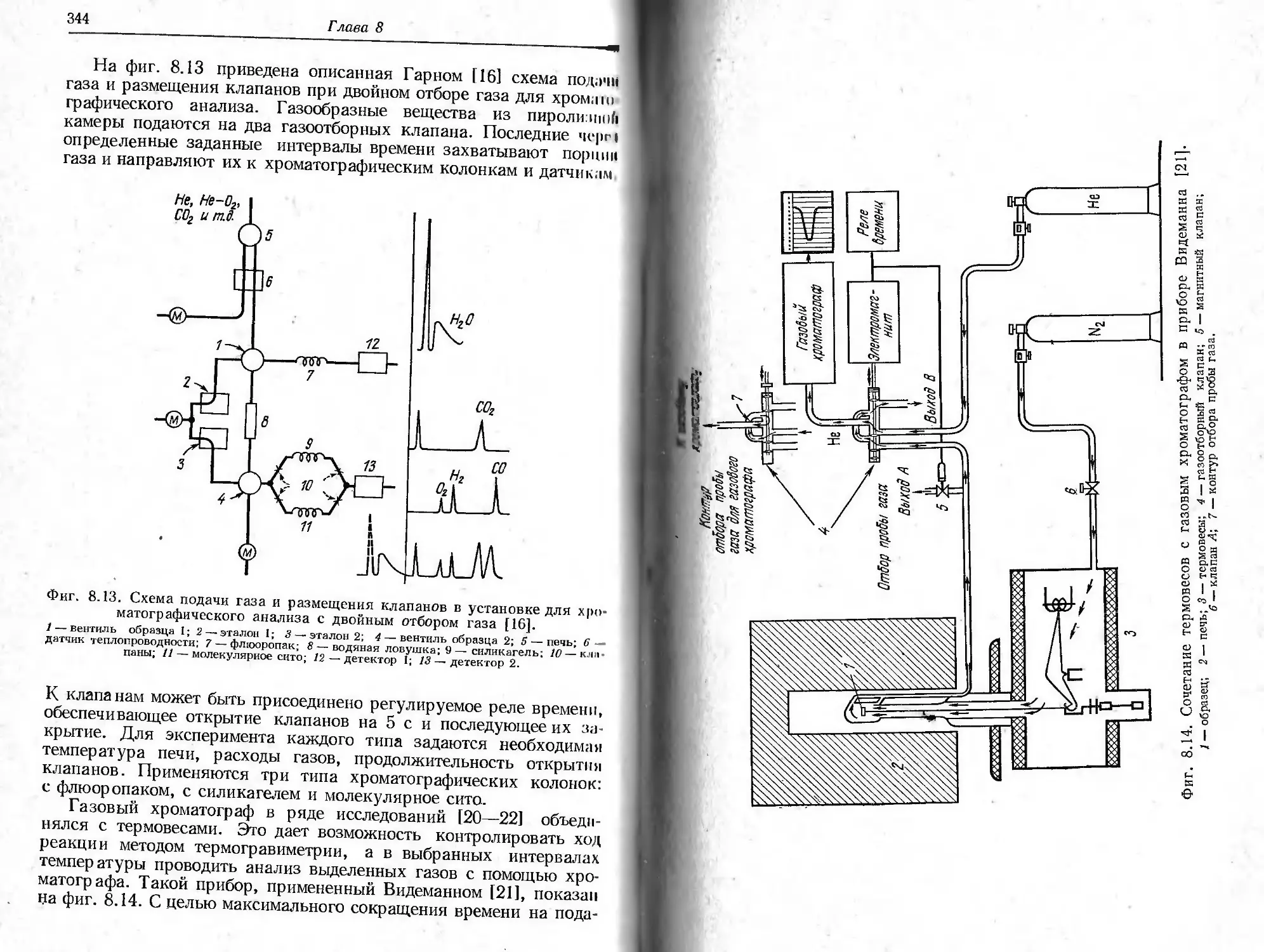
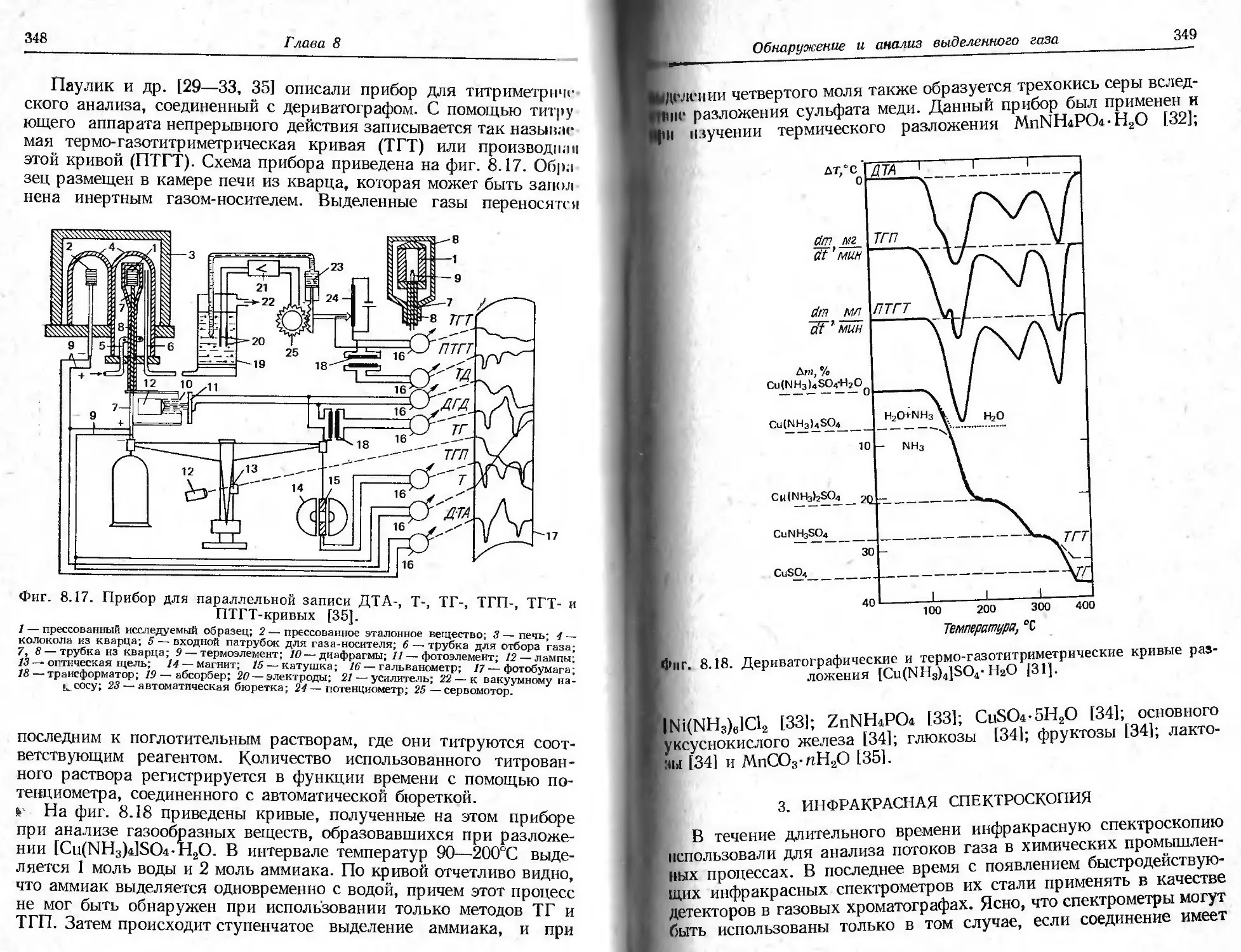
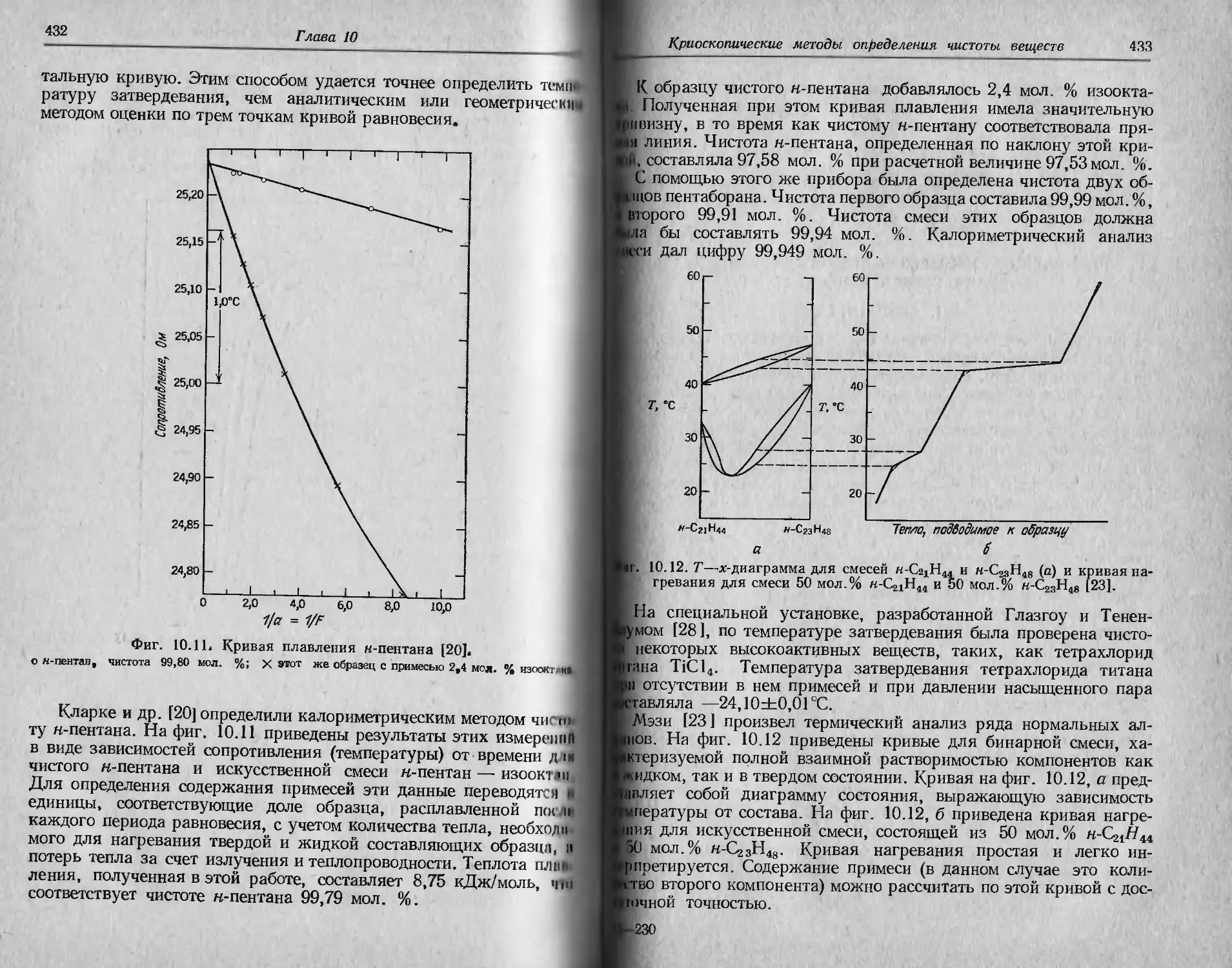
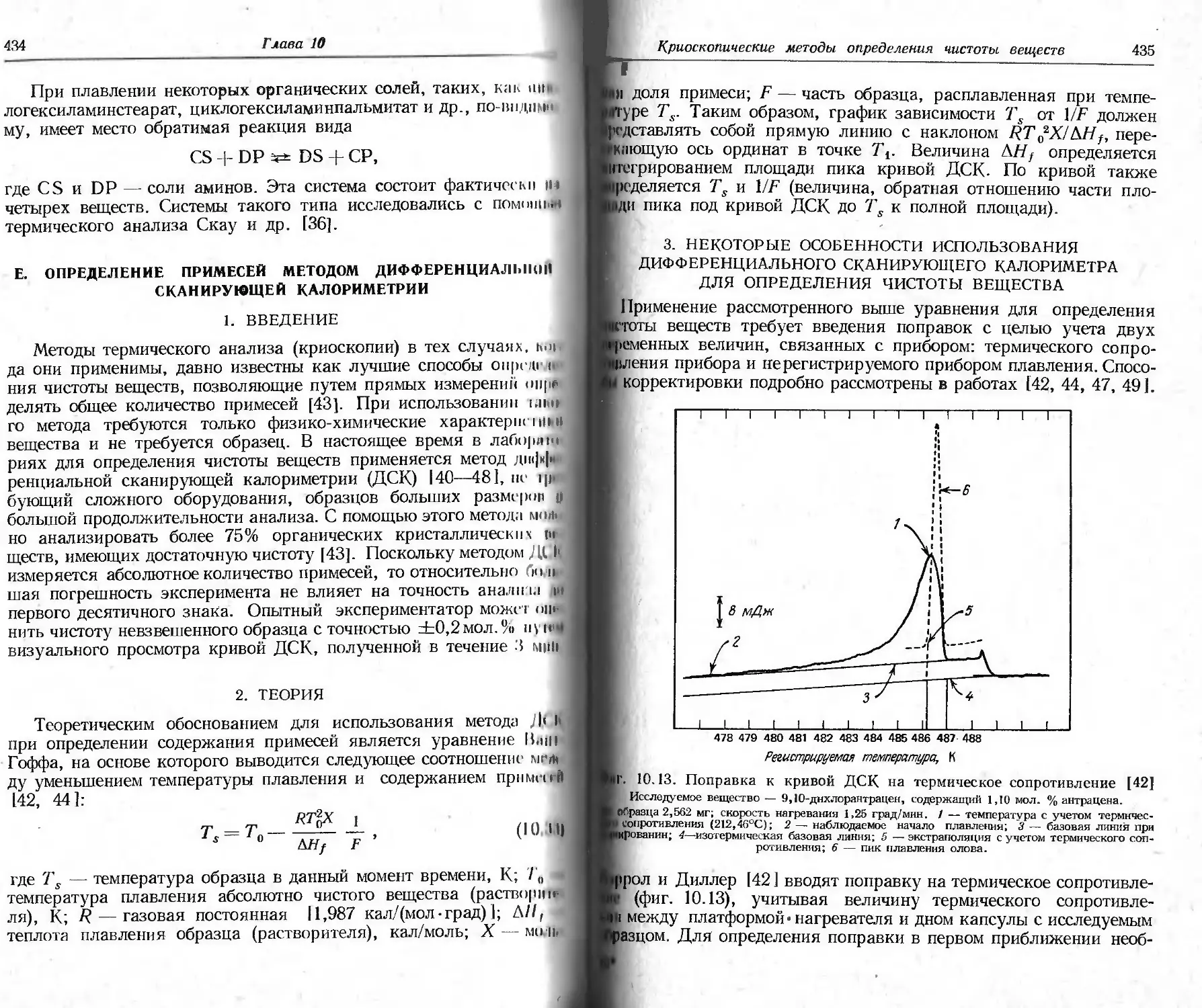
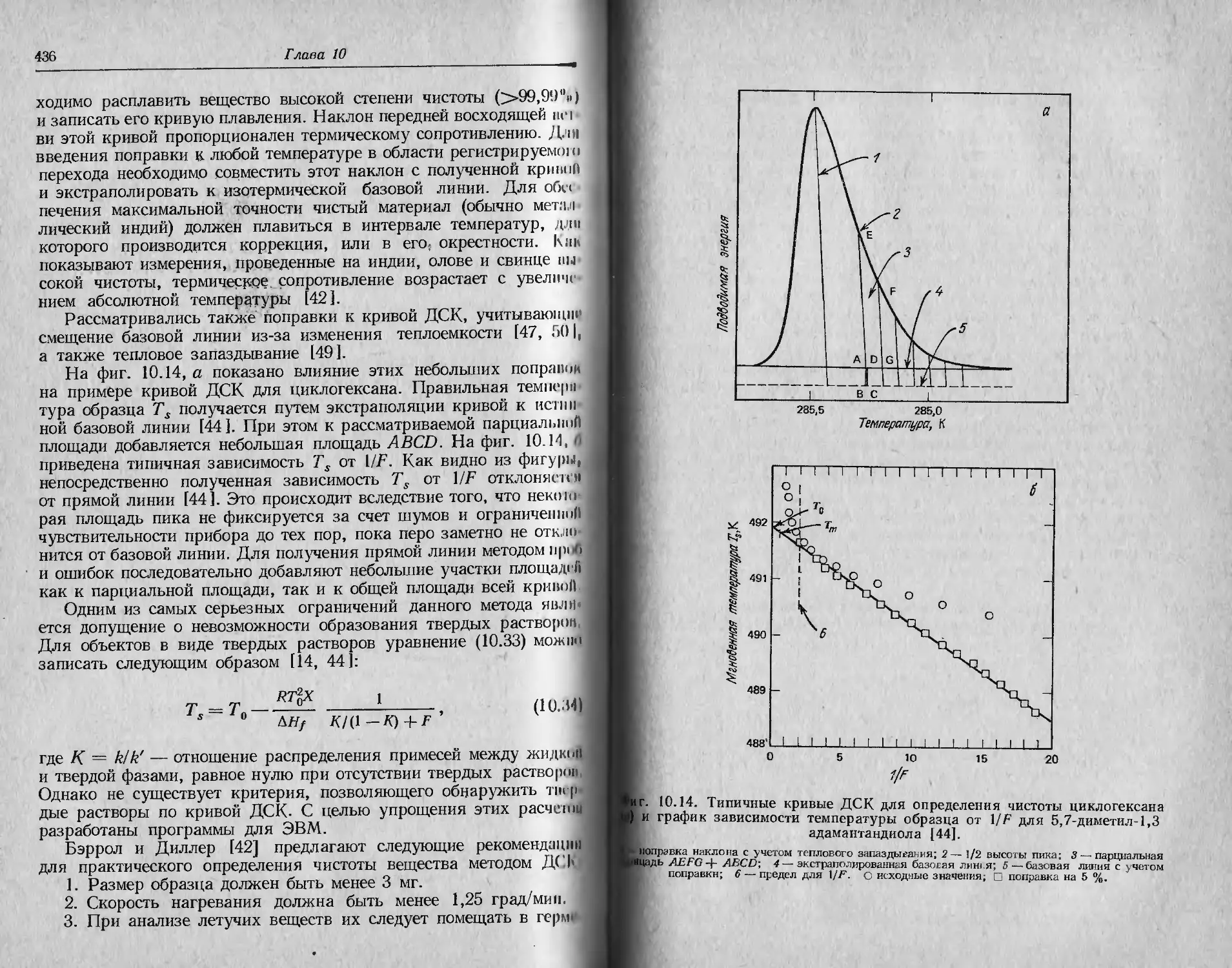
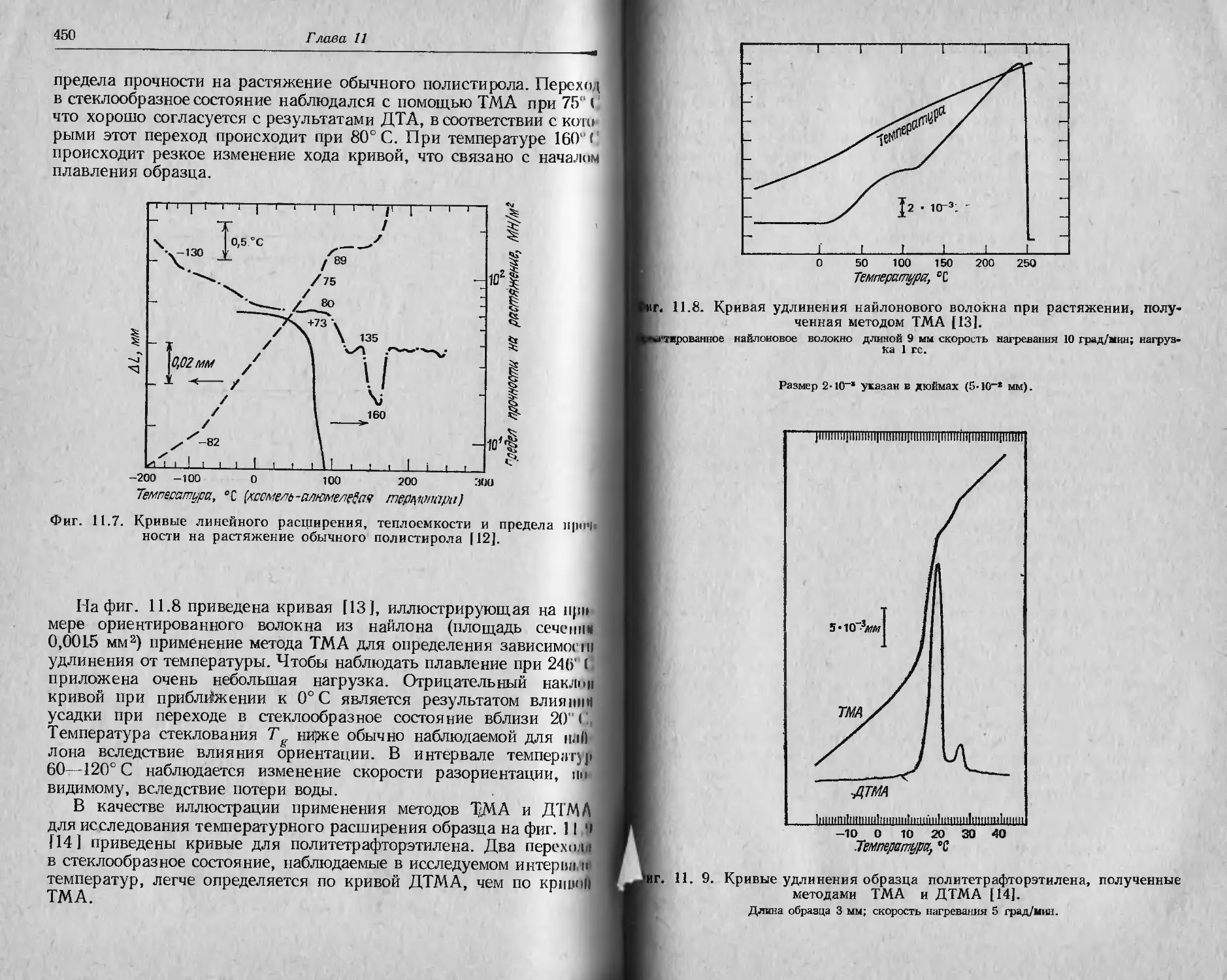
/













































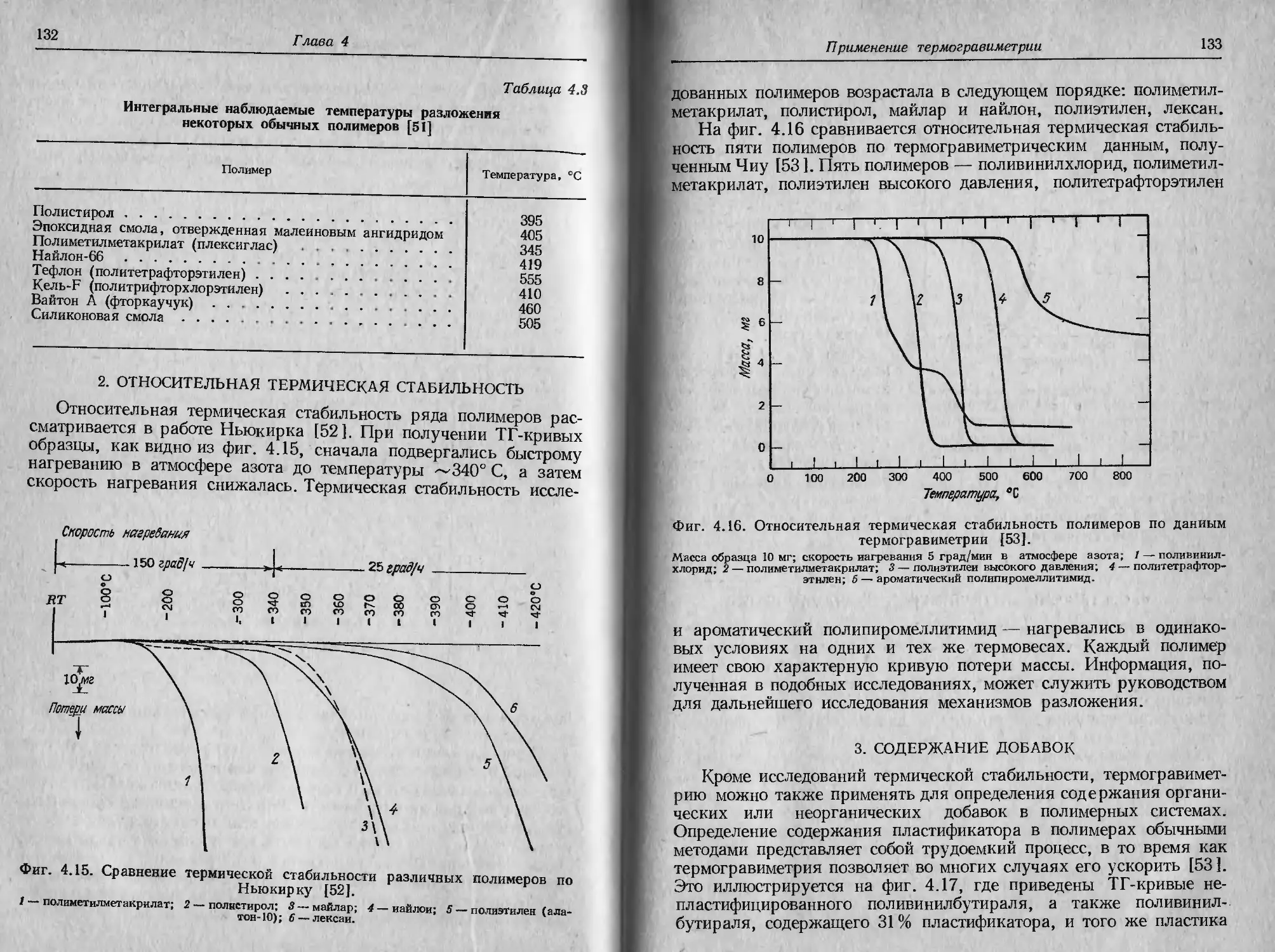
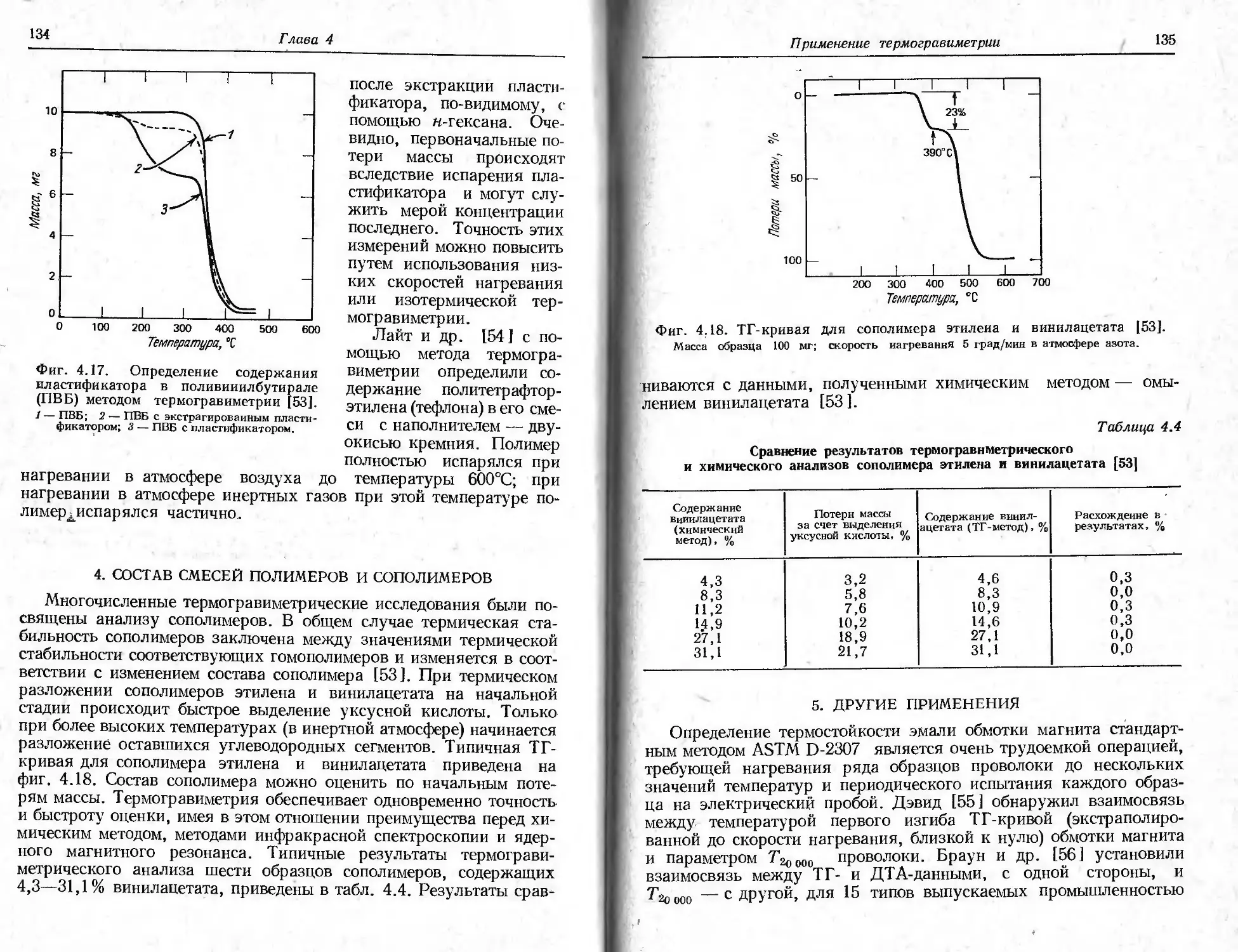













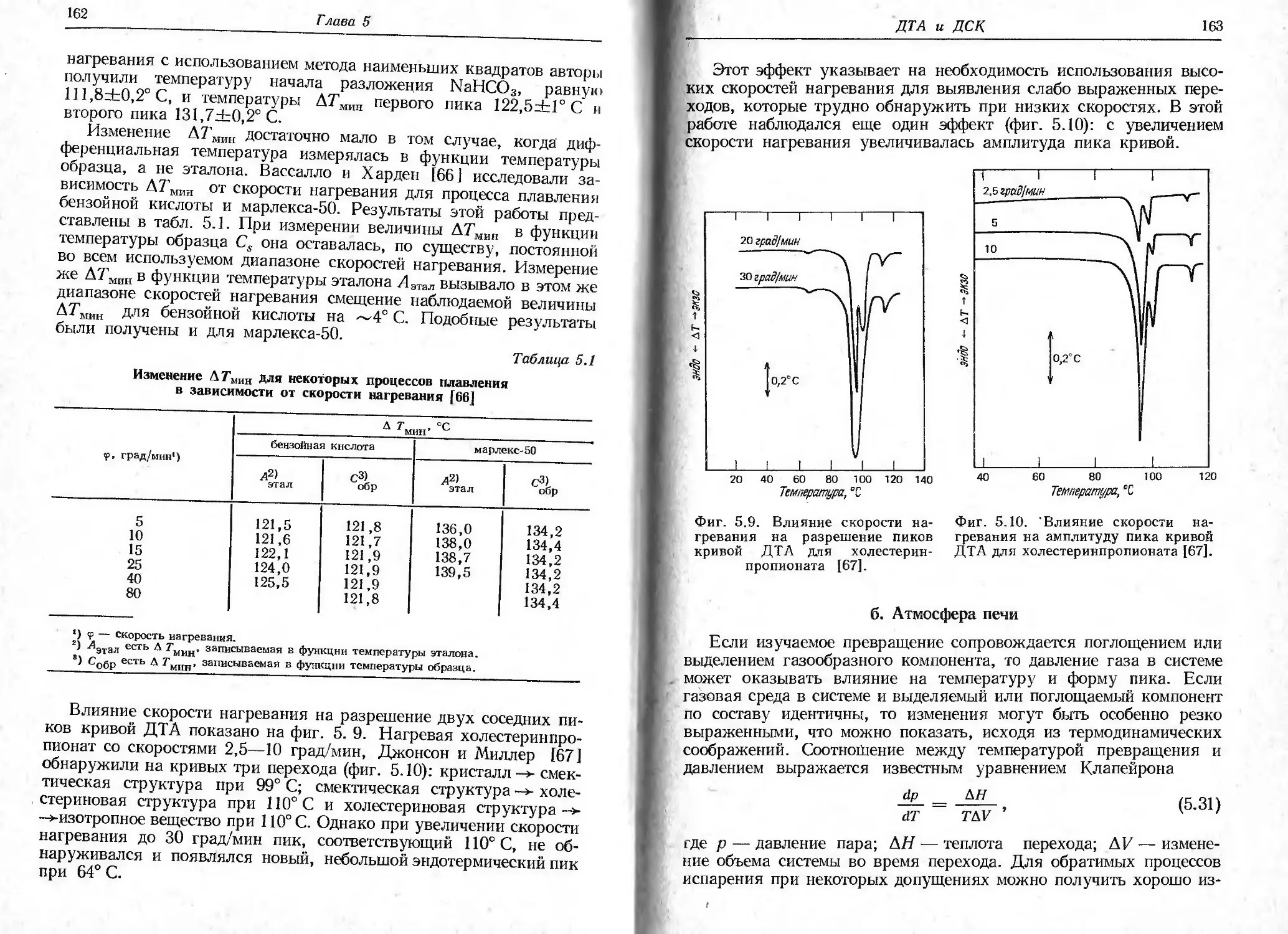

















































































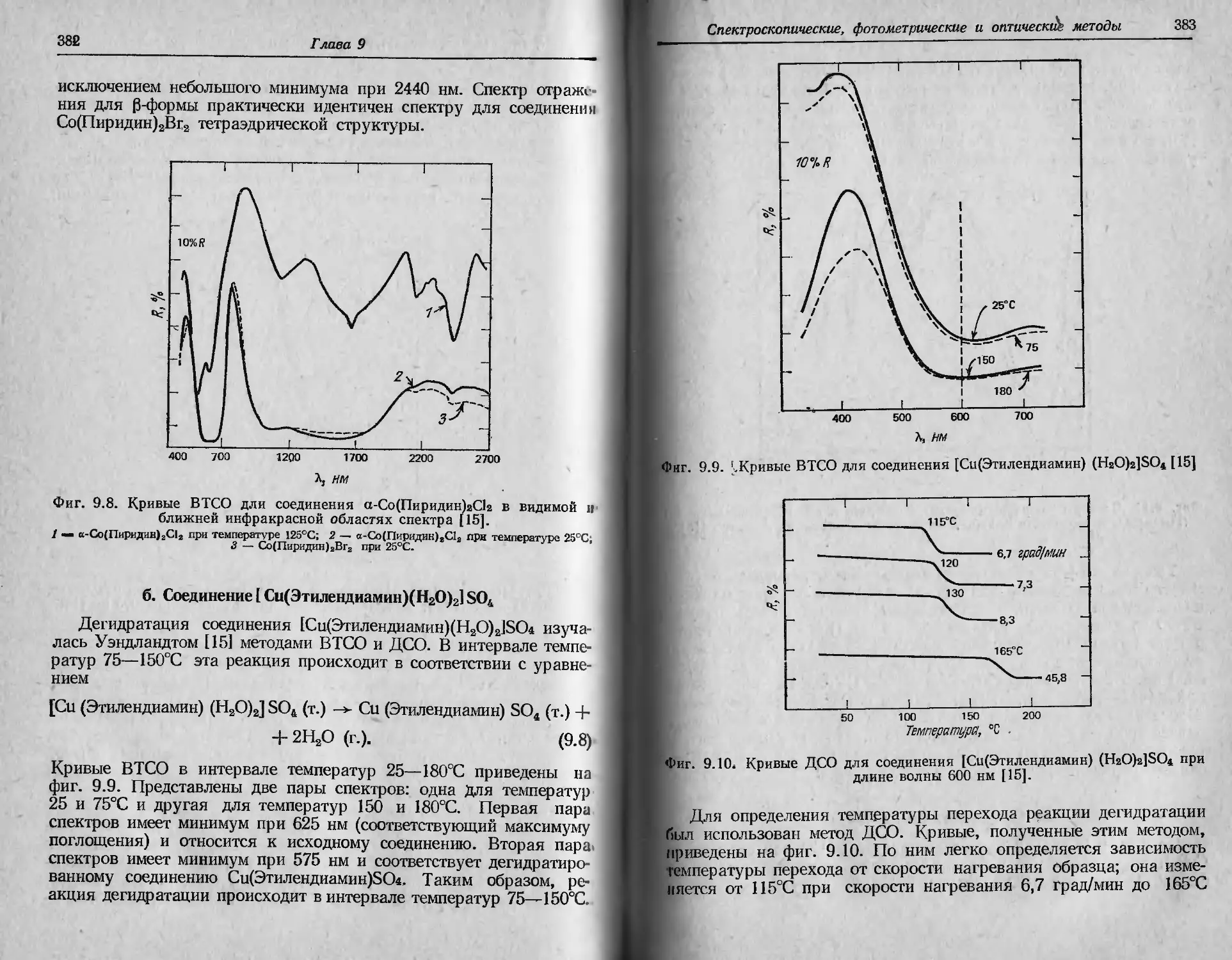
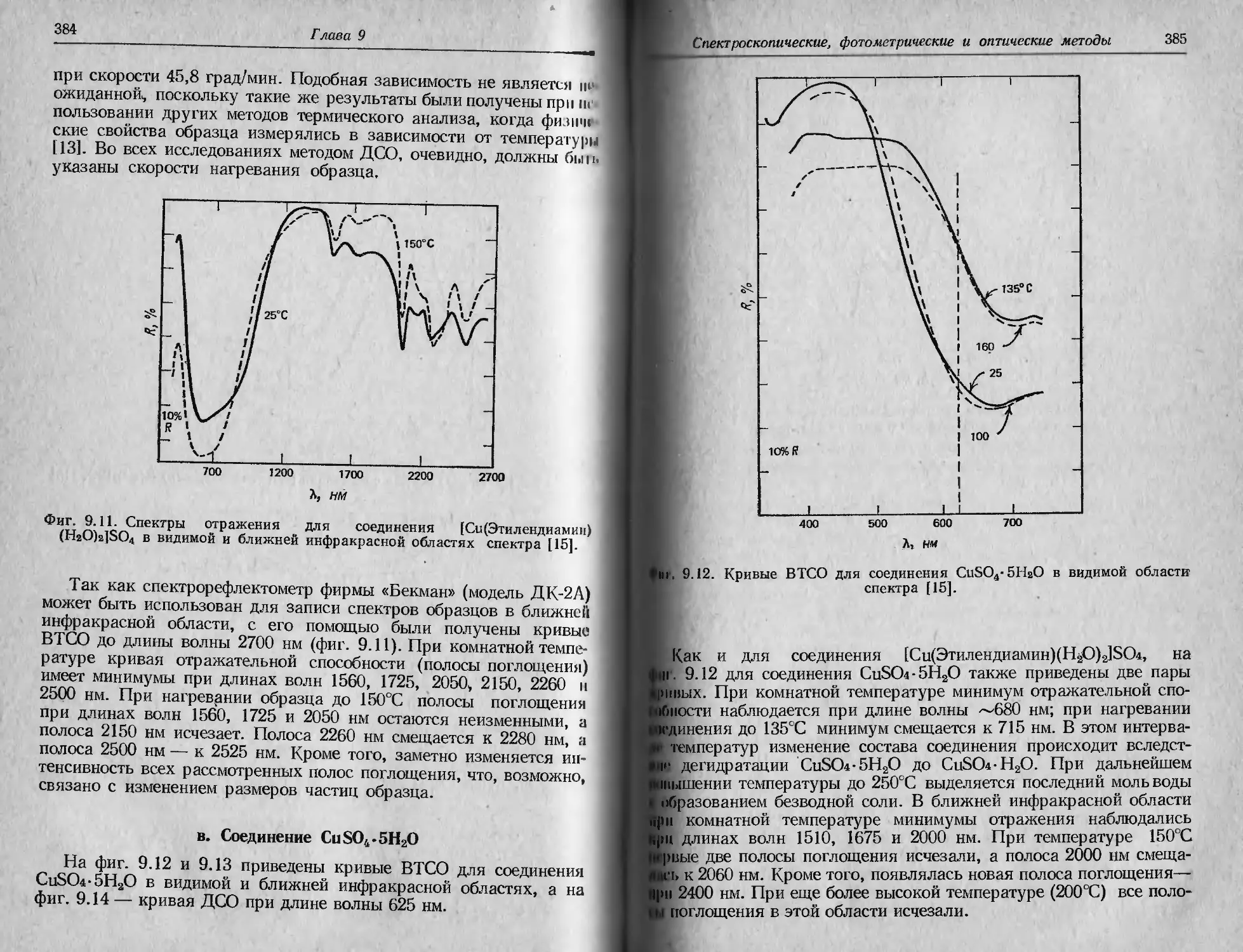
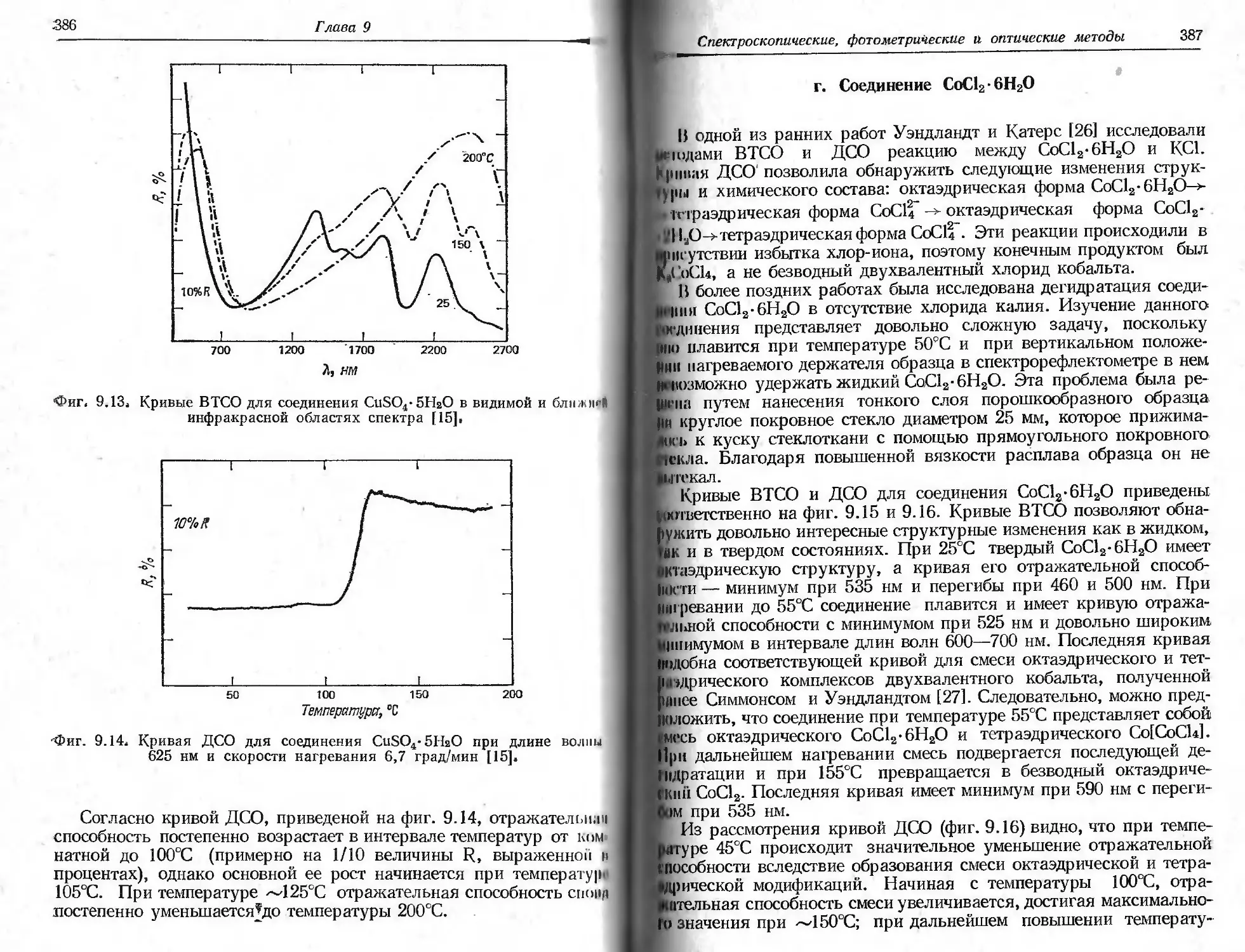


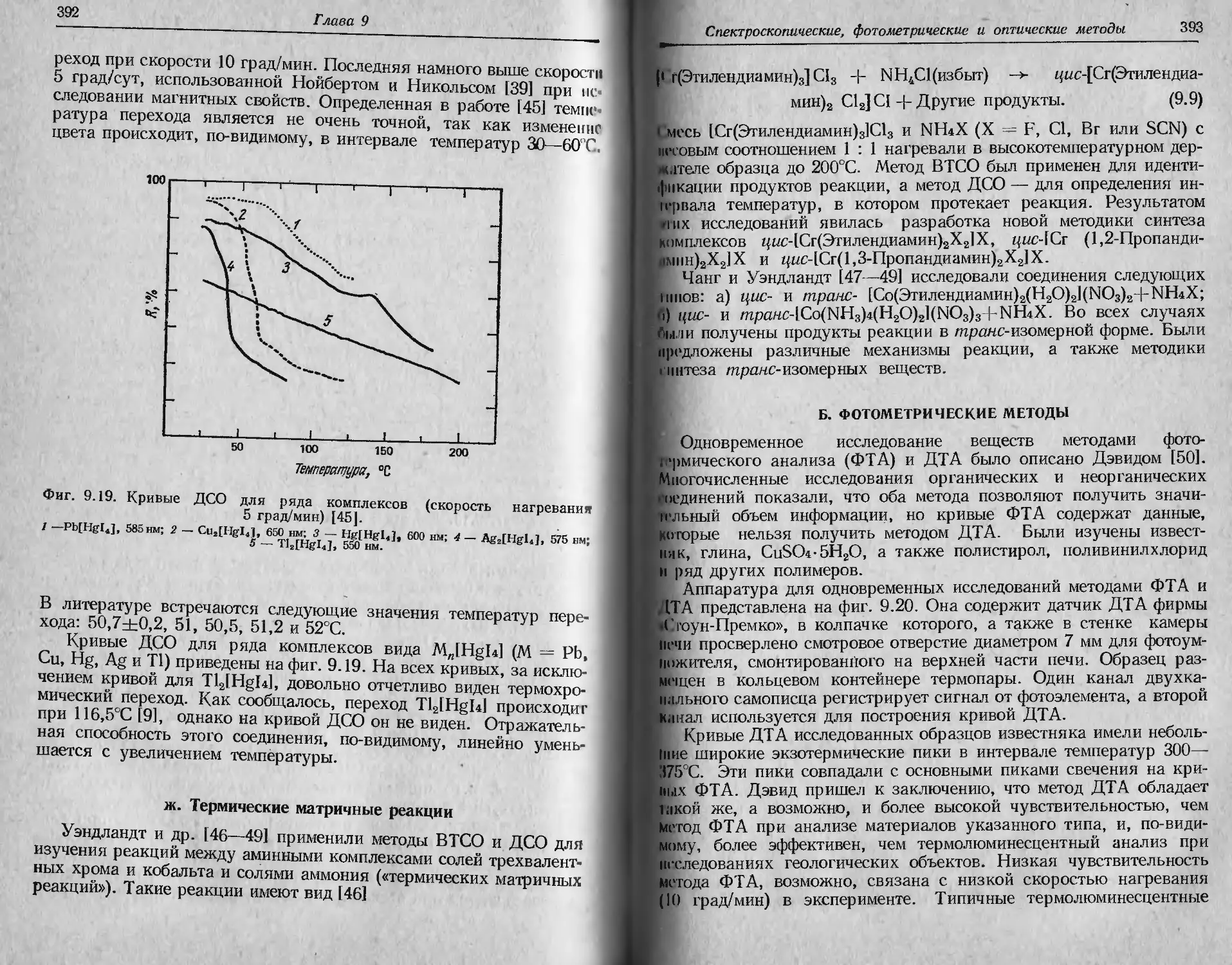





















































Автор: Уэндландт У.
Теги: химические методы анализа физика химия монография строение вещества термический анализ
Год: 1978




Текст
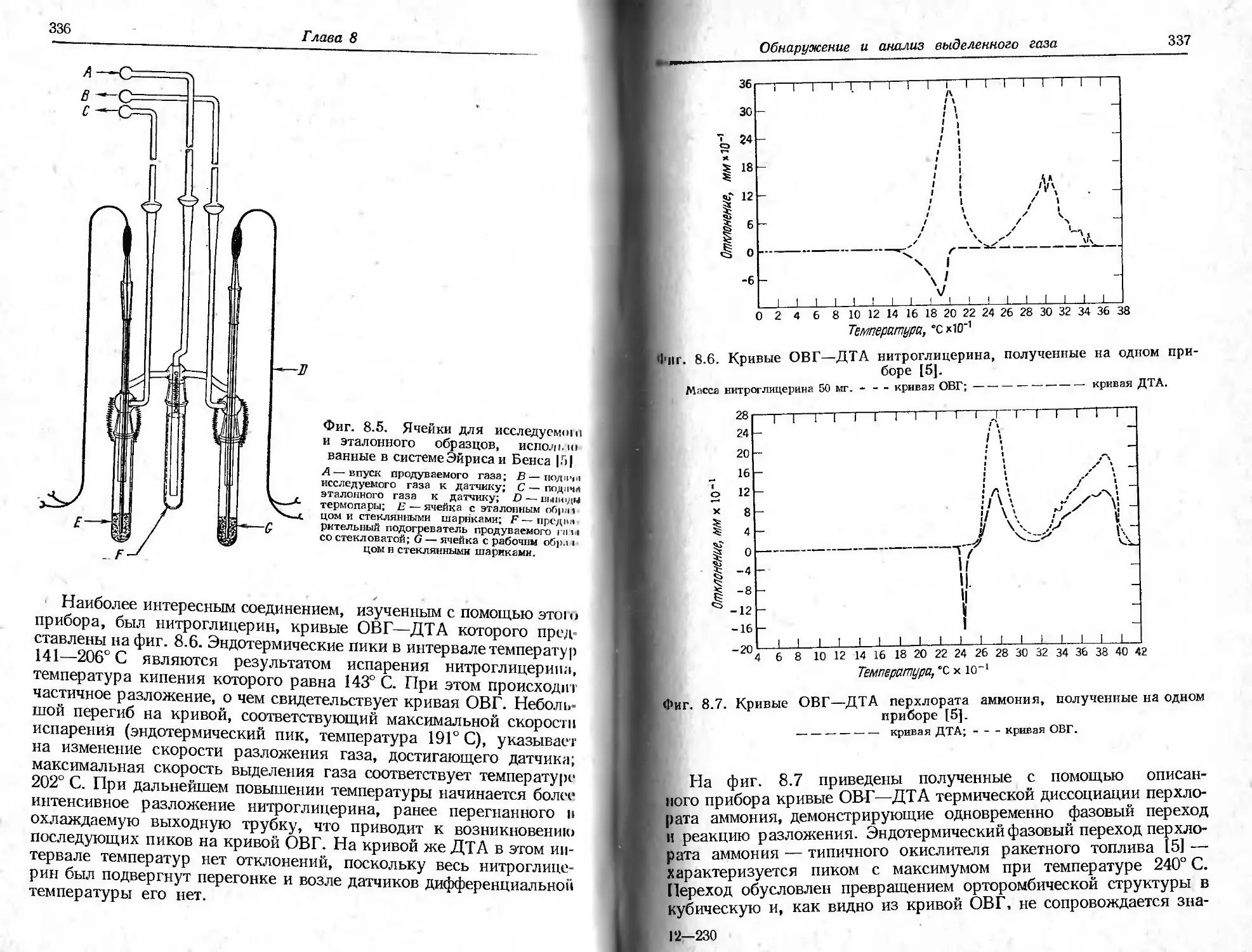
WJMUUC.
Thermal Methods
of Analysis
Second Edition
WESLEY WM. WENDLANDT
Department of Chemistry
University of Houston
Houston, Texas
A Wiley-Interscience Publication
John Wiley & Sons
New York. London. Sydney. Toronto
У. УЭНДЛАНДТ
ТЕРМИЧЕСКИЕ
МЕТОДЫ АНАЛИЗА
Перевод с английского
под редакцией В. А. Степанова
и В. А. Берштейна
ИЗДАТЕЛЬСТВО «МИР»
Москва 1978
УДК 543.2
Монография представляет собой краткий обзор теории,
экспериментальной техники и применений термического анали-
за. Дано описание новейших методов и схем приборов. Большая
часть книги посвящена основным методам термического анали-
за — термогравиметрин, дифференциальному термическому
анализу и дифференциальной сканирующей калориметрии.
Она предназначена широкому кругу специалистов — хи-
микам, физикам, биологам, материаловедам, конструкторам
научных приборов и др. Ее можно также рекомендовать в ка-
честве справочного пособия.
Редакция литературы по новой технике
Copyright© 1974, by John Wiley & Sons, Inc.
All rights reserved. Authorized translation from
English language edition published by John
Wiley & Sons, Inc.
© Перевод на русский язык, «Мнр», 1978
30100-163
041(01 )-78
163-78
ПРЕДИСЛОВИЕ РЕДАКТОРОВ РУССКОГО ПЕРЕВОДА
До недавнего времени были известны лишь некоторые класси-
ческие методы термического анализа (например, дилатометрия),
которые использовались в основном применительно к металлам
и минералам. В 60-е годы начинается бурное развитие термических
методов исследования, превратившихся в самостоятельную область
науки, границы которой весьма условны. В последние годы были
продемонстрированы большие возможности этих методов для полу-
чения ценной информации о строении, составе и свойствах твердых
тел и жидкостей различной природы, о физических и химических
процессах, протекающих в них при нагревании или охлаждении.
Настоящая книга опубликована в США вторым, значительно
расширенным изданием. В ней дан краткий обзор методов терми-
ческого анализа, в том числе разработанных в самое последнее
время. К методам этого типа в известном смысле можно отнести
все физические методы, позволяющие получить информацию типа
параметр — температура. Автор естественно не включил в книгу
(или рассматривает предельно кратко) такие классические методы,
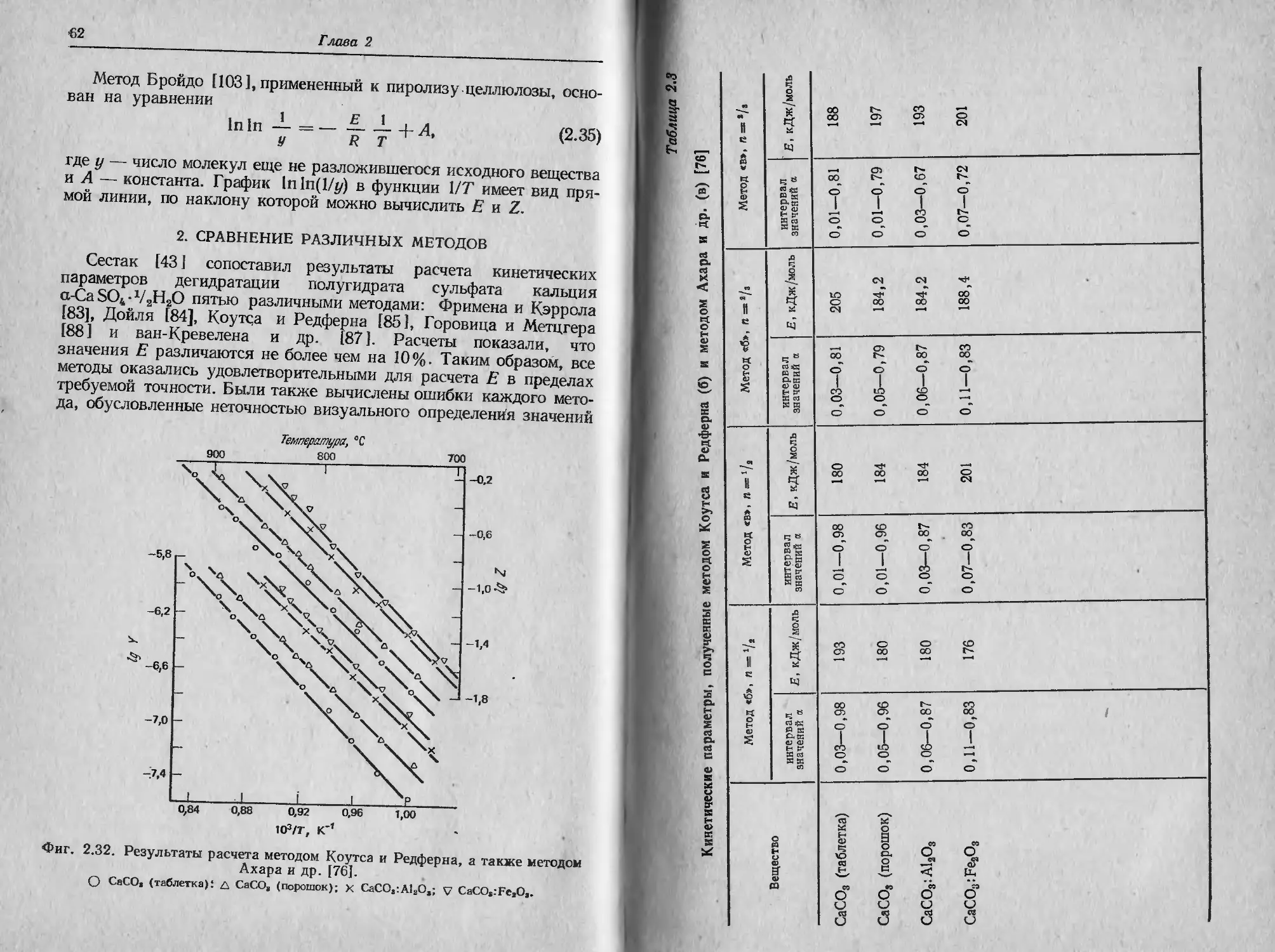
как рентгеновская дифракция, оптическая спектроскопия, ядерный
магнитный резонанс, электронный парамагнитный резонанс и т. д.
Относительно подробно рассмотрены собственно термические ме-
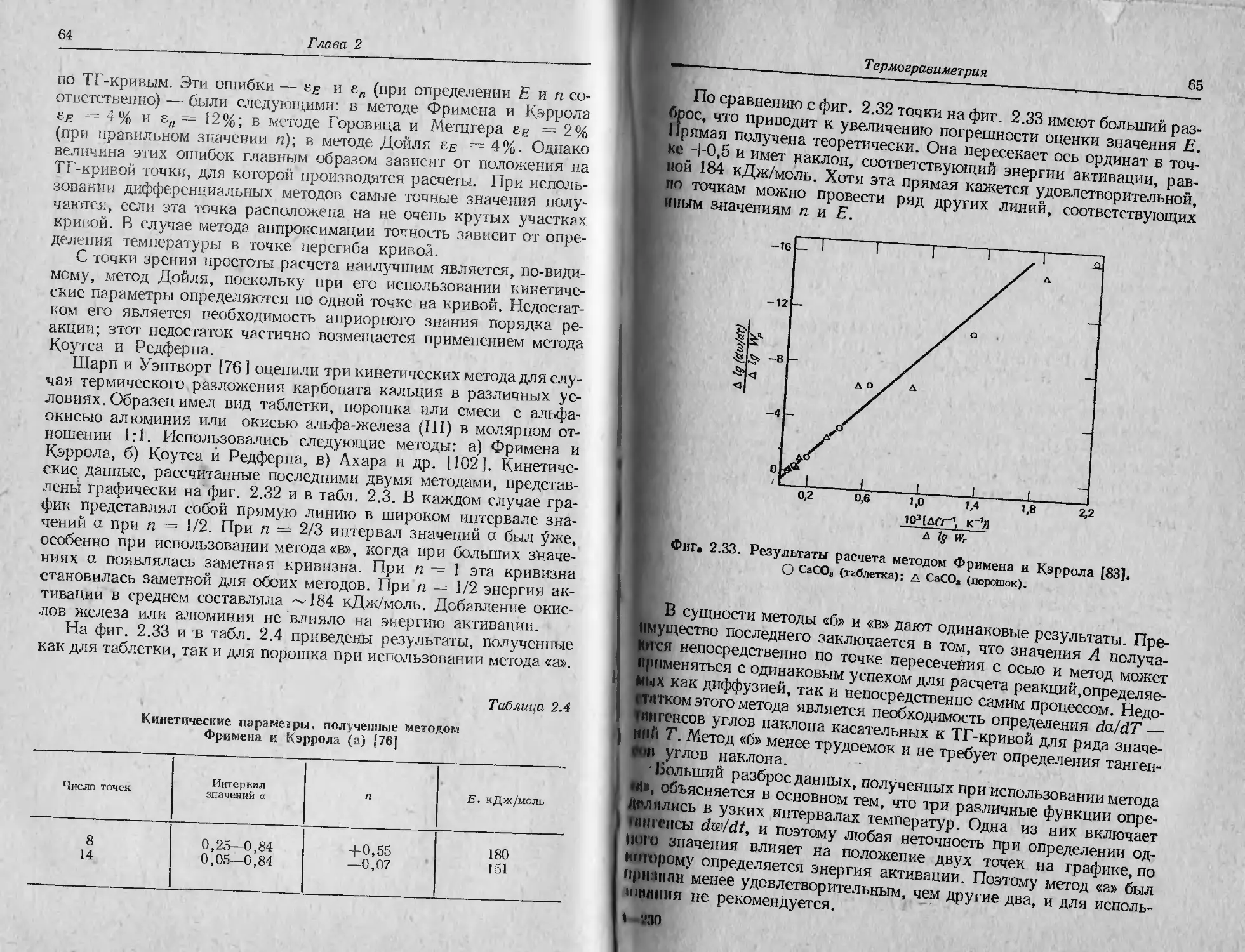
тоды: динамическая термогравиметрия (ТГ), дифференциальный
термический анализ (ДТА) и дифференциальная сканирующая
калориметрия (ДСК), а также новейшие (в том числе комбиниро-
ванные) методы.
Приведены схемы приборов, рассмотрены методические вопросы,
оценены сравнительные достоинства и недостатки отдельных мето-
дов, выявлены источники ошибок. В библиографии содержится
лишь небольшая часть оригинальных работ, но приведены важ-
нейшие обзоры. Примеры использования методов относятся глав-
ным образом к области химии, хотя применение термического ана-
лиза к физическим и другим задачам не менее многообразно.
В книгу включен перечень принятых сокращений наименований
методов; в гл. 13 некоторые термины приведены на русском и анг-
лийском языках; для разных методов, имеющих в оригинале оди-
наковое обозначение, — термомеханического и термомагнитного,
электротермического и эманационного термического — введены раз-
ные сокращения.
Перевод книги выполнен В. М. Егоровым (гл. 5, 6), И. И. Емель-
яновой (предисловия авторов, гл. 1, 2, 7), И. М. Неймарк (гл. 10—
13) и Г. В. Фирсовой (гл. 3, 4, 8, 9).
В. А. Степанов
В. А. Берштейн
ПРЕДИСЛОВИЕ АВТОРА КО ВТОРОМУ ИЗДАНИЮ
После первого издания этой книги в области термического ана-
лиза произошли огромные изменения. Девять лет между двумя из-
даниями можно считать основным периодом наиболее широкого
развития данной области. За это время было разработано много
новых методов и расширены границы их применения. О роли тер-
мического анализа свидетельствуют основание трех научных обществ
по термическому анализу и двух специализированных журналов,
а также разработка систематической терминологии. Целесообраз-
ность этих мероприятий бесспорна.
Основная цель второго издания книги — ознакомление с мето-
дами термического анализа и их применением. Книга содержит
необходимые сведения по термогравиметрии, дифференциальному
термическому анализу и другим методам, рассматриваемым в от-
дельных главах. Автор не пытался дать исчерпывающий обзор
каждого метода, иначе, как это очевидно любому, работающему
в области термического анализа, каждая глава превратилась бы
в самостоятельную монографию.
Данное издание на 90% состоит из нового материала, появив-
шегося в период 1964—1972 гг. Как и в первом издании, динами -
ческая термогравиметрия (ТГ), дифференциальный термический
анализ (ДТА) и дифференциальная сканирующая калориметрия
(ДСК) рассматриваются более подробно, чем другие методы, ввиду
их первостепенной важности: три главы посвящены ТГ и три —
ДТА (вместе с ДСК). Гл. 10 претерпела наименьшие изменения.
Больше внимания было уделено методу обнаружения (и анализа)
выделенного газа (гл. 8) и спектроскопическому, фотометрическо-
му и оптическому термическим методам (гл. 9). Введены две совер-
шенно новые главы — о применении цифровых и аналоговых вы-
числительных машин (гл. 12) и по терминологии (гл. 13). Важ-
ность этих двух глав безусловно оправдывает их включение во
второе издание. Из-за недостатка места описано лишь ограниченное
число промышленных приборов. Однако в соответствующих гла-
вах даны ссылки на литературные источники, содержащие подроб-
ное описание этих приборов.
Как всегда, при выполнении работы такого масштаба ее завер-
шение и успех зависят от поддержки и помощи многих людей. Я выра-
жаю благодарность моей жене Гэй, профессорам П. Дж. Элвингу
и И. М. Колтгофу, а также Д. А. Уайт и Л. Вашингтон. Я хотел
бы также выразить признательность Организации фонда Р. А. Уэл-
ча (Хьюстон, шт. Техас) за частичное финансирование работы.
У. У. Уэндландт
Хьюстон, штат Техас
Июнь, 1973
ПРЕДИСЛОВИЕ АВТОРА К ПЕРВОМУ ИЗДАНИЮ
Целью этой монографии является ознакомление химиков и ис-
следователей, работающих в других областях науки, с относитель-
но новыми методами измерений, которые можно объединить под
общим названием «термические методы». В прошлом многие из этих
методов были связаны с утомительной и трудоемкой процедурой
ручной записи результатов; однако в настоящее время все они пол-
ностью автоматизированы и в них используются либо аналоговые,
либо цифровые самописцы. Таким образом, благодаря автоматиза-
ции измерения можно проводить без непосредственного участия
оператора, что повышает точность и достоверность результатов
и экономит время исследователя.
Термические методы анализа открывают новые возможности
для решения существующих в химии проблем и позволяют ставить
новые задачи. Современная химическая лаборатория немыслима
без термовесов или прибора для дифференциального термического
анализа. Первый прибор позволяет путем постепенного нагревания
вещества получить информацию о его теплостойкости, составе про-
межуточных продуктов пиролиза и окончательного продукта. Вто-
рой обеспечивает информацию об изменениях энтальпии, происхо-
дящих во время термического разложения вещества, а также дает
возможность регистрировать фазовые переходы различных типов.
Эти приборы позволяют получить большое количество информации
«и очень короткий промежуток времени.
В намерения автора не входило приводить исчерпывающий об-
зор литературы по каждому термическому методу. Вследствие огра-
ничения объема эта книга является скорее критическим обзором
каждого метода. Она предоставляет исследователю сведения о пре-
имуществах и недостатках термических методов, с тем чтобы он
мог разумно их использовать.
Книга предназначена в первую очередь для исследователей,
работающих в области аналитической химии, хотя термические
методы с успехом используются и в других областях науки.
Автор благодарен профессорам П. Дж. Элвингу и И. ДЕ Колт-
। офу за их полезные совета и указания во время подготовки рукопи-
си книги; своему бывшему коллеге д-ру Э. Штурму за полезные
|<1мсчапия; профессорам Дж. Джордану и С. Т. Зенчельскому за
предоставление оттисков статей; И. Досчу и д-ру Р. Л. Стоуну за
помощь в работе, а также своим нынешним и бывшим студентам
(именно им в первую очередь книга обязана своим появлением).
Автор выражает признательность Отделению исследований Ко-
миссии США по атомной энергии, Управлению научных исследова-
ний воепно-воздушных сил США и Организации фонда Р. А. Уэлча
.'1.1 постоянную поддержку работы автора в данной области.
И наконец, автор благодарит за помощь в оформлении книги
Хардин, С. Ричмонд и К. Уайт.
Ляббок, шт. Техас у. у. Уэндландт
Январь, 1964
Книга посвящена памяти д-ра Роберта Л. Сто-
уна и проф. Ласло Эрди. Их многочисленные
работы легли в основу современного термическо-
го анализа
Глава 1
ОБЩЕЕ ВВЕДЕНИЕ
С появлением любого нового метода измерений химик получает
новый инструмент для решения проблем в области химии. Однако
иногда проходит Довольно много времени с Момента разработки
метода до его повсеместного использования. Это относится ко мно-
гим термическим методам, описанным в данной книге. Например,
первые термовесы были созданы Хонда в 1915 г., но лишь в 1947 г.
Дюваль указал на возможность их применения в гравиметрическом
анализе в неорганической химии. То же самое произошло и с мето-
дом дифференциального термического анализа (ДТА), который был
впервые описан Робертс-Остеном в 1899 г. На протяжении многих
лет ДТА был неоценимым методом определения состава минералов,
глин, сплавов металлов и т. д., но практически не использовался
химиками. Однако в последние годы метод ДТА успешно применя-
ется для решения химических задач либо сам по себе, либо совмест-
но с другими термическими методами.
11од термическим анализом в настоящей книге будут подразу-
меваться методы, в которых исследуется какой-либо физический
параметр системы в зависимости от температуры, причем этот физи-
ческий параметр регистрируется как динамическая функция темпе-
ратуры. Измерения, выполненные при постоянной температуре,
кик правило, рассматриваться не будут. В рамках этого определе-
ния основными методами термического анализа являются динами-
ческая термогравиметрия (ТГ) и дифференциальный термический
пналп.т (ДТА). К другим важным, но менее распространенным мето-
дам относятся обнаружение выделенного газа (ОВГ) и анализ вы-
деленного газа (АВГ), термомеханический анализ (ТМА), динами-
ческая спектроскопия отражения (ДСО), метод электропроводности
( >11), фототермический анализ (ФТА) и т. д. Каждый из этих мето-
доп будет подробно рассмотрен. Под указанную классификацию
не подпадают еще два важных термических метода: криоскопиче-
(Кнй анализ и термометрическая титрометрия. Первый будет рас-
гмчтреп и данной кише, поскольку он является важным методом
анализа и аналитической химии и предусматривает измерение тем-
пера 1 у ры вещества в функции нпемепи.
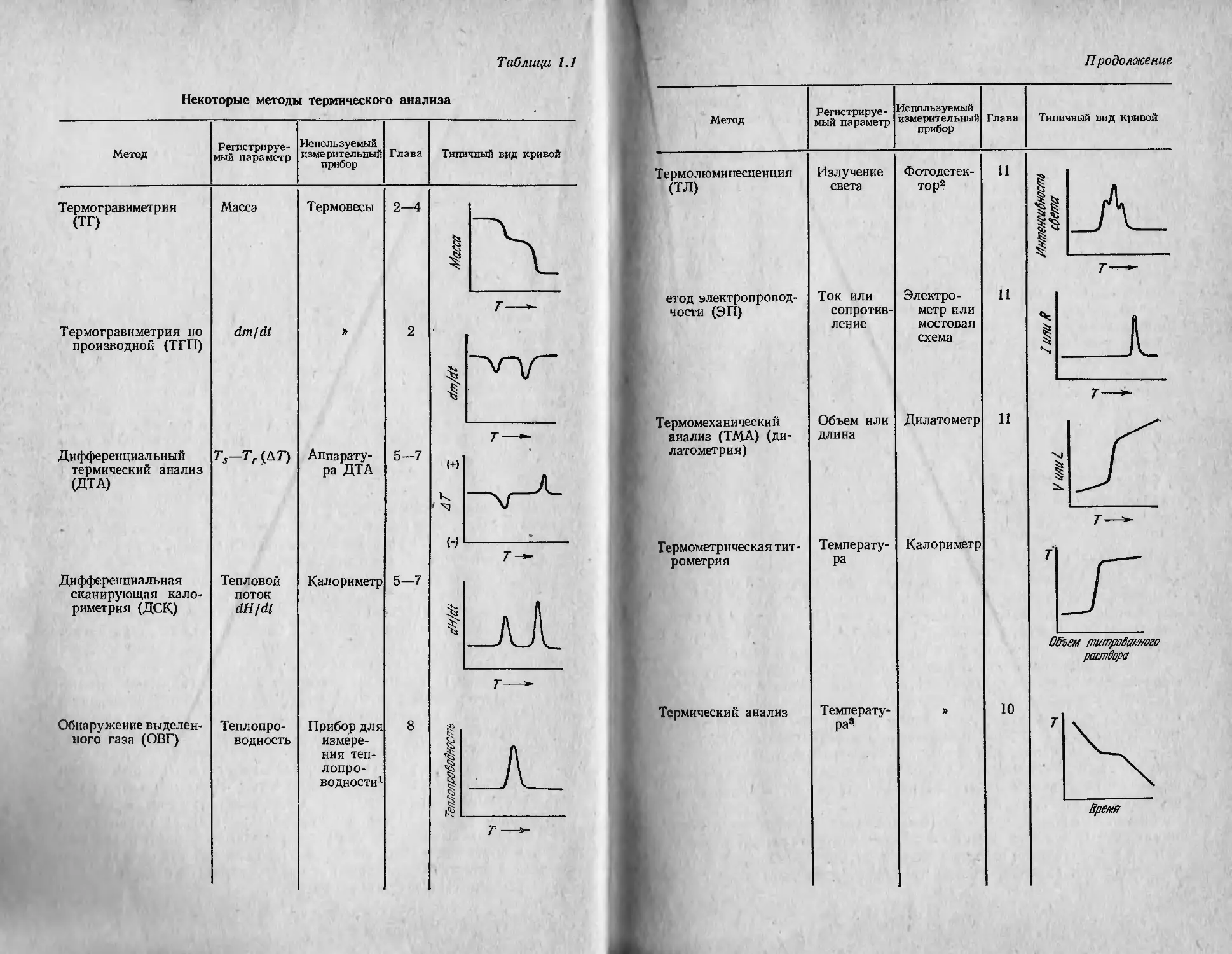
Таблица 1.1
Некоторые методы термического анализа
Метод Регистрируе- мый параметр Используемый измерительный прибор Глава Типичный вид кривой
Термогравиметрия (ТГ) Масса Термовесы 2—4 Масса
Термогравнметрия по производной (ТГП) dm/dt » 2 & VAA” < т——
Дифференциал ьный термический анализ (ДТА) Ts-Tr (AT) Аппарату- ра ДТА 5—7 (+) 1 '-q \/ (-) т—
Дифференциальная сканирующая кало- риметрия (ДСК) Тепловой поток dH/dt Калориметр 5—7 |_л£ т—*-
Обнаружение выделен- Теплопро- Прибор для 8
него газа (ОВГ) водность измере- ния теп- лопро- водности1 ТеллолробоЛт
П родолжение
Метод Регистрируе- мый параметр Используемый измерительный прибор Глава Типичный вид кривой
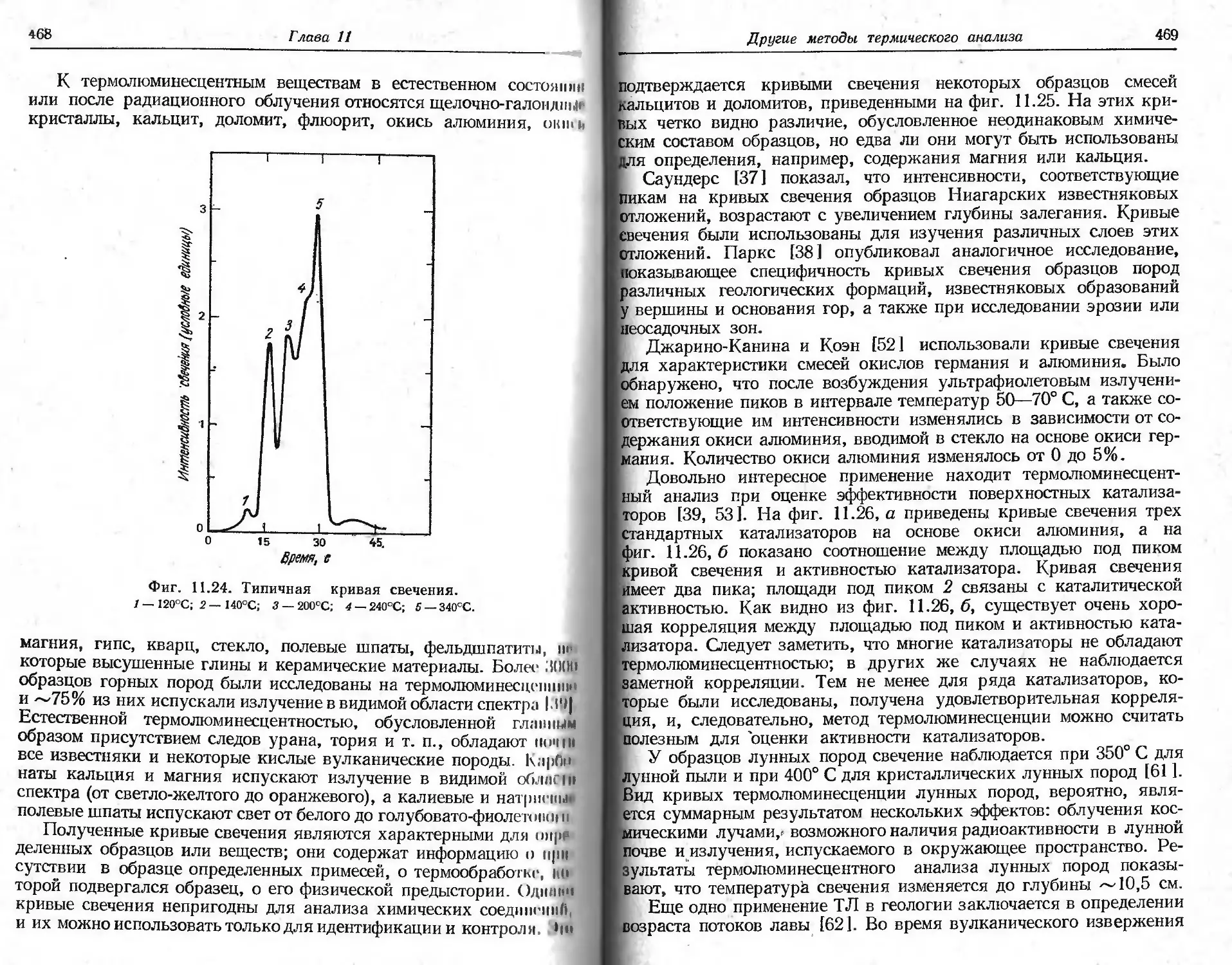
Термолюминесценция Излучение Фотодетек- и
(ТЛ) света тор2 S я /А Й / \ 1 i г-—
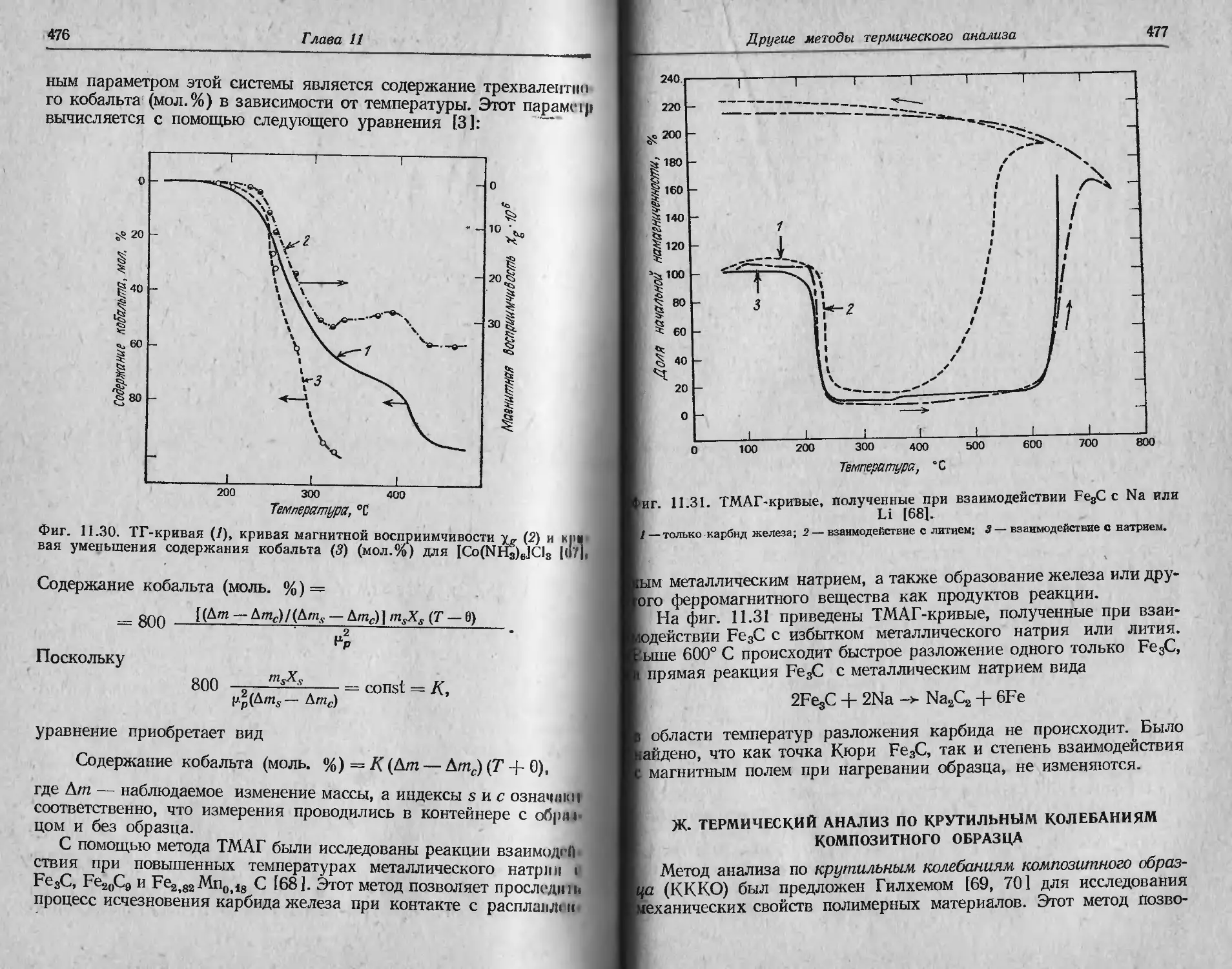
етод электропровод- Ток или Электро- 11
чости (ЭП) сопротив- ление метр или мостовая схема I им/?
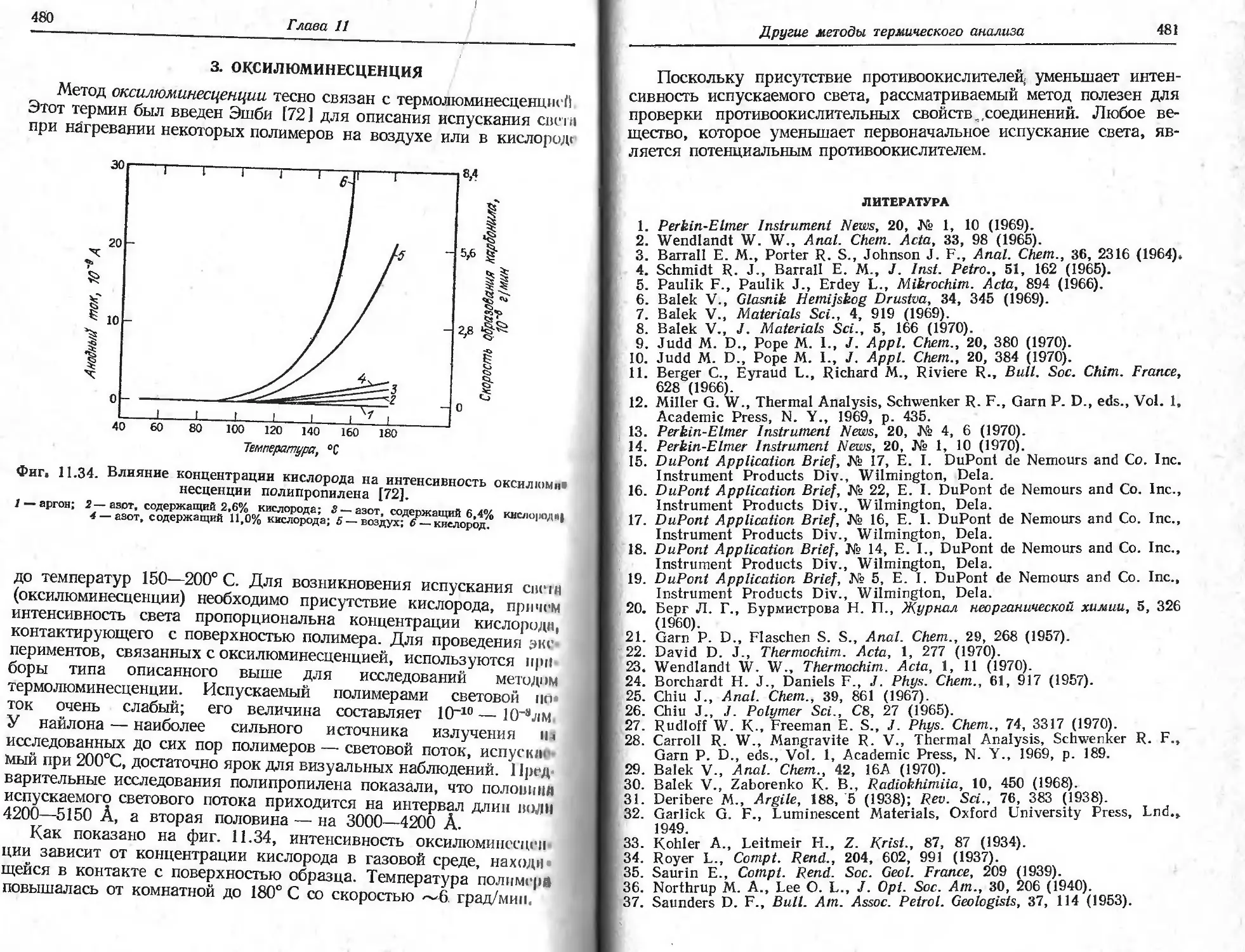
Термомеха нически й анализ (ТМА) (ди- латометрия) Термометрическая тит- рометрия Объем или длина Температу- ра Дилатометр Калориметр 11 I т—* 7 Объем титрованного раствора
Термический анализ Температу- ра5 » 10 г Время
1
Глава 1
П родолжение
Метод Регистрируе- мый параметр Нсгюл1^>тмый измеритель- ный прибор Глава Типичный вид кривой
Динамическая спект- Отража- Сиектрофо- 9
роскопия отраже- ния (ДСО) тельная способ- ность тометр т—*
Эманационный терми- Радиоак- Аппарату- II
ческий анализ (ЭТА) тивность рз ЭТА г—*
люжю испошьгювагь и другие детекторы.
1 Можно использовать фотоумножитель, ф-иодиод.
* Можно использовать также метод ДТА или ДСК
фотокамеру
или другие приборы.
В табл. 1.1 представлены различные методы термического ана-
лиза. Для каждого из них указаны регистрируемый параметр,
типичный вид кривой зависимости регистр ируемого параметра от
температуры, измерительный прибор и глава, в которой этот метод
описывается. Перечень методов в табл. 1.1, конечно, не является
исчерпывающим, поскольку почти любой аналитический метод
измерений можно считать методом термического анализа, если из-
меряемый параметр определяется как функция температуры. chr>
относится к протонному ядерному магнитному резонансу, электрон-
ному спиновому резонансу, дифракции электронов, дифракции рент-
геновских лучей, масс-спектрометрии, спектрофотометрии в ультра-
фиолетовой, видимой и инфракрасной областях и т. и. Рассмотрение
всех этих методов выходит за рамки настоящей книги, хотя возмож-
ности их применения в области высоких (или низких) температур
•очевидны и известны.
Следует отметить, что во многих случаях какой-то один метод
термического анализа может не обеспечить достаточной информа-
ции об исследуемой системе. Как и в случае применения других
аналитических методов, иногда требуются дополнительные сведе-
Общее введение
13
ия, которые могут быть получены с помощью других методов тер-
мического анализа. Например, данные ДТА или ДСК зачастую
дополняют результаты, полученные с помощью термогравиметрии.
Ерли при нагревании образца выделяются газообразные вещества,
целесообразно использовать метод анализа выделенного газа. Удоб-
но применять одновременно несколько термических методов, поз-
воляющих получить разносторонние данные при использовании
одйого и того же образца в одинаковых условиях пиролиза.
На протяжении последних десяти лет методы термического
анализа нашли широкое применение в аналитической химии. Были
Основаны три общества, занимающиеся проблемами термического
анализа: Североамериканское общество термического анализа
(NATAS). Л\еждуиародная конфедерация по термическому анализу
(ICTA) и Общество калориметрии и термического анализа (Япония).
Каждое общество уже проводило конференции и симпозиумы, ос-
новной темой которых были методы термического анализа и их при-
ложения. Публикации исследований в области термического ана-
лиза сосредоточены в двух широко признанных специализирован-
ных журналах: «Journal of Thermal AnaIysis»(E. Buzagh, J. Simon,
eds.), который начал издаваться с 1969 г., и «Thermochimica Acta»
(W. W. Wendlandt, ed.), который был основан в 1970 г. С 1972 г.
в помощь исследователям, работающим в этой области, издается
журнал «.Thermal Analysis Abstracts» (J. P. Redfern, ed.).
За последние несколько лет большое внимание уделялось вопро-
сам терминологии в термическом анализе. Рекомендации комитета
по терминологии при конфедерацппМКТА, активно работающего в
этой области, представлены в гл. 13. Однако требуется приложить
еще немало усилий для ликвидации путаницы в некоторых терми-
нах, применяемых в настоящее время.
Глава 2
ТЕРМОГРАВИМЕТРИЯ
I
А. ВВЕДЕНИЕ
Термогравиметрия (ТГ) — метод термического анализа, при ко-
тором регистрируется изменение массы образца в зависимости от
температуры. Можно выделить три вида термогравиметрии: а) изо-
термическую, или статическую, когда масса образца измеряется
на протяжении некоторого времени при постоянной температуре;
б) квазистатическую, когда образец нагревается при каждой из
ряда возрастающих температур до достижения постоянного значе-
ния массы; в) динамическую, когда температура среды, окружаю-
щей нагреваемый образец, изменяется по заданному закону (обычно
с постоянной скоростью). Исследования, описанные в этой главе,
в основном относятся к динамической термогравиметрии, которую
мы будем называть просто термогравиметрией.
Экспериментально получаемая кривая зависимости изменения
массы от температуры (называемая также кривой термолиза или
пиролиза, термограммой, термогравиметрической кривой, термо-
гравиграммой, кривой термогравиметрического анализа и т. д.)
позволяет судить о термостабильности и составе образца в началь-
ном состоянии, о термостабильности и составе веществ, образую-
щихся на промежуточных стадиях процесса, и о составе остатка,
если таковой имеется. Этот метод будет эффективным лишь при
условии, что образец выделяет летучие вещества в результате раз-
личных физических и химических процессов (гл. 4). За исключени-
ем данных об изменении массы, информация, полученная на осно-
вании термогравиметрической кривой, носит эмпирический харак-
тер, поскольку температуры переходов зависят также от парамет-
ров измерительных приборов и образца. Поэтому не имеет смысла
сравнивать термогравиметрические данные, полученные на термо-
весах разных марок, используемых в различных лабораториях.
Применение термовесов серийного производства значительно улуч-
шило положение, однако температуры переходов на термограви-
метрической кривой пока еще не являются столь фундаментальны-
ми величинами, как, например, период кристаллической решетки,
определенный на основе рентгеноструктурных данных, или интен-
сивность полосы поглощения в инфракрасной области спектра.
Т ермогравиметрия
15
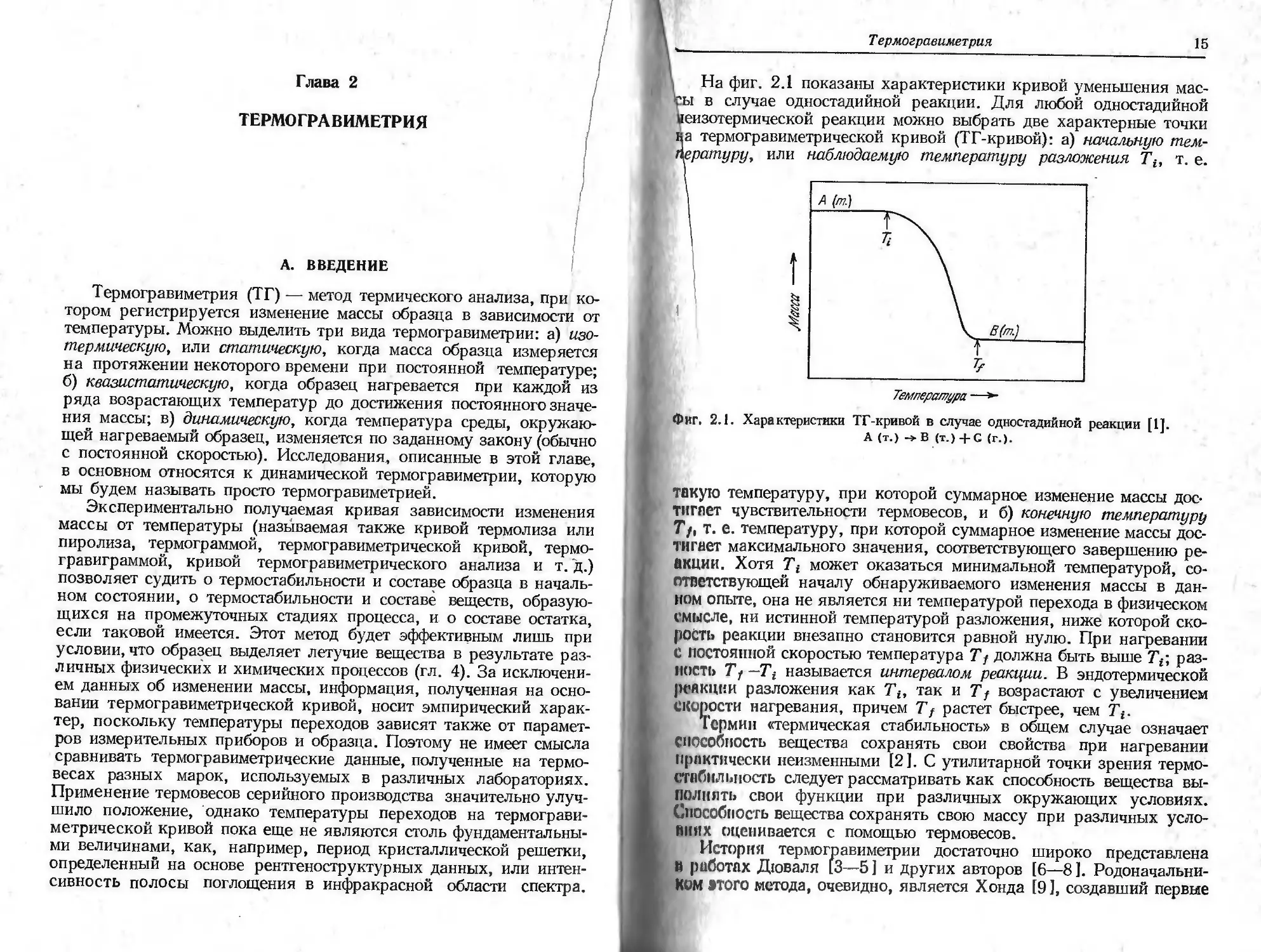
На фиг. 2.1 показаны характеристики кривой уменьшения мас-
ы в случае одностадийной реакции. Для любой одностадийной
[еизотермической реакции можно выбрать две характерные точки
а термогравиметрической кривой (ТГ-кривой): а) начальную тем-
\epamypy, или наблюдаемую температуру разложения Tt, т. е.
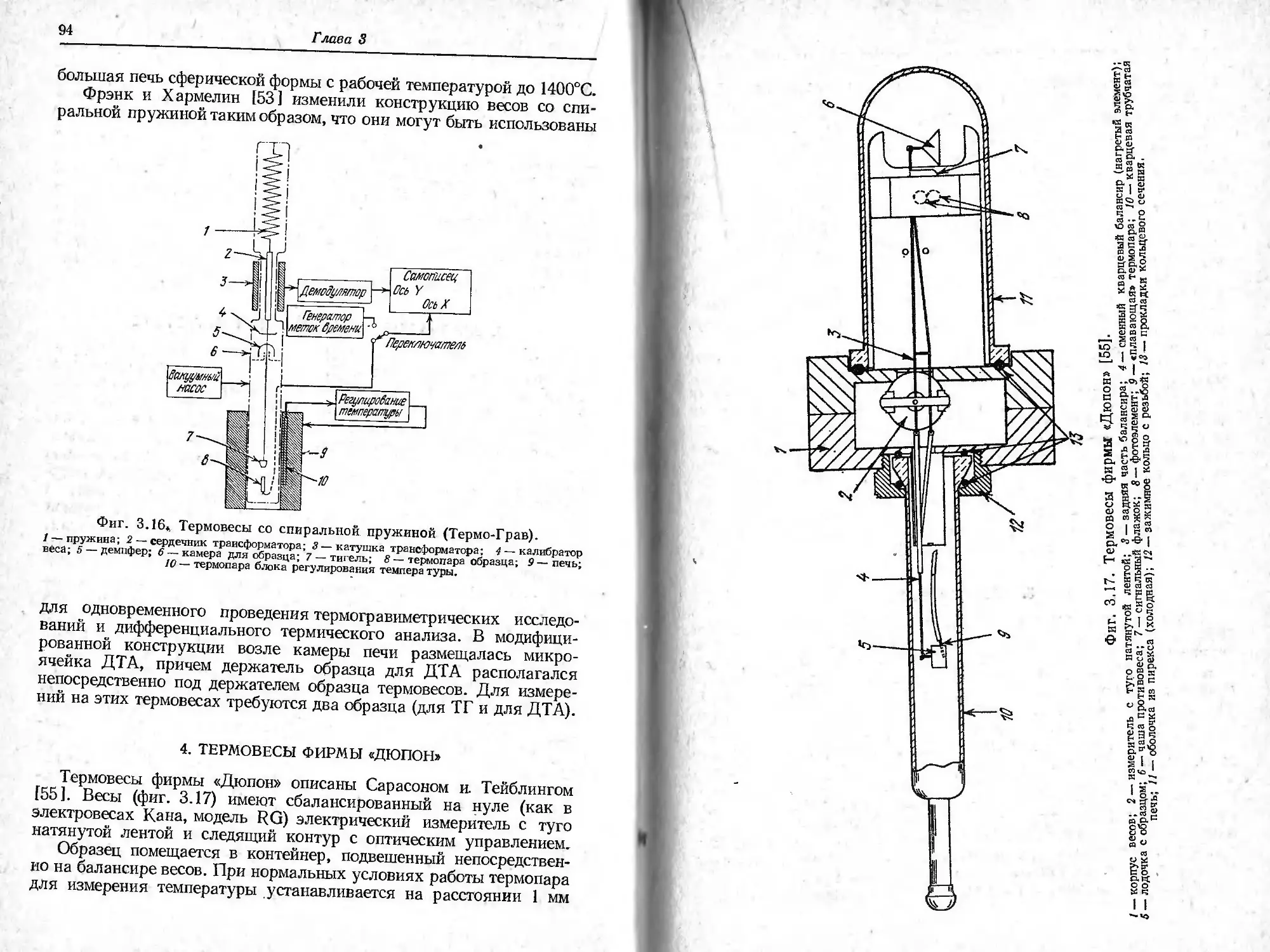
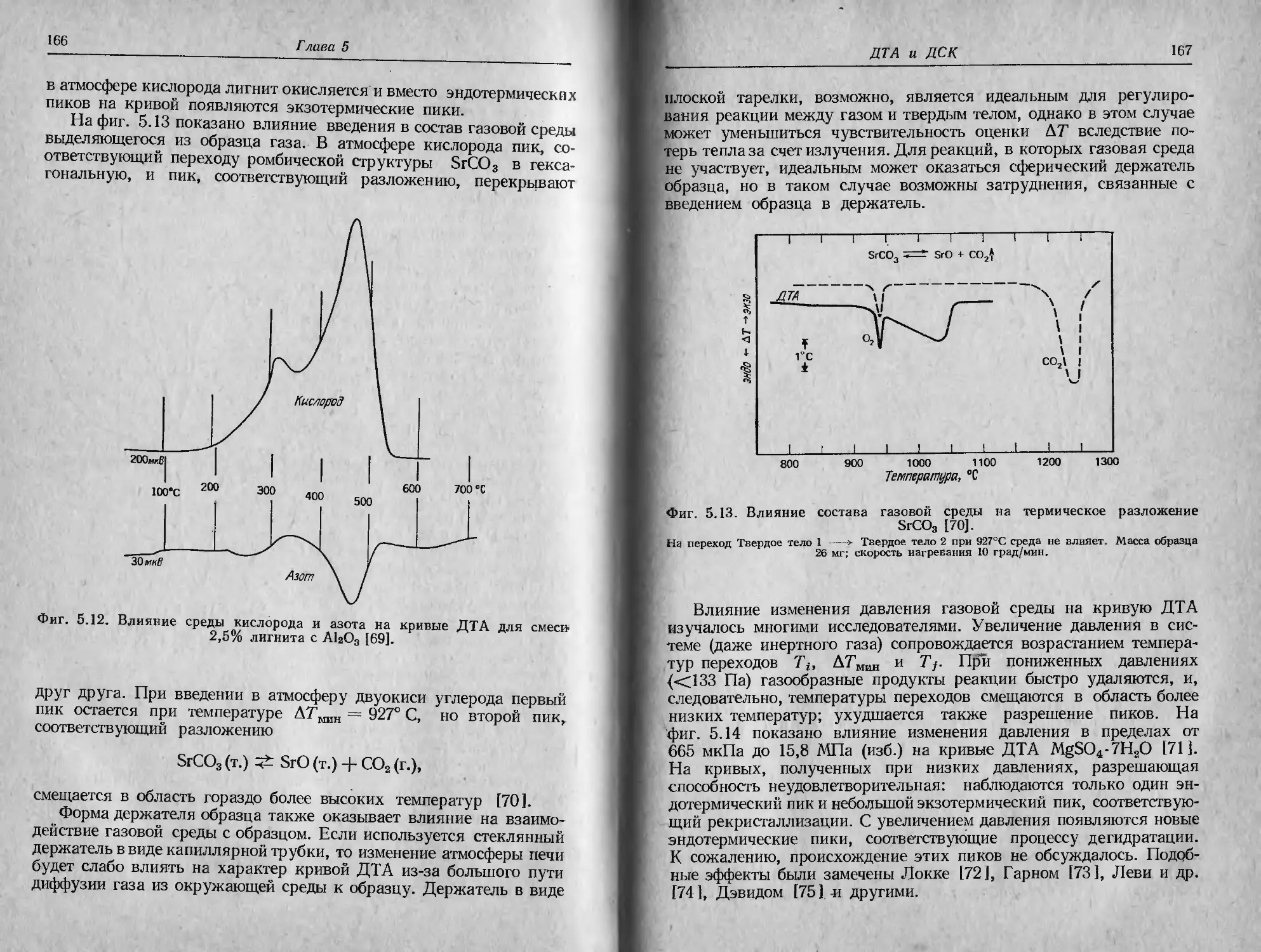
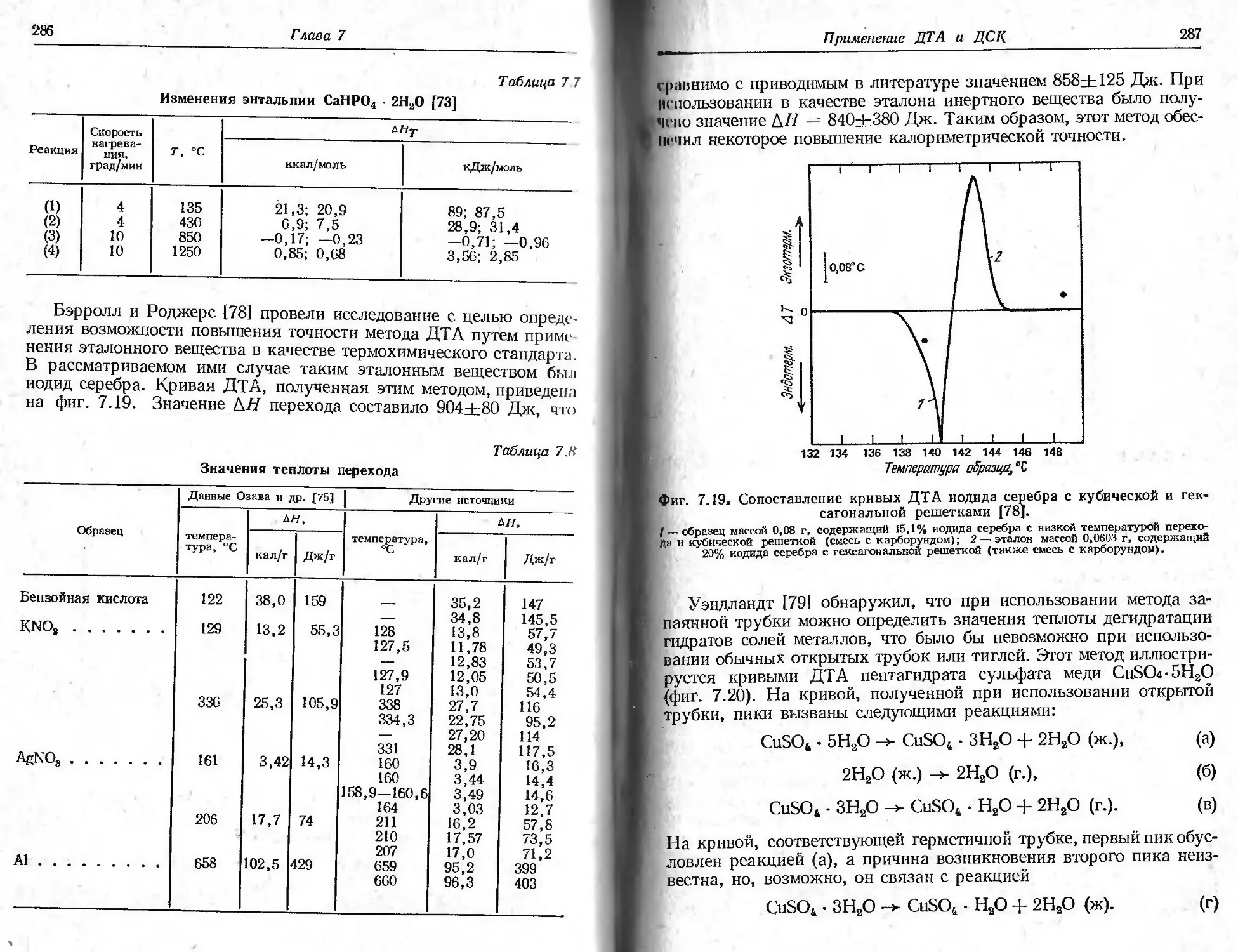
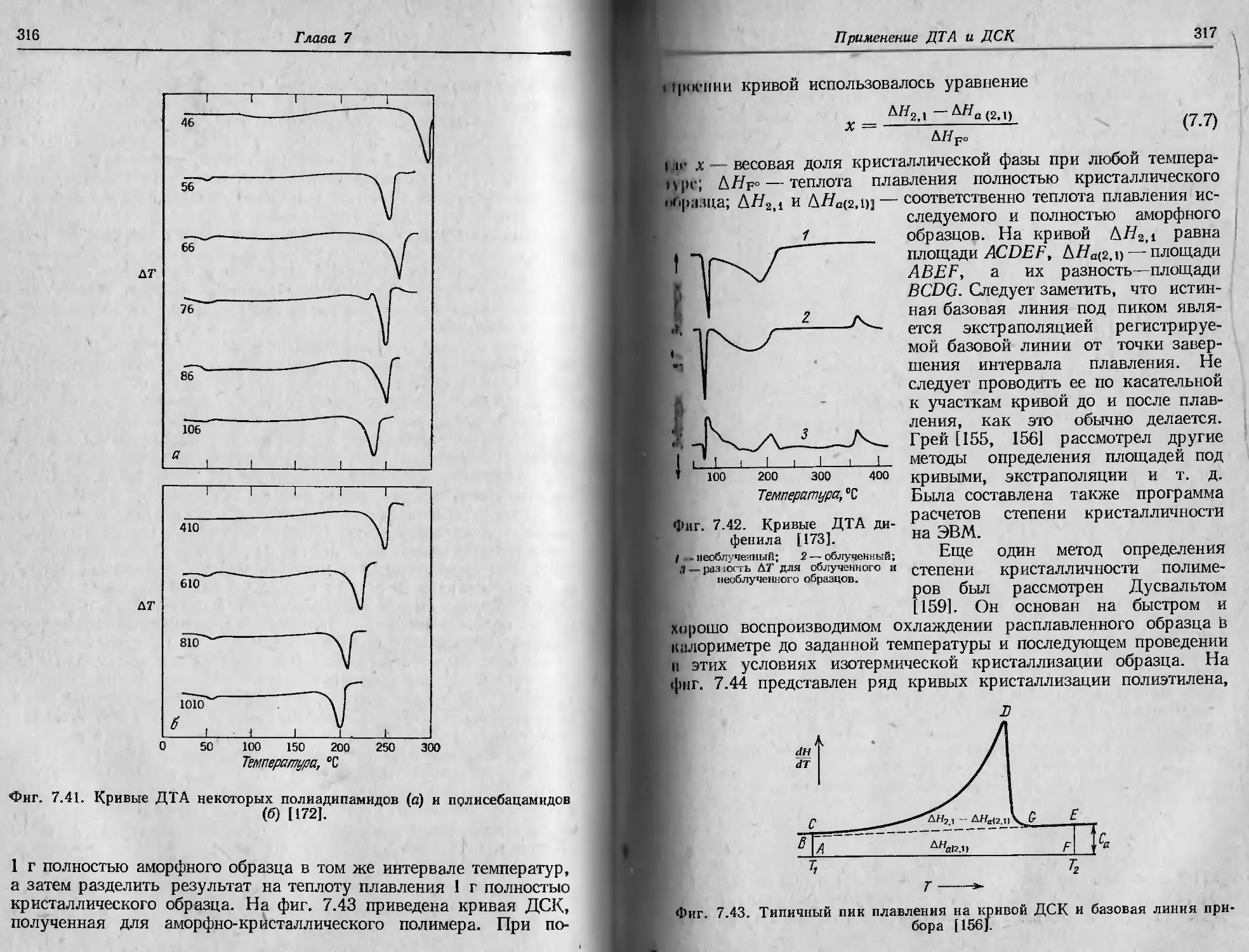
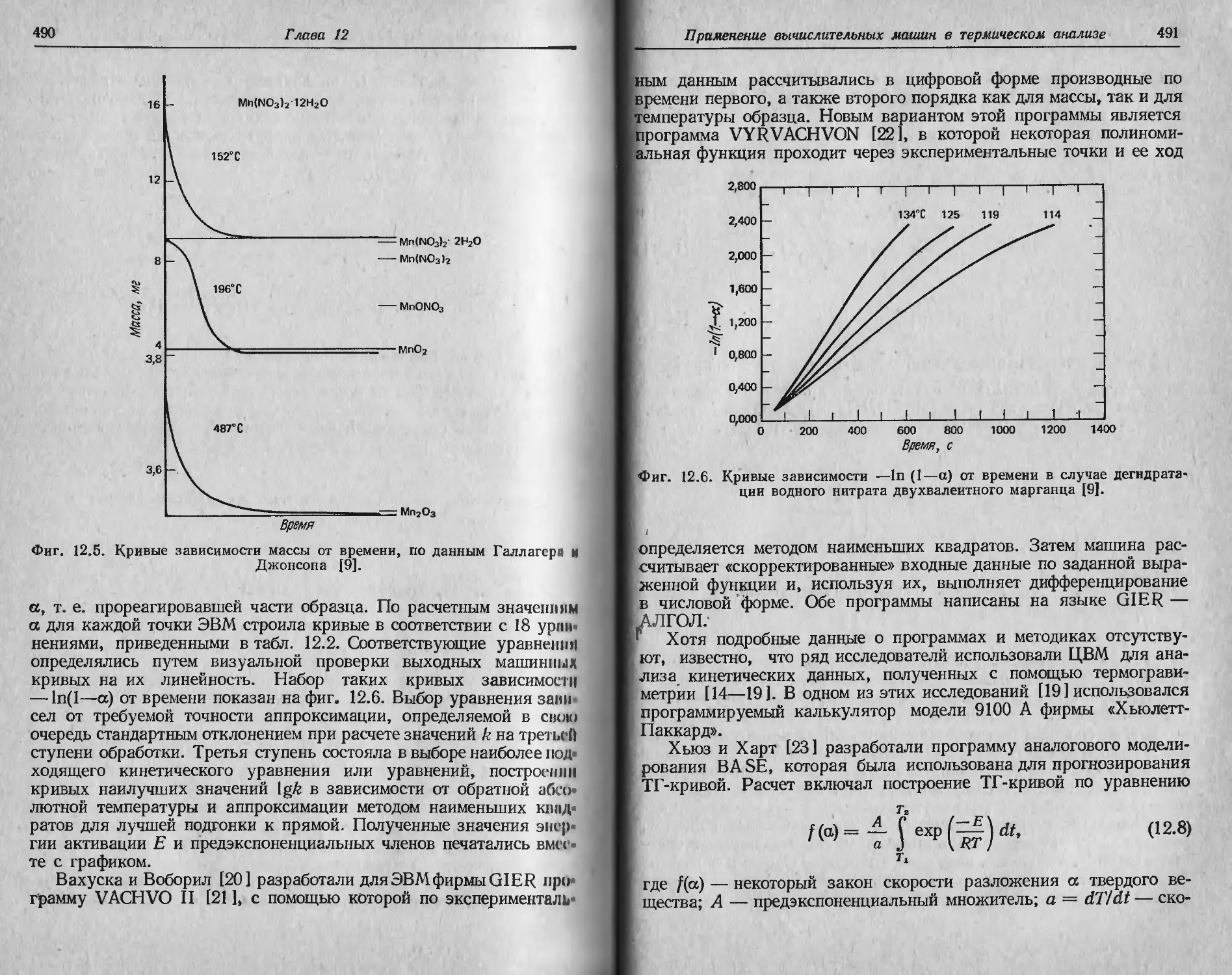
Фиг. 2.1. Характеристики ТГ-кривой в случае одностадийной реакции [1].
А (т.) ->В (т.) + С (г.).
такую температуру, при которой суммарное изменение массы дос-
тигает чувствительности термовесов, и б) конечную температуру
Т/, т. е. температуру, при которой суммарное изменение массы дос-
тигает максимального значения, соответствующего завершению ре-
акции. Хотя Tt может оказаться минимальной температурой, со-
ответствующей началу обнаруживаемого изменения массы в дан-
ном опыте, она не является ни температурой перехода в физическом
смысле, ни истинной температурой разложения, ниже которой ско-
рость реакции внезапно становится равной нулю. При нагревании
С постоянной скоростью температура Т/ должна быть выше Тр, раз-
ность Tj—Ti называется интервалом реакции. В эндотермической
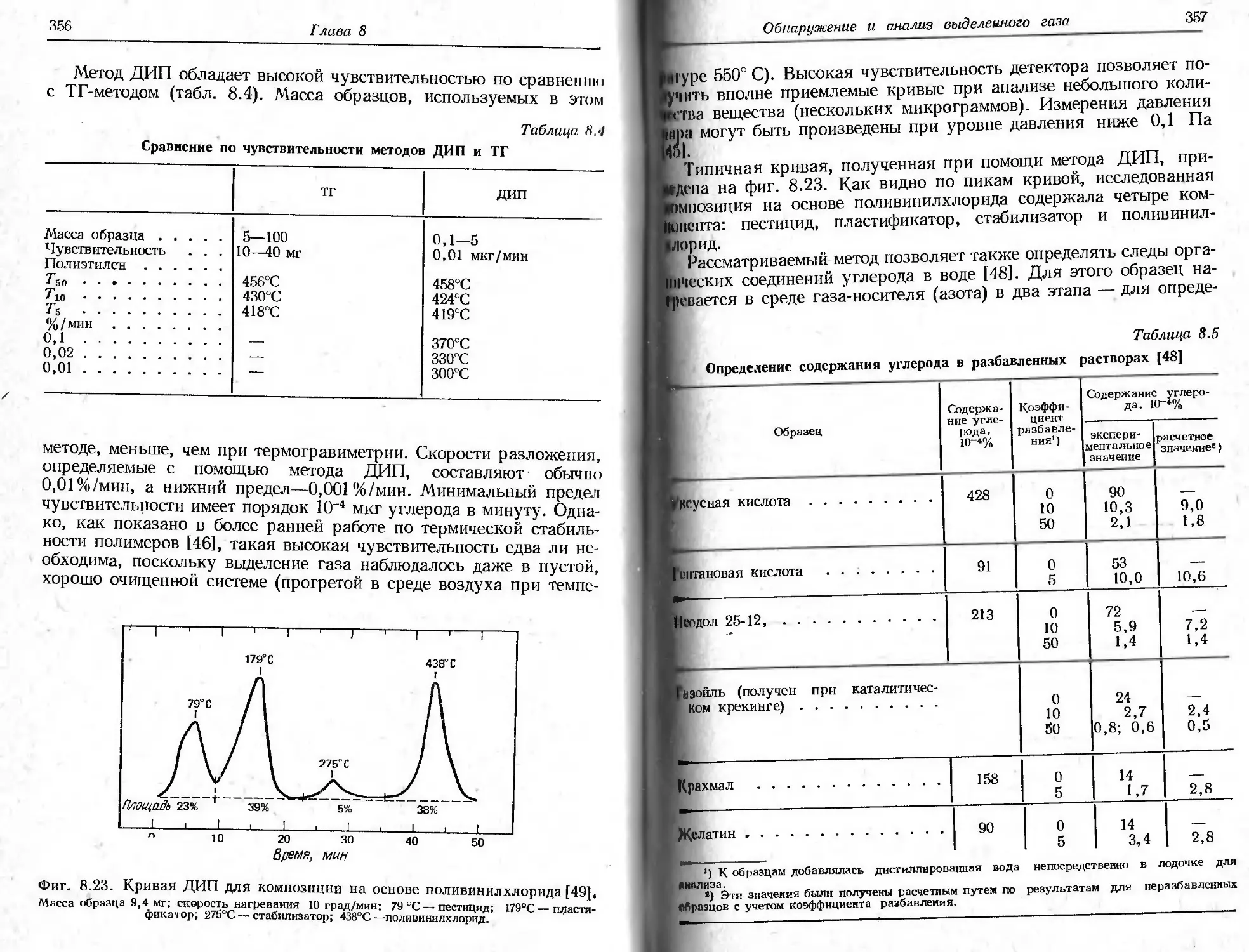
реакции разложения как Tit так и Т j возрастают с увеличением
скорости нагревания, причем Tf растет быстрее, чем Tt.
Гермин «термическая стабильность» в общем случае означает
способность вещества сохранять свои свойства при нагревании
Прпктнчески неизменными [2]. С утилитарной точки зрения термо-
стабилыюсть следует рассматривать как способность вещества вы-
полнять свои функции при различных окружающих условиях.
Способность вещества сохранять свою массу при различных усло-
виях оценивается с помощью термовесов.
История термогравиметрии достаточно широко представлена
Н работах Дюваля [3—5 ] и других авторов [6—8 ]. Родоначальни-
ком ЭТОГО метода, очевидно, является Хонда [9], создавший первые
16
Глава 2
термовесы в 1915 г. В последующие годы (1915—1920) метод был раз^
вит Гайчардом [10]. В конце 40-х —начале 50-х годов Дювалу
[3, 4 ] продемонстрировал возможности гравиметрии с помощью
термовесов. Современный этап развития термогравиметрии начался
в конце 50-х годов в связи с выпуском высококачественных про-
мышленных термовесов.
Б. НЕКОТОРЫЕ ФАКТОРЫ, ВЛИЯЮЩИЕ НА ХАРАКТЕР
ТЕРМОГРАВИМЕТРИЧЕСКИХ КРИВЫХ
Как и в любом другом методе измерений, в термогравиметрии
существует много факторов, влияющих на характер, воспроизво-
димость и точность результатов эксперимента. Термогравиметрия
отличается, возможно, даже большим числом переменных из-за
динамического характера изменения температуры образца. Дюваль
[3, 4, 11] подробно описал предосторожности, которые необходи-
мо соблюдать при работе с термовесами, а также влияние различных
факторов на результаты измерений в термогравиметрии. Ниже бу-
дут рассмотрены только важнейшие из них. Факторы, которые мо-
гут повлиять на характер термогравиметрической кривой образца,
можно разделить на две основные группы.
1) Факторы, связанные с измерительным прибором (термо-
весами):
а) скорость нагревания печи;
б) скорость записи;
в) атмосфера печи;
г) форма держателя образца и печи;
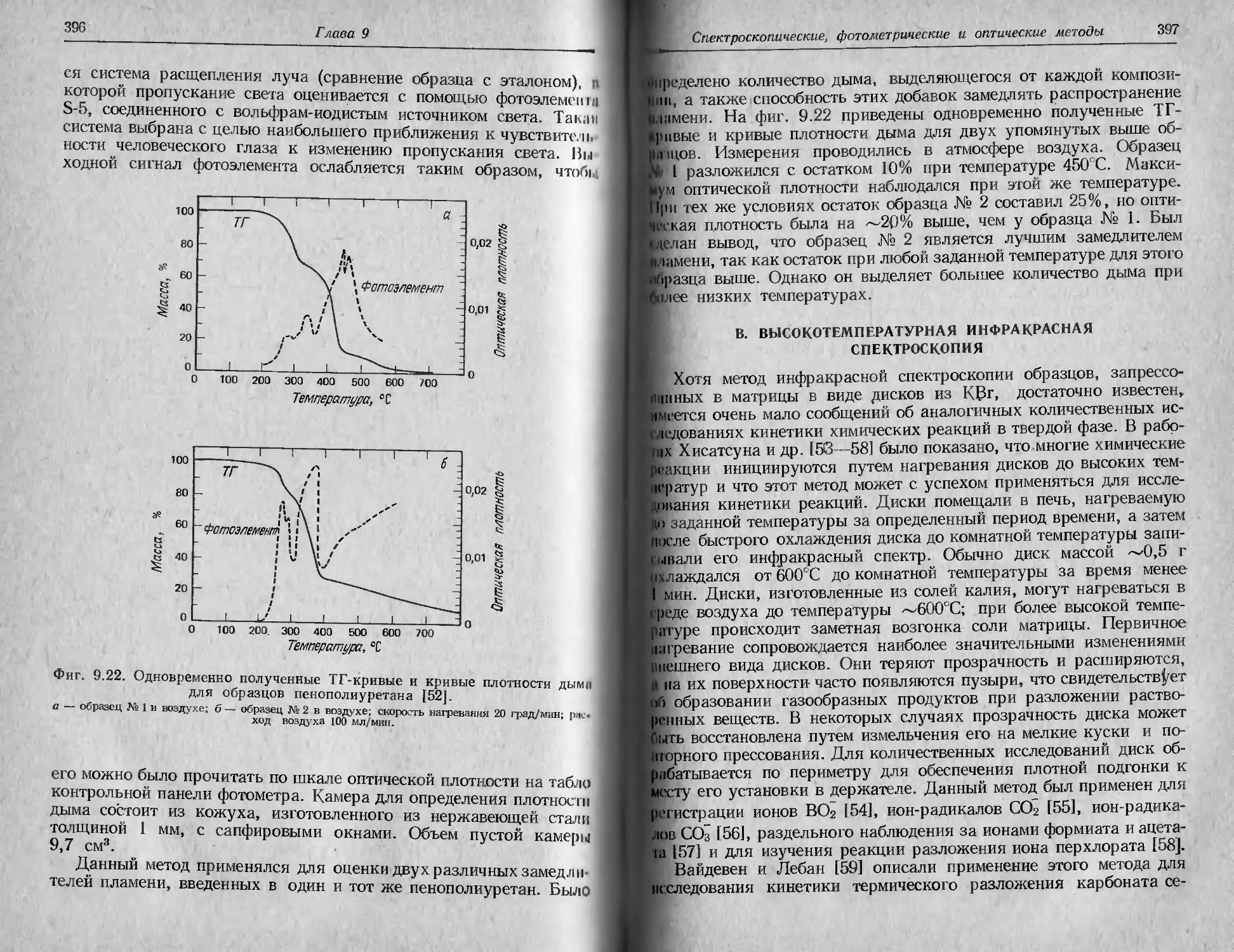
д) чувствительность записывающего устройства;
е) химический состав материала контейнера для образца.
2) Характеристики образца:
а) масса образца;
б) растворимость в образце выделяющихся из него газов;
в) размер частиц образца;
г) теплота реакции;
д) плотность упаковки частиц образца;
е) состав образца;
ж) теплопроводность.
К сожалению, роль некоторых из приведенных выше факторов
изучена недостаточно; если исследования и проводились, то их
результаты приложимы лишь к одному типу термовесов или запи-
сывающего устройства, и практически нельзя эти результаты рас-
пространять на другие типы приборов. Правда, многие из упомяну-
тых факторов, такие,как форма держателя образца, скорость запи-
си, чувствительность термовесов и выталкивающая сила воздуха
в контейнере для образца, постоянны для каждого типа термовесов.
Переменными и трудно воспроизводимыми факторами являются
Т ермогравиметрия
17
размер частиц образца, плотность упаковки его частиц, раствори-
мость выделяющихся газов в образце, конвекционные потоки в
печи и электростатические эффекты. В связи со сказанным стоит
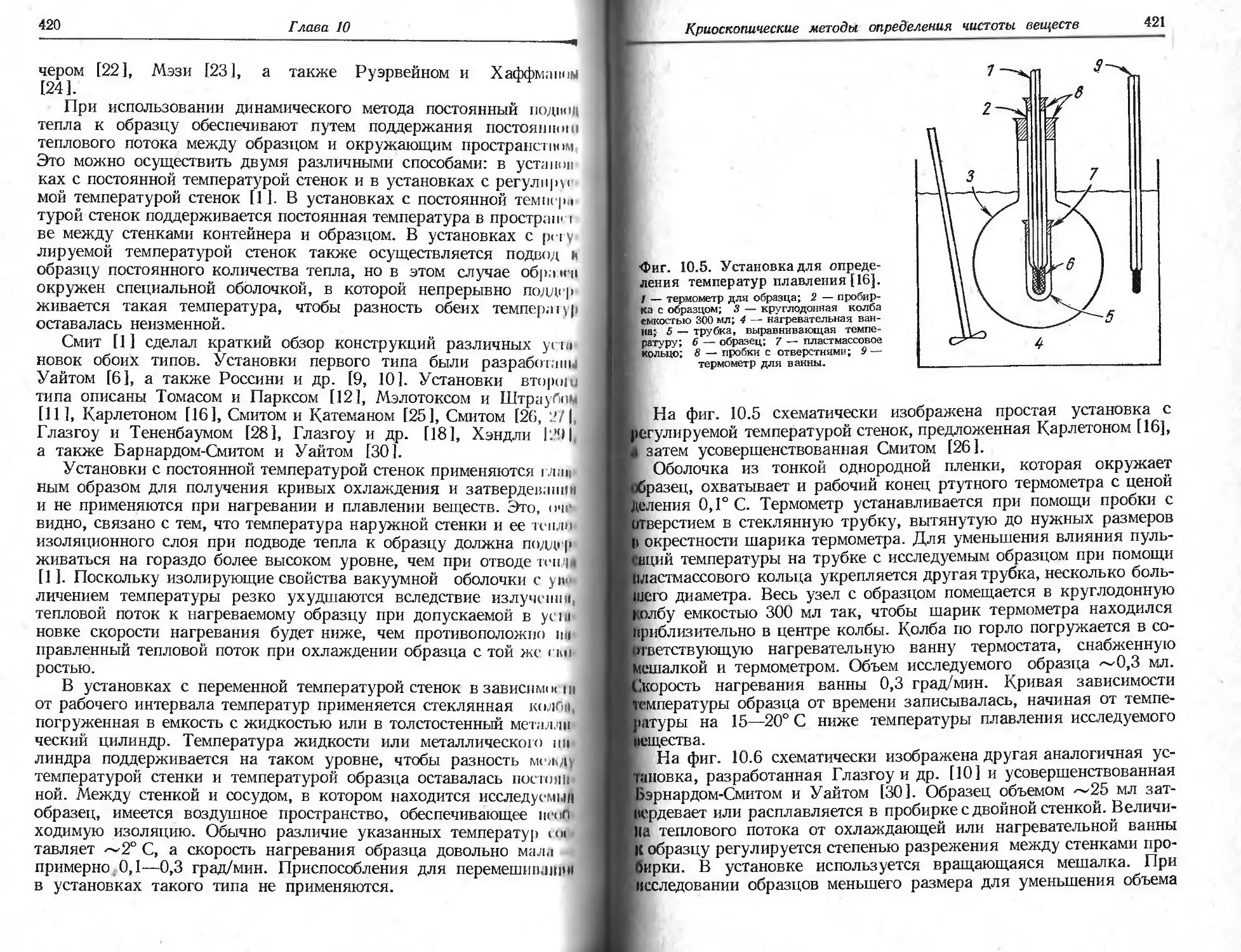
вЫразить сожаление, что отсутствует стандартный образец, позво-
ляющий сравнивать разные измерительные приборы. В гл. 3, од-
нАко, рассмотрено применение нескольких видов образцов стан-
дартного состава, предназначенных для калибровки по температуре.
