержащих веществ с, более низким молекулярным весом.
а-Олефины, спирты, альдегиды, кетоны и предшественники
карбенов, например CH2N2, включаются в растущую цепь в
ходе реакции ФТ. Обрыв цепи происходит, очевидно, в
результате ^-элиминирования с образованием а-олефинов, но такой
обрыв цепи не приводит к дезактивации каталитического
центра, поскольку скорость роста цепи остается неизменной в
течение относительно длинных периодов времени (по крайней мере
недели).
б. Механизмы реакции Фишера — Тропша. Значительные
усилия и размышления были сфокусированы на механизме
(механизмах) реакции ФТ. Многие исследования были
посвящены модельным гомогенным реакциям, свидетельствующим в
пользу отдельных стадий этих предлагавшихся механизмов
[106].
Наблюдается конкуренция трех механизмов, они заметно
отличаются друг от друга по способности формирования С = С-
связей продукта. Первый был предложен Фишером и Тропшем
в 1926 г. [107], в нем предполагалось, что СН2 (карбеновые)
фрагменты образуются и полимеризуются на поверхности
катализатора. В современном варианте, который предложил Пет-
тит [108], предполагается, что вначале происходит внедрение
СН2 в поверхностный гидрид с образованием метильной
группы, а затем на стадии роста цепи снова внедрение СН2 уже в
эту метильную группу [показано в уравнении (12.65)].
(12 65)
и см-*
I /Ч2 /°\г /С\2 - 1 /д2/^2
\\\\\\\\\\<Л \\\\\\\\N
Предполагают, что карбеновые фрагменты образуются из
поверхностных карбидов, в свою очередь являющихся
результатом диссоциативной хемосорбции СО. Эти стадии показаны в
уравнении (12.66).
с о
СО И II Но СНо
11 " * ■ ^ ■ НгО (12.66)
В пользу этого механизма свидетельствует ряд убедительных
наблюдений. Были собраны доказательства образования по-
Каталитические реакции с участием монооксида углерода 137
верхностного карбида на катализаторах ФТ, для чего вводили
сначала 13СО, а затем смесь 12СО и Н2 [1С9]. Образующаяся
смесь продуктов включала 13СН4 и углеводороды, содержащие
несколько атомов 13С в одной и той же молекуле. Предиссо-
циированные меченные 13С карбиды претерпевают
гидрирование и полимеризацию на поверхности катализатора, как
показано в уравнении (12.65).
Петтит [108] представил дополнительные доказательства в
пользу этого механизма, использовав CH2N2 с катализатором
ФТ для генерации поверхностных групп СН2. В отсутствие
водорода эти связанные с поверхностью группы СН2 димеризуют-
ся, образуя этилен; в присутствии водорода образуются
продукты ФТ, причем характерные для конкретно используемого
катализатора ФТ. Увеличение давления Н2 приводит к снижению
содержания олефинов и уменьшению среднего молекулярного
веса алкановых продуктов. В другом эксперименте 12CH2N2 был
добавлен к потоку 13СО и Н2 в ходе протекающей реакции ФТ.
Распределение меченных изотопами молекул пропилена может
быть объяснено только показанным в уравнении (12.65)
механизмом роста цепи и определенно не может быть объяснено
альтернативными механизмами, обсуждаемыми далее.
Второй механизм процесса ФТ, предложенный Андерсом и
Эмметтом [ПО], предполагает, что С—С-связи образуются в
реакции конденсации гидроксиметиленовых групп на
поверхности катализатора, как показано в уравнении (12.67). Этот
механизм не имеет прецедентов в металлоорганической химии.
Некоторые алкоксикарбеновые комплексы известны, но реакция
их конденсации неизвестна. К тому же обсуждавшиеся ранее
при рассмотрении метиленового механизма линии доказательств
трудно согласовать с гипотезой Андерсона — Эмметта.
ее и Н-.с с-он и а с- с м с-он
Третье предположение Пихлера и Шульца [111] с
последующей модификацией [112] рассматривает образование
С—С-связи в результате хорошо известной металлоорганической
реакции: миграционного внедрения алкила в карбонил с
образованием ацилов (ч. 1, разд. 6.1). Эта схема показана в уравнении
(12.68). Необходимый метальный инициатор образуется при
гидрировании метилена, возникающего из карбидного
предшественника. Таким образом, эта схема принимает в расчет
некоторую диссоциативную хемосорбцию СО и даже включает
связанные с поверхностью метиленовые частицы, но только в
качестве инициаторов цепи.
138 Глава 12
С О Н2С —О СНр Н,С С (12.68)
И иг L 11 Н2 II И* И
VA \\\\ -н2о' W ~^ V\\\
Был предложен еще один механизм, включающий комплексы
формальдегида в качестве интермедиатов [113], но он получил
слабую поддержку [106]. В настоящее время, несмотря на то
что общего согласия по поводу доминирующего пути нет,
собрано множество доказательств в поддержку карбид-метилено-
вого механизма.
в. Модельные гомогенные реакции, которые, возможно,
близки к реакции Фишера — Тропша. Сообщалось о большом числе
реакций с участием растворимых комплексов переходных
металлов, возможно, схожих с различными стадиями процессов
ФТ. Обзор этих сообщений был выполнен Херрманном [106].
Здесь будет обсуждено лишь несколько ключевых примеров.
Мостиковые метиленовые (карбеновые) комплексы в
настоящее время хорошо известны (см. ч. 1, конец разд. 3.6, в).
Модельная реакция [114], сильно напоминающая стадии роста
цепи и образования продукта в первом из предложенных
механизмов ФТ [уравнение (12.65)], приведена в уравнении (12.69).
н2 г сн3
"з?/с\?нз _}*??
\ / L ^ J L CH2J СН2*СНСН3
V» I
Но
Уравнение (12.10) [115] иллюстрирует гомогенную модель
подразумевающейся димеризации поверхностного метилена с
образованием этилена. В этом примере сильно электрофильный
восстановительный агент А1Н3 превращает СО в этилен
предположительно через бисметиленовый комплекс 15.
А!Н, - ^СИо^ -, Hn 'H (12.70)
15
Роль поверхностных карбидов была^ исследована в связи с
растворимыми кластерными комплексами карбидов [106]. Такие
Каталитические реакции с участием монооксида углерода 139
карбиды были получены из металлокарбонильных соединений
в присутствии сильных кислот Льюиса [106, 116]. Было
показано, что эти карбиды образуют связи С—Н в присутствии
водорода или протонных кислот. В других случаях кластерные
карбиды присоединяют СО в метаноле, образуя карбометокси-про-
изводные; эта реакция [уравнение (12.71)] может служить
моделью образования эфиров в условиях ФТ [117].
2+
.»#!*
(12.71)
МеОН
-Н*
J-C-OMe
М= Fe(COb
Возможная роль формильных и гидроксикарбеновых
комплексов, образующихся в процессе ФТ в результате ступенчатого
гидрирования монооксида углерода, широко обсуждалась [106];
тем не менее доказательства в пользу диссоциативной хемо-
сорбции СО с образованием поверхностных карбидов
представляются подавляющими.
Путь образования кислородсодержащих продуктов ФТ,
таких, как спирты, альдегиды и сложные эфиры, остается
неясным. Возможно, происходит конкурирующее миграционное
внедрение растущей алкильной цепи с образованием ацильного
интермедиата. Альтернативным является вариант,
иллюстрируемый сходной модельной реакцией, как показано в уравнении
(12.72). Предполагается, что мостиковый кетеновый комплекс
16 представляет собой интермедиат [118].
сн2
(0C)4Fe Ve(C0)4
(0C)4Fe-
/н2-с
Fe(C0)4
CH3OD
(12.72)
DCH2C0CH3
16
г. Гомогенно катализируемые аналоги реакции Фишера —
Тропша. Реакция ФТ является гетерогенно катализируемой.
Гомогенно катализируемые реакции между СО и Н2
обсуждались ранее в этой главе. Заметим, что в случае гомогенных
реакций существуют очень веские доказательства образования в
качестве интермедиатов металлоформильного и металлоформ-
альдегидного комплексов, что заметно контрастирует с
приведенными выше доказательствами в пользу диссоциативной хе-
мосорбции СО в случае реакции ФТ. Возможно, последняя
реакция характерна только для гетерогенных катализаторов,
140 Глава 12
хотя в присутствии сильных кислот Льюиса и
восстанавливающих агентов растворимые металлокарбонильные соединения
могут претерпевать родственное восстановление в карбид [116].
Гомогенное каталитическое гидрирование СО редко
приводит к образованию связи С—С, наиболее важным исключением
является образование гликолевого альдегида и конечного
продукта его восстановлений — этиленгликоля. В последнем случае
образование связи С—С хорошо объясняется простым
миграционным внедрением алкила в карбонил.
Появилось несколько сообщений, утверждающих, что
растворимые катализаторы — производные переходных металлов —
приводят к восстановительной полимеризации СО с
образованием углеводородов. За исключением синтеза этиленгликоля и
гомологизации спиртов, описанных в других разделах этой
главы, ни один случай гомогенно катализируемой
восстановительной полимеризации СО не был подтвержден независимо.
Например, было показано, что два ранних сообщения о
гомогенной конверсии СО в углеводороды являются ошибочными
[119, 120].
Особенно интересная реакция этого типа была связана с
пришитым к полимерной цепи производным СрСо(СО)2.
Сообщалось, что при умеренных давлениях СО/Н2 и 190 °С
суспензия этого катализатора 17 в октане приводит к образованию
ряда линейных углеводородов [уравнение (12.73)] [121].
Специальный сшитый макросетчатый полимер требовался для
получения активного катализатора, поскольку предполагалось,
что активными могут быть только пространственно
изолированные металлические центры. Это один из немногих случаев,
когда утверждается, что связанный с полимером катализатор
демонстрирует химизм, отличный от наблюдаемого в случае
соответствующего растворимого комплекса металла. Однако
очень похожие связанные с полимером кобальтовые
катализаторы впоследствии были исследованы Стилле [122] и оказались
неактивными. Эта дилемма не разрешена до настоящего
времени.
н2/со
п=1-2О
Второй интересный случай возможного гомогенного
катализа ФТ связан с использованием сильного карбонил-кластерного
предкатализатора Ir4(CO)i2 и сильнокислого расплава соли
(2AlCl3/NaCl) в качестве реакционной среды. При 180 °С смесь
СО/Н2 с давлением 1—2 атм, как сообщалось, дает легкие
углеводороды [123, 124]. Эта реакция была исследована повтор-
Каталитические реакции с участием монооксида углерода 141
но с использованием кинетических измерений в потоке и
статическом реакторе, изотопных меток и добавок предполагаемых
янтермедиатов в ходе реакции; второе исследование не дало
результатов, совпадающих с первоначальными. Результаты
второго исследования [125] соответствуют каталитической
реакции, протекающей с образованием хлорометана в качестве
первичного интермедиата [уравнение (12.74)]. Были получены
результаты, поддерживающие утверждение, что активный
катализатор является гомогенным (было обнаружено, что
гетерогенные иридиевые катализаторы дают совершенно другие
результаты), однако не удалось определить, является ли активный
катализатор кластером. Такие же результаты дал мономерный
лредкатализатор 1г(СО)3С1. Активный компонент в этой
системе — расплав исключительно реакционноспособной кислоты
Льюиса. Очевидное расхождение между этими
опубликованными результатами остается неразрешенным. Родственный пред-
катализатор в кислоте Льюиса в качестве растворителя
[Os3(CO)i2 в ВВг3], как было показано, образует метан, этан,
метилбромид и этилбромид как главные продукты [126]. Этот
результат подобен показанному в уравнении (12.74).
1Г4(СО)12
СО+ЗН2 ^[СН2О] —>■ [CHdOH] —+
AlCl3—NaCl(2 : 1), 180 °С (1—2 атм)
—>• СН3С1 —>■ СН4+С2Н6+полимерные продукты (12.74)
Очевидная неспособность растворимых комплексов
приводить к восстановительной полимеризации монооксида углерода
(химизм ФТ) соответствует представлению о том, что
диссоциативная хемосорбция с образованием карбидов, а затем
связанных с поверхностью карбенов является существенной чертой
этого химизма. Обычно только гетерогенные металлические
катализаторы могут содействовать этому курсу. В то же время
гомогенное гидрирование СО, очевидно, протекает через
образование формальдегида в качестве интермедиата. Эти
фундаментальные проблемы заслуживают дальнейшего исследования,
особенно с учетом несопоставимых результатов, о которых
сообщалось.
д. Гетерогенно катализируемое гидрирование СО в метанол.
Как было показано, метанол представляет собой
привлекательный интермедиат для производства товарных химикатов, таких,
как уксусная кислота, и для получения бензина в процессе
фирмы Mobil Oil. По этой причине синтез метанола из синтез-
газа представляет большой интерес. Хотя гомогенные
катализаторы также эффективны в этой реакции, в качестве
промышленного катализатора выбран гетерогенный. В настоящее время
это хорошо развитая технология, однако ее рассмотрение
выходит за рамки данного изложения, поэтому мы лишь кратко
142 Глава 12
опишем реакционный процесс. Этот предмет рассматривается
в обзорах [127—129]. Стандартные промышленные
катализаторы — это Си—Zn—Сг2О3 или Си—Zn—А12О3. По-видимому^
медь связывает и активирует СО, а оксид цинка активирует Н2
за счет гетеролитического расщепления, описанного в ч. 1,
разд. 5.3, в и в разд. 10.3, а. Обычно считают, что активной
формой меди является Cu(I) [127—129, 130а], хотя были
опубликованы доказательства также в пользу Cu(0) [1306].
Несмотря на то что существуют очень небольшие проблемы,
связанные с экзотермичностью этой реакции и с ингибирующими
эффектами загрязнений серой, существующая парофазная
технология хорошо себя зарекомендовала.
Формильные группы можно наблюдать методом ИК-спектро-
скопии, когда СО и Н2 адсорбируются на Си—ZnO [131].
Очевидно, образующие метанол катализаторы действуют без
диссоциативной хемосорбции СО, характерной для гетерогенных
катализаторов Фишера — Тропша.
ЛИТЕРАТУРА И ПРИМЕЧАНИЯ
1. Par shall G., Homogeneous Catalysis, Wiley-Interscience, New York, 1980.
2. Weissermel K., Arpe H. /., Industrial Organic Chemistry, Verlag Chemie,
Berlin, 1978.
3. Organic Synthesis with Metal Carbonyls, Wender I., Pino P., (Eds.), Wiley,
New York, 1968, Vol. 1; 1977, Vol. 2 (есть русский перевод первого
издания книги: Органические синтезы через карбонилы металлов. Пер. с англ./
Под ред. Уэндера И., Пино П. — М.: Мир, 1973).
4. Falbe /. Carbon Monoxide in Organic Synthesis, Springer Verlag, New York,
1970 (есть русский перевод: Синтезы на основе окиси углерода. Пер. с
нем. — Л.: Химия, 1971); New Synthesis with Carbon Monoxide, Springer
Verlag, New York, 1980.
5. См. работу [1], с. 911.
6. Laine R. M., Rinker R. G., Ford P. C, J. Chem. Soc, 99, 252 (1977).
7. Cheng С H., Hendrikson D. E., Eisenberg R.y J. Am. Chem. Soc, 99 2791
(1977).
8. King R. В., Frazier C. C, Hanes R. M., King A. D., J. Am. Chem. Soc, 100,
2925 (1978).
9. a) Yoshida Т., Veda Y., Otsuka 5., J. Am. Chem. Soc, 100, 3941 (1978);
б) Активными катализаторами этого процесса являются также комплексы,
содержащие гидрид родия: Yoshida Т., Okano Т., Veda Y.f Otshuka S.r J.
Am. Chem. Soc, 103, 3411 (1981).
10. Grice N., Kao S. C, Pettit R., J. Am. Chem. Soc, 101, 1697 (1979).
11. Sweet J. R., Graham W. A. G., Organometallics, 1, 982 (1982).
12. Banborak J. C, Cann K, Organometallics, 1, 1726 (1982).
13. Pruett R. L., Adv. Organomet. Chem., 17, 1 (1979).
14. Tolman С A., Falter J. W., Homogeneous Catalysis with Metal Phosphine
Complexes, Pignolet L. H. (Ed.), Plenum, New York, 1983.
15. Heck R. F., Breslow D. S., J. Am. Chem. Soc, 83, 4023 (1961).
16. Slaugh L. H., Mullineaux R. /)., пат. США 3239569, 3239570 (1966).
17. Руденков А. И., Менненга Г. У., Рачковская Л. Н., Матвеев К И.,
Кожевников И. В. — Кинетика и катализ, 1977, 18, с. 915—920.
Каталитические реакции с участием монооксида углерода 143
18. Bianchl M., Piacenti F., Frediani P., Matteoli U., J. Organometal. Chem ,
137, 361 (1977).
19. Whyman #., J. Organometal. Chem., 68, C23 (1974); Whyman R., J.
Organometal. Chem., 81, 97 (1974).
20. a) Mirbach M. F.t J. Organometal. Chem., 265, 205 (1984); 6) Kovdcs /.,
Ungvary F., Marko L., Organometallics, 5, 209 (1986); в) Alemdaroglu N. #.,
Penninger J. L. M., Oltay E.t Monatsh. Chem., 107, 1153 (1976); van Bo-
ven M., Alemadaroglu N. H., Penninger J. L. M.t Ind. and Eng. Chem.
(Pdt. Res.), 15, 279 (1975); r) Ungvary F.f Marko L., Organometallics, 2,
1608 (1983); д) Hoff С D., Ungvary F., King R. В., Marko L., J. Am. Chem.
Soc, 107, 666 (1985).
21. Piacenti F., Bianchi M., Bendetti M., Chim. Ind. Milan, 49, 245 (1969).
22. Это вещество идентифицировано по ИК-спектрам с фурье-преобразова-
нием: Vidal J. L, Walker W. E., Inorg. Chem., 20, 249 (1981).
23. Pruett R. L., Smith J. A., J. Org. Chem., 34, 327 (1969).
24. Fowler R.r Connor H., Bachl R. A., Chemtech, 1976, 772.
25. Yagupsky G., Brown С K., Wilkinson G.t J. Chem. Soc. (A), 1970, 1392.
26. Brown С. K-, Wilkinson G., J. Chem. Soc. (A), 1970, 2753.
27. Abatjoglou A. G., Billig E., Bryant D. R., Organometallics, 3, 932 (1984);
Garrou P., Chem. Rev., 85, 171 (1985).
28. Stefani A., Consiglio G., Botteghi C, Pino P., J. Am. Chem. Soc, 95, 6504
(1973).
29. Collman J. P.} Belmont J. A., Brauman /. /., J. Am. Chem. Soc, 105, 7288
(1983).
30. Эти данные можно обработать с помощью линейных графиков Ланвиве-
ра — Бёрка, выражающих зависимость обратных величин скорости от
концентрации субстрата. Lineweaver #., Burke D., J. Am. Chem. Soc, 56,
658 (1934).
31. Van Leeuven P. W. N. M., Roobeek C. F.t J. Organomet. Chem., 258, 343
(1983).
32. а) Этот предмет обсуждается в обзорах: Ojima I., Hirai /(., in:
Asymmetric Synthesis, Morrison J. D. (Ed.), Academic, New York, 1985, Vol. 5;
б) Приведенная ранее величина 94% (Pittman С. U., Jr., Kawabata Y.,
Flowers L. I., J. Chem. Soc, Chem. Commun., 1982, 473) другими авторами
признана ошибочной; см. работу [32а], табл. V и ссылку [1111 в ней-
33. Dang Т. P., Poulin J. С, Kagan H. В., J. Organomet. Chem., 91, 105
(1975).
34. Fritchel 5. /., Acker man I. J. H., Keyser Т., Stille J. K., J. Org. Chem., 44,
3152 (1979).
35. Schwager L, Knifton J. F., J. CataL, 45, 256 (1976).
36. Hsu C, Or chin M., J. Am. Chem. Soc, 97, 3553 (1975).
37. Whyman R.t J. Organomet. Chem., 94, 303 (1975).
38. a) Sancheez-Delgado R. A., Bradley J. S., Wilkinson G., J. Chem. Soc. Dal-
ton Trans., 1976, 399; 6) Kang H., Mauldin С H., Cole Т., Slegeir W.,
Cann K., Pettit R., J. Am. Chem. Soc, 99, 8323 (1977).
39. Senocq F., Randrianalimanana C, Thorez A., Kalck P., Choukroun R.} Ger-
vais D.t J. Chem. Soc, Chem. Commun., 1984, 1376; Kovdcs I., Hoff C. D.,
Ungvary F., Marko L., Organometallics, 4, 1327 (1985).
40. Hidai M., Fukuoka A., Koyasu Y., Uchida Y., J. Chem. Soc, Chem. Commun,
1984, 516; Koyasu Y., Fukuoka A., Uchida Y., Hidai M., Chem. Lett., 1985,
1083.
41. Bechtold M. F., Mahler W., Schunn R. Л., J. Polymer Sci., Polymer Chem.
Ed., 18, 2823 (1980).
42. Jenck /., частное сообщение; Kuntz E., франц. пат. 2314910; 2343562;
Jenck /., франц. пат. 2473504.
43. Halpern /., Proceedings of the First International Symposium on
Homogeneous Catalysis, Plenum, New York, 1977.
144 Глава 12
44. Takegami Y., Watanabe Y., Masada Я., Bull. Chem. Soc. Japan, 40, 1459
(1967).
45. Falbe /., Huppes N., Brennst.-Chem., 48, 46 (1967).
46. Brown C. K., Mowat W., Yagupski G., Wilkinson G., J. Chem. Soc. (A),
1971, 850.
47. а) Имянитов Н. С, Рудковский Д. М.—Журн. прикл. хим., 1968, 41,
т. 172—176; б) Milstein D., Huckaby J. L., J. Am. Chem. Soc, 104, 6150
(1982); в) Waller F. /., J. Mol. Catal., 31, 123 (1985).
48. Bittler K., von Kutepow N., Neubauer D., Reis Я., Angew. Chem., Int. Ed.
Engl., 7, 329 (1968); Fenton D. M.t J. Org. Chem., 38, 3192 (1973).
49. Alper H., Woell J. В., Despeyroux В., Smith D. Л Я., J. Chem. Soc, Chem.
Commun., 1983, 1270; Alper H., Leonard D., J. Chem. Soc, Chem. Commun,
1985, 511.
50. a) Knifton J. F., J. Org. Chem., 41, 2885 (1976); б) Последние
исследования каталитической активности комплексов Pt и PdSnCl3 описаны в
работе: Albinati A., Pregosin P. S., Ruegger Я., Inorg. Chem., 23, 3223
(1984).
51. Johnson R. W., Pearson R. G., Inorg. Chem., 10, 2091 (1971).
52. Weiss E., Hubel W.} Merenyi R., Chem. Ber., 95, 1155 (1962); Clarkson R.,
Jones E. R. H., Wailes P. C, Whiting M. C, J. Am. Chem. Soc, 78, 6206
(1956); Case J. R., Clarkson R.f Jones E. R. Я., Whiting M. C, Proc Chem.,
1959, 150.
53. Forster D., Adv. Organomet. Chem., 17, 255 (1979).
54. Roth J. F., Craddock J. П., Hershman A., Paulik F. £., Chemtech., 1, 600
(1971); Grove Я. D., Hydrocarbon. Proc 1972 (Nov.), 76; Paulik F. £.,
пат. США 3769329 (1973).
55. Ellwood P., Chem. Eng. News, 1969 (May 19), 148.
56. См. работу [1], стр. 81.
57. Webber К. M., Gates В. С, Drenth W.y J. Catal., 47, 269 (1977).
58. Forster D., J. Chem. Soc, Dalton Trans., 1979, 1639.
59. Webber К M., Gates B. C, Drenth №., J. Mol. Catal., 3, 1 (1977—78); см.
также: Kryzwicki A., Marczewski M.y J. Mol. Chem., 6, 431 (1979).
60. Lapporte S., Kurkov V. P., in: Organotransition Metal Chemistry, Ishii Y,
Tsutsui M. (Eds.), Plenum, New York, 1975, p. 199.
61. Подобное ингибирование, как правило, всегда наблюдается, см. [20а],
однако описаны системы, в которых отсутствует ингибирование СО, см.:
Azran J., Or chin M., Organimetallics, 3, 197 (1984).
62. Braca G., Sbrana G., Valenti G., Andrich G., Gregorio G., J. Am Chem. Soc
100, 6238 (1978).
63. a) Polichnowski S. W., J. Chem. Educ, 63, 206 (1986); Larkins Т. Н.,
Polichnowski S. W.t Justin G. C, Young D. Л., пат. США 4374070 (1983);
6) Rizkalla N., Winnick С. N., Ger. Offenleg. 2610035 (1976);
Polichnowski S. W., Larkins T. H., Jr., пат. США 4581473 (1986).
64. Tsuji /., «Decarbonylation Reactions Using Transition Metal Compounds»,
in: Organic Synthesis via Metal Carbonyls, Wender I., Pino P. (Eds.), Wi-
ley-Interscience, New York, 1977, Vol. 2, p. 595 (есть русский перевод
первого издания книги: Органические синтезы через карбонилы металлов.
Пер. с англ./Под ред. Уэндера И., Пино Р. — М.: Мир, 1970); Tsuji /.,
Ohno К., Synthesis, 157, 1 (1969).
65. Ohno К., Tsuji /., J. Am. Chem. Soc, 90, 99 (1968).
66. Doughty D. A., Pignolet L. Я., J. Am. Chem. Soc, 100, 7083 (1978).
67. Doughty D. A., McGuiggan M. F., Wang H., Pignolet L. Я., Fundamental
Research in Gomogeneous Catalysis, Tsutsui M. (Ed.), Plenum, New York,
1979, Vol. 3, p. 909.
68. Hawthorn J. O., Wilt M. Я., J. Org. Chem., 25, 2215 (1960).
69. См. работу [1], стр. 90.
70. Foglia Т. A., Ban P. A., Idacavage M. /., J. Org. Chem., 41, 3452 (1976).
Каталитические реакции с участием монооксида углерода 145
71. Domazetis G., Tarpez В., Dolphin D., James В. R.y J. Chem. Soc, Chem.
Commun., 1980, 939; Domazetis G.t James B. R.t Tarpey В., Dolphin D., ACS
Symp. Sen, 152, 243 (1981).
72. Bennett R. P., Hardy W. В., J. Am. Chem. Soc, 90, 3295 (1968).
73. UEplattenier F., Matthys P., Calderazzo F., Inorg. Chem., 9, 342 (1970).
74. Этот вопрос обсуждается в обзоре: Costa L. С.,. Catal. Rev. Sci. Eng., 25,
325 (1983).
75. Stull D. R., Westrum E. F., Jr., Sinke G. C, The Chemical Thermodynamics
of Organic Compounds, Wiley, New York, 1969.
76. Headford C. E. L., Poper W. R., in: Reactions of Coordinated Ligands,
Braterman P. S. (Ed.), Plenum.
77. a) Wayland В. В., Woods B. A., Pierce #., J. Am. Chem. Soc, 104, 302
(1982); 6) Fagan P. /., Moloy K. G., Marks T. /., J. Am. Chem. Soc, 103,
6959 (1981).
78. a) Dombek B. D., Harrison А. M., J. Am. Chem. Soc, 105, 2485 (1983);
6) Doxsee K- M., Grubbs R. H., J. Am. Chem. Soc, 103, 7696 (1981).
79. Clark G. R., Headford С E. L, Marsden K-, Poper W. #., J. Organomet
Chem., 231, 335 (1982).
80. Sweet J. R., Graham W. A. G., J. Am. Chem. Soc, 104, 2811 (1982).
81. a) Dombek B. D., J. Am. Chem. Soc, 101, 6466 (1979); 6) Vaughn G. D.,
Gladysz J. A., J. Am. Chem. Soc, 108, 1473 (1986).
82. Stimson R. E., Shriver D. F., Inorg. Chem., 19, 1141 (1980).
83. Feder H. M., Rathke J. W.t Ann. N. Y. Acad. Sci., 333, 45 (1980).
84. Feder H. M., Rathke J. W., Chen M. J., Curtis L. Л., in: Catalytic Activation
of Carbon Monoxide, Ford P. C. (Ed.), ACS Symposium Series 152,
American Society, Washington, D. C, 1981, p. 19.
85. Rathke J. W.t Feder H. M., in: Catalysis of Organic Reactions, Moser W. R.
(Ed.), Marcel Dekker, New York, 1981, p. 219.
86. Keim W., Berger M., Schlupp /., J. Catal., 61, 359 (1980).
87. Pruett R. L., Ann. N. Y. Acad. Sci., 295, 239 (1977).
88. Vidal J. L, Walker W. E.t Inorg. Chem., 19, 896 (1980) и ссылки [21—241
в этой статье.
89. Kaplan L., пат. США 4197253 (1980).
90. Vidal J. L., Walker W. E., Pruett R. L, Schoening R. C, Fiato R. C,
Fundamental Research in Gomogeneous Catalysis, Tsutsui M. (Ed.), Plenum, New
York, 1979, Vol. 3, pp. 409—518.
91. Bradley J. S., J. Am. Chem. Soc, 101, 7419 (1979).
92. Механизм действия М—H и М—CO2R при карбоалкоксилировании олефи-
нов и ацетиленов обсуждается в работе: Murray Т. F., Norton J. R., J. Amu
Chem. Soc, 101, 4107 (1979).
93. King R. В., King A. D., Tanaka K, J. Mol. Catal., 10, 75 (1981).
94. Knifton /. F., J. Am. Chem. Soc, 103, 3959 (1981).
95. a) Knifton J. F., J. Catal., 76, 101 (1982); 6) Barratt D. S.,
Cole-Hamilton D. /., J. Chem. Soc, Chem. Commun., 1981, 103, 6508.
96. Dombek B. D.t J. Am. Chem. Soc, 103, 6508 (1981).
97. a) Why man R., European Patent 33425 (1981); б) Аналогичные
результаты приведены в работе: Dombek В. D., Organometallics, 4, 1707 (1985).
98. a) Marsella J. A., Pez G. P., J. Mol. Catal., 1986; Preprints, Div. Pet. Chem.,
Soc, 31, 22 (1986); б) см. также: Knifton J. F., J. Chem. Soc, Chem.
Commun., 1985, 1412.
99. Laine R. M., Cho B. R., Wilson R. В., Jr., Cl Mol. Chem., 1, 1 (1984).
100. Fenton D. M.t J. Org. Chem., 39, 701 (1974).
101. Rivetti F., Romano U., J. Organomet. Chem., 154, 323 (1978); Rivetti F.y
Romano U.t Chem. Ind., 62, 7 (1980).
102. Nishimura К., пат. США 4229589.
103. Fenton D. M., Steinwald P. /., J. Org. Chem., 37, 2034 (1972)
104. Chem. Eng. News, 1981, Oct. 26, p. 22.
105. Rofer-DePoorter С. К., Chem. Rev., 81, 447 (1981).
10—1129
146 Глава 12
106. Herrmann W. Л., Angew. Chem., Int. Ed. Engl., 21, 117 (1982).
107. Fischer F., Tropsch #., Brennst.-Chem., 7, 97 (1926); Fischer F., Tropsch H.%
Chem. Ber., 59, 830 (1926).
108. Brady R. C.f Pettit R.t J. Am. Chem. Soc, 102, 6181 (1980).
109. Biloven P., Helle H. N., Sachtler W. M. H.. J. Catal., 58, 95 (1979).
110. a) Storch H. H., Golumbic N., Anderson R. £., The Fischer — Tropsch and
Related Synthesis, Wiley, New York, 1951; 6) Kummer /. F., Emmett P. //.,
J. Am. Chem. Soc, 75, 5177 (1953).
111. Pichler H., Schultz #., Chem. Eng. Tech., 12, 1160 (1970).
112. a) Masters C, Adv. Organomet. Chem., 17, 61 (1979); 6) Henrici-Olive G.,
Olive S., Angew. Chem., Int. Ed. Engl., 15, 136 (1976).
113. Quoted by Haggin /., Chem. Eng. News, 59, No. 43, 22 (1981).
114. Isobe K., Andrews D. G.} Mann B. E., Maitlis P., J. Chem. Soc, Chem. Com-
mun., 1981, 809.
115. Masters C, van der Woude C, van Doom J. Л., J. Am. Chem. Soc, 101,
1633 (1979).
116. a) Muetterties E. L., J. Organomet. Chem., 200, 177 (1980); 6) Tachika-
wa M., Geerts R. L, Muetterties E. L., J. Organomet. Chem., 213, 11
(1981).
117. a) Bradley J. S., Ansell G. В., Hill E. W., J. Am. Chem. Soc, 101, 7417
(1979); 6) Bradley J. S., Ansell G. В., Leonowicz M. E., Hill E. W.t J. Am.
Chem. Soc, 103, 4968 (1981).
118. Keim W., Roper W. R., Strutz Я., J. Organomet. Chem., 219, C5 (1981).
119. Doyle M. J., Kouwenhoven A. P., Schaap C. A., Van Oort B. V., J.
Organomet. Chem., 174, C55 (1979) [в статье рассматриваются работы:
Masters С, Van Doorn J. A. Offenlegungsschrift, 1977, 2644; U. K- Patent
application 75/40322 (1975)].
120. Brenner L. S., Lai Y.-H., Vollhardt K. P. C, J. Am. Chem. Soc, 103, 3609
(1981) [в статье рассматривается работа: Henrici-Olive G., Olive 5., Angew
Chem., Int. Ed. Engl., 18, 77 (1979)].
121. Perkins P., Vollhardt K. P. C, J. Am. Chem. Soc, 101, 3985 (1979).
122. Sekiya A., Siille J. K., J. Am. Chem. Soc, 103, 5096 (1981).
123. Demitras G. C, Muetterties E. L., J. Am. Chem. Soc, 99, 2796 (1977).
124. Wang H.-K-, Choi H. W., Muetterties E. L., Inorg. Chem., 20, 2661 (1981).
125. Collman J. P., Brauman J. L, Justin G., Wann G. S.t III, J. Am. Chem. Soc,
105, 3913 (1983).
126. Choi H. W., Muetterties E. L., Inorg. Chem., 20, 2664 (1981).
127. Herman R. G., Klier K.f Simmons G. W., Finn B. P., Bulko J. В., J. Catal.,
56, 407 (1979).
128. Mehta S., Simmons G. W., Klier K., Herman R. G., J. Catal., 57, 399
(1979).
129. Rung H. H., Catal Rev. Sci. Eng., 22(2), 235 (1980) (обзор).
130. a) Apai G. R., Monnier J. R., Hanrahan M. /., J. Chem. Soc, Chem. Com-
mun, 1984, 212; 6) Fleisch T. H., Mieville R. L., J. Catal., 90, 165 (1984).
131. Saussey J., Lavalley J.-C, Lamotte J., Rais Т., J. Chem. Soc, Chem. Com-
mun., 1984, 278; Lavalley J. C, Saussey /., Rais Т., J. Mol. Catal., 17, 289
(1982).
РАЗДЕЛ III
ПРИМЕНЕНИЯ В ОРГАНИЧЕСКОМ
СИНТЕЗЕ
13
ПРИМЕНЕНИЕ ГИДРИДОВ ПЕРЕХОДНЫХ МЕТАЛЛОВ
В ОРГАНИЧЕСКОМ СИНТЕЗЕ
Хотя гидриды переходных металлов находят главное
применение в каталитическом гидрировании и гидроформилировании
простых олефиновых субстратов, многие из них пригодны для
стехиометрического восстановления более сложных,
функционально замещенных систем, причем зачастую эти реакции идут
с высокой степенью регио- и стереоспецифичности. Поэтому
гидриды переходных металлов оказываются полезными
реагентами для органического синтеза, особенно когда речь идет о
получении сложных молекул.
Одним из таких реагентов является KHFe(CO)4,
генерируемый in situ при обработке Fe(CO)5 гидроксидом калия в
этаноле. Эти растворы служат эффективными восстанавливающими
системами для превращения сопряженных енонсв в насыщенные
карбонильные соединения [уравнение (13.1)].
Fe(CC% + КОН ЕЮН>- [(CO)4Fe-C-O-Hj ■ * [нРе(С0)4] " К*
о
В ненасыщенных альдегидах, кетонах, сложных эфирах,
лактонах, нитрилах и диэфирах гладко восстанавливается
двойная связь, а карбонильная группа не затрагивается [1].
Ответственным за наблюдаемые реакции восстановления является
HFe(CO)4~, хотя в системе присутствуют и другие полиядерные
карбонилгидридные комплексы железа. Стерическое напряже-
10*
148 Глава 13
ние вокруг сопряженного енона резко понижает скорость
реакции; эту каталитическую систему нельзя использовать с
субстратами, чувствительными к действию оснований. Щелочную
среду, необходимую для генерирования восстанавливающего
агента, можно с выгодой использовать. Например, при
обработке KHFe(CO)4 и различными альдегидами кетоны, содержащие
а-метильную или а-метиленовую группы, претерпевают гладкое
восстановительное а-алкилирование [2, 3]. Считается, что
реакция начинается с катализируемой основанием конденсации
кетона с альдегидом, за которой следует восстановление
образовавшегося сопряженного енона [уравнение (13.2)].
Аналогичным образом реагируют соединения с активной метиленовой
компонентой, такие, как сложные ^-кетоэфиры [4, 5], тогда как
индол подвергается гладкому восстановительному алкилирова-
нию по положению 3 [6].
^& RCH?R' (l3.2)
CH2R"
CH2R4
Тщательное изучение механизма [7], по которому
NaHFe(CO)4 восстанавливает этилакрилат, позволило
установить ключевые стадии реакции [уравнение (13.3)]. Комплекс
быстро и необратимо присоединяется к этилакрилату, давая
(в отсутствие источников протона) нерастворимый интермедиат
о-алкилжелеза [CH3CH(Fe(CO)4)CO2Et]~. Эту реакцию сильно
О
HFe(CO)4 *=* CO+HFe(CO)3+CH2=CHCOEt
О О
II н+ II
> СН2—CHCOEt > CH3CH2COEt
Н Fe(CO)4
(13.3)
ингибирует избыток СО, что указывает на диссоциативную
первую стадию. Алкилжелезо подвергается быстрому протоно-
лизу, образуя продукт восстановления. При проведении реакции
в апротонных растворителях, например в ТГФ, происходит
полное восстановление сопряженных енонов и образуются
насыщенные спирты [уравнение (13.4)] [8]. Очевидно, когда не
может осуществиться протонолиз первоначального аддукта желе-
Применение гидридов переходных металлов 149
за, происходит второй перенос гидрида, на этот раз к
карбонильной группе. Хиральные сопряженные еноны
восстанавливаются без рацемизации; например, из ( + )-карвона
получается (—)-неодигидрокарвеол.
Под действием HFe(CO)4~ восстанавливаются и некоторые
другие функциональные группы. Из хлорангидридов кислот с
прекрасным выходом получаются альдегиды [9]. Хотя об
изучении механизма реакции не сообщалось, считается, что
реакция начинается с вытеснения галогенида ионом Fe~, приводящего
к образованию RCOFe(H) (CO)4, после чего следует быстрое
восстановительное элиминирование альдегида. Этим реагентом
также легко восстанавливаются двойные связи углерод — азот.
Под действием KHFe(CO)4 и KHFe2(CO)s енамины
восстанавливаются до третичных аминов через иминиевую соль
[уравнение (13.5)] [10]. Рассмотренные химические свойства лежат в ос-
KHFe(C0)4 - N ~VR (13.5)
нове удобного метода восстановительного аминирования [11, 12],
при котором альдегиды и кетоны реагируют с первичными
аминами и Fe(CO)5 в 1 н. спиртовом растворе КОН, давая
вторичные амины [уравнение (13.6)]. Введение в эту реакцию глута-
рового альдегида [13] позволяет синтезировать N-алкилпирро-
1 н. КОН
RCHO+R/NH2+Fe(CO)5 *- [RCH = N—R'] —> RCH2NHR' (13.6)
EtOH
лидины с прекрасным выходом [уравнение (13.7)]. Все эти
реакции включают восстановление иминов, образующихся при
конденсации первичных аминов с альдегидами.
r^N
RNH2 ЫаНЫСОЦ , j (|3 ?)
OHC CHO ArNHo CO
2 R(Ar)
Другой потенциально полезный восстанавливающий агент,
разработанный Коллменом, представляет собой биядерный
кластер NaHFe2(CO)8. Этот комплекс получают при
взаимодействии Na2Fe(CO)4 с Fe(CO)5 с последующим подкислением
уксусной кислотой. В результате образуется красно-коричневый
150 Глава 13
раствор комплекса в уксусной кислоте, который
непосредственно используется для реакций восстановления. Для контроля
измеряют ЯМР-спектр раствора, в котором характерный сигнал
имеет химический сдвиг 8,47 м. д. (в сильное поле от ТМС).
Этот реагент — очень эффективный восстановитель для
сопряженных енонов всех типов. Так, с его помощью в сопряженных
кетонах, альдегидах, сложных эфирах, нитрилах, амидах и лак-
тонах гладко восстанавливается двойная углерод-углеродная
связь и не наблюдается конкурентного восстановления карбо-
нила. Эпоксиды не поддаются действию реагента, так же как
и изолированные двойные связи, которые и не восстанавливаются,
и не перегруппировываются. Затрудненные системы вступают в
реакцию, но дают продукты с очень низкими выходами
(например, в случае тестостерона выход составляет лишь 10%), к
тому же для завершения реакции требуются два или более
эквивалента NaHFe2(CO)8. Выход продуктов восстановления
повышается при добавлении NH4C1 и Fe(CO)5. Изучение
относительных скоростей реакции с различными субстратами
показывает, что двойные цис-связи (например, в енонах типа
циклогексенона) восстанавливаются быстро, что наличие
заместителей в а- или 'у-положениях к карбонильной группе
понижает скорость реакции и что не наблюдается корреляции между
скоростями восстановления и восстановительным потенциалом
енона. Последнее, вероятно, указывает на отсутствие процессов
переноса электрона [7].
На первый взгляд химия этих комплексов аналогична
таковой для NaHFe(CO)4. Однако проведенное Коллменом и
другими [7] детальное изучение механизма реакции NaHFe2(CO)8
с сопряженными енонами показывает, что два комплекса
реагируют совершенно по-разному. Напомним, что HFe(CO)4~
реагирует с этилакрилатом диссоциативным путем с быстрым
необратимым и региоспецифическим присоединением по двойной
связи и с последующим быстрым протонолизом связи железо —-
углерод [уравнение (13.3)]. В противоположность этому
NaHFe2(CO)s реагирует с этилакрилатом ассоциативным путем
с быстрым региоспецифическим, но обратимым присоединением
по двойной связи и с последующим относительно медленным
протанолизом связи железо — углерод. Биядерный комплекс
железа не фрагментирует, а интактным присоединяется к
двойной связи. Он почти в 26 раз более реакционноспособен, чем
моноядерный комплекс NaHFe(CO)4; реакция имеет первый
порядок как по димеру, так и по субстрату. Как показано в
уравнении (13.8), образующееся соединение алкилжелеза
распадается с образованием продукта двумя путями. Один из них
включает прямой протонолиз биядерной молекулы, а другой,
независимый от кислоты путь состоит в медленной фрагмента-
Применение гидридов переходных металлов 151
ции [с потерей Fe(CO)4] и последующем быстром протонолизе.
Химия этих двух систем комплементарна, поскольку
моноядерные комплексы лучше реагируют в щелочной среде, тогда как
биядерные требуют слабокислой среды. Родственный гидридо-
карбонильный кластер железа Et4N+HFe3(CO) ц~
восстанавливает ароматические нитросоединения до аминов. Реакция идет
с достаточно высокими выходами, а другие функциональные
группы (в альдегидах, эфирах, хлоридах) в реакции не
затрагиваются [14].
NaX + Fe2(C0)8 + RCH2CH2CR'
I Ил
a 1
irch=chcr'
] / (13.8)
[rCH2CHCr] + Fe(C0)4
Fe(C0)4
Эффективными восстанавливающими агентами оказались
также анионные гидридокарбонильные комплексы переходных
металлов VI группы. Так, хромдигидрокарбонильный кластер
NaHCr2(CO)io очень эффективен для восстановления двойной
углерод-углеродной связи в сопряженных кетонах, альдегидах,
сложных эфирах и нитрилах [15]. Механизм этой реакции
специально не изучался, но можно предполагать, что он
аналогичен механизму реакций с участием родственных комплексов
железа. В противоположность биядерному комплексу хрома
мономерные реагенты HCr(CO)s и HW(CO)s~ эффективно
восстанавливают органические галогениды [16]. Реакционная
способность галогенидов по отношению к вольфрамовому реагенту
уменьшается в ряду 1>Вг>С1 и первичный = третичный >
вторичный. Под действием этого реагента легко восстанавливаются
ароматические галогениды, а также 1-бромоадамантан, так что
ясно, что процесс восстановления не следует механизму SN2.
Имеются некоторые доказательства в пользу радикального
процесса, и многие особенности этой восстанавливающей
системы напоминают характерные черты хорошо изученного
комплекса CpV(CO)3H"-, который будет обсуждаться далее.
Комплекс НСг(СО)5~ также эффективно восстанавливает хлоран-
гидриды кислот до альдегидов и хорошо отличает эту
функциональную группу от аллил- или арилгалогенидов, а также от
нитрогрупп [17].
132 Глава 13
Исключительные химические свойства алкилмедных
реагентов (гл. 14) послужили моделью для разработки нескольких
родственных гидридмедных восстанавливающих агентов. Как и
в случае алкилмедных реагентов, точный состав этих «гидридов
меди» чаще всего неизвестен. Смешанные комплексы типа
[я-СзН7СЕ=ССиН]-Ы+, взятые в избытке от четырех- до
шестикратного, гладко и с прекрасным выходом восстанавливают
сопряженные кетоны и сложные эфиры до насыщенных
карбонильных соединений. Стереохимия реакции (изученная на
конденсированных циклических системах) сравнима со
стереохимией каталитического гидрирования. В полиненасыщенных
соединениях восстановлению подвергаются только сопряженные
двойные связи, а ацетиленовые сложные эфиры инертны [18].
Смешанный купрат [я-ВиСиН]~Ы+ селективно переносит гидрид
(а не алкильную группу) и также служит эффективным
агентом для 1,4-восстановления сопряженных енонов при низких
температурах (—40 °С). Реагент способен восстанавливать
даже многозамещенные субстраты, например 3,5,5-триметилцикло-
гекс-2-ен-1-он. При более высоких температурах (25 °С) он
восстанавливает альдегиды и кетоны до спиртов, а первичные,
вторичные и даже третичные галогениды, мезилаты и тозила-
ты — до соответствующих углеводородов; реакции
осуществляются с прекрасным выходом. В случае третичных галогенидов
элиминирование не наблюдается. Сложные эфиры в реакцию не
вступают [19]. Еще более активный восстанавливающий агент
для галогенидов получают при обработке иодида меди(1) двумя
эквивалентами ЫА1Н(ОМе)3 в ТГФ. Этот комплекс, структура
которого точно не определена, восстанавливает первичные,
вторичные и третичные галогениды, а также неопечтил-, аллил-,
винил-, арилгалогениды и геминальные дигалогенциклопропильные
производные до соответствующих углеводородов; для всего
набора субстратов наблюдается высокий выход продукта. Кроме
того, под действием этого реагента эпоксиды
восстанавливаются до спиртов [20].
Известны еще два очень удобных восстанавливающих
агента, хотя и их структура окончательно не ясна. Один из них
получают реакцией бромида меди(1) с двумя эквивалентами
ЫА1Н(ОМе)3 в ТГФ и называют «комплексом лития». Другой
готовят тоже из бромида меди(1), обрабатывая его одним
эквивалентом NaAlH2(OCH2CH2OCH3)2, и называют «комплексом
натрия» [21]. Оба реагента получаются в виде гетерогенных
смесей в ТГФ, которые не очищают и не характеризуют перед
употреблением (они реагируют совершенно иначе, чем LiCuH2,
получаемый альтернативным путем). Оба реагента эффективны
для 1,4-восстановления сопряженных енонов, однако для 2-цик-
логексенона и родственных систем предпочтительнее «комплекс
Применение гидридов переходных металлов 153
Li». При использовании 3 экв этого реагента циклогексенон
полностью восстанавливается до циклогексанона за 0,3 ч.
Более замещенные субстраты, 5,5-диметил- и 3,5,5-триметилцик-
логексеноны, также подвергаются восстановлению, но
несколько медленнее. В отличие от этого «комплекс натрия» более
активен по отношению к ациклическим системам, причем
восстановление идет только по 1,4-пути, тогда как с «комплексом
лития» наблюдается в значительной мере и 1,2-восстановление.
«Комплекс Na» более эффективен также для восстановления
сопряженных сложных эфиров и ацетиленовых сложных эфиров.
Во всех реакциях с «комплексом Na» наблюдается
конкурентная, катализируемая основанием конденсация сопряженных
карбонильных соединений, в результате чего наряду с
продуктами восстановления образуются олигомеры. По отношению к
другим функциональным группам «комплекс Na» также более
активен, чем «комплекс Li», специфичный для
а,^-ненасыщенных систем. Насыщенные альдегиды и кетоны, а" также алкил-
бромиды восстанавливаются «комплексом Na» почти так же
быстро, как типичные сопряженные кетоны. Сложные эфиры и
нитрилы инертны к обоим реагентам.
Хотя структура комплексов не установлена, ряд
экспериментальных данных позволяет судить о механизме реакций. При
восстановлении а,^-ненасыщенного кетона реагентом,
приготовленным из CuBr и LiAlD(OMe)3, наблюдается дейтерирование
только по ^-положению [уравнение (13.9)], что указывает на
обычный процесс 1,4-присоединения. Какой интермедиат
образуется перед обработкой водой, енолят лития или енолят меди,
не установлено. Не известна также роль процесса одноэлект-
ронного переноса в сравнении с прямым 1,4-присоединением
гидрида (являющимся формально двухэлектронным процессом).
о _ ,о н 0
2LiA!D(0Me)3 + CuBr + RCH=CHCCH3 +• [rCH-Ch'-CCH3 ] М+ —-
RCHCH2CCH3 (13.9)
D
(Этот момент более подробно обсуждается в гл. 14 при
рассмотрении родственной реакции 1,4-алкилирования алкилкуп-
ратами.) Однако тот факт, что при реакции метилциннамата с
«комплексом Na» выделяется большое количество диметил-3,4-
дифениладипината, указывает на промежуточное образование
анион-радикалов и, следовательно, на механизм, включающий
одноэлектронный перенос, по крайней мере в случае субстратов,
показанных в уравнении (13.10). Правда, в большинстве слстем
154 Глава 13
не наблюдается продуктов, которые могут образоваться из
радикалов, поэтому вопрос о значении и обобщении приведенного
выше факта остается открытым. Опять же не было показано,
что ион-радикальные интермедиа™, ответственные за димери-
зацию, ответственны также и за восстановление. х
(13.10)
PhCH=CHCOMe
PhCHCH=COMe
H-Cu-H
-CuH
le"
димери-
зацизг
PhCHCH=COMe
PhCHCH=COMe
i
0"
H+
PhCHCH2COMe
PhCHCH2COMe
6
Свойства «гидридов меди» в большой степени зависят от
способа их получения. Реагенты, полученные реакцией LiAlHU
с четырьмя эквивалентами иодида меди(1), очень эффективны
для 1,4-восстановления многих сопряженных кетонов, в том
числе и высокозамещенных по а- и (или) ^-положению. Однако
с циклогексеноном реагент не взаимодействует, в то время как
этот субстрат обычно вполне реакционноспособен по отношению
к 1,4-восстановлению соединениями типа «CuH». Однако в этом
случае, как показал Эшби [22], активным восстанавливающим
агентом служат не соединения меди, а А1Н21. Индивидуальные
гидридмедные «ат»-комплексы состава ЫяСиНп+1 (где п
меняется от 1 до 5) можно приготовить, обрабатывая LiAlH4
соответствующими литийалкилкупратами LinCu (CH3) п+ь
Химические свойства таких комплексов сильно зависят от их состава.
Например, Li2CuH3 восстанавливает сопряженные кетоны
исключительно по олефиновой связи, что приводит к насыщенным
кетонам, тогда как восстановление того же субстрата с
помощью Li4CuH5 происходит по 1,2-пути и приводит к аллиловым
спиртам. Децилбромид восстанавливается в я-декан под
действием всех упомянутых комплексов, но лучшим реагентом для
этого превращения служит Li4CuH5 [23].
Несколько отличным восстанавливающим агентом на основе
меди оказался легко получающийся комплекс (Ph3PhCuBH4.
Это белое кристаллическое вещество, устойчивое на воздухе,
растворимое во многих органических растворителях и
нерастворимое в воде и этаноле. Комплекс является очень
специфическим восстанавливающим агентом для превращения хлорангид-
ридов кислот в альдегиды. Реакцию обычно проводят в ацетоне;
ароматические, гетероароматические, а,^-ненасыщенные и про-
Применение гидридов переходных металлов 155
стые хлорангидриды алифатических кислот гладко и с
высокими выходами восстанавливаются до соответствующих
альдегидов. Сложные эфиры кетоны, нитрилы, эпоксиды, имины, алке-
ны и алкины инертны к действию этого комплекса [24, 25].
Очевидно, что комплексообразование ВН4~ с медью меняет его
восстановительную способность, ведь обычно альдегиды и
кетоны восстанавливаются под действием NaBH4.
Еще одним эффективным реагентом для восстановления
хлорангидридов кислот служит система палладий (0)—гидрид
олова. Как показано в уравнении (13.11), реакция, видимо,
происходит по пути: окислительное присоединение — переметалли-
о
ЙС-С1 +
г 9 L -1 BibSnH г ? L л
L_4Pd *• [RC-PdCIJ ■ * 1*. [RC-Pd-HJ + Bu3SnCl (13.
О
RC-H
рование — восстановительное элиминирование [26]. Система
допускает присутствие арилбромидов и нитросоединений. Однако
та же комбинация реагентов восстанавливает сопряженные
кетоны и альдегиды до насыщенных карбонильных соединений
[уравнение (13.12)] [27]. Последний процесс должен идти по
другому механизму; возможно, следует пересмотреть вывод о
преимущественном восстановлении галогенангидридов кислот
по сравнению с альдегидами.
сно
сно
Аг"
(13.12)
СНО
СНО
Bu3SnH
катализатор ЦРсС
156 Глава 13
Обнадеживающие результаты, полученные при
использовании таких восстановительных систем, как галогениды меди/
/алюминийгидрид, привели Эшби к мысли распространить этот
подход на все галогениды переходных металлов первого ряда.
Система LiAlH4/NiCl2 восстанавливает первичные, вторичные,
циклические, ароматические галогениды и тозилаты до
соответствующих углеводородов с почти количественными выходами.
Аналогичные реагенты с FeCl2, СоС12 и TiCl3 вступают в те же
реакции, но менее эффективно. Реагенты, полученные из LiAlH4
и VC13, CrCl3, МпС12 и FeCl3, неэффективны для восстановления
указанного типа [28]. Аналогичные смеси LiAlH4/MClx
восстанавливают алкены как с концевой, так и с внутренней двойной
связью, включая циклические, а также стирол до алканов. По
отношению к восстановлению алкенов реакционная способность
металлов первого ряда изменяется в следующем порядке: Со>
> Ni > Fe(II)< Fe(III)> Ti(HI)> Cr(III)> V(HI)> Mn(II)>
> Cu(I)> Zn(II). Смесь LiAlH4/NiCl2 в отношении 10:1
восстанавливает алкины с внутренней кратной связью в цис-алке-
ны, а алкины с концевой связью в алкены с концевой двойной
связью; реакция идет стереоспецифически и с количественным
выходом, причем дальнейшего восстановления алкенов в алка-
ны не наблюдается [29]. Смесь FeCl3 с тремя эквивалентами
гидрида натрия менее эффективна для этой реакции, но она
восстанавливает кетоны и альдегиды в спирты с хорошими
выходами [30]. Комплексы палладия (0) катализируют
восстановление простых аллиловых эфиров, аллилтиоэфиров, аллилтри-
алкилсилиловых эфиров и аллилсульфонов в олефины с
сохранением стерео- и региохимии [31].
Кобер [32] изучал более сложные восстанавливающие
агенты (CRA), получаемые при смешении гидрида натрия, алкокси-
да натрия и различных галогенидов переходных металлов в
разных пропорциях. Комплексы CRA гетерогенные,
нерастворимые; их реакционная способность зависит от растворителя,
соотношения реагентов, природы алкоксида и более всего от
природы соли металла. Комплекс NiCRA (4/2/1),
представляющий собой смесь гидрида натрия, тр^г-амилоксида натрия и
ацетата никеля(II) в соотношении 4:2:1 в ТГФ, оказался
эффективным восстанавливающим агентом для алкил-, арил- и
винилгалогенидов, реакционная способность которых в данной
реакции уменьшается в рядах 1>Вг>С1 и первичный >
вторичный > третичный. С помощью этой смеси можно разделить
восстановление бромидов и хлоридов, а также селективно
восстановить галогениды в присутствии карбонильных, простых
эфирных, спиртовых, нитрильных и карбоксильных групп.
В этих реакциях восстановления не наблюдается конкурентного
элиминирования НХ. Моно-и дизамещенные алкины под дейст-
Применение гидридов переходных металлов 157
вием реагента восстанавливаются до алканов. Для
восстановления кетонов и альдегидов наиболее эффективен комплекс
ZnCRA (4/4/1).
Хотя описанные выше системы находят определенное
применение в синтезе, механизм их действия в принципе не
установлен. Не известны также не только некоторые структуры, но в
большинстве случаев даже элементный состав комплексов. Ясно
только, что тем или иным образом в реакции участвуют
переходные металлы, поскольку в их отсутствие наблюдаются
совершенно другие химические закономерности. Однако в отличие
от гидридов алюминия или бора, закомплексованных с
металлом, прямое участие в реакции гидридов переходных металлов
не было однозначно продемонстрировано. Еще меньше известно
о специфической последовательности стадий в каждой
конкретной реакции восстановления и о том, каким образом субстрат
взаимодействует с восстанавливающим агентом. Вряд ли в
ближайшее время удастся выяснить все эти вопросы, если
учесть, что в реакции может участвовать множество различных
частиц. На изучение реакции Гриньяра понадобилось почти сто
лет, но до сих пор она не понята окончательно, так что нужна
запастись терпением.
В отличие от описанных комплексов, в химии которых многа
неопределенного, комплекс [(г)5-С5Н5)У(СО)3Н]~ удалось
изучить полнее. Этот комплекс, получаемый восстановлением
T]5-C5H5V(CO)4 натрием с последующим протонированием
водой, очень эффективен для восстановления практически всех
типов органических бромидов и иодидов [уравнение (13.13)].
Первичные, вторичные и третичные алкилбромиды, бензилбро-
мид, бромобензол и бромостирол восстанавливаются до
соответствующих углеводородов с выходами от хороших до очень
высоких. Хлорангидриды кислот восстанавливаются до
альдегидов, геминальные дибромоциклопропаны — до монобромоцик-
лопропанов, причем последние инертны к дальнейшему
восстановлению. Вицинальные дигалогениды подвергаются гладкому
дегалогенированию; подобно радикальным реакциям это
процесс стереоспецифический и осуществляется граяс-путем. Кето-
ны и сложные эфиры не вступают в реакцию с этим комплексом.
(13.1
I) Na/ТГФ
V(C0)4
JRX
^ RH
По некоторым признакам можно заключить, что реакции идут
по радикально-цепному механизму. Относительная реакционная
способность субстратов соответствует реакционной способности,,
наблюдаемой при восстановлении триалкилстаннанами, которое^
158 Глава 13
как известно, является радикальным процессом. Особенно
убедительный аргумент, исключающий механизм SN2 в реакциях
анионных ванадиевых комплексов, основывается на том факте,
что первичные, вторичные и третичные галогениды
восстанавливаются почти с одинаковой скоростью, а алкилтозилаты и
хлориды очень инертны. При восстановлении хиральных
вторичных галогенидов образуются рацемические углеводороды; как
из цис-, так и из граяс-винилгалогенидов получается одна и та
же смесь цис- и граяс-олефинов. Такое изменение стереохимии
характерно для процессов с участием алкильных или винильных
радикалов. Восстановление бромэтилаллилового эфира
приводит к образованию значительных количеств циклических
продуктов восстановления [уравнение (13.14)]. Этот факт можно
(13.14)
рассматривать как доказательство промежуточного
образования свободных радикалов, поскольку скорость циклизации
ненасыщенных радикалов, составляющая по крайней мере
10~~6 с"1, достаточно велика, чтобы конкурировать с
большинством внутримолекулярных процессов захвата радикального
центра. В уравнении (13.15) представлен механизм, наиболее
согласующийся со всеми изложенными данными [33]. Следует
отметить его сходство с механизмом, предложенным для
радикально-цепного окислительного присоединения (ч. 1, разд. 5.5, в)
и для реакций т]3-аллилникельгалогенидов с органическими га-
логенидами (разд. 19.5).
CpV(co)3H" + .тщиятор—*- CpV(co)3" (I3.I5)
СрУССОз1 + RX < *• CpV(CO)3X" + R*
R- ♦ CpV(C0)3H" - RH + СрУ(С0)3*
Исключительно важное значение в органическом синтезе
имеют реакции присоединения гидридов переходных металлов
к алкинам или алкенам с образованием а-алкилметаллокомп-
лексов, которые будут подробно рассмотрены в гл. 14.
ЛИТЕРАТУРА И ПРИМЕЧАНИЯ
1. Noyori R., Umeda L, Ishigami Т., J. Org. Chem., 37, 1542 (1972).
2. Cainelli G., Panunzio M., Umani-Ronchi Л., Tetrahedron Lett., 1973, 2491.
3. Cainelli G., Panunzio M., Umani-Ronchi A., J. Chem. Soc, Perkin Trans. 1,
1975, 1273.
