/
































































































































































































































































































































































































































































Автор: Крешков А.П.
Теги: спектральные методы анализа оптические методы анализа химия аналитическая химия
Год: 1970




Текст
основы
АНАЛИТИЧЕСКОЙ
ХИ М И И
ИЗДАТЕЛЬСТВО -ХИМИЯ-
1970
АПКРЕШКОВ
ИЗДАТЕЛЬСТВО-ХИМИЯ
МОСКВА • 1970
Scan AAW
А-ПКРЕШКОВ
ОСНОВЫ
Б] физико-химические
(инструментальные)
методы анализа
АНАЛИТИЧЕСКОЙ
ХИМИИ
качественный
и количественный
анализ
т
ДОПУЩЕНО МИНИСТЕРСТВОМ ВЫСШЕГО
И СРЕДНЕГО СПЕЦИАЛЬНОГО ОБРАЗОВАНИЯ СССР
В КАЧЕСТВЕ УЧЕБНИКА
ДЛЯ СТУДЕНТОВ ХИМИКО-ТЕХНОЛОГИЧЕСКИХ
СПЕЦИАЛЬНОСТЕЙ ВУЗОВ
УДК 543.4/.5(075 8)
К 79
Л. П. К р е ш к о в, Основы аналитической
химии. Физико-химические (инструментальные) методы
анализа, Изд. «Химия», 1970, стр. 472.
Книга является третьей частью курса «Основы
аналитической химии» и предназначена в качестве
учебника для студентов химических и
химико-технологических специальностей высших учебных заведений.
В книге изложены теоретические основы физико-
химических методов анализа — разнообразных
электрохимических, спектроскопических (оптических), хромато-
графических и радиометрических; описывается основная
аппаратура и техника физико-химического
эксперимента; приводится ряд типовых практических работ по
определению неорганических и органических веществ.
Подробно рассматриваются физико-химические
методы анализа неводных растворов.
В книге содержится 1& таблиц и 154 рисунка.
2-5-5
70-17
СОДЕРЖАНИЕ
Предисловие 15
Введение 17
Глава [.Основы физико-химических (инструментальных) методов анализа 19
§ 1. Особенности физико-химических методов анализа . 19
§ 2. Области применения инструментальных методов анализа 19
§ 3. Анализ веществ высокой чистоты 20
§ 4. Повышение чувствительности и точности методов определения следов
примесей 22
§ 5. Инструментальные методы титрования 23
§ 6. Применение физико-химических методов анализа для определения
индивидуальных соединений .24
§ 7. Применение физико-химических методов для анализа смесей веществ 25
§ 8. Классификация инструментальных количественных методов анализа , 25
§ 9. Электрохимические методы анализа 25
§ 10. Спектральные (оптические) методы анализа 27
§ 11. Хроматографические методы анализа 28
§ 12. Радиометрические методы анализа 29
§ 13. Масс-спектрометрнческие методы анализа 30
§ 14. Физико-химический анализ по Н. С. Курнакову 30
§ 15. Другие методы анализа 31
Глава II. Потенциометрия и потенциометрическое титрование 32
А Теоретические основы метода 32
§ 1. Зависимость величины электродных потенциалов от концентрации
(активности) 32
§ 2. Применение потенциометрического метода анализа 36
§ 3. Потенциометрическое титров ад ие 36
§ 4. Потенциометрическое титрование без тока (i = 0) 39
§ 5. Различные способы нахождения конечной точки потенциометрического
титрования .... 46
§ 6. Некомпенсационный метод потенциометрического титрования .... 49
§ 7. Потендиометр-ическое титрование под током (*=?0) 50
Б. Аппаратура и техника выполнения анализа /.52
§ 8. Измерение, электродвижущей силы .52
§ 9. Установка для поляризации электродов при потенциометрическом
титровании под током 53
§ 10. Некоторые приборы, применяемые в потенциометрическом титровании 54
§ 11. Электроды 57
В. Практические работы 62
Потенциометрическое титрование без тока (i = 0) 62
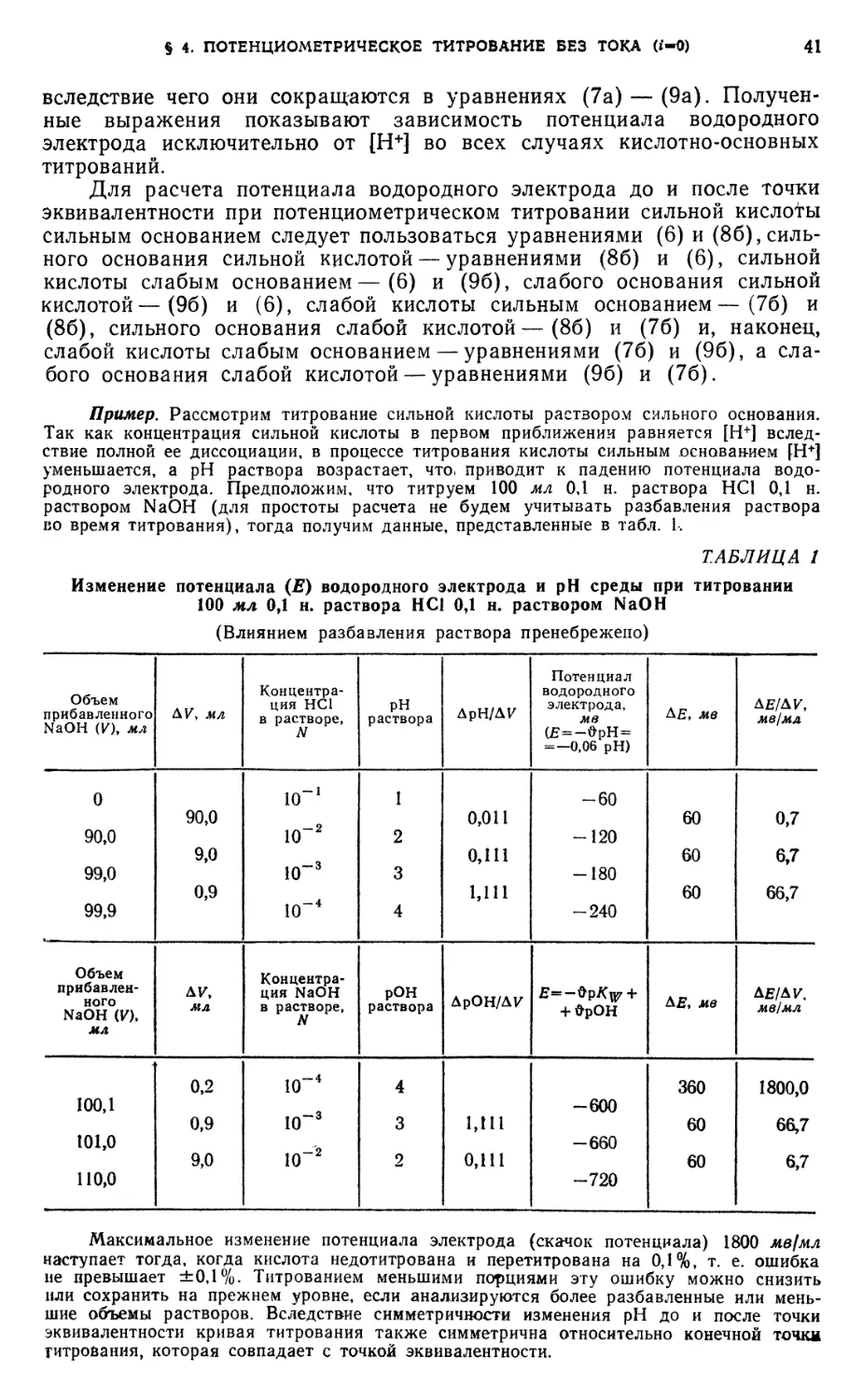
§ 12. Кислотно-основное титрование 62
Определение уксусной кислоты ..... 62
Анализ смеси хлористоводородной и борной кислот 64
§ 13. Титрование по методу окисления — восстановления 65
Определение железа (III) .65
Определение марганца (II) 66
6 СОДЕРЖАНИЕ
Определение иода 67
§ 14. Титрование по методу осаждения ! . 68
Анализ смеси иодида и хлорида 68
§ 15. Титрование по методу комплексообразования 69
Определение железа (III) 69
Потенциометрическое титрование под током (t"4=0) 70
§ 16. Титрование по методу окисления — восстановления ........ 70
Определение сурьмы (III) 70
Глава III. Кондуктометрия и кондуктометрическое титрование 72
A. Теоретические основы метода 72
§ 1. Удельная и эквивалентная электропроводность 72
§ 2. Кондуктометрические методы анализа 76
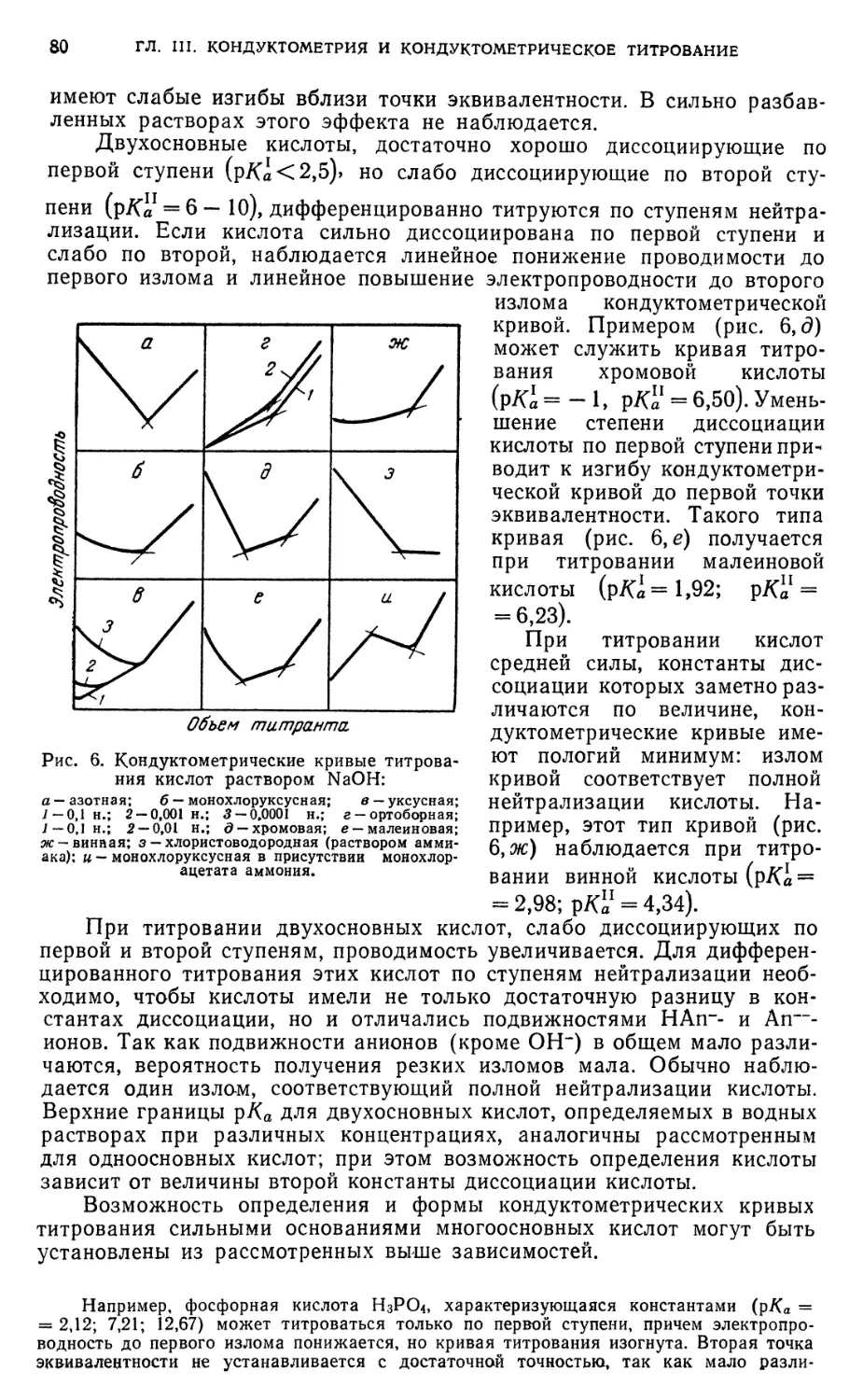
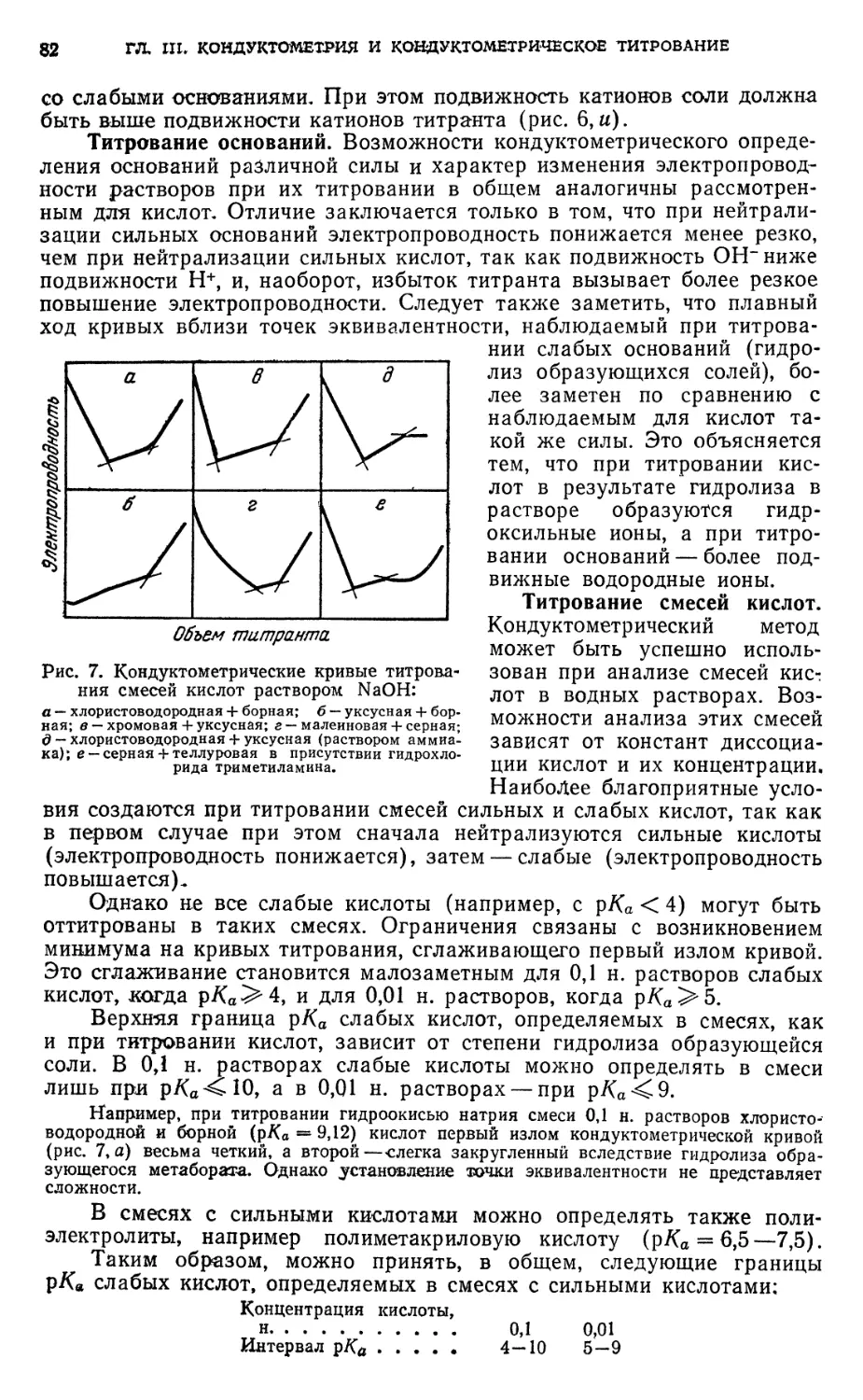
§ 3. Кислотно-основное титрование 79
§ 4. Титрование, основанное на реакциях осаждения 91
§ 5. Титрование, основанное на реакциях окисления — восстановления . . 94
§ 6. Титрование, основанное на реакциях комплексообразования 95
Б. Аппаратура и техника выполнения анализа 96
§ 7. Измерение электропроводности растворов 96
§ 8. Конструкции электролитических ячеек 1Q0
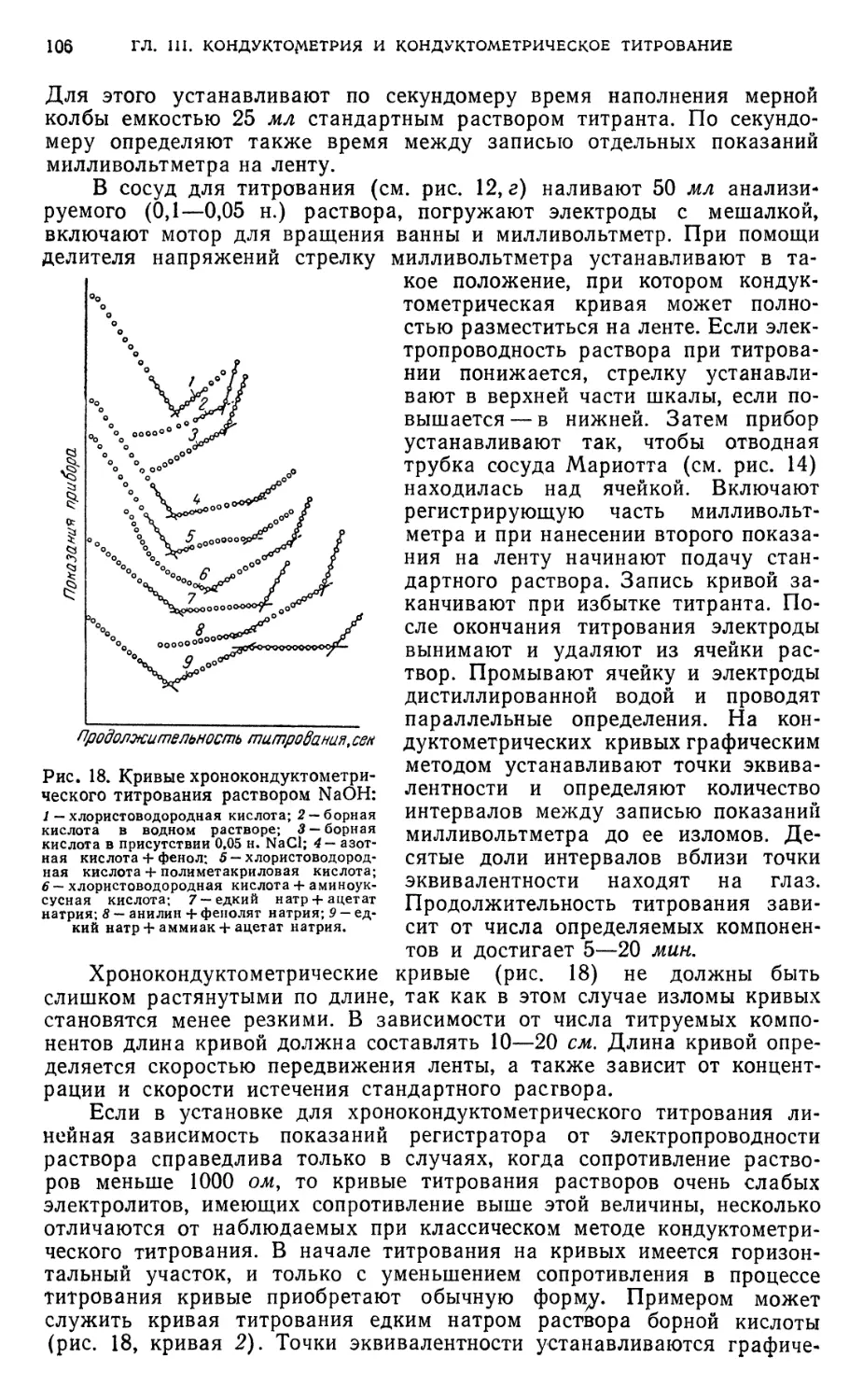
§ 9. Измерения в хроцокондуктометрическом титровании 102
B. Практические работы 103
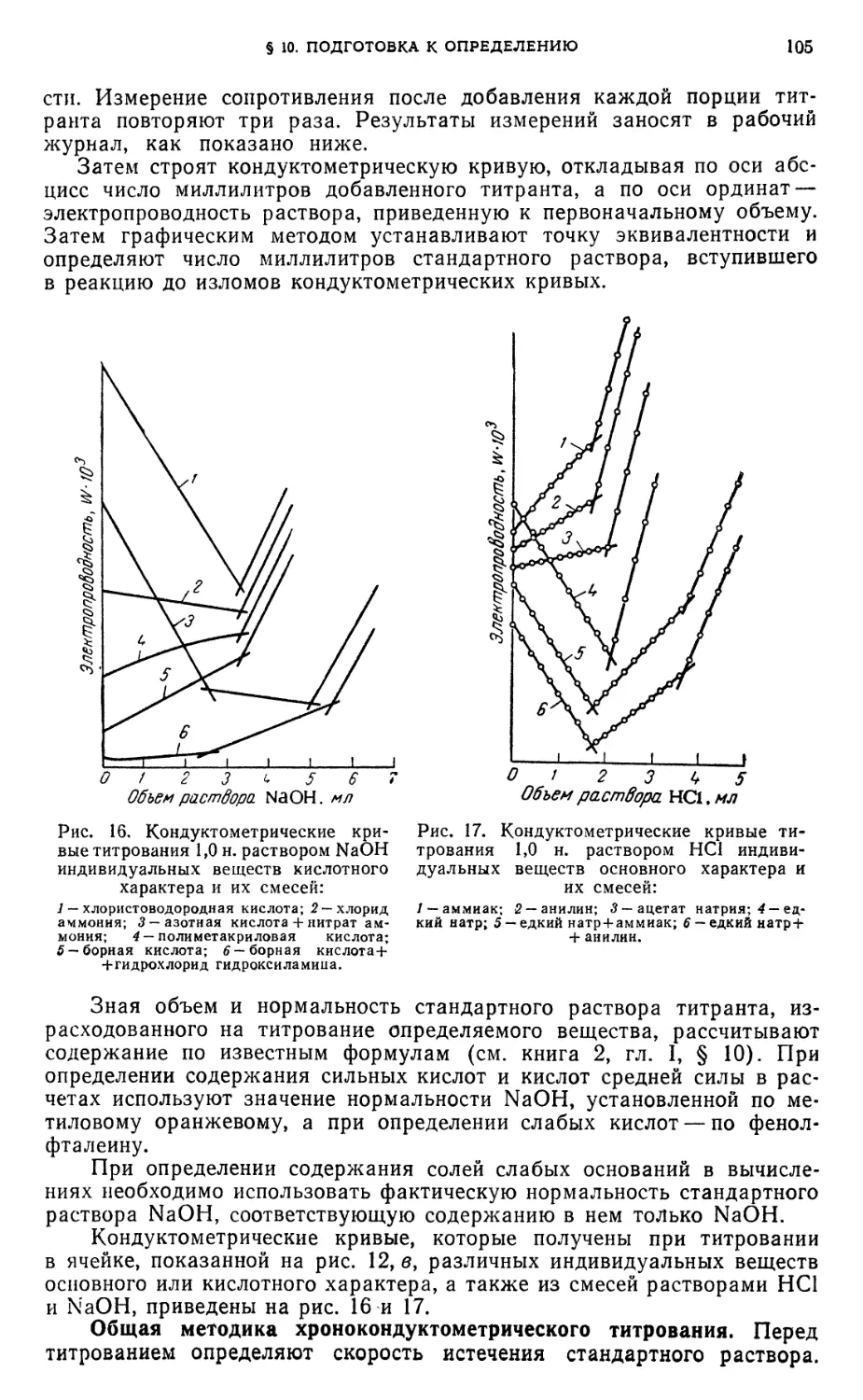
§ 10. Подготовка к определению 103
§11. Определение индивидуальных кислот методом кондуктометрического
титрования 107
Определение хлористоводородной, борной или полиметакриловой кислот 107
§ 12. Определение индивидуальных оснований методом
кондуктометрического титрования 108
Определение едкого натра, аммиака или анилина 108
§ 13. Анализ смесей оснований методом кондуктометрического титрования 108
Анализ смесей: едкий натр и аммиак; едкий натр и анилин 108
§ 14. Определение солей методом кондуктометрического титрования . . . 109
Определение ацетата натрия или хлорида аммония 109
§ 15. Анализ смесей кислот и солей слабых оснований методом
кондуктометрического титрования 109
Анализ смесей: азотная кислота и нитрат аммония; борная кислота и
гидрохлорид гидроксиламина 109
§ 16. Анализ смесей кислот методом хронокондуктометрического титрования 110
Анализ смесей: азотная кислота и фенол; хлористоводородная кислота и
полиадетакриловая кислота; хлористоводородная кислота и аминоуксусная
кислота ПО
§ 17. Анализ смесей оснований и солей слабых кислот методом
хронокондуктометрического титрования 111
Анализ смесей: едкий натр и ацетат натрия; анилин и фенолят натрия;
едкий натр, аммиак и ацетат натрия 111
Глава IV. Высокочастотное титрование 113
А. Теоретические основы 113
§ 1. Общие положения высокочастотного титрования 113
§ 2. Физические основы и погрешности метода высокочастотного
титрования 114
§ 3. Физические основы метода диэлкометрического титрования 115
§ 4. Погрешности метода диэлкометрического титрования 116
Теория измерительных ячеек емкостного типа 116
§ 5. Электрическая эквивалентная схема ячейки и физические процессы,
протекающие в ее объеме 117
§ 6. Импеданс и полная проводимость ячейки 118
§ 7. Характеристические кривые ячейки 119
§ 8. Чувствительность ячейки 121
§ 9. Критерий применимости и постоянная с-ячейки 121
Теория измерительных ячеек индуктивного типа 122
§ 10. Электрическая эквивалентная схема ячейки 122
СОДЕРЖАНИЕ
7
§ 11. Импеданс и характеристические кривые ячейки 123
§ 12. Критерий применимости и постоянная L-ячейки 123
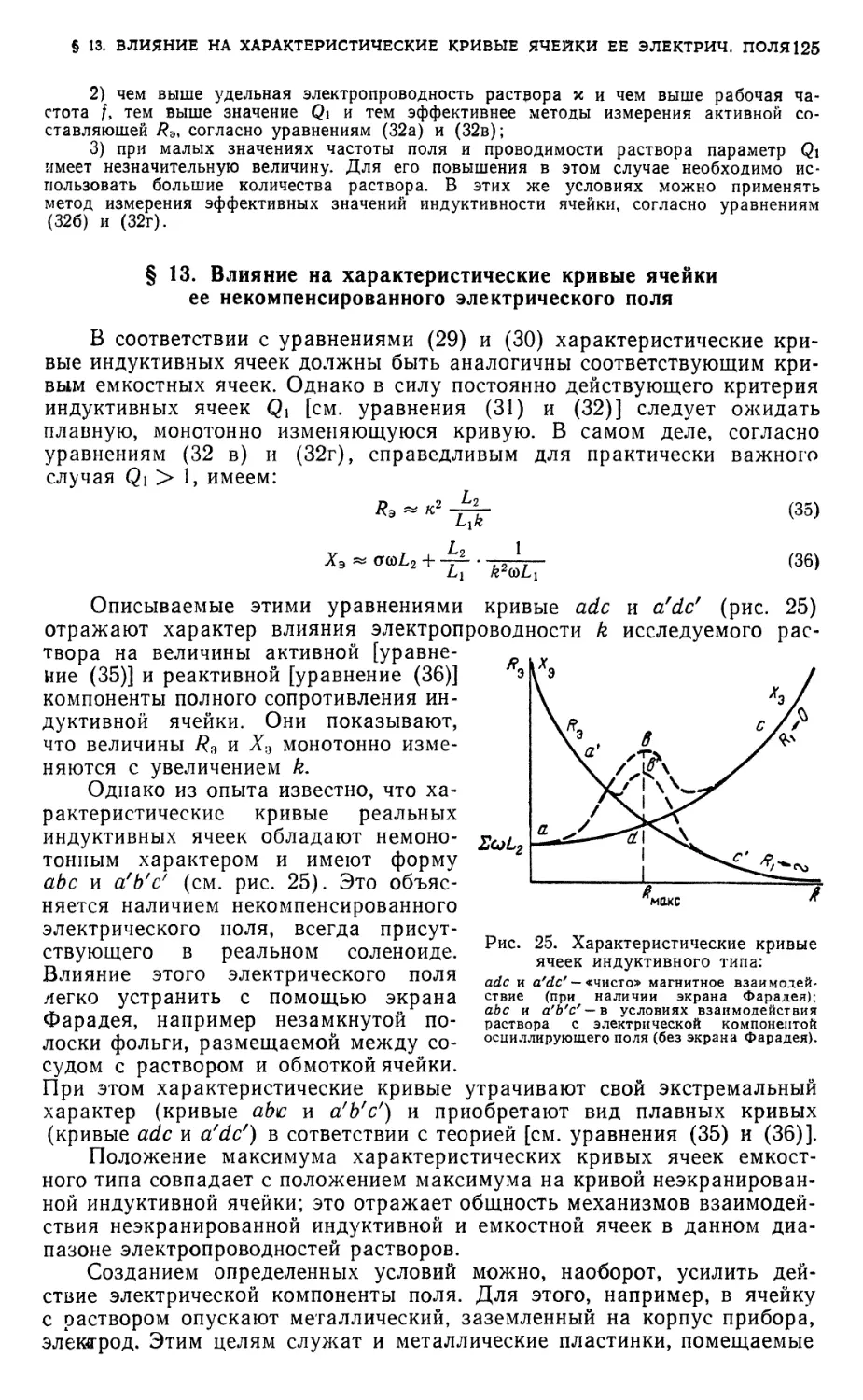
§ 13. Влияние на характеристические кривые ячейки ее некомпенсированного
электрического поля 125
Диаграммы соответствия в- 126
§ 14. Диаграмма соответствия для титрования по активной составляющей
полной проводимости 126
§ 15. Диаграмма соответствия для титрования по реактивной составляющей
полной проводимости 127
§ 16. Выбор типа диаграмм ••¦*• 128
Б. Аппаратура и техника выполнения анализа ¦*•- 128
§ 17. Измерительные ячейки (датчики) высокочастотного метода и области
их применения 128
§ 18. Классификация аппаратуры высокочастотного метода 129
§ 19. Высокочастотные титраторы 131
§ 20. Общие правила работы с приборами для высокочастотного анализа 137
В. Практические работы •¦¦••¦•*¦ 138
§ 21. Высокочастотное титрование стандартных растворов 138
Получение характеристической кривой 138
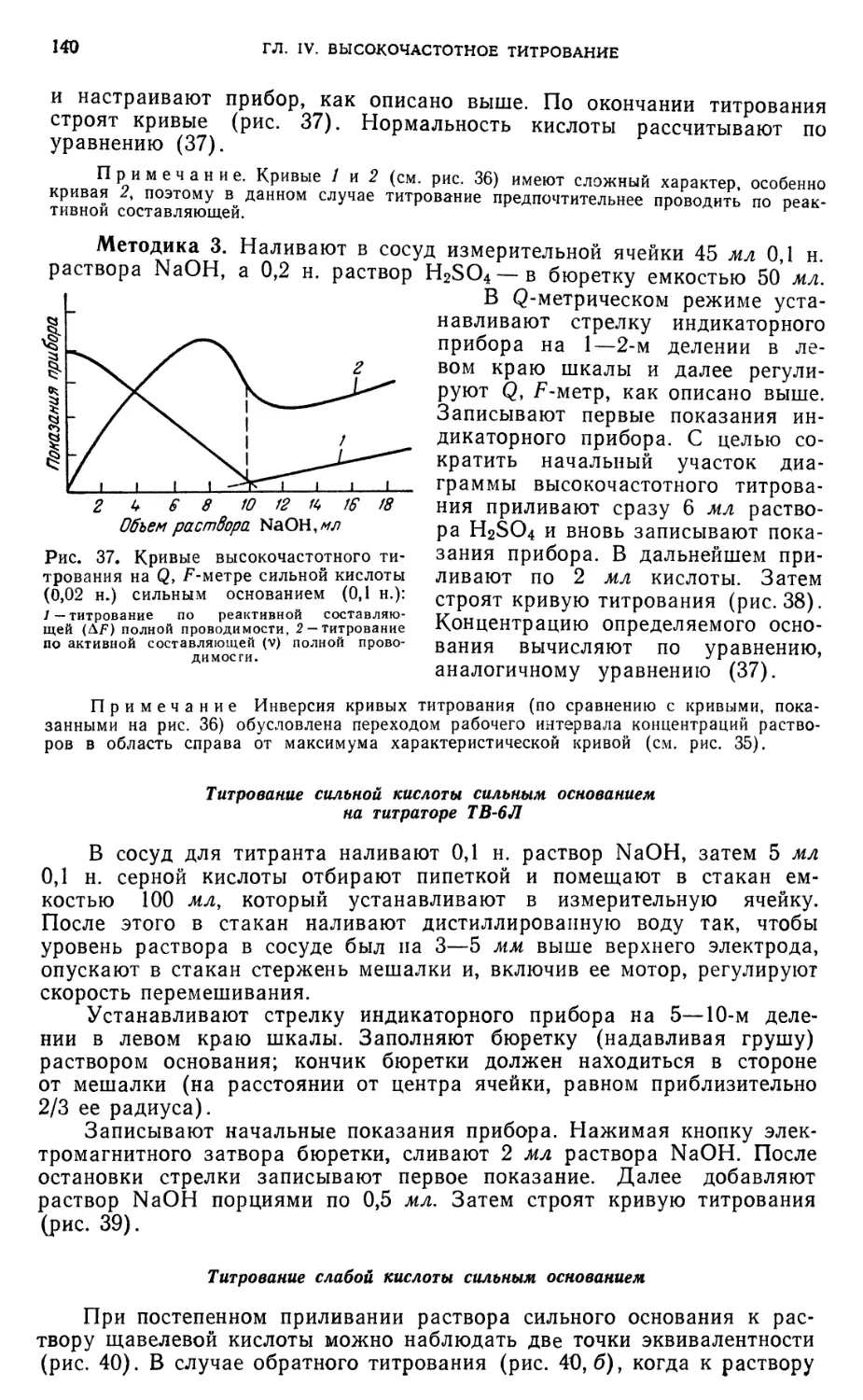
Титрование сильной кислоты сильным основанием на Q, F-метре . . . 138
Титрование сильной кислоты сильным основанием на титраторе ТВ-6Л . 140
Титрование слабой кислоты сильным основанием 140
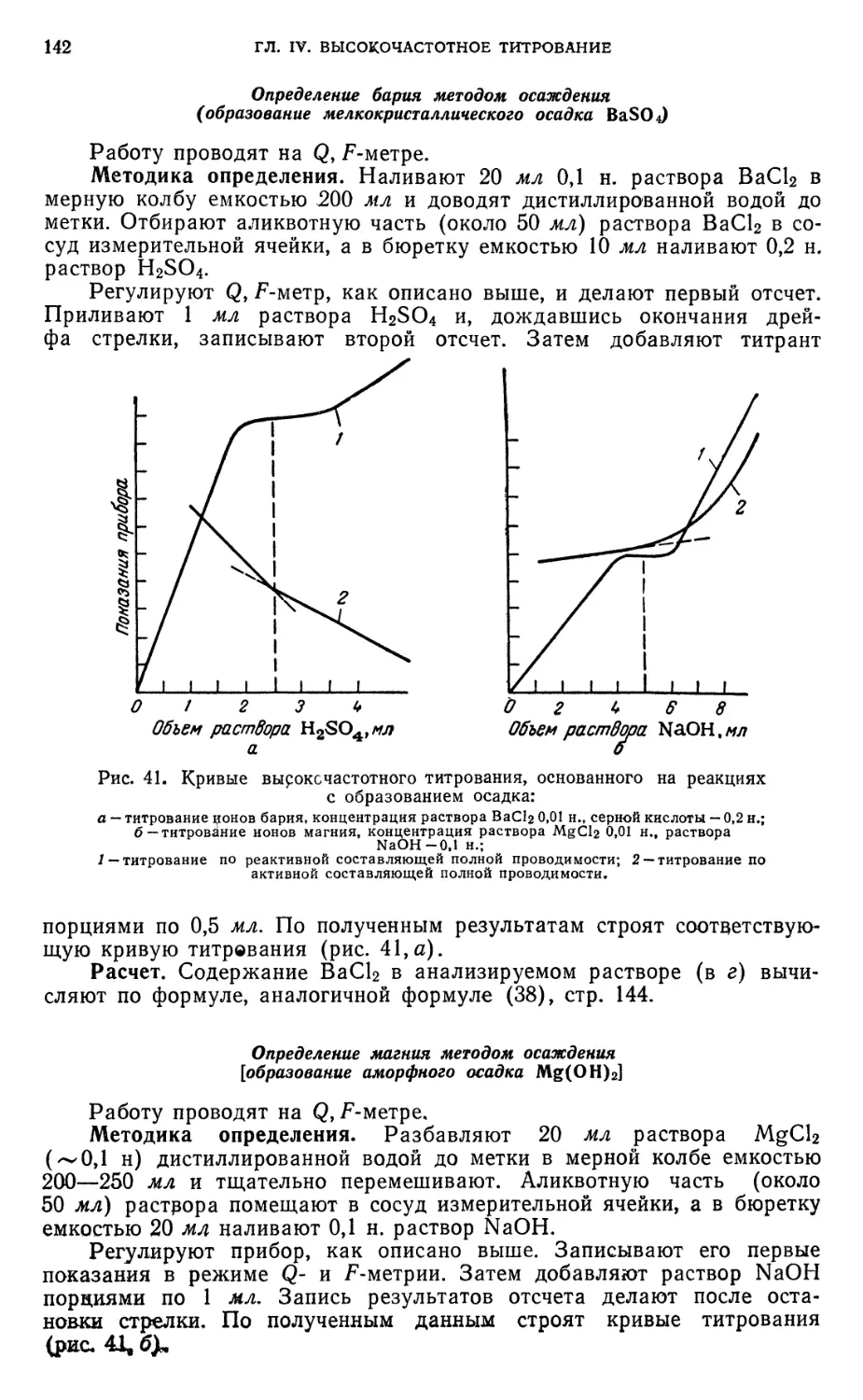
Определение бария методом осаждения 142
Определение магния методом осаждения 142
Определение железа (III) методом комплексообразования 143
§ 22. Высокочастотное титрование растворов неизвестной концентрации . . 143
Определение кислоты в растворе ЦЗ
Определение хлорид-ионов в водопроводной воде 144
Определение сульфат-ионов 144
Определение аминов 145
Определение фенолов и крезолов 145
Глава V. Полярографический метод анализа и амперометрическое титрование 147
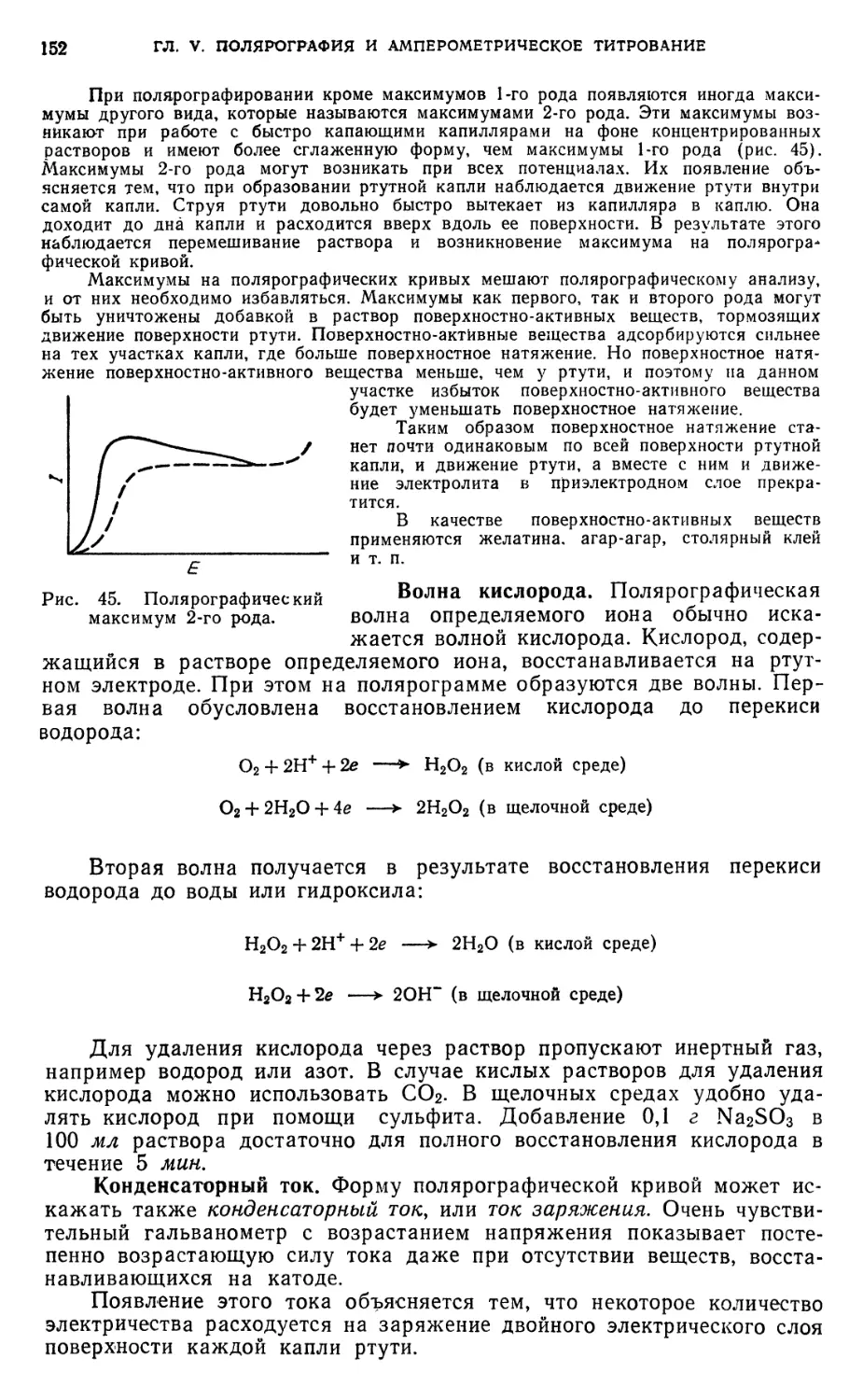
.Полярографический метод анализа 147
А. Теоретические основы ••.»«.*>.•.»««,».•«.••>»•.. .«• 147
§ 1. Предельный, или диффузионный, ток . ¦ 147
§ 2. Возникновение диффузионного тока на твердых микроэлектродах . .153
Б. Аппаратура и техника выполнения полярографического анализа . . . ¦ , 156
§ 3. Полярографы 156
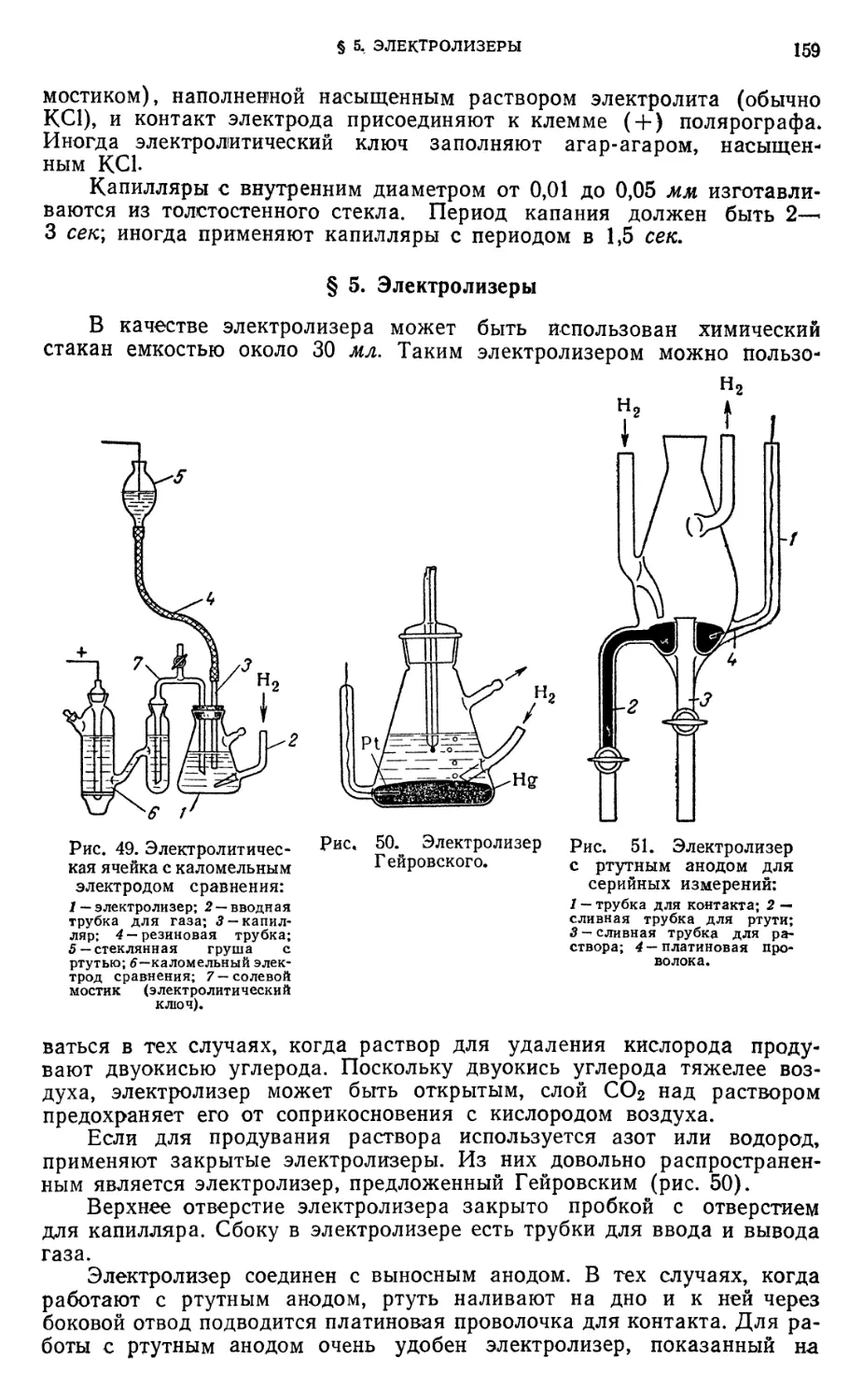
§ 4. Электролитическая ячейка 158
§ 5. Электролизеры „ 159
§ 6. Электроды сравнения 160
§ 7. Снятие полярограммы на полярографической установке с визуальным
полярографом 160
§ 8. Методы количественного полярографического анализа 161
§ 9. Правила техники безопасности при работе с металлической ртутью . . 163
Б. Новые направления в полярографии 163
§ 10. Амальгамная полярография с накоплением 164
§ 11. Осциллографическая полярография 168
§ 12. Переменнотоковая полярография ,.¦-„- 169
Г. Практические работы с применением полярографического метода анализа . 170
§ 13. Примеры полярографических определений 170
Качественное и количественное определение катионов при совместном
присутствии 170
Определение кальция 171
Определение индия (III) 172
Определение следов свинца и кадмия в металлическом цинке 173
Определение следов нитробензола в анилине 173
8
СОДЕРЖАНИЕ
Определение малеиновой и фумаровой кислот при совместном
присутствии .174
Определение альдегидов 175
Амперометрическое титрование 177
A. Теоретические основы .177
§ 14. Особенности амперометрическогО титрования 177
B. Аппаратура и техника выполнения амперометрического титрования - . - .179
§ 15. Схема установки и применяемые электроды 179
§ 16. Амперометрическое титрование с двумя индикаторными электродами 184
В. Практические работы с применением амперометрического титрования . . .187
§ 17. Примеры амперометрического титрования 187
Титрование ионов цинка или кадмия раствором гексацианоферрата (И)
калия 187
Титрование ионов свинца раствором бихромата калия 187
Титрование сульфат- и хромат-ионов при совместном присутствии ... 188
Определение хрома в минералах и рудах 189
Титрование альдегидов .190
Глава VI. Кулонометрия и кулонометрическое титрование .191
A. Теоретические основы метода 191
§ 1. Сущность и классификация кулонометрических методов 191
Прямая кулонометрия 192
§ 2. Прямая кулонометрия при постоянном потенциале рабочего электрода
(прямая потенциостатическая кулонометрия) 192
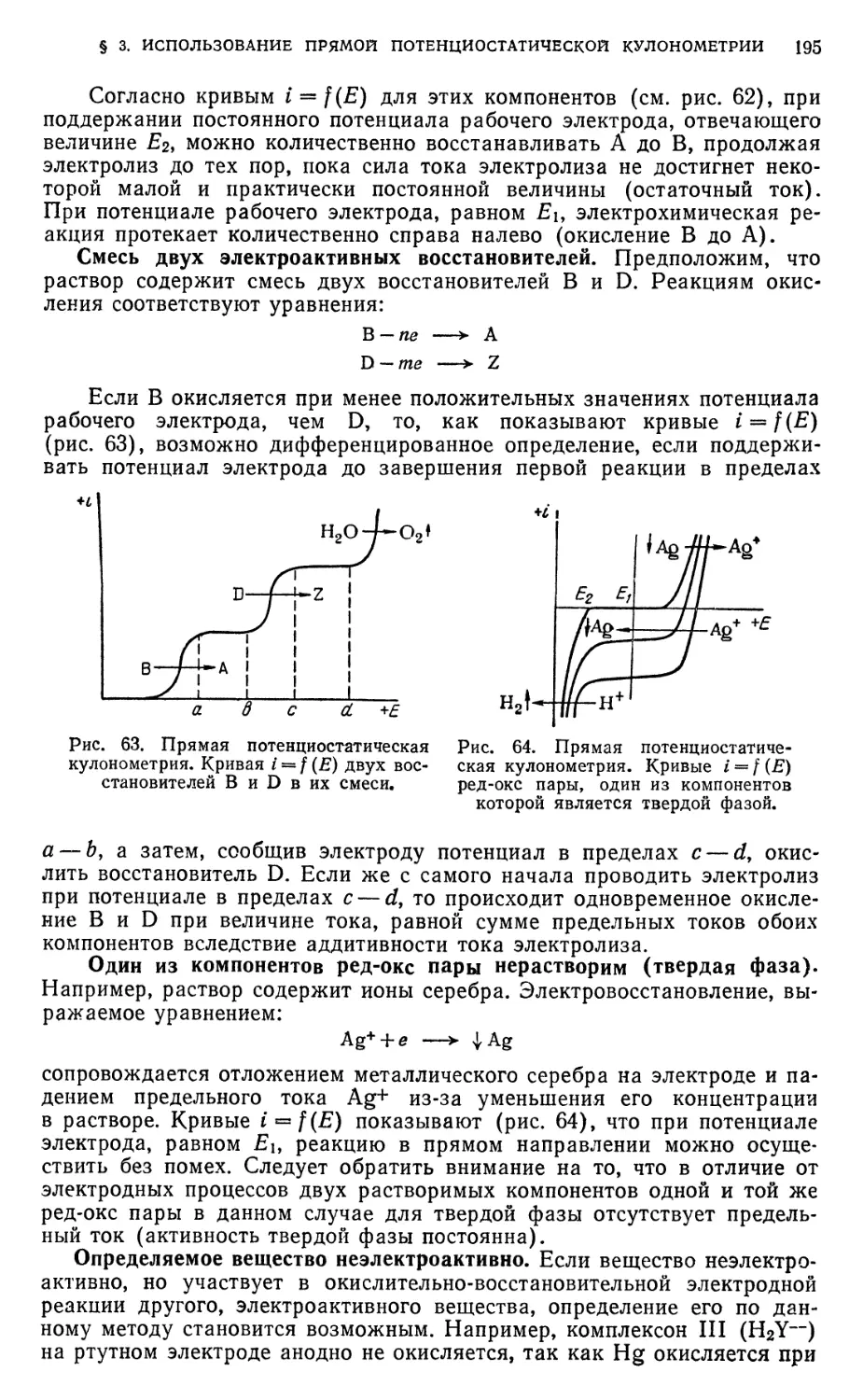
§ 3. Использование прямой потенциостатической кулонометрии 194
§ 4. Прямая кулонометрия при постоянной силе тока электролиза (прямая
амперостатическая кулонометрия) 196
§ 5. Использование прямой амперостатической кулонометрии 198
Кулонометрическое титрование 198
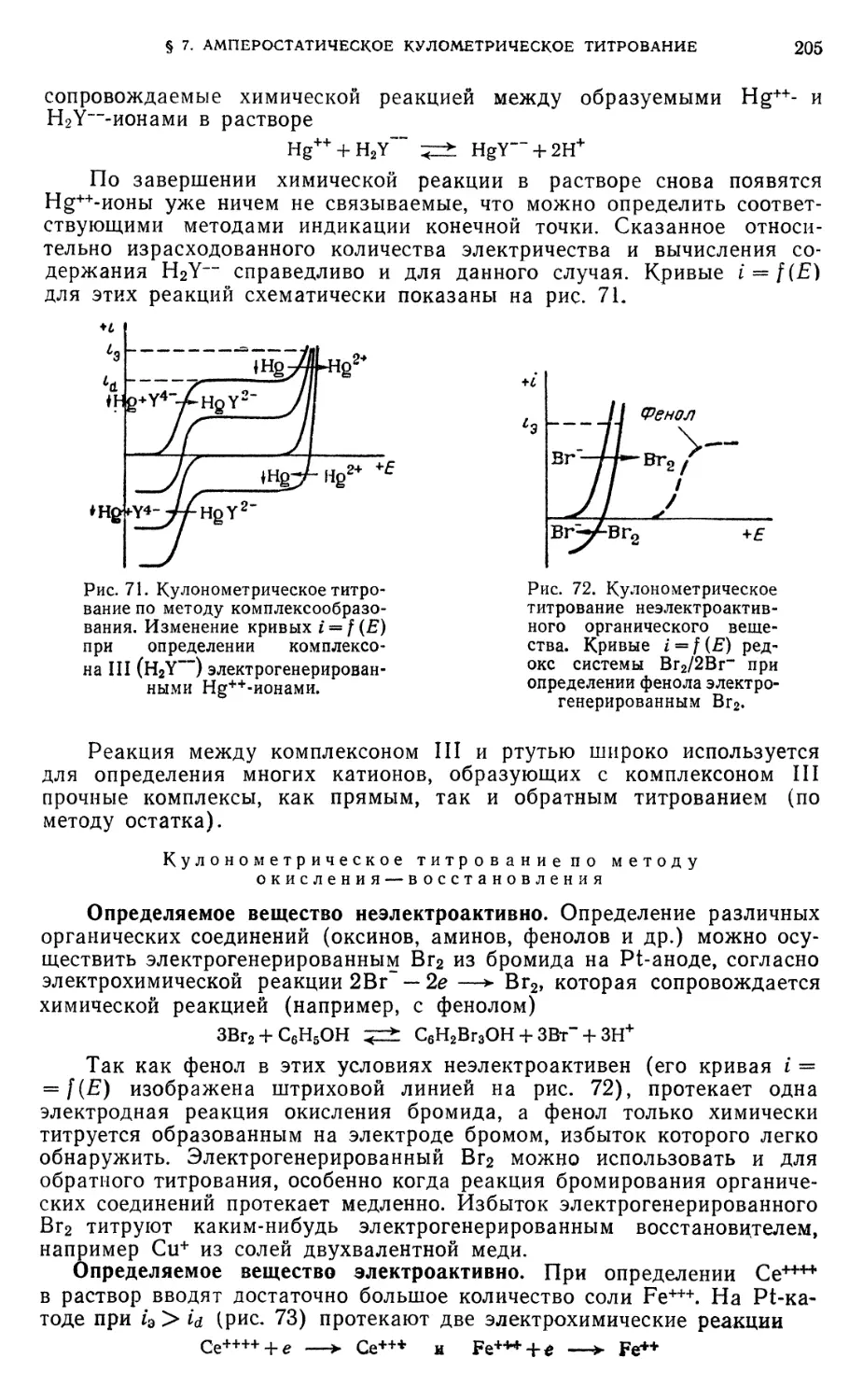
§ 6. Кулонометрическое титрование при постоянной силе тока электролиза 198
§ 7. Использование амперостатического кулонометрического титрования . . 204
§ 8. Кулонометрическое титрование при постоянном потенциале рабочего
электрода 207
§ 9. Особенности кулонометрических методов анализа 207
Б. Аппаратура и техника выполнения анализа г 208
§ 10. Электроды 208
§ 11. Электролизеры 208
§ 12. Приборы для измерения количества электричества, израсходованного
на электролиз вещества 211
§ 13. Приборы, обеспечивающие стабильность потенциала рабочего
электрода или силы тока электролиза, и установки для кулонометрического
анализа 214
B. Практические работы 216
§ 14. Работы, выполняемые методом прямой кулонометрии 216
Определение ионов железа (III) при контролируемом потенциале
рабочего электрода 216
Определение ионов меди (II) при постоянной силе тока электролиза . .218
§ 15. Работы, выполняемые методом косвенной кулонометрии, или
кулонометрического титрования, при постоянной силе тока электролиза . . .219
Определение хлористоводородной кислрты по кислотно-основному методу 219
Определение ионов церия (IV) по методу окисления — восстановления 220
Определение анилина 221
Определение иолов цинка по методу осаждения 222
СОДЕРЖАНИЕ 9
Глава VII. Спектральные (оптические) методы анализа 224
Эмиссионный спектральный анализ 224
A. Теоретические основы метода 224
§ 1. Общая характеристика метода 224
§ 2. Классификация методов эмиссионного, спектрального анализа .... 225
§ 3. Качественный спектральный анализ , 225
§ 4. Полуколичественные методы спектрального анализа 225
§ 5. Количественный спектральный анализ 226
§ 6. Основы фотографического метода эмиссионного количественного анализа 227
§ 7. Зависимость между почернением фотопластинки и интенсивностью
излучения 228
Б. Аппаратура и техника выполнения анализа * 229
§ 8. Источники возбуждения спектров 229
§ 9. Спектральные приборы . 231
§ 10. Вспомогательные приборы и принадлежности 232
§ 11. Микрофотометр МФ-2 233
B. Практические работы * 234
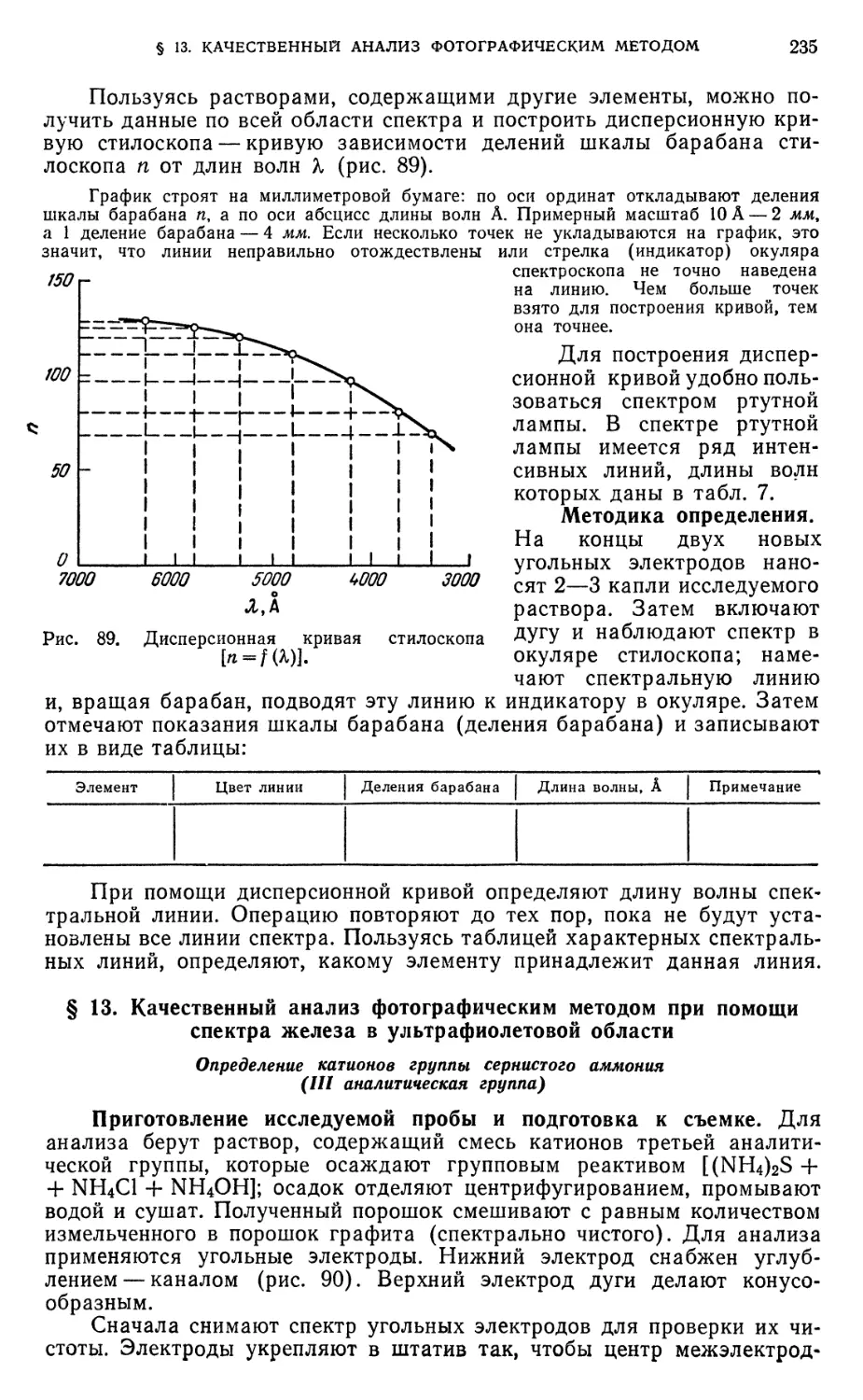
§ 12. Качественный анализ при помощи стилоскопа 234
§ 13. Качественный анализ фотографическим методом при помощи спектра
железа в ультрафиолетовой области 235
Определение катионов группы сернистого аммония 235
Расшифровка спектрограмм и идентификация элементов 237
§ 14. Полуколичественный анализ 238
Определение железа и свинца в металлическом цинке методом сравнения 238
§ 15. Количественный анализ 240
Определение железа в кварцевом песке методом трех эталонов .... 240
Спектрофотометрия пламени . . • • 241
A. Теоретические основы метода 241
§ 16. Общие характеристики метода 241
Б. Аппаратура и техника выполнения анализа 242
§ 17. Схема установки, применяемой для анализа методом спектрофотомет-
рии пламени .¦*¦ 242
B. Практические работы ¦ . . . 242
§ 18. Определения пр методу спедтрофотометрии пламени ....... 242
Определение ионов натрия в воде методом калибровочного графика . . 243
Атомно-абсорбционный спектральный анализ . , 244
§ 19, Общая характеристика метода 244
Молекулярно-абсорбционный спектральный анализ 244
А. Теоретические основы метода .„..* 244
§ 20. Общая характеристика метода 244
§ 2J. Законы поглощения света 245
§ 22. Отклонения от закона Бугера — Ламберта — Бера 246
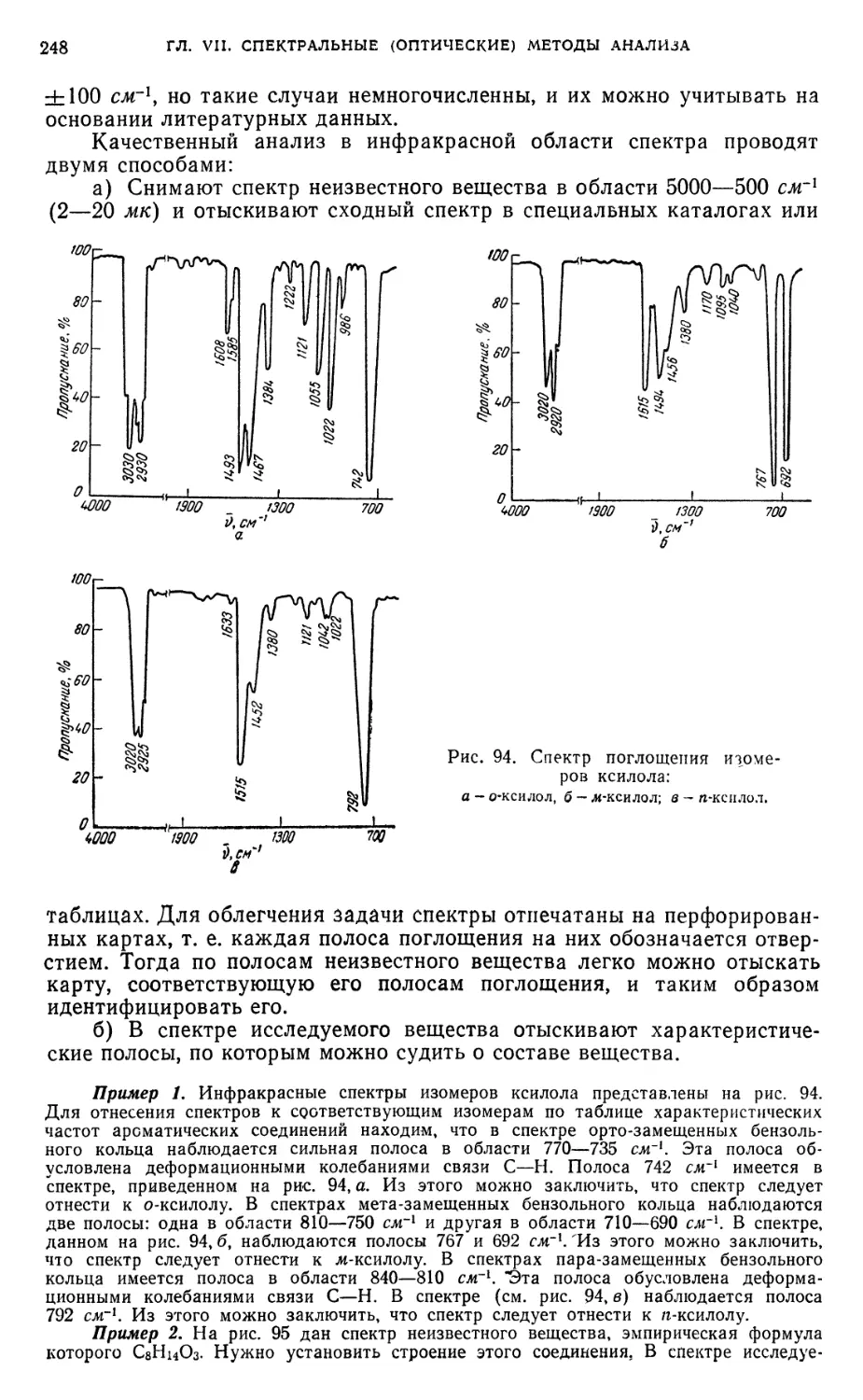
§ 23. Качественный анализ спектрофотометрическим методом 246
§ 24. Количественный анализ спектрофотометрическим методом 249
§ 25. Методы определения концентрации веществ, поглощающих в видимой
и ультрафиолетовой областях спектра 250
§ 26. Определение нескольких компонентов в растворе 252
§ 27. Определение концентрации вещества в растворе дифференциальным
методом 252
§ 28. Выбор толщины слоя и оптической концентрации исследуемого
раствора 254
§ 29. Выбор длины волны поглощаемого излучения при спектрофотометри-
ческих измерениях , . . * 254
10
СОДЕРЖАНИЕ
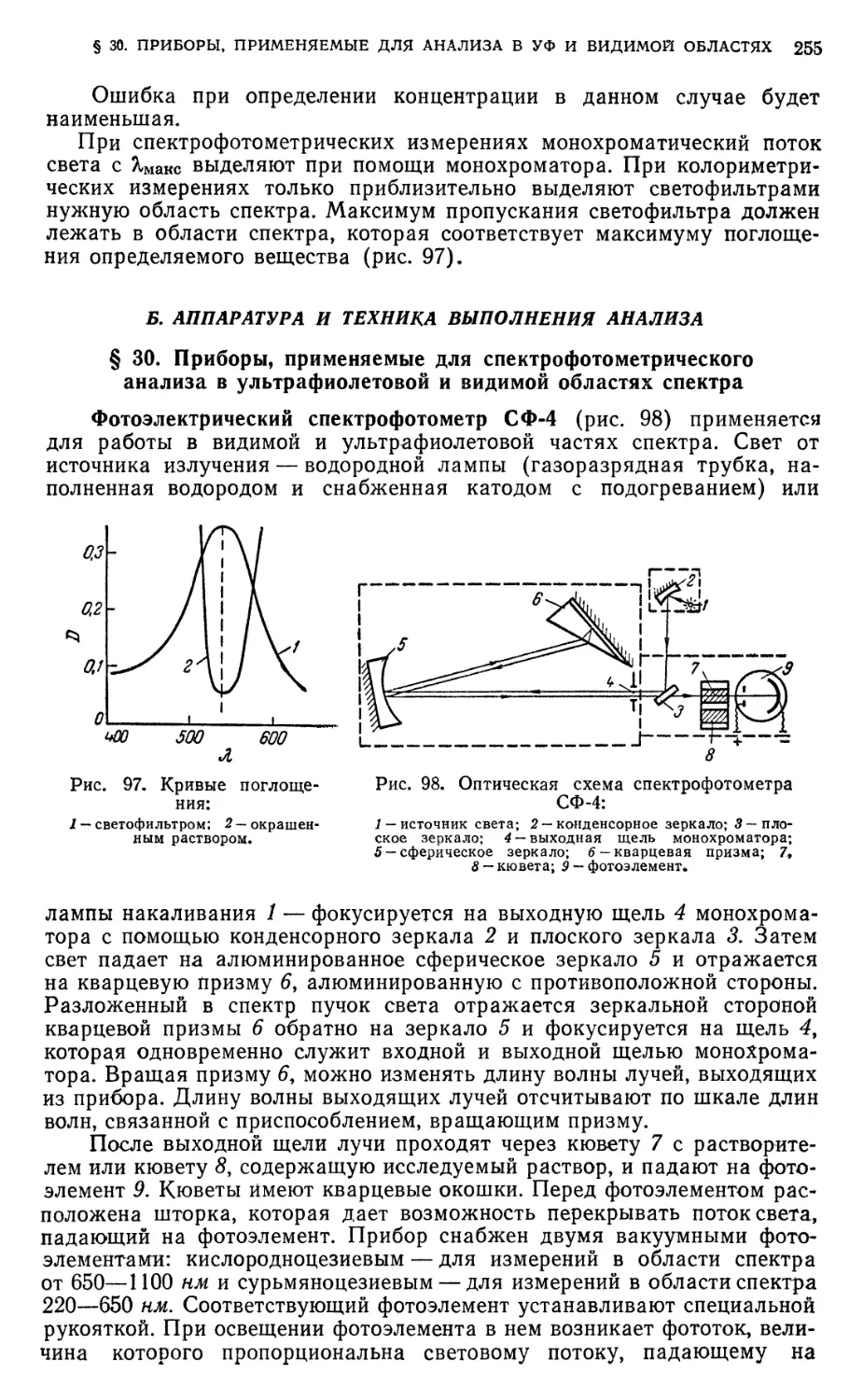
Б. Аппаратура и техника выполнения анализа 255
§ 30. Приборы, применяемые для спектрофотометрического анализа в
ультрафиолетовой и видимой областях спектра 255
§ 31. Приборы, применяемые для колориметрического анализа ..... 256
§ 32. Приборы, применяемые для спектрофотометрического анализа в
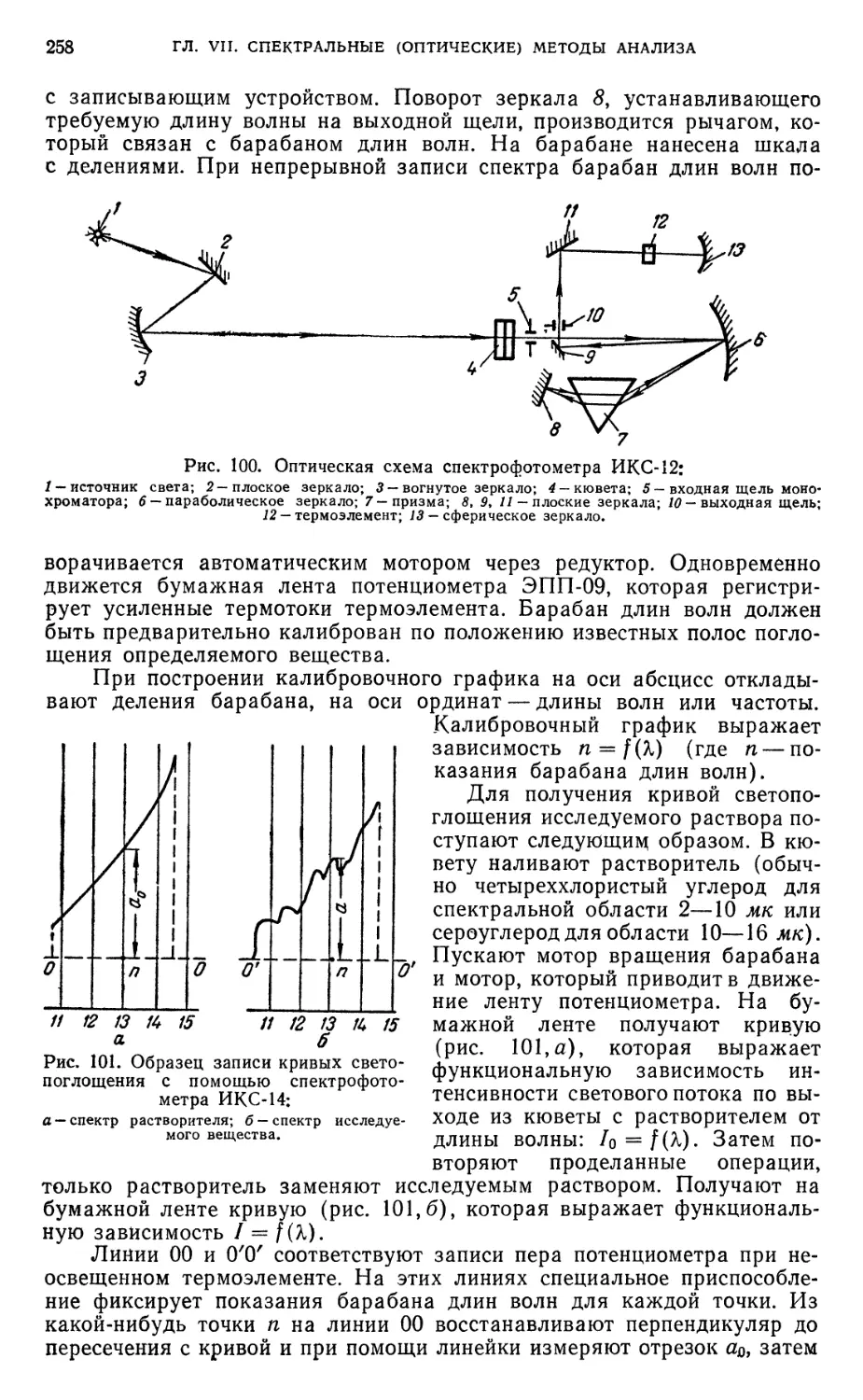
инфракрасной области спектра 257
§ 33. Нулевые растворы .*•„*_*.** 259
Б. Практические работы •••¦»••¦..¦.* 259
§ 34. Определение в ультрафиолетовой области спектра 259
Определение молибдена в стали 259
Определение титана в ильменитовых концентратах 260
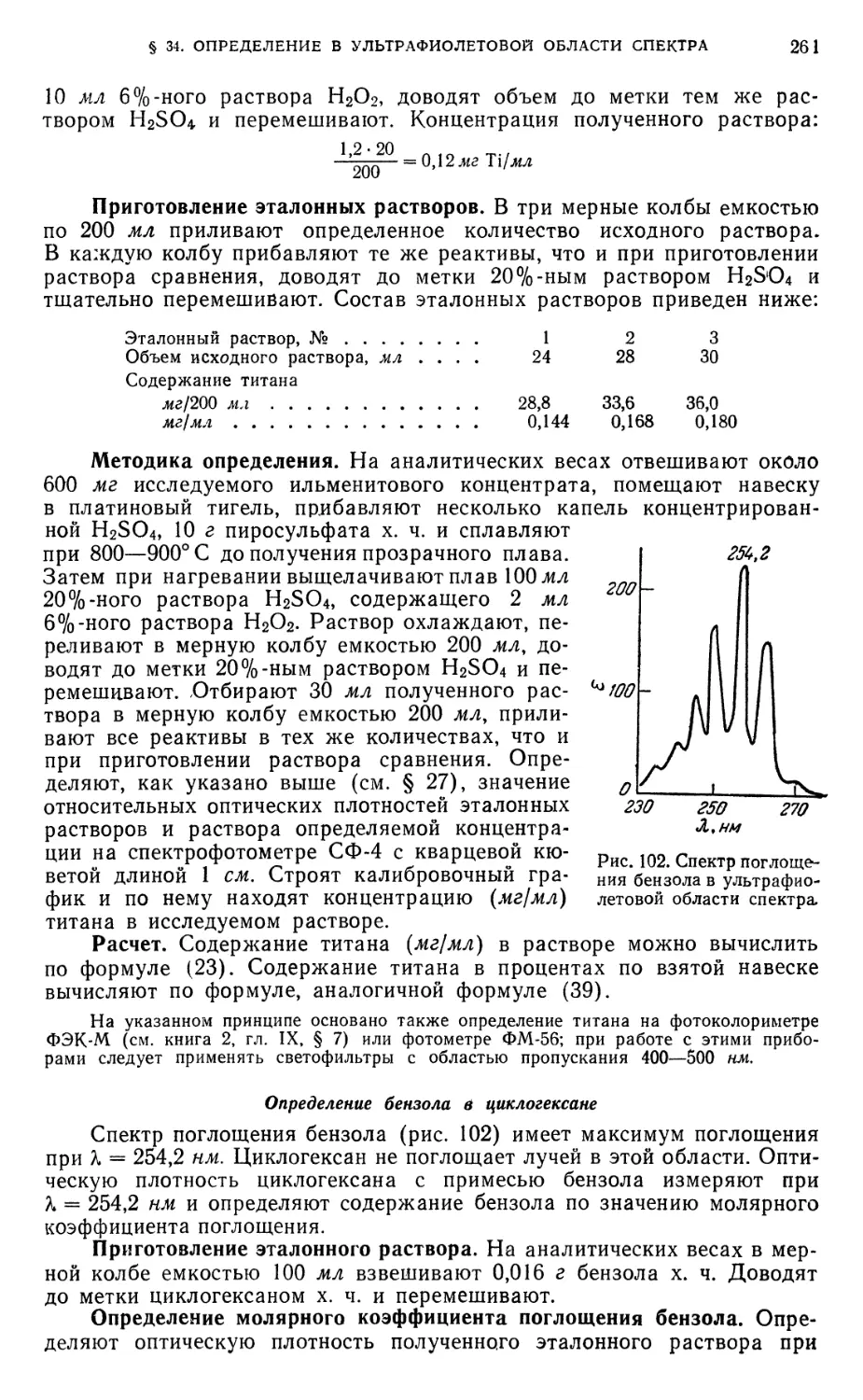
Определение бензола в циклогексане 261
§ 35. Определение в видимой области спектра 262
Определение марганца и хрома в стали при совместном присутствий . . 262
§ 36. Определение в инфракрасной области спектра 264
Определение циклогексанона в циклогексане 264
Спектрофотометрические (фотометрические) методы титрования 265
А. Теоретические основы метода 265
§ 37. Общая характеристика метода * 265
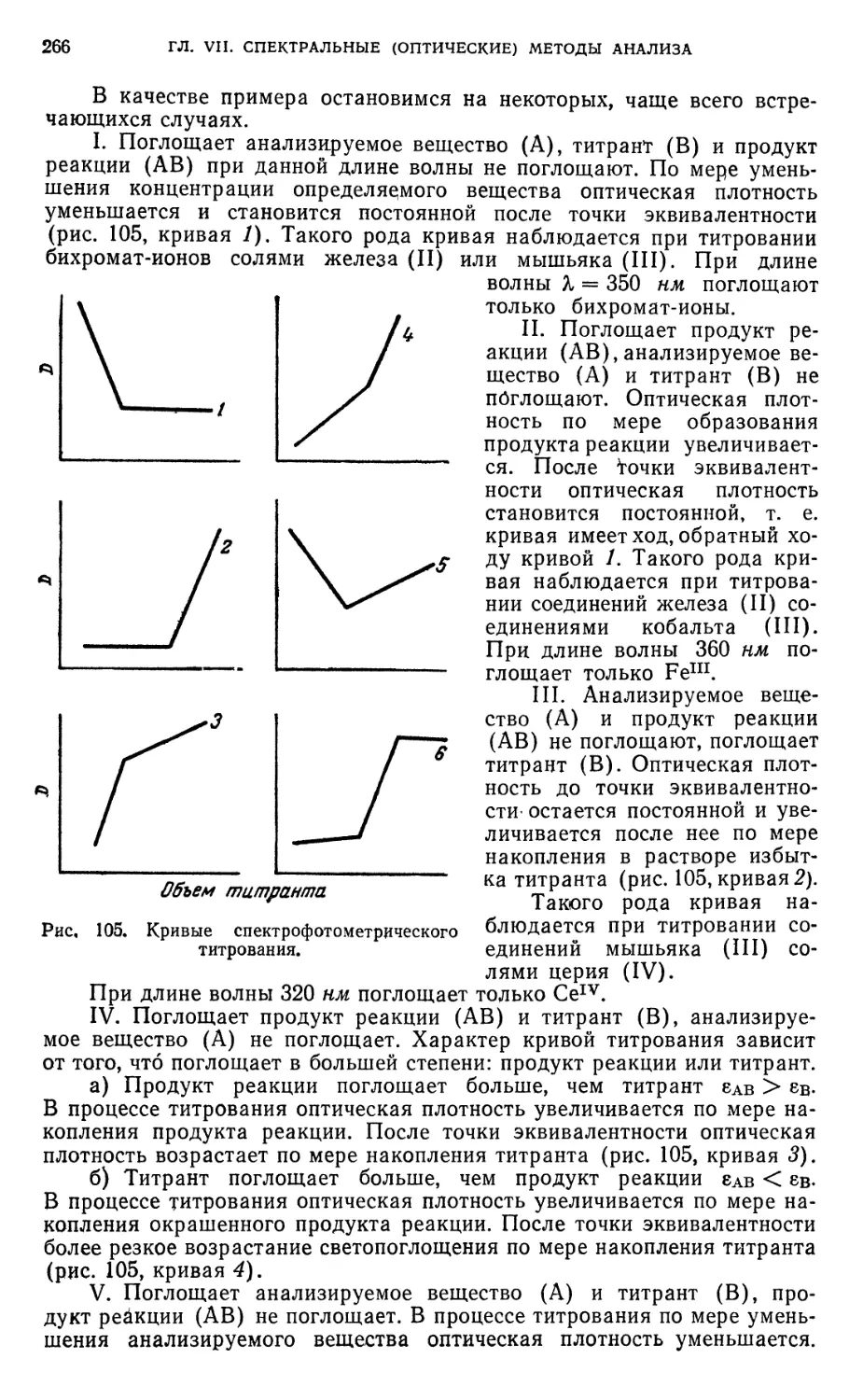
§ 38. Кривые спектрофотометрического титрования 265
Б. Аппаратура и техника выполнения титрования . . „ - 268
§ 39. Установки для спектрофотометрического титрования 268
Б. Практические работы „•«•>•¦,»•,.*¦*%* 268
§ 40. Спектрофотометрическое титрование 268
Определение арсенйтов 268
Анализ свинцово-оловянных сплавов 269
Определение хрома в сталях 269
Нефелометрический и турбидиметрический методы анализа 270
А. Теоретические основы метода .*• 270
§ 41. Общая характеристика метода 270
Б, Аппаратура и техника выполнения анализа 271
§ 42. Приборы, применяемые для нефелометрических и турбидиметрических
измерений . .••*,««« «....271
Б. Практические работы . «» • . 272
§ 43. Нефелометрические и турбидиметрические определения 272
Нефелометрическое определение хлорид-ионов 272
Турбидиметрическое определение сульфат-ионов 273
§ 44. Фототурбйдиметрическое и фотонефелометрическое титрование . . . 273
Глава VIII. Хроматографические методы анализа ..*•»*,... . 275
А. Теоретические основы .*..<>* 275
§ 1. Классификация хроматографических методов анализа 275
§ 2. Адсорбционная хроматография 276
§ 3. Разновидности газовой хроматографии 279
§ 4. Распределительная хроматография 281
§ 5. Ионообменная хроматография 284
§ 6. Константа ионного обмена . • - 286
§ 7. Осадочная хроматография 287
§ 8. Окислительно-восстановительная хроматография . 288
§ 9. Адсорбционно-комплексообразовательная хроматография 289
СОДЕРЖАНИЕ
И
Б. Аппаратура, применяемая в хроматографии 291
§ 10. Хроматографические колонки, применяемые в адсорбционио-жндкост-
ной хроматографии 291
§11. Аппаратура, применяемая в газовой хроматографии 291
§ 12. Приборы и материалы, применяемые в распределительной
хроматографии 292
§ 13. Колонки, применяемые в ионообменной хроматографии ...... 293
§ 14. Колонки, применяемые в осадочной и окислительно-восстановительной
хроматографии . 295
В. Практические работы 295
§ 15. Работы по методу адсорбционной (жидкостной и газовой)
хроматографии 295
Разделение пигментов зеленых листьев растений методом адсорбционно-
жидкостной хроматографии 295
Анализ многокомпонентной смеси газов методом газо-адсорбционной
хроматографии 297
Анализ смеси газообразных углеводородов Q — Cs методом газо-жид-
костной хроматографии . * . . 298
§ 16. Работы по методу распределительной хроматографии , 298
Анализ смеси катионов кадмия, меди и ртути (II) 298
Разделение, качественное и количественное определение аминокислот . . 299
Анализ смесей красителей , 303
Разделение катионов меди и кадмия методом тонкослойной
хроматографии 304
Определение жирных кислот методом «обращенных фаз» 304
§ 17. Работы по методу ионообменной хроматографии 305
Анализ смесей неорганических ионов 305
Подготовка ионитов для определения различных веществ 307
Определение содержания соли в растворе 308
Разделение на катионите и определение ионов меди и свинца ..... 308
Разделение на катионите и определение ионов цинка и железа (III) . .310
Разделение на анионите и определение ионов цинка и никеля 311
§ 18. Работы по методу осадочной хроматографии 312
Определение ионов никеля по величине зоны хроматограммы 312
Определение микроколичеств ионов меди 314
Концентрирование разбавленных растворов солей меди . . . . . . .315
§ 19. Работы по методу окислительно-восстановительной хроматографии . . 315
Обнаружение иодид- и бромид-ионов при совместном присутствии . . .315
§ 20. Работы по методу адсорбционно-комплексообразовательной
хроматографии 316
Разделение и определение ионов никеля и цинка; ниобия и тантала . .316
Глава IX. Радиометрические методы анализа < 318
А. Теоретические основы радиометрических методов 318
§ 1. Виды радиоактивного распада 318
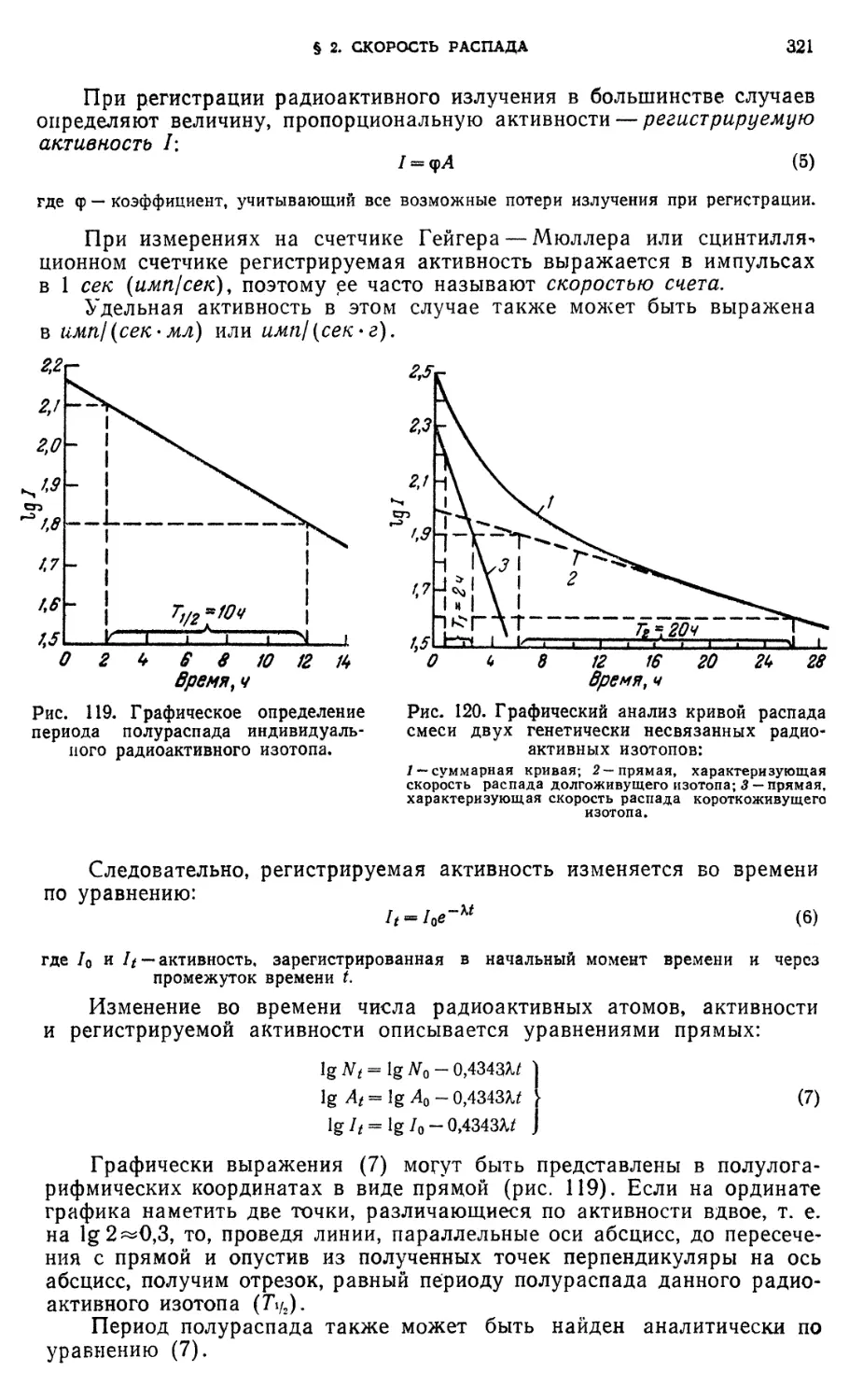
§ 2. Скорость распада 319
§ 3. Взаимодействие излучения с веществом 323
§ 4. Ошибки при измерении радиоактивности 324
Б. Элементы техники безопасности при работе с радиоактивными веществами 326
§ 5. Понятия и единицы измерения 326
§ 6. Классификация источников радиоактивных излучений и радиоактивных
изотопов . 327
§ 7. Классификация химических операций с радиоактивными- веществами 328
§ 8. Устройство лабораторий и классификация работ 329
§ 9. Специальное оборудование для работ с радиоактивными веществами ЗЗЭ
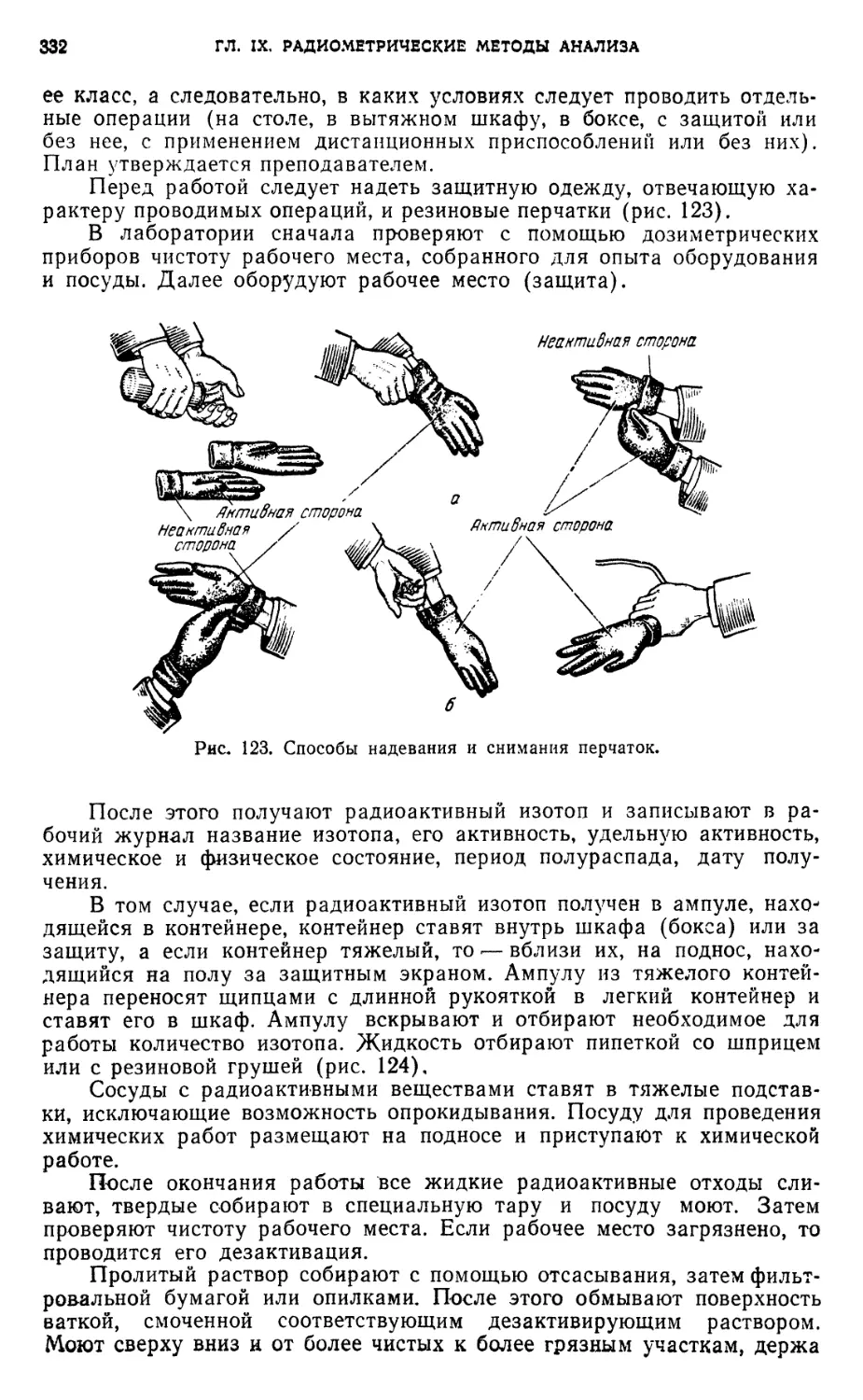
§ 10. Средства индивидуальной защиты 331
§ 11. Проведение работ в химической лаборатории с радиоактивными
веществами 331
§ 12. Правила работы с радиоактивными веществами 333
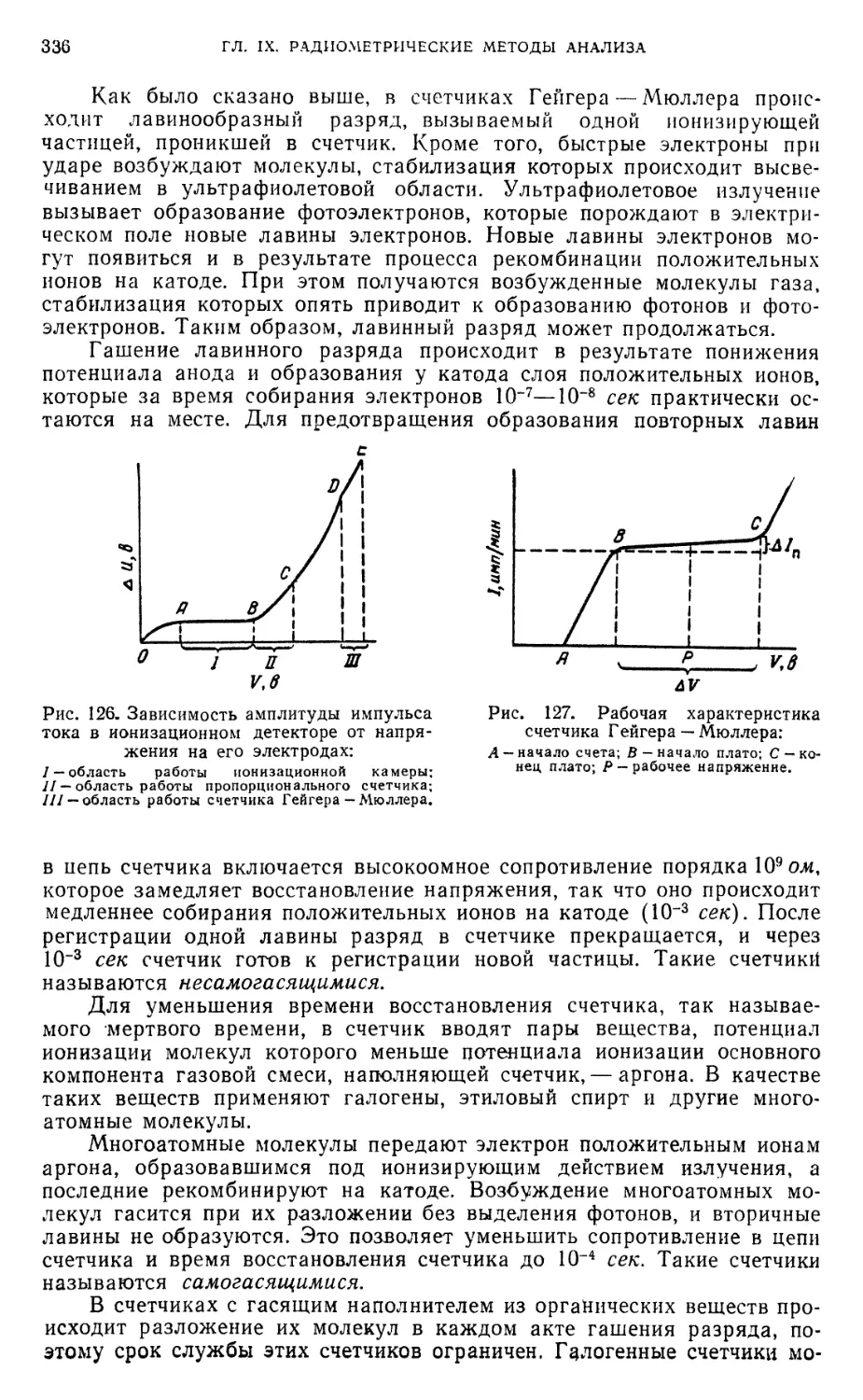
Б. Методы регистрации радиоактивного излучения 334
§ 13. Ионизационные методы 334
§ 14. Сцинтилляционный метод 338
12
СОДЕРЖАНИЕ
Г. Аппаратура и техника измерения радиоактивности 340
§ 15. Радиометрические установки 340
§ 16. Измерение радиоактивности 341
§ 17. Измерение дозы на рабочем месте, индивидуальной дозы, полученной
работающим, и загрязненности рабочих поверхностей, рук и одежды 343
§ 18. Абсолютные и относительные измерения активности 344
Д. Радиометрические методы определения 345
§ 19. Приготовление раствора с заданной активностью 346
§ 20. Прямое определение ионов химических элементов в растворе с
помощью радиоактивных реагентов 346
§ 21. Практические работы, выполняемые методом прямого определения . . 347
Определение катионов в виде фосфатов с применением 32Р 347
Определение ионов калия в виде кобальтинитрита калня с применением
60Со 347
§ 22. Метод радиометрического титрования 348
§ 23. Практические работы, выполняемые методом радиометрического
титрования 350
Определение фосфат-ионов . . 350
Определение ионов магния 351
Определение ионов цинка и меди при совместном присутствии .... 352
§ 24. Метод изотопного разбавления 352
§ 25. Практические работы, выполняемые методом изотопного разбавления 353
Определение ионов иода в присутствии ионов брома . 353
§ 26. Метод активационного анализа 354
§ 27. Практические работы, выполняемые методом активационного анализа 3$7
Определение ионов индия 357
§ 28. Фотонейтронный метод 357
§ 29. Практические работы, выполняемые фотонейтронным методом .... 360
Определение бериллия 360
§ 30. Методы определения содержания химических элементов по излучению
их естественных радиоактивных изотопов 360
§ 31. Практические работы по определению содержания химических
элементов методом измерения излучения их естественных радиоактивных
изотопов 361
Определение калия по его естественной радиоактивности 361
§ 32. Методы анализа, основанные на поглощении излучения 363
§ 33. Практические работы, выполняемые по методу анализа, основанному
на поглощении излучения 366
Определение содержания бора 366
§ 34. Метод, основанный на отражении р-излучения . 366
§ 35. Практические работы, выполняемые по методу, основанному на
отражении Р-излучения 367
Определение содержания компонента смеси с большим z по эффекту обратного
отражения р-лучей 367
Глава X. Физико-химические методы количественного определения редких
элементов 368
А. Введение 368
Б. Практические работы 369
§ 1. Работы, выполняемые методом амперометрического титрования . . . 369
Определение ванадия (IV) 369
Определение индия в концентратах 369
§ 2. Работы, выполняемые полярографическим методом 370
Определение рения в сплавах с молибденом 370
Определение индия в рудах 370
Определение таллия в кадмии 371
§ 3. Работы, выполняемые фотометрическим методом 372
Определение бериллия 372
Определение скандия в магниевых сплавах 373
Определение тория 373
Определение титана . 374
Определение циркония 375
Определение рения . . ... 376
Определение висмута 376
СОДЕРЖАНИЕ
13
§ 4. Работы, выполняемые экстракционно-фотометрическим методом . . . 378
Определение микроколичеств тория 378
Определение урана 378
Определение молибдена в сталях 378
Определение галлия 380
Определение германия 381
Определение селена 383
Определение теллура в висмуте 384
Определение теллура в селене ....... 385
§ 5. Работы, выполняемые спектрофотометрическим методом 386
Определение тантала 386
Определение таллия 386
Концентрирование и определение малых количеств таллия 387
§ 6. Работы, выполняемые флуориметрическйм методом 388
Определение индия 388
§ 7. Работы, выполняемые методом потенциометрического титрования . . 388
Определение вольфрама (VI) ¦ 388
Определение рения в сплавах . . . ; 389
Глава XI. Физико-химические (инструментальные) методы анализа
неводных растворов .391
Л. Теоретическая часть 391
§ 1. Влияние химической природы и физико-химических свойств
растворителей на свойства растворенного вещества .... ... 391
§ 2. Классификация неводных растворителей по их протонно-донорно-ак-
центорным свойствам 398
§ 3. Классификация неводных растворителей по признаку их влияния на
относительную силу электролитов 403
§ 4. Дифференцирующее действие растворителей 406
§ 5. Константы диссоциации, потенциалы полунейтрализации в неводных
средах и относительная шкала кислотности 408
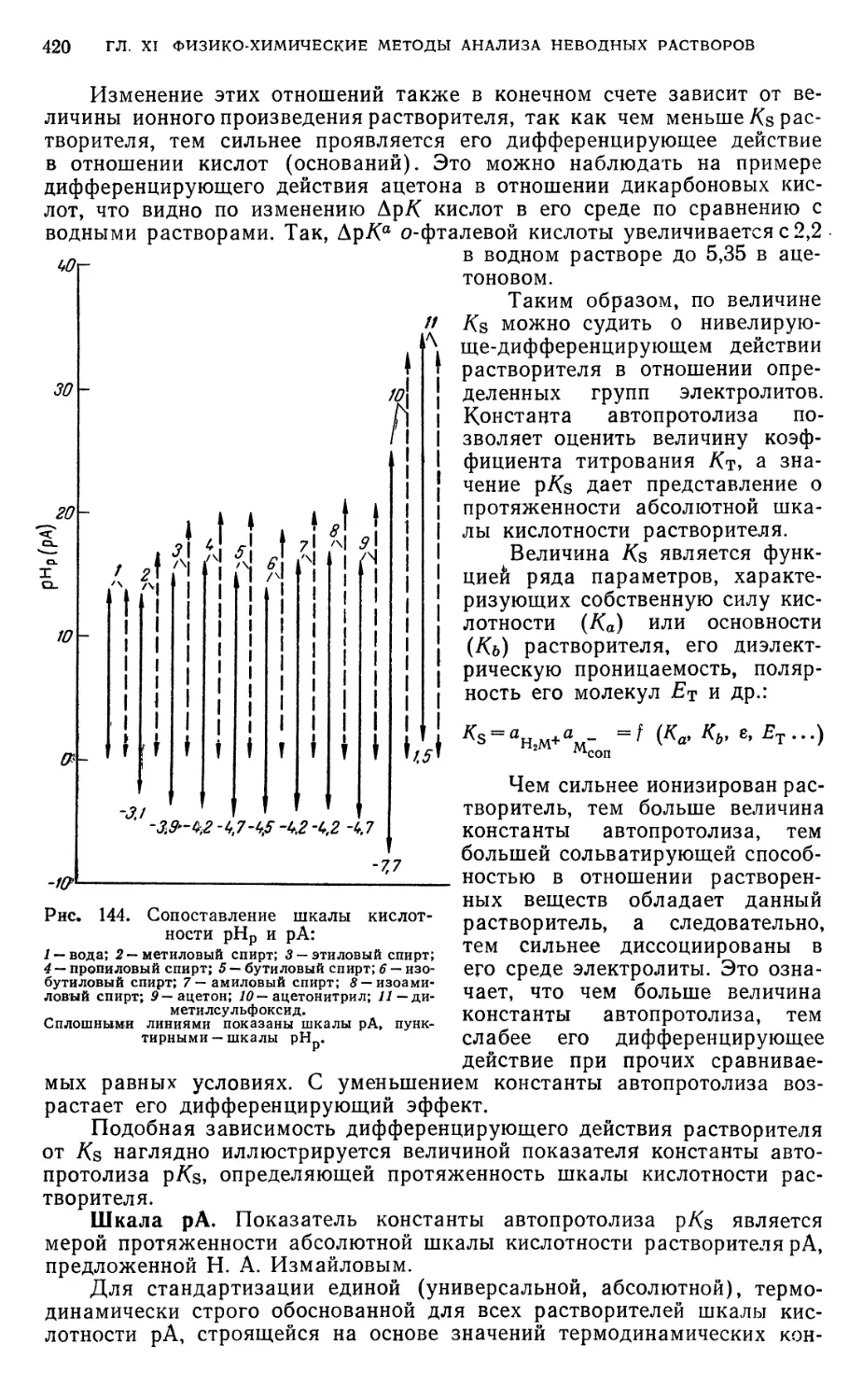
§ 6. Шкала рНр и константы автопротолиза неводных растворителей . . 414
§ 7. Абсолютная (единая) шкала кислотности растворителей 418
§ 8. Химико-аналитическое использование неводных растворителей .... 423
Б. Аппаратура и техника титрования неводных растворов 431
§ 9. Установки, используемые при амперометрическом титровании неводных
растворов 431
§ 10. Установки, используемые при потенциометрическом титровании
неводных растворов 433
§11. Установки, используемые при кондуктометрическом титровании
неводных растворов 434
§ 12. Установки, используемые при высокочастотном титровании неводных
растворов 436
§ 13. Установки, используемые при спектрофотометрическом титровании
неводных растворов 437
В. Практические работы 438
§ 14. Очистка и обезвоживание неводных растворителей 438
§ 15. Титранты, применяемые для титрования кислот и оснований в
неводных растворах 438
§ 16. Амперометрическое титрование 439
Определение нитратов в среде безводной уксусной кислоты 439
Определение ионов кадмия в среде безводной уксусной кислоты .... 440
Определение хлоридов методом амперометрического титрования с двумя
индикаторными электродами в среде изопропнлового спирта 441
§ 17. Методы прямого потенциометрического титрования 442
Определение амина в среде безводной уксусной кислоты 442
Определение бензойной кислоты в среде ацетона 443
Определение аминокислоты в среде безводной уксусной кислоты .... 443
§ 18. Методы косвенного потенциометрического титрования 444
Определение виниловых мономеров (стирола, винилтолуола, винилкси-
лола) ". 444
Анализ смесей первичных, вторичных, третичных аминов 445
Определение производных /г-фенилендиамина в смеси с я-аминодифенил-
амином 446
14
СОДЕРЖАНИЕ
§ 19. Дифференцированное потенциометрическое титрование 447
Анализ смеси аммониевых солей с аммиаком 447
Анализ смеси серной кислоты с бисульфатом натрия 448
Анализ сплава цинка с кадмием 449
Анализ смеси р-аланина и гидрохлорида гистидина 450
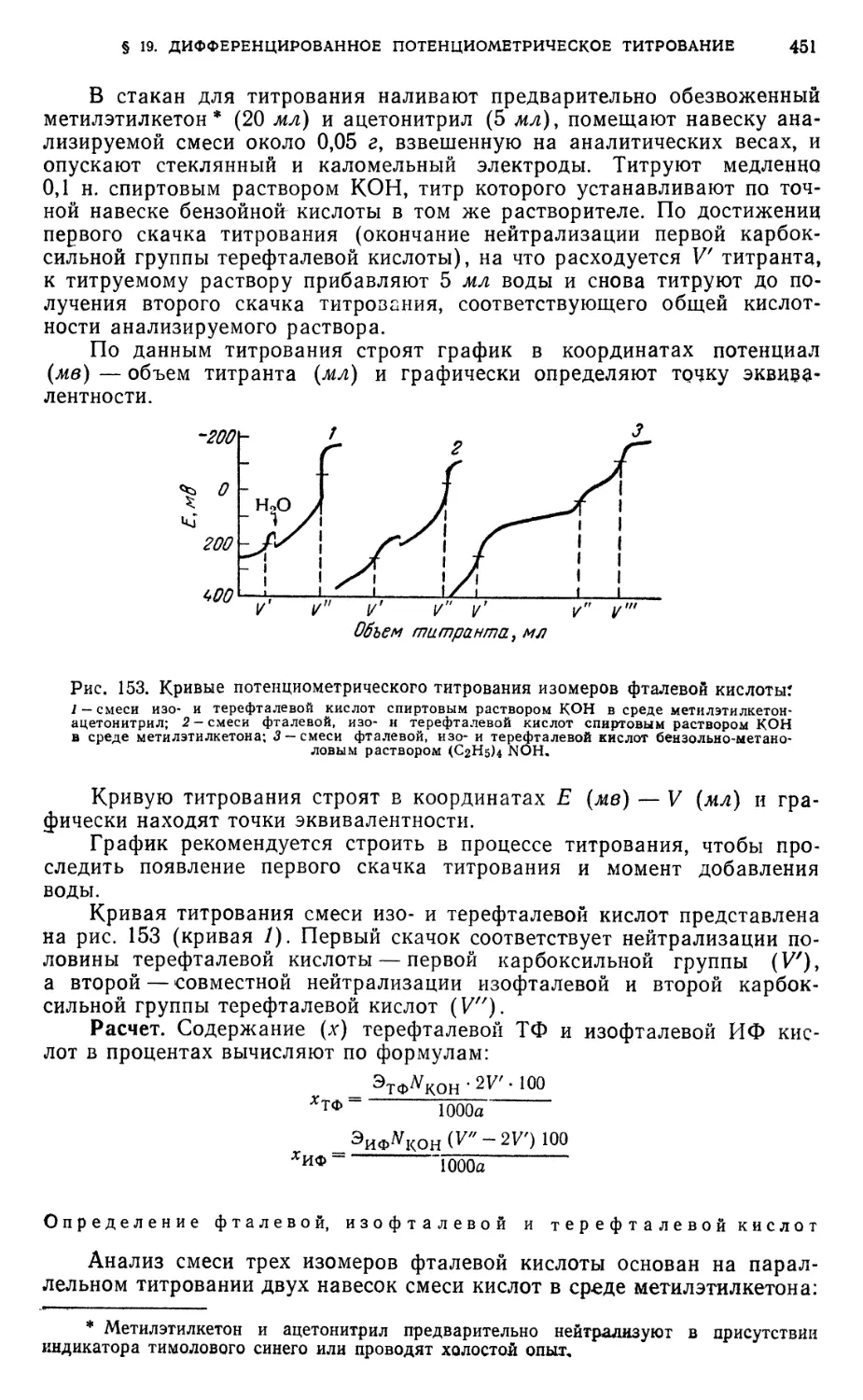
Титрование фталевой, изофталевой и терефталевой кислот 450
§ 20. Комбинированные методы потенциометрического титрования .... 452
Анализ смеси нитрита и нитрата щелочных металлов комбинированным
методом потенциометрического титрования и ионного обмена 452
Анализ смеси нитрата калия с кислотами комбинированным методом
потенциометрического титрования и ионного обмена 453
§ 21. Хронопотенциометрическое титрование 454
Анализ уксусного ангидрида и уксусной кислоты в ацетилирующих
смесях 454
§ 22. Кондуктометр ическое титрование 456
Определение анилина в среде безводной уксусной кислоты ...... 456
Определение я-фенилендиамина в среде ацетона 457
§ 23. Хронокондуктометрическое титрование 458
Определение гидрохлорида диэтиламина в среде смешанного растворителя 458
Анализ смеси хлористоводородной» уксусной и борной кислот 459
§ 24. Спектрофотометрическое титрование 459
Титрование смеси я-нитробензойной и 4-нитро-2-аминобензойной кислот 459
Анализ смеси изомеров нитроанилина 461
Безындикаторное спектрофотометрическое титрование в
ультрафиолетовой области спектра солей ароматических карбоновых кислот и их
производных 461
Предметныйуказатель 463
ПРЕДИСЛОВИЕ
Аналитическая химия как наука о методах химического анализа
является одной из основных общехимических (а в ряде учебных
заведений и профилирующих) учебных дисциплин, изучаемых студентами
химических и химико-технологических специальностей. Будущий химик
или инженер-технолог начинает формироваться уже в процессе
изучения аналитической химии, составляющей для него наряду с другими
химическими общенаучными дисциплинами фундамент
материалистического мировоззрения и прочное основание для знаний, необходимых для
последующего изучения профилирующих химических и специальных
дисциплин.
С давних пор по традиции аналитической химии в учебных планах
отводилось место вслед за курсом неорганической химии. Поэтому
аналитическая химия являлась как бы естественным продолжением курса
неорганической химии. Это обстоятельство накладывало особый
отпечаток на учебную программу по аналитической химии, представлявшей
собой теорию и практику так называемых классических (качественного,
весового и объемного) методов анализа неорганических соединений.
Все к этому привыкли, и раньше это оправдалось многими
обстоятельствами. Подлинно же современную аналитическую химию нельзя
изучать на основе только неорганической химии, поскольку на примерах
реакций, известных из курса неорганической химии, невозможно изучать
процессы, связанные с применением органических реагентов,
индикаторов, экстрагентов, органических соосадителей, ионообменных смол,
органических растворителей и т. п.
Вот почему в данной книге описываются не только методы
анализа неорганических веществ, но и методы анализа некоторых
органических соединений.
В составлении третьей книги этого учебника принимали участие
доктора химических наук:
А. П. Крешков, написавший «Введение», гл. I «Основы физико-
химических (инструментальных) методов анализа»; гл. III «Кондукто-
метрия и кондуктометрическое титрование» (совместно с Т. А.
Худяковой); гл. VII «Спектральные (оптические) методы анализа» (совместно
с Ю. Я. Михайленко и Л. П. Сенецкой); гл. VIII «Хроматографические
16
ПРЕДИСЛОВИЕ
методы анализа» (совместно с К. М. Ольшановой и Е. Н. Саюшкиной);
гл. XI «Физико-химические (инструментальные) методы анализа
неводных растворов»;
П. К. Агасян, написавший гл. II «Потенциометрия и потенциометри-
ческое титрование» и гл. VI «Кулонометрия и кулонометрическое
титрование»;
A. И. Бусев — гл. X «Физико-химические методы количественного
определения редких элементов»;
Ан. Н. Несмеянов — гл. IX «Радиометрические методы анализа»;
К. М. Ольшанова (совместно с А. П. Крешковым и Е. Н.
Саюшкиной), написавшая гл. VIII «Хроматографические методы анализа»;
Т. А. Худякова (совместно с А. П. Крешковым) —гл. III «Кондук-
тометрия и кондуктометрическое титрование»;
доценты, кандидаты химических наук:
B. А. Борк, написавшая гл. V «Полярография и амперометрическое
титрование»; В. И. Ермаков, написавший гл. IV «Высокочастотное
титрование»; Ю. Я. Мпхайленко (совместно с А. П. Крешковым и
Л. П. Сенецкой)—гл. VII «Спектральные (оптические) методы
анализа»; Е. Н. Саюшкина (совместно с А. П. Крешковым и К. М.
Ольшановой), написавшая гл. VIII «Хроматографические методы анализа».
Л. П. Сенецкая (совместно с А. П. Крешковым и Ю. Я.
Мпхайленко)— гл. VII «Спектральные (оптические) методы анализа».
Общее редактирование книги осуществлено А. П. Крешковым.
Авторы выражают глубокую благодарность чл.-корр. АН СССР,
зав. лабораторией Института физической химии АН СССР К. В. Чму-
тову, зав. кафедрой, докт. хим. наук К. Н. Мочалову и коллективу
кафедры аналитической химии Казанского химико-технологического
института им. С. М. Кирова за большой труд по рецензированию учебника
и ценные советы.
А. П. К Р Е Ш К О В
ВВЕДЕНИЕ
Управление химическим производством и выполнение всякой
научно-исследовательской работы по химии или химической технологии
основано на рационально построенной системе химико-аналитического
контроля как отдельных стадий, так и всего процесса в целом.
Исчерпывающая информация о состоянии наблюдаемой системы, ее составе
(элементном и фазовом), о свойствах получаемых продуктов, их
строении, наличии в них примесей, и т. п. возможна лишь при использовании
регистрирующих, сигнализирующих, блокирующих, вычислительных и
управляющих машин и приборов, зачастую являющихся сложными
электронными системами.
Поэтому современная аналитическая химия испытывает сильное
влияние экспериментальной физики и физической химии. Прогресс этих
наук, чрезвычайное разнообразие и точность их методов изучения
материи в значительной степени изменяют основное направление развития
аналитической химии. Все большее значение приобретают новые
физические и физико-химические (инструментальные) методы анализа,
широко применяемые в различных областях науки, техники и
промышленности, и, поскольку эти методы решают задачи химического анализа,
они составляют одну из неотъемлемых частей аналитической химии.
Физические методы анализа. Определение состава самых
разнообразных веществ можно осуществить, не прибегая к химическим или
электрохимическим реакциям (см. книга 2, «Введение», § 3). Такого
рода методы определения основываются на изучении физических свойств
или измерении физических констант исследуемого вещества, например
эмиссионных спектров поглощения, электро- или теплопроводности,
потенциала электрода, погруженного в раствор, диэлектрической
проницаемости, вращения плоскости поляризации света, показателя
преломления, флуоресценции, ядерного магнитного резонанса,
радиоактивности и т. п.
Использование физических методов анализа и исследования
основано на применении разнообразных прецизионных физических
приборов.
Физические методы анализа отличаются рядом преимуществ перед
другими методами и дают возможность решать вопросы, которые нельзя
разрешить методами химического анализа.
Физико-химические методы анализа. Для анализа веществ широко
используются химические реакции, протекание которых сопровождается
изменением физических свойств анализируемой системы, например ее
цвета, интенсивности окраски, прозрачности, флуоресценции, величины
электро- и теплопроводности, и т. д.
18
ВВЕДЕНИЕ
Все методы такого рода объединяют под общим названием «физико-
химические методы». Другцми словами, сущность физико-химических
методов анализа сводится к изучению соотношений между составом
и свойствами исследуемых систем.
Различают прямые и косвенные физико-химические методы. В
прямых методах анализа данное свойство является критерием содержания
определяемого вещества, эти методы основаны на изучении диаграммы
состав — свойство. В косвенных методах определенное свойство служит
указателем (подобно индикатору) конца реакции, т. е. в косвенных
методах используется данное свойство определяемого вещества для
фиксирования конца процесса взаимодействия (например, процесса
нейтрализации) определяемого вещества с реактивом точно известной
концентрации.
Физико-химические методы, отличающиеся высокой
чувствительностью и экспрессностью выполнения, дают возможность
автоматизировать химико-аналитические определения и являются незаменимыми при
анализе малых и ультрамалых количеств неорганических и
органических веществ. Физико-химическим методам принадлежит ведущая роль
в аналитическом контроле производства на больших предприятиях
химической промышленности, и особенно в контроле производств,
использующих в технологических процессах высокие температуры и давления,
огнеопасные, ядовитые, взрывчатые и радиоактивные вещества.
Широкое применение инструментальных методов анализа ни в
какой мере не умаляет роли классической аналитической химии, которая,
безусловно, является основой современной аналитической химии.
Поэтому на первом этапе студенты знакомятся с классическими методами
анализа и лишь с основами электрохимических, спектроскопических, хро-
матографических и некоторых других современных методов анализа
(книги 1 и 2 «Основы аналитической химии»). На втором этапе
студенты углубленно изучают и практически осваивают в лаборатории
аналитической химии потенциометрический, кондуктометрический, хро-
нокондуктометрический, высокочастотный, полярографический, амперо-
метрический, кулонометрический, эмиссионный и абсорбционные методы
спектрального анализа в видимой, ультрафиолетовой и инфракрасной
областях спектра, а также радиометрические, хроматографические и
другие методы анализа, и в том числе методы титрования неводных
растворов и методы анализа редких элементов, которые изложены в этой
книге.
ГЛАВА I
основы физико-химических
(ИНСТРУМЕНТАЛЬНЫХ)
МЕТОДОВ АНАЛИЗА
§ 1. Особенности физико-химических методов анализа
Выполнение количественных определений весовым и объемным
(титриметрическим) методами химического анализа иногда связано с
большими трудностями (см. книга 2, «Введение», § 3), главными из
них являются:
1) необходимость предварительного отделения определяемой части
от примесей;
2) сравнительно небольшая чувствительность, ограничивающая
применение классических методов для анализа малых количеств
определяемых элементов;
3) большие затраты времени (особенно в весовом методе) на
проведение полного анализа.
Физико-химические методы отличаются повышенной по сравнению
с классическими методами чувствительностью и избирательностью,
поэтому для анализа физико-химическими методами, как правило,
требуется незначительное количество анализируемого вещества, а
содержание определяемого элемента в образце может быть чрезвычайно мало.
При выполнении анализа физико-химическими методами во многих
случаях отпадает необходимость отделения определяемых компонентов
от других составных частей анализируемого вещества, а также
необходимость применения индикаторов. Для проведения анализа
физико-химическими методами иногда требуется несколько минут.
Таким образом, физико-химические методы анализа отличаются
эксирессностью, избирательностью, высокой чувствительностью.
§ 2. Области применения инструментальных методов анализа
Инструментальные методы анализа используются в различных
областях науки и техники. Можно назвать следующие примеры
применения этих методов:
химико-аналитический контроль с целью обеспечения
оптимальности химико-технологических процессов, автоматизации и сбора
необходимой информации о состоянии. отдельных звеньев физико-химических
и технологических процессов;
выполнение научно-исследовательских работ в области химии и
химической технологии с целью получения объективной информации о
течении реакций, оценки выходов продукции и чистоты получаемых
соединений, определения побочных продуктов, наличия примесей в
исходных, промежуточных и конечных продуктах реакции, изучения
свойств и строения веществ и т. п.;
экспериментальная проверка теоретических положений и
разработка новых теорий в различных областях химической науки;
20 ГЛ. L ОСНОВЫ ФИЗИКО-ХИМИЧЕСКИХ (ИНСТРУМЕНТАЛЬНЫХ) МЕТОДОВ АНАЛИЗА
анализ веществ высокой чистоты и специальных технических
материалов, применяемых в различных областях новой техники;
безындикаторное (инструментальное) титрование водных и
неводных растворов;
массовые анализы минералов, силикатов, разнообразных полезных
ископаемых неорганического и органического происхождения,
метеоритов, редких и рассеянных элементов, металлов, сплавов, неметаллов,
монокристаллов.
§ 3. Анализ веществ высокой чистоты
Развитие разнообразных областей химии, физики,
радиоэлектроники, атомной энергетики, лазерной техники и других отраслей новой
техники, в которых используются вещества высокой чистоты,
неразрывно связано с применением высокочувствительных методов анализа
металлов, неметаллов и их соединений, сплавов, интерметаллических
соединений, люминофоров, мономерных и полимерных органических
соединений и т. д.
Самые незначительные примеси (порядка Ю-5—10_8%)
посторонних элементов или их соединений делают материалы непригодными для
применения их в новой технике. Например, присутствие в специальных
сплавах миллионных долей процента примесей некоторых элементов
резко снижает их качество; незначительные посторонние включения
делают многие металлы очень хрупкими, тогда как после тщательной
очистки эти металлы становятся вязкими, ковкими и пластичными.
Содержание в полупроводниковых материалах из особо чистых элементов
и их соединений самых минимальных количеств посторонних элементов
приводит к полной непригодности их для радиоэлектроники; так
в кремщш и германии, применяемых в производстве электронных
приборов, содержание посторонних примесей не должно превышать 10~7%,
а в некоторых случаях не должно превышать одного атома примеси на
миллиард атомов кремния или германия.
Бурно развивающаяся новая техника потребовала быстрого
совершенствования методов анализа. Однако классические методы анализа
вследствие их малой чувствительности часто оказываются совершенно
непригодными для определения малых количеств примесей. Возникшая
проблема разработки методов определения ультрамалых количеств
примесей оказалась практически разрешенной широким использованием
разнообразных физических и физико-химических методов анализа:
хроматографии, ионного обмена, экстракции, спектроскопии,
люминесцентного анализа, полярографии, рентгеноскопии, масс-спектрометрии,
радиометрических, кинетических и других методов анализа, основанных
на применении прецизионных физических и физико-химических
приборов.
Чувствительность и точность определений. По чувствительности
первое место занимают масс-спектральный и радиоактивационный
методы анализа. За ними следуют широко применяемые спектральный;
спектрофотометрический и полярографический методы.
Для сравнения, приведем чувствительности определения некоторых
элементов различными методами: объемным можно легко определить
около Ю-"1 %; весовым около 10_2%; спектроскопическим и
фотоколориметрическим 10~3—10~5%; флуорометрическим 10~6—10~7%;
кинетическими 10~6—10~8%; радиохимическими Ю-8—10_9%; методом
нейтронного активационного анализа определяют многие примеси в
количествах, менее 10^8—10-9%.
§ 3, АНАЛИЗ ВЕЩЕСТВ ВЫСОКОЙ ЧИСТОТЫ
2*
Например, алюминий можно определить методом амперометрического титрования
в том случае, если его количество превышает 200 мкг/мл; методом пламенной
фотометрии— 20 мкг/мл; спектральным — 0,2 мкг/мл; спектрофотометрическим —
0,002 мкг/мл; активационкым — 0,00002 мкг/мл. Другими словами, чувствительность
указанных методов превышает чувствительность метода амперометрического
титрования приблизительно в 10; 1000; 100 000 и 10 000 000 раз соответственно.
По точности многие физико-химические методы анализа уступают
классическим, и особенно весовому методу. Нередко, когда весовым и
объемным методами достигается точность, определяемая сотыми и
десятыми долями процента, при выполнении анализа физико-химическими
методами ошибки определений составляют 5—10%, а иногда и
значительно больше.
На точность определений (помимо ошибок взвешивания и объемных
измерений) в зависимости от метода анализа оказывают влияние
различные факторы. Например, на точность эмиссионного анализа
оказывают влияние:
метод взятия средней пробы анализируемого вещества;
непостоянство источника возбуждения (электрической дуги, искры,
пламени горелки);
величина ошибки фотометрического измерения;
негомогенность фотографической эмульсии (в случае
спектрографии) и т. д.
По мере уменьшения содержания примесей в данном
анализируемом объекте точность и воспроизводимость результатов их определения
снижается.
При анализе образцов, содержащих ничтожные доли определяемых
примесей, естественно, следует прибегать к наиболее чувствительным
методам анализа, позволяющим определять малые количества
загрязнений (порядка 10~6—10~8%). Но в связи с тем, что особо
чувствительные методы, как правило, являются менее точными, при анализе
высокочистых веществ приходится мириться со снижением точности
результатов анализа.
Из этого вытекает важный вывод, что не всегда следует прибегать
к особо чувствительным методам анализа, когда в этом Hef
необходимости.
Поясним это на примере. Если требуется определить с точностью
до 1СН—10~2% содержание основного компонента (составляющего,
например, 50%) в полупроводниковом материале, то в этом случае нет
необходимости пользоваться высокочувствительными масс-спектромет-
рическими, радиоактивационными или кинетическими методами
анализа. В подобных случаях нецелесообразно применять сложное и
дорогостоящее оборудование, а вполне достаточно воспользоваться весовым
или объемным методами анализа, обеспечивающими указанную
точность анализа при большом содержании определяемого элемента. С
другой стороны, нельзя определять ничтожное содержание примесей
весовым или объемным методами, которые при высокой точности
отличаются малой чувствительностью.
Помимо относительно невысокой точности многие
физико-химические методы имеют и некоторые другие недостатки. Например,
эмиссионная спектроскопия удобна лишь при проведении массовых
анализов, так как для определения того или иного элемента в образце
требуется калибровка прибора по стандартному образцу, занимающая
много времени. Ни один из физико-химических и физических методов
анализа не является универсальным.
Необходимо отметить, что, несмотря на прогресс инструментальных
методов анализа, позволяющих решать химико-аналитические задачи,
22 ГЛ. I. ОСНОВЫ ФИЗИКО-ХИМИЧЕСКИХ (ИНСТРУМЕНТАЛЬНЫХ) МЕТОДОВ АНАЛИЗА
неразрешимые обычными методами весового или объемного анализа,
классические методы анализа не утратили своего значения,
по-прежнему играют доминирующую роль и являются основой современной
аналитической химии.
Нельзя отдать предпочтение тому или иному методу анализа, не
учитывая характер исследуемого объекта, его назначение, агрегатное
состояние, концентрацию, наличие примесей, цели анализа, требуемую
точность определения, срок исполнения анализа и т. д.
Лишь овладев самыми разнообразными методами анализа и
сочетая химические, физические и физико-химические методы анализа,
химик-аналитик сможет успешно разрешить любую поставленную перед
ним химико-аналитическую задачу.
§ 4. Повышение чувствительности и точности методов
определения следов примесей
Существует несколько путей успешного решения задач определения
следов примесей:
усовершенствование существующих инструментальных методов
анализа;
разработка новых физических и физико-химических методов;
предварительное концентрирование определяемого вещества;
сочетание и комбинирование нескольких методов, в частности
электровесового анализа и полярографии, хроматографии и
полярографии, экстракции со спектральным или спектрофотометрическим
методом и т. д.
Например, введение в практику усовершенствованных
электрохимических методов позволило на несколько порядков повысить
чувствительность. Использование более мощных нейтронных потоков дает
возможность увеличить чувствительность и селективность радиоактива-
ционного анализа; повышение разрешающей способности
гамма-спектрометров приводит к увеличению чувствительности масс-спектроскопи-
ческого метода; применение низких температур (около —180* С)
увеличивает чувствительность люминесцентного определения следов
металлов и т. д.
Предварительное концентрирование примесей электрохимическими
и хроматографическими методами, а также методами соосаждения
с органическими и неорганическими соосадителями (носителями,
коллекторами), экстракцией, дистилляцией или отгонкой позволило
повысить чувствительность определения до 10~8%. Например,
концентрирование ультрамалых количеств определяемых элементов на неподвижном
электроде, сопровождающееся последующим анодным растворением,
дает возможность увеличить чувствительность полярографического
метода от Ю-5 до 10-8%.
Несколько лет назад считали, что концентрирование в 50—100 раз
является удовлетворительным. В настоящее время решается задача
более эффективного обогащения (в 100000—1000000 000 раз) с выходом
определяемого элемента, содержащегося в виде примеси в данном
анализируемом объекте, до 95—99%. Например, И. П. Алимарин с
сотрудниками, применяя метод распределительной хроматографии с
использованием в качестве неподвижной фазы фторопластового порошка и три-
бутилфосфата, сконцентрировали следы галлия в присутствии больших
количеств алюминия, при этом коэффициент обогащения составил около
107, а выход определяемого элемента (галлия) достиг более 99%.
§ 5. ИНСТРУМЕНТАЛЬНЫЕ МЕТОДЫ ТИТРОВАНИЯ
23
Известные преимущества дает сочетание и комбинирование
нескольких методов концентрирования. Например, к большей степени
обогащения микрокомпонентов, чем концентрирование индивидуальными
методами, приводит сочетание метода концентрирования следов элементов
путем соосаждения в присутствии органических и неорганических сооса-
дителей с другими методами обогащения, особенно с экстракцией.
П. Н. Коваленко с сотрудниками показали, что комбинирование
электрохимических методов отделения (электровесового метода анализа,
внутреннего электролиза и цементации) основного компонента и
последующее определение оставшихся микропримесей полярографическим,
осииллополярографическим или другими физико-химическими методам^
является одним из перспективных направлений в аналитической химии.
Помимо описанных ранее (книги 1 и 2 «Основы аналитической
химии») методов определения элементов из очень разбавленных
растворов (1 : 1018) можно привести в качестве нового примера предложенный
Т. Г. Акимовой и О. П. Елисеевой метод концентрирования кюрия,
количественно соосаждаемого в виде комплексных соединений с осадками,
образованными реагентами арсеназо I, II и III в комбинаций с
кристаллическим фиолетовым. Этим методом можно отделять кюрий от
109-кратных количеств магния.
§ 5. Инструментальные методы титрования
Большинство физико-химических методов анализа основано на
титровании, т. е. на медленном (по каплям) прибавлении измеряемого
количества реагента к анализируемому веществу, при этом ведется
наблюдение (регистрация) тех или иных свойств, изменяющихся в течение
процесса (см. книга 2, гл. I, § 1).
В качестве реагентов чаще всего используют растворы с
определенным содержанием титрующего вещества (г-экв/кг, г-экв/г, г/л, г/мл,
г-экв/мл, мг-экв/мл, г/сек, г/капля). Иногда реагентами служат твердые,
жидкие и газообразные вещества, в том числе генерируемые
фотометрически или электрометрически. Обычно применяют такой реагент,
который вступает в химическую реакцию с определяемым веществом.
Взаимодействие смешиваемых веществ может сопровождаться не
только химическими реакциями, но и физическими явлениями
(помутнением, осветлением, расслоением на две фазы, изменением
температуры, цвета и флуоресценции раствора и т. д.).
По количеству реагента, израсходованного на реакцию до конечной
точки титрования (во многих случаях эта точка практически
соответствует точке эквивалентности), рассчитывают содержание
определяемого вещества. Если конечная точка титрования не совпадет с точкой
эквивалентности, то последнюю определяют с учетом некоторого
поправочного коэффициента, вычисляемого на основании результатов
предварительных стандартных титрований или путем теоретических расчетов.
Титрование, лежащее в основе классического объемного анализа,
является практически универсальным методом, широко применяемым
в физико-химических методах анализа. В качестве примера можно
указать йотенциометрическое, хронопотенциометрическое, кондуктометриче-
ское, кулонометрическое, фотометрическое, турбидиметрическое,
термометрическое и другие виды титрования.
Физические свойства систем, используемых при различных видах
титрования в инструментальных методах анализа, приведены ниже:
появление или исчезновение эмиссионных линий спектра
определяемого элемента;
24 ГЛ. I. ОСНОВЫ ФИЗИКО-ХИМИЧЕСКИХ (ИНСТРУМЕНТАЛЬНЫХ) МЕТОДОВ АНАЛИЗА
флуоресценция (появление, исчезновение или изменейие
флуоресценции определяемого вещества, реагента, продукта реакции или
флуоресцирующего индикатора);
оптическая активность (вращение плоскости поляризации);
рефракция;
кристаллизация (появление четко различимых под микроскопом
кристаллов);
магнитная восприимчивость (чувствительность):
диэлектрическая проницаемость;
электропроводность;
теплопроводность;
электродные потенциалы;
дифракция рентгеновских лучей и электронов;
ядерный магнитный резонанс;
абсорбция лучистой энергии (ультрафиолетовое, видимое,
инфракрасное излучение);
комбинационное рассеяние света («Раман-эффект»);
вязкость;
мутность;
плотность;
поверхностное натяжение;
радиоактивность;
теплота реакции и др.
Раздельное (дифференцированное) титрование. Когда наблюдается
несколько точек эквивалентности, соответствующих последовательным
стадиям титрования, оказывается возможным за одно титрование из
одной навески определить содержание нескольких компонентов,
входящих в состав анализируемой смеси (например, H2S04 + NaHS04,
Н3РО4 + KH2P04, Na2C03 + NaHC03, HC1 + CH3COOH, НСЮ4 + НС1 +
+ C6H5COOH, фталевая + изофталевая + терефталевая кислоты и т. д.).
В таком случае метод определения называют дифференцированным
титрованием.
§ 6. Применение физико-химических методов анализа
для определения индивидуальных соединений
Принципы определения конечной точки титрования. Определение
конечной точки титрования индивидуального соединения, как правило,
основано на резком изменении концентрации его раствора вблизи точки
эквивалентности.
В объемном классическом методе анализа конец титрования
наблюдается визуально, например по появлению или исчезновению окраски
титруемого раствора в присутствии или в отсутствие цветного
индикатора, реагирующего с определяемым веществом или с реагентом.
В инструментальных методах титрования наблюдение за
изменением концентраций реагирующих реществ основано на применении
соответствующих прецизионных физических приборов.
Способы определения конечной точки титрования. Точку
экстремальных значений соответствующей физической величины после
добавлений некоторого объема титрованного раствора принимают за
конечную точку титрования. Нередко титрование продолжают до тех пор,
пока не будет достигнута идентичность какой-либо физической
характеристики титруемой смеси и сравниваемого стандартного раствора,
в котором заранее известно содержание определяемого вещества.
В процессе титрования периодически измеряют ту или иную
физическую характеристику системы (электропроводность, подвижность
§ 7. ПРИМЕНЕНИЕ ФИЗИКО-ХИМИЧЕСКИХ МЕТОДОВ ДЛЯ АНАЛИЗА СМЕСЕЙ 25
ионов, диффузионный ток, оптическую плотность), изменяющуюся
линейно по мере изменения соотношения концентраций реагирующих
веществ.
Данные измерений наносят на график как функцию количества
добавленного раствора реагента. При этом по десяти или более
показаниям прибора строят две прямые. Точку пересечения прямых принимают
за конечную точку титрования. Чем острее угол пересечения двух
прямых, тем выше точность титрования. Помимо графического метода
нахождения конечной точки титрования существует расчетный метод,
предложенный румынским ученым Литеану.
На подобные определения сильно влияют условия титрования,
поэтому следует строго придерживаться некоторых постоянных условий.
В ряде случаев известное влияние на процесс титрования оказывают и
другие факторы — природа реагента и характер образующихся в
процессе титрования веществ.
§ 7. Применение физико-химических методов
для анализа смесей веществ
Определение индивидуальных соединений не представляет особых
трудностей, значительно сложнее определять их в йрисутствии других
вешеств. Наиболее просто анализ смесей осуществляется потенциомет-
рическим, хронокондуктометрическим й другими методами
дифференцированного титрования. Весьма сильное дифференцирующее действие
оказывают неводные растворители в отношении кислот, оснований и
солей (см. гл. XI).
Инструментальные методы анализа широко применяются для
анализа смесей разнообразных неорганических и органических веществ.
§ 8. Классификация инструментальных количественных
методов анализа
Все методы количественного анализа делятся на химические,
физические и физико-химические.
К химическим методам анализа относятся весовой, объемный и
газовый анализ (см. книга 2, «Введение», § 2).
Физические и физико-химические методы анализа подразделяются
на следующие группы (см. книга 2, гл. VI): электрохимические;
спектральные (оптические); хроматографические; радиометрические; масс-
спектрометрические.
§ 9. Электрохимические методы анализа
К группе электрохимических методов анализа относятся
следующие виды анализа.
Электровесовой анализ основан на выделении из растворов,
электролитов веществ, осаждающихся на электродах при прохождении
через растворы постоянного электрического тока. Выделившийся при
электролизе металл (или окись) взвешивают на аналитических весах
и по массе осадка судят о содержании определяемого вещества
в растворе.
Выделение на электродах различных веществ (металлов,
неметаллов, окислов и т. п.) вследствие прохождения тока объясняется
окислением восстановителей (20Н~ ~2е> 02 + 2Н+; 2СГ-=:-^-> Cl2 и т. п.)
на аноде (положительный полюс) и восстановлением окислителей
26 ГЛ. U ОСНОВЫ ФИЗИКО-ХИМИЧЕСКИХ (ИНСТРУМЕНТАЛЬНЫХ) МЕТОДОВ АНАЛИЗА
(Cu++—^->Cu; 2H+-i^->H2 и т. п.) на катоде
(отрицательный полюс).
Соотношение между количеством электричества, прошедшего через
раствор электролита, и массой образовавшихся продуктов окисления
и восстановления, которые выделяются на электродах, определяется
законом Фарадея.
Метод внутреннего электролиза (разновидность электровесового
анализа) основан на использовании электрического тока, возникающего
при погружении в раствор, например, CuS04, двух электродов,
составляющих гальваническую пару, например Zn и Pt.
В этом случае электрический ток не поступает от внешнего
источника, а возникает благодаря разности потенциалов между платиновым
электродом", на котором выделяется определяемый металл (медь), и
другим электродом, на котором происходит анодный процесс
растворения цинка.
Другими словами, процесс возникновения тока сопровождается
химическим превращением, вследствие которого на платиновом
электроде осаждается определяемый металл (например, медь) в результате
восстановления Си++-ионов, а эквивалентное количество цинка
растворяется в результате окисления с образованием Zii++-hohob.
Полярография основана на измерении силы тока, изменяющейся
в зависимости от величины напряжения в процессе электролиза, в
условиях, когда один из электродов (катод) имеет очень малую
поверхность (поляризующийся электрод), а другой (анод)—большую (непо-
ляризующийся электрод). Поляризующимся катодом являются капли
ртути, вытекающие из тонкого отверстия капиллярной трубки, а также
платиновый (вращающийся), графитовый, серебряный и другие
электроды. Неполяризующимся анодом является «донная» ртуть или
стандартные электроды сравнения с большой поверхностью. Силу тока, при
которой достигается полный разряд всех ионов анализируемого
вещества, поступающих в приэлектродное пространство вследствие
диффузии, называют предельным диффузионным током. Величина этого тока
пропорциональна исходной концентрации определяемого вещества
(ионов) в растворе.
Амперометрическое титрование (полярометрическое, вольтамперное
титрование), являющееся разновидностью полярографического анализа,
основано на изменении в процессе титрования раствора определяемого
вещества величины предельного диффузионного тока, проходящего
через раствор при постоянном напряжении между индикаторным
поляризующимся электродом и неполяризующимся электродом сравнения.
Кулонометрия основана на измерении количества электричества,
израсходованного на электролиз определенного количества вещества
при постоянном потенциале, который соответствует потенциалу
выделения данного элемента. В основе этого метода лежит закон Фарадея.
Метод титрования, в котором точка эквивалентности соответствует
моменту, когда сила тока электролиза достигает величины «фонового»
тока, называют кулонометрическим титрованием. Обычно сила фонового
тока равна нулю, так как раствор в этот момент не содержит
заряженных частиц (см. гл. VI).
Кондуктометрия основана на измерении электропроводности
анализируемых растворов, изменяющейся в результате химических реакций
и зависящей от природы электролита, его температуры и концентрации
раствора.
Метод титрования, при котором точку эквивалентности фиксируют
по пересечению двух прямых, отражающих изменение эквивалентной
§ 10. СПЕКТРАЛЬНЫЕ (ОПТИЧЕСКИЕ) МЕТОДЫ АНАЛИЗА 27
электропроводности исследуемого раствора по мере прибавления тит-
ранта в процессе титрования, называют кондуктометрическим
титрованием.
Хронокондуктометрическое титрование является разновидностью кон-
дуктометрического титрования, при котором о содержании вещества
судят по времени, израсходованному на кондуктометрическое
титрование анализируемого образца при постоянной скорости истечения
титранта.
Высокочастотное титрование (осциллометрия) является
разновидностью кондуктометрического титрования. В случае высокочастотного
титрования исследуемый раствор помещают в высокочастотное
электромагнитное поле измерительного прибора, а затем в этот раствор из
бюретки или другим способом постепенно приливают раствор титранта,
реагирующего с определяемом веществом; электроды укрепляют вне
анализируемого раствора непосредственно у стенок ячейки и повышают
частоту переменного тока до нескольких тысяч мегагерц.
Высокочастотное Титрование вследствие его особенностей иногда называют без-
контактной кондуктометрией, так как исследуемый раствор не имеет
гальванического контакта ни с электродами, ни с катушками
индуктивности— источником осциллирующего магнитного поля.
В процессе высокочастотного титрования можно измерять
электропроводность или диэлектрическую и магнитную проницаемость.
Разновидность высокочастотного титрования, при котором
измеряют диэлектрическую проницаемость растворов, называют диэлкомет-
рическим титрованием.
Потенциометрия основана на измерении потенциала электрода,
погруженного в анализируемый раствор, изменяющегося в результате
химических реакций и зависящего от температуры и концентрации
раствора.
Метод титрования, при котором точку эквивалентности
устанавливают по резкому скачку потенциала электрода, погруженного в
анализируемый раствор, называют потенциометрическим титрованием.
Хронопотенциометрическое титрование является разновидностью по-
тенциометрического титрования, при котором о содержании вещества
судят по времени, израсходованному на потенциометрическое титрование
анализируемого образца при постоянной скорости прибавления
титранта.
§ 10. Спектральные (оптические) методы анализа
К группе спектральных (оптических) методов анализа относятся
следующие методы.
Эмиссионный спектральный анализ — физический метод,
основанный на изучении эмиссионных спектров паров анализируемого вещества
(спектров испускания или излучения), возникающих под влиянием
сильных источников возбуждений (электрической дуги, высоковольтной
искры); этот метод дает возможность определять элементный состав
вещества, т. е. судить о том, какие химические элементы входят в состав
данного вещества.
Пламенная спектрофотометрия, или фотометрия пламени,
являющаяся разновидностью эмиссионного спектрального анализа, основана
на изучении эмиссионных спектров элементов анализируемого вещества,
возникающих под влиянием мягких источников возбуждения. В этом
методе анализируемый раствор распыляют в пламени. Этот метод дает
возможность судить о содержании в анализируемом образце главным
28 ГЛ. I. ОСНОВЫ ФИЗИКО-ХИМИЧЕСКИХ (ИНСТРУМЕНТАЛЬНЫХ) МЕТОДОВ АНАЛИЗА
образом щелочных и щелочноземельных металлов, а также некоторых
других элементов, например галлия, индия, таллия, свинца, марганца,
меди, фосфора.
Примечание. Кроме эмиссионной фотометрии пламени применяют
абсорбционную, называемую также атомно-абсорбционной спектроскопией или атомно-аб-
сорбционпой спектрофотометрией. Она основана на способности свободных атомов
металла в газах пламени поглощать световую энергию при характерных для каждого
элемента длинах волн. Этим методом можно определять сурьму, висмут, селен, цинк,
ртуть и некоторые другие элементы, не определяемые методом эмиссионной
фотометрии пламени.
Абсорбционная спектроскопия основана на изучении спектров
поглощения вещества, являющихся его индивидуальной характеристикой.
Различают спектрофотометрический метод, основанный на определении
спектра поглощения или измерении светопоглощения (как в
ультрафиолетовой, так и в видимой и инфракрасной областях спектра) при строго
определенной длине волны (монохроматическое излучение), которая
соответствует максимуму кривой поглощения данного исследуемого
вещества, а также фотоколориметрический метод, основанный на
определении спектра поглощения или измерении светопоглощения в видимом
участке спектра.
В отличие от спектрофотометрии в фотокцлориметрическом методе
применяют «белый» свет или «белый» свет, предварительно
пропущенный через широкополосные светофильтры.
Метод анализа по спектрам комбинационного рассеяния света.
В методе использовано явление, открытое одновременно советскими
физиками Г. С. Ландсбергом и Л. И. Мандельштамом и индийским
физиком Ч. В. Раманом. Это явление связано с поглощением веществом
монохроматического излучения и последующим испусканием нового
излучения, отличающегося длиной волны от поглощенного.
Турбидиметрия основана на измерении интенсивности света, no-
глотаемого неокрашенной суспензией твердого вещества. В турбиди-
метрии интенсивность света, поглощенного раствором или прошедшего
через него, измеряют так же, как в фотоколориметрии окрашенных
растворов.
Нефелометрия основана на измерении интенсивности света,
отраженного или рассеянного окрашенной или неокрашенной суспензией
твердого вещества (взвешенного в данной среде осадка).
Люминесцентный, или флуоресцентный, метод анализа основан на
измерении интенсивности излучаемого веществами видимого света
(флуоресценции) при облучении их ультрафиолетовыми лучами.
К оптическим методам анализа также относятся
рефрактометрический метод, основанный на измерении коэффициента преломления, и
полярометрический, основанный на изучении вращения плоскости
поляризации. Работы по этим двум методам не предусмотрены программой
курса аналитической химии, поэтому они подробно не рассматриваются
в книге.
§ 11. Хроматографические методы анализа
По механизму разделения различают несколько видов хроматогра-
фических методов анализа.
Адсорбционная жидкостная хроматография основана на
избирательной адсорбции (поглощении) отдельных компонентов
анализируемой смеси в жидкой среде. Она обусловлена различной адсорбируе-
мостью растворенных компонентов.
§ 12. РАДИОМЕТРИЧЕСКИЕ МЕТОДЫ АНАЛИЗА
29-
Адсорбционная газовая хроматография основана на использовании
различия в адсорбируемости газов и паров. В зависимости от основного
фактора, определяющего разделение, различают следующие виды
газовой хроматографии: газо-жидкостную и газо-адсорбционную.
Эти виды хроматографии приобрели очень важное значение в тех
областях промышленности, где требуется разделение сложных смесей
газов и жидкостей.
Распределительная хроматография основана на использовании
различия в распределении (сорбируемости) отдельных компонентов
анализируемой смеси между двумя несмешивающимися жидкими фазами —
подвижным и неподвижным растворителями.
Тонкослойная хроматография представляет собой разновидность
распределительной хроматографии, осуществляемой на пластинках,
покрытых тонким слоем носителя (окись алюминия, кизельгур, силикагель
и др.), который удерживает неподвижный растворитель.
Бумажная хроматография — разновидность распределительной
хроматографии, в которой носителем для неподвижного растворителя
являются полоски или листы фильтровальной бумаги, не содержащей
минеральных примесей.
Ионообменная хроматография основана на использовании
ионообменных процессов, протекающих между подвижными ионами адсорбента
и ионами электролита, содержащимися в анализируемом растворе. По
способам выполнения ионообменную хроматографию делят на
фронтальную, вытеснительную и элюентную (см. ниже).
Осадочная хроматография основана на использовании химических
реакций, сопровождающихся образованием малорастворимых веществ
(осадков). Эти реакции протекают между отдельными компонентами
анализируемого раствора со специальными реагентами, нанесенными на
поверхность носителя.
Окислительно-восстановительная хроматография основана на
использовании разных скоростей окислительно-восстановительных
реакций между реагентом, находящимся в колонке, и ионами,
содержащимися в анализируемом растворе. Разделение веществ определяется
величинами соответствующих окислительно-восстановительных
потенциалов взаимодействующих систем.
Адсорбционно-комплексообразовательная хроматография основана
на использовании реакций комплексообразования, протекающих между
комплексообразующими и хроматографируемыми компонентами.
Разделение обусловлено различием констант нестойкости образуемых
комплексных соединений.
§ 12. Радиометрические методы анализа
В анализе используются следующие радиометрические методы.
Метод прямого радиометрического определения основан на
осаждении определяемого элемента в виде малорастворимого соединения
избытком реагента заданной концентрации, меченного радиоактивным
изотопом с известной удельной активностью.
Радиометрическое титрование основано на образовании ионами
определяемого элемента с титрантом малорастворимого или легко
экстрагируемого соединения. Точку эквивалентности определяют, измеряя
радиоактивность раствора по мере прибавления титранта.
Метод изотопного разбавления основан на разбавлении раствора
соединения, меченного радиоактивным изотопом, неактивным
компонентом смеси. При этом удельная активность соединения, меченного
30 ГЛ. I. ОСНОВЫ ФИЗИКО-ХИМИЧЕСКИХ (ИНСТРУМЕНТАЛЬНЫХ) МЕТОДОВ АНАЛИЗА
радиоактивным изотопом, уменьшается. После выделения
определяемого вещества измеряют его радиоактивность. Зная начальную и
конечную удельные активности, легко вычислить содержание определяемого
вещества.
Активационный анализ основан на образовании радиоактивных
изотопов из стабильных изохопов определяемого элемента,
подвергнутого облучению ядерными частицами. При этом активность
образовавшегося радиоактивного изотопа пропорциональна числу атомов
определяемого элемента.
Фотонейтронный метод основан на образовании нейтронов под
воздействием фотонов высокой энергии на ядра химических
элементов. Выделяющиеся нейтроны регистрируют с помощью нейтронных
счетчиков.
Метод определения содержания элементов по излучению их
естественно-радиоактивных изотопов (например, 40К) основан на
сопоставлении радиоактивностей анализируемого образца и эталона или серии
эталонов с известным содержанием определяемого элемента,
§ 13. Масс-спектрометрические методы анализа
Масс-спектрометрические методы анализа основаны на
определении отдельных ионизированных атомов, молекул и радикалов
посредством разделения потоков ионов, содержащих частицы с разным
отношением массы к заряду, в результате комбинированного действия
электрического и магнитного полей (см. книга 2, гл. VI, § 5).
Масс-спектрометрические методы анализа широко применяются в
различных областях промышленности, науки и новой техники и дают
возможность установить изотопный состав и исследовать состав
продуктов реакций, содержание микропримесей в особо чистых веществах
и т. д. Но так как работы по масс-спектрометрии не предусмотрены
учебной программой по аналитической химии, в данной книге эти
методы не рассматриваются.
§ 14. Физико-химический анализ по Н. С. Курнакову
Метод, предложенный Н. С, Курнаковым, позволяет изучать
физические свойства систем в зависимости от их химического состава.
Например, для аналитических целей могут быть использованы кривые
зависимости температуры плавления от состава свинцово-оловянного
сплава. Этот метод называется физико-химическим анализом. Не
следует смешивать понятия «физико-химический метод анализа» с
понятием «физико-химический анализ».
Если в процессе нагревания или охлаждения исследуемого
вещества в анализируемом объекте не наблюдаются фазовые превращения,
связанные с выделением или поглощением тепла, то кривые нагревания
или охлаждения характеризуются плавным ходом. Если же в системе
происходят фазовые превращения, то на кривой изменения температур
в зависимости от характера этих превращений на протяжении
некоторого промежутка времени наблюдаются горизонтальные участки при
неизменной температуре или резкие перегибы кривой. Подобная кривая
охлаждения дает возможность судить о всех фазовых превращениях,
происходящих в исследуемом образце в процессе охлаждения.
Поскольку подробное описание метода Н. С. Курнакова, иначе называемого
термическим анализом, не входит в нашу задачу, отсылаем студентов
к соответствующим руководствам по физической химии.
§ 15. ДРУГИЕ МЕТОДЫ АНАЛИЗА
31
§ 15. Другие методы анализа
В последнее время наряду с известными электрохимическими,
оптическими, хроматографическими, радиометрическими, масс-спектро-
метрическими методами анализа широко применяются и другие методы;
некоторые из них описаны ниже.
Метод электронного парамагнитного резонанса (ЭПР), основанный
на использовании явления резонансного поглощения электромагнитных
волн парамагнитными частицами в постоянном магнитном поле,
успешно применяется для измерения концентрации парамагнитных
веществ, исследования окислительно-восстановительных процессов,
изучения химической кинетики и механизма химических реакций и т. п.
Метод ядерного магнитного резонанса (ЯМР) основан на
использовании резонансного поглощения электромагнитных волн исследуемым
веществом в постоянном магнитном поле, обусловленного ядерным
магнетизмом. Метод ЯМР применяется для исследования комплексных
соединений, состояния ионов в растворе, для изучения химической
кинетики и т. п.
Метод ядерной гамма-резонансной (ЯГР) спектроскопии,
основанный на наблюдении мессбауэровского эффекта. Метод ЯГР-спектроско-
пии применяется для установления характера химических связей в
веществах, а также для исследования комплексных и элементоорганиче-
ских соединений и т. п.
Ультразвуковые методы.
Модифицированные рентгеновские методы.
Эти и другие специальные методы используются не только для
анализа и исследования различных природных и технических объектов, но
и для изучения разнообразных реакций и процессов, протекающих
в экстремальных условиях (давления, температуры, радиации и т. п.).
Развитию таких методов в значительной мере способствовал прогресс
в области химической технологии производства важнейших специальных
материалов, атомной энергетики, космонавтики, биологии, биохимии
и т. п.
Внедрение в практику современных физических и
физико-химических методов анализа и исследования оказало существенное влияние
на развитие различных областей техники и науки, в том числе на раз-
витие самой аналитической химии.
ГЛАВА И
ПОТЕНЦИОМЕТРИЯ И ПОТЕНЦИОМЕТРИЧЕСКОЕ
ТИТРОВАНИЕ
А, ТЕОРЕТИЧЕСКИЕ ОСНОВЫ МЕТОДА
§ 1. Зависимость величины электродных потенциалов
от концентрации (активности)
Количественная зависимость между концентрацией (активностью)
компонентов обратимой окислительно-восстановительной ред-окс
системы и величиной электродного потенциала выражается уравнением
(см. книга 1, гл. II, § 4 и 6):
р-рО #Г [Окисл]
^-^Окисл/Восст-*- пр ш [Восст] {{)
ИЛИ
с __ dO , " 1 аОкисл , . ч
? ~ ^Окисл/Восст "*" "TF" ш ~ Ua)
Если перейти от натуральных логарифмов к десятичным и
объединить все постоянные величины, то получим:
О а
E==z ^Окисл/Воссг + ~Г ^ "^ (1б)
п аВосст
гдеО = -^- 2,303 = 0,0001983 Г = 0,0591+0,0002 (^ — 25°G); значения О
при разных температурах см. Лурье Ю. Ю., Справочник по
аналитической химии, Изд. «Химия», 1967.
В общем случае, когда в окислительно-восстановительной реакции
принимает участие более двух компонентов с различными
коэффициентами
aA + bB + dD+ ... +пе ^z± mM + qQ + ...
уравнение (16) записывается в следующем виде:
?-4.B,D..-/M,Q....+-'g A*° (2)
Л «m^q •••
Кроме ионов окислителя и восстановителя ред-окс системы в
реакциях могут участвовать ионы водорода (Н+), молекулы тех или иных
веществ (например, Н20). Участвующие в реакциях компоненты могут
представлять собой твердую фазу (металл электрода, окись металла,
малорастворимая соль, покрывающая электрод, осадок) или
газообразное вещество. Тогда в уравнение (2) не вводятся активности
(концентрации) тех компонентов, для которых они постоянны (т. е. твердой
фазы, газообразного вещества, если оно насыщает раствор при давлении
1 атм, и воды, концентрация которой в процессе реакции мало
изменяется, а поэтому может считаться постоянной).
Следует отметить, что большинство табличных величин
стандартных (нормальных) потенциалов электродных реакций вычислено из
§ I. ЗАВИСИМОСТЬ ВЕЛИЧИНЫ ПОТЕНЦИАЛОВ ОТ КОНЦЕНТРАЦИИ (АКТИВНОСТИ) 33
термодинамических данных. Они не могут быть всегда
экспериментально установлены измерением э. д. с. (электродвижущей силы)
соответствующих гальванических элементов из-за необратимости многих
окислительно-восстановительных систем. С другой стороны, хотя
величины стандартных потенциалов являются бесспорно ценной
информацией для решения ряда специфических химических задач, но в практике
аналитической химии гораздо важнее знать формальные (реальные)
потенциалы (?ф). На самом деле трудно знать с достаточной точностью
активности участвующих в электродной реакции компонентов для
вычисления потенциала по формуле (16) и суждения об
окислительно-восстановительной силе ред-окс системы в этих условиях. Обычно
известны лишь общие концентрации этих компонентов, на активности
которых сказывается влияние различных факторов (ионная сила и рН
раствора, ассоциация, комплексообразование и пр.). Поэтому если
в уравнении (16) заменить активности компонентов соответствующими
общими концентрациями, умноженными на некоторые постоянные
величины (/), отражающие любое отклонение в конкретных реальных
условиях активностей компонентов от общей их концентрации в
растворе, то получим:
F-fO ¦ » ,, Уокнсл . * . [°КИСЛ1
? - ^Окисл/Восст + п !ё ^Восст "f n lg [BoCCT]
Объединение двух первых членов правой части уравнения дает:
Ф п б [Восст]
При общей концентрации [Окисл] = [Восст] = 1 для ред-окс системы
Е=Еф. Формальные потенциалы легко поддаются экспериментальному
определению и позволяют судить об электрохимическом и химическом
поведении ред-окс систем в заданных условиях. Из сказанного
вытекает, что каждая ред-окс система характеризуется лишь одним
значением стандартного потенциала, но многочисленными формальными
потенциалами в зависимости от условий среды.
Как известно, возникновение потенциала обусловлено обменом
электронами окислителем и восстановителем на электроде. Когда
скорости этих двух процессов становятся равными, достигается
динамическое равновесие, при котором в единицу времени восстановитель отдает
столько электронов электроду, сколько окислитель отнимает у него. При
таком состоянии приобретаемый электродом потенциал называется
равновесным потенциалом. Скорости этих двух обратно направленных
процессов пропорциональны активностям участвующих в электродной
реакции компонентов. Чем больше скорость обмена электронами при
равновесном потенциале электрода, тем быстрее устанавливается и
устойчивее потенциал. Ред-окс системы, в которых скорость
электродных процессов большая, называются обратимыми. Достаточно
незначительного изменения потенциала электрода от равновесного состояния
в таких ред-окс системах (например, наложением извне некоторого
напряжения), чтобы вызвать увеличение скорости электроокисления или
электровосстановления их соответствующих компонентов.
Весьма малыми скоростями обмена электронами обладают
компоненты ред-окс систем, называемых необратимыми. Равновесные
потенциалы таких систем устанавливаются медленно (во времени) и
неустойчивы («дрейфуют»), так как они подвержены влиянию посторонних
факторов. Для того чтобы процессы окисления и восстановления
соответствующих компонентов подобных ред-окс систем протекали с доста*
34 ГЛ, II. ПОТЕНЦИОМЕТРИЯ И ПОТЕНЦИОМЕТРИЧЕСКОЕ ТИТРОВАНИЕ
точной скоростью, необходимо заметно сдвинуть потенциал электрода
от его равновесного значения. Иначе говоря, эти процессы в данном
случае протекают с более или менее большим перенапряжением, т. е.
Ai/AE (приращение силы тока, которое вызывается приращением
потенциала электрода, пропорциональное скорости этих процессов), мало,
между тем как у обратимых ред-окс систем эта величина значительна.
Практическим критерием обратимости или необратимости ред-окс
системы является точность, с которой уравнением (2) описывается
зависимость равновесных потенциалов электродов от активностей
компонентов, участвующих в электродных реакциях.
С другой стороны, электрод вообще стремится приобрести
некоторый потенциал и в том случае, когда раствор не содержит электроно-
активных веществ (т. е. таких компонентов ред-окс систем, которые
способны в данных условиях окисляться и восстанавливаться).
Например, в растворе перхлората натрия (неэлектроактивное вещество)
потенциал вызван крайне небольшим количеством электронов,
обмениваемых в единицу времени различными примесями, растворенным
кислородом, диффундирующим из воздуха, а также обусловлен медленным
разрядом ионов и молекул растворителя (обычно воды) и т. п. Так как
эти факторы непостоянны, то и приобретаемый электродом потенциал
неустойчив, медленно устанавливается, на него влияют скорость
перемешивания раствора, положение в растворе, величина, состояние
поверхности и материал электрода и т. п. Возникающий при этих
условиях потенциал называется смешанным, так как он обусловлен участием
в электродных процессах различных не поддающихся учету веществ,
в отличие от равновесного потенциала, который приобретает электрод
при участии в электрохимической реакции обоих компонентов одной и
той же ред-окс системы.
Эти факторы действуют и при возникновении равновесных
потенциалов независимо от того, обратима или необратима ред-окс система.
Видимое их влияние, однако, сказывается лишь на потенциале
необратимых ред-окс систем, так как только в этом случае скорости обоих
электродных процессов соизмеримы.
Смешанные потенциалы возникают и тогда, когда раствор
содержит в большом количестве лишь один компонент обратимой ред-окс
системы или когда вместе с ним в растворе присутствует его
сопряженная форма в весьма малых концентрациях (<Л0-5 М).
Из уравнений (1) или (2) следует, что при полном отсутствии
одного компонента ред-окс системы (гипотетический случай), потенциал
электрода должен был бы иметь величину, равную ±оо. Между тем
на практике всегда наблюдается некоторое конечное предельное
значение потенциала, которое, хотя и невозможно рассчитать по этим
формулам, но можно установить экспериментально.
В этом случае в возникновении потенциала электрода взамен
отсутствующей формы ред-окс пары принимают участие примеси,
растворитель и т. д., как и при образовании смешенных потенциалов. В конце
концов наступает равновесие, при котором скорости превращения
участвующих в данном процессе веществ становятся равными. При этом
образуется некоторое весьма малое количество отсутствующей
сопряженной формы ред-окс системы. Все это является причиной резкого
сдвига потенциала от бесконечного значения и установления некоторого
равновесного потенциала [не поддающегося вычислению по формуле
(1)], величину которого, как было сказано выше, можно найти
экспериментально. Но так как и в этом случае скорость электродной реакции
лимитирована малой концентрацией сопряженного компонента и влия-
§ 1. ЗАВИСИМОСТЬ ВЕЛИЧИНЫ ПОТЕНЦИАЛОВ ОТ КОНЦЕНТРАЦИИ (АКТИВНОСТЩ 35
нием других указанных выше факторов, то равновесный потенциал снова
устанавливается медленно и остается неустойчивым. Этот частный
случай смешанного потенциала принято называть предельным
потенциалом.
Предельный (смешанный) потенциал всегда возникает при потен-
циометрическом титровании в точке эквивалентности и вблизи нее,
когда концентрации некоторых форм двух химически реагирующих
ред-окс систем становятся исчезающе малыми (-<10""5 М), а также при
использовании таких химических реакций, в которых принимает
участие лишь одна сопряженная форма ред-окс системы [например, при
потенциометрическом титровании гексацианоферрата (II) ионами
свинца, если в растворе отсутствует гексацианоферрат (III)]. В последнем
случае достаточно внести в раствор другую сопряженную форму ред-
окс системы (при условии, что она не принимает участия в химической
реакции), чтобы электрод быстро приобрел устойчивый равновесный
потенциал. Из сказанного вытекает, что нецелесообразна попытка
измерять предельные потенциалы электродов при потенциометрических
титрованиях, особенно потенциалы в точке эквивалентности или
вблизи нее.
Стандартные (нормальные) окислительно-восстановительные
электродные потенциалы. Измерение потенциала отдельного электрода
практически неосуществимо, тогда как измерение э. д. с.
гальванического элемента, состоящего из двух полуэлементов, не
представляет сложности. Поэтому если в гальванических элементах принять
один и тот же произвольно выбранный полуэлемент, а в качестве
второго использовать электрод в различных ред-окс системах в
стандартных условиях, т. е. когда активность каждого из участвующих в
электродной реакции компонентов равна единице, то измеренные э. д. с.
позволяют судить об относительных величинах потенциалов этих
электродов (полуэлементов). Электрод, относительно которого измеряют
потенциал других электродов, принято называть электродом сравнения.
В качестве стандартного электрода сравнения принят нормальный
водородный электрод (НВЭ), стандартный потенциал (Е°) которого
условно приравнен нулю при любой температуре и давлении водорода
1 атм. В водородном электроде протекает реакция 2Н++2е ^ f H2.
Зависимость электродного потенциалу водородного электрода от
активности ионов водорода выражается уравнением:
Стандартный (нормальный) водородный электрод представляет
собой платинированный платиновый электрод*, погруженный в
насыщенный химически чистым водородом под давлением 1 атм водный
раствор, активность ионов водорода в котором равна единице (1 н. раствор
H2S04).
Таким образом, IgaH+B приведенной выше формуле равняется нулю,
а Е° принят равным нулю, поэтому Е для НВЭ также равен нулю.
Приведенные в справочниках и учебниках стандартные потенциалы
различных ред-окс систем представляют собой э. д. с. гальванических
элементов, в которых одним из полуэлементов постоянно является
НВЭ, а другим — испытуемая система, активность каждого из
участвующих в электродной реакции компонентов которой равна единице.
По предложению Международной ассоциации ученых по чистой и
* Проволока или пластинка из платины, электролитически покрытая
высокодисперсной платиной, так называемой платиновой чернью.
36 ГЛ. И. ПОТЕНЦИОМЕТРИЯ И ПОТЕНЦИОМЕТРИЧЕСКОЕ ТИТРОВАНИЕ
прикладной химии (IUPAC) стандартным электродным потенциалом
Е° следует считать потенциал полуэлементов, электрохимический
процесс в которых описывается уравнением реакции восстановления,
например:
Си++ + 2е ^=± фСа
Zn++ + 2e ^=± |Zn
Ag+ + e ^± |Ag
|AgCl+* ^z±r l-Ag + СГ
Fe+++ + e +± Fe++
МпСГ + 8Н+4-5г ^z±
4
Mn++
+ 4H20
^Cu++/Cti
*Zn++/Zn
Ag+/Ag
AgCl/Ag, СГ
?Fe+++/Fe++
MnO", Н+/Мп++, Н20
§ 2. Применение потенциометрического метода
анализа
Различают прямую потенциометрию и потенциометрическое
титрование.
Первый метод предполагает измерение абсолютного и точного
значения электродного потенциала (точнее, э. д. с. гальванического
элемента), с помощью которого можно установить активность потенциал-
определяющего иона в растворе, пользуясь уравнением (1а).
Наиболее важное значение этот метод имеет при определениях активности
иона водорода (%+), или рН растворов (рН= — lgaH+). Определение
активности других потенциалопределяющих ионов представляет большой
интерес для расчета различных термодинамических констант
электрохимических и химических равновесий; при этом предполагается, что во
всех случаях работают с обратимыми электрохимическими реакциями
и что измеренные Е соответствуют активностям
потенциалопределяющих компонентов.
Мы не останавливаемся на этих определениях, так как они
представляют собой большой самостоятельный раздел потенциометрического
метода анализа, изложенный в отдельных монографиях и учебниках.
§ 3. Потенциометрическое титрование
Потенциометрическое титрование преследует чисто прикладную
цель количественного определения данного вещества в растворе путем
его титрования стандартным раствором соответствующего реагента. При
титровании в исследуемый раствор опускают индикаторный электрод,
возникновение потенциала на котором обусловливается определяемым
веществом непосредственно (если оно электроактивно) или косвенно
(если оно неэлектроактивно) в результате химического взаимодействия
его с каким-либо другим потенциалопределяющим компонентом. В
процессе химической реакции (например, титрования) за изменением
концентрации определяемого вещества следят по изменению потенциала
индикаторного электрода.
В потенциометрическом титровании могут быть использованы
реакции нейтрализации, осаждения, комплексообразования, окисления —
восстановления и применены почти все стандартные растворы, которые
известны для визуальных титриметрических методов анализа.
§ 3. ПОТЕНЦИОМЕТРИЧЕСКОЕ ТИТРОВАНИЕ
37
Преимущества потенциометрического метода титрования. Потен-
циометрическое титрование при прочих равных условиях имеет ряд
преимуществ по сравнению с визуальными титриметрическими
методами анализа. Метод потенциометрического титрования более
чувствителен, при использовании его исключается субъективная ошибка,
возникающая при визуальном нахождении момента завершения
химической реакции, т. е. конечной точки титрования. Этот метод дает
возможность определять вещества в мутных и сильно окрашенных
растворах, дифференцированно (раздельно) титровать компоненты смеси
веществ в одной и той же порции раствора и, наконец,
автоматизировать процесс титрования, так как измеряемой величиной является
электрический параметр.
Требования к реакциям, используемым в потенциометрическом
титровании. К применяемым в потенциометрическом титровании
химическим реакциям предъявляются те же требования, что и при обычном
титриметрическом анализе:
скорость химической реакции должна быть достаточно большой;
реакция должна протекать строго стехиометрически в нужном
направлении до конца;
побочные реакции должны отсутствовать.
В тех случаях, когда химические реакции не соответствуют
перечисленным требованиям, можно пользоваться теми же приемами, что
и в обычном титриметрическом методе. Если скорость реакции
небольшая или если константа равновесия недостаточно велика (особенно
при реакциях с образованием гетерогенных фаз), можно прибавить
избыток титранта к определяемому веществу и после завершения
основной реакции титровать избыток другими подходящими титрантами
(метод обратного титрования). При недостаточно больших константах
равновесия реакций титрование может быть успешным, если создать
подходящие условия, т. е. подобрать рН среды, растворитель
(применение органических растворителей), предусмотреть удаление из сферы
реакции одного из продуктов взаимодействия и т. д. Иногда
положительный эффект дает нагревание раствора.
В отличие от обычного титриметрического метода, основанного на
применении цветных индикаторов, в потенциометрическом методе
титрования индикатором является электрод, на котором протекает
индикаторная электрохимическая реакция. В первом методе показателем
достижения точки эквивалентности служит переход окраски цветного
индикатора, во втором — резкое изменение потенциала электрода
(обычно называемое скачком потенциала), связанное с возникновением
другой электрохимической реакции на поверхности раздела электрод —
раствор.
Скачок потенциала. Возникновение скачка потенциала в точке
эквивалентности или вблизи нее дает возможность найти конечную точку
титрования по кривым титрования или сам скачок принимается как
показатель момента завершения реакции. Появление скачка
обусловлено неравномерным изменением концентрации титруемого вещества и
титранта при добавлении каждый раз одинакового объема
стандартного раствора.
Наиболее резкое изменение концентрации веществ при титровании
происходит теоретически в точке эквивалентности, где равновесная
концентрация характеризуется величиной /Сравн данной химической
реакции. Поэтому максимальное изменение потенциала электрода
(скачок потенциала) должно наступить в этот же момент вследствие
логарифмической зависимости между концентрацией (активностью)
38 ГЛ. II. ПОТЕНЦИОМЕТРИЯ И ПОТЕНЦИОМЕТРИЧЕСКОЕ ТИТРОВАНИЕ
потенциалопределяющего вещества и потенциалом электрода [см.
уравнение (1)].
Далее, чем больше концентрация титруемого раствора и титранта
и чем больше разность между величинами нормальных потенциалов
электрохимических реакций до и после точки эквивалентности (при
прочих равных условиях), тем больше скачок потенциала.
Например, в растворе осуществляется химическая реакция
А + В +± | АВ, сопровождающаяся образованием малорастворимого
осадка АВ*
где А — потенциалопределяющее вещество, способное к электровосстановлению:
А + е=ё* JD;
D — материал индикаторного электрода, способный к окислению;
В — вещество неэлектроактивное, т. е. не окисляется и не восстанавливается на
электроде, но принимает участие в электродной реакции.
До прибавления вещества В индикаторной электрохимической
реакцией является А + е *± \ D, электродный потенциал которой выражается
уравнением: п
?i=??+#lgaA (4)
По мере прибавления В активность А уменьшается и появляется
соединение АВ, которое также способно к восстановлению, согласно
электрохимической реакции \АВ + е ^ | D + В с электродным
потенциалом;
?2 = ?°-ftlgaB (4а)
Но так как D в присутствии А может окисляться при более
положительных значениях потенциала (влияние электростатических сил),
чем в присутствии В, которое связывает А — продукт окисления D в
малорастворимое соединение АВ (влияние удаления из сферы реакции),
то Е\ > еЦ- Следовательно, пока в растворе присутствует А,
равновесный потенциал электрода определяется его активностью [в соответствии
с уравнением (4)], тем более, что для второй электрохимической
реакции до точки эквивалентности в растворе практически отсутствует
компонент В. Отсюда общее правило: в смеси восстановителей электрод
принимает потенциал, обусловливаемый тем восстановителем, который
окисляется при наиболее отрицательном значении потенциала
электрода, г. е. компонентом той ред-окс системы, в которой стандартный
потенциал Е° наименьший и, наоборот, в смеси окислителей электрод
приобретает потенциалу обусловленный тем компонентом ред-окс пары,
у которой Е° наибольший. Это объясняется тем, что все компоненты в
растворе приходят в равновесие при наиболее низком значении
потенциалов в смеси восстановителей и наиболее высоком значении потен-
циалов в смеси окислителей.
Допустим, что титрование считаем законченным, когда последняя
порция титранта вызывает изменение степени оттитрованности
испытуемого раствора от 99,9% до 100,1%. Тогда, если принять начальную
активность аА = 1, то в первом случае ,аА составляет Ю-3 и,
следовательно, до точки эквивалентности электродный потенциал описывается
уравнением:
J^-^ + fllgHr8
После точки эквивалентности при появлении такого же избытка
титранта (ав = Ю-3) потенциал определяется уравнением (4а), т. е.
?2 = jE5-01gl(r3
§ 4. ПОТЕНЦИОМЕТРИЧЕСКОЕ ТИТРОВАНИЕ БЕЗ ТОКА (*-0) 39
Скачок потенциала, отвечающий изменению потенциала электрода
между этими двумя моментами титрования, равен:
Д? = Ех - Е2
(всегда следует вычитать из большего значения потенциала меньшее).