1. ФАКТОРЫ, СВЯЗАННЫЕ С ИЗМЕРИТЕЛЬНЫМ ПРИБОРОМ
(ТЕРМОВЕСАМИ)
а. Скорость нагревания печи
Влиянию изменения скорости нагревания на наблюдаемую тем-
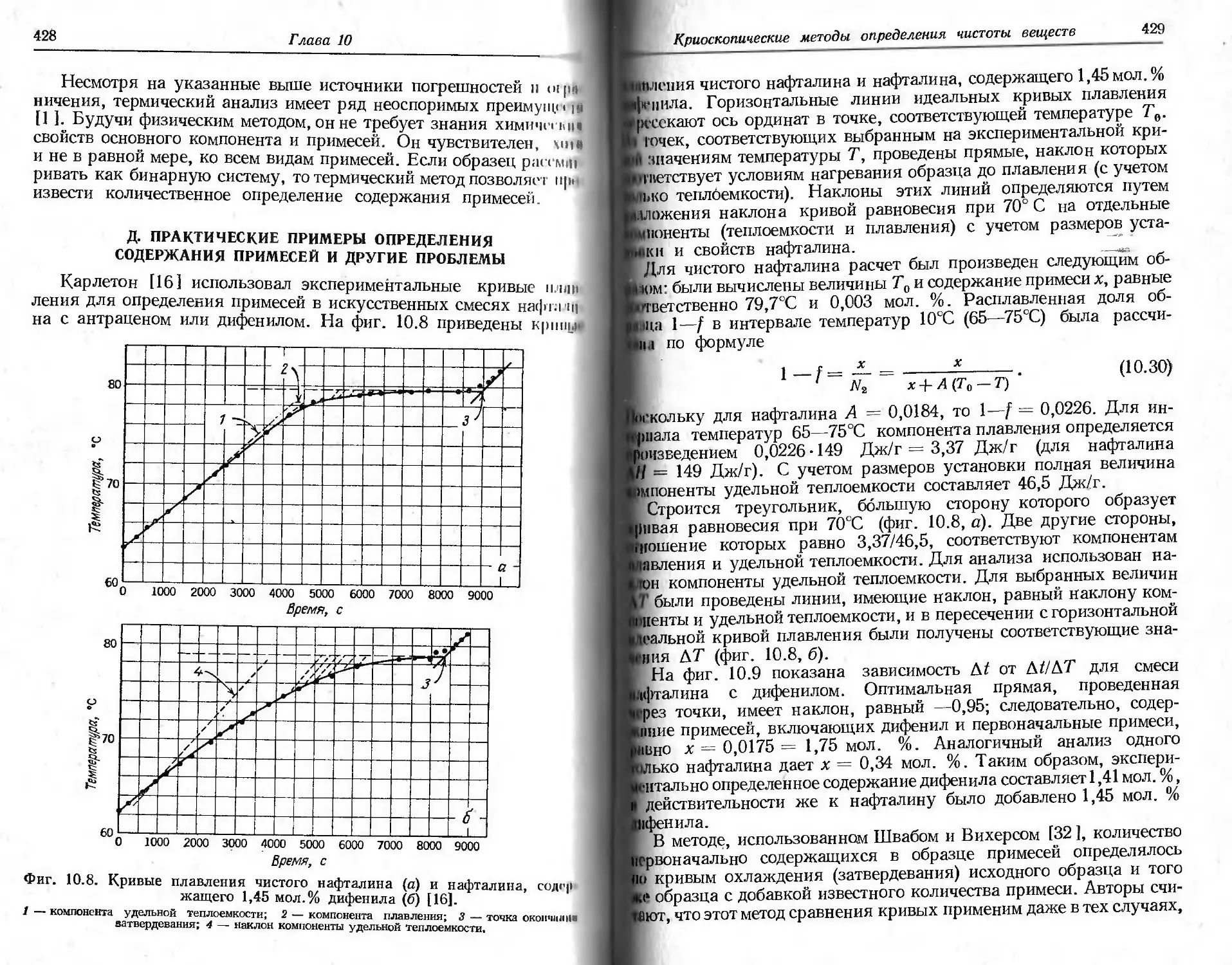
пературу разложения образца посвящено много исследований,
уступающих, возможно, по своему числу только работам, относя-
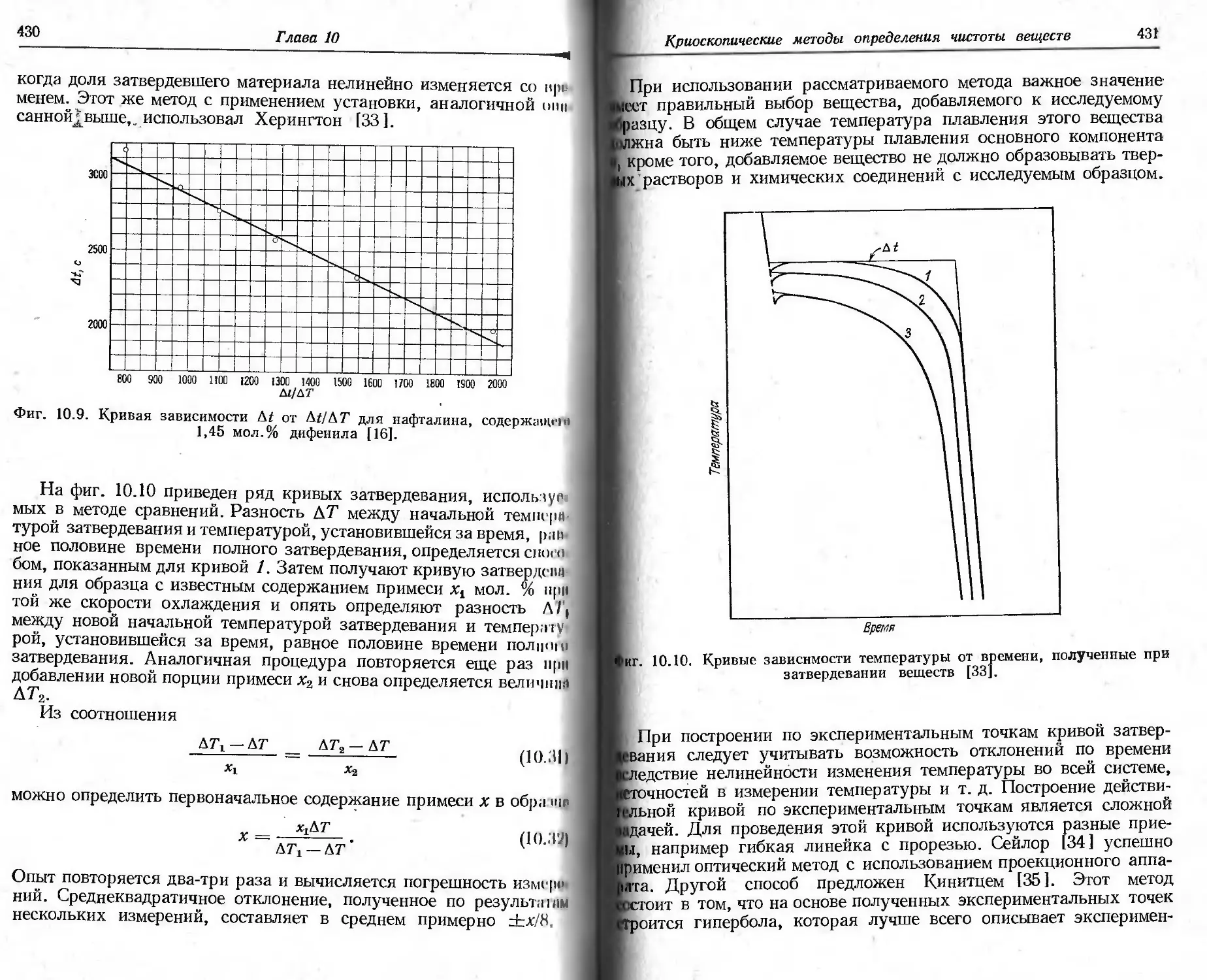
щимся к изучению влияния состава атмосферы на термогравимет-
рическую кривую. Симмонс и Ньюкирк [1 ] показали, что для Tt
и 7/ при быстром (б) и медленном (м) нагревании образца справед-
ливы следующие соотношения: для начальной наблюдаемой тем-
пературы разложения Tt
(т^х^к,
11 для конечной наблюдаемой температуры Тf
(Tf)6XTf)N.
11птервал реакции Т f—Тt изменяется в соответствии с выражением
(b-T^XTf-T^.
Для любого заданного интервала температур степень разложения
одного и того же образца при медленном нагревании больше, чем
при быстром. В случае экзотермической реакции температура об-
разца превысит температуру печи и, как показано в работе [8],
рн шость температур образца и печи в ходе реакции максимальна
при большей скорости нагревания. При протекании нескольких
последовательных реакций их удается разделить при определенных
скоростях нагревания. При увеличении скорости нагревания на
участке ТГ-кривой, имеющем вид горизонтального плато при мед-
Л ином нагревании, появляется точка перегиба.
Влияние скорости нагревания на характер ТГ-кривой образца
изучалось многими авторами. Следует отметить работы и обзоры
Дюваля [3, 4, 11 ], Ньюкирка [12], Редферна и др. [6, 8], Симмон-
са п Уэндландта [13], Деврие и Геллингса [14], Хербелла [15]
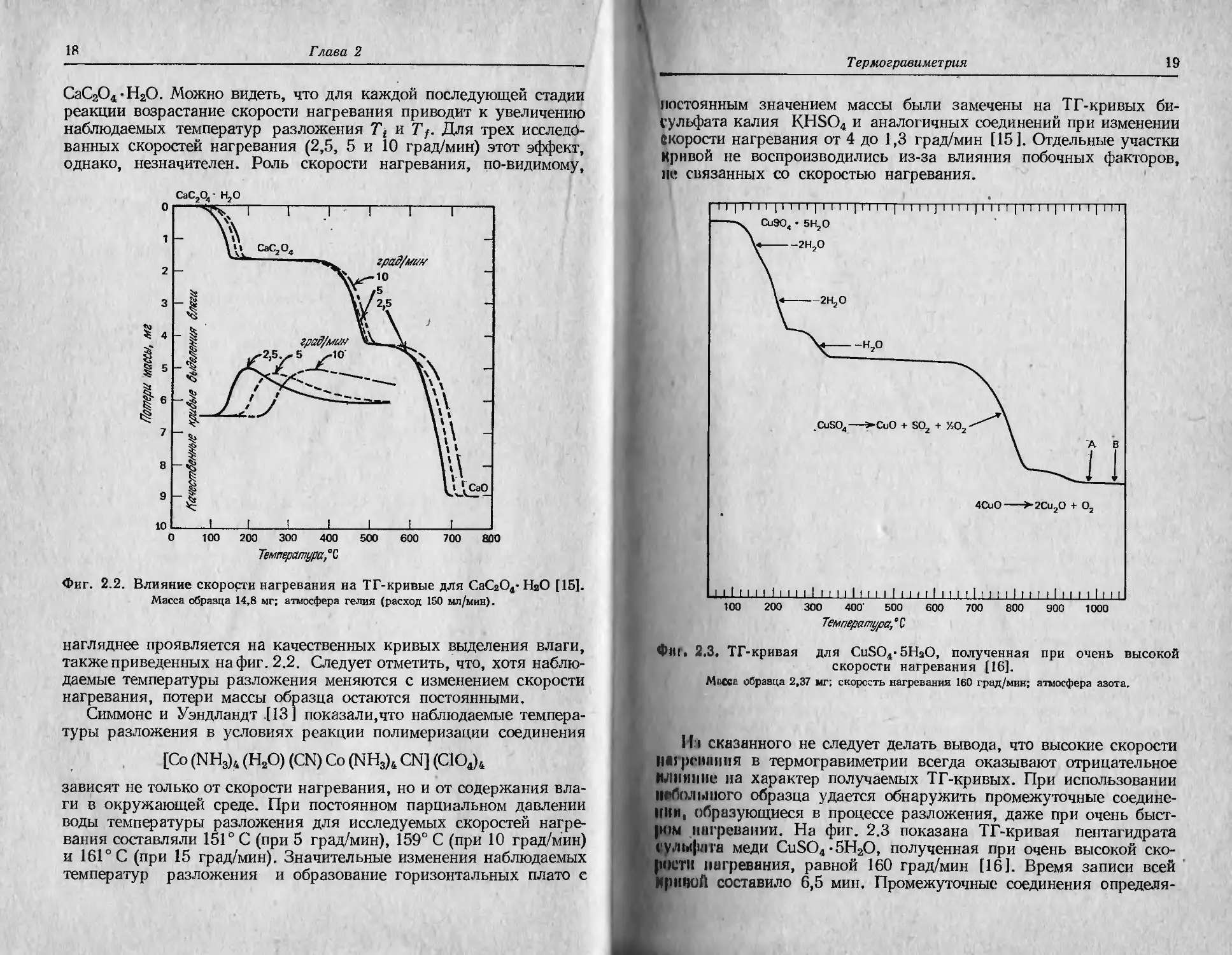
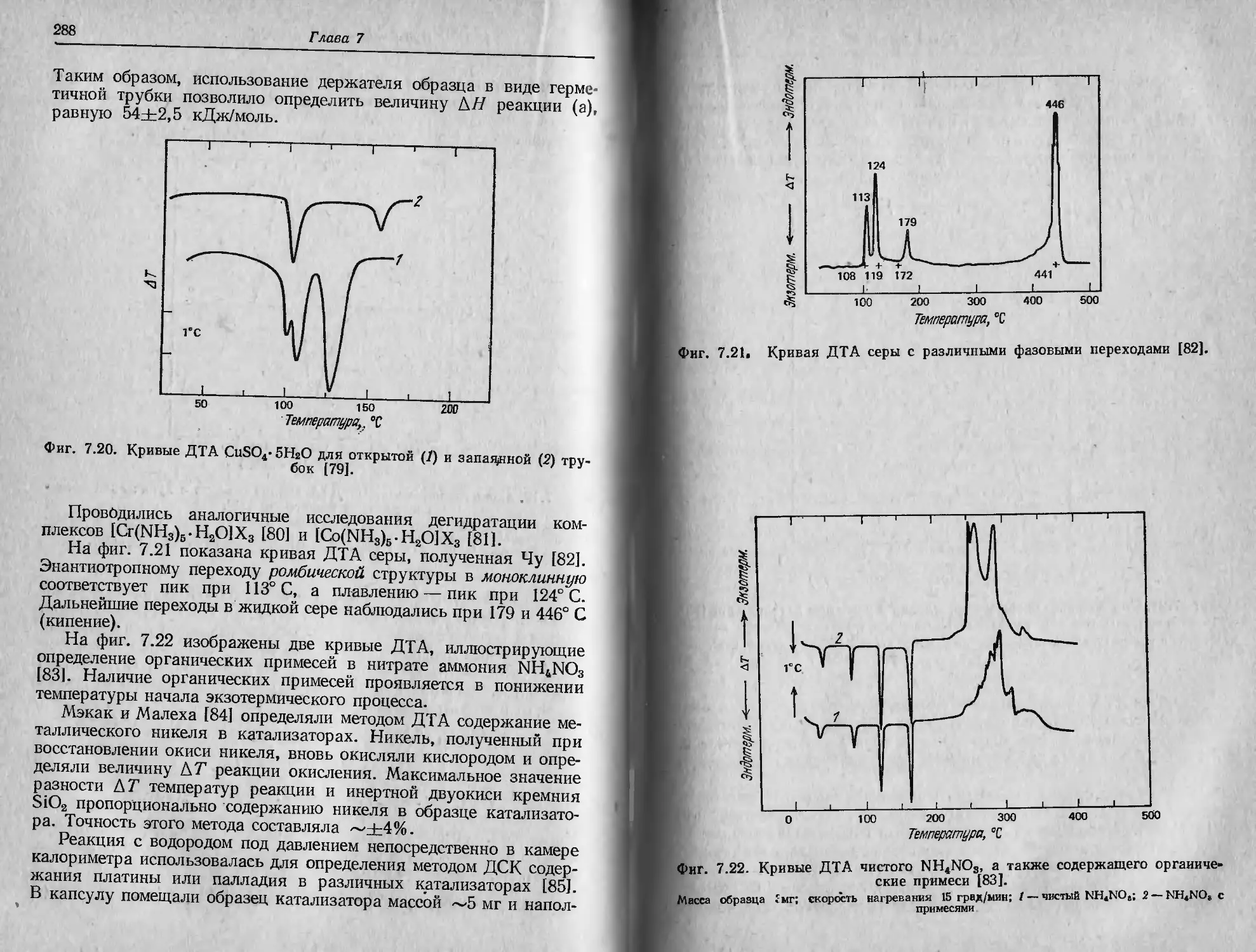
и др. Па фиг. 2.2 показаны ТГ-кривые, соответствующие трем раз-
И1ЧПЫМ скоростям нагревания гидрата оксалата кальция
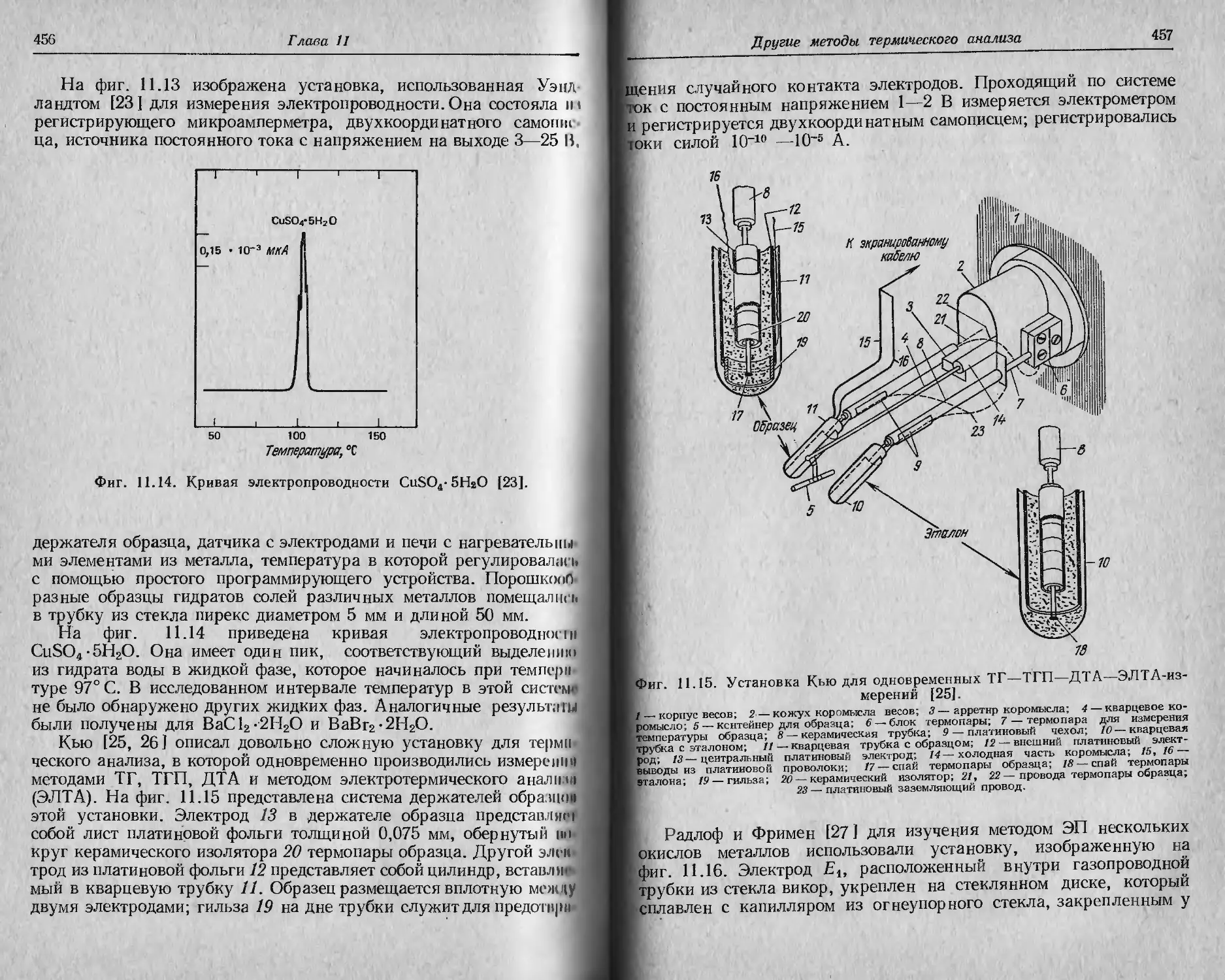
18
Глава 2
СаС2О4-Н2О. Можно видеть, что для каждой последующей стадии
реакции возрастание скорости нагревания приводит к увеличению
наблюдаемых температур разложения Tt и Tf. Для трех исследо-
ванных скоростей нагревания (2,5, 5 и 10 град/мин) этот эффект,
однако, незначителен. Роль скорости нагревания, по-видимому,
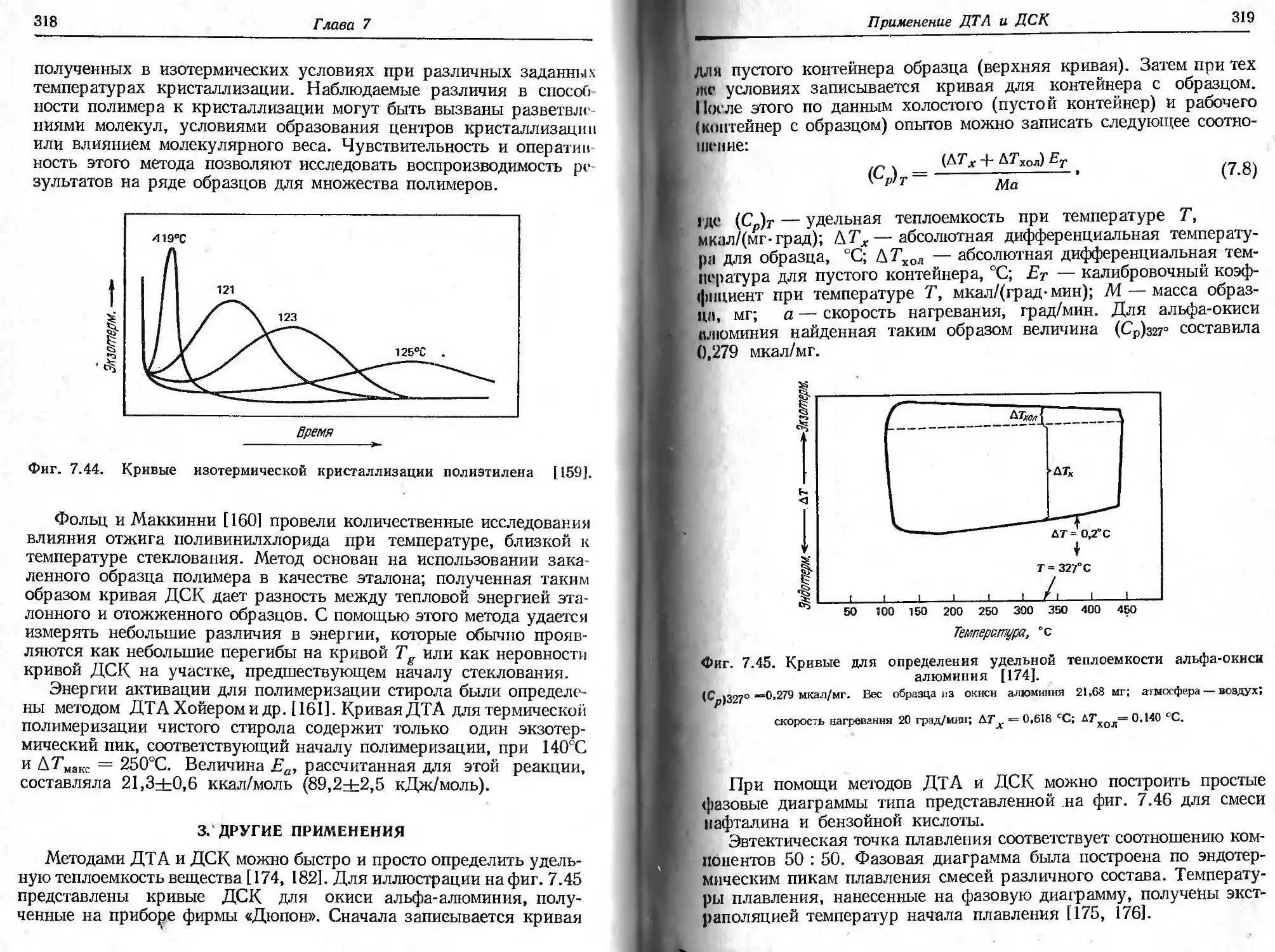
Фиг. 2.2. Влияние скорости нагревания на ТГ-кривые для СаСгО4- НаО [15].
Масса образца 14,8 мг; атмосфера гелия (расход 150 мл/мин).
нагляднее проявляется на качественных кривых выделения влаги,
также приведенных нафиг. 2.2. Следует отметить, что, хотя наблю-
даемые температуры разложения меняются с изменением скорости
нагревания, потери массы образца остаются постоянными.
Симмонс и Уэндландт [13 ] показали,что наблюдаемые темпера-
туры разложения в условиях реакции полимеризации соединения
[Со (NH3)4 (Н2О) (CN) Со (NH3)4 CN] (СЮ4)4
зависят не только от скорости нагревания, но и от содержания вла-
ги в окружающей среде. При постоянном парциальном давлении
воды температуры разложения для исследуемых скоростей нагре-
вания составляли 151° С (при 5 град/мин), 159° С (при 10 град/мин)
и 161° С (при 15 град/мин). Значительные изменения наблюдаемых
температур разложения и образование горизонтальных плато с
Т ермогравиметрия
19
постоянным значением массы были замечены на ТГ-кривых би-
сульфата калия KHSO4 и аналогичных соединений при изменении
Скорости нагревания от 4 до 1,3 град/мин [15]. Отдельные участки
Кривой не воспроизводились из-за влияния побочных факторов,
не связанных со скоростью нагревания.
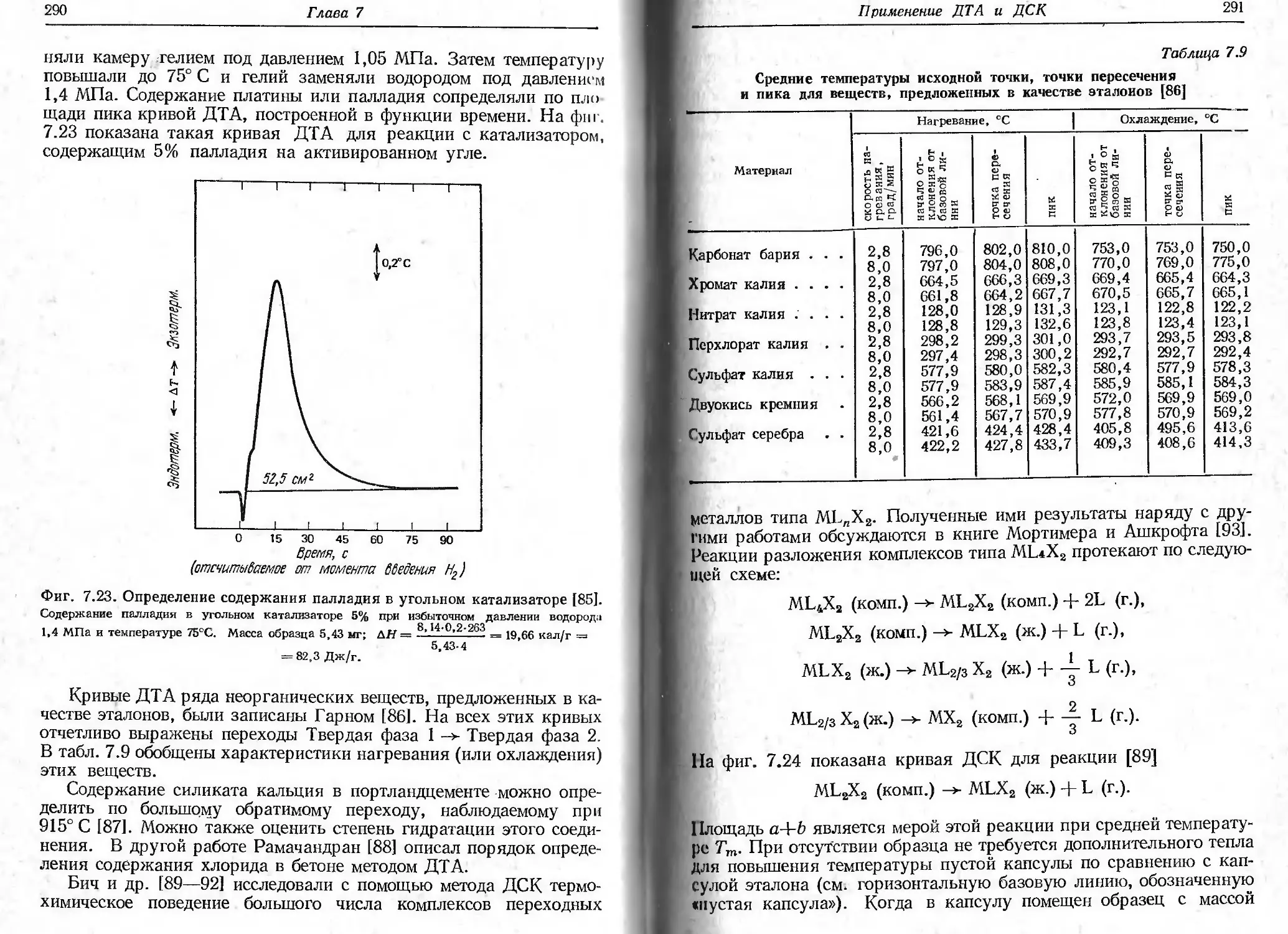
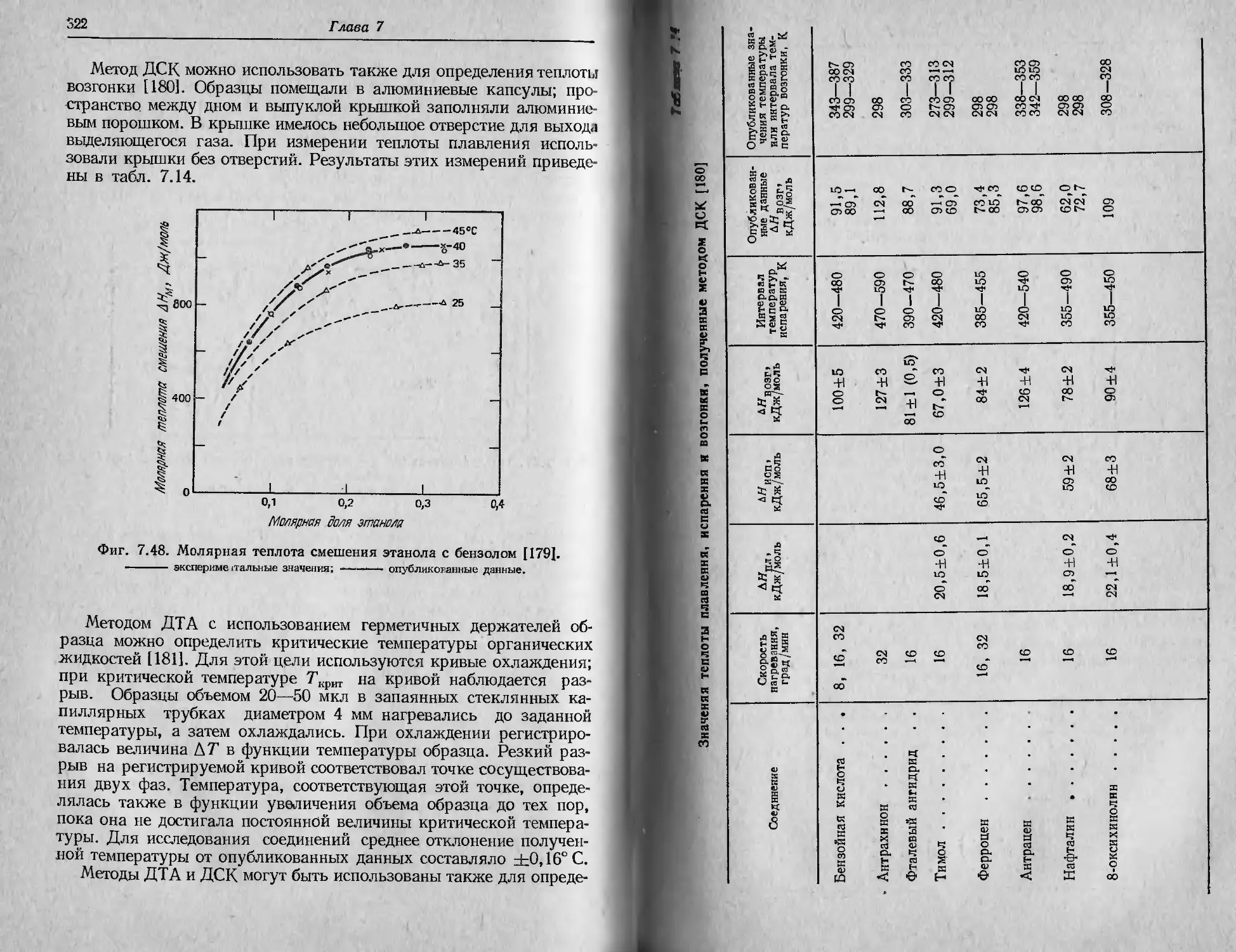
ф|П. 2.3, ТГ-кривая для CuSO4-5HaO, полученная при очень высокой
скорости нагревания [16].
Мгссс образца 2,37 мг; скорость нагревания 160 град/мин; атмосфера азота.
I I I сказанного не следует делать вывода, что высокие скорости
Н>| рсинипя в термогравиметрии всегда оказывают отрицательное
ИЛПиние на характер получаемых ТГ-кривых. При использовании
небольшого образца удается обнаружить промежуточные соедине-
нии, образующиеся в процессе разложения, даже при очень быст-
ром нагревании. На фиг. 2.3 показана ТГ-кривая пентагидрата
сульфата меди CuSO4-5H2O, полученная при очень высокой ско-
рости нагревания, равной 160 град/мин [16]. Время записи всей
Ириной составило 6,5 мин. Промежуточные соединения определи-
20
Глава 2
лись либо по точкам перегиба кривой, либо по горизонтальным
плато.
Возможность обнаружения промежуточных соединений по ТГ-
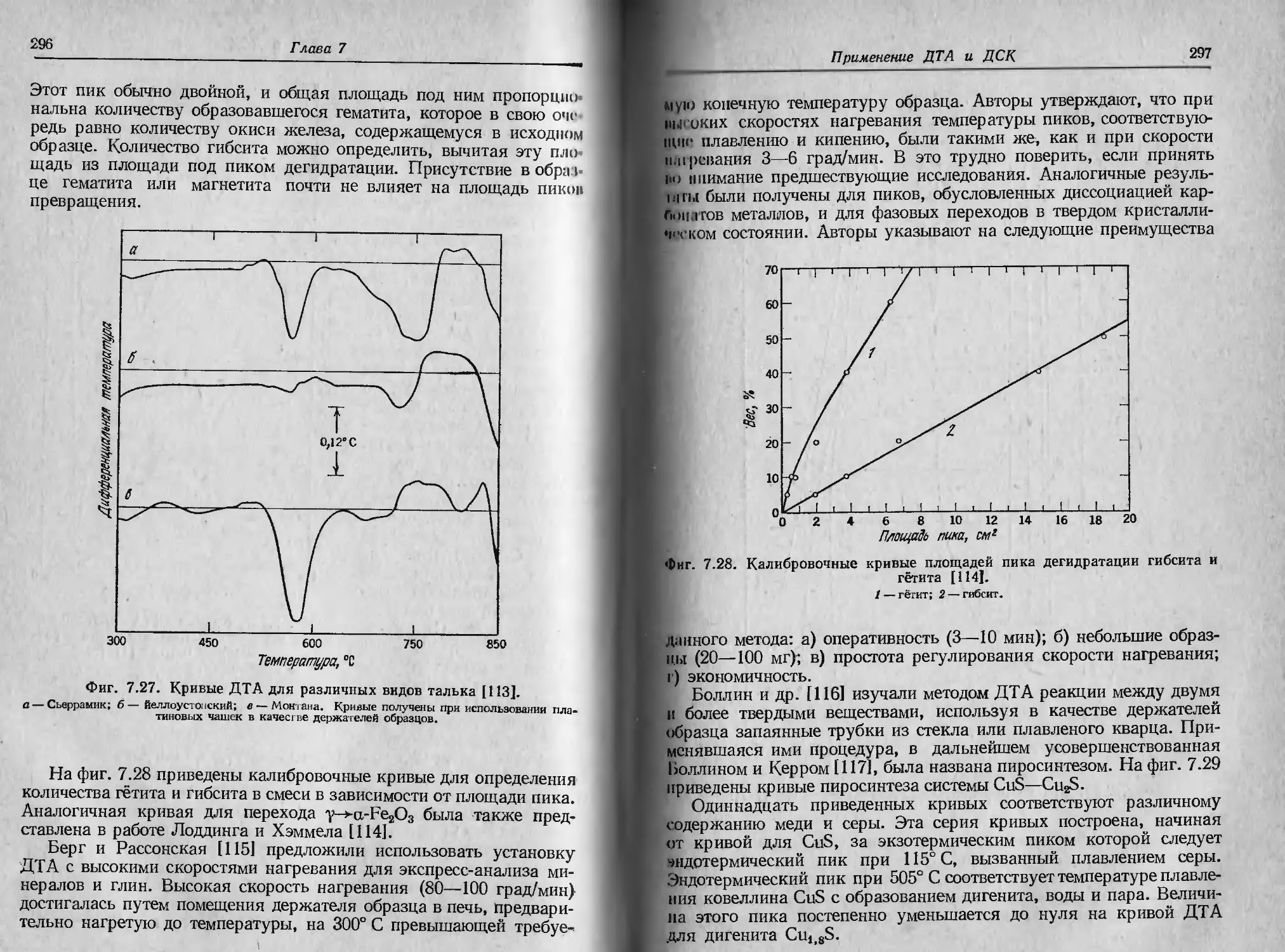
кривой, несомненно, зависит от скорости нагревания. Как видно
из фиг. 2.4,при высокой скорости нагревания кривые не имеют
перегибов, соответствующих образованию промежуточных соеди-
нений, однако такие перегибы появляются при меньшей скорости
Фиг. 2.4. Влияние скорости нагревания на образование промежу-
точных соединений [4].
а — высокая скорость нагревания; б — низкая скорость нагревания; в — низкая скорость на-
гревания н пониженная скорость записи.
нагревания [4,5]. При нагревании гептагидрата сульфата никеля
NiSO4-7H2O со скоростью 0,6 град/мин Фрухарт и Мичел [17] об-
наружили промежуточные соединения, на молекулу которых при-
ходится 1, 2, 4 и 6 молекул кристаллизационной воды. В предыду-
щем исследовании [18] при скорости нагревания 2,5 град/мин уда-
лось обнаружить только моногидрат. Аналогичные наблюдения
были сделаны при исследовании хелатного соединения (моносали-
цилальдоксинцинка) Ринасевичем и Флэггом [19], а также Деклер-
ком и Дювалем [20]. При нагревании со скоростью 300 град/ч осад-
ка, содержащего 250% влаги, в интервале температур 215—290° С
на ТГ-кривой образовалось горизонтальное плато, соответствующее
постоянному значению массы [19 ]. Деклерк и Дюваль [20], произ-
водившие нагревание со скоростью 380 град/ч, не зарегистрировали
на кривой горизонтального плато и сочли данный метод непри-
годным для определения цинка. Результаты последней работы по-
казали, что при исследовании образцов с большим содержанием
влаги нагревание должно быть медленным. Следует также заметить,
Т ермогравиметрия
21
Что неожиданный изгиб на кривой уменьшения массы может быть
обусловлен внезапным изменением скорости нагревания, и, следо-
пятельно, оказаться ложным [6]. Одним из методов, исключающих
возможные заблуждения, является непрерывная запцсь ленточным
гименшсцемтемпературы печи в функции времени. Нарушения тем-
пературного режима рассматривается в гл. 3.
Волее явно выраженные горизонтальные йлато на ТГ-кривой
можно получить при использовании квазистатического режима
нагревания, о котором упоминалось выше. Этот метод, примененный
впервые Хонда [9 J, а затем Лукашевским и Редферном [6] и Пау-
ликами [21], предусматривает прерывание линейного роста темпе-
Iштурм и продолжение опыта.при заданной постоянной температуре,
(рпвые уменьшения массы, характерные для этого метода, в общем
случае являются более крутыми, чем кривые, полученные в дина-
мическом режиме, и позволяют получить более точные данные
о конечных температурах разложения.
б. Скорость записи
Как при быстром, так и при медленном нагревании скорость запи-
си кривых уменьшения массы может заметно влиять на форму кри-
вых. На фиг. 2.5 показано влияние скорости записи на форму кри-
вых различных реакций. На фиг, 2.5. а с увеличением скорости
записи кривая реакции медленного термического разложения ста-
новится более пологой. В том случае, когда за медленной реакцией
следует быстрая (фиг. 2.5, б), при меньшей скорости записи они
разделены менее четко. Когда медленная реакция следует за быстрой
(фиг, 2.5, в), при меньшей скорости записи наблюдается такой же
«ффскт, как и в случае, представленном на фиг. 2,5, б, а именно
более короткие горизонтальные плато.
При очень высокой скорости записи наблюдается тенденция к
уменьшению до минимума различия в скоростях потери массы. Для
скорости нагревания 1—6 град/мин рекомендуется скорость про-
тнжкп ленты 15—30 см/ч [6]. У двухкоординатных самописцев
г 11Н1ИСЫО двух переменных скорость записи по оси температур
определяется чувствительностью прибора и скоростью нагревания,
II то время как скорость записи по оси массы определяется чувстви-
|гльцостью пера самописца и весов, а также скоростью термиче-
< I ого разложения. Симмонс и Ньюкирк [1 ] указали на недостатки
Использования таких самописцев для записи термогравиметр иче-
CKUX данных, поскольку неожиданные нарушения скорости, нагре-
и 1НЦЯ или чувствительности термопары могут вызвать ложные из-
менения хода кривой масса — температура, Только путем незави-
симой записи по оси температура — время удается обнаружить
•ГП нежелательные воздействия и отделить от них эффекты, связан-
на исключительно с изменением массы образца.
22
Глава 2
Фиг. 2.5. Влияние скорости движения ленты самописца на форму кривых
уменьшения массы [6].
I — низкая скорость ленты; II — высокая скорость ленты.
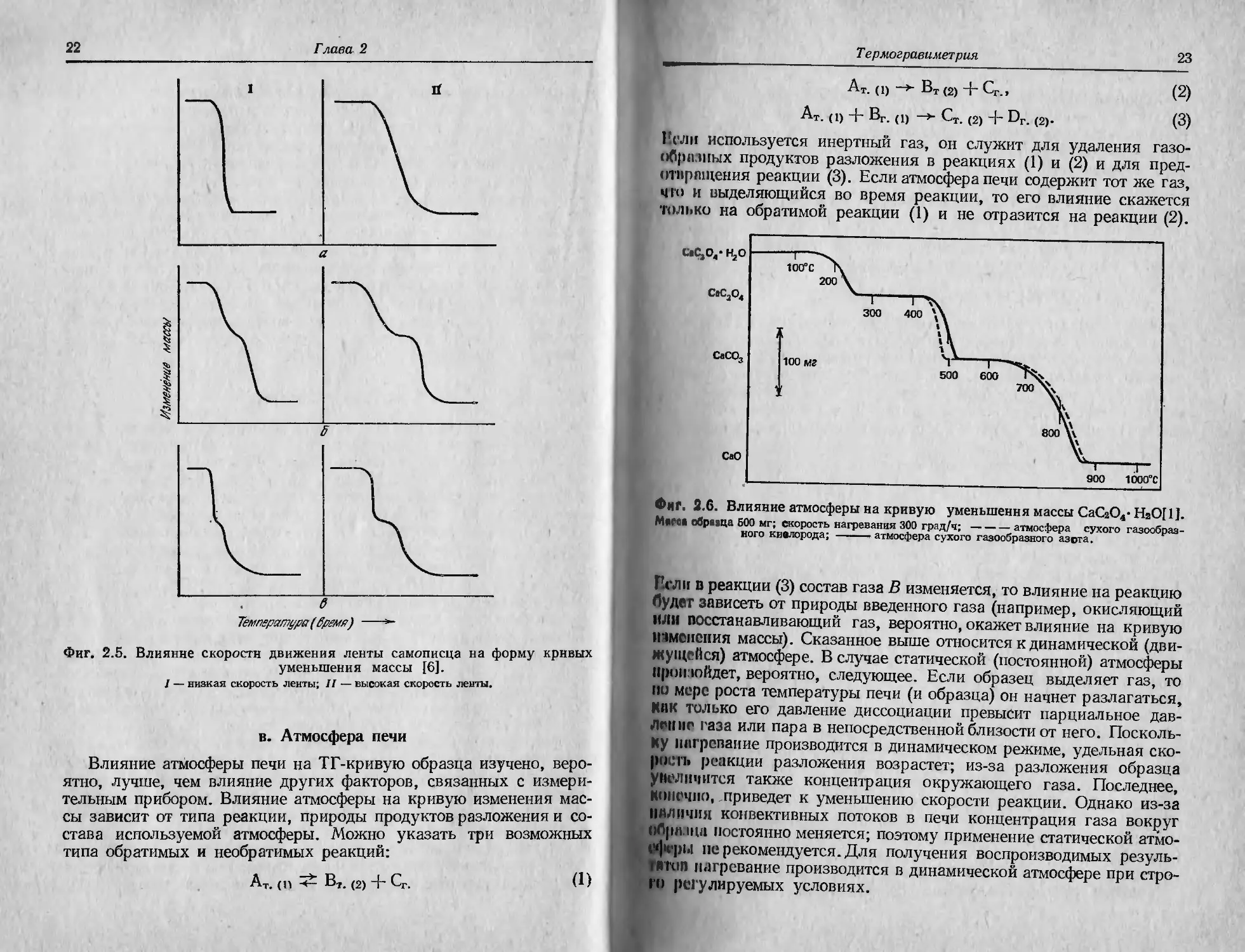
в. Атмосфера печи
Влияние атмосферы печи на ТГ-кривую образца изучено, веро-
ятно, лучше, чем влияние других факторов, связанных с измери-
тельным прибором. Влияние атмосферы на кривую изменения мас-
сы зависит от типа реакции, природы продуктов разложения и со-
става используемой атмосферы. Можно указать три возможных
типа обратимых и необратимых реакций:
Ат. (п Вт. (2) + Сг. (1)
Т ермогравиметрия
23
Ат. (1) —> Вт (2) + Сг_,
(2)
Ат. (1) + Вт. (1) —> Ст. (2) + Dr. (2).
(3)
1’слн используется инертный газ, он служит для удаления газо-
обрпзпых продуктов разложения в реакциях (1) и (2) и для пред-
отвращения реакции (3). Если атмосфера печи содержит тот же газ,
что и выделяющийся во время реакции, то его влияние скажется
только на обратимой реакции (1) и не отразится на реакции (2).
Фиг. 2.G. Влияние атмосферы на кривую уменьшения массы СаСгО4- НзО[1].
MiCCЛ обрааца 500 мг; скорость нагревания 300 град/ч;-атмосфера сухого газообраз-
ного кислорода; ---------------атмосфера сухого газообразного азота.
Гели в реакции (3) состав газа В изменяется, то влияние на реакцию
будет зависеть от природы введенного газа (например, окисляющий
или восстанавливающий газ, вероятно, окажет влияние на кривую
изменения массы). Сказанное выше относится к динамической (дви-
жущейся) атмосфере. В случае статической (постоянной) атмосферы
произойдет, вероятно, следующее. Если образец выделяет газ, то
по мерс роста температуры печи (и образца) он начнет разлагаться,
КПК только его давление диссоциации превысит парциальное дав-
ление газа или пара в непосредственной близости от него. Посколь-
ку нагревание производится в динамическом режиме, удельная ско-
рое п» реакции разложения возрастет; из-за разложения образца
увеличится также концентрация окружающего газа. Последнее,
Конечно, приведет к уменьшению скорости реакции. Однако из-за
наличия конвективных потоков в печи концентрация газа вокруг
обри ща постоянно меняется; поэтому применение статической атмо-
сферы не рекомендуется. Для получения воспроизводимых резуль-
татов нагревание производится в динамической атмосфере при стро-
го регулируемых условиях.
24
Глава 2
Хорошей иллюстрацией влияния атмосферы на обратимые и не-
обратимые реакции, такие, как (1)—(3), является фиг. 2.6 [11.
Кривые показывают влияние нагревания гидрата оксалата каль-
ция СаС2О4-Н2О в атмосферах сухого газообразного азота N2 и су-
хого газообразного кислорода О2. Обратимая реакция дегидратации
СаС2О4 Н2О (т.) СаС2О4 (т.) + Н2О (г.)
не подвержена влиянию атмосферы, так как оба газа одинаково
эффективно удаляют с поверхности образца выделяющиеся пары
воды. На второй стадии реакции разложения
СаС2О4 (т.) СаСО3 + СО (в атмосфере N2)
кривые расходятся, так как кислород взаимодействует с выделяю-
щейся окисью углерода СО, в результате чего происходит вторич-
ная реакция окисления, повышающая температуру непрореагиро-
вавшего твердого вещества. Это повышение температуры приводит
к заметному росту скорости разложения. Таким образом, разло-
жение соединения происходит быстрее и завершается при более
низкой температуре в атмосфере сухого кислорода, чем в инертной
атмосфере сухого азота.
Третья стадия реакции разложения
СаСО3 (т.) —СаО (т.) + СО2 (г.)
также обратима и поэтому не должна подвергаться воздействию
кислорода или азота. Однако можно заметить незначительную раз-
ницу в ходе кривых для указанных двух газов. Она приписыва-
лась различию в составе карбоната кальция СаСО3, образующего-
ся на второй стадии реакции разложения. СаСО3, полученный в
атмосфере кислорода, несколько отличается от СаСО3, получен-
ного в атмосфере азота. В чем состоит это отличие, в работе [1 ] не
указано, но там же установлено, что по кривым уменьшения мас-
сы нельзя обнаружить различия в размерах частиц, площадях по-
верхности, дефектах строения кристаллической решетки или дру-
гих характеристиках образца.
Для обратимых реакций, подобных описанным выше, повыше-
ние парциального давления двуокиси углерода в атмосфере печи
приводит к увеличению температуры Tt на ТГ-кривой (фиг. 2.7).
Наблюдаемая начальная температура разложения может иметь
значения в пределах примерно от 400 при пониженном давлении до
900° С в атмосфере двуокиси углерода при давлении 0,1013 МПа
(760 мм рт. ст.). Такая заметная разница обусловлена предельны-
ми‘значениями давления атмосферы печи, но она наглядно показы-
вает влияние ее атмосферы на ТГ-кривую. Паулик Дж. и др. [23]
описали подобное семейство ТГ-кривых для реакции разложения.
Многие проблемы получения воспроизводимых ТГ-кривых мо-
Т ермогравиметрия
25
гут быть решены путем использования метода «собственной» атмо-
сферы, рассмотренного в разд. Г этой главы.
Влияние паров воды в атмосфере печи изучено довольно подроб-
но HI, особенно для реакций дегидратации и гидратации. Фельдман
и Рамачандран [24] сконструировали специальные весы и контей-
нер для образца, позволившие исследовать реакции при регулируе-
мой концентрации паров воды. Многие реакции могут быть иссле-
дованы в атмосфере насыщенного водяного пара (гл. 3). Уэндландт
Температура, ° с
Фиг. 2.7. ТГ-кривые для СаСО3 при нагревании в различных атмосферах [22]
Масса образца 10 мг; скорость нагревания 10 град/мин.
П Симмонс [25] изучали термическое разложение дигидрата хлорида
бария ВаС12-2Н2О и дигидрата бромида бария ВаВг2-2Н2О в атмо-
сферах насыщенного водяного пара и сухого азота, а Хербелл [15]
исследовал восстановление окиси никеля N1O сухим и насыщенным
плитой водородом.
О влиянии пониженного давления на ТГ-кривые различных
соединений уже сообщалось. Так, Жино и Маноли [26] описали
влияние пониженного давления на реакцию дегидратации пента-
гидрата сульфата меди CuSO4 .5Н2О, а Никольсон [27 ] исследовал
воздействие пониженного давления на термическое разложение ди-
гидрата оксалата железа FeC2O4.2Н2О. Этому вопросу посвящено
много других работ.
Влияние повышенного давления на ТГ-кривые описано в работе
Ври у на и др. [281. Это влияние обратно влиянию пониженного дав-
ления; Tt реакции при повышении давления сдвигается в область
более высоких температур; интервал температур реакции Tf—Tt
>том случае также возрастает. На фиг. 2.8 показаны ТГ-кривые
( uS()4-5H2O, полученные на термовесах при повышенном давлении.
Кривая при давлении 2,07 МПа (~20 атм), после внесения поправ-
26
Глава 2
ки на выталкивающую силу воздуха, практически ие отличалась от
кривой, записанной при атмосферном давлении. Наблюдаемые
температуры разложения для этих двух кривых были почти оди-
наковыми, но, возможно, это объясняется ограниченным интерва-
лом изменения давления, в котором проводились измерения. Отри-
цательное влияние повышенного давления выражается в значитель-
ном увеличении выталкивающей силы воздуха; при этом обычно
вносятся соответствующие поправки (см. кривую 2).
Фиг. 2.8. ТГ-кривые дегидратации CuSO4- 5Н2О [28].
/ — эксперимент; 2 — с учетом поправки на выталкивающую силу воздуха.
Явное увеличение массы наблюдается иногда при термическом
разложении в высоком вакууме образцов «критической» толщины
и при использовании держателя образца специального типа. Этот
эффект иллюстрируется на фиг. 2.9, где приведены кривые дегидра-
тации СаС2О4-Н2О, полученные Видеманном [291. Пунктирная
линия соответствует реакции при нормальном атмосферном дав-
лении, а сплошная — реакции в условиях высокого вакуума. «Ска-
чок» массы образца в начале реакции происходит из-за столкнове-
ний молекул воды с содержащим образец контейнером во время
откачки воздуха.
Фридман [30] описал это явление на примере термического раз-
ложения тефлона (политетрафторэтилена) в вакууме. Он показал,
что величина этого мгновенного переходного эффекта определяется
выражением
w = tn----— av (2.1)
g \ di ) ' '
где w — показание массы образца на термовесах; т — действи-
тельная масса образца; g — ускорение силы тяжести; а — геомет-
Термогравиметрия
27
Фиг. 2.9. ТГ-кривые дегидратации СаСаО^-НаО [29].
------нормальное атмосферное давление; -- высокий вакуум.
рнчгскнй фактор, v — скорость отсасываемого газа; dm'dt — ско-
ромь изменения действительной массы.
Влияние атмосферы печи на ТГ-кривые рассматривалось также
и > мши их других работах [12, 32—37 ]. Этот вопрос нашел отраже-
iiiii и и ряде последних исследований [21, 38—41].
г. Форма держателя образца
В гл. 3 описано большое число держателей образца, используе-
мых н к'рмогравиметрии. Применяют держатели образца различ-
III формы: от ровных пластинок до глубоких тиглей различной
смкпгш. Держатели изготавливают из самых разных материалов:
г lehJiu, окиси алюминия, керамики, различных металлов и сплавов.
Симмонс и Ньюкирк [1] показали, что для гидрата оксалата
1Ы111» ('пС2О4-Н2О форма держателя образца не имеет значения
28
Глава 2
при отсутствии взаимодействия между образцом и газами, содержа-
щимися в атмосфере или продуктах разложения. Как видно из
фиг. 2.10, ТГ-кривые для СаС2О4-Н2О, нагреваемого в атмосфере
двуокиси углерода в двух разных держателях образца, при тем-
пературах выше 275° С идентичны. Как и ожидалась, дегидратация
Фиг. 2.10. Влияние формы держателя образца на ТГ-кривую для
СаСгО4- НгО в динамической атмосфере СОг [1].
< ------ плоская кварцевая тарелочка; - фарфоровый тигель.
происходила быстрее при использовании в качестве держателя об-
разца плоской кварцевой тарелочки, а не фарфорового тигля. Фор-
ма тигля не оказывала влияния на процесс разложения безводного
оксалата кальция СаС2О4, поскольку эта реакция необратима, а
в атмосфере двуокиси углерода не может протекать сколько-нибудь
существенная реакция, скорость которой определяется диффузией.
Форма держателя не влияла также на процесс разложения карбо-
ната кальция СаСО3, поскольку эта реакция обратима, а атмосфера
состояла исключительно из газа, выделившегося в процессе разло-
жения.
Когда термическое разложение происходило в динамической
атмосфере азота, от формы контейнера зависело уменьшение массы
как за счет паров воды, так и за счет двуокиси углерода. На раз-
ложение СаС2О4 форма держателя не влияла. Отмеченное влияние
формы держателя образца свидетельствует о том, что вовремя разло-
жения в тигле должно быть значительным давление паров воды и
двуокиси углерода, даже если динамическая газовая атмосфера не
содержит ни паров воды, ни двуокиси углерода [1].
На фиг. 2.11 показано различие в ТГ-кривых для пентагидрата
сульфата меди Си SO4 .5Н2О, полученных при использовании тигля
Т ермогравиметрия
29
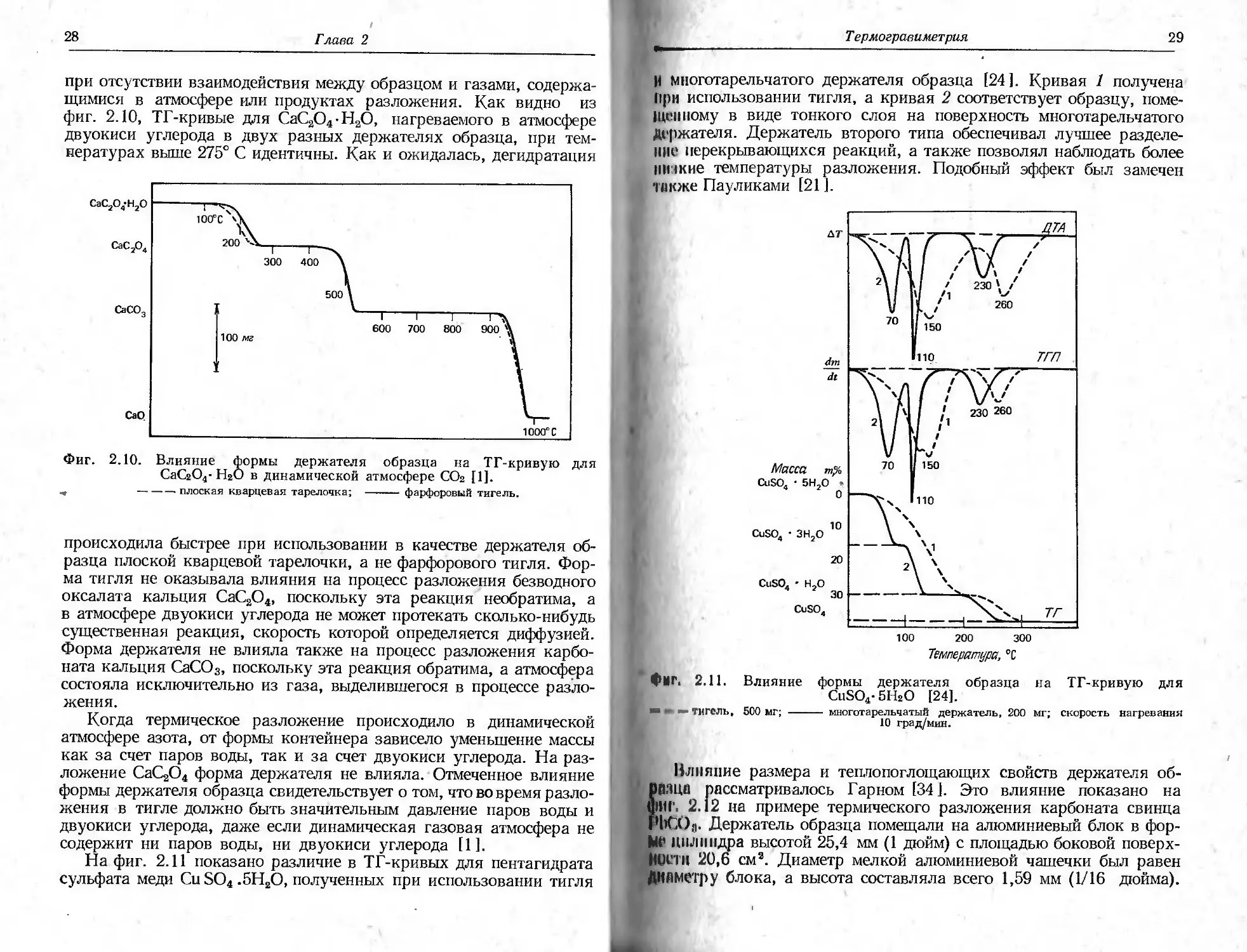
И многотарельчатого держателя образца [24]. Кривая 1 получена
При использовании тигля, а кривая 2 соответствует образцу, поме-
щенному в виде тонкого слоя на поверхность многотарельчатого
Держателя. Держатель второго типа обеспечивал лучшее разделе-
ние перекрывающихся реакций, а также позволял наблюдать более
низкие температуры разложения. Подобный эффект был замечен
также Пауликами [21 ].
Фиг, 2.11. Влияние формы держателя образца на ТГ-кривую для
CuSO4-5H2O [24].
тигель, 500 мг; --- многотарельчатый держатель, 200 мг; скорость нагревания
10 град/мин.
Влияние размера и теплоноглощающих свойств держателя об-
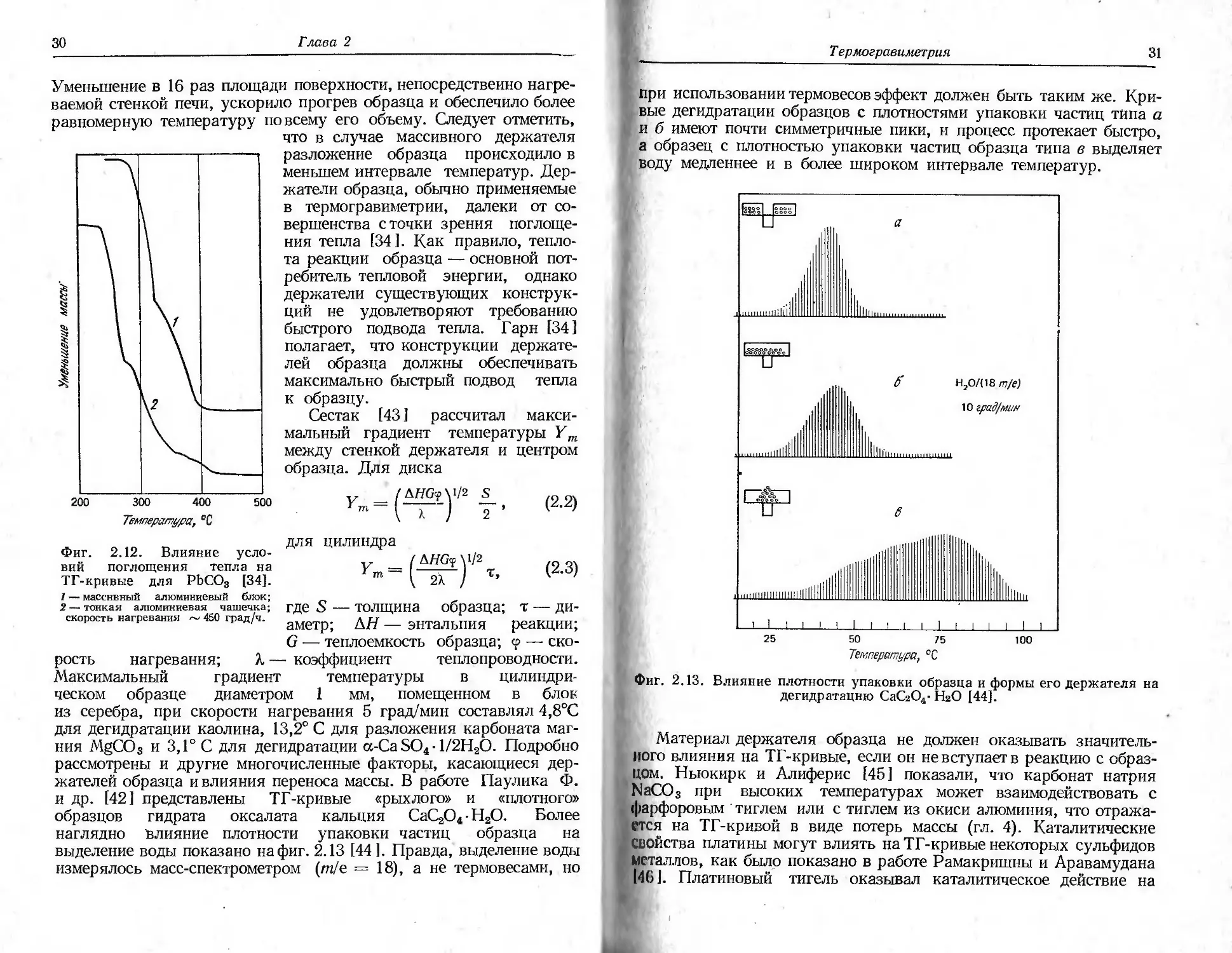
Р и. рассматривалось Гарном [34]. Это влияние показано на
ф»1Г. 2.12 на примере термического разложения карбоната свинца
PbCOg. Держатель образца помещали на алюминиевый блок в фор-
МП цилиндра высотой 25,4 мм (1 дюйм) с площадью боковой поверх-
ности 20,6 смг. Диаметр мелкой алюминиевой чашечки был равен
ДИЙМетру блока, а высота составляла всего 1,59 мм (1/16 дюйма).
30
Глава 2
Уменьшение в 16 раз площади поверхности, непосредственно нагре-
ваемой стенкой печи, ускорило прогрев образца и обеспечило более
равномерную температуру по всему его объему. Следует отметить,
что в случае массивного держателя
разложение образца происходило в
меньшем интервале температур. Дер-
жатели образца, обычно применяемые
в термогравиметрии, далеки от со-
вершенства сточки зрения поглоще-
ния тепла [34]. Как правило, тепло-
та реакции образца — основной пот-
ребитель тепловой энергии, однако
держатели существующих конструк-
ций не удовлетворяют требованию
быстрого подвода тепла. Гарн [34]
полагает, что конструкции держате-
лей образца должны обеспечивать
максимально быстрый подвод тепла
к образцу.
Сестак [43 ] рассчитал макси-
мальный градиент температуры Y,
между стенкой держателя и
образца. Для диска
т ~~ ( Л / 2 ’
т
центром
(2.2)
для цилиндра
Фиг. 2.12. Влияние уело- /ЛЯГ \1/2
вий поглощения тепла на у = ( ——(2.3)
ТГ-кривые для РЬСО3 [34]. \ 2Л / ’
I — массивный алюминиевый блок;
2~ тонкая алюминиевая чашечка; где о —ТОЛЩИНЙ обрЗЗЦсЦ Т — ДИ-
скорость нагревания ~ 450 град/ч. аметр; ДН _ энтальпия реаКЦИИ;
G — теплоемкость образца; <? — ско-
рость нагревания; А. — коэффициент теплопроводности.
Максимальный градиент температуры в цилиндри-
ческом образце диаметром 1 мм, помещенном в блок
из серебра, при скорости нагревания 5 град/мин составлял 4,8°С
для дегидратации каолина, 13,2° С для разложения карбоната маг-
ния MgCO3 и 3,1° С для дегидратации а-Са SO4 • 1/2Н2О. Подробно
рассмотрены и другие многочисленные факторы, касающиеся дер-
жателей образца и влияния переноса массы. В работе Паулика Ф.
и др. [42] представлены ТГ-кривые «рыхлого» и «плотного»
образцов гидрата оксалата кальция СаС2О4 • Н2О. Более
наглядно влияние плотности упаковки частиц образца на
выделение воды показано нафиг. 2.13 [44]. Правда, выделение воды
измерялось масс-спектрометром (т/е = 18), а не термовесами, но
Т ермогравиметрия
31
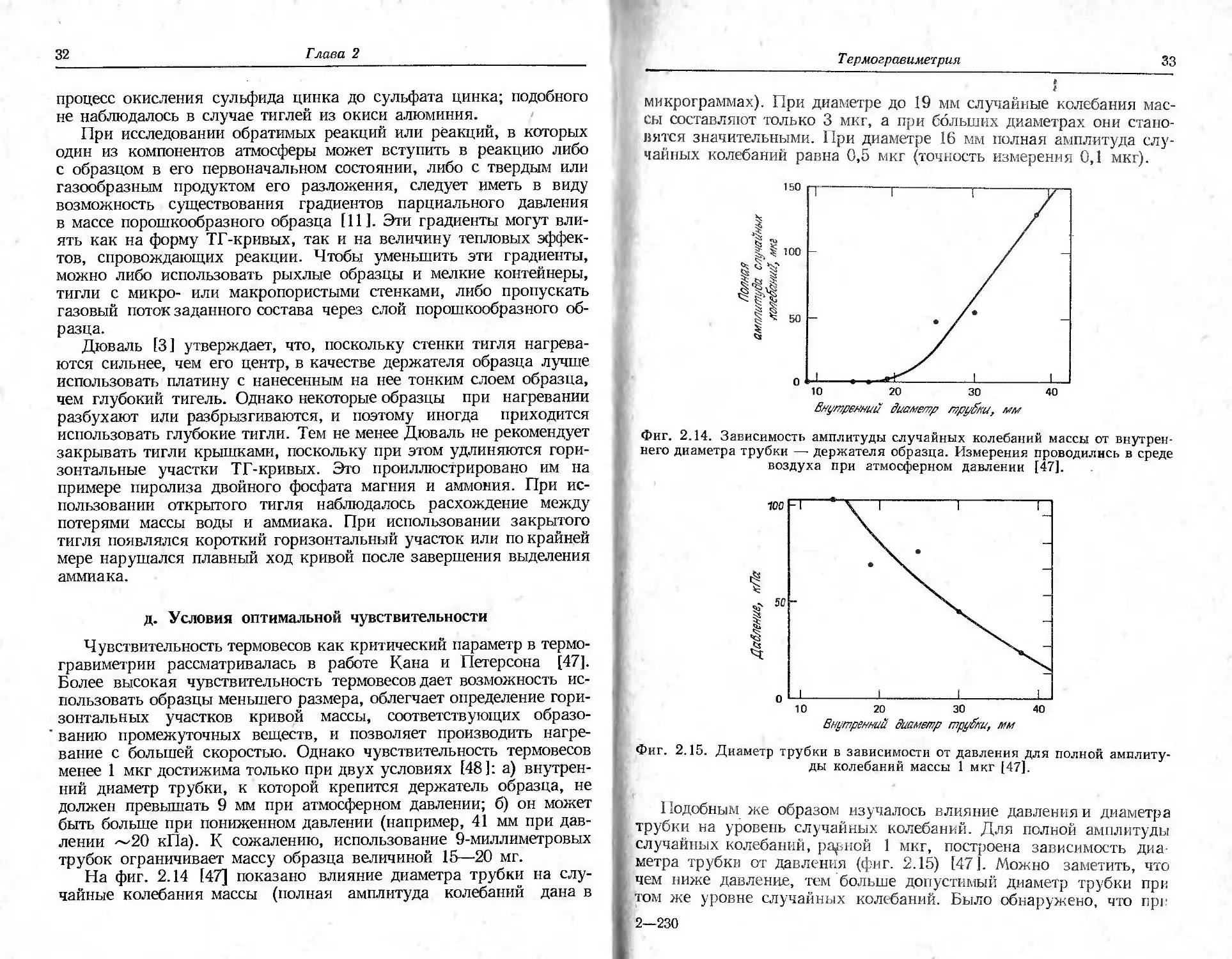
при использовании термовесов эффект должен быть таким же. Кри-
вые дегидратации образцов с плотностями упаковки частиц типа а
и б имеют почти симметричные пики, и процесс протекает быстро,
а образец с плотностью упаковки частиц образца типа в выделяет
воду медленнее и в более широком интервале температур.
& н2о/П8 т/е)
10 град/мин
. I 1__I__1_I__I__I_I__!_I__I__I_I__1__1_I__I__1_
25 50 75 100
Температура, °C
Фиг. 2.13. Влияние плотности упаковки образца и формы его держателя на
дегидратацию СаСаОд- НгО [44].
Материал держателя образца не должен оказывать значитель-
ного влияния на ТГ-кривые, если он не вступает в реакцию с образ-
цом. Ньюкирк и Алиферис [45] показали, что карбонат натрия
NaCO3 при высоких температурах может взаимодействовать с
фарфоровым тиглем или с тиглем из окиси алюминия, что отража-
ется на ТГ-кривой в виде потерь массы (гл. 4). Каталитические
Свойства платины могут влиять на ТГ-кривые некоторых сульфидов
Металлов, как было показано в работе Рамакришны и Аравамудана
I4GJ. Платиновый тигель оказывал каталитическое действие на
32
Глава 2
процесс окисления сульфида цинка до сульфата цинка; подобного
не наблюдалось в случае тиглей из окиси алюминия.
При исследовании обратимых реакций или реакций, в которых
один из компонентов атмосферы может вступить в реакцию либо
с образцом в его первоначальном состоянии, либо с твердым или
газообразным продуктом его разложения, следует иметь в виду
возможность существования градиентов парциального давления
в массе порошкообразного образца [И]. Эти градиенты могут вли-
ять как на форму ТГ-кривых, так и на величину тепловых эффек-
тов, спровождающих реакции. Чтобы уменьшить эти градиенты,
можно либо использовать рыхлые образцы и мелкие контейнеры,
тигли с микро- или макропористыми стенками, либо пропускать
газовый поток заданного состава через слой порошкообразного об-
разца.
Дюваль [3] утверждает, что, поскольку стенки тигля нагрева-
ются сильнее, чем его центр, в качестве держателя образца лучше
использовать платину с нанесенным на нее тонким слоем образца,
чем глубокий тигель. Однако некоторые образцы при нагревании
разбухают или разбрызгиваются, и поэтому иногда приходится
использовать глубокие тигли. Тем не менее Дюваль не рекомендует
закрывать тигли крышками, поскольку при этом удлиняются гори-
зонтальные участки ТГ-кривых. Эго проиллюстрировано им на
примере пиролиза двойного фосфата магния и аммония. При ис-
пользовании открытого тигля наблюдалось расхождение между
потерями массы воды и аммиака. При использовании закрытого
тигля появлялся короткий горизонтальный участок или по крайней
мере нарушался плавный ход кривой после завершения выделения
аммиака.
д. Условия оптимальной чувствительности
Чувствительность термовесов как критический параметр в термо-
гравиметрии рассматривалась в работе Кана и Петерсона [47].
Более высокая чувствительность термовесов дает возможность ис-
пользовать образцы меньшего размера, облегчает определение гори-
зонтальных участков кривой массы, соответствующих образо-
ванию промежуточных веществ, и позволяет производить нагре-
вание с большей скоростью. Однако чувствительность термовесов
менее 1 мкг достижима только при двух условиях [48]: а) внутрен-
ний диаметр трубки, к которой крепится держатель образца, не
должен превышать 9 мм при атмосферном давлении; б) он может
быть больше при пониженном давлении (например, 41 мм при дав-
лении -~20 кПа). К сожалению, использование 9-миллиметровых
трубок ограничивает массу образца величиной 15—20 мг.
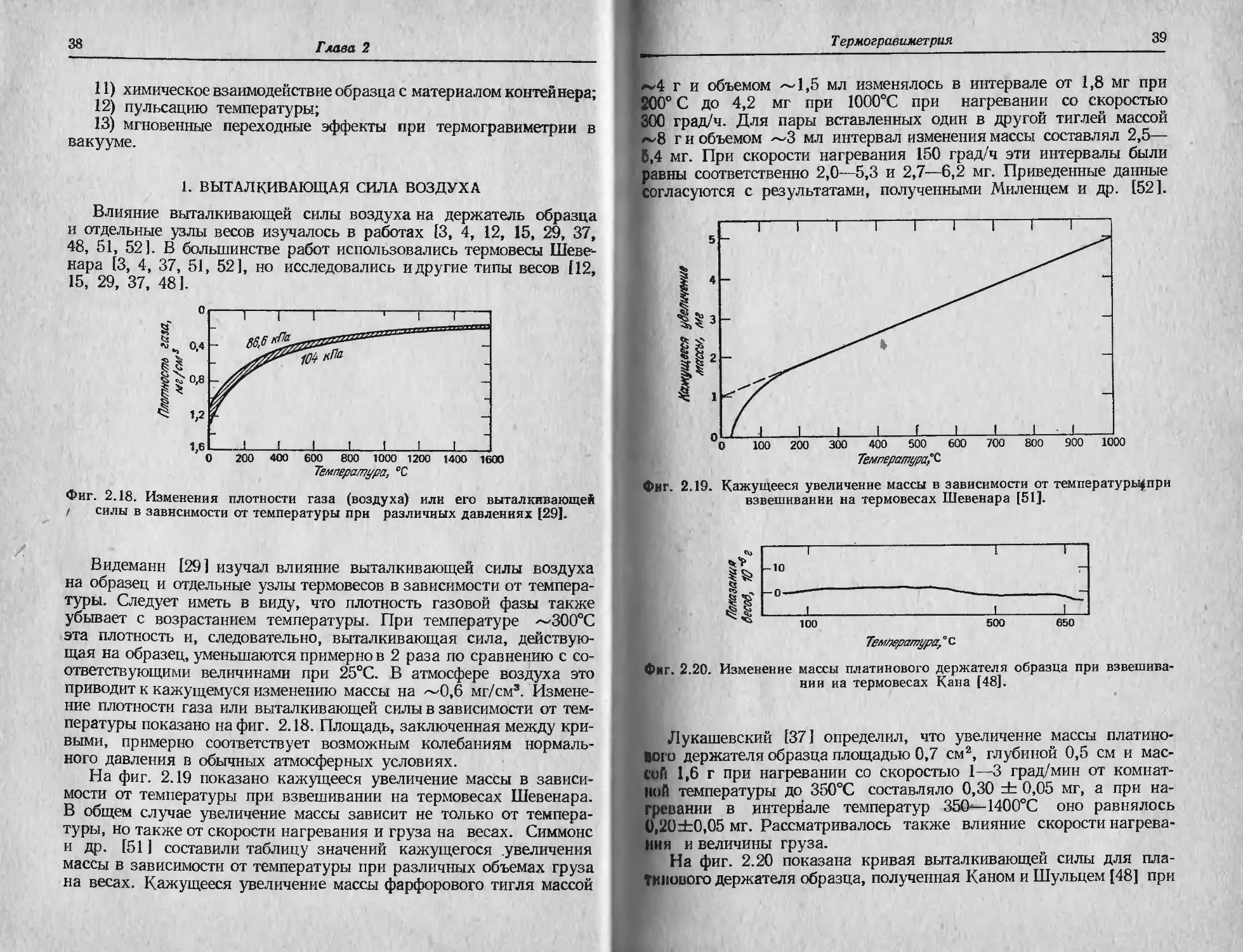
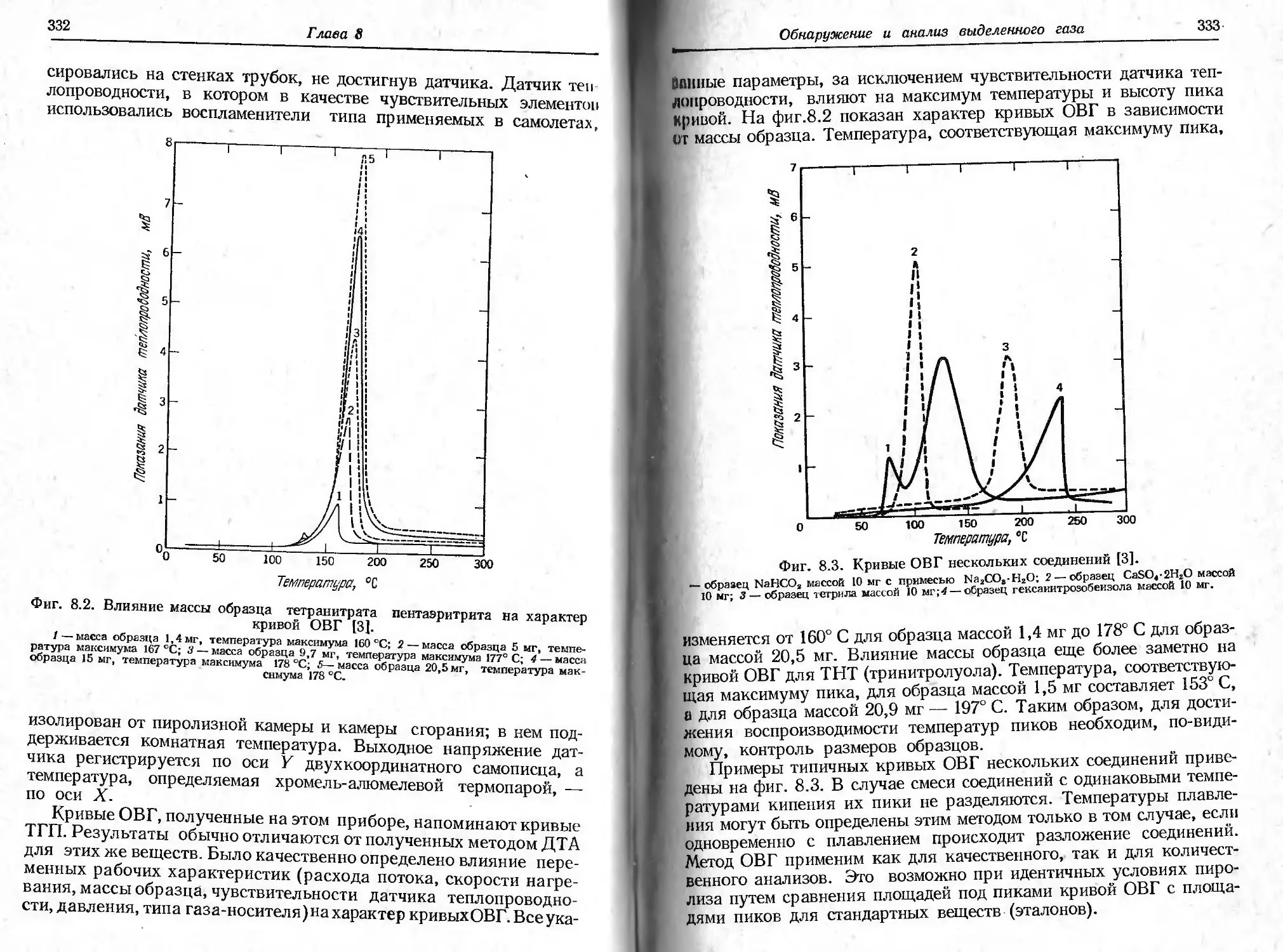
На фиг. 2.14 [47] показано влияние диаметра трубки на слу-
чайные колебания массы (полная амплитуда колебаний дана в
Т ермогравиметрия
33
микрограммах). При диаметре до 19 мм случайные колебания мас-
сы составляют только 3 мкг, а при больших диаметрах они стано-
вятся значительными. При диаметре 16 мм полная амплитуда слу-
чайных колебаний равна 0,5 мкг (точность измерения 0,1 мкг).
Внутренний диаметр труВки, мм
Фиг. 2.14. Зависимость амплитуды случайных колебаний массы от внутрен-
него диаметра трубки — держателя образца. Измерения проводились в среде
воздуха при атмосферном давлении [47].
Фиг. 2.15. Диаметр трубки в зависимости от давления для полной амплиту-
ды колебаний массы 1 мкг [47].
Подобным же образом изучалось влияние давления и диаметра
трубки на уровень случайных колебаний. Для полной амплитуды
случайных колебаний, равной 1 мкг, построена зависимость дна
метра трубки от давления (фиг. 2.15) [47]. Можно заметить, что
чем ниже давление, тем больше допустимый диаметр трубки при
том же уровне случайных колебаний. Было обнаружено, что ирг
2—230
34
Глава 2
большом диаметре трубки и заданном давлении максимальный уро-
вень случайных колебаний обычно приходится на интервал темпе-
ратур 150—650°С. При более высоких температурах амплитуда
случайных колебаний обычно меньше, чем при температурах менее
150°С. Зависимость случайных колебаний от диаметра трубки
в условиях движущегося газа (динамической атмосферы) почти не
отличается от зависимости для статических условий. В динами-
ческой атмосфере случайные колебания почти не зависели от рас-
хода газа, по крайней мере в пределах 5—500 мл/мин; при этом
максимальные значения амплитуды случайных колебаний изменя-
лись от 1 до 2 мкг. Трубка диаметром 16 мм оказалась оптималь-
ной как для статической, так и для динамической атмосферы. Та-
кой диаметр трубки хорошо удовлетворяет также условиям пони-
женного давления, за исключением тех случаев, когда необходимо
исследовать очень большие образцы.
2. ХАРАКТЕРИСТИКИ ОБРАЗЦА
а. Масса образца
Коутс и Редферн [8] показали, что масса образца может влиять
на ход ТГ-кривой:
1) вследствие отклонений изменения температуры образца от
линейного закона при эндотермической или экзотермической реак-
ции (чем больше масса образца, тем больше отклонение);
2) вследствие различий в скорости диффузии образующегося
газа через пустоты между твердыми частицами (в статических ус-
ловиях состав атмосферы, непосредственно соприкасающейся с ре-
агирующими частицами, будет до некоторой степени определяться
объемом образца);
3) вследствие существования больших градиентов температуры
внутри образца, особенно если его теплопроводность низкая.
Как видно из фиг. 2.16 [48], для обнаружения промежуточных
веществ, образующихся во время реакции, предпочтительнее ис-
пользовать небольшие образцы. На кривой, соответствующей об-
разцу массой 0,426 мг, отчетливо вырисовывается участок, указы-
вающий на образование тригидрата сульфата меди CuSO4-3H2O,
в то время как на кривой, соответствующей образцу массой 18 мг,
такой участок отсутствует. Однако, по всей вероятности, это пре-
дельный случай, так как образцы столь малой массы (0,426 мг)
обычно не используются.
Исследуя влияние массы образца на величины Т, и Tf, Ричер
и Валлет [32] обнаружили, что Tt остается практически постоянной
при изменении массы карбоната кальция от 0,25 до 1 г в атмосфере
как азота, так и двуокиси углерода. С другой стороны, начавшийся
процесс разложения [1] обычно протекает неравномерно по всей
Т ермогравиметрия
35
массе образца. Можно ожидать, что при таком неоднородном про-
цессе время, требуемое для полного разложения порошкообразно-
го образца, будет возрастать с увеличением его массы. Так как нагре-
Фиг. 2.16. Влияние массы образца
на ТГ-кривую для CuSO4- 5НаО [48].
—— масса образца 0,426 мг;-----масса
образца 18 мг. Скорость нагревания
13 град/мнн в статической атмосфере воз-
духа.
меряемая температура печи, и
вание печи происходит по ли-
нейному закону, наблюдаемая
величина Т f в результате
увеличится.
Такое изменение Tf видно
на примере термического раз-
ложения карбоната кальция
СаСО3, образующегося при
диссоциации гидрата окса-
лата кальция СаС2О4 Н2О
(фиг. 2.17) [1] При дегидра-
тации и разложении СаСО3
с увеличением массы образца
наблюдался постоянный рост
Tf. Однако, если реакция
разложения экзотермическая,
как например, разложение
оксалата кальция СаС,О4 в
атмосфере воздуха, с изме-
нением массы образца значе-
ния Tf не меняются. Б этом
случае температура образца
возрастает быстрее, чем не-
соответствующий рост удель-
ной скорости реакции может в итоге компенсировать (по крайней
мере частично) влияние массы образца. Симмонс и Ньюкирк [1]
показали, что, если реакция разложения происходит в инертной
атмосфере азота, все три значения Tf для СаС2О4-Н2О с увеличе-
нием массы образца сдвигаются в область более высоких темпе-
ратур.
б. Размер частиц образца
Влияние размера частиц образца на ТГ-кривую сравнительно
Мало изучено. Различие в размерах частиц приводит к изменению
условий диффузии образующихся газов, что влияет на скорость
реакции и, следовательно, на форму кривой. Большинство иссле-
дований, опубликованных в этой области, было связано с влиянием
размера частиц на кинетические параметры [8, 43]. Образцы в виде
крупных кристаллов могут растрескиваться, что приводит к рез-
ким спадам ТГ-кривой. Чем меньше размер частиц, тем быстрее
досгигается равновесие и тем больше для любой заданной темпера-
туры степень разложения [8].
8*
36
Глава 2
Фиг. 2.17. Влияние массы образца на ТГ-кривые для СаСгО4-НгО [1].
Скорость нагревания 300 град/ч в статической атмосфере. Масса образца: а — 126 мг;
б — 250 мг; е — 500 мг.
Сравнивая кривые термического разложения карбоната кальция
и кальцита, Ричер и Валлет [32] установили, что температуры раз-
ложения, полученные экспериментально при скорости нагревания
150 град/ч в потоке азота, составляют для порошка карбоната
кальция 783° С, для порошка кальцита 802°С и для кубика каль-
цита (массой ~350 мг) 89ГС.
Подобным образом Мартинец [491 показал, что температура
разложения хризотила падала с уменьшением размера частиц об-
разца. Для порошка наблюдалось непрерывное уменьшение массы
при нагревании от ~50 до 850°С, причем наиболее быстрое раз-
ложение происходило в интервале температур 600—700°С. При
нагревании образца в виде монолитного куска масса уменьшалась
незначительно до тех пор, пока температура не достигала 600°С.
Сходные результаты были получены при нагревании серпентина
и смеси бруцита с карбонатом. В общем случае с уменьшением раз-
мера частиц понижаются как начальная, так и конечная темпера-
туры разложения.
в. Другие факторы, связанные с образцом
Влияние теплоты реакции на кривую уменьшения массы иссле-
довано в работе Ньюкирка [12 1. Теплота реакции определяет вели-
чину разности температур образца и печи. В зависимости от харак-
тера реакции — экзотермической или эндотермической — темпе-
Т ермогравиметрия
37
ратура образца становится соответственно выше или ниже темпера-
I туры печи. Так как эти изменения температуры могут достигать
I 10°С и более (в зависимости от скорости нагревания), в расчете
' кинетических констант по ТГ-кривым неизбежны значительные
ошибки (разд. В. 3 данной главы).
Гиошон [50] показал, что наличие растворенных в образце газов
t накладывает на термогравиметрический метод серьезные ограниче-
ния. Их трудно не только исключить, но даже измерить, поэтому
I их содержание обычно неизвестно. Это видно на примере нагревания
• при 200°С в течение 3 ч твердого нитрата аммония, содержащего
1% азотной кислоты. В конце нагревания образец содержал 0,6%
азотной кислоты. Азотная кислота не является катализатором раз-
ложения образца, который сам в указанных условиях также не
выделяет азотной кислоты. Поэтому полученный результат можно
объяснить только медленным ее испарением. Концентрацию данного
растворенного вещества можно несколько уменьшить, если исполь-
зовать широкий тигель без крышки и тонкослойный образец и про-
водить нагревание в динамической атмосфере инертного газа. Со-
гласно результатам, полученным Гиошоном [50], поток инертного
газа в печи почти всегда необходим для облегчения диффузии газов
к образцу и от него.
Влияние плотности упаковки частиц образца (хотя ее и трудно
воспроизвести) рассматривалось в разд. Б.1.г настоящей главы.
В. ИСТОЧНИКИ ОШИБОК В ТЕРМОГРАВИМЕТРИИ
Возможные ошибки в термогравиметрии могут приводить к зна-
чительным неточностям при определении температуры и изменения
массы. Для повышения точности термогравиметрических данных
необходимо вводить поправки для учета ошибок, или, по крайней
мере, приблизительно определять их величину. Многие из этих
ошибок взаимосвязаны, и поэтому их нельзя рассматривать раз-
дельно. Необходимо подробно рассмотреть все источники ошибок
и их роль в термогравиметрии.
Существует много возможных источников ошибок, из которых
следует отметить следующие:
1) выталкивающую силу воздуха;
2) конвективные потоки и турбулентность в печи;
3) случайные колебания записывающего устройства и весов;
4) индукционность печи;
5) влияние электростатических сил на весы;
6) влияние окружающей среды на весы;
7) конденсацию на держателе образца;
8) измерение температуры и калибровку;
9) калибровку весов;
10) неравномерность работы лентопротяжного механизма;
38
Глава 2
11) химическое взаимодействие образца с материалом контейнера;
12) пульсацию температуры;
13) мгновенные переходные эффекты при термогравиметрии в
вакууме.
1. ВЫТАЛКИВАЮЩАЯ СИЛА ВОЗДУХА
Влияние выталкивающей силы воздуха на держатель образца
и отдельные узлы весов изучалось в работах [3, 4, 12, 15, 29, 37,
48, 51, 52]. В большинстве работ использовались термовесы Шеве-
нара [3, 4, 37, 51, 52], но исследовались и другие типы весов (12,
15, 29, 37, 48].
Фиг. 2.18. Изменения плотности газа (воздуха) или его выталкивающей
/ силы в зависимости от температуры при различных давлениях [29].
/
Видеманн [29] изучал влияние выталкивающей силы воздуха
на образец и отдельные узлы термовесов в зависимости от темпера-
туры. Следует иметь в виду, что плотность газовой фазы также
убывает с возрастанием температуры. При температуре ~300°С
эта плотность и, следовательно, выталкивающая сила, действую-
щая на образец, уменьшаются примерно в 2 раза по сравнению с со-
ответствующими величинами при 25°С. В атмосфере воздуха это
приводит к кажущемуся изменению массы на ~0,6 мг/см3. Измене-
ние плотности газа или выталкивающей силы в зависимости от тем-
пературы показано нафиг. 2.18. Площадь, заключенная между кри-
выми, примерно соответствует возможным колебаниям нормаль-
ного давления в обычных атмосферных условиях.
На фиг. 2.19 показано кажущееся увеличение массы в зависи-
мости от температуры при взвешивании на термовесах Шевенара.
В общем случае увеличение массы зависит не только от темпера-
туры, но также от скорости нагревания и груза на весах. Симмонс
и др. [51 ] составили таблицу значений кажущегося .увеличения
массы в зависимости от температуры при различных объемах груза
на весах. Кажущееся увеличение массы фарфорового тигля массой
Т ермогравиметрия
39
г и объемом ~1,5 мл изменялось в интервале от 1,8 мг при
200° С до 4,2 мг при 1000°С при нагревании со скоростью
300 град/ч. Для пары вставленных один в другой тиглей массой
~8 г и объемом ~3 мл интервал изменения массы составлял 2,5—
,4 мг. При скорости нагревания 150 град/ч эти интервалы были
равны соответственно 2,0—5,3 и 2,7—6,2 мг. Приведенные данные
согласуются с результатами, полученными Миленцем и др. [52].
Фиг. 2.19. Кажущееся увеличение массы в зависимости от температуры^при
взвешивании на термовесах Шевенара [51].
Температура, ° с
Фиг. 2.20. Изменение массы платинового держателя образца при взвешива-
нии иа термовесах Кана [48].
Лукашевский [37] определил, что увеличение массы платино-
вого держателя образца площадью 0,7 см2, глубиной 0,5 см и мас-
сой 1,6 г при нагревании со скоростью 1—3 град/мин от комнат-
ной температуры до 350°С составляло 0,30 ± 0,05 мг, а при на-
гревании в интервале температур 350—1400°С оно равнялось
0,20 ±0,05 мг. Рассматривалось также влияние скорости нагрева-
ния и величины груза.
На фиг. 2.20 показана кривая выталкивающей силы для пла-
тинового держателя образца, полученная Каном и Шульцем [48] при
40
Глава 2
использовании электровесов Кана модели RG, преобразованных
в термовесы. В интервале температур 25—650°С изменение массы
очень мало (<2-10-6 г), если диаметр трубки, к которой крепится
держатель образца, составляет 8 мм. По сравнению с весами Шеве-
нара поправка примерно в 1000 раз меньше.
Об изменениях массы образца под влиянием выталкивающей
силы воздуха в случае термогравиметр иц при высоком давлении
уже упоминалось в работе [28].
i
2. КОНВЕКТИВНЫЕ ПОТОКИ И ТУРБУЛЕНТНОСТЬ В ПЕЧИ
Кажущееся увеличение или уменьшение массы из-за конвек-
тивных потоков в печи изучалось Ньюкирком [12] и Лукашевским
[37]. Наблюдаемое «уменьшение» массы, обусловленное давлени-
Темперагт/ра, °C
Фиг. 2.21. Влияние диаметра отверстия в крышке печи на кажущееся изме-
нение массы при взвешивании на термовесах Шевенара; скорость нагревания
300 град/ч [12].
-----один фарфоровый тигель (~ 4 г);----два тигля.
и «увеличение» массы из-за турбулентности воздушной струи в боль-
шой степени определяются формой и размером тигля [12]. На
фиг. 2.21 представлено влияние диаметра отверстия в крышке печи
на кажущееся изменение массы. Видно, что для всех размеров от-
ТермогравШлетрия
41
Г~----------------------------------------------------
нсрстий (кроме самого большого) в начальной стадии нагревания
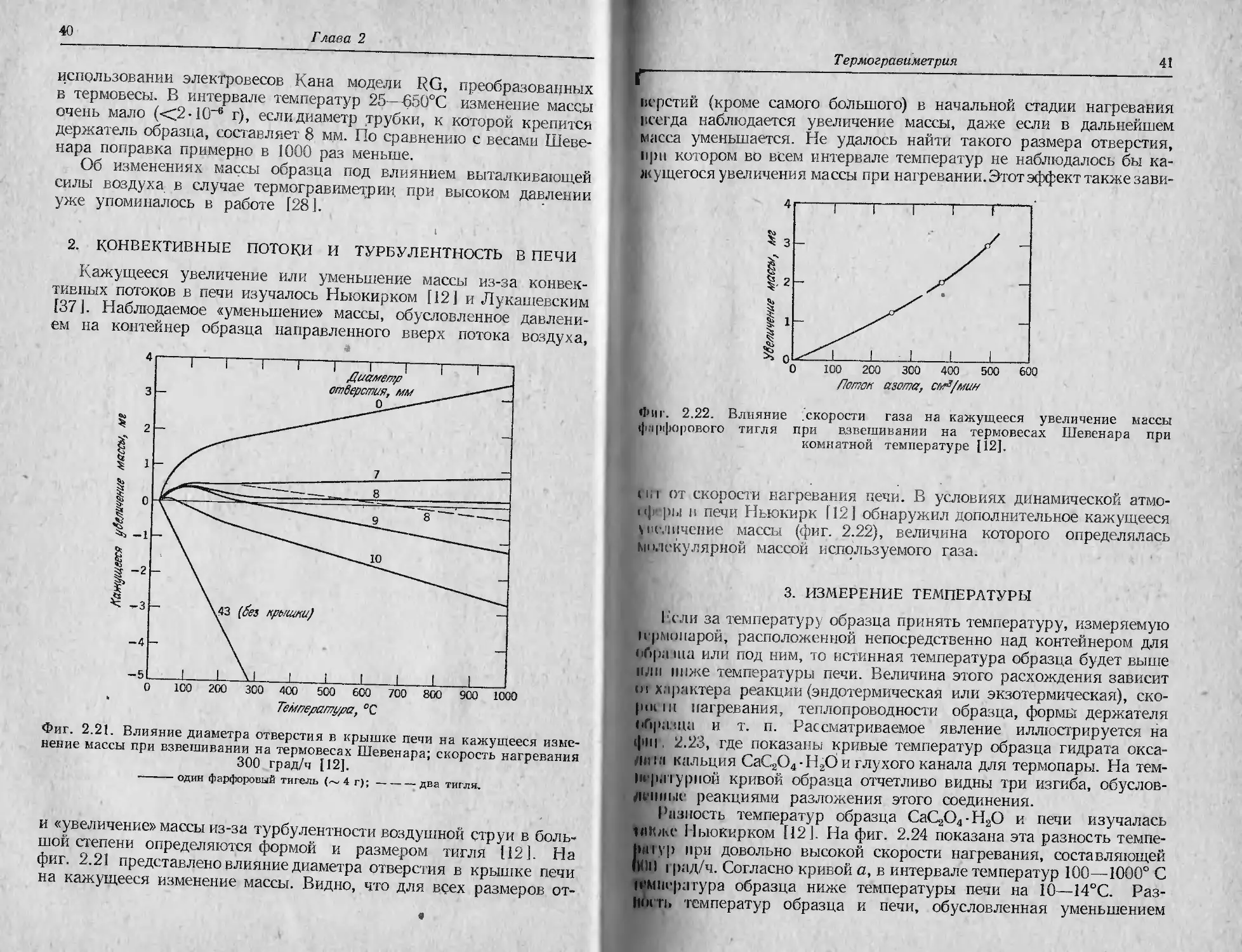
всегда наблюдается увеличение массы, даже если в дальнейшем
масса уменьшается. Не удалось найти такого размера отверстия,
при котором во всем интервале температур не наблюдалось бы ка-
жущегося увеличения массы при нагревании. Этот эффект также зави-
fl’iii. 2.22. Влияние скорости газа на кажущееся увеличение массы
фарфорового тигля при взвешивании на термовесах Шевенара при
комнатной температуре [12].
пп от скорости нагревания печи. В условиях динамической атмо-
ры II печи Ньюкирк [12] обнаружил дополнительное кажущееся
\iсличение массы (фиг. 2.22), величина которого определялась
молекулярной массой используемого газа.
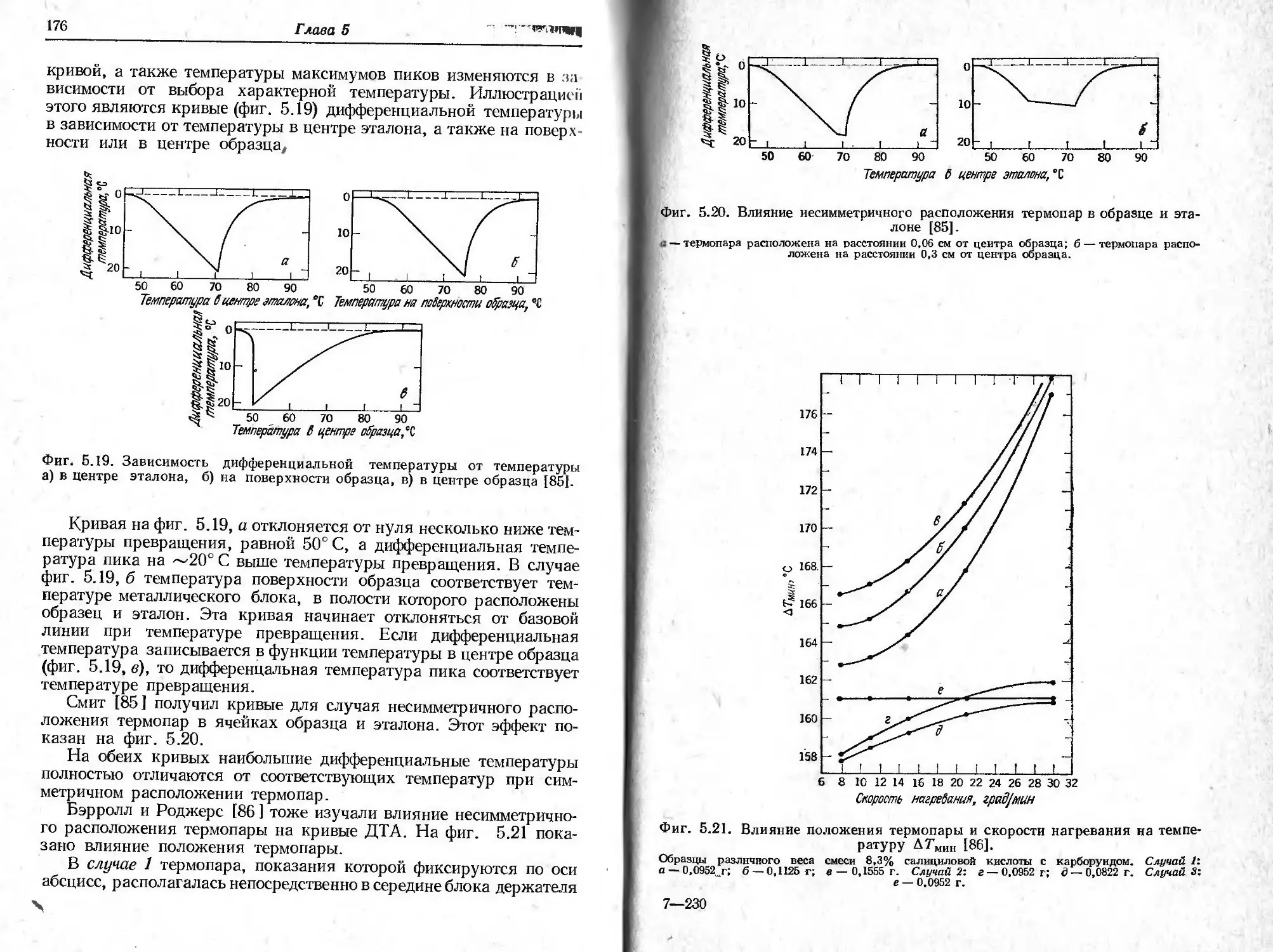
3. ИЗМЕРЕНИЕ ТЕМПЕРАТУРЫ
I ели за температуру образца принять температуру, измеряемую
црмопарой, расположенной непосредственно над контейнером для
обрата или под ним, то истинная температура образца будет выше
или ниже температуры печи. Величина этого расхождения зависит
<н характера реакции (эндотермическая или экзотермическая), ско-
рой п нагревания, теплопроводности образца, формы держателя
образца и т. п. Рассматриваемое явление иллюстрируется на
«(ни 2.23, где показаны кривые температур образца гидрата окса-
ина кальция СаС2О4 - Н2О и глухого канала для термопары. На тем-
н* pat урной кривой образца отчетливо видны три изгиба, обуслов-
/II нпы( реакциями разложения этого соединения.
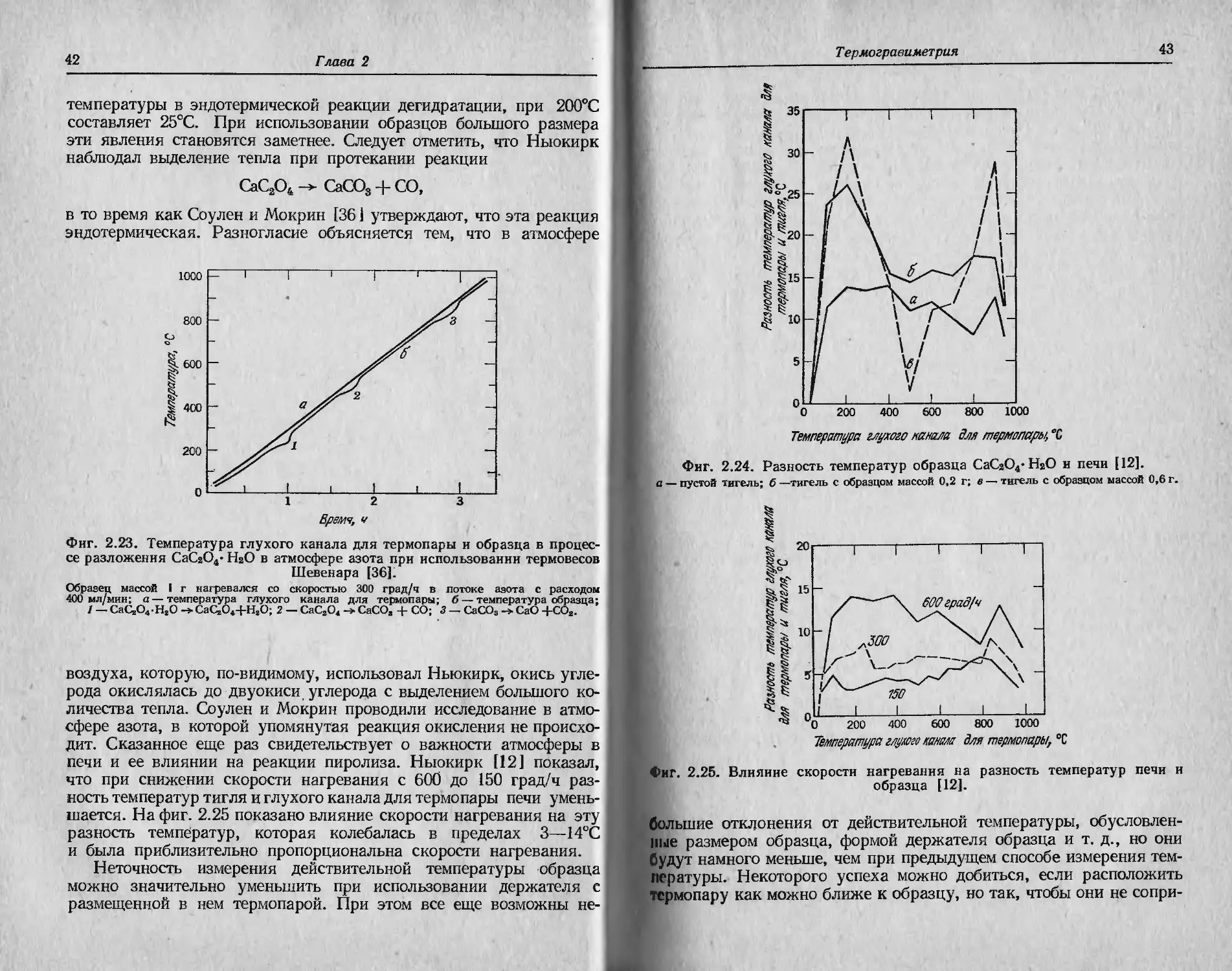
Разность температур образца СаС2О4-Н2О и печи изучалась
1ЛКже Ньюкирком [12]. На фиг. 2.24 показана эта разность темпе-
ра iyp при довольно высокой скорости нагревания, составляющей
П П । рад/ч. Согласно кривой а, в интервале температур 100—1000° С
|*Miicparypa образца ниже температуры печи на 10—14°С. Раз-
Цоси. температур образца и печи, обусловленная уменьшением
42
Глава 2
температуры в эндотермической реакции дегидратации, при 200°С
составляет 25°С. При использовании образцов большого размера
эти явления становятся заметнее. Следует отметить, что Ньюкирк
наблюдал выделение тепла при протекании реакции
СаС2О4 -> СаСО3 + СО,
в то время как Соулен и Мокрин [36 ] утверждают, что эта реакция
эндотермическая. Разногласие объясняется тем, что в атмосфере
Фиг. 2.23. Температура глухого канала для термопары и образца в процес-
се разложения СаСаО4- НаО в атмосфере азота при использовании термовесов
Шевенара [36] ‘
Образец массой I г нагревался со скоростью 300 град/ч в потоке азота с расходом
400 мл/мин; а—температура глухого канала для термопары; б — температура образца;
1 — СаСгО4.Н20 СаСЛ+НгО; 2 — СаС204 СаСОя + СО; 3 — СаСО3 СаО +СО2.
воздуха, которую, по-видимому, использовал Ньюкирк, окись угле-
рода окислялась до двуокиси, углерода с выделением большого ко-
личества тепла. Соулен и Мокрин проводили исследование в атмо-
сфере азота, в которой упомянутая реакция окисления не происхо-
дит. Сказанное еще раз свидетельствует о важности атмосферы в
печи и ее влиянии на реакции пиролиза. Ньюкирк [12] показал,
что при снижении скорости нагревания с 600 до 150 град/ч раз-
ность температур тигля и глухого канала для термопары печи умень-
шается. На фиг. 2.25 показано влияние скорости нагревания на эту
разность темпёратур, которая колебалась в пределах 3—14°С
и была приблизительно пропорциональна скорости нагревания.
Неточность измерения действительной температуры образца
можно значительно уменьшить при использовании держателя с
размещенной в нем термопарой. При этом все еще возможны не-
Термогравиметрия
43
Температура глухого каната для термопары, °C
Фиг. 2.24. Разность температур образца СаС2О4-НгО н печи [12].
а — пустой тигель; б —тигель с образцом массой 0,2 г; в — тигель с образцом массой 0,6 г.
Температура глум ханам для термопары, °C
Фиг. 2.25. Влияние скорости нагревания на разность температур печи и
образца [12].
большие отклонения от действительной температуры, обусловлен-
ные размером образца, формой держателя образца и т. д., но они
будут намного меньше, чем при предыдущем способе измерения тем-
пературы. Некоторого успеха можно добиться, если расположить
термопару как можно ближе к образцу, но так, чтобы они не сопри-
44
Глава 2
касались. Такой подход используется в термовесах фирмы «Дюпон»
1531. Термопара размещается внутри держателя образца, не касаясь
его. Как показано на примере кривых для СаС2О4-Н2О (фиг. 2.26),
этим способом можно быстро обнаружить рост температуры образ-
ца. Ранее уже отмечалось, что разложение СаС2О4 в среде воздуха
является экзотермической реакцией. Поэтому температура образ-
ца по достижении ~500° С (кривая 2) начнет очень быстро возрас-
тать до максимального значения 630° С, при котором реакция за-
канчивается. Затем температура образца падает до температуры
печи, составляющей немногим менее 500° С.
Фиг. 2.26. Определение изменения температуры образца при помощи термо-
пары, расположенной в непосредственной близости от него [53].
Скорость нагревания 15 град/мин.
Гейл и Эггер [54] показали, что режим нагревания имеет зна-
чение при оценке уменьшения массы за определенный период вре-
мени, но не оказывает влияния на скорость уменьшения массы и ки-
нетические параметры. «Симметричные» пульсации температуры
не приводят к исчезновению ошибок в том случае, когда скорость
является экспоненциальной, а не линейной функцией температуры.
Калибровка температурной шкалы в термогравиметрии описыва-
ется в гл. 3.
4. ДРУГИЕ ОШИБКИ
В правильно сконструированных термовесах некоторые из дру-
гих возможных ошибок пренебрежимо малы. Ошибки, вызванные
случайными колебаниями записывающего устройства, индукцион-
ностью печи, электростатическими силами, изменениями среды в
термовесах и т. д., могут быть исключены при правильном выборе
Термогравиметрия
45
типа термовесов, их конструкции и места установки в лаборатории.
Ньюкирк [12] обнаружил, что, если механизм взвешивания тер-
мовесов Шевенара был недостаточно хорошо теплоизолирован, масло
В гидравлических амортизаторах нагревалось, и это вызывало кажу-
щееся увеличение массы из-за увеличения выталкивающей силы
масла. В последней модели этих весов масляные амортизаторы за-
менены магнитными.
Еще одним источником ошибки является конденсация на более
холодной части стержня держателя образца. Конденсат может вновь
испариться при повышении температуры и снова осесть на еще бо-
лее удаленной от образца части стержня. Это может существенно
повлиять на результаты. Соулен и Мокрин [361 установили, что
положение усугубляется при наличии быстрого потока инертного
газа, поскольку летучие вещестйа увлекаются к стержню. Чтобы
оценить этот эффект количественно, необходимо взвешивать дер-
жатель образца, образец и поддерживающее устройство до опыта
и после него. Если их массы при этом различаются на значительную
ш личину, нужно ввести поправки. Такой метод, однако, не дает
представления о погрешности в ходе эксперимента. Соулен и Мок-
риц |36| исключили эту ошибку термовесов Шевенара, заключив
держатель тигля в керамическую или никелевую трубку. Без этой
трубки для того же вещества была получена абсолютно неверная
кривая уменьшения массы. Конечно, при исследовании соединений,
иг содержащих конденсирующихся газообразных веществ, эта
Проблема не возникает.
Кап и Шульц [55J исследовали возможность'исключения оши-
бок взвешивания в случае применения весов Кана модели RG,
Они рассмотрели влияние температуры коррозионной атмосферы
л 1нкже электрических и магнитных воздействий.
I [ерподические калибровки термовесов предупредят появление
ошибок па оси массы записывающего устройства. Многие иссле-
ДП111ПГЛП калибруют весы перед каждым опытом, устанавливая в
Конн Апере образца эталонный образец известной массы.
Г. ТЕРМОГРАВИМЕТРИЯ В СОБСТВЕННОЙ АТМОСФЕРЕ
Собе пенная («самогенерируемая») атмосфера — это атмосфера,
К iXM’ian которой входят газообразные вещества, образующиеся
11 ходе реакции, и которая благодаря специальной конструкции
Держи и ля находится в непосредственном контакте с образцом.
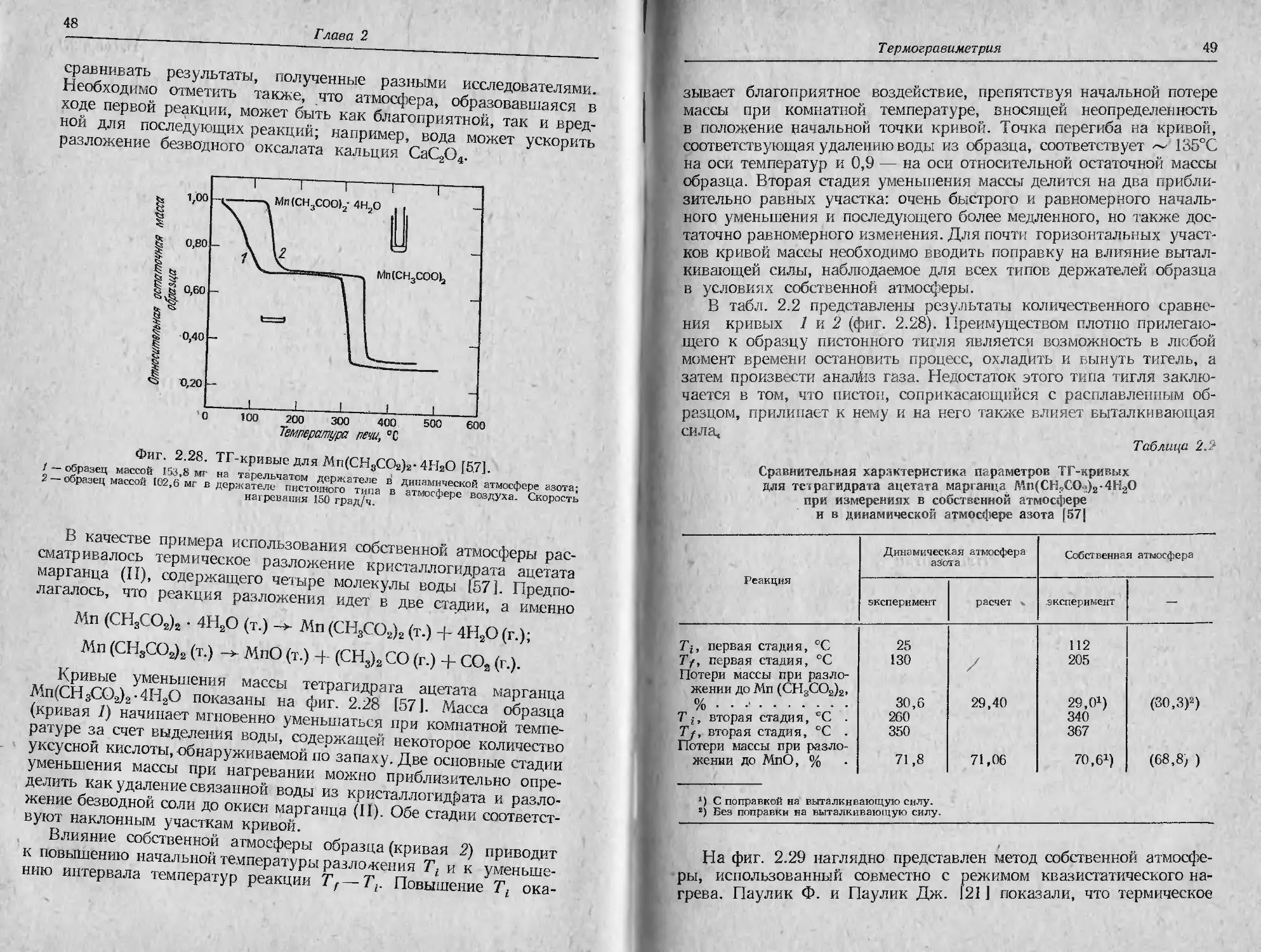
Преимущество термогравиметрирования различных соединений в
ininni атмосфере заключается в воспроизводимости ее состава и,
Сдедпиателыю, кривой уменьшения массы. Этот метод был предло-
жен в I960 г. одновременно Гарном и Кесслером [33], а также
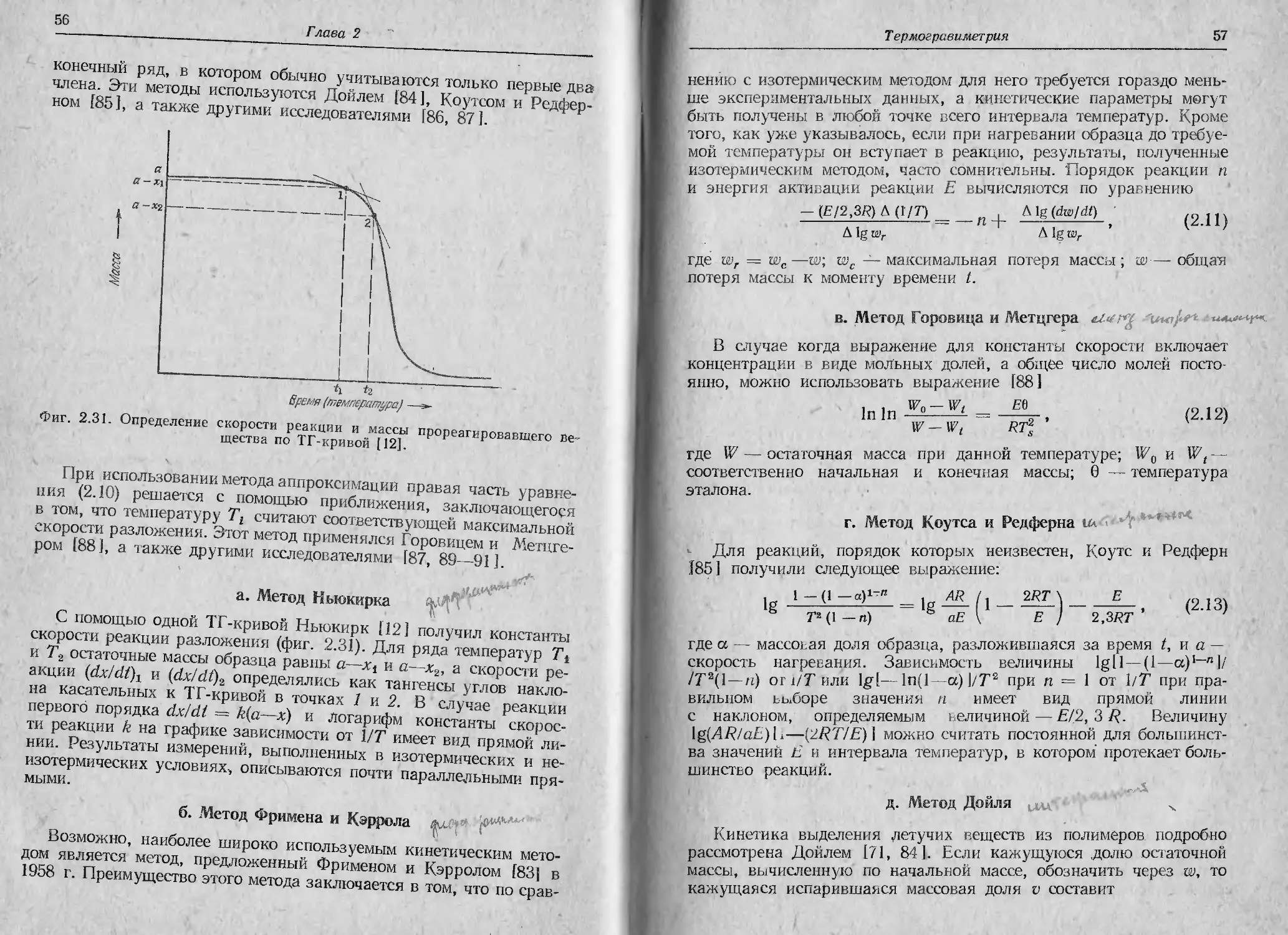
фпркелем [561. Ньюкирк [57] посвятил ему обширный обзор.
Дй|||Ц|1Й метод используется не очень широко, особенно со времени
46
Глава 2
появления высококачественных промышленных термовесов,
обеспечивающих поддержание заданной атмосферы в печи.
Важнейшей особенностью рассматриваемого метода является
конструкция держателей образца, два из которых описаны в гл. 3.
Другие типы конструкций держателя образца приводит Ньюкирк
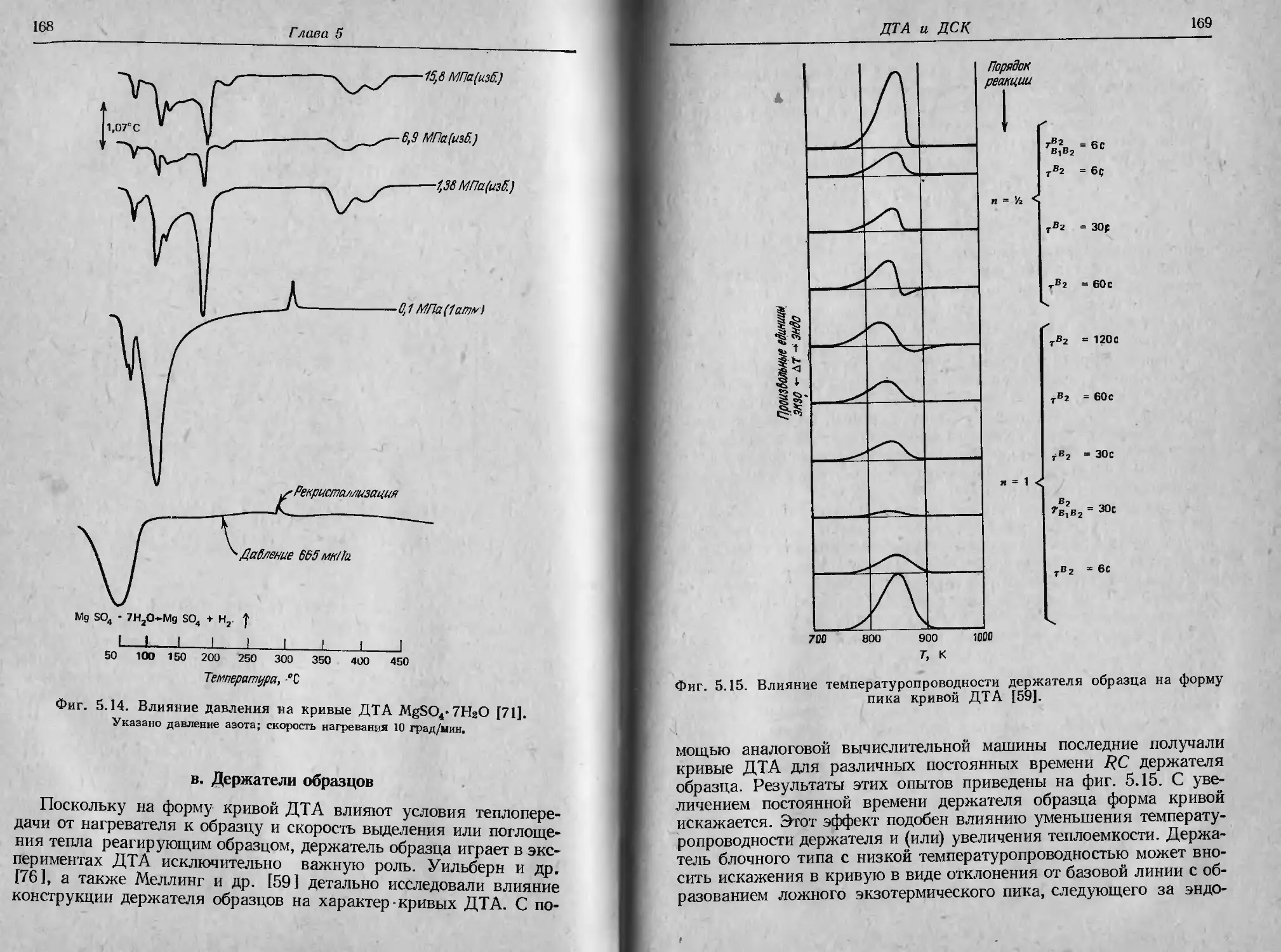
[57, 58]. На фиг. 2.27 показано, как совершенствовались держа-
тели образца, применяемые для термогравиметрии в собственной
атмосфере. В табл. 2.1 приведен перечень различных соединений,
исследованных этим методом [57].
Фиг. 2.27, Эволюция формы держателей образца, применяемых в условиях
термогравиметрии в собственной атмосфере ]57],
Основной эффект от использования термогравиметрии в собст-
венной атмосфере — это повышение давления выделяющегося газа
до 0,1 МПа (~1 атм), что приводит к благоприятным термодинами-
ческим, физическим и кинетическим условиям. Поскольку этот ме-
тод не обеспечивает точного контроля состава атмосферы, многие
исследователи применяют его только для предварительных иссле-
дований или в тех случаях, когда нет другого выхода [57]. Часто,
однако, и в других случаях точный контроль атмосферы трудно
или невозможно осуществить из-за сложности или неопределеннос-
ти состава продуктов реакции. Рассматриваемый метод хорошо
обоснован теоретически, и в ряде случаев его удобно использовать
для предварительного ТГ-исследования сложных систем твердое ве-
щество — газ.
