/






























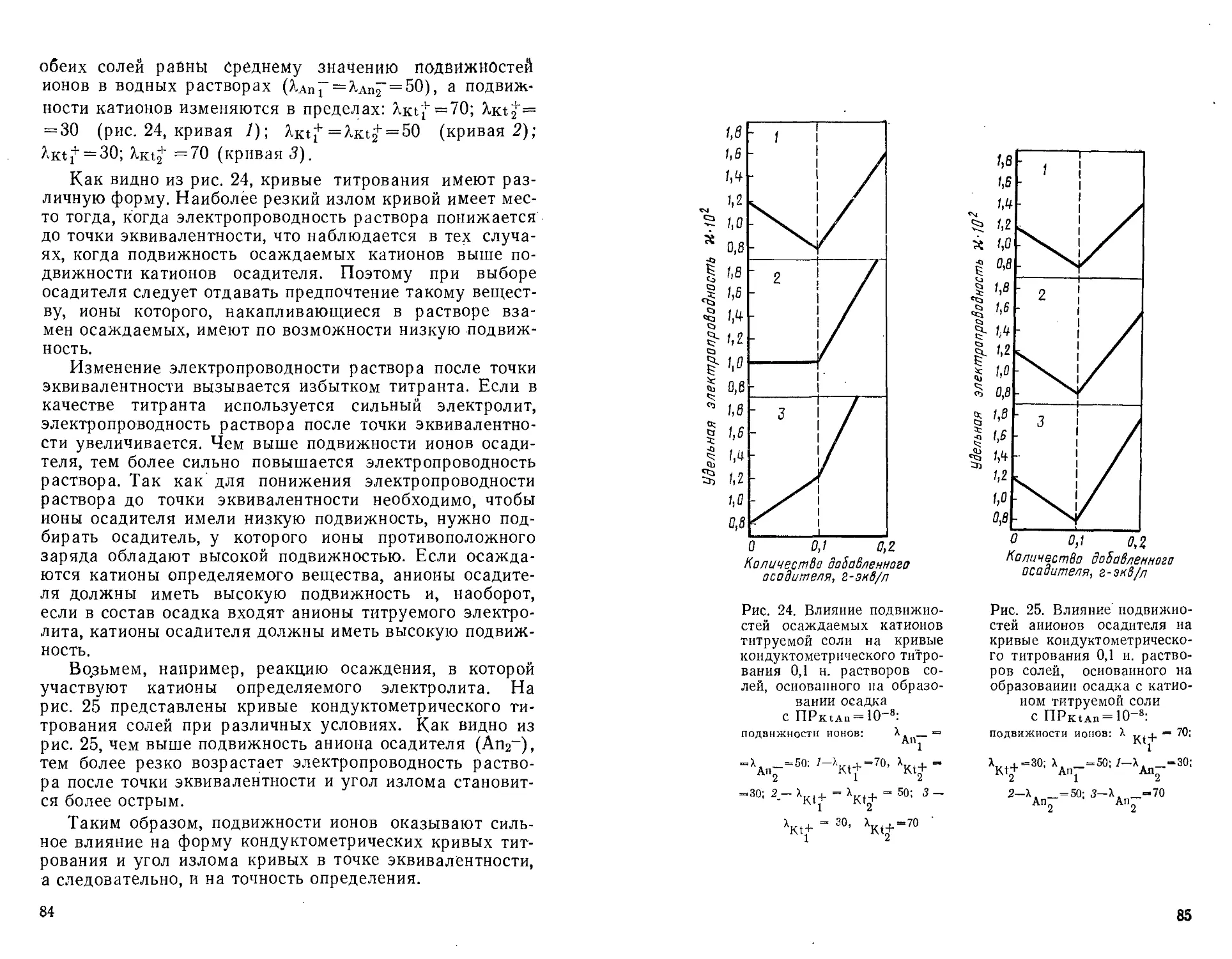








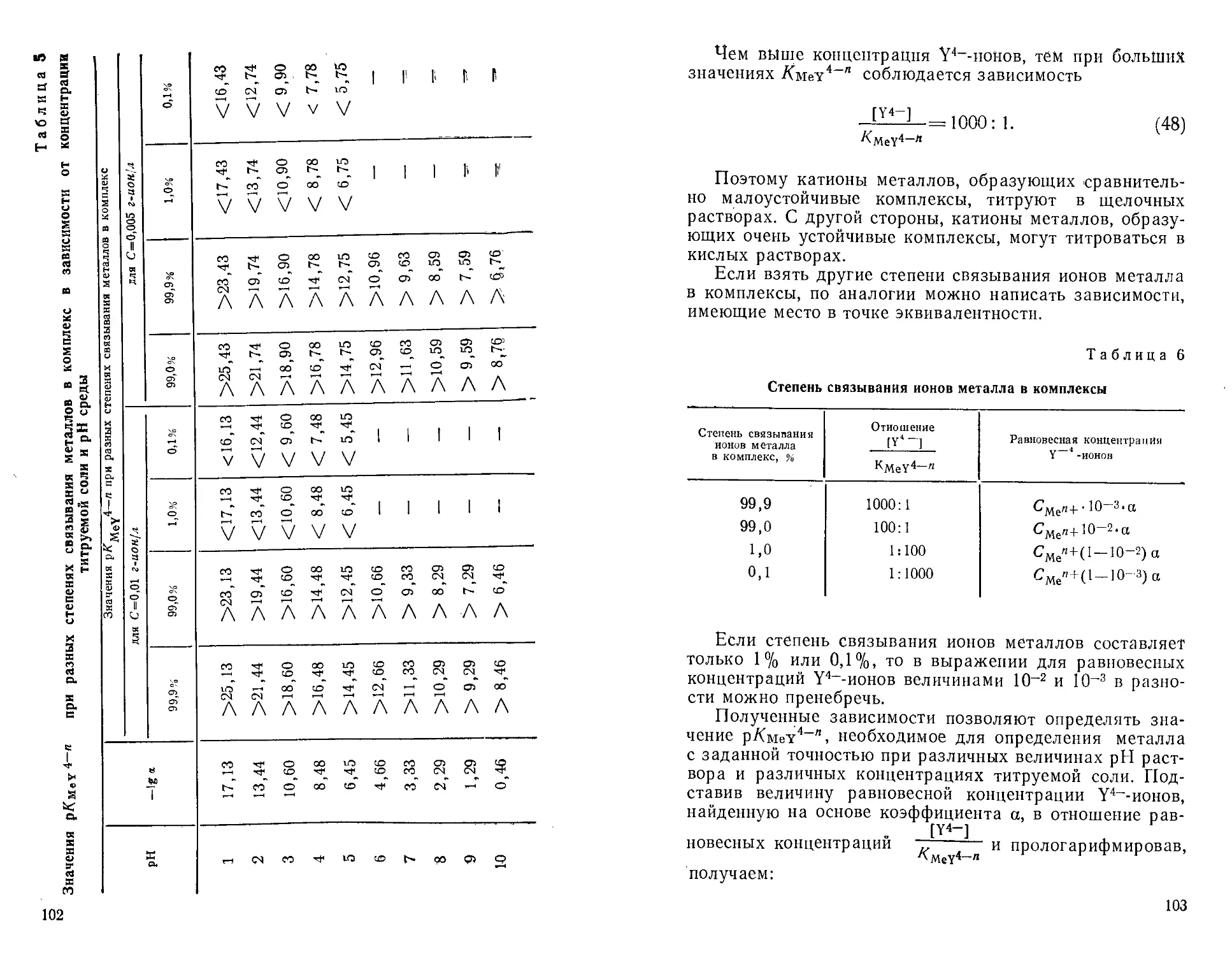


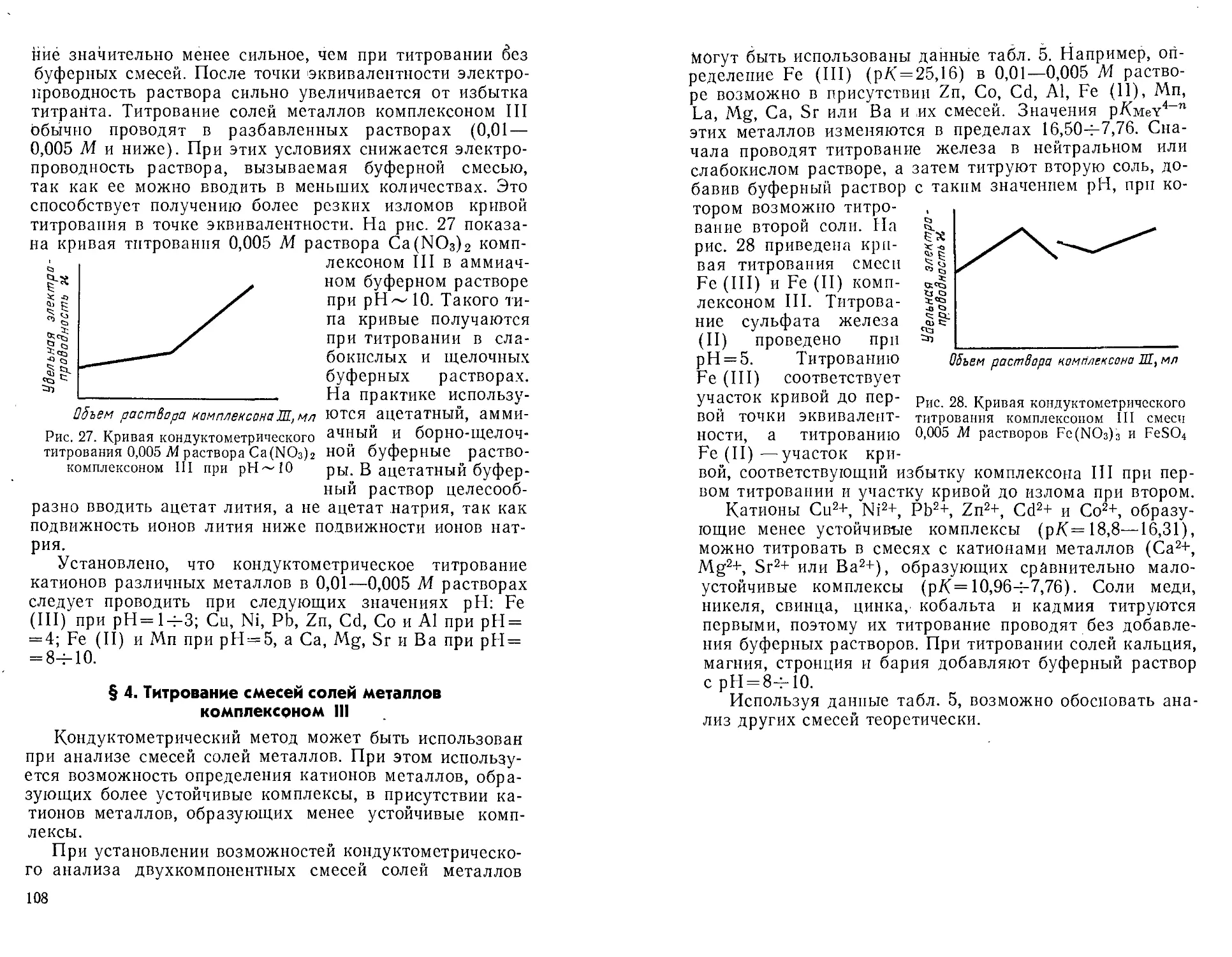

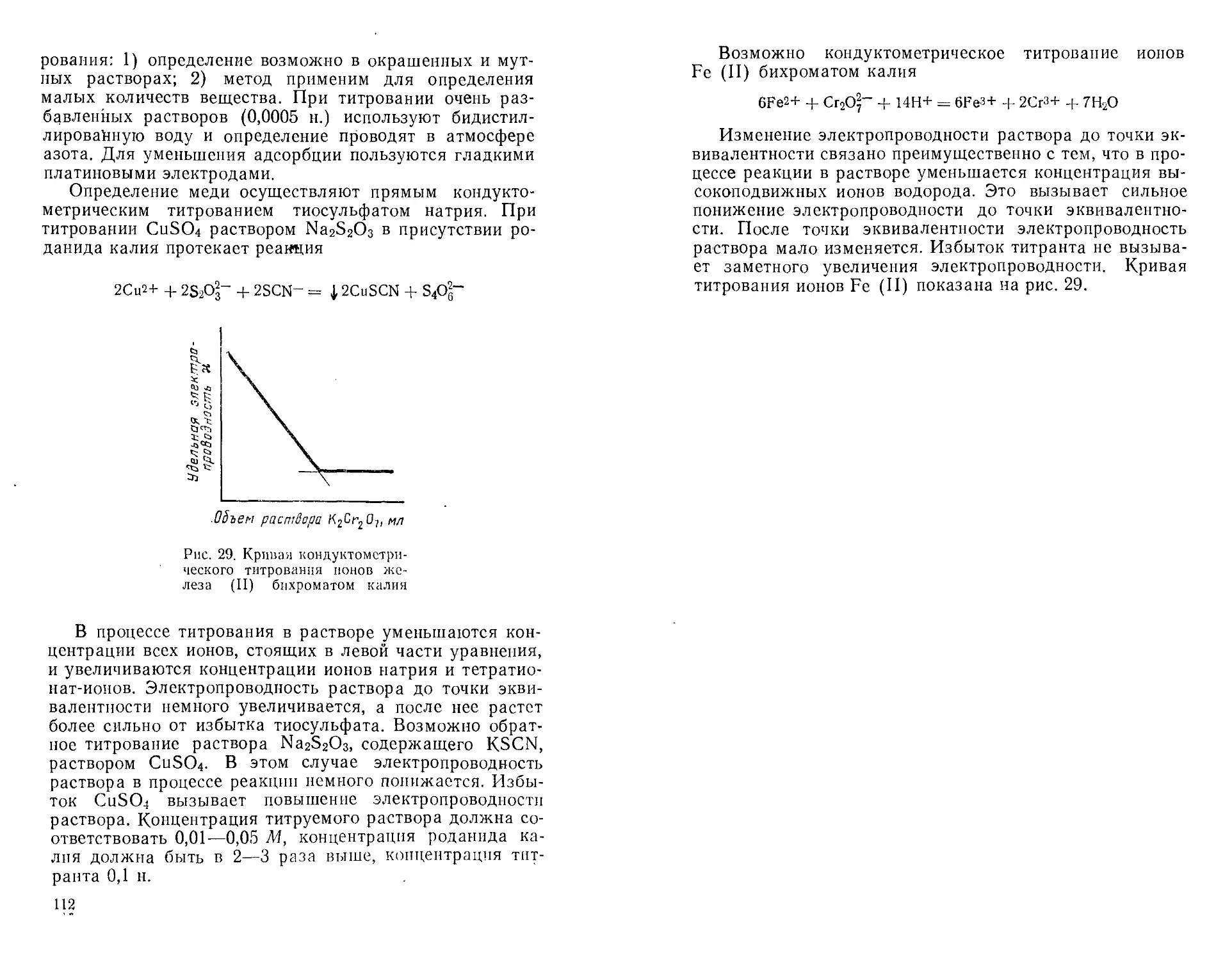













































Автор: Крешков А.П. Худякова Т.А.
Теги: химические методы анализа химия аналитическая химия
Год: 1975




Текст
Т.А.ХУДЯКОВА, А.П.КРЕШКОВ
К0НДУНТ0М1ТРИЧЕСКИЙ
МЕТОД АНАЛИЗА
©^ I
Т. А. ХУДЯКОВА, А. П. КРЕШКОВ
КОНДУКТОМЕТРИЧЕСКИЙ
МЕТОД АНАЛИЗА
Под редакцией проф. А. П. КРЕШКОВА
Допущено Министерством высшего и среднего
специального образования СССР в качестве учебного
пособия для студентов химических и
химико-технологических специальностей вузов
«вые
543
X98
УДК 543.257.5 <$У5)
Рецензенты: проф. А. М. Шкодин (Харьковский университет!
и кафедра аналитической химии Днепропетровского
химико-технологического ин-та им. Ф. Э. Дзержинского (проф. Ю. И. Усатенко]
Худякова Т. А., Крешков А. П.
Х98 Кондуктометрический метод анализа. Учеб. пособие
для вузов. М., «Высшая школа», 1975.
207 с. с ил.
Излагаются теоретические, основы, содержание и техника
кондуктометрического метода анализа неорганических и органических соединений. Особое
внимание уделено описанию аппаратуры и методов кондуктометрического
титрования, основанных на использовании реакций нейтрализации, осажде-
* ~о,т„ст „ т^иглрния — восстановления.,
ни
с
!троваНИЯ, ОСНОВаННЫХ па m,uwioJVJU- г .
,1я, комплексообразования и окисления — восстановления,
В книге приводится около 100 практических работ по кондуктометриче-
кому определению индивидуальных соединений и анализу их смесей.
20506-350_113-75 543
001(01)—75
© Издательство «Высшая школа»,
ОГЛАВЛЕНИЕ
Стр.
Предисловие 7
Глава I. Кондуктометрические методы анализа 8
§ 1. Понятие о кондуктометрических методах анализа . . 8
§ 2. Кондуктометрическое титрование 10
§ 3. Хронокондуктометрическое титрование 15
Теоретические основы кондуктометрического метода анализа 17
Глава II. Электропроводность растворов электролитов. . 17
§ 1. Понятие об электропроводности 17
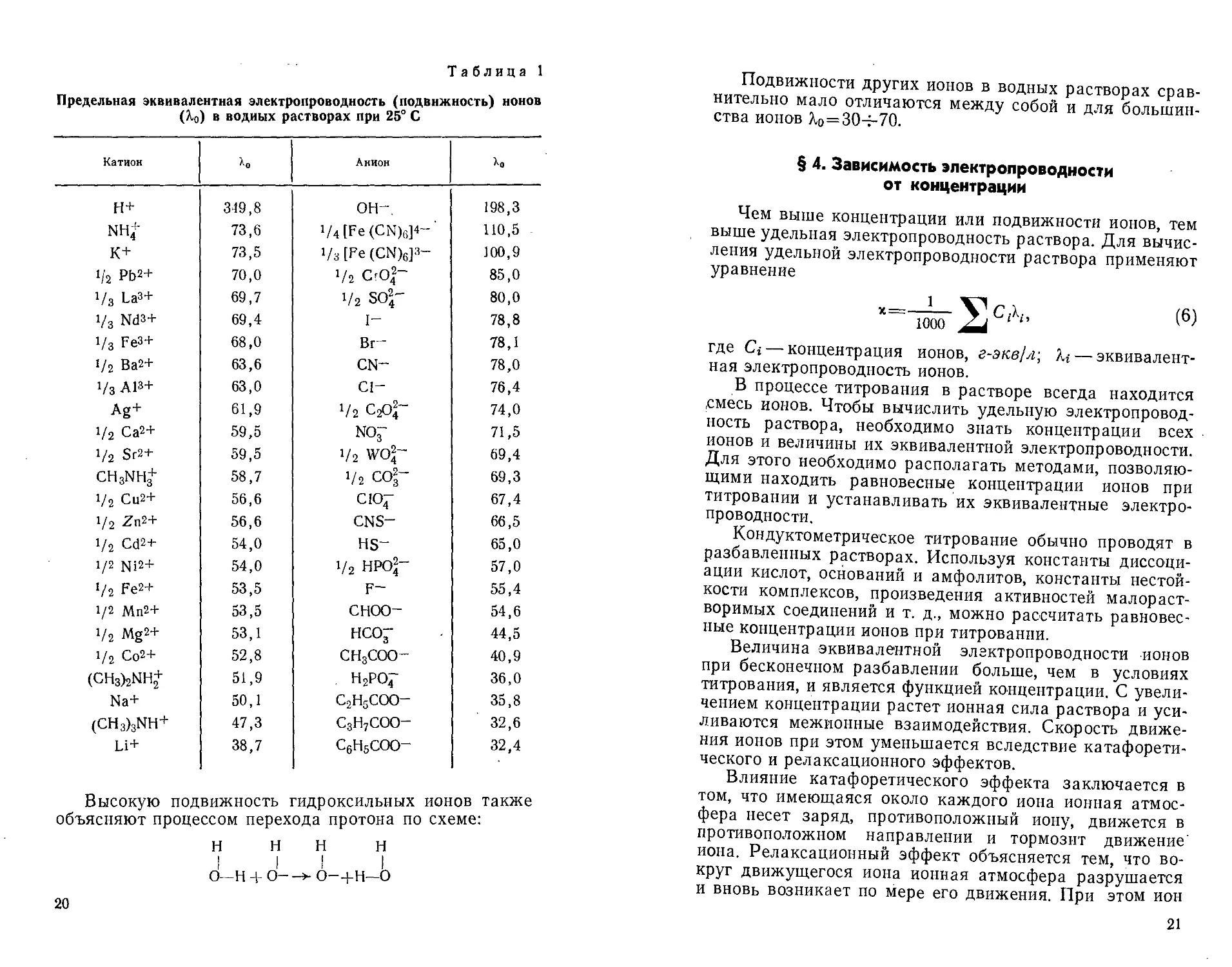
§ 2. Удельная и эквивалентная электропроводность .... 17
§ 3. Эквивалентные электропроводности (подвижности)
ионов 19
§ 4. Зависимость электропроводности от концентрации . . 21
§ 5. Зависимость электропроводности от температуры . . 24
§ 6. Зависимость электропроводности от природы
растворителя 25
§ 7. Критерии применимости кондуктометрического
метода анализа 26
Глава III. Кислотно-основное титрование 28
§ 1. Титрование кислот 28
§ 2. Титрование оснований 38
§ 3. Титрование солей 40
§ 4. Титрование смесей кислот 51
§ 5. Титрование смесей оснований 54
§ 6. Титрование смесей солей 55
§ 7. Титрование смесей кислот и солей слабых оснований 55
§ 8. Титрование смесей оснований и солей слабых кислот 64
§ 9. Титрование амфолитов 65
§ 10. Титрование смесей амфолитов с кислотами 68
§ И, Титрование смесей амфолитов <г основаниями .... 72
3
§ 12. Титрование смесей амфолитов с солями слабых
кислот нли слабых оснований
76
Глава IV. Титрование, основанное на реакциях осаждения 80
§ 1. Зависимость формы кривых кондуктометрического тит-
рования от величины растворимости осадка ..... «и
§ 2. Влияние подвижностей ионов на характер кондукто-
метрических кривых титрования od
§ 3. Требования в отношении постоянства состава осадка,
его чистоты и скорости осаждения . . во
§ 4. Определение анионов, осаждаемых в виде
малорастворимых соединений °°
§ 5. Определение катионов, осаждаемых в виде
малорастворимых соединений 92
Глава V. Титрование, основанное на реакциях комплексо-
образования . Щ
- § 1. Лиганды, используемые в кондуктометрическом
анализе . .........,•••• "7
§ 2. Влияние константы нестойкости комплекса,
концентрации раствора и рН среды иа титрование солей
металлов комплексоном III 99
§ 3. Кондуктометрическое титрование солей металлов
комплексоном III • Ю6
§ 4. Титрование смесей солей металлов комплексоном III 108
Глава VI. Титрование, основанное на реакциях окисления —
восстановления ПО
§ 1. Влияние условий проведения реакций на возможность
определения окислителей и восстановителей методом
кондуктометрического титрования ПО
§ 2. Окнслительно-восстаиовительное титрование . . . .111
Аппаратура я техиика кондуктометрического метода анализа 114
Глава VII. Измерение электропроводности растворов. . .114
§ 1. Мост Кольрауша 114
§ 2. Реохордный мост Р38 . 119
§ 3. Кондуктометр ММЗЧ-64 . .121
§ 4. Кондуктометр типа К1-4 122
§ 5. Кондуктометр «Импульс» типа КЛ1-2 123
§ 6. Установки для хронокондуктометрического титрова«
ния . . . 125
§ 7. Конструкции электролитических ячеек ..,,,, ,127
Практические работы 134
4
Стр
Глава VIII. Подготовка рабочего места, установка
аппаратуры
134
§ 1. Подготовка электролитической ячейки 134
§ 2. Установка для кондуктометрического титрования . . 135
§ 3. Методика кондуктометрического титрования 137
§ 4. Установка для хронокондуктометрического
титрования . ........ 138
§ 5. Методика хронокондуктометрического титрования . . 141
§ 6. Расчеты 142
Глава IX. Кислотно-основное титрование индивидуальных
электролитов . 148
§ 1. Приготовление стандартных растворов 148
§ 2. Титрование кислот сильными основаниями 149
§ 3. Титрование кислот слабыми основаниями 152
§ 4. Титрование оснований сильными кислотами .... . 153
§ 5. Титрование оснований слабыми кислотами 155
§ 6. Титрование солей слабых оснований сильными
основаниями 155
§ 7. Титрование солей слабых кислот сильными кислотами 157
§ 8. Титрование амфолитов сильными основаниями и
сильными кислотами ., 159
Глава X. Кислотно-основное титрование смесей
электролитов ' 162
§ 1. Титрование смесей кислот снлыЛшн основаниями . . 162
§ 2. Титрование смесей оснований сильными кислотами . . 164
§ 3. Титрование сильными основаниями двухкомпонеит-
ных смесей кислот и солей слабых оснований .... 165
§ 4. Титрование сильными основаниями
многокомпонентных смесей кислот и солей слабых оснований .... 170
§ 5. Титрование сильными кислотами двухкомпонеитных
смесей основанвй и солей слабых кислот 176
§ 6. Титрование сильными кислотами многокомпонентных
смесей оснований и солей слабых кислот 179
Глава XI. Титрование, основанное на реакциях осаждения 184
Титрование анионов , 184
§ 1. Титрование бромид-ионов нитратом серебра 184
§ 2. Определение хлорид-ионов в природных водах
титрованием нитратом ртути 185
§ 3. Определение сульфат-ионов в природных водах . . . 186
§ 4. Титрование феррогексацианид-чднов нитратом свинца 187
§ 5. Определение вольфраматов щелочных металлов . . . 188
Титрование катионов 189
§ 6. Определение ионов стронция титрованием сульфатом
лития 189
§ 7. Титрование ионов бария хроматом натрия 190
§ 8. Титрование ионов кальция оксалатом лития . . .191
§ 9. Титрование ионов марганца селенитом натрия . . . 191
§ 10. Определение ионов неодима теллуритом натрия . . - . 192
Глава XII. Титрование, основанное на реакциях комплексо-
образования 194
§ 1. Титрование солей металлов комплексоном III . . . . 194
§ 2. Титрование смесей солей металлов комплексоном III 199
§ 3. Определение фторид-ионов 203
§ 4. Определение ионов калия титрованием тетрафеннл-
боратом натрия 204
§ 5. Определение ионов меди и никеля и анализ их смеси
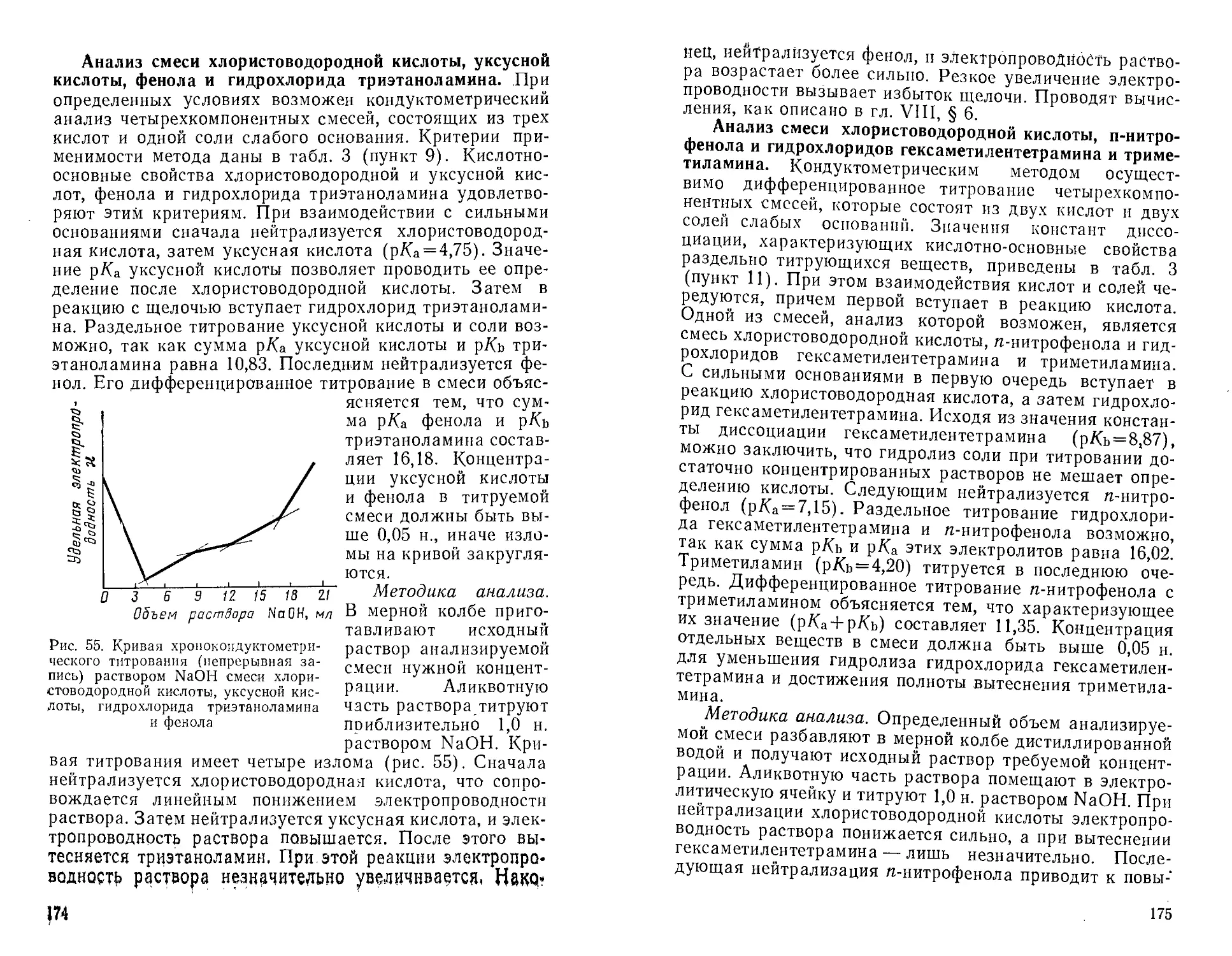
титрованием роданидом калия 206
ПРЕДИСЛОВИЕ
В связи с быстрым развитием
разнообразных областей науки, промышленности и новой
техники, наряду с классическими методами
анализа (качественным, гравиметрическим —
весовым и объемным) нашли широкое
практическое применение физико-химические
(инструментальные) методы анализа, к которым
относятся и кондуктометрические. В последнее
время благодаря работам советских ученых
кондуктометрические методы получили
большое развитие. Однако, несмотря на широкие
возможности этих методов и ряд ценных
преимуществ по сравнению с некоторыми другими
методами, в отечественной литературе нет
специального руководства по кондуктометриче-
ским методам анализа.
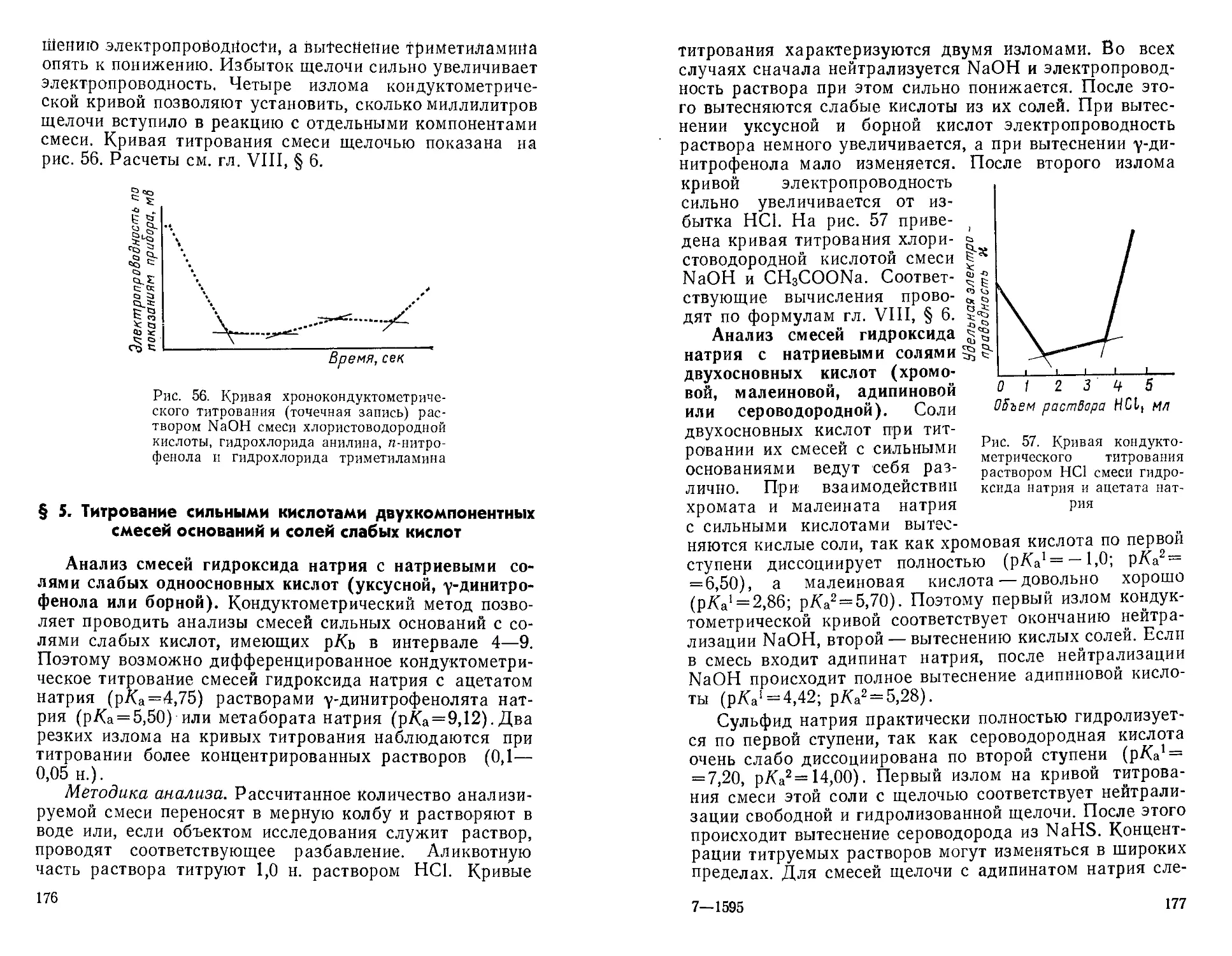
Вследствие этого студенты были
практически лишены возможности более подробно
ознакомиться с этими методами, изучаемыми в
курсе аналитической химии в разделе физико-
химических методов анализа.
Авторы настоящего руководства взяли на
себя труд восполнить указанный пробел. Оно
будет полезно не только для студентов, но и
для аспирантов и сотрудников
научно-исследовательских лабораторий.
Авторы глубоко признательны рецензентам
проф. Ю. И. Усатенко и проф. А. М. Шкодину
за ценные указания, способствовавшие
улучшению рукописи.
Все замечания, советы и пожелания по
данной книге авторы примут с
благодарностью.
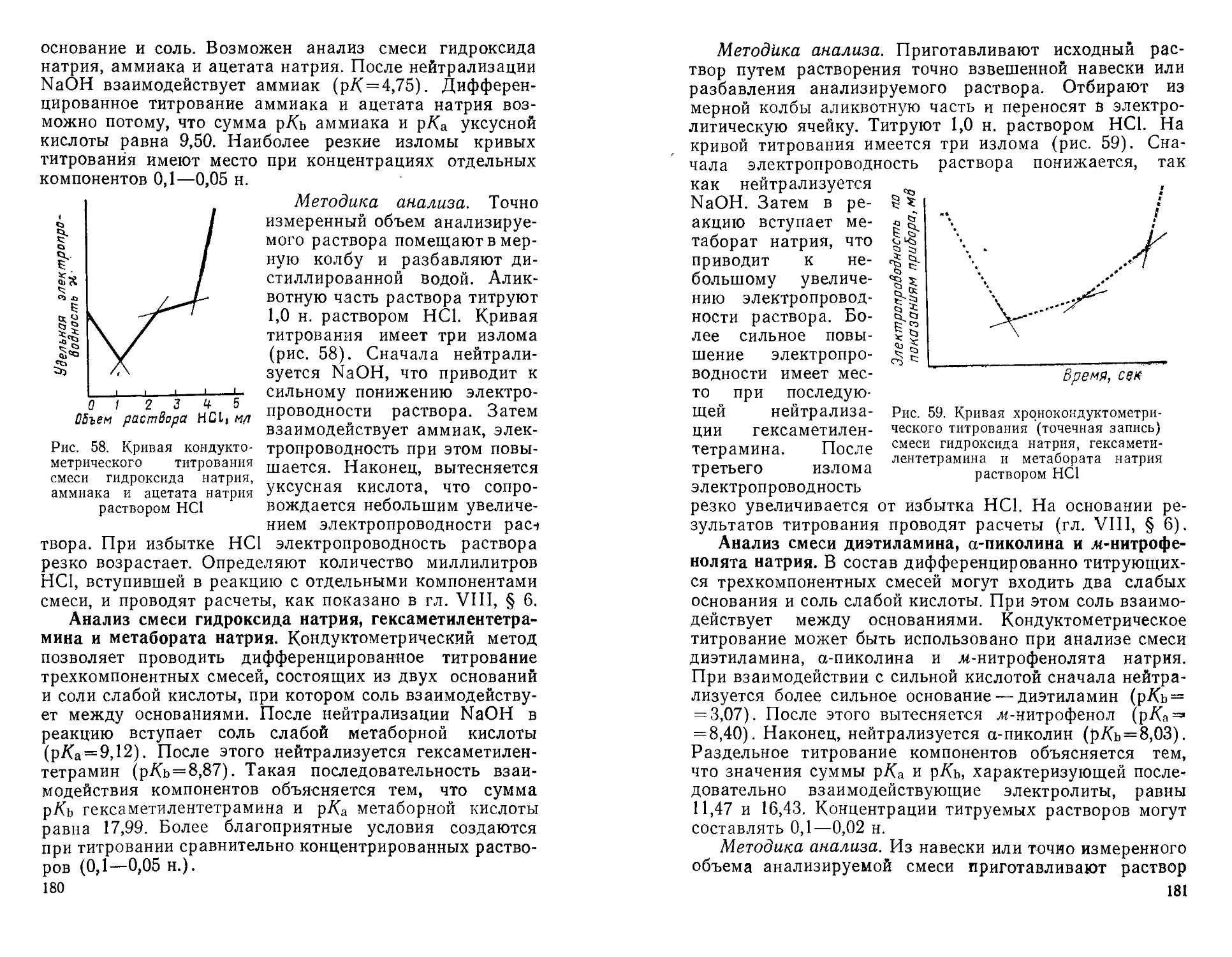
Глава I
КОНДУКТОМЕТРИЧЕСКИЕ
МЕТОДЫ АНАЛИЗА
§ 1. Понятие о кондуктометрических
методах анализа
Кондуктометрические методы анализа основаны на
измерении удельной электропроводности исследуемых
растворов.
Известно несколько методов кондуктометрического
анализа.
Прямая кон ду ктом етр ия — метод,
позволяющий непосредственно определять концентрации
электролита путем измерения электропроводности раствора
электролита с известным качественным составом.
Кондуктометрическое титрование —
метод анализа, основанный на определении содержания
вещества по излому кривой титрования. Кривую строят
по измерениям удельной электропроводности
анализируемого раствора, изменяющейся в результате химических
реакций в процессе титрования.
Хронокондуктометрическое
титрование основано на определении содержания вещества по
затраченному на его титрование времени, автоматически
фиксируемому на диаграммной ленте регистратора
кривой титрования.
Особенности кондуктометрических методов анализа.
Кондуктометрические методы по сравнению с другими
методами анализа имеют некоторые преимущества. Они
дают возможность:
1) без больших затруднений проводить определения
не только в прозрачных, но и в окрашенных и мутных
растворах, а также в присутствии окислителей и восста-
8
новителей, ограничивающих применение органических
индикаторов в других методах;
2) осуществлять определения разнообразных
неорганических и органических индивидуальных соединений;
3) анализировать не только сравнительно
концентрированные растворы, но и разбавленные до Ю-4 М;
4) проводить исследование не только водных, но и
неводных и смешанных водно-органических растворов;
5) сравнительно легко осуществлять автоматизацию
процессов титрования;
6) широко использовать разнообразные типы
реакций (нейтрализации, осаждения, комплексообразования,
окисления — восстановления, присоединения, замещения,
конденсации, омыления и т. п.), сопровождающихся
изменением электропроводности анализируемых
растворов;
7) во многих случаях избежать предварительного
отделения примесей, обычно мешающих определению
другими методами;
8) просто и точно определять конечную точку
титрования по пересечению двух прямых и соответственно
вычислять точку эквивалентности;
9) использовать переменный ток низкой частоты и
постоянный ток;
10) производить дифференцированное титрование
смесей электролитов, что невозможно осуществить
другими методами.
Как видно из изложенного выше, особыми
преимуществами отличаются методы кондуктометрического
титрования.
Области применения кондуктометрических методов
анализа. Кондуктометрические методы анализа
применяются в промышленности для осуществления непрерывного
химико-аналитического контроля производства,
определения концентрации солевых растворов, содержания солей
в минеральной, морской и речной воде, для контроля
процесса очистки и качества воды, оценки загрязненности
сточных вод, для определения следов воды в неводных
растворителях, газах, твердых солях, целлюлозе, бумаге,
зерне и т. п.
Кондуктометрические методы также применяются для
анализа окружающей среды, количественного
определения различных газов (СОг, СО, 02, NH3, БОг, H2S и др.)_.„
9
содержания вредных примесей в воздухе, воде, пищевых
продуктах, для контроля качества молока, вин,
напитков, фруктовых соков.
Кондуктометрическое титрование используется для
определения индивидуальных сильных, слабых и очень
слабых неорганических и органических карбоновых
кислот, амино-, галогено- и оксикислот, фенолов и их
производных, фармацевтических препаратов, дигуанидина,
гуминовых кислот, гидразинов, тиоцианатов, тиогликоле-
вых кислот, аминов, четвертичных аммониевых оснований
и т. д. Кондуктометрическое титрование применяется
также для анализа многокомпонентных смесей, кислот,
оснований, солей, образованных сильными кислотами
(основаниями) и слабыми основаниями (кислотами),
разнообразных катионов и анионов, окислителей и
восстановителей, комплексующихся агентов, амфолитов, смесей
минеральных, монокарбоновых и поликарбоновых кислот,
смесей оснований и солей слабых кислот, смесей кислот
и солей слабых оснований и т. п.
§ 2. Кондуктометрическое
титрование
Наиболее широко применяемым кондуктометриче-
ским методом анализа является кондуктометрическое
титрование.
Электропроводность раствора при известных условиях
пропорциональна концентрации электролита и зависит
от характера электролита, его температуры и
концентрации растворенного вещества. Пользуясь методом
титрования, основанным на измерении электропроводности,
можно с достаточной точностью построить кондукто-
метрические кривые титрования.
Точку эквивалентности фиксируют по резкому
излому кривой (пересечению двух прямых) титрования,
отражающему изменение электропроводности исследуемого
раствора по мере прибавления титранта в процессе
титрования.
Например, если при титровании определяемого
вещества А стандартным раствором реактива В образуется
нерастворимое или слабодиссоциирующее вещество,
электропроводность раствора титруемого электролита
уменьшается (вследствие уменьшения концентрации А).
Минимум электропроводности на кривой титрования наблю-
10
дается в точке эквивалентности. При избытке реагента В
электропроводность вновь возрастает. Излом,
наблюдаемый на кривой в точке эквивалентности, служит
признаком конца титрования.
При титровании сильной кислоты А сильным
основанием В концентрация кислоты уменьшается. Так как
электропроводность раствора образующейся при этом
соли (например, NaCl) значительно ниже
электропроводности титруемой кислоты и титранта, то по мере
прибавления к кислоте основания электропроводность раствора
уменьшается и в точке эквивалентности будет
наименьшей. При добавлении избытка основания
электропроводность раствора вновь начинает возрастать, что приводит
к V-образной кривой кондуктометрического титрования.
Излом кривой соответствутет точке эквивалентности.
Реакции, используемые при кондуктометрическом
титровании. При кондуктометрическом титровании
происходит химическое взаимодействие определяемого вещества
с титрантом; в процессе этой реакции изменяется
электропроводность анализируемых растворов, что
фиксируется в результате ее постоянного измерения. Необходимо,
чтобы используемые при этом реакции сопровождались
заметным изменением электропроводности. Резкое
изменение электропроводности дает излом кривой
титрования, что облегчает установление конечной точки
титрования и связанное с этим определение точки
эквивалентности.
В тех случаях, когда содержание посторонних
электролитов с большой электропроводностью в исследуемых
растворах относительно высокое, кондуктометрическое
титрование становится невозможным, так как
затрудняется установление точки эквивалентности.
В случае титрования смесей компонентов, что
связано с заметным изменением электропроводности в
нескольких точках и влечет за собой резкие изменения хода
кривой титрования, применяют дифференцированное
титрование определяемых веществ.
В кондуктометрическом титровании используются
чаще всего следующие реакции: нейтрализации;
осаждения; комплексообразования; окисления —
восстановления.
По характеру реакций классифицируют методы
кондуктометрического титрования:
11
1) кондуктометрическое кислотно-основное
титрование;
2) кондуктометрическое титрование, основанное на
использовании реакций осаждения;
3) кондуктометрическое титрование, основанное на
реакциях комплексообразования;
4) кондуктометрическое титрование, основанное на
реакциях окисления — восстановления.
Кривые кондуктометрического титрования. Кривые
кондуктометрического титрования отражают изменение
удельной электропроводности раствора при добавлениях
титранта. Для построения кондуктометрической кривой
используют значения удельной электропроводности
раствора, получаемые измерениями после добавления
каждой порции титранта. Кондуктометрические определения
возможны только при условии, что в конечной точке
титрования кривые титрования имеют четкий излом. При
титровании индивидуального вещества на кривой
титрования наблюдается один излом, а при титровании смесей
электролитов число изломов соответствует числу
определяемых компонентов. Точки эквивалентности на кондук-
тометрических кривых устанавливаются графическим
методом или рассчитываются теоретически.
Изменение электропроводности раствора при
титровании может носить различный характер. Наиболее
благоприятные условия для установления точки
эквивалентности создаются в тех случаях, когда наблюдается
линейное изменение электропроводности на ветвях
кондуктометрической кривой. Линейное изменение
электропроводности раствора, например, имеет место при
титровании сильных кислот сильными основаниями:
электропроводность раствора линейно понижается до точки
эквивалентности и линейно увеличивается после нее.
В этом случае кривая титрования представляет собой
две прямые линии, пересекающиеся в точке
эквивалентности. Линейное изменение электропроводности раствора
наблюдается при вытеснении слабых кислот или слабых
оснований из их солей сильными кислотами или
основаниями (в случаях, когда соли не подвергаются
заметному гидролизу), при выделении малорастворимых
осадков, при образовании некоторых малодиссоциированных
комплексных соединений и т. д.
Однако в ряде случаев изменение электропроводности
раствора при титровании имеет нелинейный характер,
12
что может быть вызвано различными причинами.
Нелинейное изменение электропроводности раствора может
иметь место с начала титрования на некоторой части
кривой или на всем участке кривой до точки
эквивалентности. Это объясняется тем, что в процессе реакций
смещаются ионные равновесия и изменяются
равновесные концентрации ионов определяемого вещества, еще не
вступившего в реакцию. Например, при нейтрализации
довольно сильной, но не полностью диссоциированнной
кислоты (р/<а=1) сильным основанием в процессе
реакции уменьшается степень ее диссоциации под влиянием
образующейся соли. Электропроводность раствора при
нейтрализации этой кислоты до точки эквивалентности
нелинейно понижается. Другим примером может служить
титрование солей слабых кислот (или слабых оснований),
подвергающихся сильному гидролизу. При титровании
сильными кислотами (или основаниями) этих солей
уменьшается степень их гидролиза в связи с
накоплением в растворе вытесненных слабых кислот (или
оснований). В зависимости от силы кислоты, образующей соль
(или основания), кривая титрования может быть
изогнута только в начале титрования или на всем участке до
точки эквивалентности. В некоторых случаях на кривых
титрования до точки эквивалентности может
наблюдаться пологий минимум, не имеющий аналитического
значения, что рассмотрено ниже на конкретных примерах.
Аналогичные явления наблюдаются и в других случаях.
При этом реакция может протекать количественно, а
излом кривой титрования в точке эквивалентности может
быть резко выраженным. Перед точкой эквивалентности
электроприводность изменяется линейно, если реакция
протекает до конца.
Нелинейное изменение электропроводности раствора
при титровании может быть вызвано другой причиной —
обратимостью реакций. При этом нелинейное изменение
электропроводности раствора наблюдается вблизи точки
эквивалентности. Например, при- нейтрализации очень
слабых кислот сильными основаниями образующаяся
соль подвергается гидролизу. В результате этого в
растворе увеличивается концентрация высокоподвижных
гидроксильных ионов. Электропроводность раствора в
точке эквивалентности и вблизи ее повышается. Кривая
титрования в точке эквивалентности вместо резкого из-
13
лома имеет плавный изгиб. Аналогичное явление
наблюдается в тех случаях, когда при титровании образуются
недостаточно устойчивые комплексные соединения,
сравнительно сильно растворимые осадки и т. д. Обратимость
реакций приводит к закруглению кондуктометрических
кривых вблизи точки эквивалентности.
Наиболее благоприятные условия для установления
точек эквивалентности графическим методом создаются
в тех случаях, когда наблюдается линейное изменение
электропроводности до и после конечной точки
титрования. При этом допустимо небольшое число измерений
электропроводности при титровании, а кривая
титрования в этом случае представляет собой две прямые,
пересекающиеся в точке эквивалентности.
Если изменение электропроводности раствора при
титровании носит нелинейный характер, проводят
большое число измерений электропроводности. Для
определения точки эквивалентности используют близкие к ней
участки кривой. Если реакция протекает количественно,
установление точки эквивалентности не вызывает особых
затруднений. Менее благоприятные условия создаются в
тех случаях, когда реакции обратимы. Обратимость
реакции, до определенных пределов, не мешает установлению
точки эквивалентности. При этом, используя измерения,
сделанные задолго до этой точки, прямолинейные
участки кривых продолжают до их пересечения и находят
точку эквивалентности с достаточно высокой точностью.
В случае сильно обратимых реакций нелинейное
изменение электропроводности может иметь место в
продолжение всего процесса титрования, что делает невозможным
установление точки эквивалентности графическим
методом.
В некоторых случаях кривые кондуктометрического
титрования имеют достаточно резкий излом, который,
однако, не соответствует стехиометрическим отношениям
взаимодействующих веществ. Например, это может иметь
место при образовании основных солей. Если результаты
титрования легко воспроизводятся, такие реакции могут
быть положены в основу кондуктометрических
определений. При этом стандартизацию рабочего раствора
проводят при этих же условиях кондуктометрическим
методом. Концентрацию определяемого вещества в этих
случаях удобно определять при помощи стандартных
графиков.
14
§ 3. Хронокондуктометрическое
титрование
Как было указано выше (см. § 1), наряду с
кондуктометрическим титрованием также применяют
хронокондуктометрическое титрование, основанное на
автоматической регистрации изменений электропроводности. В
этом случае концентрацию определяемого вещества
рассчитывают по времени, израсходованному на титрование,
фиксируемому автоматически на диаграммной ленте
регистратора хронокондуктометрической кривой.
Существует два способа автоматической регистрации
измерений электропроводности. В первом из них запись
хронокондуктометрических кривых производится
непрерывно. По завершении титрования с помощью
хронокондуктометрической кривой определяют длину
диаграммной ленты от начала титрования до точки
эквивалентности, а время, затраченное на титрование, рассчитывают,
зная скорость ее передвижения.
Во втором способе измерение электропроводности
осуществляется через строго определенные промежутки
времени. При этом получается точечная запись
кондуктометрических кривых. В этом случае необходимо знать
время между отдельными записями показаний
регистратора. По кондуктометрической кривой определяют
количество интервалов между точками от начала титрования
до точки эквивалентности. Время титрования равно
количеству интервалов, умноженному на время между их
записью.
Найденное тем или иным путем время титрования
позволяет определить число миллилитров стандартного
раствора, вступивших в реакцию. Для этого необходимо
знать скорость истечения стандартного раствора. Эта
величина определяется предварительно. При вычислении
концентрации титруемого раствора пользуются
соответствующими формулами.
Хронокондуктометрический метод имеет ряд
преимуществ по сравнению с обычным методом кондуктометри-
ческого титрования. Преимущества автоматической
записи кондуктометрических кривых титрования особенно
отчетливо проявляются в случаях, когда зависимость
носит нелинейный характер и требуется проводить большое
число измерений электропроводности. Более надежные
результаты получаются при анализе многокомпонентных
15
смесей, когда на кривых титрования наблюдается
несколько изломов. Расширяются практические
возможности использования реакций, которые протекают
неколичественно, или, когда вещества реагируют в нестехиомет-
рических отношениях, так как при хронокондуктометри-
ческом титровании легче воспроизвести условия
титрования. При этом в этих же условиях проводится
стандартизация рабочего раствора хронокондуктометри-
ческим титрованием. Увеличивается также точность
определений, так как можно проводить несколько
параллельных титрований, не расходуя много
времени.
ТЕОРЕТИЧЕСКИЕ ОСНОВЫ
КОНДУКТОМЕТРИЧЕСКОГО МЕТОДА АНАЛИЗА
Глава II
ЭЛЕКТРОПРОВОДНОСТЬ РАСТВОРОВ
ЭЛЕКТРОЛИТОВ
§ 1. Понятие об электропроводности
Электропроводностью называется способность
веществ (металлов, газов, жидкостей и др.) проводить
электрический ток под действием внешнего источника
электрического поля.
Электропроводность измеряется в обратных омах.
Для проводников первого рода (металлов) она
достигает 108 (ом-м)-\ для высококачественных изоляционных
материалов составляет Ю-14—Ю-16 (ом-м)~1.