Отсюда:
A? = ??-?^-f'6'lglO""6-A?0-66' (5)
Следовательно, при одинаковой активности реагирующих веществ
и степени оттитрованности скачок потенциала тем больше, чем больше
разность нормальных потенциалов электрохимических реакций до и
после точки эквивалентности. Естественно, при большей степени недо-
титрованности и перетитрованности и большей активности
реагирующих веществ АЕ также будет возрастать вследствие уменьшения
второго члена правой части уравнения.
Подобным же образом можно легко вычислить АЕ для любого типа
химической реакции и для тех случаев, когда стехиометрические
коэффициенты реагирующих соединений не равны между собой.
Б потенциометрическом титровании для оценки ожидаемой
величины скачка потенциала и построения теоретической кривой титрования
при расчете по уравнению (1а) вместо активности потенциалопреде-
ляющих веществ можно использовать их концентрации. При этом
получают достаточно правильную картину хода титрования без заметного
изменения величины скачка потенциала и, следовательно, положения
точки эквивалентности.
Гораздо более правильное представление о скачках потенциала
можно получить, если взамен Е°{ и Е?2 использовать значения Ех>ф
и Е2,фУ так как в некоторых практических условиях Е° и Еф могут
настолько различаться, что разность Д?ф = ?1,ф— Е2,ф приобретает
отрицательное значение или становится весьма малой, хотя АЕ°
показывает возможность протекания реакции с большим скачком потенциала
в точке эквивалентности.
В отличие от прямой потенциометрии при потенциометрическом
титровании нет необходимости измерять э. д. с. гальванического
элемента с большой точностью, так как некоторые отклонения от
истинного ее значения не могут заметно сказываться на скачке потенциала
(обычно равном нескольким десяткам милливольт) и на точности
нахождения точки эквивалентности. По этой же причине потенциометри-
ческое титрование нередко можно использовать для необратимых систем.
Новый вариант потенциометрического анализа, называемый потен-
циометрическим титрованием под током (1Ф0), обеспечивает лучшие
условия измерения (устойчивые потенциалы электрода и большие
скачки потенциала) и успешное использование в потенциометрическом
титровании химических реакций с участием компонентов необратимых ред-
окс систем. Этот вариант рассматривается в соответствующем разделе
данной главы.
Титрование, при котором измеряют э. д. с. гальванических
элементов, называется потенциометрическим титрованием без тока, или в от-
сутавие тока (i = 0). Оно представляет собой классический метод
потенциометрического титрования.
§ 4. Потенциометрическое титрование без тока (I = 0)
Титрование по кислотно-основному методу. Титрование сильных и
слабых кислот сильными или слабыми основаниями (и наоборот)
связано с изменением концентрации ионов водорода в растворе:
40 ГЛ. II. ПОТЕНЦИОМЕТРИЯ И ПОТЕНЦИОМЕТРИЧЕСКОЕ ТИТРОВАНИЕ
титрование сильной кислоты сильным основанием (или наоборот)
Н+ + ОН~ ^=± Н20
титрование сильной кислоты слабым основанием (или наоборот)
Н+ + КЮН ^=± H20 + Kt+
титрование слабой кислоты сильным основанием (или наоборот)
HAn + OH" ^zt Н20 + Ап~
титрование слабой кислоты слабым основанием (или наоборот)
НАп + КЮН ^z> Н20 + Kt++ Ап~
Соответствующие электрохимические реакции приведены ниже:
до точки эквивалентности (или после точки эквивалентности)
2Н+ + 2е ^± Н2 ? = fllg[H+] (4+/н2*°) <6>
2HAn + 2e +=± H2 + 2AiT ? = ?% + ft lg [^_] (7)
после точки эквивалентности (или до точки эквивалентности):
40Н--4* ^=± 2Н20 + 02 ? = ?^-dlg[OH"] (8)
4KtOH-4e ^=± 2H20 + 02 + 4Kt+ ? = ^ + ftlg jg^ (9)
Поскольку
1НАЧ_1Н1. [01Г1.^г й J^^ = [h+]^ (10)
[Anl /Снд/ [н+] [KtOH] [ОРТ] /Сг
подставив соответствующие значения из уравнения (10) в уравнения
(7) —(9), получим:
? = ^-fllg/tHAn + »!glH+] (7a)
?==?°-ftlg/^ + ftlg[H+] (8a)
?==J^ + ^lg^2H+0Ig[H+] (9a)
С другой стороны, заменив [Н+] в уравнении (6) ее эквивалентами
из уравнений (10), так как водородный электрод измеряет [Н+], и
приняв для большего удобства расчетов и графического изображения хода
кривых титрования — lg/(HAn = Р^Снап; —IgKKton = рКкюя; — lg[H+] =
= рН; —lg[OH_] = рОН и —IgKw = pKw, получим уравнения,
выражающие величины потенциала электрода:
? = *'?*нап + Ф»8-Щ^ = »18-Щ^-<>Р*на„ (76)
E = blgKw-blglOH-]--VpKw + bpOH (86)
fKt+l
(96)
Kw [Kt+] [Kt+]
Уравнение (6) в свою очередь можно переписать в следующем
виде:
Е = - ftpH (6a)
Сопоставляя уравнения (7) — (9) с уравнениями (76) — (96),
находим, что справедливы следующие равенства:
xw
vKtOH
§ 4. ПОТЕНЦИОМЕТРИЧЕСКОЕ ТИТРОВАНИЕ БЕЗ ТОКА (*-0) 41
вследствие чего они сокращаются в уравнениях (7а) — (9а).
Полученные выражения показывают зависимость потенциала водородного
электрода исключительно от [Н+] во всех случаях кислотно-основных
титрований.
Для расчета потенциала водородного электрода до и после точки
эквивалентности при потенциометрическом титровании сильной кислоты
сильным основанием следует пользоваться уравнениями (6) и (86),
сильного основания сильной кислотой — уравнениями (86) и (6), сильной
кислоты слабым основанием—(6) и (96), слабого основания сильной
кислотой—(96) и (6), слабой кислоты сильным основанием—(76) и
(86), сильного основания слабой кислотой — (86) и (76) и, наконец,
слабой кислоты слабым основанием — уравнениями (76) и (96), а
слабого основания слабой кислотой — уравнениями (96) и (76).
Пример. Рассмотрим титрование сильной кислоты растзором сильного основания.
Так как концентрация сильной кислоты в первом приближении равняется [Н+]
вследствие полной ее диссоциации, в процессе титрования кислоты сильным основан-ием fH+]
уменьшается, а рН раствора возрастает, что. приводит к падению потенциала
водородного электрода. Предположим, что титруем 100 мл 0,1 н. раствора НС1 0,1 н.
раствором NaOH (для простоты расчета не будем учитывать разбавления раствора
во время титрования), тогда получим данные, представленные в табл. U
ТАБЛИЦА 1
Изменение потенциала (Е) водородного электрода и рН среды при титровании
100 мл 0,1 н. раствора НО 0,1 н. раствором NaOH
(Влиянием разбавления раствора пренебрежепо)
Объем
прибавленного
NaOH (И), мл
0
90,0
99,0
99,9
Объем
прибавленного
NaOH (V),
мл
100,1
101,0
110,0
AV, мл
90,0
9,0
0,9
мл
0,2
0,9
9,0
Концентрация НС1
в растворе,
N
ю-
ю-2
ю-3
ю-4
Концентрация NaOH
в растворе,
N
10~4
ю-3
ю-2
РН
раствора
1
2
3
4
рОН
раствора
4
3
2
ApH/AV
0,011
0,111
1,111
ApOH/AV
1,Ш
0,111
Потенциал
водородного
электрода,
мв
(? = -#рН =
—0,06 рН)
-60
-120
-180
-240
E=-"&pKw +
+ дрОН
-600
-660
-720
Д?, мв
60
60
60
А?, мв
360
60
60
Д?/ДК,
мв/мл
0,7
6,7
66,7
АЕ/А7,
мв/мл
1800,0
66,7
6,7
Максимальное изменение потенциала электрода (скачок потенциала) 1800 мв/мл
наступает тогда, когда кислота недотитрована и перетитрована на 0,1%, т. е. ошибка
не превышает ±0,1%. Титрованием меньшими порциями эту ошибку можно снизить
или сохранить на прежнем уровне, если анализируются более разбавленные или
меньшие объемы растворов. Вследствие симметричности изменения рН до и после точки
эквивалентности кривая титрования также симметрична относительно конечной точка
титрования, которая совпадает с точкой эквивалентности.
42 ГЛ. П. ПОТЕНЦИОМРТРИЯ И ПОТЕЫЦИОМЕТРИЧЕСКОЕ ТИТРОВАНИЕ
Составляя подобным же образом таблицу изменения потенциала водородного
электрода при титровании сильных кислот слабыми основаниями, слабых кислот
сильными или слабыми основаниями (и наоборот) убедимся, что скачок потенциала
резко уменьшается с понижением степени диссоциации реагирующих кислот и
оснований. Это обстоятельство отражено в уравнениях (76) и (96), поскольку в них
входят константы диссоциации кислот и оснований.
Таким образом, если Ю-3 н. растворы сильных кислот можно
титровать сильными основаниями еще с достаточно большой точностью, то
при титровании даже 0,1 н. растворов слабых кислот слабыми
основаниями (и наоборот) приемлемо точные результаты получить
невозможно из-за незначительной величины скачка потенциала. Поэтому в
качестве титрантов следует применять наиболее сильные кислоты или
основания и избегать титрования растворами слабых кислот или
оснований.
Титрование смеси кислот или оснований.
Дифференцированное титрование смеси сильных кислот или сильных оснований
в водных растворах невозможно, так как концентрация ионов
водорода (или гидроксил-ионов) в каждый момент титрования
соответствует суммарному содержанию всех кислот или оснований в
растворе.
С другой стороны, сильную кислоту возможно определить в
присутствии слабой, если концентрация ионов водорода слабой кислоты
НАп ([Н+] = "//СнапСнап ) меньше или равна концентрации недотитро-
ванной при заданной точности сильной кислоты. Так, если следует
оттитровать 100 мл 0,1 н. раствора НС1 с точностью 0,1% (т. е. НС1
остается в концентрации 10~4 н.) в присутствии слабой кислоты, то
необходимо, чтобы /СнапСнап^ Ю~8; такое условие соблюдается,
например, в присутствии 1 н. раствора Н3В03 (/Сн3во3 «* Ю~9). Определить,
однако, содержание слабой кислоты после завершения титрования
сильной кислоты невозможно из-за очень малой величины /Снап-
Дифференцированное потенциометрическое титрование смеси слабых
кислот или многоосновных кислот либо смеси слабых оснований или
многокислотных оснований с точностью до 1% возможно в том случае,
если К\ : К2, К2 ' Дз, Кз'-К^ Ю4, где К — соответствующие константы
диссоциации кислот или оснований (см. книга 2; гл. II, § 7). В этих
условиях обеспечивается достаточно заметное изменение рН вблизи точки
эквивалентности. Примером подобного титрования может служить
последовательная нейтрализация фосфорной кислоты до Н2РО4 и HPOJ.
Потенциометрическое определение не только смеси кислот или
оснований с близкими константами диссоциации, но и смеси ряда
сильных кислот успешно можно осуществить в неводных растворах
благодаря дифференцирующим свойствам различных органических
растворителей (см. гл. XI).
Независимо от типа применяемой химической реакции при
дифференцированном титровании смеси компонентов скачки потенциала,
кроме последнего, наступают раньше точки эквивалентности, и они
заметно уменьшаются по сравнению со скачками потенциалов при
титровании индивидуальных компонентов. Это объясняется тем, что еще
до полного завершения предыдущей реакции начинается последующая
с новой электрохимической индикаторной реакцией; между тем при
отсутствии других титруемых компонентов электрохимическая
индикаторная реакция после точки эквивалентности была бы совершенно иной,
связанной с титрантом, и поэтому вызывающей большее изменение
потенциала индикаторного электрода.
§ 4. ПОТЕНЦИОМЕТРИЧЕСКОЕ ТИТРОВАНИЕ БЕЗ ТОКА (*-0) 43
Титрование по методу осаждения. Рассмотрим в качестве примера
титрование галогенид-ионов ионами серебра. Индикаторный электрод —
серебро металлическое.
Химическая реакция выражается уравнением:
Ag+ + Haf ^=± | AgHal riPAgHal = [Ag+] [НаГ] (11)
Электрохимические реакции выражаются уравнениями:
до точки эквивалентности
ф AgHal +е ^=± | Ag + НаГ Ех = Е\ - Ъ lg [НаГ] (12)
после точки эквивалентности
Ag+ + e 5=± Ag ?2 = ?^ + dlg[Ag+] (13)
ПР
Из уравнения (11) следует, что [НаГ] = f Ag^aI , подставив это
LAg J
значение в уравнение (12), получим:
?, = ?? - О lg nPAgHal + d lg [Ag+] (13a)
Поскольку Ag-электрод обратим и относительно Ag+-HOHOB, то при
одной и той же концентрации этих ионов в уравнениях (13) и (13а)
Ех = E2i следовательно:
^ = ?^ + dlgnPAgHal
Иначе говоря, Е\>Е\ на величину — Ф lg nPAgHai (lgПР —
величина отрицательная).
В процессе титрования 0,1 н. раствора галогенида, когда
концентрация его в растворе станет 10~4 н., т. е. вблизи точки эквивалентности,
потенциал составит:
Ех - ?? - О lg КГ4 - 4 + ф lg nPAgHaI -dig 1(Г4
При добавлении избытка Ag+-noHOB в таком количестве, чтобы его
концентрация в растворе стала 10"4 н., потенциал серебряного
электрода после точки эквивалентности будет:
?,2 = ?^ + 01glO-4
Разность равновесных потенциалов между этими двумя точками
титрования, отвечающая скачку потенциала, равна:
bE = E2-El = 4-&i + *leio-* = bigio-s-bigiiPAsHul
Титрование смеси ионов по м етоду о с а ж д е н и я. Если
различные анионы с одним и тем же катионом образуют
малорастворимые соединения (или различные катионы с одним и тем же анионом),
растворимости которых достаточно различаются, то возможно
дифференцированное титрование этих анионов (или катионов).
Допустим, что стехиометрические коэффициенты компонентов в
осадках равны между собой и равны их концентрации в растворе.
Соответствующие уравнения произведений растворимости этих осадков:
[Kt+] [АпГ] - ПРр [Kt+] [An^] = ПР2; [Kt+] [Anj] - ПР3 и т. д.
Если nPi < ПР2 < ПР3 и т. д., тогда при титровании раствором,
содержащим Kt+, в первую очередь взаимодействуют те ионы, с
которыми Kt+ образует наименее растворимый осадок, в данном случае
KtAni, затем KtAn2 и т. д. В качестве условия можно принять, что
44 ГЛ. II. ПОТЕНЦИОМЕТРИЯ И ПОТЕНЦИОМЕТРИЧЕСКОЕ ТИТРОВАНИЕ
титрование следует осуществить с точностью 0,1%, т. е. образование
осадка KtAn2 должно начаться не раньше, чем предыдущий ион
оттитрован на 99,9%. Когда раствор находится в равновесии со всеми
осадками, справедливо следующее равенство:
[Kt+] =
ПР, ПР2 ПР3
и т. д.
и т. д. (14)
[AnJ [An2] [Апз]
отсюда
nPi _ [AnJ"] ПР2 [An;]
"пр7~[Ап;]' "ПР7 ~ [An j]
При начальных концентрациях An]", Ап5, Апз» равных
соответственно Си Сч, С г, следует, что Апг-ионы должны образовывать
осадок тогда, когда [АпГ] = lO^/d, а ионы Апз, когда [Anj] = Ю^/Сг, и т. д.
Подставляя эти значения в уравнение (14), получим:
ПР, 1(Г8С, . ПР2 1(Г3С2
ПР2 Со ' ПР3
и т. д.
Поскольку, согласно принятому условию, Сх = С2 = С3 и т. д., из
этих равенств следует, что дифференцированное титрование ионов
возможно лишь в случае, если ПР осадков отличаются друг от друга не
менее чем на три порядка (в 1000 раз).
Титрование по методу комплексообразования. Наиболее часто
в комплексонометрическом титровании катионов в качестве титрантов
применяются комплексон III, цианиды щелочных металлов и некоторые
другие лиганды. Этим же методом возможно определение и самих ли-
гандов.
Обычно лиганды не электроактивны, однако при использовании
соответствующих электродов на их поверхности происходит
окислительно-восстановительный процесс с образованием устойчивого
потенциала.
Например, комплексон III H2Y~~ образует прочные комплексы
с большим числом катионов, в частности и с Hg++:
H.Y-+H," *± HgY- + 2H* «=|^|Й- (15)
При использовании ртутного электрода на его поверхности
протекает электрохимическая реакция:
H2Y~ + |Hg-2e Z^> HgY" + 2H+ ?1=?o + ^igjHgYjjH^2 (16)
Окисление ртутного электрода в присутствии лиганда происходит
легче, чем в его отсутствие.
После завершения реакции (15) прибавление избытка Hg++
вызывает новую электрохимическую реакцию:
Hg++ + 2e +± |Hg; Е2 - Е% + -|- lg [Hg++] (17)
Если в уравнении (17) заменить [Hg++] ее значением из уравнения
(15), то получим:
^2 = ^2~Tlg/C + Tlg р^П (18)
§ 4 ПОТЕНЦИОМЕТРИЧЕСКОЕ ТИТРОВАНИЕ БЕЗ ТОКА (t-ф
45
Поскольку ?i = Еъ из уравнений (16) и (18) следует:
т. е. ?2 больше ?? на величину -g- lg^.
Подставляя в уравнение (16) концент[ ацию недотитрованного ком-
плексона III и образованного в процессе титрования комплекса HgY~~,
можно вычислить значения Е потенциала до точки эквивалентности.
Используя уравнение (17), можно вычислить Е после точки
эквивалентности при различных концентрациях [Hg++]. По полученным значениям
Е можно представить себе величину скачка потенциала. Так как К
входит в уравнение (18), естественно ожидать, что чем прочнее
комплекс, тем больше скачок потенциала.
Титрование по методу окисления — восстановления. Если в
растворе протекает окислительно-восстановительная реакция
т ОкисЛ] -f п Восст2 Т"** т Bocctj + п Окисл2
[Восст1]т[Окисл2]/г .
А [Окисл,]т[Восст2Г К }
то ей соответствуют следующие электрохимические реакции:
до точки эквивалентности
Окисл! + пе ^z± Boccti
после точки эквивалентности
Окисл2 + те ^z± Восст2
При пфтф\ и степени оттитрованности 99,9 и 100,1% скачок
потенциала Д? определится уравнением:
Д?в?о_?о + 11 10.з.±! 103eAEoeJL0.±0eAEo^3o(^±iL\
121 /гь m ь п т \ тп )
Если же п = т = 1, то Д Е = Д?° — 6Ф.
Очень часто в ред-окс реакциях участвуют ионы водорода. Обычно они
расходуются при восстановлении кислородсодержащих окислителей, но они могут и
появляться, когда в ред-окс реакции участвует вода. Поэтому, как показано ранее (см.
книга 2, гл. III, § 4, 8, 12), ионы водорода оказывают сильное влияние на многие
реакции окисления — восстановления и, следовательно, на скачок потенциала.
Титрование смеси ионов по методу окисления —
восстановления. Потенциометрическое дифференцированное
титрование смеси окислителей или восстановителей одним и тем же
восстановителем или окислителем является наиболее успешно
применяемым методом в аналитической химии. Однако для его осуществления
необходимы два важных условия: титруемые компоненты должны
принадлежать к обратимым ред-окс системам, а Е° этих систем должны
достаточно различаться.
Поэтому, исходя из принятого выше условия (п = т = 1), можно
прийти к выводу: при дифференцированном титровании смеси
окислителей (Окисл.1, Окисл.2, ..., Окисл.п) восстановителем (Восст.)т
необходимо, чтобы Е°\ — Е\, Е\~еЦ, ..., El-i — lfn и Е°п — Е?т (каждая
разность в отдельности) были больше 6 #.
46 ГЛ. II. ПОТЕНЦИОМЕТРИЯ И ПОТЕНЦИОМЕТРИЧЕСКОЕ ТИТРОВАНИЕ
§ 5. Различные способы нахождения конечной точки
потенциометрического титрования
При потенциометрическом титровании основной задачей является
обнаружение скачка потенциала, отвечающего конечной точке
титрования. Независимо от техники измерения э. д. с. гальванического
элемента для нахождения
ТАБЛИЦА 2 этой точки классиче-
Форма записи результатов потенциометрического скими являются рас-
титрования для нахождения конечной точки четные и графические
Определяемое вещество (название, масса или объем) способы.
Титрант (название и нормальность) Расчетный способ.
При
потенциометрическом титровании, если
заранее не известно
примерное содержание
определяемого
вещества в испытуемом
растворе, проводят
ориентировочное титрование
равномерными
большими порциями
стандартного раствора
(обычно по 1 мл) и
записывают результаты
измерения э. д. с.
Например, скачок
потенциала
наблюдался после прибавления
6 мл титранта, т. е. в
первом приближении
мо>кно считать, что
конечная точка
достигается при добавлении
5,5 мл.
Титрование не
следует прекращать при
достижении
максимального значения
э. д. с, так как
необходимо убедиться в том,
что и далее Д?
продолжает уменьшаться, т. е:
в том, что этот скачок —
не случайное явление,
вызванное
неправильной работой
индикаторного электрода или
измерительного прибора.
Далее приступают
к точному титрованию
в области скачка по-
Объем
прибавленного
титранта (V),
число капель
4,5 МЛ
2
4
6
8
10
12
14
16
18
20
22
24
26
28
30
32
34
36
38
5,92 мл
Измеренная
э. д. с,
мв
624
624
622
620
617
614
610
606
601
594
584
569
550
520
441
411
392
377
367
360
АЕ, мв
0
2
2
3
3
4
4
5
7
10
15
19
30
79
30
19
15
10
7
Д?/Д V *,
мв/мл
94
135
201
254
401
1056
401
254
201
135
94
* В данном примере AV постоянно и равно объему двух
капель, г. е. 0,0743.
тенциала для нахождения конечной точки; прибавляемые порции ти:
транта должны быть возможно малыми. Обычно этот объем
обусловливается величиной константы равновесия реакции и концентрациями
титранта и титруемого раствора. Чем больше ожидаемый скачок потен-
§ 5. СПОСОБЫ НАХОЖДЕНИЯ КОНЕЧНОЙ ТОЧКИ ТИТРОВАНИЯ 47
-циала, тем меньшими объемами нужно оперировать. Наименьший объем
титранта составляет 1 каплю.
При точном титровании в новую порцию испытуемого раствора из
бюретки сразу же сливают на 1 мл меньше титранта, чем это
соответствует объему для достижения конечной точки при ориентировочном
титровании (т. е. 5,5—1 =4,5 мл). Затем ждут достижения
постоянства э. д. с. (особенно при гетерогенных реакциях), т. е. до тех пор,
пока потенциал индикаторного электрода почти перестанет изменяться
во времени (изменение в течение 1 мин должно составлять не более
3—5 мв), и начинают титровать по каплям. После достижения скачка
потенциала продолжают титрование по каплям до тех пор, пока не
убедятся в том, что далее изменение э. д. с. незначительно. Затем
записывают общий объем израсходованного титранта и по количеству
добавленных капель определяют объем капли в миллилитрах (не следует
заранее определять объем капель, так как в зависимости от скорости
сливания титранта он изменяется и в некоторых случаях это может
внести в результаты анализа заметную ошибку). Отсчет объема
титранта по бюретке проводят с точностью до сотых долей миллилитра.
Форма записи представлена в табл. 2.
Расчет результатов титрования проводят по следующим формулам:
Ъ--^-^ и vz-Vi + (b + Qv2
где V — объем титранта, прибавленного в процессе титрования (в данном примере
5,92 мл), мл;
V\ — объем титранта, прибавленного до начала титрования по каплям (в данном
примере 4,5 мл), мл;
а — число капель, прибавленное из бюретки в процессе титрования (в данном
примере 38 капель);
V2 —объем одной капли (в данном примере 0,0374 мл), мл;
F3"-объем титранта, отвечающий конечной точке (в данном примере 5,51 мл), мл;
Ь — число капель титранта, после которого наступил скачок потенциала (в
данном примере 26 капель);
с? —число капель, которое вызвало максимальное изменение потенциала
индикаторного электрода (в данном примере 2 капли).
Поскольку изменение АЕ в приведенном примере симметрично
относительно максимума, последний отвечает половине числа капель,
которое вызывает максимальное изменение потенциала электрода (d/2),
т. е. одной капле. В соответствии с характером реакции изменения
потенциала должны быть симметричными, однако нередко ход кривой
титрования из-за выбранного объема порции вносимого титранта не
симметричен. В таких случаях для точного нахождения конечной точки
следует пользоваться второй производной A2E/AV2 (табл. 3).
Как видно из табл. 3, положение максимума (90 мв) не
симметрично: до максимума АЕ равно 30 мв, а после него 70 мв,
следовательно, объем титранта, равный 11,25 мл, не точно соответствует конечной
точке.
В этом примере при объеме титранта 11,20 мл A2E/AV2 составляет
—600 мв, а при 11,30 мл +200 мв. Таким образом, добавление 0,10 мл
вызвало общее изменение на 800 мв. Чтобы найти объем титранта,
который изменяет A2E/AV2 от —600 до 0 мв, составляют пропорцию:
800-0,1 600-0,1
600 -* 800
• 0,075 мл
Для точного нахождения конечной точки титрования вместо
11,25 мл титранта следует взять 11,275 мл. Ошибка при
приближенном способе нахождения конечной точки не превышает 0,2%, поэтому
48 ГЛ. И, ПОТЕНЦИОМЕТРИЯ И ПОТЕНЦИОМЕТРИЧЕСКОЕ ТИТРОВАНИЕ
ТАБЛИЦА 3
Форма записи результатов потенциометрического титрования
для нахождения конечной точки по второй производной
Определяемое вещество (название, масса или объем)
Титрант (название и нормальность)
Объем
прибавленного
титранта (К),
мл
10,90
11,00
11,10
11,20
11,30
11,40
11,50
11,60
Измеренная
э. д. с,
мв
294
300
310
340
430
500
560
614
АЕ, мв
6
10
30
90*
70
60
54
AE/AV,
мв'мл
60
100
300
900*
700
600
540
Д2?/ДУ2
мв/мл
-40
-200
-600
4-200
+ 100
+ 60
* Скачок потенциала лежит между 11,20 и 11,30 л«л, следовательно,
конечная точка в первом приближении соответствует 11,20+0,05=11,25 мл.
на практике часто нет необходимости делать расчет второй
производной, особенно если титрование вблизи конечной точки проводят малыми
порциями титранта.
'/"
Рис. 1. Формы кривых потенциометрического титрования:
а — интегральная кривая; б —дифференциальная кривая; в— кривая по второй
производной.
Графический способ. Графический способ заключается в
построении кривой титрования в координатах значения э. д. с.
гальванического элемента (или потенциала полуэлемента)—прибавленный объем
титранта, т. е. в изображении зависимости э. д. с. от V
(интегральная кривая, построенная по данным второй и первой графы
табл. 3).
Тогда точка перегиба кривой, лежащая на середине восходящей
(или нисходящей) ее части, отвечает, конечной точке титрования. Для
нахождения точки перегиба проводят две параллельные касательные к
пологим нижней и верхней ветвям кривой и соединяют их прямой так,
чтобы точка пересечения ее с восходящей (или нисходящей) ветвью
кривой делила эту прямую на две равные части (точка Л, рис. 1,а) Точка
пересечения перпендикуляра, опущенного из точки Л, с осью абсцисс
(осью V) дает объем титранта, отвечающий конечной точке титрования.
г
А
§ 6. НЕКОМПЕНСАЦИОННЫЙ МЕТОД ПОТЕНЦИОМЕТРИЧЕСКОГО ТИТРОВАНИЯ 49
Более простым и точным способом нахождения конечной точки
является графическое изображение зависимости первой производной
от объема прибавленного титранта, т. е. AE/AV от V
(дифференциальная кривая, построенная по данным четвертой и первой графы табл. 3).
В этом случае максимум на кривой соответствует конечной точке
титрования (рис. 1, б).
Для нахождения конечной точки по кривой зависимости A2E/AV2
от У, построенной по данным пятой и первой графы табл. 3, соединяют
концы обеих ветвей кривой, которые находятся с разных сторон оси
абсцисс. Точка пересечения прямой с осью абсцисс дает объем
титранта, отвечающий конечной точке титрования (рис. 1,в).
Естественно ожидать, что формы кривых а и в должны иметь обратное
изображение, если изменение э. д. с. гальванического элемента в процессе титрования имеет
обратный ход.
Другие способы нахождения конечной точки. Для ускоренного
нахождения конечной точки предложены различные способы, которые
отличаются некоторыми преимуществами, но имеют и известные
недостатки. Эти способы в книге не рассматриваются.
§ 6. Некомпенсационный метод потенциометрического
титрования
Некомпенсационный метод потенциометрического титрования
основан на измерении в замкнутой цепи силы тока, пропорциональной
э. д. с. гальванической ячейки. Название метода не совсем удачно, так
как оно относится не столько к методу, сколько к технике измерения
электрического параметра и, кроме того, многие другие способы также
можно осуществить в некомпенсационном варианте. Однако здесь
сохранено название, под которым рассматриваемый способ широко
известен.
Э. д. с. испытуемого гальванического элемента до начала
титрования компенсируют равным, налагаемым извне, но противоположно
направленным напряжением; при этом ток в цепи отсутствует. В
процессе титрования в результате изменения э. д. с. гальванического элемента
в цепи возникает ток, который измеряют соответствующим прибором.
Так как максимальное изменение э. д. с. происходит в точке
эквивалентности, в этот момент и наблюдается максимальное возрастание тока.
После точки эквивалентности ток в цепи хотя и продолжает
возрастать, но в меньшей степени, так как величина тока пропорциональна
э. д. с. ячейки. Конечную точку титрования можно найти по
зависимости i от V или Ai/AV от У.
Для предотвращения поляризации электродов в элементе при
прохождении тока через цепь последовательно титрационной ячейке
включают большое сопротивление (несколько десятков ком). В некоторых
случаях можно настолько увеличить сопротивление, что до точки
эквивалентности, т. е. до того момента, когда э. д. с. ячейки сравнительно
мало изменяется, ток в цепи будет практически отсутствовать и
возникнет лишь в момент максимального изменения э. д. с. Такое
возникновение тока можно фиксировать как достижение конечной точки без
дальнейшей обработки результатов измерений. Величину необходимого
внешнего сопротивления находят эмпирически.
Потенциометрическое титрование с биметаллической парой
электродов. Потенциометрическое титрование с биметаллической парой
электродов основано на измерении разности потенциалов, которая
возникает между двумя электродами (чаще всего из разных материалов, а
SO ГЛ. II. ПОТЕНЦИОМЕТРИЯ И ПОТЕНЦИОМЕТРИЧЕСКОЕ ТИТРОВАНИЕ
иногда и с различной величиной поверхности), погруженными
непосредственно в титруемый раствор. В этих случаях используется то
обстоятельство» что при достижении в процессе титрования исчезающе малой
концентрации (~10~5 М) одной из сопряженных форм обратимой ред-
окс пары или же в растворе необратимой ред-окс системы (т. е. при
образовании смешанных предельных потенциалов) поведение подобных
электродов различно.
В качестве пар электродов успешно могут быть использованы,
особенно при ред-окс реакциях, платина — графит, платина — вольфрам,
платина — родий, платина — палладий и др.
Если титруемое и титрующее вещества являются компонентами
обратимых ред-окс систем, то изменение потенциала каждого
электрода до точки эквивалентности одинаково, поэтому разность потенциалов
Рис. 2. Изменение
потенциалов Pt- и
Pd-электродов при потенциометри-
ческом титровании
железа (II) раствором би-
хромата калия.
Рис. 3. Дифференциальная кривая потенциометри-
ческого титрования железа (II)— компонента
обратимой ред-окс системы — раствором бихро-
мата калия — компонента необратимой ред-окс
системы —с применением биметаллической пары
электродов (Pt — Pd).
АЕ между ними практически равна нулю. Как только концентрация
титруемого вещества изменится настолько, что возникает смешанный
потенциал, величина которого существенно зависит от материала
электрода, между электродами создается большая разность потенциалов
АЕ, указывающая на достижение конечной точки. После точки
эквивалентности с появлением другой обратимой ред-окс системы, к которой
принадлежит титрант, потенциалы электродов снова становятся
практически равными (Д?«0). Ход титрования изображается
дифференциальной кривой.
В том случае, когда титруемое вещество является компонентом
обратимой ред-окс системы, а титрующее — необратимой, до конечной
точки титрования картина аналогична описанной выше, однако после
резкого скачка потенциала АЕ остается значительной. В качестве
примера на рис. 2 приведены изменения потенциалов Pt- и Pd-электродов
в отдельности во время титрования Fe++ раствором бихромата.
Форма кривой титрования показана на рис. 3.
Преимуществом метода является простота аппаратурного
оформления эксперимента, отсутствие жидкостного диффузионного потенциала,
быстрота выполнения, но, к сожалению, измеряемые АЕ не всегда
хорошо воспроизводимы из-за недостаточной устойчивости смешанных
потенциалов.
§ 7. Потенциометрическое титрование под током (i Ф 0)
При работе с необратимыми ред-окс системами или компонентами
обратимых ред-окс систем в отсутствие сопряженных форм потенциал
индикаторного электрода устанавливается медленно, он неустойчив,
§ 8. ИЗМЕРЕНИЕ ЭЛЕКТРОДВИЖУЩЕЙ СИЛЫ
51
поэтому потенциометрическое титрование без тока (i = 0) для
определения подобных веществ малопригодно. Введение сопряженной формы
определяемого компонента обратимой ред-окс системы хотя и дает
возможность точно измерять Е в таком растворе, осуществимо лишь
в случае, если сопряженная форма не участвует в химической
реакции.
Потенциометрическое титрование под током (/ Ф 0) успешно может
быть использовано во всех указанных выше случаях. М^тод
заключается в том, что через индикаторный электрод с помощью внешнего
источника тока пропускают ток малой величины (несколько
микроампер), иначе говоря, поляризуют электрод. При этом вследствие
быстрого обмена большим количеством электронов на поверхности раздела
металл — раствор довольно скоро устанавливается разность
потенциалов, устойчивая и хорошо воспроизводимая, значение которой зависит
от величины тока поляризации.
Если в потенциометрической (электролитической) ячейке испбль-
зуется один индикаторный электрод и полуэлемент сравнения, то
первый можно поляризовать либо анодно, либо катодно в зависимости от
характера проводимой химической реакции. Можно применять также
два идентичных индикаторных электрода (из одного и того же
материала и одинакового размера) в одном и том же титруемом растворе,
но тогда поляризуют оба электрода (один — анодно, другой —
катодно). При этом не только отпадает необходимость в электроде
сравнения, но и полученные результаты измерения э. д. с.
потенциометрической ячейки соответствуют разности потенциалов АЕ между двумя
электродами. Ход титрования описывается дифференциальной кривой
в координатах AEJAV— V.
Форма криЁой аналогична получаемой при потенциометрическом
титровании в отсутствие тока (i = 0), но с той разницей, что в
зависимости от характера поляризации электрода она несколько смещена
относительно оси абсцисс в ту или иную сторону.
Этот метод (i=j=0) применим также для титрования обратимых
систем, но не имеет никакого преимущества перед потенциометрическим
титрованием без тока, не говоря уже о том, что техника эксперимента
при i Ф 0 значительно сложнее.
Б. АППАРАТУРА И ТЕХНИКА ВЫПОЛНЕНИЯ АНАЛИЗА
§ 8. Измерение электродвижущей силы
Идеальным способом измерения э. д. с. гальванического элемента
(потенциометрической ячейки) является компенсационный метод Пог-
гендорфа, в котором на электроды в потенциометрической ячейке с
помощью делителя напряжения налагают напряжение (V) от внешнего
источника постоянного тока, противоположно направленное э. д. с.
ячейки. При этом в момент, когда ток в цепи отсутствует, градиенты
э. д. с. и У' равны между собой. Задача заключается, следовательно, в
постепенном изменении напряжения до тех пор, пока через ячейку не
перестанет проходить ток, что можно проследить каким-либо
индикатором тока. Второй задачей является определение величины налагаемого
напряжения, отвечающего данному моменту, что также можно
осуществить с помощью измерителя напряжения (вольтметра). Таким образом,
когда в цепи отсутствует ток (/ = 0), согласно уравнению V = Е&—
— EK + iR (где разность Еа — Ек представляет собой э. д. с.
потенциометрической ячейки), напряжение V равно э. д. с. Если потенциал
52 ГЛ. II. ПОТЕНЦИОМЕТРИЯ И ПОТЕНЦИОМЕТРИЧЕСКОЕ ТИТРОВАНИЕ
одного электрода постоянен (электрод сравнения) и известен
относительно НВЭ, то по напряжению можно вычислить потенциал
индикаторного электрода или проследить за его изменением в процессе
титрования.
Почти все приборы для измерения э. д. с. потенциометрической
ячейки — потенциометры — имеют следующую схему (рис. 4). Один
полюс внешнего источника постоянного тока / (сухие батареи или
аккумуляторы кислотные либо щелочные с э. д. с. ~2 в) через
переключатель 2 неподвижно присоединен к одному из концов (А) делителя
напряжения 3 с равномерным
сечением проволоки и с
небольшим сопротивлением (10—
100 ом). Делитель напряжения
3 обычно снабжен шкалой с
равномерными 1100
делениями. Другой полюс источника
тока / присоединен к
переменному сопротивлению малой
величины 4У с которым второй
конец (В) делителя
напряжения 3 соединяется с помощью
подвижного контакта 5. Таким
образом, напряжение
источника / падает на постоянном
участке АВ и на некотором
участке переменного
сопротивления ав. Конец В делителя
напряжения 3 присоединяют
к одному из электродов 3i
ячейки 6У соблюдая при этом
полярность соединения, т. е.
полюс источника тока 1 и
электрод тем же знаком должны
же концу делителя 3. Второй
электрод Э2 подключают последовательно через переключатель 7,
прерыватель тока 8 и индикатор тока 9 к подвижному контакту 10,
свободно перемещаемому на делителе напряжения 3. Дополнительно к концу
В делителя напряжения 3 подключают один из полюсов стандартного
элемента Вестона 11 (соблюдая тот же порядок полярности
соединения, см. выше), другой полюс которого может быть соединен с помощью
переключателя 7 с подвижным контактом 10. Следовательно, при
одном положении переключателя 7 замыкается через прерыватель тока
8 цепь, содержащая элемент Вестона И, а при другом — цепь,
содержащая потенциометрическую ячейку 6.
Элемент Вестона 11 устанавливается для контроля цены одного
деления шкалы делителя напряжения 3 (в милливольтах), так как
э. д. с. внешних источников тока известна с недостаточной точностью и
со временем самопроизвольно падает (источник тока разряжается).
Перед работой прибор настраивают так, чтобы одно деление
шкалы делителя напряжения 3 соответствовало 1 мв. Для этого с помощью
выключателя 2 замыкают цепь внешнего источника тока. После
достижения постоянного напряжения (через 10—15 мин) переключателем 7
в цепь включают элемент Вестона 11 и фиксируют контакт 10 на
делении 1018,6 шкалы делителя напряжения 3 (точка D). Так как э. д. с,
элемента Вестона 11 равна 1018,6 мв, на участке BD напряжение его
Рис. 4. Простейшая схема установки для
измерения э. д. с. гальванических элементов
компенсационным методом при потенцио-
метрическом титровании:
/ — источник постоянного тока с малым выходным
напряжением; 2— включатель тока; 3 — делитель
напряжения; 4 — переменное сопротивление; 5, /0
—скользящие контакты; 6 — электролитическая ячейка; 7
—переключатель тока; 5 — прерыватель тока; 9 — индикатор
тока; // —элемент Вестона; 3i и Э2 — электроды
(индикаторный и сравнения).
быть присоединены к одному и тому ;
§ 9. УСТАНОВКА ДЛЯ ПОЛЯРИЗАЦИИ ЭЛЕКТРОДОВ ПРИ ТИТРОВАНИИ ПОД ТОКОМ 53
полностью падает, т. е. на каждом делении напряжение уменьшается
на 1 мв. Если при коротком замыкании цепи с прерывателем 8 через
цепь потечет ток, обнаруживаемый с помощью индикатора тока 9, то
это означает, что на участке BD падение напряжения источника тока 1
больше или меньше э. д. с. элемента Вестона 11. Передвигая контакт 5
и периодически замыкая цепь прерывателем S, находят такое
положение контакта 5 на переменном сопротивлении 4, при котором индикатор
тока 9 показывает отсутствие тока в цепи.
Для измерения э. д. с. потенциометрической ячейки ее включают
в цепь переключателем 7 и, не изменяя положения контакта 5,
передвигают контакт 10 в ту или иную сторону до тех пор, пока при
периодическом замыкании цепи прерывателем
8 гальванометр 9 снова не покажет
отсутствие тока. Величину э. д. с.
ячейки в милливольтах можно
отсчитать по местоположению контакта 10
на шкале делителя напряжения 3.
При измерениях замыкать цепь ^->^
прерывателем тока 8 можно лишь на \Sj-3
очень короткое время (не больше
нескольких секунд) во избежание
поляризации электродов. После окончания
работы следует обязательно
размыкать цепь выключателем 2 и аррети- Рис. 5. Схема простейшей установки
ровать индикатор тока 9. для поляризации электродов при по-
^ т-г „ ^ тенциометрическом титровании:
Действие почти всех потенциомет- ;_источник порстоянного Т0КГ с высоким
РОВ ОСНОВаНО На ОПИСаННОМ ПрИНЦИПе. выходным напряжением; 2 - мегомное
перетяг ,т, „~ ^~~л~ ,ттт„~Лт^л менное сопротивление; 3 — микроамперметр;
ИЗ ОТечеСТВеННЫХ ПрИООрОВ ШИрОКО *-переключатель тока; 5-электролита-
ПрИМеНЯЮТСЯ ПОТеНЦИОМеТрЫ ПОСТОЯН- ческая ячейка; Эг и Э,- электроды.
ного тока высокоомные типа ППТВ-1,
Р-300, Р-307 и другие, а также ламповые потенциометры — рН-метры
ЛП-5, ЛП-58, ЛПУ-01, ЛПМ-60М. Последние имеют двойное
назначение— измерение э. д. с. гальванических элементов и рН растворов (при
этом используют стеклянный электрод). Обычно точность измерения
э. д. с. рН-метрами меньше, чем потенциометрами. Приборы ЛП-5 и
ЛП-58 работают по компенсационному методу, а ЛПУ-01, ЛПМ-60М и
другие — по некомпенсационному. Работа с некомпенсационными
приборами проще, так как измеряемые величины (рН и Е) непосредственно
отсчитываются по положению стрелки прибора.
§ 9. Установка для поляризации электродов
при потенциометрическом титровании под током
Схема установки для поляризации одного или двух индикаторных
(рабочих) электродов при потенциометрическом титровании под током
представлена на рис. 5. Из внешнего источника постоянного тока / с
большим выходным напряжением с помощью переменного мегомного
сопротивлений 2 добиваются в замкнутой цепи небольшой, но
постоянной величины тока (от 3—10 мка), измеряемой микроамперметром 3.
В цепи последовательно с микроамперметром 3 находится
переключатель тока 4 и электролитическая ячейка 5 с электродами 3i и Эг. При
катодной поляризации индикаторный электрод, находящийся в цепи
потенциометра (см. рис. 4), с помощью переключателя 4
дополнительно подключают к отрицательному полюсу установки для поляризации,
§4 ГЛ. II. ПОТЕНЦИОМЕТРИЯ И ПОТЕНЦИОМЕТРИЧЕСКОЕ ТИТРОВАНИЕ
а при анодной поляризации электрода — к положительному полюсу
установки для поляризации. В обоих случаях к другому полюсу установки
для поляризации всегда присоединен электрод сравнения.
При дотенциометрическом титровании с двумя поляризованными
индикаторными электродами их погружают в титруемый раствор
(электрод сравнения исключается из цепи). Один из них присоединяют к
положительным полюсам поляризационной установки и потенциометра,
а второй — к отрицательным полюсам этих же установок.
Полярность присоединения лишь одного поляризованного (катодно
или анодно) индикаторного электрода не обязательно совпадает с
полярностью его в цепи потенциометра. Электрод может быть катодно
поляризован, но если его равновесный потенциал (Е) больше потенциала
электрода сравнения (?э. Ср), то он является положительным полюсом
в ячейке, следовательно, его присоединяют к положительной клемме
потенциометра и, наоборот, — к отрицательной, если Е < Еэ, ср.
Для определения примерной величины сопротивления, включаемого
в цепь для получения нужной величины тока поляризации, пользуются
формулой закона Ома:
, У-ЬЕ
R + r
где V — выходное напряжение внешнего источника тока;
Д? — разность потенциалов между электродами;
R — внешнее сопротивление;
г — внутреннее сопротивление ячейки.
Для установления i « const необходимо, чтобы величины Д? и г
были малы по сравнению с V и R. Тогда, пренебрегая величинами Д?
и г, вычисляют R, подставляя в формулу R = V/i заданную величину
тока поляризации и
§10. Некоторые приборы, применяемые
в потенциометрическом титровании
Источники постоянного тока. В потенциометрических измерениях
обычно применяют либо сухие батареи, либо кислотные или щелочные
аккумуляторы, обеспечивающие напряжение около 2 е.
Постоянный ток поляризации при потенциометрическом
титровании под током можно получить из серии сухих батарей типа БАС-80
с напряжением 80 в или из городской осветительной сети переменного
тока с помощью прибора УИП-1, который выпрямляет ток и позволяет
в определенных пределах изменять сравнительно большое выходное
напряжение. С таким же успехом можно пользоваться другими
подходящими выпрямителями и стабилизаторами тока и понижающими или
повышающими трансформаторами.
Стандартный элемент Вестонд. Это гальванический элемент с э. д. с.
1,0186 в, который можно схематически изобразить:
(-)Cd(Hg) I CdS04 • 8/ЗН20 (нас. p-p) j Hg2S04 (тв) | Hg( + )
Левосторонним электродом элемента Вестона, на котором
происходит процесс окисления при замыкании цепи, является двухфазная
(кадмиевая) амальгама (около 12% Cd по массе). Правосторонним
электродом, на котором идет процесс восстановления, является
донная ртуть. Оба электрода находятся в растворе, насыщенном
сульфатом кадмия с избытком его кристаллогидрата и сульфатом закиси
ртути в присутствии его твердой фазы. Так как активности всех этих ком-
§ 10. ПРИБОРЫ, ПРИМЕНЯЕМЫЕ В ПОТЕНЦИОМЕТРИЧЕСКОМ ТИТРОВАНИИ 55
понентов при данной температуре постоянны, то и э. д. с. элемента
постоянна. При прохождении малых токов через ячейку элемент не
поляризуется, но при сравнительно больших токах э. д. с. элемента
временно отклоняется от равновесного состояния из-за изменения
концентрации компонентов в приэлектродных пространствах. Восстановление
равновесного состояния происходит медленно, так как скорость его
ограничена скоростью диффузии ионов в растворе. Поэтому с
элементом следует обращаться бережно.
Измерители напряжения. Для измерения сравнительно малых
напряжений пользуются обычными вольтметрами или
милливольтметрами. Последние, однако, не годятся для точного измерения э. д. с.
гальванических элементов, так как невозможно исключить поляризацию
электродов.
Для точного измерения э. д. с. гальванических элементов наиболее
пригодным является описанный ранее компенсационный метод с
применением потенциометров. Существуют электронные вольтметры,
которые дают возможность непосредственно измерять с достаточной
точностью малые напряжения и э. д. с. цепей. Действие этих вольтметров
основано на принципе усиления крайне слабых токов электронными
усилителями. Такие приборы могут быть успешно использованы в по-
тенциометрическом анализе для измерения э. д. с. или для
наблюдения за ее изменением. Все измерители напряжения включают в цепь
только параллельно измеряемой системе.
Измерители тока. Для измерения поляризационных токов
пользуются микроамперметрами. Измерители тока включают
последовательно в цепь; их внутреннее сопротивление небольшое и мало
сказывается на общей силе тока. Очень чувствительным и точным методом
определения величины силы тока в какой-либо замкнутой цепи
является измерение падения напряжения (iR) через прецизионное
постоянное известное сопротивление (R), включенное последовательно в
цепь. Величину V = iR измеряют компенсационным методом с помощью
потенциометра. Зная R и V, можно вычислить i с большой точностью.
Индикаторы тока. Для индикации тока применяют гальванометры,
т. е. приборы для измерения токов небольшой величины — порядка 10-6
а/деление. На шкалах гальванометров нуль находится в середине и
с помощью таких приборов можно наблюдать за направлением тока,
В потенциометрическом анализе вполне достаточны индикаторы
тока с ценой деления шкалы около 10~6 а/деление, если измеряются
э. д. с. с точностью 1 мв. При более прецизионных работах с весьма
слабыми токами (например, при очень большом сопротивлении цепи
вследствие использования стеклянного электрода в кислотно-основном
потенциометрическом титровании) или когда требуется проследить за
изменением очень слабых токов (в полярографии, амперометрии и пр.)
успешно применяются гальванометры (зеркальные) с
чувствительностью Ю-9 aj деление и меньще. Применение электронных усилителей
тока дает возможность использовать гальванометры с меньшей
чувствительностью.
Описанные измерители и индикаторы тока и напряжения могут
быть стрелочными или зайчиковыми (со световым лучом, падающим на
шкалу прибора).
Сопротивления. Для измерения силы тока в цепи компенсационным
методом пользуются набором высокопрецизионных сопротивлений.
Для снижения тока до нужной величины при использовании
источников тока с высоким выходным напряжением необходимы
постоянные и переменные мегомные и килоомные сопротивления, включаемые
56 ГЛ. II. ПОТЕНЦИОМЕТРИЯ И ПОТЕНЦИОМЕТРИЧЕСКОЕ ТИТРОВАНИЕ
в цепь последовательно. Величину этих сопротивлений подбирают
приближенным расчетом по формуле закона Ома.
Переменные сопротивления могут быть со скользящим контактом,
с помощью которого отбирают часть общего сопротивления,
останавливая контакт в тех или иных точках проволоки постоянного сечения,
или в виде магазина сопротивления с серией катушек из проволоки
различного сечения в каждой, которые могут быть штекерами
последовательно соединены или разъединены. Так называемые курбельные
магазины сопротивления представляют собой серии катушек, соединенных
непрерывно между собой, но они имеют скользящий контакт, с
помощью которого отбирают необходимую величину сопротивления.
В качестве постоянных мегомных и килоомных сопротивлений
целесообразнее пользоваться сопротивлениями, применяемыми в
радиотехнике; они более компактны и их можно использовать при токах
большой мощности. Во избежание перегорания проволок при прохождении
слишком больших токов (или падении слишком больших напряжений
ца них) следует учитывать мощность включаемых в цепь
сопротивлений. Это особенно важно при работе с прецизионными
приборами.
Переключатели и прерыватели тока. В качестве первых могут
служить любые переключатели тока, тумблеры и пр. Прерывателем тока
является ключ Морзе, который замыкает цепь лишь при нажатии на
рычаг (как только его отпускают, цепь размыкается).
Мешалки. Для эффективного перемешивания испытуемого раствора
в процессе потенциометрического титрования применяются различного
типа мешалки: механические, вращающиеся с помощью электромотора
и магнитные. Более удобны в работе магнитные мешалки, так как они
не затрудняют доступ к титруемому раствору и потому позволяют
удобнее расположить электроды, электролитический ключ и бюретку в
ячейке. Кроме того, они обычно оснащены электрическим нагревателем,
что дает возможность проводить титрование и при нагревании.
Ячейки. Наиболее простой формой ячейки для выполнения
потенциометрического титрования является обычный химический стакан
(соответствующей емкости), внутри которого с помощью штатива
закрепляют индикаторный электрод, одно колено электролитического ключа
и мешалку (если она механическая). При перемешивании титруемого
раствора магнитной мешалкой ее непосредственно опускают в раствор.
Электрод сравнения соответствующей конструкции также может быть
погружен в испытуемый раствор, тогда отпадает необходимость "в
электролитическом ключе. В тех случаях, когда необходимо исключить ёлия-
ние газообразных соединений из внешней атмосферы, применяют
герметизированные ячейки с отверстиями для электродов, мешалки,
электролитического ключа, кончика бюретки, подачи и выхода газа (N2,
Н2, С02 и пр.).
Для проведения химических и электрохимических реакций при
постоянной температуре используют ячейку с рубашкой, внутри которой
циркулирует вода (или другая жидкость), поступающая из термостата
с заданной температурой.
При непрерывном контроле состава производственных растворов
потенциометрическим методом пользуются ячейками, конструкция
которых дает возможность проследить за изменением потенциала
индикаторного электрода в проточной жидкости.
В литературе описаны ячейки различной конструкции для той или
иной конкретной цели. Желающим ознакомиться с ними следует
обращаться к специальной литературе.
§11. ЭЛЕКТРОДЫ
57
Электролитический ключ. Для создания электрического контакта
между двумя полуэлементами гальванического элемента обычно
применяют U-образную стеклянную трубку, заполненную либо гелием агар-
агара, насыщенным соответствующим электролитом, либо насыщенным
раствором этого же электролита. Наиболее часто в качестве
электролита применяется КС1. Выбор продиктован тем, что КС1 обеспечивает
наименьший жидкостный диффузионный потенциал из-за почти
одинаковой подвижности К+- и С1~-ионов. Однако при анализе растворов
галогенидов во избежание диффузии С1_-ионов из электролитического
ключа в эти растворы следует КС1 заменить другим электролитом, не
влияющим на электрохимические и химические процессы, например
КМОз. Для того чтобы раствор электролита удержать в трубке,
необходимо концы ее плотно закрыть тампоном из фильтровальной
бумаги, смоченной тем же раствором электролита. Электролитические
ключи следует хранить в насыщенном растворе электролита, которым
они заполнены, чтобы предотвратить высыхание ключей, а также
появление в них пузырьков воздуха, препятствующих прохождению тока.
§11. Электроды
Электроды представляют собой металлические пластинки или
проволоки, которые при погружении в раствор соответствующего состава
в результате происходящего на их поверхности электрохимического
процесса и образования двойного электрического слоя приобретают
более или менее устойчивый потенциал.
Электроды можно классифицировать по роду и по назначению.
Согласно электрохимическим реакциям, протекающим на
поверхности раздела металл — раствор, различают электроды первого, второго
и третьего рода.
К электродам первого рода относятся металлы, потенциалы
которых обратимы относительно своих ионов, как, например, Ag-электрод
в растворе, содержащем Ag+-noHbi, ртутный и медный электроды в
присутствии соответственно Hg++- и Си++-ионов и т. д.
Потенциалы электродов второго рода обратимы относительно
анионов, образующих с катионами металла электрода малорастворимый
осадок; например, Ag-электрод в насыщенном растворе |AgCl в
присутствии С1"-ионов, Hg-электрод в насыщенном растворе |Hg2Cl2 в
присутствии С1--ионов и т. д.
Электроды третьего рода представляют собой металлы, которые
находятся в равновесии с раствором, насыщенном двумя
малорастворимыми электролитами с одним общим анионом и катионами, один из
которых является ионом металла электрода, а второй — посторонним,
находящимся в избытке; например, Hg-электрод в растворе,
содержащем осадок |Hg2C204 и |СаС204 и избыток Са++. Протекающая на
электроде электрохимическая реакция выражается уравнением:
2|Hg + CaC204-2e ^=± ф Hg2C204 + Са++
Величина потенциала электрода описывается формулой:
?"-
Легко доказать, что
?~4g2C204, Ca++/Hg, CaC204+ 2 lg *Са++
ПР
о .о , <N„" _н**са<>4
:Hg2C204Ca, ++/Hg,CaC204-?Hg^/Hg+ 2 Ig ПРСаС2о4
58 ГЛ. II. ПОТЕНЦИОМЕТРИЯ И - ПОТЕНЦИОМЕТРИЧЕСКОЕ ТИТРОВАНИЕ
По назначению электроды делятся на электроды сравнения и
индикаторные.
Электроды сравнения. В качестве электрода (полуэлемента)
сравнения могут служить такие электроды, на поверхности которых при
соприкосновении с подходящим раствором возникают лишь обратимые
электрохимические реакции. Благодаря этому приобретаемые ими с
большой скоростью потенциалы устойчивы. При прохождении
небольших токов в замкнутой цепи потенциалы таких полуэлементов
практически остаются постоянными, поэтому их причисляют к йеполяризуе-
мым электродам.
Наиболее часто применяемым на практике электродом сравнения
является насыщенный каломельный электрод (Нас. КЭ), состоящий из
металлической ртути, твердых фаз Hg2Cl2 и КС1 и насыщенного этими
соединениями раствора. Схематически такой полуэлемент можно
изобразить так:
Hg 1ф Hg2Cl2, КС1 (тв), Hg2Cl2 (нас. р—р), КС1 (нас. р—р)
На этом электроде происходит строго обратимая
электрохимическая реакция:
2±Hg + 2Cr~2e ^=± ф Hg2Cl2
Потенциал такого электрода определяется формулой:
Е - 4g2c.2/2Hg> 2cr " • * асг = 4^/я* + Т * ПРн.2сь - * * всг
В качестве других электродов сравнения применяются каломельные
электроды с 0,1 и 1 н. растворами хлорида калия (0,1 НКЭ, НКЭ
соответственно), меркуриодидный (МИЭ), хлорсеребряный, меркурсуль-
фатный и другие полуэлементы, состав и ?раВн которых можно найти
в химических справочниках. С таким же успехом можно использовать
стеклянный и хингидронный электроды (см. ниже).
Как видно, большинство электродов сравнения относится к
электродам второго рода.
Индикаторные электроды. Индикаторные электроды применяют для
измерения потенциалов, возникающих при погружении их в
исследуемый раствор. По величине потенциалов оценивают активности потен-
циалопределяющих веществ, а также наблюдают за изменением в
процессе химической реакции концентрации вещества» участвующего в
электрохимической реакции.
В зависимости от типа химической реакции при потенциометриче-
ском титровании применяются различные индикаторные электроды; эти
электроды универсальны только при окислительно-восстановительных
и кислотно-основных реакциях.
Так, в окислительно-восстановительных реакциях независимо от
того, какой окислитель или восстановитель титруют и какой
восстановитель или окислитель служит титрантом, могут быть использованы
электроды из одного и того же благородного металла (Pt и Аи). В
таких реакциях электроды индифферентны, т. е. в электрохимических
реакциях непосредственно не участвуют, а являются лишь
передатчиками электронов.
В кислотно-основном титровании независимо от того, какие кислота
или основание титруются и какими основанием или кислотой
химическая реакция связана с изменением [Н+], или рН, в растворе, и поэтому
для анализа достаточно иметь любой индикаторный электрод,
функционирующий как водородный, т. е. обратимый относительно Н+-ионов.
§11. ЭЛЕКТРОДЫ
50
К таковым относятся кроме водородного электрода (см. стр. 35) хин-
гидронный, металлический и стеклянный электроды.
Хингидронный электрод. Хингидрон (Н2Х2) — органическое
соединение, слаборастворимое в воде и распадающееся при этом на
эквимолекулярное количество хинона СбН602 (X) и гидрохинона
(восстановленная форма хинона) СбН4(ОН)2(Н2Х):
HV >* Y _l_ U Y. IT — [X] [Н2Х]
2л2 ч— А + п2л; д ——г^ ^ j
На погруженном в насыщенный хингидроном раствор
индикаторном электроде (Pt или Аи), обратимом относительно этой ред-окс
системы, возникает электрохимическая реакция:
Х + 2Н+ + 2е -т=> Н2Х
Потенциал электрода определяется формулой:
'X. 2Н+/Н2Х + 2 g [Н2Х]
Е = < оН+/^ + 4-^тЙт + ^1^Н+] (22)
Так как раствор насыщен относительно Н2Х2, то [X] = [Н2Х] и
— lg ггт лг1 =0;тогда уравнение (22) можно записать:
где ?^>2н+/н2х положительнее потенциала стандартного водородного
электрода на 0,6994 в (при t = 25 °С).
Уравнение (23) показывает, что потенциал электрода является в
кислотно-основной среде исключительно функцией [Н+], следовательно,
такой электрод может применяться при кислотно-основном потенцио-
метрическом титровании и измерении рН (при прямой потенциометрии)
растворов.
Хингидронный электрод очень удобен, но, к сожалению, его нельзя
применять в растворах, содержащих сильные окислители или
восстановители, так как вследствие изменения соотношения [X]: [Н2Х]
потенциал индифферентного электрода будет зависеть не только от рН
раствора. Кроме того, вследствие легкости окисления гидрохинона в
щелочкой среде кислородом воздуха искажаются показания электрода*
Поэтому на практике желательно хингидрон вносить в раствор
кислоты и титровать основанием (а не наоборот) для получения более
закономерных изменений Е от рН; однако и при обратном ходе
титрования наблюдаются достаточно большие скачки потенциала. Поскольку
хингидрон малорастворим в воде, для насыщения испытуемого рас*
твора достаточно прибавить около 50—100 мг хингидрона.
Сурьмяный электрод. В качестве металлического электрода,
обратимого относительно Н+, чаще всего используют сурьмяный
электрод. Этот металл обычно покрыт тонкой пленкой малорастворимой
окиси Sb2C>3. При погружении его в раствор, содержащий Н+*ионы,
происходит электрохимическая реакция:
2|Sb + 3H20-6* ^=± | Sb203 4- 6H*
Потенциал электрода определяется соотношением:
Е " ?sb2o3, 6H+/2Sb, зн2о + °Ig [H+1
Хотя на практике зависимость Е от [Н+] и наблюдается, нб, во-пер*
вых, величина Ф обычно не строго соответствует термодинамическому
значению при данной температуре и не постоянна, т. е. наклон кривой
60 ГЛ. II. ПОТЕНЦИОМЕТРИЯ И ПОТЕНЦИОМЕТРИЧЕСКОЕ ТИТРОВАНИЕ
зависимости Е от рН имеет разную величину в различных областях рН.
Во-вторых, величина EsbzQ H+/Sb Нз0 зависит от способов получения
чистой сурьмы и изготовления электрода, от присутствия
незначительных примесей и других влияний, поэтому значение Е° неодинаково для
различных сурьмяных электродов. Кроме того, для одного и того же
электрода при одном и том же рН раствора величины Ф и Е°
изменяются во времени, зависят от скорости перемешивания (или от его
отсутствия), а также от глубины погружения электрода в раствор.
В щелочных и сильнокислых средах показания рН сурьмяных
электродов еще более искажаются из-за окисления поверхности металлов в
первом случае и заметного растворения окисной пленки — во втором.
Несмотря на это, они успешно могут быть использованы при потенцио-
метрическом кислотно-основном титровании (но не в прямой потенцио-
метрии), так как обнаруживаемый достаточно большой скачок
потенциала в конечной точке титрования практически соответствует точке
эквивалентности или очень близок к ней.
Стеклянный электрод. Стеклянный электрод относится к
мембранным электродам, механизм действия которых все еще не
вполне установлен, однако имеется немало состоятельных объяснений
причин функционирования стеклянных электродов в качестве водородных
электродов. И хотя в данном случае отсутствуют электрохимические
реакции окисления и восстановления компонентов, обусловливающие
возникновение разности потенциала на поверхности раздела стекло —
раствор, зависимость потенциалов стеклянных электродов от рН
растворов вполне закономерно описывается уравнением, аналогичным
уравнению Нернста.
Стеклянный электрод представляет собой стеклянную трубку, на
конце которой имеется шарик с очень тонкими стенками. Шарик
заполняют соответствующим раствором, внутри его прикреплен индикаторный
электрод (например, Ag-электрод в насыщенном растворе jAgCl в
присутствии С1~). Эта часть служит в качестве полуэлемента сравнения
(El, сР). Для анализа в испытуемый раствор опускают стеклянный
электрод и измеряют потенциал относительно какого-либо другого электрода
сравнения (?э!ср). Тем самым измеряют фактически э. д. с.
гальванического элемента:
pi I стеклянная I испытуемый I pii
э- СР I мембрана | раствор | э- СР
Т&к как El.cp и El1 сР постоянны, то э.д. с. есть функция [Н+]
испытуемого раствора.
Сказанное о Е° и Ф для сурьмяных электродов в какой-то степени
справедливо и для стеклянных электродов. В сильнощелочных средах
или в слабощелочных, но в присутствии большой концентрации ионов
щелочных металлов прямолинейная зависимость э.д.с. от рН для
стеклянных электродов также заметно нарушается. С другой стороны,
измерения рН можно осуществить в сильно окислительных и
восстановительных средах (электрохимическая реакция отсутствует), что является
большим преимуществом стеклянного электрода. Вследствие огромного
внутреннего сопротивления гальванического элемента (стекло —
диэлектрик) приходится изготовлять шарики с очень тонкими стенками и
усиливать возникающий очень слабый ток цри измерении э. д. с. цепи
электронными усилителями. Поэтому усложняется конструкция и
увеличивается стоимость установок и требуется особо осторожное обращение
с такими хрупкими электродами.
§11. ЭЛЕКТРОДЫ
61
Выпускаемые рН-метры со стеклянными электродами с достаточно
толстой стенкой шариков (~0,1 мм) позволяют измерять с большой
точностью [Н+] до рН «13, но при умеренных концентрациях ионов
щелочных металлов. Эти рН-метры снабжены усилителями с большим
коэффициентом усиления тока, что дает возможность непосредственно
измерять рН раствора, не прибегая к компенсационному методу
измерения с применением очень чувствительных индикаторов тока. Поэтому
стеклянные индикаторные электроды широко используются в практике
кислотно-основного титрования и в других областях потенциометриче-
ских измерений, а кроме того, и при неводном титровании. Далее,
поскольку они химически инертны, могут быть непосредственно помещены
в титруемый раствор при использовании их в качестве электрода
сравнения. При этом увеличивается компактность гальванического элемента
(исключается электролитический ключ).
Индикаторные электроды при потенциометриче-
ском титровании по мет одам оса ждени я и комплексо-
образования. Различные осадки и комплексные соединения состоят
из самых разнообразных ионов, и потому не существует такого
универсального индикаторного электрода, который мог бы быть обратимым
относительно всех катионов и анионов. Кроме того, не всегда можно
располагать металлическим электродом, обратимым относительно своих
ионов, из-за большой электролитической упругости растворения ряда
металлов (легко окисляющихся Н+-ионами раствора) или такими
твердофазными веществами, в состав которых входит хотя бы один из
ионов, образующих в процессе титрования осадки или комплексные
соединения, но в другой степени его окисления или восстановления.
Малая селективность индикаторных электродов, казалось бы, сильно
ограничивает возможность использования потенциометрического метода
в реакциях осаждения и комплексообразования. Однако применение
электродов второго рода позволяет заметно расширить область
применения потенциометрического титрования.
Так, с Ag-электродом можно потенциометрически титровать не
только Ag+-HOHbi, но и галогениды, роданид, сульфид, гексацианоферраты (II)
и (III), цианид и другие, т. е. все те анионы, с которыми Ag+-HOHbi
образуют малорастворимые соединения или прочные комплексы.
В последние годы появились мембранные электроды,
приготовленные из смеси инертного связывающего материала и того или другого
осадка. Они обратимы относительно одного из ионов, входящих в
состав осадка мембраны, и успешно используются в прямой потенциомет-
рии и в потенциометрическом титровании для определения этих ионов
по методу осаждения и комплексообразования.
Обращение с металлическими электродами. Металлические
электроды применяют в виде пластинок, проволок, сеток, стержней или
цилиндрических палочек.
Электроды периодически подвергают обработке для регенерации
рабочей поверхности; Pt- и Au-электроды попеременно нагревают в
разбавленной (1:1) азотной и концентрированной хлористоводородной
кислотах. Перед перенесением электрода из одной кислоты в другую
необходимо тщательно промывать их водой во избежание растворения
металла в образовавшейся смеси кислот. Не следует промывать
горячие электроды холодной водой или, наоборот, опускать холодные
электроды в горячие растворы кислот, так как при этом образуются
незаметные на глаз трещины, особенно на спаях стекло — металл.
Ag-Электрод на очень короткое время опускают в теплую азотную
кислоту (1:1), содержащую несколько кристаллов KN03. Как только
62 ГЛ. П. ПОТЕНЦИОМЕТРИЯ И ПОТЕНЦИОМЕТРИЧЕСКОЕ ТИТРОВАНИЕ
на поверхности серебра появятся пузырьки газа (растворение металла),
немедленно вынимают электрод из кислоты и хорошо промывают водой.
Если Ag-электрод был использован для титрования галогенидов и
других соединений, то часть образовавшегося осадка плотно пристает к
поверхности металла и не смывается водой. Поэтому перед кислотной
обработкой электрод прополаскивают в концентрированном растворе
тиосульфата, цианида или аммиака (в зависимости от того, в чем легче
растворяется осадок). Поверхность других электродов (например,
сурьмяного и вольфрамового и т. п.) очищают тонкой наждачной бумагой
и тщательно промывают водой *.
Электроды обычно хранят в воде, слегка подкисленной несколькими
каплями хлористоводородной кислоты, периодически воду меняют.
Перед использованием электроды обязательно промывают
дистиллированной водой; сушить их нет надобности, если титрование проводят в
водной среде, так как разбавление титруемого раствора за счет
влажности электрода не может оказать существенного влияния.
В. ПРАКТИЧЕСКИЕ РАБОТЫ
ПОТЕНЦИОМЕТРИЧЕСКОЕ ТИТРОВАНИЕ БЕЗ ТОКА (i = 0)
§ 12. Кислотно-основное титрование
Определение уксусной кислоты
Потенциометрическое определение уксусной кислоты проводят
титрованием 0,1 н. раствором сильного основания, проследив
компенсационным методом за изменением э. д. с. гальванического элемента,
составленного из хингидронного индикаторного электрода (НгХ) и
насыщенного каломельного электрода сравнения.
При этом протекает химическая реакция
сн3соон + он- ^=± сн3соо~ + н2о
и соответствующая электрохимическая реакция до точки
эквивалентности
Х + 2СН3СООН + 2е ^=± Н2Х + 2СН3СОСГ
Е-рО+ *!* [Х] miir [CH»COOH1
поскольку [X] = [Н2Х] (см. стр. 59)
Это уравнение показывает, что Е до точки эквивалентности
уменьшается в соответствии с изменением соотношения концентрации недо-
титрованной кислоты и образовавшейся соли, иначе говоря, с
возрастанием рН раствора.
При появлении избытка ОН--ионов после точки эквивалентности
протекает следующая электрохимическая реакция:
Н2Х + 20Н~-2е «z± Х + 2Н20
? = ^ + ^lgJgy-^lg[OHl==^~01g[OHl (26)
т. е. потенциал электрода продолжает уменьшаться с увеличением
избытка ОН_-и6нов (по мере дальнейшего возрастания рН раствора).
Аналогично очищают поверхность графитового электрода.
§ 12. КИСЛОТНО-ОСНОВНОЕ ТИТРОВАНИЕ
63
Из уравнений (25) и (26) при введении соответствующих
значений /Снап и Kw и сопоставлении их с уравнением (23) легко доказать,
что 4, н+/н2х = Я? - <Mg #снзсоон = Е1 - * te *w> но так как ^снзсоон >
>/Cw, то J?x н+/н2х > ^i > ^2» следовательно, при небольшой степени
недотитрованности и перетитрованности указанной кислоты должен
возникать скачок потенциала, отвечающий конечной точке.
Э. д. с. потенциометрической ячейки можно выразить формулой:
э.д.с«(+)?-?Нас#кэ(-)
Однако вследствие изменения [Н+] во время титрования и согласно
приведенным уравнениям настанет момент, когда Е станет равным
?ыас кэ> а э. д. с. равной 0. С этого момента (при рН « 7,3) э. д. с. »
= ( + )?нас. кэ?(—)> поэтому необходимо переключить электроды в
полуэлементах к соответствующим клеммам потенциометра.
Методика определения. Для измерения э. д. с. потенциометрической
ячейки пользуются любым подходящим потенциометром и приводят
его в рабочее состояние, согласно приложенной к прибору инструкции.
В стакан емкостью 200 мл пипеткой помещают испытуемый раствор
уксусной кислоты, разбавляют водой до 50—80 мл, прибавляют около
50 мг хингидрона, опускают мешалку и устанавливают стакан на
площадке магнитной мешалки. В другой стакан емкостью 50—100 мл
наливают насыщенный раствор КО, опускают туда Нас. КЭ и
устанавливают рядом с титрационной ячейкой. Оба стакана соединяют
электролитическим ключом, наполненным насыщенным раствором КС1. В
ячейку с титруемым раствором погружают Pt-электрод и закрепляют с
помощью лапки и штатива так, чтобы металлическая часть была
полностью погружена в раствор, но чтобы во время вращения мешалка не
задевала электрод. Бюретку наполняют 0,1 н. стандартным раствором
щелочи, закрывают верхнее ее отверстие хлоркальциевой трубкой,
наполненной натронной известью для предупреждения проникновения
двуокиси углерода из воздуха, и закрепляют в штативе над титрационной
ячейкой. Индикаторный электрод присоединяют к положительной, а
Нас. КЭ — к отрицательной клеммам потенциометра.
Приступая к ориентировочному титрованию, сначала включают
магнитную мешалку, затем прибавляют по 1 мл щелочи и измеряют
э. д. с. гальванического элемента (см. стр. 51). Перед тем как вносить
в раствор новую порцию титранта, записывают показание
потенциометра в рабочий журнал (см. стр. 48). Титрование продолжают до тех
пор, пока не будет найден скачок потенциала, затем выключают
мешалку.
После нахождения объема титранта с точностью 0,5 мл, который
соответствует приближенному значению конечной точки титрования,
удаляют электрод, электролитический ключ и мешалку из стакана,
промывают их дистиллированной водой и высушивают фильтровальной
бумагой. В стакан снова вводят такую же порцию титруемого раствора,
так же разбавляют водой и собирают всю установку, как описано выше.
Бюретку снова наполняют титрантом до нулевой метки и, включив
мешалку, приступают к точному титрованию. Для .этого сразу вносят
объем титранта на 1 мл меньше, чем это соответствует конечной точке,
установленной при ориентировочном титровании, и, хорошо перемешав
раствор, продолжают титрование по 2 капли, измеря&я каждый раз
э. д. с. ячейки с точностью до 1 же и записывая данные, как указано
в табл. 2 (см. стр. 46). В тот момент, когда компенсация э. д. с.
становится невозможной (?<?,э.Ср), реверсируют полюса электродов к
64 ГЛ. II. ПОТЕНЦИОМЕТРИЯ И ПОТЕНЦИОМЕТРИЧЕСКОЕ ТИТРОВАНИЕ
клеммам потенциометра и продолжают титрование и измерение э. д. с.
до тех пор, цока после скачка потенциала не будет добавлено еще 0,5—
1 мл титранта. Это необходимо для того, чтобы быть уверенным в
уменьшении АЕ после достижения максимального значения и для
возможности графического изображения хода кривой титрования.
Точное положение конечной точки титрования находят расчетным
и графическим способами.
Расчет. Содержание (g") уксусной кислоты (в г) во взятом для
титрования объеме испытуемого раствора вычисляют по формуле:
_ ^NaOH^NaOH^CH3COOH
8 1000
Анализ смеси хлористоводородной и борной кислот
Дифференцированное определение НС1 и Н3В03 (в присутствии
маннита или глицерина) основано на потенциометрическом титровании
их 0,1 н. раствором щелочи до достижения значений рН раствора,
отвечающих конечным точкам титрования хлористоводородной (при рН<7)
и борноманнитовой кислот (при рН>7). Титрование проводят со
стеклянным индикаторным электродом и Нас.КЭ сравнения.
Константа диссоциации борной кислоты (/СнзвОз ^ 10 ) настолько
мала, что сильная кислота в ее присутствии практически без
осложнения может быть количественно оттитрована щелочью. По завершении
титрования НС1 титруют Н3В03 в присутствии маннита (или
глицерина). При этом борная кислота образует комплексную маннитоборную
кислоту, обладающую относительно большей константой диссоциации
(Днап^Ю-5).
Методика определения. Настройку рН-метра проводят согласно
приложенной к прибору инструкции.
Сначала экспериментально находят значения рН раствора в
конечных точках титрования НС1 и Н3В03. Для этого в титрационный
сосуд наливают пипеткой 20 мл испытуемого раствора Н3В03 и
разбавляют 40 мл дистиллированной воды. Погружают предварительно
промытые электроды и мешалку и при хорошем перемешивании измеряют
рН раствора (pHi). Найденная величина рН показывает, что в случае
титрования смеси НС1 и Н3В03 до достижения этого значения рН
борная кислота еще не вступает в реакцию со щелочью. Когда рН >• 4, НС1
окажется недотитрованной на -<0,1% (если начальная концентрация
ее ~0,1 н.). Затем в этот же раствор прибавляют 1 г маннита (или
10 мл глицерина) и титруют 0,1 н. раствором NaOH, прибавляя титрант
небольшими порциями. После каждой внесенной порции титранта
измеряют рН раствора. Результаты измерения рН записывают в журнал
и по максимальной величине ДрН/AV находят рН раствора в конечной
точке титрования борноманнитовой кислоты (рНг). Полученные
значения pHi и рНг показывают, до какой величины рН в дальнейшем
следует титровать НС1 в присутствии H3B03 (pHi) и борноманнитовую
кислоту (рНг)-
Титрант прибавляют вначале большими порциями, а затем
медленно по каплям до тех пор, пока рН раствора не достигнет ранее
определенного значения pHi раствора Н3В03. Записывают объем
титранта (Умаон), израсходованный на определение НС1. В тот же
стакан вносят 1 г маннита (или 10 мг глицерина) и после его растворения
титруют вначале большими порциями, а затем каплями титранта, следя
за показанием рН-метра, до достижения значения рН2, отвечающего
§ 13. ТИТРОВАНИЕ ПО МЕТОДУ ОКИСЛЕНИЯ - ВОССТАНОВЛЕНИЯ 65
конечной точке титрования борноманнитовой кислоты. Фиксируют в
журнале общий объем израсходованного стандартного раствора
щелочи (^шон). Разность (^шон — ^шон) дает объем титранта,
эквивалентный содержанию Н3ВО3.
Расчет. Содержание НС1 и Н3В03 во взятом для титрования объеме
испытуемого раствора рассчитывают по формулам:
_ ^NaOH^NaOH^HCl _ Г NaOH ~ ^NaOH) ^ЫаОНЭН3Вб3
?НС1 looo ; ^НзВОз ШОО
§13. Титрование по методу окисления —- восстановления
Определение железа •(/!/)
Определение железа (III) осровано на предварительном его
восстановлении до степени окисления +11 небольшим избытком
раствора хлорида олова (II) и последующем титровании стандартным
раствором бихромата калия смеси ионов железа (II) и олова (II).
Дифференцированное определение Sn++ и Fe++ в растворе при совместном
их присутствии проводят титрованием 0,05 н. раствором бихромата
калия с компенсационным методом измерения э. д. с. элемента, состоящего
из индикаторного Pt-электрода, опущенного в испытуемый раствор, и
Нас.КЭ сравнения.
Дифференцированное титрование смеси Sn++ и Fe++ возможно
благодаря тому, что значения Е° ред-окс систем SnIV/Sn++ и Fe+^/Fe4"*
достаточно сильно различаются между собой. При этом в первую очередь
происходит окисление олова (II) как более сильного восстановителя,
а затем железа (II). Кривая титрования характеризуется наличием двух
четко выраженных скачков потенциала (условия дифференцированного
титрования смеси компонентов по методу окисления — восстановления
см. стр. 43).
Химические и электрохимические реакции в различные моменты
титрования:
до первой точки эквивалентности
3Sn++ + Cr20~+14H+ +=± 3Sn++++-f 2Cr+++ + 7H20
Sn++-2e +=± Sn++++
A"i5i + Tlg"Tsn++r
после первой и до второй точки эквивалентности
6Fe++ + Cr207""+14H+ +=± 6Fe+++ + 2Cr+++ + 7H20
Fe++~e +± Fe+++
F-pQ HHg [Fe+++]
после второй точки эквивалентности
Сг20:Г+14Н+ + 6е ^=± 2Сг+++ + 7Н20
^ [Сг20Г]Гн+]14
?-*3 + -Qlg [Cr+++]2
Этот метод, как правило, предназначен для определения железа (II)
или общего содержания железа (Fe++ и Fe+++) после предварительного
восстановления железа (III). Хотя ред-окс системы SnIV/SnH и
!бб ГЛ. II. ПОТЕНЦИОМЕТРИЯ И ПОТЕНЦИОМЕТРИЧЕСКОЕ ТИТРОВАНИЕ
С^ОГ/Ст*4"* и необратимы, все же в конечных точках титрования
возникают четко выраженные скачки потенциала, особенно если проводят
титрование теплых растворов.
Методика определения. Проводят всю подготовительную работу
с потенциометром (см. стр. 63). В титрационный сосуд (стакан
емкостью 200 мл) переносят пипеткой 20 мл испытуемого раствора,
добавляют 10 мл разбавленной (1:1) хлористоводородной кислоты,
нагревают до кипения и тут же по каплям прибавляют из капельницы
раствор хлорида олова (II), содержащий 50 г SnCl2-2H20 и 200 мл
раствора НС1 (1:1) в 1 л, до полного обесцвечивания
буро-красноватой окраски комплекса хлорида железа (III). Далее добавляют 50 мл
раствора НС1 (1:1) и нагревают до 60—70° С. Опускают в раствор
мешалку, индикаторный Pt-электрод и одно колено электролитического
ключа (заполненного насыщенным раствором КС1), другой конец
которого находится в стакане (емкостью ~ 100 мл) с насыщенным
раствором КС1 и Нас. КЭ. Последний в данном случае не рекомендуется
помещать в титрационный сосуд во избежание порчи его горячим
солянокислым раствором; Pt-электрод подключают к отрицательной, а
Нас. КЭ — к положительной клемме потенциометра.
После этого приступают к титрованию 0,05 н. стандартным
раствором бихромата калия. Титрант добавляют по каплям до тех пор, пока
не наступит первый скачок потенциала, отвечающий завершению
окисления избытка олова (II). Продолжают титрование уже порциями по
1 мл раствора бихромата; когда изменение AE/AV достигнет
приблизительно 20 мв, снова вносят титрант по каплям до второго скачка
потенциала, отвечающего конечной точке титрования железа (II). После
каждой внесенной порции титранта компенсационным методом измеряют
э. д. с. ячейки, записывают результаты в журнал. По этим данным
определяют расход стандартного раствора бихромата (Ук2Сг2о7)>
вызвавшего скачок потенциала во второй конечной точке.
Расчет. Содержание железа (III) вычисляют по формуле,
аналогичной приведенной на стр. 65.
Определение марганца (II)
Определение марганца (II) проводят титрованием 0,01 н.
стандартным раствором перманганата калия бескомпенсационным методом с
применением Pt-индикаторного электрода и Нас. КЭ сравнения.
В нейтральной пирофосфатной среде марганец (II) окисляется
перманганатом до пирофосфатного комплекса марганца (III):
4Mn++ + MnOJ + 15Н2Р207" + 8Н+ ^± 5Мп (Н2Р20?)~" + 4Н20
При отсутствии пирофосфата образуется Мп02.
Электрохимическая реакция до точки эквивалентности:
Мп++ + ЗН2Р20~~е ^=± Мп(Н2Р207)™
п [Мп(Н2Р207)з""]
1 [Мп++] [Н2Р207~]3
После завершения химической реакции электродный процесс и
потенциал должны быть обусловлены появившимся избытком
перманганата:
Мп04+8Н+ + ЗН2Р207" + 4е ^=± Мп(Н2Р207)—+ 4Н20
* [Mn04"][H+]8[H2P2orf
^+7lg [мп(н2Р2о7)Г-] (28)
§ 13. ТИТРОВАНИЕ ПО МЕТОДУ ОКИСЛЕНИЯ - ВОССТАНОВЛЕНИЯ 67
Как показывают уравнения (27) и (28), скачок потенциала при
осуществлении этой химической реакции будет тем больше, чем больше
равновесная концентрация НгРгО/" в растворе, так как в уравнении
(27) она входит в знаменатель, а в уравнении (28) — в числитель и
тем самым в первом случае (до точки эквивалентности) вызывает
понижение Е, а во втором (после точки эквивалентности) — его
возрастание, т. е. увеличение АЕ в конечной точке титрования. Казалось бы,
увеличение [Н+] должно благоприятно влиять на направление реакции
[см. уравнение (28)], однако вследствие неустойчивости пирофосфат-
ного комплекса марганца (II) в кислой среде реакцию следует
проводить только в нейтральной среде. Метод успешно применяется для
определения марганца в различных сплавах.
Методика определения. Сухую батарею и микроамперметр (вместо
гальванометра) подключают к соответствующим клеммам
потенциометра. В титрационный сосуд (стакан емкостью ~200 мл) опускают
магнитную мешалку, наливают 50 мл теплого свежеприготовленного
5%-ного раствора пирофосфата натрия и ставят стакан на подставку
магнитной мешалки. При хорошем перемешивании в стакан медленно
вносят пипеткой 20 мл испытуемого раствора марганца (II). Если
образующийся белый осадок не исчезает, то раствор непригоден для
дальнейшей работы (так случается при анализе растворов сплава). В
прозрачный раствор пирофосфатного комплекса маргайца (II) опускают
индикаторный Pt-электрод и одно колено электролитического ключа,
другой конец которого находится в стакане (емкостью около 100 мл),
содержащем насыщенный раствор КС1 и Нас.КЭ. Электролитический
ключ заполнен насыщенным раствором КС1. Pt-Электрод подключают
к положительному полюсу потенциометра, а Нас. КЭ последовательно
с реостатом — к отрицательному. Сопротивление, отбираемое из
реостата и включаемое в цепь гальванического элемента, должно быть
такой величины, чтобы в момент скачка потенциала сила тока в цепи не
превышала верхнего предела показания шкалы микроамперметра.
Перед началом титрования э. д. с. гальванического элемента
компенсируют потенциометром. В этом методе нет необходимости настраивать
потенциометр стандартным элементом Вестона, так как величина
э. д. с. не имеет значения, а напряжение, взятое от потенциометра как
от делителя напряжения, сохраняется постоянным.
Раствор титруют 0,01 н. раствором перманганата калия при
перемешивании, вначале приливая по 1 мл, а вблизи конечной точки
титрования, когда изменение силы тока становится заметным, по каплям.
После каждой прибавленной порции титранта в журнале фиксируют
значения тока, измеряемые микроамперметрам. Запись проводят, как
указано в табл. 3, но с той разницей, что взамен э. д. с, АЕ, AE/AV
и т. д. измеряют значения i, At, Ai/AV и т. д. Объем титранта, при
котором достигнуто максимальное значение Ai/AV, найденное расчетными
графическим способами, соответствует конечной точке титрования.
Расчет. Содержание марганца во взятом для титрования объеме
испытуемого раствора находят по формуле, аналогичной приведенной
на стр. 64.
Определение иода
Иод определяют титрованием 0,05 н. стандартным раствором арсе-
нита натрия бескомпенсационным методом с применением
биметаллической пары электродов платина/графит. При этом 12 восстанавливается
арсенитом до иоднда.
68 ГЛ. 1Ь ПОТЕНЦИОМЕТРИЯ И ПОТЕНЦИОМЕТРИЧЕСКОЕ ТИТРОВАНИЕ
Методика определения. В титрационный сосуд (стакан емкостью
около 200 мл), в который опущена магнитная мешалка, вносят
пипеткой 20 мл испытуемого раствора 12, прибавляют 1 г NaHC03 и
разбавляют 50 мл воды. Фиксируют оба электрода (платиновый и
графитовый) в стакане таким образом, чтобы они не прикасались друг к другу.
Один из электродов подключают к скользящему контакту реостата,
неподвижный контакт которого присоединяют к ламповому вольтметру.
Второй электрод непосредственно подключают ко второй клемме
вольтметра.
Перед титрованием подбирают величину сопротивления так, чтобы
отклонение стрелки вольтметра было минимальным. Затем, не меняя
положение скользящего контакта реостата, приступают к титрованию
раствора, прибавляя небольшими порциями 0,05 н. раствор арсенита.
При этом следят за движением стрелки лампового вольтметра. Как
только 1—2 капли титранта вызовут резкое отклонение стрелки,
титрование прекращают. Объем стандартного раствора, израсходованный до
этого момента, соответствует конечной точке титрования.
С таким же успехом можно осуществить титрование
компенсационным методом, это даст возможность вычертить кривые
AE/AV—V.
Расчет. Содержание иода вычисляют по формуле, аналогичной
приведенной на стр. 64.
§ 14. Титрование по методу осаждения
Анализ смеси иодида й хлорида
Дифференцированное определение 1~ и С1~ в их смеси проводят
титрованием 0,05 н. стандартным раствором нитрата серебра с
серебряным индикаторным электродом и Нас.КЭ сравнения. Э. д. с. потенцио-
метрической ячейки измеряют компенсационным методом. Поскольку
nPAgi <C nPAgci, в первую очередь титруется иодид с большим скачком
потенциала в конечной точке, но меньшим, чем при отсутствии
хлорида. Теоретически скачок наступает несколько раньше точки
эквивалентности, но практически точка эквивалентности и конечная точка
титрования совпадают. Кривая титрования из-за присутствия хлорида
не симметрична.
Пока в растворе присутствуют ионы иодида, потенциал Нас.КЭ
больше потенциала серебряного электрода, поэтому последний должен
быть подключен к отрицательной клемме потенциометра. Обычно
завершение титрования иодида, т. е. первый скачок потенциала,
совпадает с моментом изменения знака Ag-электрода в гальваническом
элементе. Далее потенциал этого электрода медленно возрастает до
наступления второго скачка потенциала — конечной точки титрования
хлорида, после чего снова происходит замедленное повышение его.
При аргентометрическом титровании недопустимо применение
электролитических ключей, заполненных насыщенным раствором хлорида
калия; его следует заменить насыщенным раствором KN03, не
реагирующим с Ag+-HOHaMH.
Методика определения. Вся предварительная настройка
потенциометра, приемы работы и записи результатов аналогичны изложенным
выше (см. стр. 46), но перед началом титрования раствор должен
быть приблизительно 10%-ным относительно нитрата бария. Так как
система гетерогенна и происходит адсорбция осадком титрующего и
титруемого ионов, потенциал устанавливается не быстро, особенно
вблизи конечной точки титрования. Поэтому следует ждать достижения
§ 15. ТИТРОВАНИЕ ПО МЕТОДУ КОМПЛЕКСООБРАЗОВАНИЯ
69
более или менее постоянного значения Е. Обычно можно
довольствоваться данными, которые в течение 1 мин изменяются не более, чем на
3—5 мву поскольку скачки потенциала достаточно большие и такие
колебания не могут влиять на точность нахождения конечнцй точки
титрования.
Расчет. Если для достижения первой конечной точки затрачено
V мл титранта, а общий расход его при наступлении второго скачка
потенциала равен V" мл, то расчет содержания иодида и хлорида во
взятом для титрования объеме испытуемых растворов проводят по
формуле, аналогичной приведенной выше (см. стр. 65).
§ 15. Титрование по методу комплексообразования
Определение железа (III)
Определение железа (III) по методу комплексообразования
проводят титрованием 0,05 М стандартным раствором комплексона III с
индикаторным Pt-электродом и Нас. КЭ сравнения. Э. д. с. потенциомет-
рической ячейки измеряют компенсационным методом.
В кислой среде (рН около 3—4) Fe+++-HOHbi образуют достаточно
прочное комплексное соединение (FeY~) с комплексоном III (H2Y~),
вследствие чего потенциал Pt-электрода постепенно понижается. Как
только все Ре+++-ионы окажутся связанными в комплекс, наступает
резкий скачок потенциала из-за практического исчезновения окисленной
формы ред-окс системы Fe+++/Fe++. Дальнейшее прибавление титранта
почти не изменяет потенциал индикаторного электрода. Для получения
устойчивых равновесных потенциалов титруемый раствор должен
содержать сопряженную форму ред-окс пары — Fe++-HOHbi, которые в
кислой среде практически не реагируют с H2Y~~.
При этом происходят следующие химические и электрохимические
реакции:
до точки эквивалентности
Fe+++ + H2Y- +± FeY~ + 2H+ K = - [реГ~1[н+]2
[Fe+++][H2Y-]
Fe+++ + в +± Fe++ E - 2* + Ь \g ^?^ (29)
после точки эквивалентности
Fe++ + H2Y--e +± FeY" + 2H+
?2+*Ig[Fe++][H2Y-] (30)
Подставляя в уравнение (29) значение [Fe+++] из уравнения
константы равновесия, получим:
'¦"-"""^Дп <3,)
Сопоставление уравнений (31) и (30) показывает, что ??>?г на
величину fllg/C. Поскольку К величина большая, значение Д?° велико
и обусловливает большой скачок потенциала. Так как в процессе
химической реакции кислотность раствора возрастает (замещение ионов
водорода в титранте H2Y~), необходимо в титруемом растворе создать
70 ГЛ. ГГ. ПОТЕНЦИОМЕТРИЯ И ПОТЕНЦИОМЕТРИЧЕСКОЕ ТИТРОВАНИЕ
буферную среду, ибо равновесная концентрация FeY~ сильно зависит
от [Н+].
Методика определения. Вся предварительная настройка
потенциометра, приемы работы и записи результатов аналогичны изложенным
выше (см. стр. 46). В титрационный сосуд емкостью около 200 мл
вносят пипеткой 20 мл испытуемого раствора, разбавляют 50 мл воды,
прибавляют по каплям 10%-ный раствор ацетата аммония до тех пор,
пока раствор не окрасится в желто-оранжевый цвет (гидролиз соли
трехвалентного железа). Добавляют 5—10 капель 1%-ного раствора
соли Мора (не содержащей Fe+++), опускают Pt-электрод, магнитную
мешалку и один конец электролитического ключа, заполненного
насыщенным раствором КС1. Другой конец ключа опускают в стакан
емкостью около 100 мл, содержащий насыщенный раствор КС1, туда же
помещают Нас.КЭ.
Стаканы ставят на площадке магнитной мешалки и приступают, как
обычно, к титрованию 0,05 М раствором комплексона III. Вблизи точки
эквивалентности потенциал устанавливается медленно, особенно при
точном титровании по каплям, поэтому перед записью результатов
измерения э. д. с. ждут установления равновесия.
Расчет. Содержание железа вычисляют по формуле, аналогичной
приведенной на стр. 64.
ПОТЕНЦИОМЕТРИЧЕСКОЕ ТИТРОВАНИЕ ПОД ТОКОМ (i=?0)
§ 16. Титрование по методу окисления — восстановления
Определение сурьмы (Ш)
Определение сурьмы (III) по методу окисления проводят
титрованием 0,05 н. стандартным раствором КВг03. Разность потенциалов А?
двух поляризованных Pt-электродов, опущенных в испытуемый раствор,
измеряют компенсационным методом.
Сурьма (III) в сильно солянокислой среде окисляется броматом до
степени окисления +V. Бромат восстанавливается до бромид-иона,
который после завершения основной реакции взаимодействует с
избытком бромата с выделением Вг2.
При этом происходят следующие химические и электрохимические
реакции:
до точки эквивалентности
3H3Sb03 + BrOJ +± 3H3Sb04-f ВГ
H3Sb03 + Н20 - 2e ^± H3Sb04 + 2H+
о <> [Н3$Ь04][Н+]2
?-^1"*- 2 lg [H3Sb03]
после точки эквивалентности
ВгОз+5ВГ + 6Н+ ^=± ЗВг2 + ЗН20
Br2 + 2e ^zt 2ВГ
?=rf + ±j JB?i
п ^2^ 2 g [Br"]2
Первая (до точки эквивалентности) электрохимическая реакция
необратима, между тем как вторая (после точки эквивалентности)
обратима; следовательно, потенциал индикаторных электродов не может
§ 16. ТИТРОВАНИЕ ПО МЕТОДУ ОКИСЛЕНИЯ - ВОССТАНОВЛЕНИЯ 71
быть устойчивым до достижения точки эквивалентности (образование
смешанного потенциала) и годным для наблюдения за изменением
концентрации сурьмы (III) в растворе в процессе титрования.
Однако если использовать два индикаторных электрода и
поляризовать их небольшим током (несколько микроампер), то до конечной
точки титрования на одном из них в зависимости от полярности
подключения к внешнему источнику тока вероятнее всего происходит
окисление Вг" до Вг2, а на другом — восстановление Sbv до Sb111. Поскольку
эти процессы протекают при большой разнице величин потенциалов
электродов, до конечной точки титрования наблюдается большая
разность потенциалов (АЕ) между двумя поляризованными
индикаторными электродами. После конечной точки вследствие обратимости пары
Вг2/2Вг~, несмотря на ток поляризации, потенциал анода и катода
настолько близки, что АЕ резко понижается, до некоторого малого
значения и остается практически постоянной. Наблюдаемый скачок
потенциала АЕ соответствует конечной точке титрования сурьмы (III).
Методика определения. Потенциометр приводят в рабочее
состояние согласно инструкции. В титрационный сосуд (стакан емкостью
около 200 мл), в который опущена магнитная мешалка, вносят
пипеткой 20 мл испытуемого раствора SbCl3, прибавляют 0,5 г КВг,
разбавляют 50 мл воды и доливают столько концентрированной
хлористоводородной кислоты, чтобы титруемый раствор стал 4 н. относительно НС1-
Погружают Pt-электроды и подключают их к клеммам
потенциометра (в данном случае полярность подключения электродов не имеет
значения, так как они идентичны и находятся в одном и том же
растворе). Электрод, присоединенный к положительной клемме
потенциометра, одновременно подключают к положительному полюсу, а другой
электрод, присоединенный к отрицательной клемме потенциометра,
последовательно через переключатель тока, микроамперметр и мегомные
сопротивления — к отрицательному полюсу внешнего источника
постоянного тока с высоким выходным напряжением. Для получения
требуемой величины тока поляризации необходимо установить
соответствующую величину внешнего сопротивления, включаемого в цепь. Согласно
закону Ома, если требуются токи в 5—10 мка, а напряжение внешнего
источника тока около 80 в, то сопротивление должно быть 80/10-10~6-^
*<8*106 ом, или 8 мегом. Конечно, в данном приближенном расчете не
учтены внутренние сопротивления ячейки, проводников, контактов и
т. п. Однако все они вместе взятые редко составляют 3—5 ком, и
ошибка подобного расчета не превышает 1%, тем более, что имеет
значение не абсолютная величина тока поляризации, а ее постоянство
в процессе работы, которое и обеспечивается большими выходными
напряжением источника тока и внешним сопротивлением.
После включения в цепь поляризации необходимого сопротивления
замыкают цепь и контролируют микроамперметром, правильность
величины включенного сопротивления и полярность подключения
электродов. Не включая тока поляризации, приступают к ориентировочному,
а затем (при тех же условиях) с новой порцией испытуемого раствора —
к точному титрованию 0,05 н. раствором бромата калия.
Полученные результаты оформляют в виде таблиц и графика
зависимости AE/AV от V, так как в данном методе непосредственно
получают значения АЕ — разность потенциалов между двумя
поляризованными электродами.
Расчет. Содержание сурьмы (III) во взятом для титрования объеме
испытуемого раствора рассчитывают по формуле, аналогичной
приведенной на стр. 64.
ГЛАВА III
КОНДУКТОМЕТРИЯ И КОНДУКТОМЕТРИЧЕСКОЕ
ТИТРОВАНИЕ
Л. ТЕОРЕТИЧЕСКИЕ ОСНОВЫ МЕТОДА
§ 1. Удельная и эквивалентная электропроводность
Электропроводность растворов электролитов обусловливается
движением ионов под действием электрического поля. Перенос
электричества в растворах электролитов осуществляется ионами. Как и все
проводники, растворы электролитов характеризуются определенным
сопротивлением. Величина, обратная этому сопротивлению, называется
электропроводное! ью:
г=т (,)
где R — сопротивление раствора, ом;
W — электропроводность раствора, выражаемая в обратных омах, ом~К
Сопротивление раствора электролита прямо пропорционально
расстоянию / (см) между погруженными в него электродами и обратно
пропорционально их площади 5 (см2):
Я = р4 (2)
Коэффициент пропорциональности р (ом-см) называется удельным
сопротивлением. Если / = 1 см, a S = 1 см2, то р = R.
Следовательно, удельное сопротивление равно сопротивлению столба
жидкости длиной в 1 см с поперечным сечением 1 см2, т. е.
сопротивлению 1 см3 раствора.
Удельная электропроводность (к) представляет собой величину,
обратную удельному сопротивлению:
х = — (ом"1 • см"-1) (3)
Таким образом, удельная электропроводность соответствует
электропроводности 1 см3 раствора, находящегося между электродами
площадью в 1 см2, расположенными на расстоянии 1 см друг от друга.
Электропроводность электролитов удобнее относить к числу грамм-
эквивалентов растворенного вещества. Поэтому было введено понятие
эквивалентной электропроводности. Эквивалентная электропроводность
(X) равна электропроводности такого объема раствора, помещенного
между двумя параллельными электродами, расположенными на
расстоянии 1 см, который содержит один грамм-эквивалент вещества.
Эквивалентная и удельная электропроводность связаны следующей
зависимостью. Если концентрация электролита (С) выражена в грамм-
эквивалентах на 1 л, то в 1 см3 раствора содержится С/1000
грамм-эквивалентов. Объем (V) в кубических сантиметрах, содержащий 1 г-экв
растворенного вещества, называется разбавлением. Этот объем равен
§ I. УДЕЛЬНАЯ И ЭКВИВАЛЕНТНАЯ ЭЛЕКТРОПРОВОДНОСТЬ
73
1000/С. Эквивалентную электропроводность можно выразить через
удельную электропроводность и разбавление {ом~1 • см2/г-экв):
1 1000 т/
Эквивалентные электропроводности (подвижности) ионов.
Предположим, что в растворе электролита на расстоянии / находятся
электроды площадью S, к которым приложена разность потенциалов Е. Так
как в растворах электричество переносится ионами, величина удельной
электропроводности зависит от концентрации и заряда ионов, а также
скорости их движения. Допустим, что электролит образует
однозарядные ионы. Обозначим концентрацию электролита (С) в
грамм-эквивалентах, а степень его диссоциации через а. Абсолютные скорости
движения катионов и анионов при падении потенциала в 1 в на 1 см назовем
v+ и V-. Если разность потенциалов между электродами ?", а расстояние
между ними /, скорости катионов и анионов имеют значение v+E/l и
v-E/L Сила тока, проходящего через раствор, зависит от количества
ионов обоих знаков, перемещающихся в противоположных
направлениях. Через поперечное сечение S между Электродами в 1 сек пройдут
все катионы и анионы, содержащиеся в объеме (v+E/l)S и (v-E/l)S.
Сила тока, т. е. количество электричества в кулонах, перенесенное
ионами в 1 сек, равна:
где F — число Фарадея, равное 96 500 кулонов.
Согласно закону Ома W = //?, отсюда электропроводность равна:
Если S = 1 см2, а I = 1 см, то W представляет собой удельную
электропроводность (х):
аС
x = looo'F(t;+ + u-)
Поскольку эквивалентная электропроводность X = kV, a V = 1000/С,
находим:
X = aF(v+ + v-) ($)
Абсолютные скорости движения ионов очень малы, поэтому
пользуются величинами в F раз большими, называемыми подвижностями
ионов. Подвижности ионов представляют собой эквивалентные
электропроводности ионов, которые обозначают %+ и Я-. Отсюда значения
удельной и эквивалентной электропроводности могут быть выражены:
и
Я = а(Я++Я_) (8)
При бесконечно большом разбавлении а = 1 и эквивалентная
электропроводность стремится к наибольшему значению К0. В этом случае
A,°-*°. + VL (9)
Таким образом, предельная эквивалентная электропроводность
равна сумме предельных эквивалентных электропроводностей ионов или
сумме подвижностей ионов при бесконечном разбавлении. Предельные
эквивалентные электропроводности некоторых ионов приведены в табл. 4.
74 ГЛ. III. КОНДУКТОМЕТРИЯ И КОНДУКТОМЕТРИЧЕСКОЕ ТИТРОВАНИЕ
ТАБЛИЦА 4
Предельная эквивалентная электропроводность (подвижность)
ионов (Л°) в водных растворах при 25 °С
Катион
н+
NH+
К+
72 РЬ++
i/з Fe+ + +
V2 Ва+ +
V. А1+ + +
Ag+
!/2 Са+ +
!/2 Sr+ +
!/2 Си++
72 Cd+ +
V. Fe + +
V2 Mn++
V2 Mg++
Na+
Li+
K°
349,8
73,6
73,5
70
68
63,6
63
61,9
59,5
59,5
56,6
54
53,5
53,5
53,1
50,1
38,7
Анион
OH""
V4 [Fe(CN)6r =
7з tFe(CN)e]"~
V2 CrO™
V2 so4"
Г
Br""
cr
V2 cao4"
N03-
V2 co™
HS"
cio4-
F~
HCO3-
CH3COO"
C6H5COO"
K°
198,3
110,5
100,9
85
80,0
78,8
78,1
76.4
74,0
71,5
69,3
65
64,5
55,4
44,5
40,9
32,3
Подвижности ионов имеют большое значение для кондуктометри-
ческих определений, так как на основании этих величин, изменяющихся
в связи с изменением состава ионов, можно предвидеть характер
изменения электропроводности раствора в процессе титрования.
Влияние природы электролита и растворителя на
электропроводность. Природа электролита и растворителя оказывает большое
влияние на электропроводность. Как видно из табл. 4, ионы обладают
различной подвижностью. Аномально высокой подвижностью в водных
растворах обладают Н+-ионы (К0 = 349,8) и ОН--ионы (Я0 =198,3). Это
объясняется специфическим механизмом их движения в растворе.
Перемещение этих ионов к электродам осуществляется «эстафетным» путем.
Находящиеся в растворе ионы гидроксония Н30+ передают свои
протоны соседним молекулам воды, которые в свою очередь превращаются
в ионы гидроксония, а протоны перемещаются по направлению к катоду.
Ацалогичные процессы происходят и с участием гидроксильных ионов.
Другие катионы и анионы в водных растворах сравнительно мало
различаются своей подвижностью; изменение подвижности наблюдается
в пределах 30—70.
В других растворителях электропроводность электролитов
изменяется. Наибольшее влияние при этом оказывает вязкость растворителя
и его диэлектрическая проницаемость; изменяются скорости движения
ионов, степени диссоциации электролитов, а в растворителях с низкими
значениями диэлектрической проницаемости наблюдаются процессы ас-
§ 1. УДЕЛЬНАЯ И ЭКВИВАЛЕНТНАЯ ЭЛЕКТРОПРОВОДНОСТЬ
7$
социации ионов. Подвижности водородных и гидроксильных ионов
уменьшаются и в некоторых средах мало отличаются от подвижностей
других ионов.
Влияние концентрации электролита на электропроводность.
Сильные электролиты. Сильные электролиты в водных растворах
практически полностью диссоциированы и для них принимают степень
диссоциации а, равную 1. Однако абсолютные скорости движения, а
следовательно, и подвижности зависят от концентрации ионов в растворе,
что объясняется силами межионного взаимодействия. С увеличением
концентрации уменьшаются расстояния между ионами и увеличиваются
межионные взаимодействия, что приводит к торможению движения
катионов и анионов, а следовательно, к понижению их подвижности.
Поэтому эквивалентная электропроводность сильных электролитов,
имеющая максимальное значение при бесконечном разбавлении, уменьшается
с повышением концентрации.
Отношение эквивалентной электропроводности при данной
концентрации к эквивалентной электропроводности при бесконечном
разбавлении называется коэффициентом электропроводности f%. Этим
коэффициентом вносится поправка на силы межионного взаимодействия.
Величину эквивалентной электропроводности сильного электролита при
данной концентрации с учетом коэффициента электропроводности
находят по формуле:
Я = ^(А,0+ + Л°_) (10)
При малых концентрациях зависимость эквивалентной
электропроводности сильного электролита от концентрации выражается
эмпирическим уравнением: _
%~X*~aVc' (11)
где а — постоянная, зависящая от природы растворителя и температуры;
С — концентрация электролита.
Удельная электропроводность сильных электролитов с учетом
коэффициента электропроводности выражается формулой:
С увеличением концентрации удельная электропроводность сильных
электролитов сначала возрастает, а затем может понижаться, что
приводит к появлению максимума удельной электропроводности. Это
объясняется влиянием двух факторов. С одной стороны, увеличение
концентрации вызывает повышение электропроводности, так как
увеличивается содержание ионов в единице объема. С другой стороны,
увеличение концентрации снижает скорость движения ионов» что уменьшает
проводимость. Однако максимумы обнаруживаются для
концентрированных растворов, которые обычно не применяются в аналитической
практике.
Слабые электролиты. Растворы слабых электролитов имеют
невысокие концентрации ионов, и межионные взаимодействия в них
невелики. Поэтому в разбавленных растворах f^ может быть принят
равным единице, а эквивалентные электропроводности ионов можно
считать равными их предельному значению %°+ и А-. Следовательно, в
разбавленных растворах удельная и эквивалентная электропроводности
равны:
И
Я«а(Л°.+я1) (14)
76 ГЛ. III. КОНДУКТОМЕТРИЯ И КОНДУКТОМЕТРИЧЕСКОЕ ТИТРОВАНИЕ
Таким образом, большое влияние на электропроводность слабых
электролитов оказывает степень их диссоциации. С разбавлением
эквивалентная электропроводность слабых электролитов растет вследствие
увеличения степени диссоциации и принимает максимальное значение
при бесконечном разбавлении (а = 1).
При кондуктометрическом определении слабых электролитов
большое значение имеют величины констант диссоциации, так как они дают
возможность рассчитывать равновесные концентрации ионов при
данном разбавлении и предугадывать характер изменения проводимости
раствора при титровании.
Удельная электропроводность слабых электролитов незначительно
увеличивается с повышением концентрации. При высоких
концентрациях уменьшение степени диссоциации может вызвать ее понижение.
Если раствор представляет смесь электролитов, величину удельной
электропроводности разбавленных растворов вычисляют по уравнению:
Х=тш-2СЛ (15)
где Ci — концентрация ионов;
Я/ — эквивалентные электропроводности ионов.
Влияние температуры на электропроводность. Зависимость
удельной электропроводности от температуры выражают уравнением:
x/^Ml + af + p*2) 06)
где к0 — удельная электропроводность раствора при 0° С;
а и р —коэффициенты, зависящие от природы электролита и концентрации;
t — температура.
С повышением температуры электропроводность увеличивается, так
как уменьшение вязкости раствора приводит к увеличению
подвижности ионов. Увеличение степени диссоциации также может привести к
повышению электропроводности. Повышение температуры на 1 град
вызывает увеличение электропроводности раствора на 2—2,5%.
§ 2. Кондуктометрические методы анализа
Прямая кондуктометрия. Концентрация электролита может быть
определена по электропроводности раствора, так как в определенных
пределах возможна прямая пропорциональность между этими
величинами. Метод широко используют для определения индивидуальных
электролитов в растворе. Возможно также определение электролита
в смесях в случаях, когда концентрации примесей не изменяются.
Прямая кондуктометрия позволяет решать многие практические
задачи и осуществлять непрерывный контроль производства. Широко
применяется определение концентрации солевых растворов с помощью
специальных солемеров. Кондуктометрию используют для контроля
процесса очистки воды и, в частности, для контроля качества
дистиллированной воды, оценки загрязненности сточных вод, при
определении общего содержания солей в минеральной, морской и речной
воде. Методом кондуктометрии осуществляют контроль операций
промывки осадков и регенерации ионитов. Используя экстракцию
дистиллированной водой, определяют чистоту малорастворимых осадков или
органических препаратов.
Определение электропроводности — один из методов контроля
качества пищевых продуктов: молока, вин, напитков и т. д.
Прямая кондуктометрия применяется и для определения влажности
органических растворителей, газов, твердых солей, текстильных
материалов, бумаги, зерна и т. д.
§ 2. КОНДУКТОМЕТРИЧЕСКИЕ МЕТОДЫ АНАЛИЗА
77
Косвенная кондуктометрия. Наряду с прямой кондуктометрией
применяется также косвенная кондуктометрия, при которой кроме
электропроводности измеряют и другие величины (рефракцию, вязкость, рН,
плотность, массу сухого остатка и т. д.). При этом возможно
определение не только индивидуальных веществ, но и смесей. Например, по
величине электропроводности раствора и массе сухого остатка можно
провести определение двух солей—КС1 и KI в растворе.
В некоторых случаях определению электропроводности
предшествует химическое взаимодействие. Именно так проводят кондуктометри-
ческое определение различных газов: С02, СО, 02, NH3, SO2, H2S и т. д.
Например, при определении С02 измеряют электропроводность раствора
щелочи после поглощения им С02.
Для аналитических целей применяют также сочетание нескольких
методов. Примером может служить применение кондуктометрии при
хроматографическом разделении.
Кондуктометрическое титрование. Кондуктометрическое титрование
используется при определении индивидуальных веществ и анализе
разнообразных смесей. Точку эквивалентности при кондуктометрическом
титровании определяют по изменению электропроводности раствора.
Электропроводность измеряют после добавления каждой порции тит-
ранта. Зависимость электропроводности раствора от количества
добавленного титранта изображают графически. Полученный график
называют кривой кондуктометрического титрования. Кондуктометрические
кривые имеют излом, соответствующий точке эквивалентности.
Изменение электропроводности раствора до и после точки
эквивалентности может быть линейным, что, например, наблюдается при
нейтрализации сильных кислот и оснований. Кондуктометрическая кривая
в этом случае состоит из двух прямых линий, пересекающихся в точке
эквивалентности.
Однако изменение электропроводности раствора при титровании не
всегда носит линейный характер. Нелинейная зависимость наблюдается
в случаях: 1) когда реакция проходит не количественно; и 2) если
в процессе титрования изменяются степени диссоциации или степени
гидролиза веществ, участвующих в реакции, и т. д.
Когда реакции проходят не количественно (гидролиз солей,
некоторая растворимость осадков, частичная диссоциация комплексов и др.),
обратимость реакций вызывает повышение проводимости растворов.
Это становится особенно заметным вблизи точки эквивалентности
и выражается в плавности изгиба кондуктометрической кривой,
поскольку в начале титрования обратимость реакции подавляется, так как
в растворе находится избыток определяемого вещества, а в конце
титрования это же влияние оказывает избыток титранта.
Кондуктометрический метод дает возможность в определенных
пределах использовать реакции, протекающие неколичественно. Для этого
необходимо, чтобы проводимость изменялась линейно хотя бы на
отдельных участках кондуктометрической кривой до и после точки
эквивалентности. При графическом установлении точки эквивалентности
достаточно измерений, сделанных задолго до достижения этой точки.
Затем продолжают прямолинейные участки кривой титрования до их
пересечения и находят таким образом точку эквивалентности с
достаточно высокой точностью (см. рис. 6, г).
Нелинейное изменение электропроводности раствора при
титровании нередко наблюдается даже в том случае, когда реакция протекает
количественно. Излом кондуктометрической кривой при этом в точке
эквивалентности может быть резким, но сама кондуктометрическая
78 ГЛ. III. КОНДУКТОМЕТРИЯ И КОНДУКТОМВТРИЧЕСКОЕ ТИТРОВАНИЕ
кривая может иметь различные особенности (изогнутость, пологий
минимум или максимум), которые не имеют аналитического значения.
Формы кондуктометрических кривых такого типа наблюдаются, когда
в процессе титрования изменяются степени диссоциации или степени
гидролиза веществ, участвующих в реакции.
Кривые подобного типа могут быть использованы для
аналитических целей только в том случае, если перед точкой эквивалентности
наблюдается линейное изменение проводимости.
При титровании следует проводить большое число измерений
электропроводности. Для определения точки эквивалентности используют
близкие к ней участки кривых.
Хронокондуктометрическое титрование. Кондуктометрическое
титрование может быть полностью или частично автоматизировано.
В титрометрах промышленного типа, применяемых для титрования
в потоке, автоматизированы все операции: отбор пробы, добавление
растворителя и реагентов, перемешивание, фиксирование результатов
титрования, удаление анализируемого раствора из ячейки и
промывание ее.
Приборы лабораторного типа обычно полуавтоматические. Вручную
проводят отбор пробы, пуск титранта и устройства, регистрирующего
результаты титрования, удаление раствора и промывание ячейки.
Определения при помощи этих приборов обычно проводят при
постоянной скорости истечения титранта, а концентрацию определяемого
вещества рассчитывают по времени титрования. В этих приборах
осуществляется непрерывная или точечная запись кондуктометрических
кривых. В первом случае длина диаграммной ленты регистратора от
начала титрования до точки эквивалентности пропорциональна времейи
титрования. Во втором случае продолжительность титрования
устанавливают по количеству интервалов между точками до излома кондукто-
метрической кривой и временем, протекающим между записью этих
точек. Кондуктометрический метод анализа, основанный на определении
содержания вещества по времени, затраченному на его титрование,
называется хронокондуктометрическим титрованием.
При автоматической записи кривых расширяются практические
возможности применения реакций, сопровождающихся нелинейным
изменением электропроводности раствора, в том числе и реакций,
протекающих неколичественно. Воспроизводимость условий титрования позволяет
при помощи стандартных графиков проводить определения, при
которых вещества реагируют в нестехиометрических отношениях.
Область применения кондуктометрического титрования. В основу
кондуктометрических определений могут быть положены разнообразные
типы химических реакций.
К преимуществам метода кондуктометрического титрования
относится возможность дифференцированного определения веществ в
многокомпонентных смесях в водных растворах. Другим преимуществом
метода является возможность определений в окрашенных и мутных
растворах, а также в присутствии окислителей или восстановителей,
ограничивающих, например, применение кислотно-основных индикаторов.
Кондуктометрический метод позволяет проводить определения не только
в сравнительно концентрированных растворах, но и в разбавленных
до Ю-4 М.
Метод имеет ограниченное применение в случаях, когда в растворах
присутствует очень большое количество посторонних электролитов, так
как при титровании наблюдается незначительное изменение
электропроводности.
§ 3. КИСЛОТНО-ОСНОВНОЕ ТИТРОВАНИЕ
79
Точность метода. Кондуктометрическое титрование обычно
проводят без термостатирования растворов, относительные ошибки
определений индивидуальных электролитов при этих условиях составляют
±1%, а при титровании смеси электролитов ошибка не выше ±2%.
Точность определений может быть увеличена, если
электролитическую ячейку поместить в термостат и измерение сопротивления
раствора после добавления каждой порции титранта проводить после
достижения постоянной температуры.
§ 3. Кислотно-основное титрование
Титрование кислот. Титрование сильной кислоты сильным
основанием протекает согласно следующему уравнению:
H+ + An" + Kt+ + OH~" ^=± Kt+ + An" + H20
Электропроводность раствора при этом понижается, так как
высокоподвижные ионы водорода заменяются менее подвижными
катионами основания. Добавление избытка основания приводит к
повышению электропроводности. Кондуктометрическая кривая представляет
собой две пересекающиеся прямые; точка эквивалентности расположена
в месте их пересечения (рис. 6,а).
Кривые титрования кислот средней силы изогнуты и могут давать
пологий минимум, не имеющий аналитического значения. Например,
кривая титрования 0,1 ц. раствора дихлоруксусной кислоты (рКа=1,25)
сильным основанием имеет слабый изгиб вблизи точки эквивалентности,
а монохлоруксусной кислоты (рКа=2,75) — пологий минимум (рис.6,б).
С разбавлением кислоты увеличивается степень ее диссоциации и
кривые приобретают форму, характерную для более сильных кислот,
что можно видеть на примере (рис. 6, в) титрования сильным
основанием 0,1 н., 0,001 н. и 0,0001 н. растворов уксусной кислоты (рКа=4,75).
При титровании основанием ОД н. раствора СНзСООН в начале титрования
наблюдается минимум электропроводности. Однако диссоциация уксусной кислоты
быстро подавляется и на большей части кондуктометрической кривой до точки
эквивалентности происходит линейное повышение проводимости, вызываемое накоплением
СНзСОО"- и Na^-HOHOB. Между тем при титровании 0,0001 н. раствора СНзСООН
электропроводность непрерывно понижается. Образующиеся в растворе СН^СОО~-ионы
также подавляют диссоциацию СНзСООН, поэтому кондуктометрическая кривая
изогнута. Однако в связи с увеличением степени диссоциации СНзСООН в разбавленных
растворах не происходит полного подавления ее диссоциации под влиянием СНзСОО"-
ионов. Равновесная концентрация водородных ионов в этих условиях остается
достаточно высокой, и при их нейтрализации электропроводность понижается.
При титровании очень слабых кислот становится заметным влияние
гидролиза образующейся соли, которое выражается в плавном изгибе
кондуктометрической кривой вблизи точки эквивалентности.
Верхние границы \>Ка кислот, определение которых можно
проводить кондуктометрическим титрованием, имеют следующие значения:
Концентрация кислоты, н. . . ОД 0,01 0,001
рКа * <Ю <9 <8
Например, как следует из кривых титрования (рис. 6, г) ортоборной кислоты
(рКа = 9,12) сильными основаниями (образование метабората натрия), определение
этой кислоты возможно в 0,1 н. и 0,01 н. растворах. Однако кривая титрования 0,01 н.
раствора кислоты закруглена вблизи точки эквивалентности.
Кривые титрования сильных двухосновных кислот имеют V-образ-
ную форму; такие кислоты дифференцированно по ступеням
нейтрализации не определяются. Но при титровании 0,1 н. растворов даже
серной и селеновой кислот {рКа = 1.92) кондуктометрические кривые
80 ГЛ. III. КОНДУКТОМЕТРИЯ И КОНДУКТОМЕТРИЧЕСКОЕ ТИТРОВАНИЕ
имеют слабые изгибы вблизи точки эквивалентности. В сильно
разбавленных растворах этого эффекта не наблюдается.
Двухосновные кислоты, достаточно хорошо диссоциирующие по
первой ступени (рД"а<2,5)> но слабо диссоциирующие по второй
ступени (p/Ci1 = 6 — 10), дифференцированно титруются по ступеням
нейтрализации. Если кислота сильно диссоциирована по первой ступени и
слабо по второй, наблюдается линейное понижение проводимости до
первого излома и линейное повышение электропроводности до второго
излома кондуктометрической
кривой. Примером (рис. 6, д)
может служить кривая
титрования хромовой кислоты
(рК1а = - 1, р/С^1 = 6,50).
Уменьшение степени диссоциации
кислоты по первой ступени
приводит к изгибу
кондуктометрической кривой до первой точки
эквивалентности. Такого типа
кривая (рис. 6, е) получается
при титровании малеиновой
кислоты (рКа= 1,92; рК" =
= 6,23).
При титровании кислот
средней силы, константы
диссоциации которых заметно
различаются по величине, кон-
дуктометрические кривые
имеют пологий минимум: излом
кривой соответствует полной
нейтрализации кислоты.
Например, этот тип кривой (рис.
6, ж) наблюдается при
титровании винной кислоты (pKl =
-2,98; pKl1 = 4,34).
При титровании двухосновных кислот, слабо диссоциирующих по
первой и второй ступеням, проводимость увеличивается. Для
дифференцированного титрования этих кислот по ступеням нейтрализации
необходимо, чтобы кислоты имели не только достаточную разницу в
константах диссоциации, но и отличались подвижностями НАгг- и An—-
ионов. Так как подвижности анионов (кроме ОН") в общем мало
различаются, вероятность получения резких изломов мала. Обычно
наблюдается один излом, соответствующий полной нейтрализации кислоты.
Верхние границы рКа для двухосновных кислот, определяемых в водных
растворах при различных концентрациях, аналогичны рассмотренным
для одноосновных кислот; при этом возможность определения кислоты
зависит от величины второй константы диссоциации кислоты.
Возможность определения и формы кондуктометрических кривых
титрования сильными основаниями многоосновных кислот могут быть
установлены из рассмотренных выше зависимостей.
Объем титраита
Рис. 6. Кондуктометрические кривые
титрования кислот раствором NaOH:
а—азотная; б — монохлоруксусная; в— уксусная;
;-0,1н.; 2 —0,001 н.; 5 — 0.0001 н.; г — ортоборная;
I— 0,1 н.; 2 — 0,01 н.; д — хромовая; е —малеиновая;
ж — винная; з —хлористоводородная (раствором
аммиака): и — монохлоруксусная в присутствии монохлор-
ацетата аммония.
Например, фосфорная кислота Н3Р04, характеризующаяся константами (р/Са =
= 2,12; 7,21; 12,67) может титроваться только по первой ступени, причем
электропроводность до первого излома понижается, но кривая титрования изогнута. Вторая точка
эквивалентности не устанавливается с достаточной точностью, так как мало разли-
§ 3. КИСЛОТНО-ОСНОВНОЕ ТИТРОВАНИЕ
81
чаются подвижности H2POJ- и НРО~-ионов. Определение третьей точки
эквивалентности невозможно вследствие сильного гидролиза ЫазР04.
Кондуктометрическое титрование позволяет также проводить
определение полиэлектролитов, например полиметакриловой кислоты (р/Са =
= 6,5 — 7,5).
При титровании кислот сильными основаниями большое влияние
на точность определения оказывает присутствие карборатов, так как
последние взаимодействуют с кислотами по реакции вытеснения и, если
кислоты слабые, реакция проходит не количественно. Кроме того,
угольная кислота, образующаяся в растворе, может титроваться основанием,
что приводит к завышенным результатам. Поэтому следует пользоваться
растворами сильных оснований, по возможности свободными от
карбонатов. Эти ошибки устраняются, если титрование проводят раствором
Ва(ОН)2, однако в случае образования осадков формы кондуктомет-
рических кривых могут изменяться (см. § 4).
Для титрования кислот можно также использовать слабые
основания. При титровании сильных кислот слабыми основаниями вследствие
уменьшения концентрации высокоподвижных ионов водорода
электропроводность до точки эквивалентности линейно понижается аналогично
тому, как это происходит при титровании сильными основаниями:
H+ + An~ + KtOH ^z± Kt+ + ArT + H20
Однако после точки эквивалентности электропроводность остается
практически постоянной. Избыток слабого основания, диссоциация
которого подавляется находящейся в растворе солью, не может вызвать
повышения проводимости раствора. Примером может служить кривая
титрования хлористоводородной кислоты аммиаком (рис. 6, з).
Если титруют слабые или средней силы кислоты слабыми
основаниями, характер изменения электропроводности раствора до точки
эквивалентности такой же, как и при титровании сильными основаниями,
поскольку продуктами реакции и в этом случае являются хорошо
диссоциирующие соли:
HAn + KtOH ^=± Kt+ + An" + H20
При избытке слабого основания проводимость мало изменяется.
Поэтому излом в точке эквивалентности по сравнению с изломом на
кривых титрования сильными основаниями оказывается в некоторых
случаях более резким. Определения кислот слабыми основаниями (0,1 н.
растворы) возможны в случаях, когда сумма рКа титруемой кислоты и
рКь основания, используемого для нейтрализации, ^12.
Очень слабые кислоты практически не взаимодействуют со слабыми
основаниями, что может быть использовано при определении некоторых
многоосновных кислот. Например, если титровать фосфорную кислоту
раствором аммиака, то кондуктометрическая кривая имеет изломы,
соответствующие первой и второй точкам эквивалентности. При
титровании до первой точки электропроводность понижается, а до второй —
повышается. После второй точки эквивалентности электропроводность
остается постоянной, так как аммиак не взаимодействует с НРОГ"-
ионами, кислотные свойства которых выражены очень слабо.
При определенных условиях кислоты средней силы и слабые можно
титровать сильными основаниями и получать кондуктометрические
кривые с линейным повышением проводимости и точками эквивалентности,
расположенными в максимумах кондуктометрических кривых. Такого
типа кривые получаются при титровании кислот в присутствии их солей
82 ГЛ, Ш, КОНДУКТОМЕТРИЯ И КОНДУКТОМЕТРИЧЕСКОЕ ТИТРОВАНИЕ
со слабыми огагованиями. При этом подвижность катионов соли должна
быть выше подвижности катионов титранта (рис. 6,а).
Титрование оснований. Возможности кондуктометрического
определения оснований различной силы и характер изменения
электропроводности растворов при их титровании в общем аналогичны
рассмотренным для кислот. Отличие заключается только в том, что при
нейтрализации сильных оснований электропроводность понижается менее резко,
чем при нейтрализации сильных кислот, так как подвижность ОН" ниже
подвижности Н+, и, наоборот, избыток титранта вызывает более резкое
повышение электропроводности. Следует также заметить, что плавный
ход кривых вблизи точек эквивалентности, наблюдаемый при
титровании слабых оснований
(гидролиз образующихся солей),
более заметен по сравнению с
наблюдаемым для кислот
такой же силы. Это объясняется
тем, что при титровании
кислот в результате гидролиза в
растворе образуются гидр-
оксильные ионы, а при
титровании оснований — более
подвижные водородные ионы.
Титрование смесей кислот.
Кондуктометрический метод
может быть успешно
использован при анализе смесей
кислот в водных растворах.
Возможности анализа этих смесей
зависят от констант
диссоциации кислот и их концентрации.
Наиболее благоприятные
условия создаются при титровании смесей сильных и слабых кислот, так как
в первом случае при этом сначала нейтрализуются сильные кислоты
(электропроводность понижается), затем — слабые (электропроводность
повышается).
Однако не все слабые кислоты (например, с рКа < 4) могут быть
оттитрованы в таких смесях. Ограничения связаны с возникновением
минимума на кривых титрования, сглаживающего первый излом кривой.
Это сглаживание становится малозаметным для 0,1 н. растворов слабых
кислот, яогда р/Са^4, и для 0,01 н. растворов, когда р/Са>5.
Верхняя граница р/Са слабых кислот, определяемых в смесях, как
и при титровании кислот, зависит от степени гидролиза образующейся
соли. В ОД н. растворах слабые кислоты можно определять в смеси
лишь прл р/(о<^10, а в 0,01 н. растворах — при р/<а^9.
Например, при титровании гидроокисью натрия смеси 0,1 н. растворов
хлористоводородной и борной [рКа — 9,12) кислот первый излом кондуктометрической кривой
(рис. 7, а) весьма четкий, а второй—слегка закругленный вследствие гидролиза
образующегося метабораза. Однако установление точки эквивалентности не представляет
сложности.
В смесях с сильными кислотами можно определять также
полиэлектролиты, например полиметакриловую кислоту (рКа = 6,5— 7,5).
Таким образом, можно принять, в общем, следующие границы
рКа слабых кислот, определяемых в смесях с сильными кислотами;
Концентрация кислоты,
н 0,1 0,01
Интервал р/С* 4-10 5-9
Объем титранта
Рис. 7. Кондуктометрические кривые
титрования смесей кислот раствором NaOH:
а — хлористоводородная + борная; б — уксусная +
борная; в — хромовая + уксусная; г— малеиновая + серная;
Ь — хлористоводородная + уксусная (раствором
аммиака); е — серная + теллуровая в присутствии
гидрохлорида три метила мина.
§ 3. КИСЛОТНО-ОСНОВНОЕ ТИТРОВАНИЕ
83
При титровании сильными основаниями более разбавленных
растворов границы рКа для слабых кислот еще более сужаются.
При анализе смесей слабых кислот необходимо, чтобы разница
в их константах диссоциации (Ар/Са) была > 2. В тех случаях, когда
концентрации этих кислот сильно различаются, необходимо, чтобы
ДрЯа^З. Кроме того, важно, чтобы достаточно отличались и
подвижности анионов титруемых кислот, так как обе точки эквивалентности
расположены на восходящих ветвях кондуктометрической кривой.
Например, Ар7Са уксусной (р/Са = 4,75) и борной (р/Са = 9,12) кислот составляет
достаточно большую величину. Однако при титровании смеси этих кислот (рис. 7, б)
окончание нейтрализации уксусной кислоты не фиксируется четким изломом
кондуктометрической кривой, потому что мало отличаются подвижности анионов титруемых
кислот.
Сильные двухосновные кислоты, например серная и селеновая,
взаимодействуют аналогично сильным одноосновным кислотам. Они
дифференцированно титруются в смесях со слабыми одноосновными
кислотами, границы рКа которых приведены выше. Кондуктометрические
кривые при этом имеют форму, представленную на рис. 7, а.
Если в смеси с сильными кислотами входят слабые двухосновные
кислоты, возможность их определения зависит от того, находятся ли
величины их рКа в границах, допустимых для слабых кислот при данной
концентрации. При этом величина рК\ не должна выходить за нижнюю
границу, a рК1а1 — за верхнюю.
Так, в смесях с сильными одноосновными и двухосновными
кислотами определяются: янтарная (р/Са = 4,21; 5,64), глутаровая (р/Са =
= 4,34; 5,22), адипиновая (р/Са = 4,42; 5,28) и многие другие.
Титрование слабых двухосновных кислот по ступеням нейтрализации даже при
достаточной разнице в их константах диссоциации в большинстве
случаев не приводит к заметным изломам кондуктометрической кривой, так
как, в общем, подвижности НАгг- и Ап~~-ионов, образующихся в
процессе реакции, недостаточно сильно различаются. Поэтому
кондуктометрические кривые титрования смесей сильных кислот со слабыми
двухосновными кислотами аналогичны кривой, приведенной на
рис. 7у а.
Двухосновные кислоты, дифференцированно титрующиеся по
ступеням нейтрализации, можно определять в смесях с сильными и со
слабыми кислотами. Нейтрализация сильных кислот совпадает с первой
ступенью нейтрализации двухосновной кислоты, а слабых — со второй.
Если двухосновные кислоты хорошо диссоциируют по первой ступени,
при титровании их смесей электропроводность до первой точки
эквивалентности линейно понижается.
ПрИхмером могут служить кривые титрования смесей хромовой кислоты (рКа =»
= —1; 6,50) с хлористоводородной и уксусной кислотами. В первом случае сначала
совместно нейтрализуются НС1 и Н2СГО4 до HCrOJ, а затем после излома кондукто-»
метрической кривой взаимодействуют ионы HCrOJ, что приводит к повышению элек«
тропроводности. Совместно с НСгО^-ионами происходит нейтрализация СНзСООН,
что соответственно увеличивает второй участок кондуктометрической кривой. Кривая
титрования этой смеси показана на рис. 9, в.
Если двухосновная кислота дифференцированно титруется по
первой ступени, но диссоциирует по первой ступени не полностью, кондук-
тометрическая кривая титрования на первом участке изогнута.
84 ГЛ. III. КОНДУКТОМЕТРИЯ И КОНДУКТОМЕТРИЧЕСКОЕ ТИТРОВАНИЕ
Такой характер изменения проводимости наблюдается при титровании смесей,
содержащих малеиновую кислоту (р/Са = 1,92; 6,23). В смеси, содержащей малеиновую
и серную кислоты, последняя титруется на первом участке кондуктометрической
кривой (рис. 7, г)- Борная кислота в смесях с малеиновой кислотой, наоборот,
нейтрализуется на вторам участке кривой.
Для нейтрализации смесей кислот могут быть использованы слабые
основания, что создает более благоприятные условия при определении
второй точки эквивалентности, так как избыток слабого основания мало
изменяет электропроводность (рис. 7,д).
Для анализа смесей сильных и слабых кислот может быть также
рекомендован вариант, основанный на двойном титровании. Сначала
титрованием сильным основанием определяют общее содержание в смеси
сильной и слабой кислот. Затем подбирают слабое основание такой
силы, чтобы оно заметно не взаимодействовало со слабой кислотой
(сумма рКа слабой кислоты и рКь основания > 16). В этом случае при
титровании слабым основанием определяется только сильная кислота.
Еще более точные результаты могут быть получены, если при втором
титровании в качестве титранта взята соль слабой кислоты, находящейся
в смеси. При титровании слабая кислота не может взаимодействовать со
своей солью, и в реакции вытеснения участвует только сильная кислота.
Для этой цели могут быть использованы соли и других кислот,
достаточно слабых (р/Са>4), чтобы количественно участвовать в реакции
вытеснения, но более сильных по сравнению со слабой кислотой,
находящейся в смеси, чтобы не вступать с ней в реакцию.
Кондуктометрический метод при определенных условиях позволяет
проводить анализ смесей сильных кислот и слабых двухосновных
кислот, у которых p/Ci1 > 10.
На рис. 7, е показана кривая титрования гидроокисью натрия смеси серной и
теллуровой (р/Са = 7,64; 11,10) кислот в присутствии гидрохлорида триметиламина
(рКь = 6,18). При титровании сначала полностью нейтрализуется серная кислота,
затем теллуровая (до кислой соли) и, наконец, взаимодействует гидрохлорид.
Кондуктометр ическа я кривая имеет два резких излома: первый участок кривой позволяет
определять серную кислоту, второй — теллуровую.
Титрование смесей оснований. Условия кондуктометрического
титрования смесей оснований аналогична рассмотренным для смесей
кислот. Все предлагаемые варианты анализов смесей кислот могут быть
использованы при определении оснований. При этом необходимо
учитывать константы их основности.
Титрование солей. Если к раствору соли слабой кислоты, например
CH3COONa, добавить сильную кислрту, то вытеснится слабая уксусная
кислота:
CH3COO"+Na+ + H+ + Cr +± СН3СООН + Na+ + СГ
С другой стороны, слабые основания из их солей вытесняются
сильными основаниями. Соли, образованные слабыми основаниями и
слабыми кислотами, можно титровать как сильными основаниями, так
и сильными кислотами. Кондуктометрические кривые имеют достаточно
резкий излом в точке эквивалентности, когда вытесняемые слабые
электролиты имеют следующие значения р/С:
Концентрация соли, н. • 0,1 О,01
р/С >4 >5
Рассмотрим типы кондуктометрических кривых, получаемых при
титровании сильными основаниями солей, образованных катионами
слабых оснований и анионами сильных кислот (0,1 н. растворы). Эти
соли в водных растворах подвергаются гидролизу:
Kt++ АгГ + НОН ^=> КЮН + Н+ + Ап"
§ 3. КИСЛОТНО-ОСНОВНОЕ ТИТРОВАНИЕ
85
Если соль образована настолько слабым основанием, что
подвергается в растворе полному гидролизу, кривая титрования имеет
V-образную форму, так как происходит нейтрализация сильной кислоты,
образующейся при гидролизе. Такого типа кривая наблюдается при
титровании (рис. 8, а) гидроокисью натрия гидрохлорида мочевины (р/(ь =
—13,82).
С уменьшением степени гидролиза соли формы кондуктометриче-
ских кривых изменяются. Типичным примером для случая, когда
гидролиз оказывает влияние на изменение электропроводности раствора
только в начале титрования, может служить кривая титрования
(рис. 8, б) гидроокисью натрия гидрохлорида семикарбазида (р/Сь= 10,57).
Подвижность катионов этой соли выше подвижности катионов
натрия, поэтому после некоторого
понижения электропроводности раствора в
начале титрования электропроводность и
дальше продолжает понижаться, но
менее заметно. Если же подвижность
катионов соли ниже подвижности
катионов титранта, кондуктометрическая
кривая титрования соли имеет пологий
минимум, не отвечающий какой-либо
характерной для анализа точке.
Для солей, образованных
основаниями с рКь < 9 (степень гидролиза
менее 1%), влияние гидролиза
практически не обнаруживается. На всем участке
кривой до точки эквивалентности
наблюдается линейное повышение или
понижение электропроводности, что зависит от
сравнительной подвижности катионов
соли и заменяющих их в растворе
катионов титранта. Примером может служить
кривая титрования (рис. 8, в)
гидрохлорида триэтаноламина (р/Сь = 6,18)
сильным основанием.
С уменьшением значения рКь наблюдается закругление излома
кривой около точки эквивалентности вследствие обратимости реакции,
которое становится заметным, когда значение рКь = 4.
Рассмотренные типы кондуктометрических кривых характерны
также для титрования сильными кислотами солей, образованных
катионами сильных оснований и анионами слабых кислот. Отличие
заключается только в том, что понижение электропроводности в начале
титрования, вызванное гидролизом, менее резко выражено, так как
подвижность гидроксильных ионов, образующихся в растворе при
гидролизе, значительно ниже подвижности ионов водорода.
Кондуктометрический метод дает также возможность проводить
определения солей слабых многоосновных кислот или оснований.
Рассмотрим типы кондуктометрических кривых титрования сильными
кислотами солей слабых двухосновных кислот.
Если соли подвергаются в водных растворах полному гидролизу,
при титровании нейтрализуется основание, образующееся в растворе
при гидролизе, и кондуктометрические кривые имеют V-образную
форму.
При полном гидролизе соли по одной ступени и слабом гидролизе
по второй (р/Са = 4 — 9) кондуктометрические кривые имеют два излома;
Объем титранта
Рис. 8. Кондуктометрические
кривые титрования солей слабых
оснований раствором NaOH
(кривые а, б, в з, /с, л) и солей
слабых кислот раствором НС1
(кривые г, д, е, ж, и, м):
а — гидрохлорид мочевины; б —
гидрохлорид семикарбазида; в — триэтанол-
амингидрохлорид; г —сульфид натрия;
д — теллурат натрия; е — хромат натрия;
ж — малеинат натрия; з — ж-нитрофено-
лят аммония; и — уксуснокислый а-пико-
линий; к — бисульфат аммония; л— би-
тартрат натрия; м — битартрат аммония.
86 ГЛ. III. КОНДУКТОМЕТРИЯ И КОНДУКТОМЕТРИЧЕСКОЕ ТИТРОВАНИЕ
сначала нейтрализуется продукт гидролиза — сильное основание, затем
вытесняется слабая кислота из кислой соли. Примером может служить
кривая титрования (рис. 8, г) хлористоводородной кислотой сульфида
натрия (рЛ^а = 7,20; 14,00). Если же по первой ступени гидролиз
неполный, но достаточно сильный (рКа > 9), а по второй — слабый
(рКа = 4 — 9), вначале наблюдается понижение электропроводности,
связанное с нейтрализацией гидроксидьных ионов, образующихся при
гидролизе. В дальнейшем, когда в реакции участвуют преимущественно
анионы соли, сначала Агг~, затем НАгг, изменение электропроводности
зависит от сравнительной подвижности этих ионов и анионов титранта,
накапливающихся в растворе. При этом электропроводность или
продолжает понижаться, но менее сильно, чем в начале, или начинает
увеличиваться, что приводит к кондуктометрическим кривым с пологим
минимумом. Типичную кривую с минимумом (рис. 8, с?) получают при
титровании теллурата натрия (р/Са = 7,64; 11,10).
Соли кислот, имеющих рКа = 4 — 9, дают кондуктометрические
кривые с линейным повышением или понижением проводимости до
точки эквивалентности.
Соли, образованные кислотами, сравнительно хорошо диссоциирую^
щими по первой ступени (р/Сд<2,5) и слабо — по второй (р/<11>4),
количественно взаимодействуют с титрантом, образуя кислые соли. Если
кислоты по первой ступени сильно диссоциированы, при взаимодействии
их солей с сильной кислотой образуются только HArr-ионы и после
точки эквивалентности электропроводность линейно увеличивается от
избытка титранта. Например, хромат натрия (рКа = —1; 6,50)
титруется хлористоводородной кислотой до кислой соли (рис. 8, е). Если же
диссоциация двухосновной кислоты по первой ступени неполная
(значение pKl приближается к 2,5), после излома кондуктометрической
кривой в точке эквивалентности, соответствующей образованию кислой
соли, электропроводность изменяется нелинейно, появляется небольшой
изгиб кривой вследствие образования некоторого количества молекул
слабой кислоты. Однако при Ар/<а>2 в этом случае возможно
установить точку эквивалентности, соответствующую кислой соли. Такого
типа кривую получают при титровании малеината натрия (рКа =
= 1,92; 6,23) хлористоводородной кислотой (рис. 8,ж).
Рассмотрим процессы, протекающие при титровании солей,
образованных катионами слабых оснований и анионами слабых кислот,
характеризующихся (р/С>4), Такие соли можно титровать сильными
основаниями и сильными кислотами. Формы кондуктометрических
кривых также зависят от степени гидролиза соли, поскольку при гидролизе
в растворах образуются слабые кислоты и слабые основания:
Kt+ + An" + H20 ^z± KtOH + HAn
Например, при титровании гидроокисью натрия метаиитрофенолята
аммония (р/Са = 8,40, рКъ = 4,75) получается кондуктометрическая
кривая с максимумом (рис. 8,з).
Сначала в реакцию вступает образующийся в растворе при гидролизе соли
jw-нитрофенол, а затем гидролиз соли подавляется и в реакции участвуют
преимущественно ионы аммония. Так как подвижность ионов аммония выше, чем ионов натрия,
повышение проводимости сменяется понижением и кривая имеет пологий максимум.
При титровании хлористоводородной кислотой уксуснокислого а-пиколиния (рКа =з
« 4,75, рКь «= 8,03) сначала с НС1 взаимодействует а-пиколин, образующийся в
растворе при гидролизе, затем повышение проводимости становится менее сильным, так
как оно вызывается только тем, что более подвижные С1"-иояы заменяют в растворе
менее подвижные СН3СОО~-ионы (рис. 8, и), поскольку образуются мало
диссоциированные молекулы уксусной кислоты,
§ 3. КИСЛОТНО-ОСНОВНОЕ ТИТРОВАНИЕ
87
Кондуктометрическое титрование может быть использовано и при
анализе кислых солей сильных и слабых многоосновных кислот.
Кислые соли сильных двухосновных кислот могут быть образованы
катионами сильных и слабых оснований. Если основание, образующее соль,
сильное, при титровании кислой соли щелочью происходит
нейтрализация только ионов водорода, образующихся в растворе при диссоциации
соли. При этом проводимость до точки эквивалентности линейно
понижается аналогично тому, как это происходит при нейтрализации
свободных сильных кислот. Когда сильная двухосновная кислота образует
кислую соль со слабым основанием (рКъ = 4— 9), при ее титровании
сильным основанием сначала нейтрализуются ионы водорода, затем
взаимодействуют катионы слабого основания. Кондуктометрическая
кривая имеет два излома (рис. 8, к).
Если слабая двухосновная кислота образует кислую соль с катионом
сильного основания, при титровании этой соли сильным основанием в
реакцию вступают НАгг-ионы. При этом проводимость до точки
эквивалентности растет, так как в растворе образуются двухзарядные
Агг~-ионы и накапливаются катионы титранта. Подобного типа кривую
(рис. 8, л) можно получить при титровании основанием битартрата
натрия (рКа = 2,98; 4,34). В случае, когда кислая соль слабой
двухосновной кислоты образована катионами слабого основания (р/Сь=4—9),
в реакцию с основанием вступают сначала HArr-ионы, а затем катионы
слабого основания. При этом кондуктометрические кривые имеют два
излома (рис. 8, м), если {vKla+vKb)< 12.
Титрование смесей солей. При титровании сильной кислотой смеси
двух солей, образованных сильными основаниями и слабыми
кислотами, сначала вытесняется более слабая кислота, затем более сильная.
Величина Др/Са, необходимая для возможности последовательного
кондуктометрического определения солей, должна быть равна 2, если
концентрации солей близки, и 3 — если концентрации их значительно
отличаются.
Аналогичные условия необходимы при титровании сильными
основаниями смеси двух солей, образованных слабыми основаниями и
сильными кислотами. Следует обратить внимание на то, что при анализе
смесей солей должны различаться подвижности последовательно
взаимодействующих анионов или катионов.
Титрование смесей кислот и солей слабых оснований. Равновесия,
устанавливающиеся при взаимодействии сильных оснований со смесями
слабых кислот и солей слабых оснований в водных растворах, могут
быть выражены уравнениями:
HAn + OH"" ^=t AiT + H20 (17)
Kt+ + OH" ^=t KtOH (18)
Равновесная концентрация гидроксильных ионов может быть
определена из уравнений (17) и (18):
fArG Ка_ (19)
[НАп] [ОН~] К
w
ГКЮН] 1 (20)
[Kt+] [ОН"] Кь
где Ка- концентрационная константа диссоциации кислоты, образующей соль;
^-концентрационная константа дасеоииащш основания, образующего соль;
Kw — ионное произведение воды.
88 ГЛ. Ш. КОНДУКТОМЕТРИЯ И КОНДУКТОМЕТРИЧЕСКОЕ ТИТРОВАНИЕ
Равновесные концентрации гидроксильных ионов, рассчитанные по
уравнениям (19) и (20), должны быть равны:
Kw [An"] [KtOH]
Ка '[HAn] ' [Kt+]
или
[An""] . [KtOH] КаКь
[HAnJ * [Kt+] K
(21)
w
В уравнении (21) величины [Arr]/[HAn] и [KtOH]/[Kt+]
характеризуют степень нейтрализации кислоты и степень вытеснения основания
из его соли. Задаваясь различными значениями отношения этих
величин, можно определить сумму рКа и рКь, необходимую для
последовательного проведения сначала одной из реакций, а затем — другой.
В случае, если
[HAn] [Kt+]
сначала протекает реакция нейтрализации, а затем — вытеснения. При
этом если исходные концентрации кислоты и соли в смеси равны, то
99% кислоты будет нейтрализовано, когда 1% основания будет
вытеснен из его соли. Отсюда:
¦^§¦-100:1 и р1Св + р**=12 (22)
Следовательно, когда (рКа + рКь) = 12, сначала протекает
реакция нейтрализации кислоты, а затем — вытеснения основания.
В случае, если
[HAn] [Kt+]
в первую очередь вытесняется основание из его соли, а затем
нейтрализуется кислота, т. е.
i^f = 1:100 и рКа + р/С*=16 (23)
10
Таким образом, существует две области количественных
взаимодействий сильных оснований со смесями кислот и солей слабых
оснований, соответствующих различной последовательности прохождения
реакции нейтрализации и вытеснения.
При взаимодействии сильных оснований со смесями кислот и солей
слабых оснований, для которых сумма рД'а и рКь находится в
интервале 12—16, одновременно протекают обе реакции4.
Раздельное определение компонентов с ошибкой не более 0,1%
(отношение равновесных концентраций 1000 : 1 и 1 : 1000) можно
провести лишь в том случае, если значение суммы рКа и рКь составит
<11 или >17.
Заменив в уравнении (21) значения концентраций активностями,
получим:
gAn- . aKtOH ^ KgKb
aHAn aKt+ %W
или
[An-]fAn- [KtOH]fKtOH К'Хь
[HAn]fHAn- [Kt+]fKt+ K'w
(24)
§ 3. КИСЛОТНО-ОСНОВНОЕ ТИТРОВАНИЕ
89
Если коэффициенты активности недиссоциированных молекул
слабого электролита принять равными единице, а коэффициенты
активности однозарядных ионов равными среднему коэффициенту
активности и учесть соотношение концентраций кислоты и соли, то получим:
[An"] [KtOH] уХСнап
[HAn] * [Kt+] KwfxCKtAn
Равновесия, устанавливающиеся при взаимодействии сильных
оснований со смесями слабых кислот, могут быть рассмотрены более
детально с учетом влияния гидролиза. Когда участвующие в реакции
электролиты характеризуются значением (рКа + рКь) < 12, в первой
точке эквивалентности в растворе имеются анионы слабой кислоты и
катионы слабого основания, что может привести к гидролизу, который
вызовет смещение реакции нейтрализации в левую сторону, а реакции
вытеснения — в правую. Практически влияние гидролиза заметно в
случаях, когда величина (рКа + рКь) приближается к 12. Однако для
сравнительно концентрированных растворов смещение реакций
выражено в равной степени, что дает возможность устанавливать точки
эквивалентности графическим методом, если (рКа + рКь)^С 12. В этих
условиях влияние гидролиза выразится лишь небольшим закруглением
излома кондуктометрическои кривой вблизи первой точки эквивалентности.
В тех случаях, когда электролиты характеризуются значением
(рКа + рКь) > 16, в первой точке эквивалентности в растворе находится
смесь слабой кислоты и слабого основания. Протекание реакции
нейтрализации между этими электролитами вызывает смещение первой
реакции вытеснения в левую сторону и приводит к частичной
нейтрализации слабой кислоты, т. е. к смещению равновесия второй реакции в
правую сторону. Некоторое количество катионов слабого основания и
анионов слабой кислоты, которое имеется в растворе, вызывает слабое
закругление излома кондуктометрическои кривой около первой точки
эквивалентности.
Это наблюдается, когда величина (рКа + рКь) равна или немного
больше 16. При увеличении этой суммы слабое основание практически
не взаимодействует со слабой кислотой. Однако если эти
взаимодействия протекают, то в достаточно концентрированных растворах, когда
(рКа + рКь) > 16, точки эквивалентности устанавливаются
графическим методом практически всегда, так как смещение обеих реакций
выражено в равной степени.
Установлено, что можно последовательно проводить
взаимодействия, характерные для той или другой области, и осуществлять анализ
многокомпонентных смесей электролитов. При этом сначала в реакцию
вступают вещества с более сильно выраженными кислотными
свойствами, затем с более слабыми.
Условия последовательного взаимодействия при кондуктометриче-
ском титровании сильными основаниями смесей кислот и солей слабых
оснований (0,1—0,05 н. водные растворы) в двух-, трех-, четырех- и пя-
тикомпонентных смесях в зависимости от констант диссоциации
электролитов показаны в табл. 5.
При установлении возможности анализа той или иной смеси
необходимо прежде всего определить сумму рКа и рКь кислот и солей
слабых оснований. Эти данные позволят определить последовательность
взаимодействия компонентов смеси. Затем следует проверить,
находятся ли значения р/С, характеризующие отдельные кислоты и соли,
в границах, указанных в табл. 5.
ТАБЛИЦА 5
Условия количественного взаимодействия при титровании сильными основаниями смесей кислот и солей слабых оснований
{0,1—0,05 н. растворы)
(Стрелками показана последовательность титрования)
Кислота
Соль
Сильная
кислота (I)
рКа < 6
Кислота (I)
Соль (I)
Сильная
кислота (I)
Кислота (I)
рКь> 13
Соль (I)
Сильная
кислота (I)
рКь > 13
Соль (I)
Соль
ptffl = 6-10
Кислота
Кислота (II)
pit* -6-9
Соль
Кислота
рКа = 4-6
Кислота (II)
Соль (I)
р/Са = 4-б
Кислота (I)
рК~4
Кислота (II)
Кислота (I)
§ 4. ТИТРОВАНИЕ, ОСНОВАННОЕ НА РЕАКЦИЯХ ОСАЖДЕНИЯ 91
Кондуктометрические кривые титрования смесей, удовлетворяющих
условиям, приведенным в табл. 5, всегда отличаются достаточно
резкими изломами в точках эквивалентности. Это связано с тем, что
взаимодействия слабых кислот и солей чередуются и при титровании слабых
кислот электропроводность раствора всегда повышается, а при
титровании солей зависит только от сравнительной подвижности
участвующих в реакции ионов.
Титрование смесей оснований и солей слабых кислот.
Взаимодействия сильных кислот со смесями оснований и солей слабых кислот
аналогичны рассмотренным для смесей кислот и солей слабых оснований.
В основе этих взаимодействий лежат реакции нейтрализации оснований
и вытеснения слабых кислот из их солей. Возможности анализа смесей
можно установить также с помощью табл. 5, при этом значения рКъ
оснований, входящих в состав смесей, следует принять равными
значениям рКа свободных кислот, а величины рКа кислот, образующих соли,
равными (см. табл. 5) величинам рКь слабых оснований, образующих
соли.
§ 4. Титрование, основанное на реакциях осаждения
В методе кондуктометрического титрования могут применяться
реакции осаждения, например титрование нитрата серебра хлоридом
натрия;
Ag++N03 + Na+ + Cl" ^z± ф AgCl + Na++ NOJ
Ионы серебра и хлора в процессе титрования удаляются из
раствора вследствие образования осадка, а ионы натрия, вводимые при
добавлении титранта, остаются в растворе. В результате реакции Ag+-
ионы в титруемом растворе заменяются Ыа+-ионами. Изменение
состава ионов приводит к изменению электропроводности раствора.
Характер изменения проводимости при титровании зависит от сравнительной
подвижности Ag+-noHOB и заменяющих их №а+-ионов. Подвижность Ag+
(Я0 = 61,9) выше подвижности Na+ (A,0 = 50,1), поэтому при титровании
до точки эквивалентности проводимость понижается. После полного
осаждения проводимость увеличивается.
Поскольку реакции осаждения часто протекают не мгновенно,
измерение сопротивления раствора при титровании следует проводить
после достижения постоянной проводимости.
Возможность использования различных реакций осаждения при
кондуктометрическом титровании и точность определений зависят от
следующих факторов:
1) величины угла излома кондуктометрической кривой в точке
эквивалентности, обусловливаемой характером изменения проводимости
раствора при титровании;
2) растворимости осадка.
3) постоянства состава осадка, его чистоты и скорости осаждения.
Изменение проводимости раствора. При кондуктометрическом
титровании необходимо, прежде всего, чтобы излом кондуктометрической
кривой позволял устанавливать точку эквивалентности с достаточной
точностью. Чем острее угол излома, тем выше точность. Когда угол
излома очень тупой, установление точки эквивалентности затруднено.
Например, титрование нитрата серебра можно осуществлять различными
хлоридами: LiCl, NaCl или КС1. Подвижность Ag+ равна 61,9, a Li+, Na+ и К+
соответственно 38,7; 50,1 и 73,5. Для титрования Ag+ лучше применять хлорид лития, в этом
случае проводимость раствора до точки эквивалентности заметно понижается, потому
что подвижность Li+ ниже подвижности Ag+ (рис. 9,лг). Понижение проводимости
менее сильно выражено в случае титрования хлоридохМ натрия. Если же титрование
92 ГЛ. III. КОНДУКТОМЕТРИЯ И КОНДУКТОМЕТРИЧЕСКОЕ ТИТРОВАНИЕ
проводить хлоридом калия, то проводимость до точки эквивалентности повышается,
потому что подвижность К+ выше подвижности Ag+, и угол излома делается тупым.
Острота угла излома кривой зависит также от характера
изменения электропроводности раствора после точки эквивалентности. Угол
излома становится более острым, если проводимость раствора при
избытке титранта увеличивается. Повышение проводимости после точки
эквивалентности делается более сильным, если ионы титранта,
участвующие в реакции, имеют высокую подвижность.
Если сравнить кондуктометрические кривые, полученные при титровании Ag+
растворами хлорида и хромата лития (образование осадков AgCl и Ag2Cr04), более
сильное повышение проводимости после точки эквивалентности будет наблюдаться
при титровании хроматом лития, так как
подвижность CrOJ" (А,0 = 85) выше подвижности С1"
(Я° = 76,4).
Растворимость осадка. Растворимость
осадка вызывает повышение
электропроводности раствора, что практически
обнаруживается вблизи точки эквивалентности и
выражается в закруглении кондуктометриче-
ской кривой на этом участке. Влияние
растворимости незаметно на участках кривых,
удаленных от точки эквивалентности,
поскольку в начале титрования растворимость
подавляется избытком осаждаемых ионов,
а в конце — избытком титранта. Так
как точки эквивалентности можно находить
экстраполированием прямолинейных
участков кондуктометрических кривых, метод
позволяет проводить определения в
случаях, когда растворимость осадков
несколько выше, чем допускается при весовом
методе.
Для успешного применения кондуктометрического метода при
использовании реакции осаждения произведения растворимости ПР
образующихся осадков должны иметь следующие значения:
Концентрация раствора,
н 0,1 0,01 0,001
Объем титранта
Рис 9. Кондуктометрические
кривые титрования при
использовании реакции осаждения,
окисления — восстановления и
комплексообразования:
а — AgN03 раствором LiCi; б —
Na3As03 раствором 12; в — водного
0.02М раствора Fe(N03)3
комплектном III; г-0,0Ш раствора NiS04
комплексоном III ггри рН«=4.
Произведение
растворимости, ПР < 2,5 • 10 5 < 2,5-10"
< 2,5 • 10"
В разбавленных растворах влияние растворимости осадка
становится более заметным.
Растворимость осадков в ряде случаев можно понизить, если
добавить в титруемый раствор неводные растворители. С этой целью
часто рекомендуется добавление этилового спирта. Для уменьшения
растворимости осадка можно проводить титрование при пониженной
температуре.
Постоянство состава осадка, его чистоты и скорости осаждения.
Выделяющиеся в процессе титрования осадки должны иметь
определенный состав. Поэтому не все реакции осаждения могут быть
использованы в кондуктометрических определениях. Например, при титровании
сильными основаниями катионов тяжелых металлов часто образуются
осадки неопределенного состава — основные соли. Кроме того, осадки
могут загрязняться вследствие процессов соосаждения. Большое
влияние на точность определений оказывают условия титрования. Более чи-
§ 4. ТИТРОВАНИЕ, ОСНОВАННОЕ НА РЕАКЦИЯХ ОСАЖДЕНИЯ 93
стые осадки получаются при титровании разбавленных растворов, при
медленном добавлении титранта небольшими порциями и тщательном
перемешивании.
Для уменьшения соосаждения рекомендуется проводить определение
при повышенной температуре, если при этих условиях растворимость
осадка не превышает допустимых пределов.
Определение анионов. Титрование нитратом серебра.
Для кондуктометрического определения анионов широко применяется
титрование нитратом серебра. Этот реагент дает возможность
определять С1~\ В г-, I", SCN~, Сг04~> С2С>4~, тартрат, цитрат и другие
анионы. Титрование сопровождается образованием малорастворимых солей
серебра:
Na+ + Br~ + Ag+ + NC>3 —> | AgBr + Na++ NOJ
Изменение проводимости растворов при титровании до точки
эквивалентности зависит от сравнительной подвижности осаждаемых
анионов и заменяющих их в растворе Г\Юз-ионов. При титровании
CI- (Jt° = 76,4), Вг- (А,0 = 78,1), I- (Л,0 = 78,8) и СгОГ (Х° = 85)
проводимость понижается, так как подвижности этих ионов выше
подвижности NOi (АЯ = 71,5). Однако при титровании SCN~ (Х° = 57,4),
наоборот, происходит небольшое повышение проводимости, так как его
подвижность ниже подвижности NO3.
В зависимости от растворимости солей серебра изменяются
Концентрации титруемых растворов, при которых удается проводить
определения с достаточно высокой точностью. Так, титрование хлоридов
можно проводить и в очень разбавленных растворах при концентрации
С1~ 0,025 мг/мл и меньше. Это титрование используется для
определения С1~ в питьевой воде. Между тем I" можно титровать в
растворах, концентрация которых больше 0,005 н., а цитраты только при
концентрации не ниже 0,1 н.
Титрование солями бария. Соли бария используют при
титровании SOT"; обычно применяют ацетат или хлорид бария. Точные
результаты получаются только при определенных условиях, так как
возможно загрязнение осадка вследствие адсорбции. Определения
проводят при комнатной температуре в водно-спиртовой среде или" при
100° С в водном растворе.
При титровании SOI" солями бария протекает реакция:
2Na+ + SOJ" + Ва++ + 2СН3СОСГ —> | BaS04 + 2Na+ + 2СН3СОСГ
В результате этих взаимодействий БОГ" заменяется в растворе
СН3СОО~ или О-. Электропроводность растворов при титровании
понижается. Однако сравнительно сильное понижение проводимости
происходит только при титровании ацетатом бария, так как СН3СОО_- ионы
имеют низкую подвижность (А,0 = 40,9). Подвижность CI" (К0 = 76,4)
близка к подвижности SOI" (A0 = 80,0), и поэтому при титровании
хлоридом бария проводимость понижается незначительно.
Следовательно, более благоприятные условия создаются, если титрантом
является ацетат бария.
При титровании разбавленных растворов уменьшается соосажде-
ние, и результаты получаются более точными. Растворимость сульфата
бария настолько мала, что метод можно применять для определения
сульфатов в питьевой воде.
Кондуктометрическое титрование можно использовать и для анализа смеси
серной и азотной кислот. При этом проводят два титрования. Сначала титруют сильным
94 ГЛ. III. КОНДУКТОМЕТРИЯ И КОНДУКТОМЕТРИЧЕСКОЕ ТИТРОВАНИЕ
основанием и определяют общую кислотность. Затем вторую порцию раствора
нейтрализуют основанием, слабо подкисляют уксусной кислотой, добавляют спирт
(1:1) и титруют ацетатом бария. Второе титрование дает возможность определить
содержание серной кислоты.
Определение катионов. Титрованиесульфатомлития. Для
определения Ва++, Sr++ и РЬ++ используют сульфат лития. Например,
при титровании Sr(N03h протекает реакция:
Sr++ + 2NOJ + 2Li++ SOJ" —> |SrS04 + 2Li+ + 2NC>3
В результате взаимодействия определяемые катионы заменяются в
растворе 1л+-ионами. При титровании проводимость раствора
понижается, так как подвижность Ва++ (к0 = 63,6), Sr++ (А,0 = 59,5) и РЬ++
(Я0 = 70) выше подвижности Li+ (Х° = 38,7). Более точные результаты
получаются при титровании разбавленных растворов в водно-спиртовой
среде. Определение Ва++ и РЬ++ проводят в 30%-ном спиртовом
растворе, a Sr++ в 50%-ном.
Титрование хроматом натрия. Определение Ва++, Sr++ и
РЬ++ может основываться на реакциях с хроматом натрия. При этом
выделяются в осадок малорастворимые хроматы этих металлов.
Например:
Pb++ + 2NOJ + 2Na+ + CrO~ —> | PbCr04 + 2Na++ 2NO3
При титровании в растворе Ва++, Sr++ или РЬ++ заменяются Na+.
Подвижность определяемых катионов выше подвижности Na+ (^° =
= 50,1), поэтому проводимость раствора при образовании осадков
понижается. Титрование солей бария и свинца можно проводить в водных
растворах, а солей стронция в 50%-ном спирте.
Титрование оксалатом лития. Оксалат лития может
служить реагентом для определения Са++, Sr"", Ba++, Ag+, Pb4"1", Cu++ и
других. Наибольшее практическое значение имеет определение Са44",
так как его определение другими реагентами затруднено вследствие
довольно высокой растворимости осадков. При титровании солей
кальция оксалатом лития протекает реакция:
Са++ + 2Шз + 2Ы+ + С204~ —> ф СаС204 + 2Li++ 2NOJ
При этом Са++-ионы заменяются в растворе 1л+-ионами, что
приводит к понижению проводимости раствора, так как подвижность Са++
(Х° = 59,5) выше подвижности Li+. Метод дает возможность проводить
определения в очень разбавленных растворах (до 0,0001 н.) при
использовании водно-спиртовой среды (1:1)-.
§ 5. Титрование, основанное на реакциях
окисления — восстановления
Применение реакции окисления — восстановления для кондукто-
метрических определений затруднено в случаях, когда реакции
проходят в сильнокислой или сильнощелочной среде, поскольку такие
растворы имеют высокую электропроводность, которая мало изменяется в
процессе реакций. Однако если определения проводятся в умеренно
кислых или щелочных растворах, и реакция протекает с участием
водородных или гидроксильных ионов, то изменение их концентрации в
процессе титрования способствует резкому изменению проводимости
растворов. Некоторые реакции окисления — восстановления вообще
неприменимы в кондуктометрическом титровании, так как в ряде случаев
протекают с малой скоростью и время установления постоянной прово-
§ 6. ТИТРОВАНИЕ, ОСНОВАННОЕ НА РЕАКЦИЯХ КОМПЛЕКСООБРАЗОВАНИЯ 95
димости после добавления титранта оказывается слишком длительным.
Рассмотрим условия кондуктометрического титрования арсенитов
раствором иода. Это определение основано на реакции:
AsO~ + I2 + H20 ^=± AsOJ-- + 2Н+ + 2Г
Аналогично обычным объемным определениям для смещения
равновесия в правую сторону в титруемый раствор добавляется
бикарбонат. При титровании изменяется состав ионов, находящихся в
растворе, AsO™ окисляется до AsO™, и в растворе накапливается 1~,
отличающийся довольно высокой подвижностью (А,0 = 78,84).
Выделяющийся в процессе реакции Н+ взаимодействует с сравнительно мало
подвижным НСОз (А,0 = 44,50) с образованием С02 и Н20. В
результате этих процессов в растворе уменьшается концентрация менее
подвижных ионов и увеличивается концентрация более подвижных, что
приводит к повышению проводимости до точки эквивалентности. В
качестве титранта употребляют водно-спиртовый раствор иода При
работе с таким титрантом проводимость раствора после точки
эквивалентности мало изменяется, и угол излома кривой титрования
становится более острым (рис. 9,6).
Метод применим и для определения малых количеств арсенита.
При титровании очень разбавленных раствороз (< 0,0005 и.)
используют бидистиллированную воду, и определение проводят в атмосфере
азота. С целью предотвращения явлений адсорбции пользуются
гладкими платиновыми электродами.
§ 6. Титрование, основанное на реакциях
комплексообразования
Для кондуктометрических определений используют реакции
комплексообразования. Наиболее широко для титриметрических
определений применяется комплексон III (двунатриевая соль этилендиаминтет-
рауксусной кислоты).
При взаимодействии катионов металлов с комплексоном III в
водных растворах увеличивается концентрация водородных ионов, что
приводит к повышению электропроводности раствора. При избытке
титранта НгУ-'-ионы начинают взаимодействовать с находящимися в
растворе Н+-ионами, так как кислотные свойства этилендиаминтетрауксус-
ной кислоты слабо выражены (р/С« = 2,0; 2,62; 6,16; 10,26).
Концентрация Н+ в результате этих взаимодействий уменьшается,
что приводит к понижению электропроводности раствора. Таким
образом, при количественном взаимодействии катионов металлор с
комплексоном III точки эквивалентности расположены в максимумах на
кондуктометрических кривых. В водных растворах солей возможно
кондуктометрическое титрование Fein, ThIY и других ионов,
образующих очень устойчивые комплексы (рис. 9, в).
Некоторые катионы можно титровать комплексоном III в кислых
водных, растворах. В этих условиях наиболее устойчивый комплекс с
комплексоном III образует Fein (piOey- = 25,1). Кондуктометрическое
определение железа (III) возможно даже при рН = 1, но повышение
электропроводности раствора до точки эквивалентности менее резко,
выражено, чем в предыдущем случае, так как раствор с начала
титрования имеет высокую электропроводность. Однако кондуктометрическая
кривая имеет такой же характер, как и при титровании в водном
растворе соли без добавления кислоты.
96 ГЛ Ш. КОНДУКТОМЕТРИЯ И КОНДУКТОМЕТРИЧЕСКОЕ- ТИТРОВАНИЕ
Катионы, образующие менее прочные комплексы, количественно в
кислой среде не титруются, а некоторые практически не
взаимодействуют с комплексоном III в кислых растворах, и поэтому не мешают
определению железа (III). Например, при рН=1 возможно определение
Fem в присутствии ионов Zn, Cd, Al, Co, Fe11, Mn, Ca, Mg и Ва.
Кондуктометрическое титрование катионов металлов, которое
количественно не взаимодействуют с комплексоном III в описанных выше
условиях, проводят в буферных растворах. Для этих целей используют
ацетатный или борно-щелочной буферные растворы. При этом в
ацетатный буферный раствор лучше вводить ацетат лития, а не ацетат
натрия, так как подвижность Li+ ниже подвижности Na+. При
титровании в буферных растворах изменение проводимости связано с
переходом катионов в комплексные ионы, заряды которых зависят от заряд-
ности титруемых катионов. Отличаются также подвижности катионов
и образующихся комплексных ионов. Выделяющиеся в процессе
реакции ионы водорода в буферном растворе связываются, при этом
изменяется концентрация ионов, образуемых буферным раствором. Общее
влияние этих факторов приводит к тому, что электропроводность
раствора до точки эквивалентности немного повышается. При избытке тит-
ранта электропроводность раствора увеличивается более сильно.
Кондуктометрическое титрование 0,01—0,005 М растворов солей
Си и РЬ можно проводить при рН = 2, Ni, Zn, Cd, Со и Al — при рН = 4,
a Mn, Fe11 и La — при рН = 5. Металлы, образующие малоустойчивые
комплексы, такие, как Ва, Sr, Ca и Mg, определяют при рН = 9—10-
На рис. 9, г приведена кривая титрования NiS04 в ацетатном
буферном растворе при рН = 4, типичная для комплексообразования в
этих условиях.
Возможен анализ двухкомпонентных смесей солей металлов при
изменении рН среды. Например, можно проводить последовательное
титрование ионов Fe111 или ThIV и Zn, Co, Fe11, La, Mn, Ва, Ca и других.
Б. АППАРАТУРА И ТЕХНИКА ВЫПОЛНЕНИЯ АНАЛИЗА
§ 7. Измерение электропроводности растворов
Электропроводность раствора электролита может быть найдена,
если измерить активное сопротивление между погруженными в него
электродами. Для измерения сопротивления пользуются переменным
током, так как постоянный ток вызывает разложение раствора и
поляризацию электродов. Источником тока обычно служат ламповые
генераторы звуковой частоты.
Сопротивление раствора электролита определяют путем сравнения
с эталонным сопротивлением. Для этого используют мостик Уитстона
(рис. 10). Сопротивления Zb Z2, Z3 и Z4 можно подобрать так, чтобы
ток в диагонали мостика отсутствовал, т. е. сопротивление его ветвей
было пропорционально друг другу. Измеряемое сопротивление Z4
можно найти по формуле;
7—7 i*?.
Сопротивления Z\ и Z2 выбирают постоянными или сохраняют
постоянным их соотношение; Z3 может изменяться. Таким образом, при
балансировке моста регулируют сопротивление Z3 и находят
сопротивление Z4.
§ 7. ИЗМЕРЕНИЕ ЭЛЕКТРОПРОВОДНОСТИ РАСТВОРОВ
97
В качестве нуль-инструмента применяют телефоны, осциллографы,
гальванометры переменного тока или (после выпрямления переменного
тока) гальванометры постоянного тока.
Рассмотренные условия равновесия моста применимы к
переменному току, если Zi, Z2, Z3 и Z4 — активные сопротивления. Однако в
мостике переменного тока силу тока в диагонали нельзя свести к нулю,
так как к активному сопротивлению добавляется некоторое реактивное
сопротивление, обусловленное наличием емкости электролитической
ячейкц и цепи.
В электрическую эквивалентную схему электролитической ячейки
(рис. 11) кроме истинного активного сопротивления раствора /?,
зависящего от концентрации ионов и их эквивалентной электропроводности,
Рис. 10. Мостик Уитстона:
Z\, Z2, Z9, Z* — плечи мостика; с
-переменная емкость; / — звуковой
генератор; 2 — индикатор нуля; 3 —
электролитическая ячейка.
Рис. 11. Электрическая экнивалентная
схема ячейки:
R — истинное сопротивление раствора; с —
геометрическая емкость ячейки; с —емкость
двойного слоя; с и R —емкость и сопро-
п и
тивление поляризации; сх и Rx —
шунтирующие емкости, зависящие от конструкции
ячейки; с> — емкость проводов.
входят дополнительные активные и реактивные сопротивления,
возникающие в ячейке при измерении сопротивления. Электролитическую
ячейку — сосуд той или иной формы, содержащий электролит с
погруженными в него электродами, в принципе можно рассматривать как
конденсатор с электродной поверхностью 5, электродным расстоянием
/, заполненный раствором с диэлектрической проницаемостью г.
Сопротивление емкости ст двух параллельных электродов, шунтирующее
истинное сопротивление электролита в водных растворах, обычно значительно
выше истинного сопротивления раствора, и поэтому не вызывает ошибок
в измерении электропроводности. Однако при очень высоком истинном
сопротивлении электролита эти величины могут быть соизмеримы.
Возникающие ошибки уменьшаются с понижением частоты тока и
увеличением константы сосуда.
На границе металлический электрод — раствор электролита
возникает двойной электрический слой. Емкость двойного слоя влияет на
сдвиг фаз между током и напряжением, что приводит к ошибкам в
измерении истинного сопротивления раствора.
Ошибки измерений могут быть связаны с электрохимическими
процессами на электродах — разрядкой ионов, приводящей к изменению
концентрации ионов у поверхности электрода. Вследствие медленной
диффузии ионов к электроду наблюдается концентрационная
поляризация, которая создает поляризационную емкость сп и поляризационное
сопротивление /?п- Ошибки, связанные с поляризационными явлениями,
уменьшаются с повышением частоты тока и увеличением концентрации.
При частоте тока выше 1000 гц влияние поляризации незначительно.
98 ГЛ. III. КОНДУКТОМЕТРИЯ И КОНДУКТОМЕТРИЧЕСКОЕ ТИТРОВАНИЕ
Шунтирование сопротивления R емкостью сх и сопротивления Ru
возникающее при неудачной конструкции ячейки (близкое
расположение проводов, идущих от электродов, неудачное расположение
электродов по отношению к проводам и т. д.), также вызывает ошибки
измерения. Емкость проводов с2 может стать причиной емкостных
утечек тока.
Ячейки для кондуктометрических титрований, таким образом,
должны отвечать следующим основным требованиям:
1) иметь оптимальные геометрические размеры межэлектродного
пространства;
2) поляризационные явления на электродах должны быть
минимальными;
3) утечка тока, обусловленная паразитными емкостными связями,
должна быть минимальной.
Влияние емкости нельзя совершенно исключить, но емкостное
сопротивление можно компенсировать (см. рис. 10) путем включения
конденсатора с параллельно сопротивлению Z3. При этом мостик
балансируется одновременной компенсацией небаланса магазином
сопротивления Z3 и емкостью с. Однако исключить ошибки, связанные с
поляризационным сопротивлением, не удается. Эти ошибки уменьшаются при
использовании платинированных электродов, так как увеличенная
поверхность их уменьшает плотность тока.
Платинирование электродов нельзя применять, если платиновая
чернь оказывает каталитическое влияние на проводимую реакцию или
изменяет концентрацию веществ в растворе вследствие адсорбции.
В некоторых случаях удобно применять платинированные электроды,
прокаленные до красного каления (серое платинирование). Такие
электроды несколько слабее уменьшают поляризацию по сравнению с
черно-платинированными электродами, но они обладают и значительно
меньшими адсорбционными свойствами.
Установка для кондуктометрического титрования состоит из
электролитической ячейки и полум-икробюретки для титрования, звукового
генератора, мостика Уитстона и индикатора нуля. Конструкции
электролитических ячеек описаны ниже. Для подачи стандартного раствора
исцользуют полумикробюретку емкостью 10 мл, которую
устанавливают над сосудом для титрования.
Для питания системы переменным током используют генераторы
ЗГ-1, ЗГ-2, ЗГ-10, ЗГ-33 и др. Для работы используют переменный ток
частотой 1000 гц.
Мостик Уитстона собирают по схеме, показанной на рис. 10. В два
плеча мостика включают эталонные магазины сопротивлений Р-517М
(Zi и Z2). В третье и четвертое плечи включают магазин сопротивлений
Р-517М (Z3) с параллельно включенным магазином емкости Р-513 (с)
и электролитическую ячейку (Z4).
В качестве нуль-инструмента применяют осциллографический
индикатор нуля типа У2-2 (ИНО-ЗМ). При полном балансе мостика
эллипс на экране осциллографа стягивается в горизонтальную линию.
При измерений сопротивления раствора в электролитической ячейке
мостик балансируют путем одновременной компенсации небаланса
мостика при помощи магазина сопротивлений (Z3) и магазина
емкости (с).
Установка, собранная по этой схеме, имеет высокую
чувствительность и позволяет определять сопротивление растворов до 104 ом. При
измерении более высоких сопротивлений даство^ов следует увеличить
сопротивление в плечах мостика.
§ 7. ИЗМЕРЕНИЕ ЭЛЕКТРОПРОВОДНОСТИ РАСТВОРОВ
99
Для измерения сопротивления раствора при титровании может быть
также рекомендован кондуктометр марки ММЗЧ-6Ч, позволяющий
определять сопротивление до 107 ом. Питание этого прибора
осуществляется от сети переменного тока напряжением 220 в с частотой 50 гц.
Частота внутреннего генератора, питающего мостик, 1150 гц.