Держатель образца должен иметь минимально возможный «сво-
бодный» объем. В случае большого свободного объема в образце
возникают градиенты давления газа, возможны также различные
реакции и даже нестехиометрическое уменьшение массы. При боль-
шом свободном объеме труднее определить 7\ и, следовательно,
Таблица 2.1
Соединения, исследованные методом термогравиметрии
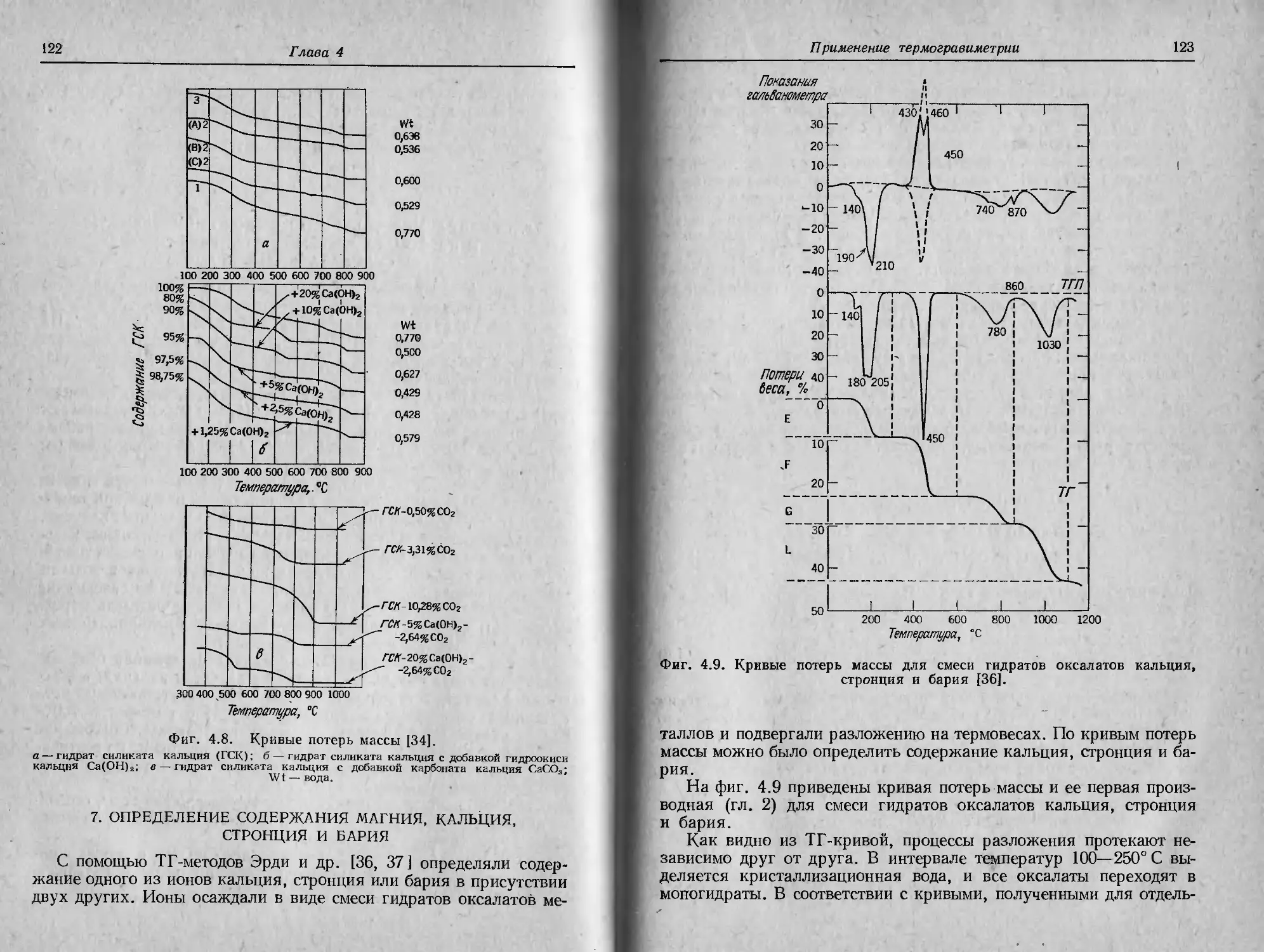
в собственной атмосфере [57]
Тип тигля1 * Вещество
п МР п к к к к п шк п шк МР п МР шк кт п шк шк п, шк к шк г II к (порошок), П, КТ КТ, к МР п кт кт, к, п г, п к к к г г п п п п п п г, шк Моногидрат карбоната аммония Антрацит Бруцит Mg(OH)2 Тетрахлорборат н-бутиламмоиия Тетрахлорборат третичнобутиламмоии я Тетрафенилборат изобутиламмония Тетрафенилборат вторичнобутиламмония Карбонат кадмия (8/3) Гидрат сульфата кадмия(П) Кальцит СаСО3 Церузит PbCOs Древесный уголь Хризотил Уголь Тетрагидрат ацетата кобальта(П) Дигидрат оксалата кобальта(Н) Гидрат оксалата кобальта CuSO4-3Cu(OH)2-H2O CuSO4-2Cu(OH)2 Пентагидрат сульфата меди Тетрахлорборат этиламмоиия Гипс CaSO4-2H2O Карбонат железа(П) Карбонат свинца(П) Карбонат свинца (II) Окись свинца Бурый уголь (лигнит) Магнезит MgCO3 Гептагидрат сульфата магния Тетрагидрат ацетата маргаица(П) Карбонат марганца(П) Тетрахлороборат н-октиламмония Тетрахлороборат «-пропиламмоиия Тетрафенилборат н-пропиламмония Родохрозит МпСО, Сидерит FeCO3 Карбонат серебра Оксалат натрия Тальк Пентагидрат нитрата тория(1У) Гексагидрат оксалата тория(1У) Гидрат уранилсульфата Гептагидрат сульфата цинка
1 Сокращение названий тиглей: Г — глубокий; К — с крышкой; КТ — капиллярный;
МР — микрореторта; П — пистонного типа; ШК — с шариковым клапаном.
48
Глава 2
сравнивать результаты, полученные разными исследователями.
Необходимо отметить также, что атмосфера, образовавшаяся в
ходе первой реакции, может быть как благоприятной, так и вред-
ной для последующих реакций; например, вода может ускорить
разложение безводного оксалата кальция СаС2О4.
Фиг. 2.28. ТГ-кривые для Мп(СН3СС>2)2-4НаО [57].
/ — образец массой 153,8 мг на тарельчатом держателе в динамической атмосфере азота;
2— образец массой 102,6 мг в держателе пистонного типа в атмосфере воздуха. Скорость
нагревания 150 град/ч.
В качестве примера использования собственной атмосферы рас-
сматривалось термическое разложение кристаллогидрата ацетата
марганца (II), содержащего четыре молекулы воды [57]. Предпо-
лагалось, что реакция разложения идет в две стадии, а именно
Мп (СН3СО2)2 • 4Н2О (т.) -> Мп (СН3СО2)2 (т.) + 4Н2О (г.);
Мп (СН8СО2)2 (т.) -> МпО (т.) + (СН3)2СО (г.) + СО2 (г.).
Кривые уменьшения массы тетрагидрага ацетата марганца
Мп(СН 3СО2)2 • 4Н2О показаны на фиг. 2.28 [571. Масса образца
(кривая 1) начинает мгновенно уменьшаться при комнатной темпе-
ратуре за счет выделения воды, содержащей некоторое количество
уксусной кислоты, обнаруживаемой по запаху. Две основные стадии
уменьшения массы при нагревании можно приблизительно опре-
делить как удаление связанной воды из кристаллогидрата и разло-
жение безводной соли до окиси марганца (II). Обе стадии соответст-
вуют наклонным участкам кривой.
Влияние собственной атмосферы образца (кривая 2) приводит
к повышению начальной температуры разложения Т\ и к уменьше-
нию интервала температур реакции Tf—T^ Повышение Ть ока-
Термогравиметрия
49
зывает благоприятное воздействие, препятствуя начальной потере
массы при комнатной температуре, вносящей неопределенность
в положение начальной точки кривой. Точка перегиба на кривой,
соответствующая удалению воды из образца, соответствует ~ 135°С
на оси температур и 0,9 — на оси относительной остаточной массы
образца. Вторая стадия уменьшения массы делится на два прибли-
зительно равных участка: очень быстрого и равномерного началь-
ного уменьшения и последующего более медленного, но также дос-
таточно равномерного изменения. Для почти горизонтальных участ-
ков кривой массы необходимо вводить поправку на влияние вытал-
кивающей силы, наблюдаемое для всех типов держателей образца
в условиях собственной атмосферы.
В табл. 2.2 представлены результаты количественного сравне-
ния кривых 1 и 2 (фиг. 2.28). Преимуществом плотно прилегаю-
щего к образцу пистонного тигля является возможность в любой
момент времени остановить процесс, охладить и вынуть тигель, а
затем произвести анализ газа. Недостаток этого типа тигля заклю-
чается в том, что пистон, соприкасающийся с расплавленным об-
разцом, прилипает к нему и на него также влияет выталкивающая
сила.
Таблица 2.2
Сравнительная характеристика параметров ТГ-кривых
для тетрагидрата ацетата марганца Мп(СН?СО..)2-4Н2О
при измерениях в собственной атмосфере
и в динамической атмосфере азота [57]
Реакция Динамическая атмосфера азота Собственная атмосфера
эксперимент расчет v эксперимент —
Tit первая стадия, °C Tf, первая стадия, °C Потери массы при разло- жении до Мп (СН3СО2)2, % Т1, вторая стадия, ®С . Ту, вторая стадия, °C . Потери массы при разло- жении до МпО, % 25 130 30,6 260 350 71,8 Z 29,40 71,06 112 205 29,01) 340 367 70,61) (30,3)2) (68,8) )
*) С поправкой на выталкивающую силу.
2) Без поправки на выталкивающую силу.
На фиг. 2.29 наглядно представлен метод собственной атмосфе-
ры, использованный совместно с режимом квазисгатического на-
грева. Паулик Ф. и Паулик Дж. [21 ] показали, что термическое
50
Глава 2
разложение гексамминоникельхлорида Ni(NH3)6Cl2 происходит
в три стадии, начало которых соответствует 180, 320 и 360°С.
Небольшой выступ на кривой в ходе первой реакции диссоциа-
ции обусловлен, по-видимому, индукционным периодом, вызванным
замедленным образованием центров разложения. Держатель об-
разца, использованный в этом эксперименте, описывается в гл. 3.
Преимущества и недостатки метода собственной атмосферы были
описаны Ньюкирком [57 ].
Ди, %
о
20
Ni(NH3>2Ci2
Ni(NH3>CI2
NlClt ад
100 200 300 400 500
Температура, °C
Фиг. 2.29. ТГ-кривые для Ni(NH3)6Cla в собственной атмосфере и квази-
статическом нагревании [21].
Недостатки
1. Поправки на выталкивающую силу изменяются в зависимости
ют молекулярной массы газа, заполняющего тигель.
2. Большие тяжелые тигли уменьшают точность определения
температуры образца.
3. При дегидратации гидратов соединений увеличивается веро-
ятность их расплавления и образования ложных горизонтальных
участков на ТГ-кривой.
4. Ухудшается разрешение в тех случаях, когда окончание
первой реакции задерживается до температуры, при которой на-
ступает следующая реакция.
5. Побочные реакции с выделяющимся газом затрудняют об-
работку результатов.
Преимущества
1. Сужается интервал реакции, улучшается разрешение пере-
крывающихся реакций, промежуточные продукты идентифициру-
ются более точно.
Термогравиметрия
51
2. Обнаруживаются новые фазы.
3. Реакции в основном протекают при постоянном давлении
газообразных продуктов, равном атмосферному. Течение реакции
(за исключением начального участка) не испытывает воздействия
переменного парциального давления.
4. Наблюдаемая начальная температура разложения более точ-
но соответствует равновесной температуре разложения.
5. Можно проводить эксперименты с веществами, подверженны-
ми окислению при повышенных температурах, без существенных
побочных эффектов от окисления.
6. Можно исследовать очень быстрые реакции без потери твер-
дого продукта.
7. Лучшие результаты получаются при изучении веществ, име-
ющих заметное давление паров при комнатной температуре. Обра-
зец можно взвесить с большей точностью, и он дает горизонтальную
базовую линию на ТГ-кривой.
8. Уменьшается влияние различий в размерах частиц и стандар-
тизируется влияние геометрии тигля. Это особенно важно при
изучении неоднородных материалов, таких, как горные породы и
минералы.
9. Облегчается рекристаллизация новых фаз из гидратов и гид-
роокисей.
10. Для необратимых реакций разложения наблюдается лучшее
разрешение и в некоторых случаях уменьшается интервал реакции,
хотя причина этих явлений неизвестна.
Рекомендуемые области применения
Термогравиметрия в собственной атмосфере полезна при иссле-
дованиях:
1) последовательных реакций, особенное участием гидроокисей,
гидратов, аммиакатов, карбонатов, ацетатов, оксалатов и сульфа-
тов;
2) неоднородных веществ;
3) растрескивающихся или взрывающихся веществ;
4) веществ, чувствительных к воздействию воздуха;
5) летучих веществ;
6) веществ, разлагающихся с образованием нескольких газо-
образных продуктов;
7) деструктивной перегонки.
Д. ТЕРМОГРАВИМЕТРИЯ ПО ПРОИЗВОДНОЙ (ТГП)
В обычной термогравиметрии масса образца m непрерывно
регистрируется в функции температуры Т или времени t:
m = f(T или /). (2.4)
52
Глава 2
Количественные определения изменений массы производятся путем
измерения расстояния между двумя точками на кривой в направ-
лении оси массы или между двумя горизонтальными уровнями мас-
сы. В термогравиметрии по производной регистрируется производ-
ная изменения массы по времени dm/dt в функции температуры или
времени, т. е.
= или/). (2.5)
Полученная кривая представляет собой первую производную кри-.
вой изменения массы. Вместо ступенчатой кривой получают ряд
пиков, площадь под которыми пропорциональна абсолютному из-
менению массы образца.
Этот метод был впервые предложен Декейзером [59, 601 в 1953 г.,
затем его применили Эрди и др. [61 ], а также Уотерс [621. Даль-
нейшие работы в этой области термогравиметрии выполнили: Эрди
[63,641, Паулик Ф. и др. [65], Уэддамс и Грей [66], Уотерс [67],
Кэмпбелл и др. [68], а также Эрди и др. [69].
На фиг. 2.30 для сравнения приведены ТГ-кривые, полученные
обычным методом (а) и методом термогравиметрии по производной
(б). Кривая б может быть получена путем обычного дифференци-
рования кривой а или путем электронного дифференцирования ТГ-
сигнала. Имеется соответствующее дополнительное оборудование
для большинства типов термовесов, и поэтому ТГП-кривую можно
легко записать вместе с ТГ-кривой. ТГП-кривая, выведенная мате-
матически или записанная непосредственно, содержит не больше
информации, чем интегральная Т1 -кривая, полученная при тех же
условиях; просто эта информация представлена в другом виде [70].
Эрди и др. [61 ] сформулировали преимущества термогравимет-
рии. по производной следующим образом:
1. Кривые можно получить одновременно с измерениями, про-
водимыми методами ТГ и ДТА (дифференциальный термический
анализ).
2. Кривые, полученные методами ДТА и ТГП, сравнимы между
собой. Однако результаты метода ДТА фиксируют даже такие из-
менения состояния образца, которые не сопровождаются уменьше-
нием массы. С другой стороны, кривые, полученные методом ТГП,
более воспроизводимы.
3. Кривые ДТА растянуты в более широком интервале темпера-
тур из-за нагревания вещества' после завершения реакции; при из-
мерениях методом ТГП точно определяются температуры начала,
максимальной скорости и завершения реакции.
4. На ТГ-кривых не удается разделить стадии, следующие не-
посредственно друг за другом. На ТГП-кривых они отображаются
острыми пиками и могут быть разделены.
5. ТГП-кривые представляют собой производные ТГ-кривых;
Термогравиметрия
53
поэтому площадь под ними точно соответствует изменениям массы.
Следовательно, ТГП может дать точную количественную информа-
цию.
6. Метод ТГП можно использовать для исследования веществ,
которые по той или иной причине нельзя анализировать методом
ДТА. Например, некоторые органические вещества при нагревании
плавятся, но исследование их методом ТГП дает удовлетворитель-
ные результаты.
Фиг. 2.30. Сравнение ТГ-кривой (а) и ТГП-кривой (б) изменения массы.
Ньюкирк и Симмонс [58, 70] подвергли метод ТГП критиче-
скому анализу. Ниже приводятся некоторые утверждения сторон-
ников этого метода и комментарии Ньюкирка и Симмонса, которые
сравнивают этот метод с методом ТГ [581.
У тверждение Комментарий
1. ТГП имеет большую точность в
начале реакции, когда изменения мас-
сы малы.
2. ТГП лучше позволяет различить
перекрывающиеся стадии реакции.
3. ТГП позволяет более точно опре-
делять температуры реакции.
4. Сравнение данных ТГП н ДТА
дает возможность отличить пики
.ДТА, сопровождающиеся изменением
массы, от пиков, вызванных только
-термическими изменениями.
1. Этим преимуществом нельзя вос-
пользоваться без применения более
точных термовесов.
2. В большинстве случаев это дей-
ствительно является преимуществом.
3. Только температуру, соответст-
вующую максимальной скорости реак-
ции.
4. Нет преимущества по сравнению
с ТГ.
54
Глава 2
Не следует отождествлять температуру, соответствующую пику
ТГП-кривой, с «температурой разложения». Первая представля-
ет собой температуру, при которой скорость изменения массы макси-
мальна, но эта температура определенно не является температурой,
при которой масса образца начинает уменьшаться, т. е. Т, [70].
Е. КИНЕТИКА РЕАКЦИЙ
1. НЕИЗОТЕРМИЧЕСКИЕ МЕТОДЫ
На протяжении последнего десятилетия динамическая термогра-
виметрия широко применяется для исследования кинетики реакций
термического разложения. Дойль [71 ] отмечает, что одна динами-
ческая кривая изменения массы эквивалентна большому числу
соответствующих изотермических кривых; кроме того, отсутствуют
ошибки, связанные с заменой образцов, поскольку всю информа-
цию получают с помощью одного и того же образца. Следует, одна-
ко, отметить некоторую ограниченность термогравиметрических
данных при проведении кинетического анализа, поскольку они поз-
воляют получать только обычные кинетические параметры про-
цесса изменения массы для определенных условий эксперимента.
Эти кинетические данные, какими бы убедительными они ни каза-
лись, все-таки остаются эмпирическими [71 ]. Как правило, не-
которые кинетические параметры могут быть оценены на основа-
нии термогравиметрических данных только в сочетании с дополни-
тельной информацией, полученной другими методами. Так, в слу-
чае полимеров полученной информации обычно недостаточно для
проведения на ее основе полного кинетического анализа. Поэтому
данные об изменении массы для этих материалов обычно подверга-
ют чисто эмпирической обработке.
Преимущества определения кинетических параметров неизо-
термическими методами по сравнению с обычными изотермическими
состоят в следующем [81: а) требуется гораздо меньше данных;
б) кинетические параметры могут рассчитываться непрерывно для
всего интервала температур; в) когда при нагревании образца до
требуемой температуры он вступает в реакцию, результаты, полу-
ченные изотермическими методами, часто сомнительны; г) требу-
ется только один образец. Определенным недостатком неизотерми-
ческих методов по сравнению с изотермическими обычно является
невозможность установления механизма реакции и определения
достоверных значений энергии активации, порядка реакции и час-
тотного фактора. Использование неизотермических кинетических
методов подвергалось сомнению [72] и критике [43, 73—79]. При-
нимая во внимание большое число критических замечаний по этому
поводу и в то же время не желая выходить за рамки этой книги,мы
лишь кратко обсудим кинетические методы. Для получения более
Т ермогравиметрия
55
исчерпывающих сведений читателю следует обратиться к другим
источникам [71, 80—821.
Расчеты кинетических параметров по ТГ-кривой основаны не-
формальном кинетическом уравнении [431
— *L = kxn, (2.6)
dt
где х — масса образца, вступившая в реакцию; п — порядок реак-
ции; k — удельная константа скорости реакции. Это уравнение
очень хорошо описывает кинетику термического разложения твер-
дых веществ, например эндотермические реакции оксалатов, пер-
манганатов, перхлоратов и азидов металлов. Зависимость удельной
константы скорости от температуры описывается уравнением Арре-
ниуса
k = Ae~E,Rr, (2.7)
где А — предэкспоненциальный множитель, Е — энергия акти-
вации, a R — универсальная газовая постоянная. Это уравнение
обычно применяется только для узкого интервала температур.
Математически для изучения кинетики разложения использу-
ется один из трех методов; а) дифференциальный, б) интегральный
или в) метод аппроксимации.
Сестак [43] показал, что соотношение между х и уменьшением
массы w описывается уравнением
-dx~-^-dw, (2.8)
“’со
где т0 — начальная масса образца и — максимальная потеря
массы. Интегрируя левую часть уравнения (2.8) от т0 до х и правую
часть от 0 до w, получим
x=^-(wm-w). (2.9)
СО
Подстановкой (2.9) и (2.7) в (2.6) и дифференцированием в лога-
рифмической форме получим требуемое выражение. Это — диффе-
ренциальный метод Фримена и Кэррола [83].
В интегральных методах используется интегральная форма
уравнения (2.6) после подстановки уменьшения массы w в уравне-
ния (2.8) и (2.9)
w Тг
((И’»—w)~"dw = ~ I e~E,RU dT- (2‘1 °)
“ 0 V г,
Правую часть уравнения можно решить различными методами, и
решение уравнения в окончательном виде представляет собой бес-
56
Глава 2
конечный ряд, в котором обычно учитываются только первые два
члена. Эти методы используются Дойлем [841, Коутсом и Редфер-
ном [85], а также другими исследователями [86, 87].
Фиг. 2.31. Определение скорости реакции и массы прореагировавшего ве-
щества по ТГ-кривой [12].
При использовании метода аппроксимации правая часть уравне-
ния (2.10) решается с помощью приближения, заключающегося
в том, что температуру Tt считают соответствующей максимальной
скорости разложения. Этот метод применялся Горовицем и Метцге-
ром [88], а также другими исследователями [87, 89—91].
а. Метод Ньюкирка ЗД"
С помощью одной ТГ-кривой Ньюкирк [12] получил константы
скорости реакции разложения (фиг. 2.31). Для ряда температур Т\
и остаточные массы образца равны а—Xi и а—х2, а скорости ре-
акции (dx/dt)r и (dx/dt)2 определялись как тангенсы углов накло-
на касательных к ТГ-кривой в точках 1 и 2. В случае реакции
первого порядка dxldt = k(a—х) и Логарифм константы скорос-
ти реакции k на графике зависимости от \/Т имеет вид прямой ли-
нии. Результаты измерений, выполненных в изотермических и не-
изотермических условиях^ описываются почти параллельными пря-
мыми.
б. Метод Фримена и Кэррола ли '
Возможно, наиболее широко используемым кинетическим мето-
дом является метод, предложенный Фрименом и Кэрролом [83] в
1958 г. Преимущество этого метода заключается в том, что по срав-
Термогравиметрия
57
нению с изотермическим методом для него требуется гораздо мень-
ше экспериментальных данных, а кинетические параметры могут
быть получены в любой точке всего интервала температур. Кроме
того, как уже указывалось, если при нагревании образца до требуе-
мой температуры он вступает в реакцию, результаты, полученные
изотермическим методом, часто сомнительны. Порядок реакции п
и энергия активации реакции Е вычисляются по уравнению
— (Е/2,3/?) Д (Г/Г) = _ п + Alg(da>/dQ ' (2 j j
Д 1g wr Д 1g wr
где wr = wc —w; wc — максимальная потеря массы ; иу —- общая
потеря массы к моменту времени I.
в. Метод Горовица и Метцгера
В случае когда выражение для константы скорости включает
концентрации в виде мольных долей, а общее число молей посто-
янно, можно использовать выражение 188]
. . Wo — W, Е6
In In -J----*- = ---5- ,
W — RT2s
где W — остаточная масса при данной температуре; Wo и Wt —
соответственно начальная и конечная массы; 6 — температура
эталона.
(2.12)
г. Метод Коутса и Редферна их
Для реакций, порядок которых неизвестен, Коутс и Редферн
185] получили следующее выражение:
,1-(1-^ = j AR_ л _ 2ет \
6 аЕ Е ) 2,
где а — массовая доля образца, разложившаяся за время t, и а —
скорость нагревания. Зависимость величины Igll — (1—а)1-"]/
/Т2(1—п) ог t/Т или lg[—1п(1— а) |/Т2 при п = 1 от \/Т при пра-
вильном выборе значения п имеет вид прямой линии
с наклоном, определяемым величиной—£72,3/?. Величину
lg(X/?/a£) I.—(2/?77£) I можно считать постоянной для большинст-
ва значений £ и интервала температур, в котором протекает боль-
шинство реакций.
д. Метод Дойля х
Кинетика выделения детучих веществ из полимеров подробно
рассмотрена Дойлем [71, 84]. Если кажущуюся долю остаточной
массы, вычисленную по начальной массе, обозначить через w, то
кажущаяся испарившаяся массовая доля v составит
58
Глава 2
v—1—w. (2.14)
Кажущаяся скорость испарения определяется умножением накло-
на ТГ-кривой —dw/dT на постоянную скорость нагревания В:
(2.15)
dt dT ' ’
На определенной стадии испарения, однако, соответствующая
доля остаточной массы является истинной h, рассчитываемой по
общей массовой доле, испарившейся на этой стадии, а не по полной
начальной массе:
Л = (2.16)
где Н — общая кажущаяся массовая доля, испарившаяся на данной
стадии, и G — кажущаяся массовая доля, оставшаяся после завер-
шения этой стадии. Из уравнений (2.14) и (2.15) следует
-5Г=-"4-' (2-I7)
Заметим, что Н и G редко бывают точно известны; во многих случаях
их определение составляет одну из основных трудностей кинетиче-
ских расчетов.
Другая проблема связана с необходимостью принимать во внима-
ние характер кинетического процесса. В общем случае
--^-=г/(Л). (2.18)
dt
где г — эмпирическая константа скорости испарения, а вид функ-
ции f(h) зависит от порядка реакции. Константа г считается эмпири-
ческой, поскольку ее величина для данного вещества не всегда опре-
деляется только температурой, но может зависеть от материала и
формы держателя образца, состава атмосферы и других факторов.
Дойль [84] выбрал символ г вместо удельной константы скорости
реакции k, чтобы подчеркнуть эмпирический характер этой констан-
ты. Уравнение Аррениуса для процесса испарения, записанное с
использованием этого символа, примет следующий вид:
r = ae~b'RT. (2.19)
Константа b имеет смысл энергии активации, по крайней мере для
части исследуемого интервала температур.
При определении констант в уравнении (2.15) обычно использу-
ется только небольшой участок ТГ-кривой, не слишком крутой и не
слишком пологий, с тем чтобы измерения были достаточно точными.
Дело в том, что в области скоростей испарения, которые малы по
сравнению со скоростью нагревания, углы наклона ТГ-кривой опре-
Термогравиметрия
59
деляются заведомо неточно, так как они значительно больше, чем
при изотермическом методе. Это происходит потому, что в термо-
гравиметрии время выдерживания при каждой температуре столь
мало, что при малых скоростях испарения последнее не удается
обнаружить.
Озава [92, 93 J разработал приближенный интегральный метод,
подобный предложенному Дойлем [841. Считается, что метод Оза-
вы имеет более широкое применение.
Жако [941 также предпринял попытку упростить метод проб
и ошибок Дойля и найти новые применения ТГ-кривым. Он исходил
из основного уравнения, приведенного в работе Дойля [84]:
g(<0= “₽(*)• (2-2°)
Rq
Логарифмирование этого уравнения дает
1g S (а) - 1g Р (к) = В, (2.21)
Rq
где В зависит только от природы исследуемого вещества и скорости
нагревания и не зависит от температуры. Значение g(a) для данной
температуры можно рассчитать по ТГ-кривой, если известна зави-
симость Да).
Метод заключается в следующем. Предполагая справедливость
зависимости Да) и используя ТГ-данные, рассчитываем значения
g(a) для различных температур. Значение кажущейся энергии акти-
вации Е находится методом проб и ошибок. Она равна величине, при
которой обеспечивается максимальное постоянство параметра
В = 1g g (a) — 1g p (x). (2.22)
e. Метод Ингрэхема и Марье
Для реакций термического разложения, описываемых линей-
ным законом, например для реакции разложения карбоната каль-
ция, константа скорости k определяется в виде
где dw — потери массы с единицы площади за время dt. Если тем-
пература образца возрастает по линейному закону, то температура
в любой момент времени t будет равна
Т = b + at, (2.24)
где а — скорость нагревания и b — начальная температура. Ис-
ходя из этого соотношения, Ингрэхем и Марье [951 показали, что
60
Глава 2
при замене dt на dT/a можно получить следующее уравнение:
lg = IgT-Iga + IgC- (2.25)
al 2,ЗиЗ/<
Энергия активации вычисляется по углу наклона кривой зависи-
мости lg(tWd7")—lg7’ -f- Iga от 1/7’. Логарифм скорости нагрева-
ния, являющийся постоянной величиной для каждого эксперимен-
та, оставлен в уравнении с целью установления корреляции между
ТГ-кривыми, полученными при различных скоростях нагревания.
ж. Метод Вахуски и Воборила
При использовании этого метода [96] кинетические постоянные
рассчитываются по ТГ-кривым с помощью дифференциального ме-
тода. Здесь также принимаются во внимание тепловые эффекты
реакций, приводящие к отклонению температуры образца от задан-
ных значений, соответствующих линейному закону. За основу бе-
рется дифференциальное уравнение термического разложения твер-
дого вещества
-g- = S(l_a)»exp(-JL),
(2.26)
где а — степень превращения. Логарифм этого уравнения имеет
вид
1п-^ = 1пй + п1п(1-а)--А. (2.27)
Пусть а и Т — функции времени. Тогда, дифференцируя (2.27)
по времени, находим
dta/di? ______ п da___________£ аТ ^gx
da/dt I — a dt Г RT* dt ( )
После перестановки получается линейное уравнение
(d^/d/2) Т2 _ da/dt Т2 £
(da/dt) (dT/dt) 1—а dT/dt R' '
Порядок реакции п можно рассчитать по углу наклона кривой,
построенной с помощью этого уравнения, а отсекаемый на оси отре-
зок этой кривой даст энергию активации.
з. Другие методы
Упрощенные методы расчета удельный констант скорости ре-
акции на основе ТГ-кривых предлагались различными авторами.
Дэйв и Чопра [97 ] рассчитывали удельную константу скорости
с помощью уравнения
Т ермогравиметрия
61
, _ (Л/т0)«-] (dx/dt)
(2.30)
(2.31)
(2.32)
(Л—а)"
где А — полная площадь под кривой ТГП; а — площадь под кри-
вой, ограниченная временем t; dx/dt — ордината кривой в момент
времени /; т0 — начальная мольная доля реагента; п — порядок
реакции.
Подобный подход был использован Папазяном и др. [98], а так-
же Адоньи [100], которые рассчитывали удельную константу ско-
рости по ТГП-кривой, используя формулу
k _ dx/dt
(а—х)п '
где а—х — масса неразложившегося реагента. Кинетические пара-
метры, полученные этим простым методом, по мнению авторов, ана-
логичны результатам, полученным при помощи изотермических
измерений.
Фармер [99] использовал выражение
1g (— ww~n) = —--— — + 1g А,
v / R In 10 T
где w — массовая доля реагента для расчета Е и А по наклону и от-
секаемому на оси отрезку параметрической кривой lg(—ww~n')
в функции 1/7.
Магнусон [101J подробно рассмотрел расчет кинетики термиче-
ского разложения высокомолекулярного полидиметилсилоксана.
Он использовал простое уравнение первого порядка
— — = А,Г,
dt р
где —dW/dt — скорость уменьшения массы образца, kp — наблю-
даемая константа скорости реакции и Д’ — масса образца.
Ахар и др. [162] разработали дифференциальный метод, при-
менимый ко всем реакциям при условии, что известен их истинный
механизм [76]. Метод основан на использовании выражения
1 А
= 1п---------,
В RT
(2.33)
1 da
(a) dT
где а — массовая доля образца, вступившая в реакцию за время t,
и В — скорость нагревания. Построив график левой части уравнения
в функции 1/7, получим прямую линию, по которой можно опреде-
делить Е и А. Форма Да) зависит от характера реакции. Например,
в случае параболического закона а2 = kt,, применимого немногим
реакциям, скорости которых определяются диффузией, имеем Да) ~
Е
(2.34)
In
62
Глава 2
Метод Бройдо [103], примененный к пиролизу целлюлозы, осно-
ван на уравнении
In In — = — -f- 4- + А, (2.35)
у R Т ' ’
где у — число молекул еще не разложившегося исходного вещества
и А — константа. График 1п1п(1/р) в функции 1/Т имеет вид пря-
мой линии, по наклону которой можно вычислить Е и Z.
2. СРАВНЕНИЕ РАЗЛИЧНЫХ МЕТОДОВ
Сестак [431 сопоставил результаты расчета кинетических
параметров дегидратации полугидрата сульфата кальция
а-Са SO4 • VaHzO пятью различными методами: Фримена и Кэррола
[83], Дойля [84], Коутса и Редферна [85], Горовица и Метцгера
[88] и ван-Кревелена и др. [87]. Расчеты показали, что
значения Е различаются не более чем на 10%. Таким образом, все
методы оказались удовлетворительными для расчета Е в пределах
требуемой точности. Были также вычислены ошибки каждого мето-
да, обусловленные неточностью визуального определения значений
Температура, °C
Фиг. 2.32. Результаты расчета методом Коутса и Редферна, а также методом
Ахара и др. [76].
Q СяСО, (таблетка)! д СаСО, (порошок); X CaCO,:Al2O,; V CaCO,:Fe,O,.
Кинетические параметры, полученные методом Коутса и Редферна (б) и методом Ахара и др. (в) [76]
64
Глава 2
по ТГ-кривым. Эти ошибки — ее и е„ (при определении Е и п со-
ответственно) — были следующими: в методе Фримена и Кэррола
= 4% и 8„ = 12%; в методе Горовица и Метцгера б£ = 2%
(при правильном значении п); в методе Дойля е£ =4%. Однако
величина этих ошибок главным образом зависит от положения на
ТГ-кривой точки, для которой производятся расчеты. При исполь-
зовании дифференциальных методов самые точные значения полу-
чаются, если эта точка расположена на не очень крутых участках
кривой. В случае метода аппроксимации точность зависит от опре-
деления температуры в точке перегиба кривой.
С точки зрения простоты расчета наилучшим является, по-види-
мому, метод Дойля, поскольку при его использовании кинетиче-
ские параметры определяются по одной точке на кривой. Недостат-
ком его является необходимость априорного знания порядка ре-
акции; этот недостаток частично возмещается применением метода
Коутса и Редферна.
Шарп и Уэнтворт [761 оценили три кинетических метода для слу-
чая термического разложения карбоната кальция в различных ус-
ловиях. Образец имел вид таблетки, порошка или смеси с альфа-
окисью алюминия или окисью альфа-железа (III) в молярном от-
ношении 1:1. Использовались следующие методы: а) Фримена и
Кэррола, б) Коутса й Редферна, в) Ахара и др. [102]. Кинетиче-
ские данные, рассчитанные последними двумя методами, представ-
лены графически на'фиг. 2.32 и в табл. 2.3. В каждом случае гра-
фик представлял собой прямую линию в широком интервале зна-
чений а при п = 1/2. При п = 2/3 интервал значений а был уже,
особенно при использовании метода «в», когда при больших значе-
ниях а появлялась заметная кривизна. При п = 1 эта кривизна
становилась заметной для обоих методов. При п — \/2 энергия ак-
тивации в среднем составляла ~184 кДж/моль. Добавление окис-
лов железа или алюминия не влияло на энергию активации.
На фиг. 2.33 и в табл. 2.4 приведены результаты, полученные
как для таблетки, так и для порошка при использовании метода «а».
Таблица 2.4
Кинетические параметры, полученные методом
Фримена и Кэррола (а) [76]
Число точек Интервал значений а п Е, кДж/моль
8 0,25—0,84 +0,55 180
14 0,05—0,84 —0,07 151
Термогравиметрия
65
По сравнению с фиг. 2.32 точки на фиг. 2.33 имеют больший раз-
брос, что приводит к увеличению погрешности оценки значения Е.
11рямая получена теоретически. Она пересекает ось ординат в точ-
ке +0,5 и имет наклон, соответствующий энергии активации, рав-
ной 184 кДж/моль. Хотя эта прямая кажется удовлетворительной,
по точкам можно провести ряд других линий, соответствующих
иным значениям п и Е.
Фиг. 2.33. Результаты расчета методом Фримена и Кэррола [83].
Q СаСОа (таблетка); д СаСО, (порошок).
В сущности методы «б» и «в» дают одинаковые результаты. Пре-
имущество последнего заключается в том, что значения А получа-
ются непосредственно по точке пересечения с осью и метод может
применяться с одинаковым успехом для расчета реакций,определяе-
мых как диффузией, так и непосредственно самим процессом. Недо-
статком этого метода является необходимость определения da/dT —
Тангенсов углов наклона касательных к ТГ-кривой для ряда значе-
ний Т. Метод «б» менее трудоемок и не требует определения танген-
e<4i углов наклона.
• Больший разброс данных, полученных при использовании метода
•М», объясняется в основном тем, что три различные функции опре-
делялись в узких интервалах температур. Одна из них включает
• пнгспсы dw/dt, и поэтому любая неточность при определении од-
ного значения влияет на положение двух точек на графике, по
которому определяется энергия активации. Поэтому метод «а» был
признан менее удовлетворительным, чем другие два, и для исполь-
чшпния не рекомендуется.
1 '.‘30
66
Глава 2
Кэррол и Манч [104] подвергли критике расчетные методы проб
и ошибок, подобные методам «б» и «в». Возникает вопрос, годится ли
критерий постоянства кинетических параметров даже для простой
химической реакции, поскольку реакция в твердой фазе очень слож-
ная. Кэррол и Манч высказали мнение, что при современном уровне
знаний постоянство кинетических параметров в общем случае для
реакций в твердой фазе является неподходящим предположением.
В том случае, когда случайные ошибки при измерениях наклона
«сглажены», особенно на начальной и конечной стадиях процесса,
можно применить метод Фримена и Кэррола. Точность этого метода
может быть при этом значительно увеличена.
3. ОПРЕДЕЛЕНИЕ МЕХАНИЗМА РЕАКЦИИ ПО
КИНЕТИЧЕСКИМ ДАННЫМ, ПОЛУЧЕННЫМ В
НЕИЗОТЕРМИЧЕСКИХ УСЛОВИЯХ
Проблема установления механизма реакции при помощи не-
изотермических кинетических методов обсуждалась Сестаком и
Берггреном [73], а также Сатавой [105]. Метод, предложенный
Сатавой, основывался на предположении, что неизотермическую
реакцию в бесконечно малом интервале времени можно рассматри- |
вать как изотермическую, скорость которой описывается выраже- ’
нием
= Ae~ElRTf(a), (2.36)
где а — массовая доля вещества, разложившаяся за время t, и f(a)
зависит от механизма реакции. При постоянной скорости нагрева-
ния dT/dt = q интегрирование уравнения (2.36) даст
а
( -^~ = ^(а)=-^р(х), (2.37)
J f («) Я?
° • !
где р(х) определяется выражением
со
л-Х р p-и р~Х
р(х) = -----—du— — + Ei(—x). (2.38)1
X J и X I
+*
Здесь и — E/RT, х — E/RT и Т — температура, при которой мае-1
совая доля образца а вступила в реакцию.
После логарифмирования уравнения (2.36) получаем
lg^(a)-lgp(x) = lg^- (2-3!))
Т ермогравиметрия
67
ПрйЬйя часть этого уравнения не зависит от температуры, а левая
•ВИМспт. В первом приближении функция lgp(x) линейно зависит
г \1Та, если величина х достаточно велика, и, таким образом,
Ч Н(х) тоже должна быть линейной функцией 1/Та. В случае точ-
|||>Ю описания механизма реакции зависимость 1g р(х) от i/Ta
Цижпа выражаться прямой линией; в другом случае это утвержде-
И" не справедливо. Чувствительность данного метода определения
Ьхйнпзма реакции невысока, но все же он дает полезную информа-
цию.
Типы наиболее часто встречающихся механизмов реакций при-
ВЮитпл в табл. 2.5, а на фиг. 2.34 указана методика определения
В»хпц)1:1ма реакции по ТГ-кривой. Из графика видно, что только
I ШВвя, соответствующая механизму Fx, представляет собой пря-
мую ЛИНИЮ.
Таблица 2.5
Наиболее часто встречающиеся механизмы реакций [105]
•ИИИИ" Уравнение Процесс, определяющий скорость реакции
X2 — kt Одномерная диффузия
Р. (1 —a) in (1 —а) + а = kt Двумерная диффузия; цилиндри- ческая симметрия
Р. —а)1/3р = ^ Трехмерная диффузия; сферичес- кая симметрия; уравнение Джандера
Р* (1—А а) — (1 — а)2/3 = kt Трехмерная диффузия; сферичес- кая симметрия; уравнение Гин- стлинга — Браунштейна
— In (1 — а) — kt Случайное образование зародышей; одно ядро на каждую частицу
* [—In (1—а)]1/2 = W Случайное образование зароды- шей; уравнение Аврами (I)
4. 1 [-1П (1— a)]V3 Случайное образование зароды- шей; уравнение Аврами (II)
1 —(1 —а)'/2 = /й Реакция на границе раздела фаз; цилиндрическая симметрия
\ 1— (1— a)l/3c=kt Реакция на границе раздела фаз; сферическая симметрия
Глава 2
69
т, К
400
D,
R
-20
430
380 390 400
т, к
Фиг. 2.34, Методика определения механизма реакции по ТГ-кривой [10|
Нижняя кривая соответствует кинетическому уравнению (см. табл. 2.5); Е=126 кДж/м<М
Z= Ю18/^1 моль-1; <7=1 град/мин. Верхние кривые представляют собой графики lg g 0
функции 1/Та, построенные по ТГ-кривой для различных кинетических уравнений. Пря«
линия, соответствующая кинетическому уравнению F,, совпадает с графиком —1g р(л)
функции 1/Га или Е = 126 кДж/моль.
2,6 •
HI.
17.
ЛИТЕРАТУРА
1. Simmons E. L., Newkirk A. E., Taianta, 11, 549 (1964).
2. Newkirk A. E., Simmons E. L., частное сообщение
3. Duval C., Inorganic Thermogravimetric Analysis, ’
Amsterdam, 1963.
4. Duval C., Inorganic Thermogravimetric Analysis, 1st ed.
sterdam, 1953.
— ТермОгравиметрия
5. Duval С., Anal. Chem., 23, 1271 (1951).
6. Lukaszewski G. M., Redfern J. P., Lab. Pract., 10, 469 (1961).
7. Jacque L., Guiochon G., Gendrel P., Bull. Soc. Chim. France, 1061 (1961).
H. Coats, A. W., Redfern J. P., Analyst, 88, 906 (1963).
'.I Honda K-, Sci. Rept. Tohoku Univ., 4, 97 (1915).
11) . Guichard M., Bull. Soc. Chim. France, 2, 539 (1935).
II. Duval C., Anal. Chim. Acta, 31, 301 (1964).
I" Newkirk A. E., Anal. Chem. , 32, 1558 (1960).
13. Simmons E. L., Wendlandt W. W., Thermochim. Acta, 2, 465 (1971).
14 De Vries K- J., Gellings P. J., J. Inorg. Nucl. Chem., 31, 1307 (1969).
Г Herbell T. P., Thermochim. Acta, 4, 295 (1972).
И* Perkin-Elmer Model TGS-1 Thermobalame Brochure, Perkin-Elmer Co.,
Norwalk, Conn.
Fruchart R., Michel A., Compt. Rend., 246, 1222 (1958).
18 Demassieux N., Malard C., Compt. Rend., 245, 1514 (1957).
| 19. Rynasiewisz J., Flagg J. F., Anal. Chem., 26, 1506 (1954).
80. De Clerq M., Duval C., Anal. Chim. Acta, 5, 282 (1951).
II. Paulik F., Paulik J., Thermochim. Acta, 4, 189 (1972).
в. Mettler Thermal Techniques Series, Tech. Bull., T-106.
3. Paulik J., Paulik F., Erdey L., Anal. Chim. Acta, 34, 419 (1966).
I 4. Feldman R. F., Ramachandran V. S., Thermochim. Acta, 2, 393 (1971).
i |. Wendlandt W. W., Simmons E. L., Thermochim. Acta, 3, 171 (1972).
I I. Guenot J., Manoli J. M., Bull. Soc. Chim. France, 2663 (1969).
7, Nicholson G. C., J. Inorg. Nucl. Chem., 29, 1599 (1967).
I, Brown H. A., Penski E. C., Callahan J. J., Thermochim. Acta, 3, 271 (1971).
I I. Wiedemann H. G., Achema Congress paper, Frankfurt, June 26, 1964.
D. Friedman H. L., Anal. Chem., 37, 768 (1965).
I. Saito Нд Sci. Rept. Tohoku Univ., 16, I (1927).
I Richer A., Vallet P., Bull. Soc. Chim. France, 148 (1953).
I. Garn P. D., Kessler J. E., Anal. Chem., 32, 1563, 1900 (1960).
I Garn P. D., Anal. Chem., 33, 1247 (1961).
I, Rabatin J. G., Card C. S., Anal. Chem., 31, 1689 (1959).
I, Soulen J. R., Mdckrin I., Anal. Chem., 33, 1909 (1961).
V Lukaszewski G. M., Nature, 194, 959 (1962).
•. Newkirk A. E„ McKee D. W., J. Catalysis, H, 370 (1968).
W, Macklen E. D., J. Inorg. Nucl. Chem., 29, 1229 (1967).
Ш Steger H. F., J. Jnorg. Nucl. Chem., 34, 175 (1972).
♦I, Newkirk A. E., Thermochim. Acta, 2, 1 (1971).
•I Paulik F., Paulik J., Erdey L., Taianta, 13, 1405 (1966).
*1, Sestak J., Taianta, 13, 567 (1966).
i< Wiedemann H. G., Thermal Analysis, Schwenker R., F., Garn P. D., eds.,
| Academic Press, N. Y., 1969, p. 229.
, IA Newkirk A. E., Aliferis I., Anal. Chem., 30, 982 (1958).
•<l Ramakrishna Udupa M., Aravamudan G., Current Sci., 39, 206 (1970).
V Cahn L., Peterson N. C., Anal. Chem., 39, 403 (1967).
I Cahn L., Schultz H., Anal. Chem., 35, 1729 (1963).
*'l Martinez E., Am. Mineralogist, 46, 901 (1961).
’0 Guiochon G., Anal. Chem., 33, 1124 (1961).
II Simmons E. L., Newkirk A. E., Aliferis I., Anal. Chem., 29, 48 (1957).
Mlelenz R. C., Schieltz N. C., King M. E., Clays and Clay Minerals
NAS-NRC, Washington, Pub. 327, 1954, pp. 289-296.
' Sarasohn I. M., Tabeling R. W., Pittsburgh Conf, on Analytical and Applied
1Ие. , шеей Spectroscopy, Pittsburgh, Pa., March 5, 1964.
2nd ed., Gayle J. B„ Egger С. T„ Anal. Chem., 44, 421 (1972).
. _ Cahn L., Schultz H. R., Vacuum Microbalance Techniques, Behrndt К. H.,
Elsevier, C(| , VoL 3> plenum> N Y_, 1963, p. 29.
l orkel W., Naturwissenschaften, 47, 10 (1960).
Глава 2
Глава 3
57. Newkirk А. Е., Thermochim. Acta, 2, 1 (1971).
58. Newkirk А. Е., Proc, of the First Toronto Symp. on Thermal Analysis,
McAdie H. G., ed., Chemical Inst, of Canada, Toronto, 1965, p. 29.
59. De Keyser W. L., Nature, 172, 364 (1953).
60. De Keyser W. L., Bull. Soc. France Ceram., 20, 1 (1953).
61. Erdey L., Paulik F., Paulik J., Nature, 174, 885 (1954).
62. Waters P. L., Nature, 178, 324 (1956).
63. Erdey L., Periodica Polytech., 1, 35, 91 (1957).
64. Erdey L., Chem. Zvesti, 12, 352 (1958).
65. Paulik F„ Paulik J., Erdey L., Z. Anal. Chem., 160, 241, 321 (1958).
66. Waddams J. A., Gray P. S., Nature, 183, 1729 (1958).
67. Waters P. L., J. Sci. Instr., 35, 41 (1958).
68. Campbell C., Gordon S., Smith C. L., Anal. Chem., 31, 1188 (1959).
69. Erdey L., Liptay B., Svehla G., Paulik F., Taianta, 9, 489 (1962).
70. Newkirk A. E., Simmons E. L., Taianta, 13, 1401 (1966).
71. Doyle C. D. Techniques and Methods of ,,Polymer Evaluation”, Slade P. E., I
Jenkins L. T., eds., Marcel-Dekker, N. Y., 1966, Ch. 4.
72. Clarke T. A., Evans E. L., Robins K. G., Thomas J. M., Chem. Comm,,
266, 1969.
73. Sestak J., Berggren G., Thermochim. Acta, 3, 1 (1971).
74. Hill R. A. W., Nature, 227, 703 (1970).
75. Simmons E. L., Wendlandt W. W., Thermochim. Acta, 3, 498 (1972).
76. Sharp J. H., Wendworth S. A., Anal. Chem., 41, 2060 (1969).
77. Sestak J„ Silikaty, 11, 153 (1967). i
78. MacCallum J. R., Tanner J., European Polymer J., 6, 1033 (1970).
79. Draper A. L., Sveum L., Thermochim. Acta, 1, 345 (1970).
80. Flynn J. H„ Wall L. A., J. Res. Natl. Bur. Stand., 70A, 487 (1966).
81. Carroll B., Manche E. P., Thermochim. Acta, 3, 449 (1972).
82. Reich L., Stivala S., Elements of Polymer Degradation, McGraw-Hill,
, N. Y., 1971.
83. Freeman E. S., Carroll B., J. Phys. Chem., 62, 394 (1958).
84. Doyle C. D., J. Appl. Polymer Sci., 5, 285 (1961).
85. Coats A. W„ Redfern J. P., Nature, 201, 68 (1964).
86. Turner R. C., Hofman J., Chen D., Can. J. Chem., 41, 243 (1963).
87. Van Krevelen W., van Heerden C., Hutjens F., Fuel, 30, 253 (1951).
88. Horowitz H. H., Metzger G., Anal. Chem., 35, 1464 (1963).
89. Fuoss R. M., Sayler I. O., Wilson H. S., J. Polymer Sci., 2, 3147 (196Ф
90. Berlin A., Robinson R. T., Anal. Chim. Acta, 27, 50 (1962).
91. Reich L., J. Polymer Sci., B2, 621 (1964).
v92. Ozawa T., Bull. Chem. Soc. Japan, 38, 1881 (1965).
93. Ozawa T., J. Thermal Anal., 2, 301 (1970).
94. Zsako J., J. Phys. Chem., 72, 2406 (1968).
95. Ingraham T. R., Marier P., Can. J. Chem. Eng., 161 (1964).
96. Vachuska J., Voboril M., Thermochim Acta, 2, 379 (1971).
97. Dave N. G., Chopra S. K-, Z. Physik. Chem., 48, 257 (1966).
98. Papazian H. A., Pizzolato P. J., Orrell R. R., Thermochim. Acta, 4, >
,, (1972).
99. Farmer R. W., Air Force Materials Lab. Tech. Rept AFML-TR-65-21
1967, pt. II.
100. Adonyi, Z., Periodica Polytech., 11, 325 (1967).
101. Magnuson J. A., Anal. Chem., 36, 1807 (1964).
102. Achar B. N. N., Brindley G. W., Sharp J. H., Proc. Int. Clay Cot
Jerusalem, 1, 67 (1966).
103. Broido A., J. Polymer Sci., Pt. A-2, 7, 1761 (1969).
104. Carroll B., Manche E. P., Anal. Chem., 42, 1296 (1970).
105. Satava V., Thermochim. Acta, 2, 423 (1971).
ТЕРМОВЕСЫ И ВСПОМОГАТЕЛЬНОЕ ОБОРУДОВАНИЕ
К НИМ
А. ВВЕДЕНИЕ
Термовесы — это прибор, позволяющий непрерывно определять
массу образца в зависимости от температуры. Образец можно на-
гревать или охлаждать с заданной скоростью или выдерживать в
изотермических условиях при постоянной температуре. Вероятно,
«наиболее распространенный способ состоит в измерении массы об-
разца при нагревании печи со скоростью 5—10 град/мин. Почти
все современные термовесы являются приборами с автоматической
записью результатов измерений, хотя в отдельных случаях при дли-
тельных изотермических измерениях запись производится вручную
(например, при работе с термовесами со спиральной пружиной).
Принципиальная схема современных термовесов представлена
па фиг. 3.1. Основными узлами термовесов являются: а) регистри-
рующие весы; б) печь; в) устройство для регулирования темпера-
туры печи по заданной программе или автоматический регулятор;
Г) ленточный самописец (с записью данных на движущуюся бу-
мажную ленту) или двухкоординатный самописец. Специфические
особенности каждого узла зависят от условий эксплуатации термо-
песов. Например, могут использоваться печи, обеспечивающие на-
грев до температур выше 2400° С и работающие в атмосфере возду-
ха, инертного газа, водорода, азота и в других средах или в вакууме.
Чувствительность регистрирующих весов также может быть раз-
личной. Например, полная шкала может быть рассчитана на интер-
валы измерений до 0,02 мг или до 100 г и более.
Лукашевский и Редферн [11 рассмотрели факторы, которые
г следует учитывать при разработке или покупке автоматических
К’рмовесов:
1. Прибор должен регистрировать уменьшение или приращение
Массы образца в зависимости от температуры и времени.
2. Температурный интервал работы печи термовесов должен
быть достаточно широким — от комнатной температуры до 1000,
1600 или 2400° С.
3. Уменьшение массы образца должно измеряться с точностью
||С менее ±0,01% при точности регистрации температуры ±1%.
4. Физические помехи, возникающие при нормальной работе
72
Глава 3
весов, такие, как радиационные и конвективные потоки или маг-
нитные поля от нагревателя печи, не должны оказывать влияния
на точность термовесов. Магнитные поля не должны также взаимо-
действовать с исследуемыми электропроводящими или магнитными
материалами.
Регулирование
атмосферы
Фиг. 3.1. Принципиальная схема современных термовесов.
5. Положение тигля внутри печи термовесов должно быть не-
изменным, чтобы регистрируемая температура соответствовала тем-
пературе образца.
6. Оборудование печи должно позволять производить нагрев
образцов в различных атмосферах.
7. Весы должны быть как можно более универсальными, чтобы
обеспечивать возможность быстрого изменения скоростей нагрева
и автоматический контроль программы изменения температуры.
8. Весы должны быть надежно изолированы от печи; необходи-
мо также принять меры по предотвращению износа кромок ножа
и других движущихся элементов для обеспечения точности взве-
шивания.
9. Необходимо дредусмотреть возможность периодической кали-
бровки весов как можно более простым способом для сохранения
требуемой точности измерений.
10. Лента, используемая для регистрации уменьшения массы!
и повышения температуры, должна иметь несколько скоростей 1
перемещения; должно быть предусмотрено устройство для отметки I
требуемых интервалов времени.
Очевидно, не все термовесы удовлетворяют одновременно v-etil
перечисленным требованиям. Однако уже существует ряд серийны' I
термовесов, в которых они удовлетворяются.
Термовесы и вспомогательное оборудование к ним
73
1. РЕГИСТРИРУЮЩИЕ ВЕСЫ
Регистрирующие весы являются, очевидно, самым важным узлом
термовесов. К ним предъявляются в основном те же требования,
что и к аналитическим весам, т. е. высокая точность, воспроизводи-
мость результатов измерений, достаточные чувствительность и до-
пустимая нагрузка, прочность конструкции и нечувствительность
к изменениями температуры окружающей среды. Кроме того, как
отмечено в работе [2 ], весы должны иметь достаточно широкий диа-
пазон измерений, обладать высокой электронной и механической
стабильностью, малой инерционностью, относительной нечувстви-
тельностью к вибрации и, наконец, иметь достаточно простую кон-
струкцию, позволяющую свести к минимуму стоимость весов и по-
требность в ремонте. Весы должны быть просты в обращении и
приспособлены для работы в различных условиях.
• • Возвратная
сит
Механическое
или электрическое
Шействие
Изменение
массы
Фиг. 3.2. Весы с уравновешивающимся балансиром [2].
Регистрирующие весы можно подразделить на два основных
типа: с неуравновешивающимся балансиром и с уравновешиваю-
щимся балансиром (с нулевой точкой).
Принцип действия автоматических весов с уравновешивающим-
ся балансиром показан на фиг. 3.2. В состав весов входит датчик,
обнаруживающий отклонения балансира весов от нулевого поло-
жения — горизонтального для рычажных весов и вертикального
для весов с электромагнитной подвеской. На балансир при этом
действует возвратная сила от электрической или механической на-
грузки, прикладываемой через соответствующие электронные или
механические связи, которая возвращает весы в нулевое положе-
ние. Возвратная сила, пропорциональная изменению массы, реги-
стрируется непосредственно или с помощью электромеханического
датчика.
Различные типы весов с неуравновешивающимся балансиром
показаны на фиг. 3.3. В этом случае отклонения балансира весов
относительно центра вращения преобразуются в соответствующие
кривые изменения массы с помощью: фотографической регистра-
ции отклонений, записи электрических сигналов от датчика смеще-
ния .и электромеханического устройства. Существуют следующие
типы отклоняющихся балансиров:
74
Глава 3
Фиг. 3.3. Весы с неуравновешивающимся балансиром [2].
а — консоль; б—спиральная пружина; в — коромысло; г — крутильная нить.
1) спиральная пружина; изменение массы вызывает сжатие или
растяжение пружины, регистрируемое с помощью соответствующих
датчиков;
2) консольный балансир, один конец которого закреплен, а
к другому, свободному концу подвешивается образец;
3) тензометр, к которому подвешивается образец; тензометр
закреплен таким образом, чтобы его сжатие и растяжение были
пропорциональны изменению массы;
4) балансир, прикрепленный к туго натянутой проволоке, слу-
жащей осью вращения; один или оба конца проволоки жестко за-
креплены; отклонения балансира пропорциональны изменениям мас-
сы образца и крутильным характеристикам проволоки.
Различные методы регистрации отклонения балансира от верти-
кального или горизонтального положения в весах с уравновешива-
ющимся балансиром описаны в работе Гордона и Кэмпбелла [2].
1) Оптические методы:
а) источник света — зеркало — фотобумага;
б) источник света — затвор — фотоэлемент.
2) Электронные методы:
а) емкостный мост;
Термовесы и вспомогательное оборудование к ним 75
1^6) взаимная индуктивность: катушка — пластина, катушка —
катушка;
в) дифференциальный трансформатор или магнитный датчик
с переменной проводимостью;
г) приемник радиации (счетчик Гейгера);
д) тензометр.
3) Механический метод: перо, электромеханически соединенное
с балансиром весов или с кулонометром.
В весах с уравновешивающимся балансиром используются сле-
дующие методы возвращения балансира в нулевое положение [2 ].
1) Механические методы:
а) снятие и добавление разновесов или перемещение гусарика
весов;
б) ступенчатое или постоянное приложение усилия от спираль-
ной или скручивающей пружины;
в) ступенчатое или непрерывное действие цепной передачи;
г) ступенчатое добавление или слив жидкости (разная выталки-
вающая сила);
д) ступенчатое увеличение или уменьшение гидравлического
давления,
2) Методы на основе электромагнитных взаимодействий:
а) катушка — сердечник;
б) катушка — магнит;
в) катушка — катушка.
3) Электрохимический метод: кулонометрическое растворение
или осаждение металла на электрод, подвешенный к балансиру
весов или кулонметру.
Существуют следующие методы записи результатов измерения
изменения веса [2J:
1) Механические методы:
а) перо, соединенное с движком потенциометра;
б) перо, соединенное с барабаном с возвратной цепью;
в) перо или электродуговой указатель, закрепленный на конце
балансира;
г) перо (перья), присоединенное к фотоэлектрическому следя-
щему механизму с сервоприводом, фиксирующему прогиб баланси-
ра.
2) Фотографический метод; Источник света — зеркало — фото-
бумага, либо сматываемая с барабана (с разметкой по времени),
либо расположенная в одной плоскости; разметка по темпе-
ратуре — с помощью зеркального гальванометра.
3) Электронные методы:
а) ток, вырабатываемый в цепи преобразователя, в качестве ко-
торого могут служить фотоэлемент, дифференциальный трансфор-
матор, тензометр, мост, радиационный детектор (счетчик Гейгера),
конденсатор, индуктор;
76
Глава S
б) ток, проходящий через катушку электромагнита.
В общем случае запись с помощью электронных устройств обес-
печивает большую гибкость и удобнее механических систем, так
как существует большое число датчиков, позволяющих получить
электрический сигнал, пропорциональный изменению массы для
весов как с неуравновешивающимся, так и с уравновешивающимся
Фиг. 3.4. Схема электровесов Кана. 1
/ — самописец; 2 — фильтр; 8— предел регистрации самописца; 4—шкала весов; 5—диа-
пазон измерения; 6 — сервоусилитель; 7 — фотоэлемент; 8 —лампа; 9 — петля В; 10 — ка-
тушка; // —ленточная подвеска; 12— магнит; 13 — петляй; /4—образец; 15—петля А.
балансиром. Аналоговые данные, непрерывно записываемые с пер-
вичной кривой, могут быть одновременно преобразованы в любую
другую удобную форму, например в производные, интегралы, лога-
рифмы и т. п., что необходимо для цифровых операций при приме-
нении компьютера или в автоматических процессах [21.
а. Весы Кана
В конструкциях приблизительно половины всех выпускаемых
промышленностью термовесов используется та или иная разновид-
ность электромагнитных весов Кана. Этот тип весов имеет высокие
чувствительность, точность и надежность, а также удобен в обра-
щении. Основной частью весов является гальванометр Дарсонваля.
К указательной игле (коромыслу) подвешивается образец. Измене-
Термовесы и вспомогательное оборудование к ним
77
ния массы образца вызывают отклонение коромысла, что приводит
к изменению тока фотоэлемента, который усиливается и подается
на катушку, прикрепленную к коромыслу. Так как катушка на-
ходится в магнитном поле, ток, проходящий через нее, вызывает
момент силы, действующий на коромысло весов и возвращающий
его в нулевое положение. Таким образом, ток катушки является
точной мерой массы образца. Принципиальная схема весов пред-
ставлена на фиг. 3.4. Весы могут быть размещены в стеклянном
корпусе, выдерживающем разрежения до 1,3-10^4 Па. Существу-
ют различные модели весов, отличающиеся чувствительностью,
пределами измерений и другими показателями.
2. ДЕРЖАТЕЛИ ОБРАЗЦОВ
Одним из важнейших узлов термовесов является держатель
образца. Его форма, размеры и материал, из которого он изго-
товлен, оказывают значительное влияние на кривую изменения
массы. В литературе описано большое число различных держателей
образцов. Наиболее характерные типы показаны на фиг. 3.5. Как
и в случае выбора держателей образцов при использовании метода
.ДТА (гл. 6), тип держателя, используемого в данном случае, зави-
сит от размера и состояния образца, а также максимальной темпера-
туры измерений. Держатели могут быть изготовлены из стекла,
кварца, алюминия, окиси алюминия, нержавеющей стали, плати-
ны и многих других материалов. Образцы могут иметь массу от 1 мг
до нескольких сотен граммов, но чаще всего масса образца состав-
ляет 5—100 мг.
В электровесах Кана широко применяется держатель образца
типа показанного на фиг. 3.5, а. Этот тип держателя изготавлива-
ется из платины или кварца и имеет форму полусферы диаметром
5—9 мм. Он используется для исследования образцов весом
1—20 мг. Держатель образца, показанный на фиг. 3.5, б, применя-
ется в горизонтальных термовесах, подобных выпускаемых фирма-
ми «Линзейс» и «Нетш». Такие держатели имеют различные разме-
ры и изготавливаются из самых разнообразных материалов. В тер-
мовесах фирмы «Меттлер» (см. [3 ]) используется много типов держа-
телей образца, подобных показанным на фиг. 3.5, в—ж. Держатель
типа показанного на фиг. 3.5, в представляет собой открытый ти-
гель емкостью 0,1—5 мл, изготовленный из платинородиевого
•сплава, окиси алюминия, кварца или графита. Держатель в виде
плоской тарелочки (фиг. 3.5, г) применяется для исследований в вы-
соком вакууме. Для предотвращения растрескивания в процессе
разложения применяются тонкослойные образцы. Многотарель-
чатый держатель (фиг. 3.5, д) позволяет увеличить удельную по-
верхность образца. Аналогичные держатели, описанные в работах
Эрди и др. [4, 5], использовались в дериватографе; каждый держа-
Глава 3
д
Фиг. 3.5. Основные
10 — 20 тарелочек, размещенных на расстоянии
типы держателей образцов для термогравиметрических
исследований.
тель состоит из
12 мм, и позволяет исследовать образцы масой 0,2—1 г. Образцы,
1при нагревании которых происходит бурное выделение газа, удобно
исследовать в держателе типа представленного на фиг. 3.5, е.
Для исследования образцов в виде полосок, например металличе-
ской фольги, применяется держатель типа показанного на фиг.
Д.5, ж. Для одновременных ТГ- и ДТА-измерений используются
держатели образца, описанные в гл. 6 (фиг. 6.2).
3’
Термовесы и вспомогательное оборудование к ним
79
Фиг. 3.6. Держатели образцов для измерений в условиях
паровой фазы под давлением [6].
образования
Для измерений в условиях образования паровой фазы под дав-
лением можно применять держатели, представленные на фиг. 3.6.
На фиг. 3.6, а схематически показано поперечное сечение держа-
теля, представляющего собой ячейку Кнудсена, описанную
Видеманном и Богэном [6]. Оболочка ячейки 1, изготовленная из
алюминия, устанавливается на термовесах с помощью керамической
трубки 7. Медь-константановая термопара 5, введенная в ячейку
через герметичное соединение, используется для измерения темпе-
ратуры образца 6 или паровой фазы. Вторая термопара 4 служит
для контроля температуры печи. Диафрагма 2 выполнена из нихро-
мовой фольги толщиной 0,01 мм и имеет небольшое отверстие. Разъ-
емное (резьбовое) соединение позволяет производить быстрое уда-
ление или замену диафрагм с отверстиями разного диаметра
(1—3 мм). Пластина 3 на другом конце ячейки закрывает отверстие,
используемое для загрузки образца. Герметизация мест соединения
концевых пластин с ячейкой осуществляется с помощью тефлоно-
вых кольцевых прокладок. Ячейку можно использовать для иссле-
дования образцов объемом не более 1,5 мл при температурах от
—100 до 200°С. Упрощенный вариант держателя со съемной крыш-
кой для аналогичных измерений в условиях образования паро-
вой фазы под давлением представлен на фиг. 3.6, б.
Фиг. 3.7. Держатели образцов для исследований в собственной атмосфере
[7-10].
— крышка, тип 2; 2— крышка, тип 1; 3— тигель; 4— образец; 5 — керамиче кий стер-
жень; 6 — термоэлемент.
Для исследований, связанных с реакциями диссоциации в собст-
венной («самогенерируемой») атмосфере [7 ], можно применять дер-
жатели образцов, изображенные на фиг. 3.7. В конструкции, пред-
ставленной на фиг. 3.7, а, зазор между поршнем и цилиндром обес-
печивает длинный диффузионный путь и эффективно предотвращает
, попадание загрязнений из атмосферы. В тигле Форкеля [8]
(фиг. 3.7, б) для надежного разделения атмосферы образца и печи
предусмотрена крышка с шаровым клапаном. Другие типы стек-
। лянных и кварцевых держателей образцов рассмотрены Ньюкирком
[9 ].Описанный Пауликом Ф. и Пауликом Дж. [10] держатель
типа представленного на фиг. 3.7, в является, вероятно, наиболее
отработанной конструкцией. Тигель, в котором находится образец,
закрывается шестью хорошо подогнанными крышками, так что
газообразным продуктам разложения приходится проходить через
, длинный и узкий лабиринт.
Термовесы и вспомогательное оборудование к ним
81
Фиг. 3.8. Вспомогательное оборудование для загрузки образцов [3].
а—вибратор cd шпателем для загрузки небольших образцов; б — тигельный вибратор для
равномерного уплотнения образцов; в — пресс для уплотнения образцов с малой плотностью
в держателе.
Оборудование для обеспечения равномерного уплотнения об-
разца показано на фиг. 3.8 [3]. Для загрузки небольших образцов
в держатель используется вибратор со шпателем (фиг. 3.8, а).
Равномерное уплотнение образца обеспечивается с помощью виб-
ратора и держателя (фиг. 3.8, б). Для уплотнения в держателе
объемистых образцов с малой плотностью применяется небольшой
пресс (фиг. 3.8, в).
Загрузку образцов, чувствительных к влаге и подверженных
окислению, в держатель термовесов фирмы «Меттлер» удобно про-
изводить с помощью колпака с регулируемой атмосферой
(фиг. 3.9) [3]. Образец вводят в закрытом держателе под колпак
4—230
82
Глава 3
Фиг. 3.9. Колпак для загруз-
ки образцов при исследовани-
ях в регулируемой атмосфере
Фиг. 3.10. Непосредственное
визуальное наблюдение за об-
разцом во время нагревания его
в печи [11].
и после создания соответствующей атмосферы подают в камеру
печи. По окончании загрузки камеру печи закрывают, а колпак сни-
мают.
На фиг. 3.10 [11 ] изображена схема устройства для непосредст-
венного визуального наблюдения за образцом в процессе его на-
гревания. Для предотвращения потерь тепла из печи и возникно-
вения градиентов температуры отверстие для наблюдения закры-
то кварцевой пластиной.
3. ПЕЧИ И УСТРОЙСТВА ДЛЯ РЕГУЛИРОВАНИЯ ИХ
ТЕМПЕРАТУРЫ ПО ЗАДАННОЙ ПРОГРАММЕ
При термогравиметрических измерениях применяют печи, по-
добные описанным в гл. 6, разд. А.4. Выбор нагревательного эле-
мента и типа печи зависит от требуемого интервала температур.
В литературе описаны печи, работающие в различных интервалах
температур — от комнатной температуры до 1000, 1600 и даже
Термовесы и вспомогательное оборудование к ним
83
2400° С. Печь может иметь горизонтальное или вертикальное рас-
положение и размещаться над механизмом весов или под ним. Разме-
щение печи над весами предпочтительней в силу ряда факторов,
главным образом из-за наличия термических градиентов в камере
печи и их влияния на температуру весов. Наибольшее распростра-
нение имеют печи с электронагревательными элементами, пределы
рабочих температур которых приведены в табл. 6.4. Только для
очень немногих термогравиметрических исследований требуются
низкотемпературные печи, хотя при измерении низкотемператур-
ной термогравиметрической чувствительности магнетиков может
быть использован криостат.
Требования тепловой симметрии печи, вероятно, менее жесткие,
чем при измерениях методом ДТА. Желательно иметь кривые рас-
пределения температур в печи, подобные показанным на фиг. 6.6.
В области расположения держателя образца должна существовать
зона с постоянной температурой.
Повышение температуры в печи регулируется программирую-
щим устройством, к которому предъявляются такие же требования,
как и к регуляторам температуры, используемым в дифферен-
циальном термическом анализе (гл. 6). Скорость нагревания долж-
на быть неизменной во время опыта и хорошо воспроизводимой, так
как нелинейное нагревание влияет на характер получаемой термо-
гравиметрической кривой, особенно если используется система ре-
гистрации по времени. Однако требования линейности изменения
температуры в данном методе все же не столь строги, как в методе
ДТА. Обычно применяется скорость нагревания 5—10 град/мин,
хотя в отдельных случаях она достигает 160 град/мин. Скорость
нагревания задается в зависимости от размера образца и характера
требуемой информации.
4. ОПРЕДЕЛЕНИЕ ТЕМПЕРАТУРЫ
И СИСТЕМЫ РЕГИСТРАЦИИ ДАННЫХ
Как отмечалось выше, в термогравиметрии изменение массы
образца записывается непрерывно в функции температуры, которая
может быть температурой камеры печи, температурой вблизи об-
разца (т. е. непосредственно у стенки контейнера с образцом) или
температурой самого образца. На фиг. 3.11 показаны эти способы
определения температуры [12]. На фиг. 3.11, а термопара разме-
щена возле контейнера с образцом, но не находится в контакте с
ним. В этом случае, конечно, существует взаимосвязь между темпе-
ратурой контейнера и температурой, определяемой термопарой,
однако температура термопары всегда ниже или выше температуры
образца в зависимости от термохимических условий реакции.
В большинстве термовесов используется этот способ определения
температуры, хотя он и не является лучшим. Особенно плохие
4*
84
Глава 3
результаты он дает при исследовании в условиях низких давле-
ний, например в глубоком вакууме; в этом случае теплопередача
происходит в основном только за счет излучения, а не конвекции
и теплопроводности.
Фиг. 3.11. Способы размещения в термовесах термопары для определения
температуры [1211
На фиг. 3.11, б термопара размещается в непосредственной
близости от образца внутри его держателя, но не имеет контакта
с ним. Этот способ определения температуры лучше предыдущего,
так как термопара в данном случае реагирует на малейшие изме-
нения температуры образца. Наиболее точные результаты дает
размещение термопары в непосредственном контакте с образцом
или с держателем образца (фиг. 3.11, в). Однако основная проб-
лема заключается в том, что провода термопары могут вызвать
ошибки при взвешивании на чувствительных регистрирующих весах
или создать помехи работе механизма весов. Единственным спосо-
бом определения фактической температуры образца без создания
помех в механизме весов является использование электронного
устройства, подвешенного около держателя образца. Это устройст-
во передает температуру образца приемнику, закрепленному около
контейнера образца. Менч и Кэррол [131 описали транзисторный
релаксационный генератор с одноконтактным транзистором и тер-
мистором для определения температуры. В этом устройстве часто-
та колебаний, являющаяся функцией температуры образца, пе-
редавалась посредством взаимной индукции двух подвесных
катушек приемнику и счетчику. Однако применение данного уст-
ройства ограничено максимальной температурой 150°С.
Вопросы калибровки печи и (или) камеры образца рассматрива-
лись Стюартом [12], а также Норемом и др. [14, 15]. Стюарт [12]
использовал обычные термовесы, оснащенные термопарой, смонти-
рованной с внешней стороны образца, а Норем и др. [14, 15] про-
Термовесы и вспомогательное оборудование к ним
85
изводили калибровку печи с использованием сопротивления для
определения температуры.
Стюарт [12 J рассмотрел три способа калибровки по температуре:
1) использование стандартных материалов с воспроизводимыми
точками уменьшения массы, соответствующими известным темпе-
ратурам;
2) использование материалов с известными воспроизводимыми
(и обратимыми) значениями температуры перехода и непосредствен-
ное измерение температуры;
3) использование материалов с магнитными переходами, кото-
рые находят отражение на кривой уменьшения массы и могут быть
связаны с определенной температурой.
Первый способ наиболее привлекателен, но изменение массы лег-
ко испаряющихся веществ зависит не только от температуры и ско-
рости ее изменения, но также от типа и состава атмосферы в печи.
Второй способ был использован Стюартом [12] в его схеме кали-
бровки по температуре, но этот метод требует, чтобы в процессе
калибровки термопара находилась в контакте с образцом или с кон-
тейнером образца. В качестве эталонных соединений были выбраны
соединения с переходами типа Твердое тело I Твердое тело II
или Твердое тело Жидкость, не зависящими от атмосферы. Та-
кими соединениями являются нитрат калия с температурами пере-
хода 129,5 и 333° С, хромат калия (665° С) и олово (231,9° С).
Норем и др. [14, 15] применили третий способ калибровки по
температуре печи и (или) держателя образца. Держатель образца
с помещенным в него ферромагнитным материалом был подвешен
в магнитном поле. По достижении температуры, соответствующей
точке Кюри материала, он перестает ощущать влияние магнитного
поля (его «эквивалентная магнитная масса» уменьшается до нуля),
и термовесы отмечают кажущуюся потерю массы. Очевидно, что для
калибровки в интервале температур от комнатной до 1000° С дол-
жен быть использован ряд ферромагнитных материалов. Идеаль-
ный эталонный материал должен отвечать следующим требованиям
[15]:
1) переход должен быть резким, т. е. происходить в небольшом
интервале температур;
2) переход должен совершаться при небольших затратах энер-
гии (в. условиях динамического сканирования в термогравиметрии
«острота» перехода обратно пропорциональна энергии перехода);
3) температура перехода не должна зависеть от состава атмосфе-
ры и давления;
4) переход должен быть обратимым, чтобы можно было повто-
рить измерения с целью выбора оптимальных условий или провер-
ки калибровки;
5) на переход не должно оказывать влияние присутствие других
эталонных материалов, что обеспечивает возможность одновремен-
86
Глава 3
кого исследования нескольких образцов и получения многоточеч-
ной калибровки в одном эксперименте;
6) переход должен легко наблюдаться при использовании эта-
лонных образцов массой в несколько миллиграммов, сопоставимой
с массой образцов, исследуемых на данном приборе.
Фиг. 3.12. Калибровка по температуре с помощью ферромагнитных этало-
нов [151.
Скорость нагревания 10 град/мин.
Пример типичной калибровочной кривой, полученной с исполь-
зованием четырех эталонных ферромагнитных образцов, приведен
на фиг. 3.12. Фактические температуры перехода указаны в скоб-
ках. Точки перехода эталонных образцов следует оценивать в ре-
жиме нагревания, так как при их охлаждении наблюдается гисте-
резис. Температуры перехода также в небольшой степени зависят
от скорости нагревания печи, но эта зависимость в интервале ско-
ростей нагревания 5—20 град/мин пренебрежимо мала. Влияние
скорости нагревания на наблюдаемую температуру начала перехода
определяется по формуле
Тнабл = Тизотерм A-RCsTn, (3.1)
Термовесы и вспомогательное оборудование к ним
87
где R — эффективное термическое сопротивление между источником
тепла и контейнером образца; Cs — эффективная теплоемкость об-
разца и контейнера; Тп — скорость нагревания. Параметр RCS
представляет собой постоянную времени прибора — важнейший
параметр, характеризующий любой прибор, в том числе и прибор
для термического анализа.
В термовесах обычно имеются два типа записывающих систем:
потенциометрический ленточный самописец с регистрацией по вре-
мени и двухкоординатный самописец. В нескольких типах выпус-
каемых промышленностью термовесов все еще используются само-
пишущие приборы, работающие по принципу «гальванометр —
световой луч — фотобумага». При использовании ленточного само-
писца, кроме кривой изменения массы, регистрируется температура
системы. В этом случае применяются либо два самописца, либо
один самописец с двумя или более перьями, либо многоточечный
самописец. Преимуществом системы этого типа является возмож-
ность наблюдения за скоростью нагревания печи и контроля ли-
нейности нагревания.
Влияние нарушений температурного режима на ход кривых
уменьшения массы при использовании обоих типов регистрирую-
щих систем показано на фиг. 3.13. На фиг. 3.13, а приведена кри-
вая, полученная на ленточном самописце с регистрацией по вре-
мени. Нарушение линейного роста температуры в точке А привело
к записи, показанной сплошной линией. Нормальная кривая, полу-
ченная при постоянной скорости нагревания, обозначена пунктир-
ной линией. Аналогично изменяется форма кривой, полученной
на двухкоординатном самописце (фиг. 3.13,6): при искажении
температурного режима кривая обозначена пунктирной линией,
а нормальная кривая — сплошной. Однако при использовании лен-
точного самописца ведется также запись температуры, что позво-
ляет сразу же отметить изменение в скорости нагревания. В случае
применения двухкоординатного самописца изменение скорости
нагревания может быть не обнаружено до тех пор, пока кривая не
будет записана несколько раз.
Большинство выпускаемых промышленностью термовесов ре-
гистрирует абсолютное, а не относительное (в процентах) изменение
массы образца; последняя форма записи результатов более удобна.
При прямой записи изменения массы для расчета ее процентного
изменения и (или) других стехиометрических расчетов необходимо
знать исходную массу образца. При записи процентного изменения
массы в эксперименте получают данные в удобном виде для различ-
ных стехиометрических расчетов. Уэндландт [72] отметил, что при
соответствующем выборе ленточного самописца можно непосред-
ственно регистрировать процентное изменение массы. Самописец
должен иметь приспособление с переменным зазором (такое, как,
например, имеет самописец Саржента, модель SRG), позволяющее
88
Глава 3
Фиг. 3.13. Влияние нарушений температурного режима на кривые умень-
шения массы.
а — ленточный самописец с регистрацией по времени (скорость нагревания падает в точке А);
б — двухкоординатный самописец.
установить перо в крайнем левом положении ленты, что соответствует
0% изменения массы. Затем любое изменение массы образца будет
регистрироваться в процентах, и нет необходимости знать точную
массу образца.
Другие характеристики регистрирующих систем и требования
к ним такие же, как и в случае самописцев, используемых в диф-
ференциальном термическом анализе (гл. 6).
Термовесы и вспомогательное оборудование к ним
89
Б. ТИПЫ ТЕРМО ВЕСОВ
1. ВВЕДЕНИЕ
Различные типы термовесов как серийных, так и лабораторных
образцов были описаны в целом ряде обзоров и книг. Следует
упомянуть обзорные работы Гордона и Кэмпбелла [2], Дюваля
Фиг. 3.14. Термовесы Хонда [30].
[16], Левина [17], Жака и др. [18], Сэйто [19], Вогэна [20], Уэнд-
ландта [21, 22] и др. Описания термовесов имеются в книгах Дюва-
ля [23, 24], Гарна [25], Китча [26], Андерсона [27], Уэндландта
[28], Сэйто [29] и др. Из-за ограниченного объема в данной работе
рассматриваются только термовесы, имеющие оригинальные конст-
руктивные элементы или существенные особенности. В этой книге
упоминаются только некоторые из множества промышленных термо-
весов, а подробное описание можно найти в недавно опубликован-
ных обзорах и книгах [21, 22, 28].
По-видимому, первыми термовесами следует считать прибор,
описанный Хонда [30] в 1915 г. Этот прибор (фиг. 3.14) содержал
весы с кварцевым балансиром. Образец был размещен в тигле из
фарфора или магнезии /, подвешенном внутри печи с электро-
нагревателем 2. На противоположном конце балансира была закреп-
лена спираль из тонкой стальной проволоки 3. Спираль была по-
гружена в сосуд Дьюара 4, заполненный маслом. Сборка спираль —
сосуд Дьюара с помощью винтового механизма регулируется та-
ким образом, чтобы поддерживать балансир весов в нулевом поло-
90
Глава 3
жении. Скорости нагревания, применяемые в этих весах, довольно
низкие: для достижения температуры 1000° С необходимо затратить
10—14 ч. Однако Хонда использовал квазиизотермический цикл
нагревания; во время перехода, сопровождающегося уменьшением
массы, температура в печи до завершения перехода поддержива-
лась постоянной. Только на одну эту процедуру иногда затрачива-
лось 1—4 ч. Как можно было ожидать, исходя из взаимного поло-
жения печи и образца, при температуре более 300° С появлялись
конвективные потоки. В исследованиях, проводимых на этих термо-
весах, применялся образец масой ~0,6 г.
Работа Хонда явилась фундаментом практически всех будущих
работ по термогравиметрии. Его термовесы позволяли исследова-
телю непрерывно взвешивать образец в процессе нагревания, а так-
же использовать преимущества квазиизотермического нагревания.
Последнее не удается выполнить автоматически даже на современ-
ных термовесах. Хонда скромно заключил свою работу словами:
«Все приведенные выше результаты не являются полностью ори-
гинальными, однако настоящее исследование, проведенное с по-
мощью термовесов, позволяет точно определить изменения струк-
туры и скорость этих изменений при соответствующих температу-
рах. Оно также обнаруживает большие преимущества применения
термовесов описанного типа для подобных исследований в химии».
Многие другие японские исследователи создали ряд модифика-
ций термовесов Хонда, а также разработали новые приборы. Ре-
зультаты их работ обобщены в монографии Сэйто [19].
Французская школа термогравиметрии была основана Гишаром
[31 ] в 1923 г. Очевидно, Гишар не был знаком с работой Хонда,
но впоследствии никогда и не претендовал на роль основополож-
ника термогравиметрии. Гишар совершенствовал методику термо-
гравиметрии, критически исследуя каждую фазу измерений, и до-
вел ее до высокой степени совершенства. Его первые термовесы
[23 ] были снабжены газовой печью, в которой дозированная подача
газа к горелке производилась с помощью устройства постоянного
уровня, состоящего из клапана, соединенного с поплавком. Попла-
вок находится в баке на поверхности воды.Вода через дозирующий
клапан перетекала в другой бак, вызывая медленное, но равномер-
ное открытие клапана подачи газа и, следовательно, постепенное
повышение температуры печи. Интересен также примененный в этих
термовесах способ измерения потери массы. Для определения по-
тери массы использовалось гидростатическое устройство, в котором
потери массы образца компенсировались добавлением небольших
количеств масла в V-образную трубку. Уменьшение массы на
100 мг соответствовало добавке 9 мл масла. Кривые потери массы,
полученные на этих весах, хорошо совпадали с результатами
Дюваля [23], полученными 40 лет спустя.
Последователями Гишара были Валле [32], Дюбуа [33] и др у-
Термовесы и вспомогательное оборудование к ним
91
гие [23]. Создание Шевенаром [34] самопишущих термовесов, по-
видимому, послужило толчком к стремительному развитию фран-
цузской школы термогравиметрии. Эти весы разрабатывались
начиная с 1936 г.; серийное их производство было налажено в
в 1945 г. Они являлись первым автоматическим фотографическим
самопишущим термогравиметрическим прибором. Благодаря Дю-
валю [23, 24 ] и другим исследователям весы Шевенара стали стан-
дартным инструментом для исследований в области термограви-
метрии. Эти термовесы будут описаны в следующем разделе.
Важными вехами развития современных термовесов оказались
также 1958 и 1964 гг., когда были опубликованы работы Пауликов
и Видеманна. Паулик Ф. и др. [351 создали многофункциональный
прибор, названный дериватографом. С помощью этого прибора,
помимо термогравиметрических кривых, можно получать кривые
их первых производных (ТГП) и дифференциального термического
анализа (ДТА). Видеманн [3] описал систему фирмы «Меттлер»,
представляющую собой по-видимому, наиболее сложные из имею-
щихся в настоящее время термовесов.
2. ТЕРМОВЕСЫ ШЕВЕНАРА
С помощью автоматических термовесов Шевенара было прове-
дено, вероятно, больше исследований, чем с помощью всех других
типов весов вместе взятых. Весы впервые были описаны Шевенаром
и др. [34] в 1944 г. и весьма успешно использованы ими для изу-
чения коррозии металлов при повышенных температурах. За по-
следние годы было разработано несколько модификаций этих термо-
весов; имеющаяся в настоящее время в продаже модель ТН-59 пред-
назначена для исследований в глубоком вакууме при температурах
до 1500° С. Модель оснащена устройством для автоматического
построения кривых масса — время или масса — температура.
Автоматическая запись производится пером, управляемым систе-
мой с фотоэлементом.
Принципиальная схема весов Шевенара не подвергалась значи-
тельным изменениям. Это весы с неуравновешивающимся балан-
сиром, подвешенным на проволоке. Отклонения балансира могут
быть зарегистрированы с помощью фотоаппарата, как в первой кон-
струкции весов, или с помощью фотодатчика. Принципиальная схе-
ма весов представлена нафиг. 3.15. Образец размещается на конце
стержня, подвешенного на одном из плеч чувствительных весов;
подвижный противовес на противоположном плече весов служит
для их настройки перед началом работы. Вредные вибрации устра-
няются магнитным демпфированием. Стержень и держатель об-
разца вводятся в печь с бифилярной обмоткой, смонтированную
над весами. Записывающий блок состоит из фотосопротивления,
установленного на подвижной тележке, и вращающегося барабана.
92
Глава 3
Фиг. 3.15. Термовесы Шевенара [34].
Луч света, отраженный от зеркала, закрепленного на балансире
весов, падает на фотоэлемент. Любое перемещение луча, вызванное
изменением массы образца, приводит к перемещению автоматиче-
ского следящего блока фотоэлемент — серводвигатель, соединен-
ного с пером, записывающим результаты на барабане. Перо соеди-
нено с фотоэлементом с помощью приспособления для проведения
тонких линий, имеющего регулируемый зазор, благодаря которому
перо проводит тонкую линию, соответствующую вертикальному
перемещению светового пятна, и не реагирует на вибрацию фото-
элемента. Скорость вращения барабана является функцией времени
или температуры. Максимальный диапазон измерений весов 50 г.
При меньших диапазонах измерений можно обеспечить чувстви-
тельность весов 1 мг на 5 мм ленты.
В литературе описано множество модификаций термовесов Ше-
венара. Гордон и Кэмпбелл (361 вместо фотографического само-
Термовесы и вспомогательное оборудование к ним
93
пишущего устройства использовали электронный самописец, в ко-
тором для превращения перемещения балансира весов в линейные
электрические сигналы применялся линейный дифференциальный
трансформатор с переменным коэффициентом трансформации. Пос-
ле демодуляции сигнал записывается на ленте регистрирующего
потенциометра. Диапазон измерений на этих весах - составляет
~400 мг при точности ±0,25% в пределах 200 мг. Время срабаты-
вания, соответствующее изменению массы 200 мг, равно ~2 с.
Система отметки температуры на самописце описана Гриффитом
[37]. Остальные изменения в этих весах относятся в основном к дер-
жателю образца и кожуху печи, усовершенствованному с целью
обеспечения лучшего регулирования атмосферы в ней.
3. ТЕРМОВЕСЫ СО СПИРАЛЬНОЙ ПРУЖИНОЙ
Имеется много различных типов самопишущих термовесов со
спиральной пружиной [38—53]. Все эти термовесы представляют
собой модификации оригинальной конструкции, предложенной
Макбэйном и Бакром [54] в 1926 г. Схема типичных термовесов
со спиральной пружиной показана на фиг. 3.16. Этот прибор, на-
зываемый Термо-Грав, состоит из весов с неуравновешивающимся
балансиром, в которых растяжение пружины измеряется с помощью
линейного дифференциального трансформатора с переменным коэф-
фициентом трансформации. Спиральная пружина размещена в
стеклянном корпусе и может работать в вакууме или в регулируемой
газовой атмосфере при атмосферном или пониженном давлении.
Деформация пружины, пропорциональная изменению массы образ-
ца, преобразуется в электрические сигналы при движении сердеч-
ника линейного трансформатора. Эти сигналы после прохождения
через усилитель и демодулятор регистрируются в виде изменения
массы по оси Y двухкоординатного самописца. По оси X записы-
вается входной сигнал, соответствующий температуре печи или
времени опыта. Система подвески состоит из чаши калибратора ве-
са, масляного демпфера и расположенных в нижней ее части колец
из плавленого кварца, служащих опорой для образца и тигля.
Весы оборудованы двумя печами для проведения последователь-
ных измерений. На этих термовесах можно измерять изменение мас-
сы от 0 до 200 мг с точностью ±1% всей шкалы при весе образца
до 10 г. Скорость нагревания печи составляет 5—100 град/ч, а ин-
тервалы по оси X регулируются из расчета на приращение темпера-
туры 200, 500 или 1000° С.
Удачная модификация рассматриваемых термовесов описана
Олсоном [50, 51], а также Фармером [45]. В описанной конструк-
ции применен самописец с осями X—У1—¥2; по оси У2 наносятся
отметки времени синхронным генератором с дистанционным управ-
лением. Кроме того, изменена конструкция печи: применяется не-
94
Глава 3
большая печь сферической формы с рабочей температурой до 1400°С.
Фрэнк и Xармелин [53] изменили конструкцию весов со спи-
ральной пружиной таким образом, что они могут быть использованы
Фиг. 3.16* Термовесы со спиральной пружиной (Термо-Грав).
1 — пружина; 2 — сердечник трансформатора; 3 — катушка трансформатора; 4— калибратор
веса; 5 — демпфер; 6— камера для образца; 7 — тигель; 8— термопара образца; 9—печь;
10 — термопара блока регулирования темпера туры.
для одновременного проведения термогравиметрических исследо-
ваний и дифференциального термического анализа. В модифици-
рованной конструкции возле камеры печи размещалась микро-
ячейка ДТА, причем держатель образца для ДТА располагался
непосредственно под держателем образца термовесов. Для измере-
ний на этих термовесах требуются два образца (для ТГ и для ДТА).
4. ТЕРМОВЕСЫ ФИРМЫ «ДЮПОН»
Термовесы фирмы «Дюпон» описаны Сарасоном и. Тейблингом
[55]. Весы (фиг. 3.17) имеют сбалансированный на нуле (как в
электровесах Кана, модель RG) электрический измеритель с туго
натянутой лентой и следящий контур с оптическим управлением.
Образец помещается в контейнер, подвешенный непосредствен-
но на балансире весов. При нормальных условиях работы термопара
для измерения температуры устанавливается на расстоянии 1 мм
96
Глава 3
от образца, и, следовательно, измеренная температура очень близ-
ка к температуре образца. Изменение массы образца в зависимости
от температуры записывается на внутреннем двухкоординатном
самописце. Чувствтельность весов составляет 2 мкг. Горизонталь-
ное расположение балансира весов в печи и контейнера образца
легко позволяет пропускать через трубку печи различные инертные
газы. Поперечное сечение образца в направлении потока газа мини-
мальное, вследствие чего крутящий момент, перпендикулярный ба-
лансиру весов, пренебрежимо мал, и поток газа не вносит сущест-
венных помех в работу термовесов. Максимально достижимая тем-
пература в печи 1200°С.
Описанная Уильямсом [56] система регулирования атмосферы
в печи рассматриваемых термовесов позволяет производить быст-
рое изменение состава динамической атмосферы (кислород, водород,
аргон).
Камеры высокого давления, в которые иногда помещают
термовесы фирмы «Дюпон», описана Брауном и др. [57], а также
Уильямсом и др. [58]. Первая из них предназначена для давления
до 2,02 МПа и температуры до 350° С, а вторая — для давления
до 6,88 МПа и температуры до 450° С. Конструкция весов Уильям-
са и др. [58] позволяет также производить одновременное измере-
ние магнитной восприимчивости образца (гл. 11). Чиу [59] описал
модификацию термовесов фирмы «Дюпон», на которой можно одно-
временно получить ТГ-, ДТА- и ЭТА-кривые (ЭТА — электротерми-
ческий анализ). Этот прибор описан в гл. 11. Другие варианты при-
боров, например сочетающих термовесы с газовым хроматографом
или масс-спектрометром, описаны в гл. 8.
5. ДЕРИВАТОГРАФ
Дериватограф, впервые описанный Пауликом Ф. и др. [35] в
1958 г., представляет собой многофункциональную систему для
термического анализа, позволяющую на одной ленте получить
ТГ-, ТГП-, ДТА- и Т-кривые образца (Т — температура). С по-
мощью дополнительных приспособлений можно также построить
TP-кривые (термического расширения), кривые производной ТР
[5], а также кривые выделенного газа (гл. 8).
Этот прибор, показанный на фиг. 3.18, включает аналитические
весы, печь, устройство для регулирования температуры печи
по заданной программе, тигли для образца и эталона, регулятор
напряжения и гальванометрический самописец, работающий по
принципу «световой луч — фотобумага». Аналитические весы с
воздушным демпфированием балансира имеют точность 20±0,2 мг
при предельном отклонении, рабочий интервал измерения массы —
от 10 мг до 10 г. ТГП-кривая получается с помощью простого при-
способления, состоящего из магнита и индукционной катушки.
Термовесы и вспомогательное оборудование к ним
97
ч
Магнит подвешивается на одном плече балансира. У обоих полю-
сов магнита размещены индукционные катушки. При изменении
массы движение магнита индуцирует в катушках ток с напряже-
нием, пропорциональным скорости изменения массы. Напряжение
О
Фиг. 3.18. Дериватограф.
1 — тигель для образца; 2— тигель для инертного вещества; 3—фарфоровая трубка;
4 — термопары; 5 — электрическая печь; 6 — нескручивающийся провод; 7 — весы; 8 — ка-
тушка; 9 — магнит; 10 — гальванометр для ТГП; И — гальванометр для измерения темпера-
туры; 12—гальванометр для ДТА; 13 — лампы; 14— оптическая щель; 15— цилиндр для
фоторегистрации; 16 — фотобумага.
измеряется одним из гальванометров со световым лучом и записы-
вается на ленту вместе с другими кривыми. Максимальная темпе-
ратура в печи 1100° С. Печь может работать в атмосфере N2, СО2,
Аг, О2 и других газов, но только при атмосферном давлении.
В литературе описаны различные держатели образцов для дери-
ватографа. Некоторые из них, показанные на фиг. 3.19, описаны
в работе 160], где рассмотрено также их применение для решения
отдельных задач. В гл. 2, разд. Г, был рассмотрен лабиринтный
держатель образца для работы в собственной («самогенерируемой»)
атмосфере.
6. ТЕРМОАНАЛИЗАТОР ФИРМЫ «МЕТТЛЕР»
Термоанализатор фирмы «Меттлер», описанный Видеманном
[3], вероятно, является наиболее совершенным универсальным
и дорогостоящим из всех типов термовесов, выпускаемых промыш-
ленностью в настоящее время. Имеются две модели этого термоана-
98
Глава 3
Фиг. 3.19. Держатели образцов для дериватографии [60].
а — держатель образца для приборов ДТА; б — тигель термовесов для определения произ-
водной; в — однотарельчатый держатель образца термо песо в для определения производной;
dj—тигель дериватографа; д — однотарельчатый держатель дериватографа; е — многота-
рельчатый держатель дериватографа. 1 — образец; 2 — эталон.
лизатора, каждая из которых представляет собой универсальный
прибор для исследований, с помощью которого можно одновремен-
но записать на одной ленте ТГ, ТГП- и ДТА-кривые одного и того
же образца, а также при необходимости давление в камере печи и
скорость потока газа.
Принципиальная схема термоанализатора фирмы «Меттлер» при-
ведена на фиг. 3.20. Весы имеют балансир из алюминия с сапфиро-
выми призмами («ножками») и плоскостями. Изменение массы об-
разца вызывает отклонение балансира весов, который передвигает
заслонку, перекрывающую луч света, падающий на два фотодиода.
Разбаланс в величине тока фотодиодов усиливается и подается на
катушку, укрепленную на балансире весов, возвращая его в нуле-
вое положение. Электрический индикатор массы имеет два диапазо-
на взвешивания обычно с тремя различными чувствительностями
(масштабами диаграммы), четвертая может быть выбрана по тре-
бованию. Запись измерений всегда ведется в двух масштабах с соот-
ношением 1:10. Один диапазон измерений 0—1000 мг с записью
в масштабе 100 мг/дюйм; второй диапазон также 0—1000 мг, но
с записью в масштабе 10 мг/дюйм. При измерениях с более высокой
Термовесы и вспомогательное оборудование к ним
99
Фиг. 3.20. Термоаиализатор фирмы «Меттлер» [3].
чувствительностью (в диапазоне 0—10 мг) масштаб диаграммы из-
меняется таким же образом. Уникальной конструктивной особен-
ностью прибора является система регулирования потока газа. При-
бор может работать в коррозионной и инертной газовых средах при
условии, что предусмотрена защита механизма весов от контакта
с коррозионной средой. Для создания вакуума в печи ~0,7 -10-8 Па
используются форвакуумный и два диффузионных насоса.
Весы оборудованы тремя печами: низкотемпературной печью
с максимальной температурой 1000° С, высокотемпературной печью
с температурой до 1600° С и печью для сверхвысоких температур
с максимальной температурой 2400° С. Схемы этих печей приведены
в гл. 6 (фиг. 6.7). Держатели образцов также описаны в гл. 6,
так как они используются как для дифференциального термического
анализа, так и для термогравиметрии.
Пени для данного прибора могут работать с различными инерт-
ными и окислительными атмосферами, а также с регулируемой
паровой атмосферой. Система регулирования паровой атмосферы
печи показана на фиг. 3.21. Такая печь применяется вместо стандарт-
100
Глава 3
Фиг. 3.21. Печь с паровой атмосферой для системы фирмы «Меттлер».
ной печи фирмы «Меттлер» с кварцевыми трубками для интервала
температур 25—1000° С. Образец помещается в держатель 10, пос-
ле чего блок печи устанавливается на место и герметизируется с
помощью стеклянного шлифа 6. Камера с образцом нагревается на-
гревательным элементом 9 с помощью отражателя 11. Жидкость
(вода или органическое соединение) нагревается до кипения в ем-
кости 1 небольшим съемным нагревателем. Газ-носитель (обычно
азот) подается по трубке 4 и переносит пар в печь через впускное
отверстие 5. Другой поток газа, продуваемый через механизм весов
7, и перегородка используются для предотвращения конденсации пара
на опорном стержне держателя образца или в камере весов. Сосуды
2 и 3 являются подпиточными резервуарами для дополнительной
подачи жидкости в емкость 1. Пар конденсируется в охлаждаемом
Термовесы и вспомогательное оборудование к ним
101
водой стеклянном холодильнике 8, и конденсат возвращается в ем-
кость 1.
Использование термовесов «Меттлер» в сочетании с газовым хро-
матографом или масс-спектрометром рассматривается в гл. 8.
7. ДРУГИЕ ТИПЫ ТЕРМОВЕСОВ
Создание термовесов на базе электровесов Кана было описано
многими исследователями. Термовесы модели RG описаны в
работах [61—63, 67, 72], термовесы модели RM — в работе [64],
а модель RTL — в работах [65, 66].
Подвесная трубка и печь, используемые в термовесах Эттера
и Смита [67 ], показаны на фиг. 3.22. Небольшой платиновый элект-
ронагреватель, размещенный внутри относительно короткой квар-
цевой трубки с образцом, служит печью. Термопара для измерения
температуры расположена внутри камеры печи у ее основания.
Подобная конструкция используется в термовесах фирмы
«Перкин—Элмер», с той лишь разницей, что в последних термопара
не используется.
8. КВАРЦЕВЫЕ ВЕСЫ
Идея использования резонаторов из кристаллов кварца для
определения изменения массы очень привлекательна из-за возмож-
ности достижения чувствительности порядка 10-12 г/см2. Опреде-
ление массы с помощью кварцевых резонаторов обсуждалось в ра-
ботах Планта [68], а также Ван-Эмпелла и др. [69]. В работе [69]
принцип определения массы основан на следующем соотношении
между резонансной частотой f, ее изменением А/ и приращением
массы Ат:
= , (3.2)
/ m
гдеУт — эффективная масса кварца между двумя электродами. Из-
мерения должны проводиться при постоянной температуре, так как
резонансная частота зависит от температуры. При относительно
высоких температурах ее изменение на 1° С вызывает относитель-
ное изменение частоты 2-10'8, что приводит к кажущемуся изме-
нению массы на 8-Ю’9 г. Однако при использовании системы с
двумя электродами, один из которых служит для измерения темпе-
ратуры, а другой для измерения изменения массы, могут быть соз-
даны резонансные кварцевые весы [69].
В другой конструкции кварцевых термовесов [68] использует-
ся кварцевое волокно диаметром 5—20 мкм и длиной около 10 мм
с тонким покрытием из золота, осажденного из паровой фазы. Во-
локно жестко закреплено на одном конце и висит между двумя па-
раллельными металлическими пластинами, к которым приложено
напряжение постоянного тока. Когда на волокно воздействует сиг-
102
-------------------------- Глава 3
Термовесы и вспомогательное оборудование к ним 103
6
S
10
~ 11
16
12
13
14
4
S
6
7
нал звуковой частоты, наблюдаются
резонансные колебания, частота ко-
торых зависит от физических свойств
волокна. Если к свободному • концу
волокна присоединяется дополни-
тельная масса, то резонансные ча-
стоты снижаются в зависимости от
величины этой массы. В работе [681
описаны также другие типы весов, в
которых использованы кварцевые
кристаллы.
17
18
Фиг. 3.22. Микропечь, исполь-
зуемая в термовесах, описан-
ных в работе [67].
— вход газа; 2 — подвесная прово-
лока весов; 3 — кварцевая трубка;
4 — скоба чашки образца; 5 — чашка
образца; 6 — рабочая камера нагре-
вателя из окиси алюминия; 7 — выход
газа; 8 — стандартная соединитель-
ная конусная трубка из кварца; 9—
выводы термопары; 10 — соединитель-
ный элемент из стекла пирекс; 11 —
высокогерметичное соединение пирек-
са с кварцем; 12 — изоляция; 13 —
термопара; 14 — нагреватель с пла-
тиновой бифилярной намоткой; 15 —
изолятор из окиси алюминия с че-
тырьмя отверстиями; 16— стандарт-
ная соединительная конусная трубка
из алюминия; 17 — герметизирующее
уплотнение; 18 — выводы питания.
9. АВТОМАТИЧЕСКИЕ ТЕРМОВЕСЫ
Современные термовесы пред-
ставляют собой автоматический
прибор, записывающий изменение
массы образца в широком диапазоне
температур. Но в них не предусмот-
рено устройства для автоматического
ввода образца в камеру печи или
для последовательного исследования
нескольких образцов. В автомати-
ческих термовесах, описанных в ра-
ботах 165, 70], производятся автома-
тическая замена образцов и регули-
рование температуры по заданной
программе. С помощью этих весов
последовательно могут исследовать-
ся восемь образцов, держателем ко-
торых является вращающийся диск.
Схема блока весов, печи и механизма
замены образцов представлена на
фиг. 3.23, а, а схема печи и форма
держателя образца — на фиг. 3.23, б.
Описываемые термовесы имеют
обычную конструкцию и состоят
из регистрирующих весов с верх-
ним нагружением (весы Кана, мо-
дель RTL), четырехканального мно-
готочечного потенциометрического
самопишущего прибора фирмы «Лидс и Нортрап» (шкала 0—5 мВ),
небольшой трубчатой печи, механизма замены образцов и
автоматического программирующего механизма для регулирова-
ния температуры печи. Отличительной особенностью этих весов яв-
ляется наличие механизма автоматической замены образцов. Заме-
па образцов производится следующим образом. Исследуемые об-
разцы помещаются в небольшие цилидрические платиновые кон-
тейнеры 11 диаметром 5 мм и высотой 2 мм. Восемь таких кон-
тейнеров располагаются в цилиндрических гнездах по периферии
алюминиевого диска 12 толщиной ~6 мм и диаметром 200 мм. Кон-
тейнер с образцом перемещается к отверстию трубчатой печи 10
Фиг. 3.23. Автоматические термовесы [70].
а — весы, печь и механизм замены образца; б — печь и держатель образца.
1 — расходомер газа; 2 — печь; 3 — диск — держатель образцов; 4 — охлаждающий вентиля-
тор; 5 — весы Кана, модель RTL; 6 — платформа весов; 7 — регулировочный винт с приво-
дом; 8—трубка подвода газа; 9— термопары; 10— иамотка и изоляция нагревателя печи;
11 — контейнер образца; 12 — диск-держатель образца; 13 — керамический штырь.
при помощи небольшого электромотора, соединенного с микровы-
ключателем, который срабатывает при совмещении отверстия
в печи с цилиндрическим гнездом в диске. Затем образец поднима-
ется керамическим штырем 13, связанным с балансиром весов. Пере-
мещение весов 5 и их платформы 6 регулируется винтом с приводом
7, расположенным в основании платформы. Применение реверсив-
ного двигателя позволяет поднимать или опускать платформу до
предельных положений, определяемых концевыми микровыключа-
телями. После размещения образца в центральной части печи в нее
подается азот или другой газ и включается механизм регулирования
температуры по заданной программе. По достижении заданной
максимальной температуры печи весы опускаются и контейнер с
образцом возвращается в диск держателя. Затем диск поворачи-
вается, перемещая следующий образец ко входу в печь. Включа-
ется охлаждающий вентилятор 4, понижающий температуру печи
104
Глава 3
Фиг. 3.24. Весы Фергюсана и др. [71] для взвешивания группы образцов,
а — держатель для переноса образца; б — схема весов; 1 — захват; 2 — держатель;
8 — образец.
до заданного нижнего уровня. Затем весь цикл повторяется с но-
вым образцом. Циклы нагревания — охлаждения печи воспроиз-
водятся для всех восьми образцов. Предварительное взвешивание
контейнеров с образцами производится на полумикроаналитиче-
ских весах «Меттлер». Вес контейнера определяется с точностью
±1 мг (вес пустого контейнера ~ 130мг). Масса каждого образца
не должна превышать 10 мг, чтобы перо не выходило за пределы
шкалы самописца. Диапазон измерений 0—10 мг с записью в мас-
штабе 0,4 мг/см. Ширина ленты 25 см, обычная скорость движения
ленты 1,7 или 4,25 мм/мин.
Термовесы и вспомогательное оборудование к ним
105
Очевидным преимуществом автоматических термовесов по срав-
нению с другими существующими конструкциями является возмож-
ность получения кривых изменения массы последовательно для
восьми образцов. Управление прибором полностью автоматизиро-
вано: от начала цикла до его завершения не требуется вмешательства
оператора. Автоматические термовесы должны найти применение
при обычных термогравиметрических исследованиях группы об-
разцов в идентичных термических условиях. Так как система пол-
ностью автоматизирована, к ней легко можно добавить небольшую
цифровую машину для обработки данных или регулирования
(гл. 12).
Несколько другой принцип положен в основу конструкции Фер-
гюсона и др. [71 ]. Описанное ими устройство для взвешивания груп-
пы образцов предназначено для исследования в течение длительного
периода времени. Образцы нагреваются в потоке смеси газов
СО2—СО—О2 в интервале температур 620—770 К- Держатель
образца, прикрепленный к карусельной балке весов, и механизм
карусели показаны на фиг. 3.24. Держатели образца изготавли-
ваются штамповкой из листового никеля и соединяются с верхней
плоской горизонтальной частью точечной сваркой. Взвешивание
образца производится после его снятия захватом с карусельной
балки. При вращении карусели держатели с образцами, лежащие
в V-образных углублениях на концах радиальных балок карусели,
проходят на соответствующей стадии цикла взвешивания через за-
хват. После того как через захват пройдут шесть образцов, карусель
останавливается. Седьмой образец находится при остановке в по-
ложении для взвешивания. Радиальные балки карусели опускаются,
и верхняя часть держателя образца повисает на захвате. В таком
положении образец находится в течение 90 с. Этого времени доста-
точно для взвешивания и записи результатов. Затем радиальные
балки поднимаются и держатель образца освобождается от захвата.
Вращение карусели возобновляется, и следующий образец пере-
носится в положение для взвешивания. Одновременно могут нагре-
ваться 20 образцов. Весь цикл взвешивания, при котором масса
каждого образца измеряется один раз, продолжается 1,5 ч.
ЛИТЕРАТУРА
1. Lukaszewski G. М., Redfern J. Р., Lab. Pract., 10, 469 (1961).
2. Gordon S., Campbell C., Anal. Chem., 32, 271R (1960).
3. Wiedemann H. G., Achema Congress, Frankfurt, June 26, 1964.
4. Erdey L , Paulik F., Paulik J., Mikrochim. Acta, 699 (1969).
5. Paulik F., Paulik J., Erdey L., Taianta, 13, 1405 (1966).
6. Wiedemann H. G., Vaughan H. P., Thermochim. Acta, 3, 355 (1972).
7. Garn P. D., Kessler J. E., Anal. Chem.., 32, 1563 (1960).
8. Forkel W-Naturwissenschaften, 47, 10 (1960).
9. Newkirk A. E., Thermochim. Acta, 2, 1 (1971).
10. Paulik F., Paulik J., Thermochim. Acta, 4, 189 (1972).
106
Глава 3
11. Erdey L-, Simon J., Gal S., Taianta, 15, 653 (1968).
12. Stewart L. N., Proc, of the Third Toronto Symp. on Thermal Analysis
McAdie H. G., ed., Chemical Institute of Canada, Toronto, Feb. 25—26
1969, p. 205.
13. Manche E. P., Carroll B., Rev. Sci. Instr., 35, 1486 (1964).
14. Norem S. D., O’Neill M. J., Gray A. P., Proc, of the Third Toronto Symp.
on Thermal Analysis, McAdie H. G., ed., Chemical Inst, of Canada, To-
ronto, Feb. 25-26, 1969, p. 221.
15. Norem S. D., O’Neill M. J., Gray A. P., Thermochim. Acta, 1, 29 (1970).
16. Duval C., Anal. Chem., 23, 1271 (1951).
17. Lewin S. Z., J. Chem. Educ., 39, A575 (1962).
18. Jacque L., Guiochon G., Gendrel P., Bull. Soc. Chim. France, 1061, 1961.
19. Saito H., Thermal Analysis, Schwenker R. F., Garn P. D., eds., Academic
Press, N. Y., 1969, p. 11.
20. Vaughan H. P., Am. Lab., Jan., 10 (1970).
21. Wendlandt W. W., Lab. Management, Oct., 26 (1965).
22. Wendlandt W. W., J. Chem. Educ., 49, 4571, A623 (1972).
23. Duval C., Inorganic Thermogravimetric Analysis, Elsevier, Amsterdam,
1953.
24. Duval C., Inorganic Thermogravimetric Analysis, 2nd. ed., Elsevier,
Amsterdam, 1963.
25. Garn P. D., Thermoanalytical Methods of Investigation, Academic Press,
N. Y., 1965, Ch. 10.
26. Keattch C., An Introduction to Thermogravimetry, Heyden, London, 1969.
27. Anderson H. C., Technique and Methods of Polymer Evaluation, Slade P. E.,
Jenkins L. T., eds., Marcel-Dekker, N. Y., 1966, Ch. 3.
28. Wendland W. W., Handbook of Commercial Scientific Instruments, Vei-
lon C., Wendlandt W. W., eds., Vol. 2, Marcel-Dekker, N. Y., в печати.
29. Saito H., Thermobalance Analysis, Gijitsu Shoin, Tokyo, 1962.
30. Honda K., Sci. Repts Tohoku Univ., 4, 97 (1915).
31. Guichard M., Bull. Soc. Chim. France, 33, 258 (1923).
32. Vallet P., Bull. Soc. Chim. France, 3, 103 (1936).
33. Dubois P., Bull. Soc. Chim. France, 3, 1178 (1936).
34. Chevenard P., Wache X., de la Tullaye R., Bull. Soc. Chim. France., 10,
41 (1944).
35. Paulik F., Paulik J., Erdey L., Z. Anal. Chem., 160, 241 (1958).
36. Gordon S., Campbell C., Anal. Chem., 28, 124 (1956).
37. Griffith E. J., Anal. Chem., 29, 198 (1957).
38. Hooley J. G., Can. J. Chem., 35, 374 (1957).
39. Извеков И. В., Труды Крымского филиала АН СССР, 4 , 81 (1953).
40. Van Norstrand R. A., U. S. Patent № 2692497, Oct. 26, 1954.
41. Rabatin J. G„ Gale R. H., Anal. Chem., 28, 1314 (1956).
42. Stephencon J. L., Smith G. W., Trantham H. V., Rev. Sci. Instr., 28, 380
(1957).
43. Fujii С. T., Carpenter C. D., Meussner R. A., Rev. Sci. Instr., 33, 362 (1962).
44. Satava V., Collection Czech. Chem. Commun., 24, 2172 (1959).
45. Farmer R. W., Thermochim. Acta, 4, 203 (1972).
46. Guenot J., Manoli J. M., Bregeault J. M., Bull. Soc. Chim. France., 2666
(1969).
47. Landsberg A., J. Sci. Instr., 41, 337 (1964).
48. Pannetier, Guenot J., Manoli J. M., Bull. Soc. Chim. France, 2829 (1964).
49. Harrison R. W., Delgrosso E. J., J. Sci. Instr., 41, 222 (1964).
50. Olson N. J., Air Force Materials Lab. Tech. Rept AFML-TR-184, Pt. 1,
June 1968.
51. Olson N. J., Thermal Analysis, Schwenker R. F., Garn P. D., eds., Vol. 1,
Academic Press, N. Y., 1969, p. 325.
52. Bremer H., Henrion G., Chem. Techn., 18, 368 (1966).
Термовесы и вспомогательное оборудование к ним
107
63. Franck R., Harmelin М., Proc, of the Third Analytical Chemistry Conf.,
Buzas I., ed., Vol. 2, Akademiai Kiado, Budapest, 1970, p. 219.
64. McBain J. W„ Bakr A. M., J. Am. Chem. Soc., 48, 690 (1926).
65. Sarasohn I. M., labeling R. W., Pittsburgh Conf, of Analytical Chemistry
and Applied Spectroscopy, March 5, 1964.
66. Williams H. W., Thermochim. Acta, 1, 253 (1970).
67. Brown H. A., Penski E. C., Callahan J. J., Thermochim. Acta, 3, 271 (1971).
58. Williams J. R., Simmons E. L., Wendlandt W. W., Thermochim. Acta,
5, 101 (1972).
69. Chiu J., Anal. Chem., 39, 861 (1967).
60. Paulik J., Paulik F., Erdey L., Anal. Chim. Acta, 34, 419 (1966).
61. Gulbransen E. A., Andrew K- F., Brassart F. A., Vacuum Microbalance
Techniques, Vol. 4, Plenum, N. Y., 1965, p. 127.
62. Feldman R. F., Ramachandran V. S., Thermochim. Acta, 2, 393 (1971).
63. Pedersen E., J. Sci. Instr., 1, 1013 (1968).
64. Scott К- T„ Harrison К. T., J. Nucl. Mater., 8, 307 (1963).
65. Bradley W. S„ Wendlandt W. W., Anal. Chem., 43, 223 (1971).
66. Cahn Electrobalance Model RTL, Instructions № 2006, Cahn Instrument Co.
67. Etter D„ Smith W. H., J. Chem. Educ., 49, 143 (1972).
68. Plant A. F., Industrial Res., July, 36 (1971).
69. Van Empel F. J., Ballegooyen E. C., Boersma F., Poulis J. A., Thermo-
chim. Acta, 5, 129 (1972).
70. Wendlandt W. W., Chimia, 26. 1 (1972).
71. Ferguson J. M., Livesey P. M., Mortimer D., Intern. Confederation of
Thermal Analysis III, Davos, Switzerland, Aug. 1971, Paper I.
72. Wendlandt W. W., Anal. Chim. Acta, 49, 185 (1970).
л
Глава 4
ПРИМЕНЕНИЕ ТЕРМОГРАВИМЕТРИИ
А. ВВЕДЕНИЕ
По своей сущности термогравиметрия является количествен-
ным методом анализа, поскольку с ее помощью можно точно опре-
делить изменение массы. Однако изменение массы происходит в
определенных интервалах температур, количественная оценка ко-
торых затруднительна вследствие зависимости температуры от
характеристик измерительного прибора и образца. При повсемест-
ном применении термовесов, выпускаемых промышленностью, ре-
зультаты термогравиметрических измерений образцов в различных
лабораториях могут быть сходными при соблюдении одинаковых
условий пиролиза.
Термогравиметрия находит широкое применение почти во всех
областях химии и в смежных областях науки и техники. В начале
50-х годов этот метод произвел революцию -в гравиметрическом
анализе в неорганической химии, а в 60-х годах подобная ситуация
повторилась в области химии полимеров. Такое же большое значе-
ние приобрели методы термогравиметрии и в решении прикладных
задач, таких, как определение характеристик различных материа-
лов, используемых в дорожных покрытиях, определение содержа-
ния влаги во многих материалах и целом ряде других задач. Как
будет показано ниже, термогравиметрия находит широкое приме-
нение для анализа в металлургии, лакокрасочной промышленности,
производстве керамических материалов, минералогии, пищевой
промышленности, органической и неорганической химии, химии
полимеров, биохимии, геохимии и др.
Применение метода термогравиметрии к конкретной задаче
возможно в том случае, если при тепловых воздействиях наблюда-
ется изменение массы. Если изменения массы не происходит, не-
обходимо использовать другие методы термического анализа, такие,
как дифференциальный термический анализ (ДТА), дифференци-
альная сканирующая калориметрия (ДСК), термомеханический ана-
лиз (ТМА) и др. Если изменения массы очень малы (<1 %), то могут
оказаться более эффективными такие методы, как обнаружение
выделившегося газа (ОВГ). Разновидности изменения (обычно по-
Применение термогравиметрии
109
тери) массы, которые можно обнаружить методом термогравимет-
рии, схематически представлены на фиг. 4.1.
Ниже перечислены некоторые из многочисленных областей при-
менения метода термогравиметрии:
1) термическое разложение органических, неорганических и
полимерных веществ;
Фиг. 4.1, Изменения массы, обнаруживаемые ТГ-методом.
2) коррозия металлов в атмосфере различных газов при повы-
I шенных температурах; .
3) реакции в твердой фазе;
4) обжиг и прокаливание минералов;
5) перегонка и испарение жидкостей;
6) пиролиз угля, нефти и древесины;
7) определение влажности, а также содержания летучих и золь-
ных компонентов;
8) определение скоростей испарения и возгонки;
9) исследования гигроскопичности и дегидратации;
10) автоматический термогравиметрический анализ;
11) термоокислительная деструкция полимеров;
12) разложение взрывчатых веществ;
13) разработка методов гравиметрического анализа;
14) исследование кинетики реакций;
15) обнаружение новых химических соединений;
16} определение давления пара и теплоты испарения.
(Многие области применения термогравиметрии рассмотрены в
литературе. Наибольшего внимания заслуживают ранние обзорные
статьи Гордона и Кэмпбелла [1 ], Дюваля [2], Коутса и Редферна
[3], Лукашевского и Редферна [4J и более новые публикации —
обзоры Мэрфи [5, 6] и Турсела [7]. Описанию метода посвящены
целые книги или отдельные главы, написанные Дювалем [8, 9],
J
по
Глава 4
Уэндландтом и Смитом [10], Уэндландтом [11], Андерсоном [12],
Дойлем [13], Бэроллом [14], Липтейем [15], Рейхом и Стайвейла
[16] и др. Справочник под редакцией Липтейя [15] содержит кри-
вые термогравиметрического (а также термогравиметрического по
производной и дифференциального термического) анализа, получен-
ные с помощью дериватографа. На прозрачных пластмассовых лис-
тах, наклеенных поверх графиков, нанесены размеры образцов и
скорости их нагревания.
Б. ПРИМЕНЕНИЕ В АНАЛИТИЧЕСКОЙ ХИМИИ
В данной главе рассматривается использование термограви-
метрии для решения аналитических задач различного типа. По-
дробно этот вопрос рассмотрен в работах Дюваля [2, 8, 9 ], а также
Палея и др. [17]. В последней работе рассматриваются следующие
области применения метода;
1) новые соединения и определение интервалов их термической
стабильности;
2) взвешивание веществ, неустойчивых при обычных темпера-
турах окружающей среды, т. е. веществ, поглощающих СО2 и Н2О,
из воздуха;
3) исследование поведения материалов в атмосферах различных
газов;
4) определение чистоты и термической стабильности аналити-
ческих реактивов, в том числе основного и примесного стандарт-
ных веществ;
5) определение состава сложных смесей;
6) систематическое исследование свойств материалов в зависи-
мости от методов их изготовления;
7) автоматический гравиметрический анализ.
Помимо приведенных выше, Дюваль [2 ] рассмотрел также сле-
дующие области применения:
1) исследование методов фильтрования (например, обугливание
фильтровальной бумаги);
2) выяснение условий сушки и прокаливания осадка;
3) использование термовесов для разработки новых методов
разделения веществ и в газометр ии;
4) исследование возгонки различных веществ;
5) исправление ошибок в аналитической химии;
6) применение в функциональном органическом анализе.
1. АВТОМАТИЧЕСКИЙ ГРАВИМЕТРИЧЕСКИЙ АНАЛИЗ
Дюваль [18] первым показал возможность быстрого определе-
ния количества ионов металлов. Операция выполнялась за 15—
20 мин с обычной для методов гравиметрии точностью, обеспечивав-
Применение термогравиметрии
111
мой независимо от квалификации оператора. Он предложил также
способ одновременного определения количеств ионов двух или трех
различных видов без предварительного их разделения. Развивая
эти идеи, Дюваль разработал метод, известный как автоматиче-
ский гравиметрический анализ, позволяющий с помощью термо-
весов быстро определить концентрацию данного иона металла или
Фиг. 4.2. Автоматический гравиметрический анализ однокомпонентиой си-
стемы [18].
состав смеси ионов. Выбор гравиметрического метода производится
на основании следующих требований:
а) быстрое количественное осаждение всех исследуемых ионов;
б) быстрое фильтрование без необходимости выдерживания осад-
ка;
в) быстрая сушка;
г) получение на термогравиметрической кривой участка по-
стоянного веса при наименьшей возможной температуре.
Сущность этого метода для однокомпонентной системы заключа-
ется в следующем (фиг. 4.2). При использовании чистого и сухого
тигля определяют базовую линию термовесов, обозначенную пунк-
тирной линией X. Затем тигель снимают с весов, заполняют его
влажным осадком и снова помещают на весы. Осадок нагревают,
и кривая потери массы записывается обычным способом. По гори-
зонтальному участку кривой ВС определяют массу te)j, а по участ-
ку DE — массу w2. Так как горизонтальные участки кривой ука-
зывают, что достигнута определенная степень стехиометрии осадка,
то умножение величин wt или w2 на соответствующий грави-
112
Глава 4
метрический коэффициент дает значение массы присутствующих
ионов металла.
Определение содержания ионов по величине вероятно, более
точно вследствие большей точности измерения массы и меньшей ве-
личины гравиметрического коэффициента осадка.
Для некоторых осадков вся операция, включая фильтрование,
сушку и запись на термовесах, занимает всего 12 мин [18].
Фиг. 4.3. Автоматический гравиметрический анализ бинарной смеси [18].
При анализе бинарных смесей применяется подобная же про-
цедура. Рассмотрим случай, представленый на фиг. 4.3. Показаны
кривые потери массы для отдельных компонентов MX и NY, вхо-
дящих в состав смеси, и для их смеси MX + NY. Компонент MX
разлагается на участке DE, а компонент NY — на участке ВС. На
кривой для смеси горизонтальные участки начинаются при тех же
значениях температур, что и на кривых для отдельных компонентов.
Таким образом, содержание компонента NY можно определить по
величине ВС на кривой для смеси, а содержание компонента MX —
по величине DE. Следовательно, с помощью одной простой опера-
ции с достаточной точностью производится анализ некоторых би-
нарных и тройных смесей.
Дюваль [18] применил этот метод при анализе бинарной смеси,
содержащей ионы кальция и магния. Ионы были осаждены в виде
оксалатов, и полученная термогравиметрическая кривая была
сопоставлена с соответствующими кривыми для оксалатов отдель-
ных металлов. Если х и у — соответственно массы кальция и маг-
Применение термогравиметрии
113
ния, a m и п — определенные по термогравиметрической кривой
массы смесей при температуре 500°С (MgO 4- СаСО3) и 900°С
(MgO + СаО), то
100х , 40,32ц :—- = т, 40 24,32 56х , 40,32ц = п. 40 24,32 (4.1) (4.2)
Следовательно,
т т~п X = . 1,1 (4.3)
Для искусственной смеси, содержащей 0,1541 г СаС2О4-Н2О
0,0453 г MgC2O4 • 2Н2О, получаем х = 0,0427 г (теоретическое
значение х — 0,0422 г). Подобные оценки были проведены при
выделении сплава медь — серебро из смеси нитратов металлов.
В. АНАЛИТИЧЕСКИЕ ОПРЕДЕЛЕНИЯ С ПОМОЩЬЮ
МЕТОДА ТЕРМОГРАВИМЕТРИИ
1. СУШКА ОСАДКОВ В ВЕСОВОМ АНАЛИЗЕ
Одним из первых применений термовесов к задачам аналитичес-
кой химии явилось определение температуры сушки и весовых ха-
рактеристик осадков. Дюваль [19] был поражен тем фактом, что
большинство авторов подробно рассматривают условия осаждения,
такие, как концентрация реагентов, их объем, показатель pH раст-
вора, время «старения» осадка и др., но очень неопределенно упоми-
нают о температурах сушки или пиролиза. Такие общие утвержде-
ния, как «прокаливать до постоянного веса» или «нагревать не бо-
лее, чем до темно-красного цвета» и т. п., совершенно неприемлемы,
когда речь идет об осадке в весовом анализе. С помощью семнадца-
ти сотрудников Дюваль выполнил термическую обработку более
1200 осадков, подготовленных для неорганического весового ана-
лиза. Только часть из них оказалась пригодной для весового опре-
деления’массы ионов различных металлов. Пригодность определя-
лась на основании скорости осаждения и оценки температур суш-
ки или прокаливания.
Одна из первых ТГ-кривых, которая была использована в весо-
вом определении кальция, приведена на фиг. 4.4 [20]. Как извест-
но, горизонтальное плато кривой в интервале температур 25—
100°С соответствует составу исходного соединения, в интервале
температур 226—346°С — составу оксалата кальция СаС2О4, в ин-
5—230
114
Глава 4
тервале 420—660°С — составу карбоната кальция СаСО3, а в ин-
тервале 840—980°С — составу окиси кальция СаО. Таким образом
можно определить температуры сушки и прокаливания, а также
состав соединения при любой температуре. Вопрос о температуре
сушки и отсутствии горизонтальных участков на ТГ-кривой рас-
сматривался многими исследователями, в том числе Симмонсом и
Ньюкирком [21]. Для многостадийной реакции термического раз-
ложения, включающей разложение большинства исследуемых ана-
литических осадков, можно сделать следующие выводы [21 ]:
Фиг. 4.4. ТГ-кривая гидрата оксалата кальция СаСгО4-Н2О [20].
1. Наличие горизонтального участка на ТГ-кривой не всегда
означает, что исследуемое соединение изотермически устойчиво
(в практическом или термодинамическом смысле) при всех значе-
ниях температур на этом участке.
2. Если на кривой, полученной для многостадийной реакции,
отсутствует промежуточный участок, на котором не происходит
изменения массы во времени в некотором интервале температур,
то можно утверждать, что реакции образования и последующего
разложения промежуточного соединения не следуют друг за другом,
а по крайней мере частично перекрывают друг друга.
3. При отсутствии горизонтального участка на кривой для по-
следовательных реакций невозможно определить точное значение
начальной или конечной температуры участка или ’Tf) или
стехиометрический состав, хотя относительно последнего часто
удается сделать определенные выводы.
Таким образом, правомерность переноса значений температуры
сушки или прокаливания, полученных по горизонтальному участ-
ку термогравиметрической кривой, на случай изотермических из-
мерений, вызывает сомнения, хотя этот метод широко используется
на практике.
Применение термогравиметрии
115
2. СУШКА И РАЗЛОЖЕНИЕ КАРБОНАТА НАТРИЯ
Сушка карбоната натрия (кальцинированной соды) является
важным процессом при установлении титра кислот, используемых,
для различых видов ацидиметрического титрования. Рекомендуе-
Фиг. 4.5. Кривые потерь массы кар-
боната натрия [23].
О 200 400 600 800 1000 1 2
Температура, “С Время, ч
или
мас-
мед-
счет
мый интервал температур
сушки составляет 250—300сС,
хотя Дюваль [22 ] утвержда-
ет, что горизонтальный учас-
ток термогравиметрической
кривой находится в интерва-
ле температур 100 — 840° С.