Материалы с электропроводностью 105 (ом-м)-1
обычно относят к проводящим телам. Электропроводность
растворов электролитов и полупроводников составляет
10-5—10* (ом-м)-1.
В растворах электролитов и ионных кристаллах
(проводниках второго рода) носителями электрического тока
являются ионы, которые движутся под действием
электрического тока, обусловливая перенос вещества,
сопровождающийся образованием новых видов вещества на
электродах. Катионы и анионы движутся в противоположных
направлениях.
§ 2. Удельная и эквивалентная электропроводность
Электропроводность раствора (W) представляет
собой величину, обратную его электрическому
сопротивлению (R):
W=±, (1)
где W — электропроводность раствора, выраженная в
обратных омах; R — электрическое сопротивление раство-
17
pa, ом. Сопротивление раствора прямо пропорционально
расстоянию между электродами / и обратно
пропорционально площади электродов S, погруженных в раствор:
R=?j-- (2)
/ выражают в см, площадь S в см2.
Коэффициент пропорциональности р (ом-см)
называют удельным сопротивлением. В случае 1=1 см, S=l см2,
p=R.
Таким образом, удельное сопротивление равно
сопротивлению столба жидкости длиной в 1 см и поперечным
сечением в 1 см2, т. е. сопротивлению 1 см3 раствора.
Различают удельную у, и эквивалентную X
электропроводность.
Удельная электропроводность к представляет собой
величину, обратную удельному сопротивлению:
*=-L. (3)
р
Следовательно, удельная электропроводность
соответствует электропроводности 1 см3 раствора, находящегося
между расположенными на расстоянии 1 см друг от
друга электродами с площадью в 1 см2.
Эквивалентная электропроводность раствора
электролита % соответствует электропроводности раствора,
содержащего один грамм-эквивалент растворенного
вещества.
Другими словами, эквивалентная электропроводность
есть электропроводность раствора, налитого в сосуд с
квадратным основанием площадью 1 см2 и такой высоты
слоя, которая соответствует содержанию одного грамм-
эквивалента растворенного электролита.
Если С — концентрация электролита (г-экв/л), то в
1 см3 раствора содержится С/1000 г-экв/л. Объем V (см3),
содержащий 1 г-экв растворенного вещества, равен
1000/С. Величина V называется разбавлением. Исходя из
этого зависимость между эквивалентной и удельной
электропроводностью выражается следующим образом:
\ = х _l°22_=xV (ом-1 ■ см2/г-экв). (4)
18
§ 3. Эквивалентные электропроводности
(подвижности) ионов
Электропроводность раствора электролита
складывается из электропроводности отдельных ионов. Свойства
ионов характеризуются эквивалентной
электропроводностью.
Эквивалентная электропроводность (подвижность)
ионов есть произведение абсолютной скорости движения
ионов на число Фарадея. Подвижность ионов зависит от
концентрации раствора. С уменьшением концентрации
ослабляются межионные взаимодействия, вязкость
раствора приближается к вязкости чистого растворителя, а
радиус ионов принимает предельное значение. В
бесконечно разбавленном растворе скорость движения ионов
становится постоянной. При этих условиях
эквивалентные электропроводности ионов принимают наибольшее
постоянное значение. Поэтому и предельная
эквивалентная электропроводность раствора электролита
представляет постоянную величину, равную сумме эквивалентных
электропроводностей ионов при бесконечном
разбавлении:
Для многозарядных ионов величину эквивалентной
электропроводности относят к одному заряду. Ионы
различной химической природы обладают разными подвиж-
ностями в водных и в неводных растворах. В табл. 1
приведены подвижности различных катионов и анионов в
водных растворах.
Как видно из табл. 1, исключительно высокой
подвижностью в водных растворах отличаются водородные
(Яо = 349,8) и гидроксильные (Яо= 198,3) ионы, что
объясняется спецификой механизма их движения в растворе:
ион гидроксония Н30+ передает свой протон соседней
молекуле воды, которая, превращаясь в ион гидроксония,
эстафетно передает протон другой молекуле Н20. Таким
образом, протон перемещается к электроду сравнительно
быстро:
Н Н Н н
Н-О— Н++0-Н->Н-0 + Н — О—Н+-> . . . и т. д.
19
Таблица 1
Предельная эквивалентная электропроводность (подвижность) ионов
(Х0) в водных растворах при 25° С
Катион
н+
NH+
К+
1/2 РЬ2+
i/з La3+
i/з Nd3+
i/з Fe3+
1/2 Ва2+
i/з А13+
Ag+
1/2 Са2+
1/2 Sf2 +
CH3NH+
1/2 СЦ2+
1/2 ^П2+
1/2 Cd2 +
1/2 Ni2+
1/2 Fe2+
1/2 Mn2+
1/2 Mg2+
1/2 Со2+
(CH3)2NH+
Na+
(CH3)3NH+
Li+
К
349,8
73,6
73,5
70,0
69,7
69,4
68,0
63,6
63,0
61,9
59,5
59,5
58,7
56,6
56,6
54,0
54,0
53,5
53,5
53,1
52,8
51,9
50,1
47,3
38,7
Анион
он-.
i/4[Fe(CN)6]4-'
i/s [Fe (CN)6]3-
1/2 CrO|~
1/2 so\-
I-
Br~
CN-
ci-
1/2 C^-
N0^
1/2 W02-
1/2 CO2-
cio^
CNS-
hs-
1/2 HP02-
F~
CHOO-
HCO7
CH3COO-
h2po^
с2н5соо-
с3н7соо-
с6н5соо-
Xo
198,3
110,5
100,9
85,0
80,0
78,8
78,1
78,0
76,4
74,0
71,5
69,4
69,3
67,4
66,5
65,0
57,0
55,4
54,6
44,5
40,9
36,0
35,8
32,6
32,4
Высокую подвижность гидроксильных ионов также
объясняют процессом перехода протона по схеме:
Н Н Н н
1111
О—Н 4- О- -»- 0-+Н—О
20
Подвижности других ионов в водных растворах
сравнительно мало отличаются между собой и для
большинства ионов Х0=30-н70.
§ 4. Зависимость электропроводности
от концентрации
Чем выше концентрации или подвижности ионов, тем
выше удельная электропроводность раствора. Для
вычисления удельной электропроводности раствора применяют
уравнение
1000 j£j ' /
где Ci — концентрация ионов, г-экв/л; Аг-—
эквивалентная электропроводность ионов.
В процессе титрования в растворе всегда находится
смесь ионов. Чтобы вычислить удельную
электропроводность раствора, необходимо знать концентрации всех
ионов и величины их эквивалентной электропроводности.
Для этого необходимо располагать методами,
позволяющими находить равновесные концентрации ионов при
титровании и устанавливать их эквивалентные
электропроводности.
Кондуктометрическое титрование обычно проводят в
разбавленных растворах. Используя константы
диссоциации кислот, оснований и амфолитов, константы
нестойкости комплексов, произведения активностей
малорастворимых соединений и т. д., можно рассчитать
равновесные концентрации ионов при титровании.
Величина эквивалентной электропроводности ионов
при бесконечном разбавлении больше, чем в условиях
титрования, и является функцией концентрации. С
увеличением концентрации растет ионная сила раствора и
усиливаются межионные взаимодействия. Скорость
движения ионов при этом уменьшается вследствие катафорети-
ческого и релаксационного эффектов.
Влияние катафоретического эффекта заключается в
том, что имеющаяся около каждого иона ионная
атмосфера несет заряд, противоположный иону, движется в
противоположном направлении и тормозит движение'
иона. Релаксационный эффект объясняется тем, что
вокруг движущегося иона ионная атмосфера разрушается
и вновь возникает по мере его движения. При этом ион
21
оказывается несимметрично расположенным в своей
ионной атмосфере. Ионная атмосфера не успевает полностью
сформироваться при движении иона и, имея заряд,
противоположный иону, тормозит его движение. Эти
эффекты усиливаются с ростом ионной силы раствора, что
приводит к снижению эквивалентной электропроводности
ионов, а следовательно, и эквивалентной
электропроводности электролита с увеличением концентрации.
Зависимость эквивалентной электропроводности
сильного электролита, диссоциирующего на два вида ионов,
от концентрации выражается уравнением Онзагера:
Х = Хо-ВУ7, (7)
где X— эквивалентная электропроводность при данной
концентрации; Хо— эквивалентная электропроводность
при бесконечном разбавлении; В — постоянная величина;
[I — ионная сила раствора:
P=-f-(Yi|Z?| + Ys|Z?|). (8)
В формуле (8) С — концентрация электролита
(моль/л); \i — число ионов данного вида, на которое
диссоциирует электролит; 1\ — валентность ионов.
Величина В в уравнении (7) равна:
M-p-fl^l+l*.!)1 (ZlZ2)e2
6№q " " ' '-*" ' ЗеДТ" 1+у~ °
X
XT/ *"*" , О)
где F — число Фарадея; е — элементарный заряд; Z\Z2—
валентности ионов; г| — вязкость растворителя (пуаз);
R — универсальная газовая постоянная; Т — абсолютная
температура; N — число Авогадро; s — диэлектрическая
проницаемость растворителя;
„_ I Z\ 1 1 zi 1 (xoi + хо2> nQ\
| Zj | + | Z2 | (Xo! I Z2 I + X02 I Z1 I ) '
где loi и Хог — эквивалентные электропроводности
отдельных ионов при бесконечном разбавлении.
Первый член в выражении (9) учитывает влияние ка-
тафоретического эффекта, а второй — релаксационного.
22
Из приведенных уравнений следует, что
эквивалентная электропроводность раствора электролита при той
или иной концентрации ниже, чем при бесконечном
разбавлении, и зависит от свойств растворенного
вещества— заряда ионов и их эквивалентной
электропроводности при бесконечном разбавлении, — а также и свойств
растворителя — диэлектрической постоянной
растворителя, вязкости, температуры.
Если подставить значения постоянных в выражение
(9), получим зависимость
Х = Х [41,25(1^1 + |Z2|) , 2,801-106
° [ iK.7")1/8 (^)3/2
X—£—-(iz.iiz^x,
Первый, катафоретический, член в выражении (9)
зависит от вязкости, а также от диэлектрической
проницаемости растворителя и температуры в степени 1/2.
Второй, релаксационный, член зависит от
электропроводности при бесконечном разбавлении, диэлектрической
проницаемости и температуры в степени 3/2. В обоих
случаях оказывают влияние заряды ионов.
Отклонение X от Хо тем больше, чем больше величина
В. Коэффициент В растет, когда понижается
диэлектрическая проницаемость, температура и вязкость. С другой
стороны, чем больше заряд ионов и величина Хо, тем
больше величина В и сильнее отклонение X от Хо.
Эквивалентная электропроводность индивидуальных
ионов в растворах, содержащих два вида ионов,
рассчитывается по аналогичным уравнениям. При этом X
заменяется на Xi и %2, а Хо на Хш или Хог- Кроме этого, в элек-
трофоретическом члене сумма (Zi+Z2) заменяется на
Z\ или Z2. Релаксационный член имеет такое же
выражение.
Указанные положения применимы для определения
эквивалентных электропроводностей очень разбавленных
растворов сильных электролитов, а также слабых
электролитов, если учитывать степень их диссоциации. При
определенных допущениях они применимы к растворам
смесей электролитов, если учитывать общую ионную
силу раствора.
Эквивалентную электропроводность ионов в более
концентрированных растворах можно рассчитать по урав-
W- (И)
23
нению Робинсона и Стокса, позволяющему находить
достаточно точные значения этих величин в водных
растворах (1:1) электролитов и их смесей, когда их ионная
сила достигает 0,1:
h = \*- fM+B*V*. (12)
(l + Bal/V)
Для водных растворов (1:1) электролитов В^О.23;
В2 = 30,32; 5 = 0,3291-108.
Параметр а называют средним расстоянием
сближения ионов и рассматривают как радиус сферы, центр
которой совпадает с центром данного иона и внутрь
которой не могут попасть центры других ионов. Для многих
ионов в водных растворах параметр а имеет порядок
3- Ю-8 см или ЗА. При этом величина Ва равняется
единице, что упрощает уравнение (10). Однако в ряде
случаев лучшее совпадение с опытом имеет место, когда
параметр а имеет другие значения. Поэтому более точные
расчетные данные могут быть получены, если параметр а
определяют экспериментальным путем в каждом
конкретном случае. Исходя из этого, существует мнение, что
величина а представляет эмпирическую поправку в
уравнении.
Уменьшение эквивалентной электропроводности ионов
с ростом концентрации приводит к нарушению
пропорциональности между удельной электропроводностью
раствора сильного электролита и его концентрацией. Если
электролит слабый, то, кроме того, с ростом
концентрации уменьшается степень диссоциации электролита, что
приводит к уменьшению равновесных концентраций
ионов.
§ 5. Зависимость электропроводности
от температуры
Удельная и эквивалентная электропроводность
раствора электролита повышается с ростом температуры.
Большое значение при этом имеет уменьшение вязкости
раствора с повышением температуры, что приводит к
увеличению подвижностей ионов Х0. Эквивалентные
электропроводности ионов при бесконечном разбавлении
обычно приводятся для температуры 25°. Подвижности
24
ионов при температуре t могут быть вычислены по
уравнению
M0 = W>[l + a('-25)], (13)
где a — температурный коэффициент
электропроводности, зависящий от природы ионов и растворителя.
Величина температурного коэффициента при средних
температурах в водных растворах для большинства ионов
изменяется в пределах 0,02—0,025. Поэтому увеличение
удельной и эквивалентной электропроводности раствора
при повышении температуры на Г составляет примерно
2-2,5%.
§ 6. Зависимость электропроводности
от природы растворителя
Удельная и эквивалентная электропроводности
раствора электролита зависят от природы растворителя.
В различных растворителях изменяются эквивалентные
электропроводности ионов при бесконечном разбавлении.
Кроме этого изменяются константы диссоциации
электролитов, что влияет на величины равновесных
концентраций их ионов.
Влияние растворителей на предельную подвижность
одних и тех же ионов (км) обычно связывали с
вязкостью растворителя.
Л.' В. Писаржевский и Вальден обнаружили, что
произведение эквивалентной электропроводности ионов при
бесконечном разбавлении на вязкость растворителя
приблизительно постоянно и не зависит от природы
растворителя:
Xoir)o^ const,
где т)о — вязкость растворителя.
Однако практика показала, что это правило
соблюдается в редких случаях и лучше оправдывается в
растворах электролитов с крупными ионами. Оно было
выведено при условии применимости закона Стокса, который
описывает движение частицы в идеальной
гидродинамической среде. Закон Стокса применим в случае отсутствия
взаимодействия между средой и движущейся частицей.
Вследствие протекания процессов сольватации в
растворах электролитов это условие не соблюдается.
25
A. M. Шкодиным установлено влияние
диэлектрической проницаемости растворителя на эквивалентную
электропроводность ионов при бесконечном разбавлении
и установлена зависимость:
— — JL
l0i%=Ae D или l0l = ^-eD, (14)
По
где А я В — постоянные величины.
Таким образом, предельная подвижность ионов
является экспоненциальной функцией величины 1/D,
обратной диэлектрической проницаемости среды.
В неводных растворах у большинства электролитов
степень диссоциации меньше, чем в водных. Даже
полностью диссоциированные в воде электролиты в
большинстве неводных растворителей диссоциируют
частично. Уменьшение силы слабых в воде электролитов в
неводных растворах может быть очень значительным.
Только в некоторых случаях удается усилить
диссоциацию слабых электролитов подбором соответствующей
среды. Напримгр, сила слабых в воде оснований
увеличивается в протогенных растворителях (уксусная
кислота), а очень слабых кислот — в протофильных
растворителях (жидкий аммиак). Величины равновесных
концентраций ионов одних и тех же электролитов при одной и
той же концентрации растворенного вещества в
различных растворителях изменяются, что влияет на ионную
силу раствора. Поэтому отклонение величины Я при
данной концентрации от Х<н может быть различным. Кроме
этого, понижение удельной электропроводности раствора
электролита при титровании в неводных растворах
может быть вызвано уменьшением равновесных
концентраций ионов, обусловленным ослаблением силы
электролитов. Поэтому удельная электропроводность растворов
одного и того же электролита в различных
растворителях и ее изменение в процессе титрования в различных
растворителях могут варьироваться в широких пределах.
§ 7. Критерии применимости
кондуктометрического метода анализа
Химические взаимодействия в растворах связаны с
образованием малодиссоциированных электролитов или
комплексных ионов, с выделением малорастворимых
соединений, окислительно-восстановительными процессами
26
и т. д. Равновесные концентрации ионов зависят от
константы автопротолиза растворителей и констант,
характеризующих различные химические равновесия, имеющие
место в растворах.
Поэтому и удельная электропроводность,
изменяющаяся в процессе химических реакций, зависит от значений
термодинамических констант, которые предопределяют
возможность применимости кондуктометрического метода
анализа конкретных соединений. Указанными,
критериями являются величины константы автопротолиза
растворителя, подвижности ионов, константы диссоциации
кислот, оснований, амфолитов и солей, константы
нестойкости комплексных соединений, произведения активностей
малорастворимых соединений и др. Установлены
значения констант, лимитирующие возможности определении
основанных на различных химических реакциях. За
пределами этих границ определения ненадежны вследствие
обратимости реакций.
Т л а в в III
КИСЛОТНО-ОСНОВНОЕ ТИТРОВАНИЕ
§ 1. Титрование кислот
Титрование кислот сильными основаниями. При
титровании различных по силе кислот сильными основаниями
на форму кривых кондуктометрического титрования
влияет концентрация кислоты, так как степень ее
диссоциации увеличивается с разбавлением.
Типичные кривые титрования соответствуют
нейтрализации полностью диссоциированных, довольно сильно
диссоциированных, слабо и очень слабо
диссоциированных кислот.
Рассмотрим типичные кривые титрования 0,1 н.
растворов указанных кислот.
Сильные кислоты в водных растворах
полностью диссоциированы. Нейтрализацию сильных кислот
сильными основаниями можно представить схемой:
Н+ + An- + Kt+ + ОН- = Kt+ + An- + Н20
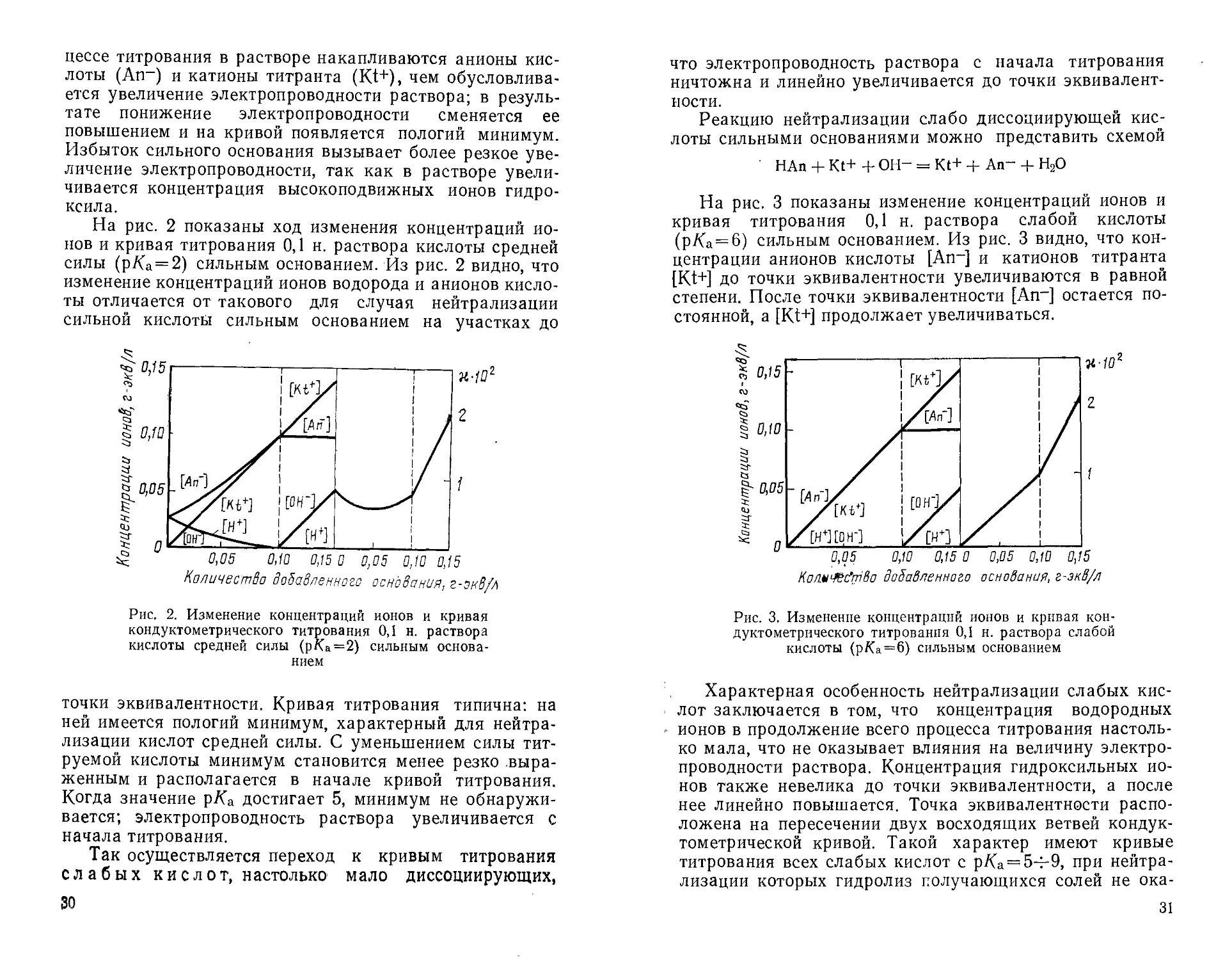
На рис. 1 показаны изменения концентраций ионов в
процессе кондуктометрического титрования и кривая
титрования 0,1 н. раствора сильной кислоты сильным
основанием. Как видно из рис. 1, концентрация ионов водоро-
0,05 0,10 0,15 0 0,05 0,10 0,15
Количества добавленного основания, г-экв/л
Рис. 1. Изменение концентрации ионов и шэивая
кондуктометрического титрования 0,1 н. раствора
сильной кислоты сильным основанием
28
да линейно понижается до точки эквивалентности, а
после нее становится ничтожно малой; концентрация
гидроксильных ионов остается незначительной до
конечной точки титрования и линейно увеличивается после
точки эквивалентности. Концентрация анионов кислоты
An- постоянна, а концентрация катионов основания Kt+
в продолжение титрования увеличивается. Кривая
титрования имеет V-образную форму, характерную для
процесса нейтрализации сильных кислот сильными
основаниями. При этом первоначальная ветвь кривой,
соответствующая уменьшению концентрации ионов водорода до
точки эквивалентности, имеет более острый угол
наклона, чем ветвь кривой, характеризующая повышение
концентрации гидроксильных ионов после окончания
процесса нейтрализации.
При нейтрализации 0,1 н. растворов кислот
средней силы (рДа^б) ход изменения концентрации
ионов носит иной характер, чем при нейтрализации сильных
кислот.
Равновесные концентрации ионов водорода и анионов
кислот средней силы вследствие неполной диссоциации
электролита до начала титрования меньше концентрации
исходной кислоты, но все же отличаются достаточно
высокими значениями и заметно влияют на
электропроводность раствора. С уменьшением силы кислоты
концентрации Н+- и Ап~-ионов все более понижаются и при
значении р/(а^5 они уже практически перестают оказывать
влияние на электропроводность раствора. При
титровании концентрация ионов водорода нелинейно понижается,
а анионов кислоты нелинейно увеличивается и в точке
эквивалентности достигает максимальной величины,
равной концентрации кислоты.
В случае титрования сравнительно хорошо
диссоциирующих кислот электропроводность раствора с начала
титрования довольно высокая и при нейтрализации
кислот до точки эквивалентности она нелинейно понижается.
На кривых титрования более слабых кислот с низкой
начальной электропроводностью до точки эквивалентности
наблюдается минимум, не имеющий аналитического
значения. Это объясняется тем, что в начале титрования
вьюокоподвижныз ионы водорода еще оказывают
влияние на электропроводность раствора, но в процессе
титрования их концентрация постепенно уменьшается и
электропроводность раствора понижается. Однако в про-
Щ
цессе титрования в растворе накапливаются анионы
кислоты (An-) и катионы титранта (Kt+), чем
обусловливается увеличение электропроводности раствора; в
результате понижение электропроводности сменяется ее
повышением и на кривой появляется пологий минимум.
Избыток сильного основания вызывает более резкое
увеличение электропроводности, так как в растворе
увеличивается концентрация высокоподвижных ионов гидро-
ксила.
На рис. 2 показаны ход изменения концентраций
ионов и кривая титрования 0,1 н. раствора кислоты средней
силы (р/Са = 2) сильным основанием. Из рис. 2 видно, что
изменение концентраций ионов водорода и анионов
кислоты отличается от такового для случая нейтрализации
сильной кислоты сильным основанием на участках до
f 0,75
* qv i""' i—■—у L" J i i : i
sg 0,05 0,10 0,15 0 0,05 0,10 0,15
Количество добавленного основания, г-эк8/л
Рис. 2. Изменение концентраций ионов и кривая
кондуктометрического титрования 0,1 н. раствора
кислоты средней силы (р/Са=2) сильным
основанием
точки эквивалентности. Кривая титрования типична: на
ней имеется пологий минимум, характерный для
нейтрализации кислот средней силы. С уменьшением силы
титруемой кислоты минимум становится менее резко
выраженным и располагается в начале кривой титрования.
Когда значение рКа достигает 5, минимум не
обнаруживается; электропроводность раствора увеличивается с
начала титрования.
Так осуществляется переход к кривым титрования
слабых кислот, настолько мало диссоциирующих,
H-1D
2
1
30
что электропроводность раствора с начала титрования
ничтожна и линейно увеличивается до точки
эквивалентности.
Реакцию нейтрализации слабо диссоциирующей
кислоты сильными основаниями можно представить схемой
' НАп + Kt+ + ОН- = Kt+ + An- + Н20
На рис. 3 показаны изменение концентраций ионов и
кривая титрования 0,1 н. раствора слабой кислоты
(р/(а = 6) сильным основанием. Из рис. 3 видно, что
концентрации анионов кислоты [Ап~] и катионов титранта
[Kt+] до точки эквивалентности увеличиваются в равной
степени. После точки эквивалентности [An-] остается
постоянной, a [Kt+] продолжает увеличиваться.
0,05 0,10 0,15 0 0,05 0,10 0,15
КоаиЧёс'тво до5авленного основания, г-зкВ/л
Рис. 3. Изменение концентраций ионов и кривая
кондуктометрического титрования 0,1 н. раствора слабой
кислоты (р/(а=6) сильным основанием
Характерная особенность нейтрализации слабых
кислот заключается в том, что концентрация водородных
ионов в продолжение всего процесса титрования
настолько мала, что не оказывает влияния на величину
электропроводности раствора. Концентрация гидроксильных
ионов также невелика до точки эквивалентности, а после
нее линейно повышается. Точка эквивалентности
расположена на пересечении двух восходящих ветвей кондук-
тометрической кривой. Такой характер имеют кривые
титрования всех слабых кислот с р/Са = 5-^9, при
нейтрализации которых гидролиз получающихся солей не ока-
31
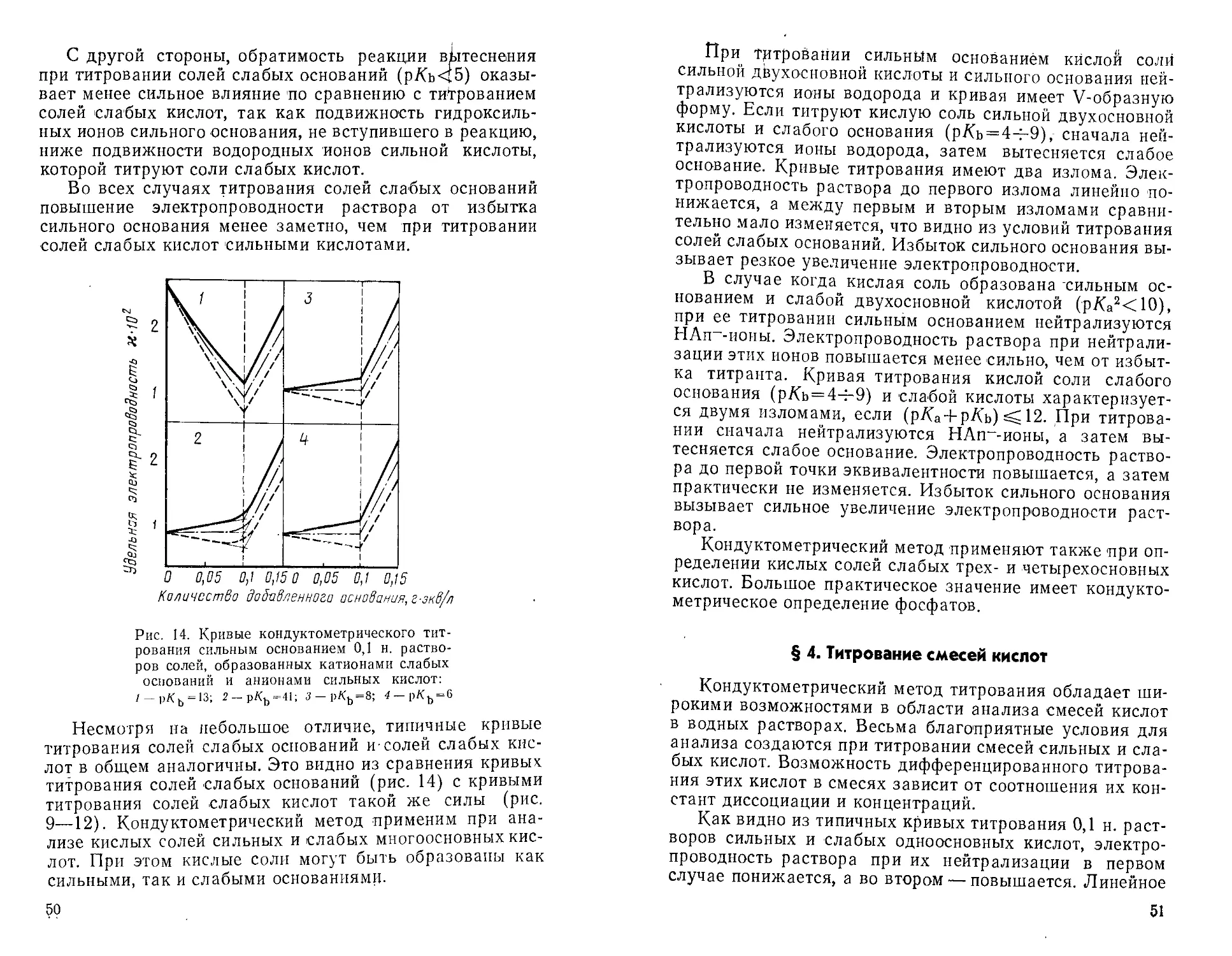
Второй, электропроводность линейно понижается до
первого излома кривой, а затем линейно повышается до
второго излома. Примером может служить кривая
титрования хромовой кислоты, которая характеризуется р/Са' =
= —1 и р/(а2 = 6,5 (рис. 6, кривая 1). Если кислота
довольно хорошо диссоциирована
по первой' ступени и слабо по
второй, электропроводность
раствора нелинейно
понижается до первого излома, а между
первым и вторым изломами
линейно увеличивается. Этот тип
кривой наблюдается при
титровании малеиновой кислоты
№• = 1,92; ptfa2 = 6,23, рис. 6,
кривая 2).
Дифференцированное
титрование двухосновных кислот
средней силы и слабых
затрудненно даже в том случае, когда
константы диссоциации кислот
по первой и второй ступени
достаточно сильно отличаются
друг от друга (Ар/Са^2). Это
объясняется тем, что первая
точка эквивалентности,
соответствующая нейтрализации
0 05 010 015 КИСЛОтЫ п0 первой ступени,
Количество добавленного Расположена на восходящих
основания, г-экб/л ветвях кондуктометрическои
кривой. Излом кривой в этой
точке может иметь место, если
подвижности ионов НАп~ и
An2- сильно отличаются.
Однако подвижности анионов
(кроме ОН-) в общем мало
отличаются и первый излом или
очень слабо выражен или совсем не обнаруживается.
Обычно кривая титрования имеет один излом,
соответствующий полной нейтрализации кислоты. Такая форма
кривой характерна для случая титрования адипиновой
кислоты (р#а1 = 4,42; р/Са2 = 5,28, рис. 6, кривая 3). В
начале титрования на кривой имеется минимум (так как
р/Са^б), после которого электропроводность увеличи-
Рис. 6. Кривые кондукто-
метрического
титрования 0,1 н. растворов
двухосновных кислот
сильным основанием:
/ — хромовой; 2—
малеиновой; 3 — адипняовой
34
вается. Излом кривой фиксирует полную нейтрализацию
кислоты.
Кондуктометрическое титрование может быть
использовано и при определении трехосновных кислот,
например фосфорной кислоты. Показатели констант
диссоциации этой кислоты по первой, второй и третьей ступени
имеют значения соответственно 2,12, 7,21 и 12,67. При
титровании фосфорной кислоты раствором щелочи
сначала нейтрализуется кислота, диссоциирующая по
первой ступени (первый кислотный эквивалент). При этом
образуются ионы Н2РО4-, и электропроводность
раствора понижается. При титровании 0,1 и. раствора кислоты
кривая титрования до первого излома слегка изогнута,
так как кислота неполностью диссоциирует по первой
ступени. При титровании более разбавленных растворов
степень диссоциации кислоты увеличивается и
электропроводность понижается линейно. Первая точка
эквивалентности фиксируется резким изломом
кондуктометрическои кривой.
Нейтрализация кислоты по второй ступени вызывает
повышение электропроводности раствора, что
характерно для нейтрализации слабых кислотных групп. В
процессе реакции нейтрализации Н2Р04~-ионы переходят в
НР042-ионы. Эта реакция протекает количественно.
Однако ее окончание не фиксируется четким изломом
кондуктометрическои кривой, так как подвижности Н2РО4--
ионов и образующихся затем НР042~-ионов мало
отличаются друг от друга. Кроме того, указанные ионы
частично гидролизуются ионами воды. При последующем
приливании щелочи нейтрализация кислоты по третьей
ступени не протекает количественно вследствие ее
обратимости в результате взаимодействия Р043_-ионов с
ионами водорода. Поэтому при определении фосфорной
кислоты используют первый излом кондуктометрическои
кривой, соответствующий нейтрализации кислоты до
Н2Р04_-ИОНОВ.
Титрование кислот слабым основанием. При
кондуктометрическои титровании кислот могут быть
использованы слабые основания. При нейтрализации сильной (а)
и слабой кислот (б) слабыми основаниями реакции
протекают по схемам:
а) Н+ + An- + КЮН --= Kt+ + An- + H2Q
б) HAn + KtOH «i Kt+ + An" + HaQ
Г Ш
В случаях, когда гидролиз получающихся при
нейтрализации солей незначителен, характер изменения
концентраций ионов и электропроводности раствора при
титровании до точки эквивалентности аналогичен
рассмотренным вышз случаям нейтрализации кислот
сильными основаниями. Отличие заключается в том, что при
титровании слабыми
основаниями электропроводность
раствора после точки
эквивалентности мало изменяется, так как
основание слабо диссоцирует.
Для рассматриваемых
случаев также имеются типичные
формы кривых титрования.
Рассмотрим процессы
нейтрализации 0,1 н. растворов
кислот слабым основанием (р/Сь =
= 5). При этом гидролиз соли
не оказывает влияния на
изменение электропроводности
раствора. На рис. 7 показана
кривая титрования сильной
кислоты слабым основанием
(кривая 1). Если в качестве титран-
та используют более слабые
основания, излом кривой вблизи
точки эквивалентности
закругляется вследствие обратимости
реакции. Основания с р/Сь>10
не могут быть использованы
при титровании 0,1 н.
растворов сильных кислот, так как
закругление кривой мешает
точному установлению точки
эквивалентности. При
нейтрализации 0,01 и 0,001 н.
растворов кислот границы р/Сь
оснований, которые позволяют
проводить определение с достаточно высокой точностью,
снижаются соответственно до 9 и 8: Условия определения
точки эквивалентности по сравнению с кривыми
титрования сильных кислот сильными основаниями менее
благоприятны, так как угол излома кривой в точке
эквивалентности становится тупым.
2 -
I-
г.
о;
х_
2 !
-И
3
А
0 0,1 0,2
Количества добавленного
основания, г-гкв/л
Рис. 7. Кривые кондуктомет-
рического титрования 0,1 н.
растворов кислот различной
силы слабым основанием
(р/Сь = 5):
/-сильная; 2 — рКа =3; 3 —
РК „-5
36
При титровании кислот средней силы
электропроводность раствора до точки эквивалентности нелинейно
понижается или на кривой появляется минимум, не
имеющий аналитического значения, как при нейтрализации
кислот сильными основаниями. Отличие в ходе кривых
наблюдается только после точки эквивалентности, так
как электропроводность остается постоянной (рис. 7,
кривая 2).
Третий тип кривых характерен для нейтрализации
слабых кислот. Электропроводность раствора до точки
эквивалентности линейно повышается, а затем остается
постоянной (рис. 7, кривая 3). Процессы нейтрализации
слабых кислот слабыми основаниями имеют следующую
характерную особенность: концентрации водородных и
гидроксильных ионов в продолжение всего процесса
титрования настолько малы, что не могут оказывать
влияния на электропроводность раствора. Изменение
электропроводности раствора вызывается только увеличением
концентраций анионов кислоты и катионов основания.
Поэтому, чем выше подвижности этих ионов, тем сильнее
увеличивается электропроводность раствора до точки
эквивалентности и тем меньше угол излома.
При титровании кислот средней силы и слабых кислот
слабыми основаниями создаются более благоприятные
условия для анализа, чем при титровании сильными
основаниями. На рис. 7 (кривые 2 и 3) показан ход
титрования кислоты средней силы (р/Са = 3) и слабой (р/Са = 5)
слабым основанием (р/Сь = 5). Излом кривых резко
выражен и точки эквивалентности устанавливаются с
высокой точностью.
При титровании слабыми основаниями в результате
гидролиза получающихся солей происходит закругление
кривых около точки эквивалентности. Критерием
возможности кондуктометрического определения кислоты может
служить сумма р/Са кислоты и р/Сь основания, так как
изменение значений р/Са и рКъ в пределах этой суммы не
вызывает существенного изменения степени гидролиза
соли при данной концентрации. Независимо от
концентрации титруемой кислоты кондуктометрические
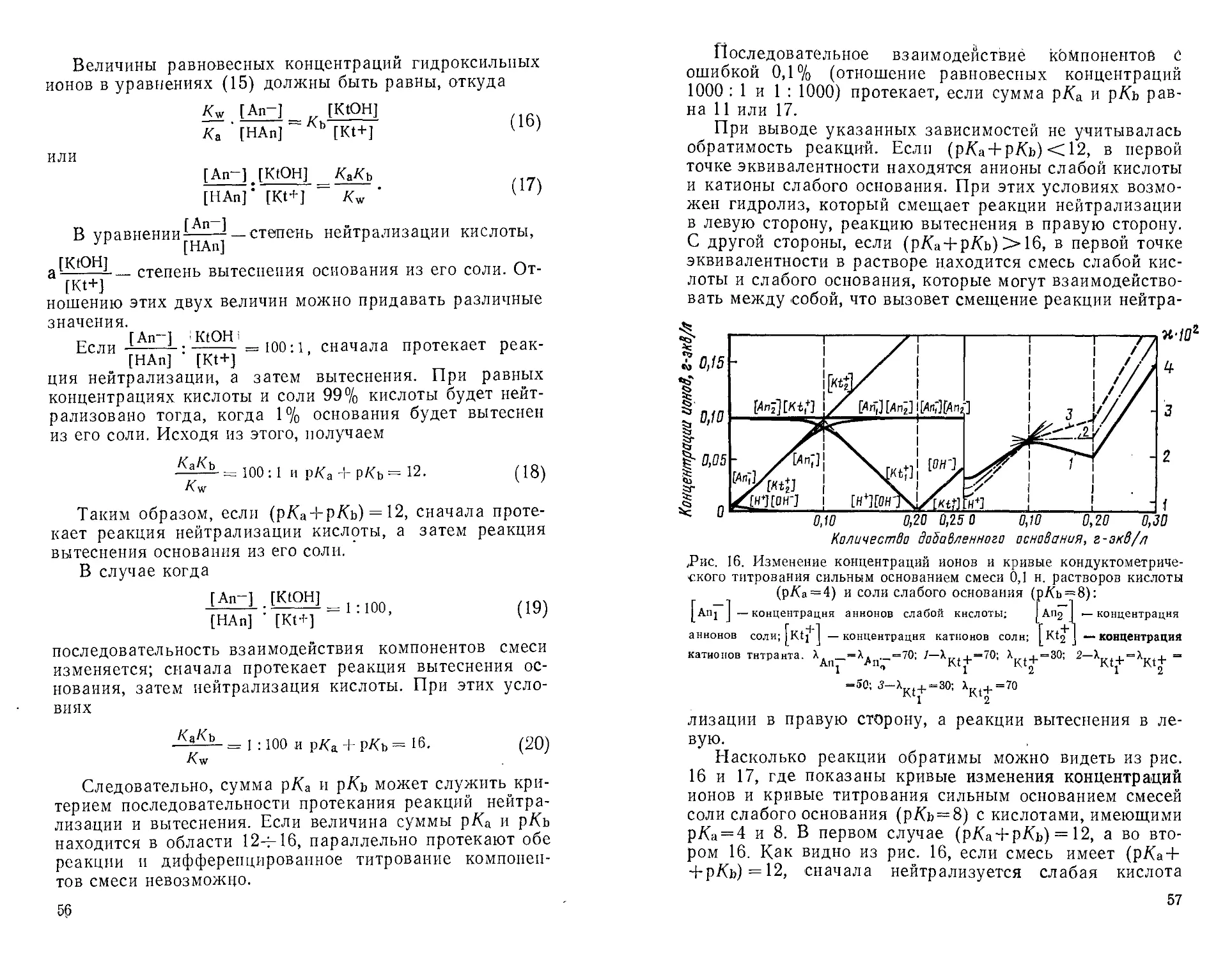
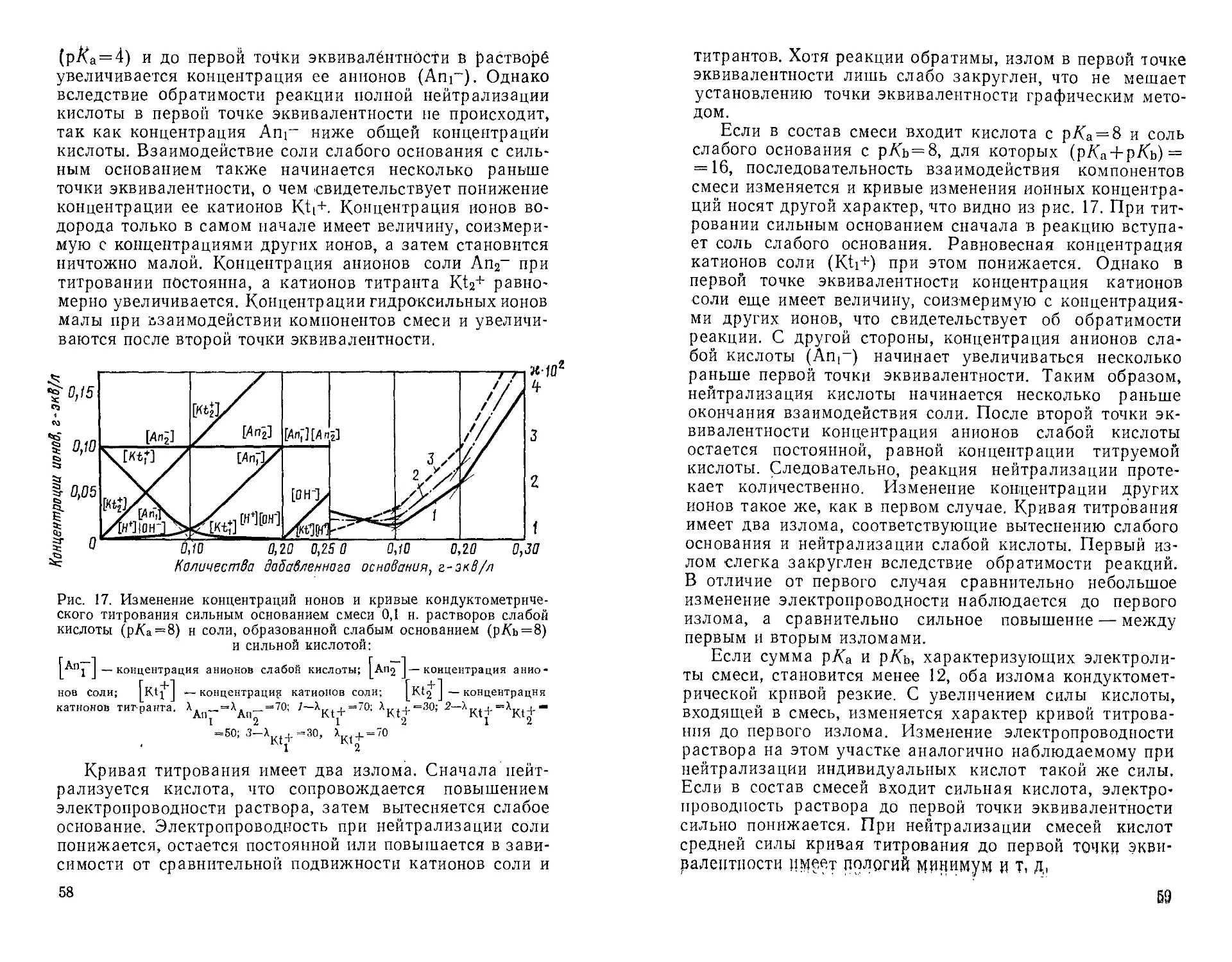
определения возможны в тех случаях, когда (р/Са + р/Сь) ** 12.
Дальнейшее увеличение суммы р/Са и р/Сь приводит к
изгибу всей кондуктометрической Кривой и определение
становится невозможным.
,37
§ 2. Титрование оснований
Титрование оснований сильными кислотами. Условия
кондуктометрического титрования оснований сильными
кислотами в общем аналогичны рассмотренным выше
условиям титрования кислот сильными основаниями (см.
§ О-
Реакции нейтрализации сильных и слабых основании
можно выразить схемами уравнений:
Kt+ + ОН- + Н+ + An- = Kt+ + An- + Н20
KtOH + Н+ + An- = Kt+ + An- + H20
Ход изменения кривых концентраций ионов при
титровании 0,1 н. растворов оснований различной силы носит
такой же характер, как и при титровании 0,1 н.
растворов кислот такой же силы. Причем все рассуждения в
отношении концентрации водородных ионов следует
отнести к концентрациям гидроксильных ионов и, наоборот,
рассуждения, касающиеся концентрации анионов
определяемой кислоты,— к концентрации катионов титруемого
основания, а концентрации катионов титранта —к
концентрации анионов кислоты, применяемой в процессе
нейтрализации основания. Однако кривые титрования
оснований и кривые титрования кислот такой же силы
несколько отличаются друг от друга.
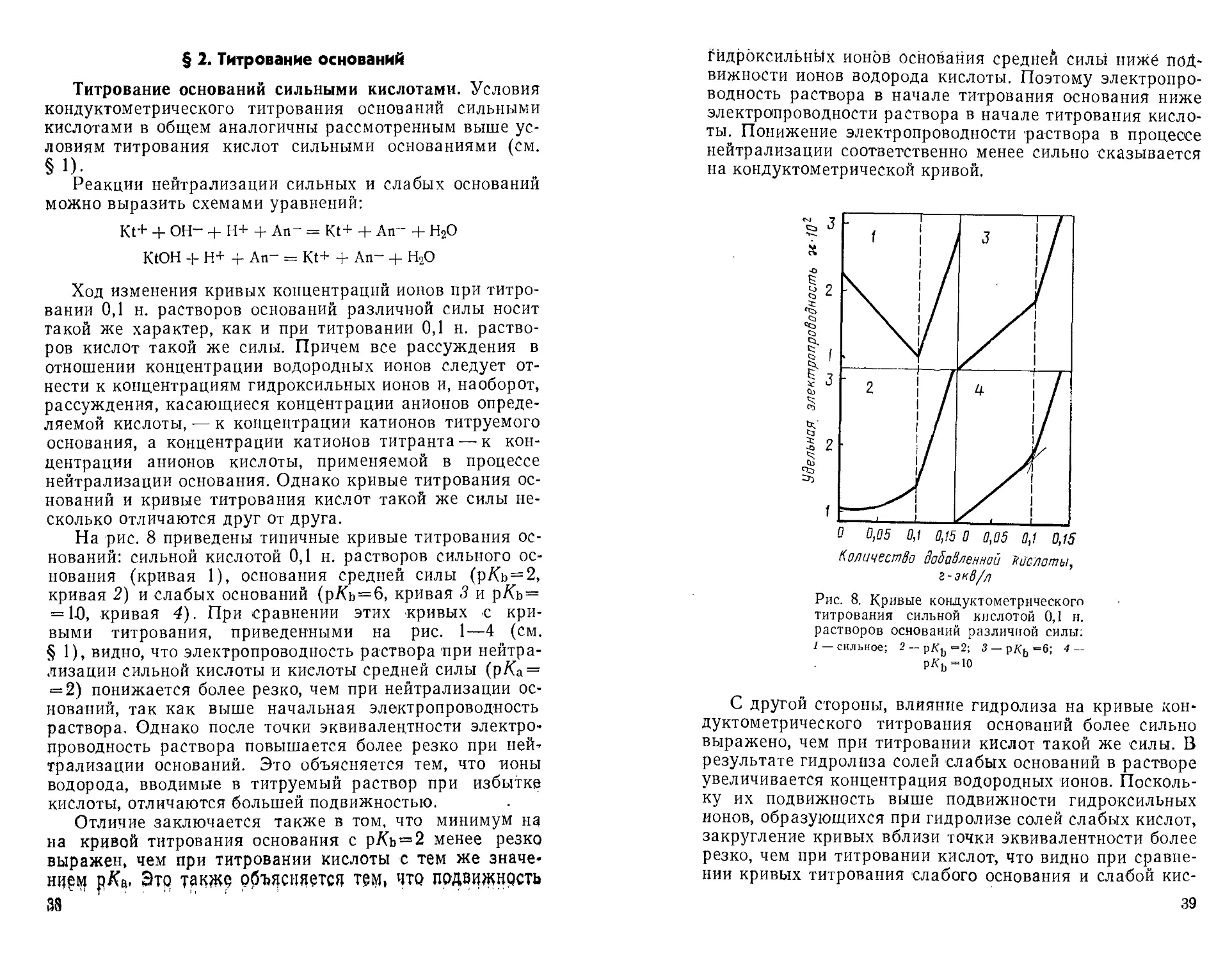
На рис 8 приведены типичные кривые титрования
оснований: сильной кислотой 0,1 н. растворов сильное
основания (кривая 1), основания средней силы (рль-2,
кривая 2) и слабых оснований (р/Сь=6, кривая S и рЛь-
= 10, кривая 4). При сравнении этих кривых с
кривыми титрования, приведенными на рис. 1— 4 ^ (см.
§ 1), видно, что электропроводность раствора при
нейтрализации сильной кислоты и кислоты средней силы (рЛа-
= 2) понижается более резко, чем при нейтрализации
оснований, так как выше начальная электропроводность
раствора. Однако после точки эквивалентности
электропроводность раствора повышается более резко при
нейтрализации оснований. Это объясняется тем, что ионы
водорода, вводимые в титруемый раствор при избытке
кислоты, отличаются большей подвижностью.
Отличие заключается также в том, что минимум на
на кривой титрования основания с р/Сь = 2 менее резко
выражен, чем при титровании кислоты с тем же значе-
ни|м рДа, Это такж9 о$щстт тем, что подвижнрсть
Гидроксильных ионов основания средней силы ниже
подвижности ионов водорода кислоты. Поэтому
электропроводность раствора в начале титрования основания ниже
электропроводности раствора в начале титрования
кислоты. Понижение электропроводности раствора в процессе
нейтрализации соответственно менее сильно сказывается
на кондуктометрической кривой.
0 0,05 0,1 0,15 0 0,05 0,1 0,15
Количество добавленной кислоты,
г-экв/л
Рис. 8. Кривые кондуктометрического
титрования сильной кислотой 0,1 н.
растворов оснований различной силы:
/ — сильное; 2 — рКъ=2; 3 — рКь = 6; 4 —
ркь-ю
С другой стороны, влияние гидролиза на кривые
кондуктометрического титрования оснований более сильно
выражено, чем при титровании кислот такой же силы. В
результате гидролиза солей слабых оснований в растворе
увеличивается концентрация водородных ионов.
Поскольку их подвижность выше подвижности гидроксильных
ионов, образующихся при гидролизе солей слабых кислот,
закругление кривых вблизи точки эквивалентности более
резко, чем при титровании кислот, что видно при
сравнении кривых титрования слабого основания и слабой кис-
39
лоты, характеризующихся значением р/С= 10 (ср. рис. 4 и
рис. 8).
Границы рД'ь оснований, допускающие их
количественные определения в растворе при различных
концентрациях, совпадают с ранее указанными границами р/Са для
кислот.
Условия кондуктометрического титрования
двухосновных оснований аналогичны рассмотренным выше
условиям титрования двухосновных кислот.
Титрование оснований слабыми кислотами. Слабые
кислоты также могут служить реаггнтами при кондукто-
метрическом титровании оснований. Реакции
нейтрализации сильного и слабого основания слабой кислотой можно
представить схематически:
Kt+ + ОН- + НАп = Kt+ + An- + Н20
КЮН + НАп = Kt+ + An- + Н20
Типичные кривые титрования слабыми кислотами
оснований различной силы до точки эквивалентности
аналогичны кривым титрования, полученным при
использовании сильных кислот. Отличие заключается в том, что
после точки эквивалентности электропроводность не
изменяется. Значения констант диссоциации слабых
электролитов, лимитирующие возможности определения,
совпадают с данными, приведенными для случаев титрования
кислот. Определения возможны, если сумма рКъ
титруемого основания и р/Са кислоты, используемой для
нейтрализации, меньше или равна 12.
Слабые кислоты используются преимущественно при
кондуктометрическом титровании слабых оснований, так
как при этом угол излома кривой в точке
эквивалентности более острый.
