/




























































































































































































































































































































































































































































































Автор: Чоркендорф И. Наймантсведрайт X.
Теги: химическая кинетика химическая технология катализ
ISBN: 978-5-91559-044-0
Год: 2010




Текст
Издательский Дом
ИНТЕЛЛЕКТ
И. ЧОРКЕНДОРФ. X. НАЙМАНТСВЕДРАЙТ
СОВРЕМЕННЫЙ КАТАЛИЗ
И ХИМИЧЕСКАЯ КИНЕТИКА
И. ЧОРКЕНДОРФ. X. НАЙМАНТСВЕДРАЙТ
СОВРЕМЕННЫЙ КАТАЛИЗ
И ХИМИЧЕСКАЯ КИНЕТИКА
Перевод с английского
В. И. Ролдугина
\
Издательский Дом
ИНТЕЛЛЕКТ
ДОЛГОПРУДНЫЙ
2010
Scan, OCR, make
Fedor Lebed
SciPower.org | Free Knowledge is Power
р#и
Издание осуществлено при финансовой поддержке Российского
Фонда Фундаментальных Исследований по проекту № 09-08-07073д
Чоркендорф И., Наймантсведрайт X.
Современный катализ и химическая кинетика: Научное издание / Чоркендорф И.,
Наймантсведрайт X. - Долгопрудный: Издательский Дом «Интеллект», 2010. - 504 с.
ISBN 978-5-91559-044-0
Второе издание известной монографии охватывает широкий круг вопросов - от
основ кинетики до переработки нефти и защиты окружающей среды.
Изложены современные методы изучения поверхностей и каталитических наночастиц. Сформулированы требования, предъявляемые к современным катализаторам. Указаны методы регулирования параметров катализаторов. Описаны
методы экспериментального изучения поверхностных реакций и теория течения
реагирующих газов в пористых каталитических слоях. Подробно рассмотрены основные технологические процессы, идущие с участием катализаторов, принципы
действия и роль катализаторов в химических топливных элементах, базирующихся
на органических и неорганических мембранах. Особое внимание уделено
каталитической нейтрализации выхлопных газов двигателей и отходящих газов
электростанций.
Для студентов и преподавателей химических и химико-технологических факультетов, исследователей и разработчиков.
I Chorkendorffi J W Niemantsverdriet
Concepts of Modern Catalysis
and Kinetics
Second, Revised and Enlarged Edition
| чИ WILEY :
WILEY-VCH Verlag GmbH &. Co KGaA
ISBN 978-5-91559-044-0
ISBN 978-3-527-31672-4 (англ.)
) 2007 WILEY-VCH Verlag GmbH &
Co. KGaA, Weinheim
D 2010, ООО «Издательский Дом
«Интеллект», оригинал-макет,
оформление
ОГЛАВЛЕНИЕ
Предисловие авторов 11
Катализ: Концептуально понятный, но далекий от зрелости 11
Глава 1
ВВЕДЕНИЕ В КАТАЛИЗ 14
1.1. Что такое катализ? 15
1.2. Катализаторами могут быть атомы, молекулы, энзимы
и поверхности твердых тел 17
1.2.1. Гомогенный катализ 17
1.2.2. Биокатализ 18
1.2.3. Гетерогенный катализ 19
1.3. Зачем необходим катализ? 21
1.3.1. Катализ и экологически чистая химия 22
1.3.2. Атомарная эффективность, is-факторы и «дружелюбие»
к окружающей среде 22
1.3.3. Химическая индустрия 24
1.4. Катализ как междисциплинарная наука 30
1.4.1. Множественность пространственных масштабов в катализе 30
1.4.2. Временные масштабы в катализе 32
1.5. Предмет книги 33
1.6. Катализ в периодических изданиях 33
1.7. Основные учебники по катализу 37
Список литературы 37
Глава 2
КИНЕТИКА ХИМИЧЕСКИХ РЕАКЦИЙ 38
2.1. Введение 38
2.2. Уравнение скорости реакций и порядки реакций 41
2.3. Реакции и термодинамическое равновесие 45
2.3.1. Пример химического равновесия: синтез аммиака 48
2.3.2. Химическое равновесие для неидеальных газов 51
2.4. Зависимость скорости реакций от температуры 52
Оглавление
2.5. Решения уравнений скорости реакций: зависимости от времени
концентраций в реакциях различного порядка 55
2.6. Взаимосвязанные реакции в проточных реакторах:
приближение стационарного состояния 58
2.7. Взаимосвязанные реакции в емкостных реакторах 63
2.8. Каталитические реакции 66
2.8.1. Приближение среднего поля 70
2.9. Изотермы адсорбции Ленгмюра 71
2.9.1. Ассоциативная адсорбция 72
2.9.2. Диссоциативная адсорбция 73
2.9.3. Конкурентная адсорбция 74
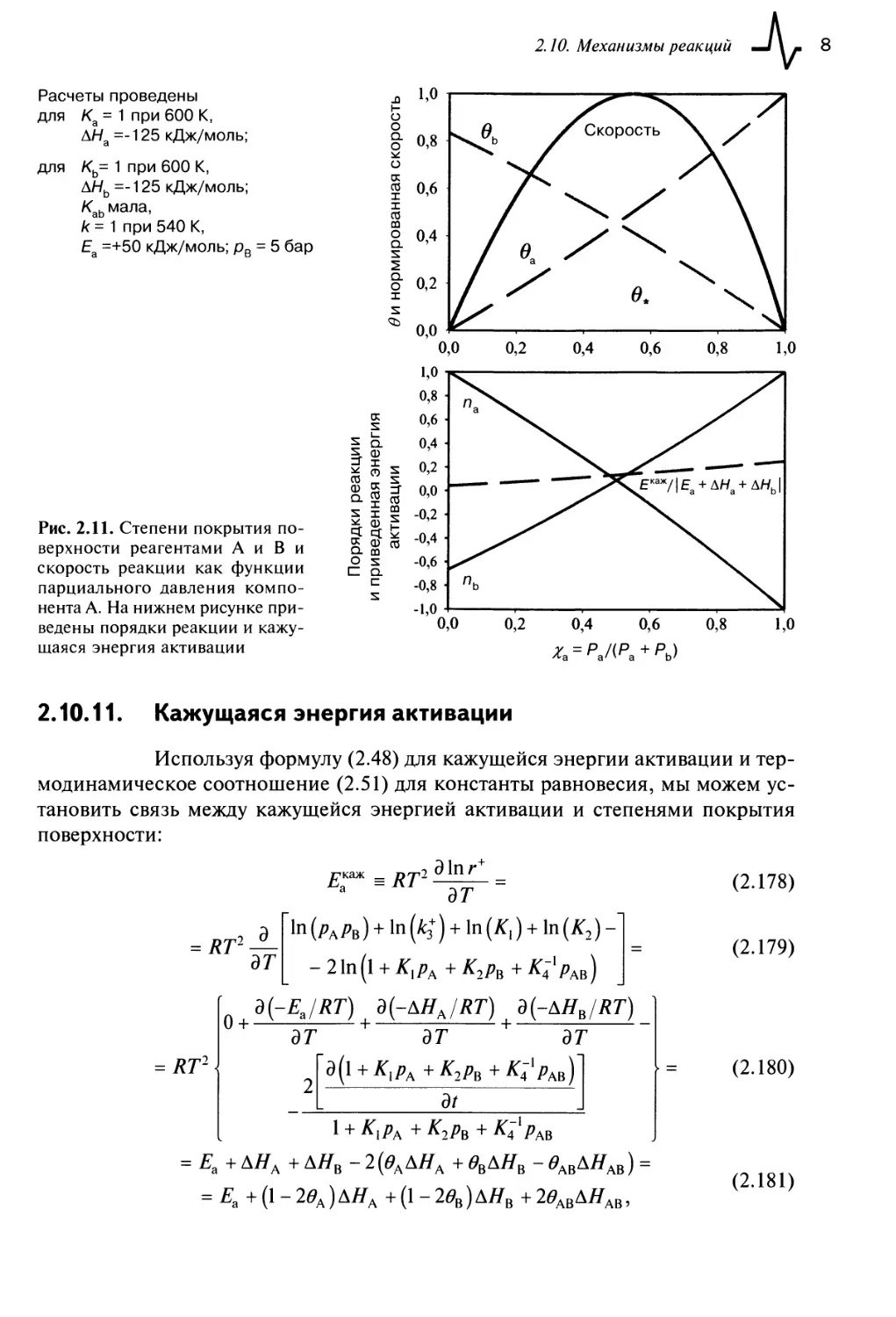
2.10. Механизмы реакций 74
2.10.1. Механизмы Ленгмюра—Хиншельвуда или Илея—Ридела 75
2.10.2. Кинетика Ленгмюра—Хиншельвуда 75
2.10.3. Полное решение 76
2.10.4. Стационарное приближение 77
2.10.5. Приближение квазиравновесия 78
2.10.6. Ступени реакций с близкими по величине скоростями 79
2.10.7. Приближение необратимых стадий 80
2.10.8. Приближение наиболее избыточного интермедиата реакции
(НИИР) 80
2.10.9. Почти свободная поверхность 81
2.10.10. Порядок реакции 82
2.10.11. Кажущаяся энергия активации 83
2.11. Энтропия, производство энтропии, автокатализ, колебательные
реакции 87
2.12. Кинетика реакций, катализируемых энзимами 92
Список литературы 97
Г л и в и 3
ТЕОРИЯ СКОРОСТЕЙ РЕАКЦИЙ 98
3.1. Введение 98
3.2. Распределение Больцмана и статистическая сумма 100
3.3. Статистические суммы атомов и молекул 103
3.3.1. Распределение Больцмана 103
3.3.1.1. Доказательство равенства Л2 и 1/Т 105
3.3.2. Распределение Максвелла—Больцмана по скоростям молекул 106
3.3.3. Полная статистическая сумма системы 107
3.3.3.1. Статистическая сумма для поступательного движения 108
3.3.3.2. Статистическая сумма колебательных движений 109
3.3.3.3. Вращательная (и ядерная ) статистическая сумма 111
3.3.3.4. Электронная и ядерная статистические суммы 112
3.4. Молекулы в равновесии 114
Оглавление jv
3.5. Теория столкновений 121
3.5.1. Частота столкновений с поверхностью 123
3.5.2. Вероятность реакции 124
3.5.3. Фундаментальные возражения против теории столкновений 126
3.6. Активирование молекул при столкновениях: теория Линдеманна 127
3.7. Теория переходных состояний 128
3.7.1. Термодинамическая форма выражения для скорости реакции
в теории переходных состояний 130
3.8. Теория переходных состояний для поверхностных реакций 134
3.8.1. Адсорбция атомов 134
3.8.1.1. Непрямая адсорбция 135
3.8.1.2. Прямая адсорбция 137
3.8.2. Адсорбция молекул 140
3.8.2.1. Непрямая адсорбция с формированием прекурсора 140
3.8.2.2. Прямая адсорбция 141
3.8.3. Реакции между адсорбированными молекулами 143
3.8.4. Десорбция молекул 145
3.9. Заключение 148
Список литературы 150
Глава 4
ОПРЕДЕЛЕНИЕ ПАРАМЕТРОВ КАТАЛИЗАТОРОВ 151
4.1. Введение 151
4.2. Рентгеноструктурный анализ (РСтА) 153
4.3. Рентгеноэлектронная спектроскопия (РЭС) 156
4.4. Спектроскопия дальней тонкой структуры рентгеновского
поглощения (СДТСРП, EXAFS) 162
4.5. Электронная микроскопия (ЭМ, ПЭМ, СЭМ) 166
4.6. Мессбауэровская спектроскопия (МС) 170
4.7. Ионная спектроскопия: масс-спектроскопия вторичных ионов,
рассеяние низкоэнергетических ионов, обратное резерфордовское
рассеяние (МСВИ, РНИ, ОРР) 174
4.8. Температурно-программируемые восстановление, окисление
и сульфидирование 177
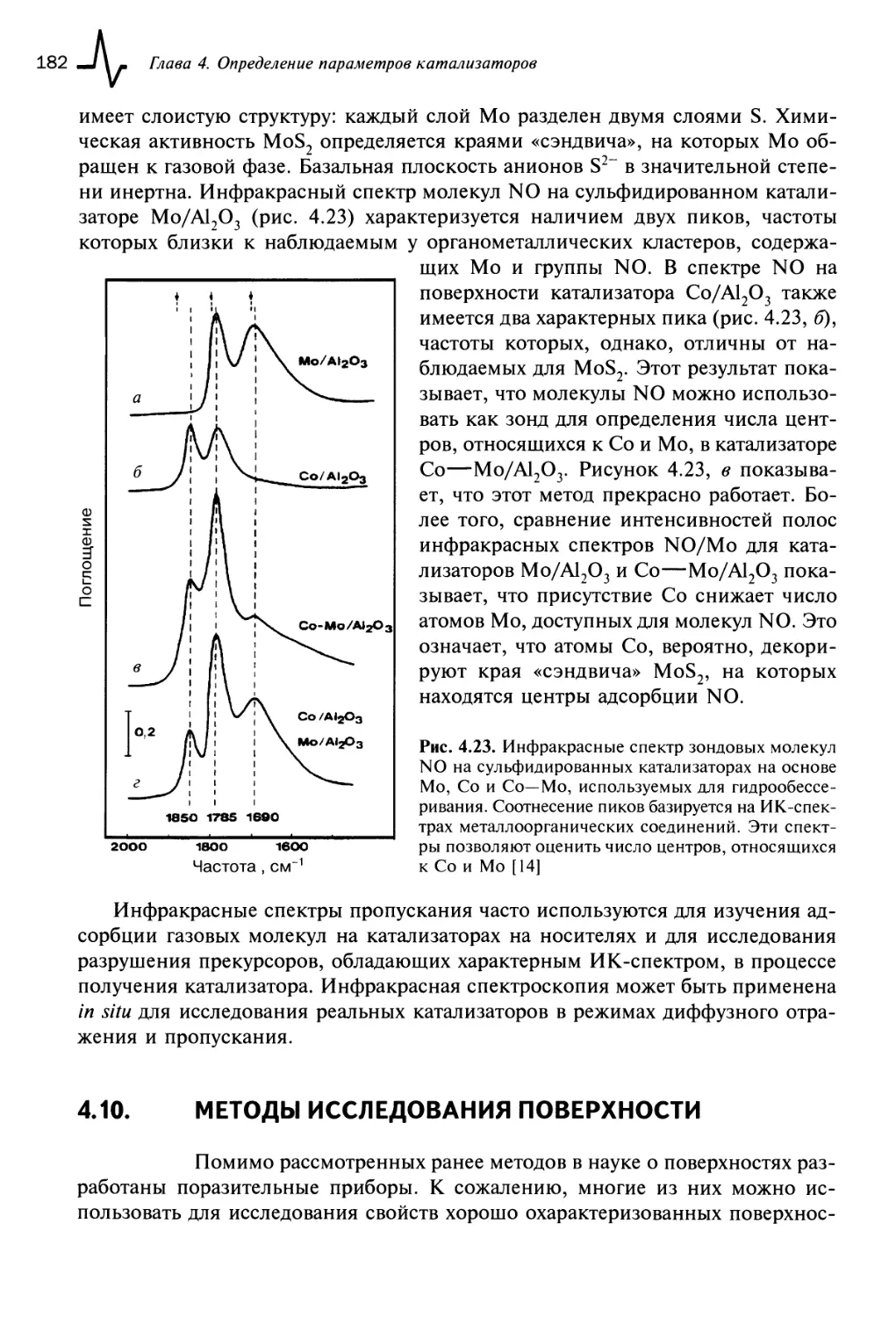
4.9. Инфракрасная спектроскопия (ИКС) 179
4.10. Методы исследования поверхности 182
4.10.1. Дифракция медленных электронов (ДМЭ) 183
4.10.2. Сканирующая зондовая микроскопия (СЗМ) 186
4.10.2.1. Сканирующая туннельная микроскопия (СТМ) 186
4.10.2.2. Атомно-силовая микроскопия (ACM) 189
4.11. Заключительные замечания 191
Список литературы 192
6 -п, Ог^швление
Глава 5
ТВЕРДЫЕ КАТАЛИЗАТОРЫ 194
5.1. Требования, предъявляемые к эффективным катализаторам 194
5.2. Структура металлов, оксидов, сульфидов и их поверхностей 197
5.2.1. Структура металлов 197
5.2.2. Кристаллография поверхности металлов 198
5.2.2.1. Поверхности кристаллов 198
5.2.2.2. Адсорбционные центры 201
5.2.2.3. Двумерная решетка 202
5.2.3. Оксиды и сульфиды 204
5.2.4. Свободная энергия поверхности 207
5.3. Характеристики малых частиц и пористых материалов 210
5.3.1. Правило Вульфа 210
5.3.2. Система пор 212
5.3.3. Площадь поверхности 213
5.4. Носители катализаторов 219
5.4.1. Кремнезем 221
5.4.2. Оксид алюминия 223
5.4.3. Углеродные носители 225
5.4.4. Формование носителей катализаторов 225
5.5. Получение нанесенных катализаторов 227
5.5.1. Соосаждение 227
5.5.2. Импрегнация, адсорбция, ионный обмен 227
5.5.3. Осаждение с отложением 230
5.6. Катализаторы без носителей 230
5.7. Цеолиты 231
5.7.1. Структура цеолита 232
5.7.2. Компенсирующие катионы и кислотность 234
5.7.3. Применение цеолитов 235
5.8. Тестирование катализаторов 236
5.8.1. Десять заповедей по тестированию катализаторов 237
5.8.2. Измерение активности 239
5.8.2.1. Транспортные ограничения и диффузионный модуль Тиле 240
5.8.2.2. Диффузия в порах 245
5.8.2.3. Следствия транспортных ограничений в тестировании
катализаторов 247
Список литературы 249
Глава 6
РЕАКЦИОННАЯ СПОСОБНОСТЬ ПОВЕРХНОСТИ 250
6.1. Введение 250
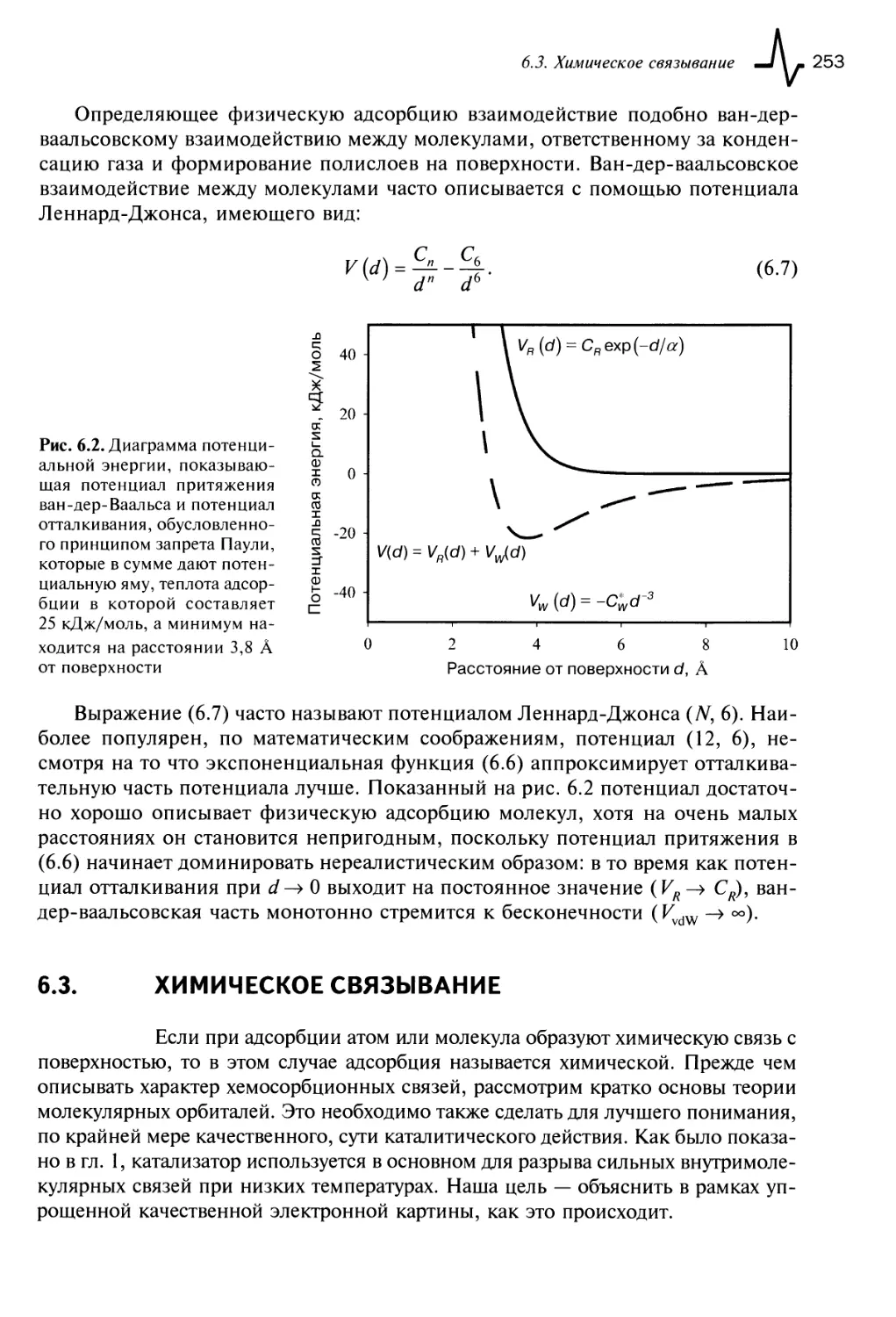
6.2. Физическая адсорбция 250
6.2.1. Взаимодействие ван-дер-Ваальса 251
6.2.2. Учет отталкивания 252
Оглавление Jv
6.3. Химическое связывание 253
6.3.1. Связи в молекулах 254
6.3.1.1. Двухатомные молекулы 254
6.3.1.2. Гомоядерные двухатомные молекулы 255
6.3.1.3. Гетероядерные системы 257
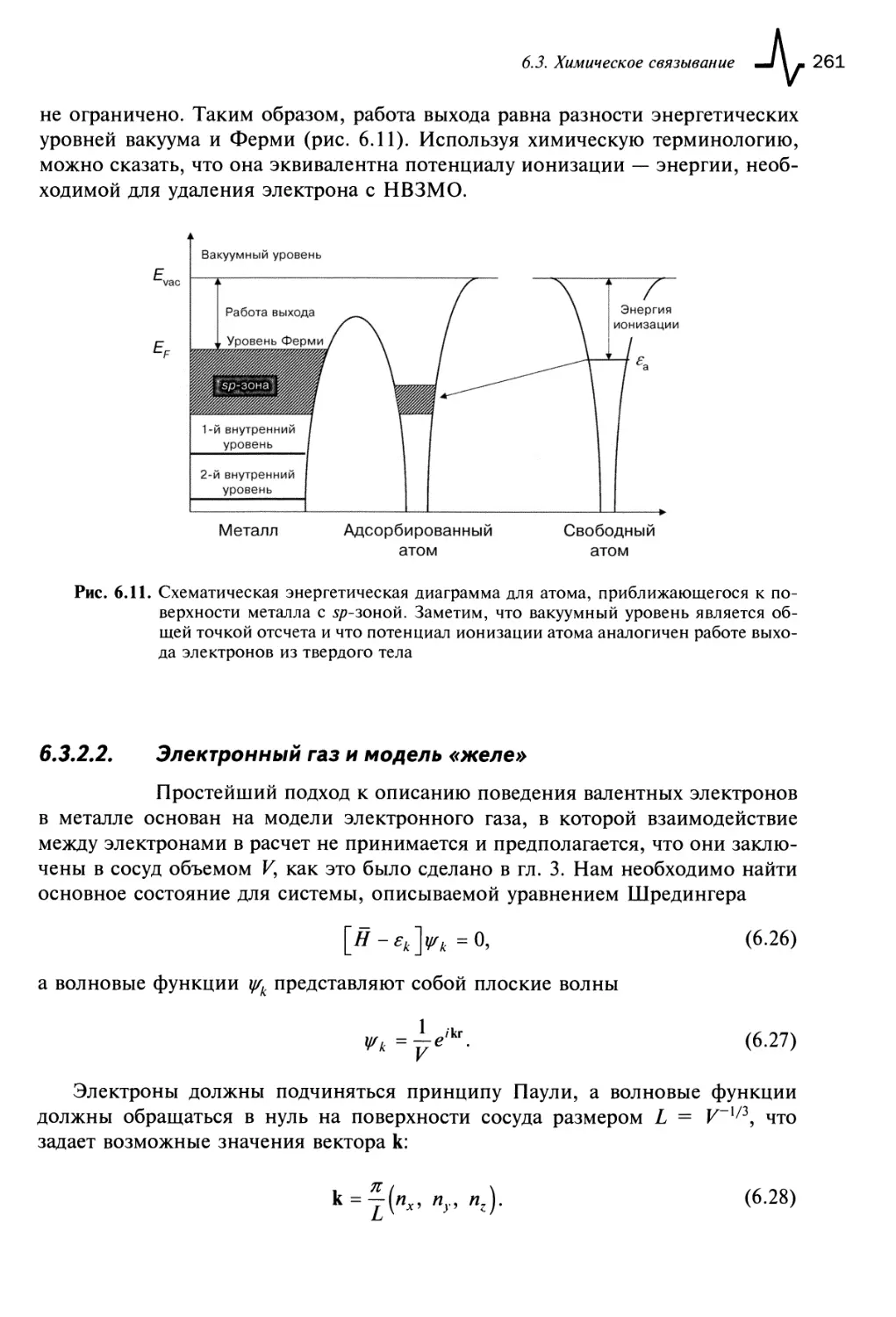
6.3.2. Поверхность твердых тел 259
6.3.2.1. Работа выхода 260
6.3.2.2. Электронный газ и модель «желе» 261
6.3.2.3. Модель сильной связи 265
6.3.2.4. Простая модель переходных металлов 269
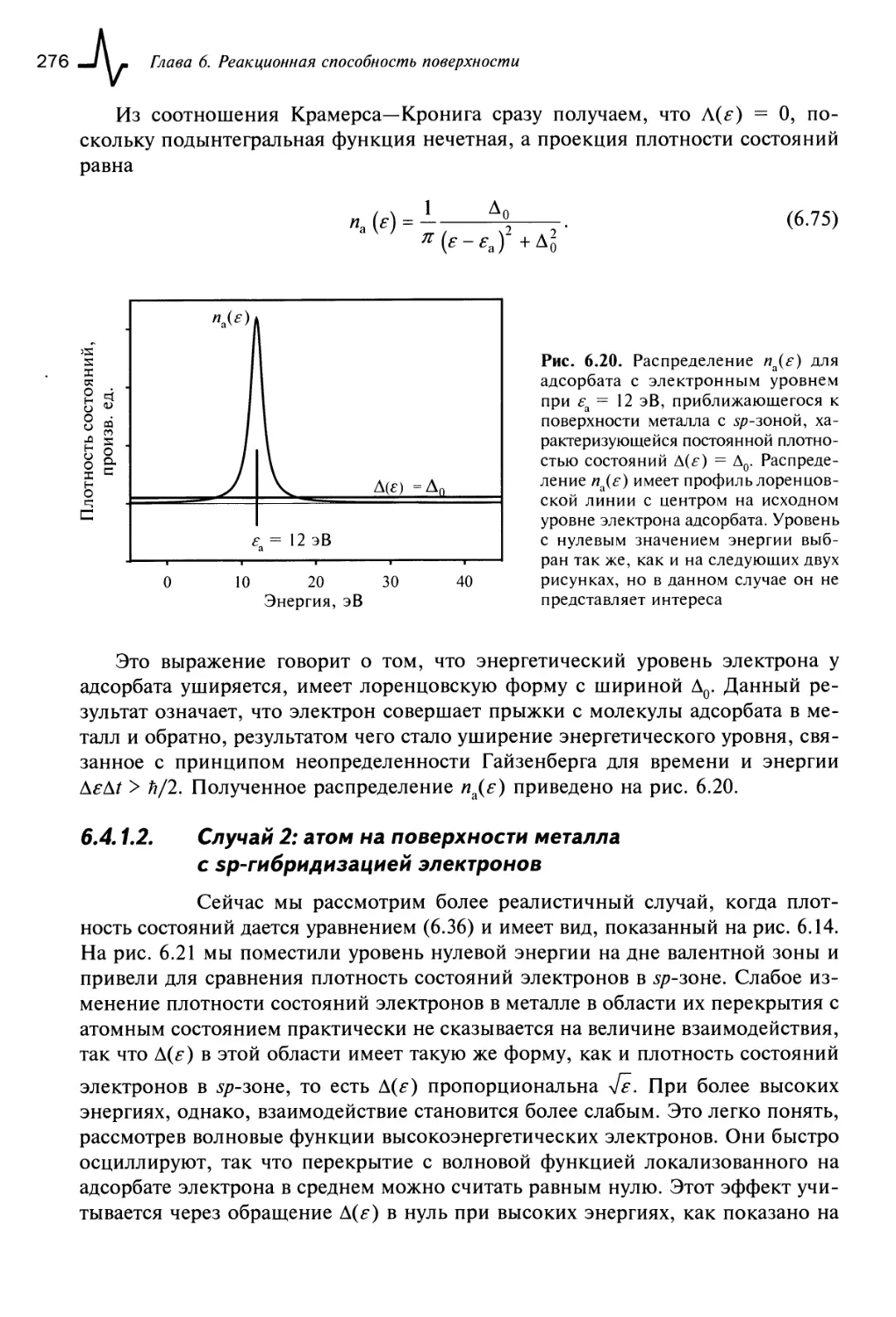
6.4. Химическая адсорбция 271
6.4.1. Модель Ньюнса—Андерсона 271
6.4.1.1. Случай 1: атом на поверхности металла с постоянной
плотностью состояний электронов 275
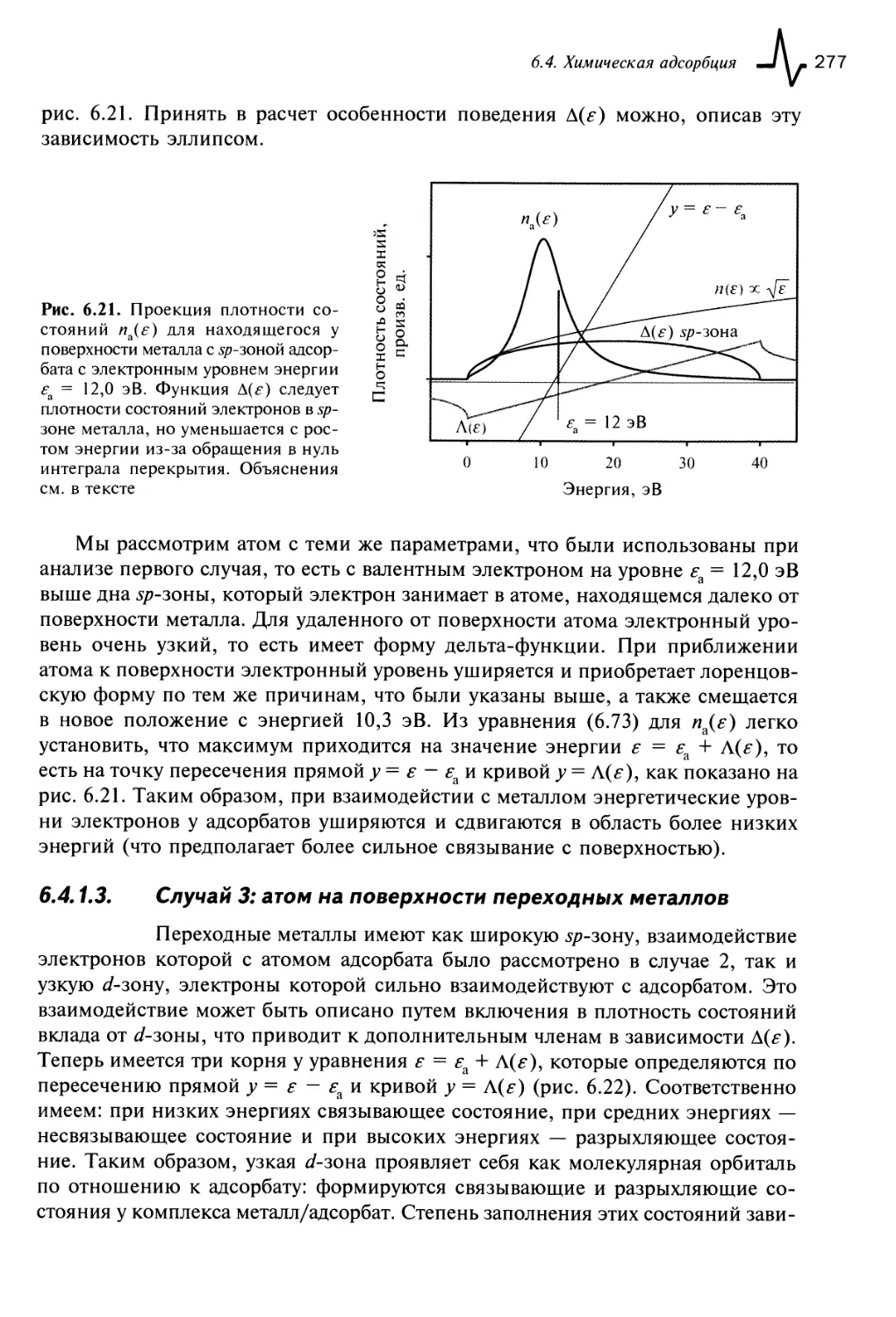
6.4.1.2. Случай 2: атом на поверхности металла с 5/7-гибридизацией
электронов 276
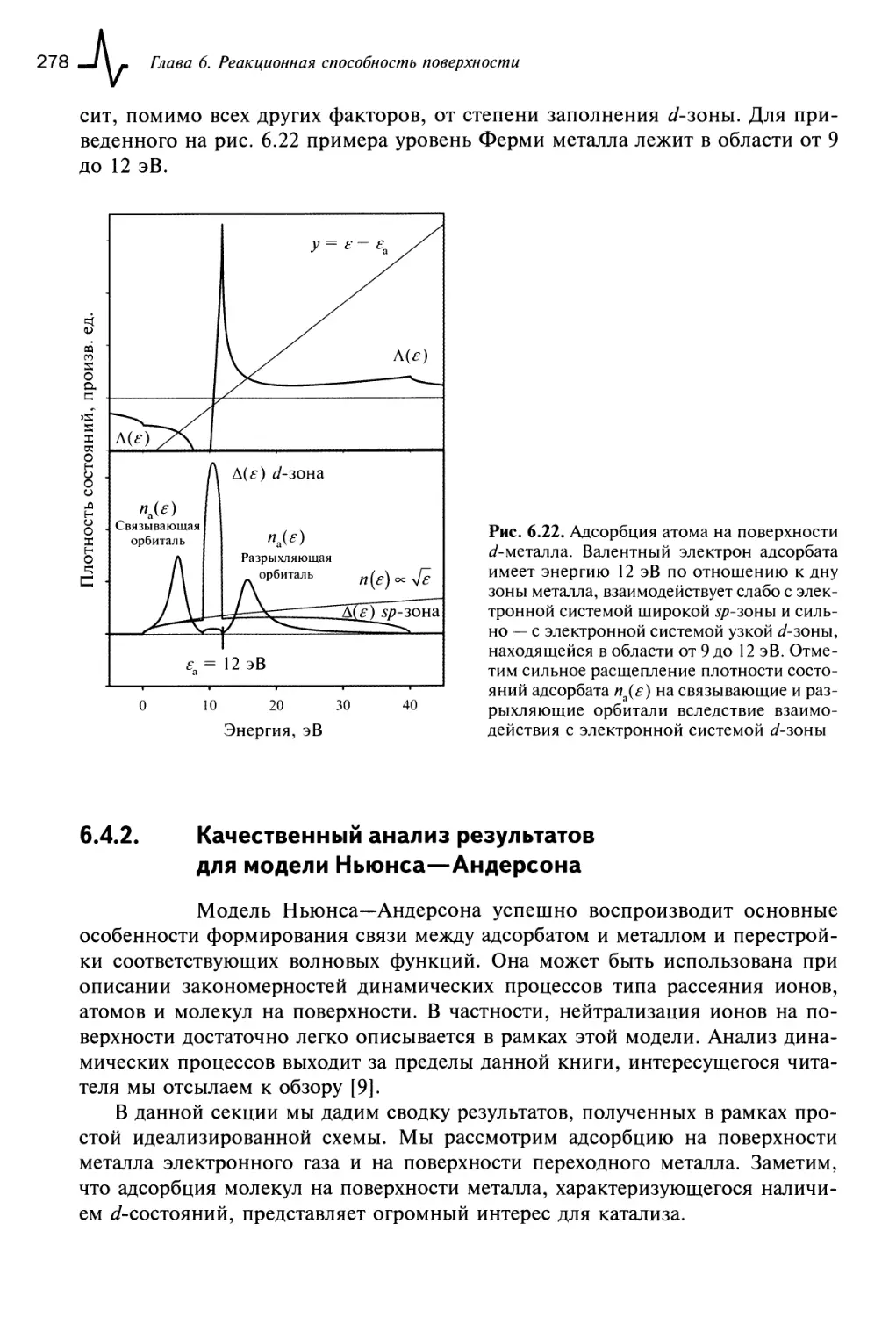
6.4.1.3. Случай 3: атом на поверхности переходных металлов 277
6.4.2. Качественный анализ результатов для модели
Ньюнса—Андерсона 278
6.4.2.1. Адсорбция на поверхности металла электронного газа 279
6.4.2.2. Адсорбция атомов на поверхности переходных
или ^/-металлов 279
6.4.2.3. Адсорбция молекул на поверхности переходных металлов 280
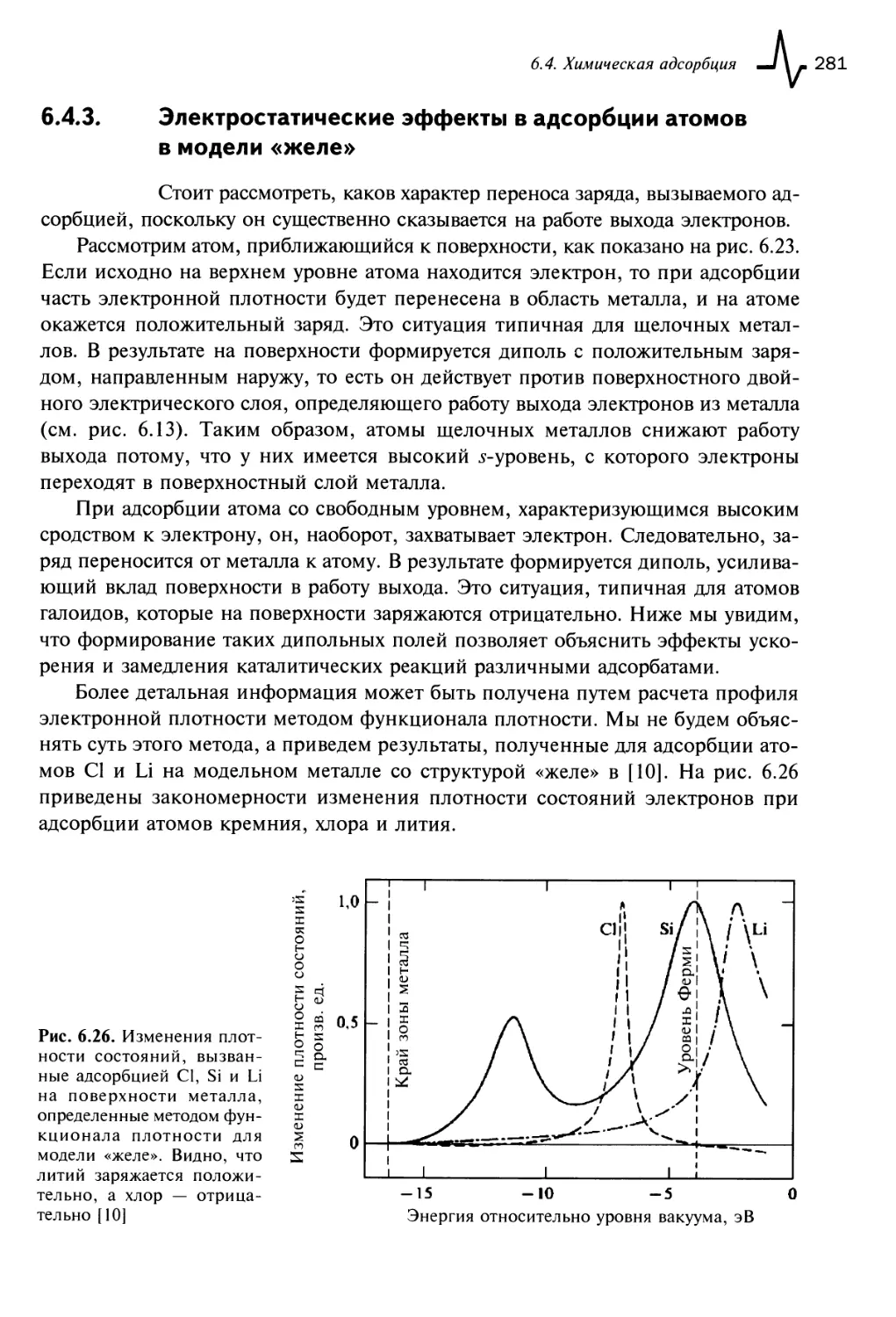
6.4.3. Электростатические эффекты в адсорбции атомов в модели
«желе» 281
6.5. Важные закономерности поведения реакционной способности
поверхности 283
6.5.1. Закономерности поведения энергии хемосорбции
атомов 283
6.5.2. Закономерности хемосорбции молекул 287
6.5.2.1. Влияние напряжений и деформации на хемосорбцию 289
6.5.3. Особенности поведения реакционной способности
поверхности 291
6.5.3.1. Физическая и химическая адсорбция, диссоциация
молекул 291
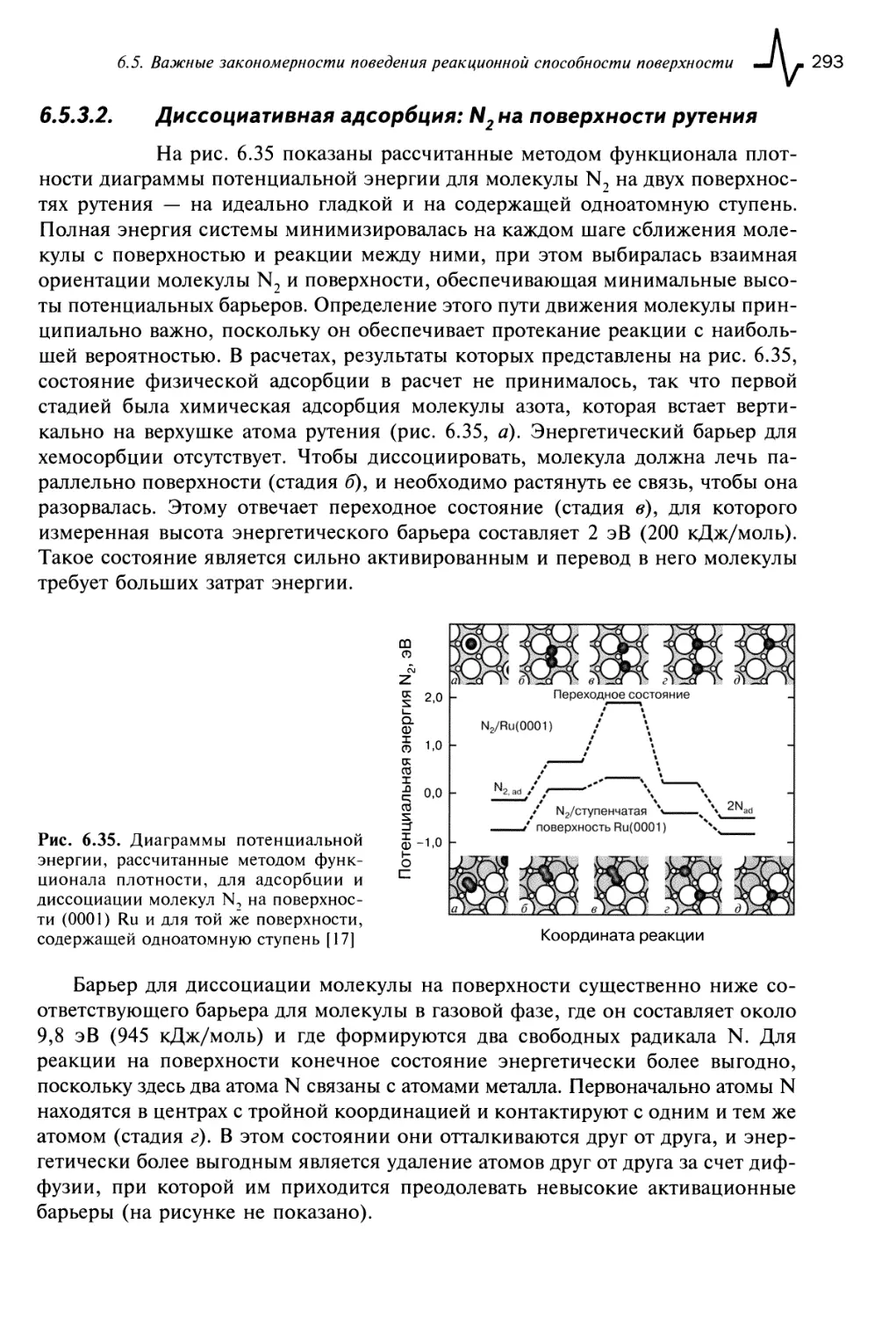
6.5.3.2. Диссоциативная адсорбция: N2 на поверхности рутения 293
6.5.3.3. Закономерности диссоциативной адсорбции 294
6.5.3.4. Переходные состояния и влияние степени покрытия
поверхности: гидрирование этилена 296
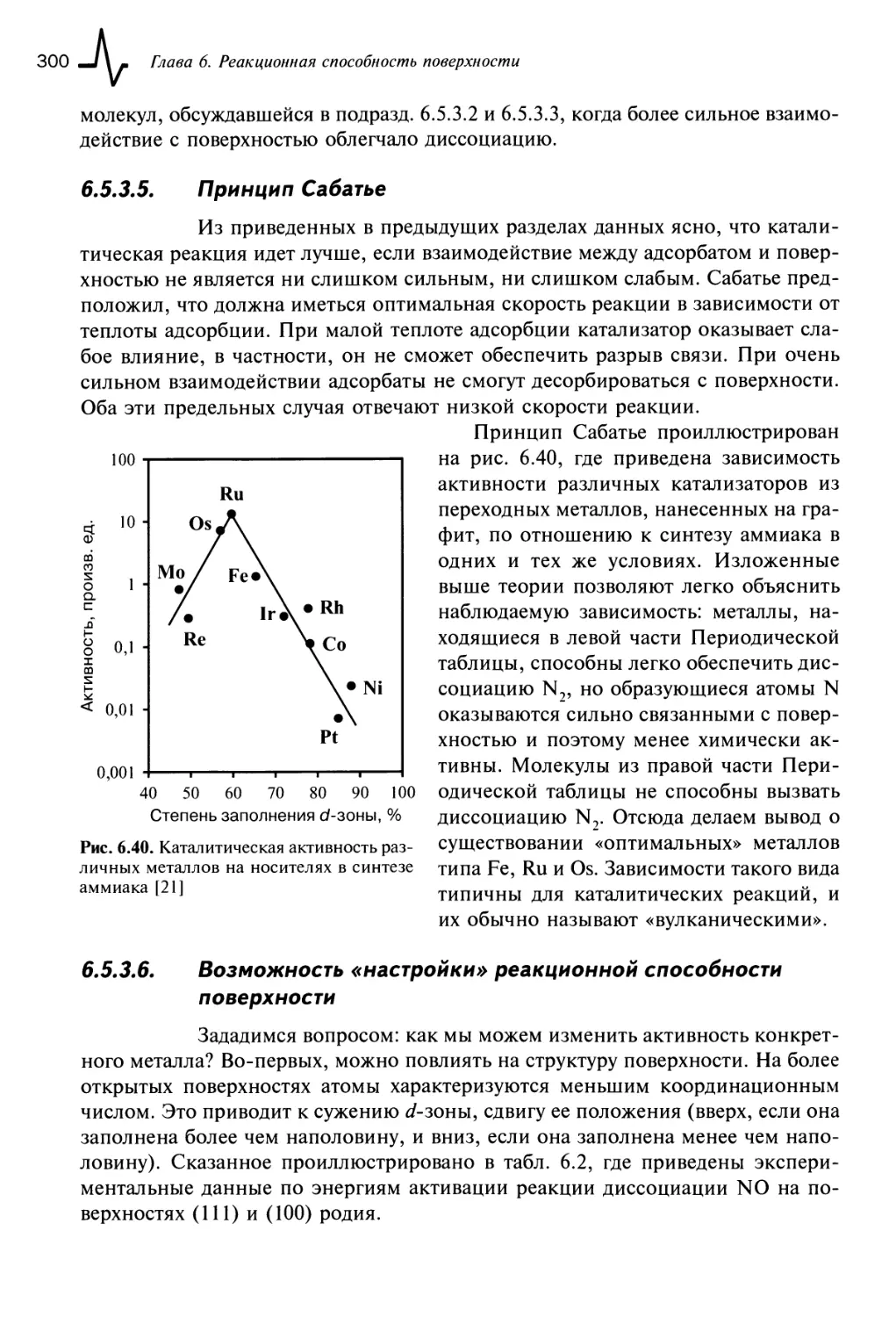
6.5.3.5. Принцип Сабатье 300
6.5.3.6. Возможность «настройки» реакционной способности
поверхности 300
6.5.4. Универсальность гетерогенного катализа 302
Приложение. Метод функционала плотности 304
Список литературы 306
Оглавление
Глава 7
КИНЕТИКА ПОВЕРХНОСТНЫХ РЕАКЦИЙ 307
7.1. Простейшие поверхностные реакции 307
7.1.1. Адсорбция и «прилипание» молекул 307
7.1.1.1. Определение коэффициента прилипания 308
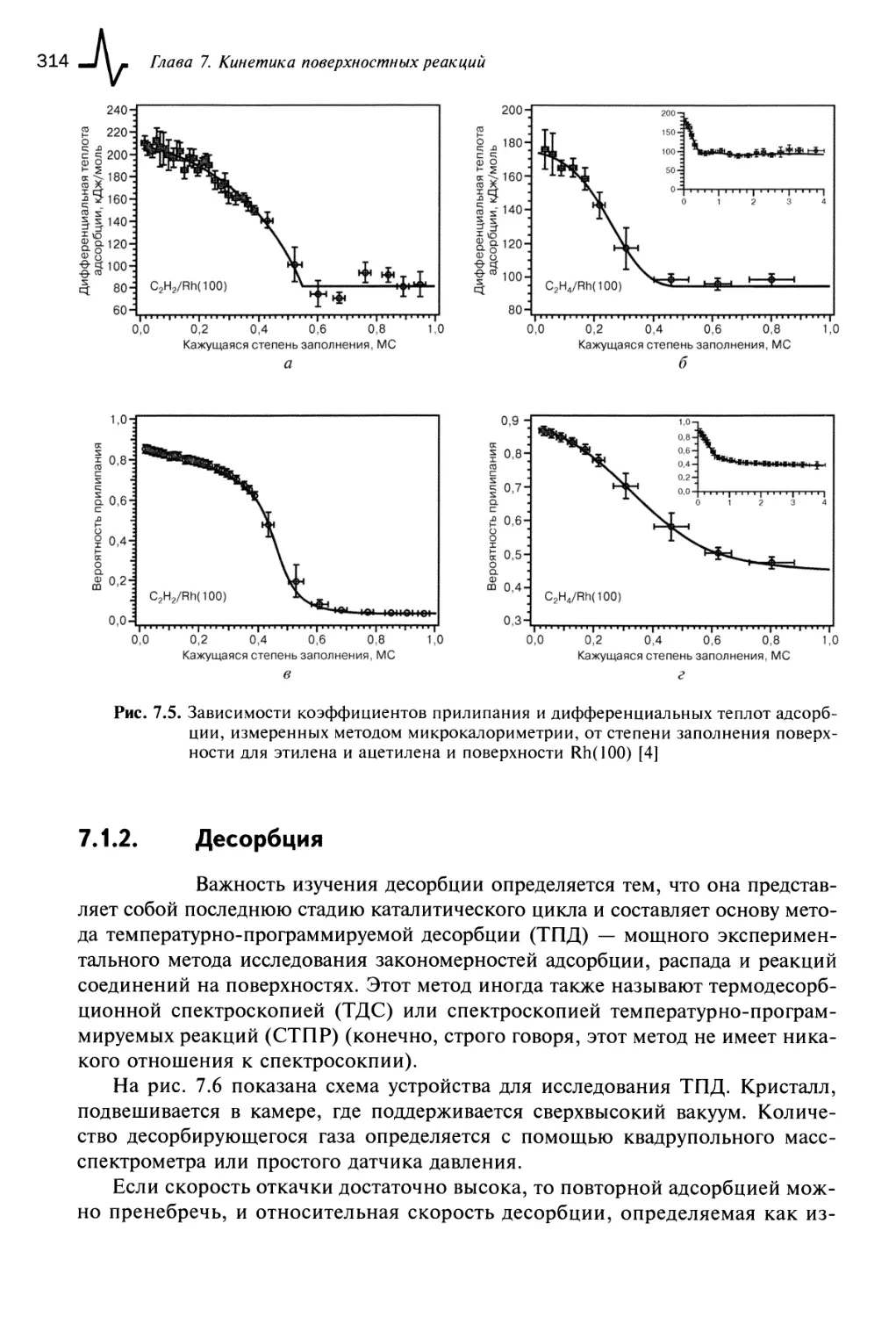
7.1.2. Десорбция 314
7.1.2.1. Количественная интерпретация данных
температурно-программируемой десорбции 317
7.1.2.2. Компенсационный эффект в температурно-программируемой
десорбции 319
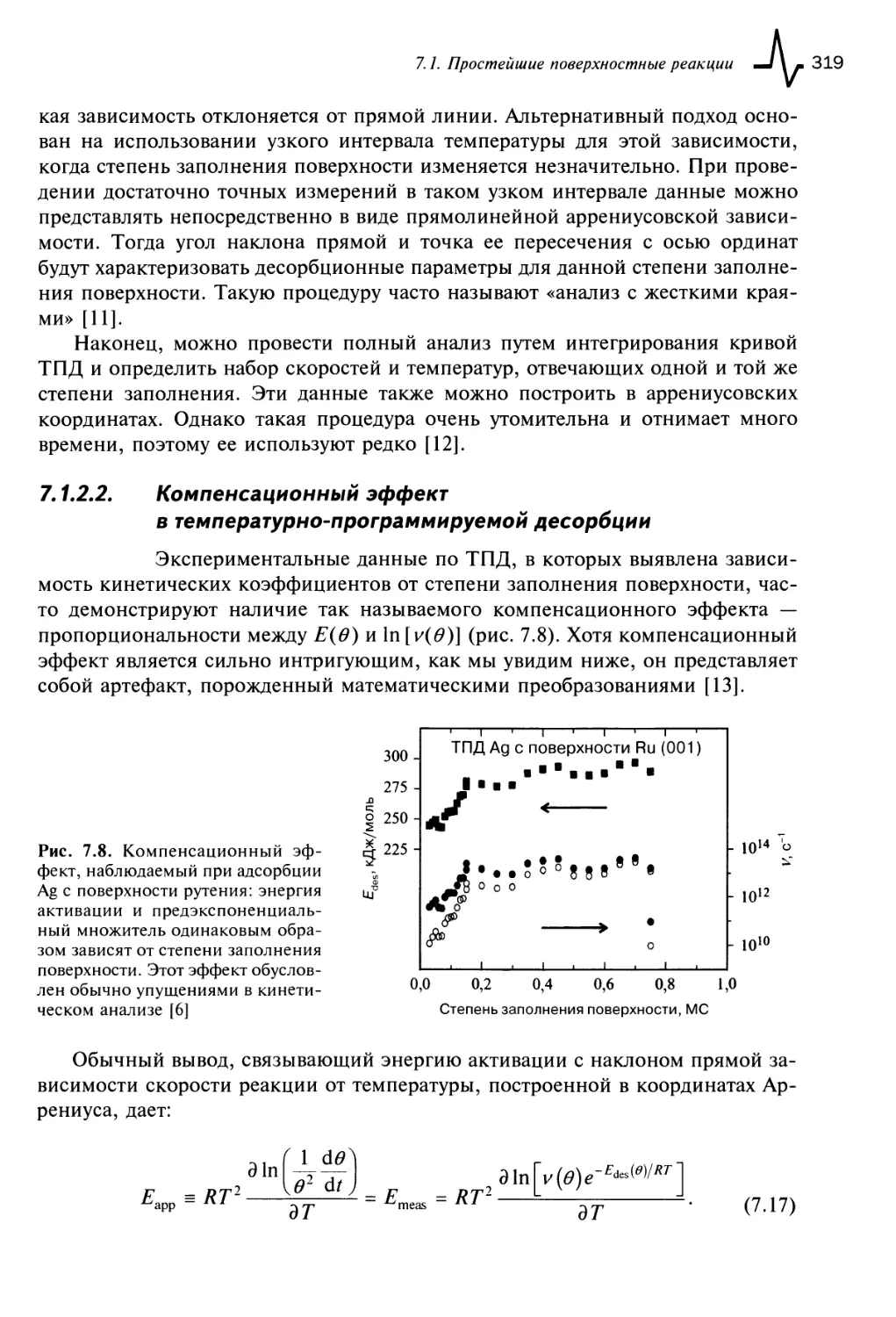
7.1.3. Роль латеральных взаимодействий в поверхностных реакциях 320
7.1.4. Диссоциативные реакции на поверхности 324
7.1.5. Интермедиаты в поверхностных реакциях 327
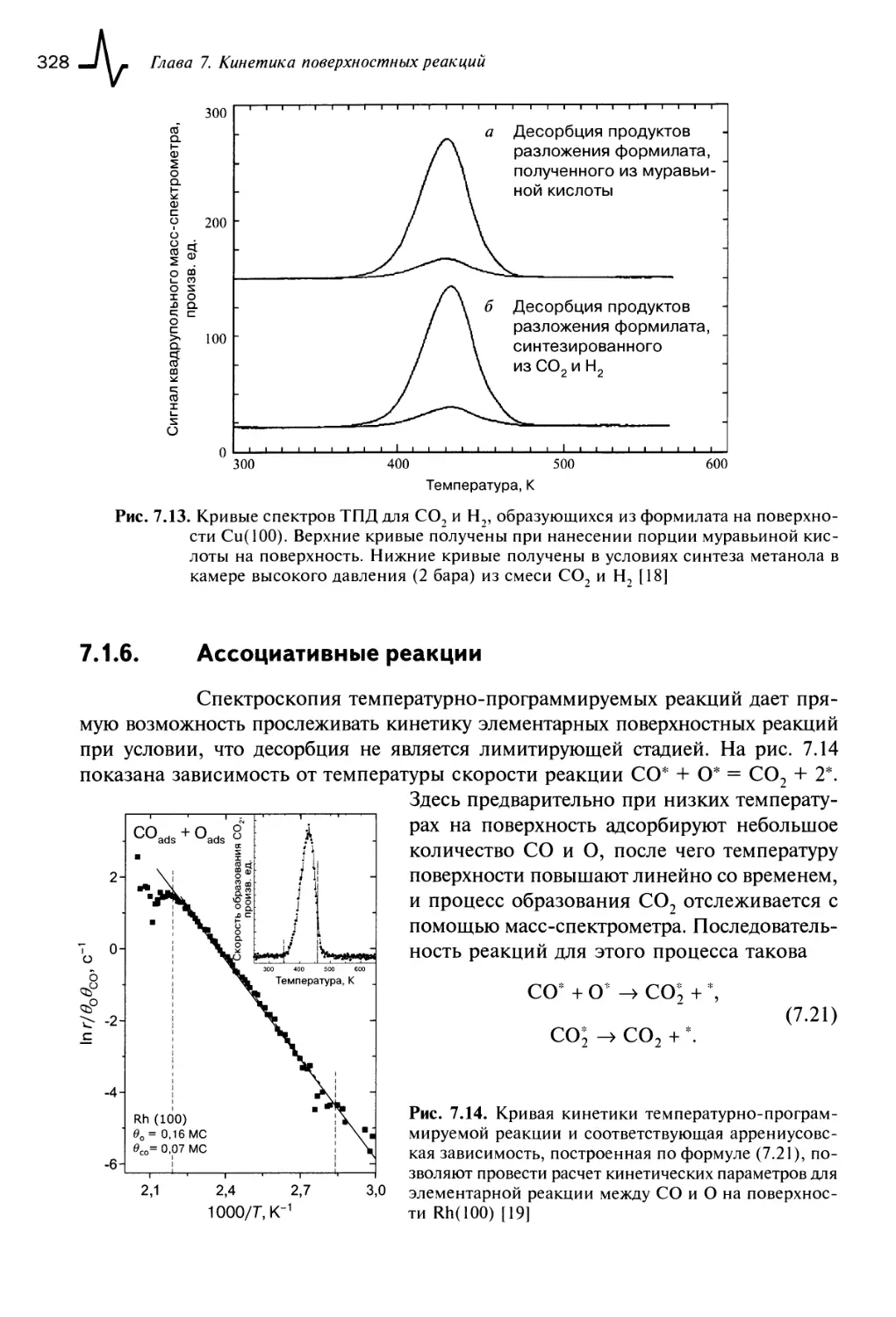
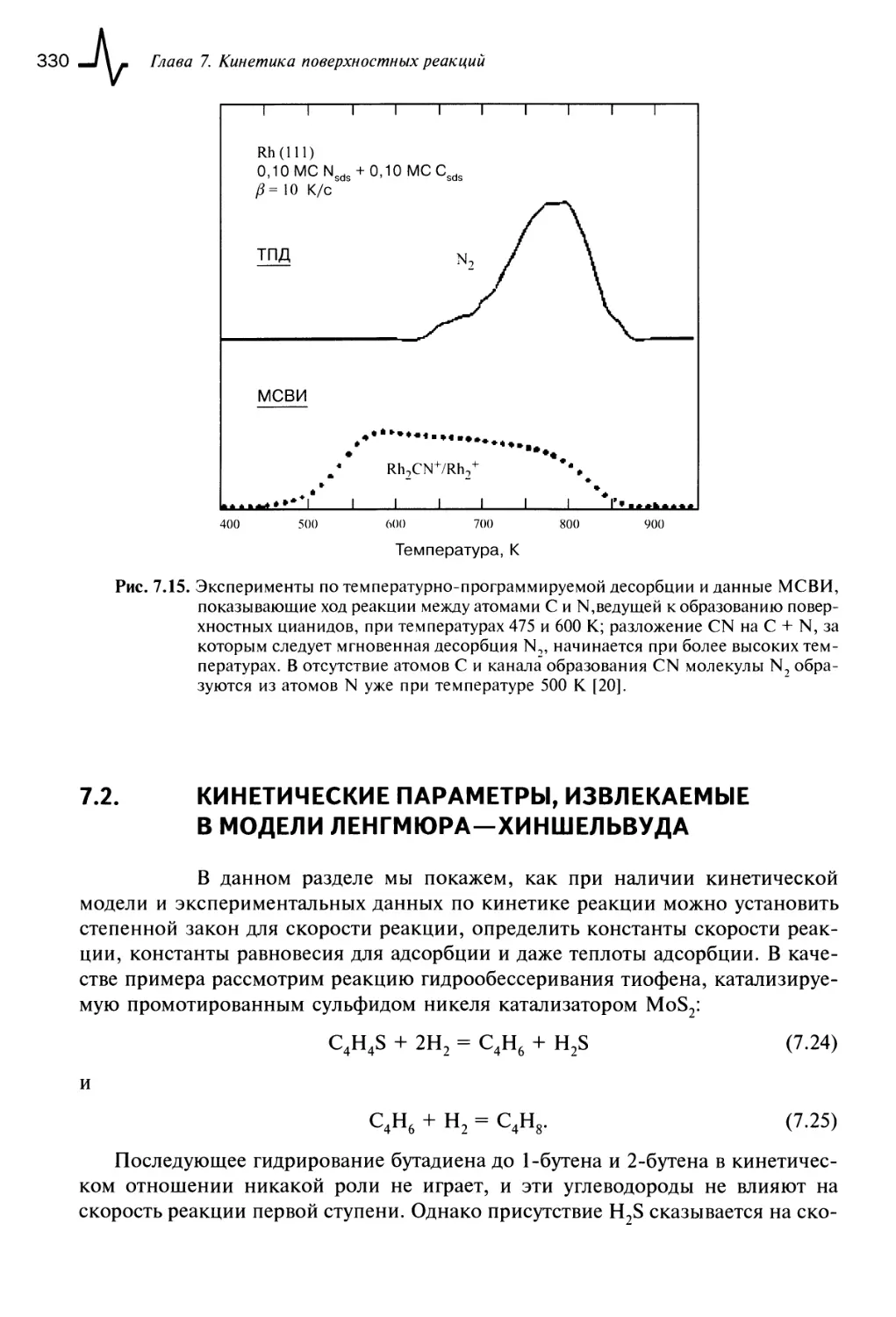
7.1.6. Ассоциативные реакции 328
7.2. Кинетические параметры, извлекаемые в модели
Ленгмюра—Хиншельвуда 330
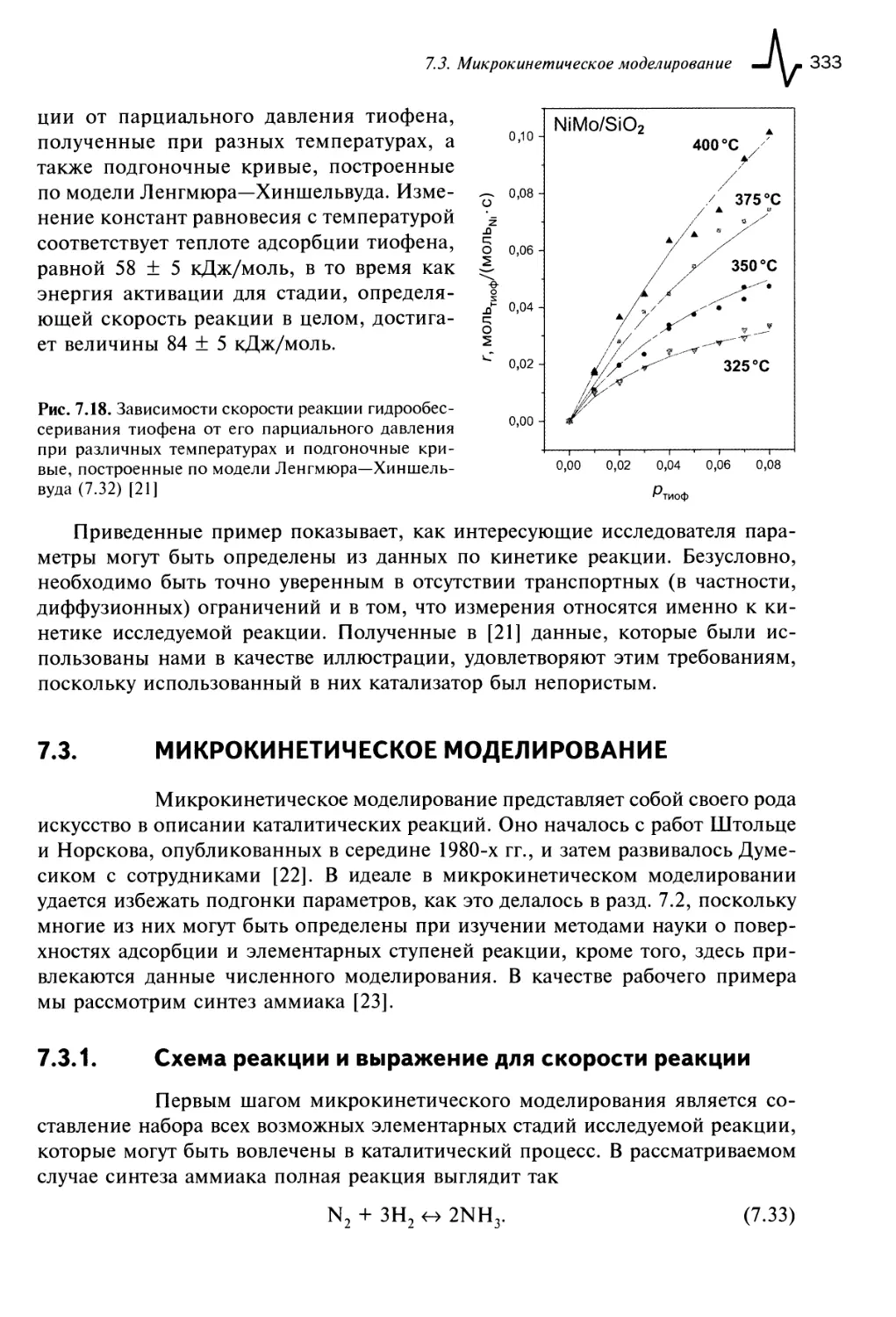
7.3. Микрокинетическое моделирование 333
7.3.1. Схема реакции и выражение для скорости реакции 333
7.3.2. Энергия активации и порядки реакции 336
7.3.3. Катализатор синтеза аммиака в рабочих условиях 340
Список литературы 342
Глава 8
ПРАКТИКА ГЕТЕРОГЕННОГО КАТАЛИЗА: ВОДОРОД 344
8.1. Введение 344
8.2. Процесс конверсии с водяным паром 344
8.2.1. Основные понятия процесса 344
8.2.2. Механистические детали конверсии с водяным паром 347
8.2.3. Проблемы в процессе конверсии с водяным паром 348
8.2.4. Пассивированный серой процесс конверсии: селективное
отравление катализатора серой 349
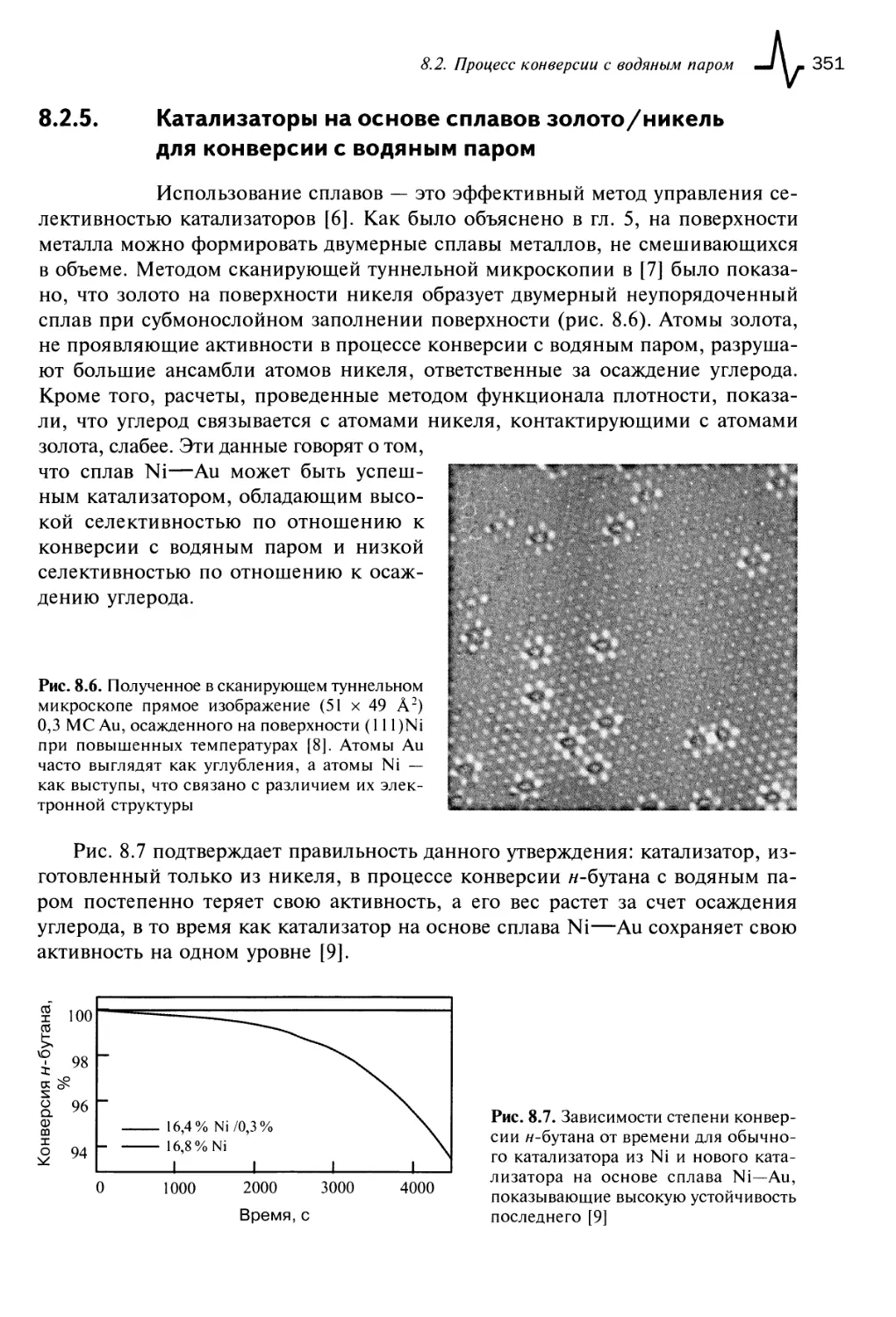
8.2.5. Катализаторы на основе сплавов золото/никель для конверсии
с водяным паром 351
8.2.6. Прямое использование метана 352
8.2.6.1. Прямое получение метанола 353
8.2.6.2. Каталитическое частичное окисление метана 353
8.3. Реакции с участием синтез-газа 354
8.3.1. Синтез метанола 354
8.3.1.1. Основные понятия процесса 354
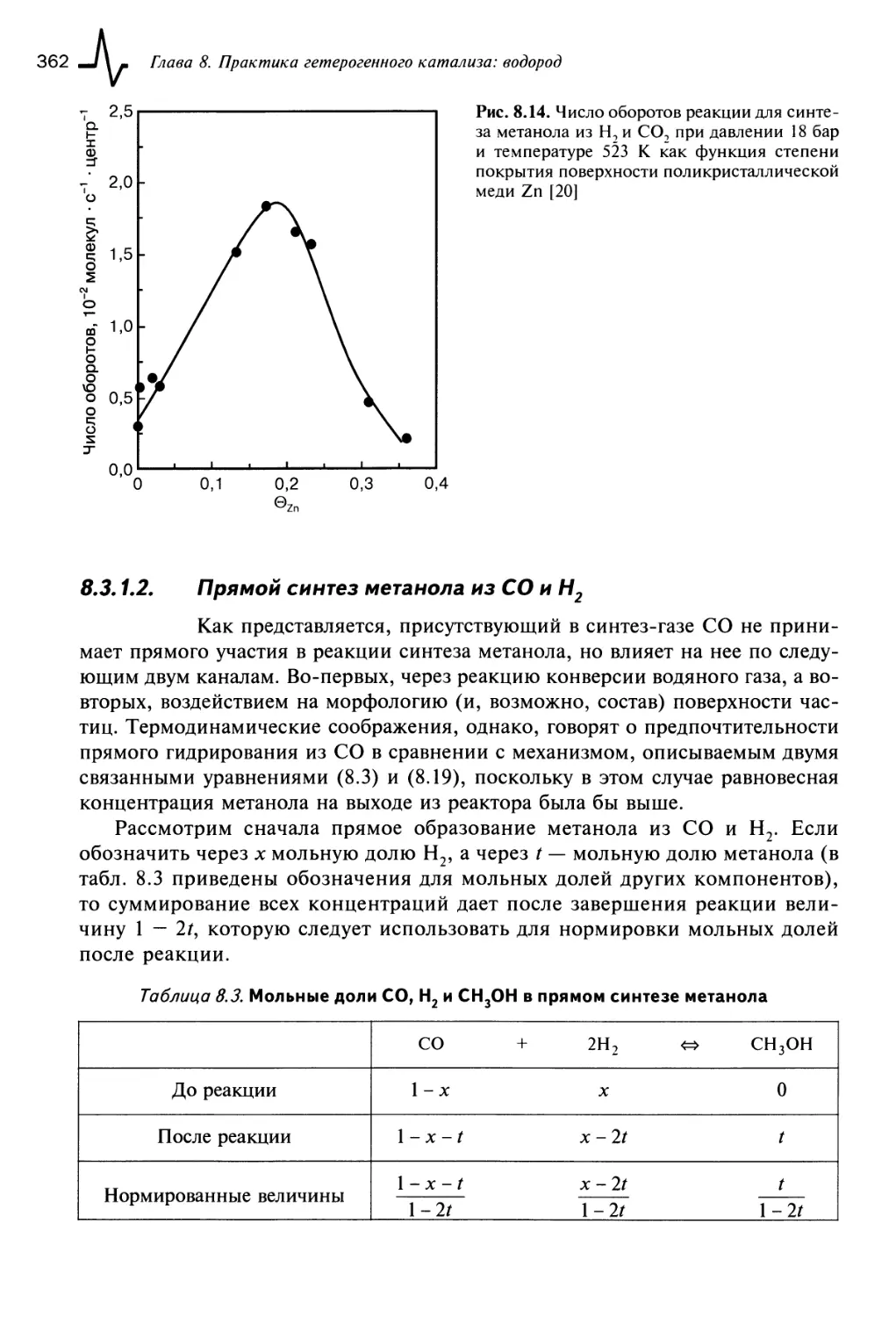
8.3.1.2. Прямой синтез метанола из СО и Н2 362
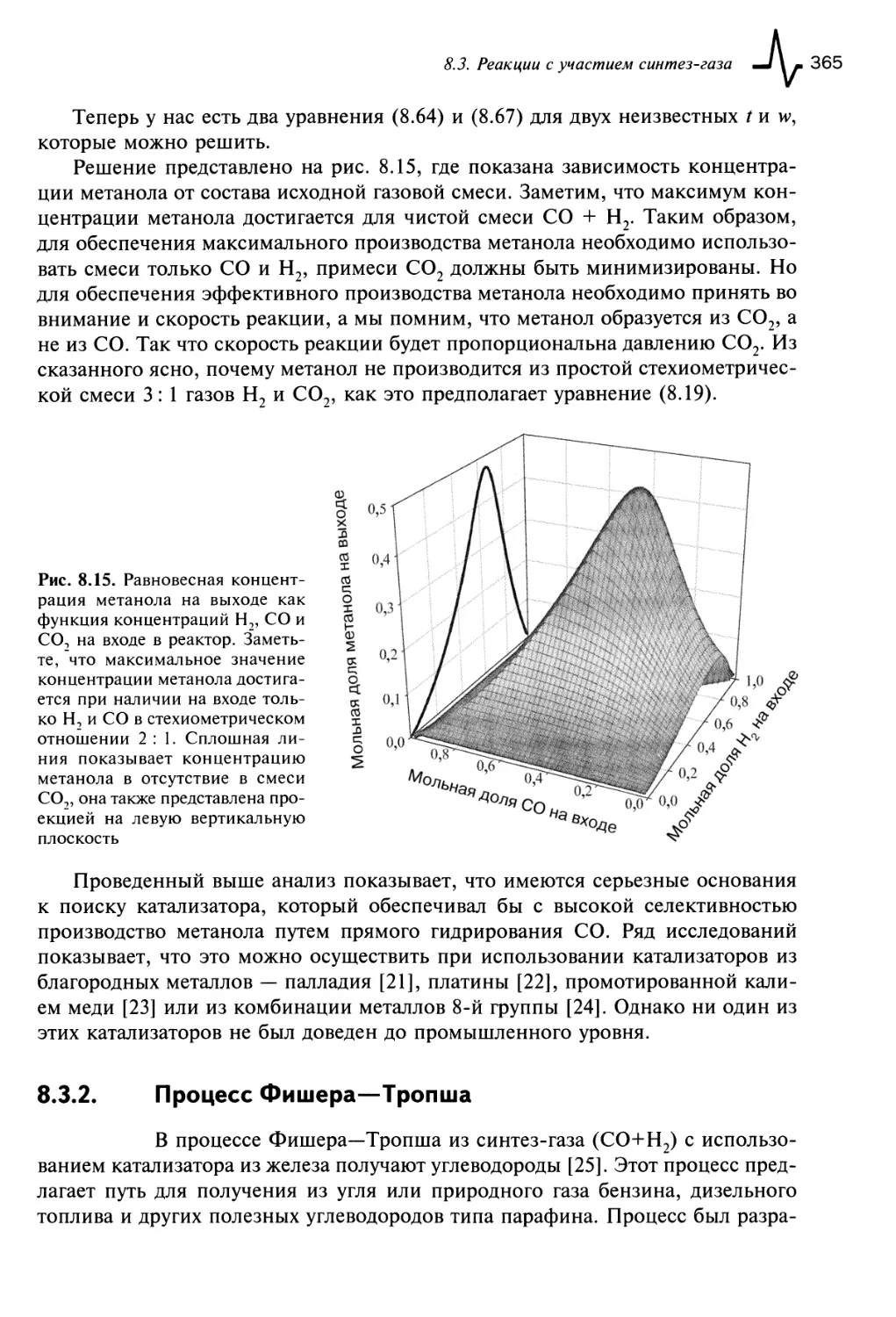
8.3.2. Процесс Фишера—Тропша 365
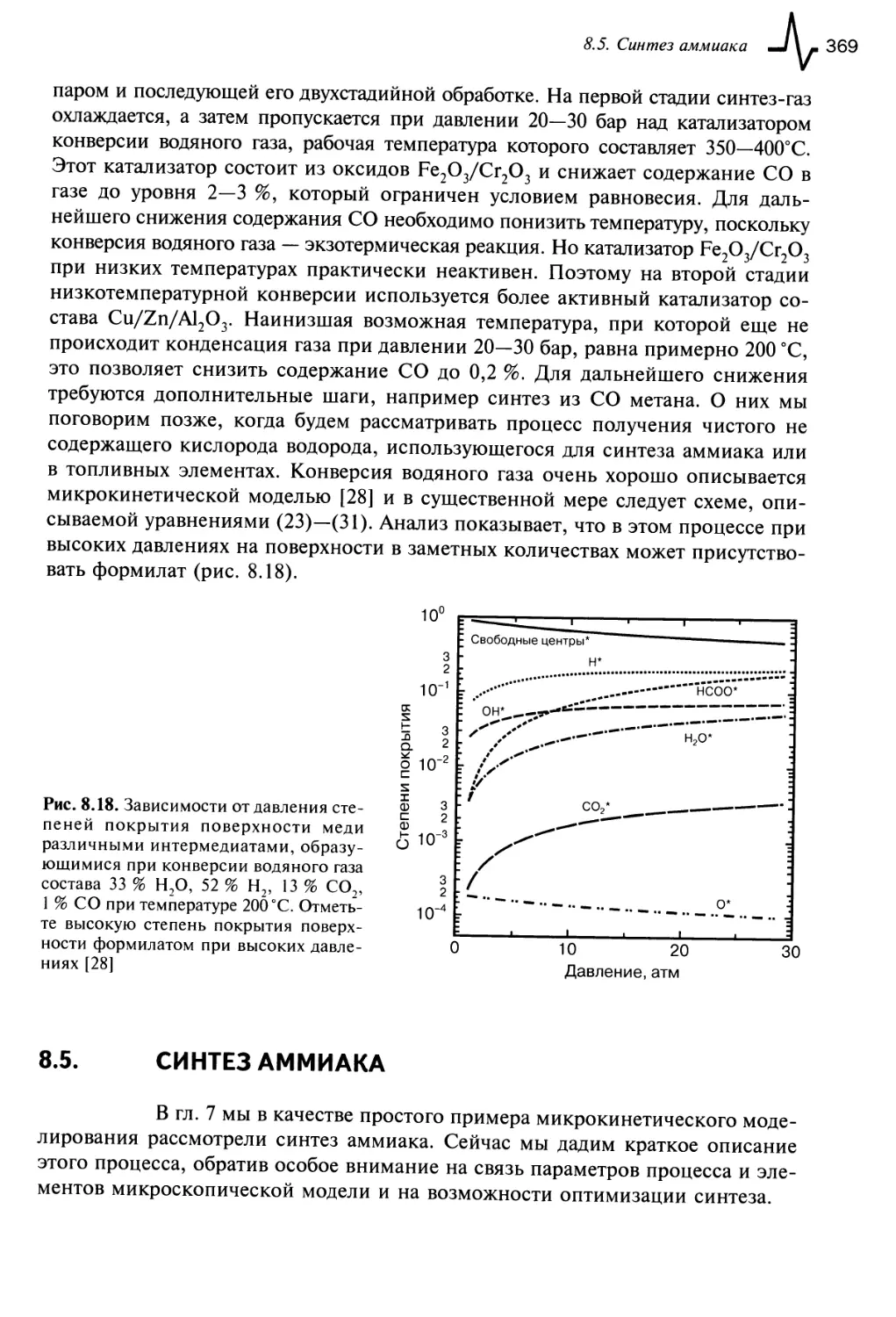
8.4. Конверсия водяного газа 368
8.5. Синтез аммиака 369
Оглавление jv
8.5.1. История синтеза аммиака 370
8.5.2. Завод по производству аммиака 372
8.5.3. Рабочий реактор 374
8.5.4. Научные предложения по повышению эффективности
катализаторов 376
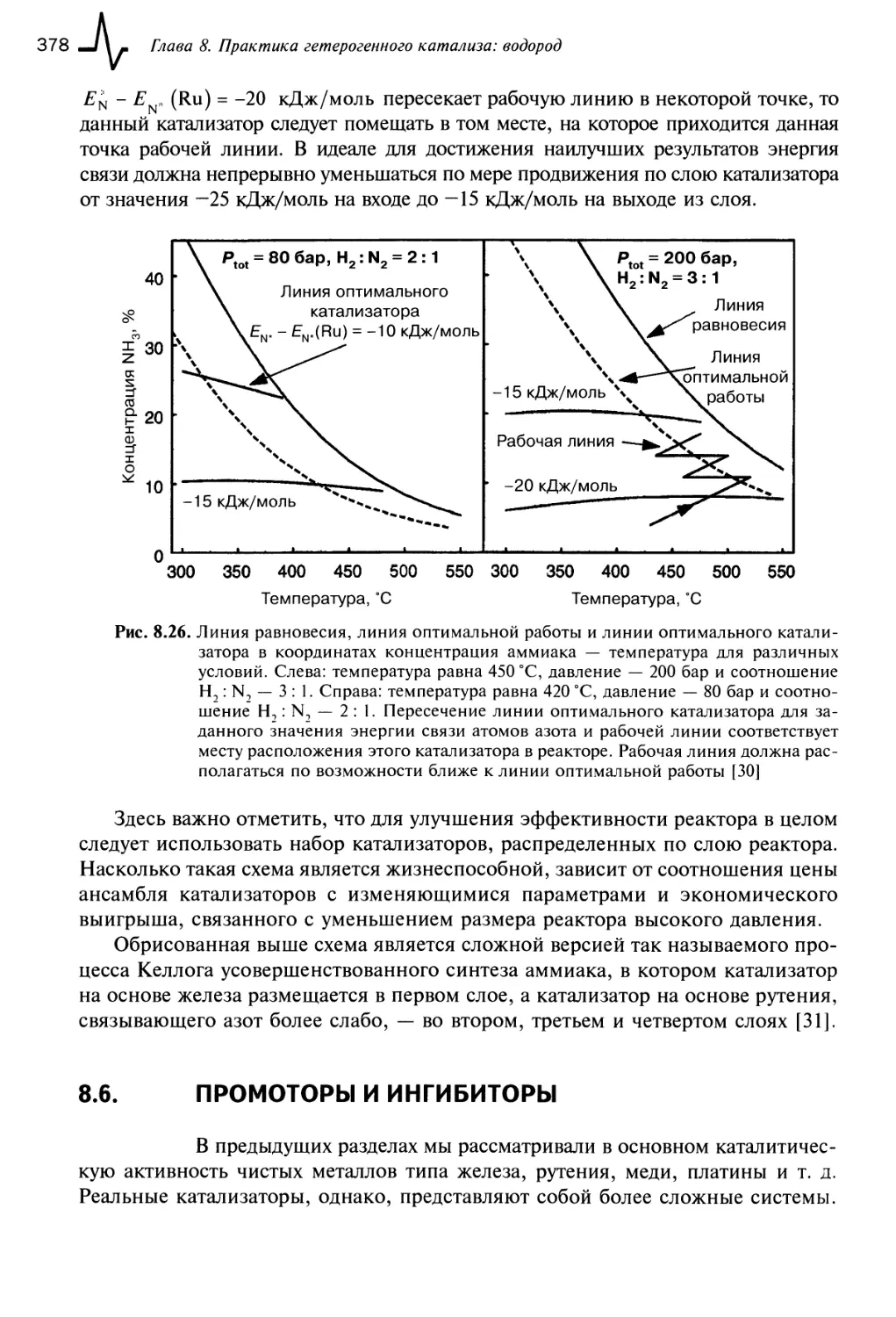
8.6. Промоторы и ингибиторы 378
8.7. «Водородное сообщество» 382
8.7.1. Потребность в возобновляемых источниках энергии 382
8.7.2. Возобновляемые источники энергии 384
8.7.3. Водород и топливные элементы 386
8.7.3.1. Топливные элементы на протонопроводящих мембранах 386
8.7.3.2. Топливные элементы на твердых оксидах 390
8.7.3.3. Эффективность топливных элементов 390
8.7.3.4. Хранение и транспортировка водорода 392
Список литературы 393
Глава 9
ПЕРЕРАБОТКА НЕФТИ И НЕФТЕХИМИЯ 395
9.1. Сырая нефть 395
9.2. Гидроочистка 399
9.2.1. Гетероатомы и нежелательные элементы 399
9.2.2. Катализаторы в гидроочистке 401
9.2.3. Механизмы реакций гидрообессеривания 403
9.3. Производство бензина 406
9.3.1. Каталитический крекинг в псевдоожиженном слое 407
9.3.2. Риформинг и бифункциональный катализ 411
9.3.3. Алкилирование 415
9.4. Нефтехимия: реакции с участием низкомолекулярных олефинов 417
9.4.1. Эпоксидирование этилена 417
9.4.2. Частичное окисление и аммоксидирование пропилена 418
9.4.3. Катализ в реакциях полимеризации 421
Список литературы 424
Глава 10
КАТАЛИЗ И ЗАЩИТА ОКРУЖАЮЩЕЙ СРЕДЫ 425
10.1. Введение 425
10.2. Каталитическая нейтрализация выхлопных газов 426
10.2.1. Катализаторы «трех процессов» 427
10.2.1.1. Каталитический нейтрализатор 430
10.2.1.2. Демонстрационные эксперименты 433
10.2.1.3. Деактивация катализаторов 433
10.2.2. Каталитические реакции с участием катализаторов
«трех процессов»: механизмы и кинетика реакций 434
Оглавление
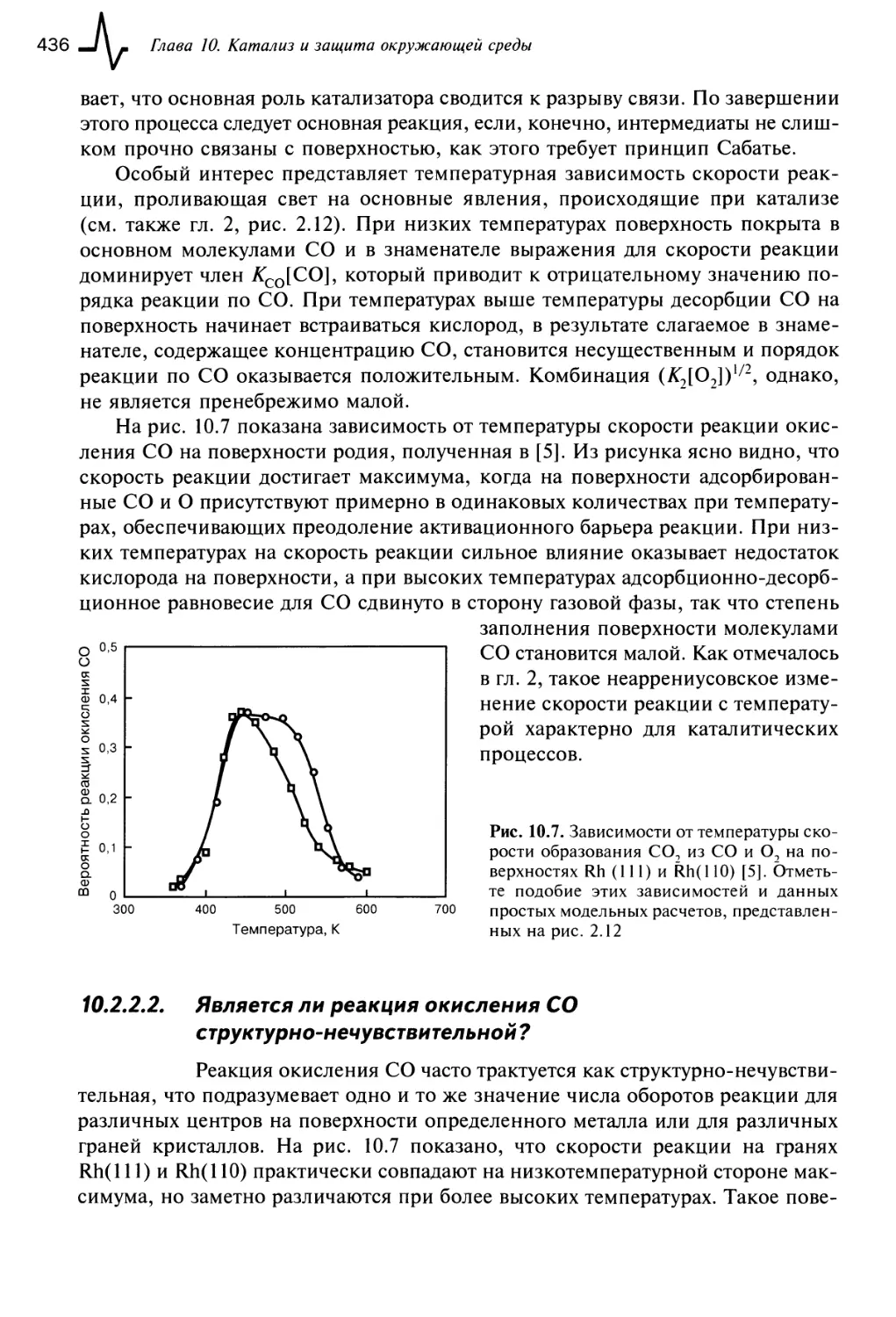
10.2.2.1. Реакция окисления СО 435
10.2.2.2. Является ли реакция окисления СО
структурно-нечувствительной? 436
10.2.2.3. Реакция СО + NO 437
10.2.2.4. Реакция СО + NO при высоких давлениях 439
10.2.2.5. Реакции с участием углеводородов 440
10.2.2.6. Катализаторы накопления/восстановления NOx в двигателях
с низким содержанием топлива 440
10.2.3. Заключительные замечания по каталитической обработке
выхлопных газов 442
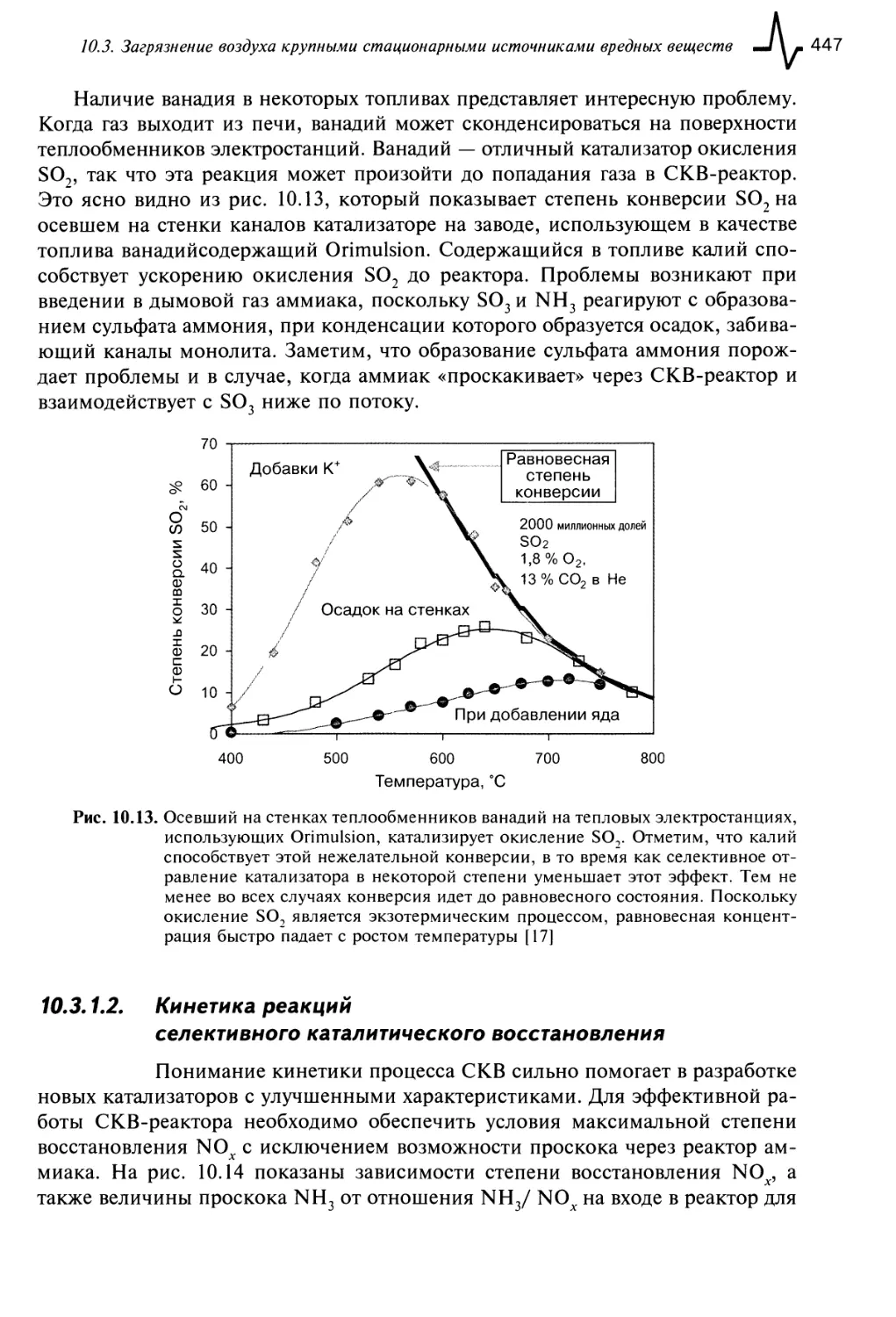
10.3. Загрязнение воздуха крупными стационарными источниками
вредных веществ 443
10.3.1. Процесс селективного каталитического восстановления 443
10.3.1.1. Катализатор для процесса селективного каталитического
восстановления 445
10.3.1.2. Кинетика реакций селективного каталитического
восстановления 447
10.3.2. Процесс селективного каталитического восстановления
для использования в транспортных средствах 451
Список литературы 451
Вопросы и упражнения 452
ПРИЛОЖЕНИЯ
Приложение А
Некоторые полезные фундаментальные постоянные 497
Коэффициенты перевода единиц энергии 498
Некоторые полезные соотношения 499
Приложение Б
Некоторые определения 500
ПРЕДИСЛОВИЕ АВТОРОВ
КАТАЛИЗ: КОНЦЕПТУАЛЬНО ПОНЯТНЫЙ,
НО ДАЛЕКИЙ ОТ ЗРЕЛОСТИ
Катализ — явление, которое в настоящее время хорошо осмыслено
на концептуальном уровне. Он был открыт как новое явление и уже использован в 1816 г. Дэви при создании безопасных шахтерских ламп. Определение
катализа было дано Берцелиусом в 1835 г., и в начале XX в. его систематическое экспериментальное исследование было выполнено Митташем. Исследования механизма каталитических реакций стали возможны после разработки Лен-
гмюром и Хиншельвудом кинетической теории. С тех пор фундаментальные
исследования каталитических реакций стали синонимом кинетического анализа. Приход в эту область спектроскопических методов, начавшийся с использования в 1950-е гг. инфракрасной спектроскопии, и последовавшее за этим развитие экспериментальной базы по определению параметров катализаторов и
свойств их поверхности открыли новые возможности в установлении связи
между каталитическими свойствами и составом и структурой материалов. Современное состояние науки о поверхностях позволяет выявить структуру адсорбционных центров и их реакционную способность. Кульминацией методов
исследования поверхности можно назвать разработанную в конце XX столетия
сканирующую туннельную микроскопию, с помощью которой удается разрешить на атомном уровне структуру поверхности и адсорбционных слоев. Существуют приемы, позволяющие проводить исследования свойств катализаторов в процессе их работы. Развитие численных методов сделало возможными
расчеты структуры адсорбционных слоев, прочности связей и даже скоростей
реакций. К 2003 г. катализ развился в наукоемкую дисциплину с прочным
концептуальным базисом. Для достаточно простых и хорошо охарактеризованных поверхностей связь между каталитической активностью при определенных
реакциях и составом и структурой поверхности хорошо осмыслена на качественном уровне.
Вместе с тем перспективы конструирования катализаторов с заданными
свойствами из первых принципов остаются еще очень туманными. Тому есть
несколько причин. Так, несмотря на то что мы можем описать каталитическую
реакцию, протекающую на хорошо охарактеризованной поверхности монокри¬
Предисловие авторов
сталла при строго определенных и «упрощенных» условиях, переход к малым
частицам катализатора, находящимся на носителях, в окружающей среде реального реактора делает это описание многократно более сложным. Миры идеальной поверхности в научной лаборатории и поверхности частицы промышленного катализатора не только разделены барьером рабочих давлений, но и
барьерами структур и используемых материалов. Сложное строение малых частиц катализаторов на носителях, которые претерпевают динамический отклик
на изменение среды реактора, только еще начали изучать. Далее, мы до сих
пор описываем кинетику каталитических реакций, базируясь на, пусть и в существенной мере усовершенствованных, теории адсорбции Ленгмюра (1915) и
кинетическом формализме Хиншельвуда (1927), в основе которых лежат представления об идеальной поверхности, эквивалентных адсорбционных центрах
и равномерно распределенных по поверхности не взаимодействующих адсорбированных молекулах.
Цель данной книги — изложить основы современного гетерогенного катализа и объяснить каталитические явления на концептуальном уровне. Кинетическая теория, являющаяся инструментом исследования и способом описания
характера проявления каталитической активности в реакторах, составляет значительную часть данной книги. В книге также подробно описана и широко
используется теория скоростей реакций, позволяющая связать скорости химических реакций со строением реагирующих молекул. Мы подробно охарактеризуем структуру твердых поверхностей катализаторов и методы их исследования. Информация о структуре положена в основу определения каталитической
Предисловие авторов
активности поверхности в рамках упрощенной теории молекулярных орбита-
лей. Все это делается для выяснения на концептуальном уровне механизма
действия катализатора и тенденций в изменении его характеристик при переходе от одной «пары» структура/поверхность к другой. Заключительная глава
посвящена промышленному катализу, что позволит читателю уяснить, как катализаторы используются на практике. Подчеркнем, что данная книга является
учебником, написана для студентов, изучающих химию, физику и химическую
технологию и желающих познакомиться с основными принципами катализа.
Много важных деталей, которые необходимо знать специалистам по катализу,
осталось за рамками данной книги; их можно найти в специальной литературе.
Эта книга основана на курсах лекций, которые авторы в течение многих лет
читали в университетах Лингби и Эйндховена. Например, гл. 1—3 составляют
базу обязательного курса «Кинетика и катализ», который студенты университета в Эйндховене проходят на втором году самостоятельного обучения, а гл. 4, 5
и 8—10 лежат в основе курса по выбору «Введение в катализ». В университете
Лингби гл. 1—7 использовались при формировании курса по выбору «Кинетика химических реакций и катализ» для магистров. В конце книги даны задачи
и упражнения по каждой главе, которые студенты могут использовать для проверки своих знаний. Все упражнения подобраны так, чтобы у студентов была
возможность приобрести навыки в моделировании кинетики реакций. Некоторые из этих задач использовались на письменных экзаменах. Решения этих
задач можно найти на веб-сайте авторов (www.cinf.dtu.dk и www.catalys.nl).
Авторы благодарны многим коллегам, занимающимся исследованием катализа и свойств поверхностей. Мы хотим выразить особую признательность Йенсу
Норскову и Рутгеру Ван-Сантену. Они оба внесли существенный вклад в теорию гетерогенного катализа. И мы хотим их поблагодарить за многочисленные
и полезные дискуссии. Наши контакты с работниками, непосредственно участвующими в каталитическом производстве, были вдохновляющими и очень
полезными. Мы хотим особо отметить Халдора Топсое из Лингби и Научный и
технологический центр компании «Шелл» в Амстердаме и Исследовательский
центр SASOL в Южной Африке. Мы также хотим поблагодарить многочисленных студентов, прослушавших наши курсы в Лингби и Эйндховене. Они научили нас многому, о чем они даже не имели представления. Мы благодарны
также нашим семьям, разрешившим нам потратить огромное количество времени на работу над этой книгой. Мы посвящаем этот труд им.
Иб Чоркендорф
Ханс Наймантсведрайт
глава ВВЕДЕНИЕ В КАТАЛИЗ
1
Спросите любого прохожего на улице, что такое катализатор, и он
или она, вероятно, скажут вам, что катализатор это то, что используют в автомобилях для очистки выхлопных газов. И действительно, каталитические нейтрализаторы выхлопных газов представляют собой очень успешное применение катализаторов, которые исполняют здесь важнейшую миссию по охране
окружающей среды. Однако катализаторы имеют более широкую область применения, чем снижение уровня загрязнения атмосферы выхлопами автомобилей. Например, биологические системы «используют» энзимы — наиболее эффективные и специфические катализаторы, которые только можно себе представить. Химическая промышленность также не может существовать без
катализаторов, которые незаменимы и в крупнотоннажном, и тонком химическом синтезе и при производстве топлива.
Катализ в промышленности
Катализ является рабочей лошадкой химических превращений в промышленности. Примерно 85—90 % продукции химической промышленности производится в каталитических процессах. Катализаторы незаменимы в:
• производстве топлива для транспорта, которое осуществляется примерно на 440 нефтеперерабатывающих заводах во всем мире;
• крупнотоннажной и тонкой химических технологиях во всех отраслях химической индустрии;
• предотвращении загрязнения среды через создание безотходных технологий (исключение производства нежелательных субпродуктов);
• снижении уровня загрязнения сточных вод, промышленных выбросов и выхлопных газов транспорта.
Для ученых и инженеров катализ — чрезвычайно интригующая междисциплинарная область деятельности. Давайте сначала посмотрим, что
представляет собой катализ, а затем узнаем, почему он так важен для человечества.
1. 1. Что такое катализ?
1.1. ЧТО ТАКОЕ КАТАЛИЗ?
Катализатор ускоряет химическую реакцию. Он не образует связей
с реагирующими молекулами, позволяя им образовывать продукт реакции, который отделяется от катализатора, оставляя его в исходном состоянии, в котором он способен принять участие в следующем акте реакции. И действительно,
мы можем рассматривать каталитические реакции как циклический процесс,
принимая участие в котором, катализатор в конце цикла возвращается в исходное состояние.
Рассмотрим каталитическую реакцию между двумя молекулами А и В, при
которой образуется продукт Р (рис. 1.1). В начале цикла молекулы А и В связываются с катализатором, затем внутри этого комплекса идет реакция с формированием продукта Р, который также связан с катализатором. На последнем
шаге продукт Р отделяется от катализатора, который при завершении цикла
возвращается в исходное состояние.
Рис. 1.1. Каждая каталитическая
реакция представляет собой последовательность элементарных
шагов, в которой реагирующие
молекулы связываются с катализатором, вступают в реакцию, находясь на нем, после чего продукт
отделяется от катализатора, высвобождая его для нового цикла
Чтобы понять, как катализатор ускоряет реакцию, давайте рассмотрим приведенную на рис. 1.2 диаграмму потенциальных энергий для каталитической и
некаталитической реакций. Для некаталитических реакций данная диаграмма
представляет собой хорошо знакомый способ наглядного объяснения уравнения Аррениуса: реакция между молекулами А и В произойдет, если они обладают энергией, достаточной для преодоления активационного барьера, показанного на рис. 1.2. Изменение свободной энергии Гиббса при переходе от
реагентов А + В к продукту Р равно A G.
Каталитическая реакция начинается с самопроизвольного связывания реагентов А и В и катализатора. Следовательно, образование такого комплекса
представляет собой экзотермическую реакцию, при которой свободная энергия системы понижается. Затем идет реакция между молекулами А и В, находящимися на поверхности катализатора. Этот процесс характеризуется собственной энергией активации, которая, однако, существенно ниже энергии актива¬
о
О г рыв
Связывание
Реакция
Глава 1. Введение в катализ
ции некатализируемой реакции. Наконец, идет отрыв продукта Р от катализатора, который является эндотермической ступенью.
Рис. 1.2. Диаграмма потенциальной энергии для каталитической
реакции, в которой реагенты и
продукт являются газами, а катализатор — твердым телом. Заметим, что энергетический барьер
существенно выше для некаталитической реакции
Энергетической диаграмме на рис. 1.2 можно сопоставить несколько важных моментов:
• катализатор предлагает альтернативный путь для реакции, который, очевидно, более сложен, но энергетически предпочтителен;
• у каталитической реакции энергия активации существенно меньше, чем у
некаталитической (этот факт будет подробно объяснен в гл. 2);
• полные изменения свободной энергии для каталитической и некаталитической реакций совпадают. Это означает, что присутствие катализатора не влияет на константу равновесия полной реакции А + В Р. То есть если реакция
не является энергетически выгодной, катализатор не может повлиять на эту
ситуацию. Катализатор изменяет кинетику, но не термодинамику реакции;
• катализатор ускоряет прямую и обратную реакции в одинаковой степени.
Другими словам, если катализатор ускоряет образование продукта Р из реагентов А и В, он то же самое делает и с разложением продукта Р на компоненты А
и В.
Становится сразу очевидным, при каких условиях присутствие катализатора не влияет на реагенты или продукт:
• при слабой связи реагентов с катализатором, когда практически невозможна конверсия А и В в продукт;
• наоборот, если связь катализатора с одним из реагентов, например, А
является слишком сильной, то все активные центры катализатора будут заняты
реагентом А, и второй компонент В не сможет с ним прореагировать с образованием продукта. Если имеется сильная связь между катализатором и реагентами А и В, то такое состояние может быть настолько устойчивым, что реакция
становится практически невозможной. В терминах рис. 1.2 это означает, что
второй уровень лежит настолько глубоко, что энергия активации для образования продукта Р на поверхности катализатора становится очень высокой. В этом
случае говорят, что катализатор отравляется одним из реагентов;
1.2. Катализаторами могут быть атомы, молекулы, энзимы и поверхности твердых тел
• аналогично, если продукт Р сильно связывается с катализатором, так что
он практически не может от него отделиться, то говорят об отравлении катализатора продуктом.
Таким образом, как это можно было интуитивно предсказать, катализатор
будет эффективен в реакции, если реагенты и продукт связываются с ним и не
слабо, и не сильно. Данное утверждение представляет собой произвольно сформулированный принцип Сабатье, который в более строгой формулировке будет
дан в гл. 2 и детально обсужден в подразд. 6.5.3.5.
До сих пор мы трактовали катализатор как абстрактное тело, давайте теперь посмотрим, какие вообще бывают катализаторы.
1.2. КАТАЛИЗАТОРАМИ МОГУТ БЫТЬ
АТОМЫ, МОЛЕКУЛЫ, ЭНЗИМЫ
И ПОВЕРХНОСТИ ТВЕРДЫХ ТЕЛ
Катализаторы обладают многообразием форм от атомов и молекул
до больших структур типа цеолитов или энзимов. Каталитические реакции могут
протекать в жидких и газообразных средах или на поверхности твердых тел.
Формирование катализатора в оптимальной для него форме, доскональное изучение его состава и формы — это важные этапы теории и практики каталитического синтеза, которые будут описаны в последующих главах.
В катализе традиционно выделяют три основных вида — гомогенный, гетерогенный и биокатализ. Мы проиллюстрируем их отдельными примерами.
1.2.1. Гомогенный катализ
В гомогенном катализе и катализатор, и реагенты находятся в одном фазовом состоянии, например, либо все они представляют собой газовые
молекулы, либо, что встречается более часто, образуют жидкую фазу. Один их
простейших примеров связан с химией атмосферы. Озон в атмосфере разлагается, помимо других возможных путей, через реакцию с атомами хлора:
С1 + 03 -> СЮ3,
СЮ3 ->С10 + 02,
СЮ + О -> С1 + 02
или суммарно
03 +0 -> 202.
Озон может разлагаться самопроизвольно или под действием света, но атомы С1 ускоряют этот процесс многократно. Поскольку атом С1 выходит из
реакционного цикла неизменным, его можно рассматривать как катализатор.
Глава 1. Введение в катализ
Так как и реагенты, и катализатор находятся в одной и той же газовой фазе, то
рассмотренный выше цикл представляет собой пример гомогенного катализа
(приведенная выше реакция сыграла определенную историческую роль в объяснении природы озоновых дыр).
В промышленности для гомогенного катализа используется большое число
соединений, с помощью которых получаются различные химические продукты. Один из многочисленных примеров — каталитическое карбонилирование
метанола до уксусной кислоты в растворе, где в качестве катализатора выступает комплекс [Rh(C0)2I2]_:
СН3ОН + СО -> СН3СООН.
Органометаллические комплексы для гомогенного катализа, с помощью
которого часто получают специфические фармацевтические соединения, синтезируют, используя управление на молекулярном уровне, так что осмысленный выбор лигандов позволяет «направить» реагирующие молекулы к желаемому продукту.
1.2.2.
Биокатализ
Энзимы являются природными катализаторами. Сейчас мы будем
трактовать энзимы как большие белковые молекулы, структурная организация
которых предопределяет появление активных центров со специфической формой (рис. 1.3). Обладая формой, оптимальным образом переводящей молекулы-реагенты (которые обычно называются субстратом) в конфигурацию, наиболее подходящую для реакции, энзимы являются в высокой степени специфичными и эффективными катализаторами. Например, энзим каталаза служит
катализатором разложения перекиси водорода на воду и кислород:
> Н20 + О-),
1ТЯ пя^я I I ’
2Н,0,
^ ^ катги
обеспечивающим неправдоподобно высокую скорость реакции, до 107 молекул
перекиси водорода в секунду!
; Субстрат 1
(^) оТрЬ1В
Субстрат 2
.о
Продукт
Энзим
Связывание
V
Реакция
о
Рис. 1.3. Схематическое представление
реакции, катализируемой энзимами.
Строение энзимов часто соответствует
форме молекул субстратов, которые они
связывают, или переходных комплексов, образуемых в катализируемых ими
реакциях. Энзимы представляют собой
высокоэффективные катализаторы и
служат мощным источником вдохновения при разработке промышленных
катализаторов
1.2. Катализаторами могут быть атомы, молекулы, энзимы и поверхности твердых тел
Энзимы обеспечивают значения скорости реакций в биологических системах, необходимые для обеспечения их жизнедеятельности. К этим реакциям
относятся формирование белков и молекул ДНК, разрушение молекул и накопление энергии в сахарах. Пример, который, возможно, имеет прямое отношение к студентам, — это разложение этилового спирта до уксусного альдегида
в организме с помощью энзима — алкогельдегидрогеназы. Некоторые люди не
переносят алкоголь (что проявляется в сильном покраснении лица после принятия даже малых доз), поскольку у них имеется недостаток энзима, ответственного за разложение уксусного альдегида.
1.2.3. Гетерогенный катализ
В гетерогенном катализе твердые тела катализируют реакции молекул в газовых смесях или растворах. Поскольку твердые тела, если они не
являются пористыми, обычно непроницаемы для молекул, то каталитические
реакции, очевидно, происходят на поверхности. Для экономии часто очень
дорогих материалов (например, платины) катализаторы приготавливают в виде
частиц нанометрового размера, осажденных на инертные пористые носители
(рис. 1.4). Твердотельные катализаторы являются рабочей лошадкой химических и нефтехимических производств, и ниже мы рассмотрим много примеров
практического использования гетерогенного катализа.
Рис. 1.4. Катализаторы представляют собой наноматериалы, а катализ — нанотехнологию.
Если мы под нанотехнологией будем понимать раздел материаловедения, посвященный регулированию свойств материалов на нанометровом уровне, то катализ можно считать отраслью, в которой наноматериалы используются на коммерческой основе уже в течение столетия. Разработано большое число методов
получения малых частиц для гетерогенного катализа, обеспечивающих их высокую устойчивость, при которой они способны выдержать жесткие условия работы в промышленных реакторах. Современный катализ, по определению, является нанотехнологией [1]
Глава 1. Введение в катализ
В качестве вводного примера мы рассмотрим ключевую реакцию в очистке выхлопных газов автомобилей — каталитическое окисление СО на поверхности частиц из благородных металлов типа платины, палладия и родия.
Чтобы описать процесс, представим, что на поверхности металла имеются
активные центры, которые мы будем обозначать звездочкой «*». Их определение будет дано несколько позже. Цикл каталитической реакции начинается с
адсорбции СО и 02 на поверхности платины, где молекулы 02 диссоциируют
на два атома О:
02 +2*
С0 + *
* 20%
СО*.
Здесь символ X* означает, что атом или молекула адсорбированы на активном
центре *.
Адсорбированные атом О и молекула СО реагируют с образованием молекулы С02, которая является достаточно стабильной и инертной, слабо взаимодействует с поверхностью платины и практически мгновенно десорбируется:
СО + О*
С02 +2\
Заметим, что на последней стадии активные центры на поверхности катализатора освобождаются и они могут участвовать в следующем реакционном
цикле. На рис. 1.5 показана диаграмма потенциальной энергии для реакционного цикла.
Рис. 1.5. Реакционный цикл и диаграмма потенциальной энергии
для каталитического окисления СО
кислородом 02
В какой момент катализатор оказывает существенное влияние на реакцию?
Предположим, что мы проводим реакцию в газовой фазе без катализатора.
Реакция пойдет, если мы сделаем температуру достаточно высокой, при которой молекулы О2 будут диссоциировать на атомы О (радикалы). Эти радикалы
мгновенно прореагируют с молекулами СО, что приведет к образованию С02.
1.3. Зачем необходим катализ?
Энергия активации газофазной реакции будет примерно равна энергии, необходимой для разрыва связи О—О в молекуле 02, то есть примерно 500 кДж/моль.
В каталитической реакции, однако, молекулы 02 диссоциируют на поверхности катализатора легко, практически с нулевой энергией активации. В этом случае энергия активации определяется в основном реакцией между молекулами
СО и атомами О и составляет величину порядка 50—100 кДж/моль. На десорбцию продукта реакции молекул С02 затрачивается только около 15—30 кДж/моль
(в зависимости от типа катализатора и его структуры). Сравнивая каталитическую и некаталитическую реакции, легко заметить, что наиболее «трудная»
стадия гомогенной газофазной реакции представляет собой разрыв связи О—О,
который под влиянием катализатора осуществляется легко. В последнем случае легкость, с которой образуются молекулы С02, в конечном итоге определяет скорость реакции по превращению СО и 02 в С02. Это — типичная
ситуация для каталитических реакций и фраза «катализатор разрывает одни
связи и позволяет образовываться другим связям» правильно отражает суть
каталитического процесса. «Полезное» действие катализатора состоит в разрыве сильных связей, тогда как последующие стадии без катализатора могут
протекать даже быстрее (это, конечно, гипотетическая ситуация). В гл. 6 мы
подробно обсудим, как поверхность способствует разрыву межмолекулярных
связей.
1.3. ЗАЧЕМ НЕОБХОДИМ КАТАЛИЗ?
Химическая промышленность 20-го столетия не перешла бы на
современный уровень, если бы она базировалась только на некаталитичских
стехиометрических реакциях. В общем случае регулировать скорость реакции
можно, изменяя температуру, концентрации, давление и время контакта реагентов. Повышая температуру и давление, можно достичь приемлемой скорости стехиометрической реакции, однако для этого потребовалось бы создавать
все более дорогие и сложные по конструкции реакторы, гарантирующие безопасность производства. Кроме того, имеются термодинамические ограничения
на условия, при которых данный продукт может быть произведен. Например,
конверсия N2 и Н2 в аммиак практически невозможна при температуре выше
600 °С. Однако для разрыва очень сильной связи N=N в молекулах N2 требуются более высокие температуры. Без катализа многие реакции, ставшие обычными для химического производства, были бы либо невозможны, либо экономически невыгодны.
Катализаторы увеличивают скорость реакции на несколько порядков и обеспечивают их проведение в более мягком термодинамическом режиме, при более низких температурах и давлениях. Эти факторы в сочетании с оптимизированными схемами реактора и производства в целом играют ключевую роль в
снижении капиталовложений и цены химического производства. Но это далеко не все преимущества каталитических процессов.
Глава 1. Введение в катализ
1.3.1. Катализ и экологически чистая химия
Технология называется экологически чистой, если в ней сырье перерабатывается эффективно, при этом исключается использование токсичных
и опасных реагентов и растворителей, а доля отходов или нежелательных продуктов минимизирована. Технологии, основанные на катализе, обычно удовлетворяют этим
критериям. Наглядным примером служит реакция селективного окисления этилена, в результате которой образуется эпоксиэтилен
(рис. 1.6), очень важный интермедиат, из которого получают этиленгликоль (антифриз),
различные полиэфиры и полиуретаны.
Старый некаталитической способ получения эпоксиэтилена (называющийся эпихлор-
гидриновым процессом) основан на трехстадийном синтезе:
С12 + NaOH -> НОС1 + NaCl, (1.1)
С2Н2 + НОС1 -> СН2С1—СН2ОН (эпихлоргидрин), (1.2)
СН2С1—СН,ОН + ± Са (ОН), -»I СаС12 + С2Н40 + Н20 (1.3)
или суммарно
С12 + NaOH +1 Са(ОН)2 + С2Н4 -> С2Н40 +1 СаС12 + NaCl + Н20.
Таким образом, на каждую молекулу эпоксиэтилена получается одна молекула соли, что создает проблему отходов, которая традиционно решалась путем
их сброса в реки. Сейчас такая практика, безусловно, является неприемлемой.
Каталитический же процесс является простым и «чистым», при нем образуется лишь небольшое количество С02. Используя серебро, активированное
небольшим количеством хлора, как катализатор, можно получить эпоксиэтилен непосредственно из С2Н4 и 02 при селективности процесса около 90 % с
превращением порядка 10 % этилена в С02. В настоящее время во всех производствах эпоксиэтилена используют катализаторы.
1.3.2. Атомарная эффективность, Е-факторы
и «дружелюбие» к окружающей среде
Большое число процессов органического синтеза основано на стехиометрическом окислении углеводородов с использованием дихромата натрия и перманганата калия или гидрировании органических соединений с
Рис. 1.6. Эпоксиэтилен — важный
интермедиат в химической промышленности
1.3. Зачем необходим катализ? л
участием щелочных металлов, борогидридов металлов или металлического
цинка. Широко распространены также процессы нитрирования ароматических соединений, в которых используются кислоты H2S04 и HN03, или ацили-
рование органических соединений в присутствии А1С13, в результате которых
в качестве побочных продуктов формируется большое количество неорганических солей.
Тонкая химическая технология преимущественно (но не исключительно!)
основана на гомогенном катализе, и растворители, задействованные в нем,
представляют дополнительную «головную боль» для окружающей среды. Согласно Шелдону [2], лучший растворитель — отсутствующий растворитель,
не если растворения избежать нельзя, то оптимальным кандидатом является
вода.
Шелдон ввел несколько показателей, позволяющих оценить эффективность
и степень воздействия на окружающую среду той или иной реакции. Атомарная эффективность равна отношению молекулярного веса целевого продукта к
полному молекулярному весу всех продуктов реакции. Например, обычное окисление вторичных спиртов
ЗС6Н5—СНОН—СН3 +2Сг03 +3H2S04 ->
-> ЗС6Н5—СО—СН3 +Cr2(S04)3 +6Н20
характеризуется атомарной эффективностью, равной 360/860 = 42 %. Каталитический же цикл
С6Н5—СНОН—СН3 +±02 с6н5—со—сн3 +Н20
имеет атомарную эффективность, равную 120/138 = 87 %, и в нем побочным
продуктом является вода. Обратный процесс каталитического гидрирования
идет со 100 %-й атомарной эффективностью:
с6н5—со—СН3 + Н2 -> с6н5—СНОН—сн3,
как и карбонилирование этой молекулы
с6н5—со—CH3 +CO -> с6н5 —сн (сн3) соон.
Другим полезным показателем безопасности для окружающей среды является is-фактор, равный отношению весов побочного субпродукта и целевого продукта. Как показано в табл. 1.1, производство, связанное с тонкой химической технологией или фармацевтикой, характеризуется большой весовой долей побочных субпродуктов. Атомарная эффективность и is-фактор могут
быть рассчитаны друг через друга, однако на практике is-фактор всегда оказывается выше теоретического, поскольку выход продукта всегда меньше оптимального, а реагенты, как правило, используются в избытке. Следует также
учесть потери растворителей и, возможно, перерасход энергии и выбросы
отработанного С02.
Глава 1. Введение в катализ
Таблица 1.1. Степень воздействия на окружающую среду в различных
сегментах химической промышленности [3]
Сегмент промышленности
Тоннаж производства
is-фактор,
кг отходов/кг продукта
Переработка нефти
i_
о
ос
<0,1
Объемное химическое
производство
0
-1^
1
ъ
<1-5
Тонкая химическая
технология
102-104
5-50
Фармацевтика
10— 1 о3
25—>100
Чтобы отразить тот факт, что отходы производства не просто составляют
некоторое количество ненужного материала, а влияют на окружающую среду;
Шелдон ввел коэффициент воздействия на окружающую среду EQ, равный
произведению Е-фактора и коэффициента «недружелюбное™» Q, которому
может быть приписано значение, характеризующее степень нежелательности
данного субпродукта. Например, Q = 0 для чистой воды, 1 для полезной соли
NaCl и 100—1000 для токсичных соединений. Очевидно, что каталитические
схемы производства, позволяющие исключить появление отходов, крайне желательны и экономические выгоды, которые, в частности, заключены в возможном снижении коэффициента недружелюбное™, являются хорошим стимулом для их развития. Предотвращение появления отходов предпочтительнее
их переработки.
1.3.3. Химическая индустрия
Каталитическое ускорение реакций позволяет осуществлять промышленно важные реакции с высокой эффективностью при приемлемых условиях. Каталитические циклы часто могут быть организованы так, что сырье
используется практически полностью при минимальном количестве отходов.
Неудивительно поэтому, что химическое производство в основной своей массе
основано на катализе: примерно 85—90 % всей продукции получается в каталитических процессах, и этот процент постоянно увеличивается.
В табл. 1.2 и 1.3 представлены наиболее важные химические продукты и
производства. В табл. 1.4 и рис. 1.7 приведены 50 основных производителей
химической продукции из разных стран.
Очевидно, что студент-химик, думающий о карьере в химической промышленности или о создании химических производств, должен иметь представление о катализе. Вот почему авторы в своих университетах обучают студентов
основам катализа на ранней стадии химического курса.
1.3. Зачем необходим катализ?
Таблица 1.2. Наболее значимые процессы в гетерогенном катализе
Реакция
Катализатор
Каталитический крекинг сырой нефти
Цеолиты
Гидроочистка нефти
Со—Mo, Ni—Мо,
Ni—W (сульфидная форма)
Конверсия тяжелого бензина в легкий
Pt, Pt—Re, Pt—Ir
Алкилирование
H2S04, HF,
твердые кислоты
Полимеризация этилена, пропилена
Cr, TiClA/MgCl2
Эпоксидирование этилена
(получение оксиэтилена)
Ag
Производство винилхлорида
(этилен + С12)
Си (хлориды)
Конверсия с водяным паром метана
в СО + Н2
Ni
Конверсия водяного газа
Fe (оксид), Cu—ZnO
Синтез метана
Ni
Синтез аммиака
Fe
Окисление аммиака до N0 и NH03
Pt—Re
Получение акрилонитрила из пропилена
и аммиака
Bi—Mo, Fe—Sb (оксиды)
Гидрирование растительных масел
Ni
Производство серной кислоты
V (оксид)
Окисление СО и углеводородов
(в выхлопных газах)
Pt, Pd
Восстановление NOx (в выхлопных газах)
Rh, оксид ванадия
Глава 1. Введение в катализ
Таблица 1.3а. Производство органических соединений в США (2005)
и средний его прирост за 10 лет (1995—2005)[4]
Органическое соединение
Производство, ктонн
% прироста
Этилен
23 974
1,2
Пропилен
15 333
2,8
Дихлорэтилен
11 308
3,7
Мочевина
5801
-2,4
Этилбензол
5251
-1,6
Стирол
5042
-0,2
Кумен
3509
3,2
Этиленоксид
3166
-0,9
1,3-Бутадиен
2046
2,1
Вин и л ацетат
1327
0,1
Акрилонитрил
1323
-0,9
Анилин
964
4,3
Бензол (1000 литров)
7574
-0,8
Таблица 1.36. Производство неорганических соединений в США (2005)
и средний его прирост за 10 лет (1995—2005) [4]
Неорганическое соединение
Производство, ктонн
% прироста
Серная кислота
36 520
-i,6
Фосфорная кислота
11599
-0,3
Хлор
10175
-1,0
Аммиак
9775
-4,7
Гидроксид натрия
8384
-2,1
Нитрат аммония
6353
-1,9
Азотная кислота
6328
-2,3
Соляная кислота
4406
2,2
Сульфат аммония
2578
0,7
1.3. Зачем необходим катализ?
Таблица 1.3в. Производство полимеров и пластиков в США (2005)
и средний его прирост за 10 лет (1995—2005) [4]
Полимеры, пластики
Производство, ктонн
% прироста
Полиэтилен
низкой плотности
3558
0,3
Полиэтилен линейный
низкой плотности
5395
8,5
Полиэтилен высокой
плотности
7328
3,7
Полипропилен
8149
5,1
Полистирол
2855
1,1
Сополимеры стирола
(АБС и т. д.)
1413
0,3
Полиамин,нейлон
568
2,1
П ол и ви н и л хл ор и д
и сополимеры
6921
2,2
Таблица 1.4. 50 ведущих компаний по производству химической продукции [4]
Рейтинг
Компания
Страна
Общий
объем
продаж,
млн $
Химическая продукция,
млн $
Химическая продукция,
%
2005
2004
2001
1998
1
1
1
4
Доу
Кемикал
США
46 307
46 307
100
2
2
3
1
БАСФ
Германия
52 271
43 682
82
3
4
7
5
Ройал Датч
Шелл
Велико¬
британия/
Нидерланды
318145
349 996
11
4
5
6
8
Эксон
Мобил
США
259 883
31 186
12
5
6
5
—
Тоталь
Франция
173713
27794
16
6
3
2
2
Дюпон
США
28 144
25 330
90
7
9
26
—
Чайна
Петролиум
&Кемикал
Китай
100 576
21 121
21
Глава 1. Введение в катализ
Продолжение табл. 1.4
Рейтинг
Компания
Страна
Общий
объем
продаж,
млн $
Химическая продукция,
млн $
Химическая продукция,
%
2005
2004
2001
1998
8
8
4
3
Байер
Германия
33 859
20654
61
9
7
9
11
Бритиш
Петролиум
Велико¬
британия
25 7838
20627
8
10
11
18
30
САБИ К
Саудовская
Аравия
20 821
18 947
91
11
13
30
46
Формоза
Пластике
Тайвань
31 775
18 747
59
12
—
50
—
JI ионделл
Кемикал
США
18606
18 606
100
13
10
14
31
Митцубиси
Кемикал
Япония
21 884
17945
82
14
12
8
35
Дегусса
Германия
14630
14630
100
15
16
13
43
Мицуи
Кеми кал з
Япония
13 372
13 372
100
16
15
12
29
Хантсман
Корпорейшн
США
12 962
12 962
100
17
32
42
—
Инеос Груп
Велико¬
британия
12 400
12 400
100
18
14
11
19
АКЗО
Нобель
Нидерланды
16 107
11 758
73
19
19
20
13
Сумитомо
Кемикал
Япония
14 146
11 458
81
20
17
22
25
Аир Ликвид
Франция
12941
11 388
88
21
20
31
24
Торей
Индастриез
Япония
12 985
11 297
87
22
24
23
47
Шеврон
Филлипс
США
10 707
10 707
100
23
18
10
6
АйСиАй
Велико¬
британия
10583
10583
100
24
—
—
—
Базелл
Нидерланды
10582
10 582
100
1.3. Зачем необходим катализ?
Продолжение табл. 1.4
Рейтинг
Компания
Страна
Общий
объем
продаж,
млн $
Химическая продукция,
млн $
Химическая продукция,
%
2005
2004
2001
1998
25
25
37
—
Шин-Этцу
Кеми калз
Япония
10 244
10 244
100
26
21
19
12
дсм
Нидерланды
10 202
10 202
100
27
23
15
17
Дайнипон
Инк&
Кеми калз
Япония
9126
9126
100
28
27
—
—
Ланкснесс
Германия
8901
8901
100
29
30
34
27
БОС
Велико¬
британия
8358
8358
100
30
28
24
—
ПиПиДжи
Индастриез
США
10210
7964
78
31
29
35
41
Асахи
Казеи
Япония
13 667
7927
58
32
40
41
26
Солвей
Бельгия
10585
7833
74
33
34
46
33
ЕНИ
Италия
86 522
7787
9
34
31
29
36
Аир
Продактс
США
8151
7743
95
35
35
36
32
Праксаир
США
7656
7656
100
36
42
—
—
Йара
Норвегия
7168
7168
100
37
41
40
44
Ром
энд Хаас
США
8027
7064
88
38
36
33
34
Эстман
Кемикал
США
7059
7059
100
39
37
38
22
Релианс
Индастриез
Индия
18661
6718
36
40
26
16
14
Дженерал
Электрик
США
165 150
6606
4
41
33
27
15
Клариант
Швейцария
6566
6566
100
42
39
—
—
Сасол
ЮАР
10912
6547
60
Глава 1. Введение в катализ
Окончание табл. 1.4
Рейтинг
Компания
Страна
Общий
объем
продаж,
млн $
Химическая продукция,
млн $
Химическая продукция,
%
2005
2004
2001
1998
43
38
21
—
Род и а
Франция
6330
6330
100
44
43
32
—
Сингета
Швейцария
8086
6307
78
45
49
44
—
Целанезе
США
6070
6070
100
46
45
48
48
Бореалис
Дания
5992
5992
100
47
46
45
21
Циба
Специалтиес
Швейцария
5955
5955
100
48
48
—
—
Нова
Кеми калз
Канада
5617
5617
100
49
47
28
—
Теджин
Япония
8486
5516
65
50
42
—
—
ЭлДжи
Кеми
Южная
Корея
7291
5468
75
2001
2005
Япония
13*
Другие
6%
Европа
48%
404,4 биллионов
$ США
Япония
13%
Другие
13%
США
29%
665,8 биллионов
$ США
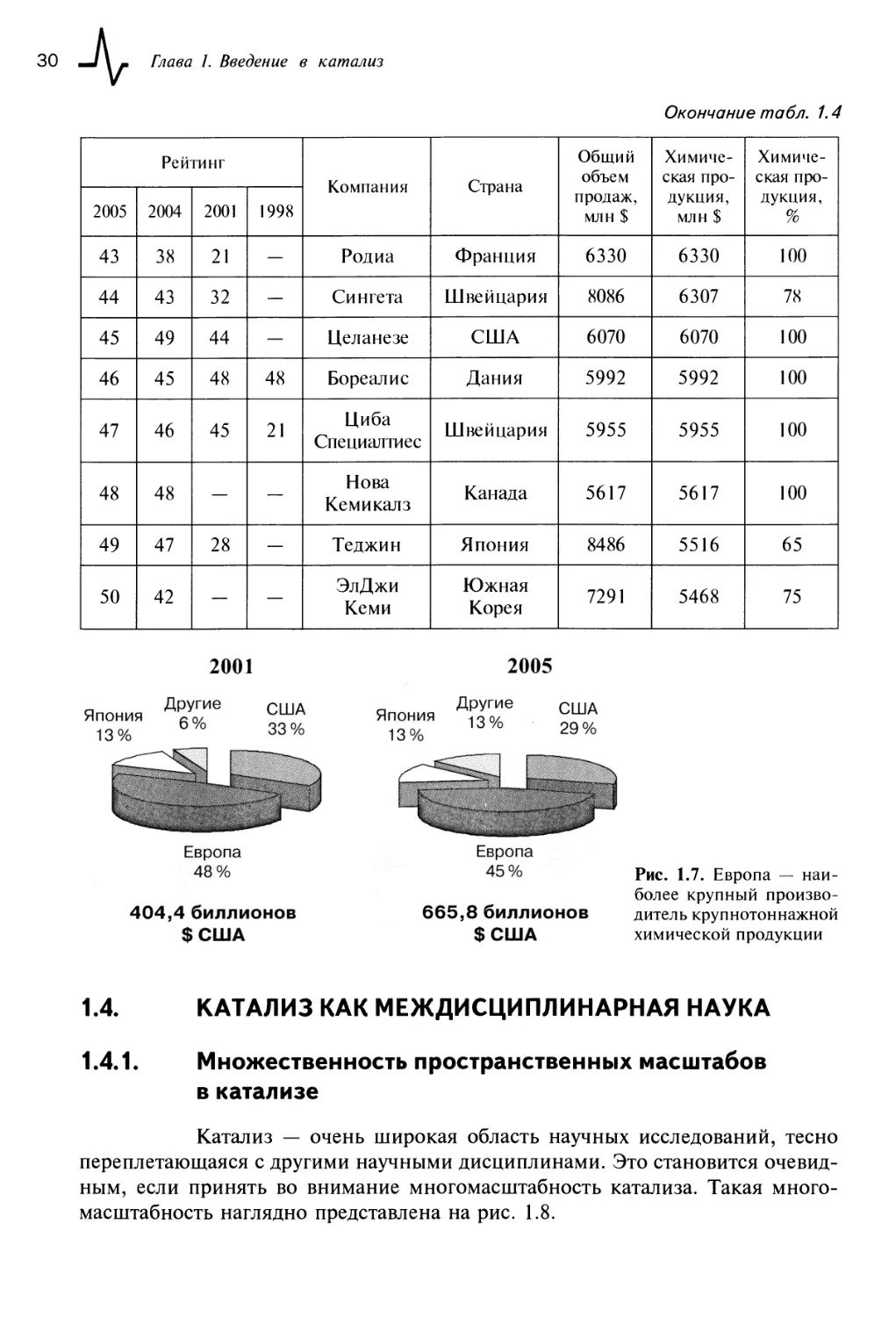
Рис. 1.7. Европа — наиболее крупный производитель крупнотоннажной
химической продукции
1.4. КАТАЛИЗ КАК МЕЖДИСЦИПЛИНАРНАЯ НАУКА
1.4.1. Множественность пространственных масштабов
в катализе
Катализ — очень широкая область научных исследований, тесно
переплетающаяся с другими научными дисциплинами. Это становится очевидным, если принять во внимание многомасштабность катализа. Такая много-
масштабность наглядно представлена на рис. 1.8.