Указателем равновесия мостика служит электронно-оптический индикатор.
Константа сосуда. При кондуктометрическом титровании измеряют
сопротивление раствора R после добавления каждой порции титранта.
Полученные данные используют для определения электропроводности
раствора W. Величину удельной электропроводности раствора %
находят из уравнения [см. также уравнения (2) и (3)]:
Если бы ток проводил раствор только в объеме между
электродами, для определения удельной электропроводности можно было бы
использовать расстояние между электродами / и их площадь 5. Однако
в электролитической ячейке ток может проводить весь находящийся в
ней раствор, так как силовые линии располагаются не только между
электродами. Кроме того, истинная поверхность электродов изменяется
при их платинировании. Поэтому удельную электропроводность
выражают следующей зависимостью:
*-*-]f (27)
где К — константа сосуда (слс1), зависящая от площади электродов и расстояния
между ними, а также от формы сосуда и объема раствора, проводящего ток.
Так как электропроводность раствора W представляет собой
величину, обратную сопротивлению (1AR), имеем:
x = KW (28)
Таким образом, для определения удельной электропроводности
раствора необходимо измеренную электропроводность умножить на
константу сосуда. Однако поскольку константа сосуда должна быть
величиной постоянной, нет необходимости при построении кондуктомет-
рической кривой пересчитывать электропроводность (W) в удельную
электропроводность (х), так как эти величины прямо пропорциональны
друг другу.
Основным требованием, предъявляемым к электролитическим
ячейкам, является постоянство константы сосуда при неизменном
объеме раствора в области тех сопротивлений, которые измеряются в
данной ячейке. Иногда наблюдается кажущееся изменение константы
сосуда, что вызывается различными рассмотренными выше
электрохимическими и электрическими явлениями. Поэтому для каждой
электролитической ячейки, используемой для аналитических целей,
предварительно проверяют постоянство константы сосуда.
Для определения константы сосуда измеряют сопротивление
стандартного раствора с известной удельной электропроводностью. В
качестве стандартов берут растворы КС1, для которых удельная
электропроводность определена с высокой точностью. Сопротивление
стандартных растворов КО нескольких концентраций (обычно 0,1 н. и 0,01 н.)
измеряют при постоянном объеме раствора, равном первоначальному
объему титруемого раствора. При особенно точных определениях
измерение проводят при постоянной температуре (в термостате). Константы
сосудов имеют различные значения от 0,1 до 10 и выше. С увеличением
100 ГЛ. I1L КОНДУКТОМЕТРИЯ И КОНДУКТОМЕТРИЧЕСКОЕ ТИТРОВАНИЕ
площади электродов и уменьшением расстояния между ними константа
сосуда уменьшается. Если измеряемое сопротивление титруемых
растворов сильно различается, необходимо иметь несколько сосудов с разными
константами.
§ 8. Конструкции электролитических ячеек
В кондуктометрическом титровании используются
электролитические ячейки разных конструкций, различающиеся по форме сосудов,
площади электродов и расстоянию между ними, а также по месту их
закрепления, способу перемешивания раствора и т. д.
Площадь электродов и расстояние между ними выбирают в
зависимости от измеряемого сопротивления. Для слабо проводящих
растворов с целью уменьшения измеряемого сопротивления увеличивают
площадь электродов и уменьшают расстояния между ними.
Критерием пригодности электролитической ячейки для
кондуктометрического титрования служит постоянство константы сосуда в
области измеряемых сопротивлений. Однако следует учитывать, что
константа сосуда измеряется при постоянном объеме жидкости в ячейке,
а при титровании объем раствора в ячейке увеличивается. Поэтому
целесообразно использовать для кондуктометрического титрования
ячейки, константа сосуда которых не зависит от объема жидкости в
ячейке. Кроме того, измеренная в процессе титрования
электропроводность раствора вследствие его разбавления всегда несколько
отличается от электропроводности, которая наблюдалась бы при
постоянном объеме. Для уменьшения ошибки в величине электропроводности
раствора вследствие разбавления в процессе кондуктометрического
титрования концентрация титранта должна быть по крайней мере в
10 раз выше, чем у титруемого раствора. Ошибку можно уменьшить,
если привести электропроводность к постоянному объему путем
использования поправочного коэффициента разбавления (Л);
л- Уо + у
где V0 — первоначальный объем электролита, ял;
V — объем прибавленного титранта, мл.
Несколько электролитических ячеек для кондуктометрического
титрования показано на рис. 12. В простейшей электролитической ячейке
(рис. 12, а) платиновые электроды 1 расположены горизонтально и
жестко закреплены в боковых стенках сосуда. В большинстве случаев
электроды в ячейках располагают вертикально, что особенно важно для
случаев титрования, сопровождающихся образованием осадков.
Константа сосуда этой ячейки зависит от объема раствора, так как весь
раствор, помещенный в сосуд, проводит ток. Раствор в такой ячейке
можно перемешивать механической или магнитной мешалкой.
Электролитическая ячейка, показанная на рис. 12,6, позволяет при
титровании проводить измерения электропроводности с более высокой
точностью. Эта ячейка отличается тем, что сосуд для титрования
значительно расширен в верхней части. Титруемый раствор (30 мл),
помещенный в ячейку, заполняет весь объем до расширения сосуда. При
титровании уровень раствора сравнительно мало поднимается, так как
заполняется расширенная часть, и константа сосуда мало изменяется.
Раствор в этой ячейке перемешивается магнитной мешалкой.
Описанная ячейка пригодна для измерения сопротивлений малопроводящих
растворов, в том числе некоторых неводных растворов; площадь
электродов этой ячейки равна 4 см2, а расстояние между ними —2 см. При
§ 8. КОНСТРУКЦИИ ЭЛЕКТРОЛИТИЧЕСКИХ ЯЧЕЕК
101
титровании неводных растворов употребляются гладкие платиновые
электроды (константа сосуда при этом равна 0,23). Если в ячейках
такого типа титруют хорошо проводящие растворы, то площадь
электродов следует уменьшить.
Электролитическая ячейка, показанная на рис. 12, в, отличается тем,
что константа сосуда не зависит от объема раствора, находящегося
Рис. 12. Конструкции электролитических ячеек:
с —ячейка с константой сосуда, зависящей от объема раствора; б —ячейка с расширенной
верхней частью сосуда; $ — ячейка с константой сосуда, не зависящей от объема раствора
(показаны разрезы сосуда и электрода); г —схема и общий вид ячейки с вращающимся сосудом
для хронокондуктометрического титрования; 1 — платиновые электроды; 2— контактные
трубки; 3 — сосуд для титрования; 4 - измерительная трубка; 5, б— краны; 7 —колодка для
закрепления электродов.
в ячейке. Крестообразные электроды / площадью 0,5 см2 впаяны в
стеклянные трубки 2. Оба конца измерительной трубки 4 переходят в
вертикальные сосуды для титрования 3 объемом 60 смг, закрытые сверху
пробками на шлифах. Сосуды соединены между собой трубкой с
краном 5. В одной из пробок сосуда имеется входной патрубок с краном 6.
Контактные трубки 2 к электродам расположены перпендикулярно к
горизонтальной измерительной трубке с целью устранения паразитных
102 ГЛ. III, КОНДУКТОМЕТРИЯ И КОНДУКТОМЕТРИЧЕСКОЕ ТИТРОВАНИЕ
емкостных связей. Пространство, в котором могут распространяться
силовые линии электрического поля между электродами, ограничено, и,
следовательно, объем раствора, проводящего ток, остается практически
постоянным. В этих условиях константа сосуда не изменяется при
различных объемах раствора.
Для защиты от двуокиси углерода воздуха и перемешивания
раствора в ячейку предусмотрена подача азота. При перемешивании
закрывают кран 5 и пропускают через раствор азот. Перемешивать можно
также при помощи груши струей воздуха, очищенного от С02.
Измерение сопротивлений при титровании в этой ячейке можно
проводить при помощи мостика Уитстона или кондуктометра ММЗЧ.
На рис. 12, г показаны погружающиеся электроды, употребляемые
при хронокондуктометрическом титровании. Электроды закрепляются
в колодке Z, с помощью которое их можно устанавливать на расстоянии
1—4 см друг от друга. Вместе с электродами закреплена трехлопастная
мешалка из фторопласта. Электроды погружают в раствор перед
титрованием. Перемешивание раствора при титровании осуществляется путем
равномерного вращения сосуда для титрования, который устанавливают
на вращающемся диске (78 об/мин). Если платинированные электроды
имеют площадь 0,5 см2 и расположены на расстоянии 4 см друг от
друга, то константа сосуда остается практически постоянной (1,12) для
растворов различной концентрации, но одного и того же объема.
§ 9. Измерения в хронокондуктометрическом титровании
Высокая точность хронокондуктометрического титрования
достигается при использовании мостиков с автоматическим
уравновешиванием и записью величин активной составляющей.
Для определений с меньшей точностью используют
неуравновешенные мостики, при этом фиксируют активное и реактивное сопротивления
растворов. Простейший метод,
используемый для
автоматизации, основан на определении
электропроводности по силе тока,
проходящего через
электролитическую ячейку в процессе
титрования при постоянном
напряжении. Силу тока определяют по
падению напряжения на
эталонном сопротивлении.
На рис. 13 показана схема
прибора с неуравновешенным
мостиком. С помощью такой
установки можно осуществлять
автоматическую запись кондуктомет-
рических кривых. Цепь состоит из сопротивлений Ri и R2,
электролитической ячейки 2, селеновых выпрямителей Зу 4 и регистратора
постоянного тока. Установка питается переменным током частотой 50 гц,
напряжением 127 в, которое стабилизируется
трансформатором-стабилизатором 1 и понижается до 8 в. Сопротивление R\ (делитель
напряжения) позволяет отбирать часть этого напряжения. Изменение силы тока
при титровании фиксируется регистратором 5. Регистратором может
служить милливольтметр постоянного тока марки МСЩ-ПР, в котором
следует увеличить скорость передвижения ленты до 2 см/мин путем
Рис. 13. Электрическая схема прибора для
хронокондуктометрического титрования:
j^ — делитель напряжения (сопротивление 200 ом)\
R2 — эталонное сопротивление (100 ом); / —
трансформатор-стабилизатор; 2 — электролитическая
ячейка; 5, 4— селеновые выпрямители или
германиевые диоды; 5 — милливольтметр.
§ 10. ПОДГОТОВКА К ОПРЕДЕЛЕНИЮ
103
замены шестерен в устройстве, протягивающем ленту. Показания
милливольтметра наносятся на ленту через 10—15 сек.
Оптимальная скорость перемещения ленты 2 см/мин; при
титровании 0,05—0,1 н. растворов время истечения 10 мл 0,3 н. стандартного
раствора должно быть около 6 мин. При увеличении концентрации тит-
ранта следует уменьшить скорость его истечения.
При работе на таком приборе может наблюдаться нелинейная
зависимость показаний прибора от электропроводности раствора в
широком диапазоне сопротивлений. Однако для успешного титрования
достаточно получить линейную зависимость только в некоторой области
сопротивления. Например, сопротивление при-
близительно 0,1 н. растворов редко бывает (Г==**Е^/=="=>
более 1000 ом, поэтому достаточно, чтобы ли- <?g5>
нейная зависимость наблюдалась только при ^^J4PL<^
сопротивлениях меньше 1000 ом. (~* И^л^-?
Для автоматизации процесса титрования ^-=
могут быть использованы и другие схемы, в
том числе электронные, а также кондуктомет-
рические приставки к стандартным
самопишущим приборам. Регистратором при этом
может служить электронный потенциометр
ЭПП-09.
Электролитическая ячейка, применяемая
в этом виде титрования, аналогична ячейке,
которая приведена на рис. 12, г.
При титровании в сосуд равномерно
каплями подается стандартный раствор. Для этой
цели используются различные дозаторы
поршневого и плунжерного действия, поплавковые,
клапанные и мембранные устройства и т. д.
Для равномерной подачи жидкости
применяют также сосуд Мариотта (рис. 14). По
мере вытекания жидкости через отводную
трубку в сосуд Мариотта пузырьками
поступает воздух через вертикальную трубку.
Воздух, поступающий в сосуд Мариотта, можно очистить от двуокиси
углерода, пропустив его через хлоркальциевую трубку, наполненную
натронной известью.
Рис. 14. Сосуд Мариотта:
а — общий вид; б — капилляр;
1 — хлоркальциевая трубка;
2 — трубка для подачи воздуха;
3 — сосуд с тубусом; 4— верхний
капилляр; 5— отводная трубка;
6 — зажим; 7 —кран; 5 —нижний
капилляр.
В. ПРАКТИЧЕСКИЕ РАБОТЫ
§ 10. Подготовка к определению
Собирают установку (см. рис. 10) для кондуктометрического
титрования, включают в нее электролитическую ячейку (см. рис. 12, в) и
подготавливают для работы; платинируют электроды и определяют
константу сосуда.
Применимы ячейки и других конструкций.
Платинирование электродов. Для платинирования электродов
используют раствор, содержащий 30 г платинохлористоводородной
кислоты и 0,3 г ацетата свинца в 1 л. Ацетат свинца способствует
образованию тонкодисперсного плотного осадка платиновой черни. Этот
раствор наливают в электролитическую ячейку (рис, 15) и пропускают
через него ток, через 1 мин меняя направление его. Плотность тока при
платинировании не должна превышать 30 мка/см2. Платинирование ведут
104 ГЛ. III. КОНДУКТОМЕТРИЯ И КОНДУКТОМЕТРИЧЕСКОЕ ТИТРОВАНИЕ
Рис. 15. Схема
установки для платинирования
электродов:
/ — электролитическая
ячейка; 2 — миллиамперметр; 3 —
реостат: 4 — источник тока.
10 мин. После платинирования электроды должны быть покрыты ровным
плотным слоем платиновой черни. Подготовленные электроды промывают
дистиллированной водой и хранят под слоем дистиллированной воды.
Определение константы сосуда. Для определения константы сосуда
готовят 0,1 н. раствор КС1, из которого путем разбавления получают
0,01н. раствор. Электролитическую ячейку промывают концентрированной
азотной кислотой, затем многократно ополаскивают дистиллированной
водой, а потом 0,01 н. раствором КО. В ячейку
помещают 25 мл 0,01 н. раствора КО и
определяют сопротивление раствора при помоши
мостика Уитстона. После этого выливают
раствор из ячейки и промывают ее 0,1 н.
раствором КС1. Переносят в ячейку 25 мл 0,1 н.
раствора КО и повторяют измерение
сопротивления.
Определение константы сосуда следует
проводить при постоянной температуре. Для этого
сосуд с раствором помещают в термостат и
выдерживают перед измерением сопротивления до
тех пор, пока он не примет температуру
термостата.
Константу сосуда вычисляют по уравнению:
K = *R (29)
где к —удельная электропроводность раствора КС1 взятой нормальности;
R — сопротивление раствора в ячейке, ом.
Значения констант сосуда (см. рис. 12, в), установленные по 0,1 н.
и 0.01 н. растворам КО, должны быть близкими, — они составляют
соответственно 6,16 и 6,07 и практически не изменяются при увеличении
объема раствора в ячейке.
Общая методика кондуктометрического титрования. Полумикробю-
ретку емкостью 10 мл наполняют стандартным раствором титранта и
устанавливают над сосудом для титрования. В электролитическую
ячейку (см. рис. 12, б) переносят 25 мл анализируемого 0,1—0,05 н.
раствора и определяют сопротивление при помощи мостика Уитстона.
Затем в ячейку добавляют титрант порциями по 0,2 мл. После
добавления каждой порции титранта раствор перемешивают. Для этого
закрывают кран 5, открывают кран 6, выходной патрубок которого
подключен к линии, подающей азот или очищенный от двуокиси углерода
воздух, и пропускают газ через раствор. После перемешивания
открывают кран 5 для равномерного распространения раствора в ячейке.
Электропроводность раствора после перемешивания обычно быстро
становится постоянной. В отдельных случаях раствор приходится
выдерживать некоторое время для достижения постоянной электропроводно-
Объем
добавленного
раствора,
мл
Сопротивление раствора (R)
1-е
измерение
2-е
измерение
3-е
измерение
Средняя
величина
Электропроводность
раствора
Поправочный
коэффициент
на
разбавление *
Электропроводность,
приведенная
к
первоначальному
объему
(W-AW)
* При работе с ячейками, константа сосуда которых не зависит от объема раствора, поправка
ке вводится.
§ 10. ПОДГОТОВКА К ОПРЕДЕЛЕНИЮ
105
сти. Измерение сопротивления после добавления каждой порции тит-
ранта повторяют три раза. Результаты измерений заносят в рабочий
журнал, как показано ниже.
Затем строят кондуктометрическую кривую, откладывая по оси
абсцисс число миллилитров добавленного титранта, а по оси ординат —
электропроводность раствора, приведенную к первоначальному объему.
Затем графическим методом устанавливают точку эквивалентности и
определяют число миллилитров стандартного раствора, вступившего
в реакцию до изломов кондуктометрических кривых.
0 1 2 3^ 56?
Объем раствора NaOH. мл
Рис. 16, Кондуктометрические
кривые титрования 1,0 н. раствором NaOH
индивидуальных веществ кислотного
характера и их смесей:
1 — хлористоводородная кислота; 2 —хлорид
аммония; 3 — азотная кислота + нитрат
аммония; 4 — полиметакриловая кислота;
5 — борная кислота; 6 — борная кислота+
+гидрохлорид гидроксиламииа.
0 1 2 3 4 5
Объем раствора НО. мл
Рис. 17. Кондуктометрические кривые
титрования 1,0 н. раствором НС1
индивидуальных веществ основного характера и
их смесей:
/ — аммиак; 2 — анилин; 3— ацетат натрия; 4 —
едкий натр; 5 —едкий натр+аммиак; 6 — едкий натр-Ь
+ анилин.
Зная объем и нормальность стандартного раствора титранта,
израсходованного на титрование определяемого вещества, рассчитывают
содержание по известным формулам (см. книга 2, гл. I, § 10). При
определении содержания сильных кислот и кислот средней силы в
расчетах используют значение нормальности NaOH, установленной по
метиловому оранжевому, а при определении слабых кислот —по
фенолфталеину.
При определении содержания солей слабых оснований в
вычислениях необходимо использовать фактическую нормальность стандартного
раствора NaOH, соответствующую содержанию в нем только NaOH.
Кондуктометрические кривые, которые получены при титровании
в ячейке, показанной на рис. 12, в, различных индивидуальных веществ
основного или кислотного характера, а также из смесей растворами НС1
и NaOH, приведены на рис. 16 и 17.
Общая методика хронокондуктометрического титрования. Перед
титрованием определяют скорость истечения стандартного раствора.
106 ГЛ. HI. КОНДУКТОД1ЕТРИЯ И КОНДУКТОМЕТРИЧЕСКОЕ ТИТРОВАНИЕ
Для этого устанавливают по секундомеру время наполнения мерной
колбы емкостью 25 мл стандартным раствором титранта. По
секундомеру определяют также время между записью отдельных показаний
милливольтметра на ленту.
В сосуд для титрования (см. рис. 12, г) наливают 50 мл
анализируемого (0,1—0,05 н.) раствора, погружают электроды с мешалкой,
включают мотор для вращения ванны и милливольтметр. При помощи
милливольтметра устанавливают в
такое положение, при котором кондук-
тометрическая кривая может
полностью разместиться на ленте. Если
электропроводность раствора при
титровании понижается, стрелку
устанавливают в верхней части шкалы, если
повышается— в нижней. Затем прибор
устанавливают так, чтобы отводная
трубка сосуда Мариотта (см. рис. 14)
находилась над ячейкой. Включают
регистрирующую часть
милливольтметра и при нанесении второго
показания на ленту начинают подачу
стандартного раствора. Запись кривой
заканчивают при избытке титранта.
После окончания титрования электроды
вынимают и удаляют из ячейки
раствор. Промывают ячейку и электроды
дистиллированной водой и проводят
параллельные определения. На кон-
дуктометрических кривых графическим
методом устанавливают точки
эквивалентности и определяют количество
интервалов между записью показаний
милливольтметра до ее изломов.
Десятые доли интервалов вблизи точки
эквивалентности находят на глаз.
Продолжительность титрования
зависит от числа определяемых
компонентов и достигает 5—20 мин.
кривые (рис. 18) не должны быть
слишком растянутыми по длине, так как в этом случае изломы кривых
становятся менее резкими. В зависимости от числа титруемых
компонентов длина кривой должна составлять 10—20 см. Длина кривой
определяется скоростью передвижения ленты, а также зависит от
концентрации и скорости истечения стандартного расгвора.
Если в установке для хронокондуктометрического титрования
линейная зависимость показаний регистратора от электропроводности
раствора справедлива только в случаях, когда сопротивление
растворов меньше 1000 ом, то кривые титрования растворов очень слабых
электролитов, имеющих сопротивление выше этой величины, несколько
отличаются от наблюдаемых при классическом методе кондуктометри-
ческого титрования. В начале титрования на кривых имеется
горизонтальный участок, и только с уменьшением сопротивления в процессе
титрования кривые приобретают обычную форму. Примером может
служить кривая титрования едким натром раствора борной кислоты
(рис. 18, кривая 2). Точки эквивалентности устанавливаются графиче-
делителя напряжений стрелку
Продолжительность титрования, сен
Рис. 18. Кривые
хронокондуктометрического титрования раствором NaOH:
1 —хлористоводородная кислота; 2 — борная
кислота в водном растворе; 3 — борная
кислота в присутствии 0,05 н. NaCl; 4—
азотная кислота 4- фенол: 5 —
хлористоводородная кислота + полиметакриловая кислота;
6— хлористоводородная кислота + аминоук-
сусная кислота; 7— едкий натр + ацетат
натрия; 8 — анилин + фенолят натрия; 9 —
едкий натр + аммиак + ацетат натрия.
Хронокондуктометрические
§ 11, ОПРЕДЕЛЕНИЕ КИСЛОТ МЕТОДОМ КОНДУКТОМЕТРИЧЕСКОГО ТИТРОВАНИЯ 107
ским методом на этих кривых с достаточной точностью, если
использовать прямолинейные участки кривых вблизи точки эквивалентности.
Для получения кривых обычного типа можно проводить титрование
на фоне сильных электролитов, например в присутствии NaCl (рис. 18,
кривая 3). Кривые титрования сильных электролитов и электролитов
средней силы (кислот, оснований, солей и смесей этих электролитов)
аналогичны получающимся в классическом методе. Поэтому
правильность выбранных параметров схемы (сопротивлений, выпрямителей)
можно проверить путем титрования основанием раствора НС1; кривая
титрования должна иметь V-образную форму (рис. 18, кривая 1).
Расчет. При вычислении результатов используют среднее значение
из двух-трех параллельных титрований. Расчеты проводят по формуле:
ntSVN
ё==-\тГ (30)
где g — содержание определяемого вещества в титруемом растворе, г;
п — количество интервалов между точками на кондуктометрической кривой до
изломов;
/ — время между записью показаний милливольтметра, сек;
Э — грамм-эквивалент определяемого вещества;
V — объем раствора, используемого для определения скорости истечения
стандартного раствора, мл;
N — нормальность стандартного раствора;
Т — время наполнения мерной колбы стандартным раствором, сек.
§11. Определение индивидуальных кислот методом
кондуктометрического титрования
Определение хлористоводородной, борной
или полиметакриловой кислоты
Определение кислот основано на реакции их нейтрализации:
H+ + Cl"" + Na+ + OH" ^z± Na+ + Cr + H20
H3B03 + Na+ + OH- ^z± Na+ + BC>2 + 2H20
CH3
-CH2—C—
I
COOH
+ nNa+ + nOH"
CH3
I
-CH2—C—
I
СОСГ
+ /zNa+ + «H20
Методика определения. Для титрования отбирают 25 мл
анализируемого раствора кислоты и переносят в электролитическую ячейку.
Проводят кондуктометрическое титрование кислоты 1,0 н. раствором
NaOH (см. § 9).
При титровании НС1 электропроводность раствора до точки
эквивалентности линейно понижается, так как нейтрализуются
высокоподвижные ионы водорода (рис. 16, кривая 1). Нейтрализация борной
кислоты сопровождается повышением проводимости раствора до точки
эквивалентности (рис. 16, кривая 5), что объясняется образованием
хорошо диссоциирующей соли. Вблизи точки эквивалентности кривая
титрования борной кислоты в более разбавленных растворах имеет
плавный изгиб вследствие гидролиза метабората натрия.
Нейтрализация слабой полиметакриловой кислоты также сопровождается
повышением электропроводности раствора, однако кондуктометрическая
кривая до точки эквивалентности слегка изогнута, что связано с
изменением степени диссоциации кислоты в процессе нейтрализации (рис. 16,
кривая 4), Избыток основания при титровании всех перечисленных
108 ГЛ. III. КОНДУКТОМЕТРИЯ И КОНДУКТОМЕТРИЧЕСКОЕ ТИТРОВАНИЕ
кислот вызывает резкое повышение электропроводности раствора. По
кондуктометрическим кривым находят число миллилитров раствора
NaOH, вступившего в реакцию с кислотами, и рассчитывают их
содержание в титруемом растворе, пользуясь известными формулами.
Грамм-эквивалент полиметакриловой кислоты принимают равным
молекулярному весу метакриловой кислоты.
§ 12. Определение индивидуальных оснований методом
кондуктометрического титрования
Определение едкого натра, аммиака и анилина
Определение основано на реакции нейтрализации оснований
раствором НС1:
Na+ + ОН" + Н+ + СГ ^z± Na+ + CF + H20
NH3 + H+ + Cr ^z± NHJ + СГ
C6H5NH2 + H+ + СГ 5=± CeH5NH3 + Cr
Методика определения. Кондуктометрическое титрование проводят
1,0 н. раствором НС1 (см. § 9).
При нейтрализации NaOH электропроводность раствора линейно
понижается, так как уменьшается концентрация высокоподвижных
гидроксильных ионов (рис. 17, кривая 4). При титровании слабых
оснований— аммиака (рис. 17, кривая /) и анилина (рис. 17, кривая 2)
происходит повышение проводимости раствора до точки эквивалентности,
вызываемое образованием хорошо диссоциирующих солей. На кривой
титрования разбавленных растворов анилина вблизи точки
эквивалентности наблюдается слабый изгиб кривой вследствие гидролиза
получающегося гидрохлорида анилина. Избыток НС1 вызывает резкое
увеличение электропроводности раствора.
На кривых титрования графическим методом устанавливают точки
эквивалентности и находят число милиллитров хлористоводородной кис-
4оты, вступившей в реакцию с основаниями. Содержание оснований
а титруемом растворе вычисляют обычным способом.
§ 13. Анализ смесей оснований методом кондуктометрического
титрования
Анализ смесей: едкий натр и аммиак; едкий натр и анилин
Определение основано на последовательной нейтрализации
оснований раствором НС1. При взаимодействии с НС1 сначала
нейтрализуется сильное основание NaOH, а затем слабые основания —аммиак
и анилин.
Методика определения. В электролитическую ячейку помещают
аликвотную часть анализируемого раствора смеси оснований и
проводят кондуктометрическое титрование 1,0 н. раствором НС1.
Кривые титрования смесей оснований имеют два излома (рис. 17,
кривые 5, 6). При нейтрализации NaOH электропроводность линейно
понижается, что вызывается уменьшением концентрации
высокоподвижных гидроксильных ионов. После первой точки эквивалентности
электропроводность начинает увеличиваться, так как при нейтрализации
слабых оснований — аммиака и анилина — образуются хорошо
диссоциирующие соли. Кривая титрования смеси NaOH с аммиаком может
§ 15. АНАЛИЗ СМЕСЕЙ КИСЛОТ И СОЛЕЙ СЛАБЫХ ОСНОВАНИИ 1Q9
иметь несколько сглаженный изгиб, соответствующий первой точке
эквивалентности; объяснение этому дано выше. При титровании смеси
NaOH с анилином, наоборот, такое явление может наблюдаться около
второй точки эквивалентности вследствие гидролиза гидрохлорида
анилина. Однако для достаточно концентрированных растворов плавность
изгибов кривых настолько слабо выражена, что не мешает
графическому установлению точек эквивалентности. По кондуктометрическим
кривым определяют число миллилитров хлористоводородной кислоты,
вступившей в реакцию с компонентами смеси, и определяют их
содержание обычными способами,
§ 14. Определение солей методом кондуктометрического
титрования
Определение ацетата натрия или хлорида аммония
Определение ацетата натрия основано на вытеснении .слабой
уксусной кислоты сильной кислотой:
Na+ + CH3COO~4-H+ + cr ^± СН3СООН + Na+ + СГ
Определение хлорида аммония основано на реакции вытеснения
аммиака сильным основанием:
NHj + Cr + Na+ + OH~ ^z± NH3 + Na+ + СГ + H20
Методика определения. Для титрования аликвотную часть
анализируемого раствора соли переносят в электролитическую ячейку. Кон-
дуктометрическое титрование ацетата натрия проводят 1,0 н. раствором
НС1, а хлорида аммония 1,0 н. растворохм NaOH (см. § 9). Изменение
электропроводности раствора при титровании солей зависит от
сравнительной подвижности ионов, завещающих друг друга в растворе
в процессе взаимодействия (см. стр. 84). При титровании
электропроводность раствора до точки эквивалентности немного увеличивается
(рис. 17, кривая 3). При титровании NH4C1 основанием, наоборот,
электропроводность раствора до точки эквивалентности немного понижается
(рис. 16, кривая 2).
По кондуктометрическим кривым определяют число миллилитров
титранта, участвующего в реакции, и рассчитывают содержание соли
в титруемом растворе, пользуясь известными формулами.
§ 15. Анализ смесей кислот и солей слабых оснований
методом кондуктометрического титрования
Анализ смесей: азотная кислота и нитрат аммония;
борная кислота и гидрохлорид гидроксиламина
Определение основано на последовательном взаимодействии с NaOH
кислот и солей слабых оснований. Последовательность взаимодействия
компонентов смеси, как рассмотрено выше (см. § 3), зависит от суммы
рКа свободной кислоты и рКъ основания, образующего соль. При
титровании раствором NaOH смеси HNO3 и NH4NO3, характеризующейся
(рКа + рКь) < 12, сначала нейтрализуется НГЮз, а затем вытесняется
аммоний (рКь = 4,75) из его соли. Если смесь образуют электролиты,
для которых (рКа + рКъ) > 16, последовательность взаимодействия
изменяется.
1 10 ГЛ. III. КОНДУКТОМЕТРИЯ И КОНДУКТОМЕТРИЧЕСКОЕ ТИТРОВАНИЕ
При титровании основанием смеси борной кислоты (рКа = 9,12)
с гидрохлоридом гидроксиламина (рКь = 8,03) сначала взаимодействует
гидрохлорид гидроксиламина, а затем нейтрализуется борная кислота.
Методика определения. В электролитическую ячейку переносят
аликвотную часть раствора смеси оснований и солей слабых кислот
и проводят кондуктометрическое титрование взятой пробы 1,0 н.
раствором NaOH (см. § 9).
При титровании сильным основанием смеси HN03 и NH4N03
сначала нейтрализуется HN03, что приводит к сильному понижению
электропроводности раствора. Когда практически полностью нейтрализована
НМОз, протекает реакция вытеснения аммиака. Так как подвижность
NHt-ионов выше подвижности Na+-noHOB, заменяющих их в растворе,
электропроводность и дальше продолжает понижаться, но менее
сильно, чем до первой точки эквивалентности (рис. 16, кривая 3). При
титровании основанием смеси борной кислоты с гидрохлоридом
гидроксиламина сначала протекает реакция с гидрохлоридом. В течение этой
реакции электропроводность раствора мало изменяется, потому что
мало различаются подвижности ионов, заменяющие друг друга в
растворе. Однако протекающая затем реакция нейтрализации борной
кислоты вызывает повышение электропроводности раствора, что и
приводит к излому кондуктометрической кривой в первой точке
эквивалентности (рис. 16, кривая 6). После второй точки эквивалентности электро-
проводность сильно увеличивается от избытка основания.
По кондуктометрическим кривым определяют число миллилитров
раствора NaOH, вступившего в реакцию с отдельными компонентами
смеси, и рассчитывают их содержание в титруемом растворе, пользуясь
соответствующими формулами,
§16. Анализ смесей кислот методом
хронокондуктометрического титрования
Анализ сцесер,: азотная кислота и фенол; хлористоводородная кислота
и полиметакриловая кислота; хлористоводородная
и аминоуксусная кислота
Определение основано на последовательной нейтрализации сильным
основанием кислот, отличающихся различной силой. При титровании
сначала нейтрализуются сильные кислоты — HN03 и HC1, а затем
слабые— фенол и полиметакриловая кислота.
Состав аминоуксусной кислоты, имеющей биполярное строение, со-
+
ответствует формуле H3NCH2COO"*. В смеси с НС1 аминоуксусная кис-
+
лота образует катионы H3NCH2COOH. Если концентрация НС1 выше
концентрации аминоуксусной кислоты, часть НС1 остается свободной.
Если же концентрация НС1 ниже, чем аминоуксусной кислоты, в
растворе не имеется свободной НС1. При взаимодействии с NaOH сначала
совместно нейтрализуются свободная НО (если она имеется в
растворе) и карбоксильные группы катионов амфолита, а затем вступают
в реакцию биполярные ионы:
Н+ + СГ + Na+ + ОН" ^=± Na+ + Cr + H20
H3NCH2COOH + Na+ + ОН" ^=± H3NCH2COO" + Na+ + Н20
H3NCH2COO" + Na+ + OH" ^=± H2NCH2C00" + Na+ + H20
Дифференцированного титрования свободной НС1 не наблюдается,
так как кислотные свойства карбоксильных групп выражены довольно
§ 17. АНАЛИЗ СМЕСЕЙ ХХНОВАНИИ И СОЛЕЙ СЛАБЫХ КИСЛОТ Ш
сильно (рКа = 2,35). Титрование Нз№"-групп амфолита протекает
количественно, потому что их основные свойства выражены достаточно слабо
(p/Cft = 4,22).
Методика определения. В сосуд для титрования наливают 50 мл
анализируемого раствора смеси кислот, погружают электроды с
мешалкой и проводят хронокондуктометрическое титрование 0,3 н. раствором
NaOH (см. § 9). При этом стрелку милливольтметра перед началом
титрования устанавливают в верхней части шкалы. Кондуктометриче-
скне кривые титрования смесей кислот имеют два излома. При
титровании смесей HN03 с фенолом (рис. 18, кривая 4) и HC1 с полимет-
акриловой кислотой (рис. 18, кривая 5) электропроводность до первого
излома линейно понижается, так как нейтрализуются сильные кислоты
HNO3 и НС1, а до второго повышается, что соответствует
нейтрализации слабых кислот — фенола и полиметакриловой кислоты. Кривая
титрования смеси, содержащей фенол, имеет очень плавный изгиб вблизи
второй точки эквивалентности вследствие гидролиза фенолята натрия.
Кривая титрования смеси НС1 и аминоуксусной кислоты (рис. 18,
кривая 6) имеет другой характер. Хотя электропроводность раствора до
первой точки эквивалентности также понижается, кривая титрования
на этом участке изогнута, что соответствует влиянию карбоксильных
групп катионов амфолита. На втором участке кондуктометрической
кривой, соответствующем взаимодействию цвиттер-ионов, происходит
линейное повышение проводимости. Участок кривой до первого излома
дает возможность рассчитать содержание в смеси НС1, а между первым
и вторым изломами — содержание аминоуксусной кислоты. Избыток
основания при титровании всех перечисленных смесей вызывает резкое
повышение электропроводности раствора.
Точки эквивалентности на кондуктометрических кривых
устанавливают графическим методом и находят количество интервалов между
показаниями милливольтметра до изломов кондуктометрических кривых.
Содержание кислот в смеси вычисляют обычным способом (см. § 9).
При этом в расчетную формулу вводят количество интервалов,
соответствующих титрованию отдельных кислот. (Грамм-эквивалент
аминоуксусной кислоты принимают равным молекулярному весу.)
§ 17. Анализ смесей оснований и солей слабых кислот
методом хронокондуктометрического титрования
Анализ смесей: едкий натр и ацетат натрия; анилин
и фенолят натрия; едкий натр, аммиак и ацетат натрия
Определение основано на последовательном взаимодействии
оснований и солей слабых кислот с хлористоводородной кислотой.
Последовательность взаимодействия может быть установлена, как рассмотрено
выше, если известны константы диссоциации электролитов, образующих
смесь. В случае когда сумма рКъ основания и рКа кислоты, образующей
соль, больше 12, сначала протекает реакция нейтрализации, а затем
вытеснения. Для смеси NaOH с CHsCOONa эти условия соблюдаются,
и поэтому при титровании хлористоводородной кислотой сначала
нейтрализуется NaOH, а затем взаимодействует CH3COONa с
образованием уксусной кислоты (р/Са = 4,75). Если компоненты смеси
характеризуются значением (рКа + рКь) > 16, последовательность
взаимодействия изменяется. При титровании хлористоводородной кислотой смеси
анилина и фенолята натрия сначала вытесняется фенол (рКа = 10,00),
а затем нейтрализуется анилин (р/Сь = 9,42). При анализе трехкомпо-
112 ГЛ. III. КОНДУКТОМЕТРИЯ И КОНДУКТОМЕТРИЧЕСКОЕ ТИТРОВАНИЕ
нентной смеси, содержащей NaOH, NH3 и CH3COONa, используются
возможности кондуктометрического анализа смесей сильных и слабых
оснований, а также смесей слабых оснований с солями слабых кислот.
При титровании этой смеси раствором НС1 сначала нейтрализуется
NaOH, затем NH3 и, наконец, вытесняется СН3СОО~ из ацетата натрия.
Методика определения. В ячейку для титрования вносят 50 мл
анализируемой смеси и проводят хронокондуктометрическое титрование
0,3 н. раствором НС1 (см. § 10). При титровании смеси NaOH с
CH3COONa сначала нейтрализуется NaOH, что вызывает резкое
понижение электропроводности раствора. Последующее вытеснение
уксусной кислоты из ацетата натрия, наоборот, сопровождается некоторым
повышением электропроводности раствора, так как подвижность
СН3СОО--ионов ниже подвижности заменяющих их в растворе
Симонов (рис. 18, кривая 7). Перед титрованием этой смеси стрелку
милливольтметра следует устанавливать в верхней части шкалы. В смеси
анилина с фенолятом натрия хлористоводородная кислота сначала
вытесняет фенол, а затем нейтрализует анилин. В том и в другом случае
в результате взаимодействия электропроводность раствора
увеличивается. Однако при титровании анилина электропроводность
повышается сильнее (рис. 18, кривая 8). Титрование этой смеси начинают,
поставив стрелку милливольтметра в нижней части шкалы.
Кривая титрования смеси NaOH, NH3 и CH3COONa имеет три
излома (рис. 18, кривая 9). Сначала происходит понижение
электропроводности раствора, а затем резкое повышение; это вызывается
последовательной нейтрализацией NaOH и NH3. После второй точки эквива-
лентаости электропроводность слабо повышается, что характерно для
процесса вытеснения уксусной кислоты из ацетата натрия.
Перед титрованием этой смеси стрелку милливольтметра
устанавливают в верхней части шкалы. Избыток титранта при титровании всех
перечисленных смесей вызывает резкое повышение электропроводности
раствора.
На кривых титрования графическим методом устанавливают точки
эквивалентности и определяют число интервалов между записями
показаний милливольтметра до изломов кондуктометрических кривых.
Содержание отдельных компонентов в анализируемых смесях вычисляют,
как описано в § 9.
ГЛАВА IV
ВЫСОКОЧАСТОТНОЕ ТИТРОВАНИЕ
А. ТЕОРЕТИЧЕСКИЕ ОСНОВЫ
§ 1. Общие положения теории высокочастотного титрования
Высокочастотное титрование применяют во всех
объемно-аналитических методах, основанных на использовании реакций нейтрализации,
окисления — восстановления, осаждения, комплексообразования и т. п.
Особенно удобен метод высокочастотного титрования, если реакцию
следует проводить в герметичной аппаратуре, при работе с
окрашенными и темными растворами, при образовании осадков и титровании
взвесей и эмульсий, а также в условиях, когда контакт электродов
с раствором недопустим вследствие возможности катализа, поляризации
и других осложняющих обстоятельств.
Метод высокочастотного титрования осуществим во всей области
практически применяемых концентраций растворов от 10"*5 до
нескольких молей. Однако этот метод неизбирателен, проведению анализа
мешают все посторонние ионы, находящиеся в растворе. Если
концентрация посторонних ионов велика, общая электропроводность раствора
оказывается настолько большой, что относительные изменения ее
становятся незначительными. Выбирая титрант, избирательно
реагирующий с определяемыми ионами, можно создать условия для анализа
и в сравнительно хорошо проводящей среде. Однако общий фон
электропроводности в растворе затрудняет установление точного положения
точки эквивалентности.
Нахождение точки эквивалентности тем точнее, чем больше
различаются тангенсы углов наклона участков кривой высокочастотного
титрования но обе стороны от этой точки. Точность определения
зависит в основном от изменения общего числа электрических зарядов,
принадлежащих находящимся в растворе ионам, от подвижности ионов,
направления процесса титрования (прямой или обратный порядок
титрования), от характера среды и ее параметров — состава,
релаксационных характеристик и т. п.
Резкие перегибы на кривых титрования могут быть получены лишь
в очень редких случаях. Значительно чаще в точке эквивалентности
образуется не излом, а плавный изгиб. В этом случае точку
эквивалентности находят путем продолжения прямолинейных участков полученной
кривой.
При высокочастотном титровании явление экранирования
электродов осадком, образующимся в процессе реакции, имеет тем меньшее
значение, чем выше рабочая частота. Это объясняется тем, что
емкостное сопротивление Хс каждой взвешенной частицы значительно
меньше сопротивления раствора. Емкостное сопротивление взвешенной
частицы, электрическая емкость которой с, при частоте поля /=10—108гц
и выше (/ и со — линейная и угловая частоты) выражается уравнением:
у __ J !_.
Ас " фс ~ 2ф
114
ГЛ. IV. ВЫСОКОЧАСТОТНОЕ ТИТРОВАНИЕ
§ 2. Физические основы и погрешности метода
высокочастотного титрования
В самом общем виде эквивалентная электропроводность раствора
Я©, измеренная при рабочей частоте ю, может быть выражена
уравнением:
где Я^— составляющая, обусловленная несимметричностью ионной атмосферы;
Яш — составляющая, возникающая вследствие электрофоретического эффекта
(встречное движение растворителя, увлекаемого противоположно
заряженными катионами и анионами);
Яоо — эквивалентная электропроводность раствора при бесконечном разбавлении.
В случае, когда частота внешнего поля со ^> 1/0 (О — время
релаксации ионного облака), распределение ионов в облаке уже не успевает
за изменениями поля, и форма облака приближается к сферически
симметричной. Внешне этот эффект проявляется в увеличении
электропроводности растворов и называется явлением дисперсии
электропроводности.
Согласно теории Дебая — Фалькенгагена, максимальному приросту
электропроводности ДА, = Яа>=:о — А,© вследствие явления дисперсии
электропроводности отвечают соответствующая длина волны L
электромагнитного поля, оптимальный размер ионного облака \/щ и время его
релаксации ©:
J- - 2,81 • 1<Г|0 1 / "°* 9 (2)
15,34 -1(Г8 2
к, - V с 2
е = ——„ ь " (з)
где С — концентрация раствора, молр(л;
е0 — диэлектрическая проницаемость растворителя;
V/ —число ионов данного знака, на которые диссоциирует одна молекула
электролита;
zt — заряд иона;
Яоо — эквивалентная электропроводность раствора при бесконечном разбавлении;
Т — абсолютная температура;
к — постоянная Больцмана.
Совместно решая уравнения (2) и (3) относительно величины С>
находим:
(2,81-1(Г10)2 15,34 -ltrySlgfl
2М)*о.в*Г
Полагая далее, что
уравнение (4) мфжцо записать в виде
8в 8° Чл *2 (5)
где /d =0,88- 10""10;
/ — линейная частота поля.
C~ConT = -?±-ef (6)
§ 3. ФИЗИЧЕСКИЕ ОСНОВЫ МЕТОДА ДИЭЛКОМЕТРИЧЕСКОГО ТИТРОВАНИЯ П5
Уравнение (6) дает возможность выявить область концентраций
раствора, в которой при данном постоянном значении / происходит
наибольший прирост электропроводности раствора. Так как длина волны
электромагнитного поля выражается формулой L = ccJf (где ссв —
скорость света, равная 3-1010 см/сек), то, объединяя это выражение с
уравнением (6), получим:
г __'св*,в -.0,2_1_ (7)
СоптЛа
Интервал длин волн, в котором проявляется эффект дисперсии
электропроводности, составляет примерно два порядка. Учитывая это,
можно найти длины волн LMaKC и LMjm (в см), отвечающие началу
дисперсионного прироста электропроводности и его окончанию:
= 26,4-
СЛ0
/-мин = 0,264-
С1а
(в)
Дисперсионный прирост, соответствующий переходу от LMaKC
к ЬМИНу не очень велик. Подбор подходящих частот дает возможность
значительно уменьшить или совсем устранить погрешности,
возникающие из-за дисперсии электропроводности [см. уравнения (8)].
Некоторые погрешности могут возникнуть при расшифровке кривой титрования
вследствие игнорирования специфичности действия высокочастотных
электромагнитных полей на исследуемый раствор.
Рассмотренные выше погрешности являются основными. Они
определяются только природой растворов и зависят главным образом от их
электрофизических свойств, являющихся в свою очередь функцией
частоты поля. Другие погрешности могут зависеть от различных
факторов (например, от типа аппаратуры, метода определения и т. д.), они
общеизвестны и здесь не обсуждаются.
§ 3. Физические основы метода диэлкометрического
титрования
Применение растворителей с низкими значениями диэлектрической проницаемости
неизбежно ведет к уменьшению степени диссоциации растворенных электролитов, а
стало быть, и к резкому снижению величины удельной электропроводности
исследуемых растворов. Это обстоятельство делает в ряде случаев малопригодным или
совершенно невозможным высокочастотное титрование, поскольку оно основано на
измерениях электропроводности *.
При анализе непроводящих или мало проводящих растворов более
целесообразным может оказаться метод измерения емкости ячейки, изменения которой
определяются диэлектрической проницаемостью раствора. Например, в бензольных и эфирных
растворах ряда органических соединений, таких, как фенол и хинолин, динитро-
фенол и анилин, пиррол и пиридин и другие, на кривых диэлектрическая
проницаемость— состав обнаруживаются характерные точки (изломы или максимумы),
отвечающие составу соединений, которые образуются в растворе в результате титрования.
Еще более четкая картина наблюдается при использовании не диэлектрических
проницаемостей, а величин поляризации Р, характеризующих степень полярности
химической связи.
Повышенные значения поляризации растворов являются следствием химического
взаимодействия растворенного вещества с растворителем или растворенных веществ
друг с другом в индифферентном растворителе. Следовательно, отклонение (АР)
экспериментально найденной поляризации от значения поляризации, вычисленной по
правилу аддитивности, является очень тонким показателем образования нестойких
химических соединений. Это дает возможность проводить качественные и количественные
* Точнее, высокочастотное титрование основано на измерении величины
рассеиваемой раствором мощности электромагнитного поля, приложенного к этому раствору.
116
ГЛ IV. ВЫСОКОЧАСТОТНОЕ ТИТРОВАНИЕ
аналитические определения. Например, применение метода поляризации позволило
обнаружить существование и установить состав комплексного соединения бромида
алюминия с нитрометаном. Четкий максимум на кривой АР — состав, точно
отвечающий эквимолярному отношению, указывает на образование соединения А1Вг3 • CH3N02.
Таким образом, метод поляризации диэлкометрического титрования состоит в том,
что к раствору вещества А в инертном растворителе последовательно добавляют
небольшими порциями титрант — второе вещество В, способное давать с А соединения
донорно-акцепторного типа.
§ 4. Погрешности метода диэлкометрическбго
титрования
На диэлектрическую проницаемость растворов оказывают большое влияние
самые разнообразные факторы: взаимодействие молекул растворенного вещества и
растворителя, ассоциация молекул, полимеризация, образование ионов, появление
загрязнений и т. д. Например, уже малые количества карбонильных групп способны сильно
«ухудшить» диэлектрические свойства раствора. Аналогичные, но более значительные
/ г
Рис. 19. Типы высокочастотных
измерительных ячеек:
а —с-тип (емкостные); б—L-тип
(индуктивные); / — погружные; // — с
внешним расположением электродов и
катушки индуктивности; 7, 2 —клеммы
включения ячейки.
б
по величине эффекты возникают вследствие электропроводности раствора, причиной
которой могут быть не только ионы, но и образование водородных связей. В
частности, электропроводность бензола при насыщении его водой возрастает примерно на
50%. Еще более заметен этот эффект для эфиров и спиртов.
Таким образом, самые различные причины могут вызвать погрешности при ди-
элкометрическом титровании. Чтобы устранить или уменьшить эти погрешности, нужно
учитывать следующие обстоятельства.
Во-первых, не существует простой функциональной зависимости между значением
диэлектрической проницаемости раствора и его составом. В особенности это относится
к спиртам, которые могут давать с водой ряд нестойких химических соединений.
Во-вторых, работа по методу диэлкометрических определений затрудняется или
становится невозможной, если растворы обладают заметной электропроводностью. Для
проводящих объектов эквивалентная емкость ячейки становится сложной функцией
диэлектрической проницаемости, что сильно усложняет, а в ряде случаев делает
невозможным получение искомых величин е и А Кроме того, приборы, предназначенные
для определения емкости, в условиях проводящих растворов, особенно при наличии
ионов, как правило, не могут работать.
ТЕОРИЯ ИЗМЕРИТЕЛЬНЫХ ЯЧЕЕК ЕМКОСТНОГО ТИПА
Для осуществления метода высокочастотного титрования
исследуемый раствор подвергается действию высокочастотного электромагнитного
поля, создаваемого внутри так называемых измерительных ячеек,
которые представляют собой электрический конденсатор или катушку
индуктивности. По этому признаку измерительные ячейки разделяются
на две большие группы: 1) емкостные ячейки, или ячейки с-типа, и
2) индуктивные ячейки, или ячейки I-типа (рис. 19).
§ 5. ЭЛЕКТРИЧЕСКАЯ ЭКВИВАЛЕНТНАЯ СХЕМА ЯЧЕЙКИ
117
*/
R,
*с,
§ 5. Электрическая эквивалентная схема ячейки
и физические процессы, протекающие в ее объеме
Простейшая измерительная ячейка емкостного типа представляет
собой стеклянный цилиндрический сосуд. На его внешней поверхности
укреплены электроды — пластины конденсатора, имеющие, например,
форму полуколец (см. рис. 19, а, II). Такая ячейка является
трехслойным диэлектриком (стенки сосуда, раствор, стенки сосуда),
заключенным между электродами. Учитывая, что каждый слой обладает
собственным значением диэлектрической проницаемости и электрическим
сопротивлением, электрическую схему с-ячейки можно изобразить так,
как показано на рис. 20, а. На
рисунке сопротивление
раствора обозначено через /?р,
емкость — через ср; для стенок
сосуда соответственно /?ст, /?ст
/ //
И Схт, ?ст*
Обычно активное
сопротивление стенок сосуда велико,
поэтому проводимостью их
(/?ст + /?ст) можно
пренебречь. С другой стороны, так
как стенки сосуда имеют
практически одинаковую толщину
по всему периметру, т. е.
сст = сСт, то общая емкость
стенок составляет с2 = (ссТ/2) = (ссТ/2). Меняя обозначения Rp на R\ и
гг, на си получаем электрическую эквивалентную схему измерительной
ячейки емкостного типа (рис. 20, б).
Активное сопротивление R\ моделирует те свойства раствора,
которые обусловливают тепловые потери. Количество энергии,
преобразуемой раствором в тепловую, определяется скоростью миграции ионов,
вызываемой градиентом потенциала в растворе. Поскольку скорость
миграции ионов, а следовательно, и активный ток IR изменяются
пропорционально градиенту потенциала, активный ток и напряжение
находятся в фазе. Таким образом, в соответствии с теоретическими
положениями электротехники имеем:
1
б
Рис. 20. Электрическая (а) и электрическая
эквивалентная (б) схемы измерительной ячейки
емкостного типа.
IR = -^- Г/макс sin Ш =* Шмакс sin со/
(9)
где ?/Макс — максимальное напряжение;
о — угловая частота;
t — время;
k — -75 электропроводность столба раствора, заключенного в измерительной ЯЧеЙ-
ке (величина k является коэффициентом пропорциональности между
совпадающими по фазе активным током, протекающим через раствор, и
приложенным к нему высокочастотным напряжением).
Обратимое смещение зарядов в толще раствора вызывает появление
реактивного тока, он моделируется емкостью с\. При устранении
градиента потенциала электрические заряды возвращаются в прежнее
положение, а энергия, затраченная на их смещение в первом полупериоде,
возвращается в систему в последующем полупериоде. В идеальном
случае этот процесс не связан с превращением электрической энергии
118
ГЛ. IV. ВЫСОКОЧАСТОТНОЕ ТИТРОВАНИЕ
в тепло. Величина реактивного тока /с, подводящего заряды к
конденсатору, выражается формулой:
lc~ dt
Подставив в эту формулу значение q, т. е. количество
электричества, накопленного в конденсаторе, которое равно произведению
емкости С\ на мгновенное значение напряжения (U = ?/макс sin со/), и
проведя дифференцирование, получим:
h = С1#макс<0 COS at = COCif/макс sin ((0* + — J (10)
Из уравнения (10) следует два вывода. Во-первых, как и в обычных
электрических цепях, содержащих емкость, реактивный ток,
протекающий через раствор, опережает приложенное напряжение на угол я/г.
Во-вторых, если в случае активного тока фактором пропорциональности,
связывающим его с высокочастотным напряжением, является величина
электропроводности k [уравнение (9)], то таким фактором
пропорциональности в случае реактивного тока является электрическая емкость С\
[уравнение (10)].
Факторы пропорциональности k и С\ в уравнениях (9) и (10)
отражают природу исследуемых веществ. Их величина зависит от
концентрации и состава растворов, но сравнительно мало меняется с
изменением частоты приложенного поля. Поэтому функциональная связь к—со
и ?i—со при высокочастотном титровании обычно не принимается во
внимание, в то время как зависимости k—С и сх — С (где С —
концентрация раствора) используются при построении характеристических
кривых (см. § 7) и должны быть учтены при постановке эксперимента
по высокочастотному титрованию.
§ 6. Импеданс и полная проводимость ячейки
Найдем импеданс (полное сопротивление) цепи (см. рис. 20,6).
Так как реактивное сопротивление емкости Zc = 1//сос, то полная
проводимость У параллельного участка (R\ и С\) составит
Y =-g- + j<*Cl (11)
Очевидно, полное сопротивление Z{} 2 ячейки между ее зажимами
1 и 2 будет равно реактивному сопротивлению стенок (/сосг)"1 и полному
сопротивлению параллельного участка Z = 1/У, т. е.
Zi, 2 = -Д- + 4- (12)
Подставляя из уравнения (11) значение У в уравнение (12),
находим:
Zi,2 L- + -r=TJ (13)
j<uc2 Rx +/coc1
Избавляясь от мнимости в знаменателе и разделяя действительную
и мнимую части полученного выражения, находим искомую величину
импеданса:
acyRf 1
7 *' -Г
+ (WCi/?i)2 ЮС2
(14)
§ 7. ХАРАКТЕРИСТИЧЕСКИЕ КРИВЫЕ ЯЧЕЙКИ
119
Обратная величина полного сопротивления Zu 2 представляет собой
полную проводимость Yh2 измерительной ячейки:
2Л2
•юс;
Уьа«-
1
¦ + /
сос2
+ со3 (схс\ + с^2)
_1
2 + СО2 (с, + С2)2
~V + co2(ci + c2)2
Учитывая далее, что -™- = й, и вводя обозначения
(15)
karc:
2Л2
? =
6 =
^2 + со2(с1 + с2)2
k2(oc2 + &3(c{cl + с?с2)
?2 + со2 (ci + с2)2
уравнение (15) можно переписать:
YU2 = g + jb
(16)
(17)
(18)
Из уравнения (18) следует, что высокочастотный ток, протекающий
в цепи ячейки через исследуемый раствор, обусловлен полной
проводимостью Y\t2, представляющей собой сумму активной g и реактивной Ъ
компонент (составляющих) проводимости, характер которых
определяется природой активного и реактивного тока.
§ 7. Характеристические кривые ячейки
При высокочастотном титровании измеряют электрические (и
магнитные) параметры исследуемых растворов в зависимости от их
состава; подобные зависимости
со,<со2<о)э
Рис. 21. Характеристическая кривая емкостной
ячейки для активной компоненты полной
проводимости:
а—общий вид; б— смещение с увеличением частоты;
/ — в прямых координатах; 2 — в полулогарифмических
координатах.
называются
характеристическими.
Две характеристические
зависимости описываются
уравнениями (16) и (17). Они
являются сложной функцией
величины k. Кривая /,
выраженная уравнением (16), имеет
максимум (рис. 21,а).
Очевидно, что аналогичные
характеристические кривые будут
получены и при использовании
значений концентрации С1/г или
удельной низкочастотной электропроводности и вместо значения &,
поскольку эти параметры связаны друг с другом зависимостью, близкой
к линейной.
Характеристическая кривая активной составляющей полной
проводимости ячейки емкостного типа. Положение максимума на кривой
может быть найдено дифференцированием уравнения (16),
приравниванием к нулю его первой производной по k и решением полученного
выражения относительно k:
&макс = О (С\ + С2) (19)
Выражения (16) и (19) позволяют сделать следующие очень
важные выводы.
1. Активная составляющая высокочастотной (полной)
проводимости измерительной ячейки с образцом, электропроводность которого
120
ГЛ. IV. ВЫСОКОЧАСТОТНОЕ ТИТРОВАНИЕ
непрерывно возрастает, изменяется не монотонно, а образует максимум.
Поэтому показания измерительного прибора сначала возрастают, а
затем начинают уменьшаться.
2. Как следует из кривой в координатах g— k (см. рис. 21,а),
величина g возрастает значительно быстрее слева от максимума
характеристической кривой, нежели справа от него, при одной и той же скорости
изменения k. Поэтому определение состава растворов, измерение
концентраций бинарных растворов и титрование могут быть проведены
с большей точностью и при большей чувствительности измерительных
устройств именно в диапазоне значений концентраций, лежащих слева
от максимума характеристической
кривой.
3. Удельная электропроводность
растворов в точке максимума прямо
пропорциональна рабочей
частоте со, т. е. характеристическая
кривая g — k с увеличением частоты
смещается вправо (рис. 21,6). Это
дает возможность путем выбора
рабочей частоты поля всегда
работать на том склоне
характеристической кривой, который отвечает
условиям и задачам эксперимента.
Вместе с тем появляется
возможность предвидеть диапазон
концентраций растворов, где расположен
максимум характеристической
кривой, и, таким образом, избежать
резкого снижения чувствительности приборов к изменению свойств
объекта в области максимума. Правильный выбор рабочей частоты позволяет
при определениях избежать двузначности отсчетов по прибору.
Характеристическая кривая реактивной составляющей полной проводимости
ячейки емкостного типа. Реактивная компонента b представляет собой произведение
угловой частоты со поля и эквивалентной емкости с0 ячейки, т. е. Ъ = сое». Поэтому на
основании уравнения (17) величина емкости эквивалентной параллельной цепи
составит:
Ь C^ + W^C^ + CjCg)
Сэ==?= ~?2 + co2(ci + c2)3 (2°)
Кривые, выражаемые уравнением (20), представлены на рис. 22. Это плавные
S-образные кривые, не проходящие через максимум подобно зависимости g — k.
Характеристическая кривая реактивной составляющей в отличие от
характеристической кривой активной составляющей не обладает максимумом, т. е.
двойственности определений, как в предыдущем случае, не возникает.
Интервал изменений емкости ячейки равен:
Рис. 22. Характеристические кривые
с9 — k для реактивной компоненты
полной проводимости емкостной ячейки
с образцом; klt k2, &з — точки перегиба
кривых.
Ac.-cJ1-"!-
ct + с2
$21)
Значение эквивалентной емкости в средней точке характеристической кривой
равно разности ее максимального значения с2 и полуразности предельных значений
6Э, т. е.
1 с2 (2ct + с2)
*-ТА*.- 2 (с1+с2)
Приравнивая это выражение уравнению (20) при к, равном kop (т. е. значению
k в средней точке характеристической кривой), получим.
с2 (2cj + с2)
*ср*2 + Ц2(с1^ + с1с2)
^p + co2(Cl + c2)2
§ 9. КРИТЕРИЙ ПРИМЕНИМОСТИ И ПОСТОЯННАЯ с-ЯЧЕЙКИ
121
откуда
?ср - со (ci + c2) (22)
Уравнение (22) идентично уравнению (19). Следовательно, положение максимума
характеристической кривой g — k для активной составляющей и положение средней
точки характеристической кривой сэ — k для реактивной составляющей полной
проводимости ячейки с образцом совпадают. Это совпадение может быть использовано
при проведении исследований, когда необходимо перейти от одной характеристической
кривой к другой.
Максимальная чувствительность измерительной установки, с помощью которой
определяют эквивалентную емкость сэ, соответствует области наиболее крутого
подъема характеристической кривой, т. е. вблизи ее точки перегиба. Положение точки
перегиба находят, приравнивая нулю вторую производную уравнения (20) и решая
полученное выражение относительно значения электропроводности к в точке перегиба
(&пер). После преобразований получаем:
knep=w^Z°' (23)
§ 8. Чувствительность ячейки
Чувствительность при измерениях активной составляющей.
Чувствительность емкостной ячейки при высокочастотном титровании по
активной составляющей полной проводимости увеличивается с возрастанием
частоты и с уменьшением отношения емкости раствора к емкости стенок,
т. е. Ci/c2. Поскольку емкость стенок характеризуется выражением
с2 « -у е2
это в свою очередь означает, что повышению чувствительности
способствует уменьшение толщины б стенок сосуда, увеличение площади S
электродов ячейки, а также применение материала для сосуда с
возможно большей диэлектрической проницаемостью ег. С другой стороны,
большей чувствительности можно ожидать при титровании в средах
с возможно низкой диэлектрической проницаемостью ei, так как с\
пропорционально 8ь
Чувствительность при измерениях реактивной составляющей.
Чувствительность емкостной ячейки при диэлкометрическом титровании по
реактивной составляющей не зависит от рабочей частоты. Вместе с тем
повышению чувствительности способствует увеличение с2 и
уменьшение с\. Это означает, что применение тонкостенных сосудов с большим
значением диэлектрической проницаемости материала сосуда более
эффективно, чем с низким значением диэлектрической проницаемости.
Однако чувствительность при измерениях реактивной составляющей
будет несколько повышаться в случае веществ с меньшим значением
диэлектрической проницаемости.
§ 9. Критерий применимости и постоянная с-ячейки
Рассмотрим физическое содержание критерия п = ®c\Ru входящего
в уравнение (14). Если частота поля со = const, то при данных
П\ <С 1 или п2 ^> 1 оказывается возможным варьировать величину R\
или С\. Так, при титровании, когда величина удельной
электропроводности исследуемого объекта (растворы электролитов малых и средних
концентраций) изменяется в широком интервале, а его диэлектрическая
проницаемость оказывается практически постоянной, т. е. Де <С Д&,
можно принять, что с\ = const. При диэлкометрическом титровании
растворов возможна обратная ситуация, т. е. Де ^> Ak и k «* const.
Величина п = <uC\Ri является в то же время критерием формы
измерительной ячейки (рис. 23). В самом деле, сопротивление
122
ГЛ. IV. ВЫСОКОЧАСТОТНОЕ ТИТРОВАНИЕ
исследуемого раствора выражается уравнением
/ 1
*1 —
У.
а соответствующая ему емкость
eS
3,6.1012я/
где / — длина столба раствора между электродами, см;
S — площадь электродов, см2;
е — диэлектрическая проницаемость раствора;
и — удельная электропроводность раствора.
С учетом этих уравнений
выражение п = cociRi может быть
записано в виде:
т
откуда
CDtfi
<х = -
3,6.1012я
/
3,6-1012яд
(24)
Рис. 23. Конструкция емкостной ячейки,
предназначенной для измерения
*,(S//«1; я-0,1):
а —общий вид; б — обозначение параметров
столба раствора.
Величина a = S/l называется
константой высокочастотной
измерительной ячейки емкостного типа. Из
этого отношения следует, что
размерность константы с-ячейки
выражается в сантиметрах.
Таким образом, определение емкости раствора при диэлкометриче-
ском титровании целесообразно проводить в ячейках с относительно
большой площадью электродов. В то же время при высокочастотном
титровании выгоднее измерять проводимость при сравнительно более
длинных столбах исследуемого раствора.
ТЕОРИЯ ИЗМЕРИТЕЛЬНЫХ ЯЧЕЕК ИНДУКТИВНОГО ТИПА
В индуктивной ячейке исследуемый образец подвергается сложному
воздействию магнитной и электрической компонент осциллирующего
поля. Механизм электрического взаимодействия уже рассмотрен.
Исследуем теперь другой идеализированный случай — «чисто» магнитное
взаимодействие раствора электролита с высокочастотным полем
индуктивной ячейки.
§ 10. Электрическая эквивалентная схема ячейки
Схема индуктивной ячейки может быть изображена в виде короткозамкнутого
витка проводника, взаимодействующего с катушкой индуктивности (рис. 24). Такой
виток наряду с определенной величиной активного сопротивления Ri обладает еще
и некоторой индуктивностью L\. Поэтому измерительную ячейку с раствором можно
Описывать как два связанных контура (рис. 24,6), взаимная индуктивность
которых М. Первичный контур L2R2 образован собственно катушкой индуктивности и
сопротивлением /?2 ее провода, а вторичный контур LiRi — исследуемым раствором.
Для удобства рассмотрения схему, приведенную на рис. 24, б, можно
преобразовать в схему последовательного типа (рис. 24,в). В ней Z,2 и /?2— по-прежнему
параметры первичного контура, а вносимая величина LBH характеризует магнитные
свойства вторичного контура — раствора. Другая вносимая величина RBB является
мерой высокочастотной энергии, поглощаемой исследуемым объектом и преобразуемой
им в тепло.
§ 12. КРИТЕРИЙ ПРИМЕНИМОСТИ И ПОСТОЯННАЯ L-ЯЧЕИКИ 123
§ 11. Импеданс и характеристические кривые ячейки
Из курса электротехники известно, что вносимые индуктивность и сопротивление
определяются следующими соотношениями:
^вн - - „9 , 9,9 Lt (25)
2,2
Я? + ©^
©Ш2
АВН -л— о ,9
#? + ©2L?
«I
(26)
гдеЛ* ==? VLXL2\
k — коэффициент связи (конструктивный параметр), определяющий количество
энергии, трансформируемой из первичного контура во вторичный.
Импеданс индуктивной ячейки составляет
Z = R2 4- Явн + /© (L2 + ?вн)
а после подстановки значений LBh и /?вн из уравнений (25) и (26) и соответствующих
преобразований
©2fc2L,Z,9 /?? + Л?сг
• + /©L2-
Z = /?2 + /?!
tf2-H©2L2
2,2
*J + ©'Lj
(27)
т. е.
Z = /?э + /«1э (28)
В этих уравнениях R3 и Ьэ — эквивалентные сопротивление и индуктивность, а
величина а — 1 — № представляет собой коэффициент рассеяния магнитных силовых
линий. Полученное уравнение аналогично уравнению (14) для емкостной ячейки,
* >
в
Рис. 24. Измерительная ячейка индуктивного типа:
а —схема; б — электрическая эквивалентная схема (трансформаторная); в
—преобразование схемы б (последовательная схема).
Поскольку сопротивление провода катушки индуктивности обычно мало (#2-*0),
получим:
&k*LxL% _ fe2©L2 *,
КЭ ~ А1 л9 . 9,9 ) D. \2 * ,Л/ 1<ЗД
л2,2
©Lt
Для реактивной составляющей (Хэ) соответственно имеем:
Хэ = ©?э j
*2 + ©2L2<t (-^Z?r)2+*
^г —о 5Т9- в ©^2 ^ —
-1Ц
tf? + A2
1 +
(30)
§ 12. Критерий применимости и постоянная 1-ячейки
Параметр Q, характеризующий относительное уменьшение высокочастотной
энергии в колебательном контуре за один период его колебаний, в радиотехнике
называется добротностью:
Q = ^ (3D
124
ГЛ IV. ВЫСОКОЧАСТОТНОЕ ТИТРОВАНИЕ
Возможны два случая: Qi < 1 и Qi > 1. В первом случае на основании
уравнений (29) и (30) имеем:
Во втором случае:
й. ~ k*<*L2 (^-) (32а)
Хэ » coL2 + a (-^V G)L2 (326)
i?.«*2^.(^7) (32в)
Хъ « acoL2 + (-^-)2 со?2 (32г)
При переходе от измерений при Q\ < 1 [уравнения (32а) и (326)] к измерениям
при Q\ > 1 [уравнения (32в) и (32г)] характеристические кривые претерпевают
инверсию, изменяясь от обратных зависимостей по /?ь т. е. от
т ¦ *-'¦(¦*•)
к прямым, т. е. к выражениям
*-'¦(-&) и *-*Ш
где F,— функциональная зависимость.
Полученные выражения показывают, что если метод измерения активной
составляющей в обоих случаях одинаково целесообразен, то титрование по реактивной
составляющей во втором случае оказывается более перспективным. Это заключение
следует из того, что величина а< 1, поэтому второе слагаемое уравнения (32г) более
«несомо» по сравнению со вторым слагаемым уравнения (326); его прирост при
изменении параметров раствора значительно сильнее отражается на измеряемой
величине Х0. С другой стороны, множитель о* снижает роль первого слагаемого уравнения
(32г), что также способствует применимости случая Q\ > 1. Кроме того, поскольку для
наиболее употребительных размеров ячейки (/ = 10 см, d — 5 см) интервал
изменений Ri составляет 102—105 ом, величина второго слагаемого в уравнении (326)
может оказываться значительно меньше, чем в уравнении (32г).
Таким образом, высокочастотное титрование с помощью измерительной ячейки
индуктивного типа более целесообразно в случае, когда параметр Q\ > 1. Выясним,
как практически можно выполнить условие Qi > 1 или Qi < 1.
Допустим, что короткозамкнутый виток проводника — раствора обладает
индуктивностью, описываемой полуэмпирическим выражением:
Г dN2 л»ч
Ll~ 50(2Z/d+l) W)
где N — число витков;
/ — толщина витка (длина сосуда);
d — диаметр витка (диаметр сосуда).
Подставляя значение L\ из уравнения (33) в уравнение (31), находим:
Qx - aLv{K (34)
Id
-Kv
21+ d
-I
6 fe
В этих выражениях % — геометрический фактор (фактор заполнения) и б —
величина, характеризующая глубину проникновения поля в раствор, являются
безразмерными величинами, а коэффициент /(v~0,l. ?— геометрический фактор (фактор
заполнения); б— относительная величина, характеризующая глубину проникновения поля
в раствор.
Величина о&г, является константой высокочастотной индуктивной ячейки; ее
размерность, как и в случае емкостной ячейки, выражается в сантиметрах.
Произведенные выкладки позволяют сделать следующие заключения о
применимости L- ячеек:
1) для повышения параметра Q\ ячейки с раствором необходимо увеличить
размеры сосуда;
§ 13. ВЛИЯНИЕ НА ХАРАКТЕРИСТИЧЕСКИЕ КРИВЫЕ ЯЧЕЙКИ ЕЕ ЭЛЕКТРИЧ. ПОЛЯ 125
2) чем выше удельная электропроводность растрора ус и чем выше рабочая
частота f, тем выше значение Q\ и тем эффективнее методы измерения активной
составляющей 7?э, согласно уравнениям (32а) и (32в);
3) при малых значениях частоты поля и проводимости раствора параметр Q\
имеет незначительную величину. Для его повышения в этом случае необходимо
использовать большие количества раствора. В этих же условиях можно применять
метод измерения эффективных значений индуктивности ячейки, согласно уравнениям
(326) и (32г).
§ 13. Влияние на характеристические кривые ячейки
ее некомпенсированного электрического поля
В соответствии с уравнениями (29) и (30) характеристические
кривые индуктивных ячеек должны быть аналогичны соответствующим
кривым емкостных ячеек. Однако в силу постоянно действующего критерия
индуктивных ячеек Qi [см. уравнения (31) и (32)] следует ожидать
плавную, монотонно изменяющуюся кривую. В самом деле, согласно
уравнениям (32 в) и (32г), справедливым для практически важного
случая Qi > 1, имеем:
Описываемые этими уравнениями кривые adc и a'dc' (рис. 25)
отражают характер влияния электропроводности k исследуемого
раствора на величины активной
[уравнение (35)] и реактивной [уравнение (36)]
компоненты полного сопротивления
индуктивной ячейки. Они показывают,
что величины /?э и Х9 монотонно
изменяются с увеличением k.
Однако из опыта известно, что
характеристические кривые реальных
индуктивных ячеек обладают
немонотонным характером и имеют форму
abc и а'Ь'с' (см. рис. 25). Это
объясняется наличием некомпенсированного
электрического поля, всегда
присутствующего в реальном соленоиде.
Влияние этого электрического поля
легко устранить с помощью экрана
Фарадея, например незамкнутой
полоски фольги, размещаемой между
сосудом с раствором и обмоткой ячейки.
При этом характеристические кривые утрачивают свой экстремальный
характер (кривые ab\c и а'Ь'с') и приобретают вид плавных кривых
(кривые adc и a'dc') в сответствии с теорией [см. уравнения (35) и (36)].
Положение максимума характеристических кривых ячеек
емкостного типа совпадает с положением максимума на кривой неэкранирован-
ной индуктивной ячейки; это отражает общность механизмов
взаимодействия неэкранированной индуктивной и емкостной ячеек в данном
диапазоне электропроводностей растворов.
Созданием определенных условий можно, наоборот, усилить
действие электрической компоненты поля. Для этого, например, в ячейку
с раствором опускают металлический, заземленный на корпус прибора,
зл'екегрод. Этим целям служат и металлические пластинки, помещаемые
Рис. 25. Характеристические кривые
ячеек индуктивного типа:
adc и a'dc' — «чисто» магнитное
взаимодействие (при наличии экрана Фарадел);
abc и а'Ь'с' —в условиях взаимодействия
раствора с электрической компонентой
осциллирующего поля (без экрана Фарадея).
126
ГЛ. IV. ВЫСОКОЧАСТОТНОЕ ТИТРОВАНИЕ
некоторыми исследователями под дно ячейки с раствором или внутрь
ее. Такие пластинки увеличивают емкостную связь исследуемого
объекта с индуктивной ячейкой, что увеличивает ее «электрическую»
чувствительность при высокочастотном титровании. Необходимо
подчеркнуть, что применение индуктивных ячеек наиболее оправдано при
работе с хорошо проводящими и концентрированными растворами
(выше 10~2 М)> а также в специальных случаях исследования
особенностей кривых титрования.
ДИАГРАММЫ СООТВЕТСТВИЯ
Задачей высокочастотного титрования является количественное
определение химического состава веществ путем бесконтактного измерения
электрических (и магнитных) параметров растворов, содержащих эти
вещества. Графики зависимостей параметров растворов (к, е, g, 6, R9,
Л'э, У, Z) от их состава называются характеристическими кривыми (см.
стр. 119). Вид характеристических кривых предопределяет вид кривых
высокочастотного титрования, которые могут иметь весьма различные
формы даже в случае применения одних и тех же реагентов. Вид
кривых титрования зависит и от типа измерительной аппаратуры, так как
реальная характеристическая кривая представляет собой сложную
функцию не только полной проводимости ячейки с раствором, но и ряда
параметров измерительного устройства.
§ 14. Диаграмма соответствия для титрования
по активной составляющей полной проводимости
Графики, связывающие изменения удельной низкочастотной
электропроводности с приростом величины определяемых активной и
реактивной компонент полной проводимости, или импеданса ячейки с
раствором, называются диаграммами соответствия. Обязательным
элементом диаграмм соответствия являются характеристические кривые.
Рассмотрим пример кислотно-основного титрования:
Na+ + OH" + H+ + CF ^z± Na+ + Cr + H20
Замена более подвижных* ионов (ОН~) менее подвижными (С1~)
приводит к тому, что на кривых низкочастотного кондуктометрического
титрования в точке эквивалентности появляется минимум (рис. 26, а);
Иное положение при высокочастотном титровании. При сопоставлении
соответствующих кривых оказывается, что одной форме кривой
низкочастотного титрования могут отвечать различные формы кривых
высокочастотного титрования. Это определяется начальной
электропроводностью титруемого раствора, причем возможны три случая. Когда
диапазон изменений низкочастотной электропроводности укладывается
между началом координат и максимумом характеристической кривой
(рис. 26,6), кривая высокочастотного титрования (abc) повторяет
форму кривой низкочастотной кондуктометрии (сравнить диаграммы айв
на рис. 26).
Если кривая низкочастотного титрования (хуг) соответствует
области справа от максимума характеристической кривой, то кривая высоко-
* При высоких частотах электромагнитного поля концепция ионных подвижностей
несостоятельна, однако она удобна для рассмотрения вопроса о виде кривых
высокочастотного титрования.
§ 15. ДИАГРАММА ДЛЯ ТИТРОВАНИЯ ПО РЕАКТИВНОЙ СОСТАВЛЯЮЩЕЙ 127
частотного титрования xyz является обратной по отношению к
низкочастотной кривой (рис. 26, в).
В третьем случае кривая низкочастотного титрования приходится
на область максимума характеристической кривой, а кривая
высокочастотного титрования (тпр) имеет два максимума (рис. 26,в).
Путем введения в титруемый раствор индифферентного
электролита, увеличивающего суммарную проводимость раствора, или
разбавлением титруемого раствора оказывается возможным искусственно
переместить рабочий интервал электропроводности на соответствующий
участок характеристической кривой и тем самым получить кривые
Рис. 26. Диаграммы соответствия емкостной
ячейки для титрования по активной
составляющей полной проводимости:
а — низкочастотное титрование; б — характеристическая
кривая; в —кривые высокочастотного титрования;
abc — в отсутствие индифферентного электролита;
тпр —в присутствии индифферентного электролита;
хyz — концентрация индифферентного электролита
возросла в сравнении со вторым случаем; Ь, п, у — точки
эквивалентности.
Рис. 27. Диаграммы соответствия
емкостной ячейки для титрования по
реактивной составляющей полной проводимости:
а —низкочастотное титрование; б
—характеристическая кривая; в — кривая высокочастотногб
титрования.
титрования желаемой формы с четкой фиксацией точки
эквивалентности. С этой же целью можно перестраивать рабочую частоту
измерительного генератора. Тогда, в соответствии с уравнением (19), максимум
характеристической кривой будет перемещаться, а рабочий интервал
проводимости титруемого раствора окажется справа или слева от него.
§ 15. Диаграмма соответствия для титрования
по реактивной составляющей полной проводимости
Кроме рассмотренного случая высокочастотного титрования,
основанного на применении построенной в координатах g — х
характеристической кривой, возможно титрование, при котором измеряемым
параметром является эквивалентная емкость сэ = bjay [уравнение (20)]. На
рис. 27 представлены диаграммы соответствия для кислотно-основного
титрования в емкостной ячейке. Рассмотрение кривых xyz показывает,
что титрованию по реактивной составляющей полной проводимости
соответствуют кривые более простой формы, чем в предыдущем случае.
Нередко это является решающим обстоятельством при выборе способа
высокочастотного титрования.
128
ГЛ. IV. ВЫСОКОЧАСТОТНОЕ ТИТРОВАНИЕ
§ 16. Выбор типа диаграмм
Сравнивая оба метода высокочастотного титрования с помощью
с-ячейки по ее активной и реактивной компоненте полной проводимости,
следует подчеркнуть, что первый из них целесообразно применять, когда
ожидается преимущественное изменение активной составляющей
импеданса ячейки и образца. С другой стороны, титрование по реактивной
компоненте может дать лучшие результаты в случае значительного
прироста диэлектрической проницаемости, т. е. при диэлкометрическом
титровании. От выбора участка характеристической кривой, в интервале
которого происходит изменение параметров исследуемого раствора,
зависит точность и чувствительность метода титрования. Последние
зависят также от однозначности положения точки эквивалентности на
характеристической кривой и от качества графических построений,
проводимых при обработке данных эксперимента.
При анализе результатов высокочастотного титрования в ячейке
индуктивного типа также составляют диаграммы соответствия
аналогично тому, как это делалось для измерительной ячейки емкостного типа.
Б. АППАРАТУРА И ТЕХНИКА ВЫПОЛНЕНИЯ АНАЛИЗА
Современные устройства для высокочастотного титрования
достаточно сложны. Чтобы понять принцип действия всего устройства и
составляющих его элементов, правильно эксплуатировать высокочастотные
установки, уметь истолковать полученные результаты, современному
химику-аналитику необходима специальная подготовка.
§ 17. Измерительные ячейки (датчики) высокочастотного
метода и области их применения
Оформление высокочастотных измерительных ячеек может быть
самым различным, оно зависит от конкретных целей и условий
применения. Однако все виды ячеек могут быть сведены к нескольким
основным типам (рис. 28).
Емкостные ячейки а — е пригодны для работы в широком
диапазоне проводимостей растворов, соответствующих изменению
концентрации хлористоводородной кислоты от 1 до 10~6 н. Ячейка типа а
удобна для титрования, когда смешение реагентов происходит внутри
самой ячейки. Ячейки б — г пригодны в условиях непрерывного
измерения параметров протекающего раствора, причем ячейку г целесообразно
применять для весьма хорошо проводящих растворов.
Области применения ячеек дне аналогичны, они не могут был?
термостатированы. Важной особенностью этих ячеек является то, что
их рабочий объем экранирован одним из электродов, соединенным
с заземленной оболочкой высокочастотного кабеля. Поэтому во время
работы можно находиться вблизи ячейки, не опасаясь вызвать
расстройку прибора.
Для исключения гальванического контакта электродов ячеек с тер-
мостатирующей жидкостью их покрывают лаком. В качестве
теплоносителя необходимо применять вещества с малым значением
диэлектрической проницаемости, иначе электроды измерительной ячейки
шунтируются слоем термостатирующей жидкости. Хорошие результаты могут
быть получены, например, при использовании в качестве теплоносителя
четыреххлористого углерода, диоксана и других веществ,
диэлектрическая проницаемость которых составляет всего несколько единиц.
Индуктивные ячейки (рис. 28, ж — л) применяются в титровании
реже. Ячейки типа ж и з принадлежат к ячейкам погружного типа.
§ 18. КЛАССИФИКАЦИЯ АППАРАТУРЫ ВЫСОКОЧАСТОТНОГО МЕТОДА 129
Ячейки типа и и к являются проточными и дают возможность
осуществлять непрерывный процесс титрования. Следует подчеркнуть, что
ячейки ж, з, и не являются чисто индуктивными, они обладают
заметным некомпенсированным электрическим полем, что усложняет их
характеристические кривые. Ячейку типа к целесообразно применять для
исследования хорошо проводящих растворов, так как трубка может
быть достаточно длинной, а ее сечение—малым. Экранирование такой
ячейки достигается двусторонним заземлением с помощью
металлических электродов, а ее связь с титратором — посредством специальной
Рис. 28. Основные типы измерительных ячеек для высокочастотного
метода:
а — е — емкостные; ж — л — индуктивные, / — электроды; 2-рубашка; 5 —полость для
анализируемого раствора; 4 — штыревой разъем; 5 —катушка индуктивности; 6 — груша.
катушки индуктивности, помещаемой внутрь спиральной трубки или
вблизи нее, а также через электроды /.
Катушка индуктивности ячейки типа л выполнена в виде
проволочной спирали и заключена в стеклянную трубку, которая целиком
погружена в исследуемый раствор. Особенность этой ячейки состоит в
наибольшем по сравнению с предыдущими ячейками взаимодействии
раствора с магнитной компонентой поля ячейки.
§ 18. Классификация аппаратуры высокочастотного метода
Существует несколько способов систематизации аппаратуры
высокочастотного метода. В одном из них высокочастотные устройства
подразделяются по виду наблюдаемых характеристических кривых и
способу включения ячейки в колебательный контур. В другом предлагается
130
ГЛ. IV. ВЫСОКОЧАСТОТНОЕ ТИТРОВАНИЕ
систематизация на основе радиотехнических признаков. Мы будем
придерживаться третьей, функциональной систематики, согласно которой
возможны три типа установок: Z-, Q- и ^-метры. Ниже приведена
общая характеристика каждого типа приборов.
Z-Метры (рис. 29, а) —приборы, позволяющие определять импеданс
исследуемого образца.
0-Метры (рис. 29, б)—устройства, широко известные в практике
радиотехнических измерений, служащие для определения добротности
колебательных контуров и значений индуктивности и емкости,
составляющих подобные контуры. При высокочастотном титровании
измерительная ячейка подключается к цепи колебательного контура. Гакое
включение может быть либо параллельным (рис. 30, а) при
сравнительно* малой электропроводности раствора, либо последовательным
(рис 30 б)—в случае хорошо проводящих объектов. При титровании
в ячейке индуктивного типа сосуд с раствором помещают в катушку
индуктивности. Если катушка электрически не экранирована от^
исследуемого раствора, такая ячейка в значительной степени взаимодействует
с раствором через электрическую компоненту (см. § 13).
Рис 29 Типы установок высокочастотного анализа:
a-Z-^rP; б-С-метр: e-F-метр, пс,„-«^^
ник питания, 4 волноме;^ь.ограничитель; 10- частотный дискриминатор.
L, С-элементы колебательного контура; <о- частота.
Важной особенностью Q-метров является то, что измерительная
ячейка подключается к системам, в которых автоматически
поддерживается резонанс токов. При этом активная и реактивная компоненты
импеданса ячейки проявляются отдельно: активная проводимость [урав-
нение (16)1 проявляется в виде изменения амплитуды колебании, а
прирост эквивалентной емкости [уравнения (17) и (20)] выражается в
раскройке колебательного контура и сдвиге его резонансной частоты
Р?-Метры- приборы, в которых происходит трансформация
изменений диэлектрической проницаемости и электропроводности исследуемого
объекта в соответствующий сдвиг рабочей частоты. Поскольку частот-
нью измерения в радиотехнике вообще проводятся с наибольшей точ-
яостью (доГ 10-7-10-* абсолютной величины), F-метры в принципе
оказываются наиболее чувствительными и точными приборами для высоко-
частотного анализа.
§ ®. высокочастотные титраторы
ш
Наиболее целесообразно использовать F-метры для измерения
диэлектрической проницаемости образца. Простейшим F-метром
является Q-метр, в состав которого вводят эталонный генератор и блок
смесителя частот (рис. 29,в). Такой прибор можно назвать F-метром
по методу биений, или Q, F-метром. Блок-схема сложного ^-метра дает
возможность преобразовывать в частотные единицы как изменения
реактивной, так и прирост активной составляющих ячейки с образцом
(рис. 29,г). Для повышения чувствительности прибора и с целью
стабилизации его режима в схему этого устройства введены каскады
усилителя-ограничителя 9 и частотного дискриминатора 10.
Рис. 30. Способы включения измерительной ячейки емкостного типа
в колебательный контур:
а — параллельно; б — последовательно; в —образец внутри катушки индуктивности
(с-ячейка образуется за счет паразитной емкости);
/ — катушка индуктивности; 2 —конденсатор колебательного контура; 3— ячейка; 4 —
паразитная емкость.
Приведенная классификация исчерпывает возможные типы
устройств для высокочастотного анализа, рассматриваемых по
функциональному признаку. Конструктивное решение той или иной установки
существенно изменяется в зависимости от диапазона частот, в которых
она должна работать.
§ 19. Высокочастотные титраторы
Титратор высокочастотный ТВ-6Л. Титратор типа ТВ-6Л
представляет собой современный прибор, применяемый в химических
лабораториях.
Упрощенный вариант электрической принципиальной схемы титра-
тора приведен на рис. 31. Титратор относится к типу Z-метров.
Особенность его состоит в том, что на четырехплечный мост 2Я> i?7, Д\, Rio, R и
подается напряжение высокой частоты, а измерение напряжения
небаланса производится на постоянном токе. Напряжение U\, величина
которого зависит от отношения RilZK, детектируется диодом Дг.
Постоянная составляющая тока диода создает на сопротивлении Rn
падение напряжения, которое сравнивается с напряжением U2. Последнее
возникает благодаря детектированию диодом Д1 высокочастотного
напряжения, получаемого от того же генератора. Это делает описываемый
прибор очень стабильным в работе. Показания его индикаторного
прибора практически не зависят от случайных колебаний амплитуды
питающего мост высокочастотного напряжения, поставляемого
генератором Г.
* На этой и последующих схемах (рис. 32, 34) сохранены обозначения, данные
в заводских описаниях приборов.
132
ГЛ. IV. ВЫСОКОЧАСТОТНОЕ ТИТРОВАНИЕ
Разбаланс четырехплечного моста, возникающий в процессе
титрования, отмечается чувствительным индикатором нуля, он представляет
собой ламповый вольтметр, сетки лампы (Лг) которого включены
в измерительную диагональ так, что на них подаются напряжения
?/, и U2.
Стрелочный прибор (ИП) с большим ходом стрелки находится
в катодной диагонали вольтметра.
Рис. 31. Упрощенный вариант электрической схемы титратора ТВ-6Л
(четырехплечный мост и его индикаторное устройство).
Для добавления порции титранта нажимают одну из двух кнопок
(«грубо» и «точно») электромагнитного затвора бюретки. При этом
возникает импульс, который при записи кривой титрования на ленте
записывающего прибора приводит к резкому отклонению пера
самописца. Все отсчеты при титровании производятся только по шкале
стрелочного прибора.
Титратор ТВ-6Л комплектуется так называемым блоком проверки,
который представляет собой электрический эквивалент измерительной
ячейки, состоящий из двух групп безындукционных сопротивлений. При
включении блока проверки вместо измерительной ячейки в
измерительное плечо моста вводится эталонная нагрузка 2500 ож + 200 ом или
200 ом + 4,7 ом. С ее помощью шкала индикаторного прибора может
быть калибрована непосредственно в обратных омах.
Титрование на приборе ТВ-6Л. До включения прибора
в сеть устанавливают штепсельный переключатель сети в нужное
положение (127 или 220 в). Включают прибор и ручкой потенциометра /?ю
устанавливают стрелку прибора на середину шкалы.
Титрант наливают в склянку, которую помещают в специальное
гнездо в корпусе прибора. Полиэтиленовой трубкой соединяют склянку
с верхним отростком автоматической бюретки. При помощи резиновой
груши создают в склянке давление и заполняют бюретку титрантом.
Блок проверки заменяют ячейкой, опускают мешалку в сосуд для
титрования и наливают в него титруемый раствор, так чтобы его
уровень был на 3—5 мм выше верхнего электрода ячейки. Включают
мешалку и устанавливают необходимую скорость вращения.
§ 19. ВЫСОКОЧАСТОТНЫЕ ТИТРАТОРЫ
133
Q,F-MeTp для физико-химического анализа и титрования растворов.
Q,F-MeTp, электрическая принципиальная схема которого приведена на
рис. 32, сконструирован в МХТИ им. Д. И. Менделеева. В прибор
включен Q-метр, образованный левой половиной двойного триода Ль
катушкой L колебательного контура и входящими в него емкостями с и с2,
междуэлектродной и монтажной емкостями и емкостью измерительной
ячейки. Этот тип генератора известен в радиотехнике под названием
генератора Эзау.
Рис. 32, Электрическая принципиальная схема Q, F-метра.
Одновременно левая половина лампы JIFi — ее внутреннее
сопротивление Rt — выполняет и вторую функцию, являясь одним из плеч
лампового моста. Второе плечо этого моста — внутреннее сопротивление
правой половины лампы R"\ третьим и четвертым плечами моста
служат анодные нагрузки R2 и 7?3-
Конденсатор с\ служит разделительной емкостью, препятствующей
попаданию анодного напряжения на рабочий контур, а конденсатор
с2 вместе с сопротивлением Rg создает отрицательное смещение на
сетке левого триода, причем величина этого смещения зависит от
амплитуды колебаний в контуре, к которому подключена измерительная
ячейка.
Для рассмотрения работы схемы предположим, что изменение
электрических параметров раствора вызывает уменьшение рассеиваемой
в нем мощности электромагнитного поля. Такой процесс происходит,
например, при падении электропроводности раствора в результате кон-
дуктометрического титрования. Уменьшение величины поглощаемой из
контура мощности вызывает нарастание амплитуды его колебаний и
соответственно увеличение отрицательного потенциала сетки левого
триода, т. е. уменьшение его анодного тока /д. Это в свою очередь уменьшает
падение напряжения на сопротивлении R2 и приводит к увеличению
потенциала анода левого триода.
С другой стороны, уменьшение l'a снижает потенциал обоих
катодов лампы (так как уменьшается падение напряжения на их общей
134
ГЛ. IV. ВЫСОКОЧАСТОТНОЕ ТИТРОВАНИЕ
нагрузке R5— R9) и вызывает соответствующее снижение
отрицательного потенциала сетки правого триода. Анодный ток правой
половины лампы 1а возрастает, это уменьшает потенциал правого анода, и
схема выходит из первоначального состояния равновесия, что
отмечается по отклонению стрелки индикаторного прибора (ИП).
Таким образом, изменение количества рассеиваемой в растворе
колебательной мощности АР проявляется в отклонении стрелки
индикатора от положения условного нуля, в то время как направление хода
стрелки отмечает знак АР — снижение или
возрастание электропроводности раствора.
Следует учитывать, что при сборке Q, F-мет-
ра необходимо соблюдать правильное включение
индикаторного прибора: при включении его
положительной клеммы к правому аноду ход
стрелки вправо указывает на возрастание потерь
высокочастотной мощности, эквивалентное
увеличению электропроводности раствора.
Установка нуля балансной схемы Л\ может
производиться скачком — при помощи
переключателя 111 и плавно путем изменения R\. Это, а
также переключение (П3) сопротивлений,
шунтирующих индикаторный прибор, дает
возможность работать в большом диапазоне проводимо-
стей исследуемых растворов.
Блок генератора сравнения и смесителя
частот f-метрической части прибора выполнен на
двойном триоде Лг, причем собственно
генератор собран на правой половине двойного триода.
Его частота может регулироваться грубо с
помощью подстроечного конденсатора сп и точно —
конденсатором сшм. Напряжения высокой
частоты измерительного (через cQ) и эталонного
генераторов подаются в цепь сетка — катод левого
триода, где происходит их смешение. Напряжение разностной частоты —
частоты биений — через конденсатор с9 подается на частотно-зависимую
цепь с8> с9, сю, Rio, Rn, Rn, Д1 и Д2, благодаря которой показания
индикаторного прибора ИП оказываются пропорциональными степени
расстройки рабочего генератора, т. е. изменению диэлектрической
проницаемости раствора при титровании.
В Q-метрическом режиме измеряют электропроводность и другие
потери высокочастотной мощности в растворах. При этом
индикаторный прибор ИП подключается (П2) к диагонали лампового моста и
фиксирует разность токов, 1а и /д. Сосуд с раствором помещают в
измерительную ячейку; стрелку индикаторного прибора устанавливают
при помощи переключателя fli и сопротивления R\ в пределах шкалы,
выбрав удобное для работы положение условного нуля. Индикаторный
прибор должен быть защищен максимальным шунтированием от
случайных бросков тока. Для этого переключатель П3 устанавливается
в положение, при котором отклонение стрелки прибора имеет удобную
для отсчета величину,
/-"-Метрический режим применяется, когда в процессе титрования
не происходит заметного изменения электропроводности и протекание
реакции титрования отражается главным образом на величине
диэлектрической проницаемости раствора, т. е. на частоте рабочего генера-
Рис. 33. Конструкция
измерительной ячейки Q,
/^-метра:
1 — плексигласовый блок;
2 - электроды; 3 — защитное
кольцо из плексигласа;
4 —стакан; 5 — проводники,
соединяющие электроды со
схемой Q, .F-мегра; 6 —
основание (подставка) — высота
5 ми (вид сверху).
§ 19. ВЫСОКОЧАСТОТНЫЕ ТИТРАТОРЫ
ТЗб
тора. Переключателем П2 присоединяют индикаторный прибор к
частотно-зависимой цепи с диодами Д1 и Д2 и максимально шунтируют,
устанавливая П3 в положение /. С помощью подстрочечного
конденсатора сп настраивают эталонный генератор — генератор сравнения Лг
в унисон с частотой рабочего генератора Ль при этом стрелка индика-»
торного прибора оказывается в нулевом положении.
Титрование проводят одним из двух возможных способов: а) по
отклонению стрелки индикаторного прибора при неизменном
положении измерительного конденсатора сИЗм; этот способ целесообразен при
малых сдвигах частоты генератора; б) по показанию Сизм- В этом
случае стрелку индикаторного прибора всякий раз устанавливают в
положение условного нуля.
Рис. 34. Электрическая принципиальная схема ссциллотитратора системы
«Пунгор»,тип ОК-302-
Измерителъная ячейка (рис. 33) в рассматриваемом приборе
представляет собой плексигласовый блок /, в котором размещены два
полуцилиндрических электрода 2. От загрязнения растворами электроды
защищены плексигласовым кольцом 3. Стакан емкостью 200 мл
вставляется внутрь ячейки, его дно упирается в специальную подставку 69
которая может быть легко вынута для чистки. Проводниками 5
электроды присоединяются к схеме Q, F-ыетра (см. рис. 32, точки А и Б).
Осциллотитратор системы «Пунгор» типа ОК-302. Принцип работы
осциллотитратора (рис. 34) аналогичен принципу работы Q, F-метра.
Осциллотитратор состоит из следующих основных блоков:
измерительного генератора Ль работающего на частоте около 140 Мгц, лампового
вольтметра Лг с индикаторным прибором на 100 мка, источника
напряжений, двух электронных стабилизаторов напряжений,
измерительной ячейки, присоединяемой к клеммам А и Б, и вибрационной
мешалки ВМ.
Измерительная ячейка подключается к колебательному контуру
через конденсаторы связи с3 и cig, причем с помощью последнего
конденсатора можно осуществить плавную регулировку связи, уменьшая
ее при больших проводимостях раствора и увеличивая в случае слабо
проводящих растворов.
136
ГЛ. IV. ВЫСОКОЧАСТОТНОЕ ТИТРОВАНИЕ
При увеличении проводимости анализируемого раствора амплитуда колебаний
в контуре уменьшается. Это вызывает рост потенциала сетки лампы генератора по
отношению к потенциалу ее катода, а стало быть и относительно линии нулевого
потенциала. Возрастание потенциала сетки Д\ фиксируется ламповым вольтметром Лг,
собранным по мостовой схеме. Первым и вторым плечом этого моста являются
внутренние сопротивления левой и правой половин двойного триода Лг; двумя другими
плечами служат катодные нагрузки Rs и Re.
Анодный ток лампы генератора, протекающий от ее катода через точки /, //
создает падение напряжения на параллельно включенных #4 и цепочке R10P2P3.
Балансировка моста осуществляется с помощью потенциометра Рг. Тонкая регулировка
нуля («компенсация») производится потенциометром Р3.
Индикаторный прибор ИП включен в диагональ лампового моста через набор
последовательных сопротивлений R21 — #26 и потенциометр Р\. При помощи
переключателя ГЪ осуществляют скачкообразную (а потенциометром Pi — плавную)
регулировку чувствительности индикаторного прибора.
Индикаторный прибор можно включать в диагональ моста в прямой и обратной
полярности. Это позволяет отрегулировать осциллотитратор таким образом, чтобы
прямое (вправо) отклонение стрелки индикатора соответствовало увеличению
проводимости раствора, а отклонение стрелки влево — возрастанию его импеданса. В
среднем положении переключателя П\ индикаторный прибор отключается от моста, что
необходимо при смене растворов, перед включением или выключением установки
и т. п. На это время для сохранения электрического режима моста неизменным вместо
микроамперметра в цепь остается включенной эталонная нагрузка /?го-
Осциллотитратор снабжен тремя типами измерительных ячеек,
которые крепятся на корпусе прибора с помощью плексигласовых винтов.
Наиболее часто применяют ячейку с круглыми электродами большого
диаметра, внутрь которых помещают химический стакан емкостью от
100 до 250 мл. В ячейке он уплотняется специальными переходными
кольцами, внешний диаметр которых соответствует диаметру
электродов ячейки, а внутренний — диаметру стакана.
Сверху стакан фиксируют металлическим кольцом, снабженным
резиновой прокладкой. Поскольку это металлическое кольцо соединено
с корпусом прибора, оно электрически экранирует ячейку сверху,
уменьшая рассеивание высокочастотной энергии в окружающее
пространство. Для снижения потерь на излучение служит и металлическая
пластина-экран, охватывающая измерительную ячейку и закрепляемая на
корпусе прибора с помощью штыревых разъемов.
Два других типа измерительных ячеек применяются для
контроля качества растворов в запаянных ампулах и при измерениях в
потоке.
Перед работой переключатель П1 устанавливают в среднее —
нейтральное положение, а переключатель чувствительности Пг — в
положение, соответствующее минимальной чувствительности прибора.
Затем прогревают прибор 20—30 мин, наливают в измерительную ячейку
титруемый раствор и переключатель П] устанавливают в левое
положение, если необходимо, чтобы отклонение стрелки индикаторного
прибора вправо соответствовало возрастанию электропроводности
раствора, и в правое положение — ее уменьшению.
Включают магнитную мешалку и делают отсчет, после этого
прибавляют первую порцию титранта и наблюдают за ходом стрелки
индикаторного прибора. Если оказывается, что отклонения стрелки малы,
то постепенно увеличивают чувствительность индикаторного прибора,
уменьшая шунтирование его. Одновременно корректируют нуль осцил-
лотитратора.
Когда исследуемый раствор является слабым электролитом или
сильным электролитом малой концентрации, то возможно, что
чувствительность прибора окажется не слишком высокой. В этом случае
целесообразно при помощи конденсатора ci9 установить величину связи,
обеспечивающую требуемую чувствительность.
§ 20. ПРАВИЛА РАБОТЫ С ПРИБОРАМИ ДЛЯ ВЫСОКОЧАСТОТНОГО АНАЛИЗА 137
§ 20. Общие правила работы с приборами
для высокочастотного анализа
Независимо от конкретного оформления, принципиальной схемы и типа приборов
для высокочастотного анализа имеются общие принципы работы, в равной мере
относящиеся ко всем высокочастотным измерительным устройствам.
Успешное применение приборов высокочастотного анализа возможно лишь при
точном знании вида характеристических кривых, форма которых зависит как от типа
и константы измерительной ячейки, так и от параметров электрической части прибора.
Поэтому перед работой с высокочастотной установкой необходимо снять ее
характеристическую кривую для растворов (КО, НС1, H2SO4 и т. п.), применяемых обычно
в качестве стандартов.
Количество раствора, помещаемого в измерительную ячейку, должно быть таким,
чтобы его мениск располагался выше верхнего электрода емкостной ячейки или
последнего верхнего витка ячейки индуктивного типа, иначе силовые линии
электромагнитного поля могут замыкаться не только через жидкость, но и через воздух. Это
приводит к резкому снижению чувствительности установки, кроме того, вследствие
изменения уровня раствора при добавлении титранта показания индикаторного
прибора титратора становится функцией и высоты столба раствора. Поскольку при
перемешивании мешалкой уровень жидкости колеблется, при недостатке раствора
возникают случайные переменные нагрузки на измерительную ячейку, что влечет за собой
хаотические броски стрелки индикаторного прибора.
С другой стороны, чрезмерное переполнение сосуда ячейки приводит к так
называемому антенному эффекту. При этом часть раствора, расположенная выше
верхнего электрода, излучает высокочастотную энергию в окружающее пространство, что
влияет на воспроизводимость результатов измерений.
Чтобы избежать излишнего увеличения объема раствора вследствие добавления
титранта, следует применять титранты с концентрацией на порядок выше
предполагаемой концентрации исследуемого раствора.
Нецелесообразно стремиться получить точные результаты в первом же
эксперименте; первая серия измерений делается для оценки приблизительного количества
титранта, необходимого для достижения точки эквивалентности. Полезно также
предварительно рассчитать требуемые объемы реагентов, исходя из концентрации титранта
и предполагаемой концентрации исследуемого раствора.
При ориентировочном определении титрант должен поступать в ячейку
непрерывной тонкой струей или равномерно по каплям. При этом наблюдают за движением
стрелки индикатора и замечают изменение направления или скорости ее движения,
район точки эквивалентности и примерный расход титранта до то^ки эквивалентности.
После этого проводят точное титрование.
Если выполняется большая серия однотипных измерений в течение длительного
промежутка времени, следует периодически, в перерывах между измерениями,
контролировать нуль прибора по эталонному раствору, например по раствору КС1,
который хранят в хорошо закрытой посуде или в запаянной ампуле.
Сосуд должен быть плотно укреплен в измерительной ячейке. Невыполнение
этого требования ведет к случайным изменениям коэффициента связи раствора с
ячейкой, т. е. в конечном счете к значительному разбросу экспериментальных точек.
Тип используемой мешалки существенной роли не играет. Для перемешивания
раствора можно применять также газ или сжатый воздух. Однако в этом случае при
ориентировочном определении необходимо убедиться, что растворение газа в
растворе не вызывает побочных реакций и существенно не изменяет его электрические
параметры.
В любом высокочастотном титраторе можно применять не только измерительные
ячейки, комплектуемые с прибором, но и другие подходящие ячейки. Основное
требование, которое при замене ячеек необходимо учитывать, состоит в том, чтобы
импеданс ячейки и определяемого раствора лежал внутри интервала величин, на которые
рассчитана измерительная схема высокочастотного устройства.
Для непрерывной записи кривой титрования к приборам высокочастотного
анализа можно подключать самопишущие приборы.
Уход за высокочастотными установками, применяемыми в аналитической практике,
состоит в следующем.
В воздухе лаборатории, в которой установлены высокочастотные приборы, не
должно содержаться паров кислот или других веществ, способных вызывать коррозию
частей и деталей прибора, а также приводить к образованию поверхностных проводи-
мостей. В особенности надо следить за чистотой клемм включения измерительной
ячейки.
В случае попадания раствора на электроды и внутрь измерительной ячейки,
необходимо прекратить работу, разобрать ячейку, тщательно протереть ее детали
фильтровальной бумагой или ватой, смоченной дистиллированной водой. Затем хорошо
просушить при комнатной температуре и вновь собрать.
138
ГЛ. IV. ВЫСОКОЧАСТОТНОЕ ТИТРОВАНИЕ
Рабочее место, на котором установлен прибор, не должно испытывать
сотрясений; его следует помещать вдали от сильноточных установок и линий электропроводок,
которые могут создавать значительные помехи.
При работе с установками высокочастотного анализа следует руководствоваться
теми же положениями техники безопасности, что и при эксплуатации обычных
радиотехнических приборов. Необходимо также учитывать повышенную опасность работы
с установками, собранными на открытых панелях, так как крепежные и контактные
клеммы этих приборов находятся под анодным напряжением, и прикосновение к ним
может повлечь за собой удар электрическим током.
В. ПРАКТИЧЕСКИЕ РАБОТЫ
§ 2L Высокочастотное титрование стандартных растворов
Получение характеристической кривой
Возможны два способа получения характеристической кривой
v = f(C) (где v — показания прибора, С — концентрация раствора).
В первом случае заранее готовят ряд растворов с различной
концентрацией, которые используют затем для измерений. Этот способ
достаточно трудоемок и требует значительного количества посуды. Второй
способ получения характеристической кривой основан на измерении
электрических параметров постепенно разбавляемого исходного
раствора.
Рассмотрим второй способ на примере серной кислоты.
Разбавление H2SO4 проводят дистиллированной водой (в общем
случае стандартный раствор разбавляют тем же растворителем).
Наливают в сосуд измерительной ячейки 15 мл 1 н. раствора H2S04. После
отсчета по прибору отбирают 5 мл серной кислоты и добавляют в тот
же сосуд 5 мл воды. Раствор тщательно перемешивают и записывают
новое показание измерительного прибора. Так поступают до тех пор,
пока не установят, что дальнейшее уменьшение концентрации
раствора H2S04 лежит за пределами чувствительности прибора и не
вызывает перемещения стрелки индикаторного прибора.
Если в сосуде ячейки находилось 15 мл раствора H2S04, то
коэффициент разбавления К = 5/15 = 0,33. Концентрация раствора H2S04
после первого разбавления С = 1(1 — 0,33) = 0,67 н.
В общем случае коэффициент разбавления К может быть
произвольным, причем, чем он меньше, тем плавнее происходит изменение
концентрации раствора. После я-кратного разбавления концентрация
раствора С = С0(1—К)п, где С0 — исходная концентрация
разбавляемого раствора.
На основании полученных данных строят характеристическую
кривую. Если исследовались растворы, концентрации которых различаются
на несколько порядков, целесообразно применять полулогарифмические
координаты v — lg С При сравнительно узком интервале
концентраций характеристическая кривая в координатах v — С может оказаться
более удобной.
Титрование сильной кислоты сильным основанием на Q, F-метре
Перед началом титрования строят характеристическую кривую
прибора (рис. 35) и по ней выбирают область рабочих концентраций
определяемых растворов и способы настройки прибора. Как следует
из рис. 35, возможны три методики определения: 1) титрование H2S04
с концентрацией до 0,01 н. — область влево от максимума
характеристической кривой; 2) титрование, соответствующее области максимума
характеристической кривой (0,005—0,015 н.); 3) титрование растворов
H2S04 с концентрацией выше 0,01 н.
§ 21. ВЫСОКОЧАСТОТНОЕ ТИТРОВАНИЕ СТАНДАРТНЫХ РАСТВОРОВ 139
Методика 1. Наливают в сосуд измерительной ячейки 50 мл
0,002 н. раствора H2S04, a 0,01 н. раствора NaOH —в бюретку
емкостью 50 мл. В Q-метрическом режиме устанавливают стрелку
индикаторного прибора вблизи правого края шкалы (положение условного
нуля) и записывают начальное показание прибора. Затем переводят
прибор в /^-метрический режим и устанавливают стрелку
измерительного конденсатора сти на нулевое деление. При помощи конденсатора
сп добиваются установления стрелки
индикаторного прибора также на
нулевом делении.
Приливают 2 мл титранта и вновь
записывают показания индикаторного
прибора в Q- и jF-метрических
режимах. Далее продолжают приливать
0,0/ 0,02 0,0Ь 0,06
Раствор H2S04,h.
Рис, 35. Характеристическая
кривая емкостной ячейки Q, /«'-метра.
О 2 U 6 8/0/2/4/6/8
Объем раствора NaOH,мл
Рис. 36. Кривые высокочастотного
титрования на Q, F-метре сильной кислоты
(0,002 н.)' сильным основанием (0,01 н):
/ — титрование по реактивной
составляющей (AF) полной проводимости; 2 —титрование
по активной составляющей (V) полной
проводимости.
раствор NaOH из бюретки порциями по 2 мл. На основании полученных
данных строят кривые титррвания (рис. 36). При необходимости
определение повторяют, а положение точки эквивалентности находят как
среднее нескольких определений.
Расчет. Концентрацию определяемой кислоты вычисляют по
формуле:
Л/ ^aOH^NaOH /Q .
N™°*= *4so4 (37)
Согласно условию эксперимента, искомая концентрация H2SO4
составляет:
N - 10,2500,01 - 0,00204 г-экв/л
что с точностью до 2% совпадает с концентрацией взятого раствора
(0,002 н.).
Примечание. Вследствие малого различия коэффициентов наклона участков
кривой (/) до и после точки эквивалентности титрование по реактивной составляющей
не дает удовлетворительных результатов.
Методика 2. Наливают 50 мл 0,02 н. раствора H2S04 в сосуд
измерительной ячейки, а 0,1 н. раствор NaOH —в бюретку емкостью 50 мл
140
ГЛ. IV. ВЫСОКОЧАСТОТНОЕ ТИТРОВАНИЕ
и настраивают прибор, как описано выше. По окончании титрования
строят кривые (рис. 37). Нормальность кислоты рассчитывают по
уравнению (37).
Примечание. Кривые / и 2 (см. рис. 36) имеют сложный характер, особенно
кривая 2, поэтому в данном случае титрование предпочтительнее проводить по
реактивной составляющей.
Методика 3. Наливают в сосуд измерительной ячейки 45 жл 0,1 н
раствора NaOH, a 0,2 н. раствор H2S04 —в бюретку емкостью 50 мл.
В Q-метрическом режиме
устанавливают стрелку индикаторного
прибора на 1—2-м делении в
левом краю шкалы и далее
регулируют Q, F-метр, как описано выше.
Записывают первые показания
индикаторного прибора. С целью
сократить начальный участок
диаграммы высокочастотного
титрования приливают сразу 6 мл
раствора H2S04 и вновь записывают
показания прибора. В дальнейшем
приливают по 2 мл кислоты. Затем
строят кривую титрования (рис.38).
Концентрацию определяемого
основания вычисляют по уравнению,
аналогичному уравнению (37).
Примечание Инверсия кривых титрования (по сравнению с кривыми,
показанными на рис. 36) обусловлена переходом рабочего интервала концентраций
растворов в область справа от максимума характеристической кривой (см. рис. 35).
Титрование сильной кислоты сильным основанием
на титраторе ТВ-6Л
В сосуд для титранта наливают 0,1 н. раствор NaOH, затем 5 мл
0,1 н. серной кислоты отбирают пипеткой и помещают в стакан
емкостью 100 мл, который устанавливают в измерительную ячейку.
После этого в стакан наливают дистиллированную воду так, чтобы
уровень раствора в сосуде был на 3—5 мм выше верхнего электрода,
опускают в стакан стержень мешалки и, включив ее мотор, регулируют
скорость перемешивания.
Устанавливают стрелку индикаторного прибора на 5—10-м
делении в левом краю шкалы. Заполняют бюретку (надавливая грушу)
раствором основания; кончик бюретки должен находиться в стороне
от мешалки (на расстоянии от центра ячейки, равном приблизительно
2/3 ее радиуса).
Записывают начальные показания прибора. Нажимая кнопку
электромагнитного затвора бюретки, сливают 2 мл раствора NaOH. После
остановки стрелки записывают первое показание. Далее добавляют
раствор NaOH порциями по 0,5 мл. Затем строят кривую титрования
(рис. 39).
2 4 S 8 10 12 Ъ 18
Объем раствора NaOH,w7
Рис. 37, Кривые высокочастотного
титрования на Q, F-метре сильной кислоты
(0,02 н.) сильным основанием (0,1 н.):
7 — титрование по реактивной
составляющей (AF) полной проводимости, 2 —титрование
по активной составляющей (v) полной
проводимости.
Титрование слабой кислоты сильным основанием
При постепенном приливании раствора сильного основания к
раствору щавелевой кислоты можно наблюдать две точки эквивалентности
(рис, 40). В случае обратного титрования (рис. 40, б), когда к раствору
§ 21 ВЫСОКОЧАСТОТНОЕ ТИТРОВАНИЕ СТАНДАРТНЫХ РАСТВОРОВ 141
основания приливают небольшими порциями определяемую
кислоту, фиксируется только вторая точка эквивалентности. Это
объясняется тем, что при внесении кислоты в раствор сильного основания,
находящегося в сосуде измерительной ячейки в заведомо избыточном
количестве, в реакцию вступает сразу вся кислота.
0 8 16 2^ 32
Раствор H2S04,/»/^
Рис. 38. Кривые высокочастотного
титрования сильного основания
(0,1 н.) сильной кислотой (0,2 н.):
/-титрование по реактивной
составляющей (Д.Р) полной проводимости;
2—титрование по активной
составляющей (V) полной проводимости.
2 3 4 5 S 7 8 9
Объем раствора NaOH, мл
Рис. 39. Кривая высокочастотного
титрования серной кислоты (0,1 н.)
раствором NaOH (0,1 н.) на ти-
траторе ТВ-6Л.
Работу выполняют на Q, F-метре.
Прямой порядок титрования. Наливают 40 мл 0,002 н. раствора
щавелевой кислоты в сосуд измерительной ячейки, а 0,01 н. раствор
NaOH — в бюретку емкостью 50 мл. Регулируют Q,F-MeTp, как
указано на стр. 139, и записывают первый отсчет. Приливают 2 мл
раствора основания и вновь записывают отсчет по индикаторному прибору
и т. д. По полученным данным строят кривые титрования (см,
рис. 40,а).
2 U 8 8 Ю 12 К 16 18
Объем раствора NslOH$mj?
а
1 2 3 4 5
Объем раствора H2C2C>4,W7
Рис. 40. Кривые высокочастотного титрования щавелевой кислоты раствором едкого
натра:
а-прямой порядок титрования, концентрация кислоты 0,002 нм основания —0,01 н.; б-обратный
порядок титрования, концентрация основания 0,001 н., кислоты —0,02 н.;
/—титрование по реактивной составляющей полной проводимости; 2 —титрование по активной
составляющей полной проводимости.
Обратный порядок титрования. Наливают в сосуд измерительной
ячейки 40 мл 0,001 н. раствора NaOH, a 0,02 н. раствор щавелевой
кислоты — в бюретку емкостью 20 мл. Регулируют Q, /''-метр, как
описано выше, делают первый отсчет и записывают его. Прибавляют 0,5 мл
раствора Н2С2О4, вновь снимают показания приборов и т. д. По
полученным данным строят кривые титрования (см. рис. 40,6).
142
ГЛ. IV. ВЫСОКОЧАСТОТНОЕ ТИТРОВАНИЕ
Определение бария методом осаждения
(образование мелкокристаллического осадка BaSOJ
Работу проводят на Q, F-метре.
Методика определения. Наливают 20 мл 0,1 н. раствора ВаС12 в
мерную колбу емкостью 500 мл и доводят дистиллированной водой до
метки. Отбирают аликвотную часть (около 50 мл) раствора ВаС12 в
сосуд измерительной ячейки, а в бюретку емкостью 10 мл наливают 0,2 н.
раствор H2S04.
Регулируют Q, F-метр, как описано выше, и делают первый отсчет.
Приливают 1 мл раствора H2S04 и, дождавшись окончания
дрейфа стрелки, записывают второй отсчет. Затем добавляют титрант
О / 2 3 4»
Объем раствора H2S04,/w
О 2 4 8 8
Объем раствора NaOH.w?
Рис. 41. Кривые высокочастотного титрования, основанного на реакциях
с образованием осадка:
а — титрование ионов бария, концентрация раствора ВаС1г 0,01 н„ серной кислоты — 0,2 н.;
б—титрование ионов магния, концентрация раствора MgCl2 0,01 н., раствора
NaOH-0,1 н.;
/ — титрование по реактивной составляющей полной проводимости; 2 —титрование по
активной составляющей полной проводимости.
порциями по 0,5 мл. По полученным результатам строят
соответствующую кривую титрования (рис. 41,а).
Расчет. Содержание ВаС12 в анализируемом растворе (в г)
вычисляют по формуле, аналогичной формуле (38), стр. 144.
Определение магния методом осаждения
[образование аморфного осадка Mg(OH)2]
Работу проводят на Q, F-метре,
Методика определения. Разбавляют 20 мл раствора MgCfo
(^0,1 н) дистиллированной водой до метки в мерной колбе емкостью
200—250 мл и тщательно перемешивают. Аликвотную часть (около
50 мл) растрора помещают в сосуд измерительной ячейки, а в бюретку
емкостью 20 мл наливают 0,1 н. раствор NaOH.
Регулируют прибор, как описано выше. Записывают его первые
показания в режиме Q- и F-метрии. Затем добавляют раствор NaOH
порциями по 1 мл. Запись результатов отсчета делают после
остановки стрелки. По полученным данным строят кривые титрования
(рис 41,6Ju
§ 22. ТИТРОВАНИЕ РАСТВОРОВ НЕИЗВЕСТНОЙ КОНЦЕНТРАЦИИ 143
Расчет. Содержание MgCl2 в анализируемом растворе (в г)
вычисляют по формуле, аналогичной формуле (38).
Примечание. Из рис. 41 следует, что параллельное титрование по активной
и реактивной составляющим полной проводимости ячейки с раствором дает возмож*
ность определять точку эквивалентности с большей достоверностью. Это в особенности
важно, когда титрование проводится в первый раз и форма кривой высокочастотного
титрования заранее неизвестна. То же относится и к опытам, целью которых является
выяснение состава образующихся в ходе реакции химических соединений.
Определение железа (Ш) методом комплексообразования
Высокочастотное титрование, основанное на реакциях
комплексообразования, является более сложным, чем описанные выше типы
определений, и дает худшие результаты.
Прямой порядок титрования. Отбирают аликвотную часть (около
1 мл) приблизительно 0,1 М раствора Fe(NH4) (S04)2 и помещают его
в сосуд измерительной ячейки осциллотитратора системы «Пунгор»,
добавляют 80—90 мл воды, так чтобы уровень раствора был не ниже
верхнего электрода ячейки, затем вводят 0,1 мл концентрированной
серной кислоты. В бюретку емкостью 50 мл наливают 0,1 н. раствор
(NH4)SCN.
Включают мешалку титратора и устанавливают стрелку
индикаторного прибора в левом крайнем положении. Титрант прибавляют
порциями по 0,5 мл.
По полученным данным строят кривую высокочастотного
титрования, она имеет вид ломаной восходящей кривой,, состоящей из двух
прямолинейных отрезков; тангенсы угла наклона ветвей этой кривой
майо различаются, поэтому определение положения точки
эквивалентности затруднено. Для проверки полученных результатов полезно
повторить операцию титрования, но уже в обратном порядке.
Обратный порядок титрования. Помещают в сосуд измерительной
ячейки 9 мл 0,1 н. раствора (NH4)SCN, наливают в нее 80—90 мл
дистиллированной воды и добавляют 0,1 мл концентрированной H2SO4.
Анализируемый раствор наливают в бюретку емкостью 10 мл. Осцил-
лотитратор регулируют, как описано выше. Сокращая начальный
участок кривой титрования, приливают сразу 2 мл раствора Fe(NH4) (S04)2,
а затем его добавляют порциями по 0,2 мл.
Кривая титрования имеет тот же вид восходящей кривой с плохо
выраженной точкой эквивалентности, но левая ветвь (до точки
эквивалентности) имеет несколько больший тангенс угла наклона, чем
ветвь после точки эквивалентности.
Расчет. Содержание Fe(NH4) (S04)2 (в г) вычисляют по формуле,
аналогичной формуле (38).
§ 22. Высокочастотное титрование растворов неизвестной
концентрации
Определение кислоты в растворе
Методика определения. Раствор определяемой кислоты доливают
дистиллированной водой до метки мерной колбы, хорошо
перемешивают и переносят аликвотную часть в сосуд измерительной ячейки.
Ставят прибор в режим Q-метрии, при котором он чувствителен
к изменению электропроводности, а переключатель направления хода
стрелки титратора системы «Пунгор» — в положение, соответствующее
144
ГЛ. IV. ВЫСОКОЧАСТОТНОЕ ТИТРОВАНИЕ
прямой зависимости изменения электропроводности исследуемого
раствора и показаний прибора. Затем включают мешалку, отмечают
показания индикаторного прибора и добавляют в сосуд ячейки
несколько миллилитров дистиллированной воды. Если стрелка индикаторного
прибора отклоняется влево, это означает, что титрование
сопровождается изменениями электропроводности, соответствующими левому
склону характеристической кривой g— х, т. е. точка эквивалентности
будет находиться в минимуме или в точке излома нисходящей кривой
высокочастотного титрования. При отклонении стрелки вправо кривая
титрования будет соответствовать интервалу электропроводности,
приходящемуся на правый склон характеристической кривой, а точку
эквивалентности следует ожидать в максимуме или в точке излома
восходящей кривой высокочастотного титрования.
Сначала проводят предварительный опыт. Прибавляют первые
порции титранта (NaOH), записывают показания индикаторного
прибора и продолжают приливать титрант такими же порциями, следя за
ходом стрелки, чтобы сделать предварительное определение
положения точки эквивалентности на кривой титрования. Затем проводят
точное титрование и строят кривую титрования. Определяют положение
точки эквивалентности и количество титранта, израсходованного на
определение.
Расчет. Содержание (g) определяемой кислоты вычисляют по
уравнению:
^NaOH^NaOH Vk /qq4
§== iooo Т7 (38)
где VK — объем исследуемого раствора;
KA — аликвотная часть пробы.
Определение хлорид-ионов в водопроводной воде
Методика определения. Пробу водопроводной воды (50—100 мл)
наливают в сосуд измерительной ячейки и дистиллированной водой
доводят уровень раствора до отметки, расположенной на 5—10 мм выше
верхнего электрода. После соответствующей регулировки аппаратуры
делают первый отсчет. Затем титруют 0,1 н. раствором AgN03
порциями по 0,1—0,2 мл. Титрование продолжают до момента резкого
возрастания электропроводности. Затем строят кривую
высокочастотного титрования, точку эквивалентности находят по пересечению двух
прямолинейных ветвей полученной кривой титрования.
Если концентрация С1~ очень мала, полезно вначале провести
предварительное титрование, наблюдая за движением стрелки
индикаторного прибора. Поскольку при замене СГ на NOJ
электропроводность раствора изменяется незначительно, при низком содержаний
хлорид-ионов кривую титрования строят следующим образом:
проводят две линии, одну — параллельно оси абсцисс через точку с
ординатой начальных показаний прибора, другую — через точки,
расположенные после излома. Точку эквивалентности находят по
пересечению этих двух прямых.
Определение сульфат-ионов
Описываемый метод титрования разработан для определения
серосодержащих соединений в различных нефтяных продуктах. После
предварительной обработки исследуемого образца переводят серу се-
§ 22. ТИТРОВАНИЕ РАСТВОРОВ НЕИЗВЕСТНОЙ КОНЦЕНТРАЦИИ 145
росодержащих соединений в форму сульфата; анализируемый раствор
должен быть 0,01—0,0001 н. по определяемому иону. Если
присутствуют карбонаты, их разрушают подкислением раствора и кипячением
его в течение нескольких минут.
Методика определения. Раствор, содержащий сульфат-ионы,
наливают в измерительную ячейку, добавляют 30—40% (по объему)
этилового спирта и нейтрализуют раствор по метиловому красному
свободным от карбонатов 0,1 н. раствором NaOH. Раствор охлаждают до
комнатной температуры и вводят в него несколько миллиграммов
сульфата бария.
После предварительной настройки титратора проводят титрование
0,1—0,02 н. раствором ВаС12. Титрант добавляется, как обычно,
затем некоторое время выжидают, чтобы система пришла к
равновесию и записывают показания прибора.
В общем случае размер пробы, количество и концентрация
титранта должны быть такими, чтобы в каждой порции титранта оказывалось
не более 5 мл и требовалось не больше 10—15 таких порций. После
окончания титрования строят кривую титрования, точку
эквивалентности определяют обычным способом.
Определение аминов
Определение сложных аминов классическими аналитическими
методами — весьма трудоемкая и длительная процедура.
Применение метода высокочастотного титрования значительно
сокращает и упрощает это определение. Например, при высокочастотном
титровании диэтилентриамина (NH2C2H4NHC2H4NH2) достаточно
провести лишь одно титрование в водной среде.
Методика определения. Около 0,2 г образца помещают в мерную
колбу емкостью 50 мл и наливают воду до метки. Для титрования
берут аликвотйую часть, содержащую около 20 мг амина, вносят ее
в сосуд измерительной ячейки и разбавляют до необходимого объема,
например до 40—50 мл. Титруют 0,1 н. раствором H2S04; отсчет по
прибору делают после добавления каждых 0,2 мл титранта.
Кривая титрования состоит из трех отрезков: первый соответствует
нейтрализации двух первичных аминогрупп, второй,
горизонтальный,— нейтрализации одной вторичной аминогруппы и третий —
избытку титранта.
При определении аминов следует учитывать, что титрование в
открытом сосуде может давать несколько заниженные результаты,
обусловленные летучестью вещества, однако опыт показывает, что
соотношение между содержанием первичных и вторичных аминогрупп
сохраняется неизменным.
Определение фенолов и крезолов
Прямой порядок титрования. В предварительно взвешенный бюкс
помещают исследуемый образец, содержащий 10—20 мг фенола, и
растворяют в 5—6 мл этилового спирта. Раствор переносят в сосуд
измерительной ячейки, обмывают стенки бюкса дистиллированной водой
и разбавляют ею раствор до необходимого объема (100—150 мл).
Содержимое сосуда ячейки продувают в течение 5 мин свободным от
кислорода азотом и титруют 0,05 М раствором гидроокиси лития. После
каждого добавления титранта перед отсчетом показаний выдерживают
ив
ГЛ. IV. ВЫСОКОЧАСТОТНОЕ ТИТРОВАНИЯ
раствор в течение 10—20 сек для того, чтобы реакция нейтрализации
прошла более полно.
По полученным данным строят кривую титрования, находят точку
эквивалентности и делают расчеты.
Обратный порядок титрования. Навеску фенола около 10 г при
соблюдении необходимых мер предосторожности помещают в мерную
цолбу емкостью 500 мл и наливают (примерно до половины)
дистиллированную воду. Добавляют в колбу 10 мл 0,1 н. раствора сильного
основания и энергично перемешивают, затем доводят дистиллированной
водой до метки. Для титрования отбирают 1 мл раствора, помещают
его в сосуд измерительной ячейки, разбавляют до необходимого объема,
например до 50 мл, и титруют 0,1 н. раствором хлористоводородной
кислоты.
Титрование крезолов проводят аналогично.
ГЛАВА V
ПОЛЯРОГРАФИЧЕСКИЙ МЕТОД АНАЛИЗА
И АМПЕРОМЕТРИЧЕСКОЕ ТИТРОВАНИЕ
ПОЛЯРОГРАФИЧЕСКИЙ МЕТОД АНАЛИЗА
Л. ТЕОРЕТИЧЕСКИЕ ОСНОВЫ
§ 1. Предельный, или диффузионный, ток
Главной особенностью процесса электролиза при
полярографическом анализе (см. книга 2, гл. VIII, § 5) является применение
индикаторного поляризующегося электрода. Вследствие очень малой
поверхности индикаторного электрода плотность тока (а/см2) на нем довольно
велика. В результате этого в части раствора, находящейся вблизи
такого электрода, концентрация определяемых ионов при электролизе
быстро уменьшается и в конце концов наступает момент равновесия,
когда все ионы, подходящие к электроду за счет диффузии, тотчас
разряжаются и благодаря этому сила тока становится постоянной
(предельный, или диффузионный, ток).
Поверхность электрода сравнения (неполяризующегося электрода)
должна быть неизмеримо больше поверхности индикаторного электрода.
На такой большой поверхности электрода сравнения плотность тока
получается довольно малой, поэтому вблизи электрода изменение
концентрации ионов очень мало и не оказывает влияния на кривую
зависимости силы тока от приложенного напряжения. В этих условиях
зависимость силы тока от напряжения выражается кривой с перегибами, или
волнами (рис. 42).
Величина силы тока. Величина силы тока на ртутном капельном
электроде в любой момент времени t жизни ртутной капли
определяется уравнением:
It = 706nDl/2rnf41/6C (1)
где If- сила тока, ма\
п— число электронов, принимаемых ионом при восстановлении или отдаваемых
при окислении;
D — коэффициент диффузии иона, см2 • сект1;
— масса ртути, вытекающей из капилляра в 1 сек, мг;
С — концентрация восстанавливающегося (или окисляющегося) иона, ммоль/л.
Из этого уравнения следует, что ток будет расти пропорционально
корню шестой степени из времени жизни капли. При данном
потенциале ток будет расти вместе с каплей и затем падать.
Если в уравнении (1) заменить t значением периода капания т,
величиной, постоянной для данного капилляра и для неизменного
уровня ртути и соответствующей максимальной величине капли, то получим
уравнение максимального тока:
/макс = 706rtD1/2m2V/6C (2)
Обычный гальванометр не дает возможности наблюдать
максимальные отклонения силы тока, так как зеркальце движется слишком
148 ГЛ. V. ПОЛЯРОГРАФИЯ И АМПЕРОМЕТРИЧЕСКОЕ ТИТРОВАНИЕ
медленно и приходится наблюдать некоторую среднюю величину тока.
Вычислено, что средний ток равен 6/7 максимального тока.
Средний ток Id называется средним диффузионным током и
определяется уравнением Ильковича:
Id = 605rtD1/2m2/3t1/6C (3)
Произведение rrtW^ называется характеристикой капилляра. При
постоянстве других факторов диффузионный ток прямо
пропорционален этой характеристике.
Величина т зависит от диаметра капилляра, давления всего
столба ртути (от верхнего уровня ртути в резервуаре до кончика
капилляра) и от температуры.
Величина периода капания ртути т зависит от давления ртути и от
диаметра капилляра. Кроме того, величина т зависит от поверхностного
натяжения и, следовательно, от состава анализируемого раствора и от
значения приложенного напряжения. Период
капания т при возрастании напряжения
сначала возрастает, проходит через максимум
при —0,56 в и затем быстро уменьшается с
увеличением отрицательного потенциала.
Произведение m2Wk изменяется с
увеличением потенциала капельного электрода
значительно меньше, чем т, и для практических
целей его можно принять постоянным в
пределах значений потенциалов от 0 до —1,0 е.
Но при более отрицательных потенциалах
уменьшение m~Wu становится заметным.
Коэффициент диффузии D зависит от при-
Рис. 42. Полярографические роды вещества, температуры и вязкости рас-
волны.
твора.
Для одного и того же иона при
одинаковых условиях (посторонние электролиты, температура,
размер капилляра, высота столба ртути) величины п, Д m, t постоянны
и сила тока прямо пропорциональна концентрации
восстанавливающегося иона:
Id = KC (4)
Эта зависимость является основой количественного
полярографического анализа: измерив силу тока, можно определить концентрацию
восстанавливающегося иона.
Фон. Для того чтобы ионы определяемого вещества перемещались
к индикаторному электроду только вследствие диффузии, а не за счет
диффузии и электростатической силы притяжения (миграционный ток),
в исследуемый раствор добавляют какой-либо индифферентный
электролит с катионом, восстанавливающимся гораздо труднее
анализируемого катиона, например КС1, KN03, NH4C1, при концентрации в 100—
1000 раз превышающей концентрацию определяемого вещества. Такой
электролит называется фоном.
Катионы электролита — фона — движутся к катоду, но не могут
разрядиться на нем при данном потенциале. Они остаются у
поверхности электрода, образуя двойной электрический слой. Электрическое
поле индикаторного электрода экранируется этими ионами фона, и
поэтому катионы определяемого вещества не притягиваются электрическим
полем катода, а двигаются к нему только за счет диффузии.
Фон имеет еще и другое значение — он увеличивает
электропроводность раствора.
§ I. ПРЕДЕЛЬНЫЙ ИЛИ ДИФФУЗИОННЫЙ ТОК
149
Процессы, происходящие на электродах. Основное преимущество
ртутного капающего электрода для полярографического анализа
катионов заключается прежде всего в том, что поверхность его постоянно
обновляется. На ней не накапливается, как на твердых электродах,
слой постороннего металла, изменяющего свойства электрода, и поэтому
условия определения остаются все время постоянными. Кроме того,
перенапряжение водорода на ртути очень велико, т. е. свободный
водород выделяется на ртуть только при больших отрицательных
значениях потенциала. Это дает возможность определять многие металлы в
нейтральных и даже кислых растворах.
Рассмотрим процессы, происходящие на ртутном капельном
электроде (катоде) и на электроде сравнения, который является анодом
(каломельный электрод).
На катоде происходит следующий процесс:
Мел+ -Ь пе < *• Me (амальгама)
Образовавшийся свободный металл диффундирует в глубь капли.
Затем капля амальгамы падает и процесс начинается снова на новой
капле. Благодаря очень малой силе тока, протекающего через
электролизер (порядка Ю"6^), концентрация вещества в растворе очень
мало изменяется и остается почти постоянной во время электролиза
(величина предельного тока не изменяется с течением времени).
Каломельный электрод, являющийся в данном случае анодом,
представляет собой сосуд, на дне которого находится металлическая ртуть,
а над ней слой каломели и насыщенный раствор КС1 (насыщенный
каломельный электрод).
Процесс на аноде заключается в том, что атомы ртути отдают
электроны, причем ионы ртути образуют с ионами хлора,
находящимися в растворе, осадок каломели:
2Hg + 2СГ = Hg2Cl2 + 2e
Потенциал ртутного анода определяется концентрацией ионов ртути в
растворе:
?=^4>* + ^1НЫ++ (5)
где ?° ,, / —нормальный потенциал системы.
Hg++/2Hg
Концентрация [Hg2]++ в свою очередь определяется концентрацией
Шгт Т++ — nPHg2Cl2 /ftv
lH?2j - —rC1-j2 w
C1-:
При электролизе концентрация хлорид-ионов несколько изменяется,
так как С1~ расходуется на образование осадка Hg2Cl2. Однако
это изменение незначительно, так как концентрация хлорида калия в
насыщенном растворе велика, а плотность тока на аноде очень мала.
Поэтому концентрация С1~ вблизи анода настолько мало изменяется,
что практически не влияет на концентрацию [Hg2]++. Вследствие этого
потенциал ртутного анода остается почти постоянным (анод не
«поляризуется»).
Согласно закону Ома:
Еш = Еа-Ек + Щ (7)
где ?Вн — напряжение, наложенное на электроды;
?а — потенциал анода;
150 ГЛ. V. ПОЛЯРОГРАФИЯ И АМПЕРОМЕТРИЧЕСКОЕ ТИТРОВАНИЕ
jE"k — потенциал катода-;
/ — сила тока в цепи;
R— сопротивление раствора.
Поскольку сила тока при полярографических определениях мала и
сопротивление раствора невелико (электропроводность раствора
увеличивается благодаря фону), падением напряжения можно пренебречь
и написать:
?вн = ?а - Ек (3)
Так как величина потенциала анода постоянна, ее можно считать
условно равной нулю *. Тогда величина потенциала ртутного катода
по отношению к нулевому потенциалу анода численно равна величине
приложенного внешнего напряжения:
?вн = - Ек (9)
Потенциал полуволны. Потенциал ртутного катода в тот момент,
когда достигнута величина напряжения разложения и начинается
электролиз, называется потенциалом выделения (или восстановления)
данного иона. Потенциал выделения зависит от природы иона, однако
на эту величину оказывает влияние концентрация
восстанавливающегося иона и некоторые другие факторы. Поэтому для качественного
определения ионов пользуются так называемым потенциалом
полуволны, который не зависит от концентрации восстанавливающегося иона.
Потенциалом полуволны называется то значение потенциала, при
котором происходит возрастание силы тока до половины предельного
значения. Потенциал полуволны можно определить с помощью
уравнения полярографической волны:
* = *•/,-^7-^7 <10>
'* п *пред *
Если откладывать по оси абсцисс значения ?, а по оси ординат
*ё 7 ZJ, T0 построенный график представляет собой прямую.
Когда i = п%ед , то lg- — =0, Е = Е\/„ и в этот момент прямая
^ 'пред 1
пересекает ось абсцисс, отсекая на ней отрезок, равный ?у2. Таким
образом, чтобы определить значение ?у2, нужно измерить расстояние от
начала координат до точки пересечения прямой с осью абсцисс.
Потенциал полуволны является величиной, характерной для
данного иона, поэтому измерив его, можно определить по
соответствующим таблицам, какой именно ион находится в растворе.
Потенциал полуволны можно определить проще, для этого на по-
лярограмме из середины волны опускают перпендикуляр на ось
абсцисс. Расстояние от точки пересечения перпендикуляра с осью абсцисс
до начала координат равш ?уа. Однако этот способ определения менее
точен.
Потенциалы полуволн ионов, указанные в таблицах, измеряются
обычно по отношению к насыщенному каломельному электроду,
взятому в качестве анода. В полярографии применяются и другие
электроды сравнения: 0,1 н. и 1 н. каломельные, меркурсульфатный,
хлорсеребряный, меркуриодидный.
При работе с ртутным анодом потенциал ртути устанавливают по
таллию.
* Вообще же потенциал насыщенного каломельного электрода, измеренный по
отношению к нормальному водородному электроду, равен +0,247 в.
§ 1. ПРЕДЕЛЬНЫЙ ИЛИ ДИФФУЗИОННЫЙ ТОК
151
Рис. 43. Максимумы 1-го
рода на полярограммах.
Максимумы на полярограммах. При полярографировании очень часто на
полярографических кривых возникают максимумы. Во многих случаях вместо нормальной
полярограммы, имеющей форму ступени, получается кривая с максимумом вследствие
того, что в некотором интервале напряжения возникает ток, значительно
превышающий ток диффузии. При дальнейшем повышении потенциала ток более или менее резко
спадает (рис. 43), достигая иногда значения предельного диффузионного тока (как
показано пунктиром на рис. 43). Однако очень часто переход максимального тока
к диффузионному происходит постепенно, и тогда определение высоты
полярографической волны становится очень затруднительным.
Согласно теории А. Н.. Фрумкина, причиной возникновения максимумов является
движение поверхности ртутной капли, вызывающее дополнительное размешивание
раствора. Это движение увеличивает поступление
восстанавливающегося вещества к катоду, а следовательно^
увеличивает силу диффузионного тока.
Движение поверхности ртутной капли объясняется
неравномерной плотностью тока на этой поверхности.
Плотность тока больше в нижних частях капли, так
как верхняя часть капли экранируется концом
капилляра. Это вызывает неодинаковое распределение
поверхностного натяжения. Если же поверхностное
натяжение в различных местах ртутной капли различно, то
происходит движение ртути вдоль поверхности от
участков с меньшим поверхностным натяжением
(поверхность расширяется) к участкам с большим
поверхностным натяжением (поверхность сжимается).
Кривая зависимости поверхностного натяжения от потенциала называется
электрокапиллярной кривой. Такая кривая представлена на рис. 44.
Поверхностное натяжение с возрастанием отрицательного потенциала вначале
возрастает, а затем убывает. При потенциале —0,56 в кривая имеет максимум.
Поверхность ртути в растворе соли ртути имеет положительный заряд.
Отрицательно заряженные ионы или дипольные молекулы притягиваются к поверхности
ртути и образуют двойной электрический слой с определенным положительным
потенциалом, которому соответствует невысокое поверхностное натяжение.
При наложении некоторого отрицательного напряжения положительный заряд
ртутной поверхности уменьшается, а поверхностное натяжение увеличивается. По мере
возрастания отрицательного потенциала электрода положительный заряд ртути еще
более уменьшается, а поверхностное натяжение увеличивается до тех пор, пока не
достигнет максимума при потенциале —0,56 в (при этом
потенциале ртуть не заряжена).
При дальнейшем увеличении напряжения ртуть
заряжается отрицательно, из раствора притягиваются
положительно заряженные ионы и снова создается
двойной электрический слой.
Теперь при увеличении отрицательного потенциала
заряд ртути уже не уменьшается, а увеличивается, а
поверхностное натяжение снижается. Таким образом,
наибольшее поверхностное натяжение наблюдается на
незаряженной ртутной капле, по мере увеличения
заряда капли (положительного или отрицательного)
поверхностное натяжение уменьшается.
Если какой-либо ион восстанавливается при
потенциале, соответствующем положительной части
электрокапиллярной кривой (ртутная капля заряжена положительно), то движение
поверхности ртутной капли и жидкости в приэлектродном слое направлено сверху вниз,
так как в этом случае поверхностное натяжение тем больше, чем больше потенциал
(см. рис. 44), а потенциал больше в нижней части капли. Если же восстановление
иона происходит при более отрицательном потенциале, чем —0,56 в (ртутная капля
заряжена отрицательно), то, хотя потенциал в нижней части капли по-прежнему
больше потенциала в ее верхней части, движение ртути и электролита направлено снизу
еверх, поскольку более высокому потенциалу соответствует меньшее поверхностное
натяжение (см. рис. 44).
В том случае, когда восстановление иона происходит при потенциале, близком
к —0,56 в (потенциал, при котором ртуть не заряжена), поверхностное натяжение
вверху и внизу капли приблизительно одинаково, и максимум не появляется.
Например, при восстановлении кадмия, потенциал полуволны которого (на фоне 1 М KNO3)
равен —0,586 в, максимум на кривой не появляется.
Рассмотренные выше максимумы носят название максимумов 1-го рода и имеют
форму пика (см. рис. 43). Максимумы 1-го рода появляютоя на фоне разбавленных
электролитов.
Рис. 44. Электрокапиллярная
кривая.
152 ГЛ. V. ПОЛЯРОГРАФИЯ И АМПЕРОМЕТРИЧЕСКОЕ ТИТРОВАНИЕ
При полярографировании кроме максимумов 1-го рода появляются иногда
максимумы другого вида, которые называются максимумами 2-го рода. Эти максимумы
возникают при работе с быстро капающими капиллярами на фоне концентрированных
растворов и имеют более сглаженную форму, чем максимумы 1-го рода (рис. 45).
Максимумы 2-го рода могут возникать при всех потенциалах. Их появление
объясняется тем, что при образовании ртутной капли наблюдается движение ртути внутри
самой капли. Струя ртути довольно быстро вытекает из капилляра в каплю. Она
доходит до дна капли и расходится вверх вдоль ее поверхности. В результате этого
наблюдается перемешивание раствора и возникновение максимума на
полярографической кривой.
Максимумы на полярографических кривых мешают полярографическому анализу,
и от них необходимо избавляться. Максимумы как первого, так и второго рода могут
быть уничтожены добавкой в раствор поверхностно-активных веществ, тормозящих
движение поверхности ртути. Поверхностно-активные вещества адсорбируются сильнее
на тех участках капли, где больше поверхностное натяжение. Но поверхностное
натяжение поверхностно-активного вещества меньше, чем у ртути, и поэтому на данном
участке избыток поверхностно-активного вещества
будет уменьшать поверхностное натяжение.
Таким образом поверхностное натяжение ста-
/ нет почти одинаковым по всей поверхности ртутной
капли, и движение ртути, а вместе с ним и
движение электролита в приэлектродном слое
прекратится.
В качестве поверхностно-активных веществ
применяются желатина, агар-агар, столярный клей
и т. п.
Рис. 45. Полярографический Волна кислорода. Полярографическая
максимум 2-го рода. волна определяемого иона обычно
искажается волной кислорода. Кислород,
содержащийся в растворе определяемого иона, восстанавливается на
ртутном электроде. При этом на полярограмме образуются две волны.
Первая волна обусловлена восстановлением кислорода до перекиси
водорода:
02 +2Н+ + 2е —> Н202 (в кислой среде)
02 + 2Н20 + 4е —> 2Н202 (в щелочной среде)
Вторая волна получается в результате восстановления перекиси
водорода до воды или гидроксила:
Н202 + 2Н+ + 2б> —> 2Н20 (в кислой среде)
Н202 + 2е —> 20Н~ (в щелочной среде)
Для удаления кислорода через раствор пропускают инертный газ,
например водород или азот. В случае кислых растворов для удаления
кислорода можно использовать С02. В щелочных средах удобно
удалять кислород при помощи сульфита. Добавление 0,1 г Na2S03 в
100 мл раствора достаточно для полного восстановления кислорода в
течение 5 мин.
Конденсаторный ток. Форму полярографической кривой может
искажать также конденсаторный ток, или ток заряжения. Очень
чувствительный гальванометр с возрастанием напряжения показывает
постепенно возрастающую силу тока даже при отсутствии веществ,
восстанавливающихся на катоде.
Появление этого тока объясняется тем, что некоторое количество
электричества расходуется на заряжение двойного электрического слоя
поверхности каждой капли ртути.
§ 2. ВОЗНИКНОВЕНИЕ ТОКА НА МИКРОЭЛЕКТРОДАХ 133
Этот ток называется конденсаторным потому, что заряжение
двойного слоя аналогично заряжению конденсатора. Чем больше
потенциал, наложенный на каплю, тем больше сила этого тока. На поля-
рограмме вместо горизонтальных участков получаются наклонные
участки.
Кроме конденсаторного тока в растворе при наложении
потенциала на электроды обычно возникает также небольшой ток в
результате восстановления примесей, например плохо удаленного кислорода.
Этот ток вместе с конденсаторным в сумме составляет так называемый
остаточный ток.
Наличие остаточного тока сильно затрудняет полярографирование»
так как изгибы полярограммы сглаживаются и определение высоты
волны на такой полярограмме весьма сложно.
Область применения полярографии с ртутным капельным
электродом. Ртутный капельный электрод может быть использован для
полярографических определений в области потенциалов от +0,3 в до —2 в
(в щелочных и нейтральных растворах) или до —1 в (в кислых
растворах) *.
На ртутном капельном электроде полярографическим методом
можно определять катионы, восстанавливающиеся при этих потенциал
лах (т. е. почти все катионы), а также некоторые анионы при
концентрации определяемых веществ в растворе до 10~4 М (в некоторых
случаях до Ю-5 М). Полярографический метод применяют также для
определения органических веществ, способных восстанавливаться на
ртутном капельном электроде.
Метод дает возможность в ряде случаев проводить определение
нескольких компонентов в смеси без их предварительного разделения
(если потенциалы восстановления соответствующих ионов достаточно
различаются между собой).
Метод обладает большой чувствительностью, его широко
применяют для определения примесей в различных веществах, особенно в
металлах и сплавах, причем зачастую без отделения основного
компонента сплава. При этом возможно определение в технических образцах
примесей металлов при содержании их до 0,001%.
§ 2. Возникновение диффузионного тока на твердых микроэлектродах
При необходимости работать в области потенциалов более
положительных, чем +0,3 в (при использовании реакций окисления
определяемых веществ на индикаторном электроде), применяют достаточно малых
размеров твердые электроды, на которых также может быть получен
диффузионный ток.
Наиболее удобным из твердых электродов является платиновый
электрод. Перенапряжение водорода на платине невелико, поэтому
водород восстанавливается при потенциале —0,1 в. Это ограничивает
использование платины в отрицательной области потенциалов. Но зато
платина не окисляется при анодной поляризации электрода до
потенциала выделения кислорода, т. е. до +1,1 Ь 1,3 в (в зависимости от
условий).
Твердые электроды могут быть неподвижными (стационарными)
или вращающимися (вибрирующими).
* При потенциалах более отрицательных, чем —2 в, выделяется водород, при
более положительных, чем +0,3 в, окисляется ртуть.
154 ГЛ. V. ПОЛЯРОГРАФИЯ И АМПЕРОМЕТРИЧЕСКОЕ^ ТИТРОВАНИЕ
Стационарные электроды. Сила тока на стационарном электроде
описывается уравнением:
I^FDSn Cs~C<> (И)
где F — число Фарадея;
D — коэффициент диффузии;
S— поверхность электрода;
п — яисло электронов, участвующих в электродной реакции;
Cs — молярная концентрация вещества в растворе;
С0 — молярная концентрация вещества у поверхности электрода;
6 — толщина диффузионного слоя.
Соответственно предельный ток выражается формулой:
Id = FDSn-^ (12)
Для того чтобы предельный ток был пропорционален
концентрации, толщина диффузионного слоя должна сохраняться постоянной.
Однако было установлено, что постоянство предельного тока на
неподвижном электроде достигается только спустя некоторое время, так
как первые несколько минут после наложения потенциала происходит
увеличение диффузионного слоя. Сначала диффузионный сЛой тонок
и сила тока быстро возрастает, а затем изменения концентраций
распространяются в глубь раствора (приэлектродный слой как бы
размывается) и толщина слоя увеличивается до тех пор, пока не достигнет
некоторой постоянной величины.
Эта особенность стационарных твердых электродов препятствует
их широкому применению в полярографии.
Вращающиеся электроды. Более широко применяются
вращающиеся микроэлектроды. Жидкость вокруг вращающегося электрода
непрерывно перемешивается, и в результате этого около приэлектрод-
ного слоя поддерживается высокая концентрация вещества.
Изменения концентрации вещества, возникающие в результате электролиза,
не распространяются в глубь раствора, а локализуются в тонком при-
электродном слое.
Нернст предположил, что внутри диффузионного слоя скорость
движения жидкости равна нулю. Тогда сила тока может быть
вычислена по той же формуле (11), что и для стационарных электродов
(жидкость неподвижна).
Однако это предположение Нернста маловероятно, так как
молекулярные силы не смогли бы удержать на поверхности электрода
слой толщиной Ю-2—Ю-3 см (по расчету он должен быть примерно
таким).
Согласно теории конвективной диффузии, разработанной Леви-
чем, у поверхности электрода имеется слой жидкости, в котором
скорость движения изменяется от нуля (непосредственно на поверхности
электрода) до v0. Этот слой называется граничным слоем (бгр).
Диффузионный слой расположен внутри этого слоя.
Концентрация в диффузионном слое изменяется с удалением от
поверхности электрода по линейному закону от 0 до С0. Диффузионный
слой значительно тоньше граничного слоя. Его можно рассчитать для
вращающегося дискового электрода, так как толщина диффузионного
слоя по всей поверхности такого электрода одинакова.
§ 2. ВОЗНИКНОВЕНИЕ ТОКА НА МИКРОЭЛЕКТРОДАХ
135
Для расчета плотности тока (?) на вращающемся дисковом
электроде Левич предложил уравнение:
* - 0,62/iFZ)2/3co1/2v,/6 (Cs - Со) (13)
где д,Р,ДС8иС0 — см. формулу (11);
а> — угловая скорость вращающегося диска;
v — кинематическая вязкость.
Обычно в полярографии используют игольчатые электроды» в
которых толщина диффузионного слоя в различных точках поверхности
электрода различна. Поэтому уравнение Левича позволяет только
качественно оценивать влияние различных факторов на величину силы
тока, получаемого на игольчатом электроде.
Факторы, влияющие на силу тока на твердом вращающемся
электроде. Из уравнения Левича следует, что, как и в случае ртутного
капельного электрода, сила тока увеличивается с увеличением числа
электронов п, коэффициента диффузии D и концентрации иона,
восстанавливающегося или окисляющегося на электроде. Кроме того, сила
тока растет с увеличением скорости вращения электрода. Однако, как
установлено опытным путем, при увеличений числа оборотов свыше
600 об/мин сила тока практически не возрастает^
Сила тока зависит и от температуры, так как при изменении
температуры изменяется коэффициент диффузии D и кинематическая
вязкость V.
Влияет на силу тока состав и концентрация фона. Это
объясняется тем, что от состава фона зависит ионная сила раствора и его
цязкость. В свою очередь ионная сила влияет на скорость переноса
ионов, а вязкость — на коэффициент диффузии ионов и на толщину
диффузионного слоя.
Сила тока тем больше, чем больше поверхность электрода. Однако
при слишком большой поверхности нарушается пропорциональность
Между силой тока и концентрацией иона, дающего электродную реакцию.
Платиновый электрод может быть использован в положительной
области потенциалов (до +1,3 в). Для определения катионоб он
применяется редко, так как поверхность его изменяется при выделении на
нем металлов. Кроме того, большинство металлов выделяется при
отрицательных потенциалах, платиновый же электрод не может быть
использован в таких условиях. Иногда для этой цели используются
твердые амальгамированные электроды, на которых перенапряжение
водорода так же велико, как и на ртути.
Из металлов на платиновом электроде могут быть определены
серебро, золото и ртуть.
Более целесообразно проводить на платиновом электроде
процессы, происходящие при положительных потенциалах и не
сопровождающиеся выделением металла на электроде. К таким процессам
относится восстановление ионов из высших валентностей в низшие
(например, СгОГ~>Сг+++) и окисление (например, 2Вг-->Вг2). Используются
также реакции окисления органических веществ.
При работе на твердых электродах отсутствуют осцилляции, что
увеличивает точность и быстроту отсчетов. Большой недостаток
твердых электродов в том, что при работе с ними воспроизводимость
определений хуже, чем при использовании ртутных электродов, так как
поверхность электрода трудно сохранить всегда в неизменном состоянии.
Особенно это относится к амальгамированным электродам.
Важным преимуществом твердых электродов является их
безвредность по сравнению со ртутными электродами.
156 ГЛ. V. ПОЛЯРОГРАФИЯ И АМПЕРОМЕТРИЧЕСКОЕ ТИТРОВАНИЕ
Б. АППАРАТУРА И ТЕХНИКА ВЫПОЛНЕНИЯ
ПОЛЯРОГРАФИЧЕСКОГО АНАЛИЗА
§ 3. Полярографы
Полярографические определения проводят на визуальных и
автоматических полярографах.
Схема полярографической установки показана на рис. 46. При
помощи внешнего источника тока / (например, батареи аккумуляторов на
4—6 в) напряжение подается на реохорд <3, с которого подвижным
контактом 4 можно снимать нужное напряжение и подавать его на
электроды 7 и 9. Сила тока в цепи контролируется чувствительным
гальванометром 5. Так как при максимальной чувствительности
гальванометра нельзя снимать полярограммы в растворах различных
концентраций, параллельно гальванометру подключают градуированное
сопротивление (шунт) 6.
Визуальные полярографы. В качестве примера рассмотрим поляро-
граф Геоприборцветмет ПВ-5. Он смонтирован в деревянном ящике.
Все ручки для управления прибором расположены на панели. Схема
полярографа аналогична схеме, показанной на рис. 46.
Аккумуляторная батарея (обычно 4—6 в) присоединяется к
клеммам полярографа. Напряжение, подаваемое на реохорд, регулируется
с помощью реостата и измеряется вольтметром.
Электроды присоединяются к соответствующим клеммам
полярографа. На электроды напряжение подается через реохорд, на шкале
которого нанесены деления (обычно 100); напряжение на электродах
постепенно увеличивают, вращая ручку реохорда. Каждое деление
реохорда соответствует 0,01 всего напряжения на реохорде (например,
0,04 в при общем напряжении на реохорде, равном 4 в). Напряжение
на электродах может быть также проконтролировано вольтметром
(после переключения).
Для измерения силы тока, проходящего через электролизер во
время полярографирования, применяют зеркальный гальванометр с
чувствительностью 10~9 а на 1 мм/м. Гальванометр установлен на
кронштейне на высоте приблизительно 1,5 м от поверхности стола, на
котором расположен полярограф. Зеркальная шкала гальванометра
укрепляется на уровне глаз работающего так, чтобы луч света,
отраженный от зеркальца гальванометра, падал на середину шкалы. Для
понижения чувствительности гальванометра (при сравнительно
больших концентрациях растворов анализируемых веществ) имеется шунт.
Включение и установка его на определенную чувствительность
проводится при помощи соответствующей ручки на панели прибора.
Для установки светового зайчика на требуемом делении шкалы
гальванометра (чаще, на «0») служит элёктрокорректор.
Электрокорректор — это устройство, с помощью которого в рамку гальванометра
подается э. д. с. того или иного направления, благодаря чему
происходит передвижение светового зайчика вдоль шкалы. Питается
электрокорректор от элемента с напряжением 1,5 в.
Для уничтожения «конденсаторных» или «остаточных» токов,
образующихся на поверхности ртутной капли, в полярографе имеется
«компенсатор»— устройство, при помощи которого на электролизер подается
ток, обратный по направлению току, протекающему в электролизере.
Для изменения поляризации электродов (катодной на анодную
или обратно) в полярографах обычно имеется соответствующий
переключатель.
§ 3. ПОЛЯРОГРАФЫ
157
Иногда заранее необходимо установить определенную высоту
волны. Для этой цели служит корректор высоты волны, позволяющий
изменять (при помощи дополнительных реостатов) силу тока,
протекающего через гальванометр.
Автоматические полярографы. Из автоматических полярографов
очень распространен полярограф системы Гейровского.
Принцип действия его основан на том, что при непрерывном
изменении напряжения на электродах световой зайчик гальванометра
движется вдоль щели вращающегося барабана со светочувствительной
бумагой и, таким образом, автоматически записывается кривая
зависимости силы тока от напряжения.
Рис. 46. Схема полярографической
установки:
/ — батарея аккумуляторов; 2 — вольтметр;
3 — реохорд; 4 — подвижной контакт; 5 —
гальванометр М-21; 6 — шунт; 7 — ртутный
капельный электрод; 8 — электролизер;
9—ртутный анод.
Рис. 47. Схема измерения тока
электролитической ячейки на электронном полярографе:
/ — эталонное сопротивление; 2 — электролитическая
ячейка; 3 — сопротивление; 4 — измерительный реохорд;
5— задающий реохорд.
Подробное описание прибора можно найти в руководстве,
прилагаемом к прибору.
В автоматическом полярографе LP-55 также производится
фотографическая запись полярограмм. Воспроизводимость определения в
этом полярографе увеличена благодаря замене подвижного потенцио-
метрического барабана неподвижным потенциометром с движком.
Скорость изменения напряжения, подаваемого на электроды, изменяется в
широких пределах.
В электронных самопишущих полярографах, например
электронном полярографе ПА-3, полярограммы автоматически записываются
чернилами на диаграммной бумаге. Схема измерения тока
электролитической ячейки на электронном полярографе приведена на рис. 47.
Для плавного подъема напряжения на ячейке электролизера
служит задающий реохорд 5. Движок реохорда с постоянной скоростью
перемещается электродвигателем. При достижении максимального
значения напряжения (1 или 2 в) на задающем реохорде электродвигатель
автоматически выключается. Одновременно с перемещением движка
реохорда барабан лентопротяжного механизма протягивает
диаграммную бумагу для записи полярограммы.
Сила тока, проходящего через ячейку электролизера, определяется
по падению напряжения на постоянном эталонном сопротивлении 19
158
ГЛ. V. ПОЛЯРОГРАФИЯ И АМПЕРОМЕТРИЧЕСКОЕ ТИТРОВАНИЕ
включенном последовательно с ячейкой. Падение напряжения на
эталонном сопротивлении измеряется компенсационным методом — по
падению напряжения на сопротивлении 3, включенном так же, как и
эталонное, последовательно с электролизером. Через это сопротивление
пропускается ток, проходящий через измерительный реохорд 4,
который соединен с электронным усилителем, в направлении, обратном
направлению тока в электролизере. Если падение напряжения на
сопротивлении 3 и эталонном сопротивлении / от тока электролизера равно
падению напряжения на сопротивлении 3 от тока, идущего с
измерительного реохорда 4, то напряжение на входе усилителя будет равно
нулю. Если ток электролизера изменится, то на входе усилителя
появится напряжение разбаланса. Оно затем сильно увеличится
усилителем и вызовет вращение электродвигателя включенного на выходе
усилителя, и перемещение движка реохорда 4
в сторЬну уменьшения разбаланса. Когда
движок реохорда дойдет до положения, при котором
напряжение разбаланса будет равно нулю,
двигатель остановится. Таким образом, каждому
значению тока в электролизере соответствует
определенное положение движка реохорда 4.
Движок реохорда 4 связан с записывающим
устройством.
Для измерения силы тока в широком
интервале с большой точностью в схему включен
набор различных эталонных сопротивлений.
Полярограф ПА-3 дает возможность
снимать также дифференциальные полярограммы,
Р 48 Л dxb показывающие зависимость производной тока
циальная'поляро??аеммНа! по напряжению от напряжения (рис. 48).
Высота пиков пропорциональна концентрации
вещества, расстояние их от начала координат характеризует природу
вещества.
Съемка дифференциальных полярограмм рекомендуется в тех
случаях, когда проводится анализ растворов, содержащих несколько
веществ с близкими потенциалами восстановления.
Электронные полярографы дают возможность повысить точность
и чувствительность полярографических определений. Эти полярографы
не чувствительны к вибрации, поскольку в их схемы не включены
гальванометры.
§ 4. Электролитическая ячейка
Схема электролитической ячейки представлена на рис. 49.
Анализируемый раствор наливают в сосуд для электролиза
(электролизер) 1. Капилляр 3 резиновой трубкой соединен со стеклянной
грушей 5, которая служит резервуаром для ртути. От высоты положения
груши зависит скорость вытекания ртути из капилляра. Грушу
закрепляют в нужном положении на штативе. В ртуть опущена стеклянная
трубка с платиновым контактом, с помощью которого ртутный катод
присоединяют к соответствующей клемме (—) полярографа. Иногда
на дно электролизера наливают ртуть и в нее опускают стеклянную
трубку с платиновым контактом для присоединения к клемме ( + )
прибора. Но чаще применяют выносные электроды сравнения:
каломельные, меркуриодидный, хлорсеребряный и др. В этом случае
электролизер соединяют с электродом сравнения стеклянной трубкой (солевым
§ 5^ ЭЛЕКТРОЛИЗЕРЫ
159
мостиком), наполненной насыщенным раствором электролита (обычно
КО), и контакт электрода присоединяют к клемме ( + ) полярографа*
Иногда электролитический ключ заполняют агар-агаром,
насыщенным ксь
Капилляры с внутренним диаметром от 0,01 до 0,05 мм
изготавливаются из толстостенного стекла. Период капания должен быть 2—<
3 сек; иногда применяют капилляры с периодом в 1,5 сек.
§ 5. Электролизеры
В качестве электролизера может быть использован химический
стакан емкостью около 30 мл. Таким электролизером можно йользо-
н2
Рис. 49.
Электролитическая ячейка с каломельным
электродом сравнения:
/ — электролизер; 2 — вводная
трубка для газа; 3 —
капилляр; 4 — резиновая трубка;
5— стеклянная груша с
ртутью; 6—каломельный
электрод сравнения; 7— солевой
мостик (электролитический
ключ).
Рис, 50. Электролизер
Гейровского.
Рис. 51. Электролизер
с ртутным анодом для
серийных измерений:
/ — трубка для контакта; 2 —
сливная трубка для ртути;
3 —сливная трубка для
раствора; 4 — платиновая
проволока.
ваться в тех случаях, когда раствор для удаления кислорода
продувают двуокисью углерода. Поскольку двуокись углерода тяжелее
воздуха, электролизер может быть открытым, слой СОг над раствором
предохраняет его от соприкосновения с кислородом воздуха.
Если для продувания раствора используется азот или водород,
применяют закрытые электролизеры. Из них довольно
распространенным является электролизер, предложенный Гейровским (рис. 50).
Верхнее отверстие электролизера закрыто пробкой с отверстием
для капилляра. Сбоку в электролизере есть трубки для ввода и вывода
газа.
Электролизер соединен с выносным анодом. В тех случаях, когда
работают с ртутным анодом, ртуть наливают на дно и к ней через
боковой отвод подводится платиновая проволочка для контакта. Для
работы с ртутным анодом очень удобен электролизер, показанный на
160 ГЛ. V. ПОЛЯРОГРАФИЯ И АМПЕРОМЕТРИЧЕСКОЕ ТИТРОВАНИЕ
рис 51. Конструкция электролизера позволяет сливать анализируемый
раствор после титрования, не выливая каждый раз ртуть, которая
служит анодом.
§ 6. Электроды сравнения
Электрод сравнения должен иметь поверхность, неизмеримо
большую, чем индикаторный электрод. Если поверхность электрода
сравнения будет мала, то плотность тока может стать настолько большой, что
потенциал электрода изменится (электрод поляризуется).
Поляризация же электрода сравнения вызовет изменение потенциала
индикаторного электрода и, как следствие, нарушение пропорциональности
между током и потенциалом.
Наиболее употребительны следующие электроды сравнения:
каломельные (децинормальный, однонормальный и насыщенный); меркур-
иодидный, меркурсульфатный и хлорсеребряный.
Устройство насыщенного каломельного электрода описано выше
(см. стр. 149). Потенциал его по отношению к нормальному
водородному электроду равен +0,247 в.
Децинормальный и однонормальный каломельные электроды
отличаются от насыщенного концентрацией раствора КО. Их потенциалы
равны соответственно +0,337 в и +0,284 в.
Меркурсульфатный электрод представляет собой сосуд, на дне
которого находится металлическая ртуть, над ней — паста из
металлической ртути и сульфата ртути (I) и 2 н. раствор серной кислоты.
Потенциал его составляет +0,682 в.
В хлорсеребряном электроде присутствует металлическое серебро,
покрытое пленкой хлорида серебра, и раствор хлорида калия.
Потенциал его равен +0,290 в.
Механизм действия перечисленных электродов и насыщенного
каломельного электрода аналогичен.
В меркуриодидном электроде металлическая ртуть находится под
насыщенным раствором КС1 (100 мл), в котором растворено 4,2 г
иодида калия и 1,3 г иодида ртути (II).
В меркуриодидном электроде нет осадка, как в других электродах
сравнения. Источником ионов ртути является комплекс K2[HgI4]. Когда
этот электрод служит анодом, образующиеся Hg-^-noHbi связываются
с иодид-ионами в комплекс [Hgl4]"~. Поскольку в растворе всегда
имеется большой избыток иодида и комплексного соединения K2[HgI4],
концентрация этих веществ заметно не изменяется, и концентрация
Hg++-HOHOB, образующихся при диссоциации комплекса, остается
практически постоянной.
Потенциал меркуриодидного электрода равен +0,02 в (по
отношению к нормальному водородному электроду).
§ 7. Снятие полярограммы на полярографической установке
с визуальным полярографом
Полярограммы снимают в следующем порядке.
Наливают в предварительно высушенный электролизер
испытуемый раствор и удаляют кислород, пропуская водород, азот или
двуокись углерода или добавляя Na2S03.
Уравнительную грушу, предварительно наполненную ртутью,
поднимают на штативе так, чтобы ртуть из капилляра начала капать, и
устанавливают скорость капания ртути 10 капель в 15 сек (скорость
вытекания ртути регулируют высотой положения груши).
МЕТОДЫ КОЛИЧЕСТВЕННОГО ПОЛЯРОГРАФИЧЕСКОГО АНАЛИЗА 161
Включают вольтметр и с помощью реостата устанавливают
необходимое напряжение на реохорде (обычно 4 в). Переключатель
вольтметра при этом установлен так, чтобы вольтметр показывал
напряжение на реохорде.
Устанавливают шунт гальванометра на наименьшую
чувствительность, включают осветитель гальванометра и сам гальванометр и при
помощи корректора ставят световой индекс (зайчик) на нулевое
деление шкалы. Постепенно увеличивают напряжение на электродах,
поворачивая ручку реохорда, и наблюдают за движением светового зайчика
по шкале. Если он пройдет лишь небольшую часть шкалы, когда
указатель реохорда покажет 100 делений, это означает, что
чувствительность гальванометра недостаточна для измерения данной
концентрации раствора. Тогда устанавливают шунт гальванометра на большую
чувствительность и снова, увеличивая напряжение, наблюдают за
движением зайчика по шкале. Так поступают до тех пор, пока не подберут
такого положения ручки шунта, при котором зайчик пробегает почти
всю шкалу при максимальном числе делений реохорда. При этом
должно наблюдаться заметное ускорение движения зайчика при
потенциале восстановления данного иона (волна).
Проверяют положение зайчика при нулевом положении указателя
реохорда, если он сместился с нуля, то устанавливают его снова на
нуль с помощью электрокорректфра.
Приступая к полярографированию (съемке полярограммы),
постепенно увеличивают напряжение поворотом ручки реохорда и
записывают показания на шкале, соответствующие каждому делению
реохорда. Так как при работе с ртутным капающим электродом всегда
наблюдаются большие или меньшие осцилляции (зайчик колеблется
между какими-либо двумя делениями шкалы), берут среднее
арифметическое из этих двух делений.
Напряжение увеличивают до тех пор, пока зайчик после скачка,
вызванного восстановлением определяемого иона, снова не начнет
двигаться по шкале медленно.
Окончив полярографическую съемку, возвращают движок
реохорда на нулевое деление, выключают гальванометр, осветитель
гальванометра и вольтметр. Опускают уравнительную грушу так, чтобы ртуть
перестала капать. Ставят на место электролизера с раствором стакан
с дистиллированной водой так, чтобы капилляр был опущен в воду.
Наносят показания гальванометра (по оси ординат) и напряжение
(по оси абсцисс) на миллиметровую бумагу. Полученные точки
соединяют плавной кривой и измеряют высоту волны.
§ 8. Методы количественного полярографического анализа
Метод расчета. Метод расчета заключается в том, что измеряют
величину диффузионного тока, а также т и т и рассчитывают
характеристику капилляра m2V/e. В уравнение Ильковича подставляют
полученные значения и значение коэффициента диффузии Д взятое из
таблиц, и вычисляют концентрацию определяемого вещества.
605/ш2/зт1/б?1/2
Однако этот метод применяется редко, он интересен главным
образом с теоретической точки зрения. Дело в том, что
определение т (взвешиванием) требует много времени, кроме того* для получе-
162 ГЛ. V, ПОЛЯРОГРАФИЯ И АМПЕРОМЕТРИЧЕСКОЕ ТИТРОВАНИЕ
ния точных результатов измерения необходимо вести при постоянной
температуре, что усложняет работу. Главным же недостатком метода
является то, что приведенные в таблицах значения D недостаточно
точны, особенно для сравнительно концентрированных растворов.
Метод калибровочных кривых. По этому методу прежде чем
приступить к полярографированию раствора определяемого вещества с
неизвестной концентрацией, готовят ряд эталонных растворов данного
вещества, концентрация которых точно известна, и снимают полярограм-
мы растворов. Строят калибровочный график, откладывая по оси
ординат высоты волн эталонных растворов, а по оси абсцисс —
соответствующие концентрации. Затем снимают полярограмму исследуемого
раствора и, определив U (или высоту волны при той же чувствитель-
йости гальванометра), находят по графику концентрацию
анализируемого раствора,
Полярограмма определяемого вещества должна быть снята в
таких же условиях, как и полярограммы эталонных растворов, т. е. с
теми же капилляром, скоростью капания, высотой столба ртути и при
той же температуре. Состав раствора должен быть по возможности
близок к составу эталонных растворов (в эталонные растворы
добавляют те же примеси).
Метод стандарта. Метод стандарта отличается от метода
калибровочных кривых тем, что здесь готовят только один эталонный раствор,
снимают его полярограмму и полярограмму анализируемого раствора
и рассчитывают концентрацию определяемого вещества по формуле,
приведенной ниже.
В отличие от условий, принятых в методе калибровочных кривых,
в этом случае эталонный раствор готовят каждый раз. Применение
метода стандарта возможно лишь тогда, когда строго соблюдается
пропорциональность между силой диффузионного тока и концентрацией
в широком диапазоне концентраций.
Если концентрация определяемого иона приблизительно известна,
то готовят стандартный раствор этого иона более или менее близкой
концентрации, причем состав и концентрация фона должны быть
такими же, как в анализируемом растворе.
Преимущество метода стандарта перед методом калибровочных
кривых в том, что здесь легче создать одинаковые условия при съемке поляро-
грамм стандартного (эталонного) и анализируемого растворов, так как
полярограммы снимают почти одновременно (тотчас же друг за другом).
Обычно снимают полярограмму стандартного раствора, а затем
при той же чувствительности гальванометра — полярограмму
анализируемого раствора. Зная концентрацию стандартного раствора (Сст)
и определив высоту волны стандартного (hCT) и анализируемого (hx)
растворов, можно рассчитать концентрацию определяемого вещества по
формуле: hxC„
ljc = —l— (15)
Пет
Метод добавок. Метод добавок заключается в том, что сначала
снимают полярограмму анализируемого раствора, затем в этот же
электролизер прибавляют по каплям стандартный раствор
определяемого иона с точно известной концентрацией, с таким расчетом, чтобы
волна возросла примерно вдвое. Снимают полярограмму при той же
чувствительности гальванометра и делают расчет по формулам:
Сх _ hx
(" СТ "СТ
И
Act = ^общ — hx (16)
§ 9. ПРАВИЛА ТЕХНИКИ БЕЗОПАСНОСТИ ПРИ РАБОТЕ -С РТУТЬЮ 163
где /гобщ ~ суммарная высота волны определяемого вещества в анализируемом и
прибавляемом растворах, мм;
Сст—концентрация добавляемого иона в электролизере.
Концентрацию определяемого иона, введенного в электролизер в
виде стандартного раствора, вычисляют по формуле:
ССТУСТ
Сст= VcT+Vx (17)
где VCt ~ количество стандартного раствора, добавленное в электролизер, мл;
Ух — объем анализируемого раствора в электролизере;
С?т — первоначальная концентрация стандартного раствора, мг{мл.
Метод добавок имеет то преимущество перед методом стандарта и
методом калибровочных кривых, что дает возможность исключить
приготовление стандартных растворов с фоном, точно совпадающим с
составом фона анализируемого раствора. Благодаря большой
концентрации стандартного раствора в электролизер прибавляют лишь несколько
капель этого раствора, причем объем раствора в электролизере и
концентрация фона практически не изменяются.
§ 9. Правила техники безопасности при работе
с металлической ртутью
Пары ртути чрезвычайно ядовиты, поэтому работав с ней нужно
очейь осторожно, так, чтобы избегать вдыхания ее паров.
Полярографическая установка должна находиться около хорошо
действующего вытяжного устройства или в вытяжном шкафу. Еще
лучше установить ее в специальном ящике из прозрачной пластмассы
(боксе) с поднимающейся передней дверцей; к задней стенке бокса должна
быть подведена вытяжная труба.
Пол в помещении, в котором работают со ртутью, должен быть
покрыт линолеумом, загнутым кверху у стен. Такой пол не имеет щелей,
и ртуть, случайно попавшая на пол, может быть собрана до последней
капли. Под полярографической установкой должен находиться
противень с загнутыми краями и гладкой поверхностью (лучше,
эмалированный).
Пролитую на пол или на стол ртуть необходимо немедленно и
тщательно собрать. Собирать ртуть можно медной амальгамированной
пластинкой или трубкой с оттянутым кончиком, соединенной шлангом с
наполненной ватой склянкой Тищенко, из которой откачивают воздух
водоструйным насосом. Можно собирать ртуть совочком из бумаги.
В случае попадания ртути в какую-либо щель рекомендуется
насыпать в нее серы. Образуя на поверхности ртути пленку, сера
препятствует испарению ртути.
Помещение, в котором работают со ртутью, необходимо часто
проветривать.
В. НОВЫЕ НАПРАВЛЕНИЯ В ПОЛЯРОГРАФИИ
Наряду с классической полярографией, которая широко
применяется в научно-исследовательских и заводских лабораториях, за
последнее время появилось много новых направлений в полярографии.
Рассмотоим важнейшие из них.
164 ГЛ. V. ПОЛЯРОГРАФИЯ И АМПЕРОМЕТРИЧЕСКОЕ ТИТРОВАНИЕ
§ 10. Амальгамная полярография с накоплением
Чувствительность классического метода полярографии во многих
случаях недостаточна. Новые отрасли техники предъявляют особые
требования к чистоте материалов и соответственно к методам ее контроля.
В связи с этим появилась необходимость в разработке методов,
позволяющих определять примеси при содержании их в анализируемом
веществе до 10~5— 10"7%.
Одним из таких методов является метод амальгамной
полярографии.
Принцип метода. Сущность метода состоит в том, что определяемое
вещество в концентрации 10~7—10~9 моль/л некоторое время
подвергается электролизу на стационарной ртутной капле при
контролируемом потенциале, несколько более отрицательном (на 0,2—0,3 в), чем
потенциал полуволны определямого иона. Определяемый элемент при
этом концентрируется в ртутной капле в виде
амальгамы. Затем выделенный элемент анодно
растворяют при потенциале, непрерывно
изменяющемся от значения, при котором проводилось
катодное выделение элемента на ртути, до более
положительных потенциалов (обычно до нуля).
Протекающий в процессе окисления амальга-
*" мы ток автоматически записывается или фотогра-
Рис. 52. Анодная по- фируется регистрирующим полярографом (напри-
лярограмма при мер полярографами Гейровского LP-55 или ПА-3).
амальгамной поляро- г» г г -г г /
графин с накоплением. При этом кривая анодного тока имеет вид
характерного зубца (пика), глубина которого
пропорциональна концентрации определяемого иона в растворе при условии
постоянства других факторов (рис. 52).
Устройство ртутного катода. Схема установки для амальгамной
полярографии подобна применяемой для обычной классической
полярографии, но ртутный катод устроен иначе.