§ 3. Титрование солей
Титрование солей слабых кислот. Титрование солей
слабых одноосновных кислот основано на реакции
вытеснения слабой кислоты сильной кислотой:
Kt+ + Aivf + Н+ + Aaf = Kt+ + Aaf + НАгц
Чем слабее кислота, образующая соль, тем более
полно протекает реакция вытеснения.
Форма кривых кондуктометрического титрования
солей зависит от степени их гидролиза, так как при этом из-
40
меняются величины равновесных концентраций анионов
соли, а также [Н+] и [ОН-]. Поскольку степень гидролиза
солей зависит от концентрации, формы кондуктометриче-
ских кривых титрования изменяются с разбавлением
раствора. Существенное влияние на электропроводность
раствора при титровании солей слабых кислот,
подвергающихся гидролитическому расщеплению, оказывают
гидроксильные ионы.
При вытеснении слабой кислоты из ее соли в растворе
уменьшается равновесная концентрация аниона соли.
Вместо этих ионов при титровании в растворе
накапливаются анионы сильной кислоты, которая вступает в
реакцию. Поэтому в случаях, когда гидролиз соли
незначителен и не оказывает существенного влияния на характер
кондуктометрических кривых титрования, а реакция
протекает количественно, изменение электропроводности
раствора до точки эквивалентности зависит от
сравнительных подвижно'стей анионов титруемой соли и
заменяющих их в растворе анионов сильной кислоты.
Если кислота, образующая соль, недостаточно слабая,
реакция вытеснения обратима. В этом случае
наблюдается изменение характера кондуктометрической кривой
вблизи точки эквивалентности. Обратимость реакции
вызывает увеличение электропроводности раствора и излом
кривой закругляется.
Исходя из этих соображений, можно выделить три
типа кривых титрования солей. К первому типу относятся
кривые титрования солей, на которые оказывает влияние
гидролиз (степень гидролиза больше 1%). Второй тип
представляют кривые титрования солей, гидролизующих-
ся незначительно (степень гидролиза менее 1%) и
образованных анионами слабых кислот, которые практически
количественно вытесняются. Третий тип — кривые
титрования солей, для которых ргакция вытеснения
обратима.
Для одной и той же соли степень гидролиза и степень
обратимости реакции вытеснения слабой кислоты
зависят от концентрации. Поэтому типичные кривые
титрования для солей, образованных анионами кислот различной
силы, следует сравнивать при постоянной
концентрации титруемой соли. При титровании более
концентрированных растворов возможности кондуктометрических
определений расширяются. Рассмотрим условия титрования
0,1 н. растворов солей.
41
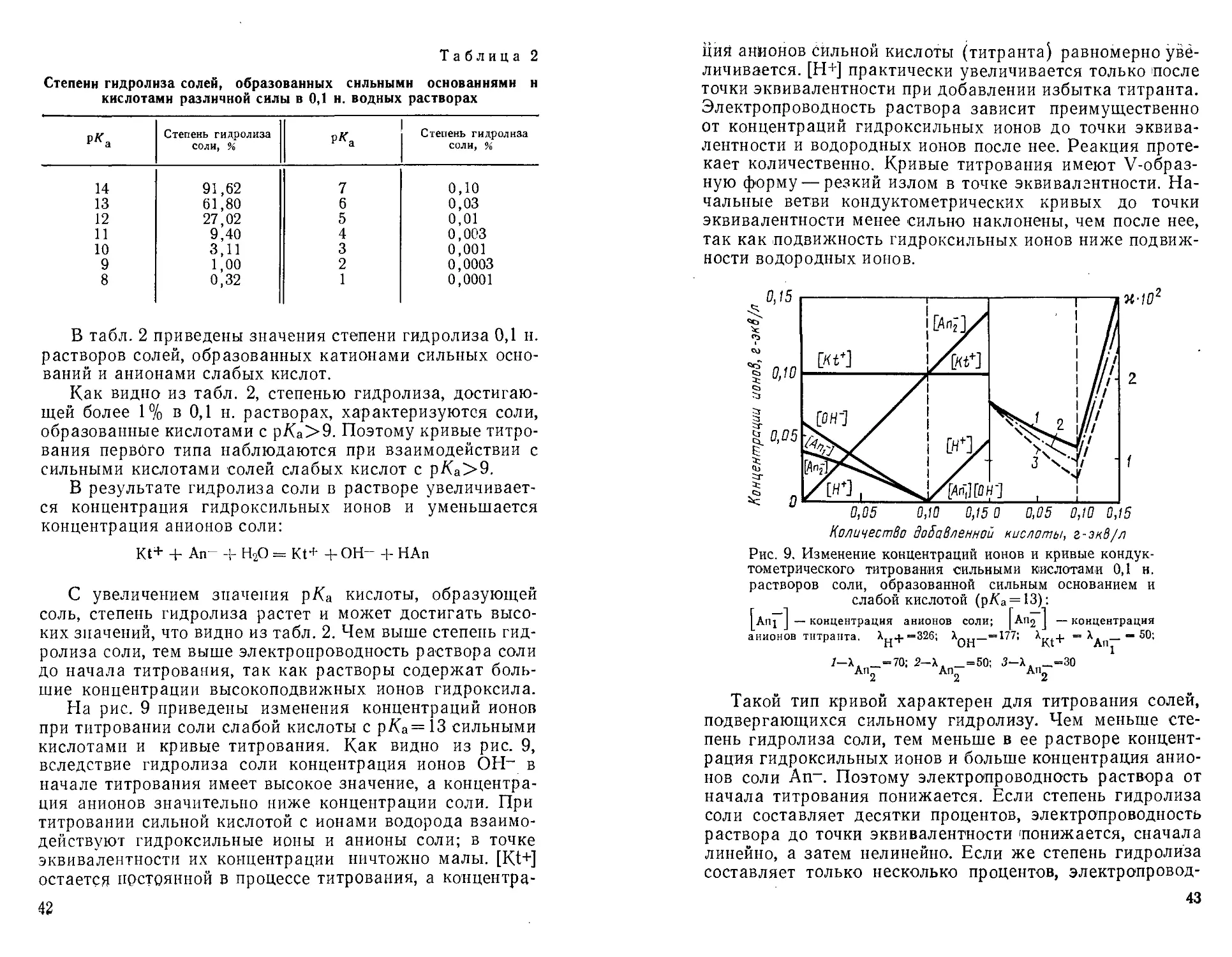
Таблица 2
Степени гидролиза солей, образованных сильными основаниями н
кислотами различной силы в 0,1 н. водных растворах
РК
v а
14
13
12
11
10
9
8
Степень гидролиза
соли, %
91,62
61,80
27,02
9,40
3,11
1,00
0,32
рк»
7
6
5
4
3
2
1
Степень гидролиза
соли, %
0,10
0,03
0,01
0,003
0,001
0,0003
0,0001
В табл. 2 приведены значения степени гидролиза 0,1 н.
растворов солей, образованных катионами сильных
оснований и анионами слабых кислот.
Как видно из табл. 2, степенью гидролиза,
достигающей более 1% в 0,1 н. растворах, характеризуются соли,
образованные кислотами с р/Са>9. Поэтому кривые
титрования первого типа наблюдаются при взаимодействии с
сильными кислотами солей слабых кислот с р/Са>9.
В результате гидролиза соли в растворе
увеличивается концентрация гидроксильиых ионов и уменьшается
концентрация анионов соли:
Kt+ + An- + Н20 = Kt+ + ОН- + НАп
С увеличением значения р/Са кислоты, образующей
соль, степень гидролиза растет и может достигать
высоких значений, что видно из табл. 2. Чем выше степень
гидролиза соли, тем выше электропроводность раствора соли
до начала титрования, так как растворы содержат
большие концентрации высокоподвижных ионов гидроксила.
На рис. 9 приведены изменения концентраций ионов
при титровании соли слабой кислоты с р/Са=13 сильными
кислотами и кривые титрования. Как видно из рис. 9,
вследствие гидролиза соли концентрация ионов ОН- в
начале титрования имеет высокое значение, а
концентрация анионов значительно ниже концентрации соли. При
титровании сильной кислотой с ионами водорода
взаимодействуют гидроксильные ионы и анионы соли; в точке
эквивалентности их концентрации ничтожно малы. [Kt+]
остается постоянной в процессе титрования, а кон центр а-
42
ция анионов сильной кислоты (титранта) равномерно
увеличивается. [Н+] практически увеличивается только после
точки эквивалентности при добавлении избытка титранта.
Электропроводность раствора зависит преимущественно
от концентраций гидроксильиых ионов до точки
эквивалентности и водородных ионов после нее. Реакция
протекает количественно. Кривые титрования имеют V-образ-
ную форму — резкий излом в точке эквивалгнтности.
Начальные ветви кондуктометрических кривых до точки
эквивалентности менее сильно наклонены, чем после нее,
так как подвижность гидроксильиых ионов ниже
подвижности водородных ионов.
Количество добавленной кислоты, г-экВ/л
Рис. 9. Изменение концентраций ионов и кривые кондук-
тометрического титрования сильными кислотами 0,1 н.
растворов соли, образованной сильным основанием и
слабой кислотой (р/(а = 13):
Ani I — концентрация анионов соли; I Ап2 J — концентрация
анионов титранта. X + =326; Х_ —=177; ^Kt+ = X — 50;
1-Х, _=70; 2-Х, _=50; 3-Х, _=30
Ап2 Ап2 Ап2
Такой тип кривой характерен для титрования солей,
подвергающихся сильному гидролизу. Чем меньше
степень гидролиза соли, тем меньше в ее растворе
концентрация гидроксильиых ионов и больше концентрация
анионов соли An-. Поэтому электропроводность раствора от
начала титрования понижается. Если степень гидролиза
соли составляет десятки процентов, электропроводность
раствора до точки эквивалентности понижается, сначала
линейно, а затем нелинейно. Если же степень гидролиза
составляет только несколько процентов, электропровод-
43
Hoci'b снижается только в начале титрования.
Дальнейший характер измэнения электропроводности зависит от
сравнительной подвижности анионов титруемой соли и
заменяющих их анионов титранта.
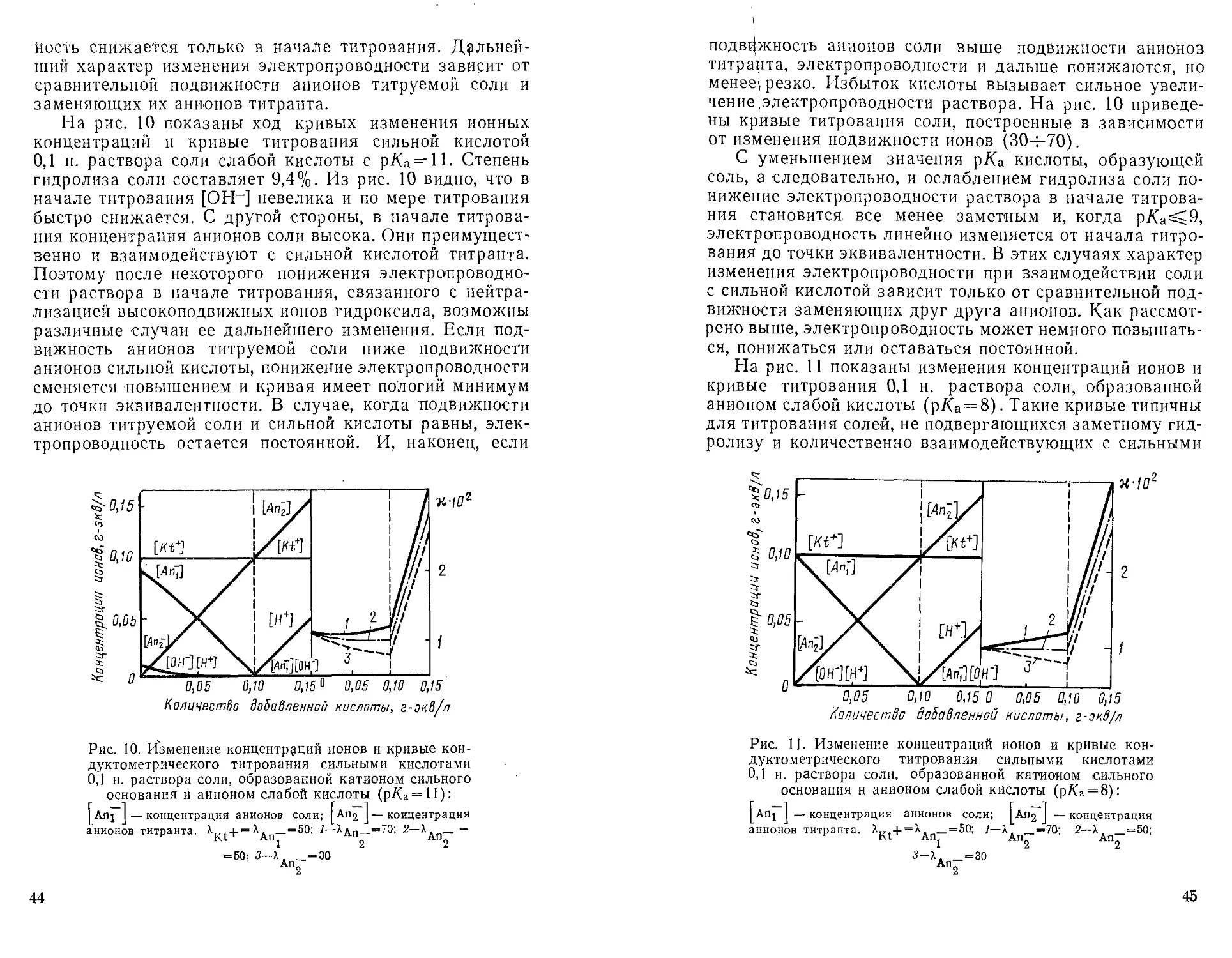
На рис. 10 показаны ход кривых изменения ионных
концентраций и кривые титрования сильной кислотой
0,1 н. раствора соли слабой кислоты с р/Са=П. Степень
гидролиза соли составляет 9,4%. Из рис. 10 видно, что в
начале титрования [ОН-] невелика и по мере титрования
быстро снижается. С другой стороны, в начале
титрования концентрация анионов соли высока. Они
преимущественно и взаимодействуют с сильной кислотой титранта.
Поэтому после некоторого понижения
электропроводности раствора в начале титрования, связанного с
нейтрализацией высокоподвижных ионов гидроксила, возможны
различные случаи ее дальнейшего изменения. Если
подвижность анионов титруемой соли ниже подвижности
анионов сильной кислоты, понижение электропроводности
сменяется повышением и кривая имеет пологий минимум
до точки эквивалентности. В случае, когда подвижности
анионов титруемой соли и сильной кислоты равны,
электропроводность остается постоянной. И, наконец, если
Количество добавленной кислоты, г-эк8/л
Рис. 10. Изменение концентраций ионов н кривые кон-
дуктометрического титрования сильными кислотами
0,1 н. раствора соли, образованной катионом сильного
основания и анионом слабой кислоты (р/Са = И):
Anj I — концентрация анионов соли; |АП2 J — концентрация
анионов титранта. X , + = ^дп—= 50; 7—Хдп_=70; 2—Хдп— ~
= 50; 3-Х, _ = 30
АП2
44
подвижность анионов соли выше подвижности анионов
титранта, электропроводности и дальше понижаются, но
менее', резко. Избыток кислоты вызывает сильное
увеличение электропроводности раствора. На рис. 10
приведены кривые титрования соли, построенные в зависимости
от изменения подвижности ионов (30-^-70).
С уменьшением значения р/Са кислоты, образующей
соль, а следовательно, и ослаблением гидролиза соли
понижение электропроводности раствора в начале
титрования становится, все менее заметным и, когда р/Са^9,
электропроводность линейно изменяется от начала
титрования до точки эквивалентности. В этих случаях характер
изменения электропроводности при взаимодействии соли
с сильной кислотой зависит только от сравнительной
подвижности заменяющих друг друга анионов. Как
рассмотрено выше, электропроводность может немного
повышаться, понижаться или оставаться постоянной.
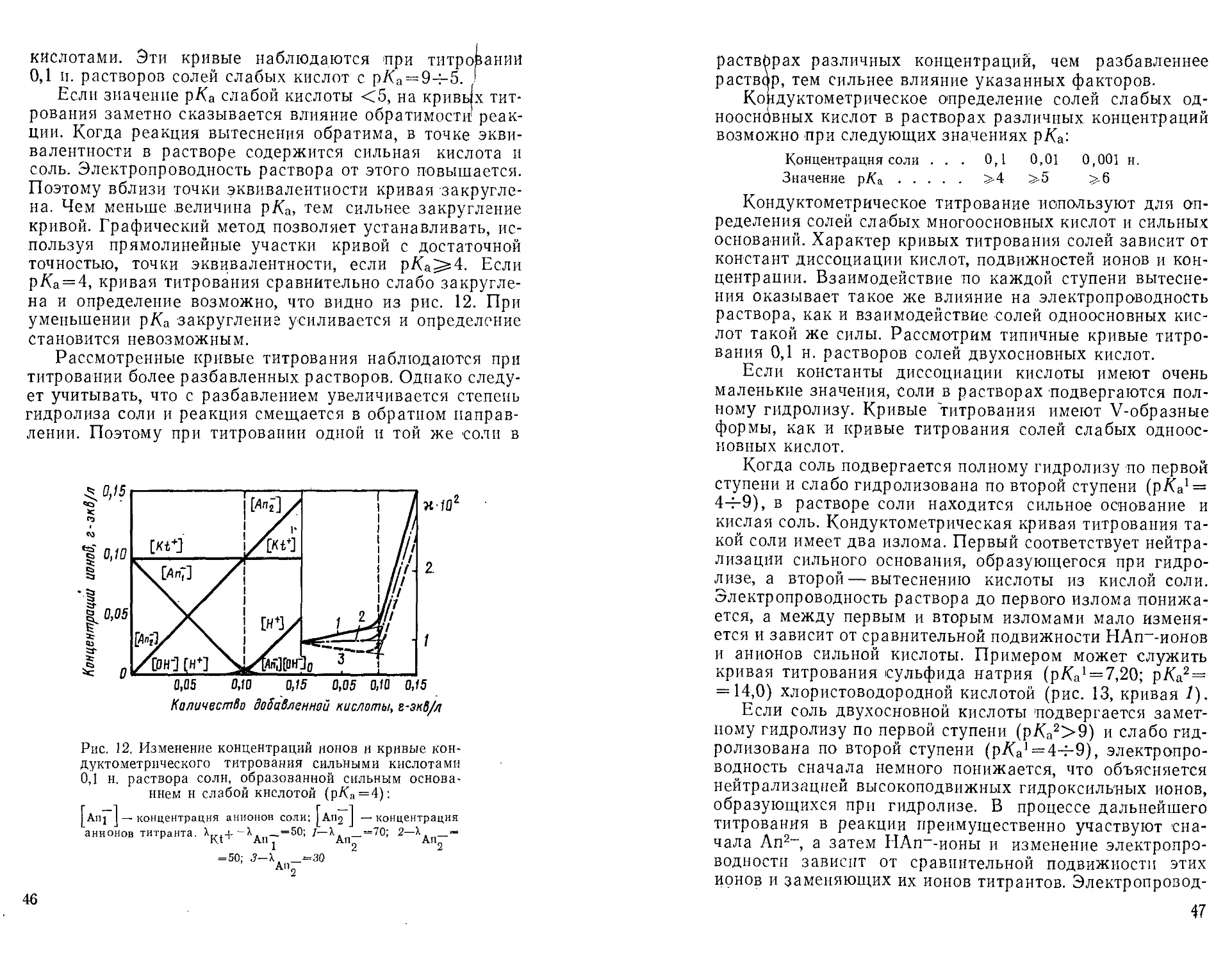
На рис. 11 показаны изменения концентраций ионов и
кривые титрования 0,1 н. раствора соли, образованной
анионом слабой кислоты (р/Са = 8). Такие кривые типичны
для титрования солей, не подвергающихся заметному
гидролизу и количественно взаимодействующих с сильными
ХоличестВо добавленной ни слоты, г-экд/л
Рис. 11. Изменение концентраций ионов и кривые кон-
дуктометрического титрования сильными кислотами
0,1 н. раствора соли, образованной катионом сильного
основания н анионом слабой кислоты (р/(а = 8):
Anj I — концентрация анионов соли; I кп^ I — концентрация
анионов титранта. Х„, + = Х _=50; 1—X, _=70; 2—X. =50;
KtT An An An~
3-1. _ = 30
An2
45
кислотами. Эти кривые наблюдаются при титровании
0,1 п. растворов солей слабых кислот с рКа = 9ч-5. I
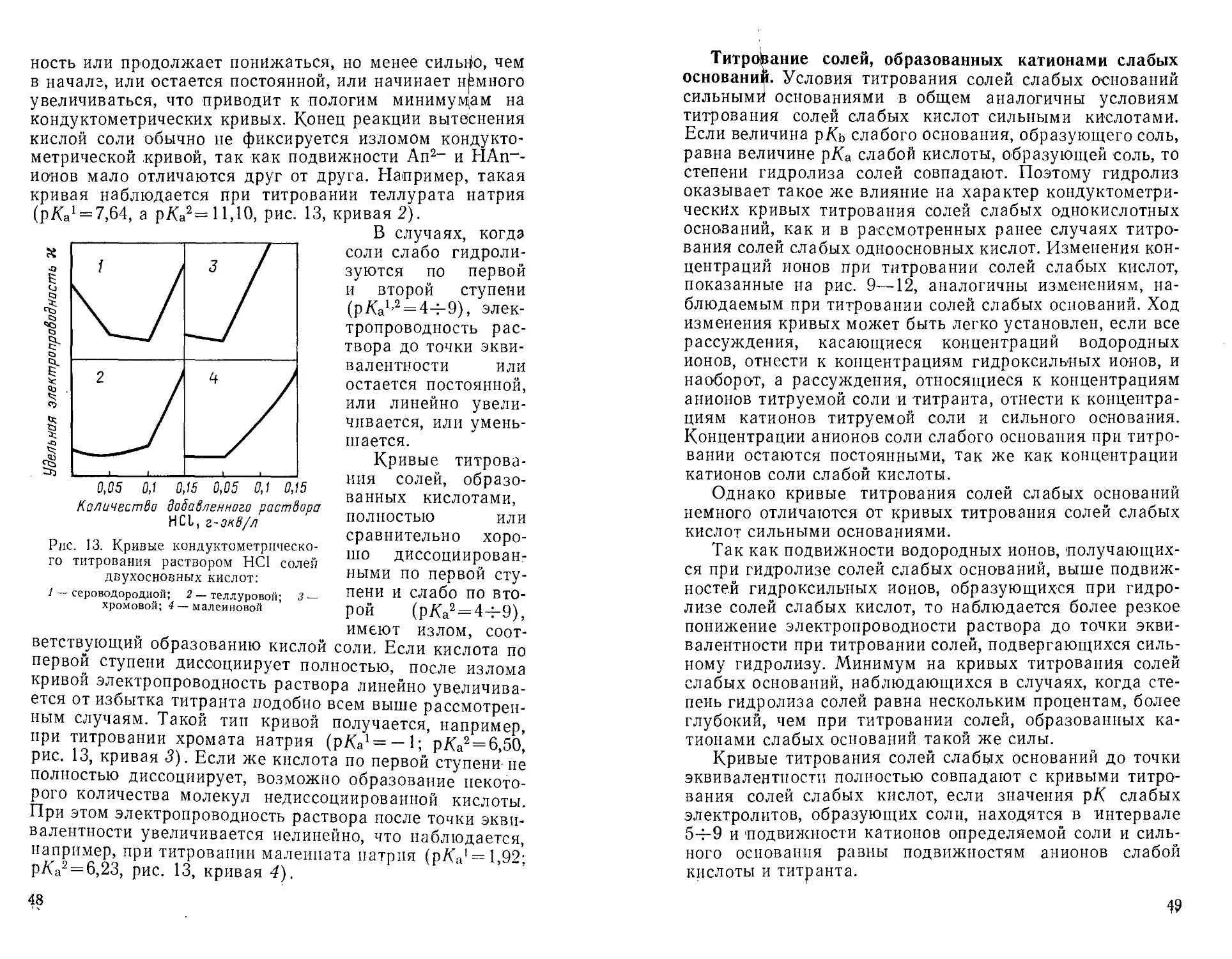
Если значение р/Са слабой кислоты <5, на кривых
титрования заметно сказывается влияние обратимости'
реакции. Когда реакция вытеснения обратима, в точке
эквивалентности в растворе содержится сильная кислота и
соль. Электропроводность раствора от этого повышается.
Поэтому вблизи точки эквивалентности кривая
закруглена. Чем меньше величина р/Са, тем сильнее закругление
кривой. Графический метод позволяет устанавливать,
используя прямолинейные участки кривой с достаточной
точностью, точки эквивалентности, если р/(а^4. Если
р/Са = 4, кривая титрования сравнительно слабо
закруглена и определение возможно, что видно из рис. 12. При
уменьшении р/Са закругление усиливается и определение
становится невозможным.
Рассмотренные кривые титрования наблюдаются при
титровании более разбавленных растворов. Однако
следует учитывать, что с разбавлением увеличивается степень
гидролиза соли и реакция смещается в обратном
направлении. Поэтому при титровании одной и той же соли в
H'Wi
0,05 0,Ш 0,15 0,05 0,10 0,15
Количество добавленной кислоты, е-экв/л
Рис. 12. Изменение концентраций ионов и кривые кон-
дуктометрцческого титрования сильными кислотами
0,1 н. раствора солн, образованной сильным
основанием н слабой кислотой (р-ЛГа =4):
[аП1 ]-,
■ концентрация а
анионов титранта. X
нионов соли; Ап2 I — концентрация
Kt
+ -ХА _-50; 1-
т An,
= 50: 3-
1
"ХАп-
Ап"
-70; 2-Х,
46
растворах различных концентраций, чем разбавленнее
раствор, тем сильнее влияние указанных факторов.
Кондуктометрическое определение солей слабых
одноосновных кислот в растворах различных концентраций
возможно при следующих значениях р/Са:
Концентрация соли . . . 0,1 0,01 0,001 и.
Значение р/(а >4 >5 >6
Кондуктометрическое титрование используют для
определения солей слабых многоосновных кислот и сильных
оснований. Характер кривых титрования солей зависит от
констант диссоциации кислот, подвижностей ионов и
концентрации. Взаимодействие по каждой ступени
вытеснения оказывает такое же влияние на электропроводность
раствора, как и взаимодействие солей одноосновных
кислот такой же силы. Рассмотрим типичные кривые
титрования 0,1 н. растворов солей двухосновных кислот.
Если константы диссоциации кислоты имеют очень
маленькие значения, соли в растворах подвергаются
полному гидролизу. Кривые "титрования имеют V-образные
формы, как и кривые титрования солей слабых
одноосновных кислот.
Когда соль подвергается полному гидролизу по первой
ступени и слабо гидролизована по второй ступени (рЛУ =
4-=-9), в растворе соли находится сильное основание и
кислая соль. Кондуктометрическая кривая титрования
такой соли имеет два излома. Первый соответствует
нейтрализации сильного основания, образующегося при
гидролизе, а второй — вытеснению кислоты из кислой соли.
Электропроводность раствора до первого излома
понижается, а между первым и вторым изломами мало
изменяется и зависит от сравнительной подвижности НАп~-ионов
и анионов сильной кислоты. Примером может служить
кривая титрования сульфида натрия (р/(а1 = 7,20; р/Са2 =
= 14,0) хлористоводородной кислотой (рис. 13, кривая 1).
Если соль двухосновной кислоты подвергается замет-
пому гидролизу по первой ступени (р/Са2>9) и слабо
гидролизована по второй ступени (р^Са1 = 4ч-9),
электропроводность сначала немного понижается, что объясняется
нейтрализацией высокоподвижных гидроксильных ионов,
образующихся при гидролизе. В процессе дальнейшего
титрования в реакции преимущественно участвуют
сначала An2-, а затем НАп~-ионы и изменение
электропроводности зависит от сравнительной подвижности этих
ионов и заменяющих их ионов титрантов. Электропрозод-
М
ность или продолжает понижаться, но менее сильно, чем
в началг, или остается постоянной, или начинает намного
увеличиваться, что приводит к пологим минимумам на
кондуктометрических кривых. Конец реакции вытеснения
кислой соли обычно не фиксируется изломом кондукто-
метрической кривой, так как подвижности Ап2_ и НАп--
ионов мало отличаются друг от друга. Например, такая
кривая наблюдается при титровании теллурата натрия
(p/fai = 7,64, а р/Са2= 11,10, рис. 13, кривая 2).
В случаях, когда
соли слабо гидроли-
зуются по первой
и второй ступени
(р/Са1-2 = 4-=-9),
электропроводность
раствора до точки
эквивалентности или
остается постоянной,
или линейно
увеличивается, или
уменьшается.
Кривые
титрования солей,
образованных кислотами,
полностью или
сравнительно
хорошо
диссоциированными по первой
ступени и слабо по
второй (рКа2 = 4^9),
имеют излом,
соответствующий образованию кислой соли. Если кислота по
первой ступени диссоциирует полностью, после излома
кривой электропроводность раствора линейно
увеличивается от избытка титранта подобно всем выше
рассмотренным случаям. Такой тип кривой получается, например
при титровании хромата натрия (р/Са1 = — 1; ptfa2 = 6,5o!
рис. 13, кривая 3). Если же кислота по первой ступени не
полностью диссоциирует, возможно образование
некоторого количества молекул недиссоциированной кислоты.
При этом электропроводность раствора после точки
эквивалентности увеличивается нелинейно, что наблюдается,
например, при титровании малеииата натрия (pAV = l 99-
рД'а2 = 6,23, рис. 13, кривая 4). ' "'
■5
I
35
<J
2 У
i i
J /
ч у
, f
0,05 0,1 0,15 0,05 0,1 0,15
Количество добавленного раствора
НС1, г-экв/л
Рис. 13. Кривые кондуктометрическо-
го титрования раствором НС1 солей
двухосновных кислот:
/ — сероводородной; 2 — теллуровой; 3 —
хромовой; 4 — малеиновой
48
Титрование солей, образованных катионами слабых
оснований. Условия титрования солей слабых оснований
сильными основаниями в общем аналогичны условиям
титрования солей слабых кислот сильными кислотами.
Если величина р/Сь слабого основания, образующего соль,
равна величине р/Са слабой кислоты, образующей соль, то
степени гидролиза солей совпадают. Поэтому гидролиз
оказывает такое же влияние на характер
кондуктометрических кривых титрования солей слабых однокислотных
оснований, как и в рассмотренных ранее случаях
титрования солей слабых одноосновных кислот. Изменения
концентраций ионов при титровании солей слабых кислот,
показанные на рис. 9—12, аналогичны изменениям,
наблюдаемым при титровании солей слабых оснований. Ход
изменения кривых может быть легко установлен, если все
рассуждения, касающиеся концентраций водородных
ионов, отнести к концентрациям гидроксильных ионов, и
наоборот, а рассуждения, относящиеся к концентрациям
анионов титруемой соли и титранта, отнести к
концентрациям катионов титруемой соли и сильного основания.
Концентрации анионов соли слабого основания при
титровании остаются постоянными, так же как концентрации
катионов соли слабой кислоты.
Однако кривые титрования солей слабых оснований
немного отличаются от кривых титрования солей слабых
кислот сильными основаниями.
Так как подвижности водородных ионов,
'получающихся при гидролизе солей слабых оснований, выше подвиж-
ностей гидроксильных ионов, образующихся при
гидролизе солей слабых кислот, то наблюдается более резкое
понижение электропроводности раствора до точки
эквивалентности при титровании солей, подвергающихся
сильному гидролизу. Минимум на кривых титрования солей
слабых оснований, наблюдающихся в случаях, когда
степень гидролиза солей равна нескольким процентам, более
глубокий, чем при титровании солей, образованных
катионами слабых оснований такой же силы.
Кривые титрования солей слабых оснований до точки
эквивалентности полностью совпадают с кривыми
титрования солей слабых кислот, если значения р/С слабых
электролитов, образующих соли, находятся в интервале
5-f-9 и 'подвижности катионов определяемой соли и
сильного основания равны подвижностям анионов слабой
кислоты и титранта.
49
С другой стороны, обратимость реакции вытеснения
при титровании солей слабых оснований (р/(ь<5)
оказывает менее сильное влияние по сравнению с титрованием
солей слабых кислот, так как подвижность гидроксиль-
ных ионов сильного основания, не вступившего в реакцию,
ниже подвижности водородных ионов сильной кислоты,
которой титруют соли слабых кислот.
Во всех случаях титрования солей слабых оснований
повышение электропроводности раствора от избытка
сильного основания менее заметно, чем при титровании
солей слабых кислот сильными кислотами.
^ L——' i 1 . 1 I
О 0,05 0,1 0,15 0 0,05 0,1 0,15
Количество до5пвленнога основания, г-эк8/л
Рис. 14. Кривые кондуктометрического
титрования сильным основанием 0,1 н.
растворов солей, образованных катионами слабых
оснований и анионами сильных кислот:
/-1>КЬ = 13; 2-pKb=41; 3-pKb = 8; 4-рКъ = 6
Несмотря на небольшое отличие, типичные кривые
титрования солей слабых оснований и-солей слабых
кислот в общем аналогичны. Это видно из сравнения кривых,
титрования солей слабых оснований (рис. 14) с кривыми
титрования солей слабых кислот такой же силы (рис.
9—12). Кондуктометрический метод применим при
анализе кислых солей сильных и слабых многоосновных
кислот. При этом кислые соли могут быть образованы как
сильными, так и слабыми основаниями.
50
При титровании сильным основанием кислой соли
сильной двухосновной кислоты и сильного основания
нейтрализуются ионы водорода и кривая имеет V-образную
форму. Если титруют кислую соль сильной двухосновной
кислоты и слабого основания (р/Сь = 4-н9),- сначала
нейтрализуются ионы водорода, затем вытесняется слабое
основание. Кривые титрования имеют два излома.
Электропроводность раствора до первого излома линейно
понижается, а между первым и вторым изломами
сравнительно мало изменяется, что видно из условий титрования
солей слабых оснований. Избыток сильного основания
вызывает резкое увеличение электропроводности.
В случае когда кислая соль образована -сильным
основанием и слабой двухосновной кислотой (р/Са2< Ю),
при ее титровании сильным основанием нейтрализуются
НАп~-иоиы. Электропроводность раствора при
нейтрализации этих ионов повышается менее сильно, чем от
избытка титранта. Кривая титрования кислой соли слабого
основания (р/Сь = 4-=-9) и слабой кислоты
характеризуется двумя изломами, если (р/Са + рД'ь)^12. При
титровании сначала нейтрализуются НАп_-ионы, а затем
вытесняется слабое основание. Электропроводность
раствора до первой точки эквивалентности повышается, а затем
практически не изменяется. Избыток сильного основания
вызывает сильное увеличение электропроводности
раствора.
Кондуктометрический метод применяют также при
определении кислых солей слабых трех- и четырехосновных
кислот. Большое практическое значение имеет кондукто-
метрическое определение фосфатов.
§ 4. Титрование смесей кислот
Кондуктометрический метод титрования обладает
широкими возможностями в области анализа смесей кислот
в водных растворах. Весьма благоприятные условия для
анализа создаются при титровании смесей сильных и
слабых кислот. Возможность дифференцированного
титрования этих кислот в смесях зависит от соотношения их
констант диссоциации и концентраций.
Как видно из типичных кривых титрования 0,1 н.
растворов сильных и слабых одноосновных кислот,
электропроводность раствора при их нейтрализации в первом
случае понижается, а во втором — повышается. Линейное
51
повышение электропроводности раствора при
нейтрализации слабых кислот наблюдается при рКа^5. Поэтому
при титровании смеси сильной кислоты со слабой
кислотой с р/Са^5 первый излом кондуктометрической кривой
всегда бывает резким. Закругление первого излома до
определенных пределов не мешает установлению точки
эквивалентности графическим методом. При титровании
смесей 0,1 н. растворов кислот определение возможно,
если рКа слабой кислоты ^4. Верхняя граница рКа
слабых кислот, которые могут входить в состав смесей с
сильными кислотами, зависит от гидролиза
получающихся солей. Как показано ранее, в 0,1 н. растворах
возможно определение слабых кислот с p/Ca^SЮ- Поэтому
общие границы рКа слабых кислот, дифференцированно
титрующихся в смесях с сильными кислотами, лежат в
пределах 4-МО.
I
"§" 0,10
1
§ 0,05
аг
5Г U
0,10 0,20 0,25 0 0,10 0,20 0,30
Количество добавленного основания, г-экВ/л
Рис. 15. Изменение концентраций ионов и кривая коидуктометриче-
ского титрования сильным основанием смеси 0,1 н. растворов сильной
и слабой (рЛ^а = 8) кислот:
J Aiij J —'Концентрация анионов сильной кислоты; J Aii2 I —концентрация
анионов слабой кислоты; [Kt ] —концентрация катионов титранта
Типичная кривая титрования смеси сильной и слабой
кислоты с рЛа = 8 и изменение концентраций ионов
представлены на рис. 15, из которого видно, что концентрация
ионов водорода линейно понижается до первой точки
эквивалентности, после чего становится ничтожно малой.
Концентрация анионов сильной кислоты (Ап~) остается
постоянной в продолжение всего процесса титрования, а
концентрация катионов титранта (Kt+) равномерно
увеличивается. До первой точки эквивалентности низкие
значения имеют концентрации анионов слабой кислоты
(Ап2~). При нейтрализации слабой кислоты концентра-
52
Ция ее анионов линейно увеличивается. После второй
точки эквивалентности концентрация этих ионов остается
постоянной. Концентрация гидроксильпых ионов
вследствие нейтрализации их кислотой остается
незначительной до второй точки эквивалентности, после которой по
мере прибавления избытка основания их концентрация
растет.
Кривая титрования смеси имеет два излома,
соответствующих нейтрализации сильной и слабой кислот. Если
значения рКз слабой кислоты лежат в пределах 4ч-5,
закругляется первый излом, а если в интервале 9-f-10,
закругляется второй излом кривой.
С уменьшением концентрации титруемых растворов
интервал рКа слабых кислот, определяющихся в смесях с
сильными кислотами, уменьшается и имеет следующие
значения:
Концентрация кислот . . 0,1 0,01 0,001 н.
Интервал р/Са 4-гЮ 5^-9 6-н8
Для дифференцированного титрования смесей слабых
кислот необходимо, чтобы разница в значениях
показателей констант диссоциации кислот была ^2, если
концентрации кислот сравнительно мало отличаются, и ^3, если
концентрации сильно отличаются друг от друга. Однако,
даже если константы диссоциации достаточно сильно
отличаются, первый излом кривой, соответствующий
нейтрализации более сильной кислоты, или слабо выражен или
совсем не обнаруживается. Это объясняется тем, что при
нейтрализации обеих кислот электропроводность
раствора увеличивается и заметный излом кривой может быть
только в том случае, если значительно отличаются
подвижности анионов титруемых кислот. Так как
подвижности анионов (кроме ОН-) в общем сравнительно мало
отличаются, кондуктометрические кривые титрования
смесей слабых кислот в водных растворах обычно имеют
один излом, соответствующий нейтрализации обеих
кислот.
Сильные двухосновные кислоты, подобно
одноосновным, дифференцированно титруются в смесях со слабыми
кислотами, имеющими значения рКа в указанных
интервалах. Типичная кривая титрования таких смесей
аналогична кривой титрования смеси сильной кислоты со
слабой одноосновной кислотой (рис. 15).
53
В смеси с сильными одноосновными и двухосновными
кислотами могут входить слабые двухосновные кислоты.
Определения возможны, если величина р/Са' не выходит
за нижнюю границу приведенных интервалов, а р/Са2 —
за верхнюю. Кривые титрования имеют обычно два
излома, фиксирующие нейтрализацию сильной и слабой
кислоты. Дифференцированное титрование слабой
двухосновной кислоты по ступеням нейтрализации в
большинстве случаев не наблюдается, что отмечено при
рассмотрении условий титрования индивидуальных слабых
двухосновных кислот. Поэтому и кривые титрования имеют
форму, показанную на рис. 15.
Двухосновные кислоты, дифференцированно
титрующиеся по ступеням нейтрализации, определяются в смесях
как с сильными, так и со слабыми кислотами.
Нейтрализация сильной кислоты совпадает с первой ступенью
нейтрализации двухосновной кислоты, а нейтрализация
слабой — со второй.
При анализе смесей кислот могут быть использованы
слабые основания, что создает более благоприятные
условия для установления второй точки эквивалентности,
так как электропроводность раствора от избытка титран-
та практически не изменяется.
§ 5. Титрование смесей оснований
Преимущества кондуктометрического метода
сохраняются и при анализе двухкомпонентных смесей оснований.
Интервал значений рКъ слабых оснований, которые могут
дифференцированно титроваться в смесях с сильными
основаниями, имеет те же значения, что и значения р/Са
слабых кислот. Типичная кривая титрования смесей
сильных и слабых оснований в общем аналогична кривой
титрования смесей кислот, приведенной на рис. 15.
Небольшое отличие заключается только в том, что понижение
электропроводности раствора при нейтрализации
сильного основания менее резкое, чем при титровании сильной
кислоты, а повышение электропроводности от избытка
титранта, наоборот, более сильное.
Условия титрования двухкислотных оснований в
смесях аналогичны рассмотренным для титрования смесей,
содержащих двухосновные кислоты.
54
§ 6. Титрование смесей солей
Для дифференцированного кондуктометрического
титрования смесей солей слабых кислот или солей слабых
оснований необходимо, чтобы разница в значениях
показателей констант диссоциации последовательно
вытесняемых слабых кислот или слабых оснований была ^2, если
концентрации солей мало отличаются, и 5?3, если
концентрации сильно отличаются друг от друга. При
титровании сначала вытесняется более слабый электролит,
затем более сильный. Однако кроме этого необходимо,
чтобы отличались подвижности анионов слабых кислот, если
титруют смесь солей слабых кислот, или подвижности
катионов слабых оснований, если проводят анализ смеси
солей слабых оснований. Поскольку подвижности анионов
и катионов (кроме ОН- и Н+) в общем мало отличаются,
достаточно четкий излом, фиксирующий окончание
взаимодействия первой соли, наблюдается только в
некоторых случаях. Например, дифференцированное титрование
компонентов имеет место в смеси гидрохлорида гидрокси-
ламина и хлорида аммония. При взаимодействии с
щелочью сначала вытесняется гидроксиламин, затем
аммиак. Подвижность ионов аммония выше, чем гидрокси-
ламмония, поэтому понижение электропроводности-
раствора при вытеснении гидроксиламина менее сильное, чем
при вытеснении аммиака. Избыток щелочи вызывает
резкое увеличение электропроводности раствора.
§ 7. Титрование смесей кислот и солей
слабых оснований
При взаимодействии сильных оснований со смесями
слабых кислот и солей слабых оснований
устанавливаются равновесия
НАп + ОН-;£Ап- + Н20
Kt++OH-^tKtOH
Используя эти уравнения, находим равновесную
концентрацию гидроксильных ионов
[An-] _ Ка , [КЮН] _ 1
[НАп] [ОН-] Kw' [Kt+] [ОН-] АГь ' j
где Ка — концентрационная константа диссоциации
свободной кислоты; Къ — константа диссоциации основания,
образующего соль; /Cw — ионное произведение воды.
55
Величины равновесных концентраций гидроксильпых
ионов в уравнениях (15) должны быть равны, откуда
Kw [An-] [KtOH]
Т>-[^ГКь[1«+] (16)
или
[An-] [KtOH] КаКь
[HAn] [Kt+] /C,
;i7)
В уоавнении^—^ — степень нейтрализации кислоты,
уу [НАп]
[КЮН] _ степень вытеснения основания из его соли. От-
[Kt+]
ношению этих двух величин можно придавать различные
значения.
Если ^А" ■* •КЮН' = 100:1, сначала протекает реак-
[HAn] ' [Kt+]
ция нейтрализации, а затем вытеснения. При равных
концентрациях кислоты и соли 99% кислоты будет
нейтрализовано тогда, когда 1% основания будет вытеснен
из его соли. Исходя из этого, получаем
^L=100:1 и р/Са + рКь=12. (18)
Таким образом, если (р/Са + р/Сь) = 12, сначала
протекает реакция нейтрализации кислоты, а затем реакция
вытеснения основания из его соли.
В случае когда
[An-] [KtOH]
[HAn] " [Kt+]
= 1:100, (19)
последовательность взаимодействия компонентов смеси
изменяется; сначала протекает реакция вытеснения
основания, затем нейтрализация кислоты. При этих
условиях
-*»**-= 1:100 и р/Са + р/Сь= 16. (20)
Следовательно, сумма рКа и рКъ может служить^кри-
терием последовательности протекания реакций
нейтрализации и вытеснения. Если величина суммы р/Са и р/Сь
находится в области 12н-16, параллельно протекают обе
реакции и дифференцированное титрование
компонентов смеси невозможно.
5§
Последовательное взаимодействие компонентой с
ошибкой 0,1% (отношение равновесных концентраций
1000 : 1 и 1 : 1000) протекает, если сумма р/(а и рКъ
равна 11 или 17.
При выводе указанных зависимостей не учитывалась
обратимость реакций. Если (рКа + рКъ) <12, в первой
точке эквивалентности находятся анионы слабой кислоты
и катионы слабого основания. При этих условиях
возможен гидролиз, который смещает реакции нейтрализации
в левую сторону, реакцию вытеснения в правую сторону.
С другой стороны, если (р/Са + р/(ь)>16, в первой точке
эквивалентности в растворе находится смесь слабой
кислоты и слабого основания, которые могут
взаимодействовать между собой, что вызовет смещение реакции
нейтрале 0,20 0,25 0 0,10 0,20 0,30
Количества добавленного основания, г-зкв/л
,Рис. 16. Изменение концентраций ионов и кривые кондуктометриче-
■ского титрования сильным основанием смеси 0,1 н. растворов кислоты
(р/(а=4) и соли слабого основания (р/(ь = 8):
JAnj I —концентрация анионов слабой кислоты; I Ап2 I —концентрация
анионов соли; Ktj I —концентрация катионов солн; [Kt2 I —концентрация
катионов титранта. X.
■•. - = хл .--70: 7-Х... ,=70; Х„,,=30;
An" .An- Kt+ Kt +
XK(+-30; XRt+=70
2_V + °XKt+
-50; 3-X„
лйзации в правую сторону, а реакции вытеснения в
левую.
Насколько реакции обратимы можно видеть из рис.
16 и 17, где показаны кривые изменения концентраций
ионов и кривые титрования сильным основанием смесей
соли слабого основания (р/Сь = 8) с кислотами, имеющими
р/Са = 4 и 8. В первом случае (р/Са + рКь) = 12, а во
втором 16. Как видно из рис. 16, если смесь имеет (р/(а +
+ рКь)=12, сначала нейтрализуется слабая кислота
57
(рКа = 4) и до первой точки эквивалентности в растворе
увеличивается концентрация ее анионов (Ani-). Однако
вследствие обратимости реакции полной нейтрализации
кислоты в первой точке эквивалентности не происходит,
так как концентрация Ani~ ниже общей концентрации
кислоты. Взаимодействие соли слабого основания с
сильным основанием также начинается несколько раньше
точки эквивалентности, о чем свидетельствует понижение
концентрации ее катионов Kti+. Концентрация ионов
водорода только в самом начале имеет величину,
соизмеримую с концентрациями других ионов, а затем становится
ничтожно малой. Концентрация анионов соли Ап2~ при
титровании постоянна, а катионов титранта Щг+
равномерно увеличивается. Концентрации гидроксильных ионов
малы при йзаимодействии компонентов смеси и
увеличиваются после второй точки эквивалентности.
-7-7"
Z/
ц-
3
г
f
И-НГ
0,10 0,10 0,25 0 0,10 0,10 0,30
Количества добавленного основания, г-экВ/л
Рис. 17. Изменение концентраций нонов и кривые кондуктометрнче-
ского титрования сильным основанием смеси 0,1 н. растворов слабой
кислоты (р/(а = 8) н соли, образованной слабым основанием (р/(ь = 8)
и сильной кислотой:
Г1! I —концентрация анионов слабой кислоты; [Ап2 J—концентрация
анионов соли; Ktj I — концентрация катионов соли; ^Kt2 J — концентрация
катионов титранта. XAn_ = XA|]_=-70; 7-XKt + =70; Хк(+=30; 2-xKt+=XKt+_
12 1 £ 1 *
= 50; 3-Х
W+
-зо, xRt + .
Кривая титрования имеет два излома. Сначала
нейтрализуется кислота, что сопровождается повышением
электропроводности раствора, затем вытесняется слабое
основание. Электропроводность при нейтрализации соли
понижается, остается постоянной или повышается в
зависимости от сравнительной подвижности катионов соли и
58
титрантов. Хотя реакции обратимы, излом в первой точке
эквивалентности лишь слабо закруглен, что не мешает
установлению точки эквивалентности графическим
методом.
Если в состав смеси входит кислота с р/*Са = 8 и соль
слабого основания с рКъ = &, для которых (р/Са + р/Сь) =
= 16, последовательность взаимодействия компонентов
смеси изменяется и кривые изменения ионных
концентраций носят другой характер, что видно из рис. 17. При
титровании сильным основанием сначала в реакцию
вступает соль слабого основания. Равновесная концентрация
катионов соли (Kti+) при этом понижается. Однако в
первой точке эквивалентности концентрация катионов
соли еще имеет величину, соизмеримую с
концентрациями других ионов, что свидетельствует об обратимости
реакции. С другой стороны, концентрация анионов
слабой кислоты (An,-) начинает увеличиваться несколько
раньше первой точки эквивалентности. Таким образом,
нейтрализация кислоты начинается несколько раньше
окончания взаимодействия соли. После второй точки
эквивалентности концентрация анионов слабой кислоты
остается постоянной, равной концентрации титруемой
кислоты. Следовательно, реакция нейтрализации
протекает количественно. Изменение концентрации других
ионов такое же, как в первом случае. Кривая титрования
имеет два излома, соответствующие вытеснению слабого
основания и нейтрализации слабой кислоты. Первый
излом слегка закруглен вследствие обратимости реакций.
В отличие от первого случая сравнительно небольшое
изменение электропроводности наблюдается до первого
излома, а сравнительно сильное повышение — между
первым и вторым изломами.
Если сумма рКа и рКъ, характеризующих
электролиты смеси, становится менее 12, оба излома кондуктомет-
рической кривой резкие. С увеличением силы кислоты,
входящей в смесь, изменяется характер кривой
титрования до первого излома. Изменение электропроводности
раствора на этом участке аналогично наблюдаемому при
нейтрализации индивидуальных кислот такой же силы.
Если в состав смесей входит сильная кислота,
электропроводность раствора до первой точки эквивалентности
сильно понижается. При нейтрализации смесей кислот
средней силы кривая титрования до первой точки
эквивалентности иэдеет пологий минимум и т, д,
Ю
Таблица 3
РКа<8 (рКа+Р^ь)<12 рКь=4-9
1. Кислота " иоль
рАГь>6 (рАГа+рА:ь)>16 р/Са=5-Ю
2. Соль ■ ^ Кислота
Сильная рКа=4-8 (р/С.+р/Сь)<12 g^f4"8
3. Кислота! Кислота2 ^оль
РК,<6 (РКа+рКь)<12 р/Сь=6-9 АР^ь>2 _ Йль=4-7
4. Кислота * Соль1
РКь>8 лРКь>2 , Р/СьГ6-9 (Р^+РКЬ)>16 Р/Са=7-Ю
5. Соль! иоль2
р*а<6 (Р^+РКь)<12 рКь=6-9 (р_/С. + Р*ь)>16 ^.=7-10
6. Кислота; ^оль
' Р*ь>8 (рКа+РКь)>16 РК.-5-8 (Р/<а+РКь)<12 РКь=4-7
7. С0ЛЬ1
—- Кислота Со.-.ьг
Сильная р/Са=4-6 (рАГа+рА:ь)<12 p/Cb=6—8 дрХь>2 р/Сь=4-6
8. Кислота! ► Кислотаг у Соль) ► Сольг
Сильная рКа=4-6 (р^а+Р^ь)<12 рКь=6-8 (рКа+рКь)>16 ^Г8Г10
9. Кислота! --Кислота2 ►Соль Кислота3
?Къ>™ др/Сь>2 РКь=8-9 (р/Са+р/Сь)>16 рАГа=7-8 (р/Са+р/СьХ^Р^4-5
10. Соль, Соль2 -Кислота ^ Соль3
рКа<4 <рАГа+рКь)<12 р/Сь=8-9 (pA:a+p/Cb)>16 рАГа=7-8 (рКа+рКь)<М Р^ь=4-5
11. Кислота! Соль) ► Кислота-2 ► иоль2
рЛГь>13 (Р^а+Р^Сь)>16 РАГа=4-6 (рАГа + рА:ь)<12 р/Съ=6-8 (рАГа+рА:ь)>16 РАГа=8-10
12. Соль, * Кислота! > с°ль1 " Кислота2
Сильная рКа~4 (рКа+рКьХ12 Р^ь~8 (рКа+рКь)>^ рКа~^ (.РКа+рКъ)<^РКъ~^
13. Кислота! ► Кислотаг * СолЫ >Кислота3 ,Соль2
14. grl(P«a+P*b)>16 К?с7ота! «*>+Ы<» gjb~8 (Р^+Р^ь)>16 ■g^V^+P^K»^^
«^Стрелки указывают последоватгльность взаимодействия компонентов.
Когда сумма рКа кислоты и рКь основания становится
больше 16, закругление первого излома пропадает.
Верхняя граница рКа кислот, которые могут определяться в
смесях, равна 10. Подобно титрованию индивидуальных
кислот, второй излом закругляется, если значения рКа
находятся в интервале 9-f-10.
Как видно из изложенного, в случаях, когда
взаимодействие кислот и солей слабых оснований в смесях
чередуется, кривые титрования независимо от подвижно-
стей ионов имеют четкие изломы, фиксирующие
титрование отдельных компонентов.
Благодаря преимуществам кондуктометрического
метода титрования смесей сильных и слабых кислот, а
также смесей кислот и солей слабых оснований удалось
разработать методы кондуктометрического анализа
многокомпонентных смесей этих соединений. В 0,1—0,05 н.
растворах возможен анализ трех-, четырех- и пятиком-
понентных смесей. Критериями осуществимости
дифференцированного титрования компонентов служат
константы диссоциации электролитов и значения суммы рКа
и рКъ, которые приведены в табл. 3.