1.4. Катализ как междисциплинарная наука
В рассмотренных выше примерах связывание реагентов с катализатором с
образованием реакционного комплекса и отрыв продукта от катализатора происходят на молекулярном уровне. В гетерогенном и гомогенном катализе и
катализе энзимами именно на этом уровне происходят химические процессы.
Для выяснения их сути необходимо понимание на элементарном уровне закономерностей разрыва связей в реагентах и их образования в продукте реакции,
которое достигается с помощью уникальных экспериментальных устройств и
современного теоретического моделирования. Эти процессы являются объектом исследования в спектроскопии, численном моделировании, кинетике и
теории элементарных процессов. Здесь все превращения происходят на субна-
нометровом уровне. Результаты научных исследований этих процессов обычно
публикуются в общетеоретических журналах по химии, физической химии и
физике.
Формованные частицы
катализатора
1 нм
Поверхность у.'
катализатора j
\
Каталитически активные
частицы на носителе
Микроскопический Мезоскопический
слои в реакторе
Макроскопический
Рис. 1.8. Различные масштабы, отражающие каталитический процесс от субнанометро-
вых размеров атомов и молекул до макроскопических масштабов промышленного реактора
Следующий уровень исследований относится к каталитически активным
частицам, характерный размер которых составляет от 1 до 10 нм, и процессам,
протекающим внутри пор частиц носителя (размер частиц около 1 мкм). На
этом уровне исследуются размер, форма, структура и состав активных частиц,
в частности, строение поверхности и предпринимаются попытки связать эти
характеристики с каталитической активностью частиц. Хотя мы будем иметь
дело в основном с гетерогенным катализом, все же отметим, что выяснение
механизма прикрепления каталитических молекул или даже энзимов к носителям также представляет огромный интерес в гомогенном и биокатализе. Эти
проблемы относятся к области формирования катализаторов, определения их
Глава 1. Введение в катализ
характеристик, тестирования в лабораторных условиях и изучения их механических свойств. На скорость формирования продукта могут оказать влияние
транспортные процессы, связанные с диффузией молекул внутри пор, поэтому
им на этом уровне также уделяется серьезное внимание. На этом уровне проводятся и академические исследования, и испытания на промышленных установках. Результаты исследований процессов, протекающих на мезоскопическом
уровне, обычно публикуются в специализированных журналах по катализу.
Следующий уровень приходится на формованные катализаторы, которые
выглядят как шарики, стерженьки или призмы с характерным размером от
нескольких миллиметров до нескольких сантиметров и иногда даже более. Создание таких катализаторов относится к области материаловедения. Здесь требуется создать образцы с определенной пористостью и высокими прочностью
и износостойкостью, способными обеспечить их продолжительную работу в
промышленных реакторах. Эта область катализа имеет дело (но не исключительно) с промышленностью, в частности с производителями катализаторов,
поэтому результаты проводимых в этом направлении исследований по большей части патентуются.
Макроскопический уровень относится непосредственно к реакторам, размер
которых в лабораторных условиях может достигать 25 см, а в промышленных
установках — 10 м. Катализатор составляет сердце реактора. Но собственно катализ представляет собой лишь часть в совокупности дисциплин, задействованных при конструировании реакторов. Так, при создании реакторов необходимо
обеспечить их работоспособность при высоких давлениях, надежное управление температурой реакции, хороший контакт между реагентами и катализатором, эффективное удаление продукта, коррозионную стойкость конструкций,
низкие потери энергии и безопасность технологического процесса. При описании кинетики каталитических реакторов на масштабе реактора в целом внешние факторы, связанные с тепло- и массопереносом реагентов и продуктов
через слой катализатора, становятся такими же внутренними, как и реакции
молекул на активном центре катализатора. При этом механическая стойкость,
чувствительность к следовым примесям в реагентах и деградация катализатора
при высоких температурах также важны, как и его активность и селективность.
Результаты исследования этих аспектов катализа отражены в научных журналах по химической технологии и патентах.
1.4.2. Временные масштабы в катализе
Характерные времена, на которых развивается каталитический процесс, изменяются в согласии с пространственными масштабами, обсуждавшимися выше. Активация молекул и разрыв связей в них происходит за пикосекунды. Завершение внутреннего цикла реакции от этапа формирования комплекса между реагентами и катализатором до отделения продукта может занимать
время от нескольких микросекунд для наиболее быстрых реакций с участием
энзимов до нескольких минут в случае сложных реакций на поверхности. На
1.6. Катализ в периодических изданиях
мезоскопическом уровне диффузия к формованным частицам, внутри них и от
них может занимать время от нескольких секунд до нескольких минут. Время
нахождения молекул в реакторе может изменяться от нескольких секунд до
(формально) бесконечности, если при реакции образуются побочные субпродукты типа кокса, которые остаются на катализаторе.
1.5. ПРЕДМЕТ КНИГИ
Данная книга акцентирована на фундаментальных аспектах каталитических реакций, рассматриваемых на молекулярном и мезоскопическом
уровнях. Поскольку катализ приводит к ускорению реакций, он относится к
явлениям кинетической природы. По этой причине мы начинаем эту книгу с
главы, посвященной кинетике и описывающей закономерности влияния внешних параметров — концентрации, давления и температуры — на скорости
реакций каталитического цикла. За ней следует глава, в которой излагаются
основы теории скоростей реакций, позволяющей установить связь между свойствами молекул и их реакционной способностью. Для выявления связи между
фундаментальными процессами, протекающими при катализе, и реальной жизнью мы показываем, как катализаторы выглядят на мезоскопическом уровне,
как их получают и как определяют их характеристики. Благодаря впечатляющим достижениям теоретической химии и развитию вычислительных методов
в последнее десятилетие стало возможным, пусть пока еще только для идеальных случаев, находить скорости реакций из первых принципов. Мы намереваемся дать читателю основы знаний, которые позволят ему почувствовать, что
представляет собой современный катализ. Для этого мы рассмотрим такие явления, как адсорбция и реакции на поверхности, а также опишем методы их
исследования. Наконец, в книге будут рассмотрены несколько промышленно
важных каталитических процессов как с прикладной, так и с фундаментальной
точек зрения. Основной акцент будет сделан на выяснение основных принципов и тенденций развития дисциплины, а не на тонких деталях, что позволит
читателю получить базовые знания, достаточные для понимания специализированной литературы по фундаментальным исследованиям катализа. Приведенная в книге литература включает основные монографии по катализу и связанным с ним дисциплинам.
1.6. КАТАЛИЗ В ПЕРИОДИЧЕСКИХ ИЗДАНИЯХ
Результаты исследований каталитических процессов обычно публикуются в общетеоретических и специализированных журналах, охватывающих
широкий спектр исследований. Это служит отражением междисциплинарности
данной области исследований. Представим периодическую печать, следуя приведенной на рис. 1.8 схеме. Результаты исследований на микроскопическом
Глава 1. Введение в катализ
уровне, связанные с фундаментальным изучением адсорбированных молекул и
элементарных реакций, обычно публикуются в общетеоретических журналах, к
которым относятся The Journal of Chemical Physics, The Journal of Physical Chemistry,
Physical Chemistry-Chemical Physics, Surface Science, Langmuir и Physical Review,
иногда подобные публикации появляются в журналах Nature и Science. Есть они
и в специализированных журналах Journal of Catalysis и Catalysis Letters.
Мезоскопический уровень исследования каталитических процессов охвачен чисто «каталитическими» журналами: Applied Catalysis, The Journal of Catalysis,
Catalysis Letters, Topics in Catalysis, Catalysis Today, Microporous Materials and Zeolite,
хотя иногда соответствующие статьи появляются в журналах The Journal of Physical
Chemistry и Physical Chemistry-Chemical Physics и ряде других изданий.
Макроскопический уровень обсуждается в журналах по химической технологии, наиболее известными из которых являются Chemical Engineering Science,
Industrial & Engineering Chemistry Research и Journal of American Institute of Chemical
Engineering (AIChE Journal).
Неожиданные результаты, которые, возможно, даже еще до конца не поняты, но представляют несомненный интерес для сообщества исследователей
катализа, публикуются в виде писем, заметок и «быстрых сообщений». Есть
специализированные журналы для «писем»: Chemical Communication, Catalysis
Letters, Chemical Physics Letters и Physical Review Letters, некоторые журналы содержат специальные рубрики писем, типа приоритетных сообщений в The Journal
of Catalysis.
Кроме того, имеются обзорные журналы высокого уровня, в которых публикуются обзорные статьи по состоянию исследований в отдельных областях.
В журналах Advances in Catalysis, Catalysis Reviews: Science & Engineering и Catalysis,
Special Periodical Reports публикуются наиболее обстоятельные обзоры. Красочный
журнал CaTTech публикует короткие обзорные статьи, а также новости сообщества исследователей катализа. Эту же важную роль исполняет рубрика
«Краткие новости» журнала Applied Catalysis.
Как и во всех научных областях, по результатам конференций издаются
сборники трудов, кратко отражающие содержание докладов. Однако такие сборники становятся все менее популярными среди исследователей. Это связано и
с менее строгим рецензированием этих работ, и с ограниченным объемом публикуемой информации, лишающими значимости эти публикации. И часто работы, опубликованные в дорогих сборниках, появляются в периодических журналах, превращая сборники в пустую трату усилий и денег.
Средним между трудами конференции, обзорами и регулярными статьями
являются тематические тома, издаваемые журналами Catalysis Today и Topics in
Catalysis. Вместо издания всех работ, представленных на конференции, главный редактор в этом случае производит отбор интересных для практики докладов и предлагает их авторам сделать не очень короткую статью, отражающую
содержание их исследований. Такие тематические тома часто несут ценную
информацию о состоянии дел в определенной области, а уровень публикаций
при этом существенно превышает средний уровень трудов конференции.
1.6. Катализ в периодических изданиях
Для отражения уровней обращения к публикациям в различных журналах и
их цитирования Институт научной информации (Филадельфия, США) ввел два
интересных показателя (Journal Citation Reports, Science Edition). Один из них —
импакт-фактор данного журнала, равный числу цитирований в течение года
статей, опубликованных в нем в предыдущие два года. Так, например, если
каждая статья, опубликованная в 1998 или 1999 г. была процитирована один
раз в 2000 г., то импакт-фактор журнала в этом году равен 1. Часто авторы
выбирают для публикации журнал, основываясь на его импакт-факторе. Для
специализированных журналов, в частности, посвященных катализу, импакт-
фактор, равный 3 или больше, рассматривается как высокий. Обзорные журналы обычно имеют высокий импакт-фактор, тогда как журналы, где публикуются письма и краткие сообщения, — более низкий.
Менее значимым, но в действительности более информативным в отношении качества журнала является параметр, характеризующий период половины
жизни цитирования. Это период, измеряемый в годах и отсчитываемый назад
от текущего года, в течение которого число цитирований опубликованных ранее статей уменьшается вдвое. Длительный период половины жизни цитирования говорит о высоком уровне публикуемых статей, которые долго сохраняют
свою значимость для научного сообщества. С данными показателями следует
обращаться осторожно, поскольку новым журналам требуется много лет до
достижения значимого периода половины жизни цитирования, показывающего, что в этих журнала публикуются статьи, долго не утрачивающие своей ценности. В табл. 1.5 приведены импакт-факторы и периоды половины жизни
цитирования для некоторых журналов, относящихся к предмету книги.
Таблица 1.5. Импакт-факторы и периоды половины жизни цитирования
для некоторых журналов
Журнал
Импакт-фактор
Период
половины
жизни цитирования
2001
2002
2003
2004
2005
2005
Catalysis Reviews
8,474
6,455
5,708
8,000
5,312
10
Journal of Catalysis
3,293
3,118
3,276
4,063
4,780
8,4
Chemical Communication
3,902
4,038
4,031
3,997
4,426
5,9
The Journal of Physical
Chemistry (B)
3,379
3,611
3,679
3,834
4,033
4,2
Applied Catalysis В
Environmental
3,643
2,866
3,476
4,042
3,809
4,6
Microporous Materials
2,497
1,990
2,701
2,093
3,355
4,6
Глава 1. Введение в катализ
Окончание табл. 1.5
Журнал
Импакт-фактор
Период
половины
жизни цитирования
2001
2002
2003
2004
2005
2005
Advances in Catalysis
6,846
10,923
7,889
9,750
2,750
10
Applied Catalysis A General
2,258
1,915
2,825
2,378
2,728
4,9
Topics in Catalysis
2,136
1,648
2,178
2,493
2,547
4,4
Catalysis Today
2,333
2,146
2,627
3,108
2,365
6,1
Journal of Molecular
Catalysis — A Chemical
1,520
1,729
2,264
2,316
2,348
4,5
Catalysis Communications
1,890
2,098
2,6
Catalysis Letters
1,852
1,559
1,581
1,904
2,088
6,5
AIChE Journal
1,793
1,626
1,667
1,761
2,036
10
Chemical Engineering Science
1,547
1,224
1,562
1,655
1,735
8,7
Journal of Molecular
Catalysis — В Enzymatic
1,408
1,451
1,475
1,547
1,685
4,2
Industrial & Engineering
Chemistry Research
1,351
1,247
1,317
1,424
1,504
6,6
Reaction Kinetics and
Catalysis Letters
0,475
0,398
0,603
0,618
0,670
7,5
Studies in Surface Science
and Catalysis
1,265
3,468
—
0,489
0,307
7,9
JACS
6,079
6,201
6,516
6,903
7,419
8,2
Phys. Rev. Lett.
6,668
7,323
7,035
7,218
7,489
6,6
Phys. Rev. В
3,070
3,327
2,962
3,075
3,185
7,7
Science
23,329
28,956
29,781
31,853
30,927
7,3
Nature
27,955
30,432
30,979
32,182
29,273
7,5
Nature Materials
—
—
10,770
13,531
15,941
2,2
Angewandte Chemie
8,255
7,671
8,427
9,161
9,596
5,2
/. 7. Основные учебники по катализу
1.7. ОСНОВНЫЕ УЧЕБНИКИ ПО КАТАЛИЗУ
1. Bowker М. The Basis and Applications of Heterogeneous Catalysis. Oxford:
Oxford Univ. Press, 1998.
2. Thomas J. M, Thomas W.J. Principles and Practice of Heterogeneous Catalysis.
Weinheim: WCH, 1997.
3. Handbook of Heterogeneous Catalysis. / Eds. Ertle G. Knozinger H., Weitkamp
J. Weinheim: WCH, 1997.
4. Somorjai G.A. Introduction to Surface Chemistry and Catalysis. New York:
Wiley, 1994.
5. Catalysis: an Integrated Approach to Homogeneous, Heterogeneous and
Industrial Catalysis / Moulijn J.A., van Leeuwen P.N.W.M., van Santen R.A.
Amsterdam: Elsevier, 1993.
6. Gates B.C. Catalytic Chemistry. New York: Wiley, 1992.
7. Campbell I.M. Catalysis at Surfaces. London: Chapman&Hall, 1988.
8. Boudart M. and Djega-Mariadasoou G. Kinetics of Heterogeneous Catalysis
Reactions, Princeton: Princeton Univ. Press, 1984.
9. Gates B.C., Katzer J.R. and Schuit G.C.A. Chemistry of Catalytic Processes.
New York: McGraw Hill, 1979.
10. Van Santen R.A. and Niemantsverdriet. Chemival Kynetics and Catalysis. New
York: Plenum, 1995.
Список литературы
1. Datye A.K., Long N.J. // Ultramicroscopy. 1988. V. 25. P. 203.
2. Sheldon R.A. // J. Chem. Tech. Biotechnol. 1997. V. 68. P. 381.
3. Sheldon R.A. // Chem. Ind. 1997. P. 12 ; 1992. P. 903.
4. Chemical Engineering News, July 10, 2006.
глава КИНЕТИКА
*Ъ ХИМИЧЕСКИХ РЕАКЦИЙ
2.1. ВВЕДЕНИЕ
В задачу кинетики входит разработка общих принципов, позволяющих определять скорости протекания химических реакций. Для этого кинетическая теория должна прежде предложить механизм реакции, то есть показать,
как из исходных реагентов через формирование промежуточных соединений
получается конечный продукт. С ее помощью определяются также зависимости от времени макроскопических параметров — концентраций компонентов,
давления и температуры. Таким образом, кинетическая теория дает нам инструмент для связи микроскопического мира реагирующих молекул и макроскопического мира технологических процессов. Для катализа кинетика является
основополагающей дисциплиной.
Исторически катализ всегда был тесно переплетен с кинетикой. В табл. 2.1
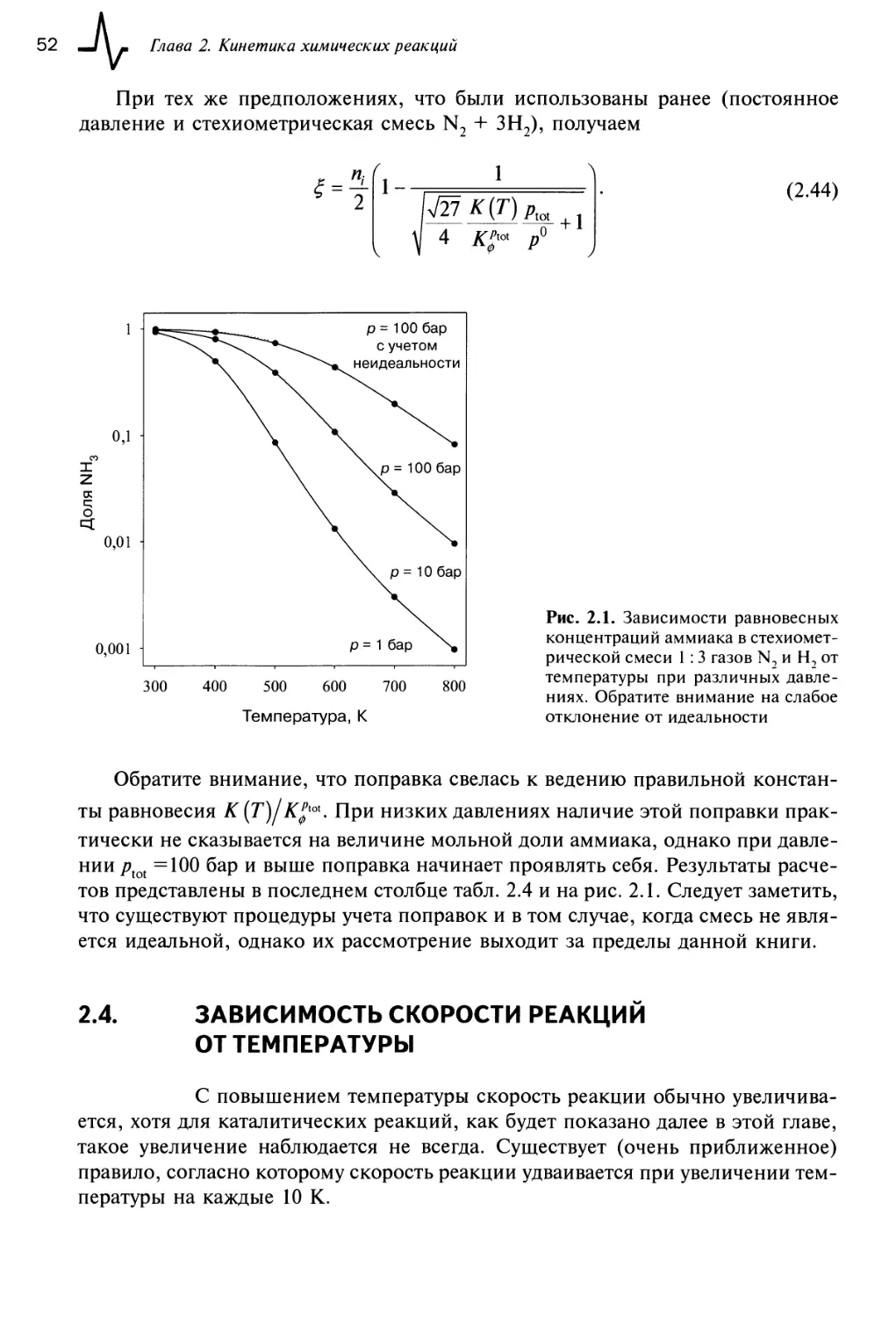
приведены основные исторические вехи развития катализа. Определять скорости химических реакций начали с 1850 г., а катализ был осознан как самостоятельное явление в первой декаде XIX столетия. Перелом в интересе к катализу возник с развитием синтеза аммиака и разработкой проточных реакторов
высокого давления. Необходимость перехода к высоким давлениям и температурам была показана Фрицем Хабером и Вальтером Нернстом, которые используя высокий уровень развития равновесной термодинамики, достигнутый
к концу XIX в. благодаря работам Вант-Гоффа, показали, что синтез аммиака в
этих условиях (при высоких давлениях и температуре) идет более легко. Необходимость выделения азота из воздуха возникала в связи с развитием индустрии производства удобрений, которые позволяли увеличить урожаи, что стало
очень актуальным при бурно растущем населении Земли. Имелся и «отрицательный» стимул к развитию синтеза аммиака: он использовался при производстве взрывчатых веществ, потребность в которых резко возросла в период
подготовки и хода Первой мировой войны.
Каталитически реакции (как и класс тесно с ними связанных цепных реакций, которые будут обсуждаться ниже) представляют собой набор взаимосвязанных химических превращений, кинетика которых находится из решения
систем дифференциальных уравнений, описывающих последовательные стадии процесса. Сложность задачи явилась стимулом к разработке квазистацио-
нарного приближения, которое было предложено Чепменом в 1913 г. и до насто¬
2.1. Введение Jv
ящего времени является ключевым элементом решения системы кинетических
уравнений.
Таблица 2.1. Историческое развитие кинетики каталитических химических
реакций [1]
1813
Те нар
Проведение разложения аммиака на некоторых металлах
1814
Кирхгофф
Осуществление гидролиза крахмала, катализируемого кислотами
1817
Гэмфри Дэви
Наблюдение, как в смеси каменноугольного
газа и воздуха проволока из платины раскаляется до бела
1818
Тенард
Измерение скорости разложения Н202
1823
Деберейнер
Селективное окисление этанола на платине
до уксусной кислоты
1834
Фарадей
Исчерпывающее описание реакции Н2 + 02,
протекающей на платине
1835
Берцелиус
Определение понятий катализа, катализатора и каталитической силы
1850
Вильгельми
Проведение первого количественного анализа скоростей реакции
1862
Гульдберг и Вааге
Формулировка закона действующих масс
1865
Харкурт и Эссон
Систематическое исследование зависимости
скорости реакции от концентрации
1884
Вант-Гофф
Издание первой фундаментальной монографии по химической кинетике
1887
Оствальд
Определение порядка реакции
1889
Аррениус
Формулировка уравнения Аррениуса
к = vexp (~Еа/квТ)
1905
Нернст
Формулировка третьего закона термодинамики
1908
Хабер
Предсказание условий синтеза аммиака
1913
Чепмэн
Разработка метода квазистационарного
приближения
1915
Ленгмюр
Создание количественной теории адсорбции газов на поверхностях
Глава 2. Кинетика химических реакций
Окончание табл. 2.1
1921
Линдеманн
Объяснение механизма мономолекулярной
реакции активацией при столкновениях
1925
Тейлор
Введение понятия о каталитически активных центрах на поверхности
1927
Хиншельвуд
Построение кинетической теории реакций в
гетерогенном катализе
1931
Онзагер
Создание неравновесной термодинамики
1935
Эйринг, Поляни
и Эванс
Разработка теории переходных состояний
или теории активированного комплекса
Исследования закономерностей разрушения вольфрамовых проволочек при
их взаимодействии с кислородом в лампах накаливания привели Ленгмюра к
созданию теории адсорбции (в 1915 г.), позволяющей связывать концентрацию
газа на поверхности с его давлением в окружающей среде. Воспользовавшись
этой теорией и гипотезой Тейлора о существовании активных центров на поверхности катализатора, Хиншельвуд в 1927 г. сформулировал основополагающие принципы кинетики каталитических реакций — теории Ленгмюра—Хиншельвуда, не утратившей своего значения до сегодняшнего дня. И до второй
половины XX столетия исследования по катализу отождествлялись с кинетическим анализом; лишь в последнее время инструментарий в изучении катализа сильно расширился, в частности за счет спектроскопических методов и численного моделирования.
Таким образом, можно сказать, что в XX столетии кинетика расширила
свои границы: начав с чисто эмпирического описания скоростей реакций, она
«присоединила» к себе множество областей научных исследований, теперь позволяющих рассматривать каталитические процессы на разных уровнях — от
молекулярного, учитывающего в том числе и электронные состояния атомов, и
строение химических связей, до крупномасштабного, оперирующего с большими количествами веществ в индустриальных реакторах.
Молекулярный уровень описания, часто называемый динамикой реакций,
принимает во внимание тонкие детали, влияющие на скорость реакции, типа
ориентации молекул при столкновении или распределения энергии по различным степеням свободы молекул. Это — фундаментальный уровень исследований, необходимый при квантово-механическом описании процессов, позволяющем выявить «правила игры» из первых принципов. Такие исследования обычно
проводятся с молекулярными пучками с привлечением лазерной спектроскопии, квантово-химических расчетов и теории активированного комплекса.
Именно на этом уровне мы можем выяснить, какие условия определяют возможность протекания реакций.
На практике нас не интересует одиночное явление, более важным представляются процессы, протекающие в большом ансамбле молекул. Поэтому
2.2. Уравнение скорости реакций и порядки реакций
нам необходимо провести усреднение полученных для частного случая результатов по всем возможным начальным состояниям и исходам, то есть по всем
координатам, ориентациям и энергиям реагирующих молекул. Это и есть предмет кинетики химических реакций. Температура представляет собой параметр,
характеризующий среднюю энергию, которой обладают молекулы, участвующие в реакции. Законы термодинамики позволяют найти изменение свободной энергии при реакции и определить предельную долю молекул, претерпевающих химическое превращение. Иначе говоря, кинетика описывает поведение
ансамбля большого числа частиц при реакции.
2.2. УРАВНЕНИЕ СКОРОСТИ РЕАКЦИЙ
И ПОРЯДКИ РЕАКЦИЙ
Рассмотрим реакцию между молекулами А и В, при которой образуются молекулы С и D, описываемую уравнением
иаА + vbB vcC + vdD, (2-1)
k
где k+ и k~ константы прямой и обратной реакций, a va, vb, vc, vd — стехиометрические коэффициенты. Скорость реакции определяется как скорость исчезновения реагентов или скорость появления продуктов реакции:
г . _± *1*1 = = J_M = J_M (2.2)
va dt vb dt vc dt vd dt
символ [X] обозначает концентрацию компонента X. Для реакций в газовой
фазе можно заменить концентрации парциальными давлениями: [X] ос рх/р°> гае
р° — нормировочное давление (например, р° = 1 бар),
г = 1 Фа = 1 Фв = 1 dPc = 1 Фр (2.3)
Vap° dt Уър° dt Vcp° dt vdp° dt
Довольно часто нормировочное давление р° не выписывают явно, а просто
полагают, что давление измеряется в относительных единицах. Реакция считается элементарной, если она идет в одну ступень, которую нельзя разбить на
промежуточные стадии, и протекает точно в соответствии с уравнением реакции (2.1). Для элементарной стадии реакции скорость равна
г = Jfc+ [Ар [Вр - Г [Ср [Dp = г+- г~. <2-4)
Здесь к+, кГ — константы скорости реакции и стехиометрические коэффициенты совпадают с порядком реакции по соответствующему компоненту.
Механизмом реакции называют последовательность ее элементарных
стадий.
Глава 2. Кинетика химических реакций
Пример
Примером элементарной реакции первого порядка является изомеризация
циклопропана в пропилен, для которой обратный процесс не идет, так что мы
можем записать скорость как
_ ^/^циклопропан _ ^/\фопилен _ п /^ сч
^ ~ — ^ — ^ /^циклопропан * \^**^/
Пример
Другим примером является равновесие
N,Q4< ** > 2NO-,, (2.6)
k~
которое представляет прямую элементарную реакцию первого порядка и обратную элементарную реакцию второго порядка. Адсорбцию молекул на или
десорбцию с поверхности также можно рассматривать как элементарные реакции, однако эти процессы часто идут в две стадии, на одной из которых молекулы находятся в состоянии прекурсора.
Константы скорости прямой и обратной реакций связаны через константу
равновесия К. Поскольку в состоянии равновесия скорость реакции г равна
нулю, имеем:
fCf [Dp k+
г = Г [Ар [Bp - Г [Ср [Dp = 0; К = 1 - Je-q 1 Jeq = —, (2.7)
L Jeq L Jeq L Jeq L Jeq [Ap [Bp k~
где [X]eq обозначает равновесные концентрации компонентов. С учетом введенной константы равновесия скорость реакции можно записать в виде
1 [Ср [Df '
/
г = к+ [Ар [В]и
1-
К [Ар [BJ
!*Ъ
= Г - г", (2.8)
где сомножитель перед скобками представляет собой скорость реакции на начальной стадии (в пределе нулевой конверсии), а выражение в скобках характеризует степень приближения реакции к равновесию. Отметим, что вблизи
равновесия скорость реакции убывает экспоненциально со временем до полного завершения реакции.
Почти все обсуждаемые в данной книге реакции являются сложными и
состоят из большого числа элементарных стадий. Это относится даже к таким,
на первый взгляд, простым реакциям, как 203 -» 302 или Н2 + С12 -» 2НС1.
В частности, реакции гетерогенного катализа всегда многоступенчаты и обязательно включают в себя стадии адсорбции на поверхности, собственно реакции
и десорбции в газовую фазу. В этой главе мы увидим, как уравнения полной
реакции конструируются по уравнениям элементарных стадий.
2.2. Уравнение скорости реакций и порядки реакций
Довольно часто нам не известны априори детали протекания сложных реакций, однако для параметризации уравнений удобно записывать скорость как
произведение концентраций или давлений в соответствующих степенях, то есть
представлять скорость степенным законом:
г = к [Ар [Bp [Cfc [Dp (2.9)
ИЛИ
Г = к+РАРвРсРо>
может принимать любые значения — целые, дробные, положительные, отрицательные или быть нулем. Полные порядки прямой
и обратной реакций равны пА + пв и пс + nD соответственно. Заметим, что
порядок реакции по определенному компоненту может быть найден по наклону прямой линии, представляющей зависимость скорости реакции от концентрации этого компонента в двойных логарифмических координатах. Следовательно, формальный путь определения порядка реакции по ее скорости состоит в нахождении следующих производных:
Э1П,- _[Х]Э1пг (210)
Э1п[Х] L J Э[Х]
Ниже мы довольно часто будем вычислять производные логарифма.
Это — очень удобный способ нахождения ряда параметров по скорости
реакции или из термодинамических уравнений. Производная логарифма
(или логарифмическая производная) функции / по переменной х равна
din / _ 1 Э/
Эх / дх
При детально известном механизме реакции можно определить зависимость
скорости полной реакции от температуры и концентрации. Обычно имеет смысл
исследовать кинетику реакции в условиях, далеких от равновесия (в пределе
нулевой конверсии), так что порядок реакции можно найти из соотношения
Э1пг+ (2.11)
х Э1п[Х]
Элементарные реакции обычно имеют порядок, выражаемый целым числом. Для полной реакции не всегда удается выразить зависимость скорости от
концентрации в виде степенного закона. При этом порядок реакции может
быть не целым числом и сохраняет свое постоянство лишь в узком интервале
изменения значений термодинамических параметров системы. Однако этого
Глава 2. Кинетика химических реакций
оказывается достаточно для описания промышленных процессов в терминах
степенных законов. В химической технологии эти законы обычно используются для предсказания поведения реакторов при варьировании температуры и
давления в ограниченных диапазонах.
Пример
В качестве примера рассмотрим реакцию образования углеводородов из
синтез-газа, где в качестве катализаторов используются металлы VIII группы:
хСО + уН2 —Fe Со > СхН2х{+2) + *Н20, (2.12)
для которой порядок по СО обычно отрицательный и лежит между —1 и 0, а по
Н2 близок к 1.
Пример
Другой интересный пример, показывающий, насколько противоречивым
может быть понятие порядка реакции, — окисление СО — важная промежуточная реакция в катализе окисления выхлопных газов
С04°2 n.M.Rh' >С02- (2.13)
При низких температурах порядки по СО и 02 равны примерно -1 и +1/2,
а при высоких температурах они становятся равными +1 и +1/2 соответственно. Таким образом, порядок полной реакции не может рассматриваться как
универсальная постоянная. Его следует трактовать как удобный параметр, значения которого определяются условиями проведения реакции. Ниже мы увидим, как при конструировании полной реакции в виде совокупности элементарных стадий значения порядков приобретают конкретное физическое содержание. Без этого вводить понятие порядка не имело смысла, поскольку всякая
модель должна оперировать с измеряемыми величинами.
Пример
Если скорость прямой реакции дается выражением
г'=т£гт- <2|4>
1 + А 9
то порядок реакции по давлению р2 равен
Э1пг+ _, К,р, ^ tcs
"j’ft д i д. г n ' (2.15)
dp2 1 + К2Р2
Из этой формулы видно, что порядок реакции зависит и от константы равновесия К2, которая изменяется с температурой, и от парциального давления р2.
Последняя зависимость, как мы увидим далее, появляется в катализе из-за
того, что реагент адсорбируется на поверхности, частично блокируя ее.
2.3. Реакции и термодинамическое равновесие яшш
2.3. РЕАКЦИИ И ТЕРМОДИНАМИЧЕСКОЕ РАВНОВЕСИЕ
В равновесии скорости прямой и обратной реакций совпадают, а
свободная энергия Гиббса G принимает минимальное значение. При постоянных температуре и давлении из условия минимума свободной энергии следует,
что производные G по числам частиц реагентов и продуктов реакции должны
быть равны нулю:
dG = +мАй пА +MBdnB + Mcdnc + ModnD = = °> (2-16)
i
где ju( — химический потенциал /-го компонента. Мы введем специальный параметр — степень полноты химической реакции £ через который изменения
чисел компонентов реагирующей смеси представляются следующим образом:
dnA = ~VAdZ> dtlB = ~VBd£’ dnC = VCd%> dnD = VDd%- (217)
Отсюда имеем равенство
d G
~7T = -VaMa -VbMb+ VqMc + VdV d = E v№ = °> (2-18)
db i
отражающее принципиально важный факт равенства нулю суммы химических
потенциалов, взятых со стехиометрическими весами. При этом мы приняли,
что стехиометрические коэффициенты для реагентов отрицательны. Химические потенциалы компонентов в смеси определяются выражением
м, = + RT in («,) = м? + RT in
Р]_
_0
кР J
(2.19)
где а — активность компонента /, которая может быть заменена либо отношением давлений р(/р°9 либо концентрацией [/] (р° — давление в стандартных
условиях, р° = 1 бар); R — газовая постоянная; Т — температура. Используя это
выражение, получаем
S = Е vit*i + RT In |П аР j = 0- (2-20)
Изменение стандартного потенциала Гиббса A G0 представляется теперь через
комбинацию стандартных химических потенциалов:
дG0 = 5>,я° = - vaMa - vbM°b + vcK + VdMd- (2.21)
/
С учетом приведенных соотношений константа равновесия химической
реакции может быть записана следующим образом:
Глава 2. Кинетика химических реакций
Активности можно приближенно заменить измеряемыми величинами, например парциальными давлениями в случае идеального газа:
к (Т) = = Щ1^(рТ+'Ъ"'С"’Л (2.23)
k Ра Ръ
или концентрациями компонентов в случае идеальных растворов
V [Ср [Dp
К (Т) = -— = Jeci —Jcq . (2.24)
[а]1; [в]-
Ниже будет показано, как фундаментальные величины типа химического
потенциала ju могут быть приближенно определены из первых принципов (через их молекулярный вес, момент инерции молекул и т. д.) и как константа
равновесия находится из условия равенства суммы химических потенциалов
нулю, уравнения (2.20). Вышеприведенные уравнения связывают термодинамические и кинетические характеристики и позволяют найти константу скорости прямой реакции по известной константе скорости обратной реакции и
термодинамическим параметрам смеси.
Чтобы упростить запись длинных уравнений с подобными членами, введем
для некоторых выражений сокращенные обозначения. Так, символом Х( будем
обозначать молекулы А, В, С и т. д., через у. — их стехиометрические коэффициенты, отрицательные для реагентов и положительные для продуктов реакции. Тогда вышеприведенные уравнения можно переписать в виде:
5>,Х,=0, (2.25)
/
^(TVnKeqf > (2.26)
/
A G°=5>/A?- (2.27)
/
Чтобы рассчитать равновесный состав смеси при данной температуре, мы
должны прежде всего найти константу равновесия из термодинамических данных, представленных, например, в табл. 2.2 и относящихся к стандартным условиям Т = 293 К, р = 1 бар. Дифференцируя уравнение (2.22) и используя
соотношение AG0 = АН° — TAS°, получаем уравнение Вант-Гоффа:
din К d
dT dT
AG
RT
A /7°
==^r. (2.28)
RT2
Интегрируя это уравнение по температуре от стандартного Т = 293 К до текущего значения Т, находим
2.3. Реакции и термодинамическое равновесие
ИЛИ
In К (Т) = In К (298) - - _Lj, (2.30)
где мы пренебрегли малым вкладом, связанным с различием теплоемкостей
реагентов и продуктов, то есть приняли, что АН° не зависит о температуры.
Уравнение (2.30) показывает, что зависимость логарифма константы равновесия от обратной температуры является линейной. Для эндотермических реакций константа равновесия возрастает с температурой, для экзотермических —
убывает. Изложенный метод пригоден лишь для оценки константы равновесия.
Более точные расчеты требуют учета температурной зависимости и А #°, и AS0.
Такие зависимости могут быть получены для широкого круга веществ из таблиц термодинамических параметров. В гл. 3 будет показано, что АН0 и AS0
достаточно слабо изменяются с температурой: это связано с особенностями
распределения энергии и энтропии по внутренним степеням свободы молекул.
В табл. 2.2 приведены значения АН0 и А5°, относящиеся к стандартным условиям, для ряда важных химических реакций.
Таблица 2.2. Термодинамические данные для ряда важных каталитических
реакций
Реакция (Г=298 К)
АН0, кДж/моль
ДG°, кДж/моль
NH3^±N2+|h2
+45,9
+16,4
^N2 +|н2 -» NH3
-45,9
-16,4
N2 + ЗН2 -» 2NH3
-91,9*
-32,8*
2NO -» N2 + 02
-182,6*
-175,2*
CH4 + H,0 -» CO + 3H,
+205,9
+142,0
CH4 + ^02 -> CO + 2H2
-35,9
-86,8
CH4 + 202 -» CO, + 2H20
-802,6
-801,0
CH4 +|o, ->CH3OH
-275,6
-111,77
C02 + 3H2 -» CH3OH + 2H20
-49,3
+3,5
CO + 2H2 -» CH3OH
-90,5
-25,1
CO + h2o -» co2 + H2
-41,2
-28,6
* На два моля NH3 или NO. Данные взяты из [2].
Глава 2. Кинетика химических реакций
2.3.1. Пример химического равновесия: синтез аммиака
В качестве примера практического использования приведенных
ранее соотношений проведем по табличным данным расчет полноты реакции
синтеза аммиака
In2+|h2^NH3> (2.31)
которую она достигает при данной температуре. Изменение стандартного потенциала Гиббса для этой реакции равно около —16,4 кДж/моль, а изменение
стандартной энтальпии составляет —45,9 кДж/моль; в обоих случаях расчет
ведется на моль NH3.
В расчетах мы примем, что газовая смесь достигает равновесия при постоянном давлении. Эта ситуация отвечает течению газа в проточном реакторе в
условиях пренебрежимо малого падения давления (заметим, что если синтез
аммиака проводится в замкнутом сосуде, то давление будет падать с ростом
степени превращения реагентов).
Введя мольные фракции компонентов Yx = px/ptot, где рх — парциальное
давление газа X, a ptot — полное давление, перепишем уравнение (2.22) в виде
К(Т) =
YVcY
1 Г 1т
I'd
C D
Yva Yvb
7 A
Ptot
p°
vc + vd~ l'a - l'b
= e~bG/RT = e-bH/RTebS/R (2.32)
Это соотношение оказывается полезным при расчетах равновесных концентраций. Из него легко видеть, что для экзотермического процесса (отрицательное АН) равновесные концентрации продуктов реакции с ростом температуры уменьшаются и увеличиваются при повышении давления, если число частиц газа в ходе реакции уменьшается (кА+ vB — vc~ vD > 0).
Применив уравнение (2.32) к синтезу аммиака и считая газ идеальным,
сразу же получаем
К (гГ\ - p-bG/RT _ ^NH3 ^NH3 о _ ^NH3 Р° п
е al/2a3/2 ~ d1/2d3/2 у1/2у3/2 п ‘
un2uh2 РщРн2 N2 7Н2 ^tot
Для получения более высокого выхода продукта, промышленный синтез
аммиака проводится при повышенных давлениях (ptot = 100—200 бар) и относительно низких температурах (Т = 400 °С). Условия равновесия обычно
записываются в терминах мольных фракций, поскольку они явно показывают происходящие в ходе реакции изменения состава. В реальных процессах
исходная смесь состоит не только из Н2 и N2, поскольку азот берется из
воздуха и в нем содержится около 1 % аргона. К тому же в водороде есть
следовые примеси метана. Учтем также возможность присутствия в исходной смеси некоторого количества аммиака. Обозначая через пх число молей
компонента X, присутствующего в реагирующей смеси, и обозначая верх-
2.3. Реакции и термодинамическое равновесие
ним индексом i исходные значения параметров, определим степень полноты реакции £ выражением
z п х ~ п1х
£-“7—• (2-34)
Конечные числа молей компонентов легко находятся по известным стехиометрическим коэффициентам, что отражено в табл. 2.3.
Таблица 2.3. Параметры, необходимые для расчета степени превращения
водорода и азота в аммиак при постоянном давлении
Компонент
V
Исходное число молей
Конечное число молей
n2
1
2
"n2
1 р
«n2 " 2^
н2
3
2
«н2
Пнг _2
NH3
+1
«NH3
«МН3 + £
Аг
0
«Аг
«Аг
Сумма
-1
«/ = «М2 + «Н2 + «NH3 + «Аг
П/ = и, - £
Поскольку мольные фракции Yx равны Yx = nx/ntot, равновесные мольные
фракции должны нормироваться на nf— число молей в смеси в конце реакции,
что приводит к уравнению
NH
+ #
К(Т) =
V „о
NH3 Р
*К2 т
п,-4
(пщ-№)^
1/2
Г «н2 -(3/2)^^
о
1
I
«;-£ J
1 щ-Z )
(2.35)
Если мы для упрощения задачи рассмотрим в качестве исходной смесь только
N2 и Н2 в стехиометрическом отношении 1: 3, то выражение (2.35) сводится к
следующему
К(Т) =
у/27 [rtj - 2gf Рш '
Решая это уравнение относительно приходим к двум корням
1
гц
g 2
1±-
№)^+1
I 4/
(2.36)
(2.37)
один из которых (со знаком +) не имеет физического смысла.
Глава 2. Кинетика химических реакций
Для парциального давления аммиака получаем выражение
£ _ (Т) ptot л/27 + 4 - 2 _
Pnh3 - eAot - I—— j= Ptot* (2.38)
«/-£ jK(T)Ptaty/Tj + 4 + 2
В табл. 2.4 приведены мольные фракции аммиака при различных значениях
температуры и давления, достигаемые в рассматриваемой системе (смесь только N2 и Н2 в отношении 1: 3). Из табл. 2.4 видно, что для достижения высокого
выхода аммиака необходимо проводить реакцию при низких температурах.
Синтез, однако, проводят при повышенных температурах, поскольку только в
этом случае разрываются связи между атомами в молекулах N2, энергия диссоциации которых составляет 940 кДж/моль. К сожалению, как видно из табл. 2.4,
использование повышенных температур приводит к очень низкой степени превращения смеси водород/азот в аммиак. Таким образом, хотя термодинамически предпочтительнее проводить синтез аммиака при низких температурах, высокий кинетический барьер блокирует его образование. Решение проблемы
состоит в использовании катализаторов, позволяющих провести диссоциацию
молекул N2 при низких температурах. Энзимы эффективно осуществляют синтез аммиака при комнатной температуре, поскольку в этих условиях практически полностью отсутствуют термодинамические ограничения. В промышленности использование катализаторов из железа позволяет проводить синтез
при давлении 200 бар и температурах 625—675 К, когда при одном проходе
через реактор достигается эффективность синтеза от 60 до 80 %. Последняя
колонка в табл. 2.4 демонстрирует влияние эффектов неидеальности (активности) газов, участвующих в реакции.
Таблица 2.4. Равновесная степень превращения N2 и Н2 в аммиак в стехиометрической смеси при постоянном давлении ptot
т, к
Мольная доля NH3
Рш = 1 баР
Р,о, = 10 бар
рш = 100 бар
Ло.= 100 бар
с учетом
неидеальности
298
6,68 х 102
0,934
0,979
0,993
0,997
400
6,04 х 10°
0,497
0,798
0,933
0,945
500
3,18 х 10_|
0,0862
0,387
0,733
0,759
600
4,18 х 10~2
0,0132
0,108
0,434
0,466
700
9,40 х 10~3
0,00303
0,0288
0,197
0,222
800
3,00 х 10“3
0,00097
0,000955
0,0821
0,097
2.3. Реакции и термодинамическое равновесие
2.3.2. Химическое равновесие для неидеальных газов
В проведенном ранее примере мы считали газы идеальными, что
обычно является достаточно хорошим приближением. Однако при высоких
давлениях и низких температурах взаимодействие между молекулами может
привести к заметным отклонениям от идеальности. В этих случаях мы можем
отождествлять активности с парциальными давлениями или концентрациями.
Для учета этих отклонений вводят [3] фугитивности (летучести)^, связанные с
парциальными давлениями соотношением:
/х = ФхРх-> (2.39)
где фх — коэффициенты фугитивности. Эти коэффициенты безразмерны, поэтому фугитивности Ух имеют размерность давления. Коэффициенты фугитивности могут быть рассчитаны через вириальные коэффициенты и определены
по табличным данным для относительных (к критическим значениям) давления и температуры газа. Коэффициент фугитивности может иметь значения
как больше, так и меньше единицы в зависимости от данных значений давления и температуры. Идеальным газам отвечает значение ф^ = 1. Ниже мы кратко изложим способ расчета равновесной степени превращения с учетом отличных от единицы коэффициентов фугитивности.
Как уже упоминалось, активности компонентов совпадают с их фугитивно-
стями (а не с парциальными давлениями), поэтому корректная форма уравнения (2.22) имеет вид
v /гг \ й'с /с /d ( o\a + b-c~d
К(Т)= —Р- = УС урц (р°) . (2.40)
nv*nvb farb ^
аА аВ J A JB
Для идеальной смеси газов (которые сами себя ведут неидеально) фугитив-
ность^ компонента X дается выражением
/х = ФхРх - 0xotYxPtof> (2.41)
где ф£ш — коэффициент фугитивности чистого газа X при давлении ptot и данной температуре. Заметим, что коэффициент фугитивности зависит как от давления, так и от температуры. С учетом определения (2.41) получаем
К(Т) =
fccfod Фс°'Фо°' У? У о*
( _ Vc + i'd-i'a-i'd
У tot
f1/3 fvb fh Рш rh Ptot YVaYVb
/А /В ГА ГВ 1A JB
Ptot
р°
к (т) = к^
V / Ф ууаууъ
1 А 2В
(2.42)
Vvc Vvd ( п Л vc + V& - l'a “ vd
1 r 1 n
p°
Применяя это выражение к случаю синтеза аммиака, находим
л
Глава 2. Кинетика химических реакций
При тех же предположениях, что были использованы ранее (постоянное
давление и стехиометрическая смесь N2 + ЗН2), получаем
1-
1
ы27 К (Г) ptc
4 р°
+ 1
(2.44)
Температура, К
Рис. 2.1. Зависимости равновесных
концентраций аммиака в стехиометрической смеси 1 : 3 газов N2 и Н2 от
температуры при различных давлениях. Обратите внимание на слабое
отклонение от идеальности
Обратите внимание, что поправка свелась к ведению правильной константы равновесия К (Т)/КРш. При низких давлениях наличие этой поправки практически не сказывается на величине мольной доли аммиака, однако при давлении ptot =100 бар и выше поправка начинает проявлять себя. Результаты расчетов представлены в последнем столбце табл. 2.4 и на рис. 2.1. Следует заметить,
что существуют процедуры учета поправок и в том случае, когда смесь не является идеальной, однако их рассмотрение выходит за пределы данной книги.
2.4. ЗАВИСИМОСТЬ СКОРОСТИ РЕАКЦИЙ
ОТ ТЕМПЕРАТУРЫ
С повышением температуры скорость реакции обычно увеличивается, хотя для каталитических реакций, как будет показано далее в этой главе,
такое увеличение наблюдается не всегда. Существует (очень приближенное)
правило, согласно которому скорость реакции удваивается при увеличении температуры на каждые 10 К.
2.4. Зависимость скорости реакций от температуры
Температурная зависимость константы скорости элементарных реакций
хорошо описывается уравнением Аррениуса:
k(T) = ve~EJRT, (2.45)
где i/так называемый предэкспоненциальный множитель и Ея — энергия активации (кДж/моль). Можно выражать энергию активации и в джоулях на одну
молекулу (очень малая величина), тогда газовую постоянную R в формуле (2.45)
необходимо заменить на постоянную Больцмана кв.
Аррениус получил уравнение (2.45), основываясь на имеющихся экспериментальных данных и результатах собственных исследований по гидролизу сахарозы,
фруктозы и глюкозы. Заметим, что предложенная Аррениусом температурная зависимость является чисто экспоненциальной и в ней предэкспоненциальный множитель является постоянным. Теории скоростей реакций, обсуждаемые в гл. 3,
показывают, что аррениусовская зависимость с высокой степенью точности является корректной. Однако постоянство (независимость от температуры) пре-
дэкспоненциального множителя не подтверждено ни одной из существующих
теорий. В теории переходных состояний предэкспоненциальный множитель имеет
степенную зависимость от температуры: Тх. Но эта зависимость незначи¬
тельна в сравнении с экспоненциальной, поэтому обычно ею пренебрегают.
ш
со
S
0
а
с
of
s
I—
CL
Ф
1
со
Рис. 2.2. Схематическое представление профиля потенциальной энергии для рекомбинации
двух атомов или диссоциации молекулы
Физическая интерпретация уравнения Аррениуса основывается на предположении о существовании энергетического барьера, с преодолением которого
связано протекание полной реакции. На рис. 2.2 эта ситуация проиллюстрирована для реакции образования молекулы из двух атомов. Когда происходит
сближение атомов по правой ветви потенциальной кривой, то сначала происходит отталкивание между ними, связанное с ростом потенциальной энергии,
идущим до достижения расстояния, на котором собственно и происходит реакция, сопровождающаяся, например, перестройкой электронной конфигурации
и образованием связи. На этом расстоянии потенциальная энергия достигает
максимума. Формирование связи вызывает уменьшение потенциальной энергии при дальнейшем сближении атомов. Сейчас мы отождествим высоту по¬
Координата реакции
или расстояние X—X
Глава 2. Кинетика химических реакций
тенциального барьера с энергией активации (ниже мы увидим, что это отождествление не вполне корректно). Отметим, что на рис. 2.2 схематически показана также энтальпия реакции.
Энергию активации можно определить по значениям скорости реакции,
измеренным при разных температурах. Зависимость логарифма константы от
обратной температуры, как видно из формулы (2.45), имеет вид уравнения прямой линии:
lnk{T) = lnv-%-±;. (2.46)
К 1
Угол наклона этой прямой определяется энергией активации, а точка пересечения с осью ординат — пердэкспоненциальным множителем. Нахождение
энергии активации, таким образом, сводится к элегантной процедуре, которая
постоянно будет использоваться ниже и выражается формулой:
Е* = ~кЧ7Т1Чл = кт2Щ^- (2.47)
а Э(1 /Т) дТ
Читатель может теперь легко рассчитать величину энергии активации, отвечающую правилу: скорость реакции удваивается при увеличении температуры на каждые 10 К.
Как уже отмечалось выше, химическая реакция достаточно редко протекает в одну стадию, так что нам необходимо приспособить данное нами определение энергии активации к многоступенчатым реакциям. Поскольку нас не
будут интересовать термодинамические эффекты, мы определим кажущуюся
энергию активации через соотношение
E^^RT1^-. (2.48)
Это определение является следствием построения экспериментальных данных для скорости реакции в аррениусовских координатах lnr+ — 1 /Т. Однако в
общем случае экспериментальные данные могут и не укладываться на линейную
зависимость. Таким образом, кажущаяся энергия активации пригодна для описания химических реакций только в ограниченном интервале температур. При
интерпретации энергии активации нужно быть особенно внимательным в отношении порядка реакции, поскольку он обычно зависит от условий проведения
эксперимента. Ниже приведен пример, в котором скорость прямой реакции зависит как от активированного процесса, так и от равновесных стадий. Этот пример отражает ситуацию, часто встречающуюся в каталитических реакциях.
Пример
Предположим, что мы имеем дело с многоступенчатой реакцией, скорость прямого процесса в которой характеризуется следующей зависимостью от давления
2.5. Решения уравнений скорости реакций: зависимости от времени концентраций
Мы примем, что параметры к и Кх могут быть представлены в виде
k(T) = ve~E*/RT (2.50)
и
Кх (Т) = e-AG*/RT = e-^Hx/RTe^x/R = Vxe-*H*IRT. (2.51)
Непосредственное применение уравнения (2.48) дает
Em = Еа + ДЯ, + ДН2 - ДН2. (2.52)
1 + К^р2
Это соотношение показывает, что кажущаяся энергия активации представляет собой истинную энергию активации, «дополненную» энтальпиями равновесных стадий, то есть в данном случае кажущаяся энергия активации зависит
как от давления, так и от температуры системы. Заметим также, что здесь мы
пренебрегли возможной неэкспоненциальной зависимостью параметров. Как
будет показано в гл. 3, величины у, Д#и AS в определенной степени являются
функциями температуры.
2.5. РЕШЕНИЯ УРАВНЕНИЙ СКОРОСТИ РЕАКЦИЙ:
ЗАВИСИМОСТИ ОТ ВРЕМЕНИ КОНЦЕНТРАЦИЙ
В РЕАКЦИЯХ РАЗЛИЧНОГО ПОРЯДКА
В общем случае скорость реакции определяется по изменению состава системы за определенный временной интервал. Для ее нахождения мы
должны установить зависимости от времени концентраций компонентов, основываясь на приведенных в предыдущих разделах дифференциальных уравнениях, связывающих скорость реакции с концентрациями реагирующих молекул.
Рассмотрим реакцию первого порядка (очевидно, реакцию изомеризации, хотя
диссоциация соединения R два фрагмента Pj и Р2, описывается точно так же)
R—i—(2.53)
скорость которой, записанная в дифференциальной форме, равна
d[R]
Г - --
dt
■ = ik[R]. (2.54)
Для получения зависимости от времени концентраций компонентов R и Р
необходимо проинтегрировать это уравнение, что легко делается методом разделения переменных:
Глава 2. Кинетика химических реакций
Пределы интегрирования были выбраны в соответствии с начальным условием [R] = [R]0 при t = 0. Из решения (2.55) легко находятся зависимости от
времени концентрации реагента и продукта реакции:
Выражение (2.56) показывает, что, построив зависимость In [R] от времени,
можно легко установить, имеет ли данная реакция первый порядок. Для реакций
первого порядка зависимость In [R] от времени должна быть линейной, а тангенс угла наклона соответствующей прямой совпадает с константой скорости.
Если реакция первого порядка действительно является необратимой, то по
времени /1/2, необходимому для превращения 50 % продукта в реагент, которое
для всех экспоненциальных зависимостей называется периодом полураспада
или полупревращения, также можно определить константу скорости:
Интегрирование кинетических уравнений для реакций второго порядка проводится чуть сложнее. Рассмотрим реакцию второго порядка следующего вида
Примем, что за некоторое время t прореагировало некоторое количество х
реагента А, тогда концентрации реагентов А и В станут равными [А]0 — х и [В]0 — х
соответственно. Используя данные обозначения, можно после разделения переменных переписать уравнение (2.59) следующим образом
Стандартный путь интегрирования таких дифференциальных уравнений
состоит в разбиении левой части на два слагаемых. То есть нам необходимо
найти коэффициенты ах и av обеспечивающие выполнение равенства
(2.56)
. In 2
к =
(2.57)
А + В—-к—> АВ,
(2.58)
для которой уравнение скорости таково
(2.59)
dx
= kdt.
(2.60)
(2.61)
Эти коэффициенты легко находятся с помощью алгебраических преобразова-
2.5. Решения уравнений скорости реакций: зависимости от времени концентраций
Данное уравнение легко интегрируется, и, с учетом начального условия л; = О
при t = 0, получаем
1
[В]0-[А](
-In
[А]0 [В]р - х
[В] [А]0-*
= kt.
(2.63)
Представляет интерес рассмотреть два частных случая. Пусть А и В являются одним и тем же компонентом, например, это могут быть радикалы О, объединяющиеся в молекулу 02. В этом случае уравнение (2.63) неприменимо, поскольку [А]0 = [В]0, и имеет место деление на ноль. Это говорит о необходимости вернуться к уравнению (2.60), которое в данном случае принимает вид
dx
(14-*г
= kdt
и интегрируется непосредственно:
1
и,-* к
= kt.
(2.64)
(2.65)
Таким образом, если мы построим зависимость величины 1/([А]0х) от времени, то получим прямую линию, тангенс угла наклона которой равен константе скорости.
Вторая интересная ситуация возникает, когда один из компонентов присутствует в значительном избытке, например, пусть [В] » [А]. Тогда [В] = [В]0 — х = [В]0,
и уравнение (2.63) принимает простую форму, отвечающую реакции псевдопервого порядка
In
[А]0
Но
= [B]0fo:
(2.66)
Для нахождения константы скорости в данном случае нужно использовать
метод, описанный при анализе реакций первого порядка, то есть необходимо
построить зависимость In [А] от времени. При этом произведение &[В]0 будет
выступать в качестве константы скорости реакции псевдопервого порядка, из
которой легко находится константа скорости реакции второго порядка.
Наконец, мы должны рассмотреть реакции нулевого порядка, хотя они встречаются достаточно редко. Порядок реакции становится псевдонулевым, если
реагент присутствует в значительном избытке и его концентрация практически
не изменяется в ходе реакции. Дифференциальная и интегральная формы уравнения скорости реакции нулевого порядка R —» Р таковы
= 1Р| = *'
(2.66)
На рис. 2.3 для сравнения показаны зависимости от времени степени превращения реагентов для реакций разного порядка. В табл. 2.5 сведены диффе-
58
\
Глава 2. Кинетика химических реакций
ренциальные и интегральные формы уравнений скорости для реакций разного
порядка.
Время, с
для реакции разного порядка
Таблица 2.5. Уравнения скорости реакции в дифференциальной
и интегральной формах
Порядок
реакции
Дифференциальная форма
d[A]_
dr
Интегральная форма
kt =
Размерность
k
0
-k
I
о
<J
моль • л-1 • с-1
1
[A]
,„[А|"
[A]
с-1
2
-k[Af
1 1
[A] [A]0
моль • л-1 • с-1
2
-k[ A][B]
1 ln [A]0 [B]
[B]0-[A]0 [A]0 [A]
моль • л-1 • с-1
2.6. ВЗАИМОСВЯЗАННЫЕ РЕАКЦИИ
В ПРОТОЧНЫХ РЕАКТОРАХ:
ПРИБЛИЖЕНИЕ СТАЦИОНАРНОГО СОСТОЯНИЯ
Как уже отмечалось выше, практически все представляющие практический интерес реакции являются многоступенчатыми. Возникает вопрос,
как кинетика полной реакции складывается из элементарных ступеней. Рассмотрим двухступенчатую реакцию, записанную в общем виде
рР/.
I I /
(2.68)
2.6. Взаимосвязанные реакции в проточных реакторах ->\г 59
Анализ кинетики таких реакций следует проводить с учетом типа используемого реактора. Мы рассмотрим реакторы двух типов — трубчатый (колончатый) и емкостной (рис. 2.4).
Рис. 2.4. Схемы трубчатого проточного и емкостного реакторов. В идеале трубчатый реактор работает,
как реактор вытеснительного типа, в котором газ толкается поршнем, движущимся по трубе. В идеальном емкостном реакторе компоненты в резервуаре
постоянно перемешиваются, так что отсутствуют градиенты концентраций компонентов. При открытых
вентилях, показанных на рисунке, идет постоянная
подача реагентов в резервуар и выгрузка продукта. В
этом случае мы имеем дело с емкостным реактором
с постоянным перемешиванием
Трубчатые реакторы обычно используются в крупномасштабных технологических процессах. Реагенты подаются в него с постоянной скоростью, а продукт
появляется на выходе также с постоянной скоростью. Говорят, что такие реакторы работают в стационарных условиях, подразумевая, что как скорости реакции,
так и концентрации компонентов в данной зоне реактора не зависят от времени
(имеются, конечно, малые флуктуации параметров около средних значений).
Емкостные реакторы используются для получения продуктов тонкого синтеза. Реагенты загружаются в них и постепенно превращаются в продукт. Очевидно, что в этом случае и скорость реакции, и концентрации компонентов
являются функциями времени. Мы начнем наш анализ с кинетики взаимосвязанных реакций, протекающих в стационарном режиме.
Без ограничения общности анализа мы можем упростить его, заменив реагенты, интермедиаты и продукты одиночными молекулами:
R < > I 4 > Р. (2.69)
*1
Уравнения скоростей реакций для каждого из участников процесса имеют вид:
ffl = -*,+ [R] + *r[I], (2.70)
= К [R] - (^Г + ^2+)[1]> (2.71)
^1 = ^[1]. (2.72)
Предположив, что процесс идет в стационарных условиях, как это имеет
место в промышленных ректорах при постоянных скоростях подачи реагентов
и отвода продуктов, мы можем концентрацию интермедиата считать постоянной, что соответствует стационарному приближению:
Труба
Резервуар
Трубчатый реактор
вытеснительного типа
Емкостной
реактор
М,о.
d t
(2.73)
Глава 2. Кинетика химических реакций
Это равенство позволяет исключить из уравнений скоростей реакций концентрацию компонента I, которая в общем случае практически не поддается
измерению:
[I] = ^ . (2.74)
1 J Jfcf + kj
Скорость образования продукта теперь определяется выражением
d[P]_*,+*i+[R]
d t kx + kj
(2.75)
Возможны два предельных случая. Если скорость второй стадии реакции
мала, мы можем пренебречь соответствующей константой в знаменателе формулы (2.75):
<2.76)
В этом случае скорость полной реакции лимитируется стадией превращения интермедиата в продукт, которая и является определяющей. Первой ступени при этом отвечает виртуальное равновесие, так что концентрация интермедиата зависит только от константы равновесия Ку
Кажущаяся энергия активации находится путем вычисления логарифмической производной, как это выражает формула (2.48):
„2э1п(ед)
дТ
Екаж = ЯТ2 ' = Я., 2 + АЯ1. (2.77)
Энергия активации для реакции как целого обычно называется кажущейся
энергией активации, и в данном случае является комбинацией параметров двух
ступеней реакции. Если энтальпия первой стадии отрицательна и велика по
абсолютной величине (то есть на первой стадии реакция экзотермична), то
кажущаяся энергия активации может стать отрицательной. Это означает, что с
увеличением температуры концентрация интермедиата падает быстрее, чем
растет константа скорости второй ступени.
Второму предельному случаю отвечает быстрая вторая стадия, когда определяющей для скорости реакции является первая стадия. В этом случае уравнение скорости принимает вид
ffl = A:,+ [R] (Л?»*,’). (2.78)
Энергия активации реакции как целого совпадает с энергией активации
первой стадии Еа г
Заметим, что быстрая стадия, следующая за лимитирующей, является кинетически несущественной. Наоборот, если быстрая стадия предшествует ли¬
2.6. Взаимосвязанные реакции в проточных реакторах
митирующей, то она влияет на полную скорость реакции, поскольку ею определяется концентрация интермедиада, который превращается в продукт на лимитирующей стадии.
Хотя рассмотренным выше примерам отвечают разные механизмы реакции, полная скорость в обоих случаях зависит от концентрации реагента одинаково. Кинетические измерения покажут, что и в том, и в другом случаях
реакция будет иметь первый порядок по концентрации [R]. Так что в общем
случае невозможно обосновать механизм реакции только на основании данных
о ее кинетике: всегда можно найти модифицированный механизм, обеспечивающий те же кинетические зависимости для скорости реакции. Тем не менее
кинетические измерения часто позволяют исключить из рассмотрения некоторые механизмы реакций.
Пример
Рассмотрим реакцию
203 ^ 302, (2.79)
которой отвечают следующие промежуточные стадии (мы пренебрегаем обратными реакциями)
03—^02 +0, (2.80)
0 + 03—^—>202. (2.81)
Уравнения скорости для каждого из компонентов таковы
<*[Оз]
dt
d[0]
dt
d[02]
dt
= -М°з]-Ыо][Оз],
= М0з]-М0][03],
= А,[03] + 2Л3[0][03].
(2.82)
(2.83)
(2.84)
Использование квазистационарного приближения дает для концентрации
кислорода выражение [О] = kjk2, и скорость полной реакции принимает вид
г = ^^ = 3Мо3]. (2-85)
Таким образом, реакция имеет первый порядок по концентрации озона.
Если бы мы полную реакцию описали одной ступенью, то она имела бы второй
порядок по концентрации озона. Кинетические измерения в этом случае позволяют определить возможный механизм реакции.
62 -l\r Глава 2. Кинетика химических реакций
Цепные реакции были открыты в 1913 г., когда Боденстейн и Дюкс обнаружили, что реакция между Н2 и С12 может быть инициирована потоком
фотонов. Их очень удивило, что на один фотон, поглощенный в реакторе
смесью, приходится около 106 (!) образовавшихся молекул НС1 (большой
квантовый выход). Объяснение этого явления было дано в 1918 г. Нерн-
стом. Он предположил, что фотон вызывает диссоциацию молекул С12 на
два радикала СГ (начальная стадия), которые начинают следующий цепной процесс:
СГ + Н2 -> НС1 + Н*,
Н* + С12 -> НС1 + СГ,
являющийся замкнутым циклом (стадии развития цепи), который длится
пока два радикала хлора не рекомбинируют с образованием молекулы хлора.
Процесс рекомбинации называют обрывом цепи. При низкой концентрации радикалов вероятность обрыва цепи составляет около 1/Ю6 [4].
Стационарное (или квазистационарное) приближение сыграло исторически
важную роль в разгадывании механизма, на первый взгляд, простой реакции
Н2 + С12 = 2НС1, которая идет по цепному механизму с образованием радикалов.
Сейчас мы рассмотрим реакцию образования N0 из молекул N2 и 02, которая
протекает, в частности, в двигателях внутреннего сгорания. В гл. 10 мы опишем,
как N0 удаляется каталитическими методами из выхлопных газов автомобилей.
Реакция начинается с диссоциации 02 на горячих частях двигателя:
02—^—>20. (2.86)
Активные атомы О вовлекаются в распространяющийся цикл формирования N0:
О + N3 —*2—» NO + N,
(2.87)
N + О, —»N0 + О.
Наконец, цикл обрывается реакциями
20—^-+02
ИЛИ
2N—^->N2. (2.88)
Мы примем, что последняя реакция является несущественной, и будем использовать парциальные давления вместо концентраций, поскольку реакция
протекает в газовой фазе. Скорость образования N0 равна
= к2РоРщ + кзРнРъ ■
(2.89)
2.7. Взаимосвязанные реакции в емкостных реакторах
X
63
Квазистационарное приближение для парциальных давлений атомов О и N
справедливо, если число циклов, совершаемых до обрыва цепи, велико. Принимая это допущение, находим:
dPu
dt
— kiPoPn, къРнРо, ~ О,
-т~ — 2^| Ро, к2РоРы-> + кзРыРо, ~к4 ра — 0.
at
Отсюда непосредственно получаем
Ро
Г, Л>/2
h
Kk4j
1/2
Ро, > Pn
Кк4 J
1/2
РоС'Рщ ■
(2.90)
(2.91)
(2.92)
Подставляя эти выражения в уравнение скорости реакции, имеем для скорости образования NO выражение:
dp]
NO
dt
= 2к,
Ч^,/2
Kk4j
РсьРн, ■
(2.93)
Порядок реакции по кислороду, равный 1/2, получился вследствие диссоциации молекул 02 на начальной ступени. С такой ситуацией мы будем сталкиваться достаточно часто при обсуждении каталитических реакций.
2.7. ВЗАИМОСВЯЗАННЫЕ РЕАКЦИИ
В ЕМКОСТНЫХ РЕАКТОРАХ
Другим важным для охраны окружающей среды примером служат
взаимосвязанные реакции, протекающие в емкостном реакторе. Мы примем,
что емкостной реактор является реактором с постоянным перемешиванием,
так что все компоненты в нем хорошо перемешаны и нет градиентов состава.
Будем также считать, что все реакции идут только в прямом направлении, так
что последовательность элементарных стадий сводится к следующей
<2'94)
Для сокращения обозначений мы опустили знаки + в константах скоростей
реакций. Уравнения скорости реакций для всех компонентов в данном процессе имеют вид
= (2.95)
= к\ [R] - к2 [I], (2.96)
^ = Ы1]- (2.97)
Глава 2. Кинетика химических реакций
Теперь нам необходимо определить зависимость концентраций от времени.
Уравнение для скорости превращения R легко интегрируется, что дает (см.
разд. 2.5)
Подставив это выражение в уравнение (2.96) и проведя разделение членов,
содержащих [I] и [R], получаем
Если бы в левой части (2.99) стояла производная произведения, например
[I ]/(/), то это уравнение можно было бы легко проинтегрировать. Чтобы при-
вести уравнение к такому виду, умножим обе части на функцию f(t), выбранную таким образом, что
Используя условие равенства нулю концентрации интермедиата в начальный момент времени, приходим к решению
Наконец, подставляя это решение уравнение для скорости реакции продукта
[Р] и используя то же начальное условие [Р] = 0 при t = 0, получаем зависимость от времени концентрации продукта:
На рис. 2.5 показаны зависимости от времени концентраций для различных
значений констант скорости. Заметим, что интермедиат появляется сразу же
после начала реакции, о чем говорит положительное значение производной по
времени от его концентрации при t = 0, тогда как скорость образования продукта в начальный момент времени равна нулю. На кривой зависимости концентрации продукта от времени имеется точка перегиба, положение которой
соответствует максимуму концентрации интермедиата.
Предположим, что мы проводим органический синтез в емкостном реакторе и наша цель — получить интермедиат, а не конечный продукт. Тогда для нас
важно определить время синтеза, при котором будет получен максимальный
[R] = [R]0 e’v-
(2.98)
(2.99)
(2.100)
В результате получаем
(2.101)
(2.102)
к* с-*2 '
к2 -к{
\
/
(2.103)
2.7. Взаимосвязанные реакции в емкостных реакторах
выход промежуточного продукта. Полагая в уравнении (2.99) равной нулю производную d[I]/d/, и подставляя в него решение (2.102), мы определяем время
tmax, при котором достигается максимум концентрации интермедиата. Подстановка этого времени в зависимость (2.102) дает нам оптимальное значение концентрации интермедиата:
1
к{ - к2
-In
к^
[I] =
L Jmax
к21(к2-к\)
[R]0-
(2.104)
S
d'
аз
о.
I-
I
0
d'
z
о
1,0
0,8
0,6
0,4
0,2
0,0
W]
к2 к,
\[1]
Рис. 2.5. Зависимости концентраций
реагента, интермедиата и продукта от
времени для последовательных реакций при различных значениях констант скорости
k,t
Пример
Сейчас мы рассмотрим другой пример, когда интермедиат — крайне нежелательная субстанция, например, она может воздействовать на организм человека, быть взрывчатым веществом или сильно загрязнять окружающую среду.
Гидрирование ароматических нитросоединений, один вариант из таких реакций приведен на рис. 2.6, часто используется в при создании красителей, отбеливателей, в агрохимической и фармацевтической промышленности. Такие реакции обычно протекают в присутствии платинового катализатора и сопровождаются образованием различных интермедиатов, среди которых есть содержащие
гидроксиламин (—NHOH) и представляющие большую опасность, поскольку
они являются канцерогенными и взрывчатыми веществами. К сожалению, при
использовании стандартного платинового катализатора такие вредные веще¬
Глава 2. Кинетика химических реакций
ства появляются в больших количествах. Модифицирование этих катализаторов так называемыми активаторами или промоторами (в данном случае оксидом ванадия) способствует существенному снижению концентрации интермедиатов. Как показано на рис. 2.6, скорость перевода гидроксиламина в амин
существенно увеличивается при использовании катализаторов с активатором.
Интермедиат практически мгновенно выводится из системы, делая реакцию и
безопасной и менее вредной для окружающей среды.
Время, мин Время, мин
Рис. 2.6. Реакция превращения замещенных нитроаренов в амины при гидрировании в
емкостном реакторе с использованием стандартного платинового катализатора.
К сожалению, промежуточный продукт — гидроксиламин — является и взрывчатым, и канцерогенным веществом. Использование модифицированного платинового катализатора, содержащего ванадиевый активатор, позволяет ускорить
вторую стадию настолько, что гидроксиламин появляется в недетектируемых
количествах (с любезного разрешения доктора Х.-У. Блазера, Новартис, Швейцария)
2.8. КАТАЛ ИТИ Ч ЕСКИ Е РЕАКЦИ И
Катализаторы ускоряют химическую реакцию, при этом их состояние практически не изменяется. Как отмечалось в гл. 1, каталитическая реакция идет, когда реагирующие компоненты связываются катализатором. В гетерогенном катализе реагенты адсорбируются на поверхности катализатора, образуя комплексы с его молекулами. Катализаторы должны быть формально
включены в кинетическую схему реакции в качестве участника, не изменяющего итоговую реакцию, как это показано на рис. 2.7, где представлен простейший каталитический цикл. Мы проанализируем кинетику этого простого
двухступенчатого процесса и сопоставим ее с кинетикой некаталитической реакции, обсуждавшейся в разд. 2.6.
Мы можем представить катализатор как набор активных центров (обозначаемых символом «*»), распределенных по поверхности. Полное число цент-
2.8. Каталитические реакции ->\г 67
ров, локализованных на поверхности, равно N и остается неизменным (при
возможной путанице с обозначением N для числа атомов мы будем использовать символ N*). Адсорбцию реагента будем трактовать как его реакцию со
свободным центром, приводящую к образованию интермедиата Г (чаще используется обозначение R*, показывающее, что это реагент, находящийся на
активном центре). Все центры адсорбции идентичны, и на них может располагаться только по одной молекуле реагента. Долю занятых молекулами реагента
R центров будем обозначать через 0R, при этом число занятых центров, очевидно, равно N6K. Доля незанятых и потому доступных для реагентов центров
определяется комбинацией 1 — вк.
Рис. 2.7. Схематическое представление
простейшей реакции в гетерогенном катализе
Реагент
Продукт
Интермедиат ^7
(
Поверхность катализатора
Нижеследующие уравнения описывают представленный на рис. 2.7 цикл:
R + < *L~» R% (2.105)
k\
R —(2.106)
Прежде чем выводить уравнения реакций, нам необходимо обсудить размерность соответствующих скоростей. В гетерогенном катализе реагенты и
продукты находятся в трехмерной газовой или жидкой среде, в то время как
интермедиат локализован на двумерной поверхности и для него мы не можем
использовать понятие концентрации, как в обычных некаталитических реакциях. На протяжении всей книги мы будем измерять скорость реакции в молях
в единицу времени. Кроме того, мы будем использовать микроскопическое
понятие «число оборотов реакции», определяемое как число молекул, претерпевших химическое превращение в единицу времени на одном активном центре. Макроскопическая скорость определяется удельной (на единицу веса или
объема) активностью катализатора и учитывает его геометрию, состав и т.д.,
тогда как число оборотов реакции является внутренней характеристикой каталитического центра.
С учетом данных определений макроскопическая скорость может быть записана в виде
(2.107)
Глава 2. Кинетика химических реакций
-^ = WV(1-0r)[R]-(£,-+*2+H,
(2.108)
(2.109)
Заметим, что скорости образования продукта и превращения реагента имеют размерность моль в единицу времени и пропорциональны числу активных
центров, то есть количеству катализатора, присутствующего в реакторе. Далее,
в случае реакций второго порядка, например, между адсорбированными компонентами А* и В*, скорость реакции в приближении среднего поля определяется выражением г = Nk0AOB. Это выражение показывает, что скорость пропорциональна числу адсорбционных центров и вероятностям нахождения на центрах молекул А и В, которые, в свою очередь, пропорциональны степеням
покрытия поверхности. В приближении среднего поля принимается, что компоненты А и В равномерно распределены по N имеющимся центрам. Это допущение близко к реальности, когда адсорбированные молекулы отталкиваются
друг от друга. Отметим также, что скорость реакции не равна г = &(М?А)(М?В),
поскольку реагирующие молекулы должны находиться на близко расположенных центрах. Важно также указать, что и для реакций второго порядка скорость обычно пропорциональна количеству присутствующего в реакторе катализатора, что отражено и в приведенной формуле г = NkOkOB.
Предположив, что каталитическая реакция осуществляется в трубчатом реакторе в стационарных условиях, мы можем использовать стационарное приближение и исключить из рассмотрения долю адсорбированного интермедиата, что дает:
В результате для скорости простейшей каталитической реакции превращения R в Р имеем
Первое из написанных выражений обычно используется в гетерогенном
катализе, а второе — типично для гомогенного и ферментативного катализа,
где константа
(2.110)
у d [р] + V ) Nk2 [R]
dt 1 + kf + k2 ) + [R]
(2.111)
k{ + k2
(2.112)
называется константой Михаэлиса (см. разд. 2.12).
2.8. Каталитические реакции
Сравнивая скорости каталитической и некаталитической реакций, мы видим, что они отличаются только знаменателем, входящим во вторую формулу
уравнения (2.111). Этот знаменатель обусловлен постоянством числа активных
центров у катализатора. Это можно легко понять, определив степень покрытия
поверхности свободными центрами в*\
То есть скорость каталитической реакции уменьшилась пропорционально
доле свободных центров.
Другой принципиальный факт, на который следует обратить внимание, связан с тем, что некаталитическая реакция превращения R в Р всегда имеет первый порядок по реагенту, тогда как в каталитической реакции порядок по R
является неопределенным и зависит от значений констант скорости, входящих
в уравнение (2.111), которые в свою очередь зависят от температуры. Мы можем только сказать, что порядок будет равен дробному числу, лежащему между
нулем и единицей, значение которого определяется условиями проведения реакции. Ранее мы определили порядок реакции пк соотношением
Взяв логарифмическую производную от уравнения (2.111), получаем следующее выражение для порядка реакции
где к' — эффективная константа скорости. Обратим внимание на сложность
выражения для порядка реакции (1 — 0R), который изменяется и с концентрацией реагента [R] и с температурой. Таким образом, говорить о порядке каталитической реакции бессмысленно, если при этом не указаны условия ее проведения.
Рассмотрим предельные случаи, исходя из общего выражения (2.111). Если
конверсия адсорбированного интермедиата в продукт или реагент происходит
быстро, то знаменатель в формуле (2.11) стремится к 1, так что поверхность
катализатора является практически свободной, вк = 0. В этом случае выражения для скоростей каталитических и некаталитических реакций практически
совпадают, а порядок реакции становится равным 1. В этих условиях целесо-
(2.113)
(2.114)
Тогда скорость реакции в форме степенного закона выглядит так:
(2.116)
Глава 2. Кинетика химических реакций
образно повышать концентрацию реагента, поскольку поверхность катализатора остается свободной и он используется неэффективно.
В другом предельном случае, когда знаменатель в формуле (2.111) становится большим, поверхность практически полностью занята интермедиатом,
поскольку скорость его превращения в продукт или реагент мала. Этому случаю отвечает приближенное уравнение вида
V^- = Nkl, (2.117)
dt
и порядок реакции становится нулевым по отношению к [R]. Заметим, однако,
что реакция имеет первый порядок по числу активных центров N. В дальнейшем нас будет интересовать активность катализатора в расчете на один центр,
так что мы можем положить N = I. Важно, однако, помнить, что активность
катализатора определяется как реакционной способностью центров, так и их
полным числом.
Кажущаяся энергия активации для реакции, описываемой уравнениями
(2.105) и (2.106), может быть найдена непосредственно по уравнению (2.111);
общее обсуждение понятия энергии активации для каталитических реакций
будет проведено ниже.
2.8.1. Приближение среднего поля
Мы уже использовали некоторые положения приближения среднего поля, когда приняли, что все компоненты равномерно распределены по
поверхности катализатора и пренебрегли взаимодействием между адсорбированными молекулами. Последнее предположение выполняется редко, поскольку
между адсорбированными молекулами всегда есть либо притяжение, либо отталкивание (рис. 2.8). Приближение среднего поля работает хорошо, когда взаимодействие между молекулами носит отталкивательный характер, а степень
покрытия поверхности невелика. В случае притяжения между молекулами приближение среднего поля может оказаться непригодным даже при низких степенях покрытия поверхности, в частности при образовании «островков» из
молекул (рис. 2.9), тогда в реакции участвуют молекулы, находящиеся на периферии островков. Вступает в игру диффузия молекул к периферийным центрам, начинается взаимодействие между процессами переноса и реакциями, и
схема полной реакции становится очень сложной. Повышение температуры
способствует однородности распределения молекул по поверхности, степень
достижения которой определяется величиной потенциала притяжения.
При высоких степенях заполнения поверхности взаимодействием между
молекулами пренебрегать нельзя, и предэкспоненциальный фактор начинает
зависеть от 0R. Ниже мы будем считать приближение среднего поля справедливым, следует, однако, помнить, что оно может стать источником заметных
ошибок. Альтернативой к данному приближению служат методы численного
моделирования, например метод Монте-Карло (см. гл. 7).
2.9. Изотермы адсорбции Ленгмюра
Рис. 2.8. Модель поверхности типа
шахматной доски при наличии адсорбированных молекул двух сортов: слева - равномерное распределение, справа показан островок из
молекул сорта В. В последнем случае возможна реакция только между молекулами А и находящимися
на периметре островка молекулами
В, так что скорость реакции уже не
пропорциональна степени покрытия
поверхности молекулами В
ш
ш
[1
т
ш
Ваг#
1
ЕЖ!
11
[в1
ш
Ц]
в
| В
ш
8
г а вА вв
г <еА ев
Рис. 2.9. Сегрегированные компоненты (черные и серые точки) на
модельной поверхности типа шахматной доски
2.9. ИЗОТЕРМЫ АДСОРБЦИИ ЛЕНГМЮРА
Первым шагом любой реакции гетерогенного катализа является
адсорбция молекул на поверхности катализатора. Здесь мы сосредоточим свое
внимание на реакциях газов на твердых катализаторах. Адсорбции газов будет
посвящена отдельная глава, тем не менее сейчас нам необходимо установить
связь между парциальным давлением газа и степенью покрытия поверхности
его молекулами. Функциональные зависимости между степенью покрытия и
давлением называются изотермами адсорбции, которые играют важную роль в
кинетике каталитических реакций.
Первая количественная теория адсорбции была разработана Ирвином Ленг-
мюром (1891—1957), исследовавшим износ вольфрамовых нитей в лампах накаливания в период его работы на фирме «Дженерал Электрике». Обобщив эти
Глава 2. Кинетика химических реакций
работы на случаи адсорбции и реакции газов на металлических поверхностях,
он внес существенный вклад в развитие кинетической теории каталитических
реакций, за что ему была присуждена в 1932 г. Нобелевская премия по химии.
Сейчас мы дадим вывод изотермы адсорбции Ленгмюра для молекулярной,
диссоциативной и конкурентной адсорбции.
2.9.1. Ассоциативная адсорбция
Вывод изотермы адсорбции Ленгмюра проводится просто. Принимаем, как и раньше, что на поверхности адсорбента имеются активные центры
одной и той же природы и адсорбированные молекулы не взаимодействуют
между собой. При наличии равновесия между газовой фазой и адсорбатом мы
можем написать уравнение реакции в виде
А + : < Ад > А • (2.118)
кА
При этом уравнение скорости реакции, очевидно, выглядит следующим
образом
d Й
^ ~ РА^А (1 ~ ) “ ^А^А 5 (2.119)
где первое слагаемое связано с адсорбцией, а второе — с десорбцией молекул.
Для равновесной ситуации мы можем написать
вА=КАрА(1-вА); КА = k+Jk-A. (2.120)
Эти два уравнения легко преобразуются в изотерму адсорбции Ленгмюра, описывающую ассоциативную адсорбцию молекул простого газа (например, СО,
NH3, NO при условии, что они не разваливаются на поверхности):
КаРа
1 + КАрА
Заметим, что доля свободных центров при этом равна
вк = . (2.121)
в‘=х-в^ = \Л „ > (2Л22)
1 + каРа
и уравнение адсорбции Ленгмюра может быть записано в форме вА= КАрАв*
(эта форма удобна при определении кинетической схемы реакции, когда доля
свободных центров не известна).
На рис. 2.10 показана зависимость степени заполнения 0Аот парциального
давления рА. При низких давлениях, когда степень покрытия поверхности еще
невелика, она линейно возрастает с давлением. Производная вА по давлению
при этом совпадает с КА. При высоких давлениях, когда поверхность насыщена
адсорбатом, степень покрытия приближается к максимальному значению, отвечающему 100 %-у заполнению активных центров.
2.9. Изотермы адсорбции Ленгмюра
Рис. 2.10. Изотермы адсорбции Ленгмюра для ассоциативной адсорбции, представленные в форме зависимости
степени покрытия поверхности от давления, для трех различных констант равновесия
к
s
н
-О
а
0
с
J2
1
Ф
С
Ф
Давление, бар
Соответствие экспериментальных данных изотерме Ленгмюра проверяется
достаточно легко: при наличии такого соответствия экспериментальные данные, построенные в координатах \/вА—\/рА, должны лечь на одну прямую,
тангенс угла наклона которой равен 1/КА.
2.9.2. Диссоциативная адсорбция
Многие молекулы типа Н2 и 02, будучи адсорбированными на твердую поверхность, практически всегда диссоциируют, поэтому в общем случае можно
считать, что равновесие устанавливается между адсорбированными атомами и
молекулами в газовой фазе. В таком случае мы имеем химическую реакцию
А2 + Г
± 2А\
которой соответствует уравнение скорости вида
AQ ,
■ = />а2*а(1-0л) -К&м
d t
приводящее к следующему условию равновесия
в\ =PAlKA2(i-eAf
и равновесным степеням заполнения поверхности
в =
1
(2.123)
(2.124)
(2.125)
(2.126)
Эти выражения можно рассматривать как изотерму Ленгмюра для диссоциативной адсорбции.
Глава 2. Кинетика химических реакций
2.9.3. Конкурентная адсорбция
Весьма важным является случай, когда компоненты А и В могут
занимать один и тот же центр, делая его недоступным для второго компонента:
А + * <—-р > А:,
В + <—-р—> В .
£
1 + КАрА + Кврв
1
1 + каРа + КъРв
(2.127)
Этой ситуации отвечают условия равновесия
@а = каРа6^ въ = КъРъ0Ф. (2.128)
Из этих выражений и условия сохранения числа центров
0А+0в+6> =1 (2.129)
получаем следующие значения степеней заполнения поверхности
q _ КаРа
А 1 + КаРа + КвРв
6>в = , „КйРв 7;- , (2.130)
Конкурентная адсорбция имеет место во многих каталитических реакциях
и существенно влияет на условия их протекания. Соотношения (2.130) показывают способ регулирования этих условий. Если, например, компонент А сильнее связывается с поверхностью (то есть КА > KQ), то увеличить концентрацию
компонента В на поверхности можно, повысив его давление /?в > /?А, что скомпенсирует разницу в силе связи молекул с поверхностью.
2.10. МЕХАНИЗМЫ РЕАКЦИЙ
Сейчас мы проведем анализ кинетики общей каталитической реакции:
А + В > АВ. (2.131)
Каталитический процесс представляет собой последовательность элементарных стадий, образующих замкнутый цикл, из которого катализатор выходит
неизменным. Идентификация каждой стадии и формирующихся на ней интермедиатов может быть сильно осложнена: для ее проведения необходимо привлечение спектроскопических методов и численного моделирования. Некоторые соображения по этому вопросу приведены в гл. 7. Здесь же мы примем, что
элементарные стадии известны, и подробно продемонстрируем, как следует
конструировать уравнения реакции для таких процессов.
2.10. Механизмы реакций -J л 75
2.10.1. Механизмы Ленгмюра—Хиншельвуда
или Илея—Ридела
В теории Лэнгмюра—Хиншельвуда принимается, что компоненты
адсорбируются на поверхность и приходят с ней в тепловое равновесие, прежде
чем они вступят в реакцию. Таким образом, компоненты реагируют, находясь
в хемосорбированном состоянии. Такое состояние реагирующих компонентов
превалирует в гетерогенном катализе.
Другой сценарий отвечает так называемому механизму Илея—Ридела, в
котором считается, что один из компонентов вступает в реакцию непосредственно из газовой фазы без предварительной аккомодации на поверхности.
Например, для реакции А + В находящаяся в газовой фазе молекула В может
приблизиться к поверхности и прореагировать с хемосорбированной молекулой А*, при этом ей не нужно адсорбироваться на поверхность. Примером
реакции, протекающей по сценарию Илея—Ридела, является реакция газофазного атомарного водорода с атомарным водородом, насытившим поверхность.
Находясь в сильно активированном состоянии, газофазные атомы водорода
(атомарный водород — радикал) легко реагируют с адсорбированными атомами водорода, и поскольку молекулы водорода слабо взаимодействуют с поверхностью, они мгновенно десорбируются.
Для кинетического описания принципиально важно, какой из сценариев —
Ленгмюра—Хиншельвуда или Илея—Ридела — реализуется в конкретной реакции, поскольку, например, во втором случае для одного из реагентов не требуется наличия свободных центров на поверхности. По механизму Илея—Ридела
реакции протекают достаточно редко, поэтому в дальнейшем мы будем рассматривать только механизм Лэнгмюра—Хиншельвуда.
2.10.2. Кинетика Ленгмюра—Хиншельвуда
Предположив, что реакция между компонентами А и В идет по
механизму Ленгмюра—Хиншельвуда, мы можем выделить следующие ее
стадии:
1)
2)
3)
4)
А +
В +
А + В
К
* А”,
± В.
± АВ + *,
АВ < 4 > АВ + \
к4
(2.132)
(2.133)
(2.134)
(2.135)
76 -l\r Глава 2. Кинетика химических реакций
Заметим, что для финального шага десорбции константа равновесия адсорбции молекул АВ равна 1 /К4, тогда как для других адсорбционных шагов она
такова
*,=f, (2.136)
К
где константа к+ описывает адсорбцию. На шаге 4 относится к десорбции.
В схеме каталитического цикла, представленной уравнениями (2.132)—(2.135),
предполагается, что все стадии реакции термодинамически и стехиометричес-
ки согласованы. Например, число центров, занимаемых на этапах адсорбции и
диссоциации, должно быть равно числу центров, освобождаемых при образовании продукта и его десорбции, что отражает основной критерий катализа,
согласно которому катализатор остается неизменным по завершении каталитического цикла.
Каждому шагу каталитического цикла соответствуют свои скорости реакции:
п = Kp&-KQa,
(2.137)
Л*2 = А'1 Р\\@ — к^О^)
(2.138)
гз = \ - kj 0АВ 0.,
(2.139)
/4= £4+6>\в -к4рАВв: ,
(2.140)
где мы для удобства исключили из рассмотрения полное число центров, так
что г характеризует скорость на одном центре катализатора или число оборотов
реакции.
Число центров на поверхности катализатора остается неименным, поэтому
сумма всех степеней покрытия всегда равна единице, и мы имеем следующее
условие баланса:
О* + + ^ав ~ 1- (2.141)
Приведенные выше уравнения постоянно будут использоваться при анализе каталитических реакций.
2.10.3. Полное решение
Для нахождения решения кинетической задачи в общем виде, например при допущении возможности изменения во времени парциальных давлений, нам нужно включить в систему и уравнения, описывающие эволюцию
степеней покрытия поверхности компонентами, участвующими в реакции:
-А = Г\-ГЪ= к;рА в, - к;0А - кз+ 6>д вв + к^0АВвг,
(2.142)
2.10. Механизмы реакций
X
d0
—— = r~, -r3 = klpB6: - k^OB - qeAeB + /c3~0AB#, (2.143)
dt
An
~~~~ ~ r3 ~ r4 = ~ ^3 ^AB^* “ ^4^AB + ^4 PaB^*» (2.144)
at
dO,
—- = -r, - Г, + r3 + r4 =
d/ “ (2.145)
= -/ч>а# + 6A - k~_pBe + + к;вАвв - k^0ABO + k+A0AB - k~ApAB6>:.
Уравнения такого типа легко решаются численными методами на персональных компьютерах без использования каких-либо приближений. Для описания кинетики переходных (зависящих от времени) процессов, характеризующих реакцию, все входящие в систему параметры, естественно, должны быть
либо известны из эксперимента, либо оценены с хорошей степенью точности.
Для зависимых от времени решений, однако, как при любых численных расчетах, могут возникнуть проблемы, связанные с их неустойчивостью. Но полное
решение не требуется, если нас интересуют только стационарные (не зависящие от времени) состояния.
2.10.4. Стационарное приближение
В промышленности, как и в лабораторных реакторах, довольно часто
реализуются процессы, при которых реагент с постоянной скоростью подается
в реактор и из него также с постоянной скоростью выходит продукт. Все переходные процессы, связанные с началом синтеза, довольно быстро вырождаются в стационарное состояние, при котором степени покрытия поверхности и
скорости принимают постоянные значения, так что производные в уравнениях
(2.142)—(2.145) обращаются в нуль:
dO
—Л- = г, - г3 = 0 => к* рлв - к{вА - к;вАвв + kj0ABe.t = 0, (2.146)
dO
—— = г, - гъ = 0 => к*рвв - квъ - к;вАвв + к^б.об, = 0, (2.147)
а/
dO
^ — гъ “ r4 = Q => к3 0А0В - къ 0АВ0,. - к4 0АВ + к4 рА^0,. = 0, (2.148)
dO,
—— = ~Г\ -Ъ + Гг +Гд = 0 =>
dt 12 3 4 (2Л49)
=> ~Кра0, + к~0А - к;рв0, + к;ов + Ц0А0В - к~ъ0АВ0„ + к\0АВ - к;рАВе = 0.
Последнее уравнение не является независимым: уравнение баланса (2.141)
позволяет получить его из трех других уравнений, так что мы всегда имеем
п —1 уравнение реакции, содержащей п элементарных ступеней. Заметим,
Глава 2. Кинетика химических реакций
что стационарное состояние не предполагает низких значений поверхностных концентраций: просто они не изменяются во времени. В стационарном
приближении мы не можем определить, как система развиваются во времени, но оно оказывается достаточным для описания многих каталитических
процессов.
2.10.5. Приближение квазиравновесия
В этом приближении предполагается, что одна из ступеней определяет скорость реакции, тогда как реакции других ступеней протекают очень
быстро и относительно них компоненты находятся в квазиравновесии. Если мы
примем, что поверхностная реакция формирования комплекса АВ* (3-я ступень,
уравнение (2.134)) является лимитирующей стадией (J1C), уравнения, отвечающие стадиям 1, 2 и 3, можно переписать в виде:
г\ = 0 к\ Рк@* ~ к\ @а ^ ft\ = К\Ра&ч (2.150)
г2 = 0 => к2рвв* = к2вв => вв = К2рвв^ (2.151)
г4 = 0 => к4вАВ = к4рАВ => вАВ = К4Х рАВ6*. (2.152)
По сути, мы пришли к уравнениям Ленгмюра для адсорбирующихся и десорбирующихся компонентов. Подставляя полученные степени заполнения поверхности в уравнение для скорости лимитирующей стадии, получаем
г - гъ ~ k~s 0А6В к3 0А^0 - кг КхК2рАрв
Г /*АВ
1
в}. (2.153)
Ра Ре ^2 Кз К\ j
Введя константу равновесия для полной реакции в газовой фазе
KG =К{К2К3К4 (2.154)
и приравнивая скорости полной реакции и лимитирующей стадии, получаем
г = г^г- =к;К,К2рАрв( 1 W. (2.155)
V PaPb^g )
Сравнивая это выражение со скоростью (2.8), легко заметить, что комбинация в скобках представляет собой степень приближения реакции к равновесию. Однако, в отличие от уравнения (2.8), здесь вместо объемных концентраций стоит ft2, то есть квадрат доли свободных центров, доступных для реаген¬
тов. Таким образом, даже если константы скорости имеют большие значения и
степень сродства реакции к равновесию велика, процесс может протекать достаточно медленно при недостатке свободных центров, то есть если поверхность
заблокирована и 0* —> 0.
2.10. Механизмы реакций
Доля свободных центров, входящая в уравнение (2.155), находится из уравнения сохранения
Ъ*'
/•=1
1,
(2.156)
где п — число различных компонентов на поверхности, включая свободные центры. Поскольку доли поверхностных центров, занятых различными атомами,
выражаются через долю свободных центров [уравнения (2.150)—(2.152)], то, решив уравнение (2.156), можно получить для последней следующую формулу:
ft: =
1
(2.157)
1 + К\Ра + KiPb + К41рАВ
Используя это равенство, можно рассчитать степени покрытия поверхности различными интермедиатами и полную скорость реакции, представив ее как
функцию констант равновесия квазиравновесных ступеней, давлений реагентов и продуктов и константу скорости лимитирующей стадии:
0В -
К\Ра
1 + к,
Ра
+ К2рв +
К~4
>
СО
К^Рв
1 + Kt
Ра
+ К2ръ +
К~4
>
со
^4 1 Рлв
1 + К\РА + К2рв + К4 /?АВ
Г = г+ - Г = к$КхК2рАрв
Р АВ
PaPbKi
G
1
1 + KiPa + К2рв +Ц рАв
(2.158)
(2.159)
(2.160)
(2.161)
Важно осознавать, что использование представлений о лимитирующей
стадии делает ограниченной применимость полученных формул. Как и в случае стационарного приближения, в рамках квазиравновесной модели нельзя
описать переходные процессы. Более того, изменив внешние условия, например, при значительном уменьшении или росте парциальных давлений, можно
сделать лимитирующей любую реакцию. Так, при проведении реакции А + В
на поверхности монокристалла в высоковакуумной камере можно реализовать такие низкие парциальные давления компонентов А и В, что скорость
адсорбции будет существенно меньше скорости поверхностной реакции.
2.10.6. Ступени реакций с близкими по величине скоростями
В случае, когда несколько стадий характеризуется малой скоростью реакции, необходимо принимать во внимание отклонения от равновесия
для каждой из этих стадий. Предположим, например, что стадии 1 и 3 в схеме,
описываемой уравнениями (2.132)—(2.135), являются медленными, а стадии 2
80 -l\r Глава 2. Кинетика химических реакций
и 4 можно рассматривать как квазиравновесные, тогда приходим к следующей
системе уравнений:
г\=к\Р&~к\вм (2.162)
АП
г, = 0 =» —— = 0 => kjрв0, = к^0в => #в = К0рБв+, (2.163)
ш
'з “ “ ^3 ft\eft ’ (2.164)
d/9
г4 - 0 => = 0 => А:40ав = к4рАВ0^ => 0АВ = K4lpABft;. (2.165)
Решение этих уравнений находится более сложно, чем в случае одной лимитирующей стадии, однако оно все равно проще полного решения.
2.10.7. Приближение необратимых стадий
Можно сделать дальнейшее упрощение квазиравновесного приближения, исключив обратные реакции на некоторых стадиях. Например, мы можем
представить ситуацию, при которой концентрация продукта АВ настолько низка,
что обратной реакцией на стадии 4 можно пренебречь. Это допущение сильно
упрощает уравнение (2.161), поскольку позволяет положить рАВ = 0, тогда
г = г=г+ =—кък\КгРкРъ (2.166)
(1 + KiPa+K2Pb)2
Следует иметь в виду, что в этом приближении нельзя описать стремление
системы к равновесию, поскольку мы считаем концентрацию продукта пренебрежимо малой. Заметим также, что уравнение (2.166) относится также к
случаю, когда в адсорбционно-десорбционном равновесии превалирует десорбция, то есть температура системы такова, что молекулы АВ не адсорбируются
на поверхности.
2.10.8. Приближение наиболее избыточного интермедиата
реакции (НИИР)
Приближение наиболее избыточного интермедиата реакции (НИИР)
также является дальнейшим упрощением квазиравновесного приближения.
Довольно часто один из интермедиатов настолько сильно адсорбируется в сравнении с другими компонентами, что он доминирует на поверхности. Этот интермедиат называется НИИР. В этом случае уравнение сохранения сводится к
следующему:
^ 0/ ~ ft: + 0Ниир - 1.
/=1
(2.167)
2.10. Механизмы реакций
X
Если в нашей схеме реакции [уравнения (2.132)—(2.135)] компонент А связывается с поверхностью сильнее, чем компоненты В и АВ, а парциальные
давления компонентов В и ВА сравнимы с давлением компонента А или меньше него, то компонент А будет НИИР, и степень покрытия поверхности для
него дается выражением
ft
_ к{Ра
0В=О, 0АВ=О, ft, = \-вА
а скорость реакции принимает вид:
г = г+ -Г = k^K[KlP аРв
Р АВ
kgPaPb
1
(2.168)
(2.169)
81
2.10.9. Почти свободная поверхность
Когда интермедиат слабо связан с поверхностью или температура очень высока, равновесие смещается в сторону газовой фазы, и поверхность остается практически свободной, что позволяет использовать приближение
ft =1. (2.170)
Выражение для скорости реакции (2.169) при этом заметно упрощается,
поскольку в нем можно пренебречь последним сомножителем. Если к тому же
принять, что лимитирующая стадия необратима или концентрация продукта
мала, то можно рассматривать только прямую реакцию, что позволяет свести
выражение для скорости реакции к следующему:
r = r+=k+K{K2pApB. (2.171)
Для определения состава реакционной смеси, отвечающего оптимальной
скорости, удобно ввести относительную концентрацию %А через соотношение:
Za=—• (2-172)
Ра + Рв Рш
Тогда
Ра=ХаРн* и Рв={1~Ха)Рш- (2.173)
Экстремум скорости легко находится путем прямого дифференциро¬
вания
^- = о^к;к,к2{1-2хА) = о^хА Л.
(2.174)
Глава 2. Кинетика химических реакций
Таким образом, в пределе нулевой степени конверсии для почти свободной поверхности скорость принимает максимальное значение при равных давлениях реагентов. Снова напомним, что при практическом использовании полученных выражений мы должны оценить, пригодно ли данное
приближение в условиях проведения реальной реакции. Тем не менее изложенная выше процедура определения зависимости скорости реакции от
мольных долей компонентов оказывается полезной при выяснении ее механизма.
2.10.10. Порядок реакции
Порядок реакции ni по отношению к компонентам А, В и АВ может быть определен путем дифференцирования скорости реакции, как это сделано в формуле (2.11). В тех немногих случаях, когда имеется численное решение кинетических уравнений, полученное по описанной в подразд. 2.10.3 схеме, порядок реакции может быть найден также численным дифференцированием.
Сейчас для проведения расчета порядка реакции мы будем считать справедливым квазиравновесное приближение, что позволяет нам использовать аналитическое выражение для скорости реакции (2.161). Имеем:
па = Ра
Э In {г*)
дрА
= 0 + 1-
= Ра
дрА
\п(к;К,К2рн)+\п(рА)
—2 In (l + К\РА + К2рв + К4 рАВ j
2К[Ра
1 + KiPa + К2рв + К41рАВ
пв = 1-20в,
ПАЪ - 20ДВ •
= 1-2*а,
(2.175)
(2.176)
(2.177)
Используем уравнение (2.161) для иллюстрации зависимости скорости реакции от парциальных давлений реагентов и температуры. Сделаем еще одно
упрощающее допущение, приняв, что продукт АВ очень быстро десорбируется
с поверхности при данной температуре реакции, так что его присутствием на
поверхности можно пренебречь на фоне поверхностных концентраций других
реагентов. Это позволяет пренебречь членом КАВрАВ в знаменателе уравнения
(2.161). В этих условиях порядок реакции по компоненту АВ является нулевым.
Результаты расчета параметров реакции приведены на рис. 2.11, где показаны
зависимости степеней покрытия поверхности компонентами А и В, порядков
реакции пА и пв от мольной доли реагента А. Следует обратить внимание, что
порядки реакции не являются постоянными, а сильно изменяются с парциальным давлением. Зависимость скорости реакции от температуры описана в следующем разделе.
2.10. Механизмы реакций ->\г 8
Расчеты проведены
для Ка = 1 при 600 К,
ДНа =-125 кДж/моль;
для Кь= 1 при 600 К,
ДНЬ =-125 кДж/моль;
КаЬ мала,
к = 1 при 540 К,
Еа =+50 кДж/моль; рв = 5 бар
Рис. 2.11. Степени покрытия поверхности реагентами А и В и
скорость реакции как функции
парциального давления компонента А. На нижнем рисунке приведены порядки реакции и кажущаяся энергия активации
о
о
Q.
О
03
I
X
03
со
о
Q.
S
Q.
0
1
S
Q.
Ф
и
к —Г
I «
Ф g
Ct *
ш 03
0Q
S
Q.
С
х3 = рлр+рь)
2.10.11. Кажущаяся энергия активации
Используя формулу (2.48) для кажущейся энергии активации и термодинамическое соотношение (2.51) для константы равновесия, мы можем установить связь между кажущейся энергией активации и степенями покрытия
поверхности:
Э In г+
дкаж s RT1
дТ
= RT2
дТ
In (РаРв ) + In (fc3+) + In (£,) + In (tf2) -
— 2 In (l + K^ph + К2Ръ + K4 />AB)
= RT2
d{-EJRT) d(-AHJRT) Э(-А HJRT)
дТ дТ дТ
Э(1 + KiPa + К2рв + a^Vab)
dt
1 + K\PA + K2pB + K41 pxu
= Ea + ДЯА + Д//в - 2 (#A Д//л + 0пАНн - #Л|!Д//ЛВ):
= £,+(!- 20А) ДЯА + (1 - 2вй) Д Нв + 20АВ Д НАй,
(2.178)
(2.179)
(2.180)
(2.181)
Глава 2. Кинетика химических реакций
где было использовано уравнение Аррениуса для скорости реакции
-е:
к; = и3+ ехр
RT
а также хорошо известное выражение для константы равновесия
К = ехр
-АС,
RT
= ехр
R
ехр
-АЯХ
RT
(2.182)
(2.183)
Мы снова приняли, что предэкспоненциальный фактор и энтропийный вклад
не зависят от температуры. Это предположение, строго говоря, не является
корректным, но, как мы увидим в гл. 3, эти зависимости являются достаточно
слабыми по сравнению с экспоненциальной зависимостью, включающей энергетические факторы. Как видно из рис. 2.11, нормированная кажущаяся энергия активации также зависит от мольных долей реагентов. Отметим, что энергия активации не совпадает с соответствующей величиной для лимитирующей
стадии. Она зависит также от энтальпий адсорбции, предшествующей лимитирующей стадии, и степеней покрытия поверхности реагентами.
Таким образом, кажущаяся энергия активации, найденная по уравнению
Аррениуса, сильно зависит от условий проведения реакции (давление, температура). Сказанное справедливо и в отношении порядка реакции по отношению к присутствующим в системе компонентам.
Пример: Окисление СО
Окисление СО — важный этап каталитического дожигания выхлопных газов двигателей внутреннего сгорания — простая реакция, которая является объектом многочисленных фундаментальных исследований. Катализаторами для этой
реакции обычно служат благородные металлы — платина, палладий, родий,
иридий и даже золото, при условии, что размер частиц катализатора очень мал.
Мы примем, что окисление при таком каталитическом процессе идет по механизму, в котором адсорбированные СО, О и С02 находятся в равновесии с
газовой фазой, то есть мы можем использовать приближение квазиравновесия.
Механизм реакции можно представить следующим образом:
1) СО + <———> со ,
2) О, + 2 < А: > 20 ,
3) со + о
4) со; <н
СО, + (лимитирующая стадия),
-) СО, + .
(2.184)
(2.185)
(2.186)
(2.187)
Здесь третья стадия, описывающая взаимодействие адсорбцированных кислорода и СО, результатом которого является образование молекулы СО,, опреде-
2.10. Механизмы реакций
ляет скорость всей реакции. Можно расписать вторую стадию более детально,
представив ее как адсорбцию 02 с образованием прекурсора 02, который подвергается последующей диссоциации. Для упрощения схемы реакции мы объединили эти два элементарных процесса в один. Интересующийся читатель может сам выяснить, к чему привело такое упрощение.
Для каждой квазиравновесной стадии мы можем либо выписать дифференциальное уравнение реакции, либо сразу же использовать изотерму Ленгмюра:
Теперь непосредственно можно найти выражение для доли свободных центров, используя уравнение баланса
Рассмотрим несколько предельных случаев. Обычно С02 настолько слабо
взаимодействует с катализатором, что его присутствием на поверхности можно
пренебречь, то есть десорбция С02 — очень быстрый процесс, так что реакцию
на четвертой стадии [уравнение (2.187)] можно считать необратимой. Тогда
комбинации, содержащие рс0з, практически равны нулю.
При низких температурах на поверхности доминирует СО, так что он является НИИР, поэтому скорость реакции можно записать в виде
^со ~
(2.188)
(2.189)
(2.190)
1
(2.191)
Скорость реакции совпадает со скоростью лимитирующей стадии:
/
\
г - к3 0СО0О к3 0СОД - к3 К{ у]К2 рсо yjPq2 1
Рсо2
(2.192)
v
/
где
KG = К{^К3К4
(2.193)
— константа равновесия полной реакции:
со + -о2< к° >со2.
2 2
(2.194)
(2.195)
Отсюда сразу видно, что в низкотемпературном пределе порядок реакции
по кислороду nQi = 0,5, по монооксиду углерода псо = —1. Отрицательное
Глава 2. Кинетика химических реакций
значение порядка говорит о том, что поверхность полностью покрыта СО и
повышение его парциального давления снижает скорость реакции вследствие
блокировки реакционных центров, исключающей возможность адсорбции кислорода и его участие в реакции.
При высоких температурах адсорбция мала, также мала и степень покрытия
поверхности реагентами, что позволяет считать ее почти свободной. Это не исключает возможности протекания реакции, но время нахождения реагентов на
поверхности до их десорбции или вступления в реакцию становится малым. Поскольку поверхность практически свободна, мы можем положить ft; =1, что дает
r = klKx4KlpC0^. (2.196)
Заметим, что порядок реакции по кислороду остается равным 0,5, по СО он
становится равным +1. Поскольку поверхность остается практически свободной,
то повышение парциальных давлений обоих компонентов приводит к увеличению
скорости реакции. Таким образом, порядок реакции сильно зависит не только от
давления, о чем говорилось в предыдущих разделах, но и от температуры.
Расчеты проведены
для Ксо = 1 при 600 К,
ЛНС0 = -135 кДж/моль;
для К= 1 при 630 К,
ДН02 = -250 кДж/моль;
Ксо?мапа,
к= 1 при 540 К,
Еа =50 кДж/моль;
Р о2~ со
Р со ~ 1
Рис. 2.12. Степени покрытия поверхности СО и О и скорость реакции
окисления СО как функции температуры (верхний рисунок). Нижний
рисунок представляет зависимости
порядков реакции и полной энергии
активации от температуры. Влияние
С02 на все процессы в расчет не
принималось
На рис. 2.12 показаны зависимости скорости реакции и ее порядков, степеней покрытия поверхности и нормированной кажущейся энергии активации от
температуры. Отметим сильное изменение этих параметров с температурой: в
о
о
Q.
О
X
03
со
о
Q.
S
Q.
О
X
S
оь
100 200 300 400 500 600 700 800 900
=г I
СВ ®
ф £
Q. Sg
S X
£ Ф
3 Ct
5 ф
Q. со
О S
1= О.
С
S
Екаж/|£а + Нсо + 0,5ДНО2|
Sj
/ \
\
ч
100 200 300 400 500 600 700 800 900
Температура, К
2.11. Энтропия, производство энтропии, автокатализ, колебательные реакции
частности, скорость реакции сначала возрастает, затем проходит через максимум, после чего начинает уменьшаться при высоких температурах. Такая зависимость ожидаема для всех каталитических реакций, но ее трудно подтвердить
на практике при использовании нанесенных катализаторов, поскольку здесь
вступают в игру диффузионные процессы на поверхности.
При низких температурах кажущаяся энергия активации является положительной, поэтому скорость реакции увеличивается с ростом температуры. При
высоких температурах, как показывает уравнение (2.181), кажущаяся энергия
активации становится отрицательной, так что скорость реакции падает с ростом температуры. Такое поведение объясняет рис. 2.12: при высоких температурах имеется дефицит адсорбированных компонентов. Скорость достигает
максимума, когда температура настолько высока, что третья стадия может протекать при значимых скоростях и в то же время на поверхности в заметных
количествах присутствуют СО и О, способные вступить в реакцию. Можно
сказать, что технология катализа состоит в выборе таких условий, когда при
достаточно высоких температурах на поверхности катализатора имеется существенное количество реагирующих компонентов.
2.11. ЭНТРОПИЯ, ПРОИЗВОДСТВО ЭНТРОПИИ,
АВТОКАТАЛИЗ, КОЛЕБАТЕЛЬНЫЕ РЕАКЦИИ
Как хорошо известно из термодинамики, изолированные системы
в состоянии равновесия обладают минимальной свободной энергией и максимальной энтропией. Если система выведена из состояния равновесия, то есть
ее энтропия стала меньше равновесной, а свободная энергия превышает равновесную, то она будет самопроизвольно релаксировать к равновесию, теряя информацию о своих предшествующих состояниях. Стремление системы к равновесному состоянию определяется необходимостью минимизации ее свободной
энергии. Примером служит реакция в емкостном реакторе, которая идет до ее
полного завершения.
Реакция, протекающая в стационарных условиях, не достигает равновесия.
В этом случае реакционная система не является изолированной, поскольку в
нее постоянно подается реагент, что делает энтропию ниже равновесной, и
отводится продукт. Степень отклонения от равновесия в этом случае характеризуется скоростью увеличения энтропии dS/dt, которая обычно называется
производством энтропии. Можно показать, что в стационарном состоянии система характеризуется минимальным значением производства энтропии. При
возмущении этого состояния система снова возвращается в него, поскольку
стационарное состояние определяется скоростями подачи реагентов и отвода
продуктов реакции in- Таким образом, можно считать, что стационарному состоянию отвечает наименьшее при заданных внешних условиях отклонение от
равновесия. Стационарные реакции, протекающие в промышленных установках, обычно осуществляются в таких условиях, и для их описания можно приме¬
Глава 2. Кинетика химических реакций
нять линейные соотношения неравновесной термодинамики. Нелинейная неравновесная термодинамика относится к режимам, когда возможны взрывные
процессы или возникают неконтролируемые осцилляции параметров системы.
Очевидно, что в промышленных процессах следует избегать таких режимов.
Стационарные состояния могут возникнуть и при сильном отклонении системы от равновесия. При превышении критической степени отклонения и
при постоянной «накачке» системы, обеспечивающей высокое значение ее свободной энергии (и низкое значение энтропии), она может перейти в неустойчивое состояние, в ней могут развиться колебательные процессы или хаотические переходы между различными стационарными состояниями.
Взрыв можно рассматривать как результат следующего автокаталити-
ческого процесса:
Взрывчатое вещество + Тепло —> Продукт + Выделение тепла.
Нагрев ускоряет экзотермические реакции, в результате которых выделяется тепло. Процесс идет до тех пор, пока взрывчатое вещество не
израсходуется полностью. Взрывные процессы могут протекать и на поверхности.
Хаотические процессы интуитивно связывают с беспорядком, но это далеко
не так. Пример из физики поясняет ситуацию: рассмотрим ламинарное течение
жидкости через прямолинейную трубку (что является аналогом стационарного
протекания реакции). Когда перепад давления на концах трубки превысит критическое значение (определяемое величиной числа Рейнольдса), течение может стать турбулентным (аналог хаотической реакции). Заметим, что при таком
турбулентном движении степень порядка остается достаточно высокой, поскольку большое число молекул движется в вихрях макроскопических размеров.
Энтропия таких самоорганизованных систем оказывается гораздо меньше энтропии текущих ламинарно или остановленных жидкостей. Читателя, заинтересовавшегося нелинейными неравновесными процессами, отсылаем к книгам
Пригожина [5] и Глейка [6].
Для возникновения хаотического или осциллирующего поведения механизм
реакции должен включать автокаталитическую стадию вида
А + Х—^—>2Х. (2.197)
Если молекула X катализирует свое собственное производство, реакция
называется автокаталитической. Если свяжем эту реакцию с двумя дополнительными стадиями
X + Y—^—»2Y,
(2.198)
Y—Ъ—*Е,
то получим колебательную реакцию, которая реально используется для моделирования природных систем и представляет интерес для химии в целом.
2.11. Энтропия, производство энтропии, автокатализ, колебательные реакции
Для конкретизации модели представим, что Y представляет популяцию лис,
символ Е означает умерших лис, символом X обозначены кролики, а символом
А — морковь. При наличии моркови кроликам живется вольготно, и они быстро размножаются. Популяция лис начинает расти, когда кролики сильно размножатся, однако это будет происходить до тех пор, пока лис станет так много,
что им не будет хватать кроликов для пропитания. Наступит голод, и популяция лис начнет уменьшаться, что приведет к новому росту популяции кроликов. Иначе говоря, величины X и Y буду колебаться противофазе. В этой схеме
имеются два принципиально важных момента — наличие автокаталитической
стадии реакции и постоянная подача реагента А, не позволяющего системе
прийти в равновесие.
Читатель может легко убедиться, что система уравнений (2.197) и (2.198)
может представлять и сбалансированную природную систему, в которой
концентрации компонентов не зависят от времени и равны [Х0] = k3/k2 и
[Y0] = k{[A]/kr Можно также показать, затратив чуть больше усилий, при значительном отклонении системы от равновесия имеется также решение вида
[X] = [Х0] + хе'ш \ со = ±sjk{k2 [A]; i = Т. (2.199)
Аналогично выглядит и решение для [Y]. Поскольку eicot = cos cot + /sin^y/,
то, как нетрудно видеть, [X] осциллирует около стационарного значения, как
это показано на рис. 2.13. Более детально кинетика осциллирующих реакций
изложена в книге [1].
ш
со
S
о
Cl
П.
оГ
s
if
03
Cl
н
Рис. 2.13. Осцилляции концентра- ®
ций X и Y в противофазе около ста- g
ционарных значений. Такой тип ^
представления временных зависимостей далее иногда будет называться дескрипторной диаграммой
Наиболее известной и подробно изученной колебательной реакцией в химии является реакция Белоусова—Жаботинского. Она была открыта российским ученым Белоусовым в 1958 г. Западным ученым она стала известна в 1970 г.,
когда механизм этой реакции был раскрыт Жаботинским [7]. Многие биологические процессы (биологические часы, биоритмы, биения сердца) основаны на
осциллирующих реакциях, для которых критическим фактором является наличие источника энергии, обеспечивающего нахождение системы в далеком от
равновесия высокоорганизованном состоянии.
Глава 2. Кинетика химических реакций
Осцилляции могут наблюдаться и на поверхности катализатора. В качестве
примера мы кратко изложим впечатляющую работу Ертля с соавторами [8, 9],
посвященную изучению окисления СО на поверхности платинового катализатора. В этой реакции осцилляции сопровождаются перестройкой поверхности
(см. также гл. 5). На рис. 2.14 показана «нормальная», а также реконструированная поверхность (110) платины. Реконструкция отвечает типу (1 х 2), который предполагает, что размер примитивной поверхностной ячейки в одном
направлении в два раза больше размера, отвечающего другому направлению.
Осцилляции возникают из-за того, что 02 с трудом диссоциирует на реконструированной поверхности. Переход от исходной к реконструированной поверхности происходит при определенной степени заполнения поверхности молекулами СО. Согласно приведенной на рис. 2.14 схеме реакции, на нереконст-
руированной поверхности (1x1) кислород присутствует в большом количестве,
где он и СО реагируют, образуя С02.
всо выше критического значения
Поверхность платины Поверхность платины
(110)—(1 х 1) (110)—(1 х 2)
02 + 2*110
^ о*110
(в0 повышается)
СО + *110
- СО*110
со*110 + о*110
-» С02 + 2*
рость
110 (высокая ско-
— 9со уменьшается)
02 + 2#1х2
- 2 0#1х2
о
й
о
со+ #1х2
- со#1х2
(0СО повышается)
со#1х2 + о#1х2
- С02 + 2#
1x2 (скорость = 0)
всо ниже критического значения
Рис. 2.14. Схема реакции окисления СО на поверхности (110) платины (левый рисунок) и
реконструированная поверхность (правый рисунок). Объяснения даны в тексте
Когда концентрация СО падает ниже критического значения, поверхность
принимает реконструкцию (1x2), при которой она становится менее активной, а СО на ней адсорбируется также хорошо. Монооксид углерода реагирует
с О, который на этой поверхности не пополняется, в результате скорость реакции падает, а концентрация СО наращивается. Когда концентрация СО превысит критическое значение, поверхность принимает исходное (1 х 1)-состояние,
и цикл начинается снова. Состояние поверхности может быть определено ме-
2.11. Энтропия, производство энтропии, автокатализ, колебательные реакции
тодом дифракции медленных электронов (он описан в гл. 4). На рис. 2.15 показано, что скорость образования С02 изменяется синхронно с интенсивностью дифракционных пиков, отвечающих двум состояниям поверхности. Заметим, однако,
что вместо одномоментной перестройки всей поверхности, формируются непрерывно перемещающиеся по поверхности зоны с разной структурной организацией. При этом формируется эффектная пространственно-временная структура,
которая не только воспроизводится в численном моделировании, но и может
наблюдаться в специально настроенном электроном микроскопе (рис. 2.16).
Рис. 2.15. Осцилляции скорости образования С02 при реакции СО + 02 на поверхности Pt(l 10), синхронные с перестройкой поверхности от реконструкции (1 х 1) к реконструкции (1x2), показанной на рис. 2.14 [10]
Рис. 2.16. Пространственно-временная картина, полученная при численном моделировании окисления СО на поверхности
платины [11]
Время, с
Черные пятна — СО
на нереконструированной
поверхности
Белые пятна — реконструированные
участки поверхности
► Серые пятна — участки» покрытые О
Осцилляции типа показанных на рис. 2.15 довольно регулярны и могут
поддерживаться в течение нескольких часов при фиксированных внешних условиях. В зависимости от скорости подачи реагентов, которая определяет степень удаления системы от равновесия, колебания могут быть более или менее
сложными и даже стать хаотическими (см., например, [12]).
92 -l\r Глава 2. Кинетика химических реакций
Насколько значимы эти явления? Во-первых, осциллирующие реакции наблюдаются в живых системах, где они играют очень важную роль. Биохимические осцилляции и неорганические колебательные системы типа Белоусова—
Жаботнинского представляют собой довольно сложную цепочку реакций. Осциллирующие поверхностные реакции более просты и служат удобной модельной
системой для изучения королевства неравновесных реакций на фундаментальном уровне. Во-вторых, как отмечалось выше, условия, при которых начинают
проявляться нелинейные эффекты, подобные вызванным наличием авто каталитической ступени, приводят к неконтролируемым состояниям системы, чего
следует избегать на практике. Это можно сделать, только имея некоторые представления о природе таких эффектов. Наконец, использование вынужденных
колебаний в некоторых случаях может обеспечить лучшую производительность
реакторов: например, когда каталитическая система «мечется» между состояниями, в одном из которых катализатор пассивирован вследствие осаждения
углерода, а в другом становится активным за счет удаления прореагировавшего
осадка.
2.12. КИНЕТИКА РЕАКЦИЙ,
КАТАЛИЗИРУЕМЫХ ЭНЗИМАМИ
Энзимы представляют собой высокоспецифические биологические
катализаторы. Они образованы из белков, содержащих различные аминокислоты, соединенные пептидными связями. Небольшой по размеру энзим инсулин, например, состоит из 51 аминокислоты. Цепочки аминокислот свернуты
в определенные трехмерные структуры (или, как говорят, обладают определенной конформацией), которые достаточно хорошо установлены для энзимов
различной природы. Внутри этой структуры случайно распределены функциональные группы, карбоксильные или аминные, которые выступают в роли активных центров.
Энзимы могут участвовать во множестве реакций, хотя обычно энзим катализирует избирательно реакцию только одного субстрата. Они называются по
типу катализируемых ими реакций или субстрата, с которым они вступают в
реакцию, путем добавления суффикса «-аза». Так, есть энзимы оксидаза, ре-
дуктаза, дегидрогеназа, гидролаза. Энзим, катализирующий разложение мочевины, называется уреаза и т. д.
Аминокислоты содержат активные группы двух типов — карбоксильные (СООН) и аминные (NH2)
Н О
I II
R - С - С - ОН
I
nh2
\
2.12. Кинетика реакций, катализируемых энзимами -J\r 93
Известно двадцать различных аминокислот. Из них составляются белки путем формирования амидных или пептидных связей между углеродом
из СООН-групп и азотом из NH2-rpynn
н о
н о
I II
NH,
I
R'
но но
I II I II
R-C-C-N-C-C-ОН
I
nh9
Н R'
Энзимы представляют собой каталитически активные белки. Их активными центрами могут служить карбоксильные или аминогруппы, с
определенным пространственным расположением.
Слабые взаимодействия нескольких типов (электростатическое, ван-
дер-ваальсовское) и водородная связь определяют в высокой степени специфический характер взаимодействия молекул субстрата с активными центрами, что делает энзимы наиболее эффективными катализаторами.
Кинетика реакций, катализируемых энзимами, схожа с кинетикой гетерогенных реакций, рассмотренных выше. Но так как на практике имеется некоторое различие в анализе кинетических уравнений, мы рассмотрим отдельно
реакции с участием энзимов.
Энзим Е участвует в реакции путем формирования комплекса с реагентом S
(обычно называемым субстратом), за которым идет образование продукта Р.
Схему реакции можно представить следующим образом:
S + Е р *' > ES, (2.200)
к\
ES——-> Р + Е. (2.201)
Поскольку энзим, субстрат и продукт распределены в одной среде, мы можем оперировать с их концентрациями. Если полная концентрация энзима
равна [E]tot, то из условия сохранения активного компонента получаем, что
[E] + [ES] = [E]toi. (2.202)
Скорость образования продукта можно, воспользовавшись схемой реакции,
сразу же записать в виде
fU[ES].
(2.203)
Глава 2. Кинетика химических реакций
Здесь нет необходимости, в отличие от поверхностных реакций, конкретизировать единицы измерения концентрации, поскольку все участвующие в реакции компоненты находятся в трехмерном пространстве реакционной среды
и все скорости измеряются в концентрациях в единицу времени. Неизвестная
концентрация не участвующих в реакции энзимов легко находится в предположении стационарности реакции:
= *+ [Е] [S] - (*Г + *j)[ES] = 0, (2.204)
откуда сразу приходим к равенству
К [E]tot [S] - *,+ [ES] [S] - (k? + к2) [ES] = 0 (2.205)
и находим
[ES] = к' ^Е]101 ^—. (2.206)
1 1 (fcf + Л2) + kf [S]
Подставив это выражение в уравнение (2.203) и введя константу Михаэлиса
[/Гм из уравнения (2.112)], получаем
d[P] = ЫЕ] [S] = к? + к2
dt Км + [S] м kl
Это — уравнение Михаэлиса—Ментена, полученное в 1913 г., характеризующее скорость реакции, катализируемой энзимами.
Сравнивая уравнение (2.207) с аналогичным соотношением, полученным
для мономолекулярной реакции на твердом катализаторе, легко установить,
что обратная величина константы Михаэлиса соответствует константе равновесной адсорбции, входящей в уравнения Ленгмюра—Хиншельвуда.
При очень высокой концентрации субстрата скорость реакции достигает
максимума:
r = k2 [E]tot при [S]» (2.208)
и энзимы используются наиболее эффективно. Поскольку к2 равна fmax/[E]tot,
то ее обычно трактуют как число оборотов реакции и часто записывают в виде
кш. С другой стороны, если концентрация субстрата меньше Км, скорость ре¬
акции принимает значение
r = -F~[EL "Ри И«*м. (2.209)
лм
а большая доля энзимов в реакции не участвует. Отношение к2/Км, называемое
константой специфичности, является удобной величиной для сопоставления
специфичности энзимов по отношению к различным субстратам. Когда энзим
2.12. Кинетика реакций, катализируемых энзимами
способен участвовать в превращениях молекул двух типов А и В, скорости этих
превращений относятся как (к2/К^)к : Существует практический вер¬
хний предел значения отношения к,/Км, равный примерно 109 моль/л • с, определяемый скоростью диффузии молекул субстрата в растворе к энзимам. Таким образом, системы, в которых достигается такое значение отношения к2/Км,
можно считать близкими к идеальным для реакций, катализируемых энзимами.
В качестве примера рассмотрим энзим—каталазу, катализирующий реакцию разложения Н202 на Н20 и 02, при числе оборотов реакции kcat = 107 с-1 и
константе специфичности к2/Км = 4 • 108 моль/л • с. Такие значения активностей на несколько порядков превышают активности гетерогенного катализа.
Следует также рассмотреть возможности управления реакциями, катализируемых энзимами. Приведенная на рис. 2.17 зависимость показывает, когда
энзимы выходят на область насыщения и скорость реакции практически не
изменяется с ростом концентрации субстрата, что делает невозможным управления реакцией за счет изменения [S]. Безусловно, наиболее эффективно управлять реакцией можно при низких концентрациях субстрата. Таким образом,
если управление реакцией желательно, то ее следует проводить в области значений [S], находящихся между 5 и ЮА"М.
катализируемой энзимами, от [S]/ATM [S]/ACM
При практическом использовании уравнения Михаэлиса—Ментена удобно
переписать его в виде
<22,0)
Таким образом, график зависимости 1/гот 1/[S] представляет прямую линию, тангенс угла наклона которой равен KM/k2[E]tot, а координата точки пересечения равна l/rmax. Такая зависимость представлена на рисунке 2.18, и она
носит название зависимости Линевивера—Бурка.
Как и для любой химической реакции, температура оказывает сильное влияние на катализ энзимами, даже если возможный интервал ее изменения ограничен. Для энзимов существует оптимальная температура (37 °С для реакций в
организме человека). При более высоких температурах их активность падает,
вследствие денатурации белков, из которых состоят энзимы.
Глава 2. Кинетика химических реакций
Важную роль здесь играет и значение pH среды, поскольку от него зависит
зарядовое состояние энзимов и, следовательно, их конформация. Имеется также оптимальное соотношение концентраций энзимов и субстрата.
[S]
Рис. 2.18. Зависимость
Линевивера—Бурка
Хотя мы здесь будем рассматривать только простейшие мономолекулярные
реакции изомеризации или распада субстрата, необходимо все же обсудить влияние на их скорость ингибиторов. Ингибиторы — это соединения, связывающиеся
с энзимами и влияющие на их активность. Конкурентные ингибиторы связываются с теми же центрами, что и субстрат; здесь имеется полая аналогия с конкурентной адсорбцией в гетерогенном катализе. Реакционная схема имеет вид:
S + Е < 4 > ES К- > Р + Е, (2.211)
*Г
I + Е < k[ > EI. (2.212)
Существуют и неконкурентные ингибиторы. Они связываются не с реакционными центрами энзимов, но через модифицирование изменяют их активность.
Конкурентное ингибирование важно для биологического управления механизмом реакции, при этом в качестве ингибитора может выступать продукт
реакции. Например, энзим инвертаза катализирует гидролиз сахарозы, при
котором она превращается в глюкозу или фруктозу. Глюкоза здесь является
конкурентным ингибитором, устанавливающим глубину протекания реакции.
Интересным практическим приложением конкурентного ингибирования
является использование этанола для торможения действия метанола — отравляющего вещества при попадании его в организм человека (например, при
употреблении дешевых спиртных напитков). Сам по себе метанол не является
токсичным, но в легких он разрушается энзимом алкогольдегидрогеназой, что
приводит к образованию высокотоксичных формальдегида и муравьиной кислоты, которые способны привести к слепоте и даже смертельному исходу пос¬
Список литературы
ле употребления небольшого количества метанола. Но так как алкогольдегид-
рогеназа связывается с этанолом сильнее, чем с метанолом, то при отравлении
метанолом следует ввести в организм этанол в количестве, достаточном для
насыщения энзимов. В течение короткого периода времени интоксикация организма метанолом будет ликвидирована. Аналогичная процедура применима и
при отравлении этиленгликолем.
Список литературы
1. Van Santen R.A., Niemantsverdriet J.W. Chemical Kinetics and Catalysis. New
York: Plenum, 1995.
2. Chase M.W. et al. JANAF Thermodynamic Tables, 3rd Edn. J. Phys. Data,
14 (suppl.), 1985.
3. Atkins P.W. Physical Chemistry. Oxford: Oxford Univ. Press, 1998.
4. Laidler K.J. Chemical Kinetics. 3rd Ed. New York: Harper&Row, 1987.
5. Prigogine I. From Being to Becoming. San Francisco: W.H. Freeman, 1980.
6. Gleick J. Chaos, Making a New Science. New York: Viking, 1987.
7. Zaikin A.N., Zhabotinsky A.M. // Nature. 1970. V. 255. P. 535.
8. Ertl G. // Adv. Catal. 1990. V. 37. P. 213
9. Imbihl R., Ertl G. // Chem. Rev. 1995. V. 95. P. 697.
10. Eiswirth M, Moeller P., Wetzl K., Imbihl R. and Ertl G. // J. Chem. Phys.
1989. V. 90. P. 510.
11. Gelten R.J., Jansen A.P.J., Van Santen R.A., Lukkien Segers J.P.L.,
Hilbers P.A.J. // J. Chem. Phys. 1998. V. 108. P. 5921).
12. Gobden P.D., Siera /., Nieuwenhuys B.E. // J. Vac. Sci. Technol. A. 1992. V. 10.
P. 2487.
глава ТЕОРИЯ СКОРОСТЕЙ РЕАКЦИЙ
з
3.1.
ВВЕДЕНИЕ
В данной главе будет изложена теория, позволяющая предсказывать значение скорости реакции, в частности величину предэкспоненци-
ального множителя. В гл. 2 мы ввели как чисто эмпирическое уравнение
Аррениуса, определяющее температурную зависимость константы скорости
реакции:
к = ve .
(3.1)
Ниже мы рассмотрим два подхода, а именно: теорию столкновений и теорию переходных состояний, строго обосновывающих уравнение Аррениуса и
позволяющих придать физический смысл входящим в нее параметрам, в частности предэкспоненциальному множителю.
О
А
С
АВ
Координата реакции
Рис. 3.1. Для вступления в реакцию комплекс должен иметь потенциальную энергию, превышающую высоту энергетического барьера
Уравнение Аррениуса соответствует реакции между двумя молекулами, схематически показанной на рис. 3.1. Молекулы вступят в реакцию, только если
их суммарная потенциальная энергия в момент столкновения будет существенно выше активационного барьера. В противном случае они разойдутся не прореагировав. Эта картина представляется правдоподобной, поскольку из нее следует правильная температурная зависимость скоростей многих процессов, и
она лежит в основе обсуждаемых в данной главе теорий скоростей реакций.
3.1. Введение
Вместе с тем для построения теории, не использующей в явном виде уравнение
Аррениуса, мы не будем отождествлять высоту энергетического барьера, обозначаемую через АЕ, и энергию активации £а, являющуюся эмпирическим
параметром. В разд. 2.4 был описан общий подход к определению энергии
активации из экспериментальных данных. Здесь подобная процедура будет
применена для нахождения энергии активации из теоретического выражения
для скорости реакции. Мы увидим, что величины АЕ и Еа в общем случае не
равны, хотя различие между ними невелико.
Обе теории скоростей реакций, обсуждаемые в данной главе, используют
предположение, что молекулы, вступающие в реакцию, приобретают дополнительную энергию за счет столкновений, переходя в активированное состояние
(на вершину активационного барьера), в котором они* уже способны образовать продукт реакции. В теории столкновений молекулы рассматриваются как
бильярдные шары или твердые сферы, для которых возможность преодоления
барьера определяет только их кинетическая энергия при столкновении. Теория
переходных состояний принимает во внимание колебательные и вращательные
движения реагирующих молекул, существенным образом влияющие на обмен
энергией между ними при столкновении, а также те степени свободы молекул,
которые могут быть возбуждены при прохождении реакционным комплексом
активационного барьера.
В книге [1] Гудстейн писал: «Людвиг Больцман, посвятивший значительную часть своей жизни развитию статистической механики, покончил
с собой в 1906 г. Пауль Эренфест, продолживший его исследования, ушел
из жизни таким же способом в 1933 г. Теперь настала наша очередь изучать статистическую механику. Наверно, будет разумно заниматься этим
предметом с осторожностью.
Для понимания сущности теории столкновений нам необходимо знать функцию распределения молекул по скоростям при данной температуре. Эта функция носит название «распределение Максвелла—Больцмана». В теории переходных состояний используется понятие статистической суммы, вытекающее
из распределения Больцмана. Таким образом, нам следует посвятить один из
разделов данной главы статистической термодинамике.
Межмолекулярные столкновения играют принципиально важную роль в
стимулировании реакционной системы к преодолению активационного барьера. Теория Линдеманна особо подчеркивает это обстоятельство и предлагает
метод для учета влияния окружающей среды на химические реакции. В ней
оговорена необходимость выполнения специальных условий, при которых будут работоспособны обсуждаемые здесь теории скоростей реакций.
В конце главы мы применим теорию переходных состояний и теорию столкновений к описанию нескольких элементарных поверхностных реакций, встречающихся в катализе.
100 -l\r Глава 3. Теория скоростей реакций
3.2. РАСПРЕДЕЛЕНИЕ БОЛЬЦМАНА
И СТАТИСТИЧЕСКАЯ СУММА
Сейчас мы кратко суммируем основные положения статистичес¬
кой термодинамики, необходимые для понимания дальнейшего изложения.
Более детально со статистической термодинамикой читатель может ознакомиться по книге [2].
Предположим, что у нас есть большой ансамбль частиц (электронов, атомов, молекул), распределенных по энергетически уровням где / = 0, 1,2, ..., N,
а N может принимать и бесконечно большое значение. Число частиц должно
быть обязательно большим, поскольку нам необходимо получить хорошо определенные средние величины. В соответствии с распределением Больцмана вероятность Pj найти систему при данной температуре в состоянии с энергией ei
дается выражением
Это распределение может быть получено сразу же, если мы воспользуемся
условием максимума энтропии для ансамбля большого числа частиц. Знаменатель в распределении Больцмана обеспечивает нормировку вероятностей Рп
при которой их сумма равна единице, то есть данным распределением охватывается 100 % состояний системы. Сумма по состояниям
называется статистической суммой. Здесь gt — степень вырождения /-го уровня, которая обеспечивает учет всех состояний, даже если они характеризуются
одной и той же энергией.
Статистическая сумма зависит от термодинамического состояния системы:
через суммирование по энергетическим уровням si она включает в себя все
свойства ансамбля частиц. Будучи безусловно корректным, такое описание, на
первый взгляд, не представляется перспективным, поскольку работать с большим числом громоздких слагаемых не очень удобно. Заметим, однако, что суммы
бесконечных математических рядов часто сводятся к простым выражениям.
Кроме того, если энергетические уровни расположены достаточно близко, то
можно рассматривать энергию как непрерывную переменную и заменить суммирование интегрированием, что обычно позволяет упростить расчеты. Важные термодинамические характеристики системы, такие как средняя энергия,
свободная энергия (химический потенциал) и энтропия, вычисляются непосредственно через статистическую сумму.
Р, =
(3.2)
у
(3.3)
/ = 0
3.2. Распределение Больцмана и статистическая сумма
X
Средняя энергия находится путем дифференцирования логарифма статистической суммы:
ё = къТ2Щ^-. (3.4)
в Э Т
Мы не всегда будем давать вывод приводимых формул, но данную можно
обосновать достаточно просто, поскольку дифференцирование сразу приводит
к выражению средней энергии через соответствующее распределение:
. (15>
= kBr- = /=» = ё.