В более позднем исследова-
нии Ньюкирка и Алифериса
[23] показано, что температу-
ра разложения безводного
карбоната натрия зависит от
типа тигля, в котором нагре-
вался образец. Результаты
этого исследования приведены
на фиг. 4.5 и в табл. 4.1.
При нагревании карбона-
та натрия в платиновых
золотых тиглях потери
сы происходят гораздо
леннее и, вероятно, за
разложения образца на окись
натрия и двуокись углерода.
Скорость потерь массы выше
в потоке сухого газообразно-
го азота (кривая 6), чем в
присутствии воды (кривая 7).
Образец карбоната натрия,
высушенный при температуре 350° С, при последующем
нагревании в платиновом тигле на воздухе при температуре 600°С
в течение 12 ч и при температуре 650° С в течение 4 ч сохранял свою
массу неизменной. Реакция карбоната натрия с крупным кварце-
вым песком быстро протекала при температуре 800 - 850°C (кривая 9)
После измельчения смеси первые признаки потерь массы наблюда-
лись при температуре ~500'С (кривая 10) или при немного более
низкой темпер ауре, чем в случае кривой 9.
Было рекомендовано сушить карбонат натрия, предназначенный
для использования в аналитических исследованиях, путем нагре-
вания в атмосфере сухого воздуха или двуокиси углерода в тигле
из платины или другого инертного материала в интервале темпе-
ратур 250—700° С.
116
Глава 4
Таблица 4.1
Кривая г) на фиг. 4.5 Материал тигля Атмосфера Образец
1 Фарфор Воздух Na2CO3
2 » Na2CO3 3)
3 » Сухой азот 3) Na2CO3
4 Окись алюминия Воздух Na2CO3
5 Платина » Na2CO3
6 » Сухой азот 3) Na2CO3
7 » Влажный азот 3) Na2CO3 Na2CO,
8 » Двуокись углерода 3)
9 » Сухой азот 3) Na2CO3+SiO2
10 » » » Na2CO3+SiO2 4)
11 Золото » » Na2CO3 5)
*) Скорость нагревания 300 град/ч, за исключением опытов 9 и 10.
2) Тигель закрыт.
3) Расход газа 250 мл/мии.
*) Скорость нагревания 300 град/ч до 520°С, а затем 50 град/ч.
в) Максимальная температура 922°С; затем образец охлаждали до 915°С и выдерживали
при этой температуре в течение 1ч. •
3. ТЕМПЕРАТУРА ПРОКАЛИВАНИЯ ОКИСИ АЛЮМИНИЯ
Хотя Дюпюи и Дюваль [24 ] еще в 1949 г. исследовали пиролиз
гидратированной окиси алюминия А12О -иН2О, полученной с по-
мощью 25 осадителей, только в более поздних работах Эрди и Пау-
лика [25 ], а также Милнера и Гордона [26 ] были затронуты вопро-
сы, касающиеся полученных при пиролизе пониженных темпера-
тур прокаливания. Минимальные температуры прокаливания
А12О3, по Дюпюи и Дювалю [24 ], заключены в интервале от 280° С
(осаждение бромом) до 1031 °C (осаждение водным раствором амми-
ака). Обнаружено плохое соответствие с упомянутыми выше ре-
зультатами Эрди и Паулика [25 ] в том смысле, что большинство
образцов все еще имело потери массы при температуре 1000° С —-
максимальной температуре использования дериватографа. Милнер
и Гордон [26 ] считают, что для осадков окиси алюминия, прока-
ливаемых и взвешиваемых обычными методами, минимальная тем-
пература прокаливания должна составлять 1200°С. Это мнение
частично подтверждается результатами, приведенными в табл. 4.2.
Как видно из таблицы, минимальная обычная температура прока-
ливания составляет 1100°С (в течение 1 ч). В случае присутствия
сульфат-иона, как, например, в методе с использованием основных
сульфатов, необходима еще более высокая температура.
Дюваль [27 ] утверждает, что если нагревание, охлаждение и
взвешивание образца производятся не на термовесах, а на обычных
Применение термогравиметрии
117
Таблица 4.2
Влияние температур прокаливания на вес осадков окиси
алюминия, полученных различными методами [26]
Температура1), °C Избыточная масса (по сравнению с конечным * значением), %
метод А (суль- фатный с исполь- зованием мочевины), % метод Б (сукци- иатный с исполь- зованием мочеви- ны), % метод В (с использова- нием моче- вины2), % метод Г (с использова- нием гидроокиси аммония), %
650 19,2 3,9 3,1 4,5
800 9,8 2,3 1,7 2,4
950 3,4 1,0 1,0 1,2
1100 0,6 0,0 0,0 0,2
1200 0,2 0,0 0,0 0,1
(2-й час) Конечное значение
*) После обугливания фильтровальной бумаги осадки прокаливали при температуре
500сС в течение 8 ч, а затем 1 ч прн указанных температурах.
2) В данном методе в отличие от сульфатного и сукцинатного методов [используются хло-
риды.
аналитических весах, то необходимо прокаливать окись алюминия
при более высоких температурах, с тем чтобы образец не впитывал
влагу при охлаждении и взвешивании. При использовании автома-
тического термогравиметрического анализа этот источник ошибок
устранен и, следовательно, допустимы более низкие температуры
прокаливания. Дюваль [27], однако, не поясняет различные кривые,
представленные Эрди и Пау ликом [25]. Последние пришли к выво-
ду, что структура гидрата окиси алюминия определяется такими
переменными, как скорость, температура осаждения и только в не-
большой степени — свойствами осадителя. Они утверждают также,
что более низкие температуры, указанные Дюпюи и Дювалем [24 ],
получены вследствие изменяющихся условий осаждения, которые
трудно воспроизводимы. Даже небольшие изменения оказывают
значительное влияние на температуру прокаливания.
4. СЛОЖНЫЕ СМЕСИ ГИДРАТОВ
Гриффит [28] применил термогравЕметрию для определения
содержания воды в смесях гидратированных и безводных солей
и обнаружил шесть фаз. В основу метода положен известный факт,
что при выборе соответствующей скорости нагревания происходит
избирательное разложение фазы с наибольшим давлением диссо-
циации. После полного разложения фазы с наибольшим давлением
диссоциации начинается разложение следующего соединения с наи-
большим давлением диссоциации и т. д. Таким образом, по массо-
118
Глава 4
вой скорости потери смесью воды можно определить содержание
воды в двух различных фазах смеси гидратов.
На фиг. 4.6, а приведена кривая термического разложения смеси
гидрата карбоната натрия (Na2CO3-H2O) и гексагидрата пиро-
фосфата натрия (Na4P2O7 • ЮН2О) при постоянной температуре
100°С. По этой кривой нельзя понять, когда полностью завершилась
дегидратация Na4P2O7- ЮН2О и началась дегидратация Na2CO3-H2O.
Фиг. 4.6. Кривые потерь массы [28].
а — кривая разложения смеси Na4P2O7• 10Н2О и Na2CO3-H2O при 100°С; б — та же смесь, но
при медленном нагревании.
Давления диссоциации этих двух соединений при высоких темпера-
турах почти одинаковы, но при более низких температурах (~ 60°С)
разница в давлениях диссоциации становится ощутимой. Если эта
смесь разлагается при температуре 60°С, то четко видно изменение
наклона кривой в точке, соответствующей стехиометрическому со-
ставу в конце дегидратации. На фиг. 4.6, б представлена кривая
для той же смеси при медленном нагревании (8,7 град/ч) в интер-
вале температур 30 100сС. В данном случае отчетливо виден из-
лом кривой, что свидетельствует о важности соответствующего
выбора скорости нагревания.
В других примерах, приведенных Гриффитом [28], были исполь-
зовны Na5P3O10-6Н2О и смесь Na5P3O10, Na5P3O10-6Н2О,
Na4P2O7, Na4P2O7 - ЮН2О, Na2CO3 и Na2CO3-H2O, а также смесь
Na2B4O7 • ЮН2О, Na4P2O7-H2O и КгСО3-1,5^0. Средняя величи-
на ошибки при анализе этих смесей составляла ±1,2%.
Применение термогравиметрии
119
5. АНАЛИЗ ГЛИН И ПОЧВ
Гоффман и др. [29, 30 ] проводили термогравиметрический ана-
лиз почв, относительно чистых глин, кристаллических карбонатов
и почв с известными добавками глин и карбонатов. На кривых раз-
ложения относительно чистых глинистых минералов при повышен-
ных температурах наблюдайся резкие изломы, что позволяет пред-
положить возможность применения термогравиметрии для изуче-
ния чистых глин и простых глинистых смесей. С помощью этого
метода можно определить также содержание воды, органических
веществ и неорганических карбонатов.
На кривых разложения большинства почв наблюдается горизон-
тальный участок (постоянный уровень массы), начинающийся при
температуре 150—180° С и заканчивающийся при 210—240° С; он
указывает на присутствие либо одной поглощенной влаги, либо
поглощенной влаги и легколетучих органических соединений. В
общем случае потери массы заключены между значениями, полу-
ченными методом Фишера и путем сушки в печи при температуре
105° С. Органические вещества воспламеняются в интервале тем-
ператур 210—240° С и обычно полностью сгорают при температуре
500° С. Анализ кривой потерь массы позволяет довольно точно опре-
делить содержание органических веществ в органических почвах
и в почвах с содержанием глины менее 15%. При содержании гли-
ны 15—40% оценка потерь массы при температуре 500° С позволяет
получить данные о содержании органических веществ, удовлетво-
рительно согласующиеся с результатами анализа сухим сожжением
и мокрым окислением. При содержании глины более 40% анализ
кривых потерь массы затруднен, поскольку не удается разделить
эффекты разложения органических веществ и выделения из глины
кристаллизационной воды. Возможно также количественное опре-
деление кристаллизационной воды в образцах чистой глины. По-
скольку выделение кристаллизационной воды из разных глин про-
исходит при различных температурах, последние могут служить
дополнительным средством идентификации глин и определения их
характеристик.
Результаты использования термогравиметрни для определения
состава смеси каолинит — гекторит приведены на фиг. 4.7 [58 ].
Штриховой и пунктирной линиями нанесены кривые для чистых
глин; каолинит выделяет в результате реакции дегидроксилирова-
ния 12,4% воды, а гекторит 20,6%. По ТГ-кривой их смеси установ-
лен ее состав (70% каолинита и 30% гекторита), согласующийся
с расчетными оценками (70 и 30% соответственно).
Малли и Кавендиш [31 ] считают, что термогравиметрию можно
применять для анализа смесей дигидрата кислого ортофосфата каль-
ция (брушита) СаНРО4-2Н2О и безводного кислого ортофосфата
кальция (монетита) СаНРО4 путем регистрации потери смесями во-
120
Глава 4
Фиг. 4.7. ТГ-кривые смеси каолинит — гекторит [58].
1 — компонент смеси каолинит, потери массы 8,9%; 2 — компонент смеси гекторит, потери
массы 6,3%; 3 — чистый гекторит, потери массы 20,6%; 4 —чистый каолинит, потерн массы
12,4%.
ды при нагревании. Потери воды при нагревании брушита соответ-
ствуют 2,5 моль на 1 моль соединения, а при нагревании монетита —
0,5 моль на 1 моль соединения. Потери массы при нагревании смеси
этих двух соединений составляют
СаНРО4 • 2Н2О = b = W&2a % (4.4)
и
СаНРО4 = а == 0,2617ft %. (4.5)
Суммарные потери массы (в процентах) равны
Т = 0,0662а + 0,2617ft, (4.6)
где ft — 100—а, и, следовательно,
Г = 26,17 —0,1955а. (4.7)
Из графика зависимости Т от а, имеющего вид прямой линии, мож-
но непосредственно определить состав смеси. Подобные расчеты
могут быть выполнены и при анализе трехкомпонентной системы.
Минералы с высоким и низким содержанием магниевого кальци-
та в смеси с арагонитом и обычным кальцитом исследовались нес-
колькими методами, включая метод термогравиметрйи [32]. Ре-
зультаты, полученные с помощью последнего метода, оказались
более точными, чем результаты изучения методом дифракции рент-
геновских лучей и жидкостным химическим анализом.
Паулик и др. [33] разработали метод определения содержания
пиритов в бокситах и глинистых минералах с помощью деривате-
Применение термогравиметрии
121
графа. Этот метод может быть использован для анализа образцов
с содержанием пиритов более 0,2%. Содержание пиритов определя-
лось по ТГП-кривой, а не по ТГ-кривой.
Анализ лунного вещества с помощью термогравиметрии описан
в гл. 8.
6. ОПРЕДЕЛЕНИЕ СОДЕРЖАНИЯ СВОБОДНОЙ ИЗВЕСТИ
И КАРБОНАТА КАЛЬЦИЯ В ГИДРАТАХ
СИЛИКАТА КАЛЬЦИЯ
Биффен [34 ] определил термогравиметрическим методом со-
держание свободной извести Са(ОН)2 и карбоната кальция СаСО3
в гидратах силиката кальция (ГСК), составляющее 1—20%. На
। фиг. 4.8 приведены кривые потерь массы ряда гидратов силиката
кальция и тех же гидратов с добавками гидроокиси кальция (из-
вести) или карбоната кальция.
Кривые для гидратов силиката кальция идентичны, не имеют
резких изломов, в интервале температур 375—650° С представляют
собой прямые линии с плавным наклоном. Изломы кривых, наблю-
даемые при температурах более 600° С, обусловлены разложением
карбоната, содержащегося в образце.
На всех кривых, полученных для искусственных смесей гидрата
силиката кальция и гидроокиси кальция, видны изломы при темпе-
ратуре ~500° С, вызванные разложением гидроокиси кальция,
что подтверждается кривыми потери массы чистой гидроокиси каль-
ция. Измеряя расстояние по вертикали от точки нарушения линей-
ности прямой (вследствие выделения кристаллизационной воды из
гидрата силиката кальция) до точки, указывающей на завершение
процесса разложения силиката кальция, можно определить потери
массы воды и рассчитать содержание гидроокиси кальция. Во всех
случаях этот метод дает хорошие результаты.
На фиг. 4.8 представлены также кривые разложения образцов
гидрата силиката кальция, содержащих карбонат кальция, а в не-
которых случаях и гидроокись кальция. На присутствие карбоната
кальция указывает излом кривой в интервале температур 700—
900° С, связанный с выделением двуокиси углерода. Содержание
последней в образце легко оценить, измерив расстояние по вертика-
ли между точкой нарушения линейности прямой и точкой начала
горизонтального участка.
Результаты определения ТГ-методом содержания воды, свобод-
ной извести и карбонатов в гидратах силиката кальция хорошо сов-
падают с данными других методов.
Рамачандран [351 определил содержание Са(ОН)2 в смесях с си-
ликатом кальция по уменьшению содержания воды в интервале
температур 450 —550° С.
122
Глава 4
100 200 300 400 500 600 700 800 900
wt
0,636
0,536
0,600
0,529
0,770
Температура,. °C
Wt
0,770
0,500
0,627
0,429
0,428
0,579
Фиг. 4.8. Кривые потерь массы [34].
а — гидрат силиката кальция (ГСК); б — гидрат силиката кальция с добавкой гидроокиси
кальция Са(ОН)2; в — гидрат силиката кальция с добавкой карбоната кальция СаСО3;
Wt — вода.
7. ОПРЕДЕЛЕНИЕ СОДЕРЖАНИЯ МАГНИЯ, КАЛЬЦИЯ,
СТРОНЦИЯ И БАРИЯ
С помощью ТГ-методов Эрди и др. [36, 37] определяли содер-
жание одного из ионов кальция, стронция или бария в присутствии
двух других. Ионы осаждали в виде смеси гидратов оксалатов ме-
Применение термогравиметрии
123
Фиг. 4.9. Кривые потерь массы для смеси гидратов оксалатов кальция,
стронция и бария [36].
таллов и подвергали разложению на термовесах. По кривым потерь
массы можно было определить содержание кальция, стронция и ба-
рия.
На фиг. 4.9 приведены кривая потерь массы и ее первая произ-
водная (гл. 2) для смеси гидратов оксалатов кальция, стронция
и бария.
Как видно из ТГ-кривой, процессы разложения протекают не-
зависимо друг от друга. В интервале температур 100—250° С вы-
деляется кристаллизационная вода, и все оксалаты переходят в
моногидраты. В соответствии с кривыми, полученными для отдель-
124
Глава 4
ных соединений, вода выделяется в следующем порядке: вначале
из оксалата бария, а затем из оксалатов стронция и кальция. Од
нако при анализе смеси осадков разложение гидратов оксалатов
стронция и кальция происходит одновременно.
После потери кристаллизационной воды на кривой в интервале
температур 250—360° С образуется горизонтальный участок, со-
ответствующий образованию безводных оксалатов металлов. Затем
разложение всех трех оксалатов происходит одновременно, и про-
цесс заканчивается при температуре ~500°С. В интервале темпе-
ратур -~500 620е С безводные карбонаты металлов устойчивы, за
исключением карбоната стронция, разложение которого полностью
завершается при температуре 1100° С. При этой же температуре на-
чинается разложение карбоната бария.
По кривым потерь массы можно получить следующие данные:
D — массу сухого остатка при температуре 100° С; Е — массу
кристаллизационной воды; F —- массу окиси углерода, образо-
вавшейся при разложении безводных оксалатов металлов;
G — массу двуокиси углерода, образующейся при разложении
карбоната кальция, и L — массу двуокиси углерода, образующей-
ся при разложении карбоната стронция. По этим данным вычис-
ляют содержание кальция С, стронция S и бария В с помощью сле-
дующих соотношений:
С = 0,91068G;
S = 1,9911L;
В = 0,586030 — 1.9457G — 2,57880
Приняв общее содержание С, S и В равным единице, можно опре-
делить ошибку анализа:
= = ^^Лк = 0Л%. (4.8)
DEFGL v ’
Одновременное определение содержания кальция и магния
с помощью кривой потерь массы образца типичного доломита было
описано Дюпюи и Дювалем (фиг. 4.10) [381.
В соответствии с принципами, описанными ранее при рассмотре-
нии автоматического гравиметрического анализа, участок EF соот-
ветствует смеси MgO+ СаСО3, а участок GH — смеси MgO+ СаО.
Разность Wj_—w2 равна массе двуокиси углерода, выделяющейся
в интервале температур 500—900° С при разложении карбоната
кальция. Содержание окиси кальция определяется по формуле
йУ(СаО) = (аУ1— w2) • — = (u)J — ay2). 1,272, (4.9)
Применение термогравиметрии
125
а содержание окиси магния — по формуле
w (MgO) = w2 — w (CaO). (4.10)
Термическое разложение чистого гидрата оксалата бария
ВаС2О4-Н2О в атмосфере азота исследовалось с помощью комбини-
рованного метода: дериватограф — термогазовая титриметрия 139]
(гл. 9).
Фиг. 4.10. Кривая потерь смеси влажных осадков карбонатов магния и
кальция [38].
8. КИСЛЫЙ ФТАЛАТ КАЛИЯ
Хотя и предполагалось, что применение термовесов исключит
путаницу, связанную с определением температур сушки и разло-
жения аналитических осадков и реактивов, в ряде случаев ситуа-
ция лишь усугубилась. В четырех термогравиметрических исследо-
ваниях, касающихся температур сушки и разложения кислого
фталата калия КНС8Н4О4, результаты оказались совершенно раз-
личными, не говоря уже о температурах сушки, рекомендуемых
в соответствии с другими методами. В работе Дюпюи и Дюваля [40 ]
впервые указывалась температура начала разложения КНС8Н4О4,
равная 172° С; в более позднем исследовании Дюваль [41] опреде-
лил, что при скорости нагревания 150 град/ч температура разло-
жения составляет 240° С, а при скорости нагревания 300 град/ч
она равна 236° С. По сообщению Белчера и др, [42], разложение
рассматриваемого соединения начинается при температуре 200° С;
интервал температур сушки, рекомендуемый в этой работе, 100—
150° С. В работе Ньюкирка и Лоэра [43] указана температура раз-
ложения ~260° С. С учетом предыдущих рассуждений о недостат-
126
Глава 4
ках термогравиметрии эти расхождения, возможно, и не удивитель-
ны. По-видимому, подобные исследования не пригодны для надеж-
ного определения температуры сушки основного стандартного ве-
щества [43].
Кривые потерь массы кислого фталата калия KHCgH4O4 в раз-
личных газовых средах приведены на фиг. 4.11.
Фиг. 4.11. Термическое разложение образцов кислого фталата калия
КНСВН4О4 весом 0,1 г при скорости нагревания 300 град/ч [43].
------ атмосфера воздуха, платиновый тигель;--- атмосфера воздуха, фарфоровый ти-
гель без теплоизоляционной втулки на опорном стержне тигля; . атмосфера азота, фар-
форовый тигель;--------атмосфера азота, предварительный подогрев образца до 300°С.
При пиролизе протекают четыре основные реакции разложения:
1 — испарение воды и фталевого ангидрида с образованием остатка
фталата калия K2CgH4O4; 2 — фталат калия разлагается с образо-
ванием карбоната калия К2СО3 и углеродсодержащего компонента;
3 — последний медленно уменьшается по массе и полностью сго-
рает, давая остаток карбоната калия; 4 — карбонат калия разлага-
ется с выделением двуокиси углерода, а образующая окись калия
К?О реагирует с фарфоровым тиглем держателя образца.
На фиг. 4.11 представлены кривые, иллюстрирующие термиче-
ское разложение вещества в различых условиях. Небольшое воз-
растание массы в интервале температур 425—450° С (штриховая
кривая) происходило вследствие испарения фталевого ангидрида
со стенок печи при возрастании температуры и его конденсации
на опорном стержне тигля.
По данным работы Ньюкирка и Лоэра [43], начальная скорость
изотермического разложения кислого фталата калия в двуокиси
углерода при температуре 235° С составляла 15 мг/(г-ч). Это зна-
чение можно сравнить с полученным экстраполяцией значением
Применение термогравиметрии
127
7 мг/(г-ч). Однако Дюваль [41] не обнаружил заметной потери
массы в изотермических условиях при температурах 150, 160 и
и 170° С. Ньюкирк и Лоэр [43] подсчитали, что при массе образца
0,5 г в экспериментах Дюваля можно было ожидать изменения мас-
сы 0,004, 0,011 и 0,03 г соответственно. Поскольку эти изменения
слишком малы, чтобы их можно было обнаружить на термовесах
Шевенара, не удивительно, что они не были замечены Дювалем.
9. ФОСФОМОЛИБДАТЫ НЕКОТОРЫХ АМИНОВ
Термическое разложение фосфомолибдатов аммония, хиноли-
на и оксина (8-хинолинола) исследовалось методом термограви-
метрии. Особенно противоречивы имеющиеся в литературе данные
о температуре сушки и прокаливания фосфомолибдата аммония.
Фиг. 4.12. Кривые потерь массы фосфомолибдата аммония [45].
« — осадок промыт азотной кислотой, сушка на воздухе; б — осадок4,7 промыт раствором
нитрата аммония, сушка на воздухе; в —• осадок промыт азотной кислотой, сушка в печи;
г — осадок промыт раствором интрата аммония, сушка в печн.
Дюпюи и Дюваль [44] установили с помощью метода Тредвелла,
что осадок имеет состав (ПН4)зРО4(МоО3)12-2HNO3-H2O. Осадок
теряет воду и азотную кислоту при нагревании до 180°С, и затем
на кривой образуется горизонтальный участок до температуры
410°С. Однако, если осадок предварительно высушен на воздухе,
потери массы до 180°С соответствуют только потере 2 моль азотной
кислоты, а не суммы (2HNO3 + Н2О). Вода, очевидно, испаряется
1’28.
Глава 4
при сушке на воздухе. Горизонтальный участок на кривой от 180
до 410°С соответствует составу (NH3)3PO4(MoO3)i2.
Используя метод осаждения Стокдейла, Уэндландт [45 ] полу-
чил кривые потерь массы, показанные на фиг. 4.12. Высушенный
на воздухе осадок начинает выделять «свободную» воду уже при
Фиг. 4.13. Кривые термического
разложения фосфомолибдата ок-
сина [49].
а — сушка и а воздухе; б — сушка в печи
при температуре 140°С.
температуре 60*С с образова-
нием в интервале температур
160—415°С участка постоян-
ной массы, соответствующего
составу (NH4)2HP(Mo3O10)4
Н2О. При температуре выше
415°С вновь наблюдаются по-
тери массы с образованием оки-
сла Р2О5-24МоО3, начиная с
температуры 500°С. Подобная
ТГ-кривая регистрируется и
при предварительной сушке
образца в печи при температуре
160°С, за исключением стадии
выделения свободной воды.
Осадок, промытый нитратом
аммония, после высушивания
на воздухе также начинает
выделять свободную воду при
55°С. В интервале температур
225 — 260° С выделяется
NH4NO3, затем в интервале
температур 260—430°С образуется участок постоянной массы, со-
ответствующий по составу (NH4)3P(Mo3O10)4. Полное разложение
начинается при температуре 435°C с образованием Р2О5-24МоО3
при 510°С, а далее уровень массы остается постоянным.
Несомненно, расхождение в результатах работ различных ис-
следователей объясняется особенностями процесса осаждения, а не
применяемыми приборами. Температура сушки осадка, промытого
азотной кислотой, по данным Уэндландта [45], равная 160—415°С,
близка к результатам (180—410° С), полученным Дюпюи и Дюва-
лем [44], хотя составы осадков различны. Но даже при несколько
отличающихся составах молекулярные веса соединений почти оди-
наковы, вследствие чего ошибка будет невелика независимо от
принятого гравиметрического коэффициента.
При исследовании фосфомолибдата оксина возникли те же проб-
лемы, что и при исследовании соответствующего соединения аммо-
ния. В работе Дюваля [46] впервые появилось сообщение о том,
что состав, соответствующий дигидрату фосфомолибдата оксина,
стабилен в интервале температур 176—225°С; в более поздней ра-
боте [47 ] сообщалось, что этот дигидрат стабилен в интервале темпе-
Применение термогравиметрии
129
ратур 236—268°С. При температуре выше 341 °C начинается его
разложение, а выше 765°С уровень массы сохраняется постоянным
и соответствует составу Р2О5-24МоО3. В работе не приведены дан-
ные и не указаны ссылки, касающиеся очевидного изменения ин-
тервала температур сушки.
Уэндландт и Брэбсон [48] исследовали термическое раз-
ложение фосфомолибдата оксина, приготовленного по методу Брэб-
сона и Эдвардса [49 ]. ТГ-кривые этого соединения приведены на
фиг. 4.13. Высушенный на воздухе осадок начинает терять массу
при температуре 60°С с установлением постоянного уровня массы,
соответствующего составу 3C9H7ON -H^PMo^O^), в интервале
температур 85—285°С. Это соединение начинает разлагаться при
температуре 375°С с образованием окисла Р2О5-24МоО3, масса
которого остается постоянной при температуре выше 470°С. Разло-
жение образца, высушенного в печи, происходит подобным образом.
Г. ПОЛИМЕРЫ
1. ВВЕДЕНИЕ
По-видимому, наиболее широкое применение термогравиметрии
за последние десять лет относится к области исследования полимер-
ных матералов. Эти исследования оказались плодотворными не
только в прикладных задачах, но и при фундаментальных исследо-
ваниях высокополимеров. Методы термогравиметрии используются
при сравнении относительной термической стабильности, исследо-
вании влияния вида и содержания различных добавок на термиче-
скую стабильность, изучении роли влаги и добавок в кинетике тер-
мической деструкции, прямом количественном анализе различных
сополимеров, исследовании устойчивости к окислению и т. д. При
изучении термоокислительной деструкции [50] с помощью термо-
гравиметрии можно выявить молекулярную структуру и относи-
тельное расположение повторяющихся мономерных звеньев, су-
ществование поперечных связей между цепями, боковые группы
гомополимеров и сополимеров и т. д. Кроме того, можно опреде-
лить константы скорости, порядок реакции, частотные факторы и
энергии активации процесса деструкции [16].
Известен ряд методов классификации полимеров по их термиче-
ской стабильности. Как указывал Фок [50], классификация очень
сложна из-за большого разнообразия возможных термических ре-
акций при повышенных температурах. Например, началом разло-
жения может служить деструкция боковой цепи, тогда как основная
цепь полимера не затрагивается; при более высоких температурах
может произойти дальнейшее разложение, в результате которого
свойства материала изменяются коренным образом. Одно вещество
может полностью разрушиться в определенных условиях нагрева-
13Э
Глава 4
ния, в то время как часть другого вещества в таких же условиях ос-
тается неразложившейся.
Поскольку значения температуры разложения, получаемые в
термогравиметрическом анализе, в значительной степени зависят
от особенностей применяемой методики, Дойль [51 ] использовал
выражение «наблюдаемая температура разложения», предостере-
гая от ошибочного толкования полученного экспериментального
значения как единственно возможного. Дойль [51 ] дает два типа
определения наблюдаемой температуры разложения. Он рассмат-
ривает «дифференциальную наблюдаемую температуру разложения»,
используемую для определения положения изломов на нормализо-
ванных термогравиметрических кривых, и «интегральную наблюдае-
мую температуру разложения», используемую для определения
формы нормализованной кривой потери массы. Значения интеграль-
ной наблюдаемой температуры определяются по кривой потерь
массы следующим образом. Как показано на фиг. 4.14, площадь
под кривой делится на небольшие квадраты. Площадь под кривой
интегрируется путем взвешивания соответствующих участков бу-
маги на аналитических весах. Масса заштрихованного участка
(фиг. 4.14), деленная на массу прямоугольника со сторонами, рав-
ными максимальным значениям по осям координат, представляет
собой полную площадь под кривой А*, нормализованную относи-
тельно остаточной массы и температуры. По значению А* опреде-
ляется температура Тд*
Т'А = 875Л* + 25. (4.11)
При определении ТА * предполагается, что все вещества испаряются
при температурах не более 900°С. Таким образом, Тд * является
скорей характеристической температурой окончания испарения,
а не интегральной наблюдаемой температурой разложения, имею-
щей практическое значение. Однако Тд * служит мерой устойчи-
вости к термическому воздействию, хотя и не особенно удовлетво-
рительной.
Чтобы сравнивать все материалы на одинаковой основе по отно-
шению к интервалу температур эксперимента, но с учетом их инди-
видуальных свойств, вводится второй параметр. Это площадь кри-
вой К*, представляющая собой отношение дважды заштрихованной
площади (фиг. 4.14) к площади прямоугольника, одна сторона ко-
торого равна характеристической температуре окончания испаре-
ния Тд*, а другая — массе остатка при температуре окончания
эксперимента 900°С.
Дойль [51 ] показал, что произведение А*К* является надежным
показателем термической стабильности для 54 полимеров совер-
шенно различных типов. Он показал, что при подстановке А*К*
вместо А* в уравнение (4.11) получаемое значение интегральной
Применение термогравиметрии
131
наблюдаемой температуры разложения имеет практический смысл
как температура испарения половины вещества. В отличие от обыч-
ной температуры, при которой испаряется половина всего вещества,
значение интегральной наблюдаемой температуры разложения по-
лучают с учетом оставшейся массы при температуре 900°С; _ этот
Фиг. 4.14. Площади под ТГ-кривой А* и К* [51].
подход может быть использован как при одностадийном, так и при
многостадийном разложении.
Интегральная наблюдаемая температура разложения, будучи
величиной, определяемой по площади под кривыми, обладает высо-
кой воспроизводимостью, и на нее оказывают лишь незначительное
влияние небольшие погрешности или систематические ошибки по-
строения кривой. Этим рассматриваемый параметр отличается от
результатов оценки только конечной точки для остаточной массы.
Даже небольшие изменения скорости нагревания не оказывают
значительного влияния на интегральную наблюдаемую температуру
разложения Значения этой температуры для некоторых полимер-
ных материалов приведены в табл. 4.3.
132
Глава 4
Таблица 4.3
Интегральные наблюдаемые температуры разложения
некоторых обычных полимеров [51]
Полимер Температура, °C
Полистирол 395
Эпоксидная смола, отвержденная малеиновым ангидридом 405
Полиметилметакрилат (плексиглас) ... 345
Найлон-66 419
Тефлон (политетрафторэтилен) 555
Кель-F (политрифторхлорэтилен) 410
Вайтон А (фторкаучук) 460
Силиконовая смола 505
2. ОТНОСИТЕЛЬНАЯ ТЕРМИЧЕСКАЯ СТАБИЛЬНОСТЬ
Относительная термическая стабильность ряда полимеров рас-
сматривается в работе Ньюкирка [52]. При получении ТГ-кривых
образцы, как видно из фиг. 4.15, сначала подвергались быстрому
нагреванию в атмосфере азота до температуры ~340° С, а затем
скорость нагревания снижалась. Термическая стабильность иссле-
Фиг. 4.15. Сравнение термической стабильности различных полимеров по
Ньюкирку [52].
1 — полиметилметакрилат; 2 — полистирол; 3 — майлар; 4 — иайлои; 5 — полиэтилен (ала-
тон-10); 6 —лексаи.
Применение термогравиметрии
133
дованных полимеров возрастала в следующем порядке: полиметил-
метакрилат, полистирол, майлар и найлон, полиэтилен, лексан.
На фиг. 4.16 сравнивается относительная термическая стабиль-
ность пяти полимеров по термогравиметрическим данным, полу-
ченным Чиу [53 ]. Пять полимеров — поливинилхлорид, полиметил-
метакрилат, полиэтилен высокого давления, политетрафторэтилен
Фиг. 4.16. Относительная термическая стабильность полимеров по данным
термогравиметрии [53].
Масса образца 10 мг; скорость нагревания 5 град/мин в атмосфере азота; 1 — поливинил-
хлорид; 2 — полиметилметакрнлат; 3 — полиэтилен высокого давления; 4— политетрафтор-
этилен; 5 — ароматический полипиромеллитимид.
и ароматический полипиромеллитимид — нагревались в одинако-
вых условиях на одних и тех же термовесах. Каждый полимер
имеет свою характерную кривую потери массы. Информация, по-
лученная в подобных исследованиях, может служить руководством
для дальнейшего исследования механизмов разложения.
3. СОДЕРЖАНИЕ ДОБАВОК
Кроме исследований термической стабильности, термогравимет-
рию можно также применять для определения содержания органи-
ческих или неорганических добавок в полимерных системах.
Определение содержания пластификатора в полимерах обычными
методами представляет собой трудоемкий процесс, в то время как
термогравиметрия позволяет во многих случаях его ускорить [53].
Это иллюстрируется на фиг. 4.17, где приведены ТГ-кривые не-
пластифицированного поливинилбутираля, а также поливинил-
бутираля, содержащего 31 % пластификатора, и того же пластика
134
Глава 4
Фиг. 4.17. Определение содержания
пластификатора в поливииилбутиралс
(ПВБ) методом термогравиметрии [53].
1 — ПВБ; 2 — ПВБ с экстрагированным пласти-
фикатором; 3 — ПВБ с пластификатором.
после экстракции пласти-
фикатора, по-видимому, с
помощью н-гексана. Оче-
видно, первоначальные по-
тери массы происходят
вследствие испарения пла-
стификатора и могут слу-
жить мерой концентрации
последнего. Точность этих
измерений можно повысить
путем использования низ-
ких скоростей нагревания
или изотермической тер-
могравиметрии.
Лайт и др. [54] с по-
мощью метода термогра-
виметрии определили со-
держание политетрафтор-
этилена (тефлона) в его сме-
си с наполнителем — дву-
окисью кремния. Полимер
полностью испарялся при
нагревании в атмосфере воздуха до температуры 600°С; при
нагревании в атмосфере инертных газов при этой температуре по-
лимер^испарялся частично.
4. СОСТАВ СМЕСЕЙ ПОЛИМЕРОВ И СОПОЛИМЕРОВ
Многочисленные термогравиметрические исследования были по-
священы анализу сополимеров. В общем случае термическая ста-
бильность сополимеров заключена между значениями термической
стабильности соответствующих гомополимеров и изменяется в соот-
ветствии с изменением состава сополимера [53]. При термическом
разложении сополимеров этилена и винилацетата на начальной
стадии происходит быстрое выделение уксусной кислоты. Только
при более высоких температурах (в инертной атмосфере) начинается
разложение оставшихся углеводородных сегментов. Типичная ТГ-
кривая для сополимера этилена и винилацетата приведена на
фиг. 4.18. Состав сополимера можно оценить по начальным поте-
рям массы. Термогравиметрия обеспечивает одновременно точность
и быстроту оценки, имея в этом отношении преимущества перед хи-
мическим методом, методами инфракрасной спектроскопии и ядер-
ного магнитного резонанса. Типичные результаты термограви-
метрического анализа шести образцов сополимеров, содержащих
4,3—31,1% винилацетата, приведены в табл. 4.4. Результаты срав-
Применение термогравиметрии
135
Фиг. 4.18. ТГ-кривая для сополимера этилена и винилацетата |53].
Масса образца 100 мг; скорость нагревания 5 град/мин в атмосфере азота.
ниваются сданными, полученными химическим методом— омы-
лением винилацетата [53].
Таблица 4.4
Сравнение результатов термогравнметрического
и химического анализов сополимера этилена и винилацетата [53]
Содержание винилацетата (химический метод), % Потерн массы за счет выделения уксусной кислоты, % Содержание винил- ацетата (ТГ-метод), % Расхождение в результатах, %
4,3 3,2 4,6 0,3
8,3 5,8 8,3 0,0
11,2 7,6 10,9 0,3
14,9 10,2 14,6 0,3
27,1 18,9 27,1 0,0
31,1 21,7 31,1 0,0
5. ДРУГИЕ ПРИМЕНЕНИЯ
Определение термостойкости эмали обмотки магнита стандарт-
ным методом ASTM D-2307 является очень трудоемкой операцией,
требующей нагревания ряда образцов проволоки до нескольких
значений температур и периодического испытания каждого образ-
ца на электрический пробой. Дэвид [55] обнаружил взаимосвязь
между температурой первого изгиба ТГ-кривой (экстраполиро-
ванной до скорости нагревания, близкой к нулю) обмотки магнита
и параметром Т2оооо проволоки. Браун и др. [56] установили
взаимосвязь между ТГ- и ДТА-данными, с одной стороны, и
Тгоооо —с Другой, для 15 типов выпускаемых промышленностью
136
Глава 4
обмоток магнитов. Образцы обмоток нагревались в динамической
атмосфере кислорода. Температура, при которой масса эмали
уменьшалась на 5%, оказывалась достаточно близкой к величине
^2о ооо» определенной стандартным методом. Определялась также
кинетика разложения [57].
Д. ПРИМЕНЕНИЕ В НЕОРГАНИЧЕСКОЙ ХИМИИ
Достоинства и универсальность термогравиметрии как анали-
тического метода исследования неорганических соединений и ма-
териалов продемонстрированы в работе Уильямса [59]. Термогра-
Фиг. 4.19. ТГ-кривая окисления NiSa для определения содержания серы [59].
Масса образца 10,41 мг; состав атмосферы О2—Аг; остаточная масса 5 мг
виметрия оказывается особенно необходимой при анализе образцов
в виде монокристаллов, имеющих слишком малые размеры для
изучения их обычными методами. Такие образцы обычно получают
в результате различных реакций, идущих с низкими выходами,
или как часть многофазного продукта малого объема, физически
отделяющаяся от смеси. В работе [59] показано, что содержание
металла и серы в монокристаллах можно определить при иссле-
довании образцов массой ~10 мг. Содержание кислорода опреде-
лялось также путем окисления SrCrOs до хромата стронция SrCrO4,
в то время как ортованадат хрома CrVO4 восстанавливался
до метаванадата хрома CrVO3.
В качестве примера определения содержания серы на фиг. 4.19
приведена ТГ-кривая окисления сульфида никеля NiS2 до окиси
никеля NiO. Сера образуется в двух интервалах температуры:
390—490 и 690—785° С. После охлаждения системы до ~100°С
Применение термогравиметрии
137
вводилась водородно-аргоновая атмосфера и NiO восстанавливалась
до металлического никеля. Суммарные потери массы серы состави-
ли 52,1% (расчетное значение 52,2%).
Результаты определения содержания кислорода в различных
соединениях представлены в табл. 4.5, а в табл. 4.6 суммированы
данные по превращению сульфидов редкоземельных металлов в
оксисульфиды.
Таблица 4.5
Результаты определения содержания кислорода
методом термогравиметрии [59]
Исходное соединение Атмосфера Содержание кислорода, (ТГ-метод), % Результаты расчета, % Температура реакции, °C
CrVO4 (орто) . . Сг2О6 ...... ТезС>4 SrCrO., рякя К5 № 1111 >>>> ** «Ч «Ч «ч 9,5 17,3 27,7 8,3 9,6 17,4 27,6 8,5 515—630 340—430 320—520 375—750
Таблица 4.6
Превращение сульфидов редкоземельных металлов
в оксисульфиды по данным термогравиметрии [59]
Исходное вещество Полученный продукт Потери массы '), %
расчет эксперимент
Nd2S3 Nd2O2S 8,4 8,3
a-Dy2S3 Dy2O2S 7,6 7,5
Er2S3 Er2O2S 7,5 7,3
Yb2S3 Yb2O3S 7,3 6,9
Pr2S3 Pr2O2S 8,5 8,5
Ho2S3 Ho2O2S 7,5 7,1
Dy2S, Dy2O2S 7,6 7,4
Ce2O2S 8,5 8,5
Y2S3 Y2O2S H,7 11,3
La2S3 La2O2S 8,6 8,4
Tm2S3 Tm2O2S 7,4 6,9
Удаляются 2 атома серн, присоединяются 2 атома кислорода.
Такой подход был использован Видеманном [60] для определе-
ния содержания титана в образце карбида титана нестехиометри-
ческого состава. Образец нагревается в атмосфере газообразного
138
Глава 4
хлора до 975е С; при этом образуется тетрахлорид титана, который
испаряется с образованием углеродного остатка. Затем путем на-
гревания образца в атмосфере воздуха в интервале температур
475—600° С определяется количество углерода в остатке. Эти про-
цессы представлены ТГ-кривой на фиг. 4.20.
Фиг. 4.20. Определение содержания титана и углерода ТГ-методом в образ-
це карбида титана нестехиометрического состава [60].
С помощью дериватографа исследовались термические свойства
чистых реактивов (ч.д.а.), предназначенных для химического ана-
лиза. Изучались соли натрия [61 ], рубидия [62 ], цезия [63 ] и ам-
мония [64]. Термогравиметрия применялась также для исследо-
вания высокотемпературных процессов плавления [65].
Е. ОПРЕДЕЛЕНИЕ ДАВЛЕНИЯ ПАРА
И ИССЛЕДОВАНИЕ ВОЗГОНКИ
С помощью метода термогравиметрии удобно определять давле-
ние пара или условия возгонки органических соединений. Эшкрофт
[66 ] определил энтальпию возгонки ряда органических соедине-
ний и неорганических хелатов, используя уравнение Лэнгмюра:
т = а
М М/2
------ р
ZrtRT )
(4-12)
Применение термогравиметрии
139
где М — молярная масса газообразных продуктов возгонки; Т —
температура в кельвинах; а — коэффициент возгонки (предполага-
емый равным единице). Применяя уравнение Клаузиуса — Кла-
пейрона к процессу возгонки, при котором площадь поверхности
образца остается постоянной, находим, что кривая зависимости
lg[m(T)'A1 от 103 /Т имеет наклон, равный —0,0522 ДЯсубл, по
которому можно вычислить ДЯсубл в кДж/моль. Скорость потерь
массы порошкообразных образцов весом 50—100 мг, помещенных
в платиновую лодочку, регистрировались при пяти—шести зна-
чениях температур с интервалом 20—30°. При выборе температуры
обеспечивающей низкие скорости потери массы и небольшие (<2%)
суммарные потери массы, были получены линейные зависимости.
Наклоны соответствующих прямых, вычисленные по методу наи-
меньших квадратов, воспроизводились с точностью —5%. Зна-
чения энтальпии возгонки, полученные этим методом, приведены в
табл. 4.7. Обнаружено хорошее совпадение между двумя группами
Таблица 4.7
Энтальпия возгонки различных соединений [66]
Соединение Л^субл* кДм< /моль Интервал температур,К ДНсубл <и3 ДРУГИХ источников), кДж/моль Интервал температур (из других источников), К
Антрахинон 105,9 335—356 112,0 298
127 470—590
106,2 428
1,4-Дигидроантрахинон 94,5 324—351 103,6 408
1,8-Дигидроантрахинон 96,5 335—356 105,9 405
1 - Аминоантрахинон 90,9 361—386 113,8 461
Бензойная кислота 89,1 299—329 91,5 343—387
100 420—480
Тимол 69,0 229—312 91,3 273—313
67 420—480
Sc (С6Н7О2)3 97,2 335—361 169 445—555
99,6 389
49,8 377—387
Ст (С6Н7О2)3 85,9 335—356 125 490—595
110,9 397
27,8 389—397
Мп (С6Н7О2)3 117,31) 335—356 77,8 383—391
Fe(C5H7O2)3 114,9 335—356 116 452—535
99,0 391
65,3 378—388
23,4 393
Со (СЬН7О2)3 86,3 335- 361 74,9 378—393
Си (С6Н7О2)2 106,1 335—361 57,3 475—560
') Вероятно, наибольшее значение.
140
Глава 4
значений для первых пяти соединений, хотя можно было ожидать
некоторого' расхождения из-за различий температурных интервалов
измерений.
Определению давления пара различных соединений посвящен
обзор Видеманна [67]. Рассмотрено определение давления пара с
Фнг. 4.21. Кривые давления
с использованием
пара, полученные методом термогравиметрии
эффузионного метода Кнудсена [67].
помощью методов термогравиметрии на основе эффузионного мето-
да Кнудсена. Держатель образца был описан в гл. 3 (фиг. 3.6).
Для некоторых измерений использовался сосуд из пирекса диамет-
ром ~15 мм. Исследовались четыре органических соединения —
пара-хлорфенил-N", N'-диметилмочевина (гербицид «монурон»),
пара-фенацетин, антрацен и бензойная кислота — в интервале
температур 250—400 К. На фиг. 4.21 приведены кривые давления
пара этих соединений в интервале давлений 133—1,33 • 10-4 Па.
Величины АДсубл, рассчитанные по этим кривым, составляют: для
монурона 104,7 кДж/моль, пара-фенацетина 115,6, антрацена 84,1
и для бензойной кислоты 86,7 кДж/моль.
Адони [68 ] разработал метод определения давления пара с по-
мощью дериватографа. Этот метод аналогичен рассмотренному
Эшкрофтом [66 J.
Применение термогравиметрии
141
Ж. ДРУГИЕ ПРИМЕНЕНИЯ
Вероятно, наиболее важными для взрывчатых веществ и твер-
дых ракетных топлив являются их характеристики в процессе хра-
нения и эксплуатации: термическая стабильность, чувствитель-
ность к ударам и трению, мощность взрыва, скорость горения или
детонации и т. д. Разработанные Мэйкоком [69 ] методы терми-
ческого анализа этих веществ являются многообещающими в плане
получения информации об упомянутых выше и других свойствах
взрывчатых веществ и твердых ракетных топлив. Термические
свойства определялись в основном с помощью методов термограви-
метрии, дифференциального термического анализа, а также изо-
термического (или адиабатического) разложения при постоянном
объеме. Физические процессы в псевдостабильных материалах,
наблюдаемые при исследованиях методом ДТА, часто можно объ-
яснить только с помощью данных термогравиметрии или по выде-
лению газа. В табл. 4.8 приведены физические процессы, протекаю-
щие в таких материалах, и методы их исследования.
Таблица 4.8
Физические процессы и методы исследования1) [69]
Физический процесс ТГ-метод ДТА-метод Скорость термического разложения
увеличение массы уменьше- ние массы эндотер- мический процесс экзотер- мический процесс
Плавление +
Возгонка + + +
Кипение + + +
Фазовое превращение — + + +
Де.сорбция + + +
Адсорбция + + +
Реакция в твердой фазе — + + —
Разложение ...... + 4- 4- +
Взрыв + + +
Окисление, восстановле-
кие + + + + +
*) + процесс исследуется указанным методом; — процесс не исследуется указанным ме-
тодом.
Непростой задачей является правильность интерпретации боль-
шого числа экспериментальных данных, полученных указанными
методами, поскольку от этого зависит выбор условий применения
данного материала. Рассмотренные методы исследования, очевид-
142
Глава 4
но, могут найти применение как при контроле качества, так и при
решении научных проблем, связанных с псевдостабильными ма-
териалами.
Фиг. 4.22. ТГ- и ДТА-крнвые для гексогена в динамической атмосфере
гелия [69].
Масса образца 15 мг; скорость нагревания 6 град/мин.
На фиг. 4.22 приведены типичные ТГ- и ДТА-кривые для взрыв-
чатого вещества (З-НМХ (гексогена). Как видно из ТГ-кривой, изо-
термические исследования массы можно производить в интервале
температур 220—280° С. При верхнем пределе температуры можно
без труда получить данные о кинетике процесса.
ЛИТЕРАТУРА
1. Gordon S., Campbell С., Anal. Chem., 32, 271R (1960).
2. Duval C„ Anal. Chem., 23, 1271 (1951).
3. Coats A. W., Redfern J. P., Analyst, 88, 906 (1963).
4. Lukaszewski G. M., Redfern J. P., Lab. Pract., 10, 552 (1961).
5. Murphy С. B., Anal. Chem., 42, 268R (1970).
6. Murphy С. B., Anal. Chem., 44, 513R (1972).
7. Toursei W., Z. fur Chemie, 7, 265 (1967).
8. Duval C., Inorganic Thermogravimetric Analysis, Elsevier, Amsterdam
1953.
9. Duval C., Inorganic Thermogravimetric Analysis, 2nd. ed., Elsevier,
Amsterdam, 1963.
10. Wendlandt W. W., Smith J. P., Thermal Properties of Transition Metal
Ammine Complexes, Elsevier, Amsterdam, 1967.
11. Wendlandt W. W., Flaschka H. A., Barnard J. A., Chelates in Analytical
Chemistry, Vol. 1, Marcel-Dekker, N. Y., 1967, Ch. 5.
12. Anderson H. C., Techniques and Methods of Polymer Evaluation, Sla-
de P. E., Jenkins L. T., eds., Marcel-Dekker, N. Y., 1966, Ch. 3.
13. Doyle C. D., Techniques and Methods of Polymer Evaluation, Slade P. E,,
Jenkins L. T., eds., Marcel-Dekker, N. Y., 1966, Ch. 4.
Применение термогравиметрии
143
14. Barrall Е. М., Guide to Modern Methods of Instrumental Analysis,
Gouw T. H., ed., Wiley-Interscience, N. Y., 1972, Ch. 12.
15. Liptay G., ed., Atlas of Thermoanalytical Curves, Vol. 1, Akademiai
Kiado, Budapest, 1971.
16. Reich L., Stivala S. S., Elements of Polymer Degradation, McGraw-Hill,
N. Y„ 1971.
17. Палей П. H., Сентюрин И. Г., Скляренко И. С., Журнал аналитической
химии, 12, 329 (1957).
18. Duval С., Inorganic Thermogravimetric Analysis, 2nd. ed., Elsevier, Am-
sterdam, 1963, p. 84.
19. Duval C., Inogranic Thermogravimetric Analysis, 2nd ed., Elsevier, Am-
sterdam, 1963, Ch. VIII.
20. Peltier S., Duval C., Anal. Chim. Acta, 1, 345 (1947).
21. Simmons E. L., Newkirk A. E., Taianta, 11 549 (1964).
22. Duval C., Anal. Chim. Acta, 13, 32 (1955).
23. Newkirk A. E., Aliferis I., Anal. Chem., 30, 982 (1958).
24. Dupuis T., Duval C., Anal. Chim. Acta, 3, 191 (1949).
25. Erdey L., Paulik F., Acta Chim. Sci. Hung., 7, 45 (1955).
26. Milner О. I., Gordon L., Taianta, 4, 115 (1960).
27. Duval C., Inorganic Thermogravimetric Analysis, Elsevier, Amsterdam,
1953, pp. 227-228.
28. Griffith E. J., Anal. Chem., 29, 198 (1957).
29. Hoffman I., Schnitzer M., Wright J. R., Chem. Ind. (London), 261 (1958).
30. Hoffman I. M., Schnitzer M., Wright J. R., Anal. Chem., 31, 440 (1959).
31. Mulley V. J., Cavendish C. D_, Analyst, 95, 304 (1970).
32. McCaleb S. B., Quest (Sun Oil Co.), 24 (1966).
33. Paulik F., Gal S., Erdey L., Anal. Chim. Acta, 29, 381 (1963).
34. Biffen F. M., Anal. Chem., 28, 1133 (1956).
35. Ramachandran V. S., Thermochim. Acta, 2, 41 (1971).
36. Erdey L., Paulik F., Svehla G., Liptay G., Z. Anal. Chem., 182, 329 (1961).
37. Erdey L., Paulik F., Svehla G., Liptay G., Taianta, 9, 489 (1962).
38. Dupuis T., Duval C., Mikrochim. Acta, 186 (1958).
39. Paulik F., Paulik J., Erdey L., Anal. Chim. Acta, 44, 153 (1969).
40. Dupuis T., Duval C., Chim. Anal., 33, 189 (1951).
41. Duval C., Anal. Chim. Acta, 13, 32 (1955).
42. Belcher R., Erdey L., Paulik F., Liptay G., Taianta, 5, 53 (1960).
43. Newkirk A. E., Laware R., Taianta, 9, 169 (1962).
44. Dupuis T., Duval C., Anal. Chim. Acta, 4, 256 (1950).
45. Wendlandt W. W., Anal. Chim. Acta, 20, 267 (1959).
46. Duval C., Inorganic Thermogravimetric Analysis, Elsevier, Amsterdam,
1953, pp. 130, 132.
47. Duval C., Inorganic Thermogravimetric Analysis, 2nd ed., Elsevier,
Amsterdam, 1963, pp. 245—246.
48. Wendlandt W. W., Brabson J. A., Anal. Chem., 30, 61 (1958).
49. Brabson J. A., Edwards O. W., Anal. Chem., 28, 1485 (1956).
50. Fock J., Some Applications of Thermal Analysis, Mettler Instrument Corp.,
1968.
51. Doyle C. D., Anal. Chem., 33, 77 (1961).
52. Newkirk A. E., Proc, at the First Toronto Symp. on Thermal Analysis,
McAdie H. G., ed., Chemical Inst, of Canada, Toronto, 1965, p. 33.
53. Chiu J., Thermoanalysis of Fiber and Fiber-Forming Polymers, Schwen-
ker R. F., ed., Interscience, N. Y., 1966, p. 25.
54. Light T. S., Fitzpatrick L. F., Phaneuf J. P., Anal. Chem., 37, 79 (1965).
55. David D. J., Insulation, 13, 38 (1967).
56. Brown G. P., HaarrD. T., Metlay M., Proc, of the 9th Electronics Inlation
Conf. IEEE 32C3-23, 1965, p. 160.
57. Brown G. P., Haarr D. T., Metlay M., Thermochim. Acta, 1, 441 (1970).
58. DuPont Application Brief, № 18, Feb. 1, 1968.
144
Глава 4
v59. Williams H. W., Thermochim. Acta, 1, 253 (1970).
60. Wiedemann H. G., Mettler Technique Series, Tech. Bull. T-103.
61. Erdey L., Simon J., Gal S., Liptay G., Taianta, 13, 67 (1966).
62. Erdey L., Liptay G., Gal S., Taianta, 12, 883 (1965).
63. Erdey L., Liptay G., Gal S., Taianta, 12, 257 (1965).
64. Erdey L., Liptay G., Gal S., Taianta, 11, 913 (1964).
65. Erdey L., Gal S., Talanta, 10, 23 (1963).
66. Ashcroft S. J., Thermochim. Acta, 2, 512 (1971).
67. Wiedemann H. G., Thermochim. Acta, 3, 355 (1972).
68. Adonyi Z., Periodica Polytech., 10, 325 (1966).
69. Maycock J. N., Thermochim. Acta, 1, 389 (1970).
Глава 5
ДИФФЕРЕНЦИАЛЬНЫЙ ТЕРМИЧЕСКИЙ АНАЛИЗ
И ДИФФЕРЕНЦИАЛЬНАЯ СКАНИРУЮЩАЯ
КАЛОРИМЕТРИЯ
А. ДИФФЕРЕНЦИАЛЬНЫЙ ТЕРМИЧЕСКИЙ АНАЛИЗ
1. ВВЕДЕНИЕ
Метод дифференциального термического анализа (ДТА) основан
на сравнении термических свойств образца исследуемого вещества
и термически инертного вещества, принятого в качестве эталона.
Регистрируемым параметром служит разность их температур, из-
меряемая при нагревании или охлаждении образца с постоянной
скоростью, которая может быть представлена в виде функции тем-
пературы образца, эталона или нагревателя. Изменения темпера-
туры образца вызываются физическими переходами или химиче-
скими реакциями, связанными с изменением энтальпии. К ним
относятся: фазовые переходы; плавление; перестройка кристалличе-
ской структуры; кипение, возгонка и испарение; реакции дегидра-
тации, диссоциации и разложения; окисления и восстановления;
разрушение кристаллической решетки и др. Эти превращения со-
провождаются поглощением или выделением тепла. В общем слу-
чае фазовые переходы, дегидратация, восстановление и некоторые
реакции разложения сопровождаются эндотермическими эффектами,
а кристаллизация, окисление и отдельные процессы разложения—
экзотермическими эффектами.
Если обозначить температуры образца и эталона соответственно
Ts и Тг, то регистрируемым параметром в дифференциальном ме-
тоде, схематически изображенном на фиг. 5.1, будет разность тем-
ператур Ts—Тг. Название метода «дифференциальный термиче-
ский анализ» несколько неудачно, так как собственно анализ, как
это понимается в исследованиях с использованием любых других
аналитических методов, может и не производиться. Более точно
содержание метода отражало бы название дифференциальная тер-
мометрия. В термическом анализе (правильнее — термометрии)
производится нагревание или охлаждение образца, а измеряемым
параметром является его температура Ts, которая регистрируется
в функции времени (гл. 10). Небольшие изменения температуры
образца с помощью этого метода обычно не удается обнаружить.
В дифференциальном методе регистрирующие термопары соединены
навстречу друг другу, и поэтому даже незначительная разница
между Ts и Тг приводит к появлению разности потенциалов, кото-
6—230
146
Глава 5
рая при соответствующем усилении сигнала может быть определена.
Повышенная чувствительнось, характерная для дифференциального
метода, позволяет исследовать образцы малого веса (вплоть до не-
скольких миллиграммов), что весьма важно.
Фиг. 5.1. Схема метода ДТА.
На фиг. 5.2 схематически изображены экспериментальные кри-
вые, полученные двумя сравниваемыми методами. В термическом
анализе образец нагревается с постоянной скоростью и температура
образца регистрируется в функции времени (фиг. 5.2, а и б). На
фиг. 5.2, а температура образца при нагревании возрастает по ли-
нейному закону. На фиг. 5.2, б наблюдаются отклонения от линей-
ной зависимости, которые обусловлены экзо- и эндотермическими
превращениями, происходящими в образце. Температура Th при
которой начинается отклонение, называется наблюдаемой темпе-
ратурой начала превращения (реакции). Превращение заканчи-
вается при температуре Тf. При дальнейшем нагревании изменение
температуры образца стабилизируется (продолжение линейной за-
висимости от времени). На фиг. 5.2, в представлена разность темпе-
ратур Ts—Тг в функции температуры системы Т (образца, эталона
или нагревателя). При Tt кривая отклоняется от горизонтального
уровня с образованием пиков, направленных в зависимости от знака
изменения энтальпии вверх или вниз. Температура окончания
превращения Тf в этом случае не соответствует ни температуре
максимума, ни температуре минимума пика, а расположена на его
высокотемпературной ветви. Точное положение Тf зависит от ряда
факторов, связанных с измерительными приборами. Таким образом,
с помощью дифференциального метода легко обнаруживаются не-
большие изменения температуры, а площадь пика кривой пропор-
циональна изменению энтальпии (±А//) и массе образца.
ДТА и ДСК
147
Типичная кривая ДТА с четырьмя видами переходов показана
на фиг. 5.3. По числу, форме и положению различных экзо- и эндо-
термических пиков относительно шкалы температур производится
качественная идентификация исследуемого вещества. Поскольку
Фиг. 5.2. Сравнение методов термического анализа и дифференциального
термического анализа.
площадь пика пропорциональна изменению теплосодержания (эн-
тальпии), метод ДТА может использоваться для полуколичествен-
ного, а в некоторых случаях количественного определения теплоты
реакции. Теплота реакции пропорциональна количеству реагирую-
щего вещества, и поэтому с помощью метода ДТА можно количест-
венно оценить массу присутствующего вещества. Итак, рассматри-
ваемый метод находит широкое применение в качественных и полу-
количественных исследованиях органических и неорганических
соединений, глин, металлов, минералов, жиров, масел, нефти, поли-
мерных материалов, каменного угля, сланцев, древесины и других
* 6*
148
Глава 5
веществ. Метод ДТА можно также использовать для определения
радиационных повреждений полимерных материалов, количества
запасенной энергии радиации, теплоты адсорбции, эффективности
катализаторов, теплоты полимеризации и др. С помощью этого
метода можно производить количественное определение концент-
рации реагирующего компонента смеси или энтальпии химических
и физических превращений.
Фнг. 5.3. Типичная кривая ДТА.
I — сдвиг базовой линии, обусловленный переходом второго рода; II— эндотермический
пик, обусловленный плавлением; Ш—эндотермический пик, вызванный реакцией разложе-
ния или диссоциацией; IV — экзотермический пик, обусловленный фазовым изменением в
кристалле.
2. ИСТОРИЧЕСКАЯ СПРАВКА
Необходимо упомянуть об истории создания метода ДТА как
интересного примера подхода к развитию новой экспериментальной
техники с позиций дисциплины, охватывающей ряд наук. Основа-
телем метода считается Ле-Шателье. В работе [1 ], опубликованной
в 1887 г., он использовал термопару для изучения термических
свойств глины и минералов. Термопара заключалась в образец и со-
единялась с гальванометром. Образец нагревался со скоростью
100 град/мин. Зеркало гальванометра освещалось вспышками от
искровых разрядов индукционной катушки, которые следовали с
интервалом 2 с. Отраженный от зеркала свет направлялся на фото-
графическую пластинку. Температура образца фиксировалась на
пластине после проявления как ряд линий, каждая из которых со-
ответствовала вспышке разряда. Для эндотермических превращений
было характерно близкое расположение линий, в то время как
экзотермические превращения вызывали увеличение расстояний
ДТА и ДСК
149
между линиями. Согласно [2 ], Ле-Шателье положил начало терми-
ческому анализу, но не ДТА.
Ле-Шателье и другие исследователи (Эшли [3], Волин [4], Рике
[5 ], Воллек [6 ], Мэллор и Хольдкрофт [7 ]) изучали термические
превращения, происходившие в веществе при нагревании, путем
регистрации с помощью термопары температуры образца в функции
времени. Изломы наблюдаемых кривых нагрева указывали на де-
гидратацию, разложение, фазовые переходы и другие реакции,
вызываемые нагреванием. Этот метод оказался мало чувствитель-
ным к небольшим тепловым эффектам и зависел от неблагоприят-
ного воздействия изменения скорости нагревания, параметров за-
писывающего устройства и других факторов.
В 1899 г. Робертс-Остен [8] предложил использовать двухтермо-
парный метод. Одна темопара размещалась в образце, а другая в
эталонном блоке, расположенном в печи. Регистрировалась разни-
ца температур в функции времени или температуры. Этот метод
оказался более чувствительным к малым изменениям температуры
в образце, чем однотермопарный. Бургесс [9] в 1908 г. обсудил до-
стоинства одно- и двухтермопарного методов для получения кри-
вых охлаждения. Данные представлялись графически в различных
функциональных зависимостях: температуры образца Т от времени
разницы в температурах образца Т и нейтрального тела Т'(Т—
— Т' = АТ) в зависимости от t\ Т ~ f(dT/dt) и7' = f^dtldT). Эти
функциональные зависимости были интерпретированы для трех
типов превращений: а) для условий, при которых вещество сохра-
няет постоянную температуру; б) для условий медленного охлаж-
дения вещества с непостоянной скоростью на некотором участке
превращения и в) для условий возрастания температуры вещества
в начале превращения. Бургесс рассмотрел также различные виды
экспериментальных установок и регистрирующих систем, извест-
ных в то время. Им были приведены также уравнения для расчета
теплоты наблюдаемых превращений.
Возможно, впервые двухтермопарный метод был использован
для изучения химических превращений Холдсвортом и Коббом [11 ],
хотя Феннер [12 ] еще раньше исследовал фазовые переходы в сили-
катных минералах. Холдсворт и Кобб изучали поведение огнеупор-
ных глин и боксита при нагревании. С момента появления этой пер-
вой работы в литературе появилось большое число публикаций
по исследованию термического разложения различных глин и мине-
ралов, в том числе работы Нортона [17], Грима [18], Беркельхаме-
ра [13], Керра и Кульпа [14], Кауфмана и Диллинга [15], Фолд-
вари-Фогла [16] и Маккензи [19]. ДТА в настоящее время являет-
ся общепринятым методом качественного и полуколичественного
исследования глин и минералов.
Первые успехи минералогов и металлургов в использовании
метода ДТА поддерживали интерес к нему как в экспериментальном,
150
Глава 5
так и в теоретическом плане. Однако возрождение метода, его
развитие и применение в области химии относится к 50-м годам,
начиная с работ Стоуна [20 ], Борхарда и Даниельса [21, 22 ]. Стоун
разработал первый высококачественный промышленный прибор
ДТА, что послужило стимулом к дальнейшим разработкам в этой
новой области техники. Борхард в докторской диссертации исполь-
зовал дифференциальный термический анализ для решения задач
в областях неорганической и физической химии. Его работа по иссле-
дованию кинетики гомогенной реакции с помощью ДТА является
классической.
Наличие серийной аппаратуры значительно расширило область
применения метода ДТА. В 60-х годах дифференциальный термиче-
ский анализ стал широко применяться в химии полимеров. Даль-
нейшее развитие метода привело к созданию в 1963 г. дифференци-
альной сканирующей калориметрии (ДСК). ДТА и ДСК стали уни-
кальными методами изучения термических свойств полимеров. В на-
стоящее время многие аспекты аналитического применения ДТА
и ДСК относятся к этой области.
Быстрое развитие и применение указанных методов вызвало
целый поток публикаций на данную тему. Здесь нет возможности
перечислить все работы, посвященные методу ДТА и его примене-
нию в области химии. Следует в первую очередь упомянуть о перио-
дических обзорах Мэрфи [25 J, которые выходили, начиная с 1958 г.,
через каждые два года; последний обзор вышел в 1972 г. [26]. В по-
следнее время опубликован ряд книг или отдельных глав, посвя-
щенных методу ДТА, в том числе Смозерса и Чианга [27, 28 ], Уэнд-
ландта [23, 24], Гарна [29], Маккензи [30], Гордона и Кэмпбелла
[32, 33], Киссинжера и Ньюмена [31 ], Бэрролла и Джонсона [34 ],
Дэвида [35], Бэрролла [36], Шульце [37], Рамачандрана [38],
Вундерлиха [39 ], Портера и Джонсона (ред.) [40, 41 ], а также
Швенкера и Гарна (ред.) [42].
3. ТЕОРИЯ
Существует ряд теорий, касающихся интерпретации кривых
ДТА. Все они различным образом устанавливают соотношения
между площадью пика дифференциальной кривой и различными
параметрами прибора и образца. Уравнения, описывающие эти
параметры, выведены на основе обычных соотношений теплопере-
дачи, геометрии образца и его держателя. Рассмотрение указанных
теорий выходит за рамки обсуждаемых вопросов, и поэтому в даль-
нейшем будут использованы только окончательные математические
выражения.
В теории Спейла и др. [10], усовершенствованной Керром и
Кульпом [14 ], площадь, ограниченная дифференциальной кривой,
ДТА и ДСК
151
определяется по формуле
= { ATdt, (5.1)
gk .)
где т — масса реагирующего вещества (образца); ЛЯ — теплота
реакции; g — геометрический фактор, постоянный для данного
прибора; k — коэффициент теплопроводности образца; А "Г — диф-
ференциальная температура (разность температур образца и эта-
лона); и /2 — пределы интегрирования. Это простое выражение
связывает с помощью констант пропорциональности g и k теплоту
реакции протекающей в образце, с площадью пика дифференциаль-
ной кривой. При этом пренебрегают членами, содержащими произ-
водные, и градиентами температуры в образце и предполагают, что
площадь пика кривой не зависит от удельной теплоемкости образ-
ца. Поэтому выражение (5.1) является приближенным.
Волд [43 ] вывел соотношение
-т? = (5-2)
Cs at at
где Cs — теплоемкость ячейки с ее содержимым; f — доля образца,
прореагировавшего за время /; у — дифференциальная температу-
ра; ys — равновесное значение дифференциальной температуры,
которое достигается за достаточно длительное время от начала
процесса ( у — у^ при t = t^\ А — константа.
Эта теория имеет следующие недостатки; предположение о по-
стоянной величине теплоемкости образца и предположение о по-
стоянстве температуры образца в любой момент времени по всему
объему. Общая теплоемкость образца складывается из теплоемкос-
ти ячейки и теплоемкости прореагировавшей и непрореагировавшей
частей образца. Эти составляющие теплоемкости изменяются в ходе
реакции. Тогда, если увеличить теплоемкость ячейки, изменение
теплоемкости образца вызовет незначительное изменение общей
теплоемкости, но при этом чувствительность уменьшится. Неодно-
родность температуры образца не является большим недостатком
метода: она влияет в большей мере на температуру превращения,
чем на тепловые эффекты. Путем уменьшения скорости нагревания,
измерения температуры на поверхности образца вблизи стенки на-
гревателя и использования различных экстраполяционных опера-
ций добиваются уменьшения ошибки, сводя ее к минимуму, но не
исключают ее полностью [43].
Для блока, выполненного из металла с предельно высоким коэф-
фициентом теплопроводности (например, никеля), в котором уста-
новлен держатель образца, имеющий форму цилиндра, Бойерсма
152
Глава 5
[44 ] нашел, что площадь пика кривой равна
f ttfdt = >
J 4Х
(5.3)
где tl и 4 — времена, соответствующие началу и концу пика; q —
теплота превращения на единицу объема; АТ — дифференциаль-
ная температура; а — радиус полости, заполненной образцом;
>. — коэффициент теплопроводности образца.
Для металлического контейнера сферической формы получено
выражение
С ttfdt = , (5.4)
J 6Х
ц
а для плоской пластины
[ATdf = ^-- (5.5)
J 2Х
Наконец, для бесконечно большого керамического блока не уда-
лось получить решений в конечном виде для одно- и двумерного
случаев, однако имеется решение для держателя сферической фор-
мы
[ + (5.6)
J 6 \ кс \s ]
ti
где Гс — коэффициент теплопроводности керамического матери-
ала и — коэффициент теплопроводности образца.
В приведенных выше выражениях, применяемых к обычным при-
борам ДТА, образец рассматривается с двух совершенно различных
позиций как генератор тепла и как терморезистор, с помощью кото-
рого измеряется дифференциальная температура. Разделяя эти две
функции, Бойерсма [44 I рекомендовал использовать металлические
чашки для образца и эталона, в которых измерялась бы разность
температур поверхностей образца и эталона. Тогда соотношение
между площадью пика кривой и теплотой реакции будет следующим:
^2
J \Tdt = . (5.7)
ц
где т — масса образца и G — коэффициент теплопередачи между
никелевой чашкой и окружающей средой. Хотя эксперименталь-
ные данные автором не были представлены, Бойерсма [44 ] конста-
тировал, что измерения, выполненные для случая дегидратации
ДТА и ДСК
153
CuSO4-5H2O, количественно подтверждают справедливость соот-
ношения (5.7). При этом использовались образцы с разной плот-
ностью упаковки частиц.
Лукашевский [45—55 1 рассмотрел комплекс проблем теплопере-
дачи в различных типах систем ДТА. Эти проблемы в целом могут
быть сведены к трем категориям [531:
а) теплопередача между источником тепла (стенкой печи или
нагревателя) и блоком калориметра путем теплопроводности, кон-
векции или излучения; .
б) теплопроводность между блоком калориметра и некоторой
средой внутри него (эталон или образец);
в) активный образец в системе может периодически подвергать-
ся превращениям, сопровождающимся поглощением тепла (эндо-
термический эффект) или его выделением (экзотермический эффект),
в зависимости от времени, температуры и положения; это усложняет
процесс теплопередачи между образцом и калориметром в условиях
быстрого изменения физических свойств образца.
Основные проблемы, относящиеся к двум последним пунктам,
можно представить математически следующим образом:
= div grad Т± AS(P, t), (5.8)
где Cs и <p5 — соответственно удельная теплоемкость и плотность
образца; член, учитывающий поглощенное или выделенное тепло,
может быть представлен в виде
А(Р, t) = Qb(l — а)", (5.9)
где Q — теплота реакции; b — константа скорости [Zexp(—Е/РГ) 1;
а — доля прореагировавшего образца; п — порядок реакции.
В предположении, что ks не зависит ни от температуры, ни от поло-
жения образца, из уравнения (5.8) следует
(—) = ds\2T ± Л*(Р’ ° , (5.10)
\ dt js sy/ Cs '
где ds — коэффициент температуропроводности образца, a Cs —
теплоемкость единицы объема образца. Для эталона, который не
поглощает и не выделяет тепло [A(P, t) = 0],уравнение (5.10)
имеет вид
(*L\==drfT, (5.11)
\ Ot Jr
где dr — коэффициент температуропроводности эталона.
Условия теплопередачи для системы ДТА, содержащей коль-
цевые термопары, рассмотрены Дэвидом [56, 57 ]. При выводе ма-
тематического выражения для теплоемкости Ср образца (или этало-
154
Глава 5
на) необходимо обратить внимание на два фактора:
влияние системы без образца на дифференциальную термопару
и влияние системы с образцом на дифференциальную термопару.
Теплоемкость держателя образца с образцом, подверженным экзо-
или эндотермическим превращениям, может быть выражена в виде
Ср, sdTz = Ks (Т3 — Т2) dt + dH, (5.12)
а для держателя эталона с эталоном
Cp,rdI\ = Kr(T3 — TJdt, (5.13)
где 1\, Т2 и Т3 — температуры, измеряемые термопарами соответ-
ственно эталона, образца и печи; CPiS и Срл — суммарные тепло-
емкости соответственно держателя образца с образцом и держателя
эталона с эталоном.
Пэкор [58] установил зависимость между площадью пика кри-
вой ДТА и общим количеством выделяемого или поглощаемого теп-
ла для держателя образца блочного типа (фирмы «Дюпон»). Для
эталона, который не участвует в химической реакции,
~^- = V2^ (5-14)
ар ot
где Тт — температура эталона, а аг — коэффициент его температу-
ропроводности. Для образца, в котором может протекать химиче-
ская реакция (или какое-либо другое превращение),
- 5 = <5Л5>
as ot к
где Ts — температура образца; as — коэффициент его температуро-
проводности; X — коэффициент теплопроводности; Q — теплота
перехода на единицу объема. Вводя дифференциальную темпера-
туру АТ, умножая обе части уравнения на dt и интегрируя по
времени в достаточно большом интервале, с тем чтобы всеми пере-
ходными процессами можно было пренебречь, получаем следующее
выражение:
f (- ----L = ATdZ + f • (5.16)
,) \as dt ar dt / J X
tl
Площадь пика кривой ДТА равна площади, заключенной между
кривой и базовой линией. Таким образом, площадь, заключенную
между нулевой и базовой линиями, следует вычесть. Тогда
б б
V2 С Д7Ч/= [ (— dt. (5.17)
.) J \ as dt аг dt /
б б
ДТА и ДСК
155
Вычитая (5.17) из (5.16) и вводя скорость нагревания а = dTrldt,
получим
^2
v2 f (AT — AT1) dt = -5- = v2 —, (5.18)
J Ла
ti
где S — площадь пика кривой. В случае цилиндра бесконечной
длины с граничным условием \2Т — 0 при г = R (т. е. 5 = 0)
и размещения термопары в центре площадь пика кривой равна
с _ a-R2 Q _ a-R2 QwP
4 Л 4 Л
(5.19)
где 7? •— радиус держателя образца; — теплота превращения
на единицу массы; р -— плотность.
Распределение температур в образце и влияние параметров об-
разца на кривую ДТА были детально исследованы Меллингом и др.
[59]. Распределение температуры в образце подчиняется известному
уравнению диффузии
ЗТ — а дд
dt ' К dt
(5.20)
где а — р — плотность; с — удельная теплоемкость; К —
коэффициент теплопроводности; dq/dt скорость выделения тепла
в единице объема. Если образец имеет форму цилиндра с радиусом
г и длиной I, то
d2T 1 dT 1 d-T d2T
dr2 r dr г2 <Э02 dt2
(5.21)
В предположении, что распределение температур по поверхности
образца не зависит от его положения (выполняется на достаточном
удалении от концов цилиндра), уравнение (5.16) преобразуется
к виду
у2Т = — I — (г — 'll. (5.22)
г L dr \ dr /J
Эти уравнения будут использованы в следующем разделе данной
главы.
Общая теория для описания кривых ДТА (а также ДСК- и ТГ-
кривых) была развита Греем [60 ], который применил те же исходные
предположения и уравнения, что и рассмотренные Волдом [43] и
Боерсма [44], а также ряд других. Основные компоненты ячейки,
рассматриваемой в термическом анализе, показаны схематически
на фиг. 5.4. Ячейка состоит из образца с контейнером при темпера-
туре Ts, источника тепловой энергии при температуре Тр и некото-
рого участка с термическим сопротивлением /?, через который теп-
156
Глава 5
ловая энергия подводится или отводится от образца со скоростью
dq/dt. При этом сделаны следующие предположения: а) тем
пература образца Ts постоянна по всему объему и равна темпе
ратуре контейнера; б) суммарная теплоемкость образца с контеп
Фиг. 5.4. Схема ячейки в терми-
ческом анализе [60].
нером Cs и термическое сопро-
тивление R постоянны во всем
исследуемом интервале темпе-
ратур; в) тепло dH/dt, выделя-
емое образцом в единицу време-
ни, положительно, а погло-
щаемое отрицательно.
Пусть в некоторый момент
времени образец выделяет теп-
ло со скоростью dH/dt. Это
тепло может либо увеличивать
температуру образца, либо рас-
сеиваться в окружающей среде.
Так как должен поддерживать-
ся тепловой баланс, сумма этих
двух эффектов должна быть рав-
ной dH/dt. Следовательно,
— = cs . (5.23)
dt s dt dt v ’
Скорость тепловых потерь в окружающую среду определяется
термическим сопротивлением и разностью температур между образ-
цом и средой. Согласно закону Ньютона,
dq Тр Ts
~dt~ ~ R
Подставляя это выражение в уравнение (5.23), получим
dH п dTs . Tp—Ts
— = -------------
dt sdt
(5.24)
(5.25)
В приборах ДТА используются две ячейки, одна из которых
служит эталоном. В эталонной ячейке dH/dt — 0. Выражение для
мгновенной скорости выделения тепла образцом можно записать
аналогично уравнению (5.25):
R (dH/dt) = (Ts — Tr)+R (Cs — Cr) (dTr/dt) +
+ flCs[d(Ts-7;)/df]. (5.26)
Предполагается, что обе ячейки имеют одинаковое термическое
сопротивление R. Теплоемкость эталона Сг не равна теплоемкости
ДТА и ДСК
157
образца Cs. Скорость нагревания эталона dTr/dt равна скорости на-
гревания образца dTs/dt и, следовательно, постоянна.
Величину R(dH/dt) в уравнении (5.26) можно представить для
любого момента времени суммой трех членов в единицах темпера-
туры (фиг. 5.5).
Фиг» 5.5. Определение R(dH/dt) по кривой ДТА [60].
Член I: Ts — Тг — дифференциальная температура, представ-
ленная регистрируемой кривой.
Член II: 7?(CS—Cr) (dTr/dt) — смещение базовой линии от ну-
левого уровня.
Член III: RCs[d(Ts—Tr )/dt] — наклон кривой в любой точке,
умноженный на постоянную RCS. Величину RCS называют постоян-
ной времени системы, и она имеет размерность времени.
В любой точке кривой
R (dH/dt) = 1 + 11+111; (5.27)
если же касательная имеет отрицательный наклон, то
R(dH/dt) = I + II — III. (5.28)
Таким образом, зная RCS, можно графически построить кривую,
отражающую термическое поведение образца в данный момент вре-
мени.
4, ФАКТОРЫ, ВЛИЯЮЩИЕ НА КРИВЫЕ ДТА
Поскольку ДТА представляет собой динамический метод, то
ход экспериментальных кривых может зависеть от большого числа
факторов. Эти факторы аналогичны рассмотренным выше для термо-
гравиметрического метода (гл. 2), но в ДТА их больше и они могут
158
Глава 5
оказывать более сильное влияние на результаты. Если кривая ДТА
используется для качественных целей, то важны форма, положение
и число эндотермических и экзотермических пиков кривой. С изме-
нением условий, например скорости нагревания или атмосферы пе-
чи, будет изменяться положение пиков кривой относительно темпе-
ратурной оси и, возможно, их число. Так, переход от среды N2 к сре-
де О2 может вызвать появление дополнительных экзотермических
Фиг. 5.6. Общий вид кривой ДТА.
пиков. Для количественного анализа представляет большой инте-
рес площадь, ограничиваемая пиком кривой, поэтому должно быть
известным влияние экспериментальных параметров на эту площадь.
Когда метод ДТА используется для измерений удельной теплоем-
кости, важны отклонения базовой линии. В этом случае для полу-
чения точных и воспроизводимых результатов необходимо учиты-
вать такие факторы, как размер частиц образца и инертного напол-
нителя, плотность упаковки частиц; местоположение в ячейке
и т. д.
Обобщенная форма ДТА-кривой, которая будет использоваться
в дальнейшем, представлена на фиг. 5.6. На показанном здесь
эндотермическом пике кривой, различают: А — участок базовой
линии до начала перехода или реакции; В — участок базовой линии
после завершения этого процесса; Тt — наблюдаемую температуру,
соответствующую началу отклонения кривой от базовой линии,
которую прибор может зафиксировать; — наблюдаемую
температуру, соответствующую минимуму пика кривой; Tf — на-
блюдаемую температуру, соответствующую окончанию отклонения
кривой. На оси температур Тп — температура эталона Тп образца
ДТА и ДСК
159
Ts и поверхности нагревателя Те. По оси У отложена дифференци-
альная температура АГ = Ts—Тг.
Как и в термогравиметрическом методе, ДТА-кривые зависят
от двух основных групп переменных: факторов, связанных с изме-
рительным прибором, и характеристик образца.
1) Факторы, связанные с измерительным прибором:
а) атмосфера печи;
б) размер и форма печи;
в) материал держателя образца;
г) геометрия держателя образца;
д) размер провода и изоляции узла термопары;
е) скорость нагревания;
ж) быстродействие регистрирующего устройства;
з) размещение термопары относительно образца.
2) Характеристики образца:
а) размер частиц образца;
б) теплопроводность;
в) теплоемкость;
г) плотность упаковки частиц образца;
д) набухание или усадка образца;
е) масса образца;
ж) влияние инертного наполнителя;
з) степень кристалличности.
а. Скорость нагревания
Несмотря на то что влияние скорости нагревания на температу-
ру АГМИН и площадь пика кривой известно уже в течение ряда
лет, детальное объяснение этого эффекта было дано только в
последнее время. Увеличение скорости нагревания, например от 2
до 20 град/мин, приводит к увеличению температур Т,, &Тмт
и Tf. В то же время влияние скорости нагревания на площадь пика
зависит от используемого параметра температуры Тп. Если Ts—Тг
рассматривать как функцию Ts, то площадь пика кривой будет
пропорциональна скорости нагревания при условии постоянства
последней в течение реакции [59]. Если Т—Тг рассматривать как
функцию времени, то площадь пика не зависит от скорости нагре-
вания. Утверждение Гарна [61] о том, что площадь пика увеличи-
вается с увеличением скорости нагревания из-за теплопереноса,
не подтвердилось [59]. Повышение скорости нагревания также
ухудшает разрешение двух соседних пиков, вследствие чего один
из них становится неразличимым. При очень низких скоростях на-
гревания площади пиков становятся настолько малыми, что на не-
которых приборах (в зависимости от типа держателя образца) эти
пики просто не удается обнаружить.
160
Глава 5
Киссинжер 162, 63] показал, что температура ДТМИН зависит
от скорости нагревания согласно выражению
=_А, (5.29)
Л (1/ЛГ..Ж,) R
где ₽ — скорость нагревания; ДТМИН — температура, соответст-
вующая минимуму пика кривой; Е — энергия активации; /? —
газовая постоянная. График зависимости 1п(₽/Д7^ин) от
должен представлять собой прямую линию с наклоном, равным
Е/R. Зависимость ДТМИП от скорости нагревания была подтверж-
дена Меллингом и др. [59 ], но на практике определение Е/R ока-
залось затруднительным.
Фиг. 5.7. Влияние скорости нагревания на пик температуры [10].
Спейл и др. [10] впервые рассмотрели истинную температуру
пика как точку, в которой скорость подвода тепла равна скорости
ДТА и ДСК
161
поглощения его образцом, и поэтому в соответствии с уравнением
(5.1)
АТ
мин
/ ан \
' at /макС
т
gk
(5.30)
Большая скорость нагревания вызовет увеличение dH/dt и ампли-
туды А7"мин, так как за один и тот же промежуток времени при
большей скорости нагревания поглотится или выделится больше
тепла. Поскольку процесс возвращения к базовой линии является
Фиг. 5.8. Экстраполяция с помощью метода наименьших квадратов темпе-
ратур пиков к нулевой скорости нагревания [64].
а — начало разложения (ф точки, рассчитанные по методу наименьших квадратов; Q экспе-
риментальные точки); б — первый эндотермический минимум ( точки, рассчитанные по ме-
тоду наименьших квадратов; □ экспериментальные точки); в — второй эндотермический
минимум (Д точки, рассчитанные по методу наименьших квадратов; Д экспериментальные
точки).
функцией времени и разности температур, с увеличением скорости
нагревания этот процесс будет происходить при более высоких тем-
пературах. Для иллюстрации Спейл и др. [10] приводили кривые
для каолина (фиг. 5.7). Несмотря на кажущуюся слабую тенденцию
к уменьшению площади пиков с понижением скорости нагревания,
площади пиков кривых различались не более чем на ±3%.
Бэрролл и Роджерс [64] определили влияние скорости нагрева-
ния на значения температур Tit A7MIIH n Tf однозамещенного
карбоната натрия NaHCO3 (фиг. 5.8). Это соединение имеет два
эндотермических пика в интервале температур 110—140° С. Путем
экстраполяции экспериментальных данных к нулевой скорости
162
Глава 5
нагревания с использованием метода наименьших квадратов авторы
получили температуру начала разложения NaHCO3, равную
111,8±0,2°С, и температуры АТМИН первого пика 122,5±]°С и
второго пика 131,7±0,2°С.
Изменение А7МШ1 достаточно мало в том случае, когда диф-
ференциальная температура измерялась в функции температуры
образца, а не эталона. Вассалло и Харден [661 исследовали за-
висимость ЛГМИН от скорости нагревания для процесса плавления
бензойной кислоты и марлекса-50. Результаты этой работы пред-
ставлены в табл. 5.1. При измерении величины АТМИН в функции
температуры образца Cs она оставалась, по существу, постоянной
во всем используемом диапазоне скоростей нагревания. Измерение
же А7МИН в функции температуры эталона ЛЭтал вызывало в этом же
диапазоне скоростей нагревания смещение наблюдаемой величины
АТМИН Д-',я бензойной кислоты на ~4° С. Подобные результаты
были получены и для марлекса-50.
Таблица 5.1
Изменение ДТМИН для некоторых процессов плавления
в зависимости от скорости нагревания [66]
<р» град/мин1) А ГМИН’ °с
бензойная кислота марлекс-50
Л2) эта л с3» обр Д2) этал с3) обр
5 121,5 121,8 136,0 134,2
10 121,6 121,7 138,0 134,4
15 122,1 121,9 138,7 134,2
25 124,0 121,9 139,5 134,2
40 125,5 121,9 134,2
80 121,8 134,4
х) ¥ — скорость нагревания.
*) Лэтал есть А Тмин, записываемая в функции температуры эталона.
•) Cogp есть Д 7'мин, записываемая в функции температуры образца.
Влияние скорости нагревания на разрешение двух соседних пи-
ков кривой ДТА показано на фиг. 5. 9. Нагревая холестеринпро-
пионат со скоростями 2,5—10 град/мин, Джонсон и Миллер [67]
обнаружили на кривых три перехода (фиг. 5.10): кристалл-* смек-
тическая структура при 99° С; смектическая структура холе-
стериновая структура при 110° С и холестериновая структура ->
->изотропное вещество при 110° С. Однако при увеличении скорости
нагревания до 30 град/мин пик, соответствующий 110° С, не об-
наруживался и появлялся новый, небольшой эндотермический пик
при 64° С.
ДТА и ДСК
163
Этот эффект указывает на необходимость использования высо-
ких скоростей нагревания для выявления слабо выраженных пере-
ходов, которые трудно обнаружить при низких скоростях. В этой
работе наблюдался еще один эффект (фиг. 5.10): с увеличением
скорости нагревания увеличивалась амплитуда пика кривой.
Температура, °C
Фиг. 5.9. Влияние скорости на-
гревания на разрешение пиков
кривой ДТА для холестерин-
пропионата [67].
Фиг. 5.10. 'Влияние скорости на-
гревания на амплитуду пика кривой
ДТА для холестеринпропионата [67].
б. Атмосфера печи
Если изучаемое превращение сопровождается поглощением или
выделением газообразного компонента, то давление газа в системе
может оказывать влияние на температуру и форму пика. Если
газовая среда в системе и выделяемый или поглощаемый компонент
по составу идентичны, то изменения могут быть особенно резко
выраженными, что можно показать, исходя из термодинамических
соображений. Соотношение между температурой превращения и
давлением выражается известным уравнением Клапейрона
dp __ А/7
~dT ~ TKV ’
где р — давление пара; АН — теплота перехода; ЛУ — измене-
ние объема системы во время перехода. Для обратимых процессов
испарения при некоторых допущениях можно получить хорошо из-
(5.31)
164
Глава 5
вестное уравнение Клаузиуса—Клапейрона, интегральная форма
которого имеет следующий вид:
1п р = — + С, (5.32)
где р — давление пара, атм; &HV — теплота испарения, кал/моль;
jR — газовая постоянная; Т — температура; С — константа, свя-
занная с энтропией перехода.
Уравнение Клаузиуса—Клапейрона можно рассматривать как
частный случай более общего уравнения В ант-Гоффа
d In Кр _ д/7
dT ~ RT2 ’
(5.33)
где Кр — константа равновесия для обратимого и идеального рав-
новесного процесса. Для процесса перехода типа твердое тело-+
-+газ, часто исследуемого методом ДТА, например
Ат> Вт. Ср.,
может быть использована
Гоффа
приближенная форма уравнения Вант-
In
(Кр)г
(АД1
1п (Рс)2 = ДН(Та-Т1)
(Рс)1 R (Т27\)
(5.34)
где (Рс)2 и (Pc)i — парциальные давления газа Сг. в системе при
температурах соответственно Т2 и 1\.
Это соотношение не только обеспечивает удобный метод опре-
деления теплоты процессов испарения, но и объясняет чувстви-
тельность пиков испарения на кривых ДТА к изменениям расхо-
да продуваемого газа, обнаруживаемую в системах ДТА с динами-
ческой газовой средой. В динамической системе, продуваемой инерт-
ным газом, который отличается по составу от газа, выделяемого
образцом, пик испарения обычно уменьшается с увеличением рас-
хода инертного газа. Если продуваемый и выделяющийся газы оди-
наковы по составу, то пик испарения в зависимости от режима про-
дувки либо перемещается в область более высоких температур, либо
остается на том же месте, что и в статических условиях. Это поведение
отражает изменения парциального давления выделяющегося газа
в непосредственной близости к поверхности образца. Если проду-
ваемый и выделяющийся газы неодинаковы по составу, то парци-
альное давление выделяющегося газа уменьшается с увеличением
расхода и пик смещается в область более низких температур. Та-
ким образом, положение пика можно изменять путем регулирова-
ДТА и ДСК
165
Фиг. 5.11. Кривые ДТА сланца,
полученные в статической и ди-
намической средах [69].
ния парциального давления выделяющегося газа у поверхности
образца с помощью продуваемого газа, используемого в качестве
«разбавителя» [68].
В атмосфере с постоянным парциальным давлением выделяю-
щегося газа не будет происходить заметного разложения образца,
пока парциальное давление газооб-
разного продукта ниже давления
диссоциации реакции разложения.
Повышение парциального давле-
ния газа вызовет повышение тем-
пературы диссоциации вещества.
Следовательно, окружающая га-
зообразная среда оказывает за-
метное влияние на кривые ДТА.
Более того, вследствие взаимо-
действия газовой среды с образ-
цом возможно также появление
пиков на кривой; например, кис-
лород воздуха вызывает реакцию
окисления и, следовательно, поя-
вление экзотермического пика.
В общем случае используют два
типа газовой среды: статическая
газовая среда (обычно в замкну-
тых системах) и динамическая газо-
вая среда, когда через камеру
печи или образец и эталон проду-
вается поток газа. Первый тип сре-
ды очень трудно воспроизводить,
поскольку состав атмосферы вбли-
зи поверхности образца непре-
рывно изменяется из-за выделения
газа образцом и конвективных
потоков. В регулируемых усло-
виях динамическую атмосферу нетрудно поддерживать и воспро-
изводить.
Стоун [69] исследовал влияние статической и динамической га-
зовых сред, а также их состава и давления на некоторые реакции
разложения. На фиг. 5.11 сравниваются результаты, полученные
при термическом разложении сланца в статической и динамической
средах. В динамических условиях температура, соответствующая
минимуму пика &Тмпп, смещается в область более низких темпера-
тур. Формы пиков кривых в обоих случаях подобны.
На фиг. 5.12 показано влияние состава газовой среды на кри-
вую ДТА для смеси лигнита с А12О3. В динамической атмосфере
азота происходит пиролиз лигнита и возгонка летучих веществ;
166
Глава 5
в атмосфере кислорода лигнит окисляется и вместо эндотермических
пиков на кривой появляются экзотермические пики.
Нафиг. 5.13 показано влияние введения в состав газовой среды
выделяющегося из образца газа. В атмосфере кислорода пик, со-
ответствующий переходу ромбической структуры SrCO3 в гекса-
гональную, и пик, соответствующий разложению, перекрывают
Фиг. 5.12. Влияние среды кислорода и азота на кривые ДТА для смеси-
2,5% лигнита с А1аО3 [69].
друг друга. При введении в атмосферу двуокиси углерода первый
пик остается при температуре АТ'мин — 927° С, но второй пик,
соответствующий разложению
SrCO3 (т.) SrO (т.) + СО2 (г.),
смещается в область гораздо более высоких температур [70].
Форма держателя образца также оказывает влияние на взаимо-
действие газовой среды с образцом. Если используется стеклянный
держатель в виде капиллярной трубки, то изменение атмосферы печи
будет слабо влиять на характер кривой ДТА из-за большого пути
диффузии газа из окружающей среды к образцу. Держатель в виде
ДТА и ДСК
167
плоской тарелки, возможно, является идеальным для регулиро-
вания реакции между газом и твердым телом, однако в этом случае
может уменьшиться чувствительность оценки АТ вследствие по-
терь тепла за счет излучения. Для реакций, в которых газовая среда
не участвует, идеальным может оказаться сферический держатель
образца, но в таком случае возможны затруднения, связанные с
введением образца в держатель.
Фиг. 5.13. Влияние состава газовой среды на термическое разложение
SrCO3 [70].
На переход Твердое тело 1 -> Твердое тело 2 при 927°С среда не влияет. Масса образца
26 мг; скорость нагревания 10 град/мин.
Влияние изменения давления газовой среды на кривую ДТА
изучалось многими исследователями. Увеличение давления в сис-
теме (даже инертного газа) сопровождается возрастанием темпера-
тур переходов Tit АТМИН и Т/. При пониженных давлениях
(•<133 Па) газообразные продукты реакции быстро удаляются, и,
следовательно, температуры переходов смещаются в область более
низких температур; ухудшается также разрешение пиков. На
фиг. 5.14 показано влияние изменения давления в пределах от
665 мкПа до 15,8 МПа (изб.) на кривые ДТА MgSO4-7H2O [71].
На кривых, полученных при низких давлениях, разрешающая
способность неудовлетворительная: наблюдаются только один эн-
дотермический пик и небольшой экзотермический пик, соответствую-
щий рекристаллизации. С увеличением давления появляются новые
эндотермические пики, соответствующие процессу дегидратации.
К сожалению, происхождение этих пиков не обсуждалось. Подоб-
ные эффекты были замечены Локке [72], Гарном [73], Леви и др.
[74], Дэвидом [75] -и другими.
168
Глава 5
I__1—_1_____I____I____I____I____I____1
50 100 150 200 250 300 350 400 450
Температура, "С
Фиг. 5.14. Влияние давления на кривые ДТА MgSO4- 7НгО [71].
Указано давление азота; скорость нагревания 10 град/мин.
в. Держатели образцов
Поскольку на форму кривой ДТА влияют условия теплопере-
дачи от нагревателя к образцу и скорость выделения или поглоще-
ния тепла реагирующим образцом, держатель образца играет в экс-
периментах ДТА исключительно важную роль. Уильберн и др.
[76], а также Меллинг и др. [59] детально исследовали влияние
конструкции держателя образцов на характер-кривых ДТА. С по-
ДТА. и ДСК
169
Фиг. 5.15. Влияние температуропроводности держателя образца на форму
пика кривой ДТА [59].
мощью аналоговой вычислительной машины последние получали
кривые ДТА для различных постоянных времени RC держателя
образца. Результаты этих опытов приведены на фиг. 5.15. С уве-
личением постоянной времени держателя образца форма кривой
искажается. Этот эффект подобен влиянию уменьшения температу-
ропроводности держателя и (или) увеличения теплоемкости. Держа-
тель блочного типа с низкой температуропроводностью может вно-
сить искажения в кривую в виде отклонения от базовой линии с об-
разованием ложного экзотермического пика, следующего за эндо-
170
Глава 5
термическим пиком. Это может привести к ошибочным заключениям
относительно кривых ДТА, поскольку наблюдаемый, эффект обус
ловлен на самом деле не свойствами образца, а применением блока
с низкой температуропроводностью.
При высоких температурах тепло будет передаваться главным
образом путем излучения в соответствии с выражением
^ = f(Tl-T2y, (5.35)
т. е. нагревание будет более быстрым. Это эквивалентно умень-
шению температуропроводности держателя.
Уильберн и др. 176] установили, что для предотвращения не-
желательных искажений кривых ДТА необходимо использовать
образцы малого размера, причем зазор между образцами и утечка
тепла должны быть минимальными, а подвод тепла к образцам дол-
жен осуществляться быстро.
Увеличение радиуса образца вызывает изменение формы кривой
ДТА вследствие возникновения разности температур в образце
в начале реакции и запаздывание возврата к базовой линии в кон-
це реакции [59 ]. С увеличением радиуса становится более выражен-
ной S-образная форма переднего фронта пика и более заметно ис-
кажение формы пика. Если форма пика важна для определения
какого-либо параметра исследуемого процесса, необходимо исполь-
зовать образцы малого размера.
По мере увеличения размера образца возрастает и температура
пика как для образца, так и для эталона. Однако увеличение темпе-
ратуры образца значительно меньше увеличения температуры этало-
на. Поэтому для обеспечения минимальных различий между образ-
цами различных размеров лучше всего при построении кривых от-
кладывать по оси абсцисс температуру образца. Таким образом,
чтобы поддерживать скорость нагревания образца во время реак-
ции по возможности постоянной, необходимо использовать неболь-
шие образцы.
Постоянная времени держателя образца уже рассматривалась
Греем [60] при анализе уравнения (5.26). Становится очевидным
преимущество держателей образцов с малым значением величины
в идеальном случае этот параметр настолько мал, что отре-
зок III на фиг. 5.5 должен быть незначительным в сравнении с
отрезком I + II. Чем меньше параметр RCS и чем более постоян-
на его величина, тем точнее прибор будет регистрировать мгновен-
ное термическое поведение образца. Однако если бы термическое
сопротивление Л? равнялось нулю, то на кривой ДТА не было бы
пиков вообще. Для высокой чувствительности требуется большое R,
что несовместимо с требованием быстродействия или высокой раз-
решающей способности, для обеспечения которых величина RCS
должна быть малой. В тех случаях, когда сам образец определяет
ДТА и ДСК
171
величину термического сопротивления R и это сопротивление не
сохраняется постоянным в течение перехода, площадь пика кривой
не будет пропорциональна теплоте превращения.
Фиг. 5.16. Гипотетический изолированный держатель образца (ДТА) и его
приближенный аналог в виде электрической цепи [77].
В обоих случаях t — время, необходимое для достижения зависимой переменной (Г или v)
величины 63,2% ее конечного значения.
Эквивалентные величины:
время /, с — время t, с; термическое сопротивление 2 — сопротивление/?, Ом; теплоем-
кость К — емкость С, Ф; мощность Р — ток /, А; температура Т — потенциал vt В.
Используя электрическую схему в качестве аналога изолиро-
ванного держателя образца (фиг. 5.16), Дош [771 разработал прос-
той метод, который можно использовать для измерения тепловой
чувствительности и временной характеристики держателя образца.
Небольшой электрический нагреватель помещался в контейнер об-
разца. Тепловая чувствительность рассчитывалась при сопостав-
лении равновесного изменения температуры с подводимой мощ-
ностью, а временная характеристика определялась путем измере-
ния постоянной времени. Последнее определяется как время, не-
обходимое для достижения сигналом, изменяющимся по экспонен-
циальному закону, величины 63,2% его равновесного значения.
Термические сопротивления £2 и постоянные времени t несколь-
ких держателей образцов приведены на фиг. 5.17. Величины со-
противлений, полученные для случаев виг, были плохо воспроиз-
водимыми из-за того, что нагреватель находился в контакте с об-
разцом. На постоянные времени, однако, это оказывало значитель-
но меньшее влияние. Известно, что тепловая чувствительность дер-
жателя типа изолированного контейнера уменьшается с возраста-
нием температуры. Этот эффект, обусловленный увеличением коэф-
фициента теплопроводности /г, приводит к уменьшению термическо-
го сопротивления.
Форма держателя образца оказывает большое влияние на интен-
сивность и площадь получаемых пиков. По формулам Бойерсма
[44] была получена величина площади пика qa2/A). для металли-
ческого цилиндрического держателя образца и qa2/Sk для сфери-
ческого держателя. Сферический держатель на практике не исполь-
172
Глава 5
зуется из-за трудности изготовления. Однако геометрия, приближа-
ющаяся по характеристикам к сферической, была предложена Леман-
ном и др. [78], использовавшими цилиндрический держатель со
Фиг. 5.17. Формы держателей образцов и значения й и t [77].
сферическим дном. При сферической форме тепловые эффекты от
всех участков образца фиксируются одновременно («в фазе») рас-
положенной в центре термопарой [79].
В общем случае держатели образцов типа блока или чашки,
изготовленные из материала с низкой теплопроводностью, обеспе-
чивают лучшее разрешение пика эндотермической реакции, чем
изготовленные из материала с высокой теплопроводностью [79—81 1;
в случае экзотермических процессов разрешающая способность
оказывается худшей при использовании материалов с низкой тепло-
проводностью. Так как реакции, изучаемые методом ДТА, явля-
ются в основном эндотермическими, то может показаться, что пред-
почтительнее держатель образца с низкой термической проводи-
ДТА и ДСК
173
мостью. В действительности же более широко используются метал-
лические держатели с высоким коэффициентом теплопроводности,
возможно, из-за легкости их изготовления и долговечности.
Керамические и металлические держатели образцов сравнива-
лись Уэббом [82], Маккензи [83], Аренсом [80] и Жерардирне
и Леми [81 ]. Уэбб установил, что в случае реакций, сопровождаю-
щихся выделением газа, керамические держатели образца дают
пики с максимумами, смещенными в область низких температур.
Однако в случае переходов кристаллической фазы, таких, как а->Р-
переход в кварце, результаты, полученные для металлических и ке-
рамических контейнеров, были идентичными. Было высказано
предположение, что это различие вызвано диффузией газообразных
продуктов разложения из керамического держателя в атмосферу
нагревателя. При этом снижалась Концентрация газа над образцом,
что приводило к ускоренному завершению реакции. При использо-
вании в керамическом держателе прокладок из кварцевого стекла
диффузия не происходила и результаты для керамического и ме-
таллического контейнеров оказывались идентичными.
Сравнение двух типов держателей образцов [82] на примере
эндотермических реакций разложения Са(ОН)2, СаСО3 и MgCO3
показало, что никелевый блок был несколько менее чувствитель-
ным, чем керамический держатель образца. При температуре
~500° С он давал площадь пика кривой, составляющую 80% пло-
щади, полученной при использовании керамического держателя,
а при ~900° С отношение площадей пиков составляло ~70%. В то
же время металлический держатель давал более отчетливые пики с
лучшим разрешением соседних пиков. Уэбб [82 ] объяснил это сле-
дующим образом. Эндотермическая реакция начинается на поверх-
ности образца вблизи стенок держателя, и в случае металлического
(например, никелевого) держателя тепло быстро поступает от боль-
шой массы металла с высокой теплопроводностью к более холодному
разлагающемуся материалу. Быстрый подвод тепла в поверхностный
слой маскирует начало реакции вследствие нейтрализции эндо-
термического эффекта прежде, чем тепло достигнет спая термо-
пары. Именно этим объясняется кажущееся запаздывание эндо-
термической реакции по температуре. В дальнейшем достигается
температура, при которой скорость разложения настолько высока,
что тепло от металла уже не может проникать в утолщенный слой
разложившегося материала с низкой теплопроводностью в коли-
честве, достаточном для компенсации эндотермического эффекта.
После этого реакция становится явно выраженной, и ее скорость
в остальной период приближается к скорости, наблюдаемой при
проведении процесса в керамических держателях.
Сравнивая керамический (пористый корунд) и металлический
(никель) держатели образцов, Маккензи [83] показал, что эндо-
термический пик для каолинита в металлическом держателе был
174
Глава 5
меньше ( ~ 75% по отношению к керамическому) и смещен на 6%
в область более высоких температур.
В табл. 5.2 дано сравнение характеристик двух основных типов
держателей образцов — блока и изолированного контейнера [84 |.
Таблица 5.2
Сравнение характеристик держателей образцов в виде блока
и изолированного контейнера [84]
Преимущества Недостатки
Блок
1. Обеспечивает однородность темпе-
ратуры
2. Обеспечивает термическое равнове-
сие
3. Обеспечивает хорошее разрешение
4. Удобен для определения темпера-
туры кипения
1. Недостаточный обмен с атмосферой
2. Невысокая калориметрическая точ-
ность
3. Затрудняет выполнение операций с
образцом
4. Чувствителен к изменению плот-
ности образца
Изолированный контейнер
1. Достаточный обмен с атмосферой
2. Достаточная калориметрическая
точность
3. Удобен для использования при вы-
соких температурах
1. Плохое разрешение
г. Расположение термопары
Смит [85 ] рассчитал распределение температур в образце и эта-
лоне (фиг. 5.18). Кривые распределения температуры эталона в лю-
бой момент времени являются параболами, уравнение которых имеет
вид
Т = Тс + at + у ах2, (5.36)
где Т — температура; t — время; х — расстояние в направлении
теплового потока; а —- скорость нагревания; а — коэффициент
температуропроводности материала; Тс — константа, имеющая
размерность температуры.
Кривые распределения температуры образца на начальной ста-
дии нагревания имеют вид параболы, но как только температура
наружных слоев достигает температуры превращения, этим слоям
требуется больше тепла, и его передача в центральную часть образ-
ЦТ А и ДСЦ
175
ца прекращается. В результате скорость нагревания в центре об-
разца уменьшается прежде, чем температура в этой зоне достигнет
температуры превращения. Как только скорость нагревания в цент-
ре уменьшится, кривая дифференциальной температуры начнет
Фиг. 5.18. Распределение температур через интервалы 18,75 с в эталоне (а)
и образце (б) [85].
отклоняться от базовой линии. В точке начала отклонения кривой
температура центральных слоев образца и эталона неравна темпера-
туре превращения. По мнению Смита [85], этой точке не следует
придавать слишком большого значения.
После завершения превращения в образце кривая распределе-
ния температуры приобретает в центральной точке острый перегиб.
Поскольку такая точка соответствует очень большому значению
второй производной в уравнении
d2T _ 1 дТ
дх2 a dt '
следует ожидать быстрого увеличения температуры в центре. За-
тем скорость возрастания температуры постепенно снижается и
через ~300 с температура образца становится равной температуре
эталона, а дифференциальная температура — снова равной нулю.
Для большей иллюстративности расчетов Смит [85] применил
способ, в котором отклонение кривой дифференциальной темпера-
туры записывалось в функции температуры эталона. В ДТА форма
(5.37)
176
Глава 5
- W-ЛИЧЦ
кривой, а также температуры максимумов пиков изменяются в за
висимости от выбора характерной температуры. Иллюстрацией
этого являются кривые (фиг. 5.19) дифференциальной температуры
в зависимости от температуры в центре эталона, а также на поверх
ности или в центре образца,
Фиг. 5.19. Зависимость дифференциальной температуры от температуры
а) в центре эталона, б) на поверхности образца, в) в центре образца [851.
Кривая на фиг. 5.19, а отклоняется от нуля несколько ниже тем-
пературы превращения, равной 50° С, а дифференциальная темпе-
ратура пика на ~20° С выше температуры превращения. В случае
фиг. 5.19, б температура поверхности образца соответствует тем-
пературе металлического блока, в полости которого расположены
образец и эталон. Эта кривая начинает отклоняться от базовой
линии при температуре превращения. Если дифференциальная
температура записывается в функции температуры в центре образца
(фиг. 5.19, в), то дифференцальная температура пика соответствует
температуре превращения.
Смит [85] получил кривые для случая несимметричного распо-
ложения термопар в ячейках образца и эталона. Этот эффект по-
казан на фиг. 5.20.
На обеих кривых наибольшие дифференциальные температуры
полностью отличаются от соответствующих температур при сим-
метричном расположении термопар.
Бэрролл и Роджерс [86 ] тоже изучали влияние несимметрично-
го расположения термопары на кривые ДТА. На фиг. 5.21 пока-
зано влияние положения термопары.
В случае 1 термопара, показания которой фиксируются по оси
абсцисс, располагалась непосредственно в середине блока держателя
Фиг. 5.20. Влияние несимметричного расположения термопар в образце и эта-
лоне [85].
а — термопара расположена на расстоянии 0,06 см от центра образца; б — термопара распо-
ложена на расстоянии 0,3 см от центра образца.
Скорость нагребания, град]мин
Фиг. 5.21. Влияние положения термопары и скорости нагревания на темпе-
ратуру ДТмин [86].
Образцы различного веса смеси 8,3% салициловой кислоты с карборундом. Случай /:
а — 0,0952_г; б— 0,1125 г; в — 0,1555 г. Случай 2: г — 0,0952 г; д — 0,0822 г. Случай 3:
е — 0,0952 г.
7—230
178
Глава 5
образца. Из кривых а, бив видно, что температура АТМИН быстро
возрастает с увеличением скорости нагревания. В случае 2 термо-
пара размещалась в центре эталона, и кривые г и д обнаруживают
менее Выраженную зависимость АТМИН от скорости нагревания.
Фиг. 5.22. Зависимость дифференциальной температуры ДТ от температуры
системы [86].
В обеих ячейках — карборунд. 1 — симметричное расположение термопар; 2 — несимметрич-
ное расположение термопар (равномерная упаковка частиц образцов); 3—несимметричное
расположение термопар (неравномерная упаковка частиц образцов).
В случае 3 в отличие от первых двух скорость нагревания не влияет
на величину ЛТмт1. Было также показано, что в случае 3 эндотер-
мический пик симметричнее, а интервал плавления уже.
Бэрролл и Роджерс [861 исследовали также влияние несиммет-
ричного расположения термопар в камерах образца и эталона на
базовую линию. Нерегулярности, наблюдаемые на базовой линии,
становились более выраженными с изменением плотности упаковки
частиц образца (фиг. 5.22).
Кривая / получена при аккуратной упаковке частиц образца
с симметричным расположением термопар относительно стенок
блока держателя образца, а также относительно верхней и нижней
поверхностей образца и эталона в обеих ячейках или камерах. Кри-
вая 2 получена для случая, когда одна термопара была смещена
на ~2 мм по отношению к стенке, верху или дну камеры образца.
Видно, что смещенная термопара нагревается быстрее, чем симмет-
рично расположенная, вызывая тем самым отклонение базовой ли-
нии на величину А. В дальнейшем дифференциальная температура
становится стабильной, и остальная часть кривой является, по су-
ществу, горизонтальной. По кривой 3 видно, что неплотная упаков-
ка частиц ведет не только к смещению кривой, но даже к появлению'
максимумов и минимумов, обусловленных, вероятно, смещением
поддерживающих частиц при их расширении в процессе нагрева-
ния.
Меллинг и др. [591 обнаружили, что положение термопары в
образце небольшого диаметра не влияет на наблюдаемые температу-
ры пиков. Смещение последних оказалось значительным в случае
ДТА и ДСК
179
образцов большого диаметра, в то время как показания термопары
эталона не изменились. Следовательно, в общем случае введение
измерительной термопары оказывает влияние на распределение тем-
пературы в образце.
Дэвид и др. [87] всесторонне исследовали влияние на кривые
ДТА способа измерения температуры, откладывая на оси абсцисс
температуру нагревателя (способ 1) или температуры образца (спо-
соб 2). Сравнение кривых ДТА для полиэтилена — материала с дос-
таточно большой удельной теплоемкостью — показало, что темпера-
тура ЛТМ[1Н в способе 2 ниже. Влияние способа оценки температу-
ры на величины температуры пиков чистых металлов показано в
табл. 5.3. В том случае, когда регистрируется температура печи,
Таблица 5.3
Величины температур пиков на ДТА-кривых в зависимости
от способа их оценки [87]
Образец1) Температура печи, °C Температура образца, °C
начало отклонения от базовой ЛИНИН максимум пика экстрапо- ляция начало отклонения от базовой лиини максимум пика экстрапо- ляция
Индий 149 152 157 151 157 151
150 152 156 152 157 153
150 150 157 151 158 151
Олово 226 234 228 226 234 229
225 234 227 229 232 231
225 233 228 229 232 231
А люминий 646 664 649 644 661 647
644 664 648 643 660 646
644 662 648 643 660 646
644 665 647 645,62) 661.62) —
-Серебро — — — 958 961 —•
1) Известные значения переходов: для индия 156,6°С, олова 231,8°С, алюминия 660,1°С н
«серебра 960,8°С.
2) Температуры определены при повышенной скорости ленты самописца.
-температуру перехода обычно определяют в точке пересечения экс-
траполируемого переднего фронта пика с базовой линией. Резуль-
таты, полученные тремя способами, показали, что в случае, когда
регистрируется температура образца, температура, соответствую-
щая максимуму пика кривой, совпадает с точкой перехода. Темпе-
ратура, полученная путем экстраполяции переднего фронта пика,
в этом случае оказывается более низкой, чем температура перехо-
да, и при ее использовании необходима соответствующая калиб-
ровка.
у*
180
Глава Б
д. Термопары
утечка через провода
термопар [44].
Путем теоретических расчетов Бойерсма [44] показал,чтй теп-
ловые потери за счет теплоотвода проводами термопары достаточно
велики и могут оказывать существенное влияние на площадь об-
разующихся пиков. Так как темпера-
тура в центре образца измеряется с
помощью термопары, то происходит
утечка части тепла из образца по
проводам термоэлемента, и измеряемая
температура образца оказывается зани-
женной.
На фиг. 5.23 показана сферическая
полость в никелевом блоке, заполняемая
образцом с введенным в него спаем
термопары радиусом г0, На участке про-
водов длиной I во время химической
реакции или иного перехода устанавли-
вается градиент температуры. Пред-
полагается, что на расстоянии /, которое
немногим больше радиуса полости а,
провода имеют температуру, равную
температуре никелевого блока. Если
А — площадь сечения проводов термо-
пары, а 60 — температура спая, то
суммарная тепловая утечка из термо-
элемента по проводам составляет
Qodt,
(5.38)
h
где \р — коэффициент теплопроводности проводов. Расчеты пока-
зывают, что для сферического держателя образца площадь пика кри-
вой равна
f Q dt = ---
J 6Х 1 4- Л/Х
(5.39)
где величина а, близкая к 1, определяется формой держателя об-
разца следующим образом:
г2
го
П = 1
а2
(5.40)
ДТА и ДСК
181
Величина Л, характеризующая тепловую утечку через провода,
определяется по формуле
д= fl — —(5.41)
р I 4w£ \ а )
Для образцов с низким значением коэффициента теплопровод-
ности (Х/А 1) площадь пика кривой становится независимой
от X, тогда как для образцов с высоким значением коэффициента
теплопроводности площадь пика будет обратно пропорциональна X
Фиг. 5.24. Характер эндотермического пика кипения, полученного при
скорости нагревания 10 град/мин, в зависимости от способа регистрации
температуры системы и диаметра проводов термопары [66].
а — диаметр провода термопары 0,079 мм; б — диаметр провода термопары 0,320 мм.
Для цилиндрических держателей площадь пика равна
f Qdt =
J 4Х
1 — ( ^/с2)[1 + 21п(с/г0)]
1 + (А/l) (Хр/Х) [In (a/r0)/2*h]
(5-42)
Для большинства образцов коэффициент теплопроводности состав-
ляет —0,3 Дж/(м-с-град) и отношение Х/А близко к 1. Поэтому,
согласно уравнению (5.39), тепловая утечка через провода термо-
пары уменьшает площадь пика не более чем на 50% ее расчетного
значения.
Хаузер [88] изучал также влияние размеров сечения проводов
и спая термопары на форму пика и пришел к заключению, что боль-
шему сечению проводов (диаметр 0,645 мм в сравнении с диамет-
ром 0,320 мм) и большему размеру спая (1,43 мм в сравнении с
182
Глава 5
0,8 мм) на кривой ДТА соответствуют более выраженные пики тер-
мического разложения. Кроме того, он установил, что провода
диаметром 0,320 мм имеют слишком высокое сопротивление для
обеспечения соответствующих электрических свойств при не
слишком большой теплопроводности. Спай диаметром 1,43 мм ока-
зался намного эффективней в поддержании э.д.с., чем спай мень-
шего диаметра, а масса спая еще не была слишком большой, т. е.
не вызывала чрезмерного поглощения тепла.
Влияние сечения проводов термопары на температуру перехода
бензойной кислоты и толуола исследовали Вассалло и Харден[66].
При использовании провода диаметром 0,320 мм при скорости на-
гревания 10 град/мин температура плавления бензойной кислоты
составляла 121,7°С. При использовании провода диаметром
0,079 мм высота пика на ~15% меньше, чем в случае провода боль-
шего диаметра. Различия в форме и высоте пиков, полученных
при различных диаметрах проводов термопары и разных способах
регистрации температур, показаны на фиг. 5.24. Из фигуры вид-
но, что для проводов разных размеров формы кривых совершенно
различны.
Тепловая утечка за счет термопары может быть выражена с по-
мощью уравнения [59]
(5.43)
где dhldt — скорость тепловых потерь: Ts — температура в центре
образца; ТА — температура окружающей среды;
(5.44)
Здесь К.н — коэффициент тепловых потерь, kw — коэффициент
теплопроводности провода, aw — площадь поперечного сечения
провода и lw — длина провода от спая до точки, в которой его тем-
пература становится равной ТА . Как и предполагалось, увеличение
тепловых потерь вдоль проводов термопары вызывает уменьшение
площади пика кривой ДТА, но интересно отметить, что уменьшает-
ся также и температура пика. Базовая линия кривой будет нулевой
линией только в том случае, когда параметры образца и эталона
идентичны и тепловые утечки в обоих случаях одинаковы. Если
тепловые утечки различны, то дифференциальная температура бу-
дет линейно увеличиваться с ростом температуры. Нелинейные из-
менения базовой линии могут быть следствием температурной за-
висимости коэффициента тепловых потерь. Тепловая утечка вдоль
проводов термопары уменьшает время достижения квазистатиче-
ского состояния и уменьшает скорость нагревания.
Калибровка температурной шкалы термопар рассматривается
в гл. 6.
ДТА и ДСК
183
5. ХАРАКТЕРИСТИКИ ОБРАЗЦА
а. Масса образца
Согласно различным теориям ДТА, площадь пика кривой про-
порциональна теплоте химической реакции или физического пере-
хода и, следовательно, массе образца. В общем случае площадь
пика прямо пропорциональна плотности <р и теплоте реакции
АД, обратно пропорциональна коэффициенту теплопроводности
k и не зависит от удельной теплоемкости [59]. Соотношение между
этими величинами может быть выражено следующим образом:
А-----(5.45)
k
где А — ДТ х Время есть площадь пика; г — радиус образца;
I — длина образца. Тогда
л = (5.46)
или
А = . (5.47)
Здесь V — объем образца, G — калибровочный коэффициент и
т — масса образца.
Влияние массы образца на площадь пика кривой будет более
подробно обсуждаться при рассмотрении количественных ДТА
и ДСК.
б. Размер и упаковка частиц образца
В ряде исследований были получены противоречивые данные
о влиянии размеров частиц и распределения частиц по размерам на
площади пиков и значения ДТМИН. Спейл и др. [10] установили,
что площади пиков реакции дегидратации каолина изменяются от
725 до 2080 мм2 при изменении размеров образцов от 0,05—0,1
до 5—20 мкм. Величина ДТМИН изменяется при этом от 580 до
625°С. Однако Нортон [89] нашел, что величина ДТМИН остается
в основном постоянной, а температура окончания реакции дегидра-
тации изменяется от 610 до 670°С при изменении размеров частиц
образца от менее 0,1 до 20—44 мкм. Гримшо и др. [90] также уста-
новили, что для образцов с частицами размером менее 1 мкм терми-
ческие свойства каолина не зависят от размеров частиц. Результаты
этого исследования представлены в табл. 5.4.
Картыо [91 ], также изучавший разложение каолинита, получил
результаты, аналогичные результатам Нортона [89] и Гримшо
[90 ]: для образцов с размерами частиц 2—1 и 0,25—0,1 мкм пло-
184
Глава 5
Влияние размера частиц образца
на термические характеристики каолина 190]
Таблица 5.4
Средний размер частиц, мкм Эндотермическая реакция Экзотермиче- ская реакция
ЛГмин' °C температура завершения, А^мин’ °C
10—44 ? 600 670 980
0,5—1,0 605 650 980
0,25—6,5 605 630 980
0,10—0,25 600 615 980
<0,10 600 610 945, 9901)
<0,10 (после диализа) 605 610 955 , 9861)
*) Два пика.
щади пиков кривой и ДТМЯЯ сохраняли практически постоянные
значения. Разногласие с работой Спейла и др. [10] было вызвано,
по-видимому, тем обстоятельством, что для получения необходимого
размера частиц каолинита его размалывали, уменьшая тем самым
степень его кристалличности.
Бэрролл и Роджерс [92 ] проводили опыты с образцами, состоя-
щими из стеклянных шариков. Они установили, что с увеличением
температуры постепенное смещение базовой линии для шариков мень-
шего размера относительно базовой линии для шариков большего раз-
мера, которое в значительной мере сохранялось при 200°С, свя-
зано с тем, что через шарики большего размера тепло передавалось
хуже, чем через меньшие шарики.
Лэнгер и Керр [65] показали, что увеличение размера частиц
каолинита приводит к увеличению температуры пика реакции де-
гидратации каолинита. Не наблюдалось сколько-нибудь заметного
смещения экзотермического пика, соответствующего фазовому пере-
ходу. Увеличение плотности упаковки частиц вызывает увеличение
теплопередачи через образец и изменение наклона базовой линии.
Было замечено [93], что на кривые ДТА нитрата серебра изме-
нение размеров частиц оказывало более сильное влияние (фиг. 5.25).
При этом изменяются не только форма пика, но и температура мини-
мума пика. АТмаа для исходного образца была равна 161 °C, а для
тонко измельченного образца сместилась к 166,5°С. После плавле-
ния образцов их кривые ДТА стали идентичными. Согласно Неги-
ши и Озава [93], влияние измельчения на переход имеет кинетиче-
скую природу и связано с возникновением барьеров (промежутков
между частицами), задерживающих плавление.
ДТА и ДСК
185
Вада и др. [94] также наблюдали резко выраженное влияние
измельчения СсЮ на кривые ДТА. Происхождение пиков, становив-
шихся более выраженными с увеличением времени измельчения,
связано с присутствием
примеси Cd(OH)2.
Из изложенного выше сле-
дует, что площадь пика кри-
вой ДТА обратно пропорци-
ональна коэффициенту теп-
лопроводности образца, ко-
торый в свою очередь зави-
сит от распределения частиц
по размерам и плотности их
упаковки [59].
Влияние плотности упаков-
ки частиц образца на кри-
вые ДТА изучалось Граве-
ром [95]. Кривая а нафиг.
5.26 соответствует случаю,
когда образец каолина поме-
щался в тигель и уплотнялся
встряхиванием. Кривая б по-
лучена в случае, когда обра-
зец утрамбовывался в тигель
маленьким стеклянным стер-
жнем. Полученные кривые
оказались идентичными, хотя
способ оценки влияния упа-
ковки здесь довольно гру-
бый.
Боллин и Бауман [96 ]
описали простую методику,
позволяющую получать вос-
производимую плотность
упаковки частиц. Образец
Время, мин
Фиг. 5.25. Кривые ДТА нитрата
серебра [93].
а — исходный образец; б — грубо измельчен-
ный образец; в — тонко измельченный образец.
укладывался вокруг термопары и уплотнялся механически
нагружаемым пуансоном. Такой способ упаковки оказался более
эффективным, чем способ «сэндвича», предложенный Баршадом
[97].
Влияние степени кристалличности образца довольно трудно
оценить из-за неопределенности этого понятия. Картью [91 ] опре-
делил степень кристалличности (для каолина) как степень совер-
шенства ориентации кристалла, не связанную с его размером. Для
пяти различных образцов каолина он нашел, что площадь эндотер-
мического пика кривой дегидратации уменьшалась с уменьшением
степени кристалличности образца. С увеличением степени крис-
186
Глава 5
талличности пики становились острее. Влияние степени кристаллич-
ности на кривые ДТА напоминает влияние размера частиц образца
и объясняется, вероятно, аналогично.
Фиг. 5.26. Влияние способа упаковки частиц образца каолина (95].
а — уплотнение встряхиванием; б — трамбование стеклянным стержнем.
в. Влияние инертного наполнителя
Если образцы «разбавить» инертным наполнителем, то их фи-
зические свойства останутся почти прежними, а площадь пика
кривой будет прямо пропорциональна теплоте реакции или пере-
хода [59]. Тем не менее разбавление уменьшает тепловой эффект
в расчете на единицу массы и вызывает уменьшение площади пика.
Наполнитель не должен, конечно, реагировать с образцом в про-
цессе нагревания.
Дин [94а] исследовал влияние добавок инертной окиси алюми-
ния на величину АТМИН пика для различных концентраций каоли-
нита и галлоизита. Для первого из этих веществ величина ДТМНЯ
колебалась в пределах от 525°C для концентрации каолинита 10%
до 570°С для концентрации 60%. Подобные результаты были полу-
чены для смесей галлоизита. Дежонг [98 ] установил линейную зави-
симость между площадью пика и весовой долей каолинита, разбав-
ленного окисью алюминия, при условии, что плотность смеси силь-
но не менялась. Для смеси иллита с окисью алюминия также на-
блюдалась тенденция к увеличению площади пика с увеличением
концентрации иллита, однако линейной зависимости обнаружено
не было.
В процессе подробного исследования Бэрролл и Роджерс [92]
определили влияние таких инертных наполнителей, как карборунд,
железо и трехокись железа, на пик, вызванный плавлением сали-
циловой кислоты (табл. 5.5). Возможно, основной причиной изме-
нения площади пика является изменение коэффициента теплопро-
водности наполнителя. Повышение теплопроводности способствует
более эффективному подводу тепла к термопаре, находящейся в
центре образца. Необходимо заметить, однако, что высокая тепло-
ДТА и ДСК
187
Таблица 5.5
Площадь пика кривой, полученного при плавлении 0,01 г
салициловой кислоты, смешанной с различными
инертными наполнителями1) [92]
Наполнитель Салициловая кислота, % Площадь пи- ка/0,01 г кислоты, мм2
Карборунд 6,87 306
Железо 8,82 710
Трехокись железа 3,40 280
Стеклянная дробь (диаметр 0,029 мм) 4,57 322
Стеклянная дробь (диаметром 0,29 мм) 5,58 289
Окись алюминия 8,60 313
Нуйол 20,00 92
* Чувствительность по Д Г 26,4 мкВ/см; скорость нагревания 7,9 град/мин.
Фиг. 5,27. «Маскирующее» действие наполнителя на пик кривой ДТА [921*
а — 8-хинолинол (6,9%) с карборундом в качестве наполнителя; б — 8-хинолииол (5,9%) о
окисью алюминия в качестве наполнителя.
проводность наполнителя может вызвать уменьшение площади
пика, когда образец с наполнителем находится в непосредственном
контакте с металлическим блоком.
В том случае, когда наполнитель не инертен, а реагирует с об-
разцом, было замечено [92 ] его «маскирующее» воздействие на не-
188
Глава 5
которые пики. Это, например, наблюдалось для 8-хинолинола с
карборундом и окисью алюминия в качестве наполнителей
(фиг. 5.27). При использовании карборунда ATMan = 76,3°С; при
использовании окиси алюминия в окрестности этой температуры
не было замечено эндотермического пика. Очевидно, окись алюми-
ния и 8-хинолинол образовывали комплекс.
6. ОСНОВНЫЕ МЕТОДИЧЕСКИЕ УКАЗАНИЯ
Основные методические параметры для ДТА (или ДСК), по дан-
ным Сарасона [84], представлены в табл. 5.6. Для образца боль-
шого размера требуются низкие скорости нагревания, при которых
Таблица 5.6
Основные методические параметры [84J
Размер образца
Большой
Малый
Повышенная
Полезен для обнаружения слабо выраженных пере-
ходов. Полезен для неоднородных образцов.