Для установления возможности
кондуктометрического анализа конкретных смесей определяют, находятся ли
значения показателей констант диссоциации,
характеризующих электролиты, в пределах указанных границ.
Анализ смесей основан на последовательном
титровании кислот и солей слабых оснований. Условия
благоприятны для анализа, так как при титровании слабых
кислот электропроводность раствора всегда довольно
сильно повышается, а при титровании солей
сравнительно мало изменяется и зависит только от подвижностей,
участвующих в реакции ионов. При титровании смесей
кислот сначала в реакцию вступают более сильные,
затем более слабые; для смеси солей, наоборот, сначала
взаимодействуют соли более слабых оснований, затем
соли более сильных. Каждая пара последовательно
взаимодействующих веществ характеризуется суммой рКа
и р/*Сь<12 или 16. В целом, при титровании всех
многокомпонентных смесей согласно схемам, приведенным в
табл. 3, происходит последовательная нейтрализация
веществ, проявляющих все более и более слабые
кислотные свойства.
При титровании 0,01 н. растворов возможности
кондуктометрического анализа значительно суживаются,
63
Максимальное значение рКа слабой кислоты такой
концентрации должно быть ^9, а рКъ слабого основания,
образующего соль, ^5. Слабые кислоты
дифференцированно титруются в смесях с сильными кислотами, если
их рКа^Ь. Степень гидролиза соли близка к 1%, если
величина рКъ около 8. Поэтому, если соль
взаимодействует после кислоты, величина рКъ должна быть ^8.
Критерии возможности титрования 0,01 н. растворов
смесей и варианты составов показаны в табл. 4.
Надежные результаты могут быть получены только
при титровании двух- и трехкомпонентных смесей.
При титровании 0,001 н. растворов возможен анализ
только двухкомпонентных смесей, если константы
диссоциации электролитов удовлетворяют следующим
условиям:
р/Са<6 (рАГа + рАГь)<12 р/Сь = 6-7
Кислота Соль
р/Сь<9 (рКа+рКь)<16 р/Са = 7 —8
Соль Кислота
§ 8. Титрование смесей оснований и солей
слабых кислот
Процессы кислотно-основного взаимодействия в
смесях оснований и солей слабых кислот можно
представить подобно процессам, рассмотренным для смесей
кислот и солей слабых оснований.
Равновесия, устанавливающиеся при взаимодействии
сильных кислот в смесях слабых оснований с солями
слабых кислот могут быть выражены уравнениями:
KtOH + H+^tKt++H20
An- + H+^±HAn
Аналогично рассмотренному выше выводятся
зависимости
[Kt+] .[HAn] _ КлКъ
[КЮНПАп-] /cw ' ( '
и /о1ч [Kt*] [HAn]
В уравнении (21) отношения — — и -
уу к ' [КЮН] [An-]
представляют собой степень нейтрализации основания и
степень вытеснения кислоты из ее соли. Если принять от-
[Kt+j [HAn] inn i i inn
ношение - -: равным 100 : 1 или 1 : 100, то сум-
[КЮН] [An-] J
64
ма р/Са и рКъ будет равна 12 или 16. В случае когда
(рКа + рКь) ^12, сначала нейтрализуется основание, а
затем вытесняется кислота из ее соли. Если же (рКа +
+ р.Кь)^16, то, наоборот, -сначала происходит
вытеснение слабой кислоты из ее соли, а затем нейтрализация
основания.
Осуществимость кондуктометрического анализа
смесей оснований и солей слабых кислот может быть
установлена из табл. 3 и 4, причем свободные кислоты
следует заменить свободными основаниями, а соли слабых
оснований— солями слабых кислот. При этом-значения
рКъ слабых оснований, входящих в смесь, следует
принять равными значениям рК& свободных кислот, а
величины рКа кислот, образующих соли, равными рКь
оснований, образующих соли.
§ 9. Титрование амфолитов
Многие вещества обладают амфотерными
свойствами и образуют биполярные ионы или цвиттерионы. Их
образование объясняется переходом протонов молекул
амфолитов от кислотных групп к основным. Для
осуществления такой внутримолекулярной нейтрализации
необходимо, чтобы кислотные свойства амфолита были
достаточно сильно выражены, а взаимное влияние двух
функциональных групп способствовало этому процессу.
С другой стороны, известен ряд амфотерных
соединений, которые не являются цвиттерионами. Примером
таких соединений могут служить о-, м- /г-аминофенолы.
Диссоциация этих соединений может быть выражена
уравнениями:
H2NC6H4OH^±H2NC6H4C- + Н+
H2NC6H4OH + Н20^±НзЫС6Н4ОН + ОН~
При взаимодействии амфолитов, не имеющих
биполярного строения, с кислотами протоны присоединяются
к основным группам, а при взаимодействии с
основаниями отнимаются от кислотных групп аналогично обычным
кислотно-основным взаимодействиям. Например,
H2NC6H4OH + Н+ = H3NC6H4OH
3—1595
H2NC6H4OH + ОН- = H2NC6H40- + Н20
65
Таким образом, ё Щелочных растворах существуют
анионы, а в кислых — катионы.
С точки зрения классической теории константы
кислотности и основности имеют значения:
+
к _[H2NC6H4Q-][H+] . у _ [H3NC6H4OH][OH-] (22)
а [H2NC6H4OH] ' b [H2NC6H4OH] ^
или в общем виде
ATa=[R-][H+]; Kb=[R+][0H~] (23)
[R] b [R] ^
Биполярное строение имеют многие амфолиты, в том
числе алифатические аминокислоты. Например, амино-
уксусная кислота (глицин), состав которой отвечает фор*
муле H2NCH2COOH, имеет строение H3NCH2COO-
Таким образом, в растворах существуют биполярные ионы,
несущие как положительный, так и отрицательный
заряды.
Взаимодействия цвиттерионов с кислотами и
основаниями существенно отличаются от наблюдаемых для ам-
фолитов, не имеющих этого строения. При
взаимодействии с кислотами протоны присоединяются к СОО~-груп-
пам, а при взаимодействии с основаниями отнимаются от
H3N+-rpynn. Однако в результате реакций, подобно
обычным амфолитам, в кислых растворах образуются
катионы, а в щелочных — анионы
H3NRCOO- + Н+ = H3NRCOOH
H3NRCOO- + ОН- = H2NRCOO- + Н20
Это — реакции вытеснения слабых кислотных и
основных групп в цвиттерионах.
Константы кислотности и основности согласно
классической теории могут быть установлены на основании
процессов диссоциации карбоксильных и аминогрупп:
H3NRCOOH^±H3NRCOO- + Н+
H2NRCOO- + H20^±H3NRCOO- + ОН-
66
Тогда
^'_[H+][H;JNRCOO-] . к> [ОН-] [H3NRCOO-] .^
' [HsNVoOH] ' "' №NRCOO-]
или в общем виде
^=ЖЧШ. g;=[QH-][R*] (25)
[R+] [R-] ^
Для многих аминокислот, характеризующихся
структурой цвиттерионов, величина р/Са' находится в
интервале 2-=-3, а р/Сь' имеет значение 4-f-5.
В справочниках иногда приводят константы
диссоциации амфолитов, рассчитанные по результатам их
титрования сильными кислотами и щелочами. При этом не
учитывается образование биполярных ионов и расчеты
основываются на том, что при титровании основаниями
нейтрализуются кислотные группы, а при титровании
кислотами — основные. Эти константы имеют
выражения, приведенные для амфолитов, не имеющих
биполярного строения, однако не характеризуют
кислотно-основные свойства биполярных ионов. Константы р/\а и рКъ
и константы диссоциации, характеризующие цвиттерио-
ны (р/Са' и р/\ь')> находятся в следующей зависимости:
р#а'=14-рКь; р/и/=14-р/уа.
Как показано выше, титрование сильными
основаниями позволяет определять в 0,1 н. растворах кислоты,
имеющие р/Са^10, и соли слабых оснований с р/\ь^4.
С другой стороны, при титровании сильными кислотами
определяются основания с р/Сь^Ю и соли слабых
оснований с р/Сь^4. Исходя из этого, амфолиты, не
имеющие биполярного строения, кислотные и основные
свойства которых характеризуются р/С^Ю, могут
титроваться как сильными кислотами, так и сильными
основаниями. К ним относятся о-, м-, /г-аминофенолы. При их
титровании кислотами и щелочами электропроводность,
раствора до точки эквивалентности увеличивается, так
как в процессе реакций в растворе образуются хррошд
диссоциирующие соли.
Кривые титрования аминофенолов кислотами и ocHq-
ваниями аналогичны. Отличие заключается только в том,
что электропроводность раствора после точки
эквивалентности при титровании кислотами, повещается более
3* 67
сильно, чем при титровании основаниями. На рис. 18
(кривая 1) для примера показана кривая титрования
щелочью л*-аминофенола. Кривая титрования
слегка закруглена вблизи точки эквивалентности, так
как кислотные свойства л«-аминофенола слабо
выражены (р/Са = 9,76).
Взаимодействие цвитте-
рионов с сильными
кислотами и основаниями представ-
- ляет собой реакции
вытеснения слабых карбоксильных и
аминогрупп в цвиттерионах.
Так как типичные цвитте-
рионы имеют значение р/Са =
= 2-^3, их титрование
сильными кислотами не может
привести к количественным
результатам вследствие
обратимости реакции. Однако
титрование щелочами,
основанное на вытеснении
слабых аминогрупп, позволяет
проводить количественное
определение цвиттерионов,
так как их рК'ь>4. На рис. 18
(кривая 2) показана кривая
титрования аминоуксусной
кислоты раствором NaOH.
При титровании до точки
эквивалентности
электропроводность раствора
увеличивается, так как
биполярные ионы (NH3CH2COO-)
переходят в анионы (NH2CH2COO-), а также потому,
что в растворе накапливаются катионы титранта',
5
I
§■
I
*:
33
о
г г 1
А
0,1 с,г
Количество добавленного
основания, г-экВ/л
Рис. 18. Кривые кондуктометри-
ческого титрования сильным
основанием 0,1 н. растворов ам-
фолитов:
/ — .м-аминофенол; 2 — глицин
§ 10. Титрование смесей амфолитов
с кислотами
В смесях амфолитов, не имеющих биполярного
строения, с кислотами происходит нейтрализация основных
"групп, что приводит к образованию соответствующих
солей. Например, в смеси ж-аминофенола (р/Са==9,76;
68
р/Сь = 9,28) с НС1 образуется хлорид состава НС1 •
•H2NC6H40H, диссоциирующий с образованием
H3NC6H4OH и С1--ИОНОВ. В Н3ЫС6Н4ОН-ионах с ще^
лочью могут взаимодействовать ОН- и H3N+-rpynnbi.
Кислотные свойства H3N+-rpynn выражены сильнее, чем
ОН--групп, и поэтому при взаимодействии катионов с
щелочью последовательно протекают реакции:
H3NC6H4OH + OH-^H2NC6H4OH + Н20
H2NC6H4OH + OH-^tH2NC6H40- + Н20
Эти равновесия характеризуются константами:
[H2NC6H4OH] 1
(26)
(27)
(28)
[H3NC6H4OH] [OH-]
[H2NC6H4Q-] _ АГа
[H2NC6H4OH] [ОН-] ~~ Kv
и, следовательно,
[H2NC6H4OH] [H9NC6H4Q-] Kv
[h3Uc6h4oh] ' [WW*] - *.«ъ •
В общем виде эта зависимость имеет выражение
JBL.[JLl = Jfw_, (29)
[R+]' [R] КаКъ
где /Са и Къ — концентрационные константы
диссоциации амфолита; Kw — ионное произведение воды.
В уравнении (29) отношения ~- и l—p
представляют собой степень смещения первой и второй реакций.
Аналогично рассмотренному выше случаю титрования
смесей кислот и солей слабых оснований во всех
приводимых ниже уравнениях для смесей, содержащих амфо-
литы, максимальная относительная ошибка разделения
компонентов взята равной 1%, учитывая, что при
графическом определении эквивалентных точек ошибка умень-
r, [R] [R]
щается. В этом случае * -——: — 100:1, и, следова-
[R+] [R-]
тельно, сумма р/Са и рДь должна быть равна 16. Так как
для многих амфолитов величина (рКа-\-рКъ)>1й, при
„взаимодействии их катионов с щелочами сначала практи-
69
чески количественно протекает реакция вытеснения, а
затем нейтрализации. При этом кондуктометрические
кривые титрования имеют два излома, фиксирующие
последовательное взаимодействие функциональных групп.
Если (рЛГат-р/Сь) = 12-f-16, обе реакции протекают
параллельно и кондуктометрические кривые титрования имеют
один излом, соответствующий окончанию обеих реакций.
При анализе следует учитывать, что для количественного
проведения реакций нейтрализации кислотных групп
амфолита необходимо, чтобы величина их р/Са была ^10.
Если смесь
содержит избыток сильной
кислоты, сначала про-
Ч\-\ ! ! ! / I исходит нейтрализация
свободной кислоты. На
рис .19 приведена
кривая титрования
щелочью смеси НС1 илг-ами-
нофенола (2:1)
(кривая 1). Кондуктомет-
рическая кривая
титрования имеет три
излома, фиксирующих
последовательное взаимо-
действие свободной
О 0,1 0,2 0,3 0,4 НС1 и двух функцио-
КолцчестВо до5аВлвиного оснований, нальных групп амфоли-
г-экВ/л
В з
§■ 5
е
Ч\У
Ч1У
Рис. 19. Кривые кондуктометрическо-
ГО титрования гидроксидом натрия
смесей амфолитов с НС1:
I — л-аминофенол + НС1 (1:2); 2 — гли-
цин+HCl (1 : 2)
та; сначала H3N-rpyn-
пы, затем ОН_-группы.
Нейтрализация НС1
сопровождается
сильным понижением
электропроводности. Элект*
ропроводность раствора после первого излома мало из-
+
меняется, при этом катионы (H3NC6H5OH) превращают?
ся в нейтральные молекулы, но в растворе увеличивается
концентрация ионов н"атрия. После второго излома
кривой электропроводность раствора увеличивается, потому
что при нейтрализации гидроксильных групп молекулы
переходят в анионы (H2NC6H50-). Для
дифференцированного определения избытка сильной кислоты в смесях
значение рКъ амфолита не должно быть значительно
выше 9, иначе первый излом кривой закругляется вследст-
70
виё гидролиза соли, образуемой сильной кислотой и ам-
фолитом. Однако анализ смеси возможен, даже если
первый излом закруглен и кривая имеет только два четких
излома, содержание амфолита может быть рассчитано
по участку кривой между первым и вторым изломами.
Общее содержание добавленной в смесь сильной кислоты
рассчитывается по участку кривой до первого излома.
Если же кривая титрования имеет три излома,
концентрация амфолита рассчитывается по участку кривой
между первым и вторым или между вторым и третьими
изломами, а общее содержание сильной кислоты — по участку
кривой до второго излома.
Если в смеси с кислотами входят цвиттерионы, в них
+
образуются катионы состава H3NRCOOH. В этих
катионах со щелочью могут взаимодействовать как СООН-,
+
так и Н31М-группы. В отличие от обычных амфолитов
кислотные свойства карбоксильных групп выражены
сильнее, чем H3N+-rpynn. Поэтому последовательно
протекают реакции нейтрализации
H3NRCOOH + ОН- = H3NRCOO- + Н20
H.NRCOO- + ОН- = H2NRCOO- + Н20
Аналогично уравнениям (28) и (29) выводятся
уравнения
[H3NRCOO-] . [H2NRCOO-] К'лК'ъ
[H3NRCOOH] [H3NRCOO-] **
принимая сокращенные обозначения,
[R±] . [R-] К'к'ь
tR+J [R4 К,
(30)
(31)
При отношении -—-:-—-=100:1 сумма р/Са' и
[R+] [R±]
рКъ' должна быть равна 12. Таким образом, область
количественных взаимодействий в смесях, содержащих
цвиттерионы, характеризуется (р/Са + р/Сь) <12, что
объясняется тем, что кислотные и основные свойства
функциональных групп цвиттерионов выражены довольно
сильно. Поэтому по сравнению с амфолитами, не имею-
71
щими биполярного строений, в кислых растворах
изменяется последовательность взаимодействия
функциональных групп амфолита: сначала нейтрализуются
карбоксильные группы, затем вытесняются аминогруппы.
Кривая титрования щелочью смееи НС1 и аминоуксус-
ной кислоты (2:1) (р/Са' = 2,35 и рДУ = 4,22) (рис. 19,
кривая 2) типична для смесей цвиттерионов с сильными
кислотами. Сначала наблюдается резкое понижение
электропроводности раствора, что связано с
нейтрализацией имеющихся в растворе ионов водорода. Кондукто-
метрическая кривая до первого излома изогнута, что
объясняется уменьшением степени диссоциации
карбоксильных групп с увеличением в растворе концентрации
цвиттерионов. После излома кондуктометрической
кривой протекает реакция вытеснения аминогрупп, что
сопровождается повышением электропроводности
раствора, так как при этом цвиттерионы переходят в анионы.
При этом избыток НС1 и аминоуксусная кислота
дифференцированно не титруются. Это объясняется тем, что
карбоксильные группы типичных цвиттерионов
характеризуются рКа/ = 2-нЗ. Участок кривой до первого излома
соответствует нейтрализации свободной НС1 и
карбоксильных групп катионов, образуемых аминоускусной
кислотой в смеси, т. е. всей НС1, добавленной в смесь.
Содержание аминоуксусной кислоты рассчитывается по
участку кривой между первым и вторым изломами.
§ 11. Титрование смесей амфолитов
с основаниями
В смесях амфолитов, не имеющих биполярного
строения, с щелочами происходит нейтрализация кислотных
групп амфолита и образование соответствующих солей.
Например, в смеси NaOH с ж-аминофенолом (р/Са=9,76;
р/Сь=9,28) образуется аминофенолят состава
H2NC6H4ONa, диссоциирующий с образованием
H2NC6H40-- и Na+-noHOB. При титровании сильными
кислотами эквимолярной смеси NaOH и ж-аминофенола
взаимодействуют анионы. При этом протекают реакции
взаимодействия оксигрупп, обладающих слабыми
кислотными свойствами, и нейтрализации аминогрупп:
H2NC6H40- + H+^tH2NC6H4OH
H2NC6H4OH + H+^tH3NC6H4OH
72
Уравнения, характеризующие эти равновесия, имеют
вид:
[H2NC6H4OH] __ 1 . [H3NC6H4OH] _ АГь (32)
[Н+] [H2NC6H40-] АГа ' [H2NC6H4OH][H+] " A'W
и тогда
[H2NC6H4OH] . [H?NC6H4OH] _ /С*
(33)
(34)
[H2NC6H40-] [H2NC6H4OH] КаКъ
или в общем виде
Jg] . [R+1 _ ^w . -
[R-] ' [R] К*КЪ *
При -I5L:EB_L—ЮО: 1 сумма р/Са и рКъ амфолита
Р [R-] [R]
должна быть равна 16. Таким образом, область, в
которой наблюдается дифференцированное титрование
функциональных групп амфолита, не имеющего биполярного
строения, характеризуется (рКа+рКь) >16. При
титровании сильными кислотами щелочных растворов
амфолита сначала вытесняются слабые кислотные группы, а
затем нейтрализуются основные группы, что
сопровождается изломами кондуктометрических кривых. Если
(рКа + рКъ) = 12-М6, обе реакции протекают
параллельно и кондуктометрические кривые имеют один излом,
соответствующий окончанию обеих реакций. Следует
отметить, что для количественного определения
основных групп амфолита необходимо, чтобы величина их
р/Сь была =^10.
Если концентрация щелочи в смеси выше, чем
амфолита, при титровании сначала происходит нейтрализация
свободной щелочи, о чем свидетельствует кривая
титрования смеси NaOH и ж-аминофенола с соотношением
концентраций компонентов 2: 1 (рис. 20, кривая /). Эта
кривая имеет три излома, соответствующих
взаимодействию NaOH и двух функциональных групп амфолита.
При нейтрализации свободной NaOH
электропроводность понижается. Затем вытесняются оксигруппы,
причем электропроводность раствора не изменяется;
наконец нейтрализуются гидроксильные группы, что
сопровождается увеличением электропроводности раствора.
Более сильное увеличение электропроводности
происходит от избытка титранта. Дифференцированное титро-
73
вание смеси свободной щелочи и амфолита возможно,
если р-Ка^Э. Если же рКа>9, происходит гидролиз
соли, образуемой амфолитом со щелочью, и первый
излом на кривой титрования закруглен. Однако общая
концентрация щелочи в смеси может быть определена и в
случае, если первый излом кривой не может быть
использован при расчетах. При этом поступают так же, как
„ описано выше для тит-
~~ рования смеси
амфолита с сильными
кислотами. Количество
сильной кислоты,
вступившей в реакцию со
щелочью, может быть
определено по участку
кривой до первого
излома, а содержание
амфолита — по участку
кривой между первым
и вторым изломами.
Если на кривой имеется
три излома, участок
кривой до первого изло-
S
г
%■
I 5
Ч)
* ч
ft
\
_
ч
1 [ 1 /
\! \Л
1 ' /
! 2 i ! /
X" \ . ,
О И,! 0,2 0,3 0,4
Количество ЗоЬаЬленнай
го титрования хлористоводородной
кислотой смесей амфолитов с NaOH:
/ — ж-аминофеиол + NaOH (1:2); 2 — гли-
цин+NaOH (1 : 2)
кислоты,г-зкВ/пма используют для оп
„ „„ „ . ределения свободной
Рис. 20. Кривые кондуктометрическо- ^ ■"• ■"•
щелочи, а участок
кривой от начала
титрования до второго
излома— для вычисления
общего содержания щелочи. Концентрацию амфолита
находят по участку кривой между вторым и третьим
изломами.
Возможен анализ смесей сильных оснований с
аминокислотами, имеющими структуру цвиттерионов. В этих
смесях, как рассмотрено выше, аминокислоты образуют
анионы состава H2NRCOO-. При титровании сильными
кислотами смесей, не содержащих избытка щелочи,
сначала нейтрализуются аминогруппы в анионах, что
приводит к образованию в растворе цвиттерионов, а затем
взаимодействуют СОО--группы по реакциям:
H2NRCOO- + H+^:H3NRCOO-
H3NRCOO- + H^tH3NRCOOH
74
Аналогично уравнениям (33) и (34) выводятся
уравнения
[H?NRCOO~] . [H3NRCOOH] :_К'як'ъ (35)
[H2NRCOO-1 ' + ~7Г~ *■ '
1 2 J [H3NRCOO-] к™
или, принимая сокращенные обозначения,
JR^.[R^]_=_kX (Зб)
[R-] '[R±] Kw
Если принять -—-:-—-=100:1, сумма рК& и
■[R~] [R±]
р/Сь' для цвиттериона должна быть равна 12. Таким
образом, если цвиттерионы характеризуются (рКа'-\-рКъ) =
= 12 или меньше, при титровании сильными кислотами
их смесей с щелочью сначала протекает реакция
нейтрализации аминогрупп в анионах, а затем вытеснения
карбоксильных групп в цвиттерионах. Однако только первая
реакция протекает количественно. Кислотные свойства
карбоксильных групп типичных цвиттерионов выражены
довольно сильно (рК&' = 2-т-З) и реакция вытеснения
обратима. Поэтому кондуктометрические кривые имеют
только один излом, соответствующий окончанию
реакции нейтрализации, после чего наблюдается нелинейное
повышение электропроводности раствора, что
свидетельствует об обратимости реакции вытеснения
карбоксильных групп.
Если титровать смесь, содержащую избыток сильного
основания, наблюдается дифференцированное титрование
свободного сильного основания. Электропроводность
раствора при этом сильно понижается. После излома
кривой нейтрализуются аминогруппы в анионах
амфолита, в результате чего образуются цвиттерионы. На
кривой титрования имеется второй излом, показывающий
окончание нейтрализации аминогрупп. После этого
электропроводность раствора нелинейно увеличивается.
Искривление кривой объясняется, как указано выше, тем,
что часть протонов связывается СОО~-группами. Такого
типа кривая (рис. 20, кривая 2) получается при
титровании смеси NaOH и аминоуксусной кислоты, в которой
концентрации компонентов относятся как 2:1.
Таким образом, индивидуальные цвиттерионы обычно
количественно с сильными кислотами не взаимодейст-
75
вуют, но в смесях, содержащих избыток сильного
основания, можно проводить их количественное
определение, так как оно основано не на реакции вытеснения
карбоксильных групп в цвиттерионах, а на реакции
нейтрализации аминогрупп в анионах, образующихся в
щелочных растворах. Содержание свободной щелочи
определяется по участку кривой до первого излома. Общее
содержание щелочи находится" по участку кривой от
начала титрования до второго излома. Концентрация ам-
фолита рассчитывается по участку кривой между первым
и вторым изломами.
§ 12. Титрование смесей амфолитов
с солями слабых кислот или слабых оснований
Кондуктометрический метод позволяет проводить
анализ смесей амфолитов, не имеющих биполярного
строения, характеризующихся рКа и р/Сь^Ю, с солями
слабых кислот или слабых оснований. При этом
необходимо создавать условия, при которых сумма р/Са и р/Сь
последовательно взаимодействующих функциональных
групп или ^12, или ^16. Поскольку кислотные и
основные свойства этих амфолитов обычно слабо выражены,
более доступны определения в области, когда (р/Са+
+ р^ь)^16.
Рассмотрим условия взаимодействия сильных
оснований в смесях, содержащих амфолиты, например, ами-
нофенолы и соли слабых оснований. Кислотные свойства
аминофенолов слабо выражены и поэтому в смесях
протекают реакции в такой последовательности:
Kt+ + OH-^tKtOH
H2NC6H4OH + OH-^tH2NC6H40- + H20
Эти равновесия характеризуются константами:
[КЮН] _ 1 /37)
[OH-][Kt+] Кь'
[Н2МС6Н40~] _К3 (38)
[H2NC6H4OH] [OH-] ATw'
где Кл — концентрационная константа диссоциации
кислотных групп амфолита; Къ — концентрационная
константа диссоциации основания, образующего соль;
76
/Cw — ионное произведение воды. Равновесные концен
трации ионов и молекул определяются из уравнения
[KtOH] . [H2NC6H4Q-] = /fw
[Kt+] ' [H2NC6H4OH] KaK
или в общем виде
(39)
[KtOH] . [R-]
*,
[Kt+] [R] К*Къ
(40)
Если отношение
[KtOH] .[R-
= 100:1, сумма р/Са
[Kt+] [R]
р/Сь равна 16. Следовательно, при данной последователь
ности взаимодействия компонентов смеси, количествен
ные определения возможны в
случаях, когда (р/Са+Р^ь) ^
^16. Таким образом, в смесях
с амфолитамТг могут
определяться соли очень слабых
оснований. При титровании этих
смесей сильными основаниями
сначала вытесняются слабые
основания из их солей, а затем
нейтрализуются кислотные
группы амфолитов. Например,
возможен анализ смеси
гидрохлорида гидроксиламина
(р/Сь = 8,03) и о-аминофенола
(рЛа = 9,87). Кривая
титрования этой смеси раствором
NaOH приведена на рис. 21
(кривая 1). При титровании
сначала вытесняется гидрокси-
ламин из его соли и
электропроводность раствора до
первого излома понижается, а затем
нейтрализуются кислотные
группы о-аминофенола, что
приводит к повышению
электропроводности после первого
излома. Избыток щелочи
вызывает резкое увеличение электропроводности раствора.
Кондуктометрические кривые титрования смесей
такого типа имеют два резких излома, так как при
взаимодействии соли изменение электропроводности раствора
О 0,05 0,1 0,15
Количество добавленного
раствора NaOH, г-зкб/л
Рис. 21. Кривые кондук-
тометрического
титрования гидроксидом натрия
смесей гидрохлорида
гидроксиламина с о-амино-
фенолом (/) и
глицином (2)
77
зависит от сравнительней подвижности катионов соли и
титранта, а при нейтрализации кислотных групп амфо-
лита электропроводность раствора всегда увеличивается,
потому что слабо диссоциирующий амфолит образует
хорошо диссоциирующую соль.
Условия титрования сильными кислотами смесей ам-
фолитов, не имеющих биполярного строения и солей
слабых кислот, в общем аналогичны рассмотренным для
смесей амфолитов и солей слабых оснований.
При титровании смесей амфолитов, например амино-
фенолов, и солей слабых кислот протекают реакции:
An- +Н+^ГНАп
H2NC6H4OH + H+^tH3NC6H4OH
Равновесные концентрации ионов и молекул
находятся из уравнения
[HAn] . [H3NC6H4OH] = К* /4П
[An-] - [H2NC6H4OH] КаКъ
или в общем виде
[НАп] . [R+] = Kw
[An-] • [R] КаКъ ' l '
где Къ — концентрационная константа диссоциации
основных групп амфолита; Ка — концентрационая
константа диссоциации кислоты, образующей соль; A'w — ионное
произведение воды.
„ [HAn] [R+] 1Г1Г1 , „ ,,
При : -—— =100:1 сумма рУСа и р/\ь должна
[An-] [R]
быть равна 16. Таким образом, в смесях с амфолитами
могут определяться соли кислот такой силы, чтобы сумма
рКъ амфолита и р/Са кислоты, образующей соль, была
^16. При взаимодействии этих смесей с сильными
кислотами сначала вытесняются слабые кислоты из их
солей, а затем нейтрализуются основные группы амфолита.
Например, возможен анализ смеси о-аминофенола
(р/Сь = 9,83) и фенолята натрия (р/Са= 10,0). При
титровании сильными кислотами сначала вытесняется фенол,
а затем нейтрализуются аминогруппы о-аминофенола.
Рассмотрим условия титрования смесей солей с цвит-
терионами. Как показано ранее, типичные цвиттерионы
количественно титруются только щелочами. Поэтому в
смеси могут входить только соли слабых оснований. Оп-
78
ределение цвиттерионов и солей основано на реакции
вытеснения слабых основных групп. Так как для многих
цвиттерионов значение рКъ находится в интервале 4—5,
соли, входящие в состав смесей, должны быть
образованы более слабыми основаниями с рКь^7. При
титровании щелочами сначала вытесняются более слабые
основания из солей, а затем взаимодействуют цвиттерионы.
Например, возможен анализ смеси аминоуксусной
кислоты (рЯ'ь = 4,22) и гидрохлорида гидроксиламина
(р/Сь = 8,03). При титровании сначала вытесняется
гидроксиламин из его соли, а затем — аминогруппы в
цвиттерионах (рис. 21, кривая 2). Электропроводность
раствора до первого излома понижается, что характерно
для вытеснения гидроксиламина. После первой точки
эквивалентности электропроводность раствора
увеличивается, так как цвиттерионы переходят в анионы. Более
сильный рост электропроводности раствора наблюдается
от избытка щелочи.
Глава IV
ТИТРОВАНИЕ, ОСНОВАННОЕ
НА РЕАКЦИЯХ ОСАЖДЕНИЯ
Кондуктометрическое титрование, основанное на
использовании реакции осаждения, в сильной степени
зависит от растворимости электролита, осаждаемого в виде
.малорастворимого осадка. Существенное влияние
оказывает также скорость образования, постоянство состава и
чистота выделяющегося осадка. В процессе химического
взаимодействия, сопровождающегося образованием
осадка, концентрации ионов в титруемом растворе сильно
изменяются. Характер кондуктометрических кривых
титрования сильно зависит от концентраций различных
ионов и их подвижностей. Ниже рассмотрено влияние
различных факторов на формы кондуктометрических
кривых титрования и возможности кондуктометрических
определений.
§ 1. Зависимость формы кривых кондуктометрического
титрования от величины растворимости осадка
Растворимость осадков ограничивает возможность
кондуктометрических определений. При титровании
разбавленных растворов растворимость осадков оказывает
более сильное влияние.
Рассмотрим взаимодействие ионов двух солей (KtiArii
и К^гАпг), при котором катионы титруемой соли (Kti+)
образуют с анионом титранта (Апг-) осадок KtiAri2:
Kt+ + Anf + Ktf + An7 = КПАп2 + Kt+ + Anf
Пусть концентрация титруемой соли равна 0,1 н., а
произведение растворимости осадка бинарного
электролита изменяется от 10-4 до 10~6. Как видно из рис. 22,
при титровании концентрация анионов титруемой соли
(Anj-) остается во всех случаях постоянной, а
концентрация катионов титранта (К1г+) равномерно увеличивается.
Равновесные концентрации катионов определяемой
соли уменьшаются в связи с выделением осадка. Однако
характер изменения концентраций этих ионов зависит от
величины растворимости осадка. Если растворимость
равна 10~2 г-ион/л, концентрация этих ионов в точке эк-
§9
вивалентности довольно высокая и реакция' не протекает
количественно. По мере уменьшения растворимости
осаждение становится более' полным, что приводит к
линейному понижению концентрации Kti+ до точки
эквивалентности, после которой их концентрация становится
ничтожно малой.
В случаях, когда реакция протекает количественно,
концентрация анионов осадителя Ап2~ ничтожно мала
до точки эквивалентности и линейно увеличивается
после нее по мере добавления избытка титранта.
Неполнота завершения реакции оказывает
существенное влияние на характер кондуктометрических
кривых титрования. На рис. 23 показаны кривые
кондуктометрического титрования указанных солей, если принять
A-Kti+=70; tactf = 50; Unf=50; ЯАп;Г = 70. Так как ПРИ
Количество добавленного
осадителя, г-ж в/л
Рис. 22. Изменение концентраций ионов
при титровании 0,1 н. растворов солей,
основанном на образовании осадков с
катионом титруемой соли:
произведение растворимости осадков: 1—10 I
2—10—Е; 3— 10—6; I Ktj I _ концентрация
катионов титруемой соли; [Ktj (—концентрация
катионов титранта; An J — концентрация
анионов титруемой соли; I д,]2 I — концентра -
иия анионов титранта
81
взятых условиях катионы оса-
дителя имеют более низкую
подвижность, чем осаждаемые
катионы, электропроводность
раствора до точки
эквивалентности понижается, а после нее
увеличивается от избытка оса-
дителя. В случае когда
произведение растворимости
бинарного электролита равно Ю-4,
кривая титрования сильно
закруглена вследствие
растворимости осадка и точка
эквивалентности не может быть
установлена. По мере уменьшения
произведения растворимости
осаждаемого электролита
излом кривой в точке
эквивалентности становится более
резким.
Так как точки
эквивалентности на кондуктометрических
кривых находят графическим
методом, закругление кривых
вблизи точек эквивалентности
до определенных пределов не
мешает их установлению. Как
видно из рис. 23, затруднения
не возникают в случае
титрования 0,1 н. растворов солей,
если произведения
растворимости бинарных электролитов
Ю-5 и 10~6. При титровании
более разбавленных растворов
солей закругление кривых
усиливается.
Для установления с
достаточной точностью положения точек эквивалентности на
кривых кондуктометрического титрования необходимо,
чтобы три четверти кривой от начала титрования до
точки эквивалентности имели прямолинейный характер. При
этих условиях, в случае образования осадков бинарных
электролитов, их произведения растворимости при титр'о-
0,2
Количество до
Пленного осадителя, г-экв/л
Рис. 23. Влияние
растворимости осадка на
кривые
кондуктометрического титрования 0,1 н.
растворов солей,
основанного на образовании
осадков с катионом
титруемой соли:
произведение растворимости
осадков: / — 10 4; 2—10~5;
3 — 10—". Подвижности
ионов:
*Kt+
■70;
XKt+ - 50:
= 50; X
Ail"
вании растворов различных концентраций должны иметь
следующие значения:
Концентрация раствора 0,1 0,01 0,001 н.
Произведение
растворимости <2,5-10-5 <2,5-10-7 <2,5-10-9
Для уменьшения растворимости осадка широко
используют метод кондуктометрического титрования
водно-органических растворов. Понижение растворимости
осадка достигается, если в качестве среды для
титрования использовать раствор, насыщенный осаждаемым
малорастворимым электролитом,- а также путем
титрования растворов при пониженной температуре (ледяная
ванна). Титрование при высоких температурах
используется только в тех случаях, когда нужно увеличить
скорость реакции, или тогда, когда при комнатной
температуре не удается получить достаточно чистые осадки.
§ 2. Влияние подвижностей ионов на характер
кондуктометрических кривых титрования
При копдуктометрическом титровании, основанном
на использовании реакций осаждения, ионы титруемого
вещества в растворе заменяются ионами осадителя.
Если в состав осадка входят катионы титруемого
вещества, они при титровании заменяются катионами
осадителя Если же в состав осадка входят анионы титруемого
электролита, они заменяются при титровании анионами
осадителя. Поэтому изменение электропроводности
раствора до точки эквивалентности в случае, когда
происходит полное осаждение, зависит от относительной
подвижности осаждаемых ионов и заменяющих их в
растворе ионов осадителя. Если подвижность осаждаемых
ионов выше подвижности заменяющих их ионов
осадителя, электропроводность раствора понижается до точки
эквивалентности. Когда подвижности ионов равны,
электропроводность остается постоянной. Если же
подвижность осаждаемых ионов ниже подвижности
заменяющих их ионов осадителя, электропроводность раствора
увеличивается до точки эквивалентности.
Рассмотрим случай количественного взаимодействия
между ионами, при котором в состав осадка входит
катион определяемого вещества.
Примем, что концентрация титруемого раствора 0,1 н.,
произведение растворимости Ю-8, подвижности анионов
83
обеих солей равны среднему значению подвйжностей
ионов в водных растворах (XAnf = ЛАп7 = 50), а
подвижности катионов изменяются в пределах: tacti" = 70; tact2"=
= 30 (рис. 24, кривая У); XKt? = taa2f = 50 (кривая 2);;
Я.кг!+ = 30; tact2+ =70 (кривая 3).
Как видно из рис. 24, кривые титрования имеют
различную форму. Наиболее резкий излом кривой имеет
место тогда, когда электропроводность раствора понижается
до точки эквивалентности, что наблюдается в тех
случаях, когда подвижность осаждаемых катионов выше
подвижности катионов осадителя. Поэтому при выборе
осадителя следует отдавать предпочтение такому
веществу, ионы которого, накапливающиеся в растворе
взамен осаждаемых, имеют по возможности низкую
подвижность.
Изменение электропроводности раствора после точки
эквивалентности вызывается избытком титранта. Если в
качестве титранта используется сильный электролит,
электропроводность раствора после точки
эквивалентности увеличивается. Чем выше подвижности ионов
осадителя, тем более сильно повышается электропроводность
раствора. Так как для понижения электропроводности
раствора до точки эквивалентности необходимо, чтобы
ионы осадителя имели низкую подвижность, нужно
подбирать осадитель, у которого ионы противоположного
заряда обладают высокой подвижностью. Если
осаждаются катионы определяемого вещества, анионы
осадителя должны иметь высокую подвижность и, наоборот,
если в состав осадка входят анионы титруемого
электролита, катионы осадителя должны иметь высокую
подвижность.
Во.зьмем, например, реакцию осаждения, в которой
участвуют катионы определяемого электролита. На
рис. 25 представлены кривые кондуктометрического
титрования солей при различных условиях. Как видно из
рис. 25, чем выше подвижность аниона осадителя (Ап2~),
тем более резко возрастает электропроводность
раствора после точки эквивалентности и угол излома
становится более острым.
Таким образом, подвижности ионов оказывают
сильное влияние на форму кондуктометрических кривых
титрования и угол излома кривых в точке эквивалентности,
а следовательно, и на точность определения.
84
Со
О
Ǥ
Q
Q
§■
О;
О
•а
сь
ив
ив
1,2
1,0
0,8
ив
UB
иг
1,0
0,6
ив
UB
ич
иг
ио
0,8
- 1 I
\'V
\
- г j /
I
-о
U8
ив
ичиг
1,0
0,8
1,8
1,6
1М
иг
ио
0,8
U
1,В
ичиг
ио
0,8,
0 0,1 0,2
Количество добавленного
осадителя, г-экВ/л
О 0,1 0,%
Количество добавленного
осадителя, г-эк8/л
Рис. 24. Влияние
подвижностей осаждаемых катионов
титруемой соли на кривые
кондуктометрического
титрования 0,1 н. растворов
солей, основанного на
образовании осадка
с ПРк4Ап = Ю-8:
подвижности ионов: X =>
-ХАпГ = 50; 7-XKt+-70' XKt+ "
XKt+ " 30'
xKt+-70
Рис. 25. Влияние'
подвижностей анионов осадителя иа
кривые
кондуктометрического титрования 0,1 и.
растворов солей, основанного на
образовании осадка с
катионом титруемой соли
сПРК4ап = 10-8:
подвижности ионов: A _j_ .
Х„, , =30; X, _
Kt+ Апх
-50; /—X. _■
АП2
70;
■ 30;
2-Х. _ = 50; 3-Х, _=>70
Ап2 АП2
85
§ 3. Требования в отношении постоянства
состава осадка, его чистоты и скорости осаждений
Кондуктометрические определения могут быть
основаны на реакциях осаждения, если осадки имеют
определенный состав, т. е. состав осадка не обязательно
должен строго соответствовать стехиометрическим
отношениям, но очень важно, чтобы он не изменялся в процессе
титрования. Эта особенность представляет большое
преимущество метода кондуктометрического титрования.
Если состав осадка не соответствует стехиометрическим
отношениям, концентрацию раствора осадителя
устанавливают кондуктометрическим методом путем титрования.
Реакции осаждения, при которых в процессе
титрования состав осадка изменяется или образуются
соединения различного состава, не могут быть использованы для
количественных определений. Изменение состава осадка
может быть связано с гидролизом, комплексообразора-
нием, выделением смешанных кристаллов и т. д.
Например, известно, что при осаждении гидроксидов тяжелых
металлов в ряде случаев образуются основные соли
различного состава. По той же причине в большинстве
случаев не удается использовать реакции осаждения
малорастворимых карбонатов и др.
Осложнения в анализе связаны также с загрязнением
осадков вследствие соосаждения и других причин. При
этом изменяются концентрации ионов, а следовательно,
и электропроводность раствора, что может привести к
искажению формы кондуктометрической кривой и
несоответствию ее излома точке эквивалентности. Следует
учитывать, что при кондуктометрической титровании, в
отличие от гравиметрического метода анализа, не всегда
возможно создать условия, при которых соосаждение
сводится к минимуму. Например, нецелесообразно под-
кисление растворов сильными кислотами с целью
получения более крупных кристаллов, так как
электропроводность раствора при титровании будет мало изменяться.
В случае необходимости изменения рН среды добавляют
слабые кислоты или основания. Как известно, более
крупные кристаллы могут быть получены при осаждении
из горячих растворов горячими растворами осадителей.
Хотя кондуктометрическое титрование горячих
растворов и осуществляется, однако его проведение связано с
определенными трудностями.
86
При выделении аморфных осадков наблюдается
адсорбция. Поэтому следует добиваться образования
крупнокристаллических осадков, для чего осаждение ведут
из разбавленных растворов при медленном добавлении
реагента и энергичном перемешивании. В этих условиях
уменьшается также окклюзия. Это следует учитывать
при кондуктометрической титровании.
В ряде случаев для уменьшения адсорбции вводят
добавки веществ, которые адсорбируются легче, чем
определяемые ионы. Например, при кондуктометрической
определении ионов магния, основанном на осаждении
его гидроксида, предложено добавлять в раствор гидро-
ксид бария (или калия), используемого в качестве
осадителя, 0,1—0,25% магнезона II. С целью уменьшения
адсорбции на осадке оксалата кальция рекомендуют
добавлять насыщенный раствор метилового фиолетового.
Этот реактив используют также при осаждении
сульфатов хлоридом бария.
При титровании могут быть использованы только
реакции, протекающие с высокой скоростью. Если
реакция протекает медленно, то кондуктометрическое
определение практически невозможно. Мешает определению
также образование пересыщенных растворов. Например,
медленное осаждение имеет место при титровании 0,01 н.
раствора сульфатов солями бария. После добавления
каждой порции осадителя электропроводность
становится постоянной через 10 мин. Для ускорения осаждения
рекомендуют сульфат бария вносить в раствор в виде
суспензии. Лучше действует добавка 25—50% спирта.
Если титрование проводят в смесях электролитов и
реакция осаждения не избирательна по отношению к
определяемому иону, применяют маскировку. Например,
кондуктометрическое определение содержания кальция в
известковых породах (осаждение оксалата кальция) в
присутствии ионов алюминия, железа, магния и марганца
возможно, если эти металлы в степени окисления +3
связать в прочные комплексы. Комплексообразование с
тиосульфатом применяют для маскировки меди при
кондуктометрической титровании цинка комплексоном III.
При кондуктометрической определении меди цитратом
лития ионы алюминия и железа (III) маскируют
фторидом аммония.
87
§ 4. Определение анионов, осаждаемых в виде
мвлорастворимых соединений
При кондуктометрическом определении анионов
используются разнообразные реакции осаждения.
Некоторые бсадители позволяют проводить определение
различных ионов. К ним относятся реакции осаждения
малорастворимых солей серебра, ртути, бария, свинца, тория и др.
Нитрат серебра. Титрование нитратом серебра
позволяет определять многие анионы С1~, В г- 1~, CN-, SCN-
Сг042-, С2042- [Fe(CN)6]3-, V033-, тартраты, цитраты, са-
лицилаты, соли янтарной и других кислот. При
титровании осаждаются малорастворимые соли серебра.
Изменение электропроводности раствора до точки
эквивалентности зависит от сравнительной подвижности
осаждаемых анионов и заменяющих их в растворе
нитрат-ионов (Яж>;Г=71,5). Более высокую подвижность
имеют некоторые анионы, в том числе С1_, Вг_, I-, Сг042-,
C2042_,[Fe(CN)6]3_ (см. табл. 1); электропроводность
раствора при взаимодействии этих ионов с нитратом серебра
немного понижается. Если подвижность определяемых
анионов ниже подвижности нитрат-ионов,
электропроводность раствора до точки эквивалентности несколько
увеличивается. Например, это имеет место при титровании
SCISh-ионов. Избыток осадителя во всех случаях
вызывает сильное повышение электропроводности
раствора.
Некоторые особенности наблюдаются при нитровании
цианидов. Сначала образуются комплексные [Ag(CN)2r"-
ионы, что приводит к понижению электропроводности
раствора. После окончания процесса комплексообразова-
ния кондуктометрическая кривая титрования имеет излом.
Затем 'происходит осаждение AgCN, что сопровождается
•повышением электропроводности раствора до точки
эквивалентности. Более сильное повышение
электропроводности наблюдается после точки эквивалентности.
Характерное поведение цианидов используется при анализе их
смесей с хлоридами. При титровании смеси сначала
образуется комплексный цианид, затем выделяется осадок
AgCN и, наконец, осаждается AgCl.
Минимальные концентрации титруемых растворов
могут быть установлены, исходя из растворимости
осадков. Например, серебряные соли салициловой, щавелевой,
лимонной и янтарной кислот довольно сильно раствори-
88
МЫ; титрование этих анионов Возможно в растворах с
концентрацией не ниже 0,1 н. С другой стороны,
определение хлоридов возможно в очень разбавленных
растворах, что используется при анализе природных вод. Метод
позволяет определять количества хлорид-ионов порядка
1—10 мкг. Титрование проводят 0,001—0,01 н. раствором
AgN03 в атмосфере азота в растворе, содержащем 90%
спирта.
Перхлорат и нитрат ртути. Из солей ртути (II) для
кондуктометрического титрования широко используется
перхлорат. Эта соль довольно хорошо растворима в воде
и является сильным электролитом. Хотя перхлорат
ртути подвергается заметному гидролизу, образующиеся при
этом соли не выпадают в осадок и раствор остается
прозрачным. В качестве осадителя на практике применяют
также нитрат ртути.
Перхлорат ртути используют для определения многих
анионов — С1-, Br- I", CN~, SCN~, [Fe(CN)6]3- ацетатов,
монохлорацетатов, формиатов, лактатов, бензоатов, солей
валериановой, салициловой и других кислот.
Как известно, меркуриат-ионы образуют с многими
анионами устойчивые комплексные ионы, которые при
дальнейшем добавлении солей ртути разлагаются с
образованием малорастворимых осадков. Комплексообразова-
ние особенно характерно при взаимодействии с Вг~~, I-,
CN-, CNS-, монохлорацетат-ионами. При титровании
солей щелочных металлов и указанных кислот
сначала образуются комплексные ионы HgBr42-, Hgl42~,
Hg(CN)42-, Hg(CNS)42- Hg(CH2ClCOO)42-, что
приводит к понижению электропроводности раствора в первой
половине кондуктометрической кривой титрования до точ«
ки эквивалентности. В некоторых случаях окончание
реакции комплексообразования фиксируется резким
изломом кондуктометрической кривой (иодид-, монохлораце-
тат-ионы). При дальнейшем добавлении реагента
выделяются осадки HgBr2, Hgl2, Hg(CN)2, Hg(CNS)2,
Hg(CH2ClCOO)2, что сопровождается повышением
электропроводности раствора. Избыток реагента вызывает
более сильное увеличение электропроводности. При этом в
некоторых случаях осадки растворяются. По-видимому,
в таких случаях образуются ионы, отличающиеся
повышенной подвижностью.
Однако в ряде случаев комплексообразование слабо
выражено или совсем не протекает; осадки выделяются
89
с начала титрования. Даже в случае значительной pacf-
воримости осадков определения возможны, так как
многие соли ртути слабо диссоциированы.
Кроме перхлората ртути в качестве осадителя
используют нитрат ртути. Эта соль нормально диссоциирует в
водном растворе. С целью предотвращения гидролиза
при растворении соли добавляют азотную кислоту.
Большое значение имеет определение хлоридов в
природных водах при помощи кондуктометрического
титрования перхлоратом или нитратом ртути. Возможны
определения в очень разбавленных растворах хлоридов
(порядка 2-Ю-4 н.).
Ацетат и хлорид бария. Соли бария используют для
кондуктометрического определения ряда анионов—■
S042~", CrC>42~, СОз2~, ТеОз2-, оксалатов, тартратов,
цитратов и т. д. Наибольшее распространение в качестве осади-
телей получили ацетат и хлорид бария. Подвижность
ацетат-ионов низка (Ясн„соо"" = 40,90), поэтому при
осаждении солей бария этим реагентом электропроводность
раствора до точки эквивалентности обычно понижается.
Преимущество ацетата бария как осадителя
выражается также в том, что устраняется отрицательное влияние
сильных кислот, если они присутствуют в растворе;
ацетат-ионы взаимодействуют с ними с образованием
уксусной кислоты. Хлорид-ионы имеют более высокую
подвижность (taa~= 76,35). Изменение электроприводности
раствора при осаждении солей бария этой солью
зависит от сравнительной подвижности ионов хлора и
осаждаемых ионов.
Широкое распространение получило
кондуктометрическое титрование сульфатов. Однако точные
результаты получаются только при определенных условиях. При
титровании водных растворов на холоду образуются
пересыщенные растворы и электропроводность медленно
устанавливается до постоянной. Мешают определению
минеральные кислоты, нитраты, ионы железа и
алюминия. Сильные кислоты нейтрализуют щелочами или
другими основаниями с теми же катионами, которые
находятся в титруемом растворе. После этого раствор
подкисляют небольшим количеством уксусной кислоты.
Титрование на холоду проводят в водно-спиртовой среде
(1:1). В присутствии нитрат-ионов даже в
водно-спиртовых растворах при титровании на холоду могут быть
получены завышенные результаты. Только в разбавлен-
90
ных растворах нитрат-ионы не мешают определению.
Титрование сульфат-ионов в присутствии нитратов в
водном растворе без добавления спирта проводят при 100°.
Электропроводность раствора при этих условиях быстро
становится постоянной. С целью уменьшения ошибок,
связанных с адсорбцией ионов при осаждении сульфата
бария, рекомендуют добавлять 0,5 мл насыщенного
раствора метилового фиолетового на 100 мл титруемого
раствора. Кондуктометрическое титрование
использовано при определении небольших количеств сульфатов в
природных водах и водных вытяжках из почвы, при
определении сульфатов в цементе и клинкере, анализе
электролитных ванн на содержание сульфатов и т. д.
Хроматы титруются солями бария даже в
разбавленных растворах. При титровании карбонатов в
разбавленных растворах необходимо добавлять спирт. Оксала-
ты, тартраты, цитраты и т. п. могут быть определены
только в водно-спиртовой среде. Для цитратов в начале
титрования наблюдается комплексообразование.
Нитрат и ацетат свинца. При помощи нитрата свинца
возможно кондуктометрическое определение различных
анионов I-, [Fe(CN)6]4-, WO2,-, оксалатов, тартратов,
солей янтарной кислоты и др.
При титровании 1~-ионов в начале образуются
комплексные ионы (РЫз)~, что приводит к понижению
электропроводности раствора. Затем выделяется осадок РЬЬ
и электропроводность раствора начинает увеличиваться.
Вследствие довольно высокой растворимости
кондуктометрическое определение возможно в растворах с
концентрацией не ниже 0,05 н.
Ферроцианид взаимодействует с нитратом свинца с
образованием осадка состава Pt>2[Fe(CN)6]. Возможно
определение ферроцианида в присутствии феррициани-
да, так как последний не дает осадка с РЬ(ЫОз)г.
При титровании других указанных анионов
выделяются соответствующие средние соли свинца. Изменение
электропроводности раствора до точки эквивалентности
при осаждении солей зависит от относительной
подвижности осаждаемых ионов и заменяющих их при
титровании нитрат-ионов (^no^ = 71,5). Например, при
титровании ферроцианид-ионов (Я[Ре(см6)]4~ = 110,5)
электропроводность понижается, а при титровании вольфрама??
ионов (Муо4~*»69,4) немного пдяышаетря,
9!
Ацетат свинца использован при определении МоО2,-"
и SiOg~-HOHOB. Титрование молибдата проводят при 100°
в растворе, нейтрализованном уксусной кислотой по
метиловому оранжевому. Ацетат-ионы отличаются низкой
подвижностью; электропроводность раствора при
осаждении солей понижается до точки эквивалентности и
увеличивается после нее.
Нитрат тория. Нитрат тория используют при кондук-
тометрическом определении фторидов, кремнефтористово-
дородной кислоты и комплексных соединений,
образованных BF3 с пиридином, диметилнитрозоамином и пиколи-
ном. Определения основаны на выделении осадка ThF-j.
Титрование фторидов проводят в 50%-ном этаноле. При
определении фторидных комплексов прибавляют
Th(NOs)4 в избытке, выдерживают несколько часов и
остаток реагента титруют раствором NaF.