D ОО J ГТ11 00
£ е-Ъ/квТ i = 0 Ч1 £ е-Ъ/къТ
1=О /=0
Ниже мы покажем, что и другие термодинамические функции — химический потенциал и энтропия — также непосредственно могут быть рассчитаны через статистическую сумму. Но прежде для пояснения физического
смысла статистической суммы мы рассмотрим два простых, но поучительных примера.
Пример. Статистическая сумма двухуровневой системы
Очень наглядно поясняет физический смысл статистической суммы двухуровневая система, например электрон в магнитном поле, когда его спин
может быть ориентирован вверх или вниз (параллельно или антипараллельно
магнитному полю) (рис. 3.2). В основном состоянии энергия е0 = 0, а возбужденное состояние характеризуется энергией Ае. Подставляя эти значения в выражение (3.3), приходим к следующей статистической сумме двухуровневой системы:
q = l + e-4/*Br. (3.6)
При низких температурах система может находиться только в основном
состоянии и статистическая сумма стремится к 1 при Т —» 0:
Ишг^0 q = 1 + lim^o е~^квТ =1 + 0 = 1. (3.7)
При высоких температурах, однако, возбужденное состояние также будет
занято. Условие максимума энтропии требует, чтобы два уровня были заселены
в равной степени. В пределе высоких температур статистическая сумма такова
101
Ншу^о q = 1 + Hmr^0 е Ае/квТ =1 + 1 = 2.
(3.8)
102 -J\, Глава 3. Теория скоростей реакций
Рис. 3.2. Статистическая сумма
и заселенности двухуровневой
системы
Температура, К
Следовательно, при высоких температурах статистическая сумма просто
равна числу доступных энергетических уровней. Значение средней энергии
находится из уравнения (3.4):
е = квТ-
dln(l + e_Af/A'Br) Аее~мккТ
(3-9)
дТ \+е-Ае/к*т’
что дает ожидаемые пределы при нулевой и бесконечно высокой температурах:
lim^o е = 0; limr^0 £ = ^Ае.
Пример. Статистическая сумма системы
с бесконечным числом уровней
Теперь мы рассмотрим систему с бесконечным числом эквидистантных
энергетических уровней, разделенных промежутком Ае. Для такой системы статистическая сумма представляет собой ряд с легко вычисляемой суммой:
-iAe/kBT _
1
1 - е
-Af/kBT
(3.10)
Читатель может легко убедиться, что статистическая сумма равна 1 и ©о для
низких и высоких температур соответственно. Средняя энергия принимает вид
£ = квТ
2 Э In [l/(l -е-д^вГ)] А£е-Ае^т
дТ
1 _ е-^/квт ’
(3.11)
и ее ожидаемые предельные значения равны нулю и бесконечности для низких
и высоких температур соответственно.
3.3. Статистические суммы атомов и молекул -J\r 103
Вышеприведенные примеры показывают, что статистическая сумма служит
индикатором числа занятых при некоторой фиксированной температуре энергетических состояний. При Т= 0, когда система находится в основном состоянии,
статистическая сумма имеет значение q = 1. В пределе высоких температур условие максимума энтропии требует одинаковой заселенности всех состояний, и
статистическая сумма совпадает с полным числом имеющихся энергетических
уровней.
3.3. СТАТИСТИЧЕСКИЕ СУММЫ АТОМОВ И МОЛЕКУЛ
Статистическая сумма играет важную роль в оценке констант равновесия и констант скоростей на промежуточных стадиях реакций. По этой
причине мы более подробно проанализируем статистические суммы атомов и
молекул. Единственной степенью свободы атомов является поступательное
или трансляционное движение. Молекулы обладают также внутренними степенями свободы, связанными с колебаниями атомов и вращением молекул
как целого.
3.3.1. Распределение Больцмана
Сейчас мы рассмотрим систему различимых частиц (заметим,
что одни и те же атомы и молекулы неразличимы, это понятие важно только
для нахождения конфигурационной энтропии). Мы примем, что различные
степени свободы независимы и полная энергия системы может быть записана как сумма энергий отдельных частиц. Наша система состоит из N частиц,
распределенных по различным уровням с энергиями на которых находится Nt частиц, при этом их суммарная энергия равна Еш. Напомним, что
число N должно быть очень большим; это условие обычно выполняется для
всех практически значимых случаев. Вероятность найти частицу с энергией ei
равна Рг.
Р =^-; УАГ=ДГ; УЛ=У^- = 1. (3.12)
' N i , , N
Полная энергия Ехох дается выражением
2(з.13)
/ / ™
где £ — средняя энергия одной частицы. Как может эта система при наложенных на нее ограничениях по полному числу частиц N и суммарной энергии Еш
придать максимальное значение энтропии S? По Больцману энтропия равна
произведению постоянной кв (постоянной Больцмана) и логарифма числа способов (W) распределить N частиц по i-м состояниям, в каждом из которых
104
\
Глава 3. Теория скоростей реакций
находится N; частиц. Таким образом, больцмановское выражение для энтропии (которое написано и на его мемориальном камне в Вене) таково
SM =fcB]n{W), (3.14)
где
“ „У
/
Величину 5tot обычно называют конфигурационной энтропией. Для больших значений N она может быть легко выражена через вероятности Р{ с помощью формулы Стирлинга:
ln(7V!) = 7Vln(7V)-7V, (3.16)
использование которой дает
S = ^f = ~KY,PiHP)r (3-17)
Имеется стандартный математический метод нахождения максимума некоторой функции при наличии ограничений. Речь идет о методе неопределенных
множителей Лагранжа. Он сводится к нахождению максимума функции
включающей энтропию и функции связи, умноженные на (пока не известные)
постоянные Лп называемые множителями Лагранжа:
f{Pi) = S = S(Pi)-A^Pi-\yA2l^eiPi-iy (3.18)
Экстремум этой функции находится из условий равенства нулю ее производных:
Ш)
ЪР; ЭР
= 0, (3.19)
которые приводят к следующим значениям вероятностей, обеспечивающим
максимум энтропии:
р. = (з 20)
Воспользовавшись условием равенства единице суммы всех вероятностей,
мы сразу же получаем
^Р _ I _ ^^-[WAb)+i]^-№/^bK- = е-[ЙДв)+1]^е-('12/^вКв (3.21)
/ / /
Из этого равенства легко находим, что
3.3. Статистические суммы атомов и молекул 105
Это уравнение позволяет выразить Л1 через Л2, но нам необходимо еще
найти Л2. Мы можем сделать это, связав входящие в уравнение (3.22) параметры с какой-нибудь термодинамической величиной. Можно показать [2, 3] , что
параметр Л2 равен \/Ти стоящая в правой части (3.22) сумма просто совпадает
со статистической суммой q. Для интересующегося читателя мы приводим в
следующем разделе доказательство равенства параметра Л2 и обратной температуры 1/Г.
3.3.1.1. Доказательство равенства А2и\/Т
ющими при расчете статистической суммы частицы, находящейся в потенциальной яме (рис. 3.3), поскольку при этом удается автоматически получить
полезное представление статистической суммы, отвечающей поступательному
движению частиц.
Рассмотрим одноатомный газ, заключенный в одномерной потенциальной
яме (обобщение на трехмерный случай проводится элементарно). Энергетические уровни атома, рассчитанные в рамках квантовой механики, равны
ти энергетических уровней, получаем яме с бесконечно высокими стенками
Полезно сопоставить равенство (3.22) с соотношениями, возника-
(3.23)
i=^,£2 = Ti2/8mL2
где /? — постоянная Планка; m — масса атома и / — ширина потенциальной я
ямы. Этот результат является следстви- 5
ем обращения в нуль простейшей вол- I
новой функции атома на границах ямы. J
о
Теперь мы можем рассчитать известную из термодинамики величину средней энергии одномерного движения, а
именно: £={квТ/2). Таким образом,
заменяя суммирование интегрированием, что допустимо вследствие близос-
Ширина ямы
Рис. 3.3. Возможные решения уравнения
Шредингера для частицы в потенциальной
О
L
106
л
Глава 3. Теория скоростей реакций
Из второго равенства (3.24) находим, что Л2= l/Т, то есть представляет собой
обратную температуру. Этот результат приводит к следующему выражению для
вероятности нахождения частицы в состоянии /:
р-е,/къ Т p-fi/kBT
Р = — = - (3.25)
1 q ’ 1
/
где q — статистическая сумма для одномерного поступательного движения:
фяк^тТ
Яшж=— Г • (3-26>
3.3.2. Распределение Максвелла—Больцмана
по скоростям молекул
Нас часто будет интересовать, как распределены скорости молекул. По этой причине нам необходимо преобразовать распределение Больцмана по энергиям в распределение Максвелла—Больцмана по скоростям молекул, совершив замену переменных или переход от энергии к скорости или,
скорее, к импульсу рх (не путать с давлением). Если уровни энергии расположены близко (как это имеет место в классическом пределе), мы можем заменить сумму интегралом:
- "о-Ъ/ЬъТ ~ p-fi/kBT
S* -1-J/Ы*. -J —-Мр=^-/, (3.27)
что дает
> e-(i2p2)/2mkBT оо е-рЦ2тквТ
L/Mdp* ‘hm^r21*1 ■ Ussst*-- <128)
Отсюда находим распределение Максвелла—Больцмана для одномерного
движения:
-рЦ2ткъТ
f(Px)dPx = -h т dAv- (3.29)
y]27rkBml
Это распределение легко обобщается на трехмерный случай, для которого в
декартовых координатах оно принимает вид:
/(л> Ру> Pz) = f{Px)f{Py)f{Pz)- (3-30)
В случае сферических координат имеем
( ™ \3/2
f(v) = 4 я
т
2 яквТ j
v2e-m?/2kBT^ (3J1)
3.3. Статистические суммы атомов и молекул ->\г 107
где v — модуль скорости молекул. В действительности распределение Максвелла—Больцмана вытекает из условия максимума энтропии системы с фиксированными числом частиц и их полной энергией.
3.3.3. Полная статистическая сумма системы
Для системы различимых частиц полная статистическая сумма представляет собой произведение статистических сумм отдельных частиц:
Q = qN (для различимых частиц). (3.32)
Если частицы неразличимы, как, например, атомы газа, число различных
конфигураций существенно уменьшается, и для ансамбля атомов или молекул
статистическая сумма равна
VN
Q = (для неразличимых частиц) (3.33)
или для смеси невзаимодействующих газов
Q = П|Ь (3.33а)
/ 1У i •
где индекс / обозначает сорт газа.
Рассмотрим частицу (атом или молекулу), у которой различные степени
свободы являются независимыми и энергия есть просто сумма энергий, приходящихся на различные степени свободы. Тогда ее статистическую сумму можно записать как произведение статистических сумм, соответствующих различным степеням свободы:
У ~ ^trans ^elec ^nucl ’ (3.34)
где #trans — статистическая сумма для поступательного движения; <?е1ес и qnud —
статистические суммы, связанные с электронами и ядром атома. Для молекул
формула (3.34) усложняется за счет добавления статистических сумм, отвечающих колебательному и вращательному движению молекул:
У ~ ^trans ?rot ^vib ^elec ^nucl * (3.35)
По этой статистической сумме можно определить ряд важных параметров,
таких как:
• химический потенциал
ц = -квТ
энергию
Е = квТ2
Э1п (6)
dN
din {Q)
д Т
V. т
N, V
(3.36)
(3.37)
108
\
Глава 3. Теория скоростей реакций
• давление
р = къТ
• энтропию
Э1п(е)
dN
yv, т
S = JL[kbT\n{Q)N v] = kB In((?) + квТ
Э1п {Q)
дТ
(3.38)
(3.39)
yv, V
Для расчета полной статистической суммы нам необходимо выяснить способ нахождения статистических сумм отдельных атомов и молекул.
3.3.3.1. Статистическая сумма для поступательного движения
Имея целью определить статистическую сумму для поступательного движения, рассмотрим частицу массой m, движущуюся со скоростью vx вдоль
отрезка длиной /, направленного по оси х. Импульс частицы равен рх = mvx, а
ее энергия — ех = р2х/2т. В фазовом пространстве, задаваемом координатой л; и
импульсом рх, доступная для частицы область движения может быть разбита на
ячейки размером А, совпадающим с постоянной Планка. Поскольку эта область чрезвычайно мала, мы можем при вычислении статистической суммы
заменить суммирование интегрированием по координатам фазового пространства л; и р . Нормируя статистическую сумму на размер ячейки А, получаем
I /
tftrans =^Jd*/ dp*e
г Рх /2тквТ
I (2ятквТ)
h
1/2
(3.40)
Эта комбинация представляет собой статистическую сумму для одномерного движения по отрезку длиной / частицы массой т. Обращаем ваше внимание
на то, что это выражение в точности совпадает с результатом квантово-механического расчета, проведенного для частицы, находящейся в одномерной потенциальной яме. Если для перемещения частицы доступна поверхность площадью А, то статистическая сумма будет равна
2D 2ятквТ
^trans j 1 5
hr
а для частицы, заключенной в объем V имеем:
<7,г1 = V
за _ 1Л2л:тквТ)312
(3.41)
(3.42)
Приведенные выше результаты позволяют сделать следующее заключение:
статистическая сумма частицы зависит от ее массы, температуры, размера доступной для движения области, а также размерности пространства, в котором
3.3. Статистические суммы атомов и молекул -J\, 109
перемещается частица. Таким образом, статистическая сумма для трансляционного движения может быть большим числом. Обычно она вычисляется для
единичного объема, если, например, определение равновесных условий равновесия соответствует данному ниже. Традиционно статистическая сумма для
поступательного движения записывается в виде
(2nmkjf1 =у_
‘/trans v j з — лЗ 5
h л J
где Л представляет собой тепловую длину волны (длину волны де Бройля) частицы в одномерном пространстве. Следует отметить, что распределение Больцмана справедливо только при выполнении условия V/A3^> 1, которое означает,
что длина волны частицы много меньше размера сосуда, в котором она заключена. Это условие предполагается выполненным для всех систем, обсуждаемых
в данной книге. Нарушения этого условия могут иметь место в экстремальных
условиях, например, при очень низких температурах (то есть для Не при температуре порядка нескольких градусов Кельвина, когда он становится сверхтекучим) или при очень высоких давлениях, типичных для звезд. При таких экстремальных условиях следует использовать распределения или Бозе—Эйнштейна, или Ферми—Дирака, в зависимости от того, является ли спин частиц целым
или полуцелым числом. За деталями мы отсылаем читателя к учебнику по статистической механике [4].
Средняя кинетическая энергия частицы находится из соотношений (3.4)
или (3.37), в которые необходимо подставить выражение (3.42):
. (2птквТ^12
f - к Т2 д
ctrans В г
= hBT. (3.44)
А3
Мы получили ожидаемый результат, поскольку в классической системе
каждая степень свободы вносит в кинетическую энергию вклад, равный 1 /2квТ.
3.3.3.2. Статистическая сумма колебательных движений
Молекулы обладают внутренними степенями свободы: в них атомы могут совершать колебательные движения, и молекула может вращаться
как целое. Энергетические уровни гармонического осциллятора, колеблющегося с частотой hv, даются выражением
*'=(/+i)M'- (3-45)
Заметим, что нулевая энергия приходится на дно потенциальной кривой, а
основное состояние — низший занимаемый уровень — лежит на 1/2йквыше.
Поскольку статистическая сумма обычно рассчитывается относительно низшего занятого уровня, мы сдвинем нулевую энергию вверх на l/2hv, что дает
е\ = ihv = £* - ^hv. (3.46)
110
л
Глава 3. Теория скоростей реакций
Таким образом, статистическая сумма, определенная относительно низшего занятого уровня, равна
a-.fyw 1 (3,47)
/ = 0 1с
в то время как статистическая сумма, рассчитанная относительно дна потенциальной кривой, имеет вид
fa = I= ‘ ^'Т. (3.48)
/' = 0 1с
Как мы увидим ниже, важны обе эти статистические суммы. Статистическая сумма q'ib во многих случаях близка к единице, если только частота колебаний не мала настолько, что hv<K квТ. При выполнении этого неравенства можно использовать разложение
е±х =\± х, (3.49)
и статистическая сумма становится близкой к классическому переделу:
?vib, class =-^7 ДЛЯ hv « квТ. (3.50)
Отметим, что двухатомная молекула в газовой фазе обладает только одной
колебательной степенью свободы, но в адсорбированном состоянии она может
приобрести еще несколько колебательных мод, частоты которых могут быть
достаточно низкими. Тогда полная статистическая сумма является произведе¬
нием индивидуальных статистических сумм:
е->,Двг
?v,b = Г1 , _ 0-hvi/kBT ’ (3.51)
/ А е
Используя одно из уравнений (3.4) или (3.36), можно найти среднюю колебательную энергию молекулы, обладающей N колебательными модами. Мы
рассчитаем ее по отношению к нулевому значению колебательного потенциала, предполагая учет всех нулевых колебаний:
^vib “
д Л e~*hVilk*T &( hv- 1 Л JL\
‘т~ ЪТ n.TTTW = + 2'4 J ■ <3'52)
Это приближенное выражение справедливо, если все моды имеют не слишком низкую частоту. В противном случае для низкочастотных мод необходимо
использовать полное выражение из (3.52).
3.3. Статистические суммы атомов и молекул
З.З.З.З. Вращательная (и ядерная ) статистическая сумма
\
Статистическая сумма для вращательного движения двухатомной
молекулы получается из квантово-механического распределения соответствующих энергетических уровней:
где j — вращательное квантовое число и / — момент инерции молекулы:
/ = //Л (3.54)
Здесь ju — приведенная масса:
111
— = — +—, (3.55)
ju ml m2
a mx и m2 — массы атомов двухатомной молекулы. Подстановка энергии (3.53)
в выражение для статистической суммы дает с учетом степени вырождения
вращательных уровней:
qrot =1^(2] + 1)е[]и + 1)н2У*ж1квТ Bljd[y(y + l)]et^ + l)*2>w*Br =18яДВД
a j = \ ® о (Т h~
„2 (3.56)
для т>шгь
Окончательное выражение в (3.56) представляет собой классический предел для статистической суммы, который можно использовать при температуре
выше критического значения, которое, однако, на практике достаточно мало
(например, 85 К для Н2 и 3 К для СО). Для гомоядерных или симметричных
линейных молекул фактор а равен 2, а для гетероядерных молекул a = 1 (табл.
3.1). Этот фактор симметрии обусловлен неразличимостью двух состояний молекул, получающихся взаимной перестановкой ядер, так что он связан с ядер-
ной статистической суммой. Фактор симметрии может быть определен из свойств
симметрии молекулы.
Таблица 3.1. Фактор симметрии для различных групп симметрии
и примеры принадлежащих им молекул
Тип симметрии
(7
Типы молекул
С,, С, и Cs
1
СО, CHFClBr, ./иезо-винная кислота СН3ОН
^2» C2v и C2h
2
Н2, Н202, Н20 и т/>я//одихлорэтилен
Сзу И ^3/7
3
NH3 и плоская молекула В (ОН)3
111
112
Л
Глава 3. Теория скоростей реакции
Средняя энергия вращательного движения легко получается дифференцированием логарифма статистической суммы:
^ =АгвГ2^1п
дТ
1 8тг1кпТ
h2
= квТ. (3.57)
Для полноты изложения приведем также общую формулу для вращательной энергии большой молекулы с главными моментами инерции /А, /в и /с:
8л-1квТ
h2
plJblc- (3-58)
В качестве упражнения мы предлагаем читателю показать, что при низкой
температуре, когда заселен только основной колебательный уровень, средняя
энергия двухатомной молекулы в газовой фазе равна 5квТ/2. А чему равна эта
энергия при высокой температуре?
3.3.3.4. Электронная и ядерная статистические суммы
Электронную статистическую сумму обычно принимают равной
просто единице, поскольку разность энергий между возбужденными состояниями, как правило, очень велика по сравнению с квТ. Но бывают и исключения:
хорошо известный пример — это молекула N0, которая имеет один неспаренный электрон на самой высокой из занятых молекулярных орбиталей. В результате орбитальный момент молекулы может иметь две противоположные
ориентации по отношению к некоторой оси (вращение по и против часовой
стрелки). Кроме того, имеются две возможные ориентации спина электрона,
что в итоге дает четыре различные конфигурации с относительно малой разностью их энергетических уровней. Все это приводит к тому, что электронная
статистическая сумма молекулы N0 изменяется от 2 до 4 в зависимости от
температуры.
Как правило, мы будем считать, что разделенным атомам, находящимся в
основном состоянии, отвечает нулевая энергия. Тогда электронная статистическая сумма равна
4е1 = а>е0е~ео/квТ + сое1е~е,/квТ + ..., (3.59)
где сое. — степень вырождения состояния с энергией и / > 0 соответствует
возбужденным электронным состояниям. Если мы, к примеру, рассмотрим
двухатомную молекулу, то ее основное состояние обычно таково:
*0 =-А=-4+у. (3.60)
Это выражение проиллюстрировано на рис. 3.4. В данном случае энергетический выигрыш при образовании молекулы равен D0 и совпадает с ее энерги-
3.3. Статистические суммы атомов и молекул ->\г 113
ей диссоциации, поскольку,
как мы уже отмечали, молекула обладает в основном
состоянии ненулевой колебательной энергией, равной
1/2/гк
Рис. 3.4. Диаграмма потенциальной энергии для диссоциирующей молекулы
Ct
ф
00
со
S
о
а
s
L.
а
ш
х
0
К
СО
X
-О
с;
со
S
ZT
X
0
I-
о
с
Расстояние, произв. ед.
Таблица 3.2. Подборка основных формул статистической механики
для газа двухатомных молекул
Статистические суммы
e~eilkBT
Pi =
^е-£'/к вг
/ = 0
ё = квТ2 dXnq
в Э Т
ju = -квТ In q
д = ^е-£‘/к*Т
/ = 0
dkBT In q
dT
К(Т) = П^'
/
Статистические суммы двухатомной молекулы (на одну степень свободы)
о
Поступательное
движение
_ [2лткъТ)и2
о
о
о
Колебания молекулы
1
0
О
Вращение молекулы
8л21квТ
^trans/^ ^
Гигантское число
для типичного значения /
Н,: 1,8 х 10ю м~‘ при 500 К
СО: 6,8 х Ю10 м"1 при 500 К
С1,: 1,1 х 1011 м_| при 500 К
^vib - j _ g-hv/kBT
Обычно равна единице,
если частота колебаний
не слишком мала
Н,: 1,000 при 500 К
СО: 1,002 при 500 К
С1,: 1,250 при 500 К
<7.°, - h2
Большое число
Н^: 2,9 при 500 К
СО: 180 при 500 К
С12: 710 при 500 К
114 -J\r Глава 3. Теория скоростей реакций
Ядерная статистическая сумма обычно не дает вклада в полную статистическую сумму молекул и, как правило, принимается равной единице. Ниже мы
будем игнорировать этот вклад.
Выражения для статистических сумм разного типа приведены в табл. 3.2.
3.4.
МОЛЕКУЛЫ В РАВНОВЕСИИ
В гл. 2 мы ввели понятия химического равновесия и констант равновесия. Сейчас мы снова вернемся к анализу химических реакций и покажем,
как константы равновесия могут быть непосредственно выражены через статистические суммы молекул, участвующих в реакции. Рассмотрим следующую
реакцию, уже обсуждавшуюся в гл. 2:
vaA + vB В
vcC + vdD.
(3.61)
Условием химического равновесия является достижение минимума свободной энергии Гиббса:
dG уг
-jj- = ~vaMa -vbMb+ vcMc +vdmd=2j viH = 0.
(3.62)
Используя уравнение (3.35), можно выразить химический потенциал непосредственно через статистическую сумму:
(3.63)
где была использована формула Стирлинга (3.16). Подставляя выражение (3.63)
в равенство (3.62), получаем
JU: = ~knT
Гэ In (C?,)l
= ~kBT
'ain (qf'/N'ij
= -kRT In
f q> 1
г I D
3N,
ЭАГ,
D
k-J
П
4; ]
= 1 =
f q° ]
«С
f 4d \
{ ЧА ]
~VA
( qB 1
U/J
l*cj
[nd)
l^J
-1-В
(3.64)
Поскольку мы по предположению имеем дело с идеальным газом при давлении рп можно воспользоваться уравнением
N,=^V-
kB Т
и преобразовать равенство (3.64) к виду
[Яс1У)РС[Чо1У]
(W
nVC nvD
vc + vD-vA-vB _ Рс PD
РааРв°
(3.65)
(3.66)
3.4. Молекулы в равновесии ->\г 115
Так как статистические суммы qt пропорциональны объему V (что задается
статистической суммой поступательного движения), отношение q.JVявляется
функцией только температуры. Тем не менее, если
константа равновесия будет иметь размерность давления в некоторой степени.
Чтобы сделать константу равновесия безразмерной, мы нормируем давление
на величину р0 = 1 бар. В результате константа равновесия приобретает следующее представление через статистические суммы q.\
Таким образом, зная детальную структуру энергетических уровней для внутренних степеней свободы молекул, участвующих в реакции, можно рассчитать
соответствующие статистические суммы, в затем, воспользовавшись уравнением (3.68), и константу равновесия. Такой способ нахождения констант равновесия является достаточно общим: после определения статистических сумм рассчитываются химические потенциалы компонентов, а затем — константа равновесия. Проиллюстрируем этот подход несколькими примерами.
Поучительно использовать для иллюстрации связи между статистическими
суммами и константой равновесия чисто гипотетический пример. Рассмотрим
равновесие в ансамбле молекул А и В, энергетические уровни которых соответствуют приведенным на рис. 3.5. Основному состоянию молекулы А отвечает
нулевая энергия, следовательно, ее статистическая сумма равна
Основной уровень молекулы В сдвинут относительно основного уровня
молекулы А на величину и. С учетом этого сдвига статистическая сумма молекулы В должна быть записана так:
VC + VD - VA - VB * °>
(3.67)
K[T)
(<7а/П>в/*Т ^ Po J
\ Vc + VD - V/K -
[PcIPoT (Рр/РоГ (3 68)
(pJp0)Va (Рв/РоГ '
Пример
(3.69)
2 2
e~(u+iAfB)/kBT _ e~u/kBT у e~iAFB/kBT
(3.70)
Воспользовавшись равенствами
116
\
Глава 3. Теория скоростей реакций
получаем следующее выражение для константы равновесия
Nr
= К=^- =
Як
-u/kBT
(■
+ е
-А?в/квТ
+ е-2А*вМв7' \
1 + е
-&еА/квТ
(3.72)
А
К
В
Авд
АеR
Аба
Рис. 3.5. Схема энергетических уровней двух гипотетических молекул
В пределе низких температур имеем К = 0 и qA = 1, что предполагает
нахождение всей системы в основном состоянии молекулы А, поскольку
ему отвечает низшее возможное значение (свободной) энергии. При высоких температурах имеем значение К = 3/2, поскольку все энергетические
уровни должны быть заселены одинаково. Этот пример показывает, что при
низких температурах основную роль играет энергия, а при высоких главенствует энтропия.
Пример: константы равновесия для диссоциации Н2, N2,02
Проанализируем теперь возможную диссоциацию Н2 при повышенных температурах. Будем считать газ идеальным и рассмотрим реакцию
2Н.
(3.73)
Проведем расчет парциального давления атомарного водорода при постоянном полном давлении и температурах Т = 1000, 2000 и 3000 К. Если полное
давление поддерживается постоянным на уровне рш= 1 бар, то в равновесии
«н2 = «initial - £ а «н = 2£ так что nfmM = п,
ния) и
Рн,-
initial
initial
zl
+ <r
Pt ot > Pu
initial+ £ (здесь £ — степень превраще-
2#
initial
+ <f
Aot •
(3.74)
В равновесии = 0, что эквивалентно равенству
/
~Мн2 + = 0*
(3.75)
3.4. Молекулы в равновесии
X
Поскольку
Ц; = -квТ
Э1п(д.)
Э N,
= -квТ
а также
N*2 Чн2
константа равновесия принимает вид
ЭТУ;
и N- =
' К т
/ Л \
~ -кйТ In
{Рн/Ро?
къТ
Ро
(Ян/V?
[Рн2/Ро)
С помощью уравнения (3.74) находим
п,
<? =
initial
л/1 + [4PjK{T)]
и Р н
= К(Т).
2 Ас
(3.76)
(3.77)
(3.78)
1 + J\ + [4Ptot/K(T)]
(3.79)
В уравнении (3.78) константа равновесия выражена через статистические
суммы, которые еще предстоит вычислить:
К(Т)
квТ
[>Г7гЦ‘]2
л /„trans /тЛ vib„rot „el
Ро J[qH2 /V)qH2qn2qn2
(3.80)
Заметим, что эта константа отвечает реакции, при которой одна молекула
водорода диссоциирует на два атома. Если примем, что в реакции из половины
молекулы Н2 образуется один атом Н, то соответствующая константа будет
равняться корню квадратному из выражения (3.80).
Статистическая сумма для поступательного движения определяется выражением
3/2
grtrans _ (27гтхкъТ)
~У~~ Тъ 5
справедливым для обоих компонентов, которое при Т= 1000 К дает:
(3.81)
^rans (2 X 3,141 X 1,66 х 1(Г27 х 1,38 х 1(Г23 X 1000)'
V (6,63x10'34)
<ns _ 23/2<7н
3/2
= 1,70 х 10 .
= 6,01 х 10 , (3.82)
(3.83)
117
118 -l\r Глава 3. Теория скоростей реакций
Электронная статистическая сумма для молекулы водорода находится легко, поскольку электронные уровни возбужденных состояний сильно удалены
от основного состояния. И при рассматриваемых температурах электронные
возбуждения можно в расчет не принимать, что дает q^ = 1. Аналогичная
ситуация имеет место для атома водорода. В основном состоянии, Нь, атом
водорода имеет один неспаренный электрон, что предполагает двукратное вырождение основного состояния, которому отвечает терм 2Sx/r Первые возбужденные электронные состояния таковы: Н2р, 2Р{/1 и H2v, 2SX/V и они оба находятся на 10,4 эВ выше основного состояния. Таким образом,
Ян “ ®е\ + а)е2е ^ В + ••• — Юе\ ~ ^
где (о означает степень вырождения.
Колебательная статистическая сумма имеет вид
<7v,b =П-Г
/' А
0-^,/kQT
. e-bvi/kBT ‘
(3.84)
Молекула Н2 имеет только одну колебательную моду с частотой 4400,39 см *,
так что hv/kB = 6332 К и
-16332/7’
< = ,е e-6332/r = °>042 при Т = 1000 К. (3.85)
Прежде чем вычислять вращательную статистическую сумму, мы должны
проверить, выполняются ли условия, при которых было получено приближенное выражение (3.56). Имеем
rot 1 8 тг1квТ /г
<7н = 7Т1- для Т»—т— = ег. (3.86)
а п~ Ъ7г~1кв
Поскольку для молекулы Н20,. = 87,6 К, при рассматриваемых температурах проблем с применением приближенного выражения (3.56) не возникает,
хотя при более низких температурах в формуле (3.56) следует использовать
сумму, а не интеграл. При Т= 1000 К можно без опасений использовать приближенную формулу (3.86), которая дает
- -
где мы приняли для а значение 2, соответствующее гомоядерной молекуле Н2.
Приписывая разделенным атомам водорода нулевую энергию, электронную статистическую сумму молекулы водорода можно представить в виде
= °>е\е°е1квТ + - = 1е°е1к*т • (3.88)
3.4. Молекулы в равновесии -J\r 119
2Н
Параметр De может быть определен по энергии диссоциации и колебательной энергии основного состояния или из термодинамических данных. Теплота
образования атомарного водорода из молекул Н2 может быть найдена в справочниках, однако оперировать с теплосодержанием атомов Н и молекул Н2 при
нормальных условиях следует, соблюдая осторожность.
Теплота образования атома Н при 298 К
(нормальные условия) равна АЯ° =
= 218,0 кДж/моль (на один атом). Проблемы отсутствуют при температуре 0 К, поскольку в этом случае теплота образования
совпадает с энергией диссоциации D0, и для
De имеем простое выражение De = D0 + hv/2,
что соответствует Т — 0 для схемы, представленной на рис. 3.6. При переходе к нормальным условиям, однако, атомы и молекулы приобретают тепловую энергию в разных количествах вследствие различия чисел
внутренних степеней свободы. При
Рис. 3.6. Энергетическая диаграмма для
определения энергии диссоциации De
молекулы Н7 из термодинамических
данных
Н = E + pV = kBT2
Э1п (Q)
дТ
+ RT
(3.89)
N, V
имеем
Нн = E + pV = kBT2
Э1п(С?н2)
эш(ен)
дТ
+ RT = -RT + RT
n, v 2
(3.90)
Ян, = Е + pV = kBT
дТ
+ RT = ^RT + RT + ^- + RT-De. (3.91)
yv, v 2 2
Поступательная энергия молекул равна 3/2RT; первый вклад, равный RT,
идет от члена pV, второй обусловлен двумя вращательными степенями свободы, есть также вклад, соответствующий нулевым колебаниям. Энтальпия атомов складывается из энергии поступательного движения и члена р V. Имеем из
определения теплоты образования
2#н = ЯНо + 2Д Н°
(3.92)
или
что дает
5 1 hv
2xlRT = jRT + ^ + 2AH°-De,
De = -^RT + ^- + 2AH° = 458,70 кДж/моль.
(3.93)
(3.94)
120 -J\r Глава 3. Теория скоростей реакций
Теперь мы можем рассчитать и константу равновесия, и парциальное давление атомарного водорода при различных температурах. При Т= 1000 К получаем
* IT км М
(1,38 х 10"25)(б,01 х Ю30 X 2)2
1,70 х 1031 х 0,042x5,71x9,38x10
23
= 5,24x10"
(3.95)
Продемонстрировав схему расчета константы Кн^ , мы можем непосредственно найти и ее, и парциальное давление рн при различных температурах
(табл. 3.3).
Таблица 3.3. Константы равновесия для реакции диссоциации молекул Н2, N2
и 02 и парциальные давления атомов при различных температурах,
рассчитанные по фундаментальным постоянным, приведенным
в табл. 3.4
т, к
К^(Т)
о
X
Кщ(Т)
Z
о
О
о
298
5,81 х 10"72
2,41 х 10“36
6,35 х Ю"160
2,52 х Ю“80
6,13 х 10~81
7,83 х 10“41
1000
5,24 х 10“18
2,29 х 10"9
2,55 х 10“43
5,05 х 10~22
4,12 х 10“19
6,42 х 10-'°
2000
3,13 х 10“6
1,76 х 10~3
2,23 х 10“18
1,80 х 10“9
1,221 х 10“5
3,49 х 10_3
3000
1,77 х 10“3
1,72 х 10н
1,01 х 10"9
3,18 х 10“5
5,04 х 10“'
5,01 х 10~'
Таблица 3.4. Данные, использованные для расчета доли диссоциировавших
молекул. Основное состояние атома водорода имеет двукратное
вырождение, а основное состояние молекулы является
невырожденным. Аналогичные допущения сделаны в отношении
молекул азота. У кислорода ситуация сложнее: основное
состояние молекулы имеет трехкратное вырождение,
а электронные состояния атома характеризуются наличием трех
близко лежащих уровней с кратностью вырождения 5, 3 и 1;
эти уровни разнесены на 1901 и 2718 Дж/моль, причем основным
является пятикратно вырожденный уровень
Молекула
Частота колебаний,
см-1
Частота вращения,
см-1
De=D0 + hv/ 2,
кДж/моль
н2
4400,39
60,864
458,7
N2
2358,07
1,9987
456,8
1580,36
1,4457
504,1
3.5. Теория столкновений -i\r 121
Как видно из табл. 3.3, значительные количества атомарного водорода
можно получить только при очень высоких температурах. Для сравнения и
дальнейших ссылок мы провели аналогичные вычисления для азота и кислорода (табл. 3.3 и 3.4). Высокая устойчивость молекул N2 (более устойчивы
лишь молекулы СО) предопределила значение параметра De, в два раза превышающее соответствующую величину для молекул водорода и указывающее на
практическую невозможность диссоциации N2 в газовой фазе. Отсюда ясна невозможность получения аммиака путем простого нагрева смеси газов N2 и Н2:
равновесие сильно сдвинуто в сторону от аммиака при высоких температурах,
требуемых для диссоциации реагентов. Попытайтесь рассчитать константу равновесия для реакции образования аммиака, используя статистические суммы
соответствующих молекул.
3.5. ТЕОРИЯ СТОЛКНОВЕНИЙ
Если мы примем, что молекулы можно рассматривать как бильярдные шары (твердые шары), не обладающие внутренними степенями свободы,
то вероятность реакции между компонентами, скажем, А и В будет зависеть от
частоты встреч молекул А и В при наличии, конечно, у сталкивающихся молекул энергии, достаточной для преодоления барьера, разделяющего реагенты А
и В и продукт реакции АВ. Сказанное говорит о необходимости расчета частоты столкновений молекул А и В.
Обозначим чрез рА и /?в числовые концентрации молекул А и В соответственно. Поскольку частота столкновений, очевидно, зависит от размеров молекул, мы введем диаметры эффективных сфер, представляющих молекулы А и В,
а именно: dA и afB.
Согласно Траутцу и Левису, первыми применившими теорию столкновений для расчета скорости реакции в 1916—1918 гг., частота столкновений (еще
не реакций) между сферами А и В и равна:
. 7i d2 и
Гсо11 “ coll Ра Р\*> ~ PkPb'i ^АВ “ 1 +^АВ> (3.96)
АВ
где nd2 — эффективное сечение столкновений; d = (dA + dQ)/2 — средний
диаметр; <тАВ — фактор симметрии, исключающий двойной учет столкновений
одинаковых молекул (£АВ = 1, если А = В, и 0, если А * В); й — средняя
относительная скорость сталкивающихся молекул.
Сущность двойного учета столкновений легко понять из следующих соображений. Предположим, что концентрации молекул равны рА = рв = Ю, а их
размеры и массы идентичны. Тогда число столкновений между молекулами А
и В равно
''сои, АВ = ^-%aAs = nd2u х 100. (3.97)
°АВ
122
\
Глава 3. Теория скоростей реакций
Если мы примем, что молекулы В тождественны молекулам А, то в тех же
условиях рА = 20. Полное число столкновений при этом не изменяется и принимает значение
л d2 и
coll, АА
Ра
ndru
х 202 = nd2u х 200,
(3.98)
'АА
то есть удвоилось по сравнению с числом столкновений между разнородными
молекулами А и В. Но при подсчете полного числа столкновений в смеси АВ
мы должны еще учесть и столкновения типа АА и ВВ, число которых таково
'coll, АА + 'coll, В В
nd~u nd~u — л ла
рА + Ръ = nd~u х 100,
(3.99)
АА
'ВВ
/
и полное число столкновений в смеси АВ становится равным 200nd2u, что
совпадает с числом столкновений молекул в объеме, содержащем только молекулы А с удвоенной концентрацией. Если бы мы не учли фактор симметрии, то
число столкновений между молекулами в системе с рА = 20 было бы сильно
завышено.
Объяснение формуле (3.96) дает рис. 3.7:
д d молекула А, имеющая площадь сечения
1/4 nd2A, движется ко второй молекуле В
с относительной скоростью и. Если центр
молекулы В лежит внутри цилиндра с поперечным сечением п2 и длиной uAt, то
за интервал времени At между молекулами обязательно произойдет столкновение.
Относительная скорость не только задает частоту столкновений молекул А и
В, но и определяет кинетическую энергию столкновения, необходимую для преодоления энергетического барьера реакции. Скорости частиц в равновесной смеси газов подчиняются распределению Максвелла—Больцмана, которое в
сферических координатах имеет вид:
У
uAt
Рис. 3.7. Между молекулами А и В происходит столкновение, только если центр
молекулы В лежит внутри цилиндра с радиусом d = (d + dB)/2 и длиной и At
f{v) = 4 п
т
3/2
2 пкъТ
v2e-mi'-/kBT.
(3.100)
Аналогичному распределению подчиняются относительные скорости частиц, нужно только заменить массу частицы m на приведенную массу ju для
конкретной пары:
3.5. Теория столкновений
\
Расчет средних абсолютной и относительной скоростей молекул для распределения Максвелла—Больцмана проводится одинаково:
л; = J xf(x)dx = J х4 л
2 пкъТ j
3/2
=
j Y3e~y2dY;
что дает
8 квТ
п m
- М д
\2квТ
* j
ч‘/2
Г8£В7Л
; й =
/
1 W J
(3.102)
1/2
(3.103)
Иногда эти скорости выражаются через молярные массы и тепловую энергию одного моля:
V -
(ттл
1 пМ )
1/2
U =
8 RT
7Щ
1/2
(3.104)
Воспользовавшись вышеприведенным выражением для средней относительной скорости, можно записать частоту столкновений молекул А и В в виде:
'coll
nd2
8квг)1/2 . _
РкРъ> ^АВ “ ^АВ *
П/Л )
(3.105)
Рассмотрим газовую смесь водорода и азота, содержащихся в пропорции 1:1,
при полном давлении 1 бар и температуре 300 К. Примем, что средний диаметр
молекул равен 3 А или 3 х Ю-10 м. Частота столкновений здесь равна
''сои = nd
ЩТ
71(Л
1/2
Pn2Ph2
{КТ)2
= 7,6 х 10 мхе.
(3.106)
Средняя скорость молекул (при Т = 300 К) равна примерно 500 м/с (476 м/с
для N2 и 1781 м/с для Н2) и число столкновений молекул за одну секунду в
одном литре газа при атмосферном давлении составляет около 1031. Отметим,
что частота столкновений сильно зависит от плотности, через рА и /9В, и достаточно слабо от температуры, как 4т.
3.5.1. Частота столкновений с поверхностью
Поскольку нас будут интересовать поверхностные реакции и катализ, нам необходимо провести расчет частоты столкновений молекул газа с
поверхностью. Чтобы молекула могла столкнуться с поверхностью площадью А,
она должна иметь компоненту скорости vx9 направленную по нормали к по-
123
124
А
Глава 3. Теория скоростей реакций
верхности (рис. 3.8). Все молекулы, имеющие скорость vx, определяемую распределением Максвелла—Больцмана в декартовой системе координат, и находящиеся на расстоянии vxAtoj поверхности, столкнутся с ней. Произведение vxAtA
= К дает некоторый объем, число молекул в котором, имеющих скорость vx,
равно f(vx)V(vx)p, где р — числовая плотность газовых молекул. Интегрируя
это выражение по vx от 0 до бесконечности, получаем полное число столкновений молекул с поверхностью площадью А за время At. Так как нас интересует число столкновений с поверхностью единичной площади за единицу времени, имеем
coll—surf
77 J Ж )V(vx)p dvx =р] vj (vx )dvx=-^-]vx [I
m
AtA
о
P
0
‘\]2 nkBT
kBT
kBT
kBT V 2nm ^lnmkBT ’
vl/2kBT
B dvv
(3.107)
где мы воспользовались связью между плотностью и давлением, установленной для идеального газа: р = {р/к^Т).
Число столкновений при давлении р = 1 бар
и температуре Т = 300 К равно
гд t
<\°
■/
✓
d А
coll—surf
= 1,08 X 1028 м"2-с_1
для водорода и 2,28 х 1027 м-2 • с-1 для азота. Так
как на одном квадратном метре поверхности
находится примерно 1,5 х 1019 атомов, каждый
из них испытает при нормальных условиях около миллиарда ударов в секунду. Это не означает, однако, что газовые молекулы прореагируют
на поверхности, поскольку реакция есть активационный процесс.
Рис. 3.8. Все молекулы, имеющие нормальную к поверхности скорость vx и находящиеся в элементе объема vx&t&A,
столкнутся с площадкой dА
3.5.2. Вероятность реакции
Возвращаясь к общему случаю реакции между двумя молекулами,
напомним, что частота их столкновений огромна — порядка 1031 л-1 • с-1. Нам
необходимо определить вероятность того, что столкнувшиеся молекулы прореагируют. Реакция произойдет, если энергия столкновений, отвечающая координате реакции и (линии, соединяющей реагирующие элементы), превышает
высоту реакционного барьера (рис. 3.9). Таким образом, нам нужно рассчитать
долю столкновений Р, у которых относительная кинетическая энергия l/2/zw2
3.5. Теория столкновений
X
больше АЕ. Введя символ wmin для относительной скорости, отвечающей условию 1/2 juu2m^ = АЕ, имеем для доли реакционных столкновений:
J uf(u)du | 47r(ju/27rkBTf12 и3е ми~/2квТйи
р _ ^min _ ^min
Juf(u)du J4л-(///2л-/:вГ)3/2 u3e~MU~^2kBTdu
= е-ЬЕ/квТ
. (3.108)
И теория столкновений дает следующее выражение для скорости реакции между компонентами А и В в единичном объеме:
'с oil
7td2 (8квТ
nil
1/2
-АЕ/квТ
Ра Рб 9 аАв - 1 + ^ав •
(3.109)
Рис. 3.9. Молекулы вступят в реакцию, если они обладают энергией,
достаточной для преодоления
энергетического барьера АЕ. Высота барьера может быть связана с минимальной относительной скоростью и . . На рисунке высота барьера АЕ = 30 кДж/моль, и только у
газа с температурой Т= 1000 К или
выше имеется заметная доля молекул, способных преодолеть барьер
с;
О
X*
5
о.
ш
I
О
Плотность
Эта скорость есть число столкновений между молекулами А и В — очень
большое число, умноженное на вероятность реакции, которая может быть очень
малой. Например, если энергетический барьер составляет 100 кДж/моль, то
при температуре 500 К вероятность реакции равна только 3,5 х 10-11. То есть
лишь малая доля столкновений заканчивается формированием продукта реакции. Так или иначе, реакция — очень редкое событие! Примеры применения
теории столкновений даны в книге [5].
Выражение (3.109) схоже с аррениусовской зависимостью, однако между
ними имеется существенное различие. В уравнении Аррениуса зависимость от
температуры — чисто экспоненциальная, тогда как в теории столкновений дополнительно возникает предэкспоненциальный множитель, пропорциональный Т1/2.
Далее мы покажем, что теория переходных состояний предсказывает для него
более сильную зависимость от Т.
125
126
\
Глава 3. Теория скоростей реакций
Энергия активации находится дифференцированием логарифма скорости
реакции:
А,
Э Т
, coll = лт Гсо11 (3.110)
Таким образом, чтобы записать скорость реакции в форме уравнения Аррениуса, мы должны заменить высоту барьера АЕ на энергию активации АЕ + \/2kQT.
Эта замена эквивалентна появлению дополнительного сомножителя е1/2 в пред-
экспоненциальном множителе:
e^nd2
"coll = —
&kRT '' 2
'АВ = 1 + <^АВ- (31П)
При обработке экспериментальных данных обычно пренебрегают температурной зависимостью предэкспоненциального множителя и определяют энергию активации, используя координаты Аррениуса, как это было сделано в гл. 2.
Из теории столкновений, однако, следует, что в этих координатах для константы скорости должна быть скорее нелинейная, чем линейная зависимость, если
энергия активации составляет величину порядка кБТ.
Насколько может быть полезной полученная в теории столкновений формула для описания адсорбции? Если адсорбция не является активированной,
£а = 0, частота столкновений совпадает с частотой ударов молекул газа о
поверхность. Частота столкновений дает верхний предел скорости адсорбции.
В общем случае скорость адсорбции гораздо ниже, поскольку для перехода в
адсорбированное состояние молекула должна, например, испытать неупругое
столкновение с поверхностью, иметь определенную ориентацию или попасть
на центр адсорбции. Все эти факторы объединяются в некоторый эффективный коэффициент, называемый коэффициентом прилипания, характеризующий
вероятность перехода в адсорбированное состояние столкнувшейся с поверхностью молекулы. В случае активированной адсорбции (например, диссоциативная адсорбция N2 и СН4 на металлах) экспоненциальная температурная зависимость включатся в коэффициент прилипания. Для неактивированной адсорбции
в теории столкновений коэффициент прилипания вводится как дополнительный подгоночный параметр. Как мы увидим далее, теория переходных состояний придает коэффициенту прилипания определенный физический смысл.
3.5.3. Фундаментальные возражения
против теории столкновений
Теория столкновений, хотя она и позволяет из первых принципов
оценить верхний предел скорости реакции, обладает существенным недостатком: с ее помощью нельзя определить изменение свободной энергии в реакции,
поскольку этот подход оперирует только с индивидуальными молекулами, а не с
их ансамблями. В результате теория столкновений вступает в серьезные проти¬
3.6. Активирование молекул при столкновениях: теория Линдеманна -1\г 127
воречия с термодинамикой. Это становится очевидным при расчете в рамках
теории столкновений константы равновесия реакции. Рассмотрим равновесие
к+
А + В ^ C + D. (3.112)
к~
Использовав теорию столкновений для расчета констант скоростей прямого и обратного процессов и взяв их отношение, получаем следующее выражение для константы равновесия
к* KdlB(8kBT!KMAB)'12 е-АЕ/квТ ^вАср -АН/квТ /о 1104
"г ndlD{UBTinnCDf-e-(AE-AH)lk*T dlDn'&
Таким образом, мы нашли связь между К и энтальпией реакции, тогда как
из термодинамического анализа равновесия следует [в частности, из уравнения
(2.32)], что константа равновесия должна быть подобным же соотношением
связана с изменением свободной энергии. Поскольку предэкспоненциальный
множитель нельзя выразить через изменение энтропии, получаем противоречие между теорией столкновений и термодинамикой.
3.6. АКТИВИРОВАНИЕ МОЛЕКУЛ ПРИ СТОЛКНОВЕНИЯХ:
ТЕОРИЯЛИНДЕМАННА
В двадцатые годы прошлого века исследователей кинетических
процессов интриговала следующая проблема. Для бимолекулярных реакций типа
«А + В = Продукт» теория столкновений дает правдоподобное описание: если
при столкновении А и В обладают достаточной энергией, то энергетический
барьер реакции может быть преодолен. Но как в рамках этой теории объяснить
мономолекулярные реакции типа переходов между изомерами (например, циклопропана в пропилен) или распада молекул (например, сульфурила S02C12 на
S02 и С12)? Были предложены в высшей степени оригинальные, но не всегда
правдоподобные объяснения для этих реакций. Так, в 1919 г. Жан Перрен предположил, что энергию для реакций поставляет излучение, идущее от стенок
сосуда. Фредерик Линдеманн, впоследствии лорд Черуэлл и министр обороны
при правительстве Черчилля периода Второй мировой войны, категорически
возражал против «радиационной теории химического действия» и в 1921 г. предложил альтернативное объяснение, которое сейчас является общепринятым.
В соответствии с гипотезой Линдеманна—Кристиансена, которая были предложена ими независимо в 1921 г., все молекулы при столкновениях с молекулами окружающей среды постоянно получают и теряют энергию. Механизм
Линдеманна может быть представлен в простой форме:
128
Л
Глава 3. Теория скоростей реакций
где мы использовали звездочку для обозначения сильно возбужденных или
обладающих большой энергией молекул, способных преодолеть барьер реакции и перейти в продукт Р, а символ М означает молекулы окружающей среды,
в число которых входят и сами молекулы А.
Применение квазистационарного приближения для высокоэнергетического интермедиата (промежуточного продукта) А* позволяет найти концентрацию этого «неуловимого» компонента:
d[As;
dt
= V [А][М] - [А*] - jfcf[A*][M] = 0, (3.116)
ГЛ„ k\ [A][Ml
[А ]= , 1 Д J ■ (3.117)
&2 + kx [MJ
Отсюда скорость реакции равна
d[Р] _ кх к^_ [А][М]
(3.118)
dt к+ + к~ [М]
При нормальном давлении, когда число столкновений очень велико (как показано в предыдущем разделе, оно равно 1031 с-1 • л-1 при давлении 1 бар), в
знаменателе основным будет второе слагаемое, и мы получаем «правильный» результат: скорость реакции пропорциональна первой степени концентрации компонента А. При очень низких давлениях, однако, доминирующим будет первое слагаемое в знаменателе. Предположим, что в системе присутствует только компонент А,
тогда скорость мономолекулярной реакции перехода А в Р принимает вид
dfPl кхк^ [А][А] 1 + Гдо , Га1 1 +
* «\м"К[] т *Г[А|**- <ЗП9)
что соответствует реакции второго порядка. Таким образом, в теории Линдеманна
чисто мономолекулярных реакций не существует, поскольку для перевода молекулы в высокоэнергетическое состояние, в котором она способна преодолеть энергетический барьер, необходимы столкновения с окружающими молекулами.
Заметим, что если мы все же запишем кинетическое уравнение для мономолекулярной реакции в форме реакции первого прядка, r= к[А], и проведем
измерения скорости в широком диапазоне давлений, то мы найдем, что константа скорости будет падать с уменьшением давления при низких его значениях. Такое уменьшение константы скорости действительно наблюдалось для
ряда реакций изомеризации и распада.
3.7. ТЕОРИЯ ПЕРЕХОДНЫХ СОСТОЯНИЙ
Базируясь на изложенной выше теории Линдеманна, Генри Эй-
ринг и независимо М.Г. Эванс и Майкл Поляни создали в 1935 г. теорию скоростей реакций, используемую до настоящего времени и называемую теорией
переходных состояний или теорией активированного комплекса.
3.7. Теория переходных состояний 129
В качественной трактовке реакция рассматривается как идущая путем формирования активированного комплекса, переходного состояния системы, при
котором она находится на вершине энергетического барьера, разделяющего
реагенты и продукты реакции. Реагирующие молекулы попадают в переходное
состояние за счет столкновений с молекулами окружающей среды. Переход
через барьер возможен только по направлению прямой реакции. Реакция описывается только одним параметром, называемым координатой реакции и обычно
совпадающим с координатой некоторого колебательного процесса. Таким образом, реакцию можно представить как путешествие по поверхности потенциальной энергии (горный ландшафт), где переходное состояние лежит в седло-
вой точке (верхней точке горного перевала).
Соответствующая схема представлена на рис. 3.10 и ей отвечает цепочка
уравнений
R-*R# -*р, (3.120)
которая показывает, что переход через барьер возможен только по направлению реакции. Мы принимаем, что реагенты R находятся в равновесии с активированным комплексом R#, характеризуемом константой К*. Теперь мы опишем реакцию с помощью подходящей координаты реакции. Если мы имеем
дело с реакцией диссоциации двухатомной молекулы, то координате реакции
отвечают валентные (растягивающие) колебания двух атомов в ней, поскольку
возбуждение колебаний ослабляет связь между атомами. Скорость формирования продукта из переходного состояния принимается равной частоте колебаний, соответствующих координате реакции, hv. Можно привести правдоподобные аргументы в подтверждение данной гипотезы: если представить, что связь
диссоциирующей молекулы растянута за пределы переходного состояния, то
она разорвется, а частота попыток осуществить такое растяжение для переходного состояния совпадает с частотой колебаний.
Рис. 3.10. Расчет q проводится относительно
общего нулевого уровня энергии. Основное состояние переходного комплекса сдвинуто вверх
на величину рассчитывается по отно¬
шению к этому уровню
Таким образом, скорость полной реакции равна
(3.121)
130
\
Глава 3. Теория скоростей реакций
Выразим скорость реакции через статистические суммы реагентов и активированного комплекса, выделив статистическую сумму, отвечающую координате реакции:
d[Pl /7# е-(ШЧк*Т) а#
= vq — [R] = v гтгт (3.122)
dt v q L J 1 - e~hv/k*T q 1 1
Поскольку частота слабо связанной системы относительно мала, то есть
kQT » hv, мы можем использовать классический предел для статистической
суммы, kBT/hv, что дает для скорости реакции в теории переходных состояний
следующее выражение:
^ = <3123>
где
kTST=^f — • (3-124)
п q
Напомним, что координата реакции была исключена из статистической
суммы активированного комплекса q'*\ ей отвечает множитель kBT/hvB выражении (3.124). Заметим, что не все вышеприведенные рассуждения в полной
мере корректны: например, довольно спорными являются рассуждения о колебаниях в окрестности точки, отвечающей переходному состоянию. Здесь сила,
действующая на частицы, становится равной нулю и ее движение можно рассматривать, скорее, как свободное по одной координате.
В формуле (3.124) статистические суммы q и qn даны относительно одного
нулевого уровня энергии, что не вполне удобно для проведения дальнейших
расчетов. Разность уровней между основным и переходным состояниями равна
высоте барьера АД уже использованной в теории столкновений. Удобно для
переходного комплекса ввести уровень, отвечающий его основному электронному состоянию (см. рис. 3.10), и уже относительно него рассчитывать статистическую сумму q%. Введя эту статистическую сумму, получаем выражение для
константы реакции, очень похожее на уравнение Аррениуса:
*TST =-^-—е’Л£ЛвГ- (3.125)
п а
3.7.1. Термодинамическая форма выражения
для скорости реакции в теории переходных состояний
Выражение (3.123) часто представляют в форме
_ к*Т #
^tst - А j
(3.126)
3.7. Теория переходных состояний -*\г 131
оговаривая, что координата реакции исключена из константы равновесия или,
иными словами, что активированный комплекс находится в равновесии с реагентами по всем параметрам, за исключением координаты реакции. Вводя связь
между константой равновесия и свободной энергией Гиббса, находим:
TST
кВТ c-AGg/RT _ КТ c-ASg/Rc-AHg/RT '
h h
(3.127)
Мы заменили постоянную Больцмана на газовую постоянную, поскольку
термодинамические величины часто выражаются в единицах кДж/моль. Рассчитаем энергию активации (кДж/моль), используя определение
К tst = RT2 ^ In Ksi = АЯ0# + RT.
Записав константу скорости в аррениусовской форме
7 ~ Е-а TST IRT
^tst _ vTSTe ’ ,
получаем предэкспоненциальный множитель в виде
t!TST =
elcBTc-ASS/R
(3.128)
(3.129)
(3.130)
Выражение (3.130) показывает, что выигрыш в энтропии при переходе от
реагентов к активированному комплексу приводит к увеличению предэкспо-
ненциального множителя и скорости реакции. Такое возрастание энтропии
типично для статистических сумм, отвечающих колебательному, вращательному и/или поступательному движению, и часто называется разрыхлением переходного состояния (рис. 3.11). Альтернативный взгляд состоит в том, что в
переходном состоянии имеется большее число уровней, которые могут быть
заняты при данной температуре, чем в основном состоянии.
Рис. 3.11. В жестком переходном состоянии энтропия ниже, чем в основном состоянии реагентов, поэтому предэкспоненциальный множитель будет меньше
стандартной величины ek^T/h ~ 1013 с-1.
Разрыхленное переходное состояние,
напротив, имеет булыиую энтропию и
больший предэкспоненциальный множитель
Жесткий
переходный
комплекс
Q* « Q
109< 10'3 с-1
AS* отрицательно
Рыхлый
переходный
комплекс
<7# » q
1013 < v< 1017с~1
AS# положительно
132 -П/- Глава 3. Теория скоростей реакций
Конечно, возможна и обратная ситуация, когда энтропия активированного
комплекса меньше энтропии реагентов (см. рис. 3.11). В этом случае говорят о
жестком переходном состоянии. Жесткость связана с тем, что вращение, колебания или движение реагентов в активированном комплексе ограничены сильнее, чем в их основном состоянии. Примером такого состояния является диссоциация молекул на поверхности, которую мы обсудим в следующем разделе.
Промежуточная ситуация, при которой энтропия переходного комплекса
отличается незначительно от энтропии реагентов в основном состоянии, отвечает стандартному предэкспоненциальному множителю hv= ekBT/h, по порядку величины равному 1011 с-1. Энтропийный фактор изменяет предэкспоненциальный множитель от 1013 до 1017 с-1. Если измеренный предэкспоненциальный множитель выходит за эти пределы, то следует либо признать формируемый
комплекс неразложимым, либо допустить, что, возможно, в реакции принимает только малое число молекул. Ниже на примере поверхностных реакций мы
покажем, что такая ситуация может встретиться на практике.
Пример
В качестве примера, иллюстрирующего применение теории переходных
состояний, рассмотрим диссоциацию двухатомной молекулы СО
СО ^ СО* -С + О. (3.131)
Координата реакции соответствует валентному колебанию вдоль связи С—О
молекулы СО, находящейся в переходном состоянии. Константа скорости такова
klsl = У е~АЕ/квТ, (3.132)
^trails ^vib ^rot
где мы опустили колебательную статистическую сумму для переходного комплекса, поскольку ей отвечает координата реакции. Учитывая очевидный факт
равенства статистических сумм поступательного движения, зависящих от массы,
температуры и доступного объема, для основного состояния и активированного
комплекса и большое значение частоты колебаний молекулы СО (2143 см-1),
которому отвечает колебательная статистическая сумма q'Vyb ~ 1 , получаем
-hv/2kBT
<злзз>
Поскольку вращательная статистическая сумма двухатомных молекул типа
СО равна
qm (СО) = 1,1= цг1_0, (3.134)
п
мы можем упростить выражение для константы диссоциации молекулы СО в
газовой фазе:
3.1. Теория переходных состояний
Отсюда получаем энергию активации
\
Еа = &Е + квТ - 4г (3.136)
и предэкспоненциальный множитель
/ # \2
о г* „
(3.137)
кпТе
v = -V-
'С-0
чгс-0/
Пример
Рассмотрим простую реакцию газов СО и 02:
С0 + 02 ±5 С02 +0, (3.138)
для которой переходному состоянию отвечает комплекс ОСОО. Определим
температурную зависимость предэкспоненциального множителя в предположении линейной структуры активированного комплекса и (для простоты) малости энергий всех колебаний по сравнению с квТ (qVlh ос kBT/hv).
В соответствии с теорией переходных состояний константу скорости реакции можно представить в виде
^sj = ^Я$хд_е-АЕ,ьт = VjSTe-^Tt (3 139)
" #о2#со
где энергии всех нулевых колебаний включены в параметр АЕ. Поскольку все
три молекулы линейны, для каждой из них мы имеем только по две вращательные моды. В результате у нас имеется 3N — 5 колебательных мод. Заметим
также, что поскольку рассматриваемые молекулы являются относительно простыми, нет необходимости анализировать эффекты типа внутреннего вращения, что могло бы сильно усложнить ситуацию. В табл. 3.5 приведены все
зависящие от температуры сомножители.
Таблица 3.5. Числа степеней свободы для поступательного, вращательного
и колебательного движений реагирующих молекул
и активированного комплекса, отвечающего реакции газов СО
и 02, и температурные зависимости вкладов от этих движений
в статистическую сумму. Заметим, что одна колебательная мода
соответствует координате реакции, поэтому приведены данные
только для шести колебательных мод
Компонент
М°Да tftrans
Мода qTOt
Мода ?vib
Итоговая q
СО
3 тщ
2 Т
1Т
j7/2
о2
зтщ
2 Т
1 т
т7/2
ОСОО
3 J-3/2
2 Т
6 т6
7-17/2
133
134 -l\r Глава 3. Теория скоростей реакций
Подставив температурные зависимости из табл. 3.5 в выражение (3.139) для
предэкспоненциального множителя, получаем температурную зависимость следующего вида:
7^17/2
(3.140)
Читатель может теперь проверить, что энергия активации, вычисляемая через
производную логарифма, содержит дополнительный к АЕвклад, равный 5квТ/2,
а для корректного сравнения выражения (3.139) с уравнением Аррениуса предэкспоненциальный множитель должен быть умножен на е5/2. Хотя предэкспоненциальный множитель достаточно сильно изменяется с температурой, отклонения зависимости \пк от 1 /Т (координаты Аррениуса) от линейной будут
незначительными, если энергия активации не слишком мала.
3.8. ТЕОРИЯ ПЕРЕХОДНЫХ СОСТОЯНИЙ
ДЛЯ ПОВЕРХНОСТНЫХ РЕАКЦИЙ
В этом разделе мы в общем виде получим выражения для скоростей элементарных поверхностных реакций: адсорбции, десорбции и диссоциации. Конкретные случаи будут рассмотрены в следующих главах. Приведенные
ниже выражения позволят глубже понять физический смысл определяемых из
эксперимента коэффициента прилипания и предэкспоненциального множителя. Мы ограничимся рассмотрением систем, в которых адсорбат равномерно
распределен по поверхности, и не будем учитывать взаимодействие между адсорбированными компонентами. Фактически мы используем приближение среднего поля, которое является корректным при малых степенях запонения поверхности и в отсутствие взаимодействия или при наличии только отталкивания между молекулами. Притяжение между адсорбированными компонентами,
как правило, приводит к образованию агрегатов и островков из молекул даже
при низких степенях заполнения. В этом случае ситуация сильно усложняется
и требует привлечения численного моделирования, например, методом Монте-
Карло.
3.8.1. Адсорбция атомов
При рассмотрении адсорбции атомов и молекул мы должны различать два случая — прямую и непрямую адсорбцию. В прямой адсорбции частица, столкнувшись с поверхностью, остается в точке удара как адсорбированный компонент. В случае более общей непрямой адсорбции частица сначала
попадает в слабо связанное состояние (прекурсор), в котором она может свободно двигаться вдоль поверхности в течение некоторого времени, пока не
будет образована ее прочная связь с некоторым адсорбционным центром.
3.8. Теория переходных состояний для поверхностных реакций
\
3.8.1.1. Непрямая адсорбция
Для непрямой адсорбции атомов переходное состояние — не что
иное, как его двумерное движение вдоль поверхности
А+*^А;*оЬИе. (3.141)
По истечении некоторого времени подвижный атом-прекурсор находит свободный центр адсорбции и формирует связь с поверхностью:
A;#obi,e^A*mobile. (3.142)
Рассматриваемая нами система состоит из объема, содержащего Ng атомов
газа, взаимодействующих с поверхностью, на которой имеется М адсорбционных центров или М/А центров на единице поверхности. Теория переходных
состояний дает следующее выражение для скорости реакции в этой системе:
А = VN = vK Ng. (3.143)
dt
Здесь мы использовали общепринятое обозначение символом # переходного состояния А^. Как мы уже видели, при рассмотрении реакций на поверхности обычно вводят понятие степени покрытия поверхности. Степень покрытия поверхности для А* дается выражением
*Л' = ~М~’ (ЗЛ44)
воспользовавшись которым, получаем
d6> d Na. K*N„ k# V
(3145)
где коэффициент kJST связывает интересующую нас степень заполнения поверхности компонентом А* и давление газа А. Заметим, что мы считали газ
идеальным, N = pV/k^T.
Нам необходимо найти выражение для константы равновесия между газом
и активированным комплексом. Координата реакции снова соответствует (очень
слабому) колебанию атома по нормали к поверхности. Колебаний вдоль поверхности нет, поскольку атом свободно движется в двух измерениях. Статистические суммы для газа и переходного комплекса таковы:
N„
J -SsL. Q si
'gas Nt,! ’ N#!
Q*=\(3.146)
'к- "ti¬
me N' и Nn — числа атомов в соответствующих двух состояниях и
qg=<i£L\ я я = €qlL- (3.147)
135
136
л
Глава 3. Теория скоростей реакций
Поскольку мы предположили наличие равновесия между газом и активированными комплексами, можно воспользоваться следующим из статистической
механики равенством jugas = jun, в котором
,Sln(Q)
// - kBT -
Из этого равенства получаем
SN
к# = N*_ = g, gp,s ^ (3.148)
^trans
и скорость захвата принимает вид
сШЛ = vN# = рЛГ» К/>А _ vql Cns ^ A(2xmkBT)/h2 КрА _
d? М М kBT М kBT Mh V (2тгтквТ^/h3 Кт
Ар к Ра (3.149)
M^2nmkBT N0.yj2KmkBT
Отсюда вытекает важный результат
Ра
d t yV0 ^2nmkBT
(3.150)
Умножив это уравнение на число центров, находящихся на единице поверхности, N0, получаем число атомов, адсорбирующихся на единице поверхности
в единицу времени
d0A М d^A 1 d^A Ра
<3,51>
Эта формула эквивалентна соотношению (3.107), полученному в теории столкновений. Это — ожидаемый результат, поскольку атомы по существу представляют собой твердые сферы, не обладающие внутренними степенями свободы.
Отметим также, что при выводе формулы (3.107) мы не накладывали каких-
либо ограничений на характер движения атомов вдоль поверхности после их
столкновения с нею.
В рассмотренной выше схеме все атомы, ударившиеся о поверхность, переходят в состояние прекурсора. Ясно, однако, что адсорбированные ранее атомы
могут блокировать центры, делая их недоступными для вновь прибывающих атомов, поэтому изложенная модель пригодна только при низких степенях заполнения поверхности. При высоких степенях заполнения мы должны более детально
проанализировать поведение адсорбированных атомов, находящихся в состоянии прекурсора. Во многих случаях атомы в состоянии прекурсора являются
слабо связанными с поверхностью. Они могут в процессе миграции вступить в
химическую реакцию с поверхностным центром и локализоваться на нем или
3.8. Теория переходных состояний для поверхностных реакций
\137
десорбироваться. Если слабо связанное состояние может формироваться и на
вершине уже адсорбированного атома, то в этом случае оно называется внешним
(в отличие от внутреннего прекурсора, связанного с чистой поверхностью), и
степень заполнения поверхности не будет оказывать влияние на скорость адсорбции. В отсутствие таких внешних состояний прекурсора скорость адсорбции
будет пропорциональна площади свободной поверхности. Заметим, однако, что
при высоких степенях заполнения предложенная модель становится непригодной, поскольку на ограниченной и фрагментированной поверхности двумерное
движение атомов газа нельзя уже считать свободным.
3.8.1.2. Прямая адсорбция
ма относится к трехмерному пространству над поверхностью (мы извиняемся
за противоречащую интуитивному ощущению терминологию, приписывающую
основное состояние молекулам над поверхностью). Переходному состоянию отвечает полностью неподвижный атом, находящийся на поверхностном центре:
Координата реакции, описывающая процесс адсорбции, связана с колебанием атома у поверхности. Строго говоря, у адсорбированного атома есть три
колебательные моды: одна нормальная к поверхности и отвечающая координате реакции и две параллельные поверхности. Две последние моды, которые
иногда называют нарушенными трансляционными модами, обычно являются
очень мягкими, и для них kET» hv. Ассоциативная (недиссоциативная) адсорбция идет в этом случае, как правило, безбарьерным образом, поэтому мы
положим АЕ = 0. Теперь мы можем в рамках теории переходных состояний
записать выражение для скорости прямой адсорбции, используя метод, примененный ранее для непрямой адсорбции.
Статистическая сумма для газа, Qgas, остается неизменной. Однако нам необходимо принять во внимание конфигурацию атомов в переходном состоянии, поскольку они сразу теряют свою подвижность на двумерной поверхности.
Снова рассмотрим поверхность, на которой имеется М центров, на каждый из
которых приходится площадь а2. Число центров на единице поверхности равно
N0 = М/А = l/а2. Не все М центров обязательно являются свободными, поскольку некоторые из них уже могут быть заняты адсорбировавшимися ранее атомами, так что число свободных центров обозначим через М' и 0* = М'/М= (1 — А).
При наличии Nn атомов, адсорбированных на этих центрах (мы снова используем символ «#» для отнесения соответствующих величин к переходному состоянию, А-^тоЫ1е), статистическая сумма системы дается выражением
При рассмотрении прямой адсорбции основное состояние для ато-
(3.152)
(3.153)
(3.154)
138 -l\r Глава 3. Теория скоростей реакций
Предполагая равенство химических потенциалов jug = jun и применяя формулу Стирлинга 1п(ЛМ) = Nln(N) — N, имеем после некоторых преобразований:
к* =^1 = (М'-ЛГ#)— ИЛИ К* = М(в, -а,)—, (3.155)
Nx Ч ' Ч
где мы ввели степень заполнения поверхности атомами, находящимися в переходном состоянии, вп = NJM. Обычно Эп «с 9^ и этой величиной можно пренебречь,
то есть — <9# = 0^ Теперь мы можем легко получить изотерму адсорбции Ленгмюра, которую подробно обсудим позже. Используя уравнение (3.145), находим
d6*A _ v^tL _ vK^Vp^ _ v(M'-N#)q# VpA _
dt ММ kBT M qg kBT
= v(Q -Й ^^2fl-vib = ^A# 42D-V\h _ ^A^ {^B^/^V2d) ^ -(AE + hv, D_vih )ДВГ _
" l‘“#j 9^ = A = A V(2nmkBTf2
a I/ tv*, ^.2 (3.156)
Pa * \квТ/Ьу21)) h ^_(AE+hv2D_vlb)/kBT _
yjlnmk^T 2 7tmkBT
-e
NqPaO* {квТ/Ьщр) h~ (Д£+^л_у.ь)двг = 0*Ра$о {T) _ (1 ^a^a^o^)
NQyj27rmkBT 2nmkBT N^-^2nmkBT NQy[27rmk^f
Здесь мы использовали уравнение (3.147) и приняли, что основное электронное состояние переходного комплекса приподнято на величину Д£ (чтобы
рассчитывать статистическую сумму переходного комплекса относительно его
собственного основного состояния) и статистическая сумма q*_D_yxh рассчитывается относительно дна потенциальной ямы, как показано на рис. 3.10. Из выражения (3.156) видно, что скорость адсорбции на единичной поверхности равна числу столкновений атомов с этой поверхностью, умноженному на так называемый коэффициент прилипания SQ(T), который всегда должен быть меньше
единицы. Коэффициент прилипания показывает, какая доля атомов, столкнувшихся с поверхностью, успешно добралась до адсорбционных центров.
Таким образом, скорость адсорбции равна
УТ),Л
dt N0p7rmkBTv A/
где
n,0
_ fT. _ Mq\D
-vib _ ^2Z)-vib _ ^2Z)-vib i ^q\
^OV7 ) ~ 2D ~ 2D /гж ~ ~TD
^trans ^trans/ ^trans-unitcell
ЛD qVL a 2ятквТ 2 InmkJ
‘/trans-unitcell M M h2 h2 ' ^
— статистическая сумма поступательного движения в ячейке площадью а2.
3.8. Теория переходных состояний для поверхностных реакций
\
Чтобы дать ощущение величины коэффициента аккомодации при прямой адсорбции, подставим в (3.158) типичные значения параметров: а = 2,5 А,
Т = 300 К, m = 40 х 1,66 х 10-27 кг для Ar, АЕ = 0 и hv = 40 см-1, получаем
с (т\ - q*D-v ib _ (bT/kV2D) hr (д£+hv2 r>-\\b )/k^T _
°0 V ) - ~2D ” TPs , e
^trans-unitcell ^ yLTlYYlk^l J
,тч2 (3.160)
В7 e-{AE+hv2D-\ib)/kBT — Q 09
hv2DJ
Изложенная модель перестает работать, когда энергия А остановится много меньше квТ, поскольку потенциал становится столь мягким, что атомы уже
не локализуются на определенном центре, а диффундируют по поверхности. В промежуточном режиме атомы совершают движение в потенциале
4,0x10"
V{x, y) = \v0
~ ,2л хЛ ( 2 л у
2-cos -cos1
(3.161)
описание которого является слишком сложным и выходит за пределы данного
рассмотрения. В некоторых случаях, когда, например, потенциал достаточно
слаб или температура очень высока, атомы перемещаются по поверхности свободно и их можно рассматривать как двумерный газ. Тогда колебательная статистическая сумма <72z>-vib должна быть заменена статистической суммой двумерного поступательного движения:
\ 2
hv2D
e-(bE+hv2D_wlb)/kBT а~ (2лткъТ) ^ (3.162)
/г
и мы приходим к формуле, отвечающей случаю непрямой адсорбции. Из приведенных выше вычислений легко видеть, что в зависимости от природы переходного состояния SQ изменяется от 1 (когда прекурсор может свободно перемещаться по поверхности) до 10_3 (когда поступательное движение атома в переходном состоянии полностью заморожено и он неподвижен) даже при АЕ = 0.
Если адсорбция является активированной, то есть АЕ > 0, то коэффициент
прилипания может быть еще меньше. Более того, при адсорбции молекул возможны также стерические препятствия, так что энергия активации начинает
зависеть от ориентации молекул по отношению к поверхностным атомам и от
положения точки столкновения молекул с поверхностью (последняя, очевидно, не является однородной). Таким образом, если адсорбция идет по прямому
активированному механизму, имеются серьезные аргументы в пользу понижения величины коэффициента прилипания. Заметим, что коэффициент прилипания атомов S0 может быть достаточно надежно аппроксимирован выражением
а* <7°#
С (т\ — ^2Z)-vib _ *f2Z)-vib „-АЕ/к^Т _
0^)-~д2D ~~2D в
^trans-unitcell ^trans-unitcell i/гоч
//0# (3.163)
_ _92^vib_ ~AE-(2hv\l/2)/kBT _ CO -AEaa/kBT
~ 2D ~ V >
^trans-unitcell
139
140 -l\r Глава 3. Теория скоростей реакций
где <?2z)-vib рассчитывается относительно основного электронного состояния
переходного комплекса, a q^-yш — относительно его основного колебательного
состояния. Мы объединили два вклада от нулевых колебаний и АЕ в энергию
активации AEact. Ниже мы объясним, как эта важная характеристика может
быть измерена в эксперименте.
3.8.2. Адсорбция молекул
Описание адсорбции молекул во многом проводится так же, как и
атомов, нужно только учесть внутренние степени свободы молекул. Фактически нам необходимо рассмотреть, как эти внутренние степени свободы изменяются при переходе молекулы из газовой фазы в переходное состояние. В теории переходных состояний скорость адсорбции в наиболее общем виде может
быть записана так
d^A, d/VAj VN# K*Ng к* V
dt Mdt M V M V M kBTPA' (• )
Координата реакции связана с колебаниями молекул по нормали к повер¬
хности и исключается из колебательной статистической суммы переходного
комплекса. Снова имеем два предельных случая.
3.8.2.1. Непрямая адсорбция с формированием прекурсора
Если молекула сначала попадает в состояние физически адсорбированного прекурсора, в котором она может свободно двигаться вдоль поверхности, а также вращаться и колебаться (пусть и с измененными частотами), то
мы имеем
Л/Г# /7#
К*=ТГ = <3165>
A' ^gas
deA vN# vVpA КЯ _ VpA <7tran.v ffrot 4vib
dt M kj м Mh q™nsq?«qTb
:S0(T).
Pa ^rot^vib _ Pa
(3.166)
Таким образом,
N0ртгткнТ <q^ N0pxmkBT
d6>A PAS0(T)
dt N0 j2nmkBT ’
(3.167)
где справа стоит частота столкновений молекул с поверхностью и некоторое
отношение, которое можно снова интерпретировать как коэффициент прилипания, то есть как вероятность захвата молекулы после ее столкновения с по¬
3.8. Теория переходных состояний для поверхностных реакций -1\г 141
верхностью. Вполне естественно, что при неизменности внутреннего состояния молекулы, когда q*ot = qf™ и q*ib = q^bs, коэффициент прилипания равен
единице, поскольку
Следовательно, в теории переходных состояний адсорбция становится более вероятной, если в состоянии подвижного физически адсорбированного
прекурсора она сохраняет способность к колебательным и вращательным движениям, присущую ей в газовой фазе. Вполне естественно, что этой ситуации
отвечают наименьшие потери энтропии при адсорбции. В общем случае переход из газовой фазы к двумерному движению всегда сопровождается потерей
энтропии, поэтому коэффициент прилипания обычно есть величина меньше
единицы.
3.8.2.2. Прямая адсорбция
торой молекула сразу же «приземляется» на адсорбционный центр с потерей
способности к перемещению вдоль поверхности. В этом случае в переходном
состоянии молекула обладает только колебательными степенями свободы, одна
из которых, отвечающая колебаниям молекулы по нормали к поверхности, является координатой реакции. Указанные обстоятельства приводят к следующему соотношению, ясно указывающему на малую величину константы скорости
адсорбции:
где мы использовали равенство <9* - 0# = 6^ = 1 — вк и выражение (3.155) для
константы равновесия. Здесь q'# — статистическая сумма молекулы в переходном состоянии с исключенной координатой реакции. В результате мы для скорости прямой адсорбции получаем
(3.168)
Другим предельным случаем является прямая адсорбция, при ко-
deA _ vNn _ vK* VpK _ {в, -вй)УрА
(3.169)
d0A _ so{t)Pa /, n ч
At ~ AT h ,, T1 У A '
d t Nn^2xmkBT
(3.170)
где
c \ _ Mg'" _ q^
a3D ag3sagas a3D ngas ngas
^trans^rot ^vib ^trans—unitcell ^ rot Tvib
(3.171)
142 -l\r Глава 3. Теория скоростей реакций
Ясно, что при прямой адсорбции коэффициент прилипания должен быть
малым, поскольку «вмораживание» молекулы в адсорбционный центр на поверхности связано со значительной потерей энтропии. В действительности адсорбция большинства молекул идет через образование подвижного прекурсора. Тем не менее прямая адсорбция также имеет место, однако она обычно
сопровождается активированной диссоциацией высокостабильных молекул.
Примером служит диссоциативная адсорбция молекул СН4, коэффициент прилипания для которой составляет величину порядка 10-8—10-6. В этом случае
коэффициент прилипания определяется не только статистическими суммами,
но и экспоненциально зависит от энергии активации, поскольку основное электронное состояние переходного комплекса поднято на величину АЕ, как это
показано на рис. 3.10:
/# _ /#0е~ЬЕ/квТ _ #0 #0 #0 -ДЕ/квТ (3 172)
Ч ~Ч е — Ч\[ь 4fms—rot^frus—trans^ * yj.Lttj
Индекс «0» указывает, что статистическая сумма рассчитывается относительно дна потенциальной ямы переходного комплекса, а не от основного состояния, благодаря чему появился экспоненциальный множитель. Статистическая сумма переходного комплекса, как обычно, может быть разбита на три
сомножителя. Первый из них, qучитывает все колебательные моды молекулы за исключением координаты реакции. Множитель qf^s_rot связан с колебаниями, обусловленными наличием предпочтительной ориентации молекулы на
поверхности при исключении возможности ее вращения. Вращение в этом случае
становится осложненным, как показано на рис. 3.12 для двухатомной молекулы СО. Последний сомножитель trans характеризует осложненное поступательное движение параллельно поверхности (см. рис. 3.12). Снова, если нам
точно известно основное состояние, мы можем вычесть энергии нулевых колебаний и перейти к измеряемой величине AEact.
Координата Осложненное Осложненное
реакции поступательное поступательное
Рис. 3.12. У адсорбированной на поверхности молекулы СО есть пять колебательных мод.
Заметим, что поверхность не обязательно симметрична по направлениям хи у,
поэтому колебания в направлениях х и у не обязательно вырождены
В общем случае нелинейная молекула, образованная из Анатомов, в газовой
фазе обладает тремя поступательными, тремя вращательными и 3N — 6 колебательными степенями свободы, которые сводятся к трем осложненным посту-
3.8. Теория переходных состояний для поверхностных реакций -*\г 143
пательным, трем осложненным вращательным и 3N — 6 колебательным модам
за вычетом одной моды, отвечающей координате реакции. У линейной TV-атомной молекулы в газовой фазе имеется три поступательные, три вращательные и
3N — 5 колебательные степени свободы, а на поверхности — три осложненные
поступательные, две осложненные вращательные и 3N— 5 колебательные моды
за вычетом моды координаты реакции. Таким образом, переходное состояние
при прямой адсорбции молекулы СО характеризуется двумя модами осложненного поступательного движения, двумя модами осложненного вращения и
одной колебательной модой. Одна мода осложненного нормального к поверхности поступательного движения исчезла, поскольку она связана с координатой реакции. У сложных по строению молекул могут быть уровни, связанные с
внутренними вращениями, что серьезно усложняет картину процесса адсорбции. Анализ таких систем выходит за рамки этой книги.
3.8.3. Реакции между адсорбированными молекулами
Сейчас мы рассмотрим адсорбированные на поверхности атомы
или молекулы, могущие вступать в химические реакции между собой. Мы оста¬
вим в стороне детали, связанные с внутренними координатами таких адсорбированных элементов, заметив лишь, что их статистические суммы могут рассчитаны изложенными выше способами. Предположим, что элемент А реагирует с
элементом В с образованием продукта АВ и промежуточного активированного
комплекса АВ#:
А:!:+В:1 АВ* , (3.173)
АВ," > АВ + . (3.174)
Снова предположим, что элементы распределены случайным образом по
поверхности. Константа равновесия для такой системы дается выражением
К* =^b1 = ^abL (3175)
#А#В
Скорость поверхностной реакции принимает вид
d0AR „ v^*k*T q'
#
-JT = = \s’K*Wb = 'У A° (3-176)
dt AB AB AB hvm„ qAqb
где символы у q'n указывают на то, что координата реакции исключена из
статистической суммы. Если мы к тому же будем рассчитывать статистическую
сумму переходного комплекса относительно его основного электронного состояния, то сдвиг энергии между двумя основными состояниями, Д£А+В_АВ#,
может быть выделен, что позволяет записать к+ в привычной форме:
144
\
Глава 3. Теория скоростей реакций
Обратным процессом для реакций (3.173) и (3.174) является диссоциация
соединения АВ на поверхности. Легко видеть, что рассмотрев реакцию
АВ, +
АВ**,
(3.178)
можно просто найти константу равновесия Кп = 0Ав#/^ав#/#ав и константу
скорости
(3.179)
где
„/0#
к- = КТ Я^ = квТ У>_е~А£АВ-АВ»/*ВГ = ^-е-Л£АВ-АВ*ЛвГ (з 180)
h Яав h Уав
На рис. 3.13 приведена диаграмма соответствующей потенциальной энергии.
АВ#**
Координата реакции
Рис. 3.13. Диаграмма потенциальной
энергии для прямой и обратной реакций
А* + В* *± АВ* + *
Если мы примем двустороннее равновесие для переходного комплекса, то
получим
de
АВ
dt
— к Ок #ab$* — 0,
что дает
к+ _ ?АВ г(А£ав-ав»~Л£а+в-ав»)/<:в7' _ AS/kB АН/квТ
(3.181)
(3.182)
Заметим, что статистическая сумма переходного комплекса исчезла из рассмотрения вследствие предположения о равновесии и константа равновесия
определяется, как это и должно быть, только параметрами начального и конечного состояний. Этот результат полезен для рассмотрения более сложных ре-
3.8. Теория переходных состояний для поверхностных реакций -i\r 145
акций. Если некоторые промежуточные стадии реакции достигли равновесия,
а нас интересует выражение итоговой скорости через статистические суммы,
то многие члены при этом сократятся. Но если равновесия на промежуточных
стадиях нет, мы можем использовать вышеприведенный подход для определения скорости реакции при условии знания энергетических уровней активированного комплекса, позволяющего рассчитать статистические суммы.
3.8.4. Десорбция молекул
Десорбция — противоположный адсорбции процесс, завершающий
каталитический цикл. Она лежит также в основе метода температурно-про-
граммируемой десорбции (ТПД), с помощью которого определяют теплоты
адсорбции и скорости реакций на поверхности в режиме, позволяющем находить энергии активации и предэкспоненциальные множители. Сказанное делает необходимым получение теоретического выражения для скорости десорбции, которое можно сопоставить с экспериментальными данными.
Десорбирующаяся молекула в переходном состоянии может быть как подвижной, что соответствует состоянию подвижного прекурсора в процессе непрямой адсорбции, так и неподвижной, когда она обладает только колебательными степенями свободы. Заметим, что при высоких температурах подвижность могут приобрести и закрепленные на адсорбционных центрах молекулы.
В качестве примера можно привести механизм десорбции молекул СО, которые
в основном состоянии на поверхности являются неподвижными, а в процессе
десорбции переходят в подвижное состояние. Такой двухступенчатый механизм
приводит к резкому возрастанию предэкспоненциального множителя по сравнению со стандартной величиной ekBT/h (которая будет найдена ниже).
Неподвижность в основном состоянии не предполагает, что молекулы не могут
диффундировать к соседним центрам. Они могут делать это, но прыжками, которые рассматриваются как отдельный канал реакции, характеризующийся собственной константой скорости. Таким образом, мы считаем, что в основном
неподвижном состоянии молекулы просто находятся очень долго на определенном центре. В таком состоянии у молекулы имеется два конкурирующих способа покинуть адсорбционный центр — перескочить в соседний центр и десорбироваться. Противоположным к неподвижному основному состоянию молекул
является их свободное движение, когда они ведут себя как двумерный газ.
Запишем скорость реакции в виде
АВ, ^ АВ?, (3.183)
АВК ' АВ I ". (3.184)
Первое уравнение соответствует формированию переходного состояния, а
второе — собственно десорбции молекул. Для нахождения константы скорости
десорбции используем изложенную выше процедуру. Выпишем выражения для
статистических сумм участвующих в реакции элементов, приравняем химичес-
146
Глава 3. Теория скоростей реакций
кие потенциалы и найдем число молекул, находящихся в переходном состоянии. Поскольку на практике удобно оперировать со степенью покрытия поверхности, сразу получаем
где отрицательный знак показывает, что в рассматриваемой реакции молекулы
АВ удаляются с поверхности. Здесь мы снова обозначаем через qAB и qAB статистическую сумму переходного комплекса с исключенной координатой реакции и статистическую сумму переходного комплекса, найденную относительно
его основного электронного состояния соответственно. Таким образом, АЕ снова
представляет собой разность энергий основного и переходного состояний молекулы.
Обычно кинетика десорбции описывается уравнением
из которого, в соответствии с определением (2.46) аррениусовской энергии,
следует:
Это соотношение не является в полной мере корректным, поскольку колебательные вклады часто выделяются из члена hv и включаются в энергию активации, которая в этом случае будет отличаться от энергии АЕ или АЕа, задаваемой
уравнением (3.188).
Заметим, что если отношение статистических сумм в выражении (3.188)
будет зависимым от температуры, то это приведет к появлению слагаемого квТ
в энергии АЕа и множителя е в комбинации hv. Таким образом, предэкспоненциальный множитель hv в существенной мере определяется природой переходного состояния.
Адсорбированные молекулы часто бывают связанными достаточно прочно
и могут рассматриваться как неподвижно сидящие на дне колебательной потенциальной ямы. Состояния переходного комплекса более разнообразны, и
прекурсор может, например, обладать высокой подвижностью по двум измерениям. Таким образом, предэкспоненциальный множитель, зависящий от статистических сумм вращательного и поступательного движений, в десорбцион-
ном процессе может существенно превосходить стандартную величину, равную
kBT/h ~ 1013 с"1. На рис. 3.14 отражены различные механизмы десорбции и
(3.187)
Еа = АЕ + квТ\ v = е
(3.188)
3.8. Теория переходных состояний для поверхностных реакций -1\г 147
приведены соответствующие предэкспоненциальные множители. В табл. 3.6
дана сводка экспериментальных данных по десорбционным параметрам молекул СО, адсорбированных на разных поверхностях. Большинство приведенных
значений предэкспоненциального множителя соответствует рыхлым переходным состояниям, о которых мы только что вкратце говорили.
Таблица 3.6. Экспериментальные данные по энергиям активации и значениям
предэкспоненциального множителя для десорбции СО и N0
с чистых поверхностей некоторых хорошо охарактеризованных
монокристаллов. Все данные получены для малых степеней
заполнения поверхности [7]
Система
Предэкспоненциальный
множитель, с-1
Энергия активации,
кДж/моль
С0/Со(0001)
1015
118
CO/Ni(l 11)
1015
130
CO/Ni(l 11)
1017
155
CO/Ni(l 11)
1015
126
C0/Ni(100)
1014
130
C0/Cu(100)
1014
67
C0/Ru(001)
1016
160
CO/Rh(l 11)
1014
134
CO/Rh( 111)
1014
134
CO/Pd(l 11)
1014
143
CO/Pd(l 11)
1015
147
C0/Pd(100)
1016
160
CO/Pd(211)
1014
147
CO/Ir(l 10)
1013
155
CO/Pt(lll)
1014
134
NO/Pt(l 11)
1015
126
NO/Pt(l 11)
1016
113
148 -l\r Глава 3. Теория скоростей реакций
Адсорбированное
состояние
Переходное
состояние
Десорбированное
состояние
Предэкспоненциальный
множитель
Подвижное
Неподвижно” ""
©
~1015 с-1
~1013 с'1
о
< >
'Р1
\
Подвижное j
О
О
со
т
О
т
О
Неподвижное у
~1013с-1
Рис. 3.14. Микроскопическая картина десорбции атома и молекулы через неподвижный и
подвижный переходный комплексы. Если переходный комплекс схож с основным состоянием, то следует ожидать, что предэкспоненциальный множитель
будет иметь значение порядка 1013 с-1. Если адсорбированные молекулы подвижны в переходном состоянии, то предэкспоненциальный множитель может
возрасти на один-два порядка. Возможность вращения десорбирующихся молекул в переходном состоянии приводит к еще большему увеличению предэкспоненциального множителя. Приведены значения предэкспоненциальных множителей для атомов и молекул типа С, N и О и СО, NO и 0?, заимствованные
из [6]
3.9. ЗАКЛЮЧЕНИЕ
В заключение данной главы отметим наиболее важные из полученных результатов. Если нам известно распределение энергетических уровней
ансамбля частиц (напомним, что статистическая механика оперирует с большим числом частиц), то мы можем рассчитать его статистическую сумму и
затем определить важные макроскопические параметры системы: энергию, давление, энтропию и химический потенциал.
Из условия равенства химических потенциалов молекул в двух системах,
находящихся в равновесии, можно по отношению статистических сумм найти
константу равновесия.
Даже если система не находится в химическом равновесии, можно определить скорость приближения к равновесию. Для этого необходимо знание детального распределения энергетических уровней молекул в переходном состоя¬
3.9. Заключение
149
нии (что дает возможность сделать основной шаг — рассчитать статистические
суммы). Заметим, что сейчас численное моделирование достигло таких успехов, когда расчет скоростей реакций стал рутинным делом. Примеры таких
расчетов будут даны в гл. 6.
Применяя аппарат статистической термодинамики, мы получили явные
выражения для величин адсорбции, скоростей реакции и десорбции молекул.
В каждом случае соответствующая константа скорости была представлена как
отношение статистических сумм переходного комплекса и реагентов. Ниже
дана сводка наиболее важных формул для элементарных поверхностных реакций. Все входящие в формулы константы (предэкспоненциальные множители,
энергии активации) могут быть в принципе рассчитаны через статистические
суммы. Их вычисление, однако, не всегда является легкой задачей, поэтому
там, где это возможно, следует использовать экспериментально найденные значения параметров.
Адсорбция
АВ+ * -» АВ ,
(3.189)
dflAB _ s0(T)pAB Л
d t N0 ^2лткъТ
(3.190)
(3.191)
Реакции
А. + В. -» АВ, + *,
(3.192)
КТ ?АВ8 _ к+р-&Еш_А_в/квТ
— ас0 ег
h ЯаЧъ
(3.193)
= ~АВМвТ
(3.194)
Десорбция
АВ, -> АВ + *,
(3.195)
d^AB _ КТ д'£ъ
(3.196)
dt
(3.197)
150
\
Глава 3. Теория скоростей реакций
Равновесие. При равновесии между адсорбционным и десорбционным процессами получаем изотерму Ленгмюра
АВ+ ^ АВ , (3.198)
^АВ _ S°^P№_e _ _ Q (3.199)
d t N0p?rmkBT
о* (3-200>
eq Р АВ + 1
где
. $о(Т) ^ A^desorption /кВ Т
^eq =
N0v^27rmkBT
Список литературы
1. Goodstein D.L. States of Matter. Dover: Prentice Hall, 1985.
2. Atkins P.W. Physical Chemistry. Oxford: Oxford Univ. Press, 1998.
3. Van Santen R.A., Niemantsverdriet J.W. Chemical Kinetics and Catalysis. New
York: Plenum, 1995.
4. McQuarrie D.A. Statistical Mechanics. New York:, Harper&Row, 1976.
5. Laidler K.J. Chemcal Kinetics. 3rd Ed. New York: Harper&Row, 1987.
6. Niemantsverdriet J.W. Spectroscopy in Catalysis. An Introduction. 2nd Ed.
Weinheim: Wiley-WCH, 2000.
7. Zhdanov VP., Pavlicek /., KnorZ. // Catal. Rev. — Sci. Eng. 1989. V. 30. P. 501.
глава ОПРЕДЕЛЕНИЕ
4 ПАРАМЕТРОВ
КАТАЛИЗАТОРОВ
4.1. ВВЕДЕНИЕ
Определение параметров катализаторов представляет собой важный раздел катализа. Методы спектроскопии, микроскопии, рентгеновской
дифракции, а также адсорбционно-десорбционные исследования или изучение
реакций в объемной фазе — все это используется для выяснения природы и
активности катализаторов. Такие исследования позволяют глубже понять природу катализа, улучшить характеристики катализаторов или создать катализаторы нового типа.
В гл. 1 мы особо подчеркивали, что свойства поверхности твердых катализаторов определяются их составом и атомной структурой. То есть с фундаментальной точки зрения было бы исчерпывающим описание характеристик катализатора, при котором состояние всех поверхностных атомов определено в условиях работы катализатора или, как говорят, определено in situ. Однако
катализаторами являются очень мелкие частицы оксидов или сульфидов металлов, находящихся на носителе. К катализаторам могут быть добавлены активаторы (промоторы), повышающие их работоспособность и/или селективность. Используются модификаторы, улучшающие механические свойства катализаторов или предотвращающие их спекание. Все говорит о достаточно
сложной структуре твердотельных катализаторов. Более того, состояние поверхности катализатора может изменяться в зависимости от условий, в которых он
работает.
Сказанное выше объясняет, почему во многих фундаментальных исследованиях сложные по структуре катализаторы заменяются упрощенными модельными системами, характеристики которых определены более надежно. В качестве таких моделей используются осажденные на носители частицы, с которых удалены все активаторы и модификаторы, хорошо охарактеризованные
частицы на плоских подложках и даже поверхности монокристаллов (рис. 4.1).
В последнем случае мы попадаем в область интересов науки о поверхностях,
где имеются разнообразные высокоинформативные методы исследования, которые, однако, не работают в случае технических катализаторов.
В промышленности основное внимание уделяется созданию активных, селективных, стабильных и механически прочных катализаторов. Для достижения этой цели необходим инструментарий, позволяющий по структурным па¬
152 -l\r Глава 4. Определение параметров катализаторов
раметрам выделять эффективные катализаторы из менее эффективных. При
этом приветствуется всякая информация, дающая возможность осуществить
такое выделение. Эмпирические соотношения между параметрами, определяемыми составом катализатора (размер и форма частиц, их пористость), и его
активностью представляют собой очень ценную информацию для развития технологии синтеза катализаторов, однако не всегда позволяют установить на молекулярном уровне механизм их работы.
чО° 0С^
<44?
Частица на плоской подложке Поверхность монокристалла
Рис. 4.1. Нанесенный катализатор, представляющий собой малые частицы на носителе с высокой
удельной поверхностью
(кремнезем или оксид
алюминия), и две простые
модельные системы, которые можно более полно охарактеризовать на
молекулярном уровне
Для исследования катализаторов используются разнообразные методы спектроскопии, электронной микроскопии и дифракции рентгеновских лучей. На
рис. 4.2 показано, что эти методы основаны на возбуждении некоторых степеней свободы катализатора (входящие стрелки) и измерении его отклика (выходящие стрелки). Например, при падении на катализатор рентгеновских лучей
Рис. 4.2. Методы определения параметров катализаторов: кружок представляет собой исследуемый образец, стрелки,
направленные внутрь, обозначают процесс возбуждения внутренних степеней
свободы, стрелки, направлены наружу,
показывают, как можно извлечь информацию о катализаторе
4.2. Рентгеноструктурный анализ (РСтА) -*\г 153
рождаются фотоэлектроны, определение параметров которых составляет сущность рентгеноэлектронной спектроскопии (РЭС) — одного из самых мощных
спектроскопических методов. Можно также нагревать катализатор и определить температуру, при которой интермедиаты реакции и ее продукты начинают
десорбироваться с поверхности катализатора (метод температурно-программи-
руемой десорбции).
ю
20
30
40
50
РСтА
Адсорбция/БЭТ
ИК-спектроскопия
РЭС/УФЭС
Температурно-программируемые методы
ПЭМ/СЭМ
ЯМР
УФ-оптическая спектроскопия
СДТСРП
ЭПР
СБТСРП
АРЭРЛ
ОЭС
дмэ1
СКР2
мс
стм
РНИ
Калориметрия
Рассеяние нейтронов
МСВИ
1 Дифракция медленных электронов;
2 Спектроскопия комбинационного рассеяния
Рис. 4.3. Число исследований, выполненных с привлечением одной
из перечисленных методик в 143 научных работах, представленных на
11 -м Международном конгрессе по
катализу в Балтиморе в 1996 г. [1].
Расшифровку сокращений см. в основном тексте
На рис. 4.3 представлены некоторые статистические данные по использованию различных методов для определения характеристик катализаторов. В данной главе будут описаны физические явления, лежащие в основе этих методов,
и приведены полученные с их помощью результаты. Более полно возможности
этих методов описаны в следующих главах и в книге [1].
4.2. РЕНТГЕНОСТРУКТУРНЫЙ АНАЛИЗ (РСтА)
Рентгеноструктурный анализ — наиболее старый метод, который
часто используется для определения параметров катализаторов. С его помощью идентифицируются фазовые состояния зерен внутри катализатора по структурным параметрам кристаллической решетки и находятся размеры частиц.
При рассеянии атомами, находящимися в кристаллической решетке, рентгеновских лучей наблюдается их дифракция. В результате интерференции интенсивность упруго рассеянных монохроматических рентгеновских лучей резко возрастает при определенной ориентации кристаллической решетки по от¬
154 -l\r Глава 4. Определение параметров катализаторов
ношению к направлению падающих лучей. Так, рис. 4.4 показывает, как по дифракции рентгеновских лучей можно с помощью соотношения Брэгга
определить расстояние между плоскостями кристаллической решетки. Здесь
Л — длина волны рентгеновского излучения; d — расстояние между плоскостями решетки; в — угол между направлением падения рентгеновских лучей и
нормалью к рассеивающей плоскости, при котором наблюдается максимум интенсивности рассеяния; п — целое число, называемое порядком отражения (или
рефлекса).
Рентгеновские лучи
Определяя значения углов 2 в, отвечающих максимумам интенсивностей
рассеянных лучей, и используя соотношение Брэгга (4.1), можно определить
расстояние между плоскостями кристаллической решетки.
При рассеянии излучения порошком дифракционная картина представляет
собой круги, поскольку частицы ориентированы случайным образом и определенные плоскости у небольшой доли частиц всегда могут быть ориентированы
под нужным углом в.
В катализе метод РСтА используется для определения фазового состава катализатора. На рис. 4.5 показана дифрактограмма, полученная для катализатора Pd/Si02, на которой выделены рефлексы, соответствующие Pd.
Метод РСтА обладает существенным ограничением: четкие дифракционные пики наблюдаются только в случае наличия у образца дальнего порядка на
значительном пространственном масштабе. Это ограничение, однако, является
информативным: по ширине дифракционного пика можно оценить размер рассеивающих плоскостей, то есть фактически размер фазового образования. Для
совершенных кристаллов дифракционые пики являются очень узкими. При¬
пЛ = 2Jsin в\ п = 1, 2, ...
(4.1)
Рис. 4.4. Рентгеновские лучи, рассеянные атомами, находящимися в
периодической решетке, интерферируют, так что рассеяние идет по
направлениям, определяемым законом Брэгга. Положение максимумов
интенсивности рассеянного излучения
позволяет определить расстояние
между плоскостями решетки и провести идентификацию фаз. Дифрак-
тограммы представляют собой зависимости интенсивности от 26. В случае поликристаллических образцов
дифракционная картина определяется только малым числом частиц. Вращение образца в процессе измерения
увеличивает число частиц, дающих
вклад в интенсивность рассеяния в
данном направлении [1]
пЛ = 2c/sin в
4.2. Рентгеноструктурный анализ (РСтА)
мер дифракционного спектра для плоскостей (111) и (200) кристалла палладия
большого размера приведен на рис. 4.5. Уширение линий начинает проявляться для кристаллов размером менее 100 нм вследствие неполного интерференционного гашения волн для углов, отвечающих минимумам интенсивности
рассеяния, когда рассеянные волны находятся в противофазе. Два дифракционных спектра для палладиевых катализаторов, представленные на рис. 4.5,
показывают, что соответствующие пики у них шире, чем у образца сравнения.
Ширина полосы связана с размером кристалла формулой Шеррера:
«-/»£*■ <4-2)
где (L) — характрный размер частицы в направлении, перпендикулярном отражающей плоскости; Л — длина волны излучения; /3 — ширина пика; в — угол
между направлением падения луча и нормалью к отражающей плоскости; К —
константа, которая часто полагается равной 1.
Рис. 4.5. Спектры дифракции рентгеновских лучей для отражения от
плоскостей (111) и (200) Pd для двух
палладиевых катализаторов, нанесенных на кремнезем, и образца сравнения [4]. Читатель может убедиться, используя уравнение (4.1), что
рефлексы для плоскостей Pd( 111) и
(200) должны наблюдаться при углах
2в = 40,2 и 46,8° для линии СиА^(по-
стоянная решетки Pd равна 0,389 нм,
din = 0,225 нм, d200 = 0,194 нм,
/ = 0,154 нм)
2в
Используя приведенные на рис. 4.5 дифрактограммы, можно получить средние размеры 4,2 и 2,5 нм для катализаторов, содержащих 2,4 и 1,1 вес. % палладия соответственно. По ширине пиков дифрактограммы можно быстро оценить размер частиц, но этот метод не всегда оказывается надежным. Более
строгим в определении размеров частиц является анализ профиля линии, проводимый с использованием преобразования Фурье.
Одно из основных преимуществ метода РСтА связано с высокой проникающей способностью рентгеновских лучей, которая дает возможность исследовать катализаторы в реальных условиях в специально сконструированных in
s/ta-реакторах. В результате удается проследить за ходом твердотельных реакций типа окисления, восстановления и сульфидирования, влияющих на активность катализатора. При этом широкие возможности открываются при исполь-
156 -l\r Глава 4. Определение параметров катализаторов
зовании синхротронного излучения, которое при большой интенсивности обладает высокой монохроматичностью, что позволяет существенно сократить
время получения дифрактограмм. Более того, для этого излучения отношение
сигнал/шум оказывается существенно выше, что дает возможность надежно
анализировать очень широкие пики, связанные с малыми частицами.
Значимость метода РСтА для катализа определяется возможностью получения ясной и недвусмысленной информации о структуре достаточно больших
частиц, а также оценки их размеров, в том числе и в условиях работы реактора.
К недостаткам этого метода можно отнести его неприменимость в случае очень
малых или аморфных частиц. То есть нельзя быть уверенным, что в системе не
присутствуют другие частицы, кроме детектируемых методом РСтА; в частности, поверхности, которые и несут каталитическую активность, не наблюдаемы
в стандартной реализации метода РСтА.
4.3. РЕНТГЕНОЭЛЕКТРОННАЯ СПЕКТРОСКОПИЯ (РЭС)
Рентгеноэлектронная спектроскопия относится к числу наиболее
часто используемых для изучения катализаторов методов. Она дает информацию об элементном составе, состоянии окисления элементов и в благоприятных случаях — о распределении одной фазы по другой [1—3].
Фотоэлектрон
Рис. 4.6. Фотоэмиссия и оже-процесс. Слева: рентгеновский фотон поглощается с испусканием фотоэлектрона. По кинетической энергии электрона можно определить
энергию связи фотоэлектрона. Испустив электрон, атом превращается в неустойчивый ион с «дыркой» на внутренней оболочке. Справа: возбужденный ион
релаксирует путем перевода электрона из внешней оболочки на свободное состояние («дырку») на внутренней оболочке. Высвобождающаяся при этом энергия передается другому электрону, оже-электрону, вылетающему из образца с
характерной для данного элемента кинетической энергией. В оже-спектроско-
пии для генерации «дырок» используется пучок электронов с энергией 2—5 кэВ
4.3. Рентгеноэлектронная спектроскопия (РЭС) ->\г 157
РЭС основана на фотоэффекте: атом, поглотивший фотон с энергией hv,
испускает электрон, находившийся на внутренней или валентной орбите и
имевший энергию связи Есв, с кинетической энергией (рис. 4.6)
Ек = hv- Eqb- ср. (4.3)
Здесь Ек — кинетическая энергия фотоэлектрона; h — постоянная Планка; v —
частота электромагнитного излучения; Есв — энергия связи фотоэлектрона по
отношению к энергии Ферми образца; ср — работа выхода электронов из него.
Традиционно в РЭС используется рентгеновское излучение линий Mg^
(hv = 1253,6 эВ) и линий А1 Ка (hv = 1486,3 эВ), для которого кинетическая
энергия фотоэлектронов попадает в диапазон 0—1 кэВ. При этой энергии длина свободного пробега электронов / в твердом теле не превышает нескольких
атомных диаметров. На рис. 4.7 показана зависимость длины свободного пробега электронов от их энергии. Из рисунка видно, что в диапазоне энергий
10—1000 эВ длина свободного пробега меньше 2 нм. Оптимальная чувствительность к поверхностному слою достигается в диапазоне энергий 50—200 эВ,
для которого Я ~ 0,5 нм, так что все электроны, попадающие в приемник,
выходят только из поверхностного слоя. В методе РЭС измеряется интенсивность N(E) потока электронов как функция их кинетической энергии. С помощью уравнения (4.3) кинетическая энергия пересчитывается в энергию связи,
которая откладывается по оси абсцисс в соответствующих спектрах. Энергии
связи электронов полностью характеризуют элемент, из которого вырываются
фотоэлектроны. На рис. 4.8 показаны рентгеноэлектронные спектры родиевого катализатора, нанесенного на оксид алюминия путем пропитки носителя
водным раствором RhCl3 и последующим восстановлением металла. Пики, связанные с Rh, Cl, А1, О, а также и С, всегда присутствующего из-за наличия
углеводородных примесей, легко идентифицируются с помощью таблиц по
энергиям связей.
Помимо пиков фотоэлектронного спектра на рис. 4.8 показаны пики, отвечающие оже-электронам. Появление этих пиков обусловлено тем, что после
вылета фотоэлектронов ион остается в сильно возбужденном состоянии, в котором на одной из внутренних оболочек имеется «дырка». Ион релаксирует по
схеме, показанной справа на рис. 4.6. Легко видеть, что оже-электроны имеют
фиксированную кинетическую энергию, которая не зависит от энергии, затраченной на отрыв фотоэлектрона и создание «дырки» на внутренней оболочке.
Тем не менее пик, отвечающий оже-электронам, относится к оси энергий связи, что, конечно, не имеет никакого физического значения. Основной оже-пик
ЛГКК-перехода в О, показанный на рис. 4.8, имеет энергию 500 эВ, но появляется при энергии связи 986 эВ, поскольку спектр был получен из линии А1 Ка
с hv= 1486,3 эВ. Оже-пик может быть идентифицирован при использовании
рентгеновского излучения двух различных длин волн: пики, отвечающие фотоэлектронам, появятся при одной и той же энергии связи, тогда как оже-пики
будут сдвинуты по энергетической шкале. Вот почему в источниках рентгеновского излучения используются бинарный анод, состоящий из Mg и А1.
Глава 4. Определение параметров катализаторов
Кинетическая энергия электронов, эВ
Рис. 4.7. Зависимость длины свободного пробега электронов от энергии. Длина свободного пробега определяет толщину слоя, из которого «извлекается» информация.
Оптимальная чувствительность к поверхностному слою достигается при энергии электронов 25—200 эВ [5]
о
со
со
S
о
Q.
С
со
0
1
о
Q.
0
СО
О
н
о
о
П.
Рентгеноэлектронный спектр
О 1s
- Система Rh/Al203
после пропитки
Оже-
пики
Rh3d
С 1s
Cl
Rh3p
А1
Рентгеноэлектронный
спектр из диапазона,
характерного для 3c/-Rh
Система RhCI3/AI203
после пропитки
|ЗсУк
Восстановленный
металл
1250 1000 750 500 250 0 320 315 310 305
Энергия связи, эВ
Рис. 4.8. Рентгеноэлектронный спектр в широком диапазоне сканирования для модельного катализатора Rh/Al203, полученного пропиткой А1203 водным раствором
RhCl3 и последующим восстановлением металла. Показаны пики фото- и оже-
электронов (слева), а также в часть спектра З^-Rh после испарения воды из
носителя и восстановления металла (справа). Видна зависимость энергии связи
от состояния окисления родия [6]
4.3. Рентгеноэлектронная спектроскопия (РЭС) -i\r 159
Пики фотоэлектронных спектров обозначаются квантовыми числами уровней, с которых вырывается электрон. Электроны с орбитальным квантовым
числом / (0, 1, 2, 3,..., обозначения s, /?, /, ...) и спином s имеют полный
момент j = 1+ s. Поскольку имеется две ориентации спина — вверх (s = +1/2)
и вниз (s = -1/2), каждому уровню с / > 1 соответствует два подуровня, разность энергий между которыми называется спин-орбитальным расщеплением.
Так, уровню 3J-Rh отвечают два фотоэлектронных пика, связанных с уровнями 3d5/2 (/=2,7 = 2+ 1/2) и 3d3/2 (/ = 2, у = 2 — 1/2). Оже-электроны, однако
обозначаются в соответствии с правилами, принятыми в рентгеновской спектроскопии. Оже-электрон, обозначаемый через KLM, появился в результате заполнения «дырки» в оболочке К электроном с оболочки L, при этом оже-элек-
трон был вырван с оболочки М. В табл. 4.1 приведена номенклатура для рентгеноэлектронной и оже-спектроскопии.
Таблица 4.1. Обозначения, используемые в рентгеноэлектронной
и оже-спектроскопии
п
/
j
Рентгеновские
термы
Электронные
уровни
1
0
1
2
К
Is
2
0
1
2
А
2s
2
1
1
2
h
2p\/i
2
1
3
2
h
2Рз/2
3
0
1
2
л/,
3s
3
1
1
2
м2
ЗР\/2
3
1
3
2
м3
ЗРз/2
3
2
3
2
м4
3^3/2
3
2
5
2
м5
3^5/2
4
3
5
2
4/5/2
4
3
7
2
4/7/2
Спин-орбитальное расщепление, а также энергия связи для конкретного
энергетического уровня возрастают с атомным номером. Отношение интенсивностей пиков дублетов, связанных со спин-орбитальным расщеплением,
определяется кратностью вырождения соответствующих уровней, которая рав¬
160 -J\r Глава 4. Определение параметров катализаторов
на 2/4-1. Так, отношение интенсивностей полос, отвечающих дублету 3J-Rh с
j = 5/2 и j = 3/2 равно 6 :4 или 3:2. Таким образом, пики фотоэлектронных
спектров, связанные с внутренними уровнями, наблюдаются в виде дублетов,
за исключением s-уровней, которые обычно дают один пик.
Энергии связей характеризуют не только элемент, но и его химическое
соединение, поскольку положение внутренних уровней хоть и слабо, но зависит от химического состояния атома. Химические сдвиги составляют обычно
0—5 эВ. Обычно энергия связи возрастает с увеличением степени окисления
атомов и для фиксированной степени окисления — с увеличением электроотрицательности лигандной оболочки. Для прояснения смысла того или иного
значения энергии связи необходимо рассмотреть эффекты, определяемые конечным состоянием атома. На практике данные рентгеноэлектронной спектроскопии привязываются к атому в состоянии, предшествующему акту фотоэмиссии. А это не совсем корректно, поскольку данные фотоэмиссии определяются и
состоянием, с которого электрон только что перешел в другое состояние. Следовательно, энергия связи фотоэлектронов содержит информацию как о состоянии атома до фотоионизации (начальное состояние), так и о состоянии атома с
«ионизованным» внутренним уровнем, в котором он находится после эмиссии
электрона (конечное состояние). К счастью, практически всегда интерпретация
энергий связи в соответствии со схемой, представленной на рис. 4.8, то есть в
терминах исходного состояния, оказывается качественно корректной. Модельный расчет создаваемого зарядами потенциала элегантно объясняет физический
механизм, лежащий в основе сдвига энергии связи, с помощью формулы
я» =*9/+2— + ^. (4.4)
J ГУ
где Е[в — энергия связи электрона в атоме /; q} — локализованный на нем
заряд; к — постоянная; q. — заряд, локализованный на соседнем атоме у; гИ —
• • Г- ref -
расстояние между атомами i и j и Ьсв — соответствующий уровень отсчета
энергии. Первое слагаемое в правой части (4.4) показывает, что энергия связи
увеличивается при росте положительного заряда, локализованного на атоме, с
которого вылетает фотоэлектрон. В твердых телах с ионной связью второе слагаемое действует противоположно первому, поскольку заряды, локализованные на соседних атомах, имеют противоположный знак. Второе слагаемое в
(4.4), в силу его сходства с решеточным потенциалом твердых телах с ионной
связью, часто называют потенциалом Маделунга.
Правая часть рис. 4.8 показывает, как состояние окисления родия в катализаторе Rh/Al203 влияет на энергию связи. Рентгеноэлектронный спектр 3J-Rh,
полученный после пропитки носителя раствором, показывает, что энергия связи для уровня 3d5/2 в Rh равна 310 эВ, что является типичной величиной для
трехвалентного состояния родия (как, например, в RhCl3). После восстановления металла с помощью тока водорода энергия связи уменьшается до 307,4 эВ,
что соответствует металлическому родию. Таким образом, рентгеноэлектронные спектры показывают и наличие родия в катализаторе, и его состояние
окисления там.
4.3. Рентгеноэлектронная спектроскопия (РЭС) -*\г 161
Практически неразрешимая проблема, с которой обычно сталкиваются при
детектировании заряженных частиц типа электронов или ионов, связана с тем,
что диэлектрические образцы могут заряжаться в процессе проведения эксперимента. Потенциал, приобретаемый образцом, определяется балансом потока
фотоэлектронов, покидающих образец, их притоком к образцу через держатели, а также потока оже- и вторичных электронов от окна источника излучения.
Поскольку образец заряжается положительно, все пики рентгеноэлектронных
спектров обычно сдвинуты на одну и ту же величину в сторону больших значений. Этот сдвиг можно легко учесть, если в образце имеются элементы, для
которых энергия связи известна. Для катализаторов на носителе из Si02, например, для этой цели можно использовать энергию связи 2/ьэлектронов Si,
равную 103,4 эВ. Если нет ничего подходящего, то можно использовать энергию связи ls-электронов С (энергия связи 284,6 эВ), который входит всегда в
присутствующие углеродсодержащие примеси.
Поправки, обусловленные однородным заряжением образца, обычно всегда удается учесть тем или иным способом. Более сложно обстоит дело с неоднородной зарядкой образца, порождающей уширение пиков и снижающей
уровень разрешения сигналов и отношение сигнал/шум. Использование специальной электронной пушки, «орашающей» образец низкоэнергетическими
электронами, и разработка оригинальных держателей типа индиевой фольги, в
которую запрессовывается порошок, способствует смягчению проблемы зарядки образцов.
Грубодисперсные
частицы
О
Высокодисперсные
частицы
Рис. 4.9. По отношению интенсивностей сигналов рентгеноэлектронного
спектра от частиц и носителя, IP/IS,
можно судить о характере распределения катализатора по поверхности
носителя in
О
Малое значение
отношения lp/ls
Частицы
Носитель
Большое значение
отношения lp/ls
Поскольку рентгеноэлектронная спектроскопия чувствительна к состоянию
поверхности, она позволяет установить, насколько хорошо частицы распределены по носителю. На рис. 4.9 схематически показаны два катализатора, содержащие одно и то же количество (по массе) частиц, обладающих различной
степенью дисперсности. У частиц малого размера почти все атомы находятся в
поверхностном слое, при этом поверхность носителя практически полностью
покрыта частицами. В этом случае поток электронов от частиц 1р оказывается
большим, а от носителя Is — малым, следовательно, отношение Ip/Is должно быть
большим. Для грубодисперсных частиц отношение Ip/Is оказывается низким.
162 -J\л Глава 4. Определение параметров катализаторов
Таким образом, по значению отношения IP/IS можно судить о степени дисперсности катализатора на носителе. Были предложены несколько моделей для
расчета степени дисперсности катализатора по отношению интенсивностей
полос рентгеноэлектронных спектров, некоторые из них оказались весьма удачными. Сказанное позволяет заключить, что рентгеноэлектронная спектроскопия предлагает альтернативный способ нахождения степени дисперсности катализаторов в условиях, когда применение обычных методов определения размеров частиц типа электронной микроскопии и хемосорбции водорода
оказывается невозможным.
В заключение отметим, что рентгеноэлектронная спектроскопия в исследовании катализаторов применяется наиболее часто. С ее помощью легко устанавливаются состав поверхностного слоя, степени окисления атомов металла и
электроотрицательности лигандов. Она также позволяет оценить характер распределения частиц катализатора по поверхности носителя в условиях, когда с
помощью электронной микроскопии или по хемосорбции водорода не удается
распознать поверхности чистого носителя и частиц, что является особо ценным для практики.
4.4. СПЕКТРОСКОПИЯ ДАЛЬНЕЙ ТОНКОЙ СТРУКТУРЫ
РЕНТГЕНОВСКОГО ПОГЛОЩЕНИЯ (СДТСРП, EXAFS)
Спектроскопия дальней тонкой структуры рентгеновского поглощения, как и рентгеноэлектронная спектроскопия, связана с процессами поглощения рентгеновских лучей и эмиссией фотоэлектронов [7]. Но в рентгеноэлектронной спектроскопии анализируется кинетическая энергия фотоэлектронов, тогда как в методе СДТСРП изучается, как эти электроны рассеиваются на
соседних атомах, что проявляется через интерференционные эффекты в спектрах
поглощения рентгеновских лучей. Этот метод позволяет получить подробную информацию о расстоянии до соседей, их числе и природе, то есть о локальной
структуре на субнанометровом масштабе. Более того, высокая проникающая способность рентгеновских лучей позволяет применять метод СДТСРП in situ.
Хотя лежащие в основе метода СДТСРП явления были известны еще 1920-е гг.,
он стал использоваться в аналитических целях только в 1970-е гг., когда появились настраиваемые синхротронные источники рентгеновского излучения. На
рис. 4.10 показана схема метода СДТСРП. Поясним ее. Рассмотрим сначала
спектр поглощения рентгеновского излучения отдельным атомом, у которого
имеется электрон с энергией связи Есв. Если направить на атом поток рентгеновских лучей с энергией hv, то их поглощение будет происходить только при
hv> Есв, вылетающий из атома электрон при этом будет иметь энергию EK = hv~ Есв.
На спектре поглощения рентгеновского излучения будут наблюдаться разрывы
(края), положение которых соответствует энергиям связи электронов в атоме.
Тонкая структура у спектра появляется при наличии у атома соседей. В этом
случае фотоэлектрон, обладающий волновыми свойствами, может рассеяться
4.4. Спектроскопия дальней тонкой структуры рентгеновского поглощения
\
Ev = hv-
/'о
163
на соседнем атоме назад (см. рис. 4.10). В силу волновой природы электрона
происходит интерференция волн, соответствующих вылетающему и рассеянному назад электрону. В зависимости от длины волны электрона, расстояния
между эмитирующим и рассеивающим атомами, а также от фазового сдвига
при рассеянии эти две волны могут либо усиливать, либо ослаблять друг друга.
В результате сечение поглощения рентгеновского излучения модулируется за
счет интерференции электронных волн, так что оно возрастает при тех значениях энергии фотонов, которым отвечает увеличение амплитуды электронной
волны. Как схематически показано на рис. 4.10, в спектре поглощения рентгеновского излучения появляется тонкая структура, простирающаяся на расстояние по энергиям в несколько сот электронвольт за край поглощения. Поглощение вблизи края связано с электронами, обладающими низкой кинетической энергией и взаимодействующими с валентными электронами. Анализ этой
части спектра, примыкающей непосредственно к краю, называют спектроскопией ближней тонкой структуры рентгеновского поглощения (СБТСРП, NEXAFS
или XANES).
h v
Рис. 4.10. Спектры поглощения
рентгеновских лучей одиночным
атомом и атомом в решетке. Тонкая структура возникает как результат интерференции волн, отвечающих вылетающему и рассеянному
назад электрону, и представляет
собой предмет СДТСРП
Полезно иметь представление о том, как межатомное расстояние, координационное число и концентрация влияют на СДТСРП-спектр: интенсивность
всплесков увеличивается с ростом числа соседей, число осцилляций обратно
пропорционально межатомному расстоянию (как и в любом эксперименте по
рассеянию или дифракции), а высота ступени края спектра пропорциональна
концентрации данных атомов в образце.
Для более четкого понимания, как структура определяется по спектру, необходимы некоторые математические преобразования. Из спектра поглощения
рентгеновского излучения сначала находится СДТСРП-функция, z(k)> которая
получается путем вычитания из представленного, например, на рис. 4.10 спектра параболического фона и ступени, то есть спектра одиночного атома. Как и
в любом эксперименте по рассеянию, сигнал представляется в виде функции
* ® •
Решетка
164 -J\r Глава 4. Определение параметров катализаторов
не энергии, а волнового вектора к. Связь между к и кинетической энергией
фотоэлектрона дается выражением:
к = ^ртеЕК = ^рте (hv - Есв). (4.5)
п п
Здесь к — волновой вектор фотоэлектрона; Ь — постоянная Планка; те — масса
электрона; ЕК — кинетическая энергия фотоэлектрона; v — частота рентгеновского излучения; Есв — энергия связи фотоэмитируемого электрона. Для студентов, не знакомых с понятием волнового вектора, поясним, что комбинация
hk/2nпредставляет собой импульс волнового кванта, a yJ(2meEK) = mev — классический импульс электрона как частицы.
В одноатомном твердом теле СДТСРП-функция, %(к), равна сумме вкладов
от рассеяния на всех соседних атомов, входящих в координационные сферы:
^ (k) = £ Ai (к)sin [2krj + <pj (*)]’ (4-6)
j
где суммирование по у относится к координационным сферам атома, испускающего электрон; А.(к) — амплитуды, равные интенсивностям рассеяния на у-й координационной сфере; г — расстояние между центральным атомом и атомами
в у-й координационной сфере; cpj(k) — полый фазовый сдвиг.
Каждая координационная сфера у вносит в функцию %(к) вклад, равный
произведению синуса и амплитуды, которая пропорциональна числу атомов Nj
в ней, представляющему наибольший интерес:
-2rjIЛ{к) ? ^
А](к) = Nj^-Sl^F^e-^. (4.7)
Другие сомножители в (4.7) учитывают экспоненциальное затухание волнового пакета электрона, движущегося в твердом теле, поправки на релаксационные эффекты в атоме, эмитирующем электрон, S0, эффекты, связанные с колебаниями решетки (последний экспоненциальный сомножитель) и, наконец,
фактор обратного рассеяния Fj на атомаху-й координационной сферы. Зависи¬
мость фактора обратного рассеяния от энергии электрона является характеристикой элемента. Следовательно, по Fj(k) иногда можно идентифицировать
рассеивающий атом. Это проиллюстрировано на рис. 4.11 на простых примерах рассеяния на димере атомов металла и тримере оксида металла. Очевидно,
что фактор обратного рассеяния определяет форму СДТСРП-вклада от конкретного соседа. Укажем, как фактор Fj(k) распознается на фоне осциллирующих функций. Как видно из схематического рис. 4.11, фактор обратного рассеяния для кислорода вносит вклад при более низких значениях волнового вектора, чем соответствующие факторы для атомов металла.
Суть анализа СДТСРП-спектра состоит в распознавании всех синусоидальных вкладов в %(к). Наиболее подходящим математическим инструментом для
этого, очевидно, является фурье-анализ. Аргумент синусоидальных функций,
4.4. Спектроскопия дальней тонкой структуры рентгеновского поглощения -J\r 165
входящих в выражение (4.8), зависит от волнового вектора к (который известен)
и от межатомных расстояний г (которые следует определить), а также от фазового
сдвига ц(к). Последний является характеристикой рассеивающего атома, находящегося в определенном окружении, и лучше всего находится по СДТСРП-спект-
ру образца сравнения, для которого известны все межатомные расстояния.
Рис. 4.11. Слева: модельный СДТСРП-спектр димера типа Си2, показывающий, что
СДТСРП-сигнал представляет собой произведение синусоидальной функции
и фактора обратного рассеяния Fj(k), поделенного на к, как это следует из
формулы (4.7). Укажем, что отношение Fj{k)/k видно как огибающая СДТСРП-
сигнала, %{к). Справа: СДТСРП-спектр Си в кластере типа Си20 представляет
собой сумму вкладов от пар Си—Си и Си—О. Математический аппарат фурье -
анализа использован для выделения вкладов от Си—Си и Си—О. Отметим, что
характеристики обратного рассеяния для Си и О проявляются в огибающей
индивидуальных СДТСРП-спектров. Фазовый сдвиг для упрощения анализа в
расчет не принимался [1]
Информация, извлекаемая из СДТСРП-спектров, становится наглядной при
переводе ее с помощью преобразования Фурье в радиальные функции распределения:
где п — целое число, выбираемое равным п = 1 или п = 2 и 3, чтобы сделать
акцент на легких или тяжелых рассеивателях, соответственно. Функция вп(г)
представляет собой вероятность найти атом на расстоянии г, умноженную на
две зависящие от расстояния г функции, учитывающие падение вклада в интенсивность от рассеяния на удаленных координационных сферах.
На рис. 4.12 показаны фурье-преобразования СДТСРП-данных для высокодисперсного катализатора Rh/Al203, полученного восстановлением родия в токе водорода при 200 и 400 °С. Из фурье-образца видно, что у атома
родия имеется соседний атом Rh на расстоянии 0,27 нм и соседний атом
кислорода на более коротком расстоянии. Эти данные говорят о том, что у
образца, восстановленного при 200 °С, имеется вклад от пары Rh—О, характеризующей наличие R203, который можно приписать невосстановлен¬
0 4 8 12 16 20
к
0 4 8 12 16 20
к
(4.8)
166
Глава 4. Определение параметров катализаторов
ным частицам. Имеющийся вклад от пары Rh—О в радиальную функцию
распределения у полностью восстановленного образца обусловлен контактами между атомами родия и атомами кислорода носителя из оксида алюминия. То есть СДТСРП позволяет получить информацию о характере взаимодействия металл/носитель.
в(г) х ю-1 в(г) х 10"1
Г, А Г, А
Рис. 4.12. Преобразование Фурье СДТСРП-спектров для Rh в катализаторе Rh/Al0O3,
полученных восстановлением родия в токе водорода при 200 и 400 °С. Виден
доминирующий вклад от соседнего атома Rh, находящегося на расстоянии
0,27 нм, и вклад от соседних атомов кислорода в Rh202 (левый рисунок) и в
области контакта металл/носитель (правый рисунок) [8]
Для использования СДТСРП в анализе катализаторов необходимо, чтобы
катализатор был монодисперсным, то есть чтобы среднее окружение каждого
атома, которое определяет СДТСРП-спектр, было одним и тем же по всему
катализатору. В случае многокомпонентных катализаторов этот метод «приобретает» существенный недостаток, связанный со все возрастающими сложностью анализа и затратами времени и необходимостью привлечения большого
объема информации, без которой теряется однозначность выводов. Тем не менее для тщательно обработанных и монодисперсных катализаторов, для которых анализ проводится с особой тщательностью, СДТСРП дает уникальную
информацию об их структуре на масштабе межатомных расстояний!
4.5. ЭЛЕКТРОННАЯ МИКРОСКОПИЯ (ЭМ, ПЭМ, СЭМ)
Электронная микроскопия позволяет непосредственно определить размер и форму частиц на носителе [9]. Характеристическая длина волны электронов составляет менее 1 А, что позволяет рассмотреть строение
частиц на атомарном уровне. На рис. 4.13 схематически показано, что происходит с первичным электроном энергией от 100 до 400 кэВ, сталкивающимся с мишенью.
4.5. Электронная микроскопия (ЭМ, ПЭМ, СЭМ)
Рис. 4.13. Результатом взаимодействия
между первичными пучком и образцом
является большое число детектируемых
«сигналов» различной природы
Пучок первичных
электронов
Рентгеновские
лучи
Электроны,
рассеянные назад
Фотоны
Оже-электроны
Вторичные электроны
Л
^ Потери
электронов
<гь
Дифрагировавшие элек
электроны ▼
Прошедшие
электроны
• Некоторая часть электронов, определяемая толщиной образца, проходит
через него без потери энергии. Поскольку уменьшение интенсивности пучка
электронов зависит от локальной плотности и толщины мишени, прошедшие
электроны создают двумерную проекцию исследуемого образца.
• Может произойти дифракция электронов на частицах при соответствующей их ориентации, что позволяет получить кристаллографическую информацию о них.
• Электроны, столкнувшись с атомами, могут рассеяться назад; рассеяние
назад идет более интенсивно при увеличении массы атомов. Если в некоторой
частице образца имеются более тяжелые атомы (например, Pt), чем у остального окружения, то на них идет более интенсивное рассеяние назад, которое
может быть зафиксировано.
• Из мишени выходят оже-электроны и рентгеновское излучение, формирующиеся при релаксации атомов, с «ионизованными» внутренними оболочками, о чем говорилось ранее при обсуждении рентгеноэлектронной спектроскопии и оже-спектроскопии.
• Электроны на определенных переходах возбуждают внутренние степени
свободы у исследуемого образца, параметры которых можно установить по
потерям энергии первичными электронами.
• Большая часть электронов теряет энергию в каскадных неупругих столкновениях. Большинство вторичных электронов, испускаемых образцом, последний теряют свою энергию в поверхностном слое.
• Идет излучение фотонов в диапазоне от ультрафиолетовых до инфракрасных волн, называемое катодолюминесценцией и связанное с рекомбинацией
электронов и дырок в исследуемом образце.
Таким образом, исследование взаимодействия первичных электронов с образцом позволяет извлечь большой объем информации по его морфологии,
кристаллографическому строению и химическому составу. Получив с помо¬
168 -l\r Глава 4. Определение параметров катализаторов
щью просвечивающей электронной микроскопии проекцию плотности образца, легко найти размер каталитических частиц.
На рис. 4.14 показаны схемы проведения исследований методами просвечивающей и сканирующей электронной микроскопии (ПЭМ и СЭМ). Устройство
ПЭМ подобно оптическому микроскопу, нужно только заменить оптические
линзы на электромагнитные. В ПЭМ пучок первичных электронов высокой энергии и плотности проходит через конденсор, в котором формируется параллельный поток электронов, падающий на образец. Поскольку поглощение электронов в образце определяется его толщиной и плотностью, то прошедшие электроны формируют двумерную проекцию массы образца, которая контрастируется
методами электронной оптики с целью получения так называемого изображения в светлом поле. Изображение в темном поле формируется с помощью
дифрагировавших электронов, направление движение которых составляет небольшой угол с пучком прошедших электронов. Типичные параметры ПЭМ
таковы: энергия электронов 100—200 кэВ, вакуум — 10-6 мбар, разрешение —
0,5 нм, увеличение от 3 х 105 до 106.
Электронная
пушка
Линзы
конденсора
£=з Образец
Апертура
К
Линзы объектива
К
1X1
Линзы
Электронная
пушка
Линзы
конденсора
Катушка
сканера
X Линзы объектива
Детектор
рентгеновских лучей
Плоскость
изображения
Образец
Детектор
электронов
тм
СЭМ/Анализатор разброса
энергии рентгеновских лучей
Рис. 4.14. Схематическое устройство просвечивающего электронного микроскопа (ПЭМ)
и сканирующего электронного микроскопа (СЭМ). СЭМ обычно содержит детектор рентгеновских лучей, позволяющий проводить анализ состава образца
Сканирующий электронный микроскоп (СЭМ) содержит растровое устройство, позволяющее перемещать узкий электронный пучок по поверхности, при
этом детектируются либо вторичные, либо рассеянные назад электроны, вылетающие с различных ее участков. Контраст изображения определяется ориентацией поверхности: поверхность, ориентированная к приемнику, выглядит
светлее, чем поверхность, направленная в сторону от него. Вторичные электро¬
4.5. Электронная микроскопия (ЭМ, ПЭМ, СЭМ)
ны имеют энергию 5—50 эВ и зарождаются в поверхностном слое образца.
Рассеянные назад электроны идут от более глубоких слоев и несут информацию о составе образца, поскольку более тяжелые элементы рассеивают электроны лучше и дают более светлое изображение.
Описанные СЭМ имеют разрешение около 5 нм. Основное различие в ПЭМ
и СЭМ состоит в том, что в СЭМ контраст определяется топологией и составом поверхности, тогда как В ПЭМ пучок электронов проецирует всю «информацию» о массе, которую он «встретит» на своем пути, на двумерное изображение, однако делает это он уже с субнанометровым разрешением.
Как ясно из рис. 4.13, электронный микроскоп имеет дополнительные потенциальные возможности в анализе образцов. Дифракционные картины (пятна
для монокристаллической частицы и круги для набора произвольно ориентированных частиц) позволяют идентифицировать различные кристаллографические
фазы, как и в методе дифракции рентгеновских лучей. Генерируемое рентгеновское излучение является характеристикой элементов, входящих в образец,
поэтому позволяет сделать определенные предположения о составе образца
или некоторой его части. Такая методика называется анализом распределения
по энергии рентгеновских лучей (АРЭРЛ).
Си{ 111),
и 0,2! н'.‘
Н,0: Н, = 1:3 5% СО в Н
Си( 111).
d = 0.21 нм
2 нм
2
2 нм
Си(1 11).
с' ■ о 1 им
==//
Си( 111).
d = 0,21 нм
Рис. 4.15. Изображения частиц Си на носителях в атмосферах различных газов, полученные с помощью ПЭМ и построенные по правилу Вульфа:
а, б — нанокристаллы Си, ограненные плоскостями (100), (110) и (111); ПЭМ-изображе-
ния получены в атмосфере Н2 при давлении 1,5 мбар и температуре 220 °С в электронном
пучке, параллельном оси |011] меди; на врезке приведены данные спектроскопии энергетических потерь медленных электронов вблизи 12 3-края Си для катализатора и медной фольги, полученные in situ; в, г — нанокристалл металлической Си в атмосфере смеси Н2
: Н20 = 3:1 при полном давлении 1,5 мбар и температуре 220 °С; д, е — нанокристалл Си
в смеси 5 % СО в Н2 при давлении 5 мбар и температуре 220 °С 110)
169
В определении параметров катализаторов просвечивающая электронная
микроскопия используется наиболее часто. В общем случае обнаружение час-
170 -l\r Глава 4. Определение параметров катализаторов
тиц на носителе возможно, если имеется значительный контраст между частицами и носителем. Это — очень серьезное ограничение, которое может препятствовать использованию ПЭМ для исследования высокодисперсных частиц
оксидов на носителях. Сейчас нахождение размеров частиц методами ТЭМ
стало обычным делом, однако оно основано на допущении, что размер изображения частицы действительно пропорционален размеру самой частицы и что
вероятность детектирования частиц не зависит от их размеров.
Хотя обычно экспериментальные методы, в которых используются пучки
электронов, предполагают проведение соответствующих исследований в высоком вакууме, появились устройства, дающие возможность изучать поведение
катализатора при низких давлениях газа. Это принципиально важный момент,
поскольку морфология малых частиц иногда сильно чувствительна к газовой
среде. На рис. 4.15 показаны полученные in situ ПЭМ-снимки катализатора,
использующегося для синтеза метанола и представляющего собой частицы меди,
нанесенные на оксид цинка. Снимки получены на ПЭМ высокого разрешения
с модифицированной камерой, позволяющей проводить исследование образцов при давлениях газа до 200 мбар и температурах до 700—1000 °С.
Из рис. 4.15 видно, что форма и морфология частиц меди радикальным
образом (и обратимо) изменяются с составом газа. Наблюдаемые изменения
морфологии вызываются сменой значений поверхностных энергий частиц Си,
подложки ZnO и межфазной энергии между ними. В присутствии водорода
или смеси паров воды и водорода частицы стремятся принять форму ограненной полусферы, в то время как в атмосфере СО частицы «растекаются» по
подложке. Полагают, что это растекание обусловлено частичным восстановлением в атмосфере СО цинка на поверхности оксида и повышением поверхностной энергии подложки. В соответствии с правилом Вульфа (правило определения равновесной формы частиц по данным о поверхностных энергиях граней,
которое будет обсуждаться в следующей главе) такое повышение поверхностной энергии действительно должно приводить к увеличению площади контакта между Си и ZnO (рис. 4.15, д, ё).
4.6. МЕССБАУЭРОВСКАЯ СПЕКТРОСКОПИЯ (МС)
Мессбауэровская спектроскопия в катализе рассматривается как
очень специфический метод. Однако она позволяет получить важную информацию о некоторых катализаторах, к числу которых относятся катализаторы на
основе железа, используемые в синтезе аммиака и в процессе Фишера—Троп-
ша, а также CoMoS-катализаторы, используемые в гидроочистке. Мессбауэровская спектроскопия позволяет получить информацию о степени окисления элементов, внутренних магнитных полях и симметрии решетки и применяется в
том числе in situ, но для ограниченного ряда элементов: железа, кобальта, иридия, рутения, сурьмы, платины и золота.