Пики широкие; низкие разрешение и точность
по температуре. Требует низких скоростей на-
гревания
Обеспечивает хорошее разрешение пиков кривой.
Пики острые. Требует достижения равновесных
значений температуры перехода при изучении
реакций нулевого порядка. Позволяет использо-
вать повышенные скорости нагревания
Скорость нагревания
Увеличивает чувствительность
Уменьшает разрешающую способность
Уменьшает точность по температуре
Атмосфера
Может реагировать с образцом
Динамическая среда предпочтительнее статической
в свою очередь уменьшаются чувствительность оценки А7 и раз-
решение пиков. Возможно, удобней всего использовать образцы
малого размера, особенно если испытания производятся с помощью
современных промышленных приборов. Образцы малого размера
позволяют увеличить скорости нагревания и обеспечивают лучшее
разрешение пиков.
ДТА и ДСК
189
Кратко общие методические требования суммированы в
табл. 5.7 [84].
Таблица 5.7
Общие методические требования [84]
Параметр Максимальное разрешение Максимальная чувствительность
Размер образца Скорость нагревания Держатель образца Отношение поверхности к объему образца Атмосфера Малый Низкая Блок Большое Высокий коэффициент k *) (Не, Н2) Большой Высокая Изолированный контей- нер Малое Низкий коэффициент k (вакуум)
*) Благодаря использованию динамической газовой атмосферы зачастую удается разрешить
конкурирующие обратимые реакции испарения.
Б. КОЛИЧЕСТВЕННЫЙ ДИФФЕРЕНЦИАЛЬНЫЙ
ТЕРМИЧЕСКИЙ АНАЛИЗ
1. ВВЕДЕНИЕ
В ДТА и ДСК широко используется метод определения теплоты
перехода или реакции и массы реагирующего образца по площади
пика кривой. Связывающая их зависимость очень проста:
АНт = КА, (5.48)
где АД — теплота превращения или реакции; т — масса реаги-
рующего образца; К — калибровочный коэффициент; А — пло-
щадь пика кривой. Калибровочный коэффициент зависит от формы
и теплопроводности держателя образца и обычно определяется при
калибровке системы с помощью соединений, для которых теплота
реакции (или перехода) известна.
Применение ДТА для определения теплоты перехода или реак-
ции рассматривали Бохон [99], Озава [100] и многие другие ис-
следователи. Основные преимущества методов ДТА и ДСК перед
классической калориметрией следующие:
а) Быстрота определения — в течение нескольких минут или
часов может быть исследован широкий интервал температур.
б) Малый вес образца — от единиц до сотен миллиграммов.
в) Универсальность — образцы могут быть жидкими или твер-
дыми.
190
Глава 5
г) Простота и легкость операций и анализа результатов.
д) Возможность измерений в процессе охлаждения и под высо-
ким давлением.
е) Возможность использования для изучения различных типов
химических реакций.
Недостатки методов ДТА и ДСК следующие:
а) Относительно низкие надежность и точность — в большинст-
ве случаев погрешность измерений составляет 5—10%.
б) Трудность применения для определения значений АД пере-
крывающихся по температуре реакций.
в) Необходимость калибровки во всем исследуемом в интервале
температур, поскольку зависит от температуры (только в ДТА).
г) Неточности в определении площадей пиков кривых из-за
изменения хода базовой линии во время перехода или реакции.
Во многих случаях исследователи проявляли, по-видимому,
несколько чрезмерный оптимизм в отношении получаемых резуль-
татов и не принимали во внимание всех переменных метода. Без
строгого контроля всех переменных (что во многих случаях трудно
осуществить) не удается получить высокой точности результатов.
Волд [43] одним из первых определил АД плавления несколь-
ких органических кислот с помощью уравнения (5.2). Величина А
была получена из графиков зависимости lg(r/—ys) от времени Д
В этом случае не потребовалось калибровки по эталону.
В ранее опубликованном исследовании Виттельса [101 ] площадь
пика кривой оказалась пропорциональной теплу, поглощенному
при разложении кальцита СаСО3. Была установлена линейная за-
висимость площади пика кривой от массы образцов в диапазоне
0,30—3,00 мг. В работе [102] Виттельс рассмотрел влияние ско-
рости нагревания и массы образца на площадь пиков, полученных
при термическом разложении тремолита Ca2Mg5 Si8O22(OH)2, и
установил зависимость
А
, (5.49)
ig[lnfl (R - с)Im] V ’
где R — скорость нагревания; А — площадь пика; тис — конс-
танты. Наибольшая чувствительность прибора была получена при
скорости нагревания 30 град/мин;, при скорости ниже 15 град/мин
она быстро падала.
В дальнейшем в этой главе будут рассматриваться более поздние
работы.
2. КАЛИБРОВКА
Калибровочный коэффициент К определяется при использова-
нии соединений с известными значениями теплоты перехода. Чаще
всего используются эталоны с известной теплотой плавления АД^
ДТА и ДСК
191
или теплотой перехода типа «твердое тело 1 -^твердое тело 2». Эта-
лоны, очевидно, должны удовлетворять определенным требованиям,
таким как химическая стабильность во время превращения, низ-
кое давление паров (во избежание вклада теплоты парообразования
в исследуемый тепловой эффект) и т. д.
. Выражение для расчета К зависит от типа используемого при-
бора и метода регистрации кривых ДТА и ДСК. Если в это выра-
жение входят такие переменные, как чувствительность оценки АТ
и скорость ленты регистрирующего устройства, то оно имеет вид
[ЮЗ]
К = , (5.50)
АкТs
где АТ/ — теплота перехода, кал/г; m — масса образца, мг;
С — скорость ленты, мм/мин; А — площадь пика, мм2; ATS —
чувствительность измерения дифференциальной температуры,
град/мм. В этом случае /С выражается в мкал/(мин-град). Подоб-
ное выражение использовал Дэвид [104]. Для систем, имеющих
большую теплоемкость, Бохон [105] рассчитал /С из выражения
K = thch, (5.51)
где th — эффективный коэффициент теплопередачи и Ch — тепло-
емкость держателя образца, причем Сй ~ chwc (ch — удельная
теплоемкость контейнера образца и wc — его масса). Коэффициент
теплопередачи можно получить из соотношения
lg(AT) = -^/ + 7. (5.52)
Во многих методах количественного ДТА калибровочный коэф-
фициент можно рассчитать математически без проведения специ-
альных экспериментов. Например, Крониг и Снуджик (см. [104])
вычислили К для цилиндрического держателя образца.
А = <р — = (5.53)
4Х 4тсЯХ
и для сферически симметричного держателя
v о2 М слх
К = 9 ---- = ------, (5.54)
т 6Х 8паК ' '
где ф — плотность образца; а — радиус держателя образца; h —
высота образца. Эти уравнения описывают разность температур
между центрами эталона и образца в предположении весьма малых
размеров спая и проводов термопары. Величины 4лЛ или 8та
соответствуют фактору формы g в теории Спейла. Подобные ре-
зультаты получил Бойерсма [44], учитывавший влияние тепловых
утечек через провода термопары. Волд [43] получил значение К
192
Глава 5
путем анализа экспоненциального падения кривой после прекра-
щения реакции. При этом время релаксации должно быть таким
большим, чтобы по окончании реакции или перехода релаксация
температуры происходила в достаточном для анализа интервале.
Другие попытки рассчитать К были предприняты Озава [100],
Пэкором [58 ] и другими авторами.
Фиг. 5.28. Зависимость коэффициента от температуры К [106].
Скорость нагревания 0,4 град/мин.
На фиг. 5.28 приведена зависимость коэффициента Д от тем-
пературы держателя образца прибора ДТА фирмы «Дюпон» [106].
Видно, что калориметрическая чувствительность прибора убывает
с температурой, т. е. на единицу площади требуется больше тепла.
В дифференциальной сканирующей калориметрии, например в
приборах фирмы «Перкин-Элмер», Д не зависит от температуры, и,
следовательно, требуется калибровка только по одной температуре.
Предложенная Уэндландтом и Уильямсом [107 ] методика также
позволяет избежать необходимых температурных калибровок в
ДТА по нескольким точкам и получить постоянную чувствитель-
ность.
Калибровочный коэффициент зависит также от других факторов,
связанных с измерительным прибором, например от атмосферы
печи. Дэвид [104] исследовал зависимость Д от температуры для
различных составов атмосферы печи (воздух, азот или гелий) при
различных давлениях (от 0,67 до 14,7 МПа). Влияние состава га-
зовой среды на величину Д показано на фиг. 5.29. Разница между
ДТА и ДСК
193
кривыми может быть обусловлена различиями в термической про-
водимости газов. Как и предполагалось, величина К не зависит от
скорости нагревания в интервале значений 2—40 град/мин.
Температура, ° с
Фиг. 5.29. Влияние состава газовой среды в печи на величину коэффициента
К [104].
1 — калориметрическая ячейка из алюминия, динамическая атмосфера азота; 2— калори-
метрическая ячейка из никеля, динамическая атмосфера азота; 3 — калориметрическая ячей-
ка из алюминия, динамическая атмосфера гелия; 4— низкотемпературная калориметрическая
ячейка из меди (температура ниже окружающей), статическая атмосфера воздуха.
Во многих случаях зависимость К от температуры выражалась
аналитически. Карелл [108] выразил эту зависимость следующим
образом:
К = — 1,3 • 10-4Т + 0,2200, (5.55)
а Озава [100] предложил следующее выражение для эмпирической
кривой:
К = 1,507259 + 8,782709 1 О^Т — 1,808468 - 1О-5?12 +
+ 6,324056 • IO’14?4. (5.56)
Видеманн и Ван-Тест [109, ПО] установили, что в качестве эта-
лона предпочтительнее использовать вещество с известной тепло-
той плавления, а не инертное вещество.
194
Глава 5
Рамачандран и Середа [111 ] описали калибровку прибора ДТА
с использованием азотнокислого серебра в качестве эталона. Ока-
залось, что для всех расчетов предпочтительнее выбирать высоту
пика, а не его площадь. Этот параметр применяли Дэвис и Холдридж
[112] для обнаружения кварца в смеси, Рамачандран [113] для ис-
следования силиката кальция, а также и другие ученые.
3. ЭТАЛОНЫ ДЛЯ КАЛИБРОВКИ
Для калибровки держателей образцов в ДТА и ДСК с целью
проведения количественного анализа предлагалось много соедине-
ний. В большинстве случаев использовались чистые металлы, хотя
применялись многие органические соединения высокой чистоты.
Для калибровки обычно используется теплота плавления; тем не
менее многими исследователями рекомендовалась калибровка по
реакциям дегидратации и разложения. Перечень эталонных мате-
риалов, используемых для калибровки, приведен в табл. 5.8.
Таблица 5.8
Эталонные материалы, используемые для калибровки в ДТА и ДСК
гЗ & н сЗ Вещество Ally
03 Е5 Я <U(J 1—< о кал/г кДж/кг кал/г кДж/кг
34,6 Азоксибензол 21,6 90,4
44,9 С2Нв 2,59 10,84
47 СВг4 4,81 20,14
48,2 Бензофенон 23,5 98,4
62,5 Пальмитиновая кислота 51,2 214,4
69 Стеариновая кислота . . 47,5 198,9
69,8 Дифенил 28,7 120,2
99,3 Фенантрен 25,0 104,7
114 орто-Динитробензол . . 32,3 135,2
121,8 Бензойная кислота . . . 33,9 141,9
125 NH4NO3 12,6 52,8
128 KNO3 12,86 53,85
130 ВаС1а-2Н2О 116,6 488,2
(—2H2O) (—2HaO)
137,2 NH4Br 882кал/моль 3,693 кДж /моль
150 CuSO2-5H2O 228,5 956,7
(-4H2O) (—4HaO)
156,4 In 6,79 28,43
177,0 KCNS 25,72 107,69
,179 4,08 17,08
180 CaSO4-2H2O 157,2 (—2H2O) 658,6 (—2H2O)
<183,1 NH4C1 1873 7,842
кал/моль кДж /моль
ДТА и ДСК
195-
СО* в Вещество д 111
кал/г кДж/кг кал/г кДж/кг
187,8 212 231,9 252 299,8 306,2 327,4 337 350 398 419,5 498 553 575 588,8 850 Пентаэритрит . 1 . . . AgNO3 Sn LiNO3 КС1О4 . NaNOs Pb KNO3 . CdCO3 К2СГ2О7 Zn PbCl2 LiBr LiSO4 Na2WO4 CaCO3 77,1 17,7 14,4 88,5 44,2 5,50 28,1 134,5 (-сад 24,4 20,9 36 62 427,1 (-сад 322,8 74,1 60,3 307,0 185,1 23,03 117,6 563,1 (- СО2) 102,2 87,5 151 260 1788,3 (-со8) 23,7 28,9 28,57 99,2 121,0 119,62
Большинство значений ДН, указанных в этой таблице, полу-
чены при постоянном давлении. Если калибровка выполнялась
при постоянном объеме, как, например, в опытах Бохона [105],
то значения А// следует пересчитать с помощью приближенного
соотношения
ДЯ(0«Д//(р) + ЯТДп, (5.57}
где AH(v) есть ДН при постоянном объеме; &Н(р) — АН при
постоянном давлении; Дп — изменение числа молей газа образо-
вавшегося в процессе реакции. Пересчитанные значения Д/7(и).
для нескольких реакций приведены в табл. 5.9.
Таблица 5.S’
Пересчитанные значения теплот реакций для постоянного объема [105]
Соединение ДН(Р) Д Н (ту) Г, °C
кал/г кДж/кг кал/г кДж/кг
MgSO4-7H2O 402 1683 398 1666 130
AgN03 210 879 122 511 400
NaNO3 776 3249 537 2248 677
СаСО3 400 1675 468 1960 787
196
Глава 5
4. РАСЧЕТ Д77
После калибровки держателя образца и определения калибро-
вочного коэффициента в интересующем исследователя интервале
температур можно вычислить АН образца с помощью уравнения
(5.8) или аналогичного выражения. Площадь пика кривой определя-
ется с помощью планиметра или иным методом. Если пики на кривой
перекрываются, то общая площадь может быть определена по час-
тям, как показано нафиг. 5.30 [106]. Если применяется метод ДТА,
Время, мин
Фиг. 5.30. Определение общей площади кривой ДТА по частям [106].
то” при расчете значений АН для каждой части площади необходи-
мо использовать соответствующее значение К- Величина АН будет
-суммой соответствующих величин, полученных для каждой части.
При использовании метода ДСК коэффициент К не зависит от тем-
пературы, и поэтому необходимо знать только одно его значение.
Это, несомненно, одно из преимуществ данного метода перед мето-
дом ДТА при количественных измерениях.
Если базовая линия в ходе реакции или перехода существенно
смещается, то определение площади пика затруднено: значения АН
в этом случае могут быть рассчитаны с большой погрешностью.
Во избежание смещения базовой линии рекомендуется применять
небольшие образцы. Повышенную стабильность базовой линии
имеют также приборы с плоскими держателями образцов термо-
электрического типа.
5. ТОЧНОСТЬ И НАДЕЖНОСТЬ ИЗМЕРЕНИЙ ДН
Несмотря на развитие статистических методов, большинство
исследователей до настоящего времени представляют численные
данные без достаточной оценки их точности и надежности. Изме-
ДТА и ДСК
197
рения при появлении нового метода или прибора обычно произво-
дят на произвольном числе образцов и рассчитывают лишь средне-
арифметические значения. Эти величины принимаются как абсо-
лютные и окончательные. Швенкер и Уайтвелл [114] использовали
статистические методы для оценки результатов, полученных мето-
дом ДСК. Проводились повторные опыты с разными скоростями
нагревания, размерами образцов, чувствительностями прибора и
металлами, используемыми в качестве эталонов. Для ограничения
числа измерений в разумных пределах в качестве эталонов при-
менялись только три металла — индий, олово и свинец.
Таблица 5.10
Точность оценки калибровочного коэффициента [114]
Число Среднеквадратичное отклонение Доверительные границы, % (степень достоверности 0,95)
образцов измерений площадей пика
г 1 1 0,0286
2 0,0277 ±5,5
2 1 0,0202 —
2 0,0196 ±3,9
4 1 0,0168 —
2 0,0139 ±2,8
4 • 0,0137 —
8 1 0,0101 —
2 0,0098 ±2,0
4 0,0096 —
Результаты расчета влияния числа измерений площади пика
на точность оценки калибровочного коэффициента показаны в
табл. 5.10 [114]. Эти данные свидетельствуют, что лучше дублиро-
вать образцы, чем измерения площади пика планиметром. Согласно
полученным значениям среднеквадратичных отклонений, при че-
тырех измерениях площади вместо двух повышение точности ре-
зультатов незначительно, тогда как с увеличением числа образцов
.точность заметно улучшается.
В табл. 5.11 показано влияние числа образцов на точность
определения коэффициента /С и значения АН некоторого перехода
(при степени достоверности 0,95). Эти результаты показывают, что
для обеспечения большей точности требуется использовать при
калибровке несколько образцов.
Известно очень мало калориметрических исследований, в ко-
торых для определения значений К и АН использовалось ~30 об-
разцов. Следовательно, погрешность измерений обычно превосхо-
дит 1% (табл. 5.11). Поскольку большинство известных измерений
198
Глава 5
Таблица 5.It
Оценка доверительных границ для Д Н [114]
Число образцов Доверительные границы, % (степень достоверности 0,95)
калиброванных неизвестных
1 2 ±6,52
4 ±6,22
8 ±5,90
2 2 ±5,56
4 ±4,80
8 ±4,36
4 2 ±4,86
4 ±3,96
8 ±3,42
8 2 ±4,40
4 ±3,46
8 ±2,80
30 30 ±1,00
относятся к первым двум типам (1—2 образца), то можно предпо-
ложить, что погрешность составляет обычно по меньшей мере 4—7%
Штурм [115] исследовал систематическую погрешность в коли-
чественном методе ДТА, выжданную изменением эффективного
коэффициента теплопередачи и теплоемкости образца, а также дер-
жателя образца. Приближенной мерой этих изменений является
логарифм площади пика; отношение логарифмов площадей эталона
и образца дает поправочный множитель для /(-
В. КИНЕТИКА РЕАКЦИЙ
Почти все кинетические методы, используемые в ДТА и ДСК^
основаны на уравнении [59]
Л. > (5.58)
где da/dt—скорость реакции, a f(a, Т) — функция количества
прореагировавшего вещества а и абсолютной температуры Т в мо-
мент времени t. Предполагается также, что скорость выделения
тепла Н прямо пропорциональна da/dt, или
Н = ВР , (5.59}
at
где В — количество тепла на единицу объема и р — плотность об-
разца. Хотя используемое уравнение кинетики реакции зависит от
ДТА и ДСК
199
ее механизма, один из вариантов этого уравнения, не применимый
для реакций, определяемых диффузией или плавлением, а также для
обратимых реакций, имеет следующий вид:
— = А (1 — а)пе~Е1КГ, (5.60)
где А — предэкспоненциальный множитель; а — доля прореаги-
ровавшего образца (0 <g а 1); п — «порядок реакции». Этот тип
уравнения был подвергнут критике со стороны Кларка и др. [116],
считающих его непригодным для реакций разложения в твердых
телах. Они установили, что понятие «порядок реакции» в этом
случае вводит в заблуждение, если оно вообще имеет смысл; кроме
того, энергия активации не очень хорошо определена, поскольку
неизвестно, к какому из протекающих процессов ее отнести.
Хотя результаты кинетических исследований методом ДТА мо-
гут вызывать определенные сомнения, было затрачено немало уси-
лий, чтобы вывести выражения, с помощью которых можно было бы
рассчитать Е и п по данным ДТА или ДСК. Обзоры по определению
кинетики реакций методом ДТА опубликованы Фридманом [117],
Шестаком и Бергреном [118], Бохоном [99], а также Мэрфи [2]
и другими.
Один из первых методов использования кривых ДТА для полу-
чения кинетических данных был предложен Марреем и Уайтом
[119]. Они разработали теорию кривых ТГП и нашли, что: а) фор-
мы кривых ДТА и ТГП аналогичны; б) наибольшая разность тем-
ператур АТмакс примерно соответствует (da/d/)MaKC. Полагая п = 1
.для ряда образцов глины, дважды дифференцируя по температуре
обе части уравнения (5.60), записанного в виде
da A —e!rt . ,
у = (1—а)’ (5,61)
и принимая d2a/dT2 — 0, получаем
—|------= — e~ElRT™*°. (5.62)
^макс
Авторы проинтегрировали также уравнение (5.60), используя
приближение
J e~E/RT dT^ у- e~E,RT (1 — . (5-63)
Го
и с помощью уравнения (5.61) получили
~ 1П (1 ~ О«) « 1 - . (5.64)
Е
200
Глава 5
Киссинжер [62] продифференцировал уравнение (5.61) и полу-
чил
е
4(1/7ыакс) R
где ₽ — скорость нагревания. Для любого значения п [63 ]
Е Л а \л-1 е~£/ЛГмакс
- U «макс) е
х макс
(5.65).
(5.66)
Это уравнение использовалось вместо уравнения (5.61) и {в случае
п #= 0 или п 1 имело вид
П(1 ~~ «макс)"1 «!+(«-!)
(5.67)
Поскольку (и—1)(2/?7’макс/£) 1, уравнение (5.65) можно пред-
ставить в приближенном виде
«а-ОмаксГ1»!. (5.68).
Подставляя (5.68) в (5.67), получаем приближенное уравнение, тож-
дественное (5.61). Отсюда Киссинжер делает заключение, что урав-
нение (5.65) не зависит от порядка реакции. Порядок реакции п
был получен исходя из показателя формы S, определенного как
абсолютное значение отношения тангенсов углов наклонаЛкривой
в точке перегиба
n«l,26S1/2. (5.69)
Рид и др. [120] использовали метод Киссинжера^при исследо-
вании кинетики реакции разложения хлористого фенилдиазония
и нашли, что полученное при этом значение Е отличалось на 42%
от соответствующих значений, полученных с помощью других ме-
тодов. Исходя из этого, они признали метод несостоятельным. К по-
добному выводу пришли Меллинг и др. [59], которые определя-
ли наклон с помощью расчетных кривых ДТА и получали занижен-
ные значения по сравнению с определенными по температуре пика
образца. Следовательно, метод с помощью которого Киссинжер рас-
считывал Е/R, оказался на практике непригодным. Пилоян и др.
1121] указали на следующие недостатки этого метода: 1) требуется
определение нескольких кривых ДТА при различных скоростях
нагревания и 2) требуется специальное устройство для регулирова-
ния температуры по заданной программе. Тем не менее Акита и
Кейз [122] пришли к заключению, что при соблюдении соответст-
вующих условий проведения опытов минимум пика кривой ДТА
совпадает с максимумом скорости реакции в соответствии с пред-
положением Киссинжера.
ДТА и ДСК
201
Пилоян и др. [121 ] разработали кинетический метод, который
также основан на уравнении (5.60). При подстановке
AT = S(daJdt), где S — площадь пика, они получили
1пАТ = С — lnf(a) — (5.70)
где С — константа. Если величина а заключена в пределах 0,05—-
0,8 (примерно у минимума пика), то величиной In/(a) можно прене-
бречь, и уравнение (5.70) упрощается
In АТ = Ci------—.
(5.71)
Ошибки полученных значений Е составляли 15—20%.
По-видимому, наиболее широкое применение получил в ДТА
кинетический метод, развитый Борхардтом и Даниельсом [22] в
1957 г. Этот метод основан на следующих допущениях: а) темпе-
ратура в веществе образца и эталона однородна (очевидно, такое
допущение можно применить только к перемешиваемым жидким,
а не к твердым телам); б) тепло передается только за счет тепло-
проводности (в обычно используемом интервале температур это
условие легко удовлетворяется для жидкостей; теплом, передавае-
мым через термопару, пренебрегают); в) коэффициенты теплопере-
дачи образца и эталона одинаковы; г) теплоемкости образца и эта-
лона должны быть также одинаковыми (это условие удовлетворя-
ется, если исследуются разбавленные растворы образца).
Действительная скорость реакции при любой температуре в за-
висимости от наклона кривой dATIdt и высоты пика АТ опреде-
ляется по формуле
— — = -^2- (с„ 4- ДАТ ), (5.72)
dt КА \ ‘ dt )
где 7V — число молей 'реагента, присутствующее в любой момент
времени и равное начальному числу молей No за вычетом числа
прореагировавших молей:
t
W = (5’73)
о
Расчетным путем получено следующее выражение для констан-
ты скорости реакции k:
. (KAV\n-i Cp(dATIdt) + KAT
k = I----- ----------------------» (5. /4)
\ Na ) [K (A — a) — CpKT]n ' ’
V — объем; A — общая площадь пика; a — площадь к моменту
202
Глава 5
времени t; п — порядок реакции. В случае реакции первого по-
рядка (и = 1)
к Cp^T/dt) + ^T
К(А-а)—СрЬТ ( ‘
В дифференциальной сканирующей калориметрии, где АТ = 0,
общее тепло равно площади пика А
АН — А. (5.76)
Из предположения о прямо пропорциональной зависимости
теплоты реакции от числа молей прореагировавшего реагента следу-
ет, что
dN _ No dH „„
а константа скорости k определяется следующим уравнением:
(5 78>
(Л —а)" V '
Установлено, что применение этого уравнения не ограничивается
областью ДСК; оно может применяться в любом методе, в котором
скорость изменения некоторого физического параметра измеряется
в функции времени и температуры в условиях переменной темпера-
туры. При этом физический параметр должен слабо зависеть от
температуры.
В многочисленных исследованиях уравнение (5.75) упрощалось.
Поскольку Cp(dATIdf) и СрАТ на порядок величины меньше вто-
рых слагаемых соответственно в числителе и знаменателе, ими
можно пренебречь, и тогда
k = -- (5.79)
А —а
для реакции первого порядка и
. _ (AV/N^AT
(А — а)п
в общем случае.
Падманабхан и др. [123], а также Агарвала и Найк [124] ис-
пользовали упрощенное выражение (5.79) для определения кине-
тики реакции термического разложения твердого порошка. С уче-
том первоначальных предположений, сделанных при выводе ис-
ходного уравнения, данное выражение, по-видимому, непригодно
для описания кинетики реакций, протекающих в твердой фазе.
Пренебрегая членом Cp(dAT/dt), Борхардт [125] вывел приб-
лиженное соотношение
(5.80)
ДТА и ДСК
203
__ d (К/Ко) __ _ da ~ АТ <5 gj,
dt dt ~ А 1 '
Кинетический метод Борхардта и Даниельса [22] был подверг-
нут обстоятельной проверке Ридом и др. [120] в 1965 г. Уравнения
'были численно проинтегрированы и получены теоретические кри-
вые ДТА, которые хорошо согласовались с соответствующими
экспериментальными кривыми. Влияние различных параметров,
таких, как скорость нагревания, порядок реакции, Е и т. д., на
кривые ДТА было также установлено численным интегрированием.
Исследуя разложение хлористого фенилдиазония, Рид и др.
[120] сопоставили результаты измерений кинетических констант
несколькими методами (табл. 5.12). Авторы сделали вывод, что ме-
тод Борхардта и Даниельса можно использовать для количествен-
ного определения кинетических параметров, если условия экспери-
мента близки к предположениям, сделанным при выводе теории, а
именно порядок реакции п относится только к одному компоненту,
отсутствуют градиенты температуры в образце и отсутствуют пере-
крывающиеся пики.
Таблица 5.12
Кинетические константы процесса разложения
хлористого фенилдиазония [120]
Метод Е 1g А (А, мин-1)
ккал/моль кДж/моль
ДТА
Рид и др. [120J, метод Борхардта
и Даниельса 28,7 120,2 18,4
Рид и др. [120], метод Киссинжера 16,7 69,9 10,8
Борхардт и Даниельс [22] 28,3 118,5 18,1
29,1 121,8
28,5 119,3
Вада [127] 30,6 128,1
Общепринятый
Кроссли и др. [128] 27,2 113,9 17,3
Мелвин-Хьюз и Джонсон [129] ... 27,025 113,154 17,26
Райх [126] видоизменил уравнение (5.81) и получил выражение
Wo) ап'
(5.82)
204
Глава 5
Фиг. 5.31. Кривые ДСК, ис-
пользованные в кинетическом
анализе Мейкоком [130].
где k — константа скорости
реакции; АТ — высота пика;
IF0 — функция исходной массы
образца;
А = J \Tdt\
о
(5.83}
со со
а = J \Tdt — J ATdt.
о 6
Был разработан разностный
метод расчета параметров Е и
п по двум кривым ДТА, полу-
ченным при двух различных
скоростях нагревания.
Используя кривые ДСК,
показанные на фиг. 5.31, и
предполагая линейную зависи-
мость в аррениусовых координа-
тах lg£—1/Т, Мейкок [130]
установил, что Е можно опреде-
лять с помощью выражения
(In d, —lnd2)
1/Л-1/Т,
4,581g (dt/d2)
1/Л-1/Т2 ’
(5.84}
где di и d2 — скорости выделения тепла при температурах соответ-
ственно Tt и Т2.
Ряд других описанных в литературе методов базируется на
приведенных выше уравнениях. Кинетические методы, описываю-
щие процессы деструкции полимеров, рассматриваются в книге
Рейха и Стайвейла [131 ].
Г. ДИФФЕРЕНЦИАЛЬНАЯ СКАНИРУЮЩАЯ КАЛОРИМЕТРИЯ’
1. ВВЕДЕНИЕ
Термин «дифференциальная сканирующая калориметрия» стал
в последние несколько лет источником терминологической путани-
цы в термическом анализе, вызванной тем, что в настоящее время
имеется два совершенно различных типа приборов, имеющих одно
и то же название. Эти два прибора имеют принципиальные различия,
и только один из них предназначен для ДТА. Имеются в виду следую-
щие приборы:
ДТА и ДСК
205
а) дифференциальные сканирующие калориметры, регистри-
рующие тепловой поток [(dq/dt)&T=o] (фирмы «Перкин-Элмер», «Дель»
татерм» и др.);
б) «дифференциальные сканирующие калориметры», регистри-
рующие на самом деле дифференциальную температуру и являю-
щиеся приборами ДТА (АТ =# 0) (фирмы «Дюпон», «Стоун», «Фи-
шер», «Линзейс» и др.)
Фиг. 5.32. Рекомендуемое представление кривой ДСК.
Термин «дифференциальная сканирующая калориметрия» (ДСК),
очевидно, впервые использовали Уотсон и др. [132] для описания
метода и экспериментальной техники, разработанной в последнее
время (1963 г.) фирмой «Перкин-Элмер». В этом методе образец
и эталон нагреваются или охлаждаются с одинаковой скоростью,
причем их температуры поддерживаются одинаковыми. Экспери-
ментальные кривые представляют собой зависимость теплового
потока dH/dt (мкал/с) от температуры. Типичные кривые ДСК по-
казаны на фиг. 5.32. Пик, направленный вверх, является эндотер-
мическим (соответствует увеличению энтальпии), а пик, направ-
ленный вниз, — экзотермическим. По внешнему виду кривая ДСК
очень похожа на кривую ДТА, за исключением принятых единиц
измерения по оси ординат. Как и в методе ДТА, площадь пика, ог-
раничиваемая кривой ДСК, прямо пропорциональна изменению
энтальпии:
Площадь = КЛНт, (5.85)
причем К в данном случае не зависит от температуры.
206
Глава 5
Основное различие приборов ДСК схематически показано на
фиг. 5.33. Схема дифференциального сканирующего калориметра
ДСК соответствует описанной О’Нейлом [133], а схема прибора
«ДСК», являющегося фактически прибором ДТА, описана Баксте-
ром [134] и Дэвидом [135]. Основное различие между двумя при-
борами заметно сразу, хотя по внешнему виду кривые похожи. Тип
Дск
„ДСК" (ДТА)
7Д77777777//7777777
Фиг. 5.33. Разница между методами
ДСК (а) и «ДСК» (ДТА) (б) [133].
информации, получаемой с помощью кривых ДСК и ДТА, сходен,
однако до недавнего времени держатели образца в методе ДСК огра-
ничивали температуру измерений величиной 500° С, в то время
как держатели в методе ДТА способны работать при гораздо более
высокой температуре. Для количественных измерений, например
определения АД, метод ДСК использовать проще, поскольку кали-
бровочный коэффициент К не зависит от температуры. Также упро-
щены расчеты в случае перекрывающихся пиков, так как использу-
ется только одно значение коэффициента К-
ДТА и ДСК.
207
Другие калориметры — фирм «Калверт», «Дельтатерм», и др. —
рассматривались в обзоре Вильхойта [136]. Опубликована библио-
графия, охватывающая период применения ДСК с 1964 по 1970 г.
[137].
2. ТЕОРИЯ
Теория калориметров фирмы «Перкин-Элмер» представлена в
работах О’Нейла [133], Грея [60] и Флинна [138]; теория калори-
метров фирм «Дюпон» и «Стоун» — в работах Бакстера [134] и Дэ-
вида [135]. Теория последних двух приборов уже рассматривалась,
в данной главе.
Фиг. 5.34. Применение уравнения (5.86) к кривой ДСК [60],
dH = + ci гс J-S-
dt dt + dt dt*
n in
Используя кривую ДСК (фиг. 5.34), Грей [601 вывел основное*
уравнение, связывающее dH/dt с измеряемыми величинами, как.
и в случае ДТА:
= + (5.86>
at at at at2
Выражение для dH/dt, как и в теории ДТА, является суммой трех
членов. Между ДСК и ДТА имеются два различия: а) термическое
сопротивление Й входит только в третий член уравнения и б) пло-
щадь пика кривой равна \q = —А77. Калибровочный коэффици-
ент служит для преобразования единиц площади в единицы коли-
чества тепла (калории) и является скорее коэффициентом преобра-
зования электрической энергии, чем термической константой, ис-
пользуемой в ДТА.
208
Глава 5
Несколько более усложненную трактовку кривых ДСК предло-
жил Флинн [138], который рассматривает три критических пара-
метра:
а) постоянный наклон кривой, вызванный линейным увеличе-
нием температуры и пропорциональный члену, обусловленному
граничной проводимостью;
б) температуру начала превращения Tit получаемую в точке
пересечения переднего фронта пика с базовой линией;
в) константу спада kt, связанную с возвратом к базовой линии
по завершении превращения.
Наклон кривой может быть описан с помощью закона охлажде-
ния Ньютона
-^- = Ah(T2 — Ti), (5.87)
at
где h — коэффициент теплопроводности границы; А — площадь
поверхности раздела; (Т2—Тt) — разность температур образца и
держателя. Температура образца описывается следующей фор-
мулой:
Л = То + — f dt, (5.88)
me J dt
где То — начальная температура; m — масса образца; с — его
теплоемкость. Т1 будет оставаться равной температуре превраще-
ния Т} для времени пребывания tt в процессе превращения, опре-
деляемого из уравнения
( дд dt = тДЯг, (5.89)
J dt
о
где AHf — теплота превращения.
Из этих уравнений при условии постоянства теплоемкости сле-
дует выражение для постоянного в ходе превращения наклона кри-
вой
Наклон = рЛД = km$cm. (5.90)
Температура начала превращения Tt будет равна
= тт + = тт+ (5.91)
nA km
.а константа спада в конце превращения
^=— (5.92)
ст
В ДСК обычно предполагается, что скорость изменения темпера-
туры эталона соответствует программируемой температуре. Бреннан
ДТА и ДСК
209
и др. 1139] рассмотрели эту проблему аналитически и нашли, что
скорость изменения температуры определяется постоянной времени
эталона. Если постоянная времени не более 1 с, то указанное пред-
1 сложение выполняется по прошествии нескольких секунд.
В противном случае такое предположение неверно.
ЛИТЕРАТУРА
1. Le Chatelier Н., Bull. Soc. Franc. Mineral, 10, 203 (1887).
2. Murphy С. B., Minerals Sci. Eng., 2, 51 (Oct. 1970).
3. Ashley H. E„ Ind. Eng. Chem., 3, 91 (1911).
4. Wholin R., Sprechsaal, 46, 749, 767, 781 (1913).
Б. Rieke R„ Sprechsaal, 44, 637 (1911).
6. Wallach H„ Compt. Rend., 157, 48 (1913).
7. Mellor J. W., Holdcraft A. D., Trans. Brit. Ceram.Soc., 10, 94 (1910—1911).
8. Roberts-Austen W. C., Proc. Inst. Meeh. Engrs (London) (1899); Metallo-
graphist, 2, 186 (1899).
9. Burgess G. K., Nat. Bur. Std. (U. S.) Bull., 5, 199 (1908—1909).
10. Speil S., Berkelhamer L. H., Pask J. A., Davis B., U. S. Bur. Mines,
Tech. Papers, 664 (1945).
11. Houldsworth H. S., Cobb J. W., Trans. Brit. Ceram. Soc., 22, 111 (1922—
1923).
12. Fenner C. N„ Am. J. Sci., 36, 331 (1913).
13. Berkelhamer L. H., U. S. Bur. Mines Rept Invest., R13762 (1944).
14. Kerr P. F., Kulp J. L., Am. Mineralogist, 33, 387 (1948).
15. Kauffman A. J., Dilling E. D., Econ. Geol., 45, 222 (1950).
16. Foldvari-Vogl M., Acta Geol. Acad. Sci. Hung., 5, 1 (1958).
17. Norton F. H., J. Am. Ceram Soc., 22, 54 (1939).
18. Grim R. E., Ann. N. Y. Acad. Sci., 53, 1031 (1951).
19. Mackenzie R. C., Differential Thermal Analysis of Clays, Central Press,
Aberdeen, Scotland, 1957.
20. Stone R. L., J. Am. Ceram. Soc., 35, 76 (1952).
21. Borchardt H. J., Daniels F„ J. Phys. Chem., 61, 917 (1957).
22. Borchardt H. J., Daniels F., J. Am. Chem. Soc., 79, 41 (1957).
23. Wendland W. W., Technique of Inorganic Chemistry, Vol. I, Jonas-
sen H. B., Waissberger A., eds., Interscience, N. Y., 1963, p. 209.
24. Wendlandt W. W„ J. Chem. Educ., 49, A623 (1972).
25. Murphy С. B., Anal. Chem., 30, 867 (1958).
26. Murphy С. B„ Anal. Chem., 44, 513R (1972).
27. Smothers W. J., Chiang Y., Differential Thermal Analysis: Theory and
Practice, Chemical Publ. Co., N. Y., 1958.
28. Smothers W. J., Chiang Y., Differential Thermal Analysis: Theory and
Practice, 2nd ed., Chemical Publ. Co., N. Y., 1966.
29. Garn P. D., Thermoanalytical Methods of Investigation, Academic Press,
N. Y., 1965.
30. Mackenzie R. C., ed. Differential Thermal Analysis, Vol. 1, Academic
Press, Lnd., 1970.
31. Kissinger H. E., Newman S. B., Differential Thermal Analysis in Analy-
tical Chemistry of Polymers, Vol. XII, Pt. II, Kline G. M., ed., Intersci-
ence, N. Y., 1962.
32. Gordon S., Encyclopedia of Science and Technology, Vol. 13, McGraw-Hill,
N. Y„ 1960, pp. 553-559.
33. Gordon S., Campbell C., Handbook of Analytical Chemistry, Meites L., ed.,
McGraw-Hill, N. Y., 1963.
8-230
210
Глава 5
34. Barrail Е. М., Johnson J. F., Techniques and Methods of Polymer Evalua-
tion, Slade P. E., Jenkins L. T., eds., Vol. 1, Marcel-Dekker, N. Y., 1966,
Ch. 1.
35. Dawid D. J., Techniques and Methods of Polymer Evaluation, Slade P. E.,
Jenkins L. T., eds., Vol. 1, Marcel-Dekker, N. Y., 1966, Ch. 2.
36. Barrall E. M., Guide to Modern Methods of Instrumental Analysis,
Gouw T. H„ ed., Wiley-Interscience, N. Y., 1972, Ch. 12.
37. Schultze D., Differentialthermoanalyze, Deutscher Verlag der Wissen-
schaften, Berlin, 1969.
38. Ramachandran V. S., Differential Thermal Analysis in Cement Chemistry,
Chemical Publ. Co., N. Y., 1969.
39. Wunderlich B., Physical Methods of Chemistry, Weissberger A., Rossi-
ter B. W., eds., Vol. 1, Pt. V, Wiley-Interscience, N. Y., 1971, Ch. VIII.
40. Porter R. S., Johnson J. F., eds., Analytical Calorimetry, Vol. 1, Plenum
Press, N. Y., 1968.
41. Porter R. S., Johnson J. F., eds., Analytical Calorimetry, Vol. 2, Plenum-
Press, N. Y„ 1970.
42. Schwenker R. F., Garn P. D., eds., Thermal Analysis, Vols. 1 and 2, Aca-
demic Press, N. Y., 1969.
43. Void M. J., Anal. Chem., 21, 683 (1949).
44. Boersma S. L., J. Am. Ceram. Soc., 38, 281 (1955).
45. Lukaszewski G. M., Lab. Practice., 14, 1277 (1965).
46. Lukaszewski G. M., Lab. Practice, 14, 40 (1965).
47. Lukaszewski G. M., Lab. Practice, 15, 75 (1966).
48. Lukaszewski G. M., Lab. Practice, 15, 82 (1966).
49. Lukaszewski G. M., Lab. Practice, 15, 187 (1966).
50. Lukaszewski G. M., Lab. Practice, 15, 302 (1966).
51. Lukaszewski G. M., Lab. Practice, 15, 431 (1966).
52. Lukaszewski G. M., Lab. Practice, 15, 551 (1966).
53. Lukaszewski G. M., Lab. Practice, 15, 664 (1966).
54. Lukaszewski G. M., Lab. Practice, 15, 762 (1966).
55. Lukaszewski G. M., Lab. Practice, 15, 861 (1966).
56. David D. J., Anal. Chem., 36, 2162 (1964).
57. David D. J., Lab. Equip. Digest, 110 (June — Aug. 1968).
58. Pacor P., Anal. Chim. Acta, 37, 200 (1967).
59. Melting R., Wilburn F. W., McIntosh R. M., Anal. Chem., 41, 1275 (1969)’.
60. Gray A- P-. Analytical. Calorimetry, Porter R. S., Johnson J. F., eds.,
Plenum Press, N. Y., 1968, p. 209.
61. Garn P. D., Thermoanalytical Methods of Investigation, Academic Press,
N. Y„ 1965, p. 60.
62. Kissinger H. E., J. Res. Nat. Bur. Standards, 57, 217 (1956).
63. Kissinger H. E., Anal. Chem., 29, 1702 (1957).
64. Barrall E. M., Rogers L. B., J. Inorg. Nucl. Chem., 28, 41 (1966).
65. Langer A. M., Kerr P. F., DuPont Thermogram, 3, № 1, 1 (1968).
66. Vassallo D. A., Harden J. C., Anal. Chem., 34, 132 (1962).
67. Johnson J. F., Miller G. W., Thermochim. Acta, 1, 373 (1970).
68. Sarasohn I. M., DuPont Thermogram, 2, № 1, 1 (1965).
69. Stone R. L., Anal. Chem., 32, 1582 (1960).
70. Mettler Thermal Technique Series, Tech. Bull. T-106.
71. David D. J., Am. Lab., 35 (Jan. 1970).
72. Locke С. E., Proc, of the Third Toronto Symp. on Thermal Analysis,
McAdie H. G., ed., Chemical Inst, of Canada, Toronto, 1969, p. 251
73. Garn P. D., Anal. Chem., 37, 77 (1965).
74. Levy P., Nieuweboer G., Semanski L. C., Thermochim. Acta, 1, 429 (I960).
75. David D. J., Anal. Chem., 37, 82 (1965).
76. Wilburn F. W., Hesford J. R., Flowers J. R., Anal. Chem., 40, 777 (1968).
77. Dosch E. L., Thermochim. Acta, 1, 367 (1970).
ДТА и ДСК
211
78. Lehmann Н., DasS. S., Paetsch Н. Н., Tenind.-Ztg. und Keram.Rundschau,
1 (1954).
79. Bayliss Р., Warne S. St., J., Am. Mineralogist, 47, 775 (1962).
80. Arens P. L., A Study of the Differential Thermal Analysis of Clays and
Clay Minerals, Excelsiors Foto-Ottset, The Hague, 1951.
81. Gerard-Hirne J., Lamy C., Bull. Soc. Franc. Ceram., 26 (1951).
82. Webb T. T., Nature, 174 686 (1954).
83. Mackenzie R. C., Nature, 174, 688 (1954).
84. Sarasohn I. M., ACS Short Course, Am. Chemical Soc., Washington, D. C.,
1969.
85. Smyth H. T., J. Am. Ceram. Soc., 34, 221 (1951).
86. Barrall E. M., Rogers L. B., Anal. Chem., 34, 1101 (1962).
87. David D. J., Ninke D. A., Duncan B., Am. Lab., 31 (Jan. 1971).
88. Hauser R. E., B. S. Thesis, N. Y., State College of Ceramics, Alfred, N. Y.,
1953.
89. Norton F. H„ J. Am. Ceram. Soc., 22, 54 (1939).
90. Grimshaw R. W., Heaton E., Roberts A. L., Trans. Brit. Ceram. Soc., 44,
76 (1945).
91. Carthew A. R., Am. Mineralogist, 40, 107 (1955).
92. Barrall E. M., Rogers L. B., Anal. Chem., 34, 1106 (1962).
93. Negishi A., Ozawa T., Thermochim. Acta, 2, 89 (1971).
94. Wada M., lida Y., Ozaki S., Japan J. Appl. Phys., 8, 1569 (1969).
94a. Dean L. A., Soil Sci., 63, 95 (1947).
95. Gruver R. M., J. Am. Ceram. Soc., 31, 323 (1948).
96. Bollin E. M., Bauman A. J., Analytical Calorimetry, Porter R. S., John-
son J. F., eds., Vol. 2, Plenum Press, N. Y., 1970, p. 339.
97. Barshad I., Am. Mineralogist, 37, 667 (1952).
98. de Jong G. J., J. Am. Ceram. Soc., 40, 42 (1957).
99. Bohon R. L., Proc, of the First Toronto Symp. on Thermal Analysis,
McAdie H. G., ed., Chemical Inst, of Canada, Toronto, 1965, p. 63.
100. Ozawa T., Bull. Chem. Soc. Japan., 39, 2071 (1966).
101. Wittels M., Am. Mineralogist, 36, 615 (1951).
102. Wittels M., Am. Mineralogist, 36, 760 (1951).
103. Collins W. E., Analytical Calorimetry, Porter R. S., Johnson J. F., eds.,
Vol. 2, Plenum Press, N. Y., 1970, p. 353.
104. David D. J., Analytical Calorimetry, Porter R. S., Johnson J. F., eds.,
Vol. 2, Plenum Press, N. Y., 1970, p. 369.
105. Bohon R. L„ Anal. Chem., 35, 1845 (1963).
106. Chiu J., Analytical Calorimetry, Porter R. S., Johnson J. F., eds., Vol. 2,
Plenum Press, N. Y., 1970, p. 171.
107. Wendlandt W. W., Williams J. R., International Confederation of Ther-
mal Analysis III, Davos, Switzerland, Paper I, Aug. 1971.
108. Currell B. 4?., Thermal Analysis, Schwenker R. F., Garn P. D., eds.,
Vol. 2, Academic Press, N. Y., 1969, p. 1185.
109. Wiedemann H. G., van Tets A., Z. Anal. Chem., 233, 161 (1968).
110. Wiedemann H. G., van Tets A., Thermochim. Acta, 1, 159 (1970).
111. Ramachandran V. S., Sereda P. J., Nature, Physical Science, 233, 134
(1971).
112. Davis С. E., Holdridge D. A., Clay Minerals, 8, 193 (1969).
113. Ramachandran V. S., J. Thermal. Anal., 3, 181 (1971).
114. Schwenker R. F., Whitwell J. C., Analytical Calorimetry, Porter R. S.,
Johnson J. F., eds., Vol. 1, Plenum Press, N. Y., 1968, p. 249.
115. Sturm E., Thermochim. Acta, 4, 461 (1972).
116. Clarke T. A., Evans E. L., Robbins K. G-, Thomas J. M., Chem. Comm.,
266 (1969).
117. Friedman H. L., Proc, of the Third Toronto Symp. on Thermal Analysis,
McAdie H. G., ed., Chemical Inst, of Canada, Toronto, 1969, p. 127.
8*
212
Глава 5
118. Sestak J., Berggren G., Svazek, 64, 695 (1970).
119. Murray P. White J., Trans. Brit. Ceram. Soc., 54, 204 (1955).
120. Reed R. L., Weber L., Gottfried B. S., I and EC Fundamentals, 4, 38
(1965).
121. Piloyan G. O., Ryabchikov I. D., Novikova O. S., Nature, 212, 1229
(1966).
122. Akita K., Kase M., J. Phys. Chem., 72, 906 (1968).
123. Padmanabhan V. M., Saraiya S. C., Sundaram A. K-, J. Inorg. Nucl.
Chem., 12, 356 (1960).
124. Agarwala R. P., Naik M. C., Anal. Chim. Acta, 24, 128 (1960).
125. Borchardt H. J., J. Inorg. Nucl. Chem., 12, 252 (1960).
126. Reich L., J. Inorg. Nucl. Chem., 28, 1329 (1966).
127. Wada G., Nippon Kagaku Zasshi, 1956 (1960).
128. Crossley M. L., Kienle R. H., Benbrook С. H., J. Am. Chem. Soc., 72,
1400 (1940).
129. Moelwyn-Hughes- E. A., Johnson P., Trans. Faraday Soc., 36, 948 (1940).
130. Maycock J. N., Thermochim. Acta, 1, 389 (1970).
131. Reich L., Stivala S. S., Elements of Polymer Degradation, McGraw-Hill.
N., Y., 1971.
132. Watson E. S., O’Neil M. J., Justin J., Brenner N., Anal. Chem., 36 1233
(1964).
133. O’Neill M. J., Anal. Chem., 36, 1238 (1964).
134. Baxter R. A., Thermal Analysis, Schwenker R. F., Garn P. D., eds.,
Academic Press, N. Y., 1969, p. 65.
135. David D. J., J. Thermal Anal., 3, 247 (1971).
136. Wilhoit R. C., J. Chem. Educ., 44, A571 (1967).
137. Perkin-Elmer Corp., Norwalk, Conn., May 1970.
138. Flynn J. H., Status of Thermal Analysis, Menis O., ed., NBS Special
Publication 338, U. S. Gov’t. Printing Office, Washington, D. C., Oct.
1970, p. 119.
139. Brennan W. P., Miller B„ Whitnell J. C., Thermochim. Acta, 2, 354 (1971).
Глава 6
ПРИБОРЫ |ДЛЯ ДИФФЕРЕНЦИАЛЬНОГО ТЕРМИЧЕСКОГО
АНАЛИЗА И ДИФФЕРЕНЦИАЛЬНОЙ
СКАНИРУЮЩЕЙ КАЛОРИМЕТРИИ
А. ДИФФЕРЕНЦИАЛЬНЫЙ ТЕРМИЧЕСКИЙ АНАЛИЗ
L ВВЕДЕНИЕ
Как и в термогравиметрии, в ДТА используется большое число
приборов различных типов. Причиной такого многообразия яви-
лось, по-видимому, то обстоятельство, что до создания надежных
промышленных приборов ДТА каждый исследователь проектировал
и создавал собственный прибор. Излишне говорить о том, что все
эти приборы различались по конструкции. В литературе имеются
описания разных типов держателей образцов, нагревателей, уси-
лительных устройств для определения дифференциальной темпера-
туры АТ, устройств для регулирования температуры по заданной
программе и регистрирующих устройств. Различия в конструкциях
приборов обусловили несоответствия кривых ДТА, полученных
Фиг. 6.1. Схема типичного прибора ДТА.
1—печь; 2— датчики температуры; 3 — образец; 4 — эталон.
214
Глава 6
для идентичных материалов в разных лабораториях. Фактически
почти невозможно было точно воспроизвести результаты, получен-
ные на другом приборе в другой лаборатории, что вызывало разно-
гласия и полемику. Появление в конце 50-х годов хороших промыш-
ленных приборов ДТА привело к некоторой стандартизации ре-
зультатов. В настоящее время стало возможным воспроизведение
данных ДТА, полученных, разумеется, при одинаковых условиях
пиролиза.
Схема типичного прибора ДТА показана на фиг. 6.1. Основны-
ми его элементами являются: печь или нагревательное устройство,
держатель образца, усилитель слабых напряжений постоянного
тока; датчик дифференциальной температуры, устройство для ре-
гулирования температуры печи по заданной программе, самописец
и аппаратура для поддержания соответствующего состава атмосферы
в печи и держателе образца. По такой схеме было разработано много
вариантов приборов ДТА, но все они основаны на измерении диф-
ференциальной температуры образца в функции температуры или
времени (в предположении линейной зависимости температуры от
времени).
2. ДЕРЖАТЕЛИ ОБРАЗЦОВ
Одйой из наиболее важных частей прибора ДТА является держа-
тель образца (и идентичный держатель эталона). Имеются описания
всевозможных держателей образцов, применяемых как в промыш-
ленных приборах, так и в установках, созданных отдельными ис-
следователями. Применение определенного типа держателя образ-
ца зависит от свойств и величины образца, а также от максималь-
ной температуры испытаний [1 ]. Держатели образцов изготавли-
вались из окиси алюминия и двуокиси циркония ZrO2, боросили-
катного стекла, стекла викор, плавленого кварца, бериллия,
нитрида бора, графита, нержавеющей стали, никеля, алюминия,
платины или платиновых сплавов, серебра, меди, вольфрама, ма-
териала образца и многих других. Некоторые типичные держате-
ли образцов, применяемые в приборах ДТА, изображены нафиг. 6.2.
На фиг. 6.2, а образец массой ~ 100 мг запрессовывался в закрытую
с одного конца трубку, в которую затем вставлялся керамический
стержень с термопарой [2]. Этот тип держателя образца позволяет
производить исследования в собственной атмосфере, создаваемой
образцом. Однако он непригоден для испытания образцов, которые
при нагревании расплавляются. Для определения теплоты взрыва
ряда взрывчатых веществ Бохон [3] использовал изохорический
держатель образца (фиг. 6.2, б), который состоял из корпуса из
нержавеющей стали и крышки, плотно соединявшейся (на резьбе
с медной прокладкой) с камерой. Внутренний объем камеры состав-
лял ~0,085 мл, а масса образца ~25 мг. Держатель образца вы-
Приборы для ДТА и ДСК
215
держивал давление до 7 кПа (изб.). Мазирес [4] создал держатель
для микрообразцов массой 1—200 мкг (фиг. 6.2, в). Образец поме-
щался в углубление, высверленное в спае термопары. Имеется опи-
сание держателя чашечной формы подобного типа для образцов
массой 0,1—10 мг. Работа с микрограммовыми навесками образцов
связана, конечно, с определенными трудностями.
Держатели образцов, используемые в приборах фирмы «Стоун»,
изображены на фиг. 6.2, г—е. Для образцов массой 10—200 мг
применялись держатели в виде маленьких чашек (фиг. 6.2, г)
из алюминия, нержавеющей стали, никеля, сплавов платины или
палладия. Для образцов массой 0,1—20 мг использовался очень
чувствительный держатель в виде кольцевой термопары (фиг. 6.2, д).
Тарелочки для образцов в последнем случае изготавливались из
алюминия, нержавеющей стали или платины штамповкой. Держа-
тель образца, показанный на фиг. 6.2, е, обеспечивает хорошее ре-
гулирование динамической газовой атмосферы. Поток газа прохо-
дит через образец и эталон; однако такой держатель непригоден
для исследования процессов плавления.
Держатели образцов, применяемые в термоанализаторах фир-
мы «Меттлер», показаны на фиг. 6.2, ж—к. В держателе, представ-
ленном на фиг. 6.2, ж, образец помещался в небольшую чашку или
тигель, которые устанавливались на небольшие круглые диски,
находившиеся в контакте со спаем термопары. Держатель образца
в виде блока из окиси алюминия показан на фиг. 6.2, з. Образец
помещался в тигель из платины или другого металла. Для больших
навесок образца можно применять держатель типа, показанного
на фиг. 6.2, и. В этом случае контейнеры изготавливали из окиси
алюминия или различных металлов. Для образцов с малой массой
можно рекомендовать держатель с микротиглями (фиг. 6.2, к).
Держатели образцов, используемые в приборах ДТА фирмы «Лин-
зейс», показаны на фиг. 6.2, л—н. Образец загружался в съемные
гильзы (фиг. 6.2, л) из металла или керамики. Держатель этого
типа удобен для очистки, поскольку гильза легко снимается с об-
разца. Его недостатком является непосредственный контакт термо-
пары с образцом, что может привести к коррозионному воздействию
материала образца на термопару и к изменению ее э.д.с. Подобный
недостаток присущ держателям образца в виде зонда (фиг. 6.2, м, о),
в которых термопара погружалась внутрь образца. Стеклянный
контейнер для образцов (фиг. 6.2, о) обычно изготавливался из
капиллярной трубки диаметром 1—2 мм. Держатель для исследо-
вания образцов, требующих горизонтального положения, показан
на фиг. 6.2, н.
Держатель образца для исследований при очень высоких темпе-
ратурах (2200° С) показан на фиг. 6.2, п. Он изготавливается из
сплава вольфрама с рением (26%) и применяется в приборах фир-
мы «Меттлер».
к
Фиг. 6.2. Держатели образцов, применяемые в” приборах’ДТА.
/ — образец; 2 — термопара; 3 — керамическая трубка; 4 — керамический стержень; 5 — резь-
бовая крышка; 6 — чашка; 7— спай эталонной термопары; 8— спай дифференциальной тер-
мопары; 9— тарелочка; 10— дифференциальная термопара; 11— эталонная термопара;
12— контейнер из стекла, металла или керамики; 13— ячейка для эталона; 14 — отверстия
для выхода газа; 15— алюминиевый блок; 16 — стенка печи; 17 — керамические изоляторы;
18 — камера третьей ячейки для измерения температуры; 19 — ячейка для образца с намо-
танным на алюминиевый цилиндр образцом; 20— спаи дифференциальных термопар;
21 — алюминиевые шайбы; 22 — керамические трубки; 23 — стеклянная или кварцевая трубка.
218
Глава 6
Уникальный держатель образца (фиг. 6.2, р) разработан Мил-
лером [5] для изучения текстильных волокон и пряжи. Образцы
волокна наматывались на алюминиевый цилиндр но канавкам, вы-
резанным на его наружной поверхности. Три одинаковых цилиндра
монтировались симметрично относительно центра вертикального
нагревателя; по одному предназна-
чались для образца и эталона, а
третий использовался для измерения
температуры печи.
Кривые ДТА образцов, запаян-
ных в трубки, определяются с по-
мощью держателей типа представлен-
ного на фиг. 6.2, с. Держатели типа
запаянной трубки будут описаны
ниже.
Для увеличения числа исследуе-
мых образцов в данный период
времени можно использовать много-
позиционные держатели, позволяю-
щие исследовать одновременно 3—6
образцов. Два держателя этого ти-
па показаны на фиг. 6.3.
Держатель (фиг. 6.3, а), разра-
ботанный Кульпом и Керром [6, 71,
имел шесть ячеек для образцов и
три для эталонов. Эти ячейки были
высверлены в никелевом блоке (глу-
бина ячеек 9,5 мм, диаметр 6,3 мм).
Для записи разности температур
каждой пары образец — эталон ис-
пользовался многоточечный само-
писец.
Фиг. 6.3. Многопозицион-
ные держатели образцов,
а —’держатель для шести образ-
цов; б — держатель для четырех
образцов; 1 — углубления для об-
разцов; 2 — блок; 3 — термопара;
4 — щели; 5 — контрольная тер-
мопара; 6 — образец, 7 —эталон
(четыре пары).
' Более современный многоэлементный держатель образца
(фиг. 6.3, б) используется в приборе фирмы «Дельтатерм». Наряду
с четырьмя изолированными парами образец — эталон применяется
контрольная термопара. Радиальные щели, прорезанные с неравно-
мерным шагом по окружности, предотвращают возникновение тер-
мических градиентов между образцом и эталоном, расположенным
в смежной радиальной части блока. Для усиления сигналов исполь-
зовались четыре усилителя постоянного тока, соединенных с че-
тырехканальным самописцем.
Кокс и Макглинн [8] также разработали многопозиционный
блочный держатель для одновременных испытаний пяти образцов.
Довольно оригинальную конструкцию предложил Бурр [9]. В его
приборе сигналы АТ от пяти образцов в алюминиевом блоке запи-
сывались с интервалами 36с многоканальным самописцем.
Приборы для ДТА и ДСК
219
3. СИСТЕМЫ ОПРЕДЕЛЕНИЯ ДТ и Т
Выбор устройства для определения температуры зависит от мак-
симальной температуры испытаний, химической реакционной спо-
собности образца, чувствительности усилителя постоянного тока
и регистрирующей аппаратуры. Чаще всего дифференциальная
температура определяется с помощью термопар, хотя применяются
также термостолбики, термисторы и сопротивления. Для исследо-
ваний при высоких температурах может быть использован также
оптический пирометр.
Сигнал термопары приблизительно пропорционален разности
температур двух спаев (эффект Зеебека), и она вполне пригодна для
измерений дифференциальной температуры [10]. Термопару можно
также использовать для измерений абсолютной и относительной
температур, если один из спаев (эталонный спай) держать при пос-
тоянной температуре. Термопары, обычно применяемые в приборах
ДТА, перечислены в табл. 6.1. Указанные в таблице температур-
Таблица 6.1
Характеристики нескольких типичных термопар при 25 °C [10]
Обозначение1) Металл № 1 (положит.) Металл № 2 (отриц.) Максимальная температура, °C Термо-э.д.с., мкВ/град
S Платина Платина — родий (10% Rh) 1600 5,5
т Медь Константан 250 40
Y, J Железо » 450 51
Е Хромель » 1000 59
К Алюмель 1000 41
Вольфрам Вольфрам — рений (26% Re) 2200 3,3
*) Приняты ISA (Международной федерацией национальных ассоциаций по стандартиза-
ции).
ные пределы относятся к достаточно точным измерениям на воздухе
при помощи термопары с диаметром провода 0,8 мм. В интервале
температур от —150 до 250° С чаще всего используется медь-конс-
тантановая термопара, имеющая при этих температурах очень ста-
бильные и воспроизводимые характеристики. Термопары из благо-
родного металла, особенно платино-платинородиевая (10% Rh)
рекомендуются для использования при измерениях, требующих
высокой точности, в интервале температур 500—1200° С. Платино-
викелевые термопары также применяются в этом интервале темпе-
220
Глава 6
ратур и имеют большую термо-э.д.с. (~40 мкВ/град). Для темпе-
ратур до 3000° С предлагалось использовать термопару карбид
тантала — графит 111].
а
Фиг. 6.4. Этапы изготовления тонкопленочной термопары [15, 16].
а — напыление золота на незакрытую часть поверхности; б — нанесение никеля гальваниче-
ским способом; в — напыление золота на незакрытую часть поверхности; г — присоединение
проводов.
С целью увеличения выходного сигнала дифференциальных тер-
мопар без применения усилителя использовались термостолбики
[8, 12, 13]. Преимущество такой системы состоит в том, что при
увеличенном сигнале уровень шумов остается низким благодаря
отсутствию электронных схем. В приборе ДТА-2000 фирмы «Меттлер»
используются термостолбики с пятью термопарами для измерения
АТ с чувствительностью 115 мкВ/град.
Обычно термопара состоит из двух проводов, соединенных то-
чечной сваркой с образованием термоспая. Предлагались и другие
типы термопар—тонкопленочные [14—16] и дисковые [17, 18].
При использовании термопар в виде тонкой пленки исключается
операция подбора равноценных спаев соответствующих проводов.
Тонкопленочные термопары можно сделать очень легкими, и их
можно изготовить с высокой точностью напылением тонких пленок
различных металлов, образующих термоспаи в местах перекрытия.
На фиг. 6.4 показаны этапы изготовления тонкопленочной термо-
Приборы для ДТА и ДСК
221
пары никель — золото [15, 16]. На кварцевую пластину в вакууме
напылялось золото, а затем гальваническим способом наносился
никель. Образцы помещались в алюминиевые капсулы и испытыва-
лись в интервале температур от —125 до 500° С. Термо-э.д.с. таких
Фиг. 6.5. Термопара дискового типа,
описанная Бакстером [18].
1 — камера образца; 2 — крышка; 8 — впускное
отверстие для продувки газом; 4 — кольцо из
серебра; 5 — капсула для эталона; 6 — спай
термопары; 7 — нагревательный блок; 8 — хро-
мелевый провод; 9 — алюмелевый провод; 10 —
термоэлектрический диск (константан); И—
капсула для образца.
термопар увеличивалась от
10 мкВ/град при 25°С до 25
мкВ/град при 200°С.
Термопара и держатель
образца дискового типа по-
казаны на фиг. 6.5 [18].
Константановый диск слу-
жил основным каналом теп-
лообмена между образцом,
нагревателем и окружающей
средой, а также являлся од-
ной из половин термопары
для измерения АГ. Хромеле-
вый провод подсоединялся
во вдавленных углублениях,
образуя таким образом хро-
мель-константановую диф-
ференциальную термопару.
Эта система применяется в
интервале температур от
—150 до 600° С. Ямамото и
др. [17] использовали в ка-
честве одной из половин тер-
мопары хромель в виде плоской гантели, состоявшей из двух
круглых дисков, соединенных узкой полоской. Провода алюмеля,
припаянные к центру каждого диска, служили второй полови-
ной термопары.
Как и во многих других аналитических методах, шкала темпе-
ратур, используемая в дифференциальном термическом анализе
и дифференциальной сканирующей калориметрии, должна быть
прокалибрована с помощью материалов, имеющих известные тем-
пературы переходов. Международная конфедерация по термиче-
скому анализу (ICTA) приняла активное участие в работе по выбору
эталонных материалов для этой цели [19] и совместно с Американ-
ским национальным бюро стандартов (NB S) содействовала их про-
мышленному выпуску [20]. Менис и Стерлинг [20] опубликовали
результаты совместной работы ICTA с 24 лабораториями. С по-
мощью десяти стандартных эталонных материалов, представлен-
ных в табл. 6.2, можно перекрыть интервал температур 125—940°С.
В работе [20 ] рассматривался прецизионный метод калибровки
температурной шкалы прибора ДТА с использованием наборов стан-
дартных эталонных материалов NBS—ICTA № 758—760. Эти
222
Глава 6
Таблица 6.2
Стандартные эталонные материалы NBS — ICTA [20]1)
Набор № 758, 125-435 °C Масса, г Набор № 759, 295—675 °C Масса, г Набор № 760. 570—940 С Масса, г
Нитрат калия 10 Перхлорат калия 10 Кварц 3
Индий 3 Сульфат серебра 3 Сульфат калия 10
Олово 3 Кварц 3 Хромат калия 10
Перхлорат калия 10 Сульфат калия 10 Карбонат бария 10
Сульфат серебра 3 Хромат калия 10 Карбонат стронция 10
В прилагаемом удостоверении (сертификате) указаны как результаты измерений тем-
ператур пика и экстраполированного начала кривой ДСК для каждого образца, так и осред-
иенные их значения.
наборы включают восемь неорганических соединений, испытываю-
щих превращение Твердое тело 1 д=ь Твердое тело 2, и два металла
высокой чистоты.
Калибровка температурной шкалы калориметра ДСК-2 фирмы
«Перкин-Элмер» была описана О’Нейлом и Файенсом [23]. Показа-
ния температуры в этом приборе в зависимости от положения пере-
ключателя могут быть получены в градусах Цельсия, кельвинах
или градусах Фаренгейта в интервале температур 100—1000 К.
Поскольку во многих промышленных процессах температура запи-
сывается в градусах Фаренгейта, часто бывает удобно использовать
в ДСК именно эту шкалу. На фиг. 6.6 приведена калибровочная
кривая, полученная при скорости нагревания 40°F/mhh.
Точность калибровки по температуре при использовании ме-
таллов, указанных на фиг. 6.6, видна из табл. 6.3. Приведенные
в таблице температуры соответствуют точкам, полученным при пере-
сечении экстраполируемого переднего фронта пика плавления и
продолжения базовой линии. Чтобы избежать влияния субъек-
тивных факторов, расчет проводился с помощью ЭВМ.
Для определения дифференциальной температуры использо-
вались термисторы с достаточно большим сопротивлением
(100 000 Ом при обычной температуре), включенные в мостовую
схему [21, 22]. В этом методе, как правило, не требуется примене-
Приборы для ДТА и ДСК.
223
Фиг. 6.6. Типичная кривая калибровки по температуре для ДСК [121].
Таблица 6.3
Точность калибровки по температуре калориметра ДСК-2
фирма «Перкин-Элмер» [23]
Эталонный материал Опубликован- ные данные по температу- ре плавления, °C Температура плавления, полученная при калиб- ровке, °C Ошибка, °C
Индий 156,63 156,79 +0,16
Олово 231,97 231,95 —0,02
Свинец 327,50 327,36 —0,14
Цинк 419,58 419,70 +0,12
Алюминий 660,37 659,53 —0,84
ния усилителя постоянного тока. Из-за быстрого уменьшения со-
противления термисторов с увеличением температуры они обычно
используются при температурах до 300° С [22 ].
4. ПЕЧИ И УСТРОЙСТВА ДЛЯ РЕГУЛИРОВАНИЯ ИХ
ТЕМПЕРАТУРЫ ПО ЗАДАННОЙ ПРОГРАММЕ
Как и в предыдущем разделе, выбор нагревательного элемента
и типа печи зависит от исследуемого интервала температур. Из-
вестны нагреватели ДТА с рабочими интервалами температур от
—190 до 2800° С. Нагреватели могут монтироваться вертикально
224
Глава 6
или горизонтально; нагревание может осуществляться с помощью
элементов сопротивления, инфракрасного излучения [24, 25], ко-
лебаний высокой частоты [11,26] или трубчатого змеевика, по ко-
торому циркулирует нагретая или охлажденная жидкость, либо
газ [27].
В конструкциях печей чаще всего применяются элементы со-
противления. Некоторые материалы элементов сопротивления и при-
близительные температурные пределы применения элементов ука-
заны в табл. 6.4. Эти температурные пределы зависят, конечно, от
конструкции и качества изоляции самой печи.
Таблица 6.4
Предельные температуры применения элементов
сопротивления нагревателей
Материал сопротивления Темпер атурная граница, °G
Нихром . 1000
Каптал . 1350
Платина 1400
Платина — родий (10% Rh) 1500
Родий 1800
Тантал 1330
Глобар (силит) 1500
Суперкантал 1600
Молибден 2200
Платина — родий (20% Rh) 1500
Хромель А 1100
Вольфрам , 2800
Типы конструкций печей для ДТА показаны на фиг. 6.7. На
фиг. 6.7, а представлена конструкция, описанная Вассалло и
Харденом [28]. Печь нагревается с помощью нагревателя патрон-
ного типа. Путем пропускания охладителя через змеевики, окружа-
ющие печь, обеспечивается быстрое охлаждение или возможность
работы при температурах ниже комнатной. Образец и эталон поме-
щены в стеклянные капиллярные трубки.
Более сложные, нагреватели, применяемые в термоанализато-
рах фирмы «Меттлер», работают в интервале температур от —150
до 2400° С (фиг. 6.7, б—д). Нагреватель, показанный на фиг. 6.7, б,
используется в интервале температур от —150 до 400° С и имеет
нагревательный элемент в виде проволочного сопротивления из
кантала. В интервале температур 25—1000° С применяется нагре-
ватель с нагревательным элементом из кантала (фиг. 6.7, в). Этот
нагреватель работает также в условиях высокого вакуума. В таких
же условиях применяется и высокотемпературный нагреватель с
б
Фиг. 6.7. Конструкции печей для ДТА.
1 — нагреватель; 2 — охладитель.
г
226
Глава 6
г
интервалом рабочих температур 25—1600° С (фиг. 6.7, г). Обмотка
его печи изготовлена из еуперкантала. Недавно была предложена
сверхвысокотемпературная печь (фиг. 6.7, д), предназначенная для
работы в интервале температур 400—2400° С. Ее нагревательные
элементы изготовлены из вольфрама. С описанными здесь печами
фирмы «Меттлер» могут использоваться держатели образца, рас-
смотренные в разд. А.2 данной главы.
Температура, °C
Фиг. 6.8. Распределение температуры в печи для ДТА [29].
Качество печей ДТА определяется способностью нагреватель-
ных элементов обеспечивать симметричное и равномерное нагре-
вание. Для получения качественных результатов распределение
температуры в зоне размещения держателя образца должно быть
однородным. Видеманн [29] получил кривые распределения тем-
пературы для печи фирмы «Меттлер» (фиг. 6.8). Заштрихованная
площадь обозначает зону однородной температуры относительно
положения держателя образца. Ямомото и др. [17] исследовали
распределение температуры в печи, используемой в приборе ДТА
собственной конструкции.
Для работы при низких температурах нагреватель можно по-
местить в сосуд Дьюара и предварительно охладить его жидким
азотом. Затем производится нагревание с помощью нагреватель-
ного элемента по заданной программе. Другой метод основан на
использовании газа в качестве теплообменной среды (фиг. 6.9)
[30]. Если охлаждение не обеспечивает температуру по крайней
мере на 30° С ниже заданной, то функционирование устройств, ре-
гулирующих температуру, не будет эффективным. Оптимальные
условия работы при низких температурах могут быть достигнуты
при использовании термоэлектрического охлаждения, однако такая
• система охлаждения еще не создана.
Приборы для ДТА и ДСК
227
S
Фиг» 6.9. Простые системы охлаждения приборов ДТА [30].
с: 1 — регулятор газа (азота); 2 — игольчатый клапан; 3 — жидкий азот; 4— медный змее-
вик 1200X6 мм; 5 — сосуд Дьюара. 6: 1 — источник постоянного тока иа 24 В; 2— реле;
3 — транзисторный тиратрон; 4 — соленоид; 5 — сосуд Дьюара; 6 — жидкий азот.
Для регулирования атмосферы внутри нагревателя и держателя'
образца применялись различные методы, в том числе: а) заполне-
ние нагревателя газовой средой [31—34]; б) вакуумные нагрева-
тели [31, 32, 35—40]; в) создание динамической среды (потока
газа) [31, 32, 41, 42]. В работе [29] описана сложная высоковакуум-
ная система для нагревателей ДТА.
Хотя большинство приборов ДТА имеет только одну печь, целе-
сообразно использовать несколько печей в комплекте с держателем
образца, усилителем и регистрирующим устройством и тем самым
увеличить число испытываемых образцов. Имеется описание [43 ]
прибора, в котором имелись четыре печи.
228
Глава 6
Скорость увеличения температуры печи задается устройством,
регулирующим температуру по заданной программе. Оно должно
обеспечивать линейное возрастание температуры в нескольких ин-
тервалах температур и, следовательно, должно быть совместимым
•Фиг. 6.10. Устройство фирмы «Дюпон», регулирующее температуру по
заданной программе.
1 — источник тока; 2 — установка на нуль; 3 — интегрирующий усилитель; 4 — источник на-
пряжения; 5 — управляющий усилитель; 6 — пусковая управляющая цепь; 7 — переключа-
тель типа SCR; 8 — счетчик; 9 — нагреватель 10 — управляющая термопара; 11 — нагрузка
(ячейка).
с различными типами термопар. Скорости нагревания должны быть
линейными и воспроизводимыми, так как нелинейность скорости
нагревания оказывает влияние на кривые ДТА. Как показал Зелл
[44 ], периодическое изменение выходной мощности программирую-
щего устройства вызывает изменение пиков и появление ложных
пиков на кривых ДТА. Другой важной характеристикой програм-
мирующего устройства является стабильность работы при измене-
нии напряжения питания и окружающей температуры. Управляю-
щая термопара программирующего устройства должна быть ском-
пенсирована либо с помощью электрической цепи, либо с помощью
холодного спая (ледяной ванны).
Приборы для ДТА и ДСК,
229
Типы устройств, регулирующих температуру по заданной про-
грамме, варьируется от несложных трансформаторов переменного
напряжения, соединенных с синхронным мотором, до более сложно-
го программирующего устройства пропорционального типа с об-
ратной связью. Программирующие устройства с прерывистым ре-
жимом работы нельзя использовать для рассматриваемой цели, по-
скольку пульсации мощности на выходе повышают термические
градиенты в нагревателе и держателе образца. Программирующее
устройство пропорционального типа на твердотельных элементах
с обратной связью, используемое в приборах термического анализа
фирмы «Дюпон», показано на фиг. 6.10. Когда разность между
командным напряжением и выходным напряжением управляющей
термопары превышает пороговое значение управляющего усилите-
ля, этот сигнал усиливается и подается к нагревателю. При этом
мощность, подводимая к нагревателю, пропорциональна сигналу
на входе управляющего усилителя. Погрешность программирующе-
го устройства в задании скорости нагревания высока и составляет
±5%, или 0,1 град/мин. Эта погрешность определяется главным
образом выходным сигналом управляющей термопары. В самом
программирующем устройстве ограничивающим фактором явля-
ются погрешность настройки и дрейф мощности управляющего тока,
которая суммируется в интегрирующем усилителе. Повторяемость
составляет 0,1 град/мин, а отклонение от линейности теплового
потока равно ±1%, или 0,01 град/мин. Повторяемость зависит
от дрейфа мощности управляющего тока и искажения, вносимого
усилителем; последнее определяется линейностью выходного сиг-
нала управляющей термопары.
В приборах фирмы «Стоун» для регулирования скорости нагре-
вания используется шаговый двигатель, у которого напряжение
на выходе имеет нелинейную характеристику, воспроизводящую
характеристики четырех наиболее употребляемых типов термопар.
Напряжение на выходе двигателя с нелинейной характеристикой
•сравнивается с напряжением на выходе термопары с такой же не-
линейностью, и разностный сигнал, или сигнал ошибки, усилива-
ется усилителем и подается на схему переключения, управляющую
напряжением нагревателя. Погрешности в линейности составляют
менее 0,25% для любого интервала в 100°С, или 0,5% полной шкалы.
Точное определение погрешности в линейности повышения тем-
пературы печи является не простой задачей. По графику зависи-
мости температуры от времени (фиг. 6.11,а) скорость нагревания
оценивается с погрешностью ±5% или менее. Для определения
незначительных отклонений от линейности кривой скорости на-
гревания используется кривая, изображенная на фиг. 6.11,6. Эта
кривая ДТА записана с камерой эталона, заполненной инертным
веществом (например, альфа-окисыо алюминия), и с пустой ^каме-
рой образца. В этих несбалансированных условиях легко различи-
230
Глава 6
мы пульсации напряжения на входе. Аналогичная картина наблю-
дается также, когда термопары для определения АТ несимметрично'
расположены в камере печи [45]. Необходимо отметить, что еще
труднее точно измерять скорости нагревания 1 град/мин и менее.
Фиг. 6.1L Кривые скорости нагревания,
а — кривая зависимости температуры от времени; б — кривая ДТА несбалансированной систе-
мы; 1 — начало; 2 — пульсации напряжения на входе.
Скорость нагревания для большинства промышленных нагре-
вателей ДТА может изменяться от 0,5 до 50 град/мин. Однако боль-
шинство кривых ДТА записывается при скоростях 10—20 град/мин.
Повышенные скорости нагревания удобно использовать для пред-
варительной проверки термического поведения образца.
Б. УСИЛИТЕЛЬ СЛАБЫХ НАПРЯЖЕНИЙ
ПОСТОЯННОГО ТОКА
И РЕГИСТРИРУЮЩИЕ СИСТЕМЫ
Выходное напряжение дифференциальной термопары составля-
ет 0,1—100 мкВ в зависимости от типа используемой термопары
(табл. 6.1) и измеряемой разности температур. Следовательно, если
Приборы для ДТА и ДСК
231
используется не очень чувствительная регистрирующая система
(полная шкала менее 100 мкВ), то сигнал КТ необходимо усилить
с помощью усилителя слабых напряжений постоянного тока. Для
применения в приборах ДТА усилитель должен иметь слабый шум,
минимальный дрейф и высокую стабильность. Нестабильность уси-
лителя ведет к нестабильности базовой линии [44], а дрейф вход-
ного напряжения или изменение температуры окружающей среды
может вызвать пульсации на выходе. Нестабильность базовой ли-
нии и шум на выходе могут быть вызваны также помехами на входе
схемы от сети питания переменного тока частотой 60 Гц.
Часто для уменьшения шума усилителя ставят емкость на вы-
ходе; иногда емкость ставят на входе [44]. Эти емкости нередко
ухудшают постоянную времени усилителя, что вызывает сдвиг пи-
ков кривой, а также ухудшение их разрешения. Поэтому для
устранения шума необходимо использовать такую емкость, которая
обеспечивала бы компромиссное соотношение между уменьшением
шума и ухудшением разрешения пиков. Неправильный выбор им-
педанса усилителя также может вызвать нелинейность выходного
напряжения и в результате искажение пиков кривой.
Имеются описания различных записывающих устройств — от
фотогальванометров до современных электронных потенциометри-
ческих самописцев. В работе Бургесса [46] содержится превосход-
ное описание некоторых из первых записывающих устройств. Со-
временные самописцы, в особенности многоточечные, по-видимому,
впервые были использованы Керром и Кульпом [7], а также Кауф-
маном и Диллингом [47]. Эти самописцы в комбинации с многопо-
зиционными держателями образцов позволили эффективней ис-
пользовать ДТА для качественной идентификации геологических
материалов. Другой метод, основанный на применении двухка-
нального самописца, состоит в записи дифференциальной темпера-
туры и температуры эталона в функции времени на одной ленте
самописца. Возможно, более удобны современные двухкоординат-
ные самописцы, позволяющие непосредственно регистрировать за-
висимость дифференциальной температуры от температуры этало-
на. В настоящее время промышленностью выпускается широкий
набор самописцев. (
6. ПРИБОРЫ ДТА
а. Введение
Прототипом современных приборов ДТА является двухтермо-
парный калориметр, разработанный Робертс-Остеном [48] в 1899 г.
С того времени было создано множество приборов, которые, однако,
лишь незначительно различались по конструкции печей, устройств,
регулирующих температуру по заданной программе, регистрирую-
232
Глава 6
щего оборудования, держателей образца и т. п. Смозерс и Чианг
[49] в 1958 г. составили подробное описание 255 известных в мире
приборов ДТА различной конструкции. Этот обзор был дополнен
новыми описаниями во втором издании их книги [50], которая
включает обширный список литературы по ДТА, содержащий
4248 наименований; во многих из этих работ также описаны исполь-
зованные приборы. В различных книгах [51—53] имеется описание
современного оборудования ДТА; промышленные приборы рас-
сматриваются отдельно [55—57].
В этой главе будут рассмотрены только те приборы, которые
обладают некоторой новизной конструкции или являются этапны-
ми в развитии техники ДТА. Вследствие недостатка места в описа-
ние будет включено только несколько промышленных приборов,
поскольку их подробные описания имеются в литературе [55—57].
б. Держатели образцов в виде герметичных трубок
Заключение образца в герметичную (иногда вакуумированную)
камеру или трубку представляет собой старый метод. Он полезен
при исследовании фазовой диаграммы [58], в особенности материа-
лов, подверженных коррозии (например, склонных к реакциям
халькогенидов), при исследованиях реакций органических соедине-
ний [59, 60], гидратов солей металлов [61], в металлургии [62], при
изучении равновесия в расплавах солей [63] и во многих других
случаях.
На фиг. 6.2,с показан простой держатель образца в виде герме-
тичной трубки. Другие типы держателей образца и эталона в ви-
де герметичных трубок представлены на фиг. 6.12 [58]. В случае,
показанном на фиг. 6.12,а, термопары герметически заделывались
непосредственно внутрь трубок, что сопряжено с трудностями обес-
печения герметичности соединения металла со стеклом. Теплопе-
редача от образца к чувствительному термоэлементу в большинстве
приведенных примеров оказывается довольно низкой. Держатель,
показанный на фиг. 6.12Д обеспечивает лучший по сравнению с
большинством других конструкций термический контакт образца
с термопарой, так как термопара имеет кольцевую форму. Вслед-
ствие термической инертности системы держатели, изображенные
на фиг. 6.12, дают довольно низкое разрешение пиков на кривых
ДТА. Имеется подробное описание подготовки к работе держате-
лей образца в виде вакуумированной запаянной трубки [66].
Во многих более поздних работах были использованы держате-
ли в виде стеклянных капиллярных трубок типа применяемых для
определения температуры плавления веществ и рассчитанных на
образцы весом в несколько миллиграммов. На фиг. 6.13 показаны
печь и держатель образца, использованные Уэндландтом [61]. Об-
разцы помещались в капиллярные трубки с внутренним диаметром
Приборы для ДТА и ДСК
233
г
Фиг. 6.12. Держатели образцов в виде герметизированных трубок, заклю-
ченных в вакуумированные контейнеры.
Конструкции: с —Робертса [64]; б —Дженсена [68]; в — Красика [65]; г —Болдина и Керра
[66]; д — Болдина [58]; е — Фактора и Хэнкса [67J.
0,9—1,1 мм, которые устанавливались в тонкостенные теплопрово-
дящие алюминиевые патроны. Идентичная пустая трубка исполь-
зовалась для эталонной термопары. После загрузки образца (мас-
сой 5—7 мг) в трубку она наглухо запаивалась с помощью кисло-
родной горелки на длине ~20 мм.
Аналогичная процедура загрузки образца, а также специаль-
ные меры предосторожности вследствие летучести образцов из ор-
ганических веществ применялась Барретом и др. [59, 60]. Капил-
234
Глава 6
лярная трубка с образцом помещалась внутрь охлаждаемого алю-
миниевого блока (охладитель — смесь сухого льда с ацетоном) и
наглухо запаивалась газовой микрогорелкой. При выборе размеров
образцов также необходимо соблюдать предосторожность: образцы
больших размеров могут созда-
вать чрезмерное давление (если
при разложении они выделяют
газообразные продукты), что
может привести к небольшим
взрывам. Размер образца дол-
жен выбираться исходя из тре-
бования, что давление газов не
должно превышать 4,5 ДШа.
Гилпатрик и др. [63] исполь-
зовали держатель ДТА в виде
герметичной трубки для изуче-
ния фазового состояния в рас-
плаве солей NaF — KF — BF3.
Фиг. 6.14. Держатель образца для
исследования фазовых переходов в-
в расплавах солей [63].
1 — ячейка; 2 — кольцо из жаропрочного ма-
териала; 3 — штуцер из жаропрочного мате-
риала; 4 — трубка из нержавеющей стали'
(диаметр 9,5 мм); 5 — контрольная термопа-
ра (диаметр ~ 1 мм); 6 — термопара образца
(диаметр ~ 1 мм); материал — никель.
Фиг. 6.13. Печь для ДТА и держа-
тель образца в виде герметичной
трубки [61].
1 — изолирующая крышка; 2 — алюминиевый
блок; 3— стеклянная капиллярная трубка;
4 — образец; 5 — термопара образца; 6 —
алюминиевый теплопроводящий патрон; 7 —
керамическая изоляционная трубка; 8 — эта-
лонная камера; 9— несущая платформа;
10 — узел крепления электровыводов.
Этот держатель (фиг. 6.14) вмещал ~4 г образца; во
избежание разделения солей и изменения состава образец в
процессе нагревания и охлаждения перемешивался. Кап-
сулы, изготовленные из никеля, откачивались и запаивались в
Приборы для ДТА и ДСК.
235
вакууме. Запаянные термопары давали воспроизводимые резуль-
таты для фазовых переходов с чувствительностью по А Т 25—100 мкВ
па 1 см ленты самописца. Подобные металлические кап-
сулы—держатели образца из танта-
ла описаны Эттером и др. [62].
Бэрролл и Роджерс [69] описали
устройство (фиг. 6.15)* которое не
связано с использованием герме-
тичной трубки, но позволяет сох-
ранять постоянное давление в про-
цессе испытания. Общий объем
ячейки составлял ~0,8 мл; капил-
лярная трубка служила резер-
вуаром для газов, выделявшихся
в процессе реакции, и предот-
вращала поступление значитель-
ного количества воздуха в атмо-
сферу образца или утечку газа.
Небольшой ртутный затвор допус-
кал расширение газов и поддержи-
вал практически постоянное дав-
ление в ячейке.
Выше уже упоминалось о ме-
таллических держателях образ-
цов, способных работать при вы-
соком давлении, которые использо-
вались Бохоном [3] для изучения
взрывчатых веществ методом
ДТА. Подобный „тип держателя
•образца из нержавеющей стали
описан Дэвидом [70].
Фиг. 6.15. Держатель образ-
ца постоянного давления для
работы в атмосфере, создавае-
мой самим образцом [69].
1 — ртутный затвор; 2 — алюминие-
вый блок; 3 — камера образца; 4 —
термопара в герметичной стеклянной
оболочке; 5 — силиконовая проклад-
ка; 6 — уплотнение из силиконового
каучука; 7 — выводы термопары (по
оси Т н ДТ); 8 — дренаж.
в. Приборы ДТА высокого давления
Краткий обзор приборов ДТА высокого давления содержится
в работе Локке [71]. Прежние исследования относились главным
образом к изучению влияния давления на равновесие систем Твер-
дое тело -> Жидкость и Твердое тело 1 -> Твердое тело 2. В прибо-
рах ДТА высокое давление (6—8 ГПа) создается с помощью порш-
невой системы, а более низкое давление — с помощью сжатых газов.
Харкер [72] описал приборы, в которых образец и эталон за-
паивались в платиновые капсулы, помещаемые в микрореактор,
находящийся под давлением. Провода, припаянные к капсулам,
выводились из микрореактора через уплотнения. Давление (опрес-
совка) создавалось сжатым азотом, хотя аргон предпочтительнее,
поскольку азот делает платиновые сплавы хрупкими. При давле-
236
Глава 6
нии 105 МПа было получено
несколько точек плавления
системы СаО — Са(ОН)2 —
Ca2SiOi.
Коэн и др. [73] описали
прибор с поршневой систе-
мой, способный работать при
давлении до 5 ГПа и темпе-
ратурах до 1200° С. Подроб-
но рассмотрены отдельные
узлы прибора высокого давле-
ния: печь, держатель образца
и система создания давления.
Нафиг. 6.16 изображен ти-
пичный узел термопары это-
го прибора. Все провода,
за исключением конца, на-
ходившегося в контакте с
контейнером образца, изоли-
ровались алундовой замаз-
кой. Капсулы для образ-
зов изготавливались из тан-
а
тала, ниобия, платины, гра-
фита или других подобных материалов в виде чашечек диаметром
1,6 мм и высотой 3,2 мм. Крышки изготавливались из того же
материала, что и капсулы. Давление к образцу передавалось через
узел печи, состоящий из деталей, изготовленных из талька,
графита, пирофиллита или нитрида бора, через трубки термо-
пары, термопары, контейнер образца и т. д. Определение
давления в образце представляет весьма сложную задачу.
Прибор ДТА, способный работать при давлении до 400 МПа и
температуре до 500°С, был описан Кубалла и Шнейдером [74].
В этом приборе (фиг. 6.17) ячейка ДТА изготавливалась из нержавею-
щей стали. В ячейке располагались два поршня Бриджмена, в ко-
торых были размещены термопары. Контейнеры с образцом и эта-
лоном были изготовлены из сплава 97% платины и 3% иридия;
они подвешивались к оболочкам спаев термопар. Верхняя и ниж-
няя поверхности держателя образца нагревались с помощью блоков
из ZrO2. Нагреватель охватывал емкости высокого давления, обес-
печивая нагревание по линейному закону со скоростями от 0,2 до
10 град/мин.
Японскими исследователями [75, 76] были описаны приборы
ДТА для исследования реакций гидрирования под высоким давле-
нием. Буске и др. [77] описали печь и держатель образца прибора
ДТА, которые могли работать при давлении до 50 МПа и максималь-
ной температуре 500° С. Эти приборы выпускаются фирмой «Нетш-
Приборы для ДТА и ДСК
237
Фиг. 6.16.
Три провода уз-
ла термопары
прибора ДТА вы-
сокого давления
[73]
6 6
Фиг. 6.17. Прибор ДТА высокого
давления Кубалла и Шнейдера [74].
а: 1 — охладители; 2 — изоляционные дис-
ки из пирофиллита; 3 — нагревательный
блок; 4 — сосуд высокого давления; 5 —
медные уплотнители; 6 — блоки из ZrO2; 7—
медный защитный кожух; 8 — калориметри-
ческий блок; 9 — термопара; 10 — корундо-
вый капилляр; И — крепежный винт; 12 —
опора; 13 — вход и выход хладагента, б —
открытый контейнер из платины и иридия:
14 — сплав платины и иридия; 15 — корунд;
/6 — стальная оболочка термопары, е — за-
крытый тефлоном контейнер с латунным дер-
жателем; 17 — тефлон; 13 — латунь.
Гератебау». Разработанный фирмой «Стоун» держатель образца
для прибора ДТА высокого давления описан Локке [71]. Этот дер-
жатель можно использовать при давлениях до 21 МПа и максималь-
ной температуре 500° С.
г. Приборы ДТА для работы при высоких
температурах
При разработке приборов ДТА для работы при температурах
выше 1200° С возникают новые проблемы. Так, становится значи-
тельной электрическая утечка из-за увеличения электропроводнос-
ти тугоплавких элементов конструкции и воздуха в нагревателе.
Предъявляются повышенные требования и к экранированию элект-
рических цепей термопар низкого напряжения. Высокая темпера-
тура часто вызывает плавление образца; расплав может разрушить
термопару и даже держатель образца. Однако даже при наличии
этих проблем удалось создать приборы ДТА, способные работать
при температурах до 3000° С и выше.
На фиг. 6.18 показан прибор ДТА, способный работать до
1575° С [78]. Нагреватель состоит из алундового сердечника, на
который намотано проволочное сопротивление из сплава платины
с родием (20% Rh). На оба конца сердечника дополнительно на-
238
Глава 6
Фиг. 6.18. Прибор ДТА Ньюкирка для использования при высоких темпе-
ратурах [78].
1 — альфа-окись алюминия; 2 — закрытые крышками платиновые тигли с карманами для
термопар; 3 — образец; 4 — провода из платины или сплава платины; 5 — медные провода;
б — холодный спай; 7 — порошкообразная изоляция из магнезии; 8 —• огнеупорный кирпич;
9 — корпус переносного нагревателя; 10 — алундовые нагревательные трубки и центральная
«опора тигля; И — алюминиевые ножки нагревателя; 12 — стальной треножник; 13 — ла-
тунные детали для выравнивания платформы
мотаны проволочные сопротивления из нихрома, используемые
при работе только в условиях очень высоких температур. Диффе-
ренциальная температура измерялась платино-платинородиевыми
термопарами (10% Rh), которые помещались во вдавленные углуб-
ления платиновых чашек образца и эталона. Для экранирования
проводов термопар их керамические изоляционные трубки были
обернуты платиновой фольгой.
Использовались промышленное устройство для регулирования
температуры по заданной программе, усилитель постоянного тока
и двухточечный самописец. Допускались скорости нагревания 0—
30 град/мин.
Недумовым [79] описаны печь и держатель образца, которые
применяются при высоких температурах без использования термо-
пары в качестве датчика температуры. Критическая оценка раз-
Приборы для ДТА и ДСК
239
личных приборов с термопарами показала-, что они не удовлетворя-
ли требованиям, возникающим при количественных высокотемпе-
ратурных исследованиях методом ДТА металлов и сплавов. Для ре-
гистрации температуры было предложено использовать вольфра-
мовые термометры сопротивления. При этом прибор мог работать
до температур выше 3000° С.
Фиг. 6.19. Универсальный прибор Руперта для использования при высоких
температурах [80].
1 — графитовый тигель; 2 — концентратор вихревых токов; 3 — оптическая система; 4 —
оптический пирометр; 5 — трубка фотоумножителя; 6 — источник питания переменного
тока; 7 — регистрация изменения температуры; 8 — осциллоскоп; 9 — ленточный самопи-
сец; 10 —• блок управления индукционным нагревом; И — индукционный нагреватель (5 кВт).
Вакуумная система; 12 — вакууметр (дюймы рт. ст.); 13 — вакууметр (мкм рт. ст); 14 —
вакуумные насосы; 15 — резервуар с аргоном.
Универсальный прибор, позволяющий проводить термический
анализ, термический анализ по производной, дифференциальный
термический анализ, а также регистрировать зависимость темпера-
туры от производной температуры по времени, был описан Рупер-
том [80—82]. Схема этого прибора представлена на фиг. 6.19 [80].
Образец помещался в тигель 1, расположенный внутри концентра-
тора вихревых токов 2. Это устройство получает энергию от индук-
ционного нагревателя 11, регулируемого с помощью блока управ-
ления 10. Свет от нагретого образца проходит через отверстие
диаметром 1,8 мм в верхней части тигля и попадает через окошко-
из пирекса или кварца в нижнюю часть оптической системы 3, раз-
деляющей световой пучок. С помощью нижнего, частично алюмини-
зированного зеркала часть светового потока отражается прибли-
* >
240
Глава 6
зительно под прямым углом к оси оптической системы и попадает
в оптический пирометр 4., который используется для измерения
температуры образца. Часть светового потока, прошедшая через
нижнее зеркало, отражается от верхнего зеркала и фокусируется
ахроматической линзой с фокусным расстоянием 27,5 см на вход-
ное отверстие диаметром 1,6 мм фотоумножителя, который исполь-
зовался при температурах выше 1400° С. При более низких темпе-
ратурах использовалась электронно-лучевая трубка 1Р21. В рабо-
те 181] описана дальнейшая модификация этого прибора.
д. Приборы ДТА для микроанализа
Получение кривых ДТА с помощью образцов массой в несколько
микрограммов уже обсуждалось в разд. А.2 [4]. Исчерпывающий
обзор приборов ДТА для микроанализа составили Зоммер и Джо-
Фиг. 6.20. Прибор ДТА для микроанализа [84].
а — держатель образца; б — электрическая схема. 1 — горячий спай; 2 — контакты; 3 —
источник переменного тока; 4 — регулятор мощности с приводом от электродвигателя; 5 —
регулятор мощности; 6 — селекторный переключатель; 7 — термопара образца; 8 — термо-
пара эталона; 9 — диод; 10 — синхронный преобразователь; 11 — измеритель температуры;
12 — измеритель дифференциальной температуры.
Приборы для ДТА и ДСК
241
хенс [83]. Они детально рассмотрели применение высокотемпера-
турной микроскопии в ДТА. В большинстве описанных приборов
спай термопары служил одновременно нагревателем, термочувстви-
тельным элементом и держателем образца.
На фиг. 6.20 изображен прибор ДТА для микроанализа, разрабо-
танный Миллером и Зоммером [84]. Электрическая цепь с регулято-
ром переменного напряжения обеспечивала скорость нагревания в
интервале от 5 град/мин до 1000 град/с. Регистрация температуры
образца и АТ производилась с помощью быстродействующего са-
мописца. После усовершенствования прибора термопары образца
и эталона устанавливались в отдельных ячейках; в оптической си-
стеме микроскопа была оставлена только ячейка образца [85].
Подобный прибор ДТА описали Проке и Златовский [86]. В их
приборе образец помещался в узел термопары. Термопара нагре-
валась током высокой частоты, амплитуда которого модулировалась
низкочастотным сигналом.
е. Автоматизация приборов ДТА
В настоящее время приборы ДТА после установки в них образ-
цов способны работать автоматически в том смысле, что повышение
температуры задается программирующим устройством, которое
выключает прибор по достижении заданной температуры. После
охлаждения печи до комнатной температуры использованный об-
разец удаляется из держателя, а на его место устанавливается но-
вый образец, и цикл нагревания повторяется.
Уэндландт и Брэдли [87, 88] описали автоматический прибор,
в котором последовательно исследуются восемь образцов. Образцы
автоматически вводятся в печь, нагреваются до заранее выбранной
температуры, после чего удаляются. После охлаждения печи до
комнатной температуры цикл повторяется. Замена образцов, на-
гревание и охлаждение печи, запись и другие операции полностью
автоматизированы.
Механизм замены образцов и платформа печи показаны на
фиг. 6.21.
Образцы в виде порошков помещаются в стеклянные капилляр-
ные трубки 4 с внутренним диаметром 1,6—1,8 мм, которые уста-
навливаются в круглую пластину держателя образцов 1. Алюми-
ниевая пластина держателя образцов диаметром 200 мм и толщи-
ной ~3 мм содержит восемь стеклянных капиллярных трубок.
Стеклянные трубки удерживаются в соответствующих положениях
упругими зажимами. Держатель приводится во вращение неболь-
шим синхронным электромотором, снабженным электромагнитной
муфтой. Вращение управляется специальным устройством с фото-
ячейкой. Возле каждого держателя образца в алюминиевой плас-
тине вырезаны щели размером 12,7x1,5 мм. Устройство с фото-
0—230
242
Глава 6
ячейкой состоит из фотоячейки и лампочки, расположенной по дру-
гую сторону пластины держателя. С помощью электромотора дер-
жатель фиксируется в тех положениях, в которых свет от лампочки
попадает через щель в пластине держателя в фотоячейку. При этом
каждая капиллярная трубка с образцом занимает строго опреде-
ленное положение относительно полости нагревателя.
После этого платформа печи 3 поднимается и стеклянная ка-
пиллярная трубка с образцом попадает внутрь алюминиевой теп-
лопередающей муфты, опирающейся на спай термопары образца.
Движение платформы нагревателя осуществляется реверсивным
электродвигателем посредством червячной передачи. Пределы пе-
ремещения платформы задаются двумя микропереключателями.
1ечь отделена от платформы 6-миллиметровым слоем изоляционно-
го материала, а в положении нагревания — втулкой 5 из марини-
та. Время перемещения держателя при замене образца составляет
15 с; для подъема платформы требуется 50 с.
После нагревания образца до верхнего температурного предела
платформа нагревателя опускается, пластина держателя образца
поворачивается в новое положение и охлаждающий вентилятор на-
правляет воздух на горячий нагреватель. Время охлаждения на-
гревателя от 450° С до комнатной температуры составляет ~20 мин.
После охлаждения нагревателя до канатной температуры цикл
повторяется с новым образцом.
Схематическое изображение печи и камеры образца приведено
на фиг. 6.22. Цилиндрическая печь 5 диаметром 38 мм и длиной 84 мм
нагревается патроном 10 из нержавеющей стали мощностью 210 Вт.
Верхний температурный предел нагревателя ~500° С. Диаметр
полостей для образца и эталона ~6 мм, а длина 38 мм. Термиче-
ский контакт с капиллярными трубками для образца и эталона /
и 7 осуществляется с помощью алюминиевых теплопередающих
муфт 3 и 8. Длина цилиндрических муфт ~18 мм. Концы муфт
просверливаются таким образом, что капиллярные трубки с образ-
цами и керамические изоляционные трубки термопар 4 и 9 диамет-
ром 1,5 мм имеют плотную посадку. В верхнюю часть нагревателя
вставляется переходная крышка 6, чтобы свести к минимуму теп-
ловые утечки из нагревателя к пластине держателя образца 2.
Поскольку автоматические приборы ДТА имеют верхний тем-
пературный предел ~500° С, их применение ограничено промежу-
точным интервалом температур, в котором можно исследовать
такие процессы, как обезвоживание гидратированных солей метал-
лов и др. Они должны найти широкое применение в обычных ис-
следованиях органических и неорганических веществ. Отличи-
тельной чертой автоматических приборов является возможность
использования компьютера, легко выполняющего расчеты темпе-
ратур реакций и площадей пиков, определения степени чистоты
веществ, расчеты Д/7 и т. д.
Приборы для ДТА и ДСК
243
Фиг. 6.21. Автоматический прибор
ДТА Уэндландта и Брэдли [87].
1 — пластина держателя образцов; 2 — печь;
3 — платформа печи; 4 — капиллярная трубка
для образца; 5 — изоляция печи; 6 — охлаж-
дающий вентилятор.
Фиг. 6.22. Печь и камера
образца.
1 — стеклянная капиллярная труб-
ка для образца; 2 — пластина
держателя образца; 3 — /тепло-
передающая муфта образца; 4 —
термопара образца; 5— блок печи;
6 — переходная крышка; 7—-капил-
лярная трубка для эталона; 8 —
теплопередающая муфта эталона;
9 — термопара эталона; 10 — на-
гревательный патрон.
ж. Другие типы приборов ДТА
Уайтхед и Брегер [36] сконструировали и изготовили один из
первых прецизионных приборов с вакуумом или инертной атмо-
сферой. Нагреватель состоял из алундового сердечника длиной
228 мм и внутренним диаметром 50,8 мм, на который наматывалось
проволочное сопротивление из хромеля А. Сердечник был защищен
четырьмя цилиндрами из листового никеля, смонтированными на
грех стойках, а весь узел был установлен внутри колпака из стек-
ла пирекс размером 305 х610 мм. Электрические соединения (вводы)
были смонтированы в нижней части стеклянного колпака. Блок
образца был изготовлен из нержавеющей стали марки 309 или 446.
Скорость' нагревания регулировалась с помощью терморегулятора
фирмы «Лидс и Нортрап». Дифференциальная температура записы-
валась на самописец фирмы «Бекман» с фотоячейкой.
Уэндландт [38] также описал простой нагреватель ДТА и дер-
жатель образца, работающие в условиях вакуума или атмосферы
заданного состава. Нагревательная трубка длиной 190 мм и диа-
9* т
244
Глава 6
Фиг. 6.23. Прибор ДТА для работы в интервале температур от —190 до
400° С [39].
1 — нержавеющая сталь; 2 — медь; 3 — провод из кантала диаметром 1,024 мм (2,4 оборо-
та/см); 4 — сосуд Дьюара из нержавеющей стали; 5 — медная капсула термопары и уплот-
нение; 6 — нержавеющая сталь; 7 — медь.
метром 25 мм была частично изготовлена из плавленого кварца.
На нижнем конце трубки были укреплены два кольца внутренним
диаметром 25 мм из стекла пирекс. Провода хромель-алюмелевой
термопары вводились в зону нагревания с помощью двухканальной
керамической трубки диаметром 3,0 мм. Чашечки для образца и
эталона диаметром 7 мм, глубиной 10 мм и объемом 0,8 мл изготав-
ливались из сплава инконель. Они были прижаты к изоляционной
трубке и находились в непосредственном контакте со спаями тер-
мопар. Нагревательным элементом служил нихромовый провод,
намотанный на изоляционную трубку из плавленого кварца или
на керамическую трубку, которая плотно насаживалась на трубку
1
Фиг. 6.24. Прибор ДТА для исследования взрывчатых и горючих веществ
1 — купол из сплава монель; 2 — чашка; 3 — термопары' в оболочках из инконеля; 4 —
стопорные планкн (4 шт.); 5 — опорный стержень; 6 — изолятор нз марннита; 7 — стопор-
нее кольцо; 8 — прокладки нз силиконового каучука (3 шт.); 9 — уплотнительные про-
кладки (2 шт.); 10 — сальник фирмы «Конекс» (2 шт.).
246
Глава 6
из кварца. Устройство для регулирования температуры нагрева-
теля по заданной программе содержало трансформатор переменного
напряжения, управляемый синхронным двигателем. Сигнал диф-
ференциальной температуры усиливался усилителем слабых на-
пряжений постоянного тока и записывался в зависимости от темпе-
ратуры двухкоординатным самописцем.
Прибор ДТА, способный работать в интервале температур
от —190 до 400° С, описан в работе Рейсмана [391. Прибор
показан на фиг. 6.23. В нем использован сосуд Дьюара, наполнен-
ный жидким азотом; скорость нагревания блока образца задавалась
увеличением напряжения в катушках нагревателя, а охлаждение
регулировалось изменением давления газа во внешней камере. Для
записи ДТА-кривых использовались промышленные усилители
постоянного тока и самописцы.
Бохон [р41 описал нагреватель ДТА и держатель образца, от-
личающиеся исключительной надежностью в работе (фиг. 6.24).
В этом приборе дифференциальные термопары изолированы от об-
разца во избежание разрушения от взрыва или химического воз-
действия образца. Это достигалось упаковкой хромель-алюмелевой
термопары в оболочку из инконеля. Нагревательная трубка и до-
полнительный нагнетательный трубопровод были изготовлены из
сплава монель. При 350° С узел нагревателя выдерживал избыточ-
ное давление до 7 МПа, а при 500° С — 2,8 МПа.
Высокочувствительный прибор ДТА, позволяющий определять
температуры фазовых превращений с погрешностью ±0,5° С в ши-
роком интервале значений скоростей нагревания, описан Вассалло
и Харденом [28]. Держатель образца и нагревательный блок пока-
заны на фиг. 6.7,а. Цепь измерения дифференциальной температуры
была обычного типа, но когда требовалась погрешность по темпе-
ратуре не более ±0,5° С, в измерительной цепи температуры нагре-
вателя использовались усилитель постоянного тока и схема подав-
ления нуля. Нагревательный блок позволял использовать капил-
лярные трубки типа применяемых для определения температуры
плавления размером (1,5-е-2) хЗО мм в качестве контейнеров об-
разца и эталона, причем термопары вставлялись в образец через
верхнюю часть трубки. Блок нагревался с помощью нагреватель-
ного патрона мощностью 30 Вт, размещенного в центре блока.
Охлаждение обеспечивалось двойным змеевиком из медной трубки.
Один змеевик погружался в охладитель, находившийся в нижней
части баллона; второй опоясывал блок. Блок охлаждался потоком
воздуха или азота, проходившим через нижний, а затем через
второй змеевик. Прибор мог работать в интервале температур от
—150 до 450° С.
Для изучения кинетики гомогенных реакций в растворах Бор-
хардт и Даниельс [901 использовали прибор из стекла (фиг. 6.25).
Ячейки состояли из двух трубок из стекла пирекс диаметром
Приборы для ДТА и ДСК
247
31,75 мм, длиной 127 мм и объемом ~60 мл. Термопары, заключен-
ные в медные трубки с покрытием из кель-F, вставлялись сверху в
содержимое трубок. Температура ванны (а следовательно, вещест-
ва образца и эталона) постепенно увеличивалась с помощью нагре-
вателя, соединенного с трансформатором переменного напряжения.
Фиг. 6.25. Схема прибора ДТА для исследования растворов [90].
1 — реагенты; 2 — растворитель; 3 — дифференциальная термопара.
Прибор ДТА, содержащий в качестве термоэлементов для опре-
» деления дифференциальной температуры термисторы, был описан
Пакулаком и Леонардом [21]. Этот прибор показан на фиг. 6.26,а.
I На фиг. 6.26,в показана мостовая схема с термистором. Подобран-
I ные попарно термисторы с сопротивлением 100 000 Ом при 25° С
I помещались в центры стеклянных трубок. Третий термистор ис-
| пользовался для определения температуры нагревателя. Все тер-
I мисторы и трубки размещались внутри печи, изготовленной из
I алюминия. Скорость нагревания регулировалась путем изменения
[ напряжения на обмотке печи с помощью трансформатора перемен-
|ного напряжения. Обычно использовалась скорость нагревания
2 град/мин.
Необходимо заметить, что в данном .случае не требовался уси-
литель постоянного тока, так как напряжение сигнала разбаланса
моста было достаточно большим для регистрации его самописцем.
Один из первых приборов ДТА с регулируемыми атмосферой и
давлением описан Стоуном в 1960 г. [41, 42]. Схема печи и держа-
! теля образца этого прибора показана на фиг. 6.27. Конструкция
держателя образца позволяет продувать поток газа через вещество
[ образца и эталона в процессе нагревания. Газ входит через трубо-
провод держателя образца и диффундирует через пористые диски,
что обеспечивает минимальную турбулентность и максимальную
1
a
6
Фиг. 6.26. Термисторный прибор ДТА Пакулака и Леонарда [2Ц.
д — держатель образца; б —« печь; в — мостовая схема с термистором; 1 — выводы; 2 —
тефлоновые оболочки; 3 — крепления электрических сопротивлений; 4 — трубки диаметром
1 мм; 5 — трубка диаметром 1,5 мм; 6 — двухканальная керамическая трубка; 7 — термис-
тор; 8 — испытательная трубка; 9 — печь; 10 — нагревательная катушка; 11 — алюминие-
вый блок; 12 — трубки с образцами; 13 — контейнеры термисторов; 14 — стеклянное кольцо
25x82,5 мм; 15 — пустая ячейка; 7\, 72, Ts — термисторы; Rlt — потенциометры на 100 Ом;
Яз» Ri — потенциометры на 1000 Ом (±1%); Re — потенциометры на 2000 Ом (±1%);
Tt, Т — термисторы с сопротивлением 100 000 Ом при 25°С; В —сухая ячейка на 1,5 В
Приборы для ДТА и ДСК
249
8
Фиг. 6.27. Схема прибора ДТА, описанного Стоуном [41, 42].
1 — камера под давлением; 2 — печь; 3 — блок образца; 4 — приспособление для быстрой
вамены держателя образца; 5 — регулируемый впуск газа; 6 — провода эталонной термо-
пары; 7 — провода дифференциальной термопары; 8 — самописец-регулятор.
однородность потока газа в образце и вокруг него. Состав газа
можно изменять в любой момент времени в процессе нагревания,
и можно совершать циклическую замену двух и более газов. Дав-
ление внутри камеры печи может изменяться от 1,33 Па до 0,7 МПа.
На фиг. 6.28 показана печь [15, 16], в которой использовались
тонкопленочные термопары, описанные выше в разд. А. 3. Это
позволило регулировать температуру в интервале от —120 до
500°С. Для работы при низких температурах предварительно ох-
лажденный азот прогонялся через змеевик, окружающий камеру
печи. Управляя потоком газообразного азота, можно было регули-
ровать изменение температуры по линейному закону.
Миллер и Вуд [91] описали калориметрическую точность и ра-
бочие характеристики высокотемпературной (1200°С) ячейки ДТА
фирмы «Дюпон». Печь и держатель образца показаны на фиг. 6.29.
Образцы загружались в платиновые чашки, помещенные над пла-
тино-платинородиевыми (10% Rh) термопарами, заключенными в
Фиг. 6.28. Печь, используемая с
тонкопленочными термопарами
[15, 16].
1 — подвешенный опорный стержень-
державка; 2 — асбест; 3 — канал для
термопары программирующего устройст-
ва; 4 — алюмниевый нагревательный
цилиндр; 5 — охлаждающий змеевик и
электрический нагреватель; 6 — съемная
крышка; 7 — чашка для образца; 8 —
кварцевая пластина; 9 — фиксирован-
ный опорный стержень.
Фиг. 6.29. Высокотемпературная (1200° С) ячейка ДТА фирмы «Дюпон»,
описанная Миллером и Вудом [91].
11 — печь; 2 — лопатка; 3 — керамический изолятор; 4 — керамическая опора; 5 — колпак;
\6 — отверстие для потока газа; 7— коническое соединение; 8 — спай термопары; 9— плати-
новая чашка; 10— вкладыш; И — образец; 12— нагревательная трубка из окиси алюминия.
Приборы для ДТА и ДСК .
251
трубку из окиси алюминия, обогреваемую нагревателем с обмоткой
из канталового провода.
Часто требуется изолировать нагреватель ДТА и держатель об-
разца от пульта управления из-за опасности взрыва, радиоактив-
ности, токсичности среды и т. п. Такие изолированные держатель
образца ДТА и нагреватель должны обеспечивать нормальную ра-
боту и не затруднять установку образца. Грейбушем и др. [92]
Фиг. 6.30. Калориметрическая ячей-
ка ДТА, описанная Бэрроллом и
др. [93].
1 — медные чашки для образцов (наружный
диаметр 4 мм и высота 6 мм); 2— медная
чашка для эталона; 3 — двухканальные ке-
рамические опоры (диаметр 3 мм н длина
50 мм); 4 — медиый колпак (диаметр 35 мм
и высота 53 мм); 5 — термопара, связанная
с программирующим устройством; 6 —• сопло
для ввода охлаждающего газа (жидкий СО2);
7 — электрический нагреватель (внутренний
диаметр 45 мм и высота 100 мм); 8 — медное
основание (диаметр 38 мм).
описана изолированная систе-
ма, в которой стандартная
ячейка ДТА фирмы «Дюпон»
была приспособлена для иссле-
дования взрывчатых веществ.
Необходимость защиты образ-
цов даже от слабой окисли-
тельной среды потребовала
вакуумирования ячеек до
0,13 мПа. Для этого был удален
пористый диск, реконструиро-
вана опора и дополнительно
уплотнены электрические и га-
зовые вводы. Для предотвраще-
ния термических градиентов в
проводах термопар были исполь-
зованы защитные стеклянные
трубки.
Несколько приборов ДТА
описано Бэрроллом и др.
[93, 94]. На фиг. 6.30 показана
калориметрическая ячейка
ДТА, в которой чувствительные
термоэлементы для определе-
ния ДТ крепились к контей-
неру образца. В этом приборе
медь-константановые термопары
припаивались к медным чашкам с наружным диаметром 4 мм, имею-
щим медные крышки. Термопары и чашки для образцов поддержи-
вались керамическими изоляционными трубками, которые прикреп-
лялись к металлическому основанию. Все чашки нагревались теп-
ловым излучением от зачерненного медного колпака, что предот-
вращало локальные перегревы от нагревательной обмотки. Ячейка
ДТА целиком помещалась под стеклянный колпак, который обес-
печивал регулируемую атмосферу при различных давлениях — от
пониженных до ~0,2 МПа.
Система с регулируемым давлением для прибора ДТА описана
Кемме и Крепсом [951. Этот прибор позволял определять давление
паров образца.
252
Глава 6
Имеются описания [96, 97] приборов ДТА, которые использова-
лись для изучения каталитических свойств вещества. Прототип
прибора описан Стоуном и Рейзом [411.
Уолд и Уиндинг [98] описали способ высокочастотного нагрева
больших образцов резины. Образец помещался между двумя па-
раллельными пластинами, соединенными с высокочастотным гене-
ратором. Температура образца определялась термопарой, располо-
женной в его центре.
Б. ДИФФЕРЕНЦИАЛЬНАЯ СКАНИРУЮЩАЯ КАЛОРИМЕТРИЯ
Из всех приборов ДСК наиболее широко использовался диф-
ференциальный сканирующий калориметр фирмы «Перкин-Элмер»
модели ДСК-1, выпущенный в 1963 г. [99, 100]. Недавно была вы-
пущена [23, 101, 102] модель ДСК-2 с расширенным интервалом
Фиг, 6.31, Конструкции держателей образцов, используемых в приборах
ДСК фирмы «Перкии-Элмер» [23].
а — схема держателя в ДСК-1; б —конструкция держателя в приборе ДСК-1В; в— конструк-
ция2 держателя в приборе ДСК-2; 1 — платиновые датчики; 2 — отдельные нагреватели;
3 — образец; 4 — эталон.
температур (до 725°С), § улучшенными воспроизводимостью и ли-
нейностью базовой линии и более высокой калориметрической чув-
ствительностью. На фиг. 6.31 сравниваются [23] держатели образ-
цов, используемые в приборах ДСК-1, ДСК-1В и ДСК-2. В ячейке
ДСК-1 держатели образца ^эталона состоят из чашки и опоры (из-
Приборы для ДТА и ДСК
253
Фиг. 6.32. Схематическая диаграмма прибора ДСК фирмы «Перкии-Элмер».
готовленных из нержавеющей стали), платинового проволочного
датчика, нихромового нагревателя и других деталей. Все эти эле-
менты соединены в очень компактный «сэндвич». Такой держатель
образца хорошо работал в интервале температур от —125 до 500°С.
В держателе образца прибора ДСК-2 в качестве конструкционных
Материалов используются платино-иридиевый сплав для корпуса
и деталей держателя, платиновая проволока для нагревателя и дат-
чика и окись альфа-алюминия для электроизоляции. Все элементы
держателя соединены точечной сваркой.
Схематическая диаграмма калориметра фирмы «Перкин-Элмер»
показана на фиг. 6.32. Прибор ДСК в отличие от прибора ДТА
поддерживает температуру образца равной температуре эталона
(или нагревательного блока), подводя тепло к образцу или эталону.
Количество тепла, требуемое для поддержания одинаковых тем-
ператур, записывается в функции времени (или температуры).
Помимо записи кривой энтальпии, при выделении образцом в про-
цессе нагревания летучих компонентов этот процесс также регист-
рируется.
254
Глава 6
Прибор содержит два «управляющих контура»: один для ре
гулирования средней температуры, а другой для регулирования
дифференциальной температуры. В первом контуре программирую
щее устройство формирует электрический выходной сигнал, про
порциональный требуемой температуре держателей образца и эта
лона. Сигнал программирующего устройства, достигая усилителя
контура средней температуры, сравнивается с сигналами, посту
пающими через вычислительный блок для определения средней
температуры с платиновых термометров сопротивления, располо
женных в держателях эталона и образца.
В контуре дифференциальной температуры сигналы с платино
вых термометров сопротивления, соответствующие температурам
образца и эталона, подаются на усилитель контура дифферент!
альной температуры через схему сравнения, которая определяет,
какая из температур — эталона или образца — выше. Для устра
нения любой разности температур между ними усилитель контура
дифференциальной температуры обеспечивает подвод соответствую-
щей по величине дифференциальной мощности к нагревателю эта
лона или образца. Сигнал, пропорциональный дифференциальной
мощности, передается также на самопишущий гальванометр, кото-
рый записывает кривую дифференциальной мощности в зависимос-
ти от времени (температуры). Площадь пика кривой в этом случае
прямо пропорциональна тепловой энергии, поглощаемой или вы-
деляемой при переходе.
Погрешность определения температуры в приборе ДСК-2 со-
ставляет ±0,1°С. Калориметрическая чувствительность изменяет-
ся в пределах 0,42—83,7 мДж/с, соответствующих всей шкале от-
клонения пера самописца 10 мВ. В нагревателе может поддержи-
ваться среда азота или аргбна (статическая или динамическая)
при давлении 50—300 кПа. Для низкотемпературных измерений
необходима среда гелия.
Имеются описания различных держателей образцов прибора
ДСК фирмы «Перкин-Элмер». Фриберг и Аллеман [103] описали
герметизированную металлическую ячейку со съемной резьбовой
крышкой. В качестве конструкционных материалов использовались
латунь, нержавеющая сталь и алюминий. Уэндландт [104] описал
держатель образца в виде стеклянной капиллярной трубки диамет-
ром 1,6—1,8 мм. Трубки помещались в алюминиевые держатели,
которые устанавливались в соответствующие ячейки калориметра.
Предлагались также держатели образцов для измерения давления
паров- жидкостей [105] и теплот смешения [106]. Мори и др. [107]
описали узел держателя образца, заключенный в вакуумную ка-
меру.
Бакстер [18] рассмотрел теорию и рабочие характеристики ячей-
ки ДСК фирмы «Дюпон». Эта ячейка, показанная на фиг. 6.5, уста-
навливается на константановом термоэлектрическом диске, который
Приборы для ДТА и ДСК
255
вляется основным каналом передачи тепла между образцом, на-
1>евателем и окружающей атмосферой, а также составной частью
?рмопары для измерения ДТ. Хромелевый провод подсоединяется
25,4
а 6
Фиг. 6.33, Ячейка ДСК, описанная Дэвидом [109].
/ — термопара для системы считывания температуры; 2 — термопара схемы переключения
при достижении заданной температуры; 5 — термопара программирующего устройства нли
термопара нагревателя; 4—входное отверстие для газа; 5—выходное отверстие для газа;
б— дифференциальная термопара со стороны образца; 7 — дифференциальная термопара со
стороны эталона; 8—керамический термоизолятор; 9— керамические опорное стержни;
10 — чашки для образцов.
к каждой платформе диска, образуя таким бразом дифференциаль-
ную хромель-константановую термопару. Температурный интер-
вал прибора от—150 до 600°С. Такая ячейка ДСКможет быть при-
способлена для работы в камере высокого давления (до 6,7 МПа)
1108].
Новая ячейка ДСК, основанная на том же принципе, что и ра-
нее рассмотренная ячейка ДСК фирмы «Дюпон», была описана Дэ-
видом [109]. Калориметрическая ячейка, показанная на фиг. 6.33,
содержит дифференциальную термопару новой конструкции в виде
тонких пластинок, изолированных от стенки и нижней части ячей-
ки для обеспечения повышенной чувствительности. Эта термопара
256
Глйва 6
а
Фиг. 6.34. Калориметр фирмы «Дельтатерм» [ПО].
а — блок-схема системы; б — сечение калориметрического узла; 1 — адиабатическая оболочка;
2 —образец; 3 —медный блок; 4—6 — медные крышки; 7—крышка камеры образца; 8— ка-
мера образца; 9—нагреватель; 10—образец; 11,12 — термопары; 13 — керамическая стойка.
состоит из положительной и отрицательной полос из платино-нике-
левого сплава. Для образцов и эталонов использовались плоские
контейнеры. Две дополнительные термопары применялись для из-
мерения температуры ячейки, что позволяло использовать их для-
Приборы для ДТА и ДСК
257
регулирования температуры по заданной программе, а также
блокировки и регистрации температуры. Максимальная температу-
ра ячейки 1000°С.
Схема динамического адиабатического калориметра фирмы
«Дельтатерм», рассмотренного Дошем и Уэндландтом [ПО], показа-
на на фиг. 6.34, а. Разность между температурами образца и адиа-
батической оболочки определялась с помощью дифференциальной
термопары Т1. Сигнал с дифференциальной термопары регулирует
подачу энергии от источника к нагревателю Н\. Мощность энергии,
поступающей к нагревателю, измерялась ваттметром (для измере-
ния использовался эффект Холла). Калориметрический узел, изоб-
раженный на фиг. 6.34, б, укреплен на керамической стойке и со-
стоит из массивного медного блока 3, закрытого медными крышка-
ми 4—6. Изготовленные нз серебра камера образца 8 и крышка 7
теплоизолированы от блока. Для определения температуры исполь-
зовались термопары И и 12 в защитной оболочке. Образцы могли
иметь вид твердых блоков, порошков или жидкостей.
Имеются описания других приборов, например микрокалори-
метра [111], подобного калориметру Кальве [112], дифференци-
альных калориметров, предназначенных для работы при высоких
температурах [114—117], и других [118—121].
ЛИТЕРАТУРА
I. Wendlandt W. W., Technique of Inorganic Chemistry, Jonassen H. B.,
Weissberger A., eds., Vol. 1, Interscience, N. Y., 1963, Ch. 6.
2. Garn P. D., Anal. Chem., 37, 77 (1965).
3. Bohon R. L., Anal. Chem., 35, 1845 (1963).
4. Mazieres C., Anal. Chem., 36, 602 (1964).
5. Miller B., Thermochim. Acta, 2, 225 (1971).
6. Kulp J. L., Kerr P. F., Science, 105, 413 (1947).
7. Kerr P. F., Kulp J. L., Am. Mineralogist, 33, 387 (1948).
8. Cox D. B., McGlynn J. F., Anal. Chem., 29, 960 (1957).
9. Burr J. T., Thermal Analysis, Schwenker R. F., Garn P. D., eds., Vol. 1,
Academic Press, N. Y., 1969, p. 301.
10. Wilhoit R. C., J. Chem. Educ., 44, A571 (1967).
11. Brewer L., Zavitsanos P., J. Phys. Chem. Solids, 2, 284 (1957).
12. Lodding W., Sturm E., Am. Mineralogist, 42, 78 (1957).
13. Joncich M. J., Bailey D. R., Anal. Chem., 32, 1578 (1960).
14. Van Tets A., Wiedemann H. G., Thermal Analysis, Schwenker R. F.,
Garn P. D., eds., Vol. 1, Academic Press, N. Y., 1969, p. 121.
15. King W. H„ Camilli С. T., Findeis A. F„ Anal. Chem., 40, 1330 (1968).
16. King W. H., Camilli С. T., Findeis A. F., Analytical Calorimetry, Por-
ter R. S., Johnson J. F,, eds., Plenum Press, N. Y., 1968, p. 261.
17. Yamamoto A., Yamada K., Maruta M., Akiyama J., Thermal Analysis,
Schwenker R. F., Garn P. D., eds., Vol. 1, Academic Press, N. Y., 1969,
p. 105.
18. Baxter R. A., Thermal Analysis, Schwenker R. F., Garn P. D., eds., Vol. 1,
Academic Press, N. Y., 1969, p. 65.
19. McAdie H. G., Thermal Analysis, Schwenker R. F., Garn P. D., eds., Vol. 1,
Academic Press, N. Y., 1969, p. 693.
258
Глава 6
20. Menis О., Sterling J. Т., Status of Thermal Analysis, Menis O., ed., Natio-
nal Bureau of Standards Special Publication 338, Washington, D. C., Oct
1970.
21. Pakulak J. M., Leonard G. W., Anal. Chem., 31, 1037 (1959).
22. Weaver E. E., Keim W., Proc. Indiana Acad. Sci., 70, 123 (1960).
23. O’Neill M. J., Fyans R. L., Eastern Anal. Symp., N. Y., Nov. 1971.
24. Hill J. A., Murphy С. B_, Anal. Chem., 31, 1443 (1959).
25. Hogan V. D., Gordon S., Anal. Chem., 32, 573 (1960).
26. Campbell C., Weingarten G., Trans. Faraday Soc., 55, 2221 (1959).
27. Clampitt В. H., Anal. Chem., 35, 577 (1963).
28. Vassallo D. A., Harden J. C., Anal. Chem., 34, 132 (1962),
29. Wiedemann H. G., Chem. Ing. Tech., 36, 1105 (1964).
30. Barrail E. M., Guide to Modern Methods of Instrumental Analysis, Gouw
T. H., ed., Wiley-Interscience, N. Y., 1972, Ch. XII.
31. Lodding W., Hammell L., Rev. Sci. Instr., 30, 885 (1959),
32. Lodding W., Hammell L., Anal. Chem., 32, 657 (1960).
33. Rudin A., Schreiber H. P., Waldman M. H., Ind. Eng. Chem., 53, 137
(1961).
34. Bohon R. L., Anal. Chem., 33, 1451 (1961).
35. Martin A. J., Edwards K- L., J. Sci. Instr., 36, 170 (1959).
36. Whitehead W. L., Breger I. A., Science, 111, 279 (1950).
37. Stone R. L., J. Am. Ceram. Soc., 35, 76 (1952).
38. Wendlandt W. W., J. Chem. Educ., 40, 428 (1963).
39. Reisman A., Anal. Chem., 32, 1566 (1960).
40. Chihara H., Seki S., Bull. Chem. Soc. Japan, 26, 88 (1953).
41. Stone R. L., Rase H. F., Anal. Chem., 29, 1273 (1957).
42. Stone R. L., Anal. Chem., 32, 1582 (I960).
43. Levandowsky J., Sacovy N., Inst, franc, petrole, XI, 818 (1956).
44. Theall G. G., Thermal Analysis, Schwenker R. F., Garn P. D., eds., Vol. 1,
Academic Press, N. Y., 1969, p. 97.