В литературе описано пять типов стандартных ртутных капельных
электродов для амальгамной полярографии.
1. Подвешенная ртутная капля. Одна-две капли ртути из обычного
капилляра с помощью лопаточки подвешиваются на металлическую
проволоку — платиновую, золотую или серебряную.
2. Лежащая капля. Получается выдавливанием определенной
порции ртути из капилляра, соединенного с резервуаром, наполненным
ртутью.
3. Висящая капля. Получается выдавливанием ртути из капилляра
с помощью винтового поршня.
4- Капля ртути электролитически выделяется на платиновой
проволоке в определенных условиях.
5. Чашеобразный ртутный электрод. В маленькую чашечку капают
определенное количество капель ртути, причем чашечку
предварительно гидрофобизпруют метилтрихлорсиланом.
Все эти виды электродов имеют достоинства и недостатки.
Для того чтобы метод мог быть использован в аналитической
химии, в первую очередь необходимо иметь электрод со строго
воспроизводимой величиной поверхности ртутной капли. Небольшая ошибка в
размере капли уже дает существенную ошибку в высоте пика. Отсюда
ясно, насколько важно стандартизировать каплю, чтобы получить
воспроизводимые результаты. Это достигается различными методами,
например, определенным углом поворота винтового поршня.
§ 10. АМАЛЬГАМНАЯ ПОЛЯРОГРАФИЯ С НАКОПЛЕНИЕМ 165
Уравнение Шевчика. Величина максимального тока в методе
амальгамной полярографии (равная глубине зубца) определяется уравнением
Шевчика:
/макс = 2175/г3/2^1/2?)1/2С (18)
где /Макс~ глубина зубца, а;
217 — постоянный коэффициент;
5 — площадь электрода, см2;
п — число электронов, участвующих в электродном процессе;
у—скорость изменения потенциала, в\сек\
D — коэффициент диффузии, см2/сек;
С—концентрация восстанавливающегося иона, моль[л.
Из уравнения Шевчика видно, что с увеличением поверхности
электрода сила тока увеличивается. Но слишком большое увеличение
поверхности ртути ведет к расширению пиков. Это явление вызывает
трудности в исследовании смеси ионов металлов вследствие возможного
слияния анодных зубцов. Обычно применяют электроды диаметром
0,8—1,5 мм.
Из уравнения Шевчика также следует, что глубина зубца должна
увеличиваться пропорционально корню квадратному из скорости
изменения потенциала.
В обычных регистрирующих полярографах скорость изменения
потенциала составляет 0,2—0,4 в/мин. Чувствительность метода
повышается с увеличением скорости изменения потенциала до 1,2 в/мин.
Однако увеличение скорости изменения потенциала
ограничивается инерционностью прибора, регистрирующего величину тока, и
особенно емкостными токами.
Влияние длительности электролиза. Очень интересным является
вопрос о влиянии длительности электролиза на величину анодного тока.
Здесь возможны два случая.
1. Объем раствора достаточно велик, радиус капли мал. Глубина
анодного пика линейно возрастает со временем электролиза, так как
при более продолжительном электролизе накапливается больше
вещества в амальгаме.
2. Объем раствора мал (2—5 мл) и капля относительно велика.
Глубина анодного зубца быстро достигает предела, так как наступает
обеднение раствора ионами восстанавливающегося вещества и дальнейшее
увеличение времени электролиза не увеличивает величину зубца.
Учитывая сказанное, работают обычно с достаточно большими
объемами растворов. В этом случае, увеличивая время электролиза,
можно повысить величину предельного тока, т. е. чувствительность
определения. Величина предельного тока при окислении амальгамы
пропорциональна концентрации вещества в амальгаме, а эта концентрация
в свою очередь зависит от концентрации ионов в растворе и времени
электролиза. Поэтому время электролиза устанавливают в зависимости
от концентрации анализируемого вещества. Для получения анодных
пиков удовлетворительной глубины при концентрации раствора около
10~7 моль/л достаточно 3—5 мин, для 10~8 моль/л—10—20 мин и для
10~9 моль/л — 40—60 мин.
Однако и при большом объеме раствора может оказаться, что
линейная зависимость тока от времени электролиза сохраняется только
до определенного времени, после чего ток достигает предельного
значения. Это бывает в тех случаях, когда выделяемый на катоде элемент
не образует амальгамы (например, никель), и увеличение времени
электролиза не оказывает влияния на величину тока. Время электролиза
166 ГЛ. V. ПОЛЯРОГРАФИЯ И. АМПЕРОМЕТРИЧЕСКОЕ ТИТРОВАНИЕ
следует при этом устанавливать опытным путем, построив кривую
зависимости анодного тока от времени электролиза.
Глубина анодного зубца при постоянстве условий опыта находится
в линейной зависимости от концентрации определяемых ионов, что дает
возможность определять их количественно.
При изменении концентрации ионов металла в растворе потенциал,
при котором начинается анодный зубец, изменяется, но потенциал
половины высоты зубца на отрицательной его ветви остается постоянным.
Значение этого потенциала близко к значению катодного потенциала
полуволны на ртутном капельном электроде, оно не зависит от
концентрации ионов металла в растворе и скорости изменения потенциала.
Измеряя потенциал половины высоты зубца, можно качественно
определить наличие тех или иных ионов в растворе.
Рассмотрим, например, методику количественного определения ионов свинца в
растворе.
Помещают в электролизер 25 мл 10~8 М раствора соли свинца на фоне I M
растворов СНзСООН и CH3COONa. Через раствор в течение 30 мин продувают очи*
щенный азот для удаления кислорода (последний окисляет амальгаму), затем в
течение 15 мин при потенциале —0,8 в и перемешивании магнитной мешалкой проводят
выделение металла на ртутном катоде. Через 30—45 сек после выключения мешалки
потенциал катода автоматически со скоростью 0,4 о/мин изменяют от —0,8 в до нуля.
Затем измеряют глубину зубца.
Такой же обработке подвергают стандартный раствор и рассчитывают концентра^
цию РЬ++ на основании пропорциональности концентрации и глубину зубца.
Преимущества амальгамной полярографии. Метод амальгамной
полярографии дает возможность определять одновременно несколько
элементов при их совместном присутствии в растворе. Если вести
электролиз при потенциале более отрицательном, чем катодный потенциал
полуволны наиболее электроотрицательного элемента, то и этот
элемент, и все более электроположительные ионы выделяются на ртути
с образованием смешанной амальгамы. На анодной полярограмме
получается несколько зубцов, глубина которых пропорциональна
концентрации каждого из элементов в растворе (рис. 53).
Если даже содержание одного из двух элементов преобладает, но
потенциалы анодных зубцов достаточно различаются между собой (на
200 мв), то возможно совместное определение этих элементов. В случае
преобладающего содержания более электроотрицательного элемента
можно вести электролиз при потенциале более положительном по
сравнению с потенциалом анодного зубца этого элемента. Тогда в
амальгаму перейдет только микропрймесь более электроположительного
элемента, который можно определять по анодному зубцу. Например, в
цинке или алюминии можно определять примеси (10^%) свинца,
сурьмы, висмута, меди и других элементов, проводя электролиз этих
металлов при потенциале более положительном, чем потенциал
восстановления алюминия или цинка.
В случае преобладающего (не очень большого) содержания более
электроположительного элемента, можно оба элемента перевести
электролизом в амальгаму и, получив зубец более электроотрицательной
примеси, прекратить съемку полярограммы перед большим зубцом
второго элемента. При меньшей чувствительности гальванометра можно
получить также зубец и второго элемента.
Иногда путем подбора комплексообразующего реагента можно
изменить потенциалы восстановления обоих ионов и создать
благоприятные условия, например, такие, в которых потенциал элемента,
находящегося в большом количестве, оказывается более отрицательным.
§ 10. АМАЛЬГАМНАЯ ПОЛЯРОГРАФИЯ С НАКОПЛЕНИЕМ
167
л
| рь+4-
in***
i
i_i
-ом
'0,6
-ав
Рис. 53. Анодная полярограм-
ма раствора, содержащего
несколько катионов.
Устранения мешающего влияния элементов можно достигнуть,
применяя электрод в виде ртутной капли, подвешенной на металлический
контакт.
Некоторые элементы — цинк, кадмий, галлий — дают с амальгамой
золота интерметаллические соединения, которые окисляются при более
положительных потенциалах, чем чистая амальгама данного металла.
Например, на электроде с золотым контактом можно определять индий
в присутствии кадмия, так как индий не образует интерметаллического
соединения с золотом, а кадмий образует.
Вообще же образование интерметаллических соединений в
амальгаме (между определяемыми элементами) может привести к
неправильным результатам анализа, поскольку при выделении таких пар
металлов, как Ni и Zn, Sn и Ni, наблюдается
понижение анодного зубца первого металла
при наличии второго. Иногда появляется
третий зубец интерметаллического
соединения. Образование интерметаллических
соединений наблюдается при концентрациях
10~3—10~4 моль)л и мало заметно при
низких концентрациях порядка Ю-*7—
10~8 моль/л. Необходимо каждый раз
опытным путем устанавливать наличие или
отсутствие взаимного влияния металлов при
их совместном выделении в амальгаму.
При определении малых примесей в
различных веществах удобнее всего
пользоваться методом добавок. Так определяют, например, примесь свинца в
ацетате цинка. Сначала измеряют глубину зубца для анализируемого
раствора. Затем, стряхнув каплю ртути, на которой проводили
определение, и подготовив новую каплю такого же размера, добавляют в тот
же раствор строго определенное количество стандартного раствора
(несколько капель из микробюретки) и снова проводят электролиз и
съемку анодного пика при всех равных условиях.
Например, пусть глубина зубца для анализируемого раствора равна 37 мм, а с
добавкой стандартного раствора (Сст) она составляет 51 мм.
Тогда расчет проводят следующим образом:
51—37 ^ 37
v-<CT *~>Х
В методе амальгамной полярографии большое внимание следует
уделять очистке реактивов и воды. Химически чистые реактивы обычно
содержат примеси тяжелых металлов — цинка, свинца, меди в
количествах, которые препятствуют определению этих элементов при
содержании 10~5—10~6%. Очистить от тяжелых металлов многие
легколетучие вещества — воду, кислоты и другие можно перегонкой в кварце*
вом аппарате.
Для извлечения микропримесей металлов из реактивов широко
применяется экстракция, ионообменная хроматография, адсорбция и
электролиз.
При работе с обычной посудой происходит выщелачивание ионов
металлов, поэтому работают с кварцевой, полиэтиленовой и тефлоно-
вой посудой.
Метод амальгамной полярографии позволяет достичь увеличения
чувствительности на 3—4 порядка по сравнению с обычным
полярографическим методом с капающим ртутным электродом. Он применим для
168 ГЛ. V. ПОЛЯРОГРАФИЯ И АМПЕРОМЕТРИЧЕСКОЕ ТИТРОВАНИЕ
определения концентраций вещества от Ю-6 до 10~9 моль/л и дает
возможность определять в особо чистых реактивах и металлах
микропримеси порядка 10~5—10~6% с точностью 5—10%.
Кроме того, метод обладает большой разрешающей способностью,
применяя его можно определять несколько элементов из одного
раствора без их предварительного химического разделения даже в том
случае, когда концентрации этих элементов значительно различаются.
Полярография с накоплением может быть не только амальгамной.
За последнее время быстро развивается так называемая пленочная
полярография с накоплением. В качестве индикаторных электродов здесь
используются твердые электроды, главным образом графитовый.
Определяемые вещества накапливаются на электроде в чистом виде
(металлы) или в виде различных соединений, а затем происходит
растворение осадка при меняющемся потенциале.
§ 11. Осциллографическая полярография
Основное отличие осциллографической полярографии от
классической заключается в том, что в осциллографической полярографии
напряжение подается на электроды со скоростью изменения до
нескольких десятков вольт в 1 сек. Это дает возможность изучать процессы,
мгновенно протекающие на электроде (до
| _ jq_7 ceK^ a также повысить чувствительность,
/| V поскольку максимальный ток на осциллограммах
/ j \^^ в несколько раз больше диффузионного тока,
^ / , ^"^ получаемого на полярограммах.
J \ Для измерений используется прибор, назы-
* ваемый катодным осциллографом.
I -| — Принцип метода. Принцип работы электрон-
макс ного осциллографа заключается в том, что тон-
Рис. 54. Осциллограмма, кий пучок электронов в катодно-лучевой трубке
проходит через две пары расположенных
перпендикулярно пластин конденсаторов. Одна пара пластин отклоняет луч
в горизонтальном направлении. Величина отклонения пропорциональна
напряжению на электродах. Другая пара пластин отклоняет луч в
вертикальном направлении, на нее подается напряжение,
пропорциональное току, протекающему в электролизере. На экране осциллографа
можно наблюдать кривую зависимости тока от напряжения (рис. 54).
Вначале кривая идет почти параллельно оси абсцисс, затем при
достижении потенциала восстановления данного иона происходит резкое
увеличение силы тока, вызванное разрядом определяемого иона на
электроде. Сила тока достигает максимума, а затем уменьшается вследствие
понижения концентрации этих ионов в приэлектродном слое в
результате электролиза.
Максимальная высота пика пропорциональна концентрации
вещества, восстанавливающегося на электроде, а потенциал максимума
характеризует его природу.
В осциллографической полярографии используют электроды в виде
стационарной ртутной капли и ртутные капельные электроды.
Но при работе с ртутным капельным электродом необходимо
создать такие условия, при которых во время съемки осциллограммы
поверхность кзпли не изменяется. Поэтому развертка напряжения
подается в конечный момент жизни капли, когда величина ее в
течение некоторого времени остается практически постоянной.
§ 12. ПЕРЕМЕННОТОКОВАЯ ПОЛЯРОГРАФИЯ
169
Максимальная сила тока (в амперах) определяется уравнением
Шевчика — Рендлса:
/макс = Kn/2Dll2m2lH2/*v42C (19)
где v — скорость изменения напряжения, в;
/(—константа. Остальные обозначения даны выше (см. стр. 147 и 165).
Чувствительность метода. Чувствительность осциллографического
метода больше чувствительности обычной классической полярографии
и дает возможность определять вещества при их концентрации 10~5 —
10"6 моль/л. Однако разрешающая способность этих методов отличается
мало. Если в растворе присутствует несколько веществ, осциллографи-
ческая полярография не дает хороших результатов, так как на
осциллограмме высоты пиков веществ более электроотрицательных,
следующих за менее электроотрицательными, могут оказаться искаженными.
§ 12. Переменнотоковая полярография
Принцип метода. Переменнотоковая полярография отличается от
классической тем, что на электроды вместе с линейно и медленно
изменяющимся напряжением подается переменное квадратно-волновое
напряжение. Это дает возможность уменьшить почти до нуля
конденсаторный ток, который сильно искажает форму полярограмм при малых
концентрациях веществ, и благодаря этому увеличить чувствительность
и разрешающую способность полярографического метода.
Ток, обусловленный электродной реакцией определяемого вещества,
не снижается до нуля при наложении переменного напряжения, так как
он иначе изменяется во времени, чем конденсаторный. Ток,
возникающий при восстановлении вещества на электроде, пропорционален /~1/2,
а конденсаторный ток — e~tlRc (где R — омическое сопротивление
электролитической ячейки, с — емкость двойного слоя). Поэтому при
наложении квадратно-волнового напряжения в конце каждого полупериода
конденсаторный ток уменьшается до нуля, в то время как
диффузионный ток имеет некоторую определенную величину.
Полярограмма представляет собой график зависимости величины
переменного тока от постоянного напряжения. Она имеет форму пика.
Высота этого пика пропорциональна концентрации определяемого
вещества. Потенциал максимума зависит от природы вещества и
совпадает с потенциалом полуволны.
Величина максимального тока определяется формулой:
/макс = Kn2Dlf2ESC (20)
где К — константа;
п — число электронов, участвующих в реакции;
D — коэффициент диффузии;
Е— амплитуда переменного напряжения, в\
S — поверхность катода, см2\
С — концентрация раствора, моль/я.
Чувствительность метода. Переменнотоковая полярография
позволяет определять вещества при концентрации их в растворе порядка
Ю-7 моль/л.
Этим методом можно определять при одновременном присутствии
в растворе ионы с довольно близким значением потенциалов полуволн.
Вместо квадратно-волнового напряжения может быть
использовано переменное напряжение, изменяющееся синусоидально, так как
при этом конденсаторный и диффузионный токи сдвигаются по фазе
на л/4. Полярографы, основанные на использовании синусоидально
170 ГЛ. V. ПОЛЯРОГРАФИЯ И АМПЕРОМЕТРИЧЕСКОЕ ТИТРОВАНИЕ
изменяющегося напряжения, называются вектор-полярографами. Они
имеют примерно такую же чувствительность и разрешающую
способность, как и квадратно-войновые полярографы.
Существуют и другие новые направления в полярографии:
дифференциальная полярография, импульсная полярография, полярография с
применением каталитических волн и т. п. С описанием этих методов
можно познакомиться в книгах по полярографии.
Г. ПРАКТИЧЕСКИЕ РАБОТЫ С ПРИМЕНЕНИЕМ
ПОЛЯРОГРАФИЧЕСКОГО МЕТОДА АНАЛИЗА
§ 13. Примеры полярографических определений
Качественное и количественное определение катионов
при совместном присутствии
Задача заключается в качественном и количественном анализе
растворов, содержащих смеси ионов цинка, никеля, меди и кадмия.
Может быть десять вариантов таких смесей:
1) Cu++ + Zn++; 2) Cu++ + Cd++; 3) Cu++ + Ni++; 4) Zn++ + Cd++;
5) Zn++ + Ni++; 6) Cd++ + Ni++; 7) Cu++ + Zn++ + Cd++; 8) Cu++ + Zn++ + Ni++;
9) Cu++ + Cd++ + Ni++; 10) Cd++ + Zn++ + Ni++.
Обнаружение ионов цинка, никеля, меди и кадмия в смесях
Качественное обнаружение ионов проводится путем снятия поляро-
грамм растворов, содержащих эти ионы, относительно насыщенного
каломельного электрода сравнения, определения потенциалов полуволн и
нахождения затем по таблице ионов, которым соответствуют
найденные потенциалы полуволн. Полярограммы снимают на фоне
аммиачного буферного раствора. Для снятия полярограмм можно пользоваться
визуальным или автоматическим полярографом.
Методика определения. Анализируемый раствор помещают в
мерную колбу емкостью 100 мл, добавляют 20 капель 0,5%-ного раствора
желатины и доливают до метки раствором фона (эквимолярная смесь
1 М растворов аммиака и NH4C1).
В сухой электролизер помещают 20 мл этого раствора, взятого
пипеткой, и удаляют из него кислород, добавив в раствор 0,5 г сухого
Na2S03 и выдержав его 5 мин.
Соединяют электролизер солевым мостиком с каломельным
электродом и снимают полярограмму. Учитывая, что в растворе находится
несколько ионов, съемку полярограммы не прекращают при первом
замедлении движения светового зайчика, а снимают ее до потенциала
—2 е. При этом должно получиться несколько волн (не менее двух).
Находят потенциалы полуволн и определяют, какие ионы присутствуют
в растворе по приведенным ниже данным:
Катион Си++ Cd++ Ni++ Zn++
Еъ, в -0,24 -0,81 -1,09 -1,36
Определение ионов цинка, никеля, меди и кадмия в их смесях
Определение может быть проведецо методом стандарта, методом
калибровочных кривых и методом добавок. Наиболее целесообразно
проводить его методом стандарта, так как для этого могут быть
использованы полярограммы, полученные при качественном обнаружение
§ 13. ПРИМЕРЫ ПОЛЯРОГРАФИЧЕСКИХ ОПРЕДЕЛЕНИЙ
171
ионов. Необходимо только приготовить стандартные растворы тех
ионов, которые были обнаружены в данном растворе, и снять их
полярограммы.
Те же полярограммы могут быть использованы и в методе
калибровочных кривых, но этот метод обычно применяют в случаях, когда
имеют в виду многократно использовать полученные калибровочные
кривые для определения тех же ионов. При проведении одного анализа
не имеет смысла строить калибровочные кривые, так как это отнимет
много времени. Ниже приведена методика определения катионов по
методу стандарта.
Методика определения. Снимают полярограмму анализируемого
раствора, как указано выше. Для приготовления стандартных
растворов ионов, присутствующих в данном растворе, в мерные колбы
емкостью по 100 мл помещают точно по 1 мл (берут пипеткой или
микробюреткой) исходных растворов этих ионов, содержащих в 1 мл 1—
5 мг иона (Cu++, Cd++, Ni++ или Zn++). Добавляют в колбы по 20 капель
0,5%-ного раствора желатины и доливают до метки тем же аммиачным
буферным раствором, который использовали при снятии полярограммы
анализируемой смеси.
В сухой электролизер помещают точно по 10 мл этих растворов
(если их два) или по 6,7 мл (при наличии трех катионов в смеси),
добавляют 0,5 г сухого Na2S03 и через 5 мин снимают полярограмму
при той же чувствительности гальванометра и тех же условиях капания
ртути, при которых снимали полярограмму анализируемой смеси.
Измеряют высоты волн определяемых катионов на полярограмме,
полученной при качественном их определении, и высоты волн этих же
катионов на полярограмме стандартных растворов и делают расчет.
Расчет. Содержание (в мг) каждого из определяемых катионов (g)
в растворе вычисляют по формуле:
где Сст — концентрация определяемого катиона в стандартном растворе в
электролизере, мг\мл\
hx— высота волны определяемого катиона в анализируемой смеси, мм\
/гст— высота волны определяемого катиона в стандартном растворе, мм;
VK— общий объем исследуемого раствора, мл.
Определение кальция
Кальций восстанавливается на ртутном капельном электроде, но
волна получается с максимумом, который не подавляется обычными
поверхностно-активными веществами. Однако если концентрацию
кальция в растворе снизить до 1,5—1,7- 10~4 М, то в 0,005 М растворе
гидроокиси тетраметиламмония можно получить хорошо выраженные (без
максимумов) волны.
Понизить максимумы на полярограмма,х кальция можно также
добавлением спирта, а устранить их совсем — введением кроме спирта
небольших количеств (5%| от содержания кальция) солей бария.
Фоном в этом случае служит 0,15 М раствор иодида тетраэтил-
аммония в 50%-ном спирте, содержащий 5% хлорида бария.
Потенциал полуволны Са++-ионов в этих условиях равен —2,09 в.
Определение Са++-ионов более целесообразно проводить методом
добавок (в целях экономии дорогостоящего иодида тетраэтилам-
мония).
Методика определения. Анализируемый раствор помещают в
мерную колбу емкостью 25 мл и доводят до метки раствором фона, 15 мл
172 ГЛ. V. ПОЛЯРОГРАФИЯ И АМПЕРОМЕТРИЧЕСКОЕ ТИТРОВАНИЕ
полученного раствора, взятого пипеткой, помещают в сухой
электролизер. Азот или водород для удаления растворенного кислорода в данном
случае не пропускают, так как кислород восстанавливается значительно
раньше Са++.
Снимают полярограмму, затем добавляют в электролизер по
каплям из микробюретки стандартный раствор соли кальция, содержащий
1 мг Са++ в 1 мл, до тех пор, пока высота волны кальция не возрастает
примерно вдвое. Предварительно следует определить объем капли,
вытекающей из микробюретки.
Снимают снова полярограмму раствора при той же
чувствительности гальванометра, при которой снимали полярограмму без добавок.
Измеряют высоты волн и делают расчет.
Расчет. Содержание (в мг) кальция (g) вычисляют по формуле:
где gcr — количество ионов кальция, введенное в электролизер в виде стандартного
раствора, мг\
hx— высота волны Са++-ионов без добавки, нм\
^общ— высота волны после добавки стандартного раствора, мм\
Va — объем анализируемого раствора в электролизере, мл\
VK — общий объем исследуемого раствора, мл.
Определение индия (Ш)
Индий восстанавливается на ртутном капельном электроде как в
нейтральных, так и в кислых растворах на различных фонах. Хорошо
выраженные волны получаются на фоне 3 н. раствора
хлористоводородной кислоты. Потенциал полуволны индия в этих условиях равен
—0,60 в (по отношению к насыщенному каломельному электроду).
Определение может быть проведено методом стандарта, методом
калибровочных кривых и методом добавок. Ниже описана методика
определения индия по методу калибровочных кривых.
Методика определения. В пяти мерных колбах емкостью по 25 мл
готовят 5 стандартных растворов соли индия (III). Для этого помещают
в колбы 1, 2, 3, 4 и 5 мл исходного раствора соли индия (с
содержанием индия 1 мг/мл) *, приливают по 10—12 мл разбавленной (1 : 1)
хлористоводородной кислоты и доливают до метки водой. Для поляро-
графирования отбирают пипеткой по 15 мл каждого стандартного
раствора и помещают в соответствующий сухой электролизер, прибавляют
по 5 капель 0,25%-ного раствора столярного клея для подавления
максимума и полярографируют при напряжении на реохорде 0,9 в и
скорости вытекания ртути 10 капель в 15 сек.
Измеряют высоты волн на полученных полярограммах и строят
калибровочный график, откладывая по оси абсцисс содержание
1п+++ (л*г/15 мл). Анализируемый раствор соли индия также помещают
в мерную колбу емкостью 25 мл, прибавляют 10—12 мл
хлористоводородной кислоты (1:1) и доливают водой до метки. Отбирают пипеткой
15 мл раствора в электролизер, прибавляют 5 капель раствора
столярного клея и полярографируют в тех же условиях, что и стандартные
растворы.
* Исходный раствор соли индия готовят, растворяя 1 г металла высокой
чистоты в 25—30 мл разбавленной (1:1) азотной кислоты. Выпаривают 3 раза с 10 мл
концентрированной хлористоводородной кислоты, к остатку приливают 50—70 мл
воды, 20 мл концентрированной хлористоводородной кислоты и доливают до метки
водой в мерной колбе емкостью 1000 мл.
§ 13. ПРИМЕРЫ ПОЛЯРОГРАФИЧЕСКИХ ОПРЕДЕЛЕНИЙ
173
Измеряют высоту волны, находят по калибровочному графику
содержание индия в 15 мл данного раствора (в мг) и вычисляют
содержание индия в данном растворе.
Определение следов свинца и кадмия в металлическом цинке
Определение проводят методом добавок. Фоном служит раствор
соли цинка, полученный при растворении навески цинка в кислоте.
Свинец и кадмий восстанавливаются раньше цинка, поэтому, несмотря на
то что содержание цинка гораздо больше, он не мешает определению.
Метод заключается в том, что сначала снимают полярограмму
раствора цинка с примесями свинца и кадмия, затем приливают в этот же
раствор, находящийся в электролизере, из микробюретки стандартный
раствор, содержащий ионы свинца и кадмия с точно известной
концентрацией, во много раз превышающей концентрацию этих ионов в
анализируемом растворе. Прибавление стандартного раствора продолжают
до тех пор, пока волна определяемого иона не возрастет примерно
вдвое.
Изменение объема при титровании не учитывают, так как оно
незначительно вследствие высокой концентрации стандартного раствора.
Методика определения. Берут навеску металлического цинка около
5 г с точностью до 0,01 г и растворяют в 40—50 мл
хлористоводородной кислоты (1:1) сначала на холоду, а затем при нагревании. После
растворения оснрвной массы цинка прибавляют 5—8 капель 3%-ного
раствора Н202 и упаривают до небольшого объема. Приливают 10—
15 мл воды, количественно переносят в мерную колбу емкостью 50 мл,
добавляют 10 капель 0,5%-ного раствора желатины и доливают водой
до метки. Для полярографирования отбирают пипеткой 15 мл
раствора, помещают в сухой электролизер и удаляют кислород пропусканием
водорода или азота в течение 20 мин.
Снимают полярограмму, затем добавляют в электролизер по
каплям из микробюретки стандартный раствор свинца, содержащий 1 мг
РЬ++ в 1 мл, до тех пор, пока высота волны свинца не возрастет
примерно вдвое. Затем по каплям добавляют в электролизер стандартный
раствор кадмия такой же концентрации до возрастания вдвое волны
кадмия.
Поскольку объемы прибавляемых стандартных растворов РЬ++ и
Cd++ очень малы, отсчет даже с помощью микробюретки будет
недостаточно точным. Поэтому лучше определить объем капли, вытекающей
из микробюретки, и, сосчитав число капель каждого из добавленных
в электролизер стандартных растворов, рассчитать объемы
прибавленных растворов.
Затем снимают полярограмму раствора при той же чувствительности
гальванометра, при которой снимали полярограмму без добавок.
Измеряют высоты волн и делают расчет.
Расчет. Содержание (в %) определяемых ионов (х) вычисляют по
формуле:
„ _ gcrhxVK - ГОР
VA(ho6*-hx)a
где а — навеска металлического цинка, мг;
?ст> hx> ^общ» ^А» ^к ~ аналогичны приведенным в формуле (22).
(23)
Определение следов нитробензола в анилине
Нитробензол восстанавливается на ртутном капельном электроде,
образуя хорошо выраженные волны; при этом форма волны, и
потенциал восстановления зависят от рН среды.
174
ГЛ. V. ПОЛЯРОГРАФИЯ И АМПЕРОМЕТрИЧЕСКОЕ, ТИТРОВАНИЕ
Реакция протекает согласно уравнению:
C6H5N02 + 6Н+ + бе —> C6H5NH2 + 2H20
Восстановление нитробензола происходит в несколько стадий, и
только конечным продуктом восстановления является анилин. Таким
образом, приведенное выше уравнение является суммарным.
В анилине с добавкой в качестве фона хлористоводородной
кислоты нитробензол образует волну при —0,45 в относительно донной
ртути. Анилин в этих условиях не образует волны и не мешает
определению. Присутствие кислорода в данном случае также не мешает поляро-
графированию, поэтому инертный газ через раствор не продувают.
Определение проводят в электролизере небольшой емкости
методом добавок.
Возможно определять полярографически в анилине нитробензол
при содержании 0,001—1%.
Методика определения. В сухой электролизер наливают 2 мл
анализируемого анилина, содержащего примесь нитробензола, и
прибавляют 0,5 мЛ хлористоводородной кислоты (плотностью 1,19 г/см3). На
дно электролизера помещают немного металлической ртути и опускают
в нее платиновый электрод, соединенный с положительным полюсом по-
лярографа.
Для подавления максимума добавляют несколько капель раствора
солянокислого нигрозина.
Снимают полярограмму примеси нитробензола в анилине. Затем
добавляют в электролизер по каплям стандартный спиртовый раствор
нитробензола с содержанием 1 мг/мл до тех пор, пока волна
нитробензола не возрастет примерно вдвое. Снимают волну нитробензола в
растворе с добавкой при той же чувствительности гальванометра. Р1змеряют
высоты волн и рассчитывают содержание нитробензола в анилине.
Расчет. Содержание (в %.) нитробензола (х) в анилине вычисляют
по формуле: , т т
х = ТГ^ЧгГ- (24)
где а — навеска анилина, введенного в электролизер, м,г;
g:t, hx, йобщ — аналогичны приведенным в формуле (22).
Определение малеиновой и фумаровой кислот
при совместном присутствии
Малеиновая и фумаровая кислоты восстанавливаются на ртутном
капельном электроде. Восстановление их происходит согласно
следующему суммарному уравнению:
НООС-СН=СН—СООН + 2Н+ + 2е —>¦ НООС—СН2—СН2—СООН
Процесс восстановления протекает необратимо. Вид полярограммы
и потенциал восстановления зависят от рН среды. Потенциалы полуволн
малеиновой и фумаровой кислот с увеличением рН возрастают, но
неодинаково. В кислых растворах обе кислоты образуют волны при
одном и том же потенциале, но с увеличением рН потенциалы полуволн их
начинают все более различаться. В нейтральном растворе на фоне
NH4C1 волна малеиновой кислоты получается при —1,38 в, а
фумаровой при —1,55 в.
Ниже дана методика определения этих кислот методом стандарта.
Методика определения. Смесь кислот помещают в мерную колбу
емкостью 100 мл. Кислоты нейтрализуют 0,1 н. раствором Са(ОН)2 по
§ 13. ПРИМЕРЫ ПОЛЯРОГРАФИЧЕСКИХ ОПРЕДЕЛЕНИЙ 175
фенолфталеину, затем доливают до метки 0,5 н. раствором NH4C1.
В сухой электролизер вносят пипеткой 15—20 мл раствора и
пропускают водород или азот в течение 30 мин. Затем снимают полярограм-
му с каломельным электродом сравнения и измеряют высоту волн
малеиновои и фумаровой кислот.
Готовят стандартный раствор, содержащий малеиновую и фума-
ровую кислоту в концентрациях 10~3 моль/л. Для этого в мерную
колбу емкостью 100 мл наливают по 10 мл t),01 M растворов малеиновои
и фумаровой кислот, нейтрализуют 0,1 н. раствором Са(ОН)2 и
доливают до метки 0,5 н. раствором NH4C1. В электролизер помещают
точно 15—20 мл этого раствора, пропускают водород или азот в течение
30 мин и снимают полярограмму при той же чувствительности
гальванометра, что и полярограмму анализируемого раствора.
Измеряют высоты волн малеиновои и фумаровой кислот и
вычисляют содержание (в мг) этих кислот в растворе.
Расчет. Вычисление делают на основании пропорциональности
концентраций и высот волн, как описано в § 8 (стр. 162).
Определение альдегидов
Почти все альдегиды восстанавливаются на ртутном капельном
электроде. В процессе восстановления одних альдегидов участвуют
два электрона на каждую молекулу (например, формальдегид, ацеталь-
дегид, масляный альдегид); в процессе восстановления других — один
электрон (бензальдегид и другие ароматические альдегиды в кислой
среде). Большинство альдегидов образует одну волну восстановления.
Алифатические альдегиды не восстанавливаются в кислых
растворах. Это их свойство используется для определения ароматических
альдегидов в присутствии алифатических.
Определение бензальдегида
Определение проводят на фоне хлористоводородной кислоты в
растворе 20—25%-ного этилового спирта. При восстановлении
бензальдегида в кислой среде происходит реакция образования гидробензоина;
2С6Н5СНО + 2Н+ + 2е —> С6Н5—СН—СН—СвНБ
I I
ОН ОН
В щелочной среде восстановление бензальдегида идет до бензи-
лового спирта:
С6Н5СНО + 2Н+ + 2е —> С6Н5СН2ОН
При рН от 2 до 6 бензальдегид образует две волны. Это
объясняется тем, что в слабокислых растворах восстановление
бензальдегида до бензилового спирта протекает в две стадии, каждой из них
соответствует определенная волна.
На фоне хлористоводородной кислоты между величиной
диффузионного тока и концентрацией бензальдегида в растворе сохраняется
прямая пропорциональность.
При работе в спиртовых растворах максимумы на кривых
устраняются, поэтому поверхностно-активных веществ не добавляют.
Определение проводят методом калибровочной кривой.
Методика определения. Анализируемый раствор помещают в
мерную колбу емкостью 25 мл и доливают до метки этиловым спиртом. В
сухой электролизер помещают 10 мл 1 н. раствора НС1 й 4 мл
анализируемого раствора бензальдегида и снимают полярограмму.
176 ГЛ. V. ПОЛЯРОГРАФИЯ И АМПЕРОМЕТРИЧЕСКОЕ ТИТРОВАНИЕ
При очень малых концентрациях альдегидов (менее 1 ммолыл)
прежде чем вносить спиртовый раствор альдегида в раствор НС1
рекомендуется продуть через последний водород или азот.
Стандартный раствор бензальдегида готовят, растворяя навеску
химически чистого бензальдегида в этиловом спирте в мерной колбе
емкостью 25 мл. Навеску берут с таким расчетом, чтобы концентрация
альдегида в полученном растворе составляла 5—7 ммолыл. В
электролизер помещают последовательно 1, 2, 3, 4 и 5 мл этого раствора и
соответственно 13, 12, 11, 10 и 9 мл 1 н. раствора НС1 и полярографи-
руют полученные растворы при той же чувствительности
гальванометра. Измеряют высоты волн и строят калибровочный график,
откладывая по оси абсцисс содержание бензальдегида в растворе в
миллиграммах, а по оси ординат — соответствующие высоты волн. Затем,
отложив по оси ординат высоту волны, полученную при полярографиро-
вании анализируемого раствора бензальдегида, находят по
калибровочному графику содержание бензальдегида в этом растворе.
Расчет. Содержание (в мг) бензальдегида {g) в данном растворе
находят по формуле:
где а — содержание бензальдегида в электролизере, мг\
VA— объем раствора бензальдегида, помещенный в электролизер, мл\
VK — общий объем исследуемого раствора, мл.
Определение формальдегида
Определение проводят в щелочной среде на фоне гидроокиси
лития с ртутным капельным электродом и каломельным электродом
сравнения.
Восстановление формальдегида на ртутном капельном электроде
может быть выражено реакцией:
НСНО + 2е + 2Н20 —> СН3ОН + 20Н~
При рН 12,7 ?./2 = —1,465 в.
Определение выполняют методом калибровочной кривой.
Методика определения. Раствор формальдегида помещают в
мерную колбу емкостью 25 мл и разбавляют приблизительно до половины
колбы 40%-ным этиловым спиртом, затем прибавляют 1 мл 0,5%-ного
раствора желатины и доливают до метки 0,2 М раствором LiOH. В
сухой электролизер помещают пипеткой 15 мл полученного раствора и
снимают полярограмму.
Готовят раствор формальдегида с известным содержанием
последнего, разбавив водой формалин до содержания формальдегида
приблизительно 1 мг/мл и определив ц нем содержание формальдегида
гидроксиламинным методом.
В четыре мерных колбы емкостью по 25 мл помещают по 1, 2, 3,
4 мл этого раствора, разбавляют приблизительно до половины колб
40%-ным этиловым спиртом, добавляют по 1 мл 0,5%-ного раствора
желатины и доливают до метки 0,2 М раствором гидроокиси лития. По
15 мл каждого раствора помещают поочередно в электролизер и
снимают полярограммы.
Строят калибровочный график, откладывая по оси абсцисс
количество формальдегида (в мг), находящегося в электролизере, а по оси
ординат — высоты волн.
§ 14. ОСОБЕННОСТИ АМПЕРОМЕТРИЧЕСКОГО ТИТРОВАНИЯ
177
Находят по графику количество формальдегида {в мг) в объеме
раствора, помещенного в электролизер (15 мл), и рассчитывают затем
его содержание во всем данном растворе.
Расчет. Содержание (в мг) формальдегида (g) определяют по
формуле, аналогичной формуле (25).
АМПЕРОМЕТРИЧЕСКОЕ ТИТРОВАНИЕ
А. ТЕОРЕТИЧЕСКИЕ ОСНОВЫ
§ 14. Особенности амперометрического титрования
Сущность метода амперометрического титрования описана во
второй книге учебника «Основы аналитической химии» (см. книга 2,
гл. VIII, § 6).
Для использования какой-либо реакции в методе
амперометрического титрования необходимо, чтобы одно из реагирующих веществ
восстанавливалось или окислялось на ртутном или твердом
вращающемся индикаторном микроэлектроде, причем величина предельного
тока была пропорциональна концентрации: Id = КС
Величина константы К зависит от того, на каком электроде
измеряется диффузионный ток: на ртутном капельном или на твердом
вращающемся (чаще, платиновом).
При работе с ртутным капельным электродом величина
диффузионного тока определяется известным уравнением Ильковича (см. § 1,
стр. 000).
При работе с твердым вращающимся электродом константа К
характеризуется следующим выражением:
Толщина диффузионного слоя б зависит от ряда условий, в том
числе от скорости вращения электрода и вязкости раствора. Для
практических целей важно, чтобы б была как можно меньше; до известной
степени это достигается увеличением скорости вращения электрода.
В методе амперометрического титрования используются реакции
осаждения, комплексообразования и окисления — восстановления.
Рассмотрим метод амперометрического титрования на примере
реакций осаждения. Титрование может быть проведено следующим
образом.
Титрование по току определяемого вещества. Этот тип титрования
применяется, когда титруемые ионы восстанавливаются или
окисляются на индикаторном электроде.
К раствору в электролизере, содержащему исследуемые ионы,
добавляют из микробюретки раствор-осадитель, переводящий
определяемые ионы в осадок. Вначале (до добавления осадителя) при
потенциале, превышающем потенциал восстановления или окисления этого
иона, гальванометр показывает наличие тока, а именно предельного
тока определяемых ионов. Если теперь, не изменяя потенциала,
добавлять к раствору осадитель, то концентрация ионов определяемого
вещества будет уменьшаться, а следовательно, будет уменьшаться и
величина предельного тока. После того как определяемые ионы перейдут
в осадок, дальнейшее добавление осадителя уже не вызовет
уменьшения тока (сила тока становится постоянной). График зависимости
показаний гальванометра от объема стандартного раствора (рис. 55,
178 ГЛ. V. ПОЛЯРОГРАФИЯ И АМПЕРОМЕТРИЧЕСКОЕ ТИТРОВАНИЕ
кривая /) имеет точку перегиба, соответствующую точке
эквивалентности.
Титрование по току титранта. В этом случае на индикаторном
электроде восстанавливаются или окисляются ионы реактива,
определяемые ионы не дают электродной реакции. К раствору определяемого
вещества добавляют стандартный раствор ионов, которые способны,
например, восстанавливаться на катоде и могут давать с определяемым
ионом осадок. Так как титруемый раствор не содержит ионов, восста-
назливающихся (или окисляющихся) на индикаторном
микроэлектроде при данном потенциале, сила тока будет постоянна до тех пор, пока
добавляемые ионы не перестанут переходить в осадок. Однако сила
тока не будет равна нулю, так как от первых
же капель титранта образуется осадок, а а
растворе над ним находятся в небольшой, но
постоянной концентрации (зависящей от
произведения растворимости осаждаемого
вещества^ в случае бинарного электролита равной
УПР ) ионы осадителя, дающие
электродную реакцию. После точки эквивалентности,
Рис. 55. Кривые титрования: когда в растворе появится избыток ионов
i- по току определяемого веще- титранта, СПОСОБНЫХ ВОССТанаВЛИВаТЬСЯ ИЛИ
ства; 2-по току титранта. г »
окисляться на индикаторном электроде, сила
тока начнет увеличиваться по мере увеличения вводимого объема
титранта (рис. 55, кривая 2). На графике точка перегиба соответствует
точке эквивалентности.
Титрование по току определяемого вещества и титранта
Реактив и определяемый ион способны восстанавливаться или
окисляться на индикаторном электроде при выбранном потенциале.
Сначала гальванометр показывает предельный ток (определяемых
ионов, уменьшающийся по мере прибавления осадителя. В точке
эквивалентности ток имеет минимальное значение (зависящее от
произведения растворимости осадка), затем в растворе начинает появляться
избыток осаждающих ионов и сила тока снова начинает увеличиваться
(рис. 56).
Аналогичные явления происходят при использовании и других
типов реакций. Во всех случаях титрования в точке эквивалентности
наблюдается перегиб кривой титрования, по которому и определяют
точку эквивалентности.
Используется также амперометрическое титрование с индикатором,
этот метод применяют, когда ни титруемый ион, ни титраит не дают в
условиях титрования диффузионного тока. В этом случае к титруемому
раствору добавляют индикатор, восстанавливающийся или
окисляющийся на индикаторном электроде и реагирующий с титрантом после
того, как прореагируют определяемые ионы.
Например, при амперометрическом титровании алюминия
раствором фторида в качестве индикатора применяют железо (III). А1+++
образует с фторидом более прочный комплекс, чем Fe+++, поэтому
сначала титруются ионы алюминия. Когда весь алюминий оттитрован, с
титрантом начинает реагировать железо. При этом происходит
уменьшение диффузионного тока Ре+++-ионов и наблюдается перегиб кривой
титрования, соответствующий точке эквивалентности.
Метод амперометрического титрования позволяет в некоторых
случаях проводить определение отдельных компонентов в смесях без пред-
§ 15. СХЕМА УСТАНОВКИ И ПРИМЕНЯЕМЫЕ ЭЛЕКТРОДЫ
179
варительного разделения. При использовании реакций осаждения это
возможно, если выполняются два условия:
а) произведения растворимости осаждаемых соединений
различаются настолько, что реакции осаждения проходят строго
последовательно;
б) электрохимическое поведение компонентов реакций позволяет
получить кривую титрования с двумя отчетливыми изломами,
соответствующими точкам эквивалентности. Кривая титрования может иметь,
например, такой вид, как показано на рис. 57.
На рис. 57 представлена теоретическая кривая титрования смеси
РЬ++ и Ва++ раствором КгСг04 при Е = —1,0 в. При титровании
последовательно происходят две реакции:
РЬ++ + К2СЮ4 —> |PbCr04 + 2K+
Ва++ + К2СЮ4 —> ф ВаСг04 + 2К+
Участок кривой АВ соответствует осаждению РЬ++, при этом ток
свинца уменьшается. На участке ВС происходит осаждение Ва++, но,
поскольку Ва++ не восстанавливается на индикаторном электроде при
—1,0 в, сила тока, остается постоянной до полного осаждения
Ва++-ионов.
w
в с
1С. 57. Кривая титро-
ния смеси РЬ++- и
1++-ионов раствором
K2Gr04.
Участок кривой CD соответствует постепенному повышению силы
тока вследствие увеличения концентрации в растворе СгОГ".
При амперометрическом титровании смеси компонентов с
использованием реакции окисления —- восстановления необходимо, чтобы
окислительно-восстановительные потенциалы значительно отличались
друг от друга.
В этом случае реакции окисления — восстановления при
добавлении реактива идут последовательно, и на кривой титрования получается
два скачка.
Б. АППАРАТУРА И ТЕХНИКА ВЫПОЛНЕНИЯ
АМПЕРОМЕТРИЧЕСКОГО ТИТРОВАНИЯ
§ 15. Схема установки и применяемые электроды
Для амперометрического титрования используется та же
принципиальная схема установки, что и при полярографических определениях.
В амперометрическую установку входит источник постоянного
тока, вольтметр, реохорд, гальванометр с шунтом, электролизер,
индикаторный электрод и электрод сравнения, соединенный с
электролизером солевым мостиком, кроме того, над электролизером
устанавливают микробюретку.
Рис. 56. Кривая
титрования по току
определяемого вещества и титран-
та.
Р
В
ГЛ. V. ПОЛЯРОГРАФИЯ И АМПЕРОМЕТРИЧЕСКОЕ ТИТРОВАНИЕ
Рис. 58. Упрощенная установка для
амперометрического титрования:
/_ ртутный капельный электрод; 2 — электрод
сравнения; 3 — гальванометр; 4 — реостат; 5 —реохорд; 6—
аккумуляторная батарея.
При использовании твердых индикаторных электродов применяют
небольшой электромотор.
Для амперометрического титрования можно пользоваться
визуальным полярографом ПВ-5 с зеркальным гальванометром М-21
(чувствительность 10~9 а/м/мм). Но в большинстве случаев установка
может быть упрощена и титрование можно проводить без полярографа
(рис. 58).
Наложение потенциала на электроды может быть осуществлено с
помощью реостата, а вместо зеркального гальванометра можно
применить стрелочный с максимальной чувствительностью 10"?—10"6 а.
При амперометрическом титровании могут быть использованы
ртутный капельный и различные твердые вращающиеся индикаторные
электроды. Твердые индикаторные электроды чаще применяются в
амперометрическом титровании,
чем в полярографии, так как
здесь не имеет значения сама
величина силы тока и ее
воспроизводимость.
При амперометрическом
титровании отмечается лишь
изменение силы тока в точке
эквивалентности, поэтому не
требуются электроды с точно
одинаковой поверхностью.
Возможность замены в
ряде случаев ртутного
капельного электрода твердыми
электродами является большим
преимуществом метода амперометрического титрования по сравнению
с полярографией, так как ртуть ядовита и работа с ней требует
специальных условий (см. выше).
Электроды. Чаще всего из числа электродов применяют
вращающийся платиновый электрод. Однако применение его ограничено тем,
что при отрицательных потенциалах на нем выделяется водород (при
О в в кислых растворах, при —0,4 в в нейтральных и при —0,8 в в
сильнощелочных). В области положительных потенциалов с
платиновым электродом можно работать до +1,1—ь 1,3 в (в зависимости от
условий). При более высоких потенциалах выделяется кислород из
воды. Кроме платинового электрода для амперометрического
титрования применяются золотой, танталовый, вольфрамовый и графитовый.
Применяют также амальгамированное твердые электроды (чаще,
серебряный).
На поверхности этих электродов имеется слой амальгамы,
поэтому перенапряжение для выделения водорода близко к значению
перенапряжения на ртути. Таким образом, амальгамированные твердые
электроды совмещают достоинства ртутного капельного электрода и
вращающегося твердого электрода.
Размеры электрода оказывают существенное влияние- на величину
силы тока (сила тока пропорциональна поверхности электрода). При
больших индикаторных электродах увеличивается чувствительность
определений, но зато возможны потери вещества за счет его окисления
или восстановления на электроде. Кроме того, наблюдается сдвиг
потенциала индикаторного электрода вследствие возрастания величины
IR. Обычно используют цилиндрические (игольчатые) твердые
электроды длиной 4—5 мм и сечением 0,3—0,5 мм. Для работы в очень раз-
§ 15. СХЕМА УСТАНОВКИ И ПРИМЕНЯЕМЫЕ ЭЛЕКТРОДЫ
181
бавленных растворах рекомендуется использовать электроды того же
сечения, но длиной 10 мм.
Обычно твердый электрод впаивают в стеклянную трубку с
двумя лопастями на нижнем конце. Лопасти служат для перемешивания
раствора. Иногда вместо лопастей для перемешивания раствора
применяют электроды, согнутые на конце почти под прямым углом.
Для вращения электрода используется небольшой мотор> причем
число оборотов его должно быть постоянным. Сила тока возрастает с
увеличением числа оборотов, но не беспредельно. Установлено, что
увеличение скорости вращения электрода свыше 300 об/мин мало влияет
на возрастание силы тока, а при скорости выше 600 об/мин изменение
скорости уже не сказывается практически на
величине силы тока. Поэтому рекомендуется поддерживать
скорость вращения электрода в пределах от 600 до
900 об/мин; небольшие изменения скорости
вращения, связанные с колебаниями напряжения в сети, не
вызывают колебаний силы тока. Если мотор делает
больше 900 об/мин, возможно разбрызгивание
раствора.
Очень удобен в работе твердый
амальгамированный электрод, изображенный на рис. 59. Для
изготовления этого электрода используют проволоку
(серебряную, медную и т. п.), диаметром окоро 0,5 мм,
которую продевают в стеклянную трубку с двумя
лопастями на нижнем конце. Пространство между
электродом и нижним концом стеклянной трубки
заливают расплавленным полиэтиленом. Длина самого
электрода составляет 4—5 мм. Длина проволоки на
несколько сантиметров больше, чем длина
стеклянной трубки. Избыточную часть проволоки
обматывают вокруг верхнего конца стеклянной трубки и
используют в дальнейшем для замены электрода
постепенно растворяющегося при амальгамировании в процессе работы.
Когда электрод становится очейь тонким после многократного
амальгамирования, слой полиэтилена и электрод срезают, вытягивают
проволоку длиной 4—5 мм и снова заливают трубку полиэтиленом.
Верхний конец проволоки припаивают к металлической муфте.
Контакт между прибором и муфтой осуществляется с помощью
металлических щеток. Стеклянную трубку соединяют с мотором резиновой трубкой.
Платиновый индикаторный электрод может быть устроен
аналогично, но так как платина очень дорого стоит, можно взять
платиновую проволоку длиной всего 2—3 см и приварить к ней медную
проволоку, верхнюю, избыточную часть которой обматывают вокруг
стеклянной трубки. Когда платиновый электрод придет в негодность от
долгого употребления, его легко обновить, срезав слой полиэтилена и
вытянув из трубки часть запасной платиновой проволоки.
Можно также впаять платиновый электрод в нижнюю часть
стеклянной трубки, на дно трубки налить для контакта немного ртути
и опустить в нее нижний конец меДной проволоки (верхний конец
которой припаян к муфте).
Если в качестве индикаторного электрода применяют ртутный
капельный электрод, то и в этом случае сила тока, а следовательно,
и чувствительность определений, зависят от величины электрода, но
вместо длины электрода необходимо рассматривать величину
характеристики капилляра (m2V/e): чем она больше, тем больше предельный
Рис. 59. Твердый
амальгамированный индикаторный
электрод:
1 — электромотор; 2 —
муфта; 5—стеклянная
трубка; 4 — электрод;
5 —щетки.
182 ГЛ. V. ПОЛЯРОГРАФИЯ И АМПЕРОМЕТРИЧЕСКОЕ ТИТРОВАНИЕ
ток. Наиболее подходящей для амперометрического титрования
является характеристика, приблизительно равная 2,5; вообще, чем
быстрее капает ртуть, тем это удобнее для титрования, так как при мелких
каплйх значительно уменьшаются осцилляции показаний гальвайометра
й отсчет может быть сделан более быстро и точно.
При работе с твердыми электродами осцилляции совершенно
отсутствуют, что является одним из главных преимуществ этих
электродов.
Ртутный электрод можно применять до потенциала —2,0 в (в
щелочной среде), а платийовый до +1,3 в (в кислой среде). В качестве
электродов сравнения при амперометрическом титрбвании применяются
каломельные электроды (насыщенный, 1,0-йормальный, 0,1-нормаль-
ный), меркуриодидный, меркурсульфатный, хлорсеребряный. При
выборе электрода сравнения обычно принимают во внимание тот процесс
окисления или восстановления, который должен происходить на
индикаторном электроде. Если между потенциалом индикаторного электрода
и потенциалом электрода сравнения получается разность потенциалов
не менее 0,3—0,4 в, то можно проводить амперометрическое
титрование без внешнего источника тока. В этом случае электрическая цепь
замыкается между индикаторным электродом и электродом сравнения
через гальванометр.
Методика титрования. Амперометрическое титрование проводится
в стаканах емкостью 25—30 мл, которые соединяются с электродом
сравнения солевым мостиком. Солевой мостик обычно заполняют агар-
агаром, насыщенным КО или KN03 либо насыщеййыми растворами
этих солей.
При проведении амперометрического титрования необходимо
учитывать то обстоятельство, что введение в раствор реактива увеличивает
объем жидкости в электролизере, Концентрация восстанавливающихся
(или окисляющихся) ионов при этом определяется двумя факторами —
количеством добавляемого реактива и объемом жидкости в
электролизере, а сила диффузионного тока не определяется уравнением Id—КС.
Поэтому при амперометрическом титровании обычно применяют
растворы титрантов с концентрацией в 10—20 раз выше концентраций
титруемых растворов, тогда изменение объема невелико и им можно
пренебречь. Так как объемы титрантов, израсходованных на титрование,
малы (вследствие большой концентрации титрайта), для титрования
пользуются микробюретками. В тех случаях, когда концентрация
стандартного раствора близка к концентрации анализируемого раствора,
все значения предельного тока, полученные при титровании,
пересчитывают на разбавление по формуле:
где / — сила тока с учетом изменения объема раствора в электролизере, а\
1т \ — измеренная сила тока, а;
V\ — исходный объем раствора в электролизере, мл;
V2 — объем добавленного титранта, мл.
Кривую титрования строят уже по пересчитанным значениям силы
тока. В большинстве случаев амперометрическое титрование проводят
без удаления кислорода из раствора и без введения
поверхностно-активных веществ, поскольку потенциал, при котором проводят титрование,
можно установить таким, чтобы он не совпадал с максимумом или
волной кислорода.
Раствор перемешивают вращением электрода, если определение
проводят с твердым электродом. При проведении титрования с ртутным
§ 15. СХЕМА УСТАНОВКИ И ПРИМЕНЯЕМЫЕ ЭЛЕКТРОДЫ 183
электродом раствор перемешивают, пропуская через него ток азота
или водорода после каждого прибавления реактива или с помощью
мешалки.
При амперометрическом титровании, как и в полярографии, в
раствор добавляют электролит-фон. Однако в некоторых случаях можно
обойтись и без фона. Вначале фоном служит избыток ионов
определяемого вещества, а затем появляющиеся в растворе ионы вещества,
образующегося в результате реакции.
Потенциал, который выбирают для проведения титрования, должен
находиться в области диффузионного тока титруемого вещества.
Потенциал обычно устанавливают на 0,1—0,3 в более отрицательным, чем
потенциал полуволны определяемого вещества. Однако при этом надо
учитывать, не будут ли при выбранном потенциале восстанавливаться
или окисляться примеси, присутствующие в растворе, и не начнется ли
выделение водорода или кислорода на индикаторном электроде.
Правильность выбора потенциала можно проверить следующим образом:
в стакан для титрования наливают раствор, аналогичный по составу
анализируемому (тот же фон, те же примеси), но не содержащий
определяемого вещества. В микробюретку наливают раствор вещества,
дающего электродную реакцию. Это может быть титрант или раствор
определяемого вещества. Установив на индикаторном электроде
выбранный потенциал, проводят титрование. При таком титровании никакой
реакции не происходит, но сила тока постепенно возрастает, так как
в растворе увеличивается концентрация восстанавливающихся (или
окисляющихся) ионов.
Затем строят кривую титрования. При правильном выборе
потенциала будет все время соблюдаться прямая пропорциональность между
силой тока и концентрацией иона, дающего электродную реакцию, т. е.
график будет прямолинейным.
Если проводить титрование при потенциале, соответствующем
потенциалу полуволны, то также будет наблюдаться прямая
пропорциональность между силой тока и концентрацией иона, дающего
электродную реакцию. В этом случае сила тока при титровании будет равна
половине силы диффузионного тока. Однако малейшее отклонение
потенциала от значения потенциала полуволны вызовет заметное
изменение силы тока, что скажется на точности определения.
Температура раствора во время титрования должна быть
постоянной, так как ее изменение может повлиять на величину коэффициента
диффузии раствора и тем самым нарушить прямую пропорциональность
зависимости сила тока — концентрация.
Преимущества амперометрического титрования. Преимуществом
амперометрического титрования перед другими физико-химическими
методами титрования (потенциометрическим, кондуктометрическйм,
а также перед объемным индикаторным) является то, что для
построения кривой титрования достаточно снять несколько точек, причем эти
точки снимают вдали от точки эквивалентности, т. е. когда в растворе
имеется избыток одного из реагирующих ионов.
При использовании в амперометрическом титровании реакций
осаждения точки для кривой титрования находят в условиях, когда
растворимость осадка меньше, чем в точке эквивалентности. Благодаря
этому применение рассматриваемого метода оказывается возможным
для определения веществ, образующих довольно хорошо растворимые
осадки, когда ни потенциометрический, ни индикаторный метод не
могут дать хороших результатов. Кроме того, в отличие от других
электрометрических методов анализа данный метод позволяет проводить
определения малых количеств веществ в сильно разбавленных растворах
184 ГЛ. V. ПОЛЯРОГРАФИЯ И АМПЕРОМЕТРИЧЕСКОЕ ТИТРОВАНИЕ
(более разбавленных, чем при полярографических определениях).
В полярографии при концентрациях определяемого вещества менее
Ю-4 моль/л волна определяемого иона становится слишком маленькой,
а увеличение чувствительности гальванометра приводит к усилению
влияния остаточного тока, затрудняющего определение. Амперометриче-
ским титрованием можно определять вещества в концентрации до
10~6 молб/л, если определение проводить по току титранта, концентрация
которого в 10—20 раз выше концентрации определяемого иона.
Поэтому после точки эквивалентности сила тока, соответствующая
восстановлению или окислению на электроде иона титранта, может
быть значительной, и перегиб кривой титрования в эквивалентной точке
будет резким, несмотря на малую концентрацию титруемого иона.
В методе амперометрического титрования могут быть широко
применены органические реактивы, использование которых при потенцио-
метрическом титровании часто ограничивается отсутствием
соответствующего индикаторного электрода.
Точность определений по методу амперометрического титрования
выше, чем точность полярографических определений, поскольку такие
факторы, как природа индифферентного электролита, характеристика
капилляра и давление ртути, не влияют на результаты определений.
§ 16. Амперометрическое титрование с двумя
индикаторными электродами
Кроме рассмотренного метода амперометрического титрования
применяют также метод амперометрического титрования с двумя
индикаторными электродами. Его называют иногда методом биамперометриче-
ского титрования.
При титровании с одним индикаторным электродом и электродом
сравнения потенциал электрода сравнения остается постоянным при
любой силе тока. Накладывая извне определенное напряжение на эти
электроды, поддерживают постоянным также и потенциал
индикаторного электрода.
При титровании с двумя индикаторными электродами оба
электрода погружены непосредственно в анализируемый раствор и между
ними создается разность потенциалов. При этом на электродах
начинают протекать соответствующие электрохимические процессы, в
результате которых в цепи появляется ток. Индикаторные электроды могут
быть одинаковыми или различными, но во всяком случае величина
поверхности одного из них не превосходит значительно величину
поверхности другого, как это имеет место в обычном, классическом методе
амперометрического титрования. Чаще всего употребляются два
одинаковых по размеру платиновых электрода.
Схема установки для амперометрического титрования с двумя
индикаторными электродами представлена на рис. 60.
Протекание тока в цепи возможно только в том случае, если
в растворе имеются вещества, восстанавливающиеся на катоде, и
вещества, окисляющиеся на аноде.
Характер кривых титрования зависит от того, обратима или
необратима титруемая система или система титранта и от приложенной
к электродам разности потенциалов.
Рассмотрим титрование гексацианоферрата (II) раствором CeIV:
CeIV+[Fe(CN)e]"" —> Се+++ + [Fe(CN)e]~
Вначале (до прибавления титранта) на аноде окисляется
[Fe^NJe]"^, а на катоде восстанавливается [Fe(CN)6]—, всегда
§ 16. АМПЕРОМЕТРИЧЕСКОЕ ТИТРОВАНИЕ С ДВУМЯ ЭЛЕКТРОДАМИ 185
имеющийся в растворе гексацианоферрата (II) в виде незначительной
цримеси. Так как примесь этих ионов мала, то и ток вначале очень мал.
При титровании количество [Fe(CN)6]—-ионов возрастает в
результате реакции и сила тока увеличивается (рис. 61, кривая 1).
Возрастание силы тока продолжается до тех пор, пока не будет оттитрована
приблизительно половина гексацианоферрата (II). После этого
концентрация ионов восстановителя начинает понижаться, что сопровождается
уменьшением силы тока. В точке эквивалентности сила тока близка
к нулю, так как концентрация [Fe(CN)6]==:=:: практически равна нулю.
После точки эквивалентности в растворе появляется избыток CeIV,
поэтому теперь на катоде восстанавливается CeIV, а на аноде окисляет-
* . .
V
Рис. 61. Кривая
титрования различных веществ
с двумя индикаторными
электродами:
/ — обратимая система
титруется обратимой системой;
2 — обратимая система
титруется необратимой
системой; 3 —необратимая
система титруется обратимой
системой.
f типу титрований, когда
и титруемая система, и система титранта обратимы, т. е. окисленная
и восстановленная формы обратимо переходят одна в другую:
[Fe(CN)<J= + е ** [Fe(CN)6] и CeIV + е ** Се+++.
Если титруемая система электрохимически обратима, а система
титранта необратима, то до точки эквивалентности кривая идет так же,
как кривая У, после точки эквивалентности ток не возрастает, а
остается практически равным нулю (рис. 61, кривая 2).
Такой тип кривой титрования получается, например, при
титровании раствора иода тиосульфатом. До точки эквивалентности в растворе
находится обратимая система 12ч=^2Г, окисленная форма которой /2
восстанавливается на катоде, а восстановленная форма I"- окисляется
на аноде. Ток возрастает до тех пор, пока не оттитрована половина
раствора иода, затем постепенно падает до нуля в точке эквивалентности.
После точки эквивалентности на аноде могут окисляться тиосульфат-
ионы, появившиеся в растворе в избытке, но тетратионат на катоде не
восстанавливается, поэтому сила тока не возрастает.
Если необратимая система титруется обратимой системой, ток
остается близким к нулю в течение всего процесса титрования до точки
ся Се-4""*-. Ток снова возрастает.
Рис. 60. Схема установки для ам-
перометрического титрования с
двумя индикаторными
электродами:
/ — платиновые электроды; 2 — стакан
для титрования; 3— мешалка;
4-бюретка; 5 - гальванометр.
Рассмотренный случай относится к
186 ГЛ. V. ПОЛЯРОГРАФИЯ И АМПЕРОМЕТРИЧЕСКОЕ ТИТРОВАНИЕ
эквивалентности. После точки эквивалентности, когда в растворе
появляется избыток титранта, сила тока возрастает (рис. 61, кривая 3).
К такому типу относится, например, титрование тиосульфат-ионов
иодом. До точки эквивалентности в растворе присутствует система
тиосульфат— тетратионат (последний в виде примеси). Поскольку тетра-
тионат не восстанавливается на катоде, ток в цепи не протекает. После
точки эквивалентности в растворе появляется система h — 21~ и в цепи
начинает протекать все увеличивающийся ток.
Величина разности потенциалов Д?, которую необходимо
приложить к электродам, чтобы в цепи протекал ток, зависит от природы
титранта и титруемого вещества. Определение этой величины основано
на предварительном полярографическом исследовании растворов,
содержащих отдельно каждый из компонентов реакции вместе с его
окисленной или восстановленной формой.
Если система обратима, как Ь — 21~, то разность потенциалов для
протекания тока требуется очень небольшая (практически, близкая
к нулю). При этом ток будет линейно увеличиваться с возрастанием
потенциала.
Если система необратима, как S2Or" — S^eT, то к электродам
требуется приложить значительную разность потенциалов. При достижении
потенциала Е на аноде будут окисляться 52ОГ""Ионы, а на катоде —
восстанавливаться ионы водорода и в цепи возникнет ток.
В трех ранее рассмотренных типах титрования участвовала хотя
бы одна обратимая система, поэтому титрование проводилось при
малой разности потенциалов (10—30 мв). Но возможно титрование
необратимой системы необратимой системой. Кривая титрования,
соответствующая этому случаю, имеет такой же вид, как кривая / на рис. 61,
но с той разницей, что титрование ведется при большой разности
потенциалов. К этому типу титрования относится, например, титрование
перекиси водорода перманганатом калия.
Метод титрования с двумя индикаторными электродами чаще всего
используется для определений, основанных на реакциях окисления —
восстановления. Однако в нем используются также реакции осаждения
и нейтрализации. Это стало возможным благодаря введению так
называемых электрометрических индикаторов. Например, для того чтобы
оттитровать раствор кислоты раствором щелочи, добавляют к
титруемому раствору несколько капель раствора иода. До точки
эквивалентности ток в цепи почти отсутствует, после точки эквивалентности,
когда в цепи появляется избыток щелочи, образуются иодид-ионы и,
таким образом, возникает пара Ь — 21", вызывающая возрастание
тока.
При использовании реакций осаждения в качестве
электрометрического индикатора обычно вводят в раствор ион, образующий пару с
одним из компонентов реакции. Например, Zn++, Ca++ и многие другие
катионы могут быть оттитрованы гексацианоферратом(П) в присутствии
гексацианоферрата (III).
Электроды в методе титрования с двумя индикаторными
электродами могут быть сделаны из различного материала. В этом случае
иногда возможно обойтись и без внешнего источника напряжения.
Электроды могут быть тогда соединены непосредственно с гальванометром.
Большим преимуществом метода титрования с двумя
индикаторными электродами перед обычным методом амперометрического
титрования является настолько резкое изменение тока в точке
эквивалентности, что не требуется вычерчивать кривую титрования. Это
значительно упрощает и ускоряет выполнение определений.
§ 17. ПРИМЕРЫ АМПЦРОМЕТРИЧЕСКОГО ТИТРОВАНИЯ
187
В. ПРАКТИЧЕСКИЕ РАБОТЫ С ПРИМЕНЕНИЕМ
АМПЕРОМЕТРИЧЕСКОГО ТИТРОВАНИЯ
§ 17. Примеры амперометрического титрования
Титрование ионов цинка или кадмия раствором
гексацианоферрата (II) калия
При титровании ионов цинка или кадмия гексацианоферратом (II)
калия на фоне K2SO4 получаются осадки K2Zn3[Fe(CN)6]2 или
K6Cd5[Fe(CN)6]4:
3Zn++ + 2K4[Fe(CN)6] —> ф K2Zn3[Fe(CN)6]2 + 6К+
5Cd+++ + 4K4[Fe(CN)6] —> ф K6Cd5]Fe(CN)6]4/+ЮК+
Титрование проводят по току реактива. Гексацианоферрат (II)
калия окисляется на вращающемся платиновом микроэлектроде, причем
диффузионный ток достигается при +0,7 в.
Методика определения. Раствор ZnS04 или CdS04 помещают в
мерную колбу емкостью 100 мл и доливают его до метки 1 М
раствором K2SO4.
В стакан для титрования наливают аликвотную часть этого
раствора и опускают индикаторный электрод и солевой мостик, соединенный
с каломельным электродом сравнения.
В микробюретку наливают 0,034 М раствора K4[Fe(CN)6].
Устанавливают на полярографе анодную поляризацию, включают мотор,
замыкают цепь (при наименьшей чувствительности гальванометра) и
устанавливают с помощью движка реохорда потенциал, равный +0,8 в.
Регулируют чувствительность гальванометра так, чтобы световой зайчик
находился примерно на середине шкалы. Приступают к титрованию
ZnS04 или CdS04 раствором K4[Fe(CN)6], замечая показания
гальванометра после прибавления каждой порции раствора по 0,2 мл. Строят
кривую титрования и определяют по ней объем титранта,
соответствующий точке эквивалентности.
Расчет. Содержание (в мг) цинка или кадмия (g) вычисляют по
формуле:
8 = Ь5СК4П?е(СК)б]7К4[Ре(сж)б)Л —- (28)
r A
где 1,5 —отношение числа молей Z11SO4 к числу молей K4[Fe(CNJe] в уравнении
реакции;
А — атомный вес цинка;
УА — объем раствора, помещенного в электролизер, мл;
VK — общий объем исследуемого раствора, мл.
Содержание кадмия рассчитывается по аналогичной формуле, ночэтношенне числа
молей CdS04 к числу молей K4[Fe(CN)e] составляет 1,25.
Титрование ионов свинца раствором бихромата калия
Определение основано на реакции:
2РЬ++ + К2Сг207 + Н20 —> 2РЬСг04 + 2К+ + 2Н+
Наиболее точные результаты получаются при титровании ионов
свинца в присутствии ацетатного буферного раствора при рН = 4,2
(1 М раствор ацетата натрия, содержащий 40 мл ледяной уксусной
кислоты в 1 л раствора).
Бихромат восстанавливается в этих условиях при потенциале,
равном нулю; титрование возможно проводить с вращающимся платиновым
188 ГЛ. V. ПОЛЯРОГРАФИЯ И АМПЕРОМЕТРИЧЕСКОЕ ТИТРОВАНИЕ
электродом, РЬ++ при этом не восстанавливается и титрование
проводится по току титранта.
Можно проводить титрование РЬ++ также по току определяемого
вещества. Если титровать при +1,3 в, то происходит окисление свинца
на индикаторном электроде:
РЬ++ + 2Н20 - 2е = РЬ02 + 4Н+
Титрование и в этом случае проводится в присутствии ацетатной
буферной смеси при рН = 4,2.
Методика определения. Титрование РЬ++ -ионов по току
бихромата. Раствор соли свинца Pb(N03)2 или РЬ(СН3СОО)2
помещают в мерную колбу емкостью 100 мл и доливают до метки
ацетатной буферной смесью. Аликвотную часть полученного раствора
наливают в стакан для титрования, опускают индикаторный электрод
и солевой мостик, соединенный с каломельным электродом сравнения.
В микробюретку наливают 0,05 М раствор К2СГ2О7. Включают
мотор, замыкают цепь и устанавливают с помощью движка реохорда
потенциал, равный нулю. Приступают к титрованию ионов свинца
раствором К2СГ2О7, приливая его порциями по 0,2 мл и замечая показания
гальванометра после прибавления каждой порции.
Строят кривую титрования и определяют по ней объем титранта,
соответствующий точке эквивалентности.
Расчет содержания свинца см. ниже.
Титрование РЬ++-ионов по току ионов свинца.
Титрование проводят так же, как описано выше, но потенциал
устанавливается равным +1,3 в. Кривая титрования в этом случае имеет другую
форму.
Расчет. Содержание (в мг) свинца {g) в обоих случаях вычисляют
одинаково, по формуле:
?^K2Cr2o/K2Cr2oHyf (29)
Y A
где .4, Ук и VA — cm. уравнение (28).
Титрование сульфат- и хромат-ионов
при совместном присутствии
Титрование основано на следующих реакциях:
Pb++ + CrOJ~ —> |PbCr04
Pb++ + SO~ —> |PbS04
Так как nPpbso4 = Ю~8, т. е. в 10 000 раз больше, чем ПРРЬсг04 =
= 10"12, осаждение этих солей при титровании должно происходить
последовательно: сначала осаждается хромат, затем сульфат свинца. Но
практически сульфат частично соосаждается с хроматом. Поэтому
титруют две пробы: в первой из них титруют только хромат, добавив
к раствору ацетат-ионы, в присутствии которых PbSC>4 не выпадает, во
второй определяют сумму сульфата и хромата.
Титрование проводят с вращающимся платиновым электродом
и насыщенным каломельным электродом сравнения по току окисления
свинца (при потенциале +1,3 в).
Методика определения. Пробу данного раствора ( — 10 мл),
содержащего хромат и сульфат натрия или калия, помещают в мерную
колбу емкостью 100 мл и доливают до метки раствором ацетатного
буферного раствора. Аликвотную часть раствора отбирают в стакан
§ 17. ПРИМЕРЫ АМПЕРОМЕТРИЧЕСКОГО ТИТРОВАНИЯ
189
для титрования и титруют 0,1 М раствором Pb(N03)2 при
потенциале + 1,3 в.
Строят кривую титрования и находят по ней объем титранта,
израсходованный на титрование хромата. Другую пробу данного раствора
объемом также 10 мл помещают в мерную колбу емкостью 100 мл,
нейтрализуют NaOH или HN03 (в зависимости от того, какой, кислой
или щелочной, была реакция данного раствора) до рН 4,8—6 и
доливают до метки 0,1 М раствором нитрата натрия.
Отбирают пипеткой аликвотную часть этого раствора в стакан для
титрования, прибавляют 5 мл этилового спирта и титруют 0,1 М
раствором Pb(N03h также при потенциале +1,3 в. Строят кривую
титрования и находят по ней объем титранта, соответствующий суммарному
содержанию сульфата и хромата, затем по разности находят объем
титранта, израсходованный на титрование сульфата.
Расчет. Содержание (g) сульфата или хромата (в мг) в данном
растворе вычисляют цо формуле:
e ^Pb(N03)2FPb(N03)29FK (3Qv
va
где VKi У а — см. формулу (28).
Определение хрома в минералах и рудах
Метод заключается в окислении хрома до CrVI и последующем
амперометрическом титровании бихромата в сильнокислом растворе
солями железа (II):
Сг20~ + 6Fe+++14H+ —> 2Cr+++ + 6Fe+++ + 7Н20
Титрование проводится с вращающимся платиновым электродом
при потенциале +1,0 в, так как Ре++-ионы дают при этом потенциале
анодный диффузионный ток, величина которого пропорциональна
концентрации.
Методика определения. Берут навеску руды или минерала,
рассчитанную таким образом, чтобы получить 100 мл приблизительно 0,002 М
раствора CrVI. Навеску образца, содержащего много кремневой
кислоты, обрабатывают серной и фтористоводородной кислотами и
нерастворимый остаток сплавляют с пиросульфатом калия. При малых
содержаниях кремневой кислоты достаточно только сплавления с
пиросульфатом калия. Сплав растворяют в воде, добавляют серную
кислоту до концентрации ОД н., несколько капель 5%-ного раствора
нитрата серебра и 0,2—0,5 г персульфата аммония, избыток которого
разрушают кипячением. В присутствии марганца прибавляют по
каплям 0,2%-ный раствор нитрита натрия до обесцвечивания раствора и
тотчас же 0,5 г мочевины.
Раствор переводят количественно в мерную колбу емкостью 100 мл
и доливают до метки 0,1 н. раствором серной кислоты. В стакан для
титрования наливают аликвотную часть полученного раствора, опускают
индикаторный электрод и солевой мостик, соединенный с каломельным
электродом сравнения.
В микробюретку наливают 0,01 н. раствор соли Мора. Включают
мотор, замыкают цепь и устанавливают с помощью движка реохорда
потенциал^ равный +1,0 в.
Раствор, содержащий СггОГ", титруют раствором соли Мора
порциями по 0,2 мл, замечая показания гальванометра после прибавления
каждой порции.
190 ГЛ. V. ПОЛЯРОГРАФИЯ И АМПЕРОМЕТРИЧЕСКОЕ ТИТРОВАНИЕ
Строят кривую титрования, определяют по ней объем титранта,
соответствующий точке эквивалентности, и рассчитывают содержание
хрома в образце в процентах.
В присутствии ванадия результаты титрования дают сумму хрома
и ванадия. В этом случае определяют отдельно ванадий и вычитают из
общего объема раствора соли Мора, израсходованного на титрование
обоих элементов, тот объем, который соответствует определенному
количеству ванадия.
Расчет. Содержание (в %) хрома (g) вычисляют по формуле:
п Fe Fe++ Cr к . ,
8 = шшу1 (31)
где a, VK и Va — см. формулу (28).
Определение альдегидов
Альдегиды реагируют количественно с 2,4-динитрофенилгидрази-
ном с образованием осадка:
N02 N02
R~C<^ + H2N-NH-~<^2/-N02 —> R-CH=N-NH-^^>-N02 + H20
Титрование проводят с ртутным капельным электродом и
каломельным электродом сравнения при потенциале от —0,4 до —0,8 е.
Методика определения. Анализируемый раствор помещают в
мерную колбу емкостью 25 или 50 мл и доливают до метки водой (если
альдегид растворим в воде) или этиловым спиртом. Отбирают оттуда
пипеткой 10 мл раствора (в которых должно содержаться 10—30 ммоль
альдегида), помещают в электролизер, прибавляют 2,5 мл
концентрированной хлористоводородной кислоты и 6—7 мл 0,05%-ного водного
раствора тимола. Если данный раствор доливали до метки спиртом, то
вместо раствора тимола добавляют в электролизер воду. Пропускают
азот в течение 5 мин и титруют 0,01 М раствором 2,4-динитрофенилгид-
разина (ДФГ) при потенциале от —0,4 до —0,8 в.
Титрант прибавляют порциями по 0,2 мл и пропускают азот через
смесь в течение 3 мин после прибавления каждой порции титранта.
Расчет. Содержание (в мг) альдегида (g) вычисляют по формуле:
й = УГК (32)
где К — отношение числа молей альдегида к числу молей 2,4-динитрофенилгидрази-
на в уравнении реакции;
М — молекулярный вес альдегида;
VK и VA — см. формулу (28),
ГЛАВА VI
КУЛОНОМЕТРИЯ И КУЛОНОМЕТРИЧЕСКОЕ
ТИТРОВАНИЕ
А. ТЕОРЕТИЧЕСКИЕ ОСНОВЫ МЕТОДА
§ 1. Сущность и классификация кулонометрических
методов
Согласно закону Фарадея, количество окисленного или
восстановленного под действием электрического тока вещества равно (см.
книга 2, гл. VII, § 1 и гл. VIII, §7):
»-* <¦>
где # — масса электрохимически превращенного вещества, г;
м — молекулярный, атомный или ионный вес вещества;
Q — количество электричества в кулонах, равное произведению силы тока
электролиза (/э) в амперах и времени электролиза (тэ) в секундах (Q^iaT^);
« — число электронов, участвующих в электрохимическом окислении или
восстановлении одного иона, атома или молекулы вещества:
F— число Фарадея, равное 96490 ± 3 (~ 96 500) кулонам, необходимым для
электрохимического превращения 1 г-экв вещества.
В зависимости от происходящих в растворе электрохимических
процессов различают прямую (первичную) кулонометрию и косвенную
(вторичную) кулонометрию, или ку лоно метрическое титрование.
Когда на рабочем электроде происходит одна из следующих
реакций;
А + пе —> В (электровосстановление) (2)
ИЛИ
В —яе —>¦ А (электроокисление) (3)
то, измеряя количество электричества, т. е. общее количество
электронов, обмененных при этих реакциях до полного их завершения, и зная
п и М вещества А или В, можно определить непосредственно их массу.
Этот способ относится к прямой кулонометрии.
Можно представить себе случай, когда и А, и В не реагируют на
электроде, тогда в их раствор вносят такое вещество, которое способно
в соответствующих условиях электроокисляться или электровосстанав-
ливаться согласно уравнениям:
D-me —> Z
или
Z + те —> D
а тот или другой продукт этих реакций (Z или D) способен химически
количественно взаимодействовать с определяемым веществом В или А.
Тогда количество электричества, затраченное на образование продукта
Z или D, эквивалентно содержанию В или А в растворе, и оказывается
возможным вычислить массу последних. Этот способ называется
косвенной кулонометрией.
Для выполнения кулонометрического определения первым
способом необходимо, чтобы определяемые вещества были электроактивными,
192 ГЛ. VI. КУЛОНОМЕТРИЯ И КУЛОНОМЕТРИЧЕСКОЕ ТИТРОВАНИЕ
между тем как во втором случае следует располагать электроактивными
вспомогательными реагентами независимо от того, электроактивны
определяемые вещества или нет.
По технике выполнения кулонометрический метод анализа делится
на потенциостатическую кулонометрию (или кулонометрию при
контролируемом потенциале) и амперостатическую (или гальваностатическую)
кулонометрию в зависимости от того, проводят ли электрохимическое
превращение веществ соответственно при постоянстве потенциала
электрода или при постоянстве силы тока электролиза. Каждый из этих
методов и техника выполнения их имеют свои ограничения,
преимущества и недостатки, но все они, без исключения, ставят перед
экспериментатором три обязательные задачи:
1) электрохимическое превращение вещества должно протекать со
100% выходом по току (100% эффективность тока), т. е. побочные
электродные реакции должны отсутствовать (только при этом условии
можно воспользоваться законом Фарадея для вычисления количества
определяемого вещества);
2) момент завершения электрохимической реакции при прямой
кулонометрии и химической реакции при косвенной кулонометрии
должен быть достаточно точно устанавливаемым;
3) количество электричества, вовлеченное в электродную реакцию,
должно быть точно определяемо подходящим способом.
Наиболее полную информацию для решения первой задачи и
частично второй (когда завершение химической реакции обнаруживают
электрохимическими методами) можно получить путем изучения
кривых зависимости ток — потенциал [/ = /(?)]. Учитывая, что в основном
кулонометрическому анализу подвергают водные растворы
определяемых веществ, следует принимать во внимание также возможность
электровосстановления или окисления самого растворителя (см. книга 2,
гл. VII, §2).
Вообще для проведения электрохимического окисления или
восстановления какого-либо вещества с 100%-ным выходом по току
необходимо электроду сообщить потенциал, отвечающий какой-либо точке на
кривой поляризации этого соединения.
Универсальным способом определения количества прошедшего
через анализируемый раствор электричества (независимо от метода куло-
нометрического анализа) является применение различных видов куло-
нометров или интеграторов тока, включенных в цепь электролиза
последовательно с ячейкой, в которой проводят кулонометрический анализ.
ПРЯМАЯ КУЛОНОМЕТРИЯ
§ 2. Прямая кулонометрия при постоянном потенциале
рабочего электрода (прямая потенциостатическая кулонометрия)
Принцип метода. В водном растворе, содержащем только ред-окс
систему А/В, оба компонента которой электроактивны, согласно кривой
I = f(E) (рис. 62), можно определить окисленную или восстановленную
форму, поддерживая постоянный потенциал рабочего электрода
соответственно отрицательнее или положительнее его равновесного
значения Яра вы.
Тогда соответствующие электрохимические реакции будут
выражены уравнениями (2) и (3). Например, при потенциале электрода Е2
произойдет первая реакция [уравнение (2)], вследствие чего уменьшится
концентрация формы А и увеличится концентрация восстановленного
§ 2. ПРЯМАЯ КУЛОНОМЕТРИЯ ПРИ ПОСТОЯННОМ ПОТЕНЦИАЛЕ 193
компонента В, а при Е\ пойдет вторая реакция [уравнение (3)],
сопровождаемая уменьшением концентрации В. В связи с этим
расположение кривых / = /(?) изменяется (см. рис. 62) до тех пор, пока
концентрации электропревращаемых веществ практически не станут исче-
зающе малыми.
Эффективность тока. Для обеспечения 100%-ной эффективности
тока для нужной реакции необходимо, чтобы испытуемый раствор не
содержал других электроактивных веществ, способных окисляться или
восстанавливаться при выбранных значениях потенциала рабочего
электрода. Поэтому перед тем как выбрать нужный потенциал рабочего
электрода, следует снять в отдельности кривые поляризации всех
веществ, присутствующих в испытуемом растворе, если, конечно, их
электрохимическое поведение в условиях про- +6
ведения анализа определяемого веще- с
ства заранее неизвестно. При наличии
мешающих компонентов следует либо
предварительно их удалить, либо, если
это возможно, изменить таким образом
условия электролиза, чтобы исключить
их мешающее влияние. Одним из
эффективных средств является связывание
мешающих компонентов в соответствующие
комплексы. Это приводит к уменьшению
равновесных концентраций примесей
настолько, что их потенциалы
электропревращения резко сдвигаются в нужную
сторону. Нередко можно достигнуть
успешных результатов изменением При- Рис. 62. Прямая потенциостатиче-
роды рабочего электрода, кислотности ^Л^льнГ^в (?) ??
среды И других факторов. ства в при потенциале рабочего
С Другой стороны, не всегда необхо- электрода Ех и вещества А при Е2.
димо удалять мешающие компоненты.
Если кривые i = f(E) определяемого вещества и мешающих
компонентов расположены достаточно далеко друг от друга, то можно
вначале провести электролиз мешающего вещества при потенциале,
отвечающем области его предельного тока (иначе говоря, предэлектролиз
для очистки растворов от примесей), затем, фиксируя потенциал
электрода, при котором происходит электропревращение определяемого
вещества, измерить количество электричества, израсходованного на его
полное восстановление или окисление.
Завершение электрохимической реакции. Скорость уменьшения
концентрации определяемого вещества пропорциональна силе тока
электролиза, а последняя в свою очередь пропорциональна концентрации
вещества. Поэтому при диффузионном переносе (см. гл. V) изменение
концентрации вещества, а следовательно, и силы тока электролиза во
времени при 100%-ной эффективности его и постоянном потенциале
рабочего электрода происходит по закону, описываемому уравнением
кинетики реакции первого порядка:
i% = iQe
-к*
(4)
где tx — сила тока электролиза в момент времени т;
/0 — сила тока электролиза в момент т = 0;
х — время электролиза;
К — коэффициент пропорциональности, равный DS(V6;
D — коэффициент диффузии;
194 ГЛ. VL КУЛОНОМЕТРИЯ И КУЛОНОМЕТРИЧЕСКОЕ ТИТРОВАНИЕ
5 — площадь поверхности электрода;
V — объем раствора;
6 т- толщина диффузионного слоя.
Таким образом, величина К зависит от соотношения S/V (D и б
более или менее постоянны для каждого опыта), т. е. тем больше, чем
больше отношение поверхности электрода к объему раствора в ячейке;
отсюда следует, что целесообразно применять большие электроды и
уменьшать объем испытуемого раствора для увеличения скорости
электролиза. Сокращению продолжительности анализа способствует и
увеличение скорости перемешивания раствора, так как при этом
уменьшается б и возрастает перенос вещества к диффузионному слою конвекцией.
Этот прием широко используется в электрогравиметрическом анализе.
Из приведенного уравнения (4) следует, что теоретически i
стремится к нулю при бесконечной продолжительности электролиза. В
действительности даже при бесконечном продолжении электролиза сила
тока по достижении некоторого малого значения перестает далее
уменьшаться и остается постоянной. Этот ток, названный остаточным током,
вызван электролизом разных примесей — деполяризаторов, например
кислорода, или медленным разрядом водорода и другими причинами.
Поэтому электролиз следует считать оконченным в момент, когда сила
тока перестает изменяться в течение некоторого времени. Однако и
в этом случае продолжительность электролиза остается довольно
большой, что несколько снижает ценность метода. Если руководствоваться
заданной точностью результатов анализа, то нет необходимости
продолжать процесс до прекращения изменения силы тока в цепи. Остановив
электролиз при величине сила тока, равной 0,01 i0 или 0ДШ0, можно
завершить электрохимическое превращение вещества с точностью 1 или
0,1% соответственно. Однако надо учесть, что для обеспечения
максимальной скорости электролиза потенциал рабочего электрода следует
поддерживать в пределах площадки предельного тока (см., например,
рис. 62), т. е. потенциал Е\ при анодной и Д2— при катодной реакциях,
так как в этих условиях наблюдается не только 100%-ный въ1ход по
току, но и максимально возможная сила тока, обусловленная переносом
вещества.
Когда электролиз проводят до достижения остаточного тока, при
заметной величине последнего следует внести поправку в полученное
значение Q, вычитая из него количество электричества, равное
произведению величины остаточного тока (*ОСт) на продолжительность
электролиза (тэ). Для снижения величины остаточного тока в ряде случаев
можно очистить электролит от электроактивных примесей проведением
предэлектролиза при том же или несколько более отрицательном, чем
Е2 (при катодных реакциях), или более положительном, чем Е\ (при
анодных реакциях), потенциале рабочего электрода (см. рис. 62).
Количество электричества, израсходованного на электролиз. Кроме
возможности использования различных кулонометров и интеграторов
тока для определения количества электричества при прямой потенцио-
статической кулонометрии эту задачу можно осуществить и другим
способом (см. специальную литературу).
§ 3. Использование прямой потенциостатической
кулонометрии
Оба компонента ред-окс системы электроактивны и растворимы.
Предположим, что водный раствор содержит только растворимую ред-
окс систему А/В (например, Fe+++/Fe++).
§ 3. ИСПОЛЬЗОВАНИЕ ПРЯМОЙ ПОТЕНЦИОСТАТИЧЕСКОЙ КУЛОНОМЕТРИИ 195
Согласно кривым i~f(E) для этих компонентов (см. рис. 62), при
поддержании постоянного потенциала рабочего электрода, отвечающего
величине Е2, можно количественно восстанавливать А до В, продолжая
электролиз до тех пор, пока сила тока электролиза не достигнет
некоторой малой и практически постоянной величины (остаточный ток).
При потенциале рабочего электрода, равном Еи электрохимическая
реакция протекает количественно справа налево (окисление В до А).
Смесь двух электроактивных восстановителей. Предположим, что
раствор содержит смесь двух восстановителей В и D. Реакциям
окисления соответствуют уравнения:
В — пе —>- А
ТУ —те —> Z
Если В окисляется при менее положительных значениях потенциала
рабочего электрода, чем D, то, как показывают кривые / = /(?)
(рис. 63), возможно дифференцированное определение, если
поддерживать потенциал электрода до завершения первой реакции в пределах
н i
,+ +?
Рис. 63. Прямая потенциостатическая
кулонометрия. Кривая / = / (Е) двух
восстановителей В и D в их смеси.
Рис. 64. Прямая
потенциостатическая кулонометрия. Кривые / = / (?)
ред-окс пары, один из компонентов
которой является твердой фазой.
а — 6, а затем, сообщив электроду потенциал в пределах с — rf,
окислить восстановитель D. Если же с самого начала проводить электролиз
при потенциале в пределах с — d, то происходит одновременное
окисление В и D при величине тока, равной сумме предельных токов обоих
компонентов вследствие аддитивности тока электролиза.
Один из компонентов ред-окс пары нерастворим (твердая фаза).
Например, раствор содержит ионы серебра. Электровосстановление,
выражаемое уравнением:
Ag+-he —> |Ag
сопровождается отложением металлического серебра на электроде и
падением предельного тока Ag+ из-за уменьшения его концентрации
в растворе. Кривые t ==/(?) показывают (рис. 64), что при потенциале
электрода, равном Ей реакцию в прямом направлении можно
осуществить без помех. Следует обратить внимание на то, что в отличие от
электродных процессов двух растворимых компонентов одной и той же
ред-окс пары в данном случае для твердой фазы отсутствует
предельный ток (активность твердой фазы постоянна).
Определяемое вещество неэлектроактивно. Если вещество неэлектро-
активно, но участвует в окислительно-восстановительной электродной
реакции другого, электроактивного вещества, определение его по
данному методу становится возможным. Например, комплексон III (H2Y—)
на ртутном электроде анодно не окисляется, так как Hg окисляется при
196 ГЛ. VI. КУЛОНОМЕТРИЯ И КУЛОНОМЕТРИЧЕСКОЕ ТИТРОВАНИЕ
более отрицательном потенциале, чем комплексон III. Однако, как
следует из кривых i — f(E) (рис. 65), Hg в присутствии комплексона III
окисляется легче, чем без него, вследствие связывания продукта
окисления (Hg++) в прочный комплекс HgY". Электрохимическая
реакция при этом выражается уравнением:
|Hg + H2Y"~-2e —> HgY""" + 2H+
Следовательно, при потенциале рабочего электрода Е можно
количественно определить комплексон III, так как электрохимическая
реакция окисления Hg при этом потенциале ограничивается скоростью
переноса из раствора к электроду молекул комплексона III. Поэтому при
полном связывании H2Y~" в комплекс HgY~~ ток в цепи стремится к
нулю. На этом же принципе основано, например, определение С1~-ионов
на Ag-электроде.
§ 4. Прямая кулонометрия при постоянной силе тока
электролиза (прямая амперостатическая кулонометрия)
Принцип метода. В этом способе прямой кулонометрии используется
в течение всего процесса анализа постоянная сила тока электролиза iB.
Метод имеет ограниченное применение, так как пригоден только для
•Hi
I -/
Рис. Ь5. Прямая потенциоста-
тическая кулонометрия.
Изменение кривых i = / (Е) при
определении неэлектроактивного
вещества.
Рис. 66. Прямая
амперостатическая кулонометрия.
Изменение кривых / = f (E) при
определении растворенных
веществ.
определения веществ, находящихся на электроде в твердом состоянии
(например, металлов, малорастворимых окисей или солей).
Действительно, если один из компонентов ред-окс пары А/В (см.
приведенный ранее пример) находится в растворе, то при анодной
реакции В — е-*А когда iQ>ia (предельного тока компонента В),
одновременно с окислением В будет происходить окисление воды: 2Н20 — Ае-+
-* f O2 + 4Н+. По мере уменьшения концентрации В (следовательно,
и id) доля окисленной воды будет возрастать, а при практически
полном исчезновении В весь так будет расходоваться на
электроокисление воды.
С некоторого момента электролиза такая же картина будет
наблюдаться и тогда, когда с самого начала i9 < id (рис. 66). До тех пор,
пока предельный ток В не станет равным току электролиза, в
электрохимической реакции участвует лишь В. При дальнейшем уменьшении
его концентрации повторятся описанные выше процессы. Таким
образом, нет возможности обеспечить 100%-ный выход по току для нужной
реакции и, хотя можно обнаружить какой-либо химической или
электрохимической реакцией момент завершения окисления В, не удастся
установить долю электричества, затраченную только на окисление В. Все
§ 4. ПРЯМАЯ КУЛОНОМЕТРИЯ ПРИ ПОСТОЯННОЙ СИЛЕ ТОКА ЭЛЕКТРОЛИЗА 197
сказанное справедливо, если в данных условиях провести катодный
процесс А + е-*В.
Если же определяемое вещество находится на электроде в
твердом состоянии (например, металл), то процесс упрощается. Как было
указано, твердые вещества не имеют предельного тока. Поэтому при
прохождении через ячейку тока электролиза ia на аноде окисляется
металл (Me — пе—*Меп+) до его полного растворения, после чего ток,
поддерживаемый постоянным, вызовет новую анодную реакцию за счет
н2о /**о2
Рис. 67. Прямая амперостатическая
кулонометрия. Изменение кривых
/ = / (Е) при определении анодным
растворением электроактивного
металла, находящегося в виде твердой
фазы на рабочем электроде.
>Н-н2о
Рис. 68. Прямая
амперостатическая кулонометрия. Изменение
кривых i = / (Е) при
последовательном восстановлении
электроактивной окиси металла Ме02,
находящейся в виде твердой фазы
на рабочем электроде.
окисления другой твердой фазы (если она также находится на
электроде), или самого электрода (если он электроактивен, например, Ag),
или находящихся в растворе более электроположительных
восстановителей, или, наконец, растворителя — обычно воды. В процессе анодного
растворения металла (рис. 67) потенциал электрода сравнительно мало
изменяется вследствие увеличения концентрации ионов растворяемого
металла, согласно уравнению Нернста. Но как только на электроде
возникает другая электрохимическая реакция, наблюдается скачок
потенциала, и тем больший, чем больше разность нормальных (на
практике— формальных) потенциалов двух ред-окс пар (?? — Е\ ),
участвующих в рассматриваемых электродных процессах. Это положение
делает понятным возможность дифференцированного
(последовательного) определения нескольких веществ, когда они находятся в твердом
состоянии на электроде и потенциалы их анодного растворения
достаточно различаются (~0,3 в и меньше в зависимости от числа
электронов п, участвующих в ред-окс реакциях).
Следовательно, для нахождения момента завершения реакции
достаточно проследить за изменением потенциала рабочего электрода во
времени и прекратить ток электролиза при наступлении скачка
потенциала или же вычертить кривые E = f(x) и по перегибу кривой найти
время завершения электрохимической реакции. Зная тэ в секундах,
соответствующих этому моменту, и величину тока электролиза i3 в
амперах, поддерживаемую в течение всего процесса постоянной, легко
вычислить количество электричества, затраченное на проводимую
реакцию (Q = bT9), и тем самым содержание определяемого вещества.
Все сказанное справедливо и для определения окисей металлов
(РЬО, РЬ02, МпОг и др.) и малорастворимых солей (например, AgCl)
на электроде, когда используются катодные токи их восстановления.
Возможно не только дифференцированное определение смеси разных
198 ГЛ. VI. КУЛОНОМЕТРИЯ И КУЛОНОМЕТРИЧЕСКОЕ ТИТРОВАНИЕ
окислов в надлежащих условиях, но и последовательное восстановление
одной и той же окиси, если металл, входящий в ее состав, обладает
несколькими степенями окисления, причем в первую очередь идет
восстановление наивысшей окиси, а затем окисей с более низкими степенями
окисления металла в них (рис. 68).
§ 5. Использование прямой амперостатической
кулонометрии
Метод имеет разнообразное применение: определение толщины
металлических покрытий и окисных пленок и, следовательно, количества
вещества в них, определение концентрации искомого вещества в
растворе предварительным выделением на электроде и последующим
анодным или катодным растворением отложенного продукта
электрохимической реакции.
КУЛОНОМЕТРИЧЕСКОЕ ТИТРОВАНИЕ
§ 6. Кулонометрическое титрование при постоянной
силе тока электролиза
Принцип метода. В методе используется сохранение заданной силы
тока электролиза iQ постоянной в течение всего процесса анализа и
измерение продолжительности электролиза тэ. Но так как в отличие от
прямой амперостатической кулонометрии в данном случае метод
применяется для определения растворенных веществ, в процессе электролиза
невозможно одновременное сохранение и силы тока, и потенциала
электрода постоянными. Поэтому, как было указано ранее, при работе
с постоянной силой тока электролиза из-за изменения потенциала
электрода неизбежны побочные электрохимические процессы и не
обеспечивается 100%-ная эффективность тока для необходимой электродной
реакции. Для предупреждения затраты электричества на побочные
электрохимические реакции в испытуемый раствор вносят электроактивное
вещество (вспомогательный реагент), которое с самого начала или
после некоторого периода электролиза (в зависимости от условий)
участвует в электрохимической реакции. При этом необходимо, чтобы
продукт реакции (промежуточный реагент) был способен количественно
химически взаимодействовать с определяемым веществом.
В кулонометрическом титровании возможны следующие случаи.
Определяемое вещество неэлектроактивно. В испытуемый раствор,
содержащий неэлектроактивное определяемое вещество А, вносят
достаточно большое количество электроактивного вспомогательного
реагента— вещества Z, способного, например, к восстановлению. Под
действием тока, сила которого поддерживается постоянной, на катоде
протекает электрохимическая реакция:
Z + e —>- D
Промежуточный реагент D, предположим, реагирует с
определяемым веществом А с образованием малорастворимого осадка:
A+.D ^z± |,AD
Суммарная реакция тогда будет:
Z + A + e ^z± jAD
Очевидно, что все электричество, прошедшее через ячейку до завершения
этой реакции, эквивалентно количеству образованного осадка и,
следовательно, содержанию А в растворе.
§ 6. КУЛОНОМЕТРИЧЕСКОЕ ТИТРОВАНИЕ ПРИ ПОСТОЯННОЙ СИЛЕ ТОКА 199
Определяемое вещество электроактивно. В испытуемый раствор,
содержащий определяемое электроактивное вещество В, вносят другое
электроактивное вещество (вспомогательный реагент) D. При заданном
анодном токе электролиза /*, если i\>id вещества В, в электродной
реакции участвуют одновременно В и D, согласно уравнениям:
В-е —> А
D-e —> Z
Если вспомогательный реагент D подобран так, что продукт его
электродной реакции Z (промежуточный реагент) способен в свою
очередь химически количественно взаимодействовать с А по типу
окислительно-восстановительной реакции, то в
растворе одновременно происходит следующая
химическая реакция:
B + Z !=? A + D
Суммарная реакция тогда будет:
В-е ^± А
т. е. как будто все вещество В электрохимически
переходит в вещество А, а вспомогательный
реагент не участвует в этой реакции, хотя в
действительности только часть В непосредственно
окисляется на электроде, а другая часть
окисляется промежуточным реагентом Z. Таким
образом, все израсходованное при этом
электричество эквивалентно содержанию В в растворе.
Та же картина наблюдается, если il3l<id
Рис. 69. Кулонометриче-
ское титрование. Кривые
анодного окисления
компонентов В, D и воды
в зависимости от
величины тока электролиза i
вещества В, с той лишь разницей, что вначале
происходит только электропревращение веще
ства В до тех пор, пока U вещества В
(вследствие уменьшения его концентрации в процессе электролиза) не станет
меньше /" (рис. 69). С этого момента процесс протекает аналогично
описанному выше. При ilu не обеспечивается 100%-ный выход по току
(окисляется также вода). Все сказанное справедливо и для катодных
процессов.
В кулонометрическом титровании обычным условием является
h > id* так как, с одной стороны, этим методом определяют малые
количества вещества (id мал), с другой, — концентрация вспомогательного
реагента должна быть достаточно большой (на практике рекомендуется
приблизительно 1000-кратная и даже выше концентрация определяемого
вещества) и, наконец, ток электролиза 1Э в основном выбирают
достаточно большим для сокращения продолжительности анализа.
Когда протекающие в растворе химические реакции относятся к
типам осаждения и комплексообразования, часть вспомогательного
реагента расходуется, но так как его концентрация значительно больше
концентрации определяемого вещества, эта часть составляет лишь
небольшую долю его и не влияет на эффективность генерации
промежуточного реагента. При окислительно-восстановительных й
кислотно-основных химических реакциях концентрация вспомогательного реагента
практически остается постоянной, так как он все время регенерируется,
т. е. действует как катализатор данной реакции, не расходуясь в
процессе анализа. При этом сколько его восстанавливается (или
окисляется) на электроде, столько же выделяется при химическом
взаимодействии промежуточного реагента с определяемым веществом.
200 ГЛ. VI. КУЛОНОМЕТРИЯ И КУЛОНОМЕТРИЧЕСКОЕ ТИТРОВАНИЕ
Необходимость вносить в испытуемый раствор вспомогательный
реагент отпадает лишь при кулонометрическом титровании в водных
растворах кислот и щелочей. Известно, что вода и ее ионы способны
восстанавливаться на катоде и окисляться на аноде. При прохождении
тока через ячейку в катодной камере восстанавливаются Н+-ионы
сильной кислоты
2Н+ + 2е > f H2
и вода
2Н20 + 2е —> f H2 + 20H"
Образовавшиеся ОН--ионы нейтрализуют Н+-ионы, не
восстановленные на электроде.
В анодной камере происходит обратный процесс — окисление
ОН--ионов щелочи
40Н" - 4е —> f 02 + 2Н20
И ВОДЫ
2Н20 - 4е —> f 02 + 4Н+
При этом Н+-ионы реагируют с ОН~-ионами щелочи с
образованием воды. В итоге в обоих случаях все электричество затрачивается
на завершение кислотно-основной реакции. Единственным необходимым
условием является прибавление в испытуемый раствор сильного
индифферентного электролита в виде нейтральной соли (K2SO4 или Na2S04)
для увеличения электропроводности раствора, иначе из-за большого
сопротивления ячейки ток электролиза будет слишком мал и это
приведет к резкому увеличению продолжительности анализа.
Таким образом, при кислотно-основном кулонометрическом
титровании вспомогательным реагентом фактически служит сам
растворитель— вода.
Роль вспомогательного реагента в кулонометрическом титровании
заключается в следующем. Когда i9 < id, электрод приобретает
потенциал, при котором возможно лишь электропревращение определяемого
вещества; но как только в процессе электролиза id становится меньше
«э (кроме того, и для работы может быть принято условие h>id), или
если определяемое вещество неэлектроактивно, то в электродную
реакцию вовлекается вспомогательный реагент, и рабочий электрод
приобретает потенциал, отвечающий электропревращению вспомогательного
реагента. Следовательно, вспомогательный реагент служит своего рода
буфером, препятствующим смещению электродного потенциала до
таких значений, когда возможны другие электрохимические процессы,
продукты которых не способны химически взаимодействовать с
определяемым веществом, а это привело бы к перерасходу электричества
(побочные процессы).
В некоторых случаях, когда электрод состоит из металла,
способного окисляться (не инертного), он сам может служить источником
(вспомогательным реагентом) электрогенерируемого промежуточного
реагента, с которым химически взаимодействует определяемое вещество.
Например, продукты окисления ртутного и серебряного электродов
Hg++- и Ag+-HOHbi широко применяются в качестве кулонометрических
титраитов.
Все приведенные примеры показывают, что при электролизе,
независимо от того, электропревращается или непосредственно
определяемое вещество или вспомогательный реагент, на каждый заряд одного
иона расходуется один электрон. Таким образом, в кулонометрии
реагентом— своего рода титрантом — фактически является электрон, а в
косвенной кулонометрии, кроме того, происходит химическая реакция,
§ 6, КУЛОНОМЕТРИЧЕСКОЕ ТИТРОВАНИЕ ПРИ ПОСТОЯННОЙ СИЛЕ ТОКА 201
аналогичная реакциям, используемым в объемном анализе, поэтому
данный метод более известен под названием кулонометрического
титрования.
В отличие от других титриметрических методов в косвенной куло-
нометрии титрант готовится электрохимически непосредственно
действием электронов, причем электрогенерацию титранта можно осуществить
в испытуемом растворе за счет внесенного в него подходящего реактива.
Такой способ называется ку лоно метрическим титрованием с внутренней
генерацией.
Электрогенерацию промежуточного реагента можно проводить и вне
испытуемого раствора, что становится особенно необходимым при
некоторых условиях: например, когда условия электрогенерации
промежуточного реагента со 100%-ным выходом по току не совпадают с
условиями количественного взаимодействия его с определяемым веществом.
Продукт электродной реакции из ячейки соответствующей конструкции
(см. рис. 78, стр. 211) поступает в титрационный сосуд, в котором
совершается химическая реакция, а конец этой реакции устанавливают
обычными, практикуемыми в титриметрических анализах способами.
Такой метод называется кулонометрическим титрованием с внешней
генерацией.
Определение эффективности тока. Для нахождения условия
обеспечения 100%-ной эффективности тока генерации промежуточного реагента
изучают кривые зависимости i = f(E) электрода в соответствующих
условиях. Однако следует учесть, что не все условия электролиза,
обеспечивающие 100%-ный выход по току, оптимальны для кулонометриче-
ского титрования, так как они могут оказаться непригодными для
протекания химической реакции в растворе и для того или иного метода
индикации конечной точки. Следовательно, необходимо обеспечить
правильное сочетание условий проведения кулонометрического титрования.
Обычно условия химической реакции известны по данным
титриметрических методов анализа, поэтому при различных плотностях тока
электролиза снимают кривые поляризации i = f(E) электродов из
различного материала (Pt, Au, Hg, W, С и др.) в растворе фона (при
соответствующей кислотности среды и температуре) в отсутствие и в
присутствии вспомогательного реагента. По полученным кривым находят
отдельные значения плотности тока, наблюдаемые в фоне (ц) и в смеси
фон и вспомогательный реагент (/0б) при одном и том же значении
потенциала испытуемого рабочего электрода. Эффективность тока X в
процентах вычисляют по формуле:
v (*'об-*ф) ЮР
л — .
'об
В основе этого способа лежит принцип аддитивности тока.
Полученные данные можно изобразить графически в координатах
выход по току (в %) — плотность тока i06 для каждой концентрации
вспомогательного реагента и других переменных, откуда и выбирают
оптимальные условия генерации промежуточного реагента. Как правило,
увеличение концентрации вспомогательного реагента приводит к
возрастанию эффективности тока. Однако не всегда целесообразно
работать при очень больших концентрациях вспомогательного реагента (см
стр.203).
В ряде случаев электрогенерацию промежуточного реагента
проводят при различных условиях, пригодных для химической реакции.
Для этого через раствор в ячейке пропускают количество
электричества (Qi) на —5% меньше, чем нш&шдимо для кулшометрического
202 ГЛЛ VI. КУЛОНОМЕТРИЯ И. КУЛОНОМЕТРИЧЕСКОЕ ТИТРОВАНИЕ
титрования заданного количества анализируемого вещества. Затем
вносят в ячейку анализируемое вещество и продолжают электролиз до
полного завершения химического взаимодействия, фиксируемого надежным
способом индикации конечной точки. На основании полученных данных
легко вычислить эффективность тока генерации промежуточного
реагента из простой пропорции.
Например, пусть для кулонометрического титрования А г вещества требуется
количество электричества, равное Q. Предварительно пропущено Qi (на ~5% меньше,
чем Q), а после внесения в ячейку А г определяемого вещества затрачено
дополнительно Q2. Если Qi + 0.2 = Q, то эффективность тока равна 100%, если же
Qi -4- Q2 > Q, то она меньше 100%. Для вычисления эффективности тока (X) в
процентах составляют пропорцию (Qi 4- Q2) : 100 = Q : X Откуда
Yr= Q-100
Qi + Q2
Естественно, в испытуемом растворе с фоном не должны
содержаться вещества, которые восстанавливаются при потенциалах
рабочего электрода более положительных (при катодном процессе
генерации промежуточного реагента) или окисляются при менее
положительных потенциалах (при анодном процессе), чем вспомогательный
реагент, иначе это приводит к перерасходу электричества ц процессе
электролиза. Кроме того, в испытуемых растворах всегда присутствует
растворенный из воздуха кислород, поэтому при катодных процессах,
если вспомогательный реагент восстанавливается при более
отрицательных потенциалах электрода, чем кислород, снова возникают
побочные электрохимические явления (восстановление кислорода),
отражающиеся на результатах анализа. В подобных случаях электрогенерацию
следует проводить в атмосфере инертного газа (например, N2),
очищенного предварительно от примесей кислорода.
Именно изучение кривых зависимости i — f{E) помогает установить
наличие соединений, мешающих правильному ходу кулонометрического
титрования. Для устранения их влияния на ход основной реакции
следует поступить так, как это указано выше (см. стр. 193). Сказанное,
однако, вовсе не означает, что в растворе всегда должны отсутствовать
другие соединения, способные восстанавливаться или окисляться
раньше, чем вспомогательный реагент (при соответствующих
электродных процессах генерации). Если продукты подобных электроактивных
веществ способны химически взаимодействовать с определяемым
веществом, то присутствие их не мешает кулонометрическому титрованию
определяемого вещества. Если же подобные примеси, кроме того,
способны в свою очередь химически взаимодействовать с промежуточным
реагентом, электрогенерированным из вспомогательного реагента, то
это позволяет дифференцированно определить примеси и искомое
вещество. Возможность последовательного кулонометрического титрования
нескольких соединений основывается, следовательно, на тех же
принципах, что и теории других электрохимических методов анализа, в
первую очередь — потенциометрического титрования. Для решения таких
задач весьма важно знать формальные потенциалы ред-окс систем,
участвующих в реакциях.
Когда выход по току несколько меньше 100%, все же можно
получить достаточно точные результаты кулонометрического титрования,
если определяемое вещество электроактивно. Поскольку электрогенери-
рованный промежуточный реагент, как мы видели в ранее приведенном
примере, взаимодействует лишь с частью определяемого вещества, а
другая часть непосредственно превращается на электроде, ошибка
сказывается только на первой доле определяемого вещества, вызывая лишь
§ 6. КУЛОНОМЕТРИЧЕСКОЕ ТИТРОВАНИЕ-ПРИ ПОСТОЯННОЙ СИЛЕ ТОКА 203
частичное завышение результатов анализа. В этом случае принято
говорить об эффективности титрования, которую находят непосредственно,
проводя кулонометрическое титрование известных количеств
определяемых компонентов в различных условиях. По величине количества
электричества Qu затраченного при кулонометрическом титровании
известного содержания вещества, и по величине Q, теоретически
рассчитанного для этого же количества вещества, вычисляют эффективность
титрования (Z) в процентах по формуле:
Z-3 .100
Эффективность титрования может быть больше эффективности тока
или равна ей.
При проведении кулонометрического анализа (как прямого, так и
косвенного) необходимо, чтобы анодная и катодная камеры ячейки
были изолированы, иначе продукты химической и электрохимической
реакции могут принять участие в электрохимической реакции на
другом электроде, что вызовет перерасход электричества, и не будет
обеспечена 100%-ная эффективность тока.
Например, при электрогенерации на катоде Fe++ из Fe+++, если бы
анод находился в одной камере с катодом, то часть Fe++,
переместившись к аноду, окислялась бы до Fe+++, не успевая взаимодействовать
с определяемым окислителем в растворе (при косвенной
кулонометрии) или, возвращаясь к катоду, повторно приняла бы участие в
катодном процессе (при прямой кулонометрии). Поэтому изоляция анодной
и катодной камер друг от друга — совершенно необходимое условие,
особенно при кислотно-основном титровании с генерированными Н+- и
ОН~-ионами вследствие их большой подвижности и малого размера.
Определение момента завершения кулонометрического титрования.
Почти все способы индикации конечной точки реакции, используемые
в титриметрических методах анализа, пригодны и при
кулонометрическом титровании. Применяются цветные индикаторы (в основном при
кислотно-основных и окислительно-восстановительных реакциях), а
также ряд инструментальных методов (потенциометрия, кондуктомет-
рия, амперометрия, спектрофотометрия, радиометрия и т. д.). Из них
наиболее часто применяют потенциометрию и амперометрию, особенно
биамперометрию. Большая концентрация вспомогательного реагента
отрицательно сказывается при использовании кондуктометрического
метода индикации конечной точки, так как электропроводность является
функцией всех ионов в растворе, и поэтому небольшое ее изменение
в процессе кулонометрического титрования трудно обнаружить.
Отметим также, что при использовании цветных индикаторов
необходимо прекратить электролиз в момент завершения титрования, т. е.
в момент перехода окраски индикатора. С другой стороны,
кулонометрическое титрование проще проводить с цветными индикаторами при
непрерывной генерации промежуточного реагента, проследив лишь за
изменением окраски раствора, между тем как при инструментальных
методах обычно приходится периодически останавливать электролиз
для измерения соответствующего параметра (?, i и пр.). Однако не
исключена возможность непосредственно следить за изменением этих
параметров, не прекращая электролиз. Нет сомнения, что
инструментальные методы, как более чувствительные, обеспечивают максимально
возможную точность результатов анализа, хотя аппаратурное
оформление метода при этом усложняется.
Определение количества электричества. Так как Q = it, то, измеряя
каким-нибудь способом величину тока электролиза h в амперах, сохра-
204 ГЛ. VI. КУЛОНОМЕТРИЯ И КУЛОНОМЕТРИЧЕСКОЕ ТИТРОВАНИЕ
няемую постоянной в течение всего процесса, и продолжительность
электролиза тэ в секундах до момента завершения химической реакции,
установленного одним из упомянутых выше методов, легко вычислить
Q. Это наиболее простой и доступный способ, хотя с таким же успехом
можно использовать в цепи генерации кулонометр любого типа или
интегра-Гор тока, подключенный последовательно к рабочей ячейке.
Тогда необходимость сохранять постоянство силы тока электролиза
отпадает.
§ 7. Использование амперостатического кулонометрического
титрования
Кулонометрическое титрование
по методу осаждения
Допустим, что требуется определить содержание ионов галогена.
Используют серебряный рабочий электрод в качестве анода. Окисление
электрода в присутствии галогенид-ионов
происходит при менее положительных
значениях потенциала, чем при их отсутствии.
При этом предельный ток пропорционален
концентрации Hal" в растворе. Если i0>id
(рис. 70), протекают две электрохимические
реакции:
Ag + НаГ — е—•> \ AgHal
Ag — е—>Ag+
Образуемые Ag+-HOHbi диффундируют
в раствор и химически взаимодействуют с
На1~-ионами:
Ag+ + HaP ^z± ф AgHal
концентрации Hal" доля тока электролиза,
отвечающая первому электродному процессу, уменьшается и
увеличивается доля тока, отвечающая второму процессу. После завершения
связывания Hal"" в малорастворимый осадок в растворе появится
избыток Ag+, что можно обнаружить каким-либо методом индикации
конечной точки. Так как количество электричества, затраченного при
второй электрохимической реакции, эквивалентно количеству
генерированных Аё+-ионов, связываемых в AgHal за счет химического
взаимодействия, общее количество электричества, прошедшее через ячейку,
соответствует содержанию На1~ в растворе независимо от того, связывается
ли он электрохимически или химически. Это позволяет вычислить
содержание На1~ в растворе, пользуясь уравнением (1).
Кулонометрическое титрованиепо методу
комплексообразования
Металлическая ртуть в присутствии H2Y-~-hohob окисляется при
потенциалах электрода менее положительных, чем в их отсутствие, a i&
в этих условиях пропорционален концентрации H2Y~-hohob в растворе,
и если h > id, то на электроде протекают две реакции
Hg + H2Y~-2e —> HgY—+ 2Н+
Рис. 70. Кулонометрическое
титрование по методу осаждения.
Изменение кривых i = / (Е) при
определении галогенидов (Hal'")
электрогенерированнымя Ag+-
ионами.
По мере уменьшения
Hg-2e —> Hg++
§ 7. АМПЕРОСТАТИЧЕСКОЕ КУЛОМЕТРИЧЕСКОЕ ТИТРОВАНИЕ
205
сопровождаемые химической реакцией между образуемыми Hg++- и
Н2У"~-ионами в растворе
Hg++ + H2Y~~ ^z± HgY~~ + 2H+
По завершении химической реакции в растворе снова появятся
Hg++-HOHbi уже ничем не связываемые, что можно определить
соответствующими методами индикации конечной точки. Сказанное
относительно израсходованного количества электричества и вычисления
содержания H2Y— справедливо и для данного случая. Кривые i = f(E)
для этих реакций схематически показаны на рис. 71.
Рис. 71. Кулонометрическое
титрование по методу комплексообразо-
вания. Изменение кривых i = / (Е)
при определении
комплексна III (H2Y~~) электрогенерирован-
ными Hg++-noHaMH.
Рис. 72. Кулонометрическое
титрование неэлектроактив-
ного органического
вещества. Кривые i=*f(E) ред-
оке системы Вг2/2Вг~ при
определении фенола электро-
генерированным Вг2.
Реакция между комплексоном III и ртутью широко используется
для определения многих катионов, образующих с комплексоном III
прочные комплексы, как прямым, так и обратным титрованием (по
методу остатка).
Кулонометрическое титрование по методу
окисления — восстановления
Определяемое вещество неэлектроактивно. Определение различных
органических соединений (оксинов, аминов, фенолов и др.) можно
осуществить электрогенерированным Вг2 из бромида на Pt-аноде, согласно
электрохимической реакции 2Вг~ — 2е —> Вг2, которая сопровождается
химической реакцией (например, с фенолом)
3Br2 + C6H5OH ^Z± СбН2Вг3ОН + ЗВт" + ЗН+
Так как фенол в этих условиях неэлектроактивен (его кривая i =
= f{E) изображена штриховой линией на рис. 72), протекает одна
электродная реакция окисления бромида, а фенол только химически
титруется образованным на электроде бромом, избыток которого легко
обнаружить. Электрогенерированный Вг2 можно использовать и для
обратного титрования, особенно когда реакция бромирования
органических соединений протекает медленно. Избыток электрогенерированного
Вг2 титруют каким-нибудь электрогенерированным восстановителем,
например Си+ из солей двухвалентной меди.
Определяемое вещество электроактивно. При определении Се"4*4"*
в раствор вводят достаточно большое количество соли Fe+++. На Pt-ка-
тоде при /э > id (рис. 73) протекают две электрохимические реакции
Се++++ + е —> Се++* и Fe+*++* —> Fe*+
206 ГЛ. VI. КУЛОНОМЕТРИЯ И КУЛОНОМЕТРИЧЕСКОЕ ТИТРОВАНИЕ
сопровождаемые химической реакцией между Fe++
Ce++++ + Fe++ ^z± Ce+++ + Fe+++
и Се
т. е. фактически все электричество расходуется на восстановление
Се++++, а содержание вспомогательного реагента остается постоянным
до конечной точки вследствие регенерации его химической реакцией
окисления Се++++-ионами электровосстановленных Регионов.
Появление после конечной точки избытка Ре++-ионов вызывает резкое падение
окислительно-восстановительного потенциала пары Fe+++/Fe++, что легко
обнаружить, например потенциометрическим методом.
Кул онометрическое титрование
по кислотно-основному методу
Кривые i = f(E) в различных стадиях титрования сильной кислоты
электрогенерированными на катоде ОН~-ионами представлены на
Рис 73. Кулонометрическое
титрование по методу окисления —
восстановления. Изменение кривых / = /(?)
при определении церия (IV)
электрогенерированными Ре++-ионами.
Рис. 74. Кулонометрическое
титрование по кислотно-основному методу.
Изменение кривых / = /(?) при
определении сильной кислоты
электрогенерированными ОН~-ионами.
рис. 74. На Pt-катоде при iB>id идут две электрохимические реакции
2Н+ + 2е —> f Н2 и 2Н20 + 2е —> f H2 + 20H"
сопровождаемые химической реакцией
ОН"ЧН+ +± Н20
Появление после окончания этой реакции избытка ОН" можно
обнаружить любыми методами индикации рН растворов (например, с
помощью кислотно-основного индикатора, рН-метром и другими
способами). Так как растворенная двуокись углерода титруется в этих
условиях, искажая результаты анализа, следует предварительно удалить ее.
При титровании слабых кислот картина аналогична.
Электрохимические реакции в данном случае выражаются уравнениями
2HAn + 2* —>f Н2 + 2Ап~ и 2Н20 + 2е —*f H2 + 20H~
а химическая реакция в растворе уравнением
01Г + НАп ^± Н20 + АгГ
Электрогенерируя на Pt-аноде Н+-ионы, можно кулонометрически
титровать таким же способом основания в анодной камере.
§ 9. ОСОБЕННОСТИ КУЛОНОМЕТРИЧЕСКИХ МЕТОДОВ АНАЛИЗА 207
§ 8- Кулонометрическое титрование при постоянном
потенциале рабочего электрода
Кулонометрическое титрование при постоянном потенциале
рабочего электрода пока еще мало практикуется. Известны лишь единичные
работы и указания о возможности использования такого метода
в анализе.
Кроме того, предложены другие варианты кулонометрического
анализа. Практическое выполнение всех этих методов имеет более или
менее специфический характер, хотя теоретические предпосылки их
основаны на тех же описанных ранее положениях. Так как изложение
этих методов выходит за рамки программы, мы не останавливаемся
на них.
§ 9. Особенности кулонометрических методов
анализа
Кулонометрические методы анализа применяются преимущественно
для определения малых количеств веществ. Учитывая, что около 105 к
(точнее ~ 96500 к) отвечает 1 г-экв электропревращенного вещества,
при измерении нескольких десятков микрокулонов (10~6 /с), что вполне
осуществимо, можно определять около 10~10 г-экв вещества.
Всем кулонометрическим способам анализа присущи, кроме
большой чувствительности, высокая правильность и воспроизводимость
анализа. Эти характеристики методов главным образом зависят от
правильности и воспроизводимости определения конца исследуемой
реакции и количества электричества. Так как в данном методе шзтользуются
электрохимические процессы, общими и основными ограничивающими
факторами являются величина остаточного тока и ее
воспроизводимость. При кулонометрическом титровании важную роль играет
чувствительность методов нахождения момента завершения химической
реакции, которая становится доминирующим фактором. В этом
отношении наиболее эффективны спектрофотометрический, радиометрический
и биамперометрический (с двумя большими поляризованными
электродами) методы.
Правильность и воспроизводимость определения количества
электричества, затраченного на нужную реакцию, обусловлены выбранным
способом измерения и качеством применяемых измерительных приборов.
В отношении селективности методов преимущество следует отдать
потенциостатической кулонометрии, так как при строгом поддержании
постоянного потенциала рабочего электрода в границах площадки
предельного тока никакие побочные электрохимические реакции не
происходят (однако следует учитывать сделанные выше замечания,
см. стр. 193).
Кулонометрическому титрованию присуща большая
чувствительность, чем всем другим известным титриметрическим методам. Кроме
того, кулонометрическое титрование обладает рядом несомненно
ценных преимуществ: исключается необходимость подготовки
стандартных растворов титрантов; могут быть использованы такие реагенты,
которые невозможно приготовить в виде стандартных растворов, или
образующие малоустойчивые растворы (С12, Вг2, Ag++, Cu+, Ti+++, Sn++
и др.); анализируемые растворы в процессе электротитрования не
разбавляются; чистота вспомогательных реагентов имеет небольшое
значение, так как предэлектролизом можно освободиться от мешающих
примесей; имеется возможность в одном и том же растворе
вспомогательного реагента многократно повторять анализ с новыми порциями
208 ГЛ. VI. КУЛОНОМЕТРЙЯ И КУЛОНОМЕТРИЧЕСКОЕ ТИТРОВАНИЕ
испытуемого раствора; в химическую реакцию вовлекаются неизмеримо
малые количества титранта; метод позволяет полностью
автоматизировать процесс анализа, что особенно ценно при работе с радиоактивными
веществами; титранты в момент генерации проявляют большую
химическую активность (зачастую реакция протекает непосредственно на
самом электроде).
Б. АППАРАТУРА И ТЕХНИКА ВЫПОЛНЕНИЯ АНАЛИЗА
§ 10. Электроды
В кулонометрическом анализе требуются два рабочих электрода.
Тот, на котором протекает необходимая электрохимическая реакция,
называется генераторным (или рабочим), а второй — вспомогательным.
Кроме того, для потенциостатической кулонометрии требуется электрод
сравнения, относительно которого контролируется потенциал
генераторного электрода. При кулонометрическом титровании, если применяются
электрохимические методы индикации завершения химических
реакций, следует дополнительно располагать соответствующими
индикаторными электродами (см. гл. II, III).
В качестве материалов для генераторных электродов могут быть
использованы платина, золото, серебро, ртуть, амальгамы, графит и
иногда вольфрам, медь, свинец, хром и пр. Наиболее часто применяются
платина и ртуть; платина более пригодна для анодных процессов,
а для катодных процессов — в тех случаях, когда электропревращение
вещества протекает при более положительных значениях потенциала
электрода, чем выделение водорода (из-за малого перенапряжения
водорода на платине). На ртутном электроде можно осуществить почти
все катодные процессы благодаря большому перенапряжению
водорода на нем. Однако из-за легкости анодного растворения ртути
проведение электролиза при несколько более положительных значениях
потенциала, чем потенциал НВЭ, недопустимо. Таким образом, эти два
электрода дополняют друг друга.
Золотой электрод подобен платиновому.
Серебряный электрод главным образом используется для
генерации Ag+ при определении галогенидов и других анионов, образующих
с Ag+ малорастворимые или комплексные соединения.
В некоторых случаях для увеличения перенапряжения водорода
благородные металлы покрывают слоем ртути либо электролитически,
либо выдержав их некоторое время в ртути; иногда пользуются
соответствующими амальгамами.
Графитовый электрод позволяет работать в несколько более
отрицательных областях потенциалов, чем электроды из благородных
металлов, но из-за пористой структуры, обусловливающей адсорбцию
веществ из раствора, он дает несколько менее воспроизводимые
результаты измерения электрических параметров и высокий остаточный ток.
Однако при соответствующей обработке (пропитка различными
составами, например смолами, парафином и пр.) графит оказывается очень
полезным генераторным (а так же индикаторным) электродом.
Металлические электроды могут иметь самые разнообразные
формы; они используются в виде пластинок, проволок, спиралей, сеток
и других форм. Графитовые электроды обычно имеют цилиндрическую
форму; ртуть для получения электродов с большой поверхностью
наливают на дно электролизера; в последнем случае электрический
контакт осуществляют проволокой из благородного металла, впаянной
§11. ЭЛЕКТРОЛИЗЕРЫ
209
в дно сосуда, другой конец которой контактирует с токопроводящеи
проволокой (медной). Иногда металлические электроды припаивают
к медной проволоке, затем спай и медную проволоку впаивают в
стеклянную трубку. Перед работой поверхность твердых электродов
подвергают обработке.
В зависимости от ранее проведенной электродной реакции ртуть
в электролизере следует или заменить новой порцией, или подвергнуть
кислотной обработке либо поляризации при соответствующих условиях.
В качестве вспомогательных электродов в основном служат
электроды из благородного металла, чаще всего из платины, иногда из
серебра (когда приходится избегать выделения на аноде газообразного
хлора, разрушающего платину). Это дает возможность помещать
вспомогательный электрод в одну камеру с генераторным электродом.
Электроды сравнения должны быть неполяризуемыми, т. е.
сохранять потенциал постоянным при прохождении через них малых токов.
Наибольшее применение в качестве электрода сравнения имеет Нас. КЭ,
хотя с успехом могут быть использованы любые другие электроды
сравнения.
§11. Электролизеры
Идеальный электролизер должен обеспечить следующие условия:
герметизацию; термостатирование; подвод газа для удаления
растворенных электроактивных газов; хорошее
перемешивание испытуемого раствора; удобное
размещение электродов, механической или
магнитной мешалки и электролитического ключа;
исключение диффузии анолита и католита из
одной камеры электролизера в другую.
Однако соблюдение всех перечисленных
условий не обязательно во всех случаях
проведения кулонометрического анализа. Например,
термостатирование требуется тогда, когда
необходимо работать при температуре выше
комнатной (если химическая реакция или
электрогенерация протекают при повышенной или
пониженной температуре). Герметизация и подвод
инертного газа необходимы, если в электроли-
зируемом. растворе содержатся С02, Ог, NH3,
H2S и т. п. или если необходимо исключить
диффундирование их из воздуха, поскольку эти
соединения могут искажать результаты анализа,
участвуя в электродных процессах или в
химических реакциях.
В качестве инертного газа удобнее всего
применять чистый азот, свободный от примесей
других газов (обычно от кислорода).
Простейшим способом очистки является
предварительное пропускание N2 через кислый раствор соли
двухвалентного ванадия или хрома.
Соблюдение всех остальных условий обязательно. Перемешивание
с помощью механической или магнитной мешалки не только
увеличивает скорость электрохимической реакции, но и обеспечивает
эффективное взаимодействие электрогенерированного продукта с определяемым
веществом.
Рис. 75. Электролизер
для прямого потенцио-
статического
кулонометрического анализа:
/ — рабочий катод (ртуть);
2 — вспомогательный анод
(платина); 3 — электрод
сравнения; 4 — механическая
мешалка; 5 - газоподводящая
трубка.
210 ГЛ. VI. КУЛОНОМЕТРИЯ И КУЛОНОМЕТРИЧЕСКОЕ ТИТРОВАНИЕ
Электроды (генераторный и индикаторные в случае
электрохимической индикации конечной точки, а также вспомогательный, если он
находится в одном растворе с остальными) должны быть так размещены
в камерах электролизера, чтобы они не соприкасались друг с другом и
не сталкивались с мешалкой.
Очень важно анодную и катодную камеры ячейки разделить таким
образом, чтобы анолит и католит не смешивались. Для этой цели можно
применять диафрагму из пористого стекла, не мешающую миграции то-
копроводящих электролитов, но препятствующую диффузии католита
и анолита. Обычно вспомогательный электрод фиксируют в стеклянной
трубке со стеклянным пористым дном (типа фильтрующего стеклянного
тигля), содержащей электролит, и все вместе погружают в испытуемый
раствор, в котором находятся рабочий (генераторный) и индикаторные
Рис. 76. Электролизер для кулоно-
метрического титрования с биамперо-
метрической индикацией момента
завершения химической реакции:
/ — рабочий электрод (платина); 2—
вспомогательный электрод (платина); 3 — индикатор?
ные электроды (платина).
Рис. 77. Электролизер для кулоно-
метрического титрования кислот и
оснований с рН-метрической
индикацией момента завершения химической
реакции:
1 — рабочий катод (платина); 2 —
вспомогательный анод (серебро); 3 — индикаторный
электрод (стеклянный); 4 — газоподводящая
трубка; 5 — электролитический ключ;
6 — магнитная мешалка.
(при необходимости) электроды и мешалка (рис. 75 и 76). Мешалка
должна энергично перемешивать (но не разбрызгивать) не только
раствор, но и поверхность ртути, если она используется в качестве
генераторного электрода (см. рис. 75). Ради простоты можно также
применять два отдельных сосуда в качестве катодной и анодной камер,
которые сообщаются между собой электролитическим ключом (U-образной
стеклянной трубкой), наполненным соответствующим раствором
электролита (обычно тот же раствор электролита, в котором находится
вспомогательный электрод) или гелем агар-агара, насыщенным пбдходящим
для данной цели электролитом, — КС1, KN03 и т. д. (рис. 77).
Все способы разделения католита и анолита вызывают сильное
увеличение внутреннего сопротивления ячейки (порядка нескольких
тысяч ом). Это сказывается на омическом падении напряжения (iR) и
при больших значениях тока в потенциостатической кулонометрии
затрудняет сохранение постоянства потенциала рабочего электрода. Кроме
того, из-за большой величины R измеряемые в процессе электролиза
предельные токи имеют малую величину. В подобных случаях
целесообразно генераторный и вспомогательный электроды помещать в один
§ 12. ПРИБОРЫ ДЛЯ ИЗМЕРЕНИЯ КОЛИЧЕСТВА ЭЛЕКТРИЧЕСТВА 211
и тот же сосуд, но необходимо применять такие вспомогательные
электроды, чтобы продукты электрохимической реакции, протекающей на
них, не мешали основной реакции.
Для этого используют вспомогательные электроды, к которым
плотно пристают образуемые на них продукты электрохимической реакции.
В результате эти продукты не могут диффундировать к генераторному
электроду. Например, при определениях с использованием катодного
процесса в солянокислой среде можно применять в качестве
вспомогательного серебряный электрод.
При проведении катодных реакций к анализируемому раствору
иногда прибавляют гидразин или гидроксиламин. На вспомогательном
аноде эти соединения окисляются
до азота
N2H4-4e —> f N2 + 4H+
и 2NH2OH-2e —> f N2 + 2H20 + 2H+
и препятствуют другим анодным
процессам; вместе с тем азот не
восстанавливается на катоде и не
мешает протеканию основной
катодной реакции. Эти приемы пригодны
в потенциостатической прямой ку-
лонометрии, но в кулонометриче-
ском титровании прибавленные
посторонние вещества могут
реагировать с участвующими в химических
и электрохимических реакциях
веществами или с их продуктами. Так
как в кулонометрическом
титровании внутреннее сопротивление цепи
имеет мало значения вследствие
фиксации величины тока генерации,
необходимость использовать
подобные приемы в основном отпадает.
Один из видов ячейки с внешней генерацией кулонометрического
титранта представлен на рис. 78,
§ 12, Приборы для измерения количества электричества,
израсходованного на электролиз вещества
В кулонометрическом анализе могут быть использованы
различные типы кулонометров, основанные на измерении количества
продуктов электрохимических реакций или на непосредственном
интегрировании тока. И в том и в другом случае эти приборы должны находиться
в цепи электролиза и быть последовательно присоединенными к ячейке
с испытуемым раствором. Так как в любой части цепи величина тока
одна и та же, через эти приборы в единицу времени протекает такой
же ток, как и через анализируемый раствор, следовательно, одно и то
же количество электричества.
Электрохимические кулонометры. Эти приборы представляют собой
соответствующей формы электролизеры, в которых тем илц иным
способом определяют количество продукта катодной, анодной или же
суммарной реакции при условии 100%-ного выхода по току для
осуществляемых электродных процессов, Определив содержание продукта реак-
Рис, 78. Электролизер для
кулонометрического титрования с внешней
генерацией промежуточного реагента:
/ — рабочий электрод; 2 — вспомогательный
электрод; 3 — механическая мешалка; 4 — титра-
ционный сосуд, в который поступает раствор
с генерированным промежуточным реагентом;
5 — вспомогательный сосуд: 6 — капиллярные
трубки, через которые растворы с продуктами
электродных реакций поступают в сосуды 4 и 5.
212 ГЛ. VI. КУЛ'ОНОМЕТРИЯ И КУЛОНОМЕТРИЧЕСКОЕ ТИТРОВАНИЕ
ции, Легко вычислить количество электричества Q на основании закона
Фарадея, если известно уравнение электрохимической реакции.
Электрогравиметрические кулонометры. К ним относятся медный,
серебряный, галогено-серебряный и другие кулонометры.
Медный кулонометр. При прохождении тока через
сравнительно концентрированный раствор сульфата меди в сернокислой среде
на Pt-катоде откладывается плотный слой металлической меди. После
промывки и сушки электрод взвешивают с достаточно большой
точностью на аналитических весах. Вследствие частичного взаимодействия
выделенной меди с находящимися в растворе Си++-ионами и
образования Си+ при измерении очень малых величин количества электричества
точность определения несколько снижается.
Серебряный кулонометр. Из раствора нитрата серебра на
Pt-катоде осаждается металлическое серебро, которое затем
взвешивают. Во избежание обеднения раствора Ag+ при измерении больших
величин количества электричества в качестве анода используют
серебряную пластинку, окисляющуюся до Ag+-noHOB, которые пополняют
убыль их вследствие катодного процесса. Преимущество этого кулоно-
метра перед медным заключается в том, что серебро имеет почти в три
раза больший электрохимический эквивалентный вес, чем медь, и не
окисляется на воздухе. Недостатком является рыхлость отложенных на
катоде кристаллов серебра, которые легко осыпаются при неудачном
промывании электрода. Плотные осадки получаются при
использовании аммиачных или цианидных растворов солей серебра.
Галогено-серебряный кулонометр. Этот кулонометр осо.-
бенно цецен для определения микроколичеств электричества. На Ag-
аноде образуется слой AgHal при электролизе растворов галогенидов
щелочных металлов. Наилучшим является иодсеребряный кулонометр.
Ё качестве анода применяется серебряная спираль; катод также
представляет собой спиралеобразную проволоку, предварительно
электрохимически покрытую слоем иодида серебра. Электролитом служит
5%-ный раствор иодида калия.
Все эти кулонометры очень несложны. Точность определения при
помощи этих приборов обусловлена не только их спецификой, но и
степенью точности взвешивания (чувствительностью аналитических весов).
Титрационные кулонометры. В электролизере таких кулонометров
при прохождении тока образуются растворимые продукты
восстановления (на катоде) или окисления (на аноде), которые затем титруют
обычным способом стандартными растворами. Естественно, в данном
случае катодная и анодная камеры электролизера должны быть
изолированы друг от друга во избежание диффузии растворов.
В подобных кулонометрах с успехом может быть использован
анодный процесс окисления, например иОдида до иода и титрование
последнего тиосульфатом, ванадила до ванадата в сернокислой среде и
титрование солью Мора, серебряного анода до Ag+ и титрование галогенидом,
или же катодный процесс восстановления, например соединения
трехвалентного железа до двухвалентного и титрование перманганатом,
воды до ОН~-ионов и титрование их какой-либо кислотой и т. д.
Во всех случаях достоверность определения количества
электричества зависит от точности установления титра и измерения
израсходованного объема стандартного раствора. Обращение с титрационными куло-
нометрами несложно; но они пригодны для определения лишь
сравнительно больших количеств электричества.
Газовые кулонометры. Принцип газовых кулонометров основан на
том, что при электролизе измеряют общий объем газа, выделенного
§ 12. ПРИБОРЫ ДЛЯ ИЗМЕРЕНИЯ КОЛИЧЕСТВА ЭЛЕКТРИЧЕСТВА 213
вследствие электрохимического разложения электролита на аноде и на
катоде. Эти приборы очень несложны, но чувствительность и точность
их малы при измерении небольших количеств электричества. Обычно
нижний предел составляет около 10 к, а верхний около 500 /с, если
емкость измерительного сосуда равняется 100 см3.
Ранее других газовых кулонометров предложен
кислородно-водородный (водяной) кулонометр. При разложении воды на двух Pt-электро-
дах, находящихся в одной камере получают на аноде 02, а на катоде Нг,
объем смеси которых измеряют в условиях термостатирования (±0,1° С),
приводят к нормальным условиям (0°С и 760 мм рт. ст.) и вычисляют
количество электричества. Для облегчения расчетов обычно пользуются
таблицами. Вода в электролизере должна содержать инертный
электролит для увеличения ее электропроводности Ц М раствор K2SO4 или
Na2S04). Теоретически 1 к соответствует 0,1741 мл смеси кислорода и
водорода. Так как некоторое количество газа растворяется в воде,
предварительно в течение 5 мин раствор насыщают им, проводя предэлектро-
лиз током 50—100 ма. При корректировании данных следует учесть
также парциальное давление водяных паров в газовой смеси. Как
правило, водяной кулонометр дает несколько заниженные результаты
вследствие образования перекиси водорода на аноде, что становится
заметным при измерении больших количеств электричества.
Во избежание этой ошибки был предложен другой газовый
кулонометр — водородно-азотный. В этом случае вода должна содержать
0,1 моль сульфата гидразина, вследствие чего взамен воды на аноде
окисляется гидразин до азота (см. стр. 211). Но так как катодный
процесс тот же, и здесь раствор остается нейтральным. При нормальных
условиях объем 0,1741 мл смеси азота и водорода отвечает 1 я.
Колориметрические кулонометры. В этих кулонометрах измеряют
с помощью электро- или спектрофотометров изменение оптической
плотности растворов, подвергающихся электролизу. Такой способ
измерения Q имеет сложное аппаратурное оформление и требует некоторых
дополнительных операций (например, построения калибровочных
графиков для нахождения концентрации определяемого вещества по
оптической плотности). Однако этот метод, не отличаясь большой точностью,
очень чувствителен, и поэтому ценен при определении весьма малых
количеств электричества (от 0,01 до 1 /с). В принципе в
колориметрических кулонометрах могут быть использованы любые электрохимические
реакции, которые вызывают изменение интенсивности окраски или цвета
в растворе. Примером может служить возрастание рН раствора в като-
лите или его падение в анолите, сопровождаемое изменением
интенсивности окраски соответствующего кислотно-основного индикатора.
Применяя подходящие светофильтры, можно проследить за изменением
интенсивности окраски кислотной или щелочной формы индикатора.
Для измерения сравнительно больших количеств электричества
можно также использовать анодную реакцию с образованием из иодида
калия иода, обладающего желтой окраской. Для измерения очень
малых количеств электричества применяют иод-крахмальную реакцию.
Кулонометрические кулонометры. Принцип действия этих
кулонометров основан на катодном выделении (в процессе электролиза
подходящего вещества) металла из концентрированного раствора его соли на
электроде из благородного металла со 100%-ным выходом по току.
После завершения основной реакции реверсированием тока анодно
растворяют отложенный металл при постоянной силе тока и определяют
с помощью электрохронометра или секундомера продолжительность
этого процесса; окончание его обнаруживается резким скачком потен-
214 ГЛ, VI. КУЛОНОМЕТРИЯ И КУЛОНОМЕТРИЧЕСКОЕ ТИТРОВАНИЕ
циала анода, измеряемого относительно какого-либо электрода
сравнения. Обычно применяют медный кулонометр, включенный в цепь
последовательно о электролизером. Так как чувствительность измерения токов
и времени очень высока (микроамперные токи, 0,01 сек), можно
определять количества электричества в широких пределах — от очень малых
(~0,01 к) до достаточно больших (—100 /с) с хорошей точностью.
Интеграторы тока. Интеграторы тока представляют собой приборы,
регистрирующие непосредственно количество электричества, прошедшее
через замкнутую цепь. Интегрирование кривых ток — время
осуществляется различными способами: графическим, электромеханическим
или электронным.
Существуют приборы, непосредственно записывающие кривые i — т,
что удобнее и надежнее, чем построение кривой вручную по данным
отдельных измерений i и т, так как первый способ дает большую
информацию. Недостатком графического способа является то, что
воспроизводимость измерения токов в начальные и конечные периоды
электролиза невелика, в первом случае из-за быстрого изменения тока, а во
втором — вследствие непостоянства величин остаточных токов.
Расчет количества электричества значительно упрощается при
использовании механических или электронных самописцев, позволяющих
вычертить прямую зависимости lg i от т.
Некоторые типы приборов оснащены приспособлениями для
непосредственного цифрового отсчета количества электричества или его
регистрации.
Действие электромеханических интеграторов основано на
применении тахометрических двигателей постоянного тока. Используя усилители
тока и систему передаточных механизмов, можно добиться
пропорциональности между скоростью вращения механизма и мгновенным током,
проходящим через цепь, т. е. число оборотов должно соответствовать
количеству электричества. Некоторые из подобных приборов снабжены
счетным механизмом, фиксирующим число оборотов (калибровкой
прибора можно определить цену оборота в кулонах) или непосредственно
количество электричества, или же, что еще более удобно, количество
вещества в миллиграмм-эквивалентах. Некоторые интегрирующие
устройства обеспечивают автоматический вычет величины остаточного тока из
величины общего тока электролиза. Эти приспособления ускоряют
определение количества электричества, но по точности уступают ряду
электрохимических кулонометров, особенно при прохождении малых токов,
из-за недостаточно строгого соблюдения линейности между скоростью
вращения и величиной тока вследствие инерционных явлений в
тахометре и передаточных механизмах. Все же некоторые из подобных
приборов обеспечивают до 0,1% воспроизводимости в широких пределах
измеряемых количеств электричества.
Более новыми и совершенными являются электронные интеграторы
тока. Предложены приборы различной конструкции, но из них
наилучшие результаты дают те, в которых интегрирование тока осуществляется
измерением полного потенциала заряжения прецизионного конденсатора
до момента завершения электролиза.
§ 13. Приборы, обеспечивающие стабильность потенциала
рабочего электрода или силы тока электролиза, и установки
для кулонометрического анализа
Для поддержания постоянства потенциала рабочего электрода
в потенциостатической кулонометрии или силы тока электролиза в ам-
§ 13. ПРИБОРЫ, ОБЕСПЕЧИВАЮЩИЕ СТАБИЛЬНОСТЬ ПОТЕНЦИАЛА 215
перостатическои кулонометрии используются в основном потенциостаты
и амперостаты (или гальваностаты) соответственно.
При отсутствии потенциостатов и гальваностатов можно
пользоваться несколько менее надежной, но простой схемой. При работе в по-
тенциостатическом режиме из внешнего источника постоянного тока
с напряжением 20—40 в с помощью делителя напряжения на электроды
(генераторный и вспомогательный)
налагают такое напряжение, чтобы
потенциал рабочего электрода
соответствовал значению, при котором
необходимо провести электрохимический
процесс. Потенциал рабочего
электрода относительно электрода сравнения
контролируют с помощью
потенциометра компенсационным методом. Для
предотвращения изменения заданного
потенциала электрода в процессе
электролиза периодически увеличивают
(или уменьшают) налагаемое извне
на электроды напряжение ручным
способом.
Для кулонометрического
титрования достаточно иметь внешний
источник постоянного тока с большим
выходным напряжением (~ 200—300 в).
Включая последовательно к ячейке
необходимые высокоомные
сопротивления, можно получить стабильный
ток электролиза требуемой величины.
В этом методе при измерении Q с
помощью кулонометра его включают в
цепь электролиза последовательно с
ячейкой, и тогда отпадает
необходимость строгого соблюдения
постоянства силы генераторного тока.
В амперостатическом методе для
определения Q = ix единственным
измеряемым переменным является т
(если i остается постоянной), поэтому
следует пользоваться секундомером с
достаточной точностью отсчета (~0,1
сек) или электрохронометром, который
приводится в движение одновременно
с включением тока генерации с
помощью одного и того же переключателя.
Прецизионные миллиамперметры обеспечивают достаточную
точность измерения тока генерации, однако при более точных работах
измеряют омическое падение напряжения iR через прецизионное
постоянное сопротивление с помощью потенциометра, подключенного к концам
этого сопротивления.
При использовании электрохимических, фотометрических или
радиометрических методов индикации момента завершения химической
реакции следует иметь соответствующие установки (см. гл. II, III, V, VII, IX).
В потенциометрическом и амперометрическом методах
индикаторный электрод помещают в камеру, где происходит химическое и электро-
Рис. 79. Блок-схема установки для
кулонометрического титрования:
/ — внешний источник постоянного тока;
2 — высокоомное переменное сопротивление
для регулировки величины тока электролиза;
3 — постоянное прецизионное сопротивление
для точного измерения силы тока
электролиза по омическому падению
напряжения iR; 4 — потенциометр для измерения
омического падения напряжения iR на
концах прецизионного сопротивления;
5 —переменное сопротивление, величину
которого подбирают равным внутреннему
сопротивлению электролизера с
испытуемым раствором; 6 — переключатель тока на
два положения; в положении а ток
электролиза протекает через электролизер 7 и
Включается электрохронометр 14, в
положении б отключается электролизер 7 и
электрохронаметр 14, ток замыкается через
переменное сопротивление 5; 7 —
электролизер с разделенными анодной и катодной
камерами; 8 — рабочий электрод; 9 -
вспомогательный электрод; 10 — диафрагма,
разделяющая катодную и анодную камеры
электролизера; 11— два индикаторных
электрода для биамперометрического
определения момента завершения химической
реакции; 12 — амперометрическая установка;
13 — внешний источник переменного тока
для питания электрохронометра; 14—
электрохронометр; 15 — миллиамперметр для
контроля силы тока электролиза в
процессе кулонометрического титрования.
216
Г Л, VI. КУЛОНОМЕТРИЯ И КУЛОНОМЕТРИЧЕСКОЕ ТИТРОВАНИЕ
химическое превращение определяемого и вспомогательного веществ, и
соединяют ее с электродом сравнения с помощью электролитического
ключа. В биамперометрическом методе два индикаторных электрода
помещают в камеру, которая содержит определяемое вещество.
В фотометрическом методе индикации генерационной камерой
служит приспособленная для этой цели кювета, помещенная в
спектрофотометр таким образом, чтобы мешалка, генераторный электрод и колено
электролитического ключа, создающего контакт с другой кюветой со
вспомогательным электродом, не рассеивали проходящий через кювету свет.
Цветные индикаторы вносят в камеру, в которой находится
определяемое вещество.
В кулонометрическом титровании нет необходимости прекращать
электролиз в момент завершения химической реакции (кроме случая
применения цветных индикаторов и кулонометров), так как при
использовании различных инструментальных методов индикации конечной
точки обычно этот момент устанавливают графически из кривых
титрования. Однако в некоторых случаях целесообразно проводить
электролиз до достижения заранее установленного значения потенциала
индикаторного электрода (при потенциометрическом методе индикации
конечной точки) или до появления или падения индикаторного тока
практически до нуля (при амперометрической индикации конечной
точки). Необходимость в таких приемах возникает при проведении
предэлектролиза.
Простая блок-схема установки для кулонометрического титрования
представлена на рис. 79.
В. ПРАКТИЧЕСКИЕ РАБОТЫ
§ 14. Работы, выполняемые методом прямой кулонометрии
Определение ионов железа (III) при контролируемом
потенциале рабочего электрода
Ионы железа (III) в кислых растворах восстанавливаются на
катоде до ионов железа (II) при потенциале рабочего электрода, равном
+ 0,3 в относительно Нас. КЭ. По завершении восстановления Fe+++TOi<
в цепи падает до некоторого малого значения, величина которого в
дальнейшем остается постоянной (остаточный ток /0). Анализ считают
законченным, когда в течение нескольких минут /о больше не изменяется.
Количество электричества Q06, протекшее через ячейку, определяют
с помощью кулонометра, включенного последовательно в цепь
электролиза. Для нахождения количества электричества Qpe, эквивалентного
содержанию железа (III), из Q06 вычитают количество электричества,
отвечающее остаточному току Q0Ct. Для этого следует установить
продолжительность электролиза в секундах (тэ) с момента замыкания цепи
до ее размыкания и вычесть произведение *0сттэ = Qoct из Q06, откуда
QFe = Qo6 — Qoct- Далее, согласно формуле Фарадея, вычисляют
содержание Fe+++ в испытуемом объеме раствора.
Методика определения. Одну U-образную стеклянную трубку
(электролитический ключ) наполняют 1 М раствором серной кислоты, а
другую — насыщенным раствором хлорида калия. Концы трубок плотно
закрывают тампонами из фильтровальной бумаги. Следят, чтобы в
трубках не оставалось пузырьков воздуха, нарушающих электрический
контакт. Два стакана емкостью 100 мл наполняют приблизительно 50 мл
1 М раствора серной кислоты. Один из этих стаканов закрывают
неплотно корковой крышкой с четырьмя отверстиями (при использовании
механической мешалки — с пятью отверстиями). В этом стакане закреп-
§ 14. РАБОТЫ, ВЫПОЛНЯЕМЫЕ МЕТОДОМ ПРЯМОЙ КУЛОНОМЕТРИИ 217
ляют катод (сетчатый платиновый полумикроэлектрод Фишера),
опускают в него магнит, конец газоподводной трубки и одно колено U-об-
разной трубки с раствором КО; ее другое колено опускают в стакан
емкостью ~ 100 мл, содержащий насыщенный раствор КО и Нас. КЭ.
В первый стакан опускают также колено U-образной трубки с H2S04,
а ее второе колено опускают в стакан с H2S04, содержащий анод
(Pt-электрод, пластинчатый, размером 1 X 1 см). Четвертый стакан
(также емкостью ~100 мл), служащий медным кулонометром,
наполняют электролитом, содержащим 1 М CuS04, 0,1 M K2SO4 и 0,1 М
H2SO4, и погружают в него два проволочных платиновых электрода
(длиной ^3 еж и диаметром 1 мм) и мешалку. Электрод, который
является в кулонометре катодом, предварительно точно взвешивают на
полумикровесах с точностью ±0,1 мг. Один из полюсов источника
постоянного тока медной проволокой присоединяют к одному из концов
сопротивления, другой конец которого подключают к одной из клемм
делителя напряжения типа мостика Кольрауша. Вторую клемму
мостика через переключатель присоединяют непосредственно ко второму
полюсу источника тока. К обоим концам мостика Кольрауша
параллельно присоединяют вольтметр. Замыкая ток переключателем тока,
регулируют величины сопротивления таким образом, чтобы напряжение
на концах мостика Кольрауша составляло приблизительно 10—15 в
(проследить за соответствием полярности подключения источника тока
и вольтметра!). К отрицательному полюсу мостика Кольрауша
присоединяют катод, а к его скользящему контакту подключают
последовательно второй переключатель тока, миллиамперметр и анод.
Потенциометр приводят в рабочее состояние (согласно приложенной к
прибору инструкции), катод присоединяют к его положительной клемме, а
Нас. КЭ — к отрицательной.
Приступают к предэлектролизу фонового раствора. Предварительно
в течение 5—10 мин и далее в течение всего электролиза через католит
пропускают очищенный от кислорода азот. Переключателем тока
замыкают цепь, подавая напряжение на концы мостика Кольрауша, и
включают мешалку. Показание шкалы э. д. с. потенциометра устанавливают
на 500 мв (разность потенциалов между катодом и Нас. КЭ) и
оставляют потейциометр постоянно включенным. Замыкают вторбй
переключатель тока и пускают в ход секундомер; из-за разбаланса
гальванометр отклоняется от нулевого положения. Быстрым перемещением
скользящего контакта мостика Кольрауша в ту или другую сторону
добиваются возвращения стрелки гальванометра к нулевому прложению.
Миллиамперметр в цепи электролиза показывает прохождение тока,
величина которого быстро падает, достигая некоторого малого значения,
не изменяющегося при продолжении электролиза. Так как в процессе
электролиза потенциал катода изменяется, периодически перемещают
скользящий контакт мостика, следя за тем, чтобы стрелка
гальванометра в потенциометрической цепи оставалась на нулевом положении.
В рабочем журнале записывают величину остаточного тока и
размыкают второй переключатель.
В цепь электролиза включают кулонометр и в катодную камеру
вносят 5 мл испытуемого раствора сульфата железа (III). Некоторое
время пропускают азот. Вторым переключателем замыкают цепь и
одновременно пускают в ход секундомер. Если стрелка гальванометра
отклоняется от нулевого деления, быстрым передвижением скользящего
контакта мостика Кольрауша возвращают стрелку в прежнее положение
и заносят в журнал начальную величину тока электролиза, отмечаемую
миллиамперметром. Электролиз продолжают до тех пор, пока сила тока
218 ГЛ. VI. КУЛОНОМЕТРИЯ И КУЛОНОМЕТРИЧЕСКОЕ ТИТРОВАНИЕ
не снизится до 0,1% первоначальной величины или до значения
остаточного тока. Следят, чтобы потенциал катода не отклонялся о*г
заданного значения (0,3 в), при необходимости манипулируя скользящим
контактом мостика Кольрауша таким же образом, как и в течение пред-
электролиза. По достижении этого момента выключают секундомер и
ток обоими переключателями и записывают в журнал
продолжительность электролиза.
Далее приступают к определению Q06, для этого вынимают из куло-
нометра катод, тщательно промывают его водой, высушивают спиртом,
а затем эфиром и взвешивают на полумикровесах. По разности массы
катода до начала и после завершения работы определяют количество
меди, выделенной в процессе электролиза, и тем самым количество
электричества Q06, прошедшее через цепь, и, следовательно, израсходованное
на восстановление Fe+++ до Fe++.
Расчет. Значение Q06 и QFe вычисляют по формуле (1). Если
электролиз прекращают при падении начальной силы тока до 0,1% ее
начальной величины, то нет необходимости вносить поправки на величину
остаточного тока, так как около 0,1% определяемого вещества остается
не восстановленным при условии, однако, что Q0 = *отэ заметно меньше
0,1 Qo6- Если же электролиз завершен до достижения остаточного тока,
то необходимо вносить поправки, как это указано выше (см. стр. 216).
Определение ионов меди (II) при постоянной
силе тока электролиза
Предварительно из испытуемого раствора количественно выделяют
медь на Pt-катоде, как при обычном электрогравиметрическом анализе,
затем анодно растворяют медь при строго постоянной силе тока
электролиза г*аи измеряют продолжительность процесса та, устанавливаемую
резким изменением потенциала анода по завершении растворения меди.
Таким образом, количество электричества Qcu, израсходованное в
анодном процессе, эквивалентно содержанию Си++ в испытуемой йорции
раствора. В этом методе расчет Q значительно проще, так как Q
соответствует произведению *ата.
Методика определения. В стакан емкостью 100 мл наливают около
45 мл раствора фона (0,1 М раствор относительно K2SO4 и H2S04) и
5 мл испытуемого раствора сульфата меди; туда же опускают
проволочные Pt-электроды (/ « 3 см и d = 1 мм) и магнитную мешалку. Один
из электродов присоединяют к отрицательному полюсу внешнего
источника тока, а второй — последовательно через переменные сопротивления,
переключатель тока и амперметр — к положительному полюсу.
Параллельно к электродам подключают вольтметр (соблюдать полярность!)
и так подбирают сопротивления, чтобы при замыкании цепи напряжение
на электродах было около 2 в. Проводят электролиз при
перемешивании раствора до тех пор, пока вся медь не выделится на катоде.
Выключают ток и прекращают перемешивание раствора. Реверсируют ток,
удаляют вольтметр, заменяют амперметр миллиамперметром и,
подбирая сопротивления, добиваются, чтобы в цепи протекал ток около 1 ма,
строго постоянный; одновременно с помощью переключателя включают
ток и запускают секундомер. При анодном процессе растворения меди
электрод должен быть подключен к клемме электронного вольтметра,
к другой клемме которого подключен Нас. КЭ, находящийся в стакане
емкостью 50 мл с насыщенным раствором КО. Этот стакан с
электролитом соединяют U-образной стеклянной трубкой, также наполненной
насыщенным раствором КС1, с электролизером.
§ 15. РАБОТЫ, ВЫПОЛНЯЕМЫЕ МЕТОДОМ КОСВЕННОЙ КУЛОНОМЕТРИИ 219
В рабочий журнал записывают величину тока электролиза и следят
с помощью электронного вольтметра за изменением потенциала анода,
покрытого медью. В момент скачка потенциала выключают ток и
останавливают секундомер. Записывают в журнал продолжительность
анодного растворения меди. Произведение силы тока электролиза /а в
амперах на время растворения та в секундах при анодном процессе дает
количество электричества Qcu, эквивалентное содержанию меди.
Расчет. Содержание меди во взятой для анализа порции раствора
вычисляют по формуле (1).
§ 15. Работы, выполняемые методом косвенной кулонометрии,
или кулонометрического титрования,
при постоянной силе тока электролиза
Определение хлористоводородной кислоты
по кислотно-основному методу
Определение хлористоводородной кислоты основано на
нейтрализации ее электрогенерированными ОН~-ионами. При электролизе
хлористоводородной кислоты на Pt-электроде (катоде) в зависимости от
величины силы тока is и концентрации кислоты могут идти следующие
процессы. Если h<ia (предельного тока Н+-ионов кислоты), вначале
восстанавливаются Н+-ионы кислоты до H2; как только в процессе
электролиза id станет меньше гэ, начинается восстановление Н20 до Н2 с
образованием ОН~-ионов, которые химически взаимодействуют с Н+-ионами
кислоты, ещ^ не восстановленными на электроде. После завершения
химической реакции избыток ОН"-ионов сообщает раствору щелочную
реакцию, что можно легко обнаружить по изменению окраски кислотно-
основного индикатора, например фенолфталеина. Если же с самого
начала 1Э > id, то процессы, происходящие в растворе, аналогичны
описанным выше, когда идет одновременное восстановление Н+-ионов кислоты
и самого растворителя — Н20.
При проведении электролиза при строгом постоянстве /э измеряют
продолжительность процесса нейтрализации кислоты тэ. По этим
данным вычисляют количество электричества Q как произведение ?этэ.
Анализируемые растворы должны содержать сильный инертный
электролит для повышения электропроводности раствора.
Методика определения. В два стакана емкостью по 100 мл
прикрепляют по платиновому пластинчатому электроду (1X1 см) и наливают
около 50 мл 10%-ного раствора сульфата калия; в о)щн из них
прибавляют 10 капель раствора фенолфталеина, опускают магнитную мешалку
и электрод и подключают его к отрицательному полюсу внешнего
источника постоянного тока, к положительному полюсу которого
присоединяют последовательно высокоомные сопротивления, миллиамперметр,
переключатель тока и второй платиновый электрод. Оба стакана
соединяют U-образной стеклянной трубкой, наполненной 10%-ным раствором
K2S04.
Проводят предэлектролиз для нейтрализации возможных в растворе
примесей с кислотными свойствами (главным образом, растворенной
двуокиси углерода). Для этого регулируют сопротивление так, чтобы
ь цепи сила тока электролиза была в пределах 5—10 ма. С помощью
переключателя замыкают ток, запускают мешалку и продолжают
электролиз до тех пор, пока в катодной камере раствор не приобретет
заметную ярко-розовую окраску. Выключают ток и в катодную камеру
220 ГЛ. VI. КУЛОНОМЕТРИЯ И КУЛОНОМЕТРИЧЕСКОЕ ТИТРОВАНИЕ
вносят 3—5 мл испытуемого раствора НС1. Снова замыкают цепь и
одновременно пускают в ход секундомер. Нейтрализацию кислоты
считают законченной, как только раствор приобретет снова ярко-розовую
окраску той же интенсивности. Тут же выключают секундомер и
одновременно ток. Повторяют опыт с новыми порциями испытуемого
раствора несколько раз, не меняя ни католит, ни анолит. В рабочем
журнале фиксируют результаты каждого эксперимента — силу тока гэ и
время тэ завершения электролиза. По средним данным (исключая
данное первого опыта) этих параметров вычисляют Q как произведение
/этэ и находят содержание НС1 во взятом объеме анализируемого
раствора по формуле (1).
Определение ионов церия (IV) по методу
окисления — восстановления
Для определения церия (IV) в анализируемом растворе создают
достаточно большую концентрацию соли железа (III) в качестве
вспомогательного реагента, из которого на катоде электрогенерируются ионы
железа (II). При силе тока электролиза i9>ia [предельный ток ионов
церия (IV) в растворе] в процессе электролиза на катоде одновременно
восстанавливаются церий (IV) до церия (III) и железо (III) до
железа (II), причем Ре++-ионы тут же взаимодействуют с ионами церия (IV),
присутствующими в растворе, восстанавливая их химически также до
церия (III). После завершения восстановления появление избытка элек-
трогенерированных Fe++ уменьшает соотношение концентрации
компонентов ред-окс пары Fe+++/Fe++, тем самым изменяется и окислительно-
восстановительный потенциал системы, что обнаруживают потенциомет-
рически по резкому скачку потенциала платинового индикаторного
электрода.
Методика определения. В стакан емкостью 100 мл (анодная камера)
наливают около 50 мл 2 М раствора серной кислоты, в другой такой же
стакан (катодная камера) вносят 50 мл 0,2 М раствора
железо-аммонийных квасцов, приготовленного в 2 М H2SO4, 2—5 мл испытуемого
раствора сульфата церия (IV) и 5 мл концентрированной фосфорной
кислоты, а в третий стакан емкостью 100 мл помещают насыщенный
раствор хлорида калия и погружают Нас. КЭ. В катодной камере
фиксируют два пластинчатых Pt-электрода (1X1 см) и мешалку, а в
анодной— третий такой же электрод. Одну U-образную стеклянную трубку
наполняют 2 М раствором H2S04, а другую — насыщенным раствором
КС1. Первую используют для создания электрического контакта между
анолитом и католитом, а вторую — между катодной камерой и
стаканом с Нас. КЭ. Катод подключают к отрицательному полюсу внешнего
источника постоянного тока, к положительному полюсу которого
последовательно присоединяют высокоомные сопротивления,
миллиамперметр, переключатель тока и анод. Второй электрод в катодной камере,
являющийся индикаторным электродом, подключают к положительной
клемме потенциометра, а Нас. КЭ — к отрицательной. Потенциометр
приводят в рабочее состояние так, как это принято при потенциометри-
ческих измерениях э. д. с. После подбора сопротивления для получения
нужной величины тока электролиза (3—10 ма) замыкают цепь
переключателем и одновременно запускают секундомер. В рабочем журнале
фиксируют величину тока электролиза /э.
Периодически, вначале через каждые 10 сек, а вблизи конечной
точки — через каждые 5 сек, размыкая цепь, измеряют потенциал
индикаторного электрода с помощью потенциометра. Электролиз продол-
§ Г5. РАБОТЫ, ВЫПОЛНЯЕМЫЕ МЕТОДОМ КОСВЕННОЙ КУЛОНОМЕТРИИ 221
жают до тех пор, пока Д? после достижения максимального значения не
начнет уменьшаться. Форма записи данных потенциометрического
измерения э. д. с, метод нахождения максимального значения АЕ и
конечной точки титрования описаны выше (гл. II), с той лишь разницей, что
вместо объема титранта V записывают время электролиза тэ, т. е.
находят максимальное значение Д? по Ат. Учитывая, что максимальное
изменение потенциала индикаторного электрода достигнуто в некотором
промежутке времени электрогенерации промежуточного реагента (в
данном случае 5 сек), в первом приближении следует считать, что половины
этого промежутка времени (2,5 сек) достаточно для достижения
максимума Д?/Дт.
Расчет. Зная общую продолжительность электролиза тэ в секундах
до момента максимального значения Д?/Дт и величину силы тока
электролиза i3 в амперах, вычисляют Q и содержание церия (IV) в
анализируемой порции раствора по формуле (1).
Определение анилина
Определение анилина основано на реакции бромирования его
избытком электрогенерированного брома и последующем кулонометри-
ческом титровании избытка Вг2 ионами электрогенерированной меди (I).
Бромирование анилина протекает медленно, поэтому
предварительно электрогенерируют избыток Вгг из бромида калия и после
завершения химической реакции образования триброманилина, реверсируя
ток электролиза, титруют избыток Вг2 катодно генерированными Си+-
ионами. Этот процесс протекает с большой скоростью, так как Вгг
восстанавливается как непосредственно на Pt-электроде, так и химически
электрогенерированными Си+-ионами.
Для обнаружения завершения второго химического процесса
наиболее пригодна амперометрическая индикация конечной точки с двумя
поляризованными электродами.
Методика определения. В один стакан емкостью 100 мл, служащий
на первом этапе анодной камербй, наливают 25—40 мл испытуемого
раствора анилина, по 5 мл 1 М раствора NaBr и 0,2 М раствора
сульфата меди (II), приготовленного в концентрированной
хлористоводородной кислоте, и доводят дистиллированной водой до объема 50 мл.
В этом стакане фиксируют три пластинчатых Pt-электрода (1X1 см)
и мешалку. В другой такой же стакан (катодную камеру) наливают
около 50 мл раствора электролита, содержащего по 0,5 моль KG1 и НС1
в 1 л, и туда же погружают четвертый аналогичный электрод. Оба
стакана соединяют U-образной стеклянной трубкой, наполненной тем же
электролитом. Один из электродов в анодной камере присоединяют
к положительному полюсу внешнего источника постоянного тока, к
отрицательному полюсу которого подключают последовательно высокоом-
ное переменное сопротивление, подобранное так, чтобы в цепи
генерации протекал ток 5—10 мау миллиамперметр, переключатель тока я
катод. Два остальных электрода в анодной камере присоединяют к
соответствующим клеммам амперометрической установки и налагают на
них напряжение « 200 мв.
Одновременно включают генерационную и индикационную цепи к
пускают в ход секундомер и мешалку. По мере генерации ток в цепи
индикации возрастает, так как Вгг медленно реагирует с анилином и
в растворе существуют оба компонента обратимой ред-окс пары Вг2/2Вг*.
Когда ток становится достаточно большим, выключают ток генерации,
останавливают секундомер и дают прореагировать Вг2 с анилином. При
этом в цепи индикации ток /и постепенно уменьшается. В случае па-
222 ГЛ. VI. КУЛОНОМЕТРИЯ И КУЛОНОМЕТРИЧЕСКОЕ ТИТРОВАНИЕ
дения in до значения остаточного тока (или до очень небольшой
величины) снова пропускают ток генерации /э и, одновременно пустив
секундомер, продолжают электрогенерацию Вг2, повторяя указанный
прием до тех пору пока *и не приобретет заметной величины и не будет
оставаться постоянным в течение нескольких минут при отключении
генерационной цепи. Фиксируют в рабочем журнале величины токов
генерации iB и индикации *и и общую продолжительность электролиза т^.
После завершения бромирования анилина (образование трибром-
анилина) реверсированием тока (простым переключением полюсов
генераторных электродов) приступают к электрогенерации Си+-ионов для
восстановления избытка Вг2. Для этого снова включают ток
электролиза i9 и секундомер, проводя электрогенерацию Си+-ионов в
небольшие промежутки времени (~10 сек). Следят за током 1И в цепи
индикации и прекращают электролиз как только ток ?и уменьшится до
значения остаточного тока или до некоторого очень малого значения.
Останавливают секундомер и размыкают цепи генерации и индикации.
Фиксируют продолжительность титрования — т" избытка Вг2
генерированными Си+~ионами. Разность между общим временем генерации Вг2и
временем титрования его избытка (т^ —т")? умноженная на величину
тока генерации /э, соответствует количеству электричества Q,
израсходованного для генерации Вг2, который необходим для окисления
анилина до триброманилина.
Если титрование избытка Вг2 генерированными Си+-ионами
доводят до некоторого малого значения *и, то в результаты анализа вносят
поправку на основании холостого опыта. Для этого в чистую камеру
с тремя электродами наливают все растворы, кроме испытуемого,
заменяя его водой, и проводят электрогенерацию Вг2 до тех пор, пока
в цепи индикации ток не приобретет величину, которая была достигнута
при титровании электрогенерированными Си+-ионами избытка Вг2
в растворе. Время, затраченное на холостой опыт, также вычитают из
общего времени генерации Вг2. Необходимо, чтобы величина ?э была
все время одинаковой.
Расчет. Для вычисления содержания анилина во взятом объеме
пробы пользуются формулой (1), с учетом что при образовании
триброманилина грамм-эквивалент анилина равен 7б его молекулярного
веса.
Определение ионов цинка по методу осаждения
Ионы цинка (II) на Pt-электроде в кислой среде неэлектроактивны,
между тем как ионы гексацианоферрата (III) восстанавливаются под
действием катодного тока до ионов гексацианоферрата (II), которые
химически взаимодействуют с ионами цинка (II) с образованием
малорастворимого осадка. Происходящая при этом электродная реакция
Fe(CN)™ + e —> Fe(CN)"
сопровождается химической реакцией, протекающей между
электрогенерированными Fe (СЩГ-ионами и 2п++-ионами
2K+ + 3Zn+++2Fe(CN)e= 5Z± | K2Zn3 [Fe(CN)6]2
Завершение химической реакции можно установить различными
электрохимическими методами, в данном случае амперометрическим
методом с двумя поляризованными электродами. По мере
образования осадка при небольшом значении налагаемого извне напряжения
на индикаторные электроды в цепи индикации не наблюдается тока
§ 15. РАБОТЫ, ВЫПОЛНЯЕМЫЕ МЕТОДОМ КОСВЕННОЙ КУЛОНОМЕТРИИ 223
из-за отсутствия в растворе одного компонента — Fe (CN)(T ред-окс пары
Fe(CN)6~~7Fe(CN)(r; при появлении избытка Fe(CN)(T оба
индикаторных электрода деполяризуются, вследствие чего в индикационной
цепи возникает ток, вначале возрастающий пропорционально
концентрации электрогенерированного гексацианоферрата (II). Построив
кривую зависимости величины тока индикации *и от времени электролиза
тэ, получим две прямые, точка пересечения которых отвечает
практически моменту полного осаждения Zn++ гексацианоферратом (II).
Найдя на графике значение тэ, отвечающее этой точке, вычисляют Q
как произведение тэ на /э.
Методика определения. В стакан емкостью 100 мл, который служит
катодной камерой, наливают по 25 мл 4 М раствора уксусной кислоты
и 0,4 М раствора гексацианоферрата (III) калия и вносят 1—5 мл
испытуемого раствора сульфата цинка. В другой стакан (анодную
камеру) вносят 50 мл 1 М раствора сульфата калия. Оба стакана
соединяют U-образной стеклянной трубкой, заполненной 1 М раствором
сульфата калия. В катодной камере фиксируют три пластинчатых Pt-элек-
трода (1 X 1см) и мешалку, а в анодной — четвертый такой же электрод.
Один из электродов в катодной камере подключают к отрицательному
полюсу внешнего источника постоянного тока, а электрод в
анодной камере — к положительному полюсу последовательно с
миллиамперметром, переключателем тока и высокоомным сопротивлением,
подобранным так, чтобы обеспечить в цепи генерации ток 5—10 ма.
Два остальных Pt-электрода в катодной камере подключают к
соответствующим клеммам амперометрическои установки и налагают на них
напряжение ~100 мв.
Одновременно включают цепи генерации и индикации и пускают
в ход секундомер и мешалку. Записывают в журнал величину тока
электролиза гэ. Электрогенерацию гексацианоферрата (II) проводят
периодически (в течение 10—20 сек) включением и отключением цепи
генерации. В промежутке между двумя периодами генерации
наблюдают за возникающим в индикационной цепи током *и, величину
которого также фиксируют в рабочем журнале. Вначале, пока в катодной
камере присутствуют 2п++-ионы, ток в цепи индикации отсутствует или
соответствует остаточному току и остается практически постоянным;
при появлении избытка электрогенерированных ионов
гексацианоферрата (II) наблюдается возрастание ги, вначале пропорциональное
количеству возникающих ионов. По данным измерения тока строят график
зависимости ?и от тэ. Точка пересечения двух прямых дает конечную
точку, отвечающую моменту завершения химической реакции. Расчет
количества Zn++ в анализируемой порции испытуемого раствора
проводят как описано выше: вычисляют Q, равное произведению силы
тока электролиза i9 на продолжительность электролиза тэ, найденную
графически.
Расчет. Содержание цинка (II) во взятой порции испытуемого
раствора находят по формуле (1).
ГЛАВА VII
СПЕКТРАЛЬНЫЕ (ОПТИЧЕСКИЕ) МЕТОДЫ
АНАЛИЗА
Спектральные (оптические) методы анализа основаны на изучении
спектров излучения, поглощения и рассеяния (см. книга 1, гл. VI, § 2).
К этой группе методов относятся (см. гл. I, § 10):
1. Эмиссионный спектральный анализ.
2. Пламенная спектрофотометрия, или фотометрия пламени,
являющаяся разновидностью эмиссионного анализа.
3. Атомно-абсорбционный спектральный анализ.
4. Абсорбционная спектроскопия.
5. Метод анализа по спектрам комбинационного рассеяния света.
6. Турбидиметрия.
7. Нефелометрия.
8. Люминесцентный, или флуоресцентный, метод анализа и др.
ЭМИССИОННЫЙ СПЕКТРАЛЬНЫЙ АНАЛИЗ
Л. ТЕОРЕТИЧЕСКИЕ ОСНОВЫ МЕТОДА
§ 1. Общая характеристика метода
В эмиссионном спектральном анализе исследуемого вещества пробу
(анализируемый объект) испаряют и возбуждают свечение паров
посредством пламени электрической дуги, высоковольтной искры или
другим источником возбуждения. Атомы каждого элемента в
возбужденном состоянии испускают волны только определенной длины, так
называемое характеристическое излучение. Благодаря этому
оказывается возможным проводить качественный эмиссионный спектральный
анализ не только простых, ной сложных веществ и их смесей.
Количественный эмиссионный спектральный анализ основан на
зависимости, существующей между интенсивностью (яркостью)
спектральных линий определяемого элемента и его концентрацией. Другими
словами, мерой концентрации данного элемента в исследуемой пробе
является интенсивность линий спектров паров определяемого элемента.
Связь между интенсивностью спектральной линии и концентрацией
элемента в исследуемой пробе выражают эмпирической формулой,
которая была выведена Б. А. Ломакиным:
где /А — интенсивность спектральной линии определяемого элемента;
С — концентрация;
а и Ъ — постоянные величины.
§ 4 ПОЛУКОЛИЧЕСТВЕННЫЕ МЕТОДЫ СПЕКТРАЛЬНОГО АНАЛИЗА 225
§ 2. Классификация методов эмиссионного
спектрального анализа
Для регистрации спектральных линий применяются визуальные,
фотографические и фотоэлектрические приборы и аппараты. В
зависимости от способа регистрации спектра различают визуальный
спектральный анализ, в котором спектр наблюдают в видимой области при
помощи стилоскопов и стилометров или при помощи флуоресцирующих
экранов, преобразующих невидимые ультрафиолетовые лучи в
видимые. Визуальный анализ применяют в качественном анализе и иногда
в количественном анализе. Если для регистрации спектров используют
фотографические пластинки, то метод анализа называется
фотографическим спектральным анализом. Особенно широко этот метод применяют
в качественном и количественном анализе. В фотоэлектрическом
спектральном анализе, который используется исключительно для
количественного анализа, спектры регистрируются фотоэлектрическими
приборами.