Эффект Мессбауэра, открытый Рудольфом Месбауэром в 1957 г., может
быть вкратце представлен как излучение и поглощение без отдачи ядрами гам¬
4.6. Мессбауэровская спектроскопия (МС)
\171
ма-квантов. В случае железа источник излучения состоит из изотопа 57Со, период полураспада которого составляет 270 дней, трансформирующегося в изотоп 57Fe (содержание в природном железе около 2 %), ядра которого при этом
остаются в возбужденном состоянии. Ядра быстро релаксируют с переходом в
первое возбужденное состояние для этого изотопа. Переход в основное состояние сопровождается излучением фотонов с энергией 14,4 кэВ, при этом естественная ширина линии является очень узкой и составляет несколько нэВ.
Поглотитель излучения содержит железо, ядра которого находятся в основном состоянии. Если ядерные уровни в поглотителе и источнике характеризуются одной и той же разностью энергий, то ядра поглотителя могут в принципе
захватывать гамма-кванты. В действительности, однако, такого точного совпадения нет, поскольку энергетические уровни ядер сдвинуты или расщеплены
за счет их сверхтонкого взаимодействия с окружением. Чтобы охватить возможные переходы между сдвинутыми или расщепленными уровнями, необходимо варьировать энергию излучаемых гамма-квантов. Это делается с помощью эффекта Допплера. Если источник движется со скоростью v к поглотителю, то энергия гамма-квантов при этом становится равной
где Е0 — энергия гамма-квантов, вылетающих из неподвижного источника; с —
скорость света. Изменения скорости в пределах от —10 до +10 мм/с оказывается достаточно, чтобы охватить имеющиеся сдвиги энергии в ядрах железа.
Гамма-кванты, участвующие в ядерных переходах, имеют очень узкий разброс по энергиям. Именно эта их спектральная «точность» позволяет зафиксировать сверхтонкие взаимодействия — незначительные изменения энергетических уровней ядер, обусловленные сменой химического состояния или электрических и магнитных характеристик окружения атомов, ядра которых участвуют
в мессбауэровском процессе. Так, рис. 4.16 представляет три типичных источника сверхтонкого взаимодействия.
1. Изомерный сдвиг S возникает как результат кулоновского взаимодействия между положительно заряженными ядрами и отрицательно заряженными ^-электронами. Он является мерой плотности ^-электронов вблизи ядер, то
есть несет информацию о степени окисления железа в поглотителе. В этом
случае мессбауэровский спектр имеет одну линию, наблюдаемую, например, в
железе с ГЦК-решеткой, характерной для нержавеющей стали или сплавов с
благородными металлами.
2. Электрическое квадрупольное расщепление является результатом взаимодействия квадрупольного момента с электрическим полем. Электрическое
поле может быть создано несимметричным распределением электронов в незаполненных оболочках самого атома или зарядами соседних ионов. Расщепляются только возбужденные уровни (см. рис. 4.16) и величина расщепления
пропорциональна напряженности поля в точке нахождения ядра. Спектр содержит в этом случае два пика, называемых квадрупольным дублетом. Спектр
такого типа часто наблюдается в высокодисперсных оксидах железа (3+).
(4.9)
172 -l\r Глава 4. Определение параметров катализаторов
3. Магнитное сверхтонкое расщепление, или так называемый эффект Зеемана, обусловлено взаимодействием ядерного магнитного дипольного момента
с магнитным полем Н в точке нахождения ядра. За счет этого взаимодействия
расщепляются оба ядерных уровня, что приводит к полному снятию вырождения. Из восьми возможных переходов разрешены только шесть (см. рис. 4.16),
и спектр содержит шесть эквидистантных пиков, называемых секстетом или
секступлетом. Расстояние между пиками пропорционально напряженности магнитного поля в точке нахождения ядра. Наиболее известным примером такого
спектра является спектр металлического железа с ОЦК-решеткой, у которого
магнитное поле, ответственное за сверхтонкое расщепление, равняется 33 Т
(330 кЭ).
4. Если на ядра действуют и магнитное, и электрическое поля, а электрическое квадрупольное взаимодействие мало, то происходит дополнительный
сдвиг возбужденных уровней и секстет может стать асимметричным, как это
наблюдается в спектре Fe203.
Движущийся Катализатор Детектор
источник
Синглет
Квадрупольный
ЛЧ/finPT
Магнитный
А 14 ГО^ГТЧ/ППйТ
и
н-
А ,цуиле1
Т I й = (у1 + у2)/2
12 ДЕ=у2-у1
wCJIvO 1 yi\J 1“ 1
123 456 H<xv6-v 1
Г
ь\
~ж
щ v2
jVYVY|
V, v2 v3 vA v5 v6
ООО
V, мм/с V, мм/с V, мм/с
Рис. 4.16. Для совмещения энергии гамма-квантов с резонансной частотой приемника
используется эффект Допплера, так что энергия излучаемых квантов равна
E(v) = Е0(\ + v/c). Для 57Fe требуемая скорость движения источника улежит в
диапазоне от -1 до +1 см/с. В мессбауэровской спектроскопии исследуемый
образец является источником гамма-квантов, а приемником служит элемент с
одной линией поглощения. Приведенные спектры являются типичными в анализе железосодержащих катализаторов. Показаны также соответствующие ядер-
ные переходы и извлекаемые из спектров мессбауэровские параметры [1]
Эффект Мессбауэра наблюдается только для атомов, входящих твердое тело,
поскольку эмиссия и поглощение излучения должны происходить без потери
энергии, связанной с отдачей. Доля событий эмиссии и поглощения фотонов,
происходящих без отдачи, называется /-коэффициентом. Она зависит от температуры и энергии колебаний решетки: / высока для жестких решеток и мала
для поверхностных атомов.
4.6. Мессбауэровская спектроскопия (МС) ->\г 173
На рис. 4.17 демонстрируется, как с помощью мессбауэровской спектроскопии можно определить состояние железа в катализаторе на носителе поле его
различной обработки. Верхний спектр относится к свежеприготовленному Fe/
ТЮ2-катализатору, то есть к катализатору, полученному пропиткой носителя раствором соли и последующей сушкой. Квадрупольный дублет имеет изомерный
сдвиг, соответствующий трехвалентному (Fe3+) железу. После восстановления
в токе Н2 при 675 К катализатор состоит в основном из металлического железа,
доказательством чего служит секстет спектра, и частично недовосстановленно-
го железа, которое дает два дублета в центральной части спектра, соответствующих Fe2+ и Fe3+.
Общая степень восстановления железа, как показывает относительная площадь под кривой секстета спектра ОЦК-железа, является высокой. В
процессе Фишера—Тропша, происходящем при
575 К в присутствии СО и Н2, металлическое
железо переходит в карбид Хэгга Fe5C2. Невосстановленное железо присутствует в форме Fe2+.
Выдержка карбонизированного катализатора в
воздушной атмосфере при комнатной температуре не изменяет карбидные фазы, но доокисля-
ет железо в двухвалентном состоянии до трехвалентного состояния.
Рис. 4.17. Мессбауэровская спектроскопия позволяет получить детальную информацию об изменении состояния
железа в катализаторе, подвергаемом различной обработке, на носителе из ТЮ2. Приведен пример для железа,
восстановленного в токе Н, при 675 К в течение 18 ч, и
железа, поучаствовавшего в процессе Фишера—Тропша
(ФТ) [И]
Типичная процедура использования мессбауэровской спектроскопии в изучении катализаторов такова: катализатор подвергается некоторой обработке,
после чего in situ при комнатной температуре снимается его спектр Мессбауэ-
ра. В случае высокодисперсного катализатора более предпочтительным является измерение спектра при криогенных температурах, поскольку доля поверхностных атомов, не испытывающих отдачи, резко возрастает при температурах
ниже 300 К. Кроме того, спектры малых частиц, которые при комнатных температурах ведут себя как суперпарамагнетики, при криогенных температурах
подвергаются магнитному расщеплению и потому несут больше информации.
В мессбауэровской эмиссионной спектроскопии в катализатор вводится
радиоактивный источник (например, 57Со), а поглотитель с узкой линией соответствующего перехода, например 57Fe, для записи спектра перемещается с
определенной скоростью. Здесь можно использовать только катализаторы, содержащие кобальт, хотя, строго говоря, получаемая информация относится к
железу, образующемуся в катализаторе за счет процесса распада 57Со —> 57Fe.
1 1 1 ! 1 1 1 1 1 1 Г"
Мессбауэровский спектр
Fe/Ti02
Г=300К
Свежеприготовленный
—1—I—I—I 1—I—I—I—I—I—Г*
-10-8 -б -4 -2 0 2 4 6 8 10
Допплеровская скорость, мм/с
174
’ Глава 4. Определение параметров катализаторов
Основное преимущество мессбауэровской спектроскопии состоит в том,
что спектры можно снимать in situ. Главным недостатком этого метода является необходимость использования в нем взаимосвязанных элементов, среди которых наиболее легко поддаются анализу железо и олово.
4.7. ИОННАЯ СПЕКТРОСКОПИЯ:
МАСС-СПЕКТРОСКОПИЯ ВТОРИЧНЫХ ИОНОВ,
РАССЕЯНИЕ НИЗКОЭНЕРГЕТИЧЕСКИХ ИОНОВ,
ОБРАТНОЕ РЕЗЕРФОРДОВСКОЕ РАССЕЯНИЕ
(МСВИ,РНИ,ОРР)
Ионная спектроскопия, хотя она используется реже, чем РЭС, позволяет получить уникальную информацию о свойствах поверхности катализатора. Когда пучок низкоэнергетических ионов (например, ионов Аг+ с энергией 0,5—5 кэВ) падает на поверхность, ионы проникают в образец и теряют
свою энергию в серии неупругих столкновений с атомами мишени. В результате с поверхности могут вылететь вторичные ионы или нейтральные частицы,
которые анализируется методом масс-спектрометрии вторичных ионов (МСВИ).
Возможно также упругое рассеяние ионов на поверхности, которое используется в методе рассеяния низкоэнергетических ионов (РНИ), или рассеяние на
глубоко расположенных атомах высокоэнергетических ионов (обратное резер-
фордовское рассеяние). Более детально с этими методами читатель может ознакомиться по книге [3].
Масс-спектрометрия вторичных ионов (МСВИ) в настоящее время является наиболее чувствительным методом исследования поверхности, данные которого, однако, с большим трудом поддаются количественной обработке. Когда на поверхность падает пучок ионов (Аг+ с энергией 0,5—5 кэВ) за счет каскада столкновений к ней подводится значительное количество энергии. За счет
этой энергии происходит выброс (десорбция) с поверхности атомов, ионов и
многоатомных кластеров. В МСВИ с помощью квадрупольного масс-спектро-
метра непосредственно детектируются положительно и отрицательно заряженные вторичные ионы.
На рис. 4.18 показан спектр положительных вторичных ионов, вылетающих
с поверхности кремнеземного носителя с прекурсором катализатора — оксида
циркония, который был осажден за счет реакции между этоксидом циркония и
гидроксильными группами носителя. Обратим внимание на выход с поверхности одноатомных ионов (Н+, Si+, Zr+) и молекулярных ионов (SiO+, SiOH+,
ZrO+, ZrOt). В спектре также ясно виден изотопный состав циркония. Учет
изотопных пиков важен для идентификации полос спектра, поскольку отношение интенсивностей пиков должно согласовываться с естественным относительным содержанием компонентов и изотопов. Кроме пиков, соответствующих оксиду циркония и кремнезему, находящимся на индиевой фольге, на
спектре, представленном на рис. 4.18, имеются пики от ионов Na+, К+ и Са+.
4.7. Ионная спектроскопия: масс-спектроскопия вторичных ионов -*\г 175
Это — типичная для МСВИ ситуация: чувствительность метода к разным элементам изменяется на несколько порядков, и элементы, присутствующие в
следовых количествах, также могут быть обнаружены этим методом.
Si
Na
МСВИ Zr(0C2H5)4/Si02
После пропитки
SiO . SiOH
Zr ZrO
IKJ | OlVjn I
-л».
Zr02
0 15 30 45 60 75 50 105 120 135
Единицы атомной массы
После
кальцинирования
ZrO
у.
In
Единицы атомной массы
90 100 110 120
Единицы атомной массы
Рис. 4.18. МСВИ-спектры катализатора Zr02/Si02, полученные после пропитки носителя
раствором Zr(OC^H5)4 и кальцинирования на воздухе. Обратим внимание на то,
как отношение интесивностей пиков ZrO/Zr отражает содержание кислорода:
стехиометрическое отношение кислорода к цирконию в этоксиде равно 4: 1, а
в оксиде 2:1 [12]
МСВИ, строго говоря, — разрушающий метод анализа. В динамической
моде, используемой для получения профиля распределения элементов по
глубине, с объекта снимается по несколько десятков атомных слоев в минуту.
В статической схеме, которая применяется при анализе адсорбированных молекул, скорость удаления поверхностных слоев не превышает значение один
монослой за час, что предполагает неизменность состояния поверхности в процессе проведения эксперимента. В этом случае молекулярные ионы несут информацию о химической структуре поверхности.
К преимуществам МСВИ можно отнести ее высокую чувствительность (предел обнаружения для некоторых элементов составляет одну миллионную долю),
способность детектировать водород и наличие эмиссии молекулярных фрагментов, которые несут в себе легко раскрываемую связь со структурой поверхности. Недостатки этого метода обусловлены плохим пониманием механизмов
формирования вторичных ионов и трудностью количественного описания экспериментальных данных. Основное препятствие здесь порождено влиянием
матрицы: при смене окружения частиц выход вторичных ионов может радикально измениться. Матричный эффект и сильное изменение чувствительное-
\
176 Глава 4. Определение параметров катализаторов
ти, которое для разных элементов периодической таблицы составляет пять порядков, делают количественную интерпретацию МСВИ-спектров реальных катализаторов чрезвычайно затруднительной.
Несмотря на имеющиеся трудности количественного описания, МСВИ при
использовании материалов сравнения может дать полезную информацию. Так,
представленные на рис. 4.18 спектры явно показывают, что по отношению
интенсивностей потоков Zr02, ZrO+ и Zr+ можно судить об химическом окружении циркония: связанный с Zr02 и ZrO+ сигнал, идущий от этоксида циркония (отношение О : Zr = 4 : 1), оказывается более интенсивным, чем соответствующий сигнал от Zr02 (отношение О : Zr = 2 : 1). При интерпретации этой
информации следует использовать спектры сравнения, полученные для чистых
Zr02 и этоксида циркония.
В методе рассеяния низкоэнергетических ионов (РНИ, которое иногда называется спектроскопией рассеяния ионов) пучок ионов атомов благородных
газов с энергией несколько кэВ упруго рассеивается на поверхности твердого
тела. Энергия рассеянных ионов определяется законами сохранения энергии и
импульса и массой атома твердого тела, на котором собственно и произошло
рассеяние. Если детектор ионов расположен под углом 90° по отношению к
падающему пучку, то легко получить следующее соотношение
F М - М.
-=г=М*\ 2™ , (4.10)
Е0 Mat+Mion
где Е0 — энергия падающих ионов; Е — их энергия после рассеяния; Mion —
масса падающих ионов; Mat — масса атома, с которым столкнулся ион. Это
соотношение показывает, что спектр энергии рассеянных ионов эквивалентен
спектру масс поверхностных атомов. На рис. 4.19 получен РНИ-спектр для
медного катализатора, нанесенного на оксид алюминия. Спектр показывает,
что в соответствии с приведенной формой первичные ионы при столкновении
с тяжелыми элементами теряют меньше энергии, чем при столкновении с легкими элементами. Дисперсность частиц Си отражена в отношении интенсивностей пиков Си : А1 в соответствующем РНИ-спектре. Здесь имеется аналогия
с РЭС-спектроскопией, однако в данном случае полученная информация относится только к поверхностному слою.
Рис. 4.19. РНИ-спектр для катализатора Си/АЦОэ, показывающий, что при столкновении с
легкими атомами ионы теряют
больше энергии, чем при столкновении с тяжелыми атомами.
Обратим внимание на ступеньку,
присутствующую в фоне, при низких энергиях. Высокий пик при
низких энергиях обусловлен распылением поверхностных ионов.
Обрыв спектра при энергии около 40 эВ указывает на наличие
О 600 1200 1800 2400 3000 Ef, эВ положительного заряда у образца
4.8. Температурно-программируемые восстановление, окисление и сульфидирование 177
Следующие факторы определяют интенсивность рассеянного пучка: сечение
рассеяния для пары падающий ион/атом мишени и вероятность нейтрализации
ионов при их взаимодействии с твердым телом. Именно последний фактор обеспечивает высокую чувствительность РНИ-спектроскопии к поверхностному слою:
так, для ионов Не с энергией 1 кэВ, прошедших через один поверхностный
слой, вероятность нейтрализации составляет 99 %. То есть большинство ионов,
попавших в детектор, рассеиваются на внешнем слое атомов. Теории, позволяющей правильно рассчитывать сечения рассеяния и вероятности нейтрализации,
в настоящее время не существует. Тем не менее имеются вполне адекватные
расчетные процедуры, позволяющие находить эти характеристики на основе данных, полученных для образцов сравнения. Способность РНИ-спектроскопии
дать количественную информацию о составе внешнего слоя многокомпонентных материалов делает ее мощным инструментом в изучении катализаторов.
Метод обратного резерфордовского рассеяния (ОРР) является эквивалентом РНИ, но в нем используются ионы высокой энергии. Здесь обычно исследуют рассеяние ионов Н+ и Не+ с энергией 1—5 МэВ на ядрах атомов мишени.
Часть ионов рассеивается назад, и в методе ОРР анализируют их распределение по энергиям. Как и в методе РНИ, спектр энергий отражает спектр масс,
но он относится к внутренней части образца. Этот метод редко используется
при изучении промышленных катализаторов, но успешно применяется в исследовании модельных катализаторов плоской формы.
4.8. ТЕМПЕРАТУРНО-ПРОГРАММИРУЕМЫЕ
ВОССТАНОВЛЕНИЕ, ОКИСЛЕНИЕ
И СУЛЬФИДИРОВАНИЕ
Методы температурно-программируемых реакций формируют класс
методик, в которых прослеживается скорость химической реакции при линейном во времени изменении температуры. Эти методы реализуются в нескольких формах, основные из которых — температурно-программируемые восстановление, окисление и сульфидирование.
Инструментальное обеспечение исследований, проводимых при программируемом изменении температуры, достаточно просто. В реактор загружается
катализатор и производится нагрев катализатора в режиме, когда температура
изменяется линейно во времени со скоростью 0,1—20 °С/мин. С помощью датчика теплопроводности или масс-спектрометра определяется состав выделяющихся газов.
Метод температурно-программируемого восстановления (ТПВ) позволяет
найти температуру, при которой происходит полное восстановление катализатора. На рис. 4.20 показана зависимость, отвечающая ТПВ железных и родиевых катализаторов, нанесенных на кремнезем. Следует обратить внимание на
три момента. Первое: существенное различие скоростей восстановления благородного металла родия и «не благородного» металла железа. Второе: площадь
178 -l\r Глава 4. Определение параметров катализаторов
под кривой ТПВ (или температурно-программируемого окисления) отражает
полный расход водорода и часто выражается в молях Н2, затраченных на моль
атомов металла, Н2/М. Это отношение равно 1,5 для родия, как показано в
правой части рис. 4.20, и данное значение говорит о том, что родий присутствует в форме оксида Rh203. Для железа отношение Н2/М оказалось существенно ниже, что говорит лишь о частичном восстановлении металла. Третье:
ход кривой ТПВ для биметаллического катализатора показывает, что он начинает восстанавливаться при более низких температурах, чем катализатор из
чистого железа, то есть родий катализирует восстановление «менее благородного» железа. Данный факт служит доказательством хорошей смешиваемости
железа и родия в свежеприготовленном катализаторе. Механизм восстановления биметаллического катализатора можно представить следующим образом:
появившийся металлический родий способствует диссоциации водорода; атомарный водород диффундирует к оксиду железа, контактирующему с металлическим родием, и мгновенно восстанавливает оксид.
Датчик Осушитель
теплопроводности
Программатор
температуры
11
1 А Л
Реактор
мл
-
luVl IL
Масс-спектрометр
500 100 200 300 400500
Температура, °С
Рис. 4.20. Схема экспериментальной установки для изучения восстановления, окисления
и десорбции. Слева вверху: реактор находится в печи, температура которой
может линейно увеличиваться со временем. Расход газа на катализаторе определяется по изменению теплопроводности смеси; здесь важно удалить следовые примеси воды и других газов, которые могут сильно повлиять на измерение теплопроводности. Слева внизу: схема аппаратуры по программированному изменению температуры в сочетании с масс-спектрометром. Справа: спектр
температурно-программируемого восстановления катализаторов из Rh, Fe и
Fe—Rh на носителе, из кремнезема, подвергнутых предварительному кальцинированию, обеспечившему полное окисление металла до начала измерений
[1, 13]
4.9. Инфракрасная спектроскопия (ИКС) -J /■ 179
Для гидрообессеривания (ГОС) и гидродеазотирования (ГДА) тяжелых фракций нефти обычно используются катализаторы на основе молибдена или вольфрама на носителе из оксида алюминия, к которым в качестве активатора добавлен кобальт или никель. Поскольку катализаторы сохраняют свою активность в сульфидированном состоянии, активация осуществляется через обработку
оксидного прекурсора катализатора в атмосфере смеси газов H2S и Н2 (или
путем выдержки катализатора в серусодержащей реакционной среде). Водород
при этом используется для предотвращения распада относительно нестабильного H2S до элементарной сферы, которая при этом накапливалась бы на поверхности катализатора. На рис. 4.21 показаны кривые температурно-программируемого сульфидирования (ТПС) катализатора
МоОз/А1203; отрицательный пик говорит о расходовании данного газа, положительный — о
его формировании. При температуре ниже
400 К H2S расходуется, а Н20 формируется,
при этом содержание Н2 практически не
изменяется. Это указывает на то, что при
низких температурах основной реакцией
является обмен кислорода на серу.
Рис. 4.21. Кривые температурно-программируемого
сульфидирования (ТПС) катализаторов Мо03/А1203
в атмосфере смеси H2S и Н2, демонстрирующие расход этих газов и формирование Н,0 с ростом температуры. Заметим, что при температуре около 500 К
с поверхности катализатора выделяется H2S, что связывается с гидрированием элементарной серы
Температура, К
При 500 К катализатор начинает потреблять водород, о чем говорит резкий
пик, при этом формируется H2S и дополнительное количество Н20, что указывает на то, что при низких температурах катализатор захватил слишком много
серы, которая сейчас высвобождается в форме H2S. При более высоких температурах катализатор продолжает обменивать кислород на серу до тех пор, пока
молибден не перейдет в форму MoS2. ТПС оказалась очень удобным инструментом в изучении реакции сульфидирования катализаторов на основе Мо03,
в том числе и активированных Со и Ni.
4.9. ИНФРАКРАСНАЯ СПЕКТРОСКОПИЯ (ИКС)
В катализе инфракрасная спектроскопия наиболее часто применяется для идентификации адсорбированных соединений и изучения механизма
их хемосорбции на поверхности катализаторов. Довольно часто инфракрасные
спектры зондовых молекул СО и N0 содержат ценную информацию о природе
адсорбционных центров на поверхности катализатора. Сперва мы рассмотрим
основы теории поглощения инфракрасного излучения.
180
Л
Глава 4. Определение параметров катализаторов
Молекулы обладают дискретными уровнями, относящимися к их вращательному и колебательному движениям. Переходы между колебательными уровнями происходят при поглощении фотонов с частотой у, лежащей в инфракрасном диапазоне (длина волны 1—1000 мкм, волновое число 10 000—10 см,
разность энергий 1240—1,24 мэВ). Для валентных колебаний молекулы С—О,
например, эта частота составляет 2143 см-1. При малых отклонениях атомов в
колеблющейся двухатомной молекуле от положения равновесия потенциальную энергию можно представить в форме, соответствующей гармоническому
осциллятору:
где к — упругая постоянная колеблющейся связи; г — расстояние между атомами и req — равновесное расстояние между ними. Соответствующие колебательные уровни равны
где п = 0, 1, 2, ...; Ь — постоянная Планка; v — частота; т{ и т2 — массы
колеблющихся атомов; jll — приведенная масса. Разрешенным переходам для
гармонического осциллятора отвечает изменение квантового числа на единицу. Общее правило отбора для поглощения фотонов требует, чтобы дипольный
момент в процессе возбуждения колебаний изменялся.
Гармоническое приближение справедливо при малых отклонениях атомов
от положения равновесия. Основной недостаток гармонического потенциала
связан с тем, что он не позволяет связи разорваться. Для физически более
реальных потенциалов типа потенциалов Леннард-Джонса или Морзе энергетические уровни не являются эквидистантными, поэтому оказываются разрешенными и переходы с Ап > 1. Такие переходы называются обертонами. Так,
один из обертонов молекулы СО в газовой фазе имеет частоту 4260 см-1 (что
чуть меньше, чем 2 х 2143 = 4286 см-1). Модель гармонического осциллятора
несет в себе определенный физический смысл. Во-первых, зная частоту, можно сразу же вычислить упругую постоянную связи. Заметим, что, как видно из
уравнения (4.11), константа к при г2 характеризует кривизну кривой потенциальной энергии, но не глубину потенциальной ямы, совпадающую с энергией
связи. Но поскольку глубина потенциальной ямы и кривизна изменяются согласованно, по частоте инфракрасных колебаний часто оценивают энергию связи.
Во-вторых, для привязки частоты к определенной связи в адсорбированных
молекулах часто используется изотопное замещение (например, D вместо Н в
адсорбированном этилене или OD вместо ОН в метаноле), поскольку связанный с таким замещением сдвиг частоты может быть легко рассчитан.
Молекулы в газовой фазе обладают вращательными степенями свободы, и
переходы между колебательными уровнями сопровождаются переходами меж¬
у{г) = \к{г-гщ)2 ’
(4.11)
(4.12)
4.9. Инфракрасная спектроскопия (ИКС) ->\г 181
ду вращательными уровнями. Для жесткого ротатора, колеблющегося как гармонический осциллятор, разрешенные уровни энергии равны
Е" =(" + l)/il/ + 8^I7'/^ + 1^ (4ЛЗ)
где j — вращательное квантовое число; I — момент инерции молекулы. Здесь
применимо третье правило отбора: переходы возможны при А/ = ±1. Заметим,
что третье правило отбора запрещает чисто колебательные переходы для молекул в газовой фазе. Вместо них появляются две боковые ветви вращательных
полос (см. рис. 4.22, где показан спектр молекулы СО).
ф
s
I
ф
ЗГ
о
Е
о
с:
Рис. 4.22. Инфракрасный спектр молекул СО в газовой фазе. Показана тонкая структура, связанная с
вращательными переходами, которая, как видно из
спектра СО на поверхности катализатора Ir/Si02,
исчезает при адсорбции [ 1 ] Волновое число, см"1
При адсорбции молекулы теряют вращательные степени свободы, поэтому
в спектре наблюдаются только колебательные переходы (но при других частотах, см. рис. 4.22). Для молекулы СО сдвиг частот определяют три фактора:
• механическая связь молекулы СО с тяжелой подложкой увеличивает частоту валентных колебаний С—О на 20—50 см-1;
• взаимодействие диполя С—О со своим изображением на проводящем поляризуемом металле уменьшает частоту валентных колебаний С—О на 25—75 см-1;
• образование химической связи между С—О и подложкой изменяет распределение электронов по молекулярным орбиталям и ослабляет связь С—О.
Таким образом, объяснять появление различия в частотах колебаний молекул С—О в газовой фазе и в адсорбированном состоянии только за счет образования химической связи, строго говоря, некорректно.
Колебательные частоты инфракрасного спектра являются специфическими
для некоторых связей, что позволяет использовать их для идентификации состояния хемосорбированных молекул. Как будет показано в приведенных далее примерах, инфракрасные спектры молекул СО и N0 можно использовать
для распознавания природы адсорбционных центров.
Сульфидированные катализаторы на основе Мо и Со—Мо, используемые
в реакциях с участием водорода, содержат Мо в форме MoS2. Это соединение
182 -J\r Глава 4. Определение параметров катализаторов
имеет слоистую структуру: каждый слой Мо разделен двумя слоями S. Химическая активность MoS2 определяется краями «сэндвича», на которых Мо обращен к газовой фазе. Базальная плоскость анионов S2- в значительной степени инертна. Инфракрасный спектр молекул N0 на сульфидированном катализаторе Мо/А1203 (рис. 4.23) характеризуется наличием двух пиков, частоты
которых близки к наблюдаемым у органометаллических кластеров, содержащих Мо и группы N0. В спектре N0 на
поверхности катализатора Со/А1203 также
имеется два характерных пика (рис. 4.23, б),
частоты которых, однако, отличны от наблюдаемых для MoS2. Этот результат показывает, что молекулы N0 можно использовать как зонд для определения числа центров, относящихся к Со и Мо, в катализаторе
Со—Мо/А1203. Рисунок 4.23, в показывает, что этот метод прекрасно работает. Более того, сравнение интенсивностей полос
инфракрасных спектров NO/Mo для катализаторов Мо/А1203 и Со—Мо/А1203 показывает, что присутствие Со снижает число
атомов Мо, доступных для молекул N0. Это
означает, что атомы Со, вероятно, декорируют края «сэндвича» MoS2, на которых
находятся центры адсорбции N0.
Рис. 4.23. Инфракрасные спектр зондовых молекул
NO на сульфидированных катализаторах на основе
Мо, Со и Со—Мо, используемых для гидрообессе-
ривания. Соотнесение пиков базируется на ИК-спек-
трах металлоорганических соединений. Эти спектры позволяют оценить число центров, относящихся
к Со и Мо [14]
Инфракрасные спектры пропускания часто используются для изучения адсорбции газовых молекул на катализаторах на носителях и для исследования
разрушения прекурсоров, обладающих характерным ИК-спектром, в процессе
получения катализатора. Инфракрасная спектроскопия может быть применена
in situ для исследования реальных катализаторов в режимах диффузного отражения и пропускания.
4.10. МЕТОДЫ ИССЛЕДОВАНИЯ ПОВЕРХНОСТИ
Помимо рассмотренных ранее методов в науке о поверхностях разработаны поразительные приборы. К сожалению, многие из них можно использовать для исследования свойств хорошо охарактеризованных поверхнос¬
Частота . см 1
4.10. Методы исследования поверхности
тей монокристаллов (см. также рис. 4.1). В следующей главе мы обсудим структуру поверхностей более детально. Вследствие сложности строения и структуры катализаторов мы часто должны для установления свойств каталитических
частиц использовать модельные системы. Мы прибегаем к этому, например,
при анализе механизма связывания, формирования упорядоченных структур и
описании реакционной способности адсорбированных частиц. С использованием монокристаллов металлов или оксидов и уникальных приборов для изучения поверхности удается получить ответ на многие из возникающих вопросов. Такая информация позволяет достичь лучшего понимания свойств реальных катализаторов. В данной книге мы не будем рассматривать все методы,
использующиеся в науке о поверхностях. Интересующийся читатель может найти
их описание в ряде учебников [2, 3, 15]. Очень чувствительные методы анализа
поверхности, такие как РЭС, ИК-спектроскопия, МСВИ, РНИ, которые применимы к исследованию катализаторов на носителях и уже были охарактеризованы нами, используются и при изучении свойств поверхностей монокристаллов. Мы завершим наше обсуждение экспериментальных методов рассмотрением дифракции медленных электронов, сканирующей туннельной микроскопии
и атомно-силовой микроскопии, которые также позволяют определить структуру поверхности.
звание метода говорит о том, что в нем вместо рентгеновских лучей используются электроны. Поскольку низкоэнергетические электроны (энергия 40—200 эВ)
не проникают глубоко в объем материала без потери энергии, то при упругом
рассеянии они несут информацию только о внешних поверхностных слоях
(см. рис. 4.7).
В методе ДМЭ используется квантово-волновой дуализм электронов, которые ведут себя и как частицы, и как волны. Электроны с начальной энергией Ер,
где-то в области минимума кривой длины свободного пробега (см. рис. 4.7),
имеют длину волны Л, которая сравнима с расстоянием между атомами в
решетке
где р — импульс электрона; h — постоянная Планка; к — волновое число;
те — масса электрона. Это соотношение можно преобразовать в следующее
4.10.1. Дифракция медленных электронов (ДМЭ)
ДМЭ является аналогом дифракции рентгеновских лучей. Само на-
(4.14)
— И
р
(4.15)
Поскольку длина волны электрона сравнима с межатомным расстоянием,
происходит дифракция упруго рассеивающихся электронов (как и рентгено¬
184 ->\r Глава 4. Определение параметров катализаторов
вских лучей в РСтА). Рассеянные назад электроны формируют определенную
дифракционную картину из пятен на флуоресцирующем экране, по которой
восстанавливается симметрия и структура поверхности.
На рис. 4.24 показана схема экспериментальной установки. Пучок электронов с энергией Ер вылетает из относительно простой электронной пушки, находящейся в центре оптической системы установки по ДМЭ, по направлению
к поверхности. Исследуемый кристалл находится в центре четырех концентрических полусферических сеток. Внутренняя сетка поддерживается при нулевом
потенциале, а две внутренние сетки имеют отрицательный потенциал, который
чуть ниже Е , так что через них могут пройти только электроны, рассеявшиеся
упруго. Между последней сеткой и внешним флуоресцентным экраном поддерживается разность потенциалов в 2—5 кВ, которая ускоряет движение электронов по направлению к экрану. Последний делается прозрачным, чтобы можно
было наблюдать дифракционную картину пятен с задней стороны без помех,
создаваемых манипулятором образца. Пятна можно сфотографировать или записать на видеокамеру. Более усовершенствованные устройства снабжены анализаторами профиля интенсивности пятен, использование которого позволяет
по распределению интенсивности внутри пятна извлечь информацию о дальнем порядке в структуре поверхности.
На рис. 4.25 приведен пример картины пятен, наблюдавшейся при ДМЭ на
поверхности (111) монокристалла Fe, после его выдержки в атмосфере азота в
условиях, когда молекулы N2 диссоциируют и поверхность насыщается атомами N. Предполагается, что поверхность подвергается реконструкции, обеспечивающей высокую степень ее заполнения атомами N.
4.10. Методы исследования поверхности -1\г 185
Рис. 4.25. Дифракционная картина,
полученная при рассеянии медленных
электронов с энергией Ер = 42 эВ на
поверхности Fe(l 11), покрытой атомами N. По оценкам степень заполнения поверхности составляет долю 0,96
от монослоя азота и реконструирована в структуру 5x5. Темное пятно в
центре картины создано электронной
пушкой [16]
Рис. 4.26. Схематическое изображение
эксперимента по ДМЭ на поверхности монокристалла с векторами элементарной ячейки а, и а2. Наблюдаемая
при рассеянии медленных электронов
дифракционная картина соответствует обратной решетке, описываемой
векторами а]' и а2
Экран
Дифрагировавшие пучки
(-1.1)
(0.1)
1.1)
(КО)
(0.0)
(0.-1)
Принципиальная схема эксперимента по ДМЭ показана на рис. 4.26. Первичный пучок электронов падает на кристалл, элементарная ячейка поверхности которого образована векторами и а2. Пучок (00) отражается прямо в
электронную пушку, и если кристалл не наклонен, он не может быть детектирован. Картина ДМЭ совпадает с обратной решеткой кристалла, которая задается векторами а] и а2. Кинематическая теория рассеивания связывает векторы обратной решетки с векторами реальной решетки с помощью следующих
выражений:
2 л
а:л = 2 71-
а2 х г
ata2 xz’
aj х z
а^2 x z5
(4.16)
(4.17)
186 ->\r Глава 4. Определение параметров катализаторов
где z — единичный вектор, направленный по нормали к поверхности. Таким
образом, по дифракционной картине можно восстановить структуру и симметрию чистой и покрытой адслоем поверхности. Заметим, однако, что в случае
упорядоченного внешнего слоя с помощью ДМЭ можно установить только размеры элементарной ячейки для комбинации субстрат/ад слой, но нельзя определить положение молекул адсорбата по отношению к атомам субстрата. При
необходимости получения этой информации следует использовать более усовершенствованную методику. Например, можно учесть, что яркость пятна, образованного на экране, зависит от энергии первичных электронов. Тогда экспериментальную зависимость яркости от Ер (называемую кривой I— V) можно
сопоставить с расчетной зависимостью, найденной для предполагаемой структуры. Путем подгонки расчетной зависимости под экспериментальные данные
можно достаточно надежно установить положения и молекул адсорбата, и атомов субстрата. Следует иметь в виду, наблюдаемая дифракционная картина
появляется как результат усреднения по всем упорядоченным структурам, сформированным на площади, на которую падают первичные электроны. Отметим
также, что прозондировать структуру методом ДМЭ можно, только если она упорядочена на достаточно большой площади: типичным является размер 30 х 30 А2.
4.10.2. Сканирующая зондовая микроскопия (СЗМ)
Сканирующая зондовая микроскопия (СЗМ) и сканирующая или
атомно-силовая микроскопия (ССМ или ACM) стали применяться в анализе
структуры поверхностей относительно недавно [17]. Эти методы позволяют
получить топографию поверхности с атомным разрешением. В сканирующей
зондовой микроскопии пьезоэлектрический элемент перемещает острие микроскопа вдоль поверхности, при этом прослеживается изменение некоторой
характеристики, отражающей взаимодействие между острием и поверхностью.
Это позволяет получить с помощью сканирующего зондового микроскопа информацию о локальной структуре поверхности.
4.10.2.1. Сканирующая туннельная микроскопия (СТМ)
Сканирующий туннельный микроскоп был изобретен Биннингом
и Рорером, которые получили за свою разработку Нобелевскую премию. Сканирующая туннельная микроскопия является вершиной сканирующих зондо-
вых методов. В ней очень тонкое острие (радиус кривизны наконечника составляет порядка 100 А) подводится близко к поверхности и небольшая разность потенциалов (напряжение смещения) поддерживается между острием и
образцом. Согласно законам классической физики между образцом и острием
ток не должен проходить, но при расстоянии меньше 0,5 нм в игру вступают
законы квантовой механики, и электроны начинают туннелировать через зазор, обеспечивая туннельный ток на уровне 1 нА, который уже поддается измерению. Схема сканирующего туннельного микроскопа показана на рис. 4.27.
4.10. Методы исследования поверхности ->\г 187
Рис. 4.27. Схематическое изображение сканирующего туннельного микроскопа. Заметим,
что на атомном уровне при радиусе кривизны 100 А наконечник выглядит
тупым, движение вдоль осей х и у обеспечивается цилиндром, образованным
из четырех пьезоэлектрических элементов, а движение по оси z — цилиндрическим пьезоэлементом, к которому прикреплен наконечник
Сканирующий туннельный микроскоп может работать в различных модах:
движение острия вдоль поверхности может совершаться при постоянном токе
или фиксированном расстоянии между острием и поверхностью. Наиболее часто эксперименты проводятся в условиях постоянства тока. Ток поддерживается
постоянным за счет изменения средствами обратной связи расстояния между
острием и поверхностью. Наконечник зафиксирован на пьезоэлектрической трубке, которая сжимается или растягивается в зависимости от приложенного к
ней потенциала. Грубая настройка расстояния между острием и поверхностью
осуществляется с помощью червячной передачи, в конструкции которой также
используются пьезоэлектрические элементы. С помощью червячной передачи
можно перемещать острие со скоростью несколько миллиметров в минуту.
Перемещение же острия в процессе сканирования вдоль оси z осуществляется
только с помощью пьезоэлектрических элементов, которые позволяют перемещать острие на расстояние до 100 нм с точностью 0,001 нм! Аналогично перемещение в плоскости х—у осуществляется за счет изменения напряжения, приложенного к четырем пьезоэлементам, образующим цилиндр. Такая конструкция позволяет осуществлять перемещение на расстояние от нескольких десятых
ангстрема до нескольких миллиметров. Вся система обычно делается миниатюрной, чтобы обеспечить высокое значение резонансных частот, и устанавливается на гасящих вибрацию подставках, чтобы по возможности исключить
внешние воздействия. При получении типичного изображения сканирующий
туннельный микроскоп проходит по 256 линиям, делая по 256 измерений на
каждой линии. Скорость сканирования достаточно высока, так что на получение изображения поверхности затрачивается от 1 до 10 с. Снимая несколько
188
Глава 4. Определение параметров катализаторов
изображений подряд, можно проследить за развитием динамических процессов на поверхности, таких как диффузия или химическая реакция.
Наконечник обычно делается из вольфрама, по крайней мере, для проведения измерений в сверхвысоком вакууме. Менее реактивные материала типа Pt
или сплавов Pt/Ir могут быть использованы при проведении исследований в
условиях окружающей среды. Имеются методы формирования очень тонких
наконечников, которые необходимы при исследовании искривленных поверхностей. При сканировании плоских поверхностей монокристаллов можно использовать и тупые наконечники, полученные путем обрезания вольфрамовой
проволоки обычными щипцами, поскольку практически весть ток будет проходить только через один выступающий над другими атом. С помощью сканирующего туннельного микроскопа получаются превосходные изображения проводящих поверхностей в реальном пространстве, при этом удается определить
положения и атомов подложки, и молекул адсорбатов.
Чрезвычайно высокая степень разрешения в СТМ связана с эффектом туннелирования, который наблюдается при перекрытии свободных уровней поверхности и занятых уровней на острие (или наоборот). Таким образом, в СТМ наблюдаются не атомы, а фактически плотности состояний в окрестности уровня Ферми.
В квантовой механике туннельный ток как функция расстояния d между
острием и поверхностью дается выражением
где Ф — работа выхода электронов. Экспоненциальная зависимость тока от
расстояния d определяется таким же перекрытием «хвостов» волновых функций, отвечающих свободным и занятым электронным состояниям, в вакууме
вне поверхностей наконечника и образца. Это обстоятельство объясняет возможность легкого приготовления подходящих наконечников. Если один из атомов выступает на 1 А над другими, то, как следует из уравнения (4.18), через
него потечет 90 % тока. Это же уравнение служит объяснением достижимости
атомного разрешения на СТМ.
I (d) ос e~m5d{A)^]
(4.18)
Рис. 4.28. СТМ-изображение поверхности
(100)PtRh. В объемной фазе количества атомов этих металлов равны, но на поверхности
находится 69 % платины (темные пятна) и 31 %
родия (светлые пятна), что соответствует с
ожидаемой степенью поверхностной сегрегации чистого сплава Pt—Rh в условиях высокого вакуума. Черные пятна связаны с присутствием примесных атомов углерода. Видно, что атомы платины и родия стремятся
объединяться в малые кластеры, состоящие
из одних и тех же элементов [18]
4.10. Методы исследования поверхности ->\г 189
На рис. 4.28 приведен пример СТМ-изображения, на котором видны разрешенные отдельные атомы металла на поверхности сплава, то есть сканирующий туннельный микроскоп позволяет получить очень важную информацию о
структуре на атомном уровне. Для применения сканирующей туннельной микроскопии не требуется создавать высокого вакуума, получать изображения поверхностей можно in situ (даже в жидких средах). К сожалению, с помощью
сканирующего туннельного микроскопа можно изучать только хорошо определенные плоские проводящие поверхности, типа поверхностей металлов или
полупроводников, но он непригоден для исследования катализаторов на оксидных носителях. Для этих поверхностей более перспективным является метод атомно-силовой микроскопии.
4.10.2.2. Атомно-силовая микроскопия (ACM)
Атомно-силовая микроскопия (ACM) или, как ее иногда называют, сканирующая силовая микроскопия (ССМ) основана на измерении слабых, но детектируемых сил порядка нескольких наноньютонов, действующих
между тонким наконечником и атомами на поверхности. Наконечник подвешивается на гибком коромысле, называемом кантилевером, и располагается на
субнанометровом расстоянии от поверхности. При сканировании образца в
плоскости х—у наконечник периодически испытывает притяжение или отталкивание от поверхности, то есть его положение на оси z изменяется. Эти отклонения могут быть измерены с помощью лазера и фотодетектора, как схематически показано на рис. 4.29. Имеются два режима работы атомно-силово-
го микроскопа.
Рис. 4.29. Схематическое изображение атомно-силово-
го микроскопа. Образец помещается на пьезоэлектрический сканер и его положение может быть зафиксировано по координатам х, у и z с точностью 0,01 нм и
выше. Острие подвешено на гибком коромысле — кан-
тилевере. Когда наконечник притягивается или отталкивается от поверхности, отклонения системы канти-
левер/наконечник измеряются следующим образом:
лазерный луч фокусируется на конце кантилевера, после
отражения от которого попадает на два фотодиода, обозначенные номерами 1 и 2. Если острие притягивается
к поверхности, то в фотодиод 2 попадает больше света,
чем в фотодиод 1. Разность интенсивностей световых
потоков, попадающих в фотодиоды, является мерой
величины отклонения кантилевера, то есть силы взаимодействия образца и наконечника
В контактной моде наконечник находится на расстоянии нескольких ангстрем от поверхности образца, и взаимодействие между ними складывается из
взаимодействий их атомов.
Вторая мода является бесконтактной и ей отвечают расстояния между наконечником и образцом, составляющие от 2 до 30 нм. В этом случае взаимо¬
Лазер
Фотодетекторы
1
Линзы
Пьезоэлектрический |
сканер
190 -l\r Глава 4. Определение параметров катализаторов
действие между наконечником и образцом можно трактовать как взаимодействие макроскопических тел. В бесконтактной моде проводится, например,
исследование методом магнитно-силовой микроскопии доменной структуры
магнетиков.
Третья мода, которая в последнее время считается стандартной в исследовании легко разрушаемых поверхностей, по сути представляет собой комбинацию контактной и бесконтактной мод и называется динамической. В этом случае кантилевер колеблется и при максимальном своем отклонении в сторону
образца только касается его. Когда колеблющийся кантилевер приближается к
положению максимального отклонения, он начинает чувствовать поверхность,
и осцилляции становятся затухающими, что детектируется методами электроники и используется в качестве основы для построения топографии поверхности сканируемого образца. В динамической моде сдвиговая сила сопротивления, связанная с горизонтальным перемещением наконечника («царапанье»),
исключается, а силы, направленные по нормали к поверхности, в существенной мере ослаблены. Эта мода стала с успехом применяться при изучении малых частиц, находящихся на плоских подложках и использующихся в качестве
модельных катализаторов.
5000 об./мин ' - ' .
7000 об./мин
9000 об./мин
(высота) = 2,5 нм
(высота) = 2,0 нм
(высота) = 1,5 нм
0 5000 нм
0 5000 нм
0 5000 нм
Рис. 4.30. АСМ-изображения частиц родия, полученных после нанесения раствора соли
на вращающуюся плоскую подложку, представляющую собой поверхность
Si(100) с слоем SiO^, испарения растворителя и восстановления металла водородом [19]
Получаемое в ACM изображение всегда несет в себе информацию о топографии изучаемой поверхности и наконечника, и исследователь обычно получает изображение, не вникая в детали его формирования. Плоские поверхности
обычно сканируют с помощью пирамидального наконечника, имеющего широкий угол расхождения граней, который, как правило, является тупым. Если
на поверхности имеются элементы, более острые, чем наконечник, то получаемое изображение соответствует, скорее, топографии наконечника, а не поверхности. На рис. 4.30 показаны АСМ-изображения частиц родия, находящихся
на плоской поверхности кремнезема. Частицы размером несколько нанометров выглядят более крупными, поскольку в данном случае кривизна наконечника определяет степень разрешения изображения. Однако измеряемая высота
частиц по отношению к подложке является правильной: когда наконечник
4.11. Заключительные замечания -J\r 191
«сползает» с частицы на подложку изменение его положения соответствует разности высот между вершиной частицы и плоскостью подложки.
В представленном на рис. 4.30 примере частицы родия формировались за
счет растекания нанесенного на быстро вращающуюся подложку из Si02/Si
раствора трихлорида родия и его последующего испарения (так называемый
метод «spin-coating»). После полного испарения растворителя проводили восстановление металла в токе водорода. На рисунке показаны изображения образцов, полученных при трех различных скоростях вращения подложки, при
этом использовались растворы с одинаковой концентрацией атомов родия.
Частицы, полученные при высокой скорости вращения, имеют меньший размер, что связано с меньшими временами испарения раствора и осаждения соли.
В этом случае нуклеация превалирует над ростом частиц, что приводит к формированию большего числа более мелких частиц соли. В результате после восстановления водородом образуются и более мелкие частицы металла, показанные на рис. 4.30. Таким образом, даже если морфология системы частица/
подложка правильно не воспроизводится, высота образований на плоской подложке находится с помощью ACM с высокой точностью.
4.11. ЗАКЛЮЧИТЕЛЬНЫЕ ЗАМЕЧАНИЯ
В данной главе мы ограничили свое рассмотрение обсуждением
наиболее распространенных методов определения параметров катализаторов.
Безусловно, имеются и применяются другие методы типа ядерного магнитного
резонанса (ЯМР), который с успехом применяется при исследовании цеолитов, электронного парамагнитного резонанса (ЭПР) или комбинационного рассеяния, оказавшиеся очень полезными в изучении некоторых оксидных катализаторов. Кроме того, практически в стандартной схеме используются методы, разработанные в аналитической химии, — элементный анализ, оптическая
и ультрафиолетовая спектроскопия, спектроскопия атомного поглощения,
калориметрия, термогравиметрия и т. д. Важными являются также адсорбционные методы, позволяющие определить внутреннюю поверхность катализаторов или поверхность металлических частиц. Они будут рассмотрены в гл. 5.
Мы уже неоднократно отмечали, что фундаментальные исследования катализаторов основаны на изучении свойств поверхности монокристаллов или
других модельных систем. Поскольку академические исследования катализаторов нацелены на определение на молекулярном уровне состава поверхности в
условиях, при которых реально работает катализатор, то можно использовать
два основных подхода. Первый из них связан с моделированием поверхности
катализатора поверхностью монокристалла. В этом случае, привлекая инструментарий, разработанный в науке о поверхностях, можно определить (в благоприятных условиях) структуру поверхности на атомном уровне. Вместе с тем в
данном подходе, несмотря на необходимость исследования каталитических
свойств таких систем в реальных условиях (давление около 1 атм и выше),
192 -J\r Глава 4. Определение параметров катализаторов
большинство имеющихся методик основано на проведении измерений в высоком вакууме, и они не могут быть реализованы в промышленных условиях
протекания реакций.
Второй подход состоит в использовании методов, с помощью которых можно проводить измерения in situ. К их числу относятся мессбауэровская спектроскопия, рентгеноструктурный анализ, спектроскопия дальней тонкой структуры
рентгеновского поглощения. Эти методы часто применяются в условиях контролируемой окружающей среды, когда реакция уже заторможена. Методики, способные работать in situ, к сожалению, не являются методами исследования поверхности и не позволяют описать ее строение на атомном уровне. В лучшем
случае они дают возможность определить состав частиц.
Таким образом, имеется дилемма: проводить исследования in situ реальных
катализаторов в производственных условиях, но при этом информация о состоянии их поверхности будет достаточно скудной, или применять очень чувствительные методы анализа поверхности, но к модельным системам и в условиях высокого вакуума. По этой причине в изучении катализаторов стоит проблема переброски мостиков между предельными условиями ультравысокого
вакуума и высокого давления и предельными системами реальных катализаторов и монокристаллов.
Наконец, хотя в настоящее время значительный объем полезной информации о состоянии катализаторов может быть получен с помощью ряда методов,
многие данные фундаментального характера все еще остаются недостижимыми. В такой ситуации плодотворной является стратегия, состоящая в комбинировании всех методов, дающих хоть какую-нибудь информацию о свойствах
катализаторов. В литературе по катализу имеется много примеров успешной
реализации данной стратегии.
Список литературы
1. Niemantsverdriet J.W. Spectroscopy in Catalysis. An Introduction. 2nd Ed.
Weinheim: Wiley-WCH, 2000.
2. Ertl G., Kiippers J. Low Energy Electrons and Surface Chemistry. Weinheim:
Wiley-WCH, 1985.
3. Feldman L.C., Mayer J.W. Fundamentals of Surface and Thin Film Analysis.
Amsterdam: North-Holland, 1986.
4. Fagherazzi G., Benedetti A., Martorana A., Giuliano S., Dganello G. // Catal.
Lett. 1990. V. 6. P. 263.
5. Samorjai G.A. Chemistry in Two Dimensions, Surfaces. Itacha: Cornell Univ.
Press, 1981.
6. Borg H.J., Van den Oetelaar L.C. A, Van Ijzendroon L.J., Niemantsverdriet J.W. //
J. Vac. Sc. Technol. A. 1992. V. 10. P. 2737.
7. Koningsberger D.C., Prince R. X-ray Absorption. New York: Wiley, 1987.
8. Koningsberger D.C., Van Zon J.B.A.D., Van’t Blik H.F.J., Visser G.J., Prins R.,
MansourA.N., Sayers D.E., Short D.S., KatzerJ.R. //J. Phys. Chem. 1985. V. 89. P. 4075.
Список литературы -J\r 193
9. Amelinckx S., Van Dyck, Кяя Landuyt J., Кяя Tendeloo G. Handbook of
Microscopy. Weinheim: VCH, 1997.
10. Hansen P.L., Wagner J.B., Helveg S., Rostrup-Nelsen J., Clausen B.S., Tops0e H. //
Science. V. 2002. 295. P. 2053.
11. Кяя Jer Kraan A.M., Nonnekens F., Stoop F., Niemantsverdriet J.W. // Appl.
Catal. 1986. V. 27. P. 285.
12. Mejers A.C.Q.M., de Jong A.M., Van Gruijthuisen L.M.P., Niemantsverdriet J.W. //
Appl. Catal. 1991. V. 70. P. 53).
13. Van’t Blik H.F.J., Niemantsverdriet J.W. // Appl. Catal. 1984. V. 10. P. 155.
14. Tops0e N.Y., Tops0e H. // J. Catal. 1983. V. 84. P. 386; J. Electron Spectrosc.
Relat. Phenom. 1986. V. 39. P. 11.
15. Woodruff D.P., Delchar T.A. Modem Techniques of Surface Science. Cambridge:
Cambridge Univ. Press, 1986.
16. Alstrup I., Chorkendorff I., Ullman S. // J. Catal. 1997. V. 168. P. 217.
17. Wiesendanger R. Scanning Probe Microscopy and Spectroscopy. Cambridge:
Cambridge Univ. Press, 1994.
18. Nieuwenhuys B.E., Schmid М., Varga P. // Surf. Sci. 1996. V. 359. P. 17.
глава ТВЕРДЫЕ КАТАЛИЗАТОРЫ
5
5.1. ТРЕБОВАНИЯ, ПРЕДЪЯВЛЯЕМЫЕ
К ЭФФЕКТИВНЫМ КАТАЛИЗАТОРАМ
Катализаторами могут быть металлы, оксиды, сульфиды, карбиды,
нитриды, кислоты, соли и в принципе материалы любой природы. Встречаются довольно разнообразные формы катализаторов: от пористых частиц до сплошных мелких частиц, находящихся на носителях. В качестве носителей выступают пористые частицы порошков, типа частиц оксида алюминия, или большие
монолитные структуры, к каковым относятся керамики, используемые при
катализе выхлопных газов автомобилей.
Процесс получения катализаторов представляет собой сочетание науки и
искусства, а также в значительной мере опыта. Хотя лежащая в его основе
химия по большей мере известна, многие технологии синтеза катализаторов
настолько сложны, что записать полную схему протекающих при этом реакций
практически невозможно.
Предположим, что нам удалось в лабораторных условиях получить образцы, характеризующиеся для некоторой реакции превосходным числом оборотов реакции на один активный центр и баснословной селективностью в отношении целевого продукта. Будут ли данные образцы эффективными катализаторами в промышленных условиях? Не обязательно. Главным образом из-за
того, что эти образцы сначала должны быть превращены в материал, обладающий следующими свойствами:
• высокой активностью на единицу объема предполагаемого реактора;
• высокой селективностью по отношению к целевому продукту при уровне
конверсии, достигаемом в предполагаемом реакторе, а также низкой селективностью по отношению к побочным продуктам, порождающим проблему утилизации отходов;
• способностью сохранять в течение длительного времени свою активность;
• при быстрой потере активности способностью к восстановлению;
• воспроизводимостью параметров при получении;
• высокой термической устойчивостью по отношению к спеканию и нарушениям структуры и низкой летучестью в окружающей среде реактора (например, когда побочным продуктом является пар);
5. /. Требования, предъявляемые к эффективным катализаторам -J\r 195
• высокой механической прочностью по отношению к дроблению (например, под давлением слоя катализатора или при формовке);
• высокой износостойкостью (сопротивлением к истиранию).
Катализаторы обычно разрабатываются для определенного процесса, то есть
для конкретной реакции, конкретного реактора и конкретных термодинамических условий. Процессы тепло- и массопереноса накладывают дополнительные
требования на морфологию и теплопроводность катализаторов. Например, важно, чтобы частицы катализатора имели определенную форму типа стержней
или таблеток, что позволяет понизить гидродинамическое сопротивление слоя
катализатора. И если наш катализатор обладает всеми перечисленными выше
достоинствами, но ему нельзя придать желаемую форму, то он никогда не будет использоваться на практике.
Наконец, чтобы катализатор был принят промышленностью, следует прежде всего рассмотреть экономические факторы, а также степень его оригинальности (то есть может ли он быть запатентован или он подпадает под патенты,
принадлежащие конкурентам).
В принципе можно считать, что гетерогенная каталитическая реакция протекает на активных центрах или на группировках молекул, где происходят элементарные превращения реагентов. Удельная активность таких центров является
основным объектом научных исследований и служит основой для сравнения
различных металлов или геометрических форм каталитических частиц. Определим число оборотов реакции (ЧОР) как число молекул реагентов, претерпевших
химические превращения на этом центре за одну секунду. В промышленности
больший интерес представляет активность на единицу объема катализатора.
Высокая степень дисперсности каталитических частиц является желаемой, если
только при этом увеличивается число нужных активных центров.
Поскольку довольно часто каталитические реакции протекают при повышенных давлениях, то для минимизации затрат на капитальное строительство
необходимо уменьшать и размер реактора. В этом контексте важным является
произведение факторов объема и времени, то есть параметра, показывающего,
как много производится продукта за единицу времени в единице объема реактора. Не имеет смысла делать катализатор, который полностью превращает
реагенты в продукт при нулевой пространственной скорости течения реагентов. Пространственная скорость течения реагентов определяется отношением
их расхода и объема реактора. При тестировании катализаторов пространственную скорость течения выбирают настолько большой, что степень конверсии
приближается к нулевому значению, но при осуществлении промышленных
процессов выбором пространственной скорости добиваются оптимального соотношения между скоростью формирования продукта и степенью приближения системы к равновесию при наименьшем количестве используемого катализатора. Стоимость катализатора также принимается во внимание, и она
может заставить сделать выбор в пользу менее активного металла, поскольку
оптимальный металл может оказаться редким и дорогим. На рис. 5.1 показаны
цены некоторых часто используемых в катализе благородных металлов. На¬
196 -l\r Глава 5. Твердые катализаторы
блюдаемые сильные колебания цен связаны с тем, что эти металлы являются
объектом спекуляций. Некоторые из указанных на рисунке металлов достаточно редки, и даже слухи о возможности использования этих металлов в качестве
катализаторов нового типа и предполагаемом в связи с этим росте спроса на
них способны повлиять на состояние рынка. Безусловно, некоторые из металлов типа никеля и железа настолько дешевы, что разработка альтернативных
катализаторов, например, для синтеза аммиака (железные катализаторы) или
конверсии с водяным паром (никелевые катализаторы) является практически
бесполезным делом.
Годы
Рис. 5.1. Изменение цен на некоторые благородные металлы за 8 лет. Заметим, что частые колебания некоторых цен связаны со спекуляцией. К концу 2006 г. цена
родия составляла 190 долларов США за грамм
Активность катализатора на единицу объема не является исключительным
значимым параметром. Важно принимать во внимание такие его характеристики, как долговечность и селективность. Нет смысла добиваться высокой степени превращения реагентов, если при этом получаются в большом количестве
ненужные продукты. В этом случае потребуется использовать затратные технологии выделения целевого продукта. Селективность катализатора, с участием
которого образуются продукты А и В, по отношению к продукту А определяется соотношением АДА + В) х 100 %. Таким образом, идеальный катализатор
должен одновременно обладать высокими степенями превращения и селективности.
Эти требования являются основными стимулами к разработке идеальной
реакции, в которой, например, метанол получается непосредственно из метана
и кислорода. Взглянув на схему реакции, можно подумать, что этот процесс
можно осуществить непосредственно, поскольку для этого нужно только добавить к метану один атом кислорода. Более того, данный процесс является строго экзотермическим (детали см. в подразд. 8.2.6.1). Проблема, однако, состоит
5.2. Структура металлов, оксидов, сульфидов и их поверхностей -1\г 197
в том, что энергетически еще более выгодным является полное окисление метана до диоксида углерода и воды. Имеются катализаторы, обладающие высокой селективностью (90 %) по отношению к окислению метана до метанола, но
для них характерна столь низкая степень превращения, что достигаемый при
их использовании выход продукта не представляет интереса для практических
приложений. Таким образом, прямое окисление метана до метанола остается
реакцией-мечтой.
5.2. СТРУКТУРА МЕТАЛЛОВ, ОКСИДОВ, СУЛЬФИДОВ
И ИХ ПОВЕРХНОСТЕЙ
5.2.1. Структура металлов
Для катализа наибольший интерес представляют металлы восьмой
группы и подгруппы Б первой группы. Для этих металлов характерны три кристаллические структуры: гранецентрированная кубическая решетка (ГЦК — Ni,
Си, Rh, Pd, Ag, Ir, Pt, Au), гексагональная плотная упаковка (ГПУ — Со, Ru, Os)
и объемно-центрированная кубическая решетка (ОЦК — Fe). На рис. 5.2 показаны элементарные ячейки этих трех структур. Заметим, что элементарная ячейка
содержит 4, 2 и 6 атомов в ГЦК, ОЦК и ГПУ-решетках соответственно. Другие
материалы типа оксидов, сульфидов и т. д. характеризуются более сложными
кристаллическими решетками, которые мы здесь рассматривать не будем. Прежде чем обсуждать свойства поверхности, сделаем несколько замечаний общего
характера.
i
L *
/
/\
А
У
У
>
/
J W
Z
Рис. 5.2. Элементарные ячейки гранецентрированной (ГЦК) и объемно-центрированной
кубических (ОЦК) и гексагональной плотноупакованной (ГПУ) решеток
Если смоделировать кристаллы плотными упаковками сфер, то ОЦК- и
ГПУ-структуры будут обладать наибольшей плотностью, при которой свободное пространство будет составлять 26 % общего объема. Каждый атом внутри
кристалла будет иметь по 12 ближайших соседей, или, иными словами, координационное число для атома внутри кристалла равно 12. У кристалла с ГЦК-решет-
кой свободное пространство составляет 32 % объем, а координационное число
для внутреннего атома равно 8.
198 -J\r Глава 5. Твердые катализаторы
5.2.2. Кристаллография поверхности металлов
Кристаллография поверхности представляет собой двумерный аналог кристаллографии объемных материалов, в котором рассматривается характер распределения атомов в различных плоскостях, формируемых атомами
объемного кристалла. Мы ограничим свое рассмотрение наиболее часто встречающимися у металлов структурами, показанными на рис. 5.2. Структуры других типов можно найти в учебниках по физике твердого тела [1—3].
5.2.2.1. Поверхности кристаллов
Структура и геометрия поверхности играет доминирующую роль в
ее реакционных, адсорбционных и каталитических свойствах. Поэтому при
анализе тех или иных явлений всегда следует конкретно указывать, с какой
структурой исследователь имеет дело. При этом необходимо использовать универсальный язык для описания разнообразных поверхностных структур. Поверхность кристалла характеризуется в первую очередь нормальным к ней вектором:
Н = Ах + ку + 1г. (5.1)
Поверхность полностью определяется набором чисел (индексов) А, к и /,
которые обычно записываются в круглых скобках (hkl), располагающихся рядом с химическим символом металла, например Си(ЮО) или Pt(lll). Атомы,
расположенные на поверхности кристалла (100), находятся в плоскости, параллельной координатной плоскости yOz. Отрицательные значения индексов А, к
и / обозначаются чертой над соответствующим числом: Си(010). Заметим, что
перестановки индексов типа Cu(100), Cu(010), Cu(001), Си(0Т0)и Cu(lOO) описывают поверхности одной и той же структуры, как показано на рис. 5.3.
Рис. 5.3. Базальные плоскости, полученные сечением элементарных
ячеек простых кристаллических
структур
Три базальные плоскости кубического кристалла имеют индексы (100), (110)
и (111). Соответствующие им сечения элементарной ячейки показаны на рис. 5.3.
Положения атомов, выходящих на поверхность при таких сечениях кристаллов, показаны на рис. 5.4.
Важным параметром, определяющим реакционную способность поверхности, является плотность находящихся на ней атомов. Согласно общему правилу, принимаемому как аксиома, чем более рыхлой является поверхность, тем
большей реакционной способностью она обладает. Это правило будет более
подробно обсуждено в гл. 6. Заметим, что поверхность (110) ГЦК-кристалла
является наиболее рыхлой, тогда как для грани (111) характерна плотная упа-
5.2. Структура металлов, оксидов, сульфидов и их поверхностей -*\г 199
ковка. Для ОЦК-кристалла ситуация противоположна: грань (111) — наиболее
рыхлая, а поверхность (110) характеризуется плотной упаковкой.
ГЦК
1/2
(100)
*"1/2 aV2
(in)
a
(НО)
ОЦК
Рис. 5.4. Структура базальных по- '“"V
верхностей ГЦК- и ОЦК-решеток.
Пунктиром показаны атомы вторых *~а~* ■■■''* *
слоев, расстояния между атомами :
даны в единицах постоянной ре-
шетки а (100) (111) (110)
Разницу между ГЦК- и ГПУ-структурами легко понять, рассмотрев последовательность расположения плотноупакованных слоев. Для ГЦК-структу-
ры плотной упаковке отвечают плоскости (111) (см. рис. 5.2 и 5.4), а для
ГПУ-структуры — плоскость (001). Геометрические расположения атомов в
этих плоскостях идентичны. Обе кристаллические решетки могут быть построены путем наложения друг на друга последующих плотноупакованных слоев.
Если атомы третьего слоя находятся непосредственно над атомами первого
слоя, то формируемая таким образом структура будет гексагональной плотной
упаковкой. Последовательность слоев в направлении (001) при этом такова:
ababab... Чтобы получить ГЦК-структуру, необходимо расположить атом каждого четвертого слоя над атомом первого слоя. Последовательность слоев в
направлении (111) выглядит так аЬсаЬсаЬс...
Более сложные поверхностные структуры, на которых имеются ступени и
даже ступени с изломами (кинками), также может быть описаны в рамках
введенных выше обозначений, например, символ n(hn kn lt) х (Av, ks, /,) означает, что на поверхности имеется п рядов атомов, формирующих террасу на
грани (hn kp lt) и одноатомная ступенька по грани (Av, ks, /,). Такого же типа
обозначения применяются при описании ступеней с кинками, делающими
ступень шероховатой. Так, структура поверхности (775) эквивалентна структуре, описываемой символом 6(111) х (11 1). Подобным же образом можно
описать поверхность произвольной структуры. Естественно, что для поверхностей произвольной структуры возникает вопрос об их устойчивости: они
могут для обеспечения минимума энергии трансформироваться в фасетиро-
ванные поверхности.
200
Л
Глава 5. Твердые катализаторы
В качестве моделей реальных катализаторов можно использовать поверхности монокристаллов, имеющие специфическую ориентацию. Такие модельные
катализаторы обычно имеют форму таблеток с характерным диаметром 5—10 мм
и толщиной 1—3 мм. Они могут быть вырезаны из монокристаллического стержня. В настоящее время большинство металлов продается в виде таких стержней.
Желаемая поверхностная структура создается путем дуговой резки макроскопических монокристаллов по нормали к выбранному направлению. Предварительно
монокристалл с использованием дифракционной картины Лауэ ориентируется
в выбранном направлении с точностью до 0,5°. Затем поверхность полируется с
помощью специальной пасты до исчезновения всех царапин и появления у
грани зеркального блеска. Иногда, в случае мягких материалов типа меди, требуется проводить дополнительную электрополировку для удаления поврежденных слоев. Наконец, кристалл выдерживается в ультравысоком вакууме (давление около Ю-10 мбар и менее) для удаления с его поверхности адсорбированных примесей, после чего поверхность подвергается многократному распылению
и закалке, с помощью которых она окончательно очищается от посторонних
компонентов. Процедура очитки поверхности варьируется от элемента к элементу, и рецепт ее эффективного осуществления можно найти в периодической
литературе. Поверхность (775) для ГЦК-кристалла, показанная на рис. 5.5, может быть легко получена их стержня, торец которого имеет ориентацию (111).
Для этого надо срезать стержень под углом 8,47° по отношению к нормали (111).
Величина этого угла находится из следующего соотношения:
cos (ср) =
щ
Рис. 5.5. Модель поверхности (775) ГЦК-кристалла. Заметим, что эта поверхность образована в основном гранями (111) со ступенями на
каждом шестом ряду
Реакционная способность поверхности зависит от концентрации ненасыщенных связей на ней. Ненасыщенные связи — это те связи, которые сформировались из исходных связей между соседними атомами, характерных для мае-
5.2. Структура металлов, оксидов, сульфидов и их поверхностей 201
сивного кристалла и разрываемых при образовании поверхности. Таким образом, для подсчета числа ненасыщенных связей нам необходимо определить
число недостающих соседей, обозначаемое через Zs, для атомов, находящихся
на поверхности. С помощью рис. 5.2 легко установить, что у атома на поверхности (111) ГЦК- или ГПУ-кристалла имеется 6 соседей в поверхностном слое
и 3 соседа в приповерхностном слое и у него отсутствуют 3 соседа из «верхнего» (отсутствующего) слоя, которые имелись бы у атома в объеме кристалла, то
есть Zs = 3. Атом на поверхности (100) ГЦК-кристалла имеет четырех соседей
в поверхностном слое, четырех в приповерхностном слое, и у него отсутствуют
четыре соседа из «внешнего» слоя, так что Zs = 4. Таким образом, у атома на
поверхности (100) ГЦК-кристалла число ненасыщенных связей на единицу
больше, чем у атома на поверхности (111), поэтому он должен быть более реакционно-активен. Используя эти же аргументы, можно установить, что атомы,
находящиеся на вершине одноатомной ступени, показанной на рис. 5.5, также
реакционно более активны, чем атомы, находящиеся на террасе. Ниже мы увидим, что такие атомы способны повысить реакционную способность поверхности на несколько порядков.
5.2.2.2. Адсорбционные центры
На рис. 5.6 показаны наиболее типичные адсорбционные центры и
даны их названия. Они называются верхушечным центром, мостиковыми центрами (короткий или длинный мостики), полостными центрами, которые могут иметь тройную или четверную координацию. В случае центра с тройной
координацией на поверхности (111) ГЦК-кристалла необходимо различать центры ГПУ- и ГЦК-структур, прямо под которыми имеется или отсутствует атом.
(100)
(111)
(110)
О
^ \ Верхушечный
* центр
©
Мост
Длинный мост
©Полостной центр
с четверной
координацией
©Полостной центр
стройной координацией
у ГЦК -решетки
©Полостной центр
стройной координацией
у ОЦК-решетки
Рис. 5.6. Адсорбционные центры на разных поверхностях
Число адсорбционных центров на поверхности непосредственно определяется ее геометрией. Рассмотрим, например, адсорбцию на полостном центре с
четверной координацией на поверхности (100) ГЦК-кристалла. Тогда концентрация адсорбционных центров просто равна числу элементарных ячеек пло-
202
\
Глава 5. Твердые катализаторы
щадью я>/2 J на одном квадратном метре, где а — постоянная ГЦК-решет-
ки. Заметим, что площадь той же элементарной ячейки на поверхности (100)
ОЦК-кристалла равна а2, где а — снова постоянная решетки.
5.2.2.3. Двумерная решетка
Поверхностные периодические структуры описываются с помощью
двумерных решеток. Любой узел этой решетки может быть получен как комбинация двух базисных векторов. Два базисных вектора задают наименьшие по
размеру ячейки, характеризующиеся одинаковым расположением атомов. Решетка кристалла строится путем перемещения элементарной ячейки на произвольные расстояния, получающиеся при линейной комбинации базисных векторов. Такие линейные комбинации базисных векторов определяют решетку
Браве, которая представляет собой набор векторов, с помощью которых можно
попасть в любой узел решетки.
В двумерном пространстве существует пять различных решеток (рис. 5.7).
Гексагональная решетка Браве служит элементарной ячейкой для поверхностей
(111) кубических структур и поверхностей (001) ГПУ-кристаллов; центрированная прямоугольная решетка является элементарной ячейкой для поверхностей
(110) ОЦК- и ГЦК-кристаллов, а прямоугольная ячейка — для поверхности (100)
кубической решетки. Переносом этих элементарных ячеек на расстояние + &а2,
где Инк — целые числа, можно получить поверхностные структуры.
Квадратная
Примитивная
прямоугольная
Центрированная
прямоугольная
t
Гексагональная
Косоугольная
Рис. 5.7. Пять различных поверхностных решеток Браве
(примитивных ячеек)
Во многих случаях число занятых адсорбционных центров не совпадает с
числом элементарных ячеек. Зачастую отталкивание между адсорбированными
молекулами или атомами не позволяет заполнить все центры, и адсорбция происходит, если только все соседние центры остаются незанятыми. Описание
адсорбционных структур проводят с учетом связи строения элементарных ячеек адсорбата и субстрата со строением исходной элементарной ячейкой подложки. На рис. 5.8 приведены примеры структур и принятая номенклатура их
описания.
5.2. Структура металлов, оксидов, сульфидов и их поверхностей \г 203
Элементарная ячейка адсорбата со структурой р(2 х 2), показанная на рис. 5.8,
по двум направлениям в два раза больше элементарной ячейки субстрата, по
этой причине она обозначается как р(2 х 2), где символ обозначает р «примитивная». Степень покрытия при этом равна 0,25 монослоя (обычно сокращается как МС).
р(2x2) с(2 х 2)
Рис. 5.8. Структуры адсорбционных слоев
типа 2 х 2 и 42 х V2(/M5 ) или с(2 х 2),
которые формируют, например, атомы
серы на поверхности Ni(100)
Такой способ обозначения поверхностных структур, называемый обозначениями Вуда, подходит для простых поверхностных структур. Он непригоден
для более сложных структур, и обычно вводят матрицы размером 2x2, которые связывают векторы и а2 подложки с аналогичными векторами адслоя.
На рис. 5.8 показана структура 72 х 72 (/М5°), характерная для атомов S на
поверхности Ni(100). Элементарная ячейка адсорбата повернута на угол 45 ° по
отношению к элементарной ячейке подложки. Такая структура часто обозначается как с(2 х 2), поскольку она может быть получена введением дополнительного атома в центр элементарной ячейки р(2 х 2). Степень покрытия поверхности у структуры с(2 х 2) составляет 0,5 МС.
Предельная степень заполнения монослоя (насыщение монослоя) зависит
от природы адсорбата. Для поверхности Ni(100) максимальной адсорбции серы
отвечает степень заполнения 0,5 МС, в то время как для молекул СО и атомов
Н она равна 0,58 и 1,0 МС соответственно. Заметим, что в обозначениях Вуда
не отражена природа центров адсорбции. Природа адсорбционных центров
определяется экспериментально методами дифракции или сканирующей туннельной микроскопии. Такие атомы, как углерод, азот, кислород и сера, предпочитают связываться с центрами, обладающими высокой степенью координации, то есть поэтому их обычно находят на центрах с тройной или четверной
координацией (см. рис. 5.6). Часто выигрыш энергии при оптимизации распределения связей с адсорбатом оказывается настолько большим, что вся поверхность перестраивается, чтобы обеспечить высокую степень координации.
Хорошо известным примером, где наблюдается реконструкция поверхности, вызванная адсорбатом, является система атомы углерода/поверхность (100)
никеля. Хотя этой поверхности присущи адсорбционные центры с четверной
координацией, дополнительный выигрыш энергии может быть получен при
так называемой «часовой» реконструкции, показанной на рис. 5.9. Конечно, на
перераспределение атомов никеля затрачивается определенная энергия, но она
оказывается ниже энергетического выигрыша, связанного с усилением связи
атомов углерода с подложкой, так что в целом при такой реконструкции энер¬
204 -l\r Глава 5. Твердые катализаторы
гия системы понижается. Подвергаются реконструкции, обеспечивающей более высокую степень координации атомов углерода, и поверхности Ni(lll), и
Ni(llO), поскольку они исходно не обладают адсорбционными центрами с высокой степенью координации.
Рис. 5.9. Слева: СТМ-изображение
так называемой «часовой» реконструкция поверхности, которая
происходит при нанесении 0,5 монослоя атомов углерода на поверхность Ni(100). Справа: модель реконструкции, построенная из твердых сфер. Пунктирные окружности
показывают позиции атомов Ni до
реконструкции [4]
Атомы (молекулы) адсорбата могут притягиваться или отталкиваться на
поверхности. Если, например, молекулы притягиваются, то они стремятся формировать островки, число и размеры которых растут с увеличением степени
покрытия поверхности; при отталкивании адсорбированных атомов их распределение по поверхности является хаотическим при малых степенях заполнения, но они начинают формировать островки при высоких степенях заполнения, когда плотная упаковка становится неизбежной. Реконструкция поверхности, характер распределения атомов адсорбата на ней, природа адсорбционных
центров — все это было предметом исследования науки о поверхностях. В настоящее время накоплен большой объем данных (см., например, [5]) по типам
реконструкции поверхностей, природе адсорбционных центров и характеру
связывания атомов адсорбата с подложкой для самых разнообразных систем.
5.2.3. Оксиды и сульфиды
Объемная структура оксидов может быть описана наилучшим образом в предположении, что они образованы из положительных ионов металла
(катионов) и отрицательных ионов О (анионов). Основной особенностью таких структур является наличие окружения из атомов О у катионов, окружения
из катионов у атомов кислорода, так что строение объемной фазы определяется в основном стехиометрией оксидов. Практических во всех оксидах анионы
О2- имеют больший размер, чем катионы металлов. Ионы кислорода не существуют в изолированной форме, а всегда окружены положительными ионами
металла.
На рис. 5.10 показаны структуры внешних поверхностей для кубической
структуры MgO, соли горькозема. Наиболее устойчивой является поверхность
(100), так что у частиц MgO наблюдаются только фасетки (100). Заметим, что у
у 0., .Q |
. i Jv4 f/:(
lip'\ :-- ,jl
i - & \ . ,© ■ .4
[ y Ш '
fe 45
5.2. Структура металлов, оксидов, сульфидов и их поверхностей -J\r 205
этого кристалла имеются поверхности (111) двух типов: на одной из которых
находятся атомы магния, а на другой — атомы кислорода. У таких поверхностей полный дипольный момент отличен от нуля, поэтому они называются полярными. На поверхностях (100) и (110) имеется равное число атомов Mg и О,
так что они в целом нейтральны или неполярны.
(ill)
Рис. 5.10. Гипотетическая частица кубического кристалла MgO с тремя различными кристаллографическими поверхностями. Поверхности (100) и (110) —
неполярные, что предполагает наличие на них одинакового числа ионов Mg и О; полярная поверхность (111) может быть образована катионами Mg
или анионами О. На практике у кристалла MgO
внешней обычно является поверхность (100)
Мд
О
J
(001)
t
("110)
МдО,
структура кристалла горькозема
Катионы оксидов переходных металлов могут находиться в различной степени окисления. Типичным примером является оксид молибдена, которому
присущи степени окисления 6+, 5+ и 4+. Поверхности оксидов, у которых
катионы находятся в низкой степени окисления, обычно являются более реакционно-активными по сравнению с оксидами высокой степени окисления. Ионы
с низкой степенью окисления могут участвовать в реакциях, вызывающих изменение их валентного состояния.
Катионы на поверхности являются льюисовскими кислотными центрами,
то есть ведут себя как акцепторы электронов. Ионы кислорода служат акцепторами протонов, то есть являются бренстедовскими основаниями. Как мы увидим ниже, эти факторы являются принципиально важными для адсорбции.
Согласно концепции основности Бренстеда, соединение, способное связывать
протон, называется основанием, в то время как бренстедовскими кислотами
называются доноры протонов. По Льюису, кислотой называется соединение,
захватывающее электрон, а доноры электронов, такие как молекулы, обладающими парами одиночных электронов, являются основаниями. Следовательно,
льюисовское основание в принципе эквивалентно бренстедовскому основанию.
Понятия же кислот у Льюиса и Бренстеда различны.
Поверхности оксидов с плотной упаковкой, такие как поверхность MgO(lOO),
по большей части неактивны, но поверхностные дефекты, в частности, связанные с кислородными вакансиями, дают центры, с которыми атомы (молекулы)
адсорбата могут связываться достаточно прочно. На рис. 5.11 показан пример
адсорбции различных молекул на дефектах поверхности ТЮ2.
Многие газы адсорбируются только после гетеролитической диссоциации;
несколько примеров приведено на рис. 5.12. С поверхностями большинства
206
Глава 5. Твердые катализаторы
оксидов водород взаимодействует слабо. Если поверхность обладает высокой
реакционной способностью, происходит гидролитическое разделение атомов
водорода. Молекулы воды на поверхности практически любых оксидов легко
диссоциируют на ионы Н+ и ОН-. В результате формируются гидроксильные
группы двух типов, одна из которых является концевой и носит основный характер, а другая, кислотная, представляет собой мостиковую группу М—(ОН)—М.
В случае углеводородов или спиртов связи С—Н и О—Н разрываются легко,
а связи С—С являются наиболее трудно активируемыми. За более детальной
информацией мы отсылаем читателя к превосходной книге Генрича и Кокса [6].
Рис. 5.11. Поверхностные дефекты, связанные
с кислородными вакансиями, создают адсорбционные центры на поверхности Ti02( 110).
Заметим, что молекула СО со своей парой
одиночных электронов «садится» на ион Ti
(льюисовский кислотный центр). Молекула 02,
диссоциирующая на дефекте, поставляет один
атом кислорода, залечивающий дефект. Молекула С07 может адсорбироваться, координируя с атомом кислорода и образуя карбонатную группу [7]
Поверхность оксида: ДД^+ - - ДД^+ - Q&- _ ДД^+
Льюисовская Бренстедовское
кислота основание
акцептор акцептор
электронов протонов
Гетеролитическая +
адсорбция: ГП Гу
Н2 Мй+ - О6- - Мй+ - О6' - Md+
Н'
г ?
н2 О мй+ - Од- - Мй+ - О6' - Md+
Бренсте- Бренсте-
довская довское
кислота основание ,
^»»з Метокси-группа
С2Н5 Н НО
Сг Н6 ; СН3 ОН дд<з+ _ q<5- _ дд<5+ _ о<5- _ дд<5+
Рис. 5.12. Многие газы гидролитически диссоциируют на поверхности оксидов
5.2. Структура металлов, оксидов, сульфидов и их поверхностей 207
Сульфиды играют важную роль в каталитических процессах, связанных с
гидроочисткой. В то время как оксиды являются ионными кристаллами, в которых катионы и анионы предпочтительно окружают друг друга с целью уменьшения отталкивания между одинаково заряженными ионами, сульфиды представляют собой кристаллы с ковалентной связью. В них отсутствует отталкивание, препятствующее формированию связей между атомами серы, так что
структура сульфидов в общем случае существенным образом отличается от структуры оксидов.
Рис. 5.13. Структура двух «пластинок», вырезанных из кристалла
MoS2 типа 2Н—MoS2 (2Н означает
наличие двух слоев серы в элементарной ячейке); вид по направлению
[010]. На верхней пластинке имеются дефекты двух типов, связанных с серными вакансиями
Наиболее важными являются сульфиды MoS2 и WS2, обладающие слоистой
структурой. В них атомы металла занимают «дырки» в трехгранных призмах в
плоскостях, расположенных между слоями серы с гексагональной упаковкой
атомов (рис. 5.13). Слои атомов сдвинуты по отношению друг к другу, что
делает необходимым включение двух слоев MoS2 в элементарную ячейку. Такая структура обозначается как 2Н—MoS2. В катализе часто встречаются частицы, состоящие из одиночных слоев MoS2 и WS2, а также пакеты таких частиц. Реакционная способность сульфидов определяется краями слоев, базальная плоскость, образованная атомами серы, является в большинстве случаев
неактивной. Края слоев могут быть образованы из атомов серы или металла,
кроме того, они могут содержать серные вакансии; края двух типов показаны
на рис. 5.13.
5.2.4. Свободная энергия поверхности
Для создания поверхности требуется затратить энергию, поскольку
при этом необходимо произвести разрыв связей. Поэтому при формировании
из одного кусочка материала двух более мелких кусочков энергия системы возрастает. Поверхностная энергия всегда положительна.
208 -J\, Глава 5. Твердые катализаторы
Свободная энергия поверхности ^связана с энергией когезии твердого тела,
A#coh, и с числом связей между атомом и его ближайшими соседями, которые
необходимо разорвать для образования поверхности:
У = A-^coh ~2 N%i (5-3)
где Zs — число недостающих ближайших соседей у поверхностного атома; Z —
координационное число объемного атома; Ns — плотность атомов на поверхности.
Металлы обладают наиболее высокой поверхностной энергией, которая
лежит в пределах от 1,5 до 3 Дж/м2, для ионных же кристаллов и оксидов она
значительно ниже — от 0,2 до 0,5 Дж/м2. Углеводороды обладают наименьшей
поверхностной энергией — от 0,02 до 0,03 Дж/м2.
Выражения типа уравнения (5.3) составляют основу расчета поверхностной
энергии в рамках модели «разорванных связей». Используя типичные для ГЦК-
металлов значения (A#coh = 500 кДж/моль, Zs/Z= 0,25, Ns = 1015 атомов/см2),
приходим к поверхностной энергии, равной примерно 2 Дж/м2. Таким образом, поверхностная свободная энергия увеличивается и уменьшается вместе с
энергией когезии, что предполагает для переходных металлов с наполовину
заполненными J-связями наибольшую величину поверхностной свободной энергии; как мы увидим ниже, это подтверждается в эксперименте.
Движущей силой многих из поверхностных процессов и явлений является
стремление системы минимизировать свою поверхностную свободную энергию. Мы отметим некоторые из них.
• Поверхности всегда покрываются компонентами, понижающими поверхностную свободную энергию системы. Металлы обычно покрываются монослоями углеводородов, оксидов и очень часто водой (ОН-группами).
• У поликристаллических образцов чистого металла поверхность образована гранями с наибольшей плотностью упаковки атомов, поскольку при формировании такой поверхности требуется разорвать минимальное число связей
(см. рис. 5.4).
• Свободные поверхности типа (110) у ГЦК-кристаллов часто перестраиваются в структуры, у поверхностных атомов которых число ближайших соседей
является максимальным.
• В сплавах компонент, обладающий меньшей поверхностной энергией,
скапливается у поверхности (поверхностная сегрегация), что делает различными составы поверхностных слоев и объемной фазы (схема 5.1).
• Примеси в металлах типа С, О или S накапливаются у поверхности, поскольку они понижают поверхностную свободную энергию и, следовательно,
полную энергию системы.
• Малые металлические частицы, находящиеся на оксидированной подложке, при повышенных температурах спекаются, поскольку уменьшение площади поверхности приводит и к уменьшению энергии системы в целом. В то
же время значительная доля поверхности носителя-оксида остается свободной, поскольку его поверхностная энергия ниже поверхностной энергии метал¬
5.2. Структура металлов, оксидов, сульфидов и их поверхностей -Л-20
лов. Однако для оксидированных подложек поверхностная свободная энергия является движущей силой растекания по поверхности, если поверхностная энергия активной фазы оказывается ниже поверхностной энергии подложки.
Добавки
Объемная фаза
Ru, Ir
Со
Fe, Ru, Rh, Ir, Pt
Ni
Ni, Pt
Cu
Fe, Co
Rh
Fe, Co, Ni, Cu
Pd
Co, Ni, Cu, Rh, Pd, Pt
Ag
Fe, Co, Ru
Ir
Fe, Co, Ni, Cu, Ru, Rh
Pt
Fe, Co, Ni, Cu, Rh, Pd, Pt
Au
Добавки
Объемная фаза
Fe, Co
Cu
Ru, Rh, Ir
Pd
Fe, Rh, Ir
Ag
Ru, Ir
Au
Формирование сплава
Фазовый распад
Атомы добавок
растворяются
в объеме
ооооооооооооо
ООООООООООООО
ООООООООООООО
ШооШошоо
ООООООООООООО
ооооооооооооо
ооооооооооооо
ооооооооооооо
ооооооооооооо
оШШЖ
Атомы добавок
накапливаются
у поверхности
ооооооооооооо
QQOOOQCSIOOQQOQ
ооооооооооооо
ооооооооооооо
ооооооооооооо
оосюооооооооо
ооооооооооооо
ооооооооооооо
©ooodfeooooooo
ооооооооооооо
ооооооооооооо
Добавки
Объемная фаза
Co, Rh, Pd
Fe
Ag, Ir, Pt, Au
Fe
Rh, Pd, Ag, Pt, Au
Co
Cu, Pd, Ag, Au
Ni
Ag, Au
Cu
Fe, Co, Ni, Pt, Au
Ru
Ni, Pt
Rh
Au
Pd
Добавки
Объемная фаза
Ni, Cu
Fe
С
Co
Cu, Pd, Ag
Ru
Cu, Pd, Ag
Rh
Ag
Pd
Cu, Pd, Ag, Pt, Au
Ir
Ag, Au
Pt
Схема 5.1. Образование сплавов и сегрегация в бинарных металлических системах, когда
один из металлов служит малой добавкой. Схема количественно предсказывает, когда два элемента формируют поверхностный сплав или твердый раствор.
Результаты соответствуют металлам, находящимся в вакууме. При наличии
адсорбирующегося газа, поверхность будет обогащаться компонентом, образующим более сильные связи с адсорбатом [8]
210 ■J Г Глава 5. Твердые катализаторы
5.3. ХАРАКТЕРИСТИКИ МАЛЫХ ЧАСТИЦ
И ПОРИСТЫХ МАТЕРИАЛОВ
Катализ относится к поверхностным явлениям, поэтому эффективные катализаторы должны иметь большую площадь поверхности, что предполагает малый размер активных частиц. Основными характеристиками при
этом являются дисперсность частиц, то есть доля поверхностных атомов в частицах, и удельная поверхность, то есть поверхность, приходящаяся на единицу
массы. Малые металлические частицы часто являются неустойчивыми, склонными к спеканию, в частности при температурах, отвечающих типичным каталитическим реакциям. По этой причине большинство твердотельных катализаторов, используемых в промышленности, представляют собой частицы, распределенные в порах инертных носителей. Материалом носителей обычно служат
кремнезем, оксиды алюминия, титана, магния, цинка, циркония, а также активированный уголь. Морфология частиц опять же определяется поверхностной
энергией как частиц самих по себе, так и носителя.
5.3.1. Правило Вульфа
Как носитель влияет на морфологию находящихся на нем частиц? Какие грани выходят на поверхность монокристалла металла? Термодинамически наиболее устойчивые конфигурации малых кристаллов определяются поверхностными свободными энергиями их
граней, а также межфазной границы кристалл/подложка. Они могут быть определены по так называемому правилу Вульфа, которое мы проиллюстрируем на примере сечения системы частица/подложка (рис. 5.14).
• В полярной системе координат откладывается вектор, длина которого принимается пропорциональной поверхностной энергии y(i, j, к) для
грани (/, у, к), перпендикулярной этому вектору.
• На концах векторов проводятся плоскости,
ортогональные векторам.
• Внутренняя огибающая этих плоскостей определяет равновесную форму частиц.
Правило Вульфа предполагает, что расстояние от грани до центра кристаллита пропорционально поверхностной энергии этой грани, то есть hi ос уг Таким образом, если грань имеет низкую поверхностную энергию, то она будет
проходить близко к центу кристаллита и «обрезать» другие грани, становясь
тем самым доминирующей в многограннике.
У металлов плотноупакованные грани в общем случае характеризуются наименьшей поверхностной энергией, поэтому они являются преобладающими у
малых частиц; к ним относятся, например, грани (111) для ГЦК- и ГПУ-крис-
Рис. 5.14. Иллюстрация правила Вульфа на двумерном кристалле, у которого поверхностные
энергии граней расположены в
следующем порядке yt < ц < ук
5.3. Характеристики малых частиц и пористых материалов 211
таллов и грани (110) ОЦК-кристаллов, впрочем, на поверхности частиц железа
хорошо представлены грани (100). Поверхностные свободные энергии для различных материалов и граней табулированы в работе [9]. Для иллюстрации зависимости поверхностной энергии от кристаллографической ориентации граней приведем несколько ее значений для палладия:
уш = 1,920 Дж/м2, ут = 2,326 Дж/м2, уи0 = 2,225 Дж/м2.
\/) 0
Отсутствие
смачивания
Полное
смачивание
/ ,
г-л- .
Yi
1 / /?
i
/ h- "
/ u
v h )
/ Vl
Ah; = 2/?.
Рис. 5.15. Иллюстрация правила Вульфа на двумерной системе, показанной на рис. 5.14,
при помещении ее на поверхность оксида. Заметим, что металлическая частица
будет растекаться по поверхности при высоких значениях /? (то есть при образовании прочных связей с поверхностью)
Рис. 5.16. Слева: равновесные формы металлических частиц-многогранников на носителе при различных соотношениях поверхностных и межфазной энергий. Справа: электронно-микроскопические снимки структур, полученных при осаждении 5—10 МС Pd на поверхности MgO [11, 12]
В случае катализаторов на носителях частицы не являются свободными,
иначе бы они мгновенно образовывали большие агломераты, стремясь уменьшить свою свободную энергию. Оксидный носитель подавляет это процесс. По
212 ■J г Глава 5. Твердые катализаторы
этой причине форма частиц определяется также межфазной энергией р на границе металл/носитель. Здесь также можно использовать правило, подобное
сформулированному Вульфом: необходимо обрезать расстояние от центра до
межфазной границы, отвечающее свободной частице, на величину A/z,. = Ph.Jy^
где /?— энергия адгезии металла к поверхности носителя (рис. 5.15). На рис. 5.16
приведены примеры равновесных форм трехмерных кристаллов.
Имеются трудности в измерении межфазной энергии Д Только недавние
достаточно аккуратные измерения формы кристаллов позволили получить ее
надежные значения. Например, для малых частиц Pd на поверхности А1203
было получено значение 2,9 Дж/м2. Эта величина существенно выше поверхностной энергии грани Pd(lll). Это означает, что на подложке будут находиться частицы, имеющие грань (111) и не растекающиеся по поверхности. И действительно, данные сканирующей туннельной микроскопии показывают [10],
что грань (111) является доминирующей.
Следует принять во внимание, что на величину поверхностной энергии
сильное влияние оказывает присутствие адсорбатов, поэтому морфология частиц зависит от состава и давления окружающего их газа. Так что форма частиц,
находящихся в вакууме, может отличаться от формы частиц в каталитическом
реакторе. Безусловно, от природы окружения зависит и химическое состояние
частиц: например, адсорбат может окислять или восстанавливать частицы. Реакции подобного типа оказывают сильное влияние на величину поверхностной
энергии, вызывая растекание каталитических частиц по подложке или, наоборот, собирание растекшейся пленки в компактные частицы. Явления подобного типа наблюдались, например, в системе Cu/ZnO, используемой при синтезе
метанола.
5.3.2. Система пор
Носители типа кремнезема, оксида алюминия и активированного
угля являются пористыми и поэтому обладают большой площадью внутренней
поверхности. Система пор в носителях обычно считается нерегулярной: она
состоит из крупных пор, связанных с межкристаллитным пространством и имеющих диаметр порядка 100 нм, и микропор с характерным размером 5—10 нм.
Носитель считается хорошим, если он обладает:
• регулируемыми площадью поверхности и пористостью,
• высокой термостойкостью,
• высокой механической прочностью и износостойкостью.
Поскольку площадь поверхности и структура пор играют ключевую роль в
свойствах катализаторов и носителей, мы сначала рассмотрим методы их определения. Для начала мы проведем полезное разграничение шероховатости и пористости. Если на поверхности имеются впадины, глубина которых больше их
ширины, то мы будем называть такую поверхность пористой (рис. 5.17). Хотя
поры удобно представлять полыми цилиндрами, следует иметь в виду, что
форма пор может быть разнообразной. Система пор цеолитов образована
5.3. Характеристики малых частиц и пористых материалов -*\г 213
микропористыми каналами и полостями, а поры в кремнеземе — это пустоты между сферическими частицами. Оксид алюминия и активированный
уголь, напротив, имеют слоистую структуру, поэтому у них поры — щелевидные. Все носители имеют микро-, мезо- и макропоры (см. вставку с определениями).
Адсорбтив:
газ, который будет адсорбироваться
Адсорбент:
материал, на который адсорбируется газ
Адсорбат:
адсорбированный газ
Микропоры:
поры размером <2 нм
Мезопоры:
поры размером 2—50 нм
Макропоры:
поры размером > 50 нм
Шероховатость:
• глубина впадин меньше их ширины;
• имеется только внешняя поверхность
Пористость:
• глубина впадин больше их ширины;
• имеются внутренняя и внешняя поверхности
Рис. 5.17. Схематическая иллюстрация различия между шероховатостью и пористостью
5.3.3. Площадь поверхности
Физические принципы, лежащие в основе измерения площади
поверхности, достаточно просты: нужно физически адсорбировать инертный
газ типа аргона или азота и определить число молекул, необходимое для формирования сплошного монослоя. Например, молекула N2 при 77 К занимает
на поверхности площадь 0,162 нм2, площадь поверхности рассчитывается непосредственно. Процедура, на первый взгляд, выглядит простой, однако часть
молекул может адсорбироваться вне монослоя и формировать полислои. Кроме
того, молекулы могут конденсироваться в малых порах с образованием жидкой фазы. Так, чем уже поры, тем легче молекулы N2 конденсируются в них.
Это явление носит название капиллярной конденсации, и его можно использовать, привлекая уравнение Кельвина, для определения типа пор и их
распределения по размерам. Но сначала нам следует более подробно рассмотреть изотермы, описывающие физическую адсорбцию. Сейчас мы дадим вывод изотермы Брунауэра—Эммета—Теллера, которая обычно называется изотермой БЭТ.
214 -l\r Глава 5. Твердые катализаторы
Разобьем, как это показано на рис. 5.18, поверхность на участки, свободные от адсорбата (доля 0О), покрытые одним монослоем (0,), двумя монослоями (62) или / монослоями (0,).
$0 в\ в2 @3
Рис. 5.18. При выводе изотермы БЭТ
поверхность разбивается на участки,
покрытые / монослоями; на каждый
участок приходится доля поверхности в.
Предположим, что на поверхности имеется N0 центров, тогда число адсорбированных атомов можно представить в виде
Nt=N^i0„ (5.4)
/ = 0
где было использовано условие
(5.5)
/ = 0
Если мы примем, что такое состояние является равновесным при температуре Г, то скорость адсорбции газа должна быть равна скорости его десорбции. Поскольку атомы (молекулы) адсорбированы физически и находятся в
слабом потенциальном поле, то можно считать, что энергетический барьер
отсутствует, а коэффициент прилипания (вероятность адсорбции молекулы
при ее ударе о поверхность) равен единице. Последнее не совсем правильно,
поскольку имеется энтропийный барьер прямой адсорбции из газовой фазы
на специфическом центре. Однако введение малого коэффициента прилипания не изменяет глобальные характеристики модели. Таким образом, в равновесии мы имеем
^ = Л?,_, - уе<!к°Те, - Fe, + ve<"!k'TeM =
= FOi-i - k,e, - F6j + кмвм = О,
где F — падающий на центр поток атомов:
г- f .
N0p7rm0kBT)
Ед — энергия десорбции атома со слоя /. Заметим, что поскольку природа
подложки формально изменяется при переходе от первого ко второму слою,
величины Е\ и ЕI могут различаться между собой. Однако для / = 2 и выше
можно рассматривать десорбцию как сублимацию полислойной пленки атомов, что позволяет положить Еj = Eld>2 и принять кх Ф к2 = к3 = ... = к^.
crrrrrf
5.3. Характеристики малых частиц и пористых материалов
X
Записывая уравнения скоростей реакций для адсорбционно-десорбцион-
ного равновесия в каждом слое, получаем
d0n Л „
dt
dfl,
"dТ
dex
~df
Ji +1
F Л
V0’
= F2 в° ,
k\ '
kx k2
-U
_ pl + l eo
k;
k\ (k2 )'
(5.6)
Полная степень покрытия поверхности, вг равна
N,
О /=1
г ж, v k k - '
Ъ = тв^'1
/=1
2 /
2)
р!кг
К °(i -F/k2y
(5-7)
где было использовано соотношение
V1 * i Х
У IX =
(i-х)2
/ = 0
Мы выразили полную степень покрытия поверхности через константы скорости и поток падающих атомов, но нам необходимо еще найти 0О:
,=1=*0+2*,=0„+:£тЧ
/=0 (=1 1=1 К1
= ва
' F'
с2 J
к 00
i+—X
/
= #0
Г, . k2 F/k2 1
U
Jfc, 1 - F/k2
(5.8)
Используя соотношение
1-х’
приводим 0О к виду
(5.9)
1 +
к2 F/k2
-1.
(5.10)
в, =
к{ 1 - F/k2
Подстановка этого выражения в (5.7) дает следующее выражение для 6t\
(k2/kt)(F/k2) Z(P/P0)
[l-F/k2 +{k2/kt)(F/k2)]{\-F/k2) [l-P/P0+Z(P/P0)]{l-P/P0)
,(5.11)
215
216
—Глава 5. Твердые катализаторы
где х = ^2/^1 — отношение констант десорбции для первого и второго слоя и
P_=F^ Р
Р0 к2
k2NQ^2nmkBT
есть отношение скоростей адсорбции и десорбции со второго и более высоких
слоев. При (Р/Рq) -> 1 полная степень покрытия поверхности 6t -> °°, поскольку
адсорбат конденсируется на поверхности с образованием полислойной пленки.
Это отвечает ситуации, когда падающий поток равен скорости десорбции с
полислойной пленки, при этом Р0 есть просто равновесное давление сконденсированного на поверхности газа, величина — приведенная в справочниках.
В реальных экспериментах образец с неизвестной площадью поверхности
помещается в небольшой объем и охлаждается (до температуры 75 К, если
используется N2). Равновесное давление (Р0) газа N2 при 75 К составляет
750 мбар. Проводится измерение количества адсорбированного газа как функции давления, а затем пересчитывается на степень заполнения монослоя
Ot =
Na PVJRT Va
N0 PVJRT V0
(5.12)
Подставляя это выражение в формулу (5.11), получаем после несложных преобразований
1 х-i Р Р
+ ^-rz—— = rj + а —
К (А ~ Р) хУ0 xv, р,
(5.13)
Теперь, если построить зависимость величины Р/[ Va(PQ — Р)] от отношения
Р/Р0, получим прямую линию, тангенс угла наклона которой равен а= (х~ 1)/х^
а координата точки пересечения с осью у равна rj = l/xV0- При этом объем газа,
заполняющего монослой, равен V0 = l/(x + rf). По этому объему можно определить число адсорбируемых в монослое молекул N0 = PVQ/kQT, и если нам
известна площадь, занимаемая одной молекулой, то полная площадь находится легко: А = А^40.
Пример: Применение метода БЭТ
В табл. 5.1 приведены данные по величинам объемов газа, адсорбированных 0,2 г катализатора.
Таблица 5.1. Давления и соответствующие значения объема N2,
адсорбированного на 0,2 г катализатора
Р, мбар
1,6
18
61
168
270
V, мкл, станд. уел.
1200
1440
1640
2100
2500
На рис. 5.19 построена зависимость, соответствующая данным табл. 5.1, в
координатах уравнения (5.13). Найденное по наклону прямой и точке ее пере¬
5.3. Характеристики малых частиц и пористых материалов -1\г 217
сечения с осью ординат значение объема газа, заполняющего монослой, равно
1616 мкл. Поскольку каждая молекула N2 занимает площадь 0,16 нм2, то полная площадь поверхности катализатора равна 6,4 м2. Поскольку вес образца
был равен 0,2 г, то удельная поверхность составляет 32 м2/г.
0,0002
CL
I
of
^ 0,0001
Рис. 5.19. Представление данных 0,0000
табл. 5.1 в координатах БЭТ, ис- 0 4
пользуемых для нахождения а и г]
В рассмотренном примере адсорбция в первом слое является доминирующей, поскольку скорость десорбции из второго слоя существенно больше скорости десорбции из первого слоя: k2/kx ~ 370. В табл. 5.2 приведены значения
степеней заполнения поверхности при давлении 61 мбар, из которой видно, что
90 % занятой поверхности приходится на монослой и лишь 8 % — на полислои.
Таблица 5.2. Доли поверхности адсорбента, приходящиеся на свободную
поверхность, монослой, два, три и четыре слоя молекул N2,
для системы, охарактеризованной в табл. 5.1 и на рис. 5.19,
при давлении 61 мбар
во
02
*3
04
0,030
0,892
0,072
0,0059
0,0005
В методе БЭТ, безусловно, есть свои ограничения, и существуют различные модификации этого метода, однако их рассмотрение выходит за рамки
данной книги. Мы лишь отметим, что изотерма БЭТ получена при следующих
допущениях:
• имеется динамическое равновесие между адсорбатом и адсорбтивом: скорости адсорбции и десорбции равны для любого слоя;
• в первом слое молекулы адсорбируются на однотипные адсорбционные
центры;
• молекулы первого слоя представляют собой центры адсорбции для второго слоя и т. д. для слоев более высокого порядка;
• взаимодействие адсорбат/адсорбат в расчет не принимается;
• для всех слоев, за исключением первого, условия адсорбции/десорбции
тождественны;
218 ■J г Глава 5. Твердые катализаторы
• энергия адсорбции второго и более высоких слоев равна энергии конденсации;
• толщина адсорбционного слоя становится бесконечной при давлении насыщения (Р = Р0).
Для металлических катализаторов на носителях метод БЭТ дает полную
площадь поверхности носителя и катализатора. При проведении измерений в
условиях, когда имеет место хемосорбция, например для Н2 и СО при комнатной температуре, адсорбция идет только на металле и это позволяет определить его дисперсность.
Площадь поверхности катализатора или носителя, определяемая по методу
БЭТ, одна из основных характеристик, интересующих исследователей, занимающихся катализом. В промышленных лабораториях, а также во многих академических лабораториях имеются установки, позволяющие проводить такие измерения.
На рис. 5.20 показана идеализированная форма изотермы, отвечающей
физической адсорбции на нанопористом или микропористом твердом теле. При
низких давлениях поверхность лишь частично заполнена молекулами газа, при
более высоких давлениях монослой заполняется (точка В на кривой), и изотерма выходит на плато. Часть изотермы от нулевого давления до точки В эквивалентна изотерме Ленгмюра. При более высоких давлениях начинает формироваться второй слой, после чего толщина адсорбционного слоя растет неограниченно, что эквивалентно конденсации, то есть формированию жидкой пленки.
По терминологии, принятой для описания физической адсорбции и одобренной ИЮПАК, такие изотермы относятся к типу II. Если адсорбент имеет преимущественно микропоры, например цеолиты или активированный уголь со
сверхвысокой удельной поверхностью (>1000 м2/г), толщина жидкой пленки
будет ограничена размером пор.
Рис. 5.20. Схематическое представление изотермы БЭТ типа И, характерной для нанопористых порошков
В этом случае вступает в игру процесс капиллярной конденсации, и адсорбция на неограниченном пористом теле описывается изотермой типа I, представленной на рис. 5.21. Высота плато при этом является мерой объема микро-
пор. При адсорбции на мезопористых материалах сначала наблюдается запол¬
5.4. Носители катализаторов -i\r 219
нение монослоя при относительно низких давлениях, как это описывается изотермой типа II, затем идет формирование многослойной пленки вплоть до капиллярной конденсации, устанавливающей предел количеству адсорбированного в данном материале газа. При удалении адсорбата из пор наблюдается
гистерезис: газ начинает выходить из пористого тела при более низких равновесных давлениях по сравнению с процессом заполнения пор, что обусловлено
необходимостью преодоления капиллярных сил. Соответствующая этой ситуации изотерма типа IV показана на рис. 5.22; она характерна для носителей на
основе кремнезема и оксида алюминия, имеющих удельную поверхность в несколько сот квадратных метров на грамм.
Рис. 5.21. Схематическое представление изотермы БЭТ типа I, характерной для цеолитов
7 J
ft
' Запол¬
Монослой
нение
пор
Точка Б
■ Субмонослой
Изотерма типа IV
Капиллярная
конденсация
(все микропоры
заполнены)
Изотерма типа I
Р/Р0
р/р0
Рис. 5.22. Схематическое представление
изотермы БЭТ типа IV, характерной для
носителей из кремнезема и оксида алюминия
5.4. НОСИТЕЛИ КАТАЛИЗАТОРОВ
Малые металлические частицы часто являются нестабильными и
склонны к спеканию при температурах, типичных для многих каталитических
процессов. По этой причине катализаторы, используемые в промышленности,
представляют собой относительно малые частицы, спекание которых подавляется тем или иным способом. Для этой цели используются различного рода
стабилизаторы структуры или частицы вводятся в тонкие поры инертного носителя. В качестве носителей могут использоваться любые материалы, являющиеся термически устойчивыми и химически инертными, но наиболее часто в
качестве носителей используются оксид алюминия, кремнезем, активированный уголь, а в отдельных случаях — оксиды магния, титана, циркония и цинка,
220
Глава 5. Твердые катализаторы
карбид кремния и цеолиты. В специальных случаях, когда требуется нестесненное течение реагирующих газов, в качестве носителей используются монолиты. В табл. 5.3 дан перечень носителей, используемых для различных каталитических реакций.
Таблица 5.3. Примеры носителей, используемых в некоторых каталитических
реакциях
Носитель
Каталитически активная фаза
Применение
CoMoS, NiMoS, NiWS
Гидроочистка
Pt, Pt—Re
Риформинг
Pt, Rh, Pd
Очистка выхлопных газов
Cu—ZnO
Синтез метанола
Cu—ZnO
Реакция конверсии
водяного газа
/-Оксид алюминия,
Ni
Конверсия
А12о3
с водяным паром
Ti02
Дегидратация
Pd, Pt, Ru, Rh
Г идрирование
Cr203, Pt
Дегидрирование
Pd
Дегидрохлорирование
CuCl2
Оксихлорирование
//-Оксид алюминия
Pt
Риформинг, реакции
изомеризации
яг-Оксид алюминия
Ni
Конверсия с водяным паром
Ag
Эпоксидирование
ею
Полимеризация
Кремнезем, Si02
H3P04
Г идратация
v2o5
Оксидирование
Оксид титана, ТЮ2
v2o5
Удаление NOx
Активированный
уголь
Pd, Pt
Гидрирование
5.4. Носители катализаторов ->\г 221
5.4.1. Кремнезем
Кремнезем является лучшим носителем катализаторов для процессов, протекающих при относительно низких температурах (ниже 300 °С), таких
как гидрирование, полимеризация и оксидирование. Его параметры, такие как
размер пор, частиц, удельная площадь поверхности, могут быть легко изменены с учетом специфических требований для конкретного процесса. В сравнении с оксидом алюминия кремнезем обладает меньшей термической стабильностью, и его склонность к образованию летучих гидроксидов в атмосфере
водяного пара при повышенных температурах также ограничивает возможности его применения в качестве носителя. Большинство кремнеземных носителей получают двумя основными способами: «золь—гель»-методом, при котором формируется ксерогель, и гидролизом в пламени, когда образуется так
называемый пирогенный кремнезем.
В «золь—гель»-методе щелочной (pH = 12) раствор силиката натрия, называемый жидким стеклом, смешивается с серной кислотой. При низком значении pH силикат-ионы превращаются в мономерные звенья Si(OH)4, которые
полимеризуются с образованием коллоидных частиц. Частицы агрегируют,
формируя трехмерную сетку из сфер — гидрогель. Сушка гидрогеля при повышенных температурах (150—200 °С) приводит к образованию так называемого
ксерогеля кремнезема — высокопористой сетки из кремнеземных сфер, способной поглощать воду из окружающей атмосферы в количестве 5—10 % от
собственного веса. Ксерогели не обладают необходимой прочностью, и у них
нет макропор, необходимых для быстрого транспорта газа в реакторе при умеренных перепадах давления. Поэтому этот материал обычно размалывают в
порошок с заданным размером частиц, из которого с помощью связующего
делают шарики или стерженьки. Структура пор при этом изначально определяется размером коллоидных кремнеземных сфер и режимом формирования из
них гидрогеля, но она может быть изменена добавлением кислоты или основания на стадии промывки гидрогеля.
Второй способ получения кремнеземных носителей, связанный с пламенным гидролизом, широко применяется при получении материалов из оксидов
с высокой удельной поверхностью. Преимущества пирогенного кремнезема по
сравнению с ксерогелями состоит в его более высоких механической прочности и чистоте. Исходным материалом для получения пирогенного кремнезема
служит SiCl4, парами которого насыщают воздух, затем смешивают его с водородом и подают в пламенный реактор, где смесь реагирует по сложной схеме с
образованием аэрозоля наноразмерных частиц Si02 и НС1. Последняя удаляется с поверхности кремнеземных частиц с помощью пара и воздуха в кипящем
слое. Полученный таким путем кремнезем обладает низкой плотностью и его
надо компактировать перед формированием из него крупных сфер. Размер первичных частиц и агрегатов можно регулировать разными способами — изменением температуры пламени, соотношения Н9/02 или содержания SiCl4 в исход-
222
\
Глава 5. Твердые катализаторы
ной смеси или времени нахождения ее в реакторе. На рис. 5.23 показана схема
потоков процесса, а в табл. 5.4 приведены некоторые свойства носителей из
пирогенного кремнезема.
SiCl4 —Ц Испаритель]
Воздух —>| СмесителТ]—^
Н2
HCI
HCI
SiCI4 + 2Н2 + 02
I
Рис. 5.23. Схема производства
кремнезема [13]
Si02
Кремнеземы с удельной площадью
поверхности до 300 м2/г образованы
плотными частицами диаметром 7 нм и
выше и не содержат микропор. Пиро-
генный кремнезем производят несколько компаний, наилучшими считаются
кремнеземы марок Aerosil® компании
Degussa и Cabosil® компании Cabot.
Рис. 5.24. Поверхность кристобалита часто используется как структурная модель поверхности кремнезема
©Sin
ОН
\
о }
Таким образом, носители из кремнезема являются аморфным материалом,
поверхность которого может обладать определенной степенью порядка, присущей, например, минералу /?-кристобалиту (рис. 5.24). На поверхности носителя из кремнезема имеются ОН-группы с плотностью примерно 4—5,5 групп на
квадратный нанометр (для кристобалита плотность поверхностных ОН-групп
равна 4,55 групп на квадратный нанометр). На поверхности кремнезема имеются только концевые ОН-группы, связанные с одним атомом Si. Нагрев приводит к дегидроксилированию поверхности, на которой остаются только отдельные ОН-группы.
5.4. Носители катализаторов jv 223
Таблица 5.4. Свойства формованного носителя из пирогенного кремнезема [13]
Удельная площадь поверхности
исходного оксида, м2/г
50
130
200
300
Удельная площадь поверхности
прессованного материала, м2Д
45
110
160
230
Удельный объем пор, м3/г
0,6
0,8
0,75
0,8
Наименьший диаметр пор, нм
8
7
6
4
Диапазон размеров пор,
охватывающий 90 % их объема
10-40
10-40
10-30
7-25
Твердость, Н
125
60
60
80
5.4.2. Оксид алюминия
Наиболее широко используемым носителем катализаторов является оксид алюминия, что связано с его высокой термической и механической
устойчивостью и многообразием химических свойств. Имеется большое число
структурных модификаций оксида алюминия, однако особый интерес вызывают только три фазы — кристаллографически упорядоченный а-А1203 с нанопо-
ристой структурой и аморфные Т]- и /-А1203 с пористой структурой. При этом
/-А1203 сам по себе является катализатором, например, процессов выделения
серы из H2S (процесс Клауса), алкилирования фенола или дегидратации муравьиной кислоты.
Носители из оксида алюминия получают путем термической дегидратации
А1(ОН)3 (гиббсит или байерит) или А100Н (бёмит) (см. схему на рис. 5.25).
Оксид алюминия распространен в природе в форме боксита (назван по местности Jle-Бо, Франция), руды, состоящей из гидроксида алюминия, кремнезема и других оксидов. В зависимости от расположения месторождения боксита
он может содержать преимущественно гиббсит (Суринам, Урал), бёмит (Франция) или диаспор (Балканы). Алюминий извлекают из руды путем ее обработки
NaOH, что дает раствор Na2Al204, из которого гиббсит выделяют кристаллизацией. Получаемый в таком процессе гиббсит всегда содержит щелочь, присутствие которой может быть нежелательным для каталитических процессов. По
этой причине оксиды алюминия, не содержащие примеси щелочи, получают
гидролизом алкоголятов алюминия, например, А1(ОС2Н5)3, в результате которого получают гелеобразный бёмит, иногда называемый псевдобёмитом. В другом способе исходным материалом служит байерит, получаемый осаждением
А1(ОН)3 из растворов солей алюминия, стимулированным добавками аммиака.
Байерит образуется при высоких значениях pH (около 12), а бёмит при нейтральных pH.
224
Глава 5. Твердые катализаторы
О 250 500 750 1000 1250
J I I I I I
О 250 500 750 1000 1250
Температура, °С
Рис. 5.25. Оксид алюминия существует в разнообразных формах. В качестве носителя
наиболее часто используется у-А\103
Оксид алюминия существует в различных формах, из которых только а-А1203
является кристаллическим. Другие формы, называемые переходными, имеют
структуру типа шпинели, в которой слои расположены в разном порядке.
Например, в структуре шпинели MgAl204 ионы кислорода распределены по
ГЦК-решетке, Mg занимает тетраэдрические, а А1 — октаэдрические центры.
Поскольку стехиометрия А1203 не полностью соответствует стехиометрии шпинели, то решетка переходных оксидов алюминия является дефицитной по катионам и содержит много дефектов. Тем не менее их дифрактограммы явно
указывают на структуру шпинели.
Используемый в качестве носителей /-А1203 обладает высокой удельной
поверхностью (50—300 м2/г), мезопорами размером от 5 до 15 нм, удельным
объемом пор около 0,6 см3/г, высокой термической стабильностью и легко
формуется с помощью экструдера в стерженьки и шарики. Термическая стабильность /-А1203 может быть существенно повышена путем введения добавок
оксида лантана, которые снижают скорость спекания и замедляют процесс трансформации /-А1203 в другие фазы. На поверхности /-А1203 имеются различные
гидроксильные группы с концентрацией от 10 до 15 ОН-групп на квадратный
нанометр, из которых линейные группы представляют собой бренстедовские ос¬
5.4. Носители катализаторов
нования (акцепторы Н+), а мостиковые — бренстедовские кислоты (доноры Н+).
После дегидроксилирования поверхность /-А1203 приобретает льюисовскую кислотность на некоординированных Als+ центрах (акцепторах электронов). В то
время как кристобалит представляет реалистическую модель поверхности носителя — кремнезема, для оксида алюминия нет аналогичной подходящей структуры. Семейство гидроксильных групп достаточно пестро: имеются линейные,
а также мостиковые с двойной и тройной координацией группы, которые легко идентифицируются методами спектроскопии. Оксид алюминия в растворах
является полианионом с положительным зарядом поверхности при pH <7 и
приобретает отрицательный заряд при более высоких значениях pH, что предопределяет возможность связывания с ним различных заряженных прекурсоров катализаторов.
Обладающий высокой термической стабильностью а-А1203 используется в
высокотемпературных процессах типа конверсии с водяным паром или в тех
случаях, когда желательна низкая удельная поверхность носителя.
5.4.3. Углеродные носители
Пористые углеродные материалы используются как носители катализаторов из благородных металлов в реакциях гидрирования органических
соединений, которые обычно осуществляются в жидких средах. Активированные угли, содержащие на своей поверхности функциональные группы,
обычно получаются пиролизом газов древесины, каменного угля, скорлупы
кокосовых орехов и т. д., который проводится в атмосфере инертных газов,
С02 или пара при температурах от 800 до 1500 °С. Активирование поверхности осуществляется путем обработки полученных пористых материалов в атмосфере кислорода. При этом достигается высокая удельная площадь поверхности до 1500 м2/г, а поры имеют размер меньше 1 нм. Обработка при более
высоких температурах приводит к формированию графитированного углерода, обладающего более низкой удельной поверхностью. Преимущество углеродных носителей предопределяется их относительно высокой химической
стойкостью и достаточно легкой процедурой извлечения дорогостоящих благородных металлов из отработанных катализаторов. Углеродные материалы
успешно применяются в качестве носителей для катализаторов из благородных металлов, используемых для проведения низкотемпературных реакций
гидрирования в жидких средах.
5.4.4. Формование носителей катализаторов
225
В описанных выше схемах получения носителей образуются порошки из малых частиц размером от нескольких микронов до нескольких миллиметров. Их использование в реакторах длиной несколько метров при фикси¬
226 -l\r Глава 5. Твердые катализаторы
рованном слое катализатора будет приводить к огромным перепадам давления
на слое. Перевод слоя в псевдоожиженное состояние приведет к выносу носителя из реактора. Следовательно, для использования катализаторов в реакторах
необходимо сформировать из них более крупные тела, обладающие достаточной механической прочностью (рис. 5.26). Уплотнение порошка дает еще один
полезный эффект — повышение удельной (на объем) активности слоя катализатора. Как правило, плотность катализатора увеличивается втрое при формовании из него таблеток. Формование носителя можно осуществлять до и после
его заполнения (путем пропитки растворами соответствующих солей и последующей сушки) активным материалом.
Рис. 5.26. Примеры формованных
катализаторов
Форма каталитических гранул, как будет видно из нижеследующего примера, определяет скорость переноса массы и перепад давления на слое катализатора. Сравнение цилиндрических стерженьков с дисками меньших длины и
диаметра показывает, что в случае дисков перепад давления меньше на 50 %, а
площадь контакта катализатора с реагирующим газом на 20 % больше, чем в
случае стерженьков.
Стерженьки или таблетки (диаметром 1,5—10 мм), диски (диаметром 6—20 мм)
и стерженьки с множественными каналами (диаметром 10—40 мм и длиной
10—20 мм) используются в тех случаях, когда требуется высокая механическая
прочность. Такие системы получаются прессованием смеси носителя со связующими (каолиновая глина, стеариновая кислота) и смазками (графит).
Во многих случаях носители прессуются в обычные цилиндры (диаметром
1—5 мм и длиной 10—20 мм) в обычном экструдере. Порошок носителя смешивается со связующим и водой, в результате чего образуется паста, которую
продавливают через тонкие отверстия желаемого размера и формы. Паста должна быть достаточно густой, чтобы полоски экструдированного материала сохраняли свою форму в процессе сушки и усадки. Высушенный материал разрезается на кусочки требуемой длины. Экструзия используется и при получении
керамических монолитных образцов типа используемых в катализе выхлопных
газов и в реакторах, в которых осуществляется удаление NOx.
5.5. Получение нанесенных катализаторов 227
5.5. ПОЛУЧЕНИЕ НАНЕСЕННЫХ КАТАЛИЗАТОРОВ
Имеется два основных способа получения нанесенных катализаторов:
1. Соосаждение каталитически активного компонента и носителя с последующими сушкой, кальцинированием (нагревом на воздухе) и восстановлением, что приводит к формированию пористого материала, обладающего высокой удельной поверхностью. Эта процедура используется в случае дешевых
материалов и при необходимости достижения оптимальной каталитической
активности на единицу объема катализатора.
2. Введение в предварительно сформированный и формованный носитель
активной фазы путем пропитки его соответствующими растворами или осаждением из растворов. Такая процедура используется в случае дорогостоящих
прекурсоров и при необходимости получения каталитически активной фазы в
виде наноразмерных частиц на носителях. Таким путем получаются катализаторы из благородных металлов.
Обсудим иллюстративные примеры обоих методов.
5.5.1. Соосаждение
В этом методе используются растворы солей каталитически активного материала и носителя, к которым добавляется осаждающий агент типа
NaOH или NaHC03. В результате происходит осаждение гидроксидов или основных солей с образованием однородной смеси, которую затем отфильтровывают. Удаление С02 и воды в процессе сушки и кальцинирования, а затем и
кислорода в процессе восстановления приводит к образованию пористого катализатора. Этим процессом, однако, трудно управлять: важно поддерживать
однородность раствора, обеспечивающую одновременность осаждения компонентов, и постоянство pH. Примерами катализаторов, получаемых соосажде-
нием, являются системы Ni/Al903 и Си — Zn/Al203, используемые при синтезе
метанола.
5.5.2. Импрегнация, адсорбция, ионный обмен
Прямой путь введения в носитель активного материала состоит в
заполнении пор раствором каталитически активного элемента и удалении растворителя сушкой. Следует иметь в виду, что в этом процессе важную роль
играет взаимодействие растворенного прекурсора с поверхностью носителя,
которое может быть использовано для эффективного распределения активного
компонента по носителю. Чтобы понять важность этого взаимодействия, необходимо более детально ознакомиться с химией поверхности гидроксилирован-
ных оксидов в растворах.
Носители из кремнезема и оксида алюминия имеют на своей поверхности
гидроксильные группы различных типов, основные из которых показаны на
228 -l\r Глава 5. Твердые катализаторы
рис. 5.27. Гидроксильные группы играют важную роль в получении катализаторов, поскольку они являются центрами на поверхности, к которым прекурсор
катализатора может прикрепиться. В то время как на поверхности кремнезема
гидроксильные группы химически неразличимы, химическая природа гидроксильных групп на поверхности оксида алюминия характеризуется большим
разнообразием. Линейные ОН-группы на поверхности оксида алюминия имеют анионный (основный) характер, на что указывает возможность их замещения ионами галогенов типа СГ и F".
Н.
ОН
I
Si
/ \
Изолированные
ОН
I
AI
/ \
Линейные
(основные)
Н Н
О О
\ /
Si
/ \
Двойные
Н
О
/ \
— AI AI —
Мостиковые
(кислотные)
/
0
1
Si
/ \ /
/
н
Si
\
Вицинальные
(с водородной связью)
Н
/?\
— AI / AI —
I Al I
/ \
Мостиковые
(кислотные)
Рис. 5.27. Примеры гидроксильных групп, присутствующих на поверхности кремнезема и оксида алюминия
В воде поверхностные гидроксильные группы вступают в реакцию с протонами и ОН_-группами, что приводит к зарядке поверхности. Реакционное равновесие может быть описано следующим образом:
М-ОН + Н+ = М — ОН2
М-ОН + ОН" = М-СГ + Н20
для основных центров, (5.14)
для кислотных центров. (5.15)
На заряд поверхности влияют pH раствора и положение изоэлектрической
точки оксида — того значения pH раствора, при котором поверхность является
нейтральной. Поверхность несет отрицательный заряд при pH ниже изоэлектрической точки и положительный заряд — выше нее. Очевидно, что зарядовое
состояние поверхности позволяет «привязать» прекурсор катализатора к ионному центру, несущему противоположный прекурсору заряд.
Например, каталитическая система Pt/Si02 обычно формируется путем
пропитки носителя из кремнезема основным (pH = 8—9) раствором, содержащим ионы тетраамина платины Pt(NH3)4+ (в раствор вводится соответствующий хлорид). Поскольку поверхность кремнезема заряжена отрицательно,
содержащие Pt ионы прикрепляются к группам SiO- и распределяются по
поверхности. Значение pH должно быть ниже 9, иначе кремнезем будет растворяться.
5.5. Получение нанесенных катализаторов -*\г 229
Для получения катализатора типа Pt/Al203 используются отрицательно заряженные ионы, содержащие платину. В водном растворе гексахлорплатино-
вая кислота диссоциирует на ионы Н+ и PtCl^ , последние легко вступают в
реакцию с основными ОН-группами. Если процесс получения катализатора
осуществляется на предварительно формованных частицах оксида алюминия,
то Pt будет преимущественно осаждаться на их поверхности, формируя так
называемый катализатор типа «яичной скорлупы». Поскольку основные группы ОН также легко замещаются другими анионами, можно использовать конкурентную адсорбцию для регулирования глубины, на которой платина осаждается в носителе. В присутствии кислот, более прочно связывающихся с поверхностью, чем гексахлорплатиновая кислота, например щавелевой или
уксусной кислоты, можно вообще подавить адсорбцию платины на внешней
поверхности носителя, как это показано на рис. 5.28. Катализаторы, в которых
активный материал находится на определенной глубине, обладают определенными преимуществами в тех случаях, когда рабочий поток газа или жидкости
содержит хорошо адсорбирующиеся элементы, отравляющие катализатор; примером является свинец в выхлопных газах двигателей внутреннего сгорания.
Рис. 5.28. Примеры внедрения во внешние слои материала носителя драгоценных
металлов
PtClg"
он он
I I
AI AI
/ \ / \
«Яичная скорлупа»
Pt/AIA
/ \2-
Cl CI
\ /
CI— Pt—Cl
/ \
0 о
1 I
AI AI
/ \ / \
+ 2HCI
«Яичный желток»
В неполярных растворителях, например спиртах, поверхностные гидроксильные группы служат центрами связывания алкоксисоединений с поверхностью
в реакциях конденсации, в которых один алкоксилиганд реагирует с протоном
поверхностной ОН-группы, что приводит к образованию соответствующего
спирта и комплекса, прикрепленного к поверхности. Примером служит процедура связывания этоксида циркония Zr(OC2H5) к поверхности кремнезема посредством реакции
Zr(OC2H5)4 + Si—ОН Si—О—Zr(OC2H5)3 + С2Н5ОН,
в результате которой на поверхности носителя образуется высокодисперсный Zr.
Затем используется кальцинирование на воздухе для удаления этоксилигандов
и превращения циркония в Zr02 [14].
230
Глава 5. Твердые катализаторы
5.5.3. Осаждение с отложением
Сущность этого метода состоит в том, что малые кристаллиты гидроксидов или карбонатов металлов осаждаются из растворов преимущественно
за счет гетерогенной нуклеации на поверхности раздела раствор/носитель. При
этом порошок носителя суспендируется в растворе соли металла, к которому
добавляется основание с целью повышения значения pH. Поскольку значение pH
должно быть постоянным во всем растворе, требуется его эффективное перемешивание, которое, однако, приводит к ряду проблем в случае реакторов больших объемов, а также пористых систем. Искусная процедура состоит в добавлении в раствор мочевины CO(NH2) [15]. После тщательного перемешивания
раствора температуру повышают до 70—90 °С, что приводит к гидролизу мочевины согласно реакции
CO(NH2), +ЗН20 -> 2NH; +С02 +20Н", (5.16)
в результате которой pH повышается равномерно во всем объеме раствора, и
при некотором его значении начинается процесс нуклеации на поверхности
носителя. Этот метод используется для получения металлических катализаторов (частиц ванадия, молибдена, магния, железа, никеля и меди), равномерно
распределенных по внутренней поверхности носителя. После осаждения твердая фаза отфильтровывается, промывается, сушится и формуется. Наконец,
катализаторы кальцинируют и далее активируют путем восстановления или
сульфидирования (см. ниже).
5.6. КАТАЛИЗАТОРЫ БЕЗ НОСИТЕЛЕЙ
Многокомпонентные катализаторы, в частности, представляющие
собой смесь оксидов, могут быть получены путем сплавления частиц в электрической дуге. Однако этот метод пригоден только для оксидов, способных
проводить электрический ток при высоких температурах. Разнообразные оксиды, например оксиды железа (магнетит, Fe304), алюминия, калия, используемые при синтезе аммиака, хорошо смешиваются в расплавах. После охлаждения сплава образец дробят, и частицы нужного размера отсеивают. Более крупные частицы подвергаются дальнейшему дроблению, а мелкие частицы подаются
в печь. Оплавленные частицы катализатора являются непористыми, однако в
них образуются поры в процессе восстановления элементов. Например, катализатор, используемый для синтеза аммиака, состоит преимущественно из Fe304,
содержит около 1 % К90 и 2,5 % А1203. Он является примером катализатора с
двойной активацией, поскольку его электронная система активирована калием, а кристаллическая структура — оксидом алюминия. Тройную активацию
этот катализатор получает при добавлении СаО. В процессе активации в потоке синтез-газа кислород удаляется из Fe304 в форме воды, в то время как металлы других оксидов не поддаются такому легкому восстановлению. В результате получается пористая структура из металлического железа, являющаяся
активной частью катализатора, используемого в синтезе аммиака.
5.7. Цеолиты -J\r 231
5.7. ЦЕОЛИТЫ
Цеолиты образуют уникальный класс оксидов, представляющих
собой микропористые кристаллические алюмосиликаты, которые встречаются
в природе и могут быть синтезированы в лабораторных условиях [16]. Решетка
цеолитов является довольно открытой и состоит из каналов и полостей, в которых катионы, молекулы воды или других адсорбированных компонентов могут
храниться или вступать в реакции. Особые адсорбционные свойства цеолитов
используются при создании моющих средств, зубных паст и осушителей с их
участием, а их кислотность делает их привлекательными для катализа.
Цеолиты были открыты в 1756 г. шведским минералогом Акселем Крон-
стедтом, обнаружившим, что минерал стильбит при нагревании теряет большое количество воды. Слово «цеолит» на греческом языке означает «кипящий
камень». Прошло более двух столетий, прежде чем цеолиты привлекли внимание химиков. В 1930-е гг. Ричард Баррер начал исследования по синтезу цеолитов, и в 1948 г. ему удалось получить первый синтетический цеолит, не имевший природного аналога. Примерно в это же время Мильтон открыл способ
синтеза цеолита А. В настоящее время цеолиты и родственные им материалы
достаточно широко синтезируются в лабораторных условиях.
Цеолиты находят широкое применение в качестве бытовых моющих средств,
осушителей и катализаторов. В середине 1960-х гг. Рабо с сотрудниками из
фирмы «Юнион Карбайд» и Планк с сотрудниками из «Мобил» показали, что
фожазитовые цеолиты являются очень перспективными твердыми катализаторами. С тех пор было открыто множество реакций с участием углеводородов,
катализируемых цеолитами. Интерес к цеолитам у исследователей фундаментальных каталитических процессов обусловлен их известной кристаллической
структурой и хорошо определенными активными центрами. Тот факт, что цеолиты обладают хорошо определенной системой пор, в которую внедрены известным путем активные центры, делает их схожими с энзимами.
Природные цеолиты могут носить имя минералов (морденит, фожазит,
ферриерит, силикалит) или иногда имя открывателя, например баррерит, в
честь профессора Баррера, или названия местности, где они были обнаружены, например бикитаит, по территории Бикита в Зимбабве. Синтетические
цеолиты обычно называют по титулу промышленной компании или университета, где они были разработаны, например название VPI-целоитов идет от
Virginia Polytechnic Institute (Политехнический институт штата Виржиния), а
название ZSM для цеолитов произошло от Zeolite Socony Mobil (цеолиты фирмы Сокони Мобил).
Для известных в настоящее время почти 600 марок цеолитов и новых, открываемых каждый год, полезно дать общую классификацию структур, предложенную ИЮПАК. В этой классификации каждой структуре ставится в соответствие три буквы, например FAU для фожазитов, MFI для ZSM-5 и MOR для
морденитов. В рамках данной структуры может быть большое число различных
цеолитов, поскольку их состав варьируется достаточно широко.
232
Глава 5. Твердые катализаторы
5.7.1. Структура цеолита
Цеолиты состоят в основном из тетраэдров (рис. 5.29), образованных группами Si04 и А104, из которых путем разнообразного «обобществления» узловых атомов О строится кристаллическая решетка (рис. 5.30).
Рис. 5.29. Тетраэдры Si04 представляют основной строительный блок
цеолитов
f
Рис. 5.30. Пример структуры цеолита. Каждый угол содержит ион
Si или А1 с ионами О между ними
(середина связывающих линий).
Приведенная структура представляет собой комбинацию из четрех-,
шести- и восьмизвенных колец
Из тетраэдров Si04 можно получить различные силикатные блоки — квадраты, шести- и восьмизвенные кольца, которые называются вторичными строительными элементами. Кристаллы цеолита строятся путем формирования из
набора строительных элементов периодических структур.
Аналогичные периодические структуры могут быть получены чередованием групп А104 и Р04. Получающиеся алюмофосфаты, однако, называются не
цеолитами, а А1РО. Цеолиты получают в гидротермальном синтезе в автокала-
вах при высоких давлениях в присутствии темплатных молекул (шаблонов) типа
тетраметил аммония, задающих структуру цеолита.
Таблица 5.5. Свойства некоторых цеолитов
5.7. Цеолиты ->\г 233
<L>
S
X
in
1
in
сч
in
о
1
Л1
Л1
AI
AI
н
О
d
о
С и.
о
m
in
о
m
m
СП
СП
СП
CN
*—
ГЧ
<и 5
o'
о"
о"
o'
o"
o"
Ю
О
№
О _
2 Х
in
T3-
MD
40
S 5
m
Ш
o'
o'
о
г-
е-ё
о"
о"
о"
о
o~
40
<^> ^
t'"-
л ^
o"
o"
S S
cf
S
s'
s'
J3
i
2
2
(U
E
О
E
Л
я
ГС
2
Q.
JS
Е
Е
Е
я
E
Os
К
fc
X
X
<D
E
t:
JS
s;
Cl
О
О
о
2
cd
s
>,
С
n
_
—
—
E PC
H
s
J3
J3
Е
<D
Е
s Л
С* d
jS
Е
1 s.
3
Е
1 S-
§ £
:S О
<u С
E S
s *-
4 X
9
1 §
&1
<D
E
=S
(U
о
u
o
E
§
s
О
О
>>
E
s
E
(U
С
a.
PC
О
с
s
H
X
о
о
>>
E
s
о
E
Cl
К
>>
о g*
(U ^
<D ^
E
S
(U
1
S
О >>
о >,
s
2
О
с
PC
1
n
со
о
§
Н &
£ £■
H u
s £
£ °
5 ^
1 i
§
E
1
о
о
E
2
tr
CS
с
s
X
(U
с
сз
о о
о к
о к
-Г я
К F—
cd
та
H
Cl
X
° -
О Л
О Л
3 E
c* °
I— t-
X
o
S
E
o
u
<D
Л
е 5*
1 3
с о
- (U
JQ ~
5 s
*: е
ч е
u
cd
cd
С
С
S
Н
0 J3
= §
1 °
R 1—
О J3
с 5
3 Е
£ о
^ и<
c9 ^
Ǥ
^ Ё
(u ja
- О
2 E
§ §
E
2
E
>,
a-
cd
ro
PC
PC
о
с
2
(U
JS
H
(U
e-
О
E
S
t5
й ю
ГС я
й СЗ
2 t=;
CS
cf
5
E
—
Е >>
Л ^
Е О.
Л Н
^ н
я &
^ F-
E «
(U
2
<D
2
E
2
E
=S
X
2
cd
X
<L>
JS
E
PC
2
tr
<D
2
<D
2
0>
2
11
E
a-
a-
(U
CJ
E
E
n
cd
2
E
2
Я
E
Е
Е
Е
В с
<d
s
Cl
CL
Я
о.
О-
о.
E( ^
<V
(U
<D
<D
<D
o
о
PC
о
2
2
E
fc
X
E
X
X
X
к
cd
(U
=5
<D
<D
<D
О
Q*
E
a.
ID
о.
о.
а.
С
Я
f-м
E
Н
Н
Н
S
sS
3
X
X
<
-J
EE
<
* 8
Н
<
<
О
H
UJ
>, *
ю
-J
ц.
U-
S
-J
S
CQ
X
<и
Q.
н
<L>
<
X
>-
H
s
-J
in
1
H
(U
S
F—
F—
н
E
H
IQ
X
S
S
S
(U
s
M
а
s
м
О
О
о
O-
о
СЛ
§
(U
я
ей
X
<D
ГГ
<D
гг
<D
O
(U
a
N
234 -М/- Глава 5. Твердые катализаторы
Цеолиты характеризуются разнообразием пористой структуры. Цеолиты
типа L (LTL) имеют одномерные параллельные каналы. Морденит (MOR)
образован каналами двух типов, которые также одномерны и параллельны.
