/
















































































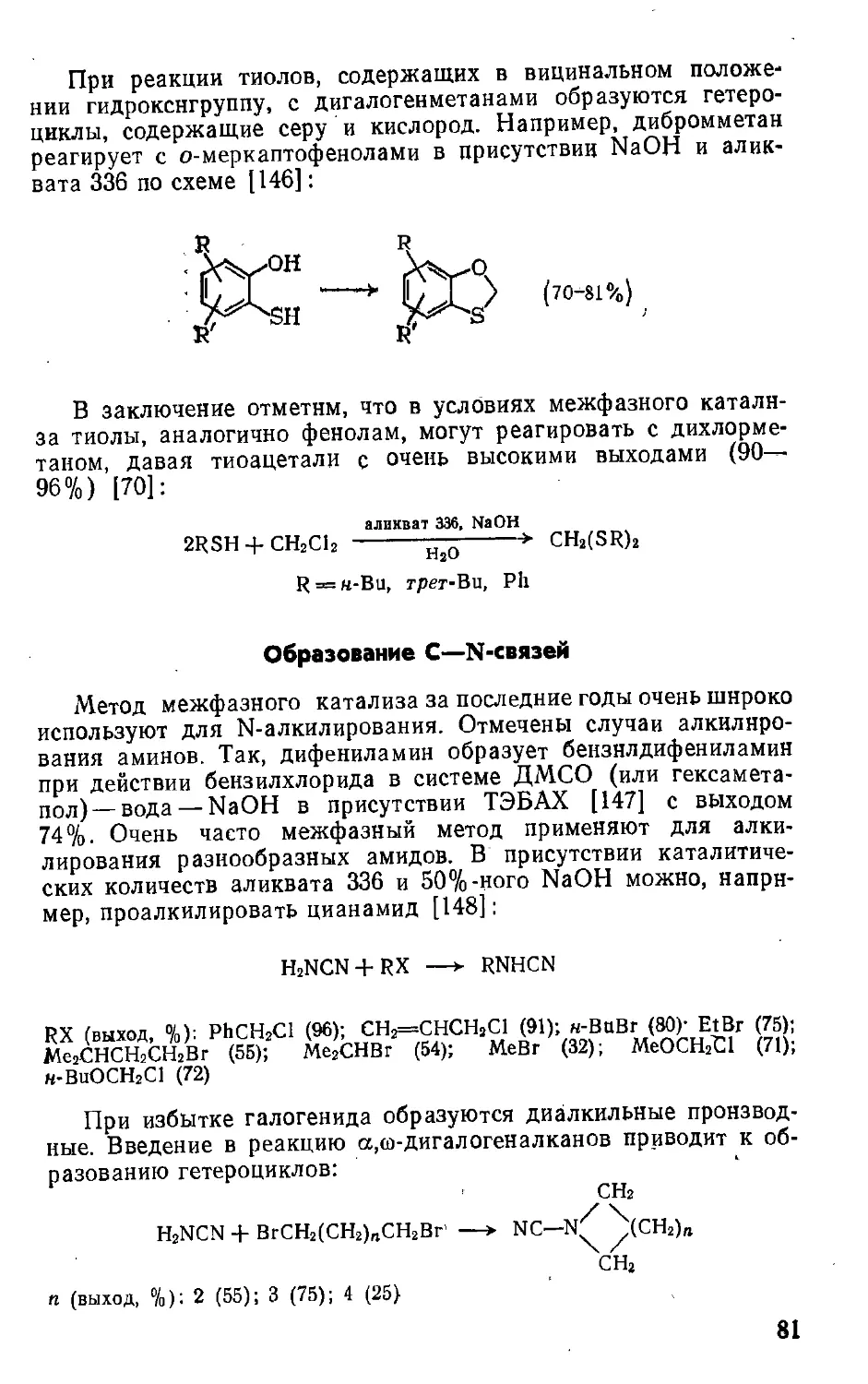









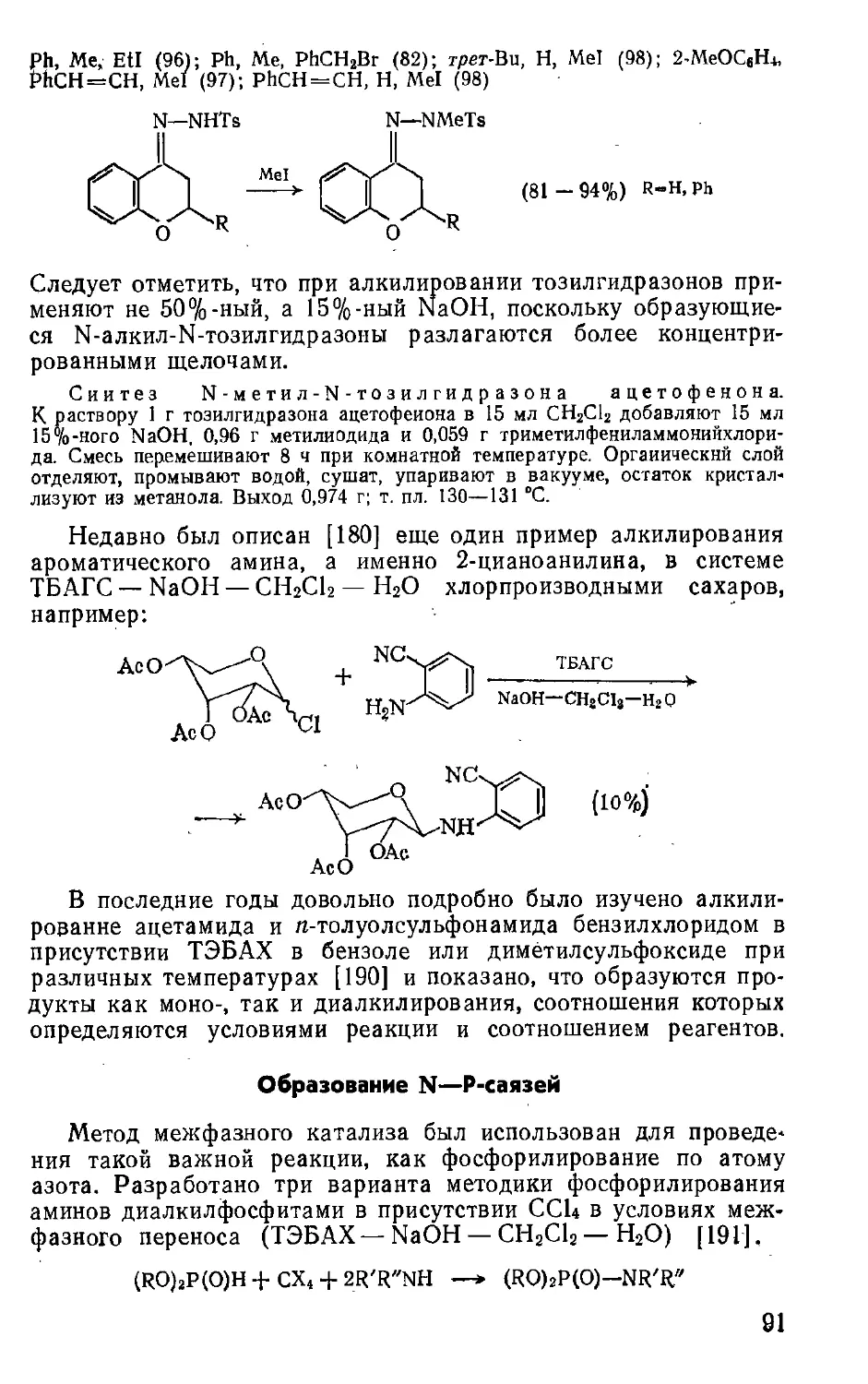

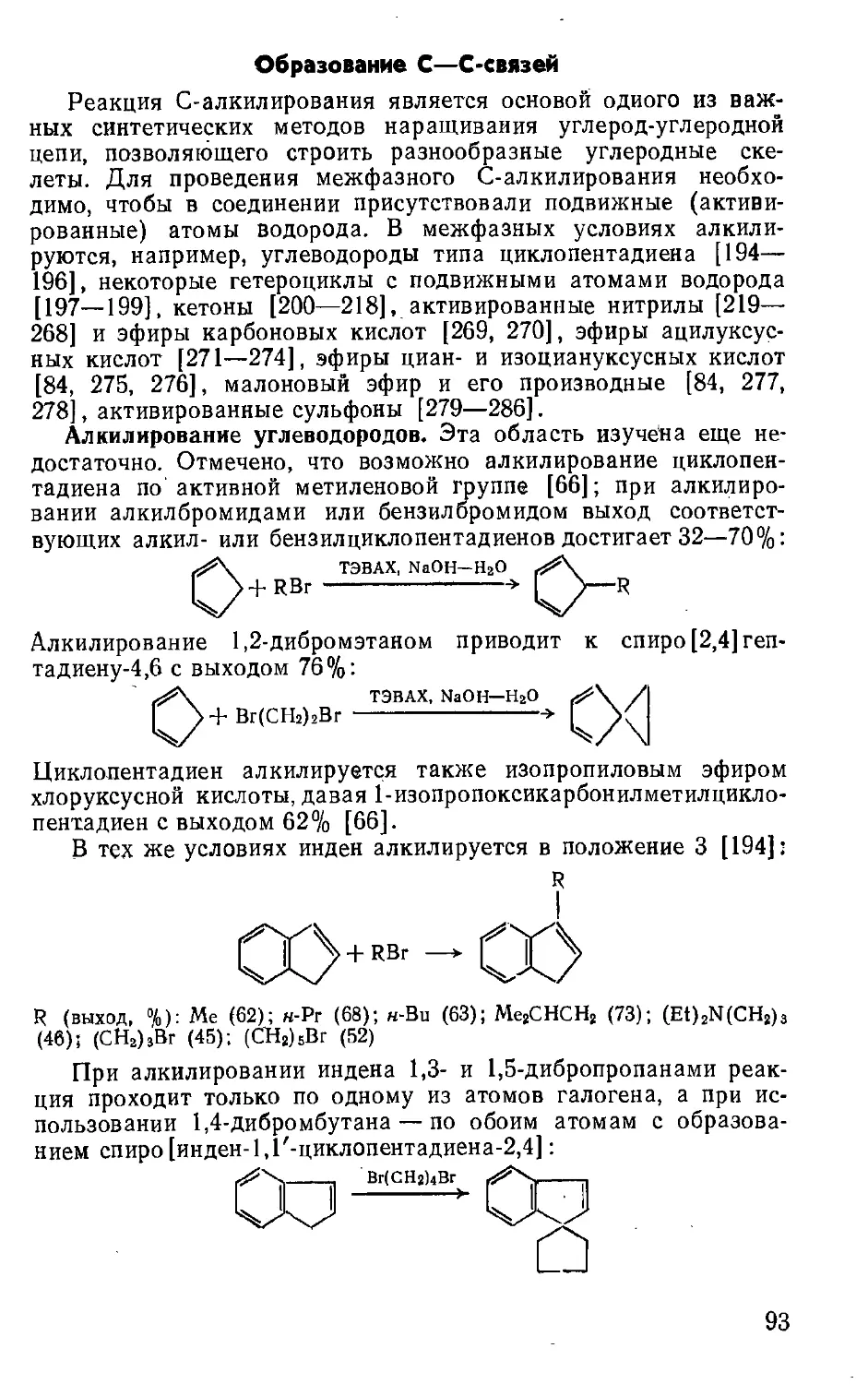








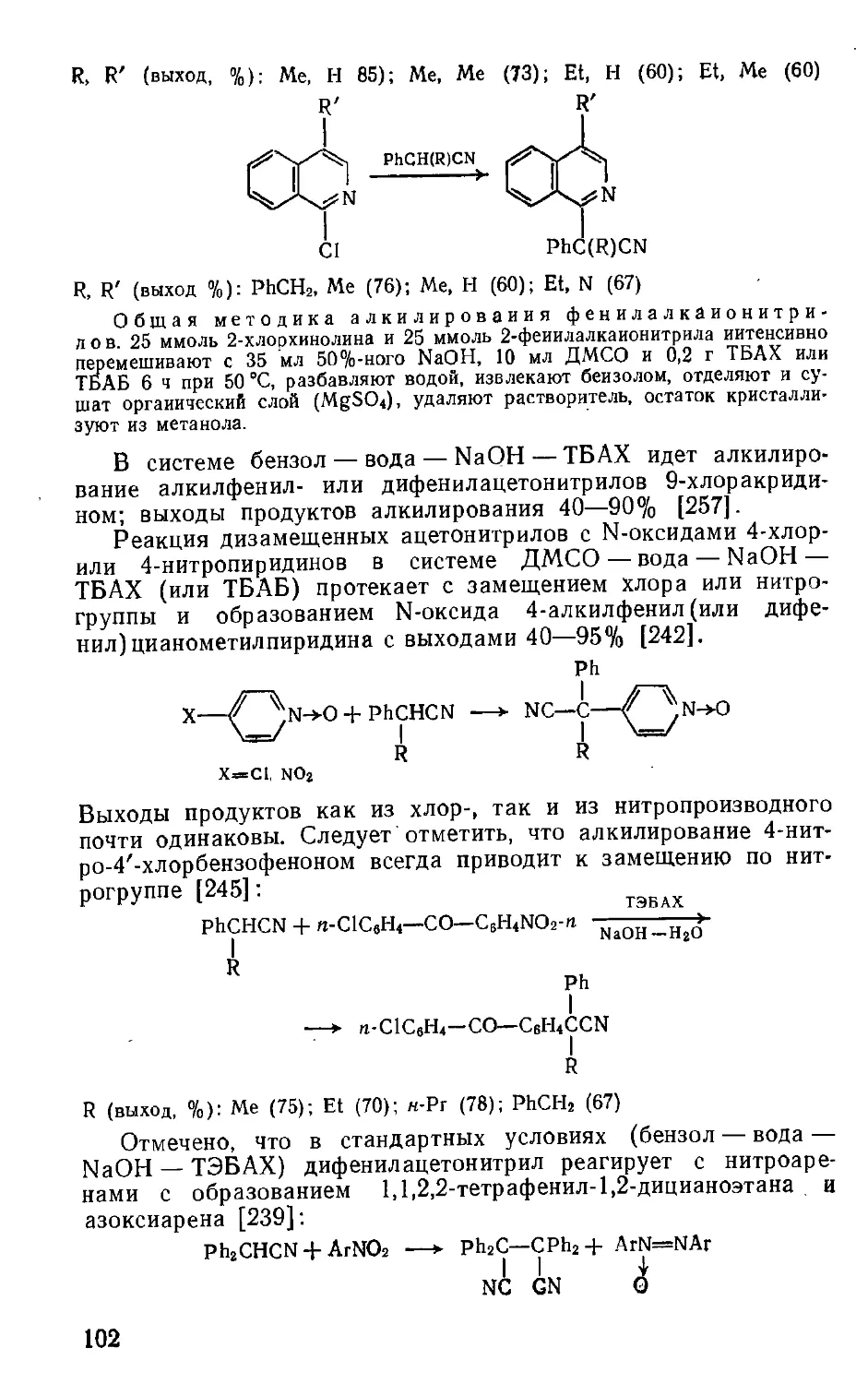





















































































Текст
УДК 547:541.012
Яновская Л. Ам Юфит С. С.
Органический синтез в двухфазных системах.—
М.: Химия, 1982.— 184 с, ил.
Книга посвящена одному из новых перспективных методов
органического синтеза — использованию катализаторов
межфазного переноса (четвертичные аммониевые или фосфонисвые соли,
краун-эфиры, криптаты и др.) в различных реакциях.
Применение этих катализаторов позволяет не только резко повысить
скорость реакций, но и использовать в качестве оснований твердые
щелочи или их водные растворы вместо алкоксидов, амидов,
гидридов щелочных металлов, самих щелочных металлов, металл-
органических соединений и т. п., устраняет необходимость
использования безводных сред даже в реакциях, очень
чувствительных к влаге. В книге приведены типичные методики
проведения разнообразных реакций, рассмотрены теоретические
вопросы межфазного катализа.
Книга рассчитана на широкий круг химиков-органиков,
работающих как в научно-исследовательских институтах, так и в
промышленности. Она будет полезна для преподавателей высших
учебных заведений, студентов и аспирантов, специализирующихся
в области органической химии и технологии.
184 с, 17 табл., 634 литературные ссылки.
Рецензенты:
докт. хим. наук проф. Е. Н. ПРИЛЕЖАЕВА,
докт. хим. наук проф. А. М. РУБИНШТЕЙН
1803000000-018
050(01)-82
© Издательство „Химия", 1982
Л. А. ЯНОВСКАЯ, С. С. ЮФИТ
ОРГАНИЧЕСКИЙ
СИНТЕЗ
в двухфазных
системах
МОСКВА, «ХИМИЯ», 1982
ПРЕДИСЛОВИЕ
В 1965 г. М. Макоша опубликовал Первую из большой серии
работ, которые положили начало использованию межфазных
катализаторов в органической химии. Следует отметить, что и
до появления в свет этих публикаций в литературе отмечались
каталитические свойства четвертичных аммониевых солей в
реакциях алкилирования и в некоторых других реакциях (работы
Жарусса, А. Т. Бабаян с сотр., Старкса и др.), однако
внедрение в практику органического синтеза межфазных катализаторов
началось с работ Макоши. Основное значение метода
межфазного катализа заключается в том, что он позволяет отказаться от
применения безводных органических сред, а также
высокочувствительных к влаге и пожароопасных щелочных металлов, их алк-
оксидов, гидридов, амидов и металлорганических соединений для
проведения многочисленных реакций нуклеофильного замещения
и присоединения, элиминирования и других превращений. Метод
основан на применении в качестве катализаторов
легкодоступных четвертичных аммониевых, фосфониевых и т. п. солей или
макроциклов типа краун-эфиров или криптатов, что позволяет
осуществить перенос анионов из водной или твердой фазы в
органическую фазу и генерировать карбанионы или карбены
действием водных растворов или измельченных твердых щелочей
(NaOH, КОН, Na2CO3, K2CO3 и др.).
Метод особенно удобен и перспективен для промышленного
использования и уже нашел применение в промышленности
тонкого органического синтеза, например, для алкилирования кето-
нов. Круг реакций, которые могут быть переведены на рельсы
межфазного катализа, широк и многообразен и практически
включает все реакции, проходящие с участием карбанионов,—
реакции Кляйзена, Кнёвенагеля, Михаэля, Виттига — Хорнера,
Кори и другие, а также различные типы реакций О-, S-, N- и
С-алкилирования, реакции нуклеофильного обмена,
элиминирования, присоединения, а также реакции, включающие
генерирование дигалогенкарбенов,
Мы убеждены, что уже настало время ознакомить с
преимуществами и перспективами метода широкие круги
химиков-органиков, работающих в научно-исследовательских институтах, в
высших учебных заведениях и, в особенности, в химической
промышленности. Предлагаемая монография рассчитана на
широкие круги химиков, работающих прежде всего в области
органического синтеза. В книге отражены все основные достижения,
перспективы и возможности дальнейшего развития метода.
Кроме того, она может послужить и непосредственным практическим
руководством, так как содержит прописи синтезов.
Авторы сочли необходимым уделить большое место
теоретическим аспектам межфазного катализа, прежде всего, механизму
процесса, поскольку знание основ межфазного катализа
позволяет выбрать наиболее подходящий вариант проведения синтеза,
решить вопрос о возможности использования условий межфаз-
пого катализа для проведения данной конкретной реакции.
Авторы
Список сокращенных названий
катализаторов
Адоген 464
Аликват 336
БГДДМАХ
БТМАХ
ГДТМАБ
ТБАБ
ТБАГС
ТБАИ
ТБАПХ
ТБАХ
ТБГДФБ
ТДТМАБ
ТМОДАБ
ТОПАХ
ТЭБА-С1, ТЭБАХ
ТЭБА-ОН
— триалкил(С8—Сю)метиламмонийхлорид
— метилтриоктиламмонийхлорид
— бензилгексадецилдиметиламмоиийхлорид
— бензилтриметнламмонийхлорид
— гексадецилтриметиламмонийбромнд
— тетрабутиламмонийбромид
— тетрабутиламмоннйгидросульфат
— тетрабутиламмонийиодид
— тетрабутиламыонийперхлорат
— тетрабутиламмонийхлорид
— трибутилгексадецилфосфонийбромид
— тетрадецилтриметнламмонийбромид
— триметилоктадециламмонийбромид
— триоктилпропиламмонийхлорид
— беизилтризтиламмонийхлорид
(триэтилбснзиламмонийхлорид)
— бензилтриэтиламмоиийгидроксид
Общие принципы и преимущества
межфазного катализа
За последние годы межфазный катализ нашел широкое
применение в самых разнообразных областях органического
синтеза. Более того, межфазный катализ уже прочно вошел в
практику промышленного синтеза ряда органических соединений.
Пожалуй, трудно назвать какой-либо иной метод, который сразу
был бы принят на вооружение синтетиками и занял такое
значительное место в органическом синтезе, вытеснив многие
старые привычные способы, включающие использование щелочных
металлов, их алкоксидов, гидридов и амидов, металлоргани-
ческих соединений и т. п. Триумфальное шествие межфазного
катализа объясняется прежде всего неоспоримыми
преимуществами и достоинствами, отличающими его от старых методов.
Как вытекает из названия метода, катализаторы
межфазного переноса используются при проведении реакций в системе,
состоящей из двух несмешивающихся фаз: жидкость —
жидкость или твердая фаза — жидкость. Одна из фаз (жидкая,
обычно водная, или твердая) включает основание и (или)
нуклеофил. Вторая фаза, как правило, является раствором
субстрата в каком-либо органическом растворителе (иногда роль
растворителя играет сам субстрат). Поскольку фаза, содержащая
основание и (или) нуклеофил, нерастворима в фазе с субстратом,
в отсутствие катализатора межфазного переноса реакция не
идет. Добавка межфазного катализатора, содержащего липо-
фильный катион, растворяющийся в обеих фазах, вызывает
обмен анионов катализатора с анионом в водной (или твердой)
фазе. Если обозначить катион катализатора межфазного
переноса Q4*, анион Х~, а катион нуклеофила в водной фазе М+ и
соответствующий анион Nu~, то ионный обмен между фазами
можно представить как равновесие
Q+ X" + М+ Nu~ ;f=fc Q+ N11" + М+ X" (водная фаза)
Далее Q4* Nu~ из водной фазы переходит в органическую фазу
с установлением равновесия
Q+ Nu" (водная фаза) ч=^ Q+ Nu~ (органическая фаза)
Перешедший в органическую фазу нуклеофил реагирует с
субстратом RX, находящимся в органической фазе. В случае
нуклеофильного замещения Q4* образует ионную пару с
уходящей группой X", и таким образом каталитический цикл
замыкается. Общую схему процесса можно представить схемой:
QNu +' RX * RNu + QX (органическая фаза)
QNu + MX ^=^: MNu + QX (водная фаза)
В системе жидкость — твердая фаза (где твердой фазой
служат NaOH, КОН, К2СО3, Na2CO3) такой обмен не идет. В этом
случае реакции, например депротонирование, по-видимому,
проходят на поверхности раздела фаз, а катализатор межфазного
переноса просто снижает энергию барьера реакции (как в
случае гетерогенного катализа). Более подробно механизм
межфазного катализа обсужден в следующем разделе.
В качестве катализаторов межфазного катализа могут быть
использопаны четвертичные аммониевые и фосфониевые соли.
Чаще всего применяют бензилтриэтиламмонийхлорид (ТЭБАХ),
тетрабутиламмонийбромид (ТБАБ), тетрабутил аммонийхлорид
(ТБАХ), метилтриоктиламмонийхлорид (аликват 336), триал-
кил (С8 — Сю) метиламмонийхлорид (адоген 464), тетрабутил-
аммонийгидросульфат (ТБАГС), бензилтриметил
аммонийхлорид, триметилоктадециламмонийбромид, трибутилгексадецил-
фосфонийбромид (ТБГДФБ); описано применение арсониевых
солей, например тетрафениларсонийхлорида. Кроме того,
предложено применять разнообразные амины и диамины (N,N'-flH-
метилпиперазин, Ы,Ы,Ы^№-тетраметилэтилендиамин, N-бутил-
пиперидин, триалкиламины и др.)- Наконец, показано, что
в системе твердая фаза — жидкость особенно удобно
использовать каталитические количества краун-эфиров (18-краун-6,
дибензо-18-краун-6, дициклогексано-18-краун-6) и криптатов
(например, 1,10-диаза-4,7,13,16,21,24-гексаоксабицикло [8.8.8] гекса-
козан). Аммониевые и фосфониевые соли могут применяться
на полимерной подложке или на силикагеле (например,
использованы N-додецил-М-метилэфедринийбромид на полимерной
подложке и трибутилгексадецилфосфонийбромид на полимерной
подложке, а также тетрабутилфосфонийбромид на силикагеле).
Можно использовать полимерносвязанные краун-эфиры
(например, 18-краун-6) или криптаты. Несомненно, что в качестве
катализаторов межфазного переноса могут служить и другие
соединения, способные к образованию ионных пар с анионами или
комплексов с катионами. Однако при выборе межфазного
катализатора в первую очередь следует предпочесть наиболее
дешевые и доступные четвертичные аммониевые соли. По всем
имеющимся экспериментальным данным эти соли не уступают,
а в ряде случаев превосходят по каталитическому эффекту
краун-эфиры и криптаты в системе жидкость — жидкость и
8
очень эффективны в большинстве случаев в системе твердая
фаза—жидкость. Различия в каталитической активности
разных четвертичных аммонийных солей хотя и существуют, но,
видимо, не столь существенны, и поэтому вряд ли можно
заметно улучшить результаты синтеза сменой катализатора.
Количество применяемого катализатора обычно составляет 1 —
10% (мол.). Однако описан метод с использованием стехиомет-
рических количеств межфазного катализатора (применяется
почти исключительно ТБАГС), разработанный Брёндстрёмом.
Этот метод называют методом «экстракции ионных пар».
Следует отметить, что в большинстве случаев реакции, проходящие
в этих условиях, идут и при использовании каталитических
количеств межфазного катализатора; лишь в редких случаях
результаты менее удовлетворительны, чем при использовании
стехиометрических количеств катализатора, которое к тому же
создает ряд неудобств, связанных с выделением и очисткой
конечных продуктов реакции, и удорожает синтез.
В нашей монографии мы уделяем основное внимание
использованию межфазных катализаторов в каталитических
количествах в системах жидкость -г- жидкость и твердая фаза —
жидкость, не затрагивая практически метода экстракции
ионных пар и реакций в гомогенных системах с использованием
стехиометрических количеств краунэфиров и криптатов.
В качестве органической фазы применяют бензол, дихлор-
метан, тетрахлорметан, 1,2-дихлорэтан, реже диэтиловый эфир,
этилацетат или ацетон. Часто реакцию ведут без растворителя,
иногда растворителем служит субстрат (например, хлороформ
при генерировании дихлоркарбена). Водной фазой служат
водные растворы солей или щелочей (NaOH, КОН, NaHCO3,
Na2CO3, K2CO3 и др., чаще всего используют 50%-ные растворы
щелочей, реже — более разбавленные, вплоть до 5—10%-ных.
В системе твердая фаза — жидкость твердой фазой служат
измельченные соли или щелочи.
Как правило, реакции проводят при интенсивном
перемешивании реакционной смеси. Очень часто реакции в условиях
межфазного катализа протекают экзотермически; в таких
случаях требуется охлаждение (обычно температуру регулируют
так, чтобы она не превышала температуры кипения
растворителя) . Иногда реакцию проводят при пониженной (0—5 °С)
или при комнатной температуре. Чаще всего реакцию проводят
при 40—60 °С.
Подводя итоги, можно сформулировать следующие
основные принципы, на которых базируется межфазный катализ:
а) изменение свойств анионов в растворе путем изменения
свойств катионов; б) введение анионов, ассоциированных с ли-
пофильными катионами, в неполярную среду, где анионы
могут легко контактировать с органическими субстратами и быть
высокоактивными,
9
Преимущества и достоинства метода заключаются в
следующем.
1. Метод позволяет исключить применение дорогостоящих
безводных растворителей. 2. Значительно повышается скорость
реакций анионов в неполярных средах. 3. Неорганические
анионы, образующиеся в процессе реакции, переходят из,
органической фазы в водную или твердую фазу. 4. Метод исключительно
удобен для промышленных процессов, его легко
автоматизировать; его применение обычно позволяет снизить промышленные
расходы. 5. Время реакции обычно невелико (по сравнению
с другими методами). 6. Выходы продуктов реакции обычно
выше, чистота их больше, чем при использовании традиционных
методик. 7. Как правило, реакции проходят более селективно.
8. В реакцию можно вводить соединения, чувствительные к
гидролизу, действию щелочей, изомеризации и пр. 9. Огромным
преимуществом является использование вместо дорогих,
чувствительных к влаге и пожароопасных щелочных металлов, их
алкоксидов, гидридов, амидов, металлорганических соединений,
водных растворов или твердых измельченных щелочей, а также
отсутствие необходимости защиты от атмосферной влаги.
10. Изменение реакционной способности органических
соединений при реакциях в межфазных условиях и возможность
осуществления реакций, не идущих в традиционных условиях.
11. Наконец, следует отметить, что использование оптически
активных межфазных катализаторов (например, N-бензил- или
Ы-додецил-Ы-метилэфедринийбромида и др.) позволяет
проводить частичный асимметрический синтез.
В заключение мы приводим список обзорных работ,
посвященных межфазному катализу и родственным методам.
Литература
Макоша М. Реакции карбаниоиов н галогенкарбенов в двухфазных
системах. — Усп. химии, 1977, т. 46, № 12, с. 1151—1166.
Вебер В., Гокель Г. Межфазный катализ в органическом синтезе. Пер. с англ./
Под ред. И. П. Белецкой. М., Мир, 1980. 257 с.
Pedersen F. /., Frensdorff И. К. Makrocyclische Polyaether und ihre Komple-
xe. —Angew. Chem., 1972, Bd. 84, № 1, S. 16—26.
Brundstrom A. Preparative Ion Pair Extraction. Apotlkarsocieteten/Hassle, La-
kemedel, Sweden, 1974. 275 p.
Christensen I. /., Eatough D. /., Izatt R. M. The Synthesis and Ion Binding
of synthetic multidentate Macrocyclic Compounds. — Chem. Rev., 1974, v. 74,
№ 2, p. 351—384.
Dehmlow E. V. Phasentransfer katalysierte Zweiphasenreaktien in der prapara-
tiven organischen Chemie. — Angew. Chem., 1974, Bd. 86, № 2, S. 187—
196; 1977, Bd. 89, № 8, S. 521—533.
Makosza M. Two-Phase Reactionen in the Chemistry of Carbanions and Halo-
carbenes — a useful Tool in Organie Synthesis. — Pure a. Appl. Chem.,
1975, v. 43, № 3, p. 439—462.
Dou H. I. M. Catalyse par transferet de phase. — Chim. Actual., 1976, № 1,
p. 41—45.
10
Gokel G. W.} Durst H. D. Principles and Synthetic Applications in Grown Ether
Chemistry. — Synthesis, 1976, № 2, p. 168—184.
Jeanne F., Trichet A. Substitutions nucleophiles a l'aide de cryptstes. — Inform.
Chim., 1976, v. 155, p. 327—346.
Knipe A. C. Crown Ethers. — J. Chem. Educ., 1976, v. 53, № 5, p. 618—622.
Makosza M. Naked Anions-Phsse Transfer. Conference Paper of the
International Workshop of Modern Synthetic Methods, 1976. Interlaken,
Switzerland, Schweizerischer Chemiker Verband, Zurich, 1976, p. 7—100.
Brandsirdtn A. Principles of Phase Transfer Catalysis by Quaternary
Ammonium Salts. —Adv. Phys. Org. Chem., 1977, v. 15, p. 267—330.
Varughese P. Quaternary Ammonium Salts. — J, Chem. Educ, 1977, v. 54, № 5,
p. 666—669.
Gokel G. W.t Weber W. P. Phase Transfer Catalysis. — J. Chem. Educ, 1978,
v. 55, № 2, p. 235—238, № 3, p. 429—433.
Izati R. M., Christensen J. J. Synthetic Multidentate Macrocyclic Compounds.
Academic Press, N. Y., 1978. 301 p.
Mclntosh J. M. Phase Transfer Catalysis using Quanternary Onium Salts.—
J. Chem. Educ, 1978, v. 55, № 3, p. 235—238.
Makosza M. Organische Reaktionen in Zweiphasen Systemen. — Chemie in un-
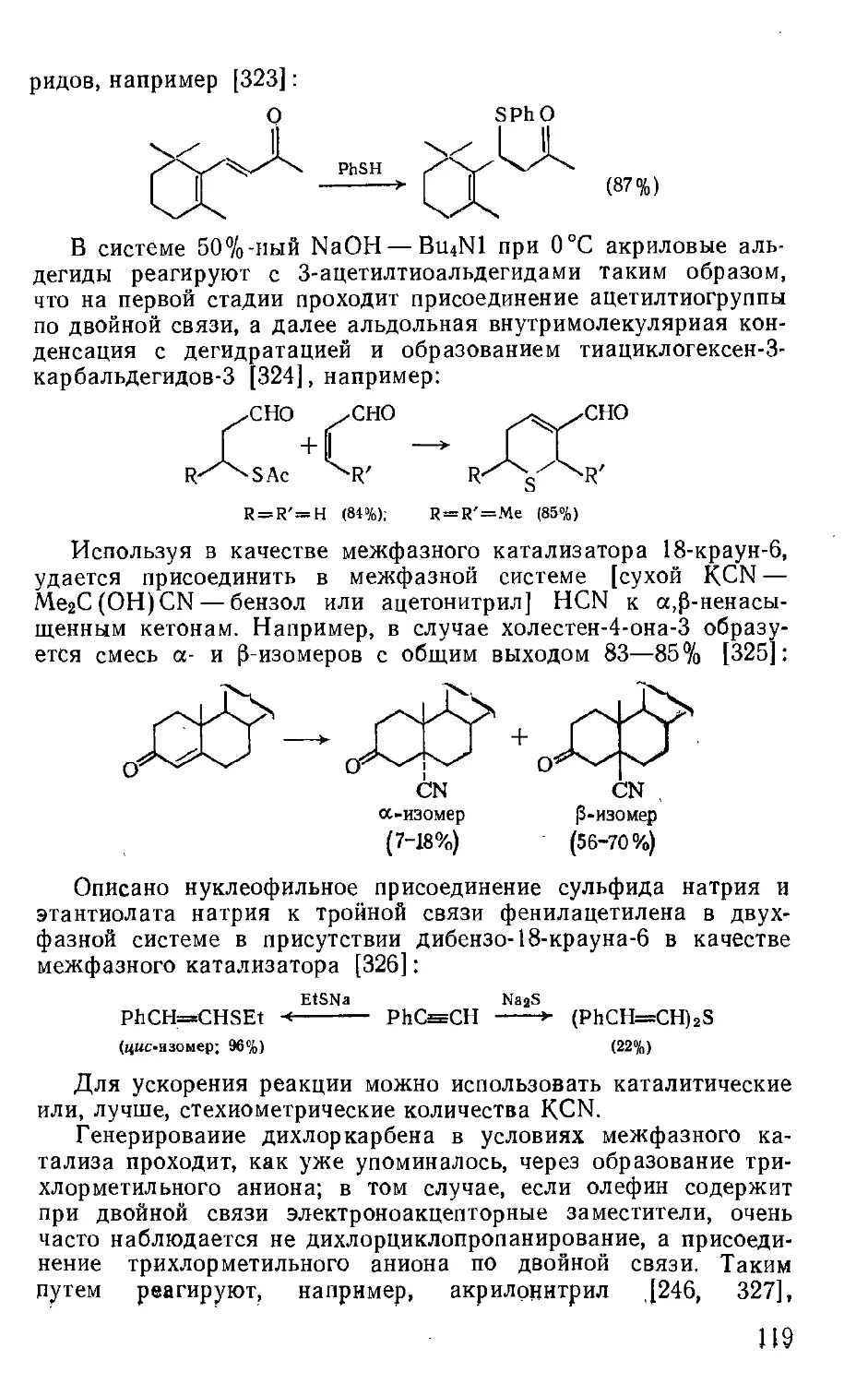
serer Zeit, 1978, Bd. 12, № 2, S. 161—168.
Dehmlow E. V., Dehmlow S. S. Phase-Transfer Catalysis. Verlag Chemie, Wein-
heim, N. Y., 1978. 368 p.
Starks C. At., Llotta С Phase-Transfer Catalysis: Principles and Techniques.
Academic Press Inc., N. Y., 1978. 264 p.
Makosza M. Two-Phase Reactions in Organic Chemistry. Survey of Progress in
Chemistry. V. 9. Academic Press Inc., N. Y., 1979. 152 p.
Jones R. С F. Phase-Transfer and Related Methods.—General Synth. Methods,
1978, v. I, p. 402—423.
Механизм межфазного катализа
Каталитические межфазные реакции могут осуществляться
как в системе жидкость — жидкость, так и в системе твердая
фаза — жидкость. По характеру водной фазы в системе
жидкость — жидкость реакции можно разделить на две группы:
реакции, в которых водная фаза представляет собою
разбавленный раствор, и реакции, в которых водная фаза
представляет собою концентрированный раствор. К первой группе
относятся, например, реакции с переносом неорганических или
органических анионов в органическую фазу: окисление ионами
М11О4 или CrCV, обмен галогена в органических галогенидах
на CN, NO2, ОСОСНз и другие функции. Ко второй группе
относятся все реакции, связанные с депротонированием под
влиянием растворов щелочей, которое ведет к образованию
органических анионов или карбенов:
~ОН ч=ь А~ + Н2О
-Н20
СНХз + 'ОН =г=а= ~СХ3 —* : СХ2 4- X"
+ Нго
Это самая обширная группа из изученных каталитических
межфазных реакций. Сюда относятся многочисленные реакции 0-,
N-, S- и С-алкилирования, конденсации, присоединения и др.
Такие реакции могут осуществляться как в присутствии
каталитических количеств катализатора межфазного переноса,
который обеспечивает транспортировку аниона А~ в
органическую фазу и тем самым дальнейший ход реакции, так и в
присутствии эквимольных количеств катализатора межфазного
переноса. В этом случае сначала проходит стехиометрическая
реакция между катализатором и субстратом с образованием
соли органического аниона и четвертичного аммониевого
катиона Q+:
АН + Q+ "ОН *=fc A~ Q+ + Н2О
Этот вариант межфазного катализа называют методом
экстракции ионных пар. В данном случае соль Q+ A~ выделяется
в виде промежуточного продукта и уже затем включается
в дальнейшие реакции. Для ознакомления с этим методом
можно рекомендовать монографию [1].
При использовании как разбавленных, так и концеитриро
ванных водных фаз метод образования активных частиц оди-
12
наков для всех реакций в данной системе и зависит только от
типа реакционной среды. Таким образом, различные реакции
можно объединить по способу генерирования активной частицы.
РЕАКЦИИ В СИСТЕМАХ С РАЗБАВЛЕННЫМИ ВОДНЫМИ ФАЗАМИ
Можно представить себе по крайней мере три типа
механизма реакций в двухфазных системах, в которых водная фаза
является разбавленным раствором. Эти механизмы различаются
фазой, в которой образуется продукт реакции. При переносе
аниона из водной фазы в органическую фазу (ОФ) реакция
будет проходить в органической фазе. Этот механизм включает
три стадии,
1. Ионный обмен:
М+ Y" + Q+ X~ =e=fc М+ X" + Q+ Y" (водная фаза)
2. Диффузия через поверхность раздела фаз:
+ Х")ВФ
3. Взаимодействие иона Y~ с субстратом в органической
фазе и образование продуктов реакции.
Скорость реакции в этом случае зависит как от скорости
диффузии через поверхность раздела фаз, так и от скорости
гомогенной реакции в органической фазе. Важными факторами
являются энергия разрушения водной оболочки аниона и
энергия пересольватации органическим растворителем. Следует
отметить, что при переходе аниона из водной в органическую
фазу наблюдается кардинальное изменение сольватации оние-
вых ионов. Анион перешедшей в органическую фазу ионной
пары Q+ Y~ крайне мало сольватирован, что даже дало повод
называть реакции таких ионных пар реакциями «голых
анионов» (см. обзор [2]). Очевидно, что для таких реакций
выгоднее всего использовать возможно более липофильные
катионы и малополярные растворители. Классическим примером
переноса анионов из водной фазы в органическую является
окрашивание бензольного слоя в малиновый цвет в системе
водный раствор КМпО4—бензол при добавлении метилтриок-
тиламмонийхлорида [3]. В настоящее время такой «малиновый
бензол» используют для окисления многих органических
соединений.
По механизму «всаливания», связанному с увеличением
растворимости органического субстрата в водной фазе при
добавлении катализатора межфазного переноса, реакция
осуществляется в водной фазе. Этот механизм также включает три
основные стадии.
13
1. Введение катализатора межфазного переноса Q+ X~
в систему; при этом повышается растворимость субстрата АН
в водной фазе и он переходит из органической фазы в водную:
АНВФ
2. Реакция субстрата с реагентом RY, образование продукта
реакции AR:
3. Переход продукта реакции в органическую фазу:
Очевидно, что в этом случае катализ связан с уменьшением
поверхностного натяжения; реакция будет идти легче при
использовании катализаторов Q+ X" с коэффициентами
распределения, близкими к единице. Повышение растворимости
органического субстрата в воде при добавлении ониевых солей
означает, что соли либо изменяют структуру воды (водного
раствора), либо взаимодействуют с органическими молекулами,
В любом случае реакционная способность молекул,
перешедших в водную фазу, должна измениться. Можно
сформулировать следующие закономерности влияния добавок солей на
растворимость неэлектролитов в воде [4—6].
1. Как правило, электролиты понижают растворимость
неэлектролитов в воде («высаливание»).
2. Чем меньше размеры иона при данном заряде, тем
большим высаливающим действием он обладает.
3. По высаливающему действию ионы могут быть
расположены в следующие ряды:
но" > sor > сог > сю; > Вго3~ > сг > сн3соо~ > ю; >
> сю; > Вг~ > г > no3"
Na+ > K+ > Li+ > Ba2+ > Са2+ > Ni2+ > Со2+ > Mg2+ > Fe2+ >
> Zn2+ > Mn2+ > Al3+ > NH4+ > H+
4. Увеличение радиуса иона (катиона или аниона) приводит
к уменьшению высаливания и переходу к всаливанию, т. е.
к повышению растворимости неэлектролита в водном растворе
соли в сравнении с водой).
5. Константа высаливания вычисляется по уравнению
Сеченова:
18 (5o/S) - К3С3
где Sq и S — растворимость неэлектролита в воде и водном растворе соли,
соответственно; С5 — концентрация соли; Ks — коэффициент высаливания.
6. Логарифм коэффициента активности неэлектролита
пропорционален концентрации соли, выраженной в моль/л или
экв/л.
14
7. Влияние добавок солей на кислые и основные
неэлектролиты различно. Основные неэлектролиты чувствительны к
заряду аниона и всаливаются солями лития.
К сожалению, в настоящее время отсутствует строгая
теория всаливания и высаливания и для практического
использования предлагается почти алхимический принцип; в с а л и -
в а ю т те электролиты, в которых неэлектролит растворяется
лучше, чем в растворителе (например, в воде), а
высаливают те, в которых неэлектролит растворяется хуже, чем
в чистом растворителе [5, 7].
В общем случае, надо помнить, что с увеличением
концентрации электролита в воде возрастает коэффициент активности
неэлектролита и с увеличением радиуса катиона растет вса-
ливание неэлектролита. Таким образом, для проведения
реакции по механизму всаливания необходимо повышать
концентрацию длинноцепочечного катиона в водной фазе. Некоторые из
немногих известных коэффициентов всаливания (для бензойной
кислоты при 25 °С [5]) приведены ниже:
Соль Ks Соль Ks
NH4I +0,021 * (C3H7)4NI -0,970
(CH3)4NI -0,256 (C4H9)4NI -1,32
(C2H5)4NI . -0,633
Простой расчет по уравнению Сеченова (см. выше)
показывает, что в результате всаливания, например под действием
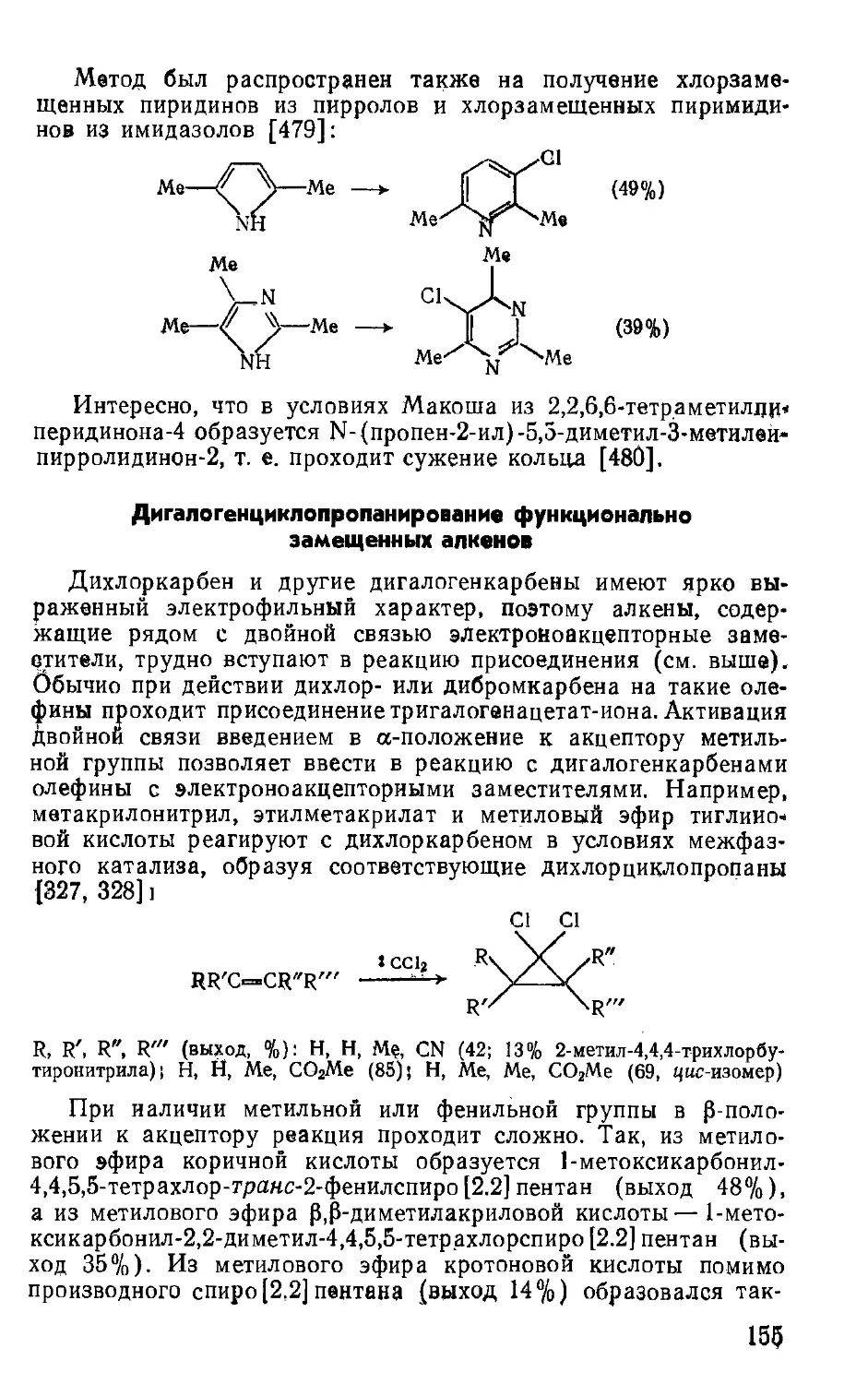
(CUHg^NI, уже при концентрации соли 1 моль/л концентрация
бензойной кислоты в водной фазе изменяется более чем на
порядок, а увеличение концентрации соли до 2 моль/л повышает
концентрацию бензойной кислоты в водной фазе еще на 1,32
порядка. Эффект всаливания заметно повышается, если в
органической молекуле есть полярные группы [4]: бензол <;
анилин < фенол <С бензойная кислота ж фталевая кислота <С
салициловая кислота. Из этого следует, что между ониевым
ионом и полярной группой в органической молекуле имеется
ион-дипольное взаимодействие, которое увеличивает
растворимость органических молекул.
Однако необходимо отметить, что переход органических
молекул в водную фазу (всаливание) может привести в
конечном итоге к так называемому мицеллярному катализу
вследствие образования мицелл под действием
поверхностно-активных веществ и повышения концентрации реагирующих веществ
внутри мицеллы. Эту возможность следует учитывать,
поскольку многие ониевые соли являются
поверхностно-активными веществами. В том случае, когда в молекуле ониевой соли
имеется одна или две длинноцепочечные группы, мицеллообра-
зование наступает довольно быстро. Так, найдено [8], что для
15
четвертичных аммониевых хлоридов с двумя длинноцепочеч-
ными радикалами критическая концентрация образования
агрегатов равна [в % (масс.)]:
(C,2H25)2N(CH3)2 СГ (I) 0,02
(CHH29)2N(CH3)2 C1" (II) 1,23
(Ci3H37)2N(CH3)2 СГ (III) 1,02
Простые жидкокристаллические фазы начинают
образовываться уже при концентрации 18% (масс.) для I, 3%—для II и
3,7% —для III.
Несмотря на большое структурное сходство катализаторов
межфазного переноса с поверхностно-активными веществами,
они весьма различаются по каталитическому действию.
Высокоэффективные катализаторы межфазного переноса обычно
являются плохими поверхностно-активными веществами.
Кинетические данные и способность ониевых солей ускорять реакции
даже в неполярных средах подтверждают предположение, что
суть их каталитического действия заключается не в
образовании мицелл, а в создании каталитического цикла, включающего
обмен ионами. Было показано [9], что реакция между 1-хлор-
октаном и цианидом натрия катализируется как анионными
поверхностно-активными веществами (например, додецилбен-
золсульфонатом натрия), так и неионными
поверхностно-активными веществами (например, продуктами реакции додеканола
и тетрадеканола с 6 моль этиленоксида); однако скорости
реакции при этом в 100—1000 раз ниже, чем при применении
четвертичных аммониевых солей. Таким образом, мицеллярный
катализ можно, конечно, рассматривать как межфазный,
однако ои обладает своей спецификой и далее не будет обсуждаться
в данной книге (см. обзоры [10—13]). Отметим, однако, что,
как правило, поверхностно-активные вещества тормозят
реакции в двухфазной системе. Это, очевидно, связано с тем, что
образование мицелл изменяет физические характеристики
системы и, кроме того, большая часть поверхности раздела фаз
занимается поверхностно-активным. веществом, что приводит
к вытеснению катализатора межфазного переноса. Именно
поэтому для каждой системы существует свой оптимальный
размер катиона, когда он еще остается катализатором межфазного
переноса, но уже не является поверхностно-активным
веществом.
Для упомянутой выше межфазной реакции между 1-хлорок-
таном и NaCN была доказана следующая схема [14]:
QCl + NaCN ^=fc QCN+NaCl (водная фаза)
QCNB(I> —* QCNO(t>
QCN+RC1 —► RCN+QC1 (органическая фаза)
16
Именно эта схема дала повод назвать катализ такого типа
«катализом межфазного переноса» [9]. Основное
доказательство справедливости этой схемы усматривается в том, что
реакция происходит в органической фазе, а не в водной, не
на поверхности раздела фаз и не в мицеллах.
В обзоре [15] приведены интересные данные о влиянии
длины цепи радикала в аммониевой соли на выход стирола при
элиминировании НВг из 1-бром-2-фенилэтана в системе
50%-ный NaOH — катализатор.
50%-ный NaOH
PhCH2CH2Br -► PhCH=CH2
Q+ X~
Q+ X- = CH3(CH2)rtN(C2H5)3 Br"
n
1
2
3
4
5
Выход, %
3
7,2
12
50
53
n
6
7
8
11
Выход, %
45,5
43,7
42,2
38,3
Хорошо видно, что при п > 5 выходы стирола снижаются.
Аналогичный результат был получен и для реакции алкилирования
фенилацетонитрила:
50%-ный NaOH
%ы
PhCH2CN + EtCl > PhCH(Et)CN
Q+ сг
В этом случае при замене, например, тетраэтиламмонийхлорида
на пропилтриэтиламмонийхлорид выход нитрила фенилэтил-
уксусной кислоты понизился с 51 до 23%, т. е. даже
незначительное увеличение одного радикала привело к торможению
реакции.
Примером реакции, проходящей по механизму «всалива-
ния», может служить бензоиновая конденсация [16], для
протекания которой необходим контакт бензальдегида с ~CN.
В двухфазной системе реакция идет плохо, однако при замене
цианида натрия в водной фазе на тетрабутиламмонийцианид
конденсация идет быстро и бензоин образуется с выходом 70%.
Это объясняется «всаливающим» эффектом аммониевой соли,
приводящим к увеличению концентрации бензальдегида в
водной фазе, где находится ион ~CN.
Помимо приведенных выше двух механизмов межфазного
катализа, типичных для систем, использующих в качестве
водных фаз разбавленные растворы, возможен еще один механизм,
при осуществлении которого реакции проходят на поверхности
раздела фазы. Рассмотрим два наиболее типичных варианта
этого механизма.
17
Первый вариант характеризуется тем, что ионная пара,
образуясь на поверхности раздела фаз, десорбируется в
органическую фазу, и реакция ионной пары с органическим
субстратом проходит в органической фазе. Реакция проходит через
ряд стадий.
1. Адсорбция катализатора и субстрата на поверхности
раздела фаз (ПРФ).
2. Ионный обмен и образование органической ионной пары
Q+ А~ на поверхности раздела фаз.
3. Десорбция ионной пары Q4* А~ в органическую фазу.
+ а")прф =«=* (Q+ а1)оф
4. Реакция ионной пары Q+ А~ в объеме органической
фазы:
Q+ A" + RY —> Q+ Y" + RA
где RY — реагент, растворимый в органической фазе.
5. Адсорбция Q+ Y~ на поверхности раздела фаз; Y~
переходит в водную фазу (замыкание каталитического цикла);
(q+ Г)оф =<=fc (0+Г)ПРФ
В этом типе механизма существенную роль играют
различия в коэффициентах распределения ионных пар. Необходимо,
чтобы коэффициенты распределения Q+ Y" и Q+ А~
существенно различались, в противном случае Q+ Y~, оставаясь в
органической фазе, будет тормозить реакцию. Именно поэтому,
например, применение активных алкилиодидов при алкилиро-
вании часто не дает удовлетворительных результатов. Иодиды
более активны на стадии 4, однако вследствие большого
коэффициента распределения тормозят процесс на стадии 5.
Принимая такую схему, не следует забывать, что роль
катализатора состоит не только в переносе аниона А~ из водной фазы
в органическую, но и в переносе аниона Y~ в водную фазу из
органической фазы. Катион катализатора Q+ может,
по-видимому, облегчить отрыв протона (или другой группы) за счет
синхронного взаимодействия или ориентации в промежуточном
комплексе:
У-— I.
ПРФ
Во втором варианте этого механизма вся реакция проходит
на поверхности раздела фаз (через ряд стадий).
1. Адсорбция субстрата АН на поверхности раздела фаз:
Q+ АН
X" г В
18
2. Образование комплекса:
ПРФ
х- н—в:
3. Образование органической ионной пары Q+ А~.
4. Реакция адсорбированной ионной пары с реагентом RY
из объема органической фазы:
4- И^оф —► (Q Y )прф 4-
б. Десорбция продукта реакции с поверхности раздела фаз:
Главной отличительной чертой такого механизма будет
успешное действие катализатора Q+ X~ с ониевым катионом
при очень малом коэффициенте его распределения и
практической нерастворимости в органической фазе образовавшейся
ионной пары Q+ А~. Это возможно в тех случаях, когда
аммониевый катион несимметричен. Так, например, бензилтриэтил-
аммониевый катион (ТЭБА+) имеет сравнительно мало
гидрофобные этильные группы, которые не мешают (а может быть,
и помогают) катиону удерживаться на водной поверхности, и
липофильиую бензильную группу, которая, однако, при
добавлении ароматических растворителей сольватируется и
становится практически нерастворимой в органической фазе [17].
Описанные выше варианты механизма в отличие от первых
двух типов его характерны не только для реакций в
двухфазных системах с разбавленными растворами в качестве
водной фазы, но и для реакций в системах, где водная фаза
представляет собой концентрированный раствор щелочи.
Интересным примером реакции, проходящей на поверхности
раздела фаз, является электрохимическое восстановление на
ртутном катоде 2,2-дихлорнорборнана I в ДОДФА в присутствии
тетраэтиламмонийбромида (система жидкость — твердая фаза)
[18]. Продукты реакции — экдо-норборнилхлорид III и нортри-
циклен IV образуются из общего промежуточного продукта —
карбаниона II. В безводных условиях, когда протон берется
из этильной группы аммониевого катиона, выход соединения III
составил 38%, а IV — 62%. При добавлении 1 моль воды
отношение продуктов III : IV становится равным 80:20. Однако
введение других доноров протонов — фенола или уксусной
кислоты — не привело к заметному изменению соотношения
продуктов реакции. Этот удивительный факт объясняется тем, что
в отличие от молекул воды молекулы фенола или уксусной
кислоты вытесняются катализатором с поверхности раздела
19
ртуть/ДМФА, где происходит образование карбаниона,
который может стабилизоваться протонами с образованием III.
Н
ш
CI
-CV
IV
Помимо увеличения концентрации органических молекул
в водной фазе добавление ониевых катионов существенно
изменяет структуру воды и тем самым активность попавших
в водную фазу органических молекул. Структура воды
нарушается вследствие разрушения гидратной оболочки вокруг
ионов ~ОН (наименьшей гидратной структурной группировкой
является группировка Н3О2, с которой прочными водородными
связями связаны другие молекулы воды) при введении R3N*,
что приводит к появлению свободных (не связанных
водородными связями) молекул воды. Это подтверждается
исследованием ИК-спектров гидратированных полимерных пленок,
содержащих группы NMe2, +NMe3 I", +NMe3 "ОН [19].
Кроме того, четвертичные аммониевые соли резко
изменяют структуру воды вследствие гидрофобных взаимодействий
углеводородных цепей с водой, что приводит к возникновению
у четвертичного аммониевого катиона плотной гидратной
оболочки («шуба»), внутри которой образуются полости («дыры»).
Это хорошо объясняет, например, увеличение растворимости
органических молекул (углеводородных газов) [4, 6], которые
могут размещаться в этих полостях, и изменение рН растворов
этих солей из-за снижения активности ионов Н+,
происходящего в результате прекращения эстафетной передачи протонов
в воде.
Структурирование воды четвертичными аммониевыми
ионами подтверждается и образованием устойчивых клатрат-гидра-
тов. Так, например, Bu4N+ F~ образует твердый гидрат, в
котором на один катион приходится 32,8 молекулы воды [20],
Bu4N+ Br~ дает гидрат состава 1:14, a Bu4N+ CI~ —гидрат
состава 1 : 18 [21, 22]. Иными словами, уже при концентрации
около 1,7 моль/л вся вода в растворе Bu4N+ F~ включается
в гидратную оболочку. Даже малые концентрации ониевых
солей (около 0,1 моль/л) вызывают резкое изменение свойств
водного раствора, поскольку в этом случае углеводородные
цепи сами входят в полости, образуемые молекулами воды
20
[23]. В случае двухфазных систем это должно приводить
к тому, что ионы Q+ стремятся находиться на поверхности
раздела фаз; при этом малые радикалы (особенно Me, Et) будут
входить в водный слой, а длинноцепочечные радикалы и
другие липофильные группы будут выталкиваться в органическую
фазу. Этому способствует также тетраэдрическая
конфигурация четвертичного атома азота [24].
Из данных по электропроводности видно [25], что ионы Q+
(симметричные катионы типа BmN+) в неводной среде не
сольватированы или сольватированы очень слабо, а ионы
щелочных металлов — сильно. В водных растворах картина
обратная. Из этого следует, что при переходе катиона Q+ из
водного слоя в органический необходимо затратить энергию на
сбрасывание водной «шубы». Процесс этот не может протекать
легко, так как необходимая энергия не компенсируется
образованием новой сольватной оболочки. Если этот процесс и
происходит в разбавленных растворах, то он крайне мало
вероятен в случае концентрированных растворов ониевых солей
и тем более щелочей. В то же время не столь гидратирован-
ные анионы могут переносить гидратную оболочку в
органическую фазу и там терять ее, насыщая органическую фазу водой.
Действительно, показано [26], что количество воды,
переносимой анионом в органическую фазу, зависит от его структуры.
Наличие этой воды может сказываться на абсолютной и
относительной скоростях реакций. Так, в системе вода — бензол
при Q+ = С1бН3зР+(С4Н9)з ион С1~ переносит в органическую
фазу 3,4 моль воды, ион Вг~—2,1 моль воды, а I—1,1 моль
воды на 1 г-ион. Следует отметить, что присутствие воды
может не только изменять скорость реакции, но иногда вообще
останавливать процесс или направлять его в другую сторону.
Характерной особенностью четвертичных аммониевых солей
является также их способность образовывать комплексы с
органическими веществами. Так, например, соли типа Bu4N+ X~
образуют с мочевиной кристаллические производные, которые
выпадают из водных растворов при комнатной температуре.
С Bu4N+ Вг~ были получены комплексы двух типов: Bu4N+
Br-.2(NH2)2CO и Bu4N+ Br--6(NH2)2CO [27]. Были получены
твердые комплексы Bu4N+ X~ с ацетонитрилом и бензолом
[28, 29]. Эти комплексы достаточно устойчивы. Например,
АЯдисс аддукта Bu4N+ NO3 ■ СбН5 при 58 °С составляет
65,27 кДж/моль [17]. С образованием комплексов связано
повышение растворимости бензола в воде в присутствии
аммониевых солей [30]. Можно считать, что подобные аддукты
образуют все ониевые соли.
При изучении катализаторов межфазного переноса следует
учитывать их сольватацию органическими растворителями.
Например, плохая растворимость ряда четвертичных аммониевых
солей в бензоле объясняется тем, что гидрофобные ионы
21
алкиламмония сильно взаимодействуют с бензолом, образуя
прочные «бензолофобные» сольваты [31]. Так, растворимость
Bu4N+ NO3 • СеНб составляет менее 0,001 моль на 1000 г
бензола.
Важным моментом является также образование ионных пар
в водной и органической фазах. В воде ионы противоположного
знака дают ионные пары только в том случае, если они
одинаково влияют на структуру воды [32, 33]. Наиболее
благоприятны два случая.
1. Оба иона имеют малые размеры; их взаимодействие при
этом имеет электростатический характер вследствие высокой
напряженности поля вокруг ионов.
2. Оба иона имеют большие размеры; в этом случае их
ассоциация приводит к уменьшению общего гидрофобного
взаимодействия.
Ассоциация ониевых катионов относится ко второму типу.
Это означает, что в водных растворах ионы Q+ будут сильнее
взаимодействовать с органическими анионами, чем катионы
щелочных металлов. Это имеет большое значение для
дальнейшей судьбы возникающих ионных пар, при образовании
которых защитная «шуба» вокруг Q+ разрушается. Хорошей
иллюстрацией влияния размеров аниона и катиона на образование
ионных пар в воде служат данные, приведенные в табл. 1.
В неводных растворах отсутствует гидрофобное
взаимодействие и, соответственно, более выгодно образование ионных пар
из малых катионов типа Li4" и органических ионов. Увеличение
донорных свойств растворителя способствует сольватации
катиона и увеличивает диссоциацию ионных пар [35].
Уменьшение размеров Q+ повышает устойчивость ионных пар и
уменьшает константу диссоциации.