§ 3. Качественный спектральный анализ
Основы качественного спектрального анализа изложены ранее (см.
книга 1, гл. III, § 9). Интенсивность линий спектра элемента зависит
от концентрации этого элемента в исследуемой пробе, поэтому с
уменьшением концентрации интенсивность многих линий настолько
уменьшается, что их нельзя различить. Для открытия элементов пользуются
так называемыми аналитическими линиями, или «последними линиями»,
которые можно обнаружить в спектре исследуемой пробы при
предельно малой концентрации открываемого элемента. Например,
последней линией в спектре натрия является линия, длина волны (к)
которой равна * 5890 А. Эта линия исчезает в спектре исследуемой пробы,
когда концентрация натрия становится меньше 10~5%. Перечень линий
всех элементов приведен в спектральных атласах.
Качественный спектральный анализ можно проводить визуальным
и фотографическим методами. Последние линии определяемого элемента
в визуальном методе отыскивают при помощи стилоскопа (см, § 12).
В фотографическом методе спектр исследуемой пробы снимают на
фотографическую пластинку при помощи спектрографа, обычно с
кварцевой оптикой, потому что последние линии большинства элементов
расположены в ультрафиолетовой части спектра. Одновременно
снимают спектр железа (спектр этого элемента содержит очень большое
число линий), который служит шкалой длин волн для идентификации
линий в спектре исследуемого элемента. Для расшифровки спектра
пользуются спектропроектором, с помощью которого проектируют на
экран небольшой участок спектра (т. е. фотографии спектра),
увеличенный примерно в 20 раз.
§ 4. Полуколичественные методы спектрального анализа
Визуальный полуколичественный анализ
Этот метод применяется главным образом для анализа сплавов.
Метод заключается в том, что интенсивность линии анализируемой
примеси сравнивается с интенсивностью линии основного элемента сплава.
* В эмиссионном спектральном анализе длины волн измеряют в ангстремах
(1 А= Ю-8 см).
226 ГЛ. VII. СПЕКТРАЛЬНЫЕ (ОПТИЧЕСКИЕ) МЕТОДЫ АНАЛИЗА
Сравниваемые линии не должны различаться по цвету и должны быть
расположены близко друг к другу. Когда интенсивности линий
приблизительно равны, примерное содержание примеси определяют по
таблицам. Эти таблицы заранее установлены для каждого сплава.
Например, для оценки содержания хрома в легированных сталях сравнивают
интенсивность некоторых линий хрома с интенсивностью некоторых
линий железа. Если интенсивность линии хрома X = 5208,4 А равна
примерно интенсивности линии железа X = 5202,3 А, то содержание хрома
примерно равно 0,10%; когда интенсивность линии хрома ^ = 4646,1 А
примерно равна интенсивности линии железа X = 4647,4 А, содержание
хрома составляет 0,7—1,1%-
Фотографические методы полу количественного анализа
Метод исчезновения линий. Число линий в спектре элемента
зависит от концентрации этого элемента в пробе. Например, если
содержание олова в исследуемой пробе равно 0,001%, в спектре пробы имеется
только слабая линия олова X = 2839,44 А. При содержании олова 0,003%,
эта линия уже хорошо заметна и появится слабая линия X = 3034,12 А.
При содержании олова 0,01 %, линии X = 2839,44 и X = 3034,12 А хорошо
видны и появляется слабая линия X = 3262,33 А. При содержании олова
0,10%, в спектре пробы появляются следующие линии олова: X = 2839,44,
X = 3034,12, X = 3262,33, X = 2426,42 А и слабая линия X = 2421,69 А.
Таким образом, если в спектре исследуемой пробы, содержащей олово,
имеются линии X = 3034,12, X = 2839,44 и X = 3262,33 А, то содержание
олова составляет примерно 0,01%.
Этот способ годится только для определения небольших
концентраций и часто используется в геологоразведочных работах.
Метод сравнений. Метод заключается в том, что при определенных
условиях с одной и той же экспозиции снимают спектр ряда проб —
стандартов с известным содержанием определяемого элемента и спектр
неизвестной пробы. Визуально сравнивают интенсивности некоторой
линии определяемого элемента в спектре исследуемой пробы и в спектрах
стандартов. Концентрация определяемого элемента в неизвестной пробе
будет примерно равна концентрации этого элемента в стандартах, когда
интенсивность некоторой линии в спектре стандарта примерно равна
интенсивности этой же линии в спектре пробы. Например,
сфотографированы на пластинке четыре стандарта с содержанием лития 0,0001;
0,001; 0,01 и 0,1%. Интенсивность линии лития X = 6708 А в спектре
пробы приблизительно равна интенсивности этой линии в спектре
стандарта с содержанием 0,0Г% лития. В исследуемой пробе содержится
примерно 0,01% лития.
§ 5. Количественный спектральный анализ
Интенсивность спектральной линии зависит от многих условий:
источника возбуждения спектральных линий, скорости испарения пробы,
освещения щели спектрального прибора и др. При случайных
изменениях этих условий меняется интенсивность спектральных линий, поэтому
количественный анализ, основанный на измерении абсолютной
интенсивности спектральных линий, недостаточно точен.
Аналитические пары линий. Для получения более точных
количественных определений обычно пользуются не абсолютными интенсивностями,
а отношением интенсивности линии определяемого элемента к интен-
§ 6. ОСНОВЫ ФОТОГРАФИЧЕСКОГО МЕТОДА ЭМИССИОННОГО АНАЛИЗА 227
сивности линии элемента сравнения. При анализах сплавов в качестве
элемента сравнения, как правило, выбирают один из компонентов
исследуемой пробы. При анализах руд и солей в пробу обычно вводят
специально подобранный элемент (метод внутреннего стандарта).
Например, при определении кобальта в сплаве на алюминиевой основе
измеряют величину относительной интенсивности линии кобальта X =
= 2307 А и линии алюминия X = 2321 А. Для определения олова
измеряют относительную интенсивность линии олова к = 2839,9 А и линии
натрия К = 2680,4 А, который (в виде хлорида натрия) вводят в
исследуемую пробу. Пару линий, используемую в количественном
спектральном анализе, — линию определяемого элемента и линию элемента
сравнения— называют гомологической, или аналитической, парой.
Для перехода от относительной интенсивности линий
аналитической пары к концентрации определяемого элемента можно
пользоваться уравнением (1).
В области небольших концентраций определяемого элемента
концентрацию элемента сравнения, который составляет большую часть
массы исследуемой пробы, считают постоянной величиной, независимой
от концентрации определяемого элемента. Действительно, в двух
образцах сплава на алюминиевой основе, содержащих 0,05 и 0,1% меди,
содержание алюминия равно 99,95% и 99,90%. Очевидно, что 99,90«
ж99,95«const Если принять концентрацию элемента сравнения за
постоянную величину, то интенсивность линии /ср элемента сравнения тоже
будет постоянной: /ср = const. В этом случае отношение интенсивности
линии определяемого элемента к интенсивности линии элемента
сравнения выражается уравнением:
/ а .
где а'=-т~- >
•'ср
а и Ъ — константы.
Логарифмирование выражения (2) приводит к следующему
уравнению:
lg-A-blgC + lgof (2а)
•'ср
Уравнение (2а) показывает, что между логарифмом относительной
интенсивности линий аналитической пары и логарифмом концентрации
определяемого вещества существует линейная зависимость. Чтобы
пользоваться уравнением (2а) для нахождения концентрации определяемого
элемента, нужно найти функциональную зависимость между lg -—• и С.
Уср
Чаще всего для этого используют фотографический метод с
последующим фотометрированием.
§ 6. Основы фотографического метода эмиссионного
количественного анализа
Плотность почернения. Для измерения относительной интенсивности
линий аналитической пары спектр исследуемой пробы фотографируют
на пластинку. На пластинке получается ряд линий, степень почернения
которых зависит от интенсивности снятых спектральных линий. Коли-
7А аС „ъ
-А=~;— = а'Сь
*ср
¦«ср
(2)
228
ГЛ. VII. СПЕКТРАЛЬНЫЕ (ОПТИЧЕСКИЕ) МЕТОДЫ АНАЛИЗА
чественно почернение фотопластинки принято измерять величиной
плотности почернения (S), которую вычисляют по уравнению:
5 = lgA (3)
где /0 — интенсивность света, проходящего через незачерненную часть фотографической
пластинки (так называемая вуаль);
/ — интенсивность света, проходящего через зачерненную часть пластинки.
Микрофотометрирование. Плотность почернения можно измерить на
микрофотометре. Принцип работы микрофотометра заключается в
следующем. Поток световых лучей пропускают через незачерненную часть
фотографической пластинки и затем направляют на фотоэлемент,
связанный с гальванометром. Определяют отклонение стрелки
гальванометра ссо при помощи миллиметровой шкалы. Затем поток световых
лучей пропускают через зачерненную часть пластинки (фотография
спектральной линии) и опять определяют отклонение стрелки
гальванометра а. Предполагая пропорциональность между количеством света,
упавшего на фотоэлемент, и отклонением стрелки гальванометра,
получаем величину почернения:
s-i*4eI*-T (4)
§ 7. Зависимость между почернением фотопластинки
и интенсивностью излучения
Для определения интенсивности спектральной линии необходимо
знать зависимость между почернением S фотопластинки и
интенсивностью / излучения, вызвавшего это почернение. Зависимость между S
и lg/ выражают кривой, которую называют кривой почернения или
характеристической кривой фотопластинки. На прямолинейном участке
этой кривой зависимость S от lg/ выражается уравнением:
де у и / ~" постоянные величины, которые зависят от сорта фотопластинки.
Если SA — почернение линии определяемого элемента, a Scp —
почернение линии элемента сравнения, то
^A-Ylg/A-/ №)
Scp-Ylg'cp-/ (7)
Вычитая уравнение (7) из уравнения (6), получаем:
SA-ScP = Ylg/A-Yte/cp (8)
Отсюда
Irr _
Ig/A=SA-^ (9)
ср
Разность почернений аналитической пары Sa — Sep обозначим
через AS; тогда уравнение (9) примет вид:
Ylgf^AS (10)
¦«ср
Подставляя в уравнение (10) значение левого члена из уравнения
(2а), получаем:
AS = yblgC + ylga' (10а)
В этом уравнении yb и уа'— величины постоянные.
§ 8. ИСТОЧНИКИ ВОЗБУЖДЕНИЯ СПЕКТРОВ
229
Уравнение (10а) показывает, что разность почернений линий
аналитической пары связана с концентрацией определяемого элемента
прямолинейной функциональной зависимостью.
Метод трех эталонов. Предложено много методов определения
функциональной зависимости разности почернений линий аналитической
пары от концентрации определяемого элемента в исследуемой пробе,
но одним из наиболее распространенных является метод трех
эталонов. Сущность метода заключается в следующем. Готовят три образца
(эталоны) исследуемого вещества, например сплава. Содержание
определяемого элемента в эталонах должно быть точно известно, и
качественный, и количественный состав эталона должен близко совпадать
с составом исследуемого вещества.
Например; если нужно определить от 0,01 до 1% меди в
алюминиевом сплаве, готовят три образца алюминиевого сплава с содержанием
меди 0,01; 0,5 и 1%. На одну пластинку фотографируют спектр
анализируемого образца и спектры эталонов. Определяют, как было указано
выше, значение разности почернения линий аналитической пары в
спектре каждого эталона ASb Д52, AS3 и значение ASa разности почернения
линий аналитической пары в спектре исследуемой пробы. На одной оси
координат наносят значение А5Ь AS2, AS3, а на другой оси — значения
логарифмов соответствующих концентраций Сь С2, С3. По полученным
точкам строят калибровочный график, который имеет вид прямой. Зная
ASX, можно по этому графику определить концентрацию Сх
определяемого элемента в исследуемой пробе.
Б. АППАРАТУРА И ТЕХНИКА ВЫПОЛНЕНИЯ АНАЛИЗА
§ 8. Источники возбуждения спектров
Пламя, В некоторых случаях для возбуждения свечения элементов
достаточно внести испытуемое вещество в ацетиленовое пламя или
пламя газовой горелки. Обычно
исследуемое вещество в виде его
хлористоводородного раствора вдувают
в пламя. Для этого применяют
распылитель (рис. 80). Исследуемую
жидкость помещают в углубление 1.
Поток воздуха, поступающий через
отросток 2, всасывает раствор через
капилляр 3 из углубления / и
увлекает мельчайшие капли жидкости
в горелку 4, газ в которую подается
через трубку 5. Этот способ
возбуждения применяют главным образом
для исследования спектров
щелочных и щелочноземельных
металлов.
Дуговой разряд. Более
совершенным является метод
возбуждения спектров при помощи дугового
разряда. При анализе тугоплавких
металлов в качестве электродов
применяются сами металлы. Для анализа минеральных солей обычно
применяют дугу между угольными электродами. Расстояние между
электродами обычно называют аналитическим или дуговым промежутком.
Дугу питают постоянным или переменным током. Дуга переменного токи
Рис. 80. Схема распылителя для
анализа по спектру пламени:
/ — углубление для исследуемой жидкости;
2 — трубка для подачи воздуха; 3 —капилляр;
4 — горелка; 5 —трубка для подачи газа.
230
ГЛ. VII. СПЕКТРАЛЬНЫЕ (ОПТИЧЕСКИЕ) МЕТОДЫ АНАЛИЗА
Рис. 81. Схема электрической дуги
переменного тока:
1 — аналитический дуговой промежуток; 2, 5
—конденсаторы; 3 — амперметр; 4-реостат; 6 — искровой
промежуток; 7 — трансформатор; 8 — реостат; 9 —
трансформатор.
в паузах тока гаснет, так как за это время катод успевает остыть
настолько, что прекращается термоэлектронная эмиссия. Поэтому дуга
переменного тока каждый раз зажигается высоковольтным импульсом
небольшой мощности, который получают при помощи так называемого
активизатора.
Схема дуги представлена на рис. 81. Она состоит из двух частей:
схема питания дуги (контур /) и схема активизатора (контур //). Обе
схемы подключены, к сети
переменного тока (220 в). В схеме
питания параллельно
аналитическому промежутку 1
подключен конденсатор 2, который
заряжается через реостат 4
сетевым током. Сила тока
контролируется амперметром 3.
В схеме активизатора
подключен конденсатор 5 и искровой
промежуток 6. Конденсатор 5
заряжается от
трансформатора 7 переменным током через
реостат 8. Когда напряжение
на электродах искрового
промежутка достигнет значения,
достаточного для пробоя,
возникает искровой разряд. При
пробое искрового промежутка в контуре // создается высокочастотный
колебательный разряд, который через повышающий трансформатор 9
передается в контур /. Прохождение высокочастотного разряда в
аналитическом промежутке 1 создает в нем проводимость, достаточную,
чтобы через промежуток пошел ток низкого напряжения и силы,
определяемой сопротивлением 4 и сопротивлением аналитического
промежутка. По этой схеме,
работают генератор АГ-1, АГ-2 и
ПС-39.
Конденсированная искра.
Для анализа металлов широко
применяют высоковольтную
конденсированную искру
между электродами из
исследуемых металлов или из
исследуемых металлов и металлическим
электродом, например медным.
Схема питания
высоковольтной конденсированной искры
приведена на рис. 82. Ток напряжением 220 в через реостат 1 попадает
в первичную обмотку трансформатора 2, повышающего напряжение до
1200—1500 в. Сила тока контролируется амперметром 3. Искра
образуется в аналитическом промежутке 4, который подключен через
катушку самоиндукции 5 к конденсатору 6. Во время зарядки
конденсатора 6 разность потенциалов образуется одновременно и на
электродах аналитического промежутка 4. Разряд конденсатора (и
образование искры в аналитическом промежутке) возникает, когда
напряжение на электродах аналитического промежутка 4 достигает значения,
достаточного для пробоя. Напряжение пробоя зависит от ряда факторов:
расстояния между рабочими поверхностями электродов и их состояния
Рис. 82.
Схема высоковольтной
конденсированной искры:
-реостат; 2 — трансформатор; 3-амперметр;
-аналитически^ промежуток; 5 —катушка
самоиндукции; 6 — конденсатор.
§ 9. СПЕКТРАЛЬНЫЕ ПРИБОРЫ
231
(чистоты, качества обработки), от состояния ионизации воздуха в нем.
Это приводит к нестабильности разряда, что вызывает нестабильность
температуры возбуждения спектральных линий.
Более стабильным является разряд в генераторе с двумя
искровыми промежутками для питания конденсированной искры (рис. 83).
Ток напряжением 220 в через реостат 1 попадает в первичную обмотку
трансформатора 2, повышающего напряжение с 220 до 1200—1500 в;
сила тока контролируется амперметром 3. Искра образуется в
аналитическом промежутке 4, который подключен через катушку
самоиндукции 5 к конденсатору 6. Последовательно с аналитическим
промежутком 4 вводится дополнительный промежуток (разрядник) 7.
Аналитический промежуток 4 шунтируется большим сопротивлением 8. Во время
зарядки конденсатора 6 от сети сопротивление 8 проводит ток, и на
электродах аналитического
промежутка 4 не образуется разности по- / t 2 _^$
тенциалов. Разрядка конденсато
ра 6 начинается пробоем проме
жутка 7. Для постоянства уело- 2Ш
вий пробоя электроды этого про- °^1
межутка делаются из вольфрама. [
После пробоя в цепи возникает
высокочастотный ток,
напряжение на шунтирующем
сопротивлении 8 быстро возрастает и
оказывается приложенные к
аналитическому промежутку 4.
Пробойное напряжение аналитического
промежутка устанавливают меньше, чем у разрядника 7, поэтому сразу
происходит его пробой. Таким образом, начало разряда и напряжение
на конденсаторе зависит только от пробойного напряжения разрядника.
Такая схема обеспечивает высокую стабильность искры. По этой схеме
сконструирован искровой генератор ИГ-3.
Рис, 83. Схема генератора с двумя
промежутками:
/ — реостат; 2 — трансформатор; 3 — амперметр;
4— аналитический промежуток; 5 —катушка
самоиндукции; 6 — конденсатор; 7 —промежуток
(разрядник); 8 — сопротивление.
§ 9. Спектральные приборы
Стилоскопы. Стилоскопы обычно (кроме стилоскопа марки СЛ-3)
снабжены преломляющим устройством, собранным по автоколлима-
ционной схеме. На рис. 84 дана оптическая схема однопризменного
автоколлимационного прибора. Поток света, проходящий через щель 1,
направляется поворотной призмой 2 на объектив 3. Затем луч падает
на преломляющую призму 4 (с углом преломления 30°), проходит ее
и отражается от грани, на которую нанесен слой алюминия,
действующий как плоское зеркало. После отражения луч вторично проходит
призму 4 и падает опять на объектив 3, который в этом случае
действует как камерный объектив, тогда как на пути света от щели 1
к призме 4 он выполнял роль коллиматорного объектива. Изображение
щели получается на фокальной плоскости 5. Спектр наблюдают
визуально при помощи окуляра. Для этого в поле зрения окуляра
выводят нужную область спектра поворотом призмы 4 при помощи
механизма, связанного с барабаном, на который нанесена миллиметровая
шкала.
В СССР применяют стилоскопы марки СЛ-3, СЛ-10, СЛ-11, СЛ-11а.
Спектрографы. Спектрографами называют приборы,
предназначенные для получения фотографий спектров. Они снабжены фотокамерой
с кассетой; конструкция прибора обеспечивает совмещение светочув-
232
ГЛ. VII. СПЕКТРАЛЬНЫЕ (ОПТИЧЕСКИЕ) МЕТОДЫ АНАЛИЗА
ствительного слоя пластинки с фокальной плоскостью объектива.
Кассету можно перемещать по вертикали при помощи механизма.
Величины перемещения отсчитываются по миллиметровой шкале. Это дает
возможность снять несколько десятков спектров на одной пластинке.
Спектрограф ИСП-51. Этот прибор предназначен для
фотографии спектров в видимой области спектра, он снабжен стеклянной
оптикой. Прибор можно использовать в
области спектра от 4000 до 10 000 А.
Спектрографы ИСП-22, ИСП-28,
ИСП-30. Оптическая схема спектрографов
ИСП-22, ИСП-28 и ИСП-30 приведена на
рис. 85. Свет, проходя через щель /, падает
на алюминированное зеркало 2, которое
выполняет функцию коллиматорного
объектива. Отраженный свет падает на призму 3.
За призмой установлен двухлинзовый
камерный объектив 4, который проектирует
спектр в плоскости эмульсии
фотопластинки 5. Фотопластинка заключена в кассету.
Призма 3 и объектив 4 этих
спектрографов изготовлены из кварца. Приборы
можно использовать в области спектра от
2000 до 6000 А.
Монохроматоры. Спектральные приборы, в которых окуляр или
фотографическая пластинка заменены узкой щелью (выходная щель),
вырезающей из спектра узкий участок, называются монохроматорами.
Рис. 84. Оптическая схема
однопризменного
автоколлимационного прибора:
/ - входная щель; 2 — призма;
3- объектив; 4 — преломляющая
Призма; 5 —фокальная плоскость.
Рис. 85. Оптическая схема спектрографа ИСП-22, ИСП-28 и ИСП-30:
/ — входная щель; 2 — алюминированное зеркало; 3 — призма; 4 — камерный
объектив; 5—фотографическая пластинка.
Они применяются для получения монохроматического излучения, т. е.
излучения определенной длины волны.
§ 10. Вспомогательные приборы и принадлежности
Осветительная система. Для освещения источником света щели
спектрального аппарата обычно применяют трехлинзовую
осветительную систему (рис. 86).
Линза 2 дает немного увеличенное изображение источника света /
на диафрагме 3, вырезающей из этого изображения среднюю зону.
В диафрагме 3 имеется набор отверстий разной высоты, которые
позволяют регулировать интенсивность светового пучка. За диафрагмой
расположена линза 4, проецирующая линзу 2 на щель 5 спектрального
прибора. При этом на щели 5 получается равномерно освещенный круг
за счет излучения части источника, приходящейся на отверстие
диафрагмы 3. Линза 6 дает увеличенное изображение щели диафрагмы 3 на
объектив коллиматора 7. В стилоскопах резкое изображение источника
§ П. МИКРОФОТОМЕТР МФ-2
233
света на щели прибора достигается посредством линзы, которая
заключена в той же оправе, что и коллиматорная линза.
Диафрагма. Очень часто необходимо рядом со спектров
исследуемого вещества снять спектр чистого железа. Для этого перед щелью
спектрографа устанавливают специальную диафрагму (рис. 87) с
окошками /—4. Перемещая диафрагму параллельно плоскости щели 5, по
Рис. 86. Оптическая схема трехлинзовой осветительной системы:
/ — источник света; 2, 4, 6— линзы; 3 — диафрагма; 5 —входная щель
спектрометра; 7 —объектив коллиматора спектрометра.
очереди открывают различные ее участки. Например, совместив с щелью
окошко 2, можно сфотографировать спектр исследуемого вещества, а
совместив окошко 3— спектр железа.
Штативы для закрепления проб и электродов
Исследуемые пробы и электроды закрепляются в штативах разно-
образной конструкции. Особенно удобен штатив ШТ-9, который
предназначен для работы с дугой постоянного
и переменного тока и с электрической вы- . п
соковольтной искрой. Штатив состоит из / J?rD~~/
плотно закрываемого металлического кожу- I ° ^сп и 6
ха, внутри которого расположены
держатели электродов. Лампа для подсветки
электродов позволяет видеть расположение
электродов по их изображению на проме- Рис- 87« Схема диафрагмы:
ЖуТОЧНОЙ ДИафраГМе ОСВеТИТеЛЬНОЙ СИСТе- i-4-окошки; 5-щель.
мы.
§11. Микрофотометр МФ-2
Оптическая схема микрофотометра МФ-2 дана на рис. 88. Свет
от лампы накаливания / направляется конденсором 2 через поворотную
призму 3 на нижний объектив 4, через который освещается небольшой
участок фотопластинки 5, расположенной на горизонтальном столике
микрофотометра. Верхний объектив 6 дает при помощи экрана 7
изображение участка спектра на экран 8. В середине экрана 8 расположена
вертикальная щель переменной ширины. Свет, пройдя через эту щель,
попадает на вентильный фотоэлемент 9, который соединен с зеркальным
гальванометром 10, установленным на задней части корпуса
микрофотометра. Гальванометр 10 служит для измерений фототоков,
возникающих при освещении фотоэлемента световыми потоками, прошедшими
через фотопластинку 5. Отсчет показаний гальванометра 10
производится таким образом. Свет от той же лампы 1 через конденсор //
падает на зеркальце 12 гальванометра 10) после отражения от него
системой зеркал 13 и 14 направляется на экран 15. Объектив 16 дает на
экране 15 изображение шкалы 17, которая расположена в средней части
конденсора 11. Смещение изображения шкалы на экране 7 относительно
индекса пропорционально углу поворота зеркальца 12 гальванометра 10.
Так как угол поворота зеркальца пропорционален интенсивности
светового потока, прошедшего через данный участок пластинки, то эта
интенсивность будет пропорциональна смещению шкалы.
234 ГЛ. VII. СПЕКТРАЛЬНЫЕ (ОПТИЧЕСКИЕ) МЕТОДЫ АНАЛИЗА
Между фотоэлементом 9 и щелью экрана 8 помещают серый
фотометрический клин 18, который дает возможность регулировать
интенсивность светового потока, падающего на фотоэлемент. Положение
клина подбирают таким образом,
чтобы с&о было равно 1000 мм, тогда:
Кроме миллиметровой шкалы
микрофотометр снабжен
логарифмической шкалой, изображение
которой получают на экране 7
поворотом зеркала 14. Логарифмическая
шкала микрофотометра получена
путем нанесения против
соответствующих делений микрометрической
шкалы вычисленных значений
почернений, умноженных на 100, т. е. 100 S.
Например, если отклонение по
логарифмической шкале составляет 84,
то почернение пластинки S = 0,84.
Фотопластинка расположена на
столике микрофотометра так, чтобы
изображения спектральных линий на экране 8 были параллельны щели
на этом экране. Смещая пластинку в направлении, перпендикулярном
спектральным линиям, достигают того, что изображение спектральной
линии совпадает с щелью. Ширина щели не должна превышать 2Д
ширины спектральной линии.
В. ПРАКТИЧЕСКИЕ РАБОТЫ
§ 12. Качественный анализ при помощи стилоскопа
1. Калибровка стилоскопа по длине волны. Сначала проверяют
чистоту угольных электродов; для этого наблюдают спектр дуги между
угольными электродами в воздухе при силе тока 2—3 а, применяя
генератор дуги типа ДГ-2 или ПС-39. В окуляре стилоскопа должен быть
виден сплошной спектр без спектральных линий. Затем выключают
дугу, дают углям остыть и на концы их наносят 1—2 капли раствора,
содержащего определенный элемент.
Если исследуют порошкообразную пробу, то ее помещают в канал,
который высверливают в одном угольном электроде.
Включают дугу и наблюдают спектр в окуляре стилоскопа. В поле
зрения окуляра стилоскопа имеется неподвижная стрелка (индикатор),
к которой поворотом барабана длин волн подводят все интенсивные
линии спектра. Отмечают показания шкалы барабана (деления
барабана). При помощи таблицы* спектральных линий (табл. 6) элементов
определяют по цвету длину волны линий взятого элемента.
Результаты записывают в виде таблицы:
Деления
барабана
Длина волны,
А
Элемент, который
соответствует
линии
*Свентицкий Н. С, Визуальные методы эмиссионного спектрального
анализа, Физматгиз, 1961,
Рис. 88. Оптическая схема
микрофотометра МФ-2:
/ — лампа; 2, 11 —конденсоры; 5 —поворотная
призма; 4, 6, 16 — объективы; 5 — фотопластинка;
7, 8, 15 — экраны; 9 — фотоэлемент;
/0—зеркальный гальванометр; 12 — зеркальце
гальванометра; 13, 14 — зеркала; 17 — пластинки с
изображением шкалы; 18 — фотометрический клин.
§ 13. КАЧЕСТВЕННЫЙ АНАЛИЗ ФОТОГРАФИЧЕСКИМ МЕТОДОМ 235
Пользуясь растворами, содержащими другие элементы, можно
получить данные по всей области спектра и построить дисперсионную
кривую стилоскопа — кривую зависимости делений шкалы барабана
стилоскопа п от длин волн К (рис. 89).
График строят на миллиметровой бумаге: по оси ординат откладывают деления
шкалы барабана п, а по оси абсцисс длины волн А. Примерный масштаб 10 А — 2 мм,
а 1 деление барабана — 4 мм. Если несколько точек не укладываются на график, это
значит, что линии неправильно отождествлены или стрелка (индикатор) окуляра
спектроскопа не точно наведена
на линию. Чем больше точек
взято для построения кривой, тем
она точнее.
Для построения
дисперсионной кривой удобно
пользоваться спектром ртутной
лампы. В спектре ртутной
лампы имеется ряд
интенсивных линий, длины волн
которых даны в табл. 7.
Методика определения.
На концы двух новых
угольных электродов
наносят 2—3 капли исследуемого
раствора. Затем включают
дугу и наблюдают спектр в
окуляре стилоскопа;
намечают спектральную линию
и, вращая барабан, подводят эту линию к индикатору в окуляре. Затем
отмечают показания шкалы барабана (деления барабана) и записывают
их в виде таблицы:
Рис. 89.
Дисперсионная кривая
стилоскопа
Элемент
Цвет линии
Деления барабана
Длина волны, А
Примечание
При помощи дисперсионной кривой определяют длину волны
спектральной линии. Операцию повторяют до тех пор, пока не будут
установлены все линии спектра. Пользуясь таблицей характерных
спектральных линий, определяют, какому элементу принадлежит данная линия.
§ 13. Качественный анализ фотографическим методом при помощи
спектра железа в ультрафиолетовой области
Определение катионов группы сернистого аммония
(Ш аналитическая группа)
Приготовление исследуемой пробы и подготовка к съемке. Для
анализа берут раствор, содержащий смесь катионов третьей
аналитической группы, которые осаждают групповым реактивом [(NH4)2S +
+ NH4C1 + NH4OH]; осадок отделяют центрифугированием, промывают
водой и сушат. Полученный порошок смешивают с равным количеством
измельченного в порошок графита (спектрально чистого). Для анализа
применяются угольные электроды. Нижний электрод снабжен
углублением — каналом (рис. 90). Верхний электрод дуги делают
конусообразным.
Сначала снимают спектр угольных электродов для проверки их
чистоты. Электроды укрепляют в штатив так, чтобы центр межэлектрод-
236
ГЛ. VII. СПЕКТРАЛЬНЫЕ (ОПТИЧЕСКИЕ) МЕТОДЫ АНАЛИЗА
ТАБЛИЦА 6
Элемент
А1
Ag
Ва
Са
Cd
Со
Сг
Си
Fe
1
Спе
Длина „
волны, А
3961,5*
3944,0
5209,1
5465,5
5535,5
4934,1
4554,0
5270,3
5265,6
5264,2
5262,2
5261,7
5260,4
5085 8
4867,9
4840,3
4813,5
5208,4
5206,0
5204,5
5218,2
5153,2 1
5105,5
5371,5
5328,0
5269,5
ктральные линии для визуального спектрального анализа
Цвет линии
Две фиолетовые линии,
появляются при
большем содержании
алюминия в пробе
Зеленый
Ярко-зеленый
Желто-зеленый
Зеленый
Фиолетовый
Группа зеленых линий
Ярко-зеленый
Группа зеленых линий
Группа зеленых линий
Группа зеленых линий
Групп! зеленых линий
Элемент
К
Mg
Мп
Na
Ni
Pb
Sn
Sr
Zn
Длина „
волны, А
5266,6
5232,9
5227,2
4047,2
4044,1
6249,9
5183,6
5172,7
4823,5
4783,4
4766,4
4762,4
4754,0
5895,9
5889,9
5476,9
5081,0
5080,5
5035,4
4980,2
4057,8
4524,7
4607,3
4810,5
4722,2
4680,1
' Цвег линии
Фиолетовый
Фиолетовый
Желтый
Синий
Синий
Ярко-синий
Яр ко-си ний
Ярко-синий
Ярко-синий
Ярко-синий
Две желтые линии
Ярко-зеленый
Три желто-зеленые
НИИ
Синий
Фиолетовый
Ярко-синий
Синий
ли-
Группа из трех голубых
линии
Полужирным шрифтом указаны наиболее интенсивные линии.
ТАБЛИЦА 7
Спектр ртутной лампы в
Цвет
Длина волны, А
6234,3
6123,4
6072,6
5740,6 *
5769,6
5460,7
4916,0 |
видимой области спектра
Цвет
Длина волны, А
4358,3
4347,5
4339,2
4108,0
4077,8
4046,7
* Полужирным шрифтом указаны наиболее интенсивные линии.
§ 13. КАЧЕСТВЕННЫЙ АНАЛИЗ ФОТОГРАФИЧЕСКИМ МЕТОДОМ 237
ТАБЛИЦА S
Наименование образца
Угольные электроды
Железные электроды .
Условия и порядок съемки спектров
Положение
кассеты, мм
10
10
10
диафрагмы
1
2
3
Экспозиция,
сек
40
40
5
Сила
тока, а
ооо
ного промежутка был на оптической оси коллиматора. Для этого при
помощи проекционного устройства, которым снабжен штатив ШТ-9,
получают изображение электродов на вспомогательном экране.
Закладывают (в темной комнате) фотографическую пластинку в кассету; при
этом эмульсия пластинки должна быть обращена к окуляру
спектрографа ИСП-22 или ИСП-28. Вставляют кассету в рамку спектрографа.
При помощи микровинта подбирают ширину щели, которая варьируется
в пределах 0,007—0,05 мм. Для правильного освещения источником
света щели спектрографа применяют трехлинзовую осветительную
систему (см. рис. 86) или другие способы освещения щели.
Фотографирование спектров. Фигурную диафрагму
помещают перед щелью спектрографа таким образом, чтобы
щель была совмещена с окошком / диафрагмы (см.
рис. 87). Закрывают затвор перед щелью спектрографа,
открывают кассету, включают генератор дуги ДГ-2 или ПС-39
(сила тока 10 а), затем открывают затвор и одновременно
включают секундомер. Через 40 сек закрывают затвор.
Когда угольные электроды дуги остынут, снимают
нижний электрод и в канал этого электрода вводят примерно
60 мг приготовленной пробы. Передвижением диафрагмы
совмещают щель с окошком 2 диафрагмы и, не меняя
положения кассеты, снимают спектр исследуемого образца,
как указано выше. Когда угли остынут, их снимают со
штатива и заменяют железными электродами.
Передвижением диафрагмы совмещают щель с окошком 3 и, как
указано выше, снимают спектр железа с экспозицией 5 сек.
В темной комнате вынимают фотографическую пластинку из кассеты,
проявляют, фиксируют и сушат спиртом. Условия и порядок съемок
приведены в табл. 8.
Расшифровка спектрограмм и идентификация
элементов
В качестве примера приводится определение алюминия.
Аналитические линии алюминия в ультрафиолетовой части спектра: К = 3082,2;
X = 3092,7; К = 3944,0 и К = 3961,5 А. На планшете № 15 атласа *
спектральных линий дан участок спектра железа в спектральной области
2930—3140 А. Фотопластинку помещают на столике спектропроектора
ПС-13 эмульсией вверх, вводят нужный участок спектра под объектив
и проецируют на экран прибора. На экран помещают планшет № 15
и совмещают линии спектра железа, спроецированные на экране, со
* Калинин С. К., Я в н е л ь А. А., Алексеева А. И., М и р з у в а н о в В. Л.,
Найма рк Л. Э., Атлас спектральных линий для кварцевого спектрографа, Госгеол^
техиздат, 1959.
н- 6мм —А
Рис. 90.
Угольный
электрод для
анализа
порошков.
238
ГЛ. VU. СПЕКТРАЛЬНЫЕ (ОПТИЧЕСКИЕ) МЕТОДЫ АНАЛИЗА
спектром железа на планшете так, чтобы совпадали линии с одной и
той же длиной волны. Если в исследуемой пробе присутствует
алюминий, то в ее спектре будет заметна линия, которая расположена между
сильными линиями железа X = 3075,7 и 3083,7 А. Эта линия является
аналитической линией алюминия Я = 3082,2 А. Линия (рис. 91, а)
алюминия, которая расположена между линиями железа X = 3091,5 и
3099,8 А, также является аналитической линией алюминия X = 3092,7 А.
Рис. 91. Открытие алюминия методом сравнения
его спектра со спектром железа:
а —сравнение с линиями планшета №15 (часть); б —
сравнение с линиями планшета № 19 (часть).
Затем заменяют планшет № 15 на планшет № 19, где дан участок
спектра железа в области 3750—4050 А, и, как описано выше, ищут в
спектре исследуемой пробы линию алюминия X = 3944,0 А, которая
располагается между линиями железа X = 3930,2 и 3950,0 А, и линию X =
= 3961,5 А — между линиями железа X = 3956,6 и 3966,0 А (рис. 91,6).
Таким же образом устанавливают в спектре пробы другие элементы
третьей аналитической группы.
§ 14. Полу количественный анализ
Определение железа и свинца в металлическом цинке
методом сравнения
Приготовление эталонных растворов. Исходный эталонный раствор.
Отвешивают 99,8 г х. ч. цинка, 0,1 г х. ч. железа и 0,1 г х. ч. свинца.
Навески растворяют в концентрированной хлористоводородной кислоте,
добавляют несколько капель HN03 и в мерной колбе доводят объем
раствора до 200 мл. Полученный раствор содержит 0,0500% Fe+++ и
0,0500% РЬ++.
§ 14. ПОЛУКОЛИЧЕСТВЕННЫЙ АНАЛИЗ
239
Разбавленные эталонные растворы. Из полученного исходного
раствора путем соответствующего разбавления готовят более разбавленные
растворы, как указано ниже;
Эталонный раствор, № 1 2 3 4 5
Объем исходного раствора, мл . 10 10 10 10 10
Объем воды, мл 0 10 40 60 90
Содержание ионов, %
Fe+++ 0,0500 0,0250 0,0100 0,0075 0,0050
Pb++ . * 0,0500 0,0250 0,0100 0,0075 0,0050
Приготовление пробы. Исследуемый образец цинка не должен
содержать больше 0,1% железа и свинца; 10 г исследуемого цинка
растворяют в концентрированной хлористоводородной кислоте и
разбавляют водой примерно до 20 ли.
Приготовление электродов. Берут шесть угольных электродов
диаметром 6 мм, в которых просверлены каналы диаметром 3 и глубиной
4 мм. В каналы пяти электродов вносят пипеткой 0,1 мл
соответствующего эталонного раствора, а канал шестого электрода пропитывают
раствором исследуемого образца цинка. Когда раствор поглотится,
электроды помещают в сушильный шкаф и приблизительно 30 мин
выдерживают при 95° С.
Фотографирование спектров. Устанавливают угольный электрод с
эталонным раствором № 1 в штатив спектрографа ИСП-22 или ИСП-28.
Перед щелью устанавливают фигурную диафрагму так, чтобы щель
была освещена окошком / диафрагмы (см. рис. 87), и снимают спектр
эталона № 1, как указано выше (ширина щели 0,01 мм, сила тока 10 а,
экспозиция 60 сек). Затем передвигают диафрагму таким образом,
чтобы щель была освещена световым потоком, проходящим через
диафрагму 2, угольные электроды заменяют на железные и снимают спектр
железа (экспозиция 5 сек), как указано в § 13. Кассету опускают на
5 мм и опять снимают пару спектров: спектр эталона № 2 и спектр
железа; таким же путем проводят фотографирование всех остальных
эталонов и исследуемого образца.
Условия и порядок съемки спектров приведены в табл. 9.
ТАБЛИЦА 9
Условия и порядок съемки спектров
Наименование образца
Положение
кассеты, мм диафрагмы
Экспозиция,
сек
Эталон № 1 ...
Железные электроды
Эталон №2 ...
Железные электроды
Эталон № 3 . . .
Железные электроды
Эталон №4 ...
Железные электроды
Эталон №5 ...
Железные электроды
Исследуемый образец
Железные электроды
10
10
15
15
20
20
25
25
30
30
35
35
1
2
1
2
1
2
1
2
1
2
1
2
60
5
60
5
60
5
60
5
60
5
60
5
240 ГЛ. VII. СПЕКТРАЛЬНЫЕ (ОПТИЧЕСКИЕ) МЕТОДЫ АНАЛИЗА
Пластинку проявляют, фиксируют и сушат. Получается шесть пар
спектров: в пяти парах спектр эталона и спектр железа, в шестой
паре — спектр исследуемого цинка и спектр железа.
Определение железа. Аналитические линии железа (в
ультрафиолетовой части спектра) X = 3020,6; X = 3581,2 и Л = 3720,0 А.
Пластинку вставляют в рамку спектропроектора ПС-18 и совмещают
изображение спектра железа со спектром железа на планшете № 15
спектрального атласа (см. стр. 237). Затем, как указано в § 13, в спектре
эталонов и в спектре образца цинка отыскивают линию железа X =
= 3020,6 А. Несколько левее этой линии расположена линия цинка
X = 3018,4 А. Визуально оценивают интенсивность линии железа X =
= 3020,6 А в спектрах эталонов и в спектре образца цинка.
Концентрация железа в исследуемом образце цинка примерно совпадает с
концентрацией железа в эталоне, когда интенсивность линии железа X =
= 3020,6 А в спектре образца цинка и в спектре эталона примерно
одинакова. Таким же образом в спектре исследуемого образца цинка
отыскивают линию железа X = 3581,2 А и линию X = 3720,0 А, пользуясь
планшетом № 18. Интенсивность этих линий в спектре исследуемого
образца цинка сравнивают с интенсивностью тех же линий в спектре
эталонов.
Определение свинца. В ультрафиолетовой части спектра
аналитические линии свинца следующие: X = 2873,0 и X = 4057,8 А. Линию
X = 2873,0 А отыскивают при помощи планшета № 13, линию Я =
= 4057,8 А — при помощи планшета № 20. Сравнивая интенсивность
этих линий в спектре исследуемого образца цинка и в спектре эталонов,
определяют приблизительную концентрацию свинца в исследуемом
образце цинка.
§ 15. Количественный анализ
Определение железа в кварцевом песке методом
трех эталонов
Метод дает возможность определить содержание железа в
кварцевом песке от 1 до 0,001%.
Приготовление эталонов
Эталон № 1. Отвешивают 9,9 г чистого кремневого ангидрида SiC>2
и 0,1 г окиси железа. Навески смешивают и тщательно растирают в
агатовой ступке. Полученный эталон содержит 1% окиси железа.
Эталон М 2. Смешивают 1 г эталона № 1 с 1 г кремневого
ангидрида. Смесь тщательно растирают в ступке. Полученный эталон
содержит 0,5% окиси железа.
Эталон М 3. Смешивают 0,1 г эталона № 2 с 0,9 г кремневого
ангидрида; смесь растирают в ступке. Эталон содержит 0,05% окиси
железа.
Приготовление электродов. Берут четыре угольных электрода
диаметром 6 мм, в которых просверлены каналы диаметром 3 мм и
глубиной 4 мм. Пробу исследуемого песка тщательно растирают в агато-
врй ступке; взвешивают 30 мг пробы и смешивают с графитовым
порошком в отношении 1:1. Затем смесь прокаливают в муфельной печи
при 300° С и без потерь вводят в канал угольного электрода.
Взвешивают по 30 мг эталонов № 1, № 2 и № 3, навески смешивают с
графитовым порошком в отношении 1:1, нагревают до 300° С и без потерь
вносят в каналы электродов.
§ 16. ОБЩАЯ ХАРАКТЕРИСТИКА МЕТОДА
241
Фотографирование спектров. Снимают спектры эталонов и спектр
исследуемой пробы кварцевого песка рядом со спектром железа
(ширина щели 0,01 мм, сила тока 10 а). Условия и порядок съемки
спектров эталонов № 1—№ 3 аналогичны приведенным в табл. 9. Спектр
пробы песка снимают при положении кассеты 25, диафрагме 2 и
экспозиции 60, соответствующую съемку спектров железных электродов
проводят при положении кассеты 25, диафрагме 2 и экспозиции 5.
Микрофотометрирование. Для определения железа в пробе
кварцевого песка берут линию спектра железа К = 2510 А. В качестве линии
внутреннего стандарта берут линию в спектре кремния X = 2503 А. При
помощи планшета № 10 атласа спектральных линий находят линию
кремния (элемент сравнения) X = 2503 А и линию железа X = 2510 А.
На микрофотометре МФ-2 определяют величину плотности почернения
SFe линии железа и величину плотности почернения Ssi линии кремния,
затем находят их разность AS: Д5 = Sj?e — Ssi-
Каждое определение повторяют 2 раза и берут среднее значение.
Таким образом получают средние значения S?e, Ssi и AS для трех
стандартов и пробы.
Калибровочный график. На миллиметровой бумаге наносят по
одной оси координат значения разности плотностей почернений линий
железа и кремния в спектре каждого эталона № 1, № 2 и № 3 (ASb AS2,
AS3). По другой оси наносят значения логарифмов соответствующих
концентраций железа. При помощи этих данных получают график,
который имеет вид прямой.
Затем по этому графику находят содержание железа в пробе.
СПЕКТРОФОТОМЕТРИЯ ПЛАМЕНИ
Л. ТЕОРЕТИЧЕСКИЕ ОСНОВЫ МЕТОДА
§ 16. Общая характеристика метода
Метод анализа по фотометрии пламени основан на измерении
интенсивности излучения атомов, возбужденных нагреванием вещества
в пламени. Для этого вводят раствор
исследуемого вещества в виде аэрозоля в
пламя газовой горелки при помощи сжатого t^
воздуха. Легко возбуждаемые элементы при |§
этом излучают лучи определенной длины д|
волны и окрашивают пламя. В некотором |^
интервале концентрации интенсивность из- |3
лучения атомов пропорциональна
концентрации атомов в растворе, который вводят в 0 Ut ог
пламя (рис. 92). На прямолинейном уча- Концентрация
стке АВ кривой зависимость интенсивности рис 92 Зависимость интен-
излучении (/) от концентрации (С) излу- сивности излучения от
кончающего элемента в растворе выражается центрации излучающего
уравнением: элемента в растворе.
I = KiC (12)
где К\ — коэффициент пропорциональности.
На зависимость интенсивности от концентрации влияет очень много
факторов, поэтому для получения сравнимых результатов необходимо
проводить измерения в стандартных условиях. Интенсивность
излучения определяют при помощи фотоэлемента. Как известно, фототоки,
возникающие при освещении фотоэлементов, прямо пропорциональны
242 ГЛ. VII. СПЕКТРАЛЬНЫЕ (ОПТИЧЕСКИЕ) МЕТОДЫ АНАЛИЗА
в определенных пределах интенсивности (/) падающего светового по-
тока: 1 = К2а (13)
где Кг — коэффициент пропорциональности;
а — сила фототока.
Из уравнения (12) и уравнения (13) находим:
К2й~К\С
К2
(14)
(На)
где /С— постоянная величина.
Источник возбуждения спектра — пламя имеет сравнительно
невысокую температуру, поэтому получаемые спектры сравнительно простые
и не содержат много линий. Простота спектров дает возможность
выделять искомые спектральные линии при помощи светофильтров или мо-
нохроматоров малой дисперсии. Метод фотометрии пламени является
разновидностью эмиссионного спектрального анализа, поэтому
приведенные выше теоретические основы эмиссионного метода анализа
в известной мере относятся и к рассматриваемому методу*,
Б. АППАРАТУРА И ТЕХНИКА ВЫПОЛНЕНИЯ АНАЛИЗА
§ 17. Схема установки, применяемой для анализа
методом спектрофотометрии пламени
Компрессор 1 (рис. 93) подает сжатый воздух в распылитель 2,
в котором распыляется исследуемый раствор. Раствор в виде аэрозоля
вводится в горелку <?, куда светильный
газ (пропан, ацетилен и т. п.)
подается через регулятор газа 7. Излучение
пламени проходит через светофильтр
4, пропускающий лучи только
определенной области спектра. Светофильтр
в некоторых установках заменен моно-
хроматором. Силу тока, возникающего
в фотоэлементе 5, измеряют
гальванометром 6 или другим прибором,
регистрирующим фототок.
Светофильтр должен быть
подобран таким образом, чтобы он
пропускал излучение определяемого
элемента и поглощал прочие излучения.
Наиболее пригодны интерференционные
светофильтры. В некоторых
установках применяются монохроматоры
наподобие используемых в
абсорбционной спектрофотометрии. В качестве
фотоэлементов обычно в простых
аппаратах применяют селеновые
фотоэлементы с запирающим слоем или сульфидно-серебряные с
запирающим слоем.
В. ПРАКТИЧЕСКИЕ РАБОТЫ
§ 18. Определения по методу спектрофотометрии пламени
Методом спектрофотометрии пламени можно определить
содержание различных элементов. Однако метод главным образом используют
Рис. 93. Схема установки,
применяемой для анализа методом
фотометрии пламени:
1 — компрессор для подачи сжатого воздуха;
2 — распылитель; 3 — горелка; 4 —
светофильтр; 5 — фотоэлемент; 6 —
регистрирующее устройство (гальванометр); 7
—регулятор газа; 8 — манометр; 9 — стакан с
исследуемым раствором.
§ 18. ОПРЕДЕЛЕНИЯ ПО МЕТОДУ СПЕКТРОФОТОМЕТРИИ ПЛАМЕНИ 243
для определения щелочных и щелочноземельных металлов, а также
некоторых других элементов (In, Tl, Pb, Мп, Си и др.). Возбуждение
атомов щелочных металлов происходит при 1200—1400° С, такую
температуру дает пламя смесей воздуха с пропаном, бутаном, светильным
газом. Для возбуждения атомов щелочноземельных металлов
необходима температура 2300°С (смесь воздуха с ацетиленом).
Определение ионов натрия в воде методом
калибровочного графика
Натрий определяют по спектральным линиям X = 5890—5896 А.
Как указано выше, в пламени светильного газа возбуждаются и
излучают только атомы щелочных металлов; поэтому натрий можно прямо
определить в присутствии прочих элементов. Определению натрия
мешают большие количества алюминия, который понижает интенсивность
излучения натрия. При определении натрия следует пользоваться
светофильтром, который пропускает излучения натрия, и селеновым
фотоэлементом, чувствительным в области 4500—7000 А.
Приготовление эталонных растворов и исследуемого раствора.
Берут на аналитических весах 0,0254 г хлорида натрия; навеску переносят
без потерь в мерную колбу емкостью 100 мл, растворяют в бидистилли-
рованной воде, доводят раствор до метки колбы бидистиллированной
водой и тщательно перемешивают; 1 мл полученного раствора содержит
0,1 мг натрия. При помощи бюретки определенные объемы этого
раствора переносят в мерные колбы емкостью 100 мл, разбавляют водой до
метки и тщательно перемешивают. Состав полученных эталонов
приведен ниже:
Эталонный раствор, №.... 1 2 3 4 5 6
Объем исходного раствора,
мл 2 3 4 5 6 7
Содержание натрия, мг/100 мл 0,2 0,3 0,4 0,5 0,6 0,7
Исследуемый раствор, содержащий от 0,1 до 1 мг натрия, помещают
в мерную колбу емкостью 100 мл, разбавляют бидистиллированной
водой, доводят до метки бидистиллированной водой и тщательно
перемешивают.
Калибровочный график. Перед фотоэлементом пламенного
фотометра устанавливают светофильтр для определения натрия. В стакан
распылителя наливают бидистиллированную воду и вводят ее в пламя
газовой горелки. Необходимо при помощи микрокранов поддерживать
давление воздуха и светильного газа постоянным; величину давления
измеряют манометром. Если при впрыскивании воды стрелка
микроамперметра отклонится, ее снова устанавливают на нуль
электрическим корректором или, если корректор отсутствует, фиксируют
показания микроамперметра. Затем в стакан распылителя наливают эталон
№ 1 и записывают показания микроамперметра. Отсчет повторяют Зраза
и берут среднее арифметическое значение. Затем распылитель и
горелку тщательно промывают бидистиллированной водой и повторяют
определения с другими эталонами.
На основании полученных результатов строят калибровочный
график, откладывая по оси ординат отсчеты по микроамперметру, а по оси
абсцисс — содержание натрия в эталонных растворах. Если
определение необходимо провести более точно, то на основании полученных
данных отсчетов по микроамперметру для эталонных растворов, пользуясь
методом наименьших квадратов, рассчитывают прямую
калибровочного графика.
244 ГЛ. VII. СПЕКТРАЛЬНЫЕ (ОПТИЧЕСКИЕ) МЕТОДЫ АНАЛИЗА
Определение содержания натрия в исследуемом растворе.
Тщательно промывают распылитель и горелку бидистиллатом, в стакан
наливают исследуемый раствор и, как указано выше, определяют
отклонение стрелки микроамперметра. Затем по калибровочному графику
находят содержание натрия в исследуемом растворе.
АТОМНО-АБСОРБЦИОННЫЙ СПЕКТРАЛЬНЫЙ АНАЛИЗ
§ 19. Общая характеристика метода
Атомы вещества в парообразном состоянии при определенных
условиях способны излучать лучи определенных длин волн (так называемое
характеристическое излучение). Вместе с тем атомы способны
поглощать то излучение, которое сами испускают. Таким образом, дл^на
волны лучей, поглощенных атомами элемента, совпадает с длиной волны
его характеристических излучений.
Методика атомно-абсорбционного спектрального анализа
заключается в том, что исследуемое вещество вводят в газовое пламя,
одновременно пламя освещают светом с непрерывным спектром, например
от лампы накаливания или от трубки с полым катодом (газоразрядная
трубка, в спектре которой наблюдаются линии элементов, входящие
в состав материала катода). В полученном спектре интенсивность света
в области характеристических частот будет меньше интенсивности
ближайших соседних участков спектра. Ослабление интенсивности
в области характеристических частот измеряют при помощи
фотоэлектрической установки. Между ослаблением интенсивности линии,
характерной для данного элемента, и концентрацией этого элемента в
исследуемой пробе наблюдается линейная зависимость.
МОЛЕКУЛЯРНО-АБСОРБЦИОННЫЙ СПЕКТРАЛЬНЫЙ АНАЛИЗ
Молекулярно-абсорбционный спектральный анализ включает: спек-
трофотометрический и фотоколориметрический анализ.
Л. ТЕОРЕТИЧЕСКИЕ ОСНОВЫ МЕТОДА
§ 20. Общая характеристика метода
Спектрофотометрический анализ основан на определении спектра
поглощения или измерении светопоглощения при строго определенной
длине волны, которая соответствует максимуму кривой поглощения
данного исследуемого вещества.
Фотоколориметрический анализ основан на сравнении интенсивности
окрасок исследуемого окрашенного раствора и стандартного
окрашенного раствора определенной концентрации.
Происхождение молекулярных спектров поглощения. Молекулы, как
и атомы, могут находиться только в определенных энергетических
состояниях, например Е0, Еи ?2> • • •, Еп. Если излучение определенной
длины волны проходит через вещество не поглощаясь, то, конечно,
энергетическое состояние молекул этого вещества останется без изменений.
Но если излучение, т. е. лучистая энергия, поглощается, то молекулы
вещества переходят из одного состояния Е^ (с меньшей энергией)
в другое состояние Е2 (с большей энергией). Согласно условию Бора
произведение волнового числа v поглощенного излучения и постоянной
Планка h равно разности энергии молекулы после поглощения и до
поглощения:
vh = E2-Ex = /S? (15)
§ 21. ЗАКОНЫ ПОГЛОЩЕНИЯ СВЕТА
245
Энергия молекулы состоит из трех составляющих: энергии
движения электронов молекулы ЕЭЛ1 энергии колебаний ?кол атомов
молекулы и энергии вращения молекулы ?вр, т. е. Е = ?Эл + ?кол + ?Вр,
таким образом:
V А = А?Эл + Л?кол + Л?вр (16)
При изменении только энергии вращения молекулы поглощенные
лучи имеют длины волн порядка 50—100 мк. Наблюдаемый спектр
называют вращательным; он лежит в далекой инфракрасной области
спектра. Если изменяется энергия колебания атомов молекул, которая
обычно связана с изменением вращательной энергии, то поглощенные
лучи тшеют длины волн порядка 2,5—20 мк. Наблюдаемый спектр
называют колебательно-вращательным; он лежит в более близкой к
видимой инфракрасной области. Наконец, если изменяется энергия
движения электронов в молекуле, то наблюдаемый спектр называют
электронным; он лежит в видимой и в ультрафиолетовой областях спектра.
В практической спектрофотометрии измерения поглощения
проводят в спектральной области, которую принято делить на 3 части:
ультрафиолетовая, видимая и инфракрасная области спектра. Единицей
измерения длин волн в ультрафиолетовой части спектра в практической
спектрофотометрии обычно служит нанометр (I нм = 10~7 см).
Ультрафиолетовая область спектра расположена в интервале длин волн 200—
400 нму видимая область — в интервале длин волн 400—700 нм.
Наконец, инфракрасная область спектра начинается примерно с 700 нм.
В инфракрасной области спектра единицей измерения длин волн
служит микрон (1 мк = К)-4 см). Очень часто инфракрасное излучение
характеризуется волновым числом v, v= IfX (где X выражено в см),
размерность v соответственно см~1. Например, длина волны 2 мк
соответствует волновому числу 5000 см~1. Имеются специальные таблицы
пересчета волновых чисел в длины волн. Наиболее доступная инфракрасная
область расположена в интервале 0,7—20 мк, более длинноволновая
область инфракрасного спектра малодоступна и практической спектрофо-
тометрией пока не используется.
§ 21. Законы поглощения света
Оптическая плотность растворов
Основной закон спектрофотометрии — закон Бугера — Ламберта —
Бера и понятие об оптической плотности раствора (D) приведены
ранее (см. книга 2, гл. IX, § 2):
Я-lg^-eC/ (17)
где /— интенсивность света, прошедшего через раствор;
/0 — интенсивность падающего на раствор света;
е — молярный коэффициент поглощения, эту величину также называют
коэффициентом погашения или экстинкцией;
С — концентрация окрашенного вещества, моль/л;
I — толщина слоя светопоглощающего раствора, см.
Отношение ///о называют пропусканием и обозначают буквой Т.
Оптическая плотность смеси. Оптическая плотность смеси (DCM)
двух или нескольких веществ в растворе аддитивно складывается из
оптических плотностей каждого компонента смеси:
DCM = Dl + D2 + D3+ ... +Dn (18)
где Di, D2} D3, ..., Dn —оптические плотности п компонентов.
246 ГЛ. VII. СПЕКТРАЛЬНЫЕ (ОПТИЧЕСКИЕ) МЕТОДЫ АНАЛИЗА
Отсюда
/>см = е1С1/+82С2/+8зСз/+ ... +епСп1 (19)
где Си Сг, Сз, .,., Сп — значения концентраций поглощающих веществ в растворе;
8i, 82, Ез, ..., еп — значения молярных коэффициентов поглощения;
/ — толщина слоя.
§ 22. Отклонения от закона Бугера—Ламберта—¦ Бера
Закон Бугера — Ламберта — Бера применим только для сред, в
которых агрегаты молекул, отдельные молекулы или ионы, которые
являются поглощающими центрами, остаются неизменными. Если
характер поглощающих центров меняется, например, в связи с
разбавлением, то показатель поглощения будет неодинаков для различных
концентраций этого вещества. Отсюда возникают отклонения от основного
закона спектрофотометрии, которые особенно заметны для
концентрированных растворов. Если в исследуемом растворе присутствуют
посторонние электролиты, то они могут вызвать деформацию молекул
окрашенных соединений и светопоглощение этих соединений изменяется.
На светопоглощение раствора влияют и многие другие факторы:
гидролиз, комплексообразование, образование промежуточных продуктов,
золей, таутомерные превращения, сольватация и др. Все эти явления
часто зависят от рН раствора.
Таким образом, колориметрические и спектрофотометрические
определения следует проводить в строго определенных интервалах значений
рН раствора. Нужный интервал рН среды создается применением
буферных растворов (см. книга 1, гл. I, § 21).
Отклонения, вызываемые не строго монохроматическим излучением.
Закон Бугера — Ламберта — Бера точно справедлив только для
монохроматического излучения. В спектрофотометрических измерениях
применяют монохроматоры, т. е. спектральные аппараты, которые
снабжены выходной щелью, вырезающей из спектра узкий участок. Но моно-
хроматор может дать строго монохроматическое излучение только
в том случае, если он снабжен бесконечно узкой щелью. В
действительности реальные аппараты снабжены щелью какой-то определенной
ширины, что вызывает некоторое отклонение от закона Бугера
—Ламберта — Бера. Особое значение немонохроматичность излучения
приобретает при измерениях в инфракрасной области спектра.
§ 23. Качественный анализ спектрофотометрическим
методом
Качественный анализ в ультрафиолетовой
части спектра
Ультрафиолетовые спектры поглощения обычно имеют две-три
иногда пять и более полос поглощения. Для однозначной идентификации
исследуемого вещества записывают его спектр поглощения в различных
растворителях и сравнивают полученные данные с соответствующими
спектрами сходных веществ известного состава. Если спектры
поглощения исследуемого вещества в разных растворителях совпадают со
спектром известного вещества, то можно с большой долей вероятности
сделать заключение об идентичности химического состава этих соединений.
Для идентификации неизвестного вещества по его спектру поглощения
необходимо располагать достаточным количеством спектров
поглощения органических и неорганических веществ. Существуют атласы, в
которых приведены спектры поглощения очень многих, в основном орга*
§ 23. КАЧЕСТВЕННЫЙ АНАЛИЗ СПЕКТРОФОТОМЕТРИЧЕСКИМ МЕТОДОМ 247
нических веществ. Особенно хорошо изучены ультрафиолетовые
спектры ароматических углеводородов.
При идентификации неизвестных соединений следует также
обратить внимание на интенсивность поглощения. Очень многие
органические соединения обладают полосами поглощения, максимумы которых
расположены при одинаковой длине волны Я, но интенсивности их
различны. Например, в спектре фенола наблюдается полоса поглощения
при X = 255 нм, для которой молярный коэффициент поглощения при
максимуме поглощения еМакс = 1450. При той же длине волны ацетон
имеет полосу, для которой еМакс = 17.
Качественный анализ в видимой части спектра
Идентификацию окрашенного вещества (например, красителя)
также можно проводить, сравнивая его спектр поглощения в видимой части
со спектром сходного красителя. Спектры поглощения большинства
красителей описаны в специальных атласах и руководствах. По спектру
поглощения красителя можно сделать заключение о чистоте красителя,
потому что в спектре примесей обычно имеется ряд полос поглощения,
которые отсутствуют в спектре красителя. По спектру поглощения смеси
красителей можно также сделать заключение о составе смеси, особенно
если в спектрах компонентов смеси имеются полосы поглощения,
расположенные в разных областях спектра.
Качественный анализ в инфракрасной
области спектра
Образование инфракрасных спектров связано с энергией колебаний
атомов молекул. Колебания могут быть направлены вдоль валентной
связи между атомами молекулы,-в таком случае они называются
валентными. Различают симметричные валентные колебания, в которых атомы
колеблются в одинаковых направлениях, и асимметричные валентные
колебания, в которых атомы колеблются в противоположных
направлениях. Когда колебания атомов происходят с изменением величины угла
между связями, они называются деформационными (обозначаются б).
Такое разделение, вообще, условно, потому что при валентных
колебаниях происходит в той или иной степени деформация углов, и наоборот.
Энергия деформационных колебаний обычно меньше, чем энергия
валентных колебаний, и полосы поглощения, обусловленные
деформационными колебаниями, располагаются в области более длинных волн.
Колебания всех атомов молекулы обусловливают полосы
поглощения, индивидуальные для молекул данного вещества. Но среди этих
колебаний можно выделить колебания групп атомов, которые слабо
связаны с колебаниями атомов остальной части молекулы. Полосы
поглощения, обусловленные такими колебаниями, называются
характеристическими полосами. Они наблюдаются, как правило, в спектрах всех
молекул, в которых имеются данные группы атомов. Примером
характеристических полос могут служить полосы 2960 и 2870 саг1. Первая полоса
обусловлена асимметричными валентными колебаниями связи С—Н
в метильной группе СН3, вторая — симметричными валентными
колебаниями связи С—Н этой же группы. Такие полосы с небольшим
отклонением (±10 см~1) наблюдаются в спектрах всех насыщенных
углеводородов и вообще в спектре всех молекул, в которых имеются СН3-группы.
Другие функциональные группы могут влиять на положение
характеристической полосы, причем разность частот может составлять до
248
ГЛ. VII. СПЕКТРАЛЬНЫЕ (ОПТИЧЕСКИЕ) МЕТОДЫ АНАЛИЗА
±100 см-19 но такие случаи немногочисленны, и их можно учитывать на
основании литературных данных.
Качественный анализ в инфракрасной области спектра проводят
двумя способами:
а) Снимают спектр неизвестного вещества в области 5000—500 смг1
(2—20 мк) и отыскивают сходный спектр в специальных каталогах или
Рис. 94. Спектр поглощения
изомеров ксилола:
а - о-ксилол, б — л-ксилол; в - я-кснлол.
таблицах. Для облегчения задачи спектры отпечатаны на
перфорированных картах, т. е. каждая полоса поглощения на них обозначается
отверстием. Тогда по полосам неизвестного вещества легко можно отыскать
карту, соответствующую его полосам поглощения, и таким образом
идентифицировать его.
б) В спектре исследуемого вещества отыскивают
характеристические полосы, по которым можно судить о составе вещества.
Пример 1. Инфракрасные спектры изомеров ксилола представлены на рис. 94.
Для отнесения спектров к соответствующим изомерам по таблице характеристических
частот ароматических соединений находим, что в спектре орто-замещенных
бензольного кольца наблюдается сильная полоса в области 770—735 см~1. Эта полоса
обусловлена деформационными колебаниями связи С—Н. Полоса 742 см~1 имеется в
спектре, приведенном на рис. 94, а. Из этого можно заключить, что спектр следует
отнести к о-ксилолу. В спектрах мета-замещенных бензольного кольца наблюдаются
две полосы: одна в области 810—750 смг1 и другая в области 710—690 сиг1. В спектре,
данном на рис. 94, б, наблюдаются полосы 767 и 692 смг1. "Из этого можно заключить,
что спектр следует отнести к ж-ксилолу. В спектрах пара-замещенных бензольного
кольца имеется полоса в области 840—810 смг1. "Эта полоса обусловлена
деформационными колебаниями связи С—Н. В спектре (см. рис. 94, в) наблюдается полоса
792 смг1. Из этого можно заключить, что спектр следует отнести к /г-ксилолу.
Пример 2. На рис. 95 дан спектр неизвестного вещества, эмпирическая формула
которого СвНнОз. Нужно установить строение этого соединения. В спектре исследуе-
§ 24. КОЛИЧЕСТВЕННЫЙ АНАЛИЗ СПЕКТРОФОТОМЕТРИЧЕСКИМ МЕТОДОМ 249
мого соединения наблюдаются следующие полосы: 2941, 1810, 1740, 1465, 1410, 1370,
1040 и 755 смг1. Данные, имеющиеся в таблицах, совпадают с полученными примерно
в интервале ±20—30 см~1. Так, полосе 2941 елг1 соответствует указанная в таблице
полоса 2960 еж-1; полосе 1370 елг1 соответствует полоса 1380 елг1 и полосе 1465 елг1—
полоса 1460 смг1. Все эти полосы связаны с различными видами колебаний связи С—Н
метильной группы. Полоса 755 см~1 обусловлена деформационными колебаниями связи
С—Н метиленовых групп (СН2)п. (Чем меньше /г, тем больше частота колебаний.)
Группа (СНг)п обусловливает по табличным
данным полосу в области 743—734 смг\ На too
основании этого можно заключить, что
исследуемое соединение содержит пропильную
группу СНз—СН2—СН2—. 80
Согласно табличным данным, колебания
в области 1950—1600 смг1 обусловлены валент- ^ I
ными колебаниями группы С=0 в ангидридах §^г
кислот —СО—О—СО—. В таблицах указаны |
две полосы: 1820 и 1760 смгх. Положение этих **
Ът
30V-
полос хорошо совпадает с полосами 1810 и
1740 смг1 в спектре исследуемого соединения.
Полоса 1810 см~1 более интенсивна, чем
полоса 1740 см~1, т. е. согласно табличным данным
эта полоса относится к ациклическим
ангидридам. По табличным данным, полоса в
области 1200—1000 смг1 обусловлена колебаниями
группы —СО—О—СО—. В спектре
исследуемого соединения наблюдается полоса 1040 еж-1,
и ее можно отнести к колебаниям группы
—СО—О—СО—.
Суммируя полученные результаты, можно сделать заключение, что исследуемое
соединение является ангидридом масляной кислоты (СНзСНгСНгСОЬО.
Рис. 95. Спектр поглощения
ангидрида масляной кислоты.
§ 24. Количественный анализ спектрофотометрическим
методом
Методы анализа веществ, поглощающих световые лучи
Концентрация определяемого вещества может быть определена
непосредственно, если в спектре поглощения раствора этого вещества
имеются ясно выраженные полосы поглощения в ультрафиолетовой,
видимой или инфракрасной областях спектра. Например, в
ультрафиолетовой части спектра поглощения бензола в циклогексане наблюдается
полоса поглощения с максимумом при X — 254,2 нм. Определив
коэффициент поглощения раствора бензола в циклогексане, можно определить
концентрацию бензола.
Особенно хорошо разработаны методы определения ароматических
углеводородов при совместном присутствии. В ультрафиолетовой части
спектра этих углеводородов наблюдаются ясно выраженные полосы
поглощения. Например, разработаны методы определения бензола, метил-
бензола, этил бензол а, 1,2-, 1,3- и 1,4-диметилбензола в бензине,
нафталине; 1,2-метилнафталина в керосиновых фракциях; антрацена в сыром
антрацене; 1,3-бутадиена в бутенах и др.
В спектре поглощения растворов редкоземельных элементов
наблюдаются хорошо выраженные полосы поглощения, которые могут быть
использованы для определения этих элементов.
Спектрофотометрическим методом можно непосредственно
определять некоторые окрашенные соединения — искусственные или
естественные красители.
Методы анализа веществ, не поглощающих световые лучи
Спектрофотометрическое определение вещества, не имеющего ясно
выраженных полос поглощения, возможно лишь после взаимодействия
этого вещества с соответствующим реактивом с образованием окрашен-
250
ГЛ. VII. СПЕКТРАЛЬНЫЕ (ОПТИЧЕСКИЕ) МЕТОДЫ АНАЛИЗА
ного продукта реакции. В спектре раствора полученного соединения
должны быть хорошо выраженные полосы поглощения в
ультрафиолетовой или в видимой частях спектра.
Например, при взаимодействии о-оксихинолина (оксин) с ионом
алюминия получается соединение, хорошо растворимое в четыреххлори-
стом углероде, в спектре поглощения которого наблюдаются полосы
поглощения при X = 260 нм (ультрафиолетовая часть спектра) и при
X = 390 нм (видимая часть).
Очень часто реакции образования окрашенных соединений
обратимы и идут при определенных условиях, которые необходимо точно
соблюдать для получения достаточно надежных результатов.
§ 25. Методы определения концентрации веществ, поглощающих
в видимой и ультрафиолетовой областях спектра
Определение концентрации вещества методом сравнения
оптических плотностей эталонного и исследуемого растворов
Для определения готовят эталонный раствор определяемого
вещества известной концентрации, которая приближается к концентрации
исследуемого раствора. Определяют оптическую плотность этого раствора
при определенной длине волны (DBT). Затем определяют оптическую
плотность исследуемого раствора (Dx) при той же длине волны и при
той же толщине слоя. Для эталонного раствора согласно уравнению
(17> ИМееМ: Дат = еСЭ1/ (20)
где е — молярный коэффициент поглощения исследуемого раствора;
/ — толщина слоя, см.
Оптическая плотность исследуемого раствора выражается такой же
формулой:
Dx^eCJ (21)
где Сх — концентрация исследуемого раствора, мг\мл.
Отсюда
(22)
Сх
Х/ЭТ ^!
эт
'СэтТГ- (22а)
а
эт
Количество (g) определяемого вещества (в мг) с учетом
разбавления раствора находим по формуле:
cxvaK
g- Vn (23)
где VK — общий объем исследуемого раствора, мл:
VA — объем окрашенного исследуемого раствора, мл;
Vn — объем аликвотной части исследуемого раствора, взятой для приготовления
окрашенного раствора, мл.
Определение концентрации вещества в растворе
по значению молярного коэффициента поглощения
Определив значение оптической плотности Дс-раствора при длине
волны X и зная значение молярного коэффициента поглощения е*,
определяемого вещества (моль/л) для лучей длины волны X, находим по
формуле (17) значение концентрации Сх исследуемого вещества
(моль 1л): п
С* = ТТ (24)
§ 25. МЕТОДЫ ОПРЕДЕЛЕНИЯ КОНЦЕНТРАЦИИ ВЕЩЕСТВ
251
Количество (g) определяемого вещества (в г) находим по формуле:
VaV
g-Cx-^-Мь (25)
где Ма — молекулярный (атомный) вес определяемого вещества (иона).
Значение молярного коэффициента поглощения ех устанавливают
следующим образом. Готовят эталонный раствор исследуемого вещества
определенной концентрации Сэт (моль/л) и измеряют значение
оптической плотности Dm этого раствора при длине волны X и значение е^,
вычисляют по формуле:
Dn (26)
8,=
/сэ
Если вещество трудно получить в чистом виде, то можно
пользоваться табличным значением е.
Определение концентрации вещества с
калибровочного графика
помощью
Функциональная зависимость D = f(C) между оптической
плотностью раствора и концентрацией поглощающего вещества может быть
установлена графически. Для этого предварительно
готовят серию растворов определяемого вещества
различной концентрации (эталонные растворы).
Измеряют значения оптической плотности этих растворов
для лучей с длиной волны Я и по полученным данным
строят кривую зависимости оптической плотности
раствора от концентрации (калибровочный график). На
ось ординат наносят значения оптической плотности
эталонных растворов фь D2, Dz, ..., Dn), а на ось
абсцисс — соответствующие значения концентраций
этих растворов (Сь С2, С3, ..., Сп). Для получения
более точных результатов рассчитывают, пользуясь
методом наименьших квадратов, уравнение для
калибровочного графика.
Определив значение Dx оптической плотности
исследуемого раствора при той же толщине слоя,
можно найти концентрацию Сх определяемого
вещества по полученному калибровочному графику. Если
раствор не подчиняется закону Бугера—Ламберта—
Бера, то прямолинейная зависимость нарушается на
некотором участке кривой или на всей кривой. В этом
случае необходимо увеличить число эталонных
растворов. Концентрацию эталонных растворов обычно выражают в мг}мл<
Количество определяемого вещества (g) в миллиграммах определяют
по формуле (23).
Рис. 96.
Оптическая схема
колориметра
погружения:
I — зеркало; 2, 3 —
кюветы; 4, 5 —
стеклянные цилиндры; 5, 7—
призмы;- S, 9— линзы.
Определение концентрации вещества методом «уравнивания»
или методом изменения толщины поглощающего слоя
Оптическую плотность исследуемого раствора Dx определяют по
формуле:
Dx = eCJx (27)
где 8 — молярный коэффициент поглощения исследуемого раствора;
Сх — концентрация определяемого вещества, мг[мл;
U — толщина слоя, cal
252
ГЛ. VII. СПЕКТРАЛЬНЫЕ (ОПТИЧЕСКИЕ) МЕТОДЫ АНАЛИЗА
Оптическую плотность эталонного раствора определяют по формуле
D3T = еСэт/эт (27а)
где Сэт — концентрация эталонного раствора, мг/мл;
/эт — толщина слоя, см.
Изменяя толщину слоя исследуемого раствора и эталонного
раствора, можно установить тождественность цвета исследуемого раствора
с цветом эталонного. Тогда оба раствора будут иметь одинаковую
оптическую плотность:
Аг=Яэт (28)
Отсюда
С*=С9Т-^ (28а)
На использрвании этого равенства основано устройство
колориметра погружения (колориметр Дюбоска), в котором тождественность
цвета достигается изменением толщины слоя растворов. Оптическая
схема колориметра погружения дана на рис. 96. Один световой поток
от зеркала 1 проходит через слой исследуемого раствора в кювете 2,
цилиндр 4, призму б, линзы 8 и 9 и попадает в окуляр, освещая правую
половину оптического поля. Другой световой поток проходит через слой
стандартного раствора в кювете 5, цилиндр 5, призму 7, линзы 8 и 9У
попадает в окуляр, освещая левую половину оптического поля. Кюветы
2 и 3 установлены на держателях, которые при помощи шестеренок и
реек передвигаются вертикально. Стеклянные цилиндры 4 и 5 с
отшлифованными концами укреплены неподвижно. Перемещая кюветы 2 и 3 по
вертикали, меняют высоту столбов раствора и добиваются исчезновения
границ раздела в окуляре оптического поля. Высоты столбов
эталонного раствора и исследуемого раствора отсчитывают по миллиметровой
шкале.
§ 26. Определение нескольких компонентов в растворе
Для определения двух компонентов в растворе устанавливают
значения оптической плотности Dx и D2 раствора смеси двух компонентов
при длинах волн К\ и Яг и составляют два уравнения, аналогичных
уравнению (19):
D. = г\СЛ + z!Cd
(29)
02Bre2Cl* + e2/<V
где I — толщина слоя, см\
С\ и С2 — концентрация определяемых веществ, моль/л;
е[, ъ2~~ молярные коэффициенты поглощения первого компонента при длине волн
%\ и Яг;
8Г» &2 ~~ молярные коэффициенты поглощения второго компонента при длине волн
%i и Яг.
Решая уравнения (29) в отношении С\ и С2, находим их значения.
Таким образом, составляя три, четыре и более уравнений типа
уравнения (29), можно определить три, четыре и более компонентов в
растворе.
§ 27. Определение концентрации вещества в растворе
дифференциальным методом
Аппараты, которые предназначены для измерения оптической
плотности растворов, снабжены двумя кюветами. В одну кювету наливают
исследуемый или эталонный раствор, в другую — растворитель, напри-
§ 27. ОПРЕДЕЛЕНИЕ КОНЦЕНТРАЦИИ ВЕЩЕСТВА В РАСТВОРЕ 253
lg
л-
h =
h
~h
= D'X
10"
10"
10"
10'
= el
¦elCo
ucx
-ElCo
=^7
'(c*-
мер чистую воду. Это дает возможность измерить оптическую плотность
исследуемого или эталонного раствора по отношению к чистому
растворителю с нулевым поглощением. Когда определение концентрации
вещества в растворе ведется по дифференциальному методу, в одну из кювет
вместо воды наливают окрашенный раствор определяемого элемента
с известной концентрацией С0, т. е. раствор сравнения. Оптическую
плотность исследуемого или эталонного раствора измеряют по отношению
к окрашенному раствору определяемого элемента. Концентрация С0
раствора сравнения должна быть меньше концентрации Сх исследуемого
раствора. Если 1\ — интенсивность светового потока, прошедшего через
слой раствора сравнения, /2— интенсивность светового потока,
прошедшего через исследуемый раствор, / — толщина поглощающего слоя, г —
молярный коэффициент поглощения определяемого вещества, то по
закону Бугера — Ламберта — Бера:
(30)
(30а)
(31)
12 10 ~'~х
или
С0) (32)
где Dx — оптическая плотность исследуемого раствора по отношению к раствору
сравнения.
Из этого уравнения следует, что между относительной оптической
плотностью исследуемого раствора D' и его концентрацией С0
существует функциональная зависимость D' = f(C), которая выражается
прямой.
Функциональную зависимость между D' и С можно установить при
помощи калибровочного графика. Для этого готовят серию эталонных
растворов определяемого вещества различной концентрации: Сь С2,
Сз, ..., Сп и раствор сравнения определяемого вещества С0 (притом С0
меньше концентрации эталонных растворов). Затем определяют
оптическую плотность эталонных растворов по отношению к раствору
сравнения (р'и Do, Аз, ..., Ол) и оптическую плотность исследуемого
раствора Drx по отношению к раствору сравнения. По полученным данным
строят калибровочный график, принимая за начало отсчета
концентрацию раствора сравнения С0, затем по калибровочному графику находят
концентрацию Сх.
Концентрацию исследуемого раствора можно определить расчетным
способом. Для этого готовят раствор сравнения с концентрацией С0,
эталонный раствор определяемого элемента с концентрацией Сэт, затем
находят оптическую плотность эталонного раствора Д>т и исследуемого
раствора Dx по отношению к раствору сравнения. Согласно
формуле (32):
Dx = et(Cx-C0)
(32а)
D'3T=el(C3T-C0)
Отсюда
^эт ^эт ~ ^0
° (33)
254
ГЛ. VII. СПЕКТРАЛЬНЫЕ (ОПТИЧЕСКИЕ) МЕТОДЫ АНАЛИЗА
Сх--~т-(Сэт-Со) + С0 (34)
Поскольку (Сэт — Со)/D'3T является постоянной величиной F, то
CX = D'XF + C0 (35)
§ 28. Выбор толщины слоя и оптимальной концентрации
исследуемого раствора
По закону Бугера — Ламберта — Бера оптическая плотность
зависит от молярного коэффициента поглощения исследуемого раствора,
концентрации раствора и толщины слоя. Теоретические расчеты
показывают, что ошибка при определении концентрации исследуемого вещества
минимальна, когда оптическая плотность исследуемого раствора равна
0,44. Практически хорошие результаты получаются при оптической
плотности от 0,2 до 1. Значение молярных коэффициентов поглощения
различных соединений меняется от долей единицы до 100 000. Оптическая
плотность раствора прямо пропорциональна молярному коэффициенту
поглощения, поэтому при толщине слоя примерно 1 см для веществ
с высоким молярным коэффициентом поглощения нужно брать
разбавленные растворы, если желательно, чтобы оптическая плотность
растворов укладывалась в пределах 0,2—1. Например, если значение
молярного коэффициента поглощения исследуемого вещества равно 100,
толщина слоя 1 см, то для получения раствора, оптическая плотность
которого примерно 0,5, нужную концентрацию (моль/л) определяют по
формуле:
0,5
100
С, = -й?=0,005
но если значение молярного коэффициента поглощения исследуемого
вещества равно 1000, тогда концентрация (моль/л) исследуемого
вещества должна быть значительно ниже, т. е.
0,5
10 000
= 0,00005
§ 29. Выбор длины волны поглощаемого излучения
при спектрофотометрических измерениях
Измерение оптической плотности раствора исследуемого вещества
нужно проводить при длине волны, которая соответствует максимуму
поглощения излучений. Если исследуемое вещество обладает
несколькими полосами поглощения, необходимо взять полосу, для которой
коэффициент поглощения наиболее высок. Эту необходимость легко
доказать. Дифференциируем уравнение (20) по С при / = 1, получаем:
dD AD
-ЗС =е*' Т-е' "ДС~ = 8* (36)
Из уравнения (36) следует, что с изменением концентрации АС
изменение оптической плотности раствора AD тем больше, чем выше
молярный коэффициент поглощения исследуемого раствора. Наибольшее
значение е находится при длине волны, которая соответствует
максимуму поглощения излучений раствором определяемого вещества;
следовательно, измерение оптической плотности раствора следует проводить
ПрИ ЭТОЙ ДЛИНе ВОЛНЫ (ЯМакс).
§ 30. ПРИБОРЫ, ПРИМЕНЯЕМЫЕ ДЛЯ АНАЛИЗА В УФ И ВИДИМОЙ ОБЛАСТЯХ 255
Ошибка при определении концентрации в данном случае будет
наименьшая.
При спектрофотометрических измерениях монохроматический поток
света с ЯМакс выделяют при помощи монохроматора. При
колориметрических измерениях только приблизительно выделяют светофильтрами
нужную область спектра. Максимум пропускания светофильтра должен
лежать в области спектра, которая соответствует максимуму
поглощения определяемого вещества (рис. 97).
Б. АППАРАТУРА И ТЕХНИКА ВЫПОЛНЕНИЯ АНАЛИЗА
§ 30. Приборы, применяемые для спектрофотометрического
анализа в ультрафиолетовой и видимой областях спектра
Фотоэлектрический спектрофотометр СФ-4 (рис. 98) применяется
для работы в видимой и ультрафиолетовой частях спектра. Свет от
источника излучения — водородной лампы (газоразрядная трубка,
наполненная водородом и снабженная катодом с подогреванием) или
Рис. 97. Кривые
поглощения:
1 — светофильтром; 2 —
окрашенным раствором.
Рис. 98. Оптическая схема спектрофотометра
СФ-4:
1 — источник света; 2 — конденсорное зеркало; 3 —
плоское зеркало; 4 — выходная щель монохроматора;
5 —сферическое зеркало; 6 — кварцевая призма; 7,
8 — кювета; 9 — фотоэлемент.
лампы накаливания 1 — фокусируется на выходную щель 4
монохроматора с помощью конденсорного зеркала 2 и плоского зеркала 3. Затем
свет падает на алюминированное сферическое зеркало 5 и отражается
на кварцевую призму 6, алюминированную с противоположной стороны.
Разложенный в спектр пучок света отражается зеркальной стороной
кварцевой призмы 6 обратно на зеркало 5 и фокусируется на щель 49
которая одновременно служит входной и выходной щелью
монохроматора. Вращая призму 6, можно изменять длину волны лучей, выходящих
из прибора. Длину волны выходящих лучей отсчитывают по шкале длин
волн, связанной с приспособлением, вращающим призму.
После выходной щели лучи проходят через кювету 7 с
растворителем или кювету S, содержащую исследуемый раствор, и падают на
фотоэлемент 9. Кюветы имеют кварцевые окошки. Перед фотоэлементом
расположена шторка, которая дает возможность перекрывать поток света,
падающий на фотоэлемент. Прибор снабжен двумя вакуумными
фотоэлементами: кислородноцезиевым — для измерений в области спектра
от 650—1100 нм и сурьмяноцезиевым — для измерений в области спектра
220—650 нм. Соответствующий фотоэлемент устанавливают специальной
рукояткой. При освещении фотоэлемента в нем возникает фототок,
величина которого пропорциональна световому потоку, падающему на
256
ГЛ. VII. СПЕКТРАЛЬНЫЕ (ОПТИЧЕСКИЕ) МЕТОДЫ АНАЛИЗА
фотоэлемент. Фототоки усиливаются усилителем постоянного тока.
В приборе фототок компенсируется током противоположного
направления, снимаемым с потенциометра. Величину компенсационного тока
контролируют с помощью индикаторного миллиамперметра, шкала
которого имеет нуль посередине. Измерительный потенциометр калиброван
в процентах пропускания от 0 до 100 и в единицах оптической плотности
от 0 до 2.
Фотоэлектрический спектрофотометр СФ-5. Для измерении в
видимой области спектра применяют спектрофотометр СФ-5, который имеет
стеклянную оптику и дает возможность проводить измерения в
спектральной области 380—1100 нм.
§ 31. Приборы, применяемые для колориметрического
анализа
Визуальные и фотоэлектрические колориметры. В визуальных
колориметрах приемником световых лучей является человеческий глаз, а
в фотоэлектрических колориметрах — фотоэлемент.
Рис. 99. Оптическая схема визуального фотометра ФМ-56:
/-источник излучений; 2 -зеркала; 3, 6 -линзы; 4 -кюветы; 5-диафрагмы;
7, 8-призмы; 9 — диск со светофильтрами; 10 — окуляр.
К визуальным колориметрам относятся колориметры
погружения. Фотоэлектрические колориметры описаны ранее (см. книга 2,
гл. IX, § 7).
Универсальный фотометр ФМ-56. Фотометр ФМ-56 относится к
визуальным колориметрам. Световой поток от источника / (рис. 99)
разделяется при помощи системы плоских зеркал 2 и линз 3 на два
параллельных пучка лучей, которые проходят через кюветы 4, диафрагмы 5
и вновь объединяются при помощи системы линз 6 и призм 7 и 8. Поле
зрения окуляра 10 разделено пополам четкой границей. Каждая
половина поля зрения окуляра освещается соответствующим пучком света,
прошедшим через соответствующую кювету 4. На диске 9 укрепляют
светофильтры, которые служат для выделения узких полос в спектре
лампы накаливания. Вращением этого диска может быть установлен
соответствующий светофильтр. Раскрытие диафрагмы 5 регистрируют при
помощи отсчетных барабанов, снабженных шкалами, калиброванными
в процентах пропускания (черная шкала) и. единицах оптической
плотности (красная шкала).
Оптическую плотность исследуемого раствора определяют
следующим образом. Перед окуляром фотометра помещают нужный
светофильтр, затем кювету с раствором устанавливают в одном пучке света,
§ 32. ПРИБОРЫ, ПРИМЕНЯЕМЫЕ ДЛЯ АНАЛИЗА В ИНФРАКРАСНОЙ ОБЛАСТИ 257
а кювету с растворителем — в другом. Барабаны, регулирующие
раскрытие диафрагм, ставят на нуль красной шкалы. Так как исследуемый
раствор больше поглощает световых лучей, чем растворитель, то обе
половины поля зрения окуляра будут освещены неодинаково. Половина поля
зрения окуляра, которая освещена световым потоком, проходящим через
растворитель, светлее половины, освещенной световым потоком,
проходящим через раствор. При помощи отсчетного барабана,
расположенного над кюветой с растворителем, уменьшают степень раскрытия
диафрагмы 5, через которую проходит световой поток, до одинакового
освещения обеих половин поля зрения окуляра. В этом случае оптическая
плотность исследуемого раствора будет равна оптической плотности,
соответствующей данному раскрытию диафрагм. Оптическую плотность
Dx раствора отсчитывают по шкале отсчетного барабана. Измерение
повторяют 3 раза и берут среднее арифметическое. После этого меняют
кюветы местами и повторяют измерения, причем барабаны снова
устанавливают на нуль красной шкалы. Определяют, как указано выше,
средний отсчет оптической плотности D2. Искомая оптическая плотность
раствора будет равна:
Фотометр ФМ-57 снабжен фотоэлементами, которые заменяют
визуальные наблюдения.
§ 32. Приборы, применяемые для спектрофотометрического
анализа в инфракрасной области спектра
Инфракрасный спектрометр ИКС-12*. Источником инфракрасных
лучей обычно служит карборундовый стержень (глобар), который
нагревают электрическим током. Для этой же цели применяют и штифт Нерн-
ста, также нагреваемый электрическим током. Штифт представляет
собой стержень из тонкоизмельченной, спрессованной и
сцементированной смеси окисей циркония, тория и церия. В холодном состоянии он не
проводит тока, поэтому его предварительно нагревают пламенем
газовой горелки для снижения сопротивления.
Оптическая схема инфракрасного спектрометра ИКС-12 дана на
рис. 100. Лучи от источника света / направляются плоским зеркалом 2
и вогнутым зеркалом 3 через кювету 4 на входную щель 5 монохрома-
тора. Выходя из щели, пучок попадает на вогнутое параболическое
зеркало 6. Далее лучи в виде параллельного пучка проходят через призму 7
из каменной соли или бромида калия (стекло сильно поглощает
инфракрасные лучи), отражаются от плоского зеркала 8 и возвращаются на
зеркало 6, от которого попадают на плоское зеркало 9 и направляются
на выходную щель 10 спектрометра. С помощью плоского /) и
сферического 13 зеркал лучи фокусируются на термоэлемент 12.
Поворачивая зеркало 6\ можно направить на выходную щель лучи с разной
длиной волны. Длину волны выходящих лучей отсчитывают на шкале
барабана, связанного с механизмом поворота зеркала.
Исследуемый раствор помещают в кювету 4 (из пластинок
каменной соли или сильвина), которая располагается перед щелью 5.
Термотоки, которые возникает в термоэлементе 12 (обычно применяют
вакуумный термоэлемент), усиливаются фотоэлектрическим усилителем
ФЭОУ-18 и регистрируются электронным потенциометром ЭПП-09
* Спектрофотометры, которые предназначены для измерений в инфракрасной
области спектра, обычно называют спектрометрами.
258
ГЛ. VII. СПЕКТРАЛЬНЫЕ (ОПТИЧЕСКИЕ) МЕТОДЫ АНАЛИЗА
с записывающим устройством. Поворот зеркала S, устанавливающего
требуемую длину волны на выходной щели, производится рычагом,
который связан с барабаном длин волн. На барабане нанесена шкала
с делениями. При непрерывной записи спектра барабан длин волн по-
Рис. 100. Оптическая схема спектрофотометра ИКС-12:
/ — источник света; 2 — плоское зеркало; 3 — вогнутое зеркало; 4 — кювета; 5 — входная щель моно-
Хроматора; 6 — параболическое зеркало; 7—призма; 8, 9, 11 — плоские зеркала; 10— выходная щель;
12 — термоэлемент; 13 — сферическое зеркало.
ворачивается автоматическим мотором через редуктор. Одновременно
движется бумажная лента потенциометра ЭПП-09, которая
регистрирует усиленные термотоки термоэлемента. Барабан длин волн должен
быть предварительно калиброван по положению известных полос
поглощения определяемого вещества.
При построении калибровочного графика на оси абсцисс
откладывают деления барабана, на оси ординат — длины волн или частоты.
Калибровочный график выражает
зависимость n = f(X) (где п —
показания барабана длин волн).
Для получения кривой светопо-
глощения исследуемого раствора
поступают следующим образом. В
кювету наливают растворитель
(обычно четыреххлористый углерод для
спектральной области 2—10 мк или
сероуглерод для области 10—16 мк).
Пускают мотор вращения барабана
и мотор, который приводит в
движение ленту потенциометра. На
бумажной ленте получают кривую
(рис. 101, а), которая выражает
функциональную зависимость
интенсивности светового потока по
выходе из кюветы с растворителем от
длины волны: /о = /(Я). Затем
повторяют проделанные операции,
только растворитель заменяют исследуемым раствором. Получают на
бумажной ленте кривую (рис. 101,6), которая выражает
функциональную зависимость / = f (Х).
Линии 00 и 04)' соответствуют записи пера потенциометра при
неосвещенном термоэлементе. На этих линиях специальное
приспособление фиксирует показания барабана длин волн для каждой точки. Из
какой-нибудь точки п на линии 00 восстанавливают перпендикуляр до
пересечения с кривой и при помощи линейки измеряют отрезок a0i затем
11 12 13 /4 15 11 12 13 1U 15
а б
Рис. 101. Образец записи кривых свето-
поглощения с помощью
спектрофотометра ИКС-14:
а— спектр растворителя; б —спектр
исследуемого вещества.
§ 34. ОПРЕДЕЛЕНИЕ В УЛЬТРАФИОЛЕТОВОЙ ОБЛАСТИ СПЕКТРА 259
из точки п на линии О'О' восстанавливают перпендикуляр до пересечения
с кривой и измеряют отрезок а. По калибровочному графику п = f (к)
определяют длину волны, которая соответствует точке п. Так как
отрезки а0 и а прямо пропорциональны термотокам в термоэлементе, а
термотоки прямо пропорциональны световым потокам световой
энергии /0 и /, то оптическая плотность исследуемого раствора при длине
волны Я будет выражаться уравнением:
Di=lg^ = lg-^ (38)
Такой расчет проводят и для других точек прямых 00 и О'О'. Чем круче
ход кривых, тем больше точек следует брать на прямых О'О' и 00. Для
каждого участка спектра выбирают такое раскрытие щели, при
котором показания потенциометра не выходилц бы за пределы шкалы.
Спектрометр ИКС-14. Этот спектрометр двухлучевой. Излучение от
источника света направляется одновременно через кювету с
растворителем и кювету с раствором. Самодисец вычерчивает на бумажной ленте
кривую функциональной зависимости пропускания от длины волны X,
т. е. прямо без предварительных расчетов определяется кривая
поглощения исследуемого раствора.
§ 33. Нулевые растворы
Если применяемые в процессе спе^рофотометрического
определения реагенты бесцветны и используются водные растворы, то в кювету
сравнения наливают дистиллированную воду. Если же применяют
окрашенные реактивы, то в кювету сравнения наливают те же реактивы
в том же количестве, но без определяемого элемента. Такой раствор
называется нулевым или раствором сравнения.
Неводные растворители, например циклогексан, сероуглерод и
другие, должны быть «оптически» чистыми, т. е. не содержать никаких
примесей, которые могут поглощать лучи той области спектра, в которой
проводят спектрофотометрическое определение. Растворители,
применяемые Для спектрофотометрических измерений в инфракрасной области
спектра, не должны содержать воды, потому что вода разрушает кюветы
из каменной соли или сильвина.
В. ПРАКТИЧЕСКИЕ РАБОТЫ
§ 34. Определение в ультрафиолетовой области спектра
Определение молибдена в стали
Для растворения стали, содержащей в своем составе примесь
молибдена, применяют смесь концентрированных хлористоводородной и
азотной кислот, при этом образуются соединения молибдена (VI).
Максимум поглощения молибдена (VI) расположен примерно в области
Я = 230 нм. Это дает возможность определять молибден в растворе
спектрофотометрическим методом в ультрафиолетовой области спектра.
Концентрацию молибдена устанавливают по значению молярного
коэффициента поглощения раствора.
Приготовление эталонного раствора. На аналитических весах
отвешивают 0,2 г молибдата аммония марки «х. ч.» и помещают его в
мерную колбу емкостью 500 мл; растворяют в воде, доводят водой до
метки и перемешивают. Молярная концентрация такого раствора равна:
ТЙ^-5Ж = °'0003236Ж0^
где 1235,9 —молекулярный вес (NH4)6Mo7024t4H20.
260 ГЛ. VII. СПЕКТРАЛЬНЫЕ (ОПТИЧЕСКИЕ) МЕТОДЫ АНАЛИЗА
Пипеткой отбирают 20 мл полученного раствора, переносят в
мерную колбу емкостью 200 мл, прибавляют 4 мл 20%-ного раствора
NaOH, доводят водой до метки и перемешивают. Молярная
концентрация полученного раствора равца 0,000032362 моль/л.
Определение молярного коэффициента поглощения. Определяют
оптическую плотность полученного эталонного раствора при X = 230 нм,
применяя кварцевую кювету длиной 1 см. Нулевой раствор готовят
следующим образом: в мерную колбу емкостью 200 мл помещают 4 мл
20%-ного раствора NaOH и разбавляют водой до метки. Оптическую
плотность эталонного раствора определяют на спектрофотометре СФ-4.
Измерения проводят 3 раза и берут среднее арифметическое
полученных результатов. По полученным значениям оптической плотности
эталонного раствора вычисляют молярный коэффициент поглощения
раствора молибдена (VI) по формуле (26).
Методика определения. Отвешивают на аналитических весах 0,4 г
стали, содержащей 2—3% молибдена. Навеску растворяют в
фарфоровой чашке в хлористоводородной кислоте (пл. 1,17 г/см3), к которой
добавляют 1—2 мл концентрированной HN03. Смесь выпаривают
досуха на водяной бане, остаток растворяют в воде, раствор фильтруют
в мерную колбу емкостью 200 мл. Осадок йромывают водой, собирая
промывные воды в мерную колбу, доводят объем до метки водой и
перемешивают. Из колбы отбирают пипеткой в стакан 20 мл раствора,
прибавляют 5 мл 20%-ного раствора NaOH и нагревают до кипения.
Получерный осадок отфильтровывают и промывают водой, собирая
фильтрат и промывные воды в другой стакан. Раствор фильтруют
вновь в мерную колбу емкостью 200 мл. Фильтр промывают водой,
собирая промывные воды в мерную колбу, доводят водой до метки и
перемешивают. Измеряют оптическую плотность полученного раствора
при X = 230 нм 3 раза и берут среднее арифметическое полученных
результатов.
Расчет. Концентрацию полученного раствора (моль/л) вычисляют
по формуле (24). Количество молибдена рассчитывают по формуле (25).
Зная содержание молибдена в исследуемой навеске, рассчитывают
процентное его содержание (х) в стали по формуле:
где а — навеска стали.
Определение титана в ильменитовых концентратах
Титан (IV) образует в сернокислой среде с перекисью водорода
комплексное соединение [ТЮ(Н202)2]++. Оптическую плотность
раствора титанового комплекса измеряют при X = 390 нм на
спектрофотометре СФ-4. Определение концентрации титана проводят
дифференциальным методом.
Приготовление исходного раствора. На аналитических весах
отвешивают 1 г Ti02, навеску помещают в коническую колбу, прибавляют
25 г х. ч. (NH4hS04 и 55 мл H2S04 (пл. 1,12 г/см3) и нагревают до
полного растворения. Когда раствор охладится, его переливают без
потерь в мерную колбу емкостью 500 мл, доводят водой до метки и
тщательно перемешивают. 1 г Ti02 содержит 0,5994 г Ti, и 1 мл
полученного раствора содержит 0,00119 г, или 1,2 мг Ti в 1 мл.
Приготовление раствора сравнения. Отбирают пипеткой 20 мл
основного раствора, приливают в мерную колбу емкостью 200 мл,
прибавляют 40 мл Н3Р04 (пл. 1,7 г/см3), 100 мл 2%-ного раствора H2S04 и
§ 34. ОПРЕДЕЛЕНИЕ В УЛЬТРАФИОЛЕТОВОЙ ОБЛАСТИ СПЕКТРА 261
10 мл 6%-ного раствора Н202, доводят объем до метки тем же
раствором H2SO4, и перемешивают. Концентрация полученного раствора:
1,2-20
200
-0,12мг Т\/мл
Приготовление эталонных растворов. В три мерные колбы емкостью
по 200 мл приливают определенное количество исходного раствора.
В каждую колбу прибавляют те же реактивы, что и при приготовлении
раствора сравнения, доводят до метки 20%-ным раствором H2SO4 и
тщательно перемешивают. Состав эталонных растворов приведен ниже:
Эталонный раствор, №....
Объем исходного раствора, мл
Содержание титана
мг/200 мл
мг/мл
1
24
28,8
0,144
2
28
33,6
0,168
3
30
36,0
0,180
Методика определения. На аналитических весах отвешивают около
600 мг исследуемого ильменитового концентрата, помещают навеску
в платиновый тигель, прибавляют несколько капель
концентрированной H2SO4, 10 г пиросульфата х. ч. и сплавляют
при 800—900° С до получения прозрачного плава.
Затем при нагревании выщелачивают плав 100 мл
20%-ного раствора H2S04, содержащего 2 мл
6%-ного раствора Н202. Раствор охлаждают,
переливают в мерную колбу емкостью 200 мл,
доводят до метки 20%-ным раствором H2S04 и
перемешивают. Отбирают 30 мл полученного
раствора в мерную колбу емкостью 200 мл,
приливают все реактивы в тех же количествах, что и
при приготовлении раствора сравнения.
Определяют, как указано выше (см. § 27), значение
относительных оптических плотностей эталонных
растворов и раствора определяемой
концентрации на спектрофотометре СФ-4 с кварцевой
кюветой длиной 1 см. Строят калибровочный
график и по нему находят концентрацию (мг/мл)
титана в исследуемом растворе.
Расчет. Содержание титана (мг/мл) в растворе можно вычислить
по формуле (23). Содержание титана в процентах по взятой навеске
вычисляют по формуле, аналогичной формуле (39).
На указанном принципе основано также определение титана на фотоколориметре
ФЭК-М (см. книга 2, гл. IX, § 7) или фотометре ФМ-56; при работе с этими
приборами следует применять светофильтры с областью пропускания 400—500 нм.
Определение бензола й циклогексане
Спектр поглощения бензола (рис. 102) имеет максимум поглощения
при X = 254,2 нм. Циклогексан не поглощает лучей в этой области.
Оптическую плотность циклогексана с примесью бензола измеряют при
X = 254,2 нм и определяют содержание бензола по значению молярного
коэффициента поглощения.
Приготовление эталонного раствора. На аналитических весах в
мерной колбе емкостью 100 мл взвешивают 0,016 г бензола х. ч. Доводят
до метки циклогексаном х. ч. и перемешивают.
Определение молярного коэффициента поглощения бензола.
Определяют оптическую плотность полученного эталонного раствора при
Рис. 102. Спектр
поглощения бензола в
ультрафиолетовой области спектра.
262 ГЛ. VII. СПЕКТРАЛЬНЫЕ (ОПТИЧЕСКИЕ) МЕТОДЫ АНАЛИЗА
% = 254,2 нм на спектрофотометре СФ-4, применяя кварцевую кювету
длиной 1 см. Нулевым раствором служит циклогексан. Измерение
повторяют 3 раза и берут среднее арифметическое. Пользуясь
полученным значением оптической плотности, вычисляют молярный
коэффициент поглощения бензола по формуле (26).
Методика определения. Определяют оптическую плотность
исследуемой пробы циклогексана при X — 254,2 нм; концентрацию бензола
(моль/л) в исследуемой пробе находят по формуле (24). Оптическая
плотность исследуемой пробы циклогексана должна быть в пределах
от 0,2 до 1. Если концентрация бензола в циклогексане очень мала, то
увеличивают длину кюветы, в которой проводят определение.
Например, предварительные опыты показали, что исследуемая проба циклогексана
содержит примерно 0,0016 г бензола в 100 мл пробы, отсюда молярная концентрация
бензола равна 0,0002048 моль/л. Согласно табличным данным молярный коэффициент
поглощения бензола при X = 254,2 нм равен 240; тогда оптическая плотность пробы
циклогексана при толщине слоя 1 см равна D = 0,0002048 -240 = 0,0491. Измерение
таких значений оптической плотности связано с очень большой ошибкой; поэтому
измерение проводят при длине кюветы, равной 10 см:
D = 0,0002048 • 240 • 10 = 0,491
Это значение оптической плотности близко к оптимальному значению (0,44).
Наоборот, если исследуемая проба циклогексана содержит много
бензола, ее предварительно разбавляют химически чистым циклогек*
саном.
Например, предварительные опыты показали, что исследуемая проба циклогексана
содержит примерно 0,16 г бензола в 100 мл пробы. Тогда молярная концентрация
бензола равна 0,02048 моль/л. Оптическая плотность такого раствора при толщине слоя
1 см равна: D — 0,02048 • 240 — 4,91. В этом случае 10 мл исследуемой пробы
разбавляют химически чистым циклогексаном до 100 мл. Тогда концентрация бензола в
циклогексане будет 0,016 г/100 мл и молярная концентрация — 0,002048 моль/л.
Оптическая плотность разбавленной пробы циклогексана при толщине слоя 1 см
составит:
? = 0,002048 -240 = 0,491
Расчет. Содержание бензола в исследуемой пробе (в г)
определяют по формуле (25).
§ 35. Определение в видимой области спектра
Определение марганца и хрома в стали
при совместном присутствии
При растворении стали в кислотах и обработке полученного
раствора окислителем хром окисляется до бихромата, а марганец до пер-
манганата. Спектры поглощения водных растворов КМп04 и К2СГ2О7
даны на рис. 103. При X = 550 нм раствор К2Сг207 почти не поглощает
световых лучей. Оптическая плотность раствора D550 обусловлена
только КМп04. Определив оптическую плотность D550 смеси КМп04 и
К2СГ2О7 при X = 550 нм, можно определить содержание КМп04 в
растворе по формуле (24), зная молярный коэффициент поглощения
КМп04. При X = 430 нм и толщине слоя 1 см оптическая плотность
раствора Z)43o аддитивно складывается из оптической плотности,
обусловленной КМп04, и оптической плотности, обусловленной К2СГ2О7, и
согласно формуле (17):
Aj30 = еКМп04С'кМп04 + 8К2Сг207 ^КгСг207 (40)
где еКМп0 — молярный коэффициент поглощения КМп04 при X = 430 нм;
СКМп04—концентрация КМп04, моль/л-^
§ 35. ОПРЕДЕЛЕНИЕ В ВИДИМОЙ ОБЛАСТИ СПЕКТРА
263
ек Сг о — молярный коэффициент поглощения К2СГ2О7 при X =» 430 нм;
Ск с 0 — концентрация К2СГ2О7, моль /л.
Расчет. Зная Скмпо4, можно по формуле (40) определить Ск2сг2о7-
Определение молярного коэффициента поглощения перманганата
калия при X = 550 и X = 430 нм. Молярный коэффициент поглощения
КМп04 при X = 550 нм примерно равен 2420 (табличные данные).
Концентрация (моль/л) эталонного раствора, который служит для
определения молярного коэффициента поглощения КМп04,— должна
быть так подобрана, чтобы оптическая плотность раствора
приближалась к 0,5 (см. § 28). Если пользоваться кюветой длиной в 1 см, тогда:
0,5 = 2420СЭТ
Рэт~
0,5
2420
= 0,0002 моль/л
Рис. 103. Спектры поглощения
водных растворов:
1-К2Сг207; 2-КМп04.
Берут на аналитических весах 0,0316 г КМп04 х. ч., пересыпают
в мерную колбу емкостью 1000 мл, растворяют в воде, прибавляют
50 мл разбавленной (1:1) H2S04,
доводят до метки и перемешивают.
Молярная концентрация полученного
раствора 0,000207 моль/л. Определяют
оптическую плотность раствора при
X = 550 и X = 430 нм, применяя
спектрофотометр СФ-4 или СФ-5 и
стеклянную кювету длиной в 1 см. Каждое
определение повторяют 3 раза и
значение молярных коэффициентов
поглощения определяют по формуле (26).
Берут среднее арифметическое
полученных результатов.
Определение молярного
коэффициента поглощения бихромата калия.
Молярный коэффициент поглощения
К2СГ2О7 при X = 430 нм равен
примерно 300 (табличные данные). Чтобы оптическая плотность раствора
бихромата была 0,5 при длине кюветы 1 см, концентрация (моль/л)
эталонного раствора должна быть:
0 5
Сэт = -щ- = 0,0017 моль/л
На аналитических весах берут 0,48842 г К2СГ2О7 х. ч. в мерную
колбу емкостью 1000 мл, растворяют в воде, прибавляют 50 мл
разбавленной (1:1) H2SO4, доводят до метки и перемешивают. Молярная
концентрация полученного раствора 0,0017 моль/л. Определяют оптическую
плотность полученного раствора при X = 430 нм, применяя стеклянную
кювету длиной 1 см, и молярный коэффициент поглощения вычисляют
по формуле (26).
Методика определения. Навеску стали подбирают таким образом,
чтобы она содержала около 0,01 г марганца и около 0,01 г хрома;
например, если сталь содержит примерно 1% марганца и 1% хрома, то
берут 1 г стали.
Навеску растворяют в 30 мл смеси кислот * при осторожном
нагревании. После полного растворения раствор количественно переносят
* К 76 мл воды приливают тонкой струей при помешивании 16 мл H2SO4
(пл. 1,84 г/см3); после охлаждения приливают 8 мл Н3Р04 (пл. 1,7 г\смг) и 55мл HNO3
(пл. 1,40 г!см3).
264
ГЛ. VII. СПЕКТРАЛЬНЫЕ (ОПТИЧЕСКИЕ) МЕТОДЫ АНАЛИЗА
II мерную колбу емкостью 100 мл, доводят объем до метки водой и
перемешивают. Помещают 10 мл приготовленного раствора в мерную колбу
емкостью 100 мл, прибавляют 10 мл 0,05 М раствора нитрата серебра,
5 г твердого персульфата аммония; смесь нагревают до кипения и
кипятят 1 мин. Затем содержимое колбы охлаждают, доводят раствор до
метки и перемешивают.
Оптическую плотность полученного раствора D430 определяют при
X = 430 нм и оптическую плотность D550 при К = 550 нм. Молярную
концентрацию КМп04 (моль/л) определяют по формуле (24), молярную
концентрацию К2СГ2О7 (моль/л) —по формуле (40).
Расчет. Количество Мп и Сг (в г) во взятой навеске определяют
по формуле (25).
Содержание марганца (хрома) вычисляют в процентах по формуле,
аналогичной формуле (39).
§ 36. Определение в инфракрасной области спектра
1UOO 1200 W00
Определение циклогексанона в циклогексане
Спектры поглощения циклогексанона и циклогексана в
инфракрасной области даны на рис. 104.
Циклогексанон сильно поглощает инфракрасные лучи при v =
= 1718 см'\ а циклогексан в этой области почти не поглощает
инфракрасных лучей, т. е. оказывается
возможным определять циклогексанон в
циклогексане спектрофотометрическим методом
в инфракрасной области спектра.
Концентрацию циклогексанона находят по
калибровочному графику.
Приготовление эталонных растворов.
В мерных колбах емкостью 50 мл
помещают циклогексанон х. ч., доводят
циклогексаном до метки и перемешивают.
Оптическую плотность полученных
растворов определяют при v = 1718 см~1
на спектрометре ИКС-11 или ИКС-12.
В качестве нулевого раствора берут
циклогексан. Сначала в кюЪету наливают
циклогексан, настраивают спектрометр
на 1718 см~\ устанавливают кювету в
спектрометре и щель спектрометра
подбирают такой, чтобы отклонение гальва;
нометра или потенциометра составляло
примерно 40% полного поглощения. Затем в ту же кювету или в
другую той же толщины наливают эталонный раствор и определяют
оптическую плотность. Толщину слоя подбирают таким образом, чтобы
оптическая плотность полученных растворов была в пределах от 0,2 до
1,0. Каждое измерение повторяют 3 раза и берут среднее
арифметическое полученных результатов. Затем строят калибровочный график.
Состав эталонных растворов приведен ниже:
О
2000 J800
1600 1400 1200 1000
1>, СМ~1
Рис. 104. Спектры поглощения:
а — циклогексанона, б — циклогексана.
Эталонный раствор, К? 1
Содержание циклогексанона, г . . . . 0,0625
Концентрация циклогексанона в
циклогексане в 50 'мл, % 0,125
0,125
3
0,250
4
0,500
5
1,000
0,250 0,500 1,000 2,000
§ 38. КРИВЫЕ СПЕКТРОФОТОМЕТРИЧЕСКОГО ТИТРОВАНИЯ 265
Определение концентрации циклогексанона в циклогексане.
Исследуемая проба циклогексана должна содержать не больше 3%
циклогексанона. Измеряют оптическую плотность пробы и по калибровочному
графику находят содержание циклогексанона.
СПЕКТРОФОТОМЕТРИЧЕСКИЕ (ФОТОМЕТРИЧЕСКИЕ)
МЕТОДЫ ТИТРОВАНИЯ
Л. ТЕОРЕТИЧЕСКИЕ ОСНОВЫ МЕТОДА
§ 37. Общая характеристика метода
Оптические методы могут служить не только для непосредственного
определения концентрации вещества, но и для определения точки
эквивалентности в процессе титрования при условии, что существует
линейная зависимость между величинами светопоглощения и концентрацией
вещества в исследуемом растворе (см. § 21).
Аликвотную часть анализируемого раствора помещают в кювету,
через которую проходит монохроматический поток света, попадающий
затем на фотоэлемент, и приступают к титрованию. В процессе
титрования отмечают значения оптической плотности.
На основании результатов титрования строят кривую спектрофото-
метрического титрования, откладывая на оси ординат значения
оптической плотности Д а на оси абсцисс — объем раствора V (в мл).
Для нахождения точки эквивалентности прибегают к методу
экстраполяции, используя такие участки кривой титрования,, в которых в
избытке находится либо определяемое вещество, либо титрант. С этой
целью проводят измерения в нескольких точках, достаточно удаленных
от точки эквивалентности, где реакция проходит еще количественно, и,
проведя через них прямые, графически находят точку эквивалентности.
Если реакция проходит количественно, кривая титрования имеет вид
двух прямых, пересекающихся в точке эквивалентности.
Метод спектрофотометрического титрования имеет ряд преимуществ
перед визуальными методами. Он более избирателен и позволяет
проводить последовательное определение нескольких компонентов из одной
пробы, дает возможность анализировать окрашенные растворы и
растворы с низкой концентрацией определяемого вещества (абсолютные
количества веществ, определяемых этим методом, лежат в пределах
1-Ю""1 — 1 • Ю-8 г), применим к реакциям, которые не заканчиваются
в точке эквивалентности (например, образование малоустойчивых
комплексов, нейтрализация слабых кислот и оснований и т. д.).
Спектрофотометрический метод титрования может быть
использован при кислотно-основном, окислительно-восстановительном, комплек-
сонометрическом и других методах титрования.
§ 38. Кривые спектрофотометрического титрования
Кривые спектрофотометрического титрования могут быть
различной формы. Характер их зависит от того, какие компоненты реакции
поглощают при выбранной длине волны.
В общем случае ход кривой титрования до и после точки
эквивалентности зависит от величины Де:
Ле = 8АВ ~ 8B ~ еА
где 8ав, ев, еА — молярные коэффициенты поглощения продукта реакции, титранта
и определяемого вещества соответственно.
266
ГЛ. VII. СПЕКТРАЛЬНЫЕ (ОПТИЧЕСКИЕ) МЕТОДЫ АНАЛИЗА
В качестве примера остановимся на некоторых, чаще всего
встречающихся случаях.
I. Поглощает анализируемое вещество (А), титрант (В) и продукт
реакции (АВ) при данной длине волны не поглощают. По мере
уменьшения концентрации определяемого вещества оптическая плотность
уменьшается и становится постоянной после точки эквивалентности
(рис. 105, кривая 1). Такого рода кривая наблюдается при титровании
бихромат-ионов солями железа (II) или мышьяка (III). При длине
волны % = 350 нм поглощают
только бихромат-ионы.
II. Поглощает продукт
реакции (АВ), анализируемое
вещество (А) и титрант (В) не
пбглощают. Оптическая
плотность по мере образования
продукта реакции
увеличивается. После ?очки
эквивалентности оптическая плотность
становится постоянной, т. е.
кривая имеет ход, обратный
ходу кривой /. Такого рода
кривая наблюдается при
титровании соединений железа (II)
соединениями кобальта (III).
При длине волны 360 нм
поглощает только Fem.
III. Анализируемое
вещество (А) и продукт реакции
(АВ) не поглощают, поглощает
титрант (В). Оптическая
плотность до точки
эквивалентности- остается постоянной и
увеличивается после нее по мере
накопления в растворе
избытка титранта (рис. 105, кривая 2).
Такого рода кривая
наблюдается при титровании
соединений мышьяка (III)
солями церия (IV).
При длине волны 320 нм поглощает только Ceiv.
IV. Поглощает продукт реакции (АВ) и титрант (В),
анализируемое вещество (А) не поглощает. Характер кривой титрования зависит
от того, что поглощает в большей степени: продукт реакции или титрант.
а) Продукт реакции поглощает больше, чем титрант 8ав > ев-
В процессе титрования оптическая плотность увеличивается по мере
накопления продукта реакции. После точки эквивалентности оптическая
плотность возрастает по мере накопления титранта (рис. 105, кривая 3).
б) Титрант поглощает больше, чем продукт реакции 8ав < ев-
В процессе титрования оптическая плотность увеличивается по мере
накопления окрашенного продукта реакции. После точки эквивалентности
более резкое возрастание светопоглощения по мере накопления титранта
(рис. 105, кривая 4).
V. Поглощает анализируемое вещество (А) и титрант (В),
продукт реакции (АВ) не поглощает. В процессе титрования по мере
уменьшения анализируемого вещества оптическая плотность уменьшается.
Рис, 105.
Объем титранта
Кривые спектрофотометрического
титрования.
§ 38. КРИВЫЕ СПЕКТРОФОТОМЕТРИЧЕСКОГО ТИТРОВАНИЯ 267
После точки эквивалентности наблюдается увеличение светопоглощения
по мере накопления избытка титранта (рис. 105, кривая 5).
VI. Поглощают все три компонента: анализируемый продукт (А),
титрант (В) и продукт реакции (АВ). Светопоглощение раствора после
достижения точки эквивалентности определяется избытком титранта.
При раздельном титровании смеси кривая титрования будет иметь
несколько изломов, число которых соответствует числу компонентов
смеси.
Например, кривая спектрофотометрического титрозания соединений висмута и
свинца (при их совместном присутствии) этилендиаминтетрауксусной кислотой (ЭДТА)
при рН = % и X = 240 нм имеет два излома, соответствующих двум точкам
эквивалентности для Bi+++ и РЬ++. В первую очередь титруется Bi++f, так как комплекс
висмута Bi — ЭДТА более устойчив, чем комплекс РЬ — ЭДТА. При этом оптическая
плотность раствора изменяется незначительно вследствие слабого поглощения
комплекса Bi — ЭДТА при этой длине волны (для Bi — ЭДТА Хмакс = 265 нм). После
того как практически весь Bi+++ оттитрован (первая точка эквивалентности), начинает
титроваться РЬ++ с образованием менее стойкого комплекса РЬ — ЭДТА. При этом
оптическая плотность изменяется более резко, так как РЬ — ЭДТА при X = 240 нм
поглощает значительно сильнее, чем Bi — ЭДТА. После второй точки эквивалентности
(для РЬ+*) оптическая плотность практически не изменяется (рис. 105, кривая 6).
Спектрофотометрическое титрование может быть применено и тогда,
когда титрант, определяемое вещество и продукт реакции не поглощают.
В этом случае используют индикаторы.
В качестве индикаторов (Ind) могут быть применены вещества,
которые сами при данной длине волны не поглощают, а образуют
соединения с определяемым веществом (AInd), титрантом (Bind) или
продуктом реакции (ABInd), поглощающие свет.
Так, при комплексонометрическом титровании Fe+++ используют
салициловую кислоту (Ind), образующую интенсивно окрашенное
соединение с Fe+++ (AInd), максимум поглощения которого % = 525 нм.
При титровании этого раствора этилендиаминтетрауксусной
кислотой (ЭДТА) происходит уменьшение поглощения, так как при рН~2,4
комплекс Fe+++ с салициловой кислотой менее устойчив по сравнению с
комплексом Fe+++ с ЭДТА, т. е. кривая титрования аналогична кривой /
(см. рис. 105). Аналогичный вид имеет кривая при титровании раствора
висмута (III) в присутствии избытка тиомочевины (Ind)
этилендиаминтетрауксусной кислотой. Тиомочевина образует с висмутом окрашенное
в желтый цвет комплексное соединение менее стойкое, чем комплекс
висмута с ЭДТА (BiY~). В качестве примера, когда индикатор образует
с титрантом окрашенное соединение, можно привести случай титрования
соли церия (IV) раствором комплексного соединения — офенантролина
с железом (II). Кривая аналогична кривой 2 (см. рис. 105).
В качестве индикаторов используются также вещества, которые в
точке эквивалентности меняют свою структуру (вследствие изменения
рН, окислительно-восстановительного потенциала системы или
концентрации ионов), что сопровождается резким изменением
светопоглощения раствора. Например, при титровании по методу нейтрализации с
кислотно-основным индикатором в точке эквивалентности содержание
определенной формы индикатора, поглощающей при выбранной длине
волны, резко возрастает. При дальнейшем прибавлении титранта
светопоглощение не изменяется (см. рис. 105, кривая 6).
Разбавление анализируемого раствора в процессе титрования
изменяет формы кривых титрования. Для уменьшения влияния разбавления
на светопоглощение применяют концентрированные растворы титранта.
При титровании разбавленными растворами надо умножить результат
каждого измерения на величину {V+V\)/V (где У —объем
анализируемого раствора, мл\ V\ — объем прибавленного титранта, мл).
268 ГЛ. VII. СПЕКТРАЛЬНЫЕ (ОПТИЧЕСКИЕ) МЕТОДЫ АНАЛИЗА
Б. АППАРАТУРА И ТЕХНИКА ВЫПОЛНЕНИЯ ТИТРОВАНИЯ
§ 39. Установки для спектрофотометрического титрования
Спектрофотометрическое титрование проводят в приборах СФ-5,
СФ-4, СФ-4А, СФД-2, в крышке кюветного отделения которых делают
два отверстия: для микробюретки и механической мешалки. Кювета
должна иметь объем 25 мл. При работе в видимой области спектра
применяют кювету из оптического стекла, в ультрафиолетовой области
спектра — кварцевую или снабженную кварцевыми окошками. Стенки
кюветы покрывают черным лаком, оставляя отверстие диаметром 1 см
на пути светового потока. Мешалка и бюретка вводятся таким образом,
чтобы их концы находились против затемненных участков кюветы.
Для фотометрических объемно-аналитических определений
применяется фотоэлектрический титрометр ФЭТ-УНИИЗ. В отличие от
спектрофотометрического титрования в фотометрическом титровании
применяется полихроматический свет (источник — лампа накаливания). В
качестве приемника световой энергии используется фотосопротивление
ФС-К1. Регистрацию фототока осуществляют микроамперметром. Для
проведения титрования стакан емкостью 150—200 мл с анализируемым
раствором помещают в гнездо титрометра, выводят стрелку
микроамперметра на правый край шкалы (90—100 делений), включают мотор
мешалки и приступают к титрованию, отмечая показания
микроамперметра после прибавления каждой порции раствора. По полученным
данным строят кривую титрования в координатах: ось ординат — показания
гальванометра, ось абсцисс — объем стандартного раствора.
Кривые при фотометрическом титровании аналогичны кривым при
спектрофотометрическом титровании.
В. ПРАКТИЧЕСКИЕ РАБОТЫ
§ 40. Спектрофотометрическое титрование
Определение арсенитов
Спектрофотометрическое определение арсенитов в
ультрафиолетовой области основано на том, что при взаимодействии слабощелочного
раствора арсенита с раствором иода происходит восстановление иода
до иодида:
As03"' + H20 + I2 ^z± As04~~ + 2r + 2H+
До точки эквивалентности в области 365 нм раствор оптически
прозрачен. После точки эквивалентности присутствие избытка Ь
вызывает увеличение оптической плотности раствора.
Методика определения. В кювету с кварцевыми окошками емкостью
25 мл помещают 5 мл ~ 0,02 н. раствора соли мышьяковистой кислоты,
5 мл 5%-ного раствора бикарбоната, около 20 капель 2 н. раствора
уксусной кислоты (рН~8) и доводят водой до объема —18 мл.
Кювету помещают в спектрофотометр и титруют 0,1 н. раствором
иода. Стандартный раствор иода прибавляют из микробюретки по
0,1 мл, перемешивают и измеряют оптическую плотность при К = 365 нм.
До точки эквивалентности, пока окисляется арсенит, не наблюдается
§ 40. СПЕКТРОФОТОМЕТРИЧЕСКОЕ ТИТРОВАНИЕ
269
изменения оптической .плотности. После точки эквивалентности
оптическая плотность раствора увеличивается. По полученным результатам
строят кривую титрования, точку эквивалентности находят графически.
Ошибка определения не превышает 0,1—0,3 отн. %. В качестве
стандартных растворов можно применять также растворы солей церия (IV).
Расчет. Зная количество добавленного из микробюретки
стандартного раствора Ь, можно вычислить содержание (g) арсенита (в г) в
анализируемом растворе:
3Na2HAs03^l2^l2 мп
g 1000 К Ч
Анализ свинцово-оловянных сплавов
Анализ свинцово-оловянных сплавов основан на растворении
сплава в HN03 и Последующем спектрофотометрическом титровании
избытка оксалата, прибавляемого для осаждения РЬ++-ионов, солями
церия (IV) в области 365 нм (Сеш и С2О4" в этой области не
поглощают).
Олово (II) определяют спектрофотометрическим титрованием
раствором сульфата церия (IV) — хлориды олова (II) и олова (IV)
прозрачны для ультрафиолетовых лучей. Поглощение, как и в
предыдущем случае, определяется только раствором сульфата церия (IV).
Методика определения свинца. Навеску сплава 0,01 или 0,1 г
(микро- или макрохимическая методика выполнения) растворяют в
1,5—15 мл разбавленной (1: 1) азотной кислоты. Раствор разбавляют
водой до 10—100 мл соответственно и нейтрализуют раствором аммиака
по метиловому оранжевому. Для уничтожения образовавшейся мути
прибавляют по каплям 78%-ную уксусную кислоту. Затем раствор
упаривают до 1 —1,5 мл (микро) или 10—15 мл (макро) и, осадив ионы
свинца известным количеством щавелевой кислоты, в фильтрате отти-
тровывают сульфатом церия (IV) ее избыток при К = 365 нм.
Методика определения олова. Навеску сплава в 0,01 или 0,1 г
растворяют в 3 или 30 мл концентрированной хлористоводородной кислоты.
Цериметрическое титрование ведут либо в токе двуокиси углерода,
либо в процессе титрования добавляют небольшие кусочки мрамора.
Ошибка определения 0,1%.
По результатам титрования строят кривые и графически
определяют точку эквивалентности.
Расчет. Содержание свинца и олова вычисляют в процентах по
следующим формулам:
= /WHAO, _ WcelL] Эрь J00
РЬ V эн2С2о4 3CeIV J at
TCe'VFCelV Q 100 ....
*Sn 3CeIV 3S" Ж <43)
Определение хрома в сталях
Определение содержания хрома в сталях основано на
спектрофотометрическом титровании при X = 350 нм солями титана (III) или
железа (II) бихромата, образующегося при окислении хрома:
Cr207~ + 6Ti+++-H4H+ —> 2Cr+++ + 6TiIV + 7H20
Cr2Oj~ + 6Fe++ + 14H+ —> 2Cr+++ + 6Fe+++ + 7H20
270 ГЛ. VII. СПЕКТРАЛЬНЫЕ (ОПТИЧЕСКИЕ) МЕТОДЫ АНАЛИЗА
Ионы железа (II) и (III), а также Сгш в этой области не
поглощают.
Методика определения. Навеску стали 0,5— I г растворяют в 10 мл
50%-наго раствора серной кислоты при нагревании и упаривают досуха
до появления паров серной кислоты. К остывшему остатку прибавляют
несколько капель азотной кислоты и выпаривают досуха. Осадок
количественно выщелачивают горячей водой, подкисленной серной кислотой,
и отфильтровывают. К фильтрату прибавляют раствор аммиака (до
слабого запаха). Осадок гидроокисей отфильтровывают и промывают
горячей водой, а затем его растворяют в серной кислоте, прибавляемой
порциями по 5 мл. В полученный раствор вносят 0,5—0,7 г сухого тар-
трата калия и добавляют избыток 15%-ного раствора NaOH и 20 мл
3%-ного раствора перекиси водорода и нагревают. Отфильтровывают
осадок гидроокиси железа (III) и удаляют кипячением избыток
перекиси. В полученный фильтрат по каплям прибавляют раствор хлорида
бария до полного обесцвечивания раствора над осадком и оставляют на
ночь. Затем осадок отфильтровывают, промывают горячей водой и
обрабатывают 50 мл 5 н. раствора серной кислоты. Фильтрат и
промывные воды переносят в мерную колбу емкостью 100 мл.
В кювету отбирают аликвотную часть и титруют при % = 350 нм
раствором соли титана (III) или железа (II). Стандартный раствор
прибавляют из микробюретки по 0,1 мл, перемешивают и измеряют
оптическую плотность раствора. По мере прибавления титранта
оптическая плотность уменьшается. После точки эквивалентности
оптическая плотность становится минимальной и не изменяется. По
полученным результатам строят кривую титрования, точку эквивалентности
находят графически.
Расчет. Сначала вычисляют количество бихромата, а затем
определяют процентное содержание хрома в процентах в стали по формуле,
аналогичной формуле (41).
НЕФЕЛОМЕТРИЧЕСКИЙ И ТУРБИДИМЕТРИЧЕСКИЙ
МЕТОДЫ АНАЛИЗА
А. ТЕОРЕТИЧЕСКИЕ ОСНОВЫ МЕТОДОВ
§ 41. Общая характеристика методов
При прохождении пучка света через дисперсные системы
наблюдается рассеяние или поглощение света твердыми частицами. Это
явление положено в основу нефелометрии и турбидиметрии.
Интенсивность светового потока, рассеиваемого небольшими
твердыми частицами взвеси, описывается уравнением Рэлея:
/=/°[FxS(1+cos2p)] (44)
где / и /0 — интенсивности рассеянного и падающего света соответственно;
F — функция, зависящая от показателя преломления частиц в растворе;
N — общее число частиц во взвеси;
V — объем частицы;
Я — длина волны падающего света;
г — расстояние до наблюдателя;
Р — угол между направлениями падающего и рассеянного света.
При нефелометрических определениях все измерения проводят при
определенных значениях F, V, г, р. Поэтому, объединяя их в одну
константу, можно записать:
I = IQKN = IQKC (45)
§ 42. ПРИБОРЫ, ПРИМЕНЯЕМЫЕ В НЕФЕЛОМЕТРИИ И ТУРБИДИМЕТРИИ 271
Отсюда интенсивность рассеянного светового потока прямо
пропорциональна числу частиц во взвесях, т. е. концентрации частиц,
находящихся в растворе. Из приведенной выше формулы следует, что
интенсивности рассеянного света в двух растворах с частицами одинаковой
формы и размеров относятся между собой, как концентрации частиц
определяемого вещества: т г
f—p- (46)
Это уравнение лежит в основе нефелометрических определений.
При нефелометрических определениях измеряют интенсивность
рассеянного света / в направлении, перпендикулярном к направлению
первичного пучка света.
При турбидиметрическом методе анализа интенсивность светового
потока уменьшается вследствие поглощения и рассеяния светового
потока и определяется уравнением:
*t-*-*tsj (47)
где / и 10 — интенсивность светового потока, прошедшего через раствор и падающего
соответственно;
С — концентрация поглощающих частиц в растворе;
/ — толщина поглощающего слоя раствора;
d — средний диаметр поглощающих частиц;
/С' и а — константы, зависящие от метода измерения и Природы суспензии;
Я — длина волны.
При аналитических турбидиметрических определениях все
измерения проводятся при определенных значениях К\ d, а и Я. Объединяя их
в одну постоянную величину, получаем:
Ig^^KCl или D^KIC (48)
Это уравнение имеет вид, аналогичный уравнению Бугера —
Ламберта— Бера, но здесь К — молярный коэффициент мутности раствора.
При турбидиметрических определениях измеряют интенсивность
света /, выходящего из кюветы в направлении падающего пучка света.
Приведенные уравнения справедливы только для очень
разбавленных суспензий (не более 100 мг на 1 л). Турбидиметрические и нефело-
метрические методы обладают высокой чувствительностью. Однако
применяются они не широко, что объясняется трудностью получения
взвесей с одинаковыми размерами частиц. Количественные нефеломет-
рические и турбидиметрические определения проводят, пользуясь
калибровочной кривой.
Для точных исследований используют метод уравнивания.
Довольно широко применяется метод фототурбидиметрического и фотонефело-
метрического титрования.
Б. АППАРАТУРА И ТЕХНИКА ВЫПОЛНЕНИЯ АНАЛИЗА
§ 42. Приборы, применяемые для нефелометрических
и турбидиметрических измерений
Турбидиметрические и нефелометрические определения проводятся
в фотоэлектрических колориметрах-нефелометрах ФЭКН-57,
нефелометре НФМ, действие которого основано на принципе уравнивания двух
световых потоков: одного от рассеивающей взвеси, другого от матового
или молочного стеклянного рассеивателя прибора. Уравнивание потоков
производится с помощью переменной диафрагмы.
272 ГЛ. VII. СПЕКТРАЛЬНЫЕ (ОПТИЧЕСКИЕ) МЕТОДЫ АНАЛИЗА
Оптическая схема нефелометра НФМ изображена на рис. 106. Свет
от лампы накаливания 1 проходит через стеклянную пластинку 2,
конденсор 3 и попадает в кювету 4, помещенную в камеру с
дистиллированной водой. Камеру с водой применяют для того, чтобы уменьшить
рассеивание света стенками кюветы. Световой поток, прошедший через
кювету, гасится в светоловушке 5, а части светового потока,
рассеянного частицами взвеси в кювете 4 и стеклянным рассеивателем 17,
собираются насадочными линзами 6 и 16. Образовавшиеся два пучка
проходят через диафрагмы 7 и 15, связанные с отсчетными барабанами и
объективами 8 и 14, направляются в ромбические призмы 9 и 13.
Бипризма 10 дает возможность наблюдать в поле зрения окуляра 12 интен-
5
Рис. 106. Оптическая схема нефелометра НФМ:
/ — лампа накаливания; 2 —стеклянная пластинка; 3 — конденсор; 4 — кювета; 5 —светоловушка;
6* 16 — насадочные линзы; 7, 15 — диафрагмы; 8, 14 — объективы; 9> 13 — ромбические призмы; /0
—бипризма; Л — светофильтры; 12 — окуляр; 17 — рассеиватель.
сивность двух пучков света. При нефелометрических определениях на
пути пучков света вводят светофильтры 11, применение которых
нивелирует разницу в оттенках двух световых потоков.
В. ПРАКТИЧЕСКИЕ РАБОТЫ
§ 43. Нефелометрические и турбидиметрические
определения
Нефелометрическое определение хлорид-ионов
Определение хлорид-ионов основано на реакции осаждения
хлоридов нитратом серебра:
Ag+ + CF —> AgCl
При малых концентрациях хлорид-ионов выпадение осадка не
происходит, а возникает помутнение раствора. Степень помутнения зависит
от концентрации хлоридов в растворе. Для стабилизации растворов
вводят стабилизирующие компоненты.
Методика определения. Навеску КО растворяют в мерной колбе
емкостью 100 мл (содержание С1~-ионов должно приблизительно
соответствовать 0,1 мг хлора/мл) и тщательно перемешивают. Из
анализируемого раствора отбирают микропипеткой 5 мл раствора и помещают
в мерную колбу емкостью 50 мл. В нее же прибавляют 10 мл 0,1 н.
раствора азотной кислоты, 2 мл 0,5%-ного раствора желатины,
дистиллированной воды до общего объема приблизительно 30 мл, 10 мл 0,005 М
§ 44. ФОТОТУРБИДИМЕТРИЧЕСКОЕ И ФОТОНЕФЕЛОМЕТРИЧЕСКОЕ ТИТРОВАНИЕ 273
раствора AgN03 и доливают водой до метки. Содержимое колбы
тщательно перемешивают. Через 5 мин раствор переносят в кювету
нефелометра и измеряют рассеивание света не менее 3 раз. Из полученных
отсчетов вычисляют среднее значение и по калибровочной кривой
определяют содержание хлорид-ионов.
Калибровочный график строят следующим образом. Из эталонного
раствора КО (вводят 0,1 г КО в мерную колбу емкостью 500 мл и
доводят водой до метки) отбирают в четыре мерные колбы емкостью по
50 мл микропипеткой соответственно 2,0; 4,0; 6,0; 8,0 мл и
приготавливают стандартные растворы, добавляя в них все реактивы, указанные
выше.
Начинают измерения с пробы, имеющей наибольшую концентрацию.
Раствор помещают в кювету. Устанавливают светофильтр, цвет которого
близок к окраске исследуемого раствора в рассеянном свете. Если
жидкость бесцветна, устанавливают зеленый светофильтр. Оба отсчетные
барабаны ставят на «0» и подбирают такой рассеиватель, при котором
в окуляре левое фотометрическое поле будет несколько светлее правого.
Вращением правого барабана уравнивают фотометрические поля по
яркости и отсчитывают «кажущуюся» оптическую плотность.
Т у рбидиметрическоё" определение сульфат-ионов
Определение сульфат-ионов основано на реакции осаждения их
хлоридом бария:
Na2S04 + ВаС12 —> BaS04 + 2NaCl
В определенных пределах концентрации сульфатов в присутствии
стабилизирующих веществ сульфат бария образует устойчивую белую муть.
Калибровочный график. Для приготовления эталонного раствора
серной кислоты в мерную колбу емкостью 500 мл из бюретки наливают
12,5 мл 0,05 н. раствора серной кислоты и водой доводят до метки. Этот
раствор используют для приготовления растворов при построении
калибровочного графика.
В мерную колбу емкостью 100 мл помещают 2 мл раствора ВаСЬ,
разбавляют немного водой и прибавляют 3 мл 0,5%-ного раствора
желатина. При помешивании вводят из микробюретки определенное
количество эталонного раствора H2S04 (от 0,135 мг до 1,0 мг БОз в 100 мл).
Через 3—5 мин заполняют кювету с толщиной слоя 20 мм и определяют
оптическую плотность на ФЭКН-57; кювету сравнения заполняют
дистиллированной водой. По полученным данным строят калибровочный
график. По оси абсцисс откладывают концентрацию сульфат-ионов вмг
на 100 мл раствора, а по оси ординат — оптическую плотность.
Методика определения. Готовят раствор анализируемого вещества.
Определяют его оптическую плотность и находят концентрацию
определяемого вещества по калибровочному графику.
§ 44. Фототурбидиметрическое и фотонефелометрическое
титрование
Наряду со спектрофотометрическим методом титрования применяют
также метод фототурбидиметрического и фотонефелометрического
титрования (см. книга 2, гл. IX, § 5).
Эти методы применяются в том случае, когда титруемое вещество
образует малораствооимое соединение с титрантом. Прибавление
274 ГЛ. VIL СПЕКТРАЛЬНЫЕ (ОПТИЧЕСКИЕ) МЕТОДЫ АНАЛИЗА
каждой порции титранта вызывает появление осадка, вследствие чего
мутность раствора увеличивается и в точке эквивалентности достигает
максимума. Дальнейшее прибавление титранта не изменяет степени
мутности, а иногда даже приводит к ее уменьшению вследствие
разбавления и слипания мелких частиц.
Максимальная мутность и максимальное поглощение световых
лучей соответствуют точке эквивалентности. Кривые турбидиметрического
и нефелометрического титрования аналогичны кривым спектрофотомет-
рического титрования.
ГЛАВА VIII
ХРОМАТОГРАФИЧЕСКИЕ МЕТОДЫ АНАЛИЗА
Л. ТЕОРЕТИЧЕСКИЕ ОСНОВЫ
§ 1. Классификация хроматографических методов анализа
Хроматографический метод, предложенный русским ученым
М. С. Цветом в 1903 г., основан на использовании сорбционных
процессов в динамических условиях. В простейшем виде эти условия создаются
при прохождении потока смеси газов, паров, жидкостей или раствора
через колонку, содержащую слой зерненого сорбента.
Простота, эффективность и универсальность хроматографического
метода дали возможность широко использовать его в различных
областях науки, промышленности и техники.
С помощью хроматографического метода возможно:
разделение сложных смесей органических и неорганических
веществ на отдельные компоненты; разделение и выделение растительных
и животных пигментов, изотопов, редкоземельных элементов и других
веществ;
очистка вещества от примесей;
концентрирование веществ из сильно разбавленных растворов;
определение молекулярной структуры некоторых соединений путем
установления связи между сорбируемостью и строением данного
вещества;
качественный и количественный анализ исследуемого вещества.
Способы выполнения хроматографического анализа. Различают
следующие способы выполнения хроматографического метода анализа:
фронтальный, вытеснительный, элюентный.
При фронтальном анализе исследуемый раствор смеси веществ
непрерывно подают в верхние части колонки и собирают отдельные
фракции фильтрата. При анализе системы, содержащей компоненты А и В,
первым из колонки вытекает чистый растворитель; затем после
насыщения сорбента менее сорбирующимся веществом, например В, из
колонки вытекает раствор, содержащий компонент В; когда же сорбент
насыщается компонентом А, в приемник поступают одновременно два
компонента А и В. Указанным способом может быть получено в чистом
виде только наименее сорбируемое вещество. Полного разделения
исследуемой смеси на составные компоненты не достигается.
При вытеснит ельном анализе в колонку вводят порцию раствора
смеси, содержащей компоненты А и В, или многокомпонентную смесь
и с помощью более сорбирующегося вещества D вытесняют ранее
сорбированные компоненты А и В. Введенное вещество D вытесняет
компонент А, который вытесняет менее сорбируемый компонент Ё. Происходит
перемещение веществ А и В вдоль слоя сорбента со скоростью, равной
скорости движения вытеснителя D. Из колонки последовательно
выходят компоненты В и А в соответствии с их избирательной
сорбируемостью на сорбенте. Между зоной первого и последующего компонента
276
ГЛ. VIII. ХРОМАТОГРАФИЧЕСКИЕ МЕТОДЫ АНАЛИЗА
образуется промежуточная зона, содержащая смесь этих компонентов.
Полнота разделения веществ зависит от условий проведения анализа.
При элюентном анализе в колонку вводят порцию исследуемой
смеси компонентов, например А, В, С. Компоненты смеси располагаются
вдоль колонки сверху вниз в перекрывающихся зонах в соответствии
с их сорбируемостью, например А > В > С. Нижняя зона хромато-
граммы в колонке содержит чистое вещество С. При промывании
сорбента растворителем вдоль колонки происходит передвижение
компонентов смеси вследствие взаимного вытеснения в соответствии с их
сорбируемостью. В фильтрате собирают компоненты в порядке повышения
их сорбируемости, т. е. вначале компонент С, затем В и А.
Главная особенность хроматографического метода состоит в том,
что при движении хроматографируемой смеси через сорбент происходит
многократное повторение процессов, обусловливающих разделение
компонентов смеси.
Методы хроматографического анализа. Хроматографические
методы различаются по следующим признакам:
по агрегатному состоянию системы, в которой проводится
разделение смеси на компоненты, — газовая, жидкостная и газо-жидкостная
хроматография;
по механизму разделения — адсорбционная (жидкостная, газовая),
распределительная, ионообменная, осадочная,
окислительно-восстановительная, адсорбционно-комплексообразовательная хроматография;
по форме проведения процесса — колоночная, капиллярная,
плоскостная (на бумаге и тонкослойная).
В ряде случаев разделение оказывается разультатом нескольких
одновременно протекающих процессов с различными механизмами. Это
приводит к образованию хроматограммы смешанного типа, однако один
из процессов всегда является доминирующим.
§ 2. Адсорбционная хроматография
К адсорбционной хроматографии относятся адсорбционно-жидкост-
ная и газовая. Основные теоретические положения этих видов
хроматографии имеют много общего, и поэтому рассматриваются в одном
разделе.
Закон адсорбционного замещения. Этот закон был открыт
М. С. Цветом и сформулирован* следующим образом:
«Вещества, растворенные в определенной жидкости, образуют
определенный адсорбционный ряд* А > В > С, выражающий относительное
адсорбционное сродство его членов к адсорбенту. Каждый из членов
адсорбционного ряда, обладая большим адсорбционным сродством, чем
последующий, вытесняет его из соединения и в свою очередь
вытесняется предыдущим».
В соответствии с теорией Ленгмюра на поверхности сорбента
находится силовое поле, которое способно притягивать молекулы
посторонних веществ, причем образуется мономолекулярный слой
адсорбированных молекул. Между поверхностью адсорбента и средой
устанавливается подвижное адсорбционное равновесие, определяемое равенством
скоростей адсорбции и десорбции молекул.
Образующееся в колонке адсорбента зональное распределение
веществ выражает относительное их положение в адсорбционном ряду.
* Цвет М. С, Хроматографический адсорбционный анализ, Изд. АН СССР, 1946.
§ 2. АДСОРБЦИОННАЯ ХРОМАТОГРАФИЯ
277
Изотерма адсорбции. Каждой концентрации адсорбируемого
вещества отвечает определенное адсорбционное равновесие при
соответствующей температуре. Зависимость количества адсорбированного
вещества от его концентрации в растворе при постоянной температуре
выражается изотермой адсорбции.
На практике встречаются три типа изотерм адсорбции: выпуклая,
вогнутая и линейная (рис. 107). Первоначальное распределение хрома-
тографируемых веществ в колонке отражают кривые 1, затем при
промывании колонки растворителем в зависимости от вида изотермы
наблюдаются три варианта распределения сорбируемого вещества: а) при
выпуклой изотерме адсорбции нижний край зоны становится более
резким, а верхний размывается, это обусловливается различной
скоростью движения хроматографируемого вещества в зависимости от его
Рис. ф7. Распределение вещества в колонке в зависимости от вида изотермы
адсорбции при промывании колонки чистым растворителем:
а, б, в-изотермы адсорбции; /, 2, 3-кривые распределения вещества в колонке;
Л1 —количество введенного в колонку хроматографируемого вещества; /-длина колонки;
С-концентрация вещества в растворе, г\ S — количество адсорбированного вещества, г.
концентрации. Такое размывание хроматограмм называют
образованием «хвостов» хроматограммы (см. рис. 107, а, кривые 2,3); б) при
вогнутой изотерме адсорбции, наоборот, верхний край зоны более
резкий, а нижний — растянутый (рис. 107,6, кривые 2, 3); в) при линейной
изотерме адсорбции концентрация веществ на колонке распределяется
симметрично вдоль зоны (рис. 107, в, кривые 2, 3).
Таким образом, вид изотермы адсорбции дает представление о
характере распределения вещества в колонке и основание для выбора
условий хроматографического разделения сложных смесей.
Изменение величины адсорбции газообразного вещества с
увеличением его концентрации выражается уравнением изотермы Ленгмюра:
ZWC
l + WC
(1)
где а — величина адсорбции;
Z7 w — величины, связанные с адсорбционной способностью поглотителя по
отношению к данному газу;
С — концентрация газа.
278
ГЛ. VIII. ХРОМАТОГРАФИЧЕСКИЕ МЕТОДЫ АНАЛИЗА
Если С <С 1, то
или
а = КС
т. е. получаем уравнение прямой, выходящей из начала координат
(рис. 108).
Если С 2> 1, то
ZWC
0= wc > или a = Z
в этом случае получаем уравнение прямой, параллельной оси абсцисс.
Анализ уравнения Ленгмюра показывает, что при малых значениях
С величина адсорбции (а) прямо
пропорциональна концентрации, а при очень
больших -— является постоянной
величиной, соответствующей насыщению
поверхности адсорбента.
Изотермы адсорбции Ленгмюра по
своему виду аналогичны адсорбционным
изотермам газов и жидкостей на
твердых поверхностях.
Однако надо учитывать, что при
поглощении молекул из жидких сред
процесс адсорбции усложняется влиянием
свойств растворителя, который,
удерживаясь на поверхности адсорбента,
уменьшает адсорбируемость растворенного вещества и искажает тип
изотерм. Поэтому в таких случаях для проведения хроматографического
процесса следует подбирать растворитель с наименьшей сорбционной
способностью йо отношению к применяемому адсорбенту.
С
Рис. 108. Изотерма адсорбции.
Рис. 109. Выходная кривая фронтального
анализа:
С—концентрация; V — объем раствора,
вытекающего из колонки.
Рис. ПО. Выходная кривая вы-
теснительного анализа:
С—концентрация; t — время, мин
(объем, мл).
Процесс последовательного извлечения веществ из колонки может
быть изображен графически. Полученная при этом кривая носит
название выходной кривой.
При проведении фронтального анализа растворов, содержащих
большое количество растворенных компонентов, на выходной кривой
возникает соответствующее количество ступеней (рис. 109). Первая
ступень соответствует наименее адсорбирующемуся веществу, вторая —
смеси двух компонентов, третья — трех компонентов, и последняя сту-
§ 3. РАЗНОВИДНОСТИ ГАЗОВОЙ ХРОМАТОГРАФИИ
279
пень соответствует всем компонентам, входящим в состав исходного
раствора.
На выходной кривой в вытеснительном методе анализа образуются
пики, по величине которых можно определить содержание отдельных
веществ (рис. НО). В случае неполного разделения пики кривой
частично налагаются один на другой. Величины площадей пиков
отвечают относительным содержаниям компонентов в смеси.
В качестве адсорбентов в адсорбционно-жидкостной хроматографии
применяют органические и неорганические вещества: сахарозу, инулин,
молочный сахар, целлюлозу, крахмал, активированную окись алюминия,
карбонат кальция, силикагель, окиси металлов, активированный уголь,
некоторые природные минералы и другие.
§ 3. Разновидности газовой хроматографии
Разделение смесей газов обусловливается различными факторами —
неодинаковой скоростью движения сорбированных компонентов вдоль
слоя, разным положением пиков в хроматограмме, воздействием
температурного поля, растворимостью в поглощающей среде, неодинаковым
отношением к вытеснителю и т. п. В зависимости от способа разделения
хроматографию газов подразделяют на несколько видов: газо-адсорб-
ционная, газо-жидкостная, хроматермография, теплодинамический метод,
капиллярная и др.
Адсорбенты
Для разделения смеси газов или соединений с низкой температурой
кипения применяют следующие адсорбенты: активированный уголь,
силикагель, окись алюминия, природные и искусственные силикаты,
а также молекулярные сита. Последние представляют собой
дегидратированные, искусственно приготовленные цеолиты с геометрической
однородностью структуры и постоянством межмолекулярных расстояний.
Так, межмолекулярное расстояние сита типа 4А, представляющего
собой кристаллический алюмосиликат натрия, составляет 4 А, а у сита
типа 5А — кристаллический алюмосиликат кальция — 5 А.
Для разделения веществ и их перемещений вдоль колонки
используют газ-носитель, т. е. подвижную фазу. Газ-носитель должен быть
инертен по отношению к разделяемому веществу и к неподвижной фазе
даже при повышенной температуре. В качестве газа-носителя
применяют: азот, аргон, воздух, двуокись углерода, гелий, водород и др.
Г а зо- адсорбционна я хроматография
В этом методе верхний слой адсорбента, помещенного в колонку,
насыщают газовой смесью, затем колонку продувают инертным газом-
носителем. При этом происходит разделение компонентов, которые
располагаются в отдельных участках колонки.
Объем газа-носителя, необходимый для вымывания
адсорбированного вещества из колонки, называется удерживаемым объемом. Время
выхода из колонки максимальной концентрации компонента принято
называть временем удерживания. Эти факторы позволяют судить о
возможности разделения анализируемой смеси на отдельные компоненты.
Г азо¦жидкости а я хроматография
Для разделения компонентов газовой смеси используется
различное распределение их между неподвижной жидкой и подвижной
газообразной фазами.
280
ГЛ. VIII. ХРОМАТОГРАФИЧЕСКИЕ МЕТОДЫ АНАЛИЗА
Пробу смеси газов или паров вводят в колонку с неподвижным
инертным носителем, на котором распределена нелетучая жидкость.
Хроматографируемые газы или пары поглощаются этой жидкостью,
затем через колонку пропускают газ-носитель, вытесняющий в том или
ином порядке компоненты разделяемой смеси. Процесс разделения
характеризуется некоторой константой, называемой коэффициентом
распределения /С, т. е. отношением концентрации вещества в жидкой
неподвижной фазе к его концентрации в газовой. В газо-жидкостной
хроматографии обычно наблюдается линейная изотерма распределения,
и разделение веществ происходит достаточно полно.
Основное различие газо-адсорбционной и газо-жидкостной
хроматографии заключается в том, что в первом случае используется
процесс адсорбции и десорбции газа или пара на поверхности твердого
вещества — адсорбента, а во втором случае — процесс растворения и
испарения газа или пара из жидкой пленки, удерживаемой твердым
инертным носителем.
Уравнение равновесной газовой хроматографии
Основным физическим фактором, определяющим поведение
адсорбированных газов, является адсорбционное равновесие. Теории,
учитывающие лишь этот фактор, рассматривают так называемую
равновесную хроматографию и не учитывают такие эффекты, как продольная
диффузия, стеночный эффект и др.
Ниже приведено основное уравнение газовой хроматографии:
где Uc — линейная скорость перемещения вещества в колонке;
со—объемная скорость газа, проходящего через слой сорбента толщиной dx;
V — объем газовой фазы, приходящейся на единицу объема слоя, при постоянной
концентрации газа;
Va — объем адсорбционного слоя;
К— коэффициент Генри (/С = Са/С, Са — концентрация компонента в слое
адсорбента или неподвижной жидкой фазе, С — концентрация компонента в
газовой фазе).
Разделение смеси газа обеспечивается различной скоростью
движения компонентов вдоль слоя адсорбента или жидкой фазы,
характеризующихся различными значениями коэффициента Генри.
Хроматермография
В процессе выполнения анализа методом хроматермографии через
колонку, содержащую сорбент, пропускают анализируемую газовую
смесь. После того как колонка отработана, т. е. обнаруживается
«проскок» наиболее слабо сорбирующегося компонента, на нее медленно
надвигают электрическую печь, создающую температурное поле, и
одновременно пропускают газ-носитель. В результате воздействия на
разделяемую смесь потока газа-носителя и перемещающегося температурного
поля происходит сжатие полосы компонента, т. е. замыкающий край
полосы будет двигаться быстрее, чем передний.
В методе хроматермографии все компоненты сложной смеси
располагаются в областях своих характеристических температур в
соответствии с теплотой адсорбции компонента на данном сорбенте.
Для разделения веществ в хроматермографии необходимо, чтобы
движение сильнее адсорбирующегося вещества происходило при более
низкой температуре, чем слабее адсорбирующегося. Только в этом слу-
§ 4. РАСПРЕДЕЛИТЕЛЬНАЯ ХРОМАТОГРАФИЯ
281
чае первое вещество будет двигаться медленнее второго и произойдет
разделение веществ. Хроматермография успешно применяется для
качественного анализа и идентификации веществ в их смесях.
Тепл одинамический метод
Теплодинамический метод представляет собой сочетание методов
непрерывного фронтального и движущегося температурного поля. Как
н во фронтальном анализе, анализируемая смесь газа подается в
колонку непрерывно, а отдельные фракции компонентов смеси для
анализа собираются периодически. Анализируемая смесь подается в
колонку с адсорбентом, вдоль которой передвигается печь. Достигнув
нижнего края колонки, она возвращается в исходное положение и снова
начинает двигаться вдоль слоя адсорбента сверху вниз. При этом зоны
компонентов, постепенно смещаясь к низу колонки, обогащаются и
достаточно четко разделяются.
Капиллярная хроматография
В капиллярной хроматографии неподвижную жидкую фазу
наносят непосредственно на стенки узкого капилляра диаметром до 0,3 мм
и длиной в несколько десятков метров.
Основные достоинства капиллярной хроматографии в том, что хро-
матографические полосы практически не размываются, поэтому
оказывается возможным исследование малых доз анализируемого вещества
и, кроме того, продолжительность анализа невелика.
Препаративная газовая хроматография
Этот метод используется при разделении больших количеств
исходной смеси. На выходе колонки помещают коллектор фракций,
с помощью которого можно получать очень чистые (99,999%)
индивидуальные вещества. Приемники коллектора связаны с
программирующим устройством так, что отбор фракций происходит автоматически
при регистрации пика того или иного компонента на ленте самописца.
Методы препаративной газовой хроматографии широко применяются
в промышленности, чаще всего для разделения двухкомпонентных
систем, например для рекуперации паров летучих растворителей, для
осушки воздуха, очистки мономеров и при других процессах.
Аналитическая реакционная газовая хроматография
В аналитической реакционной газовой хроматографии сочетаются
два метода анализа — хроматографический и химический, т. е. на всех
ступенях хроматографического анализа — от введения пробы до
детектирования— используются химические реакции. Метод реакционной
газовой хроматографии применяется в тех случаях, когда использование
обычной газовой хроматографии невозможно или связано со
значительными трудностями, например, для анализа полимеров, в элементном
анализе и т. п.
§ 4. Распределительная хроматография
В распределительной хроматографии разделение веществ
осуществляется вследствие различной и притом обратимой сорбции компонентов
смеси двумя несмешивающимися жидкими фазами — подвижным и
неподвижным растворителями.
282
ГЛ. VIII. ХРОМАТОГРАФИЧЕСКИЕ МЕТОДЫ АНАЛИЗА
Колонку наполняют носителем (силикагель, окись алюминия,
кизельгур и др.)—веществом, индифферентным к хроматографируемым
веществам и в отношении к применяемому растворителю. Носитель
удерживает на своей поверхности жидкую фазу —неподвижный
растворитель. Пробу хроматографируемого раствора, содержащего несколько
компонентов, вносят в колонку и после того, как раствор впитается,
промывают колонку подвижным растворителем. При этом происходит
перераспределение веществ смеси между двумя несмешивающимися
жидкими фазами.
Для получения четкого разделения смеси необходимо, чтобы
компоненты ее не взаимодействовали с носителем и чтобы коэффициенты
распределения их сильно различались между собой. Только соблюдение
этих условий дает возможность получать отдельные зоны чистых
веществ при промывании колонки подвижным растворителем.
Свойства подвижной и неподвижной фаз. При подборе подвижной
и неподвижной фаз, а также носителя необходимо учитывать их
свойства. Если носителем является гидрофильное вещество, то в качестве
неподвижного растворителя применяют воду, а в качестве подвиж-
ного — органический растворитель. Например, для разделения смесей
полярных веществ (аминокислот, производных пиридина и других)
применяют полярный неподвижный растворитель — воду, который хорошо
удерживается на таких гидрофильных носителях, как силикагель,
порошок целлюлозы и др. Подвижной фазой в этом случае может служить
насыщенный водный раствор фенола, я-бутанол и др. Если же
носитель— гидрофобное вещество, то неподвижным растворителем должно
быть неполярное вещество (масло, керосин, бензол, парафин), а
подвижным— полярные органические вещества и вода. Разделение
происходит вследствие различной растворимости компонентов в неподвижной
фазе.
Распределительная хроматография, выполняемая в колонках, по
характеру происходящих процессов аналогична адсорбционной, с той
разницей, что в данном случае сорбентом является неподвижный
растворитель.
Изотерма распределения веществ между двумя несмешивающимися
жидкими фазами, как правило, линейна. Однако в ряде случаев в
распределительной хроматографии наблюдается искривление линейной
изотермы распределения, что связано с процессами диссоциации и
ассоциации хроматографируемых веществ в растворителях. Например,
электролиты могут в той или иной мере диссоциировать в неподвижном,
более полярном растворителе, а если же хроматографируемое вещество
способно к ассоциации, то оно может ассоциировать в подвижном
растворителе. В обоих случаях наблюдается искривление изотермы
распределения, вследствие чего происходит образование «хвостов» (см. § 2)
и разделение веществ оказывается неполным.
Коэффициент распределения. Распределение веществ между двумя
фазами принято определять отношением количества вещества в
неподвижном растворителе к количеству вещества в подвижном
растворителе. Подобное отношение концентраций называется коэффициентом
распределения данного вещества, характерным для рассматриваемой
системы, т. е.:
* = ^= (3)
иподв
где К— коэффициент распределения;
Снеподв — концентрация определяемого компонента в неподвижной фазе, моль/л;
?подв~ концентрация того же вещества в подвижной фазе, моль/л.
§ 4. РАСПРЕДЕЛИТЕЛЬНАЯ ХРОМАТОГРАФИЯ 283
Коэффициент распределения зависит от различных факторов:
природы вещества, природы растворителя, температуры и сцособа
проведения эксперимента.
Наибольшая скорость движения наблюдается у того компонента,
который имеет наименьший коэффициент распределения между
растворителями. Скорость движения фронта зоны можно определить по
уравнению:
V Л(УПодв + К7неподв) W
где / — высота зоны, содержащей определяемый компонент;
V — объем хроматографируемого раствора, мл;
Уподв"~ объем подвижного растворителя, приходящийся на 1 мл колонки;
Унеподв "*" объем неподвижного растворителя, приходящийся на 1 мл колонки;
А— поперечное сечение колонки;
К — коэффициент распределения.
Помимо скорости движения зоны приходится учитывать скорость
смещения, т. е. величину смещения1 зоны (/?) на единицу объема
растворителя:
Уподв + А Кнёподвг
При постоянных параметрах колонки скорость смещения вещества
является функцией коэффициента распределения R = f(K)y в
соответствии с чем в колонке каждая зона двигается со своей скоростью, что
и является условием разделения компонентов смеси.
Тонкослойная хроматография
Хроматография веществ в тонких слоях (ХТС) является одним из
видов распределительной хроматографии. Разделение проводят на
пластинках, покрытых тонким слоем носителя (окись алюминия, кизельгур,
силикагель и др.), удерживающего неподвижный растворитель.Нижний
край пластинки с нанесенной на нее пробой опускают в подвижный
растворитель. При движении растворителя происходит
перераспределение веществ между двумя растворителями и перемещение их с
различной скоростью, в результате чего вещества разделяются на
составные компоненты.
Бумажная хроматография
Бумажная хроматография является разновидностью
распределительной хроматографии, в которой в качестве носителя неподвижной
фазы применяется бумага. Для этой цели используют несколько сортов
хроматографической бумаги — № 1, 2, 3, 4. С увеличением номера
плотность бумаги возрастает. От плотности бумаги зависит скорость
движения растворителя в ней, поэтому бумагу для хроматографировДния
№ 1 и № 2 называют «быстрой», а № 3 и № 4— «медленной».
Для хроматографического разделения веществ методом
«обращенных фаз» иногда используют гидрофобную бумагу*.
При колоночном варианте хроматографического разделения
методом «обращенных фаз» в качестве носитедей используют следующие
гидрофобные вещества: кизельгур, стеклянный порошок, полиэтилен.
* Для гидрофобизации бумагу пропитывают различными веществами или
подвергают химической обработке.
284
ГЛ. VIII. ХРОМАТОГРАФИЧЕСКИЕ МЕТОДЫ АНАЛИЗА
По методу получения различают следующие распределительные
хроматограммы на бумаге: одномерные и двумерные (восходящие и
нисходящие), круговые, электрофоретические.
Для одномерных хроматограмм используют бумажные полосы
шириной 4,5—5 см и длиной 30—50 см. Для получения двумерной
хроматограммы применяют листы бумаги размером примерно 20 X 25—
40 X 45 см. Восходящие хроматограммы получают при перемещении
подвижного растворителя через поры бумаги снизу вверх, а
нисходящие— сверху вниз.
Для получения круговой хроматограммы используют бумажные
круги. Подвижный растворитель перемещается от центра круга к
периферии.
Электрофоретические хроматограммы на бумаге получают путем
сочетания распределительной хроматографии на бумаге с
электрофорезом при проведении их одновременно или раздельно.
Коэффициент /?/. Нанесенные на бумагу хроматографируемые
вещества переходят в подвижную фазу и, перемещаясь с различными
скоростями по капиллярам бумаги, разделяются, концентрируясь в
различных участках бумажного листа. Однако в этом случае определить
значение коэффициента распределения, как это было указано выше, не
представляется возможным. Поэтому для количественной оценки
способности разделения веществ на бумаге введен коэффициент /?/,
представляющий отношение величины смещения зоны вещества (х) к
смещению фронта растворителя. (#/);
Коэффициент Rf и коэффициент распределения Л' связаны
следующим соотношением:
Лподв / 1 Л
к==л—b—0 (7)
лнеподв \Kf I
где Лподв и Лнеподв — соответственно поперечные сечения бумаги, удерживающие
подвижный и неподвижный растворители.
Зная содержание воды в бумаге, отношение ЛПОдвМнеподв можно
определить исходя из массы абсолютно сухой бумаги и массы бумаги,
пропитанной подвижным растворителем.
На величину Rf влияют многие факторы: природа носителей, хро-
матографируемых веществ и растворителей, условия проведения
эксперимента и др. Однако для веществ, близких по свойствам, при
постоянных условиях величина Rf является относительно постоянной и служит
показателем, позволяющим определять вещества. Чем больше различие
в величинах Rf разделяемых веществ, тем лучше разделение веществ.
§ 5. Ионообменная хроматография
Ионообменная хроматография основана на обратимом обмене
содержащихся в растворе ионов на ионы, входящие в состав ионообмен-
ника. Образование хроматограмм при этом происходит вследствие
различной способности к обмену ионов хроматографируемого раствора.
В ионообменной хроматографии в качестве элюента (вымывающего
вещества) применяют растворы электролитов.
В соответствии с принятой терминологией ионообменную
хроматографию по способам выполнения подразделяют на фронтальный, вытес-
нительный и элюентный анализы. Во всех этих видах используется мно-
§ 5. ИОНООБМЕННАЯ ХРОМАТОГРАФИЯ
285
гократное повторение процесса ионного обмена. Наибольшее
применение имеет элюентный анализ.
При осуществлении элюентной хроматографии в верхнюю часть
хроматографической колонки, наполненной, например, катионитом в
водородной форме (Н-форме), вводится смесь разделяемых ионов. Для
получения компонентов в чистом виде элюирование проводят раствором
кислоты или другим элюентом при постепенно возрастающей его
концентрации (градиентное элюирование). Если исследуют смесь элементов
разных групп периодической системы, то разделение при помощи элюи-
рования растворами электролитов облегчается тем, что элементы разной
валентности обладают различным сродством к иониту. Сорбируемость
ионов возрастает от одновалентных к двух- и трехвалентным.
Приблизительную закономерность сорбируемости ионов с одинаковой
валентностью можно представить в виде «сорбционных рядов»:
Cs+ > Rb+ >K+ > NH+ > Na+; Ba++ > Sr++ > Са++ > Mg++;
Zn++ > Cu++ > Ni++ > Co++ и т. д.
Эти ряды изменяются в зависимости от природы сорбента, хрома-
тографируемых веществ, растворов, применяемых при элюировании,
и внешних условий эксперимента.
Иониты
Применяемые в ионообменной хроматографии иониты (ионообмен-
ники) могут быть органическими или неорганическими веществами.
Наиболее часто используемыми неорганическими ионитами
являются «окись алюминия для хроматографии», пермутит, фосфат циркония
и др. В качестве органических ионитов применяют целлюлозу, сульфо-
уголь и синтетические высокомолекулярные вещества — ионообменные
смолы.
Способность к ионному обмену определяется строением ионита,
представляющего собой «каркас», на котором закреплены активные
группы. Таким образом, ионит можно рассматривать как
поливалентный ион с отрицательным или положительным зарядом, связанный
ионной связью с подвижными ионами противоположного знака.
В зависимости от обмена катионов или анионов иониты делят на
катиониты, аниониты и амфолиты.
У катионитов активными группами являются кислотные группы:
—SO3H, —СООН, —ОН и другие, которые структурно связаны со
скелетом ионита. Подвижными остаются только ионы водорода этих групп
или замещающие их катионы.
У анионитов активными группами являются основные группы:
—NH2, ==NH, 35N, четвертичные аммонийные и пиридиниевые группы.
Классификация ионитов. От степени диссоциации активных групп
зависит, насколько сильно выражены основные или кислотные свойства
ионита. В зависимости от этого различают четыре группы ионитов.
Сильнокислотные катиониты, содержащие сильнодйссоцйирующие
кислотные группы, например —SO3H. Эти катиониты способны к обмену
ионов в кислой, нейтральной и щелочной средах (КУ-1, КУ-2, СДВ,
дауэкс-50, амберлит IR-120 и др.).
Слабокислотные катиониты, содержащие слабодиссоциирующие
кислотные группы (—СООН, —ОН и др.), способны обменивать ионы
при рН>7 (КБ-2, КБ-4и др.),
Сильноосновные аниониты, содержащие четвертичные аммонийные
или пиридиниевые группировки. Эти аниониты способны к обмену ионов
286
ГЛ. VIII. ХРОМАТОГРАФИЧЕСКИЕ МЕТОДЫ АНАЛИЗА
в кислой, щелочной и нейтральной средах (АВ-17, АВ-18, амберлит
IRA-400, амберлит IRA-410 и др.).
Слабоосновные аниониты, содержащие основные группы —NH2,
=NH и в N. Обмен ионов на таких анионитах происходит при рН < 7
(АН-23, АН-2Ф и др.).
Иониты, содержащие только одинаковые ионогенные группы,
называются монофункциональными, а имеющие одновременно несколько раз-
Личных групп — полифункциональными.
В качественном анализе катионов методом ионообменной
хроматографии широко применяется «окись алюминия для хроматографии»,
отвечающая формуле [(АЬОз)* • A10j]Na+.
Для хроматографирования анионов «окись алюминия для
хроматографии» переводят в анионит обработкой ее 2 н. раствором азотной
кислоты. Полученному аниониту соответствует формула [(А120з)х-А10+] ЫОз.
Ионообменные процессы. Реакции на ионитах протекают согласно
следующим уравнениям:
Катионный обмен: R-H + Na+ ^=± R-Na + H+; R-Na + K+ ^=± R-K + Na+
Анионный обмен: R - ОН + СГ ^=± R - С1 + ОН"; R - CI + NO; ^z± R - N03 + СГ
где R — макромолекула, к которой присоединена активная группа.
Амфолитами поглощаются как катионы, так и анионы в
зависимости от рН раствора.
Поглощение ионов сорбентом зависит от природы и структуры
ионита, от природы хроматографируемых веществ и от условий
проведения эксперимента (температуры опыта, величины рН раствора и
других факторов).
§ 6. Константа ионного обмена
Реакция обмена двух однозарядных ионов А+ и В+ выражается
уравнением:
RA + B+ ^z± RB + A+
Равновесие при обмене ионов достигается в результате
одновременного действия сил различной природы, и оно может быть в первом
приближении списано законом действия масс:
[В] _ к [в+] (g.
[ХГ*А'ВИ (8)
где В и А — соответственно концентрации ионов в твердой фазе;
В+ й А+- соответственно концентрации ионов в жидкой фазе;
К а В ~~ константа ионного обмена.
Константа ионного обмена дает возможность количественно
характеризовать сорбируемость одного иона по сравнению с другим.
При этом возможны три случая:
Ка, в<1—ионы раствора имеют большее сродство к иониту, чем
ионы, входящие в состав ионита (процесс обмена ионов из раствора
протекает быстро и полно);
^Са,в>1 — ионы раствора имеют меньшее сродство к иониту, чем
ионы, входящие в ионит (процесс обмеца ионов незначителен);
Ка, в = 1 — сродство к иониту обоих ионов одинаково.
§ 7. ОСАДОЧНАЯ ХРОМАТОГРАФИЯ
287
С учетом коэффициентов активности ионов константа обмена
выражается следующим уравнением: __
/А/В
где fA и fB — коэффициенты активностей ионов в жидкой фазе;
/А и /в — коэффициенты активностей ионов в твердой фазе.
Расчет реальных систем ионного обмена сложной смеси ионов
весьма сложен. Обычно для этой цели применяют уравнение изотермы ион-
наго обмена, предложенное Б. П. Никольским:
Л1/21 a\/zi
№-*"-&; (10)
Где Л| и Л2 — активности сорбируемых ионов в ионите, мг-экв/г сорбента;
ах и а2 — активности ионов в жидкой фазе;
Z\ и z2— заряды обменивающихся ионов;
Kit 2 — константа ионного обмена.
Уравнение справедливо для следующих случаев: объем ионита не
меняется вследствие ионного обмена; «каркас» ионита не вступает в
химическое взаимодействие с ионами раствора (коэффициенты активности
сорбированных ионов равны единице); не происходит молекулярной
сорбции, а также сорбции противоположно заряженных ионов;
разделяемые ионы малы по сравнению с микропорами ионита и
беспрепятственно проникают внутрь частицы ионита.
Связь между объемом раствора, который должен быть пропущен
через колонку до появления максимума концентрации вещества на
выходной кривой, и константой обмена выражается уравнением:
гг v макс [н+]2
* = ?Zy I11)
где • Умакс — объем промывного раствора, соответствующий максимуму концентрации
вещества на выходной кривой, мл;
[Н+] — концентрация ионов водорода в промывном растворе кислоты, мг-экв/мл;
?—полная емкость ионита, мг-экв/мл;
z — заряд вытесняемого иона;
V — объем, занимаемый набухшей навеской ионита, мл.
Скорость ионного обмена между двумя фазами, а также
установление ионообменного равновесия связано с природой ионитов, хрома-
тографируемых веществ и техникой эксперимента.
Изучение кинетики ионного обмена позволяет судить о структуре
сорбентов, доступности их активных групп и об их участии в ионо-
обмене.
§ 7. Осадочная хроматография
Осадочная хроматография основана на химических реакциях
взаимодействия хроматографируемых веществ с осадителем, входящим в
состав колонки, различие в растворимости образующихся
малорастворимых осадков обусловливает их разделение.
Осадочные хроматограммы могут быть получены как в колонке на
носителе, содержащем осадитель, так и на бумаге, пропитанной
осадителем.
Носителем в осадочной хроматографии может являться
малорастворимое вещество с высокоразвитой поверхностью, обладающее
определенным сродством к применяемому осадителю или осадку и химически
индифферентное к компонентам хроматографируемого раствора, за
288 ГЛ. VIII. ХРОМАТОГРАФИЧЕСКИЕ МЕТОДЫ АНАЛИЗА
исключением случая, когда носитель одновременно выполняет и роль
осадителя, например диметилглиоксим и др. В качестве носителей
применяют силикагель, крахмал, окись алюминия, гидроокись алюминия,
сульфат бария, кварц, асбест, аниониты, катиониты, окись цинка, окись
кальция, стеклянный порошок и т. п.
При формировании осадочных хроматограмм происходит
многократное повторение элементарного акта — процесса образования и
растворения осадков. Многократность элементарного акта определяется
различием в растворимости образующихся в колонке осадков, а также
возможностью их закрепления в месте образования.
Формирование осадочных хроматограмм является результатом двух
процессов: распределения образующихся осадков в колонке в порядке
увеличения их растворимости и закрепления осадков на колонке в
месте их образования. При слабом закреплении осадков они сползают
вниз по колонке и хроматографирование оказывается невозможным.
Последовательность в распределении осадков определяется
концентрациями иона-осадителя С0:
Со= ПА г/г. (12)
где ПА — произведение активностей ионов;
Si — концентрация осаждаемого иона в осадке;
г% и zQ — заряды осаждаемого и осаждающего ионов;
Cj — исходная концентрация осаждаемого иона.
Последовательность осаждения может быть нарушена путем
изменения соотношения концентраций разделяемых ионов в хроматографи-
руемом растворе.
Для вычисления последовательности осаждения компонентов 1 и 2
используют следующее уравнение:
С0>| nAf^'(feC?)'o^
С0>2 ПА***** (ffiifof*1
Разделение осадков происходит тем лучше, чем сильнее
различаются осадки по своей растворимости. Для того чтобы разделение
веществ происходило с наибольшей эффективностью, каждый
элементарный акт должен сопровождаться значительным изменением
концентрации одного вещества по отношению к концентрации другого.
Разделение хроматографируемых веществ в колонке улучшается
при промывании хроматограммы чистым растворителем. Зоны при
этом обособляются, границы их становятся более резкими. Осадочные
хроматограммы имеют четкие нижние границы и равномерное
распределение осадка по высоте зоны. Эта особенность осадочных
хроматограмм используется для количественного определения веществ по
высоте зоны.
Осадочная хроматография в сочетании с методом предельного
разбавления может быть использована для определения микроколичеств
веществ в растворе.
§ 8. Окислительно-восстановительная хроматография
В окислительно-восстановительной хроматографии разделение
веществ обусловливается неодинаковыми скоростями
окислительно-восстановительных реакций, протекающих между окислителем или
восстановителем, которые содержатся в колонке, и ионами хроматографируемого
раствора. Направление этого процесса, а следовательно, и разделение
§ 9. АДСОРБЦИОННО-КОМПЛЕКСООБРАЗОВАТЕЛЬНАЯ ХРОМАТОГРАФИЯ 289
веществ определяются соответствующими
окислительно-восстановительными потенциалами,хроматографируемых систем. Последние
рассчитываются по уравнению Нернста.
При формировании окислительно-восстановительной хроматограм-
мы происходит повторение элементарного акта окисления —
восстановления, что определяется большой поверхностью колонки и обратимостью
протекающих процессов. Закрепление продуктов реакции происходит
вследствие проявления адгезионных свойств носителя.
Колонка в окислительно-восстановительной хроматографии
содержит два вещества — носитель и окислитель или восстановитель. В
зависимости от того, какое вещество находится в колонках, их подразделяют
на окисляющие и восстанавливающие.
На окисляющей колонке продукты реакции при хроматографиро-
вании восстановителей располагаются сверху вниз в порядке увеличения
их потенциалов, обратный порядок наблюдается при хроматографирова-
нии окислителей на восстанавливающей колонке.
Например, при хроматографировании смеси 1~, Вг~ и С1~ на
колонке с.окислителем — периодатом калия зоны располагаются сверху вниз
в порядке возрастания потенциалов: вверху зона иода (?^2/2Г=+0,53 в)>
ниже— брома {Е°Ъг212Вг„ = +1,06 в) и далее —хлора (E°cl2/2Cr=: + US5 в).