Более сложный по структуре целит ZSM (MFI) обладает прямолинейными и
синусоидальными каналами, которые перпендикулярны друг другу, то есть
двумерной пористой структурой. Цеолит А имеет полости, соединенные «окнами», которые формируют кубическую структуру. У фажезитов (FAU) типа
X и У система взаимосвязанных полостей обладает тетраэдрической структурой. В табл. 5.5 приведены характеристики некоторых наиболее известных
цеолитов.
5.7.2. Компенсирующие катионы и кислотность
Когда ионы А13+ замещают ионы Si4+ в тетраэдре, этот строительный блок приобретает общий заряд, равный —1, и, следовательно, в систему
необходимо ввести катионы типа Na+ для нейтрализации заряда. Число катионов, присутствующих во внутренней структуре цеолита, равно числу тетраэдров из оксида алюминия, входящих в решетку. Цеолиты, в которых компенсирующими являются ионы натрия, обозначаются как Na-X, Na-ZSM-5 и т. д.
Если ионы натрия заменяются протонами (получаются цеолиты Н-Х, H-ZSM-5
и т. д.), то цеолит превращается в гигантскую
Н молекулу поликислоты. На рис. 5.31 показа-
| на структура кислотного центра с протоном
^Оч О \ на мостике Si—О—А1. Поскольку этот центр
Si AI Si является донором протонов, то он называется
/ \ / \ / \ бренстедовской кислотой. Его сила зависит от
О О О О О О локального окружения протона, в частности от
Рис. 5.31. Пример твердой кислоты числа других ионов алюминия в ближайшем
окружении.
Семейство А1РО также может иметь кислотные центры, например, если
ионы А13+ замещаются двухвалентными ионами, это приводит к семейству
MeAlPO (где Me = Со, Zn, Mg, Мп), или при замене ионов Р5+ четырехвалентными ионами Si получается семейство SAPO. Безусловно, возможно провести
одновременно замену двух типов, как в случае семейства CoAPSO. Возможна и
замена тетраэдров АЮ4 и Р04 тетраэдрами Si04, что снова дает семейство цеолитов SAPO. Можно ввести в структуру цеолитов и гетероатомы, например,
при замене алюминия трехвалентным железом в цеолите ZSM-5 получается
цеолит Fe-ZSM-5.
5.7.3. Применение цеолитов
5.7. Цеолиты -J\r 235
Уникальные свойства цеолитов
• Кристалличность
• Микропористость
• Однородная система пор
• Наличие каналов или полостей
• Высокая удельная внутренняя поверхность
• Высокая термическая устойчивость
• Способность к ионному обмену
• Кислотность
• Нетоксичность
• Безопасность для окружающей среды
Высокая реакционная способность поверхности цеолитов обусловлена внедрением ионов А13+ в центры, в которых должны в обычной ситуации находиться ионы Si4+. Это их свойство, а также кристаллическая система микропор
предопределяют возможность практического использования цеолитов, например, в качестве:
• адсорбентов: при сушке, очистке, разделении. Цеолиты могут адсорбировать до 25 вес. % воды;
• ионообменников: цеолиты являются основным компонентом моющих порошков, в которых они постепенно заменяют фосфаты, использующиеся для
связывания кальция. Кальций и в меньшей степени магний, присутствующие в
воде, с помощью цеолитов А заменяются на натрий. Это — наиболее широко
распространенная на настоящий момент область применения цеолитов. Цеолиты нетоксичны и не оказывают вредного воздействия на окружающую среду.
Целиты входят также в состав зубных паст, где они снова используются для
связывания кальция и предотвращения образования налета;
• катализаторов: на поверхности цеолитов есть кислотные центры, обладающие каталитической активностью по отношению к реакциям с участием углеводородов. Более подробно это область применения цеолитов будет обсуждена в гл. 9. Система пор позволяет достигать активных центров только малым
молекулам, что предопределяет селективность цеолитов как катализаторов и
исключает из реакций как в качестве реагентов, так и продукта, большие молекулы, не способные пройти в поры.
236
Глава 5. Твердые катализаторы
На рис. 5.32 показано, как цеолит
может влиять на селективность каталитической реакции. В первом из
приведенных на рисунке случае нежелательный реагент исключается
вследствие невозможности его проникновения в цеолит. Во втором случае компонент А реагирует с формированием продуктов В и С, но размеры молекул С слишком велики, чтобы
они могли выйти из пор. В третьем
случае возможная реакция между В и
С не идет, поскольку промежуточный
комплекс не может сформироваться в
полости.
Механистические аспекты реакций, катализируемых цеолитами, кратко обсуждаются в гл. 9.
5.8. ТЕСТИРОВАНИЕ КАТАЛИЗАТОРОВ
Единственным способом узнать, действует ли данный материал как
катализатор, является непосредственное его апробирование в реакции. Определение активности катализатора не является столь прямолинейным, как может показаться на первый взгляд. На практике при работе с монокристаллами
и модельными катализаторами встречается несколько ловушек. Например, чтобы
избежать влияния термодинамических ограничений типа связанных с достижением равновесия между реагентами и продуктом, мы собираемся измерить активность в пределе нулевой конверсии. Мы также хотим получить данные в
условиях определенного состава газа и при заданной температуре. Обеспечение этих условий может стать проблематичным для достаточно быстрых и в
сильной степени эндотермических или экзотермических процессов, поскольку
в этом случае газ будет охлаждаться или нагреваться в процессе реакции.
Часто малые образцы (или даже разбавленные образцы) катализатора помещаются в трубчатый реактор вытеснительного типа (ТРВТ) или в емкостной
реактор с постоянным перемешиванием (ЕРПП), после чего проводят измерения скорости реакции. Более предпочтительным является ЕРПП, поскольку в
нем отсутствуют градиенты концентраций компонентов, что делает описание
реакции и ее кинетики более легким. Если все сделано надлежащим образом,
скорость реакции будет определяться произведением удельной активности каждого центра и числа центров, находящихся на поверхности катализатора. В случае металлов обычно принимают, что число активных центров пропорционально
площади поверхности катализатора, определяемой из данных по хемосорбции,
как это было указано выше. Поделив скорость на число центров, получаем
_У \
А 1=> 1=>А
\ /
Селективность по реагенту
в 1=> 1=>в
—v_l/—
Селективность по продукту
/Z7V
В с=> 1=>в
Реакционная селективность
Рис. 5.32. Примеры различных механизмов
селективности цеолитов в реакциях
5.8. Тестирование катализаторов -*\г 237
число оборотов реакции. Этот параметр важен при сравнении характеристической (внутренней) активности катализаторов. Однако число оборотов реакции не является подходящим параметром для оценки эффективности катализатора, используемого в промышленности, где больший интерес представляет
активность на единицу объема катализатора. В промышленности важную роль
играют многие другие факторы: смотри список в начале этой главы.
5.8.1. Десять заповедей по тестированию катализаторов
Рассмотрение всех особенностей тестирования катализаторов и
обсуждение всех возможных ловушек выходят за пределы данной книги. Вместо этого мы сформулируем «Десять заповедей по тестированию катализаторов». Это — набор правил, которыми следует руководствоваться на практике.
Они были сформулированы экспертами компании «Catalitica» [17].
1. Определи цель: какова цель тестирования? Состоит ли она, например,
в определении удельной реакционной способности или реакционного механизма или стоит задача длительного промышленного использования катализатора?
2. Используй эффективную стратегию тестирования: какие параметры
являются наиболее важными и каков путь их наиболее эффективного определения?
3. Выбери правильный тип реактора для тестирования: имеется большое
число разнообразных реакторов. Упомянутые выше трубчатый реактор и емкостной реактор с постоянным перемешиванием являются более предпочтительными для лабораторных исследований, но другие устройства также могут быть
интересны с точки зрения моделирования реальных промышленных условий.
4. Установи идеальную картину течения реагентов: она обычно считается
установленной для трубчатого реактора и емкостного реактора с постоянным
перемешиванием, но выполняются ли все другие условия, требуемые для идеального смешения компонентов? Например, твердым является правило, согласно которому диаметр d ТРВТ должен быть по крайней мере в 10 раз больше
диаметра каталитических частиц dp, что позволяет избежать влияния стенок
реактора. Количество катализатора должно быть достаточно большим, чтобы
не возникали аксиальные градиенты концентраций компонентов. Второе правило состоит в том, что толщина каталитического слоя L должна в 50 раз превышать диаметр частиц, то есть L > 50^. Более толстые каталитические слои
были бы предпочтительней, но в этом случае возникают проблемы, связанные
с появлением градиентов температуры и высоким перепадом давления.
5. Обеспечь постоянство температуры: даже малые отклонения от измеряемой температуры могут сильно сказаться на активности, поскольку скорость
реакции, как это видно из уравнения Аррениуса, экспоненциально зависит от
температуры. Градиенты температуры могут возникнуть и в каталитическом
слое, и внутри частиц из-за выделения или поглощения тепла в процессе
реакции.
238 Глава 5. Твердые катализаторы
6. Минимизируй влияние транспортных процессов: если нас интересует
внутренняя кинетическая производительность катализатора, важно устранить
транспортные ограничения, поскольку их наличие может привести к ошибочным результатам. Ниже мы обсудим, как диффузионный перенос внутри пор
катализатора может повлиять на эффективную энергию активации. Определение числа оборотов реакции при различных скоростях течения газа и размерах
каталитических частиц позволяет выявить наличие транспортных ограничений.
Полезно начинать тестирование катализатора в следующих условиях:
• используются малые каталитически частицы, что позволяет устранить
транспортные ограничения;
• исследования проводятся при низкой степени конверсии, что исключает
возникновение градиентов температуры. Такие исследования следует выбирать
при определении кинетики реакции. Может оказаться необходимым введение
в катализатор инертного материала;
• исследования проводятся при умеренных температурах, что также позволяет избежать возникновения градиентов температуры.
7. Получи значимые данные для катализатора: обычно представляет интерес
число оборотов реакции на один активный центр, определяющее кинетику процесса. Важны также и другие характеристики — селективность и выход реакции,
которые позволяют оценить промышленный потенциал катализатора. Активность
может быть охарактеризована не только числом оборотов реакции, но и
• удельной скоростью реакции на единицу массы или объема катализатора;
• пространственной скоростью (поток газа, деленный на объем слоя катализатора), при которой достигается определенная степень конверсии при данной температуре.
Не рекомендуется выражать активность через температуру, при которой
достигается определенная степень конверсии (что нередко делается в прикладных исследованиях), поскольку общая скорость каталитической реакции сложным образом зависит от температуры.
8. Определи стабильность катализатора: как быстро он теряет свою активность и в чем состоит причина его деактивации? Насколько катализатор чувствителен к различного рода примесям, которые могут присутствовать в исходном потоке в реальных условиях?
9. Следуй хорошей экспериментальной практике: всегда проводи сравнение полученных данных с результатами, установленными для идеальных условий. Проверяй воспроизводимость полученных данных и отсутствие тривиальных ограничений типа термодинамических на данный эксперимент. Для ясности и чистоты опыта проводи эксперимент в реакторе в отсутствие катализатора
и при наличии только инертного компонента, если такой используется наряду
с катализатором.
10. Формулируй результаты в однозначном смысле: описывай все детальные особенности условий, при которых тестировался катализатор, и представляй информацию об активности, селективности и конверсии в форме, обеспечивающей однозначное определение этих параметров.
5.8. Тестирование катализаторов -J\r 239
5.8.2. Измерение активности
Как отмечалось ранее, активность катализатора может быть измерена, когда он находится в трубчатом реакторе, через который прогоняют газ
при заданных составе газовой смеси и температуре. Такие измерения позволяют определить характеристическую (внутреннюю) реакционную способность
катализатора, в условиях, далеких от равновесия. Полученные данные можно
сравнивать с результатами молекулярно-кинетического моделирования. При
таком способе нахождения активности проблемы возникают только при высоких скоростях газового потока. Однако условие почти нулевой конверсии можно обеспечить и другим путем — «разбавлением» катализатора материалом носителя.
Другая серьезная проблема может возникнуть при описании переноса массы в катализаторе. Поскольку катализаторы, как правило, являются пористыми материалами, диффузия реагентов газа в поры и продуктов из них может
протекать очень медленно в сравнении со скоростью реакции на поверхности
катализатора. В этом случае диффузия начинает лимитировать скорость реакции всего процесса в целом.
Рис. 5.33. Частицы катализатора
в трубчатом реакторе находятся
в окружении газа, текущего через каталитический слой. Наличие застойной зоны у поверхности частицы может стать причиной транспортных ограничений,
если диффузия через него будет
медленной в сравнении со скоростью реакции. Но обычно наиболее лимитирующим процессом
является диффузия в порах
Довольно часто катализаторы, подвергаемые тестированию, сперссованы в
тела определенной формы, чаще всего — в таблетки. Для проведения тестирования эти тела дробят, и частицы определенного размера помещают в трубчатый
реактор с учетом всех отмеченных выше требований. Размер частиц при этом
играет существенную роль. На рис. 5.33 показано, как газовый поток обтекает
каталитическую частицу. Отметим наличие застойной зоны вблизи поверхности
частицы. Она возникает потому, что скорость газа на поверхности частицы равна нулю и формируется граничный слой, через который молекулы переносятся
только за счет диффузии. Кроме того, молекулы должны продиффундировать в
поры и выйти из них. Этот процесс протекает медленнее, чем диффузия молекул
через граничный слой. Транспортные ограничения обычно возникают только в
случае катализаторов, обладающих очень высокой активностью.
240
\
Глава 5. Твердые катализаторы
5.8.2.1. Транспортные ограничения и диффузионный модуль Тиле
Влияние транспортных ограничений обычно оценивают на примере сферической частицы, показанной на рис. 5.33. Введем безразмерную величину, называемую диффузионным модулем Тиле (Os) [18], которая характеризует степень потери эффективной активности катализатора по сравнению с
ситуацией, когда транспортные ограничения отсутствуют. В своем изложении
мы будем следовать схеме, предложенной Саттерфилдом [19], в книге которого
приведены решения и для частиц другой геометрии.
Мы начнем с рассмотрения идеальной пористой сферической частицы радиуса R. Температуру будем считать постоянной, а реагирующий газ — однокомпонентным. На макроскопическом уровне диффузионные процессы описываются
первым и вторым законами Фика, которые представляются уравнениями
j(r, t) = -DV С (г, t)
ЭС(г, t)
dt
DV2C (г, t),
(5.17)
где D — коэффициент диффузии. Заметим, что эффективный коэффициент
диффузии может быть меньше коэффициента диффузии молекул в газовой фазе
и в застойной зоне: если поры становятся очень тонкими, то молекулы чаще
буду сталкиваться с поверхностью пор, чем друг с другом. Молекулы также
могут адсорбироваться на поверхности и через некоторое время вылетать с нее.
Диффузия в таком режиме называется кнудсеновской диффузией. Обычно эффективный коэффициент диффузии характеризует промежуточный режим и
зависит от размера пор и распределения катализатора на носителе.
Скорость реакции имеет вид
Скорость
моль
2
^ surfcat
If
1
s:
Г")
S
1
C" (r, t)
моль"
.,3
/V
2
3/7
cat
_ ^ surfcat _
M
, (5.18)
где п — порядок реакции; V — объем катализатора; S — удельная площадь
поверхности на единицу объема катализатора; к — константа скорости;
С(г, t) — концентрация реагента. Нас будет интересовать только стационарное
решение. Если мы выделим внутри каталитической частицы тонкую сферическую оболочку, внутренний радиус которой равен г, а внешний — г + dr, то
легко написать уравнение баланса массы для оболочки толщиной dr (рис. 5.34).
В стационарных условиях перенос массы в оболочку через поверхность радиуса г + dr минус перенос массы из оболочки через поверхность радиуса г
совпадает с расходом массы компонента на реакцию. Иначе говоря, что вошло
внутрь и не вышло, то прореагировало. Потоки через поверхности оболочки
определяются первым законом Фика, поэтому скорость переноса компонента
равна площади поверхности А(г) = Алг2, умноженной на плотность потока j{г, t),
отсюда получаем
Скорость (г) = -A(r)jm (г) = AnrDt„dC^-,
(5.19)
5.8. Тестирование катализаторов
Рис. 5.34. Схематическое изображение сферической пористой каталитической частицы
радиуса R. Концентрация реагента вне частицы постоянна и равна С0 и снижается по мере
продвижения в глубь частицы вследствие протекающей реакции. В результате переноса
реагента внутрь частицы j.n(r) является функцией координаты г
Скорость (г + dr) = -А (г + dr) jin (г + dr) =
= А (г + dr) Z)e(T ~-+-d— = 4л{г + dr)2 Dtff
Здесь мы использовали соотношение
С (г + dr)-С (г) d С (г)
dC(,) | d’Cjr)^
dr
dr
C(r + dr) =
dr
dC(r)
dr
dr1
(5.20)
dr
+ C(r).
(5.21)
Скорость реакции равна объему оболочки 4;rr2dr [м3а1], умноженному
на удельную площадь поверхности S [M2urfcat/M3at], константу скорости к
[m3VM2urfcat х (моль)3" 1 • cj и концентрацию реагента С"(г). Таким образом, скорость реакции в оболочке равна
Скоростьоб (г) = Anr2drSkCn(r), (5.22)
что приводит к следующему уравнению баланса массы
Скорость (г) — Скорость (г + dr) = Скоростьоб (г), (5.23)
dl^ - 4* (/• + drf
dr
dr2
= 4;rr2dr^C"(r). (5.24)
Отбрасывая члены более высоких порядков по dr, получаем из этого уравнения:
242 -*\r Глава 5. Твердые катализаторы
Последнее равенство определяет безразмерную величину — диффузионный
модуль Тиле для сферической частицы:
Нижний индекс s говорит нам о том, что соотношение (5.26) пригодно
только для сферической частицы. Если мы ограничим свое рассмотрение реакцией первого порядка, то п = 1 и дифференциальное уравнение (5.25) переходит в следующее
d '-С 2d С (г) SkC(r) Ф-,,, , ч ,,,,,
+ = ~#C(r)*a-C{r), <5.27)
где
<5-28)
Введя новую переменную rj(r) = rC(r), преобразуем уравнение (5.27) в однородное дифференциальное уравнение второго порядка:
d2rj
dr
2 = а) Л, (5.29)
общее решение которого имеет вид:
rj = гС= Вхеыг + В2е~сог. (5.30)
Теперь нам надо найти функцию С(г), воспользовавшись соответствующи¬
ми граничными условиями. Эти граничные условия таковы: концентрация на
поверхности частицы считается постоянной, то есть |С(г)|г=/г = С0, в центре
частицы нет градиента концентрации, то есть \dC(r)/dr\r=0 = 0. Из последнего
условия следует, что В{ = — Bv и решение принимает вид:
C(k) = ^(e0)r -e-""-) = ^sh(юг). (5.31)
Из первого граничного условия находим постоянную Вх:
В'=2(532)
Окончательно распределение концентрации внутри частицы принимает
простой вид:
ЛСрзЬМ С0 sh [Ф5 (г//?)]
[П rsh((oR) (r/R) sh(<Ds) ’ К }
5.8. Тестирование катализаторов -J\r 243
Видно, что поле концентрации внутри частицы определяется только величиной модуля Тиле Os и нормированным радиусом r/R. Характер зависимости
концентрации от этих параметров приведен на рис. 5.35.
Рис. 5.35. Профили нормированной
концентрации для различных значений модуля Тиле. Заметим, что при
большой его величине только тонкий
слой вблизи поверхности частицы
вносит вклад в реакцию
ZT
со
Q.
2
Q.
О
X
Нормированный радиус
Зная профиль концентрации, можно найти полную скорость реакции внутри частицы, либо проинтегрировав локальную скорость, либо используя очевидное условия, что весь реагент вошедший внутрь частицы претерпевает химическое превращение. Полная скорость реакции такова:
Скорость^ = AftR2D{
eff
-d С (г)
dr
- 4/r/?'Z)effOsC0
1
1
th(Os) Os
, (5.34)
где нижний индекс diff указывает на диффузию как лимитирующую стадию процесса. В отсутствие транспортных ограничений внутреннее пространство частицы будет характеризоваться активностью, определяемой скоростью (47r/3)R3SkCQ, которая получается из формулы (5.34) при стремлении
модуля Тиле к нулю:
Фс -»0 или Фс
1
1
Л(Ф5) Ф5
Ф;
Введем фактор эффективности es, определив его как отношение скорости
реакции, лимитируемой диффузией, к скорости реакции в отсутствие диффузионных ограничений:
=
Фс
1
л(ф5)
ф<
(5.35)
Нижний индекс s указывает, что это выражение относится к сферическим
частицам. Следует также иметь в виду, что мы ограничили наше рассмотрение
необратимой реакцией первого порядка. Можно получить и более общие выражения для фактора эффективности, однако это выходит за пределы данной
книги. Несмотря на частный характер, выражение (5.35) имеет важное практи-
244 -J\r Глава 5. Твердые катализаторы
ческое значение, поскольку позволяет судить об эффективности по одному
параметру Os. На рис. 5.36 показана зависимость фактора эффективности от
параметра Ф.
Модуль Тиле Ф
Рис. 5.36. Зависимость фактора эффективности от диффузионного модуля Тиле. Эта
зависимость хорошо аппроксимируется функцией 3/Ф5
при Ф5 > 10
Заметим, что значение эффективности приближается к 1 при Ф§ 0. Диффузия не лимитирует скорость реакции при большой величине коэффициента
диффузии, малом радиусе частицы и низкой активности катализатора. С другой стороны, при большом Ф§, когда Deff мал, R велик, а скорость реакции
высока, параметр эффективности приближается к значению 3/Ф§, как это указано на рис. 5.36. Таким образом, малые частицы используются в каталитических реакциях более эффективно, чем большие.
Имеется, однако, много причин, по которым каталитические частицы делают
не слишком малыми, как это было указано в начале этой главы, одна из них
связана с необходимостью уменьшить перепад давления на каталитическом слое
реактора. Основываясь на проведенном выше анализе, можно оценить целесообразность равномерного распределения каталитической фазы по объему носителя в
конкретной ситуации. Для катализаторов из благородных металлов, возможно,
более выгодным окажется использование систем типа «яичной скорлупы», в которых благородный металл находится только на внешней поверхности частиц.
Наличие диффузионных ограничений оказывает существенное влияние на
измеряемую величину кажущейся энергии активации. Мы может представить
и константу скорости к, и энергию коэффициент диффузии Z)eff в аррениусов-
ской форме:
к = k0e-AE“ajRT и = D0e-AE««/RT.
В отсутствие диффузионных ограничений общая энергия активации равна
£арр = RT2^P- = AEacl. (5.36)
5.8. Тестирование катализаторов -i\r 245
Но при их наличии, когда Os велик, скорость реакции имеет вид:
Скорость diff = 4я-Л£)еГГФ5С0
1Ь(Ф5) Ф5
4я‘Л/)е(ГФ5С0 = 4л R С0 ^]SkDe([,
(5.37)
из которого следует следующее выражение для кажущейся энергии активации
^арр ДГ2 din (Скорость^) A£act | А Еш А £ас,^ (5-38)
дТ 2 2 2
когда
2
Видно, что кажущаяся энергия активации будет составлять примерно половину от величины, отвечающей ситуации, когда транспортные ограничения
отсутствуют. Важно помнить об этой ловушке при тестировании катализаторов. Действительно, кажущаяся энергия активации зависит от условий проведения реакции (как об этом говорилось в гл. 2) и диффузионные ограничения
могут их изменить при варьировании температуры.
5.8.2.2. Диффузия в порах
Выше мы рассмотрели диффузию в каталитической частице. Можно
также рассмотреть и транспорт газа в одиночной поре, показанной на рис. 5.37.
Описание транспорта газа в ней проводится достаточно легко. В этом случае
перенос реагента и продукта определяется только диффузией внутрь и из поры,
поскольку здесь нет конвективного течения. Рассмотрим ситуацию, когда реакция происходит на частице, находящейся внутри поры. Последнюю смоделируем цилиндром радиусом R и длиной L (см. рис. 5.37). На входе в пору
концентрацию реагента будем считать равной С0, а скорость реакции представим в виде
r= кС(х). (5.39)
dx
►
Рис. 5.37. Схематическое изображение ци- Ox L
линдрической поры С0
246
\
Глава 5. Твердые катализаторы
Рассмотрим тонкий слой цилиндра толщиной cbc и напишем для него соотношение баланса массы. Легко определить входящий в слой и выходящий из
него потоки, а также расход реагента при реакции
-7tR2D,
dC(jc)
eff '
dx
= -ttR2D(
4/in = 4/ou. “ kClnRdx,
dC(x)-(dC(x)/cbc)d*
eff
dx;
-kC(x)2nRdx, (5.40)
d 2C(x)
dx2
где
со
2k
RD,
eff
2k
Ajff R
В уравнение (5.40) время не входит, поскольку мы рассматриваем стационарное состояние; мы ввели также модуль Тиле для поры. Будем решать дифференциальное уравнение при следующих граничных условиях
1СИ„.=С«
d С(х)
dx
0,
(5.41)
второе из которых говорит о достижении определенного равновесия на конце
поры. Процедура нахождения решения подобна рассмотренной выше, так что
для решения имеем:
С (х) - С0
ch (<a)L)
сЬ(фР)
(5.42)
Читатель может легко проверить, что написанное решение действительно
удовлетворяет приведенным выше дифференциальному уравнению и граничным условиям.
Введем теперь фактор эффективности, который опять определим как отношение степеней конверсии в поре при наличии и в отсутствие ограничений,
связанных с переносом массы:
L L L
^2nRkC(x)dx |C(x)dx Jch[^y(L - jc)]dx / ч
e _ О _ О _ О _ V P /
2nRLkC^ LCq L ch (соL) Фр
Скорость реакции при наличии диффузионных ограничений равна
(5.43)
th(°p)
rm=2nRLkC,-^±.
р
(5.44)
5.8. Тестирование катализаторов
\
Снова видно, что система описывается с помощью одного параметра —
диффузионного модуля Тиле Фр. Нетрудно видеть, что имеются два предельных
случая. Диффузионные ограничения отсутствуют при D-^oo или L —»0, или к-^ О,
или R —> поскольку в этих случаях
2к1} n t
> 0 => £ —> 1 и г - кСп.
DR 0
С другой стороны, если пора длинная и/или скорость реакции высока (kL
велико), или диффузионный процесс медленный и/или пора узкая (DR мало),
эффективность будет малой и стремится к значению
£
Фр L\ 2к ’
а скорость реакции становится равной
2 nRLkC*
^diff ~ ф ■
р
2ftCoyJkR3Deff. (5.45)
Расчет кажущейся энергии активации при наличии диффузионных ограничений дает:
Еярр = RT2 д 1П s Mssl при 0. (5.46)
д T 2 2 2 2
Таким образом, рассмотрение диффузии в поре приводит к результату, подобному полученному при рассмотрении диффузии в каталитической частице.
5.8.2.3. Следствия транспортных ограничений
в тестировании катализаторов
Обычно энергия активации диффузии в газовой фазе намного меньше энергии активации каталитических реакций, поэтому, как следует из уравнений (5.38) и (5.46), полная кажущаяся энергия активации при диффузионноконтролируемом процессе составляет примерно половину величины, получаемой в отсутствие транспортных ограничений. Если мы построим зависимость
скорости от обратной температуры, то будет наблюдаться смена наклона, когда
вступят в роль транспортные ограничения.
Как показано на рис. 5.38, при низких температурах увеличение скорости
реакции с ростом температуры определяется энергией активации каталитической реакции (которая сама по себе может быть кажущейся, см. гл. 2). При
некоторой температуре скорость реакции становится настолько высокой, что
транспорт реагента не поспевает за его превращением на активных центрах.
В результате внутри частицы формируется градиент концентрации. В результате рост скорости реакции замедляется по сравнению со случаем отсутствия
247
248 -*\r Глава 5. Твердые катализаторы
транспортных ограничений. При более высоких температурах наблюдается еще
одна смена наклона температурной зависимости скорости реакции, обусловленная увеличением активности каталитической до такой степени, что лимитирующей стадией становится перенос реагента через граничный слой застойной зоны. Наконец, при очень высоких температурах реакция начинает протекать и в газовой фазе; напомним, что энергия активации газофазной реакции
намного превышает энергию активации каталитической реакции. И уже термодинамика процесса определяет, возможна ли конверсия реагента в таких
условиях. Как будет показано в гл. 8, синтез аммиака представляет систему,
для которой равновесие полностью сдвинуто в сторону реагентов при температурах, когда могла бы проявиться энергия активации газофазной реакции.
Рис. 5.38. Температурная зависимость
скорости реакции в координатах Аррениуса иллюстрирует влияние на кажущуюся энергию активации диффузии в
порах и переноса через граничный слой
застойной зоны, окружающей каталитическую частицу. Отметим, что диффузия в порах снижает кажущуюся энергию активации вдвое по сравнению с
исходным значением. При очень высоких температурах начинают играть роль
реакции в газовой фазе
Для тестирования и оптимизации параметров катализатора следует выбирать температурный диапазон, лежащий ниже области, в которой диффузия
начинает лимитировать кинетику реакции. В этом же диапазоне находится и
температура, отвечающая оптимальной работе катализатора (если условия по
термодинамическому равновесию и селективности не исключают возможности
работы при этих температурах). В принципе надо выбирать условия, отвечающие максимально возможной скорости реакции, не забывая об эффективности
работы катализатора. Для быстрых реакций типа окислительных можно принять, что рабочей является только внешняя поверхность частицы. Следовательно, для таких процессов можно использовать непористые или монолитные
катализаторы или частицы, с каталитическим материалом, находящимся только на внешней их поверхности.
Рассмотрение транспортных процессов играет важную роль в выборе формы
каталитических частиц, как это было описано в разд. 5.5 (см. также рис. 5.26).
Хотя малый размер частиц позволяет наиболее эффективно использовать каталитический материал, нельзя допускать связанных с их использованием больших перепадов давления на каталитическом слое реактора. Также надо иметь в
виду механическую прочность частиц и реактора. По этой причине необходимо объединять частицы в достаточно крупные тела, форму которых следует
оптимизировать с учетом процессов тепло- и массопереноса. Создание техно¬
\
Список литературы 249
логии каталитического процесса, обладающего высокими эффективностью и
экономичностью, требует согласованного учета многих факторов на разных его
уровнях.
Список литературы
1. Elliot S. The Physics and Chemistry of Solids. New York: Wiley & Sons, 1998.
2. Ashcroft TV., Mermin N.D. Solid State Physics. Saunder College, 1976.
3. Kittel C. Introduction to Solid State Physics. New York: Wiley & Sons, 1976.
4. Klink C., Olsen L., Besenbacher F., Steensgaard /., Laegsgaard E., Lang N.D. //
Phys. Rev. Lett. 1993. V. 71. P. 4350.
5. Samorjai G.A. Introduction to Surface Chemistry and Catalysis. New York:
Wiley&Sones, 1999.
6. Henrich V.E., Cox P.A. The Surface Science of Metal Oxides. Cambridge:
Cambridge University Press, 1994.
7. Gopel W, Rocher G., Feierabend R. // Phys. Rev. B. 1983. V. 28. P. 3427.
8. Christensen A., Ruban A. V, Stoltze P1, Jacobsen K. W, Shriver H.L., N0rskov J.K. //
Phys. Rev. B. 1996. V. 56. P. 10.
9. Vitos L., Ruban A., Shriver #., Kollar J. // Surf. Sci. 1998. V. 411. P. 186.
10. Hansen K.H., Warren Т., Stempel S., Laegagaard E., Baumer М., Freund H.-J.,
Besenbacher F., Steensgaard I. // Phys. Rev. Lett. 1999. V. 83. P. 4120.
11. www.fysik.dtu.dk/CAMP.
12. Henry C.R. // Surf. Sci. Rep. 1998. V. 21. P. 1
13. Jacobsen #., Kleinshmit P. In: Handbook of Heterogeneous Catalysis / Eds.
Ertl G., Knosinger H., Weitkamp J. Weinheim: WCH, 1997.
14. Meyers A.C.Q.M., De Jong A.M., Van Gruythuysen L.P.M., Niemantsverdriet J.W. //
Appl. Catal. 1991. V. 70. P. 53.
15. Geus J.W., Van Dillen A.J. In Handbook of Heterogeneous Catalysis /Eds.
Ertl G., Knosinger H., Weitkamp J. Vol. 1. Weinheim: WCH, 1997. P. 240.
16. Gthomas J.M., Bell R.G., Catlow C.R.A. In Handbook of Heterogeneous Catalysis /
Eds. Ertl G., Knosinger H., Weitkamp J. Vol. 1. Weinheim: WCH, 1997. P. 206.
17. Dautzenberg F.M. In Characterization of Catalyst Development: An Iterative
Approach. ACS Syposium Series. Vol. 411 / Eds. Bradley S.A., Gattuso M.J.,
Bertolacini R.J. New York: ACS, 1989.
18. Thiele W. // Ind. Eng. Chem. 1937. V. 31. P. 916.
19. Satterfield C.N. Mass Transfer in Heterogeneous Catalysis. Massachusetts:
MTI Press, 1970.
глава РЕАКЦИОННАЯ СПОСОБНОСТЬ
/С ПОВЕРХНОСТИ
6.1. ВВЕДЕНИЕ
Взаимодействие газа с поверхностью и поверхностные реакции
играют ключевую роль во многих технологически важных областях: в коррозии, адгезии, синтезе новых материалов, электрохимии и гетерогенном катализе. Эта глава посвящена описанию взаимодействия молекул газа с металлическими поверхностями в терминах химической связи. Метод молекулярных орбиталей и теория зонной структуры — основные подходы к этому описанию.
Мы ограничим свое рассмотрение только поверхностью металлов.
Численное моделирование достигло такого уровня, на котором адсорбция,
диссоциация и образование новых связей могут быть описаны с желаемой точностью. Как следствие, основные тенденции в протекании реакций могут быть
в настоящее время достаточно хорошо предсказаны. Эти теоретические исследования оказали сильное влияние на практику гетерогенного катализа, поскольку
имеется много экспериментальных данных, полученных при изучении поверхностей (например, теплоты адсорбции, структура адсорбционных слоев, частоты колебаний, энергии активации элементарных ступеней реакций), с помощью которых теоретические модели можно досконально проверить.
Как было объяснено в предыдущих главах, катализ — циклический процесс, который начинается с адсорбции реагентов на поверхности катализатора.
Обычно хотя бы один из реагентов диссоциирует, при этом идет разрыв прочной связи, в чем и состоит суть каталитического действия. Следовательно, нам
следует сконцентрироваться на физике и химии процессов адсорбции газа и
диссоциации молекул на поверхности, в частности на поверхности металла.
При приближении атома (или молекулы) к поверхности твердого тела он попадает в потенциальное поле, создаваемое атомами металла. Во взаимодействии
атомов с твердым телом обычно выделяют два режима, один из которых относится
к физической, а другой — к химической адсорбции. Мы обсудим их раздельно.
6.2. ФИЗИЧЕСКАЯ АДСОРБЦИЯ
Физическая адсорбция связана с относительно слабым взаимодействием, которое не предполагает формирование химических связей между адсорбатом и поверхностью, то есть обобществления электронов. Ответственное
6.2. Физическая адсорбция -*\г 251
за физическую адсорбцию взаимодействие обычно разбивают на две части: сильное отталкивание на малых расстояниях и ван-дер-ваальсовское взаимодействие на средних расстояниях порядка нескольких ангстрем.
6.2.1. Взаимодействие ван-дер-Ваальса
Когда атом или молекула приближаются к поверхности, то возникающие в них за счет квантовых флуктуаций дипольные моменты индуцируют
дипольные моменты в поляризуемом твердом теле. Поскольку заряды на индуцированных диполях имеют противоположные по отношению к исходному
диполю знаки, а их появление во времени коррелирует с возникновением ди-
польных моментов у атома или молекулы, то возникают силы притяжения (как
говорят, силы изображения) между атомом (молекулой) и твердым телом. Ниже
будет предложена модель, поясняющая более подробно природу этого взаимодействия.
Рассмотрим атом водорода, ядро которого находится на оси z на расстоянии d от поверхности металла, так что его положение в системе координат
(х, у, z) характеризуется вектором d = (0, 0, d). Пусть электрон имеет координаты г = (х, у, z) (рис. 6.1). И ядро, и электрон индуцируют заряды изображения
в металле, величины которых равны
q = \—-е = -е, е —> ©о. (6.1)
1 + £
Рис. 6.1. Схематическое изображение атома, находящегося на расстоянии d от
поверхности твердого тела
{х, у, Z)
Потенциал взаимодействия между зарядами дается выражением
+ 2:
2d 2 (d + z) |2d + r| ’
(6.2)
где первые два члена описывают взаимодействие между ядром и его изображением и между электроном и его изображением. Третье слагаемое появилось
как результат от перекрестного отталкивательного взаимодействия. Раскладывая уравнение (6.2) в ряд Тейлора по степеням r/d, получаем
Md, г)-
/ 1 1
х' у~
,т+т+г,
V У
(6.3)
252 -J /■ Глава 6. Реакционная способность поверхности
В результате полное взаимодействие дает потенциал притяжения, который
можно представить в виде
Vw{d)~-C^L, (6.4)
где CvdW — так называемая постоянная ван-дер-Ваальса, которая зависит от
поляризуемости атома и природы металла. Заметим, что это взаимодействие не
предполагает наличия у атома (или молекулы) постоянного дипольного момента. Следовательно инертные газы типа аргона и ксенона также могут адсорбироваться физически на поверхности. Физическая адсорбция составляют основу метода БЭТ, подробно описанного в гл. 5.
6.2.2. Учет отталкивания
Притяжение между атомом и его изображением нельзя распространить на малые расстояния, поскольку электроны атома начинают сильно
взаимодействовать с электронами твердого тела. Кинетическая энергия свободных электронов металла взрастает по мере их сближения с локализованными
электронами атома вследствие принципа Паули. Имеется некоторый выигрыш
энергии, связанный со взаимодействием электронов металла с ядром атома. Этот
выигрыш может привести к хемосорбции атома, но если это атом инертного
газа, то отталкивание будет доминирующим. Простая аппроксимация данного
взаимодействия дается экспоненциальной функцией
VR(d)~e-d'\ (6.5)
Такой выбор аппроксимирующей функции имеет физическое основание:
плотность электронов спадает экспоненциально по мере удаления от поверхности металла. Результирующий потенциал можно представить в виде суммы
V(d) = VR (d)+ Fvdw (J) = CRe-^ (6.6)
его характерный ход показан на рис. 6.2. Глубина потенциальной ямы обычно
составляет 20—25 кДж/моль и менее (табл. 6.1), положение минимума нахо¬
дится на расстоянии нескольких ангстрем от поверхности. Адсорбированные
физически молекулы не изменяют своего химического состояния под влиянием этого взаимодействия: на поверхности они обладают теми же спектроскопическими характеристиками, что и в газовой фазе.
Таблица 6.1. Характерные теплоты физической адсорбции малых молекул
Молекула
Энтальпия, кДж/моль
сн4
-21
со
-25
со2
-25
N2
-21
02
-21
6.3. Химическое связывание
\
Определяющее физическую адсорбцию взаимодействие подобно ван-дер-
ваальсовскому взаимодействию между молекулами, ответственному за конденсацию газа и формирование полислоев на поверхности. Ван-дер-ваальсовское
взаимодействие между молекулами часто описывается с помощью потенциала
Леннард-Джонса, имеющего вид:
= (6.7)
v ; d" d6 У
Рис. 6.2. Диаграмма потенциальной энергии, показывающая потенциал притяжения
ван-дер-Ваальса и потенциал
отталкивания, обусловленного принципом запрета Паули,
которые в сумме дают потенциальную яму, теплота адсорбции в которой составляет
25 кДж/моль, а минимум находится на расстоянии 3,8 А 0 2 4 6 8 10
от поверхности Расстояние от поверхности d, А
Выражение (6.7) часто называют потенциалом Леннард-Джонса (N, 6). Наиболее популярен, по математическим соображениям, потенциал (12, 6), несмотря на то что экспоненциальная функция (6.6) аппроксимирует отталкива-
тельную часть потенциала лучше. Показанный на рис. 6.2 потенциал достаточно хорошо описывает физическую адсорбцию молекул, хотя на очень малых
расстояниях он становится непригодным, поскольку потенциал притяжения в
(6.6) начинает доминировать нереалистическим образом: в то время как потенциал отталкивания при d-> 0 выходит на постоянное значение (VR-> CR), ван-
дер-ваальсовская часть монотонно стремится к бесконечности (KvdW —> °о).
6.3. ХИМИЧЕСКОЕ СВЯЗЫВАНИЕ
Если при адсорбции атом или молекула образуют химическую связь с
поверхностью, то в этом случае адсорбция называется химической. Прежде чем
описывать характер хемосорбционных связей, рассмотрим кратко основы теории
молекулярных орбиталей. Это необходимо также сделать для лучшего понимания,
по крайней мере качественного, сути каталитического действия. Как было показано в гл. 1, катализатор используется в основном для разрыва сильных внутримолекулярных связей при низких температурах. Наша цель — объяснить в рамках упрощенной качественной электронной картины, как это происходит.
253
254
Глава 6. Реакционная способность поверхности
6.3.1. Связи в молекулах
6.3.1.1. Двухатомные молекулы
Рассмотрим два атома А и В, находящиеся на достаточном удалении и характеризующиеся электронными волновыми функциями х¥а и которые являются собственными функциями атомов, отвечающими энергиям
и При сближении атомов волновые функции начнут перекрываться и перестраиваться, что приводит к образованию химической связи между атомами и в конечном итоге молекулы. Ниже мы не будем принимать в расчет
наличие спина у электронов. Образуем новую волновую функцию в виде линейной комбинации атомных орбиталей (J1KAO), которая для одного электрона имеет вид
* =
(6.8)
Гамильтониан системы выглядит так
Н
2mtJ
V2 +-
4 7t£a
_JL__L + ±
Гя Ги R
(6.9)
А
В
где га и гъ — расстояния от электрона до ядер А и В соответственно; R — расстояние между ядрами
(рис. 6.3).
Используя вариационный принцип, можно получить характеристическое уравнение
2>,(яЛ-дуЛ) = о, (6.Ю)
где Hik — матричные элементы га-
Рис. 6.3. Двухатомная молекула АВ и введенные
расстояния
мильтониана; Sik — интеграл перекрытия волновых функций. Система уравнений (6.10) имеет нетривиальное
решение, если равен нулю детерминант
aA - Е р - SE
Р -SE ав - Е
= 0,
(6.11)
где
а\=И«* =\waHwadV, ав = Ньь = jVbtf^bdF, (6.12)
Р = нл> = яьа =\VzHy/bdV, (6.13)
S = So, = \щЛщьйУ, (6.14)
6.3. Химическое связывание -l\r 255
при этом Saa = Sbb = 1, поскольку функции у/а и у/ъ нормировании. Решение
уравнения (6.11) приводит к следующему выражению для собственной энергии Е
ая +ab-2Sp±^(cca -abf -4(аа + ab)Sp + 4p2 -4aaabS2
+ _ / 0\ * (6.15)
2(l -5 )
Видно, что у двухатомной молекулы имеется два энергетических уровня, на
которых может находиться электрон; один уровень относится к связывающей
орбитали, а другой — к разрыхляющей. В рамках этого приближенного подхода
не удается получить хороших количественных результатов, но на качественном
уровне можно воспроизвести все значимые эффекты. Ниже мы рассмотрим
несколько иллюстративных примеров в пределе малых значений интеграла перекрытия (это стандартное приближение при элементарных оценках). В этом
приближении имеем
Е = а. +аь-^Р± j(ora - abf + 4Р2
6.3.1.2. Гомоядерные двухатомные молекулы
Начнем с рассмотрения гомоядерных молекул типа Н?, N2 и 02,
для которых аа = аъ = а и энергия принимает вид
Е± = а ± /3 - S/3. (6.17)
Поскольку Р < 0, a S > 0, то полученный результат говорит о том, что оба
уровня за счет отталкивания (интеграла перекрытия) сдвинуты вверх на величину S\/3\. Далее, они формируют, как показано на рис. 6.4, связывающее состояние с энергией Е+, сдвинутой вниз, и разрыхляющее состояние со сдвинутой вверх энергией Е_.
Рис. 6.4. Схематическая энергетическая
диаграмма двухатомной гомоядерной молекулы. Заметим, что расщепление уровней |2/?| пропорционально интегралу перекрытия атомных орбиталей
Если мы ограничимся рассмотрением одноэлектронного приближения, которое не вполне корректно, то можно оценить выигрыш энергии, связанный с
Е Разрыхляющая
орбиталь
|2/?| }
Е=а
/ Связывающая
орбиталь
А
256
—Глава 6. Реакционная способность поверхности
образованием химической связи, путем суммирования энергий всех занятых
орбиталей до и после образования связи и вычитания этих сумм:
АЕ = ^Е? -^El (6.18)
*осс 'осс 1ОСС
Если АЕ < 0, молекула устойчива и требуется затратить энергию на ее диссоциацию, Ediss, равную —АЕ, где
АЕ = 2E+-2a = -2\/3\ + 2S\j3\. (6.19)
Поскольку величина /? пропорциональна волновым функциям у/а и у/ъ и,
очевидно, интегралу перекрытия S, можно предположить линейную связь между S и /?: S = —у/3, что дает
АЕ = 2уР2 - 2|/?|. (6.20)
Первое слагаемое в (6.20) характеризует отталкивание, а второе представляет эффект гибридизации орбиталей, обеспечивающий формирование химической связи, как это показано на рис. 6.5.
Р
Рис. 6.5. Зависимость энергии связи от интеграла перекрытия /3
На рис. 6.6 приведена энергетическая диаграмма орбиталей для нескольких гомоядерных двухатомных молекул. Устойчивость молекул может быть
оценена по числам электронов, занимающих связывающие и разрыхляющие
орбитали (разрыхляющие орбитали иногда обозначаются символом *, например 2я*).
Из приведенной диаграммы видно, что атомы гелия не образуют устойчивых молекул, поскольку связывающая и разрыхляющая орбитали содержат по
два электрона, что приводит к отталкиванию между атомами. Молекула N2
более устойчива, чем молекула 02, поскольку у последней есть два дополнительных электрона на разрыхляющих орбиталях.
н,
1 s
""Н"'" la
£bo„d = - 4,55 эВ
(436 кДж/моль)
N2
N
\\&
!/>
А±А/ \±j_4
Лт*
чД АI
,w+.,
'-ft'
1 71
5с
^bond = - 9,84 эВ
(945 кДж/моль)
6.3. Химическое связывание
«НСо»
X
Не
Не
15
'"Н-'
£ho„d > 0 эВ
о,
ж
15
la
2р
£bond = “ 5,20 эВ
(497 кДж/моль)
Рис. 6.6. Энергетические уровни внешних орбиталей у некоторых молекул. Для реальной
ситуации эта картина слишком упрощена. Вследствие взаимодействия с 25-ор-
биталями уровни 5s располагаются выше уровня 1 р, но это не изменяет общую
устойчивость молекул
6.3.1.3. Гетероядерные системы
Для гетероядерной двухатомной молекулы мы имеем аа ф аъ. Мы
примем, что разность энергий у двух атомных уровней 6= аъ — аа > 0 и много
больше параметра взаимодействия /?. Если мы снова примем, что интеграл
перекрытия S мал, то можно разложить энергии в ряд Тейлора:
E+=a,-£- + S\0\,
(6.21)
E_=ab+tj- + S Щ.
Данные выражения показывают, что разность энергий связывающей и разрыхляющей орбиталей уменьшается с ростом разности уровней электронов в
изолированных атомах. Заметим, что сдвиги вверх и вниз снова несимметричны (рис. 6.7) вследствие наличия члена, ответственного за отталкивание, связанное с запретом для электронов занимать один и тот же уровень (отталкивание Паули).
257
258 -l\r Глава 6. Реакционная способность поверхности
<7*Е
\ I
P2/S
1
Рис. 6.7. Схематическая энергетическая
диаграмма, показывающая расщепление
уровней в гетероядерной молекуле
'$s/3
1sa
aEt
Волновые функции для связывающей и разрыхляющей орбиталей могут
быть записаны в виде
э2
(6.22)
а выигрыш энергии, обусловленный образованием связи, равен (разложение
Тейлора не использовалось):
АЕ = 2Е+ -аа -аь,
АЕ = (аЛ +аь)~ 2S0 + yj(aa - аь)2 + 4/?2 - ая - аъ = ^401 + 82 - 2Sf3.
(6.23)
Заметим, что при > О получаем результат, соответствующий гомоядер-
ным молекулам. Вводя вклад, отвечающий гибридизации,
получаем
wab = 44 p2+s\
AE = 2S\P\-W3b.
(6.24)
(6.25)
Рис. 6.8. Сводка данных метода
молекулярных орбиталей. Заметим, что устойчивость химической связи зависит не только от
силы взаимодействия, но и от
степени заполнения орбиталей
/
V
Разрыхляющая
Атомная Молекулярная Атомная
орбиталь орбиталь орбиталь
Сильная Слабая Отсутствие
связь связь связи
\
6.3. Химическое связывание 259
По сути, это все, что нам нужно для понимания механизма формирования
химической связи на поверхности и выявления реакционных путей. Более детально с методом молекулярных орбиталей можно ознакомиться по книге [1].
Основные результаты метода молекулярных орбиталей представлены на рис. 6.8.
6.3.2. Поверхность твердых тел
В данном разделе мы приведем основные результаты теории твердых тел, необходимые для понимания механизма адсорбции. Более детально
изложение теории дано в прекрасных книгах [2—4].
Механизм формирования механической
связи может быть понят на основе картины
орбиталей, обрисованной выше. Атомы металлов имеют внешние s- или /7-орбитали,
которые способны сильно перекрываться.
Механизм формирования зонной структуры
металлов показан на рис. 6.9.
Рис. 6.9. Механизм образования энергетических зон
электронов при добавлении атомов и их орбиталей.
Заметим, что энергетический зазор между связывающими и разрыхляющими орбиталями увеличивается
с ростом степени перекрытия волновых функций. При
добавлении большого числа орбиталей формируется
зона с непрерывным спектром, что показано затененной областью на нижнем рисунке
Как показано на рис. 6.9, в металлах орбитали находятся близко друг к
другу и формируют полосу почти непрерывного спектра энергий электронов. В
действительности просто физически невозможно определить расстояние между уровнями. Поведение зон во многих отношениях подобно поведению орбиталей молекул, отраженному на рис. 6.8. При слабом перекрытии волновых
функций слабым будет и взаимодействие, а зона — узкой. Такая ситуация типична для J-орбиталей (рис. 6.10), которые характеризуются ярко выраженными формой и ориентацией, сохраняющимися в металле. Следовательно, перекрытие между индивидуальными J-орбиталями атомов существенно меньше
перекрытия волновых функций внешних s- и /7-электронов. Последние в сильной степени делокализованы, то есть их нахождение не ограничено областью
вблизи какого-либо атома. В результате они формируют газ почти свободных
электронов, заполняющий все внутреннее пространство металла. Следовательно, волновые функции атомарных д/?-электронов сильно перекрываются, и
формируемая при этом зона оказывается очень широкой (см. рис. 6.10).
260 -J\, Глава 6. Реакционная способность поверхности
Понимание особенностей поведения ^/-орбиталей необходимо для выяснения природы и специфики реакционных свойств переходных металлов. Заполненные flf-зоны сужаются по мере продвижения вправо по Периодической таблице, поскольку молекулярные орбитали становятся более локализованными,
а перекрытие волновых функций уменьшается. Так, у меди d-зона заполнена
полностью и находится сразу под уровнем Ферми, в то время как у цинка она
опущена еще ниже и относится уже к внутренним уровням, локализованным
на отдельных атомах. Если мы взглянем на переходные металлы в ряду 3d, 4d и
5d, то легко обнаружим, что d-зона в этом ряду уширяется, поскольку размер
орбиталей увеличивается и степень их перекрывания возрастает.
4р :
4s ■
3d !
Атом
Металл
Рис. 6.10. Схематическое изображение энергетических уровней переходного 3^/-металла. Протяженные s-
и /ьорбитали формируют широкую
5/?-зону, показанную на рисунке
справа. Из более компактных d-орбиталей образуется узкая d-зона
Таким образом, зона — это набор молекулярных орбиталей. Нижние уровни в зоне — связывающие, верхние — разрыхляющие, а расположенные посредине уровни не являются связывающими. Металлы с наполовину заполненной
зоной, как мы увидим ниже, обладают наибольшей энергией когезии, наиболее высокими температурой плавления и свободной поверхностной энергией.
Каждый энергетический уровень в зоне носит название состояния. Важную
роль играет так называемая плотность состояний (ПС), то есть число уровней,
приходящихся на единицу длины энергетического интервала. ПС переходных
металлов часто представляют гладкой кривой (см. рис. 6.10), но в действительности кривая ПС ведет себя сложным образом, и ее ход зависит от структуры
кристалла и его симметрии. Зоны заполняются валентными электронами до
уровня Ферми. В молекуле этот уровень относится к наиболее высокой из занятых молекулярной орбитали (НВЗМО).
6.3.2.1. Работа выхода
Работой выхода называется минимальная энергия, которую необходимо затратить на отрыв электрона от твердого тела и перенос его на бесконечно большое расстояние, на котором потенциальная энергия принимается
равной нулю. Наиболее слабо связаны с твердым телом электроны, находящиеся на уровне Ферми. Все sp- и af-зоны заполняются до тех пор, пока не будут
исчерпаны все валентные электроны, наибольшее значение энергии при этом
называется уровнем Ферми. Уровень вакуума — это минимальное значение
энергии, которое может иметь свободный электрон, движение которого ничем
6.3. Химическое связывание -1\, 261
не ограничено. Таким образом, работа выхода равна разности энергетических
уровней вакуума и Ферми (рис. 6.11). Используя химическую терминологию,
можно сказать, что она эквивалентна потенциалу ионизации — энергии, необходимой для удаления электрона с НВЗМО.
Металл Адсорбированный Свободный
атом атом
Рис. 6.11. Схематическая энергетическая диаграмма для атома, приближающегося к поверхности металла с s/ьзоной. Заметим, что вакуумный уровень является общей точкой отсчета и что потенциал ионизации атома аналогичен работе выхода электронов из твердого тела
6.3.2.2. Электронный газ и модель «желе»
Простейший подход к описанию поведения валентных электронов
в металле основан на модели электронного газа, в которой взаимодействие
между электронами в расчет не принимается и предполагается, что они заклю¬
чены в сосуд объемом V, как это было сделано в гл. 3. Нам необходимо найти
основное состояние для системы, описываемой уравнением Шредингера
[H-ek]Wk= 0, (6.26)
а волновые функции у/к представляют собой плоские волны
Vk=^eikt. (6.27)
Электроны должны подчиняться принципу Паули, а волновые функции
должны обращаться в нуль на поверхности сосуда размером L = К_1/3, что
задает возможные значения вектора к:
k = f К’ ПУ> nz)-
(6.28)
262 -J\, Глава 6. Реакционная способность поверхности
Соответствующие собственные значения энергии равны
<*■*>
где т — масса электрона. Поскольку характерное число электронов N в образце металла составляет величину порядка 1023, можно считать, что возможные
значения вектора к непрерывно заполняют сферу в &-пространстве радиусом kF
и объемом VF = (An/i)k3F. Число возможных квантовых состояний равно объему VF, поделенному на объем, приходящийся на одно состояние:
(6J0)
(2л/L)
бтг 2
где двойка в правой части появилась из-за того, что в каждом состоянии может
находиться два электрона (со спинами, направленными вверх и вниз). При Т = О К
все состояния ниже kF заняты, а при более высоких температурах распределение электронов по энергиям является фермиевским
fie) de= . * , (6.31)
J V ’ е{?-м)1къТ + |
где ju — химический потенциал электронов, который при Т= О К равен juT=Q = ef.
Обычно при рассмотрении занятости энергетических уровней мы использовали распределение Больцмана. Однако это распределение пригодно лишь в
случаях, когда число доступных состояний существенно превосходит число занятых уровней, то есть случай высоких температур и низких плотностей. При
рассмотрении электронов в металле следует учесть, что при обычных температурах число состояний примерно равно числу электронов. И мы должны использовать статистические распределения для фермионов и бозонов [в случае
фермионов TV-частичная волновая функция должна изменять знак при перестановке двух частиц (или быть асимметричной), а для бозонов — не изменяться
(быть симметричной)]. К фермионам относятся электроны, протоны и соответствующие античастицы, а к бозонам — фотоны, фононы и плазмоны. Все
известные частицы попадают в один из этих классов. Существенное различие
между частицами этих классов состоит в том, что у бозонов нет ограничений
на число частиц, находящихся в данном состоянии, в то время как у фермионов в данном состоянии могут находиться только две одинаковые частицы (принцип Паули). Интересующихся следствиями из этого фундаментального различия мы отсылаем к книге [5]. Заметим, что распределение Ферми при высоких
температурах переходит в распределение Больцмана (см. рис. 6.14).
Для нахождения плотности состояний рассмотрим выражение
6.3. Химическое связывание -i\, 263
и, используя соотношение
dk 2т V т
получаем
2*!* (6.33)
Это выражение также можно записать в виде, содержащем искомую величину:
^r = ]f[e{k)]n(e)de, (6.35)
О
откуда окончательно получаем
3/2
■м-
Таким образом, плотность состояний (ПС) пропорциональна корню квадратному из энергии. Хотя приближение свободного электронного газа представляет собой простую модель, она все же с хорошей степенью точность описывает свойства электронов в ряде металлов типа Na, R, Cs, Са, Ва, Mg и
отчасти А1. В этих металлах, называемых иногда металлами свободных электронов, валентные 5- и /7-электроны «размазаны» по металлу на фоне положительно заряженных ядер и могут быть описаны в рамках модели газа свободных
электронов, заполняющих все состояния от нулевого до уровня Ферми. Для
этих металлов энергия электронов как функция волнового вектора и плотность
состояний как функция энергии показаны на рис. 6.12.
«О
о;
s
|_
Q.
I
Рис. 6.12. Энергия как функция об- СО
ратной величины волнового вектора и
плотность состояний свободного электронного газа
Гипотетическая модель «желе» для металла представляет собой сплошной
положительно заряженный фон из «размазанных» ядер, в котором распределено «море» свободного электронного газа (рис. 6.13).
Притяжение электронов положительно заряженным фоном не является
достаточно сильным, чтобы удержать электроны внутри металла. В результате
электроны частично выходят в область вакуума, то есть плотность электронов в
264 Глава 6. Реакционная способность поверхности
непосредственной близости у поверхности металла отлична от нуля. Поскольку заряд «вышедших» электронов не компенсируется положительным фоном,
то у поверхности существует двойной электрический (дипольный) слой, отрицательно заряженная часть которого находится снаружи металла. Поэтому электрон, движущийся из объема металла в вакуум, должен преодолеть потенциальный барьер высотой Ф.
Поверхность
Рис. 6.13. Распределение плотности
электронов в модели «желе» приводит к формированию двойного электрического слоя у поверхности металла, который дает поверхностный
вклад в работу выхода электронов.
Электронная волновая функция выходит в вакуум, но затухает экспоненциально, поскольку в классической механике эта область недоступна для электронов. Тем не менее
электроны в области вакуума могут
быть детектированы методами сканирующей туннельной микроскопии
Энергия, необходимая для прохождения поверхностного дипольного слоя,
представляет собой поверхностный вклад в работу выхода электронов. Она в
существенной степени зависит от структуры поверхности: для поверхности (111)
ГЦК-металлов, которая является наиболее плотно упакованной, работа выхода
имеет наибольшее значение, поскольку дипольный барьер высок. Более «разреженные» поверхности типа (110) ГЦК-металлов характеризуются меньшей
работой выхода. При наличии большого числа дефектов на поверхности работа
выхода снижается по сравнению с работой выхода для идеальной поверхности.
Для данной структуры поверхности работа возрастает при движении слева направо по Периодической таблице.
Работа выхода Ф определяется как работа, необходимая для переноса электрона из объема металла на бесконечность. Характерные значения работы выхода лежат от 2,5 до 5,5 эВ. Электрон может свободно перемещаться в пространстве, если он энергетически возбужден, по крайней мере, до уровня вакуума, как показано на рис. 6.14.
Рис. 6.14. Изменение плотности состояний с температурой. Заметим, что при повышении температуры электроны начинают занимать состояния с
более высокой энергией
6.3. Химическое связывание -1\* 265
Рис. 6.14 наглядно объясняет, почему для наблюдения заметной эмиссии из
электронной пушки необходимо нагревать нить накала до высоких температур.
Из нити накала могут вылететь только электроны, имеющие энергию выше Ф.
Заселенность энергетических состояний на уровне вакуума может быть аппроксимирована выражением е~ф/квТ, из которого вытекает хорошо известная
формула Ричардсона—Дэшмана, задающая величину тока термоэмиссии j как
функцию работы выхода Ф и температуры Т\
= 4 я те е-Ф/*вг (6 37)
{2яП)г
Нити накала делают из тугоплавких металлов типа вольфрама или иридия,
способных выдерживать нагрев до высоких температур (Т > 3000 К) в течение
длительного времени. Срок службы нити накала как источника электронов
может быть существенно продлен при нанесении на ее поверхность адсорбата, снижающего работу выхода, что позволяет понизить рабочую температуру.
В качестве такого адсорбата можно использовать торий, который частично
ионизуется на поверхности, передавая электроны нити накала, что приводит к
образованию дипольного слоя, снижающего работу выхода электронов из вольфрама. В катализе, как мы увидим ниже, для изменения влияния работы выхода металлов используются щелочные металлы.
6.3.2.3. Модель сильной связи
Сейчас для описания зонной структуры металлов мы используем
рассмотреную выше модель формирования связи в молекулах. Начнем с «цепочки» из двух атомов. Для нее может быть применен результат, полученный
для гомоядерной двухатомной молекулы: имеется два энергетических уровня,
один из которых является связывающим, а другой — разрыхляющим. При добавлении в цепочку новых атомов будут появляться дополнительные уровни
энергии по одному на каждый добавленный электрон. Этот процесс будет продолжаться до тех пор, пока не сформируется непрерывная энергетическая зона
(см. рис. 6.9). Чтобы описать структуру энергетических зон электронов в металле количественно, мы должны определить сначала гамильтониан такой системы. Для простоты мы ограничим наше рассмотрение одномерным приближением.
Каждый атом в цепочке имеет внешний электрон с энергией связи е0 и ряд
электронов, расположенных на более глубоких внутренних уровнях, которые
мы в расчет принимать не будем (рис. 6.15).
Полная потенциальная энергия для цепочки атомов, расположенных на
расстоянии а, может быть представлена суммой индивидуальных потенциалов
атомов:
V(x) = ^V(x-na), (6.38)
П
дающей периодический потенциал, соответствующий периоду решетки.
266
\
Глава 6. Реакционная способность поверхности
Рис. 6.15. Профиль потенциальной
энергии в одномерном твердом теле с
постоянной решетки а
Внутренние
уровни
Собственные значения энергии отдельного атома находятся из уравнения:
#atom ¥ =
.I V2 + V
_ * ' ' of
¥ =
(6.39)
Полная волновая функция цепочки строится в виде линейной комбинации
волновых функций, отвечающих атомным орбиталям:
У = ^сп¥{х-па),
(6.40)
что выражает собой приближение J1KAO для данной системы. Гамильтониан
цепочки аппроксимируется атомным потенциалом, к которому добавлен член,
учитывающий перекрытие орбиталей соседних атомов:
Н=Нмот + AV(x),
(6.41)
следовательно, волновая функция ^из (6.39) будет решением уравнения Шре-
дингера для цепочки атомов, если только ДК= 0. Полная волновая функция Ч7
строится с учетом теоремы Блоха, согласно которой волновая функция частицы в периодическом потенциале представляет собой произведение плоской
волны и волны той же периодичности, что и потенциал. Плотность электронов
п(х) определяется квадратом модуля волновой функции:
«(х) = |Ч'(х)|2 =YJc1„\v{x-na)\
(6.42)
Пренебрегая концевыми эффектами, мы можем записать п(х) = п(х + Са),
что подразумевает равенство с\ - с2+1 при любом целом значении числа С. Выбирая сп в виде сп = (1 /N)elkna (с / = V^T), получаем искомую волну Блоха:
(6.43)
Теперь легко найти характеристический определитель, который является
тридиагональным, поскольку у него отличны от нуля только элементы, рас¬
6.3. Химическое связывание
X
положенные на главной диагонали, и по два соседних им элемента. Следовательно,
\VlVm =«*/.*>
JV/tfatom^m = *<Л «
\y/,AV(x)yfm =-aS,
J w,AV(x)wm =~ps,
(6.44)
m± 1 •
где Sj m — символ Кронекера (S/ m = 1 для I = m и m = 0 для l Ф m). Из этих
соотношений получаем следующее выражение для энергии k-то уровня:
Ек= е0 — а — fi(elka + е~1ка) = — а — 2flcos(ka), (6.45)
представляющее, как показано на рис. 6.16, полосу непрерывных состояний
шириной 4/?.
Рис. 6.16. Зонная структура со связывающими орбиталями в нижней и
разрыхляющими орбиталями в верхней части. Ширина зоны (максимальное расстояние между связывающими и разрыхляющими орбиталями) равна 4/?. Полоса показана
только в приведенной зоне, соответствующей одной постоянной обратной решетки
'of
-G0/2 = -п/а
Gq/2 = п/а
Важный результат, вытекающий из рассмотренной модели, состоит в том,
что в одномерной периодической цепочке валентные электроны заполняют полосу непрерывных состояний, содержащую в нижней части связывающие, а в
верхней — разрыхляющие состояния, ширина которой равна четырем интегралам перекрытия для соседних атомов. Мы рассмотрели одномерное приближение, однако полученные результаты легко обобщаются на трехмерный случай.
Ширина полосы оказывается при этом равной интегралу перекрытия, умноженному на число ближайших соседей. Этот результат имеет интересное следствие:
полоса становится уже для поверхности металла, где у атомов имеются недостающие соседи. Такое сужение полос у поверхностных атомов действительно наблюдается в эксперименте, и мы в дальнейшем используем этот факт для объяснения зависимости реакционной способности поверхности от координационного числа и степени сжатия или растяжения поверхностных адслоев.
267
268 -J\, Глава 6. Реакционная способность поверхности
Изложенное выше дает полезные основы теории зонной структуры твердых
тел. В действительности зонная структура является более сложной, чем изображенная на рис. 6.16, что объясняется и трехмерностью реальных систем, и
участием орбиталей разного типа в формировании зонной структуры. Особенности электронной структуры определяют в конечном итоге физические и химические свойства твердых тел, в частности, будет ли твердое тело проводником, полупроводником или изолятором (рис. 6.17).
Металл Переходный Изолятор
электронного газа металл
Рис. 6.17. Диаграмма плотности состояний, схематически показывающая плотность состояний электронов вблизи поверхности Ферми для металлов электронного
газа, переходного металла и изолятора
Чтобы материал был хорошим проводником, необходимо обеспечить возможность возбуждения (перевода) электрона из валентной зоны (ниже уровня Ферми) в зону проводимости (свободные состояния выше уровня Ферми),
когда он может свободно перемещаться в твердом теле. Принцип Паули запрещает такое перемещение для состояний ниже уровня Ферми, где все состояния заняты. В металлах электроного газа, к которым относится рис. 6.14,
имеется множество электронов в зоне проводимости, как и дырок в валентной зоне, при любой ненулевой температуре, что обеспечивает их высокую
проводимость. К этим металлам относятся натрий, калий, кальций, магний и
алюминий.
Переходные металлы также являются хорошими проводниками, поскольку
имеют такую же д/ьзону, как металлы электронного газа, плюс частично заполненную flf-зону. Металлы группы IB, медь, серебро, золото, представляют пограничный случай, поскольку у них d-зона заполнена и расположена на несколько электронвольт ниже уровня Ферми. Наличие sp-зоны, однако, обеспечивает их хорошую проводимость.
В оксидах, например в оксиде алюминия А1203, s/ьзона алюминия гибриди-
зуется с /7-орбиталью кислорода, что приводит к образованию новой зоны, расположенной ниже уровня Ферми, в результате остается щель шириной 7 эВ,
отделяющая ее от разрыхляющей части зоны. Нижняя часть относится к валентной зоне, верхняя часть — к зоне проводимости, а щель между ними — запре¬
6.3. Химическое связывание -1\, 269
щенная зона. Этот материал является изолятором, поскольку электроны достаточно сложно возбудить с переводом их в зону проводимости, чтобы они могли
свободно перемещаться по объему оксида. Ширина запрещенной зоны обычно
превосходит 5 эВ, например для алмаза она составляет 5,4 эВ.
У полупроводников запрещенная зона существенно уже (например, для
кремния она равна 1,17 эВ). При низких температурах их проводимость падает
до нуля, но существенно возрастает при высоких температурах. Она в значительной мере зависит от наличия примесей в образце, которые, если они вводятся целенаправленно, называются легирующими добавками. Легирующие
добавки позволяют легко регулировать ширину запрещенной зоны полупроводников и придавать им желаемые характеристики, необходимые для создания электронных устройств [3, 4].
Оксиды, в частности MgO, А1203, Si02, являются хорошими изоляторами и
в высокой степени инертными материалами, поскольку электроны участвуют в
формировании химической связи и спускаются по энергии вниз далеко от уровня
Ферми. Только при наличии дефектов или примесей, поставляющих неспаренные электроны, они начинают проявлять некоторую активность. Ниже мы сконцентрируем свое внимание на более реакционно активных металлах.
6.3.2.4. Простая модель переходных металлов
В данном разделе мы, не вдаваясь в детали, опишем основные закономерности изменения реакционной способности металлов. Будет рассмотрено достаточно грубое приближение. Электронная структура многих металлов
во много сходна в отношении формирования sp-зоны, особенно это касается
металлов электронного газа. Различия связаны в основном с разной степенью
заполнения af-зоны. Мы исключим из рассмотрения лантаниды и актиниды, у
которых локализованная /-орбиталь заполнена, поскольку они не играют особой роли в катализе.
Наше представление о металлах е 4
отображает рис. 6.18, на котором в виде
блоков представлена плотность состояний для частично заполненной д/ьзоны,
отвечающей электронному газу, и заполненной до определенного уровня £
flf-ЗОНЫ. sp-Зона является широкой, A w Плотность
поскольку она сформирована в суще- состояний
ственной мере делокализованными % ~ w/2
электронами, «размазанными» по решетке. d-Зона, напротив, существен- у>-зона
но уже, поскольку перекрытие d-орбиталей существенно меньше из-за их
сильной локализации на атомах.
Если мы рассмотрим выигрыш
энергии, получаемый при образовании
flf-зона
Рис. 6.18. Плотность состояний для переходного металла
с почти заполненной */-зоной
в верхней части частично за-
п ол н е н н oil sp - зо н ы
270 -J\r Глава 6. Реакционная способность поверхности
металла из отдельных атомов, то получим, что sp-зона вносит вклад порядка 5 эВ.
Изменение энергии образования переходных металлов связано только со вкладом от d-зоны. Рассмотрим свойства металлов и вклад в энергию их образования более подробно.
Аппроксимируем, как это показано на рис. 6.18, d-зону узким прямоугольником, с параметрами
/ ч 2 w w
п\Ч = — ПРИ “у < £ < £о +~2 v£f>
W (6.46)
I \ л w w
п(е) = 0 при е < £0 - — we > £0 + — ,
где е0 — центр d-зоны; п(£) = 2/w — плотность состояний; w — ширина d-зоны,
обусловленная перекрытием атомных орбиталей. Степень заполнения /— число, лежащее между 0 и 1, определим как
г 1 Г ( \ л €F ~ £q + w/2 ,_ч
/в- J п(е)йе = -!- ^(6.47)
f0 - wj2 ^
Выигрыш энергии, получаемый при гибридизации d-орбиталей, равен
д£иуь= J H(g)(g-g0)dg = g°^ -j. (6.48)
Sq-w/I W 4
С учетом введенного определения для / имеем
Л£иуь = -/(l-/)w. (6.49)
Вспоминая о наличии ответственного за отталкивание члена, пропорционального интегралу перекрытия и числу соседей, окончательно имеем
АЕ = E0NnnS\p\ - AEhyb = EoNmS\0| -/(1 - f)w. (6.50)
Из написанного выражения видно, что энергия когезии d-металлов квадратичным образом зависит от степени заполнения зоны /
Энергия когезии равна энергии, необходимой для отрыва одного атома от
твердого тела. Или ее можно рассматривать как выигрыш энергии, приходящийся на один атом, достигаемый при образовании твердого тела из отдельных
атомов. Высокой энергии когезии соответствует высокая температура плавления. Такие металлы иногда называют тугоплавкими.
Несмотря на пренебрежение в вышеприведенном рассмотрении изменением вкладов от интеграла перекрытия и магнитных членов, полученная зависимость для энергии когезии, как видно из рис. 6.19, достаточно хорошо воспроизводит экспериментальные данные. Показанный на рис 6.19 характер изменения энергии когезии легко понять, если вспомнить, что у заполненной менее
чем наполовину d-зоны заняты только связывающие орбитали, а разрыхляю¬
6.4. Химическая адсорбция ->\г 271
щие орбитали начинают заполняться только при / > 0,5. Отсюда следует, что
максимальные значения энергии когезии должны быть у металлов с наполовину заполненной d-зоной.
1
00
0
S
S
СО
0
Рис. 6.19. Энергии когезии для трех о
групп переходных металлов. Заме- *
тим, что максимальные значения 5
энергии когезии имеют, как это §■
предсказывает уравнение (6.49), 4d- ^
и 5^-металлы, у которых d-зона заполнена примерно наполовину. Наблюдаемые отклонения для 3 J-серии
связаны главным образом с их магнитными свойствами [4]
Другая интересная особенность, которую объясняют зависимости (6.49)
или (6.50), состоит в увеличении энергии когезии в ряду 3d-, 4d, 5d-мeтaллoв
(рис. 6.19). По мере продвижения вниз по Периодической таблице размер орбиталей увеличивается и степень перекрытия возрастает. Это означает, что зоны
становятся шире и растет величина w, входящая в уравнения (6.49) и (6.50).
После небольшой экскурсии в физику твердого тела мы возвращается к
нашей исходной задаче — к описанию взаимодействия между адсорбатом и
поверхностью.
Sr Y Zr Nb Мо Тс Ru Rh Pd Ag Cd
Ba La Hf Та W Re Os Ir Pt Au Hg
6.4. ХИМИЧЕСКАЯ АДСОРБЦИЯ
В предыдущем разделе были установлены основные вехи, необходимые для описания процессов, происходящих при приближении молекулы к поверхности металла. Наиболее важные характерные черты хемосорбции хорошо
воспроизводятся моделью Ньюнса—Андерсона [6, 7], которая будет рассмотрена
в подразд. 6.4.1. Читатель, недостаточно хорошо знакомый с квантовой механикой и ее математическими методами, но желающий знать о предсказаниях этой
модели, может пропустить этот раздел и сразу перейти к подразд. 6.4.2, где дана
сводка результатов на качественном уровне. Можно также обратиться к книге
[8], с которой мы настоятельно рекомендуем ознакомиться всем читателям.
6.4.1. Модель Ньюнса—Андерсона
Рассмотрим сначала простой атом с валентным электроном, имеющим энергию £а и волновую функцию Фа, который адсорбируется на поверхности твердого тела, в котором электроны занимают набор состояний из непре¬
272
\
Глава 6. Реакционная способность поверхности
рывного спектра, характеризующихся волновыми функциями Wk и энергиями ек.
Когда адсорбат приближается к поверхности, мы должны описать состояние
всей системы гамильтонианом Я, включающим и энергии отдельных систем, и
потенциал их взаимодействия. Последний вносит вклад через матричные элементы вида V* =1 ФаНх¥к. Мы примем, что решение х¥/. задачи на собственные значения для всей системы в целом может быть представлено линейной
комбинацией волновых функций изолированных систем
у, =с,/Фа+1сЛ.
(6.51)
Ниже мы примем, что набор базисных функций является ортогональным,
то есть матричные элементы типа |ФаЧ/А; равны нулю. Решение находим из
задачи на собственные значения:
ЯЧ',= *,Ч'„
где матричные элементы таковы
= Няа = еа,
j'¥kH'¥k=Hkk=ek,
|Ф aHVk=Vak=Vka,
В результате характеристический определитель имеет вид
(6.52)
(6.53)
#аа ^
К,
Уаг
Кз -
-
К.
Н„-е
0
0
0
V2a
0
#22 -е
0
0
Ка
0
0
#зз-* •
0
К.
0
0
0
... Нт - £
= 0.
(6.54)
Как мы уже видели, у электронов в металле имеется бесконечно большое
число состояний к (п °°), и довольно сложно проследить за всеми ними.
Поэтому удобно рассмотреть проекцию новых состояний x¥i на исходное состояние адсорбата Фа. Это позволяет проявить характер изменения состояния адсорбата по мере его сближения с поверхностью и технически осуществляется
через расчет величины
6.4. Химическая адсорбция
\
в которой суммирование идет по всем собственным функциям Wr Теперь нам
необходимо использовать математический прием, связанный с введением функции Грина, что позволит преобразовать выражение (6.55) к виду
пя (е) = — lim Im
aV 1 л s->0+
Гч>,.Фа/Фау;
/ {e-£; + iS)
(6.56)
Используем тот факт, что функция Лоренца
8
f(£)
df = l,
+<52]
переходит в дельта-функцию при S 0+, то есть
+°° +°° п
-ооЯ\{£- £i) + S
f{£) -* оо при е-е{ и f(e) = 0 при е * ег
Преобразование осуществляется так
lim Im
<?-*о+
(6.57)
(6.58)
1 ]
= lim Im
i
1
id
(е-е, +iSj
_(е-е,)2+63
{e-£if+S\
<^о+
nS
lim —=
<^0+ n\^{e- e^' +^2]
n8 (e - e t).
(6.59)
Так называемая одночастичная функция Грина теперь вводится следующим образом
пя Ы = — lim Im
Л 8^ 0+
у (e-e,+iS)
Это соотношение можно представить в виде
jfj®,
(6.60)
■f{e- e, + is)'
(6.61)
где G(f) 1 — матрица, обратная e— H + id, и оно по сути является формальным
определением величины G(£)_1, то есть
(е- Н + /<У)С(£->-1 = I.
(6.62)
273
274
\
Глава 6. Реакционная способность поверхности
Последнее соотношение доказывается достаточно легко
Jy/Jy/
(е - Н + iS)G(e)~' ='£(е-Н + iS)
i
[e-Ej +iS)
= £|чур;=1,
(6.63)
/ (e- £i + IS)
и, следовательно, функция Грина в принципе определяется как обратная матрица
G(e)~x = (г - Я + iSy\ (6.64)
Перепишем теперь произведение в левой части равенства (6.62) в матричной форме и приравняем его единичной матрице
-к,
-Via е'-Ни
-V Л
г а п
о
-V..
о
... е'-Н,
пп )
Gaa
Ga\ ■
- О
f 1
0 ...
0"
Gla
G\\ ■
•• G{n
0
1 ...
0
= 1, (6.65)
=
Gna
Gn\ ■
Gnny
v0
0 0
К
(6.66)
где е' = е + iS. Отсюда сразу же получаем соотношения, содержащие Gaa:
Gaa{e-ea+iS)~YJVakGka = 1,
k
~KaGa + _ £k + l$)Gka =
которые позволяют найти Gka:
-V. G
(6.67)
r = ~VkaGaa
k° = (£-£k+isy
Подставляя это выражение в уравнения (6.66), можно исключить Gka, что дает
1
(e-ea+ iS) - £КаУ(гг -ек+18)'
(6.68)
Полагая S-) 0+, вводя комплексную функцию
?(г).Л(гИМг).£(с_^и).
и учитывая тот факт, что <7аа можно представить в виде
1
GL =
[e-ea
, <У->0+,
(6.69)
(6.70)
6.4. Химическая адсорбция
приходим к следующему выражению для А{е):
Д(*) = *£ка\S(e-ek), £->0+.
(6.71)
k
Фактически это матричный элемент переходов между состоянием адсорбата, обозначенным через «а», и состоянием металла «к». Таким образом, Д(^)
является локальной проекцией состояний металла на состояние адсорбата. Действительная часть А(е) находится с помощью соотношений Крамерса—Крони-
га. Зная мнимую часть комплексной функции, можно найти ее действительную
часть через главное значение следующего интеграла
Напомним, что интересующая нас величина — это па{е). Теперь становится
очевидной целесообразность введения функции Грина через соотношение
Иными словами, мы выразили взаимодействие между адсорбатом и металлом в терминах функций А{е) и Л(^), представляющие интеграл перекрытия
волновых функций состояний адсорбата и металла, умноженный на матричный элемент перехода. А(е) фактически есть преобразование Крамерса—Кро-
нига от А(е). Рассмотрим несколько простых случаев, для которых полученные
выражения поддаются легкой интерпретации.
6.4.1.1. Случай 1: атом на поверхности металла
электронов разной энергии имеет одно и то же значение. Конечно, таких металлов не существует, но эта ситуация легко поддается расчету. Примем также,
что матричные элементы Vak также постоянны. Это означает, что А(е) просто
пропорциональна плотности электронов в металле
(6.72)
Л
(6.73)
с постоянной плотностью состояний электронов
Рассмотрим случай, когда у твердого тела плотность состояний
Л(£) = A0-
(6.74)
276
\
Глава 6. Реакционная способность поверхности
Из соотношения Крамерса—Кронига сразу получаем, что Л(^) = 0, поскольку подынтегральная функция нечетная, а проекция плотности состояний
равна
А°г д,- <6.75)
Я{е-ея) +Д5
Энергия, эВ
Рис. 6.20. Распределение па(е) для
адсорбата с электронным уровнем
при еа = 12 эВ, приближающегося к
поверхности металла с sp-зоной, характеризующейся постоянной плотностью состояний A(s) = Д0. Распределение па(£) имеет профиль лоренцов-
ской линии с центром на исходном
уровне электрона адсорбата. Уровень
с нулевым значением энергии выбран так же, как и на следующих двух
рисунках, но в данном случае он не
представляет интереса
Это выражение говорит о том, что энергетический уровень электрона у
адсорбата уширяется, имеет лоренцовскую форму с шириной Д0. Данный результат означает, что электрон совершает прыжки с молекулы адсорбата в металл и обратно, результатом чего стало уширение энергетического уровня, связанное с принципом неопределенности Гайзенберга для времени и энергии
AeAt > Ь/2. Полученное распределение па(е) приведено на рис. 6.20.
6.4.1.2. Случай 2: атом на поверхности металла
с sp-гибридизацией электронов
Сейчас мы рассмотрим более реалистичный случай, когда плотность состояний дается уравнением (6.36) и имеет вид, показанный на рис. 6.14.
На рис. 6.21 мы поместили уровень нулевой энергии на дне валентной зоны и
привели для сравнения плотность состояний электронов в 5/7-зоне. Слабое изменение плотности состояний электронов в металле в области их перекрытия с
атомным состоянием практически не сказывается на величине взаимодействия,
так что А(е) в этой области имеет такую же форму, как и плотность состояний
электронов в s/ьзоне, то есть Д(^) пропорциональна yfe. При более высоких
энергиях, однако, взаимодействие становится более слабым. Это легко понять,
рассмотрев волновые функции высокоэнергетических электронов. Они быстро
осциллируют, так что перекрытие с волновой функцией локализованного на
адсорбате электрона в среднем можно считать равным нулю. Этот эффект учитывается через обращение А(е) в нуль при высоких энергиях, как показано на
6.4. Химическая адсорбция -J\r 277
рис. 6.21. Принять в расчет особенности поведения А(^) можно, описав эту
зависимость эллипсом.
Рис. 6.21. Проекция плотности состояний па{е) для находящегося у
поверхности металла с 5/7-зоной адсорбата с электронным уровнем энергии
еа = 12,0 эВ. Функция Д(£) следует
плотности состояний электронов в sp-
зоне металла, но уменьшается с ростом энергии из-за обращения в нуль
интеграла перекрытия. Объяснения
см. в тексте
Энергия, эВ
Мы рассмотрим атом с теми же параметрами, что были использованы при
анализе первого случая, то есть с валентным электроном на уровне еа = 12,0 эВ
выше дна sp-зоны, который электрон занимает в атоме, находящемся далеко от
поверхности металла. Для удаленного от поверхности атома электронный уровень очень узкий, то есть имеет форму дельта-функции. При приближении
атома к поверхности электронный уровень уширяется и приобретает лоренцов-
скую форму по тем же причинам, что были указаны выше, а также смещается
в новое положение с энергией 10,3 эВ. Из уравнения (6.73) для па(е) легко
установить, что максимум приходится на значение энергии е = еа + А(е), то
есть на точку пересечения прямой у = е — еа и кривой у = А(^), как показано на
рис. 6.21. Таким образом, при взаимодейстии с металлом энергетические уровни электронов у адсорбатов уширяются и сдвигаются в область более низких
энергий (что предполагает более сильное связывание с поверхностью).
6.4.1.3. Случай 3: атом на поверхности переходных металлов
Переходные металлы имеют как широкую з/?-зону, взаимодействие
электронов которой с атомом адсорбата было рассмотрено в случае 2, так и
узкую d-зону, электроны которой сильно взаимодействуют с адсорбатом. Это
взаимодействие может быть описано путем включения в плотность состояний
вклада от d-зоны, что приводит к дополнительным членам в зависимости А(^).
Теперь имеется три корня у уравнения е = еа + А(^), которые определяются по
пересечению прямой у = е — еа и кривой у = А(е) (рис. 6.22). Соответственно
имеем: при низких энергиях связывающее состояние, при средних энергиях —
несвязывающее состояние и при высоких энергиях — разрыхляющее состояние. Таким образом, узкая d-зона проявляет себя как молекулярная орбиталь
по отношению к адсорбату: формируются связывающие и разрыхляющие состояния у комплекса металл/адсорбат. Степень заполнения этих состояний зави¬
278 -J\r Глава 6. Реакционная способность поверхности
сит, помимо всех других факторов, от степени заполнения d-зоны. Для приведенного на рис. 6.22 примера уровень Ферми металла лежит в области от 9
до 12 эВ.
Рис. 6.22. Адсорбция атома на поверхности
d-металла. Валентный электрон адсорбата
имеет энергию 12 эВ по отношению к дну
зоны металла, взаимодействует слабо с электронной системой широкой sp-зоны и сильно — с электронной системой узкой d-зоны,
находящейся в области от 9 до 12 эВ. Отметим сильное расщепление плотности состояний адсорбата пй(£) на связывающие и разрыхляющие орбитали вследствие взаимодействия с электронной системой d-зоны
6.4.2. Качественный анализ результатов
для модели Ньюнса—Андерсона
Модель Ньюнса—Андерсона успешно воспроизводит основные
особенности формирования связи между адсорбатом и металлом и перестройки соответствующих волновых функций. Она может быть использована при
описании закономерностей динамических процессов типа рассеяния ионов,
атомов и молекул на поверхности. В частности, нейтрализация ионов на поверхности достаточно легко описывается в рамках этой модели. Анализ динамических процессов выходит за пределы данной книги, интересущегося читателя мы отсылаем к обзору [9].
В данной секции мы дадим сводку результатов, полученных в рамках простой идеализированной схемы. Мы рассмотрим адсорбцию на поверхности
металла электронного газа и на поверхности переходного металла. Заметим,
что адсорбция молекул на поверхности металла, характеризующегося наличием d-состояний, представляет огромный интерес для катализа.
S
о
а
с
sS
S
X
о
о
о
►Д
н
о
о
X
н
о
а
У= е-еа
А(£)
г
пМ)
Связывающая
орбиталь
Д(г) d-зона
"a(f)
Разрыхляющая
орбиталь n(e)ocyfe
ДПГ) sp-зона
£-а = 12 эВ
О 10 20 30 40
Энергия, эВ
6.4. Химическая адсорбция ->\г 279
6.4.2.1. Адсорбция на поверхности металла электронного газа
У металлов электронного газа имеется только широкая sp- юна. При
сближении адсорбата с металлом его электронные уровни уширяются и сдвигаются вниз по энергиям, что означает большую устойчивость адсорбата, находящегося на поверхности. Взаимодействие металл/атомарный адсорбат обычно
дает энергию связи около 5 эВ. Эта ситуация проиллюстрирована на рис. 6.23.
Рис. 6.23. Энергетические уровни
адсорбата уширяются и сдвигаются
вниз по энергиям при его сближении с поверхностью металла, имеющего широкую 5/7-зону. Заметим,
что первоначально свободный верхний электронный уровень адсорбата заполняется, когда из-за взаимодействия он спускается ниже
уровня Ферми металла
Металл Уровни адсорбата
6.4.2.2. Адсорбция атомов на поверхности
переходных или d-металлов
Плотность состояний переходных d-металлов определяется широкой зр-зоной и узкой, но емкой d-зоной, которые обе заполнены частично (см.,
например, рис. 6.17). Кроме того, сильное взаимодействие с электронной системой J-зоны приводит к уширению и сдвигу уровня адсорбата, как и в рассмотренном выше случае. Действительно, J-зону можно рассматривать как
энергетически широкую орбиталь, и ее гибридизация с уровнем адсорбата приводит, как показано на рис. 6.24, к формированию пары хемосорбционных
орбиталей — связывающей и разрыхляющей. Разрыхляющая орбиталь будет
заполнена, если она находится ниже уровня Ферми металла, что приводит к
частичному ослаблению адсорбционной связи.
Рис. 6.24. Взаимодействие между
атомарным адсорбатом, имеющим
один валентный уровень, и переходным металлом, обладающим широкой 5/7-зоной и узкой d-зоной, рас-
положенной у уровня Ферми. Сильное взаимодействие с электронной
системой d-зоны приводит к расщеплению уровней адсорбата на связывающие и разрыхляющие уровни.
Часть уровней адсорбата, расположенных ниже уровня Ферми, заполняется электронами
Уровни адсорбата
Расстояние от поверхности
Расстояние до поверхности
280
Л
Глава 6. Реакционная способность поверхности
6.4.2.3. Адсорбция молекул
на поверхности переходных металлов
Наконец, рассмотрим хемосорбцию молекулы, имеющей пару орбиталей, связывающую и разрыхляющую, на поверхности переходного металла
(рис. 6.25). Эта ситуация соответствует молекуле Н2, у которой на связывающей орбитали имеется два электрона, а разрыхляющая орбиталь свободна (можно
привести примеры и других молекул). В принципе мы должны повторить дважды описанную в подразд. 6.4.2.2 схему анализа: первый раз для связывающей
орбитали, а второй — для разрыхляющей. Эта процедура проиллюстрирована
на рис. 6.25.
Рис. 6.25. Связывающая <т- и разрыхляющая <т*-орбитали молекулы взаимодействуют с
орбиталями широкой 5/7-зоны и узкой d-зоны переходного металла. В первом
случае происходит уширение и сдвиг уровней, а во втором — их расщепление
с формированием связывающей и разрыхляющей орбиталей. Заметим, что если
электроны заполняют разрыхляющую орбиталь молекулы, то внутримолекулярная связь ослабевает; это может привести к диссоциации молекулы
Интересно отметить, что и связывающая, и разрыхляющая орбитали молекулы дают вклад в формирование хемосорбционной связи (см. рис. 6.25). Принципиально важным является тот факт, что заполнение исходной разрыхляющей орбитали молекулы упрочняет ее связь с поверхностью, но ослабляет внутримолекулярную связь адсорбированной молекулы. В этом лежит ключ к
пониманию механизма диссоциации молекул на поверхности. Следует заметить, однако, что значительное заполнение разрыхляющей орбитали молекулы, обеспечивающее ее диссоциацию, может произойти в переходном состоянии, например, когда молекула прогибается к поверхности, увеличивая тем
самым степень перекрытия ее орбиталей и орбиталей J-состояний. Наконец
укажем, что заполнение разрыхляющих орбиталей электронами металла часто
называют «обратным донорством».
Металл
Уровни адсорбата
vac
Расстояние от поверхности
6.4. Химическая адсорбция
6.4.3. Электростатические эффекты в адсорбции атомов
в модели «желе»
Стоит рассмотреть, каков характер переноса заряда, вызываемого адсорбцией, поскольку он существенно сказывается на работе выхода электронов.
Рассмотрим атом, приближающийся к поверхности, как показано на рис. 6.23.
Если исходно на верхнем уровне атома находится электрон, то при адсорбции
часть электронной плотности будет перенесена в область металла, и на атоме
окажется положительный заряд. Это ситуация типичная для щелочных металлов. В результате на поверхности формируется диполь с положительным зарядом, направленным наружу, то есть он действует против поверхностного двойного электрического слоя, определяющего работу выхода электронов из металла
(см. рис. 6.13). Таким образом, атомы щелочных металлов снижают работу
выхода потому, что у них имеется высокий ^-уровень, с которого электроны
переходят в поверхностный слой металла.
При адсорбции атома со свободным уровнем, характеризующимся высоким
сродством к электрону, он, наоборот, захватывает электрон. Следовательно, заряд переносится от металла к атому. В результате формируется диполь, усиливающий вклад поверхности в работу выхода. Это ситуация, типичная для атомов
галоидов, которые на поверхности заряжаются отрицательно. Ниже мы увидим,
что формирование таких дипольных полей позволяет объяснить эффекты ускорения и замедления каталитических реакций различными адсорбатами.
Более детальная информация может быть получена путем расчета профиля
электронной плотности методом функционала плотности. Мы не будем объяснять суть этого метода, а приведем результаты, полученные для адсорбции атомов С1 и Li на модельном металле со структурой «желе» в [10]. На рис. 6.26
приведены закономерности изменения плотности состояний электронов при
адсорбции атомов кремния, хлора и лития.
Рис. 6.26. Изменения плотности состояний, вызванные адсорбцией Cl, Si и Li
на поверхности металла,
определенные методом функционала плотности для
модели «желе». Видно, что
литий заряжается положительно, а хлор — отрицательно [10]
з5
S
X
05
О
н
о
о
о
S
nj
н
<и
о
о
CQ
X
го
н
5
о
о
о.
с
с
<и
S
X
<и
X
<и
£
го
S
-15 -10 -5
Энергия относительно уровня вакуума, эВ
281
282 -J\r Глава 6. Реакционная способность поверхности
Заметим, что нулевой уровень энергии выбран для вакуума и все уровни
ниже уровня Ферми заполнены. Для лития мы рассмотрим широкий 2^-уро-
вень, потенциал ионизации которого для свободного атома составляет 5,4 эВ.
Расчеты, проведенные методом функционала плотности, показывают, что при
хемосорбции этот уровень поднимается выше уровня Ферми (это противоречит изложенной выше общей картине, но не изменяет выводов), так что он
остается преимущественно свободным. Таким образом, атомы лития на поверхности металла-желе существуют в состоянии Li^, где S примерно равно 1.
При адсорбции хлора основное воздействие оказывается на незанятый 3/7-уровень, имеющий высокое сродство к электрону — 3,8 эВ. Этот уровень, сдвигаясь при хемосорбции вниз, оказывается ниже уровня Ферми, и на него переходят электроны из металла, так что заряд хлора на поверхности становится равным — 1. Кремний при адсорбции остается преимущественно нейтральным.
Картины распределений электронной плотности вблизи атомов несут важную информацию. Они показывают, на какое расстояние простирается возмущение, вносимое адатомом, или, на языке катализа, сколько адсорбционных
центров активируется или пассивируется одним адатомом. На рис. 6.27 показаны контуры плотности в виде линий постоянной электронной плотности. Заметим, что эти контуры близко отражают форму адсорбированных атомов и
электроны локализованы в близкой окрестности адсорбированных атомов и
адсорбционных центров.
Рис. 6.27. Контуры постоянной плотности заряда при адсорбции Cl, Si и Li на металле-желе:
а — полный заряд; 6 — индуцированный заряд. Сплошные линии
показывают увеличение электронной плотности, пунктирные —
уменьшение. Отметим формирование диполей с большим моментом
при адсорбции Li и С1 1101
Более интересны разностные плотности, полученные вычитанием плотности
заряда при наличии и в отсутствие взаимодействия адсорбат/металл. Сплошными линиями показано увеличение электронной плотности (избыточный отрицательный заряд), а пунктирными — убыль электронной плотности (дефицит отрицательного заряда). Эти распределения ясно демонстрируют формирование
диполей и их влияние на работу выхода.
6.5. Важные закономерности поведения реакционной способности поверхности
6.5. ВАЖНЫЕ ЗАКОНОМЕРНОСТИ ПОВЕДЕНИЯ
РЕАКЦИОННОЙ СПОСОБНОСТИ ПОВЕРХНОСТИ
Комбинируя результаты, полученные в рамках модели Ньюнса—
Андерсона и модели сильной связи, можно объяснить ряд закономерностей в
поведении реакционной способности поверхности. Это было сделано Норско-
вым с соавторами в их обзоре [11]. Здесь мы отдельно рассмотрим адсорбцию
атомов и молекул.
6.5.1. Закономерности поведения энергии
хемосорбции атомов
Когда атом с занятым уровнем с энергией ея приближается к поверхности металла, он сначала адсорбируется вследствие взаимодействия с sp-гиб-
ридизованными электронами металла. Рассмотрим, например, атом кислорода. Когда атом изолирован, на уровне 2р находится четыре электрона, но по
мере сближения с поверхностью металла 2/?-уровни уширяются и сдвигаются
вниз по энергии в результате взаимодействия с электронной ^-системой металла. Этот процесс показан на рис. 6.28 для адсорбции кислорода на металле-
желе, то есть для металла электронного газа.
Схематический Кислород Чистая
Схематический атом кислорода на поверхности поверхность
атом кислорода на металле-желе (110) Rh (110) Rh
Рис. 6.28. Схематическая иллюстрация изменения локальной электронной структуры атома
кислорода, адсорбированного на поверхности переходного металла родия, плотность состояний которого показана на правом крайнем рисунке. Гибридизация
орбиталей 2р кислорода и 5/7-зоны переходного металла определена для идеализированной модели металла-желе. Результат этой гибридизации — сдвиг вниз и
уширение 2/ьуровня. Гибридизация орбиталей этого уровня и орбиталей d-зоны
родия приводит к плотности состояний хемосорбированного атома, в которой
связывающия и разрыхляющая орбитали разделены [11]
Все уровни, находящиеся ниже уровня Ферми, заполняются электронами,
что приводит к формированию сильной связи, обусловленной электронной
^-системой, энергия которой составляет —5 эВ. Взаимодействие О — электронная sp-система металла определяет основные закономерности формирова¬
283
284 -J\, Глава 6. Реакционная способность поверхности
ния связи для переходных металлов, поскольку sp-зоны у переходных металлов
схожи. Однако мы должны взглянуть и на J-зону.
Рассмотрим взаимодействие 2/ьорбиталей О и орбиталей узкой d-зоны,
локализованных вблизи уровня ed, верхняя часть которой возмущена ^/7-элект-
ронами. Это позволяет нам трактовать систему как двухуровневую, которую
мы анализировали ранее. Результат анализа представлен на рис. 6.28, в, где
сдвинутая вниз орбиталь кислородных уровней гибридизуется с орбиталями
d-зоны родия таким образом, что орбитали адсорбата расщепляются на связывающую и несвязывающую компоненты. Это позволяет оценить выигрыш в
энергии связи по энергии гибридизации Ed_hyb:
~^d- hyb
2е+ + 2fe_ - 2fed - 2s.,
(6.76)
где/ — степень заполнения d-зоны; г. и е — энергии связывающей и разрыхляющей орбиталей адсорбата соответственно. Эти энергии можно оценить по
выражениям (6.16):
2б\ = 2
К + ed) ~ 2*ST? - yj(£a~£d) +4/?2
- £a + £d ~ 2*ST? - y/S' + 4Р~ ,
2 fe. = 2/
(fa + £d) ~ + >/(ga ~ £d)~ +
8 = £d ~ £a > °-
/(fa + £d ~ 2Sfi + tJ82 + 4/?2 j,
(6.77)
Вводя гибридизационный член
Wad = ,Js2 + 4/?2 (6.78)
и используя приведенные ранее аргументы о том, что интеграл перекрытия
пропорционален матричным элементам взаимодействия S = — у/З, получаем
^-hyb = "О -fm„ -S) + 2(1 +f)r/32. (6.79)
При £ » 4/32 имеем Wad = S + (2/32/S), что приводит к следующему выражению для энергии гибридизации
Ed-ъуь = - О - /)Щ~ + 2(1 + fW = -(1 - f)~^~ + 2(1 + f)yP2. (6.80)
° а — d
Первое слагаемое характеризует притяжение (увеличение энергии связи), а
второе — отталкивание (уменьшение энергии связи). Таким образом, в определении прочности связи атом/^-металл основную роль играют три параметра:
1) степень заполнения d-зоны;
2) матричный элемент /3 взаимодействия между волновыми функциями электронов атома и d-электронов металла;
6.5. Важные закономерности поведения реакционной способности поверхности -J\r 285
3) разность энергетических уровней электрона в исходном атоме и центра
d-зоны.
Матричный элемент взаимодействия /? изменяется с природой металла для
заданных адсорбата и адсорбционного центра на поверхности. Изменение природы адсорбата может быть учтено введением соответствующей константы пропорциональности V2md = Лах/32, где х означает различные адсорбаты. Это говорит о том, что мы можем легко установить характер влияния важных параметров /3 и S = ed — еа, рассчитав их для различных переходных металлов. Эти
данные были получены Норсковым и Хаммером [11], приведены в [12] и показаны на рис. 6.29 и 6.30.
Рис. 6.29. Матричный элемент
показывает, как d-зона металла перекрывается с 5- или /7-орбиталями
атомарного адсорбата. Отметим рост
значения матричного элемента при
движении влево. Он также возрастает при смещении вниз к колонкам групп, что связано с увеличением геометрического размера
d-орбиталей [11, 12]
з
U
2
к
X
<и
Э
о
X
Sr Y Zr Nb Мо Тс Ru Rh Pd Ag Cd
Ba La Hf Та W Re Os Ir Pt Au Hg
Sr Y Zr Nb Mo Tc Ru Rh Pd Ag Cd
Ba La Hf Та W Re Os Ir Pt Au Hg
Рис. 6.30. Положение центра flf-зоны
для трех серий переходных металлов. Заметим, что центр flf-зоны смещается вниз при движении вправо
по Периодической таблице. При
полном заполнении flf-зоны смещение ее центра вниз продолжается и
она превращается во внутренний
уровень, не влияющий на химическое поведение металла [11, 12]
Рис. 6.29 показывает, что величина /32 уменьшается с ростом степени заполнения зоны / и увеличивается при переходе от 3d- к 5d-cepnn. Это связано с тем, что размер d-орбиталей уменьшается при увеличении степени заполнения (они становятся более локализованными на атомах) и возрастает при
286 Глава 6. Реакционная способность поверхности
движении вверх по серии. Поскольку величина /3 коррелирует с шириной зоны,
это означает, что с ростом степени заполнения уровни d-зоны сужаются (перекрываются в меньшей степени из-за уменьшения геометрических размеров) и
сдвигаются вниз, пока их поведение не становится похожим на поведение внутренних уровней при полном заполнении d-зоны, как это имеет место в случае
Cu, Ag, Au, Zn, Cd и Hg.
В общем случае при подстановке реалистичных численных значений в выражение (6.80) мы получаем, что отталкивание всегда превалирует над вкладом
d-зоны в химическую связь. Поскольку ответственное за притяжение слагаемое в (6.80) уменьшается, а отталкивание увеличивается с ростом степени заполнения /, следует ожидать ослабевания связи адсорбированных атомов по мере
продвижения вправо по ряду переходных металлов. Напомним, что полная энергия
связи определяется двумя членами: вкладом, обусловленным 5/7-зоной, Е и энергией гибридизации с d-орбиталями AEd_hyb, при этом первый вклад всегда отрицателен и велик. Таким образом, рассматривая энергию связи кислорода с различными переходными металлами, легко установить, что теплота адсорбции
будет уменьшаться при движении слева направо по ряду металлов. Важно отметить, что большие энергии связи для металлов, стоящих в левой части Периодической таблицы, обусловлены отсутствием у них d-электронов, ответственных за отталкивание (рис. 6.31).
Pt Au Hg Tl Pb А
Рис. 6.31. Экспериментальные
теплоты адсорбции О, на поли-
кристаллических пленках [13]
Можно также легко понять, почему в отношении реакционной способности золото занимает особое место среди благородных металлов. Если мы применим выражение (6.80) к случаям адсорбции О на Cu, Ag и Au, то легко установим следующее. Поскольку d-зона у этих металлов заполнена, то/= 1 и ответ¬
6.5. Важные закономерности поведения реакционной способности поверхности -1\г 287
ственный за притяжение член в выражении (6.80) обращается в нуль, поэтому
остается вклад отталкивания, который дает
£bo„d = £sP + 3x2(1 +f)rP2. (6.81)
Множитель 3 обусловлен тем, что /7-орбиталь в случае кислорода имеет
трехкратное вырождение. Как указывалось выше, /?2 увеличивается при движении вниз по Периодической таблице, что предполагает наибольшее значение
вклада отталкивания для золота и наименьшую среди d-металлов устойчивость
для связи Au—О. Аналогичные аргументы можно использовать при анализе
адсорбции других атомов.
6.5.2. Закономерности хемосорбции молекул
У двухатомных молекул типа N2, 02 и СО валентные электроны,
как показано на рис. 6.6, находятся на 5(7-, 1 л- и 2я--орбиталях (отметим, что
из-за взаимодействия с 4(Т-уровнем, не показанным на рисунке, 5(Т-уровень
находится ниже 1 ^-уровня). В общем случае 1 ^-уровень заполнен и находится
низко по энергии, так что взаимодействие с металлами определяется только
5(j- и 2л--орбиталями. Заметим, что первая из них является связывающей, а
вторая — разрыхляющей для молекулярной связи. Ниже мы рассмотрим адсорбцию СО на d-металлах. Молекулы СО являются фаворитом среди тестовых
молекул у исследователей поверхности, поскольку они устойчивы и демонстрируют разнообразное химическое поведение при адсорбции, которое легко
прослеживается методами колебательной спектроскопии.
При адсорбции молекул опять же сильное взаимодействие Ьо- и 2л*-орби-
талей и 5/7-орбиталей электронов металла приводит, как это отмечалось выше,
к уширению и сдвигу вниз этих электронных уровней молекул. Кроме того,
в этом случае на величине энергии адсорбции сказывается гибридизация с
d-орбиталями металла, в результате которой происходит расщепление орбита-
лей на связывающие и разрыхляющие. Окончательный результат представлен
на рис. 6.32, который следует рассматривать как реалистическую альтернативу
качественной картине, показанной на рис. 6.25.
На рис. 6.32 сначала показан характер изменения структуры Ьо- и 2л*-уров-
ней при взаимодействии СО с алюминием — металлом электронного газа, имеющим только 5/7-зону. Показано, что оба уровня в результате такого взаимодействия уширяются и сдвигаются вниз. Появившийся дополнительный структурный элемент на сдвинутом вниз 5(Т-уровне является результатом взаимодействия
5(j- и 4(Г-орбиталей молекулы СО, которое усложняет общую картину, но не
изменяет глобального эффекта. Похожий результат получается при рассмотрении взаимодействия орбиталей СО с орбиталями 5/7-зоны платины. Гибридизация d-орбиталями приводит к формированию связывающей и разрыхляющей орбиталей, проекции которых (это следует помнить) на уровни адсорбата
мы видим на рис. 6.32. Из рисунка ясно, что при хемосорбции гибридизация
орбиталей Ъа—Ъй дает комбинацию заполненных связывающих орбиталей и
288 Глава 6. Реакционная способность поверхности
почти заполненных разрыхляющих орбиталей (на рис. 6.32 показана только
их часть), в то время как взаимодействие 2n—5d дает только связывающие
орбитали.
СО/вакуум CO/AI (111) CO/Pt (111) Pt (111)
Рис. 6.32. Результаты самосогласованного расчета электронной структуры СО, адсорбированного на А1 и Pt. Узкие 5<т- и 2п- уровни уширяются и сдвигаются вниз
при взаимодействии с электронной системой 5/7-зоны А1. При взаимодействии
с орбиталями flf-зоны Pt происходит расщепление на связывающие и разрыхляющие уровни. Приведенная для СО на Pt диаграмма показывает, что вклад
от 5<т-орбитали в энергию адсорбции мал, в то время как взаимодействие 2n—d
четко усиливает связь, поскольку здесь оказываются занятыми только связывающие орбитали [14]
Что касается энергии адсорбции, то взаимодействие 5сг- и 2л*-орбиталей с
орбиталями 5/7-зоны опять же дает большой и отрицательный (т. е. стабилизирующий) вклад в химическую связь, Esp. Вклад от гибридизации, Д£^_ЬуЬ, может
быть оценен так же, как это было сделано при адсорбции атомов.
Вклад от взаимодействия 5а— d подобен найденному для занятого состояния атома с энергией е :
A F5cr
hyb
-2(1 -/)Д
5<т
+ 2(1 +
5а •
(6.82)
с5а\
Член, описывающий взаимодействие с участием 2л--орбиталей, выглядит
иначе. На этой орбитали исходно нет электронов, так что энергия гибридизации равна
АЕ]\уЬ = 2fe+ - 2fsd = 2/
(е2л + £<1 )
2п+Ь\„ 0±
ч +Г2лР2л bd
(6.83)
Если = (е2л - sd)2 > р\п (это условие не выполняется для переходных
металлов из левой половины Периодической таблицы), то мы имеем
6.5. Важные закономерности поведения реакционной способности поверхности
откуда следует окончательное выражение
A^Vhyb ~ A-Ejfhyb + _
- V Т~Т~ + 2 (! + /) rsafih + 2 fn, Pi
£1 л £d
(6.85)
где первые два члена дают вклад в притяжение, а последние два — в отталкивание. Если нас интересуют закономерности хемосорбции молекул для последней подгруппы переходных металлов (от Fe, Ru, Os и далее вправо), у которых
d-зона узкая и для которых разность е2л — ed большая, то полученное выражение (6.85) имеет силу, но интерпретация его более сложна, чем в случае хемосорбции атомов. Тем не менее легко понять, почему СО адсорбируется на Ni,
Pd и Pt лучше, чем на Cu, Ag и Au. Для все этих металлов, относящихся к
правой части группы переходных металлов, орбитали d-зоны сильнее взаимодействуют с 2л-, чем 5сг-орбиталями, то есть р\п » p\G. Таким образом, если
мы ограничимся рассмотрением только вкладов от 2л--орбитали, то получим
Первый член здесь характеризует притяжение, поскольку до адсорбции
2л--орбиталь была свободна, а после адсорбции заполняется только ее связывающая часть. Притяжение имеет место даже и для благородных металлов из
правой части группы переходных металлов. При движении влево в Периодической таблице прочность связи увеличивается главным образом из-за смещения вверх по энергии центра d-зоны. Мы снова видим природу ослабления
катализатором внутренних связей в адсорбированной молекуле: при адсорбции
идет заполнение исходно свободной ее разрыхляющей орбитали.
6.5.2.1. Влияние напряжений и деформации на хемосорбцию
пень перекрытия электронных орбиталей атомов и поэтому сказываются на
электронном строении и реакционной активности поверхности. Хотя степень
заполнения d-зоны при этом остается прежней, ее ширина и положение центра
зоны ed изменяются при изменении степени перекрытия орбиталей. При вытягивании атома из поверхностного слоя координационное число уменьшается,
что понижает степень перекрытия сг-орбиталей и приводит к сужению зоны.
Таким образом, прикладывая напряжения или проводя деформацию твердого
тела можно изменять реакционную способность поверхности. И сейчас мы
рассмотрим, как изменяется энергия гибридизации при сдвиге положения
d-зоны и неизменной степени заполнения.
Варьируя выражение (6.86) по положению центра d-зоны, получаем
2л г 2л *
(6.86)
Деформации и напряжения в поверхностном слое влияют на сте-
290 -i\, Глава 6. Реакционная способность поверхности
^чДдслой
Подложка4^
Fe
Co
Ni
Cu
Ru
Rh
Pd
Ag
ir
Pt
Au
Fe
-0,92
0.24
-0,04
-0.05
- 0,73
0,72
-1,32
-1,25
-0,95
-1,48
-2,19
Со
-0,01
-1,17
0.20
-0.06
-0.70
-0.95
-1.65
-1,36
- 1,09
-1,89
-2.39
Ni
0,96
0,1 1
-1,29
-0.12
-0,63
- 0.74
-1.32
- 1.14
-0.86
-1,53
-2,10
Си
0,25
0.38
0,18
-2,67
-0.22
-0.27
-1.04
-1,21
0,32
-1,15
-1,96
Ru
0,30
0.37
0.29
0.30
-1,41
-0,12
-0.47
-0.40
-0,13
0,61
-0.86
Rh
0,31
0,41
0.34
0.22
0.03
-1,73
-0.39
-0,08
0,03
-0.45
-0,57
Pd
0,36
0.54
0.54
0.80
-0.11
0,25
-1,83
0,15
0,31
0,04
-0.14
Ag
0,55
0.74
0.68
0,62
0,50
0,67
0.27
-4,30
0.80
0,37
0.21
Ir
0.33
0,40
0,33
0,56
■0.01
0,03
- 0.42
-0.09
-2,11
-0.49
-0.59
Pt
0,35
0.53
0.54
0.78
0.12
0,24
0,02
0,19
0.29
-2,25
-0,08
Au
0.53 |
0.74
:
0.71
0,70
0,47
0.67
0.35
0.12
0,79
0,43
-3,56
Рис. 6.33. Закономерности изменения реакционной способности адслоев, осажденных на
подложку. На диагонали даны значения энергии для центра d-зоны чистого
металла. Другие числа указывают величину сдвига для нанесенного на подложку слоя безотносительно реальной возможности получения таких слоев. Заметим, что для систем в левом нижнем углу сдвиг d-зоны происходит вверх, что
приводит к повышению реакционной способности адслоя; противоположные
эффекты наблюдаются для систем из правого верхнего угла. Диаграммы показывают характер изменения формы d-зоны при нанесении слоя металла на
подложку, характеризующуюся большей постоянной решетки [15]
Это выражение показывает, что изменение энергии гибридизации противоположно по знаку и пропорционально величине сдвига центра d-зоны. Таким
образом, при сдвиге центра зоны вверх энергия гибридизации увеличивается, и
наоборот. Деформацию и связанный с ней сдвиг центра d-зоны можно осуще¬
6.5. Важные закономерности поведения реакционной способности поверхности -*\г 291
ствить путем выращивания данного металла на инородной подложке, характеризующейся другой постоянной решетки. Подложка может способствовать как
растяжению, так и сжатию выращиваемого металла в зависимости от соотношения их постоянных решетки.
Растяжение и сжатие приводят к сужению или расширению d-зоны и сдвигу ее вверх и вниз соответственно. Сдвиг вверх приводит к усилению взаимодействия орбиталей d-зоны и 2яг-орбитали молекулы СО и, следовательно, к
формированию более прочной связи. Сжатие приводит к противоположному
эффекту.
На рис. 6.33 продемонстрированы возможные изменения в положении центра d-зоны для монослоя одного металла, нанесенного на подложку из другого
металла, рассчитанные в [15]. Аналогичные вычисления были также проведены
при сплавлении малого количества одного металла с поверхностным слоем
другого переходного металла, при этом были получены схожие закономерности. Точность приведенных на рис. 6.33 чисел ограничена, поскольку при расчетах был сделан ряд аппроксимаций. Тем не менее они отражают основные
закономерности достаточно хорошо и проливают свет на характер изменения
реакционной способности, которое реально наблюдалось на ряде адслоев [16].
Изложенная выше теория, то есть уравнения (6.86) и (6.87), позволяет понять, почему СО и подобные ему молекулы гораздо прочнее связываются с
недокоординированными центрами типа ступеней или дефектов на поверхности. Ответ состоит в следующем. Поскольку поверхностные атомы у таких центров не имеют некоторых соседей, то перекрытие орбиталей выражено в меньшей степени, что делает d-зону более узкой. Следовательно, середина зоны
поднимается вверх, что обеспечивает более прочное связывание адсорбата.
Используя представленную на рис. 6.23 информацию, можно проводить,
как мы увидим ниже, тонкую настройку поверхности, придавая ей желаемую
реакционную способность с обеспечением заданной прочности химической
связи.
6.5.3. Особенности поведения
реакционной способности поверхности
В предыдущих двух разделах мы описали закономерности изменения энергии хемосорбции атомов и молекул на поверхности металлов. Они
относятся к финальной стадии процесса хемосорбции. Сейчас мы рассмотрим,
что происходит при приближении молекул к поверхности.
6.5.3.1. Физическая и химическая адсорбция,
диссоциация молекул
На рис. 6.34 показаны кривые потенциальной энергии для гипотетической молекулы Х2, приближающейся к поверхности с правой стороны диаграммы. Сначала молекула испытывает слабое ван-дер-ваальсовское взаимо¬
292 Глава 6. Реакционная способность поверхности
действие с поверхностью. Если молекула потеряет энергию, приобретаемую за
счет взаимодействия с поверхностью, она может быть захвачена в неглубокой
потенциальной яме и перейдет в состояние физической адсорбции. Как было
указано в гл. 5, физическая адсорбция при низких температурах используется
для определения площади поверхности катализаторов.
Рис. 6.34. Схематическое сильно упрощенное представление потенциальной
энергии взаимодействия молекулы Х2
с поверхностью металла в зависимости от координаты реакции. На больших расстояниях молекула испытывает
слабое ван-дер-ваальсовское притяжение, обеспечивающее физическую адсорбцию молекул. На следующей стадии происходит химическая адсорбция,
связанная с формированием химической связи. Если молекула способна
преодолеть барьер ЕЛ, то произойдет
ее диссоциация на два хемосорбиро-
ванных атома. Энергия, необходимая
Координата реакции, произв. ед. Для десорбции этих атомов, равна Ed
Если молекула способна перестраивать свою электронную конфигурацию,
например за счет взаимодействия с sp- и d-электронами металла, она может
перейти в состояние химической адсорбции, что мы детально обсуждали в
предыдущих разделах. В зависимости от расстояния до поверхности, прочности химической связи, молекула с большей или меньшей вероятностью может преодолеть потенциальный барьер, показанный на рис. 6.34 и разделяющий состояния физической и химической адсорбции. Обычно этот барьер
невысок, но в существенной мере зависит от природы рассматриваемой системы. Кроме того, поскольку энергия хемосорбции зависит от ориентации
молекул на поверхности, то реальный потенциал взаимодействия молекула/
поверхность выглядит белее сложно, чем это показано на рис. 6.34 для одномерного приближения.
Если мы будем дальше приближать молекулу к поверхности, она будет испытывать сильное отталкивание и ее потенциальная энергия будет повышаться. Снова заметим, что реальный ход потенциала будет более сложным, чем это
показано на рис. 6.34, поскольку он существенным образом зависит от ориентации молекул по отношению к поверхности. Для двухатомных молекул можно
предположить, что переходному состоянию к их диссоциации отвечает их ориентация параллельно поверхности. Энергетические барьеры, разделяющие состояния физической, химической и диссоциативной адсорбции, представляют
собой активационные барьеры для цепочки реакций, по которым молекула
переходит из газовой фазы в состояние диссоциативной адсорбции. Важно уметь
предсказывать и рассчитывать высоты этих барьеров, поскольку они играют
ключевую роль в определении скорости реакции.
6.5. Важные закономерности поведения реакционной способности поверхности -*\г 293
6.5.3.2. Диссоциативная адсорбция: N2 на поверхности рутения
На рис. 6.35 показаны рассчитанные методом функционала плотности диаграммы потенциальной энергии для молекулы N2 на двух поверхностях рутения — на идеально гладкой и на содержащей одноатомную ступень.
Полная энергия системы минимизировалась на каждом шаге сближения молекулы с поверхностью и реакции между ними, при этом выбиралась взаимная
ориентации молекулы N2 и поверхности, обеспечивающая минимальные высоты потенциальных барьеров. Определение этого пути движения молекулы принципиально важно, поскольку он обеспечивает протекание реакции с наибольшей вероятностью. В расчетах, результаты которых представлены на рис. 6.35,
состояние физической адсорбции в расчет не принималось, так что первой
стадией была химическая адсорбция молекулы азота, которая встает вертикально на верхушке атома рутения (рис. 6.35, а). Энергетический барьер для
хемосорбции отсутствует. Чтобы диссоциировать, молекула должна лечь параллельно поверхности (стадия б), и необходимо растянуть ее связь, чтобы она
разорвалась. Этому отвечает переходное состояние (стадия в), для которого
измеренная высота энергетического барьера составляет 2 эВ (200 кДж/моль).
Такое состояние является сильно активированным и перевод в него молекулы
требует больших затрат энергии.
ш
о
CVI
Z
§ 2,0
I—
Q.
О)
ГО 1.0
к
05
X
£ 0,0
05
S
If
Рис. 6.35. Диаграммы потенциальной ф-1,о
энергии, рассчитанные методом функ- о
ционала плотности, для адсорбции и с
диссоциации молекул N2 на поверхности (0001) Ru и для той же поверхности,
содержащей одноатомную ступень [17]
Барьер для диссоциации молекулы на поверхности существенно ниже соответствующего барьера для молекулы в газовой фазе, где он составляет около
9,8 эВ (945 кДж/моль) и где формируются два свободных радикала N. Для
реакции на поверхности конечное состояние энергетически более выгодно,
поскольку здесь два атома N связаны с атомами металла. Первоначально атомы N
находятся в центрах с тройной координацией и контактируют с одним и тем же
атомом (стадия г). В этом состоянии они отталкиваются друг от друга, и энергетически более выгодным является удаление атомов друг от друга за счет диффузии, при которой им приходится преодолевать невысокие активационные
барьеры (на рисунке не показано).
Переходное состояние
N2/Ru(0001)
KJ / .-*** \ V
1Ч2, а(I / • \ \
/ N?;/ступенчатая '> ч \
- / поверхность Ru(0001)
Координата реакции
294 -J\r Глава 6. Реакционная способность поверхности
Заметим, что диссоциация на поверхности, содержащей ступень, протекает
с существенно более низкой энергией активации. Как показано на диаграмме,
на всех стадиях молекула и ее фрагменты связаны со ступенчатой поверхностью
более сильно, чем с гладкой. Причины этого обсуждались ранее при анализе
уравнения (6.87). Однако переходное состояние подвергается существенно более
сильному воздействию, поскольку два атома N в переходном состоянии связаны
с четырьмя атомами металла в случае гладкой поверхности и с пятью — в
случае ступенчатой поверхности. Как отмечалось ранее, расположение атомов
адсорбата, при котором они связаны с разными атомами металла, является
более устойчивым по сравнению с расположением, при котором разные атомы
адсорбата взаимодействуют с одним и тем же атомом металла. Следовательно,
диссоциация идет более предпочтительно на ступенях, и если на поверхности
имеются дефекты такого типа, то они будут определять кинетику реакции диссоциации.
Чтобы пошла диссоциация, разрыхляющие орбитали молекулы N2 должны
заполниться электронами металла. Мы показали ранее, что связь молекулы с
поверхностью осуществляется через электронную систему s/ьзоны, но для диссоциации необходимо взаимодействие электронной системы d-зоны с разрыхляющей 2я--орбиталью, обеспечивающее заполнение этой орбитали. Это и есть
сущность катализа: катализатор разрывает одни связи и обеспечивает формирование других. Важное действие — разрыв сильной внутренней связи в молекуле N2. За счет взаимодействия с поверхностью потенциальный барьер диссоциации снижается и формирующиеся фрагменты молекулы становятся способными к образованию новых соединений.
6.5.3.3. Закономерности диссоциативной адсорбции
Определение переходного состояния и высоты соответствующего
активационного барьера важно для понимания хода реакции. Конечно, тонкие
детали формы потенциальной поверхности, связанные со стерическими препятствиями и энтропийными эффектами, могут не позволить системе преодолеть барьер. Высота барьера (которая, как мы объясняли в гл. 3, не сильно
отличается от энергии активации) определяется взаимодействием электронов
молекулы и подложки при заданной геометрии активационного комплекса, и в
принципе для установления характера ее зависимости от природы металла можно
привлечь те же формулы, которые были использованы при рассмотрении энергии хемосорбци.
Если мы ограничим свое рассмотрение переходными металлами из конца
периода, как это мы делали при расчете энергии хемосорбции СО, то доминирующим снова будет взаимодействие разрыхляющей орбитали и орбиталей
d-зоны, так что основной вклад равен
A^.hyb=-2/-^- + 2/r2^
Е1к £d
(6.88)
6.5. Важные закономерности поведения реакционной способности поверхности 295
Видно, что предпочтительными являются металлы с высоко расположенной d-зоной (малая величина е1л — ed)9 для которых взаимодействие будет более
сильным, а, следовательно, барьер хемосорбции — более низким. Этот результат объясняет, почему СО не диссоциирует на Си и почему реакционная способность возрастает при движении по серии переходных металлов влево. Однако можно сказать и больше.
Энергия конечного состояния также существенным образом сказывается
на высоте барьера диссоциации. Если мы рассмотрим диссоциацию СО на
поверхности металлов группы IB (Ni, Pd и Pt), то по уравнению (6.86) получим
лишь незначительное снижение высоты барьера. Хотя у платины d-зона лежит
ниже, матричный элемент для нее имеет существенно большее значение, что
компенсирует это низкое значение. Тем не менее СО диссоциирует на никеле
и не диссоциирует на платине. Это связано с тем, что конечные продукты,
атомы С и О, с никелем связываются более сильно, чем с платиной.
S
S
а
ю
Q.
О QQ
О (D
О г
х U
к ^
4,0
2,0
0,0
-2,0
-4,0
-6,0
*■ МФП: СО(а) Г
•■NO(a)
• МФП: С(а) + О(а) /
•N(a)+0(a)
□ Модель НА: СО(а)
О Модель НА: С(а) + О(а) /
■
/ ®
/° ''
п
си/
■ СО(а) jg'
• NO(a) А
[
/
■г" У
/
N(a) + О(а)
О-—^^С(а) + О(а)
Nb Мо Тс Ru Rh Pd Ag Ru Rh Pd
4,0
2,0
0,0
-2,0
-4,0
-6,0
a
о
H
a>
X
£
s
x 5
О 1
p 03
5 «
X 1
l-S
*3
2! x
О 2
о
и о
00 О
тэ
а\
£
5
5
Рис. 6.36. Рассчитанные теплоты адсорбции молекулярных СО и N0 в сравнении с теп-
лотами адсорбции продуктов диссоциации. Незаполненные символы соответствуют модели Ньюнса—Андерсона (НА), заполненные символы отвечают расчету методом функционала плотности (МФП) [11]
Анализируя закономерности изменения вероятности диссоциации в сериях
переходных металлов, можно отметить, что диссоциация становится предпочтительней при движении влево, а ассоциативная хемосробция — при движении
вправо по группам. Это прекрасно иллюстрируют данные для адсорбции СО на
переходных 4*/-металлах, приведенные на рис. 6.36, из которых видно, что молекулярная адсорбция СО на Pd и Ag более выгодна, чем адсорбция продуктов
296 -l\r Глава 6. Реакционная способность поверхности
диссоциации. Родий относится к пограничной ситуации и влево от родия предпочтительней становится диссоциация. Заметим, что теплоты адсорбции атомов С и О изменяются более резко при движении по Периодической таблице,
чем теплоты адсорбции молекул СО. Аналогичная ситуация имеет место и для
N0, который, однако, является более реакционно-активным, чем СО, и, следовательно, для него барьер диссоциации существенно ниже.
6.5.3.4. Переходные состояния и влияние степени
покрытия поверхности: гидрирование этилена
Как было отмечено в предыдущем подразделе, для понимания сущности каталитической реакции необходимо выяснить характер переходного состояния системы. К сожалению, переходное состояние сложно зафиксировать
в эксперименте, поскольку время нахождения в нем системы очень мало. Однако выяснить структуру молекул в переходном состоянии можно методами
численного моделирования, в чем мы сейчас попытаемся убедить читателя.
Ниже мы обсудим ход реакции, более сложной, чем диссоциация двухатомной
молекулы. Речь идет о каталитическом гидрировании этилена, С2Н4, до этана
С2Н6 с использованием катализатора из благородного металла. В течение длительного времени эта реакция служила в качестве тестовой для многих экспериментаторов, занимающихся катализом и исследованием поверхности (см. [18]).
Этилен, С2Н4, может адсорбироваться
двояким образом: через формирование слабой яг-связи, когда двойная связь С=С находится над атомом металла, или через
формирование более прочной двойной
сг-связи, когда два атома С этилена связываются с двумя атомами металла (рис. 6.37).
Рассмотрим поверхность (111) металла. На
ней водород адсорбируется диссоциативно
и, как полагают, находится на поверхности
металла в полостных центрах с тройной координацией.
Н2 +2* = 2Н!,
С2Н4 + ! = я--С2Н4 (я-связанный этилен),
тг-С2Н4 + = С2Н4 (этилен с двойной сг-связью с поверхностью),
*С2Н4 +Н* —» С2Н5 +2* (адсорбированный этил; лимитирующая стадия),
с2н; + н = с2н6 + 2*.
Нойрок с соавторами [19] провел вычисления методом функционала плотности структуры описываемой системы в различных состояниях на поверхности (111) палладия вплоть до образования этилового радикала. На рис. 6.38, a
Н2С=СН2 Н2С-СН2
л’-связанный Этилен с двойной
этилен сг-связью
Рис. 6.37. Различные состояния этилена, адсорбированного на поверхности
благородного металла
Схема реакции выглядит так
6.5. Важные закономерности поведения реакционной способности поверхности
показана диаграмма потенциальной энергии, начиная с момента появления
атома Н на поверхности. Как видно из диаграммы, этилен адсорбируется в
состояние с ;г-связыо, при этом теплота адсорбции составляет 30 кДж/моль, а
затем переходит в состояние с двойной сг-связью, что понижает энергию системы
еще на 32 кДж/моль.
Q.
Ф
X
CD
Q.
CD
I
CD
50 -
25
0 -
-25 -
-50
-75
50
25 -
0
-25
-50
^ Трехцентровое
Ф//ФМ
И
переходное
состояние
Н2С-СНг н
ътштяЗ?.
Многоцентровое ^
переходное н
состояние
н
с** :Н
Трехцентровое
переходное
состояние
Координата реакции
Рис. 6.38. Диаграмма потенциальной энергии процесса гидрирования этилена до интермедиата этила (С2Н5) на поверхности (111) палладия. Нулевому уровню отвечает энергия адсорбированного атома Н:
а — низкие степени покрытия поверхности: этилен адсорбирован с образованием достаточно прочной двойной ег-связи, когда два атома углерода связываются с двумя атомами
металла. В трехцентровом переходном состоянии водород и углерод связываются с одними и теми же атомами металла, что приводит к увеличению энергии системы и создает
энергетический барьер для процесса гидрирования до этиловых радикалов; б — при высоких степенях покрытия поверхности открывается еще один канал реакции, в котором
менее прочно связанный с поверхностью л—С2Н4 формирует многоцентровое переходное состояние с близлежащим атомом водорода, связанным с атомом металла. Это —
основной канал гидрирования этилена (19|
На лимитирующей стадии реакционные фрагменты должны преодолеть
значительный энергетический барьер высотой 88 кДж/моль. Наиболее вероятное переходное состояние (или, как бы мы сказали, наименее предпочтительное в данном случае) показано на рис. 6.39, а. Это состояние представляет
собой трехцентровой комплекс Pd—С—Н, в котором атом Н связан с теми же
298 -l\r Глава 6. Реакционная способность поверхности
атомами металла, с которыми связан атом углерода молекулы этилена. Действительно, как мы отмечали ранее, состояние, в котором атомы адсорбата
связываются с одними и теми же атомами металла, является менее устойчивым
по сравнению со случаем образования связи с разными атомами металла. По
этой причине трехцентровое переходное состояние, показанное на рис. 6.39, а,
лежит по энергии на 88 кДж/моль выше по сравнению с наиболее устойчивым
состоянием адсорбата. В переходном состоянии идет разрушение связей Pd—С
и Pd—Н и к СН2-группе адсорбированного этилена добавляется атом Н. При
достижении переходного состояния этиловый радикал С2Нз образуется легко и
имеет энергию адсорбции на 37 кДж/моль меньше энергии адсорбции атомов
Н, принятой за уровень отсчета энергии на рис. 6.38. Заметим, что представленная энергетическая диаграмма дает и значения энергетического барьера для
обратной реакции дегидрогенизации этиловых радикалов.
Рис. 6.39. Структура комплекса в переходном состоянии при гидрировании
этилена, соответствующая рис. 6.38:
а — трехцентровое переходное состояние;
б — многоцентровое переходное состояние 119 J
До сих пор наше обсуждение касалось только случая малых степеней покрытия поверхности, которые обычно рассматриваются при кинетическом моделировании. При высоких степенях покрытия превалирует другой механизм,
который дает альтернативу для энергетически невыгодного трехцентрового переходного состояния, представленного на рис. 6.39, а. При высоких степенях
заполнения этилен и водород вынужденно находятся ближе, что открывает новый
механизм для реакции между слабо я-связанным этиленом и близлежащим
атомом водорода. Связанный с одним атомом металла ;г-С2Н4 слегка «наползает» на близлежащий атом Н, так что этилен формирует одну сг-связь с металлом, в то время как другой атом С оказывается вовлеченным в многоцентровой
6.5. Важные закономерности поведения реакционной способности поверхности ->\г 299
переходный комплекс, как это показано на рис. 6.39, б. По длинам связей
можно четко заключить, что атом Н преимущественно связан с атомом металла, а не с молекулой этилена, что позволяет избежать отталкивания, ответственного за высокий энергетический барьер трехцентрового переходного комплекса, показанного на рис. 6.39, а. Соответствующая энергетическая схема
приведена на рис. 6.38, б, при таком механизме высота энергетического барьера составляет только 36 кДж/моль для элементарной ступени. Безусловно, при
этом не исключается и рассмотренный ранее путь реакции с участием этилена,
формирующего двойную сг-связь с металлом; здесь с увеличением степени покрытия поверхности также происходит некоторое снижение высоты энергетического барьера. Однако он не может конкурировать с процессом, идущим
через формирование многоцентрового переходного состояния.
Отметим, что данное обсуждение было проведено для отдельной ступени
процесса. Для реакции, протекающей в стационарных условиях, надо сравнить
два возможных пути по соответствующим кажущимся энергиям активации. Мы
оставляем проведение этого сравнения читателю в качестве упражнения.
Соморджай с сотрудниками [20] измерениями in situ генерации излучения
суммарной частоты дали спектроскопическое обоснование того, что гидрогенизация этилена идет непосредственно через тг-связанный адсорбат, а не через
состояния с сг-связью.
Приведенный пример иллюстрирует несколько важных моментов: во-пер-
вых, механизм реакции, адсорбированные интермедиаты и переходные состояния можно в настоящее время очень детально изучить методами численного
моделирования химии не только для двухатомных молекул, но и для более сложных систем. Получаемое при этом хорошее согласие расчетных и экспериментальных данных для энергий адсорбции и высот энергетических барьеров говорит о надежности определяемых численными методами значений. Численные
методы позволяют выявить многие особенности механизма реакции. Во-вторых,
каталитические реакции не обязательно идут через наиболее устойчивое состояние адсорбата. В случае этилена гидрирование менее прочно связанного л-С2Н4
идет гораздо быстрее гидрирования более устойчивого сг-связанного С2Н4. Действительно, на многих металлах происходит дегидрогенизация этилена до этили-
дина, =С—СН3, связанного с атомами металла. Это соединение доминирует
при низких степенях покрытия, но оно не участвует в процессе гидрогенизации.
Поэтому о нем часто говорят как о «наблюдателе» процесса. Таким образом, в
каталитическом процессе могут доминировать слабо связанные адсорбаты, для
обнаружения которых в эксперименте необходимо проводить спектроскопические исследования in situ. В-третьих, показанная на рис. 6.38 энергетическая диаграмма говорит о том, что гидрогенизация идет лучше, если углеводород связан с
поверхностью слабо (сравните высоты барьеров для реакций с участием сг-свя-
занного С2Н4 при высоких и низких степенях заполнения). Это может послужить основой для выбора металла катализатора, введения промоторов, использования сплавов, а также режима, задающего данную степень покрытия поверхности. Заметим, что здесь мы имеем отличие от случая диссоциации двухатомных
300
Глава 6. Реакционная способность поверхности
молекул, обсуждавшейся в подразд. 6.5.3.2 и 6.5.3.3, когда более сильное взаимодействие с поверхностью облегчало диссоциацию.
6.5.3.5. Принцип Сабатье
Из приведенных в предыдущих разделах данных ясно, что каталитическая реакция идет лучше, если взаимодействие между адсорбатом и поверхностью не является ни слишком сильным, ни слишком слабым. Сабатье предположил, что должна иметься оптимальная скорость реакции в зависимости от
теплоты адсорбции. При малой теплоте адсорбции катализатор оказывает слабое влияние, в частности, он не сможет обеспечить разрыв связи. При очень
сильном взаимодействии адсорбаты не смогут десорбироваться с поверхности.
Оба эти предельных случая отвечают низкой скорости реакции.
Принцип Сабатье проиллюстрирован
на рис. 6.40, где приведена зависимость
активности различных катализаторов из
переходных металлов, нанесенных на графит, по отношению к синтезу аммиака в
одних и тех же условиях. Изложенные
выше теории позволяют легко объяснить
наблюдаемую зависимость: металлы, находящиеся в левой части Периодической
таблицы, способны легко обеспечить диссоциацию N2, но образующиеся атомы N
оказываются сильно связанными с поверхностью и поэтому менее химически активны. Молекулы из правой части Периодической таблицы не способны вызвать
диссоциацию N2. Отсюда делаем вывод о
существовании «оптимальных» металлов
типа Fe, Ru и Os. Зависимости такого вида
типичны для каталитических реакций, и
их обычно называют «вулканическими».
6.5.3.6. Возможность «настройки» реакционной способности
поверхности
Зададимся вопросом: как мы можем изменить активность конкретного металла? Во-первых, можно повлиять на структуру поверхности. На более
открытых поверхностях атомы характеризуются меньшим координационным
числом. Это приводит к сужению d-зоны, сдвигу ее положения (вверх, если она
заполнена более чем наполовину, и вниз, если она заполнена менее чем наполовину). Сказанное проиллюстрировано в табл. 6.2, где приведены экспериментальные данные по энергиям активации реакции диссоциации N0 на поверхностях (111) и (100) родия.
Степень заполнения d-зоны, %
Рис. 6.40. Каталитическая активность различных металлов на носителях в синтезе
аммиака [21]
6.5. Важные закономерности поведения реакционной способности поверхности
\
Таблица 6.2. Энергии активации (кДж/моль) для реакции диссоциации N0
и десорбции N2 с двух различных граней кристалла родия
Реакция
Rh(lll)
Rh(100)
NO* + * -> N* + О*
65 ±6
37 ±5
2N* —> N2 + 2*
118 + 5
225 ± 5
Во-вторых, как мы говорили в подразд. 6.5.2.1, напряжения в монослое
металла, нанесенном на другой металл с иной постоянной решетки, также могут в существенной мере повлиять на каталитическую активность. На рис. 6.33
приведены данные по возможным величинам эффекта. Здесь результаты относятся к диссоциации метана на поверхности никеля. Сам по себе никель практически пассивен в отношении этой реакции. Как видно из уравнения (6.87),
чтобы увеличить активность металла, мы должны сдвинуть г/-зону вверх. Это
может быть достигнуто путем формирования монослоя никеля на подложке,
имеющей большую постоянную решетки. Как видно из рис. 6.33, для этой
цели в качестве подложки можно использовать рутений, у монослоя никеля на
котором d-зона поднимается вверх на 0,29 эВ. Более того, в сплаве никеля и
рутения идет сегрегация с выходом атомов никеля на поверхность, так что
монослой в данном случае обладает хорошей стабильностью. На рис. 6.41 в
качестве меры реакционной способности никеля по отношению к диссоциативной адсорбции метана использована вероятность реакционного прилипания молекулы.
Рис. 6.41. Реакционная способность монослоя Ni на поверхности (OOOl)Ru по отношению
к диссоциативной адсорбции
метана. При нулевой степени
заполнения поверхности имеем активность (коэффициент
реакционного прилипания)
чистого рутения. При нанесении сплошного монослоя никеля вероятность диссоциации
достигает максимума. При
формировании нескольких монослоев реакционная способность снижается до присущей
поверхности (lll)Ni [22]
5е-7
4е-7
Зе-7
О
Q.