4. Yamashita M., Watanabe Y.f Mitsudo T-a., Takegami Y., Tetrahedron Lett.
1975, 1867.
Применение гидридов переходных металлов 159
5. Yamashita М., Watanabe Y., Mitsudo T-a., Takegami У., Bull. Chem. Soc.
Japan, 51, 835 (1978).
6. Boldrini G. P., Panunzio M., Umani-Ronchi A., J. Chem. Soc, Chem. Com-
mun., 1974, 359.
7. Collman J. P., Finke R. G., Matlock P. L., Wahren R., Komoto R. G., Brau-
man J. /., J. Am. Chem. Soc, 100, 1119 (1978).
8. Yamashita M., Miyoshi K., Okada Y., Suemitsu R., Bull. Chem. Soc. Japan,
55, 1329 (1982).
9. Cole Т. Е., Pettit R., Tetrahedron Lett., 1977, 781.
10. Mitsudo T-a., Watanabe Y., Tanaka M., Atsuta S., Yamamoto K-, Takegami Y.r
Bull. Chem. Soc Japan, 48, 1506 (1975).
11. Watanabe Y., Yamashita M., Mitsudo Т., Tanaka M., Takegami A.,
Tetrahedron Lett., 1974, 1879; Bull. Chem. Soc Japan, 49, 1378 (1976).
12. Boldrini G. P., Panunzio M., Umani-Ronchi A., Synthesis, 1974, 733.
13. Watanabe Y., Shim S. C, Mitsuda T-a., Yamashita M., Takegami У., Bull.
Chem. Soc. Japan, 49, 2302 (1976).
14. Boldrini G. P., Umani-Ronchi A., Panunzio Л., J. Organomet. Chem., 171, 85
(1979).
15. Boldrini G. P., Umani-Ronchi A., Panunzio M., Synthesis, 1976, 596.
16. Kao S. C, Darensbourg M. У., Organometallics, 3, 646 (1984).
17. Kao S. C, Darensbourg M. У., Organometallics, 3, 1601 (1984).
18. Boeckman R. K, Michalak R., J. Am. Chem. Soc, 96, 1623 (1974).
19. Masamune S., Bates G. S,, Georghiou P. E., J. Am. Chem. Soc, 96, 3686-
(1974).
20. Masamune S., Rossy P. A., Bates G. S., J. Am. Chem. Soc, 95, 6452 (1973).
21. Semmelhack M. F., Stauffer R. D., Yamashita Л., J. Org. Chem., 42, 3180
(1977).
22. Ashby E. C, Lin J. J., Kovar R., J. Org. Chem., 41, 1939 (1976).
23. Ashby E. C, Lin J. J., Goel А. В., J. Org. Chem., 43, 183 (1978).
24. Fleet G. W. J., Fuller C. J., Harding P. J. C, Tetrahedron Lett., 1978,
1437.
25. Sorrell T. N., Spillane R. /., Tetrahedron Lett., 1978, 2473; Sorrell T. N.,
Pearlman P. S., J. Org. Chem., 45, 3449 (1980).
26. Four P., Guibe F., J. Org. Chem., 46, 4439 (1981).
27. Keinan E., Gleize P. Л., Tetrahedron Lett., 23, 477 (1982).
28. Ashby E. C, Lin J. /., J. Org. Chem., 43, 1263 (1978).
29. Ashby E. C, Lin J. /., J. Org. Chem., 43, 2567 (1978).
30. Fujisawa Т., Sugimoto K.t Ohta H., J. Org. Chem., 41, 1667 (1976).
31. Hutchins R. O., Learn /(., J. Org. Chem., 47, 4380 (1982).
32. Caubere P., Angew. Chem., Int. Ed. Engl., 22, 599 (1983).
33. Kinney R. J., Jones W. D., Bergman R. G., J. Am. Chem. Soc, 100, 7902
(1978).
14
ПРИМЕНЕНИЕ КОМПЛЕКСОВ, СОДЕРЖАЩИХ
<7-СВЯЗИ МЕТАЛЛ — УГЛЕРОД,
В ОРГАНИЧЕСКОМ СИНТЕЗЕ
Большой интерес для органического синтеза представляют
хорошо известные алкильные комплексы переходных металлов.
В ч. 1, разд. 3.5, а было показано, что химическая связь между
переходными металлами и алкильными лигандами не очень
прочная: для большинства алкильных комплексов переходных
металлов первого ряда она составляет около 30 ккал/моль, для
переходных металлов второго и третьего ряда — несколько
выше. Тем не менее гомолитический распад таких комплексов
наблюдается редко, более характерно для них элиминирование
^-водорода с образованием алкен-гидридных комплексов (ч. 1,
разд. 3.5, а и 6.3). Наблюдается также элиминирование а-водо-
рода с образованием алкилиден-гидридных комплексов (ч. 1,
разд. 6.2е), сопровождающееся иногда конкурентным
элиминированием ^-водорода (как в комплексе 59, ч. 1, разд. 3.5а).
Относительно устойчивы комплексы, в которых алкильные
группы не содержат ^-водорода, например СН3, CH2SiMe3.
В химических реакциях алкильные комплексы переходных
металлов редко проявляют карбанионный характер.
Реакционная способность алкильной группы определяется наличием ко-
валентной g-связи металл — углерод и в основном ограничена
теми реакциями, в результате которых открывается доступ к
лереходному металлу, например окислительное присоединение,
миграционное внедрение. Другими словами, переходный металл
не просто своеобразный противоион алкильной группы, а
главный детерминант ее химического поведения.
Известно несколько общих методов синтеза алкильных
комплексов переходных металлов и несколько классов реакций,
б которые они вступают. Схематически они представлены на
рис. 14.1. Благодаря разнообразию подходов практически любой
класс органических соединений может служить потенциальным
источником алкильной группы, а затем и целой
последовательности реакций, в которой участвуют алкильные а-металлоком-
плексы. Все эти подходы находят то или иное применение в
органическом синтезе; далее будут рассматриваться
соответствующие химические реакции.
Применение комплексов, содержащих а-связи М—С 161
(замещение) R(-) + MX
(замещение') М(-) + RX
(внедрение) М - r'+ RX
(внедрение) М-Н + RX
R М' + ИХ (переметаллирование)
R X + М (0) (окислительное
присоединение)
- \\ -*■ [Чис (нуклеооэильная
атака)
L + И + М (циклометалли-
робание)
(Пере- г
металлиродание) R Mv
R-C-.OMe
(внедрение)
R R' (сочетание)
R-Nuc
'(окислительное
расщепление)
R
(J3 - элиминиродание)
(бнедрение)
Рис. 14.1.
14.1. Алкилметаллы, получаемые из карбанионов и галогенидов
металлов; медьорганические комплексы
Среди производных переходных металлов, пригодных для
органического синтеза, чаще всего и охотнее химики используют
медьорганические комплексы, о чем свидетельствует появление
от 80 до 100 статей в этой области ежегодно. Столь
заслуженная популярность объясняется многими причинами, среди
которых не последнее место занимает легкость получения
реагентов и их разносторонние химические свойства. Кроме того, уже
со времен разработки катализируемых медью реакций Гринь-
яра более 30 лет назад [1] химия медьорганических соединений
легла в основу стандартных синтетических методов, поэтому
все новые достижения в этой области быстро находят
применение к решению синтетических проблем.
Учитывая интенсивность развития этой области металлоор-
ганической химии и огромное количество новых комплексов,
найденных в последние годы, мы ограничимся здесь
рассмотрением только основных положений. В табл. 14.1
классифицированы различные типы медьорганических комплексов,
используемых в синтезе [2, 3]. Большинство ранних работ в этой
области основывалось на наблюдении Хараша [4] о том, что в
присутствии каталитических количеств солей меди(1) реактивы
Гриньяра присоединяются к сопряженным енонам по положе-
11—1129
162 Глава 14
Таблица 14.1.
Типы медьорганических
реагентов
Общее наименование
RM+CuX (кат.)
RCu
RCu-Лиганд
R2CuM (M=Li, MgX)
ИгСиМ-Лиганд
RR'CuM
R(Z)CuM (Z = OR/, SR',
CN, Cl, Br)
RnCuLin-i (n>2)
RCu-BF3
RCu-BRs
Каталитическое медьорганическое соединение
Стехиометрическое медьорганическое соединение
Лигaнд=RзP, (RO)3P, R2S
Гомокупрат
Смешанный гомокупрат
Органо(гетеро)купрат, или гетерокупрат
Купраты высшего порядка
^Комплексы медьорганическое соединение — бор аи
ниям 1,4 [уравнение (14.1)]. Такой процесс представляет
препаративную ценность, но он довольно чувствителен к природе
используемой медной соли, наличию микропримесей в магнии
и (или) меди и даже к порядку добавления реагентов, поэтому
результаты часто варьируют. Хотя всегда предполагалось,
что в этих реакциях происходит промежуточное образование
медьорганических комплексов, но до середины 60-х годов не
было видвинуто убедительных подтверждений того, что стехио-
метрические медьорганические реагенты действительно
являются реакционноспособными частицами в катализируемых
медью реакциях Гриньяра. Однако, когда это стало понятным,
появилась возможность контролировать эти реакции и химия
медьорганических соединений получила бурное развитие,
большой вклад в которое внесли Кори, Хауз, Норман, Познер,
Уайтсайдс, а позднее — Эшби, Берц и Липшутц.
Наиболее изучены и широко применяются симметричные
органические купраты, образующиеся при взаимодействии двух
эквивалентов литийорганического соединения (или реактива
Гриньяра) с одним эквивалентом безводного иодида меди(1).
Такие комплексы оказались наилучшими реагентами для двух
типов превращений: алкилирования органических галогенидов
[5] и 1,4-алкилирования сопряженных енонов [6].
О О
RMgX+CH2=CHCCH3+CuX —> RCH2CH2CCH3
(ИЛ)
Органические галогениды алкилируются многими металло-
органическими реагентами, но процесс алкилирования часто
сопровождается конкурирующими реакциями, такими, как
обмен галоген — металл, элиминирование, сочетание с образова-
Применение комплексов, содержащих а-связи М—С 163
нием симметричных димеров. В отличие от этого некоторые
органические галогениды гладко алкилируются органокупрата-
ми, образуемыми in situ при взаимодействии двух эквивалентов
литийорганического комплекса с одним эквивалентом иодида
меди(1) [уравнение (14.2)]. В реакцию вступают самые
разнообразные комплексы меди; так, группа R может быть
первичной, вторичной или третичной алкильной, винильной, пропе-
нильной, арильной, гетероароматической. Можно получить и
диаллилкупраты, но они гладко реагируют только с алкилга-
логенидами и эпоксидами. Диалкинилкупраты не реагируют с
органическими галогенидами, тогда как простые ацетилиды
меди (RC=eeCCu) вступают в реакцию. В реакции с диалке-
нилкупратами сохраняется стереохимия двойной связи и
наблюдается стереоспецифическое сочетание. Эти органические
купраты значительно менее нуклеофильны, чем литийорганиче-
ские реагенты, они могут реагировать с галогенидами в
присутствии отдаленных функциональных групп, таких, как сложно-
эфирная, циано- или кетогруппа. Но если в субстрате имеется
альдегидная группа, она будет реагировать быстрее галогенида.
< О °С R'X
2RLi+CuX [R2Cu]Li —> RR' (14.2)
Реакция применима к широкому кругу галогенидов [R'X в
уравнении (14.2)]. Реакционная способность (sp3) алкилгало-
генидов уменьшается в ряду первичные > вторичные >
третичные, при этом иодиды намного реакционноспособнее бромидов
и хлоридов. Алкилтозилаты также подвергаются замещению
под действием диорганокупратов. Алкенилгалогениды очень ре-
акционноспособны по отношению к литийорганокупратам и
реагируют практически с полным сохранением конфигурации
двойной связи, что делает эту реакцию очень ценной для
органического синтеза. С помощью литийорганических купратов
можно алкилировать арилиодиды и арилбромиды, правда,
иногда при этом происходит также обмен галоген — металл, и
тогда лучше использовать реагенты типа RCu. Гладко
алкилируются также бензил-, аллил- и пропаргилгалогениды. В реакции
с пропаргилгалогенидами преимущественно образуются аллены
(по реакции типа Sn2'), однако аллилхлориды и аллилбромиды
реагируют без аллильной перегруппировки. В
противоположность им аллилацетаты реагируют в основном с аллильной
перегруппировкой. Галогенангидриды кислот превращаются в
кетоны под действием литиидиалкилкупратов, но более
эффективны для этой цели смешанные алкилгетерокупраты. Наконец,
эпоксиды претерпевают раскрытие цикла в результате алкили-
рования по менее замещенному атому углерода. Рассмотрение
множества экспериментальных данных приводит к выводу о
11*
164 Глава 14
следующем порядке реакционной способности литийдиоргано-
купратов по отношению к органическим субстратам: хлорангид-
риды кислот > альдегиды > эпоксиды, тозилаты > иодиды >
> кетоны > сложные эфиры > нитрилы. Конечно, имеется
много отклонений от этого обобщенного ряда, связанных с теми
или иными особенностями либо медьорганического реагента,
либо субстрата.
Как обычно бывает в химии металлоорганических
соединений, изучение механизма реакции сильно отстало от
синтетических исследований литийдиорганокупратов, и еще не выяснены
многие противоречия как в природе медьсодержащих
реагентов, так и в механизмах, по которым осуществляется алкили-
рование. Хотя для удобства обозначения стехиометрии реагента
его записывают формулой R2CuLi, она не указывает структуру
реагента в растворе. Исследования Пирсона [7], включающие
данные по понижению давления паров, показали, что в
растворе реагент «R2CuLi» существует в димерной форме. Величины
межатомных расстояний Си — Си, установленные по данным
рассеяния рентгеновских лучей, лучше всего согласуются с
циклической структурой 1, в которой Си и Li чередуются с
метальными группами. С помощью 71л-ЯМР-спектров Липшутц
сн3-си-сн3
U U
сн3- си-сн3
1
[8] показал, что «Me2CuLi» существует не в виде
индивидуального комплекса, а в виде равновесной смеси трех отдельных
частиц [уравнение (14.3)], что еще более усложняет вопрос.
к
[Me2CuLi]2 *=* MeLi+Me3Cu2Li (14.3)
Результаты проведенного Джонсоном [9] тщательного
изучения реакции литийдиорганокупратов с алкилтозилатами
свидетельствуют о том, что реакция идет по механизму типа SN2.
Это реакция второго порядка (скорость = k2 [RX]1[LiMe2Cu]1,
&2(Et2O) = 2,8-10-3 М-1 с"1 при — 42 °С), реакционная
способность субстрата уменьшается в ряду: первичный > вторичный >
> неопентил (тозилат); OTs > I > Вг > С1; все это согласуется
с лимитирующей стадией SN2. Примечателен тот факт, что
тозилат даже более реакционноспособен, чем иодид. Подобный
результат нельзя было бы ожидать, если бы процесс был
радикальным. В случае оптически активних вторичных тозилатов
Применение комплексов, содержащих о-связи М—С 165
наблюдается четкая инверсия конфигурации. Конечно, процесс
может осуществляться по простому механизму SN2, но тогда
трудно объяснить роль меди [уравнение (14.4)]. В качестве
альтернативы предлагалось окислительное присоединение типа
Sn2, т. е. нуклеофильная атака атомом Си [уравнение (14.5)].
R-Cu(l)-R + ^С-Х — R-C^ ♦ RCu(l) (Ц4)
R-Cu(l)-R + -C-X +- R-Cu-R + X" R-C- + RCu(l)X (!4 5)
v^v c
Главное возражение против такого пути реакции вызывает
обязательное участие в нем частиц Си (III)*. Как уже говорилось
в конце разд. 2.1, такие реакционноспособные интермедиа™
должны иметь Т-образную геометрию (трехкоординированная,
d8), что могло бы объяснить селективное кросс-сочетание.
Однако этот момент остается противоречивым. Альтернативный
механизм предусматривает окислительное присоединение типа
SN2 к интактному димеру [уравнение (14.6)]. В этой схеме
каждый атом Си димера отдает только один электрон на
окислительное присоединение, в результате чего образуются два
центра Си (II), а не один Си(III). В образующемся при
окислительном присоединении димерном аддукте алкильные группы
медьорганического реагента отличны от алкильных групп
субстрата, и поэтому наблюдается селективное кросс-сочетание.
? (14 6)
СН3"Си(1)-СН3 CH3-Cu(ll)-CH3
Li Li + RX Li L(' - RCH3 + [Li2
CH3-Cu(l)-CH3 CH3-Cu(!!)-CH3
\ CH3Ll
LiX
Механизм взаимодействия комплексов R2CuLi с винил- и
арилгалогенидами подробно не изучался [10], но очевидно, что
прямые реакции Sn2 маловероятны. Однако вполне возможно,
что механизм окислительного присоединения аналогичен
таковому в реакциях изоэлектронных комплексов Pd(0) (ч. 1, гл. 5).
Несмотря на неясность механизма, литиидиорганокупраты
широко используются для алкилирования сложных молекул
166 Глава 14
[11 —15]. Характерные примеры представлены уравнениями
(14.7) —(14.11).
(14.7)
У ОАс
NH
Me2CuLf
К Me
ОН
он
таларо-
мицин В
(14.8)
через \ к - присоединение с найме -
q/ нее затрудненной
crrwpoH6f
МеО. ОМе
MeO ОМе
CuLr
(14.9)
МеО2С
МеО^С
С02Ме
OTs
ОТНР
МеО2С
_^ МеО2С
(14.10)
СО2Ме
с4н9
ОТНР
л-с8я1
ЛГ-С3Н17.
т,иральныи
сн3
хирсигьньш
сн3 тн сн3
2R,3R,7R
Применение комплексов, содержащих g-связи М—С 167
Для синтеза сложных органических молекул широко
используется также процесс сопряженного присоединения литийдиор-
ганокупратов к сопряженным енонам [уравнение (14.12)];
группы R здесь те же, что и в реакциях алкилирования. В реакцию
вступают практически все типы диалкилкупратов с sp3-ni6pH~
дизованными группами, как с прямой цепью, так и
разветвленными первичными, вторичными и третичными алкильными
заместителями. Аналогичным образом реагируют винилкупраты,
фенилкупраты и замещенные арилкупраты. Аллильные и бен-
зильние производные тоже присоединяются к сопряженным
енонам в положения 1,4, но они не более эффективны, чем
соответствующие реактивы Гриньяра. В случае ацетиленовых
комплексов меди алкиновая группа не переносится к
сопряженному енону; эту особенность можно выгодно использовать при
работе со смешанными купратами.
О О
II II
R2CuLi+CH2=CHCCH3 —>■ RCH2CH2CCH3 (14.12)
Под действием диорганокупратов самые разнообразные
а,(5-ненасыщенные карбонильные соединения подвергаются
1,4-присоединению. Реакционная способность каждой
конкретной системы зависит от многих факторов, в том числе от
строения как комплекса меди, так и субстрата, но тем не менее
имеется достаточное количество данных, позволяющих сделать
некоторые обобщения. Наиболее реакционноспособными
субстратами оказались сопряженные кетоны: при 25 °С реакция
завершается менее чем за 0,1 с и приводит к образованию
1,4-аддуктов с прекрасным выходом. Алкильные заместители в
а-, а'- или ^-положениях енона почти не меняют скорость и
направление реакции сопряженного присоединения в случае
ациклических енонов, однако те же группы и удаленные
заместители в циклических енонах вызывают изменение
стереохимии системы. Например, в случае 4-замещенных циклогексе-
нонов преимущественно образуется траяс-аддукт, причем
количество его возрастает с увеличением пространственного объема
заместителя в положении 4 [уравнение (14.13)]. Алкилирование
1-метилокталона приводит к ^wc-продукту [уравнение (14.14)].
о о
Me2CuLi
(14.13)
Me
R = Me
RsU3O-Pr
R
72
89
Me
1
R
28
II
168 Глава 14
ОТНР
Me
ОТНР
(14.14)
Me
79%
Сопряженные сложные эфиры несколько менее реакционно-
способны по сравнению с сопряженными кетонами.
Реакционная способность а,р- и р,р-дизамещенных сложных эфиров
резко понижается, что делает эти субстраты мало пригодными для
синтетических целей. Сопряженные карбоновые кислоты не
реагируют с литийдиорганокупратами, а с сопряженными
альдегидами наблюдается конкурентное 1,2-присоединение. Реакции
сопряженных ангидридов и амидов не изучались,
Ацетиленовые сложные эфиры значительно реакционноспо-
собнее по отношению к диорганокупратам, чем соответствующие
этиленовые эфиры, реакция находит применение в синтезе.
Считается, что она происходит через винилмедный интермедиат,
образующийся в результате стереоспецифического ^wc-присоеди-
нения купрата. Дальнейшее взаимодействие этого комплекса с
различными электрофилами [уравнение (14.15)] позволяет сте-
реоспецифически синтезировать тризамещенные олефины.
Аналогичные реакции наблюдаются с ацетиленовыми кетонами,
кислотами, амидами, но они менее изучены [16].
(14 15)-
О
RCrCCOR1
4 V-c
Ru
Си
R"
R
R"
OR1
OR1
OR1
Существуют довольно разные мнения о специфике этой
реакции, поскольку изучение механизма, как это часто бывает,
значительно отстает от синтетических исследований. К 1985 г.
были сформулированы две основние точки зрения. Согласно
первой, сопряженное присоединение происходит либо путем
переноса электрона, либо путем прямого нуклеофильного
присоединения [17]; в то же время на основании данных 13С-ЯМР-
Применение комплексов, содержащих а-связи М—С 169
спектров утверждалось, что реакция идет через предварительное
комплексообразование енона с медью [18].
В пользу механизма прямого нуклеофильного присоединения
говорит тот факт, что органические купраты способны
действовать как нуклеофилы. Достаточно вспомнить, что и алкилгало-
гениды, и алкилтозилаты, и эпоксиды реагируют с
органическими купратами с обращением стереохимии, характерным для
прямого нуклеофильного замещения. С этой точки зрения
сопряженное присоединение органических купратов к енонам
подобно известному присоединению нуклеофилов по Михаэлю.
В то же время множество экспериментальных результатов
(и в первую очередь работы Хауза) согласуются с механизмом,
включающим перенос электрона [19]. Прежде всего имеется
строгая корреляция между реакционной способностью
сопряженных енонов в реакциях сопряженного присоединения и их
полярографическими потенциалами одноэлектронного
восстановления. Так, в случае легко восстанавливающихся енонов,
потенциалы восстановления которых не превышают —1,5 В,
наблюдается не алкилирование, а восстановление двойной
связи (чисто двухэлектронный процесс). Еноны, имеющие
потенциалы восстановления от —1,5 до —2,35 В, претерпевают
сопряженное присоединение, а еноны, еще более трудно
поддающиеся восстановлению (с потенциалами восстановления более
—2,35 В), не вступают в реакцию. Противники концепции
электронного переноса замечают, что потенциал восстановления
служит просто мерой электронного сродства енона и должен
коррелировать как с процессом одноэлектронного переноса,
так и с двухэлектронным процессом нуклеофильного
присоединения. Сторонники концепции электронного переноса
возражают, говоря, что другие реакции присоединения по Михаэлю
к сопряженным енонам не обнаруживают очевидной корреляции
с потенциалом восстановления. Диспут продолжается.
Хотя механизм сопряженного присоединения остается пред-
[l (СН2)6С00Ме
LiCu
RO
(14.16)
J2
ч(СН2)6СООМе
OSiFb
170 Глава 14
метом дискуссий, применение этой реакции в синтезе
продолжает расширяться. Так, на сопряженном присоединении
построен синтез простаноидов [20] [уравнение (14.16)]. Следует
отметить, что реакция идет в присутствии множества
функциональных групп как в субстрате, так и в медьорганическом комплексе.
Сопряженное присоединение органических купратов
использовалось как ключевая стадия в синтезе многих природных
соединений. С хиральными субстратами наблюдается
существенная асимметрическая индукция [уравнение (14.17)] [21].
ОМе О'
-70°С
(14.17)
диастереоселектибкость > 10:1
В случае взаимодействия ненасыщенных альдегидов с
реагентами типа R2CuLi возникает проблема конкурентного 1,2-при-
соединения. Эшби [22] разработал и изучил большое число
купратов «высшего порядка», включая Me3CuLi3, Me5Cu3Li2 и
Me3Cu2Li. Клив [23] показал, что среди этих комплексов
Ме5Сщ1л2 — наилучший реагент для сопряженного
присоединения к ^-ненасыщенным альдегидам.
Первоначальный аддукт, образующийся до тушения водой,
представляет собой енолят меди или лития [24], который может
вступать в дальнейшую реакцию с электрофилами, что было
выгодно использовано в ряде синтезов. В частности, подобным
образом было приготовлено модельное соединение дезоксипро-
стагландина Е2 [25] [уравнение (14.18)]. Впечатляющим
примером такого процесса ^-присоединения — сс-алкилирования
служит осуществленный Познером четырехстадийный синтез экви-
ленина [26] [уравнение (14.19)]. Эту реакцию с участием
комплекса меди проводят в одном сосуде, она практически
стереоспецифична и дает высокий выход продукта. Омыление
сложного эфира, защита кетогруппы и катализируемое кислотой
замыкание цикла завершают синтез.
Литийдиалкилкупраты — очень ценные реагенты, однако и
они имеют ряд серьезных недостатков, прежде всего тот, что
из двух алкильных групп только одна переходит в продукт.
Из-за неустойчивости реагент приходится вводить в реакцию в
трех-пятикратном избытке (т. е. с избытком групп R от шести-
до десятикратного), что снижает эффективность процесса,
особенно в случае объемных и сложных групп, как, например,
в химии простагландинов. Алкоксиды, меркаптиды, цианиды и
Применение комплексов, содержащих о-связи М—С 171
Me(CH^=CH)CuLi
г- О
(СН2)зСООМе
(14.18)
(СН?)3СООМе
OMe
2.
ГМФТА
MeO
Et0oC
95%
!. Н20, 0Н~
2. НО ОН, Н*
3. HF
МеО
(14.19)
Общий Выасод 52 %
ацетилиды меди(1) значительно устойчивее алкильных
производных; кроме того, они гораздо менее реакционноспособны по
отношению к тем субстратам, которые обычно используются в
реакциях с диалкилкупратами. Познер [27] разработал
смешанные алкилгетерокупраты, в которых одна группа (алкоксид,
меркаптид, цианид или ацетилид) не переносится в продукт,
стабилизируя реагент, а другая является желаемым алкилом,
что по существу решило проблему. Среди гетерокупратов
наиболее ценными оказались литийфенилтио(алкил)купраты и ли-
тий-трег-бутоксиалкилкупраты, устойчивые при температурах
от —20 до 0°С.
Эти комплексы — наиболее эффективные реагенты для
превращения хлорангидридов кислот в кетоны. При проведении
этой реакции с литийдиалкилкупратами требуется большой
избыток реагента, к тому же вторичные и третичные органокупраты
реагируют очень плохо. При проведении реакции со
смешанными купратами достаточно 10%-ного избытка реагента, чтобы
получить кетон с высоким выходом даже в присутствии
удаленных заместителей, таких, как галоген, кетогруппа или сложно-
эфирная группа. Несмотря на то что альдегиды неустойчивы к
действию литийдиалкилкупратов даже при —90 °С, взаимодей-
172 Глава 14
ствием равных количеств бензальдегида и бензоилхлорида с
одним эквивалентом литийфенилтио-(грет-бутил) купрата
удается получить пивалофенон с выходом 90% и регенерировать 73%
исходного бензальдегида. Литийфенилтио- (трег-алкил) купрат
оказался также лучшим реагентом для алкилирования
первичных алкилгалогенидов грвг-алкильными группами. Однако
вторичные и третичные галогениды в реакцию не вступают.
Возможно, самым ценным преимуществом смешанных куп-
ратов, в частности ацетилидов, является их исключительная
эффективность в реакциях сопряженного алкилирования а,р-не-
насыщенных кетонов. Поскольку этот процесс находит широкое
применение в синтезе сложных молекул, реакция со
смешанными купратами стала с успехом использоваться для полного
синтеза природных соединений, включая простагландины [28, 29]
[уравнение (14.20)].
О О'
Ph
О
il (CH2)6C02R
f\S HO
\—•! (таксисеряды Е, A, F2cJ
но
На основе дифенилфосфида (Ph2P) и дициклогексиламида
(Cy2N) в качестве непереносимых лигандов Берц [30, 31]
синтезировал смешанные купраты, отличающиеся еще большей
термической устойчивостью. Эти реагенты общей формулы
RCu(PPh2)Li и RCu(NCy2)Li устойчивы при 25 °С более часа
и достаточно реакционноспособны, так как вступают во все
типичные для органокупратов реакции.
Липшутц разработал большое число смешанных купратов
«высшего порядка», которые не только обладают повышенной
по сравнению с простыми органокупратами термической
устойчивостью, но и отличаются несколько иной реакционной
способностью. Наиболее изучен из этой серии комплекс [R2CuCN]Li2,
который получают реакцией цианида меди(1) с двумя
эквивалентами литийорганического соединения. Эти реагенты особенно
эффективны для алкилирования неактивированных вторичных
бромидов и иодидов [32, 33], которые при взаимодействии с
простыми купратами типа R2CuLi претерпевают не замещение,
а восстановление и (или) элиминирование. Как и следовало
Применение комплексов, содержащих о-связи М—С 173
R" OR ♦ [r'2CuCn]l12 ^ J^ (14 21)
R1 R"
R--Me, El, тпрет-Ъхх. R Si
^ J 3 60-90%
R'=tf-Bu, Ph, н-Рг, ^^/
ожидать, хиральные вторичные бромиды алкилируются с четкой
инверсией, но вторичные иодиды алкилируются с полной
рацемизацией под действием как (R2CuCN)Li2, так и R2CuLi [34].
Смешанные купраты высшего порядка эффективны также для
1,4-присоединения к сопряженным кетонам [35] и сложным
эфирам [36], но а,р-дизамещение енона не происходит
[уравнение (14.21)]
Разработано много других смешанных купратов, пригодных
для синтетических целей. Марино [37] изучил реакции аллил-
эпоксидов с цианокупратами и использовал их в синтезе про-
стагландинов [уравнение (14.22)]. Следует отметить, что
алкион (14 22)
0>. +
^с5нп + [tMso(ch2)7cucn]u
PGA
PGB
лирование происходит со стороны, противоположной эпоксиду,
что типично для процессов Sn2'. Медьорганические комплексы
с кислотами Льюиса RCu-BF3 особенно эффективны при 1,4-
алкилировании сопряженных енонов [38], в том числе и стери-
чески затрудненных. Под действием этих реагентов
осуществляется гладкое 1,4-алкилирование а,р-ненасыщенных кетонов,
сложных эфиров и даже некоторых карбоновых кислот. Опполь-
цер [39] изучал взаимодействие этих реагентов с хиральными
а,р-ненасыщенными сложными эфирами и наблюдал высокую
174 Глава 14
оптическую индукцию в ^-положении [уравнение (14.23)]. Как
показал Норман [40], реагенты этого класса превращают аце-
тали и кетали в эфиры, формально процесс представляет собой
замещение алкоксигруппы алкилом [уравнение (14.24)]. Хотя
механизм реакции не известен, можно предположить, что
замещению обычно нереакционноспособной алкоксигруппы
способствует комплексообразование с кислотой Льюиса,
о о
MeCuBF3-L I
^ (14.23)
Me
Н*-а:иральная кажсрора
L = CN ", РВи3
V
0R' £^L X
OR1 • OR'
Наиболее широко используемыми медьорганическими
реагентами являются диалкил- и алкилгетерокупраты, однако
простые комплексы типа RCu или RCuMX вступают во многие
такие же реакции, а в некоторых типах реакций обнаруживают
определенные преимущества, в частности в реакции
присоединения RCuMgX2 к алкинам с концевой тройной связью. При
действии R2CuLi на такие субстраты чаще всего отрывается
кислый ацетиленовый протон, комплексы RCu или RCuLiX
вообще не присоединяются, тогда как комплексы RCuMgX2,
генерируемые из реактивов Гриньяра и иодида меди(1),
претерпевают гладкое гая-присоединение к простым алкинам с концевой
тройной связью, давая винилкупраты; реакция регио- и стерео-
специфична. Следует отметить, что при этом кислая связь С—Н
не затрагивается. Винильные комплексы проявляют
реакционную способность, характерную для всех медьорганических
соединений, и реагируют с самыми разнообразными органическими
4 HZ R3
•CICH2SFT
2 3-
R2CuLi + R2-=-R3 *" )—( ] CuL! (14.25)
R Ж
Применение комплексов, содержащих о-связи М—С 175
R' H
-И
R"X
RCuMqX2
CuMgX2
С02
R' Н
\
ft"
/ \
R D
Rf
R COOH
Рис. 14.2.
/ \
субстратами (рис. 14.2) [41]. Ацетилены с внутренней тройной
связью гладко взаимодействуют с диалкилкупратами R2CuLi,
давая тризамещенные винилкупраты. В реакциях с
электрофилами образуются тетразамещенные олефины [уравнение (14.25)]
[42].
При наличии в алкинах гетероатомных заместителей регио-
химия реакции несколько усложняется и, по-видимому,
контролируется как полярностью субстрата [41], так и способностью
гетероатома координироваться с атомами меди и поставлять
его к определенному центру [43] [уравнение (14.26)]. Алкины
с хорошими уходящими группами в пропаргильном положении
+• R'C=CZ
R1 Z
А
R1 Z
В
(14.26)
Для Z«RS[44], RS0[4^ , CN [4б] , PPh2r COO", не образуется A (R'-C = C-Z)
Для Z-0Et[44], NEt2 [44], NPh2 [4 з]; не образуется Б (R-C=C-Z)
подвергаются замещению типа Sn2', давая соответствующие
аллены [47, 48]. Хиральные пропаргилсульфинаты дают хираль-
176 Глава 14
ные аллены с прекрасными выходами [49] [уравнение (14.27)].
(RCuBr)MgXLiBr ♦ НС = С-С-^Н - C=C=c" (14,27)
OSOMe
оптический Выход 88°/о>
быход 90 %
Подобное «карбокуприрование» ненасыщенных
углеводородов служит примером общего метода синтеза о-алкильных
комплексов переходных металлов, т. е. «гидрометаллирования»
или «карбометаллирования» алкенов и алкинов. Присоединение
гидридов металлов или алкилметаллов к ненасыщенным
субстратам находит все большее применение в органическом
синтезе; этому вопросу посвящен следующий раздел.
14.2. Получение алкилметаллов присоединением гидридов
металлов или алкилметаллов к алкенам и алкинам
«Карбокуприрование» алкинов, рассмотренное в
предыдущем разделе, служит частным примером более общего подхода
к синтезу о-алкильных комплексов переходных металлов,
а именно присоединения гидридов металлов или алкилметаллов
главных групп к ненасыщенным субстратам [50]. Возможность
генерировать о-алкильные металлокомплексы реакцией гидрида
переходного металла с простыми неактивированными олефина-
ми или алкинами [уравнение (14.28)] представляется
исключительно полезной. Она открывает путь к прямому введению
функциональных групп в ненасыщенные субстраты, которое
невозможно осуществить традиционными методами синтеза. Чтобы
достичь этого, необходимо сдвинуть равновесие в уравнении
(14.28) далеко вправо и тем самым добиться существенного
преобладания о-алкильного металлоорганического соединения
над гидридным олефиновым комплексом. Другой путь состоит
в том, чтобы подобрать реагент, который быстро и селективно
взаимодействует с о-алкильным металлокомплексом и не
затрагивает гидридный олефиновый комплекс. Последний путь чаще
= м—И
м-н + ^± и ^± *■ дальнейшая (14-28)
реакция
м-
н
кг
используется в каталитическом гидрировании олефинов и
алкинов, хотя в этом процессе с-алкильные металлокомплексы
обычно не обнаруживаются. Однако с реагентами, отличными от
Применение комплексов, содержащих о-связи М—С 177
водорода, этот путь не подходит. Попытки ввести
функциональную группу в олефины с использованием типичных
катализаторов гидрирования, таких, как Rh(Cl) (РРЬ3)з, оказались
безуспешными. В случае гидридных комплексов переходных
металлов с низкой формальной степенью окисления равновесие в
уравнении (14.28) сдвинуто далеко влево, по-видимому, из-за
того, что олефиновые (я-акцепторные) комплексы обогащенных
электронами металлов намного устойчивее соответствующих
алкильных (о-донорных) комплексов. Отсюда следует, что
электронно обедненные переходные металлы с формально
высокой степенью окисления должны благоприятствовать
образованию о-алкильных комплексов. Это подтверждается на
примере многих переходных металлов. Наиболее изучен (Шварцем)
с1°-комплекс циркония (IV): (r]5-C5H5)2Zr (Н)С1, который
реагирует с алкенами и алкинами, давая устойчивые алкильные
комплексы типа (т]5-С5Н5^г (R)C1, выделяемые из
реакционной смеси.
Рассмотренная реакция примечательна во многих отношениях.
Присоединение идет в мягких условиях, образующиеся
алкильные комплексы вполне устойчивы. Независимо от положения
двойной связи в исходном олефине цирконий присоединяется к
наименее стерически затрудненному положению углеводородной
цепи [уравнение (14.29)] [51]. Перемещение циркония к
концевому положению происходит путем элиминирования Zr—Н с
последующим повторным присоединением к менее
затрудненному в каждом случае положению алкильной цепи. Такая
миграция объясняется стерическими перегрузками, вызываемыми
наличием двух циклопентадиенильных колец вокруг Zr.
Реакционная способность различных олефинов по отношению к гид-
роцирконированию уменьшается в ряду: олефины с концевой
двойной связью > ^ас-олефины с внутренней двойной связью >
>граяс-олефины с внутренней двойной связью>экзоцикличе-
ские олефины > циклические олефины > дизамещенные
олефины > тризамещенные олефины. Тетразамещенные олефины с
открытой цепью и тризамещенные циклические олефины не
вступают в реакцию. К 1,3-диенам Zr—Н присоединяется по
менее затрудненной двойной связи, давая ^-ненасыщенные
алкилциркониевые комплексы.
(14.29)
12—1129
178 Глава 14
Алкины также вступают в реакцию с
при этом происходит цис-присоединение Zr—H. С
несимметричными алкинами получаются смеси винилциркониевых
комплексов, в которых преобладает менее затрудненный изомер. Его
концентрацию можно повысить с помощью небольшого избытка
Cp2Zr(H)Cl [уравнение (14.30)].
Cp22fC/"R Cp2Zr'T^R1 (14,30)
Cp«Zr(H)CI ♦ RC?CR* ** jj v N Tl
HAR' H^-R
N - Вромосукццнимид
Br,
Интерес к данной реакции связан с тем большим значением,
которое имеют превращение связи углерод — цирконий, да и
других связей углерод — металл. Например, расщепление связи
Zr — углерод N-бромосукцинимидом приводит к
соответствующему винилгалогениду с сохранением геометрии олефина [52].
Протонированием можно получить углеводороды, а бромиро-
ванием алкилбромиды [уравнение (14.31)] [53] Окисление дает
(14.31)
Cp2Zr(H)CI +
i
Вг2
Вг"
спирты подобно тому, как это происходит в процессе гидробо-
рирования/окисления. Гибсон [54] использовал
последовательность гидроцирконирование — окисление для превращения
длинноцепных олефинов с внутренней двойной связью,
образующихся при метатезисе олефинов (ч. 1, разд. 9.2) в предельные
спирты с концевой функциональной группой [уравнение (14.32)].
1) Cp2Zr(H)Cl
С14Н29СН = СНС14Н29 ^ СН3(СН2)29ОН (14.32)
2) mpem-BuOOH триконтанОЛ
Применение комплексов, содержащих а-связи М—С 1791
Представляет интерес механизм электрофильного
расщепления алкильных комплексов переходных металлов. Здесь часто
наблюдается инверсия конфигурации при атоме углерода.
Считается, что расщепление идет через окисление металла электро-
филом и последующее замещение металла по реакции SN2
(ч. 1, разд. 8.1) [55]. Однако в комплексе Cp2Zr(R)Cl цирконий
является металлом d°, и его дальнейшее окисление
затруднительно. Так что в данной системе такой механизм невозможен.
Было показано, что расщепление связи Zr—С бромом
происходит с сохранением конфигурации при атоме углерода [ч. 1, гл 8,.
уравнение (8.14)], что подтверждает предположение об
изменении в механизме [56].
Внедрение СО в комплексы Cp2Zr(R)Cl идет в очень мягких
условиях (25 °С, 1,4 атм) и приводит к соответствующим ациль-
ным комплексам с прекрасным выходом. Ацильные комплексы
реагируют с разбавленной соляной кислотой, при этом с
высоким выходом образуются альдегиды. В целом эти реакции
соответствуют «гидроформилированию» олефина, хорошо
известному каталитическому процессу, обсуждавшемуся в гл. 12..
Однако в отличие от каталитической системы в данном
процессе [уравнение (14.33)] образуются исключительно альдегиды с
концевой функциональной группой независимо от строения
исходного олефина [57]. Особый интерес представляет применение
этой реакции «гидроацилирования» к 1,3-диенам, из которых с
хорошим выходом получаются ^-ненасыщенные альдегиды
[58], как это показано в уравнении (14.34) (вторая двойная
связь, очевидно, не подвергается перегруппировке!).
В условиях окисления ацилциркониевые комплексы можно
(14.33>
Cp2Zr(H)CI
О
сно
(14.34)
ClZrCpo
/. V • 1_IJ Г. \ '
Cp2Zr(H)CI
12*
180 Глава 14
расщепить прямо до производных карбоновых кислот. Так,
в присутствии водного Н2О2 образуются карбоновые кислоты,
в присутствии Вг2 в метаноле — сложные метиловые эфиры,
а под действием N-бромосукцинимида получаются бромангид-
риды кислот. Окисление ал/шлциркониевых комплексов
различными окисляющими агентами, включая Н2О2/Н2О, трет-бутил-
гидропероксид и ж-хлоропербензойную кислоту, приводит к
соответствующим спиртам с умеренным выходом [59]. Негиси
показал, что циркониевые комплексы катализируют также
реакцию алюминийалкилов с алкинами, в результате чего
образуются винилаланы. В случае стерически затрудненных алкил-
алюминиевых субстратов, например триизобутилалюминия,
наблюдается гадроалюминирование. Конечным продуктом такой
реакции является алкилалюминиевое соединение, однако
контроль за ходом реакции с помощью ЯМР-спектров указывает на
наличие алкилциркониевых интермедиатов [уравнение (14.35)]
[60].
(14,35)
Реакция триметилалюминия с алкинами также
катализируется Cp2ZrCl2 и приводит к региоспецифическому и стереоспеци-
фическому образованию винилаланов, которые, будучи сами
довольно реакционноспособными соединениями, открывают путь
к получению разнообразных тризамещенных олефинов с
высокой степенью регио- и стереоселективности [уравнение (14.36)]
[61]. Таким способом были получены с прекрасными выходами
терпены гераниол и этилгеранат [62] [уравнение (14.37)].
Изучение механизма катализируемого цирконием карбоалю-
минирования пока не привело к окончательным выводам.
Негиси [63] показал, что реакция идет путем прямого присоединения
алюминийалшла к алкину, а не путем присоединения цирконий-
алкила, образующегося переметаллированием, однако не ясно,
как же цирконий катализирует этот процесс без участия алкил-
циркониевого реагента. Тем не менее процесс представляет
Применение комплексов, содержащих q-связи М—С 181
RC = CH + Ме3Д|
Cp2ZrCI2 \/ BuLi \ / f14"36'
Me AIMe2 Me AIMe^Bu
C02
CICOOEt/
R H R H R H
Me COOEt Me CH2OH Me COOH
,CH20H
(14.37)
COOEt
ценность для синтеза и в сочетании с процессами
окислительного присоединения и переметаллирования, которые
обсуждаются далее, дает удобный метод включения исходной
ацетиленовой компоненты в сложные молекулы.
14.3. Получение алкилметаллов переметаллированием
и внедрением
Алкилциркониевые соединения очень устойчивы; с одной
стороны, это позволяет легко их получать, но, с другой стороны,
это ограничивает их реакционноспособность и, следовательно,
их применение для дальнейших превращений. Изящное решение
проблемы было найдено с помощью реакции
«переметаллирования» цирконийалкилов, т. е. обмена циркония на другой
металл, обладающий большей реакционной способностью в
нужной реакции. В принципе это позволяет активировать олефины
путем гидроцирконирования и перенести активную алкильную
группу к другому металлу, чтобы использовать присущую ему
реакционную способность. Переметаллирование (перенос орга-
182 Глава 14
нической группы от одного металла к
другому)—общеизвестный процесс, описываемый уравнением (14.38) и уже
обсуждавшийся в ч. 1, разд. 3.5, а. Чтобы реакция шла преимущественно
в прямом направлении, необходимо, чтобы М был более
электроположительным, чем М' [64]. Но поскольку процесс
равновесный, даже в тех случаях, когда М — менее
электроположительный металл, чем М', переметаллирование может в
некоторой степени происходить. Необратимое расходование RM/ будет
сдвигать равновесие вправо и тем самым позволит использовать
процесс переметаллирования для дальнейших превращений,
которым равновесие не благоприятствует.
RM+M'X *=«= RM'+MX (14.38)
Вернемся к химии циркония. Комплексы Cp2Zr(R)Cl хотя и
реагируют с галогеноангидридами кислот с образованием кето-
нов, но реакция идет медленно и с низкими выходами. Однако
при добавлении А1С13 происходит перенос алкильной группы
от Zr к А1 и образующийся алкилалюминиевый комплекс
быстро взаимодействует с различными галогеноангидридами кислот,
давая кетоны с прекрасными выходами [65] [уравнение (14.39)].
\ С = ССН3 + Cp2Zr(H)CI
(14 39)
AlClo
CH3COCI
Поскольку конкретные алкилалюминиевые соединения
недоступны непосредственно из олефина алюминийгидрида, такой
комбинированный процесс позволяет каждому металлу проявить
присущую ему реакционную способность и осуществить
химические превращения, невозможные при использовании только
одного металлоорганического комплекса.
Цирконий способен переносить алкильную группу и к
переходным металлам нижних рядов, что позволяет сочетать
реакции цирконийорганических соединений с реакциями внедрения
(рассматриваемыми далее в этом разделе) и с реакциями
окислительного присоединения (см. следующий раздел) этих элек-
тронообогащенных металлов. Например, при гидроциркониро-
вании алкинов с концевой тройной связью образуются винил-
Применение комплексов, содержащих g-связи М—С 183
диркониевые соединения, не реагирующие с сопряженными
^нонами. Однако легкое переметаллирование с никелем
приводит к винилникелевым соединениям, вступающим с енонами
з реакцию Михаэля с образованием енолятов никеля (или цир-
хония), которые затем алкилируются электрофилами [66].
_Цварц [67] использовал эту последовательность в синтезе про-
ггагландинов [уравнение (14.40) ].
{14,40)
OSiR,
I) СН2О
2) Н30+
OS1R3
О
чсн2он
OR1
R'Cf
C5HM
(68%)
OSiR,
(11%)
Наиболее широко используется процесс переметаллирования
зт электрофильных металлов, ртути и таллия, к металлам VIII
группы, особенно к палладию (II) [68]. Это объясняется
легкостью процессов меркурирования и таллирования ненасыщенных
органических соединений и хорошо разработанной химией
(о-алкил) палладиевых комплексов.
Впервые этот процесс был использован Хеком [69] в синтезе
симметричных диарилов и заключался в сочетании арилмеркур-
галогенидов в присутствии палладия (II). Считается, что эта
реакция [уравнение (14.41)] включает переметаллирование от
ArHgX+PdX2 —> ArPdX+HgX2
ArHgX
Ar2Pd+HgX2
Ar2+Pd(0)
(14.41)
ртути к палладию с последующим восстановительным
элиминированием двух арильных групп. Ларок [70—72] применил эту
реакцию к винилмеркургалогенидам, которые легко получить из
соответствующего алкина путем гидроборирования —
меркурирования. В зависимости от условий реакции можно получить
продукты димеризации либо «голова к хвосту», либо «голова к
голове» [уравнение (14.42)] (механизм димеризации «голова
к хвосту» неясен).
184 Глава 14
(R1)
R .H(R')
H
R"oBH ♦ Hg(0Ac)2
NaCI
H(R')
(14.42)
H(R')
H /=
(R')H
ГМФТА
PhH
H HgCI
H(R')
Очень ценный с синтетической точки зрения процесс,
первоначально разработанный Хеком, основан на последовательности
переметаллирование — внедрение олефина [уравнение (14.43)].
CH2=CHY
ArHgX+PdX2 —> ArPdX+HgX2 ►•
-PdHX
►■ AiCH2—CHY >■ ArCH=CHY + Pd(0) +HX (14.43)
PdX
Эти реакции нашли широкое применение в химии нуклеозидов,
поскольку пиримидиновое кольцо легко меркурируется в
положение 5 [73, 74]. Так, Бергстром получил с неплохим выходом
ряд 5-замещенных нуклеозидов, которые обычно трудно
синтезировать другими способами [уравнение (14.44)]. Дэйвс
показал, что даже электронообогащенные олефины, такие, как
дигидропиран и другие эфиры енолов, реагируют с меркурирован-
ными пиримидинами с образованием связи С—С между
атомами С (5) пиримидина и С(1) енольного эфира или ацетата
[75, 76]. Аналогичным образом можно вводить ненасыщенные
боковые цепи в положение С(4) хинолинов [уравнение (14.45)]
[77].
J)
Li2PdCI4 HN
HN"
HgCI
2)NqBH4/
Н2
НО
ОН
(R')
(14.44)
Rf
OMe О
HN
OMe
Rl
0
„Ci
HN
•0
R1
Применение комплексов, содержащих g-связи М—С 185
Нд(ОАс).
R1
R'
НдХ
Y»CO2Me, Ph, Ar
Основываясь на последовательности меркурирование/талли-
рование — переметаллирование — внедрение, Ларок
синтезировал ряд гетероциклических систем. Таким путем получают бу-
тенолиды из пропаргиловых спиртов [уравнение (14.46)] [73],
а также кумарины [уравнение (14.47)] [79]. Таллирование
ароматических соединений происходит региоспецифично и
контролируется функциональными заместителями кольца. Таллирование
замещенных бензойных кислот осуществляется в орто-положе-
ние к карбоксилатной группе. Переметаллирование на палладий
с последующим внедрением приводит к разнообразным кума-
ринам [уравнение (14.48)] [80]. Этим методом можно получить
многие другие гетероциклы, в том числе индолы, изокарбости-
ролы, фталиды, изокумарины, бензопираны [81].
R
R-C-СгСН
ОН
CI H
у/
R HO Pd
HgCI2
NaCI
н2о
:O
R,
R'
CI
OH
R'
H
HgCI
CI
R"
CI
OH
H
V
iv
(14.46)
СО
(14.47)
с=с-сн3
Hg(0Ac)2
HOAc
NaCI
AcO
AcO
HF/HOAc
QAc
186 Глава 14
(14.48)
со2н
TKTFAb
со?н
TI(TFA)2.
т fa - rnputpmopocttfemarn
Трост [82] использовал переметаллирование оловоорганиче-
ских производных (получаемых обычно из литий- или магний-
органических соединений) палладием с последующим
внедрением тиоеноловых эфиров для построения карбоциклических
систем. Эти субстраты алкилируются по сс-положению и
претерпевают элиминирование тиоалкильной группы. Каталитическая
активность системы определяется наличием Pd(II), он
высвобождается при добавлении реагента, связывающего тиогруппы
(в данном примере HgCl2), которые в противном случае
координировали бы с Pd (II) и дезактивировали его [уравнение
(14.49)].
•SnMe3
катализатор РЫ(Ю9
НдС12 "
-PdSMe
(14.49)
\ндх2
MeSHgX + Pd(ll)
90%
Хотя во всех этих реакциях в качестве интермедиатов
участвуют палладийорганические комплексы, их редко виделяют,
обычно они образуются и используются in situ. Тем не менее для
того, чтобы эти реакции могли происходить,
палладийорганические комплексы должны обладать определенной устойчивостью.
Все рассмотренные выше реакции возможны только в тех
случаях, когда группа R не содержит водорода в ^-положении
(арил, винил, бензил, неопентил); при наличии ^-водорода в
алкильной группе быстрее любой другой реакции происходит
^-элиминирование Pd—Н, что приводит к образованию алкенов,
а не продуктов сочетания или внедрения.
14.4. Получение ал кил металлов окислительным присоединением
и переметаллированием
Один из самых распространенных подходов к о-арил- и о-ви-
нилметаллокомплексам основан на окислительном присоедине-
Применение комплексов, содержащих а-связи М—С 187
нии арил- или винилгалогенидов к низковалентным переходным
металлам, чаще всего к никелю (0) или палладию (0). В
результате образуются о-алкилметаллогалогениды*, переметаллирова-
ние которых (замена галогена на группу R) с помощью
органических производных металлов главных групп приводит к
диалкильным комплексам переходных металлов. Последние
могут подвергаться восстановительному элиминированию с
образованием связи углерод — углерод, при этом переходный металл
регенерируется в первоначальном окислительном состоянии
[уравнение (14.50)]. Этот процесс хорошо изучен и широко
применяется в органическом синтезе.
-М(О) ^ ^ RX (14.50)
R-R'
Л / X
М= Pd, Ni
М'= Mg, Zn, Zr, Sn, В, Ai, Li
R'M'
Одним из первых примеров такого процесса явилась
разработанная Кумада [83] реакция Гриньяра, катализируемая
никелем или палладием. Фосфинникелевые комплексы
катализируют селективное кросс-сочетание реактивов Гриньяра с арил-
и алкенилгалогенидами [уравнение (14.51)]. Реакция идет
гладко с первичными и вторичными алкильными, арильными, алке-
нильными и аллильными реактивами Гриньяра. В качестве
субстратов используют разнообразные арил- и алкенилгалоге-
виды, простые, конденсированные, замещенные, однако простые
алкилгалогениды не вступают в реакцию. Для простых ариль-
ных и алкильных реактивов Гриньяра наиболее эффективными
катализаторами оказались хелатирующие дифосфиновые
РЬ2Р(СН2)2РРЬ2 комплексы хлорида никеля, тогда как для
алкенильных и аллильных реактивов Гриньяра больше
подходит комплекс с лигандом Ме2Р(СН2)2РМе2, а для стерически
затрудненных реагентов и субстратов — комплекс, содержащий
две группы Ph3P.
ArX1 Ni, C| RAr
RMgX + или г г > или * MgXX' (14.51)
* Авторы включают в это название комплексы со связями арил — металл
и винил — металл. — Прим. ред.
188 Глава 14
Предполагают, что реакция идет через каталитический цикл,
представленный на рис. 14.3. Каталитически активной частицей
является L2Ni(R)X, этот комплекс получается при
окислительном присоединении галогенсодержащего субстрата к Ni(0),
образующемуся в начальной стадии. Алкилирование этого
комплекса реактивом Гриньяра приводит к несимметричному ди-
алкилникелевому комплексу, восстановительное элиминирование
последнего с образованием продукта кросс-сочетания и
регенерация L2Ni(0) завершают каталитический цикл. Если такой
механизм верен, то фосфиновые комплексы никеля (0) также
должны служить катализаторами в этой системе, а реакция
должна быть родственна некоторым реакциям сочетания с
участием Pd(0), которые будут рассмотрены ниже.
RMgX
-у \/
L2NiX2
RMgX
-'Л ...У\
МдХ2
Рис. 14.3.
Кочи [84] показал, что катализируемое никелем сочетание
арилгалогенидов с образованием диарилов (родственное
показанному на рис. 14.3) представляет собой радикально-цепной
процесс, ключевые стадии которого — окислительное
присоединение RX к соединению никеля (I) и восстановительное
элиминирование из диарильного комплекса никеля(III) ( ч. 1, разд.
5.5, г). Аналогичным образом могут происходить
катализируемые никелем реакции сочетания Гриньяра [уравнение (14.52)].
Ни один из предлагавшихся механизмов не отражает полностью
ход реакции, поскольку никак не объясняет неактивность литий-
органических реагентов в этой системе. Очевидно, что большую
роль в реакциях сочетания играет магний, но какую именно,
еще предстоит выяснить.
L2Ni(ll)X2 + RMgX - ' * » L2Ni(l)X » L2Ni(lll)X2 (14.52)
R'MgX
X
RR1 + L2Ni(l)X - L2Ni(lll)R'
R
Применение комплексов, содержащих g-связи М—С 189
Несмотря на неоднозначность механизма, процесс находит
применение в синтезе. В частности, это наилучший способ для
получения алкилированных гетероциклических соединений. Их
можно синтезировать, исходя либо из галогенированных гетеро-
циклов и алкильных, арильных или винильных реактивов
Гриньяра, либо, наоборот, из гетероциклических реактивов
Гриньяра и винилгалогенидов [уравнение (14.53)] [85, 86].
Наиболее эффективными катализаторами этих реакций
сочетания служат дифосфиновые комплексы никеля (И) или
палладия (II).
(14,53)
.MgX + RX —^ - ГнеЛ-R ■ илц (HetV-X ♦ RMgX
L2PdX2 ^-^ L2PdX2 ^"^
Рассматриваемая реакция применялась в основном для
сочетания реагентов, не содержащих р-водорода, а именно для
арильных и винильных систем. Поскольку процесс включает
промежуточное образование о-алкильных металлокомплексов,
то при наличии водорода в ^-положении происходит
конкурентное ^-элиминирование гидрида. Никель менее склонен к р-эли-
минированию, чем палладий, и потому чаще используется в
качестве катализатора. Эффективность палладия как
катализатора можно повысить, подобрав подходящий лиганд, в
присутствии которого подавляется ^-элиминирование и (или)
стимулируется сочетание. Таким лигандом оказался, в частности,
ферроценилфосфин [уравнение (14.54)], его комплекс с
палладием (И) эфективно катализирует кросс-сочетание первичных
или вторичных алифатических реактивов Гриньяра с алкилга-
логенидами, при этом совершенно отсутствует конкурентное
^-элиминирование и следующие за ним перегруппировки или
восстановление [87].
MgCI
+ RBr
MgC!
(14.54)
190 Глава 14
Поскольку в стадии образования углерод-углеродной связи
участвуют фосфиновые металлокомплексы, то используя
оптически активные (хиральные) лиганды, можно получить
оптически активные продукты сочетания. Кумада и Хаяси
использовали для реакций необычные хиральные лиганды—(аминоалкил-
ферроценил)фосфины (5—7) и аминофосфиновые лиганды,
полученные из аминокислот. Хаяси [88, 89] подробно изучил
эти реакции и показал, что комплексы этих лигандов с
никелем (II) или палладием(II) являются эффективными
катализаторами асимметрического сочетания реактивов Гриньяра с
органическими галогенидами [уравнение (14.55)]. Для получения
высокого энантиомерного избытка необходимо наличие в ли-
ганде группы Me2N; объем фосфина не имеет значения, но пла-
нарная хиральность ферроцена более важна, чем
центрированная на углероде хиральность. В случае лиганда 5, имеющего
хиральность (S, R), продукт имеет ^-конфигурацию, но с ли-
гандами 6 (R, S) и 7 (S) получается продукт S-конфигурации.
Следует отметить, что эта реакция по существу представляет
кинетическое разделение рацемического реактива Гриньяра:
один энантиомер реактива Гриньяра взаимодействует с хираль-
ным алкилникелевым интермедиатом быстрее, чем другой
энантиомер, в результате чего наблюдается оптическая индукция.
Поскольку скорость инверсии реактива Гриньяра превышает
скорость сочетания, можно получить высокий выход хирального
лродукта.
PPh2
—NMe2
Me
PPh2
6,(R),(S)
PPh2
7,(S)
MgX
R-CHCH2PPh2
NMe2
8?R=Me, изо-Ыу
Bz, U30-?vf
0/nop-Bu, Ph,
циклогексил,
трет- Вт
(14.55)
R'X
Ni1
ее до 95%
«Ph, Et,
-веке и л
:Me3SiCHPh -=\
We Г /
Ph
Негиси [90] существенно расширил применение процесса
окислительное присоединение/переметаллирование путем соче-
Применение комплексов, содержащих а-связи М—С 191
тания его с реакциями гидрометаллирования и карбометалли-
рования, которые обсуждались ранее. Винилциркониевые
реагенты, получаемые гидроцирконированием алкинов, не вступают
в реакцию сочетания с арилгалогенидами. Однако в присутствии
никелевых (0) или палладиевых(О) катализаторов наблюдается
эффективное сочетание, происходящее, по-видимому, через
окислительное присоединение — переметаллирование —
восстановительное элиминирование [уравнение (14.56)] [91]. Таким
способом были синтезированы 5-алкилпиримидины [уравнение
(14.57)] [92]. Аллилгалогениды вступают в реакцию сочетания
без аллильной перегруппировки и с сохранением геометрии
[уравнение (14.58)] [93].
(14.56х
R-C=CH
\ Pd(O)
С!
О
(14.
Me3AI
CI2ZrCp2
(14.58)
При проведении реакции с алкинами с внутренней тройной
связью продукты сочетания получаются с очень низкими выхо-
192 Глава 14
дами. Поскольку гидроцирконирование этих субстратов идет
гладко, проблема, видимо, возникает на стадии
переметаллирования. Для ее решения Негиси [94] предложил проводить
дополнительную стадию переметаллирования. После гидроциркониро-
вания осуществляют перенос алкильной группы от Zr к какому-
нибудь другому металлу, который легко вступает в данную
реакцию переметаллирования, после чего алкильную группу
переносят от этого промежуточного металла к третьему (Ni, Pd
или А1), чтобы провести нужную реакцию сочетания. Процесс
довольно сложный и выбор конкретного промежуточного
металла неочевиден. Однако экспериментально установлено, что
ZnCl2 значительно облегчает сочетание замещенных винилцир-
конатов (или аланов) с алкенил-, арил- или алкинилгалогени-
дами в условиях катализа Pd(0) или Ni(0). Поскольку в этом
процессе можно регулировать стерео- и региоспецифичность
реакций соединений циркония, никеля и палладия, на нем
основан ряд удобных синтетических методов [уравнение (14.59)].