Поскольку в обычных двухфазных системах в качестве
органической фазы используют растворители с невысокой
диэлектрической проницаемостью [бензол (2,28), хлороформ (4.7), ди-
хлорметан (8,9) и т. д.], следует ожидать, что ионы в таких
растворах будут сильно ассоциированы. Действительно, расчет
кажущейся молекулярной массы ионной пары по данным крио-
Таблица 1. Константы ассоциации некоторых четвертичных
аммониевых солей в воде при^25°С [34]
Анион
C6H5SO3
Ci2H9N2SO3" a
а Азобензол-4-сульфонат.
22
Катион
Me3N+Bu-H
Me3N+CioH2i
Me3N+Bu-w
Me3N+C10H2I
Kacc, л/моль
3
7,1
5,5
42 000
Таблица 2. Константы ассоциации (ТСасс) для пикрата
тетрабутиламмония в различных растворителях при 25 °С
Растворитель
Литература
1,2-Дихлорэтан
о-Дихлорбензол
Дихлорметан
Ацетонитрил — дноксан
Нитробензол — тетрахлорметан
гс-Нитроанилин — диоксан
10,23
10,12
10,00
10,00
10,00
10,00
4,39
58,5
22,00
24,6
12,6
1,0
37
38
37
39
40
39]
скопии и магнитной дисперсии [36] показал, что ионные пары
образуют сложные группировки. Данные по изменению
активности компонентов раствора также позволяют утверждать, что
ионные пары участвуют в каких-то сильных взаимодействиях.
В табл. 2 приведены значения констант ассоциации пикрата
тетрабутиламмония, которые подтверждают взаимодействие
ионных пар с некоторыми растворителями (см. системы: ацетонит-
рил — диоксан и /г-нитроанилин — диоксан).
Скорость реакции в органической фазе определяется
активностью ионных пар; в табл. 3 приведены значения активностей
компонентов раствора тетрабутиламмонийтиоцианата в бензоле.
Рассматривая данные по коэффициентам активности недис-
социированных ионных пар, можно предположить, что хорошо
растворимые ионные пары, концентрация которых в
органической фазе значительна, будут мало реакционноспособны из-за
агрегации. Напротив, малорастворимые ионные пары будут
легко вступать в реакцию, и скорость ее будет определяться только
Таблица 3. Активность компонентов раствора Bu4N+ ~SCN
в бензоле [17, 41]
Концентрация
соли (т),
моль/1000 г
0,00128
0,00404
0,02309
0,04895
0,07624
0,1688
Активность
соли в
бензоле
(а)
0,999
0,999
0,999
0,999
0,999
0,999
Коэффициент
активности а
бензола {f)
1,000
1,000
1,002
1,003
1,006
1,013
недиссо-
циирован-
ных иои-
ных пар
CV)
0,2500
0,1300
0,0220
0,0083
0,0053
0,0024
Концентрация
соли (тп),
моль/1000 г
0,2808
0,6327
1,6300
4,1500
7,8500
Активность
солн в
бензоле
(а)
0,999
0,998
0,967
0,916
0,914
' Коэффициент
активности а
бензола (f)
1,021
1,047
1,100
1,213
1,474
недиссо-
циироваи-
ных
.ионных пар
(■V)
0,0015
0,00047
0,00033
0,00023
0,00012
а f = а/ЛГ; у = а[т при степени диссоциации а ■< 1 (где /V —число молей бензола
в растворе данной концентрации).
23
скоростью растворения ионных пар в органической фазе. При
этом, однако, следует учесть, что в случае малорастворимых
солей будет иметь место существенная диссоциация ионных пар
на ионы, что также может сказаться на ходе реакции.
Одним из доказательств существования ионных пар в
органических растворителях являются высокие значения дипольных
моментов растворов ониевых солей. Так, значения |я в бензоле
составляют: для BmN+ Bi~ 11,6, для Bu4N+C104 14,1* для
(w3O-C5Hn)4N+ Pic~ 18,3 Д [17]. Возможно, что столь большие
дипольные моменты способствуют образованию каталитического
комплекса с другим реактантом, перестройка которого
приводит к продуктам реакции. В то же время большие дипольные
моменты ионных пар приводят к изменению диэлектрической
проницаемости раствора и тем самым к изменению скорости
реакции. Ниже приведены значения степени диссоциации (аС),
диэлектрической проницаемости (е) и среднего ионного
коэффициента активности (f±) для бензольных растворов
(rtt3o-CBHn)4N+ "SCN при 25°С [9, 17].
c-io-\
моль/л
4535
2471
960
307
157
аС
4,55- 10"3
6,42- 10~!
7,64 ■ 10"
5,80- 10",
8,00-10"7
e
4,75
4,17
3,60
2,77
2,52
lef±
3,29
3,00
2,81
3,34
3,15
моль/л
30,5
7,0
2,00
0,5
0,20
аС
3,59 ■ 10"!
5,15-10" *
8,24- 10" ,
6,65- 10"
1,90-10"
8
2,32
2,28
2,275
2,27
2,27
l£f±
2,56
1,95
1,29
0,40
0,05
Из анализа приведенных данных видно, что в бензольных
растворах, по-видимому, можно не учитывать диссоциацию
ионных пар, перешедших в органическую фазу, и использовать для
расчета концентрации и активности недиссоциированных
ионных пар.
В заключение приводим краткую сводку наиболее
существенных фактов, которые необходимо учитывать при
рассмотрении механизмов реакций в двухфазных системах с
катализаторами межфазного переноса.
1. Ониевые соли очень сильно структурируют воду, изменяя
ее свойства. Вода сильно гидратирует катионы Q+ и слабо —
органические анионы. Присутствие в воде ониевых солей
увеличивает растворимость органических молекул в водной фазе, в то
же время уже при сравнительно малых концентрациях ониевой
соли вся вода оказывается связанной с катионами.
2. Переход катионов Q+ из водной фазы в органическую
затруднен, особенно в тех случаях, когда отсутствуют еще
более сильно гидратирующиеся электролиты. Переход
органических катионов в органическую фазу может приводить к
переносу в нее гидратной воды.
24
3. Образование ионных пар в водной фазе происходит тем
легче, чем больше объемы аниона и катиона.
4. Несимметричные катионы Q+, содержащие хотя бы одну
длинноцепочечную группу, могут вести себя на поверхности
раздела фаз иначе, чем короткоцепочечные симметричные катионы.
5. Растворимость большинства ионных пар в органической
фазе невелика.
6. Диссоциация ионных пар в органической фазе (при е <
< 10—15) мала и в большинстве случаев ею можно
пренебречь.
7. Активность ионных пар в органической фазе резко падает
с увеличением концентрации, что связано с обратимым
образованием агрегатов ионных пар. Этот процесс может изменять
кинетику реакции.
РЕАКЦИИ В СИСТЕМАХ С КОНЦЕНТРИРОВАННЫМИ
ВОДНЫМИ ФАЗАМИ
К этой группе реакций относятся наиболее важные реакции
межфазного катализа. Следует отметить исключительно простой
по выполнению метод генерирования дигалогенкарбенов,
удобные методы генерирования органических анионов, в частности
карбанионов, и проведения с ними в присутствии водной фазы
таких чувствительных к влаге реакций, как реакции Вильямсо
на, Дарзана, Михаэля, Виттига — Хорнера и др., т. е.
практически всех реакций, требовавших ранее применения щелочных
металлов или их производных (алкоксидов, гидридов, амидов
и пр.) в безводных растворителях.
Изучение механизма реакции в таких системах крайне
затруднено вследствие различных дополнительных молекулярных
взаимодействий, которые отсутствуют в рассмотренных ранее
системах с разбавленными водными фазами. Именно поэтому
механизм реакций в системах с концентрированными водными
фазами изучен еще недостаточно.
Прежде чем перейти к обсуждению имеющихся данных о
механизме реакций в таких системах, необходимо рассмотреть
свойства растворов щелочей и реакции, которые происходят с
катализаторами фазового переноса в этих условиях.
Характеристика растворов щелочей
Физические свойства растворов щелочей. В двухфазных
системах, водной фазой которых являются концентрированные
растворы щелочи, плотности органической и водной фаз сильно
отличаются друг от друга. В табл. 4 приведены плотности
некоторых наиболее распространенных органических фаз, а также
50%-ных растворов КОН и NaOH. Из приведенных данных
следует, что условия перемешивания при использовании в качестве
25
Таблица 4. Плотность при 20 °С некоторых фаз, используемых
в каталитических, двухфазных реакциях [42, с. 530]
Фаза
Концентрация,
моль/л
50%-ный водный NaOH
50%-ный водный KQH
Тетрахлорметан
Хлороформ
Дихлорметан
Бензол
Ацетон
19,05
13,45
10,36
12,46
15,72
11,27
13.66
1525
1510
1595
1489
1336
879
792
растворителей хлорированных углеводородов должны
существенно отличаться от условий перемешивания при
использовании других органических растворителей. Так, при генерировании
карбенов, которое проводится в хлороформе или в смеси его
с дихлорметаном, перемешиваются два слоя с приблизительно
одинаковой плотностью, а при алкилировании, например,
ацетона в бензоле плотности слоев различаются почти вдвое.
Эффективность перемешивания [43] связана с
коэффициентом массопередачи /(масс, который численно выражают через
коэффициент молекулярной диффузии D. В случае интересующего
нас турбулентного режима
к
масс
Однако сам коэффициент диффузии является сложной функцией
плотности (р), вязкости (г|) и поверхностного натяжения (б).
Наиболее существенный вклад в эту величину вносят р и г\.
Для большинства органических веществ г\ ж 0,5—2 мПа-с.
Однако эти величины очень велики для концентрированных
водных растворов щелочей, причем здесь проявляется огромная
разница между NaOH и КОН: вязкость концентрированных
растворов NaOH существенно выше, чем у КОН (это связано с
различной мольной концентрацией их 50%-ных растворов, см.
табл. 4). При прочих равных условиях добиться хорошего
перемешивания в случае 50%-ного водного КОН существенно
легче, чем для 50%-ного водного NaOH. Таким образом,
теоретический вывод [44] о независимости скорости реакции в
двухфазной системе от интенсивности перемешивания реализуется
в случае концентрированных водных фаз только после
достижения определенного предела интенсивности перемешивания.
Химические свойства растворов щелочей. Самым
существенным свойством концентрированных растворов щелочей является
огромное сродство к протону свободных гидроксильных ионов.
Именно это позволяет проводить депротонирование различных
органических соединений, в частности генерировать карбанио-
ны. Величины сродства к протону (РА) для ионов и молекул
26
несколько отличаются по своему смыслу. Для ионов эта
величина отвечает изменению энтальпии, а не изобарного потенциала
реакции
АН —> А~Н-Н+
Поэтому в случае ионов РА одновременно характеризует
химическое сродство анионов к протону [45]. Эта величина влияет
на свойства анионов и, в частности, на взаимное вытеснение
ионов из органической фазы в водную. По данным [46], в ряду
2-
НО'
CI
Вг"
каждый ион вытесняет своего соседа слева. Это соответствует
порядку РА этих ионов (инверсия ~ОН и F~ связана,
по-видимому, с неточным определением коэффициентов распределения).
Значения сродства к протону для некоторых молекул [47] и
ионов [45] приведены в табл. 5.
Из приведенных в табл. 5 данных о сродстве к протону
различных соединений и ионов видно, что гидроксил-ион
действительно является одним из самых сильных акцепторов протона
среди одновалентных ионов, уступая только амидному иону.
Вода обладает несравненно более слабыми основными
свойствами. Твердые КОН и NaOH также имеют довольно большое
сродство к протону.
С повышением концентрации диссоциация щелочей
уменьшается незначительно. По данным [48], даже в 48%-ном
водном растворе диссоциировано не менее 71 % всех молекул.
Таким образом, учитывая огромную разницу в PA NaOH и ~ОН,
равную 5,9 эб, можно считать, что действующим началом
в концентрированных растворах щелочей являются ионы ~ОН.
Формально это выражается в значительном росте коэффициента
Таблица 5. Сродство к протону различных молекул [47] и ионов [45]
Соединение (ион)
Бензол
Толуол
Уксусная кислота
Ацетон
Н2О
NH3
NaOH
КОН
hso;
г
Сродство
эВ
7,1
8,11
8,16
8,76
7,14
9,0
10,7
11,4
12,8
13,3
к протону
кДж/моль
684,92
782,58
787,43
845,29
688,98
868,47
1032,48
1100,06
1238,46
1284,49
Соединение (ион)
Вг"
сг
НСОз
F"
НО"
H2N"
sol-
соГ
Сродство
эВ
13,66
14,00
16,04
16,7
16,6
18,17
18,77
21,59
к протону
кДж/моль
1317,96
1359,80
1451,85
1518,79
1602,47
1753,10
1811,67
2083,63
27
активности гидроксила (voh) с увеличением концентрации
щелочи [49]. Следует отметить, что в этом случае также
проявляется различие между NaOH и КОН. Хотя общий ход кривых
зависимости уон от концентрации сохраняется для обеих
щелочей, в области, близкой к насыщению, они заметно
различаются: для насыщенного раствора КОН уон —55,4, а для NaOH —
только 33,7. Возрастание коэффициентов активности, возможно,
связано с уменьшением количества «свободной» воды. Все это
приводит к тому, что при возрастании активности ~ОН
активность воды (aw) резко падает. Здесь также проявляется
различие между растворами NaOH и КОН: активность воды в
50%-ном КОН почти в два раза выше, чем в NaOH. Поэтому
для процессов, в которых вода может вызвать побочные
реакции (гидролиз и пр.), целесообразнее, по-видимому, применять
NaOH.
В любом случае чрезвычайно низкая активность воды в
50%-ных растворах щелочей вполне удовлетворительно
объясняет, почему такие растворы можно использовать для
проведения реакций, требующих безводных условий (например,
получение енолятов и пр.), или реакций, в которых участвуют крайне
чувствительные к влаге реагенты (такие, как неустойчивые илиды
фосфора или серы). Так, например, при встряхивании такого
активного галогенида, как пренилхлорид (3-метил-1-хлорбутен-2),
растворенного в смеси бензола и ацетона, с 50%-ным
водным NaOH гидролиз практически не проходит. Однако уже при
разбавлении щелочи для отделения органического слоя
пренилхлорид почти полностью превращается в 7>Т~Диметаллиловый
спирт.
Таким образом, несмотря на наличие 50% воды такие
растворы щелочей во многих случаях ведут себя аналогично
твердой щелочи, что позволяет применять их вместо неудобных в
практическом отношении твердофазных систем. В то же время
присутствие воды несмотря на ее низкую активность иногда
проявляется, особенно при наличии кислотно-основных
равновесий (например, при возможности альдольной конденсации).
Взаимная растворимость органических соединений в
концентрированных растворах щелочей. В связи с крайне
ограниченными возможностями гидратации органических молекул
растворимость их в концентрированных растворах щелочей весьма
мала. Так, ацетон довольно хорошо смешивается с водой,
однако добавление щелочи приводит к его «высаливанию» из
водной фазы, и уже в 28%-ном водном растворе щелочи
растворимость ацетона не превышает 1,1%- В 50%-ном водном растворе
щелочи растворимость ацетона составляет около 10~4 моль/л
вследствие образования енолята, который фактически
располагается на поверхности раздела фаз.
Исключительно малая растворимость органических
соединений в концентрированных растворах щелочей свойственна не
28
только ацетону. Так, в 60%-ном водном NaOH концентрация
фенилацетонитрила составляет менее 2 млн"1 [46]. Таким
образом, растворимость органических соединений в
концентрированных растворах щелочей пренебрежимо мала, что существенно
отличает эти растворы от других водных фаз, используемых в
двухфазных каталитических системах.
Растворимость неорганических солей в органических
растворителях зависит от размеров и поляризуемости анионов и
катионов. Так, растворимость NaCl в ацетоне равна 5,5-10~6 моль/л,
а для Nal она составляет 1,29 моль/л [50, с. 445]. Однако
щелочи плохо растворимы в ацетоне, что вполне естественно,
поскольку ацетон не способен разрушить прочную гидратную
оболочку у ионов (и молекул) щелочей. Имеющиеся данные [51, с.
85] указывают на ничтожную растворимость щелочи в ацетоне
при его контакте с 50%-ным водным раствором NaOH, что
проявляется химически в отсутствии реакций, характерных для
иона ~ОН. Следует отметить, что даже добавление ониевых
солей зачастую не приводит к переносу иона ~ОН в органическую
фазу.
Взаимодействие ониевых солей
с концентрированными растворами щелочей
При добавлении к достаточно концентрированному водному
раствору четвертичной соли, например ТЭБА-С1, небольшими
порциями концентрированной или твердой щелочи при
достижении концентрации щелочи 25—35% выделяется светло-желтый
органический слой, содержащий Q+~OH, который иногда
кристаллизуется. При повышении концентрации щелочи начинают
выпадать также соответствующие хлориды:
Q+ СГ-f NaOH ?=± Q+ "OH + NaCl (1)
Таким образом, на самом деле каталитическая система
Q+C1~—NaOH—Н2О является гетерофазной—два жидких не-
смешивающихся слоя и твердые NaCl и Q+ С1~. При добавлении
органического слоя (например, смеси бензола с ацетоном)
полного растворения слоя, содержащего Q+ ~OH, не происходит, и
реальная система, которую обычно называют двухфазной, на
самом деле состоит из трех жидких фаз (две органические и
одна водная) и по крайней мере одной твердой фазы. Водная
фаза кроме воды и NaOH может содержать часть органической
фазы, некоторое количество катализатора в форме Q+ С1~ и
Q+-OH, NaCl, а при наличии в системе других ионов
—соответствующие соли М+С1~ (М — металл). Содержание этих
компонентов может измениться в ходе реакции, однако в
концентрированных водных растворах щелочей растворимость любых
веществ довольно мала. В твердой фазе, образующейся при
добавлении катализатора (кроме того, иногда отвердевает слой,
включающий Q+ "ОН), могут содержаться все компоненты
29
реакционной системы, представленной уравнением (1). В ходе
реакции, проходящей с образованием новых ионов Х~ (например,
при алкилировании), кроме NaCl в этом слое появляется сте-
хиометрическое количество Na+ X". Далее происходит ионный
обмен и Q4" С1~ постепенно переходит в Q4" Х~ и Q4" "ОН,
которые включаются в каталитический цикл. Каталитическая
жидкая фаза, образующаяся по уравнению (1), не содержит
аминов. Она содержит Q+ Cl~, Q+-OH (обменная реакция идет не
полностью и выход основания не превышает 30%) и NaOH
(заметно растворяющийся в Q4" ~ОН). Органический слой
выделяется также при обработке водного раствора Q4" С1~ 50%-ным
водным раствором К2СО3, что указывает на отсутствие в нем
карбонатов Qj СОз или Q+ HCO3- Основные свойства
органической фазы — малая растворяющая способность по отношению
к электролитам, низкая константа диссоциации электролитов,
заметная ассоциация молекул и другие — были рассмотрены
выше. Для иллюстрации приведены дополнительные данные по
растворимости (табл. 6) и электрическим свойствам (табл. 7)
растворов тетраалкиламмониевых солей в некоторых
органических растворителях. Все эти данные относятся, однако, к
чистым растворителям.
Отмечено [3], что при встряхивании бензольного раствора
BiuNCl с концентрированным раствором NaOH образуется
только 0,5—2% ОН-формы; около 50% всего количества соли
остается неизменным в органической фазе, остаток соли —
в водной фазе. В системе концентрированный NaOH—СН2С12
в органической фазе находится 97% аммониевой соли. При
использовании разбавленной щелочи соль распределяется в обеих
фазах. Если органическая фаза растворяет воду или водную
щелочь (ТГФ, высшие спирты), то концентрация "ОН в ней
увеличивается.
Таблица 6. Растворимость тетраалкиламмониевых солей в различных
растворителях [51]
Соль
Растворимость а, г/100 мл
в ацето-
интрнле
в 1,2-дНмет-
окснэтане
в тетрагндро-
фураие
в диметил-
формамнде
Et4NC104
Pr4NCIO4
BU4NCIO4
Et4NBF4
Pr4NBF4
BU4NBF4
Et4NBr
Pr4NBr
Bu4NBr
26 (1,13)
21 (0,74)
70 (2,06)
37 (1,69)
36 (1,62)
71 (2,21)
7,8 (0,37)
29 (1,09)
66 (1,99)
(0,01)
(0,01)
31 (1,10)
(0,01)
(0,01)
53 (1,70)
(0,01)
(0,01)
(0,01)
(0,01)
(0,01)
50 (1,48)
(0,01)
(0,01)
65 (2,02)
(0,01)
(0,01)
4,8 (0,14)
23 (1,00)
21 (0,74)
79 (2,29)
27 (1,34)
32 (1,17)
75 (2,34)
4,1 (0,19)
18 (0,70)
52 (1,67)
a В скобках приведены концентрации (в моль/л).
30
Таблица 7. Удельное сопротивление растворов тетраалкиламмониевых солей
в различных растворителях [51а]
Соль
Et4NClO4
Pr4NC104
Bu4NC104
Et4NBF4
Pr4NBF4
BU4NBF4
Et4NBr
Pr4NBr
Bu4NBr
a В скобках
Удельное
ацетоинтрила
26 (0,60)
31 (0,60)
37 (0,60)
18 (1,69)
23 (1,0)
31 (1,0)
39 (0,60)
48 (0,60)
приведены концентрации
сопротивление (в
1,2-димет-
оксиэтана
312 (1,0)
228 (1,0)
(в моль/л).
Ом/см) в растворе а
тетрагидро-
фурана
368 (1,0)
373 (1,0)
днметилформ-
амида
52 (0,60)
64 (0,60)
77 (0,60)
38 (1,0)
51 (1,0)
69 (1,0)
88 (0,60)
106 (0,60)
Растворимость ТЭБА-С1 и ТЭБА-ОН в системе 50%-ный
водный раствор NaOH — ацетон — бензол. Растворимость
ТЭБА-С1 в бензоле очень мала и составляет всего около
2,5 ммоль/л; в ацетоне растворяется 18,2 ммоль/л ТЭБА-С1.
Таким образом, исходная концентрация катализатора в бензол-
ацетоновых смесях не может быть слишком большой. Однако
обычно ТЭБА-С1 берут около 0,04 моль/л, т. е. заведомо
больше, чем может раствориться в такой органической фазе [53].
При встряхивании бензольного раствора тетрагексиламмо-
нийхлорида с равным объемом 50%-ного водного раствора
NaOH в органической фазе остается около 99% аммониевого
катиона, при этом образуется 28% ОН-формы [3]. Такое же
количество ОН-формы образуется и в случае тетрагептилам-
монийхлорида, близкое значение было получено и для ТЭБА-ОН,
так что, по-видимому, это значение отвечает предельной
растворимости оснований Qf ~OH в бензольных растворах даже
с очень липофильными катионами [52].
С менее липофильными катионами эти значения могут быть
существенно иными. Так, при аналогичной обработке 0,014 моль
Вщ№" С1~ в 1 л бензола в органической фазе остается только
52°/о аммониевого иона и только 4,25% из этого количества
находится в ОН-форме.
Высаливающее действие концентрированных щелочей.
Большую роль играет высаливающее действие концентрированных
растворов щелочей. Так, несмотря на то что для Bu4NCl в
системе бензол —вода коэффициент экстракции Е <^ 0,1, катион
Bu4N+ довольно хороший катализатор (так же как и ТЭБА-С1)
при работе в двухфазных системах с концентрированным
водным раствором щелочи и бензольной органической фазой.
Эффект высаливания играет существенно меньшую роль для
31
Таблица 8. Экстракция ТЭБА-СХ и ТЭБА-ОН смесями ацетон — бензол
50,5%-ный раствор NaOH, 19,5 °С, 2 мин, объем органической фазы 25 мл,
объем водной фа»ы Ь мл, 1 ммоль ТЭБА-С1; максимальная концентрация иона Q+
в органической фазе 0,04 моль/л
Содержание
ацетона в
растворителе,
% (об.)
Растворимость
ТЗБА-С1,
ммоль/л
Концентрация иоиов (С),
ммоль/л
ОН
сг
"ОН + СГ
Относительная
концентрация а, %
ОН
С1"
0
30
50
65
75
90
100
а Определяется
2,6
3,1
3,7
5,3
8,2
15,6
18,2
—
П.6
23,9
29,0
30,7
30,8
31,5
по формуле (С : 0,04)
—
—
2,2
4,9
4,9
2,9
■100.
—
11,6
26,1
33,9
—
35,7
34,4
■—
29,0
59,8
72,5
76,8
77,0
78,8
—
—
5,6
12,3
—
12,3
7,3
солей, хорошо растворимых в органической фазе. Например, при
встряхивании 0,1 н. раствора гексадецилтриметиламмонийбро-
мида в дихлорметане с концентрированной щелочью в
органической фазе остается 98% соли, а при встряхивании с водой —
82% [54]. В табл. 8 приведены данные по экстракции ТЭБА-С1
и ТЭБА-ОН в зависимости от состава растворителя [55].
Расхождение между суммарными концентрациями ОН- и
С1-форм ТЭБА в органической фазе связано с протеканием аль-
дольной конденсации ацетона под влиянием щелочи, поскольку
образование диацетонового спирта резко увеличивает
растворимость обеих форм.
Интересно отметить, что с увеличением концентрации
бензола в органической фазе общая концентрация Q+ падает,
однако в разной степени для Q+ Cl~ и Q+ ~ОН. Это, по-видимому,
связано как с различиями в сольватации соли и гидроксида, так
и с различиями в скоростях ионного обмена в растворителях
разного состава [46].
Замена щелочи 50%-ным водным раствором КгСО3 приводит
к тому, что из водной фазы выделяется почти чистый Q+ С1~.
В то же время такая система является достаточно
каталитически активной при алкилировании соединений с активной
метил еновой группой; ее кинетическая основность соответствует
30%-ному водному раствору NaOH [56]. Это означает,
очевидно, что в ходе реакции почти весь Q+ С1~ постепенно
превращается в Q+ "ОН.
Следует отметить, что из-за крайне малой растворимости
солей в водной фазе данные о коэффициентах экстракции [3]
неприменимы к двухфазным системам, содержащим в качестве
водной фазы концентрированные щелочи.
32
Устойчивость межфазных катализаторов. При подборе
катализаторов межфазного переноса следует учитывать их
термическую устойчивость. Так, ТЭБЛ-С1 превращается на 72% в бен-
зилдиэтиламин при нагревании с концентрированным NaOH в
течение 20 ч; в дихлорметане при 60°С реакция идет медленнее,
и за то же время катализатор распадается только на 11,5% [3].
Фосфониевые соли более устойчивы, чем аммониевые: три-
бутилгексадецилфосфонийбромид не изменяется при нагревании
при 100°С в течение 16 ч.
Обычно используемые катализаторы устойчивы в
присутствии концентрированного NaOH при комнатной температуре в
течение нескольких дней. Повышение температуры, как и в
случае ТЭБА-С1, приводит к разложению. Так, при 60 °С катион
Bu4N+ превращается в трибутилаыин на 52% за 7 ч, а при
100°С за то же самое время — на 92%. В отличие от ТЭБА-С1
бензилтриметиламмонийхлорид распадается на равные
количества дибензилового эфира и бензилдиметиламина.
Образование катализаторов при реакции. Катализатор
межфазного переноса может образоваться в ходе реакции.
Например, при алкилировании кетонов алкилгалогенидами можно
применять в качестве катализаторов третичные амины, которые
образуют четвертичные соли с алкилгалогенидами [53].
Третичные амины можно успешно использовать также при
генерировании дихлоркарбена из хлороформа [54], который при
действии третичных аминов образует ониевые соли:
RsN + CHCb —> R3NCHCI2 СГ
В заключение этого раздела следует подчеркнуть, что
50%-ные водные растворы щелочей обладают довольно
высокими высушивающими свойствами (давление паров воды над
50%-ным водным раствором NaOH при 25°С равно всего
3,9 гПа [42]), поэтому в органической фазе вода практически
отсутствует. Это важное свойство таких двухфазных систем,
которое следует учитывать при рассмотрении механизма
реакции.
Алкилирование кетонов в системе 50%-ная щелочь —
органическая фаза
Реакция алкилирования кетонов сыла изучена на примере
алкилирования ацетона пренилхлоридом з системе бензол(или
толуол) —50%-ный NaOH (или КОН) при использовании в
качестве межфазного катализатора ТЭБА-С1 (0,001 моль на
0,25 моль ацетона) [53, 55]. В результате были установлены
основные закономерности образования 6-метилгептен-5-она-2
(метилгептснон).
1. Для достижения максимального выхода метилгептенона
требуется только 0,001 моль ТЭБА-С1 (или даже меньше) на
33
0,25 моль ацетона. Дальнейшее увеличение количеств ТЭБА-С1
(до 0,02 моль) не изменяет выхода.
2. Развитие поверхности водной фазы добавками
нейтральных поверхностно-активных веществ не увеличивает скорость
реакции и ныход метилгептенона; применение анионных
поверхностно-активных веществ тормозит реакцию.
3. Реакция имеет нулевой порядок по ацетону, что
свидетельствует об участии в реакции только молекул ацетона,
растворившихся в 50%-ной щелочи и превратившихся в №(или
К)-производные ацетона.
4. Реакция имеет первый порядок по прсннлхлориду, т. е.
лимитирующая стадия отсутствует.
5. Первый порядок реакции наблюдается также по
активности ~ОН, Поэтому при уменьшении концентрации щелочи с 50
до 28% выход метилгептенона снижается с 51 до 8,5%.
Па основании полученных результатов сделан вывод, что
ионная пара Q1 ~СН2СОСН3 образуется на поверхности
раздела фаз в результате ионного обмена между Na+ ~СН2СОСНз
и Q+ С1~. Это подтверждается также линейной зависимостью
между максимальной наблюдаемой константой скорости (при
определенном составе органической фазы) и минимальной
концентрацией Q+ С1~, при которой эта скорость достигается. Кроме
того, квантовохимическими расчетами показано, что депрото-
нирование ацетона не идет через стадию образовании енола
[56]. Предложена следующая схема образования
метилгептенона:
СНзСОСНз + NaOH *==* СН3СОСН2~ Na++ Н2О (НФ) или (ПРФ)
CH3COCII2 Na+-hQ+ СГ <=^ СНзСОСН2" Q + NaCI (ПРФ)
CH3COCHJ <2+)
—> CH3COCH2CH2CH=C(CH3)2-i- Q+ СГ (ОФ)
Q+Cr-hNaOH ч=^ Q+ OH + NaCl (ПРФ).
При этом следует помнить (см. выше), что Q+ X~ ^
QC1 + Q+OH)
Из схемы следует, что
[СНзСОСНз]°вф = [СН3СОСН3]ВФ + [СНзСОСНа ]ОФ
[СН3СОСН21оф =
где Кр =
Тогда
1ВФ
и i
те Аа.и — константа скорости анкетирования.
34
Если считать, что
[СН.СОСН3]°Вф - а [СНгСОСН3]ОФ
где а — коэффициент, связывающий исходную концентрацию ацетона с его
концентрацией в водной фазе (на ее поверхности)
то в двойных обратных координатах должна быть линейная
зависимость, что и наблюдается па самом деле.
В пользу предположения об образовании карбанионов на
поверхности раздела фаз свидетельствуют данные о
протекании ряда реакций в отсутствие катализаторов (реакции Райс-
серта, Дарзана, быстрый деитерообмен слабых С—П-кислот с
концентрированными растворами NaOH в D2O и др.) и, кроме
того, результаты проведения конкурентной реакции
присоединения трихлорметильного аниона к винилацетату и пиридиниевой
соли [46]. В двухфазной системе анионы присоединяются
только к пиридиниевому кольцу, а в гомогенной или
псевдогомогенной системе в присутствии грег-бутоксида калия образуется
исключительно 1-трихлорметилэти л ацетат:
NaOH —Н2О
О СС13
СНзСОСНСНз
Таким образом, в данном случае реакция, возможно,
протекает на границе раздела фаз, минуя стадию образования
ионной пары. В подтверждение такой точки зрения приводятся
следующие рассуждения [3]. При получении дихлоркарбена
вначале образуется двойной слой Ыа/СС1з, закрепленный на
границе раздела фаз (стадии а и б, см. ниже). Катион
катализатора взаимодействует с ~СС13, давая ионную пару,
закрепленную на поверхности раздела фаз; при этом анион ~СС13
находится в равновесии с карбеном (стадия в), которое сдвинуто
влево. В результате этого карбен находится как бы в «депо» и
может быть генерирован при добавлении акцептора даже
спустя несколько дней (стадия г). В качестве акцепторов карбена
могут быть уходящий С1~ или другие галоген-ионы,
присутствующие в системе (в этом случае можно обнаружить аддукты
олефина со смешанным карбеном :СС1Х), вода, -ОН и олефин:
они конкурируют друг с другом.
(а) NaOH-fHCCb ^=* Na+/"CC13 + Н2О
(б) Q+ + "CCI3 *=^ Q+ "CCI3
(в) "CCU ч=^ :СС12 + СГ
(г): ССЬ +
/ \
О
35
Так как вода и дихлоркарбен находятся в разиых фазах, то
гидролиз дихлоркарбена идет очень медленно: в зависимости
от нуклеофильности добавленного олефина гидролизуется от
4,1 (2-метилбутен-2) до 58% (3,3-диметнлбутен-1)
дихлоркарбена [57]. Отсутствие продуктов взаимодействия карбена с
"ОН указывает на отсутствие заметного вклада маршрута,
включающего перенос ~ОН в органическую фазу. Прн
изучении стадии депротонировання ацетона в условиях межфазного
катализа было установлено, что отрыв протона осуществляется
при воздействии NaOH на поверхности раздела фаз, а не
органическим основанием в органической фазе. При этом
скорость алкилирования линейно зависит от активности ~ОН в
водной фазе, а не от концентрации NaOH и не зависит от
концентрации ацетона.
Приведенные выше данные о реакции генерирования карба-
нионов и карбенов на .поверхности раздела фаз позволяют
предположить возможность протекания второй стадии процесса —
реакции этих частиц — также на поверхности раздела фаз, без
Перехода в органическую фазу. В этом случае роль
катализатора, находящегося на поверхности раздела фаз, будет
заключаться в координации субстрата на этой поверхности. Это
предположение подтверждается тем фактом, что алкилирование
чувствительно не к концентрации NaOH, а к активности ~ОН в
водной фазе.
Структуру комплекса, фиксированного на поверхности
раздела фаз, можно представить следующим образом:
Н Н
I
Положение иона Q+ соответствует модели замещения Sn2;
сдвиг электронной плотности в направлениях, указанных
стрелками, приведет к «<3+-производному» ацетона (субстрата). При
этом отрицательно заряженный атом углерода станет
доступным для атаки «сверху» алкилгалогенидом:
ОФ
36
Таким образом, в принципе возможно проявление каталнти
ческих свойств ониевых солей без переноса аниона в
органическую фазу, за счет комплексообразования на поверхности
раздела фаз. В этом случае ониевая соль (или гидроксид) ведет
себя аналогично гетерогенному катализатору и, сближая реак-
танты, облегчает вторую стадию реакции.
Такой «квазигетерогенный» катализ является характерной
чертой ферментативного катализа органических реакций
(следует отметить, что многие ферменты имеют в своем составе
четвертичные аммониевые группы).
Влияние структуры кетонов на направление и скорость ал-
килирования еще недостаточно изучено. Однако имеется
несколько работ, в которых эти вопросы рассмотрены (см. ниже).
Направление алкилирования сопряженных кетонов. Алкили-
рование окиси мезитила пренилхлоридом в двухфазной
системе бензол — 50%-ный водный NaOH в присутствии ТЭБА-С1
[58] приводит, как и в обычных условиях [59], к смеси а- (V)
и р- (VI) форм алкилированной окиси мезитила:
(СН3)2С=СНСОСНз + RX —>
СН3 R
—► GH2=C—CH(R)COCH3 + (СН3)2С-=С—СОСНз
V VI
Были определены при 40 °С константы прямой и обратной
реакций превращения а- и р-форм в двухфазной системе:
Оказалось, что КР = 3,15, k\ = 4,9-10~4 с-1, a A_i = 1,55-
•10~4 с"1. Таким образом, при увеличении длительности
реакции концентрация р-формы в смеси должна увеличиваться.
Однако эти результаты не дают ответа на вопрос, откуда берется
ос-форма. Устойчивой формой окиси мезитила является VII
[60, 61], которая стабильнее несопряженной формы IX и еноль-
ной формы XI. Квантовохимические расчеты показали [56],
что среди возможных анионов VIII, X и XII наиболее
устойчивыми являются VIII н X. Структуры XIII и XIV неустойчивы
и безбарьерно переходят в анион VIII. Следовательно, отрыв
протона от v-метильной группы формы VII или от гидроксиль-
ной группы енола XI приведет к одному и тому же аниону VIII.
(Как показывают квантовохимические расчеты, отрицательный
заряд в анионе VIII сильно делокализован.) Следует отметить,
что, как и в случае ацетона, образование енольной формы XI
не обязательно для получения а-формы V и что «енолят-анион»
окиси мезитила имеет структуру VIII, в которой свободная
электронная пара сопряжена с двумя я-связямн. Все эти
37
данные позволяют объяснить причины образования а-формы V
при алкилировании окиси мезитила, как это указано на схеме:
н
-Н"
С
СН
II
сад—он
XI
гСч /
С
II
сн
СН3С=О
VII
Н"
с
II
с
X
с
"СН
сн,с=о
VIII
с
сн2
I
СН3С=Ю
IX
RX
RX
с
С—R
VI
3fv ^?
С
CHR
I
V
с
II
сн
XII
СН;
с
II
сн
I
3с—о
XIII
;СН:
с
сн
CHSC—О'
XIV
Следует добавить, что для. перехода от а-формы продукта
к р-форме также необходим отрыв протона. Этот процесс
подчиняется тем же закономерностям, что и изомеризация самой
окиси мезитила. Соотношение между маршрутами VII ->■
VIII ->-V и VII -> Х-> VI будет, очевидно, определяться
различием в устойчивости анионов VIII и X, или энергией депро-
тонирования VII, приводящего к образованию этих анионов.
Для получения аниона X необходима энергия депротонирова-
ния, равная 15,23 эВ, а для VIII она составляет 14,73 эВ
(метод MINDO/З), что дает существенные преимущества
маршруту, ведущему к а-форме V.
Энергия депротонирования и скорость алкилирования кето-
нов. Если считать, что реакционная способность карбанионов
чрезвычайно велика и мало изменяется в сходных рядах
соединений, то скорость реакции с участием карбаниона будет
определяться в основном константой равновесия стадии его
образования. Это означает, что энергия депротонирования
(«кислотность») кетонов должна коррелировать со свободной
энергией их алкилирования. К сожалению, данных о кислотности
38
различных С—Н-кислот не очень много [62]. Энергия гетеро-
лптического разрыва связи С—Н (т. е. «кислотность») может
быть вычислена квантовохимнческими методами. Однако эти
методы описывают энергетику реакций в газовой фазе,
поэтому при переходе к ионам в растворе необходимо учитывать
сольватацию. Дело облегчается тем, что в ряду кетонов
порядок кислотностей в газовой фазе и в таком полярном
растворителе, как ДМСО, совпадает [63]. Кроме того, для
родственных соединений существует корреляция между энтальпией
депротонирования А//д и энтальпией сольватации Д#с.
Действительно, изучение депротонирования квантовохимическим
методом MINDO/З показало [56], что этот метод хорошо передает
порядок С—Н-кислотности, но абсолютные значения Д#л
занижаются (см. табл. 9). Естественно, что данные, приведенные
в табл. 9, могут быть использованы для определения
направления реакции и сравнительной активности кетонов только
в близких рядах соединений и в сопоставимых условиях.
Из табл. 9 видно, что замена метильной группы в ацетоне
на этильную не изменяет кислотность оставшейся метильной
группы. Наиболее кислыми оказываются атомы водорода ме-
тиленовой группы, наименее кислыми—(3-метпльной группы.
В окиси мезитила наиболее кислыми являются у-метильные
(сопряженные с двойной связью) и метинные (у двойной
связи) протоны; протоны а-метильной группы по кислотности не
отличаются от протонов метильных групп в ацетоне. Таким
образом, для этих кетонов кислотность увеличивается при
переходе от ацетона к метилэтилкетону и окиси мезитила.
Имеющиеся данные по кинетике алкилирОвания различных кетонов
[65] в общем подтверждают зависимость скорости реакции от
кислотности (табл. 10).
Таблица 9. Энтальпии депротонирования (Д//д) некоторых соединений
в газовой фазе
Исходное соединение
СН3СНО
СНзСОСИз
СН3СОС2Н5
(СН3)2С=-СНСОСНз
а По данным работы [6V
Ион
"СН СНО
"СИСОСНь
СНзСОСНСНз
"" f^ T T /""* t~*\ ^* т г
V> 1- 1 ? v> VJ \__j ^ 1. 1R
(СН3):С—СИСОСНа
(СН3),С=ССОСН3
СН2—С(СН3)СИСОСНз
ДЯД, эВ
расчетная
(MIND0'3)
15,10
15,78
14,63
15,13
15,95
15,23
14,73
экспериментальная [62]
15,91 (15,89) а
16,04 (15,99) а
15,97
16,03
—
—
39
Таблица 10. Скорости алкилирования различных кетонов
пренилхлоридом и энтальпии их депротонирования
Система бензол — 50%-ный NaOH; катализатор ТЭБА-С1; 40 СС
Кетон
Литература
6-Метилгептен-5-он-2 а> в
Метил-«-бутилкетон
Ацетон а> г
Метилэтилкетон
Циклогексанон • г
Бензилэтилкетий
б г
Циклопентанон '
4-Метилпентен-3-он-2 а
а Отношение кетон : пренилхлорнд равно 1.
б Отношение кетон : пренилхлорид равно 5.
0,905
1,0
1,03
1,24
3,9
4,64
5,1
8,4
0,27
1,15
0,21
1,18
4,48
6,19
5,93
8,26
—
368,3
368,8
368,3
—
352,5 г
—
339,7
[65]
[65]
[66]
[62]
[65]
[65]
[62]
[55]
А// группы СН2 должна быть близка Д//л СН^-группы в мстилэтнлкстонс.
Определенная экспериментально энтальпия депротонироваяня в ДМСО равна для
ацетона 62,34, для циклопентанона 64,85 и для циклогексаиоиа 64,43 кДнс/моль.
Для правильного сопоставления скоростей необходимо
учитывать вероятностный фактор — различное число способных
к замещению атомов водорода. Во всяком случае, данные
табл. 9 и 10 позволяют считать, что именно СН-кислотность
кетонов определяет направление и скорость их алкилирования
в двухфазных системах.
Тип нуклеофила. Несомненно интересен вопрос о типе ну-
клеофила, участвующего в реакции (ион А~ или ионная пара
Q+ А~). Увеличение диэлектрической проницаемости и сольва-
тирующей способности растворителей, используемых в
двухфазной системе, должно приводить к увеличению концентрации
ионов А~ и соответственно к уменьшению концентрации
ионных пар Q+ A~. В табл. 11 приведены выходы и скорости
реакции при алкилировании ацетона пренилхлоридом [55].
Сопоставление этих данных со значениями диэлектрической
проницаемости растворителей ясно указывает на определяющую
роль ионных пар, а не анионов в такого рода реакциях. В
самом деле, при переходе от бензола (е»2) к ацетонитрилу
(е^38) константа диссоциации должна увеличиваться почти
в 7 000 раз (есн3с1ч/ес6-тб)3= ^ • Поскольку ацетонитрил
является сравнительно мало сольватирующим растворителем (как и
ДМСО), то концентрация малосольватированных анионов А~
при переходе от бензола к ацетонитрилу (или ДМСО) должна
возрасти на несколько порядков, что, естественно, должно
сказаться на скорости реакции: симбатность ее изменения будет
говорить об определяющем влиянии ионов А~, а сохранение
40
Таблица П. Влияние различных растворителей на скорость алкилирования
ацетона пренилхлоридом при 40 °С
Органическая фаза (25 мл): 2 моль/л пренилхлорида 6,4 моль/л ацетона,
6 мл растворителя, 1 г С13Н28
Водная фаза: 10 г 50%-ного водного раствора NaOH, 0,23 г ТЭБА-С1
Бензол
Толуол
Хлороформ а
Ацетон б
Ацетоиитрил
Ди метилсул ьфоксид
2,28
2,37
4,8
20,7
37,5
49
6,5
7,0
1,84
7,7
3,0
5,0
а Реакция сопровождается присоединением дихлоркарбена
" Нулевой порядок по
ацетону.
3,2
3,5
0,9
3,9
1,5
2,5
к продукту
38
42
19
45
35
45
реакции.
или уменьшение скорости — о преимущественном вкладе
ионной пары Q+ A~.
Роль основности водной фазы. Изучение алкилирования
такого кислого субстрата, как ацетоуксусный эфир, позволяет
выяснить роль основности среды. Можно представить себе
следующую общую схему реакций, протекающих при алкилиро-
вании ацетоуксусного эфира, например пренилхлоридом, в
присутствии щелочей [66]:
Побочные -<—
продукты '
CO2Et
— CR2
1 1
СОМе
НС1
п
У
CO.Et
RC" К'
СОМе
н2о
кон
кон
* СОоЕГ
НС" К+
СОМе.
CChEt
CHR
СОМе
CHR
I
СОМе
КОН,
н2о
"СО2К(Н)
СН2
.СОМе J
кон, н2о
б СН3
~с°2 СОМе
Побочные
продукты
41
На схеме не указаны еще две равновесные стадии [66] :■
СН3СОСН2СОо К++ Н2О *=± СН3СОСН2СО2Н + КОН
CH3COCHRCO2" К+ + Н2О «==t CH3COCHRCO2H + КОН
Образующиеся кислоты теряют диоксид углерода и
превращаются в соответствующие кетоны. Была изучена реакция ал-
килирования ацетоуксусного эфира в следующих
конденсирующих системах [66]: А — твердый К2СО3 + ТЭБА-С1; Б —
твердый КОН; В —твердый КОН + ТЭБА-С1; Г — 50%-ный водный
раствор К2СО3; Д—50%-ный водный КгССЬ + ТЭБА-С1; Е —
50%-ный водный КОН; Ж —50%-ный водный КОН + ТЭБА-С1.
Применение твердых конденсирующих агентов (системы
А — В) приводит к образованию гетерогенной двухфазной
системы твердое вещество — жидкость, а использование водных
фаз (системы Г—Ж)—к гетерогенной двухфазной системе
жидкость — жидкость. В ходе алкилирования и декарбоксили-
рования образуются КС1, калиевая соль пренилацетоуксусной
кислоты, калиевые производные ацетоуксусного и пренилаце-
тоуксуснот эфиров, которые выпадают в осадок, образуя
третью твердую фазу.
Система Г представляет собой водный раствор К2СО3 и
КОН, образовавшегося при гидролизе соли (концентрация ~ОН
в 50%-ном растворе соли составляет 0,035 моль/л). Свойства
системы Г как основания определяются наличием в ней ионов
СОз" и ОН. Для реакций
АН+ "ОН ъ=± А" +Н2О
АН+СО!" ?=* А~+НСОз
где АН — ацетоуксусный эфир, А~ — его анион
константы равновесия соответственно равны 11700 (Кр<п) и
0,178 (Яр(2)).
Таким образом, различия в каталитических свойствах систем
Г и Е обусловлены низкой концентрацией "ОН в системе Г и
огромной разницей в величинах Кр(\) и /Ср(2) — констант
образования А~ в этих системах.
Система Д отличается от системы Г присутствием ТЭБА-С1,
что приводит к образованию органического слоя:
К2СО3 + Н2О :*=* КНСОз+
KOH+Q+cr *=>: Q+"OH
Следует отметить, что наличие не смешивающегося с
50%-ным водным раствором К2СО3 (система Д) или КОН
(система Ж) слоя, содержащего ТЭБА-ОН, позволяет считать
эти системы двухфазными жидкими системами. Добавление к
ним бензольного раствора ацетоуксусного эфира и пренилхлори-
да приводит к образованию третьего жидкого слоя, г
выпадающие в ходе реакции твердые продукты, плохо растворяющиеся
42
в водном и органическом слоях, образуют четвертую (твердую)
фазу.
Было показано, что стадии 5—7 практически не идут, а 3
и 4 развиваются по мере накопления воды за счет стадий /
и 2, причем скорость образования воды не зависит от
присутствия ТЭБА-С1. Конкурентной реакцией для образования ме-
тилгептенона из пренилацетоуксусного эфира (реакции 3 и 4)
является диалкилирование (стадии 8 я 9)\ скорость этой
реакции определяется как различием кислотностей
пренилацетоуксусного и ацетоуксусного эфиров, так и изменением в ходе
реакции соотношения концентраций ацетоуксусного эфира,
пренилацетоуксусного эфира и препилхлорида. Подробное
изучение кинетики отдельных стадий 3, 4, 8У 9, а также 5 и 6
позволило обосновать и предложить схему алкилирования
ацетоуксусного эфира:
(а) АН + КОН ч=^ К+ А" + Н2О
(б) AH+Q+"OH i=± Q+A" + H2O (органическая фаза)
(в) АН + Q+ "ОН ^=±. Q+ k~ + Н^О (поверхность раздела фаз)
(О Q+ А' + КОН ?=* К+ А" + Q+ ~OH
(д) Q+ A- + RX —^ RA+Q+ X"
(е) К+ АЧ- RX —■* RA + К+ X'
(ж) Q4 X" + КОН ^=ь Q+ "ОН + К+ X"
Согласно этому механизму, реакция алкилирования
ацетоуксусного эфира может протекать гомогенно, гетерогенно или
одновременно по обоим маршрутам. Все реакции, кроме (б),
идут гетерогенно (жидкость — жидкость или твердая фаза —
жидкость). Реакция (д) может идти как гетерогенно, так или
гомогенно в зависимости от растворимости Q+ A~. Вклад
гомогенного маршрута (перенос ~ОН в органическую фазу)
определяется растворимостью Q+ ~OH в органической фазе.
Сопоставление скоростей моиоалкилирования в системах Д,
Е и Ж позволяет предположить, что под влиянием 50%-ного
водного КОН в присутствии ТЭБА-С1 (система Ж) процесс
протекает в основном через К+ А~ и Q+ А", которые образовались
в результате гетерогенных взаимодействий с КОН и Q+ "ОН,
соответственно. Иными словами, в присутствии 50%-ного
водного КОН без катализатора реакция идет по стадиям (а — е),
в присутствии 50%-ногоК2СО3 + ТЭБА-С1 — постадиям (б—д),
а в присутствии 50%-ного водного КОН + Q+ С1~ — по всем
маршрутам.
В заключение подчеркнем, что возможность осуществления
того или иного маршрута зависит как от основности системы,
так и от кислотности органического соединения. Используя
43
различия в кислотности ацетоуксусного эфира и его
замещенного продукта и изменяя основность системы, можно проводить
реакцию с высокой селективностью.
Лльдольная конденсация в двухфазной системе
Выше был рассмотрен вопрос о переносе гидроксильного
иона из водной фазы в органическую и отмечено, что даже
такие липофильные катионы, как ТЭБА+, в паре с гидроксиль-
ным ионом очень плохо растворимы в органических
растворителях, и, как правило, то незначительное количество Q+ "ОН,
которое переходит в органическую фазу, не определяет
кинетику реакций в двухфазной системе (например, реакций алки-
лирования ацетона). Однако существуют реакции, при которых
гидроксильный ион может регенерироваться в органической
фазе. Примером такой реакции является реакция альдольной
конденсации.
При контакте ацетона или его раствора в бензоле с 50%-ной
водной щелочью в органическом слое очень быстро образуется
заметное количество продукта альдольной конденсации — диа-
цетонового спирта, который далее постепенно превращается
в окись мезитила и форон:
СН3СОСН8 + СНзСОСНз q=fc CH3COCH2C(CH3)2 ——v
I —НгО
ОН
снасосн3
СНзСОСН—С(СН3)2 ^ (СН8)2О*СНСОСН=-С(СН3)2
Было установлено, что при проведении реакции в
двухфазной системе кинетика ее (порядок реакции по ацетону, влияние
катализатора) зависит от типа применяемой щелочной системы
[67, 68] и отличается от кинетики альдольной конденсации
в гомогенной водной фазе [69, 70]. Показано также, что аль-
дольная конденсация в двухфазной системе отличается от
реакции алкилирования по отношению к интенсивности
перемешивания. Наконец, оказалось, что порядок реакции по ацетону
в бензольных растворах в присутствии 50%-ного водн. NaOH
изменялся с 2 до 3 при введении ТЭБА-С1, одновременно
изменялась и начальная скорость реакции. Уже незначительные
добавки ТЭБА-С1 резко тормозили реакцию. По-видимому, это
первый отмеченный случай торможения реакции при
добавлении незначительных количеств ониевой соли.
Если принять, что, несмотря на большую разницу в рКа
(для ацетона 20, для воды 16), ацетон и вода конкурируют
между собой в кислотно-основных равновесиях, приводящих
к диацетоновому спиртv. и если учесть, что в присутствии
50%-ного водного NaOH вода и "ОН в бензол-ацетоновом
слое отсутствуют и реакция с водой может проходить только
44
йа поверхности водной фазы, где адсорбирована ионная пара
Na+ A~, то можно представить следующую схему альдольной
конденсации ацетона (АН — ацетон, ДАС — диацетоновый
спирт, А~ — карбанион ацетона, ДАС~ — алкокси-ион диаце-
тонового спирта):
(а)
(б)
(в)
(г)
АНЧ
А~ +
ДАС
ДАС
- "ОН ^
■АН ч=*
" + Н2О
" + АН ;
> А~+Н2
ДАС"
ч^ ДАС
f=fc ДАС-
0
+ "ОН
ЬА"
Согласно этой схеме, второй порядок отвечает стадиям (а)
-»-(б)->-(в), а третий порядок, наблюдающийся в условиях
межфазового катализа, соответствует стадиям (а) ->■ (б) -*-(г),
что связано с обезвоживающим действием концентрированной
щелочи и десорбцией А~ с поверхности раздела фаз. Первый
маршрут включает гетерогенные стадии (а), (в), а может
быть, и (б), а во втором—замыкается гомогенный
каталитический цикл (б) -»- (г), не включающий гидроксильпый ион.
Существование гомогенного маршрута подтверждается тем,
что порядок реакции по ацетону в гомогенной системе
(т. е. в объеме органической фазы) под влиянием Q~h "ОН
также равен двум:
(д) Q+ "ОН + АН =f^ Q+ A" + Н2О
(е) Q+ A" + АН +=£ ДАС" Q+
(ж) ДАС" Q++AH ?=* ДАС + Q* А"
(з) ДАС" Q+ + H2O ^=± ДАС + Q+ ""ОН
Стадия (д) соответствует стадии (а), (е) — (б), (ж) — (г),
(з) — (в). Хотя в этом случае разница в концентрации ацетона
(6—12 моль/л) и воды, образующейся на стадии (д) и
включающейся в цикл (д)-+(е)-+(з) (0,01 моль/л) очень велика,
однако, как и в случае двухфазной системы без катализатора,
порядок реакции по ацетону остается равным двум, т. е.
главным поставщиком диацетонового спирта является не цикл
(( ()Ч(
Существенное уменьшение скорости реакции в гомогенных
условиях и уменьшение равновесной концентрации ДАС по
сравнению с реакцией в гетерогенных условиях может быть
сопоставлено с уменьшением скорости реакции при добавлении
катализатора. Очевидно, что эти два маршрута — гомогенный
и гетерогенный — должны взаимодействовать друг с другом
через общее промежуточное соединение. Им является,
по-видимому, ионная пара Na+ А", которая участвует в обмене
катионами, хотя концентрация ионной пары Q+ А~ в органической
фазе не очень велика. Реакция образования ДАС~ Q+ также
может проходить гетерогенно: (а)-»-(г)->-(е). Растворимость
45
этой ионной пары, в которую входит такой большой анион,
как алкокси-нон диацетоиового спирта, должна быть довольно
велика; кроме того, накопление спирта в органической фазе
должно повышать растворимость Qf ~OH в ней н еще более
ускорять гомогенный процесс. Увеличение скорости при
дальнейшем увеличении концентрации Q+ Cl~ связано, по-видимому,
с гомогенной реакцией, скорость которой возрастает с
увеличением концентрации Q+ ~ОН. Косвенным подтверждением
этого предположения является наблюдаемое монотонное
возрастание скорости реакции с возрастанием интенсивности
перемешивания. В общем, можно представить следующий цикл
реакций образования диацетонового спирта:
АН
(а)
NaOH
<Н2О
(А)
Q+ ~OH
A" NV
(и)
A" Q+
ДАС
(в)
н2о
ДАС + NaOH
н2и
(е)
АН
—АН
ДАО" Q+
ДАС + Q+ "ОН
(ж)
АН
ДАС + A" Q+
Гаким образом, несмотря на общий промежуточный продукт —
ионную пару A~Na+, реакции алкилирования и альдольной
конденсации в одной и той же системе существенно различаются
по кинетике. Перемешивание увеличивает долю гетерогенного
маршрута (а) -> (б) -> (в). Введение небольшого количества
воды в органическую фазу или некоторое разбавление водной
фазы должно подавлять гетерогенный маршрут за счет реакции
(д) и ускорять его за счет реакции (в). Факт торможения
реакции может быть объяснен следующим. На первом этапе
реакции Q+ С\~ превращается в Q+ ~ОН и происходит образование
ионных пар с анионом ацетона [стадии (а) и (д)]. Ионные
пары А~ Na+ и А~ Q+ могут обмениваться катионами
[реакция (и)]. Поскольку концентрация А~ Na+ мала и зависит от
скорости адсорбции (растворения), a Q^ А" постепенно
переходит в органическую фазу, чему в большой степени
способствует реакция (на схеме не показана)
А
ДАС
то очень быстро создается ситуация, при которой «быстрый»
гетерогенный маршрут блокируется, что приводит к
торможению реакции и изменению порядка по ацетону. При этом
предполагается, что скорость стадии (б) меньше, чем стадии (е).
46
Изучение реакции альдольной конденсации в двухфазной
системе с ониевым катализатором и в гомогенной системе
с ониевым основанием показало, что в некоторых случаях даже
незначительный перенос гидросильного иона в органическую
фазу может отразиться на кинетике реакции, вызывая
образование новой каталитической системы.
*
Изучение кинетики и механизма реакций в двухфазных
системах, прежде всего с использованием в качестве водной фазы
концентрированных растворов щелочей, еще только начинается.
Однако уже сейчас можно сказать, что реакции в двухфазных
системах представляют собой особую группу реакций со своей
спецификой, которая отличает их от аналогичных реакций в
гомогенных условиях. Влияние адсорбции органических молекул
на поверхности раздела фаз на кинетику сближает их с
гетерогенными реакциями, а образование промежуточных
комплексов субстрата с катализатором межфазного переноса и
соответственно «михаэлисовская» кинетика — с ферментативными
процессами. Таким образом, развитие этой новой области кинетики
органических реакций позволит исследовать системы,
моделирующие гетерогенные и ферментативные реакции.
Литература
1. Brdndstrom A. Preparative Ion Pair Extraction. Apotlkarsocieten/Hassle,
Lakemedel. Sweden, 1974. 275 p.
2. Makosza M.—Naked Anions-Phase Transfer. Conference Paper of the
International Workshop of Modern Synthetic Methods, 1976. Interlaken,
Switzerland, Sch'A'eizericher Chemiker Verband, Ziirieh, 1976, p. 7—100.
3. Dehmlow E. V. — Angew. Chem., 1977, Bd. 89, № 8, S. 521—534.
4. Bergen R. L., Long F. A.— J. Phys. Chem., 1956, v. 60, N 11, p. 1131—
1137.
5. Сергеева В. Ф. — Усп. химии, 1965, т. 34, № 5, с. 717—729.
■6. Long F. Л., McDevii N. — Chem. Rev., 1952, v. 51, № 1, p. 119—142.
7. Bancroft W D —Science, 1935, v. 82, № 5, p. 388-390.
8. Kuneda 11. — J. Chem. Soc. Japan. Chem. a. Ind. Chem., 1977, v. 2, № 1,
9. Starks Ch.M. — J. Am. Chem. Soc, 1971, v. 93, № 1, p. 195—199.
10. Fendier E. J., Fendler J. H. — Adv. Phys. Org. Chem., 1970, v. 8, p. 271 —
278.
11. Cordes E. H. Reaction Kinetics in Micelles. N. Y., Plenum Press, 1973.
262 p.
12. Moravetz /-/. — Adv. in Catalysis, I960, v. 20, p. 341—349.
13. Гордон Дж. Органическая химия растворов электролитов. Пер. с англ./
Под ред И. П. Белецкой. М., Мир, 1979. 712 с.
14 Starks Ch. M., Owens R. M. — J. Am. Chem. Soc, 1973, v, 95, N 9,
p. 3613—3618.
15. Dockx /. — Synhesis, 1973, N 7, p. 441—446.
16. Solodar /. — Tetrahedron Lett., 1971, N 4, p. 287—289.
17. Мищенко К. /7., Полторацкий Г. М. Вопросы термодинамики и строение
водных п неводных растворов электролитов. Л., Химия, 1968 299 с.
18. Fry A. L3 Reid R. G. — J. Am. Chem. Soc, 1973, v. 93, № 3, p. 553-558.
47
19. Цундель Г. Гидратация молекул и межмолекулярное взаимодействие. Пер.
с англ./Под ред. Ю. Н. Чиргадзе. М., Мир, 1972. 208 с.
20. McMillan R. К., Binamico Л!., Jeffrey G. Л. — J. Chem Phys 1963 v. 39,
N 12, p. 3295-3299.
21. Lindenbaum S. — J. Phys. Chem., 1966, v. 70, № 1, p. 3—9.
22. Кудрявцев И. В., Мищенко К- П., Полторацкий Г. М. — Теор и эксп.
хим., 1968, т. 4, № 1, с. 4—9.
23. Майрановский С. Г., Рубинская Т. #., Проскурякова И. В. —
Электрохимия, 1974, т. 10, № 12, с. 1502—1507.
24. Гиллеспи Р. Геометрия молекул. Пер. с англ./Под ред. Ю. А Пентина.
М., Мир, 1975. 278 с.
25. d'Arpano А. — J. Phys. Chem., 1971, v. 75, № 12, p. 3290—3294.
26. Landini D. e. a. — Chem. Commun., 1975, № 8, p. 950—951.
27. Saito S.y Lee M., Wen W.-Y. — J. Am. Chem. Soc, 1966, v. 88, № 4,
p. 5107—5112.
28. Plati Th.t Taylor II. — J. Phys. Chem., 1964, v. 68, № 12, p. 3426—3431.
29. Полторацкий Г. М, Мищенко К. П. — ЖФХ, 1965, т. 39, № 2, с. 264—269.
30. Destroyers J. E., Pelletler G. E,, Jolicoeur С. — Can. J. Chem., 1965, v. 43,
№ 12, p. 3232—3236.
31. Полторацкий Г. М. В кн.: Тезисы докладов на 1-й Менделеевской
дискуссии. М.-Л., Химия, 1967.
32. Гэрни Р. Ионы в растворах. Пер. с англ./Под ред. Л. Э. Гуревича. М.-Л.,
ГОНТИ, 1938. 210 с.
33. Glerst L., Nicolas £., Tytgat-Vanderbergen L. — Croat. Chem. Acta, 1970,
v. 42, До 1, p. 117—121.
34. Packter A., Donbrow E. — Proc. Chem. Soc, 1962, № 2, p. 220—226.
35. Krygowski T. M., Lipszajn M.t Galus Z. — J. Electroanalyt. Chem., 1973,
v. 42, № 2, p. 261—267.
36. Davies M.t Williams G. —Trans. Farad. Soc, 1960, v. 56, № 7, p. 1619—
1622.
37. fnami Y. #., Bodensen H. K., Ramsry /. B. — J. Am. Chem. Soc, 1961,
v. 83, № 12, p. 4745—4748.
38. Accasclna F. e. a. —Proc. Nat. Acad. Sci. US, 1953, v. 39, № 8, p. 917—
919.
39. d'Arpano A.t Fuoss R. M. — J. Phys. C'-em., 1963, v. 67, № 9, p. 1704—
1708.
40. Hirsch £., Fuoss R. Af. — J. Am. Chem. Soc, 1960, v. 82, № 8, p. 1018—
1023.
41. Сухотин А. М. Вопросы теории растворов электролитов в средах с
низкой диэлектрической проницаемостью. М., Госхимиздат, 1959. 96 с.
42. Справочник химика. М., Госхимиздат, 1961. Т. 3. 1005 с.
43. Кафаров В. В. Основы массопередачи. М, Высшая школа, 1979. 521 с.