Ф
Ш
ск
СО
с;
со
со
X
2е-7
Термическая диссоциация СН4
/ Ф \
при Т = 530 К
Ni/Ru (0001): отжиг при 1100 К
/ Ж
в течение 2 мин
Ru(0001)
«Ni( 111)»
#
Степень покрытия поверхности Ni, МС
Молекулы метана устойчивы, поэтому их трудно активировать. В результате вероятность реакционного прилипания оказывается малой и составляет величину порядка 10“7. Заметим, что рутений каталитически более активен, чем
никель, однако растянутый монослой, как и ожидалось, обладает большей активностью, чем рутений и никель в обычной форме.
301
302 -l\r Глава 6. Реакционная способность поверхности
6.5.4. Универсальность гетерогенного катализа
Дополнительное обоснование принципа Сабатье для реакций, в
которых диссоциация является важной ступенью реакционного механизма,
может быть получено в рамках метода функционала плотности (МФП). Такие
реакции включают два основных этапа: диссоциацию молекул с сильной связью (например, N2) и дальнейшую реакцию полученных при диссоциации атомов (например, N + ЗН —> NH3). Норсков с сотрудниками [23] показали, что
зависимость активационной энергии диссоциации Еа от энергии адсорбции
получающихся при диссоциации атомов, обозначенной как АЕ на рис. 6.42,
является линейной. Это — замечательный результат, поскольку он получен для
четырех различных двухатомных газов и большого числа ^/-металлов. Такая
линейная зависимость имеет место и для плотноупакованных, и для ступенчатых поверхностей; в последнем случае зависимость идет с тем же наклоном,
что и в первом, но со сдвигом в область меньших значений энергий активации.
Полученные данные позволяют говорить об универсальности линейной связи
между энергией активации и теплотой адсорбции продуктов диссоциации, хотя
возможно изменение параметров уравнения этой линейной зависимости для
различных поверхностных структур.
Линейная связь между энергиями активации и теплотами адсорбции или
теплотами реакции считается установленной достаточно давно. Такая связь
называется соотношениями Бренстеда—Эванса—Поляни [24, 25]. В катализе
такие соотношения были недавно найдены для реакций диссоциации, которые
приведены на рис. 6.42, а также для ряда реакций с участием короткоцепочечных углеводородных фрагментов типа реакций гидрогенизации этилена, дегидрогенизации этилена и этиловых радикалов [26].
Скомбинировав данные по диссоциации N2 на ступенчатых поверхностях и
кинетическую модель синтеза аммиака (рис. 6.42, в), можно установить, что
скорость реакции зависит от теплоты адсорбции атомов N. При это можно
сразу получить идеализированную «вулканическую» кривую типа показанной
на рис. 6.40. Следовательно, оптимальным для синтеза аммиака будет катализ
на ступенчатой поверхности, связывающей атомы N с теплотой адсорбции около
100 кДж/моль. Более сильное связывание будет способствовать диссоциации
молекул азота, но будет препятствовать участию атомов азота в последующих
реакциях. При более слабом связывании слишком большой окажется энергия
активации диссоциации. Таким образом, оптимальным является катализатор,
для которого энергия активации и теплота адсорбции достигают тщательно
сбалансированного компромисса, что находится в полном согласии с принципом Сабатье, но в действительности имеет более детальное обоснование.
Выполнимость соотношения Бренстеда—Эванса—Поляни для реакций диссоциации приводит к ряду вопросов. Почему связь между энергией активации
и теплотами адсорбции продуктов диссоциации является линейной? Почему
эта связь зависит от структуры поверхности катализатора? Почему она не зависит от природы адсорбата?
6.5. Важные закономерности поведения реакционной способности поверхности -*\г 303
-3 -2 -1 0 1 2 3
Рис. 6.42. Соотношение Бернстеда—Эвана—Поляни между энергиями активации диссоциации двухатомных молекул и теплотами адсорбции продуктов диссоциации
для ряда атомно-гладких и плотноупакованных поверхностей типа (111) ГЦК-
кристаллов, (НО) ОЦК-кристаллов и (0001) ГПУ-кристаллов (а); б — то же
самое для ступенчатых поверхностей, у которых активным является так называемый В5-центр. Отметим, что при переходе к ступенчатым поверхностям наклон прямых не изменяется, есть только небольшой сдвиг вниз по энергии
активации; в — нормированная каталитическая активность по отношению к
синтезу аммиака при различных его концентрациях [23].
Получить ответы на все эти вопросы можно, рассмотрев структуру переходного комплекса реакции диссоциации, которая практически одинакова для
всех исследованных систем. Ясно, что структура переходного комплекса для
данной геометрии субстрата не зависит от природы молекул и субстрата. Таким образом, плотноупакованные поверхности и ступенчатые поверхности,
данные по которым приведены на рис. 6.42, формируют две группы. Далее,
диссоциация характеризуется переходным комплексом, образующимся на
поздней стадии, когда два атома удалены друг от друга достаточно далеко, так
что они уже утратили свою молекулярную идентификацию. Это означает, что
изменения в характеристиках переходного комплекса следуют изменениям
конечного состояния атомов — продуктов диссоциации, которое определяется теплотами их адсорбции. Поскольку структура переходного комплекса зависит от структуры поверхности (см., например, рис. 6.35, в, где показаны
304 -l\r Глава 6. Реакционная способность поверхности
гладкая и ступенчатая поверхности рутения), для каждой из поверхностей
соотношения Бренстеда—Эванса—Поляни будет выполняться с собственными числовыми параметрами.
Установление универсальных соотношений между энергиями активации и
теплотами адсорбции для отдельных классов реакций можно рассматривать
как более точное и количественное выражение принципа Сабатье. Эти соотношения дают основу для целенаправленного поиска новых материалов для катализаторов через оптимизацию силы взаимодействия между интермедиатами и
поверхностью.
Приложение. Метод функционала плотности
В данной главе мы часто ссылались на результаты, полученные
численными методами, в частности методом функционала плотности. В настоящее время практически невозможно провести квантово-механические расчеты для макроскопического образца металла с различными адсорбированными
на нем соединениями, но можно получить реалистические результаты для более простых систем. Один из методов состоит в замене металла кластером из
3—30 атомов, на котором адсорбирована молекула, и учете в анализе всех имеющихся электронных орбиталей. Таким путем в разнообразных расчетах было
получено много полезных результатов. Очевидно, что кластер нужно брать достаточно большим, чтобы избежать различного рода артефактов, связанных, в
частности, с выбранным размером кластера; можно исключить такие артефакты, проварьировав размер кластера.
Другой подход, зародившийся в физике твердого тела, основан на усредненном описании системы многих электронов, использовании симметрии кристаллической решетки металла и учете сильного экранирования валентными
электронами внутренних уровней атома. Симметрия металла позволяет описать макроскопический образец с помощью элементарной ячейки, многократно повторенной в трех измерениях. Таким образом, описания свойств электронов только в одной ячейке может оказаться вполне достаточно для определения характеристик массивных образцов. Внутренние электроны не участвуют в
химических процессах, но экранируют заряд ядер и определяют периодический потенциал, в котором движутся валентные электроны.
Трехмерная симметрия нарушается при наличии межфазной поверхности,
но если описать систему пластинками, образованными 3—5 слоями атомов и
разделенными вакуумными зазорами такой же толщины, то симметрия может
быть восстановлена. Молекулы (атомы) адсорбата помещаются в элементарные ячейки, образованные атомами металла, размером 3x3 или 4x4. Созданная система повторяется периодически в трех направлениях. Использованный
прием позволяет снова привлечь методы физики твердого тела. Пластинки надо
брать достаточно толстыми, чтобы рассчитанные энергии сходились к предельным значениям, а расстояние между ними должно быть достаточно большим,
чтобы исключить взаимодействие между ними.
6.5. Важные закономерности поведения реакционной способности поверхности -*\г 305
Вместо отдельного рассмотрения электронов металла и адсорбата вводится
общая плотность электронов системы. Хоэнберг и Кон (Кон в 1999 г. получил
Нобелевскую премию за работы в этой области) показали, что основное состояние системы Е0 является однозначной функцией плотности электронов п0 в
основном состоянии. Без учета спина электронов энергия системы может быть
представлена в виде функционала
где Т(п) — кинетическая энергия взаимодействующих электронов. Она представляет собой сумму кинетической энергии невзаимодействующих электронов и обменной и корреляционной энергий:
где все неизвестные вклады включены в выражение для обменно-корреляцион-
ной энергии. Если вид обменно-корреляционной энергии известен, то проблема
сводится к итерационному решению так называемых уравнений Кона—Шэма,
которые представляют собой уравнения Шредингера для отдельных электронов
Написанное уравнение пригодно только для однородного электронного газа,
когда
где £хс(п) — обменно-корреляционная энергия на один электрон. Приближение, в котором используется выражение (6.93), называется приближением локальной плотности (ПЛД). Поскольку это приближение получается из первых
принципов, его часто называют подходом ab initio (то есть основанным на первых принципах). К сожалению, выражение (6.93) оказывается слишком грубым
для изменяющейся электронной плотности, поэтому его использование приводит к завышению энергий связи на несколько электронвольт. Гораздо более
точные результаты могут быть получены при учете членов, зависящих от градиента плотности электронов. Такой подход обычно называют методом функционала плотности с учетом обобщенных градиентов (МФП-УОГ)
Расчеты, выполненные в рамках МФП-УОГ, позволяют достаточно надежно предсказать путь реакции для различных молекул на поверхности металла.
Они дают детальную информацию о геометрии связей, их энергии, высоте ак¬
Е (п) = Т (и) + J п (г) v nucl (г) dr + JJ dr dr',
(6.89)
Пп) = Tks(«) + EJn),
(6.90)
(6.91)
где
(6.92)
/осс
(6.93)
306 -l\r Глава 6. Реакционная способность поверхности
тивационного барьера и структуре переходных состояний, получение которой
другими методами невозможно. Точность расчета энергетических характеристик составляет обычно несколько десятых электронвольта, что позволяет выявить практически важные тенденции в ходе химических реакций.
Список литературы
1. Atkins P.W. Molecular Quantum Mechanics. Oxford: Oxford Univ. Press. 1983.
2. Elliot S. The Physics and Chemistry of Solids. New York: Wiley&Sons, 1998.
3. Ashcroft N, Mermin N.D. Solid State Physics. New York: Holt, Rinehart and
Wintson, 1976.
4. Kittel C. Introduction to Solid State Physics. New York: Wiley&Sones, 1976.
5. McQuarrie D.A. Statistical Mechanics. New York: Harper and Row, 1976.
6. Newns D.M. // Phys. Rev. 1969. V. 178. P. 1123.
7. Anderson P.W. // Phys. Rev. 1961. V. 124. P. 41.
8. Hoffman R. Solids and Surfaces. Weinheim: WCH, 1988.
9. N0rskov J.K. // J. Vac. Sci. Technol. 1981. V. 18. P. 420
10. Lang N.D., Williams A.R. // Phys. Rev. B. 1978. V. 18. P. 616.
11. Hammer В., Nmrskov J.K. // Adv. Catal. 2000. V. 45. P. 71.
12. Andersen O.K., Jepson О., Glotzel D. in Highlights of Condensed Matter Theory
LXXXIX. Bologna: Corso Soc. Italiana di Fisica, 1985. P. 59.
13. Toyoshima /., Samorjai G.A. // Catal. Rev. Sci. Eng. 1979. V. 19. P. 105.
14. Hammer B., Morikawa K, N0rskov J.K. // Phys. Rev. Lett. 1996. V. 76. P. 2141.
15. Ruban A. K, Hammer B., Stoltze P., Jackobsen K. W, Shriver H.L., N0rskov J.K. //
J. Mol. Catal. A. 1997. V. 115. P. 421.
16. Rodriquez J.A., Goodman D.W. // Science. 1992. V. 257. P. 897.
17. Dahl S., Logadottir A., Egberg R., Larsen /., Chorkendorff /., Tornquist E.,
N0rskov J.K. // Phys. Rev. Lett. 1999. V. 83. P. 1814.
18. Samorjai G.A. Introduction to Surface Chemistry and Catalysis. New York:
Wiley, 1994.
19. Neurok M., Pallassana V., Van Santen R.A. // J. Am. Chem. Soc. 2000. V. 122.
P. 1150.
20. Cremer P.S., SuX., Shen Y.R., Somorjai G.A. //J. Am. Chem. Soc. 1996. V. 118.
P. 2942.
21. Ozaki A., Aika K. // Catalysis. V. 1. // Eds Anderson J., Boudart M. Berlin:
Springer-Verlag, 1981. P. 87
22. Egberg R.C., Chorkendorf /. // Catal. Lett. 2001. V. 77. P. 207.
23. N0rskov J.K., Bligaard Т., Logadottir A., Bahn S., Hansen L.B., Bollinger М.,
Bengaard Hammer B., Sljivancanin Z, Mavrikakis М., Xu Y, Dahl S., Jacobsen C.J.H. //
J. Catal. 2002. V. 209. P. 275.
24. Br0nsted N. // Chem. Rev. 1928. V. 5. P. 23.
25. Evans M.G., Polanyi M. // Trans. Faraday Soc. 1938. V. 34. P. 11.
26. Palassana K, Neurock M. // J. Catal. 2000. V. 191. P. 301.
ГЛАВА
7
КИНЕТИКА
ПОВЕРХНОСТНЫХ РЕАКЦИЙ
7.1. ПРОСТЕЙШИЕ ПОВЕРХНОСТНЫЕ РЕАКЦИИ
Раскрытие каталитического процесса в терминах элементарных
реакций, определение кинетических параметров соответствующих ступеней
составляют сердцевину понимания каталитических реакций на молекулярном
уровне. Как было пояснено в гл. 1 и 2, катализ — циклический процесс, содержащий несколько элементарных стадий. Поэтому для установления кинетики
каталитической реакции в целом нам необходимо знать кинетические параметры этих элементарных стадий. К сожалению, они достаточно редко поддаются
определению. Сейчас мы покажем, как коэффициенты прилипания (аккомодации), энергии активации, предэкспоненциальные множители для элементарных
ступеней могут быть найдены по данным об адсорбции, десорбции, диссоциации и рекомбинации молекул и атомов.
Зная кинетические параметры элементарных стадий, а также термодинамические характеристики типа теплот адсорбции (см. гл. 6), можно создать мик-
рокинетическую модель, описывающую реакцию в целом. Другой путь состоит
в подборе параметров, входящих в выражение для скорости реакции, полученной для определенного ее механизма. Примеры использования обоих подходов
будут даны в этой главе.
7.1.1. Адсорбция и «прилипание» молекул
Скорость адсорбции молекул газа определяется частотой их столкновений с поверхностью и коэффициентом прилипания (аккомодации):
/•ads = д, b6\TSо(Г) = (7Л)
yVq ^L7lYYlk^ 1
В равновесии скорости адсорбции и десорбции равны
г^=к~вм (7-2)
и из равенства rads и rdes можно, как это было сделано в гл. 2, получить изотерму
адсорбции Ленгмюра:
*a = 1 + A>a' (7'3)
308 -J\, Глава 7. Кинетика поверхностных реакций
Для описания кинетики адсорбции нам необходимо знать величину коэффициента прилипания. Как было показано в гл. 3, он может быть представлен
в аррениусовской форме
S = SyAE“a/kBT. (7.4)
Отметим, что энергия активации здесь может быть равной нулю. Большая
энергия активации для этого коэффициента подразумевает необходимость разрыва связи, имеющего место, например, в случае адсорбции метана или диссоциативной адсорбции N2.
7.1.1.1. Определение коэффициента прилипания
Измерение числа захваченных поверхностью молекул как функции
времени выдержки ее в атмосфере газа это — наиболее прямой путь определения коэффициента прилипания. Такие эксперименты надо проводить с особой
тщательностью, поскольку здесь требуется убедиться в установлении теплового
равновесия между газом и адсорбционным слоем. Кроме того, чтобы можно
было пренебречь десорбцией в этих адсорбционных экспериментах, необходимо знать степень заполнения поверхности, которую можно определить рядом
методов (рентгеноэлектронной, оже-электронной или инфракрасной спектроскопией) или по скорости термодесорбции.
Рассмотрим в качестве примера диссоциативную адсорбцию метана на поверхности (100)Ni. Если эксперимент проводится при температуре выше 350 К,
метан распадается на атомы углерода и водорода, который практически мгновенно десорбируется с поверхности. Следовательно, для нахождения коэффициента прилипания необходимо определить (например, методом оже-спектроскопии), как много углерода образовалось на поверхности при данной длительности выдержки поверхности в атмосфере метана. Зависимость количества
осажденного на поверхность углерода от времени выдержки представляет собой кривую захвата метана поверхностью.
В общем случае если адсорбирующаяся молекула А занимает один адсорбционный центр, то адсорбционный процесс следует кинетике реакции первого порядка:
d^ = РАв, s0(T) = Fie^ -e')S0(T), (7.5)
dt N^lnmkJ 0V ' V 531 ’ 0V '
где в' — текущее значение степени заполнения поверхности молекулами А;
0sat — предельная степень заполнения поверхности; F — поток молекул газа;
SQ(T) — зависящий от температуры коэффициент прилипания при нулевой
степени заполнения поверхности. Для разложения метана предельная степень
заполнения поверхности углеродом составляет 0,5 по отношению к числу поверхностных атомов никеля. Ниже мы будем использовать степень заполнения, нормированную на ее предельное значение
7. /. Простейшие поверхностные реакции
Теперь уравнение (7.5) можно записать в более простой форме:
dO dO'
dt e^dt
= F{\-e)S0{T).
X
(7.7)
Решение этого дифференциального уравнения легко находится методом разделения переменных:
-FS0(T)t
(7.8)
На рис. 7.1 показан набор кривых захвата, рассчитанных для нескольких
температур, а на рис. 7.2 представлены экспериментальные данные для метана
и поверхности никеля.
Рис. 7.1. Кривые захвата, соответствующие реакциям первого и второго порядков. Экспозиция дана в Па • с. В данном примере коэффициент
прилипания зависит от энергии активации, которая равна
60 кДж/моль
о
X
X
Он
<и
CQ
О
с
о?
5
X
U
1-й порядок = 0 QJ exp(-60,000/RT)
2-и порядок
200 400 600 800
Экспозиция, Па • с
1000
1200
X
<и
С
<и
н
U
1000 2000 3000
Экспозиция в СН4, произв. ед.
Рис. 7.2. Кинетика накопления на
поверхности углерода при диссоциативной адсорбции метана на поверхности (11 l)Ni, отвечающая реакции первого порядка. В эксперименте поверхность помещалась в
сверхзвуковой поток метана при
указанных на кривых температурах
газа. Температура поверхности равнялась 500 К, что обеспечивало
мгновенную десорбцию водорода
после разложения метана [1]
309
Доза газа или экспозиция обычно выражается произведением времени и
давления газа. Безусловно, физически более оправданным было бы выражать
310 -Лл Глава 7. Кинетика поверхностных реакций
экспозицию D числом молекул, столкнувшихся с адсорбционным центром в
течение эксперимента:
Мы предлагаем читателю самостоятельно провести эти простые преобразования. Заметьте, пожалуйста, что наклон кривой захвата при нулевой степени
заполнения равен S0(T), а проведенные выше выкладки сделаны в предположении отсутствия взаимодействия между молекулами адсорбата, что на практике встречается редко. Таким образом, строгий физический смысл имеет только
коэффициент захвата в пределе нулевой степени заполнения поверхности.
Довольно часто адсорбции предшествует образование подвижного прекурсора, в состоянии которого адсорбат, будучи физически адсорбированным,
диффундирует по поверхности до достижения свободного центра адсорбции.
В этом случае скорость адсорбции и коэффициент прилипания остаются постоянными вплоть до достижения определенной степени заполнения поверхности, после чего вероятность прилипания резко падает. Если прекурсор находится только на свободных поверхностных центрах, то его называют внутренним, если же он формируется на уже занятых центрах, то он называется внешним.
Здесь мы только отметим возможность наличия прекурсоров двух типов, но в
дальнейшем не будем принимать во внимание этот факт.
Для двухатомных газов, адсорбирующихся в молекулярной форме, как правило, наблюдается кинетика адсорбции, отвечающая реакциям первого порядка, в то время как диссоциативная адсорбция газов типа Н2 и N2 соответствует
реакции второго порядка. Для этого случая в пределе нулевой степени заполнения скорость адсорбции равна
Решая это уравнение методом разделения переменных, получаем уравнение
кривой захвата (см. рис. 7.1) в виде:
Начальный наклон этой кривой (t = 0) пропорционален SQ(T). Построение
аррениусовской зависимости In [^(Г)] от \/Т позволяет определить энергию
активации диссоциативной адсорбции. На рис. 7.3 приведены примеры кинетики диссоциативной адсорбции водорода и дейтерия на поверхности (100) меди
при различных температурах. Аррениусовские зависимости (врезка на рис. 7.3)
дают энергии активации, равные 48 и 56 кДж/моль для Н2 и D2 соответственно.
Проведенный выше анализ для адсорбции, отвечающей реакции первого порядка, можно перенести и на случай кинетики, отвечающей реакции второго
порядка.
de 2 рв;
S0(T) = 2FS0(l-ef.
d t N0^2nmkRT
(7.10)
(7.11)
7.1. Простейшие поверхностные реакции -J\r 311
Давления, при которых необходимо проводить эксперименты типа представленных на рис. 7.2 и 7.3, зависят от величины коэффициента прилипания.
Как правило, выдержка при давлении 10-6 Тор в течение одной секунды позволяет сформироваться одному монослою адсорбата, если вероятность прилипания составляет 100 %. Другими словами, при такой экспозиции газовые молекулы по одному разу столкнутся с каждым поверхностным атомом. Эта величина, 10_6 Тор • с, называемая ленгмюром (обозначается как J1), представляет
собой очень полезную характеристику, широко использующуюся в науке о
поверхностях. Однако соответствие между ленгмюром и монослоем не всегда
выполняется достаточно точно, в частности, при адсорбции Н2 на поверхности
(ЮО)Си для получения правильных значений коэффициента прилипания необходимо рассчитывать экспозицию более аккуратно.
Экспозиция, МС
Рис. 7.3. Кривая захвата для водорода на поверхности Си(ЮО). Экспозиция здесь выражена в числе монослоев (МС). Отметим очень низкие значения коэффициента
прилипания, поэтому эксперименты проводились при давлении 1,8 бар. На врезке
по данным экспериментов построены аррениусовские зависимости для коэффициентов прилипания Н2 и D2 [2]
В общем случае необходимо привести в равновесие газ и поверхность, чтобы они имели одну и ту же температуру. Это может быть проблематично при
низких давлениях, когда молекулы газа чаще сталкиваются с поверхностью
сосуда, а не с исследуемой поверхностью. Путем уменьшения объема и увеличения давления до диапазона в несколько миллибар за счет введения инертного газа удается обеспечить термическое равновесие между кристаллом и газом.
Проводимые в таком режиме измерения обычно называют баллонными экспериментами.
312
Глава 7. Кинетика поверхностных реакций
Таблица 7.1. Коэффициенты прилипания для диссоциативной адсорбции
ряда газов
Молекула
Поверхность
Температура, К
So
£act> кДж/моль,
источник
Н2
Cu(100)
250
5 x 10~13
48 [2]
D2
Cu(100)
250
2x 10H3
56 [2]
сн4
Ni(lll)
500
2 x 10~8
74 [1]
сн4
Ni(100)
500
7 x 10“8
59 [1]
сн4
Cu(100)
1000
8,6 x 10“9
201 [1]
n2
Ru(OOl)
400
1 x Ю"10
38 [3]
N2
Au/Ru(001)
670
5 x 10“15
>130 [3]
Загрязнения, присутствующие в газе и на поверхности, представляют еще
одну ловушку, влияющую на измеряемые значения коэффициента прилипания,
особенно когда его значения достаточно малы. Например, если коэффициент
прилипания некоторого газа составляет величину порядка 10-6, а примесный газ
присутствует в количестве одна часть на миллион, и его коэффициент прилипания велик (например, как у СО), то между этими газами будет проходить заметная конкуренция в процесс адсорбции. Чтобы избежать осложнений, необходимо проводить специальную очистку исследуемого газа.
Важную роль могут играть и неоднородности поверхности, как, например, в
случае адсорбции N2 на чистой тщательно подготовленной поверхности Ru(0001)
(рис. 7.4). Для этой поверхности коэффициент прилипания по порядку величины равен Ю-10, соответствующая энергия активации составляет 42 кДж/моль.
Известно, однако, что на любой поверхности имеются дефекты типа ступеней
между террасами. На поверхности рутения такие ступени могут быть декорированы атомами золота, которые инертны в отношении диссоциации N2. Повторение экспериментов по диссоциации N2 на поверхности рутения, у которой
все дефекты блокированы атомами золота, приводит к существенно более низким значениям коэффициента прилипания, на уровне 1СГ15—10~14 и значительно более высокой энергии активации — выше 130 кДж/моль. Таким образом,
реакционная активность поверхности может полностью определяться небольшим числом всегда присутствующих дефектов! В табл. 7.1 приведены данные
по коэффициентам прилипания для диссоциативной адсорбции, а также соответствующие энергии активации.
Коэффициенты прилипания для молекулярной адсорбции газов типа СО,
N0, С2Н4 и т. д. лежат обычно в интервале от 0,1 до 1. Коэффициенты прилипания для этилена (С2Н4) и ацетилена (С2Н2) на поверхности родия как функ¬
7. 1. Простейшие поверхностные реакции 313
ции степени заполнения поверхности показаны на рис. 7.5 [4]. Для обоих газов
при низких степенях заполнения поверхности коэффициенты прилипания высоки, но с ее ростом их значения быстро падают. Хотя такие зависимости наблюдаются довольно часто, имеются системы, у которых коэффициент прилипания остается практически постоянным до высоких степеней заполнения поверхности. В этом случае молекулы обычно попадают в состояние прекурсора
и свободно двигаются по поверхности вне зависимости от того, заняты центры
адсорбции или нет.
Чистая поверхность,
42 кДж/моль
0,0015 0,0020 0,0025 0S0030 0S0035
1 /Г, Г1
Рис. 7.4. Коэффициенты прилипания для диссоциативной адсорбции N2 на чистой и
модифицированной золотом поверхностях Ru(0001). На чистой поверхности
имеется небольшое число дефектов, ответственных за диссоциацию N,. Когда
атомы золота, декорирующие ступени, блокируют дефекты, коэффициент прилипания уменьшается на несколько порядков [3]
На рис. 7.5 приведены также зависимости дифференциальных теплот адсорбции этилена и ацетилена на поверхности родия, измеренных методом микрокалориметрии, от степени заполнения поверхности [4]. Напомним, что дифференциальная теплота адсорбции — это энергия, высвобождаемая при добавлении
малого количества адсорбата на частично покрытую им поверхность, в то время как интегральная теплота адсорбции — энергия, высвобождаемая при адсорбции данного количества газа на исходно чистой поверхности. Как видно
из рис. 7.5, и коэффициенты прилипания, и дифференциальные теплоты адсорбции убывают с ростом степени заполнения поверхности; это показывает,
что процесс адсорбции на частично заполненной поверхности идет менее эффективно, чем на чистой поверхности.
314 -l\r Глава 7. Кинетика поверхностных реакций
Кажущаяся степень заполнения, МС
Кажущаяся степень заполнения, МС
Кажущаяся степень заполнений, МС
Кажущаяся степень заполнения, МС
Рис. 7.5. Зависимости коэффициентов прилипания и дифференциальных теплот адсорбции, измеренных методом микрокалориметрии, от степени заполнения поверхности для этилена и ацетилена и поверхности Rh(100) [4]
7.1.2. Десорбция
Важность изучения десорбции определяется тем, что она представ-
ляет собой последнюю стадию каталитического цикла и составляет основу метода температурно-программируемой десорбции (ТПД) — мощного экспериментального метода исследования закономерностей адсорбции, распада и реакций
соединений на поверхностях. Этот метод иногда также называют термодесорб-
ционной спектроскопией (ТДС) или спектроскопией температурно-програм-
мируемых реакций (СТПР) (конечно, строго говоря, этот метод не имеет никакого отношения к спектросокпии).
На рис. 7.6 показана схема устройства для исследования ТПД. Кристалл,
подвешивается в камере, где поддерживается сверхвысокий вакуум. Количество десорбирующегося газа определяется с помощью квадрупольного масс-
спектрометра или простого датчика давления.
Если скорость откачки достаточно высока, то повторной адсорбцией можно пренебречь, и относительная скорость десорбции, определяемая как из¬
7. /. Простейшие поверхностные реакции -i\r 315
менение степени заполнения поверхности в единицу времени, дается выражением
где г — скорость десорбции; в — степень заполнения, измеряемая в долях монослоя; t — время; п — порядок реакции десорбции; v — предэкспоненциальный множитель; Edes — энергия активации десорбции; Т0 — температура начала
эксперимента; /3— скорость нагрева, равная dT/dt. Если адсорбция не является
активированным процессом, то Edes равна теплоте адсорбции десорбирующегося газа. Притяжение или отталкивание между адсорбированными молекулами
делает параметры Edes и v зависящими от степени заполнения поверхности.
Прежде чем мы опишем приемы извлечения кинетических параметров из данных эксперимента, рассмотрим несколько примеров.
Рис. 7.6. Схема экспериментальной установки по изучению температурно-программиру¬
емой десорбции в ультравысоком вакууме. Кристалл нагревается за счет джоу-
лева тепла, выделяемого в титановой проволочке, температура измеряется с помощью термопары, прикрепленной к тыльной стороне кристалла. Типичная скорость нагрева кристалла составляет 1—5 К с-1. За десорбцией газа следят с
помощью масс-спектрометра. Если скорость откачки достаточно высока, то амплитуда сигнала на масс-спектрометре пропорциональна скорости десорбции.
На врезке показана типичная кривая ТПД [5]
На рис. 7.7 приведены примеры кривых ТПД для процессов, соответствующих реакциям нулевого, первого и второго порядков.
Нулевой порядок имеет место, когда скорость десорбции не зависит от
степени заполнения поверхности; это имеет место для относительно больших
(7.12)
T = T0+/3t,
Датчик
температуры
Ультравысокий вакуум
Квадрупольный
масс-спектрометр
Нагревающая
проволочка
Термопара
Монокристалл
К насосу
316 -l\r Глава 7. Кинетика поверхностных реакций
островков серебра на поверхности рутения (рис. 7.7, а), когда десорбция идет в
основном с краев островков. Поскольку множитель вп в этом случае в уравнении (7.12) отсутствует, кривая имеет легко распознаваемый экспоненциальный
вид на начальной стадии. Такие ситуации встречаются редко.
Нулевой порядок
900 1000 1100
Температура, К
Первый порядок
■л—I—I—I—1—I—и—
1„ЙЛС0/Р‘(112) Г=:
Второй порядок
300 400 500 600
Температура, К
700 800 900 1000
Температура, К
Рис. 7.7. Данные температурно-программируемой десорбции, соответствующие кинети-
кам реакций нулевого, первого и второго порядков; для десорбции серебра с
поверхности рутения (а), СО со ступенчатой поверхности платины (б) и N, с
поверхности родия (в), соответственно [6—8]. Указанные степени заполнения
отвечают условиям начала эксперимента.
Приведенные на рис. 7.7, б данные отвечают реакции первого порядка,
которая наблюдается для молекул СО на ступенчатой поверхности Pt(l 12). Эта
поверхность представляет собой набор террас по граням (111) и ступеней по
граням (100). При степени заполнения ниже одной трети монослоя СО занимает центры только на ступенях, при более высоких степенях заполнения заселяются и террасы, что проявляется в наличии двух четко выраженных максимумов на кривых ТПД. Поскольку СО более прочно связан со ступенями, соответствующий максимум на кривой ТПД появляется при более высоких
температурах. Заметим, что положение максимумов не смещается при изменении степени заполнения поверхности, что является характерной чертой реакции десорбции первого порядка. Данный пример наглядно демонстрирует, как
метод ТПД позволяет выявить наличие на поверхности адсорбционных центров двух типов.
Рис. 7.7, в соответствует реакции десорбции второго порядка, имеющей
место для атомов азота на поверхности родия. Поскольку десорбция соответствует процессу N* + N* —» N2 + 2*, то, как и ожидалось, скорость реакции
изменяется как в2п. Характерной особенностью реакций десорбции второго
7. 1. Простейшие поверхностные реакции -J\r 317
порядка является сдвиг максимума в сторону более низких температур с ростом степени заполнения поверхности, что обусловлено сильной зависимостью
скорости десорбции от в.
7.1.2.1. Количественная интерпретация данных
температурно-программируемой десорбции
Площадь под кривой ТПД пропорциональна начальной степени
заполнения поверхности адсорбатом. При соответствующей калибровке этой
площади, например в сочетании с данными по дифракции медленных электронов или данными о насыщении поверхности, можно использовать ТПД для
нахождения степени заполнения поверхности. Набор кривых ТПД содержит в
себе очень ценную информацию о поверхностных концентрациях и степенях
заполнения поверхности соединениями, определение которых в сочетании с
данными о структуре, колебательных спектрах и реакционной способности делает
этот метод ценным в практическом отношении.
Кроме того, температура, при которой начинается интенсивная десорбция
молекул, показывает, насколько прочно они связаны с поверхностью (уравнение (7.12)). Входящая в уравнение (7.12) энергия активации равняется теплоте
адсорбции, если последняя идет без преодоления активационного барьера. Как
правило, это условие обычно выполняется.
В уравнение (7.12) входит также предэкспоненциальный множитель. В подразд. 3.8.4 мы описали десорбцию в терминах теории переходных состояний
(на рис. 3.14 отражены типичные для десорбции ситуации). Если переходное состояние десорбирующейся молекулы по сути является хемосорбционным, то предэкспоненциальный множитель должен иметь значения порядка ekBT/h = 10-13 с-1.
Но если молекула адсорбируется на активный центр, где она остается неподвижной, а десорбируется через состояние подвижного прекурсора, то предэкспоненциальный фактор должен быть на два-три порядка выше стандартного
значения, равного 1СГ13 с-1.
Как можно определить энергию активации по данным ТПД? К сожалению,
дифференциальное уравнение (7.12) не может быть решено аналитически. По
этой причине анализ данных ТПД представляет собой довольно сложную задачу, в частности, еще и потому, что кинетические параметры обычно зависят от
степени заполнения поверхности.
Простой, часто использующийся подход состоит в анализе кривых в терминах легко определяемых параметров типа положения максимума, Гтах, а также
ширины пика на половине его высоты.
Для десорбции, описываемой реакцией первого порядка, можно установить простое соотношение между E6cs и у, если учесть, что в точке максимума
производная скорости десорбции равна нулю:
318 -J\, Глава 7. Кинетика поверхностных реакций
откуда следует
^des ”
(7.14)
Это уравнение можно решить итерационным методом при соответствующем выборе v (обычно 1013 с-1). Схема решения такова: из эксперимента находим Тт, подставляем в правую часть (7.14) оценочное значение EdQs и рассчитываем новое значение Edes. Теперь это значение подставляем в правую часть
(7.14) и получаем уточненное значение энергии десорбции. Продолжаем до тех
пор, пока разность между двумя последовательно найденными значениями энергии десорбции не станет пренебрежимо малой; обычно хватает 3—6 итераций.
Аналогичное, но более сложное выражение можно получить для случая,
когда десорбция отвечает реакции второго порядка:
откуда следует
Следовательно, здесь необходимо также знать значение в (или оценить, например, половиной начальной степени заполнения) в момент достижения Тт.
Снова для проведения итерационной процедуры необходимо оценить и значение энергии десорбции.
Более детальные схемы включают в анализ и ширину пика десорбционной
кривой. Схема Кана—Ариса—Вайнберга позволяет надежно определить и энергию десорбции, и предэкспоненциальный множитель, при условии что имеется серия экспериментальных кривых для различных степеней заполнения и
кинетические параметры экстраполированы к нулевой степени заполнения
поверхности. Значения кинетических параметров, отвечающих высоким степеням заполнения, при этом теряют смысл, но данные для десорбции одной
молекулы с пустой поверхности оказываются вполне надежными [9, 10].
Если десорбционный процесс следует непосредственно кинетике реакций
первого порядка, то можно, поделив скорость десорбции на текущее значение
степени заполнения и построив зависимость логарифма этого отношения от
обратной температуры, получить зависимость аррениусовского типа. Эта процедура очень хорошо работает при достаточно низких степенях заполнения,
когда латеральным взаимодействием между адсорбированными молекулами
можно пренебречь. Например, для десорбции СО эта схема хорошо работает
при степенях заполнения меньше 0,3 МС (см. рис. 7.7).
При заметной роли латерального взаимодействия EdQs и v начинают зависеть от степени заполнения поверхности, тогда указанная выше аррениусовс-
d26> = Е^Р йв
dt2 kBT2 dt
(7.15)
7. /. Простейшие поверхностные реакции
\
кая зависимость отклоняется от прямой линии. Альтернативный подход основан на использовании узкого интервала температуры для этой зависимости,
когда степень заполнения поверхности изменяется незначительно. При проведении достаточно точных измерений в таком узком интервале данные можно
представлять непосредственно в виде прямолинейной аррениусовской зависимости. Тогда угол наклона прямой и точка ее пересечения с осью ординат
будут характеризовать десорбционные параметры для данной степени заполнения поверхности. Такую процедуру часто называют «анализ с жесткими краями» [11].
Наконец, можно провести полный анализ путем интегрирования кривой
ТПД и определить набор скоростей и температур, отвечающих одной и той же
степени заполнения. Эти данные также можно построить в аррениусовских
координатах. Однако такая процедура очень утомительна и отнимает много
времени, поэтому ее используют редко [12].
7.1.2.2. Компенсационный эффект
в температурно-программируемой десорбции
Экспериментальные данные по ТПД, в которых выявлена зависимость кинетических коэффициентов от степени заполнения поверхности, часто демонстрируют наличие так называемого компенсационного эффекта —
пропорциональности между Е{0) и In [v(0)] (рис. 7.8). Хотя компенсационный
эффект является сильно интригующим, как мы увидим ниже, он представляет
собой артефакт, порожденный математическими преобразованиями [13].
ТПД Ад с поверхности Ru (001)
Рис. 7.8. Компенсационный эффект, наблюдаемый при адсорбции
- 1014 'о
Ag с поверхности рутения: энергия ц/
- 1012
активации и предэкспоненциальный множитель одинаковым образом зависят от степени заполнения
поверхности. Этот эффект обусловлен обычно упущениями в кинетическом анализе [6]
0,0 0,2 0,4 0,6 0,8 1,0
Степень заполнения поверхности, МС
- Ю10
Обычный вывод, связывающий энергию активации с наклоном прямой зависимости скорости реакции от температуры, построенной в координатах Аррениуса, дает:
320
\
Глава 7. Кинетика поверхностных реакций
Однако, если предэкспоненциальный множитель и энергия десорбции зависят от степени заполнения поверхности, дифференцирование приводит к
следующему соотношению
F =Е (0\ I RT2 дв Э1п WJ 1
meas des^ ' дЕ[ дв RT
^des И
дв
(7.18)
Отсюда видно, что энергия активации включает зависящие от степени заполнения члены второго порядка, которыми обычно пренебрегают. Делать это
можно только в трех случаях. В тривиальной ситуации, когда кинетические
параметры являются постоянными или когда степень заполнения поверхности
не изменяется с температурой; последнее допущение корректно для достаточно узкого температурного интервала исследования десорбции. Третий случай
соответствует обращению в нуль выражения в фигурных скобках формулы (7.18),
то есть когда имеет место равенство
dln[v(0)]_ 1
дв ~ RT
d^des ifi)
дв
Эта пропорциональность и называется компенсационным эффектом. Важно отметить, что при пренебрежении поправочным членом в (7.18) мы как бы
«заставляем» кинетические параметры подчиняться уравнению (7.19), что выражается в появлении корреляций между предэкспоненциальным множителем
и энергией активации десорбции в форме компенсационного эффекта!
Наконец, хотя исследования температурно-программируемых десорбции и
реакций представляют собой незаменимый инструмент катализа и науки о поверхностях, имеются ограничения по их применению. Во-первых, эксперименты по ТПД проводятся в неравновесных условиях, поскольку температура постоянно повышается. Во-вторых, кинетические параметры не остаются постоянными в процессе ТПД, что связано с изменением и температуры, и степени
заполнения поверхности. В-третьих, определенное влияние на ТПД могут оказать зависящие от температуры поверхностные процессы типа диффузии и реконструкции поверхности. Таким образом, этот метод следует применять осмысленно, а обработку экспериментальных данных надо проводить с большой
степенью осторожности.
7.1.3. Роль латеральных взаимодействий
в поверхностных реакциях
Кинетические уравнения типа выражения (7.12), в которых скорость зависит от степени заполнения поверхности, представляющей собой среднюю концентрацию адсорбата на поверхности, часто оказываются непригодными, когда молекулы и/или атомы начинают чувствовать присутствие соседей. При наличии у адсорбированного атома или молекулы соседей изменяются
7.1. Простейшие поверхностные реакции
321
и сила их связи с поверхностью, и их реакционные способности. Латеральное
взаимодействие между адсорбированными молекулами сводится преимущественно к отталкиванию, и оно становится ощутимым при высоких степенях заполнения, что обычно имеет в случае каталитических реакций, протекающих при
нормальном давлении. Изучение природы латеральных взаимодействий и характера их влияния на кинетику реакций составляет важную часть так называемой стратегии «наведения мостов», в которой результаты, полученные при
изучении идеальных поверхностей, пытаются связать с процессами, происходящими в промышленных реакторах.
Надлежащий анализ характера влияния ближайших соседей на реакционную
способность молекул адсорбата должен проводиться таким образом, чтобы локальное окружение каждой реагирующей молекулы учитывалось отдельно. Это
можно осуществить в численном моделировании реакций методом Монте-Кар-
ло [14, 15]. На рис. 7.9 и 7.10 показано, как взаимодействие в адсорбционных
слоях влияет на распределение молекул по поверхности. Приближение среднего поля и средние степени заполнения поверхности не позволяют описать разнообразные реальные конфигурации молекул на поверхности. А энергия адсорбции уже зависит от числа ближайших и следующих за ними соседей:
Здесь rij — число соседей, а со~ — энергия взаимодействия между адсорбатом / и
его соседом у. При положительных значениях со~ имеет место отталкивание,
при отрицательных — притяжение между соседями. В формуле (7.20) использовано допущение, что все парные взаимодействия можно просто складывать,
а это в общем случае не является вполне корректным. При рассмотрении реакций для нахождения энергии активации можно использовать как уравнение
(7.20), так и допущение, что вклад от латерального взаимодействия с соседями
составляет постоянную долю, то есть 50 %, от изменения энергии адсорбции
реагирующих смесей, как это следует из соотношения Бренстеда—Эванса—
Поляни. Энергия латерального взаимодействия не может быть измерена экспериментально, но ее можно определить численными методами.
Представленные на рис. 7.9 данные численного моделирования относятся к
распределениям молекул одного компонента по поверхности, полученным при
различных энергиях взаимодействия ближайших и следующих за ними соседей. Представленные результаты показывают, что отталкивание между ближайшими соседями приводит к относительно равномерному распределению
молекул по поверхности и формированию упорядоченных структур при высоких степенях заполнения. Комбинированное взаимодействие — отталкивание
между ближайшими соседями и притяжение между следующими за ближайшими соседями приводит к четко выраженной структуре с(2 х 2) при высоких
степенях заполнения.
На рис. 7.10 представлены результаты, полученные для бинарной смеси
молекул А и В, распределенных по квадратной решетке однотипных центров.
(7.20)
j
2J Г Глава 7. Кинетика поверхностных реакций
Рис. 7.9. Влияние латерального взаимодействия
на распределение одного адсорбированного
компонента А по поверхности. Энергия адсорбции каждого атома А рассчитывается по уравнению (7.20), при этом энергии парных взаи-
модеиствии указаны в нижнеи части рисунков. Отрицательные значения энергии соответствуют притяжению, положительные — отталкиванию. В расчетах учитывались взаимодействия ближайших (NN) и следующих за ближайшими (NNN) соседей. Приведено также несколько упорядоченных структур (см. также гл. 5)
С( 2x2) (2x2)
Ш
(1x1)
А = 0,25 мс ^ •.!•■ -Д'Я л
г;\-у^х •• I rt:ir- •/.- Я* й 4 У. да, j
= 0,50 мс .• .v>v:
D * * Я* * I I \ Л
>ч.’>v\,
г <•т *\ «
• 0,25 МС
*ч^.," '• -’I X i **'; й. '"'Г'*
о,5о мс
нй*.-”.’ ■' ’ i¥~rD!’ "Vv*» ’J
и* ,* *VZ‘V*.*4r, X, ,n *T
Li-1’-. .. :* • ^ V-».!... r 'v .
A-A
NN
-5
-5
5
5 кДж/моль
5 кДж/моль
-5 кДж/моль
Рис. 7.10. Распределения компонентов бинарной смеси А и В по поверхности для различных комбинаций сил отталкивания и притяжения, полученные методом Монте-Карл о
7.1. Простейшие поверхностные реакции -*\г 323
Для простоты анализа было учтено только взаимодействие между ближайшими
соседями. Рассмотрим реакцию между молекулами А и В, при которой образуется соединение АВ, и проверим, действительно ли ее скорость можно представить в виде г = квА6в. Следует проанализировать два случая. Первый соответствует ситуации, когда молекулы одного типа притягиваются, а молекулы
разных типов — отталкиваются. В этом случае идет сегрегация молекул А и В,
которые формируют островки и лишь слабо смешиваются друг с другом (см.
рис. 7.10). Очевидно, что скорость реакции в этом случае будет пропорциональна не поверхностным концентрациям молекул А и В, а долям этих молекул
на периметрах островков. В другом случае, при отталкивании однотипных молекул и притяжении между молекулами разных сотов происходит почти идеальное упорядочение молекул А и В, которое является наиболее предпочтительным для осуществления поверхностной реакции между ними. Заметим,
что в случае «все отталкиваются» также происходит хорошее перемешивание
молекул А и В на поверхности.
Представленные на рис. 7.9 и 7.10 данные отвечают простой модели, в которой учитывается только взаимодействие между молекулами. Определить латеральное взаимодействие в специальных ситуациях можно только с помощью
целенаправленных измерений, которые мы рассмотрим на примере смешанной адсорбции молекул СО и атомов N.
Рис. 7.11. Влияние отталкивания между атомами N
и молекулами СО на десорбцию последних. Слева: кривые ТПД СО с чистой поверхности Rh(100),
энергия десорбции равна
135 кДж/моль. Справа: то
же самое, но когда каждая молекула СО окружена четырьмя атомами N;
энергия десорбции составляет приблизительно
60 кДж/моль. Следовательно, энергия латерального отталкивания coN_co
составляет примерно
19 кДж/моль [16]
На рис. 7.11 показаны кривые ТПД для десорбции СО с поверхности Rh(100),
покрытой упорядоченным слоем атомов N, формирующих структуру с(2 х 2).
Теплота адсорбции на 75 кДж/моль меньше теплоты адсорбции СО на чис¬
Температура, К
СО 4- C(2x2)-N/Rh(100)
Температура, К
324 -l\r Глава 7. Кинетика поверхностных реакций
той поверхности (135 ± 5 кДж/моль). Поскольку каждая молекула СО имеет
по 4 соседних атома N, то можно считать, что энергия взаимодействия (отталкивания) между СО и N составляет 75/4 = 19 кДж/моль, при условии что можно использовать приближение парного взаимодействия. Получить численное
значение энергии взаимодействия в данном случае оказалось возможным, поскольку мы точно знаем число ближайших соседей в упорядоченной структуре.
Другие способы оценки энергии парного взаимодействия состоят в ее расчете
методом функционала плотности или в подгонке данных моделирования методом Монте-Карло под реальный эксперимент, когда энергия взаимодействия
используется как подгоночный параметр.
7.1.4. Диссоциативные реакции на поверхности
Элементарные стадии, на которых происходит разрыв связей в
молекулах, составляют в катализе практически важный класс реакций. Суть
каталитического действия состоит в том, что катализатор активирует сильную
связь, которая не может быть разорвана в прямой реакции, но существенным
образом ослабляется, как это было объяснено в гл. 6, при взаимодействии молекул с поверхностью. Для наблюдения за диссоциативными реакциями необходимо использовать специальное оборудование. Метод температурно-программиру-
емой десорбции представляет прекрасную возможность мониторинга реакций,
продукты которых десорбируются с поверхности. Однако, если продукты остаются на поверхности, то необходимо использовать другие методы типа инфракрасной спектроскопии или масс-спектрометрии вторичных ионов (МСВИ).
Рассматривая в качестве примера диссоциацию N0 на поверхности родия
(100) (рис. 7.12), можно указать, что при использовании МСВИ хемосорбиро-
ванные молекулы N0 будут детектироваться в виде характеристических вторичных ионов RhNO+ или Rh2NO+, в то время как продукты диссоциации
наблюдаются как ионы Rh2N+. Из рис. 7.12 видно, что при низких степенях
заполнения поверхности диссоциация N0 на поверхности Rh(100) начинается
при температуре около 175 К и завершается при 250 К, чему соответствует
энергия активации 37 кДж/моль. Атомы N рекомбинируют и десорбируются в
форме N2 при более высоких температурах — от 600 до 800 К с энергией активации 225 кДж/моль. Атомы кислорода в исследованном диапазоне температур
остаются на поверхности. Поскольку степень заполнения поверхности исходно
была малой, а компоненты на поверхности отталкиваются при попадании в
соседние центры, то молекулы N0 избегают друг друга и атомов — продуктов
диссоциации. Следовательно, как показано в эксперименте, скорость диссоциации следует кинетике невозмущенной реакции первого порядка, а константа
скорости имеет аррениусовский вид. Картина радикально изменяется при высоких степенях заполнения.
Центральный из трех рис. 7.12 показывает ход процесса при начальном
заполнении поверхности молекулами N0, равном 0,37 МС. Если бы все молекулы диссоциировали, то полная степень заполнения поверхности равнялась
7.1. Простейшие поверхностные реакции -*\г 325
бы 0,75 МС, что является очень большой величиной. Однако полной диссоциации не происходит, поскольку отталкивание между N0 и уже образовавшимися атомами замедляет реакцию диссоциации настолько, что при 400 К десорбция начинает преобладать над диссоциацией и часть молекул N0 покидает
поверхность. Десорбировавшиеся молекулы освобождают поверхность, так что
оставшиеся на поверхности мгновенно диссоциируют, как если бы они находились на свободной поверхности.
200 400 600 800
Т,К
200 400 600 800
Т,К
200 400 600 |800
Т,К
Молекулы N0
Молекулы N0 + атомы N, О
Атомы О и N
Рис. 7.12. Влияние латеральных взаимодействий на диссоциацию NO* -> N*+0*, проявляющееся в экспериментах по ТПД и МСВИ [8] и численном моделировании
методом Монте-Карло (объяснения см. в тексте). В нижней части показано
распределение молекул N0 (серые квадраты) и атомов N или О (пятнистые
квадраты) на квадратной решетке, представляющей поверхность Rh(100)
При полном заполнении поверхности молекулами N0 процесс диссоциации
заблокирован, пока при повышении температуры не произойдет частичная десорбция молекул N0, после чего оставшиеся молекулы мгновенно диссоциируют (правая часть рис. 7.12). В данном случае диссоциация первоначально подавляется вследствие блокировки центров адсорбции, а не из-за латеральных взаимодействий.
Для описания кинетики, соответствующей представленным экспериментам,
необходимо учесть влияние локального окружения на поведение реагирующих
326 ■J; Глава 7. Кинетика поверхностных реакций
компонентов. Моделирование методом Монте-Карло, данные которого представлены в нижней части рис. 7.12, позволяет воспроизвести экспериментальные данные по скоростям реакций и поверхностным концентрациям компонентов. В моделировании поверхность Rh(100) была представлена квадратной решеткой однотипных центров. Следовательно, тонкости вроде присоединения
молекулы N0 к центру атомами N или О в расчет не принимались. Значения
кинетических параметров для диссоциации и десорбции в пределе нулевой степени заполнения поверхности были взяты из эксперимента. Они, однако, были
уточнены с учетом влияния ближайших соседей с помощью уравнения (7.20),
при этом считалось, что имеется слабое отталкивание между молекулами N0
(^no-no = ^ кДж/моль), более сильного отталкивания между N0 и атомами
{со = 17—20 кДж/моль) и очень сильного отталкивания между атомами N0
{со = 30 кДж/моль). Поскольку учитывалось взаимодействие только между ближайшими соседями, то в результате атоМы преимущественно образовывали
структуру с{2 х 2) или более рыхлые решетки, когда ближайшие к атомам центры предпочтительно оставались свободными.
Каждый численный эксперимент начинался со случайного распределения
некоторого числа молекул N0 по поверхности при выбранной степени ее заполнения. Молекулы могли диффундировать по поверхности, так что энергетически
невыгодные конфигурации (например, с несколькими молекулами на соседних
центрах) в существенной мере исключались, по крайней мере, при низких степенях заполнения поверхности. При высоких степенях заполнения поверхности
молекулы формировали и энергетически невыгодные конфигурации (рис. 7.12).
Влияние более сильного отталкивания между молекулами N0 и атомами N и О
видно из центрального рис. 7.12. Поскольку взаимодействие NO—N0 слабее
взаимодействий NO—N и N0—0, то молекулы N0 сжимаются в островки, а
атомы из-за сильного отталкивания между ними принимают оптимальную конфигурацию со структурой с{2 х 2). В таких островках все химические процессы
блокированы, пока температура не повысится до значения, при котором становится возможен разрыв связи Rh—N0 и начинается процесс десорбции. В результате появляются свободные центры, которые мгновенно используются для
диссоциации молекул N0, и поверхность заполняется атомами, формирующими
почти идеальную структуру с{2 х 2) (последний рис. 7.12).
На рис. 7.12 четко показано, как взаимодействие между ближайшими соседями на поверхности влияет на реакцию диссоциации. Такое влияние обусловлено тем, что для осуществления диссоциации требуется наличие ансамбля
центров, обладающего определенной конфигурацией. Как было показано в гл. 6
при обсуждении реакционной активности поверхности, молекула типа N0 диссоциирует при существенном перекрытии ее 2л"-орбиталей и электронной плотности металла. Для этого необходимо изогнуть молекулу, находящуюся в полостном центре, над атомом металла [17]. Ясно, что в такой процесс автоматически вовлекается несколько атомов металла.
В данном разделе мы также увидели, насколько сложной может стать кинетика поверхностных реакций, если становится необходимым принять во
7. 1. Простейшие поверхностные реакции -1\г 327
внимание латеральные взаимодействия, а также требование формирования
определенных конфигураций у ансамблей атомов и молекул. Сравните эти
требования со стандартной схемой Ленгмюра—Хиншельвуда, которая ограничена пределом низких степеней заполнения поверхности. Моделирование
методом Монте-Карло позволяет учесть локальные процессы в глобальной
картине, наблюдаемой в реальных кинетических экспериментах. С 2003 г.
такое моделирование стало возможно проводить на персональных компьютерах. Однако и это моделирование использует ряд упрощающих предположений о типе адсорбционных центров, парном аддитивном взаимодействии и
соотношении Бренстеда—Эванса—Поляни, с помощью которого из энергий
адсорбции получают энергии активации. Кроме того, практически невозможно извлечь из экспериментов энергии латерального взаимодействия, поэтому
они часто используются как подгоночные параметры, что делает их зависимыми от принятой модели. Тем не менее такое моделирование дает много
полезной информации о кинетике реакций, протекающих на заполненных
адсорбатом поверхностях; такая ситуация доминирует во многих реальных
каталитических процессах.
7.1.5. Интермедиаты в поверхностных реакциях
Если некоторое адсорбированное соединение, например интерме-
даит в каталитическом цикле, распадется на продукты, которые мгновенно десорбируются, то метод ТПД можно использовать для наблюдения за этой стадией.
Сказанное проиллюстрировано на рис. 7.13, где приведены спектры ТПД
для формила, адсорбированного на поверхности Си(ЮО). Для доказательства
того, что формила является интермедиатом в процессе синтеза метанола из
С02 и Н2, поверхность Си(ЮО) была помещена в камеру, где созданы условия,
отвечающие синтезу метанола, а затем были сняты спектры ТПД (нижние кривые на рис. 7.13). Для сравнения на этом же рисунке приведены (верхние)
кривые, относящиеся к разложению формилата, полученного при нанесении
порции муравьиной кислоты на поверхность. Поскольку и С02, и Н2 десорбируются при температурах существенно ниже того диапазона, на который приходится пик спектров ТПД, приведенных на рис. 7.13, то наблюдаемые пики
отвечают С02 и Н2, десорбция которых лимитируется реакцией разложения
формилата. Идентичность профилей спектров ТПД строго доказывает, что
формилат присутствует на поверхности в условиях синтеза метанола.
В принципе метод ТПД можно использовать для изучения катализаторов,
обладающих высокой удельной поверхностью, в трубчатом реакторе. В этом
случае, однако, максимумы оказываются сильно уширенными, что связано с
влиянием транспортных процессов. Таким образом, использование монокристаллов или частиц на плоской поверхности дает существенные преимущества в
использовании этого метода.
328
■К
Глава 7. Кинетика поверхностных реакций
300
200
100
—I 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 г~
а Десорбция продуктов
разложения формилата,
полученного из муравьиной кислоты
0 I I I I I I L—
300
б Десорбция продуктов
разложения формилата,
синтезированного
из С02 и Н2
400
500
600
Температура, К
Рис. 7.13. Кривые спектров ТПД для СО, и Н2, образующихся из формилата на поверхности Си(ЮО). Верхние кривые получены при нанесении порции муравьиной кислоты на поверхность. Нижние кривые получены в условиях синтеза метанола в
камере высокого давления (2 бара) из смеси С02 и Н2 [18]
7.1.6. Ассоциативные реакции
Спектроскопия температурно-программируемых реакций дает прямую возможность прослеживать кинетику элементарных поверхностных реакций
при условии, что десорбция не является лимитирующей стадией. На рис. 7.14
показана зависимость от температуры скорости реакции СО* + О* = С02 + 2*.
Здесь предварительно при низких температурах на поверхность адсорбируют небольшое
количество СО и О, после чего температуру
поверхности повышают линейно со временем,
и процесс образования С02 отслеживается с
помощью масс-спектрометра. Последовательность реакций для этого процесса такова
со + о -> со; + *,
(7-21)
со^ —> со^ + .
Рис. 7.14. Кривая кинетики температурно-програм-
мируемой реакции и соответствующая аррениусовс-
кая зависимость, построенная по формуле (7.21), позволяют провести расчет кинетических параметров для
элементарной реакции между СО и О на поверхности Rh(100) [19]
1000/Т, К'1
7. 1. Простейшие поверхностные реакции
Поскольку десорбция С02 идет быстрее, чем поверхностная реакция между
СО* и О*, то скорость десорбции совпадает со скоростью предшествующей
реакции
При известных начальных степенях заполнения для СО* и О* и условии
отсутствия СО на поверхности к концу эксперимента по ТПД можно определить текущие степени заполнения поверхности для СО* и О*, то есть для любой точки на кривой ТПД, показанной на рис. 7.14. Следовательно, из аррени-
усовского графика, построенного в координатах логарифм скорости десорбции, поделенной на степени заполнения, — обратная температура, можно
определить энергию активации и предэкспоненциальный множитель
Поскольку большое число экспериментальных точек для обоих экспериментов по ТПД в аррениусовских координатах ложится на прямые линии, реакция между СО* и О*, скорее всего, представляет собой элементарную ступень с энергией активации 103 ± 5 кДж/моль и предэкспоненциальным множителем, равным 1012,7±2 с-1. Проведенный анализ снова справедлив, если только
можно пренебречь зависимостью кинетических параметров от степени заполнения поверхности. Прямолинейная зависимость на рис 7.14 говорит о том,
что это условие выполнено.
Кинетику элементарных поверхностных реакций, при которых продукт остается на поверхности, можно проследить методом масс-спектрометрии вторичных ионов. На рис. 7.15 приведены данные для реакции адсорбированных
азота и углерода, при которой на поверхности образуется CN. Представленная
на рис. 7.15 кривая дает температурную зависимость отношения сигналов от
Rh2CN+ и Rh2 в спектре МСВИ. Оба сигнала измерялись одновременно в
процессе роста температуры. Кривая показывает начало образования CN при
500—600 К (Елсх = 110 ± 10 кДж/моль; п = 10п±1 с-1) и последующее его разложение при температурах 700—800 К (Eact = 210 ± 15 кДж/моль; у = 10п±1 с-1), за
которым следует мгновенная десорбция N2. Всегда при наличии методики, позволяющей определять концентрации адсорбированных компонентов в реальном времени, температурное программирование реакций открывает возможность
исследовать элементарные стадии протекающих на поверхности реакций.
Основываясь на данных науки о поверхностях и методах типа ТПД, можно
определить большинство кинетических параметров элементарных стадий, составляющих каталитический процесс. К сожалению, короткоживущие интермедиаты не могут быть зафиксированы спектроскопическими методами, поэтому остается полагаться только на методы численного моделирования или на
оценки параметров. Другая возможность, как показано ниже, состоит в подгонке кинетических параметров некоторой модели, обеспечивающей наилучшее согласие с экспериментом для скоростей полных реакций.
г = квС0в0 = ve~EMlRT всов(
(7.22)
In (r/вС0в0 ) - In V - —pj;
(7.23)
330
шт^^‘ Глава 7. Кинетика поверхностных реакций
Температура, К
Рис. 7.15. Эксперименты по температурно-программируемой десорбции и данные МСВИ,
показывающие ход реакции между атомами С и N,ведущей к образованию поверхностных цианидов, при температурах 475 и 600 К; разложение CN на С + N, за
которым следует мгновенная десорбция N,, начинается при более высоких температурах. В отсутствие атомов С и канала образования CN молекулы N2 образуются из атомов N уже при температуре 500 К [20].
7.2. КИНЕТИЧЕСКИЕ ПАРАМЕТРЫ, ИЗВЛЕКАЕМЫЕ
В МОДЕЛИ ЛЕНГМЮРА—ХИНШЕЛЬВУДА
В данном разделе мы покажем, как при наличии кинетической
модели и экспериментальных данных по кинетике реакции можно установить
степенной закон для скорости реакции, определить константы скорости реакции, константы равновесия для адсорбции и даже теплоты адсорбции. В качестве примера рассмотрим реакцию гидрообессеривания тиофена, катализируемую промотированным сульфидом никеля катализатором MoS2:
C4H4S + 2Н2 = С4Н6 + H2S (7.24)
и
С4Н6 + Н2 = С4н8. (7.25)
Последующее гидрирование бутадиена до 1-бутена и 2-бутена в кинетическом отношении никакой роли не играет, и эти углеводороды не влияют на
скорость реакции первой ступени. Однако присутствие H2S сказывается на ско¬
7.2. Кинетические параметры, извлекаемые в модели Ленгмюра—Хиншельвуда -i\r 331
рости реакции. Коротко говоря, реакция идет на тех центрах, где у катализатора недостает атомов серы (см. гл. 9, где дана более полная информация). При
высоких давлениях H2S число таких центров снижается, чем и определяется
влияние этого газа на скорость реакции.
Для установления степенного закона для скорости реакции мы должны
имерить ее значение при различных давлениях компонентов газовой смеси —
тиофена, водорода и сероводорода. На рис. 7.16 приведены эксперименталь¬
ные данные по скорости, полученные при температуре 400 °С, а также их аппроксимация степенными законами. Можно было бы построить эти зависимости в двойных логарифмических координатах и по наклону определить порядок
реакции. Как и ожидалось, получено положительное значение для порядка по
тиофену и водороду и отрицательное — по H2S (при 400 °С):
_ Фт _ /,„0,8 0,93 -0,4 /7
r ~ ~ 2 2S ’ (7.26)
где рТ — парциальное давление тиофена.
Рис. 7.16. Зависимости скорости гидрообессеривания тиофена от парциальных давлен и й
реагентов тиофена и водорода и продукта сероводорода: эксперимент и подгоночные кривые для степенного закона [21]
Теперь мы введем кинетическую схему реакции и посмотрим, соответствует ли она данным, представленным на рис. 7.16. В литературе предложены
различные трактовки кинетики гидрообессеривания. Здесь мы рассмотрим простейшую схему, в которой тиофен (Т) адсорбируется исключительно на вакансиях серы, обозначаемых через А, а Н2 диссоциативно адсорбируется на всех
центрах (обозначаемых символом *), что ведет к образованию бутадиена (В) и
H2S в лимитирующей поверхностной реакции (мы не рассматриваем малозначительную стадию гидрирования бутадиена):
332
Л
Глава 7. Кинетика поверхностных реакций
Т + 2Н -> В + SA + 2 ,
к4
SA + 2H' ±^H?S + 21 +А.
(7.29)
(7.30)
Хотя точный механизм реакции гидрообессеривания все еще обсуждается,
мы умышленно выбрали эту схему, поскольку она дает иллюстрацию кинетики
процессов, протекающих с участием активных центров двух типов. В соответствии со сказанным имеется два уравнения баланса числа центров:
(7.31)
Читатель может легко установить, что вышеприведенная схема приводит к
следующему уравнению (уравнению Ленгмюра—Хиншельвуда) для скорости
реакции
г - NAk30T0f{ -
N,kzK{K2PlP^
(7.32)
На рис. 7.17 приведены экспериментальные данные и соответствующие уравнению (7.32) оптимизированные по константам скорости зависимости для температуры 400 °С. Хорошее согласие между экспериментом и моделью Ленгмюра—Хиншельвуда говорит об ее адекватности. Найденные значения констант
скорости говорят о сильной адсорбции тиофена и сероводорода и слабой адсорбции водорода. Как следствие, членом в уравнении (7.32) можно спокой¬
но пренебречь. Порядок по Н2, как отмечалось выше, равен 0,93, что близко к
единице и является дополнительным указанием на слабую адсорбцию водорода.
2
= 0.04-
О 0,02-
а
о
U
Т = 400 °С
Кн = 0,011
0,2
КЛ
Т= 400 °С
h2s “ 35,8
М 0,02-
R = -
k'
1 + Kj[)T + ^h2s/;h2s
0,02 0.04
Ph7s
Рис. 7.17. Зависимости скорости гидрообессеривания тиофена от парциальных давлений
реагентов тиофена и водорода и продукта сероводорода: эксперимент и подгоночные кривые для модели Ленгмюра—Хиншельвуда [21]
Приведенные на рис. 7.16 и 7.17 данные относятся к одной температуре —
400 °С. При варьировании температуры будут изменяться и константы равновесия, и константа скорости. На рис. 7.18 приведены зависимости скорости реак-
7.3. Микрокинетическое моделирование -l\r 333
ции от парциального давления тиофена,
полученные при разных температурах, а
также подгоночные кривые, построенные
по модели Ленгмюра—Хиншельвуда. Изме- ~
нение констант равновесия с температурой ^
соответствует теплоте адсорбции тиофена, §
равной 58 ± 5 кДж/моль, в то время как ^
энергия активации для стадии, определя- |
ющей скорость реакции в целом, достига- g
ет величины 84 ± 5 кДж/моль. 2
с
Рис. 7.18. Зависимости скорости реакции гидрообессеривания тиофена от его парциального давления
при различных температурах и подгоночные кривые, построенные по модели Ленгмюра—Хиншельвуда (7.32) [21]
Приведенные пример показывает, как интересующие исследователя параметры могут быть определены из данных по кинетике реакции. Безусловно,
необходимо быть точно уверенным в отсутствии транспортных (в частности,
диффузионных) ограничений и в том, что измерения относятся именно к кинетике исследуемой реакции. Полученные в [21] данные, которые были использованы нами в качестве иллюстрации, удовлетворяют этим требованиям,
поскольку использованный в них катализатор был непористым.
7.3. МИКРОКИНЕТИЧЕСКОЕ МОДЕЛИРОВАНИЕ
Микрокинетическое моделирование представляет собой своего рода
искусство в описании каталитических реакций. Оно началось с работ Штольце
и Норскова, опубликованных в середине 1980-х гг., и затем развивалось Думе-
сиком с сотрудниками [22]. В идеале в микрокинетическом моделировании
удается избежать подгонки параметров, как это делалось в разд. 7.2, поскольку
многие из них могут быть определены при изучении методами науки о поверхностях адсорбции и элементарных ступеней реакции, кроме того, здесь привлекаются данные численного моделирования. В качестве рабочего примера
мы рассмотрим синтез аммиака [23].
7.3.1. Схема реакции и выражение для скорости реакции
Первым шагом микрокинетического моделирования является составление набора всех возможных элементарных стадий исследуемой реакции,
которые могут быть вовлечены в каталитический процесс. В рассматриваемом
случае синтеза аммиака полная реакция выглядит так
^тиоф
N2 + зн2 о 2NH3.
(7.33)
334 -/\л Глава 7. Кинетика поверхностных реакций
Сейчас мы составим список элементарных стадий и решим, какие из них
лимитируют скорость реакции, а какие идут в состоянии квазиравновесия. Для
синтеза аммиака считается общепринятым, что лимитирующей стадией является диссоциация N2; мы используем это предположение в нашем рассмотрении. Что касается квазиравновесных стадий, то дифференциальное уравнение
вместе с условием равновесия позволяют выразить степени заполнения поверхности участниками реакции через парциальные давления реагентов, константы равновесия и степени заполнения поверхности другими интермедиатами.
Элементарные стадии в кинетической модели синтеза аммиака выглядят так:
d в
N2 + ^ N2 ^~ Рп2^\ ~ ^n2 ~ 0 => = K\PN20,, (7-^4)
лимитирующая
щ + . стад™ > 2N. r = r+_r- = цв^в, - k;el, (7.35)
Ап Й Й
N + Н NH + = к^6п6н - к3 0NH#i: = 0 => 0N = , (7.36)
NH:I: + Н NH2 + * = к+Автвн - к;вNH Д = 0 ==> ^NH — (7.37)
nh; + н nh; +1 = к+оШ2вн -к;втъе. = о =* *NH2 = (7.38)
i
NH3 *=► NH3 + = -^6+^nh3^h + ^б/^ынз^: = 0 =» 0ЫНз = />nh3^*» (7.39)
d Й
H2 + 2* ^ 2H* - Ji- = k;pHie;- - k-^l = о => eH = jK7pHiet. (7.40)
Степени заполнения поверхности для всех адсорбированных соединений
(которые, конечно, не обязательно могут быть измерены экспериментально)
могут быть теперь выражены через константы равновесия и парциальные давления. Читатель может проверить, что результат таков
вщ=К[Рщв^а&, (7.41)
= — s агв,, (7.42)
кгк,к5к6{к1Р Н2)3/-
= к s > (7-43)
А 5 Ь 1 Р)&2
*nh, = t/ /N,HJ в. = аф., (7.44)
12 K5K6jK^~H7
^NH3 “ PnH3^i
вн = tJkjPh2 0* = aA- (7-46)
^nh3 “ g Pnh3#i (7.45)
7.3. Микрокинетическое моделирование
\
Долю свободных центров можно определить из условия, что сумма всех
степеней заполнения равна единице, следовательно,
в, = 1 - 1>Д =» 8, = ТТ^—>
т 1 + 2>,
(7.47)
где коэффициенты ai зависят только от констант равновесия и парциального
давления. Теперь доля свободных центров может быть выражена через парциальное давление и константы равновесия:
1
1 + к + ^NH3 + ^NH3 + ^NH3
K3K4K5K6J(K7pH2f K^^KiPHl K5K6jK7pH2 K,
+ Yl ^NHj + JK1Ph2
(7.48)
Возвращаясь к полной реакции, из уравнения (7.35) находим
r = r+-r~= к2вn20i: - k2 ,
Р NH,
f* — k2 p^ 0~ k2
K3K4K5K6(K7pHi)
3/2
(7.49)
в:.
Более удобная для пользования формула, показывающая, насколько реакция удалена от равновесия, получается путем введения константы К2 = k\/&J:
г = k;KtpN
1
2
Р NH,
кхк2к1к1к1к1к]рЪ2рщ
в;.
(7.50)
В равновесии скорость должна быть равна нулю, то есть
^NH3 _ V is £-2 ts2 is2 ts2 ^3 v
— - Л1Л2Л3 Л4Л5 А^Лу - Ar,.
Ph2Pn2
(7.51)
Это соотношение показывает, что произведение констант равновесия в знаменателе уравнения (7.50) равняется константе равновесия для реакции синтеза аммиака (7.33). Таким образом, скорость реакции можно записать в удобном виде
г = к;к1Рп,
1-
/^NH,
KgPh2Pn2
ей
(7.52)
где выражение в скобках показывает, насколько реакция удалена от равновесия, а множитель перед скобками равен скорости реакции при нулевой конверсии. Отметим также, что скорость реакции пропорциональна квадрату доли
свободных центров.
335
336
л
7.3.2.
Глава 7. Кинетика поверхностных реакций
Энергия активации и порядки реакции
Используя результаты гл. 2, можно также получить выражения для
таких наблюдаемых параметров, как кажущаяся энергия активации и порядок
реакции:
П: = Р: ^ и Еарр = RT
1 1 SPi ST
(7.53)
Заметим, что нас интересует только скорость прямой реакции. В кинетических исследованиях эксперименты обычно проводятся вдали от равновесия.
Проводя необходимые дифференцирования, получаем следующие выражения
для порядков:
1-20,
N-) >
пн2 = ^0N + 2^NH + #NH2 ” вн >
nNH3 = (#N + ^NH + ^NH2 + ^NH3 )>
и для энергии активации:
(7.54)
(7.55)
(7.56)
Еарр = £act + ДЯ _ 2ДЯ.0.
1 N-)
2 А#3 + АЯ4 +АЯ5 + АЯ6 +-АЯ7 ]0n -2(АЯ4 + АЯ5 + АЯ6 + АЯ7)0ын -
2[ АЯ5 + АЯ6 + ^ АЯ7 |0Nh2 2АЯ60ЫНз АЯ70н.
(7.57)
Мы снова видим, что через степени заполнения поверхности, входящие в
уравнения (7.54)—(7.57), кинетические параметры сильно зависят от условий
проведения эксперимента.
Скорость синтеза аммиака может быть легко найдена при известных (или
оцененных) значениях констант равновесия и коэффициента к2. При наличии
такой возможности следует брать экспериментальные значения констант равновесия. Например, можно измерить количество атомарного азота на поверхности Fe или Ru и тем самым определить произведение к2К{. Можно также
сравнить это значение с рассчитанным по известной диаграмме потенциальной энергии, что даст достаточно подробную информацию о переходном состоянии. Заслуживает внимания и тот факт, что хотя в модели молекулы N2
находятся в равновесии с адсорбционной фазой, состояние последней, в частности, ее статистическая сумма никакой роли не играет, поскольку
V “ КхРщО.
Яп/
?gas КТ
PnA
(7.58)
7.3. Микрокинетическое моделирование
X
2d#vM 2q'#kBT _2 q'^V
W- <™>
Следовательно, параметры, характеризующие свойства молекулярно адсорбированного N2, сокращаются, если мы объединим к2 Кх в один член, который
ответственен за образование атомарного азота. Аналогичным образом, но на более крупном масштабе сокращаются и статистические суммы, входящие в KG в
уравнениях (7.51) и (7.52). Возвращаясь к уравнению (7.59), отметим, что множитель 2 возникает из-за того, что скорость относится к атомам азота, а переходное состояние — к молекуле, при диссоциации которой появляются два атома.
Скорость прямой реакции в (7.59) можно записать обычным образом, введя
коэффициент прилипания SQ(T):
г+ = к$Кхрщв2 = =
Я gas
Р*гв* = „7V , ^чз/2 гР*2в? = (7-6°)
= 2q'„0e-AE™JkBTV вг = 2 q'e0e-&E^TVh3
Яз Z)-trans Я rot ^vibA N; V{2лткн! )';' ft
2, Р„Л(Т)
Pn 7 -!• = ~. _ с/.
N0yl2nmkBT ■ l/NQ ■(2nmkBT)qrolqVib 2 N0pnmkbT
Коэффициент прилипания в рамках теории переходных состояний рассчитывается непосредственно, как это было сделано в гл. 3. Колебательная статистическая сумма
е~Ч2к*т
9vib=J—В ’ <7-61>
поскольку hv^> квТ. Вращательная статистическая сумма равна qroi = kQT/2evoi
(erot = 0,248 мэВ). Для плотности активных центров на поверхности Fe(lll)
имеем N0 = \/\/За2 = 1/14,3 А2. Переходное состояние включает пять нарушенных мод (три поступательных и две вращательные), а шестая мода отвечает
координате реакции, по которой идет диссоциация. Все эти моды характеризуются относительно низкими частотами, но поскольку они ограничены переходным состоянием, мы примем, что hvi » &ВГ, и будем считать моды вырожденными, то есть
5 -hv,-/2/<qT /2квТ
q#0 = П \_е-^,/квт -е ' • (7-62)
В результате имеем
(-4МЧ‘Ь)/**Г ,
■?"(г) = ат/ =sSe ■ <7'63)
у]Ъа~7гт\къТ)
337
338 -l\r Глава 7. Кинетика поверхностных реакций
При 500 К предэкспоненциальный множитель равен Sfj = 1,49х10“5, что
иллюстрирует уменьшение энтропии, связанное с пространственным ограничением движения молекулы при ее переходе из газовой фазы в адсорбционное
состояние. В данном случае измерения приводят к небольшому отрицательному значению энергии активации, то есть A£measured = —0,034 эВ, которое следует
сравнить с
Д£арр = къТ
2<Яп[50(Г)]
ST
AEact-^hv + ^v,-2kBT.
Если мы примем, что
~\hv + \Yjvi = °’03 эВ>
(7.64)
(7.65)
то получим не совсем корректное значение коэффициента прилипания, если он
сопоставляется с данными эксперимента, но с правильной величиной кажущейся энергии активации для аккомодации молекул (то есть левую часть рис. 7.19).
На этом рисунке приведены экспериментальные данные для коэффициента
прилипания, а также результаты теоретических расчетов (сплошная линия). На
правой части рис. 7.19 представлены данные теоретических расчетов изменения энергии молекулы по координате реакции вблизи поверхности. Теоретический расчет (сплошная кривая) говорит о наличии энергии активации около
0,1 эВ. Таким образом, диссоциация азота лимитирует синтез аммиака не из-за
высокого энергетического барьера, а вследствие уменьшения энтропии молекул N2 в переходном состоянии, предшествующем их диссоциации!
т, к
0,5
0,0
СО о с
0
|-1,0
0
<5 -1,5
-2,0
-2,5
р
Островки
- с(2 х 2)—N/Fe( 100)
П "
! i
i I'
j i
i
1/Т, К"1
Координата реакции
Рис. 7.19. Слева: сравнение экспериментальных и полученных с учетом уравнения (7.57)
с энергией активации 0,03 эВ расчетных данных для коэффициента прилипания молекул N2 на поверхности Fe(lll). Справа: диаграмма потенциальной
энергии для молекулы азота, диссоциирующей на поверхности Fe(l 11). Отметим, что теоретическое значение энергии активации, равное 0,1 эВ, хорошо
согласуется с использованной при построении зависимостей на левом рисунке
величиной 0,03 эВ [24]
7.3. Микрокинетическое моделирование
X
Обычно энергия активации процесса диссоциации существенно превышает
квТи ситуации, подобные данной, когда кажущаяся энергии активации оказывается отрицательной, достаточно редки. Тем не менее данный пример элегантно демонстрирует, что изменения энтропии могут играть важную роль и не
обязательно энергия активации является величиной, лимитирующей скорость
процесса.
Пример
Рассчитав коэффициент прилипания азота на поверхности Fe(lll), перейдем теперь к анализу десорбции азота, для которой все кинетические параметры могут быть легко получены из экспериментов по ТПД. Комбинируя уравнения адсорбции и десорбции, можно рассчитать константу равновесной диссоциативной адсорбции азота из условия
= о = (1 _ eNf - v0e~E^re2. (7.66)
dt NapnmkJV N' 0 N
Решение этого уравнения относительно степени заполнения поверхности
азотом 6N дает следующее выражение
L, ,7.67,
где
1 + ^Kn2Pn2
A - f5>/-£des АВТ - Л£ас,\ W
Кы-,=— г— = , г— • (7.68)
N0v0j2xmkBT nm{kBT)~ v0^2nmkBT
Рис. 7.20. Зависимости степени заполнения
поверхности Fe(lll) атомарным азотом от
температуры, полученные по уравнениям
(7.67) и (7.68). Десорбция в ультравысоком вакууме проявляет себя симметричным пиком
ТПД-кривой, наблюдающимся в окрестности 740 К. Заметим, что в условиях реакции, проводимой при давлении азота 0,1 бар
(104 Па), поверхность практически полностью покрыта атомами азота
Температура, К
339
На кривой ТПД десорбция N2 проявляется как симметричный пик в
окрестности 740 К, соответствующий реакции второго порядка, с энергией
340
Глава 7. Кинетика поверхностных реакций
^des = ^0 кДж/моль и v = 1013 с-1. Используя эти данные, можно найти, привлекая уравнения (7.67) и (7.68), степень заполнения поверхности азотом как
функцию температуры. Результат расчетов показан на рис. 7.20. Из рисунка
следует, что вплоть до очень высоких температур, поверхность, контактирующая с газообразным N2, в значительной мере покрыта атомами азота. Имеет ли
это место в условиях синтеза аммиака?
7.3.3. Катализатор синтеза аммиака в рабочих условиях
Для описания синтеза аммиака нам необходимо определить степень заполнения поверхности интермедиатом и долю свободных центров. Их
нахождение требует знания индивидуальных констант равновесия. Как и ранее, часть из них удается извлечь из эксперимента, а другие могут быть рассчитаны через статистические суммы. В таком расчете снова в выражении для
степени заполнения поверхности интермедиатом некоторые статистические
суммы сокращаются.
На рис. 7.21 приведены данные, отвечающие синтезу аммиака на реальном
катализаторе в реакторе. Видно, что поверхность преимущественно покрыта
атомарным азотом и интермедиатом NH. Такое состояние поверхности приводит к торможению реакции при высоких давлениях аммиака. И действительно,
аммиак «отравляет» реакцию.
Рис. 7.21. Доли свободных центров и центров, занятых атомарным азотом и NH,
на поверхности активированного калием
железного катализатора синтеза аммиака
как функции длины реактора при температуре 673 К, давлении 100 бар и концентрации аммиака, соответствующе 68 %
от равновесного значения [25]
Происходящее в трубчатом реакторе позволяет ощутить проблемы, встречающиеся в промышленности. Рассмотрим малый объем газа, движущийся через
реактор. На входном участке реактора концентрация аммиака равна нулю (на
практике это не так, поскольку обычно используется рециркуляция аммиака),
следовательно, мы можем рассматривать данный объем как порционный реактор, в котором каталитический процесс ведет систему к равновесию. На начальном этапе реакция идет быстро, поскольку у катализатора нет заблокированных центров, но с ростом степени конверсии идет блокировка поверхности
азотом, и скорость реакции быстро падает. На рис. 7.22 показана зависимость
концентрации аммиака от расстояния до входа в реактор. На выходе из реакто¬
Длина реактора
7.3. Микрокинетическое моделирование ->\г 341
ра достигается степень конверсии в 19 %, что составляет 75 % от равновесной
степени конверсии. Эта равновесная степень конверсии достижима лишь в очень
длинных реакторах, использование которых нежелательно, и требуется оригинальное инженерное решение, позволяющее обойти это препятствие и оптимизировать степень конверсии. Другое осложнение связано с тем, что процесс синтеза — экзотермический; это вызывает рост температуры по длине реактора и
соответствующее падение максимально достижимой степени конверсии. Одно
из возможных решений связано с охлаждением реактора. Мы вернемся к этой
проблеме в гл. 8, где обсудим процесс синтеза аммиака более детально. Микрокинетическое моделирование существенно повышает степень нашего понимания процесса. Например, интуитивно можно было бы предположить, что реакционно более активная поверхность будет способствовать диссоциации азота
более эффективно. Однако при этом возрастает концентрация атомов N на поверхности, что делает более затруднительным процесс превращения реагентов в
аммиак. Более желательным было бы дестабилизировать азот на поверхности
при сохранении неизменной величины коэффициента прилипания.
Рис. 7.22. Концентрация NH3 как функция длины реактора в синтезе аммиака с
использованием активированного калием железного катализатора. Выход равен
19 % и соответствует 75 % от равновесной конверсии при давлении 100 бар и
температуре 673 К [25]
s
ц
0
се
ш
-О
1
.0
ц
о
2
се
0
X
.0
со
-О
1
I—
ш
т
о
03
0_
5
5
о
Q.
ш
со
X
о
*
.0
X
ш
и.
ш
I—
о
Длина реактора
Выход NH3 в эксперименте,
мольные доли
Рис. 7.23. Сравнение расчетных и экспериментальных значений скорости синтеза при различных давлениях. Обратите внимание на хорошее согласие теории и эксперимента, указывающее на
соответствие модели и эксперимента [23]
342 -l\r Глава 7. Кинетика поверхностных реакций
Наконец, предложенная микрокинетическая модель должна быть протестирована с использованием реальных катализаторов. На рис. 7.23 приведена
зависимость расчетного выхода аммиака из реактора от экспериментально измеренного. Видно, что микрокинетическая модель хорошо соответствует экспериментальным данным. Это не означает, что модель абсолютно правильна,
поскольку можно предложить другой набор элементарных стадий, который
приведет к такому же результату.
Подводя итоги, отметим, что промышленные каталитические процессы обычно описывают, как это было отмечено в гл. 2, с помощью степенных законов.
Однако представление скорости реакции в виде степенной функции, по сути,
является некоторой параметризацией экспериментальных данных и практически ничего не говорит о механизме процесса. Микрокинетические модели
могут хуже описывать экспериментальные данные, но позволяют исследователю сконцентрировать внимание на тех стадиях, которые являются определяющими в оптимизации процесса.
Список литературы
1. Larsen J.H., Chorkendorff I. // Surf. Sci. Rep. 1999. V. 35. P. 163.
2. Rasmussen P.B., Holmblad P.M., Christoffersen Taylor P.A., Chorkendorff I. //
Surf. Sci. 1993. V. 287/288. P. 79.
3. Dahl S., Logadottir A., Egberg R., Larsen J., Chorkendorff /., Tornquist E.,
Norskov J.K. // Phys. Rev. Lett. 1999. V. 83. P. 1814.
4. Kose R., Brown W.A., King D.A. // Chem. Phys. Lett. 1999. V. 311. P. 109.
5. Niemantsverdriet J. W. Spectroscopy in Catalysis. Weinheim: Wiley-WCH, 2000.
6. Niemantsverdriet J. W., Dolle P., Markert K, Wandelt K. // J. Wac. Sci. Technol. A.
1987. V. 5. P. 875.
7. Siddiqui H.R., Guo X., Chorkendorff I., Yates J.Т., Jr. // Surf. Sci. 1983. V. 191.
P. L813.
8. Hopstaken M.J.P., Niemantsverdriet J. W. // J. Phys. Chem. 2000. V. 104. P. 3058.
9. Chan C.M., Aris R., Weinberg W.H. // Appl. Surf. Sci. 1978. V. 1. P. 360.
10. De Jong A.M., Niemantsverdriet J.W. // Surf. Sci. 1990. V. 233. P. 355.
11. Habenschaden E., Kuppers J. // Surf. Sci. 1984. V. 138. P. L147.
12. King D.A. // Surf. Sci. 1975. V. 47. P. 384.
13. Miller J.B., Siddiqui H.R., Gates S.М., Russell J.N., Jr., Yates J. Т., Jr., Tully J.C.,
Cardillo M.J. // J. Chem. Phys. 1987. V. 87. P. 6725.
14. Lukkien J.J., Segers J.P.L., Hilbers P.A.J., Gelten R.J., Jansen A.P.J. // Phys.
Rev. E. 1998. V. 58. P. 2598.
15. Nieminen R.M., Jansen A.P.J. // Appl. Cat. A: General. 1997. V. 160. P. 99.
16. Van BavelA.P., Hopstaken M.J.P., Curulla D., Niemantsverdriet J. W. // J. Chem.
Phys. 2003. V. 119. P. 524.
17. De Koster A., Van Santen R.A. // Surf. Sci. 1990. V. 233. P. 366.
18. Taylor P. A., Rasmussen P.B., Ovensen С. V, Stoltze P., Chorkendorff I. // Surf.
Sci. 1992. V. 261. P. 1991.
Список литературы -J\r 343
19. Hopstaken M.J.P., Van Gennip W.E., Niemantsverdriet J.W. // Surf. Sci. 1999.
V. 433-435. P. 69.
20. Van Hardeveld R.A., Van Santen R.A., Niemantsverdriet J. W. // J. Phys. Chem. B.
1997. V. 101. P. 7901.
21. Borgna A., Hensen E.J.M., Van Veen J.A.R., Niemantsverdriet J.W. // J. Catal.
2004. V. 221. P. 541.
22. Dumesic J.A., Rudd D.A., Apuvicio L.A., Bekoske J.E., Trevino A.A. The
microkim\netics of heterogeneous Catalysis. Washington: American Chemical Socienty,
1993.
23. P., N0rskov J.K. // Phys. Rev. Lett. 1985. V. 55. P. 2502; J. Catal. 1988.
V. 110. P. 1.
24. Mortensen Hansen L.B., Hammer B., N0rskov J.K //J. Catal. 1999. V. 182.
P. 499.
25. Stolze P. Ц Phys. Scr. 1987. V. 36. P. 824.
глава ПРАКТИКА
8 ГЕТЕРОГЕННОГО КАТАЛИЗА:
ВОДОРОД
8.1. ВВЕДЕНИЕ
Катализ включает процессы, протекающие на разных пространственных и временных масштабах (см. также гл. 1). В предыдущих главах мы
рассматривали преимущественно явления, происходящие на молекулярном
уровне, последующие три главы посвящены масштабам, отвечающим прикладным проблемам катализа. Эти масштабы охватывают широкий диапазон размеров от огромных емкостей заводов по переработке нефти (расход до 5 двухсотлитровых бочек в секунду!) до небольших элементов, использующихся при
нейтрализации выхлопных газов. В данной главе мы рассмотрим крупномасштабные процессы конверсии с водяным паром, конверсии водяного газа и синтезы аммиака, метанола и искусственных жидких топлив. В конце главы мы
обсудим перспективы использования водорода в качестве топлива будущего.
Глава 9 посвящена проблемам переработки нефти и нефтехимии, в десятой
главе обсуждаются задачи использования катализа в защите окружающей среды. Все эти проблемы важны для ответственного обеспечения современного
стиля жизни человека.
8.2. ПРОЦЕСС КОНВЕРСИИ С ВОДЯНЫМ ПАРОМ
8.2.1. Основные понятия процесса
Конверсия с водяным паром приобрела свою значимость вследствие наличия в природе большого количества относительно недорогого метана. Она представляет собой первый шаг в большом числе крупномасштабных химических процессов, в которых используется водород. Конверсией с
водяным паром получают синтез-газ (который иногда называют сингазом),
смесь Н2, СО и С02, которую используют непосредственно в синтезе метанола и высших спиртов, для получения искусственного жидкого топлива в синтезе Фишера—Тропша. С другой стороны, в реакции конверсии водяного газа
снижение содержания СО может быть использовано для увеличения выхода
водорода.
8.2. Процесс конверсии с водяным паром
Конверсия с водяным паром идет по следующей схеме [1]
СН4 +Н20 ^ ОСО + ЗН2; Д#2°98 = +206 кДж/моль, (8.1)
CmHm+/iH20 ^ лСО + ^/1 + у^Н2; Д#298 > 0 кДж/моль. (8.2)
Одновременно идет реакция так называемой конверсии водяного газа, в
которой продуктом является С02:
С0 + Н20С02 +Н2; АЯ298 = -41 кДж/моль. (8.3)
Конверсия с водяным паром была разработана в Германии в начале XX столетия для получения водорода, необходимого для синтеза аммиака, а затем была
введена в практику в 30-е гг. прошлого века, когда природный газ и другое
углеводородное сырье типа лигроина стали доступны в больших масштабах.
Природный газ состоит в основном из метана и некоторых высших углеводородов (табл. 8.1). Присутствующая нем сера, отравляющая катализаторы, может
быть удалена до уровня 0,2 миллионных доли перед конверсией с водяным
паром. Это обычно делается каталитическим переводом серы из тиолов, тио-
фенов или COS в H2S, который затем поглощается ZnO при 400 °С на выходе из
реактора.
Таблица 8.1. Типичный состав природного газа Северного моря
Компонент
сн4
с2н6
с3н6
С4+
со2
N2
S
Об. %
94,9
3,8
0,2
0,1
0,2
0,8
4 миллионные
доли
Многие переходные металлы из концов групп типа Pd, Pt, Ru, Rh и Ir могут
быть использованы в качестве катализаторов конверсии с водяным паром, однако наиболее подходящим, в силу его дешевизны, является катализатор на
основе никеля. Реакционно более активные металлы типа железа и кобальта
также являются катализаторами этого процесса, однако они окисляются в процессе проведения реакции. Рутений, родий и другие благородные металлы обладают большей активностью, чем никель, но они менее привлекательны из-за
своей высокой цены. Типичный катализатор состоит из относительно больших
частиц никеля, нанесенных на А1203 или на шпинель AlMg04. Площадь поверхности активного металла относительно невысока, всего несколько квадратных
метров на грамм.
Процесс проводят при высоких температурах (до 1000 °С) и умеренных давлениях (25—35 бар), оптимальность таких условий легко установить из уравнений (8.1)—(8.3) и показанной на рис. 8.1 зависимости Д(7от температуры. Реакция, при которой осаждается углерод, также включена в общую схему, поскольку вклад этого процесса должен быть минимизирован.
\
345
346 -l\r Глава 8. Практика гетерогенного катализа: водород
Температура, К
Рис. 8.1. Зависимости от температуры изменения свободной энергии в конверсии с водяным паром и сопутствующих реакциях, включая процесс, при котором на катализатор осаждается углерод. Зависимости явно показывают, почему конверсию
с водяным паром надо проводить при высоких температурах
Традиционно реактор для проведения конверсии с водяным паром имеет вид
трубчатой конструкции, в которой вертикальные трубы, заполненные катализатором, окружены горелками, являющимися источником тепла для сильно эндотермического процесса (рис. 8.2). В горелках обычно сжигают природный газ.
Рис. 8.2. Сердце установки по конверсии с водяным паром состоит главным образом из
шестиярусных печей, в которых трубы реактора стоят вертикально
В последнее время стали использовать более изысканные реакторы, в которых горелки стоят на входе в реактор и нагрев катализатора осуществляется за
счет сжигания части использующегося в реакции метана.
8.2. Процесс конверсии с водяным паром -J\r 347
8.2.2. Механистические детали конверсии с водяным паром
Молекула метана устойчива и обладает высокой симметрией. Она
активируется на стадии прямой диссоциативной адсорбции, которой отвечают
высокие энергетический и энтропийный барьеры. С учетом чрезвычайно низкого значения коэффициента прилипания (см. начало гл. 7) нет ничего удивительного в том, что диссоциативная адсорбция является лимитирующей стадией процесса конверсии с водяным паром. Эта элементарная ступень, требующая значительной активации молекул, изучается достаточно интенсивно, в том
числе с учетом динамических эффектов. Так, Холмблад с сотр. [2] обнаружил,
что колебательное возбуждение молекул приводит к существенному увеличению коэффициента прилипания. В настоящее время не представляется возможным провести исследование процесса конверсии с водяным паром на хорошо охарактеризованных монокристаллах, однако обратный процесс — образование метана — был детально изучен Гудманом с сотр. [3]. Сейчас мы обсудим
последние результаты, полученные численным моделированием. Схема полной реакции выглядит так:
(8.4)
(8.5)
(8.6)
(8.7)
(8.8)
(8.9)
(8.10)
(8.11)
(8.12)
Приведенная на рис. 8.3 энергетическая диаграмма показывает, что наиболее
устойчивыми интермедиатами являются, в зависимости от природы поверхности, С или СН. Для обратной реакции образования метана наиболее высокий
энергетический барьер отвечает диссоциации СО на идеальной поверхности (111),
а на поверхности (211) эта реакция идет легко. Таким образом, центрами, на
которых происходит диссоциация СО, являются ступени на поверхности (111),
что согласуется с данными ряда других исследований. Несмотря на высокую
активность ступеней, нельзя безоговорочно принять, что именно они определяют скорость реакции, поскольку возможна их блокировка устойчивыми интермедиатами С или СН. Исследования этого процесса продолжаются, поскольку
осаждение углерода порождает ряд проблем в конверсии с водяным паром.
СН4 +Т ->СН3 t Н ,
сн, + «=» сн; + н\
сн: + «=» сн +н\
сн + ^ с + н ,
Н-,0 + 2" НО + Н ,
но + ^ о + н ,
С +0 со + ,
со ^ со + \
2Н —: Н, + 2*.
348 -l\r Глава 8. Практика гетерогенного катализа: водород
300
200
с;
о
2
' 100
UJ
-100
Рис. 8.3. Диаграмма потенциальной
энергии, полученная методом функционала плотности. Отметьте,
сколь существенно изменяется путь
реакции при наличии атомных ступеней на поверхности (lll)Ni. Ступени, прежде всего, снижают высоту
барьера для начальной диссоциации
метана. Хотя этот барьер и не самый
высокий, значительные потери энтропии приводят к очень маленькому
предэкспоненциальному множителю, в результате чего эта стадия становится лимитирующей [4]
8.2.3. Проблемы в процессе конверсии с водяным паром
Катализаторы, используемые в процессе конверсии с водяным паром, достаточно устойчивы, но на них может осаждаться углерод. Как следует
из рис. 8.1, во многих реакциях может образовываться углерод, который осаждается в форме графита на поверхности катализатора. В общем случае вероятность образования углерода увеличивается с уменьшением потенциала окисления, то есть со снижением содержания пара (которое может быть желательным
по экономическим причинам). Электронно-микроскопическая фотография,
показанная на рис. 8.4, наглядно демонстрирует, как образование углерода может
привести к разрушению катализатора и вызвать забивку каталитического слоя.
Рис. 8.4. Полученное на просвечивающем электронном
микроскопе изображение углеродных волокон, выросших на
катализаторе из Ni, нанесенного на SiO?, из газовой смеси СН4+Н? при давлении 1 бар
и температуре 763 К. Темные
пятна соответствуют частицам
Ni диаметром около 1000 А
Осадок углерода может иметь различные формы, но типичная морфология
углерода, осажденного при высоких температурах, — это «усы», представляющие собой длинные волокна графитированной сажи. Такие волокна образованы из более или менее упорядоченных искривленных графитовых слоев, упа¬
8.2. Процесс конверсии с водяным паром ->\г 349
кованных в конусы или концентрические трубки. Диаметр волокон определяется размером каталитических частиц, на которых они растут. Частицы металла при этом приобретают грушевидную форму (см. рис. 8.4). Использование
электронной микроскопии in situ позволяет проследить в реальном времени за
«прорастанием» нитей из металлических частиц. Образование нитей тесно связано с кристаллографической ориентацией частиц Ni и, например, с их модифицированием атомами Си: в последнем случае возможен рост сразу нескольких нитей из одной частицы.
Образование нитей родственно процессу синтеза одностойных углеродных
нанотрубок. В нем стоит задача целенаправленно формировать однослойные
углеродные трубки максимально возможной длины. Благодаря их чрезвычайно
высокой механической прочности и необычным электронным свойствам эти
материалы стали объектом пристального внимания ученых-материаловедов.
Рост углеродных волокон в процессе конверсии с водяным паром может
привести к катастрофической забивке реактора и, из-за большой прочности
волокон, к разрушению катализатора. Это, в свою очередь, может вызвать локальный разогрев отдельных участков трубчатого реактора, поскольку эндотермический процесс, в котором поглощается тепло, на разрушенном катализаторе
прекращается. Реактор может перегреться, что приведет к ускоренному образованию углерода, и процесс выйдет из под контроля. Таким образом, хотя снижение содержания воды дает определенные экономические выгоды, наказанием
за эту экономию может стать катастрофическое образование углерода, которого
следует избегать ради безопасности процесса и сохранения контроля за ним.
Имеется несколько способов обойти эту проблему. Очевидным является
выбор условий, при которых образование углерода оказывается термодинамически невыгодным. Это существенно ограничивает возможный диапазон рабочих параметров процесса, поскольку температура и состав газа изменяются и в
радиальном направлении, и особенно сильно по длине реактора. По этой причине особую значимость приобретает разработка катализаторов, в существенной мере тормозящих (по сравнению с типичными для конверсии с водяным
паром катализаторами) процесс образования углерода. Образование углерода
можно рассматривать как проблему придания катализатору нужной селективности, при которой конверсия с водяным паром идет эффективно, а образование углерода подавлено. Мы обсудим две успешные разработки, сделанные в
этом направлении.
8.2.4. Пассивированный серой процесс конверсии:
селективное отравление катализатора серой
Условия синтеза могут быть существенно сдвинуты в область, где
потенциал образования углерода достаточно высок, за счет пассивации катализатора серой. Этот метод был обоснован в работе [5]. В основу пассивации
катализаторов серой положены экспериментальные данные, показавшие, что
для образования углеродных волокон требуется существенно большее число
350 —*\r Глава 8. Практика гетерогенного катализа: водород
активных поверхностных атомов никеля, чем для проведения конверсии с водяным паром. Поэтому, регулируя за счет адсорбции серы число и размер невозмущенных активных центров, образованных атомами никеля на поверхности, можно управлять процессом синтеза. На рис. 8.5
проведено сравнение скорости осаждения
углерода, или «скорости коксования», и скорости конверсии для разных значений числа
не блокированных серой центров 1 — 0S на
поверхности реального катализатора. Используя простое приближение среднего поля,
можно представить скорость реакции в форме, отвечающей ленгмюровской кинетике:
r = ro(l-0s)a9 (8.13)
где г0 — скорость реакции в отсутствие атомов серы на поверхности, а параметр а указывает число атомов, участвующих в реакции. Подгоняя зависимость (8.13) под экспериментальные данные, представленные на
рис. 8.5, можно определить значение параметра а. Результат подгонки показывает, что
для образования углерода требуется существенно большее (а= 6,3) число атомов, чем
для проведения конверсии с водяным паром (а = 2,7). У обеих реакций скорость резко падает с увеличением степени
покрытия поверхности атомами серы, подтверждая роль серы как сильного яда
для катализаторов.
Важно отметить, что селективность пассивированных серой катализаторов по
отношению к конверсии с водяным паром в существенной мере возрастает, поскольку образование углерода эффективным образом подавляется. Наблюдаемое
при этом уменьшение активности может в значительной мере быть скомпенсировано либо выбором более активного катализатора, либо повышением температуры
проведения процесса. К сожалению, для устойчивого протекания пассивированного серой процесса конверсии требуется поддерживать на определенном уровне
степень заполнения поверхности катализатора серой. Это обеспечивается путем
введения в реагенты нескольких миллионных долей H2S, поскольку адсорбированная сера взаимодействует с водородом и улетает с поверхности. В результате
продукт также содержит H2S, и требуется проводить его дополнительную очистку,
поскольку катализаторы дальнейших процессов очень чувствительны к отравлению серой. Таким образом, остается крайне желательной разработка метода модифицирования катализаторов, обеспечивающего постоянное подавление процесса
образования углерода. Как показано в следующем разделе, другой способ воздействия на атомы никеля состоит в использовании сплавов.
Число свободных центров Ni,
(1-*s>
Рис. 8.5. Скорости конверсии с водяным паром и образования углерода как
функции числа свободных центров на
частично покрытой атомами серы поверхности никеля [5]
\
8.2. Процесс конверсии с водяным паром
8.2.5. Катализаторы на основе сплавов золото/никель
для конверсии с водяным паром
Использование сплавов — это эффективный метод управления селективностью катализаторов [6]. Как было объяснено в гл. 5, на поверхности
металла можно формировать двумерные сплавы металлов, не смешивающихся
в объеме. Методом сканирующей туннельной микроскопии в [7] было показано, что золото на поверхности никеля образует двумерный неупорядоченный
сплав при субмонослойном заполнении поверхности (рис. 8.6). Атомы золота,
не проявляющие активности в процессе конверсии с водяным паром, разрушают большие ансамбли атомов никеля, ответственные за осаждение углерода.
Кроме того, расчеты, проведенные методом функционала плотности, показали, что углерод связывается с атомами никеля, контактирующими с атомами
золота, слабее. Эти данные говорят о том,
что сплав Ni—Au может быть успеш- |'
ным катализатором, обладающим высокой селективностью по отношению к
конверсии с водяным паром и низкой
селективностью по отношению к осаждению углерода.
■Kg
Рис. 8.6. Полученное в сканирующем туннельном I
микроскопе прямое изображение (51 х 49 А2) ;
0,3 МС Au, осажденного на поверхности (11 l)Ni
при повышенных температурах [8]. Атомы Au
часто выглядят как углубления, а атомы Ni —
как выступы, что связано с различием их электронной структуры L. .
Рис. 8.7 подтверждает правильность данного утверждения: катализатор, изготовленный только из никеля, в процессе конверсии я-бутана с водяным паром постепенно теряет свою активность, а его вес растет за счет осаждения
углерода, в то время как катализатор на основе сплава Ni—Au сохраняет свою
активность на одном уровне [9].
Время, с
Рис. 8.7. Зависимости степени конверсии //-бутана от времени для обычного катализатора из Ni и нового катализатора на основе сплава Ni—Au,
показывающие высокую устойчивость
последнего [9]
351
352 -J г Глава 8. Практика гетерогенного катализа: водород
Заметьте, пожалуйста, что метод решения проблемы осаждения углерода
зародился на основе фундаментальных исследований поверхности и численного моделирования, которые вылились в непосредственную разработку нового
катализатора — прекрасный пример создания целевого катализатора на базе
фундаментальных знаний!
8.2.6. Прямое использование метана
Хотя это и не имеет прямого отношения к конверсии с водяным
паром, полезно рассмотреть, как можно непосредственно превратить метан в
полезный продукт. В промышленно развитых или плотно заселенных странах
метан можно легко транспортировать и распределить по газопроводу, так что он
оказывается прекрасным топливом для бытовых нужд и тепловых электростанций. Он является достаточно чистым топливом, и при его сжигании образуется
меньше диоксида углерода (газа, ответственного за парниковый эффект), чем
при сжигании нефти или угля. Однако метан является побочным продуктом
переработки нефти, осуществляемой на удаленных заводах, что делает его транспортировку экономически невыгодной. Там он просто сжигается (сжигается в
факелах, рис. 8.8). Сжигание метана в факелах представляет собой лучшее решение, чем просто выброс его в атмосферу, поскольку он примерно в 20 раз активнее
диоксида углерода в создании парникового эффекта. Строительство заводов для
полной конверсии метана с водяным паром, а также по конверсии синтез-газа в
метанол или иные жидкие углеводороды экономически оправдано только в специальных случаях. В идеале этот метан надо превратить в легко конденсирующийся продукт (с плотностью в 1000 раз больше, чем у метана), который можно
затем доставлять на кораблях или по трубопроводам до потребителя.
1
I
ШШШЯЯЯШШЯШШШШШШШШ
Рис. 8.8. Природный газ (преимущественно метан) сопутствует переработке нефти и обычно
сжигается в факелах, на удаленных заводах
8.2. Процесс конверсии с водяным паром 353
8.2.6.1. Прямое получение метанола
Прямое окисление метана до метанола представляет собой очевидную реакцию-мечту:
СН4+^02 ^СН3ОН; Д#° = -126,4 кДж/моль. (8.14)
Она конкурирует с более экзотермической реакцией окисления метана до СО
или С02:
СН4+|о2 ^ С0 + 2Н,0; ДЯ° = -519,5 кДж/моль, (8.15)
СН4 +20, «=» СО, +2Н20; ДН° = -792,5 кДж/моль. (8.16)
Достижение высокой селективности по отношению к образованию метанола сильно затруднено из-за высокой устойчивости молекулы метана. Для отрыва первого атома требуется достаточно высокая температура, при которой открыт путь к полному окислению молекулы. Имеются катализаторы окисления,
обеспечивающие выход метанола, но они обладают либо низкой селективностью, либо высокой селективностью при низкой активности. Следовательно,
для прямого окисления метана до метанола еще требуется осуществить новые
шаги, возможно подсказываемые природой. Существуют бактерии, использующие энзимы, называемые монооксигеназами, у которых имеются активные
центры на основе железа и меди, где искомая реакция идет при низкой температуре [10].
8.2.6.2. Каталитическое частичное окисление метана
Каталитическое частичное окисление метана, при котором образуются СО и Н2 по схеме
СН4 +^02 ^ СО + 2Н2; АН0 = -35,9 кДж/моль, (8.17)
является альтернативой конверсии с водяным паром, в частности, при производстве небольших количеств водорода, которые требуются, например, для
топливных элементов автомобилей. Окисление при этом осуществляется на
тонком нагретом до белого каления (1000 °С) монолите, который используется для обеспечения кратковременного (миллисекундного) контакта с ним смеси
метан—кислород. Этот метод был разработан Шмидтом с соавторами [11] и в
настоящее время рассматривается нефтяными компаниями и производителями автомобилей как способ генерации водорода непосредственно на автомобилях.
354 -J\r Глава 8. Практика гетерогенного катализа: водород
8.3. РЕАКЦИИ С УЧАСТИЕМ СИНТЕЗ-ГАЗА
Термином «синтез-газ» или «сингаз» называют смесь СО и Н2, хотя
иногда также называют и смесь N2 и Н2, использующуюся для синтеза аммиака. В зависимости от происхождения или возможных применений комбинации
СО и Н2 называют водяной газ (СО + Н2, получают при обработке паром угля),
крек-газ (СО + ЗН2 получают конверсией с паром природного газа), оксогаз
(СО + Н2 используют в реакциях гидроформилирования). Три наиболее важных области применения синтез-газа составляют синтез метанола, гидрофор-
милирование (синтез альдегидов и спиртов из олефинов) и так называемый
синтез Фишера—Тропша, с помощью которого получают искусственные жидкие топлива. Гидроформилирование осуществляют при гомогенном катализе,
поэтому оно выходит за рамки данной книги. Два других процесс будут обсуждены в следующих подсекциях.
8.3.1. Синтез метанола
8.3.1.1. Основные понятия процесса
Первоначально метанол получали перегонкой древесины. Однако
в 1923 г. на фирме BASF был получен катализатор, позволивший получать
метанол в больших количествах. Каталитический процесс идет при высоких
давлениях (300 бар) и температурах (300—400 °С), в качестве катализатора процесса первоначально использовали Zn/Cr203, впоследствии он был заменен на
более активный катализатор Cu/Zn/Al203, разработанный в компании ICI и
позволявший проводить синтез в более мягких условиях. В настоящее время
метанол производят из нестехиометрической смеси водорода, диоксида углерода и монооксида углерода (90 : 5 : 5) при давлении 50—100 бар и температуре
500—550 К с использованием катализатора Cu/Zn0/Al203. Более того, на современных производствах синтез метанола и конверсия с водяным паром ин¬
тегрированы в единую схему, так что выделяемое при экзотермическом синтезе метанола тепло используется для эндотермического процесса конверсии с
водяным паром.
Метанол используют для производства формальдегида (40—50 %), в качестве растворителя и синтеза уксусной кислоты путем гидроформилирования.
Гидроформилирование используется также для получения прекурсоров полимеров.
Механизм реакции синтеза метанола достаточно сложен, поскольку включает в себя два взаимосвязанных процесса. Формально реакцию можно рассматривать как гидрирование СО, идущее по суммарной схеме
СО + 2Н2 ^ СН3ОН; ДЯ2°98 = -91 кДж/моль, (8.18)
но в действительности она представляет собой комбинацию реакции
С02 + ЗН2 СН3ОН + Н20; Д#298 = -47 кДж/моль
(8.19)
8.3. Реакции с участием синтез-газа -J\r 355
и реакции конверсии водяного газа, описываемой уравнением (8.3). Обе реакции являются экзотермическими, поэтому процесс предпочтительно проводить
при низких температурах. Уменьшение числа молекул, связанное с образованием из реагентов продукта, делает желательным использование повышенного давления. Селективность катализатора важна для синтеза метанола, поскольку здесь
некоторые побочные реакции оказываются термодинамически более предпочтительными, чем реакция синтеза метанола, в оптимальных для синтеза условиях. Например:
СО + ЗН2 ^СН4 +2Н20,
2СО + 4Н2 » СН3ОСН3 + Н20,
2СО + 4Н2 » СН3СН2ОН + Н20.
(8.20)
(8.21)
(8.22)
В научной литературе имеется много доказательств того, что метанол синтезируется из С02 по схеме (8.19), а не из СО, как это показывает реакция
(8.18). Эксперименты с мечеными изотопами молекулами показали, что С02
является основным источником углерода в метаноле, полученном из синтез-
газа [12]. Кроме того, было показано [13], что метанол легко образуется на
поверхности монокристалла Си(ЮО) из смеси С02 и Н2 при давлении 1 бар
(рис. 8.9).
CD
О
с;
о
0
1
о
о
5
S
Т
Экспозиция, мин
Рис. 8.9. Скорость синтеза метанола на поверхности монокристалла Си(ЮО) в 1 : 1-смеси
Н2 и СО при давлении piot = 2 бар. Число оборотов реакции очень низко, и на
монокристалле образуется не больше одной миллионной доли метанола в
течение часа, что делает затруднительными количественные измерения. Тем
не менее эксперимент подтверждает, что метанол образуется непосредственно из СО., [13]
356
Глава 8. Практика гетерогенного катализа: водород
Микрокинетическая модель, включающая 13 стадий, из которых 8 относятся к реакции конверсии водяного газа, достаточно хорошо описывает процесс
синтеза метанола:
1.
Н20(г) + * «z* Н20*,
(8.23)
2.
Н20* +‘ «=! НО* +н*,
(8.24)
3.
2НО* + * ^ Н,0* +0*,
(8.25)
4.
НО* + * i=t Н* +0*,
(8.26)
5.
Н2(г) + 2* ^ 2Н*,
(8.27)
6.
СО (г) +" «=» СО*,
(8.28)
7.
со* + о ^ со; + *,
(8.29)
8.
со2(г) + * ^ со;,
(8.30)
9.
со; + н* «=* нсоо* + *,
(8.31)
10.
нсоо* + н* <=> Н2СОО* + *,
(8.32)
11.
н2соо* + н* «=» Н3СО* + 0*,
(8.33)
12.
Н3СО* + Н* ^ СН3ОН* + *,
(8.34)
13.
СН3ОН* 5Z* СН3ОН(г) + *.
(8.35)
Гидрирование диоксометилена, стадия (8.33), скоре всего, является лимитирующей реакцией, хотя в качестве кандидата на эту роль можно рассматривать и реакцию гидрирования формилата (8.32). Предполагая, что реакции (8.24),
(8.23) и (8.29) являются медленными для конверсии водяного газа, а реакция
(8.33) — медленная для синтеза метанола, приходим к следующему набору уравнений, в которых предполагается, что активный центр включает два атома меди:
К\Рн2о@* ~ ^н2о>
(8.36)
1
r2 = &2#н2сА ^ОН^Н>
(8.37)
^з^бн = ^Н20^0>
(8.38)
г4 = к4внов, - -р- в0вн,
4
(8.39)
вн = ^sPh2 #>>
(8.40)
К.РсхА = ^со ■
(8.41)
0
Г' . ^
^ \ ъ<
1
о
Г'
II
с
(8.42)
8.3. Реакции с участием синтез-газа ->\г 357
^со2 ” К$Рсо2&*’
^9^С0?^Н “ ^HCOO^i
(8.43)
9 С02 Н “^НСОО17*»
(8.44)
^ЧО^НСОО^Н _^Н2СОО^*>
(8.45)
(8.46)
^Ч2^СН3О^Н “ ^СН3ОН#=>
(8.47)
^ЧЗ^СН30 ” Рснъон&*-
(8.48)
Используя условия баланса массы для О* и ОН*, находим
(8.49)
и
(8.50)
что замыкает систему уравнений для определения различных в. Более детально
это система уравнений обсуждается в [14].
После довольно длинных вычислений для скорости образования метанола
можно получить следующее выражение
Как видно из уравнения (8.51), порядки реакции по Н2 и С02 равны 1,5 и 1,
соответственно, в условиях, когда степенями покрытия поверхности интермедиатами можно пренебречь; эти значения порядков согласуются с экспериментальными данными, из которых следует степенная зависимость от полного давления с показателем, равным 2,4. Из выражения (8.51) также можно найти, что
скорость будет максимальной для синтез-газа состава 60 % Н2 и 40 % С02. Если
лимитирующей будет стадия (8.32), описывающая гидрирование формилата, то
максимум скорости приходится на состав 50/50, как нетрудно установить из
вышеприведенных соотношений. На рис. 8.10 проведено сопоставление расчетной зависимости для скорости от мольной доли водорода в смеси и экспериментальных данных. Экспериментальные данные явно говорят в пользу реакции гидрирования диоксометилена как лимитирующей стадии.
Полный анализ выражения для скорости показывает, что данные по синтезу метанола на поверхности монокристалла Си(ЮО) описываются им, как видно из рис. 8.10, очень хорошо. Представляется особо важным тот факт, что
модель хорошо воспроизводит данные и на реальных катализаторах, получен¬
/
\
r = kllKPKsK9Kl0p3}i22pCO2 1
1 РсН30нРн20 ^2
Kg Рн2Рсо2 J
(8.51)
*58 -l\r Глава 8. Практика гетерогенного катализа: водород
ные в существенно иных условиях. Это означает, что микрокинетическая модель отражает основные особенности синтеза метанола (рис. 8.11).
Мольная доля Н2
Рис. 8.10. Скорость синтеза метанола на поверхности монокристалла Си(ЮО) как функция мольной доли Н2 в пределе нулевой степени конверсии. Сплошные линии
соответствуют кинетической модели (8.23)—(8.35), когда реакция гидрирования диоксометилена (8.33) является лимитирующей стадией. Пунктирная линия соответствует случаю, когда лимитирующей стадией является реакция гидрирования формилата [13]
Расчетное число оборотов реакции,
(центр • с)-1
Рис. 8.11. Сравнение данных, полученных в рамках микрокинети-
ческой модели и из экспериментов
по синтезу метанола на поверхности монокристалла Си(ЮО), с данными синтеза на реальном катализаторе [14]
Микрокинетическая модель позволяет также определить степени заполнения поверхности различными интермедиатами. Как показано в табл. 8.2., пред-
8.3. Реакции с участием синтез-газа ->\г 359
положение о почти свободной поверхности представляется вполне оправданным. Наибольшие степени заполнения имеют водород и формилат, но большинство центров остается свободным вплоть до давления 50 бар.
Таблица 8.2. Степени заполнения поверхности различными интермедиатами,
образующимися в процессе синтеза метанола,
для стехиометрической смеси при температуре 500 К и степени
конверсии 85%; заметьте, что при низких давлениях поверхность
почти свободна, однако при высоких давлениях она заполняется
атомами водорода и формилатом
Компонент
Рш = 2 бар
/7,0, = 50 бар
0*
8,8 х КГ1
5,2x10-'
Он
1,1x10-'
3,3x10-'
^НС(К)
1,2 х 103
7,0 х 10-2
во
2,4 хЮ"6
3,9x10-*
в\{20
4,5 х 10-5
1,7 х 10"3
вон
2,2 х1(Г3
1,7 х 10"2
всо
3,3 х1(Г3
1,6 х 10-2
всо2
3,3 х 10“3
2,8 х 10_3
вснгон
1,8 х 10“5
4,2 х 10"2
^сн3о
4,6x10-“
2,2 хЮ-8
;too
1,7 х 10-8
4,9 х 10"6
Исследования показали, что и другие вицинальные поверхности монокристалла или поликристаллической меди способны катализировать процесс образования метанола примерно в такой же степени, что и поверхность Си(ЮО)
[15, 16]. Это говорит о том, что синтез идет именно на частицах меди, а не на
ZnO, который преимущественно играет роль носителя.
Система Cu/ZnO очень динамична. Морфология частиц Си мгновенно откликается на изменение потенциала восстановления газовой смеси над ним.
Исследования методом спектроскопии дальней тонкой структуры рентгеновского поглощения показали, что изменения морфологии связаны со степенью
растекания металлических частиц по подложке [17]. По мере того как поверхность ZnO становится все более восстановленной, медь все сильнее связывает¬
360 -l\r Глава 8. Практика гетерогенного катализа: водород
ся с ней. Модельные вычисления, основанные на правиле Вульфа (см. гл. 5), в
котором учитывается межфазная энергия поверхности раздела Си—ZnO, показывают, какие морфологические изменения могут иметь место (рис. 8.12).
Частицы меди обращены гранью (111) к поверхности ZnO, обозначим соответствующую межфазную энергию через у0. Введя величину
Y ~ У interface У substrate ’
мы можем описать диапазон от полного смачивания до несмачивания с помощью соотношения у/у^ изменяющегося при этом в интервале от —1 до 1.
у/у0= 1.0
У/Уо = 0,5
-0.0
0.5
Рис. 8.12. Слева: формы частиц металла с ГЦК-структурой при различных значениях
межфазной энергии частица\подложка (как функция у/у0). При у/у0 = 1 между
частицей и подложкой нет взаимодействия и она ведет себя как свободная
металлическая частица. При у/у0 = — 1 частица стремится максимизировать площадь контакта и растекается до монослоя. Справа: площадь поверхности как
функция отношения у/у0 для различных вицинальных граней меди [18]
Данное утверждение можно обосновать с помощью следующих аргументов.
Представим сначала, что взаимодействия между поверхностями нет. Это означает, что при приведении поверхностей в контакт нет выигрыша энергии:
Р (^substrate У о) ^interface
(8.52)
Из этого равенства следует, что межфазная энергия ^nterface равна сумме
межфазных энергий разделенных поверхностей:
У (^interface ^substrate) Уо ^ ^
У о
(8.53)
и кристалл на подложке не искажается, как это показано на рис. 5.14 для случая /3 = 0 и на рис. 8.12 для у/у0 =1.
Если, однако, /?> 2у0, то будет иметь место полное смачивание, чему соответствует условие
У У interface У substrate У substrate У 0 У substrate У 0
Уо
-1
(8.54)
и соотношения А/г0 — fih^/y^ или y/yQ = —1 для рис. 5.14 и 8.12 соответственно.
8.3. Реакции с участием синтез-газа ->\г 361
Вклад различных граней в полную поверхность частицы может быть легко
вычислен; результат расчета показан на рис. 8.12, справа. Эти результаты были
включены в динамическую микрокинетическую модель синтеза метанола, что
привело к лучшему согласию расчетов с данными экспериментов на реальных
катализаторах [18].
Из изменения морфологии частиц можно извлечь определенную пользу: с
его помощью можно подавить спекание частиц меди. Как показано на рис. 8.13,
катализатор Cu/ZO со временем теряет свою активность, что связывают со
спеканием частиц. Выдерживая катализатор в течение короткого периода времени в синтез-газе, не содержащем С02, который сильно снижает поверхностную энергию, можно восстановить активность катализатора, поскольку частицы растекаются по поверхности (подобно тому, как это предсказывают модельные расчеты, данные которых представлены на рис. 8.12). Такая обработка
катализатора способствует существенному увеличению скорости производства
метанола [19].
1,5
.о
t—
о
0
1
00
? 1,0
*
03
к
03
I
I
03
GQ
а °.5
s
Q.
0
1
0,0
Рис. 8.13. Скорость синтеза метанола на катализаторе Cu/Zn0/Al203 в трубчатом реакторе как функция времени работы. Катализатор работал при температуре 494 К и
давлении 63 бар в газовой смеси, 5 % СО, 5 % С02, 88 % Н2 и 2 % N2. Отметьте
постепенное снижение активности, приписываемое спеканию частиц. После 168 ч
работы С02 перестал подаваться в газовый поток. При возобновлении синтеза
активность катализатора стала близкой к исходной, что связывают с изменением
морфологии частиц, обусловленным их растеканием по поверхности ZnO [19]
ZnO является, очевидно, подходящим носителем для частиц меди. Имеются, однако, доказательства, что он выполняет не только роль носителя. Накамура с сотр. изучали влияние Zn на синтез метанола на медном катализаторе.
Осаждая Zn на поверхность катализатора, они установили [20], что при этом
скорость реакции увеличивается примерно в шесть раз (рис. 8.14). Это говорит
о том, что цинк действует как промотор катализатора. Однако до сих про нет
единого мнения относительно утверждения Накамуры и др. о том, что в реальном катализаторе ZnO восстанавливается до такой степени, при которой идет
формирование сплава цинка и меди на поверхности частиц.
Текущее время
362
Глава 8. Практика гетерогенного катализа: водород
Рис. 8.14. Число оборотов реакции для синтеза метанола из и СО, при давлении 18 бар
и температуре 523 К как функция степени
покрытия поверхности поликристаллической
меди Zn [20]
8.3.1.2. Прямой синтез метанола из СО и Н2
Как представляется, присутствующий в синтез-газе СО не принимает прямого участия в реакции синтеза метанола, но влияет на нее по следующим двум каналам. Во-первых, через реакцию конверсии водяного газа, а во-
вторых, воздействием на морфологию (и, возможно, состав) поверхности частиц. Термодинамические соображения, однако, говорят о предпочтительности
прямого гидрирования из СО в сравнении с механизмом, описываемым двумя
связанными уравнениями (8.3) и (8.19), поскольку в этом случае равновесная
концентрация метанола на выходе из реактора была бы выше.
Рассмотрим сначала прямое образование метанола из СО и Н2. Если
обозначить через х мольную долю Н2, а через t — мольную долю метанола (в
табл. 8.3 приведены обозначения для мольных долей других компонентов),
то суммирование всех концентраций дает после завершения реакции величину 1 — 2/, которую следует использовать для нормировки мольных долей
после реакции.
Таблица 8.3. Мольные доли СО, Н2 и СН3ОН в прямом синтезе метанола
СО
+
2Н2
<=>
СН3ОН
До реакции
1-х
X
0
После реакции
\-x-t
х-21
t
Нормированные величины
\-x-t
1-21
х-21
1-21
t
1-21
8.3. Реакции с участием синтез-газа
Константа равновесия имеет вид
2
РснъонРо
\
к = ^ (8 55)
Рн2Рсо
К= [#-20]/>о = t(l-2t)p0 ^ (8 56)
[(* - 20/(1 - 20]' [(1 - X - 0/(1 - 20] (* - 20' (1 - * - 0
где р0 — нормальное давление; рх — парциальное давление компонента X. После преобразования уравнения (8.56) можно получить уравнение третьей степени относительно t, которое можно решить методом итераций. Заметим, что в
действительности в уравнении (8.55) давления следует заменить активностями,
то есть надо ввести коэффициенты фугитивности, которые играют важную роль
в процессах с участием метанола и воды. Мы оставляем проведение этой операции читателю в качестве упражнения, из которого он может оценить влияние этих эффектов, воспользовавшись связью между давлением и активностью, даваемой уравнением (8.39) гл. 2.
При прямом синтезе метанола из смеси С02, СО и Н2 обратная реакция
конверсии водяного газа усложняет систему, поскольку она конкурирует с реакцией синтеза метанола.
Мы используем ту же процедуру, что и раньше, при этом снова х —
мольная доля Н2, у — мольная доля СО и t — мольная доля метанола и
воды, получаемых в данной реакции. Сумма всех мольных долей снова
равна 1—21, и нормированные мольные доли компонентов приведены в
табл. 8.4.
Таблица 8.4. Мольные доли СО, Н2, СН3ОН и Н20 в прямом синтезе метанола
со2
+ ЗН2
сн3он + н2о
До реакции
1 - х - у
л:
0 0
После реакции
1-х-y-t
х - 31
t t
Нормированные величины
1 - х - у -t
х - 31
t t
1-21
1-21
1-21 1-21
Константа равновесия реакции равна
^ Рн^оРснъонР1 /0
Асн3он ~ з • (о.Э/)
Ри2 РС02
Мольные доли компонентов в обратной реакции конверсии водяного газа
приведены в табл. 8.5.
363
364
Л
Глава 8. Практика гетерогенного катализа: водород
Таблица 8.5. Мольные доли в реакции конверсии водяного газа
со2 + Н2 ^
± СО +
н2о
До реакции
1
1
Ч;
У
0
После реакции
1 - х- у-w х — W
у + W
W
Нормированные величины
1 - х- у-w х — W
у + W
W
Комбинируя со всеми нормированными мольными долями, получаем с учетом всех реакций
Рсо=
Рн2
1 - х - у - w -1
_ х — w — 3t
1-2/ ’
у + w
Рсо -
PH, о =
1-2/’
w +/
1-2/
Подстановка этих выражений в константу равновесия дает
К{
Р^оРсн^онРо
о-ьон
К{
Рн2Рс01
[(w + t)/{l-2t)][t/(l-2t)]p20
сн3он
[(х — w — 3t)/(l - 2t)] [(1 - X - у - W - /)/(1 - 2t)]
t(w + t)(\-2tf pi
К,
сн,он - , Лъ . л-
(х - w - it) (1 - х - у - w - t)
(8.58)
(8.59)
(8.60)
(8.61)
(8.62)
(8.63)
(8.64)
Для константы равновесия обратной реакции конверсии водяного пара имеем
Рн2оРсо
К
RWGS
К
RWGS
Рн2Рс02
[(x + t)/(l-2t)][(y + w)/(l-2t)]
[(х — w — 3/)/(1 - 2/)][(1 — х — у — w — /)/1 - 2/]
К
(w + f)(.y + w)
RWGS
(х - W - 3t) (1 - X - у - W - t)
(8.65)
(8.66)
(8.67)
8.3. Реакции с участием синтез-газа ->\г 365
Теперь у нас есть два уравнения (8.64) и (8.67) для двух неизвестных t и w,
которые можно решить.
Решение представлено на рис. 8.15, где показана зависимость концентрации метанола от состава исходной газовой смеси. Заметим, что максимум концентрации метанола достигается для чистой смеси СО + Н2. Таким образом,
для обеспечения максимального производства метанола необходимо использовать смеси только СО и Н2, примеси С02 должны быть минимизированы. Но
для обеспечения эффективного производства метанола необходимо принять во
внимание и скорость реакции, а мы помним, что метанол образуется из С02, а
не из СО. Так что скорость реакции будет пропорциональна давлению С02. Из
сказанного ясно, почему метанол не производится из простой стехиометрической смеси 3: 1 газов Н2 и С02, как это предполагает уравнение (8.19).
Рис. 8.15. Равновесная концентрация метанола на выходе как
функция концентраций Н?, СО и
С02 на входе в реактор. Заметьте, что максимальное значение
концентрации метанола достигается при наличии на входе только и СО в стехиометрическом
отношении 2:1. Сплошная линия показывает концентрацию
метанола в отсутствие в смеси
СО?, она также представлена проекцией на левую вертикальную
плоскость
Проведенный выше анализ показывает, что имеются серьезные основания
к поиску катализатора, который обеспечивал бы с высокой селективностью
производство метанола путем прямого гидрирования СО. Ряд исследований
показывает, что это можно осуществить при использовании катализаторов из
благородных металлов — палладия [21], платины [22], промотированной калием меди [23] или из комбинации металлов 8-й группы [24]. Однако ни один из
этих катализаторов не был доведен до промышленного уровня.
8.3.2. Процесс Фишера—Тропша
В процессе Фишера—Тропша из синтез-газа (СО+Н2) с использованием катализатора из железа получают углеводороды [25]. Этот процесс предлагает путь для получения из угля или природного газа бензина, дизельного
топлива и других полезных углеводородов типа парафина. Процесс был разра¬
366 -l\r Глава 8. Практика гетерогенного катализа: водород
ботан немецкими учеными Францем Фишером и Гансом Тропшем в 1923 г. в
Институте исследования угля Кайзера Вильгельма в Мюльхайме в Руре и позволял обеспечивать Германию жидким топливом в период Второй мировой
войны. В 1944 г. девять заводов путем газификации угля и с помощью процесса
Фишера—Тропша производили 600 000 тонн транспортного топлива в год [26].
В Южной Африке на заводах фирмы SASOL с 1955 г. процесс Фишера—Тропша используется для получения жидких топлив, что позволяет этой стране снизить ее зависимость от поставок нефти [27].
В настоящее время возродился интерес к процессу Фишера—Тропша как к
одному из возможных путей получения высококачественного не содержащего
серу дизельного топлива из природного газа и, возможно, как к методу утилизации природного газа на удаленных нефтеперерабатывающих заводах. Процесс представляет собой проверенную технологию и рассматривается как один
из возможных путей переработки угля и природного газа после истощения
запасов нефти. Можно также поразмышлять о его реальном применении в будущем, когда СО и Н2 будут получаться при фотокаталитической диссоциации
С02 и воды.
Поскольку получаемое в процессе Фишера—Тропша топливо не содержит
серы и ароматических соединений, оно является перспективным для рынка
товаром. Меньшее содержание серы, безусловно, прямо влияет на количество
оксидов серы в выбросах, поэтому сочетание такого топлива с разрабатываемыми сейчас методами очистки продуктов горения от СО и NOx позволит существенно снизить уровень загрязнения окружающей среды. Ароматические
соединения способствуют образованию частиц при горении дизельного топлива, поэтому их присутствие также нежелательно. Мы вернемся к этому вопросу
в гл. 10.
Синтез Фишера—Тропша используют для получения различных алканов и
алкенов:
Эти реакции в существенной степени экзотермические, например, при синтезе одного моля группы —СН2— выделяется 165 кДж. Возможен, в зависимости от использованных катализаторов условий проведения реакции, синтез углеводородов широкого диапазона — от метана до тяжелых парафинов.
Длина углеводородной цепи при данном синтезе распределена статистически по закону, носящему имена Андерсона, Шульца и Флори и имеющему вид:
где / — число атомов углерода в цепи; wt. — весовая доля цепей длиной /; а —
вероятность продолжения роста цепи, (1 — а) — вероятность прекращения
роста цепи. На рис. 8.16 показаны зависимости весовых долей продуктов реакции от параметра а. При использовании катализатора из никеля величина а
хСО + (2х + ljH', —> CxH7;c+9 + xH?0,
(8.68)
хСО + 2хН2 —> СхН2х + хН20.
(8.69)
w, = /(1 - а)2 а1 ',
(8.70)
8.3. Реакции с участием синтез-газа 367
оказывается малой, так что продуктом синтеза является в основном метан. Для
классического катализатор Фишера—Тропша — железа а = 0,65—0,70, так что
максимум приходится на бензиновую фракцию, а при использовании
катализатора из кобальта, для которого а лежит между 0,75 и 0,85
продуктом синтеза являются длинноцепочечные углеводороды. Важную роль играют и условия синтеза. При уменьшении отношения
Н2 : СО и температуры и увеличении давления также образуются более длинные углеводородные цепочки. Промотирование катализатора
калием или оксидами редкоземельных металлов приводит к увеличению параметра а. Тем не менее распределение продукта синтеза по фракциям остается достаточно широким.
Современная стратегия нацелена на увеличение значения параметра а до
единицы и проведение гидрокрекинга длинноцепочечных молекул с целью
получения фракций дизельного топлива и керосина. На рис. 8.17 показано, как
можно улучшить распределение продукта по фракциям за счет двухстадийного
процесса, в котором сначала идет синтез Фишера—Тропша, а затем — крекинг.
Рассмотрим для примера продукт, максимальные мольные доли которого приходятся на диапазон С10—С20 Эти фракции составляют около 40 % после первой стадии, а затем их содержание увеличивается до 80 % [26].
Вероятность продолжения роста а
Рис. 8.16. Зависимости весовых долей от параметра а
После гидрокрекинга
высокой жесткости
После гидрокрекинга
средней жесткости
После процесса
Фишера—Т ропша
10 20 30
Число атомов углерода
Рис. 8.17. Распределение по фракциям углеводородов, полученных в
синтезе Фишера—Тропша с кобальтовым катализатором и последующем гидрокрекинге. Видно, как
двухстадийный синтез позволяет
оптимизировать выход дизельного
топлива [26]
В настоящее время используются два технологических процесса — низкотемпературный и высокотемпературный. В первом синтез осуществляется при
температуре =220 °С и давлении 25—45 бар с использованием трубчатого реактора с фиксированным каталитическим слоем (то есть с насадкой со струйным
течением жидкости) или колонного реактора с барботером, в котором ката¬
368 -l\r Глава 8. Практика гетерогенного катализа: водород
лизатор диспергирован в жидком углеводородном парафине продукта. Высокотемпературный синтез, напротив, осуществляется при температуре =350 °С и
давлении 25 бар в реакторе с псевдоожиженным слоем циркуляционного или
нормального типов. Высокотемпературный процесс используется для получения бензина и других химических продуктов, а низкотемпературный — для
производства парафинов.
Поскольку синтез Фишера—Тропша включает в себя большое число реакций, хороший катализатор должен удовлетворять целому набору требований.
Наибольшую важность при этом имеют распределение полученного с данным
катализатором продукта по молекулярным весам, степень ветвления молекул,
содержание двойных связей окисленных молекул. Принципиальную важность
составляют низкая селективность катализатора по отношению к образованию
метана, высокая активность в конверсии водяного газа, обеспечивающая наиболее полное использование СО. Важна также низкая селективность катализатора по отношению к образованию углерода, которое приводит к снижению
активности катализатора. Благодаря легкости, с которой катализатор может
быть заменен в указанных выше реакторах в процессе их работы, важность его
стабильности по отношению к потере активности определяется в первую очередь ценой катализатора. То есть катализатор должен быть либо дешевым, либо
очень устойчивым по отношению к потере активности.
Сложилось так, что в промышленности используются в основном катализаторы на базе железа или кобальта. Рутениевые катализаторы очень активны в
синтезе Фишера—Тропша, но и слишком дороги для промышленного использования. Катализаторы на основе железа и кобальта приготовляются разными
методами, в число которых входят и осаждение металла, и пропитка носителя
(например, оксида алюминия или кремния) растворами их солей (с последующими сушкой и восстановлением). На наиболее «благородном» никелевом катализаторе синтезируется преимущественно метан, так что он используется в
основном для удаления следовых количеств СО в Н2.
Рост объема промышленных и академических исследований синтеза Фишера—Тропша, особенно быстро шедший в периоды Второй мировой войны и
нефтяного кризиса 1970-х годов, продолжается и сейчас, поскольку данный
процесс приобретает все большую значимость.
8.4. КОНВЕРСИЯ ВОДЯНОГО ГАЗА
В разделах, посвященных конверсии с водяным паром и синтезу
метанола, мы несколько раз упоминали конверсию водяного газа. Помимо того
что это — побочная реакция, конверсия водяного газа представляет и самостоятельный интерес. Она используется для улучшения состава газа, получающегося
при конверсии с водяным паром, нацеленного на уменьшение содержания СО и
увеличение доли Н2. Это особенно важно при получении Н2, использующегося
для производства аммиака, где не допускается присутствие соединений, содержащих СО или кислород. Такой водород получается при конверсии с водяным
8.5. Синтез аммиака
\
паром и последующей его двухстадийной обработке. На первой стадии синтез-газ
охлаждается, а затем пропускается при давлении 20—30 бар над катализатором
конверсии водяного газа, рабочая температура которого составляет 350—400°С.
Этот катализатор состоит из оксидов Fe203/Cr203 и снижает содержание СО в
газе до уровня 2—3 %, который ограничен условием равновесия. Для дальнейшего снижения содержания СО необходимо понизить температуру, поскольку
конверсия водяного газа — экзотермическая реакция. Но катализатор Fe203/Cr203
при низких температурах практически неактивен. Поэтому на второй стадии
низкотемпературной конверсии используется более активный катализатор состава Cu/Zn/Al203. Наинизшая возможная температура, при которой еще не
происходит конденсация газа при давлении 20—30 бар, равна примерно 200 °С,
это позволяет снизить содержание СО до 0,2 %. Для дальнейшего снижения
требуются дополнительные шаги, например синтез из СО метана. О них мы
поговорим позже, когда будем рассматривать процесс получения чистого не
содержащего кислорода водорода, использующегося для синтеза аммиака или
в топливных элементах. Конверсия водяного газа очень хорошо описывается
микрокинетической моделью [28] и в существенной мере следует схеме, описываемой уравнениями (23) (31). Анализ показывает, что в этом процессе при
высоких давлениях на поверхности в заметных количествах может присутствовать формилат (рис. 8.18).
Рис. 8.18. Зависимости от давления степеней покрытия поверхности меди
различными интермедиатами, образующимися при конверсии водяного газа
состава 33 % Н20, 52 % Н2, 13 % С02,
1 % СО при температуре 200 °С. Отметьте высокую степень покрытия поверхности формилатом при высоких давлениях [28]
8.5. СИНТЕЗ АММИАКА
В гл. 7 мы в качестве простого примера микрокинетического моделирования рассмотрели синтез аммиака. Сейчас мы дадим краткое описание
этого процесса, обратив особое внимание на связь параметров процесса и элементов микроскопической модели и на возможности оптимизации синтеза.