(14.59)
EtC = CEt
Cp2Zr{H)CI
Et
Et
Zr
Et
Et
Zn
L4Pd(0)
Br
MeO2C
ЕГ
Et
C02Me
Pd
. L Br
V
MeO2C
Et Et
После того как обнаружилось, что цинк эффективно
переносит алкильные группы к палладию и никелю, этим методом было
осуществлено множество различных реакций сочетания, все они
представляют варианты схемы, изображенной в уравнении
(14.50), отличаются только группы R. Так, на основе галогено-
фуранов, тиофенов и пиридинов, используя цинкорганические
алкилирующие агенты, можно синтезировать замещенные гете-
роароматические соединения [95]. Тем же методом получают ал-
лиларены, диарилметаны [96] и сопряженные диины [97]. Под
действием алкилцинковых реагентов в присутствии палладие-
вых(0) катализаторов хлороангидриды кислот алкилируются
Применение комплексов, содержащих g-связи М—С 193
до кетонов [98]. Метод был использован также для синтеза
мокупалида [уравнение (14.60)] [99] и гетерозамещенных
диенов [уравнение (14.61)] [iOO].
(14.60)
L2PdCI2/DIBAH
(14.60
Привлекательным источником металлоорганического
соединения с элементом «главной группы», пригодным для переме-
таллирования, служат винилбораны, легко получаемые гидро-
борированием ацетиленов. Однако долгое время не удавалось
подобрать условия, в которых бы хорошо шло переметаллиро-
вание от бора к никелю или палладию, пока Сузуки [101] не
отработал этот процесс, обнаружив, что требуется основание.
При выполнении этого требования алкилбораны являются
прекрасным источником органических групп для процесса,
описываемого уравнением (14.50). Характерные примеры применения
этой реакции представлены уравнениями (14.62) [102] и (14.63)
[103].
04.621)
OEt
+ HBV
ArX
основание^
катализатор ЦРо1
ArCH=CHOEt
ArCHgCHO
, NaOH
13—1129
(14.63)
гумулт
194 Глава 14
Стилл разработал реакцию переметаллирования от олова к
палладию на большом числе примеров. Так, приводящее к ке-
тонам алкилирование галогеноангидридов кислот комплексами
тетраалкилолова включает окислительное присоединение гало-
геноангидрида к Pd(0) с образованием комплекса ацилпалла-
дия(П) [104], алкилирование которого дает ацилалкильный
комплекс палладия (II). При восстановительном
элиминировании из последнего выделяется кетон и регенерируется
катализатор Pd(0) [105] [уравнение (14.64)]. Реакция идет с высоким
выходом и в присутствии практически всех функциональных
групп, включая альдегидную, что делает ее весьма
привлекательной с синтетической точки зрения. Дополнительные пре-
(14.64)
RC0C1
4-
L2Pd(O)
L 0
- X-Pd-C-R
L
L2Pd(O)
R*4Sn
ГМФТА
0
+ R'-C-R
R 3SnCt
L 0
R'-Pd-C-R
L
1
-—I
имущества связаны с возможностью вводить в реакцию
неустойчивые и стерически затрудненные галогеноангидриды кислот,
большой скоростью реакции и высоким числом каталитических
циклов (около 20 000 молей продукта на один моль
катализатора); кроме того, практически не наблюдается образования
побочных продуктов. Триалкилстаннаны в той же системе
реагируют в пять раз медленнее, чем R4Sn. В реакциях R2SnCl2 и
RSnCl3, взаимодействующих с ГМФТА, используются максимум
две алкильные группы. Порядок переноса алкильных групп от
олова следующий: PhC = C > PrC = C > PhCH = CH > СН2 =
= СН > Ph > PhCH2 > CH3OCH2 > Me > Bu [106]. Процесс
применим как к простым, так и к функционально замещенным
реагентам, как видно из уравнения (14.65) [107].
о о
И • 3% L2Pd(CI)(CH2Ph) (14.65)
SCI BujSn ^ Ph \
0SiR3
OSiR3 О
71%
Тот же общий механизм лежит в основе катализируемого
палладием сочетания разнообразных органических галогенидов
Применение комплексов, содержащих а-связи М—С 195
с оловоорганическими реагентами [108]. Например, при
взаимодействии аллилгалогенидов с винильными или арильными
производными олова с высоким выходом получаются
соответствующие продукты сочетания [уравнение (14.66)] [109]. Косуги [ПО]
показал возможность аминирования арилгалогенидов путем
переноса диалкиламиногрупп от олова [уравнение (14.67)]. При
0Ме Pd(dba)2
Bu3Sn
ОН
проведении реакций в присутствии монооксида углерода
наблюдается сочетание с внедрением СО. При обработке Bu3SnH и
монооксидом углерода в присутствии палладиевого
катализатора арил-, винил- и аллилгалогениды превращаются в альдегиды
[уравнение (14.68)] [111]. В тех же условиях из винилгалогени-
дов и винильных производных олова получают дивинилкетоны
[112], а из аллилгалогенидов и аллильных производных олова —
диаллилкетоны [ИЗ] [уравнение (14.69)]. Интересно, что винил-
трифлаты [уравнение (14.70) ] [114] и винилфосфанаты
[уравнение (14.71)] [115] также можно алкилировать олово- или алю-
минийсодержащими реагентами в присутствии палладиевых
катализаторов. Если учесть, что указанные субстраты
генерируются из кетонов енолизацией и реагируют далее по кислороду,
то видно, что рассматриваемый метод позволяет осуществлять
прямое превращение кетонов в замещенные олефины.
L2PdCl2
Bu3SnNEt2+ArBr * ArNEt2 (14.67)
PhCH3, 100 °C v '
40—85%
О
Pd (0) ||
RX+CO+BusSnH ► R—С—Н
50 °C
(14.68)
RX
о
катализа-
mop Pd
—^ ^
R=винил, аллил R = винил, аллил
13*
196 Глава 14
Р ffoccrna -
новление
Tf = CF3S02
OTf
(14.70)
SnBu,
L4Pd
LfC!
0P(0Ph)2
I
2} CiP(0)(0Pn).
(14,7!)
Еноляты олова вступают в реакцию сочетания с арил- [116]
и винилгалогенидами [117]i в присутствии палладиевых
катализаторов; это дает возможность алкилировать еноляты кетонов
галогенидами, которые обычно нереакционноспособны по
отношению к ним [уравнение (14.72)]. Таким образом, процесс
окислительного присоединения — переметаллирования
действительно мощный инструмент для построения углерод-углеродных
связей.
ОАс
(14.72)
Br
Pd(O)
14.5. Получение алкилметаллов окислительным присоединением
и внедрением
Еще один подход, нашедший широкое применение в
органическом синтезе, основан на реакциях окислительное
присоединение— внедрение с участием комплексов палладия (0) и в
меньшей степени никеля (0). Как и в предыдущем случае, система
ограничена органическими группами, не имеющими в
^-положении атомов водорода, поскольку иначе возникает конкурентный
процесс ^-элиминирования. Реакции, в которых окислительное
присоединение служит ключевой стадией, представлены на рис.
14.4; все они включают внедрение малых молекул по связи
Pd(II)—С с последующим элиминированием, что приводит к
образованию органического продукта и регенерации Pd(O). Так
что все эти системы в принципе можно получить
каталитическим путем [118].
Одной из первых реакций такого типа явилось
разработанное Хеком и Стиллом карбонилирование органических галогени-
Применение комплексов, содержащих а-связи М—С 197
дов в присутствии Pd(0) в качестве катализатора. Арильные,
бензильные [119] и гетероароматические галогениды [120] в
присутствии метанола карбонилируются до сложных эфиров,
в присутствии первичных аминов — до амидов, а в присутствии
Н2 — до альдегидов. В качестве катализаторов используются
Pd(PPh3)4 или PdCl2(PPh3)2, в условиях реакции под действием
СО соединения Pd(II) быстро восстанавливаются в соединения
Pd(0). Для связывания выделяющегося в реакции НХ
необходимо добавлять третичный амин. Принципиальная схема реакции
показана на рис. 14.5, однако конкретные детали процесса,
происходящего после внедрения СО, не ясны. Очевидно, что
первая стадия представляет собой окислительное присоединение
субстрата к Pd(0), после чего следует внедрение СО с
образованием комплекса Pd(II)—ацил. Способ распада этого
комплекса окончательно не ясен. Он может подвергаться восстано-
со
LnPd(O)
RX
0
RCOR'
—
♦ Pd(O) + HX
coTr'oh
L
г> пи , у
п га л
L
Рис
R'NH2
R1^
CO
. 14.4.
rcnhr'
R
RCH +
+ Pd(O) + HX
S + Pd(O) + HX
Pd(O) + HX
вительному элиминированию RCOX, который, реагируя со
спиртом (или амином), дает производное кислоты. В то же время
он может подвергаться обмену алкоксид — галогенид с
последующим восстановительным элиминированием сложного эфира.
Наконец, возможна прямая атака ацилпалладия спиртом с
образованием сложного эфира и Pd(0). Механизм превращения
ацилпалладиевого комплекса в альдегид под действием Н2 тоже
неясен, он может включать либо последовательность
окислительное присоединение — восстановительное элиминирование, либо
биядерное восстановительное элиминирование (см. ч. 1, гл. 5).
Внутримолекулярное каталитическое карбонилирование ал-
килгалогенидов открывает путь к синтезу разнообразных
гетероциклических систем. Стилл [121] показал, что пропаргиловые
спирты легко превращаются в соответствующие Z-винилиодиды,
которые в свою очередь реагируют с СО в присутствии Pd(0)
198 Глава 14
(25 °С, 2 атм), давая с прекрасным выходом Аа>р-бутенолиды
[уравнение (14.73)]. Поскольку оба процесса, и окислительное
U4.7S)
СО
HI
присоединение, и внедрение СО, происходят с сохранением
конфигурации при винильном атоме углерода, для циклизации в
лактон требуется Z-геометрия. Аналогичным образом из винил-
галогенидов можно получить сс-метиленлактоны [уравнение
(14.74)] [122]. Бэн использовал эти реакции для синтеза
различных азотсодержащих гетероциклических соединений, в том
числе ^-лактамов [уравнение (14.75)] [123], пирроло-1,4-бензо-
диазепинов [уравнение (14.76)] [124] и сс-метиленлактамов
[уравнение (14.77)] [125].
ОН
Br CO, L4Pd
MeCN,
70
Вг
HN
PPh3, Bu3N
ОМе r3
Cl
Ме-- А ^'Н^>° C0/Pd(OAc)2
Bu3N
OR"
] . CO/Pd(OAc)2
PPh3, Bu3N
'/0
0 Ph CO/Pd(OAc)2 "^p 1
Cl
OMe
3 ,p
Ph
(14..74)
(14.75)
(14.76)
(14.77)
Применение комплексов, содержащих а-связи М—С 199
Реакции карбонилирования с участием комплексов палладия
нашли широкое применение в синтезе, хотя известны и
некоторые другие переходные металлы, способные катализировать эту
реакцию. Преимущество палладия (и в меньшей степени
никеля) связано с легкостью внедрения олефина в эту систему. По
существу все эти реакции представляют варианты хорошо
изученной реакции арилирования, открытой Хеком: в палладийор-
ганический комплекс внедряется олефин и элиминируется «Pd—
Н», что в целом дает результат винильного алкилирования. Хотя
точный механизм реакции не известен, экспериментальные
данные свидетельствуют, что реакционная последовательность
включает координацию олефина с комплексом алкилпалладия(П),
^ас-внедрение олефина, а также ^ас-элиминирование «Pd—Н»
RCOR1 * L2Pd(O)
•*• нх
L2Pd(O) ♦ RX
RCOOR1 + HX
R'OH
RCX + L2Pd(O) RCOR' + HX t L2Pd(O)
О
Рис. 14.5.
[126]. Независимо от полярности олефина группа R
присоединяется преимущественно или исключительно к наименее
замещенному атому углерода двойной связи (рис. 14.6). С 1,2-диза-
мещенными алкенами реакция стереоспецифична и
осуществляется по пути ^ас-присоединения— ^ас-элиминирования с
образованием наиболее устойчивого продукта и потерей наиболее
кислого атома водорода. Если в олефине имеется хорошая
уходящая группа, такая, как галогенид или ацетат, ее
элиминирование будет предпочтительнее элиминирования гидрида.
Реакция носит довольно общий характер и используется для
получения самых разнообразных соединений. В присутствии
палладиевых катализаторов замещенные ароматические, гетеро-
ароматические и пиримидиновые галогениды реагируют с
этиленом, дизамещенными олефинами, сопряженными енонами и
N-виниламидами, давая соответствующие алкилированные оле-
200 Глава 14
фины с выходами от хороших до высоких. Если в качестве
субстрата используют аллиловые спирты, восстановительное
элиминирование происходит при спиртовом углероде, и
продуктами реакции являются карбонильные соединения [уравнение
(14.78)]. В этих случаях в качестве катализатора в реакцию
вводят соль палладия(II), например Li2PdCl4 или Pd(OAc)2.
Поскольку для катализа требуется Pd(0), очевидно, имеет место
восстановление in situ. Вероятно, восстанавливающим агентом
служит триэтиламин, который может координировать с Pd(II)
и после элиминирования ^-гидрида давать имин, НС1 и Pd(0)
[127а].
(14.78)
X2Pd(!i)
он
-PdK
Спенсер усовершенствовал этот процесс путем замены
растворителя (ацетонитрила) на ДМФ и добавления ацетата
натрия, что позволило значительно повысить число каталитических
циклов, в реакциях арилбромидов с олефинами [1276]. В этих
условиях в реакцию вступают даже арилхлориды, хотя они
реагируют несколько медленнее [128]. В системах, в которых
сами арилгалогениды нереакционноспособны, соответствующие
галогеноангидриды кислот легко вступают в реакцию, теряя СО
[уравнение (14.79)] [129]. Джеффри [130] показал, что
реакции такого типа гладко протекают при 25°С (а не при 80—
130 °С), если проводить их в ДМФ с добавлением тетрабути-
ламмонийхлорида. Подобные модификации делают арилирова-
ние «по Хеку» еще более привлекательным для синтеза
сложных и неустойчивых соединений. Внутримолекулярный вариант
этого процесса удобен для синтеза карбоциклических и
гетероциклических систем [уравнения (14.80) — (14.84)]. Региохимия
Применение комплексов, содержащих а-связи М—С 201
COCI
Pd(OAc)2
Pd(0Ac)2,
PhCH2NMe2
NaOAc
L
(14.79)
внутримолекулярных реакций менее предсказуема, и алкилиро-
вание не всегда происходит по менее замещенному положению.
катализатор
Pd (0)
CH3CN
EtO2C CO2Et
J 10
катализатор L3RhCl 10 ' I
(14.81)
«4.82)
II4.83)
$iMe3
C0/PdL4/Et3N
ТГФ
^
(14,84)
202 Глава 14
14.6. Получение алкилметаллов циклометаллированием
Во всех рассмотренных выше реакциях алкильных
комплексов переходных металлов необходимым условием образования
связи металл — углерод было наличие функционального
заместителя в органической группе, иными словами, прямое
образование такой связи в самом углеводороде невозможно.
Активация углеводородов (ч. 1, разд. 5.4), т. е. прямое внедрение
металла по неактивированной связи С—Н, интенсивно
изучается, но пока еще не стало практически приемлемым процессом.
Однако некоторые алкильные комплексы переходных металлов
сн3.
СНр*СНСООМе CH?*CHCN CH2sCHPh CH?=CPh
IQO 100 100 100
40
с | t
96 4
rv
чн
^60
H3?^t
93
H3C
"\р _
u^ к
1
'79
<
7
21
Рис.
>h
-№
14.6.
гCHCH(0M
100
OH
CH2=CHCHCH3
90 10
можно получить непосредственно из углеводородов путем «цик-
лометаллирования» [136], при котором лиганд подвергается
внутримолекулярному металлированию с образованием связи
металл — углерод как части хелатного цикла [уравнение
(14.85)] (см. также ч. 1, гл. 5).
CM * MX • м + HX
(14.85)
С точки зрения применения в синтезе много внимания
уделялось только тем комплексам палладия, которые получаются
орто-металлированием ароматических соединений, содержащих
азот в бензильном положении. Разнообразные субстраты такого
типа реагируют с Li2PdCU в мягких условиях, давая с высоким
выходом устойчивые орто-палладированные комплексы. Хотя
Применение комплексов, содержащих а-связи М—С 203
азот почти без исключений должен находиться в бензильном
положении и он несколько стерически затруднен, его химическая
природа менее важна; об этом говорит та легкость, с какой
вступают в реакцию столь разные субстраты, как диалкилбензи-
ламины, а-нафтиламины, азобензолы, 2-фенилпиридины, окси-
мы бензальдегида, имины, гидразоны [уравнение (14.86)].
Образующиеся таким способом комплексы арилпалладия(П)
подвергаются всем описанным выше превращениям, что
открывает путь к введению функциональных групп в орто-положение
упомянутых ароматических субстратов. Например, орго-палла-
Y-(N)
♦ Li2PdCI4
2 UCI
Y-(N) = -
, -CH=NOH,
. Pd \ •'
(14.86)
Pd(O) (14.87)
Et3N
дированные бензиламины присоединяются (внедряются) к
сопряженным енонам [137]; реакция [уравнение (14.87)] идет с
высоким выходом в случае енонов с незамещенной или а-заме-
щенной винильной группой, но с енонами, имеющими
заместители в ^-положении, реакция не идет совсем. Поскольку с
замещенными ароматическими субстратами орто-палладирование
часто происходит региоспецифично, с его помощью можно
селективно обрабатывать ароматические соединения [138]. Уиддоусон
[139] и Дай [140] использовали эту возможность в синтезе
изохинолиновой системы [уравнения (14.88) и (14.89)]. Для
всех этих реакций внедрения требуется присутствие триэтила-
мина. Вероятно, его роль заключается в том, чтобы вытеснить
хелатированную бензиламиногруппу, разорвать хелат и открыть
его для внедрения. орго-Палладированные соединения
подвергаются также электрофильному расщеплению. Так, реакция с
хлороангидридами кислот приводит к замещению палладия
ацильной группой и позволяет осуществить региоспецифическое
ацилирование ароматических систем [141]. В отличие от
рассмотренных ранее реакций внедрения, при которых
высвобождается металлический палладий, при электрофильном
расщеплении выделяется палладий в окислительном состоянии П.
Вследствие этого продукты расщепления проявляют тенденцию
204 Глава 14
остаться координированными с палладием, и для их выделения
часто требуется KGN [уравнение (14.90)].
XI _, (14.88)
CN
Nr
160°С
мези талек
HCN
(низкий выход)
Li2PdCf4
MeO
CI
d^
.N —
I
R
(14.90)
!)R"COCI
2) KCN
MeO
Ацетанилиды также легко подвергаются орто-палладирова-
нию с образованием шестичленных хелатных циклов, в которых
Применение комплексов, содержащих g-связи М—С 205
|шслород амидогруппы служит связующим лигандом. Эта
система очень удобна для дальнейших превращений, поскольку
палладий можно заменить множеством других групп, а также
потому, что продукты реакции, будучи амидами, не образуют
прочных комплексов с палладием. Так, внедрение СО приводит
к о-амидобензойным кислотам, внедрение олефинов — к о-ами-
достиролам [142], а при электрофильном расщеплении метилио-
дидом получаются продукты орго-метилирования [143]
[уравнение (14.91)].
(14.91)
NHCOR4-
CO2Et
Pd(0Ac)5
R3CONK
NHCOFT
сн3
Мурахаси [144] применил реакцию орго-палладирования
для селективного орго-алкилирования и арилирования бензаль-
дегидов, азобензолов и третичных бензиламинов. Обработка
различных орго-палладированных ароматических соединений
трифенилфосфином и реактивом Гриньяра или литийорганиче-
скими реагентами приводит к селективному замещению
палладия анионной R-группой. Считается, что реакция идет по пути,
показанному на рис. 14.7. Трифенилфосфин расщепляет хлорид-
ный мостик с образованием мономерного хлорофосфинового
комплекса. Замещение галогенида алкильной группой и
последующее восстановительное элиминирование завершают процесс.
Очевидно, присутствие фосфина требуется для стабилизации
промежуточного комплекса Pd—R, чтобы предотвратить как
простое восстановление Pd (II) в металлический палладий, так
206 Глава 14
и р-элиминирование из алкильных групп, содержащих водоро,
в р-положении. Алкилирование происходит только с нестабили!1
зированными карбанионами, которые преимущественно
атакуются по палладию.
До последнего времени применение циклометаллирования
было ограничено ароматическими системами. Однако
потенциально эта реакция может применяться гораздо шире.
Интересный пример введения функциональной группы в неактивирован-
4РЬ3Р
RLi
Pd(O) + 2 PPh3' '
Рис. 14.7.
ное положение неароматической системы приведен в уравнении
(14.92) [145].
(14.92)
также
14.7. Получение алкилметаллов из анионов металлов
и органических галогенидов
Многие анионы переходных металлов являются сильными
нуклеофилами и реагируют с органическими галогенидами,
давая о-алкильные металлокомплексы [уравнение (14.93)]. Боль-
\ Применение комплексов, содержащих g-связи М—С 207
шинство этих систем включают анионы карбонилов металлов и
будут рассматриваться в гл. 15.
M(CO)n-+RX -+ RM(CO)n+X- 14.93)
14.8. Получение алкилметаллов нуклеофильной атакой
олефиновых металлокомплексов
Нуклеофильная атака олефиновых металлокомплексов
приводит к а-алкильным комплексам [уравнение (14.94)].
Последние являются весьма полезными интермедиатами в
органическом синтезе и будут подробно обсуждаться в гл. 17.
(14.94)
Nuc-
ЛИТЕРАТУРА И ПРИМЕЧАНИЯ
1. См. обзор: Erdik Е., Tetrahedron, 40, 641 (1984).
2. Полезный обзор опубликован Норманом: Normant J. R., Pure Appl. Chem,
50, 709 (1978).
3. Схема адаптирована из работы: Posner G. Я., in: An Introduction to
Synthesis Using Organocopper Reagent, Wiley-Interscience, New York, 1980.
4. Kharasch M. 5., Tawney P. O., J. Am. Chem. Soc, 63, 2308 (1941).
5. Эта реакция подробно обсуждается в обзоре: Posner G. Я., Org. React.,
22, 253 (1975).
6. Эта реакция подробно обсуждается в обзоре: Posner G. Я., Org. React.,
19, 1 (1972).
7. Pearson R. G., Gregory C. D., J. Am. Chem. Soc, 98, 4098 (1976).
8. Lipshutz В. H., Kozlowski J. A., Breneman С. М., J. Am. Chem. Soc, 107,
3197 (1985).
9. Johnson C. R., Dutra G. A., J. Am. Chem. Soc, 95, 7783 (1973).
10. См. обзор по нуклеофильному замещению арилгалогенидов с участием
соединений меди: Lindley /., Tetrahedron, 40, 1433 (1984).
11. Kocienski P., Yeates С, J. Chem. Soc, Chem. Commun, 1984, 151.
12. Bevilicqua P., Roberts J. L., Synth. Commun., 13, 797 (1983).
13. Raynolds P. W., Manning M. J., Swenton J. S., J. Chem. Soc, Chem.
Commun., 1977, 499.
14. Rauscher 5., Tetrahedron Lett., 1976, 1161.
15. Mori K., Tamada 5., Matsui M., Tetrahedron Lett., 1978, 901.
16. Marino J. P., Ann. Repts. Med. Chem., 10, 327 (1975).
17. Casey C. P., Cesa M. C, J. Am. Chem. Soc, 101, 4236 (1979).
18. Hallnemo G., Olsson Т., Ullenius G., J. Organomet. Chem., 265, C22 (1984).
19. House H. O., Ace Chem. Res., 9, 59 (1976); House H. O., Wilkins J. AL,
J. Org. Chem., 43, 2443 (1978).
20. Luthy C, Konstantin P., Untch /(., J. Am. Chem. Soc, 100, 6211 (1978).
21. Roush W. #., Lesur В. М., Tetrahedron Lett., 24, 231 (1983).
22. Ashby E. C, Watkins J. /, J. Am. Chem. Soc, 99, 5312 (1977).
23. Clive D. L. J., Farina V., Beaulieu P. L., J. Org. Chem., 47, 2572 (1982).
24. В работе House H. O.f Wilkins J. M.t J. Org. Chem., 43, 2443 (1978) было
показано, что при сопряженном присоединении R2CuLi к енону должен
208 Глава 14
образовываться енолят лития, однако присутствие меди в количестве м<
нее 1% может резко менять химические свойства енолята лития, см[:
Posner G. H., Lenz С. М., J. Am. Chem. Soc, 101, 934 (1979).
25. а) Сопряженное присоединение медьорганических соединений с
улавливанием енолята обсуждается в обзоре: Taylor R. К, Synthesis, 1985, 365;
б) Posner G. Н., Sterling J. /., Whitten С. Е., Lentz С. М., Brunelle D. /.,
J. Am. Chem. Soc, 97, 107 (1975).
26. Posner G. H., Chapdelaine M. /., Lentz С. М., J. Org. Chem., 44, 3661
(1979).
27. Posner G. H., Whitten C. E., Sterling J. /., J. Am. Chem. Soc, 95, 7788
(1973).
28. Buckler R. Т., Garling D. L, Tetrahedron Lett., 1978, 2257.
29. Newton R. F., Howard C. C, Reynolds D. P., Wadsworth A. H., Cross-
land N. M., Roberts S. M., J. Chem. Soc, Chem. Commun, 1978, 662.
30. Bertz S. H., Dabbagh G., Villacorta G. M., J. Am. Chem. Soc, 104, 5824
(1982).
31. Bertz S. H., Dabbagh G., J. Org. Chem., 49, 1119 (1984).
32. Lipshutz B. H., Wilhelm R. S., Floyd D. M., J. Am. Chem. Soc, 103, 7672
(1981). См. также обзор: Lipshutz В. H., Wilhelm R. S., Kozlowski T. J,
Tetrahedron, 40, 5005 (1984).
33. Lipshutz B. H., Parker D., Kozlowski J. A., J. Org. Chem., 48, 3334 (1983).
34. Lipshutz В. П., Wilhelm R. S., J. Am. Chem. Soc, 104, 4696 (1982).
35. Lipshutz B. H., Wilhelm R. S., Kozlowski /., Tetrahedron Lett., 23, 3755
(1982).
36. Lipshutz B. #., Tetrahedron Lett., 24, 127 (1983).
37. Marino J. P., Kelly M. G., J. Org. Chem., 46, 4389 (1981).
38. Yamamoto Y., Yamamoto S., Yatagai H., Ishihara Y., Maruyama K., J. Org.
Chem., 47, 119 (1982).
39. Oppolzer W., Morretti R., Godel Т., Meunier A., Loher H., Tetrahedron Lett.,
24, 4971 (1983).
40. Ghribi A., Alexakis A., Normant J. F., Tetrahedron Lett., 25, 3075 (1984).
41. Полезно ознакомиться с обзорами: Normant J. /\, Pure Appl. Chem., 50,
709 (1978); Normant /. F., Alexakis A., Curr. Trends Org. Synth. Proc Int.
Conf. 4th, 291 (1983); Alexakis A., Commercon A., Coulentianos C,
Normant J. F.t Pure Appl. Chem., 55, 1759 (1983).
42. Germon C, Alexakis A., Normant /. F., Synthesis, 1984, 40, 43.
43. Normant J. F., J. Organometal. Chem. Libr., 1, 219 (1976).
44. Alexakis A., Cahiez G., Normant J. F.t Villieras /., Bull. Soc. Chim. Fr., 1977,
693.
45. Truce W. E., Lusch M. /., J. Org. Chem., 43, 2252 (1978).
46. Westmijze H., Kleijn H., Vermeer P., Synthesis, 1978, 454.
47. Vermeer P., Westmijze H., Kleijn H., van Dijck L. Л., Rec Trav. Chim., 97,
56 (1978).
48. Alexakis A., Commercon A., Coulentianos C, Normant J. F., Tetrahedron,
40,715 (1984).
49. Tadema G., Everhardus R. H., Westmije H., Vermeer P., Tetrahedron Lett.,
1978, 3935.
50. См. обзор по карбометаллированию алкинов: Normant /. F., Alexakis A,
Synthesis, 1981, 841.
51. Эти аргументы выдвинуты в обзорах: Schwartz J., Labinger J. A., Angew.
Chem., Int. Ed. Engl., 15, 333 (1976); Can D. В., Yoshifuji M., Shoer L. /.,
Gell K. L, Schwartz /., Ann. N. Y. Acad. ScL, 295, 127 (1977).
52. Hart D. W., Blackburn T. F., Schwartz /., J. Am. Chem. Soc, 97, 679
(1975).
53. Hart D. W.} Schwartz /., J. Am. Chem. Soc, 96, 8115 (1974).
54. Gibson Г., Tetrahedron Lett., 23, 157 (1982).
55. Bock P. L., Boschetto D. J., Rasmussen J. R., Demers J. P., Whitesides G. M.,
J. Am. Chem. Soc, 96, 2814 (1974).
Применение комплексов, содержащих g-связи М—С 209
.6. Labinger J. A., Hart D. W., Seibert W. E., Schwartz /., J. Am. Chem. Soc,
97, 3851 (1975).
$7. Bertelo C. A., Schwartz /., J. Am. Chem. Soc, 97, 228 (1975).
58. Bertelo C. A., Schwartz /., J. Am. Chem. Soc, 98, 262 (1976).
59. Blackburn T. F., Labinger J. A., Schwartz J., Tetrahedron Lett., 1975, 304L
60. Negishi E-L, Yoshida Т., Tetrahedron Lett., 21, 1501 (1980).
61. VanHorne D. E., Negishi E.-L, J. Am. Chem. Soc, 100, 2252 (1978).
62. Okukado N., Negishi E.-L, Tetrahedron Lett., 1978, 2357; Negishi E.-i., Pure
Appl. Chem., 53, 2333 (1981).
63. Negishi E.-i., Yoshida Т., J. Am. Chem. Soc, 103, 4985 (1981).
64. Эти аргументы выдвинуты в работе: Negishi E.-i., Organometallics in
Organic Synthesis, Wiley, New York, 1980, pp. 54—57.
65. Can D. В., Schwartz /., J. Am. Chem. Soc, 99, 638 (1977).
66. Loots M. J., Schwartz J., J. Am. Chem. Soc, 99, 8045 (1977).
67. Loots M. /., Schwartz /., Tetrahedron Lett., 1978, 4381.
68. См. обзор по ртутьорганическим соединениям в органическом синтезе:
Larock R. С, Tetrahedron, 38, 1713 (1982).
69. Heck R. F., Proc of the Welch Foundation, XVII, 53 (1973).
70. Larock R. C, Angew. Chem., Int. Ed. Engl., 17, 27 (1978).
71. Larock R. C, J. Organometal. Chem. Libr., 1, 257 (1976).
72. Larock R. C, Riefling В., J. Org. Chem., 43, 1468 (1978).
73. Bergstrom D. E., Ruth J. L., J. Am. Chem. Soc, 98, 1587 (1976); J. Org.
Chem., 43, 2870 (1978).
74. Bergstrom D. E., Ruth J. L., Warurck P. L., J. Org. Chem., 46, 1432 (1981);
Bergstrom D. E., Bratterani A. J., Ogawa M. K, Schweickert M. /., J. Org.
Chem., 46, 1423 (1981); Bergstrom D. L., Inoue H., Reddy P. A., J. Org.
Chem., 47, 2174 (1982).
75. Arai /., Daves G. D.f Jr., J. Org. Chem., 43, 4110 (1978); J. Am. Chem. Soc,
100, 287 (1978).
76. Lee T. D., Daves G. D., Jr., J. Org. Chem., 48, 399 (1983); Hacksell U.s
Daves G. D., Jr., J. Org. Chem., 48, 2870 (1983).
77. Dyke S. F., McCartney M. /., Tetrahedron, 37, 431 (1981).
78. Larock R. C, Riefling B.} Fellows C. A., J. Org. Chem., 43, 131 (1978).
79 Larock R. C, Harrison L. W., J. Am. Chem. Soc, 106, 4218 (1984).
80. Larock R. C, Varaprath S., Law H. H., Fellows C. A., J. Am. Chem. Soc,
106, 5274 (1984).
81. Larock R. C, Liu С L., Law H. H.t Varaprath S.y Tetrahedron Lett., 25,
4459 (1984).
82. Trost B. M., Tanigawa Y., J Am. Chem. Soc, 101, 4743 (1979).
83. Tamao K-, Sumitani K., Kiso Y., Zembayashi M., Fijioka A., Kodama S.-i.,
Nakajima I., Minato A., Kumada M.t Bull. Chem. Soc Japan, 49, 1958
(1976).
84. Tsou Т. Т., Kochi J. /£, J. Am. Chem. Soc, 101, 7547 (1979).
85. Tamao K, Kodama S., Nakajima I., Kumada M., Minato A., Suzuki К
Tetrahedron, 38, 3347 (1982).
86. Minato A., Suzuki K, Tamao K, Kumada M., Tetrahedron Lett., 25, 83
(1984).
87. Hayashi Т., Konishi M., Kobori Y., Kumada M., Higuchi Т., Hirotsu К
J. Am. Chem. Soc, 106, 158 (1984).
88. Hayashi Т., Konishi M., Fukushima M., Mise Т., Kagotani M., Tagaika M.,
Kumada M., J. Am. Chem. Soc, 104, 180 (1982); Hayashi Т., Konishi M.,
Ito H., Kumada M., J. Am. Chem. Soc, 104, 4962 (1982).
89. Hayashi Т., Konishi M.t Fukushima M., Kanehira K., Hioki Т., Kumada M,
J. Org. Chem., 48, 2195 (1983).
90. Negishi E.-i., Ace Chem. Res., 15, 340 (1982).
91. Negishi E.-i., VanHorne D. E., J. Am. Chem. Soc, 99, 3168 (1977).
92. Vincent P., Beaucourt J. P., Pichat L., Tetrahedron Lett., 23, 63 (1982)
93. Matsushita H., Negishi E.-L, J. Am. Chem. Soc, 103, 2882 (1981).
14—1129
210 Глава 14
94. Negishi E.-i., Okukado N., King A. 0., Van Horn D. E., Spiegel B. /., J. Am.
Chem. Soc, 100, 2254 (1978).
95. Negishi E.-i., Luo F. Т., Erisbee R., Matsushita #., Heterocycles, 18, 117
(1982).
96. Negishi E.-i., Matsushita H., Okukado N.t Tetrahedron Lett., 22, 2715
(1981).
97. Negishi E.-i.t Okukado N., Lovich S. F., Luo F.-T., J. Org. Chem., 49, 2629
(1984).
98. Negishi E.-i., Bagheri V., Chatterjee S., Luo F.-T., Miller /. A., Stoll A. 7\,
Tetrahedron Lett., 24, 5181 (1983).
99. Kobayashi M., Negishi E.-i., J. Org. Chem., 45, 5223 (1980).
100. Negishi E.-i., Luo F.-T., J. Org. Chem., 48, 1560 (1983).
101. Miyaura N., Suzuki Л., Chem. Lett., 1981, 879; Miyaura N., Suginome #.,
Suzuki A, Tetrahedron Lett., 22, 127 (1981).
102. Miyaura N., Maeda K., Suginome H., Suzuki A., J. Org. Chem., 47, 2117
(1982).
103. Miyaura N., Suginome H., Suzuki A., Tetrahedron Lett., 25, 761 (1984).
104. а) Катализируемые палладием реакции кросс-сочетания оловоорганиче-
ских реагентов с органическими электрофилами рассматриваются в
обзоре: Stille J. Я., Angew. Chem., Int. Ed. Engl., 25, 508 (1986); 6) Stille J.K.,
Milstein D. M., J. Org. Chem., 44, 1613 (1979).
105. Labadie J. W., Stille J. K-, J. Am. Chem. Soc, 105, 6129 (1983); Laba-
die J. W., Stille /. K-, J. Org. Chem., 48, 4634 (1983).
106. Logue M. W., Teng K., J. Org. Chem., 47, 2549 (1982).
107. Labadie /. W., Stille J. K, Tetrahedron Lett., 24, 4283 (1983).
108. Beletskaya I. P., J. Organomet. Chem., 250, 551 (1983).
109. Sheffy F. /C., Stille /. /0, J. Am. Chem. Soc, 105, 7173 (1983); Sheffy F. K.,
Godschalx J. P., Stille J. K., J. Am. Chem. Soc, 106, 4833 (1984).
110. Kosugi M., Kameyama M., Migita Т., Chem. Lett., 1983, 927.
111. Baillargeon V. P., Stille J. K., J. Am. Chem. Soc, 105, 7175 (1983).
112. Gome W. F., Wright M. E., David P. D., Labadie S. S., Stille J /C, J. Am.
Chem. Soc, 3, 1108 (1984).
114. Scott W. J., Crisp G. Т., Stille /. K. J. Am. Chem. Soc, 106, 4636 (1984).
115. Takai K-, Sato M., Oshima K-, Mozak #., Bull. Chem. Soc. Japan, 57, 108
(1984).
116. Kosugi M., Hagihara I., Sumiva Т., Migita Г., Bull. Chem. Soc. Japan, 57,
242 (1984).
117. Kosugi M., Hagihara L, Migita Г., Chem. Lett., 1983, 839.
118. Работы Хека в этой области обобщены в обзоре: Heck R. F., Pure Appl.
Chem., 50, 691 (1978).
119. Stille J. K., Kwan Wong P., J. Org. Chem., 40, 532 (1975).
120. Schoenberg A., Heck R. F., J. Org. Chem., 39, 3327 (1974).
121. Cornell A., Stille J. K-, Tetrahedron Lett., 1979, 133.
122. Martin L. D., Stille J. К„ J. Org. Chem., 47, 3630 (1982).
123. Mori M., Chiba K., Okita M., Ban У., J. Chem. Soc, Chem. Commun., 1979,
698; Tetrahedron, 41, 375 (1985). Эти химические свойства использованы
в синтезе аминоноркардициновой кислоты, см.: Chiba К., Mori M., Ban У.,
J. Chem. Soc, Chem. Commun., 1980, 770; Tetrahedron, 41, 387 (1985).
124. Ishikura M., Mori M., Ikeda Т., Terashima M., Ban У., J. Org. Chem., 47,
2456 (1982). Синтез антрамицина описан в работе: Ishikura M., Mori M.,
Terashima M., Ban У., J. Chem. Soc, Chem. Commun., 1982, 741. Mori M.,
Purvanlackas G. E., Ishikura M., Ban У., Chem. Pharm. Bull., 32, 3840
(1984); Mori M., Ishikura M., Ikeda Т., Ban У., Heterocycles, 22, 253
(1984).
125. Mori M., Washioka Y., Urayama Т., Yoshura /(., Chiba /(., Ban У., J. Org.
Chem., 48, 4058 (1983).
126. Heck R. F.t Org. React., 27, 345 (1982).
Применение комплексов, содержащих g-связи М—С 211
ll27. a) McCridle R., Ferguson G., Arsenault G. J., McAlees A. J., Stephen-
: son D. /C., J. Chem. Res. Synop., 1984, 360; 6) Spencer A., J. Organomet
Chem., 258, 101 (1983).
128. Spencer A., J. Organomet. Chem., 270, 115 (1984).
129. Spencer Л, J. Organomet. Chem., 247, 117 (1983); 265, 323 (1984).
130. a) Jeffrey Г, J. Chem. Soc, Chem. Commun., 1984, 1287; 6) Tetrahedron
Lett., 26, 2667 (1985).
131. Grigg R., Stevenson P., Worakun 7\, J. Chem. Soc, Chem. Commun., 1984,
1073.
132. Ziegler F. E., Chakraborty U. R., Weisenfeld R. В., Tetrahedron, 37, 4035
(1981).
133. Odle R., Blevins В., Ratcliff M., Hegedus L. S, J. Org. Chem., 45, 2709
(1980).
134. Harrington P. J., Hegedus L. S, J. Org. Chem., 49, 2657 (1984).
135. Negishi E.-l, Miller J. Л, J. Am. Chem. Soc, 105, 6761 (1983).
136. Циклометаллирование обсуждается в обзорах: Bruce M. /., Angew. Chem.,
Int. Ed. Engl., 16, 73 (1977); Abicht H.-P., Issleib /C, Z. Chem., 17, 1
(1977); Dehand J., Pfeffer M., Coord. Chem. Rev., 18, 327 (1976); Omae /.,
Chem. Rev., 79, 287 (1979); Coord. Chem. Rev, 32, 235 (1980); Rya-
bov A. £>., Synthesis, 1985, 233.
137. Holton R. A., Tetrahedron Lett., 1977, 355.
138. Holton R. A., Davis R. G, J. Am. Chem. Soc, 99, 4175 (1977); Bris-
don B. J., Nair P., Dyke S. F, Tetrahedron, 37, 173 (1981).
139. Girling I. R., Widdowson D. Л, Tetrahedron Lett, 23, 1957, 4281 (1985).
140. Ban N., Dyke S. F., Quessy S. N., J. Organomet. Chem, 253, 391 (1983).
141: Holton R. A., Natalie K. J., Jr., Tetrahedron Lett, 22, 267 (1981);
Clark P. W., Dyke H. J., Dyke S. F.t Perry G., J. Organomet. Chem, 253,
399 (1983).
142. Horino #., Jnoue N.. J. Org. Chem, 46, 4416 (1981).
143. Tremont S. J., Rahman H. U., J. Am. Chem. Soc, 106, 5759 (1984).
144. Murahashi S.-i., Tamba Y., Yamamura M., Yoshimura N., J. Org. Chem, 43,
4099 (1978).
145. Can K., Sutherland J. K., J. Chem. Soc, Chem. Commun, 1984, 1227.
14*
15
ПРИМЕНЕНИЕ КАРБОНИЛЬНЫХ СОЕДИНЕНИЙ
ПЕРЕХОДНЫХ МЕТАЛЛОВ В ОРГАНИЧЕСКОМ
СИНТЕЗЕ
Химия карбонильных соединений переходных металлов
началась с открытия Мондом карбонила никеля в 1890 г.
Монооксид углерода координирует практически со всеми
переходными металлами, поэтому известно огромное количество
разнообразных комплексов, иногда необычного строения. Будучи
одним из лучших л-акцепторных лигандов, монооксид углерода
стабилизирует металлы с низкой степенью окисления. Во многих
вполне устойчивых карбонильных комплексах металл имеет
очень низкую формальную степень окисления (от 0 до II или
III). Поэтому многие карбонилы металлов достаточно
обогащены электронами, чтобы реагировать как нуклеофилы и как
восстанавливающие агенты, переносящие электрон. Кроме того,
легкость реакций миграционного внедрения, при которых
карбонильные алкилметаллокомплексы превращаются в ацилме-
таллокомплексы, позволяет использовать карбонилы металлов
в качестве эффективных карбонилирующих (ацилирующих)
агентов применительно к разнообразным органическим
субстратам. На рис. 15.1 представлены основные классы реакций кар-
бонилов металлов, используемых в органическом синтезе.
Большое значение имеют каталитические реакции с участием
карбонилов металлов, которые рассматривались в гл. 12.
Настоящая глава посвящена стехиометрическим реакциям
карбонилов металлов [1], которые делятся на три главные
категории: сочетание, карбонилирование (ацилирование) и декарбони-
лирование.
15.1. Реакции сочетания
Еще в 1948 г. сообщалось о превращении аллилхлорида в
диаллил (1,5-гексадиен) под действием карбонила никеля [2].
Последующие работы Кори показали, что реакция носит общий
характер. Было разработано несколько удобных методов
синтеза, но из-за летучести тетракарбонила никеля (т. кип. 43 °С) и
его токсичности они мало использовались. Для реакции
сочетания требуются координирующие растворители, такие, как
ДМФА, ГМФТА или N-метилпирролидон; реакция легко проте-
\ Применение карбонильных соединений переходных металлов 213
кает при 25—50 °С с выделением монооксида углерода
[уравнение (15.1)]. Предполагается, что реакция идет по схеме,
Ni(C0)4
ДМФА
25-5OeC
♦ NiX2 ♦ 4С0
(15.1).
представленной на рис. 15.2 [3]. Координационно насыщенный
Ni(CO)4 довольно неустойчив; потеря одного СО создает
вакантный координационный центр, происходит взаимодействие с
аллилбромидом и образование (т]3-аллил) (бромо)карбонилни-
келя. Димеризация последнего с потерей СО дает хорошо
известный комплекс, бис-(т]3-аллил)никельбромид. В полярных
растворителях оба комплекса необратимо реагируют с
аллилбромидом, давая диаллил и бромид никеля. В неполярных
растворителях сочетание не происходит, и комплекс бис-(г)3-
М(СО)П
\
\
£-С-М(СО)п_| -«
R
Х-М-СО
. ь
^— m-cso:
\ .
Е+ \
\
Е-М(СО)П
О
?=£ X-M-C-R ^=^
А
(кислота
M-L ♦ СО
Рис. 15.1.
М-СНО:А
' M-C-Nuc
О
RC-X + М
^ внедрение
аг77- комплекс
*" карденовьгй
компле/сс
аллил) никельбромида можно выделить с прекрасным выходом
(см. гл. 19). О промежуточном образовании бис-(т]3-аллил)ни-
кельгалогенидов можно судить по характерной темно-красной
окраске, которая сначала появляется, а затем исчезает в ходе
реакции сочетания. Комплекс (т]3-аллил) (бромо)карбонилнике-
ля можно зафиксировать спектроскопически (vco = 2060 см"1),
он легко генерируется при обработке димера монооксидом
углерода в неполярных растворителях. Оба комплекса реагируют с
аллилбромидом в ДМФА, быстро образуя диаллил. В тех же
условиях при взаимодействии с СО сначала образуется
некоторое количество аллилбромида, а в конечном счете диаллил,
поэтому поддерживается равновесие, изображенное на рис. 15.2.
В отличие от подавляющего большинства других реакций ал-
лильного сочетания несимметричные аллилгалогениды
сочетаются преимущественно или исключительно по наименее
замещенному концу.
214 Глава 15
Эту особенность успешно используют для построения
циклических соединений из а,со-бисаллилгалогенидов. В ранних
работах Кори [4] описана циклизация простых 12-, 14- и 18-уг-
леродных систем с прямой цепью в соответствующие
циклические 1,5-диены; реакция идет в условиях высокого разбавления
и дает продукты с выходами от хороших до высоких. Этим
методом получено несколько макроциклических соединений, в
том числе гумулен [5], цембрен (1) [уравнение (15.2)] [6] и
макроциклические лактоны [7]. Нужно отметить, что сочетание
всегда происходит по наименее замещенному аллильному
концу. Аналогичным образом под действием карбонила никеля
Ni(C0)4
„Вг
СО + Ni(C0)3 ^=
-Вт
+ со
чсо
11
Рис. 15.2.
происходит сочетание а-галогенокетонов [8] и а-галогеносуль-
фидов [9], однако а-галогеноэфиры в реакцию не вступают.
(15.2)
N- метил-
jiuppo -
лидинок
Предыдущие реакции представляют собой
«восстановительное» сочетание в том смысле, что в ходе реакции
низковалентный никель Ni(0) окисляется до Ni(II). Нойори [10] разработал
Применение карбонильных соединений переходных металлов 215
родственную реакцию «восстановительного» сочетания а,а'-
дигалогенокетонов с разнообразными органическими
субстратами, открывающую простой путь к некоторым семичленным
циклическим соединениям, которые трудно получить другими
способами. В основе метода лежит получение продукта
взаимодействия Fe2(CO)9 с а,а'-дигалогенокетонами [уравнение
(15.3)]. Следует напомнить, что комплекс Fe(CO)5
координационно насыщен, и для инициирования реакции требуется
термический или фотохимический выброс СО с образованием Fe(CO)4-
Комплекс Fe2(CO)g представляет фотодимер комплекса
Fe(CO)5, его можно рассматривать как удобный источник
реакционноспособного Fe(CO)4, который вступает в реакцию
без инициирования нагреванием или облучением. Нуклеофиль-
ная атака (окислительное присоединение) электронообогащен-
ного Fe(0) в комплексе Fe(CO)4 на высокореакционноспособный
а-галогенокетон приводит к стабилизированному ионом Fe2+a-
бромоеноляту 2. Предполагается, что потеря бромида дает ста-
Fe(0)(CO)4
PhH
Br
Br Fe(!!)(C0}4
(15 3)
Br"
OFe(l!]Br
Br
CFe(!i3Br
билизированный железом оксаллил-катион 3, который
формально является диполярным. Однако комплексование
(координация) О~ с железом, по-видимому, приводит к комплексу,
представляющему функционально простой кислородзамещенный
аллил-катион. Хотя вполне возможно л-взаимодействие оксал-
лила с железом, его химические свойства понятны и без
дополнительного осложнения. Если учесть также, что устойчивые
катионные комплексы п3-аллилжелеза не известны, то формулу
продукта лучше записать в виде 3.
Ряд экспериментальных наблюдений [11] подтверждает
правильность предложенной схемы, а также катионный характер 3.
Первая стадия, формально представляющая двухэлектронное
восстановление субстрата, аналогична случаю восстановления
«эжЭо-сс-бромокамфоры комплексом Fe2(CO)g в водном ДМФА
216 Глава 15
[уравнение (15.4)]. Восстановление происходит стереоселектив-
но с образованием з/сзо-продуктов. Это указывает на
присутствие енолята железа, так как известно, что протонирование
Fe2(C0)9
ДМФА
OFedOBr
D2O
(15.4)
енолята камфоры осуществляется с э/сзо-стороны бициклической
системы. Вторая стадия, потеря галогена, подтверждается
результатами контроля за ходом восстановления а,а'-дибромоке-
тонов под действием 0,5 экв Fe2(CO)9 в водном ДМФА. В этих
условиях выход монобромокетона достигает максимума через
несколько часов, после чего уменьшается, тогда как выход
родоначального (небромированного) кетона постоянно растет.
Ряд экспериментальных данных подтверждает катионную
природу комплекса 3, образованного в таких непротонных
растворителях, как бензол. Реакции сса'-дибромокетонов с Fe2(CO)9
в присутствии нуклеофилов (ацетат или метанол) приводят к
продуктам, которые могут возникнуть в результате нуклеофиль-
ной атаки на катионную частицу [уравнение (15.5)]. В случае
Вг
Вг
Feg(C0)9
FeBr
О
(15.5)
МеОН
ОМе
61%
субстратов, из которых должны образовываться неопентильные
катионы, наблюдается хорошо известная перегруппировка
такого типа систем с участием карбониевых ионов, которая идет
с высоким выходом [уравнение (15.6)] В системах, содержащих
дополнительные центры, активные по отношению к катионам,
наблюдается внутреннее улавливание [уравнение (15.7)]. Нако-
Применение карбонильных соединений переходных металлов 217
нец, сам а,а'-дибромоацетон не дает продуктов, которые могут
возникнуть из оксаллильного интермедиата, поскольку в этом
случае при центрах, которые должны нести формальные
положительные заряды, отсутствуют заместители, стабилизирующие
карбокатион.
Fe2(CO)9
Вг Вг
OFeBr
(15.6)
Вг Вг
70%
Подтверждением того, что комплекс 3 имеет строение ал-
лильного катиона, служит также его поведение в реакциях
циклоприсоединения. Аллильные катионы представляют собой
трехуглеродные двухэлектронные системы, которые должны
вступать в реакции [3 + 4]-циклосочетания (формально [2Я +
+ 4Я]-циклоприсоединение) с образованием семичленных,
циклов. Действительно, комплекс 3 участвует в реакциях такого
типа, что делает его привлекательным для использования в
синтезе. Взаимодействие многих диенов с открытой цепью, в том
числе 1,3-бутадиена и изопрена, с третичными или вторичными
а,а'-дигалогенокетонами [уравнение (15.8)] приводит к
замещенным 4-циклогептенонам с выходами от хороших до высоких
[12]. Реакция открывает легкий путь к семичлеиным
циклическим системам, которые обычно трудно и долго синтезировать
218 Глава 15
другим способом. Как и следует ожидать, в реакциях циклоп-
рисоединения наиболее активны диены, имеющие высокую
равновесную концентрацию s-цис-изомера., особенно циклические
диеновые системы типа циклопентадиена, фурана, N-ацетилпир-
рола, из которых с прекрасными выходами получаются
соответствующие мостиковые бициклические системы [уравнение
(15.9)].
Feg(C0)9
Вг Вг
(15.8)
R1 О Rl
R1
(15,9)
Вг Вг
Х=СН2,О, NAc
Сам а,а/-дибромоацетон, как и дибромиды других метилке-
тонов, не образует циклоаддуктов, так что этим путем нельзя
получить незамещенные циклогептеноны. Однако проблему
можно решить, если ввести в реакцию три- или тетрабромокето-
ны, а затем провести восстановительное дегалогенирование
циклического продукта [уравнение (15.10)]. С разработкой этого
метода стал доступен полный набор циклоалкенов —
незамещенные, моно-, 1,1- ДИ-, 1,3-ди-, три- и тетразамещенные.
Рассмотренная реакция лежит в основе прямого подхода к
большому числу природных соединений, содержащих семичлен-
ные циклы. Например, из фурановых циклоаддуктов можно
получить незукон (4), ^-туйяплицин (5) и а-туйяплицин (6)
[13]. Превращения фурановых аддуктов используются также
в изящном стереоконтролируемом методе построения С-нукле-
озидов [14]. Этим методом можно получить с прекрасными
выходами разнообразные С-нуклеозиды. вводя разные субстраты
в конечной стадии конденсации. Следует отметить, что это один
из немногих способов синтеза нуклеозидов, в котором не
используются в качестве предшественников углеводы. Реакции цикло-
присоединения с участием пирролов открывают общий подход
к синтезу тропановых алкалоидов, включая тропин (7)
[уравнение (15.11)], скопин и гиосциамин [15]. Считается, что эти
реакции [3 + 4]-циклоприсоединения представляют
согласованный процесс, региоселективность которого контролируется
граничными молекулярными орбиталями циклоаддуктов [16].
Применение карбонильных соединений переходных металлов 219
(15.10)
О
R I) Fe2(C0)9
2)Zn/Cu %=S 2)FS03H
4, R-A-изо-Рг
§j R=2-0H, 4-U
JL R=2-0H, Ъ-и
(x /) 2)Zn/Cu
OH 7 OH '
Канемацу изучил реакции железооксаллил-катионов с
различными циклогептатриеновыми системами. Как и можно было
предполагать в случае циклогептатриенона, наблюдается поста-
дийное циклоприсоединение к карбонильной группе [уравнение
(15.12)] [17]. С самим циклогептатриеном вместо ожидаемого
циклоприсоединения происходит согласованный процесс «ен»-
типа [уравнение (15.13)]. Интересно, что с комплексом цикло-
гептатриена и Fe(CO)3 реакция идет совершенно другим путем,
видимо, потому, что фрагмент Fe(CO)3 способен
стабилизировать положительный заряд, возникающий в начальной стадии
[уравнение (15.14)] [18]. При нуклеофильной атаке енолята на
железопентадиенильную систему (гл. 7. 17) генерируется ц3-
аллильный комплекс железа 8, который претерпевает
окислительное внедрение СО, давая наблюдаемый продукт, хотя и с
низким выходом.
Последними примерами использования в синтезе
стабилизированных железом оксаллильных катионов служат их реакции
формального [3 + 2]-циклоприсоединения к ароматическим оле-
финам [19] и енаминам [20], приводящие соответственно к
3-арилциклопентанонам и циклопентенонам. Согласованное
термическое [3 + 2]-циклоприсоединение аллил-катиона к этилену
запрещено правилами орбитальной симметрии; множество
220 Глава 15
O-Fe
9
O-Fe
(15.12)
O-Fe
Н Н
(15.13)
O-Fe
V-Fe
(15.14)
окисление
го% в
47%
экспериментальных данных указывает, что такое циклоприсое-
динение в действительности происходит постадийно, причем
ориентация контролируется относительной устойчивостью
ионных интермедиатов [уравнение (15.15)]. Как и ранее, а,а'-бро-
0. OFeLnBr
R1. I У Fe2(CO)9 Rl jl /R2
,2 "
(15.15)
Br Br
R1 R1
Y=Art NR2
Применение карбонильных соединений переходных металлов 221
моацетон не вступает в эту реакцию; более того, даже сссс^а'-
бромоацетон не реагирует, так что этим путем нельзя получить
незамещенные продукты. Несмотря на многостадийность,
реакция стереоспецифична и идет с общим сохранением
конфигурации. По-видимому, свободное вращение в предполагаемом
интермедиате 9 предотвращается за счет сильного притяжения
между катионной частью и енолятом [21]. Считается, что
аналогичным образом происходят реакции енаминов. Даже
относительно мало нуклеофильные тозилированные енамины
подвергаются циклоприсоединению, давая трициклические продукты с
хорошим выходом [уравнение (15.16) ] [22а].
о
L Fe2(CO)9
Ts Br Вг
15.2. Реакции карбонилирования
Во всех описанных в предыдущем разделе реакциях
участвуют карбонилы металлов, но монооксид углерода служит в них
просто лигандом, обеспечивающим комплексу необходимую'
реакционную способность и (или) устойчивость,
способствующие протеканию нужной реакции. Учитывая легкость процесса?
миграции — внедрения (ч. 1, гл. 6), с помощью реакций карбо-
нилов металлов, происходящих через алкилкарбонильные комп-
лекы, можно получать соответствующие ацильные комплексы,
лишь слегка изменив условия реакции. Включение монооксида
углерода в конечный продукт служит основой ряда удобных
подходов к синтезу карбонильных соединений. В гл. 12 были
рассмотрены промышленно важные процессы
карбонилирования, здесь же внимание будет сосредоточено кга реакциях
карбонилирования, приводящих к более сложным продуктам [226].
а. Карбонилирование с помощью нейтральных карбонилов
металлов. Практически все реакции карбонилирования
включают либо внедрение монооксида углерода по о-связи металл —
углерод, либо прямую атаку нуклеофилов на связанный с
металлом монооксид углерода. И в том и в другом классе реакций
участвуют нейтральные карбонилы металлов. Некоторые из
них, особенно координационно ненасыщенные, достаточно нук-
леофильны, чтобы вступать во взаимодействие с углеродсодер-
жащими электрофилами, давая а-алкильные производные,
которые в свою очередь подвергаются реакции внедрения СО.
Типичным примером служит реакция карбонила никеля с
аллилгалогенидами и избытком монооксида углерода в
умеренно полярных растворителях, единственным продуктом которой
222 Глава 15
является карбонильное соединение (галогеноангидрид р^
сыщенной кислоты или ее сложный эфир) [23]. Аналогичные
продукты получаются при обработке предварительно
полученного (г|3-аллил)никельгалогенида с монооксидом углерода в тех
же условиях. Этот факт указывает на промежуточное
образование (г|1-аллил)никелькарбонильного комплекса, который
подвергается миграции и внедрению с последующим
восстановительным элиминированием галогенангидрида кислоты. На рис.
15.3 представлена обобщенная схема реакций карбонила нике-
о со -
i —Вг
,со J
ДМФА
II
ДМФА
.Вг
+ СО
4 со
PhH
со
'2
Рис. 15.3.
1
Вг
МеОН
ля с аллилгалогенидами, которая ясно показывает
чувствительность реакционноспособных интермедиатов к условиям реакции.
Так, при проведении реакции в бензоле (т]3-аллил)никель-
галогениды можно выделить с высоким выходом. В очень
полярных растворителях, таких, как ДМФА, в результате
сочетания получаются только диаллилы. В растворителях средней
полярности, например в Et2O, наблюдается миграционное
внедрение с образованием ацилгалогенида, который при добавлении
спирта дает ненасыщенный сложный эфир. Все перечисленные
продукты образуются из одного и того же набора реакционно-
способных интермедиатов. Другие достаточно реакционноспо-
собные органические галогениды также подвергаются карбо-
нилированию под действием карбонила никеля. В частности,
арилиодиды в спиртовых растворах дают сложные эфиры бен-
Применение карбонильных соединений переходных металлов 223
зойной кислоты с высоким выходом. По-видимому, все реакции
карбонилирования включают «окислительное присоединение»
галогенида к координационно ненасыщенному комплексу
Ni(CO)3, всегда присутствующему в равновесии с №(СО)4.
Се (IV)
Fe(C0)3
0
x
^>0
R
/
40-80%
Celv
R3NH2
кислота
Льюиса
60-88%
Рис. 15.4.
В отличие от карбонила никеля пентакарбонил железа не
столь лабилен, и для выброса СО с образованием
координационно ненасыщенного Fe(CO)4 требуется облучение. Лей
показал, что при взаимодействии последнего с аллилэпоксидами
образуются т]3-аллильные комплексы железа, вероятно, в
результате атаки Sn2/. Эти комплексы могут вступать в
дальнейшие реакции, представляющие практическую ценность. Так,
окислительное расщепление, ведущее за собой восстановитель-
224 Глава 15
ное элиминирование, приводит к р-лактонам (ч. 1, разд. 5.6)
[24]. При взаимодействии г|3-аллильных комплексов железа
с аминами наблюдается обмен сложный эфир — амин и
образование карбамоильных комплексов. (Уходящие группы в а-поло-
жении к т]3-аллильным системам, как правило, подвержены
нуклеофильному замещению, см. гл. 19.) При окислительном
расщеплении карбамоильных комплексов получаются ^-лакта-
мы с неплохими выходами [25]. Упомянутые реакции
схематически представлены на рис. 15.4.