§ 5. Определение катионов, осаждаемых в виде
малорастворимых соединений
Кондуктометрическое титрование, основанное на
реакции осаждения, позволяет проводить определение
многих катионов. В ряде случаев одни и те же осадители
могут быть использованы для определения различных
катионов. Из них наиболее распространено осаждение
сульфатов, хроматов, оксалатов, селенитов, теллуритов,
ванадатов, вольфраматов, молибдатов и др.
Сульфат лития. Сульфат лития используют для
определения различных катионов: Ва2+, Sr2+, Pb2+ и др. Он
удобен для использования в качестве осадителя потому,
что ионы лития имеют низкую подвижность (Яы+=38,7).
При титровании солей бария (А,ва2+ = 63,6), стронция
(^sr2+ = 59,5) и свинца (А,рь2+ = 70) электропроводность
до точки эквивалентности понижается, так как
подвижности этих ионов выше подвижности заменяющих их
ионов лития.
Точные результаты получаются при титровании в
водно-спиртовых растворах (1 : 1). Кондуктометрическое
титрование позволяет определять соли бария и свинца
в очень разбавленных растворах. Концентрация
титруемого раствора ВаСЬ может достигать 0,004 н., а
РЬ(гЮз)г — 0,002 н. Соли щелочных металлов и
небольшие количества минеральных кислот не мешают опредет
лению.
82
Хромат калия и натрия. Титрование хроматом натрия
применяют для определения Ва2+, Sr2+, Pb2+, Tl3+ и
других ионов. При титровании солей указанных металлов
хроматом натрия электропроводность раствора до точки
эквивалентности понижается, так как подвижность
ионов натрия (A,Na+=50,l) ниже подвижности ионов
бария (А,ва2+=63,6), стронция (?isr2+ = 59,5), свинца
(Хръ2+=70) и таллия (A,Ti3+ = 74,7). Титрование
солей бария, свинца и таллия производят в водной
среде, а солей стронция — в водно-спиртовой среде
(1:1).
Кондуктометрическое титрование солей самария
предложено проводить хроматом калия в водно-спиртовой
среде, содержащей 30% этанола. Возможно определение
разбавленных растворов, имеющих концентрацию
5-Ю-4 г-ион1л самария. Осаждение хромата самария
Sm2(Cr04)3 сопровождается небольшим увеличением
электропроводности раствора, так как подвижность
ионов калия^ (Як+=73,50) выше подвижности ионов сама-
рид. (5ism3+ =68,3). Избыток осадителя во всех случаях
вызывает сильное повышение электропроводности
раствора.
Оксалат лития и щавелевая кислота. Многие катионы
могут быть определены путем кондуктометрического
титрования оксалатом лития. К ним относятся Са2+,
Sr2+, Ba2+, Ag+, Pb2+, Cu2+ и другие ионы. В качестве
осадителя используется соль лития, потому что ионы
лития имеют низкую подвижность (Я-и*=38,7). Для кривых
титрования перечисленных катионов характерно
понижение электропроводности раствора до точки
эквивалентности; это объясняется тем, что подвижности этих ионов
выше подвижности ионов лития.
Титрование солей серебра, свинца и меди можно
проводить в водной среде, а солей кальция, стронция и
бария, особенно в разбавленных растворах, в
водно-спиртовой среде (1 : 1).
При определении кальция хорошие результаты
получаются, когда титруют нейтральные, а также аммиачные
растворы. Возможно его определение в очень
разбавленных растворах (0,0001 н.). При определении содержания
окиси кальция в известковых породах титрование
проводят в водной среде в присутствии аммиака и унитиола.
В этих условиях ионы алюминия, железа, магния и
марганца не мещавдт рпределению.
93
Определение солей тория с лучшими результатами
осуществляется при титровании щавелевой кислотой в
водных растворах.
Селенит натрия и селенистая кислота. Для кондукто-
метрического определения марганца и кобальта
применяют селенит натрия. При титровании выделяются
осадки— MnSe03 и CoSe03. Электропроводность раствора
до точки эквивалентности изменяется мало, что
объясняется незначительным отличием подвижности ионов
марганца (Ямп2+ = 53,5) и кобальта (?ico2+=52,8) от
подвижности заменяющих их ионов натрия (A,Na+ = 50,10).
Избыток селенита натрия увеличивает электропроводность.
Титрование солей марганца проводят в водно-этаноль-
ных растворах (15—20% спирта). Титрантом служит
раствор Na2Se03 в той же среде. При осаждении
солей кобальта используют смесь метанола с водой
(1:1).
Титрование селенистой кислотой позволяет проводить
кондуктометрическое определение катионов кальция, ба-
,рия, лантана, празеодима и др. Осаждение CaSe03 и
BaSe03 проводят в водных растворах. Нормальные
селениты лантана La2(Se03)3 и празеодима Pr2(Se03)3
выделяются в смеси воды с этанолом (содержание этанола
соответственно 30% и 5—10%). При титровании
селенистой кислотой электропроводность раствора до точки
эквивалентности сильно увеличивается, а пос^е нее возрас-
стает незначительно. Рост электропроводности раствора
при осаждении селенитов селенистой кислотой
вызывается тем, что в результате реакции в растворе
накапливаются высокоподвижные ионы водорода
Са2+ + 2С1- + H2Se03 = CaSe03 + 2H+ + 2С1-
Избыток осадителя немного увеличивает
электропроводность, так как селенистая кислота — слабый
электролит (р/Са' = 2,46; р/(а2 = 7,3). Кондуктометрическое
титрование селенистой кислотой применяют для
определения малых количеств лантана; концентрация
титруемого раствора может быть 4- 10~4 М.
Теллурит натрия. Кондуктометрические определения
могут основываться на осаждении малорастворимых
теллуритов. Титрование проводят теллуритом натрия
КтагТе03. Этот осадитель позволяет проводить
кондуктометрическое определение солей различных металлов —
бария, кобальта, лантана, церия, празеодима, неодиму
94
Й др. Образующиеся осадки имеют состав: ВаТе63,
СоТе03, La2(Te03)3, Ce2(Te03)3, Pr2(Te03)3, Nd2(Te03)3.
При титровании до точки эквивалентности
электропроводность раствора немного понижается, так как
подвижность ионов натрия (?iNa2+=50,10) ниже подвижно-
стей ионов бария ?iBa2+ = 63,6), кобальта (к:о2+ = 52,8),
лантана (?iLa3+ = 69,7), церия (tae3+ = 69,8), празеодима
(ЯРг3+ = 69,6) и неодима (Яш3+ —69,4). После точки
эквивалентности электропроводность раствора
увеличивается от избытка осадителя.
В качестве среды используют смесь воды с этиловым
спиртом. При определении различных катионов
рекомендуется добавлять следующие количества спирта: бария
40—50%, лантана 15—20%, церия 30%, празеодима
10%, неодима 20%.
Метод применим в случаях анализа разбавленных
растворов. Например, концентрация солей лантана,
празеодима и неодима в титруемом растворе может
составлять ю-4 м.
Ванадат натрия. Орто- и метаванадаты могут
служить осадителями при кондуктометрическом титровании
различных солей. Ортованадат натрия применяют при
осаждении солей серебра, лантана, празеодима, самария
и др. При этом образуются осадки соответствующих ма-
Лорастворимых солей. Определение солей меди проводят
при помощи метаванадата. Состав осадка соответствует
формуле Cu(V03)2.
Электропроводность раствора при осаждении солей
несколько понижается, так как подвижности осаждаемых
катионов серебра (A,Ag+ = 61,9), лантана (Яьа3+ = 69,5%),
празеодима (Хрт3+ = 69,6), самария (^sm3+=68,5) и меди
(Яси2+=56,6) ВЫше ПОДВИЖНОСТИ ИОНОВ Натрия (^Na + =
да=50,10), заменяющих" их в процессе реакции. После
точки эквивалентности электропроводность повышается от
избытка ванадата.
Титрование AgN03 проводят в водном растворе при
рН = 8—9. Смесь воды с этанолом (10—20%)
употребляют при определении других катионов.
Концентрации титруемых растворов могут быть очень
низкими. Возможно определение лантана, празеодима и
самария, если концентрация их солей в титруемом
растворе составляет 10~4 М.
Вольфрамат натрия. Осаждение малорастворимых
вольфраматов может быть положено в основу кондукто-
95
метрического определения солей различных металлов.
Титрование вольфраматом натрия Na2W04 позволяет
проводить определение солей кобальта, кадмия,
циркония, тория, самария и др. В результате реакции
выделяются осадки соответствующих малорастворимых солей.
Изменение электропроводности раствора при
выделении осадков зависит от сравнительной подвижности
осаждаемых катионов и ионов натрия. В большинстве
случаев электропроводность понижается. Избыток осади-
теля вызывает повышение электропроводности раствора.
Добавление спиртов способствует получению более
точных результатов. Например, определение ионов
кобальта проводят в смеси воды с этанолом (1:1) при
рН = 9,4. Титрование Th(N03)4 осуществляют при
температуре 70°С в присутствии 10—40% этилового спирта.
Молибдат натрия. Кондуктометрические определения
могут основываться на образовании малорастворимых
молибдатов. Титрование раствором Na2Mo04 позволяет
определять катионы кадмия, самария, празеодима,
неодима, гадолиния. При этом образуются
соответствующие средние соли CdMo04, Sm2(Mo04)3, Pr2(Mo04)3,
Ш2(Мо04)з, Gd2(Mo04)3- Электропроводность раствора
до точки эквивалентности понижается, что объясняется
тем, что подвижности осаждаемых катионов выше
подвижности заменяющих их в растворе ионов натрия. В
большинстве случаев определения проводят в смеси воды
с небольшим количеством этанола (2,5—5,0%).
Метод позволяет определить незначительные
количества редкоземельных элементов. Возможно определение
солей празеодима, неодима и гадолиния при
концентрациях титруемых растворов 10_3—Ю-4 М.
Глава V
ТИТРОВАНИЕ, ОСНОВАННОЕ
НА РЕАКЦИЯХ КОМПЛЕКСООБРАЗОВАНИЯ
Кондуктометрические определения могут быть
основаны на реакциях комплексообразования. При этом
образующиеся комплексы должны отличаться высокой
устойчивостью, иметь определенный состав с постоянным
координационным числом. Поскольку в ряде случаев
реакции комплексообразования вызывают изменение
концентрации ионов водорода в растворе, большое влияние
на возможности определений оказывают значения рН
среды при титровании. В тех случаях, когда решение
необходимо проводить только при определенных значениях
рН, добавляют буферные растворы. Влияние буферных
растворов на характер кондуктометрических кривых
титрования может быть установлено в каждом
конкретном случае. В результате реакции комплексообразования
в растворе одни ионы заменяются другими. При
добавлении буферных растворов в титруемый раствор
вводятся новые ионы. Подвижности находящихся в титруемом
растворе ионов влияют на характер кондуктометрических
кривых титрования.
§ 1. Лиганды, используемые
в кондуктометрическом анализе
При кондуктометрическом определении солей
металлов, основанном на реакциях комплексообразования,
используются различные лиганды. Например, в основу
определения ионов алюминия положена реакция с
тартрат-ионами. Образующееся комплексное соединение
растворимо в воде. Комплексные ионы [А1(С4Н406)3]3~
отличаются высокой устойчивостью. Определение ионов
алюминия возможно в присутствии ионов железа (III).
Для определения ионов алюминия используют также
реакцию с лактат-ионами. Образующееся растворимое в
воде комплексное соединение обладает большей
прочностью, чем соответствующие комплексы железа (II),
кальция и магния. Поэтому указанные ионы не мешают
определению ионов алюминия. Ионы железа (III) дают более
прочные комплексы и мешают определению. С целью
маскировки железа (III) его восстанавливают аскор-
4-1595
97
биновой кислотой до железа (II). Оксалатные комплексы
алюминия [А1(С204)3]3- также весьма устойчивы. При
определении алюминия добавляют оксалат натрия в
избытке. Избыток оксалата натрия титруют нитратом
кальция. Кондуктометрическое определение алюминия может
основываться на образовании ацетатных комплексов.
Ионы Fe (II), кальций и магния не мешают определению.
Ионы Fe (III) восстанавливают аскорбиновой кислотой.
Ионы алюминия образуют весьма устойчивые фторидпые
комплексы [A1F6]3_. Определение алюминия, основанное
на образовании этих комплексов, возможно в
присутствии ионов Fe (II) и Мп (II).
Ионы меди (II) взаимодействуют с цитратом с
образованием растворимых в воде комплексных соединений
Си3[С3Н4(ОН) (СОО)з]~, что может быть положено в
основу их кондуктометрического определения. Ионы цинка
взаимодействуют после ионов меди и не мешают
определению. Ионы алюминия и железа (III) мешают
определению ионов меди, поэтому их маскируют фторидом
аммония.
Для определения ряда ионов металлов могут быть
также использованы реакции образования
комплексных аммиакатов. Соли, содержащие [Ni(NH3)6]2+,
[Ag(NH3)2]+, [Zn(NH3)4p+ [Cd(NH3)6]2+ [Hg(NH3)4?+
можно определить путем Добавления известного
количества кислоты, взятой в избытке, и последующего
титрования избытка кислоты основанием.
Кондуктометрическое определение ртути (II) может
быть основано на образовании Hg(CN)2. Титрование
раствором цианида позволяет проводить определение
ртути в присутствии минеральных кислот.
Реакции комплексообразования используют также
при определении анионов. Примером может служить
титрование фторид-ионов при помощи хлорида алюминия.
Если проводить титрование в присутствии хлорида
натрия, образующаяся соль Na3[AlF6] выпадает в осадок,
что способствует получению резкого излома кондукто-
метрической кривой в точке эквивалентности. В
присутствии минеральных кислот титрование проводят в
ацетатном буферном растворе, содержащем 30% спирта.
Образование малорастворимых комплексных
соединений используют и при определении катионов. Прямое
титрование ионов меди 8-оксихинолином, основанное на
образовании осадка комплексной соли состава
98
Си(С9ОбМО)2, позволяет определять ионы меди.
Титрование проводят в 50%-ном этаноле. При определении
ионов калия используют образование малорастворимой
комплексной соли тетрафенилбората калия К[(С6Н5)4В].
Определение возможно в присутствии ионов натрия,
магния, кальция. Катионы кадмия, кобальта, меди, никеля
и цинка образуют малорастворимые пиридин-роданид-
иые комплексы, отвечающие составу Me(C5H5N)2(SCN)2,
что положено в основу их кондуктометрического
определения. Поскольку пиридин-роданидный комплекс меди
растворяется в тиосульфате, этот реактив используют
для маскировки меди при анализе смесей солей меди с
солями других указанных выше металлов. Для
маскировки никеля применяют диметилглиоксим.
Предварительное осаждение никеля этим реактивом позволяет
проводить анализ смеси солей никеля с солями
кобальта, кадмия и цинка. При анализе всех перечисленных
смесей сначала определяется общее содержание солей
путем титрования роданидом в присутствии пиридина.
Широкие аналитические возможности для
кондуктометрического определения металлов открываются при
использовании комплексоноз. Из них наиболее широкое
применение нашли этилендиамиптетраацетат натрия
(комплексен III) и 1,2-циклогексилендиаминотетрааце-
тат.
§ 2. Влияние константы нестойкости комплекса,
концентрации раствора и рН среды на титрование
солей металлов комплексоном III
Как известно, комплексон III (сокращенно Na2H2Y)
представляет собой двунатриевую соль этилеидиамин-
тетрауксусной кислоты (ЭДТА):
НООССН,. ,СН2СООН
>N-CH2~-CH2-N<
NaOOCCH/ 4CH2COONa
Кислотные свойства этилендиаминтетрауксусной
кислоты (H4Y) выражены слабо. Показатели констант ее
диссоциации (р/С) по четырем ступеням соответственно
равны 2,0; 2,62; 6,16; 10,16. Поэтому в растворе Na2H2Y
находятся ионы натрия и преимущественно Н2У2_-ионы.
Дальнейшая диссоциация H2Y2_-hohob на HY3~- и Y4--
ионы протекает весьма незначительно. Исходя из этого,
4* 99
реакции взаимодействия катионов металлов с
комплексном III обычно выражают уравнениями:
Ме2+ + H2Y2-^tMeY2- + 2Н+
МеЗ+ + H2Y2-^tMeY- + 2H+
Ме4+ + H2Y2-^:MeY +2H+
или в общем виде
Мел+ + H2Y2- r^MeY""4 + 2H+
Как видно из приведенных реакций, в результате
химических взаимодействий в растворах, не содержащих
буферных смесей, увеличивается концентрация ионов
водорода, что вызывает понижение рН среды. Изменение
рН среды зависит от концентрации титруемой соли, так
как выделяющиеся ионы водорода при титровании
растворов различных концентраций в различной степени
изменяют кислотность раствора.
При изменении рН раствора в нем изменяются
величины равновесных концентраций анионов ЭДТА. К тому
же в сильно кислых растворах кроме средних комплексо-
натов состава MeY4_n могут образовываться
протежированные комплексы MeHY3-™, а в сильно щелочных
растворах гидроксокомплексы MeOHY™-5, Me(OH)2Y™-6.
Рассмотрим процесс комплексообразования,
приводящий к образованию средних комплексов состава MeY4_n.
Степень связывания катионов металла в средние
комплексы зависит от константы нестойкости комплекса, рН
среды и концентрации титруемой соли.
Концентрационная константа нестойкости комплек-
соната состава Ме4~™ имеет выражение
к _ [Ме"+] [Y4-]
М Y [MeY4-"]
где MeY4-" — равновесная концентрация комплекса,
г-ион/л; [Ме"+] — равновесная концентрация катионов
металла, г-ион/л; [Y4-] — равновесная концентрация
аниона ЭДТА, г-ион/л.
Константа нестойкости комплексоиатов различных
металлов изменяется в широких пределах (р/Свду-^
^27,94, a p/W~^ 7,76).
Ш
Возьмем эквимолярную смесь соли металла и ЭДТА.
Если в этой смеси 99,9% ионов металла связано в
комплекс, то
TMeY4_"l
■±^ L = Ю00 : 1. (44)
[Ме"+]
Исходя из константы нестойкости комплекса, можно
написать
\J^T1== ГУ4"] =10oq;1. (45)
[Мел+] A:MeY4-« -
Обозначим концентрацию титруемой соли С, моль/л.
Если 99,9% катионов металла титруемой соли связаны
в комплекс, то равновесная концентрация несвязанных
в комплекс катионов металла, а следовательно, и не
вступившей в реакцию ЭДТА, составляет величину
Смел+-Ю_3. Так как при диссоциации ЭДТА получаются
ионы H3Y_, H2Y2-, HY3- и Y4~, а также остаются недис-
социированные молекулы ЭДТА, то концентрация
анионов Y4- составляет только часть общей концентрации
ЭДТА, не вступившего в реакцию в эквимолярной смеси
соли металла и ЭДТА. На основании этого получаем
[Y4-] = Сме"+ • 10-за, (46)
где а — коэффициент, показывающий, какая часть от
общей концентрации, не вступившей в реакцию ЭДТА,
находится в растворе в виде Y4_-hohob. Коэффициент а
может быть рассчитан по уравнению
а= 1 , (47)
l-|.tH+] + [Н+]8+ [Н+]3 4 [Н+]4
Ал КхКч K1K2K3 К1К2К3К4
где [Н+] — равновесная концентрация ионов водорода,
г-экв/л; /Сь /Сг, Кг, Ki—константы диссоциации ЭДТА по
четырем ступеням.
Как видно из уравнения (47), коэффициент а зависит
от рН среды. Количественная зависимость — lg а от рН
показана в табл. 5. Чем выше рН раствора, тем больше
коэффициент а. Это объясняется тем, что в щелочных
растворах происходит нейтрализация ЭДТА и в растворе
находятся преимущественно У4~-ионы. В сильно кислых
растворах, наоборот, концентрация У'Кионов очень, мала,
401
ев
о.
S
о.
S
!<
О.
s
l<S
со
■* о
t- от
И I
V V V v V
со
о
от
V V V V V
со
СМ
л
СО
СМ
л
со
V
V
л
л
со
S
от
л
г_<
СМ
л
■О"
-*
СМ
V
■О"
-*
со
V
4tf
л
«tf
^н
CN
л
-*
-*
со
о
от
со
л
о
от
оо
л
о
со
от
V
о
со
о
V
о
со
СО
1—i
Л
о
со
оо
1—1
л
о
со
о
оо
-*
л
оо
со
л
оо
ь-
V
оо
-*
оо
V
оо
-*
-*
л
-*
со
л
00
00
CM
л
-*
л
ю
ю
V
ю
СО
V
ю
^*
СМ
л
1ГЗ
-ч<
-ч<
л
Ю
со
96
о
л
СО
от
СМ
л
1
1
со
со
о
л
со
со
СМ
л
со
со
■*
63
от
л
со
со
Г-.
л
1
1
со
со
от
л
со
*-<
л
со
со
со
,59
оо
л
от
о
л
1
1
от
оо
л
OJ
о
л
от
СМ
<м
,59
ь-
л
от
от
л
1
1
от
ь-
■Л
от
от
л
а
*-,
.76
СО>
А
СО'
оо
л
1
со
со
л
со
оо
л
$
о
№
СП
102
Чем выше концентрация У4--ионов, тем при больших
значениях Кмеу4~" соблюдается зависимость
-1Х^-= 1000:1. (48)
•^MeY4-"
Поэтому катионы металлов, образующих
сравнительно малоустойчивые комплексы, титруют в щелочных
растворах. С другой стороны, катионы металлов,
образующих очень устойчивые комплексы, могут титроваться в
кислых растворах.
Если взять другие степени связывания ионов металла
в комплексы, по аналогии можно написать зависимости,
имеющие место в точке эквивалентности.
Таблица 6
Степень связывания ионов металла в комплексы
Степень связывания
ионов металла
в комплекс, %
99,9
99,0
1,0
0,1
Отношение
[Y4-]
КМеУ4-л
1000:1
100:1
1:100
1:1000
Равновесная концентрация
Y -ионов
СМе„+.10-з.а
СМе«+10~2'а
СМе"+(1-10-2)а
^"+(1-10-3)0
Если степень связывания ионов металлов составляет
только 1% или 0,1%, то в выражении для равновесных
концентраций У4~-ионов величинами 10~2 и 10~3 в
разности можно пренебречь.
Полученные зависимости позволяют определять
значение р/СмеУ4"-", необходимое для определения металла
с заданной точностью при различных величинах рН
раствора и различных концентрациях титруемой соли.
Подставив величину равновесной концентрации У4~-ионов,
найденную на основе коэффициента а, в отношение рав-
[Y4-]
новесных концентраций — и прологарифмировав,
ЛМеУ4-л
получаем:
103
Степень связывания
ионов металла
в комплекс, %
99,9
99,0
1,0
0,1
Урапнёния для р/< , при гёзличиой стейени
MeY4~n
связывания ионов металла а комплекс
PtfMeY4.-> = PCMe„+-lga + 6;
P*MeY4-"==PCMe«+-!Sa + 4;
P^MeY4-»==pC«e»+-,8a-25
В этих уравнениях величины р/СмеУ4-" и рСме"+ есть
отрицательные логарифмы константы нестойкости комп-
лекса и концентрации титруемой соли. Этими
уравнениями пользуются для установления возможности
определения катионов данного металла при помощи комплексона
III при определенных значениях рН, если известны
константа нестойкости его комплексоната и концентрация
титруемого раствора. Например, если рН раствора
равняется 4, то величина —lg a равняется 8,48. В случае
когда концентрация титруемого раствора равна 0,01 М,
а степень связывания ионов металла принята равной
99,0%, показатель константы нестойкости комплекса
должен быть равен 14,48. Металлы, образующие
комплексы с рДме4- выше этой величины, будут
взаимодействовать более полно. Если же значения рКшеУ4""
меньше 14,48, степень связывания ионов металла понижается.
При рН = 9 (—lga=l,29), той же концентрации
титруемой соли и степени связывания металлов в комплекс
величина p^MeY4-" должна быть равна 7,26. Таким
образом, в щелочных растворах могут определяться катионы
металлов, образующие сравнительно малоустойчивые
комплексы. Эти уравнения позволяют также определять
условия, при которых катионы металлов практически не
взаимодействуют с комплексоном III, что имеет большее
значение для установления возможности определения
одних металлов в присутствии других.
Возьмем для примера рассмотренные выше условия.
Если рН раствора равно 4, а концентрация титруемой
соли 0,01 М, то степень связывания будет 1%, если
рДмеу4-л =8,48. Если комплексонат металла
характеризуется величиной рДмеУ- ниже этого значения, то
степень связывания ионов металла будет еще меньше.
Поэтому металлы, имеющие p^MeY4-" ^ 14,48, могут опре-
104
деляться в 0,01 М растворах при рН = 4 в присутствии
металлов, для которых р/Смеу4_л ^8,48.
Таким образом, теоретическим путем могут быть
установлены условия, при которых возможны определения
катионов различных металлов, отличающихся
константами нестойкости комплексонатов.
Как было указано выше, для титрования солей
металлов используют ЭДТА (двунатриевую соль этиленди-
аминтетрауксусной кислоты), так как сама кислота
(ЭДТУ) плохо растворима в воде. Таким образом, два
кислотных эквивалента этой кислоты уже
нейтрализованы. Титрование солей комплексоном III иногда проводят
в растворах, не содержащих буферные смеси, а иногда
в их присутствии. Рассмотрим условия титрования солей
без буферных растворов. Как видно из приведенных
ранее реакций, при взаимодействии солей металлов с
комплексоном III катионы металла переходят в комплексные
ионы и в растворе увеличивается концентрация ионов
водорода. Если реакция протекает количественно,
концентрация ионов водорода в точке эквивалентности
практически в два раза выше концентрации титруемой соли.
Поэтому раствор имеет довольно низкие значения рН,
что может вызвать обратимость реакций. При этих
условиях могут количественно взаимодействовать только
катионы, образующие очень устойчивые комплексонаты.
К ним относятся FeY- (p/(FeY~ = 25,16), ThY-(pKThY_ =
= 23,2), TIY- (pKtiy- = 21,5), CuY2- (pKcuY-= 18,8),
NiY2- (ptf№Y2-= 18,62). PbY2^ (p#PbY2-= 18,04) и др. При
титровании без буферных растворов после точки
эквивалентности ионы водорода вступают в реакцию с H2Y2~-
ионами, которые находятся в растворе при добавлении
избытка комплексона III. При этом образуются H3Y--
ионы, а также недиссоциированные молекулы ЭДТА.
Более широкие возможности создаются при
титровании в буферных растворах. Ионы водорода,
выделяющиеся в результате реакций, взаимодействуют с
буферной смесью и рН раствора поддерживается постоянным.
При помощи приведенных выше уравнений может быть
подсчитано значение рН раствора, при котором реакция
протекает количественно для каждой конкретной соли.
Установлены также общие зависимости, показывающие,
какое значение р/СмеУ 4~п должен иметь комплекс при
титровании растворов солей различных концентраций с
заданной степенью связывания металла в комплекс при
105
различных значениях рН раствора. В табл. 5 приведены
величины рК меу4~л , которые должен иметь комплексо-
нат металла при титровании 0,01 и 0,005 М растворов
солей, если степени связывания металлов в комплекс
принять равными 99,9; 99,0; 1,0 и 0,1%, а значение рН
раствора — изменяющимся в пределах 1-М0. Данные
таблицы позволяют устанавливать значение рН,
необходимое при титровании 0,01-=-0,005 М растворов солей
катионов металлов, отличающихся константами
нестойкости комплексов, и устанавливать условия, при которых
одни катионы можно определять в присутствии других.
Эти сведения могут быть положены в основу анализа
смесей солей металлов. При этом проводят
последовательное титрование компонентов при различных
значениях рН раствора.
При титровании в буферных смесях катионы металла
переходят в комплексные ионы. Концентрация ионов
водорода при титровании сравнительно мало изменяется
вследствие буферного действия смеси. При этом в кислых
буферных смесях уменьшается концентрация соли
слабой кислоты, а в щелочных буферных смесях —
концентрация слабого основания. При избытке комплексона III
в раствор вводятся Н2У2~-ионы. Однако в кислых
буферных смесях часть этих ионов переходит в Н3У_-ионы, а
в щелочных — в НУ3_-ионы.
§ 3. Кондуктометрическое титрование солей металлов
комплексоном III
Рассмотрим условия и типичные кривые кондуктомет-
рического титрования солей металлов комплексоном III
в растворах, не содержащих буферных смесей. При
титровании до точки эквивалентности в растворе
увеличивается концентрация высокоподвижиых ионов водорода.
Кроме этого в раствор вводятся ионы натрия. Хотя
подвижность комплексонатов металлов несколько ниже
подвижности образующих их катионов металла, это не
может компенсировать повышение электропроводности
раствора, связанное с увеличением концентраций ионов
водорода и ионов натрия. Поэтому электропроводность
раствора до точки эквивалентности сильно
увеличивается. При избытке титранта Н2У2~-ионы начинают
взаимодействовать с ионами водорода. Несмотря на то что в
растворе увеличивается концентрация ионов натрия, эле.-»
106
ктропроводность раствора после точки эквивалентности'
сильно понижается. Таким образом, точки
эквивалентности расположены в максимумах на кондуктометрических
кривых.
На рис. 26 приведена кривая титрования 0,005 М
раствора NiCl2 комплексоном III, которая типична для
рассматриваемых случаев. Следует отметить, что
концентрация титруемого раствора оказывает влияние ни
форму кондуктометрической кривой после точки
эквивалентности. Чем выше кон- ,
щентрация титруемой соли,
тем выше концентрация
йотов водорода в точке
эквивалентности и тем сильнее их
влияние на понижение
концентрации Н2У2~-ионов. При
титровании очень
разбавленных растворов концентрация
ионов водорода мало
увеличивается и понижение
электропроводности раствора пос-
.ле точки эквивалентности
менее сильно выражено.
Даже могут иметь место
случаи, когда повышение
электропроводности раствора, обусловленное ионами натрия,
может частично компенсировать понижение
электропроводности раствора, связанное с понижением
концентрации ионов водорода. При этих условиях
электропроводность раствора после точки эквивалентности может
оставаться постоянной или даже несколько увеличиваться.
Кривые титрования солей металлов комплексоном III
•в присутствии буферных растворов носят другой
характер. Выделяющиеся при реакции комплексообразования
ионы водорода взаимодействуют с буферной смесью. В
кислых буферных смесях с ионами водорода
взаимодействуют анионы слабой кислоты, что приводит к
уменьшению их концентрации. В щелочных буферных смесях
ионы водорода нейтрализуют слабое основание, что
вызывает повышение концентрации их катионов. Кроме
этого, при титровании в растворе равномерно
увеличивается концентрация ионов натрия. Поэтому
электропроводность раствора до точки эквивалентности в большей
или меньшей степени повышается. Однако это повыше-
05ъем растВара
комплексона Ж, мл
Рис. 26. Кривая кондукто-
метрического титрования
0,005 М раствора №С12
комплексоном III
107
ниё значительно менее сильное, чем при титровании без
буферных смесей. После точки эквивалентности
электропроводность раствора сильно увеличивается от избытка
титранта. Титрование солей металлов комплексоном III
обычно проводят в разбавленных растворах (0,01 —
0,005 М и ниже). При этих условиях снижается
электропроводность раствора, вызываемая буферной смесью,
так как ее можно вводить в меньших количествах. Это
способствует получению более резких изломов кривой
титрования в точке эквивалентности. На рис. 27
показана кривая титрования 0,005 М раствора Ca(N03)2
комплексоном III в
аммиачном буферном растворе
при рН~ 10. Такого
типа кривые получаются
при титровании в
слабокислых и щелочных
буферных растворах.
На практике использу-
Одьем раствора комплексамиШ,мл ются ацетатный, амми-
Рис. 27. Кривая кондуктометрического ачный и борно-щелоч-
титрования 0,005 М раствора Ca(N03)2 НОЙ буферные раство-
комплексоном III при рН~10 ры. В ацетатный
буферный раствор
целесообразно вводить ацетат лития, а не ацетат натрия, так как
подвижность ионов лития ниже подвижности ионов
натрия.
Установлено, что кондуктометрическое титрование
катионов различных металлов в 0,01—0,005 М растворах
следует проводить при следующих значениях рН: Fe
(III) при рН=1ч-3; Си, Ni, Pb, Zn, Cd, Co и Al при рН =
= 4; Fe (И) и Мп при рН = 5, а Са, Mg, Sr и Ва при рН =
= 8-М0.
§ 4. Титрование смесей солей металлов
комплексоном III
Кондуктометрический метод может быть использован
при анализе смесей солей металлов. При этом
используется возможность определения катионов металлов,
образующих более устойчивые комплексы, в присутствии
катионов металлов, образующих менее устойчивые
комплексы.
При установлении возможностей
кондуктометрического анализа двухкомпонентных смесей солей металлов
108
могут быть использованы данные табл. 5. Например,
определение Fe (III) (p/C = 25,16) в 0,01—0,005 М
растворе возможно в присутствии Zn, Co, Cd, Al, Fe (11), Мп,
La, Mg, Ca, Sr или Ва и их смесей. Значения рДмеУ4-"
этих металлов изменяются в пределах 16,50^-7,76.
Сначала проводят титрование железа в нейтральном или
слабокислом растворе, а затем титруют вторую соль,
добавив буферный раствор с таким значением рН, при
котором возможно
титрование второй соли. На §.
рис. 28 приведена кри- *g
вая титрования смеси §g
Fe (III) и Fe (II) комп- «<§
лексоном III. Титрова- J1!
ние сульфата железа Ji^
(II) проведено при =»
рН = 5. Титрованию Объем раствора комплексами Ш,мп
Fe (III) соответствует
участок кривой ДО пер- Рис 28. Кривая кондуктометрического
вой точки эквивалент- титрования комплексоном III смеси
ности, а титрованию О.005 М растворов Fe(N03)3 и FeS04
Fe (II)—участок
кривой, соответствующий избытку комплексом III при
первом титровании и участку кривой до излома при втором.
Катионы Cu2+, Ni2+, Pb2+, Zn2+, Cd2+ и Со2+,
образующие менее устойчивые комплексы (р^С= 18,8—16,31),
можно титровать в смесях с катионами металлов (Са2+,
Mg2+, Sr2+ или Ва2+), образующих сравнительно
малоустойчивые комплексы (р.К= 10,96^-7,76). Соли меди,
никеля, свинца, цинка, кобальта и кадмия титруются
первыми, поэтому их титрование проводят без
добавления буферных растворов. При титровании солей кальция,
магния, стронция и бария добавляют буферный раствор
срН = 8-Ы0.
Используя данные табл. 5, возможно обосновать
анализ других смесей теоретически.
Глава VI
ТИТРОВАНИЕ, ОСНОВАННОЕ НА РЕАКЦИЯХ
ОКИСЛЕНИЯ — ВОССТАНОВЛЕНИЯ
§ 1. Влияние условий проведения реакций
на возможность определения окислителей
и восстановителей методом кондуктометрического
титрования
Полнота прохождения реакций окисления —
восстановления может быть установлена обычными методами,
исходя из величин окислительно-восстановительных
потенциалов взаимодействующих веществ. Для смещения
реакций в нужном направлении создают определенные
условия.
Особое значение для кондуктометрических
определений имеет рН среды. При больших концентрациях
водородных или гидроксильных ионов раствор имеет
высокую электропроводность, что создает неблагоприятные
условия для кондуктометрических определений, так как
в процессе титрования электропроводность сравнительно
мало изменяется. Поэтому при титровании используются
по возможности слабокислые или слабощелочные среды.
В этих условиях изменение концентрации
высокоподвижных водородных или гидроксильных ионов в процессе
реакций, наоборот, способствует изменению
электропроводности раствора при титровании.
Поскольку электропроводность раствора зависит от
концентраций и подвижностей всех ионов,
присутствующих в растворе, следует учитывать, что в случае
добавления комплексообразователей с целью изменения
окислительно-восстановительных потенциалов кривые
титрования могут носить другой характер. Изменение состава
ионов в растворе при течении сопряженных реакций
также влияет на электропроводность раствора.
Большое значение при кондуктометрическом анализе
имеет скорость реакций. Реакции окисления —
восстановления протекают с измеримой скоростью, меняющейся
в зависимости от условий их проведения. Медленно
протекающие реакции не могут быть использованы при
титровании. Скорость реакций окисления — восстановления
зависит от концентрации реагирующих веществ и рН сре-
110
ды, температуры, присутствия катализаторов и т. д. В
каждом конкретном случае следует выбирать
оптимальные для кондуктометрического анализа условия.
Увеличение концентраций реагирующих веществ с
целью повышения скорости реакций допустимо до
определенных пределов. Однако увеличение кислотности или
щелочности раствора должно быть минимальным.
Повышение температуры создает определенные трудности, так
как при титровании необходимо термостатирование. Если
в качестве катализаторов используются электролиты,
электропроводность раствора при их добавлении
увеличивается, что в общем неблагоприятно для
кондуктометрического титрования, так как вследствие этого
изменения электропроводности раствора в процессе реакции
менее резко выражены. Поэтому концентрация
катализаторов также должна быть минимальной.
Таким образом, если в основу кондуктометрического
определения положены реакции окисления —
восстановления, большое значение оказывают условия их
проведения. Изменение условий при кондуктометрическом
титровании может оказывать существенное влияние на
характер кондуктометрических кривых и возможности
кондуктометрических определений.
§ 2. Окислительно-восстановительное титрование
Ниже приводится пример кондуктометрического
титрования арсенитов. Кондуктометрическое титрование ар-
сенитов основано на окислении их иодом. Титрование
проводят в присутствии бикарбоната. В качестве титраи-
та используют водно-спиртовой раствор иода. При
титровании As033~ окисляется до As043~, и в растворе
накапливаются 1--ионы, отличающиеся довольно высокой
подвижностью (?ц- = 78,84). Выделяющиеся в процессе
реакции ионы водорода взаимодействуют со
сравнительно мало подвижными НС03_-ионами (А,нсоз~ = 44,50) с
образованием С02 и Н20. Вследствие увеличения
концентрации более подвижных ионов происходит
повышение электропроводности раствора до точки
эквивалентности. При титровании водно-спиртовым раствором иода
электропроводность титруемого раствора после точки
эквивалентности мало изменяется.
Кондуктометрпчсский метод отличается некоторыми
преимуществами по сравнению с другими методами тит-
111
рования: 1) определение возможно в окрашенных и
мутных растворах; 2) метод применим для определения
малых количеств вещества. При титровании очень
разбавленных растворов (0,0005 н.) используют бидистил-
лированную воду и определение проводят в атмосфере
азота. Для уменьшения адсорбции пользуются гладкими
платиновыми электродами.
Определение меди осуществляют прямым кондукто-
метрическим титрованием тиосульфатом натрия. При
титровании CuSC>4 раствором Na2S203 в присутствии
роданида калия протекает реавдия
2Ci^+ + 2S2O3- + 2SCN- = J, 2CuSCN + S40^_
1С <=>
.Объем раствора К2Сг2Оь мл
Рис. 29. Кривая кондуктометрн-
ческого титрования ионов
железа (II) бихроматом калия
В процессе титрования в растворе уменьшаются
концентрации всех ионов, стоящих в левой части уравнения,
и увеличиваются концентрации ионов натрия и тетратио-
нат-иоиов. Электропроводность раствора до точки
эквивалентности немного увеличивается, а после нее растет
более сильно от избытка тиосульфата. Возможно
обратное титрование раствора ЫагБгОз, содержащего KSCN,
раствором CuSC>4. В этом случае электропроводность
раствора в процессе реакции немного понижается.
Избыток C11SO4 вызывает повышение электропроводности
раствора. Концентрация титруемого раствора должна
соответствовать 0,01—0,05 М, концентрация роданида
калия должна быть в 2—3 раза выше, концентрация тпт-
ранта 0,1 н.
112
Возможно кондуктометрическое титрование ионов
Fe (II) бихроматом калия
6Fe2+ + Сг20^~ + 14H+ = 6Рез+ + 2Сгз+ + 7Н20
Изменение электропроводности раствора до точки
эквивалентности связано преимущественно с тем, что в
процессе реакции в растворе уменьшается концентрация
высокоподвижных ионов водорода. Это вызывает сильное
понижение электропроводности до точки
эквивалентности. После точки эквивалентности электропроводность
раствора мало изменяется. Избыток титранта не
вызывает заметного увеличения электропроводности. Кривая
титрования ионов Fe (II) показана на рис. 29.
АППАРАТУРА И ТЕХНИКА
КОНДУКТОМЕТРИЧЕСКОГО МЕТОДА АНАЛИЗА
Глава VII
ИЗМЕРЕНИЕ ЭЛЕКТРОПРОВОДНОСТИ
РАСТВОРОВ
§ 1. Мост Кольрауша
Для измерения электропроводности используют мост
Уитстона с питанием по методу Кольрауша —
переменным током от ламповых генераторов звуковой частоты.
Мост (рис. 30) состоит из четырех сопротивлений, одно
из которых — сопротивление раствора (Ri). Его
определяют путем сравнения с эталонным сопротивлением.
Сопротивления моста Ri, R2 и R$ можно подобрать так,
чтобы ток в диагонали моста отсутствовал, т. е.
сопротивление его ветвей было пропорционально друг другу. При
этом измеряемое сопротивление раствора Ri вычисляют
из зависимости
/?4 = /?з£-. (49)
■"■1
В качестве переменного сопротивления Rz (плечо
сравнения) берут магазин сопротивлений и
устанавливают его на величину, близкую сопротивлению раствора
электролитической ячейки. Два других плеча,
включающие R\ и R2, образованы струной реохорда. Подвижной
контакт изменяет отношение плеч и величину R2/Ri.
Струна реохорда обычно имеет длину 1000 мм. Когда
мост'уравновешен, находят длину плеча а,
соответствующего сопротивлению R2. Тогда длина плеча Ь, имеющего
сопротивление Ri, равна 1000 — а. Сопротивление
раствора вычисляют по формуле
44 V3 ЮОО — я v '
Показанные зависимости равновесия моста
применимы к переменному току, если плечи моста составлены ак-
114
тивными сопротивлениями.
При измерении
электропроводности раствора с
помощью моста переменного
тока к активному
сопротивлению добавляется
некоторое реактивное
сопротивление, вызываемое наличием
емкости электролитической
ячейки и цепи.
Рассмотрим
электрическую эквивалентную схему
ячейки, представленную на
рис. 31. Для определения
удельной
электропроводности раствора необходимо
знать истинное активное
сопротивление раствора R,
которое зависит от
концентраций ионов и их
эквивалентной электропроводности. Но
при измерении
сопротивления раствора включаются
дополнительные активные и
реактивные сопротивления.
Электролитическую ячейку,
содержащую электролит с
погруженными в него
электродами, в принципе можно
рассматривать как
конденсатор с электродной
поверхностью S и электродным
расстоянием /, заполненный
раствором с диэлектрической
проницаемостью е.
Сопротивление емкости Сг двух
параллельных электродов,
шунтирующее истинное
сопротивление электролита в
Рис. 30. Схема моста
Кольрауша:
/ — источник тока; 2—
электролитическая ячейка; 3 — нуль-инструмент;
4 — подвижной контакт; ЛВ —
струна реохорда; R3 — известное
сопротивление
-II
Сг
R
-си-
нн
я»
-CZD-
-и
Рис. 31. Электрическая
эквивалентная схема ячейки:
R — истинное сопротивление
раствора; С — геометрическая емкость
ячейки; Сд —емкость двойного
слоя; С и R—емкость и
сопротивление поляризации; С] и Ri —
шунтирующие емкость и
сопротивление, зависящие от конструкции
ячейки; Сч — емкость проводов
Рис. 32. Четырехплечий мост:
Я], #2, Яз и Rt — плечи моста; С3 —
переменная емкость; / — звуковой
генератор; 2—индикатор нуля; 3 —
электролитическая ячейка
2 ^\>
?>^
Г^)3
\Ж
1
115
вбдных растворах, обычно значительно выше истинного
сопротивления раствора и поэтому не вызывает ошибок
в измерении электропроводности. Однако при очень
высоком истинном сопротивлении раствора эти величины
могут быть соизмеримы, что может привести к ошибкам.
Ошибки уменьшаются с понижением частоты тока и
увеличением константы сосуда.
На границе металлический электрод—раствор
электролита возникает двойной электрический слой. Емкость
двойного слоя Сд влияет на сдвиг фаз между током и
напряжением, что приводит к ошибкам в измерении
истинного сопротивления раствора. Электрохимические
процессы на электродах (разрядка ионов) приводят к
изменению концентрации ионов у поверхности
электродов. Вследствие медленной диффузии ионов к электроду
наблюдается концентрационная поляризация, которая
создает поляризационную емкость Сп и поляризационное
сопротивление Rn- Эти эффекты уменьшаются с
повышением частоты тока, с уменьшением плотности тока и
увеличением концентрации электролита.
Ошибки могут быть связаны с неудачной
конструкцией электролитической ячейки; близкое расположение
проводов, идущих от электродов, неудачное расположение
электродов по отношению к проводам и т. д. При этом
сопротивление R шунтируется емкостью С\ и
сопротивлением R\. Емкость проводов С2 также может стать причиной
емкостных утечек тока.
Таким образом, различные эффекты могут быть
причиной ошибок при измерении истинного сопротивления
раствора. С целью уменьшения ошибок необходимо
выбирать оптимальные частоту и плотность тока,
концентрацию электролитов, конструкции электролитической
ячейки, материал электродов, площадь электродов и
расстояния между ними, а также состояние их поверхности.
Вследствие наличия емкости силу тока в диагонали
моста нельзя свести к нулю. Поэтому емкость
компенсируют включением конденсатора С3 параллельно
сопротивлению Rz моста (рис. 30). Схема четырехплёчего
моста с подключенным конденсатором показана на рис. 32.
Мост балансируют одновременной компенсацией
небаланса моста сопротивлением i?3 и емкостью С3. Однако
не удается полностью устранить ошибки, связанные с
поляризационным сопротивлением, следует стремиться
свести их к минимуму.
116
Большое влияние на величину поляризационного сб-
противления оказывает материал электрода. Наименьшее
поляризационное сопротивление наблюдается на
платиновых электродах, покрытых платиновой чернью.
Увеличение истинной поверхности электродов при
платинировании уменьшает плотность тока поляризации и снижает
поляризационный эффект. Несимметричное
расположение электродов увеличивает поляризационное
сопротивление и поляризационную емкость. Повышение частоты
переменного тока снижает поляризационный эффект.
При частоте тока выше 1000 гц влияние поляризации
незначительно. С увеличением концентрации величина
поляризационного сопротивления уменьшается
вследствие уменьшения градиента концентрации. Однако
величина поляризационной емкости Сп увеличивается в связи
с увеличением плотности двойного слоя. С увеличением
подвижностей ионов поляризационное сопротивление
увеличивается.
Наиболее действенными средствами для снижения
ошибок, вызываемых поляризационным сопротивлением,
являются повышение частоты и уменьшение плотности
тока и применение платинированных электродов.
Применяя платинированные электроды, имеющие достаточно
большую площадь, и электролитические ячейки с
достаточно высокой константой сосуда, а также ток высокой
частоты, можно уменьшить ошибки, вызываемые
поляризационным сопротивлением. Следует учитывать, что не
всегда можно применять платинированные электроды,
так как платиновая чернь может явиться катализатором
побочных химических реакций или вызывать изменение
концентраций веществ в растворе вследствие адсорбции.
Недопустимо использование платинированных
электродов, если при измерении электропроводности происходит
механическое удаление платиновой черни, что может
иметь место, когда объектами исследования служат
концентрированные суспензии, эмульсии, пасты и т. д.
Однако в большинстве случаев применение платинированных
электродов возможно.
Исходя из изложенного выше, для измерения
электропроводности растворов применяют мосты, схема которых
в результате регулировок приводится к состоянию
равновесия ■— ток в измерительной диагонали становится
равным нулю. При этом происходит компенсация активного
сопротивления и емкости.
117
Такие мосты могут быть Собраны из стандартных
деталей. Наиболее выгодным условием работы моста
является условие равенства сопротивлений плеч ветви, что
соответствует случаю симметричной схемы. Для
уравновешивания схемы имеются два переменных элемента
моста — R3 и С3. Если диапазон измеряемых величин
изменяется в широких пределах, желательно иметь третий
переменный параметр, позволяющий скачкообразно
изменять пределы измерений. Этим третьим параметром
является отношение вспомогательных плеч R2IRi.
Для питания системы переменным током используют
генераторы ЗГ-1, ЗГ-2, ЗГ-10, ЗГИ, ЗГ-33 и др. Для
работы обычно используют переменный ток частотой
1000 гц.
Четырехплечий уравновешенный мост собирают по
схеме, показанной на рис. 32. В два плеча моста
включают эталонные магазины сопротивлений (Rt и R2). Для
этой цели могут подойти магазины сопротивлений
Р-517 М, Р-58 и др. Вместо двух сопротивлений может
быть использовано одно — струна реохорда с подвижным
контактом. В третье плечо включают магазин
сопротивлений R3 с параллельно включенным магазином емкости
С3. Из отечественных приборов может быть использован
магазин емкости Р-513 М. В четвертое плечо моста
включают электролитическую ячейку с раствором RA.
В качестве нуль-инструмента применяют телефоны,
гальванометры переменного тока или (после
выпрямления переменного тока) гальванометры постоянного тока,
осциллографы. Когда мост уравновешен, звук в
телефоне достигает своего минимума. Однако в настоящее
время телефоны используются редко, так как работать с
ними неудобно. Также сравнительно редко применяют
гальванометры переменного тока. Гальванометры
постоянного тока используются в комплекте с выпрямителями.
Весьма удобен в работе осциллографический
индикатор нуля типа ИНО-ЗМ (У2-2) (рис. 33). Этот индикатор
обеспечивает работу с мостами, питающимися токами
частот от 50 до 1000 гц. Питающее напряжение мостов
может изменяться от 0,3 до 300 в. Прибор питается от
сети переменного тока напряжением 127 или 220 в.
Балансировку моста проводят при помощи осцилло-
графической трубки. На вертикально отклоняющие
пластины электронно-лучевой трубки через селективный
усилитель низкой частоты подается напряжение с диагонали
118
моста. Одновременно на горизонтально отклоняющие
пластины подается напряжение, питающее мост. При
этом на экране электронно-лучевой трубки
вычерчивается эллипс. Уравновешивание моста сопровождается
поворотом оси эллипса и стягиванием его в линию. Полному
балансу моста будет соответствовать прямая линия,
сливающаяся с линией горизонтальной развертки. В этом
случае напряжение на выходной диагонали моста равно
нулю.
Рис. 33. Осциллографический индикатор
нуля ИНО-ЗМ (У2-2)
В качестве нуль-инструментов могут быть также
использованы электронный индикатор нуля типа Ф-510,
индикатор нуля на транзисторах типа Ф-582 и др.
Ниже дается описание выпускаемых отечественной
промышленностью мостов переменного тока, пригодных
для измерения электропроводности растворов, а также
кондуктометров.
§ 2. Реохордный мост Р38
Реохордный мост Р38 (рис. 34) является
уравновешенным мостом со ступенчато регулируемым плечом
сравнения и плавно регулируемым отношением плеч.
Мост позволяет измерять сопротивления растворов
электролитов порядка 0,3-f-30 000 ом. Питание моста
осуществляется от сети переменного тока частотой от 50 до
500 гц, напряжением 127 или 220 в через имеющийся в
119
приборе трансформатор. Рабочая область показаний
моста делится на пять пределов измерений. Каждая из них
отличается величиной сопротивления сравнительного
плеча моста №)• Эти сопротивления могут составлять
1, 10, 100, 1000 и 10 000 ом. Чем ниже измеряемое
сопротивление, тем ниже надо установить сопротивление
сравнительного плеча моста, что осуществляется при помощи
специального переключателя. Отношение плеч (R.2lR\)
может плавно регулироваться. Мост сначала грубо урав-
Рис. 34. Реохордный мост Р38
новешивают вращением рукояток плеча сравнения и
реохорда, а затем ставят нуль-инструмент в положение
«точно» и вращением рукоятки реохорда доуравновеши-
вают мост.
В качестве нуль-инструмента используется
вмонтированный в прибор гальванометр типа М314.
Погрешность при измерении сопротивлений в
пределах 0,3—30 ом не превышает 5%, а в пределах
30—3000 ом — не более 1,5%.
120
Прибор имеет электролитическую ячейку емкостью
50 мл из стекла, отличающегося высокой химической
устойчивостью, с электродами из листовой платины,
впаянными с помощью платиновых стержней. Сосуд
закрывается притертой пробкой для предотвращения
влияния углекислого газа воздуха. Однако эта ячейка
неудобна для кондуктометрического титрования, так как
приспособлена для отдельных измерений электропроводности
раствора. При кондуктометрическом титровании
возможно применение других ячеек, которые подключаются к
соответствующим зажимам.
Кроме рассмотренного моста при
кондуктометрическом титровании могут быть использованы мост
переменного тока типа Р-556, Р.-577 и др.
§ 3. Кондуктометр ММЗЧ-64
Кондуктометр ММЗЧ-64 собран по схеме
уравновешенного моста. Внешний вид прибора показан на рис. 35.
Рис. 35. Кондуктометр ММЗЧ-64
Измерение сопротивления растворов электролитов
основано на методике уравновешивания мостовой схемы,
образованной измеряемым сопротивлением и двумя
плечами отношения. Измеряемые сопротивления растворов
могут изменяться в пределах Ю-2—107 ом.
Питание прибора осуществляется от сети переменного
тока напряжением 220 в и частотой 50 гц через
встроенный генератор звуковой частоты, генерирующий
напряжение до 1150 гц.
121
Плечо сравнения (R3) состоит из трехкаскадного
безреактивного магазина сопротивлений. Он имеет три
декады сопротивлений. Каждая из них содержит 9
сопротивлений, равных соответственно 1000, 100 и 10 ом.
Поэтому сопротивление плеча сравнения может изменяться
ступенями по 10 ом в пределах от 10 до 10 000 ом.
Величина отношения плеч R2/R1 может изменяться в широких
пределах; отношение IOR2/R1 составляет 0,014-10 000.
Изменение отношения плеч осуществляется при помощи
переключателя диапазонов. Таким образом,
переключатель диапазонов и три переключателя плеча сравнения
позволяют сбалансировать мост для измеряемых
сопротивлений в пределах от Ю-2 до 107 ом. Емкостная
составляющая датчика компенсируется с помощью
встроенного блока конденсаторов.