369
370 Глава 8. Практика гетерогенного катализа: водород
8.5.1. История синтеза аммиака
С начала XIX столетия исследователи использовали разложение
аммиака как удобную тестовую реакцию для ряда экспериментов. Им, однако,
не удалось осуществить обратную реакцию между N2 и Н2, результатом которой был бы аммиак, хотя связывание азота было задачей, в решении которой
была сильная заинтересованность. Объяснение этой неудачи было получено
после развития Вант-Гоффом равновесной термодинамики. Фриц Хабер первым показал, что для достижения значительной степени конверсии в равновесной реакции N2 + ЗН2 = 2NH3 + тепло необходимы высокое давление и низкая
температура. Поскольку при низких температурах вероятность развала молекул N2
неизмеримо мала, то для образования аммиака требуется использовать очень
активный катализатор. В 1909 г. Хаберу с сотрудниками удалось разработать
технологию получения аммиака с большой скоростью, достигавшей 2 кг в день,
с использованием катализатора на основе осмия при рабочем давлении 175 бар.
Для превращения N2 и Н2 в аммиак в приемлемых масштабах необходимо
использовать трубчатые реакторы, работающие при высоких давлениях. В то
время, однако, реакции при высоких давлениях проводили в порционных реакторах. Карл Бош, работавший на фирме BASF, разработал технологию, позволявшую получать несколько тонн аммиака в день в процессе, проводившемся при давлении 300 бар.
Хабер получил Нобелевскую премию в 1918 г., а Бош — в 1931.
Катализатор был усовершенствован Элвином Митташем, синтезировавшем более 2500 различных катализаторов и проведшим более 6500 их проверок.
В конце концов был сделан выбор в пользу плавленого катализатора на основе
железа с тройным промотированием; в качестве структурных промотеров использовались А1203 и СаО, а электронным промотером был калий. Промышленный процесс был налажен фирмой BASF, построившей первый завод в
г. Оппау в Германии, на котором производилось до 30 т аммиака в день. Сначала завод производил сульфат аммония, использовавшийся в качестве удобрения, но когда началась Первая мировая война, завод был реконструирован, и
на нем стали производить нитраты для боеприпасов. Завод существенно расширился, и в 1915 г. на нем получали в эквиваленте до 230 т аммиака в день.
Разработка промышленного синтеза аммиака послужила вехой в развитии
химической технологии, поскольку в нем было реализован крупномасштабный
непрерывный процесс в проточном реакторе, а также в промышленном катализе: многочисленные испытания, выполненные Митташем, позволили получить систематизированную информацию о каталитической активности многих
соединений.
Почему синтез аммиака так важен? Азот является важной составной частью
биологических систем, для которых аминокислоты служат строительными блоками. Хотя азот составляет 80 % атмосферы земли, входящие в нее молекулы N2
наиболее устойчивы и потому активируются с трудом.
Природа вовлекает азот в различные химические циклы тремя путями. Примерно одна треть всего азота подвергается окислению, например, при горении
8.5. Синтез аммиака
X
и в процессах выщелачивания, когда термодинамические условия (как, например, в двигателях автомобилей) благоприятствуют образованию оксидов NCK
Другим источником соединений азота являются бактерии, которые с помощь
энзима нитрогеназы в анаэробных условиях при наличии источника энергии
способны создать из N2 аммиак. Активными центрами в энзимах являются
комплексы FeMoCo. Проведенные расчеты показали [10], что в энзимах реакция идет принципиально иным путем по сравнению катализируемой металлами, которая была описана в предыдущей главе. В энзимах водород последовательно присоединяется к молекуле азота, которая начинает диссоциировать
только после присоединения к ней пяти атомов водорода. Природа, очевидно,
нашла интеллектуальный путь, позволяющий проводить процесс при комнатной температуре. Бактерии, выделяющие аммиак, живут в симбиозе со многими растениями, например со стручковым горохом или с ольхой, которые снабжают бактерии сахарами, являющимися продуктом фотосинтеза, обеспечивая
тем самым синтез аммиака, необходимый для образования аминокислот и,
следовательно, новых энзимов. Энзимы использую по крайней мере 16 молекул аденозинтрифосфорной кислоты для синтеза двух молекул аммиака, а
созданный человеком синтез почти столь же эффективен (молекулы адено-
зин-5'-трифосфорной кислоты являются «носителями» энергии в живых клетках; энергия может высвобождаться в следующем процессе аденозинтрифос-
форная кислота + Н20 аденозиндифосфорная кислота + фосфорная кислота; AG = —30,5 кДж/моль). Хотя железный катализатор синтеза аммиака не
претерпел существенных изменений, энергетическая эффективность процесса
за время его существования была повышена существенно (рис. 8.19). Сейчас
затрачивается около 400—500 кДж/моль синтезированного аммиака, что сравнимо с затратами энергии в процессе с участием энзимов.
Рис. 8.19. Затраты энергии на
производство аммиака постепенно снижаются и сейчас стали сопоставимы с затратами в
природе
1000
*
FC
Электрический разряд
100 -
Ранний синтез аммиака
Одиночный поток
Современный
синтез аммиака
10
_L
JL
-L.
I
1920 1960 2000
Годы
Синтез аммиака — второй по масштабности процесс после производства
серной кислоты (см. также гл. I). На него затрачивается 1 % всей производимой энергии. 80 % синтезируемого аммиака идет на производство удобрений
(жидкого аммиака или более удобных в обращении солей нитрата аммония,
371
372
Глава 8. Практика гетерогенного катализа: водород
сульфата аммония и т. д.), поэтому синтез аммиака является жизненно необходимым для человечества. Аммиак используется также при производстве азотсодержащих полимеров — нейлона, полиамидов, полиуретанов и взрывчатых
веществ (нитроглицерина и тринитротолуола).
8.5.2. Завод по производству аммиака
Заводы по производству аммиака строятся преимущественно в тех
районах, где имеется достаточное количество энергии (например, газа метана)
и воды и откуда возможен транспорт аммиака на кораблях. Типичный завод по
производству аммиака показан на рис. 8.20; на нем производится 2 х 1350 т
аммиака в день, при этом используется 2 х 150 т катализатора. В оборудовании
доминируют две установки по конверсии с водяным паром, которые легко можно
различить на фотографии.
■
Рис. 8.20. Завод по производству аммиака, на котором его можно синтезировать 2 х 1350 т
в день; в синтезе используется 2 х 150 т катализатора. Громадный размер завода
легко понять по размеру людей, указанных на снимке стрелкой
На рис. 8.21 показан схема производства аммиака. Сначала природный газ
обессеривается, а затем подвергается конверсии с водяным паром в первой печи,
где получается смесь непрореагировавшего метана (10—13 %), СО, С02 и Н2,
8.5. Синтез аммиака -J\ - 373
которая вместе с воздухом, содержащим необходимый для производства аммиака азот, подается во вторую печь. Здесь кислород реагирует с водородом и
метаном, выделяющееся в этой экзотермической реакции тепло идет на нагрев
газа, температура которого повышается с 800 до 1000 °С, что обеспечивает дальнейшую конверсию оставшегося метана. Затем газ охлаждается в теплообменнике, при этом отобранное тепло возвращается к первой печи, где протекает эндотермическая реакция. Газ, охлажденный до 400 °С, содержит менее 0,25 вес. %
метана. Этот газ подвергается двухстадийной конверсии, подобной рассмотренной в разд. 8.4 на примере водяного газа, которая проводится с целью уменьшения содержания СО и увеличения доли водорода.
С02 Реактор
водяного газа
Рис. 8.21. Схема заводского процесса синтеза аммиака
В синтезе аммиака присутствие кислородсодержащих молекул является
нежелательным, поскольку они способствуют окислению железа и блокируют
активные центры на поверхности катализатора. Сначала в скрубберах удаляется С02 за счет его реакции с сильным основанием. Остающийся СО (и С02)
удаляется путем проведения реакции синтеза метана, в результате которой СО
расходуется на образование метана и воды. Наконец, вода удаляется, например, с помощью молекулярных сит. Наличие метана при этом никаких проблем не представляет, поскольку он слабо взаимодействует с поверхностью
катализатора. Газовая смесь (табл. 8.6) сжимается до давления 200 бар, необходимого для синтеза аммиака, и подается в реактор.
Таблица 8.6. Типичный состав газовой смеси, подаваемой в реактор синтеза
аммиака
Газ
n2
н2
сн4
Аг
СО
%
74,3
24,7
0,08
0,03
1—2 миллионные доли
374 Глава 8. Практика гетерогенного катализа: водород
Газ вступает в экзотермическую реакцию с участием катализатора синтеза
аммиака, которая проводится при температуре 450—500 °С, в результате на выходе
из реактора аммиак составляет 15—19 об. %. Аммиак удаляется из смеси за счет
конденсации, а непрореагировавший газ подается снова в реактор. Эта часть
газа подвергается очистке, чтобы избежать накопления инертных компонентов. Аммиак конденсируется не полностью, поэтому входящий в реактор газ
уже содержит несколько процентов аммиака.
8.5.3. Рабочий реактор
Синтез аммиака является экзотермическим процессом, поэтому в
ходе реакции в реакторе выделяется тепло. Нагрев приводит к снижению равновесного парциального давления аммиака, поэтому реакцию следует проводить при наинизших возможных температурах, а скорость реакции должна быть
максимально большой. Эти противоречивые требования компромиссно удовлетворяются при температуре 450—500 °С. Реактор синтеза аммиака подвергался многократным модернизациям и стал довольно изощренной конструкцией,
позволяющей использовать небольшие количества катализатора, что снизило
его размер и инвестиционные затраты на его строительство. На рис. 8.22 показан реактор с двумя слоями катализатора, радиальным адиабатическим потоком и непрямым охлаждением.
Рис. 8.22. Схематическое изображение адиабатического
двухслойного реактора с радиальным потоком. Имеются
три входных канала и один выходной. Основная подача
исходных веществ осуществляется сверху (слева), исходные вещества следуют по (охлаждаемой) рубашке высокого давления ко дну реактора, где они нагреваются отходящими газами. Внизу к нагретому газу добавляются исходные вещества через нижний (справа) входной канал и они
протекают через центральную часть реактора, где снова в
систему добавляются исходные вещества. Затем газ проходит через первый слой катализатора (А), где он течет радиально по направлению к центру. Здесь в адиабатических
условиях проходит реакция, исходная смесь нагревается и
приближается к равновесию (В). Затем смесь охлаждается в
верхнем теплообменнике и движется ко второму слою (С),
где снова в адиабатических условиях протекает реакция,
что приводит к повышению температуры и дальнейшему
приближению системы к равновесному состоянию (D)
Сложную конструкцию показанного на рис. 8.22 реактора можно оптимизировать, используя концепцию «линии оптимальной работы» или «линию максимальной скорости» [29]. Рассмотрим зависимость концентрации аммиака от
температуры (рис. 8.23). Правая линия отвечает равновесию, она уходит моно¬
Вход
Вход
HI
Выход ■' Вход
8.5. Синтез аммиака -J\ - 375
тонно вниз, поскольку синтез аммиака — экзотермический процесс. Если мы
проведем на графике линии постоянных скоростей (для четырех различных
констант скорости) как функции концентрации аммиака и температуры, мы
получим четыре показанные на рис. 8.23 кривые. Сначала концентрация аммиака увеличивается с ростом температуры, но затем она должна начать убывать
и следовать линии равновесия. На каждой следующей приведенной линии скорости синтеза увеличивается примерно в 10 раз, причем верхней линии отвечает наименьшая скорость. Если мы теперь проведем через точки максимума
каждой кривой линию, то получим «линию оптимальной работы», которой
необходимо следовать, если поставлена задача получения наибольшего выхода
аммиака при наименьшем количестве катализатора в реакторе. Концепция «линии оптимальной работы» часто используется при синтезе, основанном на экзотермических реакциях, к числу которых относятся синтез метанола, конверсия водяного газа и окисление диоксида серы. Эта линия обычно идет «параллельно» линии равновесия со сдвигом на 30—50 °С в область более низких
температур.
о:
s
ZT
со
Q.
Рис. 8.23. Сплошная монотонно спадаю- i
щая линия справа представляет собой %
кривую равновесия. Четыре кривые со- о
ответствуют постоянным значениям скорости реакции. Поскольку желательно
работать в условиях, соответствующих
максимальному значению концентрации
аммиака, то «линия оптимальной работы» есть кривая, проходящая через точки максимума параллельно кривой равновесия [29] Температура
Принципы работы адиабатического двухслойного реактора с радиальным
потоком, показанного на рис. 8.22, схематически отображены на рис. 8.24, где
в действительности показан трехслойный реактор с непрямым охлаждением
между слоями. Линия равновесия и линия оптимальной работы показаны также на графике зависимости концентрации аммиака от температуры. Поскольку
синтез аммиака — экзотермическая реакция, то температура повышается примерно на 14—18 °С на один весовой процент синтезированного аммиака. Движению по реактору некоторого объема газа отвечает линия, показанная стрелкой на правом рисунке 8.24. При прохождении через первый слой температура
повышается, что указано направленной вверх линией в нижней части рисунка.
При охлаждении газа температура снижается, что описано сдвигом влево по
376 -l\r Глава 8. Практика гетерогенного катализа: водород
горизонтальной линии. При входе во второй слой температура снова повышается до перехода в следующую фазу охлаждения. Таким образом, двигаясь зигзагообразно через линую оптимальной работы, можно поддерживать систему в
реакторе в условиях, близких этой линии.
Рис. 8.24. Слева: схема адиабатического
трехслойного реактора с непрямым охлаждением и двумя теплообменниками.
Справа: линия равновесия (справа вверху), линия оптимальной работы и рабочая линия для реактора, показанного слева [30]
Такая процедура отражает принципы современной технологии, в которой
обычно используются двух- или трехслойные реакторы. Недавние исследования [30] показали, как такие принципы открывают новые и интересные возможности комбинирования фундаментальных основ и детального конструирования реакторов, обеспечивающего оптимальную работу слоя катализатора на
протяжении всего хода процесса.
8.5.4. Научные предложения
по повышению эффективности катализаторов
В шестой главе мы обсуждали принцип универсальности в гетерогенном катализе с целью показать, как линейная зависимость между энергией
связи и энергией активации позволяет объяснить связанный с принципом Сабатье «вулканический» вид активности катализатора как функции степени заполнения J-зоны. На рис. 6.42 было также показано, как скорость образования
аммиака коррелирует с энергией адсорбции атомов N и концентрацией газообразного аммиака. Аммиак быстрее формируется в том случае, когда атомы азота менее слабо связаны с поверхностью катализатора. Таким образом, имея
возможность непрерывно изменять энергию связи атомов азота с поверхностью (например, за счет использования сплавов), можно провести оптимизацию
параметров катализатора.
Привлекая микрокинетическую модель синтеза аммиака и соотношение
Бренстеда—Эванса—Поляни между энергией активации диссоциации N2 и энергией связи атомарного азота, можно представить теоретическое число оборотов реакции как функцию только одного параметра, а именно энергии связи
ЧК\\'
kv*"
■ ч X
t I t
350 450 550
Температура, "С
8.5. Синтез аммиака
\
атомарного азота АЕ. Для каждого набора параметров — состава газовой смеси,
давления и температуры, можно построить «вулканическую» зависимость числа оборотов реакции от АЕ\ для системы атомарного азота на рутении такие
зависимости представлены на рис. 8.25.
s
zr
*
со
CD
Q.
СО
О
ь
о
Q.
О
VD
О
О
с;
о
S
Т
En* - EN*(Ru), кДж/моль N2
- En*(Ru), кДж/моль N2
Рис. 8.25. Расчетные «вулканические» кривые для синтеза аммиака, показывающие зависимости числа оборотов реакции от энергии связи атомов азота с поверхностью
катализатора для концентраций аммиака 5, 20 и 90 %. Слева построены зависимости для температуры 420 °С, давления 80 бар и соотношения Н2: N2, равного
2:1; справа — для температуры 450 °С, давления 200 бар и соотношения Н2: N2,
равного 3:1 [30]
Рис. 8.25 показывает, как с ростом концентрации аммиака «вулканические»
кривые смещаются в сторону меньших энергий связи атомарного азота. Повторяя эти вычисления для различных значений температуры и концентрации
аммиака при фиксированном отношении N2: Н2, можно получить набор «вулканических» кривых и определить энергию связи, отвечающую максимальному
значению числа оборотов реакции для данного катализатора. Построив зависимость между концентрацией аммиака и температурой, для которых максимум
«вулканической» кривой приходится на одно и то же значение энергии связи
атомарного азота, можно получить «линию оптимального катализатора», отвечающую данной энергии связи (рис. 8.26).
Линию равновесия и линию оптимальной работы снова наносим в координатах концентрация аммиака—температура для двух наборов параметров, отвечающих рис. 8.26, но уже вместе с оптимальными линиями катализатора, которые
строятся для нескольких энергий связи. На правой части рисунка построена
также рабочая линия, и теперь можно установить, как следует распределять различные катализаторы по реактору. Так, если линия оптимального катализатора с
377
378 Глава 8. Практика гетерогенного катализа: водород
- Е * (Ru) = -20 кДж/моль пересекает рабочую линию в некоторой точке, то
данный катализатор следует помещать в том месте, на которое приходится данная
точка рабочей линии. В идеале для достижения наилучших результатов энергия
связи должна непрерывно уменьшаться по мере продвижения по слою катализатора
от значения —25 кДж/моль на входе до —15 кДж/моль на выходе из слоя.
Рис. 8.26. Линия равновесия, линия оптимальной работы и линии оптимального катали¬
затора в координатах концентрация аммиака — температура для различных
условий. Слева: температура равна 450 °С, давление — 200 бар и соотношение
Н2: N2 — 3 : 1. Справа: температура равна 420 °С, давление — 80 бар и соотношение Н2: N2 — 2 : 1. Пересечение линии оптимального катализатора для заданного значения энергии связи атомов азота и рабочей линии соответствует
месту расположения этого катализатора в реакторе. Рабочая линия должна располагаться по возможности ближе к линии оптимальной работы [30]
Здесь важно отметить, что для улучшения эффективности реактора в целом
следует использовать набор катализаторов, распределенных по слою реактора.
Насколько такая схема является жизнеспособной, зависит от соотношения цены
ансамбля катализаторов с изменяющимися параметрами и экономического
выигрыша, связанного с уменьшением размера реактора высокого давления.
Обрисованная выше схема является сложной версией так называемого процесса Келлога усовершенствованного синтеза аммиака, в котором катализатор
на основе железа размещается в первом слое, а катализатор на основе рутения,
связывающего азот более слабо, — во втором, третьем и четвертом слоях [31].
0
300 350 400 450 500 550 300
Температура, °С
350 400 450 500 550
Температура, °С
8.6
ПРОМОТОРЫ И ИНГИБИТОРЫ
В предыдущих разделах мы рассматривали в основном каталитическую активность чистых металлов типа железа, рутения, меди, платины и т. д.
Реальные катализаторы, однако, представляют собой более сложные системы.
8.6. Промоторы и ингибиторы
параметры которых оптимизированы добавками небольших количеств других
элементов, влияющих на общую активность и селективность катализатора. Рассмотрим несколько важных примеров такого влияния.
Промоторами называют материалы, усиливающие действие катализатора.
Они разделяются на структурообразующие и модифицирующие. Структурные
промоторы, как, например, небольшие добавки А1203 или СаО в катализатор
синтеза аммиака, обеспечивают сохранение высокой удельной поверхности
(20 м2/г) частиц железа в процессе работы реактора. Модифицирующие промоторы типа К или Cs (в действительности в зависимости от системы используются щелочные или щелочно-земельные металлы) усиливают действие катализатора за счет изменения электронного состояния поверхности. Обычно эти
атомы сильно поляризуются на поверхности, что приводит к формированию
дипольных полей, сильно взаимодействующих с адсорбатами и способствующих, например, диссоциации молекул азота, если они имеют в переходном
состоянии диполи, ориентированные противоположным образом, поскольку
при этом снижается энергия активации [32]. Модифицирующие промоторы
могут также влиять на равновесную степень заполнения поверхности азотом:
если, например, они взаимодействуют с молекулами аммиака, то последние
будут стремиться ориентировать свой дипольный момент по направлению, совпадающему с направлением дипольного момента, индуцированного атомами
щелочных металлов. Это приведет к отталкиванию молекул аммиака от поверхности и уменьшению степени ее отравления аммиаком. Таким образом, в
зависимости от ориентации дипольных моментов эффект может быть разный.
Влияние добавок калия ясно продемонстрировано на рис. 8.27, где показано,
что при их введении увеличивается коэффициент прилипания азота для базальной плоскости железа.
Эффект оказывается гигантским для плотноупакованной грани (110)Fe, но
следует принять во внимание, что коэффициенты прилипания здесь малы и различаются существенно для разных граней, поэтому роль калия сводится в основном к выравниванию значений коэффициента прилипания для разных граней.
1000
CNJ
Рис. 8.27. Увеличение коэффициента прилипания азота при промо-
тировании базальной плоскости
железа калием [33]
K-FW110)/Ге(110) K-Fc( 100)/Гс*( 100) K-Fe< 111 )/fe( 111)
Поверхность монокристалла
380 -J\r Глава 8. Практика гетерогенного катализа: водород
Изменение коэффициента прилипания сказывается на скорости синтеза
аммиака. На рис. 8.28 показаны значения скоростей синтеза для двух базальных плоскостей чистого железа и железа, модифицированного 0,1 МС калия.
15 -
СО
*
СО
S
2
ж *
СО CD
00
CD m
t СО 10
I!
-0 С
I—
О
О с
Q. 5
О
Условия синтеза;
Г =673 К» р = 20 бар»
стехиометрическое
соотношение реагентов,
концентрация NH3
равна 26 мбар
и
Fe(HI) • K-Fe(lll) Fe(lOO) K-fe(lOO)
Поверхность монокристалла
Рис. 8.28. Скорость синтеза аммиака для поверхностей монокристаллов железа двух типов
(с и без промотирования калием) [34]
Реакционная способность поверхностей существенно увеличивается, причем наибольший эффект наблюдается для менее плотной поверхности Fe(l 11),
которая в принципе может рассматриваться как шероховатая. В случае плотно-
упакованной поверхности реакционная способность оказывается существенно
более низкой, и имеются многочисленные указания [35], что реакционная способность здесь определяется центрами того же типа, что и у поверхности Ru(100),
обсуждавшимися в гл. 6 и 7. Таким образом, влияние калия может быть связано не только с изменением электронной структуры. Предполагают, что добавки калия приводят к появлению на поверхности более активных центров, то
есть он действует как структурообразующий промотор. Однако данное утверждение носит дискуссионный характер.
Часто считают, что щелочные металлы всегда являются промоторами катализаторов, но это неверно. Например, добавки щелочных металлов приводят к
замедлению диссоциации метана на поверхности Ni, поскольку дипольный
момент молекул метана в переходном состоянии направлен так же, как и дипольный момент атомов щелочных металлов, адсорбированных на поверхности,
что приводит к увеличению барьера диссоциации. Сказанное проиллюстрировано на рис. 8.29, где приведены также данные расчета методом функционала плотности высоты барьера диссоциации метана на чистой и модифицированной
калием поверхности монокристалла Ni. Отметим, что эффекты, связанные с
изменением электронной структуры, проявляются более сильно, поскольку
входят в показатель экспоненты, а эффекты блокировки дают линейный вклад.
Для ясности подчеркнем еще раз, что эффект промотирования обусловлен
возникновением электрического дипольного момента у молекул в переходном
состоянии. Ни молекулы азота, ни молекулы метана не имеют дипольных моментов в свободном состоянии, но он у них возникает при взаимодействии с
8.6. Промоторы и ингибиторы
электронами металла. В случае азота диполь ориентирован противоположно, а
в случае метана — по диполю адсорбированного атома щелочного металла, что
приводит к ускорению и замедлению реакции соответственно.
Рис. 8.29. Схематическое изображение действия, например, калия, осажденного на по¬
верхность металла. На атомах калия формируется дипольное поле, воздействующее на адсорбированные молекулы и на молекулы, находящиеся в переходном состоянии, что приводит к увеличению или уменьшению высоты потенциального барьера. При адсорбции метана возникающий дипольный момент
ориентирован по дипольным моментам, существующим у атомов калия, что
приводит к их отталкиванию и увеличению высоты барьера диссоциации. Сказанное подтверждается данными расчетов методом функционала плотности [36]
Другой пример использования калия в качестве промотора связан с гидрированием СО в прямом синтезе метанола, обсуждавшемся ранее [23]. Здесь
калий выступает в качестве промотора гидрирования СО, но при традиционном синтезе метанола предпринимаются серьезные усилия для удаления калия
из катализатора, поскольку он разрушает его селективность и промотирует синтез
высших спиртов, то есть в этом случае поверхность становится слишком активной. По этой причине следует соблюдать большую осторожность в использовании промоторов, обеспечивающих оптимальный эффект.
Действие яда, отравляющего катализатор, может быть связано с изменением электронной структуры или блокировкой свободных центров на поверхности. Яды можно также разбить на две группы, на адсорбирующиеся обратимо и
необратимо. Например, вода является сильным ядом для катализаторов синтеза аммиака, поскольку она способствует появлению кислорода на поверхности
железа, блокирующего активные центры подобно атомам азота. Условие равновесия для воды в рамках микрокинетической модели синтеза аммиака выписываются непосредственно: нужно просто добавить дополнительный член в
знаменатель уравнения, определяющего число свободных центров, в котором
кислород становится НИИР. Сильное влияние на скорость синтеза аммиака
оказывает содержание воды на уровне нескольких миллионных долей. К счастью, вода адсорбируется обратимо, поэтому если ее удалить из реагентов, то
адсорбированный кислород прореагирует и катализатор восстановит свою активность. Проблема оказывается более серьезной при наличии в реагентах серы
1,60
AETS = е [г = 19 кДж/моль fi = 19 эА
0,80
1,20 1,40 1,60 1,80 2,00
d(C-H), А
382 -l\r Глава 8. Практика гетерогенного катализа: водород
или хлора. Эти элементы связываются с поверхностью более прочно, поэтому
удалить их оттуда очень сложно. Сера является в высшей степени нежелательным элементом для многих катализаторов, поскольку она характеризуется очень
большой энергией адсорбции, поэтому ее надо удалять из реагентов до их поступления в реактор. Часто в качестве защитного средства используют простой ZnO,
который реагирует с серусодержащими соединениями, образуя ZnS.
В заключение отметим, что имеются также яды, которые невозможно удалить с поверхности. Хорошим примером является адсорбция Au на ступенях Ru,
однако он представляет лишь академический интерес. Более реальные проблемы связаны со свинцом, добавляемым в бензин. Если кто-то по ошибке
заполнит бак автомобиля, оснащенного катализатором, бензином, содержащим свинец, то катализатор подвергнется необратимому отравлению, поскольку свинец осядет на поверхность Pt и полностью заблокирует ее. Свинец (как
и золото) имеет низкую поверхностную энергию и поэтому всегда будет оставаться на поверхности. В общем случае следует принимать разнообразные
меры предосторожности, чтобы избежать отравления катализатора и реагенты следует очищать перед их подачей в каталитический ректор. Интересным
аспектом данной проблемы является следующий факт. В реальности каталитическая активность поверхности определяется небольшим числом центров
(например, ступенями на поверхности рутения), поэтому малые добавки могут блокировать некоторые центры, что приведет к существенному повышению селективности катализатора. Например, в случае реакций гидрирования можно получить определенный выигрыш, заблокировав наиболее активные центры (ступени), поскольку они оказывают слабое влияние на
реакцию гидрирования, но играют существенную роль в нежелательном разрыве С—С-связи. Сказанное открывает новые возможности в создании катализаторов нового типа.
8.7. «ВОДОРОДНОЕ СООБЩЕСТВО»
8.7.1. Потребность в возобновляемых источниках энергии
Наша сильная привязанность к природному топливу должна в скором времени окончиться (табл. 8.7). Это расставание может завершиться и не
так скоро, как указано в таблице, поскольку приведенные в ней цифры основаны на разведанных запасах. Идет открытие новых месторождений, улучшаются
также способы добычи нефти из уже известных месторождений. Более того,
имеются огромные резервы, связанные с наличием в природе смоляных песчаников и газогидратов, ожидающих экономически выгодных и безопасных для
окружающей среды технологий по их вовлечению в энергетический цикл. Еще
имеются большие запасы каменного угля, который может быть превращен в
газ синтезом Фишера—Тропша.
8.7. «Водородное сообщество» -J\r 383
Таблица 8.7. Известные запасы природного топлива, ежегодное потребление
и время, оставшееся до исчерпания ресурсов
Нефть
Газ
Уголь
Разведанные мировые запасы
~1,6 х 10|4л
-1,4 х 1014м3
-1,6 х 10|4т
Ежегодное потребление
-4,3 х 1012 л
-2,4 х 1012м3
-4,5 х 109 т
Время истощения ресурсов
-40 лет
-60 лет
-200 лет
Еще один важный аспект связан с тем, в какой степени выбросы С02,
вызывающего парниковый эффект, при интенсивном сжигании природного
топлива будут воздействовать на окружающую среду. В настоящее время хорошо установлено, что средняя температура на Земле постепенно увеличивается и это увеличение идет параллельно росту содержания С09 в атмосфере.
Однако остается дискуссионным вопрос, связано ли повышение температуры
только с выбросами С02. Были предложены альтернативные объяснения, в
которых предполагалось, что солнечная активность, в частности солнечный
ветер, существенно влияет на процесс образования облаков и, следовательно,
на температуру. Данные об исторически длительных периодах изменениях
климата, основанные на геологических исследованиях, показывают наличие
корреляции между солнечной активностью, сильными изменениями температуры и содержанием С02 в атмосфере. В какой мере значительны выбросы
С02, обусловленные сжиганием природного топлива, на фоне естественных
процессов? Пока имеются сомнения, следует по возможности ограничить
выбросы С02. Необходимо разработать схемы сжигания природного топлива,
обеспечивающие выделение С02 из отходящих газов и его изоляцию в стационарных хранилищах типа выработанных газовых месторождений или других
прочных геологических образований. Такая превентивная мера приведет к
удорожанию энергии на 30 %.
В чем заключаются недостатки источников энергии, использование которых не приводит к выбросам С02? Эксплуатация ядерных реакторов сложна,
имеется проблема утилизации ядерных отходов. Термоядерные реакторы более
привлекательны, но до их создания пройдет еще не один десяток лет. Реальной
альтернативой могут служить источники, использующие солнечную энергию и
энергию ветра.
Земля получает от солнца громадное количество энергии. В ясный день,
когда солнце находится в зените, поток энергии равен 1 кДж/м2. Эта величина
в 10 000 раз превышает современный уровень потребления энергии (табл. 8.8).
Поток солнечной энергии распределен по земле неравномерно, но с учетом
энергии ветра и морских волн частично неравномерность в распределении источников энергии сглаживается. Разработать методы эффективного и надежного использования этой энергии — это основная задача нашего поколения, и
она будет решена, если мы ставим целью сохранение уровня нашей жизни и
дальнейшее развитие экономики.
384
J\, Глава 8. Практика гетерогенного катализа: водород
Таблица 8.8. Поступающая солнечная энергия в сравнении с уровнем
ее потребления
Энергия, поступающая от солнца
3,8 х 1024 Дж/год
Расход энергии, связанный с деятельностью человека
3,8 х 1020 Дж/год
Потребление электрической энергии
4,6 х 1019 Дж/год
8.7.2. Возобновляемые источники энергии
В некоторых местах на Земле имеются более или менее дешевые
источники энергии. К ним относятся ветер, потоки воды, гейзеры, естественный перепад высот, позволяющий устанавливать гидроэлектростанции. Все эти
источники являются возобновляемыми, и их использование оказывает слабое
воздействие на окружающую среду. Подземные тепловые источники дают существенный вклад в энергетику Исландии, где наблюдается высокий уровень
геотермической активности.
Солнечный свет представляет собой практически неисчерпаемый источник
энергии. Имеется возможность непосредственного потребления солнечной энергии путем ее преобразования в электрическую. Здесь удается преобразовывать
до 20 % падающей энергии, однако технология создания преобразователей солнечной энергии является очень дорогой. Напротив, в фотосинтезе преобразуется около 1 % солнечной энергии, которая накапливается в зернах, соломе
или деревьях. Такая низкая эффективность устанавливает пределы потенциалу
использования биомассы, хотя, несомненно, в будущем растения будут использоваться для производства энергии.
Помимо фотосинтеза имеется два других подхода к преобразованию солнечной энергии. Один из них основан на прямом преобразовании солнечной
энергии в электрическую с помощью солнечных батарей (как иногда говорят,
фотовольтаических элементов). Второй основан на электрохимических элементах, которые в некоторой степени воспроизводят фотосинтез: в них солнечная энергия используются для проведения диссоциации воды на Н2 и 02.
Солнечная энергия переносится фотонами, энергия которых распределена в
интервале нескольких электронвольт. Хотя внутренняя часть солнца, где протекает термоядерная реакция превращения водорода в гелий, имеет высокую температуру — порядка 108 К, температура поверхности составляет всего 5800 К. Спектр солнца может быть аппроксимирован спектром излучения,
идущего от черного тела с такой температурой, что позволяет, используя закон Планка, получить для плотности падающего на Землю потока энергии
выражение
8.7. «Водородное сообщество» -J\r 385
которое связывает плотность потока энергии (Дж/м2 • с) в интервале энергий
фотонов [е, е + de] с их энергией е и температурой Т. Распределение плотности
потока по энергиям фотонов дано на рис. 8.30.
Рис. 8.30. Теоретическая плотность
потока энергии от солнца как черного тела, имеющего температуру
5800 К. Свет, достигающий поверхности Земли, искажается вследствие
поглощения инфракрасного излучения молекулами Н?0 и С02 и ультрафиолетового излучения озоном.
Интегральный поток показан на
шкале справа. Показаны оптический
диапазон, а также энергия, необходимая для образования электроннодырочной пары в Si и ТЮ2
Если для захвата фотонов в фотокаталитической реакции используется,
например, ТЮ2, то в этом процессе будет задействовано только 10 % имеющегося спектра, поскольку для создания электронно-дырочной пары в ТЮ2 требуется энергия 3,2 эВ. Фотовольтаический и фотохимический методы представляют потенциальный интерес, однако в настоящее время они слишком
дороги. Кроме того, производство полупроводников, использующихся в солнечных батареях, требует больших энергетических затрат. Тем не менее перспективы у этих элементов заманчивы. Если эффективность созданных элементов будет составлять, скажем, 10 %, то для обеспечения нынешнего уровня
потребления энергии потребуется использовать всего около 0,1 % поверхности
Земли.
В последнее десятилетие много внимания стали уделять ветряным источникам энергии. Ветряные генераторы мощностью 2 МВт и более, как полагают, могут конкурировать с традиционными тепловыми электростанциями.
В странах типа Дании ветряные генераторы поставляют существенную долю
(12 % в 2000 г.) электрической энергии, что составляет 2 % от общего потребления энергии. Имеются, однако, проблемы с использованием энергии ветра.
Например, очень сложно интегрировать нестабильный источник энергии в
общую сеть, от которой требуется высокая устойчивость по напряжению и
частоте. Имеются ограничения и по размещению ветряных генераторов. Дания планирует расположить 80 ветряных генераторов мощностью 150 МВт в
прибрежной зоне, поскольку там ветры сильнее, чем на земле. Цена строительства, поддержания генераторов в надлежащем состоянии и их обслуживания, конечно, очень высока.
Другие имеющие перспективу возобновляемые источники энергии, альтернативные существующим, как, например, источники, преобразующие энергию волн, еще далеки от экономически выгодного практического использования.
Энергия фотонов, эВ
386 -l\r Глава 8. Практика гетерогенного катализа: водород
8.7.3. Водород и топливные элементы
Основной недостаток новых возобновляемых источников энергии
обусловлен их ненадежностью. Они будут производить энергию, пока дует ветер и
светит солнце. Потребности энергии в течение суток не постоянны: имеются пиковые нагрузки. Поэтому необходимо разрабатывать способы накопления энергии и
ее высвобождения при пиковых нагрузках. Синтетическое топливо и метанол рассматриваются в качестве возможных кандидатов на роль новых источников энергии, но наибольшее внимание сейчас уделяется водороду. Он может быть получен
традиционным образом из воды под действием электричества с эффективностью
=70 %. Водород является идеальным топливом для топливных элементов.
Принцип работы топливных элементов был впервые описан Грове в 1939 г.
[37]. В настоящее время предложено несколько схем использования водорода в
электрохимических ячейках. Мы объясним механизм работы двух наиболее
важных топливных элементов — топливных элементов с ионообменной мембраной и топливных элементов на твердых оксидах.
8.7.3.1. Топливные элементы на протонопроводящих мембранах
Топливный элемент представляет собой слоистую структуру, образованную анодом, катодом и твердым электролитом (рис. 8.31). Водород вступает в
реакцию на аноде (обычно это наноразмерные частицы Pt или Pt/Ru, осажденные
на проводящую графитовую подложку), где он распадается на протон и электрон:
Рис. 8.31. Принцип работы топливного элемента с протонопроводящей мембраной. Мем¬
брана Нафион разделяет кислород и водород. Водород на поверхности анода
(слева) распадается на протон и электрон. Электроны движутся по внешней
цепи, а протоны диффундируют через мембрану, имеющую толщину приблизительно 0,1 мм. Кислород восстанавливается на катоде до отрицательно заряженных ионов, которые рекомбинируют с протонами, образуя воду. Каталитические частицы осаждены на проводящие пористые электроды, впрессованные
в мембраны для обеспечения лучшего электрического контакта
2Н2 —» 4Н+ +4е~.
(8.72)
Кластеры
Pt или Pt/Ru
/
Протоноиронодящая
мембрана
/
яг
Пористые
электроды
8.7. «Водородное сообщество»
X
Электрон уходит через внешнюю цепь, связанную с катодом, на котором
кислород восстанавливается до ионов кислорода на той же каталитической
системе, что и у анода:
02 + 4е“ —> 202~, (8.73)
а протон проходит через протонопроводящую мембрану до катода и реагирует
с ионами кислорода, что приводит к образованию воды; в результате полная
реакция выглядит так:
2Н2+02 —> 2Н20. (8.74)
Поскольку ионообменная мембрана пропускает только ионы, то электроны
вынуждены перемещаться по внешней цепи, что создает электродвижущую силу
в системе. Напряжение, создаваемое в таком элементе, определяется уравнением Нернста. Для реакции с участием водорода и кислорода имеем
/
Д(? = AG° + RT In
Рн^о
Рн, >02
(8.75)
Поскольку на каждую созданную молекулу воды приходится два электрона,
прошедшие через внешнюю цепь, а величина AG относится к одному молю, то
напряжение в цепи будет равно разности AG, поделенной на заряд, относящийся к одному молю воды, ~2eNA:
о RT ,
е = £ ;г~ж'г 1п
2eNk
Рн^о
Рн2 у]Ро2
(8.76)
Стандартный потенциал элемента для реакции водорода с кислородом определяется свободной энергией образования моля воды (газа):
о А<?° -228,582 кДж • моль"1 (R
2eNA 2х1,60х10-19 Кл х 6,02 х 1023 ’ ’
Из-за большого числа кинетических потерь на практике напряжение составляет только 0,7—0,8 В, а максимальная плотность тока равна 0,5 А/см2.
Протонообменные мембраны типа Нафион работают только во влажных условиях, то есть в системе должна присутствовать вода, что ограничивает верхний
предел рабочих температур 100 °С. Обычно ионообменные мембраны работают
при температуре около 80 °С, что делает их перспективными для применения в
автомобилях, компьютерах и мобильных телефонах. Они были разработаны в
1960-х годах для космической программы Gemini. Однако высокая стоимость
таких топливных элементов не позволят использовать их в качестве широкой
альтернативы обычным источникам энергии, основанным на сжигании природного топлива.
Значительная часть ограничений связана с материалом анода. Активным
элементом анода служит высокодисперсный металл, нанесенный на графит,
387
388
Л
Глава 8. Практика гетерогенного катализа: водород
впрессованный в мембрану. В качестве активного металла выбрана платина,
поскольку она обладает высокой активностью по отношению к диссоциации
водорода, но, к сожалению, платина очень чувствительна к следовым количествам примесей (например, СО) в газообразном водороде.
Наличие примесей составляет важную проблему для использования топливных элементов в подвижных потребителях энергии, поскольку водород для
них, по крайней мере на начальной стадии эксплуатации, предполагается получать путем разложения углеводородов или метанола на конверсионных системах, находящихся непосредственно на потребителях. Это связано с тем, что в
настоящее время нет подходящих сред для хранения водорода. В таких системах СО является обязательным побочным продуктом, и поскольку СО связывается с Pt сильнее, чем водород, а топливные элементы работают при низких
температурах, то содержание СО должно поддерживаться на уровне нескольких миллионных долей (рис. 8.32).
Рис. 8.32. Потенциал топливного
элемента с протонопроводящей
мембраной как функция плотности тока для различных значений
содержания СО в подаваемом водороде. Отметьте быстрое падение
потенциала на начальном участке
даже в случае чистого водорода. Это
падение связано с перенапряжением в системе, а более медленное
падение обусловлено внутренним
сопротивлением элемента [40]
Катализатор, находящийся на аноде, может быть сделан менее чувствительным к отравлению СО за счет использования сплавов платины с другими
металлами типа рутения, сурьмы или олова [38]. Таким образом, имеется очевидная потребность в лучших и более дешевых катализаторах. Другой способ
решения проблемы СО состоит в использовании протонопроводящих мембран, работающих при высоких температурах, когда идет десорбция СО. Такие
мембраны разработаны, но их промышленное производство не налажено.
Третий способ решения этой проблемы состоит в каталитическом удалении
СО из водорода. При этом требуется подключать процессы, отличные от рассмотренных при обсуждении синтеза аммиака, где СО удалялся путем его конверсии в метан. Такой процесс легко вписывается в крупнотоннажное производство, но его трудно совместить с компактными генераторами водорода, на¬
8.7. «Водородное сообщество» -J\r 389
ходящимися, например, на автомобиле. Возможно селективное окисление СО
кислородом в присутствии водорода, если рабочая температура будет достаточно низкой для предотвращения взаимодействия сформировавшегося С02 с Н2
в обратной реакции конверсии водяного газа. Катализаторы на основе золота
позволяют успешно провести эту процедуру при температурах, сравнимых с
температурой работы топливного элемента.
Выглядит интригующим то обстоятельство, что инертные в обычных условиях частицы золота становятся каталитически активными, когда их размер
достигает 1—3 нм. Пионерские работы Харуты [39] позволили установить, что
малые частицы золота катализируют окисление СО даже при температурах ниже
комнатной. Поскольку окисление СО идет быстрее, чем окисление водорода,
то данная реакция позволяет удерживать потери водорода на приемлемо низком уровне.
Топливные элементы наиболее эффективно работают на чистом водороде.
Его можно получать из разных топлив, причем метанол и бензин не являются
безоговорочными лидерами. Для получения водорода, не содержащего СО,
необходимо проводить риформинг или частичное каталитическое окисление
топлив. Альтернативный подход состоит в использовании топлив, в состав которых не входит углерод, например, аммиака.
В табл. 8.9 приведены данные по энергоемкости различных энергоносителей. Безопасность в использовании и простота в обращении — это наиболее
важные факторы, определяющие выбор того или иного энергоносителя. Традиционное топливо типа бензина, рассматриваемое в качестве источника водорода для автомобильного транспорта, имеет определенные преимущества, связанные с уже хорошо развитой инфраструктурой.
Таблица 8.9. Энергосодержание для различных соединений,
рассматриваемых в качестве перспективных энергоносителей
Соединение
Н2 (жидк.)
MgH2
сн3он
NH3
CO(NH2)2
Бензин
Mw, г/моль
2
26,3
32
17
60
—
AG\ кДж/моль
-228
-228
-684
-326
-654
—
г, г/мл
0,0708
1,45
0,79
0,68
1,3
—
AG°, кДж/мл
8,9
12,6а
16,9
13,1
14,1
25—30б
а Без учета затрат энергии на выделение водорода из гидрида.
6 Бензин представляет собой смесь углеводородов, поэтому энергоемкость может изменяться в некотором интервале.
Следует заметить, что топливные элементы с протонопроводящими мембранами могут работать непосредственно на метаноле. В реальности, однако,
при этом могут возникнуть проблемы, связанные, как отмечалось выше, с высокой степенью покрытия поверхности катализатора, а также с переносом ме¬
390 -l\r Глава 8. Практика гетерогенного катализа: водород
танола через мембрану. Тем не менее можно создать топливный элемент на
метаноле, обеспечивающий напряжение 0,4 В при приемлемом токе, который
можно использовать в малых переносных устройствах типа ноутбуков и мобильных телефонов, сделав их независимыми от традиционных источников
энергии. Более подробную информацию по топливным элементам читатель
может найти в обзорной статье [41].
8.7.3.2. Топливные элементы на твердых оксидах
В топливных элементах на твердых оксидах (ТЭТО) через электролитную мембрану проходит ион О2-. Для обеспечения значительной подвижности ионов требуется высокие рабочие температуры (700—800 °С). Это делает
ТЭТО практически не зависящим от природы используемого топлива, но
предъявляет определенные требования к поведению материалов, используемых
в качестве анода, например, Ni (наиболее предпочтителен), Pd, Pt и Со, при
взаимодействии с Н2 и Н20 при высоких температурах. Металл электродов
обычно диспергируется на поверхности твердого электролита, в качестве которого обычно используется стабилизированный иттрием оксид циркония. Многие фундаментальные проблемы, связанные с технологией создания ТЭТО, сходны
с встречающимися в катализе: скорость адсорбции, диссоциация, окисление
металла и восстановление оксида [42] и диффузия металла в электролит. Например, степень покрытия поверхности реагентами в рабочих условиях и влияние восстанавливающего потенциала на структуру и морфологию материала
анода до сих пор, по существу, остаются невыясненными. Эта информация
необходима для конструирования ТЭТО нового поколения, работающих при
низких температурах.
В то время как топливные элементы с ионообменными мембранами наиболее подходят для эксплуатации в подвижных и переносных устройствах, ТЭТО
удобнее использовать на стационарных объектах. Высокая рабочая температура делает их гибкими в отношении используемого топлива, в частности, позволяет использовать метанол. Выделяемое в ТЭТО тепло может быть также использовано для производства электрической энергии. По этой причине эффективность ТЭТО составляет около 60 %, в то время как для топливных элементов
с протонопроводящими мембранами даже в оптимальных условиях эта величина достигает лишь 45 %.
8.7.3.3. Эффективность топливных элементов
Топливный элемент превращает химическую энергию непосредственно в электрическую; такое превращение по своей природе является высокоэффективным процессом. Так что термодинамически достижимая эффективность составляет около 100 %. Эффективность реального топливного элемента определяется выражением
8.7. «Водородное сообщество»
в то время как эффективность в цикле Карно равна
X
<8'79>
Здесь Тс — температура холодного резервуара, то есть окружающей среды;
Th — температура процесса, то есть температура пламени. Формула (8.79) определяет эффективность обычных тепловых машин. Эффективность более 100 %,
показанная в табл. 8.10, соответствует ситуации, когда тепло окружающей среды напрямую преобразуется в электрическую энергию, что представляет лишь
академический интерес.
Таблица 8.10. Теоретические значения эффективности превращения
водорода, метана и метанола в электрическую энергию
в топливном элементе
Температура/Топливо
н2
сн4
СН3ОН
300
0,94
1,00
1,02
1000
0,78
1,00
1,08
На практике ситуация выглядит хуже, поскольку имеются потери, связанные с перенапряжением в элементе и резистивными потерями в мембране.
Перенапряжение — электрохимический термин, который, по своей сути, можно трактовать как обычный потенциальный барьер для реакции. В результате
эффективность топливных элементов на практике составляет 40—60 %. Для
сравнения, у современных турбин, используемых для генерации электрического тока, она достигает 50 %. Однако следует заметить, что эффективность тепловых машин ограничена по термодинамическим соображениям, в то время
как в топливных элементах она определяется свойствами материалов и потому
может быть повышена.
(—
о
о
Рис. 8.33. Схематическое сопоставление g
автомобилей с топливными элемента- g
ми и обычных автомобилей с двигате- ш
лями внутреннего сгорания. Отметьте, ^
что автомобили с топливными элемен- СО
тами имеют преимущества лишь при
малых нагрузках, типичных для городских условий
Автомобили с топливными элементами
Нагрузка
391
Топливные элементы как источники электрической энергии не обладают
существенно лучшей эффективностью в сравнении с тепловыми турбинами, но
они предпочтительнее двигателей внутреннего сгорания автомобилей. Послед¬
392 -l\r Глава 8. Практика гетерогенного катализа: водород
ние имеют наивысшую эффективность при очень большой частоте вращения
(до 3000 оборотов в минуту). Однако такой режим их работы достигается только на скоростных магистралях. И в городских условиях двигатель автомобилей
оказывается неэффективным. Автомобиль, стоящий перед светофором при работающем двигателе, затрачивает впустую большое количество энергии. И здесь
топливные элементы обладают явным преимуществом, поскольку генерируют
энергию только при возникновении соответствующей необходимости. Более
предпочтительная общая эффективность, отсутствие токсичных выбросов и
низкий уровень шума — основные причины выхода топливных элементов на
автомобильный рынок. На рис. 8.33 проведено сравнение эффективности автомобилей на водородном топливе, конвертируемом в электрическую энергию в
топливном элементе, и автомобилей, работающих на бензине.
8.7.3.4. Хранение и транспортировка водорода
Водород обладает высоким энергосодержанием на единицу массы
(142 МДж/кг) по сравнению с обычным углеводородным топливом, для которого оно составляет 47 МДж/кг. Однако водород представляет собой газ с очень
низкой плотностью. В стандартных условиях 1 кг водорода занимает объем 12 м3.
Перевод водорода в конденсированное состояние позволяет повысить его плотность до 71 кг/м3, но здесь возникают проблемы, связанные с необходимостью
работы с криогенными жидкостями. Рассмотрим вопрос о хранении водорода в
топливном баке при высоких давлениях. Автомобиль средних размеров при
экономичном двигателе может проехать 600 км, затратив 40 л бензина. Предположим, что аналогичная машина сконструирована с использованием технологии топливных элементов и имеет в два раза более высокую эффективность
(что является довольно оптимистическим прогнозом). Хранение 5 кг водорода,
необходимого для преодоления расстояния в 600 км, в топливном баке объемом 40 л возможно лишь при давлении 1500 бар!
Департамент Энергии США нацелен на поиск способа хранения водорода,
при котором один килограмм водорода занимал бы не больше 16 л и содержание водорода составляло бы не меньше 6,5 вес. %. Для рассмотренного выше
примера «хранилище» для 5 кг водорода тогда бы весило 77 кг и занимало
объем 80 л. В табл. 8.11 перечислены возможные кандидаты носителей энергии, которые можно использовать для генерации водорода непосредственно на
транспортном средстве. Все перечисленные кандидаты удовлетворяют требованиям, предъявляемым Департаментом Энергии. Наконец, прекрасным средством хранения водорода были бы гидриды металлов, но для развития этой
технологии потребуется еще немало лет. Например, MgH2 содержит 7,6 вес. %
водорода, то есть удовлетворяет требованиям Департамента Энергии. Проблема, однако, состоит в том, что водород не высвобождается при температуре
ниже 300 °С, то есть гидриды металлов слишком устойчивы, и потребуется еще
немало сил, чтобы гидриды металлов можно было эксплуатировать при температуре окружающей среды, а водород выделялся при давлении в несколько бар.
Список литературы -*\г 393
Прекрасный обзор работ, нацеленных на решение этой проблемы, дан в [43].
Таким образом, бензин является очевидным кандидатом на роль главного носителя энергии в первые дни существования водородного сообщества.
Таблица 8.11. Содержание водорода и объем, приходящийся на 1 кг
водорода, в потенциальных носителях энергии. Заметьте,
что при конверсии соединений предполагается использовать
воду для обеспечения максимального выхода водорода
Соединение
СН3ОН
NH3
(NH2)2co
C*HIS
Водород, вес. %
18,7
17,6
10,0
43,8
Удельный объем,
л/кг водорода
6,7
7,6
7,6
3,3
Список литературы
1. Catalysis — Science and Technology // Eds. Rostrup-Nielsen J., Anderson J.R.,
Boudart M. Berlin: Springer, 1984.
2. Holmblad P.M., Wambach J., Chorkendorf I. // J. Chem. Phys. 1995. V. 102.
P. 8255.
3. Goodman D.W. // Acc. Chem. Res. 1984. V. 17. P. 194.
4. Bengaard H., N0rskov J.K, SehestedJ., Clausen B.S., Nielsen L.P., Molenbroek A.M.,
Rostrup-Nielsen J. // J. Catal. 2002. V. 209. P. 365.
5. Rostrup-Nielsen J.R. // J. Catal 1984. V. 85. P. 31.
6. Sinfeld J.H. Bimetallic Catalysts. New York: John Wiley and Sones, 1983.
7. Nielsen L.P., Besenbacher F., Stensgaard I., Lsegsgaard E., Engdahl C., Stoltze P.,
Jacobsen K.W., N0rskov J.K. // Phys. Rev. Lett. 1993. V. 71. P. 754.
8. Jakobsen J., Nielsen L.P., Besenbacher F'., Stensgaard /., L&gsgaard E.,
Rasmussen Т., Jacobsen K.W., N0rskov J.K. // Phys. Rev. Lett. 1995. V. 75. P. 489.
9. Besenbacher F., Chorkendorf I., Clausen B.S., Hammer B., Molenbroek A.M.,
N0rskov J.K, Stensgaard I. // Science. 1998. V. 279. P. 1913.
10. Rod Т.Н., N0rskov J.K // Surf. Sci. 2002. V. 500. P. 678.
11. Hickman D.A., Schmidt L.D. // Science. 1993. V. 259. P. 343.
12. Chinchen G.C., Denny P.J., Parker D.G., Spencer M.C., Waugh КС., Whan D.A. //
Appl. Catal. 1987. V. 30. P. 333.
13. Rasmussen P.B., Kazuta М., Chorkendorf I. // Surf. Sci. 1994. V. 318. P. 267.
14. Rasmussen P.B., Holmblad P.M., Askgaard Т., Ovesen С. V, Stoltze P., N0rskov J.K,
Chorkendorf I. // Catal. Lett. 1994. V. 26. P. 373.
15. Nakano H., Nakamura I., Fujitani Т., Nakamura J. // J. Phys. Chem. B. 2001.
V. 105. P. 1355.
16. Yoshihara K, Campbell C.T. // J. Catal. 1996. V. 161. P. 776.
17. Clausen B.S., Schi0zJGrabxk L., Ovesen C.V., Jacobsen K.W., N0rskov J.K,
Tops0e H. 11 Top. Catal. 1994. V. 1. P. 367.
394 -l\r Глава 8. Практика гетерогенного катализа: водород
18. Ovesen C.V., Clausen B.S., Schi0tz Stoltze P., Tops0e N0rskov J.K. //
J. Catal. 1997. V. 168. P. 133.
19. Tops0e H., Ovesen C.V, Clausen B.S., Tops0e N. X, Nielsen P.E.H., T0rnqvist E.,
N0rskov J.K In: Dynamics of Surfaces and Reaction in Heterogeneous Catalysis /
Eds. Froment G.F., Waugh K.C. Amsterdam: Elsevier, 1997. P. 121.
20. Nakamura J., Nakamura I., Uchijama Т., Kanai Y., Watanabe Т., М.,
Fujitani T. // J. Catal. 1996. V. 160. P. 65).
21. Berlowitz P.J., Goodman D.W. // J. Catal 1987. V. 108. P. 364.
22. Fujila F., Anthony R.G., Lunsford J. // J. Catal. 1982. V. 73. P. 237.
23. Maak М., Friis-Jensen H., Sckerl S., Larsen J.H., Chorkendorf I. // Top. Catal.
2003. V. 22. P. 161.
24. Niemantsverdriet J. W., Louvers S.R.A., Кяя Grondelle J., Кяя Jer Kraan A.M.,
Kampers F.W.H., Konigsberger D.C. In: Proceedings 9th International Congress on
Catalysis. 1988. V. 2. P. 674.
25. Ропес V In: Handbook of Heterogeneous Catalysis / Eds Ertl G., Knozinger H.,
Weitkamp J. Weinheim: VCH-Wiley, 1997. V. 4. P. 1876.
26. Sfe 5.7:, Senden M.M.G., Кял ЖоЛяи tf.Mtf. // Catal. Today. 1991. V. 8. P. 371.
27. M.f. In: Catalysis / Eds. Anderson J.E., Boudant M. Heidelberg: Springer-
Verlag, 1981. V. 1. P. 160.
28. Ovesen C.V, Clausen B.S., Hammersh0j B.S., Sreffensen G., Askgaard Т.,
Chorkendorf I., N0rskov J.K, Rusmussen P.B., Stoltze P., Taylor P.J. // J. Catal. 1996.
V. 158. P. 170.
29. Dybkjsdr /., In: Ammonia: Catalysis and Manufacture / Ed. Nielsen A. Berlin:
Springer, 1995. P. 199.
30. Jacobsen C.J.H., Dahl S., Boisen A., Clausen B.S., Tops0e H., Logadottir A.,
N0rskov J.K. U J. Catal. 2002. V. 205. P. 382.
31. Czuppon T.A., Knez S.A., Schneider R.W., Woroberts G. // Ammonia Plant
Safety Relat. Facil. 1994. V. 34. P. 236.
32. Mortensen J.J., Hansen LB., Hammer B., N0rskov J.K. //J. Catal. 1998. V. 182.
P. 479.
33. Somorjai G.A., Materer M. // Top. Catal. 1994. V. 1. P. 215.
34. Stognin D.R., Somorjai G.A. // J. Catal. 1988. V. 109. P. 51.
35. Egeberg R.C., Dahl S., Logadottir A., Larsen J.H, N0rskov J.K, Chorkendorf I. //
Surf. Sci. 2001. V. 491. P. 183.
36. Bengaard H.S., Alstrup I., Chorkendorf I., Ullmann S., Rostrup-Nielsen J.,
N0rskov J.K. U J. Catal. 1999. V. 187. P. 238.
37. Grove W.R. // Phil. Mag. 1839. V. 14. P. 137.
38. Markovic N.M., Ross P.N. // CATTECH 2001. V. 4. P. 110.
39. Haruta M. // CATTECH. 2002. V. 6. P. 102.
40. Oetjen H.F., Schmidt V.M., Stimming U., Trila F. //J. Electrochem. Soc. 1996.
V. 143. P. 3838.
41. Carrette L., Friedrich K.A., Stimming U. // Fuel Cells. 2001. V. 1. P. 5.
42. KekD., Mogensen М., Pejovnik S. //J. Electrochem. Soc. 2001. V. 148. P. A878.
43. Schlapbach L., Zuttel A. // Nature. 2001. 15 Nov. P. 353.
глава ПЕРЕРАБОТКА НЕФТИ
И НЕФТЕХИМИЯ
9.1. СЫРАЯ НЕФТЬ
Сырая нефть для современного общества является наиболее важным сырьем. Примерно 450 нефтеперерабатывающих заводов во всем мире
получают из сырой нефти транспортное топливо (бензин, дизельное топливо,
керосин), смазочные масла и сырье для других химических производств. Катализаторы играют в этих процессах ключевую роль [1].
На начало III тысячелетия общие запасы нефти оценивались в 1,035 х 1012 баррелей, из которых примерно 2/3 приходится на Средний Восток. Расход нефти
в 1999 г. составил 27 х 109 баррелей; это означает, что имеющихся общих запасов нефти хватит примерно на 40 лет при сохранении нынешнего уровня ее
потребления.
Сырая нефть представляет собой смесь различных углеводородов, молекулярный вес которых достигает 2000, температура их кипения изменяется в
широких пределах — примерно от 10 до 600 °С, также в широких пределах
лежит отношение чисел атомов водорода и углерода (примерно от 3 для
низших углеводородов до 0,5 для длинноцепочечных молекул). Сырая нефть
содержит алканы (парафины), алкены (олефины) и ароматические соединения, в состав многих из них входит сера (0—3 %), азот (0—0,5 %), кислород,
ванадий и никель. Чем тяжелее молекулы, тем больше в них находится этих
нежелательных элементов. Состав сырой нефти существенным образом изменяется от одного месторождения к другому. Например, содержание бензиновой фракции в нефти, добытой в Северном море, составляет 20 % и практически равно нулю в сырой нефти, добываемой в Южной Америке. В табл. 9.1
приведен средний состав сырой нефти и основные характеристики разных
фракций.
Современный нефтеперерабатывающий завод представляет собой набор
конверсионных установок, каждая из которых нацелена на производство определенного сырья. На рис. 9.1 схематически показаны основные операции,
суть которых раскрывается частично в табл. 9.2, где представлены и соответствующие катализаторы. Прежде всего нефть перегоняют для разделения
ее на фракции — газы, жидкости (бензин, керосин и газойль) и тяжелые
фракции (так называемое «дно бочки»), остающиеся после вакуумной перегонки.
396
Глава 9. Переработка нефти и нефтехимия
Таблица 9.1. Характеристики «средней» сырой нефти
Фракция
Температура
кипения, °С
Объемный
%
Плотность,
кг/л
Содержание
серы, вес. %
Газ
—
1,5
0,5-0,6
0
Легкий бензин (<С5)
<80
6
0,66
0
Тяжелый бензин (С5—С10)
80-170
15
0,74
0,02
Керосин
170-220
9
0,79
0,1
Газойль
220-360
25
0,83-0,87
0,8
Тяжелый газойль (С20—С40)
360-530
23
0,92
1,4
Остатки (>С40)
>530
20
1,02
2,2
Рис. 9.1. Упрощенная схема переработки нефти [2]
Газофазные фракции вместе с побочными продуктами процессов крекинга
продаются как сжиженный газ, который используется в некоторых странах в
быту или как топливо для автомобилей. Все фракции, за исключением газовой,
подвергаются переработке с целью повышения их качества. С помощью гидроочистки из них удаляют гетероатомы типа серы и азота, а у бензиновых фракций повышают октановое число, то есть их детонационную стойкость.
9.1. Сырая нефть ->\г 397
Таблица 9.2. Основные элементы переработки нефти
Процесс
Цель процесса
Катализатор процесса
Г идро-
очистка
Удаление гетероатомов (S, N, О, атомы металлов)
—
Г идро-
обессе-
ривание
Удаление атомов серы, на выходе — H2S
Со—MoS2/A1203,
Ni—WS2/ А1203
Деазоти¬
рование
Удаление азота — на выходе NH3
Ni—MoS2/A1203
Деоксиге-
нирование
Удаление кислорода — на выходе Н20
Co—MoS2/A1203,
Ni—MoS2/A1203
Деарома¬
тизация
Удаление ароматических соединений
Ni—WS2/A1203
Де металлизация
Удаление металла в виде сульфидов металлов
Ni—MoS2/A1203
(скорее адсорбент,
чем катализатор)
Рифор¬
минг
Конверсия бензиновой фракции в высококачественный бензин (с высоким октановым
числом). В качестве побочного продукта получается водород, использующийся для гидроочистки, гидрокрекинга и конверсии остатков
Pt—Re/Al203,
Pt-Ir/Al203
Г идро-
крекинг
Конверсия газойля в транспортное топливо
действием водорода в сочетании с гидроочисткой и гидрированием в присутствии
бифункциональных катализаторов, в том
числе и кислотных
Ni-MO, Ni-W,
Pt-Pd,
кислотный носитель
(оксид алюминия,
цеолит)
Мягкий
крекинг
Крекинг при низком давлении
—
Депарафи-
низация
Селективный гидрокрекинг парафинов из
фракции газойля или других продуктов для
предотвращения осаждения парафина (оказывает плохое влияние на течение охлажденных потоков жидкости)
—
Каталити¬
ческий
крекинг
Конверсия тяжелого газойля в транспортное топливо
Цеолит Y
Конверсия
остатков
Конверсия остатков (действием водорода) в
мазут с низким содержанием серы и исходный материал для каталитического и гидрокрекинга
—
398 -J\r Глава 9. Переработка нефти и нефтехимия
Облагораживание тяжелых остатков составляет важную часть переработки
нефти. Эти фракции обычно содержат большое число примесей и используются в производстве асфальта. В настоящее время гидроочистка и гидрокрекинг
или каталитический крекинг жидкостей используются для конверсии тяжелых
фракций с малым содержанием S и N в легкие фракции, которые затем используются в качестве транспортного топлива, в частности бензина. В этой
крупномасштабной операции даже небольшие усовершенствования приводят к
заметному экономическому эффекту. Это иллюстрирует следующий пример:
для США увеличение выхода бензина за счет переработки тяжелых остатков на
1 % эквивалентно сокращению импорта нефти на 20 млн баррелей в год!
Добавим также, что помимо транспортного топлива, являющимся основным
продуктом переработки нефти, в этом процессе получают и исходное сырье для
других химических производств — этилен, пропилен, бутилены. Мы рассмотрим
некоторые из этих производств ниже при более глубоком анализе нефтепереработки. Объемы переработки нефти в разных странах даны в табл. 9.3
Таблица 9.3. Объемы переработки нефти [3]
Страны
Тысячи баррелей вдень
1989
1994
1999
1999,
доля,%
США
13 400
13 865
14 805
21,9
Канада
1550
1580
1720
2,5
Мексика
1420
1460
1400
2,1
Южная и Центральная
Америка
4525
4855
5640
8,3
Европа
14015
14 370
14 755
21,8
Страны бывшего СССР
9545
5075
4515
6,7
Средний Восток
4565
5225
5980
8,8
Африка
2150
2220
2405
3,6
Австралия
700
785
880
1,3
Китай
2115
2550
3125
4,6
Япония
3175
4165
4150
6,1
Другие азиатские страны
тихоокеанского побережья
4635
6315
8360
12,3
Весь мир
61 795
62 465
67735
100,0
9.2. Гидроочистка -i\r 399
9.2. ГИДРООЧИСТКА
Как указано на рис. 9.1, гидроочистка используется для удаления
нежелательных элементов и компонентов из различных фракций нефти, то есть
представляет ключевую операцию в ее переработке. Различие между гидроочисткой и гидрокрекингом состоит в следующем: хотя в обоих случаях из нефти
удаляются сера, азот и металлы и идет гидрирование соединений, при гидроочистке молекулярный вес углеводородных молекул изменяется незначительно
(то есть вследствие удаления атомов S, N или О и внедрения атомов Н), а при
гидрокрекинге молекулярный вес уменьшается в два раза и более. Гидроочистка проводится в более мягких условиях (обычно при давлении 30 бар и температуре 350 °С), чем гидрокрекинг (обычно 100 бар и 400 °С). Гидрокрекинг в
мягких условиях также иногда используют при обработке нефти, поскольку его
можно осуществить на имеющихся аппаратах гидроочистки, но при этом более
легкие продукты никогда не получаются [4].
В течение многих лет роль гидроочистки в переработке нефти все более
возрастает и предполагается, что эта тенденция сохранится и в будущем. Это
обусловлено следующими причинами:
• легкие сырые нефти (которые содержат мало примесей) становятся все
менее доступными;
• идет все более интенсивное использование тяжелой нефти («дно бочки»);
• устанавливаются все более жесткие стандарты на выбросы от транспортных средств, для обеспечения которых необходимо использовать топливо с
более низким содержанием, например, серы (ниже 10 миллионных долей).
В настоящее время, основываясь на объемах перерабатываемого материала,
можно утверждать, что гидроочистка представляет собой наиболее крупномасштабный процесс в гетерогенном катализе. По объемам продаж катализаторов
гидроочистка идет на третьем месте после каталитической обработки выхлопных газов и каталитического крекинга жидкостей [4].
9.2.1. Гетероатомы и нежелательные элементы
Сера в сырой нефти присутствует в основном в органических соединениях типа меркаптанов (R—SH), сульфидов (R—S—R') и дисульфидов
(R—S—S—R'), из которых она достаточно легко может быть удалена, а также
в тиофенах и их производных (рис. 9.2). Для обессеривания последних требуются более жестки условия, в частности использование дибензотиофенов, подобных показанным на рис. 9.2. Сера обязательно должна быть удалена из
нефти, поскольку при ее сжигании образуется серная кислота и она отравляет
катализаторы, использующиеся при нефтепереработке и дожигании выхлопных газов (в частности, в автомобилях с дизельными двигателями). Кроме того,
соединения серы в топливе способствует развитию коррозии и придают ему
неприятный запах.
400 -J\r Глава 9. Переработка нефти и нефтехимия
Ди метилдибензотиофен
Пиррол Пиридин
Индол Хинолин
Карбазол Акридин
Рис. 9.2. Типичные серу- и азотсодержащие соединения, входящие в состав сырой нефти
К числу азотсодержащих молекул относятся легко разложимые амины
(R—NH2) и более устойчивые ароматические соединения типа пиррола, пятизвенного кольца, или пиридина, шестизвенного кольца, а также их высших
производных. Молекулы, содержащие азот, отравляют кислотные катализаторы, используемые в реакциях крекинга и риформинга, а при их сжигании образуются окислы N0^.
Кислород входит в состав нафтеновых кислот, фенолов и фурана (структурный аналог тиофена и пиррола) и их высших производных. Соединения кислорода способствуют развитию коррозии и порче продуктов. Деоксигенирование
необходимо, в частности, для повышения качества биомассы.
Ненасыщенные углеводороды типа олефинов, хотя они и отсутствуют в
сырой нефти, образуются в процессе крекинга. Их присутствие нежелательно,
поскольку они объединяются в более крупные молекулы и коксуются на кислотных центрах. Олефины легко гидрируются в условиях гидроочистки, хотя
H2S и другие соединения серы замедляют эту реакцию. Полициклические ароматические соединения являются прекурсорами кокса (рис. 9.3), деактивирующего катализаторы риформинга и крекинга.
9.2. Гидроочистка -i\r 401
Рис. 9.3. Косом называют углеродные осадки,
формирующиеся на поверхности катализатора в процессе обработки углеводородов. Он
образован из ароматических соединений с
низким отношением Н:С. Показанный на рисунке графит представляет собой предельную
форму кокса. Кокс можно удалить путем его
окисления. Иногда выделяющееся при сжигании кокса тепло используется для активации других процессов
* Vw.
А А
#
Из металлических примесей наиболее нежелательными являются никель и
ванадий, присутствующие в порфириновых структурах, образующихся в растениях; они входят в основном в тяжелые остатки. Кроме того, в нефтепродуктах
может содержаться железо как продукт коррозии емкостей для хранения и транспортировки нефти. Все эти металлы могут адсорбироваться на катализаторах и
вызвать (особенно никель) усиленное осаждение углерода. Ванадий разрушает
решетку цеолитов, используемых в каталитическом крекинге жидкостей. В перерабатываемых нефтях могут присутствовать и другие элементы. Строго говоря, деметаллизация не является каталитическим процессом, поскольку металл
остается в виде сульфидов на поверхности катализатора. Разложение порфириновых структур протекает быстро и, как правило, происходит на фронтальной
части каталитического слоя на внешней поверхности каталитических частиц.
9.2.2. Катализаторы в гидроочистке
ляют собой дисульфид молибдена MoS2, промотированный кобальтом или никелем и нанесенный на ^-оксид алюминия. В качестве альтернативы может
использоваться Ni—WS2/y—А1203, но так как вольфрам стоит существенно дороже молибдена, то этот катализатор используется для проведения гидроочистки в более жестких условиях, например при гидрокрекинге. С помощью катализаторов из благородных металлов на основе платины и палладия проводится
гидрирование термически стабильных молекул. Как правило, для деазотирования и деоксигенирования используются катализаторы, промотированные Ni, а
катализаторы системы CoMoS более предпочтительны при обессеривании. Мы
рассмотрим здесь систему CoMoS, а поведение катализаторов NiMoS и NiWS
во многом будет подобным.
Благодаря большому объему исследований, выполненных за последние двадцать лет, сульфидированные катализаторы Со—Мо/А1203 в настоящее время
являются наиболее изученной системой [5]. Комбинирование методов мессбауэровской спектроскопии, спектроскопии дальней тонкой структуры рентгеновского поглощения, рентгеноэлектронной спектроскопии и инфракрасной
Использующиеся в гидроочистке катализаторы обычно представ-
402 -*\r Глава 9. Переработка нефти и нефтехимия
спектроскопии позволяет получить настолько полную информацию, что активные центры этого катализатора уже описаны на атомном уровне.
Этот катализатор получают пропиткой носителя из оксида алюминия водными растворами гептамолибдата аммония и нитрата кобальта с последующими сушкой и термической обработкой. Формирующийся оксидный катализатор переводится в сульфидную форму путем его обработки в атмосфере H2S и
Н2 или другой серусодержащей среде. Приведенные на рис. 9.4 рентгеноэлектронные спектры катализатора СоМо/А1203, полученные на разных стадиях его
формирования в процессе температурно-программируемого сульфидирования,
явно показывают превращение оксида в сульфид. Относящийся к сере пик в
спектре при 225 эВ указывает на захват серы катализатором уже при низких
температурах, и его появление связано с восстановлением Мо6+, входящего в
оксид Мо03, до Мо5+ в смешенном окружении кислорода и серы. Переход в
MoS2 происходит при температурах выше 300 °С. Кобальт в состояние сульфида трансформируется при температуре около 150 °С более или менее одновременно с образованием MoS2. Взаимодействие промотирующих элементов с
носителем из оксида алюминия играет важную роль. На носителях из оксида
кремния или углерода кобальт сульфидируется уже при низких температурах с
образованием Co8S8, который не является эффективным промотором для реакции гидрообессеривания [7].
Энергия связи, эВ
Рис. 9.4. Рентгеноэлектронные спектры (РЭС) катализатора СоМо/А1203, демонстрирующие переход от оксида к сульфиду [6]
Дисульфид молибдена имеет слоистую структуру. Каждый слой представляет собой сэндвич, образованный слоем Мо4+, заключенным между двумя слоями ионов S2- (рис. 9.5). Ионы серы образуют треугольные призмы, и половина этих призм содержит в центральной части ион молибдена. Химическая активность MoS2 определяется краями сэндвича, тогда так базальные плоскости
характеризуются низкой активностью. На этих краях находятся центры адсорбции газа и «проживает» каталитическая активность.
9.2. Гидроочистка ->\r 403
вид
сверху
С
I
Треуголная призма
Фаза СО/с 8
^Щ
Со. О О 'А
О
О.: В О
Рис. 9.5. Схематическое изображение катализатора CoMoS, а также вид сверху структуры
MoS2, построенной из треугольных призм. Кобальт может находиться в трех
состояниях: в активных соединениях CoMoS, где Со декорирует края сэндвичей
MoS2; в низкоактивных кристаллитах Co9S8; в виде ионов Со2+, внедренных в
матрицу носителя А1203
Обширные спектроскопические исследования показали, что промотирующие атомы кобальта также локализуются на краях сэндвича, в тех же местах,
где находятся атомы молибдена. Их роль сводится, скорее всего, к формированию вакансий атомов серы, которые и представляют собой активные центры
для реакции гидрообессеривания. На рис. 9.5 схематически показано, как выглядит рабочий катализатор гидрообессеривания Со—Мо/А1203. Он образован
частицами MoS2 размером в несколько нанометров; частицы декорированы
кобальтом, участвующим в формировании центров Со—Мо—S, обладающих
высокой каталитической активностью. В катализаторе присутствуют ионы кобальта, связанные с носителем из оксида алюминия, он также содержит кристаллиты устойчивого сульфида Co9S8, не обладающие каталитической активностью по отношению к реакции гидрообессеривания [5].
9.2.3. Механизмы реакций гидрообессеривания
Предполагают, что каталитические центры, ответственные за реакции гидроочистки, представляют собой вакансии атомов серы на краях слоев
MoS7 и WS2, как это показано на рис. 9.6. Следовательно, каталитическая активность зависит не только от той легкости, с которой гетероатом «сдирается»
с молекулы углеводорода, но и от скорости удаления гетероатома, обеспечивающего образование вакансии, с поверхности катализатора. Было показано, что
404
Глава 9. Переработка нефти и нефтехимия
каталитическая активность сульфидов переходных металлов коррелирует с прочностью связи сера—металл. Такая корреляция приводит к «вулканической» зависимости активности катализатора от прочности связи (рис. 9.7). Среди не-
промотированных катализаторов наибольшей активностью по отношению к
обессериванию обладают сульфиды рутения, осмия, иридия и родия. Но ни
один из них нельзя признать пригодным для использования на практике. Несмотря на большое число проведенных теоретических исследований, до сих
пор остается неясным, почему промотированные Со и Ni катализаторы MoS2
обладают высокой активностью. Объяснения включают и ослабление промоторами связи металл—сера, и образование уникального соединения сферы с молибденом и промотором.
€2Н55Н
Рис. 9.6. Схематическое представление каталитического цикла гидрообессеривания серусодержащих
углеводородов (этантиол) на вакансии серы в MoS2: молекула C2H5SH
адсорбируется, ориентируясь своим
атомом серы к молибдену. Затем
С?Н, связь С—S разрывается, и за счет
**Мо52'
удаления атома /3-Н образуется этилен, который десорбируется в газовую фазу. Каталитический центр
восстанавливается при образовании
и удалении H^S
\
'47
H2S
10001
RuS2
<
о
0
1
со
S
J 100 -
10-
1 -
MnS
Рис. 9.7. Активность переходных металлов по 0,1
отношению к реакции обессеривания подчиняется принципу Сабатье (см. подраздел 6.5.3.5).
Кривая имеет форму вулкана [8, 9]
20 30 40 50 60 70 80
Энергия связи серы с металлом,
кКал/моль
С механистической точки зрения гидроочистка циклических молекул типа
тиофена и родственных ему соединений включает гидрирование ненасыщенных колец с последующим разрывом связи между гетероатомом и соседними
9.2. Гидроочистка -*\r 405
атомами углерода. На рис. 9.8 этот процесс проиллюстрирован на примере
гидрообессеривания тиофена.
Тиофен
Тетрагидротиофен
H..S
2- бутен
^ *
бутен
Бутадиен
Рис. 9.8. Общая схема реакции для обессеривания тиофена, в которой первая стадия
включает гидрирование ненасыщенных колец, за которым следует разрыв связи
С—S, осуществляемый в два этапа. Предполагается, что первым продуктом, не
содержащим серу, является бутадиен, хотя на практике наиболее часто образуется 1-бутен. При дальнейшем гидрировании формируются изомеры 2-бутена [4]
Кинетика реакции, как отмечалось в гл. 7, описывается в рамках менее
детальной схемы, в которой переход от адсорбированного тиофена к первой не
содержащей серу углеводородной молекуле, бутадиену, осуществляется в одну
ступень. Выражение для скорости реакции имеет вид:
л NAKtKv Р'тРхЛ
г = ЫьЩвЪ = 2 2 ... ,„ч2. (91)
(l + ^ + AT^sJJl + AT^s)'
где /?т, рн и рИ s — парциальные давления тиофена, водорода и сероводорода
соответственно; к — константа скорости для стадии обессеривания тиофена и
превращения его в бутадиен; К{ — соответствующие константы равновесия;
NA — полное число вакансий серы на крае слоя MoS2, где может адсорбироваться тиофен; водород, как предполагалось, может адсорбироваться в любом
месте на крае слоя MoS2.
Принималось, что водород слабо связан с поверхностью, поэтому соответствующий вклад в знаменателе (9.1) опущен. Из выражения для скорости реакции видно, что реакция тормозится H2S. Следовательно, наиболее активные
катализаторы (находящиеся на вершине «вулкана» на рис. 9.7) должны:
• обеспечивать легкое высвобождение серы;
• быть нечувствительными к ингибированию H2S;
• сильно связывать тиофен, чтобы он легко гидрировался и обессеривался.
406
—Глава 9. Переработка нефти и нефтехимия
С ужесточением ограничений по примесям в транспортных топливах и стандартов по выбросам (табл. 9.4) роль гидроочистки возрастает с каждым днем.
Таблица 9.4. Содержание серы (миллионных долей по весу) в бензине
Годы
1994
1995
2000
2005
2010
Европа
1000
500
150
50
10?
США
—
—
—
30
10?
9.3. ПРОИЗВОДСТВО БЕНЗИНА
Бензин, продаваемый на заправочных станциях, получается на нефтеперерабатывающих заводах из различных продуктов. Как показано на рис. 9.9,
только одна треть состава бензина является продуктом каталитического крекинга. Этот продукт богат содержанием олефинов, повышающих октановое
число бензина. Однако продукты каталитического крекинга несут на себе основное содержание серы в бензине (более 95 %). Обессеривание этих продуктов возможно, но при этом гидрируются олефины, что приводит к снижению
октанового числа. Таким образом, основной задачей в переработке нефти является создание катализаторов гидрообессеривания, обладающих минимальной активностью по отношению к гидрированию углеводородной фракции,
составляющей основной компонент бензина.
7%
1 %
4%
6%
7%
11%
15%
17%
32%
Другие фракции
Бутан
Изомеризаты (ЛОЧ = 85)
Продукты гидрокрекинга
Прямогонный бензин (68)
Метил-трет-бутиловый эфир (116)
Алкилаты (95)
Продукты риформинга (99)
Продукты каталитического крекинга (93—95)
Рис. 9.9. Состав бензина и лабораторное октановое число
(ЛОЧ) для компонентов. Шкала октановых чисел устанавливается по //-пентану (ЛОЧ = 0) и 2,2,4-триметил-
пентану (ЛОЧ = 100). Значение ЛОЧ для различных фракций равно процентному содержанию 2,2,3-триметилпентана в его смеси с я-пентаном, имеющей те же детонационные характеристики, что и исследуемая фракция
9.3. Производство бензина -J\r 407
Другие составляющие бензина являются продуктами каталитического риформинга и алкилирования или насыщенными кислородом добавками (метил-
трет-бутиловый эфир), повышающими октановое число.
Мы кратко обсудим процессы, определяющие примерно 75 % состава произведенного при переработке нефти бензина.
Октановое и цетановое числа
Октановое число служит мерой детонационной стойкости бензина.
В карбюраторных двигателях воспламенение топлива инициируется искрой, зажигающейся в свечи, при этом стараются избежать самопроизвольного воспламенения (детонации) топлива. Шкала октановых чисел
устанавливается по детонационной стойкости //-пентана (ЛОЧ = 0) и
2,2,4-триметилпентана (изооктана, ЛОЧ = 100). Лабораторное октановое
число (ЛОЧ) и моторное октановое число отличаются вследствие различия
условий, при которых проводятся соответствующие измерения (скорость
вращения двигателя, температура и время горения искры). Октановое число топлива равно процентному содержанию изооктана в его смеси с я-пента-
ном, имеющей те же детонационные характеристики, что и топливо.
Характеристики топлива для инжекторных двигателей с октановым
числом выше 100 определяются из сравнения со стандартными топливами, содержащими добавки, повышающее октановое число.
Цетановое число служит мерой быстроты воспламенения дизельного
топлива. Воспламенение топлива в дизельном двигателе должно протекать быстро после его инжекции в горячий сжатый воздух. Дизельное топливо тестируется путем его сравнения со смесью цетана (гексадекана,
быстрое воспламенение) и а-метилнафталина (медленное воспламенение).
Цетановое число равно процентному содержанию цетана в смеси, имеющей ту же скорость воспламенения, что и тестируемое топливо. Следовательно, в отличие от бензина хорошее дизельное топливо должно содержать в большой доле линейные алканы и мало ароматических соединений, как это имеет место в прямогонном газойле (40—50) или газойле
гидрокрекинга (55—60). У продуктов каталитического крекинга цетановое
число мало (0—25), а октановое число велико (95). Типичное дизельное
топливо имеет цетановое число, равное примерно 50.
9.3.1. Каталитический крекинг в псевдоожиженном слое
Тяжелы фракции процесса переработки нефти (тяжелый газойль,
остатки вакуумной перегонки) с помощью крекинга переводят в более полезные бензиновые фракции [10, И]. В начале XX века крекинг проводили путем
смешения тяжелых фракций с А1С13 в емкостном ректоре. В 1930 г. Хоудри
408
\
Глава 9. Переработка нефти и нефтехимия
предложил схему каталитического крекинга с неподвижным слоем, в котором
в качестве катализатора использовались обработанные кислотой глины (Si02/
А1203). В дальнейшем были использованы синтетические катализаторы на основе аморфных кремнезема/глинозема. В настоящее время для этой цели используются цеолиты, характеризующиеся высокими кислотностью, термической стабильностью и пониженной активностью по отношению к образованию
кокса.
Крекинг представляет собой эндотермическую реакцию, что предполагает
его проведение при достаточно высоких температурах (500 °С), при этом осаждение углерода приводит к быстрой деактивации катализатора. Каталитический крекинг в псевдоожиженном слое — процесс, разработанный компанией
Standart Oil (более известной как ESSO, а сейчас — EXXON) из Нью-Джерси
(1940), как решение проблемы короткого времени жизни катализаторов. Хотя в
современных технологиях крекинг идет в быстро движущемся слое, а не в псевдоожиженном слое реактора, название «каталитический крекинг в пседвоожи-
женном слое» продолжают использовать и сейчас.
Продукты
со2, со, но.
Дымовой
газ
Регенератор
1000 К
Сепаратор
? Трубчатый
реактор
770 К
Подача сырья
Рис. 9.10. Схема реактора каталитического крекинга в псевдоожиженном слое. Крекинг тяжелых углеводородов происходит в поднимающемся слое, в
котором катализатор проводит несколько секунд и
дезактивируется из-за осаждения кокса. Сжигание
кокса в регенераторе — экзотермический процесс,
выделяющееся при этом тепло используется на регенерацию катализатора и эндотермический крекинг
Каталитический крекинг в псевдоожиженном слое используется в настоящее время более чем на 300 установках, из которых 275 расположены в Северной Америке и 70 в Европе. На рис. 9.10 показан принцип работы реактора с
псевдоожиженным слоем. Предварительно разогретые тяжелые остатки подаются в нижнюю часть вертикальной трубы, где они смешиваются с катализатором, поступающим из регенератора. В табл. 9.5 дано характерное распределение продуктов, получающихся в реакторе с псевдоожиженным слоем. Крекинг
осуществляется в процессе движения реагентов и катализатора по вертикальной трубе, в которой они находятся только несколько секунд. Этого, однако,
достаточно, чтобы катализатор полностью покрылся слоем кокса. После выхода продукта из реактора катализатор подается в регенератор, где кокс сжигается в потоке воздуха при температуре 1000 К.
9.3. Производство бензина -J\r 409
Таблица 9.5. Распределение по фракциям продуктов каталитического
крекинга в псевдоожиженном слое для катализаторов
из аморфных кремнезема/глинозема и цеолитов
Компонент
Весовой процент
Кремнезем/глинозем
Цеолит
Газ (Н2, Ср С2, С3, С4)
12-15
15-18
Бензин
30-35
45-50
Легкое масло
25
20
Остатки
20-30
10-25
Углерод на катализаторе
6-8
5
Хотя крекинг может идти и на обработанных хлором глинах и аморфных
алюмосиликатах, использование цеолитов позволило существенно повысить
выход бензина. Малый размер пор цеолита препятствует образованию крупных
молекул ароматических соединений. Эта замечательная селективность по размеру уменьшает возможность образования кокса и увеличивает время жизни
катализатора.
Цеолит в таком катализаторе обычно составляет 20 %, а остальные 80 %
приходятся на матрицу или связующее. Матрица не только определяет физические свойства катализатора, но и действует как ловушка примесей никеля,
ванадия и натрия. Удаление этих компонентов необходимо, поскольку никель
усиливает осаждение кокса на цеолите, натрий дезактивирует кислотные центры, а ванадий разрушает решетку цеолита. Кроме того, сама матрица может
проявлять активность по отношению к образованию кокса, разрушая крупные молекулы углеводородов, не способные проникнуть внутрь цеолита.
Поскольку катализатор в регенераторе проводит времени больше, чем в реакторе
(10 мин и 3 с соответственно), он должен обладать высокими прочностью,
устойчивостью к истиранию и термической стабильностью. Производство катализаторов, использующихся для крекинга в псевдоожиженном слое, является
крупномасштабным: потребность в катализаторах составляет 1000 т в день!
В мире производится, однако, более 500 000 т катализаторов для крекинга в
псевдоожиженном слое, что говорит о большой конкуренции на рынке их сбыта. На рис. 9.11 показана схема производства таких катализаторов.
Реактор с псевдоожиженным слоем и особенно регенератор поставляют
большое количество загрязняющих окружающую среду компонентов. Входящая в кокс сера при его сжигании окисляется до S02 и S03, образуются также
оксиды NOx. Кроме того, выходящий из регенератора газ содержит частицы
материала катализатора. Процесс каталитического крекинга в псевдоожиженном слое является также основным источником серы в бензине. Из всего коли¬
410 -l\r Глава 9. Переработка нефти и нефтехимия
чества серы, входящей в сырье, примерно 50 % попадает в газовую фракцию в
виде H2S, 43 % — в жидкую фракцию продукта и 7 % — в кокс, осаждающийся
на отработанном катализаторе.
Реагенты С!3, NH4CI
Промывочная жидкость
Силикат натрия
Алюминат натрия
Вода
NaOH
СО
Фильтр
Кристаллизация Сушилка
Na-цеолитэ
Источник А1203
Источник Si00
Вода
NaOH
<3*0
Материал
матрицы
Синтез
кремнезема/глинозема
Ионный
обмен
<11Ж=>
Смеситель
"/тЬч
Сушка
распылением
V.
Часiицы катализатора
для крекинга
в псевдоожиженном слое
(размер 50 -70 мкм)
Матрица'
из кремнезема/
глинозема
Час Iицы цеолита
(размер 2—10 мкм)
Рис. 9.11. Схема производства катализаторов, использующихся при крекинге в псевдоожиженном слое [11]
Механизм реакции включает в качестве интермедиатов карбониевые и кар-
бениевые ионы. Первая и наиболее трудная ступень — получение карбониевых
ионов из алканов:
Н++С„Н2я+2^[СяН2я+3]+. (9.2)
Неустойчивый карбониевый ион распадается с выделением карбениевого
иона:
[С„Н2я+3]+^[СяН2я+|]++Н2. (9.3)
Стадия крекинга описывается так:
[С„Н2я+3]+ -> [СЯ_ХН2(1_2,_,]+ -нС^Н*. (9.4)
Однажды начавшись, реакция далее идет так:
CmH2m+2 +[C„H2„+1]a+ds -» СяН2й+2 +[CmH2m+l];ds. (9.5)
9.3. Производство бензина -*\г 411
Основной канал реакции связан с разложением каребниевых ионов. Однако одновременно идет образование ароматических соединений и олигомеров, а
также кокса.
9.3.2. Риформинг и бифункциональный катализ
Ни бензиновые фракции, полученные начальной перегонкой (прямогонный бензин), ни продукты крекинга тяжелых фракций не содержат в
достаточном количестве разветвленных углеводородных молекул, за счет которых обеспечивается нужное октановое число бензина. Типичное октановое число
(ЛОЧ) получаемого непосредственно бензина лежит в диапазоне от 20 до 50, в
то время как в автомобилях используется бензин с октановым числом от 95 до 98.
Риформинг позволяет превратить линейные молекулы алканов (в прямогонном бензине) и алкенов — продуктов каталитического крекинга в разветвленные молекулы и ароматические соединения [12]. К сожалению, этот процесс
сопровождается нежелательными побочными явлениями — крекингом молекул и образованием кокса. Реакции риформинга катализируются платиновыми
катализаторами, на кислотных носителях. И катализатор, и носитель участвуют
в реакции, что позволяет говорить о них как о бифункциональном катализаторе.
Бензиновая фракция содержит много различных углеводородов в диапазоне С5—С10, продукты риформинга представляют собой сложную смесь нескольких сот различных молекул. //-Гексан — наименьшая по размеру молекула,
участвующая в реакциях всех классов риформинга (рис. 9.12).
Рис. 9.12. Бифункциональный катализатор в риформинге я-гексана [13]
%
о.
S
со
S
Vv
4К
VVV
-н4Н
Г VSA, -О Ц-
-н4|+Нг -На
-6- * #
•НаЦ+Н, -Н2 ||+Н2
■Ь 'С%
Бифункциональный
катализ в риформинге
с использованием
катализатора Pt/Al203
Изомеризация
-«аь
vj “ '?у
М
Катализируется кислотными центрами
Основные реакции таковы:
• дегидрогенизация алканов до алкенов;
• ароматизация циклических продуктов до бензола, ксилола и т. д.;
• гидрирование алкенов до алканов.
412
Л
Глава 9. Переработка нефти и нефтехимия
Для катализа этих реакций используются металлы. Катализаторами других
реакций:
• изомеризации олефинов,
• крекинга углеводородов
являются кислотные центры на поверхности носителя из оксида алюминия. Большинство этих реакций (за исключением изомеризации и гидрирования) являются в сильной мере эндотермическими, и в ходе их протекания образуется водород. По этой причине термодинамические требования предполагают использование высоких температур и низких давлений. Однако эти условия
благоприятны также для образования кокса, влияющего на время жизни катализатора. Следовательно, реальные условия проведения риформинга (температура 500 °С, давление 5—20 бар, отношение Н2: углеводороды равняется 5:10)
представляют собой компромисс между качеством продукта и его выходом, с
одной стороны, и низкой скоростью образования кокса — с другой. Заметим,
что в процессе риформинга, тем не менее, образуется водород, который используется на других стадиях переработки нефти — при гидроочистке и гидрокрекинге. При риформинге образуется также большое количество ароматических
соединений, использующихся в химической индустрии. При повышении температуры и снижении давления выход ароматических соединений увеличивается.
bPtCI,
\ CI
А|-°Х|^С|
О Pt
AI — О I CI
/ CI
О
+ 2Н++2СГ
д| — о CI
^А1 ^
°\ /РЧ
AI - О CI
I- 2Н++ 4СГ
AI — ОН
AI-OH
+СГ
^5
У-А1203
Гидроксилированный
кислотный носитель
AI — CI
AI — ОН
ОН"
С1/у-А1203
Катализатор
с повышенной
кислотностью
Рис. 9.13. Синтез бифункционального катализатора Pt/Al203. Носитель из оксида алюминия пропитывается водным раствором гексахлорплатиновой кислоты (H2PtCl6)
и НС1. Конкурентная адсорбция СГ и PtCl^ способствует равномерному распределению дисперсной платины по носителю, а адсорбированный хлор повышает кислотность носителя. После кальцинирования и восстановления получается катализатор в конечном виде
Катализатором риформинга является высокодисперсная платина, нанесенная на оксид алюминия (рис. 9.13). Наличие хлора на носителе усиливает его
9.3. Производство бензина -*\г 413
кислотность, что необходимо для проведения реакций изомеризации. Устойчивость катализатора по отношению к образованию кокса существенно повышается при введении второго металла типа рения или рутения. Такие катализаторы позволяют проводить риформинг при низких давлениях, но они очень
чувствительны к отравляющим примесям соединений, содержащих S и N, что
делает необходимым проведение эффективной гидроочистки.
Для иллюстрации принципа работы бифункционального катализатора мы
рассмотрим схему реакции изомеризации пентана [14]. Первым шагом является дегидрогенизация алкана на поверхности металла
С5Н12 ^ С5Н10 + Н2; на платине. (9.6)
Полученный алкен адсорбируется на кислотном центре на поверхности
носителя из оксида алюминия, где он подвергается изомерному превращению:
С5Н10 + Н+ -»[C5Hn]*ds; на кислотных центрах,
[C5HiiLs ['-C5H..L ; на кислотных центрах, (9.7)
[/-C5Hu]*ds -> /-С5Н10 + Н+; на кислотных центрах.
На последней стадии изомер гидрируется до алкана на поверхности металла
/-С5Н10 + Н2 -> /-С5Н12; на поверхности платины. (9.8)
Если катализатор содержит платину в количестве, достаточном для обеспечения равновесия в реакциях гидрирования-дегидрогенизации, то реакцию изомеризации можно рассматривать в качестве лимитирующей стадии, что дает
для скорости полной реакции выражение
АГас^3^2[я-С5Н|21/[Н2]
\ + К\К2 [я-С5Н12]/[Н2]
Таким образом, скорость реакции зависит только от отношения парциальных давлений водорода и я-пентана. В поддержку предложенного механизма
говорит тот факт, что скорость изомеризации я-пентана на катализаторе, не
содержащем платины, имеет во многом аналогичный вид. Суть бифункционального механизма состоит в том, что металлический катализатор способствует превращению алканов в алкены и наоборот. Это открывает возможность
осуществить изомерное превращение через карбениевые ионы, что можно провести при более низких температурах, чем превращение с участием карбоние-
вых ионов, образующихся из алканов.
В науке о поверхностях получено много ценной информации о реакциях на
поверхности монокристаллов платины с участием углеводородных молекул. За
деталями мы отсылаем читателя к книге Соморджаи [15]. Подробная информация о реакциях с участием углеводородов на кислотных центрах оксида алюминия или на цеолитах имеется также в [16].
414 -J\r Глава 9. Переработка нефти и нефтехимия
Мы завершаем этот раздел поучительным примером реакции дегидрогенизации, протекающей на поверхности металла. Особенность этой реакции состоит
в том, что она не предполагает адсорбционного равновесия, принимавшегося
ранее в этой книге для всех других реакций. Дегидрогенизация метилциклогек-
сана, С6НПСН3, до толуола, С6Н5СН3, на поверхности платины хорошо описывается последовательностью реакций, идущих только в прямом направлении:
С6НП + * —-—> С6НПСН3 >ds, (9.10)
С6НПСН3 ads —С6Н5СН3 ads + ЗН2Т, (9.11)
C6H5CH3>ads —51-» С6Н5СН3 + ’. (9.12)
Вторая ступень, конечно, не является столь элементарной, но для нашего
анализа это никакой роли не играет. Поскольку толуол менее насыщенный
углеводород, чем метилциклогексан или интермедиат, можно предположить,
что на поверхности находится в основном толуол.
Скорость реакции имеет вид:
(913)
где 0Т — степень заполнения поверхности толуолом. Условие баланса для поверхностных центров выглядит так:
вТ+вм +0, =1, (9.14)
где 0М — степень покрытия поверхности, но поскольку на поверхности доминирует толуол, можно записать:
0Т + ~ 1. (9.15)
Использование стационарного приближения для 0Т и 0М дает
d/9
J Т - - = 0, (9.16)
at
d/9
м =к1[СвНпСН3Щ-к20м=О, (9.17)
dt
(9.18)
Подстановка этого выражения в формулу (9.13) приводит к выражению
d[CtH5CH,| t,[C,H„CH,|
d t 1 + *,/*,[С,Н„СН,]'
9.3. Производство бензина ->\г 415
Высокая степень заполнения поверхности толуолом означает, что в знаменатель в правой части этого выражения есть величина, существенно превышающая единицу. В этом случае порядок реакции по метилциклогексану становится равным нулю. Мы можем сказать, что на практике реакция лимитируется десорбцией толуола с поверхности катализатора.
9.3.3. Алкилирование
Алкилаты составляют примерно 15 % бензиновой фракции (см. рис. 9.9).
Реакция алкилирования — это реакция между изобутаном [СН3—СН—(СН3)2]
и низкомолекулярными олефинами типа пропена, бутена и пентена, при которой образуются разветвленные молекулы, повышающие октановое число бензина [17, 18]. Этот процесс является экзотермическим, поэтому его следует
проводить при низких температурах; он катализируется кислотами в жидкой
фазе. Реагент изобутан получается в процессе каталитического крекинга и гидрокрекинга или производится отдельно путем изомеризации бутана. Алкилирование сопровождается нежелательной реакцией олигомеризации олефинов,
поэтому в реагентах изобутан находится в избытке. Алкилаты имеют октановое
число около 87 (ЛОЧ), поэтому они существенно повышают детонационную
стойкость бензина.
Типичные условия для процесса при использовании в качестве катализатора H2S04 таковы: температура 5—10 °С, давление 2—5 бар, отношение изобу-
танюлефины — 10, время нахождения в реакторе 20—30 мин. Расход катализатора в данной реакции высок (около 10 кг кислоты на тонну алкилированного
продукта). Во многих отношения более предпочтительным является процесс с
использованием HF: он осуществляется при температуре 25—40 °С (не нужно
проводить охлаждение), давлении 10—20 бар, реакция проводится более быстро, а расход HF составляет менее 1 кг на тонну алкилатов.
Реакция идет по цепному механизму с образованием на промежуточной
стадии карбениевых ионов. Реакция начинается с взаимодействия олефинов с
кислотой, в результате чего формируется ион С—С+—С, который затем реагирует с изобутаном, что приводит к образованию С—С+(С)—С. Этот карбе-
ниевый ион является центральным соединением в распространении реакции
до получения алкилированных продуктов, таких как 2,2-диметилпентан и связанных с его формированием соединений (рис. 9.14).
Основной недостаток процесса алкилирования связан с расходом больших количеств кислоты (до 100 кг кислоты на тонну продукта). По этой причине в качестве альтернативы рассматривается возможность использования
твердых кислот. В частности, твердая кислота типа сульфатированного циркония (S04_/Zr02) демонстрирует высокую активность в реакции алкилирования, но только на короткое время, поскольку катализатор деградирует
вследствие осаждения кокса, образующегося при олигомеризации алкенов.
Катализаторы такого типа также очень чувствительны к присутствию воды в
реагентах.
416
Глава 9. Переработка нефти и нефтехимия
Начало:
С - С = С + Н +
Пропен Протон кислоты
-> С - с+ = С
Карбениевый ион
С-С-С + С-С+=С -> С-С+-С +С-С-С
с
Изобутан
С
Распространение:
С - С+- С
С - С+- С + С - С = С -> с-с-с
с
с
с - с+- с
с-с-с
с-с+-с + с-с-с -> с-с-с + с-с+-с
с
с
с
2,2-Диметилпентан
С
Рис. 9.14. Алкилирование изобутана и пропена представляет собой цепную реакцию, идущую с образованием карбениевого иона изобутена (показан жирным шрифтом), являющимся переносчиком цепи [1]
Другим компонентом, сильно повышающим октановое число бензина,
является метил-трет-бутиловый эфир (рис. 9.15), который образуется в реакции алкилирования изобутана метанолом при использовании в качестве
катализатора кислотной полистирольной смолы, называемой «Amberlyst».
Хотя производство метил-трет-бутилового эфира шло по нарастающей в
конце XX века и он широко применяется в качестве добавок к бензину, в
настоящее время стремятся уменьшить его использование, поскольку он
был найден в подземных водах вблизи нефтеперерабатывающих заводов в
Калифорнии.
Рис. 9.15. Метил-трет-бутиловый эфир, наиболее часто использующийся для улучшения качества бензина, имеет октановое число, равное
116. Другими добавками являются этил-трет-
бутиловый эфир (ЛОЧ = 118) и трет-амилме-
тиловый эфир (ЛОЧ =111)
9.4. Нефтехимия: реакции с участием низкомолекулярных олефинов ->\г 417
9.4. НЕФТЕХИМИЯ: РЕАКЦИИ С УЧАСТИЕМ
НИЗКОМОЛЕКУЛЯРНЫХ ОЛЕФИНОВ
Низкомолекулярные олефины — в основном этилен, пропен, бутен — являются строительными элементами нефтехимической индустрии. Эти
молекулы наряду с другими образуются в процессе каталитического крекинга,
но они образуются еще и при крекинге с водяным паром бензиновых фракций.
С участием олефинов идет большое число реакций. В качестве примера мы
рассмотрим здесь эпоксидирование этилена и частичное окисление пропилена, а также полимеризацию этилена и пропилена.
9.4.1. Эпоксидирование этилена
Этиленоксид является важным интермедиатом, использующимся
для получения этиленгликоля (антифриза), полимеров, пластификаторов и
многих других продуктов [19]. В гл. 1 мы объясняли, что замена традиционных
способов производства, при которых на 1 моль эпоксидных групп приходится
1,5 моля субпродукта, на каталитический синтез с использованием катализатора из серебра является наиболее ярким примером перехода к экологически
чистым технологиям (рис. 9.16).
Рис. 9.16. Этилен плохо адсорбируется на поверхности чистого серебра, но интенсивно взаимодействует с адсорбированными атомами кислорода. При низких степенях заполнения атомы О предпочтительнее взаимодействуют со связью
С—Н этилена, что приводит к его распаду на фрагменты,
окисляющиеся до С02 и Н20. Но при высоких степенях заполнения увеличивается сродство атомов кислорода к электронам, поэтому они начинают интенсивно взаимодействовать с л’-системой этилена, что приводит к образованию эти-
леноксида [19]
!!
Ад
“Н
in i n i lit iii i
a9
Механизм реакции устанавливается непосредственно. Она начинается с
диссоциативной адсорбции кислорода на частицах серебра:
02 + 2* ^ 20*. (9.20)
Эта ступень является критической для следующей стадии, поскольку этилен адсорбируется плохо на поверхности серебра
С2Н4 + —> С2Н4.
(9.21)
418 -i\, Глава 9. Переработка нефти и нефтехимия
Поверхностная реакция между двумя адсорбированными компонентами, ведущая к образованию мгновенно десорбирующегося этиленоксида, завершает цикл:
С2н; + О -> С2Н40 + 2*. (9.22)
Из написанной схемы можно заключить, что все реагенты превращаются в
этиленоксид, но, к сожалению, это не совсем корректно. Окисление реагентов
и продуктов — это конкурирующие процессы, как показано на следующей общей схеме
О
/ \
СН2 = СН2 —► Н2С = СН2 (9.23)
С02 + Н20
Чтобы избежать превращения продукта в С02, катализатор должен иметь
низкую удельную поверхность и минимально возможную пористость, при которых время нахождения продукта в каталитическом слое достаточно мало.
Реакция эпоксидирования является экзотермической, поэтому для исключения сильных изменений температуры реактор сконструирован в виде пучка
узких трубок диаметром в несколько сантиметров, окруженных теплоносителем. Катализатор представляет собой относительно крупные частицы серебра,
нанесенные на инертный tf-Al203. Удельная поверхность катализатора меньше
1 м2Д. Введение промоторов, калия или хлора, позволяет повысить селективность катализатора с 60 до 90 % при степени конверсии этилена в 50 %.
Следовые количества хлора в катализаторе подавляют процесс окисления.
Этот эффект был обнаружен случайно, когда на промышленном предприятии
было замечено, что селективность процесса вдруг стала самопроизвольно увеличиваться день ото дня. Анализ катализатора показал наличие следовых примесей хлора, поступавшего от построенного рядом нового завода по производству хлора. В настоящее время в сырье добавляют малые количества хлорсодержащих соединений типа дихлорэтилена.
Из-за большой практической значимости и вследствие простоты реакционного механизма и катализатора было проведено значительное число фундаментальных исследований процесса эпоксидирования. Их обзор читатель может
найти в статье [19].
9.4.2. Частичное окисление и аммоксидирование пропилена
При частичном окислении пропилена образуется акролеин,
Н2С=:СНСНО, важный интермедиат производства акриловой кислоты,
Н2С^=СНСООН, или акрилонитрила H2C=:CHCN, получающегося в присутствии NH3 и являющегося мономером для полимерных акриловых волокон.
В качестве катализаторов здесь используются смешанные оксиды металлов [20].
9.4. Нефтехимия: реакции с участием низкомолекулярных олефинов
Каталитическое окисление на поверхности смешанных оксидов обычно идет
по механизму Марса—ван Кревелена [21], представленному на рис. 9.17 на
примере окисления СО. Вместо продукта поверхностной реакции между адсорбированными атомами СО и О, С02 образуется как результат взаимодействия СО и атомов О, входящих в состав решетки оксида металла. Образовавшаяся вакансия заполняется на отдельной стадии, включающей активацию 02,
которая часто идет на дефектных центрах.
Рис. 9.17. Схема механизма Марса—
ван Кревелена на примере реакции
окисления СО на поверхности оксида металла. Характерная черта механизма — использование кислорода из
решетки оксида для окисления СО,
после чего остается вакансия, заполняемая кислородом из газовой фазы
в отдельной реакции
°\ /£\ /°
/Is* ,
о о / ‘
Активирование 02
о.
м
о
Внедрение
в пропилен
oy'\
Bi
у2о
0
1
0 о
1 I
м м м м
О.
0 о о о
1 I I I
м м м м
Механизм
Марса—ван
Кревелена
СО
с
/\
0 о
1 I
м м м м
Кислород, ответственный
за отрыв Н от молекулы
пропилена
Рис. 9.18. Схематическое изображение различных кислородных центров, участвующих в частичном окислении пропилена до акролеина на
поверхности Bi2Mo06, и их предполагаемая роль
в реакции [22]
Частичное окисление пропилена идет по аналогичному механизму, хотя
структура поверхности катализатора из смешанных оксидов висмута и молибдена является существенно более сложной, чем приведенная на рис. 9.17. Как
показано на рис. 9.18, атомы кислорода, занимающие разные кристаллографические позиции, играют каждый свою роль. Атом О, образующий мостик между Bi и Мо, как полагают, ответствен за активацию связи С—Н и отрыв атома
Н от метильной группы, за которым следует адсорбция пропилена в форме
аллильной группы (H2C^^CH—СН2). Эта стадия, скорее всего, является ли-
419
420 -l\r Глава 9. Переработка нефти и нефтехимия
митирующей для данного механизма реакции. Краевой атом О, связанный с
Мо, как полагают, внедряется в углеводородную молекулу. Локализованный на
висмуте центр активирует молекулу 02, после диссоциации которой заполняется кислородная вакансия, образующаяся у атома молибдена после десорбции
акролеина.
Акролеин, в свою очередь, может быть окислен до акриловой кислоты.
Катализатором этой реакции является смешанный оксид ванадия и молибдена.
Другая важная для индустрии и родственная одной из описанных выше
реакция с участием пропилена представляет собой его частичное окисление в
присутствии аммиака, при котором образуется акрилонитрил, H2C:=CHCN.
Эта реакция аммоксидирования также катализируется смешанными оксидами
металлов типа оксидов висмута и молибдена или антимоната железа, в который вводится большое число промоторов (рис. 9.19). Из-за сильной экзотер-
мичности реакция аммоксидирования проводится в реакторе с псевдоожиженным слоем, что позволяет обеспечить ускоренный отвод тепла и постоянство
температуры (400—500 °С).
SiO
Fe Держатель
образца
К Си Мо
JwUaIjlHu
25х
Катализатор синтеза
акрилонитрила
на основе оксидов
Fe—Sb
Sb
SbO
Jl
250x
0 50 100 150 200
Атомная масса, ат. ед. массы
Рис. 9.19. Спектр вторичных ионов, вылетающих с поверхности катализатора из смешанных оксидов Fe—Sb, использующегося для селективного окисления пропилена
и аммиака до акрилонитрила. Видно наличие в катализаторе элементов Si, Си и
Мо, а также следовых количеств атомов щелочных металлов [23]
Частичное окисление с использованием сложного катализатора из смешанных оксидов металлов не является идеальным объектом для изучения на соответствующих монокристаллах как механизма реакции, так и структуры катализатора. Вместе с тем несколько каталитических циклов было (не без основания)
предложено в результате подобных исследований. Эффективный катализатор,
как полагают, должен селективно взаимодействовать с органическими реагентами в тех центрах, где имеется кислород с умеренной реакционной способностью. Это взаимодействие приводит к желаемому частичному окислению и образованию кислородной вакансии. Дефектный центр заполняется кислородом из
объемной газовой фазы, активированным на удаленных от вакансии центрах.
9.4. Нефтехимия: реакции с участием низкомолекулярных олефинов -*\г 421
9.4.3. Катализ в реакциях полимеризации
Большинство низкомолекулярных олефинов, получаемых в химической промышленности, используется для получения полимеров, полный объем
производства которых в год составляет около 100 миллионов тонн. Полимеры —
макромолекулы, молекулярный вес которых обычно лежит в пределах от 104 до 106,
имеющие линейную, разветвленную или сетчатую структуру, построенную из
небольших мономерных звеньев типа этилена, пропилена, винилхлорида, стирола и т. д. Подавляющее число полимеров получают в каталитических реакциях. Здесь мы в качестве примера рассмотрим полимеризацию этилена, протекающую с участием хромового катализатора [24].
Процесс Филлипса, полимеризация этилена с участием хромового катализатора, используется для производства примерно одной трети выпускаемого во
всем мире полиэтилена высокой плотности. Процесс был разработан в 1951 г.
и постоянно совершенствовался, так что в настоящее время имеется «семейство» различных катализаторов на основе хрома, которые можно использовать
для производства полиэтиленов около 50 сортов, различающихся молекулярным
весом макромолекул, степенью их разветвленности и типом сомономера — буте-
на, гексена или октена. Это разнообразие отражает коммерческий успех процесса Филлипса. Свойства полимерного продукта могут регулироваться путем
изменения разнообразных параметров — температуры кальцинирования катализатора, температуры и давления в процессе полимеризации, размера пор носителя из кремнезема, а также введением добавок титана или фтора, использующихся в качестве промоторов, в носитель или даже заменой носителя на оксид алюминия, что приводит к увеличению молекулярного веса макромолекул.
Хромовый катализатор, использующийся в реакциях полимеризации, отличается от катализаторов других химических реакций тем, что здесь он может
расходоваться в силу случайных причин. В процессе реакции полимерные молекулы заполняют поры и оказывают сильное внутреннее давление на носитель. В результате катализатор может разрушиться и попасть в конечный продукт в виде высокодисперсных частиц.
Из-за малого содержания хрома катализатор Филлипса достаточно сложно охарактеризовать. Типичное содержание хрома составляет 0,5 вес. % или
один атом хрома на 1 нм2. Катализатор получают пропиткой носителя водным раствором хромовой кислоты. После сушки хром присутствует в катализаторе в форме гидратированного оксида Сг(6+). Кальцинирование является
важной ступенью в получении катализатора. Для придания катализатору требуемой активности в реакции полимеризации кальцинирование следует проводить, по крайней мере, при 550 °С. Высокая температура позволяет обеспечить связь через кислородные мостики иона Сг6+ с ионами кремния (рис. 9.20).
Более того, формируются и островки чистого хрома, что позволяет рассматривать его как истинный монокристаллический катализатор. Изолированные
ионы Сг6+, однако, полностью координированы ионами кислорода, поэтому
они не проявляют активности в реакции полимеризации этилена. Восстанов¬
422
\
Глава 9. Переработка нефти и нефтехимия
ление соединений хрома этиленом при температуре 150—160 °С дает не полностью координированные ионы Сг2+ и Сг3+, которые уже являются каталитическими центрами.
575 580 585 590 595
Энергия связи, эВ
Рис. 9.20. Рентгеноэлектронные спектры
хромового катализатора, использующегося
в процессах полимеризации, а также спектры сравнения от соединения Сг(6+). Хром
им-прегнированного хромата в свежеприготовленном катализаторе имеет ту же энергию связи, что и хром в хромате щелочного
металла, дихромате или объемном СЮ3. При
кальцинировании энергия связи хрома существенно возрастает — на 1,3 эВ. Столь
необычно высокая энергия связи типична
для хроматов, образующих эфирные связи
с кремнеземом, как это имеет место в кластерах [Cr02(0Si(C6H5)20Si(C6H5)20)]2 [25]
Механизм реакции включает три основных этапа:
1. Инициирование: активация первой молекулы этилена и образование алкильного соединения на хроме.
2. Рост цепи путем последовательного внедрения молекулы этилена в связь
Cr-алкил. Рост цепи проиллюстрирован на рис. 9.21.
3. Окончание процесса через образование винильной концевой группы, отрывающейся от иона Сг. После чего образуется новая этильная группа и начинается процесс образования новой полимерной цепи.
Рис. 9.21. Рост полимерной цепи по механизму Косси—Арлмана в процессе полимеризации этилена включает стадию внедрения молекулы этилена в растущую алкильную цепь. Квадратик означает вакансию у иона хрома [26]
9.4. Нефтехимия: реакции с участием низкомолекулярных олефинов -1\г 423
Инициирование реакции идет медленно, тогда как процесс полимеризации
протекает быстро. В систему можно вводить активаторы типа триэтилалюми-
ния, которые способствуют восстановлению Сг6+ и образованию первичной
этильной группы, что позволяет существенно ускорить стадию активации.
1
Рис. 9.22. Полученные с помощью атомно-силового микроскопа изображения пленок
полиэтилена, сформированных на плоской поверхности модельного хромового катализатора. Узкие светлые полоски — ламеллярные кристаллы. Они формируют хорошо известные сферулитовые структуры при кристаллизации расплава полиэтилена. В зависимости от толщины слоя рост сферулитов останавливается на различных стадиях процесса [27]
На рис. 9.22 приведено полученное с помощью атомно-силового микроскопа изображение молекул полиэтилена, сформированных на плоском модельном катализаторе Филлипса. Чтобы понять смысл этого изображения, следует учесть, что полиэтилен образуется при температуре 160 °С, которая выше
его температуры плавления. При охлаждении полиэтилен кристаллизуется с
образованием ламеллярных (слоистых) структур, пытающихся упаковаться упорядоченно параллельно друг другу. Этому упорядочению препятствуют дефекты и сшивки между различными ламеллями. По истечении короткого времени,
связанного с протеканием реакции, на подложке формируется сплошной слой
ламеллей полиэтилена, достигающих толщины 80 нм. В этом слое наряду с полимерными молекулами присутствуют и более короткие цепи парафинов. Помимо уложенных штабелем пластинчатых элементов, относящихся к парафинам
и полиэтиленовым ламеллям, на поверхности присутствуют также кристаллические пучки молекул полиэтилена, выросшие в виде снопов. При длительном
проведении реакции слой существенно утолщается, а его морфология радикальным образом изменяется. Из рис. 9.22 видно, что на поверхности формируются упорядоченные домены из полиэтилена диаметром примерно 50 мкм.
Средняя толщина пленки составляет 250 нм. В центральной части полиэтиленовая ламелла растет в форме снопа из некоторого центра нуклеации. Действительно, ламеллы стараются уложиться параллельно друг другу, но это удается им
сделать лишь отчасти, поскольку они несовершенны из-за наличия, например,
петель полимерных молекул и разрыхленных выступающих краев. На третьей фотографии показана финальная стадия роста сферулитов в слое толщиной 420 нм.
Центры нуклеации можно еще различить, но они покрыты куполообразными структурами, как это имеет место при кристаллизации толстых слоев полиэтилена [26].
424 -J\r Глава 9. Переработка нефти и нефтехимия
Список литературы
1. Moulijn J.A., Makke М., Van Diepen A. Chemical Process Technology.
Chichester: Wiley, 2001.
2. Gosselik J.W. // Cat. Tech. 1998. V. 4. P. 127.
3. http://www.bpamoco.com.
4. Prim /?., De Beer V.H.J., Somorjai G.A. // Catal. Rev. — Sci. Eng. 1989. V. 31. P. 1.
5. Tops0e Clausen B.S., Massoth F.E. Hydrotreating Catalysis. Berlin: Springer-
Verlag, 1996.
6. Sanders A.F.H., Z)e iteer V.H.J., Кяя Кеея J.A.R., Niemantsverdriet J.W. //
Appl. Surf. Sci.. 1999. V. 144. P. 380.
7. Coulier L., Z)e iteer V.H.J., Van Veen J.A.R., Niemantsverdriet J.W. // Top.
Catal. 2000. V. 13. P. 99.
8. Pecoraro T.A., Chianelli R.R. // J. Catal. 1981. V. 67. P. 430.
9. Raybaud P., Hafner /., Kresse G., Touhoat H. // J. Phys. : Condensed Matter.
1997. V. 9. P. 11, 107.
10. Коя Ballmoos R., Harris D.H., Magee J.S. In: Handbook of Heterogeneous Catalysis.
Vol. 4. /Eds Ertl G., Knozinger H., Weitkamp J. Weinheim: Wiley-WCH, 1997. P. 1955.
11. Moulijn J.A., Makkee M., Кяя Diepen A. Chemical Process Technology.
Chichester: Wiley, 2001.
12. Sinfelt J.H. In: Handbook of Heterogeneous Catalysis. Vol. 4. /Eds. Ertl G.,
Knozinger H., Weitkamp J. Weinheim: Wiley-WCH, 19997. P. 1939.
13. Sinfelt J.H. // Adv. Chem. Eng. 1964. V. 5. P. 37
14. Van Santen R.A., Niemantsverdriet J. W. Chemical Kinetics and Catalysis. New
York: Plenum, 1995.
15. Somorjai G.A. Introduction to Surface Chemistry and Catalysis. New York:
Wiley, 1994.
16. Introduction to Zeolite Science and Practice / Eds. Van Bekkum H.,
Flanigan E.M., Jansen J.C. Amsterdam: Elsevier, 1991.
17. Weitkamp /., Traa Y. In: Handbook of Heterogeneous Catalysis. Vol. 4. /Eds.
Ertl G., Knozinger H., Weitkamp J. Weinheim: Wiley-WCH, 19997. P. 2039.
18. Corma A., Martinez A. // Catal. Rev. — Sci. Eng. 1993. V. 35. P. 483.
19. Van Satten R.A., Kuipers H.P.C.E. // Adv. Catal. 1987. V. 35. P. 265
20. Gates B.C. Catalytic Chemistry. New York: Wiley, 1992.
21. Mars P., van Krevelen D.W. // Chem. Eng. Sci. 1954. V. 3. P. 41.
22. Claeser L.C., Brazdil J.F., Hazle M.A., Mehicic M, Crasselli R.K. // J. Chem.
Soc. Faraday Trans. I. 1985. V. 81. P. 2903.
23. Niemantsverdriet J. W. Spectroscopy in Catalysis. Weinheim: Wiley-VCH, 2000.
24. McDaniel M.P. // Adv. Catal. 1985. V. 33. P. 47.
25. Thiine P.C., Verhagen C.P., Van den Boer M.J., Niemantsverdriet J.W. //
J. Phys. Chem. B. 1997. V. 101. P. 8559.
26. Arlman E.J., Cossee P. // J. Catal. 1964. V. 3. P. 89.
27. Thiine P.C., Loos P., Lemstra P.J., Niemantsverdriet J. W. // J. Catal. 1999. V. 183.
P. 1.
28. Billmeyer F. W. Textbook of Polymer Science. New York: Wiley Interscience, 1984.
глава КАТАЛИЗ И ЗАЩИТА
ю
ОКРУЖАЮЩЕЙ среды
10.1. ВВЕДЕНИЕ
В современном обществе востребованы технологии, оказывающие минимальное воздействие на окружающую среду. В идеале химические
процессы должны быть «чистыми», то есть в них не должны образовываться
вредные субпродукты и отходы. Более того, сами продукты, например топливо, при их использовании не должны создавать проблем для окружающей
среды. Водородные топливные элементы (гл. 8) и процессы гидрообессеривания (гл. 9) являются прекрасными примерами таких технологий, в которых катализ играет существенную роль. Однако выброса вредных соединений не всегда удается избежать, например на тепловых электростанциях или
на транспорте, и здесь основанные на катализе технологии очистки помогают уменьшить уровень загрязнения окружающей среды. Эти технологии являются предметом данной главы.
Транспорт и промышленные предприятия являются основными источниками загрязнения воздуха в Западном мире. Они являются поставщиками
монооксида углерода, оксидов азота (NOx), оксидов серы (SOx) и органических соединений всех типов. Катализ является незаменимым в конверсии
этих соединений в более мягкие по отношению к окружающей среде соединения типа Н2, Н20 и С02, хотя последний сейчас считается ответственным за парниковый эффект. Катализ как способ защиты окружающей среды в настоящее время используется во многих крупномасштабных
процессах: на тепловых электростанциях для очистки дымовых газов, на
транспорте, в котельных, работающих на угле, в ресторанах для удаления
ароматических соединений, образующихся при жарке мяса и гамбургеров,
в туалетах для удаления неприятных запахов, для удаления летучих органических соединений в промышленности и для разложения аммиака в сточных водах. Здесь мы сосредоточим свое внимание на основных процессах:
на каталитической нейтрализации выхлопных газов автомобилей и на системах удаления NOx из дымовых газов тепловых электростанций. Мы кратко обсудим эти процессы, использующиеся катализаторы, а также кинетику и механизмы реакций.
426
Глава 10. Катализ и защита окружающей среды
10.2. КАТАЛИТИЧЕСКАЯ НЕЙТРАЛИЗАЦИЯ
ВЫХЛОПНЫХ ГАЗОВ
Транспорт является основным источником загрязнения атмосферы крупных городов. Общее число легковых и грузовых автомобилей, автобусов и мотоциклов в мире в 1990 г. составляло около 650 миллионов. Все
эти передвижные средства являются источником выхлопных газов, и, по
оценкам, примерно 500 000 человек в мире умирает вследствие загрязнения
воздуха транспортом. На транспортные средства приходится более половины объема продуктов переработки нефти — бензина, дизельного топлива и
керосина. Кроме того, транспорт является источником примерно четверти
всех выбросов углекислого газа (порядка 200 г/км для легкового автомобиля
среднего класса).
Типичные концентрации составляющих выхлопных газов автомобилей, помимо С09 и Н20, представлены в табл. 10.1. Происхождение этих молекул
очевидно: так, СО, токсичная составляющая мгновенного действия, появляется в результате неполного сгорания углеводородов. Водород всегда присутствует в пропорции 1/3 по отношению к концентрации СО и появляется в
результате разрушения углеводородных молекул. Оксид азота (почти исключительно N0) появляется при горении топлива при высоких температурах и
ответствен за коричневатый цвет дымки, которая в солнечный день иногда
видна над городами.
Таблица 10.1. Типичные концентрации соединений в выхлопных газах [1]
Соединение
Концентрация
Углеводородыа
750 миллионных долей
NO,
1050 миллионных долей
СО
0,68 об. %
Н2
0,23 об. %
02
0,58 об. %
а) На основе С3.
Несгоревший бензин и разрушенные молекулы углеводородов типа этилена
и пропилена также составляют значительную часть выхлопных газов. Бензин
содержит различные добавки: бензол, толуол и разветвленные углеводороды,
обеспечивающие необходимое значение октанового числа. Непосредственные
выбросы этих летучих соединений, например, на заправочных станциях вносят
существенный вклад в загрязнение окружающей среды. Топлива с добавками
соединений свинца типа тетраэтилсвинца, повышающего октановое число, в
настоящее время стараются не производить, поскольку свинец является отрав¬
10.2. Каталитическая нейтрализация выхлопных газов -*\г 427
ляющим веществом для человек и катализаторов, особенно для родия, использующегося для удаления N0.
Принятый в США в 1970 г. «Закон о чистом воздухе» установил стандарты
чистоты воздуха по шести основным загрязняющим элементам — аэрозольным
частицам, оксидам серы, монооксиду углерода, оксидам азота, углеводородам
и фотохимическим окислителям. Установлены также стандарты для выбросов
автомобилей — основного источника монооксида углерода, углеводородов и
оксидов азота. Перечень стандартов дан в табл. 10.2. Предельные уровни загрязнений, установленные в Европейском Союзе (1996), легко обеспечиваются
при использовании современных катализаторов. Более жесткие стандарты, относящиеся к транспорту со сверхнизким выбросом вредных веществ, могут
быть обеспечены современными катализаторами, но транспортные средства
без вредных выбросов, вероятно, появятся, когда двигатели будут работать на
водородном топливе.
Таблица 10.2. Стандарты по выбросам (г/км) для транспортных средств,
работающих на бензине
СО
Углеводороды
NO,
Углеводород +
+ NOv
США
2,11
0,25
0,62
—
США, 1994
2,П
0,16
0,25
—
ЕС, 1996
2,2
—
—
0,5
Япония
2,1
0,25
0,25
—
Транспортные средства
с низким выбросом вредных
веществ
2,11
0,05
0,12
—
Транспортные средства
со сверхнизким выбросом
вредных веществ
1,06
0,02
0,12
—
Транспортные средства
без вредных выбросов
0
0
0
—
10.2.1. Катализаторы «трех процессов»
Катализатор «трех процессов», представляющий собой частицы Pt
и Rh, нанесенные на керамический монолит, представляет собой замечательный пример успеха технологии создания катализаторов. Он позволяет удалять
три загрязняющих среду соединения — СО, N0 и углеводороды в соответствии
со следующими полными реакциями (табл. 10.3).
428
Глава 10. Катализ и защита окружающей среды
Таблица 10.3. Реакции, протекающие в катализаторе «трех процессов»
Реакция
Наиболее эффективный катализатор
СО "Ь 02 —^ С02
Pt, Pd
СЛ+°2^С°2
Pt, Pd
NO + СО -» N2 + С02
Rh, Pd
Кроме того, N0 восстанавливается Н2 и углеводородами. Для обеспечения
одновременного протекания трех реакций (заметьте, что первые две реакции
представляют собой процессы окисления, а последняя — восстановления) необходимо обеспечить определенный состав выхлопных газов, который достигается при отношении воздух/топливо, равном 14,7 (рис. 10.1). При высоком
содержании кислорода в окислении СО расходуется слишком много монооксида углерода и его не остается на конверсию N0. При низком содержании
кислорода N0 конвертируется полностью, но углеводороды и СО не окисляются полностью. Датчик кислорода (Л-зонд) устанавливается перед катализатором, с помощью которого бортовой компьютер поддерживает необходимый
баланс топлива и воздуха, поступающих в двигатель.
Значение Л
Рис. 10.1. Эмиссии СО, NOx и углеводородов и уровень сигнала в датчике кислорода как
функции отношения воздух/топливо. Значение Л = 1 соответствует отношению
воздух/топливо, равному 14,7. Заметим, что все три загрязняющих компонента
могут быть конвертированы одновременно только в достаточно узком диапазоне изменения отношения воздух/топливо
10.2. Каталитическая нейтрализация выхлопных газов -*\г 429
Каталитическую нейтрализацию выхлопных газов стали применять в США в
легковых автомобилях с 1975 года. Первые машины с электронной системой обратной связи и катализаторами «трех процессов» были выпущены фирмой «Вольво» в 1979 г. для продажи в Калифорнии. В настоящее время все новые автомобили с бензиновыми двигателями, продающиеся на Западе, оборудованы каталитическими конверторами. Эти конверторы, безусловно, повышают качество
воздуха, но приводят к дополнительному расходу топлива, поскольку фиксированное отношение воздух/топливо снижает эффективность работы двигателей.
Принцип работы Л-зонда показан на рис. 10.2. Он представляет собой датчик кислорода, сделанный по образу и подобию топливных элементов на твердых оксидах, обсуждавшихся в гл. 8. Оксид, через который проходят ионы
кислорода, нагревается джоулевым теплом для обеспечения высокой подвижности ионов и малого времени отклика (~1 с).
Газ сравнения
Рис. 10.2. Принцип работы Л-зон-
да, используемого для регулирования системы впрыска автомобилей,
обеспечивающего нужное отношение воздух/топливо
Выхлопные газы
Керамика
Электрод из Pt
Отклик
Анализируемый газ
Содержание кислорода в выхлопных газах сопоставляется с его содержанием в газе сравнения, в данном случае в воздухе. Величина отклика определяется уравнением Нернста
RT
4 F
In
Ро
2 эталон
Ро
2выхлопы
и ее зависимость от содержания кислорода в выхлопных газах показана на
рис. 10.3. Заметьте, что сильный отклик при низком содержании кислорода
обеспечивает сильное изменение сигнала в окрестности рабочей точки, задаваемой стехиометрическим отношением. Приведенное выше соотношение показывает, что напряжение стремится к бесконечности, когда содержание кислорода с одной стороны элемента становится исчезающе малым. В реальности
этого не происходит, поскольку в этом случае оксид будет восстанавливаться и
появится электронная проводимость. Показания датчика обычно изменяются в
диапазоне от 0 до 1 В, если двигаться от нулевого парциального давления кислорода внутри датчика.
Работа А-зонда основана на диффузии кислорода через твердый электролит, и поэтому характеризуется некоторым временем задержки (или временем
430 Глава 10. Катализ и защита окружающей среды
отклика). Уменьшая толщину оксидной мембраны и увеличивая рабочую температуру, можно уменьшить время отклика, но сделать его нулевым невозможно. Например, если водитель резко нажмет на педаль газа, выхлопные газы
приобретут свойство восстановителя. Следовательно, осажденная на катализаторе сера будет гидрироваться, в выхлопных газах появится H2S, придавая им
характерный запах тухлых яиц (такой запах иногда появляется при запуске
холодного двигателя). Для избежания этого эффекта следует использовать датчики с более быстрым откликом. В идеале их следует помещать непосредственно у выхода из каждого цилиндра, поэтому они должны выдерживать высокие
температуры.
Ро2> мбаР
Рис. 10.3. Отклик /1-зонда датчика кислорода
10.2.1.1. Каталитический нейтрализатор
Типичная насадка, использующаяся для нейтрализации выхлопных газов, показана на рис. 10.4. Принципиально важная ее конструкционная
особенность состоит в том, что она не должна препятствовать течению выхлопных газов, в противном случае мотор будет глохнуть. То есть каталитический
реактор должен иметь открытую структуру. Это обеспечивается нанесением
каталитически активных частиц на многоканальную решетку, называемую монолитом. Катализаторы такого типа хорошо подходят для работы в условиях,
когда скорость реакции лимитируется транспортом реагентов вне катализатора
(см. также рис. 5.37).
На рис. 10.5 показано устройство такого катализатора. Основная несущая
конструкция образована монолитом, покрытым «отмывающим» слоем толщиной 30—50 мкм. Он представляет собой обычный носитель, состоящий
преимущественно из ^-А1203 (70—80 %) и других оксидов типа оксида церия
(10—30 %), оксида лантана или оксидов щелочно-земельных металлов (ВаО).
В некоторых конструкциях на внешнюю сторону «отмывающего» слоя наносят
NiO для поглощения серы. Более плотные оксиды яг-А1203 или Zr02 иногда
10.2. Каталитическая нейтрализация выхлопных газов
-V431
используются в качестве диффузионного барьера, препятствующего внедрению
родия в матрицу носителя при высоких температурах. Только 1—2 вес. % «отмывающего» слоя приходятся на благородные металлы (Pt, Pd, Rh). В некоторых системах используются все эти три металла, но в большинстве насадок
катализатор содержит Rh и Pt. Недавно были созданы насадки, в которых используется только палладий. Возможны различные вариации состава, но в большинстве случаев состав «отмывающего» слоя представляет коммерческую тайну. Детальное описание катализатора «трех процессов» дано в книге [2]. Здесь
мы лишь кратко опишем функции, выполняемые различными ингредиентами
катализатора.
Рис. 10.4. Типичный нейтрализатор выхлопных газов. Левый нейтрализатор
вскрыт, чтобы можно было видеть монолит. На врезке показана верхняя часть монолита, от которой отщеплен кусочек
• В катализаторе используется гамма-оксид алюминия, который обладает
большой удельной поверхностью и лучше всего подходит для осаждения благородных металлов. Типичный «отмывающий» слой из ^-А1203 имеет удельную
поверхность 150—170 м2/г. При высоких температурах, однако, гамма-оксид
алюминия переходит в альфа-фазу, поэтому следует предпринимать меры для
предотвращения такого перехода. Еще одна проблема связана с диффузией
родия в матрицу оксида алюминия, для подавления которой необходимо использовать диффузионные барьеры.
• Оксид церия подвержен частичному восстановлению (промоторами его
восстановления являются благородные металлы). Когда колебания величины
отношения воздух/топливо находятся в верхней фазе, оксид церия содержит
максимально возможное количество кислорода. Этот кислород необходим для
10 Cf/i
Частицы Pt и Rh
Рис. 10.5. Монолит, «отмывающий» слой и
частицы благородных металлов в катализаторе выхлопных газов
432 -l\r Глава 10. Катализ и защита окружающей среды
образования С02, когда система переходит в фазу обогащенной топливом смеси.
Таким образом, оксид церия противодействует осцилляциям состава смеси
воздух/топливо. Кроме того, он стабилизирует высокую удельную поверхность
^-А1203, ингибируя его превращение в альфа-оксид, подавляет агломерацию
частиц благородных металлов и уменьшает потери их массы, создавая диффузионный барьер. Вакансии кислорода, находящиеся предположительно в области контакта СеОх с благородным металлом, являются активными центрами окисления СО с участием 02 или Н20 по реакции конверсии с водяным
паром.
• Оксид лантана является валентно-инвариантным и не накапливает кислород, но успешно стабилизирует ^-А1?03. Он распределяется по поверхности
оксида алюминия и представляет барьер для растворения родия в носителе.
• Платина является катализатором окисления СО и углеводородов. Она нечувствительна к присутствию свинца и серы. Не известно, растворяется ли она
в «отмывающем» слое при высоких температурах, но ее спекание в крупные
частицы может привести к уменьшению удельной поверхности и резкому снижению каталитической активности по отношению к реакции окисления.
• Родий — основной элемент в катализаторе «трех процессов». Этот металл
является субпродуктом процесса извлечения Pt. Его месторождения находятся в
Южной Африке (2/3) и России (1/3). Однако среднее соотношение Pt: Rh = 17:1
в руде слишком велико для катализаторов. В 1991 г. 87 % всего добываемого
родия использовалось для создания катализаторов (у платины — 37 %). Несмотря на его высокую цену, родий остается практически незаменимым в катализе из-за его уникальных свойств в отношении поведения N0 на межфазных
поверхностях. Родий теряет свою активность вследствие роста частиц в восстанавливающей среде (>900 К) и из-за диффузии атомов в матрицу носителя в
окисляющей среде (>900 К).
• Палладий может быть добавкой к Rh и Pt, но может также и заменять их.
Палладий является таким же хорошим катализатором окисления, как и платина (даже в отношении окисления насыщенных углеводородов), но иногда менее активен в отношении восстановления N0. Следовательно, при использовании только Pd в катализаторе металла требуется в 5—10 раз больше, чем в
случае смешенного катализатора Pt—Rh. Палладий менее устойчив по отношению к отравлению свинцом из бензина, чем Pt, но в США используется бензин, не содержащий свинца. Для катализаторов на основе палладия требуется
более высокое содержание оксида церия, позволяющего предотвратить их деактивацию при высоких температурах.
Оптимальная рабочая температура для катализатора «трех процессов» лежит в диапазоне от 350 до 650 °С. После запуска холодного двигателя требуется
по крайней мере одна минута для установления этой температуры, что предполагает наибольшие выбросы СО и углеводородов непосредственно после запуска двигателя. Следует избегать работы катализатора при температуре выше
800 °С, поскольку в этом случае идет спекание частиц благородных металлов и
растворение родия в носителе.
10.2. Каталитическая нейтрализация выхлопных газов -1\г 433
10.2.1.2. Демонстрационные эксперименты
Выделяемую при каталитическом окислении с участием платины
энергию легко зафиксировать, нагревая спирт (несколько миллилитров), находящийся в колбе Эрленмейера, руками или с помощью горелки и поместив в
колбу нагретую до свечения фольгу толщиной 0,1 мм, раскатанную из платиновой проволоки толщиной 1 мм. Экзотермическое окисление паров спирта
будет поддерживать свечение фольги. При правильно выбранных условиях реакция может идти несколько часов. Вы, вероятно, заметите, что реакция не
является селективной по отношению к образованию С02: можно будет почувствовать запах уксусной кислоты и уксусного альдегида.
Этот простой эксперимент, показывающий возможность окисления спирта
без непосредственного горения, был придуман Дэви в 1925 году; он лег в основу первого практического применения катализа: на этом принципе работают
шахтерские лампы Дэви.
Более утонченный эксперимент может быть осуществлен при наличии насадки для каталитической нейтрализации выхлопных газов. Кусочек монолита
(длиной 5 см и диаметром 2,5 см) помещают в стеклянную трубку, служащую
каталитической насадкой. Поток выхлопных газов моделируются барботирова-
нием воздуха через спирт (например, с помощью насоса для аквариума). Содержание спирта в воздухе варьируется за счет изменения температуры. При
комнатной температуре катализатор не работает, поэтому его нагревают с помощью тепловой пушки. Начавшись, реакция продолжается сколь угодно долго. В зависимости от содержания спирта в воздухе можно вызвать и свечение
катализатора, как это имело место в рассмотренном выше примере.
10.2.1.3. Деактивация катализаторов
Разработнные в США катализаторы «трех процессов» сохраняют
высокую каталитическую активность и обеспечивают стандарты по выбросам
вредных веществ, указанные в табл. 10.2, через 5 лет или 50 ООО километров
пробега автомобиля. Поскольку катализаторы деактивируются в процессе использования, то идет их постоянное обновление, и они обеспечивают уровень
выбросов ниже заложенного в стандартах. При этом следует учитывать различие в способе эксплуатации автомобиля, связанное с особенностями стиля вождения, присущего разным водителям.
Основные причины деградации катализатора связаны термическим и механическим его разрушением, а также с отравлением различного рода примесями.