Реакции карбонилирования могут происходить также в
результате внутримолекулярной нуклеофильной атаки на
связанный с металлом монооксид углерода, после чего следует либо
внедрение, либо восстановительное элиминирование. Одним из
первых примеров такой реакции служит проведенное Стилле
[26] бискарбометоксилирование олефинов, включающее
образование палладийсодержащего карбоксилата, внедрение его в оле-
фин, миграционное внедрение СО и расщепление метанолом
[уравнение (15.17)]. Семмелхак [27] разработал
внутримолекулярный вариант этого процесса, позволивший получить
а-метиленлактоны [уравнение (15.18)]. На схеме показаны два
возможных пути реакции, но точный механизм ее не известен.
PdCI2
СО
СО
МеОН
Cl-Pd
ОМе
МеОН
Pd
i
CI
СО2Ме
чВг Ni(C0)4
^ОН основание
СО2Ме СО2Ме
внедрение
Pd CO2Me
CI
Pd(O)
(15.18)
окислительное
присоедине
ние
во сетано-
'Зительное
элиминирование
Нортон [28] изучил внутримолекулярное карбонилирование
алкинов и, исходя из р-гидроксиалкинов, получил а-метиленлак-
Применение карбонильных соединений переходных металлов 225
тоны [уравнение (15.19)]. Кинетические данные позволили
вывести уравнение скорости, согласно которому лимитирующей
стадией является координация СО (скорость = &набл [Pd][CO]),
= ССН2СН2ОН ♦ СО
а опыты с изотопной меткой показали, что спиртовый протон
оказывается целиком на концевом олефиновом углероде
[уравнение (15.20)]. Отсутствие перегруппировки экзоциклической
двойной связи в более устойчивое эндоциклическое положение,
а также легкость протекания процесса в присутствии металло-
гидрида позволили предположить механизм, представленный
уравнением (15.21). Внутримолекулярное внедрение алкина по
связи Pd—СО подтверждено независимым получением
предполагаемого интермедиата и превращением его в а-метиленлактон
[уравнение (15.22)]. Эти результаты свидетельствуют в пользу
предложенного механизма.
HC=CCH2CH20D
.о
(15.20)
L2Pd!2 5=±
W 9
LI2Pd-C
HCsCCH2CH2
. СО, медленно
L + LPdl2 J LI2PdCO
L ♦ l2Pd-C-0
HCiCCH2-CH2
forcmno
НОСН2СН2С=СК
Q
0 +
L4Pd ♦ CI-C-0CH2CH2C5CTMS
О
^ CH2CI2
L2C!Pd-COCH2CH2CsCTMS
0 0
CCA0 CF3COOH
В приведенной выше реакции лимитирующей стадией
является координация СО, и в реакцию вступает лишь небольшое
15—1129
226 Глава 15
количество Pd, координирующего с СО. Чтобы повысить
каталитическую активность системы, следует сделать более
эффективной координацию монооксида углерода. Это достигается
добавлением SnCl2, который реагирует с комплексами Pd—С1,
давая комплексы PdSnCl3. Лиганд SnCl~3 — сильный я-акцеп-
тор; находящиеся в транс-положении к нему лиганды
становятся более подвижными, они делают более доступным вакантный
координационный центр и тем самым облегчают координацию
СО. Действительно, реакционная способность новой
каталитической системы, состоящей из PdCl2, Ph3P и SnCl2 в ацетонитри-
ле, оказалась более чем в 100 раз выше по сравнению с
использовавшейся ранее системой; кроме того, новая система
обеспечила более высокие общие выходы. Однако тщательное изучение
кинетики показало, что в этой системе скорость не зависит от
концентрации СО и уравнение скорости имеет следующий вид:
скорость = &Набл [Pd]; [субстрат]. Таким образом, с изменением
катализатора меняется и механизм реакции [уравнение (15.23)].
Но каков бы ни был точный механизм реакции, она носит общий
характер; в указанных условиях циклизации подвергаются
самые разнообразные ацетиленовые и олефиновые спирты.
[L2Pd(SnCi3)(CH3CNJ+ +cy8cmpa
[L2Pd(SnCI3)(C0)(subj|+ - L2(SnCI3)PdCOCH2CH2C = CH ■
0
Ямамото разработал катализируемое палладием двойное
карбонилирование, при котором ключевой стадией также
является нуклеофильная атака на монооксид углерода,
закомплексованный с металлом. Так, обработка арил- или винилгалогени-
дов вторичными аминами и монооксидом углерода в
присутствии палладия(II) приводит к пирувамидам с высоким выходом
[уравнение (15.24)] [29, 30]. Те же продукты получаются из
моно- или диалкилпалладиевых комплексов в тех же условиях,
что подтверждает образование этих комплексов в качестве ин-
термедиатов в предыдущей реакции. Однако при
взаимодействии предварительно полученного пирувилового комплекса с
амином вместо пирувамида получается только простой амид [31],
из чего можно заключить, что в данном процессе не происходит
двойное внедрение СО. Сен [32] провел тщательное изучение
механизма и показал, что двойное карбонилирование
происходит в результате восстановительного элиминирования из ацил-
карбамоильного комплекса палладия, а не в результате двойпо-
Применение карбонильных соединений переходных металлов 227
го внедрения СО с образованием пирувилового комплекса
[уравнение (15.25)].
\Qamrt СО 9 9 9 (|5-24)
АгХ + 2С0 + 2R'2NH *- RC-C-NR'2 + RC-NR'2
IOOeC,4O ч, 80-90% 10-20%
катализа г/op 4
L Pd С Ь / ♦
г г / rUnh
Pd или Pd ™ / Rc-C-Pd-Ci
L^ X R^ "Ч R2NH ^
0 L 0 L 0 0
RC-p'd-CO + R'2NH *- RC-Pd-L • *- RC-CNR2 (15.25)
X C = 0
NR2
б. Карбонилирование с помощью анионных комплексов
металлов. Помимо рассмотренных, и другие нейтральные карбони-
лы металлов способны карбонилировать отдельные органические
субстраты, однако гораздо эффективнее для этой цели
анионные карбонильные металлокомплексы. Большинство этих
комплексов довольно сильные нуклеофилы и легко реагируют с
разнообразными субстратами, подверженными нуклеофильной
^атаке. Кроме того, будучи в высокой степени восстановленными
анионные комплексы карбонилов металлов весьма реакционно-
способны по отношению к окислительному присоединению (как
говорилось в ч. \, гл. 5, не все эти реакции идут по механизму
нуклеофильного замещения). Среди этих комплексов наиболее
изучен динатрийтетракарбонилферрат Na2Fe(CO)4, он был
открыт Куком и Коллменом [33] и нашел широкое применение в
органическом синтезе. Этот с!10-комплекс железа (II) легко
получается восстановлением продажного пентакарбонила
железа натрием с бензофенонкетилом в кипящем диоксане
[уравнение (15.26) ] [34], он также выпускается промышленностью.
диоксан
Fe(CO)5+Na+PhCOPh * Na2Fe(CO)4— 1,5-диоксан (15.26)
Удобство применения динатрийтетракарбонилферрата в
синтезе связано с его высокой нуклеофильностью и легкостью
протекания реакций миграции — внедрения (см. ч. 1, тл. 6).
Основные превращения, в которых участвует этот комплекс,
схематически представлены на рис. 15.5. Органические галогениды и
тозилаты реагируют с Na2Fe(CO)4 с образованием анионных
<18-комплексов алкилжелеза(О) (а). Реакция представляет собой
типичный процесс Sn2 с кинетикой второго порядка, обраще-
15*
228 Глава 15
нием стереохимии и соответствующим порядком реакционной
способности (CH3>RCH2>R/RCH; RI>RBr>ROTs>RCl; винил
и арил инертны). Образующиеся комплексы подвергаются про-
толитическому расщеплению (б), давая соответствующие
углеводороды, так что суммарный процесс равнозначен
восстановлению галогенида. В присутствии избытка монооксида углерода
или при добавлении трифенилфосфина наблюдается
миграционное внедрение, приводящее к ацильному комплексу (в), который
можно получить и прямым путем из Na2Fe(CO)4 и галогеноан-
гидрида кислоты (г). Ацильный комплекс реагирует с уксусной
кислотой с образованием альдегидов (д), при этом обеспечива-
RFe(CO)4
RH
RCNR'R1
О RCOOH"
RCOR1
RCHO
Рис. 15.5.
ется высокая степень превращения в альдегид как алкил-, так
и ацилгалогенида. Окислительное расщепление ацильного или
алкильного комплекса железа (0) кислородом (е) или галогеном
(ж) приводит к производным карбоновых кислот. Комплекс
ацилжелеза(О) достаточно нуклеофилен и способен
взаимодействовать с реакционноспособными алкилиодидами, давая
несимметричные кетоны с прекрасным выходом (з). Интересно, что
аналогичные несимметричные кетоны получаются и из
алкильного комплекса железа (0) в результате его реакции с
алкилиодидами и внедрения СО (з). Таким образом, рассмотренные
реакции позволяют осуществить превращения алкилгалогенидов
Применение карбонильных соединений переходных металлов 229
и галогеноангидридов кислот в алканы, альдегиды, кетоны и
производные карбоновых кислот. Реагент вполне специфичен по
отношению к галогенидам, так что реакции можно проводить
при наличии в субстрате сложноэфирной, кетонной, нитрильной
и олефиновой группировок. В случае смешанных галогенидов
(например, хлоробромосодержащих соединений) реакция идет
исключительно по наиболее реакционноспособному галогену.
Единственное ограничение в применении Na2Fe(CO)4 связано
с его высокой основностью (величина рКв близка к таковой
для группы ОН~), способствующей протеканию конкурирующих
реакций элиминирования с третичными и вторичными
субстратами. Кроме того, при использовании в качестве субстратов
аллилгалогенидов, имеющих алкильные группы в б-положении
к галогену, преимущественно образуются устойчивые 1,3-диено-
вые комплексы железа. Наконец, синтезы, включающие стадию
миграционного внедрения (в), ограничены простыми
первичными или вторичными субстратами, поскольку эта реакция не
идет при наличии в группе R электроотрицательных
заместителей.
Коллмен и Браумен провели тщательное экспериментальное
изучение представленных на рис. 15.5 реакций и обосновали их
механизмы. Комплекс алкилжелеза(О) 10 и комплекс ацилжеле-
за(0) 11 были выделены в виде устойчивых на воздухе кристал-
OC-Fe.
со
ччч СО
»со
10
R
о=с
OC-Fe,
L
II
С0
лических солей с [(Ph3P)2N]+ и полностью охарактеризованы
данными элементного анализа, ИК- и ЯМР-спектров, с ними
были проведены все индивидуальные реакции, представленные
на рис. 15.5. Кинетические исследования показали, что в
реакциях Na2Fe(CO)4 доминируют эффекты образования ионных
пар, именно этим объясняется наблюдаемое увеличение
скорости в 2-Ю4 раз при переходе от ТГФ к N-метилпирролидону (в
качестве кинетически активных частиц сравнивали контактную
ионную пару [NaFe(CO)4]~ и разделенную растворителем
ионную пару [Na+: S : Fe(C0)4]~). Уравнение скорости, порядок
реакционной способности субстратов, стереохимия при атоме
углерода, активационные параметры (особенно высокая
отрицательная энтропия активации), все эти данные согласуются с
реакцией окислительного присоединения, протекающей по типу
Sn2 без конкурентного одноэлектронного процесса [35]. На
стадию миграционного внедрения (в) также оказывает влияние
230 Глава 15
образование ионных пар, и она ускоряется при образовании
контактной ионной пары: Li+>Na+>Na-KpayH+> (Ph3PbN+.
Реакция внедрения в целом подчиняется закону скорости
второго порядка и имеет первый порядок по NaRFe(CO)4 и по
добавляемому лиганду, разница в скорости достигает
двадцатикратной величины при переходе от такого лиганда, как Ph3P (самая
медленная реакция), к лиганду Ме3Р (самая быстрая реакция)
[36].
Интересный пример использования рассматриваемых
комплексов представляет разработанное Куком [37] получение
этилкетонов из органических галогенидов, Na2Fe(CO)4 и
этилена [уравнение (15.27)]. Считается, что эта реакция происходит
через ацильный комплекс железа (0), который внедряет этилен
по связи ацил — металл и быстро перегруппировывается в а-
металлокетон, последний уже неспособен внедрить молекулу
этилена.
RX+Fe(CO)
О
II
- RCCH2CH2
L
42~ —>
Fe(CO)
О
II _
RCFe(CO)3L+CH2=CH2 —*
О О
II н+ 11
п —> RCCHCH3 —*■ RCCH2CH3
(15.27)
Fe(CO)n
Внутримолекулярный вариант внедрения олефина можно
использовать для получения циклических кетонов [уравнение
(15.28)] [33, 38]. Примером служит осуществленный Мак-Мёри
[39] полный синтез афидиколина [уравнение (15.29)].
8
2) Н*
90%
(15.29)
Под действием динатрийтетракарбонилферрата сложные
эфиры циклопропан-1,1 -дикарбоновой кислоты подвергаются
Применение карбонильных соединений переходных металлов 231
нуклеофильному раскрытию цикла с образованием
стабилизированного карбаниона в качестве интермедиата. Последний,
будучи ацильным комплексом железа, вступает во все типичные
для него реакции [уравнение (15.30)] [40].
. CO2Et 0 (15.30)
FeW + £>( _СО_ <С0>4(р1-С^^;02Е' -^-
CO2Et
67%
Еще одним удобным карбонилирующим агентом оказался
изученный Хеком [41] анионный с110-комплекс кобальта (—I),
NaCo(CO)4. Его легко получить из Со2(СО)8 восстановлением
(Na/Hg) или диспропорционированием под действием
основания. Натриевая соль представляет собой бесцветное
кристаллическое вещество, довольно неустойчивое на воздухе. Анион
проявляет умеренные нуклеофильные свойства. Он реагирует с
органическими галогенидами (первичными, вторичными, аллиль-
ными, бензильными, а также со сложными а-галогеноэфирами)
по механизму Sn2, давая алкилтетракарбонилкобальт, который
под действием монооксида углерода (1 атм) быстро
превращается в соответствующий ацильный комплекс. Последний под
действием спирта образует сложный эфир [уравнение (15.31)].
При этом в качестве второго продукта реакции получается
НСо(СО)4, который можно превратить снова в Со (СО) 4,"" и тем
самым сделать реакцию каталитической. Лучше всего проводить
реакцию при 50 °С в присутствии затрудненного третичного
амина, например дициклогексилэтиламина. В этих условиях карбо-
алкоксилирование алкилиодидов, алкилсульфатов, алкилсуль-
фонатов и алкилбромидов осуществляется с выходами от
умеренных до хороших.
О
СО II R'OH
Co(CO)4-+RX —*• RCo(CO)4 —■+ RCCo(CO)4 —+
О
II
—-> RCOR4-HCo(CO)4 (15.31)
Реакция НСо(СО)4 или NaCo(CO)4 с эпоксидами приводит
к ^-гидроксиалкилкобальткарбонильным комплексам. Они
также способны внедрять монооксид углерода, давая соответствую-
232 Глава 15
щие ацильные комплексы, которые расщепляются спиртами с
образованием сложных р-гидроксиэфиров [уравнение (15.32)].
При необходимости р-гидроксиацильные комплексы можно
выделить, но система также может работать как каталитическая,
если добавить подходящее основание. При наличии
заместителей в эпоксиде выход продукта уменьшается, что согласуется с
механизмом нуклеофильной атаки Со(СО)4~ на эпоксид. Аци-
лирование всегда осуществляется по менее затрудненной части
молекулы эпоксида.
NaCo(CO)4
но'
. Со(СО)4 СО
но
(15.32)
Со(С0}4
ROH
НО
0R ♦ Co(CO)4-
Ацилкобальткарбонильные комплексы, получаемые одним из
описанных выше методов, вступают также в реакцию с 1,3-дие-
нами, давая ацилированные (г}3-аллил) кобальткарбонильные
комплексы. Последние можно расщепить основанием до ацил-
диенов [уравнение (15.33)]. Реакцию также можно проводить
(15.33)
RC-Co(CO)3
СО
RX
Со(СО)3
н н
о
RC
Н Н
иди/и
Со (СО) 7
'Со(СО),
RC
6
Со(СО)х
X)
,Со(СОЬ
Основание
как каталитическую, получая ацилкобальткарбонильные
комплексы из Со (СО) 4 и алкил- или ацилгалогенидов в присутствии
диена, монооксида углерода и основания. Реакция, по-видимому,
происходит через 1,2- или 1,4-присоединение ацильного
комплекса кобальта к диену с последующим распадом образующегося
т^-аллильного комплекса. В случае несимметричных диенов
получается смесь продуктов. Пригодность этой системы для
синтетических целей трудно оценить, так как продукты не были
Применение карбонильных соединений переходных металлов 233
очищены, а выходы определялись по количеству поглощенного
монооксида углерода. Однако она может найти практическое
применение, если оптимизировать условия и разработать
методику выделения продуктов [41]. Как будет показано в гл. 19,
jx-аллильные комплексы кобальта, полученные аналогичным
методом, реакционноспособны по отношению к нуклеофилам,
что позволяет получать из бутадиена функционально
замещенные производные.
Нуклеофильность NaCo(CO)4 была выгодно использована
Мюреем [42] для превращения вторичных ацетатов в силило-
вые эфиры енолов [уравнение (15.34)]. В этом сложном, но
эффективном способе группа R3Si способствует замещению
ацетата.
ОАс
HSiR3' СО
(15.34,
катализатор Co2(CO)g;
50% 50 атм
НСо(СОЬ
Со(СО)4~
Т
OSiR3
ХСо(СО)4 Н Со(С°Ь
0-SiR3
Большинство рассмотренных анионных комплексов карбони-
лов металлов получают восстановлением. Однако их можно
получить и взаимодействием с гидроксидом, включающим нук-
леофильную атаку на связанный с металлом СО и последующее
декарбоксилирование с потерей СО2 (см. ч. 1, гл. 7). Этот
способ успешно применили Кассар, Альпер и дес Аббайс при
разработке аналогичных реакций карбонилирования в условиях
межфазного катализа [43]. Такой процесс в целом более
эффективен и часто обеспечивает более высокие выходы продуктов,
поэтому он находит растущее применение в химии металлоорга-
нических соединений.
в. Карбонилирование с помощью ацильных «ат»-комплек-
сов металлов. Как было показано в предыдущем разделе, химия
карбонильных производных железа и кобальта основана на
реакции аниона карбонила металла (получаемого
восстановлением нейтрального карбонила металла) с органическим галогени-
дом (реакция Sn2), приводящей к о-алкильным металлокомп-
лексам, которые внедряют СО, давая о-ацильные
металлокомплексы. Другой подход к анионным ацилкарбониль-
ным комплексам металлов основан на реакции нейтрального
карбонила металла с литийорганическим соединением [уравне-
234 Глава 15
ние (15.35)]. Образующиеся при этом ацильные «ат»-комплексы
очень реакционноспособны. При взаимодействии их с активными
электрофилами, например с реагентом Меервейна (Ме3О+Вр4~),
наблюдается О-алкилирование, приводящее к карбеновым
комплексам типа 12. Последние представляют самостоятельный
интерес и будут рассмотрены в гл. 16. При электрофильном
присоединении электрофила к металлу образуются комплексы типа
13, которые легко подвергаются восстановительному
элиминированию, в результате чего происходит ацилирование электрофила.
При реакции литийорганических реагентов с карбонилом
железа получаются те же комплексы, что и при реакции Na2Fe(CO)4
с ацилгалогенидами, их структурные характеристики хорошо
известны, а их реакции были подробно рассмотрены ранее.
Комплексы, образующиеся при реакции литийорганических
реагентов с карбонилом никеля, намного менее устойчивы и
потому хуже охарактеризованы. Им приписывается общая
формула [RCONi(CO)3]~~, с которой согласуется большинство их
химических свойств.
О О-
(-) II I
M(CO)n+RLi —* (CO)n-iM—С—R —*• (CO)n-iM=C—R
Е*
О О ОЕ
И 11 I
RCE ■<- (СО)„+1М—С—R (CO)n-iM=C—R
U+ (15.35)
13 12
Цуцуми [44] показал, что обработка ацилникелькарбонила-
тов НС1 в этаноле приводит в случае арильных комплексов к
а-дикетонам, а в случае алкильных комплексов к симметричным
кетонам [уравнение (15.36)]. Комплексы, получаемые из алкил-
О
-70 °С II НС1
RLi+Ni(CO)4 * [RCNi(CO)3]-Li+ *-
EtOH
О О О
II II I!
—->■ АгС—САг или RCR (15.36)
литиевых реагентов, значительно реакционноспособнее аналогов,
получаемых из ариллитиевых соединений. Так, первые при
взаимодействии с аллилгалогенидами при —50 °С дают
[^-ненасыщенные кетоны с высокими выходами (Хигедас) [45]. В случае
замещенных аллилгалогенидов ацилирование, как правило,
Применение карбонильных соединений переходных металлов 235
происходит по атому углерода, несущему галоген. При
использовании в качестве субстрата граяс-геранилбромида, склонного
к цис — граяс-изомеризации, конфигурация аллильной двойной
связи остается неизменной в ходе ацилирования, что указывает
на невозможность промежуточного образования производного
(т]3-аллил) никеля [уравнение (15.37)]. Арилиодиды, бензоил-
хлорид и алифатические галогениды, включая первичные иоди-
ды, нереащионноспособны по отношению к ацилникелькарбони-
латам в условиях, достаточно жестких (~Q°C) для разложения
никелевого комплекса. Однако в отличие от перечисленных
субстратов винилгалогениды вполне реакционноспособны, но
образующийся первоначально а,^-ненасыщенный кетон не
выделен, он подвергается вторичному ацилированию с образованием
1,4-дикетона [уравнение (15.38)1.
«-C4H9CNi(C0)3~Li+
Et2O
-5О°С
(15.37)
MeC-Ni(C0)3~Li+-«
(15.38)
PW
Ph О
Подобное 1,4-ацилирование сопряженных енонов
представляет самостоятельный интерес, поскольку классические методы
органического синтеза не позволяют провести прямое ацилиро-
вание такого типа; кроме того, образующиеся в качестве
продуктов 1,4-дикетоны являются важными исходными
соединениями для синтеза разнообразных гетероциклических систем. Кори
[46] показал, что реакция носит вполне общий характер.
Получаемые из метил- или я-бутиллития ацилникелькарбонилаты
взаимодействуют с бензальацетоном, метилциннаматом, 2-цикло-
гексеноном, мезитилоксидом и метилкротонатом, давая 1,4-ди-
карбонильные соединения с выходами от хороших до высоких.
Такой диенон, как 1 -фенол- 1,3-гексадиен-5-он, реагирует
исключительно по положениям 1,4 и не образует 1,6-аддуктов. Реакция
трет-бутиллития, карбонила никеля и бензальацетона приводит
к ожидаемому 1,4-диону, а также к существенным количествам
1,4,5-триона [уравнение (15.39)]. При проведении реакции в
атмосфере монооксида углерода выход триона повышается, что
указывает на весьма необычную миграцию ацильной группы к
связанному с никелем монооксиду углерода. Однако а-дикетон
может образоваться также в результате присоединения никеля
в ^-положение енона по реакции Михаэля. Реакция идет через
236 Глава 15
о-ацил-а-алкилникелевый интермедиат (например, RCONiR').
Внедрение СО по связи а-алкил— металл и последующее
восстановительное элиминирование из бисацильного соединения
[как в уравнении (15.25)] должно привести к наблюдаемому
а-дикетону.
-BuU
СО
Монозамещенные алкины также взаимодействуют с ацилни-
келькарбонилатами, давая 1,4-дикетоны. Самые высокие выходы
получены Цуцуми [47] в реакции с ароильными комплексами
[уравнение (15.40)]. Из реакции фенилацетилена с
[CH3CONi(CO)3]~Li+ были выделены небольшие количества
бензальацетона (PhCH = CHCOCH3). Это указывает на
образование сопряженных енонов в качестве интермедиатов и
связывает реакцию с 1,4-ацилированием сопряженных енонов,
рассмотренным выше. Дизамещенные ацетилены в реакцию не
вступают.
О О
II II
ArCNi(CO);Li++RC = CH —+■ [RCH = CH—С— Аг] —>•
О О
II II
—*• АгССНСН2САг (15.40)
R
Несмотря на то что ацилникелькарбонилаты пригодны для
синтеза, они практически не использовались в течение более 15
лет после их открытия. Это отчасти связано с летучестью и
токсичностью карбонила никеля и трудностями обращения с
этими веществами. В поисках решения проблемы Хегедюс [48]
разработал ацилнитрозильные комплексы кобальта, которые
вступают в те же превращения, но с которыми легче
обращаться. Комплекс (Ph3P) (СО)2 (NO) Co представляет собой
оранжевое кристаллическое вещество, нелетучее и устойчивое на
воздухе; его легко получить, использование его в эксперименте не
вызывает трудностей. При взаимодействии комплекса с литий-
органическим реагентом, по-видимому, генерируется ацильный
«ат»-комплекс, который ацилирует аллилгалогениды,
сопряженные еноны и даже карбонильную группу хинонов [уравнение
Применение карбонильных соединений переходных металлов 237
(15.41)]. Образующиеся при этом соединения кобальта, как
правило, менее реакционноспособны, чем соответствующие ацил-
никелевые комплексы. При взаимодействии карбонила никеля
Ph3P(CO)2(NO)Co
•RLi
NO 0
Ph3PCo-C-R
СО
(15.4!)
с другими нуклеофилами, отличными от литийорганических
реагентов, образуются комплексы, пригодные для дальнейшего
использования в синтезе. Так, реакция винилбромидов с
комплексами, полученными из карбонила никеля и метоксида натрия
в метаноле приводит к а^-ненасыщенным сложным эфирам с
высоким выходом (Кори) [49]. Тщательно подбирая условия
реакции, можно контролировать геометрию двойной связи.
Данная реакция может оказаться наилучшим методом карбок-
силирования в случае неустойчивых субстратов, например ви-
нилгалогенидов. Так, в разработанном Кори [50] синтезе
а-санталола превращение винилиодида в сопряженный сложный
эфир под действием Mg/CO2 осуществляется с выходом, не
превышающим 10%, тогда как при использовании CH3O~/Ni(CO)4
выход повышается до 85% [уравнение (15.42)].
Me
Ni(C0)4
(15.42)
Под действием этого комплекса легко карбоксилируются
арилиодиды и аллилбромиды, хотя субстраты подвергаются
реакции и под действием одного карбонила никеля в отсутствие
метоксида (правда, реакция идет медленнее) [49]. Как ни
странно, простые первичные алкилиодиды не реагируют с
системой метоксид натрия/карбонил никеля даже в условиях,
достаточно жестких для разложения никелевого комплекса. Более
реакционноспособен комплекс, получаемый из грег-бутилата
238 Глава 15
калия и карбонила никеля. Под его воздействием первичные
алкилгалогениды превращаются в соответствующие сложные
грег-бутиловые эфиры с вполне хорошими выходами. Очень ре-
акционноспособные винилбромиды, такие, как ^-бромостирол,
под действием карбонила никеля и пирролидина при 60 °С
претерпевают аминокарбонилирование. Однако с менее реакцион-
носпособными субстратами реакция идет медленно и не имеет
препаративной ценности. Родственная реакция (связанная
также с химией ароилникелькарбонилатов) литийдиметиламида с
карбонилом никеля приводит к комплексу, который превращает
фенилацетилен в 2-фенилтетраметилсукцинамид с умеренным
выходом [51] [уравнение (15.43)]. Однако с ^-бромостиролом
тот же реагент дает ненасыщенный амид с высоким (96%)
выходом [52].
LiNMe2 + Ni(CO)4
о о
Me2NC-CHCH2CNMe (15.43)
Ph
О
PhCH=CHCNMe2
Приведенные выше реакции ацилникелькарбонилатов
представляют определенный интерес с точки зрения синтеза, однако
ни строение промежуточно образующихся комплексов, ни
механизмы реакций не изучены. Участвующие в реакциях
комплексы могут быть димерными или полимерными, о чем
свидетельствует образование интенсивно окрашенных растворов при
взаимодействии карбонила никеля с вводимыми в реакцию
основаниями [как известно, карбонилы никеля в присутствии
оснований образуют окрашенные кластеры, например
Ni3(CO)82~] [53]. Каков бы ни был механизм реакции, он
должен существенно отличаться от механизма реакций ацилкарбо-
нильных комплексов железа, поскольку характер реакционной
способности комплексов никеля по отношению к галогенидам
(винил, арил>алил^>первичный алкил) противоположен
характеру реакционной способности комплексов железа (первичный>
>>вторичный>третичный алкил; винил- и арилгалогениды
инертны). Реакции 1,4-присоединения ацилникелькарбонилатов к
сопряженным енонам напоминают реакции литийдиалкилкупра-
тов (гл. 11), для которых был предложен, хотя и без
доказательств, механизм с переносом электрона. Реакции с
органическими галогенидами адекватно объясняются
механизмом, включающим стадию окислительного присоединения
такого же типа, как в реакции комплексов L4Pd(0) с арилиодидами,
а не механизмом типа SN2, как для реакций комплексов железа
Применение карбонильных соединений переходных металлов 239
[уравнение (15.44)]. Остается неясным вопрос, почему алкилио-
диды настолько нереакционноспособны.
(15.44)
Ni(CO)4 +•
0 R
[bcnkco)3]"
[bcni(co)2]-
RX
О
BCR +
г. Карбонилирование азиринов и азиридинов. Множество
разнообразных процессов карбонилирования функционально
замещенных органических субстратов приводит к
гетероциклическим продуктам [54]. Большинство этих процессов
представляют частные случаи и не носят общего характера, многие
процессы требуют довольно жестких условий. Однако Алпер
разработал ряд реакций ацилирования напряженных
трехчленных азотсодержащих гетероциклов, которые идут в мягких
условиях и приводят к важным гетероциклическим соединениям.
Большой интерес представляет катализируемое палладием (0)
превращение азиринов в бициклические ^-лактамы [уравнение
(15.45)] [55]. Для реакции предложен правдоподобный
механизм, и хотя он пока не подтвержден, ясно, что процесс
включает некое внедрение палладия(0) по связи С—N азирина.
Н Аг
(15.45)
Аг
CO
L4Pd
*CO
R1 H
Карбонил молибдена катализирует реакции
стабилизированных енолятов с азиринами, приводящие к циклическим имидам
[уравнение (15.46)] [56]. Механизм этого процесса тоже не
известен. Под действием карбонильных соединений родия (I)
азиридины карбонилируются до ^-лактамов [уравнение (15.47)]
[57], а а-лактамы превращаются в ацетидин-2,4-дионы
[уравнение (15.48)] [58]. По-видимому, и здесь обязательной стадией
240 Глава 15
процесса является внедрение («окислительное присоединение»)
металла по связи углерод — азот.
♦ RMCHCO2Et ♦ Мо(СО)б
Аг
о"
(15.46)
30-70%
Аг
^7
СО
Аг н
[Rh(CO)2Cl]2 Ни
бе к зол, 90сС; х
20 атм О
шН
(15,47)
/ ы-Ви-тет + СО
Ыгтрепг
[Rh(CO)2Cl];
Н-Ви-трет.
(15.48)
15.3. Декарбонилирование альдегидов и хлорангидридов
кислот [58]
Большая часть настоящей главы была посвящена
рассмотрению методов введения карбонильных групп в органические
субстраты путем «миграционного внедрения», при котором ал-
кильная или арильная группа, связанная с металлом, мигрирует
к соседнему координированному СО, что приводит к
образованию а-ацильного комплекса. Последний затем расщепляется,
давая органическое карбонильное соединение. Возможен
также и обратный процесс, при котором органические
карбонильные соединения (особенно альдегиды и хлорангидриды кислот)
претерпевают декарбонилирование. Этот процесс часто находит
применение в органическом синтезе. Многие комплексы
переходных металлов могут действовать как декарбонилирующие
агенты, но наиболее эффективен в этом отношении катализатор
гидрирования Уилкинсона (РЬзР)зИЬС1. Этот комплекс
реагирует с алкил-, арил- и винилальдегидами в мягких условиях, давая
устойчивые углеводороды и комплекс (Ph3P)2Ph(Cl) СО
[уравнение (15.49) ]. За ходом такого стехиометрического декарбони-
20-100 °С
RCHO+(Ph3P)3RhCl >■ (Ph3P)2Rh(Cl)CO+PPh3+RH (15.49)
Применение карбонильных соединений переходных металлов 241
лирования можно легко следить по изменению окраски от
темно-красной [комплекс (Ph3P)3RhCl], до канареечно-желтой
[комплекс (РЬзР)2Rh (C1) СО]. Декарбонилирование транс-а-
алкилциннамальдегидов происходит с сохранением геометрии
двойной связи и приводит к ^wc-замещенным стиролам [60]
[уравнение (15.50)]. Интересно, что хиральные альдегиды
превращаются в хиральные углеводороды с полным сохранением
конфигурации и с высокой степенью стереоселективности [61„
62] [уравнения (15.51) и (15.52)]. Используя альдегиды, дейте-
сно
(15.50)
Ph R
RhCI(PPh3)3
V=/ ♦ RhCI(CO)(PPh3)2
Ph
PPhi
Ph
Оптическая
чистота 73- $4- %
Ph
Et
С -
Me
RhCI(PPh3)3
PhCN
Phi
Et
• C-
■ H
(I5.52>
IIO°C
Me
Оптическая
чистота 81 °/o
рированные по положению С-1 [которые легко приготовить*
исходя, например, из Fe(CO4)2~] можно получить ранее
недоступные дейтерированные оптически активные углеводороды.
Механизм, предложенный для этой реакции декарбонилирова-
ния, представлен в уравнении (15.53). Первая стадия
заключается в окислительном присоединении альдегида к родиевому
комплексу, что вполне объяснимо, если учесть высокую
реакционную способность L3RhCl по отношению к окислительному
присоединению. В случае простых альдегидов первоначально
образующийся аддукт выделить невозможно. Саггс [63] вводил
в реакцию 8-хинолинкарбоксальдегид; он образует ацилгидрид-
ный комплекс родия, который стабилизируется путем хелатиро-
вания, так что его можно выделить и охарактеризовать
[уравнение (15.54)]. Следующая стадия представляет собой реакцию,
обратную внедрению карбонила, т. е. миграцию алкильной
16-1129
242 Глава 15
группы от карбонила к металлу. Хорошо известно, что
миграционное внедрение — обратимый процесс и может идти в прямом
Rh(CI)L3 к - * RhCI(L2) + L + RCHO л R-C-Rh4^
0 Cl ■ (15.53)
н
>L
RH + RhCI(CO)L2- ^Rfi>w
RhClL, - V-/ Rh>L (15.54)
или обратном направлении в зависимости от условий реакции.
Кроме того, на других примерах было показано, что такое
обратимое превращение происходит с сохранением стереохимии
мигрирующей алкильной группы, отсюда понятно сохранение
стереохимии при декарбонилировании хиральных альдегидов.
Последняя стадия заключается в восстановительном
элиминировании алкана (RH) и образовании RhCl(CO)L2. Эта стадия
необратима [алканы не подвергаются окислительному
присоединению к RhCl(CO)L2] и приводит к завершению всего процесса.
В мягких условиях, в которых проводят реакцию, RhCl(CO)L2
не реагирует с альдегидами, так как он намного менее реакци-
онноспособен по отношению к окислительному присоединению,
чем RhClL3 (я-акцепторная группа СО оттягивает электронную
плотность от металла), так что описанный процесс декарбони-
лирования является стехиометрическим. При проведении
реакции при температурах, превышающих 200 °С, оба комплекса
RhClL3 и RhCl(CO)L2 действуют как катализаторы декарбони-
лирования, по-видимому, потому, что в таких жестких условиях
RhCl(CO)L2 окислительно присоединяет альдегиды.
Эта реакция декарбонилирования находит применение в
органическом синтезе. С ее помощью можно удалить
карбонильную (альдегидную) группу, которая часто необходима, чтобы
активировать положение для функционализации, но которая не
нужна в конечном продукте. Реакция идет в присутствии многих
других функциональных групп, она применима к сложным
молекулам. Примером служит ее использование в синтезе а-свя-
занных дисахаридов [64], дезоксиподокарпата [65], оккидента-
лола [66] и грандизола [67] [уравнения (15.55) — (15.58)].
Следует отметить, что в ходе декарбонилирования может также
происходить ^-элиминирование гидрида [уравнение (15.57)].
Применение карбонильных соединений переходных металлов 243
(15.55)
3}3
Me
МеООС СН2СНО
RhCI(PPh3)3
MeCN,
кипя чение
Me
МеООС СН3
(15.56);
онс.
О'
Me
RhC|(PPh3)3
Me
ОН
(15.57>
( путем jS-элимини-
робакия)
О НС.
-ОАс
RhCKPPh
З'З
d:
-ОАс
(15.58}
г
Под действием RhCl(PPh3)3 хлороангидриды кислот также
подвергаются декарбонилированию, но в отличие от альдегидов
реакция идет более сложно. Наиболее прямой путь реакции
наблюдается в тех случаях, когда субстрат не содержит атомов
водорода в ^-положении, декарбонилирование гладко приводит
к соответствующим хлоридам [уравнение (15.59)]. Аддукт,.
образующийся в первой стадии окислительного присоединения,,
вполне устойчив, в нескольких примерах он был выделен и
полностью охарактеризован. При нагревании этого аддукта
происходит декарбонилирование, механизм которого, как
считают, совершенно аналогичен механизму декарбонилирования
альдегидов. Однако в некоторых отношениях
декарбонилирование хлороангидридов кислот отличается от декарбонилирования
альдегидов. Так, декарбонилирование оптически активного
(S) - (—) -а-трифторометилфенилацетилхлорида комплексом
ИЬС1(РРЬз)з приводит к полностью рацемическому а-трифторо-
16*
244 Тльаа 15
метилбензилхлориду [68]. Точно так же при
декарбонилировании (5)-(+)-а-дейтериофенилацетилхлорида получается бензил-
oc-di-хлорид, в котором конфигурация сохраняется только на
20—27%. Это резко отличает реакцию хлороангидридов кислот
от декарбонилирования альдегидов, при котором наблюдается
очень высокая степень сохранения конфигурации. Проведенные
Стилле [69] тщательные исследования показали, что такое
изменение стереохимии происходит в результате многих ацил-
алкильных перегруппировок, предшествующих лимитирующей
стадии восстановительного элиминирования, которая должна
быть медленной по сравнению с соответствующей стадией в
реакции альдегидов. Даже если в каждом отдельном
превращении ацил — алкил — ацил степень рацемизации очень невелика,
суммарная ее величина может оказаться значительной. При
использовании в качестве декарбонилирующего агента
Rh36Cl(PPh3)3 в реакции с бензоилхлоридом изотопная метка
36С1 равномерно распределяется между продуктом, (бензилхло-
ридом), непрореагировавшим бензоилхлоридом, и комплексом
RhCl(CO) (РРЬзЬ- Такое перераспределение изотопа хлора
указывает на то, что оба атома хлора в окислительном аддукте
эквивалентны, а также на то, что до лимитирующей стадии
потери PhCH2Cl стадия окислительного присоединения много
раз протекает и в прямом и в обратном направлении, так что
рассхматриваемая система очень динамична.
(15.59)
О L ч ci
RhCIL2 ♦ L ♦ PhCH2COCI ^ h^
V чЧч CI RhCI(CO)L2
^C! PhCHoCI
Декарбонилирование галогеноангидридов кислот,
содержащих атомы водорода в ^-положении, под действием RhCl(PPh3)3
приводит к первичным олефинам, образующимся в результате
к8-элиминирования гидрида из а-алкильного интермедиата, а не
в результате восстановительного элиминирования [уравнение
(15.60)]. При декарбонилировании э/штро-2,3-дифенилбутаноил-
хлорида получается исключительно траяометилстильбен
[уравнение (15.61)], тогда как соответствующий rpeo-изомер дает
исключительно ^wc-метилстильбен [уравнение (15.62)] [70].
Это полностью согласуется с реакцией, обратной миграционному
внедрению (ацил к алкилу) с сохранением конфигурации, после
чего следует ожидаемое cww-fj-элиминирование гидрида. Развет-
Применение карбонильных соединений переходных металлов 245
вленные хлороангидриды кислот, которые могут претерпевать
р-элиминирование в нескольких направлениях, дают смеси
продуктов, в которых преобладают наиболее замещенные олефины.
О о L с, (15-60)
RCH2CH2CCI * RhClL3 Ч RCH2CH2-C-Rh444 ^
L С|
-нг<< "Ч:. * н^Чс, + RCH=CH2
н L
Ph .
Ph
эригпро транс
- Ph Ph^ ph
трео
До недавнего времени считалось, что стехиометрическое
декарбонилирование ароилхлоридов комплексом RhCl(PPh3)3,
приводящее к хлороаренам, происходит по механизму,
представленному выше. Однако это не так, хотя литература до 1981 г.
изобилует этой ошибочной информацией. Тщательные
исследования проведенные Кампмейером [71], показали, что при сте-
хиометрическом декарбонилировании бензоилхлорида
комплексом RhCl(PPh3)3 вообще не образуется хлоробензол, а
продуктом реакции скорее является арилсодержащий комплекс
RhCl2(CO) (Ph) (РРЬзЬ- При нагревании этого комплекса
хлоробензол также не образуется. Продолжительная реакция
при 80 °С просто превращает первоначально образующийся
комплекс в смесь RhCl(CO) (PPh3)2 и RhCl2(Ph) (PPh3)2. Ни
тот, ни другой продукт не дают хлоробензола при нагревании
при 100 °С. Таким образом, стехиометрическое
декарбонилирование ароилхлоридов до хлоробензола под действием
RhCl(PPh3)3 вообще не имеет места.
Столь же неясна ситуация и с хлороангидридами а,^-ненасы-
щенных кислот. Среди буквально сотен примеров декарбонили-
246 Глава 15
рования хлороангидридов кислот не известно ни одного случая
подобной реакции с хлороангидридами а,р-ненасыщенных
кислот, нет упоминания об этих субстратах и в обширных
опубликованных обсуждениях процесса декарбонилирования. В конце
концов этот вопрос прояснил Кампмейер [72], который
показал, что хлороангидриды а,р-ненасыщенных кислот
подвергаются декарбонилированию под действием RhCl(PPh3)3 с
образованием винилфосфониевых солей в результате процесса>
представленного уравнением (15.63). Процесс включает
предполагаемое окислительное присоединение и реакцию, обратную
внедрению СО, но не включает восстановительное
элиминирование винилхлорида. Вместо этого происходит либо
восстановительное элиминирование фосфиновой соли, либо внешняя нук
леофильная атака фосфина на винильную группу с замещением
Rh(I). Имеющиеся в настоящее время данные не позволяют
различить эти два пути.
г r -: (15.63)
п !
О
CI
CU ^*ггпъ /\/\ С| с
iph3p C!
Рассмотренные выше примеры декарбонилирования
хлороангидридов кислот относятся к стехиометрическим реакциям, их
проводят в ароматических растворителях — бензоле или
толуоле— при температурах 20—100 °С. Как и в случае альдегидов,
декарбонилирование хлороангидридов кислот, включая ароил-
хлориды, можно провести каталитически в присутствии
комплекса Rh при повышенных температурах (200—300 °С). В таких
более жестких условиях реакция катализируется также
комплексами других переходных металлов, среди которых чаще всего
применяют PdCl2 или Pd/C. В то же время Пиноле [73а]
обнаружил, что катионные дифосфиновые комплексы родия,
такие, как [Rh(dppe)2]Cl и [Rh(dppp)2]Cl, каталитически де-
карбонилируют альдегиды при значительно более низких
температурах (120—150 °С). Каталитическое декарбонилирование
экономически привлекательно, однако жесткие условия не
позволяют применять его в случае неустойчивых органических
соединений, а именно такие соединения обычно служат интер-
медиатами в сложных органических синтезах. Эта проблема
Применение карбонильных соединений переходных металлов 247
частично была решена, когда был разработан метод обратного
превращения Rh(Cl) (CO) (PPh3)2 в реакционноспособный
комплекс RhCl(PPh3)3 под действием бензилхлорида и трифенил-
фосфина [736). Активный комплекс регенерируют в отдельной
стадии, поэтому при декарбонилировании больших количеств
субстрата приходится либо проводить несколько реакций с
небольшими количествами веществ, либо расходовать много
RhCl(PPh3)3, который в конечном счете будет регенерирован.
15.4. Реакции металлоациленолятов с электрофилами
Практически все рассмотренные выше реакции происходят
через ацильные комплексы металлов. Многие эти комплексы
вполне устойчивы, их нетрудно выделить и с ними легко
обращаться. В таких комплексах протоны в а-положении к
карбонильной группе кислые, их можно удалить основанием и
получить соответствующие металлоациленоляты. Либскинд [74] и
Дэвис [75] независимо изучали реакции ацильного комплекса
железа CpFe(CO) (PPh3)COCH3. При обработке этого
комплекса основанием получается соответствующий енолят, который
реагирует с разнообразными электрофилами типичным для
енолятов образом, давая продукты С-алкилирования (рис. 15.6).
CpFe(C0)(PPh3)C0CH3
Рис. 15.6.
Поскольку ацильные соединения железа окислительно
расщепляются до сложных эфиров, это дает метод гомологизации.
Наличие объемной группы CpFe(CO)PPh3 (которая сама по
себе хиральна) позволяет контролировать стереохимию реакций
с электрофилами, что представляет несомненную практическую
ценность. Это явление независимо изучали Либскинд [76] и
Дэвис [77]. Либскинд показал, что при правильном подборе
противоиона енолят CpFe(CO) (Ph3P)COCH3 реагирует с аль-
248 Глава 15
дегидами, давая альдольные продукты с высокой степенью сте-
реоселективности [уравнение (15.64)]. Так, при использовании
(15.64)
ПЛДА О ?Н О ОН
+
co^J 3)RCH0 co^J co7
PPh3 PPh3 РРЬ3
14 15
R*Et, #-Pr, изо-Рг, Ph7 транс-стирал
в качестве противоиона изо-ЪщРЛ^ в смеси продуктов
преобладает диастереомер 14 и соотношение 14: 15 достигает величины
8,2:1. Если в качестве противоиона взять SnCl+, в смеси
продуктов будет преобладать другой диастереомер, 15, и
максимальное соотношение 14 : 15 составит 1 : 12. Ту же систему
изучал Дэвис и с противоином Et2Al+ наблюдал диастереоселектив-
ность более 100:1. (Столь большое отличие в распределении
продуктов, видимо, связано с тем, что в экспериментах Дэвиса
был использован избыток алкилалюминия.) Если в качестве
противоиона берут Cu(I), получается противоположный стерео-
химический результат. Енолят меди реакционноспособен также
по отношению к кетонам, которые тоже дают альдольные
продукты [78]. Дэвис продлил этот процесс. Получив дианион
первоначального альдольного продукта и алкилировав енолят,
он выделил зр^гро-р-гидроксикислоты с очень высокой степенью
стереоселективности. Относительная конфигурация
наблюдаемых продуктов определяется алкилированием £-енолята в
аяга-конфигурации (О~ к СО) и незатрудненной поверхностью
комплекса [уравнение (15.65)].
(15.65)
СОо
R'X
ангпи- ? Е - енолягп
Либскинд [79] превращал первоначальные альдольные
продукты в а,р-ненасыщенные ацильные комплексы железа и показал,
что присоединение по Михаэлю к этим енонам происходит по р-
положению с высокой степенью диастереоселективности. Дэвис
[80] и Либскинд [81] использовали рассматриваемые реакции
в стереоселективном синтезе (5-лактамов. Дэвис исходил из ра-
Применение карбонильных соединений переходных металлов 249
цемического комплекса ацилжелеза, конденсировал его с N-фе-
нилимином бензальдегида и получил единственный диастерео-
мер, который при окислительном расщеплении давал р-лактам
[уравнение (15.66)] (химические выходы для этого процесса не
указаны). Аналогичным образом Либскинд использовал еноля-
ты лития и алюминия и конденсировал их с разнообразными
иминами. Во всех случаях получались смеси диастереомеров,
которые при окислении давали р-лактам с выходом 60—80%
[уравнение (15.67)]. Поскольку исходный хиральный комплекс
ацилжелеза можно разделить, в принципе это дает метод
асимметрического синтеза р-лактамов.
(15.67)
главный
продукт
PPh
N-R
(рацеми чес/сии)
О NHR
i. ^^^^ продцкт
OCVV* R1
PPh3
Бергман [82] продемонстрировал, что асимметрическая
индукция в системах такого типа действительно возможна и
эффективна. Он получил хиральный ацильный комплекс кобальта
16 и разделил его на энантиомерно чистые изомеры. Алкилиро-
вание енолят-иона пивальдегидом и последующее
окислительное расщепление привели к- хиральному циклобутанону,
который, по данным ЯМР с использованием хиральных сдвигающих
реагентов, оказался оптически чистым [уравнение (15.68)].
RR2P// ^ I )ЛДА
С Р тает -Ви
н он
.15.68)
Н ОН
2 ^СО J~V"777/7^-BU
трет- Ы
трет - В и "
250 Глава 15
ЛИТЕРАТУРА И ПРИМЕЧАНИЯ
1. Использованию карбонилов металлов в синтезе посвящены обзоры:
Organic Synthesis via Metal Carbonyls, Wender I., Pino P. (Eds.), Wiley, New
York, 1968, Vol. 1; 1977, Vol. 2 (есть русский перевод первого издания
книги: Органические синтезы через карбонилы металлов. Пер. с англ./Под ред.
Уэндера И., Пино П. — М.: Мир, 1970); Ryang M., Organomet. Chem. Rev.
А, 5, 67- (1970); Ryang M.} Tsutsumi S., Synthesis, 1971, 55; Alper #., J.
Organomet. Chem. Library, 1, 305 (1976).
2. Webb I. D., Borcherdt G. 7\, J. Am. Chem. Soc, 73, 2654 (1951).
3. Corey E. /., Semmelhack M. F., Hegedus L. S., J. Am. Chem. Soc, 90, 2416,
2417 (1968).
4. Corey E. J., Wat E., J. Am. Chem. Soc, 89, 2757 (1967).
5. Corey E. /., Hamanaka E., J. Am. Chem. Soc, 86, 1641 (1964); 89, 2758
(1967).
6. Dauben W. G., Beasley G. H., Broadhurst M. D., Mullet В., Peppard D. J.,
Pesnelle P., Suter C, J. Am. Chem. Soc, 96, 4724 (1974).
7. Corey E. J., Kirst H. A., J. Am. Chem. Soc, 94, 667 (1972).
8. Yoshisato E., Tsutsumi Т., J. Chem. Soc, Chem. Commtm., 1968, 33.
9. Semmelhack M. F., PhD Thesis, Harvard University, 1967, p. 74.
10. Noyori R., Ace Chem. Rev., 12, 61 (1979).
11. Noyori R., Hayakawa Y., Takaya H., Muria S., Kobayashi R., Sonada N.,
J. Am. Chem. Soc, 100, 1759 (1978).
12. Takaya H., Makino S., Hayakawa Y., Noyori R., J. Am. Chem. Soc, 100,
1765 (1978).
13. Takaya H., Hayakawa Y., Makino S., Noyori R., J. Am. Chem. Soc, 100,
1778 (1978).
14. Noyori R., Sato Т., Hayakawa Y., J. Am. Chem. Soc, 100, 2561 (1978).
15. Hayakawa Y., Baba Y., Makino S., Noyori R.y J. Am. Chem. Soc, 100, 1786
(1978).
16. Noyori R., Shimizu F., Fukuta K-, Takaya H., Hayakawa У., J. Am. Chem.
Soc, 99, 5196 (1977).
17. Ishizu Т., Mori M., Kanematsu K., J. Org. Chem., 46, 526 (1981).
18. Ishizu Т., Harano K, Yasuda M., Kanematsu /(., J. Org. Chem., 46, 3630
(1981).
19. Hayakawa Y., Yokoyama K., Noyori R., J. Am. Chem. Soc, 100, 1791 (1978).
20. Hayakawa Y., Yokoyama K., Noyori R., J. Am. Chem. Soc, 100, 1799
(1978).
21. Hayakawa Y., Yokoyama K., Noyori R.t Tetrahedron Lett., 1976, 4347.
22. a) Hegedus L. S., Holden M.t J. Org. Chem., 50, 3920 (1985); б) см. также
обзор: Narayagna С, Periasamy M., Synthesis, 1985, 253.
23. Heck R. F., Ace Chem. Res., 2, 10 (1969); Guerrieri F., Chiusoli G. P., J.
Organomet. Chem., 15, 209 (1968); Chiusoli G. P., Ace Chem. Res., 6, 442
(1973).
24. Annis G. D., Ley S. V., J. Chem. Soc, Chem. Commun, 1977, 581; Aumann R.,
Ring H., Kruger C, Goddard R., Chem. Ber., 112, 3644—3671 (1979).
25. Annis G. D., Hebblethwaithe E. M.f Hodgson S. Т., Holinshead D. M.,
Ley S. V., J. Chem. Soc, Perkin Trans. 1, 1983, 2851; Horton A. M.f Hollin-
shead D. M., Ley S. V.y Tetrahedron, 40, 1737 (1984); Hodgson S. Т., Hollin-
shead D. M., Ley S. V.y J. Chem. Soc, Chem. Commun., 1984, 495.
26. Stille J. K., Divakaruni R., J. Org. Chem., 44, 3474 (1979).
27. Semmelhack M. F., Brickner S. /., J. Org. Chem., 46, 1723 (1981).
28. Murray T. F., Norton J. R., J. Am. Chem. Soc, 101, 4107 (1979);
Murray T. F., Samseel E. G., Varma V., Norton J. R., J. Am. Chem. Soc, 103, 7520
(1981); Samsel E. G.} Norton J. R.t J. Am. Chem. Soc, 106, 5505 (1984).
29. Ozawa F., Sozama H., Yamamoto Т., Yamamoto A., Tetrahedron Lett., 23,
3383 (1982).
30. Kobayashi Т., Tanaka M., J. Organomet. Chem., 233, C64 (1982).
Применение карбонильных соединений переходных металлов 251
31. Ozawa F., Yamamoto Л., Chem. Lett., 1982, 865; Ozawa F., Sugimoto Т.,
Yamamoto Т., Yamamoto Л, Organometallics, 3, 692 (1984).
32. Chen J. Т., Sen Л., J. Am. Chem. Soc, 106, 1506 (1984).
33. Cooke M. P., J. Am. Chem. Soc, 92, 6080 (1970); Collman J. P., Ace. Chem.
Res., 8, 342 (1975).
34. Finke R. G., Son ell T. N., Org. Synth., 59, 102 (1979).
35. Collman J. P., Finke R. G., Cawse J. N., Brauman /. /., J. Am. Chem. Soc,
99, 2515 (1977).
36. Collman J. P., Finke R. G., Cawse J. N., Brauman J. /., J. Am. Chem. Soc,
100,4766 (1978).
37. Cooke M. P., Parlman R. Af, J. Am. Chem. Soc, 97, 6863 (1975).
38. Merour J. Y., Roustan J. L., Charrier C, Collin J., Benaim /., J. Organomet.
Chem., 51, C24 (1973).
39. McMurry J. E., Andrus A., Ksander G. M.f Musser J. H., Johnson M. A., J. Am.
Chem. Soc, 101, 1330 (1979); Tetrahedron, 37, 319 (1981).
40. Tamblyn W. H., Wattermere R. E, Tetrahedron Lett., 24, 2803 (1983).
41. См. обзор Хека в работе [1], т. 1, с 373—404.
42. Chatani N., Mural S., Sonoda N.t J. Am. Chem. Soc, 105, 1370 (1983).
43. См. обзор: Cassar L, Ann. N. Y. Acad. Sci., 333, 208 (1980).
44. Myeong S. K., Sawa Y., Ryang M., Tsutsumi S., Bull. Chem. Soc Japan, 38,
330 (1965).
45. Hegedus L. S., PhD Thesis, Harvard University, 1970, pp. 85—87.
46. Corey E. J., Hegedus L. S., J. Am. Chem. Soc, 91, 4926 (1969).
47. Sawa Y., Hashimoto L, Ryang M., Tsutsumi S., J. Org. Chem., 33, 2159
(1968).
48. Hegedus L. S., Perry R. /., J. Org. Chem., 50, 4955 (1985).
49. Corey E. J., Hegedus L. S.y J. Am. Chem. Soc, 91, 1243 (1969).
50. Corey E. J., Kirst H. A., Katzenellenbogen J. A., J. Am. Chem. Soc, 92, 6314
(1970).
51. Fukuoka S., Ryang M., Tsutsumi S., J. Org. Chem., 33, 2973 (1968).
52. Fukuoka S., Ryang M., Tsutsumi S.y J. Org. Chem., 36, 2721 (1971).
53. Sternberg H. W., Markby R., Wender I., J. Am. Chem. Soc, 82, 3638 (1960).
54. Синтез гетероциклических соединений путем циклизации, индуцируемой
карбонилами металлов, обсуждается в обзоре: Ohshiro Y., Hirao Г., Hete-
rocycles, 22, 859 (1984).
55. Alper H., Perera С. P., Ahmed F. R., J. Am. Chem. Soc, 103, 1289 (1981);
Alper H., Mahatantila C. P., Organometallics, 1, 70 (1982).
56. Alper H., Mahatantilla C. P., Einstein F. W. В., Willis A. C, J. Am. Chem.
Soc, 106,2708 (1984).
57. Alper H., Urso F.} Smith D. J. H., J. Am. Chem. Soc, 105, 6737 (1983).
58. Roberto D., Alper H.t Organometallics, 3, 1767 (1984).
59. Этот вопрос обсуждается в обзоре Тсуджи в работе [1], т. 2, с. 595.
60. Tsuji J., Ohno К., Tetrahedron Lett., 1967, 2173.
61. Walborsky H. M., Allen L. E.t Tetrahedron Lett., 1970, 823.
62. Walborsky H. M., Allen L £., J. Am. Chem. Soc, 93, 5465 (1971).
63. Suggs J. W., J. Am. Chem. Soc, 100, 640 (1978).
64. Iley D. E., Frasser-Reid В., J. Am. Chem. Soc, 97, 2563 (1975).
65. Trost B. M., Preckel M., J. Am. Chem. Soc, 95, 7862 (1973).
66. Sergent M., Mongrain M., Deslongchamps P., Can. J. Chem, 50, 336 (1972).
67. Hobbs P. D., Magnus P. £>., J. Chem. Soc, Chem. Commun, 1974, 856.
68. Stille /. K., Fries R. W., J. Am. Chem. Soc, 96, 1514 (1974).
69. Lau K. S. Y., Becker Y., Huang F., Baenzinger N., Stille J. K, J. Am. Chem.
Soc, 99, 5664 (1977).
70. Stille /. K, Huang F., Regan M. Т., J. Am. Chem. Soc, 96, 1518 (1974).
71. Kampmeier J. A., Rodehorst R. M., Philip J. В., Jr., J. Am. Chem. Soc, 103,
1847 (1981); Kampmeier J. A., Mahalingain S, Organometallics 3 489
(1984).
252 Глава 15
72. Kampmeier J. A., Harris S. H., Rodehorst R. M., J. Am. Chem. Soc, 103,
1478 (1981).
73. a) Doughty D. H., Pignolet L. #., J. Am. Chem. Soc, 100, 7083 (1978);
Pignolet L. #., Homogeneous Catalysis with Metal — Phosphine Complexes,
Plenum, New York, 1983; 6) Fries R. W., Stille J. K., Synth. Inorg. Met. Org.
Chem., 1, 295 (1971).
74. Liebeskind L. S.f Welker M. £., Organometallics, 2, 194 (1983).
75. Davies S. G., Baird G. /., J. Organomet. Chem., 248, Cl (1983); Aktogu N.,
Felkin #., Baird G. J., Davies S. G., Watte 0., J. Organomet. Chem., 262,
49 (1984).
76. Liebeskind L. S., Welker M. E.t Tetrahedron Lett., 25, 4341 (1984);
Liebeskind L. S., Welker M. E., Fengl R. W., J. Am. Chem. Soc, 108, 6328 (1986).
77. Davies S. G., Dordor-Hedgecock I. M., Walker Т. С, Warner P., Tetrahedron
Lett., 25, 2709 (1984); Davies S. G., Dor dor I. M., Warner P., J. Chem. Soc,
Chem. Commun, 1984, 956.
78. Ambler P. W., Davies S. G., Tetrahedron Lett, 26, 2129 (1985).
79. Liebeskind L. S., Fengl R. W., Welker M. E., Tetrahedron Lett, 26, 3075,
3079 (1985).
80. Broadly /(., Davies S. G, Tetrahedron Lett, 25, 1743 (1984).
81. Liebeskind L. S., Welker M. E., Goedken ]/., J. Am. Chem. Soc, 106, 441
(1984).
82. Theopold K- #., Becker P. N., Bergman R. G.9 J. Am. Chem. Soc, 104, 5250
(1982).