Прибор имеет электронно-оптический индикатор
баланса моста. Дисбаланс моста усиливается при помощи
двух каскадов усилителей. При балансе моста световой
сектор индикатора сужается до минимума.
Кондуктометр содержит комплект универсальных
датчиков УК-02/1. Датчики представляют собой сосуд для
раствора и трубку с двумя электродами, погружаемыми
в исследуемый раствор. При кондуктометрическом
титровании могут быть использованы и другие
электролитические ячейки.
Погрешность измерения сопротивления раствора
электролита при помощи кондуктометра ±1%.
§ 4. Кондуктометр типа К1-4
Измерительная часть прибора представляет собой че-
тырехплечий уравновешенный мост. Диапазон
измеряемых сопротивлений составляет от 100 до 90 000 ом.
Прибор питается переменным током от сети напряжением
220 в, частотой 50 гц. Выходной трансформатор
генератора, встроенный в прибор, подает переменное
напряжение частотой 1000 гц. Внешний вид кондуктометра
показан на рис. 36.
Плечами R\ и R% являются постоянные сопротивления
в 100 ом. В третьем плече R3 используется магазин
сопротивлений типа Р-33. Четвертым плечом моста
является измеряемое сопротивление электролитической ячейки.
Параллельно вторичной обмотке выходного
трансформатора включается переменное сопротивление, служащее
122
для балансировки моста по переменной составляющей.
Индикатор баланса представляет собой трехкаскад-
ный усилитель с автоматической регулировкой усиления.
Около точек баланса усилитель имеет максимальную
чувствительность, а при разбалансе моста усиление
резко падает. На выходе усилителя имеется выпрямитель с
микроамперметром типа М-494, по которому
балансируется мост. Баланс моста получается при минимальном
отклонении стрелки миллиамперметра. Кроме этого име-
Рис. 36. Кондуктометр К1-4
ются клеммы, к которым могут подключаться телефоны
или осциллограф.
Балансировку моста производят при помощи ручек
магазина сопротивлений, двигаясь от больших величин
к меньшим. Потом подстраивают попеременно ручки
магазина и ручку балансировки по реактивной
составляющей. Для увеличения чувствительности моста
увеличивают сигнал, поворачивая ручку «генератор» по часовой
стрелке. Когда мост сбалансирован, считывают величину
измеряемого сопротивления.
Погрешность измерений электропроводности
растворов составляет ±0,5%.
§ 5. Кондуктометр «Импульс» типа КЛ1-2
В кондуктометре «Импульс» использована мостовая
схема с питанием импульсным током переменной поляр-
123
ности и интегрированием синхронного выпрямленного
сигнала разбаланса. Это позволяет не проводить
уравновешивание моста по реактивной составляющей. Схема
кондуктометра включает выпрямитель, генератор
импульсного тока переменной полярности, синхронный
детектор, нуль-индикатор и измерительный мост. Общий
вид кондуктометра показан на рис. 37.
Прибор включают в сеть переменного тока
напряжением 220 в и частотой 50 гц. Отсчет показаний прибора
Рис. 37. Кондуктометр «Импульс» типа КЛ1-2
цифровой, в единицах удельной электропроводности.
Диапазон измерений от Ю-6 до 1,0 омгх • смгх.
Для питания генератора импульсного тока служит
двухполупериодный выпрямитель, собранный по
мостовой схеме на диодах. Генератор импульсного тока
переменной полярности вырабатывает напряжение питания
моста и напряжение, управляющее работой синхронного
детектора. Частота следования импульсов 450.
Нагрузкой синхронного детектора служит нуль-индикатор. В
качестве нуль-индикатора в кондуктометре использован
микроамперметр магнитоэлектрической системы.
Измерительный мост включает измерительную
ячейку, магазин сопротивлений и два сопротивления,
отношение которых может изменяться при помощи
переключателя. Диапазон измерений разбит на шесть
поддиапазонов. Величина сопротивления магазина сопротивлений
пропорциональна величине удельной электропроводности
раствора в измерительной ячейке, причем плечи моста
подобраны так, что коэффициент пропорциональности ра-
124
вен 10", где п — целое число и в зависимости от
поддиапазона измерения принимает значения от 4 до 9.
Погрешность измерения кондуктометра не превышает
0,25%, если при работе использовать термостатирование.
Прибор имеет две измерительные ячейки,
позволяющие определять электропроводности растворов,
значительно отличающиеся по величине. Константы сосуда
ячеек сильно отличаются и равны 0,12 и 120 см.-1.
Однако эти ячейки неудобны для кондукт'ометриче-
ского титрования, так как приспособлены для измерения
электропроводности раствора без его значительного
разбавления. Они применимы только в том случае, когда
используют концентрированные растворы титрантов,
которые мало увеличивают объем раствора в ячейке при
титровании. В других случаях вместо этих ячеек при
титровании подключают ячейки других конструкций с
константами сосуда, близкими к одной из двух ячеек
константы прибора. Поскольку прибор дает показания
в единицах удельной электропроводности, при
использовании других ячеек следует вводить поправочный
коэффициент:
где А — поправочный коэффициент; К\ — константа
сосуда ячейки кондуктометра «Импульс»; Кч — константа
сосуда ячейки, используемой для титрования.
§ 6. Установки для хронокондуктометрического
титрования
При хронокондуктометрическом титровании могут
быть использованы неуравновешенные мостовые схемы.
Метод основан на зависимости силы тока в цепи кондук-
тометрической ячейки от электропроводности раствора.
Для записи кондуктометрических кривых переменный
ток преобразуют в постоянный с помощью
полупроводниковых диодов. Падение напряжения на эталонном
сопротивлении, пропорциональное силе тока в цепи кондук-
тометрической ячейки, записывают, применяя
электронный потенциометр. При равномерной подаче титранта
электронный потенциометр фиксирует процесс
титрования в координатах u=f(t), которые пропорциональны
обычно принятым в кондуктометрическом титровании
KaBf(V). В §тих зависимостях ц — падение напряжения
126
на эталонном сопротивлении, t — время, %—-удельная
электропроводность и V—объем титранта.
Установка может быть собрана по схеме,
приведенной на рис. 38. Для питания установки переменным
током, стабильным по частоте и напряжению, используют
генератор типа ГЗ-33. Измерения ведут при частоте тока
600—700 гц. Цепь состоит из сопротивлений Ri, R2 и У?з>
электролитической ячейки 2, полупроводниковых диодов
(типа Д-2Е) и электронного потенциометра 3. Запись
кривой титрования можно вести на электронном потен-
/
ID
i
ТЫ
к>
к2П
5
£
Рис. 38. Схема установки для хропокондук-
тометрического титрования:
/ — генератор ЗГ-33; 2 — электролитическая
ячейка; J— электронный потенциометр ЭПП-09; Ri,
R2 н #з — стандартные резисторы с
сопротивлением, равным соответственно 91, 91 и 68 ом; 4 и
5 —выпрямители марки Д-2Е
циометре типа ЭПП-09 или КСП-4 с диапазоном 10 мв
на всю шкалу. Скорость передвижения диаграммной
ленты потенциометра 1200 мм/ч.
При хронокондуктометрическом титровании
осуществляют непрерывную равномерную подачу титранта. Для
этой цели используют различные дозаторы поршневого
и плунжерного действия, поплавковые, клапанные и
мембранные устройства. Для равномерной капельной подачи
титранта можно использовать сосуд Мариотта.
При титровании титрант подают каплями в
электролитическую ячейку, в которой раствор непрерывно
перемешивается при помощи магнитной мешалки или других
устройств. Кривая кондуктометрического титрования
наносится на диаграммную ленту пером потенциометра.
Количество миллилитров титранта, вступившего в
реакцию, находят по времени титрования до изломов кондук-
тометрических кривых.
126
§ 7. Конструкции электролитических ячеек
Конструкция электролитической ячейки имеет
большое значение в кондуктометрическом анализе, так как
электрохимические и электрические явления могут быть
источником ошибок при измерении электропроводности
растворов. Ячейка должна удовлетворять определенным
требованиям: иметь оптимальные размеры электродов и
оптимальное расстояние между ними; минимальные
Поляризационные явления на электродах; ничтожные
утечки тока вследствие паразитных емкостных связей.
Форма ячейки должна быть такой, при которой
увеличение объема раствора при титровании не вносит
существенных ошибок в измерение электропроводности.
Константа сосуда. Электролитические ячейки для
кондуктометрического титрования характеризуются
константой сосуда, которая выводится из следующих
зависимостей.
Величину удельной электропроводности раствора
определяют по уравнению
*~ S ' R '
Для вычисления удельной электропроводности
раствора кроме сопротивления раствора R необходимо знать
величину t/S, которую можно было бы определять
прямым делением расстояния между электродами / на их
площадь S, но при условии, что ток проводит только
раствор в объеме между электродами. Однако в
электролитических ячейках имеет место рассеивание силовых
линий тока, и они выходят за пределы объема
электролита между электродами. Большое значение при этом
имеет форма электролитической ячейки и место
закрепления в ней электродов. Кроме того, истинная
поверхность электродов изменяется при их платинировании.
Поэтому более точно удельную электропроводность
раствора выражают следующей зависимостью;
*=*-£-, (52)
где К — константа сосуда (см-1), зависящая от площади
электродов и расстояния между ними, а также формы
сосуда и объема раствора, проводящего ток.
127
(51)
Поскольку электропроводность раствора
W—величина, обратная сопротивлению (1/R), получаем
*=KW. (53)
Следовательно, для вычисления удельной
электропроводности раствора нужно электропроводность раствора
умножить на константу сосуда.
При определении удельной электропроводности
раствора измеряют его сопротивление (или его
электропроводность). При этом константа сосуда,
характеризующая ячейку, в которой проводится измерение, должна
быть величиной постоянной. Поэтому основным
требованием, предъявляемым к электролитическим ячейкам,
является постоянство константы сосуда в области тех
сопротивлений, которые измеряются в данной ячейке.
Иногда наблюдается кажущееся изменение константы
сосуда, что вызывается различными электрохимическими и
электрическими явлениями, связанными с неудачной
конструкцией ячейки. Учитывая это, для каждой
электролитической ячейки определяют величину константы сосуда
и ее постоянство при определенном объеме раствора в
ячейке.
Для определения константы сосуда измеряют
сопротивление стандартного раствора с известной удельной
электропроводностью. В качестве стандартного раствора
берут растворы КС1, для которых удельная
электропроводность определена с высокой точностью. Концентрации
стандартных растворов КС1 берут обычно равными 0,1 н.
и 0,01 н. Объем раствора сохраняют постоянным. При
этом исходят из первоначального объема, который
обычно берут при титровании в данной ячейке.
Константу сосуда вычисляют по уравнению
К=*Ъ ' (54)
Электролитические ячейки, используемые при кондук-
тометрическом титровании, отличаются по величине
константы сосуда, которая имеет значения от 0,1 до 100 и
выше. Константа сосуда уменьшается с увеличением
площади электродов и уменьшением расстояния между
ними. Для измерения электропроводности слабо
проводящих растворов используют ячейки с низкими
значениями константы сосуда, а при работе с хорошо
проводящими растворами, наоборот, ячейки с высокими значения-
128
ми константы сосуда. Обычно имеют несколько
электролитических ячеек, отличающихся константами сосуда,
которые могут быть использованы для различных случаев
титрования в водных и неводных растворах.
Типы кондуктометрических ячеек. Электролитические
ячейки, употребляемые при кондуктометрическом
титровании, имеют различные конструкции. Так, существует
два типа ячеек: ячейки с жестко закрепленными в
стенках электродами и ячейки «погружного» типа, в которых
электроды погружаются в раствор перед титрованием.
Для кондуктометрического титрования удобнее ячейки с
жестко закрепленными в стенках электродами, так как
даже при одном анализе приходится производить
большое количество измерений и при строго фиксированном
положении электродов результаты лучше
воспроизводятся.
Можно выделить два типа ячеек, отличающихся тем,
что в одном случае константа сосуда не зависит от
объема раствора в ячейке, а в другом зависит. Дело в том,
что константа сосуда определяется при постоянном
объеме раствора в ячейке. Однако в процессе титрования
объем раствора в ячейке увеличивается, что может
привести к изменению константы сосуда. Электролитической
ячейке можно придать такую форму, при которой
увеличение объема раствора при титровании не будет
изменять константу сосуда. Ячейки с константой сосуда, не
зависящей от объема раствора в ячейке, имеют
несомненные преимущества, так как позволяют проводить
измерения электропроводности с более высокой
точностью.
Кроме этого даже ячейки, относящиеся к указанным
типам, могут существенно отличаться по форме сосуда,
площади электродов и расстоянию между ними, месту и
способу закрепления электродов, способу перемешивания
раствора и т. д. Поэтому в практике используются
ячейки разнообразных конструкций. Усовершенствование
конструкций ставит своей целью уменьшить погрешности,
вызываемые электрохимическими и электрическими
явлениями, и сохранить постоянной константу
сосуда.
Следует также учитывать, что измеренная в процессе
титрования электропроводность раствора вследствие его
разбавления всегда несколько отличается от
электропроводности, которая наблюдалась бы при постоянном объе-
5—1595
129
ме. Для уменьшения ошибки в величине
электропроводности раствора вследствие его разбавления в процессе
титрования концентрацию титранта берут по крайней
мере в 10 раз выше, чем концентрация титруемого раствора.
Ошибку можно уменьшить, если привести
электропроводность к постоянному объему путем введения
поправочного коэффициента разбавления (А):
где Vo —■ первоначальный объем электролита, мл; V —
объем прибавленного титранта, мл.
На рис. 39 приведены несколько типов ячеек с жестко
закрепленными в стенках сосуда электродами. Наиболее
Рис. 39. Электролитические ячейки с жестко закрепленными
в сосуде электродами:
а — ячейка с константой сосуда, зависящей от объема раствора в
ячейке; б — ячейка с константой сосуда, мало зависящей от объема
раствора в ячейке; в — ячейка с константой сосуда, не зависящей от объема
раствора в ячейке (показаны разрезы сосуда и электрода); / —
платиновые электроды; 2—контактные трубки; 3 — сосуд для титрования;
4 — измерительная трубка; 5 н 6— краны
130
простой тип ячейки представляет сосуд (рис. 39, а) 3, в
боковые стенки которого жестко закреплены
электроды 1, расположенные вертикально. Диаметр и высота
сосуда могут быть разнообразны. Сверху сосуд имеет
отверстие для подачи титранта. Через это отверстие
вводится механическая мешалка. Для перемешивания
может быть использована также магнитная мешалка.
Возможно перемешивание путем встряхивания, однако
следует учитывать, что при этом сосуд может нагреваться,
что влечет за собой ошибки в измерении
электропроводности раствора.
В зависимости от измеряемого сопротивления
выбирают площадь электродов и расстояние между ними: чем
выше измеряемое сопротивление, тем больше берут
площадь электродов и меньше расстояние между ними.
Электроды обычно располагают вертикально, что особенно
важно в случаях, когда при титровании выделяются
осадки. Токоподводы 2 к электродам по возможности
удаляют от сосуда с целью уменьшения паразитных
емкостных связей. В токоподводах обычно применяют
ртутные контакты. Константа сосуда ячейки зависит от
объема раствора, находящегося в ней.
На рис. 39, б показана электролитическая ячейка
более совершенной конструкции. Она отличается тем, что
сосуд 3 значительно расширен в верхней части. Ячейку
заполняют титруемым раствором до расширенной части.
При указанных на рисунке размерах сосуда это
составляет 30 мл раствора. При титровании уровень раствора
сравнительно мало поднимается, так как заполняется
расширенная часть сосуда. Константа этого сосуда мало
изменяется с увеличением объема раствора в ячейке,
поэтому электропроводность раствора при титровании
может быть измерена с более высокой точностью.
Электроды / могут иметь различную площадь. Чем больше
площадь электродов, тем меньше константа сосуда. Если
площадь электродов равна 4 см2, константа сосуда
составляет 0,23 (при использовании гладких платиновых
электродов). В этих условиях может проводиться кон-
дуктометрическое титрование неводных растворов,
имеющих высокое сопротивление. При работе с водными
хорошо проводящими растворами применяют
платинированные электроды меньшей площадью. Перемешивание
раствора в этой ячейке осуществляется при помощи
магнитной мешалки.
5*
131
Еще более совершенную конструкцию имеет ячейка,
показанная на рис. 39, в, так как константа сосуда этой
ячейки не зависит от объема находящегося в ней
раствора. Крестообразные платиновые электроды 1 площадью
0,5 см2 впаяны в измерительную трубку 4 и контактные
трубки. Оба конца измерительной трубки 4 переходят в
вертикальные сосуды для титрования 3 объемом 60 см3,
закрытые сверху пробками. Сосуды соединены между
собой трубкой с краном 5. В одной из пробок сосуда
имеется входной патрубок с краном 6. Контактные трубки 2 к
электродам расположены перпендикулярно
горизонтальной измерительной трубке для устранения паразитных
емкостных связей. Пространство, в котором могут
распространяться силовые линии электрического поля
между электродами, ограничено, и, следовательно, объем
раствора, проводящего ток, остается практически
постоянным. Для защиты от двуокиси углерода воздуха и для
перемешивания раствора в ячейку предусмотрена подача
азота. При перемешивании закрывают кран 5 и
пропускают в раствор азот через кран 6. После перемешивания
открывают кран 5 для равномерного распределения
раствора в сосудах. Перемешивание можно также
проводить при помощи груши воздухом, очищенным от СОг.
В крайнем случае раствор можно перемешивать,
наклоняя ячейку то в одну, то в другую сторону. Так как
объем раствора, проводящего ток, мал, константа сосуда
Рис. 40, Электролитическая ячейка «погружного»
типа; схема п общий вид ячейки с вращающимся
сосудом для хропокондуктометрпческого титрования:
/ — платиновые электроды; 2 — контактные трубки; 3 — сосуд
дчя титрования; 4—колодка для закрепления электродов;
5 — мешалка
132
имеет высокое значение; при платинированных
электродах она равна 6,16. Для уменьшения константы сосуда
следует увеличить диаметр измерительной трубки и
размеры электродов.
На рис. 40 показаны электроды «погружного» типа,
применяемые при хронокондуктометрическом титровании.
Электроды / впаяны в стеклянные трубки 2,
закрепленные в колодке 4, с помощью которой можно
устанавливать их на расстоянии 1—4 см друг от друга. Вместе с
электродами в колодку закрепляется трехлопастная
мешалка 5 из стекла или фторопласта. Электроды
погружаются в раствор перед титрованием. Сосуд для
титрования представляет собой круглую стеклянную ванну.
Колодку с электродами закрепляют над ванной с
помощью съемного кронштейна. Ванну ставят на диск,
насаженный на вал электромотора. Перемешивание
раствора осуществляется путем равномерного вращения
ванны на вращающемся при титровании диске
(78 об/мин). Если платинированные электроды имеют
площадь 0,5 см2 и расположены на расстоянии 4 см друг
от друга, то константа сосуда равна 1,12. Титрант
подается в ванну каплями с постоянной скоростью. Таким
путем осуществляется титрование во вращающейся
ячейке.
ПРАКТИЧЕСКИЕ РАБОТЫ
Глава VIII
ПОДГОТОВКА РАБОЧЕГО МЕСТА,
УСТАНОВКА АППАРАТУРЫ
§ 1. Подготовка электролитической ячейки
Электролитическая ячейка является важнейшей
частью установок для кондуктометрического и хронокон-
дуктометрического титрования. Перед тем как
использовать ячейку для титрования, проверяют постоянство
константы сосуда. Поскольку в большинстве случаев
работают с платинированными электродами, сначала проводят
платинирование электродов.
Платинирование электродов. Приготавливают раствор,
содержащий 30 г платинохлористоводородной кислоты и
0,3 г ацетата свинца в одном литре. В электролитическую
ячейку наливают полученный раствор в таком объеме,
чтобы электроды были в него полностью погружены.
Пропускают через раствор ток, меняя его направление через
каждую минуту. Плотность тока не должна превышать
30 мка/см2. Продолжительность платинирования 10 мин.
После платинирования проверяют состояние электродов;
они должны быть покрыты ровным плотным слоем
платиновой чгрни. Платинированные электроды тщательно
промывают дистиллированной водой. При хранении ячейку
заливают дистиллированной водой.
Определение константы сосуда. Приготавливают 0,1 н.
раствор КС1 из химически чистого препарата. Из этого
раствора путем разбавления получают 0,01 н. раствор
КС1. Электролитическую ячейку промывают
концентрированной азотной кислотой, а затем многократно
споласкивают сначала дистиллированной водой, а после этого
0,01 н. раствором КО. Наливают в ячейку такой объем
0,01 н. раствора КС1, который обычно берут при
титровании. Помещают ячейку в термостат при 25°. После дости-
134
жения постоянной температуры измеряют сопротивление
раствора при помощи моста Кольрауша или
кондуктометра. После этого выливают раствор из ячейки и
споласкивают ее 0,1 н. раствором КС1. Помещают в ячейку 0,1 н.
раствор КО в том же объеме, что и в предыдущем случае,
и измеряют сопротивление раствора. Вычисление
константы сосуда проводят по формуле
K=*R,
(55)
где К — константа сосуда; к — удельная
электропроводность раствора КО взятой нормальности; R —
сопротивление раствора, ом.
Значения констант сосуда, найденные по 0,1 н. и
0,01 н. растворам КС1, должны быть близкими.
§ 2. Установка для кондуктометрического
титрования
Схема установки для кондуктометрического
титрования показана на рис. 41. Для подачи титранта
используют автоматические полумикробюретки емкостью 10 или
5 мл, с ценой деления 0,02-мл или полумикробюретки
емкостью 2 мл, с ценой деления 0,01 мл. Баллон для
хранения титранта обычно берут емкостью в 1 л.
Рис. 41. Установка для кондуктометрического
титрования классическим методом:
1 — автоматическая полумикробюретка; 2 — электролитическая
ячейка; 3 — кондуктометр марки ММЗЧ-64
135
Прежде чем использовать бюретки в работе, их моют
хромовой смесью, прополаскивают водопроводной и
дистиллированной водой, а затем раствором титранта.
Верхнее отвеостие полумикробюретки закрывают хлоркаль-
циевой трубкой, в которую для поглощения
СОг помещают натронную известь. В
бюретку медленно, не допуская большого
избыточного давления, нагнетают воздух
грушей, также пропуская его через колонку,
наполненную натронной известью. Избыточное
давление сбрасывают в атмосферу,
открывая на несколько секунд кран.
Можно также использовать
микробюретки (рис. 42) емкостью 5, 2 или 1 мл с ценой
деления 0,01 мл. Емкость баллонов для
хранения титранта равна соответственно 250,
200 или 100 мл.
При работе могут быть использованы
электролитические ячейки различных
конструкций. Оптимальные условия создаются
при употреблении ячейки, константа сосуда
которой не зависит от объема раствора в
ячейке (рис. 39, в). Для перемешивания
раствора в этой ячейке предусмотрена
подача азота через кран 6 или нагнетание
грушей воздуха, очищенного от СОг
пропусканием его через колонку, наполненную
натронной известью. Воздух из колонки
подают через кран 6. Если исключена
возможность взаимодействия С02 воздуха с
титруемым раствором, перемешивание можно
осуществлять, наклоняя ячейку то в одну, то в
другую сторону.
В электролитической ячейке, показанной
на рис. 39,6, шеремгшивают раствор при помощи
магнитной мешалки; ячейку устанавливают на магнитную
мешалку и перед титрованием опускают в нее магнит.
Для измерения сопротивления раствора в
электролитической ячейке ее подключают к мосту Кольрауша,
мосту Р38, Р-556, Р-577 или к кондуктометру. Если мост
Кольрауша собирают из стандартных деталей, питание
установки осуществляют от сети переменного тока через
генератор (ЗГ-1, ЗГ-2, ЗГ-10, ЗГ-11 или ЗГ-33),
используя частоту порядка 1000 гц.
Рис. 42.
Микробюретка:
/ — баллон
для титраита;
2 — бюретка
136
§ 3. Методика кондуктометрического
титрования
Полумикробюретку сначала споласкивают, а затем
наполняют титрантом и устанавливают так, чтобы кончик
бюретки находился в отверстии электролитической
ячейки для подачи титранта. В электролитическую ячейку при
помощи пипетки переносят аликвотную часть
анализируемого раствора и определяют его сопротивление. Затем
в ячейку добавляют титрант порциями по 0,1-—0,5 мл.
Величина порции зависит от концентрации титруемого
раствора. До точки эквивалентности необходимо сделать не
менее четырех измерений. В некоторых случаях для
достижения постоянной электропроводности раствор
выдерживают некоторое время, что указывается в методиках.
Измерение сопротивления после добавления каждой
порции титранта повторяют три раза. Результаты измерений
заносят в рабочий журнал, как показано ниже.
Объем
добавленного
раствора,
МЛ
Сопротивление раствора R
1-е измерение
2-е измерение
3-измерение
средняя величина
Электропроводность
раствора
tr—L
R
Поправочный
коэффициент
на
разбавление
^ Vo + V*
Уо
н
Электропроводность ра
приведенная к нервона
му обьему, W-=AW
* При работе с ячейками, константа сосуда которых не зависит от объема
раствора, поправка может ие вводиться.
Используя среднее значение электропроводности
раствора, лриведенног к первоначальному объему, строят
кривую кондуктометрического титрования. По оси абсцисс
откладывают число миллилитров добавленного титранта,
а по оси ординат — электропроводность раствора. На
кондуктометрической кривой графическим методом уста-
137
навливают точку эквивалентности и определяют число
миллилитров титранта, вступивших в реакцию с
определяемым веществом.
§ 4. Установка для хронокондуктометрического
титрования
Схема установки для хронокондуктометрического
титрования показана на рис. 43. Для непрерывной подачи
титранта с постоянной скоростью используют сосуд Ма-
Рис. 43. Установка для
хронокондуктометрического титрования:
/ — сосуд Марнотта; 2 — электролитическая ячейка; 3—-
магиитная мешалка типа ММ-1; 4 — коидуктометрический
блок- 5 — генератор ЗГ-33: 6 — электронный потенциометр
ЭПП-09
риотта или другое устройство. Сосуд Мариотта показан
на рис. 44. По мере вытекания жидкости через отводную
трубку в сосуд Мариотта через вертикальную трубку
пузырьками поступает воздух, который можно очистить от
С02 пропусканием его через хлоркальциевую трубку,
наполненную натронной известью. В отводную трубку
сосуда Мариотта вставлен капилляр 4 оттянутым концом
138
вверх для предотвращения его засорения. Изменяя
отверстие капилляра, можно добиться различной скорости
истечения стандартного раствора. Этот капилляр
закрепляется при помощи резиновой трубки. Нижний конец
капилляра при помощи резиновой трубки соединяется со
вторым капилляром 8, который расположен оттянутым
концом вниз. Второй капилляр имеет большее отверстие,
через него титрант поступает в ячейку. Скорость истече-
Рнс. 44. Сосуд Мариотта:
а — общий вид; б — капилляр; / — хлоркаль-
цневая трубка; 2 — трубка для подачи
воздуха; 3 — сосуд с тубусом; 4 — верхний капил- ■
ляр; 5 —отводная трубка; 6 — зажим; 7 —
край; 8 — нижний капилляр
ния жидкости из сосуда Мариотта устанавливают перед
титрованием путем измерения времени наполнения
пикнометра определенного объема. Емкость сосуда
Мариотта обычно составляет несколько литров.
Сосуд Мариотта промывают хромовой смесью,
споласкивают водопроводной и дистиллированной водой, а
затем раствором титранта, наполняют титрантом и
закрывают пробкой, в которую вставлена трубка для возду-
139
xa 2. Наружный конец этой трубки закрывают хлоркаль-
циевой трубкой 1, наполненной натронной известью.
Пробку, закрывающую сосуд, заливают парафином.
Заполняют титрантом отводную трубку, причем в ней не
должно оставаться пузырьков воздуха.
Заполнение можно провести следующим образом. Оба
капилляра заполняют титрантом по отдельности, а затем
их соединяют. Далее, открыв кран 7, заполняют отводную
трубку без капилляров. После этого, не прекращая
истечения жидкости из отводной трубки, вставляют в
находящуюся на ее конце резиновую трубку капилляр, который
должен быть расположен узким концом вверх. Конец
капилляра поднимают выше резиновой трубки, чтобы
можно было проверить, не остались ли в нем пузырьки
воздуха. Проверяют также, заполнен ли жидкостью нижний
капилляр. Постоянная скорости истечения жидкости из
сосуда Мариотта устанавливается тогда, когда трубка,
опущенная в сосуд Мариотта через пробку, не заполнена
жидкостью и через нее пузырьками поступает воздух.
Поэтому перед измерением скорости истечения жидкости из
сосуда Мариотта или перед титрованием сначала
спускают жидкость из трубки, подающей воздух. Для этого
открывают кран 7 на отводной трубке до тех пор, пока
воздух не начнет равномерно поступать пузырьками в сосуд
Мариотта.
Для хронокондуктометрического титрования
используют электролитические ячейки, показанные на рис. 39, б
или 40. Если для работы используется ячейка,
расширенная в верхней части, перемешивание раствора при
титровании обычно осуществляется при помощи магнитной
мешалки. Ячейку устанавливают на магнитную мешалку
и перед титрованием опускают в нее магнит. В случае,
когда титрование проводят во вращающейся ванне, ее
устанавливают на диск и закрепляют зажимами. Перед
титрованием в ванну погружают электроды с
прикрепленной к ним мешалкой. Вращение диска во время
титрования проводят при помощи электромотора, на вал
которого насаживают диск. Электроды вынимают из ванны
при помощи съемного кронштейна.
Ту или иную электролитическую ячейку перед
титрованием устанавливают так, чтобы отводная трубка
сосуда Мариотта была помещена в отверстие ячейки для
подачи титранта.
140
Для автоматической записи кондуктометг^ческой
кривой собирают установку по схеме, показание^ на рис. 38,
и подключают к ней электролитическую ячейку.
§ 5. Методика хронокондуктометрического
титрования
Сначала определяют скорость истечения стандартного
раствора (титранта). Для этого берут пикнометр
емкостью 25 мл и устанавливают по секундомеру время его
наполнения титрантом. Если проводится точечная запись
кондуктометрических кривых, то определяют по
секундомеру время между записями отдельных показаний. При
этом берут среднюю величину от общего времени
регистрации не менее 10 показаний. Если проводится
непрерывная запись кондуктометрической кривой, то на
потенциометре устанавливают необходимую скорость
передвижения диаграммной ленты.
Электролитическую ячейку ставят на магнитную
мешалку или диск для вращения ванны и переносят в нее
аликвотную часть анализируемого раствора. Когда
перемешивание проводят на магнитной мешалке, опускают в
раствор магнит и включают мешалку. Если работают с
вращающейся ванной, то в нее погружают электроды с
мешалкой и включают мотор.
Затем включают в сеть установку для измерения
электропроводности я устройства для перемещения
диаграммной ленты электронного потенциометра. На
генераторе устанавливают частоту порядка 600—700 гц (при
этом генератор работает устойчивее, если частота не
кратна 50) и ручкой регулировки входного напряжения
устанавливают перо потенциометра в таком положении,
чтобы кондуктометрическая кривая полностью разместилась
на лентг, если электропроводность при титровании будет
повышаться—в левой части, а если понижаться — в
правой части. Если характер изменения электропроводности
раствора при титровании не известен, то устанавливают
перо потенциометра в средней части шкалы.
После этого включают подачу титранта и отмечают
на диаграммной ленте точку записи, соответствующую
падению первой капли. Дальнейший процесс
титрования идет автоматически. Запись кривой титрования
заканчивают при избытке титранта. Выключают установку и
после промывания ячейки проводят повторное титрованиэ.
141
Оптимальные условия для анализа создаются, когда
кривая титрования занимает по возможности всю ширину
диаграммной ленты и не слишком растянута по длине;
при этом изломы кривых в точках эквивалентности
получаются более резкими. Скорость передвижения
диаграммной ленты следует брать 1200 мм/ч. Целесообразно
проводить повторное титрование, изменяя выходное
напряжение на генераторе, что изменяет расположение
кривой титрования по ширине диаграммной ленты. Могуг
иметь место случаи, когда кривая титрования не
размещается на диаграммной ленте и перо потенциометра еще
до окончания титрования устанавливается в правой части
шкалы. Это свидетельствует о том, что происходит
слишком сильное повышение напряжения на эталонном
сопротивлении. В этом случае повторяют титрование,
уменьшив выходное напряжение на генераторе.
На кондуктометрической кривой графическим методом
устанавливают точки эквивалентности. Если применяют
непрерывную запись кондуктометрических кривых,
находят длину диаграммной ленты от начала титрования до
излома в точке эквивалентности. В случае титрования
смесей электролитов определяют длины участков
диаграммной ленты, соответствующие титрованию отдельных
компонентов. Если применяют точечную запись
кондуктометрических кривых, находят число интервалов между
записью показаний потенциометра, соответствующих
титрованию отдельных веществ. Десятые доли интервалов
около точки эквивалентности находят на глаз.
§ 6. Расчеты
Существует несколько методов расчета концентрации
определяемого вещества при кондуктометрической
титровании. Рассмотрим методы расчетов в случае, когда
используют классический метод титрования.
Если концентрация титранта установлена
предварительно одним из химических методов, то в этом случае
по кондуктометрической кривой находят число
миллилитров титранта, вступившего в реакцию с определяемым
веществом. Общее содержание определяемого вещества
находят по формуле
^в^л.Гк. (56)
^А юоо V
А
142
где ^а — общее количество определяемого вещества, г;
Nb — нормальность титранта; Vb— объем титранта,
вступившего в реакцию, мл; Эа— грамм-эквивалент
определяемого вещества; Vk — общий объем анализируемого
раствора, мл; Va— объем аликвотной части раствора»
взятого для титрования.
Этими расчетами пользуются, когда излом
кондуктометрической кривой соответствует стехиометрическим от-
ношгниям реагирующих веществ. Однако, как
рассмотрено выше, кондуктометрические определения возможны и
в случаях, когда вещества реагируют в нестехиометри-
ческих отношениях, если излом кривой достаточно резкий,
а воспроизводимость хорошая. При этих условиях
концентрацию вступающего в реакцию рабочего раствора
определяют кондуктометрическим методом путем
титрования стандартных образцов. Для этого титруют навеску
стандартного образца или определенный объем
стандартного раствора, химический состав которых соответствует
определяемому веществу, и находят объем титранта,
вступившего в реакцию. Нормальность титранта в этом случае
находят по формулам:
при титровании навески
N„= — , (57)
где Л^в — нормальность титранта; ^а — навеска
стандартного образца, взятая для титрования, г; ЭА —
грамм-эквивалент определяемого вещества; Vb — объем титранта,
вступившего в реакцию, мл;
при титровании раствора
NB=-±-±, (58)
в Vb
где Л^в — нормальность титранта; NA — нормальность
стандартного раствора; Va — объем стандартного
раствора, взятый для титрования, мл; Vb — объем титранта,
вступивший в реакцию, мл.
Неизвестную концентрацию вещества определяют в
тех же условиях, что и концентрацию стандартного
образца. При расчетах пользуются формулой (58).
Нормальность титранта устанавливается тем же
кондуктометрическим методом.
143
Следует заметить, что даже в случаях, когда реакция
протекает количественно, часто применяют
стандартизацию титранта кондуктометрическим методом. При этом
устраняются ошибки, которые могут возникнуть при
установлении точки эквивалентности графическим методом,
так как они повторяются при титровании стандартного
образца и раствора неизвестной концентрации.
Уменьшаются ошибки, вызываемые адсорбцией ионов на
поверхности осадков, если титрование проводить в аналогичных
условиях. Меньшее влияние на точность определений
оказывают примеси, находящиеся в растворах титрантоз
(например, примеси карбонатов, находящихся в растворе
щелочей). При этом более точные результаты
получаются, если стандартизация раствора титранта проводится
по определяемому веществу. Однако в ряде случаев
стандартизацию растворов кислот, щелочей и т. д. проводят
кондуктометрическим методом, а титранты используют
для анализа разнообразных веществ.
При хронокондуктометрическом титровании в
расчетах используют другие зависимости. Расчеты
основываются на времени титрования. Если осуществляют
непрерывную запись кондуктометрических кривых, время
титрования пропорционально длине диаграммной ленты от
начала титрования до излома кондуктометрической
кривой. В случае, когда известна нормальность титранта,
пользуются формулой
LNRV9k VK
ёА mooo vA y
где g_\ — общее количество определяемого вещества, г;
L — протяженность диаграммной ленты потенциометра,
соответствующая титрованию аликвотной части
определяемого раствора, мм; NB — нормальность титранта; V —
объем пикнометра, используемого при определении
скорости истечения титранта, мл; Эа— грамм-эквивалент
определяемого вещества; / — скорость перемещения
диаграммной ленты, мм/сек; Т—время наполнения
пикнометра титрантом, сек; Vk—-общий объем
анализируемого раствора, мл; VA — объем аликвотной части, взятой
для титрования, мл.
Если вещества реагируют в нестехиометрических
отношениях или ие используют нормальность титранта,
установленную химическим анализом, концентрацию
определяемого вещества находят путем сравнения с резуль-
144
татами титрования навески стандартного образца того
же химического состава, что и определяемое вещество, по
уравнению
*л=*л--Г-~. (60)
> Act / \г
где £а — общее количество определяемого вещества, г;
£аст-"- навеска стандартного образца, г; L — протяжен
пость ленты потенциометра, соответствующая титрова
нию аликвотной части анализируемого раствора, мм
LCT — протяженность ленты потенциометра, соответ
ствующая титрованию навески стандартного образца, aim;
Vk — общий объем анализируемого вещества, мл; Va—
объем аликвотной части раствора, взятого для
титрования, мм.
В случае, когда концентрацию определяемого
вещества находят сравнением с результатами титрования не
навески, а определенного объема стандартного раствора,
расчеты проводят по формуле
(61)
LN„VCT9k VK
gh~ irT-1000 ' V
А
где gA — общее количество определяемого вещества, г;
L-—протяженность диаграммной ленты потенциометра,
соответствующая титрованию аликвотной части
определяемого раствора, мм; LCT — протяженность
диаграммной ленты потенциометра, соответствующая титрованию
стандартного раствора, мм; 7VCT ■—нормальность
стандартного раствора; 1/Ст — объем стандартного раствора,
взятого для титрования, мл; ЭА — грамм-эквивалент
определяемого вещества; Vk — общий объем
анализируемого раствора, мл; Va — объем аликвотной части раствора,
взятого для титрования, мл.
При точечной записи хронокондуктометрических
кривых в случае, когда нормальность титранта известна и
установлена скорость его истечения, при расчетах
применяют следующую формулу.
£д = L_*._JL (62
А Г. 1000 VK
где £а — общее количество определяемого вещества, г;
п — количество интервалов па кондуктометрической
кривой до точки эквивалентности; / — интервал между за-
6—1595 145
пйсыо показаний потенциометра, сек; V — объем
пикнометра, используемого для определения скорости
истечения титранта, мл; Nb — нормальность раствора титранта;
Эа — грамм-эквивалент определяемого вещества; Т —
время наполнения пикнометра титрантом, сек; Vk—
общий объем анализируемого раствора, мл; Va— объем
аликвотной части, взятой для титрования, мл.
Концентрация определяемого вещества при точечной
записи кондуктометрических кривых может быть
найдена путем сравнения с результатами титрования навески
стандартного образца, химический состав которого
соответствует определяемому. При этом концентрацию
титранта и скорость его истечения точно не устанавливают.
Однако скорость истечения должна быть постоянной.
Расчеты проводят по формуле
о- =е ZL.Zl (63)
^А ^А« пст VA'
где £а — общее содержание определяемого вещества, г;
#АСТ — навеска стандартного вещества, взятого для
титрования, г; п — количество интервалов на кондуктометри-
ческой кривой до точки эквивалентности при титровании
аликвотной части анализируемого раствора; пст —
количество интервалов на кондуктометрической кривой до
точки эквивалентности при титровании навески
стандартного вещества; Vk— общий объем анализируемого
раствора, мл; Va — объем аликвотной части, взятой для
титрования, мл.
В случае, когда в качестве стандарта берут раствор
известной нормальности, расчетная формула имеет вид
лА^с1ЭА ^
5А лст1000 Va v '
где £а — общее содержание определяемого вещества, г;
п — количество интервалов на кондуктометрической
кривой до точки эквивалентности при титровании аликвотной
части анализируемого вещества; пст — количество
интервалов на кондуктометрической кривой до точки
эквивалентности при титровании стандартного раствора; VCr —
объем стандартного раствора, взятый для титрования, мл;
ЭА—грамм-эквивалент определяемого вещества; NCT—
нормальность стандартного раствора; Vk — общий объем
146
анализируемого раствора, мл; Va — объем аликвотной
части раствора, взятого для титрования, мл.
При анализе смесей электролитов расчеты проводят
так же, по приведенным выше формулам. При этом по
кондуктометрической кривой определяют количество
миллилитров титранта, вступивших в реакцию с отдельными
компонентами смеси. В случае хронокондуктометрическо-
го титрования находят длину диаграммной ленты,
соответствующую титрованию каждого компонента или
соответствующее количество интервалов между записью
показаний потенциометра *.
* Способы и примеры вычисления в титриметрическом методе
анализа подробно изложены в книге: А. П. К р е ш к о в. Основы
аналитической химии. Изд-во «Химия», т. 2, гл. I, § 10 и т. 3, гл. II,
§ 10, 1970 г.
б*
Глава IX
КИСЛОТНО-ОСНОВНОЕ ТИТРОВАНИЕ
ИНДИВИДУАЛЬНЫХ ЭЛЕКТРОЛИТОВ
§ 1. Приготовление стандартных растворов
При кондуктометрическом титровании используют как
сильные, так и слабые кислоты и основания. Широкое
распространение в качестве титрантов получили
хлористоводородная и уксусная кислоты, гидроокись натрия и
аммиак.
Приготовление раствора хлористоводородной кислоты.
Для приготовления стандартного раствора используют
химически чистый препарат. Обычно приготавливают
1 н. раствор путем разбавления концентрированного
раствора. Концентрацию полученного раствора
устанавливают известными индикаторными методами.
Приготовление раствора уксусной кислоты. Стандрат-
ный раствор уксусной кислоты приготавливают путем
растворения рассчитанной навески химически чистого
образца в дистиллированной воде. Обычно получают
приблизительно 1 н. раствор. Для установления точной
концентрации раствора уксусной кислоты целесообразно
использовать индикаторный метод или кондуктометричз-
ское титрование раствора NaOH.
Приготовление раствора гидроксида натрия.
Приготавливают приблизительно 1 н. раствор NaOH, по
возможности свободный от карбонатов. В полученном
растворе определяют содержание щелочи и соды известным
методом титрования кислотой с двумя индикаторами.—
фенолфталеином и метиловым оранжевым. Если
определяют концентрацию сильной кислоты или кислоты
средней силы, при расчетах используют общую нормальность
раствора. В случае, когда титруют слабые кислоты,
пользуются раствором щелочи, нормальность которой
устанавливают по фенолфталеину. Когда титруют соли
слабых оснований, в вычислениях необходимо использовать
фактическую нормальность стандартного раствора,
соответствующую содержанию в нем только NaOH.
Приготовление раствора аммиака. Стандартный 1 н.
раствор аммиака приготавливают путем разбавления
концентрированного химически чистого водного раствора
148
аммиака. Стандартизацию приготовленного раствора
можно проводить индикаторным или кондуктометриче-
ским методом по раствору НС1.
§ 2. Титрование кислот сильными основаниями
Определение азотной, хлористоводородной и хлорной
кислот. Азотная, хлористоводородная и хлорная кислоты
в водных растворах полностью диссоциированы. Реакции
их нейтрализации сильными основаниями протекают
количественно. Поэтому кондуктометрическое определение
этих кислот возможно не только в сравнительно
концентрированных растворах, но и в очень разбавленных (до
Ю-4 М)
Н+ + N0^ + Na+ + ОН- = Na+ + N0^ + Н20
Методика определения. Разбавляя точно измеренный
объем определяемой кислоты в мерной колбе,
приготавливают приблизительно 0,1 н. исходный раствор. Аликвот-
ную часть этого раствора переносят в электролитическую
ячейку. Проводят кондуктометрическое титрование
кислоты 1,0 н. раствором NaOH. При титровании до точки
эквивалентности электропроводность раствора сильно
понижается, так как нейтрализуются высокоподвижные
ионы водорода. После точки эквивалентности
электропроводность раствора увеличивается от избытка щелочи.
Кривая титрования имеет V-образную форму( см. рис. 1).
Графическим методом устанавливают на кондуктомет-
рической кривой точку эквивалентности и находят
количество миллилитров NaOH, вступивших в реакцию
(расчеты см. гл. 8, § 6).
Определение муравьиной, уксусной, пропионовой и
масляной кислот. Муравьиная, уксусная, пропионовая и
масляная — кислоты средней силы относятся к
гомологическому ряду насыщенных одноосновных карбоновых
кислот. Значения р/*Са этих кислот равны соответственно
3,75; 4,75; 4,87 и 4,82. Кондуктометрическое определение
этих кислот сильными основаниями в 0,1—0,001 н.
растворах не вызывает затруднений. При титровании
протекают реакции нейтрализации. На примере нейтрализации
уксусной кислоты гидроксидом натрия уравнение реакции
изображают следующим образом:
СН3СООН + Na* + ОН- = Na* + СН3СОО- + H2q
149
-о
4:
S
а:
§
Методика определения. Навеску кислоты,
рассчитанную для приготовления приблизительно 0,1 н. раствора,
переносят в мерную колбу и разбавляют
дистиллированной водой до метки. При помощи пипетки отбирают алик-
вотную часть раствора и помещают в электролитическую»
ячейку. Проводят кондуктометрическое титрование 1,0 н..
раствором NaOH. Сначала наблюдается небольшое
понижение электропроводности раствора, так как
нейтрализуются высокоподвижные ионы водорода,
образующиеся при незначительной
диссоциации кислот. Однако1
при титровании диссоциация;
кислот быстро подавляется1
под влиянием образующихся
солей и ионы водорода уже
не оказывают существенного
влияния на
электропроводность раствора. Поскольку
концентрации катионов тит-
ранта и анионов титруемой
кислоты при титровании
возрастают, после небольшого
минимума в начале
титрования электропроводность
раствора до точки
эквивалентности увеличивается. Более
сильно минимум выражен в
случае титрования
муравьиной кислоты. После точки эквивалентности
электропроводность быстро увеличивается от избытка титранта.
Кривые титрования имеют резкий излом в точке
эквивалентности.
Кривая титрования пропионовой кислоты показана на
рис. 45. Используя прямолинейные участки кривых тит?
рования, устанавливают положение точки эквивалентно,^
сти и находят число миллилитров щелочи, вступившей в
реакцию (расчеты см. гл. VIII, § 6).
Определение о-, м- и п-нитрофенолов. Нитрофенолц
проявляют в водных растворах слабые кислотные свойству
о-нитрофенол , , . , р/Са »=■ 7,23
ж-нитрофенол . рКц*=8,40
П-ннтрофенол р/(а*=7,15
Титрование этих кислот а водных растворах с по?
мощод индикйторрв, а также пртенциометричрсш ме>
150
О 3 В 9 /2 15
v. 05ъем раствора NaOH, мл
Рис. 45. Кривая кондукто-
метрического титрования
пропионовой кислоты
раствором NaOH
Тодом затруднено. Однако кондуктометрическое
титрование нитрофенолов сильными основаниями возможно. В
связи с незначительной растворимостью нитрофенолов в
воде титрование в 0,1 н. растворе можно осуществить
только для п-нитрофенола. Максимальная концентрация
ж-нитрофенола в титруемом растворе может достигать
0,09 н., а о-нитрофенола 0,015 н. При нейтрализации
нитрофенолов протекает реакция
02NC6H4OH + Na+ + ОН- = Na+ + 02NC6H40- + Н20
Методика определения. Взвешивают на аналитических
весах навески нитрофенолов, необходимые для
приготовления исходных растворов указанных концентраций.
После растворения навесок в мерных колбах отбирают
пипеткой часть раствора и переносят в электролитическую
ячейку. Концентрация раствора NaOH, используемого а
качестве титранта, должна быть в десять раз выше
концентрации титруемого раствора. Электропроводность
раствора с начала титрования до точки эквивалентности
линейно увеличивается. Более сильное повышение
электропроводности вызывает избыток щелочи. Точка
эквивалентности расположена на пересечении двух
восходящих ветвей кондуктометрической кривой. Кривые
титрования показаны на рис. 3. Устанавливают количество
щелочи, вступившей в реакцию, и проводят расчеты, как
описано в гл. VIII, § 6.
Определение хромовой кислоты. Хромовая кислота
Н2Сг04 практически полностью диссоциирует в водных
растворах по первой ступени, а по второй ступени —
весьма незначительно (рКа.1 =— 1; р/<а2 = 6,50). Возможно
дифференцированное кондуктометрическое титрование
этой кислоты щелочью по ступеням нейтрализации.
Сначала в реакцию вступают ноны водорода, затем НСг04~-
ионы
Н+ + НСг04- + Na+ + ОН- = Na+ + HCrO^ + H20
Na+ + HCrO^ -f Na+ + OH- = 2Na+ + CrO^- + H20
Концентрация титруемых растворов может изменяться в
пределах 0,1—0,001 н.
Методика определения. Для получения исходного
приблизительно 0,1 н. раствора взвешивают на
аналитических весах навеску хромовой кислоты, переносят в
мерную колбу и растворяют в воде. Отбирают аликвотную
151
Часть полученного раствора, помещают в
электролитическую ячейку и титруют 1 н. раствором NaOH. Кривая
титрования имеет два излома. При нейтрализации
первого кислотного эквивалента электропроводность линейно
понижается. Затем в реакцию вступают HCrO-г-ионы, что
приводит к повышению электропроводности. Избыток
щелочи вызывает более сильное повышение
электропроводности раствора. Кривая титрования хромовой кислоты
показана на рис. 6 (кривая 1). По второму излому кон-
Дуктометрической кривой определяют количество
миллилитров титранта, участвующего в реакции (расчеты см.
гл. VIII, § 6).
§ 3. Титрование кислот слабыми основаниями
Определение янтарной, глутаровой и адипиновой
кислот. Янтарная, глутаровая и адипиновая кислоты
сравнительно слабо диссоциируют; между первой и второй
константами их диссоциации нет достаточного различия,
необходимого для дифференцированного титрования по
ступеням диссоциации. При кондуктометрическом
титровании этих кислот целесообразно использовать слабые
основания, потому что кривые титрования в этом случае
имеют более резкий излом.
Титрование 0,1 н. растворов слабых двухосновных
кислот слабыми основаниями возможно, если сумма р/Са2 +
+ р/Сь^12 (см. гл. III, § 1). При нейтрализации этих
кислот можно использовать раствор аммиака (р/Сь=4,75),
что видно из приведенных ниже данных:
Янтарная кислота . . . 4,21 5,64 10,39
Глутаровая кислота . . 4,34 5,27 10,02
Адипиновая кислота . . 4,42 5,28 10,03
При титровании 0,1 н. растворов излом кондуктомет-
рической кривой в точке эквивалентности весьма резкий.
Нейтрализация кислот в более разбавленных растворах
приводит к закруглению кондуктометрической кривой
вблизи точки эквивалентности. На примере янтарной
кислоты реакция нейтрализации протекает по уравнению
НООС(СН2)2СООН + 2Na+ + 20H- = 2Na+ + (СН2)2(СОО)^-+2Н20
Методика определения. Навеску анализируемой
кислоты, необходимую для приготовления определенного объе-
152
ма 0,1 н. раствора, отвешивают на аналитических весах.
Приготавливают в мерной колбе исходный раствор.
Переносят аликвотную часть приготовленного раствора в
электролитическую ячейку и проводят кондуктометриче-
ское титрование приблизительно 1,0 н. раствором
аммиака. Электропроводность раствора при титровании
сначала немного понижается, что объясняется нейтрализацией
высокоподвижных ионов водорода, образующихся при
диссоциации титруемой кислоты. Однако образующаяся
в процессе нейтрализации соль вызывает повышение
электропроводности раствора. Поэтому кривая титрования
имеет неглубокий минимум в начале титрования. После
минимума электропроводность раствора линейно
увеличивается до точки эквивалентности. При добавлении
избытка аммиака электропроводность не изменяется.
Кривая титрования имеет резкий излом в точке
эквивалентности. Определяют количество миллилитров титранта,
вступившего в реакцию, и рассчитывают содержание
кислоты в титруемом растворе, как описано в главе VIII, §6.
§ 4. Титрование оснований сильными кислотами
Определение гидроксидов лития, натрия и калия. Гид-
роксиды лития, натрия и калия являются сильными
полностью диссоциированными в воде основаниями. Сильные
кислоты количественно нейтрализуют эти основания. Кои-
дуктометрическое определение указанных щелочей
возможно не только в сравнительно концентрированных, но
и в очень разбавленных растворах. Если в качестве
титранта использовать хлористоводородную кислоту,
реакция нейтрализации протекает по уравнению
Li+ + ОН- -|- Н+ + С1- = Li+ + CI- + Н20
Методика определения. Приготавливают
приблизительно 0,1 н. раствор определяемого вещества.
Устанавливают концентрацию раствора щелочи и разбавляют его
в мерной колбе с целью получения приблизительно 0,1 н.
раствора. Если определяют содержание основного
вещества в твердом образце, раствор готовят из навески.
Аликвотную часть приготовленного раствора при помощи
пипетки переносят в электролитическую ячейку.
Проводят кондуктометрическое титрование 1,0 н. раствором
153
HC1. При титровании до точки эквивалентности
электропроводность раствора линейно понижается, а после точки
эквивалентности — линейно увеличивается. Кривая
титрования имеет V-образную форму (см. рис. 8, кривая 1).
По кондуктометрической кривой устанавливают
количество НС1 (мл), вступившей в реакцию (см. гл. VIII, §6).
Определение анилина. В водных растворах анилин
проявляет очень слабые основные свойства (р/(ь = 9,42).
При определении анилина широко используются
неводные растворители. Возможность кондуктометрического
титрования очень слабых оснований в водных растворах
может быть использована при определении анилина. Для
уменьшения гидролиза получающейся соли концентрация
титруемого раствора должна быть близка к 0,1 н. При
титровании хлористоводородной кислотой протекает
реакция
C6H5NH2 + Н+ + CI- = C6H5NH3+ + CI-
Методика определения. На аналитических весах
отвешивают навеску анилина, необходимую для
приготовления определенного объема приблизительно 0,1 н.