Высокие температуры способствуют спеканию частиц и могут вызвать реакцию
между металлом и носителем. Сильный нагрев катализатора может быть обусловлен высокой скоростью движения автомобиля или перебоями в зажигании,
приводящими к необходимости (экзотермического) окисления с участием катализатора большого количества несгоревшего топлива. Чрезмерный нагрев катализатора может привести к разрушению носителя или спровоцировать переход
гамма-оксида в альфа-оксид алюминия, который обладает более низкой удель¬
434 Глава 10. Катализ и защита окружающей среды
ной поверхностью. Удары и высокие температуры могут привести к образованию каналов, по которым выхлопные газы могут проходить, минуя катализатор.
Катализатор отравляют свинец и фосфор. Свинец в очень низкой концентрации может присутствовать и в бензине, в который не вводятся добавки соединений свинца, однако в этом случае он не представляет серьезной проблемы. Использование низкокачественного топлива, в частности, содержащего
свинец, может вызвать существенное снижение активности катализатора «трех
процессов» в отношении восстановления N0. Активность в процессах окисления, временно снижающаяся при использовании низкокачественного топлива,
может быть восстановлена до уровня, обеспечивающего стандарты по вредным
выбросам. Это легко понять, вспомнив о низкой температуре плавления свинца и его низкой энергии когезии. В результате он будет иметь низкую поверхностную энергию и проявлять склонность к декорированию поверхности, сопровождающему блокировку активных центров. Этот эффект может быть частично подавлен при наличии на поверхности адсорбированных молекул СО и
кислорода, но он может вызвать и необратимые последствия, как, например, в
случае блокировки ступеней на поверхности Rh, которые, как и при диссоциации N2 на поверхности Ru, ответственны за диссоциацию молекул N0.
Фосфор присутствует в машинном масле (~1 г/л). Он прочно связывается с
поверхностью оксида алюминия и также может блокировать активные центры
на поверхности благородного металла. Сера, хотя и является потенциальным
отравителем всех металлических катализаторов, с поверхностями платины и
родия взаимодействует относительно слабо, но доставляет неприятности для
палладиевых катализаторов. При низких температурах (ниже 300 °С) сера, окисленная до S02, блокирует поверхность благородных металлов. Небольшая доля
серы, окисленная до S03, участвует в реакции с оксидом алюминия, что приводит к образованию A12(S04)3 и снижению удельной поверхности «отмывающего» слоя. Щелочные металлы и галоиды, оставшиеся от прекурсоров катализаторов, как было установлено, сказываются отрицательно на стабильности «отмывающего» слоя и металлических частиц, по этой причине желательно в
качестве прекурсоров катализаторов использовать соединения, не содержащие
таких элементов. В бензине могут присутствовать и другие загрязняющие примеси, негативно сказывающиеся на работе катализатора «трех процессов»; к их
числу относятся, например, кремнийорганические соединения или добавки на
основе марганца. Анализ чувствительности датчика кислорода к примесям требует отдельного рассмотрения.
10.2.2. Каталитические реакции с участием
катализаторов «трех процессов»:
механизмы и кинетика реакций
Исследования окисления СО и реакции СО + N0 проводятся достаточно интенсивно. Гораздо меньше известно о процессах окисления углеводородов и их роли в восстановлении N0, анализировать которую начали отно¬
10.2. Каталитическая нейтрализация выхлопных газов 435
сительно недавно. Большой вклад в понимание этих процессов внесли исследования, выполненные на хорошо охарактеризованных монокристаллах методами науки о поверхностях. Обзор этих исследований дан в работе [3].
10.2.2.1. Реакция окисления СО
(2.184)—(2.187)]. Предполагая лимитирующей стадией рекомбинацию О и СО,
можно получить следующее выражение для скорости реакции
где V — реакционный объем; N — число каталитически активных центров;
кз+ — константа скорости прямой поверхностной реакции между СО и О;
0; — степени заполнения поверхности молекулами, указанными в индексе;
К{ — константы равновесия адсорбции; р( — парциальные давления соответствующих газов. Степень покрытия поверхности молекулами С02 считалась
пренебрежимо малой, и полагалось, что система находится далеко от равновесия, так что обратная реакция не учитывалась.
Энергетические параметры реакции окисления СО приведены на рис. 10.6.
Энергия активации для гомогенной реакции в газовой фазе между СО и 02
оценивается величиной 500 кДж/моль, затрачиваемой на разрыв связи О—О в
молекуле кислорода, и, очевидно, диссоциация 02 представляет собой лимитирующую стадию этой реакции. Катализатор, однако, облегчает диссоциацию
02 и лимитирующей становится реакция между СО* и О*, энергия активации
которой составляет 100 кДж/моль. Реакция окисления СО элегантно показы-
Реакция окисления СО подробно обсуждалась в гл. 2 [уравнения
2 ’
(10.1)
СО + 1/2 02
Рис. 10.6. Приближенная
энергетическая диаграмма для
окисления СО на палладии.
Отметьте, что самый высокий
энергетический барьер отвечает рекомбинации СО + О [4]
22
285
со2
С02
Координата реакции
436 -l\r Глава 10. Катализ и защита окружающей среды
вает, что основная роль катализатора сводится к разрыву связи. По завершении
этого процесса следует основная реакция, если, конечно, интермедиаты не слишком прочно связаны с поверхностью, как этого требует принцип Сабатье.
Особый интерес представляет температурная зависимость скорости реакции, проливающая свет на основные явления, происходящие при катализе
(см. также гл. 2, рис. 2.12). При низких температурах поверхность покрыта в
основном молекулами СО и в знаменателе выражения для скорости реакции
доминирует член Ксо[СО], который приводит к отрицательному значению порядка реакции по СО. При температурах выше температуры десорбции СО на
поверхность начинает встраиваться кислород, в результате слагаемое в знаменателе, содержащее концентрацию СО, становится несущественным и порядок
реакции по СО оказывается положительным. Комбинация (^Г2[02])1/2, однако,
не является пренебрежимо малой.
На рис. 10.7 показана зависимость от температуры скорости реакции окисления СО на поверхности родия, полученная в [5]. Из рисунка ясно видно, что
скорость реакции достигает максимума, когда на поверхности адсорбированные СО и О присутствуют примерно в одинаковых количествах при температурах, обеспечивающих преодоление активационного барьера реакции. При низких температурах на скорость реакции сильное влияние оказывает недостаток
кислорода на поверхности, а при высоких температурах адсорбционно-десорб-
ционное равновесие для СО сдвинуто в сторону газовой фазы, так что степень
заполнения поверхности молекулами
СО становится малой. Как отмечалось
в гл. 2, такое неаррениусовское изменение скорости реакции с температурой характерно для каталитических
процессов.
Рис. 10.7. Зависимости от температуры скорости образования С02 из СО и 02 на поверхностях Rh (111) и Rh(l 10) [5]. Отметьте подобие этих зависимостей и данных
зоо 400 500 600 700 простых модельных расчетов, представлен-
Температура, К ных на рис. 2.12
10.2.2.2. Является ли реакция окисления СО
структурно-нечувствительной ?
Реакция окисления СО часто трактуется как структурно-нечувствительная, что подразумевает одно и то же значение числа оборотов реакции для
различных центров на поверхности определенного металла или для различных
граней кристаллов. На рис. 10.7 показано, что скорости реакции на гранях
Rh(lll) и Rh(110) практически совпадают на низкотемпературной стороне максимума, но заметно различаются при более высоких температурах. Такое пове-
10.2. Каталитическая нейтрализация выхлопных газов ->\г 437
дение определяется тем, что при низких температурах поверхность покрыта в
основном молекулами СО, и, следовательно, скорость образования С02 определяется главным образом скоростью десорбции СО, освобождающей место
для кислорода. Поскольку теплоты адсорбции СО на указанных выше гранях
почти одинаковы, оказываются близкими и скорости окисления СО на этих
двух поверхностях. При высоких температурах, однако, когда адсорбционно-
десорбционное равновесие для СО сдвинуто в сторону газовой фазы, скорость
окисления определяется скоростью реакции между О* и СО*, и становится
явно видным различие между этими гранями (см. рис. 10.7). Кажущаяся структурная нечувствительность реакции окисления СО, как представляется, есть
результат случайного совпадения, не связанного ни с характеристиками активных центров, ни с состоянием реагентов на них.
Очень удобным в изучении характеристической каталитической активности поверхностей оказывается метод температурно-программируемой реакции,
осуществляемый при низких степенях заполнения поверхности. Такие эксперименты [6], проведенные для поверхностей (100) и (111) родия, содержащих
малые количества СО* и О*, с очевидностью демонстрируют различие активностей этих поверхностей в образовании С02. Таким образом, следует быть очень
осторожным в приписывании структурной нечувствительности реакций, выявленной в нормальных условиях высокого давления и больших степеней заполнения поверхности, характеристическим свойствам активных центров. Изложенные в гл. 6 теории хемосорбции и поверхностных реакций практически не
оставляют шансов связать эффект структурной нечувствительности со свойствами активных поверхностных центров: атомы, находящиеся в геометрически различных состояниях, всегда имеют различные энергии связи.
10.2.2.3. Реакция СО + N0
Реакция СО + N0, безусловно, зависит от структуры поверхности
катализатора и ее скорость изменяется со сменой катализирующей грани. Температурно-программируемый анализ реакции (рис. 10.8) позволяет выделить ее
отдельные ступени: СО и N0 адсорбируются примерно в одинаковых количествах на поверхностях Rh(lll) и Rh(100), и при десорбции наблюдаются пики,
соответствующие СО, С02 и N2, однако десорбция N0 при этих степенях заполнения не наблюдается. Тогда на основании экспериментов по температурно-программируемой реакции, данные которых представлены на рис. 10.8,
можно предложить следующие стадии реакции:
N0 + * <z> N0 , (10.2)
СО + ^ СО:, (10.3)
NO + * <- NOa +0,, (10.4)
СО; + О <— С02 + 2% (10.5)
N* + N* <- N2 + Т.
(10.6)
438 -l\r Глава 10. Катализ и защита окружающей среды
Основной стадией в каталитическом восстановлении N0 является разрыв
связи N—О на поверхности родия (см. также гл. 7). Хотя родий обладает достаточной активностью для осуществления этого разрыва (даже в отсутствие промоторов), тем не менее диссоциация может быть сильно заторможена при очень
высоких степенях заполнения поверхности (как это показано на рис. 7.12).
При низких степенях заполнения таких стерических ограничений нет.
0,20 МС СО + 0,26 МС N0 0,15 МС 13СО + 0,20 МС N0
Температура, К Температура» К
Рис. 10.8. Температурно-программируемая реакция NO и СО на двух различных гранях
родия. Молекулярно адсорбированный NO полностью диссоциирует при относительно низких температурах, но молекулы NO не десорбируются. Отметьте различие в селективности и реакционной способности поверхностей: на
Rh(100) большинство молекул СО окисляется до С02, а реакция начинается
уже при 300 К. На поверхности Rh(l 11) большая часть молекул СО десорбируется, не вступив в реакцию, а образование С02 не наблюдается до 400 К. Вместе с тем образование N, идет на Rh(l 11) быстрее, чем на Rh(100) [7]
В табл. 10.4 приведены кинетические характеристики элементарных стадий
реакции CO+NO, определенные в пределе низких степеней заполнения поверхности. Представленные в табл. 10.4 данные составляют важную основу для
моделирования механизма полной реакции. Кроме того, параметры элементарных стадий можно сравнивать с данными расчетов, выполненных в рамках
той или иной теории, описанной в гл. 3 и 6. Можно сказать, что кинетические
параметры элементарных стадий, наряду с данными спектроскопических исследований и численного моделирования, перекидывают мостики между молекулярными свойствами адсорбированных реагентов и их реакционной способностью. Статистическая термодинамика дает теоретический базис для выражения констант равновесия и скоростей реакций через статистические суммы
колебательных и вращательных движений молекул. Таким образом, спектро-
10.2. Каталитическая нейтрализация выхлопных газов -*\г 439
скопические исследования адсорбированных реагентов и интермедиатов дают
основу для расчета констант равновесия, а полученные расчетным путем данные о параметрах состояния молекул на различных этапах развития реакции,
начиная от структуры и основных электронного и колебательных состояний,
позволяют предсказывать кинетические параметры.
Таблица 10.4. Кинетические параметры элементарных стадий реакции
N0 + СО [7—9]
Кинетические параметры
реакции СО + NO
Rh(100)
Rh(iii)
Eact, кДж/моль
v, с 1
Eact, кДж/моль
v, с 1
Диссоциация NOads
37 ±3
10M±1
65 ± 6
10n±1
Десорбция NOads
106 ± 10*
j q1 3,5 ± 1
113 ± 10*
j o13,5 ± 1
Десорбция COads
139 ± 3
j q14 ± 0,3
155 ± 5
10l5±1
Реакция
C^ads + Oads =
103 ± 5
JQl2,7 ±0,7
67 ± 3
107,3±0,2
Реакция
N + N = N
ads ^ ads 1N2
215 ± 10
j Ql 5,1 ±0,5
118 ± 10
1010±I
10.2.2.4. Реакция СО + N0 при высоких давлениях
Реакция СО + N0 лишь частично описывается уравнениями
(10.2)—(10.7), поскольку должна быть еще одна ступень, характеризующая образование N20, которое имеет место, в частности, при низких температурах.
На рис. 10.9 в форме аррениусовских графиков показаны зависимости от температуры скоростей образования С02, N20 и N2 на поверхности (111) родия.
Сравнение с зависимостью, относящейся к более открытой поверхности Rh(llO),
показывает, что реакция является структурно-чувствительной. Поскольку N20
является нежелательным продуктом, важно выявить условия, при которых его
выход является минимальным. Заметим, что селективность по отношению к
N20, определяемая приведенным ниже отношением (10.7), резко падает при
высоких температурах, при которых обычно работает катализатор. Далее, катализатор «трех процессов» содержит частицы родия и промотор СеОх, подавляющий образование N20 [11]. Наконец, N20 вступает в реакцию с СО, также
катализируемую родием, в результате которой образуются N2 и С02
-У (N30) ■ [N^Ol^N;] Х 100 %' (l0J)
440 -l\r Глава 10. Катализ и защита окружающей среды
Рис. 10.9. Аррениусовские зависимости
для реакции NO+CO, протекающей на
поверхностях родия (111) и (110) [10]
Таким образом, механизм реакции СО + N0 при реальных давлениях очень сложен. В дополнение
к рассмотренным выше стадиям
следует учесть возможность образования интермедиатами на поверхности островков и упорядоченных
структур. Для учета этих эффектов
необходимо проводить численное
м 1,5 1,6 1,7 1,8 1,9 2,0 моделирование, например, мето-
1000/7", К1 дом Монте-Карло. Следовательно,
мы еще очень далеки от полного
кинетического описания реакции СО + N0. Интересующихся механизмом и
кинетикой этой реакции мы отсылаем к обзору [12].
10.2.2.5. Реакции с участием углеводородов
Углеводороды в выхлопных газах реагируют с кислородом и N0.
Хотя этим реакциям уделяют меньше внимания, чем окислению СО и восстановлению N0 с помощью СО, реакции с участием углеводородов важны в
определении механизма полной реакции, протекающей на катализаторе «трех
процессов», в частности и потому, что в реальных условиях реакционную смесь
нельзя считать идеально перемешанной. В результате возможно образование
зон, где, например, углеводородные фрагменты и атомы азота соадсорбируют-
ся на поверхности каталитических частиц благородных металлов, что может
привести к образованию нежелательных продуктов типа HCN. К счастью, присутствие кислорода, как оказалось, предотвращает появление таких продуктов
в реальных системах. Кинетическое описание реакции с учетом углеводородных молекул является очень сложным, поскольку их разложение на поверхности благородных металлов идет нетривиальным путем.
10.2.2.6. Катализаторы накопления/восстановления NOx
в двигателях с низким содержанием топлива
Один из непосредственных способов снижения содержания диоксида углерода в выхлопах состоит в повышении эффективности использования
топлива в двигателях. Катализатор «трех процессов», обеспечивая очистку выхлопных газов от вредных выбросов, все же диктует в качестве необходимого
условия фиксацию отношения воздух/топливо, равного 14,7 : 1. К сожалению,
10.2. Каталитическая нейтрализация выхлопных газов 441
это отношение не является оптимальным для работы двигателя. Оптимум приходится на отношение 20:1, то есть соответствует низкому содержанию топлива, когда выхлопные газы обогащаются кислородом и оксидами NOx, восстановление которых представляет довольно сложную задачу (см. рис. 10.1).
Реакция-мечта, способная немедленно решить проблему, — прямое разложение N0 на N2 и 02. Этот процесс является в сильной мере экзотермическим,
то есть A#N0 = -91 кДж/моль, и константа равновесия говорит о предпочтительном разложении N0 на молекулярные кислород и азот при низких температурах. Если бы эта реакция была осуществимой, то охлаждение газа до 500 °С
уже было бы достаточным для снижения равновесного давления N0 до уровня 1-й миллионной доли в типичных выхлопных газах, содержащих, например,
80 % азота и 20 % кислорода. В литературе имеются указания, что Си-цеолит
способен разлагать N0 непосредственно на молекулярные кислород и азот, но,
к сожалению, этот цеолит неустойчив во влажной среде [13]. Таким образом,
подходящий катализатор для этой реакции еще не найден. Металлы не являются хорошими кандидатами на эту роль, поскольку обычно поверхности, разрывающие связь N0, имеют сильное сродство к атомам О. Например, в экспериментах по температурно-программируемой десорбции N0 с поверхности родия, N0 легко диссоциировал, N2 десорбировался при разумной температуре
от 500 до 800 К (в зависимости от структуры поверхности), но кислород не
десорбировался до температуры 1000 К, которая уже выходит за пределы рабочего окна катализатора «трех процессов».
Катализатор накопления/восстановления (КНВ) NOx, разработанный в компании Тойота и ряде других фирм, представляет собой решение проблемы,
основанное на двухступенчатом процессе, в котором двигатель периодически
переключается с режима богатой топливной смеси на режим обедненной топливной смеси и наоборот. КНВ представляет собой комбинацию активного по
отношению к окислению платинового катализатора и катализатора на основе
оксида бария, накапливающего NOx. На рис. 10.10 показан принцип работы
этого катализатора.
0,10
Рис. 10.10. Принцип работы катализатора накопления NOx. При обедненной топливной смеси NO окисляется
до N02 и накапливается в ВаО в виде
нитрата бария. При насыщении накопителя кратковременное обогащение топливной смеси приводит к восстановлению нитратов и цикл можно
начинать заново. Заметим, что в реальных системах отношение длительностей периодов, отвечающих обедненной и обогащенной смесям, поддерживается на более низком уровне,
чем указано на рисунке [14]
О
z
ск
S
и
сс
Q.
0,08
0,06
о
- Богатая смесь:-
воздух/топливо = 10, 1 с
Бедная смесь:
воздух/топливо = 23,5
Входящий газ
л
0,04 -
0,02
Выходящий
газ
■Ашд! tail и/
У'
4 6
Время, мин
10
442 -l\r Глава 10. Катализ и защита окружающей среды
При обедненной смеси все компоненты выхлопных газов окисляются в катализаторе на частицах платины. В частности, N0 окисляется до NOr Последний вступает в реакцию с ВаО, что приводит к образованию BA(N03)2. При
поступлении обогащенной смеси эта стадия длится несколько секунд, в выхлопных газах имеется дефицит кислорода и присутствуют восстанавливающие
соединения типа СО, Н2 и углеводородов, которые восстанавливают нитрат
бария; в результате этой реакции образуются N2, С02 и Н20.
Оксид бария не является катализатором; все реакции с его участием идут в
стехиометрических соотношениях. Тем не менее, как показывает рис. 10.10,
даже при насыщении оксида бария как накопителя содержание NOx на выходе
из катализаторе меньше, чем на входе, что связано со способностью платины
восстанавливать NOa. в присутствии углеводородов в обогащенных кислородом
выхлопных газах.
Высокая чувствительность к примесям серы — основное препятствие к
широкому применению КНВ. Она обусловлена тем, что S02 взаимодействует с
ВаО так же, как и N02. Однако сульфат бария (BaS04) является более устойчивым соединением, чем нитрат бария, поэтому S02 деактивирует КНВ. Интересно отметить, что при высоком содержании Н2 в выхлопных газах повышается устойчивость катализатора по отношению к примесям серы. Для повышения содержания водорода в выхлопных газах в катализатор вместе с родием
вводят Zr02, который обладает активностью в реакции конверсии с водяным
паром, в результате которой из Н20 и углеводородов образуются Н9 и С02, что
и обеспечивает повышение устойчивости катализатора по отношению к примесям серы.
В настоящее время концепция КНВ востребована только на рынках тех
стран, где используется топливо с низким (30 миллионных долей и менее)
содержанием серы, например в Японии и Швеции. Впервые КНВ был апробирован компанией Тойота в 1994 году, и в настоящее время (конец 2000 г.) в
Японии такими катализаторами оборудовано около 300 000 автомобилей. Поскольку стандарты на содержание серы в топливе ужесточаются, у КНВ открываются более широкие перспективы, поскольку он позволяет бензиновым двигателям повысить эффективность их работы, при этом происходит одновременное снижение выбросов С02.
10.2.3. Заключительные замечания
по каталитической обработке выхлопных газов
Катализатор «трех процессов» и катализатор накопления/восстановления NOx являются прекрасными примерами успешных каталитических
технологий. Эти катализаторы уникальны по своей природе и могут работать
при широком изменения параметров окружающей среды, которые определяются типом автомобиля, стилем его вождения, климатическими условиями и т. д.
Этим они отличаются от промышленных катализаторов, для которых условия
проведения реакций обычно поддерживаются на определенном уровне.
10.3. Загрязнение воздуха крупными стационарными источниками вредных веществ ->\г 443
Катализатор «трех процессов» даже можно считать «сконструированным с
запасом», поскольку он не теряет своих рабочих свойств даже после многолетнего использования. При ужесточении экологических требований катализатор
«трех процессов» может быть усовершенствован с целью удовлетворения этим
требованиям. Например, высокие требования принятых в Калифорнии стандартов, связанные с необходимостью обеспечения сверхнизкого уровня выбросов, (см. табл. 10.2) могут быть легко удовлетворены при использовании катализатора «трех процессов» путем его прогрева перед запуском двигателя. Другой элегантный путь снижения уровня выбросов углеводородов в период прогрева
двигателя связан с пропусканием выхлопных газов через цеолит до их поступления в каталитический конвертор. При низких температурах углеводороды
будут адсорбироваться на внутренней поверхности цеолита. По мере прогрева
двигателя и повышения температуры выхлопных газов углеводороды будут десорбироваться из цеолита и окисляться на катализаторе. Приближение катализатора к двигателю будет способствовать более быстрому его прогреву после
запуска холодного двигателя. С новыми разработками в этой области читатель
может ознакомиться по книге [2].
10.3. ЗАГРЯЗНЕНИЕ ВОЗДУХА
КРУПНЫМИ СТАЦИОНАРНЫМИ ИСТОЧНИКАМИ
ВРЕДНЫХ ВЕЩЕСТВ
10.3.1. Процесс селективного
каталитического восстановления
Крупные стационарные электростанции представляют собой другой источник потенциально опасных выбросов в атмосферу. Предварительная
обработка топлива может способствовать снижению выбросов серы, если в качестве топлива используется нефть, но остается проблема удаления NOx, поскольку топливо сгорает при высоких температурах. Уголь, служащий топливом на некоторых станциях, в качестве основного загрязняющего компонента
содержит серу. Электростанции обычно работают в условиях обедненной смеси, что предполагает наличие избытка кислорода в дымовых газах. Как указывалось выше, существуют технологии, позволяющие очищать выхлопные газы
в автомобилях, работающих на обедненной смеси путем последовательного включения на короткое время режима обогащенной смеси между относительно длительными периодами работы с обедненной смесью. Эта технология неосуществима, по крайней мере, на гигантских электростанциях мощностью 500—1000 МВт.
Таким образом, требуется разработать каталитический процесс для стационарного режима, хотя он должен допускать возможность изменения состава дымовых газов.
В табл. 10.5 дан состав дымовых газов бойлерных, работающих на угле.
В отношении воздействия на окружающую среду наиболее серьезную пробле¬
444
Глава 10. Катализ и защита окружающей среды
му представляет S07, который в воздухе окисляется до S03, а затем приводит к
выпадению кислотных дождей. Кислотные дожди были основной проблемой
до 1960—1970-х гг. В настоящее время каталитические технологии окисления
S02 из дымовых газов до S03 (в этом процессе используется расплав V205) и
гидратирования последнего до H2S04 позволили исключить выбросы S02 и
превратили электростанции, работающие на угле, в заводы по производству
серной кислоты.
Таблица 10.5. Состав дымовых газов электростанций, работающих на угле [15]
NO,
миллионные доли
no2,
миллионные доли
so2,
миллионные доли
S03,
%
н2о,
%
°2,
%
со2,
%
Аэрозольные
частицы
(низкое
содержание
частиц), мг/м3
Аэрозольные
частицы
(высокое
содержание
частиц), г/м3
400-700
2-5
500-2000
2-20
6-8
4-5
10-12
5-20
10-20
Но удалить N0 можно только каталитически, введя в дым восстановитель —
аммиак. Такой процесс называется селективным каталитическим восстановлением (СКВ). Катализатор состоит из оксидов ванадия и титана, а его рабочая
температура лежит в интервале 600—700 К; полная реакция выглядит следующим образом:
4NH3 + 4NO + 02 -> 4N2 + 6Н20. (10.8)
СКВ-реактор
Теплообменник
Печь
начального горения
Топливо
| Смеситель
Аммиак
cz.;.s-jL'
Водяной пар
Теплообменник
Водяной пар
■110
Зола уноса
Емкость для рециркуляции кислоты J..
Конвертер S02
Чистый газ
- Воздух
Серная кислота
Охлаждающая вода
Охлаждение кислоты
Рис. 10.11. Схематическое представление процесса SNOX, использующегося для удаления SO0 и N0 из дымовых газов
10.3. Загрязнение воздуха крупными стационарными источниками вредных веществ ->\г 445
Аммиак инжектируется в чистом виде под высоким давлением или в виде
водного раствора при атмосферном давлении. Вместо аммиака можно использовать также мочевину. В этом процессе необходимо удалить по возможности
как можно больше NOx и полностью конвертировать восстановитель, поскольку выбросы NH3 из реактора были бы крайне нежелательны.
Комбинированный подход к одновременному удалению серы и NOx из дымовых газов носит называние SNOX или DESNOX. Схема установки, реализующей этот подход, показан на рис. 10.11. В нем 99 % NOx конвертируется в
реакторе СКВ, a S02 перерабатывается в серную кислоту.
10.3.1.1. Катализатор для процесса
СКВ сконструирован так, чтобы в процессе его работы при больших объемах
потока газа (например, 300 нормальных м3/с для электростанции мощностью
300 МВт) не возникали большие перепады давления. На рис. 10.12 показано
устройство ректора, объем монолитного катализатора в котором равен 250 м3,
что достаточно для электростанции мощностью 300 МВт.
Рис. 10.12. Реактор СКВ, загруженный
блоками монолита, один из которых
показан на рисунке справа. Каждый .
блок имеет размер 40 х 40 х 50 см3. Для
электростанции мощностью 300 МВт 44444
требуется более 3000 блоков [15] 4N2 + 6H20
Существуют наборы монолитных катализаторов, охватывающие весь диапазон возможных топлив, применяемых на электростанциях, — нефть, газ,
биомасса. Твердые аэрозольные частицы, представляющие определенную проблему, отфильтровываются из дымового газа с помощью электростатических
фильтров перед (при работе в условиях низкого содержания частиц) или после
СКВ-реактора (работа в условиях высокого содержания частиц).
Наносимый на монолит каталитический материал также может быть представлен в различных формах. Используются материалы на основе железа, хрома, ванадия (активные материалы), нанесенные на ТЮ2, А1203, SiO? и цеолиты.
Обзор каталитических материалов, использующихся в СКВ, дан в [16].
селективного каталитического восстановления
Как и в случае автомобильных каталитических насадок, реактор
446 -1 \r Глава 10. Катализ и защита окружающей среды
В настоящее время в качестве катализатора широко используется оксид
ванадия, нанесенный на оксид титана; этот катализатор устойчив по отношению к действию S02. Как правило, оксид титана берется в форме анатаза,
поскольку здесь удельная площадь поверхности выше, чем у рутила. Имеется
много соединений и веществ, отравляющих катализатор, например мышьяк и
калий. Последний представляет серьезную проблему при использовании в качестве топлива биомассы. Например, солома, являющаяся субпродуктом производства зерна, привлекательна как биотопливо, но содержит много калия,
который очень подвижен при высоких температурах и конденсируется в системах охлаждения газа, в частности на СКВ-катализаторе, где он может блокировать каналы монолита, снижая время его эффективной работы до нескольких
месяцев. Преодоление этой проблемы — одна из основных задач в будущем
использовании в качестве топлива биомассы. Способы восстановления и защиты СКВ-катализаторов находятся в стадии разработки.
В табл. 10.6 показаны основные виды топлив, использующихся на электростанциях. В настоящее время уголь и мазут являются основными топливами.
Перспективным является топливо Orimulsion, разработанное недавно в Венесуэле, привлекательность которого определяется высоким содержанием водорода, которое обеспечивает низкий уровень выбросов С02.
Таблица 10.6. Сравнение характеристик различных топлив, использующихся
на тепловых электростанциях. Orimulsion является тяжелым
мазутом, который извлекается путем закачки в нефтеносные
платы водяного пара, в результате чего образуется смесь мазута
и воды, которая разжижается только при повышенных
температурах
Единица измерния
Orimulsion®
Уголь
Мазут
Теплотворная
способность
МДж/кг
27,5
25
40
Содержание воды
%
30
10
0
Зола
%
0,08
13
0,05
Сера
%
2,7
1
2,5
Углерод
%
60
64
86
Водород
%
7,3
4
11
Азот
%
0,5
1
0,4
Ванадий
Миллионные доли
300
1-30
50
Никель
Миллионные доли
65
0-10
15
Магний
Миллионные доли
20
Нет данных
Нет данных
10.3. Загрязнение воздуха крупными стационарными источниками вредных веществ -*\г 447
Наличие ванадия в некоторых топливах представляет интересную проблему.
Когда газ выходит из печи, ванадий может сконденсироваться на поверхности
теплообменников электростанций. Ванадий — отличный катализатор окисления
S02, так что эта реакция может произойти до попадания газа в СКВ-реактор.
Это ясно видно из рис. 10.13, который показывает степень конверсии S02 на
осевшем на стенки каналов катализаторе на заводе, использующем в качестве
топлива ванадийсодержащий Orimulsion. Содержащийся в топливе калий способствует ускорению окисления S02 до реактора. Проблемы возникают при
введении в дымовой газ аммиака, поскольку S03 и NH3 реагируют с образованием сульфата аммония, при конденсации которого образуется осадок, забивающий каналы монолита. Заметим, что образование сульфата аммония порождает проблемы и в случае, когда аммиак «проскакивает» через СКВ-реактор и
взаимодействует с S03 ниже по потоку.
Температура, °С
Рис. 10.13. Осевший на стенках теплообменников ванадий на тепловых электростанциях,
использующих Orimulsion, катализирует окисление SO?. Отметим, что калий
способствует этой нежелательной конверсии, в то время как селективное отравление катализатора в некоторой степени уменьшает этот эффект. Тем не
менее во всех случаях конверсия идет до равновесного состояния. Поскольку
окисление SO, является экзотермическим процессом, равновесная концентрация быстро падает с ростом температуры [17]
10.3.1.2. Кинетика реакций
селективного каталитического восстановления
Понимание кинетики процесса СКВ сильно помогает в разработке
новых катализаторов с улучшенными характеристиками. Для эффективной работы СКВ-реактора необходимо обеспечить условия максимальной степени
восстановления NOx с исключением возможности проскока через реактор аммиака. На рис. 10.14 показаны зависимости степени восстановления NOx, а
также величины проскока NH3 от отношения NH3/ NOx на входе в реактор для
448 -l\r Глава 10. Катализ и защита окружающей среды
двух различных скоростей газовых потоков. Для поддержания выбросов аммиака на уровне 10 миллионных долей необходимо либо уменьшать скорость газового потока, либо увеличивать количество катализатора в реакторе.
s
о
ct
0
О
S
S
со
X
Z
*
о
*
о
о
Q.
П.
nh3/nox
Рис. 10.14. Степень восстановления NOx и величина проскока аммиака через СКВ-реактор как функции отношения NH3/ NOx для двух различных скоростей газового потока. Максимальный допустимый уровень проскока аммиака составляет
10 миллионных долей, он задает величину скорости или количество катализатора [18]
При анализе промышленных процессов кинетика реакций описывается степенными законами; порядок реакции по NH3 при этом лежит в диапазоне от
0,5 до 1,0, а по NOx — изменяется от —0,1 до 1,0. Хотя такие степенные законы
представляют интерес для практики, более глубокого понимания сущности
протекающих процессов можно достичь при использовании микрокинетичес-
кого моделирования реакции.
Думесик с соавторами [18] предложили модель, включающую шесть ступеней и основанную на общей модели окисления Марса—ван Кревелена:
1.
NH3 + V5+
-OH «zt V-ONH4,
(10.9)
2.
v-onh4.
+ V = О ^ V - ONH3 - V4+ - OH,
(10.10)
3.
NO + V - NH3 - V4+ - ОН N, + H20 + V5+ - OH,
(10.11)
4.
2V4+ - OH
<=t h,o + V3+ + V = 0,
(10.12)
5.
02 + 2V3+ —> 2V = O,
(10.13)
6.
H20 +v5+
-OH ^ v5+ -oh3o.
(10.14)
Первая ступень описывает адсорбцию аммиака, а вторая — его активацию.
Необратимая третья стадия, очевидно, не является элементарной по своей при-
10.3. Загрязнение воздуха крупными стационарными источниками вредных веществ -*\г 449
роде, но, к сожалению, информации об более элементарных ступенях здесь
нет. Четвертая ступень описывает образование воды, а пятая — повторное окисление центра V3+. На пятой ступени происходит блокировка центров адсорбированной водой. Модель, таким образом, основана на каталитическом действии частично окисленных центров и вакансий на поверхности оксида, подобном рассмотренному в гл. 9 при анализе реакции гидрообессеривания. На
рис. 10.15 реакция представлена в виде циклической схемы.
Рис. 10.15. Каталитический
цикл СКВ-процесса, идущий с участием кислотных
центров V5+—ОН и окислительно-восстановительных
центров V=0 [19]
0,6 0,7 0,8 0,9
NH3/NO
Рис. 10.16. Степень превращения NO
и величина проскока аммиака в присутствии О, и Н20 как функции отношения NH3/ NO для катализатора
V203/Ti0, при температуре 623 К.
Линии представляют результат расчета по модели (10.9)—(10.14) для представленных в табл. 10.7 параметров [18]
СКВ-катализатор имеет более сложную структуру, чем обсуждавшийся выше
металлический катализатор. Кроме того, на таком катализаторе трудно проводить исследования, методика которых разработана в науке о поверхностях.
Природа активных центров СКВ-катализатора была изучена методом темпера-
турно-программируемой десорбции N0 и NH3 и методом инфракрасной спектроскопии in situ. Это позволило определить кинетические параметры (табл. 10.7),
по которым можно рассчитать степень конверсии N0 и величину проскока
NH3 (рис. 10.16). Видно хорошее соответствие между моделью и экспериментальными данными в широком диапазоне изменения параметров системы. Это
говорит о том, что она соответствует физической реальности СКВ-процесса,
правильно отражает взаимодействие катализатора с реагирующими газами и
потому более предпочтительна в сравнении с простыми степенными законами,
при использовании которых катализатор представляет собой «черный ящик».
Вместе с тем на многие вопросы ответа еще не получено; так, не ясно, что из
450 -i\r Глава 10. Катализ и защита окружающей среды
себя представляют действительно элементарные стадии процесс и как выглядят активные центры на атомарном уровне.
СКВ-процесс был бы существенно более привлекателен, если бы удалось
полностью избежать выбросов аммиака. Предпринимались многочисленные
попытки поиска углеводородов, более удобных для использования в этом процессе, но пока еще не найдено соединений, способных конкурировать с аммиаком (или мочевиной) в СКВ-процессе.
Таблица 10.7. Кинетические параметры реакций (10.9)—(10.14),
использованные для построения зависимостей на рис. 10.16.
Предэкспоненциальные множители выражены
в с-1 или с-1 - бар-1 для реакций с участием газовой фазы
Стадия
К
е;
К
Е:
I
с-1/(с • бар)
кДж/моль
с-1/(с • бар)
кДж/моль
1
8 х 106
0
1 х 1013
84
2
1 х 10м
91
1 х 10п
133
3
1,3
23,1
—
—
4
1 х 10м
68,5
1,9 х 104
0
5
8 х 102
0
—
—
6
8 х 107
0
1 х 1013
69,3
Выхлопные газы
Сжатый воздух Vfc4—j -
Сигнал о нагрузке I ‘
двигателя Ком. : СКВ-реактор
Выходной А v
' cm нал о г NOv
Накопиюльнач ■; : I '
Раствор Очищенный
Наше восстанавливающего | газ
агента - — • *
Рис. 10.17. Принцип селективного каталитического восстановления с использованием,
например, мочевины или соединения включения аммиака и мочевины как
восстанавливающих агентов в СКВ-реакторе, установленном на дизельном
транспортном средстве типа парома или большегрузного автомобиля
Список литературы
10.3.2 Процесс селективного каталитического восстановления
для использования в транспортных средствах
Катализатор «трех процессов», обсуждавшийся ранее, можно использовать на автомобилях, работающих на бензине, но он непригоден для
дизельных двигателей, поскольку для них характерен режим обедненной топливной смеси. СКВ-технология не испытывает проблем с обогащенными кислородом выхлопными газами, поскольку восстанавливающий NOx агент добавляется в выхлопные газы. В принципе СКВ-реакторы уже используются на
подвижных средствах, выделяющих NOx, — кораблях и, в частности, паромах.
Из соображений безопасности аммиак здесь неприменим, но мочевина, которая легко превращается в аммиак и С02, является безопасным его заменителем. На рис. 10.17 показана схема работы каталитического устройства.
Следующая задача состоит в уменьшении размеров данного реактора, чтобы его можно было использовать на большегрузных автомобилях. В этом направлении сейчас ведутся интенсивные исследования, нацеленные на поиск
достойного ответа существующим вызовам в очистке выхлопных газов и защите окружающей среды.
Список литературы
1. Taylor К.С. Catal Rev. - Sci. Eng. 1993. V. 35. P. 457.
2. Heck R.M., Farrauto R.J. Catalytic Air Pollution Control. New York, Wiley, 2002.
3. Nieuwenhuys B.E. Adv. Catal. 1999. V. 44. P. 259.
4. Engel Г., Ertl G. // J. Chem. Phys. 1978. V. 69. P. 1267.
5. Bowker M, Guo Q., Joyner R.W. // Catal. Lett. 1993. V. 18. P. 119.
6. Hopstaken M.J.P., Niemantsverdriet J.W. //J. Chem. Phys. 2000, V. 113. P. 5457.
7. Hopstaken M.J.P., Niemantsverdriet J.W. //J. Vac. Sci. Technol. A. 2000. V. 18.
P. 1503.
8. Borg H.J., Reijerse /., Van Santen R.A., Niemantsverdriet J.W. // J. Chem.
Phys. 1994. V. 101. P. 10052.
9. Hopstaken M.J.P., Niemantsverdriet J. W. //J. Phys. Chem. B. 2000, V. 104. P. 3058.
10. Peden C.H.F., Belton D.N., Schmieg S.J. //J. Catal. 1995. V. 155. P. 204.
11. Oh S.H. // J. Catal. 1990/ V. 124. P. 477.
12. Zhdanov VP., Kasemo B. // Surf. Sci. Rep. 1997. V. 29. P. 31.
13. Centi G., Perathoner S. //Appl. Catal. A: gen. 1995. V. 132. P. 179.
14. Matsumoto S. CaTTech. 2000. V. 4. P. 2.
15. Tops0e N.Y. // CaTTech. 1997. V. 1. P. 125.
16. Bosh H., Janssen F.J.J.G. // Catal Today. 1998. V. 2. P. 369.
17. Rasmussen S.B. et al. // J. Power Plant Chem. 2003. V. 5. P. 360.
18. Dumesic J.A., Tops0e N.Y., Tops0e #., Chen Y., Slabiak T. //J Catal. 1996.
V. 163. P. 409.
19. Tops0e N.Y. // Science. 1994. V. 265. P. 1217.
451
ВОПРОСЫ И УПРАЖНЕНИЯ
ВОПРОСЫ ПО СОДЕРЖАНИЮ ГЛАВ
Глава 1
1.1. Насколько важна химическая индустрия для экономики вашей страны? Попробуйте определить ее вклад в общий национальный продукт.
1.2. Можете ли сказать, насколько важен катализ для химической индустрии вашей страны?
1.3. Попробуйте составить десять важнейших для химической индустрии
вашей страны каталитических процессов.
1.4. Посетите вебсайт, например, Американского химического общества
(American Chemical Society) и найдите современные версии табл. 1.3 и 1.4 и
прокомментируйте имеющиеся различия.
1.5. Дайте определение катализа.
1.6. Что составляет сущность катализа на молекулярном уровне?
1.7. Во многих учебниках сущность катализа объясняется с помощью диаграммы потенциальной энергии, на которой реагенты и продукты реакции
разделены энергетическим барьером, сопровождаемой утверждением о том, что
катализатор понижает высоту барьера. Вы согласны с этим утверждением?
Поясните свой ответ.
1.8. Поясните общие черты и различия гомогенного и гетерогенного катализа.
1.9. К каким двум введенным в разделе 1.3. категориям относятся биокатализаторы? Или их следует отнести к отдельной категории?
1.10. Почему энзимы часто являются наиболее эффективными катализаторами?
1.11. Приведите несколько примеров биокаталитических процессов, используемых в промышленности.
1.12. Объясните понятия атомарной эффективности и дружественности окружающей среде
1.13. Что такое is-фактор? Какие процессы характеризуются высоким значением is-фактора?
1.14. Объясните, какие характеристики катализаторов отражены на рис. 1.8
на различных пространственных масштабах.
Вопросы по содержанию глав -i\r 453
1.15. Используя рис. 1.2, кратко обрисуйте ситуации, отвечающие неэффективным комбинациям катализатора и реагирующих молекул.
1.16. Дайте перечень наиболее значимых научных журналов, публикующих
статьи по гетерогенному катализу.
1.17. Дайте перечень наиболее значимых журналов, в которых публикуются
статьи по гомогенному катализу. Почему исследователи гомогенного и гетерогенного катализа публикуют свои статьи в различных журналах?
1.18. Объясните разницу между письмом в редакцию, полной статьей, обзором и трудами конференции.
1.19. Что подразумевается под импакт-фактором журналов? Насколько значим этот параметр?
1.20. Что подразумевается под периодом половины жизни цитирования и
что он говорит о статусе журнала?
1.21. Классикой цитирования называются статьи, процитированные 100 и
более раз. Постарайтесь определить несколько классик цитирования в той области катализа, которой вы более всего интересуетесь. Как часто, по вашему
мнению, цитируется средняя статья по катализу?
1.22. Как бы вы наилучшим образом объяснили сущность катализа своему
приятелю, не имеющему представления о химии и химической технологии?
Глава 2
2.1. Почему кинетика является важной для катализа дисциплиной?
2.2. Насколько «старыми» научными дисциплинами являются катализ и
кинетика?
2.3. Изменяет ли катализатор состав равновесной газовой смеси?
2.4. Металлический никель успешно катализирует процесс гидрирования
двойных связей в ненасыщенных углеводородах типа пропилена и бутена. Может ли этот металл катализировать процесс дегидрогенирования алканов типа
пропана и бутана?
2.5. Чем отличаются динамика и кинетика реакций?
2.6. Чем различаются константы скорости и константы равновесия?
2.7. Поясните физический смысл химического потенциала. Как химический потенциал зависит от давления?
2.8. Укажите оптимальные условия для экзотермических реакций. Почему
они не всегда могут быть выполнены?
2.9. Укажите оптимальные условия для процессов, в которых число молекул в продукте реакции меньше числа реагирующих молекул.
2.10. Дайте определение элементарной ступени реакции и отметьте ее отличие от полной реакции.
2.11. Что такое степенной закон реакции? Каково историческое происхождение степенного закона?
2.12. Насколько степенные законы подходят для описания кинетики каталитических реакций?
454 —‘\r Вопросы и упражнения
2.13. Мы определяем порядок реакции по реагентам или продуктам?
2.14. Дайте определение фугитивности газа и скажите, где это понятие используется.
2.15. Дайте объяснение термину «эффективная энергия активации». Как
хорошо эффективная энергия активации удовлетворяет уравнению Аррениуса?
2.16. Как различаются дифференциальное и интегральное уравнения скорости реакции?
2.17. Кинетика каких промышленных процессов может быть описана в стационарном приближении?
2.18. Можно ли представить мысленную ситуацию, при которой стационарное приближение будет пригодно для описания реакций, протекающих в
емкостном реакторе?
2.19. Объясните связь между порядком каталитической реакции по определенному компоненту и степенью заполнения им поверхности.
2.20. Что означает приближение среднего поля в кинетике каталитических
реакций и когда оно становится неприменимым? В этом случае скорость реакции будет выше или ниже предсказанной в приближении среднего поля?
2.21. Дайте вывод изотермы адсорбции Ленгмюра, описывающей молекулярную адсорбцию СО на поверхности металла с адсорбционными центрами одного
типа. Проделайте то же самое для диссоциативной адсорбции Н2, а также для
случая, когда СО и Н2 адсорбируются одновременно на этой поверхности.
2.22. В чем состоит основное различие между механизмами Ленгмюра—
Хиншельвуда и Илея—Риделя? Какой из этих механизмов является наиболее
подходящим для описания каталитических реакций?
2.23. Может ли каталитическая реакция состоять из одной элементарной
стадии?
2.24. В чем состоит различие решений уравнений кинетики каталитических
реакций, полученных точно и в стационарном и квазиравновесном приближениях? В чем суть приближения НИИР (наиболее избыточного интермедиата
реакции)?
2.25. Зависит ли порядок каталитической реакции от давления и температуры?
2.26. Зависит ли кажущаяся энергия активации от давления и температуры?
2.27. По какой причине порядок каталитической реакции может изменить
знак при смене условий проведения реакции?
2.28. Может ли кажущаяся энергия активации каталитической реакции быть
отрицательной? Если да, то при каких условиях?
2.29. Почему квазиравновесное приближение становится непригодным при
малом давлении реагента?
2.30. При каких условиях скорость реакции начинает осциллировать? Приведите примеры колебательных химических реакций.
2.31. Проведите сравнение выражений Михаелиса—Ментена для скорости
реакции, катализируемой энзимами, и Ленгмюра—Хиншельвуда для скорости
той же реакции, протекающей на поверхности металла. Эквивалентны ли эти
два выражения?
Вопросы по содержанию глав -1\г 455
2.32. Предположим, что для некоторой каталитической реакции предложены два механизма. Обсудите, насколько строго можно обосновать корректность или непригодность этих механизмов, основываясь на кинетическом
анализе.
Глава 3
3.1. Какая задача стоит перед теорией скоростей реакций? Используется
ли эта теория учеными, исследующими каталитические процессы?
3.2. Формальное определение статистической суммы предполагает проведение суммирования бесконечного числа членов. Объясните, почему, тем не
менее, статистическая сумма является полезной характеристикой. Чему равно
минимальное значение, которое может принимать статистическая сумма? Почему это значение именно такое?
3.3. Обсудите основные предположения, лежащие в основе вывода распределения Больцмана по энергиям для ансамбля молекул.
3.4. Сколько степеней свободы имеется у молекулы, состоящей из N атомов?
3.5. Запишите выражение для полной статистической суммы двухатомной
гетероядерной молекулы типа СО, находящейся в газовой фазе.
3.6. При каких условиях полная статистическая сумма может быть представлена как произведение статистических сумм для отдельных степеней свободы?
3.7. Запишите выражение для статистической суммы ансамбля N молекул
СО, находящихся в газовой фазе в объеме V.
3.8. При каких условиях статистические функции поступательного, вращательного и колебательного движений адсорбированных молекул (а) близки
к единице, (б) имеют промежуточные значения и (в) велики?
3.9. По какой причине аммиак не может быть синтезирован путем нагрева
бинарного газа N2 и Н2 до температуры, при которой появляются радикалы N*?
3.10. Поясните основные положения теории столкновений. Для реакций
какого типа, по вашему мнению, пригодна теория столкновений?
3.11. Оцените, как много столкновений в единицу времени происходит в
одном литре газа при атмосферном давлении и какая доля этих столкновений
завершается реакцией (используйте характерные для реакций значения температуры и высоты энергетического барьера).
3.12. Почему теория столкновений, использующаяся для нахождения скоростей реакций, приходит в противоречие с равновесной термодинамикой?
3.13. Приведите пример элементарной мономолекулярной реакции. Как
молекула, участвующая в мономолекулярной реакции, приобретает энергию
для преодоления энергетического барьера?
3.14. Почему мономолекулярная реакция приобретает второй порядок при
низких давлениях?
3.15. Изложите кратко основы теории переходных состояний. Как можно
определить параметры, необходимые для получения количественных предсказаний на основе теории переходных состояний?
456 -l\r Вопросы и упражнения
3.16. Опишите вероятные переходные состояния для (а) диссоциации молекул в газовой фазе, (б) реакции превращения циклопропана в пропен, (в) реакции изомеризации CH3CN —» CH3NC, (г) десорбции атома с поверхности и (д)
диссоциации адсорбированной молекулы типа СО на поверхности металла.
3.17. Какие активированные комплексы называются жесткими и рыхлыми?
Как можно выявить природу активированного комплекса по величие предэкспоненциального множителя?
3.18. Объясните в рамках теории переходного состояния понятие коэффициента прилипания. Почему для прямого адсорбционного процесса коэффициент прилипания всегда меньше единицы?
3.19. Объясните, используя закономерности изменения энтропии, почему
прямая адсорбция молекул менее вероятна, чем процесс, идущий через формирование промежуточного комплекса (прекурсора).
3.20. Предложите переходное состояние для десорбции молекул, идущей с
предэкспоненциальным множителем 1016 с-1. Сделайте то же самое, когда предэкспоненциальный множитель равен 1013 с-1.
3.21. Обсудите в рамках вашего знания справедливость и полезность уравнения Аррениуса в терминах теории переходного состояния.
3.22. В чем состоит фундаментальное различие между теорией столкновений и теорией активированного комплекса?
Глава 4
4.1. Опишите морфологию типичного твердотельного катализатора на высокопористом носителе.
4.2. Какова цель определения параметров катализаторов в контексте (а)
промышленного катализа и (б) фундаментальных исследований?
4.3. Дайте для каждого (или нескольких) приведенных ниже методов описание лежащих в его основе физических принципов, характера получаемой с его
помощью информации о катализаторах, а также его преимуществ и недостатков.
Настоятельно рекомендуется использовать четкие схемы и диаграммы:
а) рентгеноструктурный анализ;
б) рентгеноэлектронная спектроскопия;
в) инфракрасная спектроскопия;
г) температурно-программируемые окисление и восстановление;
д) температурно-программируемая десорбция;
е) просвечивающая электронная микроскопия;
ж) спектроскопия дальней тонкой структуры рентгеновского поглощения;
з) комбинационное рассеяние;
и) мессбауэровская спектрскопия;
к) спектроскопия рассеяния ионов или рассеяние ионов низкой энергии;
л) масс-спектроскопия вторичных ионов.
4.4. Почему электронная и ионная спектроскопии являются чувствительными к поверхности при использовании низкоэнергетических частиц?
Вопросы по содержанию глав ->\г 457
4.5. Когда в электронном микроскопе пучок высокоэнергетических электронов падает на образец, становится доступным большой объем информации,
которую можно извлечь из процессов рассеяния, дифракции и затухания (см.
рис. 4.13). Поясните, какую дополнительную информацию об образце можно
при этом извлечь.
4.6. Для каких методов, из перечисленных в вопросе 4.3, требуется создание вакуума и почему?
4.7. Почему широко используемые в науке о поверхностях методы типа
дифракции медленных электронов, сканирующей туннельной или атомно-си-
ловой микроскопии обычно оказываются непригодными для исследования катализаторов на носителях?
4.8. Объясните физические принципы метода дифракции медленных электронов и сравните его с рентгеноструктурным анализом.
4.9. Объясните важность введения понятия «обратная решетка».
4.10. Объясните физические принципы, лежащие в основе методов сканирующей зондовой микроскопии СТМ и атомно-силовой микроскопии, и обсудите характер информации, получаемой с помощью этих методов. В чем состоит различие между этими двумя методами?
4.11. Обсудите стратегию создания модельных катализаторов, позволяющих применять методы, разработанные в науке о поверхностях, для исследования каталитических процессов.
4.12. Предложите стратегию для наведения мостов между миром адсорбции
и реакций на хорошо охарактеризованных поверхностях монокристаллов и
миром катализаторов на носителях, работающих при высоких давлениях.
4.13. Как много методов (из числа перечисленных на рис. 4.3) может быть
включено в схему, представленную на рис. 4.2?
4.14. Какие из перечисленных на рис. 4.3 методов могут быть использованы
для определения характеристик катализаторов in situ?
4.15. Если вы начинаете исследования в рамках проекта по гетерогенному
катализу и вам предложено выбрать четыре метода из числа перечисленных на
рис. 4.3, то какие методы вы выберете? Объясните свой выбор.
Глава 5
5.1. Сформулируйте основные требования, предъявляемые к твердым катализаторам, используемым в промышленности.
5.2. Изобразите структуру поверхностей (111), (100) и (110) ГЦК-кристал-
лов металла, поверхностей (110) и (100) ОЦК-кристаллов металла и поверхность (001) ГПУ-кристалла.
5.3. Выразите площадь элементарной ячейки для поверхности (100)
ОЦК-кристалла и поверхности (111) ГЦК-кристалла через постоянную решетки а.
5.4. Почему у ГЦК-кристалла поверхность (100) является более реакцион-
но-активной, чем поверхность (111)?
458 -l\r Вопросы и упражнения
5.5. Изобразите следующие структуры:
а) поверхность (110) ГЦК-кристалла с адслоем адсорбата со сверхрешеткой (2 х 1);
б) поверхность (111) ГЦК-кристалла с адслоем адсорбата со сверхрешеткой (2 х 2);
в) ту же структуру, что и в примере б), но с тремя адсорбированными
молекулами в ячейке сверхрешетки (2x2);
г) поверхность (111) ГЦК-решетки с адслоем адсорбата со сверхрешеткой
(л/з х л/з)/гзо°.
5.6. Расскажите о наиболее важных различиях между поверхностями металлов и оксидов.
5.7. Объясните различие между концепциями кислотности Люиса и Бренстеда.
5.8. Поясните роль поверхностной свободной энергии в таких явлениях,
как сегрегация сплавов, реконструкция поверхности, фасетрование и спекание
малых частиц.
5.9. Как окружение сплава (воздух, вакуум) влияет на его поверхность.
5.10. Если мы осадим на поверхность кристалла никеля небольшое количество атомов железа, то что с ними произойдет, когда система придет в равновесие? Что случится с атомами Ag на поверхности Ru, атомами Ru на поверхности Ni и атомами Со на поверхности Си?
5.11. Чем определяется форма частиц металла в вакууме? Что определяет
форму частиц металла, находящихся на подложке?
5.12. Сформулируйте основные допущения, использованные при выводе
изотермы БЭТ. Чем различаются изотермы БЭТ и Ленгмюра?
5.13. Какую информацию можно извлечь из изотермы БЭТ?
5.14. Составьте, используя каталоги и данные, представленные на веб-сайтах, обзор по коммерческим носителям катализаторов и диапазонам изменения их удельной поверхности.
5.15. Предложите простой способ получения методом восстановления катализатора с 2 вес. % Pd на Si02, используя носитель с удельной поверхностью
200 м2/г и объемом пор 0,5 мл/г. Для ускорения каких реакций может быть
использован этот катализатор?
5.16. Как можно измерить и выделить из общей площади площадь поверхности активного металла на носителе?
5.17. Расскажите о возможном применении высокодисперсного оксида титана, находящегося на поверхности оксида алюминия.
5.18. Что такое цеолиты? Чем они отличаются от носителей катализаторов?
5.19. Объясните, почему цеолиты, содержащие алюминий, имеют химически активную поверхность.
5.20. Предложите аргументы за и против использования цеолитов вместо
катализаторов на носителях.
5.21. Зачем порошок катализатора спрессовывается в тела определенной
формы?
Вопросы по содержанию глав -1\г 459
5.22. Объясните концепцию, лежащую в основе диффузионного модуля Тиле
для сферических частиц. Почему этот модуль играет важную роль в катализе?
5.23. Дайте качественное описание, используя графическое представление
аррениусовской зависимости, влияния транспортных ограничений на кажущуюся энергию активации.
5.24. Можете ли вы привести примеры, в которых скорость полного каталитического процесса контролируется переносом реагентов вне каталитических частиц?
5.25. В чем состоит разница между внутренней и внешней активностями
катализатора?
5.26. Как вы представляете себе архитектуру катализатора, который должен
использоваться в условиях, когда имеются сильные ограничения на перенос
газа внутрь каталитической частицы?
5.27. Какие из наиболее важных свойств катализаторов следует принимать
во внимание при разработке катализаторов для промышленных процессов:
а) скорость превращений на одном активном центре;
б) скорость превращений на единицу веса катализатора;
в) скорость превращений на единицу объема катализатора?
Объясните свой выбор.
5.28. Предположим, что вы приготовили катализатор из оксида железа на
носителе из оксида алюминия. Ваша цель — использовать катализатор в форме
металла, но вы желали бы иметь частицы по возможности наименьшего размера со степенью восстановления не меньше 50 %. Следовательно, вам необходимо знать размер частиц оксида железа в невосстановленном катализаторе, а
также размер частиц железа и степень их восстановления. Используя данные
гл. 4 и 5, разработайте стратегию получения этой информации (к несчастью
для вас, как нам представляется, электронная микроскопия и рентгеноструктурный анализ не позволяют получить интересующую вас информацию для
невосстановленных катализаторов).
Глава 6
6.1. В чем состоит принципиальная разница между физической адсорбцией и химической адсорбцией?
6.2. Почему атом испытывает сильное отталкивание при сильном сближении с поверхностью или другим атомом?
6.3. Объясните природу ван-дер-ваальсовского взаимодействия.
6.4. Опишите качественно, что произойдет при сближении двух атомов:
а) если их внешние оболочки заполнены частично;
б) если их внешние оболочки заполнены полностью.
6.5. Почему расщепление на связывающие и разрыхляющие молекулярные орбитали не является симметричным относительно атомарных уровней?
6.6. Объясните связь между степенями перекрытия атомарных орбиталей
и заполнения молекулярных орбиталей и прочностью связи, формируемой между
атомами.
460 -J\r Вопросы и упражнения
6.7. Почему s-электроны формируют широкую зону в металлах, а d-элект-
роны — относительно узкую? Что можно сказать о зоне, формируемой /-электронами?
6.8. Почему d-зоны у металлов из правой части Периодической таблицы
Менделеева уже, чем у металлов из левой части? Почему d-зона уширяется при
смещении в нижнюю часть Периодической таблицы по переходным металла?
6.9. Объясните понятия «уровень Ферми», «вакуумный уровень», «работа
выхода». Чему соответствуют эти понятия в терминах молекулярных орбиталей?
6.10. Напишите зависимость плотности состояний от энергии для электронного газа.
6.11. Как можно изменить работу выхода электронов через данную поверхность?
6.12. Почему d-зона у поверхности металла уже, чем в его объеме?
6.13. Изобразите в простейшей форме плотность состояний для электронных зон в металле (хороший проводник), полупроводнике и идеальном изоляторе.
6.14. Почему у изоляторов всегда практически нулевая реакционная способность? Можно ли у изолятора повысить реакционную способность?
6.15. Объясните, почему переходные металлы с заполненной наполовину
d-зоной имеют наиболее высокую температуру плавления. Почему температура
плавления повышается при переходе вниз по столбцам в Периодической таблице?
6.16. Что происходит с внешней орбиталью атома при его сближении с
металлической поверхностью? Обсудите соотношение между потенциалом ионизации атома, его сродством к электрону, работой выхода электронов из металла
и прочностью возможной связи при хемосорбции.
6.17. Почему молекулы типа СО легко диссоциируют на металлической
поверхности и не диссоциируют в газовой фазе?
6.18. Нарисуйте схематически диаграмму молекулярных орбиталей, формирующихся при адсорбции двухатомной молекулы на поверхности d-металла.
6.19. Почему при адсорбции атомов или молекул на поверхности происходит изменение работы выхода электронов?
6.20. Опишите тенденцию изменения энергии адсорбции атомов типа N и
О при переходе слева направо по переходным металлам в Периодической таблице.
6.21. Почему молекула СО связывается достаточно прочно с металлами типа
Ni, Pd и Pt и не связывается с Cu, Ag и Au?
6.22. Почему молекула СО легко диссоциирует на поверхности железа и не
диссоциирует на поверхности платины, хотя теплоты адсорбции СО на этих
металлах практически равны между собой?
6.23. Почему золото, в отличие от платины, наиболее инертно среди металлов? Почему у ртути более высокая химическая активность, чем у золота?
6.24. Как растяжение или сжатие атомов металла в поверхностном слое
влияет на энергию адсорбции и его реакционную способность?
Вопросы по содержанию глав -*\г 461
6.25. Как реакционная способность атома металла зависит от его координационного числа?
6.26. Почему многие каталитические реакции проявляют «вулканическую»
зависимость от степени заполнения d-зоны металлического катализатора?
6.27. Что случится с реакционной способностью Ag при осаждении одного
монослоя Ag на поверхность (111) Ni или Au?
6.28. Разместите следующие поверхности ГЦК-металла в соответствии с
убыванием их реакционной способности: (111), (110), (100), (001), (557). Сделайте то же самое для тех же поверхностей ОЦК-металла.
6.29. Объясните соотношение Бренстеда—Эванса—Поляни на простой диаграмме потенциальной энергии для элементарной стадии реакции.
6.30. Имеются ли другие области науки и техники, помимо катализа, в которых ценится реакционная способность поверхности?
Глава 7
7.1. Что подразумевают под коэффициентом прилипания? Как его можно
измерить?
7.2. Как можно определить энергию активации для активированного адсорбционного процесса?
7.3. В чем состоит принципиальная разница между адсорбционными процессами первого и второго рода?
7.4. Что означает явление термализации газа? Почему эксперименты по
определению коэффициента прилипания, проведенные с нетермализованным
газом, приводят к ошибочным результатам?
7.5. Перечислите адсорбционные системы, для которых характерны экстремально низкие и очень высокие коэффициенты прилипания. Можете ли вы
объяснить такие значения коэффициентов прилипания в рамках теории переходных состояний (см. гл. 3)?
7.6. Опишите экспериментальную установку для изучения температурнопрограммируемой десорбции с поверхности монокристалла.
7.7. Выведите выражение для скорости температурно-программируемой
десорбции (то есть для зависимости скорости десорбции от температуры при
постоянной скорости нагрева).
7.8. Приведите примеры десорбционных систем, подчиняющихся кинетике первого, второго и нулевого порядков. Можете ли вы дать физическую интерпретацию кинетики нулевого порядка?
7.9. Используя данные гл. 3, приведите набор предэкспоненциальных множителей, которые могут наблюдаться при десорбции газов.
7.10. Дайте краткий обзор методов, которые позволяют определить энергию
активации десорбции по данным температурно-программируемой десорбции.
7.11. Обсудите на качественном уровне, как на спектр температурно-про-
граммируемой десорбции первого порядка (например, для СО) влияют следующие факторы:
462 -*\л Вопросы и упражнения
а) наличие адсорбционных центров двух типов с сильно различающимися
теплотами адсорбции;
б) притяжение между адсорбированными молекулами;
в) отталкивание между адсорбированными молекулами;
г) коадсорбция небольшого количества промотора типа калия, стабилизирующего молекулы СО.
7.12. Можно ли использовать температурно-программируемую десорбцию
для идентификации поверхностных компонентов или путей реакции?
7.13. Предположим, что мы успешно измерили коэффициент прилипания и
энергию активации адсорбции для некоторой молекулы, а также скорость десорбции. Можно ли теперь определить константу равновесия процессов адсорбции/десорбции?
7.14. Объясните, как кинетические параметры элементарной стадии могут
быть извлечены из данных экспериментов по температурно-программируемой
десорбции с поверхности, на которой предварительно были адсорбированы
реагирующие компоненты.
7.15. Приведите, по крайней мере, две причины, объясняющие необходимость знания кинетических параметров элементарных поверхностных реакций
при установлении механизма каталитической реакции.
7.16. Объясните принципы микрокинетического моделирования и необходимость его проведения в исследовании каталитических процессов.
7.17. Почему диссоциативная адсорбция часто является лимитирующей стадией каталитических процессов?
7.18. Имеется ли связь между теплотой адсорбции молекулы, энергией активации процесса ее диссоциации и теплотами адсорбции продуктов диссоциации?
7.19. Приведите аргументы за и против проведения кинетического анализа,
основанного на подгонке параметров модели Ленгмюра—Хиншельвуда, и микрокинетического моделирования.
Глава 8
8.1. Обсудите важность процесса конверсии с водяным паром для производства водорода и синтез-газа. Является ли этот процесс эндотермическим
или экзотермическим?
8.2. Никелевые катализаторы, используемые в процессе конверсии с водяным паром, являются более стабильными по отношению к отравлению углеродом, если на их поверхности находятся атомы серы или золота. Объясните,
почему эти элементы действуют как промоторы. Что вы предпочтете в качестве
промотора: серу или золото? Объясните свой выбор.
8.3. Очень часто легкие газы типа метана, высвобождающиеся при переработке нефти, сжигают. Почему это делают? Является ли конверсия с водяным
паром одним из путей переработки этих газов?
8.4. Дайте несколько примеров производств синтез-газа, а также областей
его практического использования.
Вопросы по содержанию глав -1\г 463
8.5. Опишите потенциальные возможности процесса Фишера—Тропша как
источника топлива для транспорта.
8.6. Дайте краткое описание синтеза метанола и ответьте на следующие
вопросы:
а) Какая из ступеней является лимитирующей в данном процессе?
б) Какой из компонентов доминирует на поверхности?
в) Можно ли метанол синтезировать из СО и почему этот синтез представляет интерес?
г) Почему синтез метанола зависит от окислительно-восстановительного
потенциала реагентов?
8.7. Сформулируйте ключевые моменты реакции конверсии с водяным
газом. Какой из компонентов при этом доминирует на поверхности?
8.8. Почему синтез метанола и процесс конверсии с водяным газом всегда
взаимосвязаны?
8.9. Процесс синтеза аммиака состоит из нескольких каталитических реакций, проведение которых направлено на удаление из смеси N2 и Н2 компонентов,
дезактивирующих катализатор. Аммиак образуется только в последнем реакторе.
а) Что является источником (источниками) водорода в синтезе аммиака?
б) Перечислите стадии каталитических реакций, проведение которых необходимо для получения чистого водорода.
в) Какое соединение (элемент) является наиболее сильным ядом катализаторов и должно быть удалено?
г) Какая стадия является лимитирующей в процессе синтеза аммиака?
д) Опишите функции, которые выполняет промотор в каталитическом синтезе аммиака.
е) Объясните, почему разработка катализаторов для высокоэффективного
синтеза аммиака при низких температурах является необходимой.
ж) Объясните, почему реактор для синтеза аммиака содержит несколько
слоев катализаторов и почему его охлаждают.
8.10. Объясните понятие «линии оптимальной работы» для каталитического процесса.
8.11. В чем состоит разница между электронными и структурными промоторами?
8.12. Объясните, как электронные промоторы облегчают диссоциацию молекул типа N2.
8.13. Почему сера является ядом для синтеза аммиака?
8.14. Обсудите необходимость поиска в будущем новых источников энергии. Какие постоянные источники энергии следует принимать во внимание и
каковы перспективы их практического использования?
8.15. Объясните, почему фотокатализ, базирующийся на ТЮ2, имеет низкую эффективность при разложении воды.
8.16. Как работают топливные элементы? Что из себя представляют топливные элементы типа «топливные элементы на твердых оксидах» (ТЭТО) и
«топливные элементы на протонообменных мембранах» (ТЭПОМ)?
8.17. Объясните роль катализа в технологии топливных элементов.
464 -l\r Вопросы и упражнения
8.18. Почему ТЭПОМ чувствительны к присутствию СО, а ТЭТО — нет?
8.19. Если топливные элементы будут внедрены в автотранспорт в больших
масштабах, то какой системе распределения топлива вы отдадите предпочтение: той, где бензин останется основным источником энергии и будет преобразовываться в водород непосредственно на машине, или той, где водородом
надо будет заправляться на топливных станциях? Объясните свой выбор.
Глава 9
9.1. Объясните, как сырая нефть перерабатывается на очистительном заводе. Какой из каталитических процессов является основным?
9.2. Что является основным загрязняющим элементом сырой нефти?
9.3. Дайте краткое описание процесса гидроочистки.
9.4. Объясните, почему в процессе гидрообессеривания используются катализаторы в сульфидной форме.
9.5. Найдите причину, по которой из замещенных дибензотиофенов сера удаляется наиболее трудно в сравнении с тиофенами и простыми тиолами (см. рис. 9.2).
9.6. В зависимости от выбора катализатора процесс гидрообессеривания
сопровождается разной степени гидрированием. В каком из потоков продукта
на нефтеперерабатывающем заводе вы бы осуществляли гидрообессеривание,
сопровождаемое гидрированием, а в каком — нет?
9.7. Что является основным компонентом топлива на выходе из нефтеперерабатывающего завода? Какой из потоков продукта в наибольшей степени
повышает качество топлива?
9.8. Дайте краткое описание процесса крекинга в псевдоожиженном каталитическом слое.
9.9. Процесс крекинга на цеолитах происходит по иному механизму, чем
на поверхности металлов. В чем состоит различие и с чем оно связано?
9.10. Объясните концепцию бифункционального катализа в процессах конверсии.
9.11. Что представляет собой процесс алкилирования? В чем состоит его
значимость? Почему он является недружественным для окружающей среды?
Что можно сделать для улучшения этого процесса?
9.12. Опишите несколько процессов частичного окисления, происходящих
в потоках продукта после очистки.
9.13. Объясните механизм Марса—ван-Кревелена. В чем состоит его отличие от реакций, катализируемых металлами?
9.14. Можно ли рассматривать реакцию гидрообессеривания и как реакцию
Марса—ван-Кревелена?
9.15. Опишите механизм каталитической полимеризации этилена.
9.16. Обсудите, в какой степени катализатор полимеризации этилена Филлипса удовлетворяет всем критериям, определяющим катализатор. Сравните
этот катализатор процесса полимеризации с катализаторами крекинга в псевдоожиженном слое и платиновыми катализаторами процесса конверсии.
Вопросы по содержанию глав -1\г 465
Глава 10
10.1. Поясните, что означают превышения первой и второй степени в контексте контроля загрязнений окружающей среды.
10.2. Почему катализаторы, использующиеся для нейтрализации выхлопных газов, называются «катализаторами трех путей»?
10.3. Почему для нейтрализации выхлопных газов используются металлические катализаторы и какие реакции они катализируют?
10.4. Почему катализатор для нейтрализации выхлопных газов снабжают
системой регулировки состава смеси воздух/топливо? Как это регулирующее
устройство работает?
10.5. При каких условиях эксплуатации выхлопы автомобиля являются наиболее загрязненными?
10.6. Почему свинец является более сильным ядом, чем сера, для (части)
«катализаторов трех путей»?
10.7. Можно ли автомобиль с дизельным двигателем оборудовать «катализатором трех путей»?
10.8. Опишите, как из выхлопных газов может быть удален N0^ в условиях, когда автомобиль работает в режиме «наклоненной горелки» (то есть при
избытке кислорода). Чем привлекателен такой режим движения автомобиля?
10.9. Каков типичный состав дымовых газов тепловых электростанций?
10.10. Опишите процесс селективного каталитического восстановления,
использующийся для удаления NOx из дыма стационарных тепловых станций.
Какие реагенты обычно используются в процессе селективного каталитического восстановления?
10.11. Как можно уменьшить выбросы оксидов серы от тепловых электростанций?
10.12. Можно ли применять технологию селективного каталитического восстановления для удаления NOx из передвижных источников типа грузовиков и
кораблей?
466
Л
Вопросы и упражнения
Упражнения к главе 2
Упражнение 2.1
Реакционная способность и стационарное состояние
Какое из приведенных ниже утверждений является правильным? Исправьте некорректные утверждения.
а) В стационарном состоянии продукты реакции и реагенты находятся в
равновесии.
б) В стационарном состоянии производство энтропии при реакции принимает минимальное значение.
в) В равновесии скорости прямой и обратной реакций равны нулю.
г) Равновесная реакционная система имеет максимальную энтропию.
д) Равновесная реакционная система имеет минимальную свободную энергию.
е) Равновесная реакционная система характеризуется максимальным значением производства энтропии.
ж) Для любой реакции глобальный порядок по реагентам равен сумме стехиометрических коэффициентов.
з) Эффективный катализатор ускоряет превращение реагентов в продукты
и замедляет обратную реакцию.
Упражнение 2.2
Сродство и завершенность реакции
Для приведенной ниже реакции
константа скорости прямой реакции при комнатной температуре (300 К) равна
0,1 моль3/л • с. В момент времени t = 0 реакционная смесь состоит из компонентов, находящихся в пропорции 1: 2. При t = tx концентрация компонента А
уменьшилась до 20 % от полного числа молекул смеси, а при равновесии составляет 10%.
Сродство реакции определяется как
поскольку последнее слагаемое равно нулю в равновесии. В свою очередь это
выражение можно трансформировать к виду
означающему, что скорость реакции может быть в общем случае записана как
А + 2В ^ 2С + D
А = -Х *'//</•
Это выражение можно переписать в виде
ЖО =-£ (') +
Г
Упражнения -l\r 467
Рассчитайте:
а) константу скорости обратной реакции;
б) сродство реакции в моменты времени t = О, t = t{ и t = °° (используйте
значение RT= 2,5 кДж/моль, при Т= 300 К);
в) степень завершенности реакции в и отклонение системы от равновесия в
указанные три момента времени.
Упражнение 2.3
Разложение N2Os.
N205 разлагается по следующей реакции
2N205 —» 4N02 + 02.
Скорость реакции, как установлено, определяется уравнением
r = ^dT = *[NA]-
Покажите, что нижеследующий набор ступеней реакции приводит к данному уравнению
к
N205 *=> N02 +N03,
N02 + N03 —*—> no2 + o2 + no2,
NO + NO3 —b—> 2N02.
Упражнение 2.4
Стационарное приближение
а) Сформулируйте стационарное приближение для следующей системы взаимосвязанных реакций
R —^ Ij —^... —^ >... —> \n —> Р
и кратко обсудите, в каких случаях использование стационарного приближения является оправданным.
б) Кратко обсудите, в какой степени реакции, протекающие стационарно,
близки к равновесию. Обсуждение проведите с использованием понятий энтропии и производства энтропии.
в) Реакция дегидрогенизации метилциклогексана С6НИСН3 до толуола играет важную роль в производстве бензина. Эта реакция хорошо катализируется
платиной по следующему механизму
С6НИСН3 + —kj—> С6Н,,СНз,
с6нпсн; —*г—> с6н5сн; +зн2,
С6Н5СН; —»с6н5сн3 + .
468 -l\r Вопросы и упражнения
Получите выражение для скорости образования толуола, предполагая, что
реакция идет только в одном направлении и что степень заполнения поверхности толуолом много больше степени заполнения поверхности метилциклогек-
саном, а скорость полной реакции определяется скоростью десорбции толуола.
Упражнение 2.5
Стационарное приближение
Разложение ацетальдегида
СН3СНО—500 °с ^СН4 н-СО,
идет через образование радикалов СН*
1. СН3СО->СН‘3+СНСГ,
2. СЩ + СН3СНО -> СН4 + СН3СО‘,
3. сн3со- -> со + сн;,
4. 2СН‘з -> С2Н6.
Используя стационарное приближение, получите выражение для скорости
образования СН4.
Упражнение 2.6
Стационарное приближение в кинетике цепных реакций
Цепная реакция Н2+Вг2 -» 2НВг идет по следующим ступеням
1. Вг2 —» 2Вг*,
2. Вг‘ + Н2 -> НВг + Н-,
3. Н* + Вг2 ^ НВг + Вг*,
4. 2Вг*^Вг2.
Покажите, что скорость реакции имеет вид
d[HBr] А;[Н2][Вг2]'/2
dt ~ 1 + >t'[HBr]/[Br2] ’
где константы скорости кик' зависят от констант скоростей реакций элементарных ступеней. Подсказка: примите условие равновесия между молекулярным и атомарным бромом.
Упражнение 2.7
Гетерогенный катализ
а) Какой шаг является наиболее важным в реакциях гетерогенного катализа?
б) Используя простую энергетическую диаграмму, сравните характер изменения потенциальной энергии в каталитической и газофазной реакциях.
Упражнения -l\r 469
в) Кратко обсудите принцип Сабатье.
г) Что подразумевается под автокатализом? Дайте символический пример
автокаталитической реакции.
д) Объясните, почему реакция между адсорбированными на поверхности
родия СО и N0 при полном покрытии поверхности этими газами идет взрывным образом.
Упражнение 2.8
Профили потенциальной энергии
Энергия активации для реакции
АВ + С^А + В-50 кДж/моль
в газовой фазе составляет большую величину — 500 кДж/моль. Использование
катализатора позволяет существенно понизить энергию активации. Каталитическая реакция идет через диссоциацию молекул АВ, адсорбированных на поверхности катализатора.
а) Нарисуйте профили потенциальной энергии каталитической и газофазной
реакций на энергетической диаграмме с точным указанием ключевых значений
энергии, используя для этой цели данные из приведенной ниже таблицы.
б) Укажите, какое соединение является наиболее устойчивым интермедиатом.
в) Укажите, какая ступень скорее всего является лимитирующей.
Адсорбирующийся
атом или молекула
Atfads, кДж/моль
Нет перевода
Eact, кДж/моль
АВ
50
АВ + С -> А + ВС
500
С
75
АВ* -> А* + В*
75
А
200
СВ* -> В* + С*
100
В
125
—
—
ВС
50
—
—
Упражнение 2.9
Изотермы адсорбции Ленгмюра
Получите изотермы адсорбции Ленгмюра для следующих случаев:
а) молекулярная адсорбция СО;
б) диссоциативная адсорбция СО;
в) конкурентная адсорбция молекулярно адсорбирующегося СО и диссоциативно адсорбирующегося Н2 (без возможных реакций);
г) предположим, что каталитическое образование метанола из СО и Н2 идет
по механизму, в котором реакция между СО и первым атомом Н определяет
скорость процесса (обратными реакциями можно пренебречь), а все остальные
470
Вопросы и упражнения
стадии можно считать быстрыми, за исключением десорбции метанола, которую можно предполагать протекающей в равновесии с газовой фазой:
— предложите механизм реакции;
— получите выражение для скорости реакции;
— определите диапазон изменения порядков реакции по водороду, монооксиду углерода и метанолу в предположении, что водород адсорбируется наиболее легко в сравнении с другими газами.
Технология топливных элементов с протонопроводящими мембранами основана главным образом на диссоциации Н2 на платиновом катализаторе, после
которой атомы водорода проходят в форме протонов через мембрану и рекомбинируют с кислородом на другой стороне мембраны, образуя воду. Высвобождаемая в этом процессе энергия равна примерно 1,2 эВ на один протон. Предполагается, что такой процесс может быть реализован в портативных электрических генераторах и передвижных системах типа автомобилей. В общем случае
данный процесс идет достаточно хорошо, если используемый водород является
достаточно чистым, однако получаемый на практике Н2 содержит примесь СО.
Ниже мы рассмотрим роль СО в этом процессе. Элементарные реакции
можно записать следующим образом:
1. Определите степень заполнения поверхности водородом и СО для следующих трех газовых смесей при общем давлении рш = 1 бар, рсо = 1, 10, 100 частей
на миллион. Рабочая температура топливного элемента равна 80 °С. Имеются
следующая информация по адсорбции газов на платине
Упражнение 2.10
Проблемы топливных элементов
с протонопроводящими мембранами
1. Н,+2*<z>
2Н
2. СО + ^ СО
К
а численные значения параметров таковы
=sОсо = 1,00, кв = 1,38 X 10 23 Дж/К, mHi = 2х1,6х10~27 кг,
тсо = 28 х 1,6 х IQ 27 кг, N0 = 1,5 х 1019 м-2, v = 1 х 1013 с"1,
R = 8,31 Дж-К 1 моль о = 100 кДж/моль, Е^ = 80 кДж/моль.
Упражнения ->\r 471
2. По сравнению с реальной теплота адсорбции СО здесь занижена. Она
составляет 120—1300 кДж/моль. Предложите путь снижения чувствительности
топливного элемента к возможной блокировке его СО. Укажите, какие из приведенных параметров являются наиболее важными.
Упражнение 2.11
Синтез метанола
Последние исследования по каталитическому синтезу метанола из С02 и
Н2 с использованием медного катализатора показали, что эта реакция имеет
первый порядок по С02 , а ее порядок по Н2 равен 3/2
С02 + ЗН2 -> СН3ОН + Н20.
Предполагается, что механизм реакции включает диссоциацию молекул
водорода, который затем реагирует с С02 с образованием формилата, который
остается адсорбированным на поверхности. Адсорбированный формилат затем
гидрируется с образованием остающихся адсорбированными диоксометилена,
метокси и, наконец, метанола, который затем десорбируется. Синтез идет в
условиях, при которых поверхность остается практически свободной, а образующийся в процессе кислород быстро удаляется в форме воды. Рассматривается
только скорость прямой реакции, а сам процесс, как полагают, идет по следующим элементарным ступеням:
1.
Н, + 2' «=» 2Н',
2.
О
о
+
tl
о
о
и *
3.
со; + н = нсоо* + *,
4.
нсоо + Н ^ Н-СОО + *
5.
Н,СОО + Н «••> Н,СО + о
6.
Н3СО +Н ^ Н,СОН + *,
7.
Н,СОН J=i Н3СОН + \
1. Допишите последнюю реакцию, по которой удаляется кислород.
2. Определите, какая из ступеней, скорее всего, лимитирует процесс, если
эта простая модель объясняет наблюдаемые порядки реакции по реагентам.
3. Получите выражение для скорости реакции, приняв, что на лимитирующей стадии идет только прямая реакция, а все остальные реакции находятся в
равновесии.
4. При какой мольной доле Н2 скорость реакции имеет максимальное значение?
472 -J\r Вопросы и упражнения
Упражнение 2.12
Окисление СО
Рассмотрите процесс окисления СО кислородом 02 на поверхности катализатора из Pt (этот процесс идет при каталитическом дожигании выхлопных
газов автомобилей:
С0 + ^02 Я СО,).
а) Запишите четыре основные ступени этого процесса. Можно принять, что
в данном случае адсорбция кислорода — прямой процесс.
б) Запишите выражение для скорости реакции, приняв в качестве лимитирующей стадии рекомбинацию адсорбированных монооксида углерода и кислорода. В данном примере парциальным давлением С02 пренебрегать нельзя.
в) Покажете, что скорость реакции может быть представлена как результат
отклонения от равновесия в форме:
Г = Г* - Г = k-KC0pC0^K0ip0i
PcOj
yjPo2 Рсо
в'
где KG — константа равновесия для реакции в целом.
г) Получите выражения для порядков реакции по СО, 02 и С02, а также для
кажущейся энергии активации этого процесса.
Упражнение 2.13
Конверсия с водяным паром
Здесь мы ознакомимся поближе с процессом конверсии с водяным паром,
который используется в крупномасштабном промышленном производстве син-
тез-газа и водорода:
СН4+Н2О^СО + ЗН2.
Этот процесс проводится при высоких температурах (1000—1200 К) и умеренных давлениях. Катализатором этого процесса является Ni(Ru) на носителях из
А1203 или Al2Mg04; для порядков реакции выполняются следующие неравен-
ства: иСн4 > 0, /y0 < 0 и пщ >0.
1. Почему этот процесс проводят при умеренных давлениях и высоких температурах?
2. Предложите механизм реакции, в котором лимитирующей стадией была
бы рекомбинация адсорбированного углерода С* и адсорбированного кислорода О*, и напишите выражение для скорости реакции. Ниже мы примем, что
только один адсорбат доминирует на поверхности, так называемый наиболее
избыточный интермедиат реакции (НИИР), каковым мы будем считать кислород О*. Оправдано ли это?
Упражнения -i\r 473
3. Отвечают ли порядки реакции для данной модели наблюдаемым значениям?
Мы можем также предположить, что лимитирующей стадией является диссоциация метана. Напишите выражение для скорости реакции, предполагая,
что кислород снова НИИР.
4. Будет ли скорость реакции в этом случае соответствовать наблюдаемым
значениям?
Упражнение 2.14
Реакция гидрообессеривания
Гидрообессеривание — очень важный крупномасштабный процесс, использующийся для удаления серы из продуктов переработки нефти. В действительности это — крупнейший каталитический процесс. В качестве модели этого процесса мы рассмотрим гидрообессеривание тиофена, модельного серусодержащего
продукта переработки нефти. В качестве катализатора возьмем СоМо-сульфид
на носителе из оксида алюминия. Полная реакция выглядит так:
C4H4S + 2Н2 ^C4H6+H2S,
где продуктами являются бутадиен и сероводород. Наиболее важный итерме-
диат образуется, когда тиофеновое кольцо разрывается и интермедиат адсорбируется на поверхности в форме
Н2С = СН-СН = CH-S- \
Измеренная скорость реакции подчиняется уравнению
кРт (К'-Р»У
1 + KiPti + (K2pHj'
где рт — парциальное давление тиофена; pHi — парциальное давление водорода.
1. Почему, как кажется на первый взгляд, скорость обратной реакции пропущена в предложенной кинетике?
2. Предложите механизм реакции, в котором бы присутствовал указанный
интермедиат, и получите выражение для скорости реакции с учетом обратной
реакции. Подсказка: примите, что водород и тиофен адсорбируются на разных
центрах.
3. Покажите, что полученное вами выражение для скорости реакции сводится к написанному выше, когда процесс идет при избытке водорода.
4. Какой компонент является НИИР для данной реакции?
Скорость реакции имеет характерную зависимость концентрации водорода. Почему из этой зависимости вытекает предположение об адсорбции S* и Н:
на разных центрах?
474
Л
Вопросы и упражнения
Упражнение 2.15
Гидрирование этана
Мы примем, что гидрирование С2Н6 до СН4 идет по следующему механизму:
1. с4н6+2 ^с4н; + н ,
Лимитирующая
2. с4н; + н ^ >2сн;,
Q
3. сн; + н ^сн4 + 2%
Q
4. Н2 + 2 ^2Н%
где вторая ступень является лимитирующей, а стадии 1, 3 и 4 проходят в условиях квазиравновесия.
а) Если в систему добавить небольшое количество D2, то можно наблюдать
соединение C2H6_WDW. Объясните происхождение этого соединения.
б) Напишите уравнение для скорости реакции на второй стадии.
в) В условиях квазиравновесия можно получить следующие выражения:
Кхр в2
1 А - 1YC2H6 •'
А* &С2Н, - д J
Он
Р в:
2. 0ГН. - сщ *
К30н ’
3. вн = ^4/>Н2 ^ •
Опишите ряд критических явлений, которые не могут быть изучены при
сформулированных допущениях.
г) Запишите уравнение для скорости реакции и покажите, что в пределе
сильного различия степеней заполнения для С2Н: и СЩ она может быть записана в виде:
r = klK[Pc„
\ _.
КсРс2ньРн2 j
о
где
в « ==
1 + у]К4РИ2
Упражнения
Упражнения к главе 3
Упражнение 3.1
Средние молекулярные скорости
а) Рассчитайте среднюю скорость молекул N? при комнатной температуре
(298 К).
б) При какой температуре скорость молекул N2 будет равна скорости атомов Не при 298 К.
в) Рассчитайте среднюю поступательную энергию одного моля N2 при 100,
298 и 1000 К.
Упражнение 3.2
Столкновения в газовой фазе
Имеется смесь N2 + ЗН2 при давлении 1 бар и температуре 25 °С.
а) Рассчитайте число молекул N2 и Н2 в 1 м3 (1 моль идеального газа при
25 °С имеет объем 24,7894 л).
б) Диаметры столкновений равны для Н2 и N2 0,271 и 0,373 нм соответственно. Как много столкновений испытывают молекулы Н2 за одну секунду?
в) Как много столкновений испытывают молекулы N2 за одну секунду?
г) Как много столкновений между молекулами N2 и Н2 происходит за одну
секунду?
д) Сколько вообще происходит столкновений за одну секунду в смеси газа
объемом 1 м3?
Упражнение 3.3
Теория столкновений и скорости реакций
Рассмотрим столкновение между двумя молекулами
NO (g) + Cl2 (g) NOC1 (g) + Cl (g),
для которых реакционный диаметр равен d = 3,5 А. Определите зависимость
предэкспоненциального фактора для этой реакции от температуры.
Упражнение 3.4
Общие свойства статистической суммы
Запишите статистическую сумму для одного моля гелия и найдите выражения для:
а) энергии Е\
б) давления р\
в) химического потенциала, выраженного через стандартный химический
потенциал //° и давление, определите выражение для //°;
г) энтропии S.
475
476 Вопросы и упражнения
Упражнение 3.5
Статистическая сумма
а) Рассчитайте статистическую сумму для поступательного движения молекул азота при температуре 298 К в объеме 24,79 л (равном объему одного моля
идеального газа в этих условиях).
б) Как изменяется <7trans, если Тувеличивается, р уменьшается или Кувели-
чивается? Чему равны значения qtnns для атомов N и молекул N3 в тех же условиях, при каких находились молекулы N7 в задаче а)?
в) Частота валентных колебаний молекулы N2 равна 2330 см-1; рассчитайте
колебательную статистическую сумму молекулы N2 по отношению к основному колебательному состоянию.
г) Рассчитайте вращательную статистическую сумму молекулы N2 при температуре 298 К. Момент инерции молекулы составляет 1,407 х 10-16 кг - м2, а
фактор симметрии для нее равен 2. Как изменяется qrot с ростом температуры,
уменьшением давления и увеличением объема?
д) Рассчитайте полную статистическую сумму молекул N2.
Упражнение 3.6
Вращательная статистическая сумма
Микроволновый вращательный спектр двухатомной молекулы имеет линии поглощения 20, 40, 60, 80 и 100 см-1. Рассчитайте ее вращательную статистическую сумму при 100 К, используя значение kT/hc = 69,5 см-1, принимаемое этой комбинацией при температуре 100 К.
Упражнение 3.7
Колебательная статистическая сумма
Рассчитайте колебательную статистическую сумму по отношению к основному колебательному состоянию (т. е. относительно низшего занятого состояния)
и долю молекул, находящихся в основном состоянии при 300, 600 и 1500 К,
используя значение kT/hc = 208,5 см-1, принимаемое этой комбинацией при
температуре 300 К, для следующих молекул:
Молекула
V, см 1
h
213
С12
557
02
1556
НС1
2886
Н2
4160
Упражнения -i\r 477
Упражнение 3.8
Статистическая сумма, средняя энергия и константа равновесия
Молекула А может находиться в двух состояниях, разделенных энергией АЕ.
а) Найдите выражение для статистической суммы молекулы А и рассчитайте
ее предельные значения при низких и высоких температурах (т. е. при 0 и К).
б) Найдите среднюю энергию молекулы и рассчитайте ее предельные значения при низких и высоких температурах.
в) Предположим, что молекула А находится в равновесии с изомером В,
который обладает следующими уровнями энергии по отношению к основному
состоянию А: Д£/2, ЗАЕ/4 и АЕ. Получите выражение для константы равновесия К = [В]/[А] и рассчитайте предельные выражения для К при низких и
высоких температурах. Проанализируйте полученные выражения. Подсказка:
используйте следующие выражения:
q = ^e ^kT- I = kT2 -£=\n{q); К = Ця/.
01 j
Упражнение 3.9
Выражение константы равновесия через статистическую сумму
Два изомера А и В находятся в равновесии и обладают следующими определенными спектроскопическими методами энергетическими уровнями:
e*=iAE; £,B=(j + ljA£; / = 0,1,2...
Рассчитайте константу равновесия для реакции А <-> В при
а) низкой температуре Т= 0,\АЕ/к;
б) высокой температуре Т= 2АЕ/к\
в) очень высокой температуре Т= \0АЕ/к.
Укажите предельные значения К для этой реакции.
Упражнение 3.10
Константы равновесия для различных ступеней реакции
Рассмотрим атомы азота, адсорбированные на поверхности рутения, которая имеет ступени атомной высоты на каждые сто атомов террасы в выделенном направлении. Атомы азота связываются со ступенями сильнее, чем с террасами, на 20 кДж/моль. Колебательные вклады в статистическую сумму атомов на ступенях и террасах можно считать одинаковыми (это корректное
допущение?). Рассчитайте, как степень заполнения ступеней изменяется со
степенью заполнения террас. Подсказка: напишите статистические суммы атомов на ступенях и террасах и приравняйте соответствующие химические потенциалы.
478 -J\r Вопросы и упражнения
Упражнение 3.11
Теория переходных состояний
а) Приведите общие (в рамках теории переходных состояний) уравнения
для реакции превращения молекулы R в продукт Р, с учетом наличия переходного состояния R#; детально укажите, в каком направлении идет каждая стадия
реакции и приведите для нее соответствующие скорость реакции и константу
равновесия. Нарисуйте диаграмму энергии, ясно показывающую энергетические уровни для R, R# и Р, а также энергетический барьер АЕ.
б) Приведите общее выражение для скорости реакции, следующее из теории переходных состояний, через статистические суммы (не точное выражение) и А Е.
в) В чем состоит основное различие между выражениями для скорости реакции, получаемыми в рамках теории переходных состояний и теории столкновений?
г) Предположим, что адсорбирующаяся молекула имеет коэффициент прилипания для (недиссоциативной) адсорбции порядка 1(П3 и этот процесс не
активационный, то есть АЕ = 0. Переходное состояние какого типа можно
предложить для процесса адсорбции? Переходное состояние какого типа соответствует адсорбции, идущей с коэффициентом прилипания, равным единице?
д) Почему обычно скорость диссоциативной адсорбции существенно меньше скорости ассоциативной адсорбции?
Упражнение 3.12
Константа равновесия для адсорбции
Здесь мы будем рассматривать простую изотерму Ленгмюра, при выводе
которой предполагалось равновесие между атомами газа А и адсорбированными атомами А*:
к+
А + 1 *=► А .
к~
а) Выразите степень заполнения поверхности атомами А через константу
равновесия КА и давление газа рА.
б) Сейчас мы рассмотрим, чем определяется константа КА. Запишите
выражения для статистических сумм атома в газе и в адсорбционном слое.
Пусть адсорбированные атомы находятся на локализованных центрах с энергией адсорбции АЕ (по отношению к основному колебательному состоянию) и имеют по одной колебательной моде вдоль нормальной к поверхности координаты и по две мягкие колебательные моды по координатам вдоль
поверхности.
в) Предполагая химическое равновесие, получите выражение для вА.
Ниже следует принять, что hvJkBT= 1 и статистическая сумма двух мягких
мод близка к единице (является ли данное предположение оправданным?).
Упражнения ->\r 479
г) Используя выражение, полученное в задаче а), и следующие выражение
для к+ и к~
SA
К = ;; -и "~r=sr» кА = ^ехР
N0ftxmAkBT ’
Е
RT
получите выражение для S0А и оцените значение этого параметра в предположении, что каждый центр занимает площадь 9 А2, Т= 300 К и m = 16 ат. ед.
массы (СН4).
Упражнение 3.13
Диссоциация в газовой фазе
Рассмотрим мономолекулярную реакцию диссоциации
к +
АВ А + В.
к~
а) Приведите в общем виде выражение для скорости реакции, следующее
из теории переходных состояний, в терминах статистических сумм и энергетического барьера (явные выражения для статистических сумм записывать не
требуется).
б) Определите понятия «жесткий» и «разрыхленный» активированный комплекс и укажите, какие требования эти понятия накладывают на величину гс по
отношению к параметру го (эти величины указаны на рисунке потенциальной
кривой для процесса диссоциации молекулы А—В, где г — расстояние между
атомами А и В).
в) Скорость диссоциации для этой мономолекулярной реакции имеет второй порядок по [АВ] при низких давлениях и первый порядок при высоких
давлениях. Объясните почему.
Упражнение 3.14
Переходные состояния и предэкспоненциальные множители
Охарактеризуйте переходные состояния и дайте оценку величины предэкспоненциального множителя для следующих реакций:
480 ->\r Вопросы и упражнения
а) диссоциация этана на два метальных радикала: С2Н6 ^ 2СН*;
б) диссоциация трифторметана на радикал CF и HF: CF3H -» 2CF* + HF;
в) цис-транс-шомертация дейтерированного этилена CHD^CHD;
г) изомеризация циклопропана в пропилен;
д) изомеризация метил изоцианида в метил цианид (ацетонитрила).
Упражнение 3.15
Диссоциация молекулярного кислорода
а) Получите в рамках теории переходных состояний выражение для скорости диссоциации 02 в газовой фазе, но с использованием обозначений, приведенных на рисунке, и явных выражений для статистических сумм поступательного, вращательного и колебательного движений.
б) Перепишите полученное выражение в аррениусовской форме и найдите
энергию активации и предэкспоненциальный множитель:
qtn„=l(2pmkTf/h
на каждую степень свободы;
'Zvib = 1
для 02 и О*;
<7rot = 8p2jur2kT/h2.
Q.
CD
I
0
-0
с;
со
X
zr
I
CD
I-
o
c=
Профиль потенциальной энергии для
диссоциации О,
Упражнение 3.16
Десорбция молекулярного водорода
Водород адсорбируется диссоциативно практически на всех металлах. При
температуре 500 К водород свободно диффундирует по поверхности. Десорбция идет ассоциативно при рекомбинации двух атомов водорода, а атомарной
адсорбцией можно пренебречь.
а) Рассчитайте статистическую сумму атомов адсорбированного водорода при
500 К, предполагая, что адсорбционный центр занимает площадку 10-15 см2.
Упражнения ->\r 481
б) Выразите в рамках теории переходных состояний скорость десорбции и
константу десорбции &des через статистические суммы (точные их значения при
этом знать не требуется).
в) Считая активированный комплекс жестким, рассчитайте предэкспоненциальный множитель для десорбции Н2, приняв для полной статистической
суммы значение q'n = 3 при 500 К и положив qtnins = I(2pmkT)l/2 на одну степень
свободы.
Упражнение 3.17
Термическая десорбция серебра с поверхности рутения
Термическая десорбция серебра с поверхности (001)Ru начинается при примерно 1000 К. Для степеней заполнения от 0,15 до 1 МС скорость десорбции
хорошо описывается уравнением [Niemantsverdriet J.W., Dolle P., Markert К.,
Wandelt К. // J. Vac. Sci. Technol. A5. (1987) 875]
r = 5x 1O,30^ exp(-290,000/RT); n « 0 и Eact = 290 кДж/моль.
а) Активированный комплекс какого типа можно предложить для этого
процесса?
б) Какой физический смысл скрыт в значении п ~ 0?
Упражнение 3.18
Изомеризация циклопропана
Изомеризация циклопропана в пропилен идет со скоростью
г = 3х 10,5[циклопропан]е 274,000/лг (Еасх в кДж/моль).
а) Нарисуйте энергетическую диаграмму реакции.
б) Дайте качественное описание активированного комплекса
в) Какая из степеней свободы оказывает наиболее сильное влияние на предэкспоненциальный множитель?
г) Приведите выражения для скорости реакции, энергии активации и
предэкспоненциального множителя, следующие из теории переходных состояний.
д) Почему константа скорости этой реакции — убывание при низких давлениях?
(2 nmkTf\ 1_
hi ’ 11 j _ехр (-Мл
/кту
8 я2кТ'%П
I 1 , л^\3/2
ып
h1
где / — моменты инерции относительно трех осей.
л
482 -l\r Вопросы и упражнения
Упражнение 3.19
Десорбция молекул
Простое выражение для скорости десорбции молекулы из фиксированного
центра адсорбции имеет вид:
e-£dcs/кт'
кТ ( 2лт кТЛ S7r2jur2kT
Запишите это выражение в аррениусовской форме:
iV — 1/ act/КТ
Arr Veffe
и приведите явные выражения для предэкспоненциального множителя и энергии активации.
Упражнение 3.20
Влияние окружающей среды
Скорость газофазной реакции CH3NC —» CH3CN зависит от давления CH3NC
как реакция первого порядка при давлении 1 бар и как реакция второго порядка при давлении меньше 1 мбар.
а) Запишите выражение для скорости реакции в виде степенного закона
для этих двух случаев.
б) Объясните, почему при низких давлениях наблюдается зависимость, отвечающая реакции второго порядка, записав ступени реакции в рамках теории
переходного состояния и используя стационарное приближение.
Упражнение 3.21
Теория столкновений
Еа, кДж/моль
к, произв. ед.
25
0
35
0
40
1
45
10
50
12
60
9
75
6
Зависимость газофазной реакции А+В ^ Р от энергии была изучена при
500 К путем изменения кинетической энергии молекул А в экспериментах с
молекулярными пучками. Результаты исследования даны в таблице. Обсудите
Упражнения ->\r 483
полученные результаты в рамках теории столкновений. Какова высота энергетического барьера? Чему равна энергия активации при 500 К?
Катализатор синтеза аммиака был получен восстановлением магнетита (Fe304),
в котором растворено 3 вес. % А1203. Восстановление проведено в таких условиях, что частицы магнетита свой размер не изменяли. Восстановлению подвергалось только железо до чистого Fe, а с А1203 ничего не происходило.
1) Объясните это явление.
2) Оцените объем пор в 1 г катализатора (или относительный объем пор в %),
образующихся при восстановлении, используя следующие данные: молярный
вес MWFe = 55,85 г/моль, pF^0i = 5,18 г/см3, pFe = 7,86 г/см3 и pAUO} = 3,97 г/см3.
3) Поверхностные энергии различных граней железа следуют соотношени-
ям ^Fe(ioo) ^Fe(iio) ^Fe(i 11)* Обсудите эти соотношения и вытекающие из них
следствия для поверхности восстановленного железа. Ниже мы примем, что
восстановленное железо имеет в основном грани Fe(100) (оправдано ли это
предположение?).
4) Как было установлено методом БЭТ по изотермам адсорбции N2, восстановленный магнетит с добавками оксида алюминия имеет удельную поверхность 29 м2 на грамм катализатора. Было также найдено, что при диссоциативной адсорбции N2 на грамм катализатора садится 2,2 мл N2 (стандартные условия, т. е. температура равна 273 К, давление — 1 бар (= 100000 Па)). Определите
удельную (на грамм) площадь поверхности катализатора, приняв, что атомарный азот формирует на поверхности Fe(100) структуру с(2 х 2).
5) Чем различаются эта площадь поверхности и площадь, определенная
методом БЭТ?
В отделе химической технологии новые каталитически активные материалы получают путем сжигания летучих соединений металлов в горелке. При
этом образуются аэрозольные частицы, которые осаждают на фильтр. Если
частицы охлаждать быстро, то можно получить материал с высокой удельной
поверхностью. Ниже будем считать, что А1203 получен описанным способом.
Удельная поверхность материала определялась методом БЭТ с использованием в качестве адсорбата N2, при этом при температуре жидкого азота были
Упражнения к главе 5
Упражнение 5.1
Катализаторы синтеза аммиака
Упражнение 5.2
Метод БЭТ
484 -l\r Вопросы и упражнения
получены данные, приведенные в таблице. Равновесное давление в этих условиях Р0 = 773,81 Тор, масса образца равнялась 24,9 мг.
Р, Тор
38,65
67,61
96,67
125,75
154,62
Va, мл (стандартные условия)
0,4912
0,5407
0,5815
0,6184
0,6565
1) Определите площадь поверхности образца в предположении, что каждая
молекула N2 занимает 0,164 нм2.
2) Определите соотношение объемов, десорбирующихся из первого и второго слоев. Обоснуйте полученное значение.
3) Мы пропитали катализатор раствором соли Си и высушили его. Обсудите, как это скажется на площади поверхности.
4) Мы поместили 1 г импрегнированного катализатора в трубчатый реактор, где он был активирован в токе водорода. Примем, что вся медь восстановилась и образовались ее малые частицы, поверхность которых образована наиболее устойчивыми гранями. Мы подвергли образец слабому окислению, так что
окислился лишь поверхностный слой. При этом степень покрытия поверхности
кислородом составляет 0,5 МС по отношению к поверхностным атомам Си. Затем реактор был «промыт» Не, так что весь избыточный кислород был удален с
поверхности катализатора. Затем через катализатор пропустили водород, который прореагировал с поверхностным кислородом, в результате чего образовалась вода, количество которой было измерено. Было установлено, что количество образовавшейся воды равно 6,7 мл (в стандартных условиях газовой фазы).
Определите удельную (на грамм) активную поверхность Си в катализаторе. Си
представляет собой ГЦК-кристалл с постоянной решетки аСи = 3,61 А.
5) Теперь мы проводим эксперимент, в котором катализатор находится в токе
кислорода и водорода при температуре 400 К. Расход газа составляет 10 мл/с
(стандартные условия), а содержание воды оказалось равным 1 об. %. Определите частоту превращений для одного активного центра Си.
6) Общая энергия активации была определена по измеренной зависимости
скорости реакции от температуры. Как бы вы определили общую энергию активации?
7) По измерениям, проведенным в температурном интервале 400—600 К,
была получена энергия активации 50 кДж/моль, а для температурного интервала 600—800 К ее значение составило 27 кДж/моль. Исследователь был очень
доволен тем, что ему удалось снизить энергию активации. Как вы объясните
полученные данные и стоило ли исследователю радоваться?
Упражнение 5.3
Фактор эффективности
В данном упражнении через фактор эффективности е мы оценим влияние
транспортных ограничений на процесс тестирования катализатора синтеза амми¬
Упражнения -J\r 485
ака, подобного описанному упражнении 5.1. Мы считаем, что размер частиц
играет роль и что восстановленный и сплавленный катализатор раздроблен на
более мелкие части. В экспериментах использовалась узкая фракция со средним размером R = 0,2 мм. Будем считать частицы идеально сферическими.
Коэффициент диффузии определить достаточно сложно, но мы будем считать,
что он в 100 раз меньше коэффициента диффузии в газовой фазе, который
равен 0,4 см2/с. Тестирование проведено на стехиометрической смеси N2/H2
при давлении 4 бар в предположении, что реакция идет в условиях, далеких от
равновесия, и имеет первый порядок по азоту. Реакцию планировалось проводить при 600 К, и из данных фундаментальных исследований монокристаллов
было получено для частоты превращений значение 0,05 на один атом железа на
поверхности. Из упражнения 5.1 можно установить, что 1 г восстановленного
катализатора имеет объем 0,2 см3/г, объем пор составляет 0,1 см3/г, а полная
удельная площадь поверхности, которую мы будем считать равной удельной площади поверхности пор, равна 29 см2/г, из которой 18 см2/г приходится на поверхность (100) чистого Fe. Заметим, что природа активных центров железа все еще
обсуждается в научной литературе (мы это в расчет принимать не будем).
1. Оцените удельную (на объем) площадь поверхности пор S.
2. Оцените средний радиус пор Rp.
3. Оцените константу скорости реакции.
4. Оцените величину диффузионного модуля Тиле (Ф5).
5. Оцените фактор эффективности е и обсудите полученный результат.
6. Трубчатый реактор имеет размер 2 мм и в него загружен 1 г катализатора.
Будет ли установка удовлетворять сформулированным выше требованиям.
7. Оцените скорость течения газа, при которой можно быть уверенным, что
степень конверсии на выходе из трубчатого реактора не превышает 5 %.
Упражнения к главе 7
Упражнение 7.1
Коэффициент прилипания N2 на поверхности Fe( 100)
(продолжение упражнения 5.1)
1) Кривая ТПД с насыщенной азотом поверхности Fe(100) при нагреве со
скоростью 2 К/с является симметричной с максимумом при 740 К. Оцените
энергию активации в предположении, что десорбция описывается реакцией
второго порядка.
2) Коэффициент прилипания для N2 на поверхности Fe(100), как показали
измерения, равен
50(7’) = 500[ехр(-Д£11й/ЛвГ)],
где S0(T) = 2,5 х 10-5 при 500 К, а АЕш = 0,03 эВ. Нарисуйте диаграмму потенциальной энергии для адсорбции N2 на поверхности Fe(100) и оцените энтальпию адсорбции N на этой поверхности.
486 -l\r Вопросы и упражнения
3) Объясните наиболее просто, почему коэффициент аккомодации мал,
несмотря на малое значение энергии активации диссоциации.
4) Определите равновесную степень покрытия поверхности Fe(100) азотом
в атмосфере N2 при давлении 1 бар и температуре 800 К.
5) В экспериментах по ТПД водород десорбируется с поверхности Fe(100) при
температуре около 320 К. Обсудите на качественном уровне, какой компонент
будет наиболее избыточным интермедиатом реакции (атомарный Н или атомарный N) в условиях синтеза аммиака (700 К, Рш = 200 бар, стехиометрическая смесь).
Упражнение 7.2
Коэффициент прилипания метана на поверхности Ni( 100)
На приведенном рисунке показана степень захвата углерода на поверхности Ni(100), выдержанной в атмосфере метана при различных температурах. Метан
диссоциирует на поверхности, в результате чего водород отделяется и поверхность покрывается слоем углерода. Схема реакции имеет вид:
1. сн4 +2 ^сн; + н\
2. сн: + -> сн; + н ,
0с
3. и т. д.,
где первая ступень является лимитирующей.
1. Определите число активных центров на
поверхности Ni(100), приняв rNi = 8,90 г/см3,
молярный вес равным Mw = 58,71 г/см3 и
считая, что каждый атом Ni представляет
собой центр; Ni — металл с ГЦК-решеткой.
2. Определите абсолютное значение коэффициента прилипания при различных температурах.
3. Определите кажущуюся энергию активации для процесса аккомодации. В экспериментах для обеспечения экспозиции в
0,2 бар/с необходима выдержка образца в
0,0 0,1 0,2 0,3 течение 152 с. Определите, каково было дав-
Экспозиция, бар • с ление метана (в Тор) в этих экспериментах.
Упражнение 7.3
Адсорбция/десорбция водорода и равновесие
Коэффициент прилипания Н2 на поверхности металла был определен в экспериментах по адсорбции. Принимается, что число активных центров на поверхности равно N0= 1,5 х 1019 центров/м2 и в условиях насыщения каждый активный
центр занят одним атомом водорода. В экспериментах образец вносили в атмосферу водорода при заданном давлении и выдерживали в течение определенного
Упражнения -*\r 487
периода времени (заданная экспозиция). Затем определялась (например, методом
ТПД) величина адсорбции. Все эксперименты по адсорбции проводились при
столь низкой температуре, что десорбцией можно было пренебречь.
а) Основываясь на реакции Н2 + 2* ^ 2Н* , запишите выражение для скорости захвата атомов водорода поверхностью в терминах степени покрытия
поверхности водородом 6W
б) На приведенном ниже рисунке показаны зависимости степени покрытия
поверхности водородом от экспозиции, измеряемой в бар/с. Определите начальные коэффициенты прилипания при различных температурах.
Кривые захвата водорода
О 200 400 600 800 1000
Экспозиция, бар-с
в) Определите энергию активации для процесса аккомодации водорода на
этом металле.
2
о
к Я
s Я
н о.
о. 3
* о
о СО
Кривая ТПД для Н2
Температура, К
г) Далее принимается, что поверхность насыщена атомами водорода, и мы
оцениваем, как это обсуждалось ранее, энергию десорбции молекул водорода,
записывая спектры ТПД. Можно принять, что кривая ТПД полностью симметрична и скорость десорбции имеет максимум при 408 К (см. рис.). Приняв
488
—Вопросы и упражнения
следующие значения для предэкспоненциального фактора v= 1013 с-1 и параметра Р = 2 К/с, оцените величину энергии десорбции.
д) Катализатор был получен пропиткой носителя из А1203 раствором соли
металла с последующими сушкой и восстановлением металла. Нам необходимо определить площади поверхности носителя и активной поверхности металла. Объясните, как бы вы определили эти площади, приняв, что Н2 не реагирует с носителем, a N2 не диссоциирует на металле.
Упражнения общего характера
1. Каталитическое восстановление N0
Катализатор, используемый помимо всего прочего для очистки выхлопных
газов, представляет собой частицы Rh на носителе из А1203. Доминирующими
для частиц являются грани Rh(lll), вторичными по площади — грани Rh(100).
Rh — металл с ГЦК-структурой, постоянная решетки a = 0,381 нм.
а) Почему у частиц доминирующими являются именно эти грани? Какая из
этих граней, по вашему мнению, является более реакционно способной?
Ниже мы примем, что доминирующей является грань Rh(100). Площадь
поверхности катализатора, определенная методом БЭТ, равна 180 м2/г.
б) Дайте краткое описание природы взаимодействий, на которых основан
метод БЭТ, и укажите, площадь какой поверхности измеряется в этом методе.
Считаем теперь, что N0 хемосорбирован на частицах Rh, а температура
такова, что он не адсорбируется на А1203. Условию насыщения поверхности
Rh(100) молекулам N0 отвечает одна молекула N0, приходящаяся на два поверхностных атома родия, при этом молекулы N0 формируют структуру с(2 х 2).
После насыщения поверхности катализатора молекулами N0 были проведены
эксперименты по ТПД со скоростью нагрева 2 К/мин. Максимум скорости
десорбции N0 приходится на 460 К. Полное количество десорбированного N0
составляет 18,5 мл на грамм катализатора (р = 1 бар, Т = 300 К). Можно считать, что N0 не диссоциирует на поверхности Rh(100).
в) Оцените энергию активации десорбции N0 с поверхности Rh(100).
г) Определите площадь поверхности Rh(100), приходящуюся на грамм катализатора.
Теперь мы примем, что адсорбция N0 на Rh(100) является ассоциативной
и идет без активации. Коэффициент прилипания S(T) может быть принят равным единице для всех температур. Далее мы примем, что энергия активации
для десорбции N0 с поверхности Rh(100) равна 140 кДж/моль и два атома Rh
формируют адсорбционный центр.
д) Определите степень покрытия поверхности катализатора молекулами N0,
если он находится в атмосфере N0 при давлении PN0 = 1 мбар и Т= 900 К.
Диссоциирует ли N0 на адсорбированные атомы кислорода и азота при
этих температурах? Катализатор обычно используется в условиях, когда азот
десорбируется, а кислород удаляется за счет реакции, например, с СО. Диссоциация N0 представляет собой активационный процесс.
Упражнения ->\r 489
е) Дайте краткое объяснение, почему Rh является наилучшим катализатором среди переходных металлов второй серии: Мо, Ru, Rh, Pd, Ag.
2. Окисление водорода на Pt
Синтез воды из Н2 и 02 на поверхности платины, как полагают, идет со
следующими стадиями:
а) Можете ли вы предложить другие элементарные стадии, которые могли
бы быть учтены в синтезе воды?
б) Ниже мы будем рассматривать только пять вышеприведенных стадий и
примем, что реакции стадии 1, 2, 4 и 5 идут в условиях квазиравновесия. Напишите выражение для скорости лимитирующей стадии (3), содержащее выражения
для скорости прямой и обратной реакций. Это выражение должно содержать только
степени покрытия поверхности для соединений, входящих в уравнение (3).
в) Покажите, что эту скорость можно также представить в виде
и выразите KG через константы равновесия.
г) Мы теперь примем, что кислород является наиболее избыточным интермедиатом реакции (НИИР). Найдите 0* в этих условиях.
д) Принимая, что кислород является НИИР, определите порядки реакции
по Н2, 02 и Н20 в условиях, далеких от равновесия.
е) Перейдем к другому предельному случаю, когда в условиях реакции степень покрытия поверхности кислородом очень мала. Определите, для какого
состава смеси газов Н2 и 02 скорость реакции будет максимальной в условиях,
далеких от равновесия.
Катализатор получен пропиткой А1203 раствором соли Pt и последующими
сушкой и восстановлением металла. Активность одного грамма катализатора
определена в трубчатом реакторе в условиях, когда кислород является НИИР,
а активность катализатора очень высока. Установлено, что скорость реакции
увеличивается с температурой, но линейной зависимости в аррениусовских
координатах между In (г) и l/Т не наблюдается.
ж) Дайте, по крайней мере, одно качественное объяснение наблюдаемому
отклонению от линейной зависимости In (г) от \/Т.
1. Н2 +2 <z> 2Н
2. О, + 2 <z> 20
3. О* + Н -> НО +
4. НО +Н
5. Н20 <z> Н20 +
/
\
490
\
Вопросы и упражнения
3. Разложение N205
а) Пентаоксид диазота разлагается по следующей реакции
N,Os -> N02 + N02 +^02.
Это — реакция первого порядка, измерения константы реакции в диапазоне
температур от 273 до 338 К дали значения, приведенные в таблице:
т, к
273
298
308
318
328
338
к, с-1
7,87 х 10"7
3,46 х \0~5
13,5 х 10“5
49,8 х 10"5
150 х 10~5
487 х 10~5
Оцените предэкспоненциальный множитель и энергию активации реакции Еа.
б) Был предложен следующий механизм разложения N205:
1. N205 N02 +N03,
2. N02 + N03 NO + N02 + 02,
3. NO + NO3 -> no2 + no2.
Заметим, что реакция на первой ступени считается обратимой, в то время
как на второй и третьей ступенях реакции необратимы. Покажите с использованием стационарного приближения для интермедиатов, что скорость полной
реакции можно записать в виде:
d[N2Q5]
dt
к [N205],
и выразите величину к через константы kv k_v к2 и ку
в) Оцените в рамках теории переходных состояний температурную зависимость предэкспоненциального фактора в представленной в аррениусовской
форме константе скорости реакции NO + N03 -» N02 + N02. Иными словами,
определите п в зависимости А Тп. Можно принять, что и N03 и активированный комплекс не являются линейными. Можно также принять, что hv<z kQTи
степень вырождения всех электронных состояний равна единице.
г) Разложение N205 представляет собой реакцию, которая идет со сходными постоянными скорости в растворах при различных растворителях и в газовой фазе. Чем характеризуются реакции такого типа?
4. Каталитическое дожигание выхлопных газов
Ниже мы проанализируем модель каталитической нейтрализации СО и N0
в выхлопных газах автомобилей, при которой образуются более безопасные для
окружающей среды С02 и N2. Реакцию можно разбить на следующие элемен¬
Упражнения
\
тарные стадии, которые все, за исключением второй стадии, являющейся лимитирующей, идут в условиях квазиравновесия:
1. N0 + * N0',
2. N0* + ц -> О* +N\
3. со+
4. со1 + о* со; + \
5. со; со2 + \
6. N" + N" ^ n; + i:,
7. N2 <=> N2 + \
а) Запишите полную реакцию и выразите константу равновесия KG через
парциальные давления участвующих в реакции газов.
б) Можете ли вы привести еще хотя бы одну элементарную стадию, которая
была бы уместна для данного процесса?
в) Покажите, что при использовании вышеприведенных допущений скорость реакции может быть записана в виде:
г = к; K{pNO0;
j 1 >N. ^С°2
PnoPco
г) Выразите KG через Кх — Кг
Ниже мы примем, что адсорбированный кислород является наиболее избыточным интермедиатом реакции (НИИР), то есть ft = 1 — в0.
д) Выразите ft только через константы равновесия и парциальные давления.
е) Принимая, что кислород является НИИР, определите порядки реакции
для всех участвующих в ней газов, то есть найдите a?no, псо, и nCOj.
Пусть энергия активации была измерена в условиях почти чистой поверхности, то есть ft s 1,
ж) Получите аналитическое выражение для кажущейся энергии активации
в этих условиях, выразив ее через энергию активации лимитирующей стадии и
изменение энтальпии в квазиравновесных стадиях.
5. Емкость монослоя и реакция синтеза метана
Для создания реакционно более активной поверхности Ni на поверхности
Ru(OOOl) выращивается упорядоченный (соответствующий ГЦК-структуре) слой
Ni. Ru является металлом с ГПУ-решеткой, поверхность Ru(OOOl) имеет ту же
структуру, что и поверхность Ni(l 11), различаются только межатомные расстояния: у рутения оно больше. Элементарная ячейка поверхности Ni(lll) на рутении имеет площадь 6,36 х 1(Г20 м2, тогда как у поверхности (111) чистого
кристалла никеля ее площадь равна 5,37 х 1(Г20 м2.
491
492 -l\r Вопросы и упражнения
а) Повысится или понизится реакционная активность адслоя Ni(lll), осажденного на Ru? Объясните почему.
б) Проведем эксперименты по ТПД СО с поверхности (111) чистого кристалла Ni и с адслоя Ni( 111) на поверхности Ru(0001) при скорости нагрева 2 К/с.
В полученных спектрах ТПД максимум в скорости ТПД СО сдвигается с 500 до
550 К при переходе от поверхности (111) чистого кристалла Ni и к адслою
Ni(lll) на поверхности Ru(0001). Предполагая, что реакция десорбции имеет
первый порядок, а предэкспоненциальный множитель равен 1013 с-1, определите, какому изменению энергии связи отвечает наблюдаемый сдвиг.
Пусть у нас имеется реальный катализатор, предположим в соответствии со
сказанным выше, что все металлические частицы состоят из Ru, покрытого
слоем Ni , который имеет только одну грань (111). Нам необходимо оценить
площадь поверхности металла по данным об адсорбции СО при 300 К. Найдено, что поверхность насыщается СО при этой температуре, если 3,4 мл СО (при
давлении 1 бар и температуре 300 К) адсорбируется на 1 г катализатора.
в) Рассчитайте площадь полной поверхности металла, приходящейся на
грамм катализатора, приняв, что в насыщенном слое СО на поверхности Ni(lll),
осажденного на Ru, на один атом Ni приходится 0,5 молекул СО.
Теперь нам надо оценить степени заполнения молекулами СО поверхности
катализатора, помещенного в трубчатый реактор, где поддерживается парциальное давление рсо = 0,01 бар и температура Т= 1000 К. По оценкам энергия
десорбции составляет 147 кДж/моль, предэкспоненциальный множитель равен
1013 с-1, а коэффициент прилипания оценивается величиной 0,2 и не зависит
от температуры. Для простоты мы примем, что каждый атом Ni может адсорбировать одну молекулу СО.
г) Оцените степень заполнения поверхности молекулами СО в этих условиях.
Пусть смесь СО и Н2 проходит в реакторе над 1 г катализатора при указанных выше условиях (рсо = 1,00 бар и температура Т= 1000 К) и на выходе из
реактора доминирующим продуктом является СН4. Расход газа равен 100 мл/
мин, а концентрация метана составляет 10 %, причем измерения обоих этих
величин произведены при давлении 1 бар и температуре 300 К. Мы можем
считать газ идеальным.
д) Определите число оборотов реакции для метана на один атом Ni в предположении, что на каждый грамм катализатора приходится 15 м2 площади поверхности активного Ni(lll) на Ru.
е) В предыдущей задаче мы рассчитали число оборотов реакции, приходящееся на один атом Ni, для идеальной гладкой поверхности Ni(lll). Образование метана, однако, включает стадию диссоциации СО. Можете ли вы предложить более реалистические центры, на которых способна протекать реакция, и
что тогда следует понимать под числом оборотов реакции для этих центров?
6. Пиролиз этана
Пиролиз этана — важный промышленный процесс. Для него при температуре 1150 К и давлении 60 бар получены следующие данные:
Упражнения ->\r 493
t, с
0,06
0,15
0,36
0,62
0,79
0,98
[С2Н6], моль/м3
381
318
254
191
159
127
а) Определите порядок реакции пиролиза метана.
б) Для пиролиза С2Н6 при высоких давлениях предложен следующий простой механизм:
R1
с2н6
-> сн3 + сн3,
R2
С2Н6
+ СН3 -» сн4 +С2Н5,
R3
с,н5
^С2Н4 + Н,
R4
С2Н6
+ Н -» С2Н5 + н2,
R5
с2н5
+ С2Н5 —> С4Н10,
R6
с2н5
+ С2Н5 ->С2Н6+С2Н4
Анализ реакции, проведенный в стационарном приближении, показывает, что она имеет порядок, равный 1/2 по этану при низких степенях превращения.
При понижении давления реакция (R3) переходит от первого порядка (предел высоких давлений) ко второму (предел низких давлений). Полагая порядок
реакции (R3) вторым и предполагая, что другие зависящие от давления реакции
свой порядок не изменяют, определите выражения для d[C2H6]/d/, d[CH3]/d/,
d[C2H5]/d/ и d[H]/d/.
в) Используя стационарное приближение для радикалов, найдите порядок
реакции конверсии С2Н6 при низких ее степенях и давлениях (реакция 3 имеет
второй порядок). Можно принять, что расход этана определяется только реакцией (R4), а его образованием в реакции (R6) можно пренебречь.
г) Реакция
R7 С2Н5 + Н -> С2Н6
была предложена как важная стадия обрыва цепи в пиролизе этана, но никаких
данных по измерению скорости этой реакции нет. Используя теорию столкновений, оцените константу скорости для этой реакции при 1000 К в предположении нулевого значения энергии активации. Радиусы молекулы С2Н5 и атома
Н можно принять равными 0,4 и 0,2 нм соответственно.
7. Каталитический синтез перекиси водорода
Ниже мы рассмотрим модельную систему синтеза перекиси водорода (Н202)
с использованием твердого катализатора, содержащего гипотетический металл М. Предполагается, что реакция состоит из следующих элементарных
494
Вопросы и упражнения
стадий, из которых все, за исключением стадии (3), протекают в условиях
квазиравновесия:
Для этой реакции нет хороших катализаторов, поэтому Н202 получают в
некаталитическом химическом синтезе. Основная проблема связана с кислородом, в частности с атомарным кислородом, связывающимся слишком сильно с поверхностью гипотетического металлического катализатора.
а) Какая часть серии переходных металлов была бы хорошим кандидатом
на роль металла М?
б) Напишите правдоподобный механизм реакции, при котором стадия (2)
включала бы диссоциативную адсорбцию водорода и вода не была бы интермедиатом.
в) Энтальпия образования перекиси водорода А= -136 кДж/моль. Каковы оптимальные по температуре и давлению условия синтеза Н202 в предположении, что стадия (3) идет с конечной энергией активации?
г) Вернемся к написанной выше схеме. Покажите, что если третья стадия
является лимитирующей, то скорость реакции можно представить в виде:
и выразите KG через Кх — К5.
д) Выразите ft только через константы равновесия и парциальные давления.
Ниже мы примем, что кислород является наиболее избыточным интермедиатом реакции (НИИР), то есть в.... = 1 - 6Qi.
е) Предполагая кислород НИИР, определите порядки реакции по всем участвующим в реакции газам, то есть найдите ян , n0j и ян,0,-
Пусть каталитическая реакция происходит в пределе нулевой степени конверсии и при таких высоких температурах, что поверхность можно считать
свободной от интермедиатов, то есть ft = 1.
ж) Определите, при каком отношении концентраций Н2 и 02 скорость в
этих условиях будет максимальной.
8. Эффективность катализатора из Pt
Ниже мы рассмотрим катализатор из Pt, нанесенный на А1203, который
используется для окисления реагентов. В процессе получения катализатор
1. Н, + 2"
2.
о^ + ^ о;
з. о; + н но; +
4. но; + н ^ н,о; +
5. н,о; ^ н2о2 +
Упражнения -J\r 495
дробится и его фракция почти сферических частиц радиусом 0,1 мм используется в тестовых опытах. Плотность катализатора равна 2 г/см3, а удельная площадь поверхности по БЭТ составляет 180 м2/г. Один грамм катализатора помещался в трубчатый реактор, и площадь поверхности Pt находилась путем ее
предварительного окисления, и только площадь этой поверхности определяла
количество воды, произведенной на одном грамме катализатора из кислорода, выделяющегося в результате взаимодействия N20 с катализатором. Можно принять, что катализатор обладает только поверхностью Pt(100), а при
насыщении ее кислородом после контакта с N20 формируется структура
(л/2 х л/2)Л45° О—Pt(100). Постоянная решетки Pt равна а = 3,92 А. После
восстановления поверхности получено 21,4 мл паров Н20 при температуре
298 К и давлении 1 бар.
а) Какова площадь поверхности Pt, приходящаяся на один грамм этого катализатора?
б) Какая грань Pt наиболее устойчива в условиях вакуума и будет ли в
реальных условиях поверхность содержать только такие грани?
Теперь примем, что каждый активный центр образован четырьмя атомами
Pt и активность катализатора определяется в условиях, когда скорость реакции имеет первый порядок по концентрации кислорода. Скорость потока равна
100 мл/мин, газ содержит 21 % кислорода, температура равна 500 К, давление — 1 бар, а число оборотов реакции на один активный центр составляет
0,001 с-1. В этих условиях количество конвертированного кислорода можно
считать пренебрежимо малым.
в) Определите скорость образования и концентрацию продукта в потоке
газа в этих условиях.
Скорость образования продукта можно также записать в виде г = VSkC0,
где V — объем катализатора; S — площадь поверхности Pt, приходящаяся
на единицу объема катализатора; к — постоянная; С0 — концентрация реагента.
г) Определите значение константы к при 500 К.
В результате измерения при разных температурах (и постоянной С0) были
получены следующие значения скорости:
т, к
420
460
540
580
660
780
900
980
г, с-1
1,7 х 10"9
3,5 х 10-8
3,6 х 10-6
2,3 х 10“5
3,5 х 10“4
2,8 х 10“3
1,0 х 10“2
2,0 х 10“2
д) Постройте зависимость скорости от температуры в координатах Аррениуса и рассчитайте кажущуюся энергию активации.
е) Определите эффективность катализатора при 900 К, приняв для эффективного коэффициента диффузии значение 0,003 см2/с и считая его не зависящим от температуры. Предложите, как можно сделать катализатор более эффективным в условиях работы при высоких температурах.
496 -J\r Вопросы и упражнения
9. Неопределенности в нахождении скоростей
и энергий активации реакций
Довольно трудно оценить теоретически длины связей и частоты колебаний
для активированного комплекса и энергию его образования. Представляет интерес оценить, как неопределенность в значениях этих параметров влияет на
величину константы скорости, рассчитываемой в рамках теории переходных
состояний (ТПС). Константа скорости реакции обмена
Rl Н + Н2 ^ Н3# Н2 +Н,
определенная в рамках ТПС, равна 7,8 х 10-13 см~3/с ПРИ Ю00 К, в то время как
эксперимент дает значение 2,1 х 10-12 см_3/с. Оценки по ТПС основаны на
симметричной линейной конфигурации активированного комплекса с расстоянием Н—Н, равным 0,93 А; частота деформационных колебаний принималась равной 2193 см-1, а валентных — 978 см-1.
1. Неопределенность в оценке энергии активации реакции редко бывает меньше 8 кДж/моль. Оцените, можно ли различие между экспериментальным /гехр и
теоретическим kjST значениями констант приписать этой неопределенности.
2. Оцените, можно ли различие между кехр и kTST приписать 10 %-й неопределенности в длине связи Н—Н активированного комплекса.
3. Оцените, можно ли различие между кехр и kjST приписать неопределенности в 100 см-1 для частот колебаний активированного комплекса.
4. Найденное в эксперименте значение предэкспоненциального множителя
для реакции Н + Н2 равно 2,3 х 1014 моль-1 • см3/с-1. Приняв для молекулярных
радиусов Н и Н2 значения 0,2 и 0,27 нм, соответственно, рассчитайте вероятностный множитель Р, обеспечивающий совпадение значений констант скорости, полученных в эксперименте и в теории столкновений при 300 К.
ПРИЛОЖЕНИЯ
ПРИЛОЖЕНИЕ А
Некоторые полезные фундаментальные постоянные
Число Авагадро
"а
6,022 х 1023 моль 1
Постоянная Планка
h
6,626 х 10-34 Дж с
h/ln
h
1,055 х 10"34Дж-с
Скорость света
с
2,998 х 108 м/с
Элементарный заряд
е
1,602 х 10-19 л
Атомная единица массы а.
е. м.
1,661 х 10~27 кг
Масса электрона
те
9,110 х 10~31 кг
Масса протона
тр
1,673 х 10-27 кг
Масса нейтрона
тп
1,675 х 10-27 кг
Число Фарадея
F
9,649 х 104 л/моль
Постоянная Больцмана
кв
1,381 х 10-23 Дж/К
Газовая постоянная
R
8,314 (Дж/моль)/К
Магнитная проницаемость вакуума
Мо
1,257 х 10~6 (В • с2/Кл)/м
Электрическая постоянная
£0
8,854 х 10"12 (м ■ В)/Кл
Гравитационная постоянная
G
6,673 х 10-" м~3/с2
Коэффициенты перевода единиц энергии
Приложения
&
о
о
О
X
8
СЭ
■ч-
T
о
X
40
40^
fN
T
о
X
CN
О
40^
40
т
о
X
40
00
ON
40
т
о
X
о
00
СП
Т
о
X
о
40
СП
TJ-
Атомная
единица
энергии
о
гч
о
X
т!-
ON
CN
CN
Tj-
1
О
X
in
ON
Г"-
ch
тГ
О
X
Г"-
40^
СП
40
1
о
X
40
in
in
40
1
О
X
40
СП
—
©
О
X
Tj-
ON
CN
CN
m
О
X
CN
r-T
о
X
сп
o
CN
ъ
X
о
40
о
X
ON
СП
-
о
X
00
in
сп"
О
X
Tj-
Tj-
CN
r-T
Т
о
ГО
О
X
^1-
40
сэ
oo~
о
fN
О
X
ON
сп
сп
ГО
О
X
in
40
<о
ОС
Т
о
X
о
in
ON
4сГ
»/">
о
X
in
ON
CN
О
X
in
СП
in"
CQ
л
о
X
CN
Tj-
CN
40~
7
о
X
Г"-
сп
сэ
—
Tf
1
о
X
о
^1-
CN
m
1
О
X
г-»
40^
ос"
О
X
CN
Г"-
CN
о
X
CN
CN
4cT
кДж/моль
ГО
CM
О
X
CN
CN
<0
40~
-
о
X
Г"-
^1-
40^
ON
CTv
О
X
г-
40
ГО
1
О
X
Tf-
СП
о©~
2
X
in
СП
40^
ri
ГО
О
X
CN
CN
4cT
кДж
-
ТГ
CM
1
О
X
40
40^
fN
fN
1
О
X
CN
О
40^
Tf
1
о
X
о
TJ-
CN
40
fN
1
О
X
00
СП
fN
1
О
X
ON
in
СП
Tj-
0
1
О
X
о
о
*
3
J3
§
s
3
CQ
О
Т
о
0)
6
<
Cl
СГ)
Приложения —499
Некоторые полезные соотношения
1 бар = 760 мм рт. ст. = 105 ООО Па = 105 ООО (кг • м2)/с2
F Р = 2,632 х IО29 Р[бт м"2/с
yj27rmkB Т yjAWja е м j 7|К]
1 моль при температуре 300 К и давлении 1 бар ~ 25 л
1 мЭв = 8 см-1
квТ при 300 К = 25 мЭв = 1/40 эВ
Некоторые определения
500 -l\r Приложения
го
1
§
'i'
§
Т
о
T
о
о
о
X
8
D
1
X
^—s
ра
о
8
<D
&
X
2
ра
О
р
о
X
cd
X
'S'
cd
O-
cd
Ю
X
D
sr
Л
о.
р
Л
о.
л
X
ра
cd
X
D
^cd4
о.
о
с
ю
о
>>
О.
е
cd
in
<N
СП
i Э
X >v
сЗ
р
Р
О
£
3
со
О
fee S'
со
S
Л
со
S
н
X
D
U
ра
э5
D
cd
со
5
II
о.
о
Q.
t=
S
II
о 5
cd °
X >>
рЗ
Л
Е?
Л
X
Л
cd
a
x
л
Из
D
о.
О
Л
ю
<u
о
pa
5
К tr
^ Я
x о
$
ю
О
о
D
00
3S
D
§
О
о
|=;
о
S
О
D
ра
3S
3
II
V
II
=S
<U
О
s
(U
t0
ю
о
cd
X
in
<N
m
о
1 Я
м
s е
I
>>
t=C
о
о.
с
=5
D
§
О
5
S
§
О.
с
3S
D
§
о
5
S
гг
|=;
0
S
Т
D
1
ра
cd
X
ра
е
X
D
ра
О
е
cd
X
3
X
н
о
о
X
X
о.
D
ра
О
X
§
с
Л
i
D
О
и
О
X
ра
in
of
(N
II
и
in
<N
II
К
о
5
5
T
<
§>
§
D.
t=
=S
D
:S
2
X
X
<u
К
<u
t=C
cd*'
CO
cd
u
§
X
о
II
s
H
cd
II
Я
in
оо"
ti
ei
& °
Я *
О X
a. a
с й
н- ®
5, °
& с
2 s
§ §
Т
Л
D
О.
с:
5S
D
Л
g
S
cd
CL
(N
II
S W
о
эЯ
5
о
О
К
5
D
о
D
к
S
о
00
о
s
гг
о
t;
гг
о
о
к
S
о
Т
S
гг
л
Л
cd
Q.
S
о.
е
л
со
S
о.
1
СП
5
S
S
о
о.
D
ра
-Q
Н
О
о
Л
К
1 s
к s
>> cd
<
н о
X с
х S
К
cd
X
X
§ tS
D.
D
11
S
ST
cd
<&
О.
ра
сЗ
X
о
X
о
со
>,
CD X
"л 2
1 2
О о
g s
е
О.
О.
*Q p.
5 Е
X о.
D §
о
fS
о
о
53
РЭ
-Q
Н
О
о
53
РЭ
j3
X
D
С
2
D
с:
о
S
п
U
Л
X
5 О
CX S
S- °-
g P
Ё со
ё |
cd о
P- ^
5 °
о
D.
3 cd
X t=C
a. s
cd
о.
о
ю
о
о
S
Ё
S
Ё
и
5"
С
Э
p
к
о
S
<
<
и
гг
IP 5Sl
Л* II
ir
1 *■ ■
s§ , J
5 s sS
"l z
§s,
40
,1s ^3.
I'-a
i £
§1.
S г ^
S" ^-s*
5 НГ
sll 11
S s
£ fe
SI
? a ?s
-'i ®SS
s?£ ll
V -
- Ы '
.1.
1 § ,*
1..0Р
tU St
К ’ «-
к У'
in
1 I я*
V£§*
\ *
1 м ■
-f gS:;
.!• :t
ч' s
<St°
U =?.
л«м?;
V|I!
«II =1
I,1 gS
.is “II
? 1
^ - а -
1 £ц
sts 2 1
2 i§
V Ё
С &H
,11
Р'Ь S
sS О-*
£ ZS4
all я!
s 5 if
u* -es
к Z s
ȣS II
i;S5al
si!
M s- 11
1, *§s
**i 5i
"■Пт '
а£ rt*si
1 Рч^
s« , 5s
-.,1 s-
8|
L us
$s§ 5^
^ § s
Я- " -С "
£ £s§
la Н
8£§ = *
Научное издание
Заявки на книги присылайте по адресам:
zakaz@id-intellect.ru
solo@id-intellect.ru
тел. (495) 579-96-45
факс (495) 579-96-70
В заявке обязательно указывайте
свои реквизиты (для организаций) и почтовый адрес!
Подробная информация о книгах на сайте
http://www.id-intellect.ru
Иб Чоркендорф
Ханс Наймантсведрайт
СОВРЕМЕННЫЙ КАТАЛИЗ
И ХИМИЧЕСКАЯ КИНЕТИКА
Компьютерная верстка — Н.А. Попова
Корректор — Г.Н. Петрова
Художник — С.Ю. Биричев
Ответственный за выпуск — Л.Ф. Соловейчик
Формат 70 х 100/16. Печать офсетная.
Гарнитура Ньютон.
Печ. л. 31,5. Основной тираж 300 экз.
Доп. тираж 700 экз. Зак. № 1297
Бумага офсетная № 1, плотность 80 г/м2
Издательский Дом «Интеллект»
141700, Московская обл., г. Долгопрудный,
Промышленный пр-д, д. 14,
тел. (495) 617-41-85
Отпечатано в ООО «Чебоксарская типография № 1»
428019, г. Чебоксары, пр-т И. Яковлева, д. 15