16
ПРИМЕНЕНИЕ КАРБЕНОВЫХ КОМПЛЕКСОВ
ПЕРЕХОДНЫХ МЕТАЛЛОВ И МЕТАЛЛАЦИКЛОВ
В ОРГАНИЧЕСКОМ СИНТЕЗЕ
Карбеновые комплексы переходных металлов интенсивно
изучались уже более 20 лет, но лишь недавно стали
применяться в органическом синтезе [1, 2]. Как было показано в ч. I,
разд. 3.5, д, реакционная способность карбеновых лигандов в
принципе определяется способностью заместителей донировать
л-электроны на углерод. Карбеновые лиганды с гетероатомными
заместителями (например, О, N, C1) или с другими
заместителями, способными к я-взаимодействию с карбеновым углеродом
(например, Ph), формально рассматриваются как нейтральные
двухэлектронные доноры (ч. 1, табл. 2.2); их называют «элект-
рофильными» или «карбенами Фишера», они обычно
подвергаются нуклеофильной атаке на карбеновый углерод. Карбеновые
лиганды, не содержащие таких заместителей (например, мети-
леновые и алкилиденовые лиганды), требуют существенного л-
донирования от металла и формально считаются дианионными
четырехэлектронными донорами (ч. 1, табл. 2.2); их называют
«нуклеофильными» или «карбенами Шрока», обычно они
подвергаются электрофильной атаке по карбеновому углероду.
Однако подобная классификация может создать неверное
представление, что карбеновый лиганд одного типа никогда не
проявляет реакционноспособности, характерной для карбеновых
лигандов другого типа. В действительности реакционная
способность любого карбенового лиганда может варьировать от
комплекса к комплексу; например, обычно нуклеофильные ме-
тиленовые лиганды могут стать электрофильными в комплексах,
несущих положительный заряд.
16.1. Реакции электрофильных карбеновых комплексов
Общее представление о реакционной способности
электрофильных карбеновых комплексов дает рис. 16.1. Нуклеофиль-
ная атака на металл, приводящая к замещению в лиганде,
обсуждалась в ч. 1, разд. 4.5. Нуклеофильная атака на «электро-
фильный» карбеновый углерод, приводящая к замене гетеро-
253.
254 Глава 16
атомных заместителей у карбенового углерода, обсуждалась
б ч. 1, разд. 3.5, д, как и кислотность атомов водорода в а-поло-
жении и реакция депротонированной формы с электрофилами.
Все эти процессы открывают легкий путь к синтезу множества
карбеновых комплексов с разнообразными функциональными
заместителями. В то же время эти процессы можно
рассматривать как реакции с переносом карбена, происходящие,
по-видимому, через металлациклические интермедиаты, которые широко
применяются в органическом синтезе. Им в основном и
посвящена настоящая глава.
OR'
vc.
c_c
Рис. 16.1.
а. Реакции карбеновых комплексов переходных металлов с
алкинами. В 1975 г. Дётц [3] описал замечательную реакцию
алкинов с (алкоксиарил)-хромкарбеновыми комплексами,
приводящую к 4-метокси-1-нафтолу [уравнение (16.1)].
Впоследствии было показано, что этот процесс исключительно удобен для
синтеза органических соединений самых разных классов. Само
по себе такое превращение далеко не очевидно. Схема
связывания фрагментов показана в уравнении (16.1), а механизм будет
подробно обсуждаться позже.
ОМе
(со)5сг=с;
Применение карбеновых комплексов и металлациклов 255
(16 I)
ОМ©
Региохимия реакции зависит от характера замещения в ал-
кине. Алкины с концевой тройной связью реагируют региоспеци-
фично, давая 2-алкилнафтолы. В случае алкинов с внутренней
тройной связью и, особенно диарилалкинов региоселективность
заметно понижается [4]. Семмелхаку [5] удалось достичь
некоторого контроля региохимии в случае алкинов с внутренней
связью, сделав весь процесс внутримолекулярным [уравнение
(16.2)]. Если алкин связан с карбеновым кислородом короткой
цепью, возможен только один региохимический результат.
(16.2)
(СО)вСг-С'
Ph
in
Для образования нафтола необходимы электрофильные кар-
беновые комплексы. Порядок реакционной способности
[: C(iNR2)Ph< : C(OR)Ph< : CPh2] соответствует порядку
увеличения электрофильности карбенового углерода. Замена СО
фосфином так же резко понижает реакционную способность
карбенового комплекса [6], как и проведение реакции в
присутствии избытка СО. Для циклизации алкина, карбена и карбонильных
лигандов Дётц и Фишер [7] предложили механизм, показанный
на рис. 16.2. При потере СО возникает координационная
вакансия, которую затем занимает алкин. В результате «циклопри-
соединения» образуется металлациклобутен, внедрение СО в
последний приводит к винилкетеновым производным.
Циклизация винилкетена в арен с последующей енолизацией завершают
процесс. Хотя в целом эта схема не подтверждена, имеются
убедительные доказательства в пользу промежуточного
образования винилкетенов. Если в качестве субстрата взять такой
256 Глава 16
стерически сильно затрудненный алкин, как бис(триметилси-
лил)ацетилен, стадия циклизации будет ингибироваться,
поскольку при этом две объемные триметилсилильные группы
должны встать в орто-положение. Поэтому в результате из
реакционной смеси выдаАяется сам винилкетен [уравнение (16.3)]
[8]. Если проводить реакцию в смеси этанол — ТГФ, в качестве
ОМе
(СО)5Сг=С
Аг
-СО
♦СО
(С0)4Сг =
ОМе
Аг
R'-E-R2
(C0)4Cr = C
r'-e-r2
ОМе
(С0)4Сг—С-Аг
r'-c=c-r2
СО
ОМе
ОМе
Cr(CO)3
Рис. 16.2.
продукта также образуется кетен, который выделяют в виде
сложного эфира [уравнение (16.4)] [9].
(CO)5Cr = C + TMS-f-lMS
^ OMe
TMS
(16.3)

М
TMS ОМе
-Аг
HC=CCO2Et
EtOH/ТГФ
н Ar
EtO2C
(16.4)
ОМе
Несмотря на неоднозначность механизма, реакция широко
используется для синтеза сложных органических соединений.
Так, Дётц [10] использовал ее для получения производных
витамина К [уравнение (16.5)]; Семмелхак и Вульф [11] ис-
Применение карбеновых комплексов и металлациклов 257
пользовали эту реакцию в ключевых стадиях синтеза
антибиотиков наномицина и дезоксифренолицина [уравнение (16.6)].
Э2 n rZ'°1
Н R
ОМе
1)55°С
2)Ад2О
R1 *Н, Me
R2- Me,
ОМе ОМе
ОМе О
(16.6)
наномицим
ОМе
Подобное аннулирование заместителей у карбена не
ограничивается арилкарбеновыми комплексами. Олефиновая группа
винилкарбенов также способна к «внутреннему захвату»
возникающего винилкетена (рис. 16.2), в результате чего образуются
простые эфиры гидрохинона, а не их аналоги, конденсированные
с бензольным ядром. Этот класс реакций находит очень
широкое применение, так как винилкарбеновые комплексы легко
получаются либо взаимодействием виниллитиевых реагентов с
гексакарбонилом хрома, либо путем конденсации альдольного
типа а-анионов алкилкарбеновых комплексов с альдегидами
в присутствии кислот Льюиса [116] (см. рис. 16.1). Реакция
винилкарбеновых комплексов была подробно изучена Дётцем
[12], применившим ее в синтезе витамина Е [уравнение (16.7)]
[13], Вульф распространил реакцию на циклические
винилкарбеновые комплексы [уравнение (16.8)] [14] и разработал
изящный подход к тетрациклиновому семейству антибиотиков
[уравнение (16.9)] [15]. В случае терминально дизамещенных
винилкарбеновых комплексов енолизация невозможна, и в
качестве продуктов с хорошим выходом выделяют циклогекса-
диеноны [уравнение (16.10) ] [16].
17-1129
258 Глава 16
OMe
BBr
Me
(CO)5Cr =
X)
R A
CO
OMe
он
(16.7)
(16.8)
H +
OMe
OMe OMe
(CO)5Cr=C
(16.9)
(CO)5Cr=C
-OMe RC=CH
(16.10)
OMe
Винильные и алкинильные карбеновые комплексы являются
также очень реакционноспособными диенофилами, они
вступают в реакцию Дильса — Альдера с разнообразными диенами,
гладко давая продукты циклоприсоединения с хорошими
выходами [17]. В случае алкинильных карбеновых комплексов об-
Применение карбеновых комплексов и металлациклов 259
разующиеся аддукты Дильса — Альдера представляют собой
винилкарбеновые комплексы, которые способны вступать в
описанную выше реакцию аннулирования с алкинами. Вульф [18]
разработал комбинированную последовательность циклоприсое-
динения — аннулирования, с помощью которой в одном
реакционном сосуде можно получить довольно сложные органические
соединения из простых предшественников [уравнение (16.11)].
.ОМе
(СО)5Сг =
TMSO
' TMS
ОМе
(i6.ll)
TMS
ОМе
Рг-я
CMs
Вульф [19] разработал также родственный метод, при
котором два эквивалента алкина взаимодействуют с одним
эквивалентом монооксида углерода и одним карбена [уравнение
(16.12)]. Этот метод позволяет избежать необходимости
предварительного получения ненасыщенных карбеновых комплексов,
но он не эффективен в случае простых алкинов из-за разнооб-
MeO,
R'
чс:
о
с
ЫеО
(16.12)
= Cr, W
разия конкурентных процессов. Частично эти проблемы можно
решить, вводя в реакцию диины, что делает включение второго
алкина внутримолекулярным процессом, более
конкурентоспособным по сравнению с нежелательными побочными реакциями
[уравнение (16.13)]. Конечным продуктом этой реакции явля-
17*
260 Глава 16
ются фенолы, образующиеся in situ в результате восстановления
а-метоксидиенона соединением металла (0).
(со)5м=с
M = Cr,W
X = СН2, О, (СН2)2, C(C02Et)2
ОМе
СО
ОМе
ОМе
(16.11
ОМе
М=гС
ОМе
Несколько иначе реагируют с алкинами вольфрамкарбенс-
вые комплексы. Джофрой [20] тщательно исследовал взаимс-
действие дифенилацетилена с метоксифенилкарбеновым
комплексом вольфрама [уравнение (16.14)]. При УФ-облучении
(co)5w=c:
-ОМе
' Ph
Ph-=-Ph
ОМе
(16.14
ОМе
Ph ^ H
реагентов при низкой температуре наблюдается выброс СО и
последующее комплексование с ацетиленом. Ацетиленовый ад-
Применение карбеновых комплексов и металл ациклов 261
дукт, устойчивый при температурах ниже —30 °С, был
охарактеризован. При нагревании выше —20 °С образуется инден.
Тот же продукт получается при термической, а не
фотохимической реакции. В случае вольфрамкарбеновых комплексов,
содержащих в а-положении атомы водорода, реакция идет
другим путем. Макомбер показал, что при взаимодействии этих
комплексов с алкинами образуются сопряженные еноны;
предполагается, что при этом осуществляется следующая
последовательность: циклоприсоединение — ^-элиминирование
гидрида— восстановительное элиминирование [уравнение (16.15)].
(16.15)
(co)5w=c;
.ОМе
PhCH
3 ,
юо°с
(C0)5W~"C"~0Me
R'
Ph
H OMe
(CO)5W \y
Ph R'
MeO
Ph
Ph R
Известно много других карбеновых комплексов, но их
реакции с алкинами практически не изучались. Недавно Семмелхак
показал, что карбеновые комплексы железа реагируют с
алкинами, давая пироновые комплексы [уравнение (16.16)].
Механизм этой замечательной реакции не известен, хотя и
высказывались некоторые довольно сложные предположения,
объясняющие образование продуктов [22] [уравнение (16.17)]. Однако
взаимодействие карбеновых комплексов с алкинами еще требует
дальнейшего изучения.
б. Реакции карбеновых комплексов переходных металлов с
олефинами. Широкое изучение реакций карбеновых комплексов
с олефинами привело к разработке двух родственных процессов,
которые находят все большее применение в синтезе: это мета-
тезис олефинов и циклопропанирование. Фундаментальные
аспекты реакции метатезиса олефинов подробно обсуждались
в ч. 1, гл. 9; здесь же будет рассмотрено применение реакции в
органическом синтезе.
Карбеновые комплексы активно катализируют процесс
метатезиса олефинов [уравнение (16.18)], но на практике
предварительно полученные комплексы редко употребляют в качестве
катализаторов. Обычно в реакцию вводят смесь
предшественников катализатора, которую подбирают эмпирическим путем.
262 Глава 16
Et
V
EtO
Fe(C0)3
Fe(CO)3
. OEt
4Ph
(C0)4Fe ГГ
О
o=c
OEt
Ph
OEt
(CO)4Fe-4—Ph
(16.17)
CO
EtO
Ph
EtO
(C0)4Fe
Большинство работ в этой области выполнено на простых оле-
финах, но изучались и некоторые более сложные системы. Так,
путем метатезиса олефинов с последующим гидроцирконирова-
нием Гибсон [23] синтезировал алкилиодиды и спирты с
длинной цепью и концевыми функциональными группами [уравнение
(16.19)]. (Напомним, что гидроцирконирование сопровождается
перегруппировкой с образованием алкильных комплексов с
концевыми группами.)
(16,18)
R'
CnH2n+!CH=CH2
R'
WCI6/Me4Sn
EtOAc
м=сн2 +
Cp2ZrHCI
"w"'
(16.19)
CH2=CH2
R!
ROH
02
Применение карбеновых комплексов и металлациклов 263
Новый толчок синтетическому применению реакции
метатезиса дала разработка систем, в которых метатезису
подвергаются функционально замещенные олефины [24]. Эти системы
очень удобны для получения производных жирных кислот с
длинной цепью. Так, сложные эфиры ненасыщенных кислот с
длинной цепью претерпевают самометатезис [уравнение (16.20)]
[25] и сометатезис [уравнения (16.21) [26] и (16.22) [27]],
давая линейные продукты с неплохими выходами. Следует
отметить, что в тех же условиях ненасыщенные кетоны и кетали
не вступают в реакцию.
(16.20)
^\(cH2)gC02Me MeOgClCHgJeC^CHtCl^gCOgMe
71%
♦ сн2=сн2
(16.21)
СН3(СН2)7СН=СН(СН2)7СО2Ме ♦ СН2=СН2
8О'С 68%
(16.22)
Y=CO2Me, CH2OAc
68%
Цудзи [25] использовал внутримолекулярный вариант
эюго процесса метатезиса для синтеза макроциклических лак-
тонов [уравнение (16.23)], хотя и с низкими выходами.
Ненасыщенные нитрилы [28] и тозилаты [29] с длинной цепью
также подвергаются метатезису подобно реакциям (16.20) и
(16.21). Известен один пример метатезиса функционально
замещенных алкинов [уравнение (16.24),], [30].
о . о
(16.23)
264 Главк 16
Mo (СО) е бензол
PhC = C— (CH)Y
Y = OH, OAc, Br, CO2H, CO2Me, CN (16.24)
Предварительно полученные электрофильные карбеновые
комплексы претерпевают метатезис, если он энергетически
выгоднее конкурентного процесса циклопропанирования. Кэйзи
[31] показал, что относительно неустойчивый дифенилкарбено-
вый комплекс вольфрама вступает в реакцию метатезиса с
енольными эфирами с образованием более устойчивого меток-
сикарбенового комплекса [уравнение (16.25)]. Купер [32]
обнаружил, что устойчивость образующегося карбенового комплекса
заметно повышается даже при наличии л-метоксигруппы в фе-
нильном кольце, что способствует протеканию реакции
метатезиса [уравнение (16.26)].
Ph OMe OMe Ph (16,25)
(CO)cW=/ + =v ' - (co)5W=\ + >=•
3 \ \ . э \ /
Ph Ph Ph Ph '
(менее устой- (более
устойчивый) " "■
/Ph
(C0)5W=C
Однако гораздо чаще взаимодействие олефинов с электро-
фильными карбеновыми комплексами идет по пути
циклопропанирования. Оно наблюдается как для электронодефицитных,
так и для электронообогащенных олефинов, хотя реакции
требуют разных условий и осуществляются по разным механизмам.
В случае электронодефицитных олефинов необходима высокая
температура (выше 90 °С), ключевая стадия — потеря СО.
Реакция стереоспецифична, при наличии хиральных лигандов
хиральность переносится в циклопропан; это указывает на
участие металла в стадии образования продукта [2]. Механизм,
согласующийся со всеми этими данными и с реакцией (16.25),
был предложен Кэйзи [33] (рис. 16.3). Этот механизм включает
потерю СО с образованием вакантного координационного
центра, координацию олефина, образование металлациклобутана.
Этот интермедиат может далее претерпевать восстановительное
элиминирование с образованием продуктов метатезиса. Блестя-
Применение карбеновых комплексов и металлациклов 265
щее доказательство роли координации олефина в этом процессе
получил Кэйзи: он выделил и охарактеризовал с помощью
рентгеноструктурного анализа ключевой интермедиат цикло-
пропанирования и показал, что он превращается в циклопропан
при нагревании или при выдерживании с избытком фосфина
[уравнение (16.27)] [34].
/ \ Д или Л пе 07.
(co)4w=\ изд' PPh3 J\J^
Tol
Электронообогащенные олефины также подвергаются цикло-
пропанированию. Реакция идет при более умеренных
температурах, но требует высокого давления СО (около 100 атм) для
подавления конкурентной реакции метатезиса. Механизм реак-
ОМе / ОМе
<CO)5W=<UMe -^^V (C0)4W=( + СО ^—^ «X»4W=/
Ph ph Y^ ' Ph
MeO
=r * (C0)4W=< ^
ции совершенно иной и не включает потерю СО. Умеренные
выходы (30—60%) продуктов циклопропанирования
ограничивают практическое использование реакции. В отличие от элект-
рофильных катионные карбеновые комплексы железа
исключительно эффективны в реакциях циклопропанирования, особенно
с олефинами без функциональных групп. Из-за неустойчивости
эти комплексы генерируют и используют in situ. Хелквист
[35] получил комплекс (диметилсульфоний) метилжелеза,
являющийся устойчивым предшественником простейшего метилиде-
нового комплекса. При нагревании его с олефинами в диоксане
с прекрасным выходом образуются циклопропаны. Реакция
266 Глава 16
стереоспецифична: из цис-олефипов получаются
^ас-циклопропаны, а из траяс-олефинов — траяоизомеры [уравнение
(16.28)]. При использовании родственного (метилфенилсульфо-
ний)этильного комплекса наблюдается аналогичный перенос
этилиденовой группы [уравнение (16.29)] [36]. В этом случае
стереохимия олефина сохраняется, но в качестве продукта
получается смесь стереоизомеров за счет разного положения ме-
тильной группы в этилиденовом комплексе.
со
(16.28)
BF4
сн3
[CpFe(CO)2CHSPhj ♦ FS03CH3
CpFe(C0)9CH-S-Ph
(16.29)
со сн3
F<
[ со^ н
I; | ци с / транс
R Me
Используя родственные комплексы метоксиалкилжелеза,
Брукхарт генерировал реагенты, способные переносить этилиде-
новую [37], бензилиденовую [38а] и
циклопропилиденовуюгруппировки [386]. Они также эффективны для циклопропанирова-
ния олефинов без функциональных групп и дают
преимущественно ^^-циклопропаны с высоким выходом [уравнение
(16.30)]. Точный механизм реакции не известен, но ясно, что
металл играет определенную роль. С этилиденовым комплексом
железа, в котором вместо СО имеется хиральный фосфиновый
лиганд, наблюдается высокой степени асимметрическая
индукция [уравнение (16.31)] [39]. Исходный комплекс получают
в виде пары диастереомеров, отличающихся только конфигура-
Применение карбеновых комплексов и металлациклов 267
дией при атоме железа. Поскольку два диастереомера дают
диклопропаны с противоположной конфигурацией практически
. одинаковым энантиомерным избытком, оптическая индукция
возникает только благодаря хиральности при атоме металла.
pFe(CO)2-C-H
R
TMS0S02CF3
S CpFe(C0)2:
- Me, Ph, цикло пропил
io
(16.30)
f= Me
Ph
Ph
транс -IR,2R
35:1
Me
Ph
выход 15°/о
ее ВЪ%
(16.31)
Те же самые катионные алкилиденовые комплексы железа
можно получить протонированием а-винильных комплексов
железа под действием сильных кислот [уравнение (16.32)] [40,
41]. Однако из-за неустойчивости олефинов и циклопропанов
в сильнокислой среде выходы продуктов циклопропанирования
олефинов в этих условиях несколько понижаются.
Cp(C0)2Fe
HBF4
8F4"
(16,32)
Нейтральные карбеновые комплексы железа, получаемые
О-алкилированием анионных ацильных комплексов («ат»-комп-
лексов) активными алкилирующими агентами, реагируют
с олефинами совсем иначе. Семмелхак [42] показал, что в
результате этих реакций получают смеси енольных и аллиловых
эфиров, по-видимому, в результате циклоприсоединения и после-
268 Глава 16
дующей миграции водорода [уравнение (16.33)]. Более
подробных данных об этом процессе не имеется,
о
Fe(C0)4= + RCCI
^OEt
(C0)4Fe = C^
(16.33)
ГМФТА
Fe(CO)5
RU
X«Ph,OEt,
. SPh,
CO2Me
EtO
EtO
OEt
(CCOFe-C-R
40-60%
в. Разложение диазосоединений, катализируемое
переходными металлами. Очень многие важные реакции органических
диазосоединений катализируются переходными металлами.
С точки зрения синтеза наибольшее значение имеет реакция
циклопропанирования олефинов [уравнение (16.34)] [43]. В
качестве катализаторов используют самые разнообразные
комплексы переходных металлов, но чаще всего трифлат меди(1)
[44], соли палладия (II) [45], ацетат родия (II) [46] и
Rhe(C0)i6 [47, 48]. Наиболее успешно реакция идет с
относительно устойчивыми диазокарбонильными соединениями, менее
подверженными разложению, которое составляет главный
конкурентный процесс. Каталитическому циклопропанированию в
присутствии диазосоединений подвергаются различные олефины,
начиная от электронообогащенных эфиров енолов и кончая
электронодефицитными а,р-ненасыщенными сложными эфира-
ми, однако алкены с электронракцепторными заместителями
реагируют, вероятно, по другому механизму.
MLn
(16.34)
Дойль и сотрудники поставили два существенных вопроса,
касающихся механизма этого процесса: промежуточное
образование карбенового металлокомплекса и роль координации оле-
фина с металлом. Участие в реакции металлокарбеновых
соединений никогда не фиксировалось прямым наблюдением,
однако оно подтверждается рядом фактов. Например, если в
реакции ^ас-олефинов с этилдиазоацетатом в качестве
катализаторов используют стерически затрудненные родий(III) порфири-
ны, реакция идет стереоселективно с образованием син-цикло-
Применение карбеновых комплексов и металлациклов 269
пропанов с высоким выходом. Высокая степень стереоселектив-
ности объясняется стерическим объемом порфирина, который
контролирует направление подхода олефина к родийкарбеново-
му комплексу [49]. В присутствии хиральных катализаторов на
основе переходных металлов наблюдается высокая степень
асимметрической индукции, что указывает скорее на
ассоциацию металла с карбеновым фрагментом, чем на реакцию
«свободного» карбена [уравнения (16.35) и (16.36)] [50—52].
r_ Cu(L')
Ph
«48% (16'35)
ее 100%
(16.36)
сог
CuL* СГ-
N2CHCO2 ментил ^ Ф
С1
48% IS ТПМНО
ее 51%
чо-
(R)
Влияние замещения в олефинах и металлического
катализатора на выход и стереохимию продуктов циклопропанирования
изучено Дойлем [уравнение (16.37) ]( [53]. Полученные им ре-
х EtO2C
N2CHC02Et М П - ZS—X (16.37)
MLn B Rh2(0Ac)4, CuChP(OiPr)3. RH6(C0),e
X = BrCH2> CICH2, PhO^-Bu, OAc, OEt, 0Ви-к? изо-Рг, трет-Ы, СИ^СРЬ
CH=CHCI, CH = CHPh,
зультаты не только продемонстрировали многосторонний
характер процесса, но также показали, что регио- и стереохимия
циклопропанирования контролируется электронными факторами
металла, а это снова означает, что карбены координируются с
270 Глава 16
металлом. Более того, Дойль установил линейную корреляцию
относительной реакционной способности и стереоселективности
для катализируемых Rh2(OAc)4 реакций фенилдиазометана с
различными олефинами и стехиометрической реакции тех же
олефинов с выделенным карбеновым комплексом вольфрама
(CO)5W=CHPh [уравнение (16.38)] [54]. Это убедительно
подтверждает участие истинных карбеновых комплексов в
катализируемых металлом реакциях циклопропанирования. На
основании полученных данных Дойль [54] предложил единый
механизм, согласно которому образованию циклопропана
предшествуют образование металлокарбенов в качестве интермедиа-
тов и ассоциация нуклеофильного олефина с электрофильным
центром металлокарбенового комплекса.
PhCHN2 * >=*
Rh?(0Ac)4
Ph
(16.38)
J о3
(CO)5W=C
^ H
Олефиновые субстраты с электроноакцепторными
заместителями должны реагировать несколько иначе. В этих случаях
также образуются продукты внедрения по связи С—Н винила,
выходы продуктов ^-замещения резко понижаются, а общая
скорость реакции не зависит от используемого катализатора.
Дойль [55] высказал предположение, что олефины, содержащие
электроноакцепторные заместители, реагируют с диазосоедине-
ниями по механизму 1,3-диполярного циклоприсоединения с
последующей экструзией N2 [уравнение (16.39)]. Металлические
катализаторы только ингибируют конкурентную таутомериза-
цию образующегося циклоаддукта в 2-пиразолин, который
оказывается главным продуктом в отсутствие катализатора.
о R
RCCHN2 + •=/
Z
-N2
^перегруппировка
N-
RC
N pi
(16.39)
Применение карбеновых комплексов и металлациклов 271
Многие другие синтетически пригодные реакции диазосоеди-
нений также катализируются переходными металлами.
Примером служит внутримолекулярный процесс превращения алкинов
в циклопропаны, которые затем превращаются в трициклические
соединения [уравнение (16.40)] [56]. Амины, сульфиды, иодиды
и ацетали превращаются в илиды, которые при обработке
сложными диазоэфирами в присутствии такого катализатора,
как Rh2(OAc)4, претерпевают [2,3]-сигматропную
перегруппировку [уравнение (16.41)] [56].
(IG.40)
N2CHCO2 Ъ^-
АсО'
Rh2(0Ac)4
ОАс
АсО'
ОАс
C0CHN2
65%
Rh2(0Ac)4
ОАс
СО2Ва- трет
О
,ОАс
40%
R2CZ
(16.41)
Переходные металлы катализируют также внедрение диазо-
группы по связи С—Н и по связи гетероатом — Н (роль метал-
локарбеновых комплексов в этих процессах не известна).
Особую ценность представляют внутримолекулярные реакции
внедрения по связи С—-Н (уравнения (16.42) [57], (16.43) [58]
а
Д, оензол
N' Си
(CH2)n-COCHN2
(16.42)
i I
100%
1, 2
I) Rh2(0Ac)4
2)ТФУК
(16.43)
100%
272 Глава 16
и (16.44) [59]). Внедрение карбена по связям N—Н успешно
используется в синтезе сложных ^-лактамов [уравнение
Rh2(OAc)4 ".
6ь/ тод 60 % диссстереосеме?\ ты в* ос ^s
'S7-./3
(16.45)] [60]. Тип внедрения сильно зависит от наличия
функциональных заместителей, так что из аналогичных переходных
соединений можно получить разнообразные продукты
[уравнения (16.46), (16.47) и (16.48)] [61—63].
он
,R ОН
-0 Rh2(0Ac)4 I -R
(16.45)
N£ CO2R
О
CO2R
(16.46)
R1
R1
SEt
= PhCH2
У
0
«C-SEt.
0^
r—f
— N
0
Ph
SR1
X^N2
R' = Ar
co2r
г. Реакции карбеновых комплексов переходных металлов с
иминами и азобензолами. Высокая реакционная способность
кратных углерод-углеродных связей по отношению к электроно-
дефицитным карбеновым комплексам позволяла предположить,
что рассмотренные выше реакции можно распространить на
соединения с кратными связями углерод — гетероатом и гетеро-
атом — гетероатом, что и нашло свое подтверждение. Так, ими-
Применение карбеновых комплексов и металл ациклов 273
(Т6.47)
CO2R1
(16.48)
0
N2 c02Me
37% 24%
ны вступают в разнообразные реакции с карбенами,
стабилизированными гетероатомами VI группы. Фишер [64] показал, что
незамещенные по азоту имины проявляют свойства нуклеофи-
лов, они атакуют электрофильный карбеновый углерод и
замещают метоксигруппу, что приводит к новому карбеновому
комплексу [уравнение (16.49) ]. Магуайр и Хигедас [65] изуча-
ли N-замещенные имины (субстраты, для которых такой ход
реакции невыгоден) и получили неожиданные, но полезные
результаты. При нагревании до 30—50 °С наблюдается реакция
по сс-углероду [уравнение (16.50)], в результате отрыва кислого
сс-протона (р/Са = 8 в воде) и последующего алкилирования
образующегося при этом электрофильного иминиевого интерме-
диата получается новый карбеновый комплекс. В условиях
облучения видимым (солнечным) светом те же субстраты реагируют
совершенно иначе, давая р-лактамы; реакция идет стереоспеци-
фически и с высоким выходом [уравнение (16.51)]. Во всех
случаях в качестве продукта получается только один диастерео-
изомер. Реакция носит общий характер, большинство
замещенных иминов, в том числе хинолины, дигидроизохинолины, бензо-
тиазины, тиазины, тиазолины, оксазины, оксазолины [66],
превращаются в ^-лактамы. Оптически активный сложный эфир
D-тиазолина [уравнение (16.52)] энантиоспецифически
превращается в производное пенама, давая один энантиомер [5-лакта-
18—1129
274 Глава 16
tco)5cr=c
(CO)5Cr
(16.50)
(co)5cr=c;
' CH2-CH-Ph
NHR
(C0)5Cr=C
RNH2
Ph
(CO)5Cr=C
■ NHMe
Ph
ма. Реакция хиральных ациклических иминов идет с более
низкой степенью асимметрической индукции (около 60%).
R!
hv
Et20 или
ТГФ
MeO
(16.51)
PhCH=CH;
e, Ph, H,
{CC)5Cr-C
Me
/
ч—■
СО 2 Me
Me.
MeO1
: R,S, R)
(16.52)
Механизм этой реакции не описан, но можно представить
правдоподобную последовательность стадий, как показано на
рис. 16.4. При облучении карбеновый комплекс переходит в
возбужденное состояние, при котором карбеновый углерод
приобретает свойства нуклеофила. Атака карбенового углерода на
злектрофильный иминный углерод и атака хрома азотом приво-
\ Применение карбеновых комплексов и металлациклов 275
дят к металлациклическому интермедиату. Внедрение СО с
последующим восстановительным элиминированием дает ^-лактам
и хромосодержащий фрагмент с недостающим СО. «Циклопри-
соединение» может быть многостадийным и включать
образование связи С—С, а затем образование связи Сг—N. В то же
время азот уже может оказаться координированным с металлом
за счет своей неподеленной электронной пары и стать лигандом
вместо одной из групп СО, тогда начальная стадия образования
связи С—С станет внутримолекулярной. Вполне вероятна
некоторая ассоциация имина с карбеновым комплексом, так как
время жизни металлоорганических комплексов в
фотовозбужденном состоянии слишком короткое для того, чтобы они
участвовали в межмолекулярной реакции.
Г
(со)псг-с;
,ОМе
^Ме
.ОМе
ОМе
(co)5cr=c;
Me
(C0)4Cr=C
OMe
MeO
R3'
Рис. 16.4.
Неожиданные результаты получил Хигедас [67],
распространивший эту реакцию на азобензолы. Облучение солнечным
светом карбеновых комплексов хрома и азобензолов приводит
к смеси имидатов, состоящей из ожидаемых 1,2-диазетидинонов
и совершенно неожиданных 1,3-диазетидинонов [уравнение
(16.53)]. Соотношение продуктов зависит от условий реакции.
В мягких условиях, при 0°С и небольшой интенсивности света,
преобладающим продуктом оказывается 1,3-диазетидинон.
18*
276 Глава 16
Ph-N=N-Ph + (CO)5Cr=c;
hv
Me
OMe
Me
OMe
Me—hN'
Ph
.Ph
(16.53)
(1,2)
Me
■и-
OMe
h 0,3,
Образование 1,3-диазетидинонов и имидатов в реакции
азобензолов с карбеновыми комплексами хрома трудно было
ожидать. Вероятная последовательность стадий, в результате
которой эти продукты могут образоваться, представлена на рис.
16.5, однако она нуждается в экспериментальной проверке.
.ОМе
(СО)5Сг=С ♦ PhN=NPh
hv
(CO)nCr-C
Ph
Me
Рис. 16.5.
Для проведения реакций с азобензолами необходимо
облучение. Разумно предположить, что первая стадия заключается в
Применение карбеновых комплексов и металлациклов 277
фотоциклоприсоединении азобензола к карбеновому комплексу
хрома с образованием диазаметаллациклобутана. При большой
интенсивности света предпочтительнее внедрение СО и
восстановительное элиминирование с образованием 1,2-диазетидинона
(путь а). Подобное метатезису расщепление должно привести
к комплексу нитрен-имидат, не имеющему аналогов. С другой
стороны, повторное присоединение фрагмента иминоэфира
должно привести к металлациклу, который мог бы
подвергнуться внедрению СО и восстановительному элиминированию с
образованием 1,3-диазетидинона (путь б). Этот путь
предпочтительнее при низкой интенсивности света. Промежуточно
образующиеся комплексы могли бы подвергаться термическому
распаду с образованием имидата и «нитренового» комплекса
(путь в). Действительно, повышение температуры
благоприятствует образованию имидата, а не диазетидинона; из темного
содержащего хром остатка при подкислении выделяется анилин,
В условиях реакции оба диазетидинона устойчивы и не фрагмен-
тируют с образованием имидата.
Процесс включает мнимую реакцию метатезиса азосоедине-
ния, не имевшую аналогов ко времени ее обнаружения. Однако
Коттон [68] наблюдал аналогичную реакцию с участием димер-
ного комплекса ниобия, приводящую к устойчивым «нитрено-
вым» комплексам [уравнение (16.54)], а Вайс [69] разработал
реакцию метатезиса карбодиимидов с участием комплекса
вольфрама [уравнение (16.55)].
dk H6.54)
С!
RoS
CI CI
CI
SR2
CI
PhN=NPh
I
\l
PhN CI
CI
*С!
SRa
1 '^
20°
Ph
(CO)5W-C-Ph
I ' о
(16.55)
"Ph
I4O°C
R3N=C=NR2
(мета тезис)
16.2. Реакции нуклеофильных карбеновых комплексов
Широкое исследование химии алкилиденовых комплексов
тантала проведено Шроком (ч. 1, разд. 3.5, д) [70]. В этих
278 Глава 16
комплексах алкилиденовые лиганды стабилизированы дониро-
ванием я-электронов от электронообогащенного атома металла^
поэтому формально они представляют собой дианионные четы-
рехэлектронные доноры (ч. 1, табл. 2.2), карбеновый углерод в
таких лигандах нуклеофильный. Во многих реакциях эти
комплексы можно рассматривать как металлоорганические аналоги
классических илидов (реагентов Виттига). Так, при
взаимодействии с неопентилидентанталом многие карбонильные
соединения превращаются в олефины [уравнение (16.56)] [71]. Под
действием этих комплексов даже сложные эфиры и амиды
подвергаются олефинизации, образуя с высокими выходами
эфиры енолов и енамины соответственно, что резко отличает
их от реагентов Виттига. Отчасти это объясняется очень
высоким сродством к кислороду переходных металлов первых групп,
имеющих высокую степень окисления. Шварц [72] наблюдал
аналогичную картину со стерически затрудненными карбеновы-
ми комплексами циркония [уравнение (16.57)]. Однако, как
показал Кауфман [73] на примере очень простой системы, для
осуществления реакции совсем не обязательны столь сложные
комплексы [уравнение (16.58)].
(16.56)
СМвЗ II I To^-Hi
н , °tR '
u R - 70-90%
R= H, Me ?
R'= Ph, OEt, Me2N» Me
0
R
^ (16.57)
0 (, ^R
Ph OMe Ph
PhP 70-97%
ffrnop-Bu, циклогексил
Применение карбеновых комплексов и металлациклов 279
(16.58)
О
CI3Mo(THF)£
R1'
60-90%
По-видимому, для олефинизации карбонильных соединений
наиболее удобен реагент Теббе, образующийся при
взаимодействии CpTiCl2 с триметилалюминием [уравнение (16.59)] [74];
в присутствии пиридина этот комплекс действует как эквивалент
CpTi=CH2. Грабе [75] показал, что он превращает кетоны,
(16.59)
Cp2TiC!2 ♦ 2 А1Ме3 Cp2Ti7 ^ "° РУ
+ Ме2А1С1-ру ♦ СН4 ♦ Ме2А1С1
альдегиды, лактоны, сложные эфиры, амиды и даже карбонаты
в соответствующие метиленовые производные с прекрасными
выходами. В отличие от реагентов Виттига комплекс Теббе не
енолизует кетоны, поэтому из оптически активных кетонов
можно получить метиленовые производные без рацемизации
[уравнение (16.60) ] [76]. В реакцию с этим комплексом можно вво-
(16.60)
|f Me
м м 0"z r 0_/ -_.
0 м Et Me \ Et
Мб ~"£
70%
дез рацемизации
дить субстраты, содержащие разнообразные функциональные
заместители, что позволяет использовать его для синтеза
сложных молекул (уравнение (16.61) [77], уравнение (16.62) [78]).
Хлороангидриды и ангидриды кислот вместо ожидаемых олефи-
нов образуют еноляты [75]. Все эти данные согласуются с
механизмом, представленным в уравнении (16.63).
Под действием реагента Теббе в присутствии оснований оле-
фины дают устойчивые металлациклы [ч. 1, разд. 9.1, а и
уравнение (16.64)]. Последние легко вступают в обмен с другими
олефинами и в результате метатезиса (ч. 1, разд. 9.1, г)
образуют новые металлациклы. Комбинируя эти реакции с
обсуждавшимися выше реакциями кетонов, Грабе [79] разработал
удобный метод синтеза тетразамещенных алленов [уравнение
(16.65)]. Металлациклические интермедиа™ сами способны к
280 Глава 16
Cp2Ti=CH2
OR1
94%
(16.62)
OR1
85%
X»H,R,0RtNR2
(16.63)
дальнейшим превращениям [уравнение (16.66)]. Применение
этих реакций к более сложным системам еще ждет своего
развития.
16.3. Применение других металлациклов в органическом синтезе
Химия металлациклов применительно к метатезису олефинов
и алкинов подробно рассматривалась в ч. 1, гл. 9, процессы
полимеризации с участием металлациклов обсуждались в гл. 11.
Практически все приведенные выше реакции карбенов
происходят через металлациклические интермедиаты. Теперь
остановимся на ряде процессов, включающих металлациклы, не
являющиеся производными карбенов.
Напряженные циклические соединения подвергаются
окислительному присоединению с образованием неустойчивых метал-
лациклобутанов. Нойори [80] изучил несколько реакций цикло-
присоединения между циклопропанами и олефинами в
присутствии комплексов никеля (0); предполагается, что в качестве
интермедиатов здесь образуются металлациклобутаны. Так, би-
цикло [2.1.0] пентан в присутствии никелевого (0) катализатора
реагирует с активированными олефинами — акрилонитрилом,
метилакрилатом, диметилмалеатом, давая норборнаны (бицикло-
[2.2.1]гептаны), как показано в уравнении (16.67). Реакция,
Применение карбеновых комплексов и металлациклов 281
СН2
ос кование
Ме2А1С1
(16.64)
Cp2Ti
ср2т;
Cp2Ti
Rl
R2
H
I
(16.65)
2 R
(16.66)
видимо, включает окислительное присоединение напряженного
циклопропана к никелю (0), приводящее к металлациклобутану.
Внедрение олефина по связи углерод — никель и последующее
восстановительное элиминирование завершают процесс. Такой
механизм кажется правдоподобным, однако не имеет
достаточных экспериментальных подтверждений. В тех же условиях из
бицикло [ 1.1.0] бутана получаются другие продукты [уравнение
(16.68)] [81]. Считают, что реакция также начинается с
окислительного присоединения, однако затем гораздо более напряжен-
282 Глава 16
ный никелациклобутан фрагментирует с образованием олефин-
карбенового комплекса. Под действием последнего субстрат
(16.67}
Ni(O)
Ni
/=\
подвергается циклопропанированию, давая наблюдаемые
продукты.
(16,68)
Ni(O)
CO2R
^ COER
Ni
C02R
Нойори обнаружил также, что комплексы никеля (0)
катализируют формальное [2 + 2]-циклоприсоединение некоторых
напряженных олефинов к активированным олефинам.
Предполагают, что в этом случае реакция происходит через никелацик-
лопентан, образующийся в результате «восстановительной ди-
меризации» двух олефиновых реагентов [уравнение (16.69)]
[82].
Ni(0)
(16 69)
Z=CN,
55-72%
Применение карбеновых комплексов и металлациклов 283
Бингер подробно изучил химию родственных метиленцикло-
пропанов и разработал ряд пригодных для синтеза процессов,
которые формально можно рассматривать как [3 + 2]-цикло-
присоединение (рис. 16.6). Предложенный для них механизм
представлен в уравнении (16.70) [83]. Здесь опять реакция
начинается с «восстановительной димеризации» двух олефинов,
,._=_TMS ^ + M(0)
R = Ph
76% [84]
02R
80-90% [86]
de go 64%
Рис. 16.6.
80% §6]
приводящей к металлациклопентану. В некоторых случаях этот
интермедиат подвергается восстановительному элиминированию
с образованием бицикло [3.2.0] систем. Чаще, особенно при
использовании электронодефицитных олефиновых субстратов,
первоначально образующийся металлациклопентан
перегруппировывается в металлациклопентан, вероятно, путем ^-элимини-
рования алкила, аналогичного ^-элиминированию гидрида.
Далее восстановительное элиминирование дает формальный
284 Глава 16
(16.70)
R
[3+2]-циклоаддукт. В отсутствие олефинов метиленциклопро-
паны подвергаются самодимеризации с образованием металла-
циклов, которые далее могут подвергаться реакциям внедрения
[уравнение (16.71)] [91].
(16.71)
(BipWNKCOD, —• BipyNi ) . BipyNi^V + BipyNi^V-
60%
^LA
Еще одна важная с точки зрения синтеза реакция,
происходящая через металлациклические интермедиа™, —
катализируемая переходными металлами циклотримеризация алкинов —
будет рассмотрена в гл. 18.
ЛИТЕРАТУРА И ПРИМЕЧАНИЯ
1. См. прекрасный обзор по карбеновым комплексам в органическом
синтезе: Dotz К. Я., Angew. Chem., Int. Ed. Engl., 23, 587 (1984). Реакции кар-
беновых комплексов^ с ненасыщенными углеводородами рассматриваются
в обзоре: Заятковский Л. И., Бабицкий Б. Д. — Усп. хим., 1985, 53, с. 1152—
1171.
Применение карбеновых комплексов и металлациклов 285
2. Dotz К. Н., Fischer H., Hofmann P., Kreissl F. R., Schubert U., Weiss K.y
Transition Metal Carbene Complexes, Verlag Chemie, Deerfield. Beach,
Florida, 1983.
3. Dotz K. #., Angew. Chem., Int. Ed. Engl, 14, 644 (1975).
4. a) Dotz K. H., Muhlemeier J., Schubert U., Orama O, J. Organomet. Chem.,.
247, 187 (1983); 6) Wulff W. D., Tang P.-C, McCallum J. S, J. Am. Chem.
Soc., 103, 7677 (1981).
5. Semmelhack M. F., Bozell J. /., Tetrahedron Lett., 23, 2931 (1982).
6. Dotz К. Н., Dietz R., Chem. Rev., 110, 1555 (1977).
7. Fischer H., Muhlemeier J., Markl R., Dotz К. Я., Chem. Ber, 115, 1355
(1982).
8. Dotz K. H., Muhlemeier /, Angew. Chem, Int. Ed. Engl., 21, 929 (1982).
9. Yamashita A., Scahill Г, Tetrahedron Lett, 23, 3765 (1982).
10. Dotz K. H., Pruskil /., J. Organomet. Chem, 209, C4 (1981); Dotz K. #.,
Purskil L, Muhlemeier /., Chem. Ber, 115, 1982 (1982).
11. a) Semmelhack M. F., Bozell J. /., Sato Т., Wulff W., Spiess E., Zask Л.„
J. Am. Chem. Soc, 104, 5850 (1982); 6) Wulff W. D., Gilbertson S. R.t J. Am.
Chem. Soc, 107, 503 (1985).
12. Dotz K. H., Kuhn W., J. Organomet. Chem, 252, C78 (1983).
13. Dotz K. H.t Kuhn W., Angew. Chem, Int. Ed. Engl, 22, 732 (1983).
14. Wulff W. D., Chan K. S., Tang P. C, J. Org. Chem, 1984, 2292, 1984.
15. Wulff W. D., Tang P. C, J. Am. Chem. Soc, 106, 435 (1984).
16. Tang P. C, Wulff W. D., J. Am. Chem. Soc, 106, 1132 (1984).
17. Wulff W. D., Yang D. C, J. Am. Chem. Soc, 105, 6726 (1983).
18. Wulff W. D., Yang D. C, J. Am. Chem. Soc, 106, 7565 (1984); Wulff W. D.y
Tang P. C, Chan K. S., McCallum J. S., Yang D. C, Gilbertson S. R.y
Tetrahedron, 41, 5813 (1985).
19. Wulff W. D., Kaesler R. W., Peterson G. A., Tang P. C, J. Am. Chem. Soc,
107, 1060 (1985).
20. Foley H. C, Strubinger L. M., Targos T. S., Geoffroy G. L, J. Am. Chem.
Soc, 105, 3064 (1983).
21. Macomber D. W., Organometallics, 3, 1589 (1984).
22. Semmelhack M. F., Tamura R., Schnatter W., Springer /., J. Am. Chem. Soc,,
106, 5363 (1984).
23. Gibson Т., Tulich L, J. Org. Chem, 46, 1821 (1981).
24. См. обзор по метатезису функционально замещенных олефинов: Mol J. С.,.
J. Mol. Catal, 15, 35 (1982); CHEMTECH, 13, 250 (1983).
25. Tsuji J., Hashiguchi S, J. Organomet. Chem, 218, 69 (1981).
26. Bosma R. H. A., van der Aardweg F., Mol J. C, J. Chem. Soc, Chem. Com-
mun, 1981, 1132.
27. Villemin D., Tetrahedron Lett, 24, 2855 (1983).
28. Bosma R. H. A., Kouwenhoven A. P., Mol J. C, J. Chem. Soc, Chem. Com-
mun, 1981, 1081; van der Aardweg G. C.f Bosma R. H. A., Mol J. C.r
J. Chem. Soc, Chem. Commun, 1983, 262. '
29. Daly D. G., McKervey M. A., Tetrahedron Lett, 23, 2997 (1982).
30. Villemin D., Cadiot P., Tetrahedron Lett, 23, 5139 (1982).
31. Casey C. P., Burkhardt T. /, J. Am. Chem. Soc, 96, 7808 (1974).
32. Fong L /C, Cooper N. /, J. Am. Chem. Soc, 106, 2595 (1984).
33 Casey C. P., Tuinstra H. E., Saeman M. C, J. Am. Chem. Soc, 98, 608 (1976).
34. Casey C. P., Vollendorf N. W., Haller K. J., J. Am. Chem. Soc, 106, 3754
(1984).
35. Brandt S., Helquist A, J. Am. Chem. Soc, 101, 6473 (1979); O'Connor E. /.,
Helquist P., J. Am. Chem. Soc, 104, 1869 (1982).
36. Kremer K. A. M., Helquist P., Kerber R. C, J. Am. Chem. Soc, 103, 1862
(1981).
37. Brookhart M., Tucker J. R., J. Am. Chem. Soc, 103, 979 (1981); Brookhart ЛГ.,.
Tucker J. R., Husk G. R.y J. Am. Chem. Soc, 105, 258 (1983).
286 Глава 16
38. a) Brookhart M., Nelson G. О., J. Am. Chem. Soc, 99, 6099 (1977); Brook-
hart M., Humphrey M. В., Kratzer H. /., Nelson G. O., J. Am. Chem. Soc,
102, 7802 (1980); 6) Brookhart M., Studabaker W. В., Organometallics, 4,
943 (1985).
39. Brookhart M.f Timmers D., Tucker J. R., Williams G. D., J. Am. Chem. Soc,
105, 6721 (1983).
40. Kremer K. A. M., Kuo G.-H., O'Connor E. J., Helquist P., Kerber R. C, J. Am.
Chem. Soc, 104, 6119 (1982).
41. Casey C. P., Miles W. H., Tukuda H., O'Connor J. M., J. Am. Chem. Soc,
104, 3761 (1982).
42. Semmelhack M. F., Tamura R., J. Am. Chem. Soc, 105, 4099, 6750 (1983).
43. Катализируемые металлами реакции циклопропанирования
рассматриваются в обзорах: Doyle М. P., in: Catalysis of Organic Reactions,
Augustine R. L. (Ed.), Marcel Dekker, New York, 1985; Burke S. D., Grieco P. A.,
Org. React., 26, 361 (1979); Marchand A. P., MacBrockway N., Chem. Rev.,
7.4, 431 (1974); Wulfman D. S., Poling В., «Metal Salt Catalyzed Carbe-
noids», in: Reactive Intermediates, Plenum, New York, 1980, Vol. 1, p. 321.
44. Salomon R. G., Kochi /. /£., J. Am. Chem. Soc, 95, 3300 (1973).
45. Nakamura A., Koyama Т., Otsuka 5., Bull. Chem. Soc Japan, 51, 593 (1978).
46. Hubert A. J., Noels A. F., Anciaux A. J., Teyssie Ph., Synthesis, 1976, 600;
Doyle M. P., van Leusen D., Tamblyn W. #., Synthesis, 1981, 787.
47. Doyle M. P., Tamblyn W. H., Buhro W. E., Dorow R. L., Tetrahedron, 22,
1783 (1981).
48. См. обзоры по катализаторам на основе переходных металлов: Doyle M. Р.,
Davidson J. G., J. Org. Chem., 45, 1538 (1980); Majchrzak M. W., Kotelko A.,
Lambert J. В., Synthesis, 1983, 469.
49. Callot H. L, Metz F., Prechocki C, Tetrahedron, 38, 2365 (1982).
50. Matlin S. A., Lough W. J., Chen L., Abram D. M. H.y Zhow Z., J. Chem. Soc,
Chem. Commun., 1984, 1038.
51. Aratani Т., Yoneyoshi Y., Nagase Г., Tetrahedron Lett., 23, 685 (1982).
52. Laidler D. A., Milner D. /., J. Organomet. Chem., 270, 121 (1984).
53. Doyle M. P., Dorow R. L., Buhro W. E., Griffin J. H., Tamblyn W. H., Tru-
dell M. L., Organometallics, 3, 44 (1984).
54. Doyle M. P., Griffin /. H., Bagheri V., Dorow R. L., Organometallics, 3, 53
(1984).
55. Doyle M. P., Dorow R. L., Tamblyn W. H., J. Org. Chem., 47, 4059 (1982).
56. Doyle M. P., Tamblyn W. H., Bagheri V., J. Org. Chem., 46, 5094 (1981);
Doyle M. P., Griffin J. H., Chinn M. S., van Leusen D., J. Org. Chem., 49,
1917 (1984).
57. Jefford C. W., Johncock W., Helv. Chim. Acta, 66, 2666 (1983).
58. McKervery M. A., Tuladhar S. M.f Twohy M. F., J. Chem. Soc, Chem.
Commun., 1984, 129.
59. Taber D. F., Raman K., J. Am. Chem. Soc, 105, 5935 (1983).
60. Hirai K., Iwaro /., Fujimoto K., Tetrahedron Lett., 24, 3251 (1983); Natsu-
gari H., Matsushita Y., Tamura N., Yoshioka K., Kondo M., Okonogi K.,
Kuno M., О Mai M., J. Antibiot., 36, 855 (1983).
61. Fliri H.} Mak C.-P., Prasad K., Schulz G., Stutz P, Heterocycles, 20, 205
(1983).
62. Kametani Т., Kanaya N., Mochiyuki Т., Honda Г., Tetrahedron Lett., 24, 221
(1983).
63. Taylor E. C, Davies H. M. L., J. Org. Chem., 49, 113 (1984).
64. Fischer E. O., Hollfelder H., Kreissl F. R., Uedelhoven W., J. Organomet.
Chem., ИЗ, С31 (1976).
65. Hegedus L. S., McGuire M. A., Schultze L. M., Yijun C, Anderson O. P.,
J. Am. Chem. Soc, 106, 2680 (1984).
66. Hegedus L. S., Schiltze L. M., Того J., Yijun C, Tetrahedron, 41, 5833
(1985).
Применение карбеновых комплексов и металлациклов 287
67. Hegedus L. S., Kramer Л., Organometallics, 3, 1263 (1984).
68. Cotton F. A., Duraj S. A., Roth W. /., J. Am. Chem. Soc, 100, 4749 (1984).
69. Weiss K., Kindl P., Angew. Chem., Int. Ed. Engl, 23, 629 (1984).
70. Schrock R. R., Ace. Chem. Res., 12, 98 (1979).
71. Schrock R. R. J. Am. Chem. Soc, 98, 5399 (1976).
72. Hartner F. W., Schwartz /., Clift S. M., J. Am. Chem. Soc, 105, 640 (1983).
73. Kauffmann Т., Еппеп В., Sander /., Wieschollek R., Angew. Chem., Int. Ed.
Engl., 22, 244 (1983).
74. Tebbe F. N., Par shall G. W., Reddy G. S., J. Am. Chem. Soc, 100, 3611
(1978). Получение in situ в больших количествах описано в работе: Canniz-
zo L F., Grubbs R. #., J. Org. Chem., 50, 2386 (1985).
75. Brown-Wensley K. A., Buchwald S. L., Cannizzo L., Clawson L., Ho S.,
Meinhardt D., Stille J. R., Straus D., Grubbs R. #., Pure Appl. Chem., 55^
1733 (1983); Cannizzo L. F., Grubbs R. #., J. Org. Chem., 50, 2316 (1985).
76. Ireland R. E.t неопубликованные результаты, приведенные в [75].
77. Evans D., неопубликованные результаты, приведенные в [75].
78. Wilcox С. S., Long G. W., Suh #., Tetrahedron Lett., 25, 395 (1984).
79. Buchwald S. L., Grubbs R. #., J. Am. Chem. Soc, 105, 5490 (1983).
80. Takaya H. Suzuki Т., Kumagai Y., Yamakawa M., Noyori R., J. Org. Chem.,,
46, 2846 (1981).
81. Takaya #., Suzuki Т., Kumagai Y., Hosoya M., Kawanchi H., Noyori R., J.
Org. Chem., 46, 2854 (1981).
82. Takaya H., Yamakawa M., Noyori R., Bull. Chem. Soc Japan, 55, 582 (1982);
см. также: Ishii Y., Kawahara M., Noda Т., Ishigahi #., Ogawa M., Bull
Chem. Soc Japan, 56, 2181 (1983).
83. Binger P., Brinkmann A., Wedemann P., Chem. Ber., 116, 2920 (1983).
84. Binger P., Bentz P., J. Organomet. Chem., 221, C33 (1981); Binger P.,
Betz P., Angew. Chem., Int. Ed. Engl., 21, 1982 (1982).
85. Binger P., Schuchardt U., Chem. Ber., 114, 3313 (1981).
86. Binger P., Brinkmann A., Richter W. /., Tetrahedron Lett., 24, 3599 (1983).
87. Binger P., Wedemann P., Tetrahedron Lett., 24, 5847 (1983).
88. Binger P., Lu Q.-H., Wedemann P., частное сообщение.
89. Buch H. M., Schroth G., Mynott R., Binger P., J. Organomet. Chem.., 247^
C63 (1983).
90. Binger P., Weintz H. /., Chem. Ber., 117, 654 (1984).
91. Binger P., Doyle M. J., Benn R., Chem. Ber., 116, 1 (1983).
17
ПРИМЕНЕНИЕ АЛКЕНОВЫХ, ДИЕНОВЫХ
И ДИЕНИЛЬНЫХ КОМПЛЕКСОВ ПЕРЕХОДНЫХ
МЕТАЛЛОВ В ОРГАНИЧЕСКОМ СИНТЕЗЕ
Образовав комплексы с переходными металлами, олефины,
диены и диенильные производные меняют свою обычную
реакционную способность и 'становятся объектами нуклеофильной
атаки, в результате которой образуются новые связи между
нуклеофилом и олефиновым углеродом, а также новые связи
металл — углерод, способные претерпевать дальнейшие
превращения [уравнение (17.1)]. Этот процесс широко используется
в синтезе сложных органических соединений; его теоретические
основы обсуждались в ч. 1, гл. 7, здесь будет рассмотрено его
лрактическое применение.
Nuc
(17.1)
17.1. Нуклеофильная атака на комплексы
(т]2-олефин)палладий(П) [1]
В органических реакциях наблюдается быстрая координация
солей палладия (II) с моноолефинами, приводящая к димерным
комплексам олефин-палладий (II) галогенид. Наиболее гладко
идет координация этилена, моноолефинов с концевой двойной
связью, а затем цис- и граяс-дизамещенных олефинов. Геми-
нально дизамещенные, три- и тетразамещенные, а также элек-
трофильные олефины координируют недостаточно для того,
чтобы с комплексами можно было проводить дальнейшие реакции.
Координированные с палладием олефины, как правило,
подвержены нуклеофильной атаке; она происходит быстро и преиму-
Ш
Применение алкеновых, диеновых и диенильных комплексов 289
щественно по более замещенному положению олефина, в
результате образуется связь углерод — нуклеофил и о-связь
углерод— палладий. В большинстве случаев атака осуществляется
со стороны, противоположной металлу, без предварительной
координации нуклеофила (но последнее зависит от его природы).
Получающиеся при этом а-алкилпалладиевые -комплексы
довольно неустойчивы, они претерпевают быстрое спонтанное
^-элиминирование гидрида, давая продукт замещения ,и
«PdHCl», который разлагается на палладий (0) и НС1. В
присутствии подходящих окислителей (СиС12, бензохинон, K2S2O8,
FeCl3) палладий (0) подвергается повторному окислению до
палладия (II) и может снова участвовать в каталитическом цикле
(рис. 17.1). Все эти стадии — комплексование олефина, нук-
~ Л r Nuc
PdC,2(RCN,2 * =J *=* ^>
окисление
Nuc
HCI ♦ Pd(O) * "PdHCl"
Sb/cmpo - PdH
Рис. 17.1.
леофильная атака, ^-элиминирование и реокисление—
происходят очень быстро, поэтому соли палладия(II) служат
эффективными катализаторами во многих важных реакциях нуклеофиль-
ного замещения олефинов.
Одной из первых таких реакций, нашедшей практическое
применение, стал Вакер-процесс — окисление этилена в ацеталь-
дегид в присутствии палладиевого катализатора. Механизм
этого процесса неоднозначен, но очевидно, что ключевая стадия
включает нуклеофильную атаку водой по связанному с
палладием олефину. Как и можно было ожидать, моноолефины
с длинной цепью атакуются исключительно по более
замещенному (внутреннему) положению (см. ч. 1, гл. 7), и если нуклео-
филом служит вода, образуются кетоны [уравнение (17.2)].
Реакция в достаточной мере специфична, так что можно легко
отличить монозамещенные олефины с концевой группой от
менее сильно комплексующихся олефинов с внутренней связью.
Реакция идет в присутствии удаленных функциональных групп
[уравнения (17.3) и (17.4)]. Цудзи [2] разработал ряд
интересных синтезов, в которых рассматриваемое селективное
превращение олефинов в кетоны сочетается с известными реакциями
енолятов [уравнение (17.5)].
19-1121
290 Глава 17
PdCI2/O2/CuCI2 °
RCH=CHa RC-CH3
(17.2)
(17.3)
(17.5)
С олефинами, имеющими внутреннюю двойную связь,
реакция асли 1и идет, то очень медленно и с низкой региоселектив-
ностью. Однако в тех же условиях а,[3-ненаеыщенные сложные
эф'иры и кетоны гладко превращаются в [3-дикарбонильные
соединения [уравнение (17.6)], а простые и сложные аллиловые
эфиры — в "(-кетопроизводные [уравнение (17.7)].
(17.6)
(17.7)
В основе удобного метода синтеза кислородсодержащих ге-
тероциклов лежит внутримолекулярная атака 'нуклеофильного
кислорода на олеф'иновые 'комплексы палладия(II). Так, Мура-
хаси [4] провел циклизацию 2-аллилфенола в 2-метилбензофу-
ран с неплохим выходом [уравнение (17.8)]. В отличие от
этого фенолы с метилзамещенными аллильными группами при
Применение алкеновых, диеновых и диенильных комплексов 291
циклизации дают смеси пяти- и шестичленных циклических
продуктов, состав которых зависит от того, какая соль
палладия (II) используется [PdCb или Pd(OAc)2], а также от
количества добавляемого хлорида или ацетата [уравнение (17.9)]
[4].
Pd(OAc)z
Cu(OAc)2,
Pd
OH
(17,8)
— Pd—
-PciH
перегруппировка
(17.9)
^ - Li С! или
NaOAc
к
Отличные от фенолов олефиновые спирты циклизуются
в присутствии палладиевых катализаторов € образованием
разнообразных кислородсодержащих гетероциклов. Семмелхак [5]
объединил эту реакцию циклизации с внедрением СО в
неустойчивые промежуточные а-ал-кшшалладиевые соединения
[уравнение (17.10)]. Здесь, как >и обычно, наблюдается атака по более
замещенному концу олеф'ина. Из дизамещенных олефинов
с внутренней связью преимущественно образуются шестичлен-
ные циклы, если это в принципе возможно [уравнение (17.11)].