44. Gordon I. E., Kutina R. Е. — J. Am. Chem. Soc, 1977, у. 99, № Ю,
р. 3903—3909.
45. Измайлов Н. А. Электрохимия растворов. М., Химия, 1966. 575 с.
46. Макоша М. — Усп. химии, 1977, т. 46, Я? 10, с. 2174—2191.
47. Энергия разрыва химических связен. Потенциалы ионизации и сродство
к электрону./Под ред. В. Н. Кондратьева. М., Наука, 1974. 342 с.
48. Майоров В. Д., Либрович И. Б. —ЖФХ, 1976, т. 50, № 11, с. 2817—2820.
49. Вопросы физической химии растворов электролитов./Под ред. Г. И, Ми-
кулина. Л., Химия, 1968. 361 с.
50. Краткая химическая энциклопедия. М., Советская энциклопедия, 1967.
Т. 5. 1112 с.
51. Справочник по растворимости./Под ред. В. В. Кафарова. М.-Л., Изд. АН
СССР, 1962. Т. 2, ки. 1. 943 с.
51а. House И. О., Feng F.t Peet N. P. — J. Org. Chem., 1971, v. 36, p. 2371—
2373.
52. Аульченко И. С. и dp, — Масложировая пром., 1975, № 12, с. 2427—
48
53. Dehmlow E. V., Slopianka M., Heider. — Tetrahedron Lett., 1977, № 27,
p. 2361—2364
54. Isagawa E. V., Kimura /., Kwon S. — J. Org. Chem., 1974, v. 39, № 10,
p. 3171—3174
55. Есикова И. А. и др. — Изв. АН СССР. Сер. хим., 1979, № 7, с 1468—
1474.
56. Фаустов В. И., Юфит С. С.у Есикова И. А. — Изв. АН СССР, Сер. хим.,
1979, № 7, с. 1474—1480.
57. Dehmlow E. V.} Lissel M., Heider /. — Tetrahedron, 1977, v. 33, № 2,
p. 363—365.
58. Юфит С. С, Есикова И. Л. —Изв. АН СССР, Сер. хим., 1979, № 8,
С- 2316 232].
59. Coma I. M. — Bull. Soc. Chim. France, 1956, Nq 5, p. 1392—1404.
60. Alain В., Dubois /. E. — J. Chem. Phys., Phys.-Chem. Biol., 1972, v. 69S
№ 3, p. 623—631.
61. Izsak D., Le Feme R. J. W. — J. Chem. Soc, B, 1966, № 2, p. 251—255.
62. Camming J В., Kebarle P. — J. Am. Chem. Soc, 1976, v. 99, № 20,
p. 5818—5820.
63. Amett E. M. e. a. — Farad. Symp. Chem. Soc, 1975, v. 10, № 1, p. 20—26.
64. Bartmess J. E. — J. Am. Chem. Soc, 1979, v. 101, № 20, p. 6050—6055.
65. Есикова И. А, Юфит С. С.— Изв. АН СССР. Сер. хим., 1979, № 8,
с. 3124-3128.
66. Юфит С. С, Есикова И. Л. —Изв. АН СССР. Сер. хим., 1979, № 8,
с 2439—2446.
67. Лозанская Т. И. и др. — Изв. АН СССР. Сер. хнм., 1979, № 10, с. 3125—
3127.
68. Юфит С. С, Лозанская Т. И., Есикова И. А. —Изв. АН СССР. Сер. хим.,
1979, № 10, с. 3127—3128.
69. Дженкс В. Катализ в химии и энзимологии. Пер. с англ/Под ред.
И. В. Березина. М., Мир, 1972. 467 с.
70. Инеольд К. Теоретические основы органической химии. Пер. с англ./Под
ред. И. П. Белецкой. М., Мир, 1973. 1054 с.
Межфазный катализ
в органическом- синтезе
Катализаторы межфазного переноса особенно широко
используют в реакциях нуклеофильного замещения и
присоединения, значительно в меньшей степени — в реакциях
элиминирования. Описаны отдельные примеры использования этих
катализаторов в процессах изомеризации. Ниже последовательно
рассмотрено применение межфазного катализа в нуклеофиль-
ных реакциях замещения с участием неорганических и
органических анионов, в нуклеофильных реакциях присоединения
органических анионов по кратным связям (включая
последующие превращения продуктов присоединения, например
элиминирование и циклизацию), в реакциях присоединения дигало-
генкарбенов по простым (внедрение) и кратным связям, в
реакциях элиминирования и некоторых других превращениях.
РЕАКЦИИ НУКЛЕОФИЛЬНОГО ЗАМЕЩЕНИЯ
С УЧАСТИЕМ НЕОРГАНИЧЕСКИХ АНИОНОВ
Эти реакции достаточно широко изучены в условиях
межфазного катализа. Описаны взаимные обмены галогенов
(реакция Финкельштейна) [1 —16], замещение атомов галогенов на
тиоцианатогруппу [4, 6, 8, 17—20], циапогруппу [1, 4, 6, 7, 9,
16, 17, 19, 21—35], азидную группу [36, 37], изоцианатную
группу [1], нитрогруппу [1, 6, 35, 38], сульфидную группу [39—
42), гидроксил [1, 16]. Имеются примеры нуклеофильного
замещения при действии неорганических анионов гидроксигруппы
[43,44], мезилатного остатка [1—3, 6, 8, 9, 12, 45], нитро-
группы [46], тозилатного остатка [47, 48], диазогруппы [49].
Синтез галогенпроизводных
Синтез фторидов. Для синтеза фторидов в качестве
катализаторов межфазного переноса были использованы трибутилгекса-
децилфосфонинбромид (ТБГДФБ) [2], эфир полиэтиленгликоля
[4], тетрабутиламмонийаерхлорат (ТБАПХ) [49] и
некоторые краун-эфиры (18-краун-6, дибензо-18-краун-б, дицикло-
гексано-18-краун-6) [3, 8, 10]. Источником F~ служили KF или
KHF2 [3, 50]. Реакцию проводили в воде [2] или в смеси воды
с дихлорметаном [50] при использовании ТБГДФБ или
50
ТБАПХ, а в присутствии краун-эфиров — в ацетонитриле [10,
51], бензоле [10], гексане или пептане [12] или в сульфолане
[52]. Среди алифатических галогенидов хорошие результаты
были получены только с первичными к-алкилхлоридами, а
также бензилхлоридом. Первичные я-алкнлбромиды образуют ал-
килфториды со значительно более низкими выходами.
Совершенно не реагируют первичные разветвленные алкилбромиды,
например 1-бром-2,2-диметилпропан. Из вторичных алкилхло-
ридов фториды получаются с невысокими выходами (вторичные
алкилбромиды не дают фторидов вовсе), а циклогексилхлорид
не образует ииклогексилфторида в условиях реакции.
Помимо фторидов, даже в тех случаях, когда реакция
проходит с высокими выходами, в качестве побочных продуктов
всегда образуются олефины и спирты. Часто олефнны
становятся основными продуктами реакции; например, реакция
2-хлор- или 2-бромоктана с Кр в водном растворе в
присутствии ТБГДФБ при нагревании (160 °С) приводит к октенам
с выходом 66% или 100% соответственно; выход же 2-фторо-
ктана в случае хлорида составляет только 20%. Аналогично из
циклогексилхлорид а или циклогексилбромнда был получен
практически с количественным выходом циклогексен [2].
Следует отметить, что при использовании трмбутилгексадецилфос-
фонийбромида реакция между алкилгалогенидами и KF
проходит в довольно жестких условиях:
ТБГДФБ, Н2О, 160 °С
RC1 + KF ■> RF
R (выход, %): я-С6Н13 (80); я-С8Н17 (71); н-С12Н25 (77); PhCII2 (90)
Типичная методика проведения этой реакции приведена
ниже.
Синтез 1-фтороктана [2]. Смесь 14,9 г 1-хлороктана, 47 г
KF-2H2O и 5,1 г ТБГДФБ в 30 мл воды нагревают 7 ч в автоклаве с
магнитной мешалкой при 160°С (в бане). Органический слой отделяют,
промывают водой, концентрированной H2SO*, вновь водой, сушат над СаС12 и
подвергают разгонке. Выход 10,2 г; т. кип. 142—144 °С (760 мм рт. ст.);
п^° 1,3934 [2].
Таким образом, реакция обмена галогена на фтор проходит
только для ограниченного круга соединений, если в качестве
катализаторов межфазного переноса применять четвертичные
соли. Значительно более удовлетворительные результаты
получают при использовании мезилатов [2]. Так, ыезнлат окта-
нола-1 превращается в 1-фтороктан в приведенных выше
условиях с выходом 90%, а мезилат октанола-2 дает 2-фтороктан
с выходом 54% (выход октенов 26%) [2]. Интересно, что при
использовании оптически активного мезплата октанола-2 в
результате обменной реакции с KF (с инверсией) получают
51
оптически активный 2-фтороктан (выход 45%;
ТБГДФБ, KF
100ОСИч (+)-c6h,3chf
Me Me
Использование крауи-эфиров в качестве катализаторов
позволило расширить область применения межфазного катализа
для синтеза фторпроизводных. В частности, удалось провести
обмен хлора на фтор в некоторых арил- и гетероарилхлоридах
[10, 11, 15, 47], хлоркетонах [10, 13], сульфонилхлоридах [51]
и винилхлоридах [52]. Например, 2,4-динитрохлорбензол и
2-метил-2-хлорциклогексанон реагируют с KF в ацетонитриле
в присутствии 18-крауна-6 с образованием соответственно 2,4-
динитрофторбензола (количественный выход) и 2-метил-2-фтор-
циклогексанона (выход 31%) [10].
При нагревании (100°С, 23 ч) смеси эпихлоргидрина или
бис(хлорметил)оксетана с измельченным KF в присутствии
1,2% (мол.) 18-крауна-б хлор обменивается на фтор.
Образуются эпифторгидрин (выход 60%) и смесь З-фторметил-3-
хлорметилоксетана с 3,3-бис(фторметил)оксетапом [53].
В присутствии 18-крауна-б реакция между алкил- и арил-
сульфонилхлоридами и KF протекает экзотермически и
приводит с высокими выходами к соответствующим алкил- или арил-
сульфонилхлоридам [51]:
RSO2C1 + KF —► RSO2F
R (выход, %): Me (84); PhCH2 (89); Ph (92,5); 4-MeC6H4 (100), 4-BrCeH4
(100); 4-AcNHC6H4 (96); 5-днметнламинонафтил-1 (100)
Типичная методика проведения этой реакции приведена
ниже.
Синтез я-ацетамидофенилсульфонилфторида [51].
Смесь 117 г д-ацетамидофенилсульфоннлхлорида, 58 г KF в 200 мл ацето-
нитрила и раствора 5 г 18-крауна-б в 100 мл ацетонитрила после окончания
экзотермической реакции перемешивают в течение ночи при комнатной
температуре. Осадок отделяют, промывают водой, сушат. Выход 105 г; т. пл.
175—177 °С.
Приведенная выше методика является наилучшей для
синтеза сульфопилфторидов.
Описаны интересные реакции обмена одного атома хлора
в метиловом эфире 2-метоксикарбонил-З-хлоракриловой
кислоты и обоих атомов хлора в метиловом эфире 2-метоксикарбо-
нил-3,3-дихлоракриловой кислоты на фтор в присутствии дицикло-
гексано-18-крауна-б [52]:
ClCH=C(CO2Me)2 + KF —-> FCH=C(CO2Me)2
С12С=С(СО2Ме)2 + KF —► F2C=C(CO2Me)2
Реакцию проводят в сульфолане; выходы фторпроизводных
24—25%- Разработан [50] удобный способ синтеза бензилфто-
52
ридов, основанный на реакций KHF2 с арилдиазоалканами
в присутствии тетрабутиламмонийперхлората в системе вода —
дихлорметан при комнатной температуре. В этом случае
катализатор межфазового переноса ускоряет образование карб-
катиона из арилдиазоалканов. Такой способ генерирования
карбкатиона был осуществлен впервые.
Ar2CN2-f KHF2 —► Ar2CHF
Аг (выход, %): Ph (50); 2-BrC6H4; 4-BrC6H4
Наряду с фторидами образуются в небольших количествах
диарилкарбинолы, тетраарилэтилены, диарплкетоны и смоло-
образные продукты.
Синтез хлоридов. Межфазный катализ редко используется
для синтеза хлоридов, поскольку для их получения имеется
много удобных и простых способов. Однако следует отметить
интересный способ превращения первичных спиртов в алкил-
хлориды действием соляной кислоты в присутствии трибутил-
гексадецилфосфонийбромида [44]:
н2о
н-ROH + HCl >■ RC1
R (выход, %): w-Bu (65); w-C5HI3 (87—95), w-C8HI7 (87—94); н-С[2Н25ОН
(91—94); н~С16Н3з (91—97) (первая цифра относится к выходу продукта при
длительности реакции 3 ч, вторая — 45 ч)
Типичная методика проведения этой реакции приведена
ниже.
Синтез 1-хлордодекана. Смесь 10 г я-додеканола, 22 мл
37%-ной НС1 и 2,72 г ТБГДФБ нагревают прн 100—105°С (температура
внутри смеси) и интенсивном перемешивании до окончания реакции (контроль
с помощью ГЖХ), после охлаждения промывают 10°/с-ным раствором NaCl,
органический слой сушат над СаС12 и разгонкой выделяют 1-хлордОдекан.
Выход 8,5 г; т. кип. 86—88°С (3 мм рт. ст.); n)J 1,4419.
В отдельных случаях при проведении реакции в присутствии
водного раствора NaOH и использовании ТЭБАХ как
катализатора межфазного переноса хлорирующим агентом может
служить хлороформ. Таким путем, например, можно получить
адамантилхлорид (выход 94%) из адамантилового спирта, бен-
зилхлорид (90%) из бензилового спирта и 2-з/сзо-норборнил-
хлорид (90%) из 2-э/сзо-норборнилового спирта [44].
Естественно, что хлориды образуются при обменной
реакции между бромидами или мезилатами и хлоридами щелочных
металлов (КС1 или NaCI), причем выходы хлоридов выше
в случае мезилатов. Так, при реакции 1-бромоктана с KCI (или
NaCl) в водном растворе в присутствии дициклогексано-18-
крауна-б получают 1-хлороктан с выходом 48% [8], а в тех же
условиях из мезилата октанола-1 1-хлороктан образуется с
выходом 80% [3].
Реакция обмена между мезилатом (—)-(^)-октанола-2 и
КС1 в присутствии метилтри-к-октиламмонийхлорида в водном
53
растворе при кипячении проходит с инверсией у хирального
центра на 95% {выход (-fj-2-хлороктана 74—83%; [a]f>J
+ 33,4°; оптическая чистота 89,2% [45]}.
Интересно применение в качестве катализаторов межфаз-
ного переноса полнмерносвязаиных четвертичных солей. Так,
в системе толуол — вода при использовании катализатора
К-СбН4СН2М(Ме)Р(О) (NMe2)2 (R — полимер) при реакции
КС! с 1-бромоктаном получают 1-хлороктан с выходом 83%.
Описано хлорирование ди-трег-бутилфосфита четыреххлори-
стым углеродом при комнатной температуре в системе дихлор-
метан — вода в присутствии 20%-иого NaOH и 5% ТЭБАХ
[52]:
—> (трет-ВиО)2Р{0)С\ + СНС13
При замене ССЦ на СВг4 образуется ди-трег-бутилбромфос-
фат [54].
Синтез ди-грег-бутилбромфосфата. 9,7 г дн-трег-бугалфос-
фита в 15 мл дихлорметана добавляют по каплям при интенсивном переме-
шипании к смеси 30 мл дихлорметана, 8,3 г СВг4, 20 мл 20%-ного NaOH и
0.5 г (5%) ТЭБАХ при 20—25 °С, перемешивают 3 ч, разбавляют 50 мл
ClbCU, органический слой промывают водой (2X50 мл), сушат (Na2SO4),
удаляют растворитель в вакууме. Выход 12,2 г; п2^ 1,4490; продукт
аналитически чистый, не требует дальнейшей очистки.
К сожалению, попытка перенести этот метод на другие
диалкилфосфиты не имела успеха. Так, из диэтилфосфита даже
при 0—5 °С образовывалась смесь, состоящая из 83% тетра-
этилпирофосфата и 17% диэтилводородфосфата.
Синтез бромидов и иодидов. Опгсано несколько методик
синтеза алкилбромидов [1, 3, 6—8, 12, 46] и алкилиодидов
[1, 3, 7, 8, 12, 17, 23, 32, 46, 55—60] из соответствующих гало-
генидов или мезилатов. Наиболее общим способом синтеза
алкилбромидов и бензилбромида является использование системы
бензол — вода и полимерносвязанной тетраалкиламмониевой
соли R—C6H4NMe2Bu C1 ; в этих условиях выходы бромидов
достигают 32—72%. Следует отметить, что реакция между КВг
и мезилатом (—)-(/?)-сжтанола-2 протекает в присутствии
ТБГДФБ в водном растворе с инверсией у хирального центра
на 86%, как и в случае реакции с КС1, и приводит с выходом
a Of)
а]Ъ 29,2°; оптическая чистота
72,4 %). Интересно, что при реакции между мезилатом
(—)-(#)-октанола-2 и KI в идентичных условиях проходит
почти полная рацемизация и полученный 2-иодоктан (выход
70—74%) имеет [cc]d + 0,8° (оптическая чистота 1,7%).
Процесс рацемизации осуществляется, видимо, путем обмена
между атомами иода [4].
В присутствии ацетата калия и 18-крауна-6 арендиазоний-
фториды реагируют с бромтрихлорметаном или метилиодидом,
54
образуя соответственно арилбромиды (выходы 33—90%) или
арилиодиды (45—94%) [61]:
rt-XC6H5N2 BF4~-f BrCCl3 (Mel или I2) —-> n-XCGli4Br(mni I)
MeO, Cl, Br, NOa
Отмечено, что при кипячении (ацетоксиметил)триметилси-
лана с водным раствором NaBr или Nal в присутствии
10% (мол.) ТБАБ [или 20% (мол.) ТБАИ] в течение 11 дней
(или соответственно 2,5 ч) образуется броммстплтриметилси-
лан (выход 86%) или соответственно иодметилтриметилсилан
(выход 82%) [62]:
NaX
Me3SiCH^OAc >- Me3SiCH2X X = Вг, I
Несомненно, очень полезен способ синтеза иодхлорметана
из дихлорметана обменом с Nal в водном растворе в
присутствии ТБГД [5], одновременно образуется дииодметан.
Синтез иодхлорметана. Смесь 1 моль дихлорметана, 2,4 моль
Nal, 200 мл воды и 0.033 моль ТБГДФБ нагревают (100— 110 °С) в автоклаве
при интенсивном перемешивании в течение 18—20 ч. После охлаждения
разбавляют смесь 400 мл воды, извлекают продукты эфиром (3 X 300 мл),
промывают 5%-ным раствором бисульфита натрия (2 X 100 мл). Полученную
смесь разделяют фракционной перегонкой. Выход иодхлорметана 24 г; т. кип.
35—40°С (50 мм рт. ст.). Выход дииодметана 180 г; т. кип. 89—92°С
(50 мм рт. ст.).
Синтез тиоцианатов
Реакции алкилхлоридов, алкилбромидов или алкилиодидов,
а также бензилгалогенидов с KSCN проводят чаще всего в
водных растворах в присутствии аликвата 336 [6, 17] или
третичных аминов, например я-трибутиламина [17] (в процессе
реакции амины, по-видимому, превращаются в четвертичные
аммониемые соли), бензилтриметиламмонийхлорпда [19], тет-
рабутиламмонийбромида [18], эфира полиэтиленгликоля [4].
Можно использовать также крауи-эфиры, например дициклогек-
сано-18-краун-6 [8, 17а]. В ряде случаев выходы достигают
количественных. Так, например, проходит реакция алкилгалогени-
дов с тиоцианатом калия в присутствии аликвата 336 [17]:
RX + KSCN —► RSCN
RX (выход тноцианата, %): EtBr (92); EtI (99); я-РгВг (100); н-BuCl (100);
«-BuBr (99); я-BuI (96); н-С5НпВг (100); н-С6Н13Вг (99); я-С7Н151 (100);
H-C8Hi7Br (100)
Общая методика синтеза тноциапатов. Смесь 10 г алкил-
галогенида, двукратный избыток KSCN в виде 5%-ного водного раствора и
5% (мол.) аликвата 336 нагревают при 100 °С и перемешивают до завершения
реакции; полученные тиоцианаты извлекают подходящим растворителем и
выделяют.
Почти количественные выходы тиоцианатов получают и при
замене аликвата 336 на к-трибутнламин или н-бутиламин
55
(однако в этом случае я-бутилиодид не реагирует с KSCN)
[17]. В отсутствие катализаторов межфазного переноса реакция
либо вовсе не идет, либо выходы тиоцианатов крайне малы (1—
17%) [17].
Отличные результаты получены также при реакции алкил-
галогенидов или бензилхлорида с KSCN в присутствии ТЭБАХ
в водном растворе [19].
Синтез нитрилов
Особенный интерес представляет Р1спользование межфазного
катализа для синтеза нитрилов алифатического ряда из алкил-
галогенидов и алкилмезилатов, поскольку обычные способы
очень часто не дают удовлетворительных результатов. Этому
вопросу посвящено много работ [1, 3, 4, 6—8, 20, 22, 24, 26—
29, 31—33, 35, 53, 63, 64]. Особенно благоприятные результаты
получены при использовании в качестве катализаторов краун-
эфиров [18, 22, 26, 29], полимерносвязанных четвертичных
аммониевых солей [7, 16, 27, 28, 33] и бензилтриметиламмоний-
хлорида (БТМАХ) [20]. Были использованы также
циклические фосфониевые или арсониевые соли [31], аликват 336 [1,
6, 7, 64], эфиры полиэтиленгликоля [4]. Показано, что
реакцию между алкилбромидами и KCN катализируют также
первичные, вторичные и третичные амины, в особенности трн-к-бу-
тиламин и три-я-гексиламин [35], при использовании которых
из н-бутилбромида был получен нитрил валериановой кислоты
с выходом 100 и 91,8%, соответственно. Разветвленные
первичные алкилбромиды дают нитрилы с низкими выходами.
Разработан очень удобный способ синтеза моно- и динит-
рилов из моно- и дихлоридов или бромидов с применением
каталитических количеств 18-крауна-6:
X(CH2)nY + KCN —► NC(CH2)nCN
X, Y, п (выход, %): Вг, Вг, 3 (97,3); Вг, CI, 3 (100); Вг, Вг, 4 (100); Cl, C1,
4 (86-94)
RX + KCN —-> RCN
RX (выход, %): l-C6HI3Br (100); 1-С6Н13С1 (90,6); 2-BrBu (69,7); 2-ВгС8Н17
(56-62); 2-С1С8Н17 (77,5); PhCH2ClBr (94—100)
Типичная методика приведена ниже.
Синтез глутародинитрила. 11,7 г сухого KCN, 25 мл анетонит-
рила, 5,08 г 1,3-дихлорпропана н 1,01 г 18-крауна-6 кипятят 1,5 ч. После
окончания реакции (контроль ГЖХ) охлажденную смесь фильтруют,
разбавляют водой, извлекают дихлорметаном, сушат (MgSO4) и разгоняют после
удаления растворителей. Выход 4,1 г; т. кип. 78—82°С (0,13 мм рт. ст.) [26].
Циклогексилхлорид и циклогексилбромид в условиях
реакции образуют циклогексен (выход 32—46%), арилгалогениды
не реагируют. В аналогичных условиях при использовании в
качестве растворителя днхлорметана реагируют бензилгалогени-
Ды и их производные с образованием соответствующих нитри
лов с выходами 85—95% [36]:
ArCH2Br + KCN —> ArCHaCN
Ar — Ph, 4-O2NC6H4, 4-С1С6Н4, 3,4-(MeO),C6H3
В присутствии ТЭБАХ или гексадеиилтриметиламмонийбро-
мида (ГДТМАБ) возможно заместить хлор при двойной связи
в а-иминохлоридах на цианогруппу [65]:
KCN
XC6H4CCl=NPh —-> XCeH4C(CN)=NPh
X (выход, %): Н (86); 4-Ме (68); 4-С! (80);2-С1 (47); З-Вг (93); 4-МеО (79);
катализатор ГДТМАБ
Интересно отметить, что в некоторых случаях возможна
замена галогена в ароматическом ядре. Так, при реакции N-аце-
тил-2-хлоранилина с NaCN в водном растворе в присутствии
БТМАХ образуется нитрил 2-ацетамидобензойной кислоты с
выходом 50%. При действии CuCN на 2/-ацетамино-5-бром-2,4-
динитро-4'-диэтиламиноазобензол в присутствии трибутилоктил-
аммонийхлорида происходит замена брома на цианогруппу
(выход нитрила 80%) [66]:
NO, NHAc
NO, NHAc
02N—/ у—N=N—(x ,)—NEts
CN
Удобный способ синтеза бензоилцианидов обменной
реакцией между бензоилхлоридом и NaCN в присутствии ТБАБ
позволяет получать труднодоступные иными путями бензоилцианиды
с хорошими выходами:
п-ХСвН4СОС1 +КаС1 •—> /i-XCeH4COCN
X (выход, 7с): Н (60); Me (72); МеО (60), С1 (22)
Значительное снижение выхода в случае хлорзамещенного
объясняется протеканием конкурирующей реакции димеризации
с образованием арилпроизводных типа
4—C(CN)2—ОСОС6Н4Х-«
Выход димера в случае хлорида составляет 46%, а для
незамещенного арилпроизводного —35%.
57
Синтез бензоилцианида [66]. К смеси 51 г бензоилхлорида, 100 мг
ТБАБ п 300 мл дмх;юрметана, охлажденной до 0 °С, добавляют раствор 18 г
NaCX в 20 мл поды, перемешивают до исчезнозения бензоилхлорида
(примерно 1 ч), фильтруют, отделяют органический слой, сушат (MgSO4),
перегоняют. Выход 25 г; "т. кип. 105 °С (0,1 мм рт. ст.); т. пл. 30—32 °С. Остаток
после перегонки кристаллизуют из спирта, получают димер; т. пл. 94—95 °С.
Синтез азидов
Синтез азидов в условиях межфазного катализа изучен еще
недостаточно. Разработан общий метод синтеза алкил- и цикло-
алкилазидов из алкил- или циклоалкилгалогенидов действием
азида натрия в водном растворе в присутствии аликвата 336
[37].
RX + NaN3 —> RN3
RX (выход. %): я-BuI (89); н-BuBr (97); н-BuCl (65); «-С5НпВг (89);
я-С6Н13Вг (87): н-С7Н15Вг (92); н-С5Н17Вг (92); н-СшН21Вг (93); цикло-СсНц1
(77; одновременно образуется 23% циклогексена); цикло-С6НцВг (74 78;
одовременно образуется 12% циклогексена)
Общая методика синтеза азидов. К смеси 25%-ного водного
раствора 10,25 г азида натрия и 1,62 г аликвата 336 при хорошем
перемешивании добавляют 0,08 моль алкилгалогенида, нагревают при 100 V,,
интенсивно перемешивая. После израсходования галогеиида (контроль ГЖХ)
органический слой отделяют, сушат и перегоняют.
В присутствии тетрабутиламмонийбромида в водном
растворе или 18-крауна-б в бензоле бензилбромид также реагирует с
азидом калия, образуя бензилазид с выходом 87% [68]; если же
применять эфир полиэтиленгликоля, то выход бензилазида
повышается до количественного [4]. Достаточно гладко в
условиях межфазного катализа (ТБАБ или 18-краун-6) [68, 69]
проходит замена брома на азидную группу в этиловом эфире
бромуксусной кислоты [68]' и метиловом эфире 3-гидрокси(или
метокси)-3-фенилпропионовой кислоты [69].
Синтез нитросоединений
Синтез нитроалканов осуществляют нагреванием смеси
алкилгалогенида (или бензилгалогенида) с нитритом калия и 18-
крауном-6 (20:22:1) в ацетонитриле [36]:
RX + KNO2 —> RNO2
RX (выход, %): 1-ВгС8НГ7 (65-70); ЫС6Н17 (50-55); 2-ВгС2Н4СеН5 (32)
3-Br-l-PhC3Hfi (51): PhCH2Cl (34-51)
В качестве минорных продуктов образуются
соответствующие нитриты. Циклогексилбромид практически не дает нитро-
соединения в условиях реакции.
Отмечено, что ft-бутилмезилат образует 1-нитробутан
(выход 32%) при реакции с KNO2 в дихлорметане в присутствии
метилтриоктиламмонийхлорида [6].
58
Синтез тиолов и сульфидов
Отмечено, что в присутствии эфира полиэтиленгликоля бен-
зилбромид в бензольном растворе способен реагировать с KSH,
образуя количественно фенилметантиол [4].
PhCH2Br + KSH —> PhCH2SH
Алкилгалогениды и бензилхлорид реагируют в водном
растворе с сульфидом натрия в присутствии ТБГДФБ и образуют
диалкил (дибензил)сульфиды с высокими выходами [40, 76]:
—* RSR
RX (выход, %): н-С6Н13С1 (90); к-СвН17С1 (98); PhCH2Cl (94): «-CGHl3CH(Me)Cl
(90); н-С8Н17Вг (91); я-С8Н17СН(Ме)Вг (91); Ме3ССН2Вг (81)
Типичная методика приведена ниже.
Синтез ди-н-окгилсульфида. Смесь 14,9 г 1-хлороктана, 14,4 г
Na2S-9H2O и 5,1 г ТБГДФБ в 30 мл воды нагревают, интенсивно
перемешивая, при 70°С (контроль с помощью ГЖХ). После окончания реакции
органический слой отделяют, промывают водой, сушат (СаС12) и подвергают раз-
гоике. Выход 11,6 г; т. кип. 160—162 °С (4 мм рт. ст.), п£ 1,4690.
Аналогично проводят реакцию с другими галогенидами.
С высокими выходами сульфиды получают также при реакции
аллил- или пропаргилгалогенидов с Na2S в воде в присутствии
ТБАХ:
RCH=C(R')CH2X + Na2S —> [RCH=C(R')CH2]2S (90-96%)
R = Н, Ph, Me; R' = H, Me; X = Cl, Br
RC=CCH2X-bNa2S —> [RC=CCH2]2S (73-82%)
R = H, Ph; X = C1, Br
Синтез сульфокислот
В системе Н20 —СН2С12 —R3N (R = «-Pr, я-Bu, я-СБН1Ь
h-C6Hi3) w-C8Hi7) при действии на 2,4-динитрохлорбензол ка-
лийдисульфата K2S2O5 при О—8°С хлор замещается сульфо-
группой [71]:
O2N—^%—CI —■> O2N—( х>—SO3H (76-95%)
следует из приведенного выше материала, методики,
основанные на применении межфазного катализа в реакциях
нуклеофильного замещения, в большинстве случаев
превосходят традиционные способы по простоте выполнения, выходам и
59
качеству конечных продуктов. Эти методики можно
использовать для обмена галогенов на другие группы и их взаимного
обмена, обмена на другие группы гидроксигрупп, алкил- и арил-
сульфонатных и ацетатных групп, в некоторых случаях для
обмена нитрогрупп на галогены, тиоцианатную, нитрильную, азид-
ную (см. [72]), сульфонильную и другие группы в
насыщенных, ненасыщенных и ароматических соединениях, а также в
полимерах. Так, была осуществлена реакция нуклеофильного
замещения хлора в хлорметилполистироле в присутствии ТБАХ
или адогена 464 в о-дихлорбензоле (или в 1,2-дихлорэтане, ди-
хлорметане или бензоле) на цианогруппу (выход цианометилпо-
листирола 81 —100%), ацетоксигруппу (выход ацетоксиметил-
полистирола 98%) и др. [33]. Чрезвычайно интересны работы
по асимметрическому нуклеофильному замещению в
присутствии хиральных межфазных катализаторов; при этом удается
осуществить оптическую индукцию и получать оптически
активные продукты (см., например, [73]). Особенно ценны эти
методики для проведения реакций с малоустойчивыми,
чувствительными к влаге соединениями. Таким способом часто
можно осуществить синтез соединений, практически недоступных
иными путями. Типичным примером является синтез триметил-
силилцианида [36].
Синтез триметилснлилцианида. Смесь 6,6 г сухого
измельченного KCN, 20 мл дихлорметаиа, 12 г триметилсилилхлорида и 0,1 г
18-крауна-б кипятят 24 ч, перегоняют непосредственно из колбы, установив
колонку Вигре. Выход 3,3 г (50% от теоретического); т. кип. 114—117°С.
РЕАКЦИИ НУКЛЕОФИЛЬНОГО ЗАМЕЩЕНИЯ
С УЧАСТИЕМ ОРГАНИЧЕСКИХ АНИОНОВ
Такие реакции относятся к важнейшим реакциям
органической химии, поскольку позволяют создавать связи С—О
(синтез простых и сложных эфиров, ацеталей, гликозидов,
кислородсодержащих гетероциклов), С—S (синтез тиоэфиров), С—N
(синтез аминов), и, что особенно ценно, связи С—С (алкилиро-
вание, арилирование, ацилирование и прочие реакции
замещения по активным атомам водорода при углеродных атомах).
Образование С—О-связей
Синтез простых эфиров. Пожалуй, наиболее плодотворное
применение межфазный катализ получил в синтезе
симметричных и несимметричных простых эфиров по Вильямсону. Обычно
для синтеза алифатических эфиров или алкилбензиловых
эфиров реакцию проводят в системах дихлорметан — вода [74],
петролейный эфир — вода [75], бензол — вода [76], изопропи-
ловый эфир — вода [77]; в качестве катализаторов используют
.ТБАГС [74, 77], ТБАБ и ТБАХ [75, 76, 78, 79], ТЭБАХ [80—
60
82], аликват 336 [66, 83], краун-эфиры [84]; в специальных
случаях, например при алкилировании неполных эфиров поли-
этиленгликолей, катализаторами служат сами эти неполные
эфиры [85]. Реакция идет в интервале температур 25—70°С
(в зависимости от природы реактантов), продолжительность
реакции от 30 мин до нескольких часов. Для генерирования
RO^ используют обычно избыток NaOH (как правило, в виде
50%-ного водного раствора). Количество катализатора
составляет 3—5% (мол.); реакцию ведут при интенсивном
перемешивании. Алкилирующими агентами могут быть первичные ал-
килгалогениды (со вторичными реакциями часто идет хуже или
вовсе не проходит [74, 84]), диалкилсульфаты (чаще всего ди-
метилсульфат) [75, 85], бензилгалогениды [77, 78, 81], аллил-
галогениды [82, 85]. В реакцию алкилирования достаточно
легко входят как первичные, так и вторичные алифатические
спирты или бензиловые спирты (в случае вторичных спиртов
продолжительность реакции несколько увеличивается).
Типичная методика синтеза простых эфиров приведена ниже.
Синтез метил-2-фе и илэт илового эфира [75]. Раствор 61 г
2-фенилэтанола и 1 г ТБАИ в 200 мл петролейного эфира (т. кип. 50—70°С)
интенсивно перемешивают с- избытком 50%-ного водного раствора NaOH
в течение 15—30 мьн. Затем добавляют по каплям в течение 1 ч 63 г диме-
тилсульфата, охлаждением поддерживая температуру ниже 45 °С(
перемешивают еще 3 ч, добавляют 10 мл водного раствора аммиака и через 30 мин
смесь выливают в воду. Органический слой отделяют, продукт очищают
перегонкой. Выход 93%; т. кип. 63°С (12 мм рт. ст.).
С высокими выходами проходит алкилирование спиртов ал-
килгалогенидами в присутствии ТБАБ [74]:
ROH (или HOROH) + R'Cl —>■ ROR' (или R'O—R—OR')
R, R' (выход, %): я-Bu, PhCH2 (92); w-BuOCH2CH2, «-Bu (82); w-C8H17, w-Bu
(95); CH2 = CHCH2, w-Bu (72); — (CH2)4— Et (93; образуется
преимущественно EtO(CH2)4OEt и только 3% моноэтилового эфира);
СН3ОСН2СН(Ме)—, w-Bu (97)
Хорошие результаты дает алкилирование циангидринов в
присутствии аликвата 336 в системе СН2С12 — 50%-иый NaOH
при 0°С [81, 86]:
RR'C(OH) (CN) + R"X —> RR'CfOR") (CN) (70-74%)
R = H, Alk
(5)-3-грег-Бутил-5-гидроксиметил-2-фенилоксазолидин
гладко алкилируется 2-хлор-З-цианопиридином в обычных условиях,
давая 'соответствующий 3-цианопиридиловый эфир с выходом
84%. Однако другие спирты, например бензиловый, н-окти-
ловый, изопррпиловый, образуют эфиры при Действии 2-хлор-
3-цианопиридина только в системе твердый КОН — толуол— 18-
краун-6 (выходы 67—85%), а гр<?г-бутиловый спирт не
вступает в реакцию вовсе [87],
61
При наличии в молекуле гидроксигруппы и атома галогена
в подходящих положениях может проходить
внутримолекулярное О-алкилирование с образованием кислородсодержащих ге-
тероциклов. Например, при перемешивании 2-хлорэтаиола с
измельченным едким кали без растворителя в присутствии алик-
вата 336 образуется оксетан с выходом 25% [66]:
С1СН2СН2ОН
—О
Особенное значение межфазный катализ приобрел в химии
Сахаров, поскольку ранее существовавшие методы О-метилиро-
вання и О-бензилирования Сахаров часто приводили к
неудовлетворительным результатам. В результате систематических
исследований [88—91] было установлено, что метод
межфазного О-алкилировання ацеталированных моносахаридов с одной,
двумя и тремя свободными гидроксильными группами
значительно превосходит все ранее известные методы.
Так, О-метилирование в межфазных условиях диметилсуль-
фатом 1,2:3,4-ди-О-циклогексилиден-а-1>-галактопиранозы V,
2,3 : 5,6-ди-О-циклогексилиден-а-£)-маннофуранозы VI, 1,2 : 5,6-
ди-О-циклогексилиден-а-/)-глюкофурааозы VII (R = ОН, R' =
= Н) и 1,2 : 5,6-ди-О-циклогексилиден-а-/)-аллофуранозы VII
(R = Н, R' = OH), а также аллильного и этоксикарбонильного
VIII (R = Н, R/ = CH = CH2 или CO2Et) производных привело
к соответствующим метиловым эфирам [90], обладавшим
высокой степенью чистоты и легко кристаллизующимся (выходы
64—100%).
,о-сн2
н
ю х
V
CfiH
6 "10
VII
RO-
CH2OR
О
ОМе
О
О-
VIII
Интересно, что О-метилирование соединения VII (R — ОН,
R7~H) метилиодидом протекало очень медленно, и далее через
два дня конверсия достигла только я^70%, что можно
объяснить инпюирующим действием иодид-иона.
62
Общая методика метилирования ацеталнрованиых
моносахаридов [90]. Бензольный раствор субстрата (0,1 мг/мл),
трехкратного избытка диметилсульфата и 2% (от количества субстрата) ТЭБЛХ
встряхивают с половинным объемом 50%-ного NaOH. После завершения
реакции (контроль методом ТСХ) бензольный слой отделяют, водный
дополнительно экстрагируют половинным объемом бензола. Объединенный
бензойный раствор промывают водой, содержащей катионнт КУ-2 (Н+)
(десятикратный избыток к ТЭВАХ) для удаления катализатора, фильтруют и
упаривают. При необходимости проводят хроматографическую очистку.
Как видно из приведенных выше данных, в межфазных
условиях метилирование ацеталированных моносахаридов по
первичному, вторичным и гликозидному гидроксилам проходит с
высокими выходами. Однако продолжительность реакции
различна: в случае вторичного и гликозидного гидроксилов она
составляет 0,5—1 ч, а для первичного гидроксила — 27 ч. Следует
отметить также, что более сложные производные типа IX и X
СбП;{
,о-сн2
о
OR О-
X
•с,
метилируются значительно труднее структурно близких к ним
производных II и III (R = Н, R' = OH). Так, время
метилирования IX (R = H) достигает 7 ч, хотя выход соответствующего
метилового эфира (по интенсивности сигнала протонов мето-
ксильной группы в ПМР-спектре реакционной смеси)
достаточно высок (~70%). Выделить метиловые эфиры не удалось.
Более низкий выход О-метилового эфира из этоксикарбонильно-
го производного VIII (R = Ale, R/=CO2E() (64%) объясняют
одновременным омылением сложноэфирной группы [90]. С-за-
мещенные аиеталированные моносахариды не удалось проме-
тилировать обычными методами.
Бензилирование соединений V, VI, VII в межфазных
условиях бензилхлоридом по методике, аналогичной приведенной
выше, протекает значительно медленнее, чем метилирование:
от 10—12 ч (VII) до 34—36 ч (V, VI). Выходы
соответствующих бензиловых эфиров составляют 68—98%. Соединение VI
образует смесь соответствующих аномеров, разделенную
колоночной хроматографией, ot-аномер был получен в качестве
основного продукта (выход 81%), (3-аномер образуетси в очень
небольших количествах (10%). О-Метилированпе п О-бензплп-
рование Сахаров с двумя и тремя незамещенными гпдроксиль-
ными группами протекает значительно более сложно [90, 91].
На примерах 3-О-метил-1,2-О-циклогсксилидсн-а-£-глюкофура-
нозы XIa (R=Me, R' = H) и 5,6-ди-О-метил-1,2-О-циклогексп-
63
лиден-а-й-глюкофуранозы Х1б (R = H, R' = Me) было
найдено, что проходит ступенчатое О-алкилирование.
R'O-CH2
Метилирование Х1а диметилсульфатом при использовании
эквимольных количеств субстрата и алкилирующего агента
приводит к полиостью метилированному производному, выход
которого зависит от количеств используемого ТЭБАХ: с 2%
ТЭБАХ выход 60%, а с 10% ТЭБАХ --77%. В обоих случаях
реакция заканчивается за 10 мин. Кроме основного продукта
получается еще два минорных продукта, содержащих гидро-
ксильную группу, однако эти продукты не изучены. При
метилировании XI6 с использованием 10% ТЭБАХ выход триме-
тильного производного за 6 ч составил 85%, снижение
количества катализатора привело к уменьшению выхода до 66%,
увеличение количества диметилсульфата или
продолжительности реакции не повысило выхода. В этом случае также
образуются частично метилированные минорные продукты.
" Межфазное О-бензилированиеХ1а и XI6 по обычной методике
(см. выше) в присутствии 2% ТЭБАХ через 30 ч дало ЗДб-три-
О-бензил-1,2-О-циклогексилиден-а-£-глюкофуранозу с выходом
около 14%, одновременно образовалось два неидентифициро-
ванных продукта частичного О-бензилирования. Лишь переход
к методике экстрактивного алкилирования (метод экстракции
иоиных пар по Брёндстрёму [92]), т. е. при использовании эк-
вимольного по отношению к субстрату количества ТЭБАХ,
удалось увеличить выход до 41% при продолжительности реакции
10 ч [90].
В случае незащищенного D-маннита проходит
многоступенчатое О-метилирование с образованием сложной смеси шести
соединений. О-Бензилирование в этом случае вообще не идет.
Для некоторых моносахаридов с двумя незамещенными ОН-
группами удалось наблюдать четкие различия в реакционной
способности гидроксильных групп в зависимости от их
положения. Так, бензилирование а-метил-4,6-О-бензилиден-О-глюкопи-
раиозида бензилбромидом в присутствии ТБАГС при эквимоль-
ном соотношении реагентов приводит к 2,3-дибензильному
производному с выходом 6%, 3-бензильному производному с
выходом 2Ос/о и 2-бензильному призводному с выходом 54%,
т. е. положение 3 активнее положения 2 [91].
Обобщая изложенные выше данные, можно прийти к
заключению, что межфазный катализ следует рекомендовать для
64
О-алкилирования производных Сахаров, устойчивых к действию
щелочей и малорастворимых в воде.
Межфазный метод алкилирования очень удобен для алкили-
рования фенолов. Разработано несколько методик
алкилирования.
ArOH + RX —>- ArOR
Ar, RX (выход эфира, %): Ph; Mel (95); Ph, CH3=CHCH2Br (77); Ph,
H2C CHCH2C1 (77); Ph, н-BuBr (85); Ph, цикло-С5НцВг (73); Ph,
О
PhCH2CI (86); Ph, PhCH(Me)Br (91); Ph, BrCH2CO2Et (86); 2-PhCsH4,
Me2SO4 (81); 2-HOC6H4, Me2SO4 (78); 2-HOC6H4, Et2SO4 (83); 2-HO2CC6H4,
Me2SO4 (78); 2-HO2CC6H4, Et2SO4 (85); 2-O2NC6H4, Mel (81); 3-O2NC6H4,
Mel (79); 4-O2NC6H4, Mel (83); 4-трег-ВиСв1Ц, Mel (96); 4-rper-BuC6H4,
CH2=CHCH2Br (77); 4-трет-ВиС5Н4, PhCH2Br (83); 4-MeOC6H4, Me2SO4 (91);
l-CI0H7, H2C CHCH2C1 (42); 2-Cl0H7) Mel (92); 2-C10H7, Mel (92); 2-Ci0H7
\/
О
Me2SO4 (92); 2-Ci0H7, Et2SO4 (81); 2-C,oH7, H2C CHCH2C1 (41); 2,6-Me2C6H3,
\/
О
Me2SO4 (92); 2,6-Меа-4-т/мгг-ВиСвН2, Me2SO4 (79); 2,6-(MeO)aCeH3l Me2SO4 (91);
2,6-нз0-Рг2СвН4, Me2SO4 (84); 2,6-r/>er-Bu2CsH3 (83); 2,6-т/>ет-Ви2-4-МеСбН2,
Me2SO4 (84); 2,6-трет-ВД-4-МеОСбН2) Me2SO4 (85); 2,4,6-r/>er-Bu3CeH2,
Me2SO4 (93)
Общая методика алкилирования фенолов [93]. Смесь
50 мл дихлорметана, 50 мл воды, 10 ммоль фенола, 15 ммоль NaOH, 20—
30 ммоль алкилирующего агента, 0,1 — I ммоль катализатора перемешивают
2—12 ч в вибромиксере при комнатной температуре. Органический слой
отделяют, водный слой экстрагируют дихлормстаном. Объединенный
органический слой промывают водой, щелочным раствором и вновь водой до
нейтральной реакции. Выделяют алкилфенолы после отгонки растворителя
перегонкой или кристаллизацией. В качестве катализатора использовали бензил-
три-«-бутиламмонийгалогеиид.
Методика применима для широкого круга фенолов с элек-
троноакцепторными и электронодонорными заместителями, для
нафтолов и пространственно затрудненных фенолов; в
большинстве случаев при использовании различных алкилирующих
агентов (метилиодид, аллилбромид, эпихлоргидрин, циклопен-
тилбромид, w-бутилбромид, бензилхлорид, 1-бром-1-фенилэтан,
бромуксусный эфир, диметилсульфат, диэтилсульфат) выходы
эфиров фенолов высоки (73—95%), лишь в случае 1- и 2-наф-
тола при алкилировании эпихлоргидрином выход снижается до
41—42%.
Разработан метод метоксилирования фенолов Ди(хлормети-
ловым) эфиром, отличающийся простотой и высокими
выходами метоксимстиловых эфиров [94].
Общая методика метоксилирования фенолов. Смесь
раствора I ммоль фенола в дихлорметаие и I—2 ммоль NaOH в воде
перемешивают при комнатной температуре в присутствии 0,1—0,2 ммоль адоге-
на 464, добавляя по каплям в течение 25 мин 3—5-кратиый избыток ди(хлор-
метилового) эфира, разбавляют водой, выделяют эфир из органического слоя.
65
Таким путем получены, например:
RO О
II
R
OCH2Ph
о
(95%)
Предложен еще один метод межфазного синтеза метоксиме-
тиловых эфиров фенолов f95].
Смесь 18 ммоль фенолята калия, 2 ммоль 18-крауиа-6 в 50 мл сухого
адетонитрила перемешивают 30 мии при комиатной температуре, затем
добавляют 26 ммоль ди (хлорметилового) эфира и перемешивают 1 ч.
Таким способом с выходом 79—82% получен эфир
-СОМе
ОСН2ОМе
По аналогичной методике перемешиванием фенола с пента-
фторбензилбромидом, К2СО3 и 18-крауиом-6 в ацетонитриле или
бензоле был получен с количественным выходом
соответствующий эфир [96].
Алкилирование нафтолов, р-дикетонов и других
карбонильных соединений, способных образовывать амбидентные ионы,
может осложняться образованием не только эфиров нафтолов
Или енолов, но и С-алкильных производных. Так, при метили-
Таблица 12. Влияние растворителя и наличия катализатора (ТБАБ)
на направление метилирования нафтола-1
Р|стврритель
(% к вбдйой фазе)
Соотношение продуктов
О- и С-алкилирования
(XIII :XIV)
с ТВАБ
без ТБАБ
Общий выход (ХШ + ХГУ),
с ТБАБ
без ТБАБ
СНаСЬ (10—50%)
СНСЬ (50%)
СвН6 (50%)
100:0 а
75:25
95:5
10:90 б
100 а
85
90
70
а Аналогичные результаты получены прн использовании в качестве растворителя
С6П5Ш2 (50%).
" Аналогичные результаты получены при использовании CgHg (50%) или
(50%).
66
ровании нафтола-1 XII
О"
ОМе
хш
XIV
образуются продукты О- и С-метилирования (XIII и XIV),
соотношение которых и общий выход зависят от применяемого
растворителя и наличия катализатора [97] (см. табл. 12).
Введение катализатора межфазного переноса сдвигает
реакцию в сторону О-алкилирования вплоть до полного
исключения процесса С-алкилирования, повышает выход продуктов ал-
килирования и, кроме того, значительно сокращает время
реакции; в условиях межфазного катализа реакция заканчивается
за 20 ч вместо 5 дней.
Алкилирование 4-гидроксихинолинов диметилсульфатом, ди-
этилсульфатом или алкилбромидами (BuBr, /i-CI0H2[Br) в
присутствии 2 н. NaOH и 40%-ного раствора Bu4NOH в ССЦ или
PhCl при 40°С сопровождается изомеризацией
4-гидроксихинолинов в хинолоны-4 [98] и N-алкилированием:
\П/
R'
R
RX или
—. >
(RO}2SO2
Соотношение образующихся продуктов зависит прежде всего
от наличия и положения заместителей (см. табл. 13).
В незамещенном 4-гидроксихинолине алкилирование диэтил*
сульфатом проходит предпочтительно по атому азота с
изомеризацией в хинолон-4.
Введение заместителей в положение 3 либо не изменяет
соотношения образующихся XV и XVI (например, при R" = С1
или Вг), либо сдвигает его в пользу XV (при R// = Me, I),
причем в ряде случаев единственным направлением становится N-
алкилирование (например, при R" = CO2Et или NO2). Это
можно объяснить возникновением пространственных затруднений 0-
алкилированию или связыванию ОН-группы водородной связью.
При введении метильной группы в положение 8 или 2
единственным направлением реакции становится О-алкилирование.
Это легко объясняется созданием пространственных
затруднений у атома азота. Однако на направление алкилирования,
очевидно, влияет и природа заместителя. Так, при наличии в
положении 8 электронодонорной группы МеО преимущественно
67
ТаЗлица 13. Влияние заместителей на направление алкилирования
4-гидроксихинолинов диэтилсульфатом [98]
40%-иый раствор Bu4NOH в СС14 или PhCIj 2 н. NaOH; 40 °С
R'
Содержание
в смеси, %
XV
XVI
Выход, %
XV
XVI
н
Me
Н
н
н
н
н
н
н
н
Me
Н
Me
Н
н
Me
Н
С1
Вг
I
CO2Ef
NO2
н
С1
н
Me
Н
н
н
Me
Н
Н
Н
Н
Н
С1
н
ОМе
Н
65
0
92
0
56
59
76
100
100
0
0
76
5
35
100
8
100
44
41
24
0
0
100
100
24
95
55
0
86
0
54
58
66
98
87
0
0
75
5
32
87
8
92
43
37
26
0
0
87
94
22
70
идет N-алкилирование с изомеризацией гидрооксихинолина в
хинолон. При наличии двух метальных заместителей в
положениях 2 и 3 проходит предпочтительное О-алкилирование.
Направление алкилирования зависит, по-видимому, также
от природы алкилирующего агента. Так, алкилирование, 2,3-ди-
метил-4-гидроксихинолина диметилсульфатом дает смесь 48%
XV и 52% XVI (R = R' = Me), в то время как алкилирование
диэтнлсульфатом приводит к смеси, содержащей 5% XV и 95%
XVI (R = R' = Et). Влияние природы алкилирующего агента
при алкилировании 4-гидрокси-З-хлорхинолина показано в
табл. 14.
Алкилирование фенолов, содержащих не способные к еноли-
зации кетогруппы, проходит без осложнений, например [99]:
-COCH=CHPh
4*
PhMeaNCI
CH2CI2 — H5O,
50%-ный NaOH
(22-94%)
Алкнлирование в условиях межфазного катализа может
служить для синтеза эфиров енолов. В качестве алкилирующих
агентов могут быть использованы очень чувствительные к
щелочам а-хлорэфиры.
PhCH3COR + ClCH3OR' —► PhCH=C(R)—OCH3OR'
R, R' (выход, %): Ph, Me (55); Ph, wso-Pr (60); Pfc w-Bu (56); Me, Me (26);
Me, нзо-Рг (25); Me, k-Bu (36)
Общая методика получения эфиров енольных форм
фенилацетоиа и дезоксибензоина [100]. 0,05 моль кетона и
0,1 г ТЭБАХ в 25 мл 50%-ного NaOH интенсивно перемешивают и добавляют
по каплям (так, чтобы температура не превысила 30 °С) 0,06 моль а-хлор-
эфира. Смесь перемешивают 1 ч, разбавляют водой, извлекают бензолом
(2X16 мл). Органическую часть промывают водой, сушат (MgSO,*),
растворитель удаляют, остаток перегоняют в вакууме.
Помимо эфиров енолов в случае дезоксибензоина образуется
1,3-дибензоил-1,3-дифенилпропан: за счет деалкоксилирования
побочно образующегося продукта С-алкилирования — а-алко-
ксиметилдезоксибензоина в а-бензоилстирол и последующего
присоединения дезоксибензоина (выход 1,3-днбснзоил-1,3-дифе-
нилпропана составляет 5%).
Описан интересный случай одновременного алкилирования
по гидроксигруппам енольной формы гидроксикстона дитозила-
том в системе бензол — вода в присутствии ТБАБ и NaOH с
образованием краун-эфира [79]:
PhCHCOPh + TsO(CH2)2O(CH2)2OTs
OH
Метод межфазного катализа был успешно использован
также для синтеза некоторых полициклических фенолов.
Предложенная в работе [101] методика основана на алкилировании
циклических кетонов с одновременной их ароматизацией прн
переходе в енольную форму.
Синтез 9-метоксиантрацена. Раствор 1 г антрона и 1,9 г ди-
метилсульфата в 25 мл дихлорметана добавляют при интенсивном
перемешивании к раствору 8 г NaOH и 250 мг ТЭБАХ а 25 мл воды. Смесь
перемешивают 1 ч прн комнатной температуре, разбавляют 100 мл воды,
извлекают дихлорметаном. Органический слой промывают 10%-ной НС1, 5%-ным
NaHCOs, сушат (MgSO,i). Остаток после удаления растворителя
кристаллизуют из петролейного эфира (т. кип. 40—60°С). Выход 1,07 г (100% от
теоретического); т. пл. 97 "С.
Аналогично из акридона получают 9-метоксиакридон с
количественным выходом [101], а из 2,5-дигидро-5-метил-2-оксотио-
Таблица 14. Влияние природы алкилирующего агента на направление
алкилирования 4-гидрокси-З-хлОрхинолина [98]
400/о-иый раствор Bu4NOH в СС14 или PhCI; 2 н. NaOH; 40 °С
Алкнлирующий агент
Содержание в смеси, %
XV.
XVI
Выход, %
XV
XVI
Me2SO4
Et2SO4
w-BuBr
«-С10Н21ВГ
100
56
82
79
0
44
18
21
100
54
57
71
0
43
14
19
&9
фена в системе хлороформ — вода в присутствии ТБАГС 5-ме-
тил-2-метокситиофен с выходом 80% [102],
Использование 1,3-дибромпропана при алкилировании
позволило провести не только алкилирование енольной формы
аценафтенона, но и С-алкилирование [103].
Вг(СН2)зВг,ТЭБАХ
КаОН,ДМСО—Н2О
(41%)
Из приведенных примеров очевидно, что О-алкилирование
циклических кетонов с одновременной ароматизацией является
очень перспективным и заслуживающим внимания методом.
При реакции алифатических [104] или циклических [105]
кетонов с триметилэтилсилилацетатом [104, 105] или триме-
тилсилилхлоридом [106] в тетрагидрофуране [104, 105] или
ацетонитриле [106] в присутствии тетрабутиламмонийфторида
[104, 105] или тетраметиламмонийхлорида [106] образуются
триметилсилильные производные енолов. В случае /г-хинонов
получают производные фенолов (за счет ароматизации)»
Bu4NF
RCH=C(R')OSiMe3
ТГФ
(55-91%)
—(СН2)Л
я = 3, 4, б, 6, 10
MesSlCI
О *" Me3SiO
OSiMe§
(72-98%)
R = Н, Me, CI
Алкилкрование оксимов в бензоле в присутствии ТБАБ и
10%-ного NaOH приводит в зависимости от конфигурации ок-
сима к соответствующим эфирам оксимов или нитронам [107];-
н-ВиВг
PhCH=NOH > PhCH=NOBu-w+ PhCH=-N(O)Bu-«
сип (57,6%) (2,4%)
К-ВиВг
PhCH=NOH ■ »- PhCH=N(O)Bu+PhCH=NOBu
анти
(48%)
(12%)
Синтез сложных эфиров. Синтез сложных эфиров в условиях
межфазного катализа можно осуществить алкилированием
солей кислот галогенидами или ацилированием спиртов. Так,
70
удобная методика синтеза гс-бромфенациловых эфиров основана
на реакции а,гс-дибромацетофенона с калиевыми солями карбо-
новых кислот в ацетонитриле в присутствии каталитических
количеств дициклогексано-18-крауна-6 (твердая фаза — жидкость)
[108]:
rt-BrCsH4COCH2Br + RCO2K —► n-BrCsH4COCH3OCOR
R (выход, %): Н (93); Me (98); изо-Pr (98); к-Рг (97); w-C6HI3 (99);
трет-hn (96); Ph (93); 2-MeCsH4 (90); 2-ICeH4 (92); 2,4,6-Me3CeH2 (98);
4-rper-BuC6H4 (92)
Общая методика синтеза /г-бромфенациловых эфиров кар*
боновых кислот приведена ниже.
Синтез п-бромфенацилацетатов [101]. Перемешивают 100 мг
ацетата калия, 278 мг сс,л-дибромацетофеноиа и 15 мг дициклогексаио-18-
крауиа-6, кипятят 15 мин в 10 мл ацетонитрила. Растворитель удаляют,
остаток промывают бензолом и пропускают через колонку с силикагелем (4,6 г).
Выход л-бромфенацилового эфира уксусной кислоты 252 мг; т. пл. 85—86 °С.
Описан также стереоспецифический синтез ^мс-3,5-диацето-
ксициклопентена и синтез 4-ацетоксициклогексен-2-она реакцией
ацетата калия соответственно с ^мс-3,5-дибромциклопентеном и
4-бромциклогексен-2-оном в присутствии триоктилпропиламмо-
нийхлорида (ТОПАХ) [109].
Синтез рс-2,5-диацетоксициклопентеиа. Смесь 3 г
ацетата калия, 0,7 г ТОПАХ в 2 мл воды и 2 г ^(1С-2,5-дибромциклопеитена
в 6 мл СС14 интенсивно перемешивают при 49 °С в течение 9 ч, разбавляют
водой, извлекают эфиром. Эфирный слой высушивают и упаривают. Выход
1,33 г (75% от теоретического); т. кип. 69—70 °С (0,07 мм рт. ст.).
В присутствии аликвата 336 или 18-крауна-б и поташа в ди-
хлорметане или бензоле (т. е. в системе твердая фаза —
жидкость) перфторизогексен-3 реагирует с различными жирными
кислотами, образуя с высокими выходами соответствующие
сложные эфиры [110]:
OCOR
К2СО3 I
(CF3)2C=CF—C2F5 + RCO2H >■ (CF3)2G=C-C2F5
Одновременно образуются в небольших количествах продукты
присоединения кислот по двойной связи
OCOR
(CF3)2CH-CF-C2F5
Оригинальный метод синтеза карбонатов основан на алкн-
лировании сухого КНСО3 в смеси с К2СО3 галогенидами в
толуоле или высококипящем петролейном эфире в присутствии
аликвата 336 [ПО]:
KHCO3 + RCH2X —>■ RCH2OC(O)OCH2R
R (выход, %): K-CgHi, (72); «-CSH13 (74); «-C7H14 (71); «-C9H19 (78); h-QuH2S (в7)|
«-СцНл (83); Ph (85)1 2-MeC6H4 (81); 3-С1СбН4 (Щ
71
Общая методика алкилирования КНСО3. Смесь 10 г сухого
КНСОз, 14 г сухого КгСОз, 0,1 моль галогенида и 10 мл толуола нагревают
8—15 ч при 100 °С. Фильтрат упаривают, остаток разгоняют.
Метод позволяет получать также несимметричные эфиры
угольной кислоты. Для этого проводят сначала реакцию между
КНСОз и половинным количеством галогенида, а затем
добавляют К2СО3 и другой галогенид:
К2СО3, СбН,зВг
PhCH2Br+КНСОз —► [PhCH2OC(O)OH] ■>
—>■ PhCH2OC(O)OCeHi3
Для алкилирования солей или свободных карбоновых
кислот предложено использовать соли сульфония [111]. Например,
при перемешивании ацетата калия с «-гексилдифенилсульфоний-
перхлоратом (служит одновременно реагентом и
катализатором) в ацетонитриле при 20 °С в течение 17 ч образуется w-гек-
силовый эфир уксусной кислоты.
Ацилирование р-дикарбонильных соединений в условиях
межфазного катализа всегда приводит только к продуктам О-
ацилирования (тогда как в обычных условиях образуются, как
правило, продукты С-ацилирования или смесь О- и С-ацильных
производных) [113].
ТБАГС
RCOCHsCOR7 + R"COC1
СН2С12. NaOH
R4 /H R4 /COR'
)cc; + ;cc;
Я-изомер 2-нзомер
R, R', R" (конверсия, %; соотношение Е- н Z-изомеров): Me, Me, COMe (97;
55:45); Me, Me, COPh (99; .60:40); Me, CO2Et, COMe (83; 93:7); Me,
CO2Et, COPh (88; 96 ; 4)
Только в случае малонового эфира был получен с низким
выходом (~10%) продукт С-ацилирования. Более высокий выход
f-изомеров связан с тем, что анион в форме XVII, приводящей
к £-изомеру, более устойчив, чем в форме XVIII, приводящей
к Z-изомеру.
R R' О О
XVII XVIII
Наилучшие результаты достигнуты при использовании метода
экстракции ионных пар, т. е. при эквимольных количествах
ТБАГС и субстрата.
Имеется ряд примеров синтеза разнообразных ацетатов в
условиях межфазного катализа, в том числе алкил- и аллил-
ацетатов [66], бензилацетатов [66, 114, 115], циклоалкенилди-
72
ацетатов [116]. Наиболее интересны случаи получения
ацетатов, содержащих в эфирной группировке дополнительные
функциональные группы. Так, при действии ацетата калия на 1,2-ди-
бромэтан в присутствии каталитических количеств 18-крауна-6
в сухом ацетонитриле или бензоле (система твердая фаза —
жидкость) при 83 °С в зависимости от соотношений реагентов
образуется либо диацетат этиленгликоля с выходом 90%, либо
смесь 77% 2-бромэтилацетата и 23% диацетата этиленгликоля
[117]. В аналогичных условиях можно получить метилтиоме-
тилацетат с выходом выше 80%, а также метилтиометиловые
эфиры других кислот (выходы 85—97%) [118].
2-Гидроксиэтиловые эфиры ароматических кислот можно
получить также, используя в качестве алкилирующего реагента
2-хлорэтанол в присутствии ТБАБ [119].
В системе органический растворитель — ТБАХ — 30%-ный
NaOH первичные и вторичные спирты легко алкилируются ацил-
хлоридами, однако для алкилирования третичных спиртов
необходимо использовать систему твердая фаза — жидкость
[ТБАХ — КОН (или NaOH) — органический растворитель].