45. Barral! E. M., Rogers L. B., Anal. Chem., 34, 1101 (1962).
46. Burgess G. K., U. S. Bur. Std., Bull., 5, 199 (1908—1909).
47. Kauffman A. J., Dilling E. D., Econ. Geol., 45, 22 (1950).
48. Roberts-Austen W. C., Metallographist, 2, 186 (1899).
49. Smothers W. J., Chiang Y., Differential Thermal Analysis: Theory and
Practice, Chemical Publ. Co., N. Y., 1958, pp. 294—399.
50. Smothers W. J., Chiang Y., Differential Thermal Analysis: Theory and
Practice, 2nd ed., Chemical Publ. Co., N. Y., 1966.
51. Garn P. D., Thermoanalytical Methods of Analysis, Academic Press,
N. Y„ 1965, Ch. IX.
52. Barrail E. M., Johnson J. E., Techniques and Methods of Polymer Evalua-
tion, Slade P. E., Jenkins L. T., eds., Marcel-Dekkers, N. Y., 1966, Ch. I.
53. Mackenzie R. C., Mitchell B. D., Differential Thermal Analysis, Macken-
zie R. C., ed., Academic Press, Gordon, 1970, Ch. HI.
54. Anon, Industrial Research, 25 (Nov. 1969).
55. Wendlandt W. W., Thermal Analysis Techniques, Handbook of Commer-
cial Scientific Equipment, Veilion C., Wendlandt W. W., eds., Vol. 2,
Marcel-Dekker, N. Y. (в печати).
56. Wendland W. W., Lab. Management, 26 (Oct. 1965).
57. Wendlandt W. W., J. Chem. Educ., 49, A571, A623 (1972).
58. Bollin E. M., Differential Thermal Analysis, Mackenzie R. C., ed., Acade-
mic Press, Lnd., 1970, Ch. VII.
59. Barrett E. J., Hoyer H. W., Santoro A. V., Mikrochim. Acta, 1121 (1970).
60. Santoro A. V., Barrett E. J., Hoyer H. W., J. Thermal Anal., 2, 461 (1970).
61. Wendlandt W. W., Thermochim. Acta, 1, 419 (1970).
62. Etter D. E., Tucker P. A., Wittenberg L. J., Thermal Analysis, Schwen-
ker R. F., Garn P. D., eds., Vol. 2, Academic Press, N. Y., 1969, p. 829.
Приборы для ДТА и ДСК
259
63. Gilpatrick L. О., Cantor S., Barton С. J., Thermal Analysis, Schwenker
R. F., Garn P. D., eds., Vol. 1, Academic Press, N. Y., 1969, p. 85,.
64. Roberts H. S., J. Am. Chem. Soc., 57, 1034 (1935).
65. Kracek F. C., Trans. Am. Geophys. Un., 27, 364 (1946).
66. Bollin E. M., Kerr P. F., Am. Mineralogist, 46, 823 (1961).
67. Faktor M. M., Hanks R., Trans. Faraday Soc., 63, 1122 (1967).
68. Jensen E., Am. J. Sci., 240, 695 (1942).
69. Barrall E. M., Rogers L. B., J. Inorg. Nucl. Chem., 28, 41 (1966).
70. David D. J., Anal. Chem., 37, 82 (1965).
71. Locke С. E., Proc, of the Third Toronto Symp. on Thermal Analysis,
McAdie H. G., ed., Chemical Institute of Canada, Toronto, 1969, p. 251.
72. Harker R. I., Am. Mineralogist, 49, 1741 (1964).
73. Cohen L. H., Klement W., Kennedy G. C., J. Phys. Chem. Solids, 27, 179
(1966).
74. Kuballa M., Schneider G. M., Ber. Bunsen Gesellschaft fur physik, Chem.,
75, 513 (1971).
75. Takeya G., Ishii T., Makino K., Ueda S., Kogyo Kagaku Zasshi, 69, 1654
(1966).
76. Ueda S., Yokoyama S., Ishii T., Takeya G., Kogyo Kagaku Zasshi, 74,
1377 (1971).
77. Bousquet J., Blanchard J. M., Bonnetot B., Claudy P., Bull. Soc. Chim.
France, 1841 (1969).
78. Newkirk T. F., J. Am. Ceram. Soc., 41, 409 (1958).
79. Nedumov N. A., Differential Thermal Analysis, Mackenzie R. C., ed.,
Academic Press, Lnd., 1970, p. 168.
80. Rupert G. N., Rev. Sci. Instr., 34, 1183 (1963).
81. Rupert G. N., Rev. Sci. Instr. 36, 1629 (1965).
82. Rupert G. N., Proc, of the First International Conference on Thermal Ana-
lysis, Redfern J. P., ed., Macmillan, Lnd., 1965, p. 19.
83. Sommer G., Jochens P. R., Minerals Sci. Eng., 3, 3 (1971).
84. Miller R. P., Sommer G., J. Sci. Instr., 43, 293 (1971).
86. Sommer G., Jochens P. R., Howat D. D., J. Sci. Instr., Ser. 2, 1, 1116
(1968).
86. Proks I., Zlatovsky I., Chem. Zvesti, 23, 620 (1969).
87. Wendlandt W. W., Bradley W. S., Anal. Chim. Acta, 52, 397 (1970).
88. Wendlandt W. W., Chimia, 26, 2 (1972).
89. Wendlandt W. W., J. Chem. Educ., 38, 571 (1961).
90. Borchardt H. J., Daniels F., J. Am. Chem Soc., 79, 41 (1957).
91. Miller G. W„ Wood J. L., J. Thermal Anal., 2, 71 (1970).
92. Graybush R. J., May F. G., Forsyth A. C., Thermochim. Acta, 2, 153 (1971).
93. Barrail E. M., Gernert J. E., Porter R. S., Johnson J. F., Anal. Chem., 35,
1837 (1963).
94. Barrall E. M., Porter R. S., Johnson J. F., Anal. Chem., 36, 2172 (1964).
95. Kemme H. R., Kreps S. I., Anal. Chem., 41, 1869 (1969).
96. Macak J., Malecha J., Anal. Chem., 41, 442 (1969).
97. Dimitrov R., Compt. rend. acad. bulgare sci., 23, 1215 (1970).
98. Wald S. A., Winding С. C., Anal. Chem., 37, 1622 (1965).
99. O’Neill M. J., Anal. Chem., 36, 1238 (1964).
100. Watson E. S., O’Neill M. J., Justin J., Brenner N., Anal. Chem., 36, 1233
(1964).
101. Perkin-Elmer Thermal Analysis Newsletter, № 10 (Feb. 1972).
102. O’Neill M. J., Gray A. P., ICTA III, Davos, Switzerland, Aug. 23—28,.
1971, paper 1—24.
103. Freeberg F. E., Alleman T. G., Anal. Chem., 38, 1806 (1966).
104. Wendlandt W. W., Anal. Chim. Acta, 49, 187 (1970).
105. Farritor R. E., Tao L. C., Thermochim. Acta, 1, 297 (1970).
106. Mita I., Imai I., Kambe H., Thermochim. Acta, 2, 337 (1971).
260
Глава 6
107. Morie G. P., Powers T. A., Glover C. A., Thermochim. Acta, 3, 259 (1972).
108. Levy P. F., |Nieuweboer G., Semanski L. C., Thermochim. Acta, 1, 429
(1970).
109. David D. J., J. Thermal Anal., 3, 247 (1971),
110. Dosch E. L., Wendlandt W. W., Thermochim. Acta, 1, 181 (1970).
111. Evans W. J., McCourtney E. J., Carney W. B., Anal. Chem., 40, 262 (1968).
112. Calvet R„ Com.pt. rend., 226, 1702 (1948).
113. Sale F. R., J. Phys. E. Sci. Instr., 3, 646 (1970).
114. Thomasson C. N., Cunningham D. A., J. Sci. Instr., 41, 308 (1964).
115. Berger C., Richard M., Eyraud L., Bull. Soc. Chim. France, 1491 (1965).
116. Roux A., Recbaid M., Eyraud L., Elston J., J. Phys, et Phys. Appl., 25,
51A (1964).
117. Nicholson P. S., Lawrence Radiation Lab. Report, UCRL-17820, Sept.
1967.
118. Speros D. M., Woodhouse R. L., J. Phys. Chem., 67, 2164 (1964),
119. Speros D. M., Woodhouse R. L., J. Phys. Chem., 72, 2846 (1968).
120. Garski H., Z. angew. Chemie, 24, 206 (1968).
121. Muller W., Schuller D., Ber. Bunsen Gesellschaft fur Physik, Chem., 75,
79 (1971).
Глава 7
ПРИМЕНЕНИЕ ДИФФЕРЕНЦИАЛЬНОГО
ТЕРМИЧЕСКОГО АНАЛИЗА
И ДИФФЕРЕНЦИАЛЬНОЙ СКАНИРУЮЩЕЙ
КАЛОРИМЕТРИИ
А. ВВЕДЕНИЕ
В гл. 5 было показано, что кривые ДТА и ДСК состоят из ряда
пиков, направленных вверх или вниз относительно оси АТ или оси
теплового потока. По положению пиков на шкале температур (оси
абсцисс), их числу и форме производится качественная идентифика-
ция вещества, а площади пиков, пропорциональные энтальпии
реакции, используются для количественной оценки, например,
прореагировавших веществ или для термохимических определений.
Вследствие многообразия факторов, влияющих на ход кривых ДТА
и ДСК, температура и форма пиков являются до некоторой степени
эмпирическими. Однако в общем случае эти кривые воспроизводят-
ся на любом используемом приборе и, следовательно, могут быть
полезными в лабораторных исследованиях. Используя для калиб-
ровки прибора различные вещества, можно по заключенным между
базовой линией и термограммой пика площадям найти значения
теплоты реакции, перехода, полимеризации, плавления и т. д.
В том случае, когда теплота реакции известна, по термограмме
можно определить количество прореагировавшего вещества.
В табл.^7.1 указаны некоторые причины возникновения эндо-
термических или экзотермических пиков на кривых ДТА и ДСК.
Любое явление, которое сопровождается изменением энтальпии
пли теплоемкости (переход второго рода), можно обнаружить ме-
тодами ДТА или ДСК при условии достаточной чувствительности
прибора. Указанные явления вызываются фундаментальными из-
менениями в физическом состоянии, химическом составе, реакцион-
ной способности молекул вещества и т. д. Форма пиков, а также
их максимальная (АТмаКс) и минимальная (АТмцн) температуры
определяются в основном кинетикой реакции, хотя на них также
I влияют плотность упаковки частиц образца, его геометрические
параметры, скорость нагревания, атмосфера нагревателя и началь-
ная температура эталона. Изменение может быть связано с измене-
нием удельной теплоемкости образца; это важный параметр при
определении температуры стеклования Tg полимеров. Площадь
пика определяется изменением энтальпии, а также такими факто-
рами, как размеры, теплопроводность и удельная теплоемкость об-
разца.
262
Глава 7
Таблица 7.1
Физико-химическая природа ликов кривых ДТА и ДСК [57]
Процесс Изменение эндотермиче- ское энтальпии экзотерми- ческое
Физический
Кристаллизация...............................
Плавление....................................
Испарение....................................
Возгонка ....................................
Адсорбция....................................
Десорбция....................................
Абсорбция....................................
Переход в точке Кюри.........................
Стеклование .................................
Жидкокристаллический переход................
Немонотонное изменение теплоемкости.........
Изменение хода базовой
линии, пиков нет
+ I
Изменение хода базовон
линии, пиков нет
Химический
Хемосорбция Десольватация + 4~
Дегидратация +
Разложение + +
Окислительная деструкция +
Окисление в газовой атмосфере Восстановление в газовой атмосфере + +
Реакции окисления-восстановления + +
Реакции в твердой фазе + +
Горение +
Полимеризация +
Предварительное отверждение смол +
Каталитические реакции +
Метод ДТА использовался геологами, специалистами по кера-
мике и металлургами на протяжении многих лет. Это быстрый
аналитический метод идентификации глин и других минералов,
определения фазовых переходов и диаграмм состояния, исследова-
ния реакций, протекающих при высоких температурах, и т. д.
Обычно дифференциальный термический анализ сочетают с рент-
геноструктурным методом, дилатометрией, термогравиметриче-
ским анализом, методом электропроводности и другими методами.
Хотя ряд классических химических исследований был выполнен
методом ДТА начиная с 1930 г., химики проявили интерес к нему
Применение ДТА и ДСК
263
лишь в последние годы. Из ранних работ в этом направлении сле-
дует упомянуть работы Красека 158, 59]. В настоящее время облас-
ти применения термического анализа в химии и других науках
расширяются очень быстро. В табл. 7.2 и 7.3 указаны типичные
Таблица 7.2
Типичные области применения методов ДТА и ДСК в химии
Вещества
Тип исследования
Катализаторы
Полимеры
Смазочные вещества
Жиры и масла
Координационные комплексные соеди-
няя
Углеводы
Аминокислоты и белки
Гидраты солей металлов
Окиси металлов и неметаллов
Каменный и бурый уголь
Древесина и подобные вещества
Природные продукты
Органические соединения
Глины и минералы
I Металлы и сплавы
I |очва
Внелогические объекты
Реакции разложения
Диаграммы состояния
Кинетика реакций
Реакции в твердой фазе
Реакции дегидратации
Радиационная деструкция
Катализ
Теплота адсорбции
Теплота реакции
Теплота полимеризации
Теплота возгонки
Теплота переходов
Реакции десольватации
» »
Реакции газа с твердым веществом
Определение точки Кюри
Определение чистоты веществ
Термостабильность
Стойкость к окислению
Определение температуры стеклования
Сравнение веществ
области исследования с применением методов ДТА и ДСК. Дейст-
рительно, эти методы оказались полезными почти во всех областях
химии, однако в последнее время они находят наибольшее приме-
нение в химии полимеров для идентификации и изучения их свойств.
Ввиду широкого распространения методов ДТА и ДСК здесь
будут описаны применения, относящиеся главным образом к об-
ласти аналитической химии. В этой области методы ДТА и ДСК
могут быть использованы для контрольного или систематического
Анализа родственных, но не одинаковых веществ. В качестве конт-
рольных методов они могут применяться для экспресс-анализа с
целью выявления различий между отдельными партиями сырья в
тех случаях, когда при некоторых различиях в свойствах вещест-
ва требуется изменить технологию его обработки. В качестве срав-
нительных методов ДТА и ДСК можно использовать в некоторых
случаях для выявления материалов, дающих аномальные результа-
Некоторые примеры использования методов ДТА и ДСК в промышленности
нь’еийэхви атчкч1гихэяэх Ч—F 4—1—1—1- Ч—F4-
ГЧИЬОЦ 4-4-4- ч-
ElfNJV 4-4-4-F ч-ч- ч-ч- ч-
ESKlTUOA^QiqHl.QHPd Ч—!—F-F Ч—F 4- 4- 4-4-4-
ГТОЭЕШЭЕКЦ 4—F4—F ч-ч-ч-ч- ч- 4-4-4-
ЕЙофэсф кинэниКаоэ Ч—F-F4—!—F4- 4- Ч—F
1 141 -вЙЕпайи эияээьихаайЕКЙЕф Ч-Ч- 4-4-4—F ч-ч- ч-
ИЛЭЕЙу 4- 4- Ч—1—1—F -F Ч-Ч-
гигвдэниэд Ч-Ч-Ч- Ч-Ч- Ч-Ч- Ч-Ч-
NifiTEiaw ч—1—1- 4- ч—F Ч—1-
ГИГИНЙЭ}! Ч—F 4- Ч—1—F 4-
ОГЯЭ1Э 4- Ч-Ч- Ч-Ч-
ЕЯИП-ПОХ + Ч-Ч-Ч- -F Ч-Ч-
ЭЖ1ЭИ1ГЕНИИИЙМ S БИ ПИХ —1—1_ —1—1_ —1—-j—
вахээУпэи anxEhsiqdEg Ч—F 4- 4—F 4- 4-4-
кйэкохЭЕ1Гб Ч—F 4- Ч—1—F-F 4- 4-
ИШГЕЯИИИХ 4—1—1—1—1—1—F 4—1—F 4-
1Ч1ЭНЙЭ>1 4--F 4—F 4-4-
влнивДэ^ 4—1—F 4- 4- 4—F
Назначение Идентификация Состав (количественный) Диаграммы состояния Остаточный растворитель Гидратация — дегидратация Термостабильность Стойкость к окислению Полимеризация Отверждение Чистота Реакционная способность Каталитическая активность Стеклование Радиационная деструкция Термохимические константы
Применение ДТА и ДСК
265
гы при других испытаниях. Наконец, при подходящей калибровке
приборов эти методы применимы для количественной оценки ве-
щества или смеси веществ, а также для определения чистоты про-
дукта (гл. 10). В данной главе предпринята попытка показать лишь
возможности применения рассматриваемых методов и не ставится
В задача их исчерпывающего описания.
Б. КАТАЛОГИ КРИВЫХ ДТА
Выпущено несколько каталогов, содержащих эталонные кри-
пые ДТА. Каталог Сэдтлеровской исследовательской лаборатории
содержит более 2000 кривых ДТА, из которых 450 — термограммы
промышленных химических продуктов, 150 — фармацевтических
Фиг. 7.1, Типичная кривая ДТА (из каталога стандартных термограмм
Сэдтлеровской исследовательской лаборатории).
препаратов и стероидов, 1000 — чистых органических соединений
и 360 — чистых неорганических веществ. Предметный указатель
и указатель химических формул позволяют быстро и удобно отыс-
кать в каталоге требуемые термограммы. На фиг. 7.1 представлена
типичная кривая ДТА, полученная с помощью калориметра фир-
мы «Дюпон», модель 900. Каждая кривая этого каталога была за-
писана при одних и тех же значениях скорости нагревания и на-
чальной температуры и в одинаковых атмосферах нагревателя.
Несколько иной подход использован в указателе кривых ДТА
системы SCIFAX, выпущенном под редакцией Маккензи. Применя-
лись перфокарты (фиг. 7.2), распределенные по трем секциям: ми-
266
Глава 7
Вводные карты (20, розовые)
Карты органических соединений (311, коричневые)
кодам основного и-------------\
второго пиков(здесь 20,00) л
Карты минералов
(1012, зеленые)
Всего 1630 карт плюс 31
поисковая карта, 1 допол-
нительная кодовая карта,
2 спицы, описание на 24 стр.
Карты неорганических веществ Kaprt/ы с вырезами,
(237, голубые)соответствующими
основного пика
хесяткЬ 007 Единицы
Код вгпррого пика_________
875-925
чя.__'
См. также ।
Тчп'пнзэрои Роу
Фиг. 7.2. Перфокарта из указателя кривых ДТА системы SC1FAX.
1 — интервал температур; в котором находится основной пик; 2—код основного и второго
пиков соответственно (на кодовой карте указаны 29 стандартных интервалов); 8 — ссылки
иа другие релевантные карты; 4 — оценка величны пика, например т — средний; 5 — интер-
вал температур, в котором находится второй по величине пик (если он имеется); 6— данные
для каждой кривой, расположенные по горизонтали (все температуры в градусах Цельсия;
7* (—) — эндотермический пик; Т (-J-)—экзотермический пик); 7 — код по классификации Хэя
(минералы); 8— ссылки на литературу; 9— иомер дайной карты в алфавитном порядке
(А — первая серия); 10 — название минерала или соединения.
нералы, неорганические вещества и органические соединения. Всего
имеются 1662 карты, из которых 1012 относятся к минералам,
287 — к неорганическим веществам и 311 — к органическим соеди-
нениям, а также кодовые карты. Кривые ДТА содержатся также
в атласе кривых, полученных с помощью дериватографа 11].
В. ОРГАНИЧЕСКИЕ СОЕДИНЕНИЯ
Применение методов ДТА и ДСК для изучения органических
соединений (фиг. 7.3) исключительно многообразно. Трудно выде-
лить из них наиболее важное или наиболее широко используемое.
Применение ДТА и ДСК.
267
Фиг. 7.3. Применения методов ДТА и ДСК для изучения органических
соединений.
В производстве фармацевтических препаратов и органических со-
единений определение чистоты продукта является, пожалуй, наи-
более важным. В других областях особый интерес представляет
идентификация веществ. Только недавно благодаря использованию
герметизированных держателй образцов исследование органиче-
ских реакций методами ДТА и ДСК приобрело особую важность.
С помощью только одного опыта можно получить информацию,
извлечение которой стандартными методами потребовало бы часы
или дни.
Картина изменений энтальпии в органических соединениях
гораздо менее сложна, чем в органических полимерах. Тем не менее
е помощью ДТА и ДСК в них можно обнаружить различные поли-
морфные превращения. Изменения энтальпии в органических сое-
268
Глава 7
динениях, выражающиеся в эндотермических и экзотермических
эффектах, связаны с плавлением, испарением, переходами в твер-
дой фазе, возгонкой, дегидратацией, разложением и горением.
Применения методов ДТА и ДСК для изучения органических
соединений более подробно рассмотрены в работах [2—8]. Харак
терные для метода ДТА облас-
ти применения рассмотрены Ми-
тчеллом и Берни [2]; методы
ДСК обсуждаются Греем [41.
Термическое разложение ря-
да органических кислот было
исследовано методом ДТА Уэн-
дландтом и Хойбергом [60, 611.
Поскольку эти кислоты подвер-
гались разложению в атмосфе-
ре аргона, кривые ДТА имели
только эндотермические пики.
Эти пики вызывались такими
реакциями, как дегидратация и
декарбоксилирование, возгон-
кой, разложением и фазовыми
переходами из твердого состоя-
ния в жидкое. Температуры,
соответствующие максимумам
пиков при фазовых переходах,
были на 10—30° С выше ранее
полученных температур плавле-
ния. Кривые ДТА некоторых
органических кислот приведе-
ны на фиг. 7.4.
жой кислоты — единственной из
Фиг. 7.4. Кривые ДТА некоторых
органических кислот [60].
а — дигидрат щавелевой кислоты; б — мало-
новая кислота; в — янтарная кислота; г —
глутаровая кислота.
Кривая ДТА дигидрата
исследуемых органических кислот, содержащей кристаллизацион-
ную воду, — имела пики дегидратации со значениями АТМин, рав-
ными 110, 120 и 125° С сооответственно. Все другие пики для орга-
нических кислот были вызваны плавлением или реакциями разло-
жения. Например, второй эндотермический пик на кривой для ян-
тарной кислоты был, вероятно, вызван реакцией дегидратации,
приводящей к образованию ангидрида по следующей схеме:
о
с-он
I
СЩИагреЛмш
сн2 *
с-он
Применение ДТА и ДСК
269'
Давно известно, что меконовая кислота (I) (3-окси-4-оксо-1,4-
пиран-2,6-дикарбоновая кислота) при нагревании на воздухе до
температуры 120—220° С превращается в коменовую кислоту (II)
137]. Метод ДТА был использован для выявления наиболее подхо-
дящих условий протекания этой реакции
а—при атмосферном давлении, скорость нагре-
вания Юград/мии; б— при давлении 1,4 МПа
в атмосфере азота.
Было найдено, что коменовая кислота образуется путем декарбок-
силирования меконовой кислоты; эта реакция дает экзотермический
ПИК при ЛТмакс = 240° С.
При указанной темпера-
туре и выше нее про-
дукт возгоняется.
При использовании
ДТА в условиях высокого
давления наблюдается сме-
щение температуры кипе-
ния бензойной кислоты в
область более высоких
температур. Леви и др.
138] получили кривые ДТА
для чистой бензойной кис-
лоты при атмосферном дав-
лении и избыточном дав-
лении 1,4 МПа (фиг. 7.5).
В соответствии с кривой а
бензойная кислота пла-
иится при температуре
~122° С. При давлении
илота 1,4 МПа (кривая б) эндотермический пик температуры,
соответствующий плавлению, остается неизменным, в то время как
температура кипения повышается до 378° С. Чтобы избежать воз-
гонки и испарения и обеспечить равновесные условия, образцы ис-
пытывали в небольшой герметически закрытой алюминиевой ам-
пуле, имевшей в крышке отверстие диаметром ~50 мкм для вырав-
нивания давления.
Кривые ДТА ряда фармацевтических препаратов были описаны
Брайтоном и Феррари 19], в работе которых приводится качествен-
ная информация относительно чистоты, растворимости, структуры
п полиморфизма соединений. Кривая ДТА триамкинолондиацетата.
|!)| (фиг. 7.6) помогает установить наиболее подходящую темпера-
270
Глава 7
туру сушки этого вещества. Пики растворителя при значениях
ЛТ’мию равных 142 и 166°С, при сушке исчезают. Точное определе
ние температур плавления и кипения методом ДТА впервые под
робно рассматривалось Вассало и Харденом [10]. Они получили
Фиг. 7.6. Кривые ДТА триамкинолондпацетата [9].
Атмосфера азота; скорость нагревания 20 град/мин; 1 — перед сушкой; 2 — после сушки.
точность ±0,3 С в широком интервале плотностей тепловых пото-
ков и могли производить измерения в интервале температур от
— 150 до 450° С. Значения температур плавления Тпп и кипения
Ткип определялись по нескольким точкам на пике кривой ДТА
Увеличение температуры
Фиг. 7.7. Методы оценки тем-
ператур переходов [10].
(фиг. 7.7). Точка А находится на
пересечении базовой линии с эк-
страполированным отрезком пря-
мой низкотемпературной ветви
кривой, а точка Б расположена
на перегибе последней. Точка В
соответствует максимуму достроен-
ного методом экстраполяции пика,
а точка Г находится на пересече-
нии ниспадающей экстраполи-
рованной части кривой и базовой
линии. В табл. 7.4 приведены
температуры образца и эталона,
соответствующие указанным точ-
кам на кривой плавления бензой-
ной кислоты и кипения толуола.
За исключением точки Г, оценки
значений Тпл образцов, как пра-
вило, оказываются более близки-
ми к истинному значению. Наилуч-
Применение ДТА и ДСК
271
шее совпадение для 7ПЛ получается при использовании температу-
ры образца в точке В или температуры эталона в точке А. Темпера-
тура 7КИП, оцененная для толуола по точкам А, Б и Г, оказалась
пыше , чем при оценке по точке В, вероятно, вследствие явления
перегрева.
Таблица 7.4
Сравнение различных способов определения температур переходов
по точкам на пике кривой ДТА [10]
Бензойная кислота, температура плавления1), °C
А Б В Г
.'Налом 121,1 125,0 128,0 131,0
< )бразец 120,3 121,0 121,7 131,9
Толуол, температура кипения2), °C
А Б В г
>гплон 112,5 114,2 115,9 116,2
((бразец 114,6 113,2 111,2 118,8
*) По данным Национального бюро стандартов США, 121,8 °C.
£) По данным Мерка, 110,6 °C с погрешностью ±1°.
-150 -125 -100 -75 -50-25 0 +25
Температура.^
Фиг. 7.8. Низкотемпературная кривая ДТА н-бутана [10].
272
Глава 7
На фиг. 7.8 показана низкотемпературная кривая ДТА н-бу
тана [10]. В интервале температур от —150 до 10° С наблюдакпч и
узкие, резко выраженные, эндотермические пики, соответствуй»
щие температуре кипения (—0,5° С) и температуре плавлении
(—135° С). Температуры кипения и плавления ряда органических
-соединений указаны в табл. 7.5 [10].
Таблица 7 - >
Температуры переходов, найденные методом ДТА [10]
Соединение Температура плавле- ния, °C Температура кипе- ния, °C
* метод ДТА 1 по опублико- ! ванным данным метод ДТА по опублико- ванным данным i
.«-Бутан —135,0 —135,5 —0,5 —0,55
н-Пентан —129,5 —129,7 36,2 36,0
«-Гексан —94,5 —95,3 69,0 68,8
н-Гептан —90,3 —90,6 98,2 98,4
«-Октан —57,0 —56,8 125,6 125,6
н-Нонан — — 150,2 150,7
н-Декан — — 173,0 174,0
н-Додекан — — 215,5 216,0
Бензойная кислота 121,8 121,8 — _—
Вода 0,0 0,0 100,0 100,0
Толуол — — 111,1 110,6
Бензол 5,2 5,5 80,5 80,1
Уксусная кислота 16,5 16,6 118,4 118,1
Полиэтилен марки «алатон-10» . . . 110,5 110,5’) — -—.
Полиэтилен марки «марлекс-50» . . . 134,2 134,51) — —
Тефлон TFE 327,5 327,01) — -—
Тефлон FEP 272,0 272,5’) — —
Дельрин 170,5 171,0’) — —
’) Температуры плавления определены с
Кофлера или рентгеноструктурным методом.
помощью высокотемпературного микроскопа
Точное определение температур плавления и кипения с помощью
методов ДТА и ДСК подробно обсуждалось Бэрроллом [39]. Су-
ществует много различных методов, пригодных для таких опреде-
лений, однако Бэрролл предлагает четкий метод, легко приспосаб-
ливаемый к промышленным приборам и пригодный для изучения
широкого круга органических и неорганических соединений. Дан-
ные, получаемые с помощью этого метода, более надежны, чем из-
меренные с помощью других методов (нагретой масляной бани,
капиллярных трубок и др.). Значения температур кипения, опре-
Применение ДТА и ДСК
273
Дг ле иные методами ДТА и ДСК, гораздо легче воспроизводятся и
пбычпо лучше совпадают с результатами квазиравновесных изме-
рений, чем данные микроэбуллиометрии; кроме того, их, несомнен-
но, можно быстрее получить.
В методе ДТА требуется построение экспериментальной кривой
* Д7' в функции температуры образца, а в методе ДСК строится диф-
ференциальная кривая тепловой энергии в функции времени. Для
определения температуры плавления образец можно поместить в
Капсулу; если в образец необходимо ввести термопару, нужно
использовать смесь образца с разбавителем. В качестве капсулы
обычно используют герметически закрытый контейнер из металла
С высокой теплопроводностью. Для определения температуры ки-
пения необходимо обеспечить: равновесие жидкой и паровой фаз
и регулирование давления газовой атмосферы. Бэрролл [391 под-
робно описывает способы выполнения этих условий.
При использовании капсулы для определения температуры
плавления проводится экстраполяция с целью компенсации тепло-
вого запаздывания системы (фиг. 7.9,а). Истинная температура
плавления вещества получается путем экстраполяции низкотемпе-
ратурной ветви пика кривой до пересечения с изотермической ба-
зовой линией. Если образец находится в непосредственном кон-
такте с термопарой, истинной температурой плавления будет Ть
(фиг. 7.9,6). По фиг. 7.9,6 можно определить также минимум пика
Тт и температуру, соответствующую концу пика, Те. Расстояние
между Ть и Тт зависит от чистоты образца. В образцах с примеся-
ми Ть трудно определить из-за дрейфа базовой линии.
При измерениях температуры кипения температура Тт не имеет
большого значения. Она является лишь функцией различных ха-
рактеристик приборов, скорости диффузии паров и количества
образца в начале кипения. Температура в начале кривой Ть отве-
чает точке кипения вещества (после соответствующих поправок).
В работе Керра и Лэндиса [11] описано использование метода
ДТА для определения температур кипения и плавления микрооб-
разцов объемом 2—5 мкл, взятых на выходе газохроматографиче-
ской колонны шприцем объемом 10 мкл. Примеры определенных
таким образом температур кипения нескольких органических сое-
динений приведены на фиг. 7.10. Определение температур плавле-
ния микрообразцов мета- или пара-ксилола позволило установить
их относительную чистоту (>90%).
Определение температур перехода жидких кристаллов явилось
предметом многочисленных исследований [12—20]. Можно опреде-
лить не только температуры плавления, но также и температуры
переходов в жидких кристаллах. К первым работам в этой облас-
ти принадлежит исследование анисальдазина Бэрроллом и др. [12],
результаты которого представлены на фиг. 7.11. Необходимо обес-
печить стабильное нагревание по линейному закону, поскольку
10—230
Применение ДТА и ДСК. 275
Фиг. 7.9. Определение температуры плавления [39].
а — помещенные в капсулу образцы с чистотой 99,99999 мол. %; б — термопара погружена
непосредственно в образец; 1 — индий; 2 — олово; 8 — свинец; 4 — истинная температура
плавления; 5 — изотермическая базовая линия; 6 — сдвиг за счет погрешности прибора.
отклонение от линейности на 1° С может выразиться на кривой ДТА
в виде ложной температуры стеклования. Две кривые, представлен-
ные на фиг. 7.11, иллюстрируют «растяжение» оси температур для
разделения близко расположенных пиков. Кривая А охватывает
интервал температур 0—280° С, а кривая Б—интервал 135,80—
IK3.340 С. Температурная ось кривой Б растянута по сравнению с
Л. В интервале температур от —100 до 500° возможно «растяже-
ние» до 1000 раз любого участка в 300сС.
Фиг. 7.10. Кривые ДТА образцов различных органических соединений
объемом 2—5 мкл [И].
H2t4°C — нзопропанол; 101°С — лара-диоксан; 118—119°С —2-ацетоксибутён-2; 136,4°С — ангид-
рид уксусной кислоты; 153°С — диметилформамид: 178°С— бензальдегид; 189°С— иодбензол;
rJ'J,8’C — ор/ло-толуидин; 206°С — тетралин; 214°С — метилтиенилкетон; 234°С — фенилацето-
нитрил ; 267,5° С — изоэвгенол.
В табл. 7.6 приведены температуры переходов ряда сложных
лриров холестерина [13]. Эти температуры сравниваются с полу-
ченными Греем [21]. Температуры, определенные по минимумам
шдотермических пиков ДТА, согласуются с температурами, изме-
ряемыми визуально, в пределах ±2°. Абсолютная ошибка опреде-
ления температур во всех случаях составляла +0,1°, а воспроизво-
димость для отдельного образца при последовательных повторных
проходах интервала плавления была равна +0,05° С.
Теплота перехода для жидких кристаллов определялась рядом
исследователей [14—16]. Ее значения очень малы и в большинстве
случаев заключены в пределах 2,09—7,54 Дж/г.
Методы ДТА и ДСК широко использовались для исследования
«разовых переходов и реакций диссоциации органических взрывча-
10*
Таблица 7 Л
Температуры переходов сложных эфиров холестерина [13]
Сложный эфир холестерина ДТА1) По данным Грея») [21J
7. тг 7а S С 1
Формиат .... . —. 97,3 (60,5) 97,5
Ацетат 44 81—87 118,4 (94,5) 116,5
н-Пропионат . . 99+1 110? 115,3 102 116
я-Гептилат . . . 114,1 (<92,5) (95,5) 114
н-Нонаноат . . . 74,0 80,8 93,0 (77,5) 80,5 92
я-Деканоат . . . 85,7 91,2 (81,5) 85,5 92,5
Миристат .... 73,6 79,7 85,5 71 81 86,5
Пальмитат . . . 79,7 85,1 (78,5) 779 83
Стеарат (75,5) (79,5) 83
х) 7\ — нижний переход, промежуточный переход; Та —переход к изотропной жид
кости.
2) S — смектическая, С — холестериновая, / — изотропная жидкость.
Примечание: скобки обозначают монотропный метастабильный переход.
Фиг. 7.11. Кривые ДТА анисальдазина [12].
1 — температура плавления; 2 — температура перехода в жидком кристалле. Кривые А и Б
записаны в разном масштабе по температуре.
I
Применение ДТА и ДСК УЛ
Г
тых веществ. Холл [40] исследовал методом ДСК процесс перехода
И твердой фазе, плавление и разложение некоторых нитраминов:
/V-пикрил-Л'метилнитр амина, 1,3,5-тринитрозо-1,3,5,7-тетраазо-
циклооктана и др. Роджерс и Смит [41] изучали группу родствен-
ных веществ и определили по кривой ДСК предэкспоненциальный
множитель. Позднее Роджерс и Дайнегар [42] определили методом
ДСК теплоту плавления и другие параметры тетранитрата пента-
эритрита.
' Мюррил и др. [43—45] использовали метод дифференциальной
сканирующей калориметрии для объяснения переходов в твердой
фазе большого числа органических соединений. Переходы первого
порядка были обнаружены в соединениях тетраэдрической струк-
туры типа CR1R2R3R4, где R — метильная, метилольная, амино-,
нитро- и карбоксильная группы, а также в соединениях октаэдри-
ческой структуры. Этот метод был использован также для обнару-
жения фазовых переходов в стеаратах щелочных металлов [46],
некоторых дибензоазепинах, карбазолах и фентиазинах [16], а так-
же эфирах орто-фталевой кислоты [31]. Дор ко и др. [49] определя-
ли с помощью метода ДСК кинетику разложения в твердой фазе и
параметры активации для /V-окисей А-арил-А'-тозилоксидиимида.
Несколько работ [22—25, 311 было посвящено калориметриче-
скому определению теплоты перехода для других физических про-
цессов (плавления, кипения, процессов в твердой фазе и др.). Для
обеспечения погрешности порядка ±5% необходима тщательная
калибровка.
Чу [48] с помощью метода ДТА исследовал образование различ-
ных производных органических соединений. Он заменил традици-
онный метод приготовления производного из образца и реагента
процессом, протекающим в одну стадию. Образец нагревался вмес-
те со специальным реагентом с заданной скоростью нагревания в
определенной атмосфере. На кривой ДТА отражаются реакция
образования производного, физические переходы в образце или
избыточном реагенте и физические переходы в промежуточных сое-
динениях и продуктах реакции. В качестве держателя образца
применялись стеклянные капиллярные трубки.
На фиг. 7.12 показано образование производного гидразона
ацетона при использовании пара-нитрофенилгидразина. На кривой
а виден эндотермический пик кипения ацетона при АТмакс = 58°С.
Для пара-нитрофенилгидразина эндотермический пик при АТмакс =
= 160° С вызван плавлением вещества. В случае смеси
ацетона и /шра-нитрофенилгидразина наблюдался сложный эндо-
термический пик в интервале температур 54—80° С, обусловленный
совместным влиянием испарения избыточного ацетона, растворения
паре-нитрофенил гидразина в ацетоне и образования гидразона.
Плавление гидразона обнаруживалось по эндотермическому пику
при АТмакс — 153° С. Повторный термический анализ остатка дает
278
Глава 7
толькфодин эндотермический пик при АУицкс = 153° С. Сообщен
ное ранее значение температуры плавления производного гидразона
составляет 152°С. Сходные результаты были получены в таких
реакциях, как взаимодействие триэтиламина с пикриновой кисло-
той и декстрозы с пропиламином.
Фиг. 7.12. Кривые ДТА, показывающие образование пара-нитрофенилгид-
разона ацетона [48].
а — ацетон; б — пара-нитрофенилгидразин; в — реациопноспособная смесь ацетона и пара-
иитрофеиилгидразина; г — повторный термический анализ остатка после завершения реакция.
Описанный метод динамичен по своей природе и требует, чтобы:
а) специальный реагент быстро образовывал с образцом производ-
ное; б) полученное таким образом производное имело заметный
физический переход или характерную кривую ДТА; в) более ле-
тучий из двух реагентов был взят в избытке; г) один реагент был
растворителем для другого; д) можно было использовать катали-
затор.
Хармелин и др. [26] применили метод ДТА для проведения дие-
нового синтеза Дильса — Альдера с использованием малеинового
ангидрида и антрацена. Этот метод дает возможность определить
температуру, при которой протекает реакция, температуру плавле-
ния образующегося продукта реакции и температуру его разло-
жения.
Применение ДТА и ДСК
279
Методы ДТА и ДСК широко применяются для изучения тепло-
иых свойств взрывчатых веществ и состава ракетных топлив. Фот
147] получил кривые ДТА для некоторых пикратов, стифнатов и
сульфатов гидразина, гуанидина и гуанидиния. Найденные темпе-
ратуры разложения, как правило., были значительно ниже указан-
ных в литературе. Стэммлер [271 исследовал другие пикраты —
пикраты таллия, аммония, тетраметиламмония и тетраэтиламмония.
Фиг. 7.13. Кривые ДТА ацетанилида с различным содержанием примесей.
Эвтектический пик плавления А был записан только для образца, содержащего 2,6 мол. %
примеси; ----- содержание примеси 2,6 мол. %;-----------содержание примеси 0,8 мол. %;
-----— чистое вещество.
Дэвид [28] и Бохон [29] с помощью ДТА изучали термические ха-
рактеристики взрывчатых веществ и ракетных топлив при различ-
ных давлениях окружающей среды до 21 МПа. Определялись теп-
лоты взрыва и (или) разложения. Грейбуш и др. [30] описали опыты
по разложению основных взрывчатых веществ при использовании
камеры ДТА с дистанционным управлением.
Санторо и др. [32—35] исследовали различные органические
реакции, применяя держатели образца в виде запаянных трубок.
В качестве примера можно привести гр/с-транс-изомеризацию
стильбена и олеиновой кислоты, полимеризацию стирола, реакции
Дильса — Альдера и др. Кох [3.6] с помощью метода ДТА обнару-
жил неустойчивые промежуточные соединения в реакции с участием
органических соединений. При нагревании раствора неустойчивого
вещества можно определить температурную характеристику про-
межуточного соединения. И наоборот, отсутствие тепловых эффек-
тов означает, что неустойчивое вещество не образуется.
В гл. 10 будет описано определение относительной чистоты ор-
ганических соединений методами ДТА и ДСК- Большинство анали-
тических методов основано на методе ДСК, хотя можно использо-
вать также кривые ДТА, как это видно, например, из фиг. 7.13.
280
Глава 7
Дифференциальный термический анализ использовался для изу-
чения процессов деструкции жидких углеводородов [50—52], а
также для качественного анализа смазочных веществ [53, 54].
Ноэль [55] показал, что метод ДСК можно использовать для опре-
деления характеристик нефтепродуктов и что полученные резуль-
таты согласовывались с результатами, полученными при исполь-
зовании стандартного метода Американского общества по испыта-
нию материалов (ASTM). Метод ДСК имеет следующие преимуще-
ства перед другими методами оценки свойств нефтепродуктов:
а) результаты воспроизводимы и могут быть быстро получены;
б) требуются очень небольшие образцы (миллиграммы, а не
литры, как в обычных методах);
в) оборудование пригодно для многократного использования;
г) получаемая информация носит фундаментальный, а не узко-
эмпирический характер, поэтому она имеет научное значение, хотя
может использоваться и для контроля в исключительных ситуа-
циях.
Ноэль и Корбетт [56] использовали метод ДСК при изучении
стеклования и плавления асфальтов. Найденные ими значения Tg
хорошо согласуются с полученными ранее с помощью дилатомет-
рии. Теплота плавления асфальтовых восков составляла 109—
134 Дж/г.
Окисление автомобильной тормозной жидкости и машинного
масла изучалось методом ДСК при высоком давлении [38]. Отно-
сительная стойкость к окислению для каждого типа исследуемых
веществ оценивалась при избыточном давлении воздуха 4,2 МПа.
Г. НЕОРГАНИЧЕСКИЕ ВЕЩЕСТВА
Методы ДТА и ДСК находят столь же широкое применение для
изучения неорганических веществ, как и для изучения органиче-
ских соединений. Эндотермические и экзотермические пики обус-
ловлены фазовыми переходами (плавление, кипение, полиморф-
ные превращения), дегидратацией, диссоциацией, изомеризацией,
реакциями окисления и восстановления и др. На фиг. 7.14 указаны
возможные применения методов ДТА и ДСК, а на фиг. 7.15 —• ука-
заны классы исследованных веществ. В обзоре Маккензи и др. [62]
рассмотрены разнообразные применения метода ДТА для изучения
неорганических веществ. Как и в случае органических соединений,
приводимые ниже примеры относятся только к применениям мето-
да в аналитической химии.
Экспресс-метод определения содержания влаги в «почти сухих»
порошкообразных веществах был описан Стоуном [97]. Используя
оборудование ДТА в режиме динамического газового потока, Стоун
помещал образцы в держатель при комнатной температуре и атмо-
сферном давлении. Когда производилась откачка системы при по-
Применение ДТА и ДСК
281
Фиг. 7.14. Некоторые применения методов ДТА и ДСК для изучения не-
органических соединений.
стоянкой скорости, пик кривой ДТА начинался в той точке, где
наружное давление паров воды было меньше парциального давле-
ния паров воды образца. По высоте пика с помощью калибровоч-
ной кривой (фиг. 7.16) можно определить содержание влаги в об-
разце. Калибровочная кривая была построена при использовании
образцов с известным содержанием влаги.
Петриччиани и др. 198] исследовали влияние примеси хлората
калия на термическую стабильность перхлората аммония. Влияние
этой примеси на кривую ДТА перхлората аммония показано на
фиг. 7.17.
На представленных кривых ясно видно возрастание экзотерми-
ческого эффекта за счет перехода в кристаллической решетке пер-
хлората аммония, происходящего при температуре 244° С. При
содержании примеси КС1О3 ~0,1 % тепла, выделившегося после
завершения этого перехода, оказывается достаточным, чтобы
282
Глава 7
Фиг. 7.15. Некоторые классы неорганических соединений, исследованных
методами ДТА и ДСК.
вызвать полное термическое разложение образца. При этом про-
исходит понижение на 150° С температуры термического разложе-
ния чистого вещества, которое обычно разлагается при температуре
~400° С. Метод ДТА можно использовать для приближенного оп-
ределения количества примеси в перхлорате аммония.
Уэндландт и др. [99], изучая с помощью метода ДТА термиче-
ское разложение гидратов оксалатов тория, урана и редкоземель-
ных металлов, исследовали также смеси гидратов оксалатов ред-
коземельных металлов. На фиг. 7.18 представлены кривые ДТА
смесей гидратов оксалатов лантана—церия и празеодима—неодима.
На этой фигуре кривая 1 относится к физической смеси оксала-
тов лантана и церия состава 1 : 1, а кривая 2 — к такой же смеси,
Применение ДТЛ и ДСК.
283
Калибровочный график Кривые ДТА трех
помейобатльных партий
Фиг. 7.16. Метод определения содержания воды в сухих порошках с по-
мощью откачки и ДТА (комнатная температура) [97].
по осажденной методом гомогенного осаждения. Эндотермические
пики дегидратации физической смеси находились в соответствую-
щих температурных точках, а широкий экзотермический пик раз-
ложения (300—450° С) был сдвинут относительно пиков кривых
оксалата лантана и оксалата церия. Однако эндотермический пик
при 875° С еще присутствовал. Осажденная смесь лантана—церия
имела только один эндотермический пик дегидратации, а широкий
экзотермический пик разложения начинался при несколько более
высокой температуре. В отличие от кривой 1 кривая 2 не имела
пиков при температуре выше 500° С. Кривая 3 соответствует осаж-
денной смеси оксалатов празеодима и неодима состава 1:1. Она
имеет двойные пики дегидратации, сдвинутые в область несколько
более высоких температур. Экзотермический пик при 460° С, ха-
рактерный для оксалата празеодима, сдвинут в случае смеси к
510° С. В противоположность этому эндотермический пик оксалата
неодима при 768° С сдвинут в область более низких температур —
к 670е С, а его экзотермический пик при 675° С полностью исчез.
284
Глава 7
Из рассмотрения кривых для осажденной смеси видно, что они нс
являются простой суммой кривых отдельных оксалатов.
Эрди и Паулик [1001 изучали термическое разложение оксала-
тов бария, стронция, марганца (II), кальция, магния и цинка одно-
временно методами ДТА и ТГ в атмосферах воздуха и азота. Они
Фиг. 7.17, Кривые ДТА перхлората аммония, содержащего различные ко-
личества хлората калия [98].
обнаружили, что двуокись углерода, выделяющаяся в ходе реак-
ции, играет важную роль как ингибитор реакции и сдвигает пики
температуры в область более высоких значений.
Месмер и Ирани [73] определили изменения энтальпии при на-
гревании дигидрата гидрофосфата кальция СаНРО4-2Н2О до тем-
пературы 1300° С. Для расчета изменений энтальпии, происходя-
щих во время реакции, использовался внутренний стандарт —
карбонат кальция СаСО3. Реакция протекала по следующей схеме:
СаНРО4 - 2Н2О СаНРО4 + 2Н2О (г.). (1)
Применение ДТА и ДСК
285
СаНРО4 -> -L Са2Р2О7 (т) + А На0 (г.) (2)
А Са2Р2О7 (Т) -> А Са2Р2О7 (₽), (3)
± Са2Р2О7 (₽) -> А Са2Р2О7 (у). (4)
X £
Результаты определения изменений энтальпии приведены в
|.чбл. 7.7.
Значения теплоты перехода ряда неорганических веществ опре-
делялись с помощью нескольких новых методов количественного
ДТА [74, 75]. Результаты, полученные при использовании одного
из этих методов, приведены в табл. 7.8 [75]. Интересно отметить,
ч го для температуры плавления нитрата калия KNO3 было получе-
но значение 105,9 Дж/г, которое хорошо согласуется со значением
116 Дж/г, опубликованным ранее [761. Значение, найденное с по-
мощью прибора ДСК фиры «Дюпон», составляло 95 Дж/г; точно
1акое же значение было получено в работе [77].
Фиг. 7.18. Кривые ДТА смесей гидратов оксалатов редкоземельных ме-
таллов [99].
I — физическая смесь оксалатов лантана и церия (1:1); 2— осажденная смесь оксалатов лан-
тана и церия (1:1); 3— осажденная смесь оксалатов празеодима и неодима (1-1)
286
Глава 7
Изменения энтальпии СаНР04 • 2Н2О [73]
Таблица 7 7
Реакция Скорость нагрева- ния, град/мин Г. °C
ккал/моль кДж/моль
(1) 4 135 21,3; 20,9 89; 87,5
(2) 4 430 6,9; 7,5 28,9; 31,4
(3) 10 850 —0,17; —0,23 —0,71; —0,96
(4) 10 1250 0,85; 0,68 3,56; 2,85
Бэрролл и Роджерс [78] провели исследование с целью опреде-
ления возможности повышения точности метода ДТА путем приме
нения эталонного вещества в качестве термохимического стандарта.
В рассматриваемом ими случае таким эталонным веществом был
иоднд серебра. Кривая ДТА, полученная этим методом, приведена
на фиг. 7.19. Значение АД перехода составило 904±80 Дж, что
Значения теплоты перехода
Таблица 7.Я
Образец Данные Озава и др. [75] Другие источники
темпера- тура, °C АН, температура, °C АН,
кал/г Дж/г кал/г Дж/г
Бензойная кислота 122 38,0 159 — 35,2 34,8 147 145,5
KNOS 129 13,2 55,3 128 127,5 127,9 127 13,8 11,78 12,83 12,05 13,0 57,7 49,3 53,7 50,5 54,4
336 25,3 105,9 338 334,3 331 27,7 22,75 27,20 28,1 116 95,2 114 117,5
AgNOs 161 3,42 14,3 160 160 158,9—160,6 164 3,9 3,44 3,49 3,03 16,3 14,4 14,6 12,7
206 17,7 74 211 210 207 16,2 17,57 17,0 57,8 73,5 71,2
Al 658 102,5 429 659 660 95,2 96,3 399 403
Применение ДТА и ДСК
287
сравнимо с приводимым в литературе значением 858±125 Дж. При
Использовании в качестве эталона инертного вещества было полу-
чено значение ДД = 840±380 Дж. Таким образом, этот метод обес-
печил некоторое повышение калориметрической точности.
Температура образца,^
Фиг. 7.19, Сопоставление кривых ДТА иодида серебра с кубической и гек-
сагональной решетками [78].
/ — образец, массой 0,08 г, содержащий 15,1% иодида серебра с низкой температурой перехо-
да и кубической решеткой (смесь с карборундом); 2 — эталон массой 0,0603 г, содержащий
20% иодида серебра с гексагональной решеткой (также смесь с карборундом).
Уэндландт [79] обнаружил, что при использовании метода за-
паянной трубки можно определить значения теплоты дегидратации
гидратов солей металлов, что было бы невозможно при использо-
вании обычных открытых трубок или тиглей. Этот метод иллюстри-
руется кривыми ДТА пентагидрата сульфата меди CuSO4-5H2O
(фиг. 7.20). На кривой, полученной при использовании открытой
трубки, пики вызваны следующими реакциями:
CuSO4.5Н2О -> CuSO4 • ЗН2О + 2Н2О (ж.), (а)
2Н2О (ж.) -> 2Н2О (г.), (б)
CuSO4 - ЗН2О -> CuSO4 • Н2О + 2Н2О (г.). (в)
На кривой, соответствующей герметичной трубке, первый пик обус-
ловлен реакцией (а), а причина возникновения второго пика неиз-
вестна, но, возможно, он связан с реакцией
CuSO4 • ЗН2О -> CuSO4 • Н2О + 2Н2О (ж). (г)
288
Глава 7
Таким образом, использование держателя образца в виде герме-
тичной трубки позволило определить величину А Я реакции (а),
равную 54±2,5 кДж/моль.
Температура, . °C
Фиг. 7.20. Кривые ДТА CuSC^-SHaO для открытой (/) и запаянной (2) тру-
бок [79].
Проводились аналогичные исследования дегидратации ком-
плексов [Cr(NH3)5-H2O]X3 [801 и [Co(NH3)5-H2O]X3 [81].
На фиг. 7.21 показана кривая ДТА серы, полученная Чу [82].
Энантиотропному переходу ромбической структуры в моноклинную
соответствует пик при 113° С, а плавлению — пик при 124° С.
Дальнейшие переходы в жидкой сере наблюдались при 179 и 446° С
(кипение).
На фиг. 7.22 изображены две кривые ДТА, иллюстрирующие
определение органических примесей в нитрате аммония NH4NO3
[83]. Наличие органических примесей проявляется в понижении
температуры начала экзотермического процесса.
Мэкак и Малеха [84] определяли методом ДТА содержание ме-
таллического никеля в катализаторах. Никель, полученный при
восстановлении окиси никеля, вновь окисляли кислородом и опре-
деляли величину АТ'’ реакции окисления. Максимальное значение
разности АТ температур реакции и инертной двуокиси кремния
SiO2 пропорционально содержанию никеля в образце катализато-
ра. Точность этого метода составляла ~±4%.
Реакция с водородом под давлением непосредственно в камере
калориметра использовалась для определения методом ДСК содер-
жания платины или палладия в различных катализаторах [85].
В капсулу помещали образец катализатора массой ~5 мг и напол-
Фиг. 7.21, Кривая ДТА серы с различными фазовыми переходами [82J.
Фиг. 7.22. Кривые ДТА чистого N^NOg, а также содержащего органиче-
ские примеси [83].
Месса образца £мг; скорость нагревания 15 град/мин; / — чистый NH4N0a; 2— NH4NO8 с
примесями
290
Глава 7
няли камеру гелием под давлением 1,05 МПа. Затем температуру
повышали до 75° С и гелий заменяли водородом под давлением
1,4 МПа. Содержание платины или палладия сопределяли по пло
щади пика кривой ДТА, построенной в функции времени. На фиг.
7.23 показана такая кривая ДТА для реакции с катализатором,
содержащим 5% палладия на активированном угле.
(отсчитываемое от момента введения Н2)
Фиг. 7.23. Определение содержания палладия в угольном катализаторе [85].
Содержание палладия в угольном катализаторе 5% при избыточном давлении водорода
1,4 МПа и температуре 75°С. Масса образца 5,43 мг; ДЯ= 2-263 __ jg gg кал/г =
5,43-4
— 82,3 Дж/г.
Кривые ДТА ряда неорганических веществ, предложенных в ка-
честве эталонов, были записаны Гарном [86]. На всех этих кривых
отчетливо выражены переходы Твердая фаза 1 -> Твердая фаза 2.
В табл. 7.9 обобщены характеристики нагревания (или охлаждения)
этих веществ.
Содержание силиката кальция в портландцементе можно опре-
делить по большому обратимому переходу, наблюдаемому при
915° С [871. Можно также оценить степень гидратации этого соеди-
нения. В другой работе Рамачандран [88] описал порядок опреде-
ления содержания хлорида в бетоне методом ДТА.
Бич и др. [89—92] исследовали с помощью метода ДСК термо-
химическое поведение большого числа комплексов переходных
Применение ДТА и ДСК
291
Таблица 7.9
Средние температуры исходной точки, точки пересечения
и пика для веществ, предложенных в качестве эталонов [86]
Нагревание, °C Охлаждение, °C
Материал СО к _ £ 7. s S S = Е О от- ния от эй ли- в) Ф к к ' ° S о?'5 п д’® ф о, ф Е к
О СО"'— Ки « Ё Л ? С tr 2 СО Я СО д 5С Ф tf со 5 о S я S3 tf
- о u L Я Ь св В к й\с> я О <0 Ь Ф к со 3 СО S И КХО я gs Е
Карбонат бария . . . 2,8 796,0 802,0 810,0 753,0 753,0 750,0
8,0 797,0 804,0 808,0 770,0 769,0 775,0
Хромат калия .... 2,8 664,5 666,3 669,3 669,4 665,4 664,3
8,0 661,8 664,2 667,7 670,5 665,7 665,1
Нитрат калия .... 2,8 128,0 128,9 131,3 123,1 122,8 122,2
8,0 128,8 129,3 132,6 123,8 123,4 123,1
Перхлорат калия . . 2,8 298,2 299,3 301,0 293,7 293,5 293,8
8,0 297,4 298,3 300,2 292,7 292,7 292,4
Сульфат калия . . . 2,8 577,9 580,0 582,3 580,4 577,9 578,3
8,0 577,9 583,9 587,4 585,9 585,1 584,3
Двуокись кремния 2,8 566,2 568,1 569,9 572,0 569,9 569,0
8,0 561,4 567,7 570,9 577,8 570,9 569,2
Сульфат серебра . . 2,8 421,6 424,4 428,4 405,8 495,6 413,6
8,0 * 422,2 427,8 433,7 409,3 408,6 414,3
металлов типа МЬ„Х2. Полученные ими результаты наряду с дру-
гими работами обсуждаются в книге Мортимера и Ашкрофта [93].
Реакции разложения комплексов типа ML<X2 протекают по следую-
щей схеме:
ML4X2 (комп.) -> ML2X2 (комп.) + 2L (г.),
ML2X2 (комп.) -> MLX2 (ж.) + L (г.),
MLX2 (ж.) -> ML2/3 Х2 (ж.) + 4 L <г-)’
О
МЬ2/з Х2 (ж.) —МХ2 (комп.) + — L (г.).
3
Па фиг. 7.24 показана кривая ДСК для реакции [89]
ML2X2 (комп.) МЬХ2 (ж.) + L (г.).
Площадь а+b является мерой этой реакции при средней температу-
ре Тт. При отсутствии образца не требуется дополнительного тепла
для повышения температуры пустой капсулы по сравнению с кап-
сулой эталона (см. горизонтальную базовую линию, обозначенную
«пустая капсула»). Когда в капсулу помещен образец с массой
292
Глава 7
треаг и удельной теплоемкостью Ср>реаг, базовая линия смещает-
ся до начала перехода на величину, равную трейтСр,реаг, т. с,
произведению массы на удельную теплоемкость продукта реакции
MLX2. Лиганд L с массой mL и удельной теплоемкостью Ср,£ ш
парился во время реакции. Средняя температура Тт равна
Фиг. 7.24. Разложение комплекса MLsXa по данным ДСК [89].
—Т{). Вся теплота, поглощенная во время реакции, пред-
ставлена площадью а+6+c+d, и поэтому теплота при тем-
пературе Tt реакции определяется формулой
~ (а^~ b + с + d) — (Tf — Tt) (гппроЛСр, Прод +
+ 4" mL^”p.l )• (7-1)
Г
Перед произведением mLCp± стоит коэффициент 1/2, так как ли-
ганд выделяется из системы в интервале температур Tf—Tt приб-
лизительно с постоянной скоростью. Теплота ЬНТт при некоторой
промежуточной температуре Тт выражается формулой
Тт (T'm 7\) (/Ппрод • Ср, прод "j~ ^\Ср, L
Щреаг CPt pear )> (7-2)
поскольку
(T^m T’j) (/Ппрод Ср, прод) = — d, (7-3)
(Тт - Тг) (т£р, L) = ± <Tf - \-т£Р. ь], (7.4)
Применение ДТА и ДСК
293
(Тт - Tf) (треагСр, реаг) = ± ф + с + d), (7.5)
л Iikc. Количество тепла 1\Нтт определяется в виде
= а + Ь. (7.6)
1 т
В табл. 7.10 приведены термохимические данные для комп-
исксиых соединений Со(Пиридин)2Х2.
Таблица 7.10
Термохимические данные для комплексов типа Со(Пиридин)2Х2 [89]
Параметр
...
Температура плавления, К............
»Л/Л, ккал/моль.......................
Л //, кДж/моль......................
скорость нагревания, град/мин . . .
I Т/. К................................
К.....................
к......................
С1 Вг I
440 450
28,5+0,51) 27,3+0,9 12,3±0,32)
119,3 + 2,09 114,3+3,8 51,5+1,25
8 16 16
420 Перекры- 590
вает темпе-
ратуру пла-
вления
510 520 610
600 540 630
форма->голубая форма ДЯ==
’) Относится к голубой форме. Для перехода фиолетовая
«'3,02 ±0,07 ккал/моль.
) Теплота плавления Со(Пиридин)2/а составляет 6,3 + 0,3 ккал/моль.
Дифференциальная сканирующая калориметрия применялась
для измерения теплоты переходов некоторых соединений натрия,
калия и серебра [94]. Экспериментальные данные для роданида
калия KSCN и нитрита калия KNO2 находились в неудовлетвори-
тельном согласии с опубликованными данными.
Симчен [95] отметил, что обычно приводимая в литературе
температура разложения бикарбоната натрия NaHCO3 (270° С)
ошибочна. По данным ДСК она составляет ~100° С.
Блок [96] сообщил о методе анализа смесей хлоридов с бромида-
ми при использовании ДСК. Поскольку теплота плавления иде-
ллыюго твердого раствора типа АтХ„—ВтХ„ или АтХ„—AmY„
прямо пропорциональна концентрации растворенных ионов, метод
ДСК удается использовать для определения состава хлоридно-
бромидных смесей во всей области концентраций от 0 до 100%. При
обработке растворов, содержащих хлорид и бромид, последние
осаждаются нитратом серебра, образуя твердые растворы хлорид-
бромида серебра. Затем определяется теплота плавления образую-
294
Глава 7
щихся смешанных кристаллов, а по предварительно построен поп
калибровочной кривой процентное содержание хлорида или бро
мида.
Д. ГЛИНЫ И МИНЕРАЛЫ
Исследование глин и минералов — одна из первых областей
применения ДТА. Широкому изучению этих веществ обязаны своим
возникновением теория и техника измерения этого метода. ДТА
7.25. Применение метода ДТА для исследования глин и минералов [62].
Фиг.
использовался для идентификации глин, полученных из различных
месторождений, и широко применялся для определения содержа-
ния свободного кварца в минералах. Известно много других облас-
тей применения ДТА. Метод ДСК редко использовался для анализа
глин и минералов из-за того, что его возможности ограничены от-
носительно невысокими температурами. Большинство представляю-
щих интерес термических явлений в глинах и минералах наблю-
дается при температурах выше 500° С, а зачастую и выше 1000° С.
На фиг. 7.25 приведены области применения ДТА для исследо-
вания этих веществ [62].
Используя метод ДТА для качественного анализа, Гарн и Флэ-
шей [113,1 исследовали термическое разложение различных образ-
цов карбоната магния и талька. На фиг. 7.26 приведены кривые
Применение ДТА и ДСК
295
Температура, °C
Фиг. 7.26. Кривые ДТА промышлен-
ных образцов карбоната магния раз-
личных партий [113J.
д —партия № 6, Бэйкер; б—партия № 3,
Мерк; в — партия Кз 2, Мерк.
ДТА промышленных об-
|hi щов карбоната магния,
и на фиг. 7.27 — кривые
для талька.
Кривые образцов кар-
боната магния значитель-
но отличаются друг от
друга вследствие неодина-
ковой термической преды-
стории. Все кривые, по-
лученные для талька, име-
ют сильный экзотермичес-
кий пик, начинающийся
при температуре ~850° С.
Интенсивность процессов
по всех случаях была
примерно одинаковой, но
из-за различных примесей
кривые ДТА заметно от-
личались друг от друга.
На кривых для талька
двух видов (шт. Монтана и
Сьеррамик) наблюдается
небольшой эндотермичес-
кий пик при температуре
~570° С, а третий вид
талька (йеллоустонский)
имеет значительный эндо-
термический пик при тем-
пературе ~700° С.
Лоддин г и Хэммел
11141 исследовали методом
ДТА гётит ci-FeO-OH
и гибсит А1(ОН)3 как в
чистом виде, так и в сме-
си с другими веществами.
Если гётит нагревать в установке ДТА с регулируемой ат-
мосферой в восстановительной атмосфере водорода, он дегид-
ратируется при температуре ниже 300° С, и имеющиеся в
нем ионы трехвалентного железа немедленно восстанавливаются до
аморфной Fe3O4, которая в свою очередь рекристаллизуется в маг-
петит в интервале температур 300—360° С. Если атмосфера водоро-
да по достижении 400° С заменяется азотом, а затем воздухом, то
вследствие окисления магнетита до маггемита y-Fe2O3 возникает
экзотермический пик. Второй экзотермический пик, вызванный
превращением y-Fe2O3 в a-Fe2O3, наблюдается при 775—836°С .
296
Глава 7
Этот пик обычно двойной, и общая площадь под ним пропорции,
нальна количеству образовавшегося гематита, которое в свою оче-
редь равно количеству окиси железа, содержащемуся в исходном
образце. Количество гибсита можно определить, вычитая эту пли*
щадь из площади под пиком дегидратации. Присутствие в обрил
це гематита или магнетита почти не влияет на площадь пикон
превращения.
Фиг. 7.27. Кривые ДТА для различных видов талька [113].
а — Сьеррамик; б— йеллоустонский; в— Монтана. Кривые получены при использовании пла-
тиновых чашек в качестве держателей образцов.
На фиг. 7.28 приведены калибровочные кривые для определения
количества гётита и гибсита в смеси в зависимости от площади пика.
Аналогичная кривая для перехода у—Mi-Fe2O3 была также пред-
ставлена в работе Лоддинга и Хэммела [114].
Берг и Рассонская [115] предложили использовать установку
ДТА с высокими скоростями нагревания для экспресс-анализа ми-
нералов и глин. Высокая скорость нагревания (80—100 град/мин)
достигалась путем помещения держателя образца в печь, предвари-
тельно нагретую до температуры, на 300° С превышающей требуе-
Применение ДТА и ДСК.
297
мую конечную температуру образца. Авторы утверждают, что при
пн 'оких скоростях нагревания температуры пиков, соответствую-
щие плавлению и кипению, были такими же, как и при скорости
и.прыгания 3—6 град/мин. В это трудно поверить, если принять
но ппимание предшествующие исследования. Аналогичные резуль-
1.1 гы были получены для пиков, обусловленных диссоциацией кар-
пои.ггов металлов, и для фазовых переходов в твердом кристалли-
•вч'ком состоянии. Авторы указывают на следующие преимущества
Фиг. 7.28. Калибровочные кривые площадей пика дегидратации гибсита и
гётита [1141.
1 — гётит; 2 — гибсит.
данного метода: а) оперативность (3—10 мин); б) небольшие образ-
цы (20—100 мг); в) простота регулирования скорости нагревания;
г) экономичность.
Воллин и др. [116] изучали методом ДТА реакции между двумя
и более твердыми веществами, используя в качестве держателей
образца запаянные трубки из стекла или плавленого кварца. При-
менявшаяся ими процедура, в дальнейшем усовершенствованная
Воллином и Керром [117], была названа пиросинтезом. На фиг. 7.29
приведены кривые пиросинтеза системы CuS—C112S.
Одиннадцать приведенных кривых соответствуют различному
содержанию меди и серы. Эта серия кривых построена, начиная
от кривой для CuS, за экзотермическим пиком которой следует
эндотермический пик при 115° С, вызванный плавлением серы.
Эндотермический пик при 505° С соответствует температуре плавле-
ния ковеллина CuS с образованием дигенита, воды и пара. Величи-
на этого пика постепенно уменьшается до нуля на кривой ДТА
для дигенита Cu)i8S.
298
Глава 7
Фмг. 7.29. Кривые пиросинтеза системы CuS—C112S [117].
Исследовались также системы типа FeS—FeS2, CuFeS2, FeS1>5 и
другие сульфиды, селениды, теллуриды, арсениды и антимониды.
Определялись конструкции держателей образца, трубок для об-
разца, скорости нагревания и размеры частиц образца. Ожидается,
что пиросинтез может оказаться полезным также и в неорганиче-
ской химии.
Применение ДТА и ДСК.
299
Фиг. 7.30. Кривые ДТА необожженного гипса (а) и обожженного (штука-
турного) гипса (б) [106].
-Метод ДТА можно использовать для определения количества
необожженного гипса CaSO4-2H2O в обожженном (штукатурном)
гипсе CaSO4'0,5H2O. По кривой дегидратации гипса на фиг. 7.30,а
видно, что пик при ДТИИН = 142иС вызван дегидратацией первых
1,5 моль воды на 1 моль соли. Второй эндотермический пик (при
АТМВИ = 198°С) вызван выделением остаточной воды. Таким об-
300
Глава 7
разом, о наличии необожженного гипса в обожженном гипс»
можно судить по кривой ДТА (фиг. 7.30,6), если появляется инк
при температуре ~142'С. Площадь пика будет пропорциональна
количеству необожженного гипса в образце [106].
Кристаллический кварц при нагревании претерпевает фазовый
переход Твердая фаза 1 -> Твердая фаза 2 (а -> ₽-кварц) при тем
пературе ~573°С. Кейт и Татл [107] при исследовании 250 обр и
цов кварца обнаружили, что интервал температур перехода для
природного кварца составляет 38°С, хотя для большинства образ-
цов он не превышал 2,5°С относительно 573°С. Широкий интервал
температур перехода природного кварца приписывался присутст-
вию переменного количества посторонних ионов в кварце в твер-
дом растворе. Так как количество твердого раствора зависит от
температуры во время образования кварца, температура перехода
может использоваться в качестве критерия температуры образовп
ния (кристаллизации) образцов при аналогичном химическом
составе окружающей среды.
Фиг. 7.31. а- P-переход в кристаллах кварца [108].
Месторождения образцов: а — Бразилия; б — Корног, Панама; в — Честер, Панама; г — Хот
Спрингс, шт. Арканзас.
Применение ДТА и ДСК
301
На фиг. 7.31 представлены кривые ДСК для образцов кварца,
полученных из различных месторождений [108]. Масса образцов
составляла 32—37 мг, а скорость нагревания была 1,5 град/мин,
что позволяло свести к минимуму тепловые градиенты в образце.
Дэвис и Холбридж [109] описали количественный термический
.анализ глинистых минералов, каолинита, гибсита и гётита. Коэн
и Кренкел [ПО] определили теплоту дегидроксилирования (дегид-
ратации) двух типов алунита. Реддик [111] определил значение ДЯ
для разложения кальцита, магнезита, родохрозита, сидерита и
Анкерита. Варн и Маккензи [112] установили связь между темпе-
ратурой пика для магнезита и его концентрацией в различных сме-
сях. Снижение температуры пика, по существу, одинаково при ис-
пользовании в качестве второго компонента окиси алюминия или
карбонатов.
Т----------1----------1----------г
572’С
г
302
Глава 7
Е. БИОЛОГИЧЕСКИЕ ОБЪЕКТЫ И ПРИРОДНЫЕ
ОРГАНИЧЕСКИЕ МАТЕРИАЛЫ
Метод ДТА применительно к биологическим объектам в боль
шинстве случаев использовался для идентификации и определения
характеристик. Этого, по-видимому, следовало ожидать, принимая
во внимание сложность и неоднородность этих объектов; достаточно
сравнить, например, образец торфа с образцом бензойной кислоты
или других простых органических соединений. Кривые ДТА био
логических объектов часто имеют очень широкие пики, а во многих
случаях вообще не имеют узких эндотермических и экзотермичс
ских пиков. Многие из ранних исследований в этой области прово-
дились при отсутствии регулирования газовой атмосферы печи и
скорости нагревания, и поэтому их результаты нельзя сравнивать
с данными, полученными при использовании современной аппара-
туры.
Митчел и Бёрни [118], а также Пфайл [119] опубликовали
обзоры по применению ДТА (и других методов термического ана-
лиза) для исследования биологических объектов. Первая работа
в основном касается исследования с помощью ДТА свежего расти-
тельного материала, бактерий, частично разложившегося расти-
тельного материала, торфа и органических веществ, содержащихся
в почве. Пфайл [119] рассматривает применение ДТА для исследо-
вания тканей человеческого тела, таких, как печень, обожженные
кожные ткани, кости и др.
В табл. 7.11 представлены некоторые возможные применения
ДТА и ДСК для исследования биологических объектов и природ-
ных органических материалов.
Лабовиц [138] сообщил о фазовом переходе обезвоженного холе-
стерина при 37ГС, для которого АД — 0,66 ккал/моль (2,76 кДж/
моль) и AS = 2,1 кал/град (8,8 Дж/град). Этот переход рассматри-
вался другими исследователями.
Берлин и др. [137] определили методом ДСК теплоемкость обез-
воженного яичного альбумина и [3-лактоглобулина. Для гидрати-
рованных образцов, содержащих 0,03—0,21 г сорбированной воды
на I г белка, была обнаружена линейная зависимость между удель-
ной теплоемкостью и содержанием влаги. Берлин и др. [139] опре-
делили методом ДСК теплоту десорбции паров воды из аморфной
и кристаллической лактозы.
Олафсон и Брайан [141] с помощью метода ДСК определили
термическую стабильность 19 аминокислот. Они принимали темпе-
ратуру АТмин за температуру разложения аминокислот. В некото-
рых случаях для характеристики соединения использовалась не-
обычная форма кривых и число пиков.
Пакулак и Леонард [135] исследовали методом ДТА целлюлозу,
нитроцеллюлозу, пентаэритрит, тетранитрат пентаэритрита и другие
Применение ЦТ А и ДСК
303
Таблица 7.11
Применение методов ДТА и ДСК для исследования биологических объектов
и природных органических материалов
Объекты
Метод
Литература
Жиры, масла н воскн
Кости человека, гемоглобин
Тпбак
Полинуклеотиды
Денатурированные белки и др.
Кожа и ее составные элементы
Ьпополимеры
Дрожжи и криобиология крови
Iепатома печени человека
(дожженные кожные ткани человека
Зерно (кукуруза, пшеница, овес и др.)
Снежие растительные материалы
Ьикгерии и актиномициты
Торф и частично разложившиеся ра-
стительные материалы
Органические составные части почвы
Древесина
Целлюлоза
Лишайники
Холестерин
Лактоза
ДТА, ДСК
ДТА
ДТА
ДТА
дск
ДТА
ДТА
ДТА
ДТА,
ДТА,
ДТА,
ДТА
ДТА
ЭТА, ТГ
ТГ, МС
ТГ
ДТА
ДТА
ДТА
ДТА
ДТА
ДТА, ДСК
ДСК
[120—122, 132,
133]
123]
124
125
126,
127]
128,
130,
119
119
119'
118
118
137]
129]
131]
118
118'
134’
135
136]
138
139'
соединения этого типа. При использовании термостабилизирован-
пого прибора ДТА рабочая температура не превышала 200°С;
поэтому кривые целлюлозы и ацетилцеллюлозы не имели никаких
пиков, а нитроцеллюлоза имела экзотермический пик с АТнакс=
180°С. Аналогичные результаты были получены для ряда пен-
таэритритов.
Для нескольких типов бактериальных декстранов Морита [142]
определил связь между их молекулярным строением и пиками
кривой ДТА.
Арсено [134] изучал методом ДТА термическое разложение дре-
весины бальзамической пихты. Используя высушенную на воз-
духе древесину, а также различные ее образцы, подвергнутые экст-
рагированию несколькими реагентами, Арсено связывал каждый
пик кривой ДТА с соответствующим процессом (табл. 7.12).
Методом ДТА исследованы крахмал и другие полисахариды
1143]. На фиг. 7.32 приведены кривые ДТА, полученные для не-
скольких образцов картофельного и кукурузного крахмала. Об-
разцы приготавливали в упаковке типа «сэндвич». Для этого обра-
зец массой 150 мг помещали между двумя пластинами из обожжен-
304
Глава 7
Таблица 7 .ГЛ
Пики кривых ДТА древесины, высушенной на воздухе [134]
Пик, °C Соединение
(1) эндотермический, 145 (2) эндотермический, 163 (3) экзотермический, 210 (4) экзотермический, 265 (5) экзотермический, 285 (6) экзотермический, 300 (7) экзотермический, 330 (8) экзотермический, 360 Экстракция водно-спиртовой смесью » » » Возможно, кислотный лигнин Экстракция смесью бензола со спиртом Сумма 4 и 5 Целлюлоза То же, что и 6
ной окиси алюминия массой по 200 мг и обжимали под давлением
14 МПа.
Кривые ДТА крахмалов характеризовались эндотермическими
пиками в интервале 135—310° С, за которыми следовали два от-
четливых экзотермических пика в интервале 375—520° С. Кривые
хорошо иллюстрируют влияние предварительной обработки крах-
мала.
Фиг. 7.32. Кривые ДТА картофельного и кукурузного крахмала [143].
а— картофельный крахмал; б — картофельный крахмал (повторный цикл измерений); в — ку-
курузный крахмал: г — кукурузный крахмал, экстрагированный метанолом; д — кукурузный
крахмал, предварительно желатинизированный аммиаком.
Применение ДТА и ДСК
305
Поскольку крахмал является полимером, в состав которого
входят а-1,4- и а-1,6-глюкопир анозидные звенья, представляло
интерес исследовать термические свойства амилозы — линейной
полимерной фракции крахмала. Были получены кривые ДТА для
различных фракций амилозы, приготовленных из одних и тех же
крахмалов. Исследование трех фракций позволило выявить три
«иные особенности: эндотермические пики с ДТМиН «г 150 и 225° С
п пик с уступом при ДТмин = 315° С. Отчетливые экзотермиче-
ские пики наблюдались в интервале температур 490—510° С.
Механизм реакций термического разложения неизвестен и, ве-
роятно, довольно сложен. Кривые ДТА служат не только для опре-
деления характеристик или идентификации этих углеводов; с их
помощью можно будет получить полезную информацию для выяс-
нения связи между молекулярным составом и химическими свойст-
вами.
Морита [144] исследовал также методом ДТА несколько а- и
fi-нолиглюкозанов, а также рисовый крахмал. В этом исследовании
особый интерес представляет изучение влияния влаги на получен-
ные кривые ДТА. Были получены кривые для рисового крахмала,
хранившегося в различных условиях: в вакууме, в атмосфере с от-
носительной влажностью 100% и др. Присутствие влаги изменяло
пдотермический пик при ДТМИИ = 130° С, а пики при 275 или
310“ С оставались прежними. Эти результаты подтверждают вывод,
что первоначальный пик (130° С) обусловлен не только потерей
остаточной влаги и что процесс дегидратации не полностью обра-
тим.
Ж. ПОЛИМЕРЫ
Вероятно, наиболее широко методы ДТА и ДСК в последние
годы применялись для исследования полимеров. Обычно их ис-
пользуют для измерения температур стеклования TQ, температур
плавления Тт, степени кристалличности, теплот плавления и (или)
кристаллизации, температур разложения и многих других свойств.
Выпускается несколько серийных приборов ДТА и ДСК, предназ-
наченных главным образом для исследования полимеров.
На фиг. 7.33 показано, как различные термические процессы
Отражаются на кривой ДТА [145]. В действительности, однако,
псе эти переходы не столь четко выражены на одной и той же кри-
вой. Необходимо слегка изменять условия эксперимента, чтобы
интересующий исследователя переход наиболее четко отразился
па кривой ДТА. Например, при изучении процесса окисления ис-
пользуются образцы уменьшенного размера и опыт выполняется в
атмосфере кислорода или воздуха. Другие измерения проводятся
п атмосфере азота или при пониженном давлении. Оборудование
для ДТА и ДСК должно обеспечивать охлаждение по заданной про-
J 1-230
306
Глава 7
Температура
Фиг. 7.33. Схематическая кривая ДТА типичного полимера [145].
1 — стеклование; 2 — кристаллизация; 3 — плавление: 4 — окисление; 5 — окисления не про-
исходит; 6 — разложение.
грамме, чтобы можно было зарегистрировать температуру кристал-
лизации при охлаждении.
На фиг. 7.34 схематически показаны области применения мето-
дов ДТА и ДСК. для исследования полимеров. Прекрасные обзоры
по этому вопросу содержатся в книгах Ке [146], Слейда и Джен-
кинса [147], Швенкера [148], Портера и Джонсона [149, 150], Швен-
кера и Гарна [152] и др., а также в книге Рейха и Стайвела [1511
и обзорных статьях Мэрфи [5—8,153] и многих других авторов.
На фиг. 7.35 иллюстрируется идентификация смесей полимерон
по кривой ДТА. Чу [154] исследовал физическую смесь семи вы
пускаемых промышленностью полимеров: полиэтилена высокого
давления, полиэтилена низкого давления, полипропилена, поли-
оксиметилена, найлона-6, найлона-66 и политетрафторэтилена.
Каждый компонент имеет свою температуру плавления, которая
отражается в виде эндотермического пика соответственно при 108,
127, 165, 174, 220, 257 и 340е С. Политетрафторэтилен имеет также
низкотемпературный фазовый переход кристаллизации при темпе-
ратуре ~20° С. При идентификации этой смеси полимеров исполь-
зовался образец массой всего 8 мг, что подтверждает уникальные
возможности метода ДТА.
Андерсон [162] исследовал методом ДТА шесть различных эпок
сидных смол, как неотвержденных, так и отвержденных различными
аминами и ангидридами. Образцы массой 1—3 г смешивали с ран
ными количествами окиси алюминия. Смесь помещали в трубку,
Фиг. 7.35. Кривая ДТА смеси семи полимеров [154].
I политетрафторэтилен; 2 — полиэтилен высокого давления; 3 — полиэтилен низкого дав-
ления; 4 — полипропилен; 5 — полиоксиметилен; 6 — найлон-6; 7— найлон-66; S-политетра-
фторэтилен.
Фиг. 7.34. Применение ДТА и ДСК для исследования полимеров.
II*
308
Глава 7
которую взвешивали до и после нагревания, что позволяло опреде
лить потери массы образца.
На фиг. 7.36 приведены кривые ДТА трех отвержденных и не
отвержденных эпоксидных смол марок Epon 1310 (тетраглицидн
ловый эфир), диэпоксид AG-13E (бпс-эпоксидициклопентиловы ii
Фиг. 7.36. Кривые ДТА неотвержденных и отвержденных эпоксидных
смол [162].
МА — малеиновый ангидрид; CL — лета-фенилендиамин; скорость нагревания 2»5 град/мин.
простой эфир, или этиленгликоль) и UC Endo изомер (двуокись
дициклопентадиена). Все неотвержденные эпоксидные смолы, кро-
ме последней, имели экзотермические пики в интервале температур
300—400е С. Предполагается, что эти пики вызваны изомериза-
цией эпоксидной группы в карбонильные группы (альдегиды для
первичных эпоксидов и кетоны для вторичных эпоксидов). Появле-
ние в трубках паров свидетельствовало о том, что одновременно с
изомеризацией и полимеризацией происходит испарение и разло-
жение. Эпоксид Endo изомер имел эндотермический пик вследствие
поглощения тепла при испарении, а разложение маскировало теп-
Применение ДТА и ДСК
309
лопые эффекты, обусловленные замедленно протекающими про-
цессами изомеризации и полимеризации путем эфиризации эпок-
сидных групп. Пик при АТМИН = 184° С соответствовал температу-
ре плавления этого эпоксида.
Фиг. 7.37. Кривые ДТА смол вибрин-135 [163].
Когда три рассматриваемых эпоксида смешивали с отвердителя-
ми (малеиновым ангидридом или лщтп-фенилепдиамином), все они,
кроме AG-13E/CL и UC Endo изомер/CL, имели два экзотермиче-
ских пика и только один эндотермический пик. Последний соответ-
ствовал температурам кипения и (или) разложения как эпоксида,
так и отвердителя.
Кривые ДТА, полученные для рассмотренной выше системы,
можно использовать для определения индивидуальных характерис-
тик. Этот метод имеет редкие преимущества перед другими методами
измерений, особенно в тех случаях, когда исследуются нераствори-
мые и аморфные эпоксидные системы с поперечными связями, даю-
щие диффузное гало на рентгенограммах и вследствие непостоян-
ной физической структуры плохо воспроизводимые инфракрасные
спектры.
310
Глава 7
Мэрфи и др. [163] исследовали методом ДТА смолы вибрин-1311
(фиг. 7.37). Были взяты три образца смолы: два из них содержали
по 2% катализатора (третичнобутилпербензоата), а третий — 0,5%
Каждый образец смеси смола — катализатор нагревался (отверг
дался) в течение определенного периода времени. На кривых ДТА
1 и 2 видны два экзотермических пика в области низких температур
соответственно с АТмакс = 150 и 180е С. Первый пик исчезал после
термической обработки образца (180° С в течение 24 ч), в то врем»
как пик при 320° С сохраняется на всех трех кривых. Наличие эк
зотермического пика в области низких температур объясняется
дальнейшей полимеризацией недоотвержденной смолы, особенно
ее полиэфирного компонента. Экзотермический пик в области вы-
соких температур был вызван отверждением триаллилцианураг-
ного компонента.
Мэрфи и др. [164] в дальнейшем использовали метод ДТА для
оценки влияния различных катализаторов на процесс отверждения
смолы вибрин-135. Перекись бензоила обеспечивала наиболее
полное отверждение смолы.
С помощью Метода ДТА Клампит [165] определял содержание
линейного полиэтилена в смесях с полиэтиленом высокого давле
ния. На фиг. 7.38 приведены кривые ДТА для нескольких смесей.
Тщательное изучение кривой для образца неотожженного поли-
этилена позволило заметить пик при ДТМин = 134° С, а также
перегиб на пике. После отжига образца в течение 30 мин при 120° С
перегиб разрешался в виде двух пиков при А7МиН = 115 и 124° С
соответственно. На фиг. 7.38 представлены кривые ДТА , получен-
ные при той же процедуре отжига, но при разном процентном со
держании линейного полиэтилена в образцах. Кривые имеют эн-
дотермические пики при АТМиН = 115, 124 и 134° С соответственно.
Для чистых компонентов, однако, был получен только один пик
(полиэтилен высокого давления). Пик при = 110° С умень-
шался, а площадь под пиком 134° С возрастала с увеличением
количества линейного полимера. Пик при 115° С был связан с при-
сутствием кристаллов полиэтилена высокого давления, а пик при
134° С, по-видимому, соответствовал кр Металлам линейного поли-
этилена.
Швенкер и Бек [166] изучали методом ДТА в атмосферах возду-
ха и азота термическую деструкцию полимеров, используемых в
текстильной промышленности. Были исследованы образцы из дак-
рона, найлона-66, неопрена W и орлона. Согласно полученным ре-
зультатам по кривым ДТА можно определить и идентифицировать
такие реакции, протекающие в полимерах при термической дест-
рукции, как перегруппировка, образование поперечных связей
(«сшивка») и деполимеризация. Метод ДТА позволяет обнаруживать
незначительные изменения в составе полимера или присутствие
Фиг. 7.38. Кривые ДТА смесей линейного полиэтилена и полиэтилена вы-
сокого давления [165].
Содержание линейного полиэтилена: а —0%; б — 5%; в—10%; г — 25%; д—40%; е—100%.
|1лняиие отжига на смеси, содержащие 25% линейного полиэтилена: ж — неотожженный поли-
этилен; з — отжиг в течение 30 мин при 120° С.
312
Глава 7
Фиг. 7.39. Кривые ДТА полимерных материалов [166].
— в среде воздуха; --в среде азота; а—найлон-66; б —неопрен W.
заместителей в основной цепи, а также может оказаться полезным
для исследования механизма термической деструкции.
На фиг. 7.39 приведены кривые ДТА промышленных образцов
найлона-66 и неопрена W, полученные в атмосфере воздуха и азота.
На кривой найлона-66 при температуре ~100° С наблюдался
слабый эндотермический пик, вызванный потерей сорбированной
воды. В атмосфере воздуха происходила экзотермическая реакция,
начинающаяся при температуре ~185° С и отражающаяся на кри-
вой в виде небольшого эндотермического пика при АТМнН « 250° С.
Этот пик был вызван плавлением полимера (температура плавления
Применение ДТА и ДСК
313
Фиг. 7.40. Кривые ДТА пяти полиолефинов [167].
,i — г — полиэтилены {а — марлекс-50; б — экспериментальный; в — супердайлан; г — DYNH);
д — полипропилен.
~255°С). В атмосфере азота экзотермические пики не наблюда-
лись, что подтверждает окислительный характер реакций, проте-
кающих в атмосфере воздуха. Два эндотермических пика на кри-
вой, полученной в атмосфере азота, были обусловлены плавле-
нием полимера и реакцией деполимеризации. Очевидно, механиз-
мы термической деструкции в атмосфере воздуха и азота раз-
личны.
314
Глава Т
На обеих кривых ДТА неопрена W наблюдается экзотермши
ский пик при ЛТМИЕ « 377° С. Этот пик связан с удалением 11* I
и процессами «сшивки» в оставшемся продукте.
Температуры плавления и степень кристалличности ряда поли
олефинов были исследованы методом ДТА Ке [167]. На фиг. /. ш
представлены кривые ДТА пяти полиолефинов.
Температуры, соответствующие максимумам пиков этих кривых,
использовались для определения температуры плавления полнм<
ров. Результаты, полученные методом ДТА, согласуются с опубл и
кованными данными в пределах ±ГС, хотя некоторые полимеры
имели интервал плавления ~15° С, который оценивался по расслои
нию между началом отклонения от базовой линии и максимумом
пика. Изотактический полипропилен имел несколько более широкий
эндотермический пик при ДТМин = 169° С. Конец перехода
находился где-то за пределами максимума.
Ke [167] получил также кривую ДТА для смесей полиолефипоп
и обнаружил, что компоненты могут быть идентифицированы в том
случае, когда их температуры плавления отличаются на достагоч
ную величину. Площади пиков были пропорциональны концентр.i
ции каждого компонента в смеси.
Степень кристалличности полиэтилена рассчитывалась путем
сравнения площади соответствующего эндотермического пика <
площадью двойного пика дотриаконтана. Кривая имеет два пика,
первый из которых обусловлен вращательно-цепным переходом и
находится на несколько градусов Цельсия ниже температуры план
ления. Полученные таким образом значения степени кристаллич
кости хорошо согласуются с опубликованными данными (табл. 7.13).
Таблица 7.1.1
Степень кристалличности полиэтилена разных марок [167]
Полиэтилен Степень кристалличности, %
экспериментальные данные опубликованные данные
Марлекс-50 91 93
Супердайлан 81 65—85
Экспериментальный 86 87
DYNH 52 40—60
Кроме того, Ке [168] исследовал влияние разбавителей на тем-
пературы плавления различных видов полиэтилена. В работе [169]
сравниваются процессы плавления полиэтилена, полученного крис-
таллизацией из раствора и из расплава. Скотт [170] изучал методом
ДТА плавление и переходы второго порядка полиэтилентерефтала-
та. Рудин и др. [171] измеряли методом ДТА устойчивость различ-
Применение ДТА и ДСК
315
НМ* полимеров и резин к окислению. Сравнение кривых плавления
Н ихлпждсния до окисления и после него может служить показате-
лем степени «повреждения» или окисления полимера.
Метод ДТА применялся Ке и Сиско [172] для определения ин-
|₽|»Ш1лон плавления и стеклования промышленных найлонов, а
liih/hc гомо- и сополиамидов, полученных путем межфазной по-
ликонденсации. На фиг. 7.41 приведены кривые ДТА нескольких
иолнадипамидов и полисебацамидов.
11олиадипамиды были получены из диаминов, содержащих как
41’1 нос, так и нечетное число атомов углерода, а полисебацамиды —
ид диаминов, содержащих четное число атомов углерода. Все кри-
Вмг имели пики, обусловленные плавлением полимера, причем тем-
пература плавления понижалась с увеличением числа атомов угле-
рода в цепи диамина.
Мэрфи и Хилл [173] сообщили о применении метода ДТА для
нбпнружения изменений структуры дифенила, поливинилхлорида,
l4M|viona и пластика «версалуб» F-50. На фиг. 7.42 приведены кри-
вые для дифенила (необлученного и после воздействия облучения).
I к’облученный образец дифенила имеет кривую с двумя эндо-
термическими пиками, которые вызваны плавлением (70° С) и испа-
рен нем (175° С) этого вещества. Облученный образец, кроме этих
днух пиков, имеет экзотермический пик при температуре ~370° С.
Пик плавления наблюдался при несколько более низкой темпера-
туре. Предполагалось, что экзотермический пик при 370° С был
.нм шан окислением в среде воздуха нелетучего, образовавшегося
под действием радиации полимера дифенила, который остался в
держателе образца после испарения низкомолекулярных фрак-
ций. Понижение температуры плавления также вызывалось облу-
чением образца. Аналогичные результаты были получены и для об-
|)л щов поливинилхлорида. Отмечается, что при надлежащем вы-
боре материалов по отношению площади пика на кривой ДТА к
дозе радиации метод ДТА можно использовать в дозиметрии для
широкого интервала уровней энергии.
Метод ДСК нашел широкое применение для определения весо-
вого содержания кристаллической фазы в аморфно-кристалличе-
СНИХ полимерах. Метод основан на измерении теплоты плавления
образца полимера и правдоподобном предположении, что эта
величина пропорциональна содержанию кристаллической фазы,
рели путем экстраполяции можно определить теплоту плавления
Д/Д* гипотетического полностью кристаллического вещества, то
весовая доля кристаллической фазы будет равна [155].
Обзоры методов определения степени кристалличности полимеров
были сделаны Греем [156] и Доулем [157, 158].
Таким образом, для определения степени кристалличности об-
разца полимера можно измерить полную энергию, поглощенную
I г образца, вычесть из нее энергию, которая была бы поглощена
316
Глава 7
Применение ДТА и ДСК
317
Фиг. 7.41. Кривые ДТА некоторых полиадипамидов (а) и прлисебацамидов
(6) [172].
1 г полностью аморфного образца в том же интервале температур,
а затем разделить результат на теплоту плавления 1 г полностью
кристаллического образца. На фиг. 7.43 приведена кривая ДСК,
полученная для аморфно-кристаллического полимера. При no-
Температура, °C
Фиг. 7.42. Кривые ДТА ди-
фенила [173].
I В необлучеиный; 2 — облученный;
д— раз-1 ость AZ для облучённого и
необлученного образцов.
il роопии кривой использовалось уравнение
х = ДЯ2.1 — АЯа(2Л) (7 7)
Д// ро
I, х — весовая доля кристаллической фазы при любой темпера-
г к-; А Яр»— теплота плавления полностью кристаллического
uTip.i’uia; ДД2,1 и ДДс(2.1ц — соответственно теплота плавления ис-
следуемого и полностью аморфного
образцов. На кривой ДД2,1 равна
площади ACDEF, &На(2, i> — площади
ABEF, а их разность—площади
BCDG. Следует заметить, что истин-
ная базовая линия под пиком явля-
ется экстраполяцией регистрируе-
мой базовой линии от точки завер-
шения интервала плавления. Не
следует проводить ее по касательной
к участкам кривой до и после плав-
ления, как это обычно делается.
Грей [155, 1561 рассмотрел другие
методы определения площадей под
кривыми, экстраполяции и т. д.
Была составлена также программа
расчетов степени кристалличности
на ЭВМ.
Еще один метод определения
степени кристалличности полиме-
ров был рассмотрен Дусвальтом
[1591. Он основан на быстром и
хорошо воспроизводимом охлаждении расплавленного образца в
калориметре до заданной температуры и последующем проведении
и этих условиях изотермической кристаллизации образца. На
фиг. 7.44 представлен ряд кривых кристаллизации полиэтилена,
т
Фиг. 7.43. Типичный пик плавления на кривой ДСК и базовая линия при
бора [156].
318
Глава 7
полученных в изотермических условиях при различных заданных
температурах кристаллизации. Наблюдаемые различия в способ)
ности полимера к кристаллизации могут быть вызваны разветвле
ниями молекул, условиями образования центров кристаллизации
или влиянием молекулярного веса. Чувствительность и оператин
ность этого метода позволяют исследовать воспроизводимость ре
зультатов на ряде образцов для множества полимеров.
Фиг. 7.44. Кривые изотермической кристаллизации полиэтилена [159].
Фольц и Маккинни [160] провели количественные исследования
влияния отжига поливинилхлорида при температуре, близкой к
температуре стеклования. Метод основан на использовании зака-
ленного образца полимера в качестве эталона; полученная таким
образом кривая ДСК дает разность между тепловой энергией эта-
лонного и отожженного образцов. С помощью этого метода удается
измерять небольшие различия в энергии, которые обычно прояв-
ляются как небольшие перегибы на кривой Tg или как неровности
кривой ДСК на участке, предшествующем началу стеклования.
Энергии активации для полимеризации стирола были определе-
ны методом ДТА Хойером и др. [161]. КриваяДТА для термической
полимеризации чистого стирола содержит только один экзотер-
мический пик, соответствующий началу полимеризации, при 140°С
и АТмакс = 250°С. Величина Еа, рассчитанная для этой реакции,
составляла 21,3+0,6 ккал/моль (89,2+2,5 кДж/моль).
3. ДРУГИЕ ПРИМЕНЕНИЯ
Методами ДТА и ДСК можно быстро и просто определить удель-
ную теплоемкость вещества [ 174, 182]. Для иллюстрации на фиг. 7.45
представлены кривые ДСК для окиси альфа-алюминия, полу-
ченные на приборе фирмы «Дюпон». Сначала записывается кривая
Применение ДТА и ДСК
319
для пустого контейнера образца (верхняя кривая). Затем при тех
же условиях записывается кривая для контейнера с образцом.
После этого по данным холостого (пустой контейнер) и рабочего
(контейнер с образцом) опытов можно записать следующее соотно-
шение:
/Г, ч <ДТХ 4- АТ’хол) Ет
{р’т = Тла
(7.8)
где (Ср)г — удельная теплоемкость при температуре Т,
мкал/(мг-град); /\ТХ—абсолютная дифференциальная температу-
ра для образца, °C; ATXOJI — абсолютная дифференциальная тем-
пература для пустого контейнера, °C; Ет — калибровочный коэф-
фициент при температуре Т, мкал/(град-мин); М — масса образ-
ца, мг; а — скорость нагревания, град/мин. Для альфа-окиси
алюминия найденная таким образом величина (С’р)з27° составила
0,279 мкал/мг.
Фиг. 7.45. Кривые для определения удельной теплоемкости альфа-окиси
алюминия [174].
Вес образца из окиси алюминия 21,68 мг; атмосфера — воздух;
(Ср)327° =“0,279 мкал/мг.
скорость нагревания 20 град/мин; = 0,618 СС; &ТХОД— 0,140 СС.
При помощи методов ДТА и ДСК можно построить простые
фазовые диаграммы типа представленной на фиг. 7.46 для смеси
нафталина и бензойной кислоты.
Эвтектическая точка плавления соответствует соотношению ком-
понентов 50 : 50. Фазовая диаграмма была построена по эндотер-
мическим пикам плавления смесей различного состава. Температу-
ры плавления, нанесенные на фазовую диаграмму, получены экст-
раполяцией температур начала плавления [175, 176].
Фиг. 7.46. Фазовая диаграмма смеси нафталина и бензойной кислоты [175,
176].
Точки на фазовой диаграмме сняты по восьми кривым ДТА. а — смеси нафталина и бензой-
ной кислоты (переплавлены); вес образца 50 мг; вёс эталона 35 мг; скорость нагревания
5 град/мии; статическая ат мосфера; железо-конста тановая термопара, б—фазовая диаграм-
ма; А— все компоненты расплавлены; Б— бензойная кислота в равновесии с расплавом; В —
нафталин в равновесии с расплавом; Г — бензойная кислота и эвтектика; Д— нафталин и
эвтектика.
Применение ДТА и ДСК.
321
Методы ДТА [177] или ДСК 1178, 179] удобно использовать для
। ц рений давления пара и теплоты испарения. Метод ДСК после
»*« 1!1пчительной модификации держателя образца применялся для
i.iipcцеления теплоты испарения и теплоты смешения различных
|||цц||ческих жидкостей [1791. Металлическая крышка держателя
пн i;i вменена стеклянной со стеклянной трубкой в центре. Эта
цен питая трубка поддерживала микрошприц с жидким образцом
j 11(>1ч-печивала введение жидкого образца в держатель без нару-
пп пня температуры системы.
фиг 7.47. Последовательнее добавление пяти порций этанола (по 4 мкл)
к 40 мкл бензола [179].
В правом нижнем углу: добавление 2 мкл бензола.
11а фиг. 7.47 представлены кривые ДСК эндотермического сме-
ни пня бензола с этанолом при поддержании изотермических усло-
вий в ячейке. К 40 мкл бензола, содержащегося в образце, последо-
1ипельно добавляли порции этанола по 2—4 мкл. С повышением
। онцентрации этанола в бензоле при добавлении одинаковых пор-
ций этанола теплота смешения уменьшалась. Молярные теплоты
iмцшения
= ДДЭКСП (И! + П2)» (7-9)
|дс rii и п2 — число молей этанола и бензола, представлены на
фн1.7.48. Полученные значения теплот смешения удовлетворитель-
но согласуются с имеющимися данными.
Аналогично определялась теплота испарения органических
жидкостей [179]. Образец вводился в ячейку при помощи микро-
шприца. Площадь под кривой пропорциональна теплоте испарения.
Среднеквадратичное отклонение, полученное для пяти измерений
теплоты испарения бензола, составило ~±2% средней величины.
< >дпако данный метод нельзя использовать для определения ДЯ„
п точке кипения образца. Значения АД, можно установить путем
экстраполяции.
322
Глава 7
Метод ДСК можно использовать также для определения теплоты
возгонки [1801. Образцы помещали в алюминиевые капсулы; про-
странство между дном и выпуклой крышкой заполняли алюминие-
вым порошком. В крышке имелось небольшое отверстие для выхода
выделяющегося газа. При измерении теплоты плавления исполь-
зовали крышки без отверстий. Результаты этих измерений приведе-
ны в табл. 7.14.
Фиг. 7.48. Молярная теплота смешения этанола с бензолом [179].
" экспериме стальные значения; ——— опубликованные данные.
Методом ДТА с использованием герметичных держателей об-
разца можно определить критические температуры органических
жидкостей [181]. Для этой цели используются кривые охлаждения;
при критической температуре Ткрт на кривой наблюдается раз-
рыв. Образцы объемом 20—50 мкл в запаянных стеклянных ка-
пиллярных трубках диаметром 4 мм нагревались до заданной
температуры, а затем охлаждались. При охлаждении регистриро-
валась величина АТ в функции температуры образца. Резкий раз-
рыв на регистрируемой кривой соответствовал точке сосуществова-
ния двух фаз. Температура, соответствующая этой точке, опреде-
лялась также в функции увеличения объема образца до тех пор,
пока она не достигала постоянной величины критической темпера-
туры. Для исследования соединений среднее отклонение получен-
ной температуры от опубликованных данных составляло ±0,16° С.
Методы ДТА и ДСК могут быть использованы также для опреде-
324
Глава 7
ления точки Кюри. Как видно из фиг. 7.49, удельная теплоемкость
никеля постепенно возрастает до точки Кюри, равной 357°С, пос
ле чего наблюдается резкий излом кривой. Вес образца составлял
75 мг.
Уильямс и Чемберлен [1401 рассмотрели возможности примене-
ния ДСК для определения точек Кюри ферромагнитных материален
и точек Нееля антиферромагнитных и ферримагнитных материалов.
Большое число работ, в том числе исследования Брегера и Уай т-
хеда [183], а также Геймела и Смозерса [1841, посвящено изучению
методом ДТА угля и родственных веществ.
Фиг. 7.49. Определение точки Кюри для никеля.
Брегер и Уайтхед [183], используя вакуумный прибор ДТА,
исследовали термические свойства целлюлозы, древесины, лигнита
и различных видов каменного угля. Они обнаружили, что низко-
температурные пики лигнина исчезали, в случае торфа были замас-
кированы и вновь появлялись у лигнитов. Пики кривой разложе-
ния лигнина были подавлены в случае битуминозных углей и от-
сутствовали на кривых антрацитов.
Таблица 7.15
Значение теплотворной способности ряда арканзаских углей,
измеренной методом ДТА и стандартными методами [184]
Образец Площадь под кривой ДТА, см2 Стандартная оценка, кДж/кг
Париское месторождение, шт. Арканзас 2,23 31,045
Месторождение шт. Юта 2,97 33,67
Джеромское месторождение, шт. Аризона .... 2,67 32,55
Высококачественный уголь 3,03 33,80
Применение ДТА и ДСК.
325
Геймел и Смозерс [184] определяли содержание угля из место-
рождения шт. Юта в смеси с окисью алюминия по площадям под
пиками кривой разложения. Для концентраций угля в смесях до
12% была получена линейная зависимость площади от концентра-
ции. Было обнаружено также, что площадь под пиком кривой прямо
пропорциональна теплотворной способности угля (табл. 7.15).
ЛИТЕРАТУРА
1. Liptay G., Atlas of Thermoanalytical Curves, Vol. 1, Akademiai Kiado,
Budapest, 1971.
2. Mitchell B. D., Birnie A. C., Differential Thermal Analysis, Macken-
zie R. C., ed., Academic Press, Lnd., 1970, Ch. 22.
3. Manning M., Industrial Res., Feb., 18 (1966).
4. Gray A. P., Am. Lab., Jan., 43 (1971).
5. Murphy С. B., Anal. Chem., 38, 443R (1966).
6. Murphy С. B., Anal. Chem., 40, 380R (1968).
7. Murphy С. B„ Anal. Chem., 42, 268R (1970).
H. Murphy С. B., Anal. Chem., 44, 513R (1972).
!>. Brancone L. M., Ferrari H. J., Microchem. J., 10, 370 (1966).
10. Vassallo D. A., Harden J. C., Anal. Chem., 34, 132 (1962).
11. Kerr G„ Landis P. S„ Anal. Chem., 44, 1176 (1972).
12. Barrail E. M., Gernert J. F., Porter R. S., Johnson J. F., Anal. Chem.
35, 1837 (1963).
13. Barrall E. M., Porter R. S., Johnson J. F., J. Phys. Chem., 70, 385 (1966).
14. Barrall E. M., Porter R. S., Johnson J. F., J. Phys. Chem., 68, 2810 (1964).
IB. Johnson J. F., Miller G. W., Thermochim. Acta, 1, 373 (1970).
16. Gipstein E., Barrail E. M., Bredfeldt K., Need O. U., Thermochim. Acta,
3, 253 (1972).
17. Ennulat R. D., Analytical Calorimetry, Porter R. S., Johnson J. F., eds.,
Plenum, N. Y., 1968, p. 219.
18. Young W. R., Barrall E. M., Aviram A., Analytical Calorimetry, Por-
ter R. S., Johnson J. F., eds., Vol. 2, Plenum Press, N. Y., 1968, p. 113.
19. Barrail E. M., Analytical Calorimetry, Porter R. S., Johnson J. F., eds.,
Vol. 2, Plenum Press, N. Y., 1968, p. 121.
20. Gipstein E., Barrall E. M., Bredfeldt К. E., Analytical Calorimetry,
Porter R. S., Johnson J. F., eds., Vol. 2, Plenum Press, N. Y., 1968, p. 127.
21. Gray G. W., J. Chem. Soc., 3733 (1956).
22. Barrail E. M., Porter R. S., Johnson J. F., Anal. Chem., 36, 2172 (1964).
23. Ozawa T., Bull. Chem. Soc. Japan, 39, 2071 (1966).
24. Pacor P., Anal. Chim. Acta, 37, 200 (1967).
25. David D. J., Anal. Chim. Acta, 36, 2162 (1964).
26. Harmelin M., Duval C., Xuong N. D., Proc, of the Third Analytical Chemi-
cal Conf., Akademiai Kiado, Budapest, 1970, p. 325.
27. Stammler M., Explosivstoffe, 7, 154 (1968).
28. David D. J., Anal. Chem., 37, 82 (1965).
29. Bohon R. L., Anal. Chem., 35, 1845 (1953).
30. Graybush R. J., May F. G., Forsyth A. C., Thermochim. Acta, 2, 153
(1971).
31. Barrall E. M., Thermochim. Acta, 3, 55 (1971).
32. Barrett E. J., Hoyer H. W., Santoro A. V., Mikrochim. Acta, 1121 (1970).
33. Santoro A. V., Barrett E. J., Hoyer H. W., J. Thermal. Anal., 2, 461
(1970).
34. Barrett E. J., Hoyer H. W., Santoro A. V., Tetrahedron Letters, 5, 603
(1968). ч
326
Глава 7
35. Santoro А. V., Barrett Е. J., Hoyer H. W., Tetrahedron Letters, 19, 2297
(1968).
36. Koch E., Angew. Chem. Inter. Ed., 9, 288 (1970).
37. Atkinson G. F., Itzkovitch I. J., Anal. Chim. Acta, 49, 195 (1970).
38. Levy P. F., Nieuweboer G., Semanski L. C., Thermochim. Acta, 1, 429
(1970).
39. Barrail E. M., Thermochim. Acta, 5, 377 (1973).
40. Hall P. G., Trans. Faraday Soc., 67, 556 (1971).
41. Rogers R. N., Smith L. C., Anal. Chem., 39, 1024 (1967).
42. Rogers R. N., Dinegar R. H., Thermochim. Acta, 3, 367 (1972).
43. Murrill E., Breed L., Thermochim. Acta, 1, 239 (1970).
44. Murrill E., Breed L., Thermochim. Acta, 1, 409 (1970).
45. Murrill E., Whitehead M. E., Breed L., Thermochim. Acta, 3, 111 (1972).
46. Ripmeester J. A., Dunell B. A., Canadian J. Chem., 49, 2906 (1971).
47. Fauth M. I., Anal. Chem., 32, 655 (1960).
48. Chiu J., Anal. Chem., 34, 1841 (1962).
49. Dorko E. A., Hughes R. S., Downs C. R., Anal. Chem., 42, 253 (1970).
50. Krawetz A. A., Tovrog T., I & EC Prod. Res. Dev., 5, 191 (1966).
51. Cross С. K-, Am. Oil Chem. Soc., 47, 229 (1970).
52. Bsharah L., I & EC Prod. Res. Dev., 6, 246 (1969).
53. Vamos E., Schmierstoffe Schmierungstech., 84 (1966).
54. Trzebowski N., Freiberger Forschungsh., A367, 257 (1955).
55. Noel F., J. Inst. Petroleum, 57, 357 (1971).
56. Noel F., Corbett L. W., J. Inst. Petroleum, 56, 261 (1970).
57. Gordon S., J. Chem. Educ., 40, A87 (1963).
58. Kracek F. C., J. Phys. Chem., 33, 1281 (1929).
59. Kracek F. C., J. Phys. Chem., 34, 225 (1930).
60. Wendlandt W. W., Hoiberg J. A., Anal. Chim. Acta, 28, 506 (1963).
61. Wendlandt W. W., Hoiberg J. A., Anal. Chim. Acta, 29, 539 (1963).
62. Mackenzie R. C., ed., Differential Thermal Analysis, Academic Press,
N. Y„ 1970, Ch. 7—15.
63. Berg L. G., Differential Thermal Analysis, Mackenzie R. C., ed., Academic
Press, N. Y., 1970, Ch. 11.
64. Freeman E. S., Rudloff W. K., Differential Thermal Analysis, Macken-
zie R. C., ed., Academic Press, N. Y., 1970, Ch. 12.
65. Dollimore D., Differential Thermal Analysis, Mackenzie R. C., ed., Aca-
demic Press, N. Y., 1970, Ch. 13.
66. Webb T. L., Kruger J. E., Differential Thermal Analysis, Mackenzie R. C.,
ed.. Academic Press, N. Y., 1970, Ch. 10.
67. Mackenzie R. C., Differential Thermal Analysis, Mackenzie R. C., ed.,
Academic Press, N. Y., 1970, Ch. 9.
68. Webb T. L., Differential Thermal Analysis, Mackenzie R. C., ed., Acade-
mic Press. N. Y., 1970, Ch. 8.
69. Bollin E. M., Differential Thermal Analysis, Mackenzie R. C., ed., Aca-
demic Press, N. Y., 1970, Ch. 7.
70. Dollimore D., Differential Thermal Analysis, Mackenzie R. C., ed., Aca-
demic Press, N. Y., 1970, Ch. 14.
71. Wendlandt W. W., Smith J. P., Thermal Properties of Transition Metal
Ammine Complexes, Elsevier, Amsterdam, 1967.
72. Wendlandt W. W., Chelates in Analytical Chemistry, Flaschka H. A.,
Barnard A. J., eds., Dekker, N. Y., 1967, pp. 107—143.
73. Mesmer R. E., Irani R. R., J. Chem. Eng. Data, 8, 530 (1963).
74. Ozawa T., Momota M., Isozaki H., Bull. Chem. Soc. Japan, 40, 1583 (1967).
75. Ozawa T., Isozaki FI., Negishi A., Thermochim. Acta, 1, 545 (1970).
76. Kelley К. K., Bull. U. S. Bur. Mines, 584 (1960).
77. Соколов В. А.. Шмидт H. E., Труды Института общей и неорганической
химии АН СССР, 27, 217 (1956).
Применение ДТА и ДСК
327
78. Barrail Е. М., Rogers L. В., Anal. Chem., 36, 1405 (1964).
79. Wendlandt W. W., Thermochim. Acta, 1, 419 (1970).
80. Wendlandt W. W., D’Ascenzo G., Gore R. H., Thermochim. Acta, 1, 488
(1970).
81. Wendlandt W. WD'Ascenzo G., Gore R. H., J. Inorg. Nucl. Chem., 32,
3404 (1970).
82. Chiu J., Anal. Chem., 35, 933 (1963).
83. DuPont DTA Apparatus Bulletin, DuPont Co.
84. Macak J., Malccha J., Anal. Chem., 41, 442 (1969).
85. DuPont Application Brief, Кв 900B31 July (1970).
86. Garn P. D., Anal. Chem., 41, 447 (1969).
87. Ramachandran V. S., J. Thermal Anal., 3, 181 (1971).
88. Ramachandran V. S., Materiaux et Constructions, 4, 3 (1971).
89. Beech G., Mortimer С. T., Tyler E. G., J. Chem. Soc., (A), 925 (1967).
1)0. Beech G., Ashcroft S. J., Mortimer С. T., J. Chem. Soc., (A), 929 (1967).
91. Beech G., Mortimer С. T., Tyler E. G., J. Chem. Soc., (A), 1111 (1967).
92. Ashcroft S. J., J. Chem. Soc., (A), 1020 (1970).
03. Ashcroft S. J., Mortimer С. T., Thermochemistry of Transition Metal Com-
plexes, Academic Press, Lnd., 1970.
04. Adams J. J., House J. E., Trans. 111. Acad. Sci., 63, 83 (1970).
05. Simchen A. E., Israel J. Chem., 9, 613 (1971).
06. Block J., Anal. Chem., 37, 1414 (1965).
97. Stone R. L., Anal. Chem., 32, 1582 (1960).
08. Petricciani J. C., Wimberley S. E., Bauer W. H., Clapper T. W., J. Phys.
Chem., 64, 1309 (1960).
09. Wendlandt W. W., George T. D., Horton G. R., J. Inorg. Nucl. Chem.,
17, 273 (1961).
100. Erdey L., Paulik F., Acta Chim. Acad Sci. Hung., 7, 27 (1955).
101. Dawson J. B., Wilburn F. W., Differential Thermal Analysis, Macken-
zie R. C., ed., Academic Press, N. Y., 1970, Ch. 17.
102. Mackenzie R. C., Differential Thermal Analysis, Mackenzie R. C., ed.,
Academic Press, N. Y., 1970, Ch. 18.
103. Sudo T., ShimodaS., Differential Thermal Analysis, Mackenzie R. C., ed.,
Academic Press, N. Y., 1970, Ch. 19.
104. Vivaldi J. L. M., Hach-Ali P. F., Differential Thermal Analysis, Macken-
zie R. C., ed., Academic Press, N. Y., 1970, Ch. 20.
105. Glasser F. P., Differential Thermal Analysis, Mackenzie R. C., ed., Aca-
demic Press, N. Y„ 1970, Ch. 21.
106. Fisher DTA Instrument Bulletin, Fisher Scientific Co. Pittsburg, Pa.
107. Keith M. L., Tuttle O. F., Am. J. Sci., Bowen Vol., 203 (1952).
108. DuPont Application Brief., № 15, Jan. (1968).
(09. Davis С. E., Holbridge D. A., Clay Minerals, 8, 193 (1969).
110. Cohen A. A., Krenkel T. G., Am. Mineralogist, 55, 1329 (1970).
111. Reddick K. L., Sun Oil Quart., Na 3, 31 (1969).
112. Warne S. S., Mackenzie R. C., J. Thermal Anal., 3, 49 (1971).
113. Garn P. D„ Flaschen S. S„ Anal. Chem., 29, 271 (1957).
114. Lodding W., Hammell L., Anal. Chem., 32, 657 (1960).
115. Берг Л. Г., Рассонская И. С., ДАН СССР, 73, 113 (1950).
116. Bollin Е. М., Dunne J. A., Kerr Р. F., Science, 131, 661 (1960).
117. Bollin Е. М., Kerr Р. F., Am. Mineralogist, 46, 823 (1961).
118. Mitchell В. D., Birnie А. С., Differential Thermal Analysis, Macken-
zie R. C., ed., Academic Press, Lnd., 1970, Ch. 24.
119. Pfeil R. W., Proc, of the Third Toronto Symp. on Thermal Analysis,
McAdie H. G., ed., Chemical Institute of Canada, Toronto, 1969, p. 187.
120. DuPont Application Brief, № 20, (1968).
121. Haighton A. J., Hannewijk J., J. Am. Oil Chem. Soc., 35, 457 (1958).
122. Currell B. R., Robinson B., Taianta, 14, 421 (1967).
328
Глава 7
123. Garn Р. D., Thermoanalytical Methods of Investigation, Academic Press,
N. Y., 1965, p. 124.
124. Edmonds M. D., Core M. T., Bauley A., Schwenker R. F., Tobacco Sci ,
9, 48 (1965).
125. Hoyer H. W., Barrett E. J., Anal. Biochem., 17, 344 (1966).
126. Stein J. M., Perkin-Elmer Instrument News, 19, № 2 (1968).
127. Puett B., Biopolymers, 5, 327 (1967).
128. Hoyer H. W., J. Am. Chem. Soc., 90, 2480 (1968).
129. Hoyer H. W., Nature, 216, 997 (1967).
130. Moore R., Thermal Analysis, Schwenker R. F., Garn P. D., eds., Vol. 1,
Academic Press, N. Y., 1969, p. 615.
131. Greaves R., Davies J., Ann. New York Acad. Sci., 125, 548 (1965).
132. Luebke H. W., Breidenbach B. G., J. А. О. C. S., 46, 60 (1969).
133. Cross С. K., J. A. 0. C. S., 47, 229 (1970).
134. Arseneau D. F., Can. J. Chem., 39, 1915 (1961).
135. Pakulak J. M., Leonard G. W., Anal. Chem., 31, 1037 (1959).
136. Mitchell B. D„ Birnie A. C., Analyst, 91, 783 (1966).
137. Berlin E., Kliman P. G., Pallansch M. J., Thermochim. Acta, 4, 11 (1972).
138. Labowitz L. C., Thermochim. Acta, 3, 419 (1972).
139. Berlin E., Kliman P. G., Anderson B. A., Pallansch M. J., Thermochim.
Acta, 2, 143 (1971).
140. Williams H. W., Chamberland B. L., Anal. Chem., 41, 2084 (1969).
141. Olafsson P. G., Bryan A. M., Mikrochim. Acta, 871 (1970).
142. Morita H., J. Am. Chem. Soc., 78, 1397 (1956).
143. Morita H., Anal. Chem., 28, 64 (1956).
144. Morita H., Anal. Chem., 29, 1095 (1957).
145. Schulken R. M., Roy R. E., Cox R. H., J. Polymer Sci., Part C, 6, 1725
(1964).
146. Ke B., ed., Thermal Analysis of High Polymers, Wiley-Interscience,
N. Y„ 1964.
147. Slade P. E., Jenkins L. T., eds., Techniques and Methods of Polymer
Evaluation, Marcel-Dekker, N. Y., 1966.
148. Schwenker R. F., ed., Thermoanalysis of Fibers and Fiber-Forming Poly-
mers, Wiley-Interscience, N. Y., 1966.
149. Porter R. S., Johnson J. F., eds., Analytical Calorimetry, Vol. 1, Plenum
Press, N. Y., 1968.
150. Porter R. S., Johnson J. F., eds., Analytical Calorimetry, Vol. 2, Plenum
Press, N. Y., 1970.
151. Reich L., Stivala S. S., Elements of Polymer Degradation, McGraw-Hill
N. Y., 1971.
152. Schwenker R. F., Garn P. D., eds., Thermal Analysis, Academic Press,
Vol. 1, N. Y., 1969, Section 2.
153. Murphy C. B_, Differential Thermal Analysis, Mackenzie R. C., ed., Aca-
demic Press, Lnd., 1970, Ch. 23.
154. Chiu J., DuPont Therntogram, 2, № 3, 9 (1965).
155. Gray A. P., Perkin-Elmer Instrument News, 20, № 2, 8 (1969).
156. Gray A. P., Thermochim. Acta, 1, 563 (1970).
157. Dole M., J. Polymer Sci., Part C., 18, 57 (1967).
158. Dole M., Fortschr. Hochpolym. Forsch., 2, 221 (1960).
159. Duswalt A. A., Hercules Chemist, № 57, 5 (1968).
160. Foltz C. R., McKinney P. V., Anal. Chem., 41, 687 (1969).
161. Hoyer H. W., Santoro A. V., Barrett E. J., J. Polymer Sci., Part A-l
6, 1033 (1968).
162. Anderson H. C., Anal. Chem., 32, 1592 (1960).
163. Murphy С. B., Palm J. A., Doyle C. D., Curtiss E. M., J. Polymer Sci
28, 447 (1958).
Применение ДТА и ДСК.
329
164. Murphy С. В., Palm J. A., Doyle С. D., Curtiss Е. М., J. Polymer Sci.,
28, 453 (1958).
165. Clampitt В. Н., Anal. Chem., 35, 577 (1963).
166. Schwenker R. F., Beck L. R., Textile Res. J., 30, 624 (1960).
167. Ke B., J. Polymer Sci., 42, 15 (1960).
168. Ke B., J. Polymer Sci., 50, 79 (1961).
169. Wunderlich B., Kashdan W. H., J. Polymer Sci., 50, 71 (1961).
170. Scott N. D., Polymer, 1, 114 (1960).
17). Rudin A., Schreiber H. P., Waldman M. H., Ind. Eng. Chem., 53, 137
(1961).
172. Ke B., Sisko A. W., J. Polymer Sci., 50, 87 (1961).
173. Murphy С. B., Hill J. A., Nucleonics, 18, 78 (1960).
174. DuPont Application Brief, № 11, Jan. 15, (1968).
175. Fisher DTA Instrument Bulletin, Fisher Scientific Co.
176. Visser M. J., Wallace W. H., DuPont Thermogram, 3, § 2, 9 (1966).
177. Kemme H. R., Kreps S. I., Anal. Chem., 41, 1869 (1969).
178. Farritor R. E., Tao L. C., Thermochim. Acta, 1, 297 (1970).
179. Mita I., Imai I., Kambe H., Thermochim. Acta, 2, 337 (1971).
180. Beech G., Lintonbon R. M., Thermochim. Acta, 2, 86 (1971).
18). Hoyer H. W., Santoro A. V., Barrett E. J., J. Phys. Chem., 72, 4312 (1968).
182. Gray A. P., Perkin-Elmer Instrument News, 21, № 2 (1970).
183. Breger I. A., Whitehead W. L., Fuel, 30, 247 (1951).
184. Gamel С, M., Smothers W. J., Anal. Chim. Acta., 6, 442 (1952).
Глава 8
ОБНАРУЖЕНИЕ И АНАЛИЗ ВЫДЕЛЕННОГО ГАЗА
А. ВВЕДЕНИЕ
Обнаружение и анализ газов, выделяющихся в процессе хими
ческой реакции, в зависимости от температуры составляют суть
методов термического анализа, называемых соответственно обнару-
жение выделенного газа (ОВГ) и анализ выделенного газа (АВГ).
В соответствии с рекомендациями по терминологии Международной
конференции по термическому анализу, приведенными в гл. 13,
под методом «обнаружения выделенного газа» подразумевается ме-
тод, позволяющий определить, образовалось ли летучее вещество
при термическом анализе. Методом «анализа выделенного газа»
называется метод определения состава и количества летучих ве-
ществ, образовавшихся при термическом анализе.
Методы ОВГ и АВГ требуют применения различных приборов,
начиная от простейшего датчика теплопроводности газа до значи-
тельно более сложного масс-спектрометра высокого разрешения.
В методе ОВГ непрерывно регистрируется относительное количество
выделенного газа в зависимости от температуры. Анализ выделен-
ного газа может проводиться непрерывно или периодически. Пе-
риодический или ступенчатый анализ осуществляется обычно с
помощью газовых хроматографов. Для непрерывной регистрации
результатов анализа используются масс-спектрометры. Оба эти
способа отбора и анализа выделенного газа находят широкое при-
менение.
Области применения методов ОВГ и АВГ были рассмотрены в
книге Лоддинга [76J ив одной из глав книги под редакцией Кеньо-
на [77].
|Б. ОБНАРУЖЕНИЕ ВЫДЕЛЕННОГО ГАЗА
По-видимому, первая попытка обнаружения выделенного газа
(ОВГ) была предпринята Стоуном [1] при работе с прибором ДТА,
имеющим нагреватель закрытого типа с динамической газовой
атмосферой. К кожалению, Стоун не анализировал газы, выделя-
ющиеся из камеры нагревателя, а только регистрировал кривые
ДТА. В 1960 г. Лоддинг и Хэммел [2] установили датчик теплопро-
Обнаружение и анализ выделенного газа
331
водности на выходе газа из нагревателя прибора ДТА. Однако кри-
вые выделенного газа не были опубликованы. Упомянутые исследо-
ватели включили в систему выхода газа абсорбционные трубки, что
Позволило провести также анализ обнаруженного газа. В этом
же году Роджерс и др. 13] описали простой пиролизный прибор,
Фиг. 8.1s Блок пиролиза и вспомогательное оборудование [3].
/ — пиролизная камера; 2 — никелевая пробка; 3 — вход газа-носителя; 4 — выход газа-
поснтеля; 5 — местоположение двух патронных нагревателей; 6 — спиральная канавка на
внутренней поверхности корпуса блока; 7 — наружная оболочка блока; 8 — вход в охлаж-
дающую рубашку; 9— выход из охладающей рубашки.
Л —баллон с газом-носителем; В— регулятор давления; С — игольчатый клапан регу-
лирования расхода; D — эталон теплопроводности; Е—пиролизная камера; F — трубчатая
Камера сгорания; G — рабочая ячейка; Н — манометр; I — игольчатый клапан регулирования
давления; J — ротаметр.
который положил начало методу ОВГ. Тщательность проделанной
ими работы, особенно в части описания экспериментальных пара-
метров, позволяет считать этих авторов общепризнанными основа-
телями метода ОВГ.
На фиг. 8.1 приведен прибор Роджерса и др. [3]. Основными
частями прибора являются: пиролизная камера с электрическим
нагревателем; трубчатая камера сгорания; датчик теплопровод-
ности. В пиролизной камере размещена довольно длинная трубка
для входа газа, служащая для предварительного подогрева газа-
носителя. Подогрев производится двумя патронными электронагре-
вателями мощностью по 240 Вт, входное напряжение которых ре-
гулируется трансформатором с переменным напряжением. Трубча-
тая камера сгорания представляет собой никелевую трубку длиной
127 мм, нагреваемую электронагревателем. Трубка заполнена
смесью огнеупорного кирпича и окиси меди, поддерживаемых при
температуре 650—750° С. Назначение этой камеры — превращение
всех летучих веществ в простые газы, с тем чтобы они не конден-
332
Глава S
сировались на стенках трубок, не достигнув датчика. Датчик теп
лопроводности, в котором в качестве чувствительных элементов
использовались воспламенители типа применяемых в самолетах.
Фиг. 8.2. Влияние массы образца тетранитрата пентаэритрита на характер
кривой ОВГ [3(.
1—масса образца 1,4 мг, температура максимума 160 °C; 2— масса образца 5 мг, темпе-
ратура максимума 167 °C; 3 — масса образца 9,7 мг, температура максимума 177° С; 4 — масса
образца 15 мг, температура максимума 178 °C; 5— масса образца 20,5 мг, температура мак-
симума 178 °C.
изолирован от пиролизной камеры и камеры сгорания; в нем под-
держивается комнатная температура. Выходное напряжение дат-
чика регистрируется по оси Y двухкоординатного самописца, а
температура, определяемая хромель-алюмелевой термопарой, —
по оси X.
Кривые ОВГ, полученные на этом приборе, напоминают кривые
ТГП. Результаты обычно отличаются от полученных методом ДТА
для этих же веществ. Было качественно определено влияние пере-
менных рабочих характеристик (расхода потока, скорости нагре-
вания, массы образца, чувствительности датчика теплопроводно-
сти, давления, типа газа-носителя)на характер кривыхОВГ. Всеука-
Обнаружение и анализ выделенного газа
333
Ванные параметры, за исключением чувствительности датчика теп-
лопроводности, влияют на максимум температуры и высоту пика
Кривой. На фиг.8.2 показан характер кривых ОВГ в зависимости
От массы образца. Температура, соответствующая максимуму пика,
Фиг. 8.3. Кривые ОВГ нескольких соединений [3].
— образец NaHCOg массой 10 мг с примесью Na2COa-H2O; 2— образец CaSO4-2H2O массой
10 мг; 3 — образец тетрила массой 10 мг;4— образец гексаиитрозобеизола массой 10 мг.
изменяется от 160° С для образца массой 1,4 мг до 178° С для образ-
ца массой 20,5 мг. Влияние массы образца еще более заметно на
кривой ОВГ для ТНТ (тринитролуола). Температура, соответствую-
щая максимуму пика, для образца массой 1,5 мг составляет 153° С,
а для образца массой 20,9 мг — 197° С. Таким образом, для дости-
жения воспроизводимости температур пиков необходим, по-види-
мому, контроль размеров образцов.