(17.10)
ОН
катализатор
МеОН/СО,
CuCIo
Pd
СО, МеОН
ОН
19*
СО 2 Me
(17.11)
70 : зо
292 Глава 17
При наличии в субстрате подходящим образом
расположенной гидрокоильной группы независимо от исходной
конфигурации несущего гидроксил углерода образуются цис-лштояы
[уравнение (17.12)] [6]; это указывает на то, что стадия,
предшествующая образованию лактона, обратима.
(17.12)
Pd(OAc)2
ОН Pd
СО
6 4 % цис, 13 % транс
по отношению к
&екз иль но/чу Me
Достаточно хорошо идут 'внутримолекулярные реакции, в
которых нуклеофилом служит карбоксилат-ион; в результате
получаются ненасыщенные лактоны [уравнения (17.13) —
(17.15)]. Большинство реакций проводят со стехиометрическими
количествами палладия, но некоторые были выполнены с
каталитическими количествами. В -принципе для всех
рассмотренных реакций возможен 'каталитический процесс.
RCH=CHCH2COOH
Li2PdCI4
или ^
О.
RCH=CHCH2CH2C00H
n. = O,
R2 С-СН(
r' = H, Me, Ph
R2=H, Me, Ph
COOH
|
№.■
X
^R
Li2PdCI4
H20 " R2>
PdCI2(CH3CN)2
ТГФ, Na2CO3
0
{
X
R1
X
)
XT
= Me, Et,
= H, 6-CI
.R1
?f-U30
,7-OMe
(17.14) [в]
(17.15) [9]
Применение алкеновых, диеновых и диенильных комплексов 293
Амины также атакуют связанные с палладием олефшш, но
каталитическое "межмолекулярное аминирование олефинов
осуществить не удается, поскольку и исходный амин, и енаминовый
продукт, будучи потенциальными лигандамш палладия,
отравляют катализатор. Тем не менее стехиометрическое
аминирование связанных с палладием олефинов и последующие реакции
образующегося при этом а-алкилпалладиевого соединения были
использованы для довольно необычных превращений олефинов.
Например, Бэквалль [10] ировел окислительное расщепление
неустойчивых а-алкилпалладиевых комплексов с введением нук-
леофилов к обоим атомам углерода бывшего олефина
[уравнение (17.16)]. Реакция стереоспецифична и дает в конечном сче-
(17.16)
|| «■ PdCI2(RCN)2 ♦ 3R'2NH
NR'2
Pd-CI
NHR'2
Pb(0Ac)4
те ^г/опродукты, образующиеся в результате граяс-аминирова-
ния олефина (атака со стороны, противоположной металлу) и
последующего нуклеофильного замещения (окисленного)
палладия с инверсией конфигурации. Эффективными окислителями
в этом процессе служат бром, тетраацетат «свинца и N-бромо-
сукцини'мид. В этих условиях оксаминирование простых арило-
вых эфиров дает продукты, содержащие несколько
функциональных заместителей, с хорошими выходами [уравнение
(17.17)] [11]. При использовании в качестве лиганда хирально-
го третичного амина, а в качестве нуклеофила хирального
вторичного амина продукты оксаминирования олефинов
получаются с умеренными выходами, а величина ее составляет около
60% [12]. Если же вторичный амин (нуклеофил) ахиральный,
величина ее достигает лишь 3—11%. При проведении реакции
с простыми арилаллило'выми эфирами в качестве одной из
стадий в синтезе соединений, родственных р-блокаторам,
наблюдается умеренная оптическая индукция (величина ее порядка
11%). Использование в качестве нуклеофила первичного амина
и последующее внутримолекулярное окислительное расщепление
приводят к азиридинам с умеренным выходом [уравнение
(17.18)] [13].
294 Глава 17
Aro
I) R2NH, PdCl2(RCN)2
2)Pb(0Ac)4
ArO'
(17.17)
NR2
OAc
(17.18)
|| ♦ PdC!2(RCN)2 «■ MeNH2
NHMe
NH2Me
В отличие от межмолекулярных реакций аминирования
внутримолекулярные процессы идут без особых затруднений.
На их основе разработано множество удобных каталитических
методов построения азотсодержащих гетероциклических систем.
Так, Хегедюс [14] показал, что индолы можно легко получить
из 2-аллиланилинов в присутствии палладиевого(П)
катализатора, используя для повторного окисления палладия бензохинон
[уравнение (17.19)]. [Поскольку и анилины и индолы довольно
легко окисляются, в присутствии меди(II) наблюдается деструк-
NH2
PdCI2(MeCN)2/
Зензоссинон
II—П
(17.19)
ция продукта.] В случае олефинов с алкилированными
боковыми цепями аминирование осуществляется по наиболее
замещенному концу цепи [уравнение (17.20)]. Эти реакции также идут
через неустойчивые а-алкилпалладиевые комплексы, которые
внедряют монооксид углерода, давая соответствующий сложный
эфир. Аналогичным образом реагируют сопряженные еноны
[уравнение (17.21)] [15]. Используя последовательность
внутримолекулярное аминирование — внутримолекулярное
внедрение, Хигедас [16] прямым путем синтезировал пирролоиндоло-
хиноны, родственные митозенам [уравнение (17.22)]. В стадии
внедрения вместо обычного [З-алкилирования происходит сс-ал-
килирование; ло-видимому, это связано со стерическим
напряжением, (возникающим во внутримолекулярной реакции.
Попытки распространить гетероциклизацию на со-олефино-
вые алифатические амины столкнулись с определенными
трудностями. Основность алифатических аминов примерно в 106 раз
выше, чем у ароматических аминов, поэтому их
координационные связи с комплексами палладия(II) намного прочнее, чем
Применение алкеновых, диеновых и диенильных комплексов 295
PdCI2(MeCN)2/
бензохикон.
(17.20)
(I7.2J}
296 Глава 17
у ароматических лигандов, и комплексы олефин-амин->палла-
дий(П) вполне устойчивы. Прочно координированный с
металлом аминный азот не способен атаковать координированный
олефин. Хигедасу удалось преодолеть эти трудности, используя
вместо свободного амина его намного менее основный тозамид.
Эти кристаллические субстраты легко циклизуются в
присутствии каталитических'количеств палладия (II) и бензохинона в
качестве реоксиданта, давая необычные циклические тозилирован-
ные енамины [уравнение (17.23)] [17]. После гидрирования и
фотолитического отщепления тозильной группы получаются
с хорошим выходом свободные циклические 'амины.
(17.23)
% PdC!2(MeCN)2
бвкзоссцнон
Pd/C
NHTs
N
Ts
2) hu, ROH
В присутствии комплексов палладия(II) множество других
олефиновых 'аминов и амидов подвергаются циклизации
[уравнения (17.24) — (17.28)]. Большинство этих реакций
проводились как стехиометрические, но они без сомнения могут идти
и в каталитическом режиме.
(17.24)
CO2Et
CO?Et
R=H, Me
PdCI2(MeCN)2
NaH
(17.25) [9]
R = H, Me, Ph
CH3CN
(I J^ (17.26)
H
R1
NH^JH2
Li2PdCl4
(17.27) [20]
Применение алкеновых, диеновых и диенильных комплексов 297
NH
PdCU
NH
(17.28) [2l]
M2N - H ^0
Целый ряд проблем возникает и при использовании карба-
нионов в качестве нуклеофилов. Палладий (II)—достаточно
сильный окислительный агент, а карбавионы легко поддаются
окислению. Так что основная реакция карбанионов с олефино-
выми комплексами палладия(П) заключается в восстановлении
палладия (II) с переносом электрона до металлического
палладия; при этом в небольших количествах образуются
сопутствующие продукты окислительного сочетания карбаниона.
Глубина протекания этого нежелательного процесса зависит от
легкости восстановления соединений палладия(II), которая в свою
очередь зависит от характера лигандов, связанных с металлом,
так что в известной мере реакцию можно контролировать. Хиге-
дасу [22] удалось провести алкилирование связанных с
палладием олефинов, подбирая подходящие удаленные лиганды
палладия. Так, при взаимодействии олефин-палладийхлоридов со
стабилизированными карбаниона'ми алкилирование не
происходит. Однако если к олефин-'палладиевому комплексу
предварительно добавить 2 экв. триэтиламина, а затем добавлять карба-
нион, то ал'килирование осуществляется с высоким выходом,
причем в реакцию вступают самые разнообразные
стабилизированные карбаниюны и олефины [уравнение (17.29)]. Монооле-
(17.29)
PdCI2(CH3CN)2
фины с концевой двойной связью алшлируются с почти
количественным выходом, преимущественно по более замещенному
положению (что согласуется с внешней нуклеофильной транс-
атакой). Электронообогащенные олефины, например енамиды,
дают продукты алкилирования с высоким выходом, но электро-
нодефицитные олефины, такие, как акрилаты, вообще не
подвергаются алкилированию. Олефины с внутренней связью дают
298 Глава 17
продукты алкилирован-ия с (выходом лишь 30—40%, но изобутен
(и циклогексен в этих условиях не реагируют. При
выдерживании неустойчивых а-алкилпалладиевых интермедиатов с
монооксидом углерода в конечном счете образуются продукты «кар-
боацилиро(вания» исходного олефина, 'как это и можно было
ожидать [23]. Из олефинов с внутренней двойной связью и хи-
ральных третичных аминов в качестве лигандов можно
получить продукты адюилирования с умеренным выходом и с
величиной ее до 32% [24]. Дигидрофуран гладко алкилируется по
а-положению к кислороду, давая а-алкилпроизводные с
выходами от умеренных до хороших [уравнение (17.30)] [25]. Ни один
из этих процессов не является каталитическим, так как
невозможно найти такой окислительный агент, который 'подходил бы
для О'Кислитель'нонвосстановительных превращений с участием
палладия и одновременно был бы совместим с используемыми
в реакции карбанионами.
&
!) PdCI2(MeCN)2
2) Et3N
3) RM, -78°C
(17.30)
RM = NaC(NHCHO)(CO2Et)2, NaClNHAcKCO^,
NaC(Me)(CO2Me)2, NaO- мС /$
Недавно разработаны две новые внутримолекулярные
реакции алкилиро'вания, для которых требуются стехиометрии ее кие
количества палладия, но 'механизм обеих реакций не
установлен. Трост описал реакцию индола, имеющего олефиновый
заместитель в положении 2, с тетрафтор бор атом серебра (I) в
присутствии хлорида палладия(II). Ход реакции зависит от
положения олефина, она может идти ню пути «электрофильного
ароматического замещения» закомплексованного олефина,
находящегося ,в положении 2 издольного кольца и отличающегося
высокой электрофильностью благодаря формально катжшному
характеру [26] [уравнение (17.31)].
(17.31)
Применение алкеновых, диеновых и диенильных комплексов 299
Кенде [27] разработал циклизацию простых силиноловых
эфиров олефи'новых кетонов в присутствии стехиометрических
количеств ацетата палладия (II) [уравнение (17.32)].
Первоначально предполагалось, что реакция происходит через я-оксал-
лилпалладиевые соединения, однако более вероятным
представляется путь, включающий нуклеофильную атаку енолового эфира
на олефин-палладиевый комплекс [28]. Процесс носит
довольно общий характер и описан на множестве примеров.
В частности, он был использован в синтезе квадрона
[уравнение (17.33)] [29]. В реакцию вступают также простые
метиловые эфиры енолов, но при этом в качестве источника палладия
нужен более электрофильный Pd(CF3CO2)2 [30].
z О4 ПП (17.32)
р
(17.33)
Н -
Палладиевые комплексы хелатирующих олефинов также
могут подвергаться иуклеофильной атаке; образующиеся при этом
о-алкильные комплексы, как правило, более устойчивы, чем
комплексы, образующиеся из простых моноолефшюв. Л арок
[31а] использовал эту реакцию для превращения норборнадие-
на в аналоги простациклина [уравнение (17.34)]. Другие хела-
тираванные олефины, особенно аллильные и гомоаллильные
амины и сульфиды, также подвержены нуклеофильной атаке
(ч. 1, гл. 7), но примеры реакций с их участием ограничены
лишь очень (простыми субстратами [316].
(17.34)
PdCI2
AgOAc
Pd
CO
Ш0Н
O(CH2)5CO2H
CO2Me
76%
Соли палладия (II) катализируют разнообразные [3,3]-оиг-
матропные перегруппировки, в результате чего наблюдается об-
300 Глава 17
разевание новых о-связей С—О, С—N, С—S и С—С [32].
Часто эти перегруппировки идут в очень мягких условиях и
с высокой степенью стер-еоспецифичности, что делает их
удобными для применения в органическом синтезе. По-видимому,
перегруппировки индуцируются циклизацией [уравнение
(17.35) ] и включают внутримолекулярную нуклеофильную
атаку на комплексовшный с Pd(II) олефин.
(17.35)
Pd
Описанный метод очень удобен для перегруппировок аллил-
ацетатав [уравнение (17.36)], имидатов [уравнение (17.37)] и
тиоимидатов [уравнение (17.38)]. Они не осложняются
скелетными перегруппировками, циклизацией или реакциями
элиминирования, но применимы только к тем системам, в которых
алильная группа не замещена в положении С-2. Гриеко [33]
показал, что стереохимия процесса супраповерхностная, что
позволяет осуществить перенос хиральности в синтезе простаглан-
динов [уравнение (17.39а)].
катализатор
25°С
ОАс
ОАс
96%
NH
С3Н
3Н7
,А
'CCI,
NHCOCh
85%
катализатор
07.37)
(17.38)
0 9
Применение алкеновых, диеновых и диенильных комплексов 301
Босних тщательно исследовал катализируемые металлом
перегруппировки N-аллилимидатов в амиды. При катализе
палладием (II) наблюдается высокая стер'еоселекти'в'ность, в то время
как в присутствии палладия (0) образуются продукты и [1,3]-,
и [3,3]-перегруппировки, что свидетельствует о разных
механизмах реакций. Один из предложенных механизмов включает
jt-аллильные интер'медиаты [уравнение (17.396)].
(17.395)
О
ЧРИ
4Pti
Оверман разработал (ряд (Практически пригодных
(перегруппировок Коупа, катализируемых палладием (II). Они идут
в очень мягких условиях, с достаточно высокой стереоселектив-
ностью, а скорость их примерно в 1010 раз выше, чем у
перегруппировок без катализатора [уравнение (17.40) ] [32]. Для
перегруппировки замещенных ациклических 1,5-диенов
необходимо, чтобы в .положении С-2 был водород, а в положении
С-5 — отличный от водорода заместитель, так что нельзя
говорить об общем характере процесса. В случае хиральных
субстратов наблюдается полный перенос хиралыноеги [уравнение
(17.41)] [34]. Считается, что катализируемая реакция
происходит через ту же конформацию кресла, что и термическая
перегруппировка Коупа.
через
(17.40)
игла
302 Глава 17
17.2. Нуклеофильная атака на комплексы
(т]2-олефин)железо(Н)
Катионные комплексы г)5-цикло{пентадиенил (олефин) дикар-
боиилжелеза легко атакуются самыми разными нуклеофилами
(ч. 1, гл. 7). В отличие от комплексов палладия (см. разд. 17.1)
образующиеся при этом о-алкильные комплексы железа доволь-
ио устойчивы, и для удаления железа требуется отдельное
химическое превращение. Большинство ранних работ
ограничивалось получением о-алкильных комплексов железа из простых
олефиногв и простых нуклеофилогв, без дальнейшего изучения
продуктов. Позднее 'были исследованы более сложные системы
и найдено много /интересных с точки зрения синтеза
превращений. Примером служит изящный синтез ^-лакзтамов,
выполненный Розенблюмом [уравнение (17.42)] [35]. Комплекс Fp-изо-
бутилен, который легко получается в виде устойчивого
кристаллического вещества, без труда вступает в обмен с другими оле-
финами. Так, при взаимодействии этого комплекса с со-олефи-
'новым сложным а-кетоэфиром получается соответствующий ка-
тионный олефиновый комплекс с прекрасным выходом. Реакция
(17,42)
N-H
Ад2О
32%
с аммиаком идет по a-кетогруппе с образованием имина, который
спонтанно атакует связанный с железом олефин, давая
устойчивый о-алкильный комплекс железа с очень хорошим
выходом. При восстановлении имина боргидрадом устойчивая
группировка a-алкилжелеза не затрагивается. Внедрение СО проис-
Применение алкеновых, диеновых и диенильных комплексов 303
ходит при нагревании и приводит >к о-ащильному производному,
стабилизированному хелатирша'нием -с азотом. [J-Лактам
получается € умеренным выходом в результате окислительного
удаления железа под действием Ag2O (индуцированное
окислением восстановительное элиминирование, см. ч. 1, гл. 5).
Электрсинообогащенные олефины типа 'енольных эфиров в
результате комплексообразования с железом становятся объектом
•нуклеофиль'ной атаки. Использовав это свойство, Розенблюм
[36] разработал эффективный метод ее-винил ирования енолятов
[уравнение (17.43)]. Даже такой электронообогащенный
субстрат, как 1,2-диметоксиэтан, будучи закомплексован с
железом, вступает в реакцию с нуклеофилами. Поскольку здесь обе
метожсигруппы можно последовательно заместить нуклеофи-
(17.43)
OLi
ОМе
лами, железоолефиновый комплекс по существу является
эквивалентом винилиден-дикатиона [ (+) СН = СН (+) ]
[уравнение (17.44)] [37]. Подобные комплексы послужили основой для
синтеза дигидрофура'нов (уравнение 17.45) и а-метиленлактонов
[уравнение (17.46)] [38].
МеО ОМе Et0H OEt OEt U7.44)
чч^^ МеО ОМе ^ \ / ьтии ^ \ /
Fp [I \=J I | +
Fp Fp
OEt
HBF4 f=T 1)Nuc"
(-EtOH) EtO Fp* 2)HBF4
QNoc- *«' Я Nuc R
304 Глава 17
0Et
OLi
^-селекглрид
(Н-)
EtO
HBF4
OEt
Fp
OEt
CO2Et
°Hl.CO2Et О у |^CO2Et
(17.46)
OEt
90%
93%
Рассмотренные выше процессы привлекательны тем, что
металл участвует последовательно -в двух реакциях и в результате
минимального числа отдельных стадий осуществляется поли-
функционализация. Еще более эффективны в этом 'плане г^-ал-
лильные лиганды (полученные впервые Розенблюмом и затем
изученные Бэйкером), которые при комплексовании с железом
участвуют в трех разных реакциях [39]. Эти комплексы
получают (разными способами, но чаще всего реакцией CpFe(C0)2~
с аллилгалогенидами или аллилтозилатами. За счет комплексо-
образования олефина с железом г^-аллилжелезная группировка
проявляет свойства нуклеофила [уравнение (17.47)]. Эти
комплексы реагируют с сильными электрофилами, такими, как тетра-
цианоэтилен (TCNE), диметилацетилендикарбо'ксилат, цианаты,
сложные эфиры малеиновой и фумаровой кислот, давая цикли-
Применение алкеновых, диеновых и диенильных комплексов 30S
ческие соединения, которые формально можно рассматривать
как продукты [3+2]-циклоприсоединения. Считается, что
реакция идет чарез последовательные стадии и начинается с нуклео-
фильной атаки о-аллильной группы на акцептор. Образующийся-
при этом аддукт представляет катионный комплекс олефин —
железо, он подвергается нуклеоф'илыюй атаке отрицательно
заряженным концом акцептора, что приводит к замыканию цикла
и возникновению нейтрального алкильного комплекса железа.
Последний может уча'ство!вать (в дальнейших реакциях
расщепления или внедрения [40] [уравнение (17.47)].
(17.47)
CpFe(CO)
R3=H, CO2R't CN
В реакцию вступают различные замещенные аллильные
комплексы, а также г^-алленильные и пропаргильные
комплексы, но для нее требуются очень сильные акцепторы. /Бэйкер
изучал эти реакции «циклоприсоединения» на примере 2- и 3-меток-
сиаллильных комплексов железа [41] и использовал их для
получения саркомицина [уравнение (17.48)] и брефельдина А
[42].
(17.48)
ОМе
ОМе
CpFe
СО
еО2С
I) HCI
CN
Розенблюм [43] применил эту реакцию в сложном синтезе
гидроазуленовой циклической системы. Здесь тропилийкарбо-
нилжелезо, будучи достаточно сильным электрофилом, подвер-
20—1129
306 Глава 17
гается атаке нуклеофильным г^-аллильным 'комплексом железа
[уравнение (17.49)]. Получающийся (при этом (комплекс с двумя
атомами железа в принципе может подвергаться дальнейшей
функционализации самыми разными путями, но к настоящему
времени описано только образование сложного эфира. Розен-
блюм [44] использовал этот процесс для аннелирования пяти-
членного цикла с циклогексежшом [уравнение (17.50)].
(17.49)
Fe(CO!,
Fe(CO)3 '
/
Fe(CO),
(17.50)
0 H
AlBr,
н
45%
Как уже говорилось, связанный с металлом олефин
становится подвержен нуклеофильной атаке, по тем же причинам он
становится более устойчив к электрофильной атаке. Николас
^использовал катион циклопентадиенилдикарбонилжелеза (Fp+)
для защиты олефина от действия электрофилов. Комплекс
Рр(изобутилен)+ВР4 способен обменивать изобутилен на другие
юлефины. С диенами преимущественно координируют менее за-
мещшные или более напряженные олефины [45]. В случае
координации олефинов с енинами алкин остается
незакомплексованным [46]. Комплексы легко распадаются под действием
иодида натрия [уравнения (17.51) — (17.53)].
17.3. Нуклеофильная атака на комплексы (т]М,3-диен)железо
и (т]5-диенил)железо
Способные к хелатообразованию диолефины легче (всего
координируют с Pd (II) и Rh(I), однако сопряженные диены
образуют наиболее прочные комплексы с железом (0). Так, г14-бута-
диентрикарбонилжелезо(О) образуется просто при нагревании
тсентакарбонила железа и бутадиена в автоклаве при 140 °С
[уравнение (17.54)]. Столь жесткие условия вызваны тем, что
Fe(CO)5 координационно насыщен и относительно инертен к за-
Применение алкеновых, диеновых и диенильных комплексов 307
мещению. Тот же комплекс можно получить в более мягких
условиях, используя Fe2(CO)9 в качестве источника «Fe(CO)4».
Координационно насыщенный диеновый комплекс настолько
устойчив, что бутадиеновый лигавд можно ацилировать по Фри-
делю — Крафтсу, не разрушив комплекс (ч. 1, гл. 3).
Несопряженные диены в присутствии карбонилов железа часто
перегруппировываются, давая устойчивые сопряженные диеновые
комплексы. Франк-Нейманн использовал комплексование
1,3-диенового фрагмента а,р,ч,б-ненасыщенных альдегидов
(17.5!)
H2/Pc:
(17,52)
57%
Fe(C0)3
2C0
(17.54)
20*
308 Глава 17
(17,55)
€ Fe(CO)3 для того, чтобы осуществить реакции по альдегидной
группе в отсутствие конкурентных реакций по диеновому
фрагменту [уравнение (17.55)] [47]. В этом случае Fe(CO)3 служит
защитной группой для диена.
Бутадиеновые комплексы можно получить также реакцией
1,4-дигалогено-2-бутено'в с Fe2(CO)g [уравнение (17.56)].
Интересным примером этой реакции служит получение (г|4-циклобу-
тадиен)трика.рбонилжелеза из дихлороциклобутана [48]
[уравнение (17.57)]. Эта реакция наглядно демонстрирует
стабилизацию 'Исключительно неустойчивых органических молекул
комплексообразованием с подходящими переходными
металлами. Свободный циклобутадиен имеет очень короткое время жиз-
Fe2(CO)9 L LPvFe(C0)3
FeBr2 ♦ 6С0
(17.57)
ни и быстро димершуется, тогда как циклобутадиентрикарбо-
нилжелезо — очень устойчивый комплекс. Входящий в комплекс
циклобутадиен подвергается различным реакциям электрофиль-
ного замещения, в том числе ацилированию по Фриделю —
Крафтсу, формилированию, хлорометилированию и аминомети-
лированию. Кетогруппу ацилированного циклобутадиентрикар-
бонильного комплекса железа можно восстановить водородсо-
держащими реагентами, не разрушая комплекс [49]. В то же
Применение алкеновых, диеновых и диенильных комплексов ЗС9
время при окислении комплекса церием (IV), триэтиламинокси-
дом или пирвдин-Ы-оксидом циклобутадиеновый лиганд
высвобождается и его можно вводить в другие реакции. Петтит [50]
использовал циклобутадиентрикарбонилжелезо как надежный
источник высокореакционноспособного циклобутадиена,
пригодного для проведения различных синтезов, например кубана
[уравнение (17.58)].
♦ Се (IV)
реакция.
Дильса-
Альдера
(17.58)
кудан.
Семмелхак [51] разработал прямое алкилирование
нейтральных г|4-1,3-диеновых комплексов железа реакционноспособными
карбанионами, такими, как LiCMe2CN и LiCHPh2 (ч. 1, гл. 7).
Он показал, что преимущественно осуществляется
присоединение по незамещенному внутреннему положению, но при 0 °С
равновесие быстро смещается в сторону концевого положения.
Реакция позволяет лровести прямое алкилирование циклогекса-
диена и после подкисления получить смесь изомеров
замещенных циклогекешов [52]. В атмосфере моноокисида углерода на-
Fe(CO)3
, CHMeCN,
CH2CN, CMe2CO2Ett
CH(Me)C02Et,
CMe2CO2Li,
Ph2CH
(17.59)
главный
продукт
* 47-87%
310 Глава 17
блюдается внедрение СО и получаются «карбоадилированные»
продукты с высокими «выходами [уравнение (17.59)] [53].
Несколько сложнее обстоит дело с ациклическими диенами.
Сам закомплексованный 'бутадиен гладко превращается в
соответствующий циклопентанон\ процесс включает нуклеофиль-
ную атаку, внедрение СО и затем внедрение олефина. Однако
'наличие заместителей в диене подавляет стадию внедрения
олефина, и по мере увеличения замещения возрастает количество
продуктов «нормального» расщепления (например, альдегидов)
[уравнение (17.60)] [54]. Внедрение олефина с последующей
миграцией железа в а-положение к карбонильной группе
рассматривалось на примере внедрения этилена в комплексы
железа, генерируемые из Fe(CO)42~~ (гл. 15).
32 -i
(СОЦРе
117.60)
СО
R' J
r2=r'=h
\+- 3,
I) -HFe
По сравнению с нейтральными комплексами катионные
диеновые комплексы значительно более реакционноспособны ло
отношению к нуклеофилам. Исходя из этого, Пирсон
разработал эффективное стереоконтролируемое введение заместителей
в положения 1 и 3 молибден-циклогексадиеноеого комплекса
[55а] и в положения 1 и 5 молибден-циклогептадиеновых
комплексов [556] [уравнение (17.61)]. В реакции используется и
способность металла активировать диен, и способность
направлять входящий нуклеофил на противоположную металлу
сторону. Интересно, что с реакционноапособными катионными
диеновыми комплексами могут реагировать даже менее реакционно-
способные карбанионы, причем атаке подвергается концевое
положение диена.
Широко используются в органическом синтезе и катионные
диенильные комплексы железа, образующиеся при отрыве
гидрида от нейтральных диеновых комплексов железа. Они также
'подвергаются нуклеофиль'ной атаке, а наличие железа
определяет высокую степень регио- и стереоселективности (ч. 1, гл.7).
Применение алкеновых, диеновых и диенильных комплексов 311
Первые работы в этой области были выполнены Бёрчем, но их
практическое использование серьезно тормозилось из-за
отсутствия мягких методов дешмплексования железа из г|4-ди©новых
•комплексов. После того как были найдены мягкие окислители
(например, Me3NO) для этого процесса, он стал широко
применяться в синтезе. Большой «вклад в его разработку внесли
работы Пирсона [56], получившего ряд очень сложных
соединений.
МеМдХ
СрМо(СОЬ
Ч)
i-)CH(C02Et)2 EtO2C
EtO2C CpMo(C0)2
C02Et
Широко изучена химия диенильных комплексов,
образующихся из 4-метокситолуола. Восстановлением по Бёрчу и
последующей обработкой Fe(CO)5 получают г|4-диеновый комплекс
железа. Отрыв протона происходит исключительно из
положения, соседнего с метоксигруппой, поскольку при этом
наилучшим образом может стабилизироваться положительный заряд.
В правильно подобранных условиях полученный диенильный
комплекс будет реагировать преимущественно по метальному
концу диенильной системы ('менее богатого электронами), давая
замещенный диеновый комплекс [57]. Декомплексование и
гидролиз енолытого эфира приводят к 4-замещенному циклогексе-
нону. Таким образом, с точки зрения синтеза исходный
комплекс эквивалентен циклогексенон-'у-катиону [уравнение (17.62)].
Этот подход был использован Пирсоном в полном синтезе
многих природных соединений. Все эти методы включают
химическую активацию, а также регио- и стереоконтроль атомом
железа и затем использование фрагмента Fe(CO)3 для защиты
енольного эфира и предотвращения различных химических реак-
312 Глава 17
ОМе
ОМе
Fe(C0)c
(■OCPh
(I7.62>
Et2O, EtOH
-OMe
Fe(CO),
DMe
I MeO-
Nuc"
1
Fe(COb
MeO-
Fe(CO)2
(Защищенный
енола)
Me3NO
Nuc
н2о
OMe
Fe(C0)
OMe
MeO
Fe(C0)3
OMe
OMe
OMe
Fe(C0)
(!7.64>
NaBH4
2)S0CIE
Fe(C0)3 1)катализатору
COpR
Me
C02R
OMe
Применение алкеновых, диеновых и диенильных комплексов 313
ций, -к которым оии обычно склонны. Примерами служат
синтезы стероидных циклических систем [уравнение (17.63)] [58],
трйкотекашв [уравнение (17.64)] [59], алкалоидов лимнаспер-
мина [60] и асгшдоспер'мина [61].
Поскольку группировка трикарбонилжелеза располагается
только по одну сторону г!5-диенильного комплекса, замещенным
комплексам должна быть .присуща хиральность. Действительно,
при обработке рацемического комплекса хиральным нуклеофи-
лом с последующим разделением диастереоизомеров и
регенерацией исходного комплекса был получен единственный энантио-
мер [уравнение (17.65)] [62]. Бёрч [63] разделил г|4-диеновый
комплекс железа до отрыва водорода и использовал стереоспе-
цифич'ность последующих стадий (атака со стороны,
противоположной металлу) для синтеза оптически активного габакулина
[уравнение (15.66)].
-ОМе
(-)- ментол
(RO-)
Fe(C0)3
(♦)
. О М е |) ра з деление
(17.65)
2)НХ
е(СО)3
-ОМе =
Fe(CO)3
хтральный (+)
(разделен в Зиде
xupcuibhou соли
ампония) V°2M
Me3N0 (-
2}Ме0Н/0К"
3) НС1
(17.66)
СОгЪU-трет
(■\)-jq-докулик
ЛИТЕРАТУРА И ПРИМЕЧАНИЯ
1. См. обзоры: Hegedus L. S., Tetrahedron, 40, 2415 (1984); Akermark В.,
Bdckvall J.-E., Zetterberg К. Z., Acta Chem. Scand., B36, 577 (1982).
2. Tsuji /., Synthesis, 1984, 369; Pure Appl. Chem., 1981, 2371; Organic
Synthesis with Palladium Compounds, Springer Verlag, New York, 1980.
314 Глава 17
3. Hosokawa Т., Ohkata Н., Moritani /., Bull. Chem. Soc. Japan, 48, 1533
(1975).
4. Hosokawa Т., Miyagi S.f Murahashi S.-L, Sonoda Л, J. Org. Chem., 43, 2725
(1978).
5. Semmelhack M. F., Bodurow C, J. Am. Chem. Soc, 106, 1496 (1984).
6. Semmelhack M. F., Bodurow С, Байт Af., Tetrahedron Lett., 25, 3171
(1984).
7. Kasahara A., Izumi Т., Sato К., Маетига K-, Hayasaka Г, Bull. Chem. Soc.
Japan, 50, 1899 (1977).
8. Izumi Т., Kasahara Л, Bull. Chem. Soc. Japan, 48, 1673 (1975).
9. Korte D. E.f Hegedus L. S.t Wirth R. K-, J. Org. Chem., 42, 1329 (1977).
10. Backvall J. E., Bjorkman E. £., J. Org. Chem., 45, 2893 (1980).
11. Backvall J. E., Bystrom S. E. J. Org. Chem., 47, 1126 (1982).
12. Backvall J. E., Bjorkman E. E., Bystrom S. E., Solladie-Cavallo Л,
Tetrahedron Lett., 23, 943 (1982).
13. Backvall J. E., J. Chem. Soc, Chem. Commun, 1977, 413.
14. Hegedus L. S.f Allen G. F., Bozell /. /., Waterman E. L., J. Am. Chem. Soc,
100, 5800 (1978).
15. Hegedus L. S., Allen G. F., Olsen D. /., J. Am. Chem. Soc, 102, 3583 (1980),
16. W eider P. R., Hegedus L. S., Asada #., J. Org. Chem., 50, 4276 (1985).
17. Hegedus L. S.f McKearin J. M., J. Am. Chem. Soc, 104, 2444 (1982).
18. Hatano S., Saruwatare M., Isomura K., Taniguchi #., Heterocycles, 15, 747
(1981); Isomura K., Okada N., Saruwatare M., Yamasaki H., Taniguchi H.>
Chem. Lett., 1985, 385.
19. Kasahara A., Saito Г., Chem. Ind. (London), 1975, 745.
20. Kasahara A., Chem. Ind. (London), 1976, 1032.
21. Kasahara A., Fukuda N., Chem. Ind. (London), 1976, 485.
22. Hegedus L. S., Williams R. E., McCuire M. A., Hayashi Г., J. Am. Chem. Soc.r
102, 4973 (1980).
23. Hegedus L. S., Darlington W. #., J. Am. Chem. Soc, 102, 4980 (1980).
24. Solladie-Cavallo A., Haesslein J. L., Backvall J. £., Tetrahedron Lett., 23,
939 (1982).
25. Dunkerton L. V., Serino A. /., J. Org. Chem., 47, 2815 (1982).
26. Trost B. M., Fortunak J. M. D., Organometallics, 1, 7 (1982).
27. Kende A. S., Roth В., Sanfitippo P. /., J. Am. Chem. Soc, 104, 1784 (1982).
28. Ito Y., Aoyama H., Saegusa Т., J. Am. Chem. Soc, 102, 4519 (1980).
29. Kende A. S., Roth В., Sanfilippo P. J., Blacklock T. /., J. Am. Chem. Soc,
104, 5808 (1982).
30. Kende A. S., Battista R. A., Sandoval S. В., Tetrahedron Lett., 25, 1341
(1984).
31. a) Larock R. C, Leach D. R., J. Org. Chem, 49, 2144 (1984); 6) HoltonR.A.,
Zoeller J. R., J. Am. Chem. Soc, 107, 2124 (1985).
32. См. обзор: Overman L £., Angew. Chem, Int. Ed. Engl, 23, 579 (1984).
33. Grieco P. A., Takigawa Т., Bongers S. L., Tanaka H., J. Am. Chem. Soc, 102,
7587 (1980); Grieco P. A., Tuthill P. A., Sham H. L, J. Org. Chem, 46, 5005
(1981).
34. Overman L. E., Jacobsen E. /., J. Am. Chem. Soc, 104, 7225 (1982).
35. Berryhill S. R., Price Т., Rosenblum M., J. Org. Chem, 48, 158 (1983).
36. Chang- Т. С. Т., Rosenblum M., J. Org. Chem, 46, 4103 (1981);
Rosenblum Af., Pure Appl. Chem, 56, 129 (1984).
37. Marsi M., Rosenblum Af., J. Am. Chem. Soc, 106, 7264 (1984).
38. Chang Т. С. Т., Rosenblum M.t J. Org. Chem, 46, 4676 (1981);
Tetrahedron Lett, 24, 695 (1983).
39. Rosenblum M., Accts. Chem. Res, 7, 122 (1974).
40. Genco N., Marten D., Raghu S., Rosenblum M., J. Am. Chem. Soc, 98, 848
(1976); Abram T. S., Baker R., Exon C. Af., Rao V. В., J. Chem. Soc, Perkin
Trans. 1, 1982, 285.
Применение алкеновых, диеновых и диенильных комплексов 315
41. Baker R., Ехоп С. М., Rao V. В., Turner R. W., J. Chem. Soc, Perkin Trans. 1,
1982, 295; Abram T. S., Baker R., Ехоп С M., Rao V. В., Turner R. W.,
J. Chem. Soc, Perkin Trans. 1, 1982, 301; Baker R., Rao V. В., Erdik E.,
J. Organomet. Chem., 243, 451 (1983).
42. Baker R.t Morris M. D., Turner R. W., J. Chem. Soc, Chem. Commun., 1984,
987.
43. Watkins J. C, Rosenblum M., Tetrahedron Lett., 25, 2097 (1984).
44. Buchester A., Klemarczyk P., Rosenblum M., Organometallics, 1, 1679 (1982).
45. Boyle P. F., Nicholas К. M., J. Org. Chem., 40, 2682 (1975).
46. Nicholas К- М., J. Am. Chem. Soc, 97, 3254 (1975).
47. Frank-Newmann M., Martina D., Heitz M. P., Tetrahedron Lett., 23, 3493
(1982).
48. Emerson G. F., Watts L, Pettit R., J. Am. Chem. Soc, 87, 131 (1965).
49. Fitzpatrick J. D., Watts L, Emerson G. F., Pettit R., J. Am. Chem. Soc, 87,
3254 (1969).
50. Barobak J. C, Watts L., Pettit R., J. Am. Chem. Soc, 88, 1328 (1966).
51. Semmelhack M. F., Lee H. Т. М., J. Am. Chem. Soc, 106, 2715 (1984).
52. Semmelhack M. F., Herndon J. W., Organometallics, 2, 363 (1983).
53. Semmelhack M. F., Herndon J. W., Springer J. P., J. Am. Chem. Soc, 105,
2497 (1983).
54. Semmelhack M. F.t Herndon J. W., Liu J. /(., Organometallics, 2, 1885 (1983);
Semmelhack M. F., Le H. Т. М., J. Am. Chem. Soc, 107, 1455 (1985).
55. a) Pearson A. J., Khan M. N. /., J. Am. Chem. Soc, 106, 1872 (1984);
Tetrahedron Lett., 25, 3507 (1984); 6) Pearson A. J., Khan M. N. L, Clardy J. C,
Cin-Heng H., J. Am. Chem. Soc, 107, 2748 (1985).
56. Pearson A. /., Ace Chem. Res., 13, 463 (1980); Pure Appl. Chem., 55, 1767
(1983); Chem. Ind. (London), 1982, 741.
57. Pearson A. J., Perrior T. R., Rees D. C, J. Organomet. Chem., 226, C39
(1982); Pearson A. J., Haim P., Ong C. W., Perrior T. R., Griffin D. Л.,
J. Chem. Soc, Perkin Trans. 1, 1982, 1527; Pearson A. J., Richards I. C,
Tetrahedron Lett., 24, 2465 (1983).
58. Pearson A. J., Heywood G. C, J. Chem. Soc, Perkin Trans. 1, 1982, 2631;
Tetrahedron Lett., 22, 1645 (1981); Mincione E., Pearson A. J., Bouicelli P.,
Chandler M.t Heywood G. C, Tetrahedron Lett., 22, 2929 (1981).
59. Pearson A. J., Ong C. W., J. Am. Chem. Soc, 103, 6686 (1981); J. Chem. Soc,
Perkin Trans 1, 1981, 1614.
60. Pearson A. J., Rees D. C, J. Am. Chem. Soc, 104, 1118 (1982); J. Chem. Soc,
Perkin Trans 1, 1982, 2467; J. Chem. Soc, Perkin Trans 1, 1983, 619.
61. Pearson A. /., Tetrahedron Lett., 22, 4033 (1981).
62. Bandara B. M. R., Birch A. J., Kelly L. F., Chak Khor Т., Tetrahedron Lett.,
24, 2491 (1983); Howell J. A. S., Thomas M. J. J. Chem. Soc, Dalton Trans.,
1983, 1401; Atlon J. G., Evans D. /., Kane-Maguire L. A. P., Stephen-
son G. R., J. Chem. Soc, Chem. Commun., 1984, 1246.
63. Bandara B. M. R., Birch A. J., Kelly L. F., J. Org. Chem., 49, 2496 (1984),
18
ПРИМЕНЕНИЕ АЛКИНОВЫХ КОМПЛЕКСОВ
ПЕРЕХОДНЫХ МЕТАЛЛОВ В ОРГАНИЧЕСКОМ СИНТЕЗЕ
Практически все переходные металлы реагируют с алкина-
ми, однако лишь немногие из них образуют простые, устойчивые
комплексы (ч. 1, разд. 3.7,6), аналогичные соответствующим
комплексам металлов с олефинами (ч. 1, разд. 3.7,а и гл. 17).
Это связано с тем, что многие алкино!вые комплексы вполне
реакц'ионноелособны по отношению ,к алкинам и вступают с
ними в дальнейшее взаимодействие, давая более сложные
комплексы или устойчивые органические продукты, не пригодные для
дальнейших превращений. Очевидно, что для успешного
применения алкиновых комплексов переходных металлов в синтезе
необходимы методы, регулирующие их реакционную
способность.
18.1. Нуклеофильная атака на комплексы переходных
металлов с алкинами
В отличие от комплексов металлов с олефинами алкиновые
комплексы довольно редко 'взаимодействуют с нуклеофильными
агентами. В ч. 1, разд. 3.5,6 отмечалось, что взаимодействием
с нуклеофилами можно шолучить ванильные лиганды. Регер [1]
показал, что устойчивые комплексы Fp+ с алкинами легко
вступают в реакцию с нуклеофилами с образованием стабильных
о-виниль'ных кОхМплекшв железа [уравнение (18.1)]. Эти
прочные комплексы подвергаются внедрению СО при обработке
Ag(I) или при окислении [2], «о не вступают в дальнейшие
превращения с образованием не содержащих металла
органических соединений. Утимото [3] разработал катализируемую
палладием (II) реакцию циклизации алкинов с удаленными гидр-
оксильными или аминогруппами, приводящую к образованию
гетероциклов. Хотя механизм этого процесса не известен,
формально он включает нуклеофильную атаку на алкин. Реакция
носит общий характер, ее можно использовать для синтеза
дигидрофуранов или пиранов [уравнение (18.2)], а также
циклических иминов [уравнение (18.3)]. Исходя из субстратов
Применение алкиновых комплексов переходных металлов 317
с подходящим образом расположенными уходящими группами,,
можно получить пирролы или фураны [уравнение (18.4)].
я'п
со .
CpFe —|
RJ
Nuc-
СО
CpFe
L
(18.1)
0 R'
AgVCO CO 11 1
CpFe-^Y^^NdC
u.iu Ce(TV) L I
R
Nuc* = Ph2CuLi, Me2CuLi, (MeC = C)2CuLi,
(CH3CH = CH-)2CuLi, PhS", "CH(CO2R)2
47%
PdCI2(PhCN)2
Et2O
(транс)
(18 2)
90%
PdCU/MeCN
R-C5C-CH2CH2NH2 ——rr—Z ~^ R
OMe
hx-ch^r3
0, NH
(18.3)
40-60%
\\—d3
(18.4)
18.2. Синтетическое применение алкиновых комплексов Со2(СО)8
В отличие от большинства других карбо'нилов металлов ди-
ко'бальтоктакарбонил Co2(CO)s образует очень устойчивые
комплексы с алкинами [4]. Ал,киновые «мостики» (ч. 1, табл. 2.2)
формально являются донорами четырех электронов, т. е. в
образование координационной связи вовлечены все четыре я-электро-
на [уравнение (18.5)]. Нуклеофильность алкиновых комплексов
такого типа сильно понижена. Действительно, алкины в составе
комплекса с Со2(СО)8 защищены от электрофильных атак и
гидроборирования. Петтит [5] селективно уменьшил олефино-
вый характер енина путем комллексообразова'ния алкина
с Со2(СО)8, восстановил двойную связь гидразином и
регенерировал свободный алкин, окислительно удалив кобальт (под:
318 Глава 18
действием Fe3+). Кроме того, олефин можно гидроборировать
[уравнение (18.6)].
с—с
RC=CR + Co2(CO)8
-2СО
(СО)3Со
Со(СО)3 (18.5)
Со2(СО)а
Николас использовал тот же подход для стабилизации про-
ларгил-катионов с целью использования их в органическом
синтезе. Комплексообразование алкиновюй группы не только
защищает ее от дальнейших реакций с адаинами, но значительно
повышает устойчивость пропаргил-катионов и легкость их
образования. Та'к, комплексные проларгиловые спирты, ацетаты и
эпоксиды под действием кислот Льюиса легко расщепляются
с образованием стабилизированных металлом
пропаргил-катионов. В таком состоянии пропаргил-катионы способны
взаимодействовать с различными нуклеофилами [уравнения (18.7) и (18.8)],
давая продукты замещения при шропаргильном углероде. При
этом не наблюдается конкурентного образования алленовых
продуктов, обычного -в случае некомплексных пропаргило<вых
систем.
Смиту [11] удалось избирательно провести комллексообра-
зование алкинов с Со2(СО)8 в присутствии алкенов и
стабилизацию пропаргил-гсатионов с по'мощью Со2(СО)б, что позволило
осуществить региоспецифическое присоединение сложных эфи-
ро'в к енинам [уравнение (18.9)].
Комплексы алкинов с производными кобальта весьма
устойчивы к действию большинства электрофилов, но при высоких
температурах они реагируют с олефинами с образованием цик-
лопентеноно!в [уравнение (18.10)] [12]. Хотя механизм реакции
не известен, она находит применение в органическом синтезе.
Паусон [13] использовал эту реакцию в сочетании с описанны-
т
Z^Hj R
DNuc"
R2
I
R3
2)
окислитель
Co2(CO)Q
-2C0
R2
» 1
R'-CsC-C-Nuc
R3
Применение алкиновых комплексов переходных металлов 319
(18.7)
R2 R2
I кислота
RI с - г Г"**
, | _ Льюиса | |
' R I R3
(СО)6Со2 (СО)6Со2
Nuc-= R" {U3 AIR3) [б]
■QMe (EAS) [7]
О О
М
OTMS j-9-j
(18.8)
r'-c=c—v-o —- - r'-chc-ch Nuc" ». r'-csc-ch
'R ' ^г.ипн . I ^ снон
(C0)6Co2 (CO)6Co2 T2 (C0)6'
OAc
Nuc" = MeOH, PhOMe, ^?\, ^\^-™s jjo]
HC-C С C°2(C0)6 Hp.r ^У ^CMBF4- ^H;
HC-C-C ' HC=C-C ^"^ HCsC-C
R "2C0 I %i ' Xol
(CO»eio2 <CO)6C°2 R
HC8C-C --C-R^ ге'ш'. HCiC-C-CH2C-f
._.. ' ' 1 q окисли- I
(Cu)gCo2 R w /tz^^z-^ OR
ми выше приемами комплексования лро'паргил-катионов для
получения предшественников простагла.ндина [уравнение (18.11)].
Магнус [14] [(уравнение (18.12)] и Биллингтон [15]
[уравнение (18.13) ] использовали внутримолекулярный вариант этого
процесса для синтеза бициклических производных.
320 Глава 18
„OTHP
Со2(СО)б
;HCiCCH5OH
i d
Со2(СО)б
HC=C-CH~
I
Co2(CO)6
OTHP
(18.10)
35%
(18.11)
OTMS
OTMS R
Co2(CO)8
!!O°C
50-80%
Co2(CO)8
со, д
30-40%
(18.12)
d, I - кориоли/t
(18.13)
18.3. Катализируемая металлами циклоолигомеризация алкинов
а. В присутствии монооксида углерода. Алкины реагируют
-с многими переходными металлами в присутствии монооксида
углерода. В образующихся при этом соединениях присутствуют
как монооксид углерода, так и фрагмент лроиз'водного алкинов.
Помимо этого, часто образуются металлорганические
комплексы, лиганды которых построены из фрагментов 'производных
алкинов и -хмонооксида углерода [16]. Реакции этого типа обычно
очень сложены, и лишь немногие из них изучены в достаточной
степени, чтобы их можно было использовать в органическом
-синтезе. Одним из примеров служит предложенное Либескиндом
[17] превращение алкинов в хиноны, включающее промежуточ-
Применение алкиновых комплексов переходных металлов 321
ное образование малеоилметаллокомплексов. Известно, что хи-
ноны являются одним из продуктов взаимодействия алкинов
с монооксидом углерода, -надежно доказано промежуточное
образование малеоильных комплексов [уравнение (18.14)].
Однако реакция не имела практического значения из-за огромного
числа лобочных органических и металлоорога-нических
продуктов. Либескинд решил эту проблему, предложив эффективные
способы синтеза малеоильных и фталоильных комплексов.
Взаимодействие каждого из этих комплексов с алкинами приводит
к образованию хинонов.
R- = -R
CO ♦ MLr
Наиболее эффективен фталоильный комплекс .кобальта, об-
азующийся при взаимодействии бензоциклобутендиоиов
к ОоС1(РРЬз)з [18]. Возникающий вначале бисфосфиновый
комплекс кобальта (III) малоактивен по отношению к алки-нам,
так как для продуктивного комплексообразования с алкином
'необходим лабильный участок, расположенный аксиально по
отношению к плоскости фталоила. Однако обработка серебром (I),
приводящая к образованию ненасыщенного катионного
комплекса [19], или, что еще лучше, диметилглиоксимом в пиридине,
приводящая к образованию октаэдрического комплекса с
лабильными ^опиридиновьими лигандами (РСА), (позволяет полу-
чать активные фталоильные комплексы, эффективно
вступающие во взаимодействие с различными алкинами с образованием
нафтохинонов [уравнение (18.15)]. Внутримолекулярный
вариант этой реакции был использован для синтеза наном'ицина А
[уравнение (18.16)] [20]. Для обеспечения региоспецифического
включения несимметричного алкила необходимо связывание ал-
кина с ароматическим кольцом.
Аналопичным образом из циклобутендионов и алкинов через
их малеоильные комплексы с кобальтом получают (бензохиноны
[уравнение (18.17)] [21]. В случае несимметричных систем
в результате реакции образуется смесь региоизомеров. Главный
продукт в смеси изомеров можно предсказать на основании
представлений о поляризации алкила и комплекса (рис. 18.1).
б. С диоксидом углерода и изоцианатами.Хоберг всесторонне
изучил реакцию диоксида углерода и алкинов с комплексами
никеля (0), в результате которой образуются относительно
устойчивые, но высокореакционноспо'собные металлациклы. Они
вступают во взаимодействие с различными органическими соедине-
21—1129
322 Глава 18
CICo(PPh3)3
R=H, Me, Et
^t, OEt,
Bu, CH2NAc
Ph
(18.15)
HON NOH
PPh
3
PPh,
R1
О
70-99%
РУ
OR О
CoL2CI
(18.16)
47%
CoCIL:
R^H.Me, Et
R = Me, EfT
OEt, COzEt
CoCIL2
R3= Me
i, R4= Me, OMe
глио/ccuw
(18.17)
О
60-85%
-(CH2)4-,-(CH2)5-
Применение алкиновых комплексов переходных металлов 323
о
II Со
COoEt
S-
OEt
Рис. 18.1.
ниями, давая множество полезных продуктов, содержащих
фрагменты алкина и СО2 [уравнение (18.18)] [22].
R— =■ —R ♦ СО2 ♦ Ni(O) ♦ L
R'
R R '*
-C 0
2 СО2Ме
L-COD, ТМЭДА
CQgH-
О
2)Н>\ О R
PhN I х)—R
он
R' о''
Аналогичным образом в реакцию с алкинами и комплексами
никеля (0) ©ступают изоцианаты, давая реакционноспособные
металлациклы, претерпевающие дальнейшие превращения
[уравнение (18.19)] [23]. Хоберг обнаружил также, что комплексы
никель(О)-ТМЭДА катализируют образование 2-'Ш1ридонов или
(iB зависимости от соотношения реагентов) пуриновых
оснований из изощианатов и алкино(в [уравнение (18.20)] [24].
Волхардт [25] фундаментально исследовал катализируемую
кобальтом межмолекулярную сощжлизацию алкинов и изоциа-
нато'в; он разработал 'и применил внутримолекулярный вариант
этого процесса. Установлено, что (COD)2Ni неэффективен в
качестве катализатора внутримолекулярной реакции.
Использование в качестве катализаторов (COD)2Ni или СрСо(СО)2 в
межмолекулярном процессе приводит к образованию разных регио-
изомеров, что указывает на различие механизмов реакций
[уравнение (18.21)]. Хотя катализируемые никелем реакции
протекают, по-видимому, согласно уравнению (18.19) (конденсация
алкина и изоцианата с последующим включением второй молеку-
21*
324 Глава 18
RC=CR ♦ R'N=C»O ♦ Ni(O) ♦ L
I
R1
20-70%
2RC5CR
R'NCO
ТМЭДА/№(0)
2 R'NCO
(18.20)
R1
-20%
лы алкина), считается, что реакции, катализируемые
кобальтом, протекают через стадию димеризации алкина с
последующим включением изощианата [уравнение (18.22)], И в том и
-в другом лроцессе наиболее объемная (Ph) группа
располагается в соположении по отношению к металлу металлациклическо-
го интермедиата, тем не менее из-за различий строения этих
металлациклов наблюдаются различия в региоселективности
процессов.
Волхардт разработал также внутримолекулярную сощикли-
зацию, приводящую к индолозинонам [уравнение (18.23)].
В случае триметил'силилацет.иленов наблюдалось высокорегио-
селективное образование продукта с TMS-группой (триметил-
силильной группой) в а-положении к амидной связи. С диза-
мещенными алкинами реакция протекает несколько менее
региоселектишю. Продукт реакции (18.23) с я-пропилом (в ка(че-
'стве группы R был превращен в природный алкалоид кампто-
фецин [25].
в. Циклотримеризация алкинов. Многие переходные
металлы катализируют циклотримеризацию алкинов в ароматические
системы; один из наиболее эффективных катализаторов этого
процесса — комплекс CpCoL2. Как разъяснялось в ч. 1,
разд. 9.5, б, подобные реакции тримеризации обычно включают
координацию двух молекул алкина, образование -металлацикло-
пентадиена, координацию еще одной молекулы алкина и, нако-
Применение алкиновых комплексов переходных металлов 325
(18.21)
^^^NCO
1 СрСо(СО)2 (COD)2N,
Et
Ph
Et
Ph
e'
2 Ph— = —Et
Ph
47% (главный
продукт)
CpCo(CO)2 ♦ 2Ph—S — Et
CpCo
0
Ph
один продукт)
(18.22)
Et
Et I Ph
RNCO
ГЛ
N=C=O ♦ TMSCsC-R
! = TMS,h-Pt, n^C , ,
CpCo(CO)2
(18.23)
О
60-80%
■нец, распад на продукты с (высвобождением катализатора
(рис. 18.2). Для всех стадий, кроме последней, известны
прекрасные аналогии. Например, Колямен и Литтл [26] показали,
что азотный комплекс 1г (I) реагирует с дизамещенными алкина-
ми с образованием алюи-нового комплекса Ir(III), который
в принципе можно выделить из реакционной смеси. Последний
реагирует далее с алкином, дагвая металлациклшентадиеновый
комплекс Ir(III) (также может быть выделен в
индивидуальном состоянии), который сам каталиэирует циклотримеризацию
алкинов при температурах выше 110°С [уравнение (18.24)].
Аналогичным образом реагирует родиевый комплекс.
Добавление таких лигандов, как фосфин или СО, ингибирует
действие катализаторов обоих типов. Подобный факт установлен
Ямазаки [27], который показал, что комплекс кобальта
СрСо(РРЬз)г взаимодействует с алкина-мн с образованием
выделяемого металлацикло!пентадиенового комплекса, который
326 Глава 18
RC
СрСо
Рис. 18,2.
I
I
Ci— Ir—
L
(18.24)
L
I
C!—Ir
I
L
RC=CR
в свою очередь катализирует ци'клотримеризацию алкинов.
Реакция металлациклопентадиена с алкином, в результате которой
образуются арены, может протекать по нескольким различным
механизмам: 1) внедрение с образованием металлациклогеп-
татриена; 2) циклоприсоединение по типу реакции Дильса —
Аль'дера внутри координационной сферы металла; 3) циклопри-
соединение по Дильсу — Альдеру без предварительной
координации ал'кина. В настоящее время нет достаточных оснований
для решения вопроса в пользу того или другого механизма.
Однако Беркоу и Бергман [28] получили некоторые
доказательства прямого Ц'иклоприсоединения или внедрения (без предва-
Применение алкиновых комплексов переходных металлов 327
рительной координации) высокореакционноспособных алкинов
с кобальтациклопентадиенЬм. Тщательное кинетическое
исследование каталитического взаимодействия металлациклопентадие-
на СрОо(РРЬ3){С4Ме4} с 2-бутином, приводящего к гексаметил-
бензолу, позволило установить, что реакция включает потерю
фосфина (стадия, определяющая скорость реакции), затем
координацию алкина и, наконец, распад на продукты. Замена три-
фенилфосфина на триметилфосфин, образующий более прочные
координационные связи, предотвращает (предварительную
координацию алкина и полностью ингибирует циклотримеризацию
2-бутина (реакция не идет в течение нескольких дней при
120 °С). Однако тот же триметилфосфиновый комплекс быстро
взаимодействует при 20 °С с диметилацетилендикарбоксилатом,
давая диметиловый эфир 3,4,5,6-тетраметилфталата, причем
'процесс протекает без диссоциации триметилфосфина и
координации диметилацетилендикарбоксилата. Следует отметить, что
металлациклопентадиеновый и циклобутадиеновыи (комплексы
являются валентными таутсшерами [29]. Однако, как уже
упоминалось в ч. 1, разд. 9.5,6, принято считать, что циклобутадиено-
вые комплексы не входят в каталитический цикл реакций три-
меризации ацетилена. Циклобутадиеновые металло комплексы
обычно не катализируют реакции циклотримеризации и во
многих случаях могут быть выделены из реакционной среды в виде
малореакционноспосо<бных побочных продуктов. (Два недавних
эксперимента, подтверждающих образование т]4-циклобутадиена
в качестве интермедиата в некоторых реакциях тримеризации,
приведены в ч. 1, разд. 9.5,6).
Несмотря на то что реакция циклотримеризации была
известна достаточно давно, широкого применения в органическом
синтезе она не находила. Все попытки провести смешанную
циклотримеризацию с использованием двух различных алкинов
приводили к получению полного набора всех возможных
продуктов. Простое решение этой проблемы было найдено Волхард-
том [30], который предложил изящный метод синтеза
полициклических продуктов из простых предшественников [31]. Он
разработал соци'клотр'имеризацию 1,5-гексадиинов с бис(триме-
тилсилил) ацетиленом (BTMSA) — алкином, который сам по себе
не подвергается циклотримеризации. Первичный продукт этой
реакции, бензоциклобутан, достаточно реакционноспособен.
Термическая обработка реакционной омеси приводит к образованию
очень реакционноспособного диена, который вступает в
реакцию Дильса — Альдера с различными диенофилами [32]
[уравнение (18.25)]. Удачная особенность системы заключается
в том, что BTMSA не способен к самоконденсации, а это
облегчает социклотримеризацию. Поскольку триметилсилильная
группировка является хорошей уходящей группой в реакциях
328 Глава 18
электрофильного замещения, описанный процесс дает
возможность получать различным образом замещенные тетрагидронаф-
талины. Введение диенофила в молекулу диина (позволяет
в одну стадию получать сложные полициклические соединения
(18.25)
с
SiMe7
СрСо(СО)2
SiMe,
SiMe,
Si Me,
SiMe,
Si Me,
SiMe,
[уравнения (18.26] — (18.28)]. Этот подход был использован
для изящного синтеза рацемического эстрона [33] [уравнение
НС=С
HCiC—I J
СрСо(СО)?
Me3Si
Me3Si
80%
(18.26)
HC=C
DBTMSA
CpCo(CO)2
2) Л
I) BTMSA
60%
CpCo(CO)2
2) Д
Me3Si
OMe
N-OMe
45%
(18.27)
(18 28)
Применение алкиновых комплексов переходных металлов 329
он
I) 3BuLi, ТМ5ДА
(трианион) ^__
65%
!) TsCl
2) No!
a new oh
96%
80%
Дииновые соединения с длинной цепью также способны
к циклотримеризации; доказательством этого может служить
синтез циклической протобербериновой системы [уравнение
(18.30)] [34].
Еще одним примером соединения с тройной связью, не
способного к 'саияотримеризац'И'И, является нитрильная группировка.
Однако она вступает в реакцию сотримеризащии с алкинами
с образованием ширидиной. Взаимодействие с несимметричными
алкинами дает, к сожалению, смесь всех возможных изомеров
[35] [уравнение (18.31)]. В то же время реакция с диинами
приводит к изохинолинам [36] [уравнение (18.32)], [34]
[уравнение (18.33)].
Некоторые комплексы, катализирующие циклотримеризацию
алкинов в арены, катализируют также социклотримеризацию
22—1129
330 Глава 18
Me 0
MeO
I) TMSC=CCH2MgBr
N 2) HC-CCH2Br
MeO
MeO
MeO
I) KOH, MeOH (-TMS)
2)BTMSA
MeO'
(13.30)
TMS
•CpCo(CO)2
Rf
1 СрСо(СО)2
С _
n= 3-5
MeO.
MeO
PhCN
R"
(CH2)
CpCo(CO).