Следует отмстить, что даже в этих условиях не удается
получить бензоаты третичных спиртов [120].
Метод межфазного катализа применим не только для 0-
ацилирования производными карбоновых кислот, но и для
введения остатков других кислот, например сульфокислот или
фосфорных кислот. Описано, например, получение арилсульфонатов
с высокими выходами в системе бензол — вода или дихлор-
метан — вода в присутствии ТЭБАХ и NaOH при действии на
фенолы различных сульфонилхлоридов [121, 122]:
ArOH + RSO2CI —► RSO2OAr
Подбирая условия реакции, можно провести избирательное
сульфоиирование гидроксигруппы в 3-аминофеноле [123]. В
системе вода — PhCl — ТБАХ — NaOH (NaHCO3) образуется 3-
аминофенилбензолсульфонат с выходом 93—94%.
Однако наибольший интерес представляет межфазное О-то-
зилирование диольной группировки в моносахаридах. Как было
показано сначала на примерах ее- и р-метил-4,6-О-бензилиДен-
£)-глюкопиранозидов, при действии небольшого избытка w-to-
луолсульфонилхлорида на моносахарид в системе дихлорме-
тан — вода в присутствии ТБАГС и 5%-ного NaOH тозилиро-
вание осуществляется преимущественно по положению 2, в
результате из первого моносахарида получают 78% 2-О-тозилата
и только 7% 3-О-тозилата, а из второго — 55% 2-О-тозилата,
31% 3-О-тозилата и 7% 2,3-ди-О-тозилата [124]. В тех же
условиях из сс-метил-4,6-О-бензилиден-О-маннопиранозида
получают исключительно 2-О-тозилат с выходом 95% [124]. Было
подробно изучено тозилирование ряда О-циклогексилиденовых
производных моносахаридов с одним, двумя и тремя свобод-
73
ными гидроксилами [125]. В системе бензол — водная щелочь —
ТЭБАХ 1,2:5,6-ди-О-циклогексилиден-а-О-глюкофураноза
образует почти с количественным выходом 3-О-тозилат XIX.
В случае же 2,3 :5,6-ди-О-гексилиден-а-О-маннофуранозы
(свободный гликозидный гидроксил) образуется в качестве
основного продукта дисахарид XX, вероятно, в результате нуклео-
фильного замещения в первоначально образующемся реакци*
бнноспособном 1-О-тозилате анионом субстрата.
жпс
,О-0Н2.
о.
CqHjq
>с6ню
XX
Диолы — 3-О-метил-1,2-О-циклогексилиден-а-/)-глюкофура-
ноза и 3-О-бензил-1,2-О-циклогексилиден-а-^-глюкофураноза —
в условиях межфазного О-тозилирования дают дитозилаты с
выходами 63 и 35%, соответственно. При этом образуются
также эпоксиды XXI, вероятно, в результате межфазного
внутримолекулярного нуклеофильного замещения в первоначально
возникающих монотозилатах XXII. Монотозилат ХХПа был
получен по твердофазному варианту межфазного синтеза при
замене водной щелочи на твердый карбонат натрия [125].
Одновременно образуются дитозилат и эпоксид.
ch2ots
но—
CfiH
6ПЦ)
XXI
(а) Ц-Мв,(б) Я*
Межфазное О-тозилирование 1,2-О-циклогексилиден-а-О-
глюкофуранозы привело к образованию многокомпонентной
74
смеси, из которой с выходом 35% удалось выделить 3,5,6-три-
О-тозилат,
Общая методика межфазного О-тозилирования [125].
Бензольный раствор субстрата (100 мг/мл) встряхивают при комнатной
температуре с эквимольным количеством (по числу гидроксильных групп) л-то-
луолсульфонилхлорида, 2% (от количества субстрата) ТЭБАХ и равным
объемом 50%-ной водной щелочи. После окончания реакции (контроль с
помощью ТСХ) отделяют бензольный слой, водный слои несколько раз
экстрагируют бензолом. Объединенные бензольные слои промывают водой до
нейтральной реакции, встряхивают с равным объемом воды, содержащей
катионит КУ-2 (Н+) для удаления ТЭБАХ. После высушивания (Na2SO4)
бензольного слоя остаток хроматографируют на колонке с А13Оз или
кристаллизуют из подходящего растворителя.
Как и в реакции алкилирования, вторичные спиртовые
группы в сахарах ацилируются быстрее первичных. Скорость ацили-
рования снижается, если гидроксил участвует в образовании
водородной связи. Так, бензоилирование 1,4:3,5-диангидро/)-
глюцита в обычных условиях межфазного катализа приводит
к смеси дибензоата (12%), 5-О-бензоата (17%) и 2-О-бензоата
(40%) [126]. При тозилировании метил-3,6-ангидро-а-£-ман-
нопиранозида прежде всего реакция проходит по аксиальной
гидроксильной группе [126].
Известный интерес представляет межфазный способ фосфо-
рилирования спиртов и фенолов диалкилфосфитами в ССЦ,
который выгодно отличается от используемой стандартной
методики Этертона — Тодда высокими выходами фосфатов, чистотой
полученных продуктов, небольшим временем реакции (3 ч
вместо 12) [127].
(RO)2P(O)H + R'OH —> (ROhP(O)OR'
R, R'(b«xoa, %): Et, h-Bu(71); Et, CH2Ph (89); Et, азо-рг<37); Et, «-Bu(67);
Et, изо-Ви (56); Et, Ph (60); н-Ви, Et (71).; «-Bu, изо-Ви (62); н-Ви, PhCH2 (95);
rt-Bu, изо-Рг (36); «-Ви, Ph ($4)
Общая методика фосфорилирования спиртов. Раствор
0,25 моль (25%-ный избыток) диалкилфосфита в 60 мл ССЦ добавляют по
каплям при интенсивном перемешивании и внешнем охлаждении (баня со
льдом) к~смеси 0,2 моль спирта, 120 мл ССЦ, 60 мл 50%-ного NaOH и 1,6 г
[5% (мол.)] ТБАБ при 20—25СС. Перемешивают 3 ч при комнатной
температуре, разбавляют дихлорметаном (50 мл), фильтруют. Фильтрат
промывают 26 мл 2%-ной соляной кислоты, водой (2X25 мл), сушат (MgSO4),
растворители удаляют в вакууме (0,1 мм рт. Ст.) при 30—40 С. Получают
аналитически чистые фосфаты.
Описано алкилирование сухих калиевых еолей фосфониевых
кислот кипячением с силилгалогенидами в ацетонитриле в
присутствии 18-крауна-6 [128]:
RR'P(O)OK + R"X —> RR'PfOOR"
Синтез ацеталей. Очень удобен межфазпый катализ и для
синтеза некоторых ацеталей. Так был предложен очень
75
удобный метод метиленирования Двухатомных о-фенолов Дй-
бромметаном в условиях межфазного катализа [129]:
Этот способ пригоден не только для пирокатехина, но и для
других о-дигидроксиаренов. Методика крайне проста.
Общая методика метилинирования. Смесь 0,15 моль ди-
бромметана, 20 мл воды и 1 ммоль адогена 464 перемешивают и нагревают
до кипения, вытесняют воздух азотом и добавляют в течение 2 ч 0,1 моль
соответствующего дигидроксисоединения и 0,25 моль NaOH в 50 мл воды.
Олесь кипятят, перемешивая, еще 1 ч; выделяют продукт обычными
способами.
Таким путем из пирокатехина получен бензо-1,3-диоксолан
(выход 70%), из 2,3-дигидроксинафталина — нафто[2,3-а]-1,3-
Диоксолан (82%), из2,3-дигидроксибензальдегида — пиперональ
(80%), ' из 4-метилпирокатехина — 4-метилбензо-1,3-диоксолан
(86%) [129].
Близкая методика была разработана для получения ди(арил-
окси)метанов. Однако в системе жидкость — жидкость реакция
идет медленно; наиболее удовлетворительные результаты
получают в системе твердая фаза — жидкость в дихлорметане
(реагент и одновременно растворитель) в присутствии
измельченного КОН и катализатора межфазного переноса:
АгОН+СН2С12 —> (АгО)2СН2
g этом случае более активным является аликват 336, а не
ТЭВАХ. Реакция ведется при кипячении. Для фенола, алкил-
фенолов и р-нафтола выходы ди(арилокси)метанов составляют
80—97°/^, для пирокатехина—всего 39%, так что в этом случае
более выгодна приведенная выше методика с дибромметаном
[130]:
Метод пригоден и для получения формалей из линейных
спиртов (к-бутанол, бензиловый спирт) и циклических
спиртов (циклогексанол), выходы составляют 65—79%^ [130].
Метиленирование в условиях межфазного катализа было
распространено также на моносахариды. Таким путем
получены, например, следующие соединения [131]:
,.о-сп2
Я
Н,ОМе КО >(н,ОМе
76
Метиленирование осуществлялось перемешиванием смеси
углевода, дибромметана, катализатора (ТБАБ) и 50%-ного NaOH
прн 60—65°С в течение 0,5—2 ч. Выходы составляют 58—65%.
Аналогичная методика (катализаторы ТБАБ и ТЭБАХ)
была применена с хорошими результатами для синтеза ацеталей
из ряда гексоз и дибромметана в присутствии ТБАБ или ТЭБАХ
[132]. Например, таким путем был получен следующий ацеталь
(выход 84—86%):
"Ме2С
СМе.
СН5
Следует отметить, что природа катализатора в данном случае
не влияет на выход ацеталя.
В системе дихлорметан — вода при использовании ТБАГС в
присутствии NH4OH образуются ацилали формальдегида [133]:
2RCOOH+CH2C12 —> (RCO2)2CH2 (60-87%)
Синтез ортоэфиров. Наконец, межфазный катализ в
стандартных условиях был применен для синтеза триэтил- и три-
фторэтилортоэфиров:
CHC13 + ROH —> CH(OR)3
R = Et, CF3CH2
Общая методика синтеза ортоэфиров приведена ниже.
Синтез этилового эфира ортомуравьиной кислоты
[134]. К смеси 198 г хлороформа, 80 мл 50%-ного NaOH и 1,1 г ТЭБАХ
добавляют по каплям 23 г абсолютированного этанола, поддерживая
температуру ие выше 15—20°С (хорошее охлаждение), перемешивают при комнатной
температуре 1 ч, выливают в воду со льдом, отделяют органический слой,
водный дополнительно экстрагируют хлороформом. Объединенные хлорофор-
менные растворы промывают водой до нейтральной реакции, сушат (MgSC>4).
Выход 9 г (36% от теоретического); т. кип. 140—-145 СС.
К сожалению, другие спирты — метиловый, н-пропиловый,
изопропиловый, трег-бутиловый, вдклогексиловый— не
образуют ортоэфиров в данных условиях [134].
Таким образом, межфазный катализ может быть успешно
использован для синтеза разнообразных продуктов О-алкили-
рования (арилалкилирования, арилирования) спиртов,
карбонильных соединений и т. п.
Образование С—S-связей
В присутствии межфазных катализаторов можно получать
разнообразные сульфида взаимодействием тиолов или-их
щелочных солей с галогенидами (в случае тиолов в присутствии
77
щелочей) в двухфазной системе жидкость — жидкость или
твердая фаза — жидкость. Таким путем можно синтезировать ди-
алкилсульфиды, например [135]:
w-C8H17SH
алнкват 336, 50%-ный NaOH
«-C8Hi7SEt (95%)
Аналогично были получены арилалкилсульфиды [29, 136]
—■> w-C8H17SPh (100%)
Так же, но с использованием ТБАБ или ТЭБАХ и NaOH в
системе бензол — вода были приготовлены разнообразнейшие ал-
кил(арил- или гетероарил)сульфиды [136—139]. Для
иллюстрации можно привести синтезы 2-алкил (аллил или арил)тиотиа-
золов и бис (тиазолил-2) сульфида [137]:
R'
SH
RBr
R'-
R
—SH + Br
\/
S
N
— SR (55-90%)
'$—(/ (40%)
S
Хорошие результаты были получены также при алкилиро-
вании 2-меркаптобензотиазола (выходы достигают 70—94%),
2-меркапто-4,5-Дигидротиазола (выход 85—90%) [137], Был
осуществлен синтез 1,2-бис(бензотиазолилтио)этана с выходом
90% [137]:
SCH2—
Описано алкилированне в присутствии ТЭБАХ (NaOH,
бензол — вода) 2-меркаптопиридина в 2-алкилмеркаптопиридин
(выход 70—81%), 2-меркаптопиримидина в 2-алкилтиопирими-
дин (выход 72—95%), 2-меркаптобензоксазола в 2-алкилтио-
бензоксазол (выход 57—82%) [138].
Интересный способ синтеза хлорметилгетероарилсульфидов
основан на алкилировании соответствующих тиолов (2-меркап-
тобензотиазол и -бензоксазол, 2-меркаптопиримидин, 2-меркап-
тохинолин, 2,5-димеркаптотиадиазол, 3,5-димеркаптоизотиазол)
бромхлорметаном в присутствии КОН и ТЭБАХ (сиетема
твердая фаза — жидкость) [139], например:
SCH2CI (58-97%)
Алкилирование амбидентной гетероциклической системы,
где возможно направление реакции по двум реакционным
центрам, было проведено впервые на примере 2-тиоксо-2,3-дигидро-
имидазола и его N-метилпроизводного. Установлено, что в
межфазных условиях алкилирование проходит по атому серы [140]:
N-N
C1CH2S
SCH2CI (78%)
.— SR'
(выход, %): Et
цикло-С5Н5 (55);
(90);
C
/t-Рг (68);
5 (15);
изо-Pr (68); w-Bu (82);
2—СНСН2 (65); PhCH2
R = Me; R'
трет-Ви (0);
(54—57)
Общая методика алкилирования Ьметнл-2-тиоксо-
2,3-дигидроимидазола. К 150 мл бензола добавляют при
перемешивании 15 мл 40%-ного водного раствора NaOH, 5,7] г 1~метил-2-тиоксо-2,3-ди-
гидроимидазола, 0,05 моль RX и 0,97 г ТБАБ, перемешивают 6 ч при 60 °С.
Органический слой отделяют и сушат (молекулярные сита), растворитель
удаляют в вакууме, остаток перегоняют или кристаллизуют нз спирта.
При алкилировании 2-тиоксо-2,3-дигидронмидазола 1-бром-
бутаном (2 экв) получают 1-бутил-2-бутилтпоимидазол с
выходом 80% [140].
Интересно, что образование связи С—S возможно не только
при реакции между тиолами и галогенидами, но и при реакции
между тиоцианатами и соединениями с достаточно активными
атомами водорода. Так, в присутствии ТЭБАХ и NaOH в
системе вода — хлороформ тиоцианаты реагируют с хлороформом
или фенилметилацетонитрилом, образуя соответствующие
сульфиды [141]:
RCSN+CHC13 —> RSCCb (61-86%)
Ph, C6HnNC(CH3)3, С1(СН2)з
CN
PhSCN+PhCHCN
Me
PhC— SPh (68-77%)
Me
Алкилгалогениды способны алкилировать О-алкилдитиокар-
бонаты по атому серы в межфазных условиях с образованием
О-алкил-Б-алкилдитиокарбонатов, гидролиз которых приводит
к алкантиолам [142]. Этот способ был предложен как общий
метод синтеза алкантиолов с высокими выходами:
RX-f-KSC(S)—ОВи-трет
[RSC(S)—OBu rper]
RSH
R, методика (выход, %); С5Нц(Ме)СН, Б (72); и-С8Н:7, А (из бромида) (87),
79
Б (из хлорида) (65); С6Нп(Ме)СН, Б (из бромида) (78); м-СюН21, А (из
бромида) (88), Б (из хлорида) (60); «-Ci6H33, А (из бромида) (87]
Общая методика синтеза алкаитиоловА. Смесь 50 ммоль
алкилгалогенида, 55 ммоль К-соли О-грет-бутилднтиокарбоната, 1,5 г аликва-
та 336 и 50 мл воды перемешивают при комнатной температуре до появления
слоя желтого масла сверху и обеспвечивания водного слоя (10—15 мин).
Температуру смеси медленно повышают до 75—80 °С> перемешивают при этой
температуре 15—20 мин до окончания выделения изобутена. После
охлаждения добавляют 100 мл петролейного эфира, органический слой отделяют,
промывают водой, сушат фильтрованием через слой силикагеля, удаляют
растворитель, перегоняют остаток.
Б. Смесь 50 ммоль алкилгалогенида, 1,5 г аликвата 336, 50 мл воды
нагревают до 50 °С, добавляют малыми порпиями при перемешивании в
течение 1—2 ч 90—100 ммоль К-солн О-грет-бутилдитиокарбоната (в случае
алкилбромидов; при использований хлоридов 150—160 ммоль), каждый раз
дожидаясь обесцвечивания раствора. После введения всей солн перемешивают
еще 30 мин, далее нагревают до 75—80°С и поступают, как описано в
методике А. ,
Через О,5-диалкилдитиокарбонаты получают также
несимметричные диалкилсульфиды [143]:
аликват 336 КОН
RX + KSC(S)OR' 20иди70ОС> RSCOOR' -^ RSR'
RX, R' (выход диалкилсульфида, %): tt-CJ0H2iBr, Et (80); н-С|6Н33Вгт Et (73);
PhCHaCl, Et (71); HrCeHjsCHfMeJBr, Et (71); H-CJ2H25Br, Me (73); Mel,
«-С8Н17(34); EtI, h-C8HJ7(47); H-C10H2JBr, h-C8Hi7 (86); иэо-PrI, «-C8H17(60);
EtI, m-CJ0H2i (37); «-C8HJ7Br, h-C10H2i (85); H-C8HJ7Br, изо-Рг (25)
Типичная методика приведена ниже.
Синтез н-октилэтилсульфида [143]. Смесь 9,65 г м-октилбро-
мида, 8,33 г К-соли-О-этилднтиокарбоната, 1,68 г аликвата 336 и 50 мл воды
энергично перемешивают 5 мин при 70 °С, охлаждают до 50 °С, добавляют
14 г КОН (в виде таблеток), так чтобы температура при добавлении не пре*
вышала 80°С (2—5 мин), нагревают 30 мин при 80°С, разбавляют 100—
150 мл петролейного эфира, отделяют органический слой, сушат
фильтрованием через слой силикагеля, разгоняют. Выход октилэтилсульфида 7 г; т. кип.
108°С (16 мм рг. ст.); п" 1,4565; выход дноктнлсульфида 0,8 г; т. кип. 170—
172 DC (2 мм рт. ст.); я|,в 1,482а
Описанный выше метод был использован для синтеза самих
О.Б-дналкилдитнокарбонатов нз алкнлгалогенидов или алкил-
мезнлатов [145]. Реакцию проводят аналогично приведенной
выше методике для синтеза несимметричных диалкилсульфи-
дов, смесь обрабатывают петролеиным эфиром и выделяют O,S-
диалкнлднтиокарбонаты после отгонкн растворителя разгонкой
или кристаллизацией из смеси ацетона с метанолом.
RX + KSC(S)OR' —► RSC(S)OR'
RX, R' (выход, %): «-C8H17Br, Me (60-70); «зо-PrI, Et (88); н-BuBr, Et (91);
H-BuOSO2Me, Et (85); «-C8HJ7Br, Et (91-93); h-C8HJ7I, Et (82); w-C8HJ7CI,
Et (85-90); H-C8HJ7OSO2Me, Et (47-53); C8H13CH(Me)I, Et (80-81);
CeHisCHMeCl, Et (60-68); CH2=CHCH2CI, Et (91); PhCH2CI, Et (90); EtI,
h-C3HJ7 (81); н-С1еН33Вг, изо-Vx (100); K-C8HJ7Br, трет-Вп (93)
30
При реакции тиолов, содержащих в вицинальном
положении гидрокснгруппу, с дигалогенметанами образуются гетеро-
цнклы, содержащие серу и кислород. Например, дибромметан
реагирует с о-меркаптофенолами в присутствии NaOH и алик-
вата 336 по схеме [146]:
S
> (70-81%)
В заключение отметим, что в условиях межфазного
катализа тиолы, аналогично фенолам, могут реагировать с дихлорме-
таном, давая тиоацетали с очень высокими выходами (90—
96%) [70]:
2RSH+CH2C12
аликват 336, NaOH
Н2О
трет-Вп, Ph
CH2(SR)2
Образование С—N-связей
Метод межфазного катализа за последние годы очень широко
используют для N-алкилирования. Отмечены случаи алкилнро-
вания аминов. Так, дифениламин образует бензнлдифениламин
при действии бензилхлорида в системе ДМСО (или гексамета-
пол) — вода — NaOH в присутствии ТЭБАХ [147] с выходом
74 %. Очень часто межфазный метод применяют для алки-
лирования разнообразных амидов. В присутствии
каталитических количеств аликвата 336 и 50%-ного NaOH можно,
например, проалкилировать цианамид [148]:
H2NCN + RX
RNHCN
RX (выход, %): PhCH2Cl (96); СН2=СНСН2С1 (91); н-BuBr (80)- EtBr (75);
Ме2СНСН2СН2Вг (55); Ме2СНВг (54); МеВг (32); МеООШ (71);
м-ВиОСН2С1 (72)
При избытке галогенида образуются диалкильные
производные. Введение в реакцию а,ш-дигалогеналканов приводит к
образованию гетероциклов:
СН2
H2NCN + ВгСН2(СН2)«СНгВг*
п (выход, %); 2 (55); 3 (75); 4 (25)
NC—
СН2
81
В случае 1,2-дибромэтана и 1,3-дибромпропана основной
реакцией является элиминирование НВг. Из о-ди(бромметил)
бензола получают 2-циано-2,3-дигидроизоиндол с выходом 50%
[148]:
H2NCN
Алкилирование ацетанилидов или форманилидов гладко
проходит в системе бензол — 50%-ный NaOH — ТЭБАХ при
использовании избытка алкил(или бензил) галогенида или диал-
килсульфата (25—200%) [149] при комнатной температуре или
60—80 °С:
PhNHAc-f RX —5. PhN(R)Ac
RX (выход, %): Me2SO4 (84)г Et2SO4 (90); н-РгВг (82); н-Bul (82);
PhCH2Cl (95)
Следует отметить, что алкилирование метилформамида ди-
метилсульфатом не требует добавок ТЭБАХ [149].
Интересный метод синтеза замещенных формамидов основан
на алкилировании вторичных аминов (с одновременным
гидролизом) [150, 151] хлороформом в присутствии ТЭБАХ и
50%-ного NaOH:
RR'NH+CHCb —* RR'NCHO
R, R', методика (выход, %): Me, Ph, A (77,5); Et, Ph, A (87,5); —C5H10—
Б (79); ызо-Рг, изо-Pr, Б (45); изо-Bu, изо~Ви} Б (70); цикло-С6Н)ь цик-
ло-СвНи, А (27,6); к-С5Нц «-СБНц (90,5); Et, Et, Б (62); СН2 = СНСН2,
СН2=СНСН2, А (60); Ph, Ph, A (68)
Общие методики синтеза замещенных формамидов
приведены ниже.
А. Синтез метилфенилформамида. Смесь 10,7 г N-метилани-
лина, 50 мл хлороформа, 30 мл 50%-ного NaOH и 0,2 г ТЭБАХ при 50 .°С
перемешивают 5 я, разбавляют водой, отделяют органический слой,
промывают последовательно 2%-иой соляной кислотой и водой, сушат (MgSOJ,
разгоняют. Выход 10,5 г; т. кип. 122—123 °С (8 мм рт. ст.).
Б. Синтез N.N-диэтил формами да. Смесь 10 г диэтиламииа,
50 мл хлороформа, 40 мл 50%-ного NaOH и 0,3 г ТЭБАХ перемешивают 5 ч
при 40—50 °С, отделяют органический слой, фильтруют его через силикагель
и перегоняют. Выход 8,5 г; т. кип. 177—178 °С.
Формилирование в межфазных условиях было использовано
также для синтеза полициклических N-формильных производи
иое (см. верхние реакции на с. 83) %
82
СНС1з
r\
^5s NaOH, ТЭВАХ
C8Il9-H2O
CHCI3 (изб.)
(95%)
[152]
(60%)
NCHO
(66%) [153]
Синтез дизамещенных формамидов осуществляют алкили-
рованием монозамещенных формамидов [154].
RR'R"C—NH—CHO
R'"
RR'R"C-~ N—
R, R', R", R'" (выход, %): Me, Me, Me, Me (70); Me, Me, Me, k-Bu (92);
Me, Me, Me, PhCH2 (76); Ph, Me, H, Me (84); Ph, Me, H, h-Bu (91); Ph, Me,
H, PhCH? (87); PhCH2, Me, Me, k-Bu (98); PhCH2) Me, Me, PhCH2 (9);
— (CHa)4—, H, Me (87); — (CH2)4—, H, k-Bu (89); — (CH2)4—, H, PhCH2
(83,5)
Общая методика формилирования N-алкилформами-
дов [154]. Смесь 0,1 моль N-алкилформамида, 14 г порошкообразного NaOH,
8 г К2СО3, 3,4 г ТБАГС и .80 мл бензола перемешивают 30 мин при 35—40 °С.
Полученную кашнцу Na-соли нагревают до 60 °С и добавляют в течение 1 ч
алкилирующий агент [Me2SO4 (0,2 моль) или алкилгалогенид (0,11 моль)]
в 40 мл бензола, нагревают 4 ч при 60—70 °С. После охлаждения смесь
разбавляют 50 мл бевзола, фильтруют, осадок промывают бензолом (2X30 мл).
Беизол удаляют, кристаллический остаток сушат при 30—40 °С и 0,2 мм рт. ст.
в течение 1 ч. Получают практически чистые К^-диалкилформамиды.
Это методика очень удобна для синтеза несимметрично
замещенных формамидов.
Аналогичная методика была применена с хорошими
результатами для получения М,Ы-дизамещенных амидов из
монозамещенных амидов пропионовой или бензойной кислот [155]:
RCONHR' + R"X —-* RCONR'R"
R, R', R" (выход. %): Et, PhCH2, Et (73); Et, PhCH2) н-Bu (82); Et, PhCH2.
PhCH2 (96); Ph, Et, Et (88); Ph, Et, h-Bu (85); Ph, Et, PhCH2 (86)
83
Удобный способ синтеза а-метилен-Р-лактамов основан на
внутримолекулярном N-алкилирования З-бром-2-бромметилпро-
пионамидов с одновременным элиминированием НВг [156]:
ВгНгС^ СН2=—г=о
BrH2c
chconhr
| I
R (выход, %): Ph (86); /г-МеОС6Н4 (96); «-O2NC6H4 (83); 1,3-С12С6Н3 (78);
3,4-С12С6Н3 (92); 2,3,5-Вг3СбН3 (82); 1,3-Ме2СвН3 (92); «-NCC6H4 (76); Et (68);
м-Bu (56); цикло-С6Нц (40)
Реакция проводится в системе 40%-нын NaOH — СС14 (или
CH2CI2) в присутствии пентилтриэтиламмонийбромида при
20 °С. Если использовать в качестве растворителя хлороформ,
образуется продукт дихлорциклопропанирования метнленлакта-
ма с выходом 48% [156]:
ВгН2Сч^ . снс1з С1\/\
г 2 —NPh
С алифатическими N-алкиламидами побочно образуются
простые эфиры типа [CHf = C(CONHR)CH2]2O [156].
Незамещенные |3- н ^-лактамы в условиях межфазного
катализа могут алкилироваться по азоту. Описано, например, ал-
килирование в системе твердая фаза — жидкость (КОН и ТГФ
или ацетонитрил) в присутствии ТБАБ [157]:
В стандартных условиях по Макоше (вода — бензол, NaOH,
ТЭБАХ) проходит метилирование пирролидона-2 в 1-метилпир-
ролидон-2 с выходом 53% [158], а также алкилирование или
бензилнрование высших лактамов [158]:
(СЩа (15-73%)
NR
«=1, 3, 5; R = Alk, PhCH2
Описано алкилированне 5,5-диметилоксазолидинона-З в
системе бензол — вода — NaOH — ТБАБ [159]:
О О
'Me RBr ) \ /Me
" ^"ч X (39-78%)
О
84
Ряд работ посвящен аЛкнлированию фталимиДа калия в
системе твердая фаза — жидкость (54, 160—162].
(80-99 %)
R = Alk, PhCH2, цикло-СбНп; X = Cl, Br, I, OMs, OTs
Типичная методика алкнлирования фталимида калия
приведена ниже.
Синтез N-октилфталимида. Смесь 3,86 г 1-бромоктаиа, 4,63 г
фталимида калия, 1,01 г ТБГДФБ в 10 мл толуола перемешивают 2 ч при
100 °С (в бане) После охлаждения отфильтровывают, осадок промывают
эфиром (30 мл). Органическую фазу хроматографируют на 10 г силикагеля,
элюируя эфиром и отбирая порцию в 200 мл. Элюат промывают 10%-иым
NaOH, сушат (MgSCh), упаривают. Выход 90%; т. пл. 47—48 °С.
Аналогично при использовании (Я)-2-октшшетансульфоната
удается получить с выходом 85% (+)"2-октилфталимид [161].
При реакции фталимида калия с 1,2-дибромэтаном в случае
эквимольных количеств реагентов получают 2-бромэтилфтали-
мид (выход 80%), а при избытке алкилирующего реагента —
1,2-дифталимидоэтан (выход 81%). Лишь в случае 2,2-диметил-
пропилбромида выход соответствующего Ы-2,2-диметнлпропил-
фталимида составляет всего 26% (одновременно образуются
значительные количества продукта элиминирования). Кроме
алкилгалогенидов и тозилатов в реакции могут участвовать
этиловые эфиры хлоруксусной н 2-хлорпропионовой кислот;
выходы N-эТоксикарбонилметил- и N-2-этокснкарбонилэтилфтали-
мида составляют соответственно 91 и 90% [161]. Применение
в последнем случае в качестве катализаторов межфазного
переноса (—)- или (4-)-1-бензнлцинхонинийхлорида позволяет
получить с выходом 30—40% оптически активный N-2-этоксикар-
бонилэтилфталимид [163, 164].
Алкилирование пнрндоиа-2 алкилбромидами в системе
бензол— вода в присутствии ТБАБ и NaOH приводит к смеси
продуктов алкилнрования по азоту (И-алкилпиридои-2) и по кнс-
лороду (2-алкоксипиридин) с общим выходом 34—88% [164];
аналогично проходит алкилирование пиридона-4 [164]:
NH
Наконец, показано [165], что в системе твердая фаза —
жидкость (катализатор ТБАГС, основание NaOH — Na2CO3)
при 80°С гладко проходит моноалкилирование дифенилфосфон-
амида, причем в реакцию можно ввести вторичные галогениды:
Ph2P(O)NH2 + RX —► Ph2P(O)NHR
Имеется большое число примеров N-алкилирования (и N-
ацилирования) в различных гетероциклических системах. В
условиях межфазного катализа, правда в присутствии эквимоль-
ных количеств ТЭБАХ (т. е. методом экстракции ионных пар),
удалось даже проалкилировать азиридины [166]. Разработаны
удобные методики синтеза N-алкил- и N-бензилпроизводных пи-
разолов и имидазолов [167]:
/г-N rx, Bu4NBr /r-N
// \ _> // ^ (55-80%)
\ / №ОН, с6н6-н2о \ /
NH . NR
Х==С1, Br; R = AIk,
RX, Bu4NBr /;N (25-71%)
\ /
NaOH, C6H6-H2O
X = CI, Br; R = Alk, PhCH2
Очень эффективными оказались условия межфазного
катализа для алкилирования 1,4-дигидропиридинов, которые из-за
низкой основности атома азота менее реакционноспособны в
обычных условиях алкилирования [166],
R' R"
N
H R'"
R, %'* R*. R'"X (выход, %): Me, Me, Me, Mel (86); Me, Me, EtI (80); Me,
Me, Me, PhCH2Br (86); Me, Ph, H, EtI (60); 4-MeOC6H4, Me, Me, Mel (52);
Me, Et, Me, EtI (81); Ph, Ph, H, Mel (75)
Общая методика алкилнровання 1,4-днгндропирндн-
нов. Смесь 0,02 моль дигидропнриднна, 0,03 моль алкилнрующего агента,
0,002—0,01 моль бензнлдодецнлднметнламмонийбромнда, 5 мл 50%:ного
NaOH н 5—70 мл органического растворителя (толуол, беизол, днхлорметан)
интенсивно перемешивают 6 ч при 40— 45 °С. Органический слой отделяют,
промывают насыщенным раствором NaCl, сушат (MgSO4), удаляют
растворитель, остаток кристаллизуют из подходящего растворителя [168].
Большое внимание уделено изучение алкилировання
индола [169—171], а также его % [156] и 3-замещенных
производных [170, 172]. Было установлено, что алкилирование индолов
при соблюдении оптимальных условий (отношение катализа-
тор: субстрат =» 1 : 10, стехиометрическое количество алкили-
рующего агента, 50%-ный NaOH, проведение реакции без
растворителя или в бензоле) [171] дает N-алкилиидолы с высокими
выходами. Третичные алкилгалогениды и неактивированные
ароматические галогениды (например, бромбензол) не
реагируют с индолом. Отмечено [169], что в случае метилиодида
необходимо использовать эквимольные количества
катализатора (ТБАГС). Реакцию ведут обычно при комнатной
температуре, но в случае я-бромнитробензола реакционную смесь
приходится кипятить. Помимо N-алкилиндолов иногда образуются
1,3-диалкилиндолы, а также 1,3-Диалкил-1,4-дигндрохинолин:
NR
XXIII
XXIV
XXV
R (выход XXIII, XXIV, XXV, соответственно, %): Et (99; —; —);
Me2CH(CH2)2 (93; следы; —); Ме2С = СНСН2 (64; 21; 10); СН =СНСН2 (39;
22; 15; при использовании эквимольных количеств, катализатора 93; 5; —);
PhCH2 (93; 5; —); МеСН2СН(Ме) (42; 10; —); 4-O2NC6H4 (65; —; -)
Алкилирование 3-замещенных индолов проходит без
осложнений в положение I [170, 172] i
R R
(57-98 %)
NR'
[ = PhCH2Cl, 4-MeOOCC6H4CH2Br
R*=CH2CN, CH2COOH, (CH2)2NHAc;
R (25~94%)
Без осложнений проходит и N-алкилированне 2-замещенных
пирролов [173], например:
/~\ Me2SO4, NaOH /Г~\
NH * ' NMe
R=Me, Ac, CN, CHO
Межфазный метод пригоден также для N-сульфонидарова-
ния индола [174]:
, С6Н6—NaOH
H-BU4NHSO4
'nso2r
R (выход, %): Ph (96); 4-МеСбН4; 2,4,6-(цзо-Рг)3С6Н2 (85); Me (92)
Общая методика N-сульфоннлнрования индола [158].
К интенсивно перемешиваемому раствору 10 ммоль индола и 1 ммоль NHSO4
в 30 мл бензола добавляют 10 мл 50%-ного NaOH, через 5 мни в течение
87
120 ийн добавляют по каплям раствор 15 ммоль RSO2C1 в 15 мл бензола при
комнатной температуре, перемешивают 20 мин (в случае применения
2,4,6- («зо-Рг)зСбН25О2С1 нагревают 2,5 ч при 50°С). Органический слой
промывают водой (7 X 20 мл), фильтруют через 20 г силикагеля, сушат (MgSO*),
остаток кристаллизуют или перегоняют в вакууме.
Легко проходит алкилирование бензотриазола [175], карба-
зола [147], 2-хлорфенотиазина [176, 177], фенотиазина [177],
производных индолов [178], например:
RX
ТЭБАХ,
NaOH—СвНв—Н2О
N (52-80%)
, PhCH2; X =
BuBr
ТЭБАХ, NaOH—ДМСО—Н2О
R'X
ТЭБАХ или ТБАБ,
NaOH-CeHe-H2O
(84%)
(20-65%)
NR'
H, Ci; X = CI, Br; R' = алкил, аллил, PhCH2
Синтез М-аллил-2-хлорфенотиазина [160]. Смесь 0,01 моль
2-хлорфенотиазина, 0,025 моль аллилбромида, 20 мл бензола, 25 мл 6 н.
NaOH и 0,0025 моль ТЭБАХ перемешивают 24 ч при комнатной температуре.
Органический слой отделяют, промывают водой, сушат (Na2SO4), удаляют
растворитель, остаток очищают хроматографироваиием иа нейтральной AlgOs
(злюирование смесью СбНб — петролейный эфир, 80:20). Выход 55%; т. пл.
76 °С.
Следует отметить, что алкилирование 2-хлорфенотиазина в
некоторых случаях проходит по-разному в зависимости от
используемого межфазного катализатора. Так, алкилирование 2-
хлорфенотиазина 3-диметиламинопропилхлоридом в
присутствии ТБАБ'не проходит, в то время как в присутствии ТЭБАХ в
тех же условиях (6 н. NaOH, бензол, 80 °С) образуется N-y-
диметиламинопропил-3-хлорфенотиазин с выходом 20% [176].
Успешно проведено гликозидирование по NH-группе в 2-ме-
гилтио-4-хлор-7Я-пирроло[2,3-^] пиримидине [180] и 2-метил-
,тио-4-метокси-7Я-пирроло[2,3-й] пиримидине [181] действием
соответственно 1-бром-2,3,5-три-О-бензил-1)-арабинофуранозы и
-.D-рибозы в бензоле или дихлорметане в смеси с ацеталем в
присутствии ТЭБАХ или ТБГС и 50%-ного NaOH.
При алкилировании аминов дигалогенидами возможно
образование гетероциклов. Так, первичные амины реагируют
с 3,4,5,6-тетрабром-1,2-бис(бромметил) бензолом в системе
PhCH2Me3NOMe — хлороформ — этанол — вода с образованием
2-замещенных 4,5,6,7-тетрабром-2,3-дигидроизонндолоб с выхб-
дом 65—90% [179]:
1-Ациламино-2-гидроксибензолы реагируют с -1,2-дибромэта-
ном в системе твердая фаза — жидкость (NaOH — MeCN—
СН2С12) в присутствии аликвата 336, давая бензоксазииы [182]:
R, R', R" (выход, %): H, H, Me (7); H, H, Ph (89); H, Cl, Me (90); H, Me,
Me, (72); Me, H, Me (62); Me, H, Ph (86)
Интересно отметить, что эта реакция одновременного алки-
лирования по азоту и кислороду с образованием оксазинового
цикла не идет б дихлорметане, а при проведении в ацетонитри-
ле дает сложную смесь; только одновременное сочетание обоих
растворителей в соотношении MeCN : CH2Cl2 = 40 : 60 приводит
к успеху.
Обшая методика синтеза 3,4-дигидро-2#-1,4-бепз-
оксазинов [182]. Смесь 10 ммоль о-ациламинофенола, 1,6 г (40 ммоль)
порошкообразного NaOH, 7,52 г 1,2-дибромэтана, 0,41 г аликвата 336, 32 мл
MeCN и 48 мл СН2С12 перемешивают 24 ч при 25—30 °С в атмосфере азота,
добавляют еще 0,4 г (10 ммоль) порошкообразного NaOH, перемешивают
2 ч, фильтруют, осадок промывают эфиром. Удаляют растворители из
фильтрата, остаток очищают хроматографироваиием иа колонке с силикагелем
(элюированяе смесью эфира с петролейным эфиром с т. кип. 45—65 °С).
Алкилирование в межфазных условиях может быть успешно
использовано для алкилирования гидразинов [183, 184] и гид-
разонов [185]:
ТБАГС
PhNH=NHPh + RX „.он^н^н^ PhNH-NRPh (7-79%)
= Br, I; R = алкил, аллил, PhCH»
ГХ, ТБАХ
RR'C=NNHPh
R, R', R"X (выход, %): Ph, H, н-PrBr (70); Ph, H, PhCH2Cl (98); Ph, H,
Mel (92); Ph, H, CH2=CHCH2Br (78); Ph, H, СН^ССН2С1 (85); Ph, H,
T/?er-BuO2CCH2Br (59); Me, Me, PhCH2CI (52); Ph, Ph, н-РгВг (50); Ph, Ph,
PI1CH2CI (72); флуоролиден; н-PrCl (43); флуоролидеи, PhCH2Cl (56);
JtONGHfc.H, «-PrBr (76); n-O2NC6H4l H, PhCH2Cl (91)
89
В отсутствие катализатора межфазного переноса алкилирова-
ние гидразонов не идет или же продукты алкилирования
образуются с очень малыми выходами.
Общая методика алкилирования феиилгидразоиов.
0,02 моль фенилгидразона, 20 мл 50%-ного NaOH, 1,08 ммоль Bu4NCl и
0,02—0,04 моль алкилгалогенида перемешивают 0,5—3 ч при 30—60 °С,
разбавляют водой, отделяют продукт, промывают его водой, метанолом,
кристаллизуют. В случае жидких производных извлекают хлороформом,
органический слой промывают водой, сушат (MgSO4) и после удаления
растворителя перегоняют,
а,со-Дибромиды реагируют с двумя молекулами
фенилгидразона [185]:
PhCB=N—NHPh + Вг(СН2)ггВг + PhCH^N—NHPh —>
—v PhCH^N—N— (CH2)rt~N—N=CHPh
I I
Ph Ph
n (выход, %): 1 (70), 2 (75)
На реакции N-алкилирования гидразонов основана новая
модификация реакции Э. Фишера получения 1-алкил-, 1-алкил-
2-арил-, 1,3-диалкил- и 1,2,3-триалкилиндолов [186]:
PhNHN=CRR'
R, R', R" (выход, %)i Me, Me, Bu (50); Ph, H, Bu (54); H, H, Bu (9); H, Me,
Bu (42)
Типичная методика алкилирования гидразонов приведена
ниже.
Синтез 1-бутил-2-фенилиндола. Нагревают смесь 105 мг фе-
иилгидразоиа ацетофенона, 56 мг твердого КОН, 54 мг к-бутилбромида н
20 мг дибензо-18-крауна-6 в 5 мл сульфолаиа в течение 2 ч при 90 °С и 3 ч
при 180 °С, охлаждают, выливают в воду, извлекают эфиром. На SiO2 (гек-
сан) выделяют 67 мг 1-бутил-2-фенилиндола.
Вместо крауи-эфира можно использовать ТБАБ, а вместо
КОН - КоСОз.
Разработан метод алкилирования тозилгидразонов [187,
188], который особенно пригоден для синтеза Ы-метил-Ы-тозил-
гидразонов кетонов с низкой электрофильностью, например фла-
вонов, которые в обычных условиях не реагируют с N-Memn-N-
тозилгидразином [187]:
RR'C-N-NhTs Me3PhCH2NctXNaQH^H2o> RR'O-NNRTs
R, R', R"X (выход, %): PhCH2) Ph, Mel (90); PhCH2, PhCH2) Me (92); н-Рг,
н-Рг, Mel (90); 1е2Д4<тетрагидронафтилиден, Mel (95); Ph, Me, Mel (92)i
90
Ph, Me, EtI (96); Ph, Me, PhCH2Br (82); трет-Вп, Н, Mel (98);
PhCH=CH, Mel (97); PhCH = CH, H, Mel (98)
N—NHTs
N—NMeTs
Mel
(81-94%) R~H,Ph
Следует отметить, что при алкилировании тозилгидразонов
применяют не 5О°/о-ный, а 15%-ный NaOH, поскольку
образующиеся Ы-алкил-Ы-тозилгидразоны разлагаются более
концентрированными щелочами.
Синтез N-метил-М-тозилгидразона ацетофенона.
К раствору 1 г тозилгидразона ацетофеиона в 15 мл СН2С12 добавляют 15 мл
15%-ного NaOH, 0,96 г метилиодида и 0,059 г триметилфениламмонийхлори-
да. Смесь перемешивают 8 ч при комнатной температуре. Органический слой
отделяют, промывают водой, сушат, упаривают в вакууме, остаток
кристаллизуют из метанола. Выход 0,974 г; т. пл. 130—131 "С.
Недавно был описан [180] еще один пример алкилирования
ароматического амина, а именно 2-цианоанилина, в системе
ТБАГС — NaOH — СН2С12 — Н2О хлорпроизводными Сахаров,
например:
Ас О
ТБАГС
Ас О
Ас О
NaOH—СНгС18—Н20
(10%)
Ас О
ОАс
В последние годы довольно подробно было изучено алкили-
розанне ацетамида и я-толуолсульфонамида бензилхлоридом в
присутствии ТЭБАХ в бензоле или димётилсульфоксиде при
различных температурах [190] и показано, что образуются
продукты как моно-, так и диалкилирования, соотношения которых
определяются условиями реакции и соотношением реагентов.
Образование N—Р-саязей
Метод межфазного катализа был использован для проведе*
ния такой важной реакции, как фосфорилирование по атому
азота. Разработано три варианта методики фосфорилирования
аминов диалкилфосфитами в присутствии ССЦ в условиях
межфазного переноса (ТЭБАХ —NaOH —СН2С12 —Н2О) [191],
(RO)2P(O)H + СХ4 + 2R/R//NH
(RO)2P(O)-NR/R//
91
£, R', R , методика (выход, %): Et, H, Ph, A (77), Б (35), В (84)$ Et,
цикло-С6Ни. А (8§); Et, H, PhCH2, A (85); Et,-Et, Et, A (86), Б (85); Et,
Et, Б (81); PhCH2j H, Et, A (40), В (78); PhCH2l H, цикло-С6Н1ь А (93);
PhCHa, H, PhCH2, A (83); PhCH2, Et, Et, A (91); трег-Bu, H, цикло-С6Нп,
fc (90); трет-Вп, H, PhCH2, В (92)
Общая методика N-фосфорилирования. А. К раствору
0,1—0,125 моль диалкилфосфита и 0,1- моль амина в 30 мл дихлорметана
добавляют по каплям при перемешивании 30 мл СС14 в 30 мл СН2С12 и 40 мл
20%-иого NaOH, содержащего 1 г ТЭБАХ, перемешивают 1 ч при О—5°С
(охлаждение льдом с солью) и I ч при комнатной температуре, разбавляют
2о мл дихлорметана, отделяют органический слой, промывают его 50 мл
5%-ной НС1 и водой (2X50 мл), сушат (MgSO4), удаляют растворитель и
выдерживают 1 ч при 30—40 °С в вакууме (0,1—0,5 мм рт. ст.) или
кристаллизуют. Получают чистый препарат.
Б. К смеси 17,25 г диалкилфосфита, 0,1 моль гидрохлорида амина, 40 мл
ССЦ, 40 мл СНаСЬ н 1 г ТЭБАХ добавляют по каплям О—5°С а
интенсивном перемешивании 40 мл 30%-иого NaOH. Перемешивают 2 ч при комнатной
температуре, далее обрабатывают, как описано выше.
В. К раствору 0,05 моль диалкилфосфита и 0,05 моль амнна в 15 мл
СН2С12 добавляют при 20 °С смесь 8,3 г J0,025 моль) СВг4, 30 мл СНзСЬ,
20 мл 20%-ного NaOH и 0,5 г ТЭБАХ. Перемешивают 3 ч при комнатной
температуре, разбавляют 25 мл СН2С12, далее обрабатывают, как описано
выше.
Для фосфорилирования гидразинов, которые не удается
ввести в реакцию по разработанному для аминов методу в системе
жидкость — жидкость (см. выше), был применен метод с
использованием системы твердая фаза — жидкость [192].
(RO)2P(O)H + H2N—NH2 + CCI4 —> (RO)2P(O)—NH—NH2
R (выход, %): Et (84); н-Рг (90); изо-Рг (74); н-Bu (98); нео-С6Нц (98)
Общая методика монофосфорилирования
гидразинов. К смеси 120 мл СС14, 200 мл СН2С12, 41,4 г порошкообразного
безводного КгСОз добавляют по каплям при интенсивном перемешивании 12,5 г
80%-ного гидразиигидрата при 20—25 °С, перемешивают 15 мин и
затем-добавляют при 20—30 бС (если нужно, охлаждают) раствор 0,2 моль
диалкилфосфита в 40 мл СН2С1г, перемешивают 4 ч, фильтруют, удаляют
растворители, выдерживают 2 ч'при 100—110°С (0,5 мм рт. ст.). Получают чистый
препарат.
В системе 50%-ный КОН — ТЭБАХ —ССЦ при комнатной
температуре фосфорилируется О-алкилгидроксиламин с
образованием диалкил-М-алкоксифосфамидов [193]:
+ ссц
(EtO)2P(O)H+RONH8 СГ -"—> (EtO)2P(O)—NHOR
R (выход, %): Me (78); Et (47); н-Bu (45); PhCH2 (76)
Общая методика фосфорилироваи и я О - алкилгидро-
ксиламйнов. Добавляют по каплям 22,5 мл 50%-иого КОН при
интенсивном перемешивании и наружном охлаждении к смеси 0,1 моль
гидрохлорида О-алкилгидроксиламина, 0,125 моль диэтилфосфита, 80 мл ССЦ и 1 г
ТЭБАХ при 15—20 °С. Перемешивают 2 ч при комнатной температуре,
разбавляют 100 мл дихлорметаиа, фильтруют, отделяют органический слой,
сушат (MgSO4), удаляют растворители, остаток выдерживают 1—2 ч при 30—-
40 °С (0,1 мм рт. ст.). Получают чистый препарат.
92
Образование С—С-связей
Реакция С-алкилирования является основой одного из
важных синтетических методов наращивания углерод-углеродной
цепи, позволяющего строить разнообразные углеродные
скелеты. Для проведения межфазного С-алкилирования
необходимо, чтобы в соединении присутствовали подвижные
(активированные) атомы водорода. В межфазных условиях алкили-
руются, например, углеводороды типа циклопентадиена [194—
196], некоторые гетероциклы с подвижными атомами водорода
[197—199], кетоны [200—218], активированные нитрилы [219—
268] и эфиры карбоновых кислот [269, 270], эфиры ацилуксус-
ных кислот [271—274], эфиры циан- и изоциануксусных кислот
[84, 275, 276], малоновый эфир и его производные [84, 277,
278], активированные сульфоны [279—286].
Алкилирование углеводородов. Эта область изучена еще
недостаточно. Отмечено, что возможно алкилирование
циклопентадиена по активной метиленовой группе [66]; при алкилиро-
вании алкилбромидами или бензилбромидом выход
соответствующих алкил- или бензилциклопентадиенов достигает 32—70%:
+ RBr
ТЭВАХ, NaOH—Н2О
Алкилирование 1,2-дибромэтаном приводит к спиро[2,4]геп-
тадиену-4,6 с выходом 76%:
4- Вг(СН2)2Вг
ТЭВАХ, NaOH—Н2О
Циклолентадиен алкилируется также изопропиловым эфиром
хлоруксусной кислоты, давая 1-изопропоксикарбонилметилцикло-
пентадиен с выходом 62% [66].
В тех же условиях инден алкилируется в положение 3 [194]:
R
RBr
R (выход, %): Me (62); н-Рг (68); н-Bu (63); Me2CHCH2 (73); (Et)2N(CH2)3
(46); (СН2)3Вг (45); (CH2)sBr (52)
При алкилировании индена 1,3- и 1,5-дибропропанами
реакция проходит только по одному из атомов галогена, а при
использовании 1,4-дибромбутана — по обоим атомам с
образованием спиро [инден-1,Г-циклопентадиена-2,4]:
Вг(СН2)4Вг ^
93
Флуорен алкилируется в системе ТЭБАХ — NaOH ™
ДМСО — Н2О, образуя 9,9-диалкилпроизводные с выходом
80—83%, по реакции же с 1,4-дибромбутаном получают спи-
ро [флуорен-9,1 '-циклопентан] (выход 64 %) [ 195].
Межфазное алкилирование 2- и 4-метилпиридинов, 2,6-ди-
метилпиридина, 2,4,6-триметилпиридина избытком метилиодида
в системе Н2О —СН2С12 в присутствии NaOH и использовании
в качестве катализатора тетрабутиламмонийгидроксида
проходит по метильным группам, причем к Ме-группам в положениях
2 и 6 присоединяются по две метильные группы, а в положении
4 — три метильные группы. В результате образуются
соответственно 2-изопропилпиридин (выход 36%), 4-грег-бутилпиридин
(37%), 2,6-диизопропилпиридин (40%) и 4-грег-бутил-2,6-ди-
изопропилпиридин (20%). Метильная группа в положении 3
не метилируется в этих условиях. Образование этильной группы
из метильной .не наблюдалось [199].
Вп-трет
Mel
Алкилирование альдегидов. Описано алкилирование альде*
гидов, например изомасляного альдегида [198, 199] и 2-этил-
гексаналя [199], в системе С6Н6 — Н2О — NaOH при
использовании бензилгексадецилдиметиламмонийхлорида [198] или тет-
рабутиламмонийиодида [199]. В зависимости от активности
алкилирующего средства температура реакционной смеси
поддерживается от комнатной до 70 °С. Реакция проходит
удовлетворительно при использовании первичных активных галогени-
дов; со вторичными галогенидами образуются продукты еноли-
зации и алкилирования енолальдегидов:
RR'CH—СНО + R"X —■* RR'R"C~CHO
R, R', R"X [выход, % (температура, °С)1: Me, Me, метилиоднд [15 (42)];
Me, Me, бензилхлорид [34 (20); 75 (70)]; Me, Me, пропен-2-илхлорид [30
(20); 56 (60)]; Et, «-Bu, бензилхлорид [55 (65); образуется также бензил-2-
этилгексен-1 -иловый эфир]; Et, «-Bu, пропен-2-илхлорид [65 (65); образуется
также бутен-2-ил-2-этилгексен-1 -иловый эфир]; Et, «-Bu, З-метнлбутен-2-ил-
хлорид [60 (50), образуется также 3-метилбутен-2-ил-2-этилгексен-1 -иловый
эфир]
Недавно было показано, что в присутствии ТБАИ возможно
алкилирование изомасляного альдегида пропаргилбромидами
в дихлорметане при комнатной температуре [200]:
Ме2СНСНО + BrCH2O=CR —► Me2C(CH2C=CR)CHO (36—56%)
ТЭБАХ дает в этом случае худшие результаты.
94
Типичная методика алкилирования альдегидов приведена
ниже.
Синтез 2,2-диметил-З-фенилпропаналя [199]. К смеси
140 г NaOH, 140 г воды и 200 мл бензола добавляют 14,7 г ТБАИ, нагревают
до 70 °С и при интенсивном перемешивании добавляют в течение 5 ч смесь
288 г изомасляного альдегида и 380 г (3 моль) бензилхлорида, перемешивают
2 ч при той же температуре, органический слой отделяют, промывают водой,
сушат (MgSO4), перегоняют. Выход 364 г (75% от теоретического); т. кип.
95 °С (7,2 мм рт. ст.).
Алкилирование кетонов. Алкилирование простейших
алифатических кетонов, в частности алкилирование ацетона пренил-
хлоридом, подробно рассмотрено ранее. Следует отметить,
однако, что алкилирование простейших алифатических кетонов
пока еще не нашло широкого применения в лаборатории,
возможно потому, что реакция с ними, как правило, идет сложно
и редко приводит к удовлетворительным результатам. Так,
например, метилгептенон образуется при алкилировании ацетона
пренилхлоридом в водной системе с NaOH и ТБАБ с выходом
52% [287], что значительно ниже, чем по хорошо известному
методу синтеза метилгептенона пиролизом ацетоацетата винил-
диметилкарбинола.-Было показано, что алкилированне ряда
диалкилдикетонов в системе тетрагидрофуран — вода алкилио-
дидами в присутствии тетраэтиламмонийфторида как
межфазного катализатора приводит к алкилированным дикетонам
с высокими выходами (91—95%) [286,288):
RCOCH2COR' + R"I —► RCOCHR"— COR'
Отмечено также, что с вполне удовлетворительными выходами
(62—72%) протекает алкилирование простых кетонов изопро-
пиловым эфиром 3-хлорпропионовой кислоты [201]:
RR'CHCOCH3 + Cl(CH2)2CO2Pr-W3O _> RR'C(CH2CH2CO2Pr-W3o)COCH3
Алкилирование цикланонов (циклогексанон, 1-, 3- и 4-ме-
тилциклогексаноны и циклопентанон) в обычных условиях
(ТЭБАХ, 50%-ный NaOH, 50—60°С) по сравнению с алкили-
рованием с использованием твердого КОН практически не имеет
преимуществ. Для метилциклогексанонов выходы примерно
равны в обоих случаях (для 2-метилпроизводного 25—30%,
для 3-метилпроизводного 47—50 и 51—52%, а для 4-метилпро-
изводного 50—51 и 46—50% по межфазной методике и
методике с твердым КОН, соответственно). Для самого циклогек-
санона и циклопентанона лучшие результаты получают с
твердым едким кали [208].
Значительно больший успех достигается при использовании
межфазной методики для алкилирования активированных
кетонов. Выше уже отмечались неплохие результаты прн алкили-
95
ровании fj-дикетонов [260, 288]. Следует отметить также аЛки-
лирование 2-ацетилциклогексанона в присутствии М-бензил-N-
метилэфедринийбромида {218], что позволяет получать с
высоким выходом алкилированные продукты, обладающие
оптической активностью, например:
RX (выход продукта, %) [{ajg (CHCI3, с =1,5)]: Mel (85) [-4,6f
СН2=СНСН2Вг (90) [—23,5]
Общая методика алкилирования р-дикетонов. Смесь
20 ммоль р-дикетона,, 20 мл СН2С12 (или СНС13), 22 мл RX, 20 мл 10%-ного
NaOH и 0,2 г Ы-беизил-М-метнлэфедринийбромида интенсивно перемешивают
~ 12 ч при комнатной температуре. Органический слой отделяют, удаляют
растворитель, остаток обрабатывают эфиром, отфильтровывают катализатор,
фильтрат очищают на колонке с силикагелем (растворитель н-гексан — этил-
ацетат, 20: 1).
Интересна, что замена СН2С12 или СНСЦ на к-гексан
приводит с высокими выходами к продуктам, не обладающим
оптической активностью. Методика применима и к эфирам (3-ке-
токислот [218].
Наибольшее применение межфазный катализ нашел для
алкилирования жирноароматических кетоиов. Сравнение
алкилирования кетонов (ацетофенон, 2-метилацетофенон и 2-метокси-
ацетофенон) в присутствии твердого КОН и. реагента Макоши
(50%-ный NaOH—-ТЭБАХ) сразу обнаружило заметное
преимущество последнего [208]. Так, при алкилировании пренил-
хлоридом выход 2-метил-6-фенилгексен-2-она-6 составляет 16—•
21 % по первой методике и 34—40% по второй, в случае же
синтеза 6-п-анизил-2-метилгексен-2-она-б продукт образуется
с выходом 10% только по второй методике. Хорошие
результаты дало алкилирование фенилацетона [213], дезоксибензоина
[207] и других кетонов.
Необходимо отметить, что использование а-хлорэфиров для
алкилирования фенилацетона или дезоксибензоина в условиях
межфазного катализа приводит к О-алкильным производным
[205].
В системе ДМСО — вода (NaOH, катализатор ТЭБАХ) аце-
нафтенон гладко алкилируется изопропиловым эфиром 3-хлор-
пропионовой кислоты [204];
О=С С(СН2СН2СО2Рг-азоЬ
С1(СН2}2СО2Рг-ызо
■->
(80%)
В тех же условиях в водной среде проходит алкилирование
N-замещенных оксиндолов [205, 214], например:
R
ArCl
NMe
NMe
R, ArCI (выход, %); H, 4-ClC6H4NO2 (86); Н, 1,4-C12C6H3NO2 (70); Н,
I-Cl-3-MeOC6H3NO2-4 (89); Me, 4-ClC6H4NO2 (75); Me, l-Cl-2,4-(O2N)2C6H3 (72);
Me, I,4-Cl2C6H3NO2-2 (80); I,3-CI2C6H3NO2-4 (89); Me, l-Cl-3-MeOC6H3NO2-4 (76)
В этом случае в качестве катализатора использовали также
ТБАБ [214].
Интересный случай С-алкилирования с заменой ОН-группы
описан для 2- (S) -а-фенилэтил-3-гидрокси-3,4-дигидроизохино-
лона-1 [289]. При перемешивании этого субстрата (1,5 ммоль)
с (EtO)2P(O)CH2CN (1,20 ммоль) в СН2С12 (30 мл) с ТБАБ
(0,2 моль-экв) и 50%-ным NaOH (2,4 мл) при комнатной
температуре в течение 24 ч образуется смесь диастереомеров:
2- (S) -а-фенилэтил-2- (/?)-цианометил-3,4-дигидроизохинолона-1 и
2- (S) -а-фенилэтил-3- (S) -цианометил-3,4-дигидроизохинолона-1 с
общим выходом 80%.
При реакциях 1-метилоксиндола [205], тетралона-2 [210],
фенилацетоиа [202, 203] с 1,2-дибромэтаном под влиянием
NaOH в водном растворе в присутствии ТЭБАХ образуются
производные циклопропана:
(34-72 %)
PhCH2COMe
(34%)
СОМе (54%)
Алкилирование нитрилов. Неактивированные нитрилы, как и
следовало ожидать, не алкилируются в условиях межфазного
катализа. Активация введением в а-положение фенильного
[219, 220, 224, 253, 254, 258] или нафтильного [247]
заместителей, PhS [243], PhSe [259], Et2NCOS [260], Me2NC(S)S [290]
и других электроноакцепторных группировок приводит к
возможности алкилирования нитрилов в а-положение
разнообразными алкилирующими средствами в условиях межфазного
катализа. Так, в обычных условиях под влиянием 50%-ного NaOH
в присутствии ТЭБАХ проходит алкилирование фенилацетонит-
рила алкилгалогенидами [219, 220, 224, 261], за исключением
97
алкилиодидов, поскольку в типичной двухфазной
каталитической системе Q+ I~ преобладает над Q+ С— в органической
фазе [219, 258].
PhCH2CN + RX —► PhCH(R)CN
RX (выход, %): МеВг(89); EtBr (88); н-РгВг (78); изо-РтВт (60): w-BuBr(74);
«зо-ВиВг (47); агор-ВцВг (63); «зо-С5НИВг (72): «-С6НиВг (85); «-C7Hj5Br
(63) [220]
Гладко проходит алкилирование фенилацетоннтрила аллил-
галогенидами (2-хлорпентен-З, 3-бромциклогексен), аралкилга-
логенидамн (бензилгалогениды, 1 -фенил-1 -хлорэтан, дифенил-
метилхлорид), а также функционально замещенными галогени-
дами [220]. Интересно, что фенилацетонитрнл алкнлируется
в двухфазной системе беизилхлоридамн, активированными нит-
рогруппами (о-, м- и я-нитробензилхлориды) [250], или гете-
роарилхлоридами (5-нитро-2-хлор, З-нитро-4-хлор, З-нитро-2-
хлорпиридины) [220].
Алкилирование фенилацетоннтрила и других
активированных нитрилов широко используется для проведения
многочисленных синтезов. Так, алкилирование фенилацетонитрила
приводит к диалкилированным продуктам (даже при значительном
избытке фенилацетонитрила) — 1,3-бнс(нитрофеннл)-2-фенил-2
цианопропанам [253]:
O2NCeH4—CH2—C(CN) (Ph)—СНг—CeH4NO2
Алкилирование фенилацетонитрила нитрохлорпиридинамн
в системе бензол — вода в присутствии ТБАБ (или ТБАХ)
с NaOH при 50—55 °С приводит к соответствующим фенилцна-
нометилнитропиридинам с высокими выходами [264].
Межфазный каталитический метод был предложен для
синтеза тиогликонитрилов. Первой стадией синтеза является
алкилирование фенилтиоацетонитрила [243].
Синтез бензил-Б-фенилтиогликонитрила. 7,5 г S-фенил-
тиогликшитрила, 14,4 г бензилхлорида, 20 мл 50%-ного NaOH и 0,3 г ТЭБАХ
после прекращения экзотермической реакции перемешивают 2 ч в атмосфере
азота, разбавляют водой, продукт отделяют и кристаллизуют из метанола.
Выход 13,5 (82% от теоретического); т. пл. 152 °С.
Аналогично получают и другие Б-фенил-К-тиогликонитрнлы
[R (выход, %): Et (80), к-Bu (82), Me (75), СН2=СНСН2
(80), (СН2)зС1 (39)]. Прн избытке галогенида образуются
диалкилированные продукты, также с высокими выходами.