При хроматографировании окислителей СггОГ" и МпОГ на колонке
с восстановителем вверху образуется бурая зона марганца (^^. 0-/м ++=
= +l,52eV ниже —зеленая зона хрома (Е°Сг 0~/2Сг+++= +1,36в\.
Весьма важным моментом в формировании
окислительно-восстановительных хроматограмм является полнота протекания окислительно-
восстановительного процесса, которая обусловливает степень разделения
веществ и с количественной стороны может характеризоваться
константой окислительно-восстановительной реакции.
При проведении в колонках окислительно-восстановительных
реакций наблюдаются два случая: с одной стороны, продуктами реакции
являются осадки, с другой — растворимые вещества. В случае
образования осадка отмечается равномерное распределение осадка по высоте
зоны, наличие ровной и резкой нижней границы и равномерная
интенсивность окраски зоны по ее высоте. Зависимость высоты зоны от
концентрации раствора позволяет использовать этот фактор как критерий
для количественного определения веществ в растворах.
§ 9. Адсорбционно-комплексообразовательная
хроматография
Адсорбционно-комплексообразовательная хроматография является
одним из новых видов хроматографического метода. Разделение
веществ в адсорбционно-комплексообразовательной хроматографии
определяется различием в константах нестойкости их комплексных
соединений. В качестве носителя используют сорбент, способный удерживать
комплексообразующий реагент и продукты его реакции с катионами
металлов. Такие сорбенты получили название модифицированных
сорбентов. В отличие от осадочной хроматографии и адсорбционно-комплек-
сообразовательной хроматографии образующиеся в результате реакции
малорастворимые комплексные соединения не выделяются на носителе
или в растворе в виде новой твердой фазы, а сразу же поглощаются
носителем вследствие большей прочности связи между молекулами
комплекса и поверхностью носителя, чем между молекулами комплексного
290
ГЛ. VIII. ХРОМАТОГРАФИЧЕСКИЕ МЕТОДЫ АНАЛИЗА
соединения в его кристаллах. В качестве комплексообразующих
реагентов используют диметилглиоксим, фениларсоновую кислоту, 8-оксихино-
лин, таннин и др.
Если через слой активированного угля, помещенного в колонку
и содержащего адсорбированный комплексообразующий реагент —
диметилглиоксим, пропустить раствор солей тяжелых металлов (никеля,
железа, меди и т. п.), то последние образуют соединения, которые
располагаются вдоль колонки в порядке уменьшения их констант
нестойкости. Необходимым условием образования хроматограмм является
сорбция комплексообразующего реагента на носителе.
Схема основных процессов, приводящих к сорбции ионов (М) на
носителе-сорбенте, насыщенном комплекеообразующим веществом (А),
может быть представлена в виде следующих уравнений:
R.A„ ^=± R + nA
яА + М ^z± АяМ
R + A„M ^z± R-A„M
R-A„ + M ^=t R-A„M
где ti — координационная емкость комплексного соединения, т. е. число лнгандоЕи
связанных с центральным атомом М.
Константа равновесия К последней реакции выражается формулой:
к. [R-A„M]
Д- [R.AJIM]
Учитывая, что
[М]-С; [R.AttM]-S, a [R. AJ + [R • А„М] - Sm
получим:
K-lsJrsic или 8-ТТШ U4)
где Sm — емкость поглощения носителя;
С — концентрация поглощаемого иона;
5 — предельная сорбция.
Константа К включает константы диссоциации сорбциодных
соединений R-An и R-AnM и константу нестойкости комплексного
Соединения АПМ:
„ КПАГ [КЩ [R-A.M] %а„
[* • Ап\ ' № ™ * И [А*М1 %А„М A«M U }
Адсорбционно-комплексообразовательное поглощение катионов на
модифицированном носителе имеет внутридиффузионный механизм,
который приближенно описывается уравнением:
г0 V я
(IS)
где и — доля поглощенного вещества к моменту времени t;
г0 — радиус частицы, см\
D ¦— коэффициент диффузии, см2/сек.
Адсорбционно-комплексообразовательный метод хроматографии
используется для глубокой очистки растворов сульфата цинка, идущего
на изготовление рентгеновских экранов, от следов никеля и железа,
гасящих люминесценцию, а также применяете^ в промышленности для
получения особо чистых веществ, используемых в производстве
люминофоров и т. д.
§ И. АППАРАТУРА, ПРИМЕНЯЕМАЯ В ГАЗОВОЙ ХРОМАТОГРАФИИ 291
Б. АППАРАТУРА, ПРИМЕНЯЕМАЯ В ХРОМАТОГРАФИИ
§ 10. Хроматографические колонки, применяемые
в адсорбционно-жидкостной хроматографии
Разделение веществ методом адсорбционно-жидкостной
хроматографии проводят на хроматографическои колонке, которая представляет
собой стеклянную трубку диаметром 5—20 мм и высотой 10—100 см.
На рис. 111 представлены хроматографические адсорбционные колонки
(см. книга 2, гл. X, § 6). Колонку заполняют адсорбентом, который
может быть в виде порошка или суспензии порошка в какой-либо
жидкости.
Для ускорения фильтрации при проведении хроматографического
разделения применяют избыточное давление.
§ 11. Аппаратура, применяемая в газовой хроматографии
Для проведения газовой хроматографии применяют прибор,
называемый хроматографом. Газовый хроматограф (рис. 112) состоит из
следующих основных частей:
1) узла регулирования и измерения
скорости потока газа-носителя и системы для его
очистки;
2) хроматографическои колонки с
термостатом;
3) дозатора (шприца), применяемого для
ввода пробы;
4) детектора — прибора для обнаружения и
записи компонентов, выходящих из колонки.
Хроматографические колонки.
Хроматографические колонки готовят из металлических или
стеклянных трубок соответствующих
размеров, придавая им различные формы. Колонки
бывают прямые, V- или W-образные или
спиральные.
Диаметр аналитических колонок колеблется
в пределах 3—8 мм. Длина может достигать
размеров от 10 см до 20 м. Чаще используют
колонки длиной 2 м.
Капиллярные колонки представляют собой
спирали из стекла, полиэтилена, чаще из нержа- ^ис# 111# Хроадатогрзфи-
у т, «, v ческие колонки, приме-
веющеи стали. Внутренний диаметр колонки няемые в адсорбционно-
0,1—1,0 мм, длина 10—50 м. Для нанесения не- жидкостной хроматогра-
подвижной фазы на внутреннюю поверхность ка- фии.
пилляра жидкость растворяют в летучем
неполярном органическом растворителе и продавливают раствор через капилляр.
Затем растворитель испаряют, продувая колонку чистым сухим
инертным газом. Толщина слоя неподвижной фазы составляет обычно 3—Ьмк<
В случаях, когда необходимо пользоваться разными жидкими
фазами, применяют двухступенчатые колонки.
Дозаторы. В газо-жидкостной хроматографии величина пробы
колеблется от 0,1 до 1 мкл, а в газо-адсорбционной — от 10 до 100 мл.
Температура вводимой пробы должна быть выше температуры колонки и
детектора на 50—100 град.
Дозаторы могут быть различных типов. Для введения проб газа
обычно применяют систему, в которую отбирается известный объем
292
ГЛ. Vm. ХРОМАТОГРАФИЧЕСКИЕ МЕТОДЫ АНАЛИЗА
газа. Затем эту систему соединяют с колонкой, и проба быстро
вымывается в колонку. Часто для введения не только жидкости, но и газов
применяют микробюретку и медицинские шприцы. При использовании
автоматических хроматографов пробу жидкости предварительно быстро
испаряют и в виде газа вводят в колонку, поэтому перед колонкой
имеется испаритель с предварительным подогревом пробы, в которое
температура на 30—100 град выше температуры колонки. Пробы
твердых веществ вводят в расплавленном состоянии или растворенными в
каком-нибудь растворителе
аналогично жидким пробам.
Детекторы. Наличие и
количественное содержание хроматографируе-
мых веществ в газе-носителе
определяют с помощью детекторов, в основу
работы которых положены
физические или химические методы.
Детектор является одним из важнейших
узлов хроматографической установки.
Существующие способы
детектирования подразделяют на
дифференциальные и интегральные. Детектор,
измеряющий концентрацию раствора
в каждый данный момент, называют
дифференциальным детектором.
Интегральный детектор непрерывно
измеряет суммарное количество пробы,
вышедшее из колонки с момента
начала анализа.
К дифференциальным детекторам
относятся приборы, действие
которых основано на измерении физических величин, например
теплопроводности, плотности газа, теплоты сгорания, диэлектрической
проницаемости и т. ш Наиболее распространенным является детектор для
определения теплопроводности (катарометр).
К детекторам интегрального типа относится бюретка, наполненная
раствором поглотителя (например, едкого кали). Выходящие из
колонки в потоке газа-носителя (например, двуокиси углерода)
компоненты исследуемого вещества поступают в бюретку. Двуокись
углерода полностью поглощается щелочью, а компоненты исследуемого
вещества собираются над щелочью; объем их определяют в бюретке.
Регистрирующие приборы. Для измерения и автоматической записи
сигналов детектора применяются чувствительные регистрирующие
приборы. Чаще всего используются милливольтметры со шкалой 0—5 или
0—10 мв.
Газовые хроматографы. Для работы обычно применяются
следующие типы приборов: хроматограф универсальный УХ-1, УХ-1М;
хроматограф лабораторный ХЛ-3, ХЛ-4, ХЛ-7, ГСТЛ-3, ЛХМ-7А (рис. 113);
хроматограф промышленный ХТП-63, ХТ-63, ХПА-3-150, ХГА-4, РХ-1;
газовый хроматограф «Цвет-1».
§ Г2. Приборы и материалы, применяемые в распределительной
хроматографии
Колоночные хроматограммы получает в стеклянных колонках,
подобных тем, которые используются в адсорбционно-жидкостной
хроматографии (см. рис. 111),
Рис. 112. Схема хроматографа:
I •» регулятор давления; 2 — манометры;
3 —ротаметр; 4 — осушительная колонка;
6 —« испаритель; 6 — хроматографическая
колонка; — детектор; 8 — самописец
(регистратор); ? —конденсационные ловушки.
§ 13. КОЛОНКИ, ПРИМЕНЯЕМЫЕ В ИОНООБМЕННОЙ ХРОМАТОГРАФИИ 293
Для бумажной хроматографии применяют специальные установки.
Простейшая установка представляет собой стеклянный цилиндр, в
который помещают кювету с растворителем, опустив в нее один конец
полосы фильтровальной бумаги. Чаще всего кювету, содержащую
растворитель, располагают так, чтобы верхний край бумажной хромато-
граммы находился в этой кювете, т. е. используют нисходящий поток
растворителя. Однако применяют и восходящий поток растворителя.
В качестве герметических камер могут быть использованы также
стеклянные аквариумы. Различные камеры для получения бумажных
хроматограмм показаны на рис. 114.
Рис. 113. Общий вид хроматографа ЛХМ-7А:
/ — блок подготовки газов; 2 — термостат колонок; 3 — блок регулирования и
программирования температуры; 4 — блок управления силовыми и измерительными цепями;
5 — электросиловой блок; 6 — электронный самопишущий потенциометр; 7 — термостат
детекторов.
Камерой для получения круговых хроматограмм могут служить
эксикатор или чашка Петри.
Тонкослойная хроматографии проводится на стеклянных
пластинках размером 20 X 20 и 20 X 25 см, на которые наносится тонкий слой
носителя. Камерой для хроматографирования на закрепленном слое
носителя может служить любой подходящий по размеру стеклянный
сосуд с плоским дном. Пластинку поддерживают в вертикальном
положении при помощи подставки, сделанной из стеклянной палочки.
Сверху камеру закрывают пришлифованной крышкой или стеклом.
При работе с незакрепленным слоем носителя разделение проводят
в стеклянных кристаллизаторах (диаметр 23—25 см, высота 6—7 см),
закрытых пришлифованной крышкой. Пластинки устанавливают под
углом 15—20°.
§ 13. Колонки, применяемые в ионообменной хроматографии
Ионообменные колонки (рис. 115) бывают разнообразных типов.
Колонки, используемые в качественном анализе неорганических
веществ, представляют собой трубки длиной 100—120 мм, с внутренним
диаметром 3—6 мм. Перед использованием колонки нижний конец ее
закрывают ватным тампоном. Адсорбенты (в большинстве случаев для
294
ГЛ. VTTI. ХРОМАТОГРАФИЧЕСКИЕ МЕТОДЫ АНАЛИЗА
качественного анализа неорганических веществ используют «окись
алюминия для хроматографии») помещают в колонку при помощи
стеклянных воронок диаметром 30—50 мм, с оттянутым нижним концом.
Адсорбент вносят в колонку сразу всей порцией, после чего слой его
уплотняют на ручной центрифуге (75 об/мин) или вручную
постукиванием о твердую поверхность.
Рйс. 114. Камеры для получений хромато-
грамм на бумаге:
а-одномерной восходящей; б - нисходящей- на
* узкой полосе бумаги; в -восходящей - на широ-
коПолосе бумаги, г - нисходящей; д -двумерной
при одновременном анализе серии отдельных проб:
?i стеклянный сосуд или кювета; 2-крыщка
(пробка)- 3- зажим для бумаги; 4-бумага,
5-фронт растворителя; б-начальная линия;
7-подвижный растворитель; 8-бюкс с
неподвижным растворителем; 9 - стеклянные перекладины;
10 — рамка для листов бумаги.
После уплотнения адсорбент должен занимать приблизительно
половину объема колонки. Окись алюминия вносят в колонку в сухом
виде Растворы в колонку вносят пипетками с оттянутыми
капиллярными концами. Фильтраты собирают в пробирки, применяемые в
качественном полумикрометоде. В литературе описаны автоматические
коллекторы для сбора фракций фильтратов при хроматографическом
анализе.
§ 14. КОЛОНКИ В ОСАДОЧНОЙ И РЕД-OKC ХРОМАТОГРАФИИ
295
Для ионообменной хроматографии в количественном анализе
применяют в большинстве случаев стеклянные колонки высотой 200—
300 мм и диаметром 10—20 мм, имеющие в нижней части дренажное
устройство (стеклянная пластинка с отверстиями) и
кран для регулирования скорости вытекания жидкости.
Колонкой может служить стеклянная трубка с
оттянутым нижним концом (диаметр 5 мм), на который
надевают резиновую трубку. Скорость протекания
жидкости через колонку в этом случае регулируется
винтовым зажимом на трубке. Колонкой может
служить бюретка, применяемая в объемном анализе,
емкостью 25, 50 и 100 мл. Вместо стеклянной пластинки,
удерживающей слой зерен ионита, применяют также
стеклянную вату (см. книга 2, гл. VI, § 6). Колонку
закрепляют в металлическом штативе.
В тех случаях, когда при анализе приходится
использовать фтористоводородную кислоту, колонки
изготавливают из пластмасс.
Для регенерации и промывки ионита в колонке
ее присоединяют к склянке с нижним тубусом,
наполненной регенерирующим раствором. Скорость
протекания жидкости через ионит регулируют винтовым
зажимом.
Колонки заполняют ионитами во влажном
состоянии. Для этого сухой ионит помещают в стакан,
заливают дистиллированной водой и оставляют стоять
на 30 мин для набухания зерен, затем отмывают
декантацией от пыли и постепенно переносят ионит в
стеклянную колонку. В колонку помещают
предварительно небольшое количество воды, чтобы предотвратить
попадание пузырьков воздуха. Если в слой ионита попадают пузырьки
воздуха, то образуются каналы, понижающие эффективность действия
ионообменной колонки.
Рис. 115. Колонки
для ионообменной
хроматографии:
а— для качественного
анализа
индивидуальных веществ; б— для
хроматографического
разделения ионов.
§ 14. Колонки, применяемые в осадочной и окислительно-
восстановительной хроматографии
Колонки, используемые в осадочной и
окислительно-восстановительной хроматографии, аналогичны применяемым в ионообменной
хроматографии (см. рис. 115).
При количественном определении вещества в растворах
используют калиброванные колонки. В этом случае измеряют не высоту зоны
(мм), а ее объем (мл).
В. ПРАКТИЧЕСКИЕ РАБОТЫ
§15. Работы по методу адсорбционной (жидкостной
и газовой) хроматографии
Разделение пигментов зеленых листьев растений
методом адсорбционно-жидкостной хроматографии
Хлорофилл — зеленый пигмент растений — состоит из смеси
нескольких пигментов. При пропускании раствора хлорофилла через
колонку происходит сорбция и разделение пигментов в соответствии с их
избирательной сорбируемостью.
296
ГЛ. VIII.-ХРОМАТОГРАФИЧЕСКИЕ МЕТОДЫ АНАЛИЗА
Хроматографическое разделение пигментов зеленых листьев
растений состоит из следующих этапов: 1) экстрагирование пигментов из
растительного материала; 2) образование хроматограмм; 3) получение
растворов хлорофилла-а, хлорофилла-р и каротиноидов.
При подготовке раствора для хроматографирования пигменты
извлекают из растительного материала полярным растворителем, а затем
растворяют в смеси неполярных растворителей. Полученный раствор
хроматографируют. Первичную хроматограмму промывают полярным
растворителем и в вытекающем растворе собирают отдельные фракции
пигментов.
Методика определения. Для анализа берут навеску растительного
материала из свежих зеленых (3—5 г) или сухих листьев (1 г). Свежий
растительный материал помещают в фарфоровую ступку и тщательно
растирают. Для лучшего растирания прибавляют мелкое стекло или
кварцевый песок. Если для опыта взят сухой материал, то его
измельчают в ступке.
В полученную растертую массу добавляют 15 мл смеси бензина и
бензола (9:1) и 10 мл ацетона, снова растирают и перемешивают.
Смесь переносят на стеклянный фильтр № 2. Ступку очищают,
ополаскивая ее чистым ацетоном. Промывную жидкость также переносят на
фильтр № 2. Экстракт пигментов отфильтровывают. Для более полного
отделения пигментов оставшуюся на фильтре массу промывают
несколькими порциями ацетона (по 5 мл) до тех пор, пока
вытекающий фильтрат не станет бесцветным, т. е. не будет содержать
пигментов.
Полученный после фильтрования экстракт пигментов переносят из
приемника в делительную воронку, отделяют ацетон путем осторожного
введения в воронку 50 мл дистиллированной воды. Жидкость в
делительной воронке слегка взбалтывают. Следует избегать сильного
взбалтывания, так как может образоваться стойкая эмульсия.
После того как произойдет расслаивание двух несмешивающихся
жидких фаз, нижнюю фазу (вода — ацетон) сливают. В делительную
воронку осторожно вводят новую порцию воды (50 мл). После
расслаивания жидкостей водную фазу сливают. Операцию отмывания ацетона
повторяют 10 раз. Бензино-бензольный экстракт просушивают после
удаления всех водорастворимых веществ. Для этого экстракт переносят
в колбу емкостью 50 мл, в которую добавляют 2—3 г прокаленного
сульфата натрия. Экстракт отфильтровывают через стеклянный или
бумажный фильтр.
Хроматографирование проводят в стеклянной колонке размером
20X1 см (см. рис. 111). Внизу колонки помещают ватный тампон и
вносят небольшими порциями сорбент СаС03 или сахарный порошок.
Уплотнение сорбента проводят постукиванием трубки о твердую
поверхность.
В подготовленную колонку вводят порцию (5 мл) экстракта.
Полученную первичную хроматограмму промывают смесью бензина и
бензола (9:1). При промывании колонки происходит разделение зон,
вверху колонки образуется зеленая зона (З-хлорофилла, затем
сине-зеленая ос-хлорофилла, несколько ниже располагается зона желтого цвета
(карртиноиды).
При дальнейшем промывании колонку растворителем в фильтрат
переводятся сначала желтые пигменты (каротиноиды), затем а-хлоро-
филл и р-хлорофилл. Фракции собирают в отдельные приемники.
§ 15. РАБОТЫ ПО МЕТОДУ АДСОРБЦИОННОЙ ХРОМАТОГРАФИИ 297
Анализ многокомпонентной смеси газов
методом газо-адсорбционной хроматографии
Разделение многокомпонентной смеси газов, состоящей из
кислорода, азота, двуокиси азота, окиси углерода, закиси азота и двуокиси
углерода, достигается при десорбции хроматографируемых компонентов
смеси путем изменения температуры.
Адсорбируют компоненты смеси газов на силикагеле марки МСМ
при низкой температуре. При этой же температуре проводят десорбцию
кислорода, азота, двуокиси азота и окиси углерода. Закись азота и
двуокись углерода десорбируют при комнатной температуре.
Результаты исследований регистрируются самописцем. По хроматограмме
рассчитывают содержание каждого компонента газовой смеси в объемных
процентах.
Методика определения. Стеклянную колонку U-образной формы,
длиной 180 см, с внутренним диаметром 6 мм заполняют
предварительно высушенным в токе воздуха при
400° С измельченным силикагелем марки
МСМ (фракция 0,25—0,5 мм). Колонку
помещают в криостат с холодильной
смесью, состоящей из сухого льда и
ацетона. В качестве криостата используют
цилиндрический стеклянный сосуд Дьюа-
ра. Устанавливают температуру в крио-
стате не выше —70° С, продувают всю
систему гелием, подаваемым из баллона, ' ' объем
со скоростью 30 смъ\мин и
устанавливают на самописце нулевую ЛИНИЮ. За- Рис. 116. Выходная кривая хро-
тем вводят сухую анализируемую смесь матографического разделения,
газов, состоящую из шести компонентов
(хранить смесь газов над водой нельзя), и подсоединяют колонку к
хроматографу типа УХ-1 или ГСТЛ-3.
Наблюдают запись результатов анализа на самописце. При
температуре ниже —70° С происходит десорбция сначала кислорода,
который выходит из колонки через 3 мин, затем азота (через 4 мин),
двуокиси азота (через 10 мин) и окиси углерода (через 13 мин).
После десорбции указанных газов колонку вынимают из криостата,
нагревают до комнатной температуры и снова продувают гелием. При
этом первым на графике появляется пик, соответствующий закиси азота,
и вторым — двуокиси углерода.
Расчет. Для количественного определения состава анализируемой
смеси по хроматограммам измеряют высоту максимумов пиков или
площадь пиков.
Концентрация компонента в максимуме пропорциональна высоте
или площади пика и количеству вещества, взятого для исследования.
Высоту пика определяют, опуская перпендикуляр из максимума
пика на нулевую линию.
Площадь пика определяют с помощью планиметра или вычисляют,
умножая высоту (АВ) на ширину (CD) (рис. 116).
Расчет можно проводить по методу «нормировки», при котором
сумма высот всех пиков компонентов смеси принимается равной 100%.
Высота пика одного из компонентов, выраженная в процентах,
соответствует процентному содержанию компонента в смеси.
298 ГЛ. VUI. ХРОМАТОГРАФИЧЕСКИЕ МЕТОДЫ АНАЛИЗА
Анализ смеси газообразных углеводородов С\ — С5
методом газо-жидкостной хроматографии
Газообразные углеводороды разделяются на колонке, в которой
жидкая фаза нанесена на носитель. При пропускании газа-носителя
через колонку, содержащую анализируемую смесь, компоненты ее
последовательно вытесняются и выносятся из колонки, после чего газ
проходит через детектор; возникающий в детекторе импульс
фиксируется на ленте в виде пика.
По высоте или площади пика определяют количественное
содержание каждого компонента анализируемой газовой смеси.
Методика определения. В колонку длиной 6 му диаметром 6 мм
вносят носитель-диатомит, измельченный до величины зерна 0,25—
0,5 мм, и уплотняют. Диатомит пропитывают жидким диизоамилфтала-
том в соотношении 15% к твердой фазе.
Колонку устанавливают в хроматограф УХ-1 или ХЛ-3, проверяют
его герметичность и включают ток газа-носителя, в качестве которого
служит водород, подаваемый из баллона (осторожно!). Прибор
включают в электрическую сеть и подготавливают его к анализу, т. е.
устанавливают температуру термостата колонки (50°С), давление газа-
носителя на входе в колонку (1,5 атм) и скорость прохождения газа-
носителя (3 л/ч). После того как на самописце будет вычерчиваться
нулевая линия в виде прямой, параллельной оси объема, прибор
считают подготовленным к анализу.
Анализируемую газообразную смесь углеводородов Ci—С5 любого
состава в количестве 0,3 мл вводят шприцем в колонку, закрытую
самоуплотняющимся резиновым колпачком. Момент впуска смеси
отмечается автоматически на нулевой линии самописца. Пропускают через
колонку газ-носитель. Разделение смеси углеводородов продолжается
20—25 мин.
Порядок выхода компонентов при данных условиях опыта будет
следующий: метан, этан, этилен, пропан, пропилен, изобутан, я-бутан,
изобутилен вместе с бутиленом-1, транс-бутеп-2, цис-бутеп-2, изопентан,
З-метилбутен-1, н-пентан, пентен-1, 2-метилбутен-1, пентен-2,2-метилбу-
тен-2.
По окончании опыта отключают ток газа-носителя, выключают
прибор из электросети, вынимают ленту из самописца, расшифровывают
хроматограмму и определяют количественный состав смеси.
§ 16. Работы по методу распределительной хроматографии
Анализ смеси катионов кадмия, меди и ртути (И)
На бумагу, содержащую в порах неподвижный растворитель —
воду, наносят анализируемый раствор смеси катионов и промывают
хроматограмму подвижным растворителем. Если растворимые вещества
имеют в данной паре растворителей разные коэффициенты
распределения, то происходит разделение анализируемых катионов и выделение их
в разных частях бумажного листа. Путем проявления полученной хро-
матограммы специфическими реагентами определяют вещества,
входящие в анализируемую смесь.
Методика определения. Подготовка бумаги. Для анализа
неорганических веществ может быть использована «бумага для
хроматографии» (№ 1, 2) и фильтровальная бумага. Предварительно бумагу
обрабатывают, погружая ее на 30 мин в 0,1 н. спиртовой раствор NaOH,
затем отмывают от избытка щелочи дистиллированной водой и поме-
§ 16, РАБОТЫ ПО МЕТОДУ РАСПРЕДЕЛИТЕЛЬНОЙ ХРОМАТОГРАФИИ 299
щают на 3—4 ч в 2%-ный раствор НС1, после чего полоски бумаги
промывают дистиллированной водой и высушивают на воздухе.
Разделение катионов кадмия, меди и ртути (II). В
середину полоски фильтровальной бумаги, соответствующей размеру
камеры для получения хроматограммы, на
расстоянии 4—5 см от верхнего края
микропипеткой наносят каплю раствора
хлоридов меди, кадмия и ртути (II) в
концентрации 10—15 мг-ион/мл каждого
иона. Тот же край бумаги погружают на
глубину 2—2,5 см в растворитель — я-бу-
танол, насыщенный 1 н. раствором HCL
Когда растворитель пройдет на бумаге
расстояние, равное 20—25 см, полоску
вынимают, подсушивают при комнатной
температуре и проявляют
свежеприготовленным 10%-ным раствором (NH4bS.
В нисходящей хроматограмме
обнаруживают ионы по специфической окраске:
внизу располагается черное пятно
сульфида ртути (II), затем
темно-коричневое— сульфида меди и дальше желтое —
сульфида кадмия.
Расчет. Вычисляют Rf (рис. 117)
аналиаируемых ионов по следующей
формуле:
где х — путь, пройденный компонентом от центра
нанесения капли раствора до центра
пятна на хроматограмме» см;
Xf — путь, пройденный подвижным
растворителем от места нанесения капли раствора
до фронта движения растворителя, см.
Разделение, качественное и количественное определение аминокислот
Разделение сложной смеси аминокислот на составные части
основано на различии коэффициентов распределения аминокислот в двух
несмешивающихся растворителях. Для качественного анализа хромато-
грамму проявляют и определяют содержащиеся в растворе
аминокислоты путем подбора «свидетелей» или расчета коэффициента Rf.
Для количественного анализа проводят определения концентрации
аминокислот по интенсивности окраски или размерам пятен хромато-
грамм, а также путем элюирования аминокислот из отдельных пятен и
последующего определения их классическими количественными
методами анализа.
Если пятна на хроматограмме перекрываются или по какой-либо
причине контуры их искажаются, то данный метод не может быть
использован для количественного определения.
Качественное определение аминокислот
Подготовка бумаги. Для разделения аминокислот
используют «бумагу для хроматографии» № 1 и № 2. Бумагу тщательно
отмывают 0,3 н. раствором НС1, затем 0,5 н. раствором щелочи, избыток
Начальная
линия
/?'-¦?
3?
Л
/Э-
2&
^/ = х4
Фронт
растворителя
Рис. 117. Экспериментальное
определение R. компонентов на
бумажной хроматограмме:
xv x2, xt — путь, пройденный
соответственно первым (/) и вторым (2)
компонентами и растворителем; Л— точка
нанесения пробы.
Rf и Rf — коэффициенты
распределения
соответственно первого и второго
компонентов.
300
ГЛ, VIII. ХРОМАТОГРАФИЧЕСКИЕ МЕТОДЫ АНАЛИЗА
NaOH отмывают дистиллированной водой до отрицательной реакции
по фенолфталеину, обрабатывают 0,1%-ным фосфатным буферным
раствором (рН = 7,0 — 7,5) и высушивают.
Предварительная обработка может быть исключена, если бумага не
содержит ионов тяжелых металлов.
Растворитель. я-Бутиловый спирт — уксусная кислота — вода
(4:1: 5). Смесь встряхивают 1—2 мин и после расслаивания отделяют
слои.
Проявитель — 0,2%-ный раствор нингидрина в безводном
ацетоне (200 мг нингидрина растворяют в 100 мл чистого ацетона). Хранят
проявитель в сосуде из темного стекла.
Растворы аминокислот «свидетелей». В качестве
«свидетелей» применяют 0,01 М растворы чистых аминокислот. Навески
аминокислот, необходимые для приготовления 10 мл 0,01 М раствора
(табл. 10), берут на аналитических весах и растворяют в воде или
10%-ном изопропиловом спирте. Раствор тирозина подкисляют 0,1 н.
раствором НС1 до его полного растворения.
ТАБЛИЦА 10
Навески аминокислот* для приготовления 10 мл 0,01 М растворов
Аминокислоты
Аргинин солянокислый ....
Аспарагиновая кислота ....
Валин
Гистидин солянокислый . . .
Глицин
Глутамин
Глутаминовая кислота ....
Изолейцин (лейцин)
Навеска,
мг
8,9
21,0 !
13,3,
11,7
22,7
7,5
14,4
14,7
13,1
Аминокислоты
Лизин солянокислый . . .
Метионин
Пролин
Тирозин (навеска для
0,005 М раствора) . . .
Треонин
Фенилаланин
Цистин *
Навеска,
мг
18,1
14,9
19,0
10,5
9,0
11,9
20,4
15,1
23,8
* Перед приготовлением стандартного раствора аминокислоту высушивают в течение 48 ч в
вакуум-эксикаторе над серной кислотой.
Для приближения условий построения калибровочного графика к условиям анализа
исследуемой пробы, что значительно повышает точность анализа, поступают следующим образом: в 10 мл
воды или 10%-ного изопропилового спирта растворяют несколько аминокислот кроме определяемой
аминокислоты, кислоты выбирают с таким расчетом, чтобы их пятна не перекрывались на хрома-
тограмме при однократном пропускании растворителя.
Растворы для исследования. Для разделения берут
следующие смеси аминокислот: 1) гистидин, глицин, валин, изолейцин
(или лейцин); 2) аргинин, глутаминовая кислота, аланин, метионин;
3) лизин, серии, тирозин, фенилаланин; 4) цистин, аспарагиновая
кислота, тирозин, пролин, триптофан.
Методика определения. На листе бумаги, соответствующем размеру
камеры, на расстоянии 12 см от края, в случае применения больших
камер, или 5 см, в случае применения малых, отмечают карандашом
черту для нанесения капель раствора. Исследуемую смесь и растворы
«свидетелей» в количестве 0,01 мл наносят вдоль черты на расстоянии
2—3 см друг от друга. Капли наносят в два приема микропипеткой по
0,005 мл в одно и то же место после высушивания предыдущей капли.
Конец подсушенной бумаги, на который нанесены капли растворов,
опускают в кювету с подвижным растворителем так, чтобы пятна
исследуемых растворов не были погружены в растворитель. Хроматографи-
руют нисходящим методом. В качестве подвижного растворителя
применяют аерхний слой смеси бутанола, уксусной кислоты и воды. Нйж-
§ 16. РАБОТЫ ПО МЕТОДУ РАСПРЕДЕЛИТЕЛЬНОЙ ХРОМАТОГРАФИИ 301
ний слой используют для насыщения атмосферы камеры, помещая его
на дно. Когда фронт растворителя пройдет 2/3 длины бумаги, хромато-
грамму вынимают, высушивают и вновь пропускают через нее
подвижный растворитель. Так хроматограмму обрабатывают трижды. При
разделении смесей, содержащих небольшое число аминокислот, можно
ограничиться однократным пропусканием подвижного растворителя.
После окончания разделения хроматограмму высушивают на
воздухе и проявляют раствором нингидрина путем опрыскивания из
пульверизатора. Затем нагревают 15—20 мин при 60° С в термостате или
сушильном шкафу. Расположение аминокислот сверху вниз по
направлению движения растворителя следующее: цистин, лизин, аргинин, гис-
тидин, асиарагиновая кислота, серии (три последние аминокислоты
располагаются в виде тесно сближенных пятен); глутаминовая кислота,
треонин, аланин, пролин, тирозин, валин, метионин, триптофан, фенил-
аланин, лейцин, изолейцин (последние три аминокислоты также часто
располагаются в виде тесно сближенных пятен).
Хроматограмму проявляют 0,2%-ным раствором нингидрина в
ацетоне. Для того чтобы сохранить пятна на хроматограмме, их фиксируют
1%-ным раствором нитрата меди в ацетоне, погружая хроматограмму
в этот раствор или опрыскивая ее из пульверизатора. После
фиксации пятна приобретают красно-оранжевую окраску.
Качественный состав аминокислот в исследуемом растворе
определяют путем сопоставления положения пятен известных веществ —
«свидетелей» с положением пятен, полученных для анализируемого
раствора.
Коэффициент Rf определяют для каждой аминокислоты (см.
рис. 117). Одна и та же аминокислота при одних и тех же условиях
опыта имеет постоянную величину Rf. По полученным величинам Rf
можно в последующих опытах определить качественный состав смеси
аминокислот без применения «свидетелей».
Количественное определение аминокислот
Подготовка бумаги — см-, стр. 299.
Растворитель — см. стр. 300.
Проявитель — 0,5%-ный раствор нингидрина, который готовят
растворением нингидрина в ацетоне, ледяной уксусной кислоте и воде
(95 : 1 : 4) непосредственно перед определением.
Калибровочный график. Для построения калибровочных графиков
готовят 0,01 М растворы аминокислот. Навески аминокислот,
необходимые для приготовления 10,0 мл 0,01 М растворов, приведены выше (см.
табл. 10).
С помощью микропипетки наносят на бумагу 5, 10, 15 и 20 мкл
стандартного раствора порциями по 5 мкл с таким расчетом, чтобы
каждая точка будущего калибровочного графика соответствовала 0,05;
0,10; 0,15 и 0,20 мкг аминокислоты.
Растворитель пропускают через бумагу столько раз, сколько это
необходимо для четкого разделения аминокислот исследуемой пробы.
Хроматограмму затем высушивают, проявляют, погружая на несколько
секунд в 0,5%-ный раствор нингидрина, снова подсушивают в течение
нескольких минут на воздухе и нагревают для развития окраски
(15 мин) в сушильном шкафу при 60° С.
Контуры пятен стандартных растворов на хроматограмме осторожно
очерчивакэт карандашом и определяют их площади планиметром. Затем
строят калибровочный график в координатах: натуральные логарифмы
площадей пятен — концентрация аминокислот.
302
TIL VHL- ХРОМАТОГРАФИЧЕСКИЕ МЕТОДЫ АНАЛИЗА
Концентрацию аминокислот в анализируемом растворе можно
также вычислить па формуле:
S~alnC+b
где 5 — площадь;
С — концентрация;
а и Ъ — эмпирические коэффициенты.
Вместо площади можно определять массу пятна, для этого пятно
вырезают и взвешивают. На основании полученных данных строят
калибровочный график, откладывая на оси абсцисс натуральные
логарифмы концентраций, а на оси ординат — массу пятен.
Для достаточной точности проводят 6—10 параллельных
определений как для стандартных растворов, так и для каждой аминокислоты
неизвестной концентрации. В этом случае точность метода определяется
правильно найденными границами пятен и выбранными концентрациями,
при которых наблюдается линейная зависимость между площадью
(массой) пятна и In С.
Методика определения. Предварительно проводят калибрование
микропипетки, для этого ее заполняют дистиллированной водой, затем
на маленький кружок фильтровальной бумаги, взвешенный в бюксе,
выливают содержимое микропипетки, касаясь ее концом бумаги. Бюкс
закрывают и взвешивают на аналитических весах. Определение повторяют
5—6 раз. Результаты определений вносят в таблицу и рассчитывают
объем микропипетки (в мкл) по формуле:
Р
где р — масса воды, г;
р — плотность, г/см3.
Результаты исследования заносят в таблицу по форме:
Определение,
№ п/п
Масса, г
бюкса с
бумагой
бюкса с бумагой
и водой
воды в объеме
пипетки (р)
Объем
пипетки,
мкл
Плотность р
воды в
условиях опыта,
г/см3
За истинный объем пипетки (V) принимают среднее
арифметическое всех определений. Объем микропипетки должен быть не больше
2—5 мкл. Можно также использовать калиброванные медицинские
пипетки.
Раствор аминокислот микропипеткой наносят на полоску бумаги и
проводят хроматографирование, как указано выше (см. стр. 300).
Определив площади пятен или их массу, по калибровочным графикам
находят концентрации аминокислот в анализируемом растворе.
Определение путем элюирования аминокислот
из бумаги с последующим фотоколориметрированием
Метод основан на реакции аминокислот с нингидрином в
слабокислой среде с последующим превращением полученного в результате
реакции синего производного дикетогидринделидендикетогидриндиамина
(ДИДА) в стабильное производное меди оранжево-красного цвета,
имеющее максимум поглощения при 530 нм. Количественное определе-
§ 16. РАБОТЫ ПО МЕТОДУ РАСПРЕДЕЛИТЕЛЬНОЙ ХРОМАТОГРАФИИ 303
ние аминокислот основано на измерении оптической плотности
производного меди (ДИДА) после вымывания его из бумаги.
Линейная зависимость между содержанием аминокислоты в пятне
и оптической плотностью сохраняется в пределах 0,025—0,2 мкг
аминокислоты; оптимальное содержание аминокислоты в зоне 0,05—0,15 мкг.
Оптимальной средой для работы является слабокислая среда в пределах
рН = 5—6.
Методика определения. Хроматографическое разделение
аминокислот проводят по методике, описанной выше (см. стр. 300). После
развития хроматограммы вырезают участки пятен, занимаемые
аминокислотами; сбоку хроматограммы вырезают равные по площади опытным
контрольные участки, не содержащие определяемых веществ.
Вырезанные участки бумаги измельчают ножницами и помещают
в колбы. В каждую колбу добавляют по 10 мл 0,005%-ного этанолового
раствора сульфата меди. Объем доводят до 100 мл 96%-ным этиловым
спиртом. Синяя окраска аминокислот сменяется оранжево-красной при
образовании производного меди, которое растворимо в этиловом спирте.
При данной операции совмещается образование комплексного
соединения с медью и его экстракция из бумаги.
Колбы помещают в темное место и оставляют на 1 ч, время от
времени перемешивая их содержимое. Окрашенные растворы фотометри-
руют в фотоэлектроколориметре ФЭК-М с зеленым светофильтром
(530 нм). Затем строят калибровочный график, откладывая на оси
абсцисс концентрацию аминокислоты — мкг\лу а на оси ординат —
оптическую плотность. По калибровочному графику находят концентрацию
исследуемой кислоты.
Определение проводят в тех же условиях, в которых готовят
растворы для построения калибровочного графика.
Анализ смесей красителей
Разделение красителей методом распределительной хроматографии
основано на использовании различной величины их коэффициентов
распределения между двумя несмешивающимися жидкостями. При
проявлении хроматограммы образуются ярко окрашенные пятна составных
компонентов, и состав красителя определяют по сопоставлению этих
пятен со «свидетелями».
Методика определения. Для анализа красителей в качестве
распределительной камеры применяют небольшой эксикатор. На дно камеры
помещают стакан емкостью 50 мл, в который наливают подвижный
растворитель — 30 мл 1 н. раствора едкого натра. Вырезают круг из бумаги
для хроматографии № 1 или № 2 диаметром на 1 см меньше
внутреннего диаметра камеры. В центре бумажного круга делают отверстие,
в которое вставляют бумажный конус (диаметр внизу 1,5—2 см) для
подачи подвижного растворителя.
Бумажный круг делят карандашом на четыре сектора. На
расстоянии 2 см от центра по окружности в первый сектор наносят каплю
(объемом 5—10 мкл) 2%-ного водного раствора красителя кислотного
черного, а в последующие секторы — 2%-ные водные растворы
«свидетелей» кислотного сине-черного, кислотного оранжевого светопрочного,
кислотного бордо.
Круг переносят в камеру так, чтобы бумажный конус был погружен
в подвижный растворитель. Камеру накрывают крышкой и оставляют
для развития хроматограммы на 1—1,5 ч. Затем хроматограмму
вынимают из камеры, высушивают на воздухе и по распределению и цвету
304 ГЛ. VIII. ХРОМАТОГРАФИЧЕСКИЕ МЕТОДЫ АНАЛИЗА
пятен «свидетелей» и распределению компонентов определяют состав
исследуемого образца красителя.
Аналогично проводят анализ красителя кислотного черного Б К*.
Разделение катионов меди и кадмия методом
тонкослойной хроматографии
Разделение Си++ и Cd++ методом тонкослойной хроматографии
основано на использовании различия коэффициентов распределения этих
ионов между водой и подвижным растворителем. Хроматограмму
проявляют путем опрыскивания проявителем — раствором Na2S. В местах
расположения ионов при этом возникают пятна сульфидов, обладающие
характерной окраской.
Методика определения. На стеклянную пластинку размером 20 X 50
или 20 X 25 см помещают предварительно просеянную безводную окись
алюминия (размер частиц не должен превышать 350 меш). Окись
алюминия или носитель распределяют на пластинке металлическим валиком
до толщины слоя не более 500 мк. В качестве подвижного растворителя
применяют смесь, состоящую из 18 мл я-бутанола, 12 мл ацетона и
0,6 мл азотной кислоты (р = 1,36 г/см3). В качестве «свидетелей»
используют 0,5 н. растворы Cu(N03h и Cd(N03)2. В правый угол
приготовленной пластинки, на расстоянии 2 см от края ее, наносят
капилляром каплю исследуемого раствора смеси Си++ и Cd++, содержащего
каждый ион в концентрации 0,5 г-экв/л. Через 1,5 см по ширине
пластинки наносят еще каплю исследуемого раствора для параллельного
опыта и дальше через каждые 1,5 см — по капле раствора «свидетелей»
(солей кадмия и меди). Таким образом, наносят четыре пятна. Диаметр
наносимого пятна не должен быть более 2 мм, иначе разделение ионов
будет неполное. Пластинку помещают в камеру, на дно которой
наливают растворитель. Пластинку ставят в наклонном положении так,
чтобы слой носителя не осыпался с нее, нижний край пластинки
осторожно погружают в растворитель на 1 см.
Пятна с исследуемым раствором и «свидетелями» должны
находиться выше уровня растворителя на 1 см. Камеру закрывают и
оставляют на 50 мин для развития хроматограммы. Время развития хромато,-
граммы зависит от влажности носителя. После того как подвижный
растворитель поднимется по тонкому слою носителя на высоту не
менее 17 см, пластинку вынимают, подсушивают при комнатной
температуре и проявляют путем опрыскивания ее 1 н. раствором Na2S.
На хроматограмме проявляются два пятна: выше — желтое CdS,
ниже — черное CuS. Сравнивая окраски полученных пятен от
исследуемого раствора с пятнами «свидетелей», можно определить содержание
Си++ и Cd4** в растворе.
По полученной хроматограмме можно определить также величину
Rf для каждого катиона (см. § 16, стр. 299).
Определение жирных кислот методом «обращенных фаз»
В методе «обращенных фаз» неподвижным растворителем является
неполярное органическое вещество, а подвижным — полярное
органическое соединение. В качестве носителя применяют бумагу,
предварительно гидрофобизированную путем ацетилирования. Разделение
жирных кислот проводят «восходящим способом» с использованием метода
* Кислотный черный БК состоит из кислотного сине-прочного, кислотного
оранжевого светопрочного, кислотного алого.
§ 17. РАБОТЫ ПО МЕТОДУ ИОНООБМЕННОЙ ХРОМАТОГРАФИИ 305
«свидетелей». После разделения веществ хроматограмму проявляют и,
сопоставляя расположение пятен в хроматограмме с расположением
пятен «свидетелей», определяют состав исследуемого вещества.
Методика определения. Приготовление ацетилирован-
ной гидрофобной бумаги. Лист фильтровальной бумаги № 2
размером 10 X 35 см для удаления катионов тяжелых металлов
промывают 50%-ным растворов уксусной кислоты до обесцвечивания элюата
и высушивают. Ацетилирующую смесь готовят из уксусного ангидрида
и петролейного эфира (9 : 1 по объему), добавляя 1—2 капли
концентрированного раствора H2S04 на каждые 100 мл, и тщательно
перемешивают. Полоску бумаги опускают в ацетилирующую смесь на 40 мин,
после чего бумагу промывают 3—4 раза, в!ыдерживая ее по 15 мин
в дистиллированной воде, затем высушивают при комнатной
температуре. При дальнейшем использовании смеси время каждого
последующего ацетилирования следует увеличивать на 5—10 мин.
Получение хроматограмм. На подготовленные листы
бумаги наносит растворы смеси двух-трех кислот в количестве 20—50 мкл
каждого компонента — общее содержание кислот должно быть около
50 мкг — и растворы «свидетелей» (интервал 1,5 см), В качестве
подвижного растворителя применяют ледяную уксусную кислоту.
Подвижный растворитель наливают в стеклянную кювету и в нее помещают
нижний конец полосы бумаги. Время экспозиции 10—12 ч. После чего
бумагу вынимают, высушивают до полного удаления уксусной кислоты
(определяют по исчезновению запаха уксусной кислоты). Высушенную
бумагу выдерживают в течение 2 ч ъ сильной струе воздуха
(воздуходувка), нагретого до 30—80° С. Затем бумагу вынимают, свертывают
в виде цилиндра и проявляют.
Для этого хроматограмму помещают на 30 мин в раствор ацетата
меди [10 мл насыщенного водного раствора Си(СН3СОО)2, смешивают
с 240 мл дистиллированной воды]. Избыток ацетата меди отмывают
в проточной воде в течение 30 мин и помещают хроматограмму на
5—10 мин в разбавленный раствор гексацианоферрата (II) калия (50 мл
7,5%-ного водного раствора K4[Fe(CN)6] смешивают с 250 мл
дистиллированной воды). При этом пятна медных солей кислот окрашиваются
в интенсивный темно-красный цвет. Фон остается светло-желтым или
светло-красным.
Идентификацию отдельных пятен проводят при помощи
«свидетелей», а также по величинам Rf (см. стр. 299).
В качестве растворов «свидетелей» используют 5%-ные растворы
жирных кислот в диэтиловом эфире.
§ 17. Работы по методу ионообменной хроматографии
Анализ смесей неорганических ионов
Для проведения хроматографического разделения ионов используют
их избирательную сорбируемость на «окиси алюминия для
хроматографии». Если вещества обладают различной способностью к сорбции
(различными константами ионообмена), то они могут быть разделены на
хроматографической колонке и обнаружены в зоне их расположения
непосредственно либо после «проявления» хроматограммы.
Разделение ионов протекает тем лучше, чем дальше друг от друга
в сорбционном ряду располагаются хроматографируемые вещества.
306
ГЛ. VIII. ХРОМАТОГРАФИЧЕСКИЕ МЕТОДЫ АНАЛИЗА
На «окиси алюминия для хроматографии» неорганические катионы
располагаются по своей сорбируемое™ в следующей
последовательности:
Н+ > As111 > Sbm > Sn++ « Bi+++ > [Hg2)++ > Cr+++ - Fe+++ -
« Hg++ > UO++ > AI+++ > Pb++ > Cu++ > Ag+ > Mg++ > Zn++ > Co++ -
- Ni++ = Cd++ - Fe++ - Ba++ > Mn++ > Sr++ > Ca++ > K+ > NH+
Знак равенства между отдельными ионами в сорбционном ряду
указывает на одинаковую сорбируемость их на данном сорбенте.
Исследуемый раствор вносят в колонку, наполненную сорбентом, и
полученную первичную хроматограмму после предварительного
промывания водой проявляют специфическим реагентом. Находящиеся в
различных зонах хроматограммы сорбированные ионы, взаимодействуя
с проявителем, образуют окрашенные химические соединения.
По окраске зон, характерной для каждого иона, делают вывод о
содержании ионов в исследуемом растворе.
Методика определения. На дно хроматографической стеклянной
колонки диаметром 5—6 мм и длиной 100 мм помещают небольшой
ватный тампон. Колонку наполняют сухим сорбентом на 7г высоты.
Сорбент вносят в колонку сразу всей порцией и утрамбовывают, сначала
постукивая о твердую поверхность до прекращения усадки, а затем
уплотняя стеклянным пестиком.
Исследуемый раствор, содержащий два-три компонента,
приготавливают в виде 0,25 н. растворов различных солей, смешанных в равных
объемах (по 1 мл) в отдельном сосуде. В колонку с окисью алюминия
при помощи пипетки вносят 1—5 капель приготовленного раствора.
Ионы в образующейся хроматограмме располагаются сверху вниз
соответственно их избирательной сорбируемости. Хроматограмму
промывают 2—3 каплями воды, после чего в колонку вносят по каплям
проявитель.
Растворы, проявители и воду вводят в колонку после того, как
предыдущая жидкость полностью впитается сорбентом, иначе четкие
хроматограммы не образуются.
Обнаружение ионов висмута и ртути (I). В колонку
вносят 2 капли раствора смеси солей висмута и ртути (I), затем каплю
воды и 3 капли 10%-ного водного раствора тиомочевины. Вверху
образуется желтая зона (окрашейное соединение висмута), внизу — черная
зона (соединения ртути (I). Сорбируемость: Bi+++ > [Hg2]++.
Обнаружение ионов железа(Ш), меди и коба л ьта.
В колонку вносят 3 капли раствора смеси солей железа (III), меди и
кобальта и промывают хроматограмму 2 каплями воды.
Вверху колонки формируется бурая зона — железа, затем —
голубая — меди и внизу — розовая — кобальта. Сорбент имеет
слабощелочную реакцию (рН водной вытяжки около 8,0), поэтому образуются
гидроокиси и основные соли указанных катионов. Сорбируемость: Fe+++ >
> Cu++ > Co++.
Обнаружение роданид- ц сульфид-ионов. В колонку
вносят 1—3 капли раствора смеси роданида и сульфида аммония или
калия, промывают 1 каплей воды и проявляют 3 каплями раствора
Fe(N03h. Вверху образуется темно-красная зона, переходящая в
оранжевую (роданид железа), внизу — черная зона (сульфид железа).
Сорбируемость: SCN" > S".
Обнаружение перманганат- и хромат-ионов. В
колонку вносят 3 капли раствора смеси перманганата и хромата калия;
§ 17. РАБОТЫ ПО МЕТОДУ ИОНООБМЕННОЙ ХРОМАТОГРАФИИ 307
хроматограмму промывают тремя каплями воды. Вверху образуется
желтая зона (хромат), внизу — розовая (перманганат). Сорбируемость
СгОГ>Мп01.
Обнаружение роданид-, сульфид- и гексацианофер*
р а т (II)-и о н о в. В колонку вносят 2 капли раствора смеси роданид-,
сульфид- и гексацианоферрат (II)-ионов в виде калиевых солей,
промывают 3 каплями воды и проявляют 3 каплями 2 н. раствора Fe(N03b*
Образуются три зоны: вверху — синяя зона (берлинская лазурь),
свидетельствующая о наличии [Fe^N^p^^, затем желто-коричневая зона
(роданид железа) —SCN~ и внизу — черная (сульфид железа) — S".
Сорбируемость [Fe(CN)6]= = > SCN~ > S".
Подготовка ионитов для определения
различных веществ
Подготовка катионита. В стакан помещают 15 г воздушно-
сухого сильнокислотного катионита, заливают дистиллированной водой
и оставляют на 30 мин для набухания зерен катионита. Затем катионит
отмывают декантацией дистиллированной воды от пыли. Набухший
катионит переносят в стеклянную колонку (диаметр 12—20 мм, высота
300 мм), в которую предварительно на 7з высоты наливают воду. Над
слоем ионита все время должна находиться жидкость. В случае
попадания пузырьков воздуха в колонку ионит надо взрыхлить стеклянной
палочкой.
Для переведения катионита в Н-форму через колонку пропускают
2 н. раствор хлористоводородной кислоты. Если в колонке 15 г
катионита, то достаточно пропустить 200—300 мл 2 н. раствора НС1. Катио-
ниты обычно содержат ионы железа (III), поэтому окончание
промываний катионита устанавливают по отсутствию Fe+++ в элюате. Для этого
отбирают в пробирку или на часовое стекло 2 капли вытекающего из
колонки раствора и добавляют 2 капли раствора K4[Fe(CN)e]. В
присутствии Fe+++ выпадает синий осадок Fe4[Fe(CN)6]3- Для промывания
катионита кислотой колонку можно присоединить к сосуду с нижним
тубусом, находящемуся выше колонки и наполненному 2 н. раствором
НС1. Кислоту пропускают через ионит со скоростью 10 мл/мин
(примерно 2 капли в 1 сек). После окончания пропускания кислоты жидкость
в колонке спускают до верхнего слой катионита и промывают катионит
дистиллированной водой, также используя сосуд с нижним тубусом.
Полноту отмывания катионита от кислоты проверяют по метиловому
оранжевому. Для этого отбирают каплю вытекающего из колонки
раствора на часовое стекло и добавляют каплю индикатора. Если окраска
раствора станет желтой, то считают, что катионит полностью отмыт от
кислоты.
Подготовленный таким образом катионит готов для проведения
хроматографических разделений на катионите в Н-форме.
Подготовка анионита. В большинстве случаев для
хроматографических разделений используют сильноосновные аниониты в ОН-
или Cl-форме. Анионит в колонку вносят тем же способом, что и
катионит.
Для переведения 15 г анионита в С1-форму пропускают через
колонку 200 мл 2 н. раствора НС1 и затем отмывают от кислоты
дистиллированной водой.
Для получения анионита в ОН-форме через анионит в Cl-форме
пропускают 1 н. раствор NaOH до отрицательной реакции на С1~
308
ГЛ. VIII. ХРОМАТОГРАФИЧЕСКИЕ МЕТОДЫ АНАЛИЗА
(проба с HN03 + AgN03). Затем анионит промывают
дистиллированной водой.
Окончание отмывки анионита от щелочи проверяют по
фенолфталеину.
Определение содержания соли в растворе
При пропускании раствора соли через колонку с катионитом в
Н-форме катионы соли обмениваются на ионы водорода, при этом
выделяется кислота в количестве, эквивалентном содержанию соли в
растворе. Количество выделившейся кислоты определяют титрованием
щелочью.
В большинстве случаев ионный обмен проводят в водной среде, при
этом можно определить содержание в растворе любой соли, если соль
и соответствующая кислота хорошо растворимы в воде.
Для определения солей, подвергающихся гидролизу, применяют
ионный обмен в неводных средах. В качестве сильнокислотных катионитов
в данном случае можно использовать катиониты марки СБС, СДВ-3,
КУ-2 и другие в Н-форме.
Методика определения. Анализируемый (~0,1 н.) раствор соли из
мерной колбы отбирают пипеткой емкостью 10 мл и помещают в
колонку (диаметр 20 мм, высота 300 мм), содержащую 15 г
сильнокислотного катионита в Н-форме, подготовленного, как указано выше. Раствор
пропускают через катионит со скоростью 10 мл/мин. Вытекающий из
колонки раствор собирают в коническую колбу. Затем через катионит
пропускают дистиллированную воду, наливая ее в колонку из промы-
валки отдельными порциями по 10—15 мл. Новую порцию воды
наливают тогда, когда жидкость в колонке достигнет поверхности катионита.
Для промывания колонки пропускают около 60—100 мл воды. Полноту
вымывания выделившейся кислоты проверяют по метиловому
оранжевому; для этого отбирают на часовое стекло каплю вытекающего из
колонки раствора и прибавляют индикатор. Если при этом окраска
раствора станет желтой, то считают, что кислота полностью вымыта из
катионита. Промывные воды тщательно собирают в ту же коническую
колбу. Все содержимое конической колбы оттитровывают стандартным
0,1 н. раствором NaOH. Определение проводят 3 раза.
Расчет. Содержание соли (g) вычисляют (в мг) по общепринятой
формуле
8 ^ ^o.rm^NaOH^NaOH ~~\Г~ (16)
v A
где VK — общий объем исследуемого раствора, мл;
Vа — объем анализируемого раствора, мл.
После окончания определения катионит регенерируют. Для этого
через колонку с катионитом пропускают 200 мл 2 н. раствора НС1 для
извлечения поглощенных катионов, затем отмывают катионит от
кислоты дистиллированной водой и до следующего определения катионит
сохраняют в колонке, заполненной водой.
Разделение на катионите и определение ионов
меди и свинца
Разделение катионов меди и свинца на катионите основано на
селективном взаимодействии их с винной кислотой и аммиаком. При
добавлении винной кислоты к раствору, содержащему ионы меди и свинца,
образуются тартратные комплексные анионы меди и свинца; при
добавлении раствора аммиака тартратные комплексные анионы меди пере-
§ 17. РАБОТЫ ПО МЕТОДУ ИОНООБМЕННОЙ ХРОМАТОГРАФИИ 309
ходят в более прочные аммиачные комплексные катионы [Cu(NH3)4]++,
а тартратные комплексные анионы свинца [РЬ(С4Н20б)]"~~ остаются без
изменения.
При пропускания через катионит в Н-форме раствора, содержащего
катионы меди в виде [Си^НзЫ^ианионы свинца в виде [РЬ(С4Н2Об)]"",
комплексные катионы меди поглощаются катионитом, а комплексные
анионы свинца проходят в фильтрат. Ионы меди из катионита
извлекают 2 н. раствором хлористоводородной кислоты.
Методика разделения. В стакан емкостью 100 мл помещают смесь
из 5—10 мл 0,5 М раствора Cu(N03b и 5—10 мл 0,2 М раствора
Pb(N03)2. К полученному раствору добавляют 5 г кристаллической
винной кислоты, перемешивают до растворения и приливают 20 мл
концентрированного раствора аммиака. Полученный раствор пропускают через
колонку, наполненную 15 г сильнокислотного катионита СДВ-3 в
Н-форме (способ подготовки катионита указан выше — см. стр. 307), со
скоростью 10 мл/мин. Для вымывания комплексных анионов свинца через
колонку пропускают около 200 мл раствора, полученного добавлением
к 180 мл дистиллированной воды 6 г винной кислоты и 20 мл
концентрированного раствора аммиака. Этим же раствором споласкивают 3 раза
стакан с анализируемым раствором и выливают промывную жидкость
в колонку. Промывание колонки проводят отдельными порциями по
10—15 мл раствора. Вытекающий раствор, содержащий тартратные
комплексные анионы свинца, собирают в стакан емкостью 400 мл. Полноту
вымывания свинца из катионита проверяют реакцией с К2СГ2О7. Для
этого отбирают каплю вытекающего из колонки раствора на часовое
стекло или в пробирку, добавляют каплю раствора CH3COONa и 2 капли
раствора К2О2О7. В присутствии РЬ++-ионов выпадает желтый осадок
РЬСЮ4.
Определение свинца. Содержание свинца в растворе определяют
иодометрическим методом. Для этого к раствору, собранному в стакан,
приливают 20 мл 2 н. раствора ацетата натрия, 10 мл 2 н. уксусной
кислоты и нагревают до кипения, прибавляют 20 мл 10%-ного раствора
К2СГ2О7, кипятят несколько минут и оставляют стоять в течение 1 ч на
горячей водяной бане. Далее осадок отфильтровывают через плотный
фильтр (или через стеклянный фильтрующий тигель № 4), промывают
горячей водой, подкисленной уксусной кислотой, до полного
обесцвечивания. Осадок хромата свинца растворяют на фильтре в 150 мл горячего
раствора «хлоридной смеси»*, прибавляемой порциями по 5—10 мл
(каждой порции дают стечь, прежде чем прибавлять новую). Фильтрат
собирают в мерную колбу емкостью 200—250 мл, раствор охлаждают и
доводят до метки «хлоридной смесью». Отбирают в коническую колбу
пипеткой 25 мл полученного раствора, прибавляют 3 г К1,или
соответствующее количество раствора иодида и титруют выделившийся иод
0,05 н. раствором тиосульфата натрия до слабо-желтой окраски, затем
вводят 2—3 мл раствора крахмала и продолжают титрование до
перехода окраски из синей в зеленую.
Расчет. Содержание свинца вычисляют (в мг) по формуле,
аналогичной формуле (16).
Определение меди. После вымывания ионов свинца пропускают
через катионит 20 мл дистиллированной воды и фильтрат выбрасывают.
Для извлечения ионов меди через катионит пропускают отдельными
порциями по 10—15 мл около 200 мл 2 н. раствора хлористоводородной
* «Хлоридная смесь» — к 1 л насыщенного при комнатной температуре раствора
хлорида натрия прибавляют 150 мл воды и 100 мл концентрированного раствора HCL
310
ГЛ. VIII. ХРОМАТОГРАФИЧЕСКИЕ МЕТОДЫ АНАЛИЗА
кислоты. Вытекающий из колонки раствор, содержащий катионы меди,
собирают в мерную колбу емкостью 250 мл. Полноту извлечения
катионов меди из катионита проверяют реакцией с K4[Fe(CN)6]. В
присутствии Си^-ионов выпадает красно-коричневый осадок Cu2[Fe(CN)^].
Содержание меди в растворе определяют иодометрическим или другим
методом. Для этого хлористоводородный раствор в мерной колбе
доводят до метки дистиллированной водой. Отбирают в коническую колбу
пипеткой аликвотдую часть полученного раствора, прибавляют 3 г ио-
дида кал»ия или соответствующее количество раствора иодида.
Выделившийся иод оттитровывакж 0,05 н. раствором Na2S20^
Расчет. Содержание меди вычисляют (в мг) по формуле,
аналогичной формуле (16)*
Разделение на катионите и определение ионов
цинка и железа (НЦ
Для разделения катионов цинка и железа (III) раствор,
содержащий смесь этих катионов, пропускают через катионит в Н-форме. При
этом происходит поглощение катионов. Затем катионит промывают
раствором щелочи. Катионы цинка образуют ZnOJT-ионы и проходят
в фильтрат, ионы железа (III) остаются на катионите. Железо
извлекают из катионита 2 н. раствором хлористоводородной кислоты.
Методика разделения. В стакан емкостью 100 мл помещают смесь
из 5—10 мл 0,25 М раствора ZnS04 и 5—10 мл 0,5 М раствора FeCl3.
Полученный раствор пропускают со скоростью 10 мл/мин через колонку,
содержащую 15 г сильнокислотного катионита СДВ-3 или КУ-2 в
Н-форме. Стакан споласкивают дистиллированной водой, полученный раствор
вливают в колонку и также пропускают через катионит. Для извлечения
катионов цинка пропускают через катионит отдельными порциями по
10—15 мл около 200 мл 10%-ного раствора NaOH. Вытекающий из
колонки раствор, содержащий ионы цинка, собирают в мерную колбу
емкостью 250 мл. Полноту извлечения ионов цинка из катионита
проверяют реакцией с K4[Fe(CN)6]. Для этого отбирают на часовое стекло
или в пробирку каплю вытекающего из колонки раствора, добавляют
2 капли 4 н. раствора НС1 и каплю раствора K4[Fe(CN)6]. В присутствии
Zn++ выпадает белый осадок Zn2[Fe(CN)6].
Определение цинка. Содержание цинка в полученном растворе
определяют комплексонометрическим методом. Для этого раствор в
мерной колбе доводят до метки дистиллированной водой. Отбирают в
коническую колбу пипеткой аликвотную часть полученного раствора,
добавляют 4 н. раствор НО до рН = 1 (проверяют по индикаторной бумаге)
для перевода ZnO<T в Zn++> приливают 25 мл аммиачной буферной
смеси, 3—5 капель хром темно-синего или эриохром черного Т и титруют
0,05 н. раствором комплексона III до изменения красной окраски
раствора на синюю.
Расчет. Содержание цинка вычисляют (в мг) по формуле,
аналогичной формуле (16).
Определение железа. После извлечения ионов цинка катионит
промывают 20 мл дистиллированной воды и фильтрат выбрасывают. Для
извлечения ионов железа (III) пропускают через катионит около 300 мл
2 н. раствора НО. Вытекающий из колонки раствор, содержащий ионы
железа (III), собирают в коническую колбу емкостью 500 мл. Полноту
извлечения ионов железа (III) проверяют реакцией с K4[Fe(CN)6].
В присутствии Ре+++-ионов выпадает синий осадок Fe4[Fe(CN)6]3. Затем
в колбу вносят несколько гранул металлического цинка или стружек,
§ 17. РАБОТЫ ПО МЕТОДУ ИОНООБМЕННОЙ ХРОМАТОГРАФИИ 311
закрывают пробкой, снабженной клапаном Бунзена, и нагревают на
водяной бане до полного растворения цинка. При этом Ре+++-ионы
восстанавливаются до Ре++-ионов — проверяют с K4[Fe(CN)6]. После
растворения цинка раствор охлаждают и количественно переносят в мерную
колбу емкостью 500 мл, доводят до метки дистиллированной водой.
Содержание ионов железа (II) в растворе определяют перманганатометри-
ческим методом. Для этого отбирают пипеткой аликвотную часть
полученного раствора, приливают 15 мл раствора Циммермана — Рейнгардта
для предотвращения окисления хлорид-ионов, затем смесь разбавляют
дистиллированной водой приблизительно до 100 мл и медленно титруют
0,05 н. раствором перманганата калия до появления розовой окраски, не
исчезающей в течение 1 мин.
Расчет. Содержание ионов железа (III) вычисляют (в мг) по
формуле, аналогичной формуле (16).
Содержание Fe+++ можно также определить фотоколориметрическим
методом (см. гл. VII).
Разделение на анионите и определение ионов
цинка и никеля
Для разделения катионов цинка и никеля используют способность
ионов цинка образовывать [2пС13]~-ионы, ионы никеля не образуют
таких комплексов. При пропускании через колонку с анионитом в С1-фор-
ме раствора, содержащего катионы никеля и комплексные анионы
[2пС1з]~, происходит поглощение последних, а ионы никеля остаются
в фильтрате. Поглощение хлоридного комплекса цинка анионитом
можно представить следующим уравнением:
R—Cl+[ZnCl3r —> R—[ZnCl3] + cr
Ионы цинка извлекают из анионита промыванием
дистиллированной водой, при этом хлоридный комплекс цинка разрушается и катионы
цинка проходят в фильтрат.
Методика разделения. В стакан емкостью 100 мл помещают смесь
из 5—10 мл 0,25 М раствора ZnS04n5—10 мл 0,25 М раствора NiS04.
Анализируемый раствор разбавляют 4 н. раствором НС1 так, чтобы
раствор стал 2 н. по НС1, при этом катионы цинка образуют хлоридные
комплексные анионы. Полученный раствор пропускают со скоростью
10 мл/мин через колонку с 15 г сильноосновного анионита АВ-17
в С1-форме, предварительно промытого 50 мл 2 н. раствора НС1 для
того, чтобы раствор в колонке был также 2 н. по НС1. Стакан
споласкивают 3 раза 2 н. раствором НС1, полученный раствор вливают в колонку
и пропускают через анионит.
Вытекающий из колонки раствор, содержащий катионы никеля,
собирают в мерную колбу емкостью 250 мл. Для вымывания из анионита
ионов никеля пропускают отдельными порциями по 10—15 мл около
200 мл 2 н. раствора НС1. Полноту вымывания катионов никеля
проверяют по реакции с диметилглиоксимом. Для этого отбирают на часовое
стекло или в пробирку каплю вытекающего из колонки раствора,
добавляют 2—3 капли раствора аммиака и каплю раствора диметилгли-
оксима. В присутствии Ni++ выпадает красный осадок диметилглиокси-
мата никеля.
Определение никеля. Содержание ионов никеля в солянокислом
растворе определяют комплексонометрическим методом. Для этого
раствор в мерной колбе доводят до метки дистиллированной водой,
отбирают в коническую колбу пипеткой аликвотную часть полученного
312
ГЛ. VIII. ХРОМАТОГРАФИЧЕСКИЕ МЕТОДЫ АНАЛИЗА
раствора, добавляют концентрированный раствор аммиака (для
нейтрализации НС1) до появления запаха, прибавляют примерно 0,1—0,2 г
мурексида (готовят тщательным растиранием в ступке 0,1 г индикатора
с 10 г NaCl x. ч.) и титруют 0,05 н. раствором комплексона III до
перехода желтой окраски раствора в фиолетовую.
Расчет. Содержание никеля вычисляют (в мг) по формуле,
аналогичной формуле (16).
Определение цинка. Для извлечения ионов цинка анионит
промывают 250 мл дистиллированной воды. Вытекающий из колонки раствор
собирают в мерную колбу емкостью 250 мл. Полноту извлечения ионов
цинка из анионита проверяют реакцией с K4[Fe(CN)6]. В присутствии
Zn++ выпадает белый осадок Zn2[Fe(CN)6]. Содержание ионов цинка
в полученном растворе определяют комплексонометрическим методом
(см. стр. 310).
§ 18. Работы по методу осадочной хроматографии
Количественное определение методом осадочной хроматографии
можно проводить по величине зоны хроматограммы (макрометод) или
по способу, основанному на использовании принципа предельного
разбавления (микрометод).
Определение ионов никеля по величине зоны хроматограммы
В основу количественного определения веществ по величине зоны
положена главная особенность осадочных хроматограмм, связанная
с равномерным распределением веществ в зоне, т. е. пропорциональная
зависимость между размерами зон осадочных
хроматограмм и концентрацией исследуемого
раствора.
Сущность указанного метода состоит в
том, что для каждого определяемого вещества
предварительно строят калибровочный
график зависимости величины зон хроматограмм
от концентрации растворов, а затем при тех
Объем зоны см3 же условиях получают осадочную хромато-
грамму анализируемого раствора и по калиб-
Рис 118. Кривые зависи- ровочному графику находят его концентрацию.
rp0a^r^„4H0LXP°ZoB Последняя может быть выражена в любых
от концентрации их в рас- единицах (рцс. 118).
творе. Калибровочный график. Для определения
ионов никеля в растворе по высоте зоны
хроматограммы предварительно строят кривую зависимости изменения
размеров зон осадочных хроматограмм от концентрации растворов соли
никеля.
Построение калибровочного графика проводят следующим образом.
В колонки с одинаковыми внутренними диаметрами или
калиброванными по объему вносят смесь осадителя и носителя. Смесь готовят
тщательным перемешиванием безводной окиси алюминия с диметилглиокси-
мом в сухом виде в соотношении 100 : 1. Колонки заполняют смесью на
2/з их высоты, как указано выше (см. § 17, стр. 306). Приготавливают
серию растворов соли никеля, содержащих от 0,01 до 0,5 г-экв/л, путем
последовательного разбавления дистиллированной водой 1 н. раствора
нитрата никеля, поддерживая постоянным значение рН раствора. В
колонки вносят по 0,2 мл приготовленных растворов. Через 3—5 мин после
§ 18. РАБОТЫ ПО МЕТОДУ ОСАДОЧНОЙ ХРОМАТОГРАФИИ
313
ТАБЛИЦА 11
Состав и приготовление колонки при определении неорганических ионов
методом осадочной хроматографии по величине зоны хроматограмм
Определяемый
элемент
Си11
Си11
или Fe
Fe111
Ni"
Са
РЬП
или Ag1
Со"
Hg1
Состав колонки
осадитель
(реагент)
Рубеано-
водородная
кислота
Гексациа-
ноферрат
(II) калия
Купферрн
Диметил-
глиоксим
Дигидро-
фосфат
натрия
Хромат
калия
а-Нитрозо-
Р-нафтол
Хлорид
натрия или
хромат
калия
носитель
Безводная
окись
алюминия
Окись
алюминия в
анионной
форме
Безводная
окись алю-
, миния
Безводная
окись
алюминия
Окись
алюминия в
анионной
форме
Окись алю-
1 миния в
анионной
форме
Безводная
окись
алюминия
Окись
алюминия в
анионной
форме
содержание
осадите л я в
смеси
1%
0,1 мг-экв
на 1 г
носителя
Насыщенный
спиртовой
раствор
1%
10% в
расчете
на
безводную
i соль
0,5 мг-экв
на 1 г
носителя
i%
0,5 мг-экв
на 1 г
носителя
Приготовление
кололки
Носитель
смешивают с
осадите лем в сухом
виде
Осадитель
вносят в виде
раствора, после
чего смесь
подсушивают на
воздухе
Осадитель
вводят в колонку
с носителем
перед
определением
Носитель
смешивают с оса-
дителем в
сухом виде
Осадитель и
носитель
смешивают в сухом
виде.
Можно
осадитель
растворить в воде и
внести в
носитель. Смесь
подсушивают
при
перемешивании
То же
Осадитель
вводят в носитель
в виде
насыщенного
спиртового
раствора перед
определен и ем
Осадитель и
носитель
смешивают в сухом
виде или
осадитель вводят
в виде
раствора и смесь
подсушивают при
перемешивании
Окраска
зоны
Черная
Бордовая
(медь),
синяя
(железо)
Коричневая
Розовая
Розовая
зона в
присутствии
мурек-
сида
Желтая
| (свинец),
темно-
красная
(серебро)
Бордовая
Фиолетовая
зона в
присутствии
[дифенил-
карба-
зона
Примечание
Индикатор
вносят в
произвольном
количестве в
исследуемый
раствор
перед хрома-
тографиро-
ванием
Индикатор
вносят в
произвольном
количестве в
исследуемый
раствор
перед хрома-
тографи-
рованием
314
ГЛ. VIII. ХРОМАТОГРАФИЧЕСКИЕ МЕТОДЫ АНАЛИЗА
полного впитывания растворов проводят измерение высоты зоны ди-
метилглиоксимата никеля (мм) или ее объема (см3). Для каждой
концентрации проводят-три-четыре параллельных опыта.
Затем строях калибровочный график (см. рис. 118).
Методика определения. Для определения ионов никеля в растворе
неизвестной концентрации готовят три-четыре колонки, как указано
выше. Вносят в колонки по 0,2 мл исследуемого раствора соли никеля.
По истечении 3—5 мин проводят измерение зон хроматограмм. На
основании средней величины зоны по предварительно построенному
графику находят концентрацию исследуемого вещества в растворе.
Примечание. При использовании некалиброванных колонок необходимо
строго следить, чтобы внутренние диаметры их' были одинаковы.
Условия определения для некоторых других неорганических ионов
приведены в табл. 11.
Определение микроколичеств ионов меди
Микроколичественное определение веществ в растворах проводят
по способу предельного разбавления, применяя соответствующую
качественную реакцию. Предварительно определяют тот предел
концентрации, при котором хроматографируемое вещество практически уже не
может быть обнаружено методом осадочной хроматографии. Затем при
тех же условиях опыта исследуют анализируемый раствор и
определяют предел разбавления раствора, при котором вещество может быть
обнаружено методом осадочной хроматографии, после чего
рассчитывают по формуле концентрацию веществ в растворе.
Методика определения. Приготавливают серию колонок
одинакового диаметра (около 4—5 мм) со смесью, состоящей из безводной
окиси алюминия и руб^ановодородной кислоты (в весовом отношении
100:1). Колонку заполняют смесью на половину ее высоты, как
указано выше. Серию стандартных растворов соли нитрата меди готовят
путем последовательного разбавления 0,0005 н. раствора Cu(N03)2
дистиллированной или деионизированной водой, как указано ниже.
(Концентрацию меди в первоначальном растворе определяют
предварительно иодометрическим методом.)
В ряд колонок вносят калиброванной пипеткой точно по 0,2 мл
стандартных растворов последовательно, по мере их разбавления. В
колонке образуется серо-черная зона комплексной соли рубеаната меди.
Разбавление и исследование растворов повторяют до получения
отрицательной реакции (одновременно не менее двух параллельных
определений). Зная исходную концентрацию раствора (С) и степень
разбавления (/г), определяют предельную концентрацию исходного
раствора соли меди (В), которая может быть вычислена по формуле:
П
После этого определяют содержание соли меди в растворе
неизвестной концентрации. Исследуемый раствор также последовательно
разбавляют и исследуют методом осадочной хроматографии до
получения отрицательного результата.
Концентрацию меди в исследуемом растворе (Q) определяют по
формуле:
где Q — концентрация исследуемого раствора;
В — предельно обнаруживаемая концентрация, определенная, как указано выше;
»1 — степень разбавления раствора соли меди неизвестной концентрации.
§ 19. РАБОТЫ ПО МЕТОДУ ОКИСЛИТЕЛЬНО-ВОССТАНОВИТЕЛЬНОЙ ХР0МАТ0ГРАФИИ315
ТАБЛИЦА 11
Приготовление разбавленных растворов из 1 мл 0,0005 н. исходного раствора
пробирки
1
2
3
и т. д.
Объем
добавленной ВОДЫ, МЛ
1
2
3
и т. д.
Степень
разбавления раствора (п)
2
3
4
и т. д.
Концентрация (Q) разбавленного
раствора, г-экв/л
Q = 0,0005 : 2 = 0,000250
Q = 0,0005:3 = 0,000166
Q = 0,0005: 4 = 0,000125
и т. д.
Концентрация меди будет выражена в тех же единицах, в которых
была определена предельная концентрация. Присутствие других ионов,
за исключением серебра и ртути (I), обнаружению меди не мешает.
Техника разбавления раствора. Предельно разбавленный
раствор получают путем последовательного разбавления исследуемого
раствора чистым растворителем. Берут 10 пробирок и вносят в каждую
по 1 мл исследуемого раствора соли заданной концентрации (С),
например 0,0005 н., затем в пробирки вносят определенное количество
воды (табл. 12).
Концентрирование разбавленных растворов солей меди
При пропускании концентрируемого раствора через осадочно-хро-
матографическую колонку в результате взаимодействия содержащихся
в нем ионов с осадителем образуются малорастворимые осадки. При
последующем промывании колонки небольшим объемом растворителя
поглощенные ионы извлекаются, и вновь полученный раствор по
отношению к данному иону или группе ионов оказывается более
концентрированным, чем исходный.
Методика определения. Колонку высотой 200 мм с внутренним
диаметром 20 мм заполняют 10 г смеси безводной окиси алюминия и едкого
натра. Смесь готовят смешиванием 10 г едкого натри и 100 г безводной
окиси алюминия. Через приготовленную колонку пропускают 1 л
10~4 М раствора соли меди. Катионы меди с NaOH дают осадок
гидроокиси меди и образуют вверху колонки зону голубого цвета.
Скорость протекания раствора через колонку составляет 2—
3 мл/мин. Ионы меди из колонки извлекают путем промывания ISO-
ISO мл 2 н. раствора H2S04. Вытекающий из колонки раствор
собирают отдельными порциями по 25 мл, в которых определяют содержание
меди иодометрическим методом (см. книга 2, гл. III, § 37).
Расчет. Общее содержание меди {g) в полученном растворе
вычисляют (в мг) по формуле:
п
8 ** 2 3Cu^Na2S203^Na2Si203
1
где « — число порций раствора соли меди (по 25 мл), собранных дли титрования.
§ 19. Работы по методу окислительно-восстановительной
хроматографии
Обнаружение иодид- и бромид-ионов
при совместном присутствии
Для разделения иодид- и бромид-ионов и их количественного
определения используют различную величину
окислительно-восстановительных потенциалов их ред-окс систем. Иодид-ионы (потенциал 0,54 в)
окисляются ранее, чем бромид-ионы (потенциал 1,06 в). Выделившийся
316
ГЛ. VIII. ХРОМАТОГРАФИЧЕСКИЕ МЕТОДЫ АНАЛИЗА
иод располагается в верхней части колонки, образуя бурую зону.
Бромид-ионы окисляются до брома, зона которого располагается ниже и
может быть обнаружена с помощью проявителя. Для идентификации
иода применяют крахмал.
Методика определения. Готовят смесь, состоящую из окиси
алюминия в анионной форме, периодата калия и крахмала, в соотношении
10:1:3. Смесь тщательно перемешивают, помещают в колонки на
2/з высоты, уплотняют и вносят исследуемый раствор. В первую
колонку ввЬдят 1—2 капли 0,01 н. раствора KI. При этом наблюдается
образование синей зоны. Во вторую колонку вносят 2—3 капли 0,1 н.
раствора КВг и после полного его впитывания промывают 1—2 каплями
2 н. раствора H2S04. Затем вводят 2—3 капли проявителя —
насыщенного спиртового раствора флуоресцеина. Образуется красно-оранжевая
зона. В третью колонку вносят 2—3 капли смеси растворов KI и КВг
в тех же концентрациях и хроматограмму промывают 2 н. раствором
H2S04.
Вверху образуется синяя зона 12. После чего через хроматограмму
пропускают 5—6 капель раствора флуоресцеина. Ниже синей зоны
образуется красно-оранжевая зона, свойственная соединению брома с
флуоресцеином.
§ 20. Работы по методу адсорбционно-комплексообразовательной
хроматографии
Разделение и определение ионов никеля и цинка,
ниобия и тантала
Для разделения неорганических ионов в качестве сорбента
применяют уголь марки ДАУХ, насыщенный комплексоообразующим
агентом диметилглиоксимом, фениларсоновой кислотой и т. п. Ионы,
образующие комплексные соединения с комплексообразующим веществом,
задерживаются на колонке, другие же собирают в вытекающем из
колонки растворе. Задержанные в колонке ионы вымывают
соответствующим растворителем и в полученном растворе определяют их
концентрацию.
Методика определения. Разделение и определение
никеля и цинка. Уголь марки ДАУХ отмывают от золы горячим, а
затем холодным 5—10%-ным раствором НС1. Промывание кислотой ведут
до полного удаления ионов железа (III) — проба на K4[Fe(CN)6]. После
этого уголь промывают водой до нейтральной реакции (проба с
метиловым оранжевым) или до удаления хлорид-ионов [проба с Hg2(N03h]
и высушивают. Подготовленный уголь смешивают с диметилглиоксимом
(10% по отношению к массе угля), приливают воду для получения
кашицеобразной массы и нагревают на водяной бане при 70° С,
периодически помешивая в течение 1 ч, пока не исчезнут белые крупинки
диметилглиоксима. Затем 3 г кашицеобразного угля подогревают на
водяной бане при 70° С в течение 10 мин и вводят в колонку (диаметр
10 мм). Высота сорбента в такой колонке должна быть 23 см.
Готовят раствор сульфатов цинка и никеля с содержанием в 100 ли
10 г ZnS04 и 15 мг NiS04.
Из колонки с сорбентом выливают избыток воды и, когда над
поверхностью угля высота столба жидкости будет равна приблизительно
1 мм, в колонку вводят приготовленный раствор. В фильтрате
периодически проверяют полноту поглощения никеля (проба с
диметилглиоксимом). При появлении никеля в фильтрате прекращают приливание
§ 20, РАБОТЫ ПО МЕТОДУ КОМПЛЕКСООБРАЗОВАТЕЛЬНОЙ ХРОМАТОГРАФИИ 317
анализируемого раствора, fe тот момент, когда слой раствора над
поверхностью угля не будет превышать 1 мм, в колонку вводят воду. При
промывании водой ионы цинка удаляются из колонки, и их собирают в
отдельный приемник. Промывание проводят до полного удаления из
колонки ионов цинка — проба с (NH4)2[Hg(SCN)4]. В полученном
растворе определяют содержание Zn++ комплексонометрическим методом.
После удаления цинка извлекают из колонки никель путем промывания
5%-ным раствором НС1. Пробу на полноту извлечения никеля
проводят с диметилглиоксимом. Затем в растворе определяют никель
комплексонометрическим методом.
Разделение и определение ниобия и тантала. Для
разделения этих ионов в качестве комплексообразующего реагента
используют фениларсоновую кислоту, а в качестве носителя — уголь марки
ДАУХ, предварительно насыщенный реагентом. Раствор солей ниобия
и тантала, в котором содержание Nb и Та в пересчете на металл
должно составлять по 0,25 мг/мл, пропускают через колонку. Ниобий
проходит в фильтрат, в котором его и определяют.
Тантал задерживается на колонке и при этом количественно
отделяется от ниобия. Вымывание тантала проводят 7%-ным раствором
щавелевой кислоты при 95° С. Процесс подготовки веществ для анализа и
их разделения весьма трудоемок и длителен и поэтому здесь не
описывается.
ГЛАВА IX
РАДИОМЕТРИЧЕСКИЕ МЕТОДЫ АНАЛИЗА
Радиоактивные изотопы широко применяются в химическом
анализе. С помощью радиоактивных реагентов проводится прямое
определение радиоактивных изотопов методом осаждения, радиометрическое
титрование, анализ методом изотопного разбавления, кроме того,
применение радиоактивных изотопов дает возможность использовать ряд
физических методов анализа, основанных на поглощении, отражении
радиоактивного излучения и возникновении вторичного излучения, а также
проводить так называемый активационный анализ.
Каждый радиометрический метод имеет свои специфические
особенности и аппаратуру. Общим для всех методов является
использование радиоактивных изотопов, которое требует знания их основных
свойств и техники безопасности применения, а также необходимость
измерения радиоактивности и, следовательно, умения применять
соответствующую аппаратуру.
Радиоактивные изотопы используются также для установления
полноты и чистоты разделения многокомпонентных смесей. Контроль
разделения при электрофорезе, электролизе, хроматографии, экстракции,
разгонке и т. п. легко осуществляется с помощью радиоактивных
изотопов, входящих в состав компонентов смеси. При этом можно
установить загрязнение одной фракции разделенной смеси другим
компонентом и степень разделения вещества, а следовательно, и проверить
методику обычного химического анализа.
А. ТЕОРЕТИЧЕСКИЕ ОСНОВЫ РАДИОМЕТРИЧЕСКИХ МЕТОДОВ
§ 1. Виды радиоактивного распада
Самопроизвольное превращение одних атомных ядер в другие
(радиоактивный распад) происходит путем испускания а-, р~-, р+-частиц*,
Y-лучей или захватом ядром электрона /С-, а иногда L- и Af-оболочек
атома.
Y-Лучи являются результатом перехода ядра атома данного
элемента с одного возбужденного уровня энергии на другой или в
невозбужденное состояние.
Если возбужденное ядро имеет небольшую энергию, то его переход
в невозбужденное состояние может происходить излучением фотона
или электрона конверсии. Электрон конверсии срывается с /(-оболочки
атома. Вследствие этого у*излУчение низкой энергии сопровождается
* ос-Частицы — ядра атомов гелия, Р"-частицы — электроны, р+-частицы —
позитроны.
5 2. СКОРОСТЬ РАСПАДА
319
испусканием электронов конверсии. Переход электронов с высших на
освободившуюся внутреннюю оболочку атома вызывает
характеристическое рентгеновское излучение элемента, испускающего у-лУчи-
При а-распаде ядро атома изотопа материнского элемента
переходит в ядро атома изотопа дочернего элемента, стоящего в
периодической системе на две клетки влево от материнского элемента. Если
переход совершается на возбужденный уровень энергии ядра изотопа
дочернего элемента, то за излучением а-частицы следует испускание одного
или нескольких фотонов с энергией, в сумме равной разности
уровней энергии возбужденного и невозбужденного ядер. ос-Частицы,
испускаемые при распаде ядер материнского изотопа, вследствие перехода
на дискретные уровни энергии ядра дочернего элемента имеют
дискретный (линейчатый) спектр по энергии.
Р-Распад ядра атома изотопа материнского элемента ведет к
образованию ядра атома изотопа дочернего элемента, стоящего в
периодической системе на одну клетку вправо (р~) или влево (р+) от
материнского элемента. Он также часто сопровождается образованием ядра
атома дочернего изотопа в возбужденном состоянии и, следовательно,
-у-излучением.
Спектр энергий р-частиц данного радиоактивного изотопа
непрерывный и изменяется от нулевой до некоторой максимальной энергии,
соответствующей разности энергий ядер атомов материнского и
дочернего изотопов в одном из возможных его энергетических состояний.
Отсутствие постоянства энергии р-частиц связано с механизмом их
испускания. Ядро состоит из нейтронов и протонов, появление р-частиц
(электронов или позитронов) связано с переходом в ядре нейтрона (п) в
протон (р) или протона в нейтрон:
п —> /? + fT + v
р —> « + p+ + v
где v — нейтрино.
Энергия радиоактивного распада распределяется статистически
между р-частицей и нейтрино, и только при нулевой энергии нейтрино
р-частица имеет максимальную энергию.
Электронный захват ведет к переходу ядра атома изотопа
материнского элемента в ядро атома изотопа дочернего элемента в одном из
его энергетических состояний, стоящего в периодической системе на
одну клетку влево от материнского элемента. Электронный захват
сопровождается характеристическим рентгеновским излучением дочернего
элемента, а в ряде случаев — у"излУчением-
При распаде одного ядра атома данного изотопа может
испускаться только одна ос- или р-частица и несколько ^-квантов, несколько
электронов конверсии и квантов рентгеновского излучения. Это необхо-*
димо учитывать при определении количества распавшихся ядер по
зарегистрированному числу -у-квантов.
§ 2. Скорость распада
Скорость распада индивидуального радиоактивного изотопа
подчиняется закону, по которому число радиоактивных ядер, распадающихся
за бесконечно малый промежуток времени, пропорционально числу
наличных радиоактивных ядер:
-™L = XNt
(1)
320
ГЛ. IX. РАДИОМЕТРИЧЕСКИЕ МЕТОДЫ АНАЛИЗА
где Nf — число радиоактивных ядер, имеющихся в данный момент времени t;
Я —радиоактивная постоянная (константа распада) данного вида ядер (сект1),
т. е. доля ядер, распадающихся за единицу времени, при условии, что
средняя продолжительность жизни ядер велика по сравнению с выбранной
единицей времени.
Интегрирование этого выражения приводит к экспоненциальной
зависимости изменения числа радиоактивных ядер во времени:
Nt = N0e-u (2)
где iVo — начальное количество радиоактивных ядер.
Число ядер (М), распадающихся за время t, равно разности
начального числа ядер и имеющихся в момент времени t:
М = N0 - N0e~Kt - No (l - е~и) (3)
Скорость распада равна:
Л=^=^(1-е-«) (4)
Число актов распада ядер радиоактивного вещества в единицу
времени (скорость распада)—Л называют радиоактивностью
(активностью) .
За единицу активности принято кюри — активность источника,
в котором происходит 3,7-1010 распадов в 1 сек. Производные от кюри
единицы: милликюри (мкюри = Ю-3 кюри), микрокюри (мккюри —
= 10-6 кюри), нонакюри (нкюри = 10~9 кюри), пикокюри (пкюри =
= 10~12 кюри), мегакюри (Мкюри = 106 кюри), килокюри (ккюри —
= 103 кюри). В международной системе СИ за единицу радиоактивности
принят распад в 1 сек (1/сек).
Скорость радиоактивного распада, отнесенная к единице массы или
объема радиоактивного вещества, называется удельной активностью.
Удельная активность обычно выражается в кюри/г, кюри/мл или
кратных кюри единицах на единицу массы или объема вещества.
Скорость радиоактивного распада характеризуется также
периодом полураспада (Тг/2), который представляет собою промежуток
времени, в течение которого распадается половина взятого числа
атомов, т. е.
'No 2
Следовательно [см. уравнение (2)]
е
щ2=
ТЧ2-
-м«
:1П2 =
In 2
X
Y
= 0,693
0,693
X
и
т. е.
Убыль активности во времени также подчиняется основному закону
радиоактивного распада. Это ясно, если уравнение (2) умножить на X
и принять во внимание уравнение (1), по которому скорость распада
равна XN:
At = Aoe~u
где А0 и At — соответственно начальная и активность через промежуток времени L
§ 2. СКОРОСТЬ РАСПАДА
321
При регистрации радиоактивного излучения в большинстве случаев
определяют величину, пропорциональную активности — регистрируемую
активность I:
7-фЛ (5)
где ф — коэффициент, учитывающий все возможные потери излучения при регистрации.
При измерениях на счетчике Гейгера — Мюллера или сцинтилля-»
ционном счетчике регистрируемая активность выражается в импульсах
в 1 сек (ими) сек), поэтому ее часто называют скоростью счета.
Удельная активность в этом случае также может быть выражена
в имп/(сеК'Мл) или имп/(сек*г).
2,2Г
О 2 Ь 6 в 10 12 ^
время, ч
Рис. 119. Графическое определение
периода полураспада
индивидуального радиоактивного изотопа.
12 16
Время, ч
Рис. 120. Графический анализ кривой распада
смеси двух генетически несвязанных
радиоактивных изотопов:
7 —суммарная кривая; 2— прямая, характеризующая
скорость распада долгоживущего изотопа; 3 — прямая,
характеризующая скорость распада короткоживущего
изотопа.
Следовательно, регистрируемая активность изменяется во времени
по уравнению:
¦•10е
-и
(6)
где /о и // — активность, зарегистрированная в начальный момент времени и через
промежуток времени t.
Изменение во времени числа радиоактивных атомов, активности
и регистрируемой активности описывается уравнениями прямых:
lg At=lgA0-
•0,4343М ]
-0,4343*,/ }
(7)
lg // = lg /о - 0,4343Я/
Графически выражения (7) могут быть представлены в
полулогарифмических координатах в виде прямой (рис. 119). Если на ординате
графика наметить две точки, различающиеся по активности вдвое, т. е.
на lg 2^0,3, то, проведя линии, параллельные оси абсцисс, до
пересечения с прямой и опустив из полученных точек перпендикуляры на ось
абсцисс, получим отрезок, равный периоду полураспада данного
радиоактивного изотопа (Ту2).
Период полураспада также может быть найден аналитически по
уравнению (7).
322
ГЛ. IX. РАДИОМЕТРИЧЕСКИЕ МЕТОДЫ АНАЛИЗА
Очень большие периоды полураспада определяются путем
измерения абсолютной активности (А) известного количества изотопа:
1 Чг - А (8)
где N — число атомов взятого изотопа.
Изменение активности во времени при радиоактивном распаде
смеси двух генетически несвязанных изотопов в полулогарифмических
координатах выражается кривой. Если эти изотопы имеют сильно
отличающиеся периоды полураспада, то кривую легко разложить на две
пересекающиеся прямые, каждая из которых характеризует скорость
распада одного радиоактивного изотопа (рис. 120).
Накопление дочернего радиоактивного изотопа при распаде
материнского можно выразить дифференциальным уравнением, в котором
первый член правой части равенства представляет собой скорость
распада материнского изотопа, а второй—скорость распада образующегося
дочернего изотопа:
^±=XlNl^X2N2 (9)
где М{ н N2 — соответственно число ядер атомов материнского и дочернего изотопов.
Решение этого уравнения при условии, что в начальный момент
времени дочерний изотоп отсутствует — N2, о = 0 и h <С Я2, т. е. скорость
распада материнского изотопа практически постоянна, приводит к
выражению:
tf2eML(i_e-W) (10)
i\»2
Уравнение (10) применяют также для расчета накопления
радиоактивного изотопа при постоянной скорости его образования по ядерной
реакции. В этом случае вместо произведения *k\N\ — скорости
образования дочернего радиоактивного изотопа — используют величину Q,
характеризующую скорость образования изотопа по ядерной реакции:
JV-.3 (1-в-М) (11)
где N — число образовавшихся радиоактивных атомов;
Q- FLa
F — поток частиц (сек'1 • см~2); L — число частиц в мишени; сг — коэффициент
пропорциональности, имеющий размерность площади (см2), который называют
эффективным сечением ядерной реакции. (Он характеризует вероятность
протекания реакции при падении одной ядерной частицы на тонкую мишень
площадью 1 см2);
Я — константа распада радиоактивного изотопа.
При /->оо уравнение (10) переходит в следующее соотношение:
М/-, = Я2ЛГ2 (12)
Это выражение носит название векового равновесия.
При соизмеримых периодах полураспада, но больших для
материнского изотопа, уравнение (9) при #2, о = 0 и Кх^СХц имеет вид:
N2
*l5!i-(е-V _*-**)
При достаточно больших значениях t величиной е можно прене-
-Kit
бречь, так как она мала по сравнению се , и считать отношение чисел
атомов дочернего и материнского изотопов постоянным. Однако это
§ 3. ВЗАИМОДЕЙСТВИЕ ИЗЛУЧЕНИЯ С ВЕЩЕСТВОМ
323
отношение уменьшается со скоростью, соответствующей скорости
распада материнского изотопа:
Ni Х2 — М
Уравнение (13) характеризует так называемое подвижное
радиоактивное равновесие.
§ 3. Взаимодействие излучения с веществом
При прохождении излучения радиоактивных изотопов через
вещество энергия излучения расходуется главным образом на ионизацию и
возбуждение атомов и молекул вещества.
а-Частицы характеризуются сильным ионизирующим действием и
малым пробегом (длиной траектории в данном веществе). Траектории
а-частиц прямолинейны. Неоднородность поглощающего вещества
приводит к некоторому разбросу величин их пробега при одинаковой
энергии. Пробеги а-частиц в воздухе составляют несколько сантиметров.
Даже тонкая металлическая фольга полностью поглощает ос-частицы.
р-Лучи оказывают меньшее ионизирующее действие, и их пробег
в веществе значительно больше. Траектории р-частиц вследствие
рассеивания не прямолинейны. Максимальной энергии р-частиц
соответствует максимальный пробег их в веществе.
Ослабление потока р-частиц в веществе приближенно подчиняется
экспоненциальному закону:
/-/ов""1** (14)
где /0 и /—число Р-частиц, соответственно падающих на поглотитель и прошедших
через слой поглотителя толщиной х, см;
jx— линейный коэффициент ослабления, см~К
Коэффициент jm зависит от максимальной энергии р-лучей и свойств
поглощающего материала. Так как основные потери энергии р-части-
цами происходят при их взаимодействии с электронами среды, то |л = ятг,
где п — число электронов в 1 см3 вещества; к — константа, не
зависящая от природы вещества.
Величина п описывается уравнением:
NApz
- АН (15)
где
Р-
Z —
Ав-
• число Авогадро;
¦ плотность вещества;
заряд ядра атома;
• атомный вес.
Следовательно;
"~ Ав
NApz
11 = К. :
(161
Для большинства химических элементов z/AB ^ 0,5, поэтому ji/p
является приблизительно постоянной величиной для р-излучения
данной энергии (jx/p = \ia — называют массовым коэффициентом
ослабления, см2/г). Закон ослабления р-излучения при введении \ха принимает
вид:
/ = /0<Г^ (17)
где d — толщина слоя поглотителя, е[см2.
Закон ослабления является приближенным, поэтому для
нахождения пробега р-частиц в веществе определяют изменение активности
324
ГЛ. IX. РАДИОМЕТРИЧЕСКИЕ МЕТОДЫ АНАЛИЗА
препарата р-излучателя с изменением толщины поглотителя до тех пор,
пока она не сравняется с величиной фона. Затем строят график в
координатах логарифм активности — толщина слоя и по нему находят
максимальный пробег.
Эмпирически найдена зависимость максимального пробега р-лучей
Ямакс (г/см2) от их энергии ?Макс (Мэв):
для ?Макс<0,8 Мэв
*макс = - 0Л1 + ^0,012 +0,27?*акс (18а)
и для EHUKt > 0,8 Мэв
Ямакс = 0,542?макс - 0,133 (186)
Максимальный пробег можно также найти измерением слоя
вещества, ослабляющего интенсивность р-излучения вдвое (dy2):
Зная величину di/2, можно также найти \ia из соотношения:
*А = 0,693/|1в
При прохождении у-лучей через вещество может происходить
полная передача энергии -у-квантов электронам внешних оболочек атомов
(фотоэффект), частичная передача энергии у-квантов электронам (комп-
тон-эффект) и аннигиляция Y-KBaHT0B (образование пар электрон —
позитрон). Первые два процесса непосредственно приводят к
ионизации атомов и молекул, последний процесс связан с ионизацией среды
образовавшимися электронами и позитронами. Ионизационный эффект
от у-лучей сравнительно мал, а их проникающая способность велика.
Ослабление потока ^-квантов точно отвечает экспоненциальной
зависимости [формулы (14) и (17)Ь Коэффициент ослабления ^-излучения
является суммой коэффициентов ослабления вследствие фотоэффекта,
комптоновского рассеяния и образования пар.
§ 4. Ошибки при измерении радиоактивности
При измерении радиоактивности возможны систематические и
случайные ошибки. Первые связаны с неточностью работы аппаратуры,
методов взятия проб, приготовления образцов для измерения и т* п. Эти
ошибки подлежат устранению.
Случайные ошибки, обязательные при измерении любой величины,
связаны с чувствительностью метода и аппаратуры. Наряду с обычными
случайными ошибками, при измерении радиоактивности имеют место
случайные ошибки, обусловленные статистической природой
радиоактивного распада. Практически можно найти лишь суммарную ошибку,
связанную со статистическим характером радиоактивного распада и
пределами точности аппаратуры.
Обычно оценку точности измерений радиоактивности проводят
исходя только из статистического характера радиоактивного распада,
т. е. оценивают предел точности измерения (или величину суммарной
ошибки).
Разброс результатов измерения числа распадов характеризуется
величиной ожидаемого стандартного отклонения от среднего значения.
Квадрат ожидаемого отклонения (дисперсия) выражается следующим
уравнением;
°"=42><-^)2 (19)
§ 4. ОШИБКИ ПРИ ИЗМЕРЕНИИ РАДИОАКТИВНОСТИ 325
где п — число измерений;
Яг — измеренная величина;
Р — среднее значение измеренных величин.
Отсюда среднее квадратичное отклонение:
**-1/4~/~1><-р)2
(20)
В случае, если число измерений меньше 20, вместо величины п
лучше принимать число измерений на единицу меньше действительного,
т. е. п — 1.
Для характеристики меры точности измерения активности
пользуются также так называемым абсолютным статистическим
отклонением ДР:
др= у р « у рг (21)
Абсолютное статистическое отклонение величин регистрируемой
активности Д/ может быть найдено, если принять во внимание, что
АР = Ш (22)
где t — продолжительность отдельного радиометрического измерения.
Из соотношений (21) и (22) следует:
"-/i-/^
(23)
Средняя квадратичная ошибка величин регистрируемой активности
равна:
В этом случае также при числе измерений меньше 20 вместо п берется
п — 1.
Приближенная оценка абсолютного статистического отклонения
отдельного измерения может быть проведена по формуле (23). Для
определения а/ необходимо провести не менее пяти измерений.
Результат измерения указывается в виде среднего арифметического.
При этом отклонение от среднего арифметического должно быть в УТГраз
меньше ошибки отдельного измерения, т. е.
Средняя квадратичная ошибка среднего арифметического:
Относительную точность б находят делением абсолютной точности
(абсолютного статистическбго отклонения) на среднее арифметическое:
6 e &J_ e &JP_ в 1 ^ _1 I 1
7 р УЪ VT ~ V17 ~ Vp~i
Измерение радиоактивности образца заключается в
последовательном измерении радиоактивности образца с фоном и одного фона.
326
ГЛ. IX. РАДИОМЕТРИЧЕСКИЕ МЕТОДЫ АНАЛИЗА
Ошибка определения радиоактивности образца без фона
складывается из ошибок измерения образца с фоном и фона:
ДГ= V№ + W-\/'±+-^ (28)
а-]Лос)2 + (аф)2 (29)
Индексы с и ф использованы для обозначения величин, относящихся
к образцу с фоном и фону.
Оптимальная продолжительность измерения образца и фона
определяется выражениями (30) и (31).
б2/2
б2/2
"А- pj2 (30)
(31)
где б — заданная величина относительного статистического отклонения в долях
единицы;
/ — разность /с — /ф, т. е. регистрируемая активность препарата без фона.
Б. ЭЛЕМЕНТЫ ТЕХНИКИ БЕЗОПАСНОСТИ ПРИ РАБОТЕ
С РАДИОАКТИВНЫМИ ВЕЩЕСТВАМИ
§ 5. Понятия и единицы измерения
Радиоактивное излучение в дозах, превышающих предельно
допустимые, вредно действует на организм людей. В связи с этим при
использовании радиометрических методов анализа необходимо точно и
неукоснительно соблюдать правила техники работы с радиоактивными
веществами.
Понятия доза и мощность дозы введены для определения величины
эффекта, производимого ионизирующим излучением.
Для оценки действия рентгеновских или ^-лучей в воздухе введено
понятие экспозиционная доза.
Экспозиционная доза — энергия рентгеновского или у-излучения,
израсходованного на ионизацию воздуха, она измеряется в
рентгенах (р).
Рентген — доза рентгеновского или у-йзлучейия в воздухе, при
которой сопряженная корпускулярная эмиссия в 1 смъ при 0° С и
760 мм рт. ст. производит ионы, несущие 1 CGSE электричества каждого
знака. В единицах СИ 1 р =* 2,57976-10~4 к/кг.
Для количественной оценки действия любого ионизирующего
излучения в любом облучаемом веществе введено понятие поглощенной дозы.
Поглощенная доза — энергия ионизирующего излучения,
поглощенная в единице массы облучаемого вещества. Измерения поглощенной
дозы ведутся в радах.
Рад — поглощенная доза излучения, равная 100 эрг/г облучаемого
вещества. В единицах СИ 1 рад = 10~2 дж/кг.
Измерять поглощенную энергию трудно, поэтому чаще в
практике в качестве единицы измерения применяется рентген. При
этом поглощенная доза приближенно может быть рассчитана по
формуле:
Dn = 0,88?э -?-
где ?>п— поглощенная доза;
?)э — Доза, поглощенная воздухом;
jx и ц-о — коэффициенты истинного поглощения у-пучъ& в веществе и воздухе.
§ б.. КЛАССИФИКАЦИЯ РАДИОАКТИВНЫХ ИЗОТОПОВ
327
В живой ткани одинаковые количества энергии различных видов
излучения оказывают различное биологическое действие, поэтому введены
понятия биологического эквивалента рентгена (бэр) и биологического
эквивалента рада (бэрад).
Биологический эквивалент рентгена — количество энергии,
поглощенной живой тканью и вызвавшее такой же биологический эффект,
как поглощенная доза в 1 рентген рентгеновских или у-лучей.
Мощность дозы — доза в единицу времени.
Предельно допустимая доза — доза, систематическое облучение
которой в течение многих лет не должно вызывать у человека
необратимых изменений в организме в течение всей его жизни.
В настоящее время в СССР установлена предельно допустимая
доза облучения 0,1 бэр в неделю, 0,017 бэр в день и 5 бэр в год.
В работе при необходимости допускается однократная доза
внешнего облучения 3 бэр в любые 13 последовательных недель при условии,
что годовая доза не будет превышать 5 бэр. Для кистей рук предельно
допустимая доза устанавливается в 10 раз больше для (3-частиц и
в 5 раз больше для других видов излучения при условии, что на все
тело падает излучение, не превышающее предельно допустимой дозы.
Суммарная доза Dc за все время работы с радиоактивными
изотопами не должна превышать величины:
Z)C = 5(W- 18)
где N — возраст работающего, годы.
Облучение организма может происходить не только внешними
источниками излучения, но и при попадании радиоактивных веществ
внутрь организма. В связи с этим введены понятия и нормы предельно
допустимых концентраций (ПДК) радиоактивных изотопов в воздухе
рабочих помещений и воде.
§ 6. Классификация источников радиоактивного излучения
и радиоактивных изотопов
Источники радиоактивного излучения делят на закрытые и
открытые. Закрытые — должны быть герметичны. Открытые — любые
негерметичные источники излучения, которые могут создавать радиоактивное
загрязнение воздуха, аппаратуры, поверхностей столов, стен и т. п.
При работе с закрытыми источниками необходимые меры
предосторожности сводятся к предохранению от внешнего облучения.
Закрытые источники излучения активностью выше 0,2 г-экв радия
должны быть помещены в защитные устройства с дистанционным
управлением и устанавливаться в специально оборудованных помещениях.
При работе с закрытыми источниками меньшей активности следует
применять экраны, соответствующие по толщине и материалу роду и
энергии излучения радиоактивного источника, а также дистанционные
инструменты, применение которых должно снижать дозу до предельно
допустимой. Лаборатории при работе с закрытыми источниками могут
быть обычными.
При работе с открытыми источниками необходимо учитывать:
относительную радиотоксичность изотопа, которая зависит от его периода
полураспада, вида и энергии излучения; активность на рабочем месте;
физическое состояние вещества; особенность работы.
Для каждого радиоактивного изотопа установлена предельно
допустимая концентрация (ПДК) в воздухе рабочих помещений *.
* Санитарные правила работы с радиоактивными веществами и источниками
ионизирующих лучей, Гссатомиздат, 1963.
328
ГЛ. IX. РАДИОМЕТРИЧЕСКИЕ МЕТОДЫ АНАЛИЗА
По убывающей степени радиотоксичности радиоактивные изотопы
делятся на четыре группы предельно допустимых концентраций:
Группа А — изотопы особо высокой радиотоксичности (ПДК не
более 1 • Ю-13 кюри/л): 90Sr, 226Ra, 239Pu и др.
Группа Б — изотопы высокой радиотоксичности (ПДК от Ы0~13
до МО-11 кюри/л): 22Na, 45Са, 60Со, 89Sr, 1I0Ag, 131I, 137Cs, 144Ce, 210Pb,
U (ест.) и др.
Группа В — изотопы средней радиотоксичности (ПДК от ЫО"11
до МО"9 кюри/л): 24Na, 32Р, 35S, 36C1, 42K, 56Mn, 55>59Fe, 69Zn, 76As, 82Br,
124, 125Sb? 140Ba и др
Группа Г — изотопы наименьшей радиотоксичности (ПДК от
1 • 10~9 кюри/л) 3Н, 14С и др.
§ 7. Классификация химических операций
с радиоактивными веществами
Все лабораторные операции делят на пять групп:
1. Хранение.
2. Простые операции с растворами (переливание, разбавление,
отбор проб, вскрытие ампул и т. п.).
3. Обычные операции с растворами (растворение, осаждение,
фильтрование, промывание осадков, титрование, экстрагирование,
центрифугирование и т. п.).
4. Сложные химические операции с растворами (нагревание,
выпаривание, перегонка, горячее экстрагирование и фильтрование, реакции
с газовыделением и т. п.).
5. Операции с сыпучими пылеобразующими и сухими веществами
(отбор проб для взвешивания, измельчение, смешивание, прессование
порошков, прокаливание, возгонка и т. п.).
Хранение радиоактивных веществ допустимо только в закрытых
сосудах из стекла, кварца, пластмассы или металла за соответствующей
их активности защитой. При хранении в открытых сосудах в результате
испарения, поверхностной диффузии, явления отдачи при а-излучении
может происходить загрязнение воздуха.
Простые и обычные операции с растворами связаны
лишь со случайными механическими загрязнениями аппаратуры,
поверхностей столов, пола и т. п. и требуют аккуратной работы. Для
уменьшения опасности загрязнений работу ведут в эмалированных кюветах
или на подносах из нержавеющей стали за соответствующей защитой
и, если необходимо по величине активности, под тягой.
При проведении ряда операций, связанных с падением капель,
возможно распыление растворов радиоактивных веществ, поэтому в таких
случаях необходимо принимать некоторые меры предосторожности —
переливать растворы по палочке, фильтровать и титровать по стенке
в закрытый сосуд с отводной трубкой, закрытой ватным тампоном,
и т. п. Пробы следует отбирать пипеткой с шприцем или с резиновой
грушей.
Сложные операции и операции с сыпучими
веществами требуют особой предосторожности, так как при проведении их
неизбежно попадание радиоактивных веществ в воздух. Все такие
работы следует вести в вытяжных шкафах или боксах. В ряде случаев
необходимо применять герметичную аппаратуру. Реакции с
газовыделением, возгонка и работа, при проведении которой распыляются
твердые образцы, представляют особую опасность, поэтому их можно
проводить только в герметичных боксах.
§ 8. УСТРОЙСТВО ЛАБОРАТОРИЙ И КЛАССИФИКАЦИЯ РАБОТ
329
§ 8. Устройство лабораторий и классификация работ
По степени радиационной опасности работы с открытыми
радиоактивными веществами делятся на три класса в зависимости от
активности на рабочем месте, группы радиотоксичности радиоактизного
изотопа и характера выполняемой работы.
Классификация работ, проводимых с открытыми радиоактивными
веществами при обычных химических операциях, приведена в табл. 13.
ТАБЛИЦА 13
Классификация работ, проводимых с открытыми
радиоактивными веществами
Группа
ради отокси ч -
ноет и
А
Б
В
Г
Класс работ
1
10
100
1000
10 000
по активности (в милликюри) изотопов
2
0,01-10
о,1-юо
1 -1 000
10-10 000
3
0,0001-0,01
0,001-0,1
0,01-1,0
0,1-10
Для установления класса работ с другими видами химических
операций при использовании изотопов данной группы токсичности (см.
стр. 328) следует соответствующую величину, приведенную в табл. 13,
умножить на поправочный коэффициент, значения которого указаны
ниже:
Характер процесса
Хранение
Простые операции с растворами . . .
Обычные химические операции . . . .
Сложные операции с растворами . . .
Операции с сухими пылеобразующими
и порошкообразными веществами .
Поправочный
коэффициент
100
10
1
0,1
0,01
В соответствии с классом работ лаборатории должны иметь
соответствующее оборудование, и их также делят на три класса.
При работах третьего класса к планировке лабораторий особых
требований не предъявляется, но их лучше проводить в отдельных
помещениях. Оборудование лабораторий должно соответствовать требованиям,
предъявляемым к обычным химическим лабораториям. Простые и
обычные операции третьего класса можно выполнять на лабораторных
столах, а более сложные — в обычных вытяжных шкафах с соблюдением
соответствующих мер предосторожности.
Работы второго класса проводятся в отдельных, специально
оборудованных помещениях. В такой лаборатории должно быть особое
хранилище изотопов, отдельные помещения для расфасовки, для химической
работы, для измерений, душевая, пункт дозиметрического контроля,
помещение для приема и хранения пищи. В лабораториях устанавливается
повышенный (5 — 10-кратный) обмен воздуха, специальные шкафы,
боксы и защитные камеры. Особые требования предъявляются к
покрытию стен, полов, оборудованию.
Хранилище радиоактивных веществ должно быть размещено в
отдельном помещении и оборудовано сейфами для хранения
радиоактивных веществ, средствами для перемещения тяжелых защитных
контейнеров, местом предварительной обработки веществ с дистанционным
333
ГЛ. IX. РАДИОМЕТРИЧЕСКИЕ МЕТОДЫ АНАЛИЗА
оборудованием. В сейфы радиоактивные вещества помещаются в
контейнерах.
В лаборатории должны быть защитные экраны (стационарные или
разборные), средства дистанционной работы (ручные и механические
манипуляторы, щипцы и т. п.).
Все работы второго класса выполняются в вытяжных шкафах или
боксах.
Описание лабораторий для работ первого класса не входит в нашу
задачу, так как такие работы проводятся только при активациониом
анализе, с использованием ядерного реактора.
§ 9. Специальное оборудование для работ
с радиоактивными веществами
Вытяжные шкафы и боксы изготовляются из легко моющихся
материалов. Присоединение их к вентиляционной системе
осуществляется через специальный фильтр. В передней стенке шкафов и
боксов вмонтированы перчатки или ручные манипуляторы. Передняя стенка
шкафов подъемная или раздвижная, боксов — стационарная.
Аппаратуру и рабочие растворы в бокс вносят через форкамеру, соединенную
с боксом шлюзом. Все управление подачей газа, электроэнергии, воды
Рис. 121. Контейнеры для радиоактивных изотопов:
а — для транспортировки; б — сборники;
1 -свинцовый КИЗ-8; 2 -свинцовый КИЗ-17; 3 - свинцовый КИЗ-38: 4-чугунный КИ 3-35;
5-дюралюминиевый КИЗ-42; 6 -свинцовый КИЗ-29; 7- свинцовый С-30; 8 -для твердых v-активных отходов;
9- для ЖИДКИХ Y-активных отходов; /0-для жидких а- и ^-активных отходов; 11 -для твердых а- и
^-активных отходов.
и сжатого воздуха находится вне шкафа и бокса на передней панели.
Стенки боксов для работ с высокоактивными р- и ^-излучателями
должны иметь достаточную толщину для поглощения излучения.
Защитные экраны бывают стационарные и разборные. Для
защиты от р-излучения применяют экраны из органического или силикат-
§ 11. ПРОВЕДЕНИЕ РАБОТ С РАДИОАКТИВНЫМИ ВЕЩЕСТВАМИ 331
ного стекла, для работ с v-излучателями — из свинцового стекла, чугуна
или свинца.
Контейнеры — защитные емкости, предназначенные для
транспортировки и хранения радиоактивных веществ (рис. 121). Они
изготовляются из свинца и чугуна для у-излучателей, алюминия и карболита
для р- и а-излучателей. Для хранения нейтронных источников
контейнеры изготовляются из парафина, содержащего бор. Для сбора и
хранения твердых и жидких радиоактивных отходов имеются специальные
контейнеры.
Дистанционные приспособления (рис. 122),
предназначенные для работ с радиоактивными изотопами, могут быть в виде щип-
Рис. 122. Приспособления для дистанционной работы.
цов и специальных ручных и механических захватов. Они дают
возможность захватывать и манипулировать с предметами и посудой
различной формы.
§ 10. Средства индивидуальной защиты
При работе с радиоактивными изотопами необходимо надевать
халаты (полукомбинезоны или комбинезоны), шапочки и резиновые
перчатки. При проведении работ второго класса следует пользоваться
специальной обувью или надевать поверх обычной резиновую или
матерчатую предохранительную обувь. Для защиты глаз применяют очки со
свинцовыми стеклами. Если есть необходимость, то пользуются
респиратором, противогазом, шлемом или пневмокостюмом.
§11. Проведение работ в химической лаборатории
с радиоактивными веществами
Прежде чем приступить к работе с радиоактивными веществами,
необходимо тщательно ознакомиться с соответствующими правилами.
Далее составляют план (инструкцию) проведения работы, определяют
332
ГЛ. IX, РАДИОМЕТРИЧЕСКИЕ МЕТОДЫ АНАЛИЗА
ее класс, а следовательно, в каких условиях следует проводить
отдельные операции (на столе, в вытяжном шкафу, в боксе, с защитой или
без нее, с применением дистанционных приспособлений или без них).
План утверждается преподавателем.
Перед работой следует надеть защитную одежду, отвечающую
характеру проводимых операций, и резиновые перчатки (рис. 123).
В лаборатории сначала проверяют с помощью дозиметрических
приборов чистоту рабочего места, собранного для опыта оборудования
и посуды. Далее оборудуют рабочее место (защита).
Рис. 123. Способы надевания и снимания перчаток.
После этого получают радиоактивный изотоп и записывают в
рабочий журнал название изотопа, его активность, удельную активность,
химическое и физическое состояние, период полураспада, дату
получения.
В том случае, если радиоактивный изотоп получен в ампуле,
находящейся в контейнере, контейнер ставят внутрь шкафа (бокса) или за
защиту, а если контейнер тяжелый, то —вблизи их, на поднос,
находящийся на полу за защитным экраном. Ампулу из тяжелого
контейнера переносят щипцами с длинной рукояткой в легкий контейнер и
ставят его в шкаф. Ампулу вскрывают и отбирают необходимое для
работы количество изотопа. Жидкость отбирают пипеткой со шприцем
или с резиновой грушей (рис. 124),
Сосуды с радиоактивными веществами ставят в тяжелые
подставки, исключающие возможность опрокидывания. Посуду для проведения
химических работ размещают на подносе и приступают к химической
работе.
После окончания работы все жидкие радиоактивные отходы
сливают, твердые собирают в специальную тару и посуду моют. Затем
проверяют чистоту рабочего места. Если рабочее место загрязнено, то
проводится его дезактивация.
Пролитый раствор собирают с помощью отсасывания, затем
фильтровальной бумагой или опилками. После этого обмывают поверхность
ваткой, смоченной соответствующим дезактивирующим раствором.
Моют сверху вниз и от более чистых к более грязным участкам, держа
§ 12 ПРАВИЛА РАБОТЫ С РАДИОАКТИВНЫМИ ВЕЩЕСТВАМИ 333
вату пинцетом. Загрязненную вату складывают в емкость для твердых
отходов.
При рассыпании радиоактивного порошка выключают
вентиляционные устройства, закрывают загрязненное место чашкой до
подготовки к дезактивации. Порошок сначала собирают сухим способом, а
затем смоченной фильтровальной бумагой. Работу проводят в респираторе.
Далее поступают, как и с раствором.
Эффективность дезактивации проверяют дозиметрическим
прибором.
Рис. 124. Пипетки с приспособлением для отбора жидкостей:
а —со шприцем; б —с грушей; в —с поршневым наконечником.
После этого перчатки, не снимая с рук, моют и проверяют их чистое
ту и чистоту защитной одежды дозиметром, снимают перчатки и
защитную одежду, проверяют чистоту рук и затем уходят из
лаборатории.
§ 12. Правила работы с радиоактивными веществами
1. Прежде чем приступить к работе с радиоактивными
веществами, необходимо определить класс работы и проводить ее в
соответствии с этим.
2. На рабочих местах необходимо оградить защитными экранами
сосуды и приборы с радиоактивными веществами так, чтобы
работающий за защитой получал не более 0,017 бэр в день.
3. В лаборатории должны находиться только те радиоактивные
вещества, которые необходимы в данное время.
4. Работающий должен быть в защитной одежде, соответствующей
классу проводимых работ.
5. Стирку спецодежды проводят в специально отведенном месте.
6. Для защиты глаз рекомендуется надевать специальные очки.
7. Все операции с открытыми радиоактивными препаратами,
кроме простых операций с растворами при уровне активности,
соответствующем работе третьего класса, следует выполнять в вытяжных
шкафах или боксах.
8. Работы, связанные с возможностью распыления радиоактивных
веществ, ведутся в боксах с применением приспособлений,
уменьшающих распространение радиоактивных, веществ.
9. Если активность соответствует работе второго класса, сложные
операции с растворами и операции с пылеобразующими веществами
334
ГЛ. IX. РАДИОМЕТРИЧЕСКИЕ МЕТОДЫ АНАЛИЗ*
проводят в шкафах или боксах, снабженных фильтром для очистки
удаляемого воздуха.
10. При работе необходимо применить дистанционные
инструменты — захваты, щипцы, шпатели, пинцеты и т. п.
11. Заполнение пипеток всасыванием ртом запрещается,
необходимо пользоваться шприцем, грушей, насосом. Промывалка снабжается
грушей.
12. Для предотвращения попадания растворов на стол и пол
работу следует проводить в кюветах.
13. В лаборатории запрещается курить, хранить пищевые
продукты, принимать пищу и пить.
14. Необходим контроль загрязненности рабочих поверхностей:
столов, вытяжных шкафов, приборов, пола, спецодежды и рук до и
после работы.
15. При работе следует пользоваться индивидуальным
дозиметром.
16. Запрещается работа в лаборатории, если на лице или руках
имеются порезы. В случае ранения необходимо тщательно промыть
порез струей воды.
17. Радиоактивные вещества хранят в контейнерах, которые ставят
в металлические сейфы, снабженные необходимой защитой от
излучения.
18. Изотоп, взятый для работы, должен быть зарегистрирован в
рабочем журнале, далее провидится регистрация всех операций с
радиоактивным веществом, его расход и отходы.
19. Запрещается сливать радиоактивные отходы в раковину.
Необходимо собирать жидкие и твердые отходы в соответствующие емкости,
короткоживущие изотопы отдельно от долгоживущих.
20. Литература и рабочие журналы во избежание загрязнения не
должны находиться на рабочих столах. Записи ведут в отдельном месте.
21. Все работающие с радиоактивным изотопом должны
проходить инструктаж.
22. Все систематически работающие с радиоактивными изотопами
должны подвергаться медицинскому освидетельствованию.
23. В каждом случае должна быть составлена инструкция по
работе, дезактивации и ликвидации аварий.
В. МЕТОДЫ РЕГИСТРАЦИИ РАДИОАКТИВНОГО ИЗЛУЧЕНИЯ
Существует много методов регистрации радиоактивных излучений.
Остановимся лишь на тех, которые наиболее широко применяются в
практике, и в частности в работах, описанных в этой главе.
§ 13. Ионизационные методы
На ионизационном эффекте, производимом радиоактивным
излучением, основан принцип работ следующих типов детекторов:
ионизационной камеры, пропорционального счетчика и счетчика Гейгера —
Мюллера. Все эти детекторы представляют собой наполненные той
или иной газовой смесью сосуды, которые имеют два электрода. Схема
включения детектора показана на рис. 125. Механизм ионизации газов
излучением различного типа и энергии не одинаков, но энергия,
затрачиваемая на образование пары ионов во всех случаях составляет около
34 эв. Величина первичной ионизации, т. е. ионизация, производимая
ядерной частицей непосредственно, зависит только от доли энергии,
§ 13. ИОНИЗАЦИОННЫЕ МЕТОДЫ 335
которую ядерная частица (фотон) теряет на пути в детекторе.
Вторичные эффекты зависят от величины напряжения, приложенного к
электродам детектора. Величина напряжения обусловливает также
механизм регистрации излучения и тип детектора.
Изменение величины импульса тока от напряжения, приложенного
к электродам детектора, выражается кривой, приведенной на рис. 126.
При отсутствии напряжения большая часть образовавшихся под
действием излучения ионов рекомбинирует. С приложением
напряжения ионы начинают направленно перемещаться к катоду, а электроны —
к аноду. Часть их, не рекомбинируя, доходит до электродов, в цепи
протекает ток, импульс которого появляется на сопротивлении 4 при
разряде и заряде емкости 3 (см. рис. 125). По мере возрастания
напряжения на участке ОА (см. рис. 126) все большая доля ионов
достигает электродов и амплитуда импульса тока растет. На участке АВ все
порожденные излучением ионы достигают электродов, поэтому
амплитуда импульса остается постоянной; ионизационный ток, протекающий
, I *'—I т-
Рис. 125. Схема включения ионизационных детекторов
излучения:
1 — детектор; 2— источник напряжения; 3 — емкость; 4 — высокоом-
ное сопротивление.
в детекторе, называется током насыщения. В области напряжений,
при которых получается ток насыщения, работают ионизационные
камеры. С помощью ионизационных камер измеряется ионизационный
ток, который пропорционален интенсивности излучения, а следовательно,
активности радиоактивного вещества.
При напряжениях, лежащих выше точки В, электроны,
образовавшиеся под действием радиоактивного излучения, разгоняются в
электрическом поле до таких скоростей, что производят вторичную
ионизацию молекул газа. В области ВС вторичная ионизация
пропорциональна величине первичной ионизации. Эта область напряжений, в которой
работает пропорциональный счетчик.
При увеличении напряжения выше точки D каждая попавшая в
детектор частица вызывает лавинный разряд. В этой области,
называемой областью Гейгера, работают счетчики Гейгера — Мюллера. В
области Гейгера величина вторичной ионизации не зависит от величины
первичной ионизации, амплитуда импульса не зависит от рода
ионизирующих частиц, но зависит от напряжения на электродах детектора.
Влияние величины напряжения на работу счетчика Гейгера—-Мюллера
па иллюстрируется кривой, представленной на рис. 127. Эта кривая
называется рабочей или счетной характеристикой счетчика Гейгера —
Мюллера. При измерении активности счетчиками Гейгера —Мюллера
пользуются участком BD (амплитуда импульса почти постоянна), это
так называемое плато счетчика. Считается, что счетчик нормально
работает, если наклон плато (Д) не превышает 0,15% на 1 в:
__ Л/п-ЮО
(32)
336
ГЛ. IX. РАДИОМЕТРИЧЕСКИЕ МЕТОДЫ АНАЛИЗА
Как было сказано выше, в счетчиках Гейгера — Мюллера
происходит лавинообразный разряд, вызываемый одной ионизирующей
частицей, проникшей в счетчик. Кроме того, быстрые электроны при
ударе возбуждают молекулы, стабилизация которых происходит
высвечиванием в ультрафиолетовой области. Ультрафиолетовое излучение
вызывает образование фотоэлектронов, которые порождают в
электрическом поле новые лавины электронов. Новые лавины электронов
могут появиться и в результате процесса рекомбинации положительных
ионов на катоде. При этом получаются возбужденные молекулы газа,
стабилизация которых опять приводит к образованию фотонов и
фотоэлектронов. Таким образом, лавинный разряд может продолжаться.
Гашение лавинного разряда происходит в результате понижения
потенциала анода и образования у катода слоя положительных ионов,
которые за время собирания электронов 10~7—Ю-8 сек практически
остаются на месте. Для предотвращения образования повторных лавин
Рис. 126. Зависимость амплитуды импульса
тока в ионизационном детекторе от
напряжения на его электродах:
/ — область работы ионизационной камеры;
// — область работы пропорционального счетчика;
/// — область работы счетчика Гейгера — Мюллера.
Рис. 127. Рабочая характеристика
счетчика Гейгера — Мюллера:
Л —начало счета; В - начало плато; С —
конец плато; Р — рабочее напряжение.
в цепь счетчика включается высокоомное сопротивление порядка 109 ом,
которое замедляет восстановление напряжения, так что оно происходит
медленнее собирания положительных ионов на катоде (10*3 сек). После
регистрации одной лавины разряд в счетчике прекращается, и через
10~3 сек счетчик готов к регистрации новой частицы. Такие счетчики
называются несамогасящимися.
Для уменьшения времени восстановления счетчика, так
называемого мертвого времени, в счетчик вводят пары вещества, потенциал
ионизации молекул которого меньше потенциала ионизации основного
компонента газовой смеси, наполняющей счетчик, — аргона. В качестве
таких веществ применяют галогены, этиловый спирт и другие
многоатомные молекулы.
Многоатомные молекулы передают электрон положительным ионам
аргона, образовавшимся под ионизирующим действием излучения, а
последние рекомбинируют на катоде. Возбуждение многоатомных
молекул гасится при их разложении без выделения фотонов, и вторичные
лавины не образуются. Это позволяет уменьшить сопротивление в цепи
счетчика и время восстановления счетчика до 10~4 сек. Такие счетчики
называются самогасящимися.
В счетчиках с гасящим наполнителем из органических веществ
происходит разложение их молекул в каждом акте гашения разряда,
поэтому срок службы этих счетчиков ограничен. Галогенные счетчики мо-
§ 13. ИОНИЗАЦИОННЫЕ МЕТОДЫ
337
гут работать неопределенно долгое время, так как процесс разложения
галогена на атомы обратим.
При регистрации а- и (3-частиц счетчиком Гейгера — Мюллера
каждая частица, попавшая в счетчик, дает лавинный разряд и
регистрируется. Ионизация газа внутри счетчика у-лучами маловероятна, более
вероятно выбивание электронов фотоном из стенок счетчика, поэтому
эффективность счетчика по отношению к у-лучам составляет 0,5—2%.
Счетчики Гейгера — Мюллера имеют различную конструкцию и
назначение. Основные виды счетчиков показаны на рис. 128. По форме
различают цилиндрический (128, а) и торцовый (128,6) счетчики.
Цилиндрический счетчик с толстыми стеклянными стенками, покрытый
изнутри металлом, с натянутой по его оси изолированной металлической
нитью служит для регистрации -улУчей. Такой же счетчик с тонкими
^ -Ф- $
6
Рис. 128. Схема счетчиков Гейгера — Мюллера:
а—цилиндрический для регистрации р- или Y-излучения: б —торцовый для регистрации
мягкого (3-излучения; в —4л-счетчик; г — цилиндрический счетчик с рубашкой; д — счетчик
погружения; е — проточный счетчик для измерения активности газов.
металлическими стенками предназначен для регистрации р-частиц.
Торцовый счетчик имеет тонкое слюдяное окошечко и вертикально
расположенную нить. Он позволяет регистрировать (3-излучение малой
энергии и при очень малой толщине окна даже а-частицы. В отдельных
случаях такой счетчик делается без слюдяного окна и в него
непрерывно подается струя наполняющего газа. Для регистрации активности
жидкостей служит счетчик погружения (рис. 128, д) и счетчик с
рубашкой (рис. 128, г). Для полной регистрации активности служит 4я-счет-
чик (рис. 128, в), который представляет собой как бы два соединенных
торцами торцовых счетчика, Активность газов регистрируется
проточным, или туниковым, счетчиком (рис. 128, е). При этом радиоактивный
газ вводится внутрь его.
Для регистрации активности счетчик Гейгера—Мюллера включают
в схему, в которой импульс тока под действием напряжения,
создаваемого высоковольтным выпрямителем, поступает на усилитель, не только
усиливающий малый ток импульса, но и формирующий его для
дальнейшей регистрации. С усилителя импульс тока подается на
пересчетное устройство и затем на электромеханический счетчик импульсов.
Назначение пересчетного устройства пропускать на механический счетчик
лишь малую, определенную долю импульсов тока, так как
электромеханический счетчик не может регистрировать большие скорости счета.
338
ГЛ. IX. РАДИОМЕТРИЧЕСКИЕ МЕТОДЫ АНАЛИЗА
§ 14. Сцинтилляционный метод
Большое значение имеет сцинтилляционный метод регистрации
радиоактивных излучений. Сцинтилляционный метод основан на
испускании световых квантов так называемыми фосфорами
(люминофорами) под действием ионизирующих излучений. В качестве фосфоров
в настоящее время используют кристаллы ряда солей, иногда
активированные включением примесных катионов, например ZnS(Ag), Nal(Tl),
Lil(Sn), CaW04, антрацен, нафталин, стильбен, терфенил в
полистироле и т. п. Разные люминофоры служат для регистрации различного
вида излучений.
В фосфоре часть энергии ионизирующего излучения затрачивается
на возбуждение молекул (атомов), которые переходят в нормальное
энергетическое состояние, испуская фотоны в видимой и
ультрафиолетовой областях спектра. Регистрация этих фотонов проводится с
помощью так называемого фотоэлектронного умножителя (ФЭУ).
Рис. 129. Схема фотоэлектронного умножителя и его включения:
1-фосфор в кожухе; 2-фотокатод; 3~диноды; 4- анод (коллектор); 5-источник высокого
Напряжения; #-сопротивление делителя напряжения; RA - сопротивление цепи схемы.
Принцип работы фотоэлектронного умножителя основан на
явлении вторичной эмиссии электронов. На рис. 129 приведена схема ФЭУ.
Он представляет собой стеклянный баллон, в котором создан глубокий
вакуум. В баллоне расположены фотокатод 2, диноды 3 и анод
(коллектор) 4. Фотокатод примыкает непосредственно к фосфору 1. На ди-
нодах создается последовательно возрастающий по отношению к катоду
положительный потенциал.
Возникающие под действием ионизирующей частицы или у-фотона
световые кванты выбивают из фотокатода электроны. Электроны под
действием разности потенциалов устремляются к первому диноду, из
которого каждый электрон выбивает несколько большее число
электронов. Последние разгоняются под действием разности потенциалов и
попадают на второй динод, выбивая из него большее количество
электронов. Процесс повторяется вплоть до попадания электронов на анод.
Каждой вспышке света в фосфоре соответствует импульс напряжения
на аноде, который регистрируется электронной схемой.
Фосфор помещается в кожухе, имеющем на одном из торцов окно.
С помощью этого окна осуществляется контакт его с катодом ФЭУ,
причем ФЭУ также помещается в светонепроницаемый кожух, чтобы
исключить влияние видимого света. Принципиальные схемы
конструкций сцинтилляционных детекторов представлены на рис. 130.
§ 14. СЦИНТИЛЛЯЦИОННЫЙ МЕТОД
339
При измерении радиоактивности твердых и жидких препаратов
с помощью фосфоров и ФЭУ могут применяться твердые и жидкие
фосфоры. Для регистрации а-излучения обычно используется кристалл
ZnS(Ag), при этом кристалл может быть тонким, с торцом,
защищенным фольгой или тонкой пленкой. р-Излучение измеряют при помощи
пластмассовых фосфоров, иногда кристаллов солей. Фосфор для
регистрации р-излучения должен иметь толщину, приблизительно равную
максимальному пробегу в нем (З-лучей. При этом условии
эффективность счета р-лучей равна единице, а у"лУчи регистрируются очень
мало. Регистрация р-лучей может осуществляться и с помощью
жидких фосфоров. Особенно удобно измерять активность р-излучателей
с малой энергией (14С, 3Н, 35S и т. п.), растворяя образец в фосфоре.
Y-Лучи регистрируются с помощью фосфора Nal(Tl), при этом берут
толстые и по возможности большие монокристаллы, эффективность
регистрации в которых достигает 30%.
Рис. 130. Схемы сцинтилляционных детекторов:
/ — измерение радиоактивности твердых препаратов; а —по (3-лучам; б— по
Y-лучам; // — измерение радиоактивности жидких веществ: а—на торце фосфора;
б — в кристалле с колодцем;
2 — фосфор; 2 — ФЭУ; 3 — радиоактивное вещество.
Число квантов, возникающих под действием ионизирующей частицы
в фосфоре, пропорционально энергии ионизирующей частицы;
соответственно амплитуда импульса также пропорциональна энергии
излучения. Это позволяет регистрировать не только число ионизирующих
частиц, но и определять их энергетический спектр.
Следует сказать, что наряду с фоном космического излучения и
загрязнений в ФЭУ появляются паразитные импульсы (фон) вследствие
выбрасывания фотокатодом тепловых электронов и выбивания
электронов с динодов положительными ионами. Уменьшение фона достигается
с помощью дискриминатора, пропускающего сигналы только с
достаточно большой амплитудой.
Скорость счета радиоактивного образца и фона зависит от
напряжения на ФЭУ, величины усиления линейного усилителя и порога
дискриминации. Так как сцинтилляционный счетчик не имеет плато,
подобного счетчику Гейгера — Мюллера, то необходима надежная
стабилизация напряжения, подаваемого на ФЭУ и линейный усилитель.
Оптимальные условия измерения на сцинтилляционном счетчике
меняются в зависимости от рода и энергии излучения, и при переходе
от измерений одного вида или энергии к другим необходимо заново
снять характеристику счетчика.
340
ГЛ. IX. РАДИОМЕТРИЧЕСКИЕ МЕТОДЫ АНАЛИЗА
Измерения радиоактивности сцинтилляционными детекторами и
счетчиками Гейгера—Мюллера могут проводить на различного типа
радиометрах, установке Б, ПС-5М («Волна») и т. п.
Г. АППАРАТУРА И ТЕХНИКА ИЗМЕРЕНИЯ РАДИОАКТИВНОСТИ
Регистрация радиоактивности с помощью того или иного вида
электронной счетной аппаратуры с детекторами ядерных излучений
(сцинтилляционными, Гейгера—Мюллера и т. п.) является обязательным
элементом любой практической работы в радиометрических методах
анализа. Наиболее употребительными являются установки Б и ПС-5М
(«Волна»).
§ 15. Радирметрические установки
Установки Б. Все установки этого типа состоят из счетчика
Гейгера—Мюллера, помещенного в защитный домик, блока гасящего
сопротивления (БГС), пересчетного устройства (ПС), высоковольтного
Рис. 131. Установка Б со счетчиком Гейгера —Мюллера:
1 — счетчик; 2— блок гасящего сопротивления; 3 —защитный домик; 4
—пересчетов устройство.
выпрямителя (ВСЭ) и электромеханического счетчика (СБ). Эти блоки
могут быть в виде отдельных приборов (Б-1) или собраны в один (Б-2,
Б-3, Б-4). Пересчетное устройство пропускает на электромеханический
счетчик в зависимости от кратности пересчета 64-й, 16-й, 4-й или
каждый импульс.
Установка Б-4 состоит (рис. 131) из блока счетчиков БГС и
пересчетного прибора ПП-16. Установка предназначена для регистрации
импульсов тока от низковольтных газовых счетчиков (галогенных).
Счетчик присоединяется к БГС и для уменьшения фона располагается
в защитном домике (экране). БГС двумя кабелями соединяется с
пересчетным прибором. Прибор включается в сеть (220 в), при этом на
счетчик поступает напряжение 390 в.
Напряжение, подаваемое на счетчик, является рабочим
напряжением галогенных счетчиков СТС-5,6; СБТ-7,9. Максимальная скорость
счета прибора равна 15 000 имп/мин, при большей скорости счета
необходимо вводить поправки на разрешающее время, которое для ПП-16
равно 70 мксек.
На передней панели пересчетного прибора под стеклом
расположены шесть декатронов, по которым ведется отсчет прошедших
импульсов. Правый декатрон отсчитывает единицы, следующий — десятки,
затем сотни, тысячи, десятки и сотни тысяч импульсов.
§ 16. ИЗМЕРЕНИЕ РАДИОАКТИВНОСТИ
341
Установка ПС-5М («Волна»). Установка (рис. 132) дает
возможность регистрировать активность счетчиком Гейгера—Мюллера (блок
УГС-1) или сцинтилляционным детектором (блок УСС-1). Детектор
(блок УГС-1 или УСС-1) подключается к пересчетному прибору (ПСТ-
100). Высокое напряжение на детектор подается с высоковольтного
Рис. 132. Установка ПС-5М («Волна») со сцинтилляционным
счетчиком:
1 — фосфор и фотоумножитель в кожухе; 2 — стабилизатор напряжения; 3 —
пересчетный прибор.
выпрямителя (ВСВ-2). В блок ПСТ-100 вмонтирован электронный
секундомер с автоматической установкой времени измерения. Кроме того,
в нем есть стрелочный прибор, показывающий интегральную скорость
счета. В приборе нет дискриминатора — уровень дискриминации в нем
постоянен. Сцинтилляционный детектор включает в себя
фотоумножитель и сменные фосфоры.
§ 16. Измерение радиоактивности
Измерение счетчиком Гейгера — Мюллера
Подготовка установки к работе. Прежде чем приступить к
измерению, по паспорту проверяют правильность включения всех блоков
установки. Тумблеры, кроме тумблера на электромеханическом
счетчике, ставят в положение «выключено», а ручку регулятора напряжения
на высоковольтном выпрямителе поворачивают против часовой стрелки
до упора. В установку ПС-5М включают блок УГС-1.
Проверка прибора (см. инструкцию). Включают питание установки
от сети и выжидают 2—3 мин, пока прогреются лампы пересчетного
прибора. Контрольные лампы блоков установки должны гореть.
Правильность работы пересчетного прибора устанавливают в
режиме проверки, измеряя число прошедших импульсов тока в течение
1 мин от сети переменного тока. Скорость прохождения импульсов от
сети определяют при всех кратностях пересчета по 3 раза. Берут
среднее арифметическое полученных результатов. Порядок работы прибора
при измерении должен соответствовать указанному в инструкции.
Полученный результат сравнивают с частотой сетевого тока (3000 имп/мин).
Отклонение не должно превышать 2%.
Снятие счетной характеристики счетчика Гейгера — Мюллера.
После прогревания высоковольтного выпрямителя в течение 15 мин можно
342
ГЛ. IX. РАДИОМЕТРИЧЕСКИЕ МЕТОДЫ АНАЛИЗА
приступить к снятию счетной характеристики счетчика Гейгера —
Мюллера.
Радиоактивный препарат (500—1000 имп/мин) помещают в
фиксированном положении в защитном домике счетчика. Устанавливают
максимальную кратность пересчета и переключают прибор для работы.
Затем, включив прибор, медленно вращают ручку регулировки
высоковольтного выпрямителя по часовой стрелке до положения, при котором
неоновые лампы пересчетного прибора начинают сигнализировать о
прохождении импульсов (напряжение начала счета). Измеряют
активность препарата при напряжении начала счета в течение 2 мин.
Повышают напряжение на счетчике на 50 в и повторяют измерение
активности препарата. Снова повышают напряжение на 50 в и измеряют
активность. Так поступают до тех пор, пока регистрируемая активность не
возрастет на 20—30%. Не следует повышать напряжение настолько, что
счетчик резко повысит регистрируемую ак-*
/ тивность. Это связано с непрерывным
разрядом в счетчике и ведет к его порче. Абсолют-
-— ное статистическое отклонение отдельного
измерения находят по формуле (23) и строят
график зависимости активности от
напряжения на счетчике. Наклон плато рассчитывают
по формуле (32). Величину рабочего напря-
О д жения Р выбирают в положении Уз—У2 рас-
Рис 133. Зависимость ско- стояния от начала горизонтального участка
рости счета на счетчике на графике.
Гейгера - Мюллера от вели- Измерение фона. После выбора рабочего
чины активности. напряжения счетчика из защитного домика
вынимают радиоактивный препарат и
измеряют фон счетчика при выбранном рабочем напряжении в течение 5 мин.
Определяют скорость счета фона в имп/мин и абсолютное статистическое
отклонение.
Определение разрешающего времени счетной установки. Поочередно
измеряют активность серии эталонов с известным содержанием
радиоактивного вещества. По полученным результатам строят график (рис.
133) в координатах измеренная активность / — содержание
радиоактивного вещества А. От начала координат по прямолинейному участку
графика проводят прямую. Для нахождения активности из
соответствующей точки на оси абсцисс восстанавливают перпендикуляр до
пересечения с этой прямой. При этом точка 1\ соответствует числу
сосчитанных импульсов, а точка /о — числу, которое было бы получено, если
работа установки не ограничивалась величиной разрешающего времени
установки.
Разрешающее время находят по формуле:
т /о-Л
Измерение сцинтилляционным счетчиком
Если работу прободят на установке ПС-5М, то к блоку ПСТ-100
подключают блок УСС-1, предназначенный для регистрации излучения
сцинтилляционным методом.
Подготавливают установку к работе по инструкции.
Выбор оптимального режима установки для работы на сцинтилля-
ционном детекторе. В держатель сцинтилляционного счетчика помещают
радиоактивный препарат (500—1000 имп/мин). Так как регистрируемая
сцинтилляционным счетчиком активность зависит от напряжения на
§ 17. ИЗМЕРЕНИЕ ДОЗЫ НА РАБОЧЕМ МЕСТЕ И ИНДИВИДУАЛЬНОЙ ДОЗЫ 343
ФЭУ, усиления линейного усилителя и порога дискриминации, то для
установления оптимального режима необходимо подобрать эти
параметры.
Усиление линейного усилителя установки ПС-5М
заводом-изготовителем устанавливается равным 100, но может быть изменено на 50
и 20. Порог дискриминации в удтановке равен 50 мв. Поэтому выбор
оптимального режима сводится к выбору рабочего напряжения
на ФЭУ.
Для этой цели, постепенно увеличивая напряжение на ФЭУ,
находят положение, при котором сигнальные лампы декатронов начнут
сигнализировать о прохождении импульсов. Затем измеряют скорость
прохождения импульсов. После записи показаний прибора вынимают
препарат и измеряют фон. Снова вставляют препарат, напряжение на
ФЭУ повышают на 50 в и измеряют скорость прохождения импульсов.
Препарат вынимают и измеряют фон. Далее еще увеличивают
напряжение на 50 в и измеряют скорость прохождения импульсов от
препарата и фона. Так поступают до тех пор, пока активность препарата и
фона не будут близки по порядку величин. После этого снижают
напряжение на ФЭУ на 50—100 в.
Результаты измерений наносят на график в координатах (У"/с —
— V?<b f ~~ напряжения на ФЭУ, где /с и /ф — скорость прохождения
импульсов от препарата и фона.
Максимум кривой на графике соответствует оптимальному и,
следовательно, рабочему напряжению на ФЭУ. Пределы рабочих
напряжений ФЭУ для различного рода излучений указываются в его
паспорте.
§ 17. Измерение дозы на рабочем месте, индивидуальной дозы,
полученной работающим, и загрязненности рабочих поверхностей,
рук и одежды
Для измерения дозы на рабочем месте применяется дозиметр с
ионизационной камерой, например МРМ-1. Ионизационная камера через
усилитель связана со стрелочным прибором, калиброванным в
рентгенах.
Для измерения дозы, получаемой работающим в течение всего
рабочего дня, служат индивидуальные дозиметры КИД-1 или ДК-02. Они
представляют собой миниатюрные ионизационные камеры, которые
перед употреблением заряжают от зарядного устройства; во время
облучения они разряжаются. Электроскоп показывает полученную дозу.
Определение загрязненности рук, одежды и рабочих поверхностей
в лаборатории обычно проводят радиометрами «Тисе» и «Луч».
Работа с радиометром «Тисе»
Радиометр «Тисе» имеет три сменных детектора: для измерения
загрязненности поверхностей р-йзлучающими веществами (блок «ТЧ»);
для измерения загрязненности малых поверхностей а-излучающими
радиоактивными веществами (блок «ТИ») и для определения
загрязненности больших поверхностей а-излучающими радиоактивными
веществами (блок «ТЮ»). Для практических работ, приведенных в этой
книге, необходим только блок «ТЧ», который состоит из трех
параллельно включенных счетчиков Гейгера—Мюллера типа СТС-6. Блок
«ТЧ» через длинный кабель и катодный повторитель, предназначенный
для усиления сигнала, поступает в основной блок. Электронный блок,
формирующий и регистрирующий импульсы тока, имеет электромехани-
344
ГЛ. IX. РАДИОМЕТРИЧЕСКИЕ МЕТОДЫ АНАЛИЗА
ческий счетчик и стрелочный прибор, шкала которого калибрована на
шесть диапазонов в имп/мин. Питание прибора осуществляется от сети
переменного тока через стабилизированный выпрямитель,
расположенный в основном блоке.
Включение и подготовка к работе. Подключают к установке
блок «ТЧ». По инструкции проводят, подготовку прибора к работе.
Для работы выбирают шкалу, по которой значение у-фона в
помещении не превышает 4/s всей шкалы.
Помещают на блок «ТЧ» предмет, не загрязненный радиоактивным
веществом, и проводят измерение; при этом стрелка должна
установиться на нуль, а на табло загорится надпись «Чисто». Затем на блок
«ТЧ» помещают предмет, загрязненный р- или у-излучающим
радиоактивным веществом; при включении прибора стрелка сначала
переместится к нулевому делению, а затем плавно двинется по шкале в
сторону возрастания значений и остановится у деления, соответствующего
измеряемой радиоактивности. Прибор дает показания суммарной Р- и
у-радиоактивности загрязнений поверхности.
Устанавливают порог срабатывания сигнального устройства. Для
этого помещают на блок «ТЧ» р-радиоактивный эталон, регулятор
чувствительности должен быть выведен до отказа влево. Через 12 сек на
табло загорится сигнал «Чисто», тогда вращают регулятор
чувствительности вправо до тех пор, пока не загорится сигнал «Грязно».
После подготовки прибора к работе его оставляют включенным
в течение всего времени работы в лаборатории и по мере необходимости
проверяют загрязненность рук, перчаток, одежды и поверхностей
столов, пола, аппаратуры.
Работа с радиометром «Л у ч - А>>
Переносной универсальный радиометр «Луч-А» предназначен для
обнаружения радиоактивных загрязнений поверхности аппаратуры,
одежды, пола, мебели и т. п. Он может питаться от сети переменного
тока или от сухих батарей. В качестве детекторов излучения применяют
торцовый счетчик Гейгера — Мюллера для измерения мягкого р-излуче-
ния (с энергией не ниже 0,15 Мэв), цилиндрический счетчик Гейгера —
Мюллера для измерения жесткого р-излученйя и у-излучения. Шкала
прибора калибрована на четыре диапазона в имп/сек.
§ 18. Абсолютные и относительные измерения активности
Регистрируемая счетной установкой активность равна
произведению активности на коэффициент счета ср [см, формулу (5)].
Коэффициент счета представляет собою произведение целого ряда
коэффициентов, учитывающих поправки на величину телесного угла
(геометрический коэффициент счета), — т), эффективность регистрации данного
вида излучения — е, поглощение — /с, обратное рассеяние — q,
самоослабление — |:
Ф = r\\Kqz (33)
В том случае, когда изотоп имеет сложную схему распада,
например изотоп испускает при каждом распаде радиоактивного ядра
несколько квантов разной энергии, или когда изотоп распадается с
испусканием частиц, обладающих разной энергией, то для каждого кванта
или частицы коэффицценты е, к, | и q будут различны. В этом случае
для вычисления активности необходимо учесть схему распада:
/ = А [ц (iiittfieiPi + 62^2^282^2 + •..)] (34)
§ 18. АБСОЛЮТНЫЕ И ОТНОСИТЕЛЬНЫЕ ИЗМЕРЕНИЯ АКТИВНОСТИ 345
где р — число квантов, излучаемых при распаде одного ядра изотопа, или доля частиц
(квантов) данной энергии. Индексы «1, 2 ...» относятся к отдельным
компонентам излучения.
Выражение (34) может быть использовано также для вычисления
активности в распад/сек смеси изотопов по активности,
зарегистрированной в имп/сек.
Введение большого числа поправок сложно и уменьшает точность
результатов измерений, поэтому стремятся, чтобы поправочные
коэффициенты были близки к единице.
Активность твердых препаратов р- и у-излучателей (в распад/сек)
определяют с помощью 4я-счетчиков Гейгера — Мюллера (см. рис. 128),
в которых препарат с очень малой толщиной помещают на тонкую
пленку внутри счетчика так, что поглощение, отражение и
самопоглощение практически отсутствуют. Определение активности (в распад/сек)
твердых и жидких препаратов р-излучателей можно проводить также
торцовым и цилиндрическим счетчиком Гейгера — Мюллера, но в этом
случае необходимо учесть геометрический коэффициент счета, что
снижает точность определения с 1—2% до 10—15% в первом и 25—30%'
во втором случае. Сцинтилляционный метод дает возможность
определять абсолютную активность не только а- и р-излучателей, но и у-излу-
чателей.
Однако в лабораторной практике проще проводит^ относительные
измерения, когда активность исследуемого препарата сравнивается с
активностью эталона с известным содержанием радиоактивного
изотопа [распад/(сек*г) или имп/(сек-г)] или активностью другого
исследуемого препарата. Если активности препаратов и эталона измеряют в
строго одинаковых условиях (один и тот же детектор и прибор для
измерения, одинаковое положение по отношению к детектору, одно и то же
вещество, подготовленное для измерения одинаковым способом и
равномерно распределенное по объему препарата, слой которого имеет
одинаковую толщину и нанесен на одинаковую подложку), то все
поправочные коэффициенты сокращаются, и из отношения измеренных
активностей можно найти по формуле (5) абсолютную активность препарата.
Для измерения а- и р-излучения лучше пользоваться «бесконечно»
тонкими (т. е. практически не поглощающими излучения) или
насыщенными (толщина больше максимального пробега) слоями. Для у-лучей
условие строгой одинаковости толщины слоя и подложки не
обязательно.
Измерение активности более точно при большем удалении
препарата от детектора, таю как при этом меньше сказывается
неравномерность распределения радиоактивного вещества по поверхности
препарата. Однако удаленность препарата ограничивается величиной
активности препаратов. Можно также улучшить результаты измерения,
связанные с неравномерностью распределения радиоактивного вещества
по поверхности препарата, повторив измерение после поворота
препарата на 90, 180, 270° и взяв среднее из четырех полученных результатов
измерений.
#. РАДИОМЕТРИЧЕСКИЕ МЕТОДЫ ОПРЕДЕЛЕНИЯ
Все операции с открытым радиоактивным веществом, описываемые
в этой книге, соответствуют работе третьего класса. Перед проведением
работы третьего класса надевают халат, шапочку и резиновые перчатки.
Сосуды с радиоактивными веществами ставят в эмалированную кювету.
346
ГЛ. IX. РАДИОМЕТРИЧЕСКИЕ МЕТОДЫ АНАЛИЗА
По окончании работы ненужные радиоактивные растворы сливают
в специально предназначенные для этого сосуды (тяга!); твердые
радиоактивные отходы помещают в специальные контейнеры. Чашечки
для измерения активности с радиоактивным препаратом помещают в
специальный сосуд. Не снимая перчаток, моют посуду и перчатки. Про-
веряют дозиметром чистоту посуды, рабочего места и перчаток.
Снимают перчатки, моют руки и проверяют чистоту их и одежды
дозиметром, после чего покидают лабораторию.
§ 19. Приготовление раствора с заданной активностью
Перед проведением любой работы с радиоактивными изотопами
необходимо приготовление исходного раствора с заданной активностью.
Радиоактивные изотопы поставляются в стеклянных запаянных
ампулах или пробирках с притертыми пробками. Эти сосуды находятся
в специальных контейнерах, толщина стенок которых достаточна для
создания вблизи контейнера безопасных условий работы. К каждому
препарату прилагается паспорт, в котором указывается химическое
соединение радиоактивного изотопа, его общая и удельная активность на
определенное время, масса химического соединения или объем раствора.
Для приготовления раствора с необходимой для работы общей и
удельной активностью прежде всего рассчитывают, исходя из
паспортных данных, необходимое разбавление и количество неактивного
соединения, которое следует добавить.
Пример. Расчет разбавления, необходимого для приготовления 0,1 М раствора
гидрбфосфата натрия с удельной активностью 105 имп/(мин • мл). По паспорту в
ампуле находится 10 мкюри Ъ2Р с удельной активностью 10 мкюри/мл и с содержанием
фосфора 5 мг\мл. Коэффициент счета р-излучения на выбранном приборе составляет
0,05. Удельная активность 5 исходного препарата в имп/сек может быть найдена по
формуле, аналогичной формуле (5):
S = 3,7 -107- 10- 0,05- 1,85 • 107
Требуемое разбавление равно:
1,85* 107-60
105
Для получения такого разбавления необходимо промежуточное разбавление.
Сначала под тягой, за защитой, достаточной для поглощения излучения в такой
степени, чтобы доза не превышала предельно допустимой, вскрывают ампулу. Из
ампулы пипеткой со шприцем отбирают 0,5 мл исходного раствора и выливают его в
мерную колбу емкостью 50 мл. Затем остаток раствора из ампулы переливают в
пробирку с притертой пробкой, снова помещают в контейнер и уносят из лаборатории.
Колбу до метки заполняют 0,1 М раствором ЫагНРО.*. Затем из этой колбы мерной
пипеткой со шприцем берут 0,5 мл раствора и помещают его в другую мерную колбу
емкостью 50 мл. Снова доводят объём раствора до метки 0,1 М раствором Na2HP04.
§ 20. Прямое определение ионов химических элементов
в растворе с помощью радиоактивных реагентов
Метод основан на осаждении ионов определяемого элемента в виде
нерастворимого осадка избытком осадителя (реагента) известной
концентрации, меченного радиоактивным изотопом. Удельная активность
осадителя (реагента) должна быть известна. Метод применим во всех
случаях селективного образования определяемыми ионами
нерастворимого осадка с меченым реагентом.
Для определения количества искомого элемента можно
воспользоваться двумя способами.
§ 21. РАБОТЫ, ВЫПОЛНЯЕМЫЕ МЕТОДОМ ПРЯМОГО ОПРЕДЕЛЕНИЯ 347
1. После осаждения определяют активность осадка. Количество
(тх) искомого элемента (в г-экв) находят по формуле:
'о
где 1\ — активность осадка, имп[мин;
/о— удельная активность реагента-осадителя, имп/(мин• г-экв).
2. Осаждение из известного объема исследуемого раствора Vz ведут
определенным объемом реагента V\. Перед осаждением определяют
объемную удельную активность реагента-осадителя /о в имп/(мин«мл).
После осаждения определяется активность 1 мл фильтрата h
в имп/(мин • мл).
Количество шх искомого элемента (в г-экв) вычисляют по формуле:
/0^1-/2(^1 + ^2) _,
'ОМ
где тй — число грамм-эквивалентов взятого для осаждения реагента;
ть — число грамм-эквивалентов избытка осадителя.
Второй способ, хотя и требует большего числа измерений, по
существу, проще, так как проводится измерение определенного объема
раствора, а измерение растворов значительно легче стандартизовать,
чем измерение твердых препаратов.
§ 21. Практические работы, выполняемые методом
прямого определения
Определение катионов в виде фосфатов с применением 32Р
В работе используется осаждение радиоактивным фосфатом
катионов и определение активности избытка осадителя.
Методика определения. Работа проводится по третьему классу
(см. стр. 329). Сначала определяют удельную активность реагента —
раствора Na2HP04 с точно известной концентрацией (0,1—0,3 М),
содержащего 32Р. Для этого на лабораторном столе с помощью пипетки
емкостью 1—5 мл, снабженной шприцем, отбирают 1 мл раствора и
переносят его в чашечку для измерения активности, выпаривают досуха
под лампой, стоящей под тягой, и измеряют активность сухого остатка
с помощью счетной установки с детектором р-излучения в стандартных
условиях (измерение активности — см. стр. 341).
На лабораторном столе к 10 мл анализируемого раствора из
бюретки постепенно приливают раствор меченого фосфата до прекращения
образования осадка. Осадок коагулирует при нагревании раствора на
водяной бане, затем его отфильтровывают. Переносят 1 мл фильтрата
пипеткой со шприцем в чашечку для измерения активности, выпаривают
досуха под лампой (тяга!) и измеряют активность остатка в условиях,
в которых производилось измерение удельной активности.
Содержание искомого элемента в растворе вычисляют по фо?*
муле (36).
Определение ионов калия в виде кобальтинитрита калия
с применением 6flGo
Работа основана на осаждении калия в виде кобальтинитрита
(60Со) с последующим определением активности образовавшегося
осадка.
348
ГЛ. IX. РАДИОМЕТРИЧЕСКИЕ МЕТОДЫ АНАЛИЗА
Методика определения. Работа проводится по третьему классу
(см. стр. 329).
Определяют удельную активность раствора Co(N03h с точно
известной концентрацией (~0,3 М), содержащего 60Со. Для этого 1 мл
раствора с помощью пипетки емкостью 1—5 мл, снабженной шприцем,
переносят в мерную колбу емкостью 25 мл. Эти операции проводят на
лабораторном столе в кювете. Раствор доводят до метки, отбирают из
него чистой пипеткой со шприцем 2 мл и переносят в чашечку для
измерения активности. Выпаривают раствор досуха под лампой (тяга!)
и измеряют активность остатка с помощью счетной установки с
детектором мягкого р- или у-излучения в стандартных условиях (измерение
активности— см. стр. 341). Рассчитывают удельную активность
кобальта в имп/ (мл • г-же).
К анализируемому раствору хлорида калия (5—10 мг) в
центрифужной пробирке емкостью 10 мл, с делениями добавляют 5 мл
20%-ного раствора LiN02, чистой пипеткой емкостью 2—5 мл со
шприцем приливают 2 мл исходного раствора меченого нитрата кобальта и
подкисляют 2 н. раствором уксусной кислоты. Содержимое пробирки
перемешивают и оставляют стоять на 2 ч. Затем осадок
центрифугируют, раствор декантируют и осадок промывают 5%-ным раствором
нитрата калия, подкисленного уксусной кислотой. Промытый осадок
растворяют в 2 н. соляной кислоте в центрифужной пробирке при
нагревании (тяга!), доводят до объема 10 мл, отбирают пипеткой 1 мл в
чашечку для измерения активности и выпаривают под лампой (тяга!).
Активность остатка измеряют в стандартных условиях.
Содержание калия в растворе вычисляют по формуле (35.)
§ 22. Метод радиометрического титрования
Метод основан на образовании определяемым ионом с реагентом
малорастворимого или легко экстрагируемого соединения, т. е.
соединения, легко удаляемого из титруемого раствора. Индикатором служит
изменение радиоактивности раствора по мере введения реагента. Точку
эквивалентности определяют по излому на кривой титрования объем
введенного реагента — радиоактивность титруемого раствора. Кривые
титрования для различных случаев приведены на рис. 134. Концентра*
ция реагента (г-экв/л) должна быть известна.
Возможны три варианта метода.
1. Радиоактивным изотопом метят один из элементов, входящих в
состав титруемого раствора. В процессе титрования измеряют
изменение активности раствора над осадком или экстракта образовавшегося
соединения. Кривые титрования для этого случая представлены на
рис. 134, а.
Для практического определения эквивалентного объема
титрованного раствора при титровании этим способом сначала находят
начальную активность раствора /о и добавляют такой объем реагента V\,
чтобы в реакцию вошла приблизительно половина определяемого вещества.
Затем измеряют активность полученного раствора /ь Расчет ведут по
формуле:
Jo — h
При измерении активности органической фазы /i = /o — /i в случае
применения экстракционного метода объем У0 вычисляют по формуле:
Vo-V^ (38)
§ 22. МЕТОД РАДИОМЕТРИЧЕСКОГО ТИТРОВАНИЯ
349
2. Титрование ведут реагентом, содержащим радиоактивный
изотоп. Измеряется увеличение активности титруемого раствора после
точки эквивалентности, т. е. после окончания образования осадка
определяемого иона с реагентом или соединения, удаляемого экстракцией.
Кривые титрования представлены на рис. 134,6.
Для практического определения эквивалентного объема необходимо
измерить активность /2 раствора после прибавления небольшого избытка
реагента объемом V2 и активность /3 после добавления новой порции
реагента объемом 1/3:
™-™,. (39)
Уо=-
h-h
3. Титрование раствора, содержащего радиоактивный изотоп
определяемых ионов, ведут реагентом, также содержащим радиоактивный
Рис. 134. Кривые титрования:
«—активный раствор; б —активный реагент; в — активный реагент и раствор; г—активный
второй компонент анализируемой смеси; д — активный первый и третий компоненты смеси;
1 — осаждение; 2 — экстракция.
изотоп. Измеряется изменение активности по мере введения реагента
в раствор. Точка эквивалентности отвечает минимуму активности
раствора над осадком или максимуму активности экстракта. После точки
эквивалентности начинается нарастание активности титруемого
раствора над осадком за счет добавляемого реагента. Кривые титрования
представлены на рис. 134, е. Этот способ целесообразно применять
только, наряду с графическим методом нахождения точки
эквивалентности.
Для определения точки эквивалентности в первых двух способах
не следует снимать всю кривую титрования. Достаточно провести два-
три измерения и вычислить точку эквивалентности расчетным путем.
Если объем прибавляемого к раствору реагента относительно велик
и измеряют активность лишь аликвотной части раствора, то
измеренную активность раствора (водного) необходимо умножить на
поправочный множитель Д, связанный с изменением объема:
л=Пач + 7до1 (40)
350
ГЛ. IX. РАДИОМЕТРИЧЕСКИЕ МЕТОДЫ АНАЛИЗА
Описанные методы титрования применимы при отсутствии
мешающих элементов, т. е. ионов, образующих с реагентом в тех же условиях
осадок или экстрагируемое комплексное соединение. Если условия
образования осадков или экстрагируемых комплексных соединений у
разных типов ионов различны, то с помощью одного радиоактивного
изотопа возможно последовательное определение этих ионов. Например,
для определения ионов цинка и ртути в смеси с применением
радиоактивного изотопа 65Zn титрование ведут дитизоном при рН = 4,7.
Сначала образуется только комплексное соединение ртути, при этом
хлороформный экстракт неактивен, а водный
раствор имеет постоянную активность.
После первой точки эквивалентности нач-
К водоструйному нется образование комплекса цинка с ди-
^ насосу тизоном, активность хлороформного
экстракта возрастает, а активность водного
слоя падает до второй точки
эквивалентности (см. рис. 134, г).
Этот способ применим и для
последовательного определения трех типов ионов
в растворе. Радиоактивным изотопом
метят ионы, образующие наиболее прочное и
наименее прочное комплексное соединение.
Кривая титрования имеет вид, показанный
на рис. 134, д.
Радиометрическое титрование
экстракционным способом обычно осуществляется
в пробирках с притертой пробкой. После
добавления реагента и экстрагента,
перемешивания и расслаивания фаз измеряют
активность одной из фаз.
Радиометрическое титрование методом
осаждения обычно проводится в приборе,
изображенном на рис. 135. Реагент из
бюретки поступает в сосуд для титрования.
Счетчик для измерения активности
окружен рубашкой, которая соединена с
сосудом для титрования пористым фильтром, а двухходовым краном — с
водоструйным насосом и системой сжатого воздуха. Для измерения
активности раствора с помощью водоструйного насоса его всасывают в
рубашку счетчика. После измерения раствор сжатым воздухом
вытесняют в сосуд для титрования. Воздух, вытесняющий раствор,
одновременно хорошо его перемешивает.
Рис. 135. Прибор для
радиометрического титрования:
/ — счетчик Гейгера— Мюллера;
2 — стеклянная рубашка; 3 —
стеклянный фильтр; 4 — бюретка;
5 — трехходовой кран; 6 —
титруемый раствор.
§ 23. Практические работы, выполняемые методом
радиометрического титрования
Определение фосфат-ионов
Определение фосфат-ионов методом радиометрического титрования
проводят путем осаждения меченых 32Р фосфат-ионов магнезиальной
смесью:
Mg++ + NH4+ + PO™ —> |MgNH4P04
Методика определения. Работа проводится по третьему классу
(см. стр. 329). Исследуемый раствор, содержащий фосфат-ионы
§ 23. РАБОТЫ, ВЫПОЛНЯЕМЫЕ МЕТОДОМ РАДИОМЕТРИЧЕСКОГО ТИТРОВАНИЯ 351
( — 5*10"*3 моль), помещают в мерную колбу емкостью 25 мл, стоящую
в кювете, добавляют 1 мл раствора Na2HP04, содержащего 1 мккюри
32Р с удельной активностью не ниже 1 мккюри/мг фосфора, и
разбавляют раствор водой до метки. Из колбы в стаканы емкостью 200 мл
пипеткой со шприцем отбирают две пробы раствора по 10 мл9
добавляют к каждой пробе по 5 мл 25%-ного раствора аммиака и б г
твердого хлорида аммония и затем дистиллированной водой раствор
разбавляют до объема 50 мл. Все эти операции проводят в кювете на
лабораторном столе.
В кристаллизатор со льдом ставят стакан е раствором и вводят
в него трубку с пористым фильтром прибора для радиометрического
титрования (см. рис. 135). Измеряют фон счетчика в течение 1 мин
(измерение активности — см. стр. 341). Всасывают пробу раствора
через фильтр в рубашку счетчика и измеряют активность пробы в
течение времени, достаточного для регистрации 3—5 тысяч импульсов.
Затем раствор спускают в стакан и повторяют измерение фона
счетчика.
Титрованный 0,1 М раствор хлорида магния наливают в бюретку.
Затем приливают 1 мл раствора MgCl2 в стакан с раствором фосфата,
смесь перемешивают и раствор просасывают через пористый фильтр
в рубашку счетчика. Снова проводят измерение активности. Раствор
спускают в стакан и прибавляют еще 1 мл стандартного раствора MgCl2.
Измеряют фон счетчика. Повторяют операции измерения активности и
наносят результаты на график в координатах активность — объем
добавленного реагента. Добавление раствора MgCl2 ведут до тех пор, пока
на графике не получится четкий излом прямой. Определяют
графически точку эквивалентности. Повторяют определение со второй
параллельной пробой раствора и рассчитывают содержание фосфат-ионов
в исследуемом растворе.
Определение ионов магния
Определение ионов магния методом радиометрического титрования
проводят путем осаждения их фосфат-ионами, содержащими 32Р.
Уравнение реакции аналогично приведенному в методике «Определение
фосфат-ионов» (см. стр. 350).
Методика определения. Работа проводится по третьему классу (см.
стр. 329). Прежде чем приступить к ее выполнению, надевают халат,
шапочку и резиновые перчатки. Работу проводят на лабораторном
столе, в кювете.
Исследуемый раствор, содержащий ионы магния (~5-10~3 моль),
помещают в мерную колбу емкостью 25 мл и доводят раствор
дистиллированной водой до метки. Переносят пипеткой в два стакана емкостью
200 мл пробы раствора по 10 мл. В каждый стакан добавляют по 5 мл
25%-ного раствора аммиака и 5 г хлорида аммония. Полученный
раствор дистиллированной водой доводят до ~50 мл, помещают в
кристаллизатор со льдом и затем вводят в него трубку с пористым фильтром
прибора для радиометрического титрования (см. рис. 135).
В бюретку емкостью 25 мл помещают титрованный 0,1 М раствор
Na2H32P04 с удельной активностью 0,5 мккюри!мл. Измеряют фон
счетчика в течение 1 мин. Затем из бюретки прибавляют 1 мл
титрованного раствора радиоактивного фосфата, смесь перемешивают и
раствор просасывают через пористый фильтр в рубашку счетчика.
Активность измеряют в течение времени, достаточного для регистрации
5 тысяч импульсов. Раствор спускают из рубашки счетчика в стакан и
352
ГЛ. IX. РАДИОМЕТРИЧЕСКИЕ МЕТОДЫ АНАЛИЗА
прибавляют еще I мл раствора радиоактивного фосфата из бюретки.
Снова измеряют активность раствора.
Данные титрования наносят на график в координатах активность
раствора — объем добавленного реагента. Добавление радиоактивного
фосфата повторяют до тех пор, пока на графике не выявится излом и
наклон прямой после излома. Повторяют титрование с параллельной
пробой. Графически находят точку эквивалентности и рассчитывают
содержание ионов магния в исследуемом растворе.
Определение ионов цинка и меди при совместном присутствии
Гексацианоферрат (II) калия осаждает из раствора ионы меди и
цинка:
2Cu+++ [Fe(CN)6r= —> | Cu2[Fe(CN)6]
2Zn++ + [Fe(CN)6]== —> ф Zn2[Fe(CN)G]
Так как растворимость гексацианоферрата (II) меди много меньше,
чем гексацианоферрата (II) цинка, то до завершения полного
осаждения Cu2[Fe(CN)6] ионы цинка остаются в растворе. Это дает
возможность дифференцированно титровать ионы меди и цинка в присутствии
одного индикатора.
Методика определения. Работа проводится по третьему классу (см.
стр. 329). Прежде чем приступить к работе, надевают халат, шапочку
и резиновые перчатки. Работу с радиоактивным раствором проводят
в кювете, на лабораторном столе.
Анализируемый раствор помещают в мерную колбу емкостью
100 мл, добавляют 2 мл концентрированного раствора НС1 и пипеткой
со шприцем 1 мл раствора, содержащего радиоактивный изотоп 65Zn
с удельной активностью порядка 105 имп/(мин- мл) и не менее
1 мкюри/г. Дистиллированной водой доводят объем раствора до метки.
Пипеткой со шприцем переносят по 5 мл исследуемого раствора из
мерной колбы в восемь центрифужных пробирок и добавляют в них из
микробюретки 0,15; 0,30; 0,45; ...; 1,2 мл 0,2 М раствора K4[Fe(CN)6].
Выделившиеся осадки отделяют центрифугированием. Из каждой
пробирки пипеткой емкостью 1 мл со шприцем отбирают пробы
прозрачного раствора по 1 мл в чашечки для измерения активности.
Последовательность отбора проб — от восьмой пробирки к первой. Затем берут
пробу в 1 мл из мерной колбы. Все растворы в чашечках упаривают
досуха под лампой для выпаривания [тяга!). Измеряют фон счетчика.
Затем измеряют активность остатка в каждой чашечке в течение
времени, достаточного для регистрации 3—5 тысяч импульсов. После
каждого измерения определяют фон счетчика. Результаты измерения
активности за вычетом фона наносят на график в координатах активность —
объем реагента. Точку эквивалентности определяют графически.
Повторяют определение ионов меди и цинка и вычисляют их содержание
в исследуемом растворе.
§ 24. Метод изотопного разбавления
Метод основан на тождественности химических реакций изотопов
данного элемента. Он применим для количественного определения
компонентов трудно разделяемых сложных смесей.
Для его осуществления к анализируемой смеси, содержащей
tnx г-экв определяемого вещества, добавляют та г-экв того же
вещества, содержащего в своем составе радиоактивный изотоп с актив-
§ 25. РАБОТЫ, ВЫПОЛНЯЕМЫЕ МЕТОДОМ ИЗОТОПНОГО РАЗБАВЛЕНИЯ 353
ностыо /о. Удельная активность S0 введенного радиоактивного вещества
должна быть известна. После равномерного распределения
радиоактивного вещества в системе любым доступным способом выделяют часть
{ш\ г-экв) определяемого вещества в чистом состоянии и измеряют ее
активность /j или весовую удельную активность S\. Удельная
активность выделенной части соединения равна удельной активности
определяемого вещества в растворе после добавления изотопного
разбавителя:
Sieii- = Ь . (41)
nil rtio + тх
Отсюда /0 ,...
тх = -~- Шх — Шо (42)
или, принимая во внимание, что ——=^S0i находим:
mx = mJ^--l) (43)
Если содержание добавляемого вещества т0 «С тх, то
И /
(44)
лц (45)
-л л
Следует отметить, что в анализируемой смеси не должно быть
веществ, способных к изотопному обмену с введенным радиоактивным
веществом.
Для выделения некоторой части искомого соединения применяют
любые методы, позволяющие выделить чистое соединение:
экстрагирование, хроматографию, перегонку, осаждение, электролиз,
электрофорез и т. п. Определение содержания выделенного вещества выполняется
колориметрическим, спектрофотометрическим, весовым, объемным и
другими методами. Можно также рассчитать содержание выделенного
вещества, исходя из объема и концентрации реагента, примененного
для осаждения.
§ 25. Практические работы, выполняемые методом
изотопного разбавления
Определение ионов иода в присутствии ионов брома
Количественное определение ионов иода, брома и хлора при их
совместном присутствии путем осаждения галогенидов серебра
затруднено вследствие почти одинаковой растворимости галогенидов серебра.
Однако в аммиачном растворе иодид серебра может быть частично
осажден в чистом состоянии без примесей бромида и тем более хлорида
серебра. Это позволяет определять ионы иода в присутствии ионов
брома и хлора методом изотопного разбавления.
Методика определения. Работа проводится по третьему классу (см.
стр. 329). Прежде чем приступить к выполнению работ, надевают халат,
шапочку и резиновые перчатки. Все операции с радиоактивным
раствором проводят в кювете, на лабораторном столе.
Анализируемый раствор, содержащий смесь ~0,3 г иодида и
~0,2 г бромида натрия, помещают в колбу емкостью 50 мл и доводят
до метки дистиллированной водой. В два стакана емкостью 100 мл
берут пипеткой две пробы точно по 10 мл и в каждый добавляют
пипеткой со шприцем по 1 мл раствора 1311 с точно известной удельной
354
ГЛ. IX. РАДИОМЕТРИЧЕСКИЕ МЕТОДЫ АНАЛИЗА
активностью [5000 имп/(мин»мл) без носителя], 2 мл
концентрированного раствора аммиака и разбавляют растворы 10 мл воды. Если
удельная активность радиоактивного раствора неизвестна, то в третий стакан
из бюретки наливают 6 мл 0,05 М раствора неактивного иодида натрия,
пипеткой со шприцем вводят 1 мл активного раствора, 2 мл
концентрированного раствора аммиака и также разбавляют 10 мл
дистиллированной воды. Затем проводят частичное осаждение иодида серебра
в двух первых и полное в последнем стакане добавлением из бюретки
при помешивании в первом случае 4 мл 0,05 М раствора AgN03, а во
втором случае 6 мл того же раствора. Для лучшей коагуляции осадка
к раствору добавляют несколько кристалликов нитрата аммония и
осторожно нагревают растворы с осадком почти до кипения (тяга!). Чтобы
избежать выделения свободного иода, к раствору перед кипячением
добавляют несколько кристалликов сульфита натрия. Осадки
количественно переносят посредством холодного 0,01 М раствора HN03 на
плотный фильтр, в специальной воронке для приготовления осадков для
измерения, промывают небольшим количеством дистиллированной воды и
спиртом или ацетоном, сушат на воронке при открытом водоструйном
насосе, закрыв воронку крышкой от тигля для предохранения от
разложения осадка под действием света. Активность осадков на фильтре
измеряют на счетчике в течение такого времени, чтобы число
подсчитанных импульсов равнялось 3—5 тысячам.