Примеры типичных кривых ОВГ нескольких соединений приве-
дены на фиг. 8.3. В случае смеси соединений с одинаковыми темпе-
ратурами кипения их пики не разделяются. Температуры плавле-
ния могут быть определены этим методом только в том случае, если
одновременно с плавлением происходит разложение соединений.
Метод ОВГ применим как для качественного, так и для количест-
венного анализов. Это возможно при идентичных условиях пиро-
лиза путем сравнения площадей под пиками кривой ОВГ с площа-
дями пиков для стандартных веществ (эталонов).
334
Глава 8
Приборы ОВГ, предназначенные для пиролиза различных поли
меров, были описаны Вассало [4]. Система обнаружения газа и
этих приборах состоит из термисторного датчика теплопроводно» i и
с термисторами сопротивлением 100 000 Ом, включенными по м<н
товой схеме при токе 10 мА. Между пиролизной камерой и ячейкой
датчика размещена заполненная окисью меди камера сгорании,
Фиг. 8.4. Совмещенный прибор ОВГ—ДТА Лоддинга и Хэммела [2J.
/ — трубчатый нагреватель; 2— кольцевая прокладка; 3 — игольчатые клапаны; 4— про-
вода к мостовой схеме для измерения сопротивлений; 5 — датчик теплопроводности; 6 — аб-
сорбционные трубки; 7 — эталон; 8—образец.
в которой достигается температура 800° С; конструкция камеры
подобна описанной выше [3]. Для регистрации кривых ОВГ вместо
ленточного самописца применен двухкоординатный самописец.
В противоположность приведенным выше утверждениям автор счи-
тает, что метод ОВГ не может заменить метода ТГ, однако при изу-
чении полимеров со сходной структурой метод ОВГ обладает боль-
шей селективностью и является более быстрым. Так, при исполь-
зовании данного метода потеря массы в 2 мг может быть обнаружена
при значительно более высоких скоростях нагревания, чем в тер-
могравиметрии.
На фиг. 8.4 показан совмещенный прибор ОВГ—ДТА, ис-
пользуемый Лоддингом и Хэммелом [2]. Печь изготовлена из ре-
кристаллизованной окиси алюминия и может быть использована
при избыточных давлениях до 3,5 МПа и температуре 1200° С. Газы,
поступающие в камеру печи, проходят через исследуемый и эталон-
Обнаружение и анализ выделенного газа
335
цый образцы и затем раздельно выводятся из камеры. По утвержде-
нию авторов, система обнаружения и анализа газов позволила оп-
ргделить природу пиков кривой ДТА. Однако в их работе не при-
ведены примеры таких определений.
Предполагается, что в общем случае особенно существенные
результаты можно ожидать от комбинации метода ОВГ и какого-
либо другого метода термического анализа, например ДТА или
ТГ, так как такое сочетание дает возможность наилучшим образом
Использовать преимущества каждого метода. Примером может
Служить комбинированный прибор ОВГ—ДТА, при использова-
нии которого все измерения проводятся на одном образце при оди-
наковых скоростях нагревания, атмосфере печи,, размерах частиц
образца и других параметрах эксперимента. В табл. 8.1 сопостав-
лены возможности ОВГ и ДТА для исследования химических и фи-
зических изменений в образцах.
Таблица 8.1
Применение методов ОВГ и ДТЛ для исследования физических
и химических процессов [4]
ОВГ
Процесс
ДТА
(эндо-
терм.)
да нет
Разложение....................................
Плавление ....................................
Фазовый переход в кристаллах .........
Десорбция.....................................
Испарение:
десольватация..............................
кипение............................... . .
возгонка . . . ............................
1) Возможен также экзотермический процесс.
2) Возможна конденсация до достижения газом датчика.
Одна из первых систем для одновременного получения кривых
ОВГ и ДТА была описана Эйрисом и Бенсом [5]. Эта система
(фиг. 8.5) позволяет производить непрерывный контроль выделяе-
мых газов, а также выборочный отбор любой пробы газов
И одновременно получать кривую ДТА. Держатели исследуе-
мого и эталонного образцов были изготовлены из стекла пирекс и
присоединены к стеклянному газопроводу с помощью стандартных
стеклянных соединительных шлифов. Обнаружение выделяемых
газов производится датчиком теплопроводности. В качестве «раз-
бавителя» исследуемого образца и инертного эталонного образца
использовались стеклянные шарики диаметром 0,1 мм.
336
Глава 8
Фиг. 8.5. Ячейки для исследуемой!
и эталонного образцов, исполыо
ванные в системе Эйриса и Бенса |.г>|
А—впуск продуваемого газа; В—подпчл
исследуемого газа к датчику; С — подпчл
эталонного газа к датчику; D — выводи
термопары; Е — ячейка с эталонным об|»н
цом и стеклянными шариками; F — предвп
рительный подогреватель продуваемого гл ы
со стекловатой; G — ячейка с рабочим обрд i
цом н стеклянными шариками.
Наиболее интересным соединением, изученным с помощью этой»
прибора, был нитроглицерин, кривые ОВГ—ДТА которого пред
ставлены на фиг. 8.6. Эндотермические пики в интервале температур
141—206° С являются результатом испарения нитроглицерина,
температура кипения которого равна 143° С. При этом происходи т
частичное разложение, о чем свидетельствует кривая ОВГ. Неболь-
шой перегиб на кривой, соответствующий максимальной скорости
испарения (эндотермический пик, температура 191° С), указывает
на изменение скорости разложения газа, достигающего датчика;
максимальная скорость выделения газа соответствует температуре
202° С. При дальнейшем повышении температуры начинается более
интенсивное разложение нитроглицерина, ранее перегнанного и
охлаждаемую выходную трубку, что приводит к возникновению
последующих пиков на кривой ОВГ. На кривой же ДТА в этом ин-
тервале температур нет отклонений, поскольку весь нитроглице-
рин был подвергнут перегонке и возле датчиков дифференциальной
температуры его нет.
Обнаружение и анализ выделенного газа
337
Температура, ’С хЮ’1
Фиг. 8.6. Кривые ОВГ—ДТЛ нитроглицерина, полученные на одном при-
боре [5].
Масса нитроглицерина 50 мг. - — кривая ОВГ;--------------кривая ДТА.
Фиг. 8.7. Кривые ОВГ—ДТА перхлората аммония, полученные на одном
приборе [5].
-----------кривая ДТА;------кривая ОВГ.
На фиг. 8.7 приведены полученные с помощью описан-
ного прибора кривые ОВГ—ДТА термической диссоциации перхло-
рата аммония, демонстрирующие одновременно фазовый переход
и реакцию разложения. Эндотермический фазовый переход перхло-
рата аммония — типичного окислителя ракетного топлива [5] —
характеризуется пиком с максимумом при температуре 240° С.
Переход обусловлен превращением орторомбической структуры в
кубическую и, как видно из кривой ОВГ, не сопровождается зна-
12—230
338
Глава 8
ОВГ ДТА
и
Фиг. 8.8. Совмещенный прибор для исследований методами ОВГ и ДТА [7].
1 — эталон; 2— никелевая трубка; 3—трубки изолятора с двумя отверстиями; 4— чашки
из инконеля; 5 — образец; 6 — обмотка печи; 7 — кольцевая уплотнительная прокладка;
8 — расходомер; 9 — датчик теплопроводности; 10—печь.
чительным выделением газа. Следующий экзотермический пик п
соответствующий ему пик на кривой ОВГ при температуре 258° С
приписываются началу разложения NHaClOr, стадия окончатель-
ного разложения начинается при температуре 320°С. Результаты
этого исследования не совпадают с другими данными ДТА [1, 61,
вероятно, вследствие различия в скоростях нагревания и в ис-
пользуемых «разбавителях» образцов.
Обнаружение и анализ выделенного газа
33«
Фиг. 8.9. Кривые ОВГ—ДТА CuSO4- 5НгО, полученные на одном приборе [7].
Эйрис и Бенс [51 отмечали некоторые трудности эксплуатации
предложенной ими системы, возникающие вследствие ускоренного
испарения образца из-за наличия потока газа и неконтролируе-
мой конденсации пара в холодной части выходной трубки. Испаре-
ние образца при температуре ниже температуры кипения затрудня-
ет определение последней, а его конденсация до прохождения через
датчик нежелательна, так как впоследствии поток газа может на-
греться до достаточно высокой температуры, чтобы вызвать разло-
жение конденсата.
Сходный прибор ОВГ—ДТА был описан Уэндландтом (фиг. 8.8)
171. Прибор имеет более прочную конструкцию, чем прибор, опи-
санный в работе [5]. Кроме того, при использовании прибора Уэнд-
лаидта не требуется «разбавитель» в виде стеклянных шариков.
Образец и эталон помещаются в небольшие чашки из инконеля
емкостью 0,27 мл, расположенные непосредственно на спаях диф-
ференциальной термопары. В качестве газа-носителя используется
гелий. Газы, выделяющиеся при разложении, обнаруживаются тер-
мистором в датчике теплопроводности обычной конструкции. На-
пряжение на выходе мостовой схемы термистора и усиленное на-
пряжение от А71 регистрируются по осям Y двух двухкоординатных
<амописцев; одновременно регистрируется температура печи по
осям X этих самописцев.
На фиг. 8.9 приведены кривые ОВГ—ДТА, полученные при де-
гидратации CuSO4-5H2O. Пики кривых соответствуют следующим
реакциям:
12*
340
Глава 8
а) пик при 85°С
CuSO4 5Н2О (т.) -> CuSO4 ЗН2О (т.) + 2Н2О (ж.),
2Н2О(ж.) -> 2Н2О (г.) (из насыщенного раствора);
б) пик при 115°С
CuSO4 • ЗН2О (т.) -> CuSO4 • Н2О (т.) 4- 2Н2О (г.);
в) пик при 230сС
CuSO4 - Н2О (т.) -> CuSO4 (т.) + Н2О (г.).
Каждому пику на кривой ДТА соответствует пик на кривой OBI,
обозначающий выделение летучего продукта разложения — париц
воды.
При термической диссоциации этилсульфата калия (фиг. 8.1Щ
фазовый переход в кристалл, отмеченный пиками на кривой ДТА,
не вызывает появления пиков на кривой ОВГ [8]. Переход а >р
для KC2Hf)SO4 сопровождается образованием пика на кривой Д1 Л
при температуре 93СС. При дальнейшем нагревании этого соедн
нения появляется пик плавления (207° С), который также не iimcci
соответствующих пиков на кривой ОВГ. При новом повышении
температуры наблюдается эндотермический пик разложения, <н
ражающийся на кривой ОВГ. Реакция разложения имеет вид
р-КСДГДО, (т.) -> K2S2O7 (т.) + QH, (г.) + Н2О (г.).
Такой же ряд кривых ДТА—ОВГ был получен для метилсу.н.
фата калия.
Аппаратура для одновременного отбора газа и ДТА была опт .1
на Мэрфи и др. [9]. Газы, выделенные из системы, находящейся
вначале под вакуумом, собирали в стеклянную колбу. Затем сое i .in
выделенных газов определяли с помощью масс-спектрометра, ('.и
стема данного типа может быть применена только в том случае,
когда на кривой ДТА существует один пик.
Преимущества аппаратуры для одновременных исследований м<
годами ДТА и ОВГ очевидны. С помощью этой аппаратуры легко
обнаруживается начало кристаллического или другого фазовою
перехода, что обеспечивает дополнительные данные для интерщн
тации кривой ДТА. По результатам анализа выделенных газон г
помощью любых физических методов можно определить стехно
метрические условия реакции, вызывающей образование пикон,
Появляется также возможность исследования термического поводе
ния образца в изотермических условиях, в том числе в различных
газовых средах при разных давлениях.
Как указывалось ранее [51, требуются лишь незначительные
изменения в стандартной аппаратуре ДТА для присоединения при
Обнаружение и анализ выделенного газа
341
бора ОВГ. Затраты на переоборудование полностью окупаются
дополнительной информацией, получаемой при исследовании одно-
1о образца. Некоторые фирмы — «Коламбиа сайентифик индаст-
риз», «Текникел Эквипмент» («Дельтатерм»), «Перкин-Элмер» и
др. — при выпуске ряда приборов ДТА предусматривают возмож-
ность присоединения к ним аппаратуры ОВГ.
Фиг. 8.10. Кривые ОВГ—ДТА этилсульфата калия, полученные на одном
приборе [8].
Томас и др. [10] описали прибор для исследования реакций дис-
социации в динамической газовой атмосфере. Для проведения реак-
ций диссоциации и спекания при температурах до 1000°С обравец
помещали в платинородиевую трубку. Для контроля выделяемых
газов применяли датчик теплопроводности. Площади пиков кри-
вой ОВГ интегрировали с помощью механического интегратора,
соединенного с самописцем. Основное назначение прибора — ис-
следование условий спекания различных минералов и одновремен-
ное определение их площадей поверхности.
Уэр [11] предложил материал, температура термического пере-
хода которого может быть использована для калибровки по темпе-
ратуре в процессе выделения газа. Было установлено, что-сульфат
калия, содержащий небольшое количество растворенного углекис-
лого газа, выделяет его при плавлении, поэтому пик на кривой ОВГ
этого образца совпадает с пиком плавления на кривой ДТА и не
зависит от скорости нагревания и содержания кислорода в атмо-
342
Глава 8
1
Фиг. 8.11. Держатель об-
разца, применяемый при
калибровке по темпера-
туре для ОВГ [11].
1 — платиновая крышка; 2 —
платиновый чехол; 3 — окись
алюминия; 4—тигель для об-
разца; 5 — термопара.
Вход Выход
газа газа
сфере. Во время калибровки применялся держатель образца, по
казанный на фиг. 8.11. Он состоял из платинового тигля, помещен
ного в держатель из окиси алюминия, закрывающийся платиновой
крышкой. Между платиновой крышкой и держателем образца и i
окиси алюминия пропускался газ-по» и
тель, который у верхней части держан
ля контактировал с образцом, затем омы
вал стенки тигля и через небольшое ы
верстие в центре держателя образца н<п
тупал к датчику плотности газа. Согл.п
но расчетам, при расходе газа 30 мл/мп и
время прохождения газа от образна ло
датчика плотности составляет менее (> <
Предварительные испытания этой системы
показали, что на выделение газа не вл и я
ют ни изменение скорости нагревания не
чи от 3 до 10 град/мин, ни изменение со
держания кислорода в газовой атмосфере
Предложенный метод следует рассмагрп
вать как возможный вариант калибровки
по температуре для метода ОВГ; однако,
прежде чем сделать окончательный выбор,
необходимы испытания в широком дна
пазоне режимов.
Держатель образца для одновременных
исследований методами ОВГ и ДСО был
описан Уэндландтом и др. [12, 13]. Эгог
в гл. 9, был применен для изучении
термической диссоциации гидратов солей и координационных
соединений.
Методы ОВГ были использованы разичными исследователями
[3, 14, 15, 18] для получения кинетических и термодинамических
данных. Эти методы находят такое же применение, как методы ТГ
и ДТА, но их преимущество заключается в простоте аппаратуры.
Критическая оценка метода ОВГ и соответствующих проблем
анализа газов была сделана Гарном [16], а также Финдейсом и
ДР- [17].
В. АНАЛИЗ ВЫДЕЛЕННОГО ГАЗА (АВГ)
Возможность определения состава выделенного газа значительно
расширяет область применения метода ОВГ. Анализ газов можем
производиться непрерывно или периодически с помощью разлил
ных методов (фиг. 8.12). Чаще всего применяются методы масс-
спектрометрии, газовой или тонкослойной хроматографии, методы
с использованием химических детекторов и инфракрасной спект-
Обнаружение и анализ выделенного газа
343
Ьргкоппи. В большинстве исследований производится непрерывный
Ш1ЙЛИЗ газов, так как это требует меньших затрат времени и труда.
fVlihiKO аппаратура для непрерывного анализа обычно более доро-
Н»1, особенно если применяется ЭВМ. Камерой для пиролиза об-
|ц|ош могут служить обычная печь, описанная в разд. Б данной
Фиг. 8.12. Методы анализа выделенных газов.
Главы, прибор для ДТА или термовесы. Здесь не рассматривается
анализ радиоактивности газов, выделяемых в процессе химической
реакции, поскольку этот метод описывается отдельно в гл. 11,
МВД. Г.
1. ГАЗОВАЯ ХРОМАТОГРАФИЯ
Для определения состава газов, выделяемых в процессе химиче-
ской реакции, используется мощный аналитический метод газовой
хроматографии. К пиролизной камере с помощью газоотборного
клапана присоединяется газовый хроматограф. Клапан может
иметь ручное [19] или автоматическое управление [20, 21]. В от-
дельных случаях газы после хроматографа подаются на масс-
спектрометр [22] для точной идентификации состава газообразных
продуктов.
344
Глава 8
На фиг. 8.13 приведена описанная Гарном [16] схема подачи
газа и размещения клапанов при двойном отборе газа для хром.но
графического анализа. Газообразные вещества из пироли:пю(|
камеры подаются на два газоотборных клапана. Последние черм
определенные заданные интервалы времени захватывают порции
газа и направляют их к хроматографическим колонкам и датчикам
Фиг. 8.13. Схема подачи газа и размещения клапанов в установке для хро-
матографического анализа с двойным отбором газа [16].
1 — вентиль образца 1; 2 — эталон 1; 3 — эталон 2; 4 — вентиль образца 2; 5 — печь; 6 —
датчик теплопроводности; 7 — флюоропак; 8 — водяная ловушка; 9— силикагель; 10 — кла-
паны; 11 — молекулярное сито; 12 — детектор 1; 13 — детектор 2.
К клапанам может быть присоединено регулируемое реле времени,
обеспечивающее открытие клапанов на 5 с и последующее их за-
крытие. Для эксперимента каждого типа задаются необходимая
температура печи, расходы газов, продолжительность открытия
клапанов. Применяются три типа хроматографических колонок:
с флюоропаком, с силикагелем и молекулярное сито.
Газовый хроматограф в ряде исследований [20—22] объеди-
нялся с термовесами. Это дает возможность контролировать ход
реакции методом термогравиметрии, а в выбранных интервалах
температуры проводить анализ выделенных газов с помощью хро-
матографа. Такой прибор, примененный Видеманном [21], показан
На фиг. 8.14. С целью максимального сокращения времени на пода-
Koww
отбора правы
газа Вря газового
хроматографа
Фиг. 8.14. Сочетание термовесов с газовым хроматографом в приборе Видеманна
/ — образец; 2 — печь; 3 — термовесы; 4 — газоотборный клапан; 5 — магнитный клапан;
6 — клапан А; 7 — контур отбора пробы газа.
346
Глава 8
чу газов объем печи был уменьшен до 35 см3. Газы вводились в хрн
матограф с помощью газоотборного клапана, открывающегося ин
определенные промежутки времени. ТГ- и ОВГ-кривые, получен
ные с помощью этого прибора, приведены на фиг. 8.15. При р.п. в,
женин СаС2О4-Н2О выделяются вода, окись и двуокись углероы,
но поскольку эксперимент проводился в атмосфере воздуха, окшц
углерода сразу же окислялась до двуокиси углерода. По сооопи
нию Видеманна, масс-спектрометр является более быстродействии
щим прибором, чем газовый хроматограф, при приблизительно одн
наковой точности полученных результатов.
Фиг. 8.15. ТГ- и ОВГ-кривые разложения СаСгО4-НгО [21].
Другие комплексные системы, сочетающие обнаружение выдс
ленного газа (ОВГ) и его хроматографический анализ, описаны
Вистом [23], Дорси [24], Франком и Пуром [25, 27], а также Сарне
ром и др. [26]. Термин «термический анализ реакции» использую
ся в работах [25, 27] для обозначения метода, при котором образец
вступает в реакцию с различными веществами, а продукты распаде
идентифицируются с помощью метода газовой хроматографии
Для точного определения состава выделенного газа, а также дли
других исследований проводится пиролиз паровой фазы — регулп
руемые реакции термодеструкции образца [26].
Хроматограф для анализа выделенного газа, разработан и и 11
Воллином [28], использовался для анализа атмосферы и поверх
ности планеты Марс. Назначение этого хроматографа — опредг
лить по пикам кривой АВГ, имеют ли органические вещества пи
Марсе упорядоченное распределение по молекулярным весам (чш
указывало бы на их биологическое происхождение) или распредели
Обнаружение и анализ выделенного газа
347
line является случайным и, следовательно, происхождение веществ
пебиологическое. Особый интерес представляют специальные хи-
мические детекторы для отдельных газов, в которых проба газа
вступает в экзотермическую реакцию с материалом детектора. В де-
биторе воды в качестве такого материала применялись гидриды
Фиг. 8.16. Миниатюрный совмещенный прибор (ДТА—АВГ—газовый хро-
матограф), сконструированный Воллином [28].
/ — образец; 2 — печь ДТА; 3 — колонна нагревателя; 4 — бифилярная хроматографическая
колонка; 5— эталон (ДТА); 6—термопара: 7 — электролитический газогенератор; 8 — вход-
ное отверстие вакуумного аккумулятора; 9 — соленоид; 10—фотоионизационный детектор.
кальция или лития, а в детекторе двуокиси углерода могут исполь-
зеваться гидроокиси натрия или лития. Схема прибора Боллина
приведена на фиг. 8.16. Следует отметить, что прибор для ДТА—
АВГ и газовый хроматограф имели диаметр всего лишь 25 мм!
2. ТИТРИМЕТРИЧЕСКИЕ МЕТОДЫ
Количество выделенного газа можно определить методом непре-
рывного титрования. Газ-носитель вытесняет выделенный газ из
камеры печи и переносит его в водный поглотительный раствор,
где он непрерывно титруется. Титрованный раствор выбирается
в зависимости от вида выделенного газа. Например, аммиак титру-
ется разбавленной соляной кислотой, а вода определяется по ме-
тоду Фишера. Методом титрования можно количественно опреде-
лять содержание воды, хлористого водорода, аммиака, двуокиси
серы, двуокиси углерода и хлора [29].
348
Глава 8
Паулик и др. [29—33, 35] описали прибор для титриметрнчг
ского анализа, соединенный с дериватографом. С помощью тигру
ющего аппарата непрерывного действия записывается так называй
мая термо-газотитриметрическая кривая (ТГТ) или производнаи
этой кривой (ПТГТ). Схема прибора приведена на фиг. 8.17. Обр.т
зец размещен в камере печи из кварца, которая может быть загюл
йена инертным газом-носителем. Выделенные газы переносятся
Фиг. 8.17. Прибор для параллельной записи ДТА-, Т-, ТГ-, ТГП-, ТГТ- и
ПТГТ-кривых [35].
1 — прессованный исследуемый образец; 2 — прессованное эталонное вещество; 3 — печь; 4 —
колокола из кварца; 5— входной патрубок для газа-носителя; 6 — трубка для отбора газа;
7, 8 — трубка из кварца; 9 — термоэлемент; 10 — диафрагмы; 11 — фотоэлемент; 12— лампы;
13 — оптическая щель; 14 — магнит; 15— катушка; 16— гальванометр; 17-—фотобумага;
18 — трансформатор; 19 — абсорбер; 20— электроды; 21 — усилитель; 22 — к вакуумному на-
k сосу; 23 — автоматическая бюретка; 24—потенциометр; 25 — сервомотор.
последним к поглотительным растворам, где они титруются соот-
ветствующим реагентом. Количество использованного титрован-
ного раствора регистрируется в функции времени с помощью по-
тенциометра, соединенного с автоматической бюреткой.
г На фиг. 8.18 приведены кривые, полученные на этом приборе
при анализе газообразных веществ, образовавшихся при разложе-
нии [Cu(NH3)4]SO4-H2O. В интервале температур 90—200°С выде-
ляется 1 моль воды и 2 моль аммиака. По кривой отчетливо видно,
что аммиак выделяется одновременно с водой, причем этот процесс
не мог быть обнаружен при использовании только методов ТГ и
ТГП. Затем происходит ступенчатое выделение аммиака, и при
Обнаружение и анализ выделенного газа
349
цдглснии четвертого моля также образуется трехокись серы вслед-
fiiic разложения сульфата меди. Данный прибор был применен и
Ф" изучении термического разложения MnNHiPO4-H2O [32];
дг/с
о
ДТА
dm мг_
dt 'мин
dm мп
Дж, %
Cu(NH3)4SO4-H2O0
Си (NHabSO, _
10
NH3
cunh3so,i
CuS04
Cu(NHj)2SO4 _ 2Q ____
ДИ
h2o+nh3 у \у н2о
Температура, °C
Фиг. 8.18. Дериватографические и термо-газотитриметрические кривые раз-
ложения [Cu(NHs)4]SO4-Н2О [31].
lNi(NH3)e]Cl2 [33]; ZnNHiPCh [33]; CuSO4-5H2O [34]; основного
уксуснокислого железа [34]; глюкозы [34]; фруктозы [34]; лакто-
зы [34] и МпСО3-пН2О 135].
3. ИНФРАКРАСНАЯ СПЕКТРОСКОПИЯ
В течение длительного времени инфракрасную спектроскопию
использовали для анализа потоков газа в химических промышлен-
ных процессах. В последнее время с появлением быстродействую-
щих инфракрасных спектрометров их стали применять в качестве
детекторов в газовых хроматографах. Ясно, что спектрометры могут
быть использованы только в том случае, если соединение имеет
350
Глава 8
по меньшей мере одну полосу поглощения в инфракрасной обл.к i и
Выделившиеся из образца газы с помощью газа-носителя подаю к н
из пиролизной камеры в кювету инфракрасного анализатора. I !
пользуемые системы инфракрасной спектроскопии могут со'о р
жать: а) недиспергирующий анализатор; б) обычный спектромшр,
Фиг. 8.19. Кривые выделения воды (или С2Н2) при дегидратации CuSO.-
• 5Н2О [37].
Количество С2Н2 пропорционально концентрации воды. Массы образцов; 1 — 60 Мг; 2— 40 мс |
5 — 20 мг.
в котором диспергируется ИК-излучение; в) прибор типа полосо-
вого фильтра и г) интерференционный спектрометр. Все эти при-
боры достаточно подробно рассмотрены в обзоре Лоу [36].
Кисс [37, 38] объединил термовесы Шевенара со спектрофото-
метром типа UR 10; выделенные газы подавались из термовесов в
кювету спектрофотометра длиной 10 см. Был разработан метод оп-
ределения содержания аммиака или воды (по образованию С2Н2)
в выделенных газах. Содержание воды не измерялось непосредст-
венно, так как присутствие водородных связей в молекуле значи-
тельно снижало чувствительность измерения поглощения. Однако
при пропускании выделенной воды через карбид кальция образо-
вывался ацетилен, который обнаруживали по полосе поглощения
728 см-1. В качестве примера на фиг. 8.19 приведены кривые выде-
ления воды из CuSO4-5H2O. Из других соединений были исследова-
ны парамолибденовокислый аммоний и паравольфрамовокйслый
аммоний. В более позднем исследовании с помощью данного мето-
да определялся состав двойных смесей аммиака и воды [38].
Обнаружение и анализ выделенного газа
351
4. ТЕРМИЧЕСКИЙ АНАЛИЗ ПО ВЫДЕЛЕННЫМ
ЧАСТИЦАМ (ТАЧ)
Термический анализ по выделенным частицам заключается в
обнаружении частиц в выделенных газах в зависимости от темпера-
1уры. В присутствии пересыщенного водяного пара частицы прев-
ращаются в ядра конденсации для воды и могут быть обнаружены
Вход газа
Вход
азота
Фиг. 8.20. Схема прибора ТАЧ [41].
методом рассеяния света. Капли конденсата на частицах быстро
увеличиваются и достигают размеров, достаточных для рассеяния
света. Интенсивность рассеянного света, регистрируемая электро-
вакуумным фотоэлементом темнопольной оптической системы, про-
порциональна числу первоначально образовавшихся ядер конден-
сации. Чувствительность этого метода очень высока — одна часть
вещества в 10ls частях воздуха. Метод был впервые использован
Дойлем [39] и подробно рассмотрен в обзорах Мэрфи и др. [40, 41].
На фиг. 8.20 дана общая схема прибора ТАЧ. Образцы полиме-
ра размером 25x3x0,35 мм нагревали в медной трубчатой печи
с заданной скоростью 50 град/ч. Над образцом пропускали водород
с расходом 3 мл/с, но поскольку требуемый расход газа для счет-
чика ядер конденсации составлял 100 мл/с, после печи и теплооб-
352
Глава 8
менников в газовую среду дополнительно вводился азот. В табл
8.2 указаны некоторые вещества, исследованные данным методом
Таблица 8.'J
Температуры разложения полимеров, полученные методом ТАЧ [40]
Полимер Атмосфера Скорость нагревания, град/ч Температуря разложенн я, °C
Полиметилметакрилат Н2 50 310-320
Воздух 50 242
» 180 237
Полистирол н2 50 337
Полистирол с наполнителем SiO2 . . н2 50 337
Фенолформальдегидная смола .... Н2 50 432
Полихлортрифторэтилен N2 180 340
Фталоцианин меди n2 180 230
Цинк-4,4' -бпс-тиопиколинамидоди- бензофенон n2 180 255
Воздух 180 350
Мэрфи [40] описал конвертер, создающий ядра конденсации из
реагента, который обычно не обнаруживается методом ТАЧ. На-
пример, аммиак можно обнаружить при пропускании его через
колбу с небольшим количеством соляной кислоты. Газообразный
НС1, находясь над раствором, вступает в реакцию с аммиаком, об-
разуя ядра конденсации из хлорида аммония. В табл. 8.3 указа-
Таблица 8.3
Газы, обнаруживаемые методом ТАЧ [40]
Соединение Процесс превращения Легко обнару- живаемая концентрация, ю-« %
Аммиак Кислотно-основный 0,005
Бензол Фотохимический 2
Двуокись углерода Электрохимический 5
Окись углерода Химический 1
Хлор » 1
Этиловый спирт Обратимый фотохимический . . 5
Фреон 12-21 Пиролиз 2
Хлористый водород Кислотно-основный 0,5
Углеводород Фотохимический 0,1 х
Метил меркаптан Окислительно-фотохимический 0,01
Этиламин Кислотно-основный 0,5
Ртуть Фотохимический 0,01
Двуокись азота Г идролиз 0,5
Обнаружение и анализ выделенного газа
353
I НМ другие газы и используемые процессы их химического превра-
|цспия. Метод ТАЧ является непрерывным и позволяет производить
I инилиз газа при использовании приборов для химического превра-
щения путем обнаружения ядер конденсации. Это единственный
Метод, предоставляющий такую возможность.
5. ТОНКОСЛОЙНАЯ ХРОМАТОГРАФИЯ (ТСХ)
|Хотя тонкослойная хроматография и не находит широкого при-
менения, она используется иногда для анализа выделенного газа,
I также для кинетических исследований. Конечно, обычные газы
(IC могут быть исследованы этим методом, но соединения с высоким
I Молекулярным весом, которые трудно идентифицировать другими
способами, могут быть разделены и охарактеризованы с помощью
'ICX. Следует также отметить, что оборудование для ТСХ значи-
К'льно дешевле, чем для других методов анализа.
В методе, применяемом Роджерсом [42, 431, образец нагревает-
(' Си в потоке газа-носителя с заданной скоростью или в изотермиче-
ких условиях. Поток газа-носителя, содержащий продукты раз-
ложения, ударяется о поверхность активированной пластины ТСХ.
Пластина перемещается у отверстия выхода потока из пиролизной
i Кимеры в зависимости от температуры образца. Следовательно,
В любое положение вдоль зоны столкновения пластины с потоком соот-
ветствует определенной температуре образца. Пластина затем обра-
батывается обычными способами хроматографии для выделения
' Отдельных продуктов реакции. После окончательной обработки
f Пластина содержит данные двух типов по составу и соответствую-
щей ему температуре.
Результаты хроматографического анализа тринитротолуола
(ТНТ) [42] и соответствующая кривая ДТА показаны на фиг. 8.21.
Тит и летучие примеси начинают испаряться и обнаруживаются на
пластине ТСХ в интервале температур 125—135° С, соответствую-
щем первому появлению газа на пиролизной кривой. Основная
Часть ТНТ испаряется и накапливается, не разлагаясь, так как это
соединение обладает относительно высокой термической стабиль-
ностью. В интервале температур, в котором происходит экзотерми-
ческое разложение ТНТ (отражающееся на кривой ДТА), появля-
ются следующие вещества: 2,4,6-тринитробензиловый спирт, 4,6-
динитроантранил, 1,3,5-тринитробензол, 2,4,6-тринитробензойная
кислота, а также следы неидентифицированного соединения. Со-
четание точности метода ТСХ и характерной окраски распыляемо-
ю реагента позволяет сравнительно просто идентифицировать все
основные компоненты.
Роджерс и Смит [43] применили метод ТСХ для исследования
Кинетики реакций.
Сталь [44] описал процесс пиролиза, в котором для идентифика-
ции продуктов разложения также использовался метод ТСХ. Обра-
354
Глава 8
Фиг. 8.21. Графическое представление данных термического разложении
тринитротолуола (ТНТ) (42].
1 — кривая ДТА; 2 — кривая пиролиза; 3 — зона ТНТ; 4 — зона 2,6-ДНТ и 3,5-ДНТ; 5 - .пин»
2,4-ДНТ; 6 — зона 1,3,5-тринитробензола; 7— зона 4,6-дннитроаитранила; 8 — зона 2,4,6- г|>и
нитробензилового спирта; 9— зона неиде^тифицироваиного соединения; 10 — зона 2,4,6-трппи
тробензойной кислоты. Вес образца 0,284 мг; скорость нагревания 11 град/мин; газ-носите л I.
воздух.
зец помещали в стеклянный патрон с коническим наконечником и
быстро нагревали на короткий промежуток времени. Выделенные
газы осаждались в виде пятна на пластине ТСХ, которая затем <><>
рабатывалась соответствующими реактивами. Автор рассмотрел
несколько вариантов процесса и продемонстрировал его примет'
ние для анализа лекарств, добавок к пищевым продуктам, осадком
и реагентов в фотохимии.
Свое исследование рассматриваемого метода Роджерс [42) за
ключил предположением, что этот метод найдет наиболее широкое
применение при изучении полимеров. Его преимущества наиболее
полно проявляются при исследовании больших молекул с повторяю
щимися звеньями, а не при анализе простейших конечных иродук
тов пиролиза. Поскольку в этом методе не используются датчики
Обнаружение и анализ выделенного газа
355
теплопроводности, ионизации в пламени и др., нет необходимости
в особых газах-носителях. Процессы и взаимодействия могут иссле-
доваться в любой среде газа или пара, например в кислороде, воз-
духе, азоте, гелии, водороде, двуокиси углерода или в смесях, со-
держащих водяные пары, кислоты, аммиак и т. д.
6. МЕТОД ИОНИЗАЦИИ В ПЛАМЕНИ
Детектор ионизации в пламени (ДИП) был использован Эггерт-
сеном и др. 145—49] для обнаружения углеродсодержащих соедине-
ний в газообразных продуктах пиролиза. Прибор состоит из не-
большой печи для образца, объединенной с детектором ионизации
в высокотемпературном пламени. Поток газа-носителя переносит
Фиг. 8.22. Схема детектора ионизации в пламени [49].
/ — коллектор; 2 — нагреватель; 3 — электрометр; 4 — печь; 5— нагреватель; 6— сдвоенный са-
мописец; 7 — датчик; 8 — регулятор температуры.
выделенные газы из пиролизной камеры печи к детектору. Основные
блоки прибора показаны на фиг. 8.22. Печь и сопло детектора объе-
динены в один узел, изготовленный из стеклянных трубок (стекло'
викор). Такая конструкция и высокая температура пламени (обыч-
но 500° С) позволяют получить все летучие продукты. Печь нагре-
вается электрическим нагревателем, температура которого регу-
лируется с высокой точностью по заданной программе. Сопло на-
гревается с помощью блока из нержавеющей стали. Для удобства
калибровки детектора предусмотрена возможность вдува образцов,
эталонного газа через герметичный ввод с помощью шприца. Ло-
дочка для образца, изготовленная из алюминия, золота или плати-
ны, закреплена на проволочной рамке из нержавеющей стали, при-
крепленной к термопаре в защитной оболочке.
356
Глава 8
Метод ДИП обладает высокой чувствительностью по сравнению
с ТГ-методом (табл. 8.4). Масса образцов, используемых в этом
Таблица 8.4
Сравнение по чувствительности методов ДИП и ТГ
ТГ Дип
Масса образца 5—100 0,1—5
Чувствительность . . . 10—40 мг 0,01 мкг/ми н
Полиэтилен
т5п 456СС 458°С
Ло 430'С 424°С
Т5 418°С 419ГС
%/мин
0,1 — 370°С
0,02 — 330°С
0,01 — ЗОО'С
методе, меньше, чем при термогравиметрии. Скорости разложения,
определяемые с помощью метода ДИП, составляют обычно
0,01 %/мин, а нижний предел—0,001 %/мин. Минимальный предел
чувствительности имеет порядок 10"4 мкг углерода в минуту. Одна-
ко, как показано в более ранней работе по термической стабиль-
ности полимеров [46], такая высокая чувствительность едва ли не-
обходима, поскольку выделение газа наблюдалось даже в пустой,
хорошо очищенной системе (прогретой в среде воздуха при темпе-
10 20 30 40 50
Время, мин
Фиг. 8.23. Кривая ДИП для композиции на основе поливинилхлорида [49],
Масса образца 9,4 мг; скорость нагревания 10 град/мин; 79 °C —- пестицид; 179°С — пласти-
фикатор; 275°С— стабилизатор; 438°С—поливинилхлорид.
Обнаружение и анализ выделенного газа
357
Ви гуре 550° С). Высокая чувствительность детектора позволяет по-
дучить вполне приемлемые кривые при анализе небольшого коли-
Ьства вещества (нескольких микрограммов). Измерения давления
й|><1 могут быть произведены при уровне давления ниже 0,1 Па
иг>|.
Типичная кривая, полученная при помощи метода ДИП, при-
едена на фиг. 8.23. Как видно по пикам кривой, исследованная
Ьмпозиция на основе поливинилхлорида содержала четыре ком-
Иоиспта: пестицид, пластификатор, стабилизатор и поливинил-
[ |Лорид.
[ Рассматриваемый метод позволяет также определять следы орга-
нических соединений углерода в воде [48]. Для этого образец на-
гревается в среде газа-носителя (азота) в два этапа — для опреде-
Таблица 8.5
Определение содержания углерода в разбавленных растворах [48]
| Образец Содержа- ние угле- рода, 10~*% Коэффи- циент разбавле- ния1) Содержание да, 1 экспери- ментальное значение углеро- о~*% расчетное значение’)
’»КС ус на я кислота 428 0 10 50 90 10,3 2,1 9,0 1,8
I сптановая кислота 91 0 5 53 10,0 10,6
]Нсодол 25-12 213 0 10 50 72 5,9 1,4 7,2 1,4
Тпзойль (получен при каталитичес- ком крекинге) 0 10 50 24 2,7 0,8; 0,6 2,4 0,5
Крахмал 158 0 5 14 1,7 2,8
Желатин 90 0 5 14 3,4 2,8
*) К образцам добавлялась дистиллированная вода непосредственно в лодочке для
анализа.
£) Эти значения были получены расчетным путем по результатам для неразбавленных
пЛрпзцов с учетом коэффициента разбавления.
358
Глава 8
ления летучих (<15ОСС) и нелетучих (150—550° С) органическим
соединений углерода. Испарение воды на первом этапе до некотороИ
степени изменяет чувствительность детектора, ио этот недостапн»
легко устраняется соответствующей калибровкой прибора. Ни/к
ний предел чувствительности детектора составляет ~2-10“5%. I'i
зультаты анализа содержания углерода в различных органических
соединениях приведены в табл. 8.5. Этот метод особенно эффект
вен при определении следов органических веществ, которые упг
тучиваются или перегоняются с водяным паром в условиях экси<-
римента. Исследование нелетучих веществ не всегда дает удовле!
верительные результаты, особенно в случае природных органшн
ских материалов, например углеводов и белков. В некоторых
случаях, однако, этот недостаток может быть устранен соотве rc i
вующей калибровкой.
7. МАСС-СПЕКТРОМЕТРИЯ (МС)
Вероятно, наиболее широкое применение при анализе выделен
ных газов находит масс-спектрометрия. Масс-спектромеры обеспс
чивают четкую идентификацию газов, выделившихся в процессе
термического воздействия, и во многих случаях позволяют опрел*
лить количественное содержание каждого компонента. Наряду с
другими методами термического анализа, например ТГ и ДТА,
Фиг. 8.24. Системы термического анализа, в которых применяется масс-
сиектрометрия.
Обнаружение и анализ выделенного газа
359
мисс-спектрометрия является, по-видимому, наиболее совершен-
ным методом исследования реагирующей системы.
Системы термического анализа, в которых используется масс-
спектрометрия, показаны схематически на фиг. 8.24. Выделяющиеся
газы из пиролизной камеры, прибора ДТА или термовесов могут
непосредственно вводиться в масс-спектрометр. Если выделенный
газ является сложной смесью соединений, то для предварительного
разделения можно использовать газовый хроматограф, а затем
масс-спектрометр. Вследствие большого объема и разнообразного
характера данных, получаемых при использовании масс-спектро-
метра, может потребоваться большая ЭВМ для обработки данных
пли небольшая ЦВМ для контроля за работой прибора и обработки
поступающих с него данных. Применение масс-спектрометрии в
термическом анализе подробно рассмотрено Фридманом 150], а
также Ланжером и Голке [51].
а. Совмещенный метод МС—АВГ
Простейший случай применения масс-спектрометра для анали-
за выделенных газов — это нагревание образца с постоянной ско-
ростью в соответствии с заданной программой в вакуумной камере
масс-спектрометра. Анализ выделенных газов производится не-
прерывно в процессе сканирования по массе. Регистрация ведется
путем построения графика зависимости ионного тока от времени или
температуры для каждой рассматриваемой массы ионов, либо путем
построения графика зависимости одной массы ионов от времени
или температуры пиролиза. Оба эти метода находят широкое приме-
нение [50].
В 1963 г. Ланжер и Голке [521 впервые применили новый метод
МС—АВГ для анализа выделяющегося при нагревании газа. В раз-
работанной ими установке образец нагревался небольшой печью
в вакуумной камере времяпролетного масс-спектрометра. Запись
масс-спектра продуктов разложения производилась через заданные
интервалы времени. Исследовались BeSO4-4H2O, CaSO4-2H2O,
CuSOi-5H2O и другие соединения.
Фридман [50] первоначально планировал использовать совме-
щенный метод МС—АВГ для получения дополнительных данных
для кинетического анализа, выполняемого ТГ-методом, но пришел
к выводу, что масс-спектрометрия дает значительно лучшие резуль-
таты при анализе сложных смесей газов, чем ТГ. В его установке
пиролиз производился в трубчатой печи, расположенной на рас-
стоянии ~1 м от электронного пучка масс-спектрометра. Проводить
пиролиз можно было после предварительного обжига трубчатой
печи при очень высоких температурах. В основном были исследова-
ны полимеры типа полибензимидазолов, полиэфиров, полиамидов
и др.
360
Глава 8
Шульман [531, а также Шульман и Лохте [544 55J использовали
аналогичную систему, в которой времяпролетный масс-спектро-
метр применялся для анализа газов, выделяемых из ячейки Кнуд-
сена. Другие совмещенные системы МС—АВГ описаны Остином и
др. [56], Брауном и др. [57], Греем и др. [58], а также Голином и
Др. [59].
Фиг. 8.25. Прибор, разработанный Уэндландтом и Саутерном, для измере-
ний методом ОВГ—МС [60].
а — схема прибора; б — пиролизная камера; MS — масс-спектрометр; R — самописцы; S —
образец; FU — печь; OU — выход. 1 — печь; 2 — термопара; 3 — образец; 4 — блок образца.
Обнаружение и анализ выделенного газа
361
б. Совмещенный метод ОВГ—МС
Уэндландт и Саутерн [601 описали прибор для одновременного
получения кривых ОВГ и МС образца. Этот прибор (фиг. 8.25)
позволяет производить нагревание образца при атмосферном дав-
лении в инертных или каких-либо других средах вместо обычно
Фиг. 8.26. Кривые ОВГ и МС для [Cu(NHs)4]SO4- НгО [60].
применяемого в масс-спектрометрии вакуума [52]. Пиролизная ка-
мера состоит из трубчатой печи, нагреваемой при использовании
программирующего устройства — регулятора температуры. Вы-
деленные газы уносятся из пиролизной камеры потоком газа-носи-
теля (обычно гелия) и регистрируются датчиком теплопроводнос-
ти. По короткой капиллярной трубке из нержавеющей стали часть
выделенных газов подается к масс-спектрометру для анализа. Масс-
спектрометр может быть использован для записи т/е в функции
температуры или для регистрации полного масс-спектра через оп-
ределенные интервалы времени.
Данные, полученные на этом приборе, иллюстрируются на при-
мере термического разложения [Cu(NH3)dSO4-H2O (фиг. 8.26).
Метод термогравиметрии не позволяет получить ответа на вопрос,
какое вещество — вода или аммиак — выделяется раньше в на-
чальной стадии термического разложения, поскольку молекуляр-
ные массы этих веществ различаются незначительно (соответствен-
но 18 и 17). Применение масс-спектрометрии легко разрешает во-
прос. Как видно из фиг. 8.26, кривая ОВГ имеет пять пиков — при
температурах 145, 175, 285, 380 и 460е С. Кривая МС обеспечивает
интерпретацию этих данных с помощью следующих реакций:
362
Глава 8
[Cu(NH3)4]SO4 - Н2О (т.) -> [Cu(NH3)4]SO4 (т.) + Н2О (г.),
[Cu(NH3)4]SO4 (т.) -> Си (NH3)2SO4 (т.) + 2NH3 (г.),
Cu(NH3)2SO4 (т.) -> Cu(NH3) S04 (т.) + NH3 (г.),
Cu(NH3) SO4 (т.) -> CuSO4 (т.) + NH3 (г.),
2CuSO4 (т.) -> СиО CuSO4 (т.) + SO2 (г.) + О2 (г.).
в. Совмещенный метод ДТА—ОВГ—МС
Возможности масс-спектрометрии возрастают при введении еще
одного термического параметра — данных ДТА. Ланжер и др.
[61], а также Голке и Ланжер [62] первыми использовали модифп
цированную камеру ДТА от промышленного прибора в совмещен
ной установке ДТА—ОВГ—МС. Через определенные интервалы
времени выделенные газы подавались для анализа в масс-спектро
метр. Позднее к установке была добавлена внутренняя камера
ДТА для работы в высоком вакууме [63].
Уэндландт и др. [64] изменили пиролизную камеру в своей
системе МС—ОВГ, с тем чтобы наряду с измерениями двумя други
ми методами производить измерения методом ДТА.
Другие системы ДТА—ОВГ—МС описаны Редферном и др. [651.
а также Голином и др. [59].
г. Совмещенные методы ТГ—ОВГ—МС
и ТГ—ДТА—ОВГ—МС
Первое сообщение о совмещении термовесов и масс-спектромет-
ра появилось в работе Зитомера [66], который объединил термо-
весы фирмы «Дюпон» с времяпролетным масс-спектрометром. Он
применил этот прибор для исследования термического разложения
полиметиленсульфида и сополимера малеиновокислого гидразина
и метилвинилового эфира. Уильсон и Хэмейкер [67] соединили вы-
соковакуумные термовесы с квадрупольным масс-спектрометром.
В качестве источника тепла они использовали кварцевые лампы
с заданной скоростью нагревания 10 град/мин до максимальной
температуры 435° С. Исследовалось термическое разложение полн-
метилметакрилата, полиоксиметилена, полистирола и других
полимеров.
По-видимому, наиболее совершенной системой для измерении
ТГ—ДТА—ОВГ—МС-методом является система, включающая тер-
мовесы фирмы «Меттлер» и квадрупольный масс-спектрометр,
которая была описана Видеманном [68], а также Гебсоном и Джон-
Обнаружение и анализ выделенного газа
363
niiuM 169]. Схема этого прибора приведена на фиг. 8.27. Образец
Имсег исследоваться как в вакууме (~0,001Па), так и при давле-
нии до 1 атм (~0,1 МПа). Реакционная камера / размещена в печи
И отделена от термовесов диффузной перегородкой. Выделенные
fUhir. 8.27. Система, состоящая из термовесов фирмы «Меттлер» и квадруполь-
ного масс-спектрометра [68, 69].
Гизы поступают непосредственно к анализатору массы 2, соединен-
ному через контрольную панель масс-спектрометра с самописцем
|Л Общее давление определяется по датчику ионизации 4, который
Иикже используется для получения кривой ОВГ (по изменению
давления в системе). Связь между измеренным общим давлением
и ионным током эталонного газа позволяет провести калибровку
Масс-спектрометра в единицах абсолютного парциального давления
Или в А/мм рт. ст.
ТГ- и МС-кривые для CaC2Oi-H2O, полученные с помощью этого
прибора, приведены на фиг. 8.28. Образец нагревался в высоком
вакууме; в процессе нагрева парциальное давление непрерывно
регистрировалось масс-спектрометром. Величины молекулярной
массы 18 (Н2О+), 28 (СО* и N*) и 44 (СО*) периодически регистриро-
Ьались самописцем. В ходе процесса требовалось изменять чувст-
вительность прибора из-за быстрого возрастания количества вы-
деляющихся СО и СО2.
364
Глава 8
Гибсон и др. [69—72] применили описанную выше аппаратуру
для исследования выделения газов из геохимических образцов h
образцов лунного грунта. Примером этих исследований могут слу
жить приведенные на фиг. 8.29 ТГ—МС-кривые для образца луп
него грунта, доставленного космическим кораблем «Аполлон-15»,
Вся обработка масс-спектроскопических данных была выполнен»
мини-ЭВМ.
Фиг. 8.28. Термическое разложение СаСаО4- НаО, описанное Видеман-
ном [68].
Смит и Джонсон [73, 74] описали многоцелевой прибор, поз-
воляющий одновременно получать ТГ-, ДТА-, ОВГ- и МС-кривые
образца. Прибор использовался для исследования горючих слан-
цев. Ченг и Мид [75] описали установку, включающую термовесы,
газовый хроматограф и масс-спектрометр с высокой разрешающей
способностью, и рассмотрели ее применение для анализа термо-
деструкции полимеров. На фиг. 8.30 приведены три кривые для
образца пенополистирола, полученные на этой установке [751:
а) ТГ-кривая для образца весом 3 мг, нагреваемого со скоростью
15 град/мин в атмосфере гелия, б) хроматограмма выделенных га-
зов и в) масс-спектр низкого разрешения участка хроматограммы,
включающего пик № 15. Выделенные газы представляют co6oi’i
co,n2
Обнаружение и анализ выделенного газа 367
полученные на одной
>иг. 8.30. Кривые для образца пенополистирола,
установке [75].
ми н примерно 20 компонентов, 16 из которых были легко иденти-
I'liii прованы с помощью масс-спектрометра с высокой разрешаю-
Hiri'l способностью.
8. ТЕРМОБЛРОГРАВИМЕТРИЯ (ТБГ)
Метод термобарогравиметрического анализа был впервые вве-
и п в 1965 г. Бэнкрофтом и Гессером 178], разработавшими прибор
Ain непрерывного измерения массы, давления и температуры.
Бог прибор, обеспечивающий наилучшие результаты при невысо-
кие начальных давлениях и температурах до 800° С, был исполь-
iiiH.ui для изучения термического разложения броматов металлов.
Более совершенная аппаратура для термобарогравиметрии опи-
oiiia Мэйкоком и Бай Вернекером [79], которые измеряли измене-
ние давления в системе термоанализатора фирмы «Меттлер» с по-
мощью термопары и датчика давления «Баратрон». Схема прибора
||>ш>едена на фиг. 8.31. Датчик «Баратрон» имеет чувствительность
in :| мм рт. ст. (~0,13 Па) и применяется до давления 760 мм рт. ст.
Фиг. 8.31. Прибор Мэйкока и Вернекера для термобарогравиметрических
измерений [79].
[механический насос; 2 — весы; 3—-диффузионные насосы; 4—вакуумная ловушка
с жидким азотом; 5— датчик «Баратрон».
100 кПа). Пример данных, получаемых с помощью этого прибо-
ра, приведен на фиг. 8.32. Одновременные измерения уменьшения
массы образца и увеличения общего давления газообразных про-
дуктов диссоциации производились с шагом 10° С в интервале тем-
ператур 80—150° С. Эти данные позволили получить информацию
о процессах возгонки и разложения перхлората нитрония. Кроме
ц>го, Мэйкок с соавторами исследовали с помощью этого метода
'нрывчатые вещества [80] и перхлорат аммония [81].
368
Глава 8
Обнаружение и анализ выделенного газа
369
Фиг. 8.32. Одновременное измерение уменьшения массы, увеличения дав»
ления и профиля температуры для перхлората нитрония [79].
IN.
1*1
VI
I П
н.
II
iS.
|(>
ЛИТЕРАТУРА
1. Stone R. L., Anal. Chem., 32, 1582 (1960).
2. Lodding W., Hammell L., Anal. Chem., 32, 657 (1960).
3. Rogers R. N., Yasuda S. K., Zinn J., Anal. Chem., 32, 672 (1960)
4. Vassallo O. A., Anal. Chem., 33, 1823 (1961).
5. Ayres W. M., Bens E. M., Anal. Chem., 33, 568 (1961)
6. Gordon S., Campbell C., Anal. Chem., 27, 309 (1955).
7. Wendlandt W. W., Anal. Chim. Acta, 27, 309 (1962).
8. Wendlandt W. W., Sturm E., J. Inorg. Nucl. Chem., 25, 535 (1963).
9. Murphy С. B., Hill J. A., Schacher G. P., Anal. Chem., 32, 1374 (1960)
10. Thomas J. Hieftje G. M., Orlopp D. E., Anal. Chem., 37, 762 (1965)
11. Ware R. K-, Thermochim. Acta, 3, 49 (1972).
12. Wendlandt W. W., Dosch E. L., Thermochim Acta., 1, 103 (1970).
13. Wendlandt W. W., Bradley W. S., Thermochim. Acta, 1, 143 (1970).
14. Ingraham T. R., Proc, of the Second Toronto Symp. on Thermal Analysis,
McAdie H. G., ed., Chemical Institute of Canada, Toronto, 1967, p. 21.
15. Wist A. O., Thermal Analysis, Schwenker R. F., Garn P. D., eds. Vol. 2,
Academic Press, N. Y., 1969, p. 1095.
16. Garn P. D., Taianta, 11, 1417 (1964).
17. Findeis A. F., Rosinski K. D. W., Petro P. P., Earp R. E. W. Thermo-
chim. Acta, 1, 383 (1970).
.in,
;i'i.
4<>.
II.
-12.
43.
44.
-15.
4b.
Ingraham T. R., Fraser D., Proc, of the Third Toronto Symp. on Thermal
Analysis, McAdie A. G., ed., Chemical Institute of Canada, Toronto, 1969,
p. 101.
('hiti J., Anal. Chem., 40, 1516 (1968).
Chiu J., Thermochim. Acta, 1, 231 (1970).
Wiedemann H. G., Thermal Analysis, Schwenker R. F., Garn P. D., eds.,
Vol. 1, Academic Press, N. Y., 1959, p. 229.
Chang T. L., Mead T. E., Anal. Chem., 43, 534 (1971).
Wist A. O., J. Cas. Chromatog., March, 157 (1967).
Dorsey G. A., Anal. Chem., 41, 350 (1969).
Franc J., Pour J., Anal. Chim. Acta, 48, 129 (1969).
Sarner S. F., Pruder G. D., Levy E. J., Am. Lab., Oct., 57 (1971).
Franc J., Pour J., J. Chromatog., 32, 2 (1968).
Bollin E. M., Thermal Analysis, Schwenker R. F., Garn P. D., eds., Aca-
demic Press, N. Y., 1969, p. 255.
Paulik J., Paulik F., Thermochim. Acta, 4, 189 (1972).
Paulik J., Paulik F., Erdey L., Mikrochim. Acta, 886 (1966).
Paulik J., Paulik F„ Taianta, 17, 1224 (1970).
Paulik J., Paulik F., Proc, of the Third Analytical Chemistry Conference,
Budapest, 1970, p. 225.
Paulik J., Paulik F., Proc, of the Third Analytical Chemistry Conference,
Budapest, 1970, p. 231.
Gal S., Simon J., Erdey L., Proc, of the Third Analytical Chemistry Con-
ference, Budapest, 1970, p. 243.
Paulik F., Paulik J., Thermochim. Acta, 3, 13, 17 (1971).
Low M. J. D., Gas Effluent Analysis, Lodding W., ed., Marcel-Dekker,
N. Y., 1967, Ch. 6.
Kiss A. B., Acta Chim. Acad. Sci. Hung., 61, 207 (1969).
Kiss A. B., Acta Chim. Acad. Sci. Hung., 63, 243 (1970).
Doyle C. D., WADD Tech. Rept. 60-283, U. S. Air Force, Wright-Patterson
Air Force Base, Ohio, May 1960.
Murphy С. C., Gas Effluent Analysis, Lodding W., ed., Marcel-Dekker,
N. Y., 1967, Ch. 7.
Murphy С. B., Van Luik F. W., Pitsas A. C., Plastics Design and Processing,
July, 16 (1964).
Rogers R. N., Anal. Chem., 39, 730 (1967).
Rogers R. N., Smith L. C., J. Chromatog., 48, 268 (1970).
Stahl E., Analyst, 723 (1969).
Eggertsen F. T., Stross F. H., J. Appl. Polymer Sci., 10, 1171 (1966).
Eggertsen F. T., Joki H. M., Stross F. H., Thermal Analysis, Schwen-
ker R. F., Garn P. D., eds., Academic Press, N. Y., 1969, p. 341.
4,'. Eggertsen F. T., Seibert E. E., Stross F. H., Anal. Chem., 41, 1175 (1969)
I 48. Eggertsen F. T., Stross F. H., Anal. Chem., 44, 709 (1972).
j-i. Eggertsen F. T., Stross F. H., Thermochim. Acta, 1, 451 (1970).
fill Friedman H. L., Thermochim. Acta, 1, 199 (1970).
I fil. Langer H. G., Gohlke R. S., Gas Effluent Analysis, Lodding W., ed.,
' Marcel-Dekker, N. Y., 1967, .Ch. 3.
52. Langer H. G., Gohlke R. S., Anal. Chem., 35, 1301 (1963).
I 63. Schulman G. P., Polymer Letters, 3, 911 (1965).
64. Schulman G. P., Lochte H. W., J. Appl. Polymer Sci., 10, 619 (1966).
65 Schulman G. P., Lochte H. W., J. Macromol. Sci. (Chem.), Al, 413 (1967).
6(i. Austin F., Dollimore J., Harrison B., Thermal Analysis, Schwenker R. F.,
Garn P. D., eds., Vol. 1, Academic Press, N. Y., 1969, p. 311.
В t>7. Brown J. G., Dollimore J., Freedman С. M., Harrison В. H., Thermochim.
Acta, 1, 499 (1970).
68 Gray D. N., Schulman G. P., Conley R. T., J. Macromol.
Al, 395 (1967).
Sci. (Chem.),
13—230
370
Глава 8
59. Gaulin С. A., Wachi F., Johnston T. H., Thermal Analysis, Schwcn
ker R. F., Garn P. D., eds., Vol. 2, Academic Press, N. Y., 1969, p. 14Ы1
60. Wendlandt W. W., Southern T. M., Anal. Chim. Acta, 32, 405 (1965).
61. Langer H. G., Gohlke R. S., Smith D. H., Anal. Chem., 37, 433 (19(>l>)
62. Gohlke R. S., Langer H. G., Anal. Chem., 38, 530 (1966).
63. Langer H. G., Brady T. P., Thermal Analysis, Schwenker R. F., Garn P. 1> ,
eds., Vol. 1, Academic Press, N. Y., 1969, p. 295.
64. Wendlandt W. W., Southern T. M., Williams J. R., Anal. Chim. Achi,
35, 254 (1966).
65. Redfern J. P., Treherne B. L., Aspimal M. L., Wolstenholme W. A., 1711»
Conference on Mass Spectrometry and Allied Topics, Dallas, Texas, May
1969.
66. Zitomer F., Anal. Chem., 40, 1091 (1968).
67. Wilson D. E., Hamaker F. M., Thermal Analysis, Schwenker R. I ,
Garn P. D., eds., Vol. 1, Academic Press, N. Y., 1969, p. 517.
68. Wiedemann H. G., Thermal Analysis, Schwenker R. F., Garn P. D., eds.,
Vol. 1, Academic Press, N. Y., 1969, p. 229.
69. Gebson E. K-, Johnson S. M., Thermochim. Acta., 4, 49 (1972).
70. Gibson E. K., Thermochim. Acta, 5, 243 (1973).
71. Gibson E. K., Johnson S. M., Proc, of the Second Lunar Science Confereii
ce, Vol. 2, MIT Press, Cambridge, Mass., 1971, p. 1351.
72. Gibson E. K., Moore G. W., Thermochim. Acta (в печати).
73. Smith J. W., Johnson D. R., Thermal Analysis, Schwenker R.
Garn P. D., eds., Vol. 2, Academic Press, N. Y., 1969, p. 1251.
74. Smith J. W., Johnson D. R., Am. Lab., Jan., 8 (1971).
75. Chang T. L., Mead T. E., Anal. Chem., 43, 534 (1971).
76. Lodding W., Gas Effluent Analysis, Marcel-Dekker, N. Y., 1967.
77. Kenyon A. S., Techniques and Methods of Polymer Evaluation, Marcel
Dekker, N. Y„ 1966, Ch. 5.
78. Bancroft G. M., Gesser H. D., J. Jnorg. Nucl. Chem., 27, 1537 (1965).
79. Maycock J. N., Pai Vernecker V. R., Anal. Chem., 40, 1935 (1968).
80, Maycock J. N., Pai Vernecker V. R., Thermochim. Acta, 1, 191 (1970).
8L Pai Vernecker V. R., McCarty M., Maycock J. N., Thermochim. Acta, 3, 37
(1971),
Глава 9
СПЕКТРОСКОПИЧЕСКИЕ, ФОТОМЕТРИЧЕСКИЕ
И ОПТИЧЕСКИЕ МЕТОДЫ ТЕРМИЧЕСКОГО АНАЛИЗА
А. ВЫСОКОТЕМПЕРАТУРНАЯ И ДИНАМИЧЕСКАЯ
СПЕКТРОСКОПИЯ ОТРАЖЕНИЯ
1. ВВЕДЕНИЕ
Измерение излучения, отраженного от матовой поверхности,
|рсдставляет собой область спектроскопии, известной как спектро-
) метода с методом
(копия диффузного отражения. Сравнение этого
пектроскопии пропускания дано на фиг. 9.1.
Отраженное излучение определяется отноше-
нием ЦПГ, где Ц — интенсивность падающе-
ft), а 1Г — интенсивность отраженного из-
лучения. Отражение излучения возможно в
ультрафиолетовой, видимой и инфракрасной
областях электромагнитного спектра. Суммар-
ное излучение, отраженное от матовой по-
верхности, RT включает две составляющие:
Направленную составляющую R (иногда называ-
емую зеркальной) и диффузную составляющую
[1, 21. Первая составляющая наблюдается,
например, при отражении от поверхности мо-
нокристаллов, а вторая —- в результате про-
никновения излучения в тело и его выхода
После многократного рассеяния.
Согласно теории Кубелка и Мунка [3],
диффузное отражение от слоя порошкообразно-
го образца толщиной 1—3 мм (увеличение
Толщины сверх этого значения не оказывает
илияния на отражение) при заданной длине
9.1.
Фиг.
роскопия
кания (а)
троскопия
б
Спект-
пропус-
и спек-
отра-
жения (б).
полны определяется по формуле
1 - [fe/(fe + 2s)]1/2 ,
/о‘ l + [fe/(fe + 2s)]1/2
(9.1)
где I — интенсивность отраженного излучения; /0 — интенсив-
ность падающего излучения; k — коэффициент поглощения; s —
.коэффициент рассеяния. Коэффициент поглощения является тем
же самым, что и определяемый по известному закону Ламберта —
Бэра Т — е~ы. Зеркальное отражение определяется по формуле
13*
372
Глава 9
R _L = (П-1)2 + ^№ 9
Io (n+l)2 + «2№ ’ '
где n — показатель преломления и Л — показатель поглощения,
определяемый по закону Ламберта
, , / — 4т. Kd \ ...
/ = /0 ехр —------ . (9.3)
\ \> /
Здесь Хо — длина электромагнитной волны в вакууме и d — тол-
щина слоя.
После ряда алгебраических преобразований уравнение (9.1)
можно привести к более известной формуле
(1-^)2 = А (94)
2Яоо s' v ' ’
Левая часть этого уравнения обычно называется функцией ослаб
ления, или функцией Кубелка — Мунка, и обозначается /(Коо).
В экспериментах редко определяется абсолютная диффузная отра-
жательная способность образца. Чаще определяется относитель-
ная отражательная способность образца по сравнению с соответст
вующим белым эталоном. В этом случае в исследуемой области
спектра k = 0, К«>, этаЛ= 1 [из уравнения (9.4)1 и используется
соотношение ,
(9.5)
этал
Из соотношения (9.5) с помощью функции ослабления можно полу-
чить отношение k/s:
/(гто)=(-Ц_М1^ А. (9.6)
Взяв логарифм функции ослабления, получим
lg/(roo) = lg& —Igs. (9.7)
Таким образом, кривая зависимости 1g/ (гет) от длины волны, или
волнового числа, для данного образца должна соответствовать
спектру поглощения соединения со смещением по оси ординат на
величину —lg s. Кривые, полученные путем таких измерений отра-
жательной способности, называются обычно характеристическими
цветовыми кривыми или типичными цветовыми кривыми. В об-
ласти более коротких волн иногда наблюдается небольшое систе-
матическое отклонение (несовпадение со спектром поглощения)
из-за слабого возрастания коэффициента рассеяния.
При использовании современны^ двухлучевых спектрофото-
метров, оборудованных приставкой отражения, автоматически за-
Спектроскопические, фотометрические и оптические методы
373
писывается кривая зависимости гте от длины волны. Многие иссле-
(ователи строят кривую отражательной способности в процентах
(Д’, %) или используют табличную функцию ослабления (4]/(гоо)
пли k/s в зависимости от длины волны или волнового числа. По-
ппдимому, наиболее распространенным является первый метод
регистрации данных. ,
В приведенном здесь кратком введении в спектроскопию отра-
жения рассмотрены самые элементарные принцийы этого метода.
Как и следует ожидать, спектроскопия отражения находит наибо-
'и е широкое применение при изучении твердых или порошкообраз-
ных образцов, хотя она может быть также применена и при иссле-
довании жидких и пастообразных материалов. Метод дает возмож-
ность быстрого определения «цвета» образца и удобен для исполь-
зования, поскольку в продаже имеются соответствующие серийные
приборы. В связи с тем что поглощение и отражение падающего
н (лучения происходят только на поверхности образца, этот метод
широко используется при исследовании химии и физики поверх-
ностей. [5].
2. ВЫСОКОТЕМПЕРАТУРНАЯ СПЕКТРОСКОПИЯ
ОТРАЖЕНИЯ
Практически все исследования с применением спектроскопии
отражения (следует отметить, что этот термин относится здесь ис-
ключительно к спектроскопии диффузного отражения) проводятся
при температуре окружающей среды или в некоторых случаях при
более низких температурах. Последние, по-видимому, могут найти
применение в исследованиях монокристаллов для выяснения при-
чин возникновения «горячих полос», т. е. переходов, возникающих
при колебательном возбуждении основных состояний. Однако во
многих случаях довольно значительная дополнительная информа-
ция о химической системе может быть получена по спектру отраже-
ния при повышенных температурах. Обычно исследования проводят
при температурах 100—300° С, хотя допустимы и более высокие
температуры.
При исследованиях методом высокотемпературной спектроско-
пии отражения используют два способа измерений: первый — это
и .мерение спектра образца при различных постоянных, или изо-
шрмических, температурах; второй — измерение изменения отра-
жательной способности образца при повышении температуры. Пер-
цый способ является статическим и называется высокотемператур-
ной спектроскопией отражения (ВТСО) [6]; второй является дина-
мическим и называется динамической спектроскопией отражения
(ДСО) [7, 8]. Результаты, полученные с помощью этих двух мето-
дов, схематически представлены на фиг. 9.2. На фиг. 9.2,а приве-
дены кривые ВТСО, т. е. спектры образца, зарегистрированные
374
Глава 9
при возрастающих фиксированных температурах (от Ti до Та).
Как видно из фигуры, максимум кривых при длине волны Xj умень
шается с увеличением температуры с образованием нового макси-
мума при длине волны Х2. Путем измерения спектров при малых
б б
Фиг. 9.2. Кривые ВТСО (а) и кривые ДСО при длинах волн Xf (б) и Х2 (я)
II].
приращениях температуры можно определить минимальную тем-
пературу, при которой начинается термический переход. Более
точно температуры переходов определяются динамическим мето
дом (фиг. 9.2,6 и в). Как видно из фиг. 9.2,6, при построении отража-
тельной способности в зависимости от температуры образца,, уве-
личиваемой с небольшой заданной скоростью, при заданной длине
волны Х4 можно получить нисходящую кривую. При длине волны
Х2 можно получить восходящую кривую ДСО (фиг. 9.2,в), свиде-
тельствующую о возрастании отражательной способности с увели-
чением температуры. Эти кривые с постоянными значениями X
Спектроскопические, фотометрические и оптические методы
375
Позволяют определить температуры начала и конца термических
переходов, а также исследовать отдельно каждый переход; умень-
шение массы и изменение энтальпии не оказывают влияния на ре-
» зультаты измерений. Метод ДСО применим для определения тер-
мической устойчивости вещества, а также изменений структуры
« Образца в зависимости от температуры. Этот метод является много-
обещающим в качестве дополнительного метода термического ана-
лиза, используемого наряду с термогравиметрией, дифференциаль-
ным термическим анализом, высокотемпературным рентгепострук-
|турным методом и др. [45].
3. АППАРАТУРА
(Конструкция нагреваемого держателя образца (температурной
кюветы) описана многими исследователями. Асмуссен и Андерсон
|9] изучали спектры отражения нескольких комплексов M2[Hgl4]
при повышенных температурах с целью исследования термохроми-
чсского перехода (3-формы в a-форму. Применяемая ими темпера-
| турная кювета представляла собой никелированный латунный
I блок диаметром 60 мм и высотой 85 мм, в нижней части которого
• размещалась камера с небольшой лампочкой накаливания. Тем-
пература блока регулировалась путем изменения тока накала лам-
почки. В верхнем конце блока находилась камера для образца
диаметром 35 мм и глубиной 0,5 мм. Для определения температу-
ры образца использовалась медь-констаптановая термопара, по-
груженная в порошкообразный образец.
Кортум [2] изучал спектр отражения двухвалентного иодида
I ртути при температуре 140° С, но не привел описания конструкции
температурной кюветы и особенностей эксперимента. Другая кон-
| Струкция температурной кюветы рассмотрена в работе Хэтфилда
[и др. [10]. Кювета представляла собой металлический блок с за-
крепленным в нем нагревательным элементом. Детальное описание
I способа измерения температуры в этой работе отсутствует. Не ука-
I зана также толщина образца.
В 1963 г. Уэндландт и др. [11] описали первую из разработан-
I пых ими температурных кювет для высокотемпературной спектро-
ft скопни отражения. Основной алюминиевый блок кюветы диаметром
I 60 мм и толщиной 11 мм был получен механической обработкой.
Образец размещался в круглой лунке диаметром 25 мм и глубиной
I мм, выточенной на наружной поверхности блока кюветы. В лунке
Па равных расстояниях были вырезаны два кольцевых ребра для
| увеличения площади поверхности держателя и для предотвраще-
ния выпадения образца из него при вертикальном положении. Тем-
пературная кювета нагревалась спиралью из нихромовой проволо-
ки сопротивлением 15 Ом, намотанной на асбестовую доску и покры-
' тую сверху тонким слоем асбестовой бумаги. Температура образца
376
Глава 9
определялась хромель-алюмелевой термопарой, размещенной
в двухканальной керамической трубке. Спай термопары соприка
сался с алюминиевым блоком непосредственно за лункой образца
Для предупреждения передачи тепла от держателя образца к окщ
жающей сфере был изготовлен теплоизоляционный кожух
из гнутой алюминиевой трубки диаметром 6,35 мм и измельченною
влажного асбеста. После высыхания теплоизоляционный кожух
скреплялся цементным раствором со сферой, и кювета прикрепли
лась к нему металлическим пружинным зажимом.
Модификация описанного выше держателя образца рассмотрен.!
в работах Уэндландта и Джорджа [121, а также Уэндландта 1131
Круглый алюминиевый диск держателя обогревался патронным
нагревателем, ввинченным' непосредственно за лункой для обра ।
ца. В блоке устанавливались две хромель-алюмелевые термона
ры: одна вблизи нагревателя, а другая у дна лунки образца дли
обеспечения более тесного контакта с уплотненным образцом. Тер
мопара блока использовалась для управления устройством для ре
гулирования температуры по заданной программе, а термопара
образца — для определения температуры образца.
Еще одна конструкция температурной кюветы описана Уэнд
ландтом и Хечтом [1]. Она состоит из алюминиевого блока диаме г
ром 50 мм и толщиной 25 мм, в котором выточено углубление для
образца диаметром 25 мм и глубиной 1 мм. Для нагревания образца
использовался патронный нагреватель мощностью 35 Вт в защш
ном кожухе из нержавеющей стали, установленный в блоке кюветы
Как и в предыдущем случае, были использованы две термопары:
одна для регулятора температуры, другая для измерения темпе-
ратуры образца. При исследовании образцов, выделяющих газо
образные продукты, применялись покровные пластины из стекла
пирекс или кварца для предотвращения загрязнения окружи
ющего пространства.
В недавно опубликованной работе Уэндландта [151 был описан
нагреваемый держатель образца, предназначенный для исследова
ния небольших образцов, в основу конструкции которого был по
ложен держатель Фрея и Фродима [14]. Образец тонким слоем
укладывается на ткань из стекловолокна, прикрепленную к на-
греваемому алюминиевому блоку стеклянной крышкой с металли-
ческим зажимом. Размеры алюминиевого блока 40 х50 мм. Блок
обогревается круглым нагревательным элементом, размещенным
внутри держателя. Провода к нагревателю и термопаре подводятся
с помощью выводных шин, находящихся на верхней части блока.
Алюминиевый блок и выводы смонтированы на основании из
теплоизоляционного материала размером 50 х50 мм.
Лишь для нескольких из описанных выше температурных кю-
вет были предприняты попытки создать регулируемую атмосферу
вокруг образца в процессе его нагревания. Этой цели до некоторой
Спектроскопические, фотометрические и оптические методы
377
Фиг. 9.3. Схема нагреваемого дер-
жителя образца (температурной
кюветы) [16].
/ — блок из серебра и нагреватель образ-
ца; 2— стеклянная нли кварцевая покров-
мня пластина; 3— круглая прокладка; 4 —
Кит-барашек (один из четырех); 5 —
подача газа в камеру образца; 6 — труб-
ки для вывода газа; 7 — трубка для по-
дачи газа; 8 — соединительный кабель.
• лепени служили пластины из стекла пирекс или кварца, но их ос-
новное назначение заключалось в предотвращении случайного
впадения образца в пространство спектрорефлектометра. На
фиг. 9.3 представлен держатель Уэндландта и Доша [16], позво-
нющий регулировать атмосферу около образца.
Образец размещен в лунке глубиной 1 мм и диаметром 10 мм,
«точенной на поверхности нагревательного блока из серебра.
Влок диаметром 25 мм обогревается двумя нагревателями из ни-
Ьомовой проволоки сопротивлением по 2,6 Ом. Блок находится
корпусе квадратного сечения 55x55 мм и высотой 13 мм. Нагре-
> л гель отделен от основного блока держателя образца тонким слоем
цюляции из керамических волокон. Держатель со стороны образ-
уй прикрыт кварцевой пластиной размером 50 x50 мм и толщиной
I мм, прижатой двумя металлическими полосами. Каждая полоса
Прикреплена к держателю двумя барашковыми винтами. Винты, а
кЛедовательно, и крышки легко снимаются, что облегчает загрузку
I удаление образца. Между пластиной и держателем образца уста-
новлена газонепроницаемая кольцевая прокладка внутренним диа-
метром 44 мм. В верхней части держателя размещены две алюми-
ниевые трубки диаметром 3 мм для регулирования входа и выхода,
нза из камеры образца.
К нагреваемому держателю образца с регулируемой атмосфе-
рой можно легко присоединить датчик теплопроводности термн-
ирного типа, что позволяет при использовании многоканального
Чймописца одновременно получать кривые ДСО и ОВГ [17]. Схема
tuKoro модифицированного прибора приведена на фиг. 9.4. Датчик
(исдинен с микродетекторной системой «Карл» модели 1000 метал-
Ическими и резиновыми трубками. Датчик теплопроводности
размещен в алюминиевом блоке, нагреваемом до 100°С с помощью
интронного нагревателя. Блок присоединен к камере предваритель-
ного подогрева, в которой также поддерживается температура
КЮ°С. Эта камера предназначена для подогрева гелия перед по-
378
Глава 9
Выход газа
Вход газа
Фиг. 9.4. Схема совмещенного прибора ДСО—ОВГ [17].
1 — регулятор температуры; 2 — датчик ДСО; 3 — датчик теплопроводности; 4 — регулятр
датчика теплопроводности; 5 — сфера; 6 — многоканальный самописец; 7 — камера предвари
тельного подогрева; 8 — нагреватель.
дачей его к датчику. Выход с мостовой схемы датчика подается ii.i
один из четырех каналов (О—5 мВ) многоточечного ленточного пи
тенциометрического самописца фирмы «Лидз энд Нортрап». Дли
обеспечения регулируемого повышения температуры датчика ДСО
используется регулятор температуры прибора ДТА фирмы «Дель-
татерм», модель III. Выходной сигнал от спектрорефлектомецш
фирмы «Бекман» (модель ДК-2А) также подается на мвогоканаль»
ный самописец, как и выходной сигнал термопары, размещенной
в блоке нагревателя ДСО.
Спектроскопические, фотометрические и оптические методы
379
Фиг. 9.5. Схема совмещенного прибора ВТСО—ДСО [l]s
— двухкоординатный самописец; 2 — ленточный самописец; 3 — устройство для регулирова-
ния температуры по заданной программе; 4 — спектрорефлектометр.
Блок-схема системы ВТСО—ДСО представлена па фиг. 9.5.
Высокотемпературный держатель образца объединен с регулятором
температуры и двумя самописцами. Один самописец1 регистрирует
температуру образца в зависимости от времени; второй (двухкоор-
динатный) используется для регистрации отражательной способ-
ности образца в зависимости от температуры, как это и требуется
для исследований методом ДСО. Спектральные измерения произ-
водятся спектрорефлектометром фирмы «Бекман» (модель ДК-2А),
Или фирмы «Бош энд Ломб» (модель «Спектроник 505»).
4. ПРИМЕНЕНИЕ МЕТОДОВ ВТСО И ДСО
ДЛЯ ИССЛЕДОВАНИЯ НЕОРГАНИЧЕСКИХ СОЕДИНЕНИЙ
а. Переход октаэдрической структуры соединения
Со(Пиридин)2С12 в тетраэдрическую
Переход октаэдрической структуры бис-пир идин-кобальт-
Хлорида Со(Пиридин)2С12 в тетраэдрическую является предметом
ряда исследований [12, 22]. Уэндландт и Джордж [22] изучали этот
переход методами высокотемпературной спектроскопии отражения
(ВТСО) и динамической спектроскопии отражения (ДСО). Было
380
Глава 9
обнаружено, что термический переход начинается при температуре
~100°С и завершается при ~135°С. Процесс перехода сопроводи
дается изменением цвета от фиолетового (октаэдр) до синего (тчч
раэдр). Было установлено, что при охлаждении до комнатной тем
пературы обратного изменения цвета не происходит.
Фиг. 9.6. Кривые ВТСО для соединения Со(Пиридин)гС12 при различных
температурах [18].
В более поздней работе Уэндландта [18] сообщается, что в дей
ствительности структурные изменения имеют обратимый характер
Голубая форма после выдержки при комнатной температуре в гс-
чение 24 ч вновь превращается в фиолетовую. Кривые ВТСО для
Со(Пиридин)2С12 при 20 и 120сС приведены на фиг. 9.6. При 20 С
минимумы отражения наблюдались при 520 и 620 нм; кривая
имеет перегибы при 500 и 550 нм. Сильное отражение происходи!
в интервале длин волн 350—450 нм и при. 580 нм. Наибольшая
поглощающая способность голубой тетраэдрической формы при 120"С
наблюдается в интервале длин волн 500—700 нм; кривая имеет
перегибы при 425, 480 и 510 нм. После выдержки при комнатной
температуре в течение 24 ч голубой формы соединение вновь полу-
чало спектр отражения при 20°С. Как видно из фиг, 9.6, новый
спектр отражения почти полностью совпадает со спектром исход-
ного соединения, имеющего октаэдрическую структуру. Таким об-
Спектроскопические, фотометрические и оптические методы
381
pl юм, установлено, что переход тетраэдрической структуры в ок-
ьтщрическую совершается очень медленно при стационарных ус-
кишях; тетраэдрическая структура не переходит в октаэдрическую
сразу же после охлаждения до комнатной температуры.
Влияние скорости нагревания на переход октаэдрической струк-
1Уры в тетраэдрическую показано с помощью кривых ДСО на
фиг. 9.7. Скорость нагревания изменялась от 1,25 до 10 град/мин;
Фиг. 9.7. Кривые ДСО для соединения Со(Пиридин)2С1г при различных
скоростях нагревания; кривые записаны при длине волны 675 нм [18].
последняя величина считается довольно высокой для исследований
методом ДСО. Как ни странно, наблюдаемая температура пере-
хода оказалась наибольшей (107°С) при скорости нагревания
1,25 град/мин и наименьшей (95СС) при самой высокой скорости
нагревания. Однако с увеличением скорости нагревания возрастает
интервал температур перехода от 107—123°С для самой низкой
скорости нагревания до 95—145°С для скорости нагревания
10 град/мин. Совершенно обратное явление наблюдается при ре-
акциях диссоциации с выделением летучих продуктов.
На фиг. 9.8 приведены спектры отражения для соединения
Со(Пиридин)еС12 в видимой и ближней инфракрасной областях
115]. Минимумы отражения при комнатной температуре для а-
формы наблюдались при длинах волн 1140; 1670, 2150 и 2440 нм.
При нагревании соединения до 125°С все минимумы исчезали, за
382
Глава 9
исключением небольшого минимума при 2440 нм. Спектр отраже-
ния для fj-формы практически идентичен спектру для соединении
Со(Пиридин)2Вг2 тетраэдрической структуры.
Фиг. 9.8. Кривые ВТСО дли соединения а-Со(Пиридин)гС12 в видимой и
ближней инфракрасной областях спектра [15].
1 — а-Со(Пиридив)2С12 при температуре 125°С; 2 — а-Со (Пиридин) 8С12 при температуре 25°С;
3 — Со( Пиридин )2Вг2 при 25°С.
б. Соединение [ Си(Этилендиамин)(Н2О)2] S04
Дегидратация соединения [Си(Этилендиамин)(Н2О)215О4 изуча-
лась Уэндландтом [15] методами ВТСО и ДСО. В интервале темпе-
ратур 75—150°С эта реакция происходит в соответствии с уравне-
нием
[Си (Этилендиамин) (Н2О)2] SO4 (т.) Си (Этилендиамин) SO4 (т.) +
+ 2Н2О (г.). (9.8)
Кривые ВТСО в интервале температур 25—180°С приведены на
фиг. 9.9. Представлены две пары спектров: одна Для температур
25 и 75°С и другая для температур 150 и 180°С. Первая пара
спектров имеет минимум при 625 нм (соответствующий максимуму
поглощения) и относится к исходному соединению. Вторая napa>
спектров имеет минимум при 575 нм и соответствует дегидратиро-
ванному соединению Си(Этилендиамин)ЗО4. Таким образом, ре-
акция дегидратации происходит в интервале температур 75—150°С
Спектроскопические, фотометрические и оптический методы 383
Фиг. 9.9. [.Кривые ВТСО для соединения [Си(Этилендиамин) (HaO)2]SO4 [15]
Фиг. 9.10* Кривые ДСО для соединения [Си(Этилендиамин) (HaOhJSO* при
длине волны 600 нм [15].
Для определения температуры перехода реакции дегидратации
был использован метод ДСО. Кривые, полученные этим методом,
I приведены на фиг. 9.10. По ним легко определяется зависимость
В температуры перехода от скорости нагревания образца; она изме-
няется от 115°С при скорости нагревания 6,7 град/мин до 165°С
384
Глава 9
при скорости 45,8 град/мин. Подобная зависимость не является ш
ожиданной, поскольку такие же результаты были получены при in
пользовании других методов термического анализа, когда физн'н
ские свойства образца измерялись в зависимости от температуры
[13]. Во всех исследованиях методом ДСО, очевидно, должны бып.
указаны скорости нагревания образца.
Л, нм
Фиг. 9.11. Спектры отражения для соединения (Си(Этилендиамин)
(H2O)2]SO4 в видимой и ближней инфракрасной областях спектра [15].
Так как спектрорефлектометр фирмы «Бекман» (модель ДК-2А)
может быть использован для записи спектров образцов в ближней
инфракрасной области, с его помощью были получены кривые
ВТСО до длины волны 2700 нм (фиг. 9.11). При комнатной темпе-
ратуре кривая отражательной способности (полосы поглощения)
имеет минимумы при длинах волн 1560, 1725, 2050» 2150, 2260 и
2500 нм. При нагревании образца до 150°С полосы поглощения
при длинах волн 1560, 1725 и 2050 нм остаются неизменными, а
полоса 2150 нм исчезает. Полоса 2260 нм смещается к 2280 нм, а
полоса 2500 нм — к 2525 нм. Кроме того, заметно изменяется ин-
тенсивность всех рассмотренных полос поглощения, что, возможно,
связано с изменением размеров частиц образца.
в. Соединение CuSO4-5H2O
На фиг. 9.12 и 9.13 приведены кривые ВТСО для соединения
CuSO«-5HaO в видимой и ближней инфракрасной областях, а на
фиг. 9.14 — кривая ДСО при длине волны 625 нм.
Спектроскопические, фотометрические и оптические методы
385
tin, 9.12. Кривые ВТСО для соединения CuSO4-5H2O в видимой области
спектра [15].
Как и для соединения [Си(Этилендиамин)(Н2О)2]5О4, на
ши. 9.12 для соединения CuSO4-5H2O также приведены две пары
кривых. При комнатной температуре минимум отражательной спо-
•обпости наблюдается при длине волны ~680 нм; при нагревании
• •единения до 135°С минимум смещается к 715 нм. В этом интерва-
Ь температур изменение состава соединения происходит вследст-
р' дегидратации CuSO4-5H2O до CuSC>4-H2O. При дальнейшем
пышении температуры до 250°С выделяется последний моль воды
В образованием безводной соли. В ближней инфракрасной области
ри комнатной температуре минимумы отражения наблюдались
Кщ длинах волн 1510, 1675 и 2000 нм. При температуре 150°С
Пгрвые две полосы поглощения исчезали, а полоса 2000 нм смеща-
лись к 2060 нм. Кроме того, появлялась новая полоса поглощения—
При 2400 нм. При еще более высокой температуре (200°С) все поло-
К| поглощения в этой области исчезали.
386
Глава 9
Фиг. 9.13, Кривые ВТСО для соединения CuSO^-SHaO в видимой и блнжн»*
инфракрасной областях спектра [15].
Фиг. 9.14. Кривая ДСО для соединения CuSO4-5HsO при длине волны
625 нм и скорости нагревания 6,7 град/мин [15].
Согласно кривой ДСО, приведеной на фиг. 9.14, отражателы1ли
способность постепенно возрастает в интервале температур от ком
натной до 100°С (примерно на 1/10 величины R, выраженной и
процентах), однако основной ее рост начинается при температур
105°С. При температуре >-^125оС отражательная способность спои/,
постепенно уменьшается^до температуры 200°С.
Спектроскопические, фотометрические и оптические методы
387
г. Соединение СоС12-6Н20
В одной из ранних работ Уэндландт и Катере [26] исследовали
мподами ВТСО и ДСО реакцию между СоС12-бН2О и КС1.
♦ кривая ДСО позволила обнаружить следующие изменения струк-
цры и химического состава: октаэдрическая форма СоС12-бН2О->
к» тетраэдрическая форма C0CI4 -> октаэдрическая форма СоС12-
•'.’|1аО->тетраэдрическая форма C0CI4 • Эти реакции происходили в
гшсутствии избытка хлор-иона, поэтому конечным продуктом был
jl’oCU, а не безводный двухвалентный хлорид кобальта.
В более поздних работах была исследована дегидратация соеди-
IH'Iiidi СоС12-бН2О в отсутствие хлорида калия. Изучение данного
||нгдннения представляет довольно сложную задачу, поскольку
[ню плавится при температуре 50QC и при вертикальном положе-
нии нагреваемого держателя образца в спектрорефлектометре в нем
возможно удержать жидкий СоС12-6Н2О. Эта проблема была ре-
f ш?иа путем нанесения тонкого слоя порошкообразного образца
ini круглое покровное стекло диаметром 25 мм, которое прижима-
Ш<К1> к куску стеклоткани с помощью прямоугольного покровного
пекла. Благодаря повышенной вязкости расплава образца он не
щтекал.
Кривые ВТСО и ДСО для соединения СоС12-6Н2О приведены
Соответственно на фиг. 9.15 и 9.16. Кривые ВТСО позволяют обна-
ружить довольно интересные структурные изменения как в жидком,
Клк и в твердом состояниях. При 25°С твердый СоС12-бН2О имеет
1 ЭКТаэдрическую структуру, а кривая его отражательной способ-
ности — минимум при 535 нм и перегибы при 460 и 500 нм. При
Mui реванш! до 55°С соединение плавится и имеет кривую отража-
||»л|цюй способности с минимумом при 525 нм и довольно широким
I минимумом в интервале длин волн 600—700 нм. Последняя кривая
подобна соответствующей кривой для смеси октаэдрического и тет-
Ьвадрического комплексов двухвалентного кобальта, полученной
pliuee Симмонсом и Уэндландтом [27]. Следовательно, можно пред-
положить, что соединение при температуре 55°С представляет собой.
1месь октаэдрического СоС12-бН2О и тетраэдрического CofCoCW.
11рн дальнейшем нагревании смесь подвергается последующей де-
гидратации и при 155°С превращается в безводный октаэдриче-
ский СоС12. Последняя кривая имеет минимум при 590 нм с переги-
бом при 535 нм.
Из рассмотрения кривой ДСО (фиг. 9.16) видно, что при темпе-
ратуре 45°С происходит значительное уменьшение отражательной
способности вследствие образования смеси октаэдрической и тетра-
эдрической модификаций. Начиная с температуры 100°С, отра-
цительная способность смеси увеличивается, достигая максимально-
го значения при ~150'С; при дальнейшем повышении температу-
Фиг. 9.15. Кривые ВТСО для соединения СоСЬ-бНаО [15].
Температура, °C
Фиг. 9.16. Кривая ДСО для соединения СоС12-6Н2О при длине волны 700 и
и скорости нагревания 6,7 град/мин [15].
Спектроскопические, фотометрические и оптические методы
389
। l>i отражательная способность несколько снижается. На этой кри-
jioii отражаются также структурные изменения, рассмотренные
м .нпе.
д. Соединение N i( Пиридин),. С12
Янг [28] применил метод ВТСО для исследования дезаминиро-
*м1Н1Я №(Пиридин)4С12. Полученные им кривые представлены на
•|ш. 9.17,а. При температуре 25°С приведен спектр исходного со-
фит. 9.17. Кривые ВТСО для соединения МЦПиридин)4С12 в матрице из
А(аО3 (50%) (а) и кривые ДСО для того же образца при длине волны 450 нм
(б) [28).
390
Глава 9
единения М1(Пиридин)4С12. В интервале температур 125—!/!••
выделяются 2 моль пиридина на каждый моль соединения. Cneinu
при 175°С соответствует соединению №(Пиридин)2С12. В интерп.-i.'i»
температур 175—275°С выделяется еще 1 моль пиридина, в резулг
тате чего спектр при 275°С соответствует соединению Ni(Ilnpn
дин)С12. Потеря пиридина и изменение отражательной способы и ш
исходного соединения при 450 нм видны на кривой Д(ы
(фиг. 9.17,6). Переход №(Пиридин)аС12->-ЬЩПиридин)2С12 начи||,и
ся при 145°С и завершался при 160°С; дополнительная потери
пиридина начиналась при 210°С и заканчивалась при 220°С. Уш
личение наклона кривой ДСО было обусловлено возрастанием гем
пературы образца.
е. Термохромизм AgjHglJ
Термохромизм AgJHgh] привлекал большой интерес исследи
вателей с тех пор, как соединение было впервые получено Кавешу
и Виллмом [29] в 1870 г. Впервые этот переход был подробно изуч< в
Кетеларом [30—33] с применением рентгеновских методов, а таили
по измерениям удельной теплоемкости и электропроводное i и
В других работах были получены дополнительные данные, касаю
щиеся цветовых изменений [34,36, 39], дилатометрии [35], Кристал
лической структуры [37, 38], магнитной восприимчивости |!1|,
электропроводности [39, 40] и термической устойчивости [41] эки и
соединения. Было предложено использовать его как индикатор тем
пературы [36, 42] и как пигмент для термочувствительных красок
[42—44].
Термохромизм AgJHgh] является следствием разупорядочениц
структуры, проходящего не менее чем в три фазы. Согласно Кете
лару 133], обе модификации — желтая низкотемпературная р-фор
ма и красная высокотемпературная а-форма — содержат ионы
иодида в плотноупакованной кубической решетке, а ионы серебри
и ртути расположены в отдельных тетраэдрических пустотах,
[3-форма имет тетрагональную симметрию, причем ионы ртути
размещены в вершинах элементарной кубической ячейки, а ионы
серебра — в центрах вертикальных граней. При возрастании тем
пературы ионы серебра и ртути получают возможность меняться
местами в решетке, а также занимать два новых положения в цеш
рах верхней и нижней граней элементарного куба, свободных при
более низких температурах. При температуре выше 52°С пололи*
ние ионов серебра и ртути становится полностью неупорядоченным,
Поэтому a-форма обладает гранецентрированной кубической сим
метрией. Более поздние измерения магнитной [39] и диэлектриче-
ской поляризации [37, 39] подтвердили существование третьей
фазы — |3'-формы. С увеличением температуры ионы серебра при
р—>-р'-переходе произвольно занимают 2/3 центров граней кубов.
Спектроскопические, фотометрические и оптические методы
391
Фиг. 9.18. Кривые ВТСО для соединения Ag2[HgI4] [45].
11|>и^₽'-»-а-переходе происходит дальнейшее разупорядочение, а
попы ртути и серебра произвольно размещаются в 3/4 вершин и
центров граней элементарной ячейки [37], соответствующей двум
кубическим (но не изотропным) ячейкам, размещенным одна на
другой. Функция Паттерсона позволяет предположить, что часть
IIтомов серебра размещена неупорядоченно, переместившись из
узлов тетраэдров, где они находились в окружении ионов иодида,
н междоузлия октаэдров. Следует ожидать, что ионы серебра в
междоузлиях будут довольно подвижны, так как октаэдрические
пустоты значительно больше тетраэдрических. Данное предпо-
ложение подтверждается также низкой энергией активации проводи-
мости p-p-Ag2[Hgh] (50,2 кДж/моль при температуре ниже20°С) [371.
На фиг. 9.18 приведены кривые отражательной способности
для AgatHgh] в интервале температур 23—100°С. Желтая (3-форма
обладает довольно высокой отражательной способностью при дли-
нах волн более 500 нм. При переходе в красную a-форму максимум
кривой смещается в сторону больших длин волн. Изменение цвета
зависит от скорости нагревания. При скорости 2,5 град/мин пере-
ход завершается при несколько более низкой температуре, чем пе-
392
Глава 9
реход при скорости 10 град/мин. Последняя намного выше скорости
5 град/сут, использованной Нейбертом и Никольсом [39] при нс»
следовании магнитных свойств. Определенная в работе [45] темпе
ратура перехода является не очень точной, так как изменение
цвета происходит, по-видимому, в интервале температур 30—60 С.
Фиг. 9.19. Кривые ДСО для ряда комплексов (скорость нагревания
5 град/мин) [45].
1 —Pb[HgI4], 585нм; 2 — Cu2[HgI4J, 650 нм; 3 — Hg(Hgl4], 600 нм; 4 — Ag2[Hgl4], 575 им;
5 — Tl2[HgI4], 550 нм.
В литературе встречаются следующие значения температур пере-
хода: 50,7±0,2, 51, 50,5, 51,2 и 52°С.
Кривые ДСО для ряда комплексов вида Mn[Hgh] (М = РЬ,
Си, Hg, Ag и Т1) приведены на фиг. 9.19. На всех кривых, за исклю-
чением кривой для TlJHgld, довольно отчетливо виден термохро-
мический переход. Как сообщалось, переход Tl2[Hgh] происходит
при 116,5°С [9], однако на кривой ДСО он не виден. Отражатель-
ная способность этого соединения, по-видимому, линейно умень-
шается с увеличением температуры.
ж. Термические матричные реакции
Уэндландт и др. [46—49] применили методы ВТСО и ДСО для
изучения реакций между аминными комплексами солей трехвалент-
ных хрома и кобальта и солями аммония («термических матричных
реакций»). Такие реакции имеют вид [46]
Спектроскопические, фотометрические и оптические методы
393
|*[г(Этилендиамин)3] С13 + NH4C1 (избыт) -> гщс-[Сг(Этилендиа-
мнн)а Cl2] CI Другие продукты. (9.9)
। месь [Сг(Этилендиамин)3]С13 и NHiX (X = F, Cl, Вг или SCN) с
штовым соотношением 1 : 1 нагревали в высокотемпературном дер-
теле образца до 200°С. Метод ВТСО был применен для иденти-
фикации продуктов реакции, а метод ДСО — для определения ин-
ициала температур, в котором протекает реакция. Результатом
них исследований явилась разработка новой методики синтеза
комплексов г|«с-[Сг(Этилендиамин)2Х2]Х, цис-1Сг (1,2-Пропанди-
imiih)2X2]X и щ/с-[Сг(1,3-Пропандиамин)2Х2]Х.
Чанг и Уэндландт [47—49] исследовали соединения следующих
пшов: а) цис- и транс- [Со(Этилендиамин)2(Н2О)2](НО3)2+№Н4Х;
6) цис- и mpaHC-[Co(NH3)4(H2O)2](NO3)3+NH4X. Во всех случаях
были получены продукты реакции в транс- изомер ной форме. Были
предложены различные механизмы реакции, а также методики
। пнтеза /иранс-изомерных веществ.
Б. ФОТОМЕТРИЧЕСКИЕ МЕТОДЫ
Одновременное исследование веществ методами фото-
гермического анализа (ФТА) и ДТА было описано Дэвидом [50].
Многочисленные исследования органических и неорганических
ос ди нений показали, что оба метода позволяют получить значи-
тельный объем информации, но кривые ФТА содержат данные,
оторые нельзя получить методом ДТА. Были изучены извест-
няк, глина, CuSO4-5H2O, а также полистирол, поливинилхлорид
и ряд других полимеров.
Аппаратура для одновременных исследований методами ФТА и
1ТА представлена на фиг. 9.20. Она содержит датчик ДТА фирмы
•(.Тоун-Премко», в колпачке которого, а также в стенке камеры
печи просверлено смотровое отверстие диаметром 7 мм для фотоум-
ножителя, смонтированного на верхней части печи. Образец раз-
мещен в кольцевом контейнере термопары. Один канал двухка-
вльного самописца регистрирует сигнал от фотоэлемента, а второй
канал используется для построения кривой ДТА.
Кривые ДТА исследованных образцов известняка имели неболь-
шие широкие экзотермические пики в интервале температур 300—
175°С. Эти пики совпадали с основными пиками свечения на кри-
П1.1Х ФТА. Дэвид пришел к заключению, что метод ДТА обладает
Гнкой же, а возможно, и более высокой чувствительностью, чем
Метод ФТА при анализе материалов указанного типа, и, по-види-
мому, более эффективен, чем термолюминесцентный анализ при
исследованиях геологических объектов. Низкая чувствительность
метода ФТА, возможно, связана с низкой скоростью нагревания
(1(1 град/мин) в эксперименте. Типичные термолюминесцентные
394
Глава 9
кривые получают при скоростях нагревания 10—16 град/с. Длим
[50] обнаружил, что при исследовании полимерных образцов плав
ление и стеклование не обнаруживались методом ФТА, хотя пн
может быть применен для изучения реакций разложения. Следи
вательно, совмещение методов ДТА—ФТА позволило разделнн
Фиг. 9.20. Прибор для одновременных исследований методами ФТА и Д ГЛ
[50].
1 — фотоумножитель; 2 — печь; 3 — держатель образца; 4 — дифференциальная термопара)
5 — образец; 6 — смотровое отверстие для фотоумножителя.
эти процессы. Пик на кривой ФТА появлялся при температуре,
приблизительно равной температуре начала окисления, подобно
тому как это наблюдалось при оксилюминесценции. То же само
происходило даже в тех случаях, когда нагревание производилось
в потоке азота.
Руперт [51] применил более сложную аппаратуру для наблюди
ния фазовых переходов в раскаленных материалах. В приборе
Руперта для наблюдения за изменениями температуры нагретого
образца использован фотоумножитель. Фотоэлемент фиксирует ни
тенсивность свечения образца, которая пропорциональна темпера
туре. При этом не требуется знать точного соотношения между вы
ходным сигналом с фотоэлемента и температурой образцаДтак кап
Спектроскопические, фотометрические и оптические методы
395
ропзводится калибровка по температуре с помощью калиброван-
ию оптического пирометра, получающего часть света от образца.
Ц1|>азец размещен в тигле, расположенном внутри концентратора
ши. Последний получал энергию от индукционного нагревателя,
1цг. 9.21. Прибор Лоэра и Леви для одновременных термогравиметриче-
ских исследований и измерения плотности дыма [52].
1 — фотометр; 2 — аттенюатор; 3 — образец; 4 — самописец (х — у — у').
Мощность которого регулировалась специальным регулятором,
fciier от образца проходил через отверстие диаметром 1,8 мм в верх-
ней части тигля и попадал через окно из кварца или стекла пирекс в
Ьнжнюю часть разделителя луча. Часть света отражалась от час-
тршо алюминированного зеркала примерно под прямым углом к
Ьси разделителя луча и попадала на оптический пирометр, исполь-
иуемый для измерения температуры образца. К типичным кривым
хлаждения, полученным этим методом, относятся кривая затвер-
К’вания смеси карбида молибдена и углерода при температуре
54О°С, кривая охлаждения эвтектической смеси карбида цирко-
ния и углерода при 2855°С и переход в твердой фазе дикарбида
урана при 1800°С.
Прибор для одновременного определения плотности дыма и
кермогравиметрических исследований был описан Лоэром и Леви [52].
Этот прибор (фиг. 9.21) состоит из термогравиметрического анализа-
тора фирмы «Дюпон» (модель 950), соединенного с прецизионным
фотометром фирмы «Дюпон» (модель 410). В фотометре использует-
396
Глава 9
ся система расщепления луча (сравнение образца с эталоном), п
которой пропускание света оценивается с помощью фотоэлемента
S-5, соединенного с вольфрам-иодистым источником света. Такай
система выбрана с целью наибольшего приближения к чувствите.п.
ности человеческого глаза к изменению пропускания света. Вы
ходной сигнал фотоэлемента ослабляется таким образом, чтобы
Фиг. 9.22. Одновременно полученные ТГ-кривые и кривые плотности дыми
для образцов пенополиуретана [52].
а — образец № 1 в воздухе; б — образец №2 в воздухе; скорость нагревания 20 град/мин; рас*
ход воздуха 100 мл/мин.
его можно было прочитать по шкале оптической плотности на табло
контрольной панели фотометра. Камера для определения плотности
дыма состоит из кожуха, изготовленного из нержавеющей стали
толщиной 1 мм, с сапфировыми окнами. Объем пустой камеры
9,7 см3.
Данный метод применялся для оценки двух различных замедли-
телей пламени, введенных в один и тот же пенополиуретан. Было
Спектроскопические, фотометрические и оптические методы
397
Определено количество дыма, выделяющегося от каждой компози-
। in, а также способность этих добавок замедлять распространение
। 1амени. На фиг. 9.22 приведены одновременно полученные ТГ-
црпвые и кривые плотности дыма для двух упомянутых выше об-
ращен. Измерения проводились в атмосфере воздуха. Образец
Bi 1 разложился с остатком 10% при температуре 450'С. Макси-
мум оптической плотности наблюдался при этой же температуре.
При тех же условиях остаток образца № 2 составил 25%, но опти-
ческая плотность была на —20% выше, чем у образца № 1. Был
«делан вывод, что образец № 2 является лучшим замедлителем
пламени, так как остаток при любой заданной температуре для этого
•разца выше. Однако он выделяет большее количество дыма при
Вплее низких температурах.
В. ВЫСОКОТЕМПЕРАТУРНАЯ ИНФРАКРАСНАЯ
СПЕКТРОСКОПИЯ
Хотя метод инфракрасной спектроскопии образцов, запрессо-
шиных в матрицы в виде дисков из К₽г, достаточно известен,
имеется очень мало сообщений об аналогичных количественных ис-
следованиях кинетики химических реакций в твердой фазе. В рабр-
। six Хисатсуна и др. [53—58] было показано, что-многие химические
реакции инициируются путем нагревания дисков до высоких тем-
ператур и что этот метод может с успехом применяться для иссле-
дования кинетики реакций. Диски помещали в печь, нагреваемую
о заданной температуры за определенный период времени, а затем
после быстрого охлаждения диска до комнатной температуры запи-
тывали его инфракрасный спектр. Обычно диск массой --0,5 г
охлаждался от 600°С до комнатной температуры за время менее
I мин. Диски, изготовленные из солей калия, могут нагреваться в
реде воздуха до температуры —600'С; при более высокой темпе-
ратуре происходит заметная возгонка соли матрицы. Первичное
нагревание сопровождается наиболее значительными изменениями
вешнего вида дисков. Они теряют прозрачность и расширяются,
| па их поверхности часто появляются пузыри, что свидетельствует
।) образовании газообразных продуктов при разложении раство-
ренных веществ. В некоторых случаях прозрачность диска может
ыть восстановлена путем измельчения его на мелкие куски и по-
Нторного прессования. Для количественных исследований диск об-
рабатывается по периметру для обеспечения плотной подгонки к
месту его установки в держателе. Данный метод был применен для
регистрации ионов ВО2 [54], ион-радикалов СО2 [55], нон-радика-
лов СОз [56], раздельного наблюдения за ионами формиата и ацета-
и 157] и для изучения реакции разложения иона перхлората [58].
Вайдевен и Лебан [59] описали применение этого метода для
исследования кинетики термического разложения карбоната се-
398
Глава 9
ребра. Применение подогреваемой кюветы позволяет производии,
непрерывный количественный анализ ИК-активных реагентов ц
продуктов реакции их разложения непосредственно в процессе их
образования. Температурная кювета изготавливается из нержа
веющей стали и может нагреваться до температуры 500°С. Оки >
кюветы из кристалла KRS-5 поддерживаются при комнатной тем
пературе путем охлаждения водой. Такой же метод был применен
Уэндландтом [60].
Танака и др. [61] применили нагреваемую кювету с регулируй
мой температурой по заданной программе для исследования терми
ческого разложения ряда аминных комплексов трехвалентнсно
кобальта. В качестве материала матрицы диска использовался l\( I
или КВг. Сообщалось, что при повышенных температурах эти ма
териалы часто становились непрозрачными для инфракрасного
излучения. Нагреваемая кювета для ИК-спектроскопии при темпе-
ратурах до 200°С была также описана Леру и Монтано [62].
Г. МЕТОДЫ ТЕРМИЧЕСКОЙ ОПТИЧЕСКОЙ
МИКРОСКОПИИ
1. МИКРОСКОПИЯ РАСПЛАВА
Термин «микроскопия расплава» применяется для обозначения
методов исследования, при которых нагревание соединения пл и
смеси производится на предметном стекле микроскопа [64]. Этим
методом изучаются все процессы, происходящие при нагревании
образца до расплавления (начальное состояние, возгонка, разло
жение, начало плавления и т. п.), в самом расплаве (показатель
преломления, точка кипения, критическая температура раствор.!
и т. п.), при затвердевании (углы кристаллов, двойное лучепрелом
ление, скорость роста кристалла и т. д.) и охлаждении (полиморф
ные превращения, ортоскопические и коноскопические наблюдении,
диаграммы состава и др.). По-видимому, наиболее важными обл.к
тями применения данного метода являются следующие: а) харакге
ристика и идентификация чистых соединений; б) определение чис
тоты соединения; в) анализ бинарных смесей; г) определение дна
грамм состава бинарных и трехкомпонентных систем; д) объяснены'
диаграмм состояния; сюда также относятся исследования: е) поли
.морфизма; ж) кинетики роста кристаллов; з) деформации кристал-
лической решетки [63; 64]. Метод позволяет быстро произведи
идентификацию плавких соединений, требует небольшого количе
ства исследуемого материала, несложного оборудования и относи
тельно простой подготовки обслуживающего персонала. Во многих
случаях удается очень быстро определить чистоту расплавленного
соединения, так как примеси обычно видны в виде нерасплавни
шихся включений, жидкой эвтектики или легкоплавких включений
Спектроскопические, фотометрические и оптические методы 399*
Г---------------------------------------------------------------------------Г
Лиг. 9.23. Нагреваемый столик микроскопа фирмы «Меттлер» (модель FP-2).
• — блок-схема системы регулирования; б — схема нагреваемого столика; 1 — держатель
рлзца; 2 — тепловой фильтр; 3,5 — нагревательные пластины; 4 — вентилятор; 6 — внут-
- ренняя стенка; 7 — наружная стенка.
Плавление смеси является быстрым и надежным способом опреде-
ления идентичности состава двух образцов. Полные диаграммы
'состава бинарных и трехкомпонентных смесей можно получить в
речение нескольких часов. Из последних работ, посвященных это-
му методу, следует упомянуть обзоры Богена [65], Смита [66],.
Соммера [67], Соммера и Джохенса [58], а также Маккрона [63].
Различные конструкции нагреваемых столиков микроскопа были
рассмотрены Маккроном [63, 64]. Наилучшей является, по-види-
мому, конструкция Кофлера, впервые описанная в 1930 г. Начиная
С 1940 г. столик Кофлера выпускается промышленностью. Основным.
400
Глава 9
достоинством этого прибора является его надежность; темпера) у pl
указываемая термометром, находится в точном соответствии < к»
пературой исследуемого препарата. Более сложный по кот ipvi
ции нагреваемый столик фирмы «Меттлер» (модель FP-2) (фи. Ч М
выпускается промышленностью с 1968 г. Определение темш|<(
туры производится платиновым термометром сопротивлении!
размещенным в непосредственной близости к образцу. Эиер! ни,
подводимая к нагревательным элементам, определяется разит nJ
между сопротивлением термометра сопротивления и сопропш
нием потенциометра с приводом от электродвигателя. Пока фаю и
ческая температура и температура, определяемая программнИ
равны, частота вращения ротора двигателя, вырабатывающего ни
налы в соответствии с программой, является линейной фуиьпнш
температуры. Считывание температуры производится путем при
соединения двигателя к системе зубчатых колес счетчика. МоЦ|
быть заданы три скорости нагревания: 0,2, 2,0 и 10 град/мин и ни
тервале температур от —20 до 300°С. Погрешность измерении И
регулирования температуры составляет ±0,ГС при темпера и р»
ниже 100°С и ±0,1% при температуре выше 100°С. Нагрев,чемии
столик, который можно присоединить к любому стандартному
микроскопу, состоит из регулируемого держателя образца, рц»
металлических нагревательных пластин с заключенными меи,1>
ними проводами высокого сопротивления и платиновым терм<>м< i
ром^ а также компактного вентилятора для обеспечения цирку hi
ции воздуха в камере. Конструкция столика позволяет производна
одновременное нагревание образца сверху и снизу, т. е. поддержи
вать равномерное распределение температуры.
Подробное описание применения микроскопии расплава дли
решения химических и физических задач содержится в рабонн
Маккрона [63, 64].
2. ИНТЕНСИВНОСТЬ ДЕПОЛЯРИЗОВАННОГО СВЕТА
И ФОТОМЕТРИЧЕСКАЯ ТЕРМИЧЕСКАЯ МИКРОСКОПИЯ
В микроскопии расплава термические изменения в образце ни
блюдаются визуально; для непрерывной регистрации этих измеш
ний можно применить микрофотографирование. Так как чувстпн
тельность глаза в восприятии незначительных изменений, прош \и
дящих за несколько минут, сравнительно мала, для обнаружении
таких изменений используются автоматические устройства. Один
из возможных способов заключается в применении фотоэлемент
для измерения изменения пропускания света образцом или иитеи
сивности двойного лучепреломления [45, 65, 69—78]. Другим ено
собом является использование камеры сканирования в ИК-обласгн,
подобной описанной Хайзером [79]. Первый способ используек и
преимущественно для точного определения точек плавления, поли
Спектроскопические, фотометрические и оптические методы
401
Ффпых переходов и скоростей кристаллизации, а второй, более
। пикиый способ позволяет обнаружить малейшие изменения тем-
крптуры исследуемого участка образца.
11рибор для регистрации пропускания света образцом имеет
ниычцо следующие узлы [65, 69, 73]: а) фотодетектор, в качестве
второго может быть применен фотоэлемент, фотоумножитель или
ртосопротивление; б) нагреваемый столик микроскопа; в) устрой-
ll цю для регулирования температуры по заданной программе;
|) микроскоп; д) держател£> образца; е) самописец (обычно двух-
1шифдинатный). По мере нагревания образца со скоростями 0,2—
• ) град/мин выходной сигнал фотодетектора записывается в функ-
I НИН температуры образца. При измерении пропускания света обра-
тен, помещается либо между двумя покровными стеклами, если он
приготовлен в виде расплава, либо под покровное стекло, если он
• приготовлен в виде суспензии в силиконовом масле [69]. Масло
Уменьшает преломление света и позволяет более эффективно про-
ииюдить измерения двойного лучепреломления. Для исследования
фнсталлических или частично кристаллических полимеров можно
•Применять методику погружения. Полимерный образец погружают
к тонкий слой (0,5 мм) высоковязкого полидиметилсилоксана, на-
<итенного на предметное стекло микроскопа. Еще один способ со-
Ьгоит в применении стеклянных капиллярных трубок для разме-
щения образца [65]. Порошкообразные образцы набиваются в ниж-
1|1Кно часть трубки; жиры, масла, воскообразные вещества легко
•вливаются в трубки в расплавленном состоянии при помощи шпри-
1111 с длинной иглой.
( Г Кривые нагревания и охлаждения нитрата аммония, полученные
^фотометрическим методом [73], приведены на фиг. 9.24. На кривых
«•по видны четыре полиморфных перехода в интервале температур
М -200иС. Переход орторомбической структуры в моноклинную
К При температуре 42*С проявляется в возрастании интенсивности
Света, а переход моноклинной в тетрагональную при температуре
НЬ'С сопровождается уменьшением интенсивности. Резкое сниже-
нию интенсивности света (~125°С) указывает на переход тетраго-
ппльной структуры в кубическую форму, и, наконец, при темпера-
туре 169°С происходит плавление кубической структуры. При
Охлаждении расплав кристаллизуется с образованием кубической
структуры при температуре 175°С. Несколько резких изменений
интенсивности света указывают на различные фазовые переходы
Цитрата аммония. Все измерения были выполнены в поляризован-
ном свете.
Метод микроскопических измерений интенсивности деполяри-
зованного света (ИДС) был предложен Магиллом [80] в 1960 г.
КОсновными элементами прибора для измерения ИДС являются
Источник света, поляризаторы, держатель образца, анализатор и
, соответствующая система регистрации данных. Бэрролл и Джон-
114—230
402
Глава 9
Фиг. 9.24. Фотометрические кривые для нитрата аммония [73].
а — кривая нагревания; б — кривая охлаждения.
сон [74], а также Миллер [75, 76] описали применение этого методд
для исследования полимеров. Миллер [75] предпочитает называв,
этот метод термическим поляризационным анализом.
Прибор для измерения ИДС, используемый Кэрроллом и Джои
соном [74], показан на фиг. 9.25. Наиболее подходящей для этого
Спектроскопические, фотометрические и оптические методы
403
♦иг. 9.25. Схема прибора для из-
* рения интенсивности деполяри-
зованного света [74].
i микроскоп, модель «Юнитрон MPS»; 2 —
I. ► стина анализатора; 3—пластина поляризато-
Е 4 — подвижный конденсор Аббе; 5 —
. и.фрамовая лампа; 6 — подвижный нагрева-
HtiH столик микроскопа; 7 — нагреватель; 8—
опара устройства для регулирования тем-
уры по заданной программе; 9— термопа-
для измерения температуры образца; 10—
положение образца; 11 — штепсельный
ъсм, прикрепленный к раме микроскопа;
• вкранированный провод блока регистрации
Пых; 13 — фотоэлемент; 14 — подвижное
жало; 15 — окуляр; 16 — модульный блок
'А фирмы «Дюпон» (модель 900); 17—про-
ирующее устройство; 18 — двухкоорди-
натный самописец.
15
рибора оказалась система регистрации данных прибора ДТА
Ирмы «Дюпон» (модель 900). Интенсивность деполяризованного
гнета измерялась фотоэлементом, соединенным с мостовой схе-
мой Уитстона. Нагреваемый столик микроскопа, изготовленный из
дно го бруска, использовался в интервале температур от —40 до
П0()°С при скорости нагревания ~5 град/мин или в изотермических
ловиях. При больших скоростях нагревания применялась тер-
Iкнчески малоинерционная печь с платиновым нагревателем, рабо-
Г адющая при температурах до 800° С.
Уэндландт [45] применил метод микроскопии для определения
отражательной способности образца. Разработанный им прибор
(фиг. 9.26) состоит из микроскопа 1 отражательного типа с неболь-
пим увеличением (обычно 100) и с освещением от монохроматора 2.
Ограженное излучение детектируется фотоумножителем 3 и уси-
лителем 4, а затем регистрируется двухкоординатным самопис-
цем 5 или ленточным самописцем 6. Для нагревания образца до
I гсмпературы 250°С используется нагреваемый столик 7 фирмы
•Меттлер» (модель FP-2). С помощью этого устройства можно соз-
дать изотермические условия (±ГС) или нагревать образец с за-
шной скоростью. Образец перемещается через освещенное опти-
ческое поле микроскопа с помощью реверсивного двигателя 8.
Через определенные интервалы времени реле с датчиком времени 9
меняет направление вращения двигателя на обратное. Это позво-
ляет производить сканирование отраженного излучения от образ-
на, представляющего собой монокристалл или порошкообразную
14*
404
Глава 9
смесь. Порошкообразный образец можно непосредственно ц<№ц
щать на нагреваемое предметное стекло микроскопа или в стеклим
ные капиллярные трубки с внутренним диаметром до 1,1 мм. В и
леднем случае можно получить кривую отражения образца и
паянной трубке.
Фиг. 9,26. Прибор для микроизмерений отражательной способности |4Л|
1 — микроскоп; 2 — лампа и монохроматор; 3 — фотоумножитель; 4 — усилитель н
питания; 5 — двухкоординатный самописец; 6 — ленточный самописец; 7 — нагреваемый (г
лик фирмы «Меттлер» (модель FP-2); 8 — реверсивный двигатель; 9 — реле и датчик bjxwih.
Возможны два режима работы прибора: а) режим разверпи
(сканирования), при котором регистрируется отражательная спи
собность поверхности образца в функции пути сканирования лум
при комнатной температуре или повышенных температурах; б) ре»
жим, при котором изменение отражательной способности обра ни
записывается в функции температуры. Первый режим работы па
вается методом высокотемпературного сканирующего микроотра^Л
ния (СМО), а второй — методом динамического микроотражешц
(ДМО). Применение этих двух методов иллюстрируется на пример!
дегидратации соединения CuSO4-5H2O.
Кривые СМО для монокристалла CuSO4-5H2O [78] при комн/п
ной температуре приведены на фиг. 9.27. Они представляют cofrjfl
отражательную способность при сканировании в точках 1, 2 и 1
поверхности кристалла. Применялась такая геометрия отражении
(90790°), при которой максимум кривой соответствует максимуму
зеркального отражения от поверхности кристалла. Таким обрм
зом, поверхности, расположенные перпендикулярно лучу падаю
Спектроскопические, фотометрические и оптические методы
405
Г, 10 света, обеспечивали наибольшую интенсивность отражения,
рииые для разных кристаллов отличаются не очень хорошей вос-
»i пнводимостью, поскольку поверхности отдельных кристаллов
Н' одинаковы.
Кривые СМО для того же монокристалла CuSO«-5H2O при раз-
умных температурах приведены на фиг. 9.28. В интервале темпе-
и1 9.27. । Кривые СМО для монокристалла CuSO4-5НаО при длине
волны 450 нм и температуре 30° С (увеличение 100) [78].
1________1--------1--------1-------1--------1______-
0,1 0,3 0,5 ММ
♦иг. 9.28. Кривые СМО для монокристалла CuSO4-5H2O при различных
пмпературах и длине волны падающего света 450 нм (увеличение 100) [78].
406
Глава 9
ратур 30—50°С кривые почти не изменяются. Но при температуре
70°С наблюдается общее уменьшение максимумов зеркалыкнп
отражения, которое становится еще более заметным в интервал'
температур 80—100°С. Такое ослабление зеркального отражении
объясняется образованием поверхностного слоя CuSO4-3H2O с Он
лее высокой светопроницаемостью, чем исходное соединение. 1 а
ким образом, образование CuSO4-3H2O легко обнаруживается mi
тодом СМО.
50 100 150 «с
Фиг. 9.29. Кривые ДМО образцов CuSO^-SlbO в запаянной (/) и OTKpu'inft
(2) трубках [78].
Скорость нагревания 10 град/мин; длина волны падающего света 450 нм (увеличение 80).
На фиг. 9.29 приведены кривые ДМО, характеризующие пы
деление воды при дегидратации порошкообразных образцов CuSOi
X 5Н2О, помещенных в запаянную и открытую капиллярные трубки
Отражательная способность образца, помещенного в открытую ки
пиллярную трубку, уменьшается при возрастании температуры
В запаянной трубке выделенная вода сохраняется, и в итоге при
дальнейшем нагревании отражательная способность не увеличц
вается.
ЛИТЕРАТУРА
1. Wendlandt W. W., Hecht Н. G., Reflectance Spectroscopy, Intersciem
N. Y., 1966, Ch. 3, 4.
2. Kortum G., Trans. Faraday Soc., 58, 1624 (1962).
3. Kubelka P., Munk F., Z. Techn. Physik, 12, 593 (1931).
4. Wendlandt W. W., Hecht H. G., Reflectance Spectroscopy, Interscieii'H,
N. Y., 1966, pp. 275—279.
5. Wendlandt W. W., Hecht H. G., Reflectance Spectroscopy, Intersciiiun,
N. Y., 1966, Ch. 8.
6. Wendlandt W. W., Franke P. H., Smith J. P„ Anal. Chem., 35, 105 (19M|
7. Wendlandt W. W„ Science, 140, 1085 (1963).
Спектроскопические, фотометрические и оптические методы 407
Лпоп., Chem. Eng. News, April 15, 62 (1963).
HL Asmussen R. W., Anderson P., Acta. Chem. Scand., 12, 939 (1958).
fj. Hatfield W. E., Piper T. S., Klabunde U., Inorg. Chem., 2, 629 (1963).
I, Wendlandt W. W., Franke P. H., Smith J. P., Anal. Chem. 35, 105 (1963).
, Wendlandt W. W., George T. D., Chemist-Analyst, 53, 100 (1964).
H. Wendlandt W. W., Thermal Methods of Analysis, Interscience, N. Y., 1964,
Ch. 10.
Ц4, Frei R. W., Frodyma M. M., Anal. Chim. Acta, 32, 501 (1965).
И, Wendlandt W. W., Modern Aspects of Reflectance Spectroscopy, Wend-
landt W. W., ed., Plenum Press, N. Y., 1968.
|l Wendlandt W. W., Dosch E. L., Thermochim. Acta, 1, 103 (1970).
II*. Wendlandt W. W., Bradley W. S., Thermochim. Acta, 1, 143 (1970).
iH Wendlandt W. W„ J. Thermal Anal., 1, 469 (1970).
I», Cox E. G., Shorter A. J., Wardlaw W., Way W. J. R., J. Chem. Soc., 1556
(1937).
HD, Dunitz J. D., Acta Cryst., 10, 307 (1957).
I II. Mellor D. P., Coryell C. D., J. Am. Chem. Soc., 60, 1786 (1938).
I Й, Wendlandt W. W., George T. D., Chemist-Analyst, 53, 71 (1964).
I |. Ocone L. R., Soulen J. R., Block В. P., J. Inorg. Nucl.Chem., 15, 76 (1960).
I 4. Beech G., Mortimer С. T., Tyler E. G., J. Chem. Soc., 925 (1967).
tMurgulescu I. G., Segal E., Fatu D., J. Inorg. Nucl. Chem., 27, 2677 (1965).
Wendlandt W. W., Gathers R. E., Chemist-Analyst, 53, 110 (1964).
I I. Simmons E. L., Wendlandt W. W., J. Inorg. Nucl. Chem., 28, 2187 (1966).
I L Yang W. Y., неопубликованные результаты. ।
I I, Caventou E., Willm E., Bull. Soc. Chim. Fr., 13, 194 (1870).
I 0. Ketelaar J. A. A., Z. Krist., 87, 436 (1934).
I I, Ketelaar J. A. A., Z. Physik. Chem., B26, 327 (1935).
I j. Ketelaar J. A. A., Z. Physik. Chem'., B30, 35 (1938).
I |, Ketelaar J. A. A., Trans. Faraday Soc., 34, 874 (1938).
I 4 Suchow L., Keck P. H., J. Am. Chem. Soc., 75, 518 (1953).
I B. Thomas D. G., Staveley L. A. K-, Cullis A. F., J. Chem. Soc., 1727 (1952).
I I, Bachman С. H., Maginnis J. B., Am. J. Phys., 19, 424 (1951).
I T. Olsen С. E., Harris P. M., Phys. Rev., 86, 651 (1952); U. S. Department
of Commerce, Office, Tech. Ser., PB Dept. 156, 106, 61 pages, 1959.
№. Hahn H., Frank G., Klinger W., Z. Anorg. Allgem. Chem., 279, 271 (1955).
№ Neubert T. J., Nichols С. M., J. Am. Chem. Soc., 80, 2619 (1958).
10. Rothstein J., Phys. Rev., 98, 271 (1955).
II. Heintz E. A., J. Inorg. Nucl. Chem., 21, 64 (1961).
№. Andrews W. S., Gen. Elec. Rev., 29, 521 (1926).
4'1. Perez H. G., Quim Ind. Sao. Paulo, 4, 137 (1936).
14. Horiguchi, Funayama Y, T., Nakanishi T., Sci. Papers Inst. Phys. Chem.
Res. Tokyo, 53, 274 (1959).
№. Wendlandt W. W., Pure and Appl. Chem., 25, 826 (1971).
Hi. Wendlandt W. W., Stembridge С. H., J. Inorg. Nucl. Chem., 27, 575 (1965).
H''. Chang F. C„ Wendlandt W. W., J. Inorg. Nucl. Chem., 32, 3535 (1970).
4 Chang F. C., Wendlandt W. W., Thermochim. Acta, 2, 293 (1970).
№. Chang F. C., Wendlandt W. W., Thermochim. Acta, 3, 69 (1971).
№). David D. J., Thermochim. Acta, 3, 277 (1972).
II. Rupert G. H., Rev. Sci. Instr., 34, 1183 (1963).
B, Loehr A. A., Levy P. F., Am. Lab., Jan., 11 (1972).
B. Hisatsune I. G., Perkin-Elmer Instrument News, 16, № 2, 2 (1965).
tl. Hisatsune I. C., Suarez N. H., Inorg. Chem., 3, 168 (1964).
i. Hartman К- O., Hisatsune I. C., J. Chem. Phys., 44, 1913 (1966).
BG. Hisatsune I. C., Adi T., Beahm E. C., Kempf R. J., J. Phys. Chem., 74,
3225 (1970).
J7, Hisatsune I. C., Beahm E. C., Kempf R. J., J. Phys. Chem., 74, 3444 (1970).
J, Hisatsune I. C., Linnehan D. G., J. Phys. Chem., 74, 4091 (1970).
408
Глава 9
59. Wydeven Т., Leban М., Anal. Chem., 40, 363 (1968).
60. Wendlandt W. W., Smith J. P., Thermal Properties of Transition Mel
Ammine Complexes, Elsevier, Amsterdam, 1967, p. 35.
61. Tanaka N., Sato M., Nanjo M., Sci. Reports Tohoku Univ., 48, 1 (191И1
62. LeRoux J. H., Montano J. J., Anal. Chem., 38, 1808 (1966).
63. McCrone W. C., Mettler Technical Information Bulletin, № 3003, 1901,
64. McCrone W. C., Fusion Methods in Chemical Microscopy, Interscieiii.
N. Y., 1957.
65. Vaughan H. P., Thermochim. Acta, 1, 111 (1970).
66. Smith R. V., Am. Lab., Sept., 85 (1969).
67. Sommer G., Instr. Tech. Southern Africa, 2, 10 (1965).
68. Sommer G., Jochens P. F., Minerals Sci. Eng., 3, 3 (1971).
69. Kolb A. K., Lee C. L., Trail R. M., Anal. Chem., 39, 1206 (1967).
70. Vaughan H. P., Microscope, 17, 71 (1969).
71. Reese D. R., Nordberg P. N., Ericksen S. P., Swintosky J. V., J. Phor*.
Sci., 50, 177 (1961).
72. Hock C. W., Arbogast J. F., Anal. Chem., 33, 462 (1961).
73. Faubion B. ., Anal. Chem., 43, 241 (1971).
74. Barrail E. M., Johnson J. F., Thermochim. Acta, 5, 41 (1972).
75. Miller G. W., Thermochim. Acta, 3, 467 (1972).
76. Miller G. W., Analytical Calorimetry, Porter R. S., Johnson J, F., edi,|
Vol. 2, Plenum Press, N. Y., 1970, p. 397.