Применение а, со-дигалогенпроизводных приводит к фенилтиоцнано-
циклоалканам:
/SPh
Н2С с;
п (выход, %): 1 (47), 3 (69), 4 (50)
98
Алкилирование фенилацетонитрила эфирами галогенуксус-
ной кислоты в водном растворе NaOH в присутствии ТЭБАХ
позволяет получать с высокими выходами эфиры 2-феннл-2-циа-
нопропионовой кислоты [234]:
CH2CO2R
PhCH2CN4-XCH2CO2Ph -—► PhCHCN+ PhC(CH2CO2R)2CN (76—93%)
Реакция идет при 20—25 °С, побочно образуются диалкилиро-
ванные производные. Аналогично получают динитрил-3-фенил-
3-цианоглутаровой кислоты и динитрилы фенилцианозамещен-
ных высших кислот [228, 231]:
T3BAX-NaOH-H2O
PhCH2CN + CI(CH2)nCN ■>
—> PhC(CN) (CH2)nCN (41—45%)
Из фенилацетонитрила и 1,2-дибромэтана был получен фе-
нилциклопропан [291].
Из фенилселеноацетонитрила и 1,2-дибромэтана был
получен 1-фенилселено-1-цианоциклопропан с выходом 80% [259],
а из фенилацетонитрила и 1,4-дибромбутана— 1-фенил-1-циано-
цнклопентан с выходом 88% [229]:
PhSeCH2CN + Br(CH2)2Br
'CN
PhCH2CN+Br(CH2)4Br —- / Vх
Аналогично получено н следующее производное циклопен
тана [260]:
.SCONEt2
Et2NCOSCH2CN+ Вг(СН2)4Вг
\
CN
Использование для алкилирования фенилацетоннтрила р, р'-
дихлордиэтилового эфира (система 50%-ныйЫаОН — ТЭБАХ —
вода) позволило замкнуть тетрагидропирановый цикл [225] 'j
PhCH2CN+(CICH2CH2)2O —► О()( (69%)
Замыкание цикла может быть осуществлено введением
в реакцию с замещенными фенилацетонитрилами бис(Р-хлор-
этнл)тозиламнна [238]:
ТЭВАХ—NaOH —Н2О " / у /СбН4 X
n-XC6H4CH2CN + TsN[(CH2)2CI]2 ■> TsV Y
\*
:=H. Cl (68-72%j
99
Дизамещенные фенилацетонитрилы чрезвычайно легко алки-
лируются. Это позволяет вводить в молекулу не только алкиль-
ные, аллильные или бензильные радикалы, но и арильные, и
гетероарильные.
Ph2CHCN + RX —»- Ph2C(R)CN
RX (выход, %): MeBr (94); EtBr (92); я-BuBr (94); я-С5НцВг (94); я-СБН,3Вг
(92); СН2=СНСН2Вг (95); PhCH2Cl (98); PhCHClMe (96); Ph2CHCl (96);
Et2NCH2CH2Cl (85)
Общая методика алкилирования дифенилацетонитрила (и
других дизамещенных активных ацетонитрилов) очень проста.
Синтез 2,2-дифенил-4-диэтиламинобутиронитрила.
9,7 г дифеиилацетоиитрила, 6,8 г 2-хлорэтилдиэтиламнна, 0,13 г ТЭБАХ и
15 мл 50%-ного NaOH перемешивают до окончания реакции (контроль ТСХ)
и выделяют после обычной обработки продукт алкилирования. Выход 12,4 г;
т. кип. 165 °С (0,8 мм рт. ст.); п$ 1,5528.
Аналогично получают другие производные [222]. Следует
отметить, что алкилирование дифенилацетонитрила (или ал-
килфенил ацетонитрилов) диалкиламиноалкилхлор идами при
60—70 °С в присутствии 70%-ного раствора NaOH проходит
без катализатора [230] за счет образования четвертичных
аммониевых солей в процессе реакции.
Алкилирование же Ы,Ы-диметиламинофенилацетонитрила
требует присутствия ТЭБАХ.
PhCH(NMe2)CN+RX —> PhC(NMe2) (R)CN
RX (выход, %): EtBr (56); изо-РтЪт (61,5); я-BuBr (75); СН2=СНСН2С1
(75,5); PhCH2Cl (82)
Общая методика алкилирования N.N -диметиламино-
фснилацетонитрила. Растирают 20 г 50%-ного NaOH и 13,3 г
измельченного твердого NaOH до гомогенизации, добавляют 0,05 моль Ы,Ы-ди-
метиламииофеиилацетонитрила, 0,055 моль RC1 (или RBr) и 0,1 г ТЭБАХ,
нагревают 4 ч при 65—70 °С, разбавляют водой, извлекают бензолом, после
высушивания и удаления растворителя очищают перегонкой в вакууме или
кристаллизацией.
В то время как фенилацетонитрил не реагирует с галоген-
бензолами, даже с активированными, дифенилацетонитрил и
алкилфенилацетонитрилы алкилируются нитрохлорбензолами
[236, 240, 245, 252, 253], 2-хлорхинолинами [266], 1-хлоризо-
хинолинами [266], а также 9-хлоракридином [257]. Для того
чтобы реакция проходила, нитрогруппа должна находиться
в орто- или пара-положении к хлору в бензольном кольце. При
наличии в бензольном кольце двух атомов хлора в орто- и
/га/?а-положениях к нитрогруппе проходит преимущественно
реакция по /ш/?а-положению, однако в некоторых случаях от-
100
мечается появление продуктов реакции по орто-положению и
продуктов диалкилирования [252]:
NO2
Г
т
X
NO2
] + PhCH(R)CN
NO2
1
ТЭБАХ
NaOH—H2O
NO2
C(R)GN
PhC(R)CN
xxvi
X
xxva
PhC(R)CN
XXVIII
X, R (выход XXVI, %): Cl, Me (82); Br, Me (93); Cl, Et [77,6; 2,4 (XXVII)];
Br, Et (76); Cl, Ph [76,4; 3,6 (XXVII)]; Cl, CH2Ph [51,7; 15,6 (XXVII);
10,7 (XXVIII)]; Cl, CHPh2 (94)
Иногда наблюдается нуклеофильное замещение не хлора,
а нитрогруппы. Так, 4-нитро-З-хлорбензофенон алкилирует ал-
килфенилацетонитрилы и дифенилацетонитрил (вода — NaOH —
ТЭБАХ), давая алкилфенил (или дифенил)-4-бензоил-2-хлор-
фенилацетонитрилы [255]:
PhCO
R
NO2 + PhCHCN
PhC
(R)CN
R (выход, %): Me (71); Et (61); Ph (78)
Следует отметить, что реакция алкилфенил- или дифенилацето-
нитрилов с 6-нитро-2-хлорбензофеноном в тех же условиях
приводит к продуктам замещения хлора — 2-алкилфенил- или ди-
фенил-6-нитробензофенонам (выход 57—70%) [255].
Алкилирование алкилфенил- или дифенилацетонитрилов
2-хлорхинолинами или 1-хлоризохинолинами с 50%-ным NaOH
в системе ДМСО — вода в присутствии ТБАБ или ТБАХ
проходит нормально с хорошими выходами [266]:
R' R'
PhCH(R)CN
C(R)CN
101
R> R' (выход, %): Me, H 85); Me, Me (73); Et, H (60); Et, Me (60)
R' R'
PhCH(R)CN
Cl PhC(R)CN
R, R' (выход %): PhCH2, Me (76); Me, H (60); Et, N (67)
Общая методика алкилироваиия фенилалкаионитри-
дов. 25 ммоль 2-хлорхинолина и 25 ммоль 2-феиилалкаионитрила интенсивно
перемешивают с 35 мл 50%-ного NaOH, 10 мл ДМСО и 0,2 г ТБАХ или
ТБАБ 6 ч при 50 °С, разбавляют водой, извлекают бензолом, отделяют и
сушат органический слой (MgSO^, удаляют растворитель, остаток
кристаллизуют из метанола.
В системе бензол — вода — NaOH—ТБАХ идет алкилиро
вание алкилфенил- или дифенилацетонитрилов 9-хлоракриди*
ном; выходы продуктов алкилирования 40—90% [257].
Реакция дизамещенных ацетонитрилов с N-оксидами 4-хлор-
или 4-нитропиридинов в системе ДМСО — вода — NaOH —
ТБАХ (или ТБАБ) протекает с замещением хлора или нитро-
группы и образованием N-оксида 4-алкилфенил (или дифе-
нил)цианометилпиридина с выходами 40—95% [242],
Ph
X—/^N-K) + PhCHCN —► NC—С
R R
X—Cl, NO2
Выходы продуктов как из хлор-» так и из нитропроизводного
почти одинаковы. Следует отметить, что алкилирование 4-нит-
ро-4'-хлорбензофеноном всегда приводит к замещению по нит-
рогруппе [245]:
ТЭБАХ
PhCHCN + га-С1СбН4—СО—CBH4NO2-rt м ^ц ц >■
I NaOH — Н2О
R
Ph
I
—► rt-ClC6H4-CO—C6H4CCN
R
R (выход, %): Me (75); Et (70); я-Рг (78); PhCH2 (67)
Отмечено, что в стандартных условиях (бензол — вода —
NaOH — ТЭБАХ) дифенилацетонитрил реагирует с нитроаре-
нами с образованием 1Д,2,2-тетрафенил-1,2-дицианоэтана и
азоксиарена [239]:
Ph2CHCN + АгДОа —► Ph2C—CPh2+ ArN=NAr
NC GN О
102
Сложные превращения наблюдались также при реакции
фенилацетонитрила и его замещенных с 2,4-дихлорнитробензо-
лом (бензол — 50%-ный NaOH — ТЭБАХ, 50—60 °С) [252]:
наряду с нормальным продуктом алкилирования по орто-пояо-
жению образовывались продукты димеризации, циклизации
и пр.:
NO2 NO2 NO2
PhCH2CN
CHCN
COPh
Cl
Cl
N
\
О
О + Cl-
"=N
COPh COPh
Использование замещенных галогенидов позволяет ввести
в молекулу нитрилов другие заместители. Так, реакция дифе-
нилацетонитрила с а,со-дибромидами приводит к галогеннитри-
лам [226]:
Ph2CHCN + Вг(СН2)лВг — + Ph2C(CN)(CH2)nBr (82—91%)
При использовании фенилэтилацетонитрила выходы
снижаются до 47—65% из-за частичного элиминирования НВг. Алкили-
рование фенилэтилацетонитрила 1,3-дибромпропаном дало
помимо (3-бромпропил) фенилэтилацетонитрила аллилфенилаце-
тонитрил с выходом 38% [226].
Алкилирование а-хлорэфирами в зависимости от
соотношений реагентов и строения исходного фенилацетонитрила дает
алкоксинитрилы или динитрилы:
PhCHCN + R'CHOR"
A
R'CHOR"
I
PhC—CN
R
CN-,
PhC—
R, R', R" (выход А, %): Me, Me, Me (60); Me, Me, изо-Рт (75); Ph, H, Me
(80); Ph, H, изо-Рт (88); Ph, Me, Me (50). R, Y (выход Б, %): H, CH2 (88);
Ы, СНМе (70); Me, СН2ОСН2 (84); Ph, СН2ОСН2 (90); ыэо-РгОСН2, СН2ОСН5
(68)
Общая методика синтеза алкоксиннтрилов. К 0,1 мол>
нитрила, 0,3 моль 50%-ного NaOH, 10—30 мл бензола и 0,25 г ТЭБАХ дб-
бавляют при хорошем перемешивании при 20—30 °С постепенно 0,1—0,15 моль
103
ct-хлорэфира, перемешивают 30 мин; отделяют продукт, очищают перегонкой
или кристаллизацией.
Типичная методика синтеза динитрилов приведена ниже.
Синтез динитрила 2,4-дифенилглутаровой кислоты
[204]. К смеси 11,7 г фенилацетонитрила, 24 г 50%-ного NaOH и 0,23 г
ТЭБАХ добавляют по каплям при 30 °С 8 г ди(хлорметилового) эфира,
перемешивают до окончания реакции, обрабатывают разбавленной соляной
кислотой и отделяют динитрил. Выход 10,8 г; т. пл. 89—91 °С. Кристаллизацией из
метанола и воды выделяют изомер с т. пл. 65—67 °С.
Аналогично проходит реакция и в присутствии 20 мл бензола.
Алкилирование бромнитроалканами позволяет получать нит-
ронитрилы [235]:
R NO2 R NO2
| | ТЭВАХ | |
PhCHCN + Me2C(CH2)2Br ■ ^ц ц > PhC(CH2)2CMe2 (20—82%)
CN
Зфиры а-галогенкислот алкилируют фенил ацетонитрилы,
давая производные, содержащие одновременно нитрильную и
алкоксикарбонильную функции [233]:
GN
PhCH(R)CN + XCH2CO2R' —> PhC(R)—CH2CO2R'
R, R', X (выход, %): Me, изо-Рг, Br (74); Me, трет-Вп, Cl (76); Me, цик-
ло-СвНц, Вг (78); Et, трет-Ъи, Cl (77); н-Рг, трет-Ци, Cl (74); изо-Pr, трет-Ъи,
Br (56); н-Bu, трет-Bu, Br (69); н-С5Нц, трет-Ви, Cl (64); н-С5Ни, цик-
ло-СбНп, Cl (26); Me2N(CH2)2( rper-Bu, Br (80); Ph, трет-Ви, Cl (97); PhCH2>
rper-Bu, Cl (95)
Рекомендуется использовать трет-бутиловые эфиры галоген-
кислот, поскольку онн трудно гидролизуются.
Для получения функционально замещенных нитрилов можно
использовать и алкилирование функционально замещенных
фенил ацетонитрилов. Так, а-алкоксифенилацетонитрнл алкили-
руется в условиях межфазного катализа с образованием а-ал-
коксинитрилов [242}:
PhCHCN+R'X —> PhC(R/)CN
I I
OR OR
Rf R'X (выход, %): Me, EtBr (75); Me, н-PrBr (73); Me, мзо-РгВг (45); Me,
CH2=CHCH2C1 (73); Me, PhCH2Cl (70); Ph( CH2Br2 (70); Ph, Br(CH2)2Br (64);
Ph, Br(CH2)3Br (15); CH2Ph, EtBr (71); CH2CH=CH2, EtBr (44); Me,
4-O2NCbH4CI (42); CH2CH2C1, 4-O2NCBH4CI (50); Me, Me2C=CHCH2Cl (78);
Me, BrCH2Br (70); Me, Br(CH2)2Br (12); Ph, EtBr (63); Ph, н-РгВг (66); Ph.
Me2CHBr (30); Ph, CH2=CHCH2Cl (71); Ph, Me2C=CHCH2Cl (69)
а-(р-Хлорэтокси)фенилацетонитрил циклизуется при
действии водного раствора NaOH в присутствии ТЭБАХ до
соответствующего оксетана [242]:
-О
PhCH(CN)O(CH2)2Cl —> I/CN (50%)
104
Интересная реакция протекает при алкилировании алкил-
фенилацетонитрилов в присутствии серы в условиях
межфазного катализа (ДМСО — вода — NaOH — ТЭБАХ). В
результате этой реакции образуются тиоэфиры нитрилов [267]:
PhCH(R)CN + R'X-{-S —> PhC(R)(CN)SR' (49—72%)
Алкилирование замещенных арилацетонитрилов о-нитробен-
зилхлоридом (или бромидом) в системе бензол — 50%-ный
NaOH —ТЭБАХ при 40— 55 °С привело к 2-арил-2-(о-нитробен-
зил)алканкарбонитрилам, которые были использованы для
дальнейшего синтеза замещенных 3,4-дигидрохинолинов путем
восстановительной циклизации [256].
В заключение можно отметить еще алкилирование
соединений Райссерта, которое легко проходит с разнообразными ал-
килгалогенидами, л-нитрохлорбензолом и другими активными
галогенидами [237]:
Описано бензоилирование дифенилацетонитрила бензоилхлори-
дом с образованием бензоилдифенилацетонитрила с выходом
64% [241]:
Ph2CHCN + PhCOCl —> Ph2C(CN)COPh
Алкилирование амииов. Алкилирование в а-положение К
аминогруппе можно осуществить, превращая амины
предварительно в основания Шиффа, которые далее легко алкилируются
в условиях межфазного катализа [263, 292, 293]:
+ R'"X __► RR'C=NCHR"R'"
Этот прием лежит в основе синтеза а-аминокислот: полученное
из бензофенона и аминоацетонитрила шиффово основание (R =
= R' = Ph, R" = CN) алкилируют и затем подвергают
кислотному гидролизу [263]. Алкилированные шиффовы основания
получали со следующими выходами: 85% (R"' = Me), 90%
(Et), 79% (аэо-Рг), 76% (шю-СБНп), 82% [МеСН2СН(Ме)1
75% (PhCH2).
Типичная методика алкнлирования оснований шиффа
приведена ниже.
Синтез Ph2C=NCH(Et)CN. К охлажденной (вода со льдом) смеси
1 г основания Шиффа, 0,1 г ТЭБАХ и 1,1 г 50%-ного NaOH добавляют по
каплям в течение 1—2 ч 0,4 мл этилбромида, перемешивают 2 ч при 0°С;
через 24 ч (комнатная температура) разбавляют смесь 20 мл дихлорметаыа
и 40 мл воды. Водный слой извлекают дихлорметаном (3 X Ю мл).
Объединенные органические слои промывают водой (3 X Ю мл), 10 мл насыщенного
раствора NaCl, высушивают (MgSO4>, удаляют растворитель. Выход 1,08 г
(адаслр; 90% о? теоретического).
№
Интересно, что при использовании метода экстракции
ионных пар в данном случае выход конечного продукта заметно
ниже (всего 77%).
Алкилирование шиффовых оснований было использовано
также для синтеза разветвленных аминов [292]. С этой целью
получают основание Шиффа из бензальдегида и бензиламина,
затем алкилируют в системе твердая фаза — жидкость (КОН +
+ К2СО3 — ацетонитрил) алкилгалогенидом и полученное ал-
килированное основание Шиффа подвергают кислотному
гидролизу; выходы аминов 52—80% (считая на исходный нераз-
ветвленный амин).
Алкилирование арилуксусных кислот. Эфиры кислот, не
активированные электроноакцепторными заместителями, не алки-
лируются в условиях межфазного катализа. Алкилирование
арилуксусных кислот осуществляется обычно в жидком
аммиаке в присутствии амида натрия или М-изопропил-М-циклогекси-
ламида лития (в последнем случае используют в качестве
растворителя ТГФ) и Дает, как правило, плохие результаты в
условиях межфазного катализа. Однако комплексообразование
арилуксусных эфиров типа ArCH (R)CO2R' с трикарбонилом
хрома повышает кислотность эфиров до такой степени, что
позволяет осуществить алкилирование даже их метиловых
эфиров, которые в обычных условиях межфазного катализа омы-
ляются [247]. Так, алкилирование комплекса трикарбояилхро-
ма с метиловым эфиром алкилфенилуксусной кислоты легко
проходит в присутствии гексадецилтриметиламмонийбромида
'(40% катализатора от массы субстрата) в системе 50%-ный
NaOH — бензол при комнатной температуре [247]. Общая
схема реакции для метиловых эфиров арилуксусных кислот Дана
Ниже:
R'Br
PhCH(R)CO2Me > (СО)3Сг ■ PhC(R) (R')CO2Me
R (вцхол^- %J_: Me, Me (70); PhCHk PhCH2 (100); CH2 = CHCH2,
CH CHCH (100); СНезССН2, СНезССН2 (100)
Химическим или фотохимическим окислением образующихся
комплексов трикарбонилхрома можно количественно выделить
лиганды. Также легко проходит алкилирование 1-метоксикарбо-
нил-1-#:индана [270]:
Следует отметить, что алкилирование комплексов
циклических эфиров приводит исключительно к з/сзо-изомерам {по от-
106
ношению к группе Сг(СО)3]:
(СО)аСг
(СО)3Сг
В значительной степени стереоспецифически алкилируются
также линейные эфиры, например:
CH2CO2R
tr(CO)3
В"
А В
(общий выход 45%; А:Б=72?28)
Это указывает на сходство межфазного катализа с SN2-peaK-
циями в диполярных растворителях. В обоих случаях
возникают тесные ионные пары между карбанионом и противоионами.
Описано алкилирование 9-этоксикарбонилфлуорена фена-
цилхлоридом в системе твердая фаза — жидкость (К2СО3 —
MeCN) в присутствии 18-крауна-6 [84]:
PhCOCH2Cl
CO2Et
(89%)
EtO2C
Алкилирование эфиров ацилуксусных кислот. Предметом
многочисленных исследований было алкилирование эфиров аце-
тоуксусной кислоты. Первое из них относится еще к 1954 г.
[294], когда было замечено, что алкилирование этилового
эфира ацетоуксусной кислоты 1,3-дихлорбутеном-2 ускоряется
в присутствии аминов (фактически катализатором служила
четвертичная соль, образующаяся при реакции аминов с
хлоридом— алкилирующим агентом). Выход моноалкильного
производного составил в оптимальных условиях 57% [294—296].
Позже алкилирование метилового эфира ацетоуксусной
кислоты было подробно исследовано [271]. Показано, что в системе
107
бензол — вода — NaOH в присутствии аликвата 336 алкилиро-
вание этого'эфира активными алкилгалогенидами, такими, как
бензилхлорид, аллилхлорид, аллилбромид, пренилхлорид, ге-
ранилбромид, проходит с удовлетворительными выходами [271]:
СН3СОСН2СО2Ме + RX —> CH3COCH(R)CO2Me
RX (выход, %): СН2==СНСН2С1 (30)- СН2=СНСН2Вг (85); Ме2С=СНСН2С1 (30);
PhCH2Cl (85); Ме2С=СНСН2СН2С(Ме)==СНСН2Вг (85)
Для насыщенных неактивированных алкилгалогенидов
использовали методику экстракции ионных пар [215, 273, 297].
Алкилирование ацилуксусных эфиров, прежде всего эфиров
типа ацетоуксусного эфира, в системе жидкость — жидкость
ограничено тем, что наряду с С-алкилированием в этой системе
возможно, и иногда проходит, О-алкилирование [292, 297].
Кроме того, в ряде случаев скорость реакции С-алкилирования
меньше скорости гидролиза сложного эфира, и в результате
во время реакции образуется ацетоуксусная кислота, которая
Далее распадается. Наконец, если даже алкилирование успевает
осуществиться, в водной системе в присутствии NaOH
возможен распад алкилированного продукта до кетона. Это
наблюдается, например, в случае алкилироваиия этилацетоацетата
геранилбромидом или геранилхлоридом в системе бензол —
50%-ный NaOH — ТЭБАХ, когда наряду с геранилацетоуксус-
ным эфиром (выход 25—30%) образуются заметные
количества (до 30—40%) геранилацетона. Для уменьшения
возможности гидролиза эфиров ацетоуксуснои кислоты предложено
использовать более трудно гидролизуемые эфиры — грег-бутило-
вый, изопропиловый или, хотя бы, метиловый.
Проблема алкилироваиия ацетоуксусных эфиров без
осложнений решается при переходе к системе твердая фаза —
жидкость. Недавно было показано, что в системе NaOH —
бензол в присутствии бензилгексадецилдиметиламмонийхлорида
(БГДДМАХ) при 50—60 °С метил- и этилацетоацетаты легко
алкилируются как активированными (бензилгалогениды), так
и неактивными (метилиодид, N, N-диметиламиноэтилхлорид)
алкилгалогенидамн, образуя с удовлетворительными выходами
моноалкилпроизводные [198]:
CH3COCH2CO2R + R'X —у CH3COCH(R')CO2R
R, R'X (выход, %): Me, Mel (42); Me, PhCH2Cl (71); Et, PhCH2Cl (72); Me,
PhCH2Br (80); Me, 4-7-р£7--ВиСБН4СН2Вг (62); Me, Me2N(CH2)2Cl (25); Et
Me2N(CH2)2CI (45)
Типичная методика алкилирования ацетоацетатов приведена
ниже.
Синтез метилового эфира 2-6ензилацетоуксусной
кислоты. К взвеси 60 г порошкообразного NaOH в 80 мл бензола, содержащего
3,9 г БГДДМАХ, добавляют при интенсивном перемешивании 174 гметилацето-
ацетата, через 15 мин добавляют по каплям при 50—60 °С 307 г бензилбро-
мида, перемешивают при той же температуре 2 ч, фильтруют, фильтрат
подвергают разгонке.
108
Алкилирование ацетоуксусного эфира этилтозилатом в
двухфазной системе показало, что в системе твердый КОН —
бензол образуется исключительно продукт С-алкилирования, в то
время как в системе КОН—1,2-диметоксиэтан или КОН — ди-
этиловый эфир в присутствии каталитических количеств дицик-
логексано-18-крауна-6 образуется 46—54% продукта С-моноал-
килирования и 52—41% продукта О-алкилирования [297].
Описан пример алкилирования этилацетоацетата 1,2-дибром-
этаном с образованием 1-ацетилциклопропанкарбоновой-1
кислоты, однако реакция проводилась в условиях экстракции
ионных пар, т. е. в присутствии эквимольного количества
четвертичной аммониевой соли ТЭБАХ [248].
Отмечен также случай алкилировання циклического Р-ке-
тоэфира — 1-этоксикарбонилциклопентанона. При проведении
реакции в системе бензол (или хлороформ)—вода — NaOH
с использованием в качестве катализатора (1,3-диметил-2-этил-
гексадецил)пентилпиридинийтетрафторбората продукты
С-алкилирования образуются с выходом 57—66% [272]:
RBr / \ А
О О
R = СН2=СНСН2) PhCH2
Алкилирование производных малоновой кислоты. При
использовании системы жидкость — жидкость для реакции
алкилирования эфиров малоновой кислоты существуют те же
ограничения, что и для ацетоуксусного эфира (см. выше). Для
предотвращения гидролиза по эфирной группе рекомендуется вводить
в реакцию трег-бутиловый эфир малоновой кислоты, а в случае
применения малоактивных алкилирующих агентов, например
tt-бутилбромида, добавлять к реакционной смеси ДМСО [277].
При использовании избытка алкилирующего средства
образуются диалкилпроизводные.
RX
CH2(CO2Bu-rper)2 >■ RCH(CO2Bu-rper)2 [или R2C(CO2Bu-7-per)2]
RX (выход моноалкилпроизводного, %): PhCH2CJ (70); СН2=СНСН2С1
(46,5); EtBr (53); н-BuBr (45); Br(CH2)3Cl (55); С1(СН2)2СО2Рг-изо (70);
у-пнперидилпропилбромид (16,5); Br(CH2)4Br (7)
Общая методика алкилирования гр^т-бутилмалоиа-
тов. Смесь 0,025 моль грет-бутилмалоната, 15 мл 50%-ного NaOH и 0,2 г
ТЭБАХ смешивают с эквимольным количеством (или избытком)
алкилирующего агента (в случае малоактивных галогенидов добавляют немного ДМСО),
нагревают 2 ч при 40—45 °С при интенсивном перемешивании, разбавляют
водой, извлекают бензолом, органический слой сушат (MgSO4), растворитель
удаляют, выделяют алкил-грег-бутилмалонат разгонкой в вакууме или
кристаллизацией нз петролейного эфира.
Использование в этом случае системы твердая фаза —
жидкость позволяет обойти все затруднения [84]. Так, при нагре-
109
йании (110°С, 1 ч) смеси эквимольных количеств этилмало-
ната и w-бутилбромида в присутствии эквимольного количества
поташа и ТБАБ образуется диэтиловый эфир я-бутилмалоновой
кислоты с выходом 93%- Нагревание смеси этилмалоната с ал-
лилхлоридом (1:1) с КгСО3 и дициклогексано-18-крауном-6
при 90 С в течение 1 ч дает диэтиловый эфир алл ил малоновой
кислоты с выходом 94%.
Алкилирование малонового эфира 1,2-дибромэтаном по
методу экстракции ионных пар (50%-ный NaOH — ТЭБАХ)
приводит к циклопропандикарбоновой-1,1 кислоте (выход 75%)
[225]. Описано алкилирование ацилзамещенного этилмалоната
аллилиодидом в присутствии ВщЫОН в отсутствие
растворителя [278].
(CH2)2COCH(CO2Et)2 —> \У \cH2)2COC(CO2Et)2
Удовлетворительные результаты дает также метод
экстракции ионных пар [298].
Очень активен в реакциях алкилирования циануксусный
эфир. Активные галогениды (аллилбромид, бензилхлорид)
реагируют с циануксусным эфиром с большой легкостью, давая
диалкилпроизводные. Например, метиловый эфир циануксусной
кислоты в системе твердая фаза — жидкость алкилируется бен-
зилхлоридом в отсутствие растворителя, образуя с выходом
87% метиловый эфир дибензидциануксусной кислоты [242]:
ТЭБАХ, Na2CO3
NCCH2CO2Me + PhCH2Cl -> (PhCH2)2C(CN)CO2Me
В системе хлороформ — вода (NaOH, ТБАГС) удается проал-
килнровать метиловый эфир циануксусной кислоты я-гексили-
одидом с образованием моноалкилпроизводного [250]:
NCCH2CO2Me-bH-CeH13I —> NCCH(C6Hl3-H)CO2Me (62%)
Можно осуществить также алкилирование алкилзамещенных
циануксусных эфиров, например [275]:
R'Br. ТЭВАХ —50%-ный NaOH —СН2С12
RCH(CN)CO2Bu-rper +
—j- RR/C(CN)CO2Bu-7-per
R( Rf (выход, %): Me, СН2=СНСН2 (67); MeSCH2CH2l MeSCH2CH2 (87;
этиловый эфир)
я-Бутилхлорид и изопропнлхлорид не реагировали с этими
субстратами в условиях межфазного катализа.
Описано алкилирование динитрила малоновой кислоты ал-
килбромидами в присутствии аликвата 336 в системе вода —
ПО
NaOH [1]:
H2C(CN)2 + RBr —>- R2C(CN)
R (выход, %): н-Bu (87); h-C6Hi3 (90)
Хорошие результаты были получены при алкилировании этого
динитрила полимерносвязанными алкилгалогенидами [268],
например (R — полимер):
BuNOH, NaOH
R-CH2C1 + CH2(CN)2 оС[2СбН4НгО-» R-CH2CH(CN)2 (96%)
В заключение стоит упомянуть, что активирование субстрата
может быть достигнуто не только сочетанием двух
карбонильных групп, карбонильной и цианогрупп или двух цианогрупп,
но и сочетанием цианогруппы с фосфамидной группой в р-по-
ложении [249]:
Bu4NOH или ТЭБАХ
(Me2N)2P(O)CH(R)CN+R'X
—► (Me2N)2P(O)C(R)(R')CN (71—100%)
X = C1, Br, I; R = H, Me; R' = Alk, аллил, PhCHj
Алкилирование сульфонов. В условиях межфазного
катализа возможно алкилирование разнообразных а л кил ар и л
сульфонов. Так, в присутствии ТЭБАХ в системе NaOH — йода МО-
гут быть проалкилированы арилсульфонилиодметаны [282]'*
/~\ R'x уГ~\
R—К У—SO2CH2I > R—f N)—SOaCHIR'
R, R'X (выход, %); H, EtBr (71); Н, н-BuBr (74); Н, PhCH2CI (89); Me,
EtBr (85); Me, CH2=CHCH2Br (60); Me, к-СаН1БВг (55); Me, PhCH2Cl (78)
Реакция очень чувствительна к пространственным
затруднениям и вторичные галогениды (например, 2-бромпропан) не
алкилируют сульфоны в приведенных выше условиях. При
реакции тех же сульфонов с а, (о-диброалканами образуются 1-иод-
1-фенилсульфонилциклоалканы и дисульфоны:
—SO2CH2I + (ВгСН2)2А —>
1 СН2
^ ^A + (rt-RC6H4—SO2CHICH2)2A
сн2
A; R (выход сульфонилциклоалкаиа и дисульфоиа, соответственно, %): —,
Me, (68, 23);.—, Н (54, 46); СНгСН2, Н (37); СеН4(о), И (39)
Общая методика алкилироваиия арил-1-иодалкил-
сульфонов. Смесь 5 ммоль арил-1-иодалкилсульфоиа, 7,5 мл 50%-ногб
NaOH, 0,05 г ТЭБАХ и 7,5 ммоль RX (5,5 ммоль в случае PhCH2Cl)
перемешивают 2—3,5 ч (реакция проходит экзотермически) при 25—30 СС, смесь
разбавляют водой, извлекают хлороформом (4X15 мл), органический слой
промывают водой, сушат (MgSOO, после упаривания отделяют кристаллы.
сушат на воздухе.
Ml
Аналогично проводят реакцию и с а, о^-дибромалканами.
В близких условиях проходит также алкилирование арил-1-
хлор(или бром)метилсульфонов и арил-1,1-дихлор(или ди-
бром)метилсульфонов [281]:
RZ
ArSO2CHXY -—► ArSO2C(R)XY
Аг, X, Y, RZ (выход, %): 4-МеСбН4, Н, Вг, EtBr (67); 4-МеСБН4, Н, Вг,
«-ВиВг (68); 4-МеСбН4, Н, Cl, PhCH2Cl (60); Ph, Cl, Cl, PhCH2Cl (84); Ph,
Cl, Cl, EtBr (72); Ph, Br, Br, PhCH2Cl (75)
В том случае, если рядом с галогенсодержащим звеном
имеется —СНг-группа, проходит не алкилирование, а
элиминирование галогенводорода с образованием алкена. Реакция
между арил-1,1-ДИХлорметилсульфонами и а,о-дибромидами
(например, 1,4-дибромбутаном) идет по обоим атомам с
образованием соединения
ArSO2CCl2(CH2)4CCl2SO2Ar
Тозилметилизоцианиды, которые в обычных условиях алки-
лирования образуют смеси моно и диалкилпроизводных, в
условиях межфазного катализа дают только монопроизводные с
высоким выходом [262, 280]. Возможно и дальнейшее
алкилирование, но с небольшими выходами. Реакция проводится в
системе СН2С12 —Н2О (NaOH, Bu4NI):
—► rt-MeCBH4SO2CH(R)N=C (40—95°/0)
R = Me, Et, CH2=GHCH2, изо-Pr, я-Рг, PhCH2
Описано алкилирование в межфазных условиях этилового
эфира 3-метил-4-фенилсульфонилбутен-2-овой кислоты [283]:
Me R Me
i RX | I
PhSO2CH2G=CHCO2Et *■ PhSOsCH—C=«CHCO2Et (90%)
X = Br, I; R = Me, Me2G=CHCH2
Наконец, было показано [284], что в условиях межфазного
катализа алкилируются не только сульфоны, но и сульфонами-
ды. Реакция в обоих случаях идет медленно, но ее ускоряют
добавки небольших количеств гексаметапола (гексаметилфос-
фортриамид).
PhCHR— SO2X+ R'Y —у PhCRR'-SO2X
R, X, R'Y (выход, %); H, 4-MeC6H4, EtBr (80); Н, 4-МеСбН4, «-BuBr (84);
Н, 4-МеСбН4) СН2=-СНСН2Вг (63); Н, 4-МеС6Н4, PhCH2Cl (90), Н, 4-МеСбН4,
Вг(СН2)4Вг (56); Н, морфолиио, EtBr (61); Н, морфолиио, «-РгВг (64); Н, мор-
фолиио, СН2=СНСН2Вг(71); Н, Et2N, PhCH2Cl (66); Me,4-MeCGH4, MeBr (66);
Me, 4-МеСбН4) PhCH2Cl (63); Ph, Me, PhCH2Cl (62); F, Ph, EtBr (62); Cl,
Ph, EtBr (77); Cl, морфолиио, EtBr (81)
Общая методика ал к и лироваиия сульфоиов и суль-
фонамидов. Смесь 0,01 моль сульфоиа или сульфоиамида, 0,011—0,02 моль
алкилгалогеиида, 0,2 г ТБАБ, 1 мл гексаметапола и 15 мл 50%-иого NaOH
перемешивают 2—5 ч при 35—60 °С, разбавляют 50 мл воды, отделяют
продукт фильтрованием или извлекают бензолом (3X25 мл).
И?
Прочие реакции замещения
В условиях межфазного катализа (50%-ный NaOH, ТЭБАХ)
четыреххлористый углерод хлорирует некоторые соединения с
подвижными атомами водорода, например морфолид фенилме-
тансульфокислоты [284]:
PhCH2SO2—N. \)+СС14 —► PhCCl2SO2—N. ,0
В аналогичных условиях из фенилацетилена получают хлор-
фенилацетилен с выходом 45% [299]:
PhC=CH + CCl4 —> PhOsCCl
Описан межфазный каталитический синтез бром- и иодаре-
нов. Реакция идет при действии на раствор арена в
хлороформе избытка галогенирующего агента (бромтрихлорметан, иод-
метан, молекулярный иод) в присутствии каталитических
количеств 18-крауна-6 [61]. Реакция основана на генерировании
арил-радикалов.
В присутствии 18-крауна-6 арендназонийборфториды
реагируют в бензоле с ацетатом калия, превращаясь в биарилы
[300]. Реакция проходит через образование диазоацетата,
дальнейшие превращения которого приводят к арил-радикалу:
—> я-С1СбН4№ОАс —>
rt-ClC6H4N=N—О—N=NCeH4CI-n —>■
n-ClCBH4N+
rt-ClC6H4N=N—О • + N2 +
. + СБНБ —► п-С1СбН4— СБНБ
Синтез 4-хлорбифеиила. К смеси 1,37 г 4-хлорбеизолдиазоиий-
борфторида и 0,08 г 18-крауиа-6 в 60 мл бензола добавляют в темноте при
комнатной температуре в атмосфере азота 1,2 г ацетата калия (в один
прием), Смесь перемешивают 1,5 ч, фильтруют, промывают насыщенным
раствором NaCl, водой, сушат (Na2SC>4), удаляют растворитель, хроматографией
на А12Оз (растворитель н-гексаи) выделяют 4-хлорбифеиил. Выход 0,92 г
(80% от теоретического); т. пл. 76—77 °С.
Аналогично получены и другие бифенилы ХС6Н4 —Ph
X (выход, %): 4-МеО (80); 4-Ме (73); З-Ме (58); Н (62); 4-Вг (81); 4-Н (60);
4-NO2 (85)
а также 2,4,6-триметил-4/-хлорбифенил (55%), 4-(тиенил-2)-Ь
хлорбензол (62%) и 1-нитро-4-(тиенил-2)бензол (38%). Таким
образом, реакции? Гомберга — Бахмана—Хея также можно
проводить в условиях межфазного катализа.
Недавно [301] обнаружена новая необычная реакция
замещения ацетатной группы в ацилалях на трихлорметильную
в условиях генерирования дихлоркарбена по Макоша (см.
с. 147 [441]). Оказалось, что ацилали акролеина, кротоно-
вого альдегида, 3,3-диметилакролеина, коричного альдегида и
т
3,3-дихлоракролеина реагируют с хлороформом при действии
50%-ного водного раствора едкого натра в присутствии
каталитических количеств ТЭБАХ по схеме:
СС13
СНС1з, 50%-ный NaOH I
RR'C=CHCH(OCOCH3)2 такд 9П0Г * RR'C=CHCHOCOCH8
(50-75%)
Среди других реакций замещения следует отметить реакции
изотопного обмена. В условиях межфазного катализа (CH2CI2 —
D2O — NiOD—ТЭБАХ) проходит дейтерирование индена в
положения 1 и 3 (выход 95%) и флуорена в положение 9 (98%)
[302]. В присутствии триметилоктадециламмонийбромида в
системе твердая фаза—жидкость (бензол — NaOD) тиазол дей-
терируется в положения 2 и 5 (93—96%), 2-замещенные тиазо-
лы — в положение 5 (выход 80—90%), за исключением 2-трет-
бутилтиазола, в случае которого выход 5-замещенного
составляет лишь 20%» а в основном проходит обмен на дейтерий в
а-положении в алкильной группе; 5-этилтиазол дейтерируется
в положение 2 (92%), а 4-метил- и 4-трет-бутилтиазолы — в
положения 2 (93 и 83% соответственно) и 5 (20 и 8,5%,
соответственно) [303]. В октаноне-2 [1], ацетофеноне и фенилацето-
нитриле [304] дейтерирование идет по метиленовой группе
(90-95%),
Отмечено, что в условиях межфазного катализа 1-метилин-
ден легко изомеризуется в 3-метилинден [302].
ПРИСОЕДИНЕНИЕ К ПРОСТЫМ И КРАТНЫМ СВЯЗЯМ
В этом разделе рассмотрены реакции присоединения в
условиях межфазного катализа по простым связям (реакции
внедрения), по различным кратным связям, а также реакции
окисления и восстановления.
Реакции внедрения
Описан ряд реакций внедрения дихлоркарбена,
генерированного в межфазных условиях в системе жидкость — жидкость в
присутствии ТЭБАХ. Так, дихлоркарбен внедряется в метил-
циклогексан и метоксициклогексан, образуя соответственно 1-
метил-1-дихлорметилциклогексан (выход 4%) и 1-метокси-Г-ди-
хлорметилциклогексан (13%) [205], в цис- и rpawc-декалины,
давая цис- и rpawc-9-дихлорметилдекалины (29 и 4%,
соответственно) [305], в этилбензол, приводя к 2-фенил-1,Ьдихлорпро-
пану (2%) [276]. Дихлоркарбен реагирует также с кумолом
[305, 306] с образованием 2-метил-2-фенил-1,1 -дихлорпропана
(18—22%) и тетралином, давая 1-дихлорметилтетралин (21%)
[306]. Отличные результаты были получены при внедрении
дихлоркарбена по С—Н-связи адамантана, 1-метил- и 1?3-дим$-
114
тилаДамантанов в системе бензол — вода — NaOH — ТЭБАХ
[307]:
(91-100%)
СНС12
Во всех случаях внедрение проходит по связи С-1—Н с
образованием 1-дихлорметилпроизводных, 1-бром-, 1-метокси- и
1-карбоксиадамантаны практически не реагируют с дихлоркар-
беном [307]. В случае диамантана реакция с дихлоркарбеном
проходит по положениям 1 и 4, приводя к 1-дихлорметил- и 4-
дихлорметилдиамантанам с выходами 63 и 37%, соответственно
[308]. 1-Метил-1,2-дигидрохинолин также реагирует с
дихлоркарбеном. При этом образуются 1-метил-2-дихлорметил-1,2-ди-
гидрохинолин (30%) и 1-метил-4-дихлорметил-1,4-дигидрохино-
лин (20%) [308].
В качестве типичного примера приведем методику получения
1-дихлорметиладамантана [277].
Синтез 1-дихлорметнладамантана. Смесь 13,6 г адаман-
тана, 20 мл 50%-ного NaOH, 20 мл бензола н 0,4 г ТЭБАХ интенсивно
перемешивают прн 43 °С н добавляют по каплям (около 6 ч) 80 мл хлороформа.
Перемешивание продолжают 30 мин, отделяют 1-дихлорметиладамантан.
Выход 54% (на взятый и 91% на израсходованный адамантан).
Дибромкарбен, генерированный в межфазных условиях
(CH2Cl2 — 50%-ный NaOH— ТЭБАХ), реагирует с адаманта-
ном, образуя 1-дибромметиладамантан с выходом всего лишь
35% [309].
Внедрение дихлоркарбеиа по С—Н-связям простых
линейных эфиров, например диизопропилового эфира [305], или
циклических эфиров, например тетрагидрофурана [306], дает
низкие выходы продуктов внедрения (5—18%). Дибромкарбен
внедряется в диизопропиловый эфир, образуя изопропил(2-ди*
бромметилизопропиловый) эфир с выходом 43% [309].
Однако 2-замещенные 1,3-диоксоланы реагируют с
дихлоркарбеном в условиях межфазного катализа селективно,
исключительно по связи С-2—Н, образуя с высокими выходами 2-ди-
хлорметил-1,3-диоксоланы [311, 312]. Аналогично проходит
реакция с 1,3-диоксанамн с образованием 2-дихлорметил-1,3-ди-
океанов. Применение бромоформа вместо хлороформа позволяет
получать 2-дибромметильные производные 1,3-диоксанов и 1,3-
диоксоланов [311,312].
■vS
п=\. 2
115
Ёыходы составляют 84—-<НЙ/О на вступивший в реакцию диоксолан йЛй Дй-
оксан, однако при наличии в положении 2 электроноакцепторных
заместителей конверсия субстрата снижается с 55—74% (R = Me, Et, н-Pr, изо-Pr,
w-Bu, трет-Ъи н-С5Н1Ь h-C6Hi3, «-С7Н15, h-C8H17i h-C9Hi9, h-Ci0H2i, цик-
ло-СвНи, Ph, 4-MeOC6H4) до 12—37% (R =- PhCH2, PhCH2CH2, C1CH2, BrCH2,
ClCH2CH2l CH2CH2CN, 3- или 4-МеС6Н4, 3- или 4-С1С6Н4). При R = СС13 или
4-О2МСбН4 реакция не идет. То же наблюдается и в случае внедрения ди-
бромкарбена.
Общая методика синтеза
2-дихлорметил-1,3-диоксолан ов и -лиоксанов [281]. Смесь 0,#> моль диоксолана (или диоксаиа),
500 г 50%-ного NaOH, 500 мл хлороформа и 1 г ТЭБАХ интенсивно
перемешивают (800—1000 об/мин) в течение 24 ч при охлаждении льдом, добавляют
300 г Na2SC>4, перемешивают, декантируют, остаток извлекают эфиром (3 раза
по 200 мл). Объединенные органические фазы сушат (Na2SO4), растворитель
удаляют, остаток перегоняют в вакууме.
Реакцию с дноксанами ведут в течение 40 ч. Для генерирования дибром-
карбеиа из 375 г бромоформа в качестве растворителя добавляют "500 мл
СН2С12.
Реакция дихлоркарбена с некоторыми спиртами приводит к
хлоридам:
R—ОН + : СС12 —* R—О—СС12 —> R—О—С—С1 —> RC1 + СО
I
н
=o c
Низшие спирты (метиловый, я-пропиловый, изопропиловый,
г/?ет-бутиловый) и циклогексанол дают сложную смесь
продуктов; этанол и 2,2,2-трифторэтанол, как отмечалось выше,
образуют ортоэфиры [133]. С хорошими выходами были получены
(адамантил-1) хлорид (94%) из 1-адамантилового спирта и бен-
зилхлорид (90%) из бензилового спирта в системе 50%-ный
NaOH — СНС13 — ТЭБАХ [313]. 2-э/сзо-Норборниловый спирт
превращается в этих условиях в 2-э/сзо-норборнилхлорид с
выходом 90%, однако 2-эмЭо-норборниловый спирт дает смесь 2-
экзо- и 2-эндо-норборнилхлоридов с выходами 47 и 44%,
соответственно. (Адамантил-1) метиловый спирт образует смесь
(адамантил-1) хлорида (выход 40%), гомоадамантилхлорида
(13%) и (адамантил-1)метилформиата (35%).
Реакция дихлоркарбена с первичными аминами начинается
с координации электрофильного дихлоркарбена со свободной
парой электронов атома азота. В образующемся цвиттер-ионе
происходит перенос протона от азота к углероду с образованием
N-замещенного аминодихлорметана, из которого образуется изо-
нитрил последовательным р- и а-элиминированием HCI [314—
316]. Такой межфазный вариант карбиламинного синтеза изо-
нитрилов по Гофману значительно проще, чем двухстадийный
метод с использованием фосгена. Для синтеза метил- и этили-
зоцианидов удобнее использовать дибромкарбен [314].
Типичная методика синтеза изоцианидов приведена ниже.
116
Синтез фенилизоциаиида [283]. Смесь 18,6 г анилина. 16 мл
хлороформа, 60 мл 50%-иого NaOH, 0,5 г ТЭБАХ и 50 мл СН2С12 интенсивно
перемешивают, примерно через 10 мин смесь вскипает, через 1 ч смесь
разбавляют водой, из органического слоя после высушивания выделяют
разгонкой фенилизоцйанид. Выход 11,7 г (57% от теоретического); т. кип. 52—53 °С
(12 мм рт. ст.)
Аналогично получены: MeNC (выход 50%), h-BuNC (60%), h-Ci2H25NC
(41%), цикло-CeHnNC (48%); rper-BuNC (50%), сс-нафтилизоцианид (20%),
PhCH2NC (40%).
Другой способ синтеза изоцианидов основан на реакции
N-сульфиниламинов с дихлоркарбеном [317]. Реакция
проходит в системе твердая фаза — жидкость (бензол или циклогек-
сан—КОН) с использованием в качестве катализаторов дибен-
зо-18-крауна-б, дициклогексано-18-крауна-6, ТБАХ или ТЭБАХ:
R—N=S=O —-> R—N=C
R (выход, %): Ph (85—96); 4-МеС6Н4 (80); 4-МеОС6Н4 (80); 4-С1С6Н4 (75);
мезнтил (75); цикло-СеНц (75)
Описанная выше реакция вторичных аминов с хлороформом
в условиях межфазного катализа [150, 151], очевидно, идет как
реакция внедрения дихлоркарбена по связи С—N с
последующим гидролизом дихлорметильной группы [318]:
RR'N—Н + :СС12 —► RR'N—СНС12 —* RR'N—CHO
Отмечено, что при реакции аллилбромида с бромоформом
в условиях межфазного катализа (50%-ный NaOH — ТЭБАХ)
наряду с 2,2-дибром-1-бромметилциклопропаном (44%)
образуется продукт нуклеофильного замещения брома в аллилбро-
миде на трибромметильный анион — 4,4,4-трибромбутен-1
(28%), а также тетрабромметан и пербромэтилен [319]:
СН2=СНСН2Вг
Вг\/\
£^СН2Вг + СН2=СНСН2СВгБ + СВг4
Интересно, что при использовании в качестве катализатора
межфазного переноса гексадецилтриметиламмонийбромида из
металлилбромида образуется только 2,2-дибром-1-бромметил-1
метилциклопропан (40%), в то время как в присутствии ТЭБАХ
получают только 4,4,4-трибром-2-метилбутен-1 (38%) [320].
Аналогично в присутствии ТЭБАХ нз аллилбромида, 4-бром-2-
метилбутена-2 и 4-бромбутена-2 образуются соответственно
4,4,4-трибромбутен~1 (50%) t 5,5,5-трибром-2-метнлпентен-2
(58%) и 5,5,5-трибромпентен-2 (40%); из первых двух
соединений образуются одновременно диены, а из последнего — 2,2-дн-
бром-1-метил-3*(2,2,2-трибромэтил) циклопропан. Хлороформ
образует с аллилбромидами только циклопропаны независимо от
природы катализатора,
117
Присоединение анионов к углерод-углеродным
кратным связям
Следует отметить, что реакции присоединения по углерод*
углеродным связям в условиях межфазного катализа изучены
недостаточно и в этой области еще открыто широкое поле
деятельности.
Межфазный катализ был применен для получения дихлори-
дов и дибромидов из алкенов действием галогеноводородных
кислот и пероксида водорода в присутствии ТЭБАХ. Метод
удобен тем, что устраняет необходимость работы с элементными
галогенами—потенциальными источниками опасности.
RC№=CHR' + 2HX + Н2О2 —►* RCHX—CHXR'
Общая методика галогеиирования алкенов [321].
К охлажденной льдом смеси 50 ммоль алкеиа, 50 ммоль СаСЬ, 10 мл
концентрированной НС1, 10 мл ССЦ и 100 мг ТЭБАХ добавляют по каплям 6 мл
30%-ного Н2О2, смесь оставляют нагреваться до комнатной температуры,
перемешивая 20 мин, разбавляют петролейным эфиром, промывают водой, су*
шат, удаляют растворитель, остаток перегоняют.
Этим путем получены: 1,2-дихлорциклогексан (76%), 1,2-
дибромциклогексан (95%), 1,2-дихлорциклогептан (75%), 1,2-
Дибромциклогептан (95%), 1,2-дихлорцйкЛооктан (77%), 1,2-
Дибромциклооктан (96 %), 1,2-дихлороктан (56%), 1,2-дибром-
октан (92%).
Разработана удобная методика синтеза вицинальных иод-
тиоцианатов и иодизотиоцианатоВ действием иодтиоциана на
алкены в системе вода — хлороформ в присутствии адогена 464
[322]:
NCS
Ph(R)C=CHR' -—> Ph(R)C—CHIR' + Ph(R)C—CHlR'
XXIX XXX
R, R' (выходы XXIX fi XXX, соответственно, %): H, H (55, 14); Me, H (71);
H, Me (31, 2)
Одновременно образуются иодгидрины (1—4%).
Общая методика синтеза тиоцианатов и изотиоциа-
и а т о в. К раствору 0,61 г тиоцианата калия в 1 мл воды добавляют 0,76 г
йода И 0,03 г адогена 464 в хлороформе (не содержащем спирта). Добавляют
по каплям 0,103 г цйклогексеиа, перемешивая 2 ч при комнатной температуре,
избыток иода удаляют, встряхивая с раствором NaHSCb, извлекают смесь
СНгСЬ, проводят через небольшую колонку с А12О3 для удаления адогеиа,
растворитель удаляют, остаток разделяют методом препаративной ТСХ.
Описана межфазная методика присоединения тиолов к а,Р-
непредельным кетонам в присутствии тетраалкиламмонийфто-
118
ридов, например [323]:
О
SPhO
PhSH
(87%)
В системе 50%-иый NaOH— Bu4NI при 0°С акриловые
альдегиды реагируют с 3-ацетилтиоальдегидами таким образом,
что на первой стадии проходит присоединение ацетилтиогруппы
по двойной связи, а далее альдольная внутримолекулярная
конденсация с дегидратацией и образованием тиациклогексен-3-
карбальдегидов-3 [324], например:
-С НО ^СНО ^ ^СНО
SAc
= R'=H (84%);
= R'=Me (85%)
Используя в качестве межфазного катализатора 18-краун-6,
удается присоединить в межфазной системе [сухой KCN —
МегС(ОН)СЫ — бензол или ацетонитрил] HCN к а,р-ненасы-
щенным кетонам. Например, в случае холестен-4-она-З
образуется смесь а- и fi-изомеров с общим выходом 83—85% [325]:
CN ,
р-изомер
(56-70%)
Описано нуклеофильное присоединение сульфида натрия и
этантиолата натрия к тронной связи фенилацетилена в
двухфазной системе в присутствии дибензо-18-крауна-6 в качестве
межфазного катализатора [326]:
ос-изомер
(7-18%)
PhCH==CHSEt
(цис-изомер; 96%)
EtSNa
PhteCH
Na2S
(PhCH=CH)2S
(22%)
Для ускорения реакции можно использовать каталитические
или, лучше, стехиометрические количества KCN.
Генерирование дихлоркарбена в условиях межфазного
катализа проходит, как уже упоминалось, через образование три-
хлорметильного аниона; в том случае, если олефин содержит
при двойной связи электроноакцепторные заместители, очень
часто наблюдается не дихлорциклопропанирование, а
присоединение трихлорметильного аниона по двойной связи. Таким
путем реагируют, например, акрилрнитрил ,[246, 327],
119
кротононитрил [328], нитрил транс-коричной кислоты [328],
метилакрилат [327], изопропилакрилат [328], изопропилкрото-
нат [323] и винилфенилсульфон [327]:
ТЭБАХ — 50%-ный NaOH
RCH^CHA + СНС13 ■> RCH(CCla)CHaA
R, А (выход, %): Н, CN (72); Me, CN (17); Н, СО2Ме (43); Pb, CN (14);
Н, СО2Рг-мзо (43); Me, СО2Рт-изо (20); Н, SO2Ph (70)
Если в а-положении к электроноакцепторной группе находится
метильная группа (алкилметакрилаты, метиловый эфир цис-2-
метилкротоновой кислоты и пр.), то образуются в основном
циклопропаны [327,328].
Аналогично алкенам, содержащим электроноакцепторные
группировки, реагирует и винилацетат, что указывает на
высокую электрофильность винильной двойной связи в винилаце-
тате [327, 329]:
СНС13 —50%-ный NaOH—ТЭБАХ
СН2-=СНОСОМе ■> CHsCH(CCl3)OCOMe (65%)
Кроме трихлорметильного аниона в условиях межфазного
катализа (50%-ный NaOH —ТЭБАХ) к винилацетату
присоединяются анионы, генерированные из алкилфенил- или алкокси-
фенилацетонитрила [330]:
Ph(R)CHCN+CH2=-CHOCOMe —>. Ph(R)C(CN)CH(Me)OCOMe
R (выход, %): Me (64); Et (70), Ph (15); OMe (56); ОРт-изо (53)
Продукты реакции образуются в виде смеси двух диастереоме-
ров, которые могут быть легко разделены препаративной ГЖХ.
Аналогично в тех же условиях реагирует с винилацетатом
соединение Райссерта (М-бензоил-1-циано-1,2-дигидроизохинолин),
давая а-(изохинолил-1)этилбензоат (выход 73,5%). Интересно,
что дифенилацетонитрил образует в основном динитрил тетра-
фенилянтарной кислоты — продукт электронного переноса. Фе-
нилацетонитрил дает 2-фенилкротононитрил и 3-метил-2,4-дифе-
нилглутаронитрил.
Трихлорметильный анион способны присоединять также ви-
вилбензоат, винилпивалат и 1-ацетоксибутадиен-1,3:
СН2=СНСН=СНОСОМе + СНС1а —► СН3СН=СНСН(ССЬ)ОСОМе
(20%) -
Енолацетаты кетонов, за исключением циклических кетонов,
не присоединяют трихлорметильный анион [330]. •
Общая методика проведения реакции между алкил-
или алкоксифенилацетонитрилом и винилацетатом [330].
К интенсивно перемешиваемой смеси 0,1 моль нитрила, 0,1 г ТЭБАХ, 15 мл
бензола или ацетонитрила и 5 мл (в случае алкоксипроизводных 10 мл)
50%-ного NaOH добавляют по каплям 10,4 г (0,12 моль) внннлацетата,
поддерживая температуру 15 °С охлаждением, перемешивают 1,5—2 ч при 20—
25 °С, разбавляют водой, отделяют органический слой, нейтрализуют, промы*
вают, высушивают (MgSO4). Продукт очищают разгонкой в вакууме.
Остается пока еще недостаточно изученной реакция
Михаэля, часто используемая для синтеза соединений с различными
функциональными группами из алкенов, содержащих электро-
ноакцепторные заместители. Впервые осуществление этой
реакции в условиях межфазного катализа было описано [331]
на примере присоединения 2-нитропропана к этилакрилату:
CH2=CHCO2Et + Me2CHNO2 —> Me2C(NO2)CH2CH2CO2H
Синтез 4-метил-4-нитровалериаковой кислоты [331 ].
100 г этилакрилата добавляют по каплям при интенсивном перемешивании
(температура не выше 50 °С) к смеси 89 г 2-китропропана, 10 мл 40%-ного
NaOH и 2 г ТЭБАХ, нагревают 2 ч при 50—60 °С, растворяют в бензоле,
промывают водой, разбавленной соляной кислотой, насыщенным раствором
NaHCOa, высушивают, подвергают разгонке. Выход 142 г (75% от
теоретического); т. кип. 135—137 °С (18 мм рт. ст.).
Позднее было описано присоединение СН-кислот к а,Р-не~
предельным нитросоединениям на примере метил-4,6-О-бензили-
ден-2,3-дезокси-3-нитро-а(или Р)-й-эрг/гро-гексен-2-пиранозида
[332, 333]. Интересно, что в случае а-изомера образуются
менее устойчивые лшяяо-изомеры [332].
Типичная методика синтеза приведена ниже.
Синтез метил- 4,6 -О-беизилиден-2-бис (этоксикарбо-
нилметил) - 2,3 - дидезокси - З-нитро-а-й-маннопиранози-
д а [332]. Смесь 105,9 мг метил-4,6-О-беизилиден-2,3-дезокси-3-нитро-а-О-
эрагро-гексен-2-пиранозида, 69 мг диэтилмалоната, 24 мл бензола, 9,6 мл 0,2 н.
NaOH и 12 мг ТБГДФБ перемешивают 2 ч при комнатной температуре.
Смесь промывают водой (3X5 мл), высушивают, удаляют растворитель,
полученный сиропообразный продукт очищают на колонке с силикагелем
(вымывание бензолом). Выход 88,4 мг (65% от теоретического); [а]2р —33,4°
(с=1, СНС1а).
Следует отметить, что ацетоуксусный эфир дает два манно-
изомера. В случае малодинитрила получают как глюко-, так и
лшяяо-изомер в соотношении 1 : 1,3 с общим выходом ~80%.
Соотношение манко- и глкжо-изомеров зависит от времени
проведения реакции; через 6 ч оно снижается до 0,3, через 24 ч —
до 0,1 и наконец весь лшяяо-изомер переходит в глкжо-изомер.
Присоединение СН-кислот к р-изомеру всегда приводит кглюко-
изомерам [333] (выходы 77—83%).
Для получения нормальных продуктов реакции из диацил-
метанов реакцию, проводят в присутствии следов NaOH. При
использовании 0,2 н. NaOH (см. общую методику синтеза) в
случае ацетилацетона образуются продукты дезацилирования
(выходы 81—83%).
Стандартная методика межфазного катализа (50%-ный
NaOH — ТЭБАХ) оказалась непригодной для реакции
Михаэля с а,р-непредельными альдегидами: наблюдалось глубокое
осмоление этих соединений. Однако применение системы
твердая фаза — жидкость (Na2CO3— бензол) позволило ввести в
реакцию Михаэля с малоновым и ацетоуксусным альдегидом
121
акролеин, кротоновый альдегид, Диметилацеталь фумарового
диальдегида, 3-(3-винилформил)-1-метил-2,2-дихлорциклопро-
пан и получить с удовлетворительными выходами нормальные
продукты присоединения [334].
RCH2CO2Et + R'CH=CHCHO —v RCHCO2Et
R'CHCH2CHO
R, R' (выход, %): CO2Et, H (50); CO2Et, Me (60); CO2Et, (MeO)2CH (44);
CO2Et, Ph (65); CO2Et, 1-метил-2,2-днхлорциклопропнл-3 (75); COMe, H (47);
COMe, Me (46); COMe, (MeO)2CH (64); COMe, 1-метил-2,2-дихлорциклопро-
пил-3 (54)
Общая методика проведения реакции Михаэля. К сме-
сн эквимольных количеств (по 0,05 моль) малонового или ацетоуксусного
эфира, соды и каталитических количеств ТЭБАХ в 20 мл бензола добавляют
при интенсивном перемешивании 0,05 моль альдегида. Смесь нагревают при
40—50 °С (диметилацеталь фумарового диальдегида выдерживают при 15 °С)
в течение 0,5—4 ч, отфильтровывают, фильтрат разбавляют эфиром,
промывают водой, сушат (MgSO4), разгонкой выделяют продукты присоединения.
При реакции коричного альдегида с ацетоуксусным эфиром
образуется 5-фенилциклогексенон-З с выходом 55%:
PhCH=CHCHO -f- CH3COCH2CO2Et —*
В этих же условиях илиденмалодинитрилы гладко реагируют с
бромциануксусным эфиром, образуя с высокими выходами (82—
95%) 1,1,2-трициано-2-этоксикарбонил циклопропаны [335].
Илиденцианацетаты взаимодействуют с броммалоновым и
бромциануксусным эфирами в системе ДМФА — КгСО3 — ТЭБАХ с
образованием транс-1,1,2-триэтоксикарбонил-2-циаиоциклопро-
панов с выходами 35—77% [336].
Циануксусный эфир дает нормальный продукт реакции Мн-
хаэля с выходом 20% только с акролеином при —15 °С, с
остальными альдегидами идет реакция Кнёвенагеля (см. ниже).
Смесь продуктов реакции Михаэля и Кнёвенагаля дают 5,5-ди-
хлорпентадиен-2,4-аль и 7,7-дихлоргептатриен-2,4,6-аль при
взаимодействии с малоновым или ацетоуксусным эфиром:
RCHCO2Et
I
—► С12С=СН(СН=СН)„-1СНСН2СНО
ХХХГ XXXII
ft, R (выходы XXXI и XXXII, соответственно, %): 1, CO2Et (75; 25); I, COMe
(45; 55); 2, CO2Et (18; 82)
Конденсация а,|3-ненасыщенных альдегидов и полиеновых
альдегидов с броммалоновым эфиром с использованием
стандартной методики (50%-ный NaOH —СН2,С12 — ТЭБАХ) [337,
338] при 5—7°С позволила получить ряд 3-1?-2-формил-1,1-ДИ"
122
(этоксикарбонил) циклопропанов. Реакция проходила стереоспе-
цифично и образовывались исключительно траяс-изомеры. В
случае полиеновых альдегидов реакция осложняется образованием
1,1,2,2-тетраэтоксикарбонилэтилена. Ниже приведена типичная
методика.