Исходя из объема и концентрации раствора AgN03, рассчитывают
удельную активность исходного раствора радиоактивного иодида
натрия. Из полученных значений удельной активности, активности проб и
их массы рассчитывают содержание иода в анализируемом растворе.
§ 26. Метод активационного анализа
В основе метода лежит образование радиоактивного изотопа из
определяемого элемента под воздействием облучения анализируемого
образца ядерными частицамр.
Активность образовавшегося радиоактивного изотопа
пропорциональна числу атомов определяемого элемента, интенсивности потока
ядерных частиц и сечению ядерной реакции этих частиц с определяемым
элементом. В соответствии с законом накопления радиоактивного
изотопа при облучении какого-либо вещества активность (распад/сек)
к концу облучения может быть вычислена по формуле:
л *по 1п2Ч tnKOFo [\ / 0,693/, 41
А = 6,02 • 1023 А 1 - ехр / - '^ Ч (46)
где m — количество определяемого элемента, г;
к — относительное содержание активируемого изотопа в элементе;
а — эффективное сечение ядерной реакции, см2;
F0 — интенсивность потока бомбардирующих частиц, частиц/(см2 • сек);
tx — время облучения, сек\
Лв—атомный вес элемента, из которого образуется радиоактивный изотоп;
Ту — период полураспада образующегося изотопа, сек.
Для определения величины тп необходимо, следовательно, знать
величину а, /с, Лв, 7\/2 и найти величину Л0, зная t\. На практике
измерение активности ведется через некоторое время t2 после прекращения
облучения, вследствие чего следует в формулу (46) ввести поправку на
распад радиоактивного изотопа. В этом случае
§ 26. МЕТОД АКТИВАЦИОННОГО АНАЛИЗА
355
где At — активность образовавшегося при облучении радиоактивного изотопа из
определяемого элемента к моменту времени /2 после окончания облучения.
Этот так называевый абсолютный метод может дать правильные
результаты при условии постоянства интенсивности потока ядерных
частиц в течение всего времени облучения и во всем объеме мишени,
постоянства энергетического спектра частиц, от которого зависит
величина а, и постоянства числа активируемых атомов в мишени, т. е. число
их не уменьшается в процессе облучения мишени.
Постоянство интенсивности потока нейтронов нарушается, если
основной материал облучаемой мишени имеет высокое сечение ядерной
реакции с нейтронами и последние сильцо поглощаются поверхностными
слоями мишени.
Абсолютный метод обладает малой точностью (ошибка достигает
40—50%), так как соблюдение перечисленных выше условий
затруднительно, а сечение ядерных реакций определено с невысокой точностью.
Поэтому на практике пользуются относительным методом, в котором
одновременно с исследуемым образцом облучению подвергается один
или несколько эталонов с известным содержанием определяемого
элемента. Состав эталонов должен быть близок к составу анализируемых
образцов. В этом случае величина сечения ядерной реакции и
изменение интенсивности потока ядерных частиц, вносящие основные ошибки
в определение, не играют роли. Расчет проводится по формуле:
где тх и тэ — содержание элемента в анализируемом образце и эталоне;
/*'И 1Э ~ активность анализируемого образца и эталона, приведенных к одному
времени измерения.
При наличии серии эталонов пользуются графическим методом.
Точность относительного метода около 10%.
Измерение активности облученных образцов можно проводить
непосредственно после их переведения в раствор, а также после
предварительного химического. выделения определяемого элемента в осадок,
экстракт или раствор любыми доступными методами.
В большинстве случаев в результате облучения вследствие
параллельных ядерных реакций образуется смесь радиоактивных изотопов
ряда элементов из элементов, входящих в состав анализируемого
образца.
В отдельных случаях возможно образование и того изотопа, по
которому ведется определение, но не по основной, а по параллельным
реакциям. Так, при облучении кремния по реакции
30Si(n,Y)3,Si -^-> 31Р
образуется фосфор, и, следовательно, определению фосфора мешает
кремний. Это сильно снижает точность определения фосфора в кремнии.
Поэтому прямое измерение радиоактивности мишени, как правило,
неприменимо. Если основной материал мишени активируется мало, может
применяться измерение излучения определенной энергии, отвечающей
искомому радиоактивному изотопу, с помощью ^-спектрометров. Однако
чувствительность такого метода мала.
Предварительное разделение химическими методами дает
возможность отделить изотоп, образовавшийся при облучении определяемого
элемента, от мешающих определению активности радиоактивных
компонентов смеси частично (для спектрометрических определений) или
полностью (в случае дальнейшего измерения с помощью счетчиков).
356
ГЛ. IX. РАДИОМЕТРИЧЕСКИЕ МЕТОДЫ АНАЛИЗА
Химическое разделение также позволяет определять несколько
элементов из одной облученной мишени.
По типу частиц, применяемых для облучения, различают:
нейтронный активационный анализ (облучение нейтронами), фотоактивацион-
ный анализ (облучение фотонами высокой энергии) и активационный
анализ с помощью заряженных частиц.
Наиболее важным явится нейтронный активационный анализ,
который проводится с применением тепловых (~0,001 Мэв) или быстрых
(1 —14 Мэв) нейтронов. В первом случае образование радиоактивного
изотбпа идет по реакции захвата нейтрона с испусканием фотона (л, у).
Во втором случае протекают главным образом реакции захвата
нейтрона с выбрасыванием протона (я, /?), а-частицы (/г, а) или двух
нейтронов (я, 2п).
Для осуществления нейтронного активационного анализа необходимы
потоки нейтронов высокой интенсивности, поэтому он проводится в
ядерном реакторе или иногда с помощью нейтронного генератора.
Чувствительность нейтронного активационного анализа в
реакторах с замедлителем нейтронов при потоке 1012 нейтронов/Ъи2 • сек
показана ниже:
Определяемые элементы
Са, Si, Bi, Fe
Sr, Ag, Hg, Nd, Tl, Zr, Sn, Cr, Mo, Ru, Pt . . .
K, Cs, Ba, Zn, Cd, Y, Ce, Er, Ge, Pb, Hf, P, Se,
Co, Ni, Os
Na, Cu, Au, Al, La, Pr, As, Sb, Se, I
Tb, Tm, Yb, Та, W
In, Sm, Ho, Mn, Re
Eu, Dy
С увеличением времени облучения чувствительность определения
повышается.
Таким образом, нейтронный активационный анализ обладает
невысокой точностью, но очень высокой чувствительностью. Поэтому он
применяется главным образом для определения ультрамалых примесей
в чистых материалах.
Активационный анализ обладает высокой специфичностью, так как
радиоактивный изотоп, образующийся из определенного элемента или
одного из изотопов изотопной смеси данного элемента, обладает только
ему присущим периодом полураспада, характером и энергией излучения.
Применение активационного анализа связано с необходимостью
принятия мер предосторожности при работе. Для его проведения
необходима специально оборудованная лаборатория.
При наличии реактора, оборудованного пневмопочтой, для актива-
циоиных определений используют реакции образования короткоживу-
щих радиоактивных изотопов. В этом случае проводятся измерения
активности облученных образцов без химической обработки с
использованием у-спектРометРов- В отдельных случаях использование для
активационного анализа короткоживущих изотопов возможно и не в
реакторе. Так, для определения кислорода образцы облучают фотонами с
энергией 10—30 Мэв с помощью бетатрона или линейного ускорителя
электронов, при торможении которых на мишени появляется поток
тормозного излучения — фотонов высокой энергии. Из кислорода по
реакции 160 (у, п) 150 образуется радиоактивный изотоп кислорода с
Гуа = 2,1 мин и энергией позитронов 1,6 Мэв.
Чувствительность
определения,
10"
10"
10"
10"
10"
10"
10"
-6
-7
-8
-9
-10
-и
-12
г
§ 28. ФОТОНЕЙТРОННЫЙ МЕТОД
357
§ 27. Практические работы, выполняемые методом
активационного анализа
Определение ионов индия
Естественная смесь изотопов индия состоит из двух изотопов: 1131пи
П51п. Эффективное сечение реакции захвата медленных нейтронов пз1п
равно 2,5 барн и И51п— 138 барн. При захвате нейтронов идут реакции:
п51п+,„ -> 1,6in + V; U6m s^> ,16sn
I,aln + i» -> "4mIn + Y; IM*In shs* 1UI" -rfe- U4Sn
При кратковременном облучении благодаря высокому
эффективному сечению реакции захвата нейтронов 1151п и малому сечению 1131п
основная активность облученного ийдия будет зависеть от 1161п.
В работе используется относительный метод нейтронного
активационного анализа раствора, содержащего индий. Активация проводится
лабораторным источником нейтронов с интенсивностью потока не ниже
10б нейтронов в 1 сек.
Методика определения. Работа выполняется по третьему классу
(см. стр. 329). Облучение нейтронами ведут в специальном помещении
согласно инструкции. Все операции с растворами осуществляются в
кювете, на лабораторном столе.
В шесть пробирок из кварца или плексигласа помещают по 10 мл
эталонных растворов, содержащих индий, и анализируемый раствор.
Пробирки вставляют в ячейки парафинового блока и облучают с
помощью лабораторного источника нейтронов, обеспечивающего
интегральный поток ~1010 нейтронов. Для этой цели парафиновый блок
помещают вблизи места хранения источника нейтронов. Источник вносят,
соблюдая меры предосторожности против облучения, в центральное
отверстие парафинового блока. По прошествии необходимого времени
(^2 ч) источник перемещают в место хранения (см. инструкцию по
использованию нейтронного источника), вынимают образцы и эталоны
и проводят измерение их активности, опуская поочередно пробирки
в колодец кристалла сцинтиллмционного счетчика. Определяют
активность к моменту прекращения облучения образцов и эталонов.
Строят график зависимости активности эталонов от содержания
в них индия. Используя эталонный график, по величине активности
анализируемого раствора находят в нем содержание индия.
§ 28. Фотонейтронный метод
Метод основан на образовании нейтронов под действием фотонов
высокой энергии на ядра химических элементов:
Количество нейтронов регистрируют с помощью нейтронных
счетчиков.
Для осуществления этой реакции необходимы фотоны, энергия
которых больше энергии связи нуклонов в ядре. Энергия связи нуклонов
в ядре большинства химических элементов составляет около 8 Мэв.
Для получения фотонов такой энергии необходимы специальные
установки, которые затрудняют измерение интенсивности потока
нейтронов. Энергия Y-квантов радиоактивного распада не превышает ЗМзя,
358
ГЛ. IX. РАДИОМЕТРИЧЕСКИЕ МЕТОДЫ АНАЛИЗА
С помощью у-квантов радиоактивных источников возможны лишь
реакции с дейтерием (энергия связи нуклонов — 2,226 Мэв) и бериллием
(энергия связи нуклонов — 1,666 Мэв).
В настоящее время фотонейтронный метод применяется только для
определения бериллия: 9Ве (у, п) 8Ве. Если в анализируемом образце
наряду с бериллием находятся атомы дейтерия, необходимо исключить
образование нейтронов по реакции 2Н (у, п) *Н, т. е. следует
использовать источник фотонов с энергией у-квантов в пределах 1,7—2,2 Мэв.
Таким источником является 124Sb, период полураспада которого равен
60,1 дня, энергия фотонов— 1,7 и 2,1 Мэв и средняя энергия
фотонейтронов, образующихся при реакции с бериллием, 0,024 Мэв. Недостатком
этого источника является
сравнительно короткий период
полураспада, вследствие чего
интенсивность его
уменьшается в 2 раза в течение
двух месяцев.
Установка для
проведения анализа (рис. 136)
состоит из источника 5,
помещенного в свинцовый корпус 1
для поглощения у-излучения с
целью защиты
экспериментатора. Источник крепится на
штанге 3\ в верхнем
положении штанги источник нахо*
дится вне кольцеобразного
сосуда для исследуемого
образца б и в нижнем рабочем
положении — в сосуде. Сосуд
для исследуемого образца
находится на выдвижном
столике 7 и выдвигается наружу
при помощи рукоятки 8.
Интенсивность источника
у-излучения контролируется
счетчиком 12, соединенным с
регистрирующим прибором //. На средней части свинцового корпуса коакси-
ально расположен контейнер 14, заполненный парафином, для
замедления нейтронов. Контейнер имеет несколько вертикальных каналов,
отстоящих на равных расстояниях от центра источника. В каналах
расположены борные пропорциональные счетчики нейтронов СНМО-5
13. Счетчики нейтронов соединяются параллельно с выходом на один
регистрирующий прибор с амплитудным дискриминатором. При
четырех борных счетчиках, использовании в качестве источника
у-излучения 100 мкюри l24Sb и массе исследуемой пробы 100 г чувствительность
установки составит 4« 10"4%^
Вся установка для безопасности работы окружена наружной
свинцовой защитой 9.
При определении последовательно измеряют интенсивность потока
нейтронов от эталонного и анализируемого образцов, затем содержание
бериллия находят графически или вычисляют по формуле;
Рис. 136. Установка для фотонейтронного
определения бериллия:
/ — свинцовый корпус; 2 — корпус подшипника;
3 — штанга; 4 — стопор, закрепляющий штангу; 5
—источник Y-излучения; 6 — рабочий сосуд с исследуемым
веществом; 7 — выдвижной столик; 8 — рукоятка;
9 — защитная свинцовая стенка; 10, /5 —блоки
электропитания счетчиков; // —внешний регистрирующий
прибор; /2 —счетчик Y-излучения; 13 — счетчик
нейтронов; 14 — контейнер; 16-прибор для регистрации
нейтронов.
С -Г Iх-
(49)
§ 28. ФОТОНЕЙТРОННЫЙ МЕТОД
359
где Сх — концентрация бериллия в исследуемом растворе;
Сэ — концентрация бериллия в эталоне;
/х — активность анализируемого образца;
/э — активность эталона.
Для правильного проведения анализа сыпучих образцов также
необходимо соблюдение одинаковых условий облучения. Формула (49) для
этого случая преобразуется в следующую:
»-*-ёг (50)
где рх и рэ — содержание бериллия соответственно в образце и эталоне, %;
тх и тэ— масса анализируемого и эталонного препаратов.
Для повышения точности измерений необходимо, чтобы эталоны и
анализируемый образец имели близкий состав и плотность. Жидкие
эталоны готовят растворением навесок окиси бериллия в
концентрированной серной кислоте, твердые — смешением окиси бериллия с
наполнителем, которые размельчают до 200—400 меш. и тщательно
перемешивают.
При проведении анализа возможно поглощение тепловых нейтронов
в исследуемом образце в том случае, если в нем присутствуют элементы
с высоким эффективным сечением захвата тепловых нейтронов, такие,
как литий, бор, кадмий, редкоземельные элементы. Для учета этой
потери нейтронов проводят измерение потока нейтронов, возникающего
при анализе эталонного препарата. Затем повторяют измерение,
поместив эталонный препарат за кадмиевый экран толщиной 1 мм,
который полностью поглощает тепловые нейтроны. Разница двух
выполненных измерений, отнесенная к первому измерению, представляет
собой максимально возможную ошибку, которая колеблется в
пределах 4—16%.
С уменьшением количества анализируемого вещества указанная
ошибка уменьшается, однако падает и чувствительность определения.
При больших образцах чувствительность повышается, но так как
поглощение у-излУчения анализируемым образцом и эталоном
неодинаково, вследствие их различной плотности возникает ошибка,
составляющая несколько процентов. Для учета поглощения у-излучения в образце
проводят два определения содержания бериллия в сосудах разной
толщины.
Содержание бериллия (рх) в процентах вычисляют по формуле:
р*~Ри + 1?=г?а" (51)
где рм и /7б — содержание бериллия (концентрация) в малом и большом сосудах, %;
du и d& — толщина слоя вещества в малом и большом соосудах.
Ошибки метода при правильном проведении анализа слагаются из:
1) ошибки за счет поглощения нейтронов и ^"излУчения
образцом—2%;
2) ошибки анализа эталонов — 2%;
3) ошибки, связанной с определением поглощения образца в
приборе,-2%;
4) ошибки измерения—1%.
Чувствительность метода при источнике 100 мкюри 124Sb, времени
измерения 25 мин и фоне 1 имп/мин — 1 мг ВеО в образце*
360
ГЛ. ГХ. РАДИОМЕТРИЧЕСКИЕ МЕТОДЫ АНАЛИЗА
§ 29. Практические работы, выполняемые фотонейтронным
методом
Определение бериллия
Методика определения. В работе используется закрытый источник
Y-излучения с активностью 100 мкюри. Эта работа требует специального
помещения, в котором располагается установка для анализа.
Перемещение источника в установке должно осуществляться в соответствии
с инструкцией. Перед работой надевают халат и шапочку.
Включают счетную установку в электрическую сеть и прогревают
ее в течение 30 мин.
Устанавливают на детекторах паспортное рабочее напряжение.
Регулируя порог дискриминации (см. инструкцию), добиваются фона
в 1 имп/мин на каждый включенный счетчик. Измеряют фон установки.
В кольцеобразный рабочий сосуд (см. рис. 136) с внешним
диаметром 19 мм, с внутренним диаметром 5 мм и высотой 70 мм помещают
эталонный раствор с известным содержанием бериллия (порядка 0,5—
0,8 г-атом/л). Вдвигают столик с рабочим сосудом внутрь установки и
опускают штангу с источником в рабочее положение. Измеряют
интенсивность потока нейтронов в течение 3 мин. Поднимают источник у-из-
лучения в верхнее положение. Вынимают рабочий сосуд с раствором из
установки. Затем в рабочий сосуд такого же диаметра наливают
анализируемый раствор и проводят измерение, как указано выше. Затем
снова в течение трех минут измеряют фон установки. Содержание
бериллия в анализируемом образце вычисляют по формуле (49), вычтя
из полученных значений активности величину фона установки.
Повторяют все операции измерения с рабочими сосудами,
внешний диаметр которых меньше 8 мм, и снова рассчитывают содержание
бериллия в растворе по формуле (49).
По формуле (51) рассчитывают содержание бериллия с учетом
поглощения уизлучения анализируемым и эталонным материалами.
Закрыв рабочий сосуд кадмиевым экраном, повторяют измерение
нейтронного потока от образца. По полученным данным рассчитывают
максимальную ошибку от поглощения нейтронов анализируемым
материалом. Оценивают ошибку метода и его чувствительность.
§ 30. Методы определения содержания химических элементов
по излучению их естественных радиоактивных изотопов
Многие химические элементы являются радиоактивными, т. е. все
их изотопы радиоактивны. К ним относятся технеций, прометий и все
естественные и искусственные элементы, стоящие в периодической
системе элементов после висмута. Кроме того, ряд нерадиоактивных
химических элементов в естественной смеси изотопов содержит
радиоактивные изотопы.
Следует сказать, что на практике из нерадиоактивных элементов
только калий определяется этим способом.
Для определения содержания радиоактивных элементов или
нерадиоактивных, содержащих в естественной смеси радиоактивные изотопы,
можно использовать два способа.
В первом способе определяется активность анализируемого образца
(распад/сек) и по формуле (1) рассчитывается количество искомого
элемеат^
§ 31. ОПРЕДЕЛЕНИЕ ЭЛЕМЕНТОВ ПО ИЗЛУЧЕНИЮ ИХ ИЗОТОПОВ 361
Величина X должна быть известна; Этот способ сложен, так как
определение активности (распад/сек) в большинстве случаев
представляет собой трудную задачу (см. стр. 344).
Практически более доступно относительное измерение, т. е.
измерение активности анализируемого образца и эталона или серии эталонов
с известным содержанием определяемого элемента. Необходимо, чтобы
была достигнута полная идентичность условий измерения образца и
эталонов: положение их относительно детектора излучения, толщина
слоя и плотность. В этом случае содержание элемента в анализируемом
образце может быть найдено по калибровочному графику в
координатах активность — содержание элемента или по формуле:
рх = Рэ-^- (52)
где рх и рэ — содержание искомого элемента в образце и эталоне;
1Х и /э — активность анализируемого образца и эталона.
Третий способ основан на определении количества дочернего
радиоактивного изотопа, накапливающегося за определенный промежуток
времени из материнского:
ЯдЛГд
^ = / -л*ч <53>
кЬ-е V)
где ЛГд и ЛГМ — количества атомов дочернего и материнского радиоактивных изотопов;
Яд и Ям — константы распада дочернего и материнского изотопов;
/ — время накопления дочернего изотопа из чистого материнского.
Этот способ применяется для определения содержания тех изотопов,
которые распадаются с образованием других радиоактивных изотопов,
например радия по дочернему радону, урана по UXi и т. п.
В этом способе, как и в предыдущем, можно измерить абсолютную
активность дочернего элемента и по формуле (46) вычислить число
атомов материнского элемента и его содержание в анализируемом
образце. Более применим относительный метод, в котором активность
дочернего элемента в анализируемом образце определяют сравнением ее
с активностью эталонного образца, содержание материнского элемента
в котором известно. Накопление дочернего элемента в образце и
эталоне должно осуществляться за одно и то же время, и материнский
элемент перед накоплением должен быть тщательно химически очищен от
дочернего. В этом случае для расчета применима формула (52).
Если продолжительность накопления дочернего элемента в образце
и эталоне неодинакова, то необходимо вносить поправку на время
накопления и расчет проводить по формуле:
ix i-rv»
где /ih/2- время накопления дочернего элемента в анализируемом образце и эталоне.
§ 31. Практические работы по определению содержания
химических элементов методом измерения излучения
их естественных радиоактивных изотопов
Определение калия по его естественной радиоактивности
В работе проводится определение калия по его естественной
радиоактивности абсолютным и относительным методами.
Естественная смесь изотопов калия содержит: 39К — 93,08%, 40К —
0,119% и 41К—6,91%. 40К является радиоактивным изотопом
362
ГЛ. IX. РАДИОМЕТРИЧЕСКИЕ МЕТОДЫ АНАЛИЗА
с периодом полураспада 1,2 • 1010 лет, испускающим р~-лучи с
максимальной энергией 1,33 Мэв, в 12% случаев имеет место Д"-захват. Пе^
риод полураспада этого изотопа столь велик, что 1 мг естественного
калия имеет всего около- двух распадов в 1 мин и для регистрации
излучения калия необходимо брать большие навески. В слое вещества про-*
исходит поглощение р~-частиц, максимальный пробег которых может
быть вычислен по формуле (18а), по их максимальной энергии —
1,33 Мэв. Он равен приблизительно 0,55 г/см2. При плотности
порошкообразных проб около 2 г/см3 слой полного поглощения ^'-излучения
40К составит приблизительно 0,3 см. Слои с толщиной более 0,3 см
имеют активность по р~-лучам при измерении в одинаковых
условиях, пропорциональную содержанию калия в образце. ^'Излучение
Рис. 137. Кювета сцинтилляционного
датчика для измерений активности
40К:
1 —исследуемый образец; 2 —экран из
тонкой алюминиевой фольги; 3 — пленка из
сцинтиллятора; 4 — светопровод из
плексигласа; 5 — фотоумножитель; 6 —
светозащитный чехол; 7 — резиновая прокладка.
Рис. 138. Кювета счетчика
Гейгера—Мюллера для измерения
активности 40К:
/ — счетчик; 2 — крышка и дно кюветы;
3 — полиэтиленовая пленка; 4 — исследуемое
вещество; 5 —корпус кюветы.
(1,45 Мэв) у сопровождающее i^-захват, увеличивает скорость счета
активности приблизительно на 20%, но она пропорциональна содержанию
40К в образце и при близких плотностях образца и эталона при
относительных измерениях не искажает результатов.
В случае относительных измерений строят калибровочный график
в координатах активность — содержание калия в эталонах. Затем
определяют активность анализируемого образца и по графику находят
содержание в нем калия.
Метод применим для определения калия в минералогических
образцах. Он дает возможность определить содержание калия около 3—5%
с ошибкой до 10% (относительных).
Активность (распад/сек) можно также определить путем измерения
активности слоя, толщина которого больше пробега |3~-частиц в данном
образце. В этом случае активность образца вычисляют по формуле:
г _ sSK
(55)
где /н— активность насыщенного слоя образца, зарегистрированная счетчиком,
имп/мин;
s— поверхность 1 г образца, см2/г;
Sk — удельная активность, имп/(мин • г);
|л — коэффициент самопоглощения в материале образца, г/см2.
По значению SK вычисляют удельную активность в абсолютных
единицах SK, о распад/(сек • г) по формуле (34) с заменой Л на SK,0 и /
на SK.
§ 32. МЕТОДЫ АНАЛИЗА, ОСНОВАННЫЕ НА ПОГЛОЩЕНИИ ИЗЛУЧЕНИЯ
Методика определения. Работа проводится без особых
предосторожностей.
В кювету сцинтилляционного датчика (рис. 137) из тонкой
органической пленки (1—2 мг/см2) или кювету счетчика Гейгера —
Мюллера (рис. 138) помещают образец исследуемого вещества. В первом
случае кювета помещается в колодец светопровода, выложенного сцин-
тиллятором в виде пленки (/г-терфенил в полистироле) и закрытого от
света тонкой алюминиевой фольгой (2 мг/см2). Во втором случае
кювета окружает р-счетчик Гейгера — Мюллера. При таких положениях
кюветы можно пренебречь поглощением излучения на пути к счетчику
и отражением р-излучения, а геометрический коэффициент счета
считать равным 1 и учесть лишь коэффициент самопоглощения, который
для 40К равен 8,9 см2/г.
Проводится измерение фона с образцом NaCl. Затем
последовательно проводится измерение серии образцов смесей NaCl и КС1 с
известным содержанием калия. По этим данным строят калибровочный
график зависимости активности от содержания калия в образцах.
Измерив активность анализируемых образцов — солей калия или их
смесей, находят по калибровочному графику содержание в них калия,
затем вычисляют содержание калия в анализируемых образцах по
формулам (1) и (55).
§ 32. Методы анализа, основанные на поглощении
излучения
Методы основаны на регистрации изменения интенсивности потока
ядерного излучения того или иного вида, прошедшего через слой
анализируемого материала.
Метод, основанный на поглощении Р - л у ч е й
Ослабление потока р-лучей в веществе подчиняется
экспоненциальному закону (4), который, принимая во внимание формулу (16), может
быть записан следующим образом:
/ = /0е в (56)
Используя это выражение при постоянном значении z/AB9 можно
определить р при известной толщине слоя (х) или х при известном р,
т. е. определить толщину или плотность материала.
В то же время при постоянном значении произведения рл: можно
определить отношение г/Ав. Для большинства элементов оно близко
к 0,5, а для водорода z/AB = 1. Это различие позволяет определить
отношение числа атомов водорода к числу атомов углерода.
Определение проводят в приборе (рис. 139). Излучение от
источника попадает в две ионизационные камеры, соединенные
противоположными полюсами через индикатор нулевого тока 3. В одну
ионизационную камеру излучение попадает через поглотитель постоянйой
толщины, а в другую —через испытуемый образец 5 и подвижный клин 4.
Положение клина калибруется в единицах отношения Н: С. В результат
измерения вносится поправка на изменение р. Точность определения
составляет 0,03 %.
364
ГЛ. IX. РАДИОМЕТРИЧЕСКИЕ МЕТОДЫ АНАЛИЗА
Методы, основанные на поглощении у"излУчения
Узкий пучок Y-квантов поглощается в веществе по
экспоненциальному закону. Так же, как и для р-лучей, в случае определения изменения
потока у-лучей после прохождения через определенный слой вещества
можно воспользоваться уравнением (14) и по известной толщине
поглощающего слоя найти плотность данного материала или по известной
плотности вычислить его толщину. Величина линейного коэффициента
ослабления ц зависит от состава анализируемого вещества.
Рис. 139. Схема прибора для определения отношения
количества водорода к углероду в органических соединениях:
1 — ионизационные камеры; 2— источник ^-излучения; 3 —индикатор
нулевого тока; 4 — подвижный клин; 5— образец; 6 — поглотитель.
Для определения состава сначала строят калибровочный график
зависимости коэффициента \х от содержания анализируемого элемента
в смеси (сплаве), затем определяют \i анализируемого образца и по
графику находят содержание интересующего компонента.
Методы, основанные на поглощении характеристического
рентгеновского излучения радиоактивных
изотопов
Поглощение рентгеновского излучения подчиняется
экспоненциальному закону. Если этот закон записать в форме, в которой используется
массовый коэффициент поглощения [л/р, получим формулу:
/-/„*
р
(57)
где d — толщина поглощающего вещества, г/см2.
Отсюда по ослаблению потока характеристического рентгеновского
излучения можно определить \х/р.
Если поглощение происходит в смеси веществ, то полный массовый
коэффициент поглощения равен сумме коэффициентов отдельных
компонентов;
I —П.
(58)
где rii — доля /-того компонента в смеси.
Определив на опыте полный массовый коэффициент поглощения и
зная коэффициенты поглощения отдельных компонентов, можно
установить содержание того или иного компонента смеси. Этим путем опре-
§ 32. МЕТОДЫ АНАЛИЗА, ОСНОВАННЫЕ НА ПОГЛОЩЕНИИ ИЗЛУЧЕНИЯ 365
деляют содержание серы в углеводородах, если известно соотношение
количества атомов углерода и водорода в них. Значение jx/p для серы
равно 200 смг1г, для углерода—10 см2/г и для водорода —0,5 см2/г,
что позволяет определять малые содержания серы.
Если пучок характеристического рентгеновского излучения
направить на сплав двух соседних в периодической системе элементов,
например излучение, получающееся при захвате электрона /(-оболочки
67Ga, на сплав меди и никеля, то поглощение излучения будет зависеть
от состава сплава. Это связано с тем, что никель поглощает
характеристическое рентгеновское излучение 67Zn сильнее меди, так как энергия
перехода 67Ga->67Zn -f ftv, равная 8,7 кэву недостаточна, чтобы вызвать
/(-переход у меди (энергия перехода 9,0 кэв), и достаточна для
/(-перехода у никеля (8,4 кэв). Это дает возможность анализировать сплавы
меди с никелем по поглощению излучения 67Ga.
Методы, основанные на поглощении нейтронов
Ряд изотопов обладает высоким эффективным сечением реакции
захвата тепловых нейтронов. Поглощение нейтронов при их
взаимодействии с ядрами элементов, входящих в состав поглощающего вещества,
подчиняется экспоненциальному закону:
j^j^-olx
(59)
где 10 и J — поток нейтронов, соответственно падающих на образец и прошедших
через слой поглощающего вещества толщиной х;
L — число атомов поглощающего вещества в 1 смг;
о — эффективное сечение ядерной реакции, см2.
Определив опытным путем /0 и / — величины, пропорциональные /о
и /, и зная величину о, можно найти Lx— число атомов определяемого
изотопа в образце. Однако значение о известно недостаточно точно,
а экспоненциальный закон выполняется не строго, поэтому на практике
прибегают к относительному методу.
Сначала измеряют поглощение
нейтронов в образцах с известным содержанием ; 4 *
искомого элемента, затем поглощение
^
ш
г4-т 3
-к
* *
'* *#
i
I
* *# ** *|
Рис. 140. Разрез установки для
определения бора:
/ — анализируемый образец;
2—парафиновый блок; 3 —нейтронный
источник; 4 — детекторы нейтронов.
нейтронов в анализируемом образце. По |#*j
калибровочному графику находят
содержание искомого элемента в
анализируемом образце. Таким образом можно
определить содержание бора, кадмия,
лития, ряда редкоземельных элементов.
Схема установки для определения
бора и других элементов по поглощению
нейтронов представлена на рис. 140.
Детектирование потока нейтронов можно проводить: во-первых,
непосредственным измерением борными счетчиками; во-вторых, по
количеству образующегося в детекторе (родий, марганец и т. п.) при
ядерной реакции с нейтронами радиоактивного изотопа; в-третьих, по
возбуждаемому в результате ядерной реакции вторичному
корпускулярному излучению, например, определение лития по вторичным а-ча-
стицам и тритонам, образующимся по реакции 61л(я, а)3Н. Подобно
этому, по а-лучам или протонам, образующимся при реакциях
1СВ (n, a)7Be, 14N(/i, p) ,4С и т. п., можно определить содержание бора,
азота и других элементов.
Збб
ГЛ. IX. РАДИОМЕТРИЧЕСКИЕ МЕТОДЫ- АНАЛИЗА
§ 33. Практические работы, выполняемые по методу анализа,
основанному на поглощении излучения
Определение содержания бора
Определение содержания бора по поглощению нейтронов основано
на высоком сечении реакции (а = 755-10~24 см2) пВ(д, а)71л.
Поток нейтронов через анализируемое вещество направляется на
детектор нейтронов, в качестве которого применяют Мп02. По реакции
55Мп (я, y) 56Mn образуется 56Мп с периодом полураспада 2,5 ч, по его
активности находят изменение потока нейтронов. В работе используют
относительный метод, по которому определяют активность детекторов
после прохождения нейтронами эталонных и анализируемых образцов.
Методика определения. Работу выполняют в специальном
помещении, где расположен источник нейтронов с интегральным потоком
~ 106 нейтрон/сек. Пользование источником нейтронов проводится в
соответствии с инструкцией. Перед работой надевают халат, шапочку и
резиновые перчатки.
Анализируемый образец, содержащий 0,03—0,3 г бора, делят на две
части и взвешивают на аналитических весах. Каждую часть истирают
в ступке с 50 смг кварцевого песка, взятого с помощью цилиндра.
Приготовленные смеси помещают в две кюветы. Другие четыре кюветы
заполняют эталонными образцами с известным содержанием бора (0,01—
0,5 г). Во внутреннюю часть всех кювет помещают равные количества
сухой двуокиси марганца (детектор нейтронов). Шесть кювет с
эталонными и анализируемыми образцами помещают в гнезда парафинового
блока (анализируемые образцы напротив друг друга). Вносят источник
в центральное отверстие парафинового блока и облучают образцы и
эталоны в течение 2 ч.
По окончании облучения удаляют источник из парафинового блока
за защиту, вынимают кюветы, из них вынимают сосуды с двуокисью
марганца, служащую детектором нейтронов, и измеряют активность
детекторов нейтронов на сцинтилляционном счетчике (кристалл с
колодцем). Измерение активности каждого детектора проводят в течение
4 мин. Исходя из измеренных активностей, вычисляют активности ко
времени окончания облучения и строят график зависимости
активности от содержания бора в эталонах. Затем по активности
анализируемых образцов по графику находят содержание в них бора и вычисляют
процентное содержание бора в анализируемом образце.
§ 34. Метод, основанный на отражении р-излучения
р-Излучение, отраженное от того или иного вещества, меняет свою
интенсивность и энергию. Изменение интенсивности потока р-лучей
зависит от заряда ядра атомов отражающего материала и определяется
соотношением:
*~*z2t* (60)
где /— коэффициент отражения;
к — коэффициент пропорциональности;
z — заряд ядра атомо* отражающего вещества.
Энергия отраженного р-излучения ?макс связана с энергией
падающего излучения .?wc выражением:
Ег *»0 127г°&Е" (61)
^макс u»i^/'* смакс v '
§ 35. РАБОТЫ ПО МЕТОДУ ОТРАЖЕНИЯ ^ИЗЛУЧЕНИЯ
367
где ?макс"" максимальная энергия отраженного Р-излучения;
-^макс~~ максимальная энергия падающего Р-излучения.
В соответствии с этим уравнением энергия р-излучения,
отраженного от легких элементов, много меньше энергии р-частиц, отраженных
от тяжелых элементов. Это позволяет определять содержание
элементов с большим z в смеси с элементами с малым г, например вольфрама
в сталях, железа в рудах и т. п.
При анализе поток р-частиц от источника направляется на
анализируемый материал. Поток отраженного р-излучения попадает на
детектор излучения через фильтр, толщина которого достаточна для
поглощения излучения, отраженного от легких элементов.
Последовательно измеряют потоки отраженного р-излучения от эталонов, содержащих
известное количество определяемого элемента и анализируемого
образца. Затем строят калибровочный график в координатах
интенсивность излучения — содержание определяемого элемента в эталоне. По
графику находят содержание определяемого элемента в образце.
Точность определения около 0,4% при строгом соблюдении
постоянства условий измерения, интенсивности источника и толщины образцов
и эталонов.
§ 35. Практические работы, выполняемые rto методу,
основанному на отражении р-излучения
Определение содержания компонента смеси с большим z no эффекту
обратного отражения $-лучей
Целью работы является определение содержания химического
элемента с большим z в смеси с элементами с малыми г, например
определение содержания железа в рудах по эффекту обратного рассеяния
р-лучей.
Методика определения. В работе используются закрытые источники
р-излучения с активностью 5—10 мкюри (204Т1, 90Sr). Необходимо
предохранение только от их внешнего излучения. Перед работой надевают
халат и шапочку.
Источники р-излучения хранят в специальных контейнерах, за
стенками которых доза не превышает предельно допустимой.
Для измерения отраженного р-излучения с помощью пинцета
помещают р-излучающий препарат в защитный домик счетчика, под него —
кассету с эталонным образцом и над ним — алюминиевый экран с
толщиной, достаточной для поглощения основной части р-излучения,
отраженного от легких компонентов анализируемого материала.
Измеряют интенсивность отраженного р-излучения серии эталонов.
Строят калибровочный график в координатах интенсивности
регистрируемого излучения — содержание тяжелого компонента в эталоне.
Измеряют интенсивность р-излучения, отраженного от
анализируемого образца, и по графику находят содержание тяжелого компонента
в нем.
ГЛАВА X
ФИЗИКО-ХИМИЧЕСКИЕ МЕТОДЫ
КОЛИЧЕСТВЕННОГО
ОПРЕДЕЛЕНИЯ РЕДКИХ ЭЛЕМЕНТОВ
Л. ВВЕДЕНИЕ
К редким элементам (РЭ) условно относят примерно 60
элементов, промышленное получение и более или менее широкое практическое
использование которых началось сравнительно недавно:
1. Легкие металлы — литий Li, рубидий Rb, цезий Cs и бериллий Be.
2. Редкоземельные металлы — лантан La, все лантаноиды (Се, Pr, Nd,
Pm, Sm, Eu, Gd, Tb, Dy, Ho, Er, Tm, Yb, Lu), скандий Sc и иттрий Y.
3. Рассеянные элементы — галлий Ga, индий In, таллий Т1,
германий Ge (гафний Hf и рений Re).
4. Тугоплавкие металлы — титан Ti, цирконий Zr, гафний Hf,
ванадий V, ниобий Nb, тантал Та, молибден Мо, вольфрам W и
рений Re.
5. Радиоактивные элементы — радий Ra, полоний Ро, актиний Ас
и все актиноиды (Th, Pa, U, Np, Pu, Am, Cm и другие трансурановые
элементы).
6. Так называемые малые элементы — висмут Bi.
7. Благородные металлы — платина Pt и платиновые металлы (Ru,
Os, Rh, Ir, Pd), золото Au и серебро Ag.
8. Неметаллы — бор В, селен Se и теллур Те.
9. Инертные газы — гелий Не, неон Ne, аргон Аг, криптон Кг,
ксенон Хе.
Для определения РЭ наиболее часто применяют фотометрические и
спектральные, а для определения сравнительно больших количеств —
титриметрические методы (главным образом комплексонометрические
и редоксметрические с установлением точки эквивалентности
индикаторными, амперометрическими, потенциометрическими и другими
способами). Небольшие количества РЭ определяют также полярографическим
методом. Ряд элементов (литий, рубидий, цезий, индий) определяют
методом фотометрии пламени. Другие методы (радиоактивационный, рент-
геноспектральный, масс-спектральный) применяются сравнительно
редко. Кроме того, для определения РЭ применяется атомно-абсорбци-
онная спектроскопия.
Редкие и обычные элементы разделяют осаждением с широким
использованием различных органических реагентов и маскирующих
веществ. Для этих же целей применяют также хроматографические,
экстракционные и некоторые другие методы. Аналитическая химия РЭ
подробно представлена в ряде фундаментальных руководств. В данную
главу включены лишь несколько примеров физико-химических
инструментальных методов определения важнейших РЭ.
§ 1. РАБОТЫ, ВЫПОЛНЯЕМЫЕ МЕТОДОМ АМПЕРОМЕТРИЧЕСКОГО ТИТРОВАНИЯ 369
Б. ПРАКТИЧЕСКИЕ РАБОТЫ
§ 1. Работы, выполняемые методом амперометрического
титрования
Определение ванадия (IV)
Метод основан на титровании соединений VIV стандартным
раствором сульфата церия (IV). Конечную точку находят по току
восстановления избыточных ионов CeIV при потенциале +0,5 в платинового
вращающегося электрода относительно насыщенного каломельного
электрода. При титровании VIV в присутствии железа необходимо, чтобы
часть последнего находилась в двухвалентном состоянии. В этом
случае вначале титруют Fe п раствором Ce(S04)2 при потенциале от +0,8
до +0,9 в, а затем VIV при потенциале +0,5 в. Метод используют для
определения ванадия в феррохроме.
Методика определения. Навеску' феррохрома (0,5—1,0 г)
растворяют при нагревании в 30—40 мл H2S04 (1 :4), добавляют после
растворения несколько капель азотной кислоты для окисления большей
части ионов железа (II) и кипятят раствор до удаления окислов азота.
Раствор переносят в мерную колбу емкостью 100 мл, разбавляют водой
до метки. Аликвотную часть (25 мл) помещают в электролизер и
устанавливают разность потенциалов 0,8—0,9 в. Отсутствие диффузионного
тока указывает, что в растворе не содержится ионов железа (П). В атом
случае необходимо добавить (по каплям) немного, приблизительно
0,02 н. раствора соли Мора. Затем титруют 0,004 н. раствором Ce(S04)2
сначала при потенциале от +0,8 до +0,9 в до тех пор, пока
гальванометр не покажет отсутствия тока. После этого устанавливают разность
потенциалов +0,5 в и титруют VIV раствором Ce(S04)2 До появления
диффузионного тока от избытка ионов CeIv.
Определение индия в концентратах
Метод основан на титровании индия (III) при рН 1,0 раствором
динатриевой соли этилендиаминтетрауксусной кислоты (комплексон III).
Точку эквивалентности устанавливают по исчезновению диффузионного
тока восстановления 1пш-иона на ртутном капельном электроде при
потенциале от —0,7 до —0,8 в относительно насыщенного каломельного
электрода. Определению не мешают многие элементы, с которыми
обычно приходится встречаться при анализе индийсодержащих
продуктов, а именно Zn, Mn, Cd, Co, A1. Титрованию не мешают также
значительные количества Fe++ (^ 10 мг). Железо (III) восстанавливают
до Fe++. Влияние олова (<? мг) и сурьмы (<2 мг) устраняют введем
нием винной кислоты. Определение возможно в присутствии небольших
количеств (^0,5 мг) ионов меди, если их замаскировать тиомочевиной,
и ионов свинца, а также мышьяка (<С2 мг). Большие количества этих
элементов затрудняют установление точки эквивалентности вследствие
того* что медь, свинец и мышьяк дают диффузионный ток. Однако эти
элементы легко отделяются от индия в ходе анализа: мышьяк и свинец
удаляются при разложении пробы смесью хлористоводородной и
серной кислот и упаривании раствора до появления паров H2S04; медь —
при осаждении гидроокиси индия избытком аммиака. Определению
мешает висмут.
Методика определения. Анализируемый материал (около 0,5 г)
разлагают смесью хлористоводородной и серной кислот (по 5 мл) и
упаривают до выделения паров H2S04, затем разбавляют водой до 50 мл и
370 ЕЛ. Xt ФИЗИКО-ХИМИЧЕСКИЕ МЕТОДЫ ОПРЕДЕЛЕНИЯ РЕДКИХ ЭЛЕМЕНТОВ
отфильтровывают выпавший осадок. Из фильтрата осаждают
гидроокиси избытком аммиака. Осадок отфильтровывают и растворяют в
хлористоводородной кислоте, переводят раствор в мерную колбу емкостью
50—100 мл и объем доводят водой до метки. Для титрования отбирают
аликвотную часть раствора (10—20 мл). Вводят 0,5—1 г винной
кислоты, 1—2 мл 4%-ного раствора аскорбиновой кислоты и 0,2 мл
5%-нбго раствора тиомочевины. Раствор нейтрализуют аммиаком по
тропеолйну 00 до перехода окраски из красной в желтую. Затем
прибавляют 15—20 мл буферного раствора. ДаЛее титруют из полумикробю-
ретки емкостью 5 мл, прибавляя раствор порциями по 0,5 мл. Точку
эквивалентности находят графически по изменению диффузионного
тока индий в зависимости of количества прибавленного стандартного
раствора.
§ 2. Работы, выполняемые полярографическим методом
Определение рения в сплавах с молибденом
Метод оснойан на переведении рения в виде перрената в раствор
и отделений большей части молибдена в виде малорастворимого молиб-
дата кальция. В полученном растворе рений определяют
полярографическим методом при потенциале от —0,3 до —0,4 в на фоне 5 н.
раствора H2S04.
Методика определения. Навеску сплава (0,1 г) смешивают с
трехкратным количеством окиси кальция и спекают в муфельной печи при
700—750° С. Охлажденный плав Помещают в стакан, содержащий 50 мл
воды, затем тигель вынимают из стакана, а раствор нагревают до
кипения в присутствии 5 мл 10%-ного раствора персульфата аммония.
Осадок отфильтровывают, промывая его 2—3 раза горячей водой, на
воронке Бюхнера. Фильтрат переносят в мерную колбу емкостью 100 мл
и доводят водой до метки. Отбирают аликвотную часть раствора (10 мл)
в мерную колбу емкостью 100 мл и доводят до метки 5 н. раствором
H2S04. Пропускают ток азота для удаления кислорода и полярографи-
руют при потенциале от —0,3 до —0,4 в относительно насыщенного
каломельного электрода.
Определение индия в рудах
Метод основан на удалении всех элементов, мешающик
полярографическому определению индия, цементацией их цинковой амальгамой
в присутствии не менее 20% сульфатов. Индий при этом остается в
растворе в виде комплексного сульфатного аниона [In(S04)]"~, который не
восстанавливается цинковой амальгамой. Соединения таких элементов,
как As, Sb, Bi, Си, Те, Se, Sn, Tl, Cd и некоторые другие, энергично
восстанавливаются цинковой амальгамой, растворяясь при этом в ртути
(Cd, Sn, T1, Си) или выделяясь в виде рыхлого, черного осадка.
Элементы высших валентностей Fe+++, Vv, CrVI, TiIV) восстанавливаются
до низших. В полученном растворе индий определяют полярографиче-
ски после введения 10% хлорида натрия от массы раствора. Метод
может быть применен для определения индия в производственных
продуктах и отходах. Потенциал полуволны для индия —1,0 в относительно
насыщенного каломельного электрода.
Методика определения. Навеску анализируемого материала (1—Зг)
разлагают при нагревании в смеси азотной и серной кислот и
нагревают до выделения паров H2S04. Остаток обрабатывают горячей во-
§ 2. РАБОТЫ, ВЫПОЛНЯЕМЫЕ ПОЛЯРОГРАФИЧЕСКИМ МЕТОДОМ 371
дой и при наличии заметного осадка последний отфильтровывают. К
полученному раствору прибавляют столько серной кислоты (или
эквивалентное ей количество какого-либо сульфата), чтобы концентрация ее
составляла не менее 20%. Затем выделяют элементы цементацией
цинковой амальгамой при перемешивании раствора мешалкой со скоростью
350—400 об/мин в течение 45 мин. По окончании цементации раствор
фильтруют, добавляют к раствору около 10% по массе хлорида натрия
и полярографируют, определяя индий по методу добавок.
Определение индия в сульфидных рудах
Индий определяют полярографическим методом в 3 н. растворе
НС1 при потенциале от —0,4 до —0,8 в относительно насыщенного
каломельного электрода. Определению мешают соединения Си, Bi, Cd, Sn.
Индий отделяют осаждением в форме гидроокиси с применением в
качестве коллектора гидроокиси железа (III). Осаждение проводят из
горячего сильнощелочного раствора в присутствии комплексона III. После
растворения осадка в хлористоводородной кислоте восстанавливают
кислород и железо (III) металлическим железом.
Методика определения. Навеску анализируемой руды 0,1—1,0 г при
нагревании разлагают 3 мл хлористоводородной кислоты (пл. 1,19 г/сж3)
и несколькими миллилитрами хлорной кислоты. Упаривают раствор
досуха. К остатку прибавляют 40—50 мл горячей воды и нагревают до
кипения. Нерастворимый остаток отфильтровывают, промывают
горячей водой и отбрасывают. К кислому фильтрату прибавляют 20—25 мл
5%-ного раствора комплексона III и нейтрализуют до щелочной
реакции 10%-ным раствором NaOH и затем добавляют 15—20 мл избытка
его. Разбавляют раствор до 150—200 мл, медленно нагревают и
кипятят 3—5 мин, затем ставят на кипящую водяную баню до полной
коагуляции осадка (3d—40 мин). Осадок отфильтровывают через бумажный
фильтр, предварительно промытый горячим 5%-ным раствором NaOH.
Осадок промывают горячим 1%-ным раствором NaCl, 1—2 раза
горячей водой и растворяют в горячем 3 н. растворе НС1. Упаривают
раствор, если есть необходимость, и переводят в мерную колбу емкостью
25 мл, доводя до метки 3 н. раствором НС1. Заполняют раствором
электролизер, добавляют на кончике шпателя 0,2—0,3 г металлического
железа и через 40—45 мин полярографируют. Количество индия находят
методом добавок.
Определение таллия в кадмии
Ионы таллия (I) обратимо восстанавливаются на капельном
ртутном электроде при потенциале около —0,50 в относительно
насыщенного каломельного электрода. Потенциал полуволны не зависит от
состава основного электролита. Как на фоне NH4OH, так и на фоне НС1
высота волны пропорциональна концентраций Т1+-ионов в растворе.
В аммиачной среде Т11 в отсутствие ионов меди хорошо определяется
в металлическом кадмии и его солях. На фоне хлористоводородной
кислоты потенциалы полуволн Tl1, Sn11 и РЬП одинаковы. Для разделения
волн Sn и Т1 вводят тартрат, подавляющий волну олова, а для
разделения волн РЬ и Т1 вводят комплексон III, смещающий в слабокислой
среде потенциал полуволны РЬ в сторону более отрицательных
значений (—1,1 в), что может быть использовано также при определений
Т1Т в свинце. В этих условиях медь восстанавливается при потенциале
372 ГЛ. X, ФИЗИКО-ХИМИЧЕСКИЕ МЕТОДЫ ОПРЕДЕЛЕНИЯ РЕДКИХ ЭЛЕМЕНТОВ
—0,3 в. Полярографическое определение обычно проводят после
выделения таллия экстракционным методом.
Методика определения. Эфирные вытяжки Т11 (после отделения
таллия из 1 М раствора НВг) упаривают на водяной бане (отгоняя
эфир). К остатку приливают 3 мл H2S04 (1:1), выпаривают раствор до
появления паров H2S04 и прибавляют 3—5 капель 30%-ного раствора
Н202 до обесцвечивания раствора. После охлаждения обмывают стенки
колбы водой (5 мл) и снова нагревают до выделения H2S04. Затем
к остатку прибавляют 10—15 мл воды, вводят 0,2—0,3 г Na2S03 и
кипятят 5 мин до исчезновения запаха S02. Раствор переносят в мерную
колбу емкостью 25 мл, нейтрализуют по конго красному 25%-ным
раствором NH3 и добавляют 2,5 мл его избытка. Затем раствор
охлаждают, доводят его объем до метки и тщательно перемешивают.
Отбирают в стакан 15 мл раствора, вводят около 1 г Na2S03, 5 капель
0,25%-ного столярного клея, перемешивают и полярографируют.
§ 3. Работы, выполняемые фотометрическим методом
Определение бериллия
Метод основан на образовании ионами бериллия с алюминоном
комплексного соединения красного цвета с максимумом светопоглоще-
ния 530 нм. Метод пригоден для определения малых количеств
бериллия. Максимальная окраска развивается при рН 4,6—5,4, коэффициент
молярного поглощения е равен 9200 ± 200. Для образования
окрашенного соединения при концентрации 2—50 мкг бериллия в 50 мл
раствора необходимо прибавлять 2 мл 0,4%-ного раствора алюминона.
Элементы, образующие в слабокислой среде устойчивые комплек-
сонаты, не мешают определению (медь, никель, алюминий и др.). При
определении бериллия в сплавах на ниобиевой основе ниобий маскируют
тартратом, а другие ионы — комплексоном III. В этих условиях
окрашенное соединение с алюминоном дают только ионы бериллия.
Калибровочный график. Для построения калибровочного графика
в ряд мерных колб емкостью 50 мл вводят по 2 мл 0,4%-ного раствора
алюминона, от 5 до 25 мкг бериллия с интервалом 5 мкг в виде раствора
хлорида бериллия с содержанием 5 мкг/мл Be, 2 мл 5%-ного раствора
комплексона III, 10 мл ацетатного буферного раствора с рН 5,1—5,3 и
разбавляют до метки водой. Измеряют с зеленым светофильтром
оптическую плотность серии растворов относительно холостого раствора,
содержащего все компоненты в тех же количествах, но без бериллия.
Методика определения. Для определения бериллия 0,1 г сплава
нагревают в платиновом или кварцевом тигле в муфельной печи при
700—800° С, затем окислы сплавляют с 1,5—2,0 г пиросульфата калия
до получения прозрачного плава. В стакане емкостью 100—150 мл
выщелачивают плав 10 мл 10%-ного раствора винной кислоты, осторожно
вынимают тигель стеклянной палочкой, ополаскивают 10—15 мл
горячей воды, нейтрализуют аммиаком (1 : 3) до рН 6 по универсальной
индикаторной бумаге, фильтруют в мерную колбу емкостью 100 мл и
разбавляют водой до метки.
В мерную колбу емкостью 50 мл помещают 2 мл 0,4%-ного
раствора алюминона, 2 мл 5%-ного раствора комплексона III, затем
отбирают пипеткой аликвотную часть, содержащую 5—25 мкг бериллия,
переносят в ту же мерную колбу, прибавляют 10 мл ацетатного
буферного раствора с рН 5,1—5,3, разбавляют до метки водой и измеряют
§ 3. РАБОТЫ, ВЫПОЛНЯЕМЫЕ ФОТОМЕТРИЧЕСКИМ МЕТОДОМ 373
с зеленым светофильтром оптическую плотность относительно
холостого раствора.
Содержание бериллия в аликвотной части находят по
калибровочному графику.
Определение скандия в магниевых сплавах
При взаимодействии ионов скандия с ксиленоловым оранжевым
при рН 1,5—5 образуется растворимое в воде соединение
красно-фиолетового цвета. Сам реагент окрашен в кислой среде в желтый цвет, а
при рН 5,5 окрашивается в красно-фиолетовый цвет.
Стабильность окраски достигается через 10 мин после сливания
растворов, интенсивность окраски растворов устойчива 2 суток.
Оптическая плотность растворов скандия пропорциональна его концентрациям
в широком интервале. Чувствительность 0,1 мкг/мл скандия.
Определение скандия при помощи ксиленолового оранжевого
проводят при рН 1,5. В этих условиях не мешают ионы щелочноземельных
элементов, лантана, празеодима, неодима, самария, церия (III), иттрия,
цинка, кадмия, алюминия, марганца, железа (II). Поэтому метод можно
применять для фотометрического ъпределения скандия в
металлическом магнии и магниевых сплавах без отделения компонентов сплава.
Мешают ионы циркония, тория, галлия и висмута, образующие с
ксиленоловым оранжевым окрашенные соединения. Соединения железа (III)
и церия (IV) предварительно восстанавливают аскорбиновой кислотой.
Калибровочный график. Для построения калибровочного графика
в пять мерных колб емкостью 50 мл вводят от 10 до 50 мкг скандия
с интервалом 10 мкг в виде раствора его соли с содержанием Sc
10 мкг/млу 5 мл буферного раствора с рН 5; 5 мл 0,05%-ного раствора
ксиленолового оранжевого и разбавляют водой до метки. В этих же
условиях готовят раствор сравнения, содержащий все компоненты в
указанных количествах и не содержащий скандия. Через 20 мин измеряют
оптическую плотность растворов на фотоэлектроколориметре
относительно раствора сравнения и по полученным данным строят
калибровочный график.
Методика определения. Для определения скандия в магниевых
сплавах навеску 1 г (при содержании 0,002—0,005% скандия)
растворяют в 10—20 мл соляной кислоты (1 : f) в стакане емкостью 100 мл.
Раствор выпаривают до объема 10 мл, количественно переносят в
мерную колбу емкостью 50 мл, споласкивая стенки стакана небольшими
порциями воды, прибавляют 5 мл свежеприготовленного 2%-ного
раствора аскорбиновой кислоты, 50%-ный раствор ацетата натрия (пока не
появится сиреневое окрашивание бумаги, смоченной конго красным),
5 мл буферного раствора с рН 1,5; 5 мл 0,05%-ного раствора
ксиленолового оранжевого и разбавляют водой до метки. Через 20 мин
измеряют оптическую плотность раствора в тех же условиях, как при
построении калибровочного графика.
Содержание скандия в растворе сплава находят по
калибровочному графику.
Примечание. При содержании больше 0,005% скандия навеску сплава
соответственно уменьшают. Для определения скандия аликвотная чаеть раствора сплава
должна содержать 20—40 мкг скандия.
Определение тория
Ионы тория с арсеназо III образуют окрашенное и прочное
соединение. Это позволяет определять торий в сильнокислых растворах без
отделения сульфатов, фосфатов, фторидов, оксалатов и других ионов.
374 ГЛ. X. ФИЗИКО-ХИМИЧЕСКИЕ МЕТОДЫ ОПРЕДЕЛЕНИЯ РЕДКИХ ЭЛЕМЕНТОВ
Реакция очень чувствительна. Светопоглощение растворов (565—
665 нм) сильно зависит от природы кислоты и других факторов.
При помощи этого реагента можно определять торий в цирконе,
ниобийсодержащих и других продуктах. Для определения
микрограммовых количеств тория окрашенное соединение тория с арсеназо III
экстрагируют органическим растворителем.
Калибровочный график. Для построения калибровочного графика
в ряд мерных колб емкостью 25 мл вводят 1—5 мл стандартного
раствора нитрата тория с содержанием Th 5 мкг/мл, затем по 1 мл
0,05%-ного водного раствора арсеназо III и по 8 мл концентрированной
соляной кислоты (пл. 1,18 г/см3). Растворы разбавляют до метки водой,
хорошо перемешивают и измеряют оптическую плотность на фотоэлек-
троколориметре или универсальном фотометре относительно
холостого раствора. Для его приготовления берут те же растворы, но торий
не вводят.
Методика определения. Для определения тория в пробе проводят
те же операции, как и при построении калибровочного графика. Если
в пробе присутствует титан (не более 100 мкг) или цирконий (не более
1 мг), в анализируемый раствор перед разбавлением вводят 1 мл
0,5%-ного раствора щавелевой кислоты.
Определение титана
Ионы титана в кислой среде с диантипирилметаном образуют
растворимое в воде соединение желтого цвета с максимумом светопогло-
щения 380 нм. Изменение концентрации кислоты в пределах 0,5—4 н.
не влияет на величину оптической плотности. Чувствительность
определения 0,01 мкг Ti в 1 мл, коэффициент молярного поглощения е
равен 18 000. Окраска соединения титана с диантипирилметаном
развивается в течение 45 мин и устойчива несколько месяцев, оптическая
плотность титана в широком интервале пропорциональна его
концентрациям.
Определению титана при помощи диантипирилметана не мешают
ионы магния, алюминия, цинка, кадмия, марганца, меди, циркония,
редкоземельных элементов, молибдена, ниобия и тантала, поэтому
метод можно применять для определения титана в легких, черных и
цветных сплавах. Ионы никеля, хрома и кобальта не реагируют с
диантипирилметаном, но мешает собственная окраска ионов; поэтому раствор
сравнения должен содержать все компоненты, кроме
диантипирилметана. Ионы железа (III) и ванадия (V)предварительно
восстанавливают гидроксиламином.
Калибровочный график. Для построения калибровочного графика
в пять мерных колб емкостью 25 мл вводят от 1 до 5 мл раствора соли
титана с содержанием Ti 5 мкг!мл, по 5 мл 5%-ного раствора
диантипирилметана и по 5 мл соляной кислоты (пл. 1,17—1,19 г/см3).
Растворы разбавляют водой до метки и измеряют оптическую плотность
на фотоэлектроколориметре по отношению к воде. Строят
калибровочный график.
Методика определения. Навеску анализируемой пробы,
содержащую 100—200 мкг титана, растворяют при нагревании на песочной
бане в 10 мл серной кислоты (1:1), упаривают до 1—2 мл и
растворяют в воде. Раствор разбавляют до 15 мл водой, прибавляют 15 мл
хлористоводородной кислоты (пл. 1,17—1,19 г/см3), 10 мл 5%-ного
раствора диантипирилметана и 10 мл хлороформа. Экстрагируют железо
5—6 мин, органический слой отделяют, а затем вновь прибавляют хло-
§ 3. РАБОТЫ, ВЫПОЛНЯЕМЫЕ ФОТОМЕТРИЧЕСКИМ МЕТОДОМ 375
роформ, 10 мл 5%-ного раствора диантипирилметана и еще раз
экстрагируют.
Водный слой переносят в мерную колбу емкостью 50 мл и
разбавляют водой до метки. Отбирают пипеткой 10 мл полученного раствора
в колбу емкостью 25 мл, прибавляют 5 мл 5%-ного раствора
диантипирилметана и разбавляют водой до метки. Измеряют оптическую
плотность раствора в тех же условиях, как и при построении
калибровочного графика.
Содержание титана в аликвотной части находят по
калибровочному графику.
Определение Циркония
Определение основано на использовании реагента арсеназо III. Ар-
сеназо III является высокочувствительным и селективным реагентом
для фотометрического определения циркония. Окрашенный в красный
цвет в кислой среде раствор арсеназо III при введении соли циркония
быстро изменяет окраску на синюю или сине-фиолетовую в
зависимости от соотношения реагента и циркония. Интенсивность окраски
растворов соединения постоянна в интервале кислотности,
соответствующей 1—3 н. растворам НС1, но сильно изменяется во времени. Для
стабилизации окрашенного раствора вводят желатину. Для получения
устойчивых результатов цирконий предварительно переводят в
постоянную ионную форму (цирконил-ион) кипячением с 2 н. раствором НС1.
Оптические плотности растворов соединения циркония с арсеназо III
пропорциональны концентрациям в интервале 5—30 мкг циркония
в 50 мл раствора.
Определение микроколичеств циркония возможно в присутствии
следующих ионов — более 100 мг алюминия, до 20 мг олова (II, IV), до
10 мг бериллия, никеля, титана, до 5 мг хрома,' редкоземельных
элементов и ниобия, до 50 мг тартратов, до 100 мг сульфатов. При введении
хлорида олова (II) определение возможно в присутствии 15 мг железа
и 5 мг меди.
Мешают определению ионы урана, тория, реагирующие с
арсеназо III в кислой среде с образованием окрашенных соединений, а
также фториды, фосфаты и оксалаты, образующие прочные
комплексные соединения с цирконием и разрушающие окрашенное соединение
циркония с арсеназо III.
Метод применим для определения циркония в рудах и побочных
продуктах (шламы, фракции с высоким содержанием титана, железа,
алюминия, олова и т. п.).
Калибровочный график. Для построения калибровочного графика
в ряд мерных колб емкостью 50 мл вводят 1—5 мл раствора оксихло-
рида циркония в 2 и. хлористоводородной кислоте с содержанием Zr
5 мкг/мл и разбавляют до 10 мл 2 н. раствором НС1. Растворы
нагревают до кипения на песочной бане, охлаждают, добавляют по 3 мл
1%-ного раствора желатины и по 2 мл 0,05%-ного раствора арсеназо III
и разбавляют до метки 2 н. раствором НС1. В раствор сравнения
вводят в тех же количествах все реактивы, кроме циркония.
На универсальном фотометре или фотоэлектроколориметре
измеряют оптическую плотность растворов по отношению к раствору
сравнения при максимуме светопоглощения и строят калибровочный график.
Методика определения. Навеску 0,1—0,2 г анализируемого
материала с содержанием 0,1—0,5% циркония помещают в платиновую
чашку, в которую предварительно вносят 5 г смеси карбоната и тетра-
бората натрия (3:2), перемешивают и сплавляют в муфельной печи
376 ГЛ. X, ФИЗИКО-ХИМИЧЕСКИЕ МЕТОДЫ ОПРЕДЕЛЕНИЯ РЕДКИХ ЭЛЕМЕНТОВ
при 900° С в течение 7—10 мин. Распределяют плав вращением чашки
так, чтобы он застыл на стенках тонким слоем, заливают плав водой
и нагревают на песочной бане до растворения его. Раствор с осадком
переносят в стакан емкостью 300 мл, разбавляют водой до 100—150 мл
и нагревают для лучшей коагуляции осадка. Осадок отфильтровывают,
промывают горячей водой и растворяют на фильтре 2 н. раствором
НС1. Фильтрат собирают в мерную колбу емкостью 50 мл при навеске
0,2 г и содержании циркония в объекте 0,1% или в колбу емкостью
100 мл при навеске 0,1 г и содержании циркония 0,5%, разбавляют до
метки 2 н. раствором НС1.
Для определения циркония отбирают аликвотную часть раствора
(5 мл или около 10—15 мкг циркония), переносят в мерную колбу
емкостью 50 мл и далее поступают, как описано при построении
калибровочного графика.
Содержание циркония находят по калибровочному графику.
Определение рения
Метод основан на способности рения каталитически ускорять
реакцию восстановления теллурата натрия до элементного теллура
хлоридом олова (II). Выделяющийся теллур в присутствии защитного
коллоида (желатины) окрашивает раствор в черно-коричневый цвет.
Определение 0,1—0,001 мкг рения возможно в присутствии более 100 мкг
следующих ионов: меди, ртути, германия, олова, свинца, сурьмы,
висмута, мышьяка, рубидия и осмия. Мешающее влияние молибдена и
вольфрама устраняют связыванием их винной кислотой. Метод может
быть применен для определения рения в горных породах после
выделения его в виде сульфида.
Методика определения. Для исключения возможных из-за
неодинакового состава контрольного и стандартного растворов ошибок
целесообразно применять метод добавок. В три калиброванные пробирки
помещают равные объемы анализируемого раствора (по 1 мл), затем
в одну из них вводят раствор, содержащий 0,05 мкг Re, а в другую
0,005 мкг Re, в третью 1 мл дистиллированной воды и в каждую из
пробирок добавляют точно по 1 мл реакционной смеси, содержащей
Na2Te04. Растворы тщательно перемешивают и оставляют стоять от
1—2 до 16—18 ч до появления в растворе, содержащем только
определяемое количество рения, достаточно интенсивной окраски. Измеряют
оптическую плотность этого раствора на фотоэлектроколориметре с
синим светофильтром относительно того из двух стандартов, в котором
развивпГаяся окраска наиболее близка к окраске анализируемого
раствора.
Расчет. Содержание (х) вычисляют (в мкг/мл) по формуле:
aDx
х =
Dx+а ~" Аг
количество Re (мкг), добавленного к анализируемому раствору (мл)\
оптическая плотность анализируемого раствора;
оптическая плотность анализируемого раствора с добавкой.
Определение висмута
Висмут с тиомочевиной образует окрашенное соединение. Этому
соединению приписывают различный состав: Bi(CSN2H4)3Cl3;
Bi(CSN2H4) (N03)2OH; Bi(CSN2H4)Cl3.
Оптимальный интервал кислотности, создаваемой азотной
кислотой, 0,4—1,2 н., концентрация тиомочевины в конечном объеме 1%. Оп-
где а —
Dx-
?>х+а -
§ 3. РАБОТЫ, ВЫПОЛНЯЕМЫЕ ФОТОМЕТРИЧЕСКИМ МЕТОДОМ 377
тические плотности растворов подчиняются закону Бера в интервале
концентраций 2—16 мкг/мл Bi. Окраска устойчива около 1,5 ч. Метод
применим для определения висмута в свинцовых и алюминиевых
сплавах.
Калибровочный график. В ряд мерных колб емкостью 50 мл вводят
0,05—0,25 мг висмута в виде раствора нитрата висмута с содержанием
Bi ~ 0,1 мг/мл, прибавляют 7 мл раствора азотной кислоты (1:1) при
определении висмута в алюминиевых сплавах или 7 мл азотной кислоты
(1 :4) при определении висмута в свинце, 10 мл насыщенного раствора
тиомочевины, разбавляют до метки водой и перемешивают. Измеряют
оптическую плотность растворов на фотоэлектроколориметре и строят
калибровочный график.
Определение висмута в алюминиевых сплавах
Метод пригоден для определения висмута в алюминиевых сплавах,
содержащих 3,5—6% меди и 0,2—0,6% свинца.
Методика определения. Навеску алюминиевого сплава 0,1 г обра*
батывают без подогревания 5 мл хлористоводородной кислоты (1:1)
в стакане емкостью 100—150 мл. При этом алюминий, магний и другие
элементы переходят в раствор, весь же висмут, а также большая часть
свинца и меди остаются в остатке. По окончании растворения
немедленно прибавляют 5 мл дистиллированной воды и нерастворившийся
остаток отфильтровывают на маленьком бумажном фильтре, промывая
его 2 раза небольшими порциями горячей воды. Отфильтровывание и
промывание остатка следует проводить возможно быстро, иначе для
висмута получаются заниженные результаты. Промытый осадок
растворяют на фильтре в 5—10 мл горячей азотной кислоты (1:1), собирая
жидкость в мерную колбу емкостью 50 мл. Фильтр промывают
небольшими порциями азотной кислоты (1:10), а затем водой. Промывные
воды собирают в ту же колбу. В колбу вводят 10 мл насыщенного
водного раствора тиомочевины и раствор разбавляют водой до 50 мл.
Измеряют оптическую плотность раствора на фотоэлектроколориметре
с синим светофильтром.
Содержание висмута находят по калибровочному графику.
Определение висмута в свинце
Определение висмута в Свинце основано на том, что свинец
количественно осаждается в виде основной соли из разбавленного раствора
нитрата свинца при рН 6, а висмут количественно осаждается из
слабоазотнокислого раствора при рН 4,1 ± 0,2.
Определению не мешают цинк, кадмий, кобальт, никель, медь,
мышьяк (-<Л%), олово (-<0,1%), железо (III) (^0,05%) и небольшие
количества хлорида. Мешает определению сурьма, образующая с тио-
мочевиной окрашенное соединение. Влияние сурьмы устраняют
введением в раствор винной кислоты.
Методика определения. Навеску свинца (1 г при содержании
0,005—0,05% висмута и 10 г при содержании 0,0005—0,005% висмута)
растворяют в азотной кислоте (1:4) при подогревании. Избыток
кислоты выпаривают, остаток растворяют в небольшом количестве
дистиллированной воды и нейтрализуют 2 н. раствором карбоната натрия до
появления мути, вызываемой образованием основных солей, после чего
прибавляют еще 4—5 мл 2 н. раствора карбоната натрия и кипятят 1 —
2 мин. Полученный осадок содержит весь висмут и немного свинца.
378 ГЛ. X. ФИЗИКО-ХИМИЧЕСКИЕ МЕТОДЫ ОПРЕДЕЛЕНИЯ РЕДКИХ ЭЛЕМЕНТОВ
По возможности полностью отфильтровывают осадок на
маленьком фильтре и, не промывая, переносят фильтр вместе с осадком в
стакан, в котором проводили растворение свинца.
Осадок растворяют в 5—10 мл азотной кислоты (1 :4). Если
предполагается наличие в свинце значительных количеств сурьмы, то вводят
1—2 г винной кислоты. К полученному раствору, перенесенному
в мерную колбу емкостью 50 мл, прибавляют 10 мл
насыщенного раствора тиомочевины, разбавляют водой до метки и измеряют
оптическую плотность на фотоэлектроколориметре с синим
светофильтром.
Количество висмута находят по калибровочному графику.
§ 4. Работы, выполняемые экстракционно-фотометрическим
методом
Определение микроколичеств тория
Для ойределения очень малых количеств тория окрашенное
соединение тория с арсеназо III экстрагируют изоамиловым спиртом в
присутствии хлорида дифенилгуанидиния и монохлоруксусной кислоты.
Метод сочетает собственно определение тория с его экстракционным
концентрированием и дает возможность определять торий при
разбавлении 1 :5-108 (0,02 мкг Th/мл).
Калибровочный график. Для построения калибровочного графика
в делительные воронки емкостью 25—30 мл вводят 1—5 мл
стандартного раствора ТЬ(ЫОз)4 с содержанием Th 1 мкг/мл, по 1 мл
0,025%-ного раствора арсеназо III, 4,0 мл 0,1 н. раствора азотной
кислоты, по 1 г монохлоруксусной кислоты, по 10 мл 20%-ного раствора
хлорида дифенилгуанидиния и по 10 мл изоамилового спирта. Смесь
встряхивают 2 мин, дают разделиться фазам, органический слой
фильтруют через бумажный фильтр в сухую кювету и измеряют оптическую
плотность по отношению к раствору сравнения, полученному аналогично
и не содержащему торий.
Методика определения. Анализируемый раствор проводят через все
описанные выше операции. Содержание тория находят по
калибровочному графику.
Определение урана
Арсеназо III образует с уранил-ионом комплексное соединение
зеленого цвета с максимумом светопоглощения 655 нм. Чувствительность
определения 0,01—0,02 мкг урана, коэффициент молярного поглощения
е равен 75 500. Оптимальная область рН 1,7—2,5. Определению не
мешают сульфаты, фториды, оксалаты, фосфаты. Из катионов мешают
только торий, цирконий, алюминий, хром (III) и редкоземельные
элементы, однако их можно замаскировать введением подходящих веществ
(сульфосалициловая кислота в 0,05 н. хлористоводородной кислоте для
алюминия, щавелевая кислота для циркония и гафния и т. д.).
Восстанавливая уран (VI) до урана (IV), можно резко повысить
селективность определения, проводя его в 5—6 н. хлористоводородной
кислоте в присутствии щавелевой кислоты. В качестве восстановителя
лучше всего применять гранулированный цинк в присутствии
аскорбиновой кислоты, предотвращающей частичное окисление урана (IV)
кислородом воздуха.
Экстракционно-фотометрический вариант определения урана (VI)
основан на способности соединения урана с арсеназо III
экстрагироваться бутиловым спиртом из растворов, содержащих комплексон III и
§ 4. РАБОТЫ, ВЫПОЛНЯЕМЫЕ ЭКСТРАКЦИОННО-ФОТОМЕТРИЧЕСКИМ МЕТОДОМ 379
хлорид дифенилгуанидиния. Комплексон III маскирует катионы,
образующие устойчивые комплексонаты. Определению не мешают хлориды,
нитраты, фториды (при содержании до 1 мг)\ сульфат и фосфат
осаждаются хлоридом дифенилгуанидиния и после насыщения ими
растворителя флотируются. Осадок отделяют фильтрованием экстрактов;
влияние тория устраняют прибавлением фторида (5 мг фторида калия на
5 мл раствора).
Калибровочный график. Для приготовления серий стандартов в ряд
пробирок вводят 5—50 мкг урана с интервалом 10 мкг, в виде раствора
нитрата уранила с содержанием U 5 мкг/мл и погружают их в
кипящую водяную баню, раствор выпаривают досуха, пропуская через него
воздух, сухой остаток растворяют в 2,0 мл 0,05 н. хлористоводородной
кислоты. Вводят 2,5 мл 5%-ного раствора комплексона III, 1,0 мл
0,05%-ного раствора арсеназо III, 0,5 мл 20%-ного раствора хлорида
дифенилгуанидиния и 5,0 мл бутилового спирта. Экстрагируют,
энергично встряхивают пробирки около 30 сек и после разделения фаз
измеряют оптическую плотность органического слоя на фотометре или
фотоэлектроколориметре по отношению к воде.
Методика определения. В образце определяют уран аналогично
после разложения пробы любым подходящим способом.
Примечание. Можно предварительно экстрагировать уран 20%-ным
раствором трибутилфосфата в хлороформе с применением нитрата аммония в качестве
рысаливателя и комплексона III для удержания мешающих элементов в водной фазе,
а затем реэкстрагировать уран раствором арсеназо Ш и определять уран измерением
оптаческой плотности водной фазы.
Определение молибдена в сталях
Молибден (V) образует с роданидами окрашенные соединения,
состав которых зависит от кислотности среды и концентрации роданида.
Соединения молибдена (VI) восстанавливают до пятивалентного
состояния хлоридом олова (II), иодидом калия, аскорбиновой кислотой, тио-
мочевиной в присутствии солей меди (II) и другими восстановителями.
Наиболее надежные результаты получаются при использовании
последних трех восстановителей. На процесс восстановления молибдена (VI)
сильно влияет кислотность раствора.
В зависимости от концентрации роданида могут образовываться
соединения с мольным отношением Mo : SCN =1:1 до Mo : SCN =1:6.
Наиболее интенсивно окрашены соединения с мольным отношением
Mo:SCN = l:5 (коэффициент Молярного поглощения е равен 15000)
й Mo : SCN = 1:6 (коэффициент молярного поглощения е равен 12600).
Определению молибдена роданидным методом не мешают ионы
алюминия, кобальта, урана, тантала, натрия, калия, кремния, кальция,
магния, титана, ванадия, хрома, марганца, никеля, цинка, мышьяка,
серебра, олова, сурьмы и ртути. Соединения железа (III) и меди
усиливают интенсивность окраски, вероятно, вследствие образования много-
йдерных комплексов, содержащих молибден, железо (или медь) и
роданид. Мешающее влияние вольфрама устраняют введением винной
кислоты, препятствующей образованию роданидных комплексов вольфрама.
Известйо большое число различных вариантов фотометрического
роданидного метода определения молибдена, разработанных с целью
достижения высокой чувствительности и получения надежных и
воспроизводимых результатов. Одним из них является экстракционно-фото-
метрический вариант определения молибдена в сталях.
380 ГЛ. X. ФИЗИКО-ХИМИЧЕСКИЕ МЕТОДЫ ОПРЕДЕЛЕНИЯ РЕДКИХ ЭЛЕМЕНТОВ
Калибровочный график. В пять делительных воронок емкостью
50 мл взодят по 3 мл 8%-ного раствора сульфата железа (III), от 0,5
до 2,5 мл раствора молибдата натрия (аммония) с содержанием Мо
0,1 мг/мл, 5 мл серной кислоты (1 : 1), 5 мл 5%-ного раствора роданида
калия и энергично встряхивают 2—3 мин. Затем прибавляют 5 мл
35%-ного раствора хлорида олова (II) и вновь энергично встряхивают
1—2 мин. При этом растворы постепенно окрашиваются в янтарный или
красновато-бурый цвет.
К растворам прибавляют по 10—12 мл диэтилового эфира,
экстрагируют соединение молибдена 2—3 мин, эфирный слой сливают в сухие
мерные колбы емкостью 25 мл и повторяют экстрагирование. Эфирные
экстракты разбавляют диэтиловым эфиром до 25 мл и измеряют их
оптическую плотность на фотоэлектроколориметре в кюветах, закрытых
крышками для предотвращения испарения эфира. По полученным
данным строят калибровочный график.
Методика определения. Навеску 0,05—0,25 г стали с содержанием
0,01—0,05% молибдена помещают в стакан емкостыа 100 мл, прибавляют
10 мл серной кислоты (1:4) и нагревают до 60—70° С. По окончании
растворения прибавляют 2 мл 30%-ной перекиси водорода, раствор
кипятят несколько минут для окисления карбидов, затем фильтруют через
бумажный фильтр для удаления углерода, нерастворимый остаток
промывают водой и отбрасывают.
Фильтрат и промывные воды объединяют, упаривают в стакане до
объема 10—Г5 мл. При анализе простых углеродистых и легированных
сталей или сталей, содержащих вольфрам, остаток не отфильтровывают.
Прибавляют 0,5 г винной (лимонной) кислоты, если сталь содержит
вольфрам, затем нейтрализуют 10%-ным раствором едкого натра до
рН 8—9 по универсальной индикаторной бумаге, прибавляют 5 мл
серной кислоты (1:1) и переносят в делительную воронку емкостью 50 мл.
Прибавляют 5 мл 5%-ного раствора роданида калия и далее поступают,
как описано при построении калибровочного графика. Содержание
молибдена в навеске находят по калибровочному графику.
Определение галлия
Принцип метода. Метод основан на взаимодействии ионов галлия
с родамином С в среде 6 н. хлористоводородной кислоты,
сопровождающемся доследующей экстракцией хлоргаллата родамина подходящим
органическим растворителем. В качестве экстрагентов могут быть
использованы: бензол, смесь его с диэтиловым эфиром (3:1), с бутил
ацетатом (4: 1),а также смесь хлорбензола с четыреххлористым углеродом.
Молярный коэффициент поглощения 8 бензольного экстракта при 565 нм
составляет 60000, а при экстракции смесью хлорбензола и четыреххло-
ристого углерода (3:1) молярный коэффициент поглощения е экстракта
при 562 нм равен 78900.
Определению мешают ионы железа (III), сурьмы (V), мышьяка (V),
молибдена (VI), таллия (III), теллура (IV), селена (IV), алюминия
(>0,3 мг), меди (>2 мг). Соединения алюминия и меди отделяются
при экстракции галлия бутилацетатом из 6 н. раствора НС1. Влияние
Fe, Sb, As, Mo, Те, Se и Т1 устраняют введением TiCl3.
Калибровочный график. В делительные воронки емкостью 25—50 мл
помещают растворы, содержащие 1—5 мкг галлия, с интервалом 1 мкг
Ga, добавляют до объема 5 мл 6 н. раствора НС1, затем вводят 0,2 мл
5%-ного раствора треххлористого титана, 10 мл смеси бензола и бутил-
ацетата и 0,5 мл 0,5%-ного раствора родамина С. Взбалтывают 2 мин.
§ 4. РАБОТЫ, ВЫПОЛНЯЕМЫЕ ЭКСТРАКЦИОННО-ФОТОМЕТРИЧЕСКИМ МЕТОДОМ 381
После окончания разделения фаз нижний слой отбрасывают, а
окрашенный экстракт фильтруют через слой стеклянной ваты. Измеряют
оптическую плотность экстрактов на спектрофотоколориметре ФЭК с
зеленым светофильтром в кюветах с толщиной слоя 1 см относительно того
же органического растворителя и строят калибровочный график.
Методика определения. В анализируемом растворе определяют
галлий, как описано выше, и содержание его находят по калибровочному
графику.
Определение галлия в алюминии
Наилучшим методом отделения галлия от сопутствующих элементов
для последующего фотометрического определения является экстракция
его из 6 н. раствора НС1 диэтиловым эфиром или менее летучим бутил-
ацетатом при соотношении фаз 1:1. Коэффициент распределения в
последнем случае приблизительно равен 400. Реэкстрагируют галлий
водой. Определение галлия проводят родаминовым методом.
Методика определения. Навеску 0,25 г образца металлического
алюминия растворяют при умеренном нагревании в 20—30 мл 6 н.
растворе НС1. К полученному раствору прибавляют по каплям 5%-ный
раствор TiCl3 до фиолетовой окраски раствора. Выдерживают 2—3 мин и
переводят в делительную воронку емкостью 100 мл, ополаскивая стакан
3—5 мл 6 н. хлористоводородной кислотой. К раствору прибавляют
равный объем бутилацетата и энергично встряхивают 1 мин. После
разделения фаз нижний слой отбрасывают, экстракт промывают 2 раза 6 н.
хлористоводородной кислотой (по 2—3 мл). Реэкстрагируют галлий
10—15 мл воды, взбалтывая в течение 1 мин. После расслаивания
нижнюю фазу переводят в фарфоровую чашку. Повторяют реэкстракцию
галлия водой, сливая реэкстракт в ту же чашку, прибавляют 0,1 aNaCl,
упаривают на водяной бане досуха. Сухой остаток растворяют в 6 н.
хлористоводородной кислоте, раствор переводят в мерную колбу
емкостью 25 мл и объем доводят до метки этой же кислотой. Отбирают
5 мл раствора, переносят в делительную воронку емкостью 25—50 мл и
поступают далее, как указано в предыдущей методике.
Определение германия
Метод основан на взаимодействии германия (IV) с фенилфлуоро-
ном в кислом растворе с образованием красного осадка. При малом
количестве германия появляется суспензия, которую можно стабилизовать
добавлением защитного коллоида. Соединение германия с фенилфлуо-
роном не экстрагируется органическими растворителями, но
флотируется. Изменение концентрации кислоты в сравнительно широких
пределах (0,3—1,5 н.) не влияет на образование этого соединения. При
меньшей кислотности выпадает осадок реактива. Максимум
поглощения окрашенных растворов фенилфлуората германия находится при
500 нм, однако при измерениях лучше пользоваться светофильтром
с максимумом светопропускания 530 нм. Молярный коэффициент
поглощения (е) комплекса 38500 ±800.
С фенилфлуороном реагируют также титан, цирконий, гафний,
олово (IV), ниобий, тантал, сурьма (III), теллур, молибден, вольфрам.
Окислители ванадий (V),xpoM (VI), марганец (VII) и церий (IV)
окисляют реагент. Ионы галлия и мышьяка в кислых растворах не реагируют
с фенилфлуороном. Не мешают определению фторид (<1 мг в 10 мл)
и железо (III) (100 мкг в 10 мл).
382 ГЛ. Х4 ФИЗИКО-ХИМИЧЕСКИЕ МЕТОДЫ ОПРЕДЕЛЕНИЯ РЕДКИХ ЭЛЕМЕНТОВ
Калибровочный график. В ряд мерных колб емкостью 25 мл вводят
5—25 мкг германия с интервалом 5 мкг (в виде раствора с содержанием
Ge 5 мкг/мл), оставляя одну колбу пустой. Прибавляют во все колбы
по 4 мл 6 н. раствора НС1, воду до 20 мл, 1 мл 1%-ной желатины и
перемешивают. Затем вводят по 1,5 мл 0,05%-ного раствора фенилфлуо-
рона, перемешивают и доводят водой до метки. Оставляют стоять 30 мин.
Измеряют оптическую плотность ра фотоэлектроколориметре или
фотометре, пользуясь светофильтром с максимумом пропускания при 530 нм,
относительно раствора сравнения, полученного в холостом опыте.
Методика определения. При анализе различных объектов германий
отделяют от мешающих ионов экстракцией четыреххлористым
углеродом из 9 М раствора НС1 или дистилляцией GeCl4 (в присутствии
окислителя).
Для определения германия в угле 1 г пробы помещают в
платиновую чашку, прибавляют 0,5 г СаО, перемешивают, приливают 6 мл
насыщенного раствора Ca(N03b (50 мг в 100 мл общего объема )и
выпаривают сначала на водяной бане, а потом на песочной. После этого
озоляют в муфельной печи, медленно нагревая до 400—450° С для
сгорания основной массы углистых частиц, затем повышают температуру
печи до 700—800° С. Слишком резкое нагревание в начале сжигания
приводит к вспышке и распылению сжигаемой массы. После сгорания
основной массы угля смесь 2—3 раза перемешивают шпателем для
ускорения выгорания углистых частиц. Сжигание продолжают до
получения белого или буроватого порошка. Охлаждают, приливают по кац-
лям при перемешивании 5 мл азотной кислоты и выпаривают на родя-
ной бане досуха. К остатку прибавляют 5 мл фтористоводородной
кислоты и снова упаривают досуха. Прибавляют 5 мл фтористоводородной
и 10 мл фосфорной кислоты и выпаривают сначала на водяной бане,
а затем на песочной до удаления HF и получения сиропообразного
остатка, который смывают 25 мл воды в стакан и нагревают до
распадения комков.
При анализе железных окисных руд германий может быть
извлечен разложением навески фтористоводородной, азотной и фосфорной
кислотами. Навеску 0,5—1,0 г помещают в платиновую чашку,
прибавляют 5 мл азотной и 5 мл фтористоводородной кислоты и выпаривают
на водяной бане досуха, затем добавляют 5 мл фтористоводородной и
5 мл фосфорной кислоты и выпаривают до удаления HF.
Сиропообразный остаток смывают 25 мл воды в стакан и нагревают до растворения
остатка.
При анализе силикатов их разлагают смесью фтористоводородной,
серной и азотной кислот (или смесью HF, Н3Р04 и HN03). Навеску
0,5 г помещают в платиновую чашку, прибавляют 3 мл серной кислоты
(1:1), 0,5 мл концентрированной азотной ц 5 мл фтористоводородной
кислоты. Выпаривают до появления паров H2SO4. Добавляют 5 мл зоды
и нагревают до растворения осадка.
Экстракция германия. Полученный тем или иным способом раствор
(объем 25 мл) охлаждают до комнатной температуры, прибавляют
75 мл концентрированной хлористоводородной кислоты (на возможное
помутнение не обращают внимания), охлаждают, переносят в
делительную воронку емкостью 200 мл, вводят 20 мл четыреххлористого
углерода и экстрагируют 2 раза по 2 мин. Экстракт промывают 3 раза
2%-ным раствором солянокислого гидроксиламина в 9 н.
хлористоводородной кислоте (каждый раз по 10 мл), взбалтывая в течение 1 мин.
Реэкстрагируют германий 6 мл воды, повторяя операцию 2 раза,
взбалтывая каждый раз в течение 1 мин.
§ 4. РАБОТЫ, ВЫПОЛНЯЕМЫЕ ЭКСТРАКЦИОННО-ФОТОМЕТРИЧЕСКИМ МЕТОДОМ 383
Водные вытяжки переносят в мерную колбу емкостью 25 мл и
далее поступают так же, как при построении калибровочного графика.
Определение селена
Принцип метода. Селен (IV) образует с 3,3'-диаминобензидином
в нейтральной, щелочной или кислой среде окрашенное в желтый цвет
соединение, плохо растворимое в воде.
Образующийся дифенилдипиазоселенол хорошо экстрагируется
толуолом при рН 5, это используется при экстракционно-фотометрическом
определении. В разбавленных водных растворах циазоселецол образует
окрашенные растворы, оптические плотности которых пропорциональны
концентрациям селена в интервале 0,25—2,5 мкг/мл. Молярный
коэффициент поглощения & составляет 10 200 при 347—349° С и 0,1 н.
концентрации кислоты. Окраска растворов развивается в этих условиях через
50 мин и сохраняется в течение 5 ч.
Определению не мешают алюминий, барий, кальций, кадмиц,
кобальт, калий, магний, марганец, молибден (VI), никель, теллур (IV),
натрий, цинк, аммоний, бромид, хлорид, нитрат, фосфат, сульфат,
цитрат, оксалат и тартрат.
Железо (III) можно маскировать фторидом, оксалатом.
3,3'-ДиамиНобензидин применяется для фотометрического
определения селена в чистом индии, мышьяке и их полупроводниковых
соединениях.
Калибровочный график. В ряд стаканов емкостью 50 мл вводят
стандартный раствор, содержащий 1,0—7,0 мкг селена, с интервалом
2,0 мкг, в один стакан стандартный раствор не вводят. Добавляют по
40 мл воды, 1 мл 2,5%-ного раствора комплексона III, 2 мл муравьиной
кислоты (1:9) и нейтрализуют аммиаком (1:1) по крезоловому крар-
ному до рН 2—3 (желтая окраска индикатора). Затем вводят 2 мл
0,5%-ного свежеприготовленного раствора 3,3'-диаминобензидина в
хлористоводородной кислоте и оставляют на 30 мин. Прибавляют аммиак
(1:1) до рН 8 (фиолетовая окраска раствора индикатора), переносят
в делительные воронки емкостью 75—100 мл, добавляют 11 мл толуола
или бензола и экстрагируют окрашенное соединение 1 мин. Экстракты
фильтруют через сухой бумажный фильтр в кювету с толщиной слоя
3 см и измеряют оптическую плотность на фотоэлектроколориметре
ФЭК-Н-54 при 415—-420 нм и строят калибровочный график.
Методика определения селена в мышьяке. Навеску 1,0 г мышьяка
растворяют при умеренном нагревании в 6 мл смеси равных объемов
азотной и хлористоводородной кислот, избегая бурной реакции.
Упаривают почти досуха, прибавляют 15—20 мл воды, нагревают до
получения прозрачного раствора, который после охлаждения разбавляют
водой до 40 мл, вводят комплексон III и муравьиную кислоту и далее,
как описано выше (см. Калибровочный график).
Методика определения селена э индии. Навеску 0,5 г индия
растворяют при умеренном нагревании в 3 мл азотной кислоты, раствор
упаривают почти досуха. Остаток растворяют в 20 мл соляной кислоты
(1:7), прибавляют растрор, содержащий 50 мг мышьяка, 1 мл раствора
хлорида олова (II), 10 мл 10%-ного раствора гцпофосфита натрия
в хлористоводородной кислоте (1:7) и немного мацерированной
бумаги. Раствор нагревают до кипения, кипятят 5—7 мин до появления
осадка, разбавляют хлористоводородной кислотой (1:7) до 50 жл и
оставляют на ночь.
На следующий день осадок отфильтровывают через тампон из
мацерированной бумаги и промывают 8—10 раз хлористоводородной
384 ГЛ. X. ФИЗИКО-ХИМИЧЕСКИЕ МЕТОДЫ ОПРЕДЕЛЕНИЯ РЕДКИХ ЭЛЕМЕНТОВ
кислотой (1 : 7). Промытый осадок вместе с тампоном переносят в
стакан, в котором проводили осаждение, прибавляют 6 мл
хлористоводородной кислоты (пл. 1,17—1,19 г/см3) и 3—5 капель азотной кислоты
(пл. 1,5 г/см3). Стакан погружают в водяную баню и выдерживают при
70—80° С до растворения осадка. Прибавляют 10—15 мл горячей воды,
2 мл 5%-ного раствора мочевины для разрушения избытка окислителя,
хорошо перемешивают и отфильтровывают на фильтре «белая лента»,
фильтр промывают 2—3 раза теплой водой. К фильтрату добавляют
4 мл 25%-ного растворе лимонной кислоты, охлаждают и далее, как
описано выше (см. Калибровочный график).
Методика определения селена в арсениде и антимониде индия.
Навеску 0,5 г арсенида или антимонида индия растворяют при умеренном
нагревании в 6 мл смеси равных объемов концентрированных
хлористоводородной и азотной кислот и воды и упаривают почти досуха.
При анализе арсенида индия остаток растворяют при нагревании
в 20 мл хлористоводородной кислоты (1 :7), прибавляют 1 мл раствора
хлорида олова (II), 10 мл раствора гипофосфита натрия и немного ма-
церированной бумаги, в холостой опыт вводят дополнительно 1 мл
раствора арсената натрия (50 мг мышьяка).
При анализе антимонида индия остаток растворяют при
нагревании в 5 мл 25%-ного раствора лимонной кислоты, затем вводят 20 мл
хлористоводородной кислоты (1:7), 0,5 мл раствора арсената натрия,
1 мл раствора хлорида олова (II), 10 мл раствора гипофосфита натрия
и немного мацерированной бумаги.
Далее в обоих случаях раствор нагревают до кипения, кипятят 5—
10 мин до появления осадка, разбавляют хлористоводородной кислотой
(1 :7) до объема 50 мл и оставляют на ночь. Далее, как описано при
определении селена в индии, но при анализе антимонида индия вместо
2 мл вводят 4 мл раствора 3,3'-диаминобензидина.
Определение теллура в висмуте
Хлоридный комплекс теллура (IV) с диантипирилпропилметаном
количественно экстрагируется дихлорэтаном из сильносолянокислых
растворов. Таким путем удается легко отделять любые количества
теллура от селена, меди, свинца и висмута (от последних двух в
присутствии комплексона III).
Для определения теллура хлоридный комплекс переводят в бромид-
ный комплекс с диантипирилпропилметаном или диантипирилметаном и
измеряют оптическую плотность экстракта. Молярный коэффициент
поглощения е при 450 нм равен 4200.
Метод очень прост в выполнении, дает хорошо воспроизводимые
результаты, отнимает немного времени и может быть использован для
определения Те в товарном селене, висмуте, меди и свинце.
Калибровочный график. В серию делительных воронок емкостью
50 мл вводят от 10 до 100 мкг Те в виде раствора теллуристой кислоты
с содержанием Те 0,01 мг/мл. Прибавляют воду до объема 10 мл,
затем 5 мл бромистоводородной кислоты и оставляют на 1—2 мин. Вводят
1 мл 1%-ного раствора диантипирилпропилметана и 5 мл дихлорэтана,
встряхивают 1 мин. Отделяют экстракты, фильтруют их через сухой
фильтр в кювету с толщиной слоя 1 см, измеряют оптическую плотность
на фотоэлектроколориметре (или спектрофотометре) относительно
дихлорэтана и строят калибровочную кривую.
Окраска экстрактов устойчива длительное время.
§ 4 РАБОТЫ, ВЫПОЛНЯЕМЫЕ ЭКСТРАКЦИОННО-ФОТОМЕТРИЧЕСКИМ МЕТОДОМ 385
Методика определения. Навеску 0,2—0,5 г свинца или висмута
растворяют в 20—25 мл азотной кислоты (1 :3). Раствор выпаривают
почти досуха, обмывают стенки колбы водой и вновь выпаривают до*
суха. Сухой остаток растворяют в 15—40 жл 0,1 М раствора
комплексна III при нагревании. Добавляют концентрированную
хлористоводородную кислоту в объеме, равном объему введенного раствора
комплексна III. Раствор переносят в делительную воронку емкостью 1Q0 ида
200 мл, добавляют 2 мл 5%-ного раствора диантипирилпропилметана
в уксусной кислоте (1:1), перемешивают и добавляют 5 мл
дихлорэтана. Встряхивают 1 мин.
После разделения фаз экстракт сливают в чистую делительную
воронку емкостью 50 мл. К оставшейся водной фазе прибавляют еще 5 мл
дихлорэтана и снова встряхивают 1 мин. Дихлорэтанные экстракты
объединяют. Водные растворы отбрасывают. Экстракты промывают
двумя порциями по 10 мл хлористоводородной кислоты (1:1).
Теллур извлекают, встряхивая экстракт с 10 мл 0,02 М раствора
комплексона III. Эту операцию проводят 2 раза. После разделения фаз
сливают дихлорэтан. Водные растворы объединяют, переводят в
мерную колбу емкостью 50 или 100 мл и доводят до метки 0,02 М
раствором комплексона III.
Отбирают аликвотную часть раствора 5 мл, добавляют 5 мл 0,02 М
раствора комплексона III, 10 мл бромистоводородной кислоты, 1 мл
1%-ного раствора диантипирилпропилметана, 5 мл дихлорэтана и
встряхивают 1 мин.
После разделения органическую фазу фильтруют через сухой
фильтр в кювету с толщиной слоя 1 см. Измеряют оптическую
плотность на фотоэлектроколориметре ФЭК-Н-57 или на спектрофотометре'
относительно дихлорэтана.
Содержание теллура находят по калибровочному графику.
Определение теллура в селене
Хлоридный комплекс теллура (IV) с диантипирилпропилметаном
количественно экстрагируется дихлорэтаном из солянокислых
растворов, а тот же комплекс селена (IV) при этом полностью остается
вводной фазе (вне зависимости от концентрации кислоты). Таким путем
удается легко отделять любые количества теллура от селена.
Для определения теллура хлоридный комплекс переводят в бро-
мидный комплекс с диантипирилпропилметаном и измеряют оптическую
плотность полученного раствора.
Калибровочный график. В серию делительных воронок емкостью
50 мл вводят от 10 до 100 мкг Те в виде раствора теллуристой кислоты,
добавляют воду до объема 10 мл, по 5 мл перегнанной
бромистоводородной кислоты и оставляют на 1—2 мин, затем добавляют по 0,5 мл
раствора аскорбиновой кислоты, 1 мл 1%-ного раствора
диантипирилпропилметана, 5 мл дихлорэтана и встряхивают 1 мин.
После разделения фаз экстракты отделяют, фильтруют через сухие
фильтры в кювету с толщиной слоя 1 см и измеряют оптическую
плотность на фотоэлектроколориметре или спектрофотометре относительно
дихлорэтана.
Окраска экстрактов устойчива длительное время.
Методика определения. Навеску 0,5 г металлического селена
растворяют в 15—20 мл концентрированной азотной кислоты, добавляют
5 мл серной кислоты (1:1) и выпаривают до появления паров H2S04.
После охлаждения осторожно прибавляют 5 мл воды и снова
386 ГЛ. Х9 ФИЗИКО-ХИМИЧЕСКИЕ МЕТОДЫ ОПРЕДЕЛЕНИЯ РЕДКИХ ЭЛЕМЕНТОВ
выпаривают до появления паров H2S04, охлаждают и остаток переводят
в мерную колбу емкостью 50 мл хлористоводородной кислотой (1:1).
Отбирают аликвотную часть 5 или 10 мл (в зависимости от содержания
теллура) полученного раствора в делительную воронку емкостью 100 мл,
вводят 1—2 мл 5%-ного раствора диантипирилпропилметана в уксусной
кислоте (1:1), прибавляют хлористоводородную кислоту (1:1) до
объема 20 мл, 5 мл дихлорэтана и встряхивают 1 мин.
После разделения фаз экстракт сливают в чистую делительную
воронку емкостью 50 мл. К оставшейся водной фазе добавляют еще 5 мл
дихлорэтана и снова встряхивают 1 мин, экстракт отделяют. Дихлор-
этанные экстракты объединяют (водный раствор отбрасывают) и
промывают 2 раза хлористоводородной кислотой (1:1) порциями по 10 мл.
Теллур извлекают из объединенного экстракта встряхиванием
с 10 мл 0,02 М раствора комплексона III. После разделения фаз
дихлорэтан сливают. К оставшемуся водному раствору добавляют 5 мл
бромистоводородной кислоты и дают постоять 1 мин, затем прибавляют
0,5 мл раствора аскорбиновой кислоты, 1 мл 1%-ного раствора дианти-
пирилпропилметана и 5 мл дихлорэтана, встряхивают 1 мин.
Объединенные экстракты фильтруют через сухой фильтр в кювету с толщиной
слоя 1 см. Оптическую плотность измеряют относительно дихлорэтана
на фотоэлектроколориметре ФЭК-Н-57 или спектрофотометре.
Количество теллура находят по калибровочному графику.
§ 5. Работы, выполняемые спектрофотометрическим
методом
Определение тантала
Тантал с пирогаллолом образуют комплекс в среде 4 н. раствора
НС1 и 0,0175 М оксалата. Молярный коэффициент поглощения
комплекса е в этих условиях составляет 4775. Оптическая плотность
растворов пропорциональна концентрациям тантала до 40 мкг/мл.
Определению мешают молибден (VI), вольфрам (VI), уран (VI), олово (IV).
Влияние ниобия, титана, циркония, хрома, ванадия (V), висмута, меди
не существенно, и его можно учесть введением их в холостой раствор.
Определению тантала мешает фторид, платина, поэтому сплавление
анализируемых проб нельзя проводить в платиновой посуде.
Калибровочный график. В ряд мерных колб емкостью 50 мл вводят
0,2; 0,4; 0,6; 0,8 и 1,0 мг Та205 (в виде раствора), прибавляют 25 мл 8н.
раствора НС1, вводят 10 мл раствора пирогаллола и разбавляют
раствором оксалата аммония до метки. Оптическую плотность раствора
измеряют на спектрофотометре СФ-4 при 325 нм.
Методика определения. В анализируемом растворе определяют
тантал, как описано выше. Содержание тантала находят по
калибровочному графику.
Определение таллия
Метод основан на образовании йодистого комплекса
таллия (III) с диантипирилметаном или диантипирилпропилметаном
(C23H2402N4'HTll4 или C26H30O2N4 * НТП4) и последующей их
экстракции бензолом. Молярный коэффициент поглощения е бензольного
экстракта при 400—405 нм составляет 12 000 и не зависит от природы
основания. В присутствии комплексона III как маскирующего средства при
рН 2—3 определение таллия возможно в присутствии ионов меди (II),
железа (III), висмута, а также кадмия, свинца, цинка, галлия (III),
индия (III).
§ 5. РАБОТЫ, ВЫПОЛНЯЕМЫЕ СПЕКТРОФОТОМЕТРИЧЕСКИМ МЕТОДОМ 387
Калибровочный график. В маленькие стаканы помещают раствор
соли таллия (I) с содержанием Т1 40, 80, 120, 160, 200 мкг, добавляют
2—3 мл бромной воды, нагревают до кипения, избыток брома удаляют
кипячением. После охлаждения растворы переносят в делительные
воронки емкостью 25—50 мл, ополаскивая стаканы 2—3 мл 1 н. серной
кислоты, затем вводят 2—3 мл 0,1%-ного раствора иодида калия и 1—
2 мл 0,05%-ного раствора диантипирилметана или диантипирилпропил-
метана в уксусной кислоте (1 : 10). Экстрагируют 10 мл бензола, после
разделения слоев нижний сливают, а верхний фильтруют через сухой
бумажный фильтр. Оптическую плотность экстракта измеряют
относительно бензола при 400 нм в кювете с толщиной слоя 1 см на
спектрофотометре СФ-4.
Методика определения. В анализируемом растворе определяют
таллий, как описано выше. Содержание Т1 находят по калибровочному
графику.
Концентрирование и определение малых количеств таллия
Метод основан на соосаждении таллия сульфидом висмута Bi2S3.
Осадок сульфида висмута затем растворяют, таллий восстанавливают
гидроксиламином и вновь выделяют с коллектором КВ(С6Нб)4 при
рН 5—6 в присутствии комплексона III. Осадок растворяют в 2 н.
хлористоводородной кислоте, таллий окисляют бромом до Т1ш, добавляют
родамин Б и экстрагируют комплекс бензолом. Таким образом таллий
отделяется от Ga, Sb, Fe, мешающих дальнейшему определению.
Методика определения. Навеску 1 г анализируемой породы
обрабатывают в платиновой чашке 3 раза 8—10 мл фтористоводородной
кислоты, выпаривая раствор каждый раз досуха, затем 2 раза смесью 8—
10 мл фтористоводородной и 0,5 мл серной кислот и нагревают на плитке
до полного удаления паров H2SO4. Остаток растворяют при нагревании
в 100 мл 1 н. хлористоводородной кислоты, добавляют 1 мл 0,1 М
раствора BiCl3 (20 мг Bi), 5 мл 2 н. раствора Na2S и центрифугируют.
Промывают осадок 5—6 раз промывной жидкостью (0,5 мл 2 н.
хлористоводородной кислоты и 0,5 мл 2 н. раствора Na2S на каждые 100 мл воды),
растворяют в 1—2 мл смеси ССЦ + Вг2 и переносят в стакан емкостью
250 мл, используя для этого 5—6 мл азотной кислоты. Раствор
выпаривают до 1—2 мл, разбавляют водой до 50 мл, добавляют 2 г
солянокислого гидроксиламина, 20 мг хлорида калия, 25 мл насыщенного
раствора комплексона III и по каплям свежеприготовленный раствор тетра-
фенилбората натрия. Осадок центрифугируют и промывают 5—6 раз
бидистиллятом, растворяют при нагревании в хлористоводородной
кислоте, которую добавляют порциями по 2—3 мл, переносят в стеклянную
чашку и выпаривают на водяной бане досуха. Остаток растворяют
в 4 мл 6 н. хлористоводородной кислоты и переносят в делительную
воронку емкостью 10 мл. Добавляют 4 мл бензола и проводят экстракцию
органических веществ дважды. Бензольный слой промывают 2—3 раза
6 н. хлористоводородной кислотой. Объединенные кислотные вытяжки
переносят в стеклянную чашку, добавляют 1 мл бромной воды, каплю
10%-ной сульфосалициловой кислоты и встряхивают до полного
обесцвечивания раствора. Затем добавляют 0,4 мл 0,5%-ного раствора
родамина Б, 4 мл бензола и экстрагируют. Бензольный слой переносят
в кювету и измеряют оптическую плотность на спектрофотометре СФ-4.
Одновременно ставят холостой опыт и так же измеряют оптическую
плотность бензольного раствора. Количество таллия находят по
калибровочному графику, построенному с использованием аналогичной
методики.
388 ГЛ. X. ФИЗИКО-ХИМИЧЕСКИЕ МЕТОДЫ ОПРЕДЕЛЕНИЯ РЕДКИХ ЭЛЕМЕНТОВ
§ 6. Работы, выполняемые флуориметрическим методом
Определение индия
Метод основан на взаимодействии бромидного комплекса индия
с родамином 6Ж. Образующееся соединение экстрагируют бензолом из
15 н. серной кислоты и определяют концентрацию индия по
интенсивности флуоресценции экстракта. Мешающие ионы железа (III), меди (II),
олова (IV), сурьмы (III), таллия (III), золота (III),ртути (II) удаляют
при экстракции индия бутилацетатом с последующей реэкстракцией
хлористоводородной кислотой. Возможен ускоренный вариант отделения
мешающих элементов с применением двукратного осаждения
аммиаком и цементации на металлическом железе.
Калибровочный график. В ряд пробирок емкостью 10 мл
прибавляют 0,1—1,0 мкг соединения In с интервалом 0,1 мкг, разбавляют до
10 мл 15 н. азотной кислотой, вводят 100 мг металлического железа
(восстановленного водородом) и выдерживают 30 мин. Отбирают пипеткой
аликвотную часть (5 мл) и через ватный тампон переводят раствор
в делительную воронку емкостью 25 мл, добавляют 0,1 мл 0,25%-ного
водного раствора родамина 6Ж, 6 мл бензола и 0,5 мл 40%-ного
раствора бромида калия. Сразу же после добавления проводят экстракцию
в течение 30 сек. Дают раствору отстояться, переводят слой
органического растворителя в сухую пробирку и измеряют интенсивность
флуоресценции на флуорометре.
Методика определения. При определении индия в рудах навеску
0,3 г растворяют в 25—30 мл смеси хлористоводородной и азотной
кислот при нагревании на песочной бане. По окончании разложения
раствор упаривают до небольшого объема (5 мл) и разбавляют водой до
25—30 мл. Из полученного раствора осаждают гидроокиси добавлением
небольшого избытка 25%-ного раствора аммиака. Осадок
отфильтровывают через плотный фильтр и промывают 2—3 раза 1%-ным
раствором сульфата аммония. Осадок на фильтре растворяют горячей серной
кислотой (1 :2), промывают фильтр 2—3 раза водой, собирая фильтрат
и промывные воды в стакан, в котором проводилось осаждение. Затем
вновь осаждают гидроокиси. Отфильтровывают осадок, промывают 3—
4 раза 1%-ным раствором сульфата аммония и 1—2 раза водой.
Осадок на фильтре растворяют 15 н. серной кислотой, собирая фильтрат
в пробирку и разбавляя его до 10 мл той же серной кислотой. После
охлаждения добавляют 0,15—0,20 г железа, восстановленного
водородом, и перемешивая, выдерживают в течение 1 ч. Затем раствор
переводят в другую пробирку, добавляют 100 мг железа и вновь
выдерживают 30 мин. Из пробирки отбирают аликвотную часть (5 мл) и
далее, как описано при получении стандартной шкалы. Сравнивают
интенсивность анализируемого раствора с серией стандартных растворов
визуально или «а флуорометре.
§ 7. Работы, выполняемые методом потенциометрического
титрования
Определение вольфрама (VI)
Метод основан на восстановлении соединений шестивалентного
вольфрама до пятивалентного солями хрома (II). Точку
эквивалентности устанавливают потенциометрическим методом. По окончании
восстановления вольфрама (VI) наблюдается отчетливый скачок потен-
ииала. При титровании потенциал платинового электрода устанавли-
§ 7. РАБОТЫ, ВЫПОЛНЯЕМЫЕ МЕТОДОМ ПОТЕНЦИОМЕТРИЧЕСКОГО ТИТРОВАНИЯ 389
вается тотчас же после добавления соли хрома (II). Титрование
проводят в среде концентрированной хлористоводородной кислоты (на
5,0 или 10 мл 0,1 М раствора вольфрамата натрия необходимо 100 или
150 мл концентрированной хлористоводородной кислоты). Лимонная,
винная, щавелевая и муравьиная кислоты не влияют на ход титрования.
В присутствии ионов железа (III), меди (II), хроматов и молибдатов
на кривой титрования наблюдается два скачка. Окончанию
восстановления вольфрама соответствует второй скачок (в присутствии
молибдена второй скачок также соответствует восстановлению
пятивалентного молибдена до трехвалентного). Поэтому в присутствии элементов,
более легко восстанавливающихся, чем вольфрам, целесообразно
вначале оттитровать WVI раствором соли хрома (II) до соответствующего
вольфраму скачка потенциала, а затем потенциометрическим методом
титровать образовавшийся Wv раствором бихромата калия. Этот метод
очень удобен для определения вольфрама в шеелитовом концентрате.
Методика определения. Навеску 0,25 г тонкорастертого концентрата
в небольшой фарфоровой чашке обрабатывают при нагревании 4—5 мл
концентрированной хлористоводородной кислоты. После разложения
шеелита и упаривания почти сухой остаток смачивают 3 мл 20%-ного
раствора едкого натра. К полученному раствору прибавляют 10 мл
насыщенного раствора щавелевой кислоты и содержимое чашки смывают
в сосуд для титрования, содержащий 100 мл концентрированной
хлористоводородной кислоты. Сосуд закрывают резиновой пробкой, в
которой имеются отверстия для подвода и отвода С02, для кончика
бюретки, солевого мостика и платинового индикаторного электрода. Для
удаления кислорода через титруемый раствор пропускают в течение
30 мин из аппарата Киппа двуокись углерода. Титруют 0,1 н. раствором
CrS04 при перемешивании магнитной мешалкой и пропускании С02 до
получения скачка потенциала, соответствующего окончанию
восстановления вольфрама (VI), после чего оттитровывают вольфра*м (V)
раствором бихромата калия.
Определение рения в сплавах
Метод основан на восстановлении солями хрома (II) перрената до
четырехвалентного рения. Титрование проводят при 60—70° С в среде
4 н. серной кислоты в присутствии небольших количеств иодида калия
как катализатора. Точку эквивалентности устанавливают с помощью
компенсационного потенциометра, применяя в качестве индикаторного
электрода платиновую пластинку, а в качестве электрода сравнения —
насыщенный каломельный полуэлемент. Определение возможно в
присутствии небольших количеств молибдена (RerMo = 1 : 1), а также
железа, титана, хрома, ванадия, никеля, кобальта, ниобия и меди.
Последние легко отделяются в виде гидроокисей путем осаждения аммиаком
или щелочью перед титрованием.
Методика определения. Навеску сплава 0,5 г растворяют при
нагревании в смеси 5 мл хлористоводородной и 5 мл азотной кислот. Раствор
выпаривают на водяной бане до 2—3 мл, добавляют немного воды, 2 мл
концентрированной серной кислоты и выпаривают вновь сначала на
водяной бане, а затем на электрической плитке при слабом нагревании
до появления паров H2S04. Остаток растворяют при нагревании в 50 мл
воды, затем осаждают гидроокиси аммиаком (для сплавов Re с Fe, Cr,
V, Ti, Nb) или 5%-ным раствором NaOH (для сплавов Re с Со, Ni, Си).
Фильтрат выпаривают до небольшого объема, прибавляют серную
кислоту до нейтральной реакции по лакмусу, затем добавляют 7 мл
390 ГЛ. X, ФИЗИКО-ХИМИЧЕСКИЕ МЕТОДЫ ОПРЕДЕЛЕНИЯ РЕДКИХ ЭЛЕМЕНТОВ
концентрированной серной кислоты на 50 мл конечного объема и
разбавляют водой точно до 50 мл. Для титрования в зависимости от
содержания рения отбирают аликвотные части (5; 10 или 20 мл). Раствор
помещают в герметически закрытую ячейку для потенциометрического
титрования, через которую пропускают ток азота. Раствор нагревают
до 80° С, вводят 1 мг иодида калия и титруют 0,1 н. раствором CrS04,
добавляя его вначале по 1 мл> а при приближении скачка потенциала —
по 0,1 мл. В присутствии молибдена титрование продолжают до
получения второго скачка потенциала, определяя объем раствора,
израсходованный на титрование рения, как разность объемов раствора CrS04
между вторым и первым скачком потенциала. 1 мл 0,1 н. раствора
CrS04 эквивалентен 6,210 мг рения.
ГЛАВА XI
ФИЗИКО-ХИМИЧЕСКИЕ (ИНСТРУМЕНТАЛЬНЫЕ)
МЕТОДЫ
АНАЛИЗА НЕВОДНЫХ РАСТВОРОВ
Новые методы анализа, основанные на использовании реакций,
которые протекают в неводных растворах, имеют большое значение.
Прогресс, наблюдаемый в аналитической химии неводных
растворов, объясняется многими их особенностями. Основное преимущество
использования неводных растворов состоит в том, что в среде неводных
растворителей можно дифференцированно (раздельно) титровать
многокомпонентные смеси веществ, которые нельзя оттитровать в водных
растворах. Например, в неводных средах можно дифференцированно
титровать смеси электролитов, характеризующихся близкими
значениями рК, смеси соединений, относящихся к одному и тому же гомоло*
гическому ряду, смеси изомеров и т. п. (см. книга 2, гл. II, § 34).
Л. ТЕОРЕТИЧЕСКАЯ ЧАСТЬ
§ 1. Влияние химической природы и физико-химических свойств
растворителей на свойства растворенного вещества
Развитие теории аналитической химии неводных растворов.
Успешное применение методов титрования неводных растворов оказалось
возможным благодаря развитию теории аналитической химии неводных
растворов, достигшей уже такого развития, которое позволяет с
известной степенью достоверности предвидеть поведение растворенного
вещества в данном растворителе, теоретически объяснить процессы,
протекающие при титровании разнообразных веществ в неводных средах,
предопределить выбор растворителя и титранта для данного
конкретного случая титрования, произвести соответствующие количественные
расчеты и т. д.
В растворах различных веществ в жидких неводных растворителях
и сжиженных газах помимо ионов, предсказываемых теорией
электролитической диссоциации, имеются разнообразные ионы и молекулы,
вызывающие аномалии в поведении истинных растворов, которые не
могут быть объяснены ни гипотезой С. Аррениуса, ни современными
теориями Дебая — Хюккеля и Л. Онзагрра, поскольку предметом их не
является изучение влияния растворителей на свойства электролитов.
Следует отметить, что теория Бренстеда и другие теории, предметом
которых было исследование влияния растворителей на силу кислот и
оснований, также не объясняют аномалий д поведении электролитов
в неводных растворах. Как показывают исследования, указанные
аномалии обусловливаются взаимодействием растворенного вещества с
растворителем.
392 ГЛ. XI. ФИЗИКО-ХИМИЧЕСКИЕ МЕТОДЫ АНАЛИЗА НЕВОДНЫХ РАСТВОРОВ
Влияние индивидуального характера растворителя сказывается на
электропроводности, на состоянии ионов в растворах, на
преимущественном направлении реакций, на химических свойствах
растворенного вещества и т. п.
Кислотно-основное взаимодействие растворенного вещества с
растворителем. Особый интерес для аналитической химии представляет
необычное поведение в неводных растворах многих веществ. Например*
СН3СООН, обладающая в водных растворах свойствами слабой
кислоты, ведет себя в жидком аммиаке как сильная кислота, а в среде
жидкого фтористого водорода — как основание.
Столь необычное поведение СН3СООН в различных
растворителях объясняется тем, что вода является слабым, а жидкий аммиак —
сильным акцептором по отношению к протонам СН3СООН, т. е. в
реакции
СН3СООН + Н20 ^±: СНзСОСГ + Н30+
протолитическое равновесие смещено справа налево, а в реакции
CH3COOH + NH3 ^=5, СН3СОСГ -f- NHJ
протолитическое равновесие смещено слева направо.
Безводный жидкий фтористый водород обладает донорными
свойствами и в реакции:
CH3COOH+H2F2 ^=± CH3COOH?-bHFJ
протолитическое равновесие смещено слева направо. Поэтому уксусная
и муравьиная кислоты и фенолы, проявляющие в водных растворах
кислые свойства, ведут себя в среде жидкого аммиака как сильные
кислоты, а в среде жидкого фтористого водорода — как основания.
В растворителях типа безводной уксусной кислоты кислые свойства
проявляют лишь те минеральные кислоты, которые являются сильными
кислотами в водных растворах. Все органические кислоты, как
правило, не проявляют в протогенных растворителях кислых свойств. В еще
более протогенных растворителях типа безводной серной кислоты или
жидкого фтористого водорода они в раде случаев обнаруживают
свойства оснований. Это специфическое действие кислых растворителей на
кислоты в обычном смысле понимания связано со сродством к протону
молекул и анионов кислот (НАп), растворенных в протогенном
растворителе (НМ).
Аналогично в основных растворителях типа безводного аммиака
основные свойства проявляют лишь те из оснований, которые обладают
сильноосновным характером в водных растворах.
Следовательно, сила основания возрастает с увеличением
кислотности, а сила кислоты — с увеличением основности растворителя. Сила
основания или кислоты связана также с величиной диэлектрической
проницаемости е растворителя.
А. М. Шкодин показал, что рК кислот, оснований и солей в кислых
растворителях одной химической природы является линейной функцией
1/е — величины, обратной диэлектрической проницаемости среды.
Таким образом, сила данной кислоты или основания, а также
характер кривых титрования зависят не только от природы
растворенного вещества, но и от свойств растворителя и величины его
диэлектрической проницаемости.
Самой сильной кислотой в растворителе (НМ) является сольвати-
рованный протон НгМ+ (ионы лиония) и сильнейшим основанием ионы
лиата М~*
§ 1. ВЛИЯНИЕ РАСТВОРИТЕЛЕЙ i*A РАСТВОРЕННОЕ ВЕЩЕСТВО 393
Количественная зависимость силы кисЛот и оснований от свойств
растворителя и растворенного вещества. Для характеристики изменения
свойств электролитов под влиянием растворителей можно
воспользоваться выведенным Н. А. Измайловым основным уравнением,
характеризующим влияние растворителей на силу электролита:
Е А -А
** пх л\юл
*о6-*диссв RT (1)
где Коб — константа диссоциации, измеренная обычными методами и выражающая
отношение произведения активностей свободных ионов к сумме активностей
недиссоциированных частиц электролита;
/Сдисс —для кислот эта величина определяется константами двух равновесий —
константой собственной кислотности кислоты (/(а), характеризующейся
сродством аниона кислоты к протону и константой собственной кислотности
ионов лиония растворителя Ка ц м+, характеризующейся сродством
молекулы растворителя к протону или обратной ей величиной — константой
собственной основности растворителя Кь,нм, т. е.
^диссe ~F aHM= Kcfib, HM аНМ (2)
а, Н2М+
Для оснований величина /СДИСс определяется константами собственной основности
основания (Къ) и константой собственной основности ионов лиата растворителя К _
или обратной ей величиной — константой собственной кислотности растворителя
Ка, НМ, Т. е.
Кдиес = -j? «HM = Kb^a, HM %М &)
ХЬ,М~
где ^jAx —сумма энергий сольватации ионов (У A —A Kt+ + ^ А ~)>
Амол —энергия сольватации молекул.
Для кислоты величина А + представляет собой энергию сольватации протона,
X, Кд
складывающуюся из энергии присоединения протона к молекуле растворителя НМ,
т. е. образования иона лиония НгМ^, и энергии его сольватации.
Для основания величина А + выражает энергию сольватации ВН+-иона, обра-
X, Кд
зующегося в процессе взаимодействия молекулы основания с ионами водорода
растворителя;
А — энергия образования и сольватации иона лиата М~.
Частными случаями приведенного выше уравнения (1) являются:
для кислот
^HAn = _
is
а, Н2М+
для оснований
К п Ах~Аи.оп
kb^Vhm,—тг- (5)
ь, м~
Пользуясь этими уравнениями, можно проследить влияние прото-
генных и протофильных растворителей на силу электролитов.
Зависимость значения Ах от величины, обратной диэлектрической
проницаемости среды, характеризует влияние диэлектрических свойств
растворителей на силу электролитов.
В растворителях с высокой диэлектрической проницаемостью, на-
примерь в воде, для кислот устанавливается следующая система
равновесий:
HAn + H20 ^z± Н30+-АгГ ^=± Н30+ + АгГ
СА"= « ¦ НМ е Ш (4)
394 ГЛ. XI. ФИЗИКО-ХИМИЧЕСКИЕ МЕТОДЫ АНАЛИЗА НЕВОДНЫХ РАСТВОРОВ
Ионная пара Н30+*Ап- в таких растворителях является сильным
электролитом и практически полностью диссоциирует на ионы Н30+ и
An~. Поэтому
к _ [НзОЧ [An"] ^
ДнАп [HAn] (6)
Величина Кяап является количественной мерой силы кислоты.
В растворителях, подобных безводной уксусной кислоте, т. е.
обладающих низкой диэлектрической проницаемостью, даже самые сильные
электролиты лишь очень слабо диссоциируют на ионы. В таких средах
преобладают ионные пары с малыми константами диссоциации.
Образование ионных пар можно рассматривать по Кольтгофу как
процесс ионизации, а распад ионных пар на ионы — как процесс
диссоциации. Эти процессы в среде безводной уксусной кислоты можно
выразить следующими уравнениями:
для сильной кислоты (НАп)
HAn + HAc ^z± H2Ac+-Air ^=± Н2Ас+ + Агг
для сильного основания (В):
В + НАс ^=± ВН+.Ас~ +± ВН+ + Ас~
т. е.
а„
+ &«¦,"*
дгНАп _ Н2Ас «An ,-.
^иоя а *7'
"НАп
В , gBH+.Ac- (
где Д"н^п и /СВ —константы равновесия, отражающие процессы ионизации кислоты
и основания;
а — активности.
Константы диссоциации кислоты или основания можно выразить
следующими уравнениями:
а А.а
ьгНАп _ НгАс^ An /m
хдисс а
+ .А«"~
НгАс^.Ап"
а л.а -
„в _ вн+ Ac nm
ВН+-Ас
НАп
Константа диссоциации Коь может быть представлена в виде
следующего уравнения
.___т_ - л
аНАп
уНАп, Н2Ас An" (И)
где afHAn — активность недиссоциированных молекул растворенного вещества,
представляющая собой сумму активностей свободных молекул кислоты анап
и активностей ионных пар alii^.pLXi-
Или
tf-HAn H2Ac An
НАп Н2Ас+ «An
Из уравнения (7) находим:
в1адс+.Аа-"^^вИАа
§ I. ВЛИЯНИЕ РАСТВОРИТЕЛЕЙ НА РАСТВОРЕННОЕ ВЕЩЕСТВО 395
Следовательно
CL JL.CL _ CL J.CL
ь^НАп НгАст An НгАст An"
Д об = ——
аНАп + ^ионПаНАп аНАп I1 + ^ион")
Подставляя значение аНАп из уравнения (7), получим:
а +а _КНАп
ь-НАп НгАст An ион
ан2Ас+.Ап-(1+К^П)
Отсюда [см. уравнение (9)] получим:
iv-HAn w'HAn
ьгНАп_ лДИСс^ион ¦ ,10ч
Доб ~ - „HAn W
1 + ^ион
Аналогично можно получить общую константу диссоциации
основания Ков:
а +а Кв Кв
егВ ВН Ас дисс ион /10\
Коб = = в (13)
В ВН Ас ион
Величины К^п и К*ш, по мнению Кольтгофа и Брукеястейна,
дают лучшее представление о силе кислоты или основания в
неводном растворителе, чем обычные константы равновесия. Однако
уравнения, описывающие К^^1 и /Сион, представляют собой лишь
частный случай общего уравнения Н. А. Измайлова.
Согласно Измайлову, диссоциация кислот, солей и оснований на
ионы в водных и неводных растворах зависит от ряда сопряженных
динамических равновесий: образования сольватов — продуктов
присоединения электролита к молекулам растворителя, диссоциации сольватов
с образованием сольватированных ионов лиония и лиата, ассоциации
сольватированных ионов с образованием ионных пар, или двойников.
Соотношения между активными концентрациями продуктов этих
реакций зависят от свойств растворенного электролита и растворителя, а
также от их концентраций.
Констацта диссоциации (Коб), определяемая обычными методами,
эквивалентна общей константе диссоциации и выражается по
Измайлову следующим образом (см. книга 2, гл. II, § 33):
v ^дисс
Аоб^ ¦ (Н)
А "т" ^нест "*~ *^пр
где К дисс — константа равновесия, отражающего процесс диссоциации образовавшегося
продукта присоединения на сольватированные ионы;
Кнест — константа нестойкости, отражающая процесс образования продуктов
присоединения молекул электролита к молекулам растворителя;
Дпр — константа, отражающая процесс превращения недиссоциированного
продукта в ионные пары; Кпр = /Сдисс Касс = К^п
Обратная величина константы ассоциации по Измайлову
эквивалентна константе диссоциации по Кольтгофу:
т*—\ ._ ifHAn
*\асс ^дисс
В ряде случаев уравнение (14) принимает частные значения.
Например, при условии, когда /Снест значительно меньше 1:
К JC K~~^ lx-HAn ifHAn
rr ^дисс ^пр^асс *Удисс ^ион /1Kv
д0? as —_ = « HAll \*&)
i+/enp 1+/спр i+^HOH
396 ГЛ. XI. ФИЗИКО-ХИМИЧЕСКИЕ МЕТОДЫ АНАЛИЗА НЕВОДНЫХ РАСТВОРОВ
Пользуясь этими уравнениями, можно решать многие вопросы,
интересующие химиков-аналитиков подобно тому, как применение закона
действия масс в его классической форме дает врзможность решать
задачи, связанные с использованием водных растворов.
Реакции титрования. Вследствие малой диэлектрической
проницаемости некоторых неводных растворителей типа безводной уксусной
кислоты все известные кислоты и основания мало диссоциированы в них.
Наиболее сильной кислотой в среде безводной уксусной кислоты
является хлорная кислота (р/( = 4,87). Серная кислота в безводной
уксусной кислоте проявляет себя более слабой кислотой (р/С = 7,24),
чем сама уксусная в водном растворе (р/С = 4,74). Поэтому для
титрования слабых оснований в неводных растворах очень часто применяют
растворы хлорной кислоты в безводной уксусной кислоте и диоксане.
Как показали наши исследования, лучшим растворителем для хлорной
кислоты является метилэтилкетон или смесь растворителей: безводная
уксусная кислота — уксусный ангидрид. В качестве титрантов
оснований широко используются также n-толуолсульфокислота и
хлористоводородная кислота. Процессы, протекающие при титровании
органических оснований R(Ar)NH2 в среде протогенных растворителей, можно
представить в виде уравнений:
нсю4-ьсн3соон ^± сн3соон?. cioj ^=± сн3соон? 4- cioj
R(Ar)NH2 + CH3COOH ^=± R(Ar)NH? • СН3СОСГ ^Z± R(Ar)NHj + СН3СОСГ
СН3СООН? + СН3СОСГ ^z± 2CH3COOH
R(Ar)NHj + C10J ^z± R(Ar)NH2-HC104
R(Ar)NH2 + HC104 —> R(Ar)NH2 • НСЮ4
Для титрования кислот в неводных растворах применяют
преимущественно органические основания, имеющие малые значения р/С.
Большей частью применяют гидроокиси тетраалкиламмония, например
гидроокись тетраэтиламмония.
Процессы, протекающие при титровании кислот в неводных
растворителях, можно представить в виде уравнений:
о он он
II II II
НАп + СН3-С-СН2СНз ^=± [СН3-С~СН2СН3] • Ап- ^=± [СН3—С—СН2СН3] + Atr
ОН О"
I I
(C2H5)4NOH + CH3~C = CHCH3 :^± (C2H5)4N+ +[СН3-С=СНСН3] + Н20
ОН О"" О
II I II
[СН3—С—СН2СН3] + [СН3—С = СНСН3] ^=± 2СН3—С—СН2СН3
(C2H5)4N+ + An" ^=t (C2H5)4NAn
НАп + (C2H5)4NOH —> (C2H5)4NAn + H20
Кислотные или основные свойства электролитов в неводных
растворах объясняются в известной мере тем, что соответствующие
продукты реакции растворенного электролита с молекулами растворителя,
подобно аквакомплексным соединениям {а), сообщают растворам кис-
§ 1. ВЛИЯНИЕ РАСТВОРИТЕЛЕЙ НА РАСТВОРЕННОЕ ВЕЩЕСТВО 397
лую или щелочную реакцию среды вследствие диссоциации по типу
кислот (б) или по типу оснований (в):
[CrPy2(H20)„]+++ ^± [CrPy2(H20)n-,OH]++ + H+ (a)
[СгРу2(Н20)^,ОН]++ ^=± [СгРу2(Н20)„_2(ОН)2]+ + Н+ и т. д.
RNH2 + rtHAc ^=± [RNH2(HAc)„] (б)
[RNH2(HAc)„] ^=± H+^RNH^HAc^Ac]-
[RNHa(HAcV,Acr 5=± Н+ + [RNH2(HAc)„.2Ac2]"" и т. д.
[RNH2(HAc)„] ^=± [RNH3(HAc)n^]+ + Ac- (в)
Существование в неводных растворах кислот и оснований, не
содержащих в своем составе ионов водорода или гидроксил-ионов, может
быть объяснено свойствами соединений, склонных акцептировать
молекулы и ионы, являющиеся носителями кислых и основных свойств:
Н30+ и ОН- — в водных растворах; NHt и NH<> — в среде жидкого
аммиака; СН3СООН2+и СНзСОО- — в среде безводной уксусной кислоты
и т. д.
В процессе титрования под воздействием кислот или оснований
динамические равновесия смещаются, а координированно связанные
молекулы растворителя, являясь акцепторами или донорами протонов
или соответствующих анионов, реагируют с образованием продуктов
нейтрализации.
Изменение в соотношении силы электролитов. Под влиянием
растворителей изменяется не только сила электролитов, но и соотношение
в их силе. Указанное положение особенно применимо к кислотам и
основаниям различной природы, растворимым в растворителях с низкой
диэлектрической проницаемостью. Это дает возможность осуществлять
дифференцированное титрование смесей кислот или оснований, а также
многоосновных кислот и оснований.
С другой стороны, в зависимости от избранного растворителя
меняется отношение:
где /Cs — ионное произведение данного растворителя.
Возможность осуществления того или иного конкретного случая
титрования в неводных растворах основана на выборе соответствующих
растворителей.
Интересно, что во многих случаях, когда в неводных растворителях
титруют слабые электролиты, изменение рН кислот и оснований
происходит аналогично изменениям, наблюдающимся при нейтрализации
сильного основания сильной кислотой в водных растворах, и
получаемые кривые нейтрализации имеют резкие скачки титрования. Это
объясняется большой протяженностью шкалы рН неводных растворителей,
характеризующихся очень малым значением /Cs- Поэтому в среде не*
водных растворителей с p/Cs, сильно превышающем pAV, возможно
дифференцированно титровать слабые и очень слабые кислоты (или
основания) и соединения, нерастворимые в воде, разлагаемые водой и
образующие с водой стойкие нерасслаивающиеся эмульсии.
398 ГЛ. XI. ФИЗИКО-ХИМИЧЕСКИЕ МЕТОДЫ АНАЛИЗА НЕВОДНЫХ РАСТВОРОВ
§ 2. Классификация неводных растворителей
по их протонно-донорно-акцепторным свойствам
По характеру участия в процессах кислотно-основного
взаимодействия все растворители делят на два класса:
протолитические — кислые (или протогенные), основные (или про-
тофильные), амфипротные (или амфотерные);
апротонные — нейтральные (или инертные).
Протолитические растворители. К протолитическим растворителям
относятся такие химические соединения, молекулы которых способны
быть либо донорами, либо акцепторами протонов.
Кислые, или протогенные, растворители проявляют
в отношении оснований протонно-донорные свойства, к этой группе
относят: H2F2, H2SO4, НСООН, СНзСООН, СН3СН2СООН, (СН3СО)20,
НОСН2СН2ОН, C6H5N02, CH3N02, СН3ОН и др.
Схему взаимодействия такого растворителя (НМ) с растворенным
в нем основанием (В) можно представить следующим образом:
в + нм ^=> вн+ + м~
ониевый анион
катион растворителя
Например
(CH3)3N + CH3COOH ^z± (CH3)3NH+ + CH3COO~
Однако молекулы протогенных растворителей могут присоединять
чужие протоны от кислот, обладающих более выраженным протоген-
ным характером. Например, уксусная кислота принимает протоны от
молекул жидкого фтористого водорода.
Основные, или протофильные, растворители
проявляют в отношении кислот протонно-акцепторные свойства.
К этой группе относят: NH3, N2H4, H2NCH2CH2NH2,
CH3CH2CH2CH2NH2, C5H5N, (СН2)402 и др.
Схему взаимодействия такого растворителя (НМ) с растворенной
в нем кислотой (НАп) можно представить следующим образом:
HAn + HM ^=± МН+ + АгГ
ониевый анион
катион кислоты
Например
СН3СООН + C5H5N Z?± C5H5NH4* + СН3СОО"
Однако молекулы таких растворителей могут и отдавать свои
протоны основаниям, обладающим более сильным сродством к протону.
Например, этилендиамин отдает свои протоны МЩ-ионам:
H2NCH2CH2NH2 + NHJ ^=± H2NCH2CH2NH" + NH3
Другими словами, протолитические растворители принимают
непосредственное участие в протонно-донорно-акцепторном, т. е. кислотно-
основном, взаимодействии,
Амфипротные, или амфотерные, растворители
являются химическими соединениями амфотерного характера. Эти
растворители проявляют свойства доноров протонов по отношению к
основаниям и акцепторов протонов по отношению к кислотам.
К этому типу растворителей относят воду, этиловый, пропиловый,
бутиловый спирты, ацетон, метилэтилкетон и некоторые другие
органические растворители.
§ 2. КЛАССИФИКАЦИЯ РАСТВОРИТЕЛЕЙ ПО ИХ СВОЙСТВАМ 399
Апротонные растворители. К апротонным растворителям относят
химические соединения нейтрального характера, практически не
способные ни отдавать, ни присоединять протоны вследствие того, что
молекулы апротонного растворителя не ионизированы.
Апротонные растворители либо совсем не встуйают в процессы
протолитического (кислотно-основного) взаимодействия с
растворенным веществом, либо слабо проявляют кислотно-основные свойства.
В их среде кислотно-основное равновесие осуществляется без заметного
протонно-донорно-акцепторного участия молекул растворителя. К этому
типу растворителей относят жидкие углеводороды (бензол, толуол, гек-
сан и др.) и жидкие галогенпроизводные углеводородов (хлороформ,
четыреххлористый углерод, дихлорэтан и др.).
Апротонные диполярные растворители. К растворителям
апротонного характера относят еще одну группу веществ, которые называют
апротонными диполярными растворителями. Представителями этой
группы являются ацетонитрил CH3CN, диметилформамид HCON(CH3h,
диметилацетамид CH3CON(CH3)2, диметилсульфоксид (CH3hSO, гек-
саметилфосфортриамид OP[N(CH3)2]3, К-метил-2-пирролидон и другие.
Такие вещества не содержат в своем составе активных атомов
водорода, слабо проявляют склонность к образованию водородных связей,
но вступают в побочные реакции гомо- и гетероконъюгации за счет
водородных связей конъюгирующих веществ (см. ниже).
Количественное выражение сродства к протону. Количественная
зависимость силы электролита от свойств вещества и растворителя
может быть выражена уравнением (1), в котором /СДИсс электролита
определяется сродством к протону ионов или молекул диссоциирующего
вещества в вакууме. Если электролит является кислотой, то для него
значение /СДИсс определяется сродством к протону аниона этой кислоты,
и может быть охарактеризовано константой собственной кислотности
электролита в вакууме Ка [см. уравнение (2)].
Сродство к протону ионов в газообразном состоянии в вакууме
можно представить следующим уравнением реакции:
Н?аз + Ап7аз ^ НАпгаз + л; (16)
где Нраз — протон в газообразной фазе в вакууме;
Ап~аз ~ анион в газообразной фазе в вакууме;
НАпГаз — молекула кислоты в газовой фазе в вакууме;
тс — протонное сродство.
Величину протонного сродства к протону вычисляют, пользуясь
следующим уравнением:
я - - Д#° + Д#°А _ + Л#° (17)
HAn An Нт х '
где Д#НАп — изменение энтальпии при образовании газообразной кислоты НАпгаз;
А^дп- — изменение энтальпии при образовании аниона Ап~аз;
Д^н+ ~" изменение энтальпии при образовании протона #газ-
Другими словами, сродством к протону называют максимальное
количество энергии, выделяемое при присоединении протона молекулой
или ионом.
Если речь идет об основаниии, то для него значение /СДИсс
[уравнение (1)] определяется сродством, к протону его молекулы (например,
NH3) или иона (например, NHj) и может быть охарактеризовано
константой собственной основности электролита Кь [см. уравнение (3)],
являющейся величиной, обратной константе кислотности ВН+-ионов.
400 ГЛ. XI. ФИЗИКО-ХИМИЧЕСКИЕ МЕТОДЫ АНАЛИЗА НЕВОДНЫХ РАСТВОРОВ
Величины сродства к протону (табл. 14), представляющего собой
энергию присоединения протона, выражают в ккал/моль (ккал/г-ион)
или кдж/моль (кдж/г-ион). Критерием силы основания является
сродство к протону.
ТАБЛИЦА 14
Величины сродства к протону
Основание
(молекула
или ион)
Уравнение
Протонное сродство
ккал/моль
(ккал/г-ион)
кдж/моль
{кдж/г-ион)
н2о
NH3
сю4-
hso4-
I-
Вг-
N03~
С1-
hs-
СНдСОО-
с6н5соо-
нсоо-
CN-
F-
он-
NHJ
SO"
С03"
s—
о—
Н+ + Н20 ^zt H30+
:NH+
Н+ + СЮ? ^=± НСЮ4
Н+ + HS04~;
H+ + I-:
Н++Вг~
Н+ + N0^ ;
Н+ + С1-:
H+ + HS-:
Н+ + СНаСОО-;
Н+ + СбН5СОО- :
Н+ + НСОО- :
H+ + CN-:
H+ + F-:
Н+ + ОН-;
Н+ + NHJ
H+ + SO":
Н+ + СО":
H+ + S~-:
Н+ + 0—:
: h2so4
*hi
*HBr
:HN03
:HCl
:H2s
: CH3COOH
: C6H5COOH
:HCOOH
:hcn
*HF
!H20
tNH3
: hso4-
:hco^"
::HS-
:он-
184
212
285
296
307
315
320
325
343
343
345
347
348
363
383
419
433
498
541
615
773
890
1193
1239
1285
1319
1340
1361
1436
1436
1444
1453
1457
1520
1604
1754
1803
2085
2265
2575
Как следует из табл. 14, ярко выраженным сродством к протону
обладают ЫНг-ионы, превосходящие в этом отношении ОН~-ионы,
известные как носители сильноосновных свойств. Это означает, что
носителями наиболее сильных основных свойств являются не щелочи (NaOH,
КОН и др.) в водных растворах, а амиды щелочных металлов (NaNH2,
KNH2) в среде безводного жидкого аммиака.
Сильные в воде кислоты отличаются меньшими значениями величин
сродства к протону. Однако следует иметь в виду, что эти величины во
всех случаях остаются высокими. Это означает, что диссоциация в
вакууме электролитов по типу кислот, сопровождающаяся отрывом
протонов, может протекать лишь при условии затраты большого количества
энергии (1>300 ккал/моль). Относительно высокая диссоциация
некоторых кислот в воде или других растворителях объясняется
взаимодействием протонов и анионов кислот с молекулами растворителей
(сольватация), при которой выделяется энергия, достаточная для отрыва
протонов. Так, суммарная энергия образования иона гидроксония и его
дальнейшей сольватащш равна 258 ккал. Эта энергия на 100—150 ккал
превосходит энергию сольватации других катионов.
§ 2. КЛАССИФИКАЦИЯ РАСТВОРИТЕЛЕЙ ПО ИХ СВОЙСТВАМ 401
Сродство к протону молекулы воды jtHa0 составляет 184 ккал/моль.
Следовательно, вода является более слабым основанием по сравнению
с соединениями, сродство к протону которых значительно выше
(например, сродство к протону аммиака составляет 212 ккал/моль, а амид-
иона — 419 ккал/г-ион).
Естественно, что существуют более слабые основания, чем вода,
которые проявляют свои протонно-акцепторные свойства по отношению
к очень сильным кислотам. Существуют и более сильные основания,
чем NHjrHOHbi (см. табл. 14), такого рода основания проявляют свои
протонно-акцепторные свойства по отношению к очень слабым кислотам.
Все анионы кислот в той или иной мере проявляют основные
свойства, причем анионы органических, слабых в воде неорганических и
полностью депротонизированных многоосновных кислот (СН3СОСГ, CN~»
S~~, СО-Г, РОГ"~ и т. п.) проявляют наиболее четко выраженные
основные свойства.
Из минеральных кислот самой сильной является хлорная, анион
которой отличается наименьшей величиной сродства к протону (см.
табл. 14); наиболее сильными из оснований являются О—-ионы,
реагирующие с Н+-ионами с образованием ОН~-ионов и характеризующиеся
максимальным сродством к протону среди известных оснований.
Взаимодействие протолитических растворителей с молекулами
растворенного вещества. Наглядное представление о взаимодействии
протолитических растворителей с растворенным веществом дают
следующие уравнения:
R2NCONH2 + CH3COOH ^z± R2NCONH? + СН3СОСГ
основание донор
протона
R2NCONH2 + NH3 ^=± R2NC0NH~4-NHJ
кислота акцептор
протона
о о- он
II I II
2СНз-С-СН2СН3 *=* [СН3—С = СНСН3] + [СН3—С-СН2СН3]
растворитель
О"
I
R2NCONH2 + [CH3—C = CH—СНз] ^=± R2NCONH" + СН3СОСН2СН3
кислота акцептор протона
ОН
II
R2NCONH2 + [CH3—С—СН2СН3] ^=± R2NCONH? + CH3COCH2CH3
основание донор протона
Образование продуктов присоединения. Переход протона от
кислоты к протофильному растворителю или от протогенного растворителя
к основанию следует рассматривать как конечную стадию их протонно-
донорно-акцепторного взаимодействия, которому предшествует, согласно
данным Измайлова, образование молекулярных соединений.
Таким образом, кислоты диссоциируют не по схеме Бренстеда
HAn + MH ^z± МН+ + АгГ
а по такой схеме:
HAn + лНМ ^z± НАп-НМл ^z± МН+ . Ап^л ^=± MHj + An^
402 ГЛ. XI. ФИЗИКО-ХИМИЧЕСКИЕ МЕТОДЫ АНАЛИЗА НЕВОДНЫХ РАСТВОРОВ
Другими словами, на ионы диссоциируют не сами кислоты, а
продукты присоединения их молекул к молекулам растворителя.
Молекулярные соединения часто возникают за счет связи между
водородом кислоты и электроотрицательными атомами (кислородом,
азотом, фтором и др.), которые обладают необобществленной парой
электронов. Например, молекулы карбоновых кислот преимущественно
ассоциированы в димеры:
н3с-с^ ^с-сн3
х)-.-но/
или
СН3
0=СЧ /0Н
I
СН3
Молекулярные соединения возникают также при взаимодействии
кислот и фенолов с кетонами:
R\ />
N С = О •.. НОС6Н5; СН3С^ Ж
К'/ ХОН-.-0=С
уксусной кислоты с диоксаном:
/>
СНзС ^СН2—СН2ч
хон • • • о хо •. • но—с—сн3
^СНг-СН/" II
О
триметиламина с муравьиной кислотой:
Н—С и т. д.
XOH...N(CH3)3
Границы применимости классификации. Деление растворителей на
протолитические и апротонные носит условный характер, так как
некоторые вещества, известные как типична нейтральные соединения, в ряде
случаев ведут себя как основания или кислоты; некоторые амфипрот-
ные растворители могут быть отнесены к апротонным диполярнымит. д.
Кислотные и основные свойства проявляют даже углеводороды. Их
реакции с едкими щелочами (а), металлорганическими соединениями
(б), кислотами (в), щелочными металлами (г),хлоридом алюминия (д)
и т. п. можно рассматривать как процессы кислотно-основного
взаимодействия:
CH=CH + NaOH ^z± CH=CNa + H20 (a)
C6H6 + C2H5Na —> C6H5Na + C2H6 (б)
C10H8 + HF —> CI0H9F (в)
(C6H5)3CH + K+ —> (C6H5)3CK + H+ (г)
С6Нв + НА1С14 —> СбН7А1С14 (д)
Приведенные реакции свидетельствуют о проявлении
углеводородами протонно-донорных, т. е. кислых, свойств (реакции а, б, г) и про-
тонно-акцепторных, т. е. основных, свойств (реакции в, д).
§ 3. КЛАССИФИКАЦИЯ РАСТВОРИТЕЛЕЙ ПО ИХ ВЛИЯНИЮ НА ЭЛЕКТРОЛИТЫ 403
Донорными свойствами объясняются многие известные реакции
углеводородов, сопровождающиеся замещением их атомов водорода
атомами металлов. Акцепторными свойствами объясняется проявление
электропроводности растворов углеводородов в жидких галогенводо-
родах, сильно увеличивающейся по мере добавления в раствор галоге-
нидов бора, алюминия, бериллия, сурьмы и других соединений,
склонных образовывать комплексные ионы типа: [BF4]~, [А1СЦ]"", [SbCU]—,
[BeF4]"~ и т. д.
Вследствие того что углеводороды проявляют слабые основные
свойства, продукты их присоединения к кислотам, например:
R(Ar)H + HAn ^=± R(Ar)H-HAn
являются слабыми электролитами, растворы их не проводят
электрический ток. Они, как правило, слабо распадаются с образованием карбо-
ний-ионов согласно уравнению:
С6Н6Н An +± С6Н? + An"
Тем не менее углеводороды в указанном смысле можно
рассматривать как основания, которые называют карбооснованиями.
Таким образом, чем сильнее кислота НАп и чем выше
диэлектрическая проницаемость среды, тем вероятнее ионизация продукта
присоединения, сопровождающаяся образованием R (Ar) H? -ионов, т. е. тем
более отчетливо углеводороды проявляют основные свойства.
Следовательно, классификация растворителей по их протонно-ак-
цепторным свойствам условна и распространяется лишь на те
соединения, которые явно ве^ут себя как кислоты, основания или проявляют
типично амфотерный характер.
§ 3. Классификация неводных растворителей по признаку
их влияния на относительную силу электролитов
Нивелирующее и дифференцирующее действие растворителей. По
признаку влияния на относительную силу кислот, оснований и солей
и по способности изменять соотношение в силе электролитов все
растворители обычно делят на два класса: нивелирующие и
дифференцирующие.
К нивелирующим относят растворители, в среде которых сила
кислот, оснований и солей уравнивается; или, строго говоря,
нивелирующими растворителями являются такие, в среде которых соотношения
в силе электролитов (т. е. Кнап, и : ^Снап, i и КкгАп, и: Kkiah, i),
характерные для водных растворов электролитов, сохраняются (см. книга 2,
гл. II, § 34).
К дифференцирующим относят растворители, в среде которых
проявляются значительные различия в силе кислот, оснований и солей.
В среде такого рода растворителей соотношения в силе электролитов
сильно меняются по сравнению с их соотношением в водных растворах.
Однако многочисленные экспериментальные исследования
показали, что каждый растворитель в той или иной степени обладает и
дифференцирующими, и нивелирующими свойствами с той лишь разницей:
одни растворители обладают большим дифференцирующим, чем
нивелирующим, действием в отношении веществ одной и той же группы.
Отсюда следует очень важное теоретическое положение,
заключающееся в том, что деление растворителей на две стабильные группы
нивелирующих и дифференцирующих растворителей принципиально
неверно, условно и относительно, так как в зависимости от целого ряда
404 ГЛ. XI. ФИЗИКО-ХИМИЧЕСКИЕ МЕТОДЫ АНАЛИЗА НЕВОДНЫХ РАСТВОРОВ
факторов один и тот же растворитель может оказывать и
нивелирующее, и дифференцирующее действие.
Даже вода, которая, по мнению многих исследователей, является
нивелирующим растворителем, оказывает дифференцирующее действие
в отношении кислот, обладающих малым отношением второй константы
диссоциации к первой (Д'нап, иАКнап, i ^ 10~4).
Поэтому целесообразно говорить о нивелирующе-дифференцирую-
щем действии растворителей в отношении определенных электролитов.
Нивелирующие и дифференцирующие свойства растворителей следует
рассматривать как две стороны одного и того же явления, подобно
тому, как окислительно-восстановительные свойства веществ
рассматриваются в неразрывной связи при реакциях окисления —
восстановления.
Влияние диэлектрической проницаемости на диссоциацию
электролитов. Один и тот же электролит под влиянием разнообразных
неводных растворителей, характеризующихся различными протолитическими
свойствами и разными значениями диэлектрической проницаемости,
может быть сильным или слабым электролитом и даже совсем потерять
электролитические свойства. Так, а-нафтиламин является слабым
основанием (рЛв = 10,01) в водной среде, очень сильным р/(в = 0,88 вереде
безводной муравьиной кислоты (протогенный растворитель), а в среде
жидкого аммиака и других протофильных растворителей совсем не
проявляет основных свойств.
В протофильном растворителе, каким является гидразин,
обладающий высокой диэлектрической проницаемостью (е = 52), слабые кислоты
(р/С[Н20]3—9) полностью диссоциированы, т. е. проявляют
сильнокислые свойства. В то же время в среде протофильного растворителя —
пиридина (е = 12,3) сильные в воде хлорная, хлористоводородная, бро-
мистоводородная и другие кислоты проявляют слабокислые свойства
(р/( = 3-5).
Таким образом, слабые основания (типа а-нафтиламина) в среде
протогенных растворителей, имеющих высокую диэлектрическую
проницаемость, проявляют сильноосновные свойства, а в среде протофильных
растворителей ведут себя как неэлектролиты.
Точно так же сила кислот в протофильных растворителях сильно
зависит от величины диэлектрической проницаемости растворителя.
Чем выше диэлектрические проницаемости двух сравниваемых,
например, протофильных растворителей, тем сильнее диссоциированы
кислоты.
В растворителях, имеющих очень малую диэлектрическую
проницаемость, большинство электролитов практически не диссоциируют.
Поведение кислот и оснований в некоторых растворителях. Сила
кислот и оснований в среде различных растворителей прежде всего
определяется природой самого растворенного электролита. Свойства и
структура электролита определяют величину константы диссоциации
данного электролита в избранном растворителе. Относительная сила
электролитов одной и той же природной группы (например, карбоновые
кислоты) в различных растворителях в большинстве случаев
сохраняется, и соотношение р/С в данном растворителе к р/С в воде или к р/С
в другом растворителе выражается линейной функцией.
Наиболее всесторонне исследовано поведение в среде безводной
уксусной кислоты оснований (Конант и Холл) и кислот (Измайлов, Шко-
дин, Кольтгоф и Брукенстейн).
В среде безводной уксусной кислоты большее число веществ
проявляют свойства оснований, чем в воде. При этом в уксусной кислоте
§ 3. КЛАССИФИКАЦИЯ РАСТВОРИТЕЛЕЙ ПО ИХ ВЛИЯНИЮ НА ЭЛЕКТРОЛИТЫ 405
слабые основания усиливают свои основные свойства, а ряд веществ,
не проявляющих основные свойства в воде (кофеин, теобромин), ведут
себя в уксуснокислых растворах как основания; сильные основания
вследствие малой диэлектрической проницаемости уксусной кислоты
становятся слабее.
В то же время в среде безводной муравьиной кислоты,
проявляющей ясно выраженные протогенные свойства и обладающей высокой
диэлектрической проницаемостью (е = 57,9), все основания (за редким
исключением) являются сильными.
В среде безводной уксусной кислоты число веществ, проявляющих
кислотные свойства, меньше, чем в воде. Так, карбоновые кислоты
в среде уксусной кислоты не проявляют кислых свойств; кислоты,
имеющие в воде сильнокислые свфйства, становятся в уксуснокислой среде
слабыми.
Ослабление силы кислот в среде безводной уксусной кислоты
обусловлено не только ее протогенным характером, но и низкой
диэлектрической проницаемостью (е = 6,19).
Муравьиная кислота, несмотря на более сильные протогенные
свойства по сравнению с уксусной кислотой, почти не ослабляет силу
минеральных кислот, так как обладает высокой диэлектрической
проницаемостью.
Сопоставление значений р/С некоторых электролитов в
муравьиной, уксусной и пропионовой кислотах показывает, что сила
электролитов в ряду протогенных растворителей одной химической природы
линейно зависит от обратной величины диэлектрической
проницаемости их.
В жидком фтористом водороде и других жидких галогенводородах
вследствие их сильно выраженных протогенных свойств не только
основания, а также спирты, альдегиды, кетоны, фенолы, карбоновые
кислоты и даже углеводороды проявляют основные свойства.
В среде протофильных растворителей увеличивается число веществ,
проявляющих кислые свойства, и уменьшается число соединений,
проявляющих основные свойства. Сила слабых кислот увеличивается;
многие слабые кислоты становятся сильными. Однако сильные кислоты
в среде протофильных растворителей с малой диэлектрической
проницаемостью становятся слабыми.
Возможность дифференцированного титрования. В водной среде
силы многих солей, кислот и оснований нивелированы, поэтому их не
удается дифференцированно оттитровать в присутствии друг друга.
Например, при титровании смеси НС1 + H2S04 + СНзСООН получают
представление об общей кислотности раствора, обусловливаемой
содержанием всех трех кислот. В соответствующем неводном растворителе,
под влиянием которого сила кислот возрастает или понижается и
соотношения в их силе изменяются, оказывается возможным
дифференцированное титрование, т. е. удается установить не только общую
кислотность раствора, но и содержание каждой кислоты, входящей в состав
анализируемой смеси.
То же можно сказать в отношении оснований, дифференцирующихся
под влиянием растворителей. У растворителей, в среде которых
оказывается возможным осуществлять дифференцированное титрование,
превалируют дифференцирующие свойства в отношении определенных
групп электролитов. Дифференцирующим действием могут обладать
протогенные, протофильные, амфипротные, апротонные диполярные и
смешанные растворители.
406 ГЛ. XI. ФИЗИКО-ХИМИЧЕСКИЕ МЕТОДЫ АНАЛИЗА НЕВОДНЫХ РАСТВОРОВ
§ 4. Дифференцирующее действие растворителей
Классификация случаев дифференцирующего действия
растворителей. Измайлов установил три типа дифференцирующего действия
растворителей по отношению кислот и оснований:
1) дифференцирующее действие кислых растворителей с малой
диэлектрической проницаемостью (например, СН3СООН, е = 6,19);
2) дифференцирующее действие основных растворителей с низкой
диэлектрической проницаемостью (например, бутиламин, е = 5,3);
3) дифференцирующее действие, вызывающее изменение
относительной силы электролитов различных природных групп под влиянием
любых растворителей.
Предложенная Измайловым * классификация случаев улучшения
условий титрования в неводных растворах, основанная на изменении
соотношений Ks/Кнап и Ks/Kb; Kuau, и/^Снап, i и Kb,ii/Kb,i> представ-
лена в табл. 15.
ТАБЛИЦА 15
Классификация случаев улучшения условий титрования в неводных растворителях
Случай I. Уменьшение
соотношения -Ks/^HAn или
Ks/Kb пРи титровании
Случай II. Уменьшение
соотношения /СнАп, Il/^HAn, I
в дифференцирующих
растворителях при титровании
Случай III. Уменьшение
соотношения Кв, ll/Кв, I B
дифференцирующих растворителях
при титровании
а) сильных кислот; К$
уменьшается
б) очень слабых кислот в
основных растворителях;
/СНАп увеличивается
в) очень слабых оснований
в кислых растворителях;
/Св увеличивается
г) амфотерных веществ,
если под влиянием
неводного растворителя
*i
HAn
К»
*в
^HAn
а) смеси минеральных
кислот с сильными
органическими кислотами
б) смеси органических
кислот различной природы
в) смеси сильных
минеральных кислот
г) двухосновных кислот по
ступеням диссоциации
д) солей сильных
органических кислот по
вытеснению кислотой
е) смеси солей
а) смеси минеральных
оснований с сильными
органическими основаниями
б) смеси органических
оснований различной природы
в) смеси сильных
минеральных оснований
г) двухосновных оснований
по ступеням диссоциации
д) солей сильных
органических оснований по
вытеснению основанием
Причины, обусловливающие дифференцирующее действие
растворителей. Дифференцирующее действие растворителей на электролиты
обусловливается взаимодействием их, сопровождающимся образованием
продуктов присоединения с различной прочностью. При этом природа
и физико-химические свойства растворителей оказывают
многообразное влияние на силу электролитов, которая изменяется в растворах
дифференцирующих растворителей в разной степени, а также зависит
от природы растворенного вещества.
Например, протогенные растворители нивелируют основания,
которые в их среде, как правило, становятся одинаково сильными и
дифференцируют минеральные кислоты.
* Измайлов Ы. А., Электрохимия растворов, гл, 11, Изд. «Химия», 1967,
§ 4. ДИФФЕРЕНЦИРУЮЩЕЕ ДЕЙСТВИЕ РАСТВОРИТЕЛЕЙ 407
Протофильные растворители, наоборот, нивелируют сильные и
слабые кислоты и дифференцируют сильные основания и очень слабые
кислоты.
Амфипротные и апротонные диполярные растворители, например
метилэтилкетон, ацетонитрил, нитрометан, изопропиловый и изобутило-
вый спирты и т. п., обладают высокими дифференцирующими
свойствами. Смеси амфипротных и апротонных диполярных растворителей
с инертными (апротонными) растворителями, характеризующимися
малой диэлектрической проницаемостью (бензол, хлороформ, дихлорэтан
и др.), также обладают высокими дифференцирующими свойствами.
Дифференцирующее действие растворителей зависит в основном от
тех же факторов, которые определяют силу кислот и оснований в
растворе того или иного растворителя. Например, оно зависит от кислотно-
основных свойств, диэлектрической проницаемости, дипольного момента
молекул растворителя, его сольватирующей способности, образования
водородных связей, комплексообразования и т. п. Зачастую свойства
растворителей не только разных природных групп, но и одной
природной группы изменяются скачкообразно. Поэтому разные растворители
оказывают различное дифференцирующее действие на кислоты,
основания и соли.
Зависимость дифференцирующих свойств растворителей от их
кислотно-основных свойств. В протогенных растворителях происходит
дифференцирование кислот вследствие того, что большое количество
веществ, проявляющих в водных растворах кислотные свойства, в прото^
генных растворителях их не проявляют.
В протофильных растворителях происходит дифференцирование
оснований вследствие того, что большое количество веществ,
проявляющих в водных растворах основные свойства, в протофильных
растворителях их не проявляют.
Амфипротные растворители оказывают дифференцирующее
действие вследствие того, что они практически в одинаковой степени влияют
на поведение в них веществ и с кислотными, и с основными свойствами.
Этим, например, объясняются высокие дифференцирующие свойства ме-
тилэтилкетона в отношении кислот и оснований.
Зависимость дифференцирующих свойств растворителей от их
диэлектрической проницаемости. Дифференцирующее действие
растворителей одной природной группы в ряде случаев тем выше, чем ниже их
диэлектрическая проницаемость.
Например, протогенные растворители: безводная уксусная (в = 6,19),
трихлоруксусная (е = 4,5) и масляная кислота (е = 2,4) — в отличие от
муравьиной кислоты (е = 57,9), нивелирующей кислоты и основания,
дифференцируют не только сильные минеральные кислоты, но и очень
слабые в воде основания (рК = 10—14).
Протофильные растворители с малой диэлектрической
проницаемостью, например пиридин (е = 12,3), в отличие от протофильных
растворителей с высокой диэлектрической проницаемостью, например
гидразина (е = 52), дифференцируют сильные основания и очень слабые
в воде кислоты.
Следовательно, дифференцирующее действие некоторых
протогенных и протофильных растворителей обусловлено низкими
диэлектрическими проницаемостями.
Проявление растворителями различного дифференцирующего
эффекта на различные типы кислот. Сила нейтральных кислот (типа
Н2С03, H2S04, H3PO4 и т. п.) по мере ступенчатой диссоциации
постепенно падает вследствие образования анионных кислот (типа
408 ГЛ. XI. ФИЗИКО-ХИМИЧЕСКИЕ МЕТОДЫ АНАЛИЗА НЕВОДНЫХ РАСТВОРОВ
НСОз, HSO4, НгРО^, HPOi"" и т. п.), которые обладают меньшими
константами диссоциаций, поэтому дифференцирующее действие
растворителей в отношении этих кислот проявляется в различной
степени.
Например, в водном растворе Н3Р04 диссоциирует по типу
кислоты, Н2РО4 проявляет преимущественно кислые свойства, HPCV"
реагирует как основание (см. книга 1, гл. XII, § 9). Дифференцирующий
эффект неводных растворителей проявляется иначе в отношении
нейтральных и анионных кислот. Так, в ряде неводных растворителей не только
HPOJ", но и Н2РО4 имеет преимущественно основной характер. То же
самое можно сказать и о различном дифференцирующем действии
неводных растворителей по отношению катионных кислот (типа
[А1 (Н20) б]"*, [А1 (Н20) 5ОН]++, [А1 (Н20) 4 (ОН) 2]\ [СгРу2 (Н20) п]+++,
[СгРуИ-НгОК-чОН]-"", [СгРу2/(Н20)п-2(ОН)2]+ и т. д.), нейтральных
(NH3, C6H5NH2) и анионных оснований ([А1 (ОН)6Г~ [А1 (Н20)(ОН)5]~),
катионных оснований (Н2ШН?, [Zn (Шз)6Г) И Т. П.
§ 5. Константы диссоциации, потенциалы полунейтрализации
в неводных средах и относительная шкала кислотности
Определение Кнап и Kb в неводных растворах. По величинам
констант диссоциации /Снап (и р/СнАп), Кв (и р/Св) можно судить о
нивелирующем и дифференцирующем действии данного растворителя в
отношении определенных групп кислот или оснований.
А. М. Шкодиным и Л. П. Садовничей установлено, что основания,
рК которых в воде отличаются друг от друга на несколько порядков, в
безводной уксусной кислоте нивелируются и по силе становятся
одинаковыми с ацетатами щелочных металлов, являющихся в уксусной
кислоте самыми сильными основаниями.
А. М. Шкодиным и Л. И. Куркузаки были определены также
константы диссоциации кислот в безводной уксусной кислоте. Оказалось,
что константы диссоциации минеральных кислот на 2—3 порядка
больше (а р/СнАп на 2—3 порядка меньше), чем это считалось раньше. По
силе кислоты располагаются в ряд НСЮ4 > H2S04 > HC1. Из
установленных значений рК следует, что сильные в воде минеральные кислоты
в безводной уксусной кислоте имеют среднюю силу или становятся
слабыми, т. е. уксусная кислота оказывает дифференцирующее действие по
отношению к минеральным кислотам.
Определение методом электропроводности констант диссоциации ди-
фенилгуанидина, изомеров фенилендиамина и других оснований в
среде метилэтилкетона и смешанного растворителя хлороформ — метил-
этилкетон и методом потенциометрического титрования констант
диссоциации ряда неорганических и органических кислот в среде метилового,
этилового, я-пропилового, я-бутилового и изопропилового спиртов, а
также констант ионизации триметилацилоксисиланов и некоторых других
соединений показало, что по сравнению с данными, полученными для
водных растворов, в метилэтилкетоне рК этих соединений уменьшается
на 2 единицы, а в смешанном растворителе на 3 единицы. Такое
действие этих неводных растворителей связано с их меньшей диэлектрической
проницаемостью.
Константы диссоциации исследуемых кислот и оснований
вычисляются на основании данных, полученных для стандартных растворов.
В рассматриваемых случаях в качестве стандартов для кислот
использовались растворы хлористого водорода, а для оснований — метилэтил-
§ 5. КОНСТАНТЫ, ПОТЕНЦИАЛЫ И ШКАЛА КИСЛОТНОСТИ.
409
кетоновые растворы дифенилгуанидина. Константы диссоциации
вычислялись по следующим формулам.
При титровании спиртовым раствором НС1:
РКХ - р/Сст o^gg (18)
где Еу,х— потенциал полунейтрализации исследуемого основания, Ь
?уз> ст — потенциал полунейтрализации стандартного основания, Ь.
При титровании кислот метилэтилкетоновым раствором
дифенилгуанидина:
VKX - рКст + $щ (19)
Оценка относительной силы кислот и оснований по потенциалам
полунейтрализации. Влияние растворителей на силу кислот и оснований
можно оценить методом определения относительной кислотности
электролитов по потенциалам полунейтрализации (?i/2). В момент, когда
нейтрализовано 50% определяемой слабой кислоты или слабого
основания [см. уравнения (23) и (24)], рН = рК. Следовательно, величина
потенциала полунейтрализации определяется величиной р/С титруемой
кислоты или основания и может служить критерием их относительной
силы в неводных растворах. Обычно потенциалы полунейтрализации ис-*
следуемых кислот и оснований измеряют по отношению какого-либо
стандартного вещества. Часто в качестве стандарта при определении
потенциалов полунейтрализации кислот используют бензойную кислоту,
а при определении потенциалов полунейтрализации оснований — дифе-
нилгуанидин. Относительное значение потенциала полунейтрализации
исследуемого электролита (&Еу2) вычисляется как разность значений
потенциалов полунейтрализации исследуемого электролита,
определяемого экспериментальным путем, и стандартного вещества:
Потенциалы полунейтрализации раствора находятся в прямой
зависимости от силы растворенных кислот или оснований.
Стрейли показал, что зависимость между потенциалами
полунейтрализации в уксусном ангидриде и рК (Н20) разная для нейтральных
и анионных оснований. Анионы по сравнению с нейтральными
соединениями проявляют себя более сильными основаниями в уксусном
ангидриде, чем в воде, вследствие чего возможно их дифференцированное
определение в этом растворителе.
Наряду со структурой растворенного вещества при определении ?у2
играет значительную роль и природа растворителя. Так, в среде
уксусной кислоты наблюдается линейная зависимость Д?у2 от рК (Н20) для
многих оснований. В среде ацетона, ацетонитрила и нитрометана,
обладающих хорошими дифференцирующими свойствами, для каждой
группы оснований существует своя зависимость. Изопропиловый спирт
оказывает нивелирующее действие на амины.
Измерение потенциалов полунейтрализации производных
бензойной кислоты, фенола, дикарбоновых кислот, аминов и других
соединений в среде спиртов, кетонов, ацетонитрила, диметилформамида и
смешанных растворителей по отношению к бензойной кислоте и дифенил-
гуанидину показало, что для этих растворителей зависимость Д?у2 от
р/С (Н20) является линейной. Каждой природной группе соответствует
своя линейная зависимость. Для дикарбоновых кислот эта зависимость
выражается двумя пересекающимися прямыми, соответствующими
410 ГЛ. XI. ФИЗИКО-ХИМИЧЕСКИЕ МЕТОДЫ АНАЛИЗА НЕВОДНЫХ РАСТВОРОВ
диссоциации первых и вторых карбоксильных групп (рис. 141).
Заметное отклонение наблюдается для щавелевой и фумаровой кислот.
Обработка экспериментальных данных методом математической
статистики позволила вывести уравнения, характеризующие эти
зависимости.
Дифференцирующее действие растворителей в отношении данной
группы электролитов характеризуется наклоном прямой к оси абсцисс:
чем круче идет прямая, тем больше различаются между собой
потенциалы полунейтрализации электролитов и тем больше возможность
раздельного определения их в смеси.
Рис. 1,41. Относительная сила дикарбоновых кислот в среде спиртов:
/ — метилового; // — этилового; /// — пропилового; IV — изопропилового; V — бутилового;
VI — изобутилового; 1 — щавелевая; 2 — малоновая; 3 — янтарная; 4 — глутаровая; 5 — фталевая;
tf — 3-нитрофталевая; 7 — 4-нитрофталевая; 8 — малеиновая; 9 — фумаровая, 10 — итаконовая;
11 — яблочная; 12 — винная.
Из рис. 141 также видно, что изопропиловый спирт проявляет
большее дифференцирующее действие в отношении вторых карбоксильных
групп дикарбоновых кислот, чем в отношении первых.
Значение потенциалов полунейтрализации при выборе растворителя»
Потенциалы полунейтрализации могут служить полезными
характеристиками при выборе растворителя для данного конкретного случая
титрования. С этой целью при титровании слабых кислот (или слабых
оснований) сопоставляют относительную шкалу кислотности Es (см.
ниже) избранного растворителя с потенциалом полунейтрализации
определяемого электролита. Основный предел относительной шкалы
кислотности растворителя определяет возможность титрования в его среде
слабых кислот, так как их потенциалы полунейтрализации по мере
ослабления кислых свойств электролитов смещаются в основную
область относительной шкалы кислотности. Кислотный предел
относительной шкалы кислотности растворителя определяет возможность
титрования в его среде слабых оснований, так как их потенциалы
полунейтрализации по мере ослабления основных свойств электролитов
смещаются в кислую область.
Чем выше протяженность относительной шкалы растворителя, тем
больше возможность дифференцированного титрования смесей кислот
(или оснований). При этом предпочтительно титровать слабые кислоты
в тех растворителях, положение шкал кислотности которых
соответствует большему основному пределу относительной шкалы кислотности,
что обеспечивает четкие скачки титрования. Слабые основания предпоч-
§ 5. КОНСТАНТЫ, ПОТЕНЦИАЛЫ И ШКАЛА КИСЛОТНОСТИ
411
тительно титровать в растворителях с большими кислотными
пределами относительной шкалы кислотности.
Например, для титрования смеси слабых кислот с р/СнАп(Н20) ^5
необходимо остановить свой выбор на растворителе, относительная
шкала кислотности которого смещена в основную область или имеет
одинаково большие основный и кислотный пределы относительной шкалы
кислотности. В случае титрования смесей, содержащих сильные
кислоты, для которых условия титрования определяются большой величиной
кислотного предела относительной шкалы кислотности, пользуются
растворителями с относительной шкалой кислотности, смещенной в
кислую область.
Для титрования смесей слабых оснований с рХв(Н20)^5
необходимо применять растворители, относительная шкала кислотности
которых смещена в кислотную область или имеет одинаково большие
кислотный и основный пределы относительной шкалы кислотности.
При титровании смесей, содержащих сильные основания, для
которых условия титрования определяются большой величинай основного
предела относительной шкалы кислотности, применяют растворители с
относительной шкалой кислотности, смещенной в основную область.
Эмпирическая (относительная) шкала кислотности растворителей.
Иногда выбор растворителя для данного конкретного случая
титрования делается на основе эмпирической (относительной) шкалы
кислотности растворителя Es и потенциалов полунейтрализации электролита
Еу2 в данном растворителе.
Величина Es представляет собой разность потенциалов
полунейтрализации стандартных растворов кислоты и основания, например
хлорной кислоты и гидроокиси тетралкиламмония
Es = ЕЧ*. НС104 - Е% R4NOH (21)
или в общем виде:
(22)
"/з. НАп"
¦Ех
'/2, в
Другими словами, эмпирическая шкала кислотности представляет
собой область значений потенциалов, выраженную в милливольтах,
которая отражает максимальный предел ее использования при кислотно-
основном титровании электролитов стандартными растворами кислоты
и основания (например, хлорной кислоты и гидроокиси
тетралкиламмония).
ТАБЛИЦА 16
Относительная шкала кислотности некоторых растворителей
№
Растворитель
Протяженность шкалы
Es, мв
540
616
690
775
780
790
800
810
850
910
980
990
№
13
14
15
16
И?
18
19
20 |
21
22
23
24
Растворитель
Протяженность шкалы
1
2
3
4
5
6
7
8
9
10
11
12
Вода
Этиленгликоль .
Метиловый спирт
Гексиловый спирт
Амиловый спирт
Этиловый спирт
Бутиловый спирт .
Пропиловый спирт
Изоамиловый спирт .
Изобутиловый спирт .
Изопропиловый спирт
Пиридин . . .
вгор-Бутиловый спирт
Нитробензол . . .
Метилбензилкетон
грет-Бутиловый спирт
Нитрометан ....
Диметилформамид .
Диметилсульфоксид
Ацетофенон ....
Метилбутилкетон .
Ацетон
Метилэтилкетон . .
Ацетонитрил . . . ,
1000
1070
1080
1100
1140
1240
1275
1280
1300
1360
1410
1445
412 ГЛ. XI. ФИЗИКО-ХИМИЧЕСКИЕ МЕТОДЫ АНАЛИЗА НЕВОДНЫХ РАСТВОРОВ
Величина потенциала полунейтрализации определяется
кислотностью (основностью) титруемой кислоты (основания). В момент
нейтрализации 50% слабой кислоты или слабого основания, т. е. в тех
случаях, когда отношение концентраций кислоты (основания) и соли
в буферном растворе равно единице, величина [Н+] численно
приближается к величине /Снап или Kb-
Q
[Н+] = КНАп-т; « #нап (23)
рН
чНАп с
Р^НАп
KtAn
ИЛИ РН ~ Р#В
(24)
Относительная шкала кислотности некоторых растворителей
приведена в табл. 16 и рис. 142.
Рис. 142. Относительная шкала кислотности растворителей*
Цифры, указанные на рисунке, соответствуют номерам растворителей в табл. 16.
Как следует из табл. 16, низшие спирты и этиленгликоль
характеризуются относительно небольшой величиной шкалы кислотности.
Изопропиловый и в особенности трет-бутиловый спирт, пиридин,
нитробензол, диметилсульфоксид, ацетонитрил, метилэтилкетон отличаются
большой протяженностью относительной шкалы кислотности и,
следовательно, обладают высокими дифференцирующими свойствами.
Нитрометан и диметилформамид характеризуются достаточно
большой протяженностью шкалы кислотности: шкала диметилформамида в
большей своей части расположена в основной, а нитрометана — в
кислой области. Пиридин, отличающийся более ярко выраженными
основными свойствами по сравнению с диметилформамидом, имеет
относительно меньшую шкалу кислотности.
Добавление апротонных растворителей к протолитическим
приводит к изменению Es смешанных растворителей. Апротонные растворите-
§ -5. КОНСТАНТЫ, ПОТЕНЦИАЛЫ И ШКАЛА КИСЛОТНОСТИ. 413
ли имеют малую диэлектрическую проницаемость, поэтому НСЮ4 и
(C2H5)4NOH в среде смешанных растворителей диссоциированы
меньше, чем в среде протолитических растворителей. Это приводит к тому,
что в одних случаях смеси апротонных растворителей с протолитиче-
скими растворителями имеют относительные шкалы кислотности
больше, чем у протолитических растворителей, а в других случаях — меньше.
Так, добавление апротонного растворителя к спиртам приводит к
увеличению шкалы кислотности, а добавление его к кетонам и нитрилам,
которые обладают большими шкалами, приводит к их уменьшению.
На основании этих положений можно сделать выводы о том, что
для дифференцированного титрования смеси электролитов в качестве
сред следует использовать кетоны, ацетонитрил, нитрометан,
нитробензол, диметилформамид, диметилсульфоксид и смеси бензола и
хлороформа с кетонами, ацетонитрилом и другими амфипротными и апротон-
ными диполярными растворителями с большой протяженностью шкалы
кислотности (высокошкальные). В среде этих растворителей
получаются наиболее резкие скачки титрования и дифференцированно
титруются многокомпонентные смеси кислот. Добавление углеводородов к
спиртам (в особенности с изо-строением) способствует увеличению их
дифференцирующего действия.
Методом определения потенциалов полунейтрализации
пользовались различные исследователи. Особенно большую работу в этом
направлении проделали голландские ученые Ван-дер-Хейде и Дамен,
определившие эмпирическую (относительную) шкалу потенциалов 12
растворителей.