R1
R"
MeO
MeO
(18.31)
(18.32)
(18 33)
TMS
алкшнов и алкенов, приводящую к циклогексадиенам
[уравнение (18.34)]. Процесс может протекать с промежуточным
образованием металлациклопентадиенов или металлациклопентенов
в зависимости от относительной координационной способности
участвующих ненасыщенных соединений. Ямазаки [37] показал,
что комплекс CpCo(PPh3)2 'взаимодействует последовательно
Применение алкиновых комплексов переходных металлов 331
с двумя различными алкинами с образованием смешанного цик-
лопентадиенового комплекса кобальта, который при добавлении
R
R^ I .R' ^34>
-R1
2RC=CR
R'CH = CHR'
олефина дает циклогексадиеновые комплексы или циклогекса-
диены [уравнение (18.35)] в зависимости от природы
заместителей. Предложенный О/бщий механизм реакции представляется
вполне реальным, поскольку каждый из промежуточно
образующихся комплексов был выделен и характеризован. Сомнение
Со
(18.35)
R'Cp
Со
окисление
R3CH=CHR3
R2
вызывает только (последняя стадия. Из кинетических данных
явствует, что реакция ингибирует'ся при добавлении фосфина,
а это указывает на предварительную координацию олефина
с металлациклопентадиеном. Однако остается не ясным, какая
стадия следует за этим—внедрение с образованием металла-
циклогептадиена или присоединение по Дильсу — Алыдеру внутри
координационной сферы металла. Несмотря 'на
неопределенность механизма, реакция находит применение в синтезе.
Внутримолекулярный вариант социклизации алкинов и алкенов
позволил Волхардту [38] разработать новый подход к синтезу
эстрона [уравнение (18.36)].
СрСо(СО)2
МеО
(18.56)
1) FeCI3
2) Н+, Н20
СоСр
65%
МеО
22*
332 Глава 18
ЛИТЕРАТУРА И ПРИМЕЧАНИЯ
1. Reger D. L., Belmore К. Л., Mintz E., McElligot P. /., Organometallics, 3,
135 (1984).
2. Reger D. L., Mintz E., Organometallics, 3, 1759 (1984).
3. Utimoto K.t Pure Appl. Chem., 55, 1845 (1983).
4. Dickson R. S., Fraser P. /., Adv. Organomet. Chem., 12, 323 (1974).
5. Nicholas К- М., Pettit R., Tetrahedron Lett., 1971, 3475.
6. Padmanabhan S., Nicholas K. M., Tetrahedron Lett., 24, 2239 (1983); J.
Organomet. Chem., 212, 115 (1981).
7. Padmanabhan S., Nicholas К. М., Tetrahedron Lett., 23, 2555 (1982); Lock-
wood R. F., Nicholas К- М., Tetrahedron Lett., 1977, 4163.
8. Hodes H. D., Nicholas K. M.t Tetrahedron Lett., 1978, 4349.
9. Nicholas K. M., Muluaney M.f Bayer M., J. Am. Chem. Soc, 102, 2508 (1980).
10. Saha M., Nicholas К. M, J. Org. Chem., 49, 418 (1984).
11. Schegolev A. A., Smit W. A., Kalyan Y. В., Krimer M. Z., Caple R,
Tetrahedron Lett., 23, 4419 (1982); Smit W. A., Schegolev A. A., Gybin A. S., Mi-
kaelain G. S., Synthesis, 1984, 887; Mihaelian G. S., Gybin A. S., Smit W.A.,
Caple R., Tetrahedron Lett., 26, 1269 (1985).
12. Billington D. C, Pauson P. L., Organometallics, 1, 1560 (1982).
13. Jaffer H. /.; Pauson P. L, J. Chem. Res. (S), 1983, 244.
14. Exon C, Magnus P., J. Am. Chem. Soc, 105, 2477 (1983).
15. Billington D. C, Tetrahedron Lett., 24, 2905 (1983); Billington D. C, Wil-
lison C, Tetrahedron Lett., 25, 4044 (1984).
16. Pino P., Braca G., «Carbon Monoxide Addition to Acetylenic Substrates», in:
Organic Synthesis via Metal Carbonyls, Wender I., Pino P. (Eds.), Wiley,
New York, 1977, Vol. 2, pp. 420—516; Hubel W., «Organometallic Derivatives
from Metal Carbonyls and Acetylene Compounds», in: Organic Synthesis via
Metal Carbonyls, Wender I., Pino P. (Eds.), Wiley, 1968, Vol. 1, pp. 273—
340 (есть русский перевод первого издания книги: Органические синтезы
через карбонилы металлов. Пер. с англ./Под ред. Уэндера И., Пино П. —
М.: Мир, 1970).
17. Liebeskind L. S., Baysdon S. L., South M. S., Iyer S., Leeds J. P.,
Tetrahedron, 41, 5839 (1985).
18. Liebeskind L. S., Baysdon S. L., South M. S., Blount J. F., J. Organomet.
Chem., 202, C73 (1980).
19. Liebeskind L. S.f Baysdon S. L., South M. S., J. Am. Chem. Soc, 102, 7397
(1980); Baysdon S. L., Liebeskind L. S., Organometallics, 1, 771 (1982).
20. South M. S., Liebeskind L. S., J. Am. Chem. Soc, 106, 4181 (1984).
21. Liebeskind L. S.f Leeds J. P., Baysdon S. L., Iyer S., J. Am. Chem. Soc, 106,
6451 (1984); Liebeskind L. S., Jewell C. F., J. Organomet. Chem., 289, 305
(1985).
22. Hoberg #., Schaefer D., Burkhardt G., J. Organomet. Chem., 228, C21 (1982);
Hoberg H., Schaefer D., Burkhardt G., Kruger C, Ramao M. /., J.
Organomet. Chem., 266, 203 (1984); Hoberg H., Aapotecher В., J. Organomet. Chem.,
270, C15 (1984).
23. Hoberg H., Oster B. W., Synthesis, 1982, 324; J. Organomet. Chem., 234,
C35 (1982).
24. Hoberg H., Oster B. W., J. Organomeet. Chem., 252, 359 (1983).
25. Earl R. A., Vollhardt K. P. C, J. Am. Chem. Soc, 105, 6991 (1983); J Org
Chem., 49, 4786 (1984).
26. Collman J. P., Ace Chem. Res., 1, 136 (1968); Collman J. P., Kang J. W,,
Little W. F.f Sullivan M. P., Inorg. Chem., 7, 1298 (1968).
27. Wakatsuki Y., Kiramitsu Т., Yamazaki H., Tetrahedron Lett., 1974, 4549.
28. McAllister D. R., Bercaw J. E., Bergman R. G., J. Am. Chem. Soc, 99, 1666
(1977).
29. Yamazaki H., Hagihara N., J. Organomet. Chem., 7, P22 (1967).
30. Vollhardt K. P. C, Ace. Chem. Res., 10, 1 (1977).
Применение алкиновых комплексов переходных металлов 333
31. Vollhardt К. Р. С., Angew. Chem., Int., Ed. Engl., 23, 539 (1984) и
цитированные в этой статье работы.
32. Aalbersberg W. G. L., Barkovich A. J., Funk R. L, Hillard R. L,
Vollhardt K. P. C, J. Am. Chem. Soc, 97, 5600 (1975).
33. Funk R. L., Vollhardt K. P. C, J. Am. Chem. Soc, 99, 5483 (1977); 101, 215
(1979); 102, 5253 (1980).
34. Hillard R. L., Parnell C. A., Vollhardt К- Р. С, Tetrahedron, 39, 905 (1983).
35. Bonnemann #., Angew. Chem., Int. Ed. Engl., 17, 505 (1978); 24, 248
(1985).
36. Naiman A., Vollhardt К. Р. С, Angew. Chem., Int. Ed. Engl., 16, 708
(1977).
37. Wakatsuki Y., Yamazaki H., J. Organomet. Chem., 139, 169 (1977).
38. Sternberg E. D., Vollhardt K. P. C, J. Org. Chem, 47, 3447 (1982); Cli-
net J.-C, Dunach E., Vollhardt K- P. C, J. Am. Chem. Soc, 105, 6710 (1983);
Sternberg E. D., Vollhardt K. P. C, J. Org. Chem, 49, 1564, 1574 (1984);
Dunach E., Halter man R. L.} Vollhardt K. P. C, J. Am. Chem. Soc, 107,
1664 (1985).
19
ПРИМЕНЕНИЕ Г13-АЛЛИЛЬНЫХ КОМПЛЕКСОВ
ПЕРЕХОДНЫХ МЕТАЛЛОВ В ОРГАНИЧЕСКОМ
СИНТЕЗЕ
Несмотря на то (что т]3-аллильные комплексы—весьма
распространенные металлоорганические соединения, лишь немногие
из них нашли применение в качестве реагентов органического
синтеза. Настоящая глава посвящена химии наиболее
изученных г|3-аллильных комплексов палладия, а также использованию
в органическом синтезе аналогичных комплексов никеля,
железа, кобальта, молибдена и вольфрама. Структура и природа
связи г}3-аллильных металлокомплексов обсуждались в ч. 1,
разд. 3.7, д, а их взаимодействие с нуклеофилами описано в ч. 1,
разд. 7.4,-в. Просмотр этих разделов облегчит понимание
материала данной главы.
19.1. Реакции теломеризации сопряженных диенов,
катализируемые палладием
Взаимодействие сопряженных диенов с нуклеофилами в
присутствии такого катализатора, как ацетат палладия/трифенил-
фосфин [Pd(0) генерируется in situ], приводит к образованию
димеров, включающих один эквивалент нуклеофила. Этот
процесс, называемый теломеризацией [уравнение (19.1)], впервые
был описан Цудзи [1] ив настоящее время подробно изучен.
В реакции теломеризации активно участвуют такие нуклеофилы,
как вода, амины, спирты, соли карбоновых кислот, енамины,
нитроалканы и стабилизированные карбанионы [уравнение
(19.2)] [2]. Механизм процесса подробно не изучался;
предполагают, что он включает катализируемую палладием (0)
«восстановительную» димеризацию бутадиена с образованием
бис-т]3-аллильного комплекса палладия. Нуклеофильная атака
на этот промежуточный комплекс и последующее
восстановительное элиминирование из г]3-аллилпалладийгидрида приводят
к регенерации катализатора — палладия (0).
Возможность каталитической сборки функционально
замещенных ненасыщенных линейных углеводородных цепей,
впервые описанная Цудзи, была использована для синтеза
разнообразных природных соединений. Так, селективное окисление по
Вакеру (*ч. 1, гл. 11) олефина с концевой двойной связью в те-
Применение г)3-аллильных комплексов переходных металлов 335
(19.1)
^1 \У^ _ Г~\У\ YH
+ Pd(O)
С"
♦ Pd(O) ♦
досстано-
бительное
злы
минирование
минорный
продикт
Pd(OAc)2
РЬ,Р:
= Y=CO2Et, A = H
= CO2Et,
= SO2Ph, Y =
=Y = CO2Et, A = NHAc
Х=СОМе, Y=NHAc, A=R
X = COR, Y = OH, A = R
X=NO2, Y=CH3, A=H
(19.2)
(♦ изомер)
ломере уксусная кислота — бутадиен (приводит к образованию
кетоацетата, который превращают в макроциклический лактон
[3]. Его минорный изомер превращают в 1,5-дикетон и
используют для бисаннелирования в синтезе стероидов
[уравнение (19.3) [4]. Если в качестве нуклеофила взять аце-
тамидомалонат, можно (получить аминокислоты с длинной
цепью [уравнение (19.4)] [5]. Во многих других синтезах, где
в качестве исходных веществ используют бутадиеновые теломе-
ры, реакции с участием металлоорганических соединений
аналогичны описанным выше, так что синтезы отличаются друг от
друга только дальнейшими превращениями телом еров.
Замещенные 1,3-диены также подвергаются теломеризации, однако
при этом часто получаются сложные смеси стерео- и региоизо-
меров. Множество продуктов образуется также в случае, когда
нуклеофилом является аммиак или первичные амины,
поскольку о'ни 'могут атаковать более чем одну октадиенилыную
боковую цепь. Подобные -сложные процессы довольно редко
используются в синтезе.
336 Глава 19
ОАС Н2О, 02
(-)C(CO2Et)2
NHAc
(19.4)
CO2Et
91% NHAc
Образующийся в ходе этого процесса теломеризации
бис (т]3-аллил)!палладиевый интермедиат можно подвергнуть
карбонилироеанию. Уэйд [6] получил таким путем диены,
необходимые для синтеза бревикомина [уравнение (19.4)].
♦ СО ♦ EtOH
Pd(OAc)o
(19.5)
sCO2Et
(79:21)
экзо- и эндо~&ре5икомин
19.2. Использование в синтезе предварительно полученных
г)3-аллилпалладиевых комплексов
г]3-Аллил!палладийгалогениды — желтые кристаллические
вещества, устойчивые на воздухе, обычно получают
непосредственно из олефинов и 'солей палладия(II) в различных условиях
Применение г]3-аллильных комплексов переходных металлов 537
(ч. 1, гл. 3). Эти -комплексы довольно инертны к большинству
химических реагентов и по этой причине 'были мало 'исследова-
ны. Однако Цудзи [7] обнаружил, что в присутствии сильно
координирующих лигандов, та'ких, как фосфины или ДМСО, нук-
леофилы легко атакуют г}3-аллильный лиганд [уравнение
(19.6)]. Реакция носит довольно общий характер, в нее
вступают разнообразные г}3-аллилпалладийгалогениды и
стабилизированные кар'банионы. Подробное изучение реакции пробел
Трост. В случае несимметричных т]3-аллилпалладиевых
комплексов регио'селектшвноеть атаки зависит от структуры т]3-ал-
сильного комплекса и от природы карбаниона, хотя обычно
доминирует алкилированше менее замещенного конца г}3-аллиль-
ной системы [уравнение (19.7)]. Еноляты гсетонов и амины
также реагируют с г]3-аллилпалладиевы)ми комплексами
(см. ниже).
(19,6)
CH(CO2R)2
Pd
/\
Ph3P PPH3
SO2Me
СО о Me
MeSO2CHCO2Me
Ph^P, ТГФ
CH(CO2R)2
CH(CO2R)2
(I)
(19,7)
CH(CO2R)2
Ph3P, ТГФ
To обстоятельство, что т]3-аллилпалладийгалогениды
образуются .преимущественно с юесопряженными двойньими связями,
а стабилизированные серой карбаиионы атакуют эти комплексы
исключительно по менее замещенному т]3-аллильному
положению, позволяет провести «пренилирование» [8] [введение «пре-
нильной» группы Ме2С = СНСН2, уравнение (19.8)],
необходимое для 'синтеза природных полиизопренов.
Эти реакции нашли применение во многих интересных
синтезах, например в алкилировании холестанона и тестостерона
338 Глава 19
CO2Me
SO2Ph (19.8)
COoMe
CO? Me
в положение 6 [уравнение (19.9)] [9], синтезе стероидов с
необычной стереохимией при С-20 [уравнение (19.10) [10],
синтезе витамина А и родственных соединений [уравнение (19.11)]
R=C8M|3,0H
ЛУ
i)CH(CO2Me)2f
2) Lil, ДПФА
(19.9)
70% 6:1
СН2СО2Ме
ХСНСО2Ме
Ph2P(CH2)2PPh2
(19.10)
ОМе
X = C02Me 8!%
X=PhS02 83%
SOoPh
ОАс
Ph3P
ДМФА
Ph (19.1
АсО
Сочетая образование т]3-аллилпалладиевых комплексов при
присоединении ртутьорганических соединений к диенам (внед-
Применение г)3-аллильных комплексов переходных металлов 339
рение) (см. ч. 1, разд. 3.7, д) с внутримолекулярной нуклео-
фильной атакой на т]3-аллилпалладиевые комплексы, Ларок [12]
разработал общий подход к синтезу разнообразных
гетероциклических систем [уравнения (19.12) и (19.13)].
OH LiPdCI3
(переметал-
Н9СI дарование }
HgCf
71%
PdCI J
(внедрение)
119.12)
Pd-CI
76%
(19,13)
основание
По-видимому, все описанные процессы включают внешнюю
нуклеофмльную атаку со стороны, противоположной металлу.
Кроме того, многие углеродсодержащие нуклеофилы
первоначально атакуют металл, а затем переносятся к аллильной
группе в направляемой лигандом стадии «восстановительного
элиминирования». Шварц [13] показал, что алкенилциркониевые
реагенты, получаемые гидроцирнонированием алкинов (гл. 14),
переносят свои алкильные группы к т]3-аллилпалладиевым
(комплексам в стадии переметаллирова'ния от более электроположи-
телыного циркония \к 'менее электроположительному палладию.
В присутствии хороших ^-акцепторных лигандо'в, в частности
малеинового ангидрида, наблюдается реакция сочетания
[уравнение (19.14)]. Региоселективность отчасти зависит от природы
jc-акцепторного лиганда. Макмарри [14] использовал эти
реакции для получения длинноцепных ненасыщенных <кетоальдеги-
дов с целью их дальнейшей восстановительной циклизации
в м акр ациклические полиены [уравнение (19.15)]. Аналогичный
подход применил Шварц [15] для разработки избирательного
340 Глава 19
кросс-сочетания аллильных реактивов Гриньяра с т]3-аллилпал-
ладийгалогенидами [уравнение (19.16)].
(19.141
О
(главный OR
продукт)
96%
OHC
(стерои
V
i
■ъ . <
^i О
?'
96%
(19.15)
(19.16)
Нуклеофильная атака на г]3-аллилпалладиевые комплексы
практически всегда происходит по одному из аллильных концов,
однако Хигедас [16] обнаружил, что а-разветвленные еноляты
сложных эфирое в присутствии ГМФТА атакуют центральный
атом углерода в т]3-аллилпалладийгалогенидах, в результате
чего образуются циклопропаны [уравнение (19.17)]. Посколь-
Применение т]3-аллильных комплексов переходных металлов 341
ку и а-раэветвление, и ГМФТА способствуют О-алкилированию
енолятов сложных эфиров, образование циклопропана было
объяснено О-палладированием енолята с последующим
внутримолекулярным алкилированием т]3-аллильной группы. Однако
экспериментальных подтверждений такого механизма не
имеется.
СО2Ме
ГМФТД/ТГФ
МеО
(19.17)
и
бание
Кроме реакций с нуклеофилами, т]3-аллилпалладийхлор|иды
легко окисляются, давая сопряженные еноны или аллиловые
спирты, ацетаты или хлориды в зависимости от природы
окислителя. Например, стероидные т]3-аллильные комплексы
превращаются в сопряженные еноны при окислении ж-хлорпербензой-
ной кислотой [17], оксидом хрома (VI) в ДМФА [18] или
кислородом при облучении [19] [уравнение (19.18)]. В то же
время грег-бутилгидро'пероксид — МоО2(асас)2 окисляет более
замещенный атом углерода, в результате чего образуются
аллиловые спирты [уравнение (19.19)] [20], а под действием солей
меди получаются аллилхлориды, аллилбромиды или аллилацета-
ты в зависимости от природы медной соли [21]. Аллильное окис-
леиие олефинов в аллилацетаты катализируется солями
палладия (II) и, очевидно, протекает через окисление т]3-аллильного
интермедиата [уравнения (19.20) и (19.21)] [22,23].
19.3. Реакции аллильных субстратов, катализируемые палладием
Реакции, обсуждавшиеся в разд. 19.2, пригодны для прямого
алкил'Ирования или ам'иниро'вания олефинов в аллильное
положение, но недостатком их является необходимость вводить
в реакцию стехиометрические (Количества дорогих солей
палладия. Родственный и [потенциально более удобный процесс
основан на катализируемом палладием (0) нуклеофилыюм
замещении аллильных соединений. В реакцию вступают соединения
разных классов, дающие аллильные уходящие группы: ацетаты
и другие сложные эфиры, простые эфиры, спирты, амины, суль-
342 Глава 19
(19.18)
ZO°C7 АсОН ОАс
(19.19)
{19.20}
О (19.21)
1.94: Г
85%
ОАс
финаты, эпоксиды и нитросоединения. В качестве нуклеофилов
чаще в'сего применяются стабилизированные кар(банионы и
амины. С хиральными аллильными субстратами процесс в конечном
счете протекает с сохранением конфигурации, очевидно,
благодаря двойному обращению конфигурации (см. ч. 1, гл. 7).
Считается, что реакция идет через окислительное присоединение
аллильного субстрата к комплексу палладия (0) с образованием
того же катионного т]3-аллильного комплекса, что <и в стехио-
метрических реакциях, обсуждавшихся выше. Нуклеофильная
атака на этот аллильный комплекс приводит к образованию
наблюдаемого продукта аллильного замещения и регенерации
палладиевого(О) катализатора [уравнение (19.22)] [24]. Такой
механизм хорошо согласуется со стереохимией реакции
(сохранение конфигурации), являющейся результатом одного обраще-
(19.22)
Применение Г13-аллильных комплексов переходных металлов 343
ния конфигурации в стадии окислительного присоединения и
второго обращения <в стадии нуклеофильной атаки Однако при
взаимодействии аллилацетата с [РсЦРЬзРЬ] образования т]3-ал-
лилпалладийацетата не наблюдалось, хотя более поздние
исследования показали наличие 1,3-сдвига ацетата. Та.к что если
первая стадия предложенного механизма верна, она должна
быть легко обратима и т]3-аллилпалладиевые соединения
должны присутствовать в чрезвычайно малых концентрациях (более
основный палладиевый комплекс [(СузР)4Рс1] пр'И обработке
аллил ацетатами действительно образует т]3-аллилпалладиевые
комплексы) [25].
Каким бы ни был механизм реакции, она широко
используется в органическом синтезе и представляет один из наиболее
общих и полезных процессов, 'катализируемых переходными
металлами. Здесь будут приведены отдельные примеры из
множества имеющихся литературных данных. В качестве аллиль-
ных субстратов чаще всего используют аллилацетаты, что
объясняется как их доступностью, так и тем фактом, что они были
изучены первыми. В качестве нуклеофилов в реакциях с т]3-ал-
лилпалладием наиболее эффективны амины. Так, Бэквалль [26]
с помощью этого процесса получил 3-пиридилаллиламины
[уравнение (19.23)]; Трост синтезировал макроциклические
азотсодержащие гетероциклы [уравнение (19.24)] [27] и оснОВ-
ОАс
о-
С —Аг
Pd (acac)
Ar
(19.23)
dip hoc, ТГФ, 55%
Зру, 4ру, 2ру
40-80%
NM62
АсО
(19.24)
AcHN
70-80%
NHAc
ные циклические системы алкалоидов актинаболина, мезембри-
на и ибогамина [28]. Путем сочетания рассматриваемой
реакции с процессом образования углерод-углеродной связи,
который ускоряется системой Pd(II)/Ag(I) и механизм которого не
установлен [29], был осуществлен синтез ибогамина [30] и ка-
тарантина [уравнение (19.25)] [31]. Эти примеры хорошо
иллюстрируют как разнообразие структур, доступных в результате
344 Глава 19
аллильного аминирования, так -и толерантность процесса к
наличию различных функциональных групп.
О н (19.25)
OC-R .Ns
\\ /Г ^)
PdCI2
ибо гам
Широкое применение нашло также аллильное алкилирова-
ние аллилацетатов, катализируемое палладием. С помощью этой
реакции можно легко получить спироциклы [уравнение (19.26)]
[32], макроциклы [уравнение (19.27)] [33] и циклопропаны
[уравнение (19.28)] [34].
ОАС
CO2Et
CO2Et
NaH, 7%, L4Pd
ТГФ , 65%
66%
(19.26)
EtO
основание
84%
U9.27)
=\ .OAc
OH
AcO
NaH
L4Pd
Ac2O
(19.28)
60-90%
X*CN,
VC0zBu.-m/?e/n7 CN^ C02Et, COz?h
70%
Применение т]3-аллильных комплексов переходных металлов 345
Вводя в реакцию с ненасыщенными аллилацетатам'и
ненасыщенные карбанионы в качестве нуклеофилов, Трост [35]
разработал эффективный подход к синтезу трициклических систем
[уравнение (19.29)]. Обычно эта реакция алкилирования
применима только .к стабилизированным карбанионам, еноляты ке-
тонов вступают в нее довольно плохо. Однако Негиси [36]
обнаружил, что еноляты цинка или бора легко подвергаются ал-
лильному алкилированию, что свидетельствует о
чувствительности реакции к -природе противоиона [уравнение (19.30)].
(19.29)
ОАс
CO2Et
ТГФ,
кипячение
35%
96%
(19.30)
OZnCI
5%
АсО
В присутствии палладиевых катализаторов множество
других нуклеофилов взаимодействует с аллилацетатами. Так, на-
гтример, если в качестве нуклеофила используется трифенилфос-
фин, образуются аллилфосфониевые соли, которые затем
непосредственно можно ввести в реакцию Виттига [уравнение
(19.31)] [37]. Трибутилстаннан восстанавливает аллилацетаты
ОАс
5%L4Pd
PPh-,
I) BuLr
2) RCHO
70-80%
до олефинов в присутствии палладиевого(О) катализатора [38].
Используя в качестве нуклеофила натриевую соль /г-толуолсуль-
23—1129
^346 Глава 19
фокислоты, аллилацетаты можно превратить в аллилтозилаты
[39]. Атака обычно происходит преимущественно по наименее
замещенному положению г]3-аллильной группы, хотя регио'селек-
тивность процесса зависит от условий реакции [40]. Из аллил-
ацетатав можно щолучить аллилсиланы [уравнение (19.32)] [41]
и аллилстаяяаны [уравнение (19.33)] [42], используя алюми-
нийсодержащие реагенты в качестве нуклеофилов и палладий
в качестве катализатора. Все эти примеры 'иллюстрируют
большой набор нуклеофилов, участвующих в катализируемом
палладием процессе.
(I9.32)
(Me3Si)3AI O£t2 I i TMS
L4Pd
827c
OAc
(19,33)
70%
Описываемый .класс химических процессов, катализируемых
палладием, не ограничен реакциями простых, функционально не
замещенных ацетатов. Так, цианогидринацетаты алкилируются
стабилизированными карбанионами в присутствии
Pd(O)-катализатора до акрилоеитрилов [уравнение (19.34)] [43]. Атака
происходит по внутреннему положению, по-видимому, с
образованием сопряженного нитрила. Реакция идет в присутствии три-
метилсилильной группы как в аллильно!М, так и в винильном
положении, что позволяет получить аллил- или винилсиланы,
пригодные для дальнейших превращений [уравнения (19.35) и
(19.36)] [44, 45]. Атака и здесь осуществляется по наименее
замещенному положению.
Трост [46] разработал катализируемые палладием реакции
бифункционального аллилацетата с активированными олефина-
ми и на их основе .предложил эффективный метод синтеза цик-
лопентаноидных систем [уравнение (19.37)]. Считают, что
реакция начинается с окислительного присоединения с
образованием г]3-аллильного комплекса палладия (II). Отщепление три-
Применение г]3-аллильных комплексов переходных металлов 347
метилсилильной группы приводит к цвитерионному триметилен-
м становому комплексу, <в котором (все три группы СН2 химически
эквивалентны. Присоединение этого комплекса <к электроно-
деф'ицитному олефину протекает, по-видимому, ступенчато.
В 'первой стадии анионная часть т]3-аллильного лиганда присо-
ОАс . рн МвО2С СО2Ме (19.34)
.СОоМе L"Pd ^^
+ {-)<Г с
*CN
^ОАс
TMS
TMS
IMS
СО2Ме
97%
СО2Ме
СО2Ме
TMS
(окислительное
присоединение)
(19.35)
(19.36)
(19.37)
L M
CO2Me
CO2Me
СОрМе
единяется по Михаэлю к енону, затем образовавшийся енолят
улавливается электрофильным катиошшм г]3-аллилпалладиевым
комплексом [уравнение (19.38)]. Этот процесс оказался
исключительно важным для синтеза. Его внутримолекулярный
вариант позволяет получить бициклические системы [уравнение
(19.39)] [47]. Используя эту реакцию, Трост синтезировал
альбен [уравнение (19.40)] [48] и логанин [уравнение (19.41)]
[49].
Аса
TMS
Y X
W
(19.38*
CO2R, COR, SO2Ph, Ph
23*
£48 Глава 19
.TMS
СО2Ме
ОАс
TMS
I ♦
CO2Et
Pd(OAc)2
со Et (U30'?rQh
(19.39)
МеО2С н
(19,40)
CO2Et
ОАс
TMS
L4Pd
(19.41)
60%
Катализируемые палладием реакции аллильных соединений
весьма разнообразны, «и практически в них может участвовать
любая система, содержащая аллильную уходящую группу, так
что в реакцию можно вводить большой набор органических
субстратов. Например, Трост и Цудз-и 'изучили реакции аллилэпо-
ксидов. Эти соединения региоспецифично взаимодействуют с ну-
клеофилами ига олефиновой, а не по эпоксидной группировке.
Кроме того, раскрытие эпоксидного кольца в начальной стадии
приводит к образованию алкоксида, который в свою очередь,
действуя как основание, способствует образованию активного
карбаниона in situ [уравнение (19.42)] [50]. Эти реакции были
использованы для синтеза линейных полиизо'преноидных
соединений [51], макроциклов [52] [уравнение (19.43) ] и
построения боковых цепей стероидов [53]. На примере стероидов Цуд-
зи [54] показал, что процесс представляет собой cuh-SnZ'
присоединение, причем атакующая и уходящая группировки
располагаются исключительно в 1,4-смя-тюложении [уравнение
(19.44)]. Это соответствует окислительному присоединению
Pd(O) со стороны, противоположной эпоксиду, с последующим
алкилированием т]3-аллильного комплекса со стороны,
противоположной палладию (двойное обращение конфигурации).
Применение г]3-аллильных комплексов переходных металлов 349
SO2Ph
74%
(19.44)
TMSO
В этом процессе аллильного замещения могут участвовать и
другие кислородсодержащие уходящие группы; так, были
проведены реакции с аллилфениловыми эфирами [55], аллилфос-
фатами [56] и даже четвертичными аллиламмониевыми солями
[57]. При (использо)вании хирального палладиевого
катализатора наблюдается умеренная асимметрическая индукция
[уравнение (19.45)] [58]. В реакцию вступают даже аллиллитросоеди-
нения, но процесс несколько осложняется легкой
перегруппировкой в нитроолеф'ины б тех случаях, когда в а-положении
присутствуют кислые протоны. Однако Тамура и Хигедас [59]
показали, что 'сопряженные нитроолефины можно подвергнуть
аллильному аминирова'нию, по-видимому, за счет образования
аллилнитросоединений in situ [уравнение (19.46)].
Одной из наиболее сложных задач органического синтеза
является контроль стереохимии ацильных систем. Стереохими-
ческая природа образования т]3-аллилпалладиевых комплексов
350 Глава 19
о о
Pd(OAcb
dipftos *
бензол:
(19.45)
diphos* (SKR)BPPFA
Вшад 82%, ее 48%
(19.46*
и их реакций с карбанионами была -использована Тростом [60]
для передачи асимметрии от одного хирального центра конфор-
мационноподвшкных систем к другому, более удаленному. Такг
реакции органических соединений палладия использованы для
передачи хиральности виниллакто'нового участка к удаленному
винильному атому углерода, что приводило к образованию ин-
термедиата с двумя хиральными центрами в положениях 1,5
[уравнение (19.47)]. Для успешного протекания этого процесса
(19.47)
Me
СН(СО2Ме)2
Me,
со2н
СН(СО2Ме)2
Me
Н *'
т]3-аллилпалладиевый комплекс должен образовываться только
из показанной на рисунке конформации, т]3-аллильный комплекс
должен сохранять свою стереохимию, а нуклеофильная атака
должна происходить региошецифично по концевому атому
углерода т]3-аллильной системы. Все эти требования 'выполняются,
и реакция происходит с 90%-ным выхо'дом и стереоселектив.но-
стью, превышающей 95%. Эти реакции были использованы для
синтеза боковых цепей витамина Е [уравнение (19.48)] [61].
Аналогичная передача хиральности осуществлена в процессе
Применение т]3-аллильных комплексов переходных металлов 351
катализируемой палладием (0) 'перегруппировки винилогов лак-
тонов в циклопентаноны или щжлогептено>ны [уравнение
(19.49)] [62].
~СН(СО2Ме)2
(PdL4)
OR
Н Me Н Me
95% О
диастереомерно
чистый
119.4Q)
R = Н, Me
R'= NMe2, OBu
катализатор \ \
Pd(O) R' ' '■
о или R'c
93-98%
R О
(19.49)
да 98%
(в зависимости от к
катализатора)
Цудзи разработал удобный подход к реакциям с участием
г|3-аллилпалладия, (использовав в качестве исходных аллилкар-
бо'наты. Они легко претерпевают окислительное присоединение
к комплексам палладия(0), давая т]3-аллилпалладиевые интер-
медиаты, которые затем отщепляют СО2 и образуют т]3-аллил-
палладийалко1ксиды. Далее, в зависимости от природы неал-
лильной группы R в карбонате можно провести ряд
превращений, важных с точки зрения синтеза; см. уравнения (19.50)
[64], (19.51) [65], (19.52) [66].
Цудзи [67] разработал также формальное [3+2]-щгкл(шр|и-
соединение с использованием аллилкарбонатов, несущих
удаленные анионстабилиЗ'Ирующие заместители [уравнение (19.53)]
[см. также уравнение (19.37)].
Наконец, Ноза'ки [68] разработал ^катализируемую
(палладием (0) перегруппировку циклопропанов, содержащих электро-
ноа'кцепторные (анио'истабилизирующие) группы и диеновые
боковые цепи. Реакция, по-видимому, происходит через
окислительное присоединение циклопропанового кольца к Pd(O) с
образованием т]3-аллилпалладиевого комплекса и
стабилизированного карбаниона. Внутримолекулярное алкилирование
удаленного аллильного ко'нца приводит к образованию наблюдаемого
352 Глава 19
4 (19.50)
kV
0
A,
Pd(O)
(19.52)
J3-элиминирование
Pd(O)
Применение г^-аллильных комплексов переходных металлов 353
(19.53)
Pd(O)
= CN, ArSO2
продукта [уравнение (19.54)]. Первая стадия этого процесса
аналогична реакциям аллилэшксидов [уравнение (19.41)].
Ц9.54)
СО? Me
Pd(O)
С02Ме-
PdT
МеОоС
C02Me
С02Ме
19.4. Реакции 1,2- и 1,3-диенов с участием
г)3-аллилпалладиевых(П) интермедиатов
Среди ненасыщенных углеводородов, легко превращаемых
в т]3-аллил!палладиевые соединения, наиболее распространены
олеф'ины, однако весьма реакционноспособны и такие
субстраты, 1как 1,2- и 1,3-диены. Замещением \ио центральному
атому углерода аллен (1,2-диен) можно превратить (в т]3-аллил-
палладиевый комплекс. Цудзи [69] использовал внедрение ал-
лена в а-алкшшалладиевые(П) комплексы для получения т]3-ал-
лилпалладиевых производных, пригодных для дальнейшей функ-
ционализац.ии посредством реакций, обсуждавшихся в разд. 19.3
АгХ ♦ L4Pd
L
ArPdX
L
Ar
Cnh
G
Ar
(19.55)
70-90%
354 Глава 19
[уравнение (19.55)]. Хлоропалладирование аллена в среде бен-
зонитрила дает интересный г)3-аллилпалладиевый комплекс
с двумя алленовыми фрагментами [70]. Хигедас [71]
установил, что аллилхлорид можно заместить в отсутствие трифенил-
фосфина, но при добавлении трифенилфосфина т]3-аллильный
лиганд взаимодействует с нуклеофилами [уравнение (19.56)].
(19.56)
PdCI2
-Pd-
внедрение
4l
NuH
NuH
NuH2= H2C(CO2Et)2, CH2(SO2Ph)(CN)t BuNH2, MeNHNHMe
Если вводить в реакцию нуклеофилы, способные снова
реагировать после депротонирова'ния, можно получить необычные экзо-
циклические диены. Используя каталитические количества
хлорида палладия и окислительно расщепляя т]3-аллилпаладиевый
комплекс in situ хлоридом меди(II) (разд. 19.2), можно
получить 2,3-бис ^хлорометил) бутадиен с высоким выходом
[уравнение (19.57)]. Последний превращается в те же экзоцикличеокие
диены; таким образом, открывается возможность получения
диенов с помощью лишь каталитических количеств дорогих
солей палладия(II) [72].
(19.57)
===== + PdClp
CuCIo
PdCU
CI CI
1,3-Диены также легко превращаются в г|3-аллилпалладие-
вые комплексы путем хлоропалладирования. Как и в случае ди*
мерных алленовых комплексов в уравнении (19.56), хлороме-
тильная группа весьма реакционноспособна по отношению к ну-
клеофилам и в присутствии добавляемого лиганда
взаимодействует с концом т]3-аллильной группы. Акермарк [73]
использовал этот процесс для 1,4-диалкилирования 1,3-диенов
[уравнение (19.58)]. Бэквалль [74] интенсивно исследовал эту систему
и разработал эффективный метод стерео- и региоселективной
Применение т]3-аллильных комплексов переходных металлов 355
1,4-функционализацИ'И 1,3-|диенов. При (введении в реакцию
ацетата палладия начальное ацетоксилирование олефина
происходит с образованием граяс-изомера, что согласуется с
внешней нуклеофильной атакой. В отсутствие хлорида
окислительное расщепление т]3-аллилпалладиевого комплекса при-
(19.58)
PdCl
ТМЭДА
водит к цис-миграции координированного ацетата к т]3-ал-
лильному лиганду, в результате чего образуется транс-
диацетат и регенерируется катализатор — ацетат
палладия (II). Напротив, в 'присутствии каталитических количеств
хлорида окислительное расщепление приводит к атаке
некоординированным ацетатом со стороны, противоположной
палладию, в результате чего образуется только цш:-диацетат
[уравнение (19.59) ] [75]. Процесс носит общий характер, он изучен
Pd(OAc)2 __
(19.59)
Pd(OAc)2
ОАс
ОАС
Pd{OAc)2
АсО
Ас О"
катализатор .
LtCL pd
LlOAc /
окислитель
ОАс
Pd(OAc)2
на примере более двадцати циклических и ациклических диенов.
Введение в реакцию больших избытков хлорида позволяет с
-высокой эффективностью синтезировать 1-ацетокси-4-хлоро-2-алке-
356 Глава 19
ны. Реакция также стереоселективна и происходит через транс-
ацетоксйпалладироваиие с последующим окислительным
замещением палладия хлоридом со стороны, противоположной
металлу. Процесс имеет практическое значение, поскольку
позволяет химическим путем дифференцировать два конца диена,
а также получить субстраты, в которых два положения
потенциально доступны для реакций с участием т]3-аллилпалладие-
овых соединений (аллилгалогениды более реакционноспособны,
чем аллилацетаты [76]). Таким методо'м Бэквалль [77]
синтезировал (/?*/?*)- и (/?*5*) -изомеры феромона пчелы Карпенте-
ра [уравнение (19.60) ].
(19.60)
Pd<QAc)2, LiCI ^
Li О Ас, 5ензо-
осикон, НОАс ■ 2) ОН
19.5. Использование в синтезе г]3-аллильных комплексов
других металлов
Анион карбонила кобальта [Со(СО)4]~ представляет собой
слабое основание с умеренными нуклеофильньгми свойствами.
Он взаимодействует с активными органическими галогенидами,
давая алкилкарбо-ниль'ные комплексы кобальта, которые легко
внедряют монооксид углерода и превращаются в ацилкарбо-
нильные комплексы кобальта. Хек [78] обнаружил, что ацилко-
бальтовые производные легко внедряют диены, что приводит
vk ацилированным т]3-аллильньш комплексам ко'бальта. Под
действием сильных оснований последние отщепляют протон и дают
свободный ацилдиен. Хигедас [79] установил, что аналогичные
т]3-аллильные комплексы кобальта легко алкилируются
стабилизированными кар-банионами, в результате чего в конечном
счете получаются продукты 1,4-ацилирова'ния — алкилирования
диенов [уравнение (19.61)].
Применение Т13-аллильных комплексов переходных металлов 357
(19.61)
Со(СО)4- ♦ RX
RCo(CO)4
СО
Трост показал, что многие реакции аллилацетатов € нукле-
офилами, катализируемые комплексами палладия (0),
катализируются также (комплексами молибдена (0) -и вольф(рама(0), но>
региоселектив'ность реакций для всех трех металлов различна.
Палладий направляет нуклеофил к наименее стерически
затрудненному концу аллильного субстрата. Молибден направляет*
реакцию по более затрудненному концу, если в качестве
атакующего нуклеофила используют малонат, и по менее
затрудненному концу ери использовании нуклеофилов, более
чувствительных к стер'ическим факторам [80]. Вольфрамовый
катализатор направляет нуклеофил в наиболее стерически
затрудненные участки независимо от структуры .нуклеофила [81] (ч. 1,
разд. 3.4,6). Основываясь на экспериментальных данных в
сочетании с расчетами молекулярных орбиталей, Трост пришел
к выводу, что в катализируемых вольфрамом реакциях
существенную роль играют электронные свойства системы, тогда как
в реакциях, катализируемых палладием, доминируют стериче-
ские факторы. Молибден в этом ряду занимает промежуточное
положение [82]. Предполагают, что все эти реакции включаюг
образование *?]3-аллильных металлокомплексов.
(19.62)'
358 Глава 19
Цудэи обнаружил, что подобные реакции аллилкарбонатов
катализируются и другими переходными металлами помимо
палладия. Так, молибденовые, родиевые и никелевые комплексы
катализируют перегруппировки сложных аллил-^-кетоэфиров в
а-аллилкетоны [уравнение (19.63)] [83]. Фосфиновые
комплексы родия катализируют алкилирование аллилкарбодатов угле-
родсодержащими нуклеофилами [уравнение (19.64)] [84].
Предполагают, что оба процесса протекают через образование
соответствующих т]3-аллильных металлокомплексов.
с-ск
6
MLn (-CO2)
(М=Мо, Rh, Ni)
(19.63)
(19.64}
.ОСОМе
LnRhX
.RhO-C-OMe
X
RhOMe
NucH
, Nuc
-MeOH
у)3-Аллилникельгалогениды также находят применение в
органическом синтезе [85]. Хотя они изоструктурны и изоэлект-
ронны с т]3-аллилпалладийгалогенидами, комплексы никеля
отличаются по химическим свойствам. Т13-Аллилникельгалогениды
получают с высоким выходом при взаимодействии аллилгалоге-
нидов с комплексами никеля (0), обычно с кар'бонилом никеля
или бис (циклооктадиен) никелем -в неполярных растворителях,
таких, как бензол [уравнение (19.65)]. Эта реакция допускает
Ni(CC)4
или
Ni(COD)2
PhH
(19.65)
наличие самых разнообразных заместителей в аллильной цепи
и (позволяет получать множество важных с точки зрения
синтеза комплексов с различными функциональными группами. В
отличие от соответствующих комплексов палладия т]3-аллилникель-
галогениды невозможно получить непосредственно из олефинов.
Фактически не известны комплексы никеля(II) с олефинами, не
известны пути активации аллильного положения для
отщепления протона. т]3-Аллил»никельгалогениды представляют собой
твердые кристаллические вещества от темно-красного до крас-
Применение Т13-аллильных комплексов переходных металлов 359s
но-коричневого цвета; в растворе комплексы чувствительны
к -воздуху, но без доступа воздуха устойчивы в течение
нескольких лет.
В отличие от т]3-аллилпалладийгалогенидов т]3-аллилникель-
галогениды «не (подвергаются иуклеофильной атаке. Они сами
ведут себя как нуклеофилы, реагируя с органическими галоге-
нидами, альдегидами и кетонами с переносом аллильной
группы. Далее будет показано, что эти реакции идут по радикально-
цепному механизму, а не по механизму .нуклеофильного
замещения. Тем не 'менее по химическим свойствам т]3-аллилникель-
галогениды существенно отличаются от соответствующих
комплексов палладия.
Наиболее изучена «и широко применяется в синтезе реакция
т]3-аллилникельгалогенидов с органическими галогенидами, в
которой галоген замещается на аллильную группу [уравнение
(19.66)]. Эта реакция, открытая Кори, протекает только в по-
\у, ♦ R'X -=*- R \ + N«BrX (19.66)
лярных координирующих растворителях, таких, как ДМФА,
ГМФТА или N-метилпирролидон. Арил-, винил-, алкилбромиды:
(первичные и вторичные) и соответствующие иодиды
реагируют с высокими выходами, причем арил- и овинилгалогениды
более реакционноспосо'бны, чем алкилгалогениды. Хлориды
реагируют значительно медленнее, чем бромиды и иодиды. Эта
реакция не затрагивает большинство функциональных групп,
таких, как гидроксильная, сложноэфирная, амидная и нитриль-
ная. Комплексы никеля взаимодействуют преимущественно
с бро'мидами, а не с хлоридами, если они одновременно
присутствуют в молекуле и не реагируют с кетонами и альдегидами.
С несимметричными т]3-аллильными группами сочетание
происходит исключительно по наименее замещенному концу
молекулы в отличие от большинства других аллильных металлооргани-
чесиих соединений. Это свойство было успешно использовано
для синтеза а-санталена и э/ш-р-санталена [86] [уравнение
(19.67)]. Хотя аллилгалогениды очень реакционноспособны по^
отношению к комплексам никеля, в результате сочетания
обычно образуются все возможные продукты, что объясняется
быстрым обменом т]3-аллильного лигаада с аллилбромидом [уравнение-
(19.68)]. Е'сли две аллилыные группы отличаются электро'Н-
ным строением, то в значительной мере происходит
селективное кросс-сочетание [87, 88] [уравнение (19.69)]. Следует
отметить, что 2-метоксиаллилникелевый комплекс служит удобным
источником ацетонильной группы (СН3СОСН2~) для введения"
в такие субстраты, как арил- и винилгалогениды, которые обыч-
360 Глава 19
но не взаимодействуют с енолятами ацетона. Интересно, что
стереохимия олефина в т]3-алл'ильной системе утрачивается
в реакции переноса аллила, однако стереохимия олефина в ви-
нилгалогенидных субстратах обычно сохраняется.
■Л
Вг | быстро Вг
' V * ^ ^Вг (19.68)
(19.69)
CO2Et
ОМс
Для синтеза разнообразных изопреноидных хинонов из
соответствующих защищенных бромогидрохинонов Сато
воспользовался способностью т]3-аллилникельгалогенидов
взаимодействовав с ароматическими галогенопроизводными в очень
мягких условиях, не затрагивая большинства других
функциональных групп. В уравнении (19.70) приведена общая
последовательность превращений для получения простейшего аналога
витамина К [89]. Способность этих комплексов никеля
реагировать с полностью замещенными ароматическими галоген-идами
в мягких условиях, когда сочетание идет только ш наименее
<ггерически затрудненному аллильно'му положению и при этом
не затрагиваются ацетатные защитные группировки, делает их
весьма привлекательным инструментом синтеза. Для получения
витамина К (с более длинными боковыми цепями) требуются
Применение т^-аллильных комплексов переходных металлов 361
длинноцепные аллилгалогенидные комплексы. Они были
получены -из соответствующих бромидов в ©иде масел и
использовались без дополнительной очистки. Для получения серии кофер-
ОАс
ОАс
(19.70*
ОАс
I) КОН, NaHS03
2)FeCI3
ментов QT аналогичные превращения проведены с защищенным
(бромо) диметоксиметилбензохиноном [90]. Примеры других
синтезов с участием арилгалогенидов, проведенных Хигедасом
[87, 91], показаны © уравнениях (19.71), (19.72) и (19.73).
МеО
МеО
MeOv
(19.71)
ОМе
ОМ»
ОМе
ДМФА
соон
Pd(E)
0^ О
(19.72).
COOMe
NaH
(19.73).
362 Глава 19
Несмотря на то что при взаимодействии т]3-аллилникельгало-
генидоъ с органическими галогенидами образуются простые
продукты, процесс их образования весьма сложен. По-видимому, он
представляет собой радикально-цепную реакцию, которая
.инициируется облучением или добавлением восстанавливающих
агентов. Реакция полностью ингибируется при добавлении
менее одного мольного процента
ж-динитробензола—эффективного акцептора анион-радикалов. Хиральные вторичные галогениды
рацемизуются при аллилировании вследствие
промежуточного образования «свободных» радикалов с неспаренным
электроном на центральном атоме углерода. В то же время винил-
галогениды не меняют своей стереохимии, поскольку в этом
случае подобные свободные радикалы не образуются. Наконец,
в полярных растворителях наблюдается интенсивное
перераспределение г)3-аллилникельбромидного комплекса, которое
приводит к равновесной смеси, содержащей существенные
количества бис-у)3-аллилникеля, который нереакционноспособен по
отношению к органическим галогенидам [уравнение (19.74)].
ДМФА
<? Ni )) ♦ NiBro (19 74)
Для объяснения всех экспериментальных наблюдений Хиге-
дас [92] предложил механизм, показанный на рис. 19.1.
Согласно этому механизму, радикально-цепные особенности
процесса обусловлены исключительно той ролью, какую играют
производные никеля(I), генерируемые в стадии «инициирования
[например, за счет внутримолекулярного переноса электрона
во внутренней сфере димерного (т]3-аллил)никельгалогенида,
который затем распадается с образованием соединения (т]3-ал-
лил) никеля(I) (3)], а также в стадии восстановительного
элиминирования (стадия в). Эти соединения никеля (I) служат
ключевыми интермедиатами (передачи цепи. Ингибиравачше процесса
наблюдается в тех случаях, когда эти соединения
захватываются ловушками радикалов.
Для субстратов всех классов «двухэквивалентное»
окислительное присоединение органического галогенида к комплексу 3
приводит к образованию соединения никеля(III) 5 (стадия а).
В результате восстановительного элиминирования (стадия в)
образуются продукт сочетания и галогенид никеля(I).
Последний реагирует с исходным комплексом 1 (стадия г), давая
снова соединение (т]3-аллил) никеля (I) 3, обеспечивающее передачу
цепи. В случае винилгалогенидов обе стадии — окислительное
присоединение (стадия а) и восстановительное элиминирование
(стадия в) — протекают с сохранением геометрии олефина, что
определяет сохранение конфигурации в продукте сочетания.
(инициирование) RNi(II)BrBrNi(II) ^^ RNi(III)BrBrNi(I)R ^RNi(I) +RNi(III)Br2
A 3
(a)
(окислительное присоединение) RNi(I)+R'X —>• RNi(III)R'X
3 5
RNi(II)Br RNi(I) *
RNi(III)R'X (-RNi(FI)Br +=± RNi(III)R'X <==^ RNi(I) + RNi(III)R'X "
()
б' 5 б
(в)
обмен
рацемизация
(г)
Ni(I)X+RNi(II)Br —^ NiBrX+RNi(I) носитель цепи
Рис. 19.1. Общий механизм реакции
364 Глава 19
С алкилгалогенидами стадия окислительного присоединения
происходит, 'по-видимому, с обращением конфигурации в
процессе типа SN2. Восстановительное элиминирование должно идти
с сохранением конфигурации, так что в целом в реакции
наблюдается изменение стереохимии. Однако быстрый обмен 5 (с
переносом алкильной группы) либо с 3 (луть б), либо с 1
(путь б'), происходящий с обращением конфигурации, должен
приводить к рацемическому комплексу 5, так что в результате
во'сстановительного элиминирования получается рацемический
продукт. Пусть б соответствует реакции «самообмена» ал килыного
'комплекса металла со своим предшественником, имеющим более
низкую степень окисления. Этот процесс хорошо известен для
систем Rh (I) —Rh (III) и Со (III). Такой механизм предложен
для наблюдавшейся рацемизации хиральных бензильных'
комплексов Pd(II). Путь б' соответствует переносу алкильной
группы от соединения никеля (III) 5 к исходному комплексу
никеля (II) 1. Из этих двух процессов б' представляется более
вероятным из-за более высокой концентрации 1 по сравнению с 3.
Таким образом, рацемизация может происходить без участия
свободных радикалов.
Л
(19.75)
(19.76)
т^-Аллилникельгалогениды лучше реагируютс органическими
галогшидами, 'чем с альдегидами или !кетонами, но в несколько
более жестких условиях (при 50 °С вместо 20 °С) карбонильные
соединения вступают в реакцию [уравнение (19.75)], давая го-
моаллиловые спирты [86, 93]. Наиболее реакционноспособны
а-дикетоны, из которых получаются сс-кетогомоаллиловые
спирты. Легко идут реакции с альдегидами и циклическими кетона-
ми, включая холестанон, прогестерон, 5-сс-андростан-3,17-дион
('при этом стероиды реагируют по наиболее реакционноспособ-
ной карбонильной группе), однако простые алифатические и
Применение т]3-аллильных комплексов переходных металлов 365
«^-ненасыщенные кетоны весьма инертны. В отличие от других
аллильных металлоорганических соединений реакция идет по
менее замещенному концу аллильной группы. Взаимодействие
1]3-(2-карбоэтоксиаллил)никельбромида с альдегидами и кето-
нами приводит к опиро-а-метилен-^-бутиролактонам [уравнение
(19.76)].
ЛИТЕРАТУРА И ПРИМЕЧАНИЯ
1. См. обзоры: Tsuji /, Organic Synthesis with Palladium Complexes, Sprin-
ger-Verlag, Berlin, 1980; Top. Curr. Chem., 91, 30 (1980); Pure Appl. Chem.,
53 2371 (1981); 54, 197 (1982); Ace. Chem. Res., 6, 8 (1973); Adv. Organo-
met. Chem., 17, 141 (1979); Ann. N. Y. Acad. ScL, 333, 250 (1980).
2. Hegedus L. S., in: Comprehensive Carbanion Chemistry, Buncel E., Durst T.
(Eds.), Elsevier. Amsterdam, 1984, pp. 1—64.
3. Takahashi Т., Minami /., Tsuji /, Tetrahedron Lett., 22, 2651 (1981).
4. Tsuji /, Pure Appl. Chem., 53, 2371 (1981).
5. Haudegoud J.-P., Chauvin Y., Commerluc D., J. Org. Chem., 44, 3063 (1979).
6. Byron N. Т., Grigg R., Kongkathip В., Ramer G., Wade A. R., J. Chem. Soc,
Perkin Trans. 1, 1984, 1643.
7. Tsuji J., Takahashi H., Morikawa M.f Tetrahedron Lett., 1965, 4387; Tsuji /.,
Bull. Chem. Soc. Japan, 46, 1896 (1973); Trost В. М., Weber L., Strege P. A.,
Fullerton T. I., Dietsche T. /., J. Am. Chem. Soc, 100, 3416 (1978).
8. Trost B. M., Strege P. A., Weber L., Fullerton T. J., Dietsche T. /, J. Am.
Chem. Soc, 100, 3426 (1978).
9. Collins D. /., Jackson W. R.t Timms R. N.t Tetrahedron Lett., 1976, 495.
10. Trost B. M., Verhoeven T. R., J. Am. Chem. Soc, 100, 3435 (1978).
11. Marchand P. S., Wong H. S., Blount J. F.t J. Org. Chem, 43, 4769 (1978).
12. Larock R. C.t Harrison L. W., Hsu M. Я, J. Org. Chem, 49, 3662 (1984).
13. Riediker M., Schwartz /, Tetrahedron Lett, 22, 4655 (1981); Hayasi Y.,
Riediker M., Temple J. S., Schwartz /., Tetrahedron Lett, 22, 2629 (1981);
Temple J. S., Reidiker M., Schwartz /, J. Am. Chem. Soc, 104, 1310 (1982).
14. McMurry J. E., Matz J. R.t Tetrahedron Lett, 23, 2723 (1982); McMurry J. Ё.,
Matz /. R., Kees K. L.t Bock P. A., Tetrahedron Lett, 23, 1777 (1982).
15. Goliaszewski A., Schwartz /., J. Am. Chem. Soc, 106, 5028 (1984); Organo-
metallics, 4, 417 (1985).
16. Hegedus L. S., Darlington W. H., Russell C. E.t J. Org. Chem, 45, 5193
(1980).
17. Mahe C, Patin H., vanHulle M.-T., Barton D. H., J. Chem. Soc, Perkin
Trans. 1, 1981, 2504.
18. Satoh J. Y., Horiuchi C. A., Bull. Chem. Soc. Japan, 54, 625 (1981).
19. Muzart I., Pale P., Pete J. P, Tetrahedron Lett, 24, 4567 (1983).
20. Jitsukawa K., Kaneda K., Teranishi S, J. Org. Chem, 48, 389 (1983).
21. Castanet Y., Petit F., J. Chem. Res. (S), 1982, 238.
22. Horiuchi C. A., Satoh J. У, J. Chem. Soc, Perkin Trans. 1, 1982, 2595.
23. McMurry J. E., Kocovsky P, Tetrahedron Lett, 25, 4187 (1984).
24. Trost B. M., Verhoeven T. R.t J. Am. Chem. Soc, 102, 4730 (1980) и
цитированные в этой статье работы; Trost В. М., Асе Chem. Res, 13, 385 (1980);
Pure Appl. Chem, 53, 2357 (1981); Tetrahedron, 33, 2615 (1977); Tsuji /.,
Pure Appl. Chem, 54, 197 (1982); Takahashi K., Miyaki A., Hata G, Bull
Chem. Soc. Japan, 45, 230 (1972); Atkins K. E.t Walker W. E., Manzik R. M.t
Tetrahedron Lett, 1970, 3821.
25. Yamamoto Т., Saito O., Yamamoto A., J. Am. Chem. Soc, 103, 5600 (1981).
26. Backvall J. E., Nordberg R. E.f Nystrom J. E., Hogberg T.} Ulff В J Org
Chem., 46, 3479 (1981).
366 Глава 19
27. Trost В. М., Cossy /., J. Am. Chem. Soc, 104, 6881 (1982).
28. Trost В. Л1, Genet J. P., J. Am. Chem. Soc, 98, 8516 (1976).
29. Trost B. M., Fortunak J. M. D., Organometallics, 1, 7 (1982).
30. Trost B. M., Godleski S. A., Genet J. P., J. Am. Chem. Soc, 100, 3930
(1978).
31. Trost В. Л1, Godleski S. A., Belletire J. L, J. Org. Chem., 44, 2052 (1979).
32. Godleski S. A., Valpez R. S, J. Org. Chem., 47, 381 (1982).
33. Trost B. M., Brickner S. /., J. Am. Chem. Soc, 105, 568 (1983).
34. Genet J. P., Prau F., J. Org. Chem., 46, 2414 (1981).
35. Trost B. M., Lautens M., Hung M.-H., Carmichael C. S., J. Am. Chem. Soc.,.
106, 7641 (1984); Trost B. M.t Lautens Л1, J. Am. Chem. Soc, 107, 1781
(1985).
36. Negishi EA., John R. A., J. Org. Chem., 48, 4098 (1983).
37. Tsukahara Y., Kinoshita H., Inomata K., Kotake #., Bull. Chem. Soc. Japan,.
57, 3013 (1984).
38. Keinan E., Greenspoon N., Tetrahedron Lett., 23, 241 (1982); J. Org. Chem.,
48, 3545 (1983); J. Org. Chem., 47, 4380 (1982).
39. Inomata K-, Yamamoto Т., Kotake #., Chem. Lett., 1981, 1357.
40. Julia M., Mel M., Riglini A., Uguen D., J. Organomet. Chem., 235, 113 (1982).
41. Trost B. M.t Yoshida J.-L, Lautens M., J. Am. Chem. Soc, 105, 4494 (1983).
42. Trost B. M., Herndon J. W., J. Am. Chem. Soc, 106, 6835 (1984).
43. Tsuji J., Ueno #., Kobayashi Y., Okumoto #., Tetrahedron Lett., 22, 2573
(1981).
44. Hirao Т., Endo J., Ohshiro Y.f Agawa Г., Tetrahedron Lett., 22, 3079
(1981); Trost B. M., Self C. R.t J. Org. Chem., 49, 468 (1984).
45. Trost B. M., Self C. R.t J. Am. Chem. Soc, 105, 5942 (1983).
46. Trost B. M., Chem. Soc. Rev., 11, 141 (1982); Trost B. M., Chan D. M. Г.,
J. Am. Chem. Soc, 105, 2315, 2326 (1983); Trost В. Л1, Nanninga T. N.,
Satoh Г., J. Am. Chem. Soc, 107, 121 (1985); Trost В. М., Angew. Chem.,
Int. Ed. EngL, 25, 1 (1986).
47. Trost B. M., Chan D. M. Т., J. Am. Chem. Soc, 104, 3733 (1982).
48. Trost В. М., Renault P., J. Am. Chem. Soc, 104, 6668 (1982).
49. Trost B. M., Nanninga T. N.t J. Am. Chem. Soc, 107, 721, 1075 (1985).
50. Trost B. M., Molander G. A., J. Am. Chem. Soc, 103, 5969 (1981).
51. Tsuji J., Kataoka H., Kobayashi У., Tetrahedron Lett., 22, 2575 (1981).
52. Trost В. Л1, Warner R. W., J. Am. Chem. Soc, 104, 6110 (1982); 105, 5940
(1983); Takahashi Т., Kataoka #., Tsuji /., J. Am. Chem. Soc, 105, 147
(1983).
53. Wicha J., Kabat M. M., J. Chem. Soc, Chem. Commun., 1983, 984.
54. Takahashi Т., Ootake A., Tsuji /., Tetrahedron Lett., 25, 1921 (1984).
55. Kobayashi Y.t Tsuji /., Tetrahedron Lett., 22, 4295 (1981); Tsuji J., ShimizuL,
Minami L, Ohashi У., Tetrahedron Lett., 23, 4809 (1982).
56. Tomagawa Y., Nishimura K-, Kawasaki A., Murahashi S.-i., Tetrahedron Lett.,
23, 5549 (1982).