Синтез 2,2-ди (этоксикарбонил) циклопропанкарбаль-
д е г и д а. К смеси 23,9 г броммалонового эфнра, 8,4 г свежеиерегнанного
акролеина, 20 мл СНгСЬ и небольшого количества ТЭБАХ добавляют при
5—7 °С при энергичном перемешивании 6 мл 50%-ного NaOH, перемешивают
30 мин прн комнатной температуре, промывают водой, сушат (Na2SO4),
подвергают разгонке. Т. кип. 87—89°С (0,4 мм рт. ст.).
В близких условиях проходит реакция броммалонового
эфира с винилметилкетоном, а-хлоракрилонитрилом, винилтрифе-
нилфосфонийгалогенидами и а-диэтоксифосфонометил акрил а-
том [339].
Общая методика. К смеси 40 мл СН2С1г и 4,1 мл 9,5 н. водного
раствора NaOH добавляют 100 мг ТЭБАХ и затем при 0°С в атмосфере Ыг
за 10 мин при интенсивном перемешивании добавляют смесь 0,4 моль
броммалонового эфира н 0,4 моль непредельного соединения, перемешивают 1 ч
при 0°С и 2 ч при комнатной температуре, разбавляют 35 мл воды,
отделяют органический слой, сушат (Na2SO4), растворитель удаляют, остаток
обрабатывают эфиром, отделяют катализатор; полученный продукт прн
необходимости очищают на АЬОз.
Следует отметить, что в этих условиях при реакции
броммалонового эфира с изопропенилметилкетоном, метил-р,|3-диметок-
сивинилкетоном, коричным альдегидом и диметилвинилтрифенил-
фосфонийгалогенидом образуется только 1,1,2,2-тетраэтоксикар-
бонилэтан, а из кротонового альдегида, кроме того,— смесь ди-
астереомеров 1,1-ДИЭтоксикарбонил-3-[2/,2/-диэтоксикарбонилок-
сиранил-3] -2-метилциклопропанов [339].
Таким путем, варьируя методику, можно получать
функционально замещенные циклопропаны:
ЕЮгСх /CO2Et
BrCH(CO2Et)2+RCH=CR'R" —> /\ '
R— г—^
R, R', R" (выход, %): H, H, CHO (48-70); Me, H, CHO (46); Ph, H, CHO (44);
CH(OMe)2, H, CHO (43); H, H, COMe (97); H, Cl, CN (75); H, CO2Me,
P(O)(OEt)2 (65)
Фосфорсодержащие производные циклопропана образуются
при реакции фосфониевых солей со стабилизованными илидами
серы в системе, содержащей ТЭБАХ и 50%-ный NaOH [340]:
PPh3 Br"
Вг" Ph3PCH=CHY + Me2S—CHX —> /\
X ' Х Y
123
Описано присоединение карбанионов, генерированных из фе-
нилацетонитрила, к этоксиацетлену и я-бутилтиоацетилену в
системе твердая фаза — жидкость (NaOH или КОН — ДМСО,
ТЭБАХ) [341].
Ph(R)CHCN + CH=CR' —> Ph(R) (CN)CCH=CHR'
R = OEt; R' (выход, %): Me (64); Ph (77); PhCH, (74); PibCH (100).
R = SBu; R' (выход, %): Me (74); н-Рт (68); изо-Рх (74); PhCH2 (78);
PhC(Me)H (64); Ph2CH (96); Ph (55)
Общая методика. К смеси 0,025 моль нитрила, 0,03—0,05 моль
этоксиацетилена или н-бутилэтинилсульфида и 5 мл ДМСО в атмосфере N2
добавляют 3 г порошкообразного- NaOH или КОН, перемешивают 3 ч при
40—50 °С, после охлаждения (в случае этоксиацетилена оставляют в
закрытом сосуде иа 24—36 ч) промывают эфиром, извлекают бензолом и выделяют
продукты разгонкой или кристаллизацией из бензола.
Изучено влияние природы хиральных катализаторов и
условий реакции (температура, растворители) на оптическую
индукцию при присоединении нитроалканов, тиофенола и других
нуклеофилов к а,|3-непредельным кетонам, сульфоксидам и
сульфонам. Среди большого числа хиральных четвертичных
аммониевых солей наилучшими оказались Л^-(о-ннтробензил)хи-
нинийхлорид (в его присутствии тиофенил присоединяется к
циклогексен-2-ону-1, давая З-фенилтно-5-фенилциклогексанон с
оптическим выходом 35,6%), М-додецил-й-метилэфедринийбро-
мид и N-бензил-М-метилэфедринийбромид (при их
использовании нитрометан присоединяется к транс-халкояу с оптическим
выходом 24 и 26,4%, соответственно). Установлено, что
повышение температуры, а также увеличение диэлектрической
постоянной растворителя снижает оптический выход [342].
Присоединение анионов к связям углерод — гетероатом
К этой группе относится большое число важнейших реакций
органического синтеза. Многие из них уже осуществляют в
условиях межфазного катализа: альдольная конденсация,
реакция Кнёвенагеля, бензоиновая конденсация, реакция Дарзана,
реакция Виттига — Хорнера и другие.
Альдольная конденсация. Описано довольно много примеров
альдольной конденсации альдегидов н кетонов с
разнообразными компонентами, содержащими активный водород. Так,
ароматические альдегиды конденсируются с 2-метилбензоксазолом
(или 2-метилбензотиазолом), образуя в качестве главных
продуктов 2-(р-арил-р-гидроксиэтил) бензоксазолы (или -тиазолы)
[343]:
N N Аг
^ч ArCHO (i^V^ ^ I
^>—СНз ►■ I] ^>—СН2СНОН
X X
Аг, X (выход, %): Ph, 0(50); 4-С1СвН4, О (26); Ph, S (49); ^"MeCeH*. S (62);
4-С1С6Н4 (80)
124
При реакции с я-метоксибензальдегидом образуется смесь
гидроксипроизводного и 2- (л-метоксифенилвинил) бензоксазола
(или -тиазола) (кротоновая конденсация).
Общая методика. Смесь эквимольных количеств 2-метилбензоксазо-
аа (или -тиазола) и ароматического альдегида, (по 10 ммоль), 3 мл 50%-иого
NaOH и 0,23 г ТЭБАХ перемешивают 1—2 ч при комнатной температуре
(в случае реакции я-С1С6Н4СНО и я-МеС6Н4СНО с метилбензотиазолом —
24 ч), отделяют полученный продукт.
Недавно было впервые показано, что ароматические
альдегиды вступают в альдольную конденсацию с N-бензилиденбен-
зиламином в условиях межфазного катализа:
ArCHO-f-PhCH=NCH2Ph —► ArCH(OH)CH(Ph)N=CHPh (26—84%)
Ar = Ph, нафтил-2, 2-CIC6H4, 4-CICeH4) 4-MeC6H4, 4-МеОС6Щ
Общая методика [344]. К смеси эквимольных количеств (по
10 ммоль) ароматического альдегида и N-бензилидеибеизиламииа и 0,5 ммоль
ТЭБАХ добавляют по каплям 3 мл 50%-иого NaOH. Смесь перемешивают или
оставляют стоять до затвердевания (20—60 мин).
Получают смесь диастереомеров. При проведении реакции
в дихлорметане или ацетонитриле выходы продуктов
конденсации уменьшаются.
Конденсация N-бензилиденбензиламина с бензилиденацето-
феноном приводит к смеси изомеров 1-бензилиденамино-З-бен-
зоил-1,2-дифенилпропана (продукты реакции Михаэля) с
выходом 44% независимо от того, проводится реакция без
растворителя или в растворителе (дихлорметан, ацетонитрил):
Ph Ph
PhCH=CHCOPh+PhCH=NCH2Ph —>■ PhCOCH2CH—CHN=CHPh
При реакции N-бензилиденбензиламина с нитрилом коричной
кислоты в отсутствие растворителя образуются два изомера
тетрагидро-3-циано-2,4,5-трифенил пиррола (8% изомера с
т. пл. 163—165°С, 14% изомера с т. пл. 123—125°С), а в
растворителе (дихлорметан) кроме производного пиррола (7%)
получают с выходом 25% смесь диастереомеров продукта
присоединения по Михаэлю:
ТЭБАХ—50%-ный NaOH
PhCH=CHCN + PhCH=NCH2Ph ; ►
CH2CI2
Ph Ph
I I
PhCH=NCH—CHCH2CN
При низких температурах альдегиды и кетоны реагируют с
хлороформом в условиях межфазного катализа с образованием
а-(трихлорметил) карбинолов [345, 346].
_► RR'C(OH)CC13
R, R' (выход, %): Ph, Н (80): 4-МеСБН4, Н (62); изо-Pr, Н (34); Me, Me (69);
Et, Me (13); -(CH2)4— (33); -(CH2)5- (23)
125
Общая методика синтеза а- (трихлормети л)
карбинолов. К раствору 1 моль альдегида (или кетона) и 2,3 г ТЭБАХ в 250 мл
СНС13 (на 1 моль кетона берут 0,5 моль СНС13), охлажденному до 0°С,
добавляют при интенсивном перемешивании (0—5°С) 64 мл 50%-ного NaOH
(для кетоиа — 0,6 моль NaOH), перемешивают 30—120 мин при 0°С,
выливают на лед и 120 мл 1 н. НС1; выделяют продукты обычным путем.
Ароматические кетоны не реагируют в этих условиях.
Если проводить реакцию между хлороформом и альдегидами
при повышенной температуре в тех же условиях (см. выше)
образуются а-гидроксикислоты [345, 346], На этой основе
разработан простой и удобный метод синтеза миндальных кислот
[347].
n-RC6H4CHO4-CHCl3 —> n-RCBH4CH(OH)CO2H
R (выход, %): Н (75); Me (80); ОМе (83)
Общая методика синтеза миндальных кислот. К
раствору 0,1 моль альдегида и 1,23 г (0,005 моль) ТЭБАХ в 16 мл хлороформа
добавляют со скоростью 1—2 капли в 1 мин 25 мл 50%-ного NaOH при
56 ± 2 °С, перемешивают 1 ч, после охлаждения выливают в воду так, чтобы
осадок растворился, раствор промывают эфиром, подкисляют 50%-ной H2SO4,
извлекают непрерывно (12 ч) эфиром (перколятор), из экстракта выделяют
гидроксикислоту.
я-Нитробензальдегид осмоляется в этих условиях, а из ке-
тонов получают смесь а-гидроксикислоты, а-хлоркислоты и а,р-
непредельной кислоты [345]:
i *> ?> Г
RCH2CR' 4- СНСЬ —> RCH2C^ 4- RCH2C( + RCH==CCO2H
ХЮ2Н
XXXIII XXXIV XXXV
R, R' (выходы XXXIII, XXXIV, XXXV, соответственно, %): H, Me (55;
следы; 10); _(CH2)3— (27; следы; 23); -(СН2)4- (29; 7; 8); -(CH2)S- (13;
4; 5)
Варьируя температуру, в случае циклических кетонов иногда
удается направить реакцию с хлороформом в определенную
сторону. Например, при реакции циклогексанона с
хлороформом в системе 50%-ный NaOH — ТЭБАХ при 20°С удается
получить 1-хлорциклогексанкарбоновую-1 кислоту с выходом
45%, в то время как при 55—60°С кроме этой кислоты (выход
33—42%) образуется 1-гидроксициклогексанкарбоновая-1
кислота (6—10%) [348].
Оригинальный метод синтеза а-аминоарилуксусных кислот
основан на реакции ароматических альдегидов с хлороформом в
межфазных условиях в присутствии аммиака:
АгСНО+СНС1з4-Ш3 —> ArCH(NH2)CO2H
Аг (выход, %): Ph (66); 4-С1СБН4 (81); 4-FCBH4 (50); 3-FC6H4 (59)
Общая методика синтеза а-амииоарилуксусных
кислот [349]. Через смесь 6,72 г КОН, 1,69 г СНСЬ и 11,2 мл 33%-ного NH4OH
и раствора 0,46 г ТЭБАХ в 10 мл дихлорметана пропускают в течение 5 мин
при 0°С ток NH3, а затем в непрерывном токе NH3 добавляют по каплям
126
te течение 1 ч 0,02 моль альдегида и 2,55 мл (6,03 моль) хлороформа в 10 мл
дихлорметана, поддерживая температуру реакционной смеси около 0°С,
перемешивают 6 ч при 0°С и 12 ч при комнатной температуре, добавляют 30 мл
воды н 10 мл CH2CI2. Водный слой обрабатывают 4 раза дихлорметаном,
затем водный слой упаривают в вакууме до 30 мл, фильтруют, доводят рН
до 6—7 концентрированной НС1, охлаждают до 0°С. Осадок отделяют,
промывают водой, спиртом и эфиром, сушат.
Возможность использования межфазного катализа для циан-
гидринного синтеза была показана на примере реакции 1,2:5,6-
ди-О-циклогексилиден-а-£-/?ш5о-гексофуранозулозы-3, которая
сильно осмоляется при обычном варианте циангидринного
синтеза. В условиях межфазного катализа при встряхивании
бензольного раствора кетозы с водным раствором цианида калия
в присутствии ТЭБАХ с выходом 85,6% образуется 1,2:5,6-ди-
О-циклогексилиден-3-С-циано-а-/)-аллофураноза XXXVI (R =
CN [350]:
С5Н
о
CN, CH2NO2
Конденсация 1,2:5,6-ди-О-циклогексилиден-а-£)-/?«бо-гексо-
фуранозулозы-3 с нитрометаном лучше всего проходит в
твердофазном варианте межфазного синтеза при использовании в
качестве катализатора дибензо-18-крауна-б [351]:
Синтез 1,2:5,6-ди-О-циклогексилиден-3-С-нитроме-
тил-а-О-аллофуранозы XXXVI (R = CH2NO2). Смесь 1 г гидрата
кетозы, 0,2 мл нитрометаиа, 10 мг дибензо-18-крауна-6 и 0,5 г измельченного
в порошок карбоната калия в 10 мл бензола встряхивают 3 ч при комнатной
температуре, фильтруют, фильтрат выпаривают досуха, остаток дважды
кристаллизуют из спирта. Выход 0,48 г (43% от теоретического); т. пл. 97—98°С.
Ацетонитрил реагирует с ароматическими альдегидами и ке-
тонами, а также с циклогексаноном в межфазных условиях
(КОН—18-крауи-б) по типу кротоновой конденсации с
образованием а,р-непредельных нитрилов [352]:
MeCN + RCOR' —► RR'C=CHCN
R, R' (выход, %): R Ph (82); 4-МеСБН4, Н (61); 4-МеОСБН4, Н (81); 3,4-ме-
тилендиоксифенил, Н (86); 4-С1С6Н4, Н (57); Ph, Ph (84); — (СН2)5— (50)
В заключение отметим работы, в которых проведено
присоединение в межфазных условиях по связям C=N [353] и
Cs=N [354]. Показано, что тозилметилизоцианид реагирует с
сероуглеродом в системе жидкость — жидкость (СНОз —
127
10%-ный NaOH) в присутствии ТБАБ, образуя 4-тозил-5-мер-
каптотиазол (выход 94%) [353]:
/ТЗ
n-MeCBH4SO2CH2NC+CS2 —> ^ \—SH
S
Разработан метод синтеза тиоамидов из нитрилов и
сероводорода в присутствии межфазных катализаторов (аликвата 336,
ТБАХ или дибензо-18-крауна-б) [354].
ssN + HjS —+ RC(S)NH2
R (выход, %): 4-МеС6Н4 (86); 3-пирнднл (96); 4-С1С6Н4 (88); PhCH2 (58);
н-Рт (46)
Синтез тиопнкотннамнда [319]. Перемешивают смесь 14,8 г
нитрила никотиновой кислоты в 75 мл бензола, 0,54 г Na2S-9H2O в 20 мл
воды и 1 г аликвата 336 в автоклаве и обрабатывают прн 70 йС
сероводородом до постоянного давления 0,2 МПа (2 атм), продолжают интенсивное
перемешивание в течение 1 ч, после охлаждения отделяют тиоамид, промывают
водой, бензолом. Выход 18,8 г; т. пл. 121—122 °С.
Из приведенного выше материала вполне очевидны
перспективы использования межфазного катализа в реакциях
конденсации карбонильных соединений с разнообразными
компонентами, содержащими активный атом водорода.
Реакция Кнёвенагеля. Как отмечалось выше, в условиях
межфазного катализа (Na2CO3 — бензол — ТЭБАХ) циануксус-
ный эфир реагирует с разнообразными а,Р-непредельными
альдегидами, за исключением акролеина, давая продукты
конденсации по Кнёвенагелю [334]:
t —* RR'C^CHCH===C(CN)CO2Et
R, R' (выход, %): Me, H (15);- (MeO)2CH, H (14); Ph, H (64); Cl, CI (QO);
PhMeN, H (85); Me,N, H (44); C12C=CH, H (75); PhCH=CH, H (86);
H, C12C=CHCH=CH (75); Me2C=CHCH2CH2, Me (91)
Межфазный метод в ряде случаев значительно удобнее и
проще обычного варианта проведения реакции Кнёвенагеля, а
для полиеновых альдегидов является единственно приемлемым.
Следует отметить, что альдегиды, разветвленные в р-положении,
дают продукты реакции Кнёвенагеля также при конденсации с
малоновым и ацетоуксусным эфирами. В случае ацетоуксус-
ного эфира из цитраля образуется смесь открытого и
циклического продукта:
MeCOCH2CO2Et + Ме2С=СНСН2СН2С(Ме)=СНСНО —►
CO2Et Me EtO2G
—> МеСОС=СН=СНС(СН2)2СН=СМе2 + II U
Бензоиновая конденсация. В присутствии каталитических
количеств lS-крауна-б цианид-ион в межфазных условиях катали-
128
зует бензоиновую конденсацию [355]:
2АгСНО > АгСОСН(ОН)Аг
Аг (выход, %): Ph (73,9—78); 4-МеСбН4 (44,3—99); фурил (35,6—66,2)
Синтез бензоина. К смеси 20 ммоль KCN, 1,22 ммоль 18-крауна-6
н 2,7 мл воды прн 60 °С добавляют бензальдегид, перемешивают 15 мнн.
Осадок отделяют, раствор дополнительно обрабатывают хлороформом,
выделяют беизонн.
Реакция Дарзана. Возможность использования условий
межфазного катализа для проведения реакции Дарзана была
продемонстрирована на примере конденсации альдегидов и кето-
нов с хлорацетонитрилом (жидкость — жидкость) [356] и этил-
хлорацетатом (твердая фаза — жидкость) [84]:
RR'CO + C1CH2CN —v RR'C CHCN
\/
О
R, R' (выход, %): Ph, Н (75);- Me, Me (60); Ph, Me (80): -(CH5)4- (65)
—(CH2)4CH(Mc)— (78); Ph, Ph (55)
Типичная методика проведения этой реакции приведена ниже.
Синтез 1-оксаспнро [2.5] октанкарбоннтрила-2 [356].
К смесн 10,8 г цнклогексанона, 20 мл 50%-ного NaOH н 0,4 г ТЭБАХ
добавляют по каплям прн 15—20 °С 7,6 г хлорацетонитрнла, интенсивно
перемешивают 30 мнн; выделяют продукт обычным способом. Выход 10,8 г; т. кип.
87 °С (5 мм рт. ст.).
В реакции были использованы также а-хлорсульфоны [357—
359], фенилхлорацетонитрил [360, 361] и другие замещенные
хлорацетонитрилы [361], хлоруксусный эфир [84], хлорацетофе-.
нон [362].
1-Хлоралкансульфономорфолиды конденсируются с
альдегидами стереоселективно, образуя только транс-изомеры оксира-
нов, в то время как в случае несимметричных кетоиов
образуется смесь цис- и транс-изомеров в отношении 1:1. Предложены
две методики проведения реакции [323].
Методика А. Растворяют 10 ммоль 1-хлорметансульфономорфолида,
10 ммоль альдегида илн кетона в 4 мл" ацетоннтрила и- добавляют по каплям
прн 10—15 °С к Ю мл интенсивно перемешиваемого 50%-ного раствора NaOH,
содержащего 0,06 г ТБАХ, через 1 ч выделяют окснраны.
Методика Б. Смесь 10 ммоль 1-хлоралкансульфономорфолнда, 10 ммоль
альдегида илн кетона в 10 мл бензола добавляют по каплям в течение
30 мин при 10—15°С к смеси 10 мл 50%-ного NaOH, 0,06 г ТБАХ и 1 мл
гексаметапола (добавка диполярного апротонного растворителя очень актнвн-
рует реакцию в двухфазной системе), перемешивают 2 ч прн комнатной
температуре, выделяют оксираны обычным путем.
Таким путем получены следующие соединения [323]:
n( ;
/2( ;o
с—с; N /
129
R, R', R", методика (выход, %): изо-Рт, Н, Н, А (82); Ph, Н, Н, А (62);
нафтил-2, Н, Н, А (90); Me, Me, Н, А (90); — (СН2)б—, Н, А (91); Ph, Ph, H,
А (89); Ph, Me, H, А (85); Ph, H, Ph, Б (69); Ph, Ph, Ph, Б (74)
Необходимо отметить, что применение хиральных
четвертичных аммониевых солей при реакции карбонильных соединений с
я-толилхлорметилсульфоном приводит к оптически активным
оксиранам (максимальный оптический выход 2,5%). Для
достижения оптической индукции важно присутствие гидрокси-
группы в ^-положении ониевой соли. Индукция повышается
также при связывании катализатора на полимерной матрице.
При использовании полимерносвязанного катализатора удается
достичь оптических выходов порядка 23%. Такой выход был
получен при реакции метилэтилкетона с я-толилхлорметилсуль-
фоном в присутствии
R—CH2N(Me)2CH(Me)CH(OH)Ph Br"
в качестве катализатора ^R — полимер) в системе 50%-ный
NaOH — дихлорметан [310]. Образующийся 2-метил-1-п-тозил-
2-этилоксиран (выход 40%) представлял собой смесь Z- и Е-
изомеров в отношении 32 : 68.
Таким образом, реакция Дарзана в межфазном варианте
может быть успешно использована для синтеза эпоксикетонов
[362], эпоксинитрилов [356, 360, 361, 363—365], эпоксисульфо-
намидов [358], эпоксисульфонов [357, 360, 366], эфиров эпокеи-
карбоиовых кислот [84].
Реакция Кори. Как известно, ароматические альдегиды и ке-
тоны реагируют с сульфилидами, образуя оксираны. Обычно
эта реакция требует безводных условий. Распространение на
иее метода межфазного катализа явилось большим успехом.
Показано, что в обычных условиях межфазиого катализа (15 н.
NaOH — бензол, комнатная температура, перемешивание) при
действии додецилдиметилсульфонийиодида (который
одновременно служит катализатором межфазного переноса) на бензаль-
дегид образуется фенилоксиран с выходом 81% [367]:
PhCHO + Ci2Hi5SMe2 Г —► Ph—НС СН2
v
Предложен и другой вариант реакции Кори. Реакцию
осуществляют в трехфазной системе: полимерносвязанный сульфо-
нийметилид — СНгСЬ — водный раствор NaOH. Катализатором
служит тетрабутиламмонийиодид или тетрабутиламмонийгидро-
ксид; реакцию проводят при комнатной температуре при
интенсивном перемешивании в течение 1—4 дней [368].
Ph
S(CH2R'h X-^-PhCOR'' —> ;С CHR'
130
X = FSO3", MeSO;, MeCH2SOI; R = полимер
R'f R" (выход, %): H, H (97); Me, H (94); Ph, H (96); H, Ph (100; смесь
цис- и гранс-изомеров в отношении 4 : 1); Ph, Me (96)
Помимо ароматических альдегидов и кетонов (ацетофенон,
бензофеыон) в реакцию могут входить алициклические кетоны
и а,р-иепредельные альдегиды [369]. Например, циклогексанон
реагирует с додецилдиметилсульфонийхлоридом в системе
бензол — вода — NaOH, образуя бициклический оксид с выходом
88%:
0 + «-Ci2H2BSMe2 СГ
Коричный альдегид взаимодействует с триметилсульфоний-
иодидом (в этом случае катализатор необходим, используют
ТБАИ) в системе СН2С12—вода — NaOH и дает р-фенилвинил-
оксиран с выходом 83%:
PhCH=CHCHO4-Me3S+ I" -^> Н2С СН—CH=CHPh
О
Для введения аллильной группы в оксиран можно
использовать реакцию между альдегидом и илидом, полученными из
соответствующей соли [370], например:
О
PhCHO + CH2=CHCH2CH2SMe2 Х~ —» Ph—^ ^—СН2СН=СН2
(86%)
В этом случае катализатором межфазного переноса также
является сульфониевая соль (реакцию проводят в системе
толуол— вода — NaOH).
Как и в случае реакции Дарзана, при проведении реакции
Корн с хиральным катализатором можно провести энантиосе-
лективный синтез оксиранов [371, 372]. Так, в присутствии (—)-
Ы^-диметилэфедринийбромида бензальдегид реагирует с три-
метилсульфонийиодидом (в системе CH2CI2 — вода — NaOH) в
атмосфере азота при 28 °С в течение 48 ч, образуя с выходом
77% 2-фенилоксиран, содержащий 67% энантиомера [371].
Позже было выяснено, что 2-фенилоксиран загрязнен 2-метил-З-фе-
нилоксираном [372].
Реакция Виттига — Хорнера. Один из основных недостатков
реакции РО-олефинирования заключается в том, что для
выделения неустойчивых фосфоранов или для выделения фосфона-
тов необходима безводная среда. Оказалось, что в условиях
межфазного катализа возможно выделение малоустойчивых
фосфоранов из их солей. Реакцию можно проводить в присутст
вии четвертичных аммониевых солей, например ТБАИ, или в
отсутствие специальных катализаторов, поскольку сами фосфо-
ниевые соли могут играть эту роль [373].
RCHO + Rh3P—CHR'R" X" —* RCH=CR'R"
131
R, R', R" (выход, %; соотношение цис- и транс-изомеров): 4-МеСбН4, PhCHj,
Н (88; 44:56); 4-O2NC6H4, PhCH2, H (78; 56:44); 3-O2NCeH4, PhCH2, H (80;
53:47); PhCH=CH, PhCH2, H (87; 36:64); антранил-9, PhCH2, H (65;
только тракс-изомер); Ph, Me, Me (30); Ph, H, Me (20); PhCH=CH, H, Me (51)
Ниже приведена типичная методика проведения этой
реакции.
Синтез стильбена. К смеси 2,06 г бензальдегида, 7,86 г бензил-
трифенилфосфонийхлорида и 10 мл дихлорметаиа при интенсивном
перемешивании добавляют 10 мл 50%-ного NaOH (при этом развивается
слабоэкзотермическая реакция). Через 30 мин органический слой отделяют, промывают
водой, сушат, удаляют растворитель, добавляют 15 мл спирта, охлаждают
льдом, отделяют кристаллический транс-стшьбен. Выход 1,2 г (33% от
теоретического); т. пл. 123—124 °С.
Из фильтрата разгонкой выделяют цис-стшъбеп. Выход 1,7 г (47% от
теоретического); т. кип. 60—62 °С (0,01 мм рт. ст.); «^ 1,6215.
Таким образом, реакция Виттига в межфазных условиях
приводит к высоким выходам алкенов при использовании
ароматических альдегидов и фосфоииевых солей на основе бензил-
или алкнлгалогенидов. Как и в обычных условиях,
образуются смеси цис- и r/шяс-изомеров, за исключением стерически
затрудненных альдегидов. Кетоны не реагируют в этих
условиях.
Для проведения реакции Виттига чрезвычайно удобно
применять полимерносвязанные фосфониевые соли, которые в
условиях межфазного катализа дают достаточно высокие выходы
при реакции как с насыщенными альдегидами (формалин,
н-гептаналь), так и с ароматическими и гетероароматическими
альдегидами (замещенные бензальдегиды, фурфурол) и а,|3-не-
предельными альдегидами (коричный альдегид [374]).
Межфазный катализ может быть применен также для
выделения самого неустойчивого из фосфоранов — метилентрифенил-
фосфорана и для проведения реакции Виттига не только с
ароматическими, но и с алифатическими альдегидами, причем в
последнем случае выходы алкенов также высоки [375].
Общая методика проведения реакции Виттнга. Смесь
3 ммоль алкилтрифенилфосфопнйгалогенида, 1,5 ммоль альдегида, 3 мл
бензола и 9 мл водного раствора щелочи (концентрация зависит от типа фосфо-
ниевой соли; для метилтрифенилфосфонийхлорида илн -иодида используют
5 н. NaOH, для аллилтрифенилфосфонийбромида—0,5 н. NaOH, для этил-
или «-пентилтрифенилфосфонийбромида — 15 н. NaOH), перемешивают
несколько дней (от 24 до 90 ч) при комнатной температуре; выделяют алкены
обычным путем.
По этой методике получены алкены состава RCH=CHR/.
R, R' (выход, %): Н. Ph (80—99); Н, 4-С1СбН4 (95); Н, 4-МеСбН4 (55);
Н, 4-МеОСбН4 (38); PhCO, Ph (36); Н, «-C7Hi5 (73); Н, а-фурил (63 г
Н, PhCH = CH (68); Ph, Ph (81)'; Ph, к-С7Н15 (82); Ph, PhCH^CH (71);
HH h ) CH CH С (6 CH CH фл (72)
, () () ()
CH2=CH, Ph (60); CH2 = CH, к-С7Н,5 (64); CH2 = CH, а-фурил (72);
CH2^=CH, PhCH=CH (30); Me, Ph (46); «-C5H11, Ph (41)
Отмечено, что при проведени i реакции Виттига с
выделением неустойчивого трифенилэтиленфосфорана з присутствии
133
Таблица 15. Влияние растворителя на соотношение цис- и транс-изомеров
в реакции Виттига
R
Ph
Ph
Et
R'
Ph
Me
Ph
Растворитель
ТГФ
CH2CI2
ТГФ
CH2C12
ТГФ
CH2C12
цис : транс
22:78
30:70
85:15
22:78
25:75
46:54
Общий выход, %
96
97
96
93
82
—"~
К2СО3 и каталитических количеств 18-крауна-б в ТГФ
(кипячение, 18 ч) образуются предпочтительно г{ис-алкены, в то
время как в дихлорметаие (кипячение, 12 ч)—граяс-алкены. При
реакции с более устойчивыми фосфоранами, например с бензи-
лидентрифенилфосфораном, растворитель не влияет иа
стереохимию реакции [376]. В отсутствие 18-крауна-б реакция не
идет.
Влияние растворителя на соотношение цис- и гранс-изомеров
образующихся алкенов RCH=CHR/ показано в табл. 15.
Было показано, что в присутствии дибензо-18-крауна-б
можно провести реакцию между я-толилтрифторметилкетоиом и
различными солями бензилтрифенилфосфония в ацетонитриле при
избытке KF вместо NaOH.
CF3
n-MeCeH4COCF3-f-Ph3PCH2Ar СГ —>
Аг (выход, %; соотношение цис'.транс или цис-цис : цис -транс : транс-транс^
изомеров): Ph (100; 17:83); 4-O2NC6H4 (88; 31:69); сс-тненил (100; 15:85);
—СеН4— (п) (100; 5 : 32 : 63); 2,5-(МеО)2СвН3 (88; 3 : 25 : 72)
Общая методика [377]. Смесь 1 ммоль дибензо-18-крауна-б,
200 ммоль сухого KF и 75 мл абсолютного ацетонитрила перемешивают
20 мин при 70—80 °С в атмосфере азота, добавляют 10 ммоль я-толилтри-
фторметилкетона, 10 ммоль моно- или 5 ммоль бис(фосфониевой) соли в 50 мл
ацетонитрила, перемешивают 2 ч, фильтруют, выпаривают досуха, остаток
растворяют в хлороформе и выделяют алкен хроматографией на SiO2.
Двухфазный метод в системе дихлорметан — вода— NaOH
был использован для синтеза 1,4-дипиридил- и 1,4-дихинолилбу-
тадиенов-1,3 из глиоксаля и соответствующих пиридилметил-
или хинолилметилтрифосфониевых солей [378]; наилучшие
выходы достигались при проведении синтеза в атмосфере азота.
RCH2PPh3 Х" + ОНС—СНО —> R(CH=CH)2R
X = С1, Вг; R == пирндил-2 (выход 18%), хииолил-2 (выход 18%)
RCH2PPh3 X" + R'CH«CHCHO —► R(CH=CH)2R'
X s= Cl, Вг; R = R' = пиридил-2 (выход 71 %)
133
В системе вода — хлороформ — Na2CO3 был осуществлен
синтез 2-винилбензимидазола из формальдегида и
соответствующей фосфониевой соли [379].
В системе бензол — 25%-пый NaOH — ТЭБАХ ферроценаль-
дегид реагирует с метилентрифенил- и бензилидентрифенилфос-
форанами, давая с хорошими выходами (80—85%) винилфер-
роцен и смесь цис- и т/шяс-р-фенилвшшлферроценов [380].
Недавно [381] предложен упрощенный метод проведения
реакции Виттига для ароматических альдегидов, использование
которого позволяет получать олефины с преимущественным
содержанием (70—84%) Z-формы (в обычных методиках
образуются смесь Z- и Я-изомеров примерно в равных количествах).
С высокими выходами алкены были получены из аль-форм
моносахаридов по реакции Виттига при генерировании фосфора-
нов из арилидентрифецилфосфониевых солей с катализатором
ТЭБАХ в системе бензол— 50%-ный NaOH (алкены XXXVIIIa
и XXXVIII6; выходы соответственно 85 и 75%) или в системе
бензол — ДМСО — твердый КгСО3 (алкены XXXVIIIb —
XXXVIIIe; выходы соответственно 55, 60, 45 и 65%) [382]:
сно
СН=СШ?
PhaP=CHR
СН2О
XXXVII
СН2О
XXXVIII
(a) R = нафтил-1; (б) R = 4-O2NC6H4CH = CH; (в) R = 1 - метилбензо-
имидазолил-2; (г) R = бензоимидаяолил-2; (д) R = 1,5-диметилбензоимида-
золил-2; (е) к = 1-метил-5-иитробензоимидазолнл-2
Хорнеровский вариант речкции Виттига с использованием
фосфонатов также удалось легко осуществить в условиях
межфазного катализа [345, 346]:
(EtO)2P(O)CH2R//
RR/C=CHR"
R, R', R" (выход, %; соотношение Z- и Е-изомеров): Ph, H, CN (77;'24: 76);
Me, H, CN (51; 39:61); Me, Me, CN (62); Ph, H, CO2Et (56; только Е-изо-
мер); Me, H, CO2Et (54; только Е-изомер); Ph, H, пиридил-2 (71; только
Е-изомср); PhCH = CH, H, пиридил-2 (68)
Общая методика [383]. Раствор 35 ммоль фосфоната и 35 ммоль
альдегида или кетона в 5 мл дихлорметана добавляют по каплям при
перемешивании к смеси 20 мл 50%-ного NaOH, 35 мл дихлорметана и 0,7 г
ТБАИ. В случае циано- и этоксикарбонилфосфонатов реакция протекает экзо-
134
термически и заканчивается через 15 мин, в случае (пирндил-2)метилфосфо*
ната смесь кипятят около 3 ч. Из органического слоя после отгонки
растворителя выделяют продукт и кристаллизуют из петролейного эфира.
Таким путем было приготовлено большое число 1-арил-4-
фенилбутадиенов-1,3:
А
PhCH=CHCHO+(EtO)2P(O)CH2Ar —
Б
ArCHO + PhCH=CHCH2P(O) (OEt)2 —
PhCH=CHCH=CHAr
Аг (выход по схемам А и Б, %); Ph (72; 70); 4-ВгС6Н4 (81; —); пиридил-2
(75; 44); 4-O2NC6H4 (—; 57); пнридил-3 (—; 55); пиридил-4 (—; 12); фурил-2
(—; 84); PhCH=CH (—; 80)
Следует отметить, что при реакции с фурфуролом
применяли 75%-ный раствор NaOH. Интересно, что для 4-формилпири-
дина значительно более высокий выход 1- (пиридил-4)-4-фенил-
бутадиена-1,3 (52% по сравнению с 12% в условиях
межфазного катализа) получают при реакции в спирте с трет-бутоксидом
калия [384].
Отмечено, что реакция с фосфонатами может идти без
катализатора, как и в случае фосфоранов (см. выше); при этом
образуются исключительно или преимущественно £-изомеры
(несомненно, что и в этом случае катализаторами являются сами
фосфонаты) [385]. Реакция идет при комнатной температуре
или кипячении в системе дихлорметан — 50%-ный NaOH;
(EtO)2P(O)CH2R + R'CHO —> RCH=CHR'
R, R' (выход, %; соотношение Е- и Z-изомеров): CN, Ph (80; 74:26); PhCO,
Ph (55; не определено); PhCO, 4-С1С6И4(65; не определено); PhCO, 4-ВгСвН4
(63; не определено); СО2Н, Ph (95; только Е-изомер); PhS, Ph (87; 74:26);
PhSO2, Ph (53; 82:18); MeSO, Ph (51; 77:23); MeSO2, Ph (65; только
Е-изомер); (EtO)2P(O), Ph (79; только Е-изомер); (EtO)2P(O), 4-BrC6H4 (74;
только Е-изомер); (ЕЮ)гР(О), 4-Me2NCBH4 (76; только Е-изомер);
(EtO)2P(O), PhCH^CH (70; не определено)
Межфазный катализ был использован для синтеза ряда а,Р*
ненасыщенных сульфидов, сульфоксидов и сульфонов реакцией
соответствующих фосфонатов с альдегидами в системе
дихлорметан — 50%-ный NaOH — ТЭБАХ (комнатная температура или
кипячение) [386]:
(EtO)2P(O)CH2S(O)ftR + R'CHO —> RS(O)nCH^=CHR'
п, R, R' (выход, %; соотношение Е- и Z-изомеров): О, Me, Ph (59; 81 : 13);
О, Ph, Ph (81; 48:52); О, Ph, 4-Me2NCBH4 (40; 80:20); I, Me, Ph (51;
70:30); I, Ph, Ph (54; 83: 17); I, Ph, 4-ClC6H4 (57; только Е-изомер); I, Ph,
4-MeOCeH4 (48; только Е-изомер); 2, Me, Ph (85; только Е-изомер); 2, Me,
4-ClCeH4 (71; только £-изомер); 2, Me, 4-MeOCflH4 (73; только Е-изомер).
13S
Изучена [348] также зависимость соотношения £- и Z-изо-
меров от природы межфазного катализатора при реакции
между бензальдегидом и диэтилфенилтиометилфосфонатом:
Катализатор
BxZ
Me4NCl 59:41
н-ВщЖМ 43:57
Me3PhCH2NCl 47:53
Et3PhCH2NCl 48:52
H-Bu3PhCH2NCl 44:56
Me4NBr 77:23
Катализатор
E:Z
Et3PhCH2NBr 50 : 50
H-Bu4NBr 81 : 19
Me4NI 83:17
Et4NI 72; 28
h-BuJ 83: 17
Как видно из полученных данных, увеличение объема
аниона в соли приводит к увеличению количества £-изомера,
природа алкильных заместителей мало влияет на соотношение
изомеров. По-видимому, имеет значение и природа реагентов. Так,
изучение влияния природы катализатора и растворителя на
стереоселективность реакции на примере реакции бензальде-
гида с (EtO)2P(O)CHMeCN [387] показало, что природа
растворителя (бензол, гексан, дихлорметан, хлороформ, тетра-
хлорметан, диэтиловый эфир) и катализатора {«-Bu4NCl,
H-Bu4NBr, w-ВщШ, PI14PCI, Ph4AsCl,C16H33NMe2[(CH2)2OH]Br,
Ci6H33NMe(CH2OH)2Br} не влияет существенно на
стереоселективность реакции, но влияет на ее скорость.
РЕАКЦИИ ОКИСЛЕНИЯ
Известно много методов окисления. Однако большое
значение этой реакции побудило исследовать применимость
межфазных катализаторов в реакции окисления в тех случаях,
когда органический субстрат нерастворим в водных средах,
а окислитель в свою очередь нерастворим или плохо растворим
в органических растворителях. Принципиальная возможность
использования межфазных катализаторов для окисления
нерастворимых в воде субстратов в апротонном растворителе была
показана еще в 1965 г. [388]. В качестве катализатора была
использована четвертичная арсониевая соль — метилтрифенил-
арсонийхлорид; при этом происходил обмен хлор-аниона на
анион перманганата из водного раствора перманганата калия
и перенос этого аниона в хлороформенный раствор субстрата
(октен-1, пропанолы-1 и -2, гептанол-4 и др.), где и проходило
окисление. В таких условиях пропанол-2, например, на 100%
превращался в ацетон. г/?ег-Бутиловый спирт, толуол, этилаце-
тат, диэтиловый эфир, ацетон или дипропилкетон не
окислялись. Позже было установлено, что октен-1 и децен-1
превращаются соответственно в гептановую кислоту (количественный
выход) и нонановую кислоту (выход 91%) при окислении нейт-
136
ральным водным раствором КМпО4 в присутствии
каталитических количеств ТБАБ. Реакция проходит экзотермически [1].
Была изучена экстрагируемость иона перманганата из водного
раствора в бензол и показано, что при содержании в водном
растворе 1 ммоль МпОГ и использовании 3,12 ммоль ТБАБ
в бензол переходит 0,97 ммоль МпОГ» которые и могут
расходоваться на окисление. Аналогичные результаты были
получены и с бензилтриэтиламмонийхлоридом, тетрабутилфосфоний-
бромидом, гексадецилтриметиламмонийбромидом и аликватом
336; в присутствии тетраметиламмонийхлорида и додецилсуль-
фата натрия реакция не идет. Малиновые растворы
перманганата в бензоле в дальнейшем были довольно широко
использованы для окисления [389—402]. Помимо четвертичных
аммониевых солей для переноса МпО4 в органическую фазу были
успешно использованы краун-эфиры (дициклогексано-18-краун-б
[8, 390, 398] и 18-краун-6 [397]). Кроме бензола в качестве
растворителей применяли пентан [396], дихлорметан [393, 394,
399, 401] и пиридин [354]. Во многих случаях протеканию
реакции благоприятствовала добавка уксусной кислоты [393,
396,399,401].
Среди наиболее интересных примеров следует отметить
окисление метилциклогексана в 1-гидрокси-1-метилциклогексан
[393} и окисление цнс-декалина в ^мс-декалол:
PhCH2Et3NMn04
(44%)
АсОН
PhCH2Et3NMn04
AsOH
В тех же условиях из фенилалканов, содержащих рядом
с фенильным остатком метиленовую группу, образуются алкил-
фенилкетоны [393]:
Ph(CH2)3Me —> PhCO(CH2)2Me (44%)
PhCH2CHMe2 —► PhCOCHMe2 (26%)
Окислением бициклического алкена в присутствии дицикло-
гексано-18-крауна-6 была получена (3-ацетил-2,2-диметилцикло-
бутил-1) уксусная кислота [390].
Предложен удобный метод окисления алкилбензолов в бен-
золкарбоновые кислоты в системе углеводород (твердый
углеводород растворяют в дихлорэтане) — вода — ТДТМАБ при
75—85 °С в течение 1—4,5 ч; выходы кислот достигают 90—
95% [402].
137
На основе межфазного катализа разработан улучшенн
метод синтеза цнс-1,2-гликолей окислением олефинов перман-
ганатом [394]. Типичная методика приведена ниже.
Синтез цис-циклооктандиола-1.2. К" 11 г tfwc-циклооктена
в 100 мл дихлорметана добавляют 100 мл 40%-ного NaOH с 1 г ТЭБАХ,
охлаждают до 0°С ив течение 2 ч при интенсивном перемешивании
добавляют небольшими порциями 15,8 г КМпО*. Смесь оставляют на ночь при 0 °С,
затем обрабатывают SO2 для связывания МлОг, добавляют 500 мл диэти-
лового эфира, отделяют органический слой, водный дополнительно
экстрагируют эфиром (3 X 150 мл). Объединенные органические слон сушат (MgSO4),
фильтруют, удаляют растворитель в вакууме, остаток кристаллизуют из
«-гептана. Выход 7,8 г (50% от теоретического); т. пл. 76—78°С.
Аналогично из r/кшс-циклодецена получают транс-циклоне-
кандиол-1,2, а из циклогексена — г{Ис-циклогександиол-1,2
(однако выход всего лишь 15%).
Удобный метод синтеза 2,2-дибромциклопропанкарбоновых
кислот основан на окислении 2,2-дибром-1-винилциклопропанов
КМпО4 в системе бензол — вода н присутствии ТЭБАХ или
ТБАБ [395]:
(70-79%)
Вг'
Наконец, хороший метод синтеза а-дионов основан на
окислении ацетиленов КМ11О4 в системе дихлорметан — вода — адо-
ген464 [401].
RC=CR' —> RCOCOR7
R, Ц' (выход, %): Ph, Ph (93); Ph, w-Pr (56—81); Ph, к-Bu (41); к-СБН13,
к-СБН13 (54-57); к-С7Н15, h-C7H1g (68-80)
Типичная методика синтеза а-дионов приведена ниже.
Синтез гексадекандиона- 8,9. К раствору 5,85 г КМпО4 в 100 мл
воды добавляют смесь 2 г гексадецииа-8, 100 мл дихлорметана и 1,5 г адо-
геиа 464. Раствор перемешивают и кипятят 6 ч, добавляют 2 г NaHSOs,
перемешивают 15 мин, подкисляют концентрированной НС1, осадок МлОг
восстанавливают, добавляя небольшими порциями NaHSCb- Водную фазу
насыщают NaCl, извлекают дихлорметаном (3 Х75 мл). Органические длои
объединяют, промывают 5%-ным NaOH (3X75 мл), сушат (MgSO*),
растворитель удаляют, остаток кристаллизуют из метанола. Выход 1,55 г (68% от
теоретического); т. пл. 51—52°С.
Установлено, что условия межфазного катализа непригодны
для окисления ди-я-октил- и дибензилсульфидов, а также ди-
бензилсульфоксида (помимо КМпО4 были испытаны О2, NaOCl
HNaIO4) [403].
Дихромат калия оказался эффективным окислителем
спиртов до альдегидов в межфазных условиях. Разработаны три
методики проведения окисления.
гг? или г*
RCH2OH ■> RCHO
R, методика (выход, %): Ph, A (82); Б (92); PhCH2, Б (62; 91 при
использовании 4,1 моль К2СГ2О7); Ph(CHa)2) Б (80—90); PhCH==CH, A (91), Б (0),
138
В (80); 4-МеСБН4, Б (95); 4-МеОСБН4, Б (1), В (98); 4-O2NC6H4, Б (55); Me,
Б (5); н-Pr, Б (34), В (19); н-С5Н„, В (90); м-С7Н15, Б (82), В (91); С9Н1е,
А (6); «-СиНа8. В (98); «-СиД», Б (90), В (95); СН2 = СН(СН2)3, Б (95);
2-гидрокси-7-тетрагидрофурилгептил, В (83); 3-(2,2-диметил-1,3-дноксаннл-4)-
пропил, В (10); 8-(2,2-диметил-1,3-диоксанил-4)октил, В (78).
А [367]. Спирт окисляют эквимольиым количеством КгСг2О7 в бензоле
при 55 С в присутствии эквимольных количеств адогена 464 (методика
экстракции ионных пар).
Б [403]. К Ю ммоль спирта в 25 мл дихлорметана добавляют 0,9 ммоль
ТБАГС и кратковременно встряхивают со стехиометрнческим количеством
(3,3 ммоль) К2СГ2О7 в 25 мл 9 М серной кислоты.
В [406]. К интенсивно перемешиваемому раствору спирта в хлороформе
или дихлорметане, содержащему каталитические количества ТБАГС (0,1 моль
на 1 моль субстрата), добавляют постепенно при —5 ~0°С стехиометриче-
скос количество (0,66 моль на 1 моль субстрата) насыщенного раствора
КгСгОч в 30%-ной H2SO<.
Методики А и Б были использованы также для окисления
вторичных спиртов в кетоны:
К2Сг207
RCH(OH)R' >
R, R', методика (выход, %): Ph, Me, A (80); rt-CBHi3, Me, A (33), Б (95);
-(СН2)8- Б (65); -(СН2)8- А (43)
Методика А непригодна для окисления алифатических
первичных спиртов, методика Б — для а,|3-ненасыщенных спиртон
(см. также [407]). Наиболее универсальной является методика
В, которая применима как к алифатическим (кроме первых
членов ряда), так и к а,Р-непредельным и бензиловым спиртам.
Для окисления первичных и вторичных спиртов предложено
использовать также HCrOi", связанный с закрепленным на
полимере ионом триметиланилиния [408]. Реакцию ведут в
бензоле или гексане, например (R — полимер):
R—CeH4NMe3 HCrO" ^
он
Отмечено, что первичные бромиды можно окислить до
альдегидов действием К2Сг04 в гексаметаполе в присутствии ди-
циклогексано- или дибензо-18-крауна-6 [409]. Таким путем,
например, фариезилбромид был окислен до фарнезаля с выходом
80%, а геранилбромид —до гераниаля с выходом 82% [409],
Для окисления бромидов в альдегиды пригоден полимерно-
связанный триметиланилинийхромаТ [410], например:
B43 НСгО;
R7CH2Br ► R'CHO
бензол
R'(выход, %): — С(Ме)=*СНСО2Н (95); Ph (98); гераннл (95)
Можно использовать также хлориды. Например, бензилхло-
рид окисляется в этой системе в бензальдегид с выходом 95%.
139
Окисляются и вторичные спирты: из дифенил метил бромида был
получен бензофенон (выход 95%), а из 9-бромфлуорена —
флуоренон-9 (97%). Для окисления первичных и вторичных
спиртов в альдегиды и соответственно кетоны предложено
использовать комплексы СгОз с четвертичными аммониевыми
солями. Эти комплексы растворимы в дихлорметане, и для
окисления можно применять каталитические количества СгОз.
Продолжительность реакции составляет от 5 мин до 2 ч. Таким
путем были приготовлены октанон-2 (выход 70—95%) из ок-
танола-2, коричный альдегид (выход 85%) из коричного
спирта, октаналь (выход 65%) из октанола-1 и др. [411].
Эффективным окислителем спиртов и аминов в
карбонильные соединения в условиях межфазного катализа является ги-
похлорит-ион [412]. Метод был использован для превращения
арилкарбинолов и алициклических спиртов в карбонильные
соединения [7, 412, 413], а также для превращения вторичных
аминов в кетоны [412].
NaCl
RCH2OH *• RCHO
R (выход, %); Ph (76); 2-MeOC6H4 (47); 4-MeOC6H4 (79); 4-С1С6Н4 (82)
NaCIO
RCHOHR' * RCOR'
R, R'(выход, %); Ph (82); -(CH2)6~ (89)
Типичная методнка превращения приведена ниже.
Синтез п - то лу и лов о г о альдегида [412]. К раствору 1,25 г
ге-метилбензилового спирта и 0,18 г ТБАГС в 25 мл дихлорметана добавляют
25 мл 10%-иого NaCIO (2,95 г). Смесь перемешивают при ~24°С,
контролируя ход реакции с помощью ГЖХ. Через 135 мин спирт, конвертируется иа
90%. Выход 85% (по ГЖХ).
Кроме дихлорметана в качестве растворителя можно
использовать бензол, тетрахлорметан, хлороформ и этилацетат.
Последний особенно благоприятен при окислении аминов;
промежуточно образующиеся N-хлорамины можно прогидролизовать
в кетоны или использовать в других реакциях. Межфазный
метод приготовления N-хлораминов и продуктов на их основе
превосходит все известные методы, используемые с этой целью.
В случае аминов, содержащих в а-положении водородные ато-
Таблица 16. Окисление аминов гипохлоритом
Амин
Продукт окисления
Выход, %
Циклогексиламин
2-Норборниламин
Бензгидриламин
N-Метилбензила мин
Циклогексилметиламин
я-Октиламин
Циклогексанои
Норборнанон-2
Бензофеион
Ацетофенои
Циклогексанкарбонитрил
1-Цианогептаи
98
84
94
98
76
60
140
мы, получают не карбонильные соединения, а нитрилы [312].
Продукты окисления некоторых аминов приведены в табл. 16.
Окисление аренов в межфазных условиях приводит к
оксидам аренов с выходами 10—90% [314].
Система вода —NaCIO — ТБАГС (или ТЭБАХ) была
применена для окисления гидрохинонов (при 20 °С) и пирокатехинов
(при —10 ч-—5°С) в соответствующие хиноны (выходы 70—
90%) [415].
Для окисления вторичных стероидных спиртов аллильного
типа была использована HsIOe в системе бензол — вода в
присутствии 2,3-Дихлор-5,6-дицианобензохинона [416]:
(58-86%)
В этой же системе окисляются до кетонов эфиры аллильных
спиртов [416]:
PhCH=CHCH(OR)R —» PhCH=CHCOR (80—91%)
R = H, Me, Ph
В присутствии аликвата 336 и сокатализатора (OsO4, RuO2
и др.) в системе бензол — вода НЮ6 окисляет алкены-1 до
альдегидов или кислот [66], например:
Me(G8Hi7)sNCl
Ме(СН2)5СН=СН2 — >- Ме(СН2)5СНО, (86%)
Me{C8H17)8NCl
Ме(СН2)5СН=СН2 —-—>~ Me(CHz)5CO2H (90-100%)
RuO
/
Межфазный катализ с использованием хиральных
катализаторов был применен для окисления халконов пероксидом
водорода в щелочных средах с целью получения оптически
активных оксидов [417—419]. Оказалось, что в присутствии N-бен-
зилхининийхлорида удается получать избыток одного из энан-
тиомеров (до 25%).
Окисление халкоиа. Раствор 20,4 г халкона в 125 мл толуола
интенсивно перемешивают с раствором 7,5 г NaOH в 90 мл 30%-иого Н2О2,
содержащим 750 мг N-бензилхининийхлорида. Продукт выделяют
хроматографией иа SiO2 (растворитель дихлорметан). Выход 21,8 (95% от
теоретического); [a] d —51 °.
В аналогичных условиях с высокими выходами был получен
ряд оптически активных соединений — л-хлор- и л-нитрохалко-
ны, нафтил-2-фенилвинилкетон, (бензотиенил-2)винилфенилке-
тон и др.
Соли пероксидов, например пёроксид калия, нерастворимы
в неполярных средах, а в гидроксилсодержащих средах
неустойчивы; КО? немного растворяется в диполярных апротонных
Hi
растворителях, например в ДМСО. Вследствие этого до
появления метода межфазного катализа КОг в органическом
синтезе почти не применяли. Несколько лет назад было
обнаружено, что в присутствии краун-эфиров (18-краун-6- [420,
423], дициклогексано-18-краун-б [388, 389]) возможна солю-
билизация КО2 в ДМСО, бензоле, ТГФ, ДМФА, диметоксиэтане
и диэтиловом эфире [420]. С этого момента начались поиски
синтетических путей его использования. В настоящее время
установлено, что К.О2 может быть применен для окисления
9,10-дигидроантрацена в антрацен в присутствии 18-крауна-б
в ДМСО [423]:
(93%)
Окисление алкил- или бензилгалогенидов в тех же условиях
приводит к спиртам (выходы 41—75%) [424].
Из 3-бромхинолина получают 3-гидроксихинолин с выходом
63% [425]. 2,4-Динитрофенилгалогениды и 2- или 4-нитрофе-
нилбромиды легко окисляются КО2 в бензоле в присутствии
дициклогексано-18-крауна-б с образованием соответствующих
фенолов [429]:
JL (Y (94-95%)
Первичные и вторичные алкилбромиды, мезилаты и тозн-
латы превращаются, при действии КО2 в бензоле в присутствии
дициклогексано- 18-крауна-6 в диалкилпероксиды (выходы 42—
77%) [430], в небольших количествах образуются спирты и
олефины. X
По-виднмому, образование пероксидов является реакцией
замещения типа 5N2, поскольку реакция с (—)-(#)-2-бромок-
таном протекает более чем на 94% с инверсией конфигурации
и образованием (+) - (5, S) -ди (октил-2) пе^оксида [430].
В присутствии 18-крауна-б сложные эфиры под действием
КО2 превращаются в кислоты (после обработки водой) [380,
383]:
RCO2R' —* RCOOH
R, R' (выходД): Me, 2-С8Н17 (89); трет-Ъи, Me (81); Ph, Et (88); m-C7Hi5,
Me (98); м-С7Н15, изо-Рт (98); «-C7H15, трет-Ви (96); «-C7H15l PhCH2 (100);
m-C9Hi9, Ph (84)
Реакция проходит с разрывом связи ацил — кислород; это
подтверждается тем, что при реакции (i?)- (октил-2) ацетата
с КО2 образуется (#)-октанол-2 (сохранение конфигурации на
99%).
а-Кето-, а-гидрокси- и а-галогенкетоны, сложные эфиры или
кислоты расщепляются КО2 (бензол, 18-краун-б) с образова-
142
нием кислот [426, 427]:
RCHXCOR' —> RCO2H-f R'CO2H
RCOCO2H —> RCO2H (42—930/0)
RC6H4COCH====CHC6H4R' —* RC6H4CO2H + R/C6H4CO2H (88—100%)
При действии КО2 в бензоле, толуоле или ДМСО в
присутствии 18-крауна-б арилгидразины превращаются в арены
(выходы 54—81%) [422], а 1,2-диарилгидразины — в азосоедине-
ния (выходы 85—98%) [422].
N-Замещенные N-хлорамины реагируют с КО2 в эфире
в присутствии 18-крауна-6, давая азины [428]:
RCH2N(C1)R/ —> RCH=NR' (49—88%)
R = A1K, Ph; R'^AIK, PhCH2
Недавно разработан удобный метод расщепления кетонов
с образованием кислот действием КО2 в бензоле или системе
бензол — вода в присутствии аликвата 336. Метод особенно
удобен для получения а,ш-дикарбоновых кислот из циклических
кетонов [431]. Кетоны, не содержащие а-водородного атома,
не окисляются КО2-
:(CH2)ft —► HO2C(CH2)rtCO2H (89—94%)
п = 4, 5, 9
Аналогично реагирует камфара с образованием ^амфарной
кислоты (выход 93%). Линейные кетоны о§р«3уют Две кислоты
в отношении 1 : I, например: ■--''" ^
RCOCH2CH2Me —> RCO2H + Mej3K2CP2H (общий выход 96%)
За последние годы отмечено несколько случаев
использования межфазного катализа в других реакциях окисления. Так,
установлено, что в присутствии аликвата 336 флуорен
окисляется кислородом воздуха в системе бензол — вода — NaOH,
образуя с количественным выходом флуоренон. Аналогично
проходит окисление 9-окса- или 9-тиадигидроантраценов с
выходами соответственно 84 и 99% [391]:
О
В этих условиях речь идет об окислении анионов,
генерированных в межфазных условиях. В близких условиях анионы,
генерированные из активированных нитрилов, превращаются
в кетоны [433], например:
NaOH, Et4NCl, O2
PhCH(CN)CH2CH2NMe2 -* PhCOCH2CH2NMe2
4 ' дноксан—вода
143
В системе хлороформ — вода — NaOH 2,4,6-три {4-трет-бу-
тилфенил) фенол (катализатор) трикалиигексацианоферрат
окисляет гидразины в азосоединения (выходы 63—98%) [434].
Бензилхлориды можно окислить до альдегидов 2-нитропро-
паном в системе дихлорметан — вода — Bu4NOH [435]:
ArCH2Cl + Me2CHNO2 —» АгСНО (63—80<>/0)
Ar = Ph, 2- или 3-O2NC6H4
Реакция окислительного сочетания проходит в системе
дихлорметан — КОН — 18-краун-б, при этом из анилина
получают азобензол с выходом 35% [66].
Наконец, отмечено, что в присутствии 18-крауна-6 или алик-
вата 336 удается генерировать синглетный кислород (!О2)
фотохимическим путем с использованием в качестве
сенсибилизаторов эозина или розы бенгальской в дихлорметане или
сероуглероде. Таким путем удалось получить пероксид из
антрацена и 3-гидропероксид из 2,3-диметилбутена-1 [436]:
(86%)
Ме2С«=СМе2 ► CH2^C(Me)—C(Me)2— ООН (71-74%)
Как вдгно. из всего изложенного выше, межфазный катализ
в ряде случаев Может быть успешно использован для
проведения многих реакщшЪкисления обычными и специальными
реагентами. Особенно интересно и перспективно применение
межфазных условий для окисления органических соединений пер-
манганатом калия («малиновый бензол») и дихроматом калия
(«оранжевый бензол»).
РЕАКЦИИ ВОССТАНОВЛЕНИЯ
Межфазный катализ имеет ограниченное применение в
реакциях восстановления. Алюмогидриды — очень
распространенные агенты восстановления — разлагаются водой, а борогид-
риды натрия и калия достаточно хорошо растворимы и могут
быть использованы в спиртовых растворах, в которых
растворимы многие органические субстраты. Однако в некоторых
специальных случаях межфазная методика может представить
определенный интерес и имеет преимущества по сравнению с
обычными способами. Это прежде всего асимметрическое
восстановление борогидридами калия [437] и натрия [438, 439]
в присутствии асимметрических катализаторов межфазного
переноса— (5)-М-метил-Ы-додецил- или Ы-метил-Ы-гексадецил-
эфедринийбромида [395—397], (—) - (R) -]М-додецил-Ы,Ы-диме-
тиламифетаминийбромида [396] и др. Таким путем в системе
144
1,2- дихлорэтан — вода — NaBH4 — катализатор (Ы-додецил-Ы-
метилэфедринийбромид) при восстановлении ацетофенона был
получен 1-фенилэтанол (оптический выход 39%), а из 4-метил-
пентанона-2 — 4-метилпентанол-2 (оптический выход 1,71 —
4,55%) [398]. Восстановление ряда кетонов КВН4 в
присутствии ^-додецил-Ы-метилэфедринийбромида привело с высокими
выходами к соответствующим спиртам [439]:
квн4
RCOR' >- RCH(OH)R'
R, R' (выход, %): rt-C6HiS, Me (100); мзо-Рг, изо-Рг (86); PhCH2, PhCH2
(100); Ph, Me (97); Ph, Et (100); Ph, Ph (100); 4-C1C6H4j H (95); Ph, H (95);
4-г/?£т-бутшщиклогексаион (100; транс: цис = 85: 15)
Однако при восстановлении октанона-2, 1-фенилпропанона-1
и ацетофенона не было обнаружено оптической индукции.
Восстановление альдегидов проводилось при 0°С, кетонов — при
25 °С.
Описано также [438] восстановление борогидридом натрия
в системе бензол — вода— (—)-^<]-додецил-^<]-метилэфедриний-
бромид при 0°С. Типичная методика приведена ниже:
Синтез З-фенилбутанолов-2. Смесь 10 ммоль 1-фенилбутано-
на-2, 6 ммоль NaBH*, 10 мл воды, небольшое количество бензола и 0,5 ммоль
катализатора интенсивно перемешивают 5 ч прн 0 °С. Органический слон
отделяют, водный слой извлекают дихлорметаном, объединенные органические
слои сушат (Na2SO4); на колонке с SiO2 (растворитель легкий петролейный
эфир — диэтиловый эфир, 5:3) выделяют смесь трео- и э/зиг/ю-изомеров
З-феннлбутаиола-2 (65:35); с помощью препаративной хроматографии
выделяют грео-изомер; [а^1 = 0,6 .
Аналогично с помощью NaBH4 получают и другие
вторичные спирты RCH(OH)R'.
R, R' (выход, %; оптический выход, %): Ph, изо-Рт (100; 3,6); Ph, трет-Ви
(77; 13,9); мезитил, Me (50; 2,3 при 25 °С); Ph(Me)CH, Me [65 (трео-юо-
мер), 35 (эрытро-изомер); 2,8]; Ph(Et)CH, Me [80 (т/?ео-изомер), 20 [эритро-
изомер)]
Описано [399] гидрирование углерод-углеродной двойной
связи в системе бензол — вода — NaOH— ТЭБАХ в
присутствии в качестве катализатора Кз[Со(СЫ)БН].
Синтез 2-метилбутена-2 [440]. В атмосфере Н2 2,2 ммоль
катализатора (CN~:Co —5,2) вносят в 40 мл воды содержащей 2,2 ммоль
NaOH и 2,2 ммоль ТЭБАХ, добавляют 20 мл бензола и 5 ммоль изопрена.
Гидрируют, контролируя течение реакции с помощью ГЖХ. Реакция
заканчивается через 12 ч. Выход 87%.