Они установили, что кислотно-основные свойства растворителя
определяются областью значений потенциалов, которая может быть
измерена экспериментальным путем в рамках кислотного и основного
пределов шкалы растворителя, обусловливаемой кислотностью сольватиро-
ваиного протона Н2М+ и основностью сольватированного аниона Мёож.
Указанными авторами приводится схема, иллюстрирующая
использование шкалы потенциалов, приводятся таблицы и даны рекомендации,
позволяющие химику-аналитику подобрать среду для титрования
большинства кислот и оснований и их смесей, часто встречающихся в
аналитической практике.
Протяженность и положение эмпирической (относительной) шкалы
кислотности может в известной мере служить некоторым практическим
критерием выбора в ряде случаев растворителя или смеси растворителей
при титровании данных электролитов.
Однако обоснование выбора растворителя для данного конкретного
случая титрования на основе эмпирической (относительной) шкалы
кислотности нельзя считать строго научно обоснованным по следующим
причинам.
1. Поскольку уравнение (23) является приближенным, то и
определение эмпирической шкалы кислотности по потенциалам
полунейтрализации носит тоже приближенный характер.
Значение ?\. зависит от степени электролитической диссоциации
кислоты (или основания), но константа диссоциации этой кислоты (или
основания) в данном растворителе может в значительной степени
отличаться от /(нап слабой кислоты (или слабого основания), для смеси
которой с ее солью в буферном растворе, содержащем равные
концентрации кислоты (основания) и соли, применимо уравнение (23), поэтому
для данцого случая (когда рН Ф рК) уравнение (23) оказывается
несостоятельным.
414 ГЛ. XL ФИЗИКО-ХИМИЧЕСКИЕ МЕТОДЫ АНАЛИЗА НЕВОДНЫХ РАСТВОРОВ
2. Указанные выше титранты — хлорная кислота и гидроокись тетр-
алкиламмония — не являются лучшими и единственными реагентами,
применяемыми для титрования оснований и кислот; вместо них могут
быть в зависимости от необходимости и целесообразности использованы
другие титранты.
3. В реальных условиях титрования при определении потенциалов
полунейтрализации указанным методом вместе с титрантами в
титруемый раствор, как правило, неизбежно вводятся посторонние
растворители (обычно уксусная кислота с НС104 и метиловый или этиловый
спирт и бензол с R4NOH, в которых растворены реагенты),
осложняющие условия титрования и изменяющие эмпирическую шкалу
кислотности избранного растворителя, так как титрование в этом случае
ведется не в среде чистого растворителя. В результате сказывается
отрицательное влияние примесей на процесс кислотно-основного титрования
и данные получаются не точные.
4. В растворах хлорной кислоты, приготовленных даже на основе
растворителя, применяемого для исходной титруемой смеси, всегда
содержится вода. В процессе кислотно-основного титрования гидроксил-
содержащих оснований, а при титровании гидроокисью тетралкиламмо-
ния карбоксильных и фенольных групп в качестве продукта
нейтрализации также образуется вода, резко изменяющая основной и кислотный
пределы относительной шкалы кислотности высокошкальных
растворителей.
5. На величину относительной шкалы кислотности оказывают
сильное влияние различные факторы (концентрация, образование
нерастворимых соединений, комплексообразование, типы применяемых
электродов и т. п.), в зависимости от которых протяженность и положение
относительной шкалы кислотности данного растворителя может сильно
меняться. Например, в случае выпадения в осадок в процессе потенцио-
метрического титрования образующейся соли, в момент
полунейтрализации Снап Ф CKtAn и рН Ф р/С, что приводит к неверным результатам.
6. На относительную шкалу кислотности также оказывают
заметное влияние посторонние примеси, содержащиеся в исходном
растворителе (в том числе и вода), которые в зависимости от их
кислотно-основных свойств, положения и протяженности собственных шкал
кислотности, отличающихся от шкалы кислотности растворителя среды, могут
оказывать очень сильное влияние, искажающее результаты
исследования. Причем для резкого изменения шкалы кислотности исходного
растворителя иногда достаточно даже следов посторонних примесей.
Отсюда вытекает очень важный в теоретическом и практическом
отношении вывод, что суждение о нивелирующе-дифференцирующем
эффекте растворителя или смеси растворителей по относительной шкале
кислотности Esy которую используют иногда исследователи неводных
растворов, имеет лишь узкопрактическое значение и носит
приближенный характер.
§ 6. Шкала рНр и константы автопротолиза неводных
растворителей
Оценка кислотности неводных растворов. При оценке кислотности
неводных растворов возникает необходимость сравнения кислотности
различных веществ в одном и том же неводном растворителе, как это
делается при оценке кислотности водных растворов. С другой стороны,
большое значение имеет сравнение кислотности растворов электролитов,
растворенных в различных растворителях.
§ 6. ШКАЛА рНр И КОНСТАНТЫ АВТОПРОТОЛИЗА РАСТВОРИТЕЛЕЙ 415
Определение относительной кислотности раствора электролита в
данном неводном растворителе принципиально не отличается от
определения ее для водных растворов. Тем не менее при определении рН в
неводных растворах допускается большое количество ошибок.
Например, при измерении рН неводных растворов по отношению к
насыщенному водному каломельному электроду возникают наиболее серьезные
ошибки. Величины рН неводных растворов не могут быть правильно
измерены, если пользоваться для измерения рН неводных растворов рН-
метром, откалиброванным по водным стандартам.
При определении рН неводных растворов необходимо заменять
насыщенный водный раствор КС1 в каломельных электродах не метаноло-
вым раствором КС1 (как это обычно делают), а соответствующими
растворами КС1 в неводных растворителях, в среде которых проводят
титрование. Только при этом условии измеренное значение рН неводного
раствора может быть сопоставлено с точно известным значением рН
стандартного неводного раствора.
Для оценки кислотности неводных растворов Н. А. Измайловым
предложена шкала рНр, специфичная для данного растворителя, и
шкала рА, универсальная для всех растворителей.
Подобно тому как рН в водных растворах рассматривается как
величина отрицательного логарифма концентрации ионов водорода [Н+]
(точнее активности ионов водорода ан+), рН в данном неводном
растворителе (рНр) представляет собой отрицательный логарифм активности
ионов лиония НгМ"1*:
HAn + МН ;^± Н2М+ + Ап~
pHp = -lg[H2M+]
или
рНр — Ig аНгМ+ — lg тН2МУНаМ+ (25)
*
где yh M+ — коэффициент активности в бесконечно разбавленном растворе;
т — моляльная концентрация электролита.
Активность ионов лиония в неводном растворителе, выражающая
эффективную концентрацию электролита в данном растворе и
отражающая в целом влияние взаимного притяжения и отталкивания ионов,
сольватацию, неполную диссоциацию и т. п., и коэффициент активности
YH2M+ стандартизуются по отношению к бесконечно разбавленному
раствору (обозначается индексом*) в той же среде, т. е. в любой среде при
т~^0, а* = т, у* = 1.
Протяженность шкалы рНр различных растворителей. При
сопоставлении шкалы рН воды со шкалой рНр неводных растворителей
(рис. 143) легко заметить, что протяженность шкал рН всех
растворителей различна.
Подобно тому как ионное произведение воды (см. книга 1, гл. I,
§11) выражается уравнением:
[н3о+][он-] = ^
так для растворителей, способных диссоциировать, подобно воде,
ионное произведение (константа автопротолиза Ks) может быть выражено:
[Н2М+][М"] = К5 (26)
Следовательно, абсолютная протяженность шкалы кислотности
предопределяется величиной ионного произведения растворителя Ks
4t6 ГЛ. XI. ФИЗИКО-ХИМИЧЕСКИЕ МЕТОДЫ АНАЛИЗА НЕВОДНЫХ РАСТВОРОВ
(константы автопротолиза) и измеряется величиной показателя
константы автопротолиза p/Cs (табл. 17).
Как следует из табл. 17 и рис. 143, значения p/Cs для различных
растворителей не совпадают, это указывает на то, что и значение рН,
отвечающие точке нейтрализации разных растворов, тоже не совпадают.
Состояние кислотно-основного равновесия самоионизации
растворителя определяется его природой и способностью его молекул
акцептировать или отдавать протоны под влиянием сольватации,
сопровождающейся образованием ионов типа гидроксония: NHt, СН3СООН2,
C2H5OHJ, H3SOJ, H2NOJ, H3F2, H2CN+ и т. п.
Рис. 143. Протяженности шкал рНр различных растворителей.
Цифры, указанные на рисунке, соответствуют номерам растворителей в табл. 17.
Относительное положение шкалы кислотности различных раствори
телей* В случае растворения в воде сильной кислоты устанавливается
следующее равновесие:
HAn + H20 z?± H30++ArT
Вода, акцептируя протон сильной кислоты, проявляет свойства
основания. Сила растворенной в воде кислоты определяется ее
действием на воду, обусловленным состоянием приведенного выше
равновесия. Например, карбоновые кислоты оказывают слабое влияние на
состояние этого протолитического равновесия, т. е. являются слабыми
кислотами.
Чем сильнее действие кислоты на воду и чем в большей степени
молекулы воды склонны сольватировать протоны, тем больше указанное
равновесие сдвигается в правую сторону и тем сильнее растворенная
кислота.
В общем виде приведенное выше уравнение выражается;
НАп + НМ
Н2М+ + АгГ
где НМ — молекула растворителя.
§ б ШКАЛА рНр И КОНСТАНТЫ АВТОПРОТОЛИЗА РАСТВОРИТЕЛЕЙ 417
ТАБЛИЦА 17
Показатели констант автопротолиза pKs и диэлектрические проницаемости е
некоторых неводных растворителей
Растворитель
8
_
16,9
37,4
18,50
52
12,3
1 14,7
—
18,3
1 16,35
14,4
19,7
26,5
17,7
vKs
33,3
32,52
32,2
25,70
24,70
22,80
21,40
21,10
20,84
20,80
20,50
20,40
20,30
20,10
№
15
16
Г/
18
19
20
21
22
23
24
125
26
27
Растворитель
P*s
1
2
3
4
5
6
7
8
9
10
11
12
13
14
Диметилсульфоксид
Аммиак (жидкий) . . .
Ацетонитрил
Метилэтилкетон . . .
Гидразин
трет-Бутиловый спирт
Изоамиловый спирт .
Ацетон (енольная
форма)
Изопропиловый спирт
вгор-Бутиловый спирт
Амиловый спирт . . .
Пропиловый спирт . .
Ацетон (90%-ный) +
вода (10%)
«-Бутиловый спирт . .
Этиловый спирт ....
Диметилформамид . . .
Диоксан (70%-ный) . .
Пропиленгликоль . . .
Формамид
Метиловый спирт . . .
Этиленгликоль
Уксусный ангидрид . .
Уксусная кислота
(безводная)
Фтористый водород
(жидкий)
Муравьиная кислота . .
Серная кислота ....
Вода
25,2
37,6
25
29,46
109,5
32,65
37,7
20,5
6,19
83,6
57,9
84,0
78,3
19,14
18,00
17,86
17,76
17,00
16,70
15,60
14,50
14,45
10,00
6,30
5,0
14,0
Константа /С, выражающая состояние динамического равновесия
указанного процесса, определяется константами двух равновесий:
НАп
Н+ + An"
(а)
^НАп~
к
аНАп
Н^ + НМ
ан+анм
(б)
Н2М+ "
а
н2м+
или
гг ^нАп ан^М+аАп-
Д = -fr =
ДН2М+ аНАпаНМ
(27>
Следовательно, в неводном растворе /Снап, или сила кислоты, тем
больше, чем больше равновесие сдвигается вправо, т. е. чем сильнее
растворитель проявляет основные свойства.
Естественно, когда растворитель проявляет очень сильные основные
свойства и растворенная кислота сильная, то она нацело нейтрализуется
самим растворителем, а равновесие полностью сдвигается вправо.
В таких случаях кислые свойства растворенных сильных кислот
проявляют сольватированные протоны, образующиеся в процессе
диссоциации молекул НАп.
Из этого следует, что шкала кислотности ограничивается
основностью данного растворителя: в растворителях (например, пиридин)
более основных, чем вода, шкала кислотности сдвигается в основную
сторону, в менее основных растворителях (например, этиленгликоль) в
кислую сторону и, наоборот, в растворителях более кислых, чем вода
(например, уксусная кислота), шкала кислотности сдвигается в
кислотную, а в менее кислых (метилпиридин) в основную сторону.
418 ГЛ. XI. ФИЗИКО-ХИМИЧЕСКИЕ МЕТОДЫ АНАЛИЗА НЕВОДНЫХ РАСТВОРОВ
Взаимодействие растворенного основания с водой, когда вода,
являясь донором протона, проявляет свойства кислоты, можно выразить
уравнением
В + НОН ^z± ВН+ + ОН"
или в общем виде:
В + НМ ^=± ВН+ + М"
Константа, выражающая состояние динамического равновесия этого
процесса (К), определяется константами двух равновесий:
(а)
(б)
или
(28)
где К"в — константа основности основания, растворенного в неводном растворителе;
К - — константа основности ионов лиата — обратная величина константе диссоциа-
м
ции растворителя.
В сильнопротогенных растворителях (например, в НСООН) самые
щелочные растворы кислее самых кислых водных растворов; самые
кислые растворы в протофильных растворителях (например, NH3) щелоч-
нее самых щелочных водных растворов.
Таким образом, кислотные пределы протогенных растворителей
превосходят кислотный предел воды, а основные пределы протофильных
растворителей превосходят ее основной предел. Наконец, некоторые ам-
фипротные растворители отличаются тем, что их кислотные и основные
пределы либо относительно мало отличаются от таковых для воды
(С2Н5ОН), либо сильно превышают их (метилэтилкетон).
Другими словами, большинство растворителей отличается друг от
друга не только протяженностью, но и относительным положением шкал
рНр, что имеет большое значение в аналитической химии неводных
растворов.
§ 7. Абсолютная (единая) шкала кислотности растворителей
Характеристика кислотно-основных свойств растворителя. Для
каждого растворителя характерными являются следующие
кислотно-основные константы.
1. Константа автопротолиза (ионное произведение) растворителя
Ks'
НМ ^z± H+ + M"
К* = ан+аж- (29)
2НМ ^z± H2M+ + M"
*s = <WaM~ <29а>
§ 7. АБСОЛЮТНАЯ (ЕДИНАЯ) ШКАЛА КИСЛОТНОСТИ РАСТВОРИТЕЛЕЙ 419
2. Константа кислотности растворителя Ка, нм-
НМ ^=> Н+ + М"
о, ,а
^,нм=^1 (30)
"нм
3. Константа основности ионов лиата растворителя Кь м- —
обратная величина константы кислотности растворителя
Н+ + М" ^=± НМ
*а. нм ан+ам~
4. Константа кислотности ионов лиония растворителя Ка Нзм"1"-'
Н2М+ ^z± Н+ + НМ
ан+анм
«а.М+—1Г-Г (32)
Н,М+
5. Константа основности растворителя Кь, нм— обратная величина
константы кислотности ионов лиония растворителя:
н+ + нм ^=± н2м+
К l - °Н»М+ М\
Да, Н2М+ ан+аНМ
Константы с индексами а и Ь не зависят от среды и носят название
констант собственной кислотности (растворителя или ионов лиония) или
собственной основности (растворителя или ионов лиата) в стандартных
условиях (в среде с бесконечно большой диэлектрической
проницаемостью, в вакууме или в водном бесконечно разбавленном растворе, в
котором а = а* уо). Соответствующие им константы с индексами НAn
и В зависят от среды.
Константа автопротолиза является важнейшей термодинамической
характеристикой растворителя. Она играет существенную роль при
обосновании выбора растворителя для той или иной цели.
В неводных растворителях, отличающихся различными значениями
Ks (и соответственно pKs), наблюдается изменение в силе кислот
(оснований) в связи с увеличением или уменьшением отношений константы
автопротолиза к константе диссоциации кислоты или основания:
Кт==~К^Г; P^T = P^s-P^HAn (34)
iVHAn
/ет = -^— ; р/Ст = p/Cs - p/CB (35)
АВ
где/Ст—константа (коэффициент) титрования.
При этом скачки титрования увеличиваются в тех случаях, когда
Кнап (или Kb) уменьшаются в меньшей степени, чем /Cs, или когда
константа диссоциации увеличивается, а константа автопротолиза
уменьшается. Также изменяются отношения Кт констант диссоциации
кислот и отношения Кт констант диссоциации оснований:
тт > А^ ~ ТГ » АТ Т?
АНАп, I дН2Ап А В, I
Р#Т - ДрЯ" = Р^НАп, II - Р^НАп. 1 (Щ
420 ГЛ. XI ФИЗИКО-ХИМИЧЕСКИЕ МЕТОДЫ АНАЛИЗА НЕВОДНЫХ РАСТВОРОВ
Изменение этих отношений также в конечном счете зависит от
величины ионного произведения растворителя, так как чем меньше Ks
растворителя, тем сильнее проявляется его дифференцирующее действие
в отношении кислот (оснований). Это можно наблюдать на примере
дифференцирующего действия ацетона в отношении дикарбоновых
кислот, что видно по изменению АрД" кислот в его среде по сравнению с
водными растворами. Так, АрКа о-фталевой кислоты увеличивается с 2,2
в водном растворе до 5,35 в
ацетоновом.
Таким образом, по величине
// Ks можно судить о нивелирую-
\\ ще-дифференцирующем действии
-/#><
»
растворителя в отношении
определенных групп электролитов.
Константа автопротолиза
позволяет оценить величину
коэффициента титрования /Ст, а
значение p/Cs Дает представление о
протяженности абсолютной
шкалы кислотности растворителя.
Величина Ks является
функцией ряда параметров,
характеризующих собственную силу
кислотности (Ко) или основности
(Къ) растворителя, его
диэлектрическую проницаемость,
полярность его молекул ?т и др.:
1*1 **"ан,м+вм- =/ (К*Кь,е9Ет...)
fjr^T mcon
Чем сильнее ионизирован
растворитель, тем больше величина
константы автопротолиза, тем
большей сольватирующей
способностью в отношении
растворенных веществ обладает данный
растворитель, а следовательно,
тем сильнее диссоциированы в
его среде электролиты. Это
означает, что чем больше величина
константы автопротолиза, тем
слабее его дифференцирующее
действие при прочих
сравниваемых равных условиях. С уменьшением константы автопротолиза
возрастает его дифференцирующий эффект.
Подобная зависимость дифференцирующего действия растворителя
от Ks наглядно иллюстрируется величиной показателя константы
автопротолиза pKs, определяющей протяженность шкалы кислотности
растворителя.
Шкала рА. Показатель константы автопротолиза p/Cs является
мерой протяженности абсолютной шкалы кислотности растворителя рА,
предложенной Н. А. Измайловым.
Для стандартизации единой (универсальной, абсолютной),
термодинамически строго обоснованной для всех растворителей шкалы
кислотности рА, строящейся на основе значений термодинамических кон-
-Z7
Рис, 144. Сопоставление шкалы
кислотности рНр и рА:
1 —вода; 2—метиловый спирт; 3 — этиловый спирт;
4 — пропиловый спирт; 5 — бутиловый спирт; 6 — изо-
бутиловый спирт; 7—амиловый спирт; 8 — изоами-
ловый спирт; 9— ацетон; 10— ацетонитрил; Л—ди-
метилсульфоксид.
Сплошными линиями показаны шкалы рА,
пунктирными — шкалы рН .
§ 7 АБСОЛЮТНАЯ (ЕДИНАЯ) ШКАЛА КИСЛОТНОСТИ РАСТВОРИТЕЛЕЙ 421
стант автопротолиза, Н. А. Измайлов воспользовался средними
коэффициентами активности сильных кислот в среде неводных растворителей.
В отличие от шкалы рНр шкала рА различных растворителей
начинается не с нуля, а с разных значений рА (рис. 144). Как видно из
рис. 144, шкала рА позволяет представлять не только протяженность,
но и ее положение по отношению шкал других растворителей,
например воды.
Показатель единой шкалы кислотности, который Измайлов
обозначил через рА, отнесенный к единому стандартному состоянию
(бесконечно разбавленный водный раствор), определяется величиной
обратного логарифма активности ионов водорода в данном растворителе НМ:
РА—'««нчнм) (37)
где а + — абсолютная активность ионов лиония НгМ+, отнесенная к активности
Н (НМ)
протона в разбавленном водном растворе: а + = а +у +
Следовательно, рА можно выразить в виде следующего уравнения:
РА = ->еан2м+-1^о,н+ <38>
В этом уравнении %2м+ отнесена к бесконечно разбавленному
неводному раствору лиония, а единый коэффициент активности (Y0f н+)
отнесен к водному раствору протона. Отрицательный логарифм
коэффициента активности (— lgу0, н+) служит мерой перехода от шкалы рНр
к шкале рА.
Мерой коэффициентов активности является энергия сольватации
протона, т. е. энергия переноса Н+ из вакуума в среду данного
растворителя. Изменение энергии сольватации протона обусловливается
главным образом изменением основности растворителя.
Величину — lg#H2M+ можно выразить следующим образом:
"lBVapHpB"lgmY*
где рНр — обратный логарифм активности ионов лиония в среде данного растворителя
НМ, т. е. отрицательный логарифм произведения концентрации ионов
лиония т на концентрационный коэффициент активности у*.
Следовательно
pA = pHp-lgY0H+ (39)
Значение рНр может быть измерено для любого неводного
раствора по отношению стандарта в том же самом неводном растворе.
Величина lgYo, н+ более точно оценивает изменение кислотности,
чем другие величины, например функция кислотности Гаммета Н0 (Н_Ь
Следовательно, при пользовании абсолютной шкалой кислотности
отпадает необходимость в эмпирической (относительной) шкале
кислотности, не являющейся строгой научной основой для суждения о
кислотности или основности растворителя.
Из сказанного можно также заключить, что понятия о силе кислоты
и кислотности принципиально отличаются друг от друга. В то время
как сила кислоты в любом растворителе обусловливается ее
константой диссоциации, кислотность определяется активностью ионов лиония,
связанной с основностью данного растворителя, их концентрацией и их
концентрационными и едиными коэффициентами активности.
Например, слабая в воде кислота в среде основного растворителя становится
сильной, но ее неводный раствор может быть менее кислым, чем в воде,
422 ГЛ. XI. ФИЗИКО-ХИМИЧЕСКИЕ МЕТОДЫ АНАЛИЗА НЕВОДНЫХ РАСТВОРОВ
что обусловлено, с одной стороны, увеличением числа ионов лиония,
а с другой стороны, уменьшением активности Н+-ионов.
Критерий нивелирующе-днфференцирующего действия
растворителя. Нивелирующе-дифференпирующее действие растворителя зависит
от ряда факторов, к числу которых (наряду с сольватирующей
способностью растворителя, склонностью к образованию водородных связей,
способностью молекул и ионов растворителя и растворенного вещества
к ассоциаций! и комплексообразованию, диэлектрической
проницаемостью, полярностью, дипольным моментом растворителя и т. п.) в
первую очередь относятся следующие.
1. Величина константы автопротолиза (ионного произведения)
растворителя /Cs, являющаяся важнейшей термодинамической константой
растворителя.
2. Протяженность и положение абсолютной (единой) шкалы
кислотности растворителя, которые определяются величиной показателя
константы автопротолиза pKs и кислотно-основными характеристиками:
^а, НМ> *^Ь, М~"> ^а, Н2М+> ^Ьу НМ* "
Чем больше величина pKs, тем больше протяженность шкалы
кислотности, а следовательно, тем сильнее проявляется
дифференцирующее действие растворителя в отношении определенных веществ,
отличающихся в данной среде кислотно-основным характером.
3. Величины констант диссоциации кислот и оснований в данной
среде, так как сила кислоты или основания в любом растворителе (НМ)
характеризуется константой диссоциации /Снап или Kb или их
показателями р/СнАп и рКв- Указанные величины позволяют с большой
точностью решать вопрос о нивелирующе-дифференцирующем действии
растворителя в отношении данной смеси электролитов.
Выбор растворителя. Основополагающие выводы в отношении
выбора неводного растворителя или смеси растворителей для данного
конкретного случая титрования могут быть сформулированы
следующим образом:
1. Исходный растворитель или соответствующая смесь
растворителей, прежде всего должны обладать достаточной растворяющей
способностью в отношении определяемого соединения или титруемой
смеси веществ.
2. Избираемый растворитель должен обладать большой
протяженностью абсолютной шкалы кислотности (обусловливаемой значением
его константы автопротолиза Ks) и соответствующим ее положением,
в рамки которой должны укладываться все скачки титрования
определяемых веществ многокомпонентной смеси.
3. В ряде случаев исходные растворители не должны содержать
даже незначительных количеств мешающих примесей, проявляющих
четко выраженные кислые или основные свойства (НгО, С02, СН3ОН и
т. п.), которые во многих случаях оказывают отрицательное влияние
на титрование неводных растворов, приготовленных на основе
растворителей с очень малыми значениями констант автопротолиза.
4. Избираемый растворитель должен обеспечивать наилучшие
соотношения констант автопротолиза и констант диссоциации титруемых
электролитов.
5. Растворитель, в котором растворяется анализируемое вещество,
по возможности должен соответствовать растворителю, в котором
растворен реагент, или растворитель, применяемый для приготовления тит-
ранта, должен соответствовать растворителю, используемому для
растворения анализируемого продукта. В процессе смешения титруемой
§ 8 ХИМИКО-АНАЛИТИЧЕСКОЕ ИСПОЛЬЗОВАНИЕ НЕВОДНЫХ РАСТВОРИТЕЛЕЙ 42$
смеси с титрантом не должно происходить нежелательных побочных
процессов и явлений, искажающих результаты анализа.
6. В случае выбора вместо индивидуального растворителя смеси
растворителей или в случае титрования раствора, приготовленного на
основе индивидуального растворителя, титрантом, приготовленным на
основе других растворителей, необходимо иметь в виду, что при
смешивании растворителя с относительно высоким значением pKs
(например, метилэтилкетона, диметилсульфоксида, ацетонитрила) с протоли-
тическим растворителем, характеризующимся низким значением рК&
(например, водой, метиловым спиртом, уксусной кислотой),
наблюдается резкое уменьшение абсолютной шкалы кислотности по сравнению о>
шкалой избранного растворителя. Поэтому состав смешанного
растворителя в случае осуществления дифференцированного титрования
смеси электролитов должен соответствовать необходимой протяженности
абсолютной шкалы кислртности.
7. При использовании в качестве растворителя смеси амфипрот-
ного (или диполярного апротонного) растворителя с протогенным или
с протофильным наблюдается в ряде случаев не только резкое
уменьшение протяженности шкалы кислотности по сравнению со шкалой
избранного растворителя, но и смещение кислотного или основного-
пределов исходного растворителя, что может оказывать существенное
влияние на условия титрования электролитов.
8. Величины констант диссоциации кислот и оснований, зависящие
от физико-химических свойств растворителей, также определяют выбор
растворителя. Например:
а) при увеличении Ks константа диссоциации кислоты (или
основания) в данной среде Кяап (или Kb) должна увеличиваться в большей
степени, чем константа автопротолиза;
б) при уменьшении Ks значение Кяап (или Kb) не должно
увеличиваться;
В) ^НАгГ (ИЛИ ^HAn, II) И /СнАп (ИЛИ КкАп, i) *, Д'в, II И Кв, I
ДОЛЖНЫ по возможности больше отличаться друг от друга. При этом
титрование с точностью до 0,1% оказывается возможным при р/Ст ~ 3—4.
г) при титровании кислот (при постоянном значении Ks) Кт
уменьшается по мере увеличения Кнап с уменьшением К&, нм, т. е. с
уменьшением кислотности растворителя или увеличением его основности Кь, нм.
При титровании оснований условия титрования улучшаются в связи
с увеличением константы собственной кислотности ионов лиония
растворителя Ка, НгМ+, т. е. с увеличением кислотности растворителя Ка, нм-
§ 8. Химико-аналитическое использование неводных
растворителей
Случаи применения неводных растворителей. Изменение физико-
химических свойств растворенных соединений, происходящее под
влиянием растворителей, широко используют в химическом анализе.
Обычно неводные растворители применяют вместо воды в техслу-*
чаях, когда требуется:
растворить анализируемое вещество, нерастворимое в воде и
вводных растворах кислот, щелочей, комплексующих агентах и т. п., или
когда необходимо понизить растворимость вещества, хорошо раствори-»
мого в воде;
усилить или ослабить силу электролитов, изменяющуюся в
зависимости от природы избранного растворителя;
424 ГЛ. XI. ФИЗИКО-ХИМИЧЕСКИЕ МЕТОДЫ АНАЛИЗА НЕВОДНЫХ РАСТВОРОВ
перевести вещество из неионизированного состояния в
ионизированное и наоборот;
изменить соотношение в силе кислот, оснований и солей;
уменьшить или увеличить отношение между ионным
произведением Ks растворителя и константой диссоциации растворенной кислоты
Анап или основания /Св;
затормозить или свести к минимуму гидролиз, оказывающий
отрицательное влияние на состояние динамического равновесия некоторых
реагирующих веществ;
обеспечить дифференцирующее действие среды для раздельного
титрования отдельных компонентов многокомпонентных смесей;
осуществить исследование химико-аналитических процессов при
заданных величинах диэлектрической проницаемости среды, имеющей
очень важное значение для определения характера кривых кислотно-
основного титрования;
измерить константы диссоциации соединений, нерастворимых в
воде;
усилить или ослабить электропроводность растворов;
изменить протяженность и положение относительной шкалы
кислотности и т. д.
Возможности кислотно-основного титрования слабых электролитов
в неводных средах. В неводных растворах можно титровать очень
слабые (в воде) кислоты и основания, при титровании которых в водных
растворах не удается получить резкого скачка титрования вследствие
гидролиза солей слабых кислот или слабых оснований, образующихся
в водных растворах. Причиной гидролиза вещества в водном растворе
является собственная диссоциация воды, например:
СНзСОСГ + НОН ^=± СНзСООН + ОРГ
Точно так же собственная диссоциация неводных растворителей
вызывает сольволиз образующихся при титровании солей:
СНзСОСГ + С2Н5ОН ^=± СНзСООН + С2Н5СГ
Чем сильнее собственная диссоциация растворителя, т. е. чем
больше ионное произведение растворителя (константа автопротолиза), тем
в большей степени происходит сольволиз. Например, в водном растворе
(Kw = 10~14)соли азотной кислоты не подвергаются гидролизу, а
вереде безводной серной кислоты (Ks = 2,4* Ю-4) происходит сольволиз этих
солей:
NC>3 + H2S04 ^z± HNO3 + HSO4
Константа автопротолиза многих неводных растворителей, как
правило, меньше, чем у воды, поэтому в неводных растворах соли в
значительно меньшей степени подвергаются сольволизу. Однако разница
в численных значениях ионных произведений различных растворителей
весьма велика, и это дает возможность производить такие виды
титрования, которые практически неосуществимы в водной среде.
Реакции, протекающие в процессе титрования слабых электролитов.
При титровании в водном растворе сильной кислотой слабого
основания или сильным основанием слабой кислоты рН мало изменяется
вблизи точки эквивалентности (см. книга 2, гл. II, § 5 и 6); кривые
титрования имеют пологий характер; скачок титрования у точки
эквивалентности уменьшается с увеличением р/С титруемого основания или
кислоты.
При титровании слабого основания в водной среде сильной
кислотой протекают две реакции.
§ в. ХИМИКО-АНАЛИТИЧЕСКОЕ ИСПОЛЬЗОВАНИЕ НЕВОДНЫХ РАСТВОРИТЕЛЕЙ 425
Основная реакция — взаимодействие титруемого основания В с
кислотой:
В + Н+ ^z± BH+
Побочная реакция — взаимодействие воды как основания с
кислотой:
Н20 + Н+ ^=± Н30+
В результате основная реакция протекает не количественно, так
как значительная часть кислоты наряду с основной реакцией может
параллельно реагировать с молекулами воды.
Чем слабее титруемое основание и чем сильнее проявляет
основные свойства избранный растворитель по отношению к кислоте-титран-
ту, тем в меньшей степени протекает основная реакция нейтрализации
слабого основания.
Например, ароматические амины с рК (НгО) = 10, проявляющие
слабые основные свойства, невозможно, как правило, количественно
оттитровать в водном растворе вследствие протекания побочной
реакции. Если в качестве растворителя использовать химическое
соединение, обладающее более слабыми основными свойствами по отношению
кислот, чем вода, то основная реакция протекает количественно, а
побочная— в незначительной степени или совсем не идет. Благодаря
этому многие основания, не титруемые в водной среде, могут быть
успешно оттитрованы в апротонном, протогенном, апротонном диполярном и
амфипротном растворителях или в их смесях.
С другой стороны, при титровании слабой кислоты в водной среде
сильным основанием протекают следующие реакции.
Основная реакция — взаимодействие титруемой кислоты с сильным
основанием:
HAn + B ^z± ВН+ + Ап"
Побочная реакция — взаимодействие воды как кислоты с сильным
основанием:
НОН + В ^± ВН+ + ОН"
В результате основная реакция протекает не количественно, так
как значительная часть оснозания реагирует с молекулами
растворителя.
Чем слабее титруемая кислота и чем сильнее проявляет кислые
свойства растворитель по отношению к основанию-титранту, тем в
меньшей степени протекает основная реакция нейтрализации слабой
кислоты.
Слабые кислоты, характеризующиеся рК (Н20) « 10, невозможно
количественно оттитровать в водном растворе вследствие протекания
побочной реакции.
Если в качестве растворителя использовать химическое
соединение, обладающее более слабыми кислыми свойствами по отношению
оснований, чем вода, то основная реакция протекает количественно, а
побочная — в незначительной степени или совсем не идет. Благодаря
этому многие кислоты, не титруемые в водной среде, могут быть
успешно оттитрованы в апротонном, протофильном, апротонном
диполярном, амфипротном растворителях или в их смесях.
Из изложенного выше вытекает очень важный вывод, что апро-
тонные растворители оказываются весьма полезными во многих
случаях титрования слабых оснований или слабых кислот, которые
невозможно оттитровать в водной среде. При введении апротонных
растворителей уменьшается величина Ks среды, изменяется диэлектриче.
426 ГЛ. XI. ФИЗИКО-ХИМИЧЕСКИЕ МЕТОДЫ АНАЛИЗА НЕВОДНЫХ РАСТВОРОВ
екая проницаемость смешанного растворителя, ослабляется кислотность
или основность протолитического растворителя, сдвигаются процессы
кислотно-основного титрования в нужном направлении и т. п.
Реакции конъюгации. Наряду с описанными побочными реакциями
нейтрализации в апротонных диполярных растворителях в
присутствии конъюгирующих оснований, а также кислот могут протекать
другие побочные реакции — гомо-(а) и гетероконъюгации (б):
ВН+ + В ^± ВН+-В (а)
ВН+ + В' +± ВН+-В' (б)
обр [вн+-в]
Авн+.в [ВН+] [в] ^
AiT + HAn ^=± Ап"-НАп (а)
АгГ + HAn' ^z± An"-HAn' (б)
кобр = [An" • HAn] (
AAn~.HAn [An"] [HAn] к '
Комплексы, образующиеся в результате этих реакций, достаточно
устойчивы, чем и объясняется разница между кислотно-основными про-*
цессами, протекающими в апротонных диполярных растворителях
(например, ацетонитрил) и воде.
Образованию этих побочных продуктов способствует относительно
высокая концентрация растворов. Поэтому во избежание протекания
реакций конъюгации следует проводить титрование разбавленных
растворов.
Влияние апротонных растворителей. Ранее существовало мнение,
что прибавление апротойных растворителей к протолитическим всегда
улучшает условия кислотно-основного титрования вследствие
уменьшения /Cs смешанных растворителей по сравнению с /Cs протолитических
растворителей. Однако, как показали исследования, прибавление
апротонных растворителей к протолитическим в одних случаях
действительно приводит к улучшению, а в других случаях к ухудшению условий
кислотно-основного титрования. Так, прибавление бензола к
метиловому спирту приводит к увеличению относительной шкалы кислотности
по сравнению со шкалой кислотности метилового спирта, а
прибавление бензола к ацетонитрилу и ацетону — к уменьшению относительной
шкалы кислотности.
Это явление можно объяснить следующим образом. При
добавлении апротонных растворителей к протолитическим, с одной стороны,
происходит уменьшение К$ среДы, что ведет к увеличению
относительной шкалы кислотности, с другой стороны, наблюдается уменьшение
диэлектрической проницаемости дреды, что ведет к уменьшению
относительной шкалы кислотности, так как потенциалы полунейтрализации
НС104 и (СгНбЬЬЮН при титровании их в присутствии апротонных
растворителей сближаются. Следовательно, в зависимости от того,
какой из указанных выше факторов оказывает наибольшее влияние,
«относительная шкала кислотности смешанных растворителей либо
увеличивается, либо уменьшается по сравнению со шкалой кислотности
протолитических растворителей. В случае смешанного растворителя
метиловый спирт — бензол решающую роль играет уменьшение /Cs среды,
-а в случае ацетонитрил — бензол и ацетон — бензол главное значение
имеет уменьшение диэлектрической проницаемости среды (при
прибавлении бензола к ацетонитрилу в отношении 4:1 от 37,5 до 9,3, а в
случае ацетона — от 20,7 до 6,0).
§ 8. ХИМИКО-АНАЛИТИЧЕСКОЕ ИСПОЛЬЗОВАНИЕ НЕВОДНЫХ РАСТВОРИТЕЛЕЙ 427
В большинстве случаев при добавлении, значительных количеств
апротонного растворителя к протогенному (или протофильному)
растворителю получается смешанный растворитель, характеризующийся
высокими дифференцирующими свойствами вследствие уменьшения
кислотно-основной активности избранного растворителя.
Условия титрования. Исходя из того что под влиянием
растворителей изменяется не только сила кислот и оснований и соотношения
между ионным произведением и силой кислот или оснований, условия
титрования кислот и оснований будут определяться соответственно
соотношениями /Cs/Хнап и Ks/Кв- В зависимости от того, как изменяются
эти отношения, могут улучшаться или ухудшаться условия титрования
кислот и оснований. Если это отношение уменьшается, то условия
титрования будут улучшены, и наоборот. Например, в воде для
хлористоводородной кислоты — Ig(KslKHAn) = 13, а в среде 50%-ного ацетона
это отношение увеличивается до 18; для бензойной кислоты в водной
среде — lg((Ks/KuAii) =9,8, а в среде 50%-ного ацетона —10,5.
Условия титрования дикарбоновых кислот также улучшаются при
переходе от водной среды к неводной, например к этиловому спирту.
Так, для малоновой кислоты в водном растворе при диссоциации ее по
первой ступени — \g{Ks/KH2An):= 10,7, а при диссоциации по второй
ступени — \g(Ks/KHAn-) = 7,3; в среде этилового спирта эти отношения
увеличиваются до 11,9 и 9 соответственно.
Как уже говорилось выше, влияние неводных растворителей
сказывается не только в изменении К кислот и оснований, но и в
изменении соотношения в силе кислот или оснований. Это обстоятельство
значительно расширяет возможность кислотно-основного титрования, так
как в неводных растворах можно дифференцированно титровать смеси
электролитов, К которых в водном растворе очень близки. Возможность
раздельного титрования смеси кислот или оснований определяется
соотношением Кнап, и/^Снап, i или Кв, п/Кв, ь В среде дифференцирующих
растворителей эти соотношения оказываются значительно меньше, чем
в водных растворах.
В табл. 18 приведены Лр/С бензойной кислоты и 2,6-динитрофе-
нола в воде и различных неводных растворителях. В неводных
растворителях Ар/С этих кислот больше, чем в воде, следовательно, в среде
неводных растворителей улучшаются условия дифференцированного
титрования смеси 2,6-динитрофенола и бензойной кислоты.
ТАБЛИЦА 18
Изменение А рК кислот в среДе различных растворителей
Растворитель
Бензойная кислота, р/С
2,6-Динитрофенол, р/С
Др#
Вода
4,2
4,02
0,18
Форм-
амид
6,23
4,55
1,68
Ацето-
нитрил
8,5
6,6
1,9
Метиловый
спирт
9,4
7,85
1,55
Этиловый
спирт
10,13
8,21
1,92
Ацетон,
90°/а-ный
9,8
6,45
3,35
Ацетон
11,95
8,78
3,17
Влияние воды. Присутствие воды в неводных растворителях (в
особенности высокошкальных), как правило, оказывает неблагоприятный
эффект на процессе титрования. Это объясняется тем, что кислоты
реагируют с водой как с основанием, а основания— как с кислотой (см.
стр. 425). В результате многие реакции нейтрализации протекают не
428 ГЛ. XI. ФИЗИКО-ХИМИЧЕСКИЕ МЕТОДЫ АНАЛИЗА НЕВОДНЫХ РАСТВОРОВ
количественно, так как титранты реагируют не только с определенным
веществом, но и с ионами воды.
Степень влияния воды зависит от соотношения шкал кислотности
неводного растворителя и воды и их расположения. Например, вода
практически не оказывает влияния на кислотные пределы шкалы
низших спиртов, но значительно изменяет основные пределы шкалы изопро-
пилового и трет-бутилового спиртов (рис. 145), так как основные
пределы их шкалы значительно больше, чем у воды.
Вода оказывает влияние как на кислотный, так и на основный
пределы шкал кислотности кетонов и ацетонитрила. Присутствие воды в
основных растворителях типа диметилформамида и диметилсульфоксида,
шкалы кислотности которых смещены в основную область, не вызывает
iuu-ia ou >uuva /00% 50 Ю0% 100% 50 100%
Диметил- Этилен- Пиридин Метило- трет-бути- Вода
срормамид гликоль вый спирт лоВый спирт
Рис. 145. Влияние сорастворителей на протяженность и
положение шкалы кислотности растворителей.
изменения кислотного предела шкалы, но основные пределы их шкал
сильно изменяются под влиянием относительно кислого по сравнению
с ними растворителя (воды).
Следует заметить, что влияние воды на кислотно-основной
характер других растворителей можно просто объяснить реакцией
нейтрализации более кислым растворителем другого растворителя,
характеризующегося более выраженными основными свойствами. Поэтому
наблюдаемое изменение кислотно-основных свойств растворителей, смешанных
с водой, можно рассматривать лишь как частный случай влияния
сорастворителей на положение и протяженность шкалы кислотности.
Влияние других сорастворителей. Присутствие в данном
растворителе других сорастворителей (помимо воды) может оказывать
существенное влияние на его шкалу кислотности в зависимости от
содержания, природы и характера растворителя (см. рис. 145). Разумеется, это
влияние может иметь большие последствия в процессе
кислотно-основного титрования. Успешное титрование индивидуальных электролитов и
возможность осуществления дифференцированного титрования их
смесей прежде всего зависит от изменения кислотно-основных свойств
избранного растворителя, происходящего в процессе смешивания
растворителей— намеренно добавляемых к данному растворителю (спиртов,
кетонов, уксусного ангидрида и т. п.), вводимых с титрантами (воды.
§ 8. ХИМИКО-АНАЛИТИЧЕСКОЕ ИСПОЛЬЗОВАНИЕ НЕВОДНЫХ РАСТВОРИТЕЛЕЙ 42>
спирта, уксусной кислоты) и, наконец, образующихся в процессе
нейтрализации (воды, спиртов, уксусной кислоты и т. п.).
Наиболее глубокие изменения кислотно-основных свойств исходного
растворителя наблюдаются при добавлении первых порций другого
растворителя, отличающегося от данного растворителя более
выраженным кислотным или основным характером. Последующее добавление
сорастворителя оказывает меньшее влияние на относительную шкалу
кислотности смешанного растворителя. Например, прибавление даже
незначительных количеств кислого растворителя к апротонному дипо-
лярному или к амфипротному вызывает резкое увеличение кислотных
свойств смешанного растворителя, обусловливающее сильное
ослабление основных свойств смеси по сравнению с исходным растворителем.
Прибавление протофильного сорастворителя к указанным
растворителям приводит к увеличению основных свойств и резкому ослаблению
кислотных свойств данного растворителя.
Все многообразие такого рода влияния кратко можно свести к
следующим случаям.
1. Если кислотно-основной характер смешанных растворителей
примерно одинаков и протяженности их шкал кислотности pKs совпадают,
то кислотно-основные свойства смеси практически не изменяются. Это
явление характерно для смесей: метиловый спирт — этиловый спирт;
метиловый спирт — этиленгликоль; этиловый спирт — пропиловый спирт;
ацетон — метилэтилкетон и т. д.
2. Если протяженности шкал кислотности pKs каждого
индивидуального растворителя одинаковы, а кислотно-основной их характер
различен, например один из растворителей является амфипротным,
а другой проявляет кислый характер, то положение шкалы
кислотности смеси по мере прибавления протогенного растворителя смещается
в кислую область за счет уменьшения основного предела амфипротного
растворителя. Такое явление наблюдается при смешивании: изопропи-
ловый спирт — нитрометан; трег-бутиловый спирт — нитробензол.
В случае прибавления к амфипротному растворителю
протофильного, проявляющего основной характер, положение шкалы кислотности
смеси по мере прибавления основного растворителя смещается в
основную область за счет уменьшения кислого предела амфипротного
растворителя. Например, такой процесс протекает при смешивании: изопро-
пиловый спирт — пиридин; ацетон — диметилсульфоксид и др.
3. Если протяженности и положение кислотной шкалы pKs
смешиваемых индивидуальных растворителей различны, то по мере
прибавления к растворителю, характеризующемуся большой протяженностью
шкалы кислотности, растворителя с малой шкалой происходит резкое
уменьшение шкалы кислотности смешанного растворителя. Это
наблюдается при смешении: ацетон — метиловый спирт; метилэтилкетон —
этиловый спирт; ацетонитрил — этиленгликоль; диметилсульфоксид —
уксусная кислота; пиридин — метиловый спирт и т. п.
При прибавлении к низкошкальному растворителю с малой
шкалой кислотности значительных количеств растворителя с большой
шкалой происходит увеличение шкалы кислотности смешанного
растворителя. Причем наибольшее увеличение протяженности шкалы
кислотности наблюдается при сильном разбавлении первого растворителя
вторым. Это явление наблюдается при смешивании: вода — ацетонитрил;
метиловый спирт — метилэтилкетон; уксусная кислота — грет-бутиловый
спирт и т. д.
Особенно резкое изменение протяженности и положения шкалы
кислотности наблюдается при смешении растворителей, сильно
430 ГЛ. XI. ФИЗИКО-ХИМИЧЕСКИЕ МЕТОДЫ АНАЛИЗА НЕВОДНЫХ РАСТВОРОВ
отличающихся Друг от друга кислотно-основными свойствами,
например уксусная кислота с пиридином; метиловый спирт с диметилформ-
амидом; этиленгликоль с диметилсульфоксидом и т. д.
Выбор метода. При оценке и выборе методов кислотно-основного
титрования необходимо учитывать влияние растворителя не только на
свойства определяемого вещества, но и на свойства продуктов
взаимодействия титруемого соединения с реактивом. Условия
кислотно-основного титрования определяются рядом факторов, которые можно разбить
на три группы.
К первой группе относятся факторы, обусловливающие выбор
растворителя как среды для проведения кислотно-основного
титрования; ко второй — факторы, связанные с выбором титранта; к третьей —
факторы, предопределяющие выбор способа определения конечной
точки титрования. Только при учете всех этих факторов титрования можно
выбрать наиболее эффективный метод определения индивидуальных
соединений и дифференцированного титрования многокомпонентных
смесей.
Титрование в неводных растворах по методу осаждения.
Применение неводеных растворителей для титрования по методу осаждения
представляет большой интерес, так как под влиянием растворителя
сильно изменяется растворимость веществ. Соединение, хорошо
растворимое в воде, может оказаться малорастворимым в каком-либо
неводном растворителе, и наоборот, соединение, нерастворимое в воде —
хорошо растворимым в органическом растворителе. Например,
сульфат и оксалат натрия хорошо растворимы в воде, а в среде безводной
уксусной кислоты эти соединения настолько мало растворимы, что
становится возможным весовое определение ионов натрия осаждением их
в виде оксалата или сульфата. В среде жидкого аммиака AgCl
реагирует с Ba(N03b с образованием осадка ВаСЬ — соли, хорошо
растворимой в воде, и т. д.
Применение неводных растворителей для титрования по методу
осаждения позволяет: определить такие ионы, определение которых
в водных растворах методом осаждения невозможно; селективно
определять одни ионы в присутствии других; заменить дефицитные и
дорогостоящие реактивы (например, нитрат серебра) более дешевыми и
доступными.
Возможность осуществления с достаточной точностью титрования
по методу осаждения зависит при прочих равных условиях от
растворимости осаждаемой соли в данной среде.
Теоретическими исследованиями Н. А. Измайлова и др.
установлено, что растворимость вещества в органическом растворителе зависит
в первую очередь от величины диэлектрической проницаемости
растворителя, а логарифм растворимости (lgP) соединения в данном
растворителе определяется уравнением:
lg P = А + — <
8
где А и В — постоянные величины;
8 — диэлектрическая проницаемость растворителя.
Экспериментальным путем найдено три типа зависимости
растворимости соединения от диэлектрической проницаемости е растворителя:
растворимость уменьшается с уменьшением е; растворимость
увеличивается с уменьшением е; растворимость проходит через максимум.
В. Н. Семенченко считает, что два первых типа зависимости
растворимости от диэлектрической проницаемости растворителя являются
лишь частными случаями третьего типа, но в первом случае практиче-
§ 9. УСТАНОВКИ ДЛЯ АМПЕРОМЕТРИЧЕСКОГО ТИТРОВАНИЯ
431
ски нельзя подобрать растворитель с настолько высоким значением
диэлектрической проницаемости, а во втором — с настолько низким, чтобы
кривая прошла через максимум.
Растворимость вещества по В. Н. Семенченко и М. И. Шахпаро-
нову зависит от соотношения полярностей растворимого вещества и
растворителя. Растворимость максимальна в тех случаях, когда
отношение обобщенных дипольных моментов \i' растворяемого вещества и
растворителя jx" приблизительно равняется единице:
JL
«1;
I* вТ
где fi — дипольный момент;
V — объем молекулы.
Растворимость таких полярных веществ, как неорганические соли,
увеличивается с возрастанием диэлектрической проницаемости и для
них справедливо уравнение Измайлова.
Б. АППАРАТУРА И ТЕХНИКА ТИТРОВАНИЯ НЕВОДНЫХ
РАСТВОРОВ
§ 9. Установки, используемые при амперометрическом
титровании негодных растворов
Аппаратура. Для амперометрического титрования применяют
установку (рис. 146), в которой используется визуальный полярограф ПВ-5
с зеркальным гальванометром М-21 и система двух индикаторных элек-
Рис. 146. Установка для амперометрического титрования неводных
растворов:
/ — электролизер; 2 — вращающийся электрод; 3 — неподвижный электрод:
4 — бюретка; 5 —визуальный полярограф ПВ-5.
тродов — вращающегося и неподвижного или одного индикаторного
электрода и электрода сравнения (обычно насыщенного каломельного).
Электролизер 1, в который наливают титруемый раствор,
представляет собой химический стакан емкостью 25—30 мл с опущенными в него
индикаторными электродами 2 и 3. При титровании с одним
индикаторным электродом вместо электрода 3 в электролизер опускают один
конец электролитического ключа, заполненного раствором хлорида или
432 ГЛ. XI. ФИЗИКО-ХИМИЧЕСКИЕ МЕТОДЫ АНАЛИЗА НЕВОДНЫХ РАСТВОРОВ
нитрата калия в агар-агаре, другой конец которого опущен в стакан
с электролитом, в который погружен также кончик каломельного (или
какого-либо другого) электрода сравнения. Индикаторный электрод 2
приводится во вращение мотором, число оборотов которого
регулируется при помощи реостата. Над электролизером укрепляется в
штативе бюретка 4 емкостью 10 мл с ценой деления 0,02 мл.
Напряжение постоянного тока на полярограф 5 подается от
аккумуляторной батареи (обычно на 4 в).
Сила тока измеряется гальванометром ГМ-21 (на рисунке не
показан), чувствительность гальванометра 10~9 а*мм\м.
Рис. 147. Схема установки для потенциометрического титрования:
/ — автоматическая полумикробюретка емкостью 10 мл (цена деления 0,02 мл); 2 — стакан для
титрования; 3 —электроды; 4— потенциометр; 5—магнитная мешалка; 6 — осушительные системы для
воздуха и азота; 7 —барботер для азота; 8 — буферная склянка; 9 — баллон с азотом; 10 — стеклянный
кран для сбрасывания избыточного давления воздуха из сосуда, в котором содержится титрант;
// —резиновая груша.
Приемы работы с полярографами описаны выше (Сл. гл. V, § 3).
Техника титрования. В бюретку, предварительно тщательно
вымытую и высушенную, наливают титрант (например, 0,1 н. раствор
роданида калия в изопропиловом спирте) и устанавливают мениск на
нуль.
Анализируемый раствор доливают до метки мерной колбы
соответствующим растворителем. Аликвотную часть помещают в электролизер
/ (также тщательно вымытый и высушенный), добавляют растворитель
до общего объема 15—20 мл и опускают электроды.
Включают полярограф 5 и приливают из бюретки в электролизер
титрант небольшими порциями (по 0,05—0,1 мл), записывая
показания гальванометра после прибавления каждой порции.
Кривую титрования строят в координатах объем титранта (мл) —
показания гальванометра и находят точку эквивалентности по перегибу
кривой.
§ 10. УСТАНОВКИ ДЛЯ ПОТЕНЦИОМЕТРИЧЕСКОГО ТИТРОВАНИЯ 433
§ 10. Установки, используемые при потенциометрическом
титровании неводных растворов
Аппаратура. Для потенциометрического титрования используют
установку (рис. 147), в которой применяются потенциометры ЛП-5,
ЛП-58 или ЛПМ-60 М и система индикаторного электрода и электрода
сравнения (обычно стеклянного и насыщенного каломельного
электродов). Стакан для титрования емкостью 50—100 мл закрывается
крышкой, в которой имеются отверстия для кончика бюретки,
электродов и трубки для азота.
С целью предотвращения попадания воды из
каломельного электрода в неводный раствор определяемого
вещества насыщенный раствор хлорида калия в нем
заменяют насыщенным неводным раствором электролита,
например метаноловым раствором хлорида калия.
Титрование в неводных растворах также
осуществляют, используя и другие системы электродов. Например, в
качестве индикаторных электродов применяют хингидрон-
ный, водородный, сурьмяный, графитовый, платиновый, ок-
сиплатиновый и некоторые другие, а в качестве электродов
сравнения — хлорсеребряный и стеклянный.
Техника титрования. Перед употреблением бюретку /
тщательно моют (см. книга 2, гл. I, § 7). Верхнее
отверстие вымытой и высушенной бюретки закрывают хлоркаль-
циевой трубкой, наполненной натронной известью и
плавленым хлоридом кальция; при работе воздух, нагнетаемый
резиновой грушей 11, должен проходить через систему
осушительных колонок 6, заполненных также натронной
известью и плавленым хлоридом кальция. Кран бюретки
смазывают какой-либо неводной (органической или крем-
нийорганической) смазкой, которая не растворяется в
используемых для приготовления титрантов растворителях.
Нагнетание в сосуд с титрантом воздуха для поднятия
титранта в бюретку следует проводить медленно,
равномерно, накачивая воздух так, чтобы не создавать
большого избыточного давления. После заполнения бюретки
титрантом избыточное давление сбрасывают, открывая кран
10 на короткий промежуток времени.
При титровании по микрометоду можно применять
бюретку, изображенную на рис. 148. Емкости таких
бюреток составляют 5; 2 или 1 мл с ценой деления 0,01 мл,
емкость баллона для хранения титранта — соответственно
250, 200 или 100 мл.
При титровании кончик бюретки должен находиться вблизи уровня
жидкости или быть опущенным в титруемый раствор (см. рис. 147). Во
избежание поглощения раствором двуокиси углерода из воздуха очень
слабые кислоты титруют в токе сухого азота, который подают из
баллона через осушительные колонки с натронной известью и хлоридом
кальция; трубку, подводящую азот, располагают вблизи уровня раствора
или опускают в раствор. Перемешивание титруемого раствора обычно
осуществляют магнитной мешалкой или током сухого азота.
Визуальное титрование можно проводить в конической колбе, перемешивая
раствор вращательным движением руки.
При работе с сухими веществами навеску анализируемого вещества
или смеси веществ, содержащую приблизительно 0,2—0,5 мг-экв
Рис. 148.
Микробюретка:
/ — стеклянный
баллон для
хранения
титранта;
2—бюретка с ценой
деления 0,01
мл.
434 ГЛ XI ФИЗИКО-ХИМИЧЕСКИЕ МЕТОДЫ АНАЛИЗА НЕВОДНЫХ РАСТВОРОВ
определяемого компонента, взвешивают на аналитических весах с
точностью 0,0002 г; при меньших количествах определяемого вещества
следует брать навеску на полумикровесах с точностью 0,00002 г. При
использовании полумикробюретки емкостью 10 мл навеску желательно
рассчитывать таким образом, чтобы объем титранта составил не менее
2 мл; большие навески слабых кислот и оснований, требующие расход
8—9 мл титранта, часто вызывают снижение резкости конечной точки
титрования.
При использовании метода пипетирования, особенно в случае
работы с жидкими веществами, рассчитанную навеску анализируемого
образца взвешивают на аналитических весах и помещают в мерные
колбы емкостью 50, 100 или 250 мл. Раствор желательно готовить в том
растворителе, в среде которого проводят титрование.
Потенциометрическое титрование. В сухой стакан
наливают 25—40 мл растворителя и нейтрализуют примеси в растворителе
по соответственно выбранному индикатору (или проводят холостой
опыт), затем помещают навеску анализируемого вещества или аликвот-
ную часть исследуемого раствора и опускают электроды, которые
предварительно обмывают дистиллированной водой, ополаскивают
спиртом и обтирают фильтровальной бумагой. Титрованный раствор из
бюретки добавляют по 0,1 мл, а вблизи точки эквивалентности по 0,02—
0,05 мл\ перемешивание осуществляют магнитной мешалкой; после
каждого добавления титранта дают установиться потенциалу и снимают
показания потенциометра. Титрование продолжают до установления
постоянного потенциала.
Показания потенциометра удобнее записывать по шкале,
выраженной в милливольтах, однако используют и шкалу рН. По данным
титрования строят график в координатах Е (мв) —объем титранта (мл) или
рН — объем титранта (мл) и определяют точку эквивалентности по
скачку титрования. При наличии нескольких определяемых веществ или
смеси электролитов, титруемых ступенчато, на кривой титрования
наблюдается соответствующее число скачков титрования, отвечающих
нескольким точкам эквивалентности.
§11. Установки, используемые при кондуктометрическом
титровании неводных растворов
Аппаратура. Для кондуктометрического титрования неводных
растворов может быть использована аппаратура, описанная в главе IIL
Обычно для кондуктометрического титрования неводных растворов
применяют установку, показанную на рис. 149.
Техника титрования. Титрование проводят в ячейке емкостью 25 мл
с платинированными платиновыми электродами (1X1 см2), впаянными
в пришлифованную крышку ячейки и отстоящими друг от друга на
расстоянии 5 мм.
После каждого титрования поверхность электродов тщательно
очищают путем многократного споласкивания электродов растворителем.
В ячейку для титрования наливают около 20 мл растварителя и вносят
навеску определяемого вещества. Титрованный раствор реактива
прибавляют приблизительно по 0,2 мл при перемешивании магнитной
мешалкой. По данным измерения строят кривую титрования в
координатах сопротивление (ом) — объем титранта (мл) и графически находят
точки эквивалентности.
Хронопотенциометрическое безбюреточное титрование. Характерной
особенностью этого метода является постоянная скорость подачи
Рис. 149. Схема установки для кондуктометрического титрования:
/ —осциллографический индикатор ИНО-314; 2 — звуковой генератор ЗГ-1; 3 — реохордный
мост сопротивления Р-38; 4 — магнитная мешалка; 5—платиновые электроды; 6 — ячейка для
титрования; 7—автоматическая полумикробюретка; 8 — осушительные системы для воздуха
и азота; 9 — баллон с азотом; 10 — буферная склянка; 11 — барботер для азота; 12 — резиновая
груша.
Рис. 150. Схема установки для полуавтоматического хрвнопотенциометри-
ческого безбюреточного титрования в неведных раеяъорах:
/—сосуд Мариоа>та; 2 — штатив; 3 — стакан для титрования; 4 — стеклянный электрод;
5 — электролитический ключ, заполненный метаноловым раствором КС1; 6 — стакан
с насыщенным метаноловым раствором КС1; 7 — хлорсеребряный электрод; 8 —
установка для отбора титранта; 9 — магнитная мешалка; 10 — рН-метр; 11 — магазин
сопротивлений; 12 — автоматический потенциометр.
436 ГЛ XI. ФИЗИКО-ХИМИЧЕСКИЕ МЕТОДЫ АНАЛИЗА НЕВОДНЫХ РАСТВОРОВ
титранта в анализируемый раствор и непрерывная запись показаний
рН-метра в процессе титрования. Для подачи титранта с постоянной
скоростью используют сосуд Мариотта, а для автоматической записи кривых
титрования — самопишущий потенциометр, диаграммная бумага
которого равномерно протягивается электродвигателем, синхронно с
подачей титранта. Поэтому если при обычном потенциометрическом
титровании мерой количества определяемого вещества, служит объем
стандартного раствора, расходуемый на титрование, то в описываемом
методе о количестве вещества судят по соответствующей длине
диаграммной ленты самописца.
Рис. 151. Схема установки для высокочастотного титрования
I — осциллограф; 2 — стакан для титрования; 3 — электроды; 4 — автоматическая по ммикробюретка;
5 —осушительные системы для азота и воздуха; 6 — баллон с азотом; 7—буферная склянка; 8 — бар-
ботер для азота; 9 — резиновая груша.
Аппаратура. Установка для проведения полуавтоматического хро-
нопотенциометрического титрования показана на рис. 150.
Емкость сосуда Мариотта 1 л\ впаянные в пришлифованную пробку
трубки имеют диаметр 5—6 мм. Длина горизонтальной части напорной
трубки составляет около 10 см, а общая длина вертикальной части,
включая соединительную трубку и капилляр, около 50 см. Скорость
подачи титрантов из сосуда Мариотта приблизительно 0,5 мл/мин
устанавливается путем подбора диаметра капиллярного отверстия
подающего капилляра.
Скорость движения диаграммной ленты составляет 1440 мм/ч.
§12. Установки, используемые при высокочастотном
титровании неводных растворов
Аппаратура. Для высокочастотного титрования неводных растворов
может быть использована аппаратура, описанная в главе IV. Наиболее
распространенным прибором, используемым для высокочастотного
титрования, является высокочастотный титратор Пунгора (типа ОК-302)
§ 13 УСТАНОВКИ ДЛЯ СПЕКТРОФОТОМЕТРИЧЕСКОГО ТИТРОВАНИЯ 437
с прилагаемыми ячейками емкостью 250, 150 и 100 мл. Схема
установки для высокочастотного титрования показана на рис. 151.
Техника титрования. В тех случаях, когда при титровании
изменения электропроводности раствора незначительны, возникает
необходимость увеличения чувствительности прибора. Для этого последовательно
включают звенья делительной цепочки, увеличивая включением
последующего звена чувствительность микроамперметра в 2 раза.
На 100 мл растворителя берут навеску анализируемого вещества
0,3—0,6 мг-экв, причем используют 0,1—0,2 н. растворы титрантов.
Известно, что во избежание влияния на результаты титрования фактора
разбавления концентрация титрующего раствора должна в 10—100 раз
превышать концентрацию титруемого раствора.
В сухой стакан для титрования наливают 100 мл растворителя,
пускают ток сухого азота для перемешивания титруемого раствора и
вносят навеску определяемого вещества. При непрерывном перемешивании
током сухого азота прибавляют раствор титранта по 0,5—1,0 мл (в
соответствии с рекомендацией, приложенной в описании к прибору) и
снимают показания прибора (мка). Кривую титрования строят в
координатах сила тока (мка)—объем титранта (мл) и графически находят
точки эквивалентности на пересечениях прямых отрезков; после
последней точки эквивалентности следует снять еще пять или шесть
показаний прибора. Расчет ведут обычным способом, учитывая в случае
необходимости объем титранта, израсходованного на нейтрализацию
примесей в растворителе.
§ 13. Установки, используемые при спектрофотометрическом
титровании неводных растворов
Аппаратура. Для спектрофотометрического титрования используют
приборы СФ-5, СФ-4, СФ-4А, СФД-2, устройство которых описано
в гл. VII. В крышке кюветной камеры применяемого спектрофотометра
просверливают два отверстия: одно для кончика бюретки, другое для
механической мешалки. При титровании используются кюветы
объемом 25 мл.
При работе в видимой области спектра применяют кювету из
оптического стекла, в ультрафиолетовой области — кварцевую или
стеклянную кювету, снабженную кварцевыми окошками. Стенки кюветы
покрывают черным лаком, оставляя окошки диаметром 1 см по пути
светового потока. Бюретку и мешалку укрепляют таким образом, чтобы
их концы находились против затемненных участков кюветы.
Схема установки для спектрофотометрического титрования
показана на рис. 152.
Техника титрования. Метод спектрофотометрического титрования
основан на измерении оптической плотности исследуемого раствора, из^
меняемой в процессе титрования. Для уменьшения влияния
разбавления на светопоглощение применяют относительно концентрированные
растворы титранта или вводят поправку на разбавление. Для работы
готовят приблизительно 2,5-Ю-2—1 • Ю-3 н. растворы анализируемого
вещества (навеску 20—100 мг растворяют в 20—40 мл неводного
растворителя, отбирают аликвотную часть исходного раствора и
разбавляют в кювете до концентрации 2-Ю-3—1 • 10~4 н. Титрование проводят
без кюветы сравнения. Кювету с исследуемым раствором помещают в
кюветную камеру спектрофотометра. Техника работы со
спектрофотометром описана в гл. VII.
При определении кислот, титрующихся с увеличением оптической
плотности, исходное значение оптической плотности выбирают в
ГЛ. XI. ФИЗИКО-ХИМИЧЕСКИЕ МЕТОДЫ АНАЛИЗА НЕВОДНЫХ РАСТВОРОВ
пределах 0,05—0,1, при определении кислот, титрующихся с
уменьшением оптической плотности, и ароматических аминов —0,6—0,8.
Титрант желательно готовить в том же растворителе, в котором
растворяют исследуемое вещество. Концентрация его должна быть
2-Ю"2 —2-10"3 н. Титрование проводят, прибавляя из полумикробюрет-
ки по 0,1—0,2 мл раствора; после добавления очередной порции титран-
та раствор перемешивают и измеряют оптическую плотность.
Рис. 152. Схема установки для спектрофотометрического титрования:
/ - спектооФотометр; 2-кюветная камера; 3-кювета для титрования; 4- механическая мешалка;
Ч- гибкий привод мешажи; 6- автоматическая полумикробюретка (аналогичная приведенной на
рис. 15Г); 7-осушительная система для воздуха; 8 - резиновая груша.
Кривую титрования строят в координатах оптическая плотность —
объем титранта (мл). Точку эквивалентности находят путем
экстраполяции двух прямолинейных участков кривой титрования до
пересечения.
В. ПРАКТИЧЕСКИЕ РАБОТЫ
§14. Очистка и обезвоживание неводных растворителей
В качестве растворителей, для неводного титрования чаще всего
применяют: муравьиную и уксусную кислоты, уксусный ангидрид,
метиловый, этиловый, изопропиловый, втор- и грет-бутиловый спирты,
ацетон, м'етилэтилкетон, метилизобутилкетон, пиридин, диметилформамид,
ацетонитрил, нитробензол, хлороформ и др. Для очистки и
обезвоживания неводных растворителей применяют методы, описанные в
специальных руководствах*.
Необходимо иметь в виду, что все органические растворители, как
правило, ядовиты. Поэтому отбирать пробы необходимо автоматической
пипеткой или пипеткой с грушей; ни в коем случае не засасывать
растворители при помощи пипеток, применяемых при работе с водными
растворами. Установку для титрования в неводных растворах следует
располагать вблизи тяги, а при использовании сильнопахнущих
растворителей— в вытяжном шкафу.
При работе с неводными растворителями нужно соблюдать и
другие меры особой предосторожности во избежание отравления, пожара и
взрывов, руководствуясь инструкциями по технике безопасности.
§ 15. Титранты, применяемые для титрования кислот
и оснований в неводных растворах
Для титрования соединений, проявляющих в неводных растворах
свойства оснований, используют неводные растворы неорганических и
* К р е ш к о в А. П., Быкова Л. Н., К а з а р я н Н. А., Кислотно-основное
титрование в неводных растворах, Изд. «Химия», 1967.
§ 16. АМПЕРОМЕТРИЧЕСКОЕ ТИТРОВАНИЕ
439
органических кислот. Наиболее сильной из минеральных кислот в не-
водршх растворах является хлорная кислота. Поэтому стандартные
неводные растворы ее широко применяются для титрования оснований.
Для тех же целей используют неводные растворы хлористоводороднай
кислоты, спиртовые растворы я-толуолсульфокислоты и фторсульфоно-
вой кислоты, являющейся более сильной кислотой по сравнению с
хлорной, но разлагающейся (медленно) при действии воды:
HSO3F + HOH ^=± H2S04 + HF
Для титрования соединений, которые проявляют в неводных
растворах свойства кислот, используют неводные растворы гидроокисей,
ацетатов и алкоголятов щелочных металлов, гидроокисей четвертичных
аммониевых оснований, дифенилгуанидина и т. п.
Приготовление и установка стандартных неводных растворов
хлорной и хлористоводородной кислот. Наиболее часто в лабораторной
практике применяют уксуснокислые, диоксановые, ацетоновые, метилэтилке-
тоновые и спиртовые растворы хлорной кислоты. Нередко рассчитанное
количество НС104 или НС1 растворяют в том органическом
растворителе, в котором предполагается титровать определяемое вещество.
Титры неводных растворов кислот устанавливают на той же
аппаратуре, на которой проводят титрование определяемых веществ, и при
той же температуре, при которой титруют анализируемые образцы.
Во многих случаях перед установкой титров кислот (и оснований)
проводят холостой опыт для того, чтобы учесть содержание основных
(и кислых) примесей в растворителе. В ряде случаев вместо холостых
опытов растворитель предварительно нейтрализуют кислотой (или
основанием) в присутствии соответствующего индикатора.
Для установки титров приготовленных стандартных растворов
кислот применяют: бифталат калия, карбонат натрия, дифенилгуанидин и
некоторые другие основания марки «х. ч.».
Техника установки титров описана ранее (см. книга 2, гл. II, § 36).
В каждом отдельном случае применяют либо индикаторный, либо
соответствующий инструментальный методы установления точки
эквивалентности.
Приготовление и установка стандартных неводных растворов
оснований. Для титрования кислот наиболее часто применяют спиртовые
растворы гидроокисей щелочных металлов; бензольно-метаноловые (эта-
ноловые) или изопропаноловые растворы алкоголятов щелочных
металлов; уксуснокислые растворы ацетата натрия; бензольно-метаноловые
Хили этаноловые) растворы гидроокисей тетраметил (этил) аммония;
спиртовые растворы дифенилгуанидина.
Для установки титров приготовленных стандартных растворов
оснований применяют: бензойную, салициловую и другие кислоты марки
«х. ч.».
§ 16. Амперометрическое титрование
Определение нитратов в среде безводной уксусной кислоты
Все соли азотной кислоты растворимы в воде. Между тем многие
нитраты нерастворимы в некоторых неводных растворителях.
Вследствие этого становится возможным применять для их определения методы
осаждения.
Предлагаемый метод определения нитратов основан на осаждении
Ba(N03)2, который, как известно, хорошо растворим в воде, но
малорастворим в среде безводной уксусной кислоты.
440 ГЛ. XI. ФИЗИКО-ХИМИЧЕСКИЕ МЕТОДЫ АНАЛИЗА НЕВОДНЫХ РАСТВОРОВ
Нитраты титруют стандартным уксуснокислым раствором ацетата
бария в среде безводной уксусной кислоты по току восстановления
нитрат-ионов.
Установка для амперометрического титрования описана выше (см.
рис. 146).
В качестве индикаторного электрода используют медный
вращающийся микроэлектрод (см. гл. V, § 15), в качестве электрода
сравнения — насыщенный каломельный электрод, который соединяют с
электролизером электролитическим ключом, заполненным насыщенным
раствором хлорида калия в агар-агаре.
Нитрат-ионы восстанавливаются на медном вращающемся
индикаторном электроде на фоне 0,05 н. уксуснокислого раствора LiCl,
образуя хорошо выраженную полярографическую волну с Еу2 = —0,45 в.
Диффузионный ток достигается при Е = —0,8 в\ в соответствии с этим
амперометрическое титрование проводят при потенциале —1,0 в.
Кривые титрования имеют два излома. Первый излом соответствует
примерно половине оттитрованного вещества, второй — точке
эквивалентности.
Методика определения. Навеску анализируемого вещества,
содержащего 0,04—0,07 г нитрата, взвешенную на аналитических весах в
стакане емкостью 30 мл, растворяют в 1—2 каплях воды и прибавляют 20 мл
0,05 н. уксуснокислого раствора хлорида лития.
В микробюретку наливают стандартный 0,2 н. уксуснокислый
раствор ацетата бария.
В стакан опускают медный индикаторный электрод и соединяют
электролизер с каломельным электродом при помощи
электролитического ключа, затем включают мотор, скорость вращения электрода
должна быть около 800—900 обIмин. Устанавливают потенциал —1,0 в,
включают гальванометр и титруют нитрат-ионы раствором ацетата
бария, прибавляя его порциями по 0,2 мл. После прибавления каждой
порции стандартного раствора отмечают положение светового зайчика
на шкале.
На основании полученных данных строят график зависимости
показаний гальванометра (мм) от прибавленного объема титранта и
графически находят объем титранта (мл), соответствующий точке
эквивалентности.
Расчет. Содержание нитрата в процентах вычисляют по формуле:
3MeN03^a++FBa++'1QQ
*MeN03 ' lOOOS
Определение ионов кадмия в среде безводной уксусной кислоты
Метод основан на образовании осадка CdBr2. Ионы кадмия
титруют стандартным уксуснокислым раствором бромида калия в среде
безводной уксусной кислоты по току окисления бромид-ионов. В
процессе титрования выпадает осадок CdBr2, который как известно, хорошо
растворим в воде, но малорастворим в безводной уксусной кислоте.
Установка для амперометрического титрования (см. рис. 146)
описана выше.
В качестве индикаторного электрода используют платиновый
вращающийся микроэлектрод (см. гл. V, § 6), в качестве электрода
сравнения — насыщенный каломельный электрод, который соединяют с
электролизером электролитическим ключом, заполненным насыщенным
раствором нитрата калия в агар-агаре.
§ 16. АМПЕРОМЕТРИЧЕСКОЕ ТИТРОВАНИЕ
441
Бромид-ионы окисляются на платиновом вращающемся электроде
на фоне различных электролитов (KN03, LiN03, CH3COOLi, H2S04),
образуя хорошо выраженные полярографические волны. В зависимости
от фона Еу2 колеблется от + 0,93 до + 1,45 в.
Амперометрическое титрование ионов кадмия раствором бромида
проводят без фона, так как в присутствии постороннего электролита
задерживается образование осадка CdBr2 (вследствие увеличения ионной
силы раствора). Фоном в данном случае служит вначале титруемая
соль кадмия, а затем электролит, образующийся при титровании.
Титрование проводят при потенциале 4-1,0 &.
Кривые титрования имеют три излома, последний из которых
соответствует точке эквивалентности.
Метод позволяет определять кадмий в присутствии многих других
катионов, так как последние не осаждаются бромид-ионами в среде
безводной уксусной кислоты.
Этим же методом можно определять бромид-ионы.
Методика определения. В стакан для титрования емкостью 30 мл
наливают 1 —15 мл анализируемого раствора, содержащего 5—25 мг
Сс1++-ионов, и прибавляют безводную уксусную кислоту до объема 20 мл.
В микробюретку наливают стандартный 0,1 н. уксуснокислый раствор
КВг. В стакан опускают платиновый индикаторный электрод и
соединяют электролизер с каломельным электродом с помощью
электролитического ключа, затем включают мотор, скорость вращения электрода
должна быть около 800—900 об/мин. Устанавливают потенциал, равный
+ 1,0 в, включают гальванометр и титруют ионы кадмия уксуснокислым
раствором КВг, прибавляя его порциями по 0,2 мл. Положение
светового зайчика на шкале отмечают после прибавления каждой порции.
На основании полученных данных строят график зависимости
показаний гальванометра (мм) от прибавленного объема титранта (мл) и
графически находят объем титранта (мл), соответствующий точке
эквивалентности.
Расчет. Содержание (g) кадмия (в г) вычисляют по формуле:
9Cd++^KBr^KBr
g~ Гооо
Определение хлоридов методом амперометрического титрования
с двумя индикаторными электродами в среде изопропилового спирта
Хлориды титруют в среде изопропилового спирта стандартным
изопропаноловым раствором роданида калия. В процессе титрования
выпадает осадок КС1, который, как известно, хорошо растворим в воде,
но малорастворим в изопропиловом спирте.
Установка для амперометрического титрования (см. рис. 146)
описана выше.
В качестве катода используют Ёращающийся медный электрод,
приводимый во вращение мотором (1000—1200 об/мин). Подробное
описание устройства электрода дано выше (см. гл. V, § 15). В качестве
анода используют пластинку из медной фольги толщиной 0,3 мм, согнутую
в форме цилиндра, ее опускают в стакан для титрования.
Особенностью метода является то, что ни определяемые вещества,
ни титранты не дают электродных реакций на медном катоде и аноде.
При возрастании напряжения наблюдается увеличение силы тока, но
предельный ток не достигается. Возникновение тока в цепи объясняется
переходом в раствор ионов меди с анода и восстановлением их на
катоде. Роль определяемых ионов состоит в том, что в процессе титрования
442 ГЛ. XI. ФИЗИКО-ХИМИЧЕСКИЕ МЕТОДЫ АНАЛИЗА НЕВОДНЫХ РАСТВОРОВ
происходит изменение электропроводности раствора, а
следовательно, и силы тока, вследствие уменьшения концентрации определяемых
С1~-ионов по мере их осаждения в виде KG (ионы хлора в растворе
по мере титрования заменяются ионами титранта). Наиболее резко
меняется концентрация титруемых ионов вблизи точки эквивалентности.
Методика определения. В мерной колбе емкостью 25 мл
приготовляют приблизительно 0,05 М изопропаноловый раствор определяемого
хлорида. В стакан для титрования емкостью 30 мл помещают 2 мл этого
раствора, 15 мл изопропилового спирта и опускают электроды, затем
включают мотор, скорость вращения катода должна быть 1000 —
1200 об/мин. Устанавливают потенциал, равный —2,0 в, включают
гальванометр и титруют хлорид-ионы 0,05 М изопропаноловым раствором
KSCN, прибавляя его из микробюретки порциями по 0,1—0,2 мл. При
этом образуется мелкокристаллический осадок КС1.
На основании полученных данных строят кривую титрования в
координатах показания гальванометра (мм)—объем титранта (мл),
эта кривая имеет V-образную форму, по излому кривой находят объем
титранта (мл), соответствующий точке эквивалентности.
Расчет. Содержание хлорида в процентах вычисляют по формуле:
3MeCl%SCN^KSCN^K ' 1QQ
*MeCI 10<WAa
§ 17. Методы прямого потенциометрического титрования
Определение амина в среде безводной уксусной кислоты
Амины, как правило, являются слабыми основаниями. Так,
показатель константы основности анилина в воде (рКв) равен 9,42. Кроме
того, анилин малорастворим в воде. Поэтому его определение в водной
среде прямым индикаторным или потенциометрическим методом
оказывается невозможным. Как указано ранее (см. книга 2, гл. II, § 36),
использование в качестве сред для титрования неводных растворителей:
уксусной кислоты, кетонов, спиртов нитрилов и их смесей с
углеводородами — дает возможность определить анилин методом
нейтрализации, используя визуальный способ обнаружения точки эквивалентности
в присутствии кристаллического фиолетового.
Описываемая методика потенциометрического определения амина
(анилина, толуидина, пиридина и т. п.) основана на растворении
определяемого вещества в среде безводной уксусной кислоты и
последующем титровании его уксуснокислым раствором хлорной• кислоты.
Установка для потенциометрического титрования (см. рис. 147)
описана выше.
Методика определения. Навеску исходного амина (адалина,
толуидина, пиридина) 0,2 г, взвешенную на аналитических весах, переносят
в мерную колбу емкостью 50 мл, растворяют в безводной уксусной
кислоте и доводят тем же растворителем до метки. Затем отбирают точно
откалиброванной пипеткой аликвотную часть раствора (10 мл),
переносят ее в стакан для титрования, добавляют 15—20 мл уксусной кислоты
и титруют потенциометрическим методом 0,1 н. уксуснокислым
раствором НСЮ4 (V). Титрование проводят со стеклянным и каломельным
электродами.
Для определения основных примесей, содержащихся в уксусной
кислоте, проводят холостой опыт. В стакан для титрования наливают
уксусную кислоту в количестве, равном аликвотной пробе анализируе*
§ 17. МЕТОДЫ ПРЯМОГО ПОТЕНЦИОМЕТРИЧЕСКОГО ТИТРОВАНИЯ 443
мого раствора, и титруют уксуснокислым раствором НСЮ4. Этим путем
определяют объем титранта, израсходованный на нейтрализацию
основных пр,имесей (V).
Расчет. Содержание (g) анилина В (в г) вычисляют по формуле:
Эв*нсю«<Г-ПГк
8 1000FA
По описанной методике можно определять различные производные
анилина и другие основания, р/С которых в воде не более 10.
Определение бензойной кислоты в среде ацетона
В качестве сред для титрования бензойной кислоты используют
органические основания, спирты, кетоны, нитрилы и их смеси с
углеводородами.
Ниже описывается метод потенциометрического определения
бензойной кислоты, основанный на растворении навески кислоты в среде
ацетона и титровании ее бензольно-метаноловым раствором гидроокиси
тетраэтиламмония (ГТЭА) или этаноловым раствором КОН.
Методика определения. Перед титрованием нейтрализуют
содержащиеся в ацетоне кислые примеси по тимоловому синему. Для этого в
стакан для титрования приливают 20—25 мл ацетона, добавляют 2—
3 капли тимолового синего и несколько капель 0,1 н. бензольно-метано-
лового раствора ГТЭА или этанолового раствора КОН до перехода
окраски индикатора от желтой к синей. Затем в стакан с
нейтрализованным растворителем переносят взятую на аналитических весах навеску
бензойной кислоты приблизительно 0,05—0,06 г и титруют тем же
раствором (V), которым предварительно нейтрализовали растворитель.
Титрование проводят со стеклянным и каломельным электродами.
По данным титрования строят график в координатах Е (мв) —
V (мл) и определяют точку эквивалентности. По графику находят объем
раствора основания, необходимый для нейтрализации определяемой
кислоты, и рассчитывают ее содержание.
Вместо предварительного титрования кислых примесей,
содержащихся в ацетоне, можно проводить холостой опыт, и тогда при
вычислении содержания бензойной кислоты в анализируемом растворе
учитывают объем стандартного раствора основания, израсходованный на
титрование холостой пробы (V).
По описанной методике можно также определять алифатические
и ароматические карбоновые кислоты и их окси-, галоген- и нитропро-
изводные.
Расчет. Содержание (g) бензойной кислоты Б. К (в г) вычисляют
по формуле:
ЭБ. К^ГТЭА^ГТЭА
g 1000
или
_ ЭБ. кА^ГТЭА ГГТЭА - ^ГТЭа)
g~* 1000
Определение аминокислоты в среде безводной уксусной
кислоты
Метод определения основан на титровании метилзтнлкетоцовым
раствором хлорной кислоты аминокислоты в среде безводной уксусной
444 ГЛ, XU ФИЗИКО-ХИМИЧЕСКИЕ МЕТОДЫ АНАЛИЗА НЕВОДНЫХ РАСТВОРОВ
кислоты, в которой аминокислоты проявляют себя как основания:
HOOC(CH2)rtCH2NH2+€H3COOH ^=± НООС(СН2)л CH2NH? + СН3СОСГ
НСЮ4 + СН3СОС2Н5 ^z± [СН3СОС2Н5]Н+ + С104
[СН3СОС2Н5]Н+ + СН3СОО- —> СНзСОС2Н5 + СН3СООН
HOOC(CH2)n CH2NH? + СЮ" —> НООС(СН2)п CH2NH+CIOJ
HOOC(CH2)nCH2NH2 + HC104 —> НООС(СН2)л CH2NH+C104
Методика определения. Навеску аминокислоты 0,03—0,04 г, взятую
на аналитических весах с точностью до 0,0002 г, переносят в стакан для
титрования емкостью 50 мл и растворяют в 25 мл безводной уксусной
кислоты при непрерывном перемешивании. Затем в стакан для
титрования опускают предварительно тщательно промытые дистиллированной
водой и спиртом и осушенные каломельный и стеклянный электроды.
В качестве титранта применяют 0,1 н. метилэтилкетоновый раствор
хлорной кислоты. Снимают показания прибора, прибавляя титрант по
0,1 мл. Затем строят кривую титрования в координатах Е (мв) —
V (мл) и определяют точку эквивалентности.
Расчет. Содержание (х) определяемой аминокислоты (А. К) в
процентах вычисляют по формуле:
ЭА. К^НСЮ/НСЮ, • 10°
Х ' 1000а
§ 18. Методы косвенного потенциометрического титрования
Определение виниловых мономеров (стирола, винилтолуола,
винилксилола)
Потенциометрический метод определения виниловых мономеров
основан на использовании реакции присоединения ацетата ртути (II) по
месту двойной связи виниловых мономеров в среде метилового спирта:
R—CH=CH2 + Hg(OCOCH8)2-h€H3OH —> R—CH-CH2 + CH3COOH
избыток | |
СНаО HgOCOCH3
Реакция идет с достаточной скоростью в присутствии
катализаторов — нитратов щелочных металлов. Избыток ацетата ртути связывают
в виде мало диссоциированного хлорида ртути (II):
Hg(OCOCH3)2 + 2СГ —> HgCl2 + 2СН3СОСГ
Уксусную кислоту, выделяющуюся при реакции в количестве,
эквивалентном содержанию мономера, титруют стандартным спиртовым
раствором щелочи.
Приготовление спиртового раствора ацетата ртути. В мерную колбу
емкостью 1 л переносят 24 г Hg(OCOCH3h, добавляют 2 мл ледяной
уксусной кислоты и доливают до метки 4,5 М метанолового раствора
LiN03. Содержание уксусной кислоты в растворе ацетата ртути
(холостой опыт) устанавливают методом потенциометрического титрования.
Для этого отбирают 20 мл раствора ацетата ртути, прибавляют 25 мл
2 М метанолового раствора LiCl и титруют 0,1 М метаноловым
раствором КОН (W).
Методика определения. Навеску анализируемого вещества 2—3 г,
взвешенную в закрытом бюксе на аналитических весах, переносят
в мерную колбу емкостью 100 мл, растворяют в метиловом спирте и
доводят тем же растворителем до метки. Точно калиброванной
пипеткой отбирают аликвотную часть (обычно 5 мл) полученного раствора
§ 18. МЕТОДЫ КОСВЕННОГО ПОТЕНЦИОМЕТРИЧЕСКОГО ТИТРОВАНИЯ 445
мономера и переносят в стакан для титрования. Затем в стакан вносят
20 мл приготовленного раствора ацетата ртути и закрывают часовым
стеклом.
Реакционную смесь перемешивают с помощью магнитной мешалки
в течение 10 мин. Затем, не прекращая перемешивания, в стакан
добавляют 25 мл 2 М метанолового раствора LiCl для связывания
избытка ацетата ртути. Сразу после этого в стакан опускают стеклянный и
каломельный электроды и титруют смесь 0,1 н. метаноловым раствором
КОН (V). Во время титрования смесь перемешивают магнитной
мешалкой.
Кривую титрования строят в координатах Е (мв) — V (#л).
Расчет. Содержание (х) виниловых мономеров в процентах
вычисляют по формуле:
Эв.мЛГкон(К-ПУПЮ
х 1000УАа
Анализ смесей первичных, вторичных и третичных аминов
Для анализа смесей первичных, вторичных и третичных аминов
применяют метод ацетилирования первичных и вторичных аминов уксусным
ангидридом и связывания первичных аминов салициловым альдегидом
или фталевым ангидридом. Фталевый ангидрид имеет ограниченное
применение, так как при повышенном содержании в анализируемых смесях
вторичных аминов фталевый ангидрид реагирует с ними, образуя
нейтральные соединения.
В последнее время в качестве ацетилирующего реагента применяют
пиромеллитдиангидрид в среде диметилсульфоксида. При
использовании этого реагента реакция идет быстрее. Для связывания первичных и
вторичных аминов применяют также пиридиновый раствор янтарного
ангидрида.
Большинство методов определения смесей первичных, вторичных и
третичных аминов основано на проведении трех титрований: 1)
сначала определяют суммарное содержание аминов титрованием смеси
неводным раствором хлорной или хлористоводородной кислот; 2) смесь
аминов обрабатывают уксусным ангидридом, при этом происходит аце-
тилироваиие первичных и вторичных аминов:
RNH2 + (СН3СО)20 —> RNHCOCH3 + СНзСООН
xNH + <CH3CO)20 —* xNCOCH3 + CH3COOH
R"/ WX
поскольку третичные амины не ацетилируются, содержание их
определяют титрованием кислотой; 3) смесь аминов обрабатывают
салициловым альдегидом, при этом первичные амины реагируют с образованием
оснований Шиффа:
RNH2 + ОСНС6Н4ОН —> RN = CHC6H4OH + H20
Наконец, титрованием кислотой определяют суммарное содержание
вторичных и третичных аминов.
При анализе смесей первичных, вторичных и третичных аминов
в качестве растворителей используют уксусную кислоту, гликоли,
спирты, кетоны и т. п. Для определения вторичных и третичных аминов
нельзя применять растворы хлорной и хлористоводородной кислот,
приготовленные на основе безводной уксусной кислоты, содержащей
446 ГЛ. XI. ФИЗИКО-ХИМИЧЕСКИЕ МЕТОДЫ АНАЛИЗА НЕВОДНЫХ РАСТВОРОВ
уксусный ангидрид, так как он реагирует с первичными и вторичными
аминами.
Методика определения. Навеску анализируемой смеси около 1 г,
взятую на аналитических весах, переносят в мерную колбу емкостью
50 мл, растворяют в смеси этиленгликоль — изопропиловый спирт (1:1)
и доводят этим же растворителем до метки. Для анализа используют
три аликвотные порции по 10 мл. Одну порцию переносят в стакан для
титрования, приливают 15—20 мл смеси этиленгликоль —
изопропиловый спирт (1:1) и титруют 0,1 н. раствором хлористоводородной или
хлорной кислот (V"), приготовленных в среде того же растворителя.
Вторую порцию обрабатывают 5—10 мл уксусного ангидрида, через
30 мин к пробе приливают смесь этиленгликоль — изопропиловый спирт
(1:1) и титруют раствором кислоты (V).
К третьей пробе прибавляют 3 мл салицилового альдегида и
выдерживают на водяной бане при 40—50° С в течение 10 мин, после
охлаждения приливают 15—20 мл растворителя и титруют 0,1 н. раствором
кислоты (V).
Расчет. Содержание (g) третичного амина (Т. А) (в г) вычисляют
по формуле:
Эт. А*нсю4ГГ.
g 1000 VA
Содержание (g) вторичного амина В. А (в г) вычисляют по
формуле:
g 1000 VA
Содержание (g) первичного амина П. А (в г) вычисляется по
формуле;
Эп.а*нс1о4(^-ПГ,
g IQ0QVA
Определение производных п^фенилендиамина в смеси
с п-аминодифениламином
Ряд производных типа RHNC6H4NHR синтезируют, исходя из /г-фе-
нилендиамина (ПФДА) H2NC6H4NH2, а некоторые производные — типа
RHNC6H4NHC6H5 — получают из м-аминодифениламина (ПАДФА)
H2NC6H4NHC6H5. Поэтому в процессе синтеза указанных соединений
очень важно следить за составом промежуточных продуктов,
содержащих смеси исходных и синтезируемых веществ.
Прямое, раздельное титрование смесей производных ПФДА или
ПАДФА с исходными продуктами невозможно ни в неводных, ни тем
более в водных растворах, поскольку указанные соединения ведут себя
как слабые однокислотные основания.
Однако, пользуясь тем, что ПАДФА при титровании в неводных
растворах ведет себя как первичный амин (титруется одна первичная
группа, в то время как вторичная не реагирует), а при титровании
производных ПФДА реагирует лишь вторичная аминогруппа, можно
дифференцированно оттитровать смесь указанных аминов.
Методика определения. Навеску анализируемого образца,
содержащую около 0,1—0,2 г производного ПФДА и от 0,1—0,2 до 0,01—0,02 г
ПАДФА, взвешенную на аналитических весах, помещают в стакан
емкостью 100 мл% наливают 10 мл ацетона к 3 мл салицилового альдегида
§ 19. ДИФФЕРЕНЦИРОВАННОЕ ПОТЕНЦИОМЕТРИЧЕСКОЕ ТИТРОВАНИЕ 447
и выдерживают на водяной бане при 40—50° С в течение 10 мин.
Реакция протекает согласно уравнению:
НОСбН4СНО + H2N—^""^-NH—<^"^ —>
—> НОС6Н4СН= N-^^~NH~<^~^ + Н20
Затем полученную смесь охлаждают, прибавляют к ней еще 15 мл
ацетона и титруют ацетоновом 0,1 н. раствором НСЮ4 потенциометриче-
ским методом.
Кривая титрования характеризуется наличием двух скачков.
Первый скачок соответствует нейтрализации производного ПФДА (V)> а
второй— нейтрализации основания Шиффа (V')> т. е. соответствует
содержанию ПАДФА.
Определение можно проводить также в среде других неводных
растворителей, например в метилэтилкетоне и смеси его с хлороформом
(4:1).
Расчет. Содержание (g) производного л-фенилендиамина (ПФДА)
в процентах вычисляют по формуле:
эПФДА^нсю/-Ю0
6 1000а
Содержание (g) n-аминодифениламина (АДФА) в процентах
вычисляют по формуле:
Эадфа^нсюЛ^-Ю'ЮО
g 1000a
§ 1Й. Дифференцированное потенциометрическое титрование
Анализ смеси аммониевых солей с аммиаком
Определение основано на взаимодействии в среде метиловый
спирт — ацетон анализируемой смеси с гидроокисью тетраэтиламмония
(ГТЭА):
NHj + NH3 + rtR4NOH —> NH4OH + NH3+R4N+ + R4NOH
избыток
Смесь оснований: избыток гидроокиси тетраэтиламмония (не
вошедшей в реакцию с компонентами анализируемой смеси), гидроокись
аммония, выделенная из аммониевой соли, и аммиак, содержащийся
в анализируемой смеси, потенциометрически титруют ацетоновым
раствором хлорной кислоты:
NH4OH + NH3 + R4NOH + 3HC104 —> 2NH4C104 + R4NC104 + 2H20
Методика определения. Навеску анализируемой смеси 0,03 г
взвешивают в маленьком бюксе на аналитических весах, переносят в стакан
для титрования, бюкс промывают 1—2 мл метилового спирта. После
растворения к полученному раствору добавляют избыточное количество
(Угтэа) стандартного бензольно-метанолового раствора гидроокиси
тетраэтиламмония (ГТЭА), доливают до 20 мл ацетоном.
Титрование проводят на установке (см. рис. 147), описанной выше.
В качестве титранта применяют 0,1 н. ацетоновый раствор хлорной
кислоты, которую прибавляют порциями по 0,1 мл. Во время титрования
записывают объем израсходованного титранта {мл) и показания йотен-
циометра (мв).
44S ГЛ. XI. ФИЗИКО-ХИМИЧЕСКИЕ МЕТОДЫ АНАЛИЗА НЕВОДНЫХ РАСТВОРОВ
Кривые титрования строят в координатах Е (мв) — V (мл); они
характеризуются двумя скачками: первый скачок титрования
соответствует титрованию избыточного количества ГТЭА (1/нсю4), второй —
титрованию суммарного количества гидроокиси аммония, выделенной из
нитрата аммония при его взаимодействии с ГТЭА, и свободного
аммиака, содержащего в анализируемой смеси (^нсю4)«
Расчет. Содержание (g) аммониевой соли (в г) вычисляют по
формуле
_ 9NH4An (^ГТЭА^ГТЭА "" ^НС1оДнСЮ4)
8 1000
или в пересчете на NH3, содержащийся в аммониевой соли (g'):
, _ ЭГ\ГН, (УУтЭА^ГТЭА ~~ ^НСЮ4^НСЮ4)
ъ 1000
Содержание (g) свободного аммиака (в г) вычисляют- по формуле:
ЭМН3^НС104^НСЮ4 ,
8 1000 8
Анализ смеси серной кислоты с бисульфатом натрия
Метод основан на прямохМ дифференцированном титровании
стандартным спиртовым раствором дифенилгуанидина (ДФГ) смеси серной
кислоты с бисульфатом натрия в среде смешанного растворителя
ацетон— этиленгликоль (2: 1). В этих условиях серная кислота
нейтрализуется последовательно по двум ступеням диссоциации. Кривая потен-
циометрического титрования характеризуется двумя скачками: первый
скачок соответствует нейтрализации ионов водорода, образующихся в
процессе диссоциации серной кислоты по первой ступени, второй —
нейтрализации ионов водорода, образующихся в процессе ее
диссоциации по второй ступени, т. е. бисульфат-ионов.
Процесс нейтрализации указанной смеси можно представить в виде
следующих уравнений реакций:
H2S04 + CH3COCH3 ^=± HSGJ + [СН3СОСН3] Н+
(C6H5NH2)C = NH + [СН3СОСН3] Н+ —* (C6H5NH2)C- NH2 + CH3COCH3
HSOJ + CH3COCH3 +± SOJ" + [СН3СОСН3] Н+
(C6H5NH2)C = NH + [СН3СОСН3] Н+ —* (C6H5NH2)C«NH2 + CH3COCH3
2(C6H5NH2)C = NH2 + S04~ —> [(C6H5NH2)C-NH2]2SOr
Если в растворе присутствует только серная кислота, на
титрование ионов водорода, образующихся в процессе ее диссоциации по
первой и второй ступени, расходуются равные объемы стандартного
раствора основания {V).
В случае титрования смеси H2SO4 + HSOJ на вторую стадию
титрования, разумеется, идет больший объем раствора основания (V").
Методика определения. Пробу анализируемого образца,
содержащую около 0,3—0,5 г исходного продукта, точно взвешивают на
аналитических весах, помещают в мерную колбу емкостью 50 мл, доливают
сначала немного этиленгликоля и затем смесь ацетона с этиленгликолем
(2:1) до метки. Д,ля титрования отбирают пипеткой аликвотную часть
исходного раствора и переносят в стакан для титрования. Затем добав-
$ 19. ДИФФЕРЕНЦИРОВАННОЕ ПОТЕНЦИОМЕТРИЧЕСКОЕ ТИТРОВАНИЕ 449
ляют тот же смешанный растворитель до объема 20 мл при
непрерывном перемешивании. Полученную смесь титруют 0,1 н. изопропаноло-
вым (метаноловым или этаноловым) раствором дифенилгуанидина.
Титрование проводят на установке (см. рис. 147), описанной выше.
Каломельный электрод заполняют насыщенным спиртовым раствором
хлорида калия.
Титрант добавляют порциями по 0,1 мл, а вблизи точки
эквивалентности по 0,02 мл. Кривую титрования строят в координатах Е (мв) —
V (мл).
Расчет. Содержание (х) серной кислоты в процентах вычисляют по
формуле:
ЭН25О^ДФГ2^.100
* 1000а
Содержание (xf) бисульфата в процентах вычисляют по формуле:
hsoj ДфГ '
Анализ сплава цинка с кадмием
Определение основано на растворении сплава, содержащего цинк и
кадмий, в концентрированной азотной или бромистоводородной кислоте:
Zn + Cd + 4HN03 + 5HN03 —> Zn(N03)2 4- Cd(N03)2 + 4N02 + HN03 + 4H20
избыток избыток
Полученный раствор упаривают, остаток растворяют в этиловом
спирте, смесь разбавляют ацетоном и титруют стандартным бензольно-
метаноловым раствором гидроокиси тетраэтиламмония:
Zn(N03)2 + Cd(N03)2 + HN03 + 5R4NOH —> Zn(OH)2 + Cd(OH)2 + 5R4NN03 + Н20
избыток
Методика определения. Навеску сплава около 0,03 г взвешивают
на аналитических весах, растворяют в азотной кислоте в стакане для
потенциометрического титрования, полученный раствор осторожно
упаривают на электрической плитке почти досуха, охлаждают и остаток
при перемешивании магнитной мешалкой растворяют в 10 мл ацетона.
Титрование проводят на установке (см. рис. 147), описанной выше.
В качестве титранта применяют 0,1 н. бензольно-метаноловый раствор
гидроокиси тетраэтиламмония (ГТЭА), который прибавляют порциями
по 0,1 мл из полумикробюретки.
Кривую титрования строят в координатах Е(мв) — V (мл), она
характеризуется тремя скачками.
Первый скачок соответствует титрованию избыточного количества
азотной кислоты (V), второй — титрованию нитрата цинка (V"),
третий— титрованию нитрата кадмия (V").
Расчет. Содержание (х) цинка в процентах вычисляют по формуле:
Э2п^ГТЭАУ'-100
Х ЮООа
Содержание (х) кадмия в процентах вычисляют по формуле:
*в ЮООа
450 ГЛ. XI. ФИЗИКО-ХИМИЧЕСКИЕ МЕТОДЫ АНАЛИЗА НЕВОДНЫХ РАСТВОРОВ
Анализ смеси $-аланина и гидрохлорида гистидина
Метод определения основан на титровании метилэтилкетоновым
раствором хлорной кислоты смеси аминокислот в среде смешанного
растворителя (безводная уксусная кислота — ацетонитрил), в котором
аминокислоты ведут себя как основания различной силы.
Методика определения. Навеску смеси 0,06—0,08 г, взятую на
аналитических весах с точностью до 0,0002 г, переносят в стакан для
титрования емкостью 50 мл и растворяют в 5 мл безводной уксусной
кислоты при перемешивании, добавляют 20 мл безводного ацетонитрила
и снова перемешивают. Затем опускают в раствор тщательно промытые
дистиллированной водой и спиртом и осушенные электроды
(каломельный и стеклянный). Перемешивая раствор магнитной мешалкой,
титруют его 0,1 н. метилэтилкетоновым раствором хлорной кислоты
порциями по 0,1 мл.
Кривую титрования строят в координатах Е (мв) — V (мл) и
определяют точки эквивалентности. Кривая титрования характеризуется
двумя скачками, первый из которых соответствует нейтрализации р-ала-
нина {V), второй — гидрохлорида гистидина (V"), который в среде
смешанного растворителя ведет себя как довольно слабое основание.
Расчет. Содержание (х) р-аланина (Ал) в процентах вычисляют
по формуле:
Х 1000а
Содержание (х) гидрохлорида гистидина (Г) в процентах в смеси
вычисляют по формуле:
Х 1000а
Титрование фт а левой, изофталевой и терефталевой кислот
Из трех изомеров фталевых кислот наиболее сильной кислотой
является фталевая кислота (К\ = 1,3 • 10~3; /С2 = 3,9 • 10~5). Константы
диссоциации изофталевой и терефталевой кислот примерно одинаковы
(К\ = 2,4 • 10*4 и /С2 = 2,5 • Ю-5), поэтому раздельное титрование смесей
этих кислот в воде невозможно.
Определение изо- и терефталевой кислот
Определение основано на потенциометрическом титровании 0,1 н.
спиртовым раствором КОН смеси изомеров в среде смешанного
растворителя метилэтилкетон — ацетонитрил (4: 1).
Титрование происходит ступенчато: сначала нейтрализуется первая
карбоксильная группа терефталевой кислоты, проявляющей себя в
смешанном растворителе более сильной кислотой по сравнению с
изофталевой:
HOOC—СбН4—СООН + КОН —> НООС—С6Н4—СООК + Н20
Затем нейтрализуются более слабые кислоты: изофталевая и вторая
карбоксильная группа терефталевой, проявляющие себя в неводной
среде как кислоты, равные по своей силе:
ноос~с6н4-~соок + кон —> коос—с6н4—соок + н2о
Методика определения. Для того чтобы избежать попадания воды
в титруемый раствор, водный насыщенный раствор КС1 в каломельном
электроде заменяют насыщенным метаноловым раствором КС1.
§ 19. ДИФФЕРЕНЦИРОВАННОЕ ПОТЕНЦИОМЕТРИЧЕСКОЕ ТИТРОВАНИЕ 451
В стакан для титрования наливают предварительно обезвоженный
метилэтилкетон * (20 мл) и ацетонитрил (5 мл), помещают навеску
анализируемой смеси около 0,05 а, взвешенную на аналитических весах, и
опускают стеклянный и каломельный электроды. Титруют медленно
0,1 н. спиртовым раствором КОН, титр которого устанавливают по
точной навеске бензойное кислоты в том же растворителе. По достижениц
первого скачка титрования (окончание нейтрализации первой
карбоксильной группы терефталевой кислоты), на что расходуется V титранта,
к титруемому раствору прибавляют 5 мл воды и снова титруют до
получения второго скачка титрования, соответствующего общей
кислотности анализируемого раствора.
По данным титрования строят график в координатах потенциал
(мв) — объем титранта (мл) и графически определяют точку
эквивалентности.
Рис. 153. Кривые потенциометрического титрования изомеров фталевой кислоты:
1 —смеси изо- и терефталевой кислот спиртовым раствором КОН в среде метилэтилкетон-
ацетонитрил; 2 — смеси фталевой, изо- и терефталевой кислот спиртовым раствором КОН
в среде метилэтилкетона; 3 — смеси фталевой, изо- и терефталевой кислот бензольно-метано-
ловым раствором (СгНбЬ NOH.
Кривую титрования строят в координатах Е (мв) — V (мл) и
графически находят точки эквивалентности.
График рекомендуется строить в процессе титрования, чтобы
проследить появление первого скачка титрования и момент добавления
воды.
Кривая титрования смеси изо- и терефталевой кислот представлена
на рис. 153 (кривая 1). Первый скачок соответствует нейтрализации
половины терефталевой кислоты — первой карбоксильной группы (V"),
а второй — совместной нейтрализации изофталевой и второй
карбоксильной группы терефталевой кислот (Vй).
Расчет. Содержание (х) терефталевой ТФ и изофталевой ИФ
кислот в процентах вычисляют по формулам:
_ Этф^кон-^-КЮ
*тф-
^иф-
1000а
ЭиФ^кон(у"-2Н100
~1000а
Определение фталевой, изофталевой и терефталевой кислот
Анализ смеси трех изомеров фталевой кислоты основан на
параллельном титровании двух навесок смеси кислот в среде метилэтилкетона:
* Метилэтилкетон и ацетонитрил предварительно нейтрализуют в присутствии
индикатора тимолового синего или проводят холостой опыт.
452 ГЛ. XI. ФИЗИКО-ХИМИЧЕСКИЕ МЕТОДЫ АНАЛИЗА НЕВОДНЫХ РАСТВОРОВ
одной навески — спиртовым раствором КОН и другой — бензольно-
метаноловым раствором (C2H5)4NOH.
Методика определения. В стакан для титрования наливают
предварительно обезвоженйый метилэтилкетон (20 мл), помещают навеску
анализируемой смеси около 0,07—0,08 г, взвешенную на аналитических
весах, и титруют 0,1 н. спиртовым раствором КОН. По достижении
первого скачка титрования к титруемому раствору прибавляют 5 мл воды
и снова титруют до получения второго скачка титрования. Другую
навеску растворяют в метилэтилкетоне (25 мл) и титруют 0,1 н. бензоль-
но-метаноловым раствором (C2H5)4NOH, не прибавляя воду в процессе
титрования.
По данным титрования вычерчивают две кривые (рис. 153,
кривая 2) в координатах Е (мв) — V (мл). Кривая 2 характеризует
процесс титрования анализируемой смеси 0,1 н. спиртовым раствором КОН,
а кривая 3— бензольно-метаноловым раствором (C2H5)4NOH (ГТЭА).
Первый скачок титрования на кривой 2 соответствует совместной
нейтрализации первых карбоксильных групп фталевой и терефталевой
кислот (V), а второй — совместной нейтрализации вторых
карбоксильных групп фталевой и терефталевой кислот и всей изофталевой кислоты
(V"). Общий расход титранта составляет V + Vй = Vkoh. На кривой
3 (см. рис. 153) первый скачок титрования соответствует
нейтрализации первой карбоксильной группы фталевой кислоты (Угтэа), второй —
совместной нейтрализации изо- и терефталевой кислот (Угтэа), а
третий — нейтрализации второй карбоксильной группы фталевой кислоты
(Wtsa).
Расчет. Содержание изофталевой кислоты (ИФ) в процентах
вычисляют по формуле:
Эиф^конКон-2Укон)^0
*ИФ 1000а!
Содержание фталевой (Ф) и терефталевой (ТФ) кислот в процентах
вычисляют по формуле:
, _, ч ЭфмКон-2^оН-100
(*ф + *тф) ШОО^
Содержание фталевой кислоты в процентах вычисляют по формуле:
Эф^гТЭА-^ГГЭА-lQQ
Хф 1000а2
Содержание терефталевой кислоты (ТФ) в процентах вычисляют
по разности:
*ТФ ~ \ХФ "^ *Тф) ~" ХФ
§ 20. Комбинированные методы потенциометрического
титрования
Анализ смеси нитрита и нитрата щелочных металлов комбинированным методом
потенциометрического титрования и ионного обмена
Определение основано на прямом потенциометрическом
титровании стандартным спиртовым раствором хлористоводородной кислоты
нитрита щелочного металла, проявляющего основные свойства в среде
метанола или этанола:
Ж)?+Шз + Н+-ЬСГ —> HN02 + NOJ + CF
§ 20. КОМБИНИРОВАННЫЕ МЕТОДЫ ПОТЕНЦИОМЕТРИЧЕСКОГО ТИТРОВАНИЯ 453
Затем проводят ионный обмен на анионите АВЧ7 в QH-форме нитрата,
являющегося в данной среде нейтральным соединением, и хлорида,
получающегося в процессе взаимодействия нитрита с
хлористоводородной кислотой:
HN02+N03 + Cr+3R—ОН —> H20+R—N02+R—C1+R—NO3+20H~
Ионы ОН~, образующиеся в результате ионного обмена, титруют
потенциометрически:
20НГ + 2Н+ —> 2Н20
Методика определения нитрита калия. Навеску анализируемого
вещества около 0,03 г взвешивают в маленьком бюксе на аналитических
весах, переносят в стакан для титрования, бюкс промывают
метиловым спиртом и полученный раствор доливают этим же спиртом до 20 мл.
Раствор при перемешивании титруют 0,1 н. метаноловым
раствором хлористоводородной кислоты. Заканчивают титрование тогда,
когда в процессе прибавления кислоты происходит резкое изменение
потенциала.
Кривую титрования строят в координатах Е (мв)— V (мл), она
характеризуется одним скачком.
Расчет. Содержание, (g) нитрита калия (в г) вычисляют по
формуле:
= 9kno?^hci^hci
8 moo
Методика определения нитрата калия. Полученную после
титрования смесь подвергают ионному обмену на анионите АВ-17 в ОН-фарме.
Для этого раствор пропускают через анионит со скоростью 4 капли
в 1 сек. После окончания реакции обмена анионит промывают малыми
порциями метилового спирта, заканчивают промывание, пропустив
через колонку 50 мл раствора спирта. Полученный раствор, содержащий
эквивалентное количеству С1~- и ЫОз-ионов количество ОН~-ионов,
получаемых в результате анионного обмена нитрата и хлорида, оттитро-
вывают стандартным метаноловым раствором хлористоводородной
кислоты потенциометрическим методом в тех же условиях, как и при
определении нитрита калия.
Кривую титрования строят в координатах Е (мв) — V (мл), она
характеризуется одним скачком.
Расчет. Содержание (g) нитрата калия (в г) вычисляют по
формуле:
_ ЭКЫО^НС1Г HCl~^HCl)
е 1000
где VHCi — объем стандартного раствора хлористоводородной кислоты,
израсходованной на титрование нитрата калия, мл;
V^ci ~~ объем стандартного раствора хлористоводородной кислоты,
израсходованной на титрование основания, выделенного в количестве, эквивалентном
содержанию нитрата и хлорида калия, мл.
Анализ смеси нитрата калия с кислотами комбинированным методом
потенциометрического титрования и ионного обмена
Определение основано на прямом потенциометрическом
титровании свободной кислоты стандартным метаноловым раствором едкого
кали в среде метанола:
ЫАп + КОН —* К+-ЬАгГ + Н30
454 ГЛ. XI. ФИЗИКО-ХИМИЧЕСКИЕ МЕТОДЫ АНАЛИЗА НЕВОДНЫХ РАСТВОРОВ
Затем проводят ионный обмен на катионите СДВ-3 в Н-форме
нитрата, содержащегося в анализируемой смеси наряду с анионами кислот
(Ап~) и проявляющего в данной среде нейтральные свойства:
2K+ + N03 + An" + 2R—H —> HN03 + HAn + 2R—K
Продукты ионного обмена титруют потенциометрически основа-"
нием:
HN03 + HAn + 2KOH —> KN03 + КАп + 2Н20
Методика определения кислоты. Навеску анализируемого вещества
около 0,03 г взвешивают на аналитических весах в маленьком бюксе,
переносят в стакан для титрования, приливают 10 мл метанола.
Перемешивая магнитной мешалкой, титруют 0,1 н. метаноловым раствором
едкого кали, который прибавляют порциями по 0,1 мл. Заканчивают
титрование, когда в процессе прибавления едкого кали происходит
резкое изменение потенциала. Кривую титрования строят в
координатах Е (мв) — V (мл), она характеризуется одним скачком.
Расчет. Содержание (g) кислоты (в г) вычисляют по формуле:
3hno3^koh^koh
8 1000
Методика определения нитрата калия. Полученную после
титрования смесь подвергают ионному обмену на катионите СДВ-3 в Н-форме.
Для этого раствор пропускают через катионит малыми порциями со
скоростью 2 капли в 1 сек. После окончания реакции обмена катионит
промывают малыми порциями метанола, заканчивают промывание,
пропустив через колонку 50 мл метилового спирта. Полученцый
раствор, содержащий эквивалентное свободной кислоте и ЫОз-ионам ко«
личество кислоты, оттитровывают стандартным метаноловым
раствором едкого кали. Титрование проводят в тех же условиях и на той
же установке, как и при определении свободной кислоты. Кривую
титрования строят в координатах Е (мв) — V (мл), она характеризуется
одним скачком титрования.
Расчет. Содержание (g)' нитрата калия (в г) вычисляют по
формуле:
^ экяо,%он (vкон - ^кон)
8 1000
объем стандартного раствора КОН, израсходованного на титрование
свободной кислоты, содержащейся в анализируемом образце, мл;
объем стандартного раствора КОН, израсходованного на титрование
кислоты, выделенной в результате ионного обмена в количестве,
эквивалентном свободной кислоте и NOJ-ионам, мл.
§ 21. Хронопотенциометрическое титрование
Анализ уксусного ангидрида и уксусной кислоты
в ацетилирующих смесях
Метод анализа смеси уксусного ангидрида и уксусной кислоты
основан на реакции ангидридов с первичными аминами (см. стр. 445).
Уксусный ангидрид реагирует с анилином согласно следующему
уравнению:
(СН3СО)20 + H2NC6H5 —> C6H5NHCOCH3 + CH3COOH
Ацетилирующую смесь обрабатывают определенным количеством
анилина. Избыток анилина обратно оттитровывают диоксановым рас-
У кон
§ 21, ХРОНОПОТЕНЦИОМЕТРИЧЕСКОЕ ТИТРОВАНИЕ
455
твором хлорной кислоты и определяют содержание уксусного
ангидрида (и одновременно количество уксусной кислоты, образовавшейся
из ангидрида). Титрованием метаноловым раствором едкого кали
устанавливают общее количество уксусной кислоты (содержащейся в
исходной смеси и выделившейся в результате взаимодействия ангидрида
с анилином). Содержание уксусной кислоты в исходной смеси
рассчитывают по разности.
Приготовление растворов. Стандартный раствор. В мерную колбу
емкостью 50 мл, содержащую около 1,5 мл метиленхлорида, помещают
500—800 мг (СН3СО)20 и 100—150 мг СН3СООН (взвешивают на
аналитических весах), затем приливают с помощью пипетки 10 мл
10%-ного раствора анилина в диоксане, смесь
и выдерживают 10 мин, после чего доводят
объем в колбе до метки метиловым
спиртом.
Раствор ацетилирующей смеси. В
мерную колбу емкостью 50 мл отбирают
пипеткой 1,5 мл ацетилирующей смеси, приливают
10 мл 10%-ного раствора анилина в
диоксане, тщательно перемешивают и
выдерживают 10 мин, после чего доводят объем в
колбе до метки метиловым спиртом.
Раствор для холостого опыта. В мерную
колбу емкостью 50 мл помещают 10 мл
10%-ного раствора анилина в диоксане и
доводят объем в колбе до метки метиловым
спиртом.
Растворы титрантов. 0,5 н. растворы тит-
рантов — диоксановый раствор хлорной
кислоты и метаноловыи раствор едкого кали — готовят, не устанавливая
точных концентраций.
Методика определения. Перед началом работы дают
стабилизироваться току титранта. Пока не начато титрование, титрант собирается
в резервуар для отбора титранта (см. рис. 150).
В стакан для титрования емкостью 50 мл помещают с помощью
пипетки испытуемый раствор и приливают растворитель. Ставят
стакан на столик магнитной мешалки под концом капилляра сосуда Ма-
риотта так, чтобы расстояние между концом капилляра и столиком
мешалки составляло 10 см, а капилляр находился ближе к левой
стенке стакана, на расстоянии 0,5 см от нее. Погружают в раствор
стеклянный электрод и электролитический мост и перемешивают содержимое
стакана. Включают ход диаграммной ленты самописца. Одновременно
с падением в анализируемый раствор первой капли титранта
замыкают ток в цепи, отмечая начало съемки. Дальнейшее титрование
протекает автоматически, в то же время вычерчивается кривая
титрования.
На полученной кривой находят конечную точку титрования. Для
этого к вертикальной части кривой проводят касательную и находят
середину отрезка касания. Расстояние от точки начала титрования до
точки конца титрования называют «длиной ленты» (рис. 154).
Для определения содержания уксусного ангидрида в
ацетилирующей смеси последовательно титруют описанным способом дшзксановым
раствором хлорной кислоты 10 мл стандартного раствора, 10 мл
раствора ацетилирующей смеси и 10 мл раствора для холостого опыта с
10 мл метанола.
тщательно взбалтывают
Длина ленты, мм
Рис. 154. Кривая
полуавтоматического хронопотенцио-
метрического безбюреточ-
ного титрования
метаноловым раствором КОН
уксусной кислоты в среде
метилового спирта.
456 ГЛ. XI. ФИЗИКО-ХИМИЧЕСКИЕ МЕТОДЫ АНАЛИЗА НЕВОДНЫХ РАСТВОРОВ
Для определения общего содержания уксусной кислоты
последовательно титруют метаноловым раствором едкого кали 10 мл
стандартного раствора и 10 мл раствора ацетилирующей смеси с 10 мл метанола.
Расчет. Содержание уксусного ангидрида (х) в процентах
вычисляют по формуле:
(l'o-l'o)VA-lOOOa
где аа — содержание (СН3СО)гО в Va мл стандартного раствора, мг;
—-^ЙГ — «титр миллиметра», выражаемый в мг (СНзСО^О на 1 мм;
10 10
^о» %' *1"~ Длина бумажной ленты при титровании анилина в холостом опыте в
стандартном и исследуемом растворах, мм;
VA~объем аликвотной пробы, мл;
а = Fp — навеска ацетилирующей смеси [V — объем ацетилирующей смеси, мл; р —
плотность ацетилирующей смеси (18°С), г/см*].
Расчет. Содержание (х) в ацетилирующей смеси уксусной кислоты
в процентах вычисляют по формуле:
*0 10 """ *0 ~а
Х~ 1000FAa
где ак — содержание уксусной кислоты в УА стандартного раствора, мг;
1оу h ~ длина бумажной ленты при титровании СНзСООН в стандартном и
исследуемом растворах, мм;
Эк и Эа — грамм-эквиваленты уксусной кислоты и уксусного ангидрида.
§ 22. Кондуктометрическое титрование
Определение анилина в среде безводной уксусной кислоты
Определение анилина основано на реакции нейтрализации его в
среде безводной уксусной кислоты, в которой усиливаются основные
свойства аминов. В процессе нейтрализации анилина
электропроводность раствора изменяется; в точке эквивалентности наблюдается
резкий скачок титрования, свидетельствующий об окончании
нейтрализации и появлении в титруемом растворе избытка титранта.
Реакция протекает согласно следующему уравнению:
C6H5NH2 + СНзСООН ^=± 'C6H5NH3 + CH3COO"
НС104 + СН3СООН ^=± СН3СООН+ + СЮ4
CH3COOHj + СН3СОО- —> 2СН3СООН
C6H5NH3+ClOJ —> C6H5NH3C10J
C6H5NH2 + HC104 —> C6H5NH2 • HCI04
Методика определения. Навеску анилина около 0,2 е, взятую на
аналитических весах, переносят в мерную колбу емкостью 50 мл,
растворяют в безводной уксусной кислоте и доводят тем же растворителем
до метки. Отбирают точно откалиброванной пипеткой аликвотную
часть раствора (10 мл), переносят в сосуд для титрования и добавляют
уксусной кислоты столько, чтобы платиновые электроды были полностью
покрыты раствором.
При титровании анилина, являющегося слабым основанием,
стрелку гальванометра устанавливают у начала шкалы (на 25—30 делений).
§ 22. КОНДУКТОМЕТРИЧЕСКОЕ ТИТРОВАНИЕ
457
После настройки схемы навеску анилина титруют 0,1 н. уксуснокислым
раствором НС104, приливая реагент при непрерывном перемешивании
по 1 мл, а вблизи точки эквивалентности — по 0,2 мл (V).
Кривую титрования строят в координатах удельная
электропроводность (ом~1 -см~1)—объем титранта (мл). Излом нач кондуктомет-
рической кривой соответствует точке эквивалентности.
Для определения основных примесей, содержащихся в уксусной
кислоте, проводят холостой опыт. В сосуд для титрования наливают
уксусную кислоту в количестве, равном взятому для титрования
навески анилина, и титруют уксуснокислым раствором НС104. Таким путем
определяют объем титранта (V), израсходованный на нейтрализацию
основных примесей.
По описанной методике можно определять различные производи
кые анилина, толуидины, пиридин и другие основания, рК которых
в воде не более 10.
Расчет. Содержание (g) анилина (в г) рассчитывают по формуле:
?нсю4(^НУк
8 1000FA
Определение п-фенилендиамина в среде ацетона
Определение n-фенилендиамина (ПФДА) основано на реакции
нейтрализации его n-толуолсульфокислотой (ПТСК) в среде ацетона.
В этих условиях происходит дифференцированное титрование
п-фенилендиамина по ступеням нейтрализации. При титровании до первой
точки эквивалентности электропроводность повышается, а до второй —
понижается, это объясняется тем, что образующаяся средняя соль
выпадает в осадок. При избытке титранта электропроводность снова
повышается.
Собирают установку с мостиком Уитсона по схеме, показанной на
рис. 10 (см. гл. III, § 8). Для работы используют электролитическую
ячейку, изображенную на рис. 12,6 (см. гл. III, § 7), с гладкими
платиновыми электродами площадью 4 см2, расположенными на
расстоянии 2 см друг от друга. Титрантом служит 0,3 н. ацетоновый раствор
я-толуолсульфиновой кислоты.
Методика определения. Навеску /г-фенилендиамина (1—3 мг-экв)
растворяют в 30 мл ацетона и переносят в электролитическую ячейку,
заполняя ее до расширенной части. Ячейку закрывают крышкой с
двумя отверстиями (для хлоркальциевой трубки и кончика полумикробю-
ретки). Полумикробюретку заполняют титрантом и устанавливают над
электролитической ячейкой. Определяют сопротивление раствора в
ячейке. Это измерение повторяют 3 раза. Затем из полумикробюретки
добавляют титрованный ацетоновый раствор п-толуолсульфокислоты
порциями по 0,2 мл. После добавления каждой порции титранта
раствор перемешивают при помощи магнитной мешалки и измеряют
сопротивление раствора. Измерения заканчивают тогда, когда
наблюдается уменьшение сопротивления раствора от избытка титранта. На
основании полученных величин сопротивлений (R) рассчитывают
электропроводность раствора (1/#).
Для повышения точности определения вводят поправочный
коэффициент на разбавление А = (Vq + V)/Vq и определяют
электропроводность раствора, приведенную к первоначальному объему (A'1/R).
После этого строят кондуктометрическую кривую в координатах
электропроводность раствора (ом~]*см~1)—объем титранта (мл).
458 ГЛ. XI. ФИЗИКО-ХИМИЧЕСКИЕ МЕТОДЫ АНАЛИЗА НЕВОДНЫХ РАСТВОРОВ
Кондуктометрическая кривая титрования n-фенилендиамина харак*
теризуется двумя изломами, соответствующими двум ступеням
нейтрализации /i-фенилендиамина. По кондуктометрической кривой
определяют объем я-толуолсульфокислоты, вступившей в реакцию до второго
излома.
Расчет, Содержание (g) ПФДА (в г) вычисляют по формуле:
эпфда^птск^птск
е 1000
§ 23. Хронокондуктометрическое титрование
Определение диэтиламингидро хлорид а в среде
смешанного растворителя
Определение основано на реакции вытеснения ионами натрия
диэтиламина из его соли:
(C2H5)2NH • НС1 + NaOH —> (C2H5)2NH + NaCl + Н20
или
(C2H5)2NH2 + ОН* —> (C2H5)2NH + Н20
Для хронокондуктометрического титрования в ацетоно-водной
среде используют описанную выше установку и электролитическую ячейку,
показанную на рис. 12,г (см. гл. III, § 8).
Для титрования веществ кислотного характера готовят 0,3 н. аце*
тоно-водный (1:1) раствор NaOH. Раствор щелочи должен быть по
возможности свободным от карбонатов. Концентрацию стандартного
раствора устанавливают по метиловому оранжевому и фенолфталеину.
Используя полуденные результаты, рассчитывают нормальность, соот*
еетствующую содержанию в растворе только NaOH.
Методика определения. В ячейку для титрования наливают 25 мл
раствора гидрохлорида диэтиламина и 25 мл ацетона, погружают
электроды и мешалку, включают мотор для вращения ячейки и
милливольтметр. Раствор перемешивают несколько минут для достижения
постоянной электропроводности и при помощи делителя напряжений стрелку
милливольтметра устанавливают в верхней части шкалы. Включают
регистрирующую часть милливольтметра и при нанесении второго
показания на ленту начинают подачу раствора NaOH. После окончания
титрования поднимают кронштейн с электродами и вынимают их из
ячейки. Электроды, мешалку и ячейку тщательно промывают
дистиллированной водой и проводят параллельное определение. На кондукто-
метрических кривых графическим методом устанавливают точки
эквивалентности, определяют количество интервалов между точками до
излома кондуктометрической кривой и берут среднее значение. Отсчет
начинают со второй точки, десятые доли интервала вблизи точки
эквивалентности определяют на глаз. Электропроводность сначала
понижается, а после точки эквивалентности быстро возрастает.
Кондуктометрическая кривая титрования диэтиламингидрохлорида
характеризуется одним изломом.
Расчет. Содержание (g) гидрохлорида диэтиламина ДЭАГХ (в г)
рассчитывают по формуле:
_ ГС^ДЭАГХ^ЦаОН^
g 1000*i
где п — количество интервалов между точками на кондуктометрической кривой до
излома;
t — время между записью показаний милливольтметра, сек;
§ 24. СПЕКТРОФОТОМЕТРИЧЕСКОЕ ТИТРОВАНИЕ
459
V — емкость мерной колбы, используемой для определения скорости истечения
стандартного раствора NaOH, мл;
ty— время наполнения мерной колбы раствором NaOH, сек.
Анализ смеси хлористоводородной, уксусной и борной кислот
Определение основано на последовательной нейтрализации кислот,
обладающих различной силой. В процессе нейтрализации сначала
нейтрализуется наиболее сильная кислота НС1, затем СН3СООН и, нако-
нец, наиболее слабая Н3В03.
В работе используют описанную выше установку и электролитиче-
скую ячейку изображенную на рис. 12, г (см. гл. III, § 8).
Для титрования готовят 0,3 н. ацетоно-водный раствор NaOH, по
возможности не содержащий карбонатов. Концентрацию раствора
NaOH устанавливают по метиловому оранжевому и фенолфталеину.
При определении НС1 и СИ3СООН в расчетах используют
нормальность, установленную по метиловому оранжевому, а при определении
Н3В03 — по фенолфталеину.
Методика определения. В ячейку для титрования наливают 25 мл
раствора смеси кислот и 25 мл ацетона, погружают электроды и
мешалку, включают мотор для вращения ячейки и милливольтметр.
Раствор несколько минут перемешивают для достижения постоянной
электропроводности. Стрелку милливольтметра при помощи делителя
напряжений устанавливают в верхней части шкалы. Включают
регистрирующую часть милливольтметра и при нанесении второго показания на
ленту начинают подачу раствора NaOH. Когда титрование закончится,
поднимают кронштейн с электродами и вынимают их из ванны. Элект->
роды, мешалку и ячейку тщательно промывают дистиллированной
водой и проводят параллельное определение.
Кондуктометрическая кривая титрования смеси НС1, СН3СООИ и
Н3В03 раствором NaOH имеет три излома. Сначала нейтрализуется
НС1, при этом электропроводность сильно понижается. Кривая титро^
вания до первой точки эквивалентности имеет слабый изгиб, что
объясняется уменьшением силы НС1 в ацетоно-водной среде. Затем
последовательно нейтрализуются СН3СООН и Н3В03; повышение
электропроводности раствора при нейтрализации СН3СООН более сильно
выражено, что приводит к появлению второго излома кондуктометриче-
ской кривой. При избытке щелочи наблюдается резкое повышение
электропроводности раствора. На кондуктометрических кривых
графическим методом устанавливают точки эквивалентности и определяют
количество интервалов между изломами. Отсчет начинают со второй
точки. Десятые доли интервалов вблизи точек эквивалентности
определяют на глаз.
Расчет. Содержание кислот в смеси определяют по формуле,
приведенной в § 21 (см. стр. 458). При этом в расчетную формулу вводят
число интервалов, соответствующих титрованию отдельных кислот, их
грамм-эквиваленты и нормальность NaOH, как указано выше, по
метиловому оранжевому или фенолфталеину.
§ 24. Спектрофотометрическое титрование
Титрование смеси п-нитробензойной и 4-нитро-2-амино-
бензойной кислот
Обычно для проведения спектрофотометрического титрования при-
менякгг спектрофотометр СФ-4, снабженный разборной крышкой кю-
ветной камеры. В крышке имеются отверстия для полумикробюретки
и механической мешалки.
460 ГЛ. XI. ФИЗИКО-ХИМИЧЕСКИЕ МЕТОДЫ АНАЛИЗА НЕВОДНЫХ РАСТВОРОВ
Приготовление раствора титранта. Металлический натрий 0,05—
0,07 г, тщательно очищенный от окиси, растворяют в колбе с притертой
пробкой в 100 ж л смеси абсолютного изопропилового и обезвоженного
метилового спиртов (2:1). После полного растворения натрия колбу
закрывают и содержимое колбы тщательно перемешивают. Затем
приблизительно определяют нормальность полученного раствора
титрованием его аликвотной части 0,01 н. раствором хлористоводородной
кислоты по метиловому красному. В случае, рели концентрация спиртового
раствора метилата натрия сильно отличается от 0,02 н., добавляют или
еще металлического натрия, или раствор разбавляют той же смесью
спиртов. Полученный раствор выдерживают в течение суток в колбе,
закрытой пробкой. За это время из раствора выпадает карбонат
натрия. Раствор осторожно сливают с осадка и точно устанавливают
нормальность титранта, титруя им навеску салициловой кислоты при Я =
= 330 нм таким же образом, как это описано ниже для смеси
кислот.
Методика определения. Навеску 0,15—0,17 г смеси /г-нитробензой-
ной (ПНБ) и 4-нитро-2-аминобензойной (НАБ) кислот, взвешенную на
аналитических весах, растворяют в колбе с притертой пробкой в
смеси изопропилового и метилового спиртов (2: 1). После полного
растворения кислот в растворителе пипеткой переносят 3 мл раствора в
кювету и добавляют 17 мл той же смеси спиртов. Кювету
устанавливают в кюветную камеру таким образом, чтобы пучок света проходил
ниже поверхности раствора. Кюветную камеру закрывают крышкой.
В бюретку наливают приготовленный спиртовой раствор алкоголята
натрия, кончик бюретки пропускают в отверстие в крышке так, чтобы
он не пересекал пучка света. Спустя 20 мин после включения прибора
приступают к титрованию. По шкале длин волн устанавливают длину
волны 460 нм, при закрытой шторке приводят стрелку гальванометра
к нулю. Если не наблюдается скольжения стрелки влево или вправо,
то можно приступить к работе. На шкале оптических плотностей
устанавливают исходную оптическую плотность. При непрерывном
перемешивании раствора из бюретки добавляют спиртовой раствор алкоголята
натрия порциями по 0,1—0,2 мл. После добавления каждой порции
титранта измеряют оптическую плотность раствора, устанавливая
стрелку гальванометра на нуль рукояткой барабана оптических
плотностей.
Для установления точки эквивалентности строят график в
координатах оптическая плотность — объем добавленного титранта {мл).
Пересечение прямолинейных ветвей графика дает положение точки
эквивалентности. Положение первого перегиба на кривой титрования
определяется содержанием /г-нитробензойной кислоты, второго — 4-нитро-
2-аминобензойной кислоты.
Расчет. Содержание (*)п-нитробензойной кислоты в процентах
вычисляют по формуле:
1000FAa
Содержание (х) 4-нитро-2-аминобензойной кислоты в процентах
вычисляют по формуле:
х=*
10(WAa
§ 24. СПЕКТРОФОТОМЕТРИЧЕСКОЕ ТИТРОВАНИЕ
461
Анализ смеси изомеров нитроанилина
Метод спектрофотометрического титрования дает возможность
определять в среде неводных растворителей слабые основания с
р/С(Н20) = 11 — 14, а также их двух-, трех- и четырехкомпонентные
смеси.
Для титрования используют спектрофотометр СФ-4. Стеклянную
кювету с толщиной слоя 17 мм устанавливают в кюветной камере так,
чтобы кончик бюретки и мешалка не попадали в световой пучок.
Определения проводят в видимой области спектра при одной
длине волны, при которой поглощают все анализируемые изомеры. Средой
для титрования служат: безводная уксусная кислота, ее смесь с
ацетоном или метилэтилкетоном (1:4). В качестве титранта применяют
0,04—0,08 н. раствор хлорной кислоты в том же растворителе, титр ее
устанавливают визуально (по кристаллическому фиолетовому), потен-
циометрически по бифталату калия или спектрофотометрически
поточной навеске ж-нитроанилина.
Методика определения. Навеску изомера (или смеси изомеров)
нитроанилина 0,0400—0,600 г, взятую на аналитических весах,
растворяют в смеси уксусная кислота — кетон (1:4). В кювету отбирают
8—10 мл (с точностью до 0,01 мл) полученного раствора и помещают
ее в кюветное отделение прибора. Раствор перемешивают и
регулировкой щели устанавливают начальную оптическую плотность.
В процессе определения титрант добавляют из полумикробюретки
порциями по 0,05—0,10 мл, раствор перемешивают и определяют его
оптическую плотность.
По результатам титрования строят график в косфдинатах
оптическая плотность раствора — объем добавленного титранта (Унсю4, мл).
Точку эквивалентности находят по излому кривой титрования.
Метод спектрофотометрического титрования может быть использо*
ван также для анализа двух-, трех- и четырехкомпонентных смесей:
анилина, о-толуидина, я-толуидина, о-анизидина с соответствующими
нитропроизводными изомерами.
Расчет. Содержание (g) основания (в г) вычисляют по формуле:
8 1000FA
Безындикаторное спектрофотометрическое титрование
в ультрафиолетовой области спектра солей ароматических карбоновых
кислот и их производных
Метод безындикаторного кислотно-основного
спектрофотометрического титрования основан на последовательном изменении светопогло-
щения раствора в процессе титрования вследствие различия в спектрах
поглощения молекулярной и ионной форм соединений.
В среде амфипротных растворителей — кетонах, нитрилах и смесях
их со спиртами —- прямым ацидиметрическим титрованием
определяются следующие соли:
Салицилат натрия (X = 340 нм) в среде 90%-ного ацетона.
Фенолят лития (к =» 315 нм) в среде этанола.
о-Нитробензоат калия (К = 340 нм) в среде 90%*-ного ацетона.
льНитробензоат кадия (К = 310 нм) в среде этанола.
о-Хлорбензоат калия (К = 290 нм) в среде этанола.
В качестве среды для титрования могут быть использованы: 90% -ный
ацетон, ацетонитрил. Титрант —0,02—0,1 н. этаноловый раствор хло-
462 ГЛ. XI. ФИЗИКО-ХИМИЧЕСКИЕ МЕТОДЫ АНАЛИЗА НЕВОДНЫХ РАСТВОРОВ
ристоводородной кислоты, титр его устанавливают либо потенциомет-
рическим методом по бифталату калия, либо спектрофотометрическим
методом по салицилату натрия при X = 340 или К = 320 нм.
Методика определения. Навеску определяемой соли (~0,001 г-экв)у
взвешенную на аналитических весах, растворяют в 50 мл этилового
спирта или 90%-ного ацетона, отбирают 1—5 мл раствора в кювету для
титрования, разбавляют до 20 мл соответствующим растворителем,
перемешивают и устанавливают начальную оптическую плотность. После
добавления каждой последующей порции титранта (0,05—0,10 мл)
раствор перемешивают и записывают оптическую плотность. Титрование
проводят без кюветы сравнения. В процессе титрования первоначала
ный объем не должен увеличиваться более чем на 5—10% (во избежав
ние ошибок, связанных с изменением оптической плотности вследствие
разбавления).
Кривую титрования строят в координатах оптическая плотность
раствора — объем добавленного титранта (мл). Точку эквивалентности
определяют по излому на кривой титрования.
Расчет. Содержание {g) соли (в г) вычисляют по формуле:
ЭСДГНС11/НС1^к
ё 1000FA
ПРЕДМЕТНЫЙ УКАЗАТЕЛЬ
Автопротолиз, константа 414 ел., 418, 424
Адипиновая кислота, титрование кондукто-
метрическое 83
Адсорбенты хроматографические 279
Адсорбция 276 ел.
изотерма 277 ел.,.
Азотная кислота
титрование кондуктометрическое с СеН5ОН
ПО
H2S04 93
NH4NO3 109
Активационный метод анализа 30, 354 ел.
нейтронный 356
Активность ионов, влияние на электродные
потенциалы 32
Э-Аланин, потенциометрическое титрование в
неводных растворах 450
Альдегиды
определение полярографическое 175
титрование амперометрическое 190
Алюминий, титрование кондуктометрическое 96
д-Аминодифениламин, потенциометрическое
титрование 446
Аминокислоты
разделение и определение 299
титрование потенциометрическое 443
Аминоуксусная кислота, титрование хроно-
кондуктометрическое 111
Амины
титрование амперометрическое 442
— высокочастотное 145
— потенциометрическое 445
Аммиак
титрование кондуктометрическое 108
— потенциометрическое 447
— хроноконду кто метрическое 111
Аммоний
титрование кондуктометрическое метанит-
рофенолята 86
нитрата 109
хлорида 109
— потенциометрическое 447
Амперометрическое титрование 26, 147, 177 ел.,
187 ел , 369 ел.
аппаратура 179 ел.
в неводных растворах 431 ел., 439 ел.
методика 182 ел.
необратимой системы 185
обратимой системы 185
по току определяемого вещества 177
по току определяемого вещества и тит-
ранта 178
редких элементов 369 ел.
с двумя индикаторными электродами
184 ел.
с индикатором 178
Амперостаты 215 ел.
Амфолиты 285 ел.
Анилин
титрование кондуктометрическое 108
в неводных растворах '456
в смеси с едким натром 108
— кулонометрическое 221
— потенциометрическое 443
— хронокондуктометрическое 111
Аниониты 285 ел.,
подготовка для анализа 307
Аннигиляция 324
Анодная реакция 194, 196
Арсениты
титрование кондуктометрическое 95
ь= спектрофотометрическое 268
Ассоциации константа 395
Атомно-абсорбционный спектральный анализ
224, 244
Барий
титрование высокочастотное 142
— кондуктометрическое 94, 96
соли, применение в кондуктометрическом
титровании 93
Бензальдегип. определение полярографическое
175
Бензойная кислота, титрование
потенциометрическое 443
Бензол, определение спектрофотометрическое
261
Бериллий
определение фотометрическое 372
— фотонейтронным методом 358
Биамперометрическое титрование 184 ел., 216
Бисульфаты, титрование потенциометрическое
448
Боксы для работы с радиоактивными
веществами 330
Бор, определение по поглощению нейтронов
366
Борная кислота
титрование кондуктометрическое 107
в смеси с гидрохлориДом гидроксил-
амина 109
малеиновой кислотой 84
уксусной кислотой 83
хлористоводородной кислотой
64, 82
титрование хронокондуктометрическое 459
"Бром, определение методом изотопного
разбавления 353
Бромиды
определение хроматографическое 315
титрование амперометрическое 441
— кондуктометрическое 93
Бугера — Ламберта ~~ Вера закон 245, 253, 271
отклонения 246
Бумага
гидрофобная 283
Хроматографическая 283
Бэр 327
Бэрад 327
Ванадий (IV), титрование амперометрическое
369
Весовой метод анализа, чувствительность 20
Вестона стандартный элемент 54
Вещества высокой частоты, анализ 20 ел.
Винилксилол, титрование потенциометрическое
444
Винилтолуол, титрование потенциометрическое
444
Винная кислота, титрование
кондуктометрическое 80
Висмут
обнаружение хроматографическое 306
определение фотометрическое 376 ел.
Вода, влияние на реакции титрования в
неводных растворах 427
Водород
перенапряжение на электроде 149
амальгамированном 155, 180
платиновом 153
Вольтамперное титрование см.,
Амперометрическое титрование
464
ПРЕДМЕТНЫЙ УКАЗАТЕЛЬ
Вольтметры 55
Вольфрам (VI), титрование потенциометриче-
ское 388
Высокочастотное титрование 27, 113 ел.,
138 ел.
аппаратура 129 ел.
в неводных растворах 436 ел.
ошибки 114, 116
Газ-носитель 279
Газы, хроматографический анализ 297 ел.
Галлий, определение экстракционно-фотомет-
рическое 380 ел.
Гальванометры 55, 97
Гальваностаты 215 ел.
Гаммета функция кислотности 421
Гейгера область 335
Гейгера — Мюллера счетчики 321, 334 ел.
измерение радиоактивности 341, 345
снятие характеристики 341
Гейровского электролизер 159
Гексацианоферрат (II)
обнаружение хроматографическое 307
титрование биамперометрическое 184
Генерация реагента 199, 208
Генри коэффициент 280
Германий, определение экстракционно-фото-
метрическое 381
Гетероконъюгации реакции 426
Гидроксиламина гидрохлорид, титрование
кондуктометрическое 109
Гистидина гидрохлорид, титрование потенцио-
метрическое 450
Глобар 257
Глутаровая кислота, титрование кондуктомет-
рическое 83
Гомоконъюгации реакции 426
Датчики см. Ячейки
Двойники см. Ионные пары
Дебая — Фалькенгагена теория 114
Детекторы
в хроматографии 292
ионизационные 334 ел.
сцинтилляциониые 338 ел.
Диаграммы соответствия 126 ел.
выбор 128
для титрования по активной
составляющей 126
реактивной составляющей 127
Диафрагма в спектральном анализе 233
Дисперсия
отклонения 324
электропроводности 114
Диссоциация, электролитов
в неводных растворителях 394 ел.
влияние диэлектрической проницаемости
404
константа 393, 400, 408 ел.
Дистанционные приспособления 331
Дистилляция 22
Диффузия
конвективная, теория 154
коэффициент 148
Дихлоруксусная кислота, титрование кондук-
тометрическое 79
Диэлектрическая проницаемость 27
влияние на диссоциацию электролитов 404
измерение 115 ел., 131
растворителей 393. 407, 417
Диэлкометрическое титрование 27, 115 ел.
ошибки 116
Диэтиламина гидрохлорид, титрование хроно-
кондуктометрическое 458
Добротность 123
Доза радиоактивного излучения
измерение 343
мощность 326, 327
поглощенная 326
предельно допустимая 327
экспозиционная 326
Дозаторы 291
Дозиметр с ионизационной камерой 343
Дуговой разряд 229
Дюбоска колориметр 252
Едкий натр
титрование кондуктометрическое 108
— хронокондуктометрическое 111
Емкость ячейки, метод измерения 115
Железо
обнаружение хроматографическое 306
определение кулонометрическое 216
— методом эмиссионного спектрального
анализа 238, 240
— хроматографическое 310
титрование высокочастотное 143
— кондуктометрическое 95 ел.
— потенциометрическое 65, 69
Закон(ы)
адсорбционного замещения 276
Бугера — Ламберта — Вера 245 ел., 253, 271
Ома 54
ослабления 323
поглощения света 245
Фарадея 191
Замещения адсорбционный закон 276
Защита от радиоактивности 331
Идентификация элементов 237
Излучение
интенсивность 228
поглощаемое, выбор 254
радиоактивное 318 ел.
взаимодействие с веществом 323
ионизационный эффект 334
отражение, методы определения 366 ел.
поглощение, методы определения
362 ел.
регистрация 334 ел., 339 ел.
характеристическое 244
Измайлова уравнение 393
Изотерма
адсорбции 277 ел.
ионного обмена 287
Изотопы радиоактивные 318 ел., 327
определение по излучению 30, 360 ел.
Изофталевая кислота, титрование
потенциометрическое 450 ел.
Ильковича уравнение 148, 177
Импеданс 118 ел.
определение 130
с-ячейки 118 ел.
L-ячейки 123 ел.
Индий
определение активационным методом 357
— полярографическое 172, 370 ел.
— флуориметрическое 388
титрование амперометрическое 369 ел.
Индикаторы электрометрические 55, 98, 186
Интеграторы тока 214
Интерметаллические соединения 167
Иод
определение методом изотопного
разбавления 353
— потенциометрическое 67
применение в кондуктометрическом
титровании 95
Иодиды
обнаружение хроматографическое 315
титрование кондуктометрическое 93
— потенциометрическое 68
Ионизация
вторичная 335.
первичная 334
Иоииты 285 ел.
классификация 285 ел.
подготовка для анализа 307
Ионное произведение растворителя 418, 424
Ионные пары 394 ел.
Ионный обмен
изотерма, уравнение 287
константа 286
Ионообменники 285
Искра конденсированная 230
Кадмий
определение в цинке 173
— полярографическое 170
— хроматографическое 298, 304
титрование амперометрическое 187, 440
— кондуктометрическое 96
— потенциометрическое 449
Калий
битартрат, титрование кондуктометриче-
ческое 87
определение по естественной
радиоактивности изотопов 361
— с применением 60 СО 347
соли, применение в амперометрическом
титровании 187
ПРЕДМЕТНЫЙ УКАЗАТЕЛЬ
465
Кальций
определение полярографическое 171
титрование кондуктометрическое 94, 96
Камеры
для бумажных хроматограмм 293 ел,
ионизационнные 334
Капилляр, характеристика 148
Карбооснования 403
Катарометр 292
Катиониты 285
Катод ртутный 164
Катодная реакция 194, 196
Кванты 319
Кинетический метод анализа 20
Кислород, полярографическая волна 152
Кислотно-основные реакции
в титровании биамперометрическом 186
высокочастотном 138 ел., 143
кондуктометрическом 79 ел., 87, 107,
109
— *-¦ кулонометрическом 199 ел., 206
потенциометрическом 39 ел., 42, 62 сл„
хронокондуктометрическом ПО ел.
Кислотности
функция Гаммета 421
шкала абсолютная 418 ел.
— единая 418 ел.
— относительная 40S, 411
положение 416
-— эмпирическая 411
— рА 415; 420
— рН 397
— рНр 414 ел.
Кислотность неводных растворов, оценка
414 ел.
Кислоты
диссоциация 395, 404
определение методом обращенных фаз 304
— спектрофотометрцческое 461
сила 392 ел., 404
зависимость от свойств растворителя
393
оценка по потенциалам
полунейтрализации 409
сродство к протону 399 ел.,
титрование в неводных растворителях
396 ел., 424, 438
— высокочастотное 138 ел.
— кондуктометрическое 79 ел., 107
— '—в смеси с солями 87, 109
— кулонометрическое 199 ел., 206
— потенциометрическое 39 ел., 42, 62 ел..
453
— хронокондуктометрическое 110 ел.
Классификация инструментальных методов
анализа 25
Ключ электролитический 57
Кобальт
обнаружение хроматографическое 306
радиоактивный изотоп, применение в
анализе 347 ел.
титрование кондуктометрическое 96
Колебания атомов молекул 247
Коллекторы 22
Колонки
двуступенчатые 291
для хроматографии адсорбцнонно-жидко-
стной 291
газовой 291
ионообменной 293
— •— окислительно-восстановительной 295
рсадочной 295. 314
капиллярные 291
приготовление 313
Колориметр (ы)
визуальные 256 ел.
Дюбоска 252
погружения 251 ел.
фотоэлектрические 256 ел.
Колориметр-нефелометр 271
Комбинирование методов анализа 23
Комплексон III
применение в титровании
кондуктометрическом 95
потенциометрическом 44
Комплексообразования реакции
при титровании амперометрическом 177
— — высокочастотном 143
— •-» кондуктометрическом 95
— *~ кулонометрическом 199. 204
потенциометрическом 43 ел., 61, 69
Комптон-эффект 324
Кондуктометр 99
Кондуктометрические методы анализа 76 ел.
Кондуктометрическое титрование 26, 72, 77 ел.,
103 ел.
аппаратура 96 ел.
в неводных растворах 434 ел., 456 ел*
методика 104
точность 79
Кондуктометрия 26г см. также
Кондуктометрическое титрование
аппаратура 96 ел.
бесконтактная 27
косвенная 77
прямая 76
Конетанта(ы)
автопротолиза 414 ел., 418, 424
ассоциации 395
диссоциации 393, 400, 408 ел.
ионного обмена 286
кислотности ионов лиония 419
— растворителя 419
нестойкости 395
основности ионов лиата 418 ел.
— основания 418
— растворителя 419
собственной кислотности 393, 399
— основности 393
сосуда 97, 99, 104
титрования 419
«с»-ячейки 121
«?»-ячейки 123
Контейнеры для радиоактивных веществ 330
Концентрация оптимальная, в спектрофото-
метрическом методе анализа 254
Концентрирование примесей 22
Конъюгации реакции 426
Коэффициент(ы)
Генри 280
диффузии 148
ослабления 323
поглощения, молярные 254
разбавления 100, 138
распределения 280, 282, 284
счета 344
титрования 419
электропроводности 75
Rf 284
Красители, анализ 303
Крезолы, определение 145
Кривая (ые)
выходная фронтального анализа 278
дисперсионная стилоскопа 235
полярографические 151
почернения 228
прямой потенциостатической кулономет-
рии 195
рабочая счетчика Гейгера — Мюллера 335
титрования амперометрического 182
— биамперометрического 184 ел.
— высокочастотного 115
— характеристическая 138
— кондуктометрического 78 ел., 83, 105
— кулонометрического 204 ел.
— нефелометрического 274
— потенциометрического 48 ел.
— спектрофотометрического 265 ел.
— турбидиметрического 274
— хронокондуктометрического 106
ток — потенциал 192
характеристическая фотопластинки 228
— «с»-ячейки 123, 125
— «L» -ячейки 119 ел.
электрокапиллярная 151
Критерий применимости ячейки 121, 123
Ксилолы, определение 248
Кулонометр(ы) 211 ел.
водородно-азотный J213
водяной 213-
газовые 212
галогено-серебряный 212
кислородно-водяной 213
колориметрические 213
кулонометрические 213
медный 212
серебряный 21?
титрационные 212
электрогравиметрические 212
электрохимические 211 ел.
Кулонометрическое титрование 26, 191, 198 ел.
амперостатическое 198 ел., 204 сл.;
аппаратура 208 ел.
при постоянной силе тока 198 ел., 219 ел.
при постоянном потенциале 207
реагенты 198
466
ПРЕДМЕТНЫЙ УКАЗАТЕЛЬ
Кулономётрическое титрование
с внешней генерацией 201
с внутренней генерацией 201
чувствительность 207
эффективность 203
Кулонометрия 26, 191 ел.
амперометрическая 192, 196 ел.
аппаратура 208 ел.
вторичная 191
гальваностатическая 192
косвенная см. КулОнометрическое
титрование
первичная 191
потенциосхатическая 192 ел.
при контролируемом потенциале 216 ел.,
при постоянной силе тока 196 ел., 218
при постоянном потенциале 192
прямая 192 ел., 216 ел.
реагент 200
чувствительность 207
Курнакова метод анализа 30
Кюри 320
Лантан, определение 96
Левина
теория конвективной диффузии 154
уравнение 155
Ленгмюра
изотермы адсорбции 278
теория 276 ел.
Лиата ионы 392
константа основности 418 ел.
Линии спектральные
аналитические, пары 225, 227 ел.
гомологические, пары 227
для визуального спектрального анализа
234
исчезновение, метод 226
интенсивность 224
«последние» 225
яркость 224
Лиония ионы 392
активность 415
константа кислотнбети 419
Литий
соли, применение в кондуктометрическбм
титровании 94
фенолят, титрование спектрофотометриче*
ское 461
Ломакина формула 224
Люминесцентный метод анализа 28, 224
Люминофоры 338
Магний
титрование высокочастотное 142
— кондуктометрическое 96
— радиометрическое 331
Максимумы на полярограммах 151 ел.
Малеиновая кислота
титрование кондуктометрическое 80, 84
— полярографическое 174
Манипуляторы 331
Марганец
определение спектрофотометрическое 262
титрование кондуктометрическое. 96
— потенциометрическое' 66
Мариотта сосуд 103
Масс-спектральный метод анализа 20, 30
Медь
обнаружение 306
определение кулонометрическое 218
*- методом хроматографии ионообменной
306, 308
осадочной 314
распределительной 298
— . тонкослойной 304
— полярографическое 170
титрование кондуктометрическое 94, 96
— радиометрическое 352
Метилэтилкетон, растворитель 396
F-Метры 130
но методу биений 131
рН-Метры 53, 61
Q-Метры 130
Q, F-Метры 131, 133
Z-Метры 130
Мешалки 56
Микрофотометр 233
Микрофотометрирсвание 228
Микроэлектроды 153
индикаторные 177
спектральный
Милливольтметры 55
Молекулярно-абсорбционйый
анализ 244 ел.
Молекулярные соединения 402
Молибден
определение спектрофотометрическое 259
— экстракционно-фотометрическое 379
Монохлоруксусная кислота, титрование конг
дуктометрическое 79
Монохром аторы 232, 246
с малой дисперсией 242
Мостик Уитстона 97
Мочевины гидрохлорид, титрование
кондуктометрическое 85
Напряжение, приборы для измерения 55
Насыщения ток 335
Натрий
определение методом спектрофотометрии
пламени 243
титрование кондуктометрическое ацегата
109
малеината 86
теллурата 86
хромата 86
— спектрофотометрическое салицилата 461
— хронокондуктометрическое ацетата 111
фенолята 111
хромат, применение в титровании кон-
дуктометрическом 94
Неводные растворители
амфипротные 398
амфотерные 398
апротонные 398 сл„
— диполярные 399
взаимодействие с растворенным вёщест*
вом 392
влияние на реакции титрования 424 ел*
свойства растворенного вещества 391
¦ силу электролита 393
выбор 422
дифференцирующее действие 403, 406 ел
диэлектрическая проницаемость 393, 407,
417
инертные 398
кислотно-основные свойства 418
кислые 392, 398
классификация по влиянию на силу
электролита 403
1 дифференцирующему действию 406
протонно-донорно-акцепторным
свойствам 398 ел.
методы анализа 391
нейтральные 398
нивелирующе-дифференцирующее
действие 422
нивелирующие 403
обезвоживание 438
основные 392, 398
очистка 438
применение 421 ел.
протогенные 392, 398, 407
протолитические 398, 401
протофильные 398, 407
смешанные 412
Неводные растворы, реакции титрования
396 ел., 424 ел.
Нейтрино 319
Нейтронный активационный метод анализа
20, 365 ел.
Нейтроны 319
поглощение, методы определения 365 ел,
Нернста штифт 257
Нефелометр 272
Нефелометрический метод анализа 28, 224,
270 ел.
Нефелометрия 28, 224, 270 ел.
Никель
определение полярографическое 170 ел.
— хроматографическое 311 ел, 316
титрование кондуктометрическое 96
Ниобий, определение хроматографическое
316
Нитраты
титрование амперометрическое 439 ел.
— потенциометрическое 452 ел.
Нитриты, титрование потенциометрическое
452 ел.
4-Нитро-2-аминобензойная кислота,
титрование спектрофотометрическое 459 ел*
Нитроанилин, титрование
спектрофотометрическое 461
ПРЕДМЕТНЫЙ УКАЗАТЕЛЬ
467
Нитробензоат калия, титрование
спектрофотометрическое 461
Нитробензойная кислота* титрование
спектрофотометрическое 459
Нитробензол, определение полярографическое
173
Носители 22
в хроматографии 282 ел.
Объем удерживаемый 279
Объемный метод анализа, чувствительность
20
Окисления — восстановления реакции
при титровании амперометрическом 177 ел.
— г- биамперометрическом 186
> кондуктометрическом 94
кулонометрическом 199, 205, 219 ел,
потенциометрическом 44 ел., 65, 70
Оксалаты, титрование кондуктометрическое 93
Олово, титрование спектрофотометрическое 269
Ома закон 54
Оптические методы анализа 27, 224 ел.
Ортоборная кислота, титрование
кондуктометрическое 79
Осадки, растворимость 92
Осаждения реакции
при титровании амперометрическом 177
биамперометрическом 186
в неводных растворах 430
высокочастотном 142
* кондуктометрическом 91
¦ кулонометрическом 199, 204, 222
¦ потенциометрическом 43, 61, 68
Основания
диссоциация 395, 404
константа основности 418
приготовление и установка растворов 439
сила 392, 404
зависимость от свойств растворителя
393
оценка по потенциалам
полунейтрализации 409
сродство к протону 399
титрование в неводных растворах 396, 424,
438
— высокочастотное 141 ел.
— кондуктометрическое 82, 84, 108
в смеси с солями 91
— кулоноМетрическое 199 ел., 206
— потенциометрическое 42
— хронокондуктометрическое 111
Осциллографы 97, 168
Осциллометрия 27
Осциллотитратор «Пунгор» 135
Осцилляции 155, 182
Отгонка 22
Отклонение
абсолютное статистическое 325
квадратичное 325
Ошибка(и)
измерения радиоактивности 324
систематические 324
случайные 324
средняя квадратичная 325
Пары ионные 394 ел.
Переключатели тока 56
Перманганат-ионы, обнаружение хроматогра-
фическое 306
Пигменты, разделение хроматографическое
295
о-Пиколиний уксуснокислый, титрование
кондуктометрическое 86
Пиридин, титрование амперометрическое 442
Пламя 229, 242
Платинирование электродов 103
серое 98
Плотность растворов оптическая 245
вычисление 251
Поглощения света
законы 245
полосы 247
Подвижность ионов 73
Позитроны 318
Полиметакриловая кислота
титрование кондуктометрическое 81 ел.,
107
— хронокондуктометрическое 110
Полосы поглощения характеристические 247
Полуволны потенциал 150
Полунейтрализации потенциал 408 ел.
Полураспада пепиод 320 сл4
Полуэлементы 35
Поляризация
измерение 115 ел.
электродов 53 ел.
Полярограммы
дифференциальные 158
максимумы 151
снятие 160 ел.
Полярографический анализ 147 ел.* 170 ел.,
370 ел.
аппаратура 156 ел.
метод добавок 1$2
— калибровочных кривых 162
— расчета 161
— стандарта 162
чувствительность 20, 153
Полярография 26 см. также
Полярографический анализ
амальгамная с накоплением 164, 167
дифференциальная 170
импульсная 170
осциллографическая 168 ел.
переменнотоковая 169
пленочная с накоплением 168
с применением каталитических волн 170
Полярографы 156 ел.
Полярометрический метод анализа 28
Полярометрическое титрование см.
Амперометрическое титрование
Постоянная
«с»-ячейки 121
«L»-ячейки 123
Потенциал (ы)
восстановления 150
выделения 150
зависимость от концентрации
(активности) ионов 32
нормальные 32
окислительно-восстановительные 35, 289
полуволны 150
полунейтрализации 408 ел.
предельный 35
равновесный 33
реальные 33
скачок 37, 41
смешанный 34
стандартные 32, 35, 38
формальные 33
Потенциометрическое титрование 27, 1Й2.
36 ел., 62 ел., 388 ел.
аппаратура 51 ел.
без тока 39 ел.
в неводных растворах 433, 442 ел.
дифференцированное 447 ел,
комбинированными методами 452 ел.
косвенными методами 444 ел.
прямое 442 ел.
некомпенсационный метод 49
определение редких элементов 388 ел.
под током 39, 50 ел.
с биметаллической парой электродов 49
с двумя поляризованными электродами 54
Потенциометрия 27, 32 ел.
аппаратура 51 ел.
прямая 36
Потенциометры 53
Потенциостаты 215 ел.
Почернение
линий аналитической пары 227 ел.
фотопластинки 228
плотность 227
Предэлект^лиз 193, 216
фона 217
Прерыватели тока 56
Примеси
концентрирование 22
определение 22
Присоединения продукты 401
Проницаемость
диэлектрическая см. Диэлектрическая
проницаемость
магнитная 27
Протоны 319
энергия сольватации 393
количественное выражение сродства 399
рА шкала 415, 420
рН шкала неводных растворителей 397
рНр шкала 414 ел.
Равновесие радиоактивное
вековое 322
подвижное 323
Рад 326
биологический эквивалент 327
468
ПРЕДМЕТНЫЙ УКАЗАТЕЛЬ
Радиоактивационный метод анализа 20
Радиоактивность 320
абсолютная 322
естественная изотопов 361
измерение 341 ел., 344
раствора 346
регистрируемая 321
удельная 320
Радиоактивные вещества 331
классификация 329
хранение 327, 329
Радиоактивные реагенты, применение для
определения ионов 346 ел.
Радиоактивный распад, виды 318 ел.
Радиометр
Луч-А 344
Тисе 343
Радиометрические методы анализа 29, 318 ел.,
327 ел., 345 ел.
Радиометрические установки 340 ел,
Б 340
Б-4 340
ПС-5М («Волна») 341
разрешающее время 342
Радиометрическое титрование 29, 348 ел.
Радиометрия 215
Разбавление 72
изотопное, метод 29, 352 ел.
коэффициент 100, 138
Распада скорость 319
Распределения коэффициент 280, 282. 284
Растворимость в неводных растворителях 431
Растворитель(и)
неводные см. Неводные растворители
неподвижный 281
подвижный 281
Растворы нулевые 259
Реагенты для титрования 23
Регенерация электродов 61
Ред-окс системы, необратимые и обратимые
33
Редкие элементы, физико-химические методы
определения 368 ел.
Рений
определение полярографическое 370
— фотометрическое 376
титрование потенциометрическое 389
Рентген 326
биологический эквивалент 327
Рентгеновские методы анализа
модифицированные 31
Рентгеновское излучение, методы
определения, поглощение 364
Рефрактометрический метод анализа 28
Роданиды
определение хроматографическое 306 ел.
титрование кондуктометрическое 93
Ртуть
металлическая, техника безопасности при
работе 163
обнаружение хроматографическое 306
определение хроматографическое 298
Рэлея уравнение 270
Ряды сорбционные 285
Света поглощение, законы 245
Светофильтры 242
Свинец
определение методом эмиссионного
спектрального анализа 238
амальгамной полярографии с
накоплением 166
— полярографическое 173
— хроматографическое 308
титрование амперометрическое 187
— кондуктометрическое 94, 96
— спектрофотометрическое 269
Селен, определение экстракционно-фотомет-
рическое 383
Селеновая кислота, титрование
кондуктометрическое 79
Семикарбазида гидрохлорид, титрование
кондуктометрическое 85
Серебро
нитрат, применение в кондуктометриче-
ском титровании 91
титрование кондуктометрическое 94
Серная кислота
титрование кондуктометрическое 79
с азотной кислотой 93
с малеиновой кислотой 83
с теллуровой кислотой 84
титрование потенциометрическое 448
Скандий, определение фотометрическое 373
Слой
граничный у электрода 154
диффузионный 154, 177
толщина в спектрофотометрическом ана«
лизе 254
электрический двойной 97, 148
Смолы ионообменные 285
Соли
диссоциация в неводных растворах 395
определение хроматографическое 308
титрование кондуктометрическое 84, 87,
96, 109
— "— в смеси с кислотами 87, 109
— хронокондуктометрическое 111
Сольватация протона, энергия 393
Сольваты 395
Соосадители 22
Соосаждение 22
Сопротивление растворов
активное 117
емкостное 113
измерение 96 ел.
полное 118
удельное 72
Сорастворители 428
Сорбент модифицированный 289
Спектр(ы)
возбуждения 229
инфракрасные 247
комбинационного рассеяния света 28. 224
лампы ртутной в видимой области 236
поглощения, молекулярные 244
фотографирование 237
Спектральные методы анализа 27, 224 ел.
Спектральный анализ 27, 224 ел.
визуальный 225
качественный 225
количественный 226
полуколичественный 226
смеси катионов группы сернистого
аммония 235 ел.
фотографический 225, 226
чувствительность 20
Спектрограммы, расшифровка 237
Спектрографы 231
Спектрометр инфракрасный 257 ел.
Спектроскопия 20, 28, 224
Спектрофотометрические методы титрования
265 ел.
аппаоатура 268
Спектрофотометрический анализ 28, 244 ел.,
386 ел.
аппаратура 255 ел.
в видимой области 262
в инфракрасной области 264
в неводных растворах 437, 459
безындикаторный 461
в ультрафиолетовой области 259
веществ, непоглощающих свет 249
веществ, поглощающих свет 249 ел.
дифференциальным методом^252
качественный 246 ел.
количественный 249 ел.
методом изменения толщины
поглощающего слоя 251
методом уравнения 251
определение редких элементов 386 ел.
по значению молярного коэффициента
поглощения 250
с помощью калибровочного графика 251
чувствительность 20
Спектрофотометрия пламени 27 ел., 224,
241 ел.
аппаратура 242
Спектрофотометры фотоэлектрические 255 ел.
Сравнения элемент 227
Сродство к протону 399 ел.
Стандарта внутреннего метод 227
Стилометры 225
Стилоскоп 225, 231
дисперсионная кривая 235
калибровка по длине волны 234
Стирол, титрование потенциометрическое 444
Стронций, титрование кондуктометрическое
94, 96
Сульфаты
определение турбидиметрическое 273
титрование амперометрическое 188
— высокочастотное 144
— кондуктометрическое 93
Сульфиды, обнаружение хроматографическое
306 сл4
ПРЕДМЕТНЫЙ УКАЗАТЕЛЬ
469
Сурьма (НГ), определение потенциометриче-
ское 70
Сциитилляционный метод регистрации
радиоактивных излучений 338
Счета скорость 321
коэффициент 344
Счетчик(и)
Гейгера — Мюллера 321, 334 ел., 341, 345
иесамогасящиеся 336
плато 335
пропорциональный 334
самогасящиеся 336
сциитилляционный 321, 342
Таллий
концентрирование 387
определение полярографическое 371
— спектрофотометрическое 386
Тантал
определение спектрофотометрическое 386
— хроматографическое 316
Тартраты, титрование кондуктометрическое 93
Теллур, определение экстракционно-фотомет-
рическое 384 ел.
Теллуровая кислота, титрование
кондуктометрическое 84
Терефталевая кислота, титрование потенцио-
метрическое 450 ел.
Термический анализ 30
Тетраэтиламмония гидроокись, титрант 306
Титан
определение спектрофотометрическое 250
— фотометрическое 374
Титр неводных растворов кислот 439
Титранты для титрования кислот и
оснований в неводных растворах 438 ел.
Титраторы высокочастотные 131 ел.
правила работы 137 ел.
Титрование
дифференцированное 24, 405, 413, 447
инструментальные методы 23
конечная точка 24
коэффициент 419
раздельное 24
Титрометры 78
Ток
активный 117
диффузионный 26, 147
на микроэлектродах 153
средний 148
заряжения 152
конденсаторный 152
насыщения 335
остаточный 153, 194
постоянный, источники 54
предельный 147, 154, 177
площадка 194
реактивный 117
эффективность 193, 201
Толуидин, титрование потенциометрическое
442
«-ТолуолсульфоКйслота, титрант 396
Торий
определение фотометрическое 373
— экстракционно-фотометрическое 378
титрование кондуктометрическое 96
Точность определения 20, 325
повышение 22
Триэтаноламина гидрохлорид, титрование
кондуктометрическое 85
Турбидйметрический метод анализа 270 ел.
аппаратура 271
Турбидиметрия 28, 224, 270 ел.
Углеводороды
акцепторные свойства 402
"газообразные, анализ хроматографйческий
298
доиорные свойства 402
Удерживания время 279
У иг стона мостик 97
Уксусная кислота
безводная 392
диэлектрическая проницаемость 394
титрование кондуктбметрическое 79, 83
— потенциометрическое 62
— хронокондуктометрическое 459
— хронолотенциометрическое 454
Уксусный ангидрид, титрование хронопотсн-
циометрическое 454
Ультразвуковые методы анализа 31
Умножители фотоэлектронные 338
Уран, определение
экстракционно-фотометрическое 378
Фаз(ы)
неподвижная 282
обращенные 283
подвижная 279, 282
Фарадея
закон 191
экран 125
/г-Фенилендиамин и его производные,
титрование потенцио~метрическое 446, 457
Фенол
титрование высокочастотное 145
— хронокондуктометрическое 110
Физико-химический анализ no H. С. Курна-
кову 30
Флуоресцентный метод анализа 28, 224
Флуоресценция 28
Флуориметрический метод 20, 388 ел.
Фон
в ам пирометрическом титровайии 183
в полярографии 148
предэлектролиз 217
радиоактивный, измерение 342
Формальдегид, определение
полярографическое 176
Фосфаты
определение методом радиометрического
титрования 350
радиоактивный, использование в
радиометрическом анализе 347
Фосфорная кислота, титрование
кондуктометрическое 80 ел.
Фосфоры 338
Фотоактивационный анализ 356
Фотоколориметрический анализ 28, 244 ел*
Фотометр универсальный 256 ел.
Фотометрические методы анализа 216, 372 ел.
см. также Спектрофотометрический
анализ
Фотометрические методы титрования см.
СпектрофотСметрические методы
титровайия
Фотометрия пламени см. Спектрофотометрия
пламени
Фотон 335
Фотонейтронный метод анализа 30, 357 ел.
Фотонефелометрическое титрование 273
Фотопластинка
плотность почернения 227 ел.
характеристическая кривая 228
Фототурбидиметрическое титрование 273
Фотоэффект 324
Фталевая кислота, титрование
потенциометрическое 450
Фумаровая кислота, определение полярогра-
фическое 174
Хингидрон 59
Хлор, определение методом изотопного
разбавления 353
Хлорбензоат калия, титрование
спектрофотометрическое 461
Хлориды
определение нефелометрическое 272
— потенциометрическое 68
титрование амперометрическое 441
— высокочастотное 144
— кондуктометрическое 93
Хлористоводородная кислота 396
определение потенциометрическое 64
приготовление и установка растворов 439
титрование кондуктометрическое 81 ел., 86,
107
с НзВОз 82
с Н2Сг04 и СНзСООН 83
с полиметакриловой кислотой НО
титрование кулонометрйческое 219
— хронокондуктометрическое 110, 459
Хлорная кислота 396
приготовление и установка раствора 439
Хром
определение спектрофотометрическое 262
титрование амперометрическое 189
— спектрофотометрическое 269
Хроматермография 279 ел.
Хроматогпаммы
йдсорбциойно-комплексообразовательные
290
бумажные 293
восходящие 284
470
ПРЕДМЕТНЫЙ УКАЗАТЕЛЬ
Хроматограммы
двумерные 284
колоночные 292
круговые 284
нисходящие 284
одномерные 284
окислительно-восстановительные 289
осадочные 287
смешанного типа 276
электрофоретические 284
Хроматографические методы анализа 28 ел.,
275 ел.
аппаратура 291 ел.
Хроматография
адсорбционная 276 ел., 282 ел.
газовая 29, 295
жидкостная 28, 291, 295
адсорбционно-комплексообразовательная
29, 276, 289, 316
бумажная 29, 283
вытеснительная 284
газо-адсорбционная 279 ел., 297
газо-жидкостная 29, 276, 279 ел., 298
газовая 276, 279, 291
аналитическая реакционная 281
теплодинамический метод 279
жидкостная 276
ионообменная 29, 276, 284 сл„ 293, 305 ел.
капиллярная 276, 279, 281
колоночная 276
окислительно-восстановительная 29, 276,
288, 295, 315 ел.
осадочная 29, 276, 287, 295, 312, 314
плоскостная 276
равновесная 280
распределительная 29, 276, 281 ел., 298 ел,
теплодинамический метод 281
тонкослойная 29, 283
фронтальная 284
элюентная 284
Хроматографы газовые 291 ел.
Хроматы
обнаружение хроматографическое 306
титрование амперометрическое 188
— кондуктометрическое 93
Хромовая кислота, титрование
кондуктометрическое 80, 83
Хронокондуктометрическое титрование 27, 78,
102 ел., ПО ел.
в неводных растворах 458 ел.
методика 105 сл„
электроды 102
Хронопотенциометрическое титрование в
неводных растворах 434 ел., 454 ел*
Церий (IV), титрование кулонометрическое
222
Циклогексанон, определение спектрофотомет-
рическое 264
Цинк
определение хроматографическое, адсорб-
ционно-комплексообразовательное
316
— на анионите 311
г— катионите 310
титрование амперометрическое 187
— кондуктометрическое 96
— кулонометрическое 222
— полярографическое 170
¦— потенциометрическое 449
— радиометрическое 352
Цирконий, определение фотометрическое 375
Цитраты, титрование кондуктометрическое 93
а-Частицы 323
З-Частицы 319
Чернь платиновая 35
Чувствительность определения 22
Шевчика уравнение 165
Шевчика — Рендлса уравнение 169
Шкала кислотности
абсолютная 418 ел.
единая 418 сл„
относительная 408, 411
относительное положение 416
эмпирическая 411
рА 415, 420
рН 397
рНр414 ел.
Шкафы для радиоактивных веществ 330
Штативы для спектрального анализа 233
Штифт Нернста 257
Щипцы 331
Экран(ы) 330
Фарадея 125
Экранирование электродов ИЗ
Экстракционно-фотометрические методы
анализа 378 ел.
Экстракция 22
Электровесовой метод анализа 26, 194
Электрогравиметрический метод анализа 26.
194
Электрод (ы)
амальгамированные твердые 155, 180
водородный 58
нормальный 35
потенциалы 41
вольфрамовый 180
вращающиеся 153, 155, 180
вспомогательные 208 ел.
второго рода 57
генераторный 208
графитовый 62, 180, 208
для амперометрического титрования
179 ел.
для кулонометрии 208
для полярографического метода анализа
147
для потенциометрического титрования 57
для спектрального анализа 233, 237
для хронокондуктометрического
титрования 102
золотой 180, 208
индикаторные 58, 168. 181
поляризующийся 26
каломельные 58, 149, 160, 182
мембранные 61
меркуриодидный 58, 160, 182
меркурсульфатный 58, 160, 182
металлические 208
обращение с ними 61
пары 50
первого рода 57
платинированные 98, 103
платиновые 153. 155, 181, 208
платинированный 35
поляризация 49, 53 ел
рабочий 208
регенерация 61
ртутные 155, 208
капельные 149, 153, 164, 168, 1?0 ел.
серебряный 208
сравнения 35, 58, 160, 209
неполяризующиеся 26
стационарная капля 167 ел.
стационарные 153
стеклянный 53, 58, 60
сурьмяные 59 ел.
танталовый 180
третьего рода 57
хингидронный 58 ел.
хлорсеребряный 58, 160, 182
Электродвижущая сила, измерение 51 ел.
Электролиз внутренний, метод 26
длительность 165
Электролизеры 159, 209 ел.
Электролиты
реакции титрования 424 ел.
сила, влияние растворителей 393, 397 ел.
Электронный парамагнитный резонанс, метод
(ЭПР) 31
Электроны 318
Электропроводность растворов 27, 72
активная 130
влияние растворителя и электролита 74 ел.
— температуры 76
— фона 148
дисперсия 114
измерение 96 ел*
коэффициент 75
удельная 72, 99
эквивалентная 72 ел., 114
предельная 73
Электрохимические методы анализа 25 ел*
Элюент 284
Элюирование градиентное 285
Эмиссионный спектральный анализ 27, 224 ел.,
234 ел.
аппаратура 229 ел-,
точность 21
ПРЕДМЕТНЫЙ
Эффект
антенный 137
ионизационный 334
Эффективность тока 193
Ядерная гамма-резонансная спектроскопия,
метод (ЯГР) 31
Ядерный магнитный резонанс, метод (ЯМР)
31
Ядро атома 319
Янтарная кислота, титрование кондуктомет-
рическое 83
Ячейки
для высокочастотного титрования 116 ел.,
128 ел.
УКАЗАТЕЛЬ 471
Ячейки
для кондуктометрии 97, 100
для кулонометрии 209 ел.
для полярографии 158
для потенциометрий 51, 56, 63
емкостные ПС, 118, 121, 128
емкость, метод измерения 115
измерительные 116 ел.
индуктивные 116, 122 ел., 128
критерий применимости 121, 123
проводимость полная 118 ел.
«с»-типа 116, 118. 121, 128
чувствительность 121
«1»-типа 116, 122 ел., 128
чувствительность 126
электролитические 100 ел., 158 ел.
эффект антенный 137
АНАТОЛИЙ ПАВЛОВИЧ КРЕШКОВ
ОСНОВЫ АНАЛИТИЧЕСКОЙ ХИМИИ
Издательство „Химия", М., 1970 г.
472 с. УДК 54/.S (075.8)
Редактор Л. Н. Овсяникова
Технический редактор В. В. Коган
Художник А. Ф. Серебряков
Корректоры С. Л. Федотова, Г. А Артемова
Т-11099. Подписано к печати 4/VIII 1970 г. Формат бумаги 70Xl08Vie.
Печ. л. 29,5. Усл. печ. л. 41,3. Уч.-изд. л. 38,43. Тираж 70000 экз.
Типогр. бум. № 2. Цена 1 р. 53 к. Тем. пл. 1970 г., № 17. Зак. № 659.
Ордена Трудового Красного Знамени
Ленинградская типография № 2 имени Евгении Соколовой
Главполиграфпрома Комитета по печати при Совете Министров
СССР. Ленинград, Измайловский проспект, 29.