77. Bruckner H. P., Heide K-, Z. fur Chemie, 10, 125 (1970).
78. Wendlandt W. W., Thermochim. Acta, 1, 419 (1970).
79. Hyzer W. G., Research Development, 61, Feb. 1972.
80. Magill J. H., Nature, 187, 770 (1960).
Глава 10
КРИОСКОПИЧЕСКИЕ МЕТОДЫ ОПРЕДЕЛЕНИЯ
ЧИСТОТЫ ВЕЩЕСТВ
А. ВВЕДЕНИЕ
Чистоту органических и неорганических соединений можно
циределить многими методами: от простейших физических методов,
« пованных на определении точек кипения и плавления, до более
•/южных методов, таких, как метод абсорбции и эмиссионная спектро-
Цопия, требующих применения специальных приборов. В табл. 10.1
представлены основные методы определения чистоты (или при-
Таблица 10.1
Методы определения примесей в химических соединениях [1]
Метод L Чувстви- тельность* 1 Соединение
органи- ческое неоргани- ческое
(игктроскопия излучения и рентгеновская | псктроскопия 10-4—10-6 X
Мрнвационный анализ 10-1—10-8 X X
Яплярографический анализ 10-8 X X
JLcr-спектрометр и я 10-3—10-6 X X
Коматог рафия 10-5 X X
тгктроскопия поглощения 10-3 X
Кмерение электропроводности 10-8 X
Комический анализ 10"5 X X
* Минимальное содержание примеси, обнаруживаемое с помощью данного метода.
гссй) с помощью измерительных приборов. Приведенные в таблице
|ниые являются лишь оценочными и могут изменяться в широких
ределах в зависимости от вида основного компонента и присутст-
||фцих в нем примесей. Первые шесть методов позволяют опреде-
ить в одном эксперименте число примесей и содержание каждой
I них. С помощью метода электропроводности определяется содер-
Ьиие ионов примесей в водных и безводных растворах, а также
шных компонентов в полупроводниках. Несмотря на то что пре-
лы применения этого метода довольно ограничены, для ряда
шмесей он дает оптимальные результаты.
410 Глава 10
Наиболее широкими потенциальными возможностями для oiipi
деления чистоты химических веществ обладает метод термическим
анализа [11. Он применим ко всем веществам, достаточно устончи
вым при температуре плавления, и позволяет определить обннч
количество примеси, не растворимой в твердой фазе. Термический
анализ можно определить как метод, устанавливающий количсг i mi
примеси (или примесей) в веществе на основе зависимости темнгрп
тура—время, или температура—теплосодержание в окрестности
точки плавления вещества. Глазгоу и Росс [38] считают более upc i
почтительным общий термин «криоскопия», которым они назыпакн
науку определения точек затвердевания жидкостей или точек план
ления твердых веществ в условиях равновесия между твердой и
жидкой фазами и использование этих измерений для аналитически и
целей. Общепринятым является определение «точки затвердевании»
и «точки плавления» как температуры, при которой бесконгчии
малое количество твердого вещества находится в состоянии ранне)
весия с жидкостью при проведении измерений в условиях равнин»
сия с воздухом при давлении 1 атм (~0,1 МПа).
Для определения зависимости температура — время или гем
пература — теплосодержание вещества используются различи!*
методы. К ним относятся:
1. Термометрические методы (их не следует путать с термомстрн
ческим титрованием), при которых в различные моменты прощчти
плавления вещества получают кривые температура—-время. Вы
деление или поглощение тепла происходит непрерывно и пренму
щественно с постоянной скоростью. В термометрических метода»
не производится прямого измерения количества тепла, поглощен
ного или выделенного в единицу времени, но его можно рассчи r;»iii
как часть полной теплоты плавления вещества.
2. Калориметрические методы, при которых получают записи
мости теплосодержания от температуры. Для анализа в этом слу
чае применяется адиабатический калориметр или дифференциала
ный сканирующий калориметр. Последний, обеспечивая почти
такую же точность, как и адиабатический калориметр, горл «и
удобнее в работе.
3. Дилатометрический метод (определение зависимости обыМй
от температуры) и метод определения диэлектрической постоянной
вещества. Последний метод, по-видимому, не уступает в точное п]
другим методам, а также в силу своей физической природы не in
висит от количества материала, используемого для анализа. Uni
два метода здесь рассматриваться не будут.
Обзоры по применению термического анализа для оценки ши
тоты органических соединений были опубликованы Стётеваншм
[2], Сайном [3], Мэтью [4], Смитом [1, 5], Глазгоу и Россом [ЗЯ],
Скау и Артуром [39] и другими [40—42].
Криоскопические методы определения чистоты веществ
411
Б. ТЕОРИЯ
С теоретической точки зрения зависимость теплосодержания от
Чгмпературы рассматривалась многими исследователями, начиная
Т Уайта [6] в 1920 г. Другие ранние работы в этой области принад-
сжат Эндрюсу и др. [7], Скау [8], Мэйру и др. [9], Глазгоу и др.
ПО], Мэлотоксу и Штраубу [11], Томасу и Парксу [12]. Более
поздние исследования были выполнены Россини [13], Мастранело
и Дорнтом [14], Бэдли [15] и Смитом [1]. Для получения более под-
робной информации, чем в данной книге, следует обратиться к
Чтим источникам.
Фиг. 10.1. Кривая плавления для двухкомпонентной системы [16].
реальная кривая для равновесного состояния; ---кривая для идеализированного
<f|4iH,ecca нагревания без плавления (компонента удельной теплоемкости); 1 —нагревание твердо-
in образца; 2—плавление в эвтектической точке; 3 — нагревание н плавление образца; 4— на-
гревание жидкого образца.
Чтобы проанализировать зависимость температуры от времени,
[рассмотрим кривую, представленную на фиг. 10.1. Фундаменталь-
' пый анализ этой кривой сделан Уайтом [6] и дополнен Карлетоном
116]. Кривая зависимости температуры от времени в окончательном
чиде основана на линейном соотношении между затраченным теп-
лом и временем и на уравнении
^V2 = ~(T0-T) = A(T0-T)t (10.1)
где N2 — мольная доля растворенного вещества; ДД •— теплота
Плавления растворителя; То — температура затвердевания чис-
того растворителя; Т — равновесная температура. Это уравнение
применимо только в том случае, если рассматривается почти чис-
412
Глава 10
тое вещество, в котором примеси при затвердевании основного ком
понента остаются в растворе.
Линейное соотношение между подводимым теплом и временем
следует из существования постоянной разницы температур между
образцом и ванной. Подводимое тепло при плавлении имеет дп»
составляющие: а) составляющую, определяемую удельной тепли
емкостью и затрачиваемую на повышение температуры образна и
термометра; б) составляющую, непосредственно затрачиваемую ня
плавление вещества. На фиг. 10.1 показано разделение этих дпуд
составляющих во времени. Прямой участок PQ соответствует пл
греванию твердого образца и термометра. В случае двухкомш|
нентной системы образуется горизонтальный участок QR, соогш i
ствующий эвтектической температуре. За этим участком плавления
и нагревание происходят одновременно в соответствии с кривой Д'Л
Кривая сначала изгибается, а затем выравнивается, пока не р.к
плавится все твердое вещество; затем, когда начинается нагрева и пр
расплавленной жидкости, кривая резко изменяет наклон (участи
ST).
Штриховая линия соответствует идеализированному процег< у(
при котором весь твердый образец нагревается до точки плавлений
и затем изотермически плавится. Линии QO и OS отвечают двум
компонентам реального процесса — определяемым удельной тепли
емкостью и плавлением. Горизонтальная линия OS фактически
соответствует плавлению двух веществ, растворителя и растворен
ного вещества с различными значениями теплоты плавления. 0,1
нако для большинства исследуемых веществ количество расткн
репного вещества настолько мало, что при анализе центральной ч.ц
ти кривой можно считать, что доля расплавленного вещества при
порциональна соответствующему участку на линии OS.
Анализ кривых можно проводить несколькими способами, пн
пример по отрезку XY между реальной кривой и идеальной гори
зонтальной линией или по наклону прямой PQ, который можно
определить с ничтожно малой погрешностью. Однако измерении
обычно начинаются при температуре, при которой уже происходи!
плавление вещества, и поэтому наклон участка кривой, соответ
вующего компоненте, зависящей от теплоемкости, неизвестен. В тех
случаях, когда свойства исследуемого образца известны, наклон
отрезка XY может быть приближенно определен с учетом размер
ного фактора, удельной теплоемкости и теплоты плавления обр.п
ца, а также на основании грубой оценки количества примесей и
предположении идеального раствора. Однако обычно эти свойства
неизвестны, но либо сами свойства, либо наклон начального учли
ка кривой могут быть предварительно оценены с достаточной дли
дальнейшего анализа точностью.
Если через х выразить мольную долю примесей в исходном обри ।
це, а через Т!—температуру затвердевания образца с примесями (гем
Криоскопические методы определения чистоты веществ
413
пгратура в точке S), то из уравнения (10.1) можно получить следую-
Кре:
N2 = A(T0-T), (10.2)
х = Л(70-Л), (10.3)
ыкуда
N^x + A^-T). (10.4)
|’сли f — мольная доля растворителя, затвердевающего при темпе-
Ирнтуре Т, то
f — Nz х __ -4 (^i Т) __ АКТ (10 5)
' ДГ2 х+Д(7’1-Г) х + АКТ' ' * ’
Если At — время плавления, соответствующее отрезку KS, а
A/j — полное время плавления, представленного горизонтальным
[участком OS на идеальной кривой плавления, то
f=~- (Ю-6)
е
Уравнения (10.5) и (10.6) преобразуются в выражение
I д/ = "‘--7 7Г- <'°'7>
Таким образом, из теории следует, что график зависимости
At от АН АТ будет иметь вид прямой линии с тангенсом угла нак-
лона —х!А. Член уравнения А, равный
А = , (10.8)
КТ2
I характеризует свойства основного компонента образца. Если
I величина АН неизвестна, то А можно определить путем дополни-
тельных измерений с использованием образца, содержащего из-
I вестную мольную долю добавленного растворенного вещества. Дру-
гими словами, АН может быть определена путем сравнения кривой,
Полученной для исследуемого образца, с кривой для эталонного
вещества с известной теплотой плавления, полученной на этой же
установке. В таком случае х является произведением А и x!At при-
I чем последний член определяется из наклона кривой.
Аналогичное выражение можно получить для кривых, представ-
I ляющих зависимость теплосодержания от температуры. Россини [ 13]
I показал, что термодинамическое соотношение для равновесия меж-
ду жидкой фазой основного компонента и примеси, с одной стороны,
и кристаллической фазой основного компонента с другой,
выражается уравнением
In Nt = In (1 - А2) = - А (То - 7) [1 + В (То-Т) + ... ], (10.9)
414
Глава 10
где Ni и N2 — соответственно мольные доли основного и примеспо
го компонентов в жидкой фазе; То — температура затвердевании
чистого основного компонента (Л\ = 1); Т — равновесная темне
ратура смеси. Величина А определяется по уравнению (10.8), а Н
другая криоскопическая константа, определяемая по формуле
где Ср — молярная теплоемкость жидкости, меньшая теплоемкости
в твердом состоянии. Для образцов высокой чистоты значение /’
приближается к То а величина N2 — к нулю; поэтому уравнение
(10.9) можно записать в следующем виде:
N2 = A(T0-T). (10.11)
Если — мольная доля примеси в жидкой фазе для расплав
ленной части образца F, то
Af2 = 7v;_l, (10.12)
г
где N2* — мольная доля примеси в образце. Совместное рассмотрс
ние уравнений (10.11) и (10.12) дает
Л£ 1
Т = Т0-^-- (10.13)
Л F
График зависимости Т от 1/F представляет собой прямую липши
с наклоном —N2*IA. Пересечение этой прямой с осью ординат при
1/F = 0 соответствует температуре То. Таким образом, по наклону
прямой можно определить чистоту образца (содержание примеси
в образце). На фиг. 10.2 представлена кривая зависимости !//'
от температуры для бензотрифторида.
Приведенные выше расчеты основаны на следующих предполо
жениях [17]: а) Т — температура термодинамического равновесия,
б) в жидкой фазе образуется идеальный раствор; в) примеси иг
растворимы в твердой фазе и г) N2* 1 - Отклонение зависимости
Т—\/F от линейной может означать, что не все эти предположения
выполняются полностью.
Ганн [37] предложил для количественного определения содср
жания примесей другой метод, который может быть также исполь
зован для определения удельной теплоемкости и теплоты плавле-
ния образца.
В соответствии с законом Ньютона уравнение для передачи тепли
к образцу имеет вид
^L = k(Tb-Ts), (io.li)
at
Криоскопические методы определения чистоты веществ
415
Фиг. 10.2. Кривая плавления бензотрифторида [17].
где Ть — температура нагревательного блока; Ts — температура
образца; k — коэффициент теплопередачи. Скорость изменения
температуры образца определяется следующим образцом:
dTs _ k(Tb — Ts)
dt nCs + Cg
(10.15)
где n — число грамм-молекул в образце; Cs — молярная тепло-
емкость; Cg — теплоемкость стеклянной колбы, упаковки образ-
ца и части термопары. Если Ть возрастает с постоянной скоростью
г, то Ts будет асимптотически приближаться к кривой зависимости
температуры от времени и следовать параллельно ей, так что
dTs _ dTb
dt dt
(10.16)
и смещение по температуре h в данный момент времени за счет
подвода тепла будет равно
h = Тъ - Ts = г (nCs + Cg)/k, (10.17)
а смещение по времени I при данной температуре равно
I = Tb-Ts = nCs-Cg _ (10.18)
г k
Таким образом, I зависит только от теплоемкости и k, a h явля-
ется также функцией скорости нагревания г:
h — rl. (10.19)
В методе определения чистоты образца, использованном Ган-
пом [37 ], предполагается, что k — постоянная, но нет необходи-
мости знать ее численное значение, а также нет необходимости знать
численное значение величин п, Cs и С .
416
Глава 10
На фиг. 10.3 для иллюстрации расчета этим методом приведет
идеализированная кривая плавления. Кривая АВ отражает изме
нение температуры нагревательного блока, возрастающей примерив
с постоянной скоростью г. Кривая CGF отражает изменение темпе
ратуры образца t на том участке кривой, где она отклоняется ш
Фиг. 10.3. Идеализированная кривая плавления 137].
линии, параллельной АВ. Из уравнения (10.14) следует, что тепло,
переданное образцу при нагревании его от температуры Т, до Tf,
будет равно
Hf — Ht = k f (Tb — Ts)dt = k(ABFGC). (10.20)
it
Если бы плавление не сопровождалось поглощением тепла, то об-
разец нагревался бы по закону, описываемому кривой CDEF,
отрезок которой DE соответствует То — температуре плавления,
а участки CD и EF расположены на различных расстояниях 1С и /j
от линии АВ, отражающих различие теплоемкостей образца в твер-
дом и жидком состоянии, причем теплоемкость твердого образца
обычно меньше. Поглощение тепла определяется произведением
k(ABFEDC)-, следовательно, молярная теплота плавления образна
АН будет равна
пАН = k (ABFGC) — k (ABFEDC) = k (CDEFG). (10.21)
Криоскопические методы определения чистоты веществ
417
II ющад ь CDEFG, обозначаемая буквой Z, на практике обычно
фнределяется графическим интегрированием путем деления ее на
Сколько приблизительно равных треугольников, площадь кото-
йХ легко измерить.
Тепло, подводимое к образцу для нагревания его от температу-
ы Tt до температуры Тп, равно произведению k(AHIC) и обозна-
Кп'ся буквой Ж, Тепло, которое потребовалось бы для нагревания
нч'рдого образца от Tt до Тп в отсутствие плавления равно k(AJRC)
обозначается буквой X. Величина X определяется не графическим
к утрированием, а из выражения
X = rlc ( tn — it) = hc ( tn — /г). (10.22)
> личество тепла, которое потребовалось для плавления части
Оразца за время tn, равно k(W—X); величина W—X обозначается
грез Y. Величина, обратная расплавленной доле образца, F~*
типа
(10.23)
е F"1 может быть вычислена для любой точки кривой плавления.
Г Для идеальных или достаточно разбавленных растворов закон
нит-Гоффа, описывающий понижение температуры затвердевания,
•моет вид
То — температура плавления чистого вещества и N2 — моль-
пя доля примеси. Следовательно, график зависимости Тп от F~l
дет представлять собой прямую линию, тангенс угла наклона
порой, умноженный на криоскопическую константу RT2!AH,
•вен N2.
В. ЭКСПЕРИМЕНТАЛЬНОЕ ОБОРУДОВАНИЕ
I Для термического анализа статическим методом требуется пре-
^вионный адиабатический калориметр. В литературе описано
ИЬльшое число адиабатических калориметров. На фиг. 10.4 схема-
ически изображен калориметр, использованный Глазгоу и др. [18]
Идя определения чистоты бензола и других веществ в интервале
Г^гмператур 10—300 К-
I Внутри калориметра на тонкостенной трубке подвешен медный
Контейнер емкостью 106 мл с исследуемым образцом. К наружной
гонке контейнера радиально от центрального глухого канала для
Нагревательного элемента и термометра сопротивления, прикреп-
цялись луженые медные лопасти. Лопасти припаивались тонким
Иоем олова. Для обеспечения условий, близких к изотермическим,
418
Глава 10
к верхней части контейнера была прикреплена тонкая медная ни
ловая оболочка. Для уменьшения потерь тепла путем излучешн|
внешняя поверхность контейнера, внутренняя и внешняя поверх,
ности медной оболочки и внутренняя поверхность адиабатическом»
экрана были покрыты слоем электролитически осажденного золою
и отполированы. В пространстве, окружающем контейнер с обр.п
цом и адиабатический экран, поддерживался глубокий вакуум
10-6 мм рт. ст. (133 мкПа).
Сопротивление платинового термометра измерялось с помощью
моста Мюллера. По времени нагревания, силе тока и напряжении»
на нагревателе сопротивлением 100 Ом из константановой пронп
локи определялась электрическая мощность на входе прибора
Сила тока и напряжение в нагревателе измерялись потенциометром
Веннера, соединенным с катушкой сопротивления и переключай
лем напряжения. Время нагревания определялось точным счетчн
ком времени.
Расчеты, необходимые для определения удельной теплоемко» i и
образца при измерениях на адиабатическом калориметре, рассмо!
рены Сталлом [19]. При напряжении а, приложенном к контамам
нагревателя в течение t с во время нагревания образца, по нация
вателю проходит ток I А. Следовательно, количество тепла Н в дню
улях будет равно
H = Iet. (10.25)
Это количество тепла вызовет подъем температуры образца »ц
первоначальной Tt до конечной постоянной температуры Тf, уст а
новившейся в образце в результате нагревания. Таким образом,
AT — Tf—Tt — подъем температуры за счет количества тепля
Н и 1/2(Т/ + Tt) = Та— средняя температура нагреваемого отн.»
ема.
Подводимое тепло поглощалось контейнером массой w г с удели
ной теплоемкостью Срс при температуре Та и образцом массой U i
с удельной теплоемкостью Cps при той же температуре. Матемаги
чески это выражается формулой
Д = (®Срс + №Ср,)ДТ (10.211)
и в сочетании с уравнениями (10.14) и (10.25) дает
— — — Cpc=Cps. (10.27)
WT W р р '
Уравнение (10.27) является основным уравнением, используемым
для расчета теплоемкости образца. После небольших преобразим
ний его можно использовать для расчетов на ЭВМ.
Другие калориметры, используемые для исследования плавле-
ния, рассмотрены Кларке и др. [20], Астоном и Финком [21 ], 11нл
11
К вь!соковакуумному
насосу
К высокоВакуумной
системе
/г Взвешивающему
устройству
•иг. 10.4. Адиабатический калориметр для анализа летучих соединений
[18].
J — выводы; 2 — тонкостенная трубка из монель-металла; 3 — сосуд Дьюара; 4 — термообра
готанное кольцо; 5 — выводы с нагревателями; 6 — термопары для измерения температуры
Чрубки; 7 — экран с нагревателями; 8 — нагреватель калориметра; Р — термометр сопротив-
ления; 10 — термопары для измерения температуры экрана; 11 — зажимная колодка.
420
Глава 10
чером [22], Мэзи [23], а также Руэрвейном и Хаффманом
[24].
При использовании динамического метода постоянный подпои
тепла к образцу обеспечивают путем поддержания постоянно)и
теплового потока между образцом и окружающим пространством
Это можно осуществить двумя различными способами: в уста поп
ках с постоянной температурой стенок и в установках с регулпр\<
мой температурой стенок [1 ]. В установках с постоянной темпера
турой стенок поддерживается постоянная температура в простри)и i
ве между стенками контейнера и образцом. В установках с р<ту.
лируемой температурой стенок также осуществляется подвод и
образцу постоянного количества тепла, но в этом случае обра ten
окружен специальной оболочкой, в которой непрерывно поддер
живается такая температура, чтобы разность обеих темпера)ур
оставалась неизменной.
Смит [1] сделал краткий обзор конструкций различных уши
новок обоих типов. Установки первого типа были разработаны
Уайтом [6], а также Россини и др. [9, 101. Установки второю
типа описаны Томасом и Парксом [12], Мэлотоксом и Штраубом
[11 ], Карлетоном [16], Смитом и Катеманом [25], Смитом [26, 2/|,
Глазгоу и Тененбаумом [28], Глазгоу и др. [18], Хэндли Г.'9|,
а также Барнардом-Смитом и Уайтом [30].
Установки с постоянной температурой стенок применяются i лиц
ным образом для получения кривых охлаждения и затвердев.)пни
и не применяются при нагревании и плавлении веществ. Это, <>че
видно, связано с тем, что температура наружной стенки и ее тепл»
изоляционного слоя при подводе тепла к образцу должна подд» |>
живаться на гораздо более высоком уровне, чем при отводе тенД|
[1 ]. Поскольку изолирующие свойства вакуумной оболочки с уп
личением температуры резко ухудшаются вследствие излучении!
тепловой поток к нагреваемому образцу при допускаемой в уст
новке скорости нагревания будет ниже, чем противоположно и«
правленный тепловой поток при охлаждении образца с той же < ни
ростью.
В установках с переменной температурой стенок в зависимо) tn
от рабочего интервала температур применяется стеклянная колбп,
погруженная в емкость с жидкостью или в толстостенный металл и
ческий цилиндр. Температура жидкости или металлическою пи
линдра поддерживается на таком уровне, чтобы разность мгжД|
температурой стенки и температурой образца оставалась посклП)
ной. Между стенкой и сосудом, в котором находится исследуем мн
образец, имеется воздушное пространство, обеспечивающее иеап
ходимую изоляцию. Обычно различие указанных температур c.ot
тавляет ~2° С, а скорость нагревания образца довольно мала
примерно 0,1—0,3 град/мин. Приспособления для перемешивании
в установках такого типа не применяются.
Криоскопические методы определения чистоты веществ
421
Фиг. 10.5. Установка для опреде-
ления температур плавления [16].
I — термометр для образца; 2 — пробир-
ка с образцом; 3 — круглодонная колба
емкостью 300 мл; 4 — нагревательная ван-
на; 5 — трубка, выравнивающая темпе-
ратуру; 6 — образец; 7 — пластмассовое
кольцо; 8 — пробки с отверстиями; 9 —
термометр для ванны.
На фиг. 10.5 схематически изображена простая установка с
регулируемой температурой стенок, предложенная Карлетоном [16],
В затем усовершенствованная Смитом [26].
Оболочка из тонкой однородной пленки, которая окружает
образец, охватывает и рабочий конец ртутного термометра с ценой
Деления 0,1° С. Термометр устанавливается при помощи пробки с
отверстием в стеклянную трубку, вытянутую до нужных размеров
п окрестности шарика термометра. Для уменьшения влияния пуль-
саций температуры на трубке с исследуемым образцом при помощи
1Ластмассового кольца укрепляется другая трубка, несколько боль-
шего диаметра. Весь узел с образцом помещается в круглодонную
КПлбу емкостью 300 мл так, чтобы шарик термометра находился
приблизительно в центре колбы. Колба по горло погружается в со-
ответствующую нагревательную ванну термостата, снабженную
Мешалкой и термометром. Объем исследуемого образца ~0,3 мл.
Скорость нагревания ванны 0,3 град/мин. Кривая зависимости
температуры образца от времени записывалась, начиная от темпе-
ратуры на 15—20° С ниже температуры плавления исследуемого
кещества.
На фиг. 10.6 схематически изображена другая аналогичная ус-
гиновка, разработанная Глазгоу и др. [10] и усовершенствованная
Вэрнардом-Смитом и Уайтом [30]. Образец объемом ~25 мл зат-
вердевает или расплавляется в пробирке с двойной стенкой. Величи-
на теплового потока от охлаждающей или нагревательной ванны
К образцу регулируется степенью разрежения между стенками про-
Оирки. В установке используется вращающаяся мешалка. При
исследовании образцов меньшего размера для уменьшения объема
422
Глава 10
камеры образца в нее вставляется алюминиевая трубка. Темпера
тура образца измеряется платиновым термометром сопротивлении
и мостом Мюллера. Эту установку можно также использовать дли
определения теплоты плавления. В последнем случае в ванну пи
мещают ряд небольших алюминиевых лопастей, которые обеспсчи
вают равномерное распределение тепла по образцу.
Фиг. 10.6. Установка для определения температур затвердевания [30|
1 — выводы к мосту Мюллера; 2 — платиновый термометр сопротивления; 3 — вата; 4
охлаждающая трубка; 5 — сосуд Дьюара; 6 — охлаждающее вещество; 7 — теплоизоляции;
8 — алюминиевая лопасть; 9 — вращающаяся мешалка.
Г. ПОГРЕШНОСТИ, НЕДОСТАТКИ И ДРУГИЕ ФАКТОРЫ,
ВЛИЯЮЩИЕ НА РЕЗУЛЬТАТЫ ИЗМЕРЕНИЙ
Погрешности, возникающие при определении зависимости тем
пературы от теплосодержания или времени, подробно проаналп»
зированы Смитом [311 и Маккалофом и Уэддингтоном [17]. Cmiii
рассмотрел качественные соображения, касающиеся скоростей фл
зовых переходов, скоростей диффузии и разностей температур при
исследованиях «тонкопленочным» методом. Маккалоф и Уэддиш
тон на основе результатов оценки точек плавления 125 вещеетп
проанализировали недостатки калориметрического метода.
Криоскопические методы определения чистоты веществ
423
1. НЕДОСТАТКИ ДИНАМИЧЕСКОГО МЕТОДА
а. Переход Твердая фаза—Жидкость
При подводе тепла к системе ее температура возрастает до тех
• нор, пока суммарная скорость поглощения тепла при плавлении
i III станет равной скорости ее подвода. При отводе тепла от системы
гс температура понижается до тех пор, пока скорость выделения
тепла при кристаллизации не станет равной скорости охлаждения.
Ъремя, мин
«фиг. 10.7. Кривые нагревания (верхняя) и охлаждения (нижняя) образца
антипирина, содержащего 0,1 мол.% ацетанилида [31],
Оба эти состояния возможны при температуре ниже температуры
термодинамического равновесия Тт. Однако в окрестности этой тем-
пературы скорость выделения тепла может быть малой и лишь не-
много возрастать с понижением температуры. Следовательно, по-
лученная температура может значительно отличаться от равновес-
ной. Конечная температура остается постоянной до тех пор, пока
скорость тепловыделения равна скорости охлаждения. Это иллюст-
рируется кривыми нагревания и охлаждения антипирина с при-
месью 0,1 мол. % ацетанилида, приведенными на фиг. 10.7. Кривая
нагревания чистого антипирина имела область постоянной темпе-
ратуры при 110,45° С, не зависящую от скорости нагревания. Кри-
вая охлаждения, полученная при скорости, сравнимой со скоростью
нагревания, имела небольшой пик переохлаждения, после которо-
го температура возрастала всего до 109° С. В этом случае максималь-
ная температура зависела от скорости охлаждения. Кривые, полу-
ченные при нагревании и охлаждении смеси антипирина и 0,1 мол. %
ацетанилида, близки к кривым нагревания и охлаждения чис-
того антипирина.
424
Глава 10
Аналогичный характер кривых наблюдается для азобензол», I
бензилбензоата и нарн-ксилола; соответствующие кривые для n.n|<
талина несколько отличались по виду.
б. Переходы в твердой фазе
Обычно кривая нагревания отражает переходы в твердой фл ю, I
или энантиотропные переходы. Если плавление начинается до in
вершения перехода в твердой фазе, то кривая плавления будет п« I
I достоверной. Смит [31 ] рекомендует в таких случаях перед опреде I
। лением кривой плавления выдерживать образец в течение некоторн
го времени при температуре выше температуры перехода.
в. Скорости диффузии
Когда твердая фаза и жидкость в многокомпонентной chcicmo I
находятся в состоянии термодинамического равновесия, соснш
твердей фазы обычно отличается от состава жидкости. Если ешт I
г ма подвергается дальнейшему плавлению или кристаллизации, id
состав, по крайней мере, одной из фаз вблизи границы раздела с дру
гой фазой будет изменяться. Это будет стимулировать процесс днф
фузии, стремящейся уравнять концентрации компонентов в твердой I
и жидкой фазах.
г. Влияние перемешивания
Перемешивание способствует образованию однородной жидкий
фазы и не оказывает влияния на неоднородность твердой фа пл
Поэтому даже при перемешивании толстые слои могут оказании |
неоднородными. Перемешивание выгодно в тех случаях, когда чцр
| может вызвать размельчение твердых частиц, способствующее ort I
щему ускорению кристаллизации. Смит [31 [ вообще выражш-г
сомнение в целесообразности перемешивания, поскольку его мши
но использовать только при определенных соотношениях жидкий
и твердой фаз.
д. Тепловые потоки
I п
При подводе или отводе тепла возникают перепады температур
во всех частях калориметрической системы, включая стенки, обря
зец и даже термометр. Эти перепады являются источником rioi|w
шностей, величина которых зависит от тепловых потоков, ......
элементов системы и теплопроводности конструкционных материй
лов. Расчет величины этих погрешностей приведен в работе (’мши
[311.
Криоскопические методы определения чистоты веществ
425
е. Перепады температур при плавлении
I Плавление исследуемого образца начинается у внутренних сте-
нок контейнера и постепенно распространяется в направлении
шарику термометра. С началом плавления тепловой поток к термо-
Ьгру заметно уменьшается. Он не понижается до нуля, поскольку
Ьмнература термометра ниже температуры в зоне плавления. Раз-
ь ть между температурой, показываемой термометром, и темпера-
>урой зоны плавления дает погрешность измерения, постепенно
Уменьшающуюся до нуля. Важно знать, в течение какого времени
•nt погрешность уменьшится до величины, лежащей в пределах
>ц пости измерений. Смит предпринял попытку точно рассчитать
»Г<| время [31 ].
ж. Влияние контакта между поверхностями
I Между поверхностями стеклянных стенок контейнера, образцом
• шариком термометра не существует абсолютного контакта. Это
•>||)нподит к дополнительному перепаду температур. Тепловой по-
из окружающего пространства может через держатель посту-
Ьп> к шарику термометра и затем к образцу. При несовершенном
ш такте между образцом и шариком термометра последний будет
называть более высокую температуру.
2. НЕДОСТАТКИ СТАТИЧЕСКОГО МЕТОДА
Недостатки статического метода в большей или меньшей степени
<*|')Йственны любому методу определения температуры плавления.
Ниже рассмотрены эти ограничения [17 J.
а. Неопределенность в оценке концентрации примесей
Неравномерное распределение примеси в жидкой фазе может
iR|iiihccth к уменьшению наклона кривой плавления и в результате
I сниженным значениям N2*. Еще более серьезный источник оши-
бок — образование твердых растворов — уже давно признавался
«пн возможное ограничение всех методов определения чистоты ве-
гства по его точке плавления, но лишь в последнее время выясни-
лось, что это явление имеет общий характер.
б. Образование твердых растворов
Образование твердых растворов возможно даже в веществах
высокой степени чистоты, так как примеси часто бывают изомерами
яг ионного компонента. По данным Маккалофа и Уэддингтона [17],
примерно половина кривых плавления исследованных ими веществ
Импт заметные отклонения от линейности в координатах Т—1/F,
426
Глава 10
что указывает на образование твердых растворов. Фактически
кривые плавления, сохраняющие линейность на любой станин
плавления образца, встречаются редко. В большинстве случ.н и
наименьшую ошибку дает расчет содержания примесей по тангсш у
угла наклона кривой плавления, когда большая часть обра щч
уже расплавлена.
в. Применение теории твердых растворов
Если кривая плавления четко указывает на образование тигр
дого раствора В заметных количествах, то для точного расчета <п
держания примесей может потребоваться применение теории ик р
дых растворов [14, 15], хотя Смит [1 I подверг критике такой мены
[14]. К сожалению, этот метод зачастую оказывается неудовлспи*
рительным. В ряде случаев расчеты на основе теории твердых ран
воров хорошо согласуются с экспериментальными данными, п<>
высокая чувствительность метода к малым термометрическим
ошибкам делает результаты расчета содержания примесей недосю
верными. Например, разность температур при расплавлении /I)
и 90% образца может быть определена с погрешностью ±0,0005 <
В расчетах, основанных на теории твердых растворов, такая опшб
ка будет соответствовать погрешности 500% при определении со
держания примесей в сверхчистых веществах с нормальными крио
скопическими константами. Если же предположить, что раствори
мость в твердом состоянии отсутствует, то эта же ошибка (0,00051 )
дает погрешность в оценке содержания примеси, равную 150%,
3. СРАВНЕНИЕ РЕЗУЛЬТАТОВ, ПОЛУЧЕННЫХ
СТАТИЧЕСКИМ И ДИНАМИЧЕСКИМ МЕТОДАМИ
Следует отметить, что содержание примесей, оцениваемое era
тическими методами, систематически ниже результатов, получен
Таблица 10.
Сравнение результатов, полученных статическим и динамическим
методами [18]
Образец Чистота, мол, %
Расчет по известно- му содержанию примеси Динамический метод Статический метод
А 1001) 99,994 + 0,002 99,9937+0,0010
В 99,9964 90,970 + 0,004 99,958+0,005
С 99,9610 99,940 + 0,002 99,947 +0,005
1) Образец, чистота которого находится за пределами чувствительности применяемых
методов анализа.
Криоскопические методы определения чистоты веществ 427
цых на том же образце с помощью динамических методов [3, 4].
Однако весьма тщательные измерения, проведенные Глазгоу и др.
,||Н| для образца бензола высокой степени чистоты с добавкой из-
Ьстиого количества примеси н-гептана, показали, что различие
Ш'жду этими двумя методами не столь велико, как считалось ранее.
К табл. 10.2 приведены результаты такого исследования. Предпо-
йгается, что в данном случае причиной расхождения результатов
Гмализа статическим и динамическим методами является сорбиро-
Кйиная вода.
4. РЕКОМЕНДАЦИИ
Смит [31 ] предложил следующие рекомендации для проведения
|грмического анализа:
1. При использовании статического метода после каждого пе-
риода нагревания вещества должен следовать период адиабатиче-
ской выдержки достаточной продолжительности для достижения
необходимой степени равновесия.
2. При использовании динамического метода предпочтительнее
проводить исследования при нагревании, а не при охлаждении.
3. Перед началом измерения кривых нагревания образец следует
I не менее часа выдерживать при температуре, несколько меньшей
емпературы начала плавления.
4. При определении кривых нагревания не рекомендуется при-
менять перемешивание.
5. Скорость нагревания образцов, имеющих небольшую тепло-
му плавления, должна быть по возможности меньшей.
6. Каждую кривую следует подвергать проверке и выбирать
на ней участок, наиболее подходящий для определения содержания
примесей. Помимо экспериментальных проверок, касающихся ме-
тодики, следует проверить, удовлетворяет ли кривая уравнению
131]
CfCm
Cf + Y(Cm-Cf)
(10.28)
т = т
1 у 1 а
LITie Ту — температура, при которой расплавилась часть Y образца;
Та — температура плавления абсолютно чистого вещества; Cf и
i,’m—-константы; р — содержание примеси в образце, мол. %.
Уравнения (10.17) можно преобразовать к виду
Т =Ci--------, (10.29)
у Cs + Y
где Cit С2 и С3 — константы, которые можно определить алгебраи-
I чески путем выбора трех пар соответствующих значений Ту и Y.
График зависимости Ту от 1/(С3 Г Y) должен представить собой
прямую линию с наклоном, равным С2.
428
Глава 10
Несмотря на указанные выше источники погрешностей и oip*
ничения, термический анализ имеет ряд неоспоримых преимупи <
[1 ]. Будучи физическим методом, он не требует знания химичи пи»
свойств основного компонента и примесей. Он чувствителен, мне
и не в равной мере, ко всем видам примесей. Если образец pat < мц|
ривать как бинарную систему, то термический метод позволяет ирн
извести количественное определение содержания примесей.
Д. ПРАКТИЧЕСКИЕ ПРИМЕРЫ ОПРЕДЕЛЕНИЯ
СОДЕРЖАНИЯ ПРИМЕСЕЙ И ДРУГИЕ ПРОБЛЕМЫ
Карлетон [16] использовал экспериментальные кривые и ыи
ления для определения примесей в искусственных смесях нафта hi
на с антраценом или дифенилом. На фиг. 10.8 приведены крнньи»
время, с
Фиг. 10.8. Кривые плавления чистого нафталина (а) и нафталина, содгр
жащего 1,45 мол.% дифенила (6) [16].
1 — компонента удельной теплоемкости; 2 — компонента плавления; 3 — точка окончил мн
затвердевания; 4 — наклон компоненты удельной теплоемкости.
Криоскопические методы определения чистоты веществ
429
Мтсния чистого нафталина и нафталина, содержащего 1,45 мол. %
фенила. Горизонтальные линии идеальных кривых плавления
11'Секают ось ординат в точке, соответствующей температуре То.
। точек, соответствующих выбранным на экспериментальной кри-
м .значениям температуры Т, проведены прямые, наклон которых
»иЬетствует условиям нагревания образца до плавления (с учетом
. ъко теплбемкости). Наклоны этих линий определяются путем
изложения наклона кривой равновесия при 70° С на отдельные
I люненты (теплоемкости и плавления) с учетом размеров уста-
«мки и свойств нафталина. '~"а
Г Для чистого нафталина расчет был произведен следующим об-
Мзом: были вычислены величины То и содержание примеси х, равные
Ответственно 79,7°С и 0,003 мол. %. Расплавленная доля об-
мана 1—/ в интервале температур 10°С (65—75°С) была рассчи-
11 i по формуле
х
(10.30)
1 — f = ~
' N2 х+А(Т0-Г)
Поскольку для нафталина А = 0,0184, то 1—f — 0,0226. Для ин-
ь риала температур 65—75°С компонента плавления определяется
pi выведением 0,0226 • 149 Дж/г = 3,37 Дж/г (для нафталина
А// = 149 Дж/г). С учетом размеров установки полная величина
Ьмпоненты удельной теплоемкости составляет 46,5 Дж/г.
Строится треугольник, большую сторону которого образует
Иивая равновесия при 70°С (фиг. 10.8, а). Две другие стороны,
ношение которых равно 3,37/46,5, соответствуют компонентам
давления и удельной теплоемкости. Для анализа использован на-
шим компоненты удельной теплоемкости. Для выбранных величин
Т были проведены линии, имеющие наклон, равный наклону ком-
1(«>иенты и удельной теплоемкости, и в пересечении с горизонтальной
сальной кривой плавления были получены соответствующие зна-
. ния А74 (фиг. 10.8, б).
Г На фиг. 10.9 показана зависимость А/ от А//АТ для смеси
ыфталина с дифенилом. Оптимальная прямая, проведенная
' рез точки, имеет наклон, равный —0,95; следовательно, содер-
> нние примесей, включающих дифенил и первоначальные примеси,
юно х = 0,0175 = 1,75 мол. %. Аналогичный анализ одного
лько нафталина дает х = 0,34 мол. %. Таким образом, экспери-
>-цтально определенное содержание дифенила составляет 1,41 мол. %,
г действительности же к нафталину было добавлено 1,45 мол. %
дифенила.
В методе, использованном Швабом и Вихерсом [321, количество
первоначально содержащихся в образце примесей определялось
<io кривым охлаждения (затвердевания) исходного образца и того
। *i* образца с добавкой известного количества примеси. Авторы счи-
»иют, что этот метод сравнения кривых применим даже в тех случаях,
430
Глава 10
когда доля затвердевшего материала нелинейно изменяется со ирг
Дг/ДТ
Фиг. 10.9. Кривая зависимости AZ от kt/кТ для нафталина, содержащст
1,45 мол.% дифенила [16].
На фиг. 10.10 приведен ряд кривых затвердевания, использус
мых в методе сравнений. Разность А7 между начальной темпера-
турой затвердевания и температурой, установившейся за время, рай
ное половине времени полного затвердевания, определяется спот
бом, показанным для кривой 1. Затем получают кривую затвердепи
ния для образца с известным содержанием примеси х1 мол. % при
той же скорости охлаждения и опять определяют разность А
между новой начальной температурой затвердевания и темпера! у
рой, установившейся за время, равное половине времени полной!
затвердевания. Аналогичная процедура повторяется еще раз при
добавлении новой порции примеси х2 и снова определяется величина
ДТ2.
Из соотношения
\ТХ — АТ _ АТ2 — АТ
Xi Xg
(10.31)
можно определить первоначальное содержание примеси х в обра in
хх\т
АТ1! — АТ ’
(1()..I'J)
Опыт повторяется два-три раза и вычисляется погрешность измерь
ний. Среднеквадратичное отклонение, полученное по результ.тгиМ
нескольких измерений, составляет в среднем примерно ±х/8.
Криоскопические методы определения чистоты веществ
431
При использовании рассматриваемого метода важное значение
.ест правильный выбор вещества, добавляемого к исследуемому
Вразцу. В общем случае температура плавления этого вещества
Кжна быть ниже температуры плавления основного компонента
I, кроме того, добавляемое вещество не должно образовывать твер-
I.IX растворов и химических соединений с исследуемым образцом.
• иг. 10.10. Кривые зависимости температуры от времени, полученные при
затвердевании веществ [33].
I При построении по экспериментальным точкам кривой затвер-
вания следует учитывать возможность отклонений по времени
Вследствие нелинейности изменения температуры во всей системе,
неточностей в измерении температуры и т. д. Построение действи-
тельной кривой по экспериментальным точкам является сложной
дачей. Для проведения этой кривой используются разные прие-
мы, например гибкая линейка с прорезью. Сейлор [34] успешно
применил оптический метод с использованием проекционного аппа-
рата. Другой способ предложен Кинитцем [35]. Этот метод
остоит в том, что на основе полученных экспериментальных точек
Троится гипербола, которая лучше всего описывает эксперимен-
432
Глава 10
тальную кривую. Этим способом удается точнее определить темпе-
ратуру затвердевания, чем аналитическим или геометрический
методом оценки по трем точкам кривой равновесия.
Фиг. 10.1L Кривая плавления н-пентаиа [20].
о н-пентан, чистота 99,80 мол. %; X Этот же образец с примесью 2,4 мол. % изоокт-к»
Кларке и др. [20] определили калориметрическим методом чш п>
ту н-пентана. На фиг. 10.11 приведены результаты этих измерений
в виде зависимостей сопротивления (температуры) от времени д im
чистого н-пентана и искусственной смеси н-пентан — изоокт.in
Для определения содержания примесей эти данные переводятся а
единицы, соответствующие доле образца, расплавленной пос/ц
каждого периода равновесия, с учетом количества тепла, необходп
мого для нагревания твердой и жидкой составляющих образца, п
потерь тепла за счет излучения и теплопроводности. Теплота пл на»
ления, полученная в этой работе, составляет 8,75 кДж/моль, что
соответствует чистоте н-пентана 99,79 мол. %.
Криоскопические методы определения чистоты веществ
433
К образцу чистого н-пентана добавлялось 2,4 мол. % изоокта-
Полученная при этом кривая плавления имела значительную
пвизну, в то время как чистому н-пентану соответствовала пря-
чя линия. Чистота н-пентана, определенная по наклону этой кри-
>и, составляла 97,58 мол. % при расчетной величине 97,53 мол. %.
Г С помощью этого же прибора была определена чистота двух об-
ft,i шов пентаборана. Чистота первого образца составила 99,99 мол. %,
« второго 99,91 мол. %. Чистота смеси этих образцов должна
ft ia бы составлять 99,94 мол. %. Калориметрический анализ
Кгси дал цифру 99,949 мол. %.
1Г. 10.12. Т—х-диаграмма для смесей я-С^Нм и я-С2зН48 (а) и кривая на-
гревания для смеси 50 мол.% н-С21Н44 и 50 мол.% я-С23Н4в [231.
На специальной установке, разработанной Глазгоу и Тенен-
цумом [28], по температуре затвердевания была проверена чисто-
I некоторых высокоактивных веществ, таких, как тетрахлорид
liana TiCl4. Температура затвердевания тетрахлорида титана
рн отсутствии в нем примесей и при давлении насыщенного пара
Цтавляла —24,10+0,01 “С.
Мэзи [23] произвел термический анализ ряда нормальных ал-
|пов. На фиг. 10.12 приведены кривые для бинарной смеси, ха-
иктеризуемой полной взаимной растворимостью компонентов как
• идком, так и в твердом состоянии. Кривая на фиг. 10.12, а пред-
I тляет собой диаграмму состояния, выражающую зависимость
Ьмпературы от состава. На фиг. 10.12, б приведена кривая нагре-
,|ция для искусственной смеси, состоящей из 50 мол. % н-С21//44
эО мол.% н-С2зН48. Кривая нагревания простая и легко ин-
|рпретируется. Содержание примеси (в данном случае это коли-
тво второго компонента) можно рассчитать по этой кривой с дос-
рочной точностью.
-230
434
Глава 10
При плавлении некоторых органических солей, таких, как ши
логексиламинстеарат, циклогексиламинпальмитат и др., по-нпднми
му, имеет место обратимая реакция вида
CS + DP DS + СР,
где CS и DP — соли аминов. Эта система состоит фактически IH
четырех веществ. Системы такого типа исследовались с помонпчн
термического анализа Скау и др. [36].
Е. ОПРЕДЕЛЕНИЕ ПРИМЕСЕЙ МЕТОДОМ ДИФФЕРЕНЦИАЛЬНОМ
СКАНИРУЮЩЕЙ КАЛОРИМЕТРИИ
1. ВВЕДЕНИЕ
Методы термического анализа (криоскопии) в тех случаях, мп
да они применимы, давно известны как лучшие способы онредг и
ния чистоты веществ, позволяющие путем прямых измерений опрг
делять общее количество примесей [43]. При использовании ыкн
го метода требуются только физико-химические характерце цшн
вещества и не требуется образец. В настоящее время в лаборти
риях для определения чистоты веществ применяется метод дп<|и|»
ренциальной сканирующей калориметрии (ДСК) 140—48], не ц>
бующий сложного оборудования, образцов больших размером i
большой продолжительности анализа. С помощью этого метода мшк.
но анализировать более 75% органических кристаллических мг
ществ, имеющих достаточную чистоту |43]. Поскольку методом Д1 h
измеряется абсолютное количество примесей, то относительно бо н
шая погрешность эксперимента не влияет на точность аналн ia i
первого десятичного знака. Опытный экспериментатор може т <>ш
нить чистоту невзвешенного образца с точностью ±0,2мол. % и\
визуального просмотра кривой ДСК, полученной в течение 3 мни
2. ТЕОРИЯ
Теоретическим обоснованием для использования метода /1< I
при определении содержания примесей является уравнение В.нц
Гоффа, на основе которого выводится следующее соотношение мел
ду уменьшением температуры плавления и содержанием примиrft
[42, 441:
Ts = Т0------°— — ,
5 АЯ/ F
(Ю И|
где Ts — температура образца в данный момент времени, К; / 0 <
температура плавления абсолютно чистого вещества (раствории*
ля), К; R — газовая постоянная 11,987 кал/(мол• град) I; А//,
теплота плавления образца (растворителя), кал/моль; X - - мои.
Криоскопические методы определения чистоты веществ
435
I
мн доля примеси; F — часть образца, расплавленная при темпе-
Пуре Ts. Таким образом, график зависимости Ts от 1/F должен
(надставлять собой прямую линию с наклоном RT02Х/&Нf, пере-
дающую ось ординат в точке 7\. Величина AHf определяется
нитрированием площади пика кривой ДСК. По кривой также
проделается Ts и 1/F (величина, обратная отношению части пло-
1пди пика под кривой ДСК до Ts к полной площади).
3. НЕКОТОРЫЕ ОСОБЕННОСТИ ИСПОЛЬЗОВАНИЯ
ДИФФЕРЕНЦИАЛЬНОГО СКАНИРУЮЩЕГО КАЛОРИМЕТРА
ДЛЯ ОПРЕДЕЛЕНИЯ ЧИСТОТЫ ВЕЩЕСТВА
I Применение рассмотренного выше уравнения для определения
стоты веществ требует введения поправок с целью учета двух
it ременных величин, связанных с прибором: термического сопро-
иления прибора и не регистрируемого прибором плавления. Спосо-
М корректировки подробно рассмотрены в работах [42, 44, 47, 49 J.
478 479 480 481 482 483 484 485 486 487 488
Регистрируемая температура, К
)Г. 10.13. Поправка к кривой ДСК на термическое сопротивление [42]
| Исследуемое вещество — 9,10-днхлорантрацен, содержащий 1,10 мол. % антрацена.
Образца 2,562 мг; скорость нагревания 1,25 град/мин. 1 — температура с учетом термнчес-
I» сопротивления (212,46°С); 2 — наблюдаемое начало плавления; 3 — базовая линия при
(Жрованин; 4—изотермическая базовая линия; 5 — экстраполяция с учетом термического соп-
ротивления; 6 — пик плавления олова.
врол и Диллер [421 вводят поправку на термическое сопротивле-
(фиг. 10.13), учитывая величину термического сопротивле-
|н между платформой < нагревателя и дном капсулы с исследуемым
разцом. Для определения поправки в первом приближении необ-
436
Глава 10
ходимо расплавить вещество высокой степени чистоты (>99,99"ii)
и записать его кривую плавления. Наклон передней восходящей шч
ви этой кривой пропорционален термическому сопротивлению. Дли
введения поправки к любой температуре в области регистрируемо! о
перехода необходимо совместить этот наклон с полученной кривой
и экстраполировать к изотермической базовой линии. Для обы
печения максимальной точности чистый материал (обычно метал-
лический индий) должен плавиться в интервале температур, дли
которого производится коррекция, или в его; окрестности. Кик
показывают измерения, проведенные на индии, олове и свинце ны
сокой чистоты, термическое сопротивление возрастает с увелнчс
нием абсолютной температуры [42].
Рассматривались также поправки к кривой ДСК, учитывающие
смещение базовой линии из-за изменения теплоемкости [47, 501,
а также тепловое запаздывание [49].
На фиг. 10.14, а показано влияние этих небольших поправок
на примере кривой ДСК для циклогексана. Правильная темпер»
тура образца Ts получается путем экстраполяции кривой к истин
ной базовой линии [44]. При этом к рассматриваемой парциальной
площади добавляется небольшая площадь ABCD. На фиг. 10. М,
приведена типичная зависимость Ts от \JF. Как видно из фигуры,
непосредственно полученная зависимость Ts от 1/F отклоняете»
от прямой линии [44]. Это происходит вследствие того, что некою
рая площадь пика не фиксируется за счет шумов и ограничен null
чувствительности прибора до тех пор, пока перо заметно не otk.hi!
нится от базовой линии. Для получения прямой линии методом пры>
и ошибок последовательно добавляют небольшие участки площад! в
как к парциальной площади, так и к общей площади всей кривой
Одним из самых серьезных ограничений данного метода явлн
ется допущение о невозможности образования твердых растворов
Для объектов в виде твердых растворов уравнение (10.33) можно
записать следующим образом [14, 44]:
_ „ RTlX 1
Ts = То-------------------
* 0 ДЯ/ Л/(1- К) +F
(10 И1
где К = k/k' — отношение распределения примесей между жидкоп
и твердой фазами, равное нулю при отсутствии твердых растворов
Однако не существует критерия, позволяющего обнаружить тт р
дые растворы по кривой ДСК- С целью упрощения этих расчеши
разработаны программы для ЭВМ.
Бэррол и Диллер [42] предлагают следующие рекомендации
для практического определения чистоты вещества методом ДСК
1. Размер образца должен быть менее 3 мг.
2. Скорость нагревания должна быть менее 1,25 град/мин.
3. При анализе летучих веществ их следует помещать в герм* |
I
Температура, К
7/5
г. 10.14. Типичные кривые ДСК для определения чистоты циклогексана
и график зависимости температуры образца от 1/F для 5,7-диметил-1,3
адамантандиола [44].
поправка наклона с учетом теплового запаздывания; 2— \j‘2 высоты пика; 3 — парциальная
Щадь AEFG-}- ABCD; 4 — экстраполированная базовая лине я; 5—'базовая линия с учетом
поправки; 6 — предел для 1/F. о исходные значения; □ поправка на 5 %.
438 Глава 10
тичную капсулу и необходимо обеспечить хороший термичссмш
контакт.
4. Ось температур должна быть точно откалибрована. Jlp11 |‘*
счете 1/F площадь следует измерять от первой отмечаемой npiifiit
ром температуры (начала) плавления до эндотермического миниму-
ма. Должно быть определено не менее шести точек.
5. Размеры образца и скорость нагревания должны быть но/цЛ
раны таким образом, чтобы наклон эндотермической кривоп |ь
превышал наклона кривой чистого эталонного вещества, котп|....
она имеет на половине высоты пика.
6. Запаздывание по температуре должно быть измерено г пн
мощью эталонного вещества с температурой плавления, блпзьпП * ।
температуре плавления исследуемого вещества. .
Все эти шесть рекомендаций связаны с тепловым запаздывании»,
временной характеристикой калориметра и необходимостью пр<т<«
дить исследование в условиях, наиболее близких к равновее ныМ
Образование эвтектической смеси с температурой плавления, б/НМ
кой к температуре плавления основного вещества, уменьшает м>ч (
ность метода ДСК, но при низких концентрациях вы л»,
можно получить удовлетворительные результаты. В неком >ри«
случаях методом ДСК можно исследовать вещества, образукиИН»
смешанные кристаллы, однако расчеты, основанные на уравш iii|i
Вант-Гоффа, не дают в этих случаях удовлетворительных реп и-
татов. При содержании примесей менее 5 мол. % калибровочпйя
кривая, построенная на основе зависимости эндотермичы мин
минимума от концентрации примесного компонента, позволив
с высокой точностью оценить концентрацию примеси.
Таблице /•< ||
Сравнение чистоты некоторых углеводородов, очищенных зонной кристаллизацией, по данным ГХ и
дск pi 1
Соединение Марка ГХ, % площади дск. МП-'ll
Аценафтен и НС 322 99,99(170) 99,99 99,
Антрацен Дибензил Дифенил U НС 323 U НС 324 99,99(220) 98,24 (125) 99,99(170) 99,95 99,97 99,99 99,99 99,И 9'1,Щ ЧЧ.П» 99 , *4
Дурол ....... Нафталин U НС 325 99,99(75) 99,96 99,«
UHC 326 99,99(140) 99,96 99, *11
U НС 327 99,99(140) 99,99 99, W
Фенантрен 99,99(210) 99,99(210) 99,75 99,73 •19 99.Н 99 4
Пирен U НС 328 99,97(250) 99,94
U НС 329 99,99(250) 99,97 ‘14 *•
/пранс-Стильбен . 99,77(200) 99,97 99 91
поро-Трифеннл и НС 330 99,99(240) 99,97 9'» .Ни
Криоскопические методы определения чистоты веществ 439
I
I Джой и др. [41] сравнили результаты определения чистоты ве-
Иеств, полученные методом ДСК, газовой хроматографии (ГХ)
I титрометрическим методом. В табл. 10.3 приведены результаты
нализа методами ДСК и ГХ ряда полициклических углеводородов,
ищейных путем многократной зонной очистки с последующей воз-
мпкой. Как видно из таблицы, несмотря на различную физическую
В|’иРОДу методов, полученные результаты дают хорошее совпадение,
педует, однако, отметить, что значение 99,99% площади простав-
лю в таблице для газовой хроматографии в тех случаях, когда не
рло обнаружено пика примеси.
4. ПРИМЕНЕНИЕ МЕТОДА ДСК
ДЛЯ ОПРЕДЕЛЕНИЯ ЧИСТОТЫ ВЕЩЕСТВ
11 Дифференциальная сканирующая калориметрия была примене-
для определения чистоты большого числа веществ [41]. Список
pi ледованных этим методом веществ включает: алифатические угле-
{Ьороды [44], амиды, амины и карбаматы [43, 51], производные
Ьзола [43, 51, 52], галогензамещенные соединения [41, 43, 44],
личную кислоту [52], органические фосфаты [43], пестициды [43]’
рмацевтические препараты [51, 52], стероиды [52], бензойную
Ьк-лоту [41], полициклические углеводороды [41], мочевину [41],
пестерин [41], вещества, образующие жидкие кристаллы
, 53, 54 ], и др. В одной из работ [43] приведены результаты опре-
|.1ения содержания примесей и теплоты плавления методом ДСК
В 95 сверхчистых органических соединений.
5. ОЦЕНКА МЕТОДА
IВ связи с тем что метод ДСК позволяет производить быстрое
Следование веществ чиСтотой от 98,0 до 99,95 мол. % при ис-
шьзовании образцов массой в несколько миллиграммов, он явля-
к» в настоящее время наиболее ценным методом оценки чистоты
Салических соединений [41]. Тем не менее следует отметить, что
I термически стабильного соединения низкая степень чистоты,
ученная этим методом при достаточном числе измерений, явля-
В>1 достоверным свидетельством, что вещество действительно не
Ист высокой чистоты. И наоборот, высокая степень чистоты, по
иным ДСК, еще недостаточное подтверждение, что соединение
клвительно является таковым. При чистоте веществ выше
ВО мол.%, у которых ход кривых ДСК до температуры плавле-
I корректируется расчетом, величина определяемой чистоты
Вно зависит от принятых при расчете допущений. Таким обра-
В в совРеменных приборах практическим верхним пределом для
шлютных измерений методом ДСК является чистота 99,95мол.%.
° же время возможно определение разницы в содержании при-
440
Глава 10
месей до 0,005 мол.%. При многократном повторении анализ
может быть определена относительная чистота двух партий вещей
ва до 99,98 мол.%.
ЛИТЕРАТУРА
1. Smit W. М., Z. Е lektrochem., 66, 779 (1962).
2. Sturtevant J. М., Calorimetry, Techniques of Organic Chemistry, 2nd < d
Weissberger A., ed., Vol. 1, Interscience, N. Y., 1949, Part 1, p. 731.
3. Cines M. R., Solid-Liquid Equilibria of Hydrocarbons, Physical Cheiululu
of Hydrocarbons, Farkas A., ed., Vol. 1, Academic Press, N. Y., HKhI
Ch. 8.
4. Mathiew M. P., Acad. Roy. Belg. Classe Sci. Mem., 28, № 2 (1953)
5. Smit W. M., Thermal Analysis, Elsevier Press, Amsterdam, 1959.
6. White W. P., J. Phys. Chem., 24, 393 (1920).
7. Andrews D. H., Kohmann G. T., Johnston J., J. Phys. Chem., 20, ‘l|‘
(1925).
8. Skau E. L., J. Am. Chem. Soc., 57, 243 (1935).
9. Mair B. J., Glasgow A. R., Rossini F. D., J. Res. Natl. Bur. Std. (U. Л.1
26, 591 (1941).
10. Glasgow A. R., Streiff A. J., Rossini F. D., J. Res. Natl. Bur. Std. (U. ' Л
35, 355 (1945).
II. Malotaux R. N. M. A., Straub J., Rec. Trav. Chim., 52, 275 (1933).
12. Thomas S. B., Parks O. S., J. Phys. Chem., 35, 2091 (1931).
13. Rossini F. D., Chemical Thermodynamics, Wiley, N. Y., 1950.
14. Mastrangelo S. V. R., Dornte R. W., J. Am. Chem. Soc., 77, 6200 (lOM
15. Badley J. H., J. Phys. Chem., 63, 1991 (1959).
16, Carleton L. T., Anal. Chem., 27, 845 (1955).
17. McCullough J. P., Waddington G., Anal. Chim. Acta, 17, 80 (1957).
18. Glasgow A. R., Ross G. S., Horton A. T., Enagonio D., Dixon II I
Saylor С. P., Furukawa G. T., Reilly M. L., Henning J. M., Anal. C'/id
Acta, 17, 54 (1957).
19. Stull D. R., Anal. Chim. Acta, 17, 133 (1957).
20. Clarke J. T., Johnston H. L., De Sorbo W., Anal. Chem., 25, 1156 (HIM
21. Aston J. G., Fink H. L., Anal. Chem., 19, 218 (1947).
22. Pilcher G., Anal. Chim. Acta, 17, 144 (1957).
23. Mazee W. M., Anal. Chim. Acta, 17, 97 (1957).
24. Ruehrwein R. A., Huffman H. M., J. Am. Chem. Soc., 65, 1620 (IIHfl
25. Smit W. M., Kateman G., Anal. Chim. Acta, 17, 161 (1957).
26. Smit W. M., Chem. Weekblad, 36, 750 (1939).
27. Smit W. M., Rec. Trav. Chim., 75, 1309 (1956).
28. Glasgow A. R., Tenenbaum M., Anal. Chem., 28, 1907 (1956).
29. Handley R., Anal. Chim. Acta, 17, 115 (1957).
30. Barnard-Smith E. G., White P. T., Anal. Chim. Acta, 17, 125 (1957) I
31. Smit W. M., Anal. Chim. Acta, 17, 23 (1957).
32. Schwab F. W , Wichers E., Temperature — Its Measurement and CmilM
in Science and Industry, Reinhold, N. Y., 1941, p. 256.
33. Herington E. F. G., Anal. Chim. Acta, 17, 15 (1957).
34. Saylor С. P., Anal. Chim. Acta, 17, 36 (1957).
35. Kienitz H., Anal. Chim. Acta, 17, 43 (1957).
36. Skau E. L., Magne F. C., Mod R. R., Anal. Chim. Acta, 17, 107 (l|Hl|
37. Gunn S. R., Anal. Chem., 34, 1292 (1962).
38. Glasgow A. R., Ross G. S., Treatise on Analytical Chemistry, Kolthoff I I
Elving P. J., eds., Vol. 8, Interscience, N. Y., 1968, Part I, p. 4'Н1|Л
39. Skau E. L., Arthur J. C., Physical Methods of Chemistry, Weissbcrgri
Криоскопические методы определения чистоты веществ
441
Rossiter В. Weds., Vol. 1, Wiley-Interscience, N. Y., 1971, Part V,
p. 105.
(I. Barrail E. M., Johnson J. F., Purification of Inorganic and Organic Mate-
rials, Zief M., ed., Marcel-Dekker, N. Y., 1969, p. 77.
I. Joy E. F., Bonn J. D., Barnard A. J., Thermochim. Acta, 2, 57 (1971).
L Barrall E. M., Diller R. D., Thermochim. Acta, 1, 509 (1970).
I. Plato C., Glasgow A. R., Anal. Chem., 41, 330 (1969).
Й. Driscoll G. L., Duling I. N., Magnotta F., Analytical Calorimetry, Por-
ter R. S., Johnson J. M., eds., Vol, I, Plenum Press, N. Y., 1968, p. 271.
I Driscoll G. L., Duling I. N., Magnotta F., Sun Oil Quart. Rev., №3, 24
F (1969).
I'. Barrall E. M., Vogel M. J., Thermochim. Acta, 1, 127 (1970).
I Heuvel H. M., Lind К. C. J. B., Anal. Chem., 42, 1044 (1970).
K. Sondack D. L., Anal. Chem., 44 , 888 (1972).
B. Gray A. P., Perkin-Elmer Instrument News, 16, № 3, 9 (1966).
fl. Brennan W. P., Miller B., Whitwell J. C., Ind. Eng. Chem. Fundam., 8,
314 (1969).
I Reubke R., Mollica J. A., J. Pharm. Sci., 56, 822 (1957).
I. De Angelis N. J., Papariello G. J., J. Pharm. Sci., 57, 1868 (1968).
I Barrail E. M., Johnson J. F., Porter R. S., Mol. Cryst., 8, 27 (1969).
I Ennulat R. D., Mol. Cryst., 8, 247 (1969).
I
Глава 11
ДРУГИЕ МЕТОДЫ ТЕРМИЧЕСКОГО АНАЛИЗА
А. ВВЕДЕНИЕ
Хотя основными методами термического анализа являются пр
могравиметрия и дифференциальный термический анализ, cyiip 11
вует ряд других термических методов, весьма полезных при ни и
довании некоторых типов химических соединений. Многие из шн
методов разработаны недавно, но имеют потенциальные возмо/н*
ности для использования в будущем. Некоторые из них исполни*
ются в дополнение к методам ТГ или ДТА и, следовательно, д.иы
возможность еще глубже изучить рассматриваемый вопрос.
Почти все аналитические методы, позволяющие получать Н'М
пературные зависимости параметров, можно отнести к методам п-р
мического анализа. Сюда можно было бы включить спектроскопии!
в ультрафиолетовой, видимой и инфракрасной областях спскгрц
ядерный магнитный резонанс, электронный спиновый резонам,
методы дифракции рентгеновских лучей и электронов и многие др)
гие. Ограниченный объем книги не дает возможности столь niiipn
ко представить термический анализ. Поэтому в данной главе буду!
рассмотрены только такие общеизвестные термические методы, кик
дилатометрия (термомеханический анализ), термолюминесцепппн,
электропроводность, эманационный термический анализ и i 1
К сожалению, каждый из этих методов будет описан кратко, н i
время как одной лишь дилатометрии можно было бы посвяпиь
целую монографию.
Б. ДИЛАТОМЕТРИЯ (ТЕРМОМЕХАНИЧЕСКИЙ АНАЛИЗ)
1. ВВЕДЕНИЕ
Метод, называемый дилатометрией, состоит в определении
изменения длины или объема образца в зависимости от темпера ц
ры. Этот метод уже давно используется при исследовании керамики
и металлов и недавно стал применяться для исследования полиме р
ных материалов. В случае полимеров этот метод может быть при
менен не только для оценки изменений длины или объема. При п>
пользовании соответствующих датчиков и контрольно-измерп irJI*
ной аппаратуры можно определять температуру размягчения, Ц’М
Другие методы термического анализа
443
ературу стеклования, деформацию при нагревании под нагрузкой,
ОДуль упругости и другие параметры. Поэтому термин «дилато-
ггрия» используется не часто; обычно применяется более широкое
,более определенное название метода — термомеханический анализ
ГМА). Дилатометрию твердых тел называют также термодилато-
Мприческим анализом (ТДА) и термическим анализом размеров
ГАР). В данной книге используются два термина •— «дилатомет-
тя» и «термомеханический анализ».
Классическая дилатометрия обычно применяется для обнару-
Ц'иия изменений объема или длины, вызванных различного рода
(новыми переходами. Чаще всего определяется фазовый переход
| твердом состоянии: Твердая фаза 1 -> Твердая фаза 2; хотя
ржно определять и переходы Твердая фаза -> Жидкость и Твер-
дя фаза -> Газ. Дилатометрия может быть также использована для
пределения усадки и спекания образца при нагревании, для опре-
снения линейного коэффициента теплового расширения образца,
^пературы стеклования Tg, температуры размягчения, темпера-
и), при которых наблюдается рост деформации, и т. д. Исследова-
Ия проводятся в интервале температур от —150 до 2200° С как при
Ьгревании, так и при охлаждении образца.
Грей 11] рассмотрел метод регистрации производной кривой
МА, т. е. метод дифференциального термомеханического анализа
1ТМА). Этот метод особенно ценен нри измерениях теплового
рсширения. Изменение длины образца в зависимости от темпера-
кры обьгчно выражается уравнением
/ = /0(1+аТ), (11.1)
|с I — длина образца при температуре Т; 10 — длина образца
ри 0° С; Т — температура, ° С; а — линейный коэффициент теп-
лого расширения. При использовании метода ТМА / записыва-
ли в виде функции температуры. При отсутствии фазового пере-
ада и в предположении, что а — величина постоянная, зависи-
рсть будет выражаться прямой линией с наклоном, равным а.
ели уравнение (11.1) продифференцировать по времени, то будет
олучено следующее выражение:
„е dT/dt — скорость нагревания, являющаяся постоянной вели-
)цной. Таким образом, при измерениях теплового расширения
^особом ДТМА перемещение пера самописца прямо пропорцио-
льно коэффициенту теплового расширения. При соответствую-
щей градуировке коэффициент а может быть получен непосредст-
iiiHo с кривой. Кроме того, фазовый переход первого рода отра-
Н’гся на кривой в виде пика, площадь которого будет соответство-
Вть изменению длины А/.
444
Глава 11
2. ИЗМЕРИТЕЛЬНАЯ АППАРАТУРА
Измерительная аппаратура, применяемая в дилатометрии, u.iii
в методе ТМА, весьма проста. Изменения объема AV или длины А/
образца определяются с помощью механического, оптического или
электрического датчика и регистрируются в функции темпера rypi
или времени. В качестве примера на фиг. 11.1 представлен просш
прибор [2]. Изменения длины образца измеряются с помощью
Фиг. 11.1. Установка для дилатометрических измерений [2].
а — узел дилатометра; б — схема установки в целом; /—трансформатор; 2— сердс’пиц
трансформатора; 3— нажимной щуп; 4 — стеклянная трубка; 5 — образец; 6 — печь; 7 —
гулятор трансформатора; 8 — термопара; 9 — демодулятор; 10 — трансформатор; 11 — пкчьо
12 — регулятор температуры; 13— самописец.
нажимного щупа линейным дифференциальным трансформатором,
Сердечник трансформатора соединен непосредственно с нажимны ч
щупом, линейное перемещение которого создает на выходе трат
форматора напряжение, пропорциональное этому перемещен ши
Перемещение порядка ±0,1 мм вызывает выходной сигни i
±4,37 мВ. Таким образом, можно измерить весьма малые изменсн1П|
длины образца.
Несколько более сложный прибор, в котором дилатометрическим
измерения проводились параллельно дифференциальному термит'
скому анализу, описан Бэрролом и др. [3, 4 |. Этот прибор изобрд
жен на фиг. 11.2. Изменение длины образца измеряется трансфер
матором с подвижным сердечником, который соединен с рычажными
весами. Чтобы получить двукратное механическое усиление, плечи
рычага со стороны трансформатора в два раза длиннее плеча со сто
роны образца. При максимальной рабочей чувствительности при
Другие методы термического анализа
445
6
I цг. 11.2. Установка Бэрролла и др. для измерения теплового расширения
[3, 4].
К—весы и держатель образца; б — схема установки; -/—подвижный сердечник трансфор-
Ьгора. связанный с зубчатым колесом и рейкой,; 2— нониус, 4 витка/мм; 3 — кварцевый
Куп; 4г—алюминиевый нагревательный блок, помещенный в сосуд Дьюара с жидким азо-
юм; 5 —кварцевая базовая плита; 6 — нониус положения образца; 7 —термопара устройст-
для регулирования температуры по заданной программе; 8 — термопара для измерения
Кпературы образца; 9— кварцевое основание образца; 10—первичная обмотка; 11— вто-
рая обмотка; 12— подвижный сердечник трансформатора; 13—термопара образца; 14—
эталонная термопара.
Гора удлинение образца на ~2,5-10-5 мм соответствует 1 мм шка-
лы самописца. Согласно утверждению Бэрролла, предел чувстви-
шльности прибора лимитируется вибрацией помещения.
• На фиг. 11.3 изображен один из стандартных термомеханиче-
сьих анализаторов. При измерении растяжения образца или глу-
446
Глава 11
Фиг. 11.3. Термомехани-
ческий анализатор.
1 — подставка для гирь; 2—погру-
женный поплавок; 3 — трансфор-
матор; 4 — сердечник трансформа-
тора; 5— соединение датчика с сер-
дечником; 6 — трубка для образца;
7 — поглотитель тепла; 8 — ох-
лаждающий агент; 9 — изоляция;
10 — печь.
Фиг. 11.4. Типы датчиков в уста-
новках ТМА.
а — датчик для измерения теплового расшире-
ния; б — сжатия; в — изгиба; е, д — растяже-
ния; 1—трубка для образца; 2— край трубки;
3—зажимы для образца; 4—образец; 5—датчик.
бины вдавливания в него индентора образец помещается на iijiai
форму кварцевой трубки. Соответствующий кварцевый датчик со
единен с сердечником линейного регулируемого дифференциалыкн о
трансформатора. При любом изменении положения сердечника iu>
Другие методы термического анализа
447
(меняется выходное напряжение трансформатора, которое регист-
рируется самописцем. Узел датчика имеет подставку для гирь, что
[позволяет заранее задавать нагрузку на поверхность образца.
Весь узел поддерживается пластмассовым поплавком, жестко ук-
репленным на оси и полностью погруженным в жидкость высокой
плотности. Преимущество такой опоры состоит в том, что в диапазо-
не нагрузок, при которых Ъоплавок полностью погружен в жидкость,
[истинная нагрузка на образец совершенно не зависит от положения
датчика. Чувствительность прибора составляет 4 • 10“s мм на 1 мм
шкалы самописца со шкалой на 10 мВ. Для получения температур
П интервалах от —150 до 325 и 25—700° С используются две печи.
На фиг. 11.4 изображены датчики, применяемые в ТМА. Дат-
чик, изображенный на фиг. 11.4, а, используется для измерений
теплового расширения, на фиг. 11.4, б — для определения глуби-
ны вдавливания индентора в образец, температур размягчения
hi т. п. Для определения температуры размягчения по Вика или де-
формации при нагревании под нагрузкой используется датчик, изо-
браженный на фиг. 11.4, в. На фиг. 11.4, г и д представлены
[специальные датчики для текстильных волокон и пленок. В датчи-
ке, изображенном на фиг. 11.4,0, образец помещается в прорези
жимов, после чего последние обжимаются. С помощью такого
(датчика облегчается обращение с очень тонкими образцами и изу-
чение их под действием равномерного натяжения.
В литературе описано также одновременное исследование мето-
дом дилатометрии и другими методами термического анализа. Так,
например, известно сочетание дилатометрии с дериватографией
15] (фиг. 8.17), с эманационным термическим анализом [6—8],
к- методами ДТА и ЭП [9, 101 и другими методами [3, 4, 11].
3. ПРИМЕНЕНИЕ
Кривые, приведенные на фиг. 11.5, иллюстрируют использова-
ние дилатометрии для определения фазовых переходов различных
типов ]2.] Для соединений KAsFe, KC2H5SO4 и Со(Пиридин)2С12
показаны фазовые переходы в твердом состоянии Твердая фаза 1->
1*Твердая фаза 2, на примере дегидратации ВаС12 -2Н2О— проявле-
ние реакции разложения, а на примере плавления ацетанилида —
игроявление фазового перехода типа Твердая фаза-> Жидкость.
На фиг. 11.6 [9] приведены дилатометрическая кривая и кри-
вые ДТА и ЭЛТА для соединения Sr(OH)2-8Н2О. Можно было ожи-
дать, что потеря 8 моль воды каждым молем этого вещества вызовет
шачительное сокращение объема исходной гидроокиси;
Sr(OH)2 • 8Н2О имеет плотность 1,88 г/см3, в то время как
Sr(OH)2 — 3,62 г/см3. При температуре —-220° С скорость сокра-
щения объема образца заметно замедляется, что, по-видимому, свя-
зано с образованием и последующим разложением Sr(OH)2-H2O.
448
Глава 11
Фиг. 11.5. Типичные дилатометрические кривые различных веществ |2|.
1—KAsFe; Г — метилсульфат калия (KCHaSO«); 3 — ацетанилид; 4—ВаС1,-2НгО; 5 — темп
сатура плавления; 6 — Со(Пиридин)гС|2; 7— этилсульфат калия (КС.гНе8О4).
Недавно этим комбинированным методом были исследованы такие
соединения, как Са(ОН)2 и Ва(ОН)2 [9]; карбонаты кальция, строн-
ция и бария [10]; СаС2О4-Н2О, CaCO3-MgCO3, Са SO4 • 2Ш),
каолин и NH4NO3 [5]; ZnO + Fe2O3[6]; реСгО4-2Н2О и основной
карбонат железа [8] и многие другие соединения.
Шмидт и Бэррол [4] опубликовали подробное исследование, про-
веденное методом внедрения индентора при нагревании асфальтом
различного состава. За температуру размягчения была принят
Другие методы термического анализа
449
иг. 11.6. Полученные на одном приборе кривая ДТА (о); дилатометриче-
ская кривая (б) и кривая ЭЛТА для Sr(OH)2-8Н2О (в) [9].
емпература, при которой кривая отклонялась от прямой линии.
1о своим характеристикам при нагревании асфальты похожи на
ростые линейные вязкоупругие полимеры с низким молекулярным
есом.
Более поздние сведения касаются применения дилатометрии
ТМА для исследования полимерных материалов. Подробное ис-
Педование интервалов стеклования в полимерах было выполнено
филлером [12]. Была предложена механическая модель перехода
олимеров в стеклообразное состояние на основе приведенных в ли-
ературе данных и корреляции между объемом, теплоемкостью
механическими свойствами неорганических стекол с соответству-
ющими характеристиками органических полимеров. На фиг. 11.7
12] приведены кривые линейного расширения, теплоемкости и
450
Глава 11
предела прочности на растяжение обычного полистирола. Переход
в стеклообразное состояние наблюдался с помощью ТМА при 75" (
что хорошо согласуется с результатами ДТА, в соответствии с кото-
рыми этот переход происходит при 80° С. При температуре 160" (
происходит резкое изменение хода кривой, что связано с началом
плавления образца.
Фиг. 11.7. Кривые линейного расширения, теплоемкости и предела проч*
ности на растяжение обычного полистирола [12].
Нафиг. 11.8 приведена кривая [13], иллюстрирующая на при
мере ориентированного волокна из найлона (площадь сечении
0,001,5 мм2) применение метода ТМА для определения зависимо» hi
удлинения от температуры. Чтобы наблюдать плавление при 24(>'
приложена очень небольшая нагрузка. Отрицательный наклон
кривой при приближении к 0°С является результатом влиянии
усадки при переходе в стеклообразное состояние вблизи 20' t
Температура стеклования Tg нцже обычно наблюдаемой для ii.il)
лона вследствие влияния ориентации. В интервале температур
60—120° С наблюдается изменение скорости разориентации, па
видимому, вследствие потери воды.
В качестве иллюстрации применения методов ТМА и ДТМЛ
для исследования температурного расширения образца на фиг. 11 '<
[14] приведены кривые для политетрафторэтилена. Два переходi
в стеклообразное состояние, наблюдаемые в исследуемом интерна, |г
температур, легче определяется по кривой ДТМА, чем по криво!!
ТМА.
11.8. Кривая удлинения найлонового волокна при растяжении, полу-
ченная методом ТМА [13].
1рованное найлоиовое волокно длиной 9 мм скорость нагревания 10 град/шш; нагруз-
ка 1 гс.
Размер 2'10-1 указан в дюймах (5-W* мм).
-10 0 10 20 30 40
Температура, ’С
11. 9. Кривые удлинения образца политетрафторэтилена, полученные
методами ТМА и ДТМА [14].
Длина образца 3 мм; скорость нагревания 5 град/мин.
452
Глава И
Метод ТМА находит также практическое применение при оире»
делении условий сварки полиэтиленовых пленок при нагревании
[15], для контроля качества пластмассовой футеровки [16], дли
определения характеристик полихлорвиниловых пластизолей 1171
и композиции поливинилхлорида и дивинилнитрильного каучуки
[18], для определения коэффициента линейного расширения эпок-
сидных смол [19] и во многих других случаях.
В. ЭЛЕКТРОПРОВОДНОСТЬ
Удобным способом изучения температур полиморфных фазовым
переходов является непрерывное измерение изменения электрик
ского сопротивления или электропроводности в зависимости от гем
пературы. В некоторых случаях этот метод можно использован»
в дополнение к дифференциальному термическому анализу и калия
риметрии с большей пользой, чем другие описанные выше методы
Измерительная аппаратура обычно проще, чем при ДТА, но нот
можности применения все же более ограничены. Комбинированны!!
прибор, сочетающий ДТА и измерение электропроводности, описи!)
Бергом и Бурмистровой [20].
При фазовых переходах изменение температурного коэффинп
ента электрического сопротивления происходит вследствие изме
нения ионной подвижности в решетке (т. е. плавления) или вследгр
вие изменения в электронных энергетических уровнях [21 ]. Измен»'*
ние сопротивления в зависимости от температуры легко измеряете и
с помощью мостовой схемы постоянного или переменного toki и
соответствующей регистрирующей аппаратуры.
На фиг. 11.10 приведены мостовая схема и высокотемператур
ный держатель образца, описанные Гарном и Флашеном [21 ]. Mori
имеет довольно простую схему, в которой с каждой ячейки можии
получить напряжение 0,0015—1,5 В. Выходной сигни i
усиливается микровольтовым усилителем постоянного тока н
записывается одним каналом двухканального ленточного самописца
Температура образца, определяемая термопарой, регистрирует!
вторым каналом.
Образец помещают между двумя дисковыми платиновыми элек-
тродами. Электроды диаметром 9 и 12 мм и толщиной 0,25 мм дли
улучшения электрического контакта прижаты к образцу специаль-
ным нажимным контактом. Температура образца измеряется плм ।
тино-платинородиевой термопарой, приваренной к верхнему элек|
роду.
Существует заметное различие между кривыми электросопрп-
тивления чистого кристаллического ниобата калия KNbO3 и смеси
порошков состава KNbo.9Tao.1O3 (фиг. 11.11) [21]. Чистый KNbOij
имеет острый пик фазового перехода в кристаллической фазе в ин-
тервале температур 200—220° С, в то время как для смеси порши
Самописец
а
О 1 2 г
I___I____I
S
г. 11.10. Установка Гарна и Флашена для измерения электропроводности
[21].
мостовая схема; б — высокотемпературная ячейка; 1 — платиновые диски; 2— плати-
новый провод; 3 — платинородневый провод (I0%Rh); 4 — алунд; 5 — образец.
454
Глава 11
Фиг. 11.11. Фазовые перехо-
ды, определенные по измене-
нию электрического сопротив-
ления (21].
а—KNbOg gTag j О3 (керамика);
б — KNbOa (монокристалл).
Фиг. 11.12. Прибор дли сов-
местных ДТА—АТА-измере-
ний |22].
/, 2—выводы к электрометру; 3— вы
воды термопары; 4 — щтуцер для по
дачи охлаждающей воды; 5 — Капил
лярный изолятор нз стекла пирекс,
6 —чашка для образца; 7 — диффсрсн
циальная термопара; 8 — платиновый
электрод; 9 — крышка держателя об
разца; 10 — соединительная муфт и
платинового электрода.
ков характерно постепенное изменение сопротивления, начип;цп
щееся около 150° С.
Как показали Берг и Бурмистрова [20], комбинированные ш
следования изменения электропроводности образца и методом 711 А
полезны при изучении реакций, протекающих в твердой фазе. В пи
чале таких реакций наблюдается резкое возрастание электропрп
водности. Электропроводность измеряется также при исследовании
плавления чистых веществ и эвтектических смесей, а также дли
определения полиморфных переходов в солях. Кроме того, по элгк i
ропроводности расплавленной соли можно определить ее чиспну
[20].
Прибор для совместных ДТА—АТА-измерений (АТА — амиерн
метрический термический анализ) был описан Дэвидом [22]. Энн
Другие методы термического анализа
455
прибор (фиг. 11.12) состоял из электрода 8, изготовленного из пла-
тковой проволоки, и небольшой чашки из нержавеющей стали 6
платиновым покрытием, являющейся вторым электродом в изме-
рительной электрической цепи. В чашку помещали образец такого
же типа, как и в случае кольцевых термопар. Поскольку фазовые
5
|>иг. 11.13; Установка, использованная Уэндландтом для измерения элект-
ропроводности [23].
сдатчик; 2 — трубка для образца; 3 — образец; 4 — печь; 5 — двухкоординатный са-
рписец; 6 — микроамперметр; 7 — источник постоянного тока; 8 —- дифференциальный
компьютер.
переходы первого рода часто сопровождаются уменьшением объе-
ма образца, для поддержания постоянного контакта с ним верхний
клектрод 8 был подвижным. К образцу прикладывали соот-
ветствующее напряжение (1— 1000 В) и проходивший по цепи по-
стоянный ток измеряли электрометром, соединенным последова-
:льно с источником питания. Этим методом определяли количество
еды, выделившейся при дегидратации CuSO4-5H2O, а также изу-
ми процесс плавления нитрата калия и некоторых полимерных
атериалов.
Уэндландт [23] впервые использовал измерения электропровод-
эсти (ЭП) для определения четверных точек на диаграммах состоя-
ния гидратов солей различных металлов. До этого четверные точки
определялись методом ДТА [24 ]. При использовании ДТА четвер-
ная точка обнаруживается обычно по небольшому пику на плече
Гюлее интенсивного эндотермического пика. В случае CuSO4-5H2O
и четверной точке присутствуют следующие четыре фазы:
1 uSO4-5H2O, CuSO4-3H2O, Н2О (жидкость) и Н2О (пар).
456
Глава II
На фиг. 11.13 изображена установка, использованная Уэпл-
ландтом [23] для измерения электропроводности. Она состояла in
регистрирующего микроамперметра, двухкоординатного самопш
ца, источника постоянного тока с напряжением на выходе 3—25 В,
Фиг. 11.14. Кривая электропроводности CuSO4- 5НаО [23].
держателя образца, датчика с электродами и печи с нагревательны,
ми элементами из металла, температура в которой регулировалась
с помощью простого программирующего устройства. ПорошкопП.
разные образцы гидратов солей различных металлов помещались
в трубку из стекла пирекс диаметром 5 мм и длиной 50 мм.
На фиг. 11.14 приведена кривая электропроводное i и
CuSO4-5H2O. Она имеет один пик, соответствующий выделению
из гидрата воды в жидкой фазе, которое начиналось при темпер»
туре 97° С. В исследованном интервале температур в этой система
не было обнаружено других жидких фаз. Аналогичные результаты
были получены для ВаС12-2Н2О и ВаВг2-2Н2О.
Кью [25, 26] описал довольно сложную установку для термп
ческого анализа, в которой одновременно производились измерении
методами ТГ, ТГП, ДТА и методом электротермического аналп.ш
(ЭЛТА). На фиг. 11.15 представлена система держателей образной
этой установки. Электрод 13 в держателе образца представлял
собой лист платиновой фольги толщиной 0,075 мм, обернутый ufll
круг керамического изолятора 20 термопары образца. Другой элем*
трод из платиновой фольги 12 представляет собой цилиндр, вставляй*]
мый в кварцевую трубку 11. Образец размещается вплотную между
двумя электродами; гильза 19 на дне трубки служит для предотврн»
Другие методы термического анализа
457
щения случайного контакта электродов. Проходящий по системе
ок с постоянным напряжением 1—2 В измеряется электрометром
и регистрируется двухкоординатным самописцем; регистрировались
токи силой 10-10 —10~s А.
Фиг. 11.15. Установка Кью для одновременных ТГ—ТГП—ДТА—ЭЛТА-из-
мерений [25].
/ — корпус весов; 2— кожух коромысла весов; 3—арретир коромысла; 4 — кварцевое ко-
ромысло; 5— контейнер для образца; 6 —блок термопары; 7 — термопара для измерения
температуры образца; 8 — керамическая трубка; 9 — платиновый чехол; 10— кварцевая
трубка с эталоном; 11 — кварцевая трубка с образцом; 12 — внешний платиновый элект-
род; 13—центральный платиновый электрод; 14—холодная часть коромысла; 15, 16—
выводы из платиновой проволоки; 17—спай термопары образца; 18— спай термопары
эталона; 19— гильза; 20— керамический изолятор; 21, 22—провода термопары образца;
23 — платиновый заземляющий провод.
Радлоф и Фримен [27 ] для изучения методом ЭП нескольких
окислов металлов использовали установку, изображенную на
фиг. 11.16. Электрод Ei, расположенный внутри газопроводной
трубки из стекла викор, укреплен на стеклянном диске, который
сплавлен с капилляром из огнеупорного стекла, закрепленным у
458
Г лава 11
одного конца трубки. Аналогично укреплен и второй электрод /я,
Для обеспечения надежного электрического контакта служит нр\
жина, создающая равномерный прижим электродов к исследуемому
образцу — монокристаллу или таблетке, спрессованной из порош
ка. Газопроводная трубка может быть помещена в трубчатую печь
Для устранения влияния помех от окружающей среды установки
должна быть тщательно экранирована.
Фиг. 11.16. Установка Радлофа и Фримена для измерения электропровод
ности |27].
1 — платиновый провод; 2—платинородневый провод (10% Rh); 3—пружина; 4—тефл»»
новая трубка; 5 — платиновый провод.
Джад и Поуп [9] описали прибор для одновременного провед»
ния электротермического анализа (ЭЛТА) и дилатометрических
исследований. Этот прибор состоит из нагреваемой трубки, изгп
товленной из глиноземистого цемента, укрепленной горизонтальнн
в трубчатой печи с проволочным нагревателем из кантала, макси
мальная рабочая температура которой 1250° С. Трубка с одного
конца заканчивается узкой шейкой, присоединенной к вакуумной
линии. На другом конце трубки закреплена металлическая скосы,
плотно соединенная двумя кольцевыми уплотнителями, образуют»!
ми вакуумно-плотное уплотнение. Гильза фиксирует образец прнб
лизительно в центре печи. Электрический контакт с образцом осу
ществляется с помощью двух платиновых дисковых электродон,
прижатых к образцу с противоположных сторон.
Кэррол и Мэнгрейвит [28 ] описали установку, позволявшую
проводить измерения методами ЭП и ДСК на одном и том же обра i
це.
Г. ЭМАНАЦИОННЫЙ ТЕРМИЧЕСКИЙ АНАЛИЗ
Метод эманационного термического анализа (ЭТА) основан ни
введении в твердое вещество инертных радиоактивных газов н н i
мерении последующего их выделения при нагревании этого веще»|
ва [7, 29]. Выделение радиоактивного газа дает возможность р»
Другие методы термического анализа
459
Фиг. 11.17. Реакционная ка-
мера для одновременных из-
мерений методами ЭТА, ДТА
и дилатометрии (по Бэлеку)
16].
/ — активированный образец; 2 — эта-
лон для ДТА; 3 — обоазец для дила-
тометрических измерений; 4—биметал-
лическая термопара; 5 — кварцевый
стержень для дилатометрических из-
мерений; 6 — кварцевые сосуды; 7 —
опорные трубки; 8 — блок из металла;
9 — наружный сосуд из кварца; 10 —
стеклянный шлиф; 11—трубка для ох-
лаждения жидкости.
гистрировать различного рода изменения, происходящие в вещест-
ве нри нагревании: химические реакции, такие, как дегидратация,
термическая диссоциация и синтез; полиморфные превращения; плав-
ление; превращение метастабильных аморфных структур в кристал-
лические соединения; изменение концентрации дефектов в кристал-
лической решетке. По сравнению с обычными методами ТГ и ДТА
метод ЭТА имеет некоторые преимущества. Он позволяет изучать
структурные изменения в соединениях в динамических условиях
даже в тех случаях, когда эти изменения не сопровождаются тепло-
выми эффектами (например, фазовые переходы второго рода). В дру-
гих случаях, когда образуются мелкокристаллические или аморф-
ные фазы, метод ЭТА оказывается более чувствительным, чем рент-
геновские методы.
На фиг. 11.17 и 11.18 показаны установки ЭТА, которые поз-
воляют одновременно регистрировать также кривые ДТА и дилато-
метрические кривые [6 J. В нагревательную камеру помещаются
образец (обычно массой 100 мг), в который введен радиоактивный
газ, эталон для ДТА (А12О3) и образец для дилатометрических из-
мерений. Температура измеряется с помощью термопар, введенных
460
Глава 11
непосредственно в образцы. Скорость нагревания обычно состав, in
ет 8—10 град/мин, поскольку эта скорость является оптимально!)
для измерений методами ДТА и ЭТА. Выделившийся из обра щц
радиоактивный газ переносится потоком газа-носителя с постоя.im
расходом в камеры для измерения радиоактивности (фиг. 11.18)
Фиг. 11.18. Установка ЭТА [6].
1— электронный потенциометр; 2—интегратор для подсчета а-частиц; 3 — интшрпнр
для измерения 3-излучения; 4— дилатометрический датчик; 5 — термопара; осушиi ц;
газа; 7 — фотоумножитель; 8 — сцинтилляционная камера; 9— катодный повтори гил» '
10— камера для измерения 3-излучения; 11— электронагреватель; 12—реакционный сосуд
из кварца; R — реометр.
ИЛИ
Прибор одновременно регистрирует а-распад радона и ₽-излучепн«
ксенона, предварительно введенных в образец путем ионной бом-
бардировки. Кривая ЭТА записывается одновременно с кривой Д| Л
и дилатометрической кривой на ленту многоточечного само
писца.
Число атомов, выделившихся из небольшого кристаллика, мож
но представить в виде суммы двух членов [30 ]
Е = Ег + Еа,
р r0S . Dq2 S
Е = —— с -j- --- ---ф exp
4m X. m
где Er — число высвобожденных радиоактивных атомов за сч«ч
отскоков; Ed — диффундирующая часть ослабленных радиоактпи
Q
2RT
Другие методы термического анализа 461
Температура, °C
Фиг. 11.19. Эманационная кривая РсгО3.
— в координатах Е — Т; б — в кбординатах lg(£—Ег) — 1/Т, где Ег—величина Е при
комнатной температуре; фэр — физический эквивалент рентгена.
1ых атомов; г0 — длина пробега отскакивающих атомов; S — удель-
1ая поверхность; m — масса кристаллика; Do — предэкспоненци-
льный множитель в выражении
D = Do exp
— V
RT J ’
3 — постоянная диффузии; Q — энергия активации диффузии
радиоактивных атомов в твердом теле; /? — газовая постоянная;
в — плотность вещества; к— константа радиоактивного распа-
да; Т — абсолютная температура.
462
Глава 11
Фиг. 11.20. Термическое разложение ТЬ(СгО4)г-6НгО [71.
1 — кривая ДТА; 2 — зависимость скорости выделения ксенона от температуры; 3 — крц
вая ЭТА.
На фиг. 11.19, а приведена зависимость скорости эманации
от температуры для Fe2O3. Кроме того, как показано им
фиг. 11.19, б, можно построить кривую вида Ed — в полу
логарифмических координатах, где Ed определяется по величине
Е (мощности эманации при рассматриваемой температуре) и /
(величины Е, измеренной при комнатной температуре). Участки
кривой, изображенной нафиг. 11.19, б, имеют два наклона: низко
температурная область с малым значением 2\gEd/2T ивысокотемш
ратурная область с большим значением 21gEd/27. При температуре
693° С кривая имеет излом. Эта температура составляет 0,53 темпера
туры плавления Fe2O3. Для порошков других кристаллических
неорганических веществ изломы кривых эманации наблюдаются
при температуре, составляющей 0,5—0,6 их абсолютной темпера
туры плавления. Эта температура связана с началом достаточке
интенсивного движения атомов или ионов в кристаллической решет
ке, способного вызвать заметную скорость диффузии в твердом
теле.
Другие методы термического анализа
463
Таким образом, с помощью классического эманационного метода
[можно определить этот интервал температур, имеющий очень боль-
шое практическое значение, поскольку при более высоких темпера-
турах процессы в твердой фазе могут происходить за счет диффузии.
Наклон отдельных участков кривой можно использовать для опре-
деления энергии активации эманационного процесса в твердом ве-
иестве для данного интервала температур. Вычислено, что энергия
| пффузии радона в Fe2O3 составляет Q ~ 62,8 ± 12,6 кДж/моль
I интервале температур 600—700° С и Q = 167,5 ± 20,1 кДж/моль
> интервале температур 850—1100° С.
На фиг. 11.20 приведены кривые ЭТА и ДТА [7] для
ГЬ(С2О4)2-6Н2О. Кривая ЭТА позволяет выявить три процесса:
I) ступенчатую дегидратацию в интервале температур 40—220° С
(кривая ДТА в этом интервале температур имеет только два эндо-
рермических пика, соответствующих переходам 6Н2О -> 2Н2О и
и 2Н2О-> Н2О); 2) разложение Th(C2O4)2 • Н2О с соответствующим
[ диком в интервале температур 300—400° С; 3) переход аморфного
ThO2 в кристаллический с соответствующим пиком при 400—500° С.
[Последовательность реакций, определенная методом ЭТА, пол-
ностью потверждается кривой скорости выделения ксенона, вве-
денного в образец путем ионной бомбардировки.
Д. ТЕРМОЛЮМИНЕСЦЕНЦИЯ
Метод термолюминесценции (ТЛ) состоит в измерении выделяе-
мой образцом световой энергии в зависимости от температуры при
[медленном нагревании образца с постоянной скоростью до темпе-
ратур, не превышающих температур свечения. Полученная таким
бразом кривая, называемая кривой свечения, состоит из ряда пиков,
Вызванных испусканием световой энергии при различных повы-
иенных температурах. По интенсивности свечения и температурам,
гоответствующим пикам на кривых, можно охарактеризовать или
[идентифицировать образец. Можно также использовать эти данные
[для других целей, например для изучения геологической актив-
ности, определения возраста древней глиняной посуды, исследова-
ния лунных пород, оценки катализаторов, а также в радиационной
дозиметрии твердых тел и др. Обнаружено также, что метод тер-
молюминесценции очень полезен в геохимических исследованиях
известняковых и доломитовых осадочных пород и вулканической
шавы.
Самые первые исследования по применению термолюминесцен-
ции для анализа и идентификации горных пород были выполнены
Дерибером [311, Гарликом [32], Колером и Лейтмайером [33],
Ройером [34], Сорином [35], а также Нортрапом и Ли [36]. Более
'поздние исследования проведены Саундерсом [371, Парксом [38 ],
[аниельсом и др. [391, Льюисом [401, а также Боузом и др. [41].
464
Глава 11
Ингерсон [421 и Целлер [43] рассмотрели применение термолюмп
несценции к проблемам геологической термометрии и определении
возраста геологических пород. Даниельс и др. [39], Боуз и др. |41 |
и ряд других авторов [44—48, 57 ] составили превосходные обзоры,
посвященные термолюминесценции в целом.
Зона проводимости
Рентгеновское
излучение
%gennoma
Электронные
ловушки
-Дырочные ловушки-Ъ-
V
а
Валентная зона
Рентгеновское
излучение
;-----*> Зона проводимости
Лплота\
Уровни примесей
'v
Валентная зона
-центр
У^центр
—.Зона проводимости
"1--------Г”
-ъ-Высшии уровень
Валентная зона
Фиг. 11.21. Схематические зониые модели термолюминесценции [57|.
а — простейшая модель; б — модель термолюминесценции в присутствии примеси; в мА
дель с электронными и дырочными ловушками и центрами рекомбинации на приметим*
нонах.
Способность к термолюминесценции приобретают кристалличе-
ские вещества, например щелочно-галоидные кристаллы, шил*»
радиоактивного облучения или воздействия рентгеновских лучей.
Радиация вызывает дислокацию электронов в кристалле, в резулм
тате которой может образоваться f-центр. Дальнейший персхоц
F-центров или электронных ловушек в состояние с более низкой
энергией за счет повышения температуры кристалла сопровождай-
ся выделением световой энергии. Величина последней (интснени
ность свечения) зависит от изменений кристаллической структуры,
Другие методы термического анализа 465
концентрации примесей в кристалле и характера предварительной
[шзической обработки образца.
I На фиг. 11.21 [57] приведена простая модель процесса термо-
иоминесценции. В случае, представленном на фиг. 11.21, а, иони-
зирующее излучение создает в кристалле электрон и дырку. Элект-
рон блуждает в зоне проводимости до тех пор, пока не попадет в элек-
тронную ловушку, образуя F-центр, а дырка перемещается в ва-
нтной зоне до тех пор, пока не попадет в дырочную ловушку, об-
разуя У-центр. При нагревании электрон (дырка) возбуждается и
ереходит в зону проводимости (валентную зону), по которой бес-
порядочно перемещается до тех пор, пока не рекомбинирует с
дцркой (электроном), испуская свет. Однако в этой простой модели
к учитывается доминирующее влияние примесей на процесс термо-
юминесценции [57 ]. Например, в обычно используемых фосфоре и
IJF для получения хорошо ощутимого эффекта термолюминес-
тенции необходимо наличие примесей.
I На фиг. 11.21,6 схематически изображен процесс термолюми-
несценции в веществе, содержащем двухвалентную примесь. В этом
r.iynae под действием ионизирующего облучения, образуются элек-
троны и дырки. Затем дырка может попасть в ловушку, образован-
ию катионом примеси. Когда при нагревании высвобождается
Ьлектрон, он рекомбинирует с дыркой на примесном ионе, возбуж-
тея характерную люминесенцию. Примесные ионы могут Также
Кразовывать комплексы типа Z-центров, способных захватывать
Иектроны. При нагревании кристалла ловушки высвобождают
ектроны, создавая пики термолюминесценции, причем сначала
вобождаются ловушки с низкой энергией, потом глубокие, с бо-
е высокой энергией.
Ряд исследователей рассмотрели также другие модели и теории
термолюминесценции [39, 59, 60].
| Бойд [49] исследовал кинетику изотермического затухания
термолюминесценции монокристаллов фтористого лития. С помощью
модели, аналогичный используемой в теории абсолютных скоростей
имических реакций, он показал, что интенсивность термолюми-
несценции I может быть выражена в функции времени следующим
уравнением:
7= [К/(К+1)] ~
ШК/(К+1)Р+1/^}2 ’ 1
где Ei — средняя энергия перехода; k3 — константа скорости;
В= n^/rii, щ, п2 и п3— числа молекул соответственно в перво-
Ьнальном, возбужденном и конечном состояниях.
Различные приборы для измерения термолюминесценции твер-
дых образцов описаны многими авторами: Урбахом [50], Рэндаллом
м Уилкинсом [511, Бойдом [49], Даниельсом и др. [39], Саундерсом
h.—230
466
Глава 11
Электропитание
110В, 60 Гц
Фиг. П.22. Схема установки для измерений методом термолюминесценции
[40].
1 — заслонка; 2— светящийся образец; 3 — светонепроницаемая печь; 4 — фотоумпи н
те ль; 5 — термопара.
[371, Парксом [38] и Льюисом [401. В основном в такие устанонкп
входят следующие части: нагреваемый блок образца; устропс i но
для регулирования температуры образца или электропитание ин
заданной программе; фотоумножитель с источником питания и лги
точный самописец с двумя перьями или двухкоординатный самоппп ц
На фиг. 11.22 изображена схема одного из таких приборпн,
описанного Льюисом [401. Обычно кривые свечения получают при
скорости нагревания ~1 град/мин. С увеличением температура
образец излучает свет, измеряемый фотоумножителем, выходной
ток которого пропорционален интенсивности испускаемого cm 11
После преобразования в соответствующее выходное напряжении
выходной сигнал фотоумножителя регистрируется одним из канн
лов ленточного самописца. Другой канал самописца соединен с ipp
мопарой, спай которой расположен в камере образца, и записыка»»
температуру последнего. Таким образом, обе величины — пн пн
сивность испускаемого света и температура образца — зашн ы
ваются в функции времени. Интервал рабочих температур ycniiioh
ки — от температуры окружающей среды до 500° С. Кривые (п»>
Другие методы термического анализа
467
Ь:ния можно получить для тонкоизмельченного образца массой
II мг.
На фиг. 11.23 приведена схема прибора ТЛ, используемого
|1я радиационной дозиметрии. Установка разделена на два отдель-
ных блока — детектор термолюминесценции (показан на фигуре)
автоматический интегрирующий пикоамперметр. Последний при-
«•няется для преобразования сигналов, поступающих с детектора,
200 TL).
В — зеркало; 2 — оптические фильтры; 3 — выдвижной держзтель образца; 4—планшет;
источник света; 6 —линия для подачи инертного газа; 7 — стальной корпус, толщи-
Ив стенки 1,6 мм; 8 — изоляция толщиной 38 мм; 9— магнитный экран; 10—электроста-
тическое защитное устройство; 11 — специальный фотоумножитель с низким уровнем шу-
••ов; 12—основание, обеспечивающее малую утечку; 13— вентилятор; 14 — теплообмен-
ник; 15 — термоэлектрическое охлаждающее устройство; 16 — охлаждающая рубашка;
17 — линзы.
и более удобную для оператора форму. Рабочий интервал темпера-
кур детектора 50—400° С; температура возрастает линейно с за-
ранее установленной скоростью до максимальной заданной темпе-
ратуры, которая поддерживается до включения цепи реле времени.
Кривые свечения в этой установке получаются при довольно высоких
(скоростях нагревания; например, температура 240° С достигается
ва 50 с.
На фиг. 11.24 показана типичная кривая свечения. Исследуе-
1мое вещество представляет собой легированный фторид лития
TLD-100, поставляемый фирмой «X аршоу кемикл». Интенсивность
испускаемого света измерена в условных единицах (при необходи-
мости их можно перевести в микроламберты или люмены). По оси
X отложены единицы времени (указанное время пропорционально
изменению температуры).
16*
468
Глава 11
К термолюминесцентным веществам в естественном состоянии
или после радиационного облучения относятся щелочно-галондпцг
кристаллы, кальцит, доломит, флюорит, окись алюминия, ок ши
Фиг. 11.24. Типичная кривая свечения.
1 — 120°С; 2 — 140°С; 3 — 200°С; 4 — 240°С; 5 — 340сС.
магния, гипс, кварц, стекло, полевые шпаты, фельдшпатиты, иг
которые высушенные глины и керамические материалы. Более 3()(М»
образцов горных пород были исследованы на термолюминесценцию
и ~75% из них испускали излучение в видимой области спектра I 1'»|
Естественной термолюминесцентностью, обусловленной главным
образом присутствием следов урана, тория и т. п., обладают ikmiih
все известняки и некоторые кислые вулканические породы. КарПп
наты кальция и магния испускают излучение в видимой облип и
спектра (от светло-желтого до оранжевого), а калиевые и натрш ним
полевые шпаты испускают свет от белого до голубовато-фиолепнннн
Полученные кривые свечения являются характерными для пир
деленных образцов или веществ; они содержат информацию о при
сутствии в образце определенных примесей, о термообработке, пи
торой подвергался образец, о его физической предыстории. Однпин
кривые свечения непригодны для анализа химических соединений,
и их можно использовать только для идентификации и контроля *|и
Другие методы, термического анализа
469
[Подтверждается кривыми свечения некоторых образцов смесей
кальцитов и доломитов, приведенными на фиг. 11.25. На этих кри-
вых четко видно различие, обусловленное неодинаковым химиче-
ским составом образцов, но едва ли они могут быть использованы
|для определения, например, содержания магния или кальция.
Саундерс [37] показал, что интенсивности, соответствующие
Пикам на кривых свечения образцов Ниагарских известняковых
отложений, возрастают с увеличением глубины залегания. Кривые
свечения были использованы для изучения различных слоев этих
отложений. Паркс [38] опубликовал аналогичное исследование,
доказывающее специфичность кривых свечения образцов пород
(различных геологических формаций, известняковых образований
у вершины и основания гор, а также при исследовании эрозии или
шеосадочных зон.
Джарино-Канина и Коэн [52] использовали кривые свечения
для характеристики смесей окислов германия и алюминия. Было
обнаружено, что после возбуждения ультрафиолетовым излучени-
ем положение пиков в интервале температур 50—70° С, а также со-
ответствующие им интенсивности изменялись в зависимости от со-
держания окиси алюминия, вводимой в стекло на основе окиси гер-
мания. Количество окиси алюминия изменялось от 0 до 5%.
Довольно интересное применение находит термолюминесцент-
ный анализ при оценке эффективности поверхностных катализа-
торов [39, 53]. На фиг. 11.26, а приведены кривые свечения трех
[стандартных катализаторов на основе окиси алюминия, а на
фиг. 11.26, б показано соотношение между площадью под пиком
кривой свечения и активностью катализатора. Кривая свечения
имеет два пика; площади под пиком 2 связаны с каталитической
активностью. Как видно из фиг. 11.26,6, существует очень хоро-
шая корреляция между площадью под пиком и активностью ката-
лизатора. Следует заметить, что многие катализаторы не обладают
термолюминесцентностью; в других же случаях не наблюдается
заметной корреляции. Тем не менее для ряда катализаторов, ко-
торые были исследованы, получена удовлетворительная корреля-
ция, и, следовательно, метод термолюминесценции можно считать
полезным для оценки активности катализаторов.
У образцов лунных пород свечение наблюдается при 350° С для
лунной пыли и при 400° С для кристаллических лунных пород [61 ].
Вид кривых термолюминесценции лунных пород, вероятно, явля-
ется суммарным результатом нескольких эффектов: облучения кос-
мическими лучами/ возможного наличия радиоактивности в лунной
почве и излучения, испускаемого в окружающее пространство. Ре-
зультаты термолюминесцентного анализа лунных пород показы-
вают, что температура свечения изменяется до глубины ~10,5 см.
Еще одно применение ТЛ в геологии заключается в определении
возраста потоков лавы [62]. Во время вулканического извержения
205г
Фиг. 11.25. Кривые свечения некоторых образцов смесей кальцитов и доло-
митов в естественном состоянии и после воздействия у-излучения [40].
Цифра I указывает иа предварительное воздействие ^-излучения.
Образец Доломит, % Кальцит, %
1а 89,7 10,3
2а 92,2 7,8
За 0,0 100.0
4а 48,6 51.4
Другие методы, термического анализа
471
Катализатор А
Температура
Фиг. 11.26. Кривые свечения катализаторов (о) и связь термолюминесценции
с активностью катализатора (6) [39].
горячая лава утрачивает прошлую способность к термолюминес-
ценции, а после охлаждения она определенный период времени
подвергается воздействию облучения вследствие естественной радио-
активности окружающей среды. Поэтому интенсивность термолю-
472
Глава 11
Время, год
Фиг. 11.27. Кривые свечения черепка глиняной посуды (а) и отношение
естественной термолюминесценции к чувствительности прибора (ГЬмпт /*»|
в зависимости от возраста глиняной посуды [63].
А — термолюминесценция, индуцированная рентгеновским облучением; Б — Термолюмингч им
ция типичного черепка; 1—Сехин; 2— Салинас; 3— Патачи; 4— Гуалаи; 5 — Кагупк
минесценции может дать исходные данные для определения волрт
та вулканической лавы, расчет.которого производится с учек>М
ряда факторов.
Измерения термолюминесценции черепков глиняной посуды и<
пользовались также для определения времени ее изготовлсш1|1
Глиняная посуда’после придания ей формы подвергалась обжигу
процессу, который также ликвидирует накопленную способность и
термолюминесценции в кристаллах кварца, содержащихся в глнин
при изготовлении посуды [57]. Затем на протяжении определении
го периода времени эти кристаллы кварца накапливают потепцн
альную способность к термолюминесценции под действием облу*
чения, вызванного небольшой естественной радиоактивностью глн
Другие методы термического анализа
473
кы и радиоактивностью почвы. По интенсивности термолюминес-
ценции можно определить дату обжига и, следовательно, возраст
'посуды [63, 64].
На* фиг. 11.27 [631 приведены кривые свечения черепка гли-
няной посуды; приведена также зависимость отношения естествен-
|й термолюминесценции к чувствительности прибора. (TLM;1KC/S)
швисимости от возраста посуды. Это отношение является доволь-
точным индикатором археологического возраста, особенно если
я его определения проводится серия повторных опытов на череп-
х, относящихся к одному и тому же периоду времени, а затем
ределяется среднее значение.
По-видимому, одним из наиболее важных применений термо-
)минесценции является измерение ионизирующей радиации
7, 60]. Поскольку интенсивность термолюминесценции пропор-
циональна интенсивности радиации, предварительно воздейство-
вавшей на твердое тело, кристаллы и порошки могут служить в ка-
I естве дозиметров радиации. Преимущество такого метода измере-
ния радиации заключается в небольших размерах дозиметров и
способности их воспринимать большие дозы облучения. Небольшой
|кристалл термолюминесцентного материала заделывается в значок,
I который носит персонал. Периодически этот кристалл помещается
В детектор (термолюминесцентный прибор) и нагревается. По ин-
егральной интенсивности испускания света можно определить на-
копленную дозу ионизующего излучения.
Термолюминесценция использовалась также для излучения ра-
диационных повреждений [541 и радиоактивности некоторых мине-
ралов [33, 38, 55, 56].
Е. ТЕРМОМАГНИТНЫЙ АНАЛИЗ
Измерение магнитной восприимчивости соединения в функции
|гемпературы часто используется для определения изменений со-
стояния окислов, изучения реакций восстановления и окисления, ис-
следования ферромагнитных и антиферромагнитных свойств веществ
н т. д. Без исследований зависимости от температуры определение
Висла неспаренных электронов, предсказание состояния окислов
I стереохимические оценки магнитно-концентрированных систем
|б5] не могут быть достаточно обоснованными.
Описано большое число разнообразных приборов для регулиро-
вания температуры образца при исследовании магнитных свойств.
В одном из новых приборов применены вспомогательные устройст-
ва для регулирования температуры в ЯМР- и ЭПР-спектрометрах
1651 при исследованиях методом Гойи или Фарадея. Малей и Кэйс
166] описали микровесы со спиральной пружиной, которые могут
быть использованы как для измерения магнитной восприимчивости
методом Фарадея, так и для исследования адсорбции и ТГ-измерений
474
Глава 11
Таблица II I
Результаты, полученные методами ТГ и ТМАГ для CuSO4-5H2O [66]
Температура, °C Предполагаемая формула Магнитная восприимчивость
10е ед. СГС 10" ед. СГС *т. Ю’ ед ОЧ1
25 100 р=133 мПа CuSO4-5H2O CuSO4-H2O 5,860 6,632 1129,6 1178,4 —491)
’) Соответствует 4 моль воды, если х (НгО) = — 12,9-10*‘.
при дегидратации CuSO4-5H2O. В табл. 11.1 приведены данные,
иллюстрирующие применение этих весов для термогравиметрнч,
ского (ТГ) и термомагнитного (ТМАГ) анализов. При температур!!!
25 и 100° С и давлении 133 мПа из молекулы этого соединения вы
деляется четыре молекулы воды. При этом изменение магнитной
восприимчивости связано не только с потерей молекул воды, пи
также и с изменением парамагнетизма образца. Последний факщ|1
Фиг. 11.28. Установка Симмонса и Уэндландта для ТГ—ТМАГ [67],
1 — регистрирующие полумикровесы Айнсворта; 2— трубчатаи печь; 3— контейнере р«<
цом; 4 — термопара, измеряющая температуру печи; 6 — электромагнит; 6 — гидравлики |
поршень; 7 — двухкоординатный самописец; 8— программирующее устройство для р«|улир<
вания температуры печи; 9— регулятор напряжении; 10 — источник питания электрон!гин>
Другие методы термического анализа
475
Фиг» 11.29. ТГ—ТМАГ-кривая для [Co(NH3)6]CIs [67].
легко объясняется с помощью теоретического расчета магнитной
восприимчивости Си SO4-5H2O при 100° С на основании закона
Кюри—Вейса. Этот же прибор был использован для исследования
адсорбции кислорода на т-окиси алюминия по изменению магнит-
ной восприимчивости во время этого процесса. Результаты измере-
ний свидетельствуют об образовании димера О4 с очень слабой маг-
нитной восприимчивостью.
Симмонс и Уэндландт [67] описали фарадеевскую установку,
на которой можно производить ТГ- и ТМАГ-измерения в интервале
температур от комнатной до 500° С. Схема этого прибора приведена
на фиг. 11.28. Для регистрации результатов эксперимента исполь-
зовался двухкоординатный самописец, один канал которого записы-
вает температуру, а другой ТГ-кривую. На ТГ-кривых наблюдались
отклонения, вызванные периодическим приложением к образцу
магнитного поля. На фиг. 11.29 приведена ТГ—ТМАГ-кривая для
соединения [Co(NH3)e]CI3. Как видно из фиг. 11.30, наиболее цен-
476
Глава 11
ным параметром этой системы является содержание трехвалептпп
го кобальта (мол.%) в зависимости от температуры. Этот парамор
вычисляется с помощью следующего уравнения [3]:
Температура, °C
Фиг. 11.30. ТГ-кривая (/), кривая магнитной восприимчивости (2) и кие
вая уменьшения содержания кобальта (3) (мол.%) для [Co(NH3)6]Cls |U7|«
Содержание кобальта (моль. %) ==
_ 800 — bmc)/(bms — Дтс)| msXs (Т — 6)
(4
Поскольку
800 д msXs-------= const = К,
— Ьтс)
уравнение приобретает вид
Содержание кобальта (моль. %) = К (Ат — Атс) (7 + 0),
где Ат — наблюдаемое изменение массы, а индексы s и с означал it
соответственно, что измерения проводились в контейнере с обрш
цом и без образца.
С помощью метода ТМАГ были исследованы реакции взаимодгй
ствия при повышенных температурах металлического натрии t
Fe3C, Fe20C9 и Fe2i82MnOil8 С [68]. Этот метод позволяет проследи i и
процесс исчезновения карбида железа при контакте с расплаши и
Другие методы термического анализа
477
•иг. 11.31. ТМАГ-кривые, полученные при взаимодействии Fe3C с Na или
Li [68].
/ —только карбид железа; 2 — взаимодействие с литием; 3 — взаимодействие с натрием.
ым металлическим натрием, а также образование железа или дру-
эго ферромагнитного вещества как продуктов реакции.
На фиг. 11.31 приведены ТМАГ-кривые, полученные при взаи-
одействии Fe3C с избытком металлического натрия или лития.
Ыше 600° С происходит быстрое разложение одного только Fe3C,
прямая реакция Fe3C с металлическим натрием вида
2Fe3C + 2Na -> Na2C2 + 6Fe
области температур разложения карбида не происходит. Было
айдено, что как точка Кюри Fe3C, так и степень взаимодействия
магнитным полем при нагревании образца, не изменяются.
Ж. ТЕРМИЧЕСКИЙ АНАЛИЗ ПО КРУТИЛЬНЫМ КОЛЕБАНИЯМ
КОМПОЗИТНОГО ОБРАЗЦА
Метод анализа по крутильным колебаниям композитного образ-
а (КККО) был предложен Гилхемом [69, 70] для исследования
:еханических свойств полимерных материалов. Этот метод позво-
1
478
Глава 11
7
2
3
4
5
6
7 Фиг. 11.32. Установка для
термического анализа по
g крутильным колебаниям
композитного образца [71].
S 1 — реверсивный соленоид; 2 —
W рычаг; 3 — тефлоновая трубка;
™ 4 — поддерживающий стержень;
5 — композитный образец; 6—тер-
77 мопара; 7 — удлинитель; 8—элек-
тровакуумный фотоэлемент; 9 —
12 круглый диск; 10 — источник све-
та; 11 — магнитный стабилизатор;
xjf 12 — прокладка; 13 — резиновая
** опора диска.
ляет определять термомеханическим способом переходы в полиме-
рах в регулируемой атмосфере в интервале температур от — РИ1
до 500° С. Образец изготавливается путем пропитки стекляшки и
шнура раствором исследуемого полимерного материала, последую*
щего испарения растворителя и переводом полимера в твердое <п
стояние. Готовый образец в виде пропитанного шнура нагревает»
и при этом подвергается свободным крутильным колебаниям. Полу
чаемый при этих колебаниях параметр — относительная жесткое и.
1/р2 (р — период колебаний) — используется как мера модул*
сдвига. Параметр механического затухания колебаний 1/п, где п
число колебаний между двумя произвольными, но фиксированны
ми граничными положениями в серии колебаний, служит в качен
ве меры логарифмического декремента затухания. Изменения и-
носительной жесткости и затухания связываются, насколько мн
возможно, с изменениями, происходящими в полимерах. С помощь!"
метода КККО нетрудно определить основные и вторичные персхо
ды, например температуры плавления и стеклования, а также оце-
нить влияние многих химических и деструктивных реакций. Этому
методу посвящен обзор, сделанный Гилхемом [71).
На фиг. 11.32 показан прибор, используемый в методе KKKU
Частота колебаний (менее 1 Гц) и затухание свободных колебании
маятника дают информацию о модуле и внутреннем трении в исслф»
дуемом полимере. Электрический аналог затуханий колебаний ма-
ятника получается путем ослабления светового луча круглим
диском; пропускание света последним линейно связано с углом на
ворота относительно луча.
Другие методы термического анализа
479
На фиг. 11.33 приведены кривые, полученные методом КККО
(ля триацетата целлюлозы [69]. Композитный образец в виде стек-
[янного шнура, пропитанного триацетатом целлюлозы, изготавли-
1ался из 7%-ного раствора этого полимера в метиленхлориде. Перед
анализом образец нагревался до 150° С для удаления растворителя,
i затем охлаждался до 25° С. Кривые указывают на переход в
Фиг, 11.33, Кривые, полученные методом КККО для триацетата целлюлозы
[69, 71].
Скорость нагревания 2 град/мин в атмосфере азота.
стеклообразное состояние при температуре ~190°С, где происхо-
дит резкое уменьшение жесткости с образованием максимума за-
тухания. При плавлении исследуемого полимера при 290° С про-
исходит быстрое уменьшение жесткости с образованием еще одного
। максимума затухания. Дальнейшее увеличение жесткости и умень-
шение затухания связывают с образованием поперечных связей и
(или) процессом упрочнения цепей.
Рассматриваемый метод использовался также для исследования
других соединений [711, в том числе желатины, синтетических
смол, полибензимидазола и др.
480
Глава 11
3. ОКСИЛЮМИНЕСЦЕНЦИЯ
Метод оксилюминесценции тесно связан с термолюминесценцией
Этот термин был введен Эшби [72 ] для описания испускания свети
при нагревании некоторых полимеров на воздухе или в кислороде
Фиг. 11.34. Влияние концентрации кислорода на интенсивность оксилюмн»
несценции полипропилена [72].
1 — аргон; 2—азот, содержащий 2,6% кислорода; 3 — азот, содержащий 6,4% кислородщ
4 — азот, содержащий 11,0% кислорода; 5 — воздух; С — кислород.
до температур 150—200° С. Для возникновения испускания спет
(оксилюминесценции) необходимо присутствие кислорода, причем
интенсивность света пропорциональна концентрации кислородц,
контактирующего с поверхностью полимера. Для проведения экс
периментов, связанных с оксилюминесценцией, используются при
боры типа описанного выше для исследований методом
термолюминесценции. Испускаемый полимерами световой по»
ток очень слабый; его величина составляет 10-10 — 10~8лм
У найлона — наиболее сильного источника излучения iii
исследованных до сих пор полимеров — световой поток, испускп»
мый при 200°С, достаточно ярок для визуальных наблюдений. 11ред
варительные исследования полипропилена показали, что половине
испускаемого светового потока приходится на интервал длин волн
4200—5150 А, а вторая половина — на 3000—4200 А.
Как показано на фиг. 11.34, интенсивность оксилюмипссцеп
ции зависит от концентрации кислорода в газовой среде, находи
щейся в контакте с поверхностью образца. Температура полимере
повышалась от комнатной до 180° С со скоростью ~6 град/мин.
Другие методы термического анализа
481
Поскольку присутствие противоокислителей уменьшает интен-
сивность испускаемого света, рассматриваемый метод полезен для
проверки противоокислительных свойств, .соединений. Любое ве-
щество, которое уменьшает первоначальное испускание света, яв-
ляется потенциальным противоокислителем.
ЛИТЕРАТУРА
1. Perkin-Elmer Instrument News, 20, № 1, 10 (1969).
2. Wendlandt W. W., Anal. Chem. Acta, 33, 98 (1965).
3. Barrall E. M., Porter R. S., Johnson J. F., Anal. Chem., 36, 2316 (1964).
4. Schmidt R. J., Barrall E. M., J. Inst. Petro., 51, 162 (1965).
5. Paulik F., Paulik J., Erdey L., Mikrochim. Acta, 894 (1966).
6. Balek V., Glasnik Hemijskog Drustva, 34, 345 (1969).
7. Balek V., Materials Sci., 4, 919 (1969).
8. Balek V„ J. Materials Sci., 5, 166 (1970).
9. Judd M. D„ Pope M. 1., J. Appl. Chem., 20, 380 (1970).
10. Judd M. D„ Pope M. I., J. Appl. Chem., 20, 384 (1970).
11. Berger C., Eyraud L., Richard M., Riviere R., Bull. Soc. Chim. France,
628 (1966).
12. Miller G. W., Thermal Analysis, Schwenker R. F., Garn P. D., eds., Vol. 1,
Academic Press, N. Y., 1969, p. 435.
13. Perkin-Elmer Instrument News, 20, № 4, 6 (1970).
14. Perkin-Elmer Instrument News, 20, № 1, 10 (1970).
15. DuPont Application Brief, № 17, E. I. DuPont de Nemours and Co. Inc.
Instrument Products Div., Wilmington, Dela.
16. DuPont Application Brief, № 22, E. I. DuPont de Nemours and Co. Inc.,
Instrument Products Div., Wilmington, Dela.
17. DuPont Application Brief, № 16, E. 1. DuPont de Nemours and Co. Inc.,
Instrument Products Div., Wilmington, Dela.
18. DuPont Application Brief, № 14, E. I., DuPont de Nemours and Co. Inc.,
Instrument Products Div., Wilmington, Dela.
19. DuPont Application Brief, № 5, E. I. DuPont de Nemours and Co. Inc.,
Instrument Products Div., Wilmington, Dela.
20. Берг Л. Г., Бурмистрова H. П., Журнал неорганической химии, 5, 326
(1960).
21. Garn Р. D., Flaschen S. S., Anal. Chem., 29, 268 (1957).
22. David D. J., Thermochim. Acta, 1, 277 (1970).
23. Wendlandt W. W., Thermochim. Acta, 1, Il (1970).
24. Borchardt H. J., Daniels F., J. Phys. Chem., 61, 917 (1957).
25. Chiu J., Anal. Chem., 39, 861 (1967).
26. Chiu J., J. Polymer Sci., CS, 27 (1965).
27. Rudloff W. K-, Freeman E. S„ J. Phys. Chem., 74, 3317 (1970).
28. Carroll R. W., Mangravite R. V., Thermal Analysis, Schwenker R. F.,
Garn P. D., eds., Vol. 1, Academic Press, N. Y., 1969, p. 189.
29. Balek V., Anal. Chem., 42, 16A (1970).
30. Balek V., Zaborenko К. B., Radiokhimiia, 10, 450 (1968).
31. Deribere M., Argile, 188, 5 (1938); Rev. Sci., 76, 383 (1938).
32. Garlick G. F., Luminescent Materials, Oxford University Press, Lnd.,
1949.
33. Kohler A., Leitmeir H„ Z. Krist., 87, 87 (1934).
34. Royer L., Compt. Rend., 204, 602, 991 (1937).
35. Saurin E., Compt. Rend. Soc. Geol. France, 209 (1939).
36. Northrup M. A., Lee O. L., J. Opt. Soc. Am., 30, 206 (1940).
37. Saunders D. F., Bull. Am. Assoc. Petrol. Geologists, 37, 114 (1953).
482
Глава 11
38. Parks J. M., Bull. Am. Assoc. Petrol. Geologists, 37, 125 (1953).
39. Daniels F., Boyd C. A., Saunders D. F., Science, 117, 343 (1953).
40. Lewis D. R., J. Phys. Chem., 60, 698 (1956).
41. Bose S. N., Sharma J., Dutta В. C., Trans. Bose Res. Inst. (Calcutta), 20,
117 (1955).
42. Ingerson E., Econ. Geol. 50th Anniv. Volume, 341 (1956).
43. Zeller E. J., Congr. Geol. Intern. Compt. Rend. 19e Algiers, 12, 365 (1952).
44. Mott N. F., Gurney R. W., Electronic Processes in Ionic Crystals, Oxfoid
University Press. Lnd., 1940.
45. Fonda G. R., Seitz F., Cornell Symp. of the American Physical Sociely,
Wiley, N. J., 1948.
46. Kroger F. A., Some Aspects of the Luminescence of Solids, Elsevier, Am
sterdam, 1948.
47. Leverenz H. W., An Introduction to the Luminescence of Solids, Wiley,
N. Y„ 1950.
48. Pringsheim P., Fluorescence and Phosphorescence, Interscience, N. Y ,
1949.
49. Boyd C. A., J. Chem. Phys., 17, 1221 (1949).
50. Urbach F., Sitzber. Akad. Wiss. Wein. Math. Naturw. Rl., Abt. Ila, 139,
363 (1930).
51. Randall J. T., Wilkins M. H. F., Proc. Roy. Soc (London), A184, 317
(1945).
52. Garino-Canina V., Cohen S., J. Am. Ceram. Soc., 43, 415 (1960).
53. Boyd C. A., Hirschfelder J., U. S. Patent № 2573245, Oct. 30, 1951.
54. Morehead F. F., Daniels F., J. Phys. Chem., 56, 546 (1960).
55. Ellsworth H. V., Canad. Dept. Mines Geol. Survey. Econ. Geol. Ser., № II,
55 (1932).
56. Alt M., Steinmetz H., Z. Angew. Mineral., 2, 153 (1940).
57. DeWard L. A., Stoebe T. G., Am. Scientist, 60, 303 (1972).
58. Mayhugh M. R., J. Appl. Phys., 41, 4776 (1970).
59. Christy R. W., Johnson N. M., Wilbarg R. R., J. Appl. Chem., 38, 20'JU
(1967).
60. Cameron J. R., Suntharalingam N., Kenney G. N., Thermoluminescent
Dosimetry, University of Wisconsin Press, Madison, 1968.
61. Dairymaple G. B., Doell R. R., Science, 167, 715 (1970).
62. McDougall D. J., ed., Thermoluminescence of Geological Materials, Ain
demic Press, N. Y., 1968.
63. Mazess R. B., Zimmerman D. W., Science, 152, 347 (1966).
64. Zimmerman D. W., Archeometry, 13, 29 (1971).
65. Hyde К. E., J. Chem. Educ., 49, 69 (1972).
66. Mulay L. N„ Keys L. K., Anal. Chem., 36, 2383 (1964).
67. Simmons E. L., Wendlandt W. W., .Anal. Chim. Acta, 35, 461 (1966)
68. Charles R. G., Yannopoulas L. N., Haverlack P. G., J. Inorg. Nucl. Client ,
32, 447 (1970).
69. Gillham J. K., Appl