раствора. Отбирают аликвотную часть из полученного в
мерной колбе раствора и переносят в электролитическую
ячейку. Титруют кондуктометрическим методом 1,0 н.
раствором НО.
Электропроводность раствора при титровании до
точки эквивалентности линейно
увеличивается. Избыток титранта
вызывает более сильное
повышение электропроводности раствора.
Вблизи точки эквивалентности
кривая титрования слегка
закруглена. На рис. 46 приведена
кривая кондуктометрического
титрования 0,1 н. раствора анилина
хлористоводородной кислотой.
При установлении точки
эквивалентности используют
прямолинейные участки на ветвях
кондуктометрической кривой.
Определяют количество кислоты,
вступившей в реакцию, и рассчитывают
содержание анилина (гл. VIII,
§6). '
0 \ 2 3 Ч-
Ойъем растбора
НСЬ, мл
Рис. 46. Кривая
кондуктометрического
титрования анилина раствором
на
154
§ 5. Титрование оснований слабыми кислотами
Определение моно-, ди- и триэтаноламинов. Моно-,
ди- и триэтаноламины отличаются по силе. При переходе
от моно- к триэтаноламину основные свойства
ослабляются. Как было установлено выше, наиболее
благоприятные условия для кондуктометрического титрования
слабых оснований создаются, когда в качестве титрантов
Используют слабые кислоты. При этом должны
соблюдаться следующие условия: сумма рКъ основания и р/Са
кислоты должна быть ^12. Для определения этанолами-
нов можно использовать уксусную кислоту (р/Са = 4,75).
Значения суммы р/Са и рД'ь удовлетворяют
предъявляемым требованиям:
Моноэтаноламин .... 4,50 9,25
Диэтаноламии 5,12 9,87
Триэтаноламин 6,18 10,88
При титровании протекает реакция нейтрализации,
которая для моноэтаноламина выражается уравнением
(C2H5)NH2 + СН3СООН = (C2H5)NH^ + CH3COO-
Методика определения. Для приготовления исходного
приблизительно 0,1 н. раствора моно-, ди- или триэта-
ноламииа на аналитических весах взвешивают
соответствующие навески. После приготовления раствора в
мерных колбах отбирают при помощи пипетки аликвотную
часть и помещают в электролитическую ячейку.
Титруют кондуктометрическим методом 1,0 н. раствором
СНзСООН. При титровании до точки эквивалентности
электропроводность раствора повышается, а после нее
остается постоянной. При титровании моноэтаноламина
в начале кривой титрования наблюдается небольшой
минимум, характерный для нейтрализации основания
средней силы. Кривая титрования триэтаноламина
слегка закруглена вблизи точки эквивалентности. По
кондуктометрическим кривым находят количество миллилитров
участвующей в реакции кислоты. Расчеты см. гл. VIII,
§6.
§ 6. Титрование солей слабых оснований
сильными основаниями
Определение солей аммония. Метод основан на
вытеснении аммиака из солей аммония сильными основания-
Ми. При этом не имеет значения, какие кислоты образу-
155
ют аммонийную соль. Возможен анализ хлорида,
нитрата, сульфата, ацетата и других солей неорганических и
органических кислот. Концентрацию титруемого
раствора лучше брать близкой к 0,1 н., так как при титровании
очень разбавленных растворов усиливается обратимость
реакции и излом кондуктометрической кривой вблизи
точки эквивалентности закругляется. Для примера
приводим реакцию взаимодействия нитрата аммония с гид-
роксидом натрия
NHJ
Ь N0~ -|- Na+ + ОН --= Na+ + N0"
,3 +NH3 + H20
Методика определения. На аналитических весах
отвешивают навеску определяемой соли аммония,
необходимую для приготовления
определенного объема
приблизительно 0,1 н.
раствора. После растворения
соли в воде из мерной
колбы пипеткой отбирают
аликвотную часть и
переносят в
электролитическую ячейку. Титрование
проводят 1,0 н. раствором
NaOH кондуктометриче-
ским методом. При
титровании до точки
эквивалентности
электропроводность раствора
понижается, так как подвижность
ионов аммония выше
подвижности заменяющих их в растворе ионов натрия.
Избыток щелочи вызывает сильное повышение
электропроводности раствора. Кривая кондуктометрического
титрования хлорида аммония щелочью показана на рис. 47.
На кондуктометрической кривой устанавливают точку
эквивалентности и определяют, сколько миллилитров
щелочи вступило в реакцию (расчеты см. гл. VIII,
§ 6).
Определение гидрохлоридов гидразина и карбамил-
гидразина (семикарбазида). Метод основан на
вытеснении сильными основаниями гидразина и карбамилгидра-
зина из их солей. Хотя основные свойства гидразина в
водных растворах (р/Сь = 5,90) выражены значительно
сильнее, чем карбамилгидразина (р/Сь= 10,57), кондук-
156
3 В 9 11 15
Объем pacmSapa ИаОН,мл
Рис. 47. Кривая
кондуктометрического титрования NH4C1
раствором NaOH
тометрическое титрование позволяет проводить оиреде*
ления в обоих случаях. Для уменьшения обратимости
реакции титрование гидрохлорида гидразина лучше
проводить в 0,1 -—0,01 н. растворах. Концентрация
титруемого раствора гидрохлорида карбамилгидразина может
быть более низкой. Возможно кондуктометрическое dnpe^
деление не только гидрохлоридов указанных оснований,
ио и их солей с другими кислотами. Вытеснение
гидразина щелочью протекает по реакции
NoHjJ" + С1- -\- Na+ + ОН- ■--■ N2H4 + CI- + Na+ + Н20
Методика определения. Навеску определяемой соли,
рассчитанную для приготовления приблизительно 0,1 н.
раствора, отвешивают на аналитических весах,
переносят в мерную колбу и растворяют в воде. Пипеткой
отбирают аликвотную часть раствора и переносят в ячейку
для титрования. Проводят кондуктометрическое
титрование 1,0 н. раствором NaOH. При титровании
гидрохлорида гидразина электропроводность раствора до точки
эквивалентности линейно понижается. Характер кривой
титрования гидрохлорида карбамилгидразина на этом
участке несколько отличается. Так как основные
свойства карбамилгидразина выражены очень слабо, соль
подвергается гидролизу и раствор в начале титрования
содержит довольно высокую концентрацию водородных
ионов. Поэтому в начале титрования электропроводность
раствора сильно понижается, так как уменьшается
концентрация водородных ионов. При дальнейшем
титрования гидролиз соли подавляется и электропроводность
понижается менее сильно. Понижение
электропроводности раствора до точки эквивалентности
свидетельствует о том, что подвижности катионов титруемых солей
выше подвижности катионов натрия, заменяющих их в
процессе титрования. После точки эквивалентности
электропроводность сильно увеличивается от избытка щелочи.
Находят число миллилитров щелочи, прореагировавшей
с солями (расчеты см. гл. VIII, § 6).
§ 7. Титрование солей слабых кислот
сильными кислотами
Определение ацетатов. Метод основан на вытеснении
уксусной кислоты из ее солей сильными кислотами.
Катионы, входящие в состав соли, могут быть различной
157
химической природы. Возможно кондуктометрпческое
определение ацетатов лития, натрия, калия, магния,
кальция и других металлов, а также ацетатов,
образованных разнообразными органическими основаниями. С
целью уменьшения обратимости реакций, титрование
ацетатов следует проводить в 0,1 н. растворах. При
титровании ацетатов хлористоводородной кислотой
протекает реакция
Na+ + СН3СОО - + Н+ + CI- = Na+ + CI- -j- CH3COOH
Методика определений. Приготавливают исходный
приблизительно 0,1 и. раствор соли. Отбирают пипеткой
из мерной колбы аликвотную
часть раствора и переносят в
сосуд для титрования. Проводят
кондуктометрпческое титрование
1,0 н. раствором НС1>. С начала
титрования до точки
эквивалентности электропроводность
раствора немного увеличивается, так
как подвижность ацетат-ионов
ниже подвижности заменяющих
их ионов хлора. Избыток НС1
вызывает сильное повышение
электропроводности раствора.
Вблизи точки эквивалентности
кривая титрования слегка
закруглена вследствие обратимости
реакции. На рис. 48 показана
кривая хронокондуктометрического
титрования ацетата натрия
хлористоводородной кислотой. При
определении точки
эквивалентности используют прямолинейные участки на ветвях кон-
дуктометрической кривой. Устанавливают количество
миллилитров кислоты, вступившей в реакцию (расчеты
см. гл. VIII, § 6).
Определение фенолятов. Фенол в водных растворах
проявляет очень слабые кислотные свойства (р/Са =
= 10,00). Поэтому при взаимодействии фенолятов
различных металлов с сильными кислотами реакции
вытеснения фенола протекают практически количественно. На-
например, при взаимодействии фенолята натрия с HCI
Na+ + С6Н50- + Н+ + С1- = С6Н5ОН + Na+ + С1~
0 1 2 3 Ц- 5 В
Время, мин
Рис. 48. Кривая
нокондуктометрического титрования
(непрерывная запись)
СНзСОСЖа раствором
НС1
158
Возможно также кондуктометрпческое определение
солей, образованных различными производными фенола:
нитрофенолятов, хлорфенолятов, бромфенолятов, иодфе-
нолятов и др. Концентрации титруемых растворов не
лимитированы.
Методика определения. На аналитических весах
отвешивают навеску фенолята натрия, рассчитанную для
приготовления приблизительно 0,1 н. раствора.
Переносят навеску в мерную колбу и растворяют в воде. Часть
раствора помещают в электролитическую ячейку.
Титруют кондуктометрическим методом 1,0 н. раствором
НС1. В начале титрования электропроводность раствора
незначительно понижается, так как нейтрализуются
высокоподвижные гидроксилыше ионы, имеющиеся в
растворе вследствие гидролиза соли. Однако в процессе
титрования гидролиз соли быстро подавляется и
электропроводность раствора начинает увеличиваться. Это
объясняется тем, что в растворе фенолят-ионы заменяются
ионами хлора, имеющими более высокую подвижность.
Большая часть кривой до точки эквивалентности носит
линейный характер. Минимум на кривой в начале
титрования очень слабо выражен. При избытке НС1
электропроводность раствора сильно увеличивается.
Устанавливают число миллилитров НС1, вступившей в реакцию,
и определяют концентрацию соли в растворе, как
описано в гл. VIII, § 6.
§ 8. Титрование амфолитов сильными основаниями
и сильными кислотами
Определение амфолитов, имеющих биполярное
строение: глицина, а-аланина, валина, лейцина и серина.
Глицин, а-алаиин, валии, лейцин и серии имеют биполярное
строение, так как вследствие внутримолекулярной
нейтрализации протоны переходят от кислотных групп к
основным. Например, глицин (аминоуксусная кислота)
имеет строение H3NCH2COO-. Определение этих
амфолитов основано на вытеснении слабых основных групп
сильными основаниями
NH3CH2COO- + Na+ + ОН~ = H2NCH2COO- + Na+ + Н20
Концентрацию титруемых растворов указанных
амфолитов, если позволяет растворимость, следует брать
159
близкой к 0,1 н. При этом излом кривой в точке
эквивалентности резко выражен.
Методика определения. При определении используют
отдельные навески анализируемых аминокислот. Исходя
из объема титруемого раствора, рассчитывают навеску
определяемого вещества, необходимую для получения
приблизительно 0,1 н. раствора. Пробу растворяют в
воде в небольшом стакане и переносят в сосуд для
титрования. Проводят кондуктометрическое титрование 1,0 н.
раствором NaOH. При титровании до точки
эквивалентности электропроводность раствора увеличивается, так
как в растворе накапливаются катионы титранта, а
биполярные ионы переходят в анионы. При избытке
щелочи наблюдается еще более сильное повышение
электропроводности раствора. Точка эквивалентности
расположена на пересечении двух линейных участков кондукто-
метрической кривой. Кривая титрования 0,1 н. раствора
глицина, показана на рис. 18 (кривая 2). Находят
количество щелочи, вступившей в реакцию, и рассчитывают
концентрацию определяемого вещества, как описано в
гл. VIII, § 6.
Определение амфолитов, не имеющих биполярного
строения: о-, м и п-аминофенолов. Аминофенолы не
имеют биполярного строения. При взаимодействии аминофе-
нолов с сильными основаниями происходит
нейтрализация кислотных групп, а при взаимодействии с сильными
кислотами —■ основных
H2NC6H4OH + Na+ + ОН- zg. H2NC6H40- + Na+ + Н20
H2NC6H4OH + H+ + CI- ;£ H3NC6H4OH + Cl-
Кислотиыс и основные свойства аминофенолов
выражены очень слабо:
о-аминофенол 9,71 9,28
ж-аминофенол 9,87 9,83
п-аминофенол 10,30 8,50
Однако метод кондуктометрического титрования
позволяет проводить определение аминофенолов,
основываясь на реакции их нейтрализации сильными
основаниями или сильными кислотами. Для уменьшения
гидролиза получающихся солей концентрацию титруемых
растворов следует брать близкой к 0,1 н,
Ш
Методика определения. На аналитических весах
отвешивают навеску аминофенола и приготавливают
приблизительно 0,1 н. раствор. Аликвотную часть раствора
помещают в сосуд для титрования. Проводят
кондуктометрическое титрование 1,0 н. раствором NaOH или НС1.
При титровании электропроводность раствора до точки
эквивалентности увеличивается, так как в растворе
образуются хорошо диссоциирующие соли. Избыток NaOH
или НС1 вызывает более сильное повышение
электропроводности раствора. Кривые титрования закруглены
вблизи точки эквивалентности, что особенно заметно в случае
титрования п-аминофенола. На рис. 18 (кривая /) для
примера приведена кривая титрования щелочью м-аыи-
нофенола. При установлении точки эквивалентности
используют прямолинейные участки кондуктометрической
кривой. Находят число миллилитров щелочи или
кислоты, вступившей в реакцию, и проводят расчеты (см. гл.
VIII, §6).
Главв X
КИСЛОТНО-ОСНОВНОЕ ТИТРОВАНИЕ
СМЕСЕЙ ЭЛЕКТРОЛИТОВ
§ 1. Титроввние смесей кислот сильными основвниями
Анализ смесей хлористоводородной кислоты с
слабыми кислотами (метакриловой, уксусной, п-нитрофенолом,
л-нитрофенолом или борной). Метод основан на
последовательной нейтрализации сильной хлористоводородной
кислоты и слабых кислот. Если концентрации
компонентов смеси находятся в интервале 0,1—0,05 п., то в смесях
с сильными кислотами определяются все слабые
кислоты с рКа = 4-М0, а в 0,01 н. растворах — имеющие рКа =
= 5-г-9.
Значения р/Са метакриловой и уксусной кислот, п-нит-
рофенола, л-нитрофенола и борной кислоты равны
соответственно: 4,36, 4,75, 7,15, 8,40 и 9,12*. Поскольку эти
значения р/Са достаточно высокие, кондуктометрический
метод позволяет проводить анализ смесей сильной
хлористоводородной кислоты с этими слабыми кислотами.
При титровании более разбавленных растворов
затруднен анализ смесей, содержащих метакриловую и борную
кислоты. В первом случае закругляется первый излом
кондуктометрической кривой вследствие усиления
диссоциации метакриловой кислоты в разбавленном растворе.
Во втором случае закругляется второй излом
кондуктометрической кривой, что связано с гидролизом метабора-
та натрия.
Методика анализа. Точно отмеренный объем
анализируемой смеси кислот помещают в мерную колбу и
разбавляют дистиллированной водой до метки.
Концентрация отдельных компонентов смеси после разбавления
должна составлять 0,1—0,05 н. Аликвотную часть
раствора переносят в ячейку. Титруют кондуктометрический
методом 1,0 н. раствором NaOH. Кривые титрования
перечисленных смесей аналогичны: на них наблюдается
два излома. Сначала нейтрализуется
хлористоводородная кислота, что сопровождается понижением злектро-
* При нейтрализации борной кислоты Н3ВОз образуется мета-
борат натрия. Поэтому приводится показатель константы
диссоциации метаборной кислоты.
163
проводностн раствора вследствие нейтрализации высоко»
подвижных ионов водорода. Затем взаимодействуют
слабые кислоты, что приводит к повышению
электропроводности раствора, так как при этом образуются хорошо
диссоциирующие соли. Избыток щелочи вызывает
сильное повышение электропроводности раствора.
Для примера на рис. 49 приведена кривая кондукто-
метрического титрования смеси хлористоводородной и
уксусной кислот. Устанавливают количество
миллилитров щелочи, вступившей в реакцию с отдельными
кислотами (расчеты см. гл. VIII, § 6):
Анализ смесей
хромовой кислоты с сильной
(хлористоводородной) и
слабой (уксусной)
кислотами. Хромовая кислота
диссоциирует по двум
ступеням. Первая константа
диссоциации значительно
отличается от второй
константы ее диссоциации.
Поэтому при титровании
хромовой кислоты
раствором щелочи наблюдается
два скачка на кривой
титрования, благодаря чему
хромовая кислота может
быть оттитрована по двум
ступеням
электролитической диссоциации. Кроме того, возможно титрование
смеси хромовой кислоты с другими кислотами, в том числе
и с хлористоводородной кислотой.
Концентрация компонентов в смесях хромовой
кислоты с хлористоводородной кислотой может изменяться в
широких пределах. Титрование смесей хромовой и
уксусной кислот проводят в 0,1—0,05 н. растворах.
Методика анализа. Определенный объем
анализируемой смеси разбавляют дистиллированной водой в
мерной колбе с таким расчетом, чтобы концентрация
отдельных компонентов смеси соответствовала 0,1—0,05 н.
При помощи пипетки часть раствора переносят в
электролитическую ячейку. Титруют кондуктометрический
методомц1,0 н. раствором NaOH. При титровании смеси
хромовой кислоты с хлористоводородной кислотой пер-
J В 9 12 -15
Объем раствора NaOH, мл
Рис. 49. Кривая кондуктомет-
рического титрования смеси
хлористоводородной и
уксусной кислот раствором NaOH
163
вый излом кривой соответствует нейтрализации
Хромовой кислоты по первой ступени и хлористоводородной
кислоты, а второй — полной нейтрализации хромовой
кислоты. Уксусная кислота в смесях с хромовой
кислотой нейтрализуется совместно с нейтрализацией
второго кислотного эквивалента хромовой кислоты. В
обоих случаях электропроводность раствора до первого
излома кривой сильно понижается, что характерно для
нейтрализации сильных кислот. Между первым и вторым
изломами происходит некоторое увеличение
электропроводности раствора, характерное для нейтрализации сла-
бокислотпых групп. Сильное повышение
электропроводности раствора наблюдается от избытка титранта.
Находят число миллилитров щелочи, вступившей в реакцию
с отдельными кислотами. Количество миллилитров
щелочи, участвующей в реакции нейтрализации
хлористоводородной кислоты, находят по разности между первым
и вторым изломами, а взаимодействующих с уксусной
кислотой, наоборот, — между вторым и первым изломами
(расчеты см. гл. VIII, § 6).
§ 2. Титрование смесей оснований сильными кислотами
Анализ смесей гидроксида натрия со слабыми
основаниями (аммиаком, триэтаноламином, а-пиколином или
анилином). Метод кондуктометрического титрования
позволяет проводить анализ смесей сильных оснований
со слабыми (с р/Сь==4-=-10 в 0,1—0,05 н. растворах и с
рКъ = 5^-9 в 0,01 и. растворах).
При титровании смесей сильными кислотами сначала
нейтрализуется сильное основание (гидроксид натрия),
а затем слабые основания. В состав смесей могут
входить слабые основания различной силы: аммиак (рКъ =
= 4,75), триэтаноламин (р/Сь = 6,18), а-пиколин (рКъ =
= 8,03) или анилин (р#ь = 9,42). Более благоприятные
условия для кондуктометрического титрования
создаются, если концентрация индивидуальных компонентов в
смесях соответствует 0,1—0,05 н. Титрование очень
разбавленных растворов при анализах смесей, содержащих
аммиак и анилин, затруднено, так как увеличение
степени диссоциации аммиака в разбавленных растворах
обусловливает закругление первого излома кондуктометри-
ческой кривой, а кривая кондуктометрического титрова-
164
ний очень разбавленного раствора смеси, содержащей
анилин, имеет сильно закругленный второй излом, что
объясняется гидролизом солей анилиния.
Методика анализа. В мерной колбе разбавляют такой
объем анализируемой смеси оснований, чтобы
концентрация отдельных компонентов смеси находилась в
пределах 0,1—0,05 и. Аликвотную часть раствора переносят в
электролитическую ячейку и проводят кондуктометриче-
ское титрование 1,0 н. раствором НС1. Кривые
титрования типичны для нейтрализации смеси сильного и
слабого оснований. Они имеют
два излома. Первый
соответствует нейтрализации
сильного (NaOH), второй —
слабого основания.
Электропроводность раствора при
титровании до первого излома
линейно понижается, а
между первым и вторым
изломами линейно увеличивается.
Сильное повышение
электропроводности раствора
вызывает избыток НСк Кривая
кондуктометрического
титрования смеси 0,075 н.
растворов гидроксида натрия и
анилина раствором НС1
приведена на рис. 50.
Определяют число миллилитров НС1,
вступившей в реакцию с отдельными основаниями, и
определяют концентрации оснований в смеси (см. гл. VIII,
§ 6).
§ 3. Титрование сильными основаниями
двухкомпонентных смесей кислот и солей слабых
оснований
Анализ смесей солей аммония с кислотами. Анализ
смесей кислот и солей слабых оснований возможен, если
сумма р/Са кислоты и рКъ основания, образующего соль,
^12. При этих условиях сначала нейтрализуется
кислота, а затем взаимодействует соль. Так как значение рКъ
аммиака равно 4,75, в состав смесей с солями аммония
могут входить все кислоты, имеющие рКа^7.
Концентрации компонентов в смесях при этом могут изменяться
165
/2345
Объем раствора Ш, мл
Рис. 50. Кривая
кондуктометрического титрования
смеси растворов NaOH и
анилина раствором
хлористоводородной кислоты
В пределах 0,1—0,05 и. Например, возможен анализ
смесей хлорида аммония с хлористоводородной, три-, ди- и
монохлоруксусной, муравьиной, уксусной и другими
кислотами. Кроме хлорида аммония в состав смесей могут
входить аммонийные соли других кислот. Возможность
анализа указанных смесей видна из следующих данных:
Кислота рКа рКа + рКъ
Хлористоводородная . . —7 —2,25
Трихлоруксусная . .
Дихлоруксусная . .
2,85 7,60
3,75 8,50
4,75 9,50
Монохлоруксусная .
Муравьиная . . . .
Уксусная
0,70 5,45
1,25 6,66
Методика анализа. В мерную колбу помещают точно
измеренный объем анализируемой смеси хлорида
аммония с одной из указанных кислит. Концентрация
отдельных компонентов смеси после разбавления должна
составлять 0,1-—0,05 н. Аликвотную часть раствора
переносят в электролитическую ячейку. Титруют кондуктомет-
рическим методом 1,0 н. раствором NaOH. При
титровании сначала нейтрализуется кислота, затем вытесняется
аммиак из его соли. Кривые титрования имеют два
излома. Характер изменения электропроводности при
нейтрализации кислоты зависит от силы кислоты. При
нейтрализации хлористоводородной, трихлоруксуснои и
дихлоруксусной кислот электропроводность раствора
понижается. Кривая титрования смеси, содержащей моно-
хлоруксусную кислоту, имеет до первого излома резкий
минимум, не имеющий аналитического значения, что
характерно для нейтрализации этой довольно сильной
кислоты. Менее резко минимум выражен при
нейтрализации более слабых муравьиной и уксусной
кислот. На большей части кривых титрования этих
кислот наблюдается повышение электропроводности
раствора до точки эквивалентности. При взаимодействии
хлорида аммония с щелочью происходит понижение
электропроводности раствора, так как подвижность ионов
аммония выше подвижности заменяющих их ионов
натрия. Кривая титрования смесей муравьиной кислоты и
NH4CI показана на рис. 51. Находят количество
миллилитров NaOH, вступившего в реакцию с кислотой и
хлоридом аммония (гл. VIII, § 6).
Анализ смесей гидрохлорида гидроксиламина с
кислотами. Гидроксиламин в водных растворах проявляет
очень слабые основные свойства (р^ь=8,30) и количест-
166
венно вытесняется сильными основаниями из его солеи.
В смесях гидрохлорида гидроксиламина с кислотами
последовательность взаимодействия компонентов может
быть различной. При (рЯа + рЯь) =^12. сначала
взаимодействует кислота, затем соль. Если же (р/Са+Р^Сь)^16,
то, наоборот, сначала взаимодействует соль, а затем
кислота. Эти зависимости соблюдаются в пределах
концентраций компонентов 0,1—0,05 н. Поэтому в смесях
гидрохлорида гидроксиламина с кислотами, имеющими
р^Са<3,7, последовательно взаимодействуют кислота и
чЗ
,х...
^ с Время, сек
Рис. 51. Кривая хронокондуктометрического
титрования (точечная запись) смеси
муравьиной кислоты и NH4CI раствором NaOH
соль. Когда значение р/Са кислоты ^7,7, наоборот,
сначала взаимодействует соль, затем кислота. В первом
случае в состав смесей могут входить, например,
хлористоводородная и сульфаниловая кислоты, а во втором
случае — борная кислота и фенол. Сумма рКа. и рКъ для
смесей разных электролитов с гидроксиламином имеет
значения:
Хлористоводородная
кислота
Сульфаниловая кислота
Борная кислота ....
Фенол
Методика анализа. Взвешивают на аналитических
весах навеску анализируемой смеси кислоты и соли.
Растворяя навеску в воде в мерной колбе, приготавливают
.исходный 0,1—0,05 н. раствор. Если для анализа берут
раствор смеси, его разбавляют в мерной колбе до
указанной концентрации. Аликвотную часть раствора
переносят в электролитическую ячейку. Титруют кондуктомет-
рическим методом 1,0 н. раствором NaOH.
W
—7
3,25
9,12
10,00
1,30
11,55
17,15
18,03
При титровании смесей, содержащих
хлористоводородную и сульфаниловую кислоты, сначала
нейтрализуются кислоты, затем вытесняется гидроксиламин.
Кривые титрования имеют два излома. Нейтрализация
хлористоводородной кислоты сопровождается сильным
понижением электропроводности раствора. При
нейтрализации сульфаниловой кислоты электропроводность
раствора проходит через минимум. Вытеснение гидроксиламина
из его соли вызывает
небольшое понижение
электропроводности раствора; это
показывает, что
подвижность катионов титруемой
соли выше подвижности
ионов натрия.
Кривые титрования
смесей, содержащих борную
кислоту и фенол, имеют
другой характер. Сначала
вытесняется гидроксиламин,
что приводит к некоторому
понижению
электропроводности раствора, затем
нейтрализуются слабые
кислоты, что вызывает
повышение электропроводности.
Избыток титранта сильно
увеличивает
электропроводность раствора. По кривым титрования" находят
количество миллилитров щелочи, вступившей в реакцию с
отдельными компонентами смеси, и рассчитывают их
концентрации (гл. VIII, § 6).
Определение фосфатов аммония и анализ смеси
Н3Р04 с NH4H2P04. Одно- и двухзамещепные фосфаты
аммония и их смеси нашли широкое применение в
качестве удобрений. Эти соли в сочетании с другими солями
аммония и калия образуют ряд сложных удобрений. При
контроле производства фосфорных удобрений применяют
метод анализа смеси однозамещенного фосфата аммония
и фосфорной кислоты. Фосфаты аммония являются
солями слабого основания (рЛл> = 4,75) и слабой
многоосновной кислоты (рКа1 = 2,\2, р/Са2 = 7,21; р/Са3= 12,67). Такие
солн можно титровать как щелочами (вытеснение
слабого основания), так и сильными кислотами (вытеснение
«о
с _
EJ5
* "а.
eta с
О. О;
Cud
<5^
••
-
'X""
i
-■<:ы/
•v^V
rS
Время, сек
Рис. 52. Кривые хронокон-
дуктометрического
титрования (точечная запись)
раствором NaOH фосфатов
аммония и их смесей с Н3Р04:
/ —(NH4)3P04; 2 — (NH^HPOi
3 — NH4H2PO<; 4 - H3PO1 +
+ NH4H2PO4
Ш
слабой кислоты). На рис. Ъй приведены кривые
титрования (NH4)3P04; (NH4)2HP04; NH4H2P04 и смеси H3P04
с NH4H2P04 раствором NaOH.
Трехзамещенный фосфат аммония подвергается в
водных растворах гидролизу практически до конца:
(NH4)3P04 + H20 ^t (NH4)2HP04 + NH4OH
При титровании (NH4)3P04 щелочью вытесняется
аммиак и электропроводность раствора понижается, так
как более подвижные ионы аммония заменяются менее
подвижными ионами натрия. Однако такое понижение
электропроводности наблюдается только при
прибавлении двух грамм-эквивалентов щелочи, после чего
электропроводность раствора начинает увеличиваться, так
как находящиеся в растворе НР042~-ионы практически
со щелочью не взаимодействуют.
Кривая титрования щелочью (NH4)2HP04
аналогична кривой титрования трехзамещенного фосфата. При
титровании щелочью NH4H2P04 сначала протекает
реакция нейтрализации: Н2Р04~-ионы переходят в НР042_-
ионы и электропроводность раствора увеличивается.
Затем взаимодействуют ионы аммония, что приводит к
понижению электропроводности раствора.
Дифференцированное титрование этих ионов возможно, так как сумма
р/Сь аммиака и р/Са2 фосфорной кислоты равна 11,96.
При дальнейшем добавлении щелочи
электропроводность раствора увеличивается, как при титровании двух-
и трехзамещенных фосфатов аммония.
Метод кондуктометрического титрования позволяет
проводить анализ смеси Н3Р04 и NH4H2P04. При
титровании щелочью смеси сначала нейтрализуется первый
кислотный эквивалент кислоты (р/Са1 = 2,12), что
вызывает понижение электропроводности раствора. Затем в
реакцию вступают Н2Р04~-ионы, которые находятся в
растворе в результате нейтрализации фосфорной кислоты по
первой ступени, а также вследствие диссоциации соли.
При этом образуются НР042~-ионы и
электропроводность раствора увеличивается. После этого
взаимодействуют ионы аммония и электропроводность раствора опять
понижается. Окончание этой реакции фиксируется
изломом кондуктометрической кривой, так как при
дальнейшем добавлении щелочи происходит повышение
электропроводности. Таким образом, кривая титрования имеет
три излома, позволяющих определить концентрации ком-
169
понентбй смеси. Участок кривой до первого излома
Используют для определения концентрации фосфорной
кислоты, а между вторым и третьим изломами — для
определения концентрации NH4H2PO4.
§ 4. Титрование сильными основаниями
многокомпонентных смесей кислот и солей
слабых оснований
Анализ смеси хлористоводородной, уксусной кислоты
н хлорида аммония. Метод кондуктометрического
титрования применим для анализа трехкомпонентных смесей,
в состав которых входят сильная и слабая кислоты и
соль слабого
основания. Критерии
применимости метода
приводятся в табл. 3 и 4
(пункт 3). Смесь
хлористоводородной и
уксусной кислот и
хлорида аммония
удовлетворяет требованиям в
отношении
кислотно-основной силы
электролитов. При титровании
сильными основаниями
сначала
нейтрализуется хлористоводородная,
а затем уксусная кислота, что возможно, поскольку
значение р^а уксусной кислоты равно 4,75. После кислот
взаимодействует хлорид аммония. Дифференцированное
титрование уксусной кислоты и хлорида аммония
допустимо, потому что сумма рКа уксусной кислоты и рКъ
аммиака равна 9,50. Концентрации компонентов в смеси
не должны быть меньше 0,02 н.
Методика анализа. Анализируемый раствор
разбавляют в мерной колбе и берут для титрования аликвот-
ную часть. Титрование ведут стандартным 1,0 н.
раствором NaOH. Кривая титрования имеет три излома в
результате последовательной нейтрализации
хлористоводородной и уксусной кислот и вытеснения аммиака.
Избыток NaOH сильно повышает электропроводность
раствора. Кривая титрования смеси показана на рис. 53.
Расчеты см. гл. VIII, § 6.
0 1 23 Ч 5 Б 7 в 9 10 11 12 13 Щ
Время, мин
Рис. 53. Кривая хронокондуктометри-
ческого титрования (непрерывная
запись) раствором NaOH смеси
хлористоводородной кислоты, уксусной
кислоты и хлорида аммония
170
Анализ смеси монохлоруксусной кислоты, борной
кислоты и гидрохлорида гидразина. При кондуктометриче-
ском титровании трехкомпонентных смесей, содержащих
две кислоты и соль слабого основания, при
определенных условиях соль может взаимодействовать между
кислотами (см. табл. 3 и 4, пункт 6 и 4). При этом обе
кислоты могут быть слабыми. Если смесь состоит из
монохлоруксусной и борной кислот и гидрохлорида гидрокси-
ламина, кислотно-основные свойства электролитов
удовлетворяют предъявляемым требованиям. Сначала с
сильным основанием взаимодействует монохлоруксусная
кислота (рКа = 2,85), затем гидрохлорид гидроксилами-
на. Последовательное титрование этих соединений
возможно потому, что сумма р/Са монохлоруксусной
кислоты и р/Сь гидроксиламина равна 11,15. Затем
нейтрализуется борная кислота (р/Са = 9,12). Дифференцированное
титрование двух последних компонентов объясняется тем,
что сумма рКь гидроксиламина и р/Са борной кислоты
равна 17,35. Концентрация борной кислоты не должна
быть ниже 0,01 н.
Методика анализа. В мерной колбе разбавляют
соответствующий объем раствора анализируемой смеси или
растворяют взятую на аналитических весах навеску
исследуемого образца. Аликвотную часть полученного
раствора титруют 1,0 н. раствором NaOH. Кривые титрования
характеризуются тремя изломами. Сначала
нейтрализуется монохлоруксусная кислота, затем вытесняется гид-
роксиламин и, наконец, нейтрализуется борная кислота.
На кривой титрования до первого излома имеется
пологий минимум, не имеющий аналитического значения, что
характерно для нейтрализации монохлоруксусной
кислоты. После первого излома электропроводность раствора
понижается, что всегда имеет место при вытеснении
гидроксиламина. Нейтрализация борной кислоты вызывает
повышение электропроводности раствора. Избыток тит-
ранта увеличивает электропроводность раствора более
сильно. Вычисляют количество миллилитров щелочи,
вступившей в реакцию с отдельными компонентами
смеси, и определяют их концентрации, как описано в гл. VIII,
§6.
Анализ смеси п-нитрофенола с гидрохлоридами ееми'
карбазида и триметиламина. При анализе
трехкомпонентных смесей, в состав которых входят две соли слабых
рснований-и ода слабая, кислота, ШЩЩ быть использо^
Ш
вано кондуктометрическое титрование. Интервалы
значений констант диссоциации, лимитирующие возможности
определения электролитов, показаны в табл. 3 (пункт 7).
Согласно табл. 3 возможен анализ смеси п-нитрофенола
с гидрохлоридами семикарбазида и триметиламина. При
титровании сильными основаниями последовательно
взаимодействуют гидрохлорид семикарбазида, п-нитрофенол
и гидрохлорид триметиламина. Семикарбазид проявляет
слабые основные свойства (р/Сь= 10,57). Сумма рКъ
семикарбазида и рКа n-нитрофенола равна 17,15. Поэтому
в первую очередь взаимодействует соль. Триметиламии
является более сильным основанием (р/Сь = 4,20). Сумма
рКщ n-нитрофенола и рКъ триметиламина равна 11,35,
что позволяет проводить дифференцированное
титрование кислоты и соли в указанной последовательности. При
титровании очень разбавленных растворов затруднено
количественное вытеснение триметиламина. Поэтому
концентрация гидрохлорида триметиламина должна быть
>0,05 н.
Методика
анализа. Приготавливают
исходный раствор
путем растворения в
мерной колбе
взвешенной на
аналитических весах навески
анализируемой
смеси. Аликвотную
часть раствора
переносят в
электролитическую ячейку и
титруют 1,0 и.
раствором NaOH. На
кривой титрования
имеется три излома
(рис. 54). Гидрохлорид семикарбозида в водных
растворах подвергается заметному гидролизу. Поэтому
электропроводность при титровании этой солн более сильно
понижается в начале титрования, так как
нейтрализуются высокоподвижные ионы водорода, концентрация
которых выше в начале титрования. В процессе титрования
гидролиз соли подавляется и электропроводность
раствора линейно понижается до первого излома кривой.
Вторым вступает в реакцию я-нитрофенол. При его нейтра-
5 в.
Is
X..
X
-^
Время, сек
Рис. 54. Кривая хронокондуктометрн-
ческого титрования (точечная запись)
раствором NaOH смеси
п-нитрофенола с гидрохлоридами семикарбазида
и триметиламина
172
лизации наблюдается повышение электропроводности
раствора. После этого вытесняется триметиламии из его
соли. Реакция приводит к понижению
электропроводности раствора. От избытка щелочи электропроводность
резко увеличивается. Третий излом кривой слегка
закруглен вследствие некоторой обратимости реакции. Расчеты
см. гл. VIII, § 6.
Анализ смеси серной кислоты, сульфата гидроксила-
мина и сульфата аммония. Кондуктометрическое
титрование может быть использовано при анализе трехкомпо-
нентных смесей, состоящих из одной кислоты и двух
солей слабых оснований. При этом сначала нейтрализуется
кислота, а затем последовательно вытесняются слабые
основания из их солей. Условия, при которых возможны
эти определения, показаны в табл. 3 (пункт 4).
Основываясь на этих условиях, возможен анализ смеси серной
кислоты, сульфата гидроксиламина и сульфата аммония.
При титровании сильными основаниями сначала
нейтрализуется серная кислота. После этого вытесняется гидро-
ксиламин. Если концентрация этой соли выше 0,01 н.,
гидролиз не мешает определению и дифференцированное
титрование серной кислоты и соли возможно. Последним
вступает в реакцию сульфат аммония. Значения
констант диссоциации гидроксиламина (р/Сь = 8,03) и
аммиака (р/Сь = 4,75) сильно отличаются, разница в значении
рКъ составляет 3,28. На этом основывается
дифференцированное титрование солей. Реакция вытеснения
аммиака в сильно разбавленных растворах обратима. Поэтому
концентрация сульфата аммония в титруемом растворе
не должна быть ниже 0,02 н.
Методика анализа. Взвешивают на аналитических
весах навеску анализируемой смеси, переносят в мерную
колбу и растворяют в дистиллированной воде.
Аликвотную часть раствора титруют приблизительно 1,0 н.
раствором NaOH. Дифференцированное титрование
компонентов смеси фиксируется тремя изломами кондук-
тометрической кривой. Сначала нейтрализуется серная
кислота, электропроводность раствора при этом сильно
понижается. Затем вытесняются гндроксиламин, а после
него — аммиак. При вытеснении оснований
электропроводность раствора немного понижается; более сильное ее
понижение наблюдается при вытеснении аммиака.
Избыток щелочи вызывает рост электропроводности. Расчеты
проводят по известным формулам (гл. VIII, § 6).
173
Анализ смеси хлористоводородной кислоты, уксусной
кислоты, фенола и гидрохлорида триэтаноламина. При
определенных условиях возможен кондуктометрический
анализ четырехкомпонентных смесей, состоящих из трех
кислот и одной соли слабого основания. Критерии
применимости метода даны в табл. 3 (пункт 9). Кислотно-
основные свойства хлористоводородной и уксусной
кислот, фенола и гидрохлорида триэтаноламина
удовлетворяют этим критериям. При взаимодействии с сильными
основаниями сначала нейтрализуется
хлористоводородная кислота, затем уксусная кислота (р/Са = 4,75).
Значение рКа уксусной кислоты позволяет проводить ее
определение после хлористоводородной кислоты. Затем в
реакцию с щелочью вступает гидрохлорид
триэтаноламина. Раздельное титрование уксусной кислоты и соли
возможно, так как сумма рКа уксусной кислоты и рКъ
триэтаноламина равна 10,83. Последним нейтрализуется
фенол. Его дифференцированное титрование в смеси объяс-
ясняется тем, что
сумма рКа фенола и рКъ
триэтаноламина
составляет 16,18.
Концентрации уксусной кислоты
и фенола в титруемой
смеси должны быть
выше 0,05 н., иначе
изломы на кривой
закругляются,
j Б з 11 15 18 и Методика анализа.
05ъем растдора NaOH, мл В мерной колбе прип>
Рис. 55. Кривая хронокондуктометри-
ческого титрования (непрерывная
запись) раствором NaOH смеси
хлористоводородной кислоты, уксусной
кислоты, гидрохлорида триэтаноламина
и фенола
тавливают исходный
раствор анализируемой
смеси нужной
концентрации. Аликвотную
часть раствора титруют
приблизительно 1,0 н.
раствором NaOH.
Кривая титрования имеет четыре излома (рис. 55). Сначала
нейтрализуется хлористоводородная кислота, что
сопровождается линейным понижением электропроводности
раствора. Затем нейтрализуется уксусная кислота, и
электропроводность раствора повышается. После этого
вытесняется триэтаноламин. При этой реакции электропро
водность раствора незначительно увеличивается. HaKQr
|74
нец, нейтрализуется фенол, п электропроводность
раствора возрастает более сильно. Резкое увеличение
электропроводности вызывает избыток щелочи. Проводят
вычисления, как описано в гл. VIII, § 6.
Анализ смеси хлористоводородной кислоты, п-нитро-
фенола и гидрохлоридов гексаметилентетрамина и триме-
тиламина. Кондуктометрический методом
осуществимо дифференцированное титрование
четырехкомпонентных смесей, которые состоят из двух кислот и двух
солей слабых оснований. Значения констант
диссоциации, характеризующих кислотно-основные свойства
раздельно титрующихся веществ, приведены в табл. 3
(пункт 11). При этом взаимодействия кислот и солей
чередуются, причем первой вступает в реакцию кислота.
Одной из смесей, анализ которой возможен, является
смесь хлористоводородной кислоты, я-нитрофенола и
гидрохлоридов гексаметилентетрамина и триметиламина.
С сильными основаниями в первую очередь вступает в
реакцию хлористоводородная кислота, а затем
гидрохлорид гексаметилентетрамина. Исходя из значения
константы диссоциации гексаметилентетрамина (р/Сь = 8,87),
можно заключить, что гидролиз соли при титровании
достаточно концентрированных растворов не мешает
определению кислоты. Следующим нейтрализуется
«-нитрофенол (р/Са = 7,15). Раздельное титрование
гидрохлорида гексаметилентетрамина и n-нитрофенола возможно,
так как сумма рКъ и рКа этих электролитов равна 16,02.
Триметиламин (рЛо> = 4,20) титруется в последнюю
очередь. Дифференцированное титрование n-нитрофенола с
триметиламином объясняется тем, что характеризующее
их значение (рКа + рКъ) составляет 11,35. Концентрация
отдельных веществ в смеси должна быть выше 0,05 н.
для уменьшения гидролиза гидрохлорида
гексаметилентетрамина и достижения полноты вытеснения
триметиламина.
Методика анализа. Определенный объем
анализируемой смеси разбавляют в мерной колбе дистиллированной
водой и получают исходный раствор требуемой
концентрации. Аликвотную часть раствора помещают в
электролитическую ячейку и титруют 1,0 н. раствором NaOH. При
нейтрализации хлористоводородной кислоты
электропроводность раствора понижается сильно, а при вытеснении
гексаметилентетрамина — лишь незначительно.
Последующая нейтрализация я-нитрофенола приводит к повы-"
175
шеншо электропроводности, а вытеснение триметиламина
опять к понижению. Избыток щелочи сильно увеличивает
электропроводность. Четыре излома кондуктометриче-
ской кривой позволяют установить, сколько миллилитров
щелочи вступило в реакцию с отдельными компонентами
смеси. Кривая титрования смеси щелочью показана на
рис. 56. Расчеты см. гл. VIII, § 6.
§1
eft i>
§.*
С: о;
§-1
Is
..^
Время, сек
Рис. 56. Кривая хронокондуктометриче-
ского титрования (точечная запись)
раствором NaOH смеси хлористоводородной
кислоты, гидрохлорида анилина,
«-нитрофенола и гидрохлорида триметиламина
§ 5. Титрование сильными кислотами двухкомпонентных
смесей оснований и солей слабых кислот
Анализ смесей гидроксида натрия с натриевыми
солями слабых одноосновных кислот (уксусной, у-динитро-
фенола или борной). Кондуктометрический метод
позволяет проводить анализы смесей сильных оснований с
солями слабых кислот, имеющих р/Сь в интервале 4—9.
Поэтому возможно дифференцированное кондуктометри-
ческое титрование смесей гидроксида натрия с ацетатом
натрия (рла=4,75) растворами у-динитрофенолята
натрия (р/(а = 5,50) или метабората натрия (р/Са=9,12). Два
резких излома на кривых титрования наблюдаются при
титровании более концентрированных растворов (0,1—
0,05 н.).
Методика анализа. Рассчитанное количество
анализируемой смеси переносят в мерную колбу и растворяют в
воде или, если объектом исследования служит раствор,
проводят соответствующее разбавление. Аликвотную
часть раствора титруют 1,0 н. раствором НС1. Кривые
176
После второго излома
титрования характеризуются двумя изломами. Во всех
случаях сначала нейтрализуется NaOH и
электропроводность раствора при этом сильно понижается. После
этого вытесняются слабые кислоты из их солей. При
вытеснении уксусной и борной кислот электропроводность
раствора немного увеличивается, а при вытеснении у-ди
нитрофенола мало изменяется,
кривой электропроводность
сильно увеличивается от
избытка НС1. На рис. 57
приведена кривая титрования
хлористоводородной кислотой смеси
NaOH и CH3COONa.
Соответствующие вычисления
проводят по формулам гл. VIII, § 6.
Анализ смесей гидроксида
натрия с натриевыми солями
двухосновных кислот
(хромовой, малеиновой, адипиновой
или сероводородной). Соли
двухосновных кислот при
титровании их смесей с сильными
основаниями ведут себя
различно. При взаимодействии
хромата и малеината натрия
с сильными кислотами
вытесняются кислые соли, так как хромовая кислота по первой
ступени диссоциирует полностью (р/Са1 = —1,0; рКа2 =
= 6,50), а малеиновая кислота — довольно хорошо
(р/Са1 = 2,86; р/(а2 = 5,70). Поэтому первый излом кондук-
тометрической кривой соответствует окончанию
нейтрализации NaOH, второй — вытеснению кислых солей. Если
в смесь входит адипинат натрия, после нейтрализации
NaOH происходит полное вытеснение адипиновой
кислоты (р/Са1 = 4,42; р/Са2 = 5,28).
Сульфид натрия практически полностью гидролизует-
ся по первой ступени, так как сероводородная кислота
очень слабо диссоциирована по второй ступени (р/Са1 =
= 7,20, рКа2= 14,00). Первый излом на кривой
титрования смеси этой соли с щелочью соответствует
нейтрализации свободной и гидролизованной щелочи. После этого
происходит вытеснение сероводорода из NaHS.
Концентрации титруемых растворов могут изменяться в широких
пределах. Для смесей щелочи с адипинатом натрия сле-
0 1 2 3 4 5
Объем pacmBopa HClt мл
Рис. 57. Кривая кондукто-
метрического титрования
раствором НС1 смеси
гидроксида натрия и ацетата
натрия
7—1595
177
дует брать при титровании более концентрированные
растворы.
Методика анализа. Отвешивают на аналитических
весах нужное количество анализируемой смеси, переносят
в мерную колбу и растворяют в дистиллированной воде.
Если определяют концентрации компонентов в
анализируемом растворе, проводят соответствующее
разбавление исследуемой смеси. Аликвотную часть раствора
помещают в электролитическую ячейку и титруют 1,0 н.
раствором НС1. Кривые титрования имеют два излома. При
титровании до первого излома электропроводность
раствора во всех случаях понижается, так как
нейтрализуется NaOH. Когда в состав смеси входят хромат и малеи-
нат натрия, участок кривой титрования между первым и
вторым изломами соответствует вытеснению кислой соли.
При взаимодействии малеината с НС1
электропроводность раствора незначительно увеличивается, а при
взаимодействии хромата немного уменьшается.
В смесях, содержащих адипинат натрия, после
нейтрализации щелочи полностью вытесняется адипиновая
кислота, что сопровождается небольшим повышением
электропроводности раствора. В смесях щелочи с
сульфидом натрия первый излом кривой указывает на
окончание нейтрализации свободного NaOH и щелочи,
образующейся при гидролизе ЫагЭ до NaHS, а второй излом
показывает окончание реакции вытеснения H2S из NaHS.
На втором участке кривой электропроводность раствора
немного понижается. Избыток НС1 во всех случаях
вызывает сильное увеличение электропроводности раствора.
Нелинейное повышение электропроводности раствора
после второго излома имеет место только в случае
титрования смеси, содержащей малеинат натрия, вследствие
взаимодействия с кислотой кислой соли. Устанавливают
количество миллилитров НС1, вступившей в реакцию с
отдельными компонентами смеси, и проводят расчеты,
как описано в гл. VIII, § 6.
Анализ смесей ацетата натрия с основаниями
различной силы (NaOH, пиперидином, моноэтаноламином или
гидразином). Метод кондуктометрического титрования
может быть использован при анализе смесей ацетатов
различных металлов с основаниями, отличающимися по
силе. Поскольку уксусная кислота характеризуется
величиной р/Са=4,75, в смеси с ацетатами могут входить все
основания, имеющие р/Сь^7. При этом во всех случаях
178
сначала нейтрализуется основание, затем
взаимодействует ацетат, что всегда имеет место, если сумма р/Сь
основания и р/Са кислоты, образующей соль, <;12. Исходя из
этого, возможен, например, анализ смесей ацетата
натрия с гидроокисью натрия, диэтиламином,
моноэтаноламином или гидразином. Кислотно-основные свойства
электролитов, входящих в состав смесей,
характеризуются следующими константами:
Гидроксид натрия
Диэтиламии . . .
Моноэтаноламин .
Гидразин
Р*Ь
0,77
3,07
4,50
5,90
Р*а + Р*Ъ
3,96
7,82
9,25
10,65
Для полученя резких изломов на кондуктометриче-
ских кривых концентрация отдельных компонентов в
смеси должна составлять 0,1—0,05 н.
Методика анализа. Приготавливают исходный
раствор смеси указанной концентрации. Аликвотную часть
раствора титруют 1,0 н. раствором НС1. Кривые
титрования имеют два излома, соответствующие
взаимодействию оснований и соли. Характер кривых титрования
отличается только до первого излома кривой. Если в смесь
входит гидроксид натрия, электропроводность раствора
до первого излома линейно понижается.
Нейтрализация слабых оснований — диэтиламина,
моноэтаноламина и гидразина, наоборот, вызывает
повышение электропроводности раствора. В случае
титрования смесей, содержащих диэтиламин и моноэтаноламин в
начале титрования на кривой наблюдается небольшой
минимум. На участке кривой между первым и вторым
изломами электропроводность раствора немного
увеличивается, что характерно для взаимодействия ацетата с
хлористоводородной кислотой. Избыток титранта
вызывает сильное увеличение электропроводности растгюра.
Расчеты см. гл. VIII, § 6.
§ 6. Титрование сильными кислотами многокомпонентных
смесей оснований и солей слабых кислот
Анализ смеси гидроксида натрия, аммиака и ацетата
натрия. Кондуктометрический метод позволяет
проводить анализ трехкомпонентных смесей, состоящих из
двух оснований и соли слабой кислоты. При этом
последовательно взаимодействуют сильное основание, слабое
7* 179
о ; 2
Объем растВ
основание и соль. Возможен анализ смеси гидроксида
натрия, аммиака и ацетата натрия. После нейтрализации
NaOH взаимодействует аммиак (р/С = 4,75).
Дифференцированное титрование аммиака и ацетата натрия
возможно потому, что сумма рКъ аммиака и рКа уксусной
кислоты равна 9,50. Наиболее резкие изломы кривых
титрования имеют место при концентрациях отдельных
компонентов 0,1—0,05 н.
Методика анализа. Точно
измеренный объем
анализируемого раствора помещают в
мерную колбу и разбавляют
дистиллированной водой. Алик-
вотную часть раствора титруют
1,0 н. раствором НС1. Кривая
титрования имеет три излома
(рис. 58). Сначала
нейтрализуется NaOH, что приводит к
сильному понижению
электроде НС1,м/1 проводности раствора. Затем
взаимодействует аммиак, элек-
Рис. 58. Кривая кондукто- трОПрОВОДНОСТЬ При ЭТОМ ПОВЫ-
метрического титрования шается. Наконец, вытесняется
смеси гидроксида натрия,
аммиака и ацетата натрия уксусная кислота, ЧТО СОПро-
раствором НС1 вождается небольшим
увеличением электропроводности pac-i
твора. При избытке НС1 электропроводность раствора
резко возрастает. Определяют количество миллилитров
НС1, вступившей в реакцию с отдельными компонентами
смеси, и проводят расчеты, как показано в гл. VIII, § 6.
Анализ смеси гидроксида натрия, гексаметилентетра-
мина и метабората натрия. Кондуктометрический метод
позволяет проводить дифференцированное титрование
трехкомпонентных смесей, состоящих из двух оснований
и соли слабой кислоты, при котором соль
взаимодействует между основаниями. После нейтрализации NaOH в
реакцию вступает соль слабой метаборной кислоты
(рЛа = 9,12). После этого нейтрализуется гексаметилен-
тетрамин (р/(ь = 8,87). Такая последовательность
взаимодействия компонентов объясняется тем, что сумма
рКъ гексаметилентетрамина и рКа метаборной кислоты
равна 17,99. Более благоприятные условия создаются
при титровании сравнительно концентрированных
растворов (0,1—0,05 н.).
180
Методика анализа. Приготавливают исходный
раствор путем растворения точно взвешенной навески или
разбавления анализируемого раствора. Отбирают из
мерной колбы аликвотную часть и переносят в
электролитическую ячейку. Титруют 1,0 н. раствором НС1. На
кривой титрования имеется три излома (рис. 59).
Сначала электропроводность раствора понижается, так
как нейтрализуется
NaOH. Затем в
реакцию вступает ме-
таборат натрия, что
приводит к
небольшому
увеличению
электропроводности раствора.
Более сильное
повышение
электропроводности имеет
место при
последующей
нейтрализации
гексаметилентетрамина. После
третьего излома
электропроводность
резко увеличивается от избытка НС1. На основании
результатов титрования проводят расчеты (гл. VIII, § 6).