Без ТЭБАХ реакция идет крайне медленно с побочным образованием
2-метилбутена-1 и 3-метилбутана.
В аналогичных условиях пентадиеи-1,3 дает гранс-пентен-2
(выход 80 %), 2,3-диметилбутадйен-1,3 — смесь 2,3-диметилбу-
тена-2 (80%) и 2,3-диметилбутена-1 (20%). Успешно проходит
также гидрирование а,|3-непредельных карбонильных
соединений в насыщенные карбонильные соединения. Так, получены
бутанон-2 (выход 60%) из бутеи-З-она-2, З-метилбутанон-2
(85%) из З-метилбутен-З-она-2, масляный альдегид (выход
145
всего 20% из-за полимеризации) из кротонового альдегида,
ментен-8-он-2 (смесь транс-цис-изоыеров в отношении 6:1) из
ментадиен-1,8-оиа-2.
Таким образом, принципиально межфазиый катализ может
быть использован как для восстановления гидридами, так и
для чисто каталитического восстановления.
ПРИСОЕДИНЕНИЕ ДИГАЛОГЕНКАРБЕНОВ
Циклопропанирование дигалогенкарбенами не только
является одним из наилучших способов синтеза циклопропанового
кольца, но и позволяет получать как моногалогенциклопропа-
ны, так и циклопропаны, не содержащие атомов галогена в
кольце. Следует отметить, что циклопропаны интересны как
сами по себе, так н как отличные синтоны для получения
большого числа разнообразных циклических и алифатических
соединений, трудно доступных иными путями. Особенно бурное
развитие метода дигалогенциклопропанирования наблюдается
в последние 15 лет, после того как для генерирования дигало-
генкарбенов было предложено использовать действие на три-
галогенметаны концентрированных растворов едкого натра
в присутствии ТЭБАХ (реакция Макоша) или других
четвертичных аммониевых или фосфониевых солей, а также краун-
эфиров. Эти межфазные методы генерирования дигалогенкар-
бенов в настоящее время практически вытеснили прочие
способы и сделали дихлоркарбен (и другие дигалогенкарбены)
наиболее доступными нз всех известных циклопропанирующих
агентов.
Предложен следующий механизм катализа генерирования
дигалогенкарбенов солями тетраалкиламмония в двухфазной
системе:
+ 50%-ныЙ NaOH +
PhCH2NEt3 Cl" ►• PhCH2NEt8 "ОН
CHC1S + EtsNCH2Ph "OH —► C13C~ Et3NCH2Ph -f H2O
C13C" Et8NCH2Ph —► tCCU + EteNCHaPh СГ
Этот механизм подтверждается тем, что в случае
генерирования дихлоркарбена в присутствии олефина, содержащего
электроноакцепторные заместители, часто наблюдается
присоединение трихлорацетат-аниона, образующегося на первой
стадии реакции.
Что касается механизма присоединения дигалогенкарбенов,
генерированных в условиях межфазного катализа, т. е.
химическим путем, то в результате многочисленных исследований,
выполненных в основном на непредельных углеводородах, было
установлено два факта: 1) присоединение дигалогенкарбенов
к олефннам проходит стереоспецифично как ^«^-присоединение
и 2) дигалогенкарбены ведут себя в этой реакции как электро*
фильные реагенты. Отсюда следует, что дигалогенкарбены на-
146
ходятся в синглетном состоянии, т. е. два свободных электрона
в карбене имеют противоположные спины.
Далее мы рассмотрим синтетическое применение дигалоген-
карбенов для дигалогенциклопропанирования различных
классов соединений.
Дигалогенциклопропанирование ненасыщенных углеводородов
Существует несколько методов генерирования дигалогенкар-
бенов, которые разрабатывались в основном на примере ди-
хлоркарбена. Самый важный и простой из них метод
Макоша— генерирование дихлоркарбена из хлороформа действием
50%-ного раствора NaOH в присутствии каталитических
количеств ТЭБАХ (см., например, [441]) в присутствии алкена.
Синтез 1-фенил-2,2-дихлорциклопропаиа. Смесь 12 г
хлороформа, 20 мл 50%-ного NaOH, 0,4 г ТЭБАХ и 10,4 стирола
перемешивают 4 ч при 40 °С, разбавляют водой, отделяют органический слой,
разгоняют. Выход 15 г (80% от теоретического).
Аналогично получают 1,2,2-триметил-3,3-дихлорциклопропан
из триметилэтилеиа (выход 60%), 7,7-дихлорбицикло
[4.1.0]гептан из циклогексена (72%) и др.
Другой метод генерирования дихлоркарбена заключается
в кипячении мелко раздробленного трихлорацетата натрия
в хлороформе в присутствии 5—10% (мол.) тетра-я-гептилам-
монийбромида, аликвата 336, ТБАГС или ТЭБАХ и алкена,
подлежащего днхлорциклопропанированию [442]. Этот вариант
относится к межфазной системе твердая фаза — жидкость.
Таким путем были приготовлены 7,7-дихлорбицикло [4.1.0] гептан
(выход 57%), 9,9-дихлорбицикло [6.1.0] нонан (выход 73%),
6,6-дихлорбицикло [3.1.0] гексан (выход 16%), 1,1,2,2,3-пента-
хлорпропан (выход 9%), 2,3-дифенил-1,1-Дихлорциклопропан
(граяс-изомер, 7%), 2-н-бутил-1,1-дихлорцнклопропан (7,5%)..
Этот способ позволяет получать дихлоркарбен в нейтральных
условиях и может быть использован для дихлорциклопропани-
рования соединений, чувствительных к щелочам, или
соединений, не реагирующих в условиях Макоша (см. выше), например
для трихлорэтилена.
Еще один метод генерирования дихлоркарбена также
осуществляется в системе твердая фаза — жидкость [443]. С его
помощью было осуществлено дихлорциклопропанирование а-пи-
неиа;
Дихлорциклопропаннрование а-пинена. 100 мл хлорофор-
, 20,4 г «-шшена, 20 г порошкообразного NaOH и 0,3 г ТЭБАХ перемеши-
147
вают 15 мии, охлаждают льдом, осадок отфильтровывают, промывают 100 мл
хлороформа, кристаллизуют из метанола. Выход 20,3 г; т. пл. 64—-65 *t.
Помимо четвертичных аммониевых солей для генерирования
дихлоркарбена можно применять также краун-эфиры,
например дибензо-18-краун-6 [444].
Синтез 7,7-дихлоркарана. Перемешивают 6,1 г циклогексеиа,
40 мл 50%-ного NaOH, 20 мл хлороформа и 0,1 г дибензо-18-крауна-6 в
течение 9 ч при 40 °С, выливают в воду, органический слой отделяют, сушат,
перегоняют. Выход Юг (61% от теоретического, т. кип. 70 °С (8 мм рт. ст.);
п2£ 1,5088.
Чаще всего применяется методика Макоша.
Эти четыре метода генерирования дигалогенкарбенов
используют для выделения и других дигалогенкарбенов — ди-
бромкарбена [445, 446], дииодкарбена [447], фторхлоркарбена
[448], бромфторкарбена [448, 449], иодфторкарбеиа [450],
бромхлоркарбена [451] и иодхлоркарбена [447].
В реакцию с дихлоркарбеном входят самые разнообразные
алкены: бутен-2 [441], гексен-1 [452], З-метилбутен-2 [441,
452], 3,3-диметилбутен-1 [452]. Соответствующие дихлорцикло-
пропаны образуются с высокими выходами (60—97%). Лишь
в случае пространственно затрудненного 3,3-диметилбутена-1
выход снижается до 33% [452].
1-Фенилэтилен (стирол), как уже отмечено выше, гладко
реагирует в условиях Макоша, образуя 1-фенил-2,2-дихлорцик-
лопропан с выходами 80—95% [441, 453] (см. также [64]).
1,2-Дифенилэтилен (стильбен), который не реагирует в системе
хлороформ — трет-ВиОК, образует с реагентом Макоша 1,2-ди-
фенил-3,3-дихлорциклопропан с выходом 96%. Так же хорошо
проходят превращения других малореакциоиноспособных фенил-
замещенных этилеиов, например, 1,1-дифенил-, 1,1,3-трифенил-
этиленов и 1,1-Дифенилпропена-1, приводящие к
соответствующим дихлорциклопропанам [454] с выходами 79—96%. Тет-
рафенилэтилен не реагирует в межфазных условиях.
а,ш-Дифенилполиены не реагируют с дихлоркарбеном,
генерированным по методу Макоша. Однако перфторполиены типа
полностью rpaftc-RC6H4(CF=CF)«C6H4R (R=Me, ОМе; п =
= 1—3) присоединяют дихлоркарбен. В случае триенов
реакция проходит по концевым связям. а,оо-Перфтортетраен не
присоединяет дихлоркарбен [455].
Гладко проходит реакция с циклическими моноолефинами,
такими как циклогексен [441—443], циклогептен [456], а-пи-
нен [443], циклопентен [443]. Выходы продуктов дихлорцикло-
пропаиирования достигают 80—98%.
1'48
Циклопропанирование циклоалифатических диенов,
содержащих двойные связи в цикле и боковой цепи, не сопряженные
друг с другом, проходит по обеим двойным связям, как,
например, в случае лимонена [443].
С1
С1
С1
а
Однако если использовать вместо ТЭБАХ бензил (р-гидрок-
сиэтил)диметиламмонийхлорид, то можно провести реакцию по
одной из двойных связей лимонена, а именно по двойной связи
в цикле [457]:
(62%)
Аналогично из 4-винилциклогексена получают 4-винил-7,7-
дихлорбицикло [4.1.0] гептан с выходом 93% [457].
Близкая картина наблюдается при действии дихлоркарбена
на 1 Д9-4мс,гр£Шс,77?сшс-циклододекатриен: в присутствии гек-
садецилтриметиламмонийбромида получают продукт циклопро-
панирования по всем трем двойным связям, а в
присутствии бензил (Р-гидроксиэтил) диметиламмонийхлорида — про-»
дукт ииклопропанирования по одной из связей [415]:
С1
С1
С1
С1
X
X
X
CJ
X
CI
Довольно подробно было изучено присоединение
дихлоркарбена, генерированного по Макоша, к норборнадиену—стери-
чески затрудненному несопряженному диену [458—460]. Было
установлено, что реакция протекает с образованием трех
продуктов, образующихся за счет перегруппировки первоначально
возникающих моноаддуктов:
CL
(77-80°,$}
(7-12%)
сг а
149
С перегруппировкой проходят также реакции норборнена
[461] и метилнорбориена [462] с Дихлоркарбеном. Во втором
случае помимо расширения цикла наблюдается дегидрохлори-
рование;
:cci
(10%)
Весьма сложно протекает реакция 7-т/?ег-бутоксинорборна-
Диена с дихлоркарбеном [460]. Присоединение проходит почти
исключительно в сия-положение к грег-бутоксигруппе, что,
возможно, объясняется координацией электрофильного дихлоркар-
бена со свободной парой электронов атома кислорода до или
во время присоединения по двойной связи (общий выход
продуктов составляет 40%):
(38%)
jper-BuO
(7%)
Bu-jper
Cl CI
(30%)
Сложно, по-видимому, через присоединение дихлоркарбена
по двойной связи с последующим раскрытием циклопропано-
вого кольца с образованием хлорзамещенного аллильного кар-
боннй-нона и внутримолекулярным присоединением гидроксиль-
ной группы идет реакция зядо-5-гидроксйметилнорборнена-2
с дихлоркарбеном. В результате образуется 3-хлор-5-окСатри-
цикло[5.2.1.04-8]децен-2[463]. :,
Присоединение дихлоркарбена к дьюаровскому гексаме||р-
беизолу также проходит сложно и приводит к трем продуктам
150
Cl
Дихлоркарбен достаточно легко присоединяется к
сопряженным диенам и полиенам. Так, в зависимости от соотношений
реагентов из бутадиена-1,3 можно получить в условиях Макоша
предпочтительно либо моно-, либо бисаддукт [454]. При
соотношении бутадиен: хлороформ, равном 2:1, образуется 42%
моно- и 6% диаддукта, при соотношении 1:2—13% моно- и
38% диаддукта.
\/\ У \ У\ У\ У
В случае 1,4-дифенилбутадиена-1,3 образуется смесь
изомерных 3,ЗЧдифенил-1,1,^1'-тетрахлорбициклопропилов [458]
с общим выходом 48%, из которой удается кристаллизацией
выделить жезо-форму (выход 41 %).
Циклопентадиен образует в условиях Макоша как
хлорбензол, так и бисаддукт (выход 10%) [458]:
л—n
XI
Циклооктатетраен образует 9,9-дихлорбицикло [6.1.0] нона-
триен-2,4,6 с выходом 24% и бисаддукт с выходом 16% [454].
Дихлорциклопропаиирование моноаддукта дает бисаддукт с
выходом 32%, а при использовании избытка дихлоркарбена —
3,3,6,6,11,11-гексахлортетрацикло [8.1.0.02'4.05'7]-ундецен-8 [465].
С высоким выходом (67%) моноаддукт можно получить при
использовании для генерирования дихлоркарбена системы
вода — CI3CO2Na — аликват 336 [466].
ci Cl
х
Cl (
^Cl (
В условиях межфазиого катализа был получен ряд дихлор-
циклопропаиов из алленов. Из алкилалленов, как и из самого
аллена, образуются главным образом бисаддукты [467],
151
cl
однако в случае 1,1-диметил- и трифенилалленов удается
задержать реакцию на стадии образования моноаддуктов. Из 1-фе-
нилаллена образуется также спиро[2.3]гексан [468],
RCH=C=CH2
PhCH=C=CPh2
С12С
R-HC
СС12
СН2 (23—90%)
ССЬ
Ph
—Н(>—С=
CPh2 (40%)
Тетрафенилбутатриен-1,2,3 в условиях Макоша дает два
бисаддукта [454]:
ci a
Ph
Ph
,Ph
4Ph
Cl CI
или
Cl Cl Ci Cl
Plu A A /Ph
>
Ph
Cl C1C1C1
Ph4
Ph/
1,4-Ди-г/?ег-бутил-1,4-дифенилбутатриен образует моноад-
дукт, а в присутствии большого избытка дихлоркарбена — би-
саддукт [469]:
Ph
\/
cci.
,Ph
трет-Bw
Ph,
трег-Ви'
СС12
\
CCI
Ви-трег
Ph
Винилацетилены реагируют с дихлоркарбеном по двойным
связям [454, 470]. Так, из 2,5-диметилгексадиен-1,5-ина-3
образуется бисаддукт с сохранением тройной связи [470]:
СС12 СС12
:cci2 / \ / \
Me
Me
Me
(62%)
Me
Если в молекуле диенина одна из двойных связей входит
в цикл, реакция с дихлоркарбеном приводит только к моноад-
дукту по двойной связи в цикле [454, 470].
152
Ацетилены, например толан, реагируют с дихлоркарбеном
в межфазных условиях с образованием циклопропенов,
например [454]:
О
jcci2 \
phOsCPh > Ph £=-Ь—ph (23%)
Однако при реакции уже октина-4, а также дифенилбутадиина-
1,3 с дихлоркарбеном выходы соответствующих циклопропено-
нов очень низки; продукты реакции можно обнаружить только
по спектрам [454].
Аналогично дихлоркарбену в условиях межфазного катализа
реагируют с разнообразными алкеиами (гексены-1 и -2, геп-
тен-1, октены-1 и -2, децен-1, бутен-2, г/?ег-бутил-3,3-диметил-
бутен-1, 2-метил- и 2,3-ДИметилбутены-2, циклопентен, цикло-
гексен, стирол, изопропенилбензол, циклооктадиен-1Д нонен-4,
октатетраен и многие другие) дибромкарбен [445, 446, 471,
472], фторхлоркарбен [448] (в этом случае из-за низкой
температуры кипения фтордихлорметана реакцию проводят прн
—40°С), бромхлоркарбен [451] (в присутствии дибеизо-18-
крауна-6) и иодхлоркарбен (447] (катализатор ТЭБАХ).
Таким путем был осуществлен синтез ряда незамещенных и
замещенных 1-фтор-1-хлорциклопропанов (выходы 45—60%) [448,
471 ], 1 -бром-1 -фторциклопропанов (выходы 30—88 %) [448,
473, 474], 1-иод-1-фторциклопропанов [450], 1-бром-1-хлорцик-
лопропанов (выходы 44—76 %) [451] и 1 -иод-1 -хлорциклопро-
панов (выходы 49—65%) [448].
Общая методика синтеза дибромциклопропаиов [445].
К смеси 0,4 моль бромоформа, 0,2 моль алкена, 0,4 г ТЭБАХ и 0,8 мл
этилового спирта в течение 10 мин при 40—45°С добавляют 100 мл 50%-иого
раствора NaOH, перемешивают 2,5—3 ч при 40—45 °С, выливают в воду,
извлекают дихлорметаиом, из органического слоя выделяют дибромциклопропан.
Вместо ТЭБАХ удобно использовать амины [446].
Смесь 0,1 моль алкена, 50,6 г бромоформа в 25 мл дихлорметаиа, 0,4 мл
три-я-бутиламииа и 40 мл 50%-ного NaOH интенсивно перемешивают 4 ч
при 40—45 °С. Смесь разбавляют 200 мл воды и 50 мл дихлорметана,
отделяют органический слой, водный слой дополнительно дважды экстрагируют
дихлорметаиом, объединенные органические слои промывают водой,
разбавленной соляной кислотой, водой, сушат, удаляют растворитель. Остаток очи*
щают разгонкой в вакууме или кристаллизацией."
В случае фторхлоркарбена всегда образуются смеси син-
анпг-изомеров. Изучение стереоизомериого состава продуктов
присоединения бромфторкарбена к арилэтиленам [449]
показало, что для монозамещенных арилэтиленов, как правило,
независимо от природы заместителя R в арильном остатке (Н,
4-Ме, 4-МеО, 3-МеО, 4-Br, 4-O2N и др.) в образующейся смеси
несколько преобладает awrw-изомер. Однако в случае 2,6-диме-
токсифенилэтилена, 2,4,6-триметилфенилэтилена и пентафтор*
153
фенилэтилена в смеси образующихся изомерных бромфторцик-
лопропанов преобладают син-кзоиеры [449].
Следует отметить, что реакция между алкенами и дииод-
карбеном, генерированным в условиях Макоша из йодоформа,
приводила к дииодциклопропанам только в случае арнлалке-
иов; при этом выходы были, как правило, невысоки:
максимальный выход (59%) был получен в случае гс-хлорстирола.
Реакция со стиролом привела к 2,2-дииод-1-фенилциклопропану
с выходом 21%, из стильбена был получен 3,3-дииод-1,2-дифе-
нилциклопропан с выходом всего 2% [447].
Среди других реакций присоединения дигалогенкарбенов
к углеводородам, содержащим кратные связи, можно
упомянуть присоединение в межфазных условиях к пирену двух
молекул дихлоркарбена, к бульвалену трех молекул и, наконец,
к циклооктатетраену четырех молекул дихлоркарбена (однако
выходы соответствующих аддуктов были низкими) [475];
С1 Cl
Взаимодействие дихлоркарбена в обычных условиях
Макоша с нафталином и антраценом приводит к образованию
продуктов расширения колец с последующим дегидрохлорирова-
нием, и лишь из фенантрена удается получить с выходом 65%
нормальный продукт присоединения дихлоркарбена по связи
9,10 [476]. Сложные превращения проходят также при
реакции дихлоркарбена с 1,2-диметоксибифениленом. [477J.
Способность дихлоркарбена к внедрению и расширению
колец была положена в основу метода синтеза 3-галогенхииоли*
нов из индолов [478, 479Ji
tcx5
„ R', X (выход, %): Me, Н, Cl (53); Н, Ph, Cl (68)j Me, Me» CI (65); H, Me,
1 (45); Me, H, Br (24); H, Ph, Br (37)
154
Метод был распространен также на получение хлорзаме-
щенных пиридинов из пирролов и хлорзамещенных пиримиди-
нов из имидазолов [479]:
(49%)
Me
\ N
Me—/ V-Me —> j| J (39%)
Интересно, что в условиях Макоша из 2,2,6,6-тетр.аметилод*
перидинона-4 образуется К-(пропен-2-ил)-5,3-диметил-3-метилеи-
пирролидинон-2, т. е. проходит сужение кольца [480],
Дигалогенциклопропанирование функционально
замещенных алкенов
Дихлоркарбен и другие дигалогенкарбены имеют ярко
выраженный электрофильный характер, поэтому алкены,
содержащие рядом с двойной связью электроноакцепторные
заместители, трудно вступают в реакцию присоединения (см. выше).
Обычно при действии дихлор- или дибромкарбена на такие оле-
фины проходит присоединение тригалогенацетат-иона. Активация
двойной связи введением в а-положение к акцептору метиль-
ной группы позволяет ввести в реакцию с дигалогенкарбенами
олефины с электроноакцепторными заместителями. Например,
мвтакрилонитрил, этилметакрилат и метиловый эфир тиглиио*
вой кислоты реагируют с дихлоркарбеном в условиях
межфазного катализа, образуя соответствующие дихлорциклопропаны
{327, 328] i
С1 С1
RR'O
R" \R'"
R, R', R", R'" (выход, %): H, H, M?, CN (42; 13% 2-метил-4,4,4-трихлорбу-
тиронитрила); Н, Й, Me, CG2Me (85); H, Me, Me, CO2Me (69,
При наличии метальной или фенильной группы в (3-поло-
жении к акцептору реакция проходит сложно. Так, из
метилового эфира коричной кислоты образуется 1-метоксикарбонил-
4(4,5,5-тетрахлор-г/7аяс-2-фенилспиро [2.2] пентан (выход 48%),
а из метилового эфира р,§-диметилакриловой кислоты—1-мето-
ксикарбонил-2,2-диметил-4,4,5,б-тетрахлорспиро [2.2] пентан
(выход 35%). Из метилового эфира кротоновой кислоты помимо
производного спиро[2,2]пентана (выход 14%) образовался так-
165
же метиловый эфир З-(трихлорметил)масляной кислоты (выход
10%) [327,328]:
С1 СО2Ме
^ С1—
: cci2 I ^
МеСН=СНСО2Ме *• С13ССНСН2СО2Ме +
С1—
Cl Me
Следует отметить, что спиропентан, полученный из метилкро-
тоната, представляет собой смесь двух стереоизомеров в
отношении 45:55. Промежуточными продуктами в этой реакции
являются, по-видимому, 2,2-дихлор-Ьэтоксикарбонилциклопропаны,
поскольку при действии реагента Макоша в стандартных
условиях на 3-фенил-2,2-дихлор-1-этоксикарбонилциклопропан и 3,3-
диметил-2,2-дихлор-1-этоксикарбонилциклопропан образуются
спиропентаны.
Этиловый эфир 4-метилпентадиен-2,3-овой кислоты
реагирует с дихлоркарбеном по связи, удаленной от этоксикарбониль-
ной группы, образуя 3,3-диметил-2,2-дихлор-1-этокснкарбонил-
метиленциклопропан с выходом 69% [296, 297].
Менее электрофильный дибромкарбен, генерированный в
условиях Макоша, более активно реагирует с алкенами,
содержащими электроноакцепторные заместители. Так, метиловый
эфир |3,р-диметилакриловой кислоты образует при действии
бромоформа и 50%-ного NaOH в присутствии ТЭБАХ 2,2-ди-
бром-3,3-диметил-1-метоксикарбонилциклопропан [481];
Вг Вг
t СВг2 Ме\ /\
Ме2О~СНС02Ме > У^—^—СО2Ме(89%)
В аналогичных условиях из а,Р-непредельных кетонов
образуются 1-ацил-2,2-дибромциклопропаиы [482]. Подробно
изучена также реакция между различными а,|3-непредельными ке-
тонами и дихлоркарбеном [482, 483]. Было установлено, что
все а-алкилвинилметилкетоны, за исключением 3-метилбутен-
З-она-2, а также все 3,4-диалкил- или 2,3,3-триалкилбутеноны-2
гладко реагируют с дихлор- и дибромкарбенами с
образованием соответствующих ацилдигалогенциклопропанов:
:сх2
MeCOCfR')—CR'R'"
R, R', Rff, X (выход, %): Me, H, H, Cl (0); Me, H, H, Br (85); н-Pr, H, H,
Cl (25); tt-Pr, H, H, Br (50); изо-Рг, H, H, Cl (35); изо-Рг, H, H, Br (90);
трет-Ви, H, H, Cl (35); Me, H, Me, Cl (35); Me, H, Me, Br (70); Me, H, Et,
Cl (50); Me, H. Et, Cl (60); Me, H, изо Pr, Cl (50); Me, H, изо-Рг, Br (30);
Me, Me, Me, Cl (50); Me, Me, Me, Br (30)
Выход циклопропанов, как правило, выше при использовав
нии дибромкарбена, за исключением тех случаев, когда
увеличиваются стерические затруднения.
При действии галогенкарбенов на непредельные кетоны, не
имеющие алкильных групп в а-положении, не получены
удовлетворительные результаты, однако эти кетоны можно ввести
в реакцию в виде дноксоланов [482]:
Х2С p
0 CRR' /У
J^rV—V
R, R', X (выход, %): Н, Me, C1 (35); Н, Me, Вг (30); Me, Me, Cl (70); Me,
Me, Br (80)
Ацетальная защита пригодна также для альдегидов,
которые полимеризуются при действии дихлоркарбена [484—487].
Однако в стандартных условиях Макоша при использовании
эквимольного количества или даже двухкратного избытка
хлороформа и 50%-ного NaOH выходы ацеталей формилциклопро-
панов, например из диэтилацеталей кротонового или винилук-
сусного альдегида, оказались низкими (21—23%), а в
некоторых случаях (тетраметилацеталь глутаконового диальдегида и
1,1,3-триэтоксигексен-4) ацетали вообще не реагировали. Лишь
увеличение избытка хлороформа н 50%-ного NaOH до
четырехкратного позволило значительно повысить выход ацеталей и
ввести в реакцию ацетали глутаконового диальдегида и 1,1,3-три-
этоксигексен-4:
ссь
:cci2 / \
RCH=CHCH(OR')2 > R—с -—CH(OR')2
R, R' (выход, %): Н, Et (47); Me, Et (93); Ph, Et (45); (MeO)2CHCH2,
Me (48); (MeO)2CHCH2) Me (24)
Общая методика синтеза ацеталей 2-формил-1,1-дн-
хлорциклопропаиов [485, 486]. Смесь 0,04 моль ацеталя
непредельного альдегида, 0,16 моль хлороформа и 0,5 г ТЭБАХ нагревают до 40 °С л
при интенсивном перемешивании добавляют в течение 10 мии 24 мл 50%-ного
раствора NaOH, перемешивают 3 ч при 40—45 °С, разбавляют -загустевшую
смесь 40 мл дихлорметана и 100 мл воды, извлекают дихлорметаном
(3 X150). Объединенные органические слои промывают водой, сушат
(Na2SO4), удаляют растворитель, остаток перегоняют в вакууме..
Несоблюдение оптимальных условий реакции приводит к
резкому снижению выхода. Так, применяя 2,5-кратный избыток
хлороформа и 50%-ного NaOH при реакции с диметилацета-
лем р-этилакролеина, получили [487] диметилацеталь 2-фор-
мил-1,1-Дихлор-3-этилциклопропана с выходом 19,3%.
Необходимость использования большого избытка хлороформа при
генерировании дихлоркарбена по Макоша подчеркнута и в
работе, специально, посвященной выяснению оптимальных условий
157
реакции дихлорциклопропанирования [488]. При реакции с
малоактивными соединениями иногда необходимо применять и
еще больший избыток хлороформа и 50%-ного NaOH. Так,
тетраметилацеталь фумарового диальдегида не реагирует с ди-
хлоркарбеном даже при использовании четырехкратного
избытка реагентов, однако при 10-кратиом избытке хлороформа и
50%-иого NaOH удалось получить бис(диметилацеталь)1,2-ди-
формил-3,3-дихлорциклопропана с выходом около 50% [489].
Дихлорциклопропанироваиие ацеталей сопряженных полие-
новых альдегидов проходит по-разному в зависимости от
природы концевого заместителя и числа двойных связей в
молекуле. Так, диэтилацеталь сорбинового альдегида в стандартных
условиях образует аддукт по концевой двойной связи [486,
490], а при 8-кратном избытке реагентов — бисаддукт по обеим
двойным связям [486, 491]:
СС12
г cci2 / \
Ме(СН—CH)2CH(OEt)2
СН=СНСН(ОШ)2
(44%)
2:cci2
l
[486, 487]
(50%)
Диэтилацеталь октатриен-2,4,6-аля при 4-кратном избытке
реагента Макоша образует смесь двух бисаддуктов [486, 491] i
ССЬ
Me(CH«CH)3CH(OEth —► Me—с \~Р—СН=»СНСН(ОБ*)2 +
СС12
ССЬ ССЬ
+ Me ■ ^—СН=»СН—^—^—CH(OEt)2
Суммарный выход обоих бисаддуктов, соотнощеиие между
которыми равно 2:3, достигает 44%. При действии же да аце*
таль триенового альдегида 12-кратного избытка реагента
Макоша был получен ацеталь терциклопропанкарбальдегнда с
выходом 18%:
з: ccij I / \ I
Me(CH=CH)8CH(OEt)2 > MeL—с —J8CH(OEt)2
Ацетали ш,ш-дихлорполиеналей при реакции с дихлоркарбе-
иом в условиях Макоша образовывали циклопролановое кольцо
по двойной связи, соседней с ацетальной группой [486, 490].
Это можно объяснить тем, что атомы хлора за счет сильного
'—/-эффекта значительно обедняют электронную плотность
двойной связи именно на конце цепи. Ацеталь р,р-дихдоракро-
леина вообще не реагирует с дихлоркарбеиом; удлинение же
ненасыщенной цепочки ослабляет елияаде хлора и реакция по
двойной связи у ацетальной группы становится возможной.
Выходы продуктов реакции увеличиваются по мере удлинения
цели:
:coi2
С12С—CH(CH=«CH)ftCH(OEt)2 *■
ССЬ
—» СЬС—СЩСН—СН)л-1—*—*—CH(OEt)2
п (выход, %)i 0 (0)\ 1 (18); 2 (40)
Дихлорциклопропанирование избытком дихлоркарбена аце-
талей муконового альдегида [489] и октатриен-2,4,б-аля [491]
приводит соответственно к ацеталям би- и терциклопропанди-
Карбальдегидов (п = 2, 3)j
[ССЬ I
—*—^—J«CH(OEt)«
При действии 4-кратного избытка реагента Макоша иа ди-
Этилацеталь цитраля дихлорциклопропанирование проходит
исключительно по более активной концевой двойной связи,
однако увеличение количества реагента до 8-кратного избытка
позволяет получить продукт дихлорциклопропанирования по
обеим связям [492] i
М
|
CH2CH2G«GHCH(OEt)
t
Me
\
1
СН2СН»—ч—7—СН(°ШЬ
ссь ecu
Следует отметить, что ацеталь оксида цитр а ля также при
соединяет дихлоркарбен [493]:
Me
Me2G=*CHCH2CH2C CHCH(OEt)a —*•
О
Me
Ме2С СНСН2СН2С CHCH(OBt)j
V, V
Хорошие результаты получены в реакции Макоша с алли-
ловыми спиртами. Сам аллиловый спирт осмоляется при
действии дихлоркарбена [494], а при реакции с фторхлоркарбеном
(в автоклаве) образует 1-фтор-1-хлорциклопропилметанол с
выходом всего лишь около 15% [495]. Зато замещенные алли-
ловые спирты легко превращаются в замещенные дихлорцикло-
пропилметанолы [495—498] или дибромциклопропилметанолы
(при действии дибромкарбена) [498]:
С1
:сх2 R
с=с с
|\R
ОН
R, R1, R3, R8, R4, X (выход, %): Н, Н, Me, Н, Н, С1 (70); Н, Н, Me, Н, Н,
Вг (34); Н, Н, Me, H, Me, Cl (75); Н, Н, Me, H, Me, Br (62); Me, H, Me, H,
Н, Вг (45); Me, Me, H, Me, Н, Cl (90); Me, Me, H, Me, H, Br (70); H, H, Me,
Me, Me, Cl (69); Me, Me, H, Me, Me, Cl (88); Me, Me, H, Me, Me, Br (88),
Me, Me, Me, H, H, Cl (81); Me, Et, H, H, H, Cl (92); Me, Me, H, H, H, H,
Ci (74); JV^e, Afc, H, я-Bu, H, Cl (79); Me, Me, H, я-СвН1ь Н, Cl (60); Me, Me,
H, CH3=CHCH2, H, Cl (57)
Незамещенный аллиловый спирт может быть
предварительно превращен в ацеталь ацетальдегида (реакцией с винилэти-
ловым эфиром — в аллилэтилацеталь, с ацетальдегидом — в ди-
аллилацеталь). Взаимодействие этих ацеталей с дихлоркарбе-
ном дает соответственно (1,1-дихлорциклопропил-2)метилэтил-
и бис[(1,1 -дихлорциклопропил-2) метил] ацетали ацетальдегида
с высокими выходами [494]:
OR OR' CCl2
I ■ :cci2 I / \
MeCH—OCH2CH^CH2 ** MeCH-OCH2— *
CCU
R « R'_ Et (60%); R = CH=CH2, R' =* CH2 ' ^ (80%)
Дихлор- и дибромциклопропилметанолы образуются также
при реакции линалоола с дихлор- или дибромкарбенами с
выходами соответственно 89 и 93% [498]. Первичные спирты,
например гераниол, дают продукты циклопропаиирования с очень
низкими выходами.
В условиях межфазного катализа достаточно хорошо
реагируют простые виниловые эфиры [499—506]. Дивиниловый эфир
дает при этом смесь моно- и бисаддуктов:
СС12
:cci2 / \
■осн=сн2
[CCU 1
—^—J2o
Простые эфиры вииидцетиленовых. спиртов образуют при
160
действии дихлоркарбена моиоаддукты по двойной связи. [501,
507]:
ОМе СС12 ОМе
I : cci2 / \ I
sO~CR"R"' >• RR'C CHCseCR"R'"
Фторхлоркарбеи, генерированный в двухфазной системе по
методу Макоша, легко присоединяется к простым виниловым
эфирам, давая алкоксифторхлорциклопропаны [495, 508—510],
например;
:cfci
Ме2С=СНОМе »-
CFC1
—*» Ме2С СН—ОМе (смесь цис~ и гра«с-изомеров, 79%)
При действии фторхлоркарбена на 2,6-диметил-1-метокси-
гептадиен-1,5 образуется смесь продуктов присоединения по
двойным связям в положениях 1,2 и 5,6 с преобладанием
первого изомера (каждый из изомеров является смесью четырех
диастереомеров [510].
ОМо
I
•ОМе
ОМе
L Cl F
1
(42%)
2-Хлор-1,4-диоксен реагирует с дихлоркарбеном,
полученным по Макоша, необычным путем [505]:
0 ° ,СС12
о о C1
Бромфторкарбен, генерированный по Макоша, реагирует с
винйлэтиловым и бутилвиниловыми эфирами [501, 508],
образуя наряду с продуктами дихлорциклопропанирования (выход
28—Зо%) непредельные соединения. Следует отметить, что
воегда получают только один из возможных изомеров:
CH2=-CHOR
QR
Фторхлоркарбен реагирует в системе 50%-иый NaOH —
CHCbF — ТЭБАХ с линейными и циклическими ацеталями,
содержащими двойные связи [495]:
F С!
cfci
161
R, R't R" (выход, %): (EtO)2CHCH2CH2, Me, H (43); (~\—OMe, Me,
О
H (47) {ситанти, 1:1); >—CH2CH2; Me, H (49) (один изомер)
При нагревании этилеиоксида с хлороформом в
присутствии четвертичных аммониевых [511] или фосфониевых [512]
солей генерируется дихлоркарбен, который способен
присоединяться к олефинам, давая геж-дихлорциклопропаны. Однако
при реакции с виниловыми эфирами образуются не 2-алкокси-
1,1-дихлорциклопропаны, а ацетали а-галогензамещеиных а,р-
неиасыщенных альдегидов. Лишь при реакции вииилэтилового
эфира с хлороформом и этиленоксидом (соотношение 1:1:2)
в присутствии ТЭБАХ при 150 °С наряду с этил-§-хлорэтил-
ацеталем 2-хлоракролеина (выход 65%) был выделен 2,2-дихлор-
1-этоксициклопропан (выход 5%). В тех же условиях из ди-
фторхлорметана был генерирован дифторкарбен и получены
ге.м-дифторциклопропаны [513, 514].
В условиях Макоша 1-этоксибутадиен-1,3 и З-метил-1-этокси-
бутадиен-1,3 присоединяют дихлоркарбен по концевой двойной
связи, образуя с выходом 52—59% соответствующие виниловые
эфиры с преобладанием транс-фориы [515] s
СН-СНОШ
Аллилгалогениды образуют при действии галоформов и
50%-ного NaOH в присутствии ТЭБАХ смесь производного цик-
допрапана, галогеналкена и галогеналкадиена [516]:
CH2==*CHCPJ2X + CHYZ2 —►
■СН2Х
X
Y Z
z
Выше отмечено, что эфиры непредельных кислот плохо всту-
рают в реакцию дихлорциклопропанирования в межфазных ус-
Ловиях. Однако эфиры сорбиновой кислоты и октатриен-2,4,6-
ойой кислоты реагируют с дихлоркарбеном в обычных условиях
Макоша с образованием циклопропановых колец по наиболее
удаленным от карбалкоксигруппы двойным связям [502]:
ссь
:ссь / \
Me(CH«CH)nCO2R > Me— -—(CH«CH)n-iCO3R
/г, R (выход, %J: 2, Me (22); 2, Et (50); Зг Et (35),
В работе [517] показано, что бие(пентадиен-1,3-ил)ацеталъ
ацетальдегида, полученный ацетализацией ацетальдегида сор-
бииовым спиртом, гладко реагирует с дихлоркаобеном,
генерированным в межфазных условиях, образуя бисаддукт, который
после гидролиза дает смесь двух изомерных спиртов:
а:са2
МеСН[ОСН2(СН=СН)2Ме]2 +
ССЬ Me CCh
—у Мв—С ^—СН=СНСН2ОСНОСН2—С -*—СН«СНМв -*-*
(94%)
ссь ecu
—v Me—^—*—СН=СНСН2ОН + МеСН=»СН * »—СН2ОН
Таким образом, дихлорциклопропанирование ацеталя,
полученного из ацетальдегида и сорбинового спирта, идет иеселек-
тивно. При использовании 8-кратного избытка реагента Мако-
ша присоединение дихлоркарбена проходит по всем четырем
двойным связям с выходом 76% [517}.
Отмечено также ]518], что аллиловый спирт можно
защитить ацетилированием; получающийся аллилацетат гладко
реагирует с дихлоркарбеном, давая 1-ацетокеиметил-2,2-дихлор-
циклопропан с выходом 56%. Используя ацетальную защиту,
можно получить из этил(3-фенилпропен-2-ил)- и этил (2,2-Диме-
тилпропен-2-ил) ацеталя ацетальдегида соответствующие ди-
хлорциклопропаны с хорошими выходами:
OEt OEt ССЬ
Me—CHOR ^ MeCH—OCRa"—c ^—R"
R'=*H, R"=~Ph (выход 62%); R ~ CMe2CH=.CH2,
£' = Me, R" = H (выход 67%)
Еще одиа интересная возможность использования дихлор-
карбена заключается в реакции его с основаниями Шиффа,
которая в ряде случаев приводит к азиридинам [519], например:
Г1 NPh
t cci2 ы\ / \
PhN=CHCeH4NO2-rt > V—^—CeH4NO2-/t (62%)
С К
Аналогично из л-метокси-Л^-(«-нитробензилидеи) анилина
получают 1-я-метоксифенил-2-п-нитрофенил-3,3-днхлоразиридин с
выходом 59%. Однако алифатические основания Шиффа,
например грег-бутил-N- (2-метилпропилиден) амин, или основания
Шиффа из циклических аминов, например К-(я-нитробензили-
деи)циклогексиламин, образуют соответственно N-грег-бутила-
мид 4-метил-2-хлормаслянои кислоты (выход 10%) и N-цикло-
гексил-«-иитрофенил-а-хлорацетамид (выход 74%) [519].
В тех же условиях изопропил (2,2-диметшшропилиден) амин
163
превращается в 2-грег-бутил-1-изопропил-3,3-дихлоразиридин
(выход 75%).
Недавно [520] установлено, что винилнитрамины способны
присоединять дихлор- и дибромкарбены в двухфазной системе,
образуя алкил [2,2-дихлор(или дибром)циклопропил]нитрамины
с выходами 25—67%:
СС1
RN(NO2)CH=CH2 —► RN(NO2)—^—s
R=-Me, MeCHCl, PhCH2, MeOCH2i EtOCH2
Выход уменьшается с повышением электроноакцепторных
свойств нитрамина.
МЕЖФАЗНЫЙ КАТАЛИЗ В МЕТАЛЛОРГАНИЧЕСКОЙ ХИМИИ
Описано немного примеров использования межфазного
катализа для синтеза и превращений металлорганических
соединений. Однако этих примеров достаточно, чтобы видеть
перспективность более широкого внедрения межфазного катализа в
эту область. Так, было показано, что в системе 50%-ныйводный
раствор NaOH — галоформ — ТЭБАХ — KF фенилртутьхлорид
реагирует с хлороформом или бромоформом, образуя
соответствующие фенил ртутные производные с выходом 54—72%
[521]:
PhHgCl + СНХ3 —> PhHgCXs
Межфазный катализ оказался очень удобным для орто-ме-
таллирования тиобензофенонов действием Fe3(CO)i2 в системе
бензол — водный раствор NaOH— ТЭБАХ [522]:
Ar H
В
Mcoh
е(СО)3
Выходы составили 70% для самого тиобеизофенона, 80% Для
4,4'-диметокситиобензофенона, 76% для тиокетона Михлера и
36% для 4,4/-диметилтиобензофенона.
В условиях межфазного катализа (бензол — 5 и. NaOH —
ТЭБАХ) аллилгалогениды реагируют с дикобальтоктакарбони-
лом, давая я-аллильные комплексы [523]:
RCH=C-CH2Br
R, R' (выход, %): H, H, (80); H, Me (73); Me, H (80); Ph, H (72)
164
В аналогичных условиях три- и тетрагалогенпроизводные
также реагируют с дикобальтоктакарбонилом, образуя
кластерные соединения [523].
С6Нб, ТЭБАХ t У X.
(со)8с -^ х
*Со(Сб)
Реагент, R (выход, %): ССЦ, С! (42); CBr4, Br (11); PhCCl3l Ph (53);
СС13СО2Ви-трет, СО2Ъъ~трет (30); СС13СН2ОН, СН2ОН (следы)
В системе бензол — водный раствор NaOH в присутствии
BU4NI карбонилы металлов легко образуют комплексы с
различными лигандами (РРЬз, AsPh3, дипиридил, дифосфин) [524,
525]:
M(CO)6 + L —> M(CO)nL (51-94%)
М = Cr, Mo, W; п = 4, 5
Наконец, межфазный метод был успешно применен для
восстановления нитробензолов в анилины посредством додекар-
бонилжелеза [526—528]:
С6ИВ ТЭБАХ
водн
R (выход, %): Me (85);MeO (92); Cl (88); СОМе (60)
В заключение приведем еще один интересный пример
применения межфазного катализа в металлорганической химии —
карбонилирование арилгалогенидов в системе водный раствор
NaOH —ксилол в присутствии ТЭБАХ и РЦРИ)
RX-f CO-f 2NaOH — > RCO2Na + NaX + H2O
В этих условиях получены: фенилуксусная кислота (выход
83%) и 4-бромбензойная кислота из я-дибромбензола
(конверсия 90%, селективность 95%) [529].
Дибромкарбен внедряется в боковую цепь комплекса три-
карбонилжелеза с диенами-1,3 при проведении реакции в
системе СН2С12 — 50%-ный NaOH—аликват 336 с СНВг3 [530]:
СНВг2
Fe(CO)3 Fe(CO)3
(27-36,6%)
В присутствии 18-крауна-6 замещенные циклопентадаены
165
легко реагируют с FeCl2 в системе КОН — ТГФ, давая
ферроцены [531]$
R
*
* *
Введение в практику межфазного катализа означает новый
этап в развитии органического синтеза. Выше мы постарались
осветить наиболее важные стороны синтетического
использования межфазного катализа. Однако его возможности далеко
еще не исчерпаны, свидетельством чему являются
многочисленные новые публикации, посвященные различным аспектам
применения межфазного катализа. Так, расширяется применение
межфазного катализа в реакциях элиминирования из дигало-
геиалкенов с образованием алкенов [7] и алкинов [533,
534], в реакции стереоспецифического образования алкенов из
оксидов [535]. Приведены новые интересные примеры
окислительного гидролиза вторичных нитрилов в кетоны [536] и
окисления нитрилов в амиды щелочным раствором пероксида
водорода в присутствии ТБАГС [537], окисления метиленкетонов
до оксидов [538], изомеризации аллилбензола в стирол в
системе ксилол — водный NaOH —адоген 464 [532]. Описаны
новые примеры генерирования реакционных частиц, например фе-
нилтиохлоркарбена [540] и этоксикарбонилнитрена [541].
Наконец, межфазный катализ применен для облегчения нуклео-
фильного замещения в системе пар — твердая фаза при
проведении газообразного алкилгалогеиида через колонку с солью
(реагент) и катализатором межфазного переноса, нанесенным
иа твердый носитель [542].
Bee это указывает на то, что предстоит еще большая работа
по переведению на условия межфазного катализа многих ну-
клеофильных реакций. Несомненно, предстоит еще многое
сделать и в области изучения механизма межфазного катализа*
Литература
1. Starks С. М.~ J. Am. Chem. Soc, 1971, v. 93, № 1, p. 195—199.
2. Landini D., Rolla F., Montanari F. — Synthesis, 1974, № 6, p. 428—430.
S. Landini A, e. a.— Chem. Commun., 1974, Jsfe 11, p. 879—880.
4. lehmkuhel H.t Rabet F., Hauschied K. — Synthesis, 1977, № 3, p. 184—186.
5. Landini D.t Rolta F. — Chem. Ind., 1974, Nfe 13, p. 533—534.
6. lanczyk A, e. a. — Angew. Chem., 1978, Bd. 90, Ш 1, S. 58.
7. Regen S. R.~ J. Org. Chem., 1977, v. 42, № 5, p. 875—879.
8. Landini D. e. a. — Gazz. Chim. Ital, 1975, v. 105, № 7/8, p. 863—864.
9. Cinquinl M. e. a.—Chem. Commun., 1975, v. 130, № 10, p. 393—Ш.
166
10. Liotta С. L e. a —J. Am. Chem., Soc, 1974, v. 96, № 7, p. 2250—2252.
11. Gross M., Peter f. — Bull. Soc. chim. France, 1975, № 3—4, p. 871—874.
12. Cainelli G., Manescalchi F., Panunzio M. — Synthesis, 1976, № 7, p. 472.
13. Fitjer L — Synthesis, 1977, K° 3, p. 189—191.
14. Fedoryfiski M. e. a, — Synth. Commun., 1977, v. 7, № 4, p. 287—292.
15. Watanabe J., Mukaijama Г. —Chem. Lett., 1978, Ks 4, p. 349—352.
16. Regen S. L., Nigam Л., Bese J. /. — Tetrahedron Lett., 1978, № 31,
p. 2757—2760.
17. Reeves W. A e. a. — Synth. Commun., 1976, v. 6, № 7, p. 509—514.
18. Cinquini M.t Tundo P. — Synthesis, 1976, Ks 8, p. 516—519.
19. Reeves W. P., White M. R.t Bier £>. — J. Chem. Ed. 1978, v. 55, № 1,
p. 56—62.
20. Sugimoto N. e. a.~ Chem. Farm. Bull., 1962, v. 10, Mb 2, p. 427—430.
21. Cainelli G.r Manescalchi F.t Panunzio M. — Synthesis, 1979, № 2, p. 141—
144.
22. Cinquini M. e. a. — Chem. Commun., 1976, J\fe 11, p. 394—396.
23. Cinquini M.y Montanari F., Tundo P. — Gazz. Chim. Ital., 1977, v. 107,
№12, p. 11—14.
24. Ktmura C, Kashiwaga K., Murai К. — Асахи гарасу когё гидзюцу сорей-
хай кенкю хокаку, 1974, Mb 24, р. 59—67; С. А., 1976, v. 84, 121027g.
25. Chveds В. М., Weber W. P. —J. Org. Chem., 1976, v. 41, № 21, p. 3486—
3487.
26. Cook F. L, Bowers C. W., Liotta C. L — 3. Org. Chem., 1974, v. 39, № 23,
p. 3416—3418.
27. Regen S. L. — J. Am. Chem. Soc, 1975, v, 97, № 20, p. 5956—5957.
28. Simchen G., Kobler H. — Synthesis, 1975, № 9, p. 605—607.
29. Tundo P. — Synthesis, 1978, N° 4, p. 315—316.
30. White B. A.} Baizer M. M. — J. Chem. Soc, Perkin I, 1973, № 19,
p. 2230—2236.
31. Samaan S., Rolla F. — Phosphorus a. Sulfur, 1978, v. 4, № 2, p. 145—148.
32. Tomai M.t Ikeda M., Kakiuchi H. — Tetrahedron Lett., 1978, № 39,
p. 3757—3758.
33. Frechet J. M.t de Smet M. Z>., Farall M. /. —J. Org. Chem., 1979, v. 44,
№ П, p. 1774—1779.
34. Kobler H. Schuster K.-H.y Simchen G. —Ann., 1978, Ks 12, S. 1946—1962.
35. Reeves W. P., White M. R. — Synth. Commun., 1976, v. 6, Ns> 3, p. 193—
197.
36. Zubrick G. W., Dunbar B. I., Durst H. £. — Tetrahedron Lett., 1975, № 1,
p. 71—74.
37. Reeves W. P,, Bahr M. .L — Synthesis, 1976, № J2, p. 823.
38. Tamura e. a. — Chem. Pharm. Bull., 1978, v. 26, № 9, p. 2874—2879.
39. Gelbard G., Colonna S. — Synthesis, 1977, Kq 2, p. 113—116.
40. Landini D., Rolla F. — Synthesis, 1974, Mb 8, p. 565.
41. Landini D., Rotia F. — Org. Synthesis, 1978, v. 58, p. 143—146.
42. Martinetz В., Hiller A. — Z. Chem., 1978, Bd. 18, Hi 2, S. 61—62.
43. Kim 1. K, Noh J. S. — Taehan Hwahak Haechi, 1974, v. 18, № 6, p. 421—
422; С. А., 1975, v. 82, 15563y.
44. Landini /)., Montanari F.t Rolla F. — Synthesis, 1974, Ns 1, p. 37—38.
45. Tabuschi /., Yoshida Z.t Takahashi N. — L Am. Chem. Soc, 1971, v. 93,
№ 7, p. 1820.
46. Landini D., Quid S., Rolla F. — Synthesis, 1975, JNb 6, p. 430—431.
47. Markezich N. e.a. — J. Org. Chem., 1977, v. 42, Ks 21, p. 3435—3436.
48. Henbest H. В., Jackson W. R. — J. Chem. Soc, 1962, № 3, p. 954—959.
49. Kent P. 1#\, Joung R. С — Tetrahedron, 1971, v. 27, № 17, p. 4057—4064.
50. Bethell D.t McDonald /(., Rao K. S. — Tetrahedron Lett., 1977, № 17,
p. 1447—1448.
51. Bianchi Th. Л., Cate L. Л. —J. Org. Chem. 1971, v. 42, № 4, p. 2031—
2032
52. Ykman P., Hall H. K. — Tetrahedron Lett., 1975, № 29, p. 2429—2432.
167
53. Kawakami Y., Yamashita Y. — J. Org. Chem., 1980, v. 45, № 19, p. 3930—
3932.
54. Gajda Т., Zwierzak Л. — Synthesis, 1976, № 4, p. 243—244.
55. Tundo P. —Chem. Commun., 1947, № 18, p. 641—642.
56. Fornasier R. e. a. — Tetrahedron Lett., 1976, № 17, p. 1381—1384.
57. Tipson R. S., Clapp AT. A.t Cretcker L. £/. —J. Org. Chem., 1947, v. 12,
№ 1, p. 133—137.
58. Rolia /\, Roth W., Homer L. — Naturwiss., 1977, Bd. 64, № 6, S. 337—
338.
59. Tomoi M. e. a. — Tetrahedron Lett., 1978, № 33, p. 3031—3034.
60. Tundo P.—Tetrahedron Lett., 1978, Ks 47, p. 4693—4696.
61. Korzeniowski S., Gokel G. W. — Tetrahedron Lett., 1977, № 40, p. 3519—
3522.
62. Ambasht S. e. a. — Synthesis, 1980, № 4, p. 318—320.
63. Colonna 5., Fornasier R. — Synthesis, 1975, № 8, p. 531—532.
64. Wewnan ЛГ. S. e. a. — J. Org. Chem., 1975, v. 40, № 20, p. 2863—2870.
65. Smith J. p., Irwin D. С — Synthesis, 1978, № 12, p. 894—895.
66. Compendium of Phase-Transfer Reactions and Related Synthetic Methods/
Ed. by W. E. Keller. Fluka AG, CH-9470 Buchs, Switzerland, 1979. 165 p.
67. KSnig K. E., Weber W. P. — Tetrahedron Lett., 1974, № 26, p. 2275—2278.
68. Nakajima Y. e. a. — Bull. Chem. Soc. Japan, 1977, v. 50, p. 2025—2027.
69. Nakajima Y., Oda Л, Inouye Y. — Tetrahedron Lett., 1978, № 4, p. 3107—
3110.
70. Herriot A. W.t Picker D. — Synthesis, 1975, № 7, p. 447—448.
71. Gisler M., Zoiiinger Я. —Angew. Chem., 1981, Bd. 93, № 2, S. 184.
72. Koenig K> E., Weber W. P. — Tetrahedron Lett, 1974, № 24, p. 2275—
2278.
73. Julia S. e. a, —Tetrahedron Lett., 1980, v. 21, № 38, p. 3709—3712.
74. Freeman H. Я., Dubois R. A. — Tetrahedron Lett., 1975, № 38, p. 3251—
3254.
75. Merz Л. —Angew. Chem., 1973, Bd. 85, № 14, S. 868.
76. Viliieras /., Bacquett C, Normant J. E. — J. Organomet. Chem., 1975,
v. 97, № 3, p. 355—359.
77. Rosenthal A. /\, Vargas L. A.t Dixon J. F. — Chem. Phys. Lipids, 1977,
v. 20, № 3, p. 205.
78. Qzernecki S., Geqrgoulis C, Provelengiou C. — Tetrahedron Lett., 1976,
№ 39, p. 3535—ЗВЗб.
79. Merz Л. —Angew. Chem., 1977, Bd. 89, № 7, S. 484—485.
80. DiCesare P., Gross В. —Carbohyd. Res. 1976, v. 48, № 2, p. 271—275.
81. Merz A., Tomahogh R. — J. Chem. Res. (S), 1977, № 11, p. 273 (M3070).
82. Андреев В. ЛГ., Бибичева А. И., Лапикова Л. С. — Журн. ВХО
им. Д. И. Менделеева, 1978, т. 23, № 2, с. 231—233.
83. Gases В., Juiia /. — Tetrahedron Lett, 1974, № 20, p. 2077—2078.
84. Fedorynski M. e. a, — J. Org. Chem., 1978, v. 43, № 24, p. 4682—4684.
85. Zupancic B. G., Sopcic M. — Synthesis, 1979, № 2, p. 123—124.
86. Cazus В., Julia S. — Tetrahedron, 1979, v. 35, № 22, p. 2655—2660.
87. Serio Dungaan J., Grabowski E. J. J., Russ M. /(. — Synthesis, 1980, № 7,
p. 573—575.
88. Жданов Ю. А. и др. — ДАН СССР, 1977, т. 237, № 5, с. 1104—1106.
89. Жданов Ю. А. и dp. — RAH СССР, 1978, т. 238, № 5, с. 1102—1104.
90. Жданов Ю. А. и др. —ЖОХ, 1978, т. 48, № 11, с. 2614—2621.
91. Garegg Р. Л, Iversen Г, Oscarsen S. — Carbohyd. Res., 1976, v. 50, № 2,
C12—C14.
92. Brandstrom A. Preparative Ion Pair Extraction. Apotlkarsocieteten/Hassle,
Lakemedel, Sweden, 1974. 275 p.
93. McKiliop A., Fiaud J.-C, Hug R. P. — Tetrahedron, 1974, v. 30, № 11,
p. 1379—1382.
94. van Heerden F. R. e. a. — Tetrahedron Lett., 1978, № 7, p. 661—662.
95. Rail G.-F. e. a. — Tetrahedron Lett, 1976, № 13, p. 1033—1036.
96. Davis B, — Anal. Chem., 1977, v. 49, № 6, p. 832—834.
163
97. D'Inkan E., Viout P. — Tetrahedron, 1975, v. 31, № 2, p. 159—164.
98. Renault J. u.-C.r., 1977, v. 285C, № 5, p. 199—201.
99. Caccki S., La Tovre F., Misltl D. ~ Chem. Ind., 1977, № 23, p. 961.
100. Jonczyk A., Fedorynski M.t Makosza M. — Rocz. Chem., 1974, v. 48, №10,
p. 1713—1722.
101. Wiiiner /., Haipern M. — Synthesis, 1979, № 3, p. 177.
102. Cederiund £., Jesnerson A.t Hornfeid A. — Acta Chem. Scand., 1971, v. 25,
№ 10, p. 3056.
103. Jonczyk A., Serafin В., Skuiimawska E. ~~ Rocz. Chem., 1973, v. 47, Ks 1,
p. 89—98.
104. Nakamura E., Hashimoto K., Kuwajima 1. — Tetrahedron Lett, 1978, № 24,
p. 2079—2082.
tt)5. Dei Cima T.y Biggi G., Pietra F. — J. Chem. Soc, Perkin II, 1979, Ms 1,
p. 55—58.
106. Miller L. L. — L Org. Chem., 1978, v. 43, № 15, p. 3078—3080.
107. Shinozaki #., Yoshida N.t Tatjima M.— Chem. Lett., 1980, № 7, p. 869—
870.
108. Durst D. — Tetrahedron Lett., 1975, № 28, p. 2421—2424.
109. Тоги Т. e. a. — Synthesis, 1974, № 12, p. 867.
110. Yanagida S., Noji Y.t Okahara M. — Tetrahedron Lett, 1977, Mb 33,
p. 2893—2894.
111. Dehmiow E. V., Lissei Af. —Chem. Ber., 1981, Bd. 114, № 3, S. 1210—
1215.
112. Budet В., Julia M.t Ramirez-Munoz M. — Synthesis, 1980, № 11, p. 926—
929.
113. Jones R. A.t Nokheo S., Singu S. — Synth. Commun., 1977, v. 7, № 3,
p. 195— IS?.
114. Toke /.., Szabd G. Г. —Acta Chim. Acad. Sci. Hung., 1977, v. 93, № 3/4,
p. 421—424.
115. Cainelii G., Manescalchi F. — Synthesis, 1975, № 11, p. 723—724.
116. Kurozumi S. e. a. — Bull. Chem. Soc. Japan, 1971, v. 50, № 6, p. 1357—
1360.
117. Liotte C. e. a. — Tetrahedron Lett., 1974, Na 28, p. 2417—2420.
118. Wadi L., Gerdes J. M.t Wirth R, P. — Tetrahedron Lett, 1978, № 8, p. 731 —
732.
119. Yoshino Y.e.a.—^. Chem. Soc, Perkin 1, 1977, № 11, p. 1266—1272.
120. Szeja W. — Synthesis, 1980, № 5, p. 402—403.
121. Schwarz S., Weber G. — Z. Chem., 1975, Bd. 15, № 4, S. 270—272.
122. Schwarz S. e. a. — Z. Chem., 1976, Bd. 16, № Ц, S. 439—441.
123. Tappo H.— Synthesis, 1980, № 7, p. 577—578.
124. Garregg P. /., Ikersen 7\, Oscarsen S. — Carbohyd. Res., 1977, v. 53, № 1,
C5 Q7
125. Жданов Ю, А. и др. — ДАН СССР, 1979, т. 245, № 4, с. 846—848.
126. Szeja W. — Chem. Commun., 1981, № 5, p. 215—216.
127. Zwierzak A. — Synthesis, 1976, № 5, p. 305—306.
128. Golubski Z. Я. — Synthesis, 1980, № 8, p. 632—634.
129. Bashail A. P., Collins /. ^.—Tetrahedron Lett., 1975, Кэ 40, p. 3489^
3490.
130 Dehmiow E. V.y Schmidt /. — Tetrahedron Lett., 1976, № 2, p. 95—96,-
131. Kim K. S., Szarek V. A. — Synthesis, 1978, № 1, p. 48—50.
132. Cesare P. D.t Gross В. —Carbohyd. Res., 1976, v. 48, № 2, p. 271—275
133. Holmberg K., Hansen B. — Tetrahedron Lett., 1975, № 27, p. 2303—2306.
134. Makosza M.t Jerzak В., Fedorynski M. ~ Rocz. Chem., 1975, v. 49, № 8,
p, 1783—1785.
135. Kimura C, Kashiwaya K.t Murai JO — Юки госэй кагаку кёкайси, 1977
v. 35, Ks 8, p. 669—671; С. А., 1977, v. 87, № 23, 183911m.
136. Herriott A. N.> Picker D.~ J. Am. Chem. Soc, 1975, v. 97, № 9, p. 2345—
2349.
137. Dou H. J. M. e.a. — Helv. Chim. Acta, 1978, v. 61, № 8, p. 3143—314a
169
138. Goralski С. Т., Burk G. Д. —J. Org. Chetn., 1977, v. 42, № 18, p. 3094—
3095.
139. Dou H. J. M. e. a. — Phosphorus a. Sulfur, 1977, v. 3, Кз 2, p. 355—358.
140. Hassanaly P. e. a —Synthesis, 1977, № 4, p. 253—254.
141. Makosza M.t Fedorynski M. — Synthesis, 1974, № 4, p. 274—275.
142. Degani /., Fochi R., Santi M. — Synthesis, 1977, № 12, p. 873—874.
143. Degani /., Fochi R., Regandi V. — Synthesis, 1979, № 3, p. 178—181.
144. Degani /., Fochi R. — Synthesis, 1978, № 5, p. 365—368.
145. Degani 1., Fochi R., Rogandi V. — Synthesis, 1980, № 5, p. 375—378.
146. Cabbydu 5., Macciono A., Seed M. — Synthesis, 1976, № 12, p. 797—798.
147. Jouczyk A., Makosza Af. —Rocz. Chem., 1975, v. 49, № 6, p. 1203—1205.
148. Johczyk A., Ochal Z., Makosza M. — Synthesis, 1978, № 12, p. 882—883.
149. Brehtne & —Synthesis, 1976, № 2, p. 113—114.
150. Graeme /., Fronlich /., Muhlstadt M. — Z. Chem., 1974, Bd. 14, Ns 11,
S. 434.
151. Makosza Af., Kasprowicz A, — Rocz. Chem., 1975, v. 49, № 9, p. 1627—
1631.
152. Kawashima K. e. a — Chem. Pharm. Bull., 1978, v. 26, № 3, p. 942—950.
153. Sasaki 7\, Eguchi S., Toi N.—J. Org. Chem., 1978, v. 43, № 20, p. 3810—
3813.
154. Gajda T. e. a. — Synthesis, 1979, № 7, p. 549.
155. Koziara Л., Zawadski S.y Zwierzak A. — Synthesis, 1979, № 7, p. 527—529.
156. Fletcher S. R.t Kay I. T. — Chem. Commun., 1978, № 20, p. 963—964.
157. Reuschling D., Pietsch Я., Linkies A. — Tetrahedron Lett., 1978, № 7,
p. 615—618.
158. Palecek J., Kuthan /. —Z. Chem., 1977, Bd. 17, № 7, S. 260—261.
159. Ly B. e.a. —J. Heterocycl. Chem., 1977, v. 14, № 12, p. 1275—1281.
160. Makosza M.t Serafin fi. —Rocz. Chem., 1965, v. 39, № 12, p. 1799—1803.
161. Landini D.t Rolla F. — Synthesis, 1976, № 6, p. 389—391.
162. Julia 5., Oinebreda A., Guixer J, — Chem. Commun., 1978, № 6, p. 742—
744.