57. Hirao Т., Yamada N., Ohshiro Y.f Agawa Т., J. Organomet. Chem., 236, 409
(1982).
58. Yamamoto K., Tsuji J., Tetrahedron Lett., 23, 3089 (1982).
59. Tamura R., Hegedus L. 5., J. Am. Chem. Soc, 104, 3727 (1982); Tamura R.,
Hayashi /(., Kai Y., Oda D.y Tetrahedron Lett., 25, 4437 (1984).
60. Trost B. M., Klun T. P., J. Am. Chem. Soc, 101, 6756 (1979).
61. Trost B. M., Klun T. P., J. Am. Chem. Soc, 103, 1864 (1981).
62. Trost B. M., Runge T. A, J. Am. Chem. Soc, 103, 2485, 7550, 7559 (1981).
64. Tsuji /., Minami /., Shimizu /., Tetrahedron Lett, 24, 1793 (1983); Shimi-
zu I., Ohashi Y., Tsuji /, Tetrahedron Lett, 24, 3865 (1983).
65. Tsuji J., Minami I., Shimizu /., Tetrahedron Lett, 24, 5635, 4713, 5639, 1325
(1983).
Применение т]3-аллильных комплексов переходных металлов 367
66. Tsuji L, Minami L, Shimizu /., Tetrahedron Lett., 25, 2791 (1984); Tsuji J.,
Shimizu I., Minami I., Ohashi Y., Sugeura Т., Takahashi /(., J. Org. Chem.,
50, 1523 (1985).
67. Shimizu I., Ohashi Y., Tsuji /., Tetrahedron Lett., 1985, 26.
68. Morizawa Y., Oshima K., Nozaki #., Tetrahedron Lett., 23, 2871 (1982);
Israel J. Chem., 24, 149 (1984).
69. Shimizu I., Tsuji /., Chem. Lett., 1984, 233.
70. Schultze R. G., Tetrahedron Lett., 1964, 301; Tetrahedron, 20, 2809 (1964);
Lupin M. S., Shaw B. L., Tetrahedron Lett., 1965, 883.
71. Hegedus L. S., Kambe N., Tamura R.f Wood gate P., Organometallics, 2, 1658
(1983).
72. Hegedus L. S., Kambe N., Ishii Y., Mori A, J. Org. Chem., 50, 2240 (1985).
73. Akermark В., Ljunguist A., Panunzio M., Tetrahedron Lett., 22, 1055 (1981).
74. Backvall J. £., Accts. Chem. Res., 16, 335 (1983); Pure Appl. Chem., 55,
1669 (1983).
75. Backvall J. E., Bystrom S. E.3 Nordberg R. E., J. Org. Chem., 49, 4619
(1984).
76. Backvall /. E., Nystrom J. E., Nordberg R. E., J. Am. Chem. Soc, 107, 3676,
6892 (1985).
77. Backvall J. E., Bystrom S. E., Nystrom J. £., Tetrahedron, 41, 5761 (1985).
78. Heck R. F.t in: Organic Synthesis via Metal Carbonyls, Wender I., Pino P.
(Eds.), Wiley, New York, 1968, vol. 1, pp. 379—384 (есть русский перевод
первого издания книги: Органические синтезы через карбонилы металлов.
Пер. с англ./Под ред. Уэндера И., Пино П. — М.: Мир, 1973); Heck R. F.,
Breslow D. S., J. Am. Chem. Soc, 85, 2779 (1963).
79. Hegedus L. S., Inoue Y., J. Am. Chem. Soc, 104, 4917 (1982); Hegedus L. S.>
Perry R. /., J. Org. Chem., 49, 2570 (1984).
80. Trost B. M., Lautens M., J. Am. Chem. Soc, 104, 5543 (1982); 105, 3343
(1983); Organometallics, 2, 1687 (1983).
81. Trost B. M.f Hung M. H., J. Am. Chem. Soc, 105, 7757 (1983).
82. Trost B. M., Hung M. #., J. Am. Chem. Soc, 106, 6837 (1984).
83. Tsuji J., Minami /., Shimizu /., Chem. Lett., 1984, 1721.
84. Tsuji J., Minami I., Shimizu /., Tetrahedron Lett., 25, 5157 (1984).
85. См. обзоры: Semmelhack M. F., Org. React., 19, 115 (1972); Baker R., Chem.
Rev., 73, 487 (1973); Jolly Wilke (в работе [2]; Hegedus L. S., J. Organimet.
Chem. Lib., 1, 329 (1976); Billington D. C, Chem. Soc Rev., 14, 93 (1985).
86. Corey E. J., Semmelhack M. F., J. Am. Chem. Soc, 89, 2755 (1967).
87. Hegedus L. S., Stiverson R. /(., J. Am. Chem. Soc, 96, 3250 (1974).
88. Guerrieri Fr, Chiusoli G. P., Merzoni S., Gazz. Chim. Ital., 104, 557 (1974).
89. Saito K.f Inoue S., Sato /(., J. Chem. Soc, Chem. Commun, 1972, 953; J.
Chem. Soc, Perkin Trans. 1, 1973, 2289.
90. Inoue S.} Yamaguchi R., Saito K., Sato K.t Bull. Chem. Soc. Japan, 47, 3098
(1974); Sato K., Inoue S.} Yamaguchi R., J. Org. Chem., 37, 1889 (1972).
91. Hegedus L. S., Korte D. E.t Wirth R. /(., J. Org. Chem., 42, 1329 (1977).
92. Hegedus L. S., Thompson D. H. P., J. Am. Chem. Soc, 107, 5663 (1985).
93. Hegedus L. S., Wagner S. D., Waterman E. L., Siirala-Hansen K., J. Org.
Chem., 40, 593 (1975).
20
ПРИМЕНЕНИЕ АРЕНОВЫХ КОМПЛЕКСОВ ПЕРЕХОДНЫХ
МЕТАЛЛОВ В ОРГАНИЧЕСКОМ СИНТЕЗЕ
Как было показано в ч. 1, разд. 3.7, в, некоторые переходные
металлы, например Cr, Mo, W, Fe, V, Mn, Rh, образуют с
аренами устойчивые комплексы, которые можно охарактеризовать.
Уже начато изучение ареновых комплексов железа и марганца,
но пока только ареновые комплексы хрома нашли (Применение
« органическом синтезе, что имеет свои причины. Арентрикар-
бонильные комплексы хрома легко получить просто при
нагревании Сг(СО)б с ареном. Эти комплексы представляют собой
кристаллические вещества, устойчивые на воздухе, что
облегчает работу с ними. Они вступают во многие синтетически ценные
реакции. Ареновая группа легко отделяется от комплекса при
мягком окислении металла водным Се(IV) или иодом, а также
при выдерживании комплекса на воздухе при облучении
солнечным светом. Последний 'момент очень важен для органического
синтеза, так как присутствие металла в конечном продукте
обычно нежелательно.
Комплексообразование арена с фрагментом Сг(СО)3 во
многих отношениях меняет его химические свойства. Схематически
это показано на рис. 20.1 [1]. По отношению к ароматическому
преимущественно нуклео-
доильное замещение -^ увеличение сольболива
\ Г)Г>Р
кобышекие ^cEETV— с-с
кислотности" "*" ~^ ' < ■ ■
Л (Jjjii
стерическое У zx^ повышение кислотности
затруднение \
Рис. 20.1.
ядру Сг(СО)3 служит электроноакцепторным фрагментом,
вследствие чего закомплексованный арен активируется по
отношению к нуклеофильному замещению и дезактивируется к элек-
трофильному замещению. Включенное в комплекс «элвктро-
дефицитное» ареноеое 'кольцо (Способно лучше
стабилизировать отрицательный заряд. Ареновые «и бензильные
протоны становятся более кислыми и (потому легче поддаются
368
Применение ареновых комплексов переходных металлов 369
литиированию. Наконец, хромоюодержащий фрагмент
блокирует одну -поверхность арена и тем «самым
направляет входящий реагент «на (противоположную
(незакомплексованную) сторону. На всех этих эффектах основано
применение ареновых комплексов в органическом синтезе, которому
и посвящена настоящая глава.
20.1. Присоединение углеродсодержащих нуклеофилов
к ареновым комплексам хрома [2]
Семмелхак подробно изучил прямое взаимодействие арен-
трикарбонильных комплексов хрома с карбанионами. Этот
процесс подробно обсуждался в ч. 1, гл. 7, схематически он
представлен уравнением (20.1). Нуклеофильная атака карбаниона
R^ ^H
ГО]
R(-)
Сг(СОЬ
(20.1)
Cr(C0)3 Cr(CO)3
осуществляется со стороны, противоположной металлу, что
приводит к образованию анионного г]5-циклогексадиенильного
комплекса. При окислительном расщеплении генерируется алкилиро-
ван'ный арен, при протонолизе — циклогексадиен; при реакции
с электрофилами получаются либо арентрикарбонильный
комплекс хрома, либо ацилироъанные соединения (ом. ниже). При
наличии заместителей в ароматическом кольце региоселекти'в-
ность соответствует ожидаемой для нуклеофильного
ароматического замещения (в жегднположение к электронодонорным
группам), и ее можно предсказать на основании допущения о том,
что граничные молекулярные орбитали центрированы
относительно арена [3]. Как мож'но предположить для реакций
нуклеофильного ароматического замещения, лиганды,
повышающие электронную плотность на атоме металла, как, например,
фосфи'ны, лонижают общую реакционную способность ареновых
комплексов по отношению к нуклеофильной атаке [4а, 46].
Кроме того, ряд экспериментальных данных указывает на то,
что 'предпочтительным 'местом нуклеофильной атаки является
24—1129
370 Глава 20
углерод, заслоненный карбонильной группой фрагмента Сг (СО)з
IB наиболее устойчивом конформере [4в].
Оба эффекта комплексования арена с хромом,
активирующий и направляющий, .можно(выгодно использовать в
синтетических целях. Такие электронообогащенные ароматические
соединения, как индолы и бензофураны, обычно инертны в
реакциях нуклеофильного замещения. Более того, они подвергаются
электрофильному замещению по электронообогащенным
положениям 1, 2 и 3, но положение 4 не затрагивается. Как показал
Семмелхак [5], шмплексообразовавие этих аренов с хромом
позволяет провести алкилирование даже по положению 4, если
только в 'положении 3 нет объемного заместителя,
направляющего реакцию по положению 7 [уравнение (20.2)]. Это
существенно расширяет синтетические возможности, поскольку в
обычных условиях очень трудно ввести функциональные заместители
в 'положения С-4 и С-7.
R
0 ft
2)1,
(20.2)
NH, NMe, NBz, NSiR3> NC02
Hf Met CH2SiMe3
CH2CN, V-CN, У-С02Ви- трет, (^ ),
Представляет также интерес внутримолекулярный вариант
ароматического алкилирования, гладко проходящий с карбанио-
нами, стабилизированными цианогруппой (в случае сложноэфир-
ной группы внутримолекулярная реакция не имеет
препаративной ценности). Образование того или иного продукта зависит
от условий реакции, длины цепи -и наличия других заместителей
в ароматическом кольце. Так, реакция между комплексом 5-фе-
нилвалеронитрила и литийтетраметилпиперидидом (с
образованием аниона) и последующее окисление иодом приводят к
производному 1-цианотетрагидронафталина с высоким выходом
[уравнение (20.3)]. Следующий высший гомолог в тех же уело-
-(CH2)3CH2CN
I) UTMP, -78°С
Сг(СО)3
(20.3)
Применение ареновых комплексов переходных металлов 371
виях дает смесь 1,2- и 1,1-опирот1родуктов. Если проводить
реакцию при более высокой температуре (О °С вместо —78 °С)
и до окисления обработать трифтороуксусной кислотой, то
образуются почти исключительно спиросоединения [уравнение
(20.4)]. Такое изменение продуктов свидетельствует об
обратимой природе процесса алкилирования (ч. 1, гл. 7), а также
о кинетическом контроле при низких температурах и
термодинамическом контроле при более высоких температурах.
Следующий низший гомолог не циклизуется в индановую систему,
а претерпевает димеризацию с образованием метациклофана
[уравнение (20.5)] [6]. Очевидно, ход реакции очень зависит
от структурных особениоютей субстрата.
Сг(СО)3
\) LiTMF, ~78»С
2)Н*
3)!2
(20.4)
28 (72%)
1) LiTMP, p°C,
24 ч,
2) Н +
3) 12
97
Cr(C0)3
2)12
CN
U0.5)
Как было показано ранее, при окислительном расщеплении
промежуточного г]5-циклогексадиенильного комплекса хрома
регенерируется арен. Однако при расщеплении тех же комплексов
кислотой образуются свободные циклогексадиены [уравнение
(20.6)]. Процесс носит общий характер, на его основе из
(г|6-анизол) трикарбонилхрома можно получить 3-замещенные
циклогексеноны [уравнение (20.7)] [7]. Если протолитическое
расщепление (проводить в присутствии избытка шдаоо'ксида
углерода, можно регенерировать Сг(СО)б, исходное соединение
24*
372 Глава 20
для аренового комплекса [8].
1) RW
Сг(С0)з
-ОМе
Сг(СО)3
ъ\ окисление
i)R(-)
3) NH4OH
оме
(главный изомер) (го.6)
(20.7)
н2о
Семмелхак [9] использовал внутримолекулярное алкилиро-
©ание аренов в сочетании с протолитическим расщеплением
в 'изящном синтезе акоренона В [уравнение (20.8)].
Алкилирование закомплексованного >с хромом арена защищенным циа-
ногидрином (эквивалент ацил-аниона), последующее декомп-
лексование и гидролиз цианогидрина приводят к единственному
региоизомеру тризамещенного арена. При модификации боковой
цепи с введением хирального центра получается рацемическая
смесь продуктов. При комплексовании их с хромом возникает
еще один хиральный центр (см. ч. 1, гл. 3) «и образуется смесь
диастереомеров в соотношении 3 : 2. Разделение этой смеси и
последующее внутримолекулярное алкилирование
соответствующего диастереомера дают спироциклический предшественник
акоренона В.
Хотя г]5-циклогексадиенильные комплексы хрома можно
погасить кислотами, реакции с целым рядом других электрофилов
приводят к г]6-арено'вым комплексам [уравнение (20.1)]. Дело
в том, что для большинства (карбанионов алкилирование
обратимо и анион, находящийся в равновесии с алкилированным
аренам, преимущественно реагирует с добавляемым электрофи-
лом. Эту обратимость отметил Сем-мелхак ори алкилировании
хлоробензольных комплексов хрома (ч. 1, гл. 7). Кюндиг [10]
показал, что (1,4-диметоксинафталин)трикарбо[нилхром
подвергается кинетическому алкилиро'ванию по ^-положению, но
с большинством карбанионов алкилирование обратимо, поэтому
термодинамически более выгоден продукт, образующийся в
результате а-алкилирования [уравнение (20.9)].
Однако оказалось, что дитиан-анион, а также фениллитий
и гр£т-бутиллитий необратимо присоединяются в ^-положение.
Кюндиг выгодно использовал это обстоятельство в двух
случаях. Окислением ^-аддукта дитиана церием (IV) был получен
с хорошим !выходом интермедиат для синтеза дауномицинона
[уравнение (20.10)] [10].
Применение ареновых комплексов переходных металлов 37:
NC
МеО
П Li
0«C, 0,5 ^
(C0)3Cr
2)
3) -OH
4) H +
Сг(С0)6
XZS \—. диоксан.
(рацеми ч ее кий)
(20.8)
только один изомер
92%
, Зиастереомер
'" (рацемически
ЛДА
диастереомер А —
(рацемический) -78° С
Т'н
90%
{+)-ахоренон В
ОМе
ОМе
ОМе Сг(С0)3
()
374 Глава 20
оме
ОМе
о
(20.10)
!
ОМе Сг(С0)3
91%
ОМе
Се (ЕЛ
0Ме Сг(С0)-т
()
62%
ОМе
да у но пи ц и нон
ОМе
65%
Еще более интересна проведенная Кюндигом [И] электро-
филь'ная атака на г]5-циклогексадиенильный комплекс хрома,
необратимо образующийся из дитиан-карбаниона. В результате
внедрения монооксида углерода в продукт вместо ожидаемого
алкилирования наблюдается ацилирование ароматического
соединения. Процесс носит общий характер [уравнения (20.11) —
(20.13)] и может использоваться в синтезе. Выделение СО из
комплекса не представляет труда, при этом исходный Сг(СО)б
регенерируется. Следует также отметить, что электрофил
(ацильная группа) оказывается с той же самой стороны,
которая занята металлом, так что реакция идет по пути,
включающему атаку электрофила на металл, внедрение СО, а затем
восстановительное элиминирование [уравнение (20.14)].
20.2. Литиирование арентрикарбонильных комплексов хрома
Семмелхак (показал, что в (т]6-арен)трикарбонильных
комплексах хрома -связи С—Н ароматического кольца отличаются
•повышенной кислотностью. При обработке комплексов я-бутил-
литием происходит металлирование кольца; образующиеся при
этом литио(т]6-арен)трикарбо»ильные комплексы хрома
способны взаимодействовать с разнообразными электрофилами.
Металлирование комплексов хрома с анизолом, фторобензолом и
хлоробензолом всегда осуществляется по орто-положению [12]
Применение ареновых комплексов переходных металлов 375
1 V
Cr(C0)3
Гл
(20.11)
RX
СО
, Et,PhCH2,
R
H
70-90%
0
OMg
I) RLi
Cr(C0)3
MeO NvN—R1
2) R'Xf CO
OMe
42-76%
(20.12)
(CO)3Cr
I) RLi
2) R'X, CO
3) PPh3
OMe
R1
(20.13)
(20.14)
376 Глава 20
[уравнение (20.15)]. В результате получаются устойчивые
(т)6-арен) трикарбонильные комплексы хрома, которые можно
подвергнуть дальнейшей функционализацйи путем нуклеофиль-
(20Л5)
Cr(COh
Сг(СО)3
Y«H, ОМе, F, CI
Е^С02, МеХ, PhCHO, TMSCI, MeCOMe
Я*СООМе, Me, PhCHOH, TMS, Me2C0H
ного замещения. Семмелхак [13] использовал преимущество
обоих процессов в синтезе френолицина [уравнение (20.16)].
Одной из ключевых стадий в этом изящном синтезе является
катализируемое палладием гидроксиацилирование (гл. 17).
ОМе
TMS
(СОКСг
TMS
DBuLi
2)Cul
Cr(CO)3
96%
(20.16)
2) l2
3) Н+,
CN
63%
ОМе
ОН
PdCI2/CuCI2
СО, МеОН
срр с ноли ц а /1
Сочетая реги'оспецифическое литиирование фторобензольного
«комплекса хрома с легким вытеснением фтора из
ароматического кольца этой системы, Уиддоусон [14] синтезировал
разнообразные гетероциклы [см., например, уравнение (20.17)].
На основе литиирования закомплексованных с хромом аренов
он разработал метод введения функциональных заместителей
в обычно нереакционно'способное положение С-4 индолов [урав-
Применение ареновых комплексов переходных металлов 377
нение (20.18)] [15]. Изучена также региохимия литерования
многих других замещенных арентр>икарбонильных комплексов
хрома [16].
BuL!
Cr(COh
Сг(С0)3
\dQ.\7)
Сг(С0)3
(СО)3Сг О
75%
(PhNCO, Ph2CC0, PhNCS
\
Si(iPr),
(CO)3Cr
I) BuLi-ТМЗДА
2) EX '
3) hv
= TMS.COgEt, CO^Me,
\
(20.18)
20.3. Активация бензильного положения арентрикарбонильных
комплексов хрома
Еще одним проявлением «электроноакцепторных» свойств
группы Сг(СО)з, входящей в комплекс с ароматическим
соединением, служит ее способность стабилизировать отрицательный
заряд в положениях С-1 и С-2 боковой алкильной цепи. Это
было продемонстрировано на примере двойного алкилирования
закомплексованного метилфенилацетата по бензильному
углероду. Реакция легко протекает в условиях, при которых
свободный лиганд не реагирует [уравнение (20.19)]. Интересно, что
(20.19)
-СНаСООМе
Сг(СОЬ
СООМ0
диалкилирование преобладает даже при проведении реакции
с метилиодидом в мягких условиях, в то время как при замене
378 Глава 20
одной группы СО трифенилфосфитовым лигандом (обладающим
лучшими донорными свойствами) реакционная способность
системы понижается и наблюдается только специфическое моно-
алкилирование бензильного положения с умеренным выходом
[17]. Декомплексование ареновых лигандов осуществляют при
выдерживании на солнечном свету с доступом воздуха.
Еще более наглядным примером активации бензильного
положения в ареновых комплексах с Сг(СО)3 служит реакция
т]6-этилбензолтрикарбонилхрома с метилиодидом и грег-бутила-
том калия в ДМСО, приводящая после декомплексования к изо-
пропилбензолу (71%) и грег-бутилбензолу (8%). Аналогичным
образом бензальдегид [18] и этилоксалат [19] вступают в
конденсацию альдольного типа по бензилыному положению
закомплексованных аренов [уравнение (20.20)].
Сг(СО)3
EtOOC-OEt
i! H
О О
ДМСО
(20-20)
Способность Сг(СО)3 стабилизировать отрицательный заряд
в бензильном шоложении была использована Семмелхаком для
введения функциональных заместителей в ^-положение стиро-
лов. Алкилирование по ^-положению смещает отрицательный
заряд на сс-углерод (бензильный), где он стабилизируется хромо-
содержащим фрагментом. Улавливание этого аниона электро-
филом приводит к образованию продукта, содержащего две
функциональные группы в боковой цепи [уравнение (20.21)]
[20]. Объединив ^-алкилирование боковой цепи и литиирование
кольца, Уемура [21] получил нужные интермедиа™ в синтезе
дезоксиантрациклинонов [уравнение (20.22)].
(20.2!)
€*= H*, Melt PhCOCI
Сг(СО)3
, C(CN)(Me)(OR), w-Bu, Ph, f )—
S
Применение ареновых комплексов переходных металлов 379
(СО)3С
(СО)3Сг
О-^ OEt
(20.22)
20.4. Стерические эффекты в арентрикарбонильных
комплексах хрома
Еще одной особенностью г]6-арентрикарбонильных
комплексов хрома, на которой основано их применение в органическом
синтезе, являются стерические препятствия для подхода
реагентов к арену со стороны, занятой группой Сг(СО)з. Так,
комплекс на основе о-метоксиацетофенона подвергается атаке
реактивами Гриньяра преимущественно со стороны, противоположной
прутше Сг(СО)3 [уравнение (20.23)]. Как правило, стерео-
специфичность реакции достаточно высока, но она зависит от
природы аниона [22]. Более высокая стереоспецифичность на-
ОМе
ОМе
(20.23)
RMgX
Cr(CO),
Cr(CO)3
(d,l) ч (♦ другой диастереомер)
блюдается в циклических системах. Реакция хрома с
рацемическим 1-инданолом происходит исключительно со стороны гидр-
оксилыной группы; образующийся'при это'м арентрикарбонильный
комплекс хрома 'можно (разделить на энантио'меры.
Окисление любого из энантиомеров приводит к инданоновому комплек-
380 Глава 20
су, который оптически активен только благодаря тому, что
с хромом закомплексована определенная сторона арена. Алки-
лирование оптически активного комплекса метилиодидом/гидри-
дом натрия происходит исключительно со стороны,
противоположной хрому, и дает оптически активный комплекс с 2-метил-
инданоном. Последний высвобождает лиганд под действием
света и воздуха. Восстановление оптически активного
комплекса с 2-'метилинданоном (под действием борогидрида калия
также осуществляется исключительно со стороны,
противоположной хрому и приводит к оптически активному 2-метил-1-ин-
данолу. Аналогичная картина наблюдается с реактивами Гринь-
яра [23]. Подобным же о>бразом можно получить и подвергнуть
дальнейшим превращениям комплексы с тетралоном [24].
Реакции схематически представлены на рис. 20.2. Дес Аббайс [25]
[один, энантиомер)
Рис. 20.2.
(один энан-
гпиомео)
наблюдал родственные направляющие эффекты при а-алкилиро-
вании метиларилацетато'в.
Возможности использования направляющих стерических
эффектов и активирования бензильного положения при
комплексовании аренов с хромо'м показаны на рис. 20.3 [26]. Исходя из
той же концепции, Жауен [27а] провел стереоспецифическое
ацилирование ^-кольца стероидов [уравнение (20.24)]. Дэвис
Применение ареновых комплексов переходных металлов 381
воспользовался упомянутыми двумя эффектами для стереоспе-
цифического введения функциональных групп в бензильное
положение закомплексованных с хромом дигидроизохинолино!в
[276] и р-фенетиламинов [27в].
0 Me
90%
87%
Рис. 20.3.
(20.24)
Сг(СО)
Cr(C0)3
Е^МеХ, Me2CHX,«-C,2H25X» CH^O
Комплексообразование с хромом позволяет также
контролировать стереохимию внутримолекулярных реакций
апеллирования по Фриделю — Крафтсу [уравнение (20.25)] [28],
восстановление тетралонов бррогидридом [уравнение (20.26)] [29]
и реакцию Риттера с участием тетралолов [уравнение (20.27)]
[30].
Выполненный Уемура [31] стереоспецифический синтез
производных тетрагидронафталина [уравнение (20.28)] оанован на
сочетании ряда свойств арено1вых комплексов хрома. Соллади-
Кавалло разделил комплекс о-метилбензальдегида с хромом и
провел его алкилирование с высоким выходом « высокой
степенью энантиоапецифичности [уравнение (20.29)] [32].
Приведенные примеры иллюстрируют широкие возможности
управления стереохимией аренов за счет их комплексообразования
с хромом.
382 Глава 20
-СН-СНоСООН
лолифосфсрная
кислота
Сг(СО)3
/
<СО)3Сг
(20.25)
/ н главный
продукт
R = H, Me, Et, изо-Pv
минорный
/ /Т"""^ 'R продукт
О
NaBH4/изо-PrОН или
,3 (20.26)
(СО)3Сг
Me, Et,шо-Рг,Н R2 = H, Me, Et, азо-Pr
>■ Me, Et, шо-Рг, R4= H, Mg, Et, изо-рг, трё/я-BU
OK
Cr(C0)3
DH2so4
f
Cr(CO)3
:NCMe
(20.27)
Me
H CO
Cr(C0)3
единстбенньш
продукт
'(20.28)
I) BuLi
2)H
85%
(CO)3Cr
Ы IS
Et2O
87%
I
(C0)3Cr
W ISS ее
(20.29)
Применение ареновых комплексов переходных металлов 383
20.5. Реакции ареновых комплексов других переходных
металлов
Под действием стабилизированных карбанионов дикатионы
бис-(г|6-арен) железа подвергаются нуклеофильной атаке по
одному ароматическому кольцу, а в случае более реакционноспо-
собных карбаиионов атаке подвергаются оба кольца. Жесткие
условия, необходимые для получения исходного аренового
комплекса, не позволяли до настоящего времени применить эту
реакцию к более сложным системам. Нуклеофильной атаке по
ароматическому кольцу подвержены также более доступные моно-
катио;ны г|6-арен-г|5-ци1кло|пентадиенилжелеза. Под действием
разнообразных гетероатомных соединений можно провести
классическое нуклеофильное ароматическое замещение галоге-
нида в соответствующем комплексе с хлоробензолом [33].
Применение комплексов железа в синтезе сложиых органических
соединений представляется (весьма многообещающим.
(20.30)
ОМе
OLi
ОМе
Мп(С0)с
= Н,Вг
I )
Мп(СОЬ
Z)окислитель
ОМе
= OMe, H
(20.31)
R ♦ Mn(CO)3(CH3CN)3
Взаимодействие (т]3-арен)трикарбонильных комплексов
марганца с нуклеофилами протекает различными путями в
зависимости от природы нуклеофила (ч. 1 гл.7).Эти реакции
потенциально интересны, хотя пока мало применялись ib синтетических
целях. Пирсон [34] показал, что енолят-анианы гладко алкили-
руют арено'вый фрагмент таких комплексов [уравнение (20.30)].
Швайгарт [35] установил, что после алкилирования марганец-
384 Глава 20
содержащий фрагмент можно выделить путем расщепления
кислотой .и снова ввести в реакцию [уравнение (20.31) ]. В данной
области ожидаются новые интересные результаты.
ЛИТЕРАТУРА И ПРИМЕЧАНИЯ
1. Рисунок взят из обзора по химии комплексов аренов с трикарбонилами
металлов: Semmelhack M. F., Organomet. Chem. Libr., 1, 361 (1976).
2. См. обзоры: Semmelhack M. F., Pure Appl. Chem., 53, 2379 (1981);
Semmelhack M. F., Cleak G. R., Garcia J. L, Harrison J. J., Thebtaranonth У.,
Wulff W., Yamashita У., Tetrahedron, 37, 3957 (1981).
3. Semmelhack M. F., Garcia J. L., Cortes D., Hong R., Carpenter В. К-,
Farina R., Organometallics, 2, 467 (1983); Jackson W. R., Rae J. D., Wong M. G.,
Semmelhack M. F., Garcia J. N., J. Chem. Soc, Chem. Commun, 1982, 1359.
4. a) Semmelhack M. F., Seufert W., Keller L., J. Organomet. Chem., 226, 183
(1982); 6) Desobry V., Kundig E. P., Helv. Chim. Acta, 64, 1288 (1981);
в) Boutonnet J. C, Mordenti L., Rose E., Le-Martret O., Precigoux G., J.
Organomet. Chem., 221, 147 (1981) и цитированные в этой статье работы.
5. Semmelhack M. F.t Wulff W.t Garcia J. L., J. Organomet. Chem., 240, C5
(1982); Boutonnet J.-C.f Levisalles J., Rose E., Precigoux G., Courseille C,
Platzer N., J. Organomet. Chem., 255, 317 (1983).
6. Semmelhack M. F., Thebtaranonth V., Keller L., J. Am. Chem. Soc, 99, 959
(1977).
7. Semmelhack M. F., Harrison J. J., Thebtaranonth У., J. Org. Chem., 44, 3275
(1979).
8. Boutonnet J. C, Levisalles J., Normant J. M., Rose E., J. Organomet. Chem.,
255, C21 (1983).
9. Semmelhack M. F., Yamashita Л., J. Am. Chem. Soc, 102, 5926 (1980).
10. Kundig E. P., Desobry V., Simmons D. P., J. Am. Chem. Soc, 105, 6962
(1983).
11. Kundig E. P., Simmons D. P., J. Chem. Soc, Chem. Commun., 1983, 1320;
Kundig E. P., Pure Appl. Chem., 57, 1855 (1985).
12. Semmelhack M. F.t Bisaha J., Czarny M., J. Am. Chem. Soc, 101, 768
(1979).
13. Semmelhack M. F., Zask A.t J. Am. Chem. Soc, 105, 2034 (1983).
14. Ghavshou M., Widdowson D. A., J. Chem. Soc, Perkin Trans. 1, 1983,
3065.
15. Nechvatal G., Widdowson P. A., J. Chem. Soc, Chem. Commun., 1982,
467.
16. Masters N. F.f Widdowson D. A., J. Chem. Soc, Chem. Commun., 1983, 955;
Beswick P. J., Leach S. J., Masters N. F., Widdowson D. A., J. Chem. Soc,
Chem. Commun., 1984, 46; Fukui M.t Ikeda Т., Oishi Г., Tetrahedron Lett.,
23, 1605 (1982); Uemuera M., Nishikawa N., Take K., Oknishi M., Hirotsu K.,
Higuchi Т., Hayashi У., J. Org. Chem., 48, 2349 (1983); Uemura M.t Take #.,
Hayashi У., J. Chem. Soc, Chem. Commun., 1983, 858.
17. Jaouen G., Meyer A., Simmoneaux G., J. Chem. Soc, Chem. Commun., 1975,
813.
18. Brocard J., Lebibi J., Couturier D., J. Chem. Soc, Chem. Commun., 1981,
1264.
19. Halet J. F., Saillard J. Y., Car о R., LeBihan J. Y., Top S., Jaouen G., J.
Organomet. Chem., 267, C37 (1984); Car о В., LeBihan J. У., Guillot J. P.f Top S.,
Jaouen G., J. Chem. Soc, Chem. Commun., 1984, 602; Brocard J., Laconi A.,
Couturier D., Top S., Jaouen G., J. Chem. Soc, Chem. Commun., 1984, 475.
20. Semmelhack M. F.} Suefert W.f Keller L., J. Am. Chem. Soc, 102, 6586
(1980).
Применение ареновых комплексов переходных металлов 385
21. Uemura М„ Minami Т., Hayashi У., J. Chem. Soc, Chem. Commun., 1984,
1193.
22. Besancon J., Tiroflet J., Card A., Dusausoy Y., J. Organomet Chem 59
267 (1973).
23. Meyer A., Jaouen G., J. Chem. Soc, Chem. Commun., 1974 787
24. Jaouen G., Meyer A, J. Am. Chem. Soc, 97, 4667 (1975).
25. des Abbayes H., Boudeville M. A., Tetrahedron Lett., 1976 2137- J Org
Chem., 42, 4104 (1977). ' '
26. Jaouen G., Meyer A., Tetrahedron Lett., 1976, 3547.
27. a) Top S., Vessiers A., Abjean J. P., Jaouen G., J. Chem. Soc, Chem.
Commun., 1984, 428; Jaouen G., Top S., Laconi A., Couturier D., Brocard J
J. Am. Chem. Soc, 106, 2207 (1984); 6) Blagg J., Davies S. G., Mobbs В. Е.,
J. Chem. Soc, Chem. Commun., 1985, 619; в) Blagg J., Davies S. G., J. Chem.
Soc, Chem. Commun., 1985, 653.
28. Саго В., Jaouen G., J. Organomet. Chem., 228, 87 (1982).
29. Саго В., Jaouen G., J. Organomet. Chem., 220, 309 (1981).
30. Top S., Jaouen G., J. Org. Chem., 46, 78 (1981).
31. Uemura M., Isobe /(., Take K., Hayashi Y., J. Org. Chem., 48, 3855 (1983);
Uemura M., Isobe /(., Hayashi Y., Chem. Lett., 1985, 91.
32. Solladie-Cavallo A., Sufferth /., Tetrahedron Lett., 25, 1897 (1984).
33. Применение оловоорганических комплексов ароматических соединений
рассматривается в обзоре: Astruc D., Tetrahedron, 39, 4027 (1983).
34. Pearson A. J., Richards I. С, J. Organomet. Chem., 258, C41 (1983).
35. Chung Y. K., Willard P. G., Sweigart D. A., Organometallics, 1, 1053 (1982),
25-1129
СПИСОК СОКРАЩЕНИЙ
А
Ас
асас
AIBN
Аг
BD
bipy (Bipy)
BTMSA
Bz
CIDNP
ChgH
chiraphos
COD (cod)
COT (cot)
Cp
Cp*
€y (cy)
DCC
diars
dien
diglyme
DIOP (diop)
DIP AMP
DIPHOL
DIPHOS (diphos, dppe)
DMF
dmgH
DMSO
Angstrom unit, 10~8 cm — ангстрем (10~8 см)
acetyl, CH3CO — ацетил
acetylacetonate anion — ацетилацетонат-анион
azobis(isobutyronitrile) — азобис(изобутиронит-
рил)
aryl — арил
1,3-butadiene— 1,3-бутадиен
2,2'-bipyridyl — 2,2'-дипиридил
bis (trimethylsilyl) acetylene — бис(триметилси-
лил)ацетилен
benzyl — бензил
chemically induced dynamic nuclear polarization —
химически индуцированная динамическая
поляризация ядер (ХПЯ)
the monoanion formed by removing one proton
from dicyclohexylglyoxime — моноанион,
образующийся при удалении одного протона из дицикло-
гексилглиоксима
Ph2PC(H) (Me)C(Me)Ph2 —хиральный лиганд
(рис. 10.3)
1,5-cyclooctadiene — 1,5-циклооктадиен
1,3,5,7-cyclooctatetraene — 1,3,5,7-циклооктатетраен
cyclopentadienylide anion, CsHs" — циклопентадие-
нилид-анион
pentametyhlcyclopentadienylide anion — пентаме-
тилциклопентадиенилид-анион
cyclohexyl — циклогексил
dicyclohexylcarbodiimide — дициклогексилкарбоди-
имид
o-phenylenebis (dimethylarsine), о-СбН4 (AsMe2) 2 —
о-фениленбис (диметиларсин)
diethylenetriamine — диэтилентриамин
diethylene glycol dimethyl ether — диэтиленгликоля
диметиловый эфир
хиральный бидентантный фосфиновый лиганд
(рис. 10.3)
хиральный бидентантный фосфиновый лиганд
(рис. 10.3)
хиральный фосфин
l,2-bis(diphenylphosphino) ethane — 1,2-бис(дифе-
нилфосфино) этан
N,N-dimethylformamide — г\[,г\[-диметилформамид
(ДМФА)
the monoanion formed by removing one proton
from dimethylglyoxime — моноанион,
образующийся при удалении одного протона из диметилглиок-
сима
dimethylsulfoxide — диметилсульфоксид (ДМСО)
формальная d-электронная конфигурация
Список сокращений 387
dppe
EDTAH4
Е°
ее
en
ESCA
esr, epr
EXAFS
fac
Fp
FT
glyme
Hb
hfa
HMPA
HOMO
L
LUMO
M
Mb
Mes
MO
NHE
NMP
Np
Nuc
OAc
OEP
Pc
phen
pn
PPIX
РУ
R
Rf
S
l,2-bis(diphenylphosphino) ethane— 1,2-бис(дифе-
нилфосфино)этан
ethylenediaminetetraacetic acid (the tetraanion is
EDTA) — этилендиаминотетрауксусная кислота
(тетраанион обозначается EDTA)
полуволновой потенциал, в вольтах
per cent enantiomeric excess — энантиомерный
избыток в процентах
ethylenediamine — этилендиамин
electron spectroscopy for chemical analysis —
электронная спектроскопия для химического анализа
(ЭСХА)
electron spin (or paramagnetic) resonance —
электронный спиновый (парамагнитный) резонанс
(ЭПР)
extended x-ray absorption fine structure —
развернутая тонкая структура рентгеновского
поглощения
facial — фасиальный (поверхностный)
FeCp(CO)2
Fischer — Tropsch process — процесс Фишера —-
Тропша (ФТ)
ethylene glycol dimethyl ether, MeOCH2CH2OMe —
этиленгликоля диметиловый эфир
hemoglobin — гемоглобин
hexaf luoroacetylacetonate anion — гексафтораце-
тилацетонат-анион
hexamethylphosphoric triamide, (Me2N)3PO — гек-
саметилфосфортриамид (ГМФТА)
highest occupied molecular orbital — высшая
заселенная молекулярная орбиталь (ВЗМО)
любой монодентатный лиганд, часто трифенилфос-
фин
lowest unoccupied molecular orbital — низшая
свободная молекулярная орбиталь (НСМО)
центральный атом металла в молекуле
myoglobin — миоглобин
mesityl, 1,3,5-trimethylphenyl — мезитил, 1,3,5-три-
метилфенил
molecular orbital — молекулярная орбиталь (МО)
normal hydrgen electrode (reference) —
нормальный водородный электрод (эталон)
N-methylpyrrolidinone — N-метилпирролидинон
naphthyl — нафтил
nucleophile — нуклеофил
acetate anion — ацетат-анион
octaethylporphyrin dianion — октаэтилпорфирин-
дианион
phthalocyanine dianion — фталоцианин-дианион
1,10-phenanthroline — 1,10-фенантролин
1,2-propylenediamine — 1,2-пропилендиамин
protoporphyrin IX dianion — дианион протопорфи-
рина IX
pyridine — пиридин
алкильная группа
перфторалкильная группа
solvent — растворитель
25*
388 Список сокращений
salen
TCNE
THF
TMED (tmeda)
IMP
TMS
TPP
TPPTS
TTP
X
the diimine of salicylaldehyde and ethylenediamine —
диимин салицилового альдегида и этилендиамина
tetracyanoethylene — тетрацианоэтилен
tetrahydrofuran — тетрагидрофуран (ТГФ)
NX^NMetramethylethylenediamine — N,N,N'N'-
тетраметилэтилендиамин
meso-tetramesitylporphyrin dianion — жезо-тетраме-
зитилпорфирин-дианион
tetramethylsilane — тетраметилсилан (ТМС)
meso-tetraphehylporphyrin dianion — жезо-тетрафе-
нилпорфирин-дианион
trl (m-sulf onatophenyl) phosphine — три- (ж-сульфо-
натофенил) фосфин
meso-tetra (p-totyl) porphyrin dianion — жезо-тетра-
(я-толил) порфирин-дианион
frequency in cm"1 — частота, см"1
micron (10~6 m) or magnetic moment in Bohr
magnetons— микрон (10~6 м); магнитный момент
в магнетонах Бора
галоген
I, ПРЕДМЕТНЫЙ УКАЗАТЕЛЬ
Адипиновая кислота 54
Адиподинитрил 52
Азетидин-2,4-дионы 239
Азиридины 293
Азирины 239
Акоренон В 372
Акриловая кислота 117
Алкалоиды 311, 343
Алкилирование 148, 162, 170, 186,
234, 247, 249, 297, 309, 337
5-Алкилпиримидины 191
N-Алкилпирролидины 149
Алкилсиланы 50
Аллены 175, 279
Аллил-катион 215, 217, 219
Аллилсиланы 346
Аллилстаннаны 346
Аллилтозилаты 346
Алиллэпоксиды 348
Альбен 347
Алюминия комплексы 182
Аминирование 149, 195, 293, 344
(Аминоалкилферроценил) фосфины
190
Аминокарбонилирование 238
Аннулирование 257
Антибиотики тетрациклиновые 257
Арилирование 200
Аспидоспермина алкалоиды 311
«Ат»-комплексы 154, 234, 236, 267
Афидиколин 230
N-Ацетилфенилаланина сложные эфи-
ры 22, 24
1-Ацетокси-4-хлоро-2-алкены 355
Ацетонцианогидрин 52
(2)-а-Ациламидокоричной кислоты
производные 20, 22
Ацилирование 203, 212, 235, 239, 374
Ацилникелькарбонилаты 234
Бензохиноны 321
Берна восстановление 26, 311
Бицикло[2.2.1]гептаны 280
Бревикомин 336
Брефельфин А 305
1-Бромоадамантан 151
т]4-Бутадиентрикарбонилжелезо (0)
306
Бутенолиды 198
Вакер-процесс 13, 42, 289, 334
Ванадия комплексы 157
Винилаланы 180
Винилбораны 193
Ви'нилиден-дикатион 303
Винилиндолы 197
Винилкарбены 257
Винилкетен 256
Винилкупраты 174
Винилфосфониевые соли 246
Витамин А 338
Витамин Е 257, 350
Витамин К 257, 360
Виттига реакция 278, 345
Внедрение миграционное 9, 17, 61,
63, 102, 119, 129, 137, 222, 240
Водорода
активация 8, 56
перенос 47
Вольфрама комплексы 151, 260, 264,
270, 357
Восстанавливающие агенты 147, 156
Габакулин 313
Гексаметилбензол 327
Гераниол 180
Гидрирование
альдегидов 11, 40
ангидридов 41
аренов 33
асимметрическое 21, 40
1,4-дигидробензолов 26
диенов 30
кетонов 40
а-метилстирола 10
монооксида углерода 125, 141
нитрилов 41
390 Предметный указатель
нитроолефинов 27
нитросоединений ароматических
42
олефинов 5, 7, 21, 135
полистирола 39
сложных эфиров 41
стероидов 28
циклогексанонов 47
циклогексена 17
эргостеролацетата 26
Гидроалюминирование 180
Гидрометаллирование 191
Гидросилилирование 42, 48
Гидроформилирование 40, 42, 78, 100,
ПО, 179
Гидрохинон 118
Гидроцианирование 52
Гидроцирконирование 177, 262
Гиосциамин 218
Гликолевый альдегид 125, 129
Глутаровый альдегид 149
Гомологизация 247
Грандизол 242
Грина механизм 64
Гриньяра реакция 161, 187
Гумулен 214
Дауномицинон 373
Дезоксиантрациклины 378
Дезоксиподокарпат 242
Дезоксипростагландин Ег 170
Дезоксифренолицин 257
Декарбонилирование 122, 240
Диазетидиноны 275
Диазосоединения 268
Диаллилы 212, 222
Дигидроизохинолины 381
Дигидрофураны 303, 316
Дикобальтоктакарбонил 317
Дильса — Альдера реакция 258, 326
Диментил(изопропил)фосфин 80
Диметиладипинат 115
(2,2-Диметил-4-пентен-1-ил) платина
64
Динатрийтетракарбонилферрат 227
Дисахариды 242
Дифенилметандиизоцианат 124
Ибогамин 343
Изотопный
обмен 36, 50, 53, 103, 244
эффект 17, 65
Изохинолины 329
Инден 261
Индолозиноны 324
Индолы 294, 376
Ионообменные смолы 44
Иридия комплексы 111, 120, 140, 32S
Камптофецин 324
Карбиды 138
Карбоалкоксилирование 113, 231
Карбоалюминирование 180
Карбоксилирование 237
Карбокуприрование 176
Карбометаллирование 191
Карбонилирование 100, 117, 122, 132,
196, 221, 239, 336
( + )-Карвон 149
Катализ
гомогенный 5
межфазный 233
«обратный» межфазный 112
Катализаторы
гомогенные 6, 9, 12, 26, 30, 34Г
45, 112
иммобилизованные 42
Катарантин 343
Квадрон 299
Кинетический анализ 25, 109, 225,
327
Кобальта комплексы 36, 67, 101, 112,
120, 140, 231, 236, 249, 317,
324, 356
Конверсия водяного газа 99, 114, 135
Кори реакция 359
Косее механизм 61
Коста механизм 126, 129
Коупа перегруппировка 301
Кремния
гидриды 49
диоксид 44
Кросс-сочетание 165, 187, 189, 340,
359
Крэбтри катализатор 28, 46
Кубан 309
Кумарины 185,
Железа
комплексы 147, 151, 215, 219,
227, 247, 266, 302, 316, 383
пентакарбонил 223, 227, 306
Р-Лактамы 198, 224, 239, 248, 272
Лактоны 224, 292
макроциклические 214, 263, 335
Предметный указатель 391
Лантанорганические соединения 9
Лимнаспермина алкалоиды 311
Литийкупраты 170, 238
Лития комплексы 152, 154
Логанин 347
Мак-Куиллина катализатор 42, 46
Марганца комплексы 129, 383
Меди комплексы 152, 154, 161, 170,
173
Меервейна реагент 234
Меркурирование 183, 185
Металлациклы 80, 94, 253, 264, 275,
280, 321
Металлоациленоляты 247
Металлокарбены 91
Металлокарбины 91
Метанол 127, 141
Метатезис
алкинов 91
олефинов 73, 178, 261, 280
Метациклофан 371
2-Метилбензофуран 290
а-Метиленлактамы 198
а-Метиленлактоны 198, 224, 303
2-Метил-1-инданол 390
Метилстильбены 244
N-Метилформамиды 132
4-Метокси-1 -нафтол 254
Митозены 294
Михаэля реакция 183, 235, 248, 347
Молибдена комплексы 310, 357
Найлон-6,6 55
Наномицин 257, 321
Натрия комплекс 152
Незукон 218
(—)-Неодигидрокарвеол 149
Никеля
карбонилы 212, 221, 238
комплексы 55, 76, 82, 87, 187,
236, 280, 358
Ниобия комплексы 277
Нуклеозиды 184, 218
Оксалаты 133
Оксаллил-катион 219
Оксо-катализаторы 11, 101, 104, 110
Оксо-реакция 11, 100, 111
Олигомеризация 60, 76, 81, 83, 91
Олова
гидриды 155
хлориды 116, 226, 248
Оловоорганические реагенты 195
Ортометаллирование 12
Осмия комплексы 128
Остановленной струи метод 15
Палладия комплексы 75, 84, 116, 133,
155, 165, 183, 187, 199, 226,
288, 334
Пентахлоротиофеноксид 120
Переметаллирование 155, 180, 181,
186, 190, 339
Переходных металлов
анионы 206
гидриды 147, 152
карбонилы 212, 221
комплексы аллильные 11, 81, 84,
158, 213, 219, 232, 306, 334,
353
— алкеновые 288
— алкильные 61, 148, 160, 176,
206, 229, 262, 299
— алкиновые 316
—• ареновые 368
— ацильные 103, 109, 114, 120,
179, 221, 228, 234, 247
— диенильные 311
— диеновые 306, 311
— карбеновые 253, 261, 272, 277,
282
Пивалофенон 171
Пираны 316
2-Пиридоны 323
Пирроло-1,4-бензодиазепины 198
Пирролоиндолохиноны 294
Пирролы 317
Пластификаторы 101
Платины комплексы 111
Полиизопрены 337
Полимеризация
алкинов 75, 90
ацетиленов 75
диенов 81
лактонов 75
норборнена 74
олефинов 60
циклоалкенов 74
Полипропилен 69
Политиолы 46
Полиэтилен 67
Предкатализатор 6, 30, 49, 119, 261
Пренилирование 337
392 Предметный указатель
Присоединение окислительное 8, 50,
53, 120, 155, 165, 181, 186, 190,
196, 223, 240, 281, 351, 362
Пропаргил-катионы 318
Простагландины 172, 183, 300, 319
Простаноиды 169
Простациклин 299
Региоселективность 47, 50, 55, 115,
218, 255, 310, 324, 337, 346
Реппе реакция 100
Риттера реакция 381
Родия
карбонилы 239
комплексы 18, 22, 24, 34, 40, 46»
87, 101, 104, 119, 120, 130, 241,
268, 358
Рутения комплексы 34, 36, 40, 47,
111, 124, 128
Р-Фенетиламины 381
2-Фенилтетраметилсукцинамид 238
Феромоны 356
Ферроценилфосфин 190
Фишера — Тропша процесс 98, 134
Флеш-фотолиз 16
Флори — Шульца распределение 77„
135
Френолицин 376
Фриделя— Крафтса реакция 307, 381
Фураны 317
Хемосорбция диссоциативная 139, 141
Химически индуцированная
динамическая поляризация ядер 10
Хинолины 184
Хиноны 321, 360
Хрома комплексы 32, 95, 151, 254,.
368
Сантален 359
а-Санталол 237
Саркомицин 305
Скопин 218
Сочетания реакция 212
Спиро-а-метилен-'у-бутиролактоны
365
Стероиды 311, 335, 348, 364
Таллирование 185
Тантала комплексы 66, 277
Теббе реагент 279
Теломеризация 334
Тетрагидронафталины 328, 381
Тетракис (трифенилфосфит) никель 54
Титана комплексы 68
Толуилендиизоцианат 124
Трикотеканы 311
3,5,5-Триметилциклогекс-2-ен-1-он 152
а-Трифторометилбензилхлорид 244
Тропин 218
Туйяплицин 218
Цембрен 214
1-Цианотетрагидронафталин 370
Циглера катализаторы 67, 73, 76, 79,
81
Циглера — Натта катализатор 70, 91
Циклоалкены 218
(т]4-Циклобутадиен)трикарбонилже-
лезо 308
Циклогексадиены 330
Циклододекатриен 81
Циклометаллирование 202
Циклооктатетраен 94
Циклоолигомеризация 87, 94, 320
Циклопентадиенилкарбонилжелеза
катион 306
Циклоприсоединение 218, 258, 261,
282
Циклопропанирование 261, 268, 282
Циклопропаны 266
Циклотримеризация 324
Циркония комплексы 177, 278
Углерода монооксид 98, 212, 221, 259,
298, 320, 356, 374
Уилкинсона катализатор 12, 18, 26,
123, 240
Уксусная кислота 118, 141
Эквиленин 170
Эластомеры 73, 124
Элиминирование восстановительное
8, 10, 17, 50, 53, 119, 134, 155,
187, 222, 234, 242, 261, 275,
281, 339, 362
Предметный указатель 393
Энантиоселективность 25, 40, 52, ПО, Этилгеранат 180
190, 273, 293 Этиленгликоль 126, 129, 140
Эстрон 328
2-Этилгексанол 102 Янтарная кислота 117
ОГЛАВЛЕНИЕ
РАЗДЕЛ II. КАТАЛИТИЧЕСКИЕ ПРОЦЕССЫ
10. ГОМОГЕННЫЕ КАТАЛИТИЧЕСКИЕ ПРОЦЕССЫ:
ГИДРИРОВАНИЕ, ГИДРОСИЛИЛИРОВАНИЕ, ГИДРОЦИАНИРОВАНИЕ . . 5
10.1. Введение 5
10.2. Общее представление о гомогенном каталитическом
гидрировании олефинов 5
10.3. Механизмы гидрирования олефинов в условиях гомогенного
катализа 7
10.4. Примеры применения дигидридных катализаторов гидрирования
олефинов 26
10.5. Гомогенное каталитическое гидрирование аренов 33
10.6. Гомогенное гидрирование других функциональных групп ... 39
10.7. Иммобилизация гомогенных катализаторов гидрирования . . 42
10.8. Тест на гомогенность катализаторов гидрирования .... 45
10.9. Гомогенный катализ в реакциях с переносом водорода ... 47
10.10. Гомогенное каталитическое силилирование 48
10.11. Гомогенное каталитическое гидроцианирование олефинов и
ацетиленов 52
Литература и примечания 56
11. КАТАЛИТИЧЕСКАЯ ПОЛИМЕРИЗАЦИЯ ОЛЕФИНОВ И
АЦЕТИЛЕНОВ 60
11.1. Введение 60
11.2. Механизм(ы) полимеризации/олигомеризации моноенов ... 60
11.3. Механизм стадии роста цепи 63
11.4. Полиэтилен 67
11.5. Полипропилен 69
11.6. Этилен-пропиленовые сополимеры 72
11.7. Полимеризация, включающая метатезис олефинов с раскрытием
цикла 73
11.8. Катализируемая металлом электрофильная полимеризация . . 75
11.9. Олигомеризация алкенов 76
11.10. Олигомеризация и полимеризация сопряженных диенов ... 81
11.11. Олигомеризация и полимеризация алкинов 90
Литература и примечания 96
394
Оглавление 395
12. КАТАЛИТИЧЕСКИЕ РЕАКЦИИ С УЧАСТИЕМ МОНООКСИДА
УГЛЕРОДА 98
12.1. Введение 98
12.2. Конверсия водяного газа и связанные с ней реакции .... 99
12.3. Гидрокарбонилирование олефинов (оксо-реакция) .... 109
12.4. Каталитическое карбоалкоксилирование олефинов 113
12.5. Карбонилирование ацетиленов 117
12.6. Карбонилирование спиртов: процесс получения уксусной кислоты
фирмы Monsanto # 118
12.7. Родственные процессы карбонилирования, прсмотируемые иоди-
дом 121
12.8. Декарбонилирование 122
12.9. Восстановительное карбонилирование ароматических нитросоеди-
нений 124
12.10. Гомогенно катализируемое гидрирование СО 125
12.11. Восстановительное карбонилирование аммиака 132
12.12. Окислительное сочетание монооксида углерода 133
12.13. Гетерогенно катализируемые реакции водорода с монооксидом
углерода 134
Литература и примечания 142
РАЗДЕЛ III. ПРИМЕНЕНИЯ В ОРГАНИЧЕСКОМ СИНТЕЗЕ . . 147
13. ПРИМЕНЕНИЕ ГИДРИДОВ ПЕРЕХОДНЫХ МЕТАЛЛОВ В
ОРГАНИЧЕСКОМ СИНТЕЗЕ 147
Литература и примечания 158
14. ПРИМЕНЕНИЕ КОМПЛЕКСОВ, СОДЕРЖАЩИХ с-СВЯЗИ
МЕТАЛЛ—УГЛЕРОД, В ОРГАНИЧЕСКОМ СИНТЕЗЕ 160
14.1. Алкилметаллы, получаемые из карбанионов и галогенидов
металлов; медьорганические комплексы 161
14.2. Получение алкилметаллов присоединением гидридов металлов
или алкилметаллов к алкенам и алкинам 176
14.3. Получение алкилметаллов переметаллированием и внедрением . 181
14.4. Получение алкилметаллов окислительным присоединением и
переметаллированием 186
14.5. Получение алкилметаллов окислительным присоединением и
внедрением 196
14.6. Получение алкилметаллов циклометаллированием 202
14.7. Получение алкилметаллов из анионов металлов и органических
галогенидов 206
14.8. Получение алкилметаллов нуклеофильной атакой олефиновых ме-
таллокомплексов 207
Литература и примечания 207
15. ПРИМЕНЕНИЕ КАРБОНИЛЬНЫХ СОЕДИНЕНИЙ
ПЕРЕХОДНЫХ МЕТАЛЛОВ В ОРГАНИЧЕСКОМ СИНТЕЗЕ 212
15.1. Реакции сочетания 212
15.2. Реакции карбонилироаания ... 221
396 Оглавление
15.3. Декарбонилирование альдегидов и хлороангидридов кислот . . 240
15.4. Реакции металлоациленолятов с электрофилами 247
Литература и примечания 250
16. ПРИМЕНЕНИЕ КАРБЕНОВЫХ КОМПЛЕКСОВ ПЕРЕХОДНЫХ
МЕТАЛЛОВ И МЕТАЛЛАЦИКЛОВ В ОРГАНИЧЕСКОМ СИНТЕЗЕ
16.1. Реакции электрофильных карбеновых комплексов 253
16.2. Реакции нуклеофильных карбеновых комплексов 277
16.3. Применение других металлациклов в органическом синтезе . . 280
Литература и примечания . . . 284
17. ПРИМЕНЕНИЕ АЛКЕНОВЫХ, ДИЕНОВЫХ И ДИЕНИЛЬНЫХ
КОМПЛЕКСОВ ПЕРЕХОДНЫХ МЕТАЛЛОВ В ОРГАНИЧЕСКОМ
СИНТЕЗЕ 288
17.1. Нуклеофильная атака на комплексы (т]2-олефин)палладий(П) 288
17.2. Нуклеофильная атака на комплексы (т]2-олефин)железо(Н) . . 302
17.3. Нуклеофильная атака на комплексы (т]4-1,3-диен) железо и (т]5-дие-
нил) железо 306
Литература и примечания 313
18. ПРИМЕНЕНИЕ АЛКИНОВЫХ КОМПЛЕКСОВ ПЕРЕХОДНЫХ
МЕТАЛЛОВ В ОРГАНИЧЕСКОМ СИНТЕЗЕ 316
18.1. Нуклеофильная атака на комплексы переходных металлов с алки-
нами 316
18.2. Синтетическое применение алкиновых комплексов Со2(СО)8 . . 317
18.3. Катализируемая металлами циклоолигомеризация алкинов . . 320
Литература и примечания 332
19. ПРИМЕНЕНИЕ г]3-АЛЛИЛЬНЫХ КОМПЛЕКСОВ ПЕРЕХОДНЫХ
МЕТАЛЛОВ В ОРГАНИЧЕСКОМ СИНТЕЗЕ 334
19.1. Реакции теломеризации сопряженных диенов, катализируемые
палладием 334
19.2. Использование в синтезе предварительно полученных т]3-аллилпал-
ладиевых комплексов 336
19.3. Реакции аллильных субстратов, катализируемые палладием . . 341
19.4. Реакции 1,2- и 1,3-диенов с участием т]3-аллилпалладиевых(Н)
интермедиатов 353
19.5. Использование в синтезе г]3-аллильных комплексов других
металлов 356
Литература и примечания 365
20. ПРИМЕНЕНИЕ АРЕНОВЫХ КОМПЛЕКСОВ ПЕРЕХОДНЫХ
МЕТАЛЛОВ В ОРГАНИЧЕСКОМ СИНТЕЗЕ 368
20.1. Присоединение углеродсодержащих нуклеофилов к ареновым
комплексам хрома 369
20.2. Литиирование арентрикарбонильных комплексов хрома . . . 374
20.3. Активация бензильного положения арентрикарбонильных
комплексов хрома 377
397
20.4. Стерические эффекты в арентрикарбонильных комплексах хрома 379
20.5. Реакции ареновых комплексов других переходных металлов . . 383
Литература и примечания 384
Список сокращений 386
Предметный указатель 389
УВАЖАЕМЫЙ ЧИТАТЕЛЬ!
Ваши замечания о содержании книги, ее
оформлении, качестве перевода и другие просим присылать
по адресу: 129820, Москва, И-ПО, ГСП, 1-й
Рижский пер., д. 2, издательство «Мир».
Научное издание
Джеймс Коллмен, Луис Хигедас, Джек Нортон,
Ричард Финке
МЕТАЛЛООРГАНИЧЕСКАЯ ХИМИЯ
ПЕРЕХОДНЫХ МЕТАЛЛОВ.
ОСНОВЫ И ПРИМЕНЕНИЯ
В 2-х частях
2
Заведующий редакцией академик О. А. Реутов
Зам. зав. редакцией Н. А. Козырева
Старший научный редактор Г. Б. Шкляева
Младший редактор И. С. Ермилова
Художник В. А. Медников
Художественный редактор М. Н. Кузьмина
Технический редактор А. Л. Гулина
Корректор Н. А. Гиря
ИБ № 6895
Сдано в набор 30.05.89. Подписано к печати 19.09.89.
Формат 60X90Vi6. Бумага книжно-журнальная. Печать высокая.
Гарнитура литературная. Объем 12,50 бум. л. Усл. печ. л.
25,00. Усл. кр.-отт. 25,00. Уч.-изд. л. 25,65. Изд. № 3/6426.
Тираж 1650 экз. Зак. 1129. Цена 5 р. 20 коп.
Издательство «Мир» В/О «Совэкспорткнига»
Государственного комитета СССР по печати. 129820, ГСП, Москва, И-110,
1-й Рижский пер., 2.
Московская типография № 11 Государственного комитета
СССР по печати. 113105, Москва, Нагатинская ул., д. 1.