Анализ смеси диэтиламина, а-пиколина и ж-нитрофе-
нолята натрия. В состав дифференцированно
титрующихся трехкомпонентных смесей могут входить два слабых
основания и соль слабой кислоты. При этом соль
взаимодействует между основаниями. Кондуктометрическое
титрование может быть использовано при анализе смеси
диэтиламина, а-пиколина и ж-нитрофенолята натрия.
При взаимодействии с сильной кислотой сначала
нейтрализуется более сильное основание — диэтиламин (р/Сь =
= 3,07). После этого вытесняется ж-нитрофенол (р/Са =■
= 8,40). Наконец, нейтрализуется а-пиколин (рКъ = 8,03).
Раздельное титрование компонентов объясняется тем,
что значения суммы р/Са и рКъ, характеризующей
последовательно взаимодействующие электролиты, равны
11,47 и 16,43. Концентрации титруемых растворов могут
составлять 0,1—0,02 н.
Методика анализа. Из навески или точно измеренного
объема анализируемой смеси приготавливают раствор
181
§1
II
оз
S
Время, сек
Рис. 59. Кривая хронокондуктометри-
ческого титрования (точечная запись)
смеси гидроксида натрия,
гексаметилентетрамина и метабората натрия
раствором НС1
для титрования. Аликвотную часть раствора титруют
1,0 н. раствором НС1. При титровании
электропроводность раствора все время увеличивается. Однако кривая
титрования имеет три излома, так как нейтрализация
оснований вызывает более сильное повышение
электропроводности раствора, чем нейтрализация соли. При
титровании последовательно реагируют — диэтиламин,
ж-нитрофенолят натрия, а-пиколин.
В начале титрования на кривой имеется неглубокий
минимум. Наиболее сильное повышение электропровод-
^^о
С:
-X»
£
^
>
<ti
<о
тропра
!Г
01
е-
«о
ci
Cl
заниям
tl
V
13
t
^■'■-^
мя, сек
Рис. 60. Кривая хронокондуктометрического
титрования (точечная запись) раствором
НС1 смеси гидроксида натрия,
моноэтаноламина, анилина и 5,5-диэтилбарбитурата
натрия
ности наблюдается при добавлении избытка НС1.
Расчеты см. гл. VIII, § 6.
Анализ смеси гидроксида натрия, моноэтаноламина,
анилина и 5,5-диэтилбарбитурата натрия. Возможно
дифференцированное кондуктометрическое титрование
четырехкомпонентной смеси, состоящей из трех
оснований и соли слабой кислоты. Константы диссоциации,
характеризующие кислотно-основные свойства
электролитов, должны при этом удовлетворять условиям,
изложенным выше. Этим условиям соответствуют степени дис-
соцации гидроксида натрия, моноэтаноламина, анилина
и 5,5-диэтилбарбитуровой кислоты. При титровании
сначала нейтрализуется NaOH, затем моноэтаноламин.
После этого взаимодействует диэтилбарбитурат натрия.
Последовательное взаимодействие моноэтаноламина и
соли возможно, так как сумма р/Сь моноэтаноламина и
р/Са диэтилбарбитуровой кислоты равна 11,93. Послед-
182
Ним Вступает в реакцию анилин. Дифференцированное
титрование диэтилбарбитурата натрия и анилина
протекает потому, что сумма р/Са диэтилбарбитуровой
кислоты и рКъ анилина равна 16,85. Анализ затруднен, если
концентрации компонентов смеси <0,03 н.
Методика анализа. Приготавливают исходный
раствор путем растворения точно взвешенной навески
анализируемой смеси или путем разбавления пробы
анализируемого раствора. Аликвотную часть раствора
титруют 1,0 н. раствором НС1. Кривая титрования
характеризуется четырьмя изломами (рис. 60). Первым вступает в
реакцию гидроксид натрия, электропроводность раствора
при этом понижается. Затем нейтрализуется
моноэтаноламин, что приводит к повышению электропроводности
раствора. При последующем вытеснении
диэтилбарбитуровой кислоты электропроводность раствора
незначительно понижается. Нейтрализация анилина вызывает
увеличение электропроводности раствора. Избыток НС1
резко увеличивает электропроводность раствора.
Определяют число миллилитров НС1, вступивших в реакцию
с каждым компонентом смеси. Расчеты см. гл. VIII, § 6.
Глава XI
ТИТРОВАНИЕ, ОСНОВАННОЕ
НА РЕАКЦИЯХ ОСАЖДЕНИЯ
ТИТРОВАНИЕ АНИОНОВ
§ 1. Титрование бромид-ионов
нитратом серебра
Бромиды образуют малорастворимый осадок с
ионами серебра (ПРа§вг = 4,0- Ю-13). При взаимодействии
бромида натрия с нитратом серебра протекает реакция
Na+ + Br- + Ag+ + NOjf = AgBr + Na+ + NOf
Определения возможны не только в сравнительно
концентрированных растворах, но и в очень разбавленных,
порядка Ю-4 н. и ниже.
Приготовление стандартного раствора нитрата
серебра. 0,1 н. раствор нитрата серебра получают растворением
рассчитанного количества химически чистого AgNOe в
определенном объеме воды. Концентрацию стандартного
раствора нитрата серебра устанавливают по химически
чистому хлориду натрия
методом осаждения,
используя в качестве
индикатора хромат натрия*.
Стандартизацию раствора
AgNC>3 можно провести
кондуктометрически по
раствору химически
чистого КВг.
Методика
определения. Рассчитывают
навеску анализируемой соли,
необходимую для
приготовления определенного
объема 0,01 н. раствора.
Навеску переносят в мерную колбу и растворяют в
дистиллированной воде. Аликвотную часть раствора
помещают в элекролитическую ячейку и титруют 0,1 н. раство-
йВъвм раствврд /4jjN0j, M/7
Рис. 61. Кривая кондуктометри-
ческого титрования бромида
натрия нитратом серебра
А. П. К р е ш к о в. Основы аналитической химии т 2 М
«Химия», 1970. > • • •.
184
ром AgN03. При титровании электропроводность
раствора до точки эквивалентности понижается, так как
подвижность ионов брома (Хвг~=78,1) выше подвижности
заменяющих их нитрат-ионов (Xno3~=71,5). Избыток
нитрата серебра вызывает повышение
электропроводности раствора.
Кривая титрования бромида натрия нитратом
серебра показана на рис. 61. По кондуктометрической кривой
определяют число миллилитров AgN03, вступивших в
реакцию, и рассчитывают концентрацию бромида натрия
в титруемом растворе (см. гл. VIII, § 6).
§ 2. Определение хлорид-ионов в природных водах -
титрованием нитратом ртути
Определение хлорид-ионов в природных водах
основано на их взаимодействии с Hg(N03h
2CI- + Hg2+ -+ HgCl2
Хлорид ртути (II) слабо диссоциирует и сравнительно
мало растворим в воде. Кондуктометрический метод
применим, если количество хлорид-ионов в титруемой
пробе >0,2 мг.
Метод отличается высокой избирательностью.
Определение ионов хлора возможно в присутствии многих
других ионов К+, Na+, NH4+ Mg2+, Са2+, Al3+, Fe3+ N08-,
СОз2-, НСОз-, S042- и Si032-, если их концентрация не
превышает концентрацию ионов хлора более чем в 25
раз.
Приготовление раствора нитрата ртути (II). При
титровании используют 0,1 н. раствор Hg(NOs)2-
Приготовление раствора нитрата ртути (II) и установка его
нормальности детально описаны в литературе*.
Стандартизацию раствора Hg(NOs)2 можно провести
кондуктометрический методом, используя стандартный раствор
химически чистого NaCl.
Методика определения. Точно отмеренный объем
анализируемой воды помещают в электролитическую
ячейку. Содержание хлорид-ионов в пробе должно быть
более 0,2 мг. При анализе растворы с высокой
концентрацией ионов хлора разбавляют до 0,01—0,001 н. К
* См.: А. П. Крешков. Основы аналитической химии, т. 2.
М., «Химия», 1970.
185
раствору добавляют одну каплю метилового оранжевого
и нейтрализуют 1 н. раствором HN03. Прибавляют 2—3
капли раствора HN03 на 25 мл титруемого раствора. В
присутствии S042--hohob количество добавляемой HNO3
увеличивают до 6—7 капель. Проводят титрование
приблизительно 0,1 н. раствором Hg(NC>3)2-
Электропроводность раствора до точки эквивалентности мало
изменяется, так как подвижности ионов хлора Скс\~ =76,4) и
заменяющих их нитрат-ионов (%wof =71,3) близки.
Избыток нитрата ртути (II) вызывает повышение
электропроводности. Расчет см. гл. VIII, § 6.
§ 3. Определение сульфат-ионов
в природных водах
Сульфат бария мало растворим в воде (ПРва504 =
= 1,08-Ю-10), поэтому реакцию осаждения сульфата
бария используют для кондуктометрического определения
сульфат^ионов в природных водах. Содержащиеся в воде
сульфат-ионы осаждают хлоридом бария, добавленным
в избытке. Избыток хлорида бария титруют стандартным
раствором сульфата натрия. С целью ускорения
кристаллизации осадка добавляют сухой растертый BaS04) a
для уменьшения адсорбции одноименных с осадком ионов
вводят метиловый фиолетовый. При титровании
протекает реакция
Ва2+ + 2С1- + 2Na+ + SO^_ = BaS04 + 2Na+ + 2C1-
Приготовление стандартных растворов ВаС12 и
Na2S04. Для приготовления стандартных растворов
используют химически чистые препараты ВаС12-2Н20 и
Na2S04. Концентрация раствора ВаС12 должна быть
приблизительно 0,01 н., a Na2S04—0,05 н. Концентрацию
стандартных растворов устанавливают весовым или
объемным методом*. Для стандартизации растворов может
быть использовано кондуктометрическое титрование
растворов сульфата натрия и хлорида бария с известной
концентрацией.
Методика определения. Отмеривают определенный
объем воды, содержащий 2—10 мг сульфат-ионов, и
переносят в электролитическую ячейку. В раствор добавля-
* См.: А. П. Крешков. Основы аналитической химии, т. 2.
М., «Химия», 1970.
186
Ют 1—2 капли метилового красного и по каплям 0,1 н.
НС1 до нейтральной реакции. Вносят в раствор сухой
растертый BaS04 (—100 мг) и 0,5 мл насыщенного
раствора метилового фиолетового. После перемешивания
добавляют 10 мл 0,01 н. раствора ВаС12 и
дистиллированную воду до 100 мл. Избыток хлорида бария оттитровы-
вают кондуктометрически 0,05 н. раствором Na2S04. При
титровании в растворе ионы бария заменяются ионами
натрия. Подвижность ионов бария (Хва2+ =63,6) выше
подвижности ионов натрия (XNa+ = 50,l). Поэтому
электропроводность раствора до точки эквивалентности
немного понижается. После точки эквивалентности
электропроводность увеличивается от избытка титранта. По кон-
дуктометрической кривой определяют число миллилитров
Na2S04, вступивших в реакцию. Поскольку определение
проводят по остатку, находят количество миллилитров
ВаС12, вступивших в реакцию с сульфат-ионами,
находящимися в анализируемой воде. При расчетах пользуются
формулами гл. VIII, § 6.
§ 4. Титрование феррогексацианид-ионов
нитратом свинца
Нитрат свинца взаимодействует с феррогексацианида-
ми с образованием малорастворимого осадка феррогекса-
цианида свинца по реакции
4K++[Fe(CN)6]4- + 2РЬ2+ + 4NO^ = Pb2[Fe(CN)6] + 4K+ + 4NO^
Феррицианиды не мешают определению, так как не
образуют осадка с нитратом свинца.
Приготовление стандартного раствора нитрата
свинца. Приготавливают 0,1 н. раствор нитрата свинца. При
этом используют химически чистый препарат РЬ(1\Ю3)2-
Концентрацию приготовленного раствора проверяют
одним из описанных в литературе методов. Для этой цели
может быть использован метод комплексонометрического
титрования в аммиачной среде с применением эриохрома
черного Т в качестве индикатора.
Методика определения. Навеску, рассчитанную для
приготовления определенного объема 0,01 н. раствора
феррогексацианида калия, отвешивают на аналитических
весах и растворяют в мерной колбе в дистиллированной
воде. Аликвотную часть раствора переносят в
электролитическую ячейку и проводят кондуктометрическое титро-
187
вание 0,1 н. раствором Pb(N03)2- При титровании до
точки эквивалентности электропроводность раствора
понижается, что объясняется тем, что подвижность.
[Fe(CN6]4--HOHOB (^[Fe(CN)*4- =110,5) выше подвижности
заменяющих их при титровании гЮз--ионов (Xno^" =
= 71,5). После точки эквивалентности
электропроводность растет от избытка титранта. На кривой кондукто-
метрического титрования устанавливают точку
эквивалентности и определяют, сколько миллилитров РЬ(гЮз)г
вступило в реакцию с феррогексацианидом калия. При
расчете используют формулы гл. VIII, § 6.
§ 5. Определение вольфраматов щелочных металлов
Определение основано на образовании
малорастворимого осадка вольфрамата свинца. При титровании воль-
фрамата натрия нитратом свинца протекает реакция
2Na+ + W02- + РЬ2+ + 2NCV = PbW04 + 2Na+ + 2NO^
В качестве
стандартного раствора используют
0,1 н. раствор Pb(N03)2,
приготовление которого
рассмотрено выше.
Методика
определения. Рассчитывают
навеску анализируемого
вольфрамата натрия,
необходимую для приготовления
Шёъем раствора РЬ(Ы03), мл приблизительно 0,01 и.
раствора. Навеску
растворяют в определенном
объеме дистиллированной
воды. Аликвотную часть
анализируемого раствора
пипеткой переносят в сосуд для титрования и титруют
0,1 н. раствором РЬ(г>Юз)2. Подвижность WC>42--hohob
(tavo4- =69,4) немного ниже подвижности гТО3~"-ионов.
Поэтому электропроводность раствора до точки
эквивалентности несколько увеличивается. После точки
эквивалентности наблюдается сильное увеличение
электропроводности от избытка осадителя. Кривая титрования
вольфрамата натрия нитратом свинца показана на рис.
|
Рис. 62. Кривая кондуктомет-
рического титрования
вольфрамата натрия нитратом свинца
188
62.^ Точка эквивалентности на кондуктометричеекой
кривой устанавливается легко, так как характер изменения
электропроводности раствора до и после нее сильно
отличается. Находят число миллилитров РЬ(гЮз)2,
вступивших в реакцию, и проводят расчеты, как показано в
главе VIII, § 6.
ТИТРОВАНИЕ КАТИОНОВ
§ 6. Определение ионов стронция титрованием
сульфатом лития
Сульфат стронция сравнительно мало растворим в
воде (flPsrS04 = 2,8-Ю-7). В основу кондуктометрического
определения солей стронция положена реакция
осаждения сульфата стронция 0,1 н. раствором сульфата лития
Sr2+ + 2ДО^ + 2Li+ + SO\~ = SrS04+2Li + + 2NO!f
Для уменьшения растворимости осадка и адсорбции
ионов титрование проводят в водно-спиртовой среде
(1 : 1). При этом используют соли лития, потому что его
ионы отличаются низкой подвижностью, что позволяет
получить более благоприятную форму
кондуктометричеекой кривой до точки эквивалентности —
электропроводность раствора при выпадении осадка понижается.
Концентрация титруемого раствора должна быть ~ 0,01 н.
При титровании более разбавленных растворов излом
кривой закругляется вследствие растворимости осадка.
Приготовление стандартного раствора сульфата лития.
Стандартный (приблизительно 0,1 н.) раствор
приготавливают из химически чистого Li2S04. Концентрацию
приготовленного раствора устанавливают весовым
(осаждением сульфата бария), комплексонометрическим или
другими методами *.
Методика определения. Исходный 0,02 н. раствор
анализируемой соли стронция готовят путем растворения в
дистиллированной воде навески, отвешенной на
аналитических весах. Концентрацию титруемого раствора
доводят до 0,01 н. Аликвотную часть раствора разбавляют в
два раза этиловым спиртом и титруют кондуктометриче-
ским методом 0,1 н. раствором сульфата лития. Электро-
* А. П. К р е ш к о в. Основы аналитической химии, т. 2. М.,
«Химия», 1970.
189
проводность раствора до точки эквивалентности
понижается, так как подвижность ионов стронция (Xsr2+ = 59,5)
выше подвижности заменяющих их ионов лития (Ки+ —
= 38,7). После точки эквивалентности
электропроводность увеличивается от избытка титранта. На кондукто-
мзтрической кривой графическим методом устанавливают
точку эквивалентности и определяют количество
миллилитров осадителя, вступивших в'реакцию. Расчеты см.
гл. VIII, § 6.
§ 7. Титрование ионов бария хроматом натрия
Ионы бария взаимодействуют с хромат-ионами
с образованием малорастворимого хромата бария
(ПРвасго4=1,6- Ю-10). На этой реакции основано кон-
дуктометрическое определение ионов бария. Вследствие
незначительной растворимости осадка определение
возможно не только в сравнительно концентрированных
растворах, но и в очень разбавленных, порядка 0,001 н.
При титровании нитрата бария хроматом натрия
протекает реакция
Ва2+ + 2NO;f + 2Na+ +.CrO^ = BaCr04 + 2Na+ + 2NO^
Приготовление стандартного раствора хромата
натрия. В качестве стандартного раствора используют 0,1 н.
раствор КтагСг04, приготовленный из химически чистого
препарата. Стандартизацию раствора осуществляют
иодометрическим методом *.
Методика определения. Навеску анализируемой соли
рассчитывают для приготовления 0,01 н. раствора. Из
приготовленного раствора отбирают аликвотную часть и
помещают в электролитическую ячейку. Проводят
кондуктометр ическое титрование 0,1 н. раствором NagCrO,}.
Подвижность ионов бария (^ва2+ = 63,6), переходящих
в осадок, выше подвижности накапливающихся при
титровании ионов натрия (>,Na+=50,l). Поэтому
электропроводность раствора при выделении осадка понижается.
Избыток осадителя вызывает сильное увеличение
электропроводности раствора. По кондуктометрической
кривой определяют расход стандартного раствора хромата
натрия. При расчетах пользуются формулами гл.
VIII, §6.
* А. П. К р е ш к о в. Основы аналитической химии, т. 2. М.,
«Химия», 1970.
190
§ 8. Титрование ионов кальция оксалатом лития
Оксалат кальция мало растворим в воде (ПРсасуэ,—
= 2,6-Ю-9). Взаимодействие ионов кальция с оксалат-
ионами протекает согласно уравнению
Са2+ + 2NOJ- + 2Li+ + С20^_ = СаС204 4- 2Li+ + 2NOJ"
При титровании ионов кальция обычно употребляют
оксалат лития, так как ионы лития отличаются низкой
подвижностью. Если использовать водно-спиртовые
растворители, определение возможно в очень разбавленных
растворах (0,001 н.). Стандартные растворы оксалата
лития берут в десять раз более концентрированными, чем
титруемый раствор.
Приготовление стандартного раствора оксалата лития.
Стандартный 0,1 н. раствор готовят из химичэски
чистого Ы2С2О4. Концентрацию приготовленного раствора
проверяют перманганатометрическим методом *.
Методика определения. Исходный приблизительно
0,02 н. раствор анализируемой соли кальция получают
растворением рассчитанного количества соли в
определенном объеме дистиллированной воды. В
электролитическую ячейку переносят аликвотную часть раствора и
разбавляют в два раза этиловым спиртом. После того
как раствор достигнет комнатной температуры, титруют
0,1 н. раствором оксалата лития. В процессе титрования
до точки эквивалентности электропроводность раствора
понижается; это вызывается тем, что ионы кальция,
характеризующиеся более высокой подвижностью (^са2+ =
= 59,50), заменяются ионами лития, обладающими
более низкой подвижностью (Ки+ =38,7). При добавлении
избытка осадителя электропроводность раствора
увеличивается. По кондуктометрической кривой находят
количество миллилитров оксалата лития, вступивших в
реакцию, и проводят расчеты, как описано в гл. VIII, § 6.
§ 9. Титрование ионов марганца селенитом натрия
Для кондуктометрического определения ионов
марганца используют реакцию их осаждения селенитом натрия
Mn2+ + 2CI- + 2Na+ + SeO2- = MnSe03 + 2Na+ + 2CI-
* А П. Крешков. Основы аналитической химии, т. 2. М.,
«Химия», 1970.
191
Для уменьшения растворимости осадка титрование
проводят в водном растворе, содержащем 20% этилового
спирта. В этом же водно-спиртовом растворителе готовят
стандартный раствор селенита натрия. .
Приготовление стандартного раствора селенита
натрия. Для приготовления 0,1 н. стандартного раствора
используют химически чистый селенит натрия.
Растворителем служит смесь воды с этиловым спиртом (l-r-4).
Концентрацию приготовленного раствора проверяют
известными методами: перманганатометрическим, бромато-
метрическим и др.
Методика определения. Отвешенную на аналитических
весах навеску анализируемого вещества, необходимую
для приготовления приблизительно 0,01 н. раствора,
переносят в мерную колбу и растворяют в смеси воды и
этилового спирта (20% этилового спирта). Аликвотную
часть раствора титруют 0,1 н. раствором КагБеОз.
Подвижности ионов марганца (^мп2+ =53,5) и заменяющих
их при выделении осадка ионов натрия (^Na+ =50,1)
близки. Поэтому электропроводность раствора в
процессе титрования до точки эквивалентности мало
изменяется. Только избыток осадителя вызывает увеличение
электропроводности раствора. Построив кривую кондуктомет-
рического титрования, устанавливают на ней точку
эквивалентности и находят расход осадителя. Проводят
расчеты, как описано в гл. VIII, § 6.
§ 10. Определение ионов неодима
теллуритом натрия
При титровании нитрата неодима теллуритом натрия
ионы неодима образуют малорастворимый в воде
теллур ит Ш2(Те03)з:
2ШЗ+ + 6NO7 + 6Na+ + STeOf- = Nd2(Te03)3 + 6Na+ + 6NO7
Точные результаты получаются в водно-этанольных
растворах. Содержание этилового спирта в растворе при
определении неодима должно составлять 25 вес. %.
Концентрации титруемых растворов следует брать 10-3—Ю-4
н., а концентрацию титранта — порядка 0,01 н.
Приготовление раствора теллурита натрия.
Приготавливают 0,01 н. стандартный раствор Na2Te03 из
химически чистого реактива. Стандартизация раствора теллу-
192
рита натрия может быть проведена при помощи периода-
та калия, перманганата калия и другими методами.
Концентрация титранта может быть определена кондук-
тометрическим титрованием растворов солей неодима с
известной концентрацией.
Методика
определения. Приготавливают в
мерной колбе исходный
приблизительно Ю-3 н.
раствор нитрата неодима.
Раствор готовят из точно
отвешенной на
аналитических весах навески.
Отбирают аликвотную часть
раствора и переносят в
электролитическую
ячейку. Добавляют этиловый
спирт в количестве 25%.
Проводят титрование
0,01 н. раствором теллу-
рата натрия. При
титровании до точки эквивалентности электропроводность
раствора понижается, так как более подвижные ионы
неодима (XNd3+=69,4) заменяются менее подвижными ионами
натрия (^Na+ = 50,1). Избыток титранта вызывает
увеличение электропроводности раствора. Кривая кондукто-
метрического титрования нитрата неодима показана на
рис. 63. По кондуктометрической кривой определяют
число миллилитров КагТеОз, вступивших в реакцию, и
определяют содержание неодима, пользуясь формулами
гл. VIII, § 6.
ОЬъем раствора Naz Te03, мл
Рис. 63. Кривая кондуктометри-
ческого титрования нитрата
неодима теллуритом натрия
Глава XII
ТИТРОВАНИЕ, ОСНОВАННОЕ
НА РЕАКЦИЯХ КОМПЛЕКСООБРАЗОВАКИЯ
§ 1. Титрование солей металлов комплексоном III
Концентрации солей многих металлов в растворах
могут быть установлены путем кондуктометрического
титрования комплексоном III. В зависимости от константы
нестойкости образующегося комплекса титрование
проводят в присутствии буферных растворов или без них.
Приготовление стандартного раствора
комплексна III. Для приготовления стандартного раствора
комплексна III применяют двунатриевую соль этилендиамин-
тетрауксусной кислоты. Эта соль кристаллизуется с
двумя молекулами воды и имеет состав ЫагСюНиОвКг^НгО.
Продажный препарат предварительно высушивают при
80° С. Для получения 1 л 0,1 н. раствора требуется взять
18,6126 г этой соли. Концентрацию приготовленного
раствора устанавливают по хлориду цинка или по фиксана-
лу соли магния.
Приготовление 0,1 н. раствора хлорида цинка.
Навеску металлического цинка 3,269 г, содержащего не менее
99,99% цинка, растворяют в мерной колбе емкостью 1 л
в 50 мл НС1 (1:1). После того как раствор охладится,
его разбавляют дистиллированной водой до метки. При
этом получается точно 0,1 н. раствор хлорида цинка.
Приготовление аммиачного буферного раствора
(рН=8). 54 г NH4CI растворяют в 200 мл
дистиллированной воды и смешивают с 350 мл концентрированного
аммиака. Полученный раствор разбавляют до 1 л.
Стандартизация раствора комплексона III. 25 мл
раствора соли цинка помещают в коническую колбу
емкостью 250 мл и нейтрализуют раствором аммиака до
пожелтения раствора в присутствии метилового красного.
Добавляют 2 мл аммиачной буферной смеси.
Полученный раствор разбавляют дистиллированной водой до
100 мл. Вносят индикатор эриохром черный Т и титруют
комплексоном III до синего окрашивания.
Приготовление буферных растворов для
кондуктометрического титрования. Ацетатные буферные
растворы (рН=4 и 5). 50 мл 1,0 н. раствора
194
CH3COOLi смешивают соответственно с 40 или 15 мл
1,0 н. раствора НС1 и разбавляют дистиллированной
водой до 250 мл.
Борно-щелочные буферные растворы
(рН = 8 и 10). 50 мл 0,1 М раствора Н3В03 смешивают
соответственно с 3,97 или 43,9 мл 0,1 н. раствора NaOH
и разбавляют дистиллированной водой до 1000 мл.
Аммиачный буферный раствор (рН = 10).
Смешивают 1,0 н. растворы хлорида аммония и аммиака
в объемных отношениях 1 : 5.
Определение солей железа (III). Катионы железа
(III) образуют весьма устойчивые комплексы с
комплексоном III. При взаимодействии этих соединений
протекает реакция
Fe3+ + H2Y2-^tFeY- + 2H+
Образующийся комплексонат характеризуется
величиной p/CpeY"" =25,16. Титрование катионов железа (III)
можно проводить в водных растворах солей, а также в
кислых растворах. Если концентрация титруемой соли
составляет 0,01—0,02 н., определения возможны даже при
рН = 1. При этом следует учитывать, что концентрация
ионов водорода увеличивается при титровании и в точке
эквивалентности в два раза выше концентрации
титруемой соли, независимо от того, присутствугт в растворе
кислота или нет. При титровании солей железа (III) в
кислых растворах кислотность раствора следует брать по
возможности более низкой, так как при этих условиях
угол излома кривой становится более острым.
Определению не мешают катионы следующих металлов: Zn, Co,
Cd, Al, Fe (II), Мп, La, Mg, Ca, Sr и Ва. При титровании
первыми в реакцию вступают катионы железа (III). Эта
реакция протекает количественно; в точке
эквивалентности раствор имеет кислую реакцию. При этих условиях
указанные катионы практически с комплексоном III не
взаимодействуют.
Методика определения. В мерной колбе
приготавливают приблизительно 0,01 н. раствор анализируемой
смеси. При этом используют навеску, взятую на
аналитических весах. Переносят пипеткой 50 мл приготовленного
раствора в электролитическую ячейку. Проводят кондук-
тометрическое титрование 0,01 н. раствором
комплексона III. При титровании до точки эквивалентности
электропроводность раствора сильно повышается, так как в
195
результате реакции в растворе накапливаются ёы-
сокоподвижные ионы водорода, а также катионы
натрия. После точки эквивалентности электропроводность
раствора сильно понижается, потому что ионы водорода
взаимодействуют с Н2У2--ионами, поступающими
в.раствор при избытке титранта. По кондуктометрической
кривой определяют количество миллилитров комплексона III,
вступившего в реакцию, и проводят расчеты, как
показано в гл. VIII, § 6.
Определение ионов цинка. Определение ионов цинка
проводят в буферных растворах, имеющих рН = 4. При
титровании солей цинка комплексоном III протекает
реакция
Zn2+ + H2Y2- ^tZnY2- + 2Н+
Выделяющиеся в результате реакции ионы водорода
взаимодействуют с буферной смесью. Если буферная
смесь состоит из ацетата лития и хлористоводородной
кислоты, в реакцию вступают ацетат-ионы и
концентрация этих ионов в растворе уменьшается. Когда раствор
содержит достаточное количество буферной смеси,
величина рН раствора поддерживается постоянной.
Концентрацию титруемой соли берут 0,01—0,02 н. Определению
не мешают катионы Са, Mg, Sr и В а, которые в растворе
с рН = 4 практически с комплексоном III не
взаимодействуют.
Методика определения. Растворяют в мерной колбе,
отвешенной на аналитических весах, навеску соли цинка
и приготавливают приблизительно 0,01 н. раствор. 50 мл
этого раствора переносят в электролитическую ячейку и
добавляют 20 мл ацетатного буферного раствора (рН =
= 4). Проводят кондуктометрическое титрование 0,1 н.
раствором комплексона III. При титровании до точки
эквивалентности электропроводность раствора немного
увеличивается. Более сильное повышение
электропроводности происходит при добавлении избытка титранта. На
кондуктометрической кривой устанавливают точку
эквивалентности и определяют число миллилитров
комплексона III, вступивших в реакцию. Расчеты см. гл. VIII, § 6.
Определение жесткости воды. Кондуктометрический
метод позволяет проводить анализ природных вод.
Возможно определение общей, карбонатной и магниевой
жесткости воды. Преимущество метода кондуктометриче-
ского титрования заключается в том, что возможен
анализ мутных и окрашенных вод.
196
Определение общей жесткости воды.
Общую жесткость воды выражают числом
миллиграмм-эквивалентов кальция и магния в одном литре.
Определение основано на образовании комплексоиатов кальция
(p#caY2~= 10,96) и магния (p/CMgY2-=8,69) при
взаимодействии с комплексоном III. При титровании протекают
реакции
Са2+ + H2Y2-r£CaY2- + 2H+
Mg2+ + H2Y2-^tMgY2- + 2Н+
Образующиеся при реакции ионы водорода
связываются буферной смесью. Титрование проводят в
аммиачном буферном растворе при рН = 10. Определению не
мешают ионы алюминия, если их концентрация не
превышает 20 мг/л, и ионы железа (III) при их концентрациях
не более 100 мг/л. В присутствии железа возможно
образование гидроокиси, которая не мешает определению.
Приготовление раствора комплексона
III. При титровании используют 0,05 н. раствор
комплексона III. Приготовление и стандартизация раствора
комплексона III в присутствии индикаторов описаны выше.
Концентрацию приготовленного раствора
комплексона III можно также установить кондуктометрический
титрованием 0,01 н. раствора, состоящего из смеси
растворов хлорида кальция и сульфата магния, взятых в
объемных отношениях 3:1. Для приготойления такого
раствора в мерную колбу емкостью 100 мл помещают
при помощи пипетки 7,5 мл 0,1 н. раствора хлорида
кальция и 2,5 мл 0,1 н. раствора сульфата магния и раствор
доводят до метки дистиллированной водой (при
определении общей нормальности смеси учитывают
поправочные коэффициенты нормальности отдельных солей).
0,1 н. раствор хлорида кальция готовят растворением
высушенного при 110° химически чистого карбоната
кальция. 0,1 н. раствор сульфата магния может быть
приготовлен из фиксанала соли. Поскольку в продажу
поступают 0,01 и 0,05 н. фиксаналы сульфата магния, объэм
дистиллированной воды при растворении соли
соответственно уменьшают.
Для стандартизации раствора проводят
кондуктометрическое титрование приготовленной смеси по методике
определения жесткости воды. Для титрования берут
5 мл смеси. По кондуктометрической кривой определяют
число миллилитров комплексона III, участвующего в ре-
197
акции. Нормальность раствора комплексона III
определяют, пользуясь формулами гл. VIII, § 6.
Методика определения. В сосуд для титрования
помещают при помощи пипетки от 2 до 10'мл исследуемой
воды (в зависимости от ее жесткости). Затем добавляют
0,5 мл аммиачного буферного раствора и
дистиллированную воду до объема 50 мл. Аммиачный буферный раствор
приливают после добавления некоторого количества
воды, так как иначе в водах, богатых солями магния, может
выпасть осадок его гидроокиси, что увеличивает
продолжительность титрования. Полученный раствор титруют
комплексоном III. При титровании до точки
эквивалентности электропроводность раствора немного
увеличивается, а после нее возрастает более сильно от избытка тит-
ранта. Некоторое повышение электропроводности до
точки эквивалентности объясняется тем, что в раствор
вводятся ионы натрия и увеличивается концентрация ионов
аммония в результате взаимодействия буферной смеси с
выделяющимися при реакции ионами водорода. По
кондуктометр ической кривой устанавливают количество
миллилитров раствора комплексона III, вступившего в
реакцию. Вычисление жесткости воды проводят по формуле
,y_o,o5vy<:iooo_5ovv<: ,б5^
^Н,0 ^Н,0
где Ж — жесткость воды, мг-экв/л; VT — объем
израсходованного раствора комплексона III, мл; К —
поправочный коэффициент 0,05 н. раствора комплексона III;
I^h,o —объем взятой для исследования воды, мл.
Определение магниевой ж е с тк о с т и.
Титрование ионов магния комплексоном III проводят после
предварительного осаждения ионов кальция оксалатом
натрия. Образующаяся при этом взвесь оксалата
кальция не мешает определению. Если в воде присутствуют
ионы алюминия и железа, в присутствии оксалата они
образуют комплексы, но при действии аммиачной
буферной смеси возможно выделение их гидроокисей, которые
также не мешают определению.
Методика определения. В стакан отмеривают 5—
25 мл воды, добавляют одну каплю метилового
оранжевого, а затем 0,1 н. раствор НС1 до появления розовой
окраски воды. Раствор кипятят и выпаривают до 5—
10 мл. После охлаждения его до 80° прибавляют 1 каплю
насыщенного раствора оксалата натрия и по каплям
198
0,5 мл аммиачной буферной смеси с рН=10.
Охлажденный раствор вместе с взвесью оксалата кальция переносят
в электролитическую ячейку, разбавляют
дистиллированной водой до 50 мл и титруют 0,05 н. раствором
комплексона III. Характер кривой титрования аналогичен
получающейся при определении общей жесткости воды. По
кривой титрования находят число миллилитров
комплексона III, вступившего в реакцию, и определяют
содержание магния (в мг-экв/л), пользуясь формулой (65).
Определение кальциевой жесткости.
Кальциевую жесткость находят по разности между
общей жесткостью и магниевой жесткостью.
§ 2. Титрование смесей солей металлов
комплексоном III
Анализ смесей солей железа (III) и железа (II).
Комплексонаты железа (III) и железа (II) сильно
отличаются по устойчивости (p/(peY_ = 25,16; pA'peY2""^ 14,33).
Поэтому возможно последовательное титрование солей
сначала железа (III), а затем железа (II), причем
протекают реакции:
Fe3+ + H2Y2-^tFeY- + 2H+
Fe2+ + H2Y2-^tFeY2- + 2H+
Объектами анализа могут быть смеси водных
растворов солей или слабо кислые растворы. Сначала без
добавления буферного раствора проводят титрование
комплексоном III железа (III). При титровании Ре3+-ионов
электропроводность раствора до точки эквивалентности
увеличивается, так как при реакции выделяются
высокоподвижные ионы водорода и вводятся ионы натрия.
Титруемые растворы берут в 0,01—0,02 н. концентрациях.
Величина рН раствора в точке эквивалентности,
соответствующей окончанию титрования железа (III), при этом
может быть менее 2. В этих условиях катионы железа
(II) с комплексоном III не взаимодействуют. После
достижения первой точки эквивалентности добавляют
несколько порций титранта и заканчивают титрование.
Первая точка эквивалентности легко устанавливается, так
как повышение электропроводности сменяется
понижением. После этого добавляют буферный раствор с рН = 5
и продолжают титрование. В реакцию с железом (II)
вступает избыток комплексона III, добавленного при
первом титровании, а также поступающий при втором. Таким
199
образом, дотитровывают железо (II). Титрование
заканчивают при сильном увеличении электропроводности от
избытка титранта.
Методика определения. Взвешивают на
аналитических весах навеску анализируемой смеси, необходимую
для получения определенного объема раствора с
концентрацией компонентов, равной приблизительно 0,01—0,02 н.,
50 мл полученного раствора переносят в
электролитическую ячейку. Проводят кондуктометрическое титрование
железа (III) 0,1 н. раствором комплексона III.
Электропроводность раствора до первой точки эквивалентности
сильно увеличивается, а после нее сильно понижается,
так как ионы водорода взаимодействуют с H2Y2~ = ионами,
поступающими в раствор при избытке титранта.
Когда достигнуто заметное понижение
электропроводности, заканчивают первое титрование и в раствор
добавляют 20 мл ацетатной буферной смеси. После этого
продолжают титрование. Электропроводность раствора до
второй точки эквивалентности мало изменяется. Хотя
концентрация ацетат-ионов в растворе при титровании
уменьшается, а катионы Fe2+ переходят в менее
подвижные комплексные ионы, в растворе накапливаются ионы
натрия, что не может вызвать сильного изменения
электропроводности. После второй точки эквивалентности
электропроводность раствора опять сильно увеличивается
от избытка титранта.
Кривая титрования смеси Fe(N03)3 и FeS04
приведена на рис. 28. Титрованию Ре(ЫОз)з соответствует
участок кривой до первого излома, а титрованию FeSC>4 —
участок кривой, соответствующий избытку комплексона
III при первом титровании, и участок кривой до излома
при втором. Расчеты см. гл. VIII, § 6.
Анализ смеси солей свинца и магния. Комплексонат
свинца значительно устойчивее комплексоната магния
(р/(рьу2_ =18,04; p/CMgY2" = 8,69). Поэтому возможно
дифференцированное титрование этих солей в смесях
комплексоном III. Определения основаны на реакциях:
РЬ2+ + H2Y2- ^tPbY2- + 2H+
Mg2+ + H2Y2-^t!VlgY2- + 2H+
Для титрования берут водные 0,01—0,005 М растворы
.солей. Титрование катионов свинца проводят без
добавления буферного раствора, а титрование катионов магния
проводят в буферном растворе (рН = 8). Сначала титру-
200
ют соль свинца в растворе смеси. Когда повышение
электропроводности раствора сменится ее понижением,
заканчивают первое титрование. Затем добавляют в
титруемый раствор буферную смесь (рН = 8) и дотитровывают
соль магния.
Методика определения. Растворяют отвешенную на
аналитических весах навеску анализируемой смеси,
приготавливают исходный раствор с концентрацией
компонентов порядка 0,01—0,02 н. В электролитическую
ячейку переносят 50 мл приготовленного раствора и проводят
кондуктометрическое титрование 0,1 н. раствором
комплексона III. Сначала в реакцию вступают ионы цинка.
Электропроводность раствора до первой точки
эквивалентности сильно увеличивается за счет накопления
высокоподвижных водородных ионов и ионов натрия. После
точки эквивалентности электропроводность раствора
падает, так как снижается концентрация водородных ионов,
взаимодействующих с Н2У2~-ионами. Когда
электропроводность раствора начинает понижаться, добавляют
несколько порций титранта и заканчивают титрование.
После этого добавляют 20 мл борно-щелочного буферного
раствора (рН = 8) и проводят второе титрование. При
добавлении буферной смеси избыток комплексона III,
находящийся в растворе в результате первого титрования,
вступает в реакцию с катионами магния. Поэтому при
втором титровании дотитровывают оставшиеся в растворе
ионы магния. Электропроводность раствора при
титровании до второй точки эквивалентности мало изменяется,
что объясняется тем, что в растворе уменьшается
концентрация борат-ионов, но увеличивается концентрация
ионов натрия. При избытке титранта электропроводность
сильно увеличивается. Количество миллилитров
комплексона III, вступившего в реакцию с РЬ(гЮз)2,
определяют по участку кривой до первого излома. Число
миллилитров комплексона III, взаимодействующего с MgSO,},
находят по участку кривой, соответствующему избытку
титранта при первом титровании и участку кривой до
излома при втором титровании. Расчеты проводят, как
описано в гл. VIII, § 6.
Анализ смеси солей железа (III) и кобальта (II).
Комплексонат железа (III) —один из самых устойчивых
комплексонатов (рК?е\~ =25,16). Комплексонат
кобальта также отличается высокой устойчивостью (p/CcoY2~ =
= 16,31) и мешает титрованию железа. Дифференцирован-
201
ное определение компонентов смеси возможно только й
случае, если титрование железа (III) проводить в
растворе, содержащем такое количество кислоты, при котором
катионы кобальта не взаимодействуют с комплексо-
ном III. При титровании железа (III) добавляют
некоторый избыток титранта. Затем для понижения
концентрации ионов водорода в титруемый раствор добавляют
ацетат лития. Находящийся в растворе комплексон III при
этих условиях взаимодействует с катионами кобальта.
Проводят второе титрование, при котором оставшиеся в
растворе катионы кобальта дотитровываются комплексо-
ном III. При указанных условиях количественно
протекают реакции
Fe3+ + H2Y2- = FeY- + 2Н+
С02+ + H2Y2- = CoY2- + 2H+
Методика анализа. Приготавливают исходный раствор
смеси; концентрация солей должна соответствовать
приблизительно 0,01 н. раствору. Для этого отвешивают на
аналитических весах нужное количество смеси. В
электролитическую ячейку переносят 25 мл приготовленного
раствора и добавляют 25 мл 0,1 н. раствора НС1.
Титрование ионов железа (III) проводят комплексоном III.
Когда повышение электропроводности раствора сменится
понижением, заканчивают первое титрование. Затем в
раствор добавляют 10 мл 1,0 н. раствора CH3COOLi и
проводят титрование ионов кобальта. При титровании
ионов железа (III) комплексоном III в присутствии
кислоты электропроводность раствора до точки
эквивалентности повышается менее сильно, чем при титровании
раствора, не содержащего кислоту. Также менее резко
выражено понижение электропроводности раствора после
первой точки эквивалентности. Это объясняется тем, что
раствор с начала титрования имеет высокую
электропроводность и повышение электропроводности от
выделяющихся при реакции ионов водорода менее сильно
сказывается. С другой стороны, на фоне
высокоэлектропроводного раствора менее сильно сказывается понижение
концентрации ионов водорода в связи с их
взаимодействием с Н2У2~-ионами после точки эквивалентности. При
втором титровании до точки эквивалентности,
соответствующей окончанию титрования катионов кобальта,
электропроводность раствора немного понижается, а после нее
немного увеличивается. Подобно рассмотренному выше,
202
участок кривой до излома при первом титр
ветствует взаимодействию нитрата железа
миллилитров комплексона III, вступившего
нитратом кобальта, может быть найдено по
вой, соответствующему избытку титранта
титровании и участку кривой до излома
титровании. Концентрацию отдельных "солей
ют, как показано в гл. VIII, § 6.
ювании соот-
(III). Число
в реакцию с
участку кри-
при первом
при втором
рассчитыва-
§ 3. Определение фторид-ионов
Определение фторидов основано на их взаимодействии
с хлоридом алюминия по реакции
6Na+ + 6F- + AI3+ + 3Cl-->Na3[AIF6] + 3Na+ + 3CI-
Образующийся комплекс [A1F6]3_ отличается высокой
устойчивостью (р/([А1Р.]з- =19,84). Если в титруемый
раствор добавить при этом хлорид натрия, то Na3[AlF6]
выпадает в осадок. В
случае выпадения осадка
получается более благоприятная
форма кондуктометрической
кривой, так как
электропроводность раствора до точки
эквивалентности довольно
сильно понижается.
Определению не мешают С!--,
N03-- и S042_-HOHbi.
Возможно определение
фторидов в очень разбавленных
растворах. Титрование очень
разбавленных растворов
проводят в присутствии 30—50% спирта.
Приготовление стандартного раствора А1С13.
Приготавливают 0,05 н. раствор хлорида алюминия (3aici„ =
=М/6). Концентрацию раствора устанавливают весовым
или объемным методом. Комплексонометрическии метод
определения алюминия детально описан *.
Методика определения. На аналитических весах
отвешивают такое количество анализируемого фторида,
которое требуется для приготовления приблизительно 0,01 н.
раствора. В электролитическую ячейку помещают 25 мл
05ъем раствора AI Clj, м
Рис. 64. Кривая кондуктометри-
ческого титрования фторида
натрия хлоридом алюминия в
присутствии хлорида натрия
* А.
«Химия»
П. Крешков.
1970,
Основы аналитической химии, т. 2. М.,
203
приготовленного раствора, добавляют 5 мл 0,1 н.
раствора NaCl и 15 мл этилового спирта. Раствор
разбавляют дистиллированной водой до 50 мл. Проводят кондук-
тометрическое титрование 0,05 н. раствором А1С1з.
Электропроводность раствора до точки эквивалентности
понижается. Это объясняется тем, что при
взаимодействии шесть ионов фтора заменяются тремя ионами хлора.
Выпадение осадка Na3AlF6 также способствует
понижению электропроводности раствора. При добавлении
избытка титранта электропроводность увеличивается.
Кривая титрования NaF раствором А1С1з показана на рис. 64.
По кондуктометрической кривой определяют число
миллилитров А1С1з, вступивших в реакцию, и проводят
расчеты, как описано в гл. VIII, § 6.
§ 4. Определение ионов калия титрованием
тетрафенилборатом натрия
Ионы калия образуют малорастворимый в воде
осадок комплексной соли с тетрафенилборатом К[(СбН5)4В].
Произведение растворимости тетрафенилбората калия
равно 2,25- Ю-8. При титровании КС1 протекает реакция
К+ + CI- + Na+ + [(С6Н5)4В]~ = К[(С6Н5)4В] + Na+ + CI-
Титруемый раствор берут в 0,005 н. концентрации. В
качестве титранта используют 0,1 н. раствор Ыа[(СбН5)4В].
Определение калия возможно при рН = 5-М0. Поэтому
проводят титрование в присутствии уксусной кислоты
(КС1: СН3СООН=1 : 5). Минеральные кислоты мешают
определению. С другой стороны, титрование в щелочных
растворах приводит к положительным результатам.
Крупное преимущество данного метода заключается в
возможности определения калия в присутствии ионов
натрия, магния и кальция. При этом отношение
эквивалентов КС1: посторонняя соль может достигать 1 : 10. При
определении калия допустимо также присутствие
железа (III). Однако отношение эквивалентов КС1: FeCl3
может изменяться лишь в пределах 1—2.
Ионы аммония мешают определению калия, так как
образуют с реагентом аналогичный осадок. Для
удаления ионов аммония можно нагревать исследуемый
раствор с щелочью. Если отношение эквивалентов NH4C1:
NaOH равняется 1 : 1 и 1 :4, продолжительность нагрева-
204
ния на водяной бане должна составлять соответственно
30 и 15 мин. При этом происходит полное выделение
аммиака. Если рН раствора после удаления ионов аммония
менее 10, можно проводить титрование в щелочном
растворе. При более высоком значении рН раствора перед
титрованием проводят его нейтрализацию уксусной
кислотой.
Приготовление раствора Na[(C6H5)4B]. Около 3,5 г
тетрафенилбората натрия растворяют в 100 мл
дистиллированной воды при размешивании. К мутному
раствору добавляют приблизительно 1 г А1(ОН)3,
перемешивают 5 мин и фильтруют. Прозрачный раствор используют
при титровании. Концентрация раствора не изменяется в
течение нескольких недель. Концентрацию титранта
проверяют путем кондуктометрического титрования
стандартным раствором КС1, приготовленным из фиксанала.
Методика определения. Приготавливают в мерной
колбе исходный приблизительно 0,005 н. раствор КС1. Для
этого отвешивают на аналитических весах рассчитанную
навеску хлорида калия. Если объектом анализа служит
раствор, проводят
соответствующее
разбавление. Аликвотную
часть раствора (50 мл)
переносят в
электролитическую ячейку и
титруют 0,1 н. раствором
Na[(C6H5)4B]. После
добавления каждой
порции выжидают две
минуты. При
титровании до точки
эквивалентности
электропроводность раствора
понижается, так как
подвижность ионов калия (^к+ = 73,5) выше подвижности
заменяющих их ионов натрия (^Na+ = 50,1). После точки
эквивалентности электропроводность раствора
увеличивается в связи с ростом концентрации Na+- и [(СбН5)4В]_-
-ионов. Кривая титрования показана на рис. .65. Если
определение калия проводят в присутствии Na+-, Mg2+,
Са2+ или Ре3+-ионов, понижение электропроводности до
точки эквивалентности и повышение после нее менее
резко выражены, так как эти ионы увеличивают общую элек-
205
Объем раствора Ыа [(С6 Нд^В], М/7
Рис. 65. Кривая
кондуктометрического титрования хлорида натрия
тетрафенилборатом натрия
тропроводность раствора. По кондуктометрической
кривой титрования определяют количество миллилитров
титранта, вступившего в реакцию, и проводят
вычисления, как описано в гл. VIII, § 6.
§ 5. Определение ионов меди и никеля и анализ
их смеси титрованием роданидом калия
Ионы меди и никеля образуют малорастворимые пи-
ридин-роданидные комплексы состава CuCCsHsN^*
•(CNS)2, Ni(C5H5N)2(CNS)2
Cu2+ + 2NO^ + 2C5HSN + 2K+ + 2SCN~ =
= Cu(C6H5N)2(SCN)2 + 2K+ + 2NCV
Ni2+ + 2NO^ + 2C5H5N + 2K+ + 2SCN~ =
= Ni(C5H5N)2(SCN)2 + 2K+ + 2NO^
Титрование ведут стандартным раствором KSCN.
Концентрация пиридина не оказывает влияния на
точность определений. Однако она должна быть выше
концентрации ионов меди и никеля. Содержание меди в
20 мл титруемого раствора может изменяться в пределах
0,3—5,4 мг, а никеля — в пределах 1,2—7,0 мг, что
соответствует примерно 0,002—0,01 н. раствора.
При анализе смеси ионов меди и никеля проводят
маскировку Си2+-ионов. Пиридин-роданидный комплекс
меди растворяется в тиосульфате натрия. Сначала
определяют общее содержание ионов меди и никеля путем
титрования пиридинового раствора роданидом калия. Затем
определяют содержание ионов никеля путем титрования
смеси в присутствии тиосульфата натрия. Его
концентрация должна быть в 10 раз выше, чем концентрация ионов
меди.
Метод отличается хорошей избирательностью: А13+-,
Cr3+-, Fe3+-, Fe2+-, Bi3+-, As3+-, Sn4+-, Pt2+-, Pd2+-, Ru3+-,
Ir3+, OsIV-, Rh3+-HOHbi не мешают определению.
Присутствие ионов марганца допускается в небольших
количествах. Затруднения вызывают Au3+-, Ag+- и РЬ2+-ионы.
Ag+- и РЬ2+-ионы предварительно могут быть удалены
осаждением соответственно в виде хлорида и сульфата.
Приготовление растворов роданида калия, пиридина
и тиосульфата. Приготавливают 0,1 н. раствор роданида
206
калия растворением его навески. При этом могут быть
использованы фиксаналы. Стандартизация раствора
роданида может быть проведена титрованием нитратом
серебра *.
При анализе используются также приблизительно
0,2 М раствор пиридина и 1,0 М раствор тиосульфата
натрия. Эти растворы готовят растворением в воде
соответствующих навесок этих солей.
Методика анализа. При определении концентрации
индивидуальных солей меди и никеля приготавливают
исходные растворы этих солей. Для этого отвешивают на
аналитических весах навески анализируемых образцов,
переносят в мерную колбу и растворяют в
дистиллированной воде. Аликвотную часть раствора переносят в
электролитическую ячейку. На каждые 20 мл титруемого
раствора добавляют 1—2 мл 0,2 М раствора пиридина.
Титрование проводят 0,1 н. раствором KSCN.
Если объектом анализа служит смесь солей меди и
никеля, приготовленный раствор титруют дважды.
Первое титрование позволяет определять общее содержание
солей. При этом титровании на каждые 20 мл исходного
раствора добавляют 2—3 мл 0,2 М раствора пиридина.
При втором титровании кроме пиридина в раствор
вносят 2—3 мл 1,0 М раствора тиосульфата натрия. В этих
условиях титруются только ионы никеля.
Электропроводность раствора до точки
эквивалентности во всех случаях немного увеличивается, так как
подвижности ионов меди (А,си2+=56,6) и никеля (Kw2+ =54)
ниже подвижности заменяющих их в растворе при
титровании ионов калия (А,к+ = 73,5). После точки
эквивалентности электропроводность более сильно увеличивается от
избытка титранта. Количество миллилитров KSCN,
вступивших в реакцию с ионами никеля, находят по
результатам второго титрования. Количество титранта,
вступившего в реакцию с ионами меди, находят по
разности между первым и вторым титрованием. Расчеты
проводят, как указано выше (см. гл. VIII, § 6).
* А. П. Крешков. Основы--аналитической химии, т. 2. М.,
«Химия», 1970.
207
Татьяна Александровна Худякова,
Анатолий Павлович Крешков
Кондуктометрический
метод анализа
Редактор Г. С. Гольденберг. Художник Ю. Д. Федичкин
Художественный редактор Т. М. С к в о р ц о в а. Технический редактор
Т. Д. Гарина. Корректор М. М. Малиновская
Сдано в набор 18/IX—74 г Подп. к печати 23/VI—75 г. Формат 84Х1087э2
Бум. тип. № 2 Объем 6,5 печ. л. 10,92 усл. п. л. 10,25 уч.-изд. л.
Изд. № Хим-505 Тираж U000 экз. Зак. 1595 Цена 36 коп.
План выпуска литературы издательства
«Высшая школа» (вузы и техникумы) на 1975 г. Позиция № 113
Москва, К-51, Неглинная ул., д. 29/14,
Издательство «Высшая школа»
Московская типография № 8 Союзполнграфпрома
при Государственном комитете Совета Министров СССР
по делам издательств, полиграфии и книжной торговли,
Хохловский пер., 7Ц