163. Julia S. e. a. — J. Chem. Soc, Perkin I, 1891, № 3, p. 574—577.
164. Dou H. J. M,t Hassondly A, Metzger J. — J. Heterocycl. Chem., 1977, v. 14,
№ 2, p. 321—323.
165. Slusarska E., Zwierzak A. — Synthesis, 1980, № 9, p. 717—719.
166. Maurette M.-T. e. a. —C.r., 1976, v. 282C, № 13, p. 599—602.
167. Dou H. J. M., Metzger /. — Bull. Soc. chim. France, 1976, № 11/12,
p. 1861—1864.
168.' Palecek /., KuThan /. — Synthesis, 1976, № 7, p. 550—551.
169. Barco A., Benetti S., Pollini G. P. — Synthesis, 1976, № 2, p. 124—125.
170. Суворов H. H. и др. — Хни. гетероцикл. соед., 1976, № l, c. 191—193.
171. ВоссЫ V. е.а. — Synthesis, 1976, № 6, p. 414—416.
172. de Silva S. O.f Snieckus V. — Can. J. Chem., 1978, v. 56, № 12, p. 1621—
1625.
173. Wang N. С, Тео К. Е., Anderson //. /. — Can. J. Chem., 1977, v. 55,
№23, p. 4112—4116.
174. llli V. O. — Synthesis, 1979, № 2, p. 136.
175. Bohm R. — Pharmacie, 1978, v. 33, № 2, p. 83—85.
176. Masse /.-Synthesis, 1977, № 5, p. 341—342.
177. Rylski Lt Senczuk L.t Ligier W. — Acta Polon. Pharm., 1974, № 7,
p. 550—556.
178. Rucman R.y Stres /., Jurgec M. — Vestn. Slov. Kern, Drus., 1978, v. 25,
JSTe 3, p. 283—300; С. А., 1978, v. 89, № 25, 215637m.
179. Kreher R., Herd K. /. — Z. Naturforsch., 1974, Bd. 29b, № 9/10, S. 683—
687.
180. Winkler H. Z>., Seela F, — Chem. Ber., 1980, Bd. 113, № 6, S. 2069—2080.
181. Hasselmann D., Seela F, —Chem. Ber., 1980, Bd. 113, № 10, S. 3389—
3393.
182. Coudert G., Gullaumet G.t Loubinoux B. — Synthesis, 1979, Кг 7, p. 541—
543. f
Щ
183. Berg-Nielsen #., Bemdten E. — Acta Chem. Scand., 1972, v. 26, X° 10,
p. 4130—4138
184. Малхасян А Ц. и др. — Арм. хим. журн., 1980, т. 33, № 2, с. 152—155.
185. Johczyk A., Wostowska Л, Makosza M. — Synthesis, 1976, № 12, р. 795—
796.
186. Суворов Я. Я., Плутицкий Д. Н., Смушкевич Ю. И. —Ж. орг. хим., 1980,
т. 16, с. 872—876.
187. Cacchi S., La Torre F.} Misti D. — Synthesis, 1977, № 5, p. 301—303.
188. Jonczyk A.t Wostowska /., Makosza M. — Bull. Soc. chim. Belg., 1977,
v. 86, № 9, p. 739—740.
189. Penis J. P., Singu 5., Newton T. A. — Synthesis, 1979, № 3, p. 178—180.
190. Малхасян А. Ц. и др. — Арм. хим. жури., 1979, т. 32, № 4, с. 223—226.
191. Zwierzak Л. — Synthesis, 1975, № 8, р. 507—509.
192. Zwierzak A., Sulewska А. — Synthesis, 1976, № 12, р. 835.
193. Zwierzak A,, Brylikowska Л —Synthesis, 1975, № Ц( р. 712—714.
194. Makosza М. — Tetrahedron Lett., 1966, № 38, p. 4621—4624.
195. Makosza M. — Bull. Acad. Polon. Sci., 1967, v. 15, № 4, p. 165—167.
196. Малхасян А. Ц. и др. — Арм. хим. журн., 1980, т. 33, № 4, с. 335—337.
197. Hart L S., Killen Ck. R. /., Saunders К. О.— Chem. Commun., 1979, № 1,
p. 24—25.
198. Purchlt V. W., Subramanlan R. — Chem. Ind., 1978, № 18, p. 731—732.
199. Diete H. K., Brannock К- С — Tetrahedron Lett., 1973, № 15, p. 1273—
1276.
200. Шустрова Т. А- и др.—Ж- орг. хим., 1981, т. 17, № % с. 329—332.
201. JoAczyk Д., Serafin B.t Makosza M. — Rocz. Chem., 1971, v. 45, № б,
p. 1027—1039.
202. Jouczyk A., Serafin В., Makosza M. — Tetrahedron Lett., 1971, № 17,
p. 1351—1354.
203. Jonczyk A., Serafin B.t Makosza M. — Rocz. Chem., 1971, v. 45, № 12,
p. 2097—2105.
204. Jouczyk Л., Serafin В., Skulimowska E.~ Rocz. Chem., 1971, v. 45, № 7/8,
p. 1259—1266.
205. Makosza Л1, Fedorynski M. — Rocz. Chem., 1971, v. 45, № 11, p. 1861 —
1869.
206. Jonczyk A., Serafin В., Czyzewsky Л —Rocz. Chem., 1973, v. 47, № 3,
p. 529—536.
207. Makosza M. ел. — Rocz. Chem., 1973, v. 47, № 1, p. 77—88.
208. Андреев В. Л1, Бабичева А. Я., Журавлева М. Я. — Ж. орг. хнм., 1974,
т. 10, № 7, с. 1470—1475.
209. JoAczyk A., FedoryAski M., Makosza M. — Rocz. Chem., 1974, v. 48, № 10,
p. 1713—1722.
210. Jonczyk A., Pytlewski Г—Rocz. Chem., 1975, v. 49, № 7—8, p. 1425—
1429.
211. Samuelsson В., Lamm В. —Acta Chem. Scand., 1971, v. 23, № 5, p. 1555—
1558.
212. Mikolajczyk M. ем. — Tetrahedron Lett. 1975, № 43, p. 3757—3760.
213. Reeves N. P., Hilbnch R.-G. — Tetrahedron, 1976, v. 32, № 18, p. 2235—
2237.
214. Makosza M.y Wojciechowski #., Jawdosiuk Л1 — Polish J. Chem., 1978,
v. 52, №6, p. 1173—1178.
215. Burgstahler e. a. — Synthesis, 1977, N2 6, p. 405—407.
216. Clam J. Я., Miller J. M. — Chem. Commun., 1977, № 2, p. 64—65.
217. Jouczyk Д., Ludwikaw M.t Makosza M. — Rocz. Chem., 1977, v. Si, № 1,
p. 175—179.
218. Fiaud J. C. — Tetrahedron Lett., 1975, № 40, p. 3495—3496.
219. Makosza M.t Serafin B. — Rocz. Chem., 1965, v. 39, № 9, p. 1223—Ц31.
220. Makosza Л1, Serafin B. — Rocz. Chem., 1965, v. 39, № 10, p. 1401—1469.
221. Makosza M.t Serafin B. — Rocz. Chem., 1965, v. 39, № 11, p. 1595—16Q2.
222. Makosza M.t Serafin В. —Rocz. Chem., 1965, v. 39, № 12, p. 1799—18<)3.
223. Makosza M., Serafin B. — Rocz. Chem., 1965, v. 39, № 12, p. 1805—1816.
171
224. Makosza M., Serafin В., Urbanski Т. — Chim. Ind. (Paris), 1965, v. 93,
№ 5, p. 537—538.
225. Makosza At, Serafin S. —Rocz. Chem., 1966, v. 40, № 11, p. 1647—1658.
226. Makosza M., Serafin £.— Rocz. Chem., 1966, v. 40, № 12, p. 1966—1971.
227. Makosza Ai, Serafin В., Jawdosiuk Ai — Rocz. Chem., 1967, v. 41, № 6,
p. 1037—1046.
228. Lange J., Makosza Л1 — Rocz. Chem., 1967, v. 41, № 7—8, p. 1303—1309.
229. Makosza M., Serafin B. — Przem. Chem., 1967, v. 45, № 3, p. 393—395.
230. Makosza M.t Serafin В., Bolesawska Т. —Rocz. Chem., 1968, v. 42, Ns 5,
p. 817—823.
231. Lunge /. — Rocz. Chem., 1968, v. 42, № 10, p. 1619—1630.
232. Makosza Ai, Jawdosiuk M.— Bull. Acad. Polon. Sci., 1968, v. 16, № 5,
p. 597—602.
233. Makosza Ai — Rocz. Chem., 1969, v. 43, №1, p. 79—86.
234. Makosza M. — Rocz. Chem., 1969, v. 43, № 2, p. 333—339.
235. Makosza M., Serafin B.y Gajos Л — Rocz. Chem., 1969, v. 43, № 4,
p. 671-676.
236. Makosza M. — Tetrahedron Lett., 1969, № 9, p. 673—676.
237. Makosza M. — Tetrahedron Lett., 1969, Mb 9, p. 677—678.
238. Ryisi L, Gafewski F. — Acta Polon. Pharm., 1969, v. 26, № 2, p. 115—117.
239. Makosza Ai, Jagustryn-Grochowska J. M.t Jawdosiuk M. — Rocz. Chem.,
1971, v. 45, № 5, p. 851—858.
240. Makosza Ai, Ludwikow Ai — Butt. Acad. Polon. Sci., 1971, v. 19, № 4,
p. 231—236.
241. Yaies P. e.a. — Can. J. Chem., 1971, v. 49, № 17, p. 2850—2860.
242. Makosza M.. Goetzen R. — Rocz. Chem., 1972, v. 46, № 7, p. 1239—1248.
243. Makosza Ai, Bialecka E., Ludwikow M. — Tetrahedron Lett., 1972, JVb 23,
p. 2391—2394.
244. Jonczyk A., Fedorytiski M.y Makosza M. — Tetrahedron Lett., 1972, № 23,
p. 2395—2396.
245. Makosza M. e.a. — Tetrahedron, 1974, v. 30, № 20, p. 3723—3735.
246. Makosza M., Ludwikow Л1 — Angew. Chem., 1974, Bd. 86, Mb 8, p. 744.
247. Makosza M., Ludwikow M., Vrniaz A. — Rocz. Chem., 1975, v. 49, № 2,
p. 297—306.
248. Singh R. /C., Danishefsky S. — J. Org. Chem., 1975, v. 40, № 20, p. 2969—
2970.
249. Blanchard J. e. a. — Synthesis, 1975, Mb 10, p. 655—657.
250. Vittorelii P. V. e.a. — Helv. Chim. Acta, 1975, v. 58, № 5, p. 1379—1425.
251. Makosza Л1, Johcyk A. — Org. Synth., 1976, v. 55, p. 91—95.
252. Makosza M., Jagustzyn-Grochowska J. M., Jawdosiuk M. — Rocz. Chem.,
1976, v. 50, № 11, p. 1841—1858.
253. Makosza M., Jaguslzyn-Grochowska J. M. — Rocz. Chem., 1976, v. 50,
№ 11, p. 1859—1866.
254. Takahashi К- е. а —Ниппон кагаку кайси, 1976, № 1, p. 144—147; С. А.,
1976, v. 84, № 15, 105162s.
255. Makosza M., Ludwikow M. — Rocz. Chem., 1977, v. 51, № 4, p. 829—833.
256. Makosza M. Kmiotek-Skarzynska J.y Jawdosiuk M. — Synthesis, 1977, № 1,
p. 56—58.
257. Wiiczynski W., Jawdosiuk Л1, Makosza M. — Rocz. Chem., 1977, v. 51,
№ 9, p. 1643—1650.
258. Makosza M., Biaiecka Я. — Tetrahedron Lett, 1977, № 2, p. 183—186.
259. Masuyama Л, Ueno J., Okawara M. — Chem. Lett., 1977, № 7, p. 835—838.
260. Masumoto У., Ueno Y.t Okawara M.— Bull. Chem. Soc. Japan, 1977, v. 50,
Mb 11, p. 3071—3072.
261. Chieiiini E., Solaro i?. —Chem. Commun., 1977, № 7, p. 231—232.
262. Possei O., van Leusen A. M. — Tetrahedron Lett., 1977, № 48, p. 4229—
4232.
263. Bonnet M. J., Eckrich 7 M, — Tetrahedron Lett., 1978, № 47, p. 4625— '
4628.
264. Jawdosiuk M. e.a.—Polish. J. Chem., 1978, v. 52, № 11, p. 2189—2195.
172
265. lawdosiuk M. e.a.— Polish. J. Chem., 1979, v. 53, № 3, p. 617—621.
266. Jawdosiuk M., Ludwikow M., Bednarska B. — Polish. J. Chem., 1979, v. 53,
№ 4, p. 805—810.
267. Jonczyk A.— Angew Chem., 1979, Bd. 91, Mb 3, S. 228.
268. Frechet Л M. J., De Smet M.t Farrall M. /. — Tetrahedron Lett., 1979, №2,
p. 137—138.
269. O'Donnel M. Л, Bonice Л Л1, Earp S. E. — Tetrahedron Lett., 1978, № 30,
p. 2641—2644.
270. Abbayes des H., Boudeviiie MM.— J. Org. Chem., 1977, v. 42, № 25,
p. 4104—4108. -
271. Durst H. A, Liebeskind /.. — J. Org. Chem., 1974, v. 39, № 22, p. 3271—
3273
272. Tanaka Т., Mukayama Г. —Chem. Lett., 1976, M \\t p. 1259—1262.
273. Brandstrom Д., Junggern U. — Ada Chem Scand., 1969, v. 23, Ns 6,
p. 2204—2205.
274. Brandstrom A., Junggern U. — Acta Chem. Scand., 196^ v. 23, № 7,
p. 2536—2541.
275. Schollkopj Я., Норре Д., Jentch P. —Chem. Ber., 1975, Bd. 108, Ks 5,
S. 1580—1592.
276. Brandstrom A., Junggern {/. — Acta Chem. Scand., 1969, v. 23, Ks 6,
p. 2203—2204.
277. Jonczyk A.t Luduwikow M., Makosza M.— Rocz. Chem., 1973, v. 47, Mb 1,
p. 89—98.
278. Shiovri Г, Hatnada Y. — J. Org. Chem., 1978, v. 43, Ks 43, p. 3631—3632.
279. Koutek B.y Paviiekova L, Soucek M. —Coll. Chem. Commun., 1974, v. 39,
Ks 1, p. 192—203.
280. van Leusen A. M., Bouma R. /., Possel O. — Tetrahedron Lett, 1975, №40,
p. 3487—3488.
281. Jonczyk A., Banko K., Makosza M.—J. Org. Chem., 1975, v. 40, № 2,
p. 266—267.
282. Johczyk A.t Pytlewski T. — Synthesis, 1978, Mb 12, p. 883—889.
283. Cardilio G. e.a. — Chem. Ind., 1977, № 22, p. 873.
284. Goiinski Л, Jonczyk A., Makosza M. — Synthesis, 1979, № 6, p. 461—463.
285. Малхасян А. Ц. и др.— Арм. хим. журн., 1979, т. 32, № 6, с. 465—470.
286. Малхасян А. Ц. и др. — Арм. хим. журн., 1979, т. 32, № 8, с. 678—682.
287. Kise Я. е.а.~ Юкагаку, 1977, v. 26, № 8, р. 474—478; С. А., 1977, v. 87,
133818г.
288. Clark J. Я., Miller J. M. — J. Chem. Soc, Perkin I, 1977, № 14, p. 1743—
1745.
289. Wakabayoshi Т., Waianabe K. — Tetrahedron Lett., 1978, № 4, p. 361—362.
290. Masuyama Y., Ueno У., Okawara M. — Tetrahedron Lett., 1976, № 34,
p. 2967 2970.
291. Сычкова JI. Д., Шабаров Ю. С —Ж. орг. хим., 1980, т. 16, № 10,
с. 2086—2091.
292. Asai Т., Аоуата Г, По Т. — Synthesis, 1980, П 10, p. 8H—812.
293. Малхасян Д. Я. и др. — Арм. хим. жури., 1979, т. 32, № 12, с. 952—956
294. Бабаян А. Г, Гамбарян Я.— ЖОХ, 1954, т. 24, Ms 10, с. 1882—1887.
295. Babayan А. Г., Indjikyan Sh. Я. — Tetrahedron, 1964, v. 20, № 6, p. 1371—
1376.
296. Бабаян А. Г., Инджикян Ш. Г. — ЖОХ, 1957, т. 27, № 5, с. 1201—1206.
297. Курц А. К. и др. —Ж- орг. хим., 1973, т. 9, Jsfe 7, с. 1313—1318.
298. Brandstrom Л., Junggern (/.—Tetrahedron Lett., 1972, Kb 6, p. 473—476.
299. Makosza M., Fedorynski M. — Rocz. Chem., 1975, v. 49, K« 10, p. 1779—
1781.
300. Korzeniowski S. H., Blum Lt Gokel G. W. — Tetrahedron Lett., 1977, Ns>22,
p. 1871—1874.
301. Крышталь Г. В. и др. — Изв. АН СССР. Сер. хим., 1981, № 12, с. 2870—
2874.
302. Wiliner /., Halpern M.t Rabinowitz M. — Chem. Commun., 1978, № 4,
p. 155—156.
173
303. Spiltane W. /., Dou H. J.-M., Metzger /. — Tetrahedron Lett., 1976, № 26,
p. 2269 2272.
304. Spillane W. /., Hassandly P., Dou H. J. M. — С. г., 1976, v. 283C, № 6,
p. 289—290.
305. Dehmlow E. V. — Tetrahedron, 1971, v. 27, № 17, p. 4071^4075.
306. Makosza M., Fedorynski M. — Rocz. Chem., 1972, v. 46, JMb % p. 311—313.
307. Tabushi /., Toshida Z., Takahashi N. — J. Am. Chem. Soc., 1970, v. 92,
№ 22, p. 6670—6672.
308. Tabushi 1. ел. — Tetrahedron Lett, 1973, № 2, p. 107—110.
309. Goh S. H. e.a. — Ansl J. Chem., 1975, v. 28, № 2, p. 381—384.
310. Steinberg K. — Tetrahedron Lett., 1980, v. 21, № 22, p. 2149—2150.
311. Steinbeck K. — Tetrahedron Lett, 1978, Ks 13, p. 1103—1106.
312. Steinbeck X. — Chem. Ber., 1972. Bd. 112, № 7, S. 2402—2412.
313. Tabushi /., Yoshida Zen-ichiy Takahachi N. — S. Am. Chem. Soc, 1971,
v. 93, № 7, p. 1820—1823.
314. Weber W. P., Gokel G. N., Vgi J. K. — Angew. Chem., 1972, Bd. 84, № 12,
S. 587.
315. Weber W. P., Gofef G. №. —Tetrahedron Lett., 1972, № 17, p. 1637—
1640.
316. Gokel G. W., Widera R. P., Weber W. P. —Org. Synth., 1975, v. 55,
pm 96 99,
317. Domschke G., Beckeri R.t Mayer B. — Synthesis, 1977, Jft 4, p. 275—276.
318. Graefe /., Fronlich /., Muheistadt M. — Z. Chem., 1974, Bd. 14, JMe 6,
S. 434—438.
319. Лайбеш //. H. и др. — ЖОХ, 1978, т. 14, № 4, с. 878.
320. Baird М. S. е.а. — Chem. Commun., 1979, № 5, p. 210—211.
321. Г^е Lok Hot Gupta В. G. В., Olah G. А. — Synthesis, 1977, № 10, р. 676—
677.
322. Woodgate Р. О., Lee Н. Я., Ruttedge P. S. — Synthesis, 1977, Nb 7,
p. 462—464.
323. Kuwajima /., Murotushi 7\, Nakamura E. — Synthesis, 1976, № 9, p. 602—
604.
324. Mclntosh J. M.t Khalil #. — J. Org. Chem., 1977, v. 42, JVe 12, p. 2123—
2126.
325. Liotia Ch. L, Dabdoub A. M., Zalkow L H. — Tetrahedron Lett., 1977,
№ 13, p. 1117—1120.
326. Трофимов Б. Л., Амосова С. В., Носырева В. В, —Ж. орг. хим., 1976,
т. 12, № 6, с. 1366—1367.
327. Dehmlow E. V. —Ann., 1972, Bd. 758, S. 148.
328. Makosza M., Gajos /. — Bull. Acad. Pollon. Sci., 1972, v. 20, № 1, p. 33—
36.
329. Makosza M., Fedoryuski M. —Rocz. Chem., 1972, v. 46, № 3, p. 533—535.
330. FedoryAski M., Gorzkewaka /., Makosza M. — Synthesis, 1977, № 2,
p. 120-122.
331. Makosza M., Jawdosiuk M. — Bull. Acad. Polon. Sci., 1968, v. 16, № 11—
12, p. 597—606.
332. Sakabara T.t Sudoh R. — J. Org. Chem., 1975, v. 40, № 19, p. 2823—2826.
333. Sakabara T.t Yamada M., Sudoh tf. — J. Org. Chem., 1976, v. 41, № 4,
p. 736—737.
334. Krychtat e.a. — Synthesis, 1979, J\fe 2, p. 107—109.
335. Штеменко И. И., Крышталь Г. В., Яновская Л. А. — Изв. АН СССР. Сер:
хим., 1980, № 12, с. 2831—2836.
336. Штеменко Н. И., Крышталь Г. В.} Яновская Л. А. — Изв. АН СССР. Сер.
хим. 1980, № 10, с. 2420—2423.
337. Штеменко Н. Я., Кучеров В. Ф., Яновская Л. А. — Изв. АН СССР. Сер.
хим., 1978, № 6, с. 1444—1445.
338. Штеменко Н. Я., Крышталь Г. В., Яновская Л. А. — Изв. АН СССР. Сер.
хим., 1981, № 3, с. 547—552.
339. Mclntosh /. M., Khalil Я. —Can. J. Chem., 1978, v. 56, JVe 16, p. 2134—
2138.
174
340. Hammerschmidt /\, Zbiral E.— Ann., 1977, № 6, S. 1026—1038.
341. Makosza M., Jawdosiuk Al —Bull. Acad. Polon. Sci., 1968, v. 16, № 11—
12, p. 589—593.
342. Colonna S., Re H., Wynberg //. —J. Chem. Soc, Perkin I, 1981, № 3,
p 547 552
343. Dryanska V., Ivanov C. — Tetrahedron Lett., 1975, № 41, p. 3519—3520.
344. Dryanska V., Popandova-Yamboiieva K., Ivanov C. — Tetrahedron Lett.,
1979, № 5, p. 443—446.
345. Merz A.t Tomahogh R. — Chem. Ber., 1977, Bd. 110, Mb 1, s. 96—106.
346. Martz /. Т., Gokel G. W., Olafson R. A. — Tetrahedron Lett, 1979, Mb 17,
p. 1473—1476.
347. Merz A. — Synthesis, 1974, № 10, p. 724—725.
348. Кипе P., Muhlstadt M., Grael /. — Synthesis, 1976, № 12, p. 825—826.
349. landini D., Mantanari /\, Rolla F. — Synthesis, 1979, № 1, p. 26—27.
350. Жданов Ю. А. и dp.— ДАН СССР, 1976, т. 231, № 4, с. 868—869.
351. Жданов Ю. А и др.— ДАН СССР, 1978, т. 238, № 6, с. 1355—1357.
352. Gokel G. W., Di Biase S. A.y lipisko B. A. — Tetrahedron Lett., 1976 №39,
p. 3495—3496.
353. van Leusen A. M.t Wildeman /. — Synthesis, 1977, № jt p. 501—502.
354. Cassar L, Panosqian 5., Giordano C. — Synthesis, 1978, № 12, p. 217—
219.
355. Akabori S., Ontomi M.y Arai /(. — Bull. Chem. Soc. Japan, 1976, v. 49,
№ 3, p. 746—747.
356. Makosza Л1, Bialecka E., Ludvikow M. — Tetrahedron Lett., 1972, № 23,
p. 3391—2394.
357. Reinach-Hirizbach F., Durst T. — Tetrahedron Lett., 1976, № 41, p. 3677—
3680.
358. Golinski /., Makosza M. — Synthesis, 1978. № 11, p. 823—825.
359. Durst Г. e.a. — Can. J. Chem., 1979, v- 57, № 3, p. 258—266.
360. Colonna S., Fomasler R.t Rfeifler U. — J. Chem. Soc, Perkin I, 1978, № 1,
p. 8—11.
361. d'Incan E., Seyden-Penne /. — С. г., 1975, v. 28lC, JVb 23, p. 103—133.
362. Hummelen L C, Wynberg //. — Tetrahedron Lett., 1978, Ш 12, p. 1089—
1092.
363. Johczyk Л., Fedoryhski M,, Makosza M. — Tetrahedron Lett., 1972, № 23,
p. 2395—2398.
364. Makosza M., Ludvikow Л1 — Angew. Chem., 1974, Bd. 86, № 20, S. 744—
749.
365. lonczyk Л., Kwast A., Makosza M. — Chem. Commun., 1977, JNJb 24,
p'. 902—903.
366. Johczyk A., Banko K., Makosza M.—S. Org. Chem., 1975, v. 40, Ш 2,
p. 266—267.
367. Yam Y. ел. —Chem. Commun., 1973, № 15, p. 527—528.
368. Ferrall M. /., Durst 7\, Frecht J. M. /. — Tetrahedron Lett., 1979, N° 3,
p. 203—206.
369. Merz A., Markl G. —Angew. Chem., 1973, Bd. 85, № 19, S. 867.
Я70. Hatch M. /. — J. Org. Chem., 1969, v. 34, K° 7, p. 2133—2137.
37L Hi'yama Г e, a. — S. Am. Chem. Soc, 1975, v. 97, Да 6, p. 1626—1627.
372. Hiyama T. e.a. — J. Am. Chem. Soc, 1976, v. 98, № 2, p. 641—643-
373. Murkl G., Merz A. — Synthesis, 1973, Nq 5, p. 295—297.
374. Clark S. D.t Harrison S. R.t Hodge P. — Tetrahedron Lett., 1980, v. 21,
Jjfe 14, p. 1375—1378.
375. Tagakt W. e.a. — Tetrahedron Lett, 1974, № 30, p. 2587—2590.
376. Boden R. M. — Synthesis, 1975, ,№ 12, p. 784.
377. Kopmehe G., Nuck #. — Chem. Ber., 1979, Bd. 112, № 6, S. 2342—2346.
378. Humig S., Stemmler /. — Tetrahedron Lett., 1974, № 36, p. 3151—3154.
379. I7onQ$ И. Я., Симонов А. М., Зубенко A. A. — Хим. гетероцикл. соед.,
1976, № 8, с. 1145.
380. Боев В. Я., Домбровский А. В. — ЖОХ, 1980, т. 50, JSfe 1, с. 121.
38Ь Delmas М. е.а. — Synth. Comm., 19$l, у. 11, № 2, p. 125.
382. Жданов Ю. А. и др. —ДАН СССР, 1979, т. 244, Nb 5, с. 1122—1124.
383. Piechucki С. — Synthesis, 1974, № 12, р. 869—870.
384. Piechucki С. — Synthesis, 1976, № 3, р. 187—188.
385. Mikolajczyk M. е.а. — Synthesis, 1976, № 6, р. 396—398.
386. Mikolajczyk M. е.а. — Synthesis, 1975, Пч 4, р. 278—280.
387. D'Inkan Е. — Tetrahedron Lett., 1977, Кя 9, p. 951—954.
388. Gibson N. A., Hosking /. N.— Aust. J. Chem., 1965, v. 18, № 1, p. 123—
128.
389. Herriot A. W., Picker D. — Tetrahedron Lett., 1974, № 16, p. 1511—1514.
390. Sam D. /., Simmons H. E. — J. Am. Chem. Soc, 1972, v. 94, № 11,
p. 4024—4025.
391. Menger F. M., Rhee J. U.t Rhee H. /(. —J. Org. Chem., 1975, v. 40, M> 25,
p. 3803—3805.
392. Sala 7., Sargent M. V, — Chem. Commun., 1978, № 6, p 253—254.
393. Schmidt W. P., Schafer H. /.— Angew. Chem., 1979, Bd. 91, № 1, S. 77.
394. Weber W. P., Shepherd /. P. — Tetrahedron Lett., 1972, Kb 48, p. 4907—
4908.
395. Helm K- H., Lee D. G., Shattebl L.— Acta Chem. Scand., 1978, v. 32B,
№ 9, p. 693—695.
396. Krepcho P. A., Larson J. R.t Edridge J. M. — J. Org. Chem., 1977, v. 42,
№ 23, p. 3749—3753.
397. PannelK. #., Mclntosk Л — Chem. Ind., 1977, № 22, p. 872.
398. Foglia T. A., Ban P. A., Malloy A. /. —J. Am. Oil Chem. Soc, 1977, v. 54,
№ 11, p. 858A—861A.
399. Lee D. G., Chang V. S. —J. Org. Chem.T 1978, v. 43, № 8, p. 1532—1536.
400. Newman M. 5., Dati H. M., Hing W. M. — J. Org. Chem., 1975, v. 40, № If
p. 262—263.
401. Lee D. G., Chang V. S. — Synthesis, 1978, № 6, p. 462—463.
402. Артамашкина Г. А., Гринфенльд А. А., Белецкая И. П. — Ж. орг. хим.,
1980, т. 16, № 4, с. 698—702.
403. Dou H. J.-M., Komeili-Zadeh H., Crozet M. — C. г., 1977, v. 284C, К« 17,
р. 685—688.
404. Hutchins R. О. е.а. — Tetrahedron Lett., 1977, JVe 48, p. 4167—4170.
405. Pletcher A, Tait S. /. D. — Tetrahedron Lett., 1978, № 18, p. 1601—1602.
406. Landini D., Montanari F., Rolla F. — Synthesis, 1979, № 2, p. 134—136.
407. Pletcher D.t Tait S. Л D. — Tetrahedron Lett, 1978, № 18, p. 1601—1604.
408. Cainelti G. e.a. — J. Am. Chem. Soc., 1976, v. 98, № 21, p. 6737—6738.
409. Cardillo G., Orena M.y Sandri S. —Chem. Commun., 1976, № 6, p. 190.
410. Cardillo G., Orena M.f Sandri S. — Tetrahedron Lett., 1976, № 44,
p. 3985—3988.
411. Gelbard G., Brunelet Th.t Joketteau C. — Tetrahedron Lett., 1980, v. 21,
№ 47, p. 4650—4654.
412. /о" G. A.y Freedman H. //. — Tetrahedron Lett., 1976, Nb 20, p. 1641—
1644.
413. Fletcher D., Tomov N. — J. Appl. Electrochem., 1977, v. 7, № 11, p. 1501—
1509.
414. Krishnan S., Kuhn D. G., Hamilton G. A. — J. Am. Chem. Soc, 1977, v. 99,
№ 24, p. 8121—8123.
415. Ishii F., Kishi K. — Synthesis, 1980, № 9, p. 706—708.
416. Caccht S.t La Torre F.t Paolucci G. — Synthesis, 1978, № 11, p, 848—849.
417. Wynberg H.f Greijdanus В. —Chem. Commun., 1978, № 5, p. 427—429.
418. Hammelen J. C, Wynberg H. — Tetrahedron Lett., 1978, № 12, p. 1089—
1092.
419. Helder R. e.a. — Tetrahedron Lett., 1976, № 21, p. 1831—1834.
420. San Filippo /., Valentine L 5. —J. Org. Chem., 1975, v. 40, № 12,
p. 1678—1680.
421. San Filippo J. e.a. — J. Org. Chem., 1976, v. 41, Nb 3, p. 586—588.
422. Chem C. /., San Filippo /. —J. Org. Chem., 1977, v. 42, Mb 1, p. 178—iSi.
423. Moro-oka У. e.a. — Chem. Lett., 1976, JVe 12, p. 1293—1294.
424. Corey E. /. e.a, — Tetrahedron Lett, 1975, № 3/, p. 3183—3186.
176
42В. Yamaguchi 7\, van der Plas И. С — Rec. Trav. Chim., 1977, v. 96, Mb I,
p. 59—90.
426. Rosenthal /., Primer A. — Tetrahedron Lett., 1976, № 32, p. 2805—2808.
427. San Filippo /., Chern C. /., Valentine I. S. — J. Org. Chem., 1976, v. 41,
№ 6, p. 1077—1078.
428. Scully F. E., Davis R. C. — J. Org. Chem., 1978, v. 43, № 7, p. 1467—
1468.
429. Frimer A.> Rosenthal /. — Tetrahedron Lett., 1976, N!> 32, p. 2809—2812.
4.40. Jonson R. A, Nidy E. G.— J. Org. Chem., 1975, v. 40, № 12, p. 1680—
1688.
431. Ussel M.t Dehmlow E. V. — Tetrahedron Lett., 1978, № 39, p. 3689—3690.
432. Alneri Я., Bottaccio G., Carletti V. — Tetrahedron Lett., 1977, Mb 24,
p. 2117—2120.
4?3. Jarrousse /. /., Rautin /. С —С. г., 1977, v. 284C, № 13, p. 503—504.
434. Dimroth #., Tuncker W. — Synthesis, 1977, № 5, p. 339—340.
435. Butt B. L e.a.— Tetrahedron Lett., 1977, № 35, p. 3063—3066.
436. Boden R. M. — Synthesis, 1975, № 11, p. 78S—784.
437. Qolonna 5., Fornasier R. — Synthesis, 1975, № 8, p. 531—532.
438. Balcells /., Colonna S., Fornasier R. — Synthesis, 1976, № 4, p. 266—267.
439. Masse J. P., Parayre E. R. — Chem. Commun., 1976, № 12, p. 438—439.
440. Reger D. L, Habib M. M., Fauth D. /. — Tetrahedron Lett, 1979, Nb 2,
p. 115—116; J. Org. Chem., 1980, v. 45, № 19, p. 3860—3865.
441. makosza M.y Waw'rzyniewicz At— Tetrahedron Lett., 1969, № 53, p. 4659—
4662.
442. Dehmlow E. V. — Tetrahedron Lett., 1976, № 2, p. 91—94.
443. Jubla S., Ginebreda A. — Synthesis, 1977, № 10, p. 682—683.
444. Костиков Р. Р., Молчанов Л. Я. — Ж. орг. хим., 1975, т. 11, № 8, с. 1767.
445. Makosza M., Fedorynsk't M.-~ Synth. Commun., 1973, v. 3, № 4, p. 305—
309.
446. Makosza M., Fedorynski M.~ Rocz. Chem., 1976, v. 50, № 12, p. 2223—
2226.
447. Mathies jR., Weyerstaht P.—Angew. Chem., 1974, Bd. 86, № 1, S. 42—43.
448. Weyerstahl P., Blume G., Mailer Ch. — Tetrahedron Lett., 1971, Nb 42,
p. 3869—3872.
449. Аксенов В. С, Терентьев F. Л. —Изв. АН СССР. Сер. хим., 1977, № 3,
с. 623—628.
450. Weyerstahl P., Mathias /?. —Tetrahedron Lett., 1973, № 8, p. 611—612.
451. Fedorynski M. — Synthesis, 1977, № 10, p. 923—925.
452. Dehmlow E. V., Lissel M. — l Chem. Res. (S), 1978, № 12, p. 310^311.
453. Gokel A. e.a. — J. Org. Chem., 1973, v. 38, № 10, p. 1913—1918.
454. Dehmlow Ё. V., Schonefeld /. — Ann., 1971, v. 744, S. 42—50. .
455. Кремлев М. М., Фиалков К). А., Ягупольский JJ. М.—Ж- орг. хим., 1981,
т. 17, № 2, с. 332—337.
456. Joshi G. С, Singh N.t Paude L M. — Tetrahedron Lett., 1972, № 15,
p. 1461—1664.
457. Hiyama T. e.a, — Tetrahedron Lett., 1975, № 34, p. 3013—3016.
458. Dehmlow E. V. — Tetrahedron, 1972, v. 28, № 1, p. 175—179.
459. Jefford С W., Heros de los V., Burger (/. — Tetrahedron Lett., 1976, № 9,
p. 703—706.
460. Kwantes P. M., Klumpp G. W. — Tetrahedron Lett., Л976, № 9, p. 707—710.
461. Makosza M. — Pure Appl. Chem.. 1975, v. 43, № 3/4, p. 436—462.
462. Jefford C. W.y Heros de los V., Delay f. —Helv. Chim. Acta, 1972, v. 55,
№ 6, p. 2214—2237.
463. Sasaki 71., Eguchi S., Щпуата Г. —J. Org. Chem., 1973, v. 38, № 12,
p. 2230—2234.
464. Bart #., Nitta M. — Tetrahedron Lett., 1974, № 24, p. 2109—2112.
465. Dehmlow E. V., Klabuhn Я., Hass E. Ch. — Ann., 1973, № 3, S. 1063—
1066.
466. Dehmlow E. V., Remmler Т. — J. Chem. Res. (S), 1977, № 1, p. 72—75.
467. <ireibrokk 7\ — Acta Chem. Scand., 1973, v. 27, № 9, p. 3207—3210.
177
468. Dehmlow E. V. — Tetrahedron Lett., 1976, № 3, p. 203—206.
469. fochims I. C, Karich G. — Tetrahedron Lett, 1974, Mb 48, p. 4215—4218.
470. Тищенко И. Г., Глазков Ю. В., КуЛинкович О. Г.— Ж. орг. хим., 1973,
т. 9, № 12. с. 2510—2513.
471. Новокрещеных В. Д., Мочалов С. С, Шабаров Ю. С.—Ж- орг. хим.,
1979, т. 15, № 3, с. 465—492.
472. Dehmlow Е. V., Ezimera G. С. — Tetrahedron Lett., 1970, № 47, p. 4047—
4050.
473. Mailer Ch., Stler F.t Weyerstahl P.— Chem. Ber., 1977, Bd. 110, № 1,
S. 124—137.
474. Mailer Ch.. Weyerstahl P. — Tetrahedron, 1975, v. 31, № 16, p. 1787—
1789.
475. Dehmlow E. V., Lissel Л1 —Ann., 1979, № 2, S. 181—193.
476. Blame G., Neumann 7\, Weyerstahl P. —Ann., 1975, № 2, S. 201—2(&
477. Sato e.a. — Tetrahedron Lett, 1977, № 25, p. 2151—2154.
478. Kwon S. e.a. — Synthesis, 1976, № 4, p. 249.
479. De Angelis F., Gambacorta A., Nlcoletti #, —Synthesis, 1976, № 12,
p. 798—800.
480. Und #., Winkler T. — Tetrahedron Lett., 1980, v. 21, № \} p. tl§—120.
481. Makosza M., Fedoryfiski M. — Synth. Commun., 1973, № 4, p. 305—ЗЙ9.
482. Sydnes L, Skaitebol L. — Tetrahedron Lett., 1975, № 52, p. 4603—4606.
483. Barlet R.~~ Bull. Soc. СЫт. France, 1977, K« 5/6, p. 543—550.
484. Аксенов В. С. и др. — В ки.: Сборник трудов IV Всесоюзной
конференции по химии ацетиленов. Т.1. Алма-Ата. Изд. АН Казах. ССР, 1972,
с. 223 226.
485. Хусид А. X. и др. — Изв. АН СССР. Сер. хим., 1975, № 12, с. 2787—2789.
48J3. KhUsid е. а. — Tetrahedron, 1977, v. 33, № 1, p. 77—84.
487. Bollus W._ £., Gross I. H., Blankenly — 3, Org. Chem., 1975, v. 40, № 12,
p. 1848—1849.
488. Qehmlow Ё. 1Л, Lissel M. — Tetrahedron Lett., 1976, № 21, p. 1783—1786.
489. Крышталь Г. Вп Кучеров В. Ф., Яновская Л. Л. —Изв. АН СССР, Сер.
зшм., 1978, В \Х с 2803—2805.
490. Крышталь Г. В. и др. — Изв. АН СССР. Сер. хим., 1976, № 2, с. 424—
426.
491. Хусид А. X. и др. — Изв. АН СССР. Сер. хим., 1977, № 9, с. 2135—2138.
492. Хусид А. X. и др—Шзк. АН СССР. Сер. хим., 1977, № 3, с. 709—711.
493. Глушко Л. Я. и др. — Изв. АН СССР, Сер. хим., 1980, № 5, с. 1048—
1051.
494. Khusld А. К* е. а. — Synthesis, 1977, № 6, р. 428—430.
495. Schiqsser M., le Van Chau — Helv. Chim. Acta, 1975, v. 58, № 8,
p. 2595—2564.
496. niyama Т., Tsukanaka M.t Mozaki H. — 3. Am. Chem. Soc, 1974, v. 96,
№ 11, p. 3713—3714.
497. Hiydma e.a.— Tetrahedron Lett, 1976, № 34, p. 3013—3016.
498. Kleveiand K., Skattebol L, Sydnes L K. —Acta Chem. Scand., 1977,
v. B31, № в^р. 463—468
499. Fedoryhski M. — Wiadom. Chem., 1976, v. 30, № 9, p. 575—597.
500. Шостаковский С. M.t Ретиуский А. А., Бобров А. В. — Изв. АН СССР.
Сер. хим., 1974, Ш 8, с. 1818—1823.
601. Савииых Л?, В., Аксенов В. С, Цхай Л. Э. — Изв. Сиб. отд. АН СССР.
Сер. хим., 1974,,№2, вып. 1, с. 112—114.
502. Ретинский А. Д. —Изв. АН СССР, Сер. хим., 1974, № 7, с. 1613—161S.
503. Савиных Ю. В., Аксенов В. С —Изв. Сиб. отд. АН СССР. Сер. хим.,
1977, №2, вып. 1, с 92—94.
504. Савиных Ю. В., Аксенов В. С, Богатырева Т. А. — Изв. Сиб. отд. АН
СССР. Сер. хим., 1975, Jslb 14, вып. 6, с. 111—114.
505. Кузнецов И. В., Красавцев И. И. — Укр. хйм. жури., 1976, т. 42, № 10,
с. 968-972.
S05a. Бредихин A. A.t ПлШНКОв В, 3,тЖ- орг, хйМ-i 1976, т, 12,
с. 1001—1004.
178
S06. Хусид Л. X., Яновская Л. Л.— Изв. АН СССР, Сер. хим., 1981, № 9,
q 2135 2137.
607. Тищенко И. 'Г. и др. —Ж. орг. хим., 1975, т. 11, Jsfe 3, с. 576—581.
508. Schlosser М., he Vau Chau — Hely. Chim. Acta, 1967, v. 58, № 8, p. 2596—
2604.
509. Schlosser M.t Bessier У. — Helv. Chim. Acta, 1977, v. 60, Ks I, p. 590—
595.
510. Bessier Y., Dang Ngoc-Huo Savary —Helv. Chim. Acta, 1977, v. 60, № 5,
p. 1739—1746.
511. Nerdel F. e.a. — Ann., 1967, Bd. 710, S. 36—58.
512. Weyerstahl P. e.a. — Chem. Ber., 1967, Bd. 100, № 6, S. 1858—1864.
513. Kamel Af., Kimpenhaus W. /., Buddrus /. — Chem. Ber., 1976, Bd. 109,
jsjg 7 s 2351 2364.
514. Kimpenhaus W., Buddrus /. — Chem. Ber., 1976, Bd. 109, № 7, S. 2370—
2381.
515. Хусид A. X., Яновская Л. Л. —Изв. АН СССР. Сер. хим., 1980, № 12,
с. 2790—2794.
516. Мандельштам Г. В. и др. —Ж- орг. хим., 1980, т. 16, № 12, с. 2513—
2518.
517. Крышталь Г. В., Кучеров В. Ф., Яновская Л. Л.— Изв. АН СССР. Сер.
хим., 1976, № 4, с. 929—930.
518. Крышталь Г. В., Хусид А. X. — Изв. АН СССР. Сер. хим., 1979, № 12,
с. 2381—2383.
519. Makosza M.t Kacprowicz A. — Rocz. Chem., 1974, v. 48, № 12, p. 2129—
2135.
520. Ившин В. П., Кемешен М. С, Велик Н. /7. — Ж- орг. хим., 1980, т. 16,
№ 8, с. 980—984.
521. Fedoryfiski Af., Makosza M. — J. Organomet. Chem., 1973, v. 51, № 1,
p. 89—91.
522. Alper #., Des Roches D. — 3. Organomet. Chem., 1976, v. 117, № 2,
p. C44—C46.
523. Alper #., Abbayes de tf., Des Roches D. — 3. Organomet. Chem., 1976,
v. 121, № 2, p. C31—C34.
§24. Shukla А. Я., Preetz W. — Angew. Chem., 1979, Bd. 91, № 2, S. 160—161.
625. Hui K. Y., Shaw B. £,. —J. Organomet. Chem., 1977, v. 124, № 2, p. 262—
264.
5?0. Alper #., Des Roches D. — Angew. Chem., 1977, Bd. 89, № I, S. 43.
527. Abbayes des H.t Alper #. — J. Am. Chem. Soc, 1977, v. 99, № 1, p. 98—
104.
52§. Alper #., Path H.-N. — Nouv. J. Chim. 1978, v. 2, № 3, p. 245—247.
529. Cassar £., Foa M., Cardlano A.—-J. Organomet. Chem., 1976, v. 121, № 3,
p. 065.
530. Alper H.> Armaratungs S. — Tetrahedron Lett., 1980, v. 21, № 17, p. 1589—
1592.
531. SaUsova M, Alper Я. — Angew. Chem., 1979, Bd. 91, № 12, S. 858—859.
&32. Landlni D., Quid S., Rolla F. — Synthesis, 1975, Ks 5, p. 397—398.
533. Dehmlow E. V., ttesel M. —Ann., 1980, № 1, S. 1—13.
034. Dervan P. В., Shippey M. A. — J. Am. Chem. Soc, 1976, v. 98, № 3,
p, 1265—1268.
565. Gorgues Л., £.<? C04 Л. — Tetrahedron Lett., 1976, № 50, p. 4723—4726.
SS6. Donetti Л., Boniardi O., Ezhaya Л. —Synthesis, 1980, № 12, p. 1009—
101!.
537. Cacchi S., Misiti Z>., La Torre F. — Synthesis, 1980, № 4, p. 243—244.
538. Hemin F., Pete J. P. — Synthesis, 1980, № 11, p. 995.
539. Halpern M. e.a. — Tetrahedron Lett., 1981, v. 22, № 7, p. 703—704.
540. Makosza M., Bialecka E. — Tetrahedron Lett, 1971, № 47, p. 4517—4518.
541. Seno M., Namba Т., Hise H. — J. Org. Chem., 1978, v. 43, Mb 17, p. 3345—
3348.
543. TunAo Л —J. Org. Chem. 1979, v. 44, № 12, p. 2048-2049,
178
Предметный указатель
Адамантан 114
замещенные 53, 114—116
Азиды, получение 59
Азиридин, производные 163, 164
Азобензол 144
Акридин, замещенные 69, 100, 102
Акриловая кислота, эфиры 120,
121
Акрилонитрил 119, 123
Акролеин 113, 122, 128
производные 113, 114, 157, 158
Алкантиолы, получение 80
Аллен 151
производные 152
Аллилбромид 117
Аллиловый спирт 160, 163
О-Аллоза, производные 62, 127
Альдольная конденсация 44 ел.,
124 ел.
Амиды, алкнлироваиие 81 ел.
Амнны, алкилирование 81 ел.
Анилин 15, 144
замещенные 57, 91
Антрацен 142, 144, 154
производные 69, 142, 143
Ацеиафтеиои 70, 96
Ацетали, получение 75—77
Ацетальдегид 39
Ацетамид 91
Ацетилен, замещенные 113, 119, 124
Ацетон 24, 28, 33, 34, 36, 39, 40, 44,
95, 136
фенил- 96, 97
Ацетонитрил 56, 125
диметиламино- 100
днфенил- 100—103, 105, 120
селеио- 99
фенил- 17, 29, 97 ел:, 103, 114,
120, 124
фенилхлор- 129
фенил(р-хлорэтокси)- 104
феиилэтил- 17, 103
хлор- 129
Ацетоуксусная кислота
метиловый эфир 107, 108
этиловый эфир 41—43, 107—109,
121, 122
Ацетоуксусный альдегид 121
Ацетофенон 96, 114, 131, 140, 145
производные 91t 96, 125, 129
Ш)
Бензальдегид 17, 129—132, 136, 139
замещенные 15, 76, 125, 126
Бензгндрнламнн 140
Бензнлазнд 58
Бензиламин, N-метил- 140
Бензилбромид 54, 58, 59
Бензиловый спирт 53, 61, 76, 116
4-метил- 140
Бензилхлорид 53, 116, 139
нитро- 98
Бензнлэтилкетон 40
Бензонлцнаннды 57, 58
Бензоин 17, 129
дезокси- 68, 69, 96
Бензоиновая конденсация 128, 129
Бензойная кислота 15
4-бром- 165
эфнры 12%
Бензол 15
бромнитро- 142
1,4-дибром- 165
1,2-ди(бромметил)- 82
2,4-диннтро-1-сульфо- 59
2,4-диннтрофтор- 52
2,4-динитрохлор- 52, 59
изопропенил- 153
1-интро-2,4-дихлор- 103
этил- 114
(Бензотиенил-2) винилфенилкетон 141
Бензофенон 105, 131, 140
производные 101, 102, 164
1,2-Бис(беизотиазолилтно)этан 78
Бис (тназолнл-2) сульфид 78
Бифеннл, замещенные ИЗ
Бифенилен, 1,2-диметоиси- 154
Бицикло[3.1.0]гексан, 6,6-дихлор- 147
Бицикло [4.1.0] гептан
4-вииил-7,7-дихлор- 149
7,7-днхлор- 147
Бицикло [6.1.0] ионан, 9,9-дихлор- 147
Бицикло[6.1.0]нонатриен-2,4Д
9,9-дихлор- 151
Бульвалеи 154
Бутадиен-1,ЗЛ51
2,3-диметил- 145
1,4-дипиридил- 133
1,4-дихииолил- 133
З-метил-1-этоксн- 162
1-этокси 162
Бутадиин-1,3, дифенил- 153
Бутан
1-бром- 56
иод- 56
мезилоксн- 58
1-нитро- 58
1-тиоциано- 55
1-фенил- 137
1-хлор- 53
Бутанол-1 53, 76
Бутанол-2, 3-фенил- 145
Бутанон-2 39, 40, 130, 145
3-метнл- 145
1-фенил 145
Бутатриен, производные 152
Бутен-1
грег-бутнл-3,3-днметил- 153
2,3-днметил- 144, 145
4,4,4-трибром- 117
4,4,4-трибром-2-метнл- 117
Бутен-2 148
4-бром- 117
4-бром-2-метнл- 117
диметил- 145, 148, 153
метил- 145, 148, 153
3-метил-1-хлор- 28, 33, 34, 40—43
Бутен-З-он-2 145
3-метил- 145, 156
Бутнлвиннловый эфир 161
гдег-Бутиловый спирт см. Пропанол-
2, 2-метил-
Бутиронитрил, замещенный 100
Валериановая кислота, производные
56, 121
Виннлацетальдегнд 157
Винилдиметилкарбинол 95
Виннлметнл кетой 123
Винилэтнловый эфир 161, 162
ВиттигаХорнера реакция 131 ел.
Внедрения реакции 114 ел.
Восстановления реакции 144 ел.
Х)-Галактоза, производные 62
Галогенпроизводные, получение 50 ел.,
118 ел.
Гексадекан, 1-хлор- 53
Гексадекандион-8,9 138
Гексадекаиол-1 53
Гексадецин-8 138
Гексадиен-1,5-ин-3, 2,5-диметил- 152
Гексаи, 1-хлор- 53
Гексаналь, 2-этил- 94
Гексанол 53
Гексен-2-он-6, производные 96
Гексены 148, 153, 157
Гептаднен-1,5, замещенный 161
Гептан, 1-циано- 140
Гептаналь 132, 141
Гептановая кислота 136Л 141
Гептанод-4 136
Гептен-1 153
Гептен-5-он-2, 6-метил- 33, 34, 40, 95
Гераниаль 139
Геранилацетон 108
Гераннлацетоуксусцый эфир 108
Гераннлбромид 108, 133
Геранилхлорид 108
Гераниол 160
Глиоксаль 133
Глутаконовый диальдегид 157
Глутаровая кислота, дииитрил 56
замещенные 99, 104
О-Глюкоза, производные 64, 73
О-Глнцит, производное 75
Гомберга-Бахмана-Хея реакция 113
Дарзана реакция 129
Дезоксибензоин 68, 69, 96
Декалин 114, 137
Декалол 137
Децен-1 136, 153
Диамантан 115
Днацетоновый спирт 45
Дибензнлсульфид 138
Дибензилсульфоксид 138
Днвиннловый эфир 160
Диизопропнловый эфир 115
1,3-Диоксан, 2-замещениые 116
1,4-Диоксеи, 2-хлор- 161
1,3-Диоксолан 116
замещенные 76
Ди-«-октилсульфид 59, 138
Дипропилкетон 136
Дифеинламни 81
Диэтиловый эфир 136
Додекан, 1-хлор- 53
Додеканол-1 53
Дьюаровский бензол, гексаметил- 150
Изоиидол, производные 82, 89
Изомасляный альдегид 94
Изопропенилметилкетон 123
Изопропиловый спирт см. Пропанол-2
Изотиазол, 3,5-димеркапто- 78
Изохннолон-1, производные 97
Изоцианнды, получение 116, 117
Имидазол 86
производные 79, 134
Ииден 93, 114
Иидол 86, 87
замещенные 90
Камфора 143
Камфорная кислота 143
Каран, 7,7-днхлор- 148
Карбазол 88
Карбены 35, 36, 114 ел., 146 ел.
Катализаторы межфазных реакций
7 ел., 16 ел.
Кневенагеля реакция 122, 128
Кори реакция 130, 131
181
Коричная кислота, производные 120,
125, 165
Коричный альдегид 113, 123, 132, 140
Коричный спирт 140
Кротоиовая кислота, производные 120,
155, 156
Кротоновая конденсация 125
Кротоноэый альдегид 113, 122, 123,
146, 157
Кумол .114
Лимонен 149
Линалоол 160
Макоша реакция 146, 147
Малоновая кислота
динитрил ПО, 121
диэфир днбутиловый 109
днэфир диэтиловый 72, 93, ПО
замещенные 110, 122, 123
Малоновый альдегид 121
Масляный альдегид 145
D-Манноза, производные 62, 73—75,
121
Мезнтила окись 37, 39, 44
Метаириловая кислота
производные 155
Метан
бромдифенил- 140
дихлор- 55
иодхлор- 55
нитро- 124
^етанол 77
Метантиол, фенил- 59
Метил-«-бутилиетои 40
Метилгептенон см. Гептен-5-он-2, 6-
метил-
Метил-р,р-диметоксивинилкетон 123
Метилнзоциаиид 116
Метил-2-фенилэтиловый эфир 61
Метилэтилкетон см. БутанОн-2
Механизм межфазных реакций 12 ел.
Михаэля реакция 121 ел.
Михлера тнокетои 164
Мочевина 21
Муконовый альдегид 159
Нафталин 154
2,3-дигндрокси- 76
Цафтил-2-фенилвинилкетон 141
Нафтолы 65, 76
Цикотинамид, тио- 128
Нитрилы, получение 56—58
Нитросоединения, получение 58
Нонановая кислота 136
Нонен-4 153
Норборнадиен 149
Норбориан, замещенны* 19, 53, 116,
140
Норборнен 150
182
Окисления реакции 136 ел,
Оксазол, производные 78, 84, 124
Оксетан, замещенные 52
Оксиндол, 1-метил- 97
Оксиран 162
производные 130, 131
Октан
амино- 140
бром- 51, 53, 54, 142
1,2-дибром- 118
1,2-дихлор- 118
2-иод- 54
мезнлокси- 51, 53
фтор- 51
1-хлор 17, 53, 54, 59
2-хлор- 51, 53
Оитаналь 140
Оитанол-1 51, 53, 61, 140
Октанол-2 51, 140, 142
Октанон-2 11, 140, 145
Октатетраен 53
Октатриен-2,4,6-аль 158, 169
Октатриен-2,4,6-овая кислота 163
Октены 51, 136, 141
«-Октилэтилсульфид 80
Октин-4 153
Ортомуравьиная кислота, эфиру 77
Пентадиен-1,3 145
Пентадиен-2,3-овая кислота, 4-метнл-
156
Пентанол-2, 4-метил- 145
Пентанон-2, 4-метил- 145
Пентен-2 145
Пентеи-З-он-2, 4-метил- 40
а-Пинен 147, 148
Пиперазин, ^Г,Ы-диметил-8
Пиперидин 8
Пиперональ 76
Пиразол 86
Пиреи 154
Пиридин, производные 8, 78, 85, 94»
98, 102, 135
Пиримидин, 2-меркапто- 78
Пирокатехин 76
Пиррол, замещенные 87
Пирролидон-2 84
Пирролопирнмидин, производное 88
Полистирол, замещенные 60
Пренилхлорид см. Бутен-2, З-метил-1-
хлор-
Пропаи
1-бром-2,2-диметил- 51
2-метил-1 -фенил- 137
пентахлор- 147
1-тиоцианато- 55
Пропаналь, 2,2-днметцл-З-фенил- 95
Пропаиол-1 77, 116, 137
Пропаиол-2 61, 77, 116, 136
2-метил- 61, 77, 116, 136
Пропанон-1, 1-феийл- 145
Щюпен, 1,1-дифёнил- 148
Пропиловый спирт см. Пропанол-1
Пропионовая кислота, замещенные,
эфяры 58, 99
Рейссерта соединения 105, 120
£-Рй#о-гексофуранозулоза,
производные 127
Салициловая кислота 15
Силан, замещенные, 55, 60
Сорбнновая иислота, эфнры 162
Сорбиновый альдегид 158
Сорбиновый спирт 163
Стильбен 132, 148
Стирол 17, 147, 148, 153, 154
хлор- 154
Сульфиды, получение 59, 77 ел.
Сульс юкислоты, получение 59, 60, 73
Сульс юиамиды, алкилирование 112
Сульфоиы, алкилированне 111, 112
Тетралии 114
Тетралон-2 97
Тиадиазол, замещенные 78
Тиазол 114
производные 78, 114, 124, 128
Тиолы, получение 59
Тиофеи, производные 69, 70
Тиофенол 124
Тиоцнаиаты, получение 55, 56
Толаи 152
я-Толилтрифторметилкетон 133
я-Толуиловый альдегид 140
Толуол 136
я-Толуолсульфонамид 91
Трназол, бензо- 88
Уксусная кислота
бром-, эфиры 58
бромциано-, эфиры 122
нитрил см. Ацетонитрнл
фенил- 165
хлор-, эфиры 129
циано-, эфиры ПО, 120, 128
эфиры 35, 136
Фарнезаль 139
Фариезилбромид 139
Фенантреи 154
Фейнлизоцианид 117
Фейилртутьхлорид 164
Фенол 15, 76
амино- 73
1>енотнГазЙй, замещенные 88
Феррацен; йаййцеииые 134
Финкельштейна реакция 60
Флуорен 94, 114, 143
замещенные 107, 140, 143
Формальдегид 132, 134
Формамнды 82, 83
Фор он 44
Фталевая кислота 15
Фталимид, замещенные 85
Фумаровый диальдегид 122, 158
Фуран, тетрагидро- 115
Фурфурол 132, 135
Хинолин, замещенные 67—69, 78,115,
142
Холестен-4-он-З 119
Циангидринный синтез 127
Циклогексан, замешенные 51, 56, 58,
114, 118, 126, 140
Циклогександиол-1,2 138
Циклогеисанол 76, 77} 116
Циклогексанон 40, 95, 126, 131, 140,
143
замещенные 52, 95, 96, 137
Цнклогексен 51, 56, 138, 147, 148, 153
4-винил- 149
Циилогексен-2-он-1 124
замещенные 71
Циклогептан, замещенные 118
Циклогептанон 143 .
Циклогептеи 148
Циклодекаидиол-1,2 138
Циклодецен 138
Циклододекатриеи 149
Циклооктаднеи-1,5 153
Циклооктан, замещенные 118
Циклооитандиол-1,2 138
Циклооктатетраен 151, 154
Циклооктен 138
Циклопеитадиеи 93, 151
Циклопентанон 40, 95, 109
Циклопентен 148, 153
замещенные 71, 99
Циклопропан, замещенные 99, 109,
ПО, 117, 146 ел., 154
Циклопропанирование 146 ел.
Цитр а ль 128, 159
Эпихлоргидрнн 52
$пифторгидрии 52
Этан
1-бром-2-фенил- 17
1,2-дибром- 73
1,2-дифталимидо- 85
тиоцианато- 55
Этанол 77, 116
2,2,2-трифтоги 116
1-фенил- 145
183
Этанол
2-фенил- 61
2-хлор- 62
Этилен
2,6-днметокснфеннл- 153
1,1-днбеннл- 148
1,2-днфенил- см. Стильбен
пентафторфенил- 153
2,4,6-триметилфеннл- 153
Стилей
1,1.2-трнфеннл-
1-феннл- см. Стирол
Этнленглнколь ацетаты 73
Этнлендиамнн, тетраметнл- 8
Этнленокснд см. Оксиран
Этилизоцианнд 116
Эфиры простые, получение 60 ел.
Эфиры сложные, получение 70 ел.
ЛЮДМИЛА АЛЕКСАНДРОВНА ЯНОВСКАЯ,
СЕРГЕЙ САМОЙЛОВИЧ ЮФИТ
ОРГАНИЧЕСКИЙ СИНТЕЗ
в двухфазных системах
ИБ
1059
Редактор М. Н. ПАСТУШЕНКО
Художественный редактор Н. В, НОСОВ
Художник А. К. МАЛКИН
Технический редактор В. М. СКИТИНА
Корректоры Л. В. Гаврилина, П. А. Иванова
Сдано в наб. 27.01.82. Подп. в печ. 24.06.82. Т. 11 800. Формат бумаги бОХОД*
Бумага тип. № 2. Гари, литературная. Печать высокая. Усл. печ. л. 11,5.
Усл. кр.-отт. 11,75. Уч.-изд. л. 12.90. Тираж 3 400 экз. Заказ № 79. Цена 2 р.
Изд. № 1928.
Ордена «Знак Почета» издательство «Хнмня», 107076, Москва,
Стромынка. Д. 13.
Ленинградская типография № 2 головное предприятие ордена Трудового
Красного Знамени Ленинградского объединения «Техническая книга» ик1.
Евгений Соколовой Саюзполиграфпрома при Государственном комитете СССР
по делам издательств, полиграфии н книжной торговли» 193052, г .Ленинград,
Л*5?, Измайловский проспект, 29.
СОДЕРЖАНИЕ
Предисловие . . ' 4
СПИСОК СОКРАЩЕННЫХ НАЗВАНИИ КАТАЛИЗАТОРОВ .... 6
ОБЩИЕ ПРИНЦИПЫ И ПРЕИМУЩЕСТВА МЕЖФАЗНОГО
КАТАЛИЗА . 7
Литература 10
МЕХАНИЗМ МЕЖФАЗНОГО КАТАЛИЗА 12
Реакции в системах с разбавленными водными фазами .13
Реакции в системах с концентрированными водными фазами ..... 25
Характеристика растворов щелочей 25
Взаимодействие ониевых солей с концентрированными растворами
щелочей .29
Алкилирование кетонов в системе 50%-ная щелочь — органическая
фаза 33
Альдольная конденсация в двухфазной системе 44
Литература 47
МЕЖФАЗНЫИ КАТАЛИЗ В ОРГАНИЧЕСКОМ СИНТЕЗЕ 50
Реакции нуклеофильного замещения с участием неорганических анионов 50
Синтез галогенпроизводных 50
Синтез тиоцианатов 55
Синтез нитрилов 56
Синтез азидов 58
Синтез нитросоедииений 58
Синтез тиолов и сульфидов 69
Синтез сульфокислот . 59
Реакции нуклеофильного замещения с участием органических анионов 60
Образование С—О-связей 60
Образование С—S-связей '77
Образование С—N-связей 81
Образование N—Р-связей 91
Образование С—С-связей ( 93
Прочие реакции замещения . .'. .' . ."""* 113
Присоединение к простым и кратным связям . ■ 114
Реакции внедрения . 114
Присоединение анионов к углерод-углеродным кратным связям . . .118
Присоединение аншэнов к связям углерод — гетероатом 124
Реакции окисления 136
Реакции восстановления 144
Присоединение дигалогенкарбенов 146.
Дигалогенциклопропаиирование ненасыщенных углеводородов . . . 147
Дигалогенциклопропанирование функционально замещенных алкенов 155
Межфазный катализ в металлоргаиической химии 164
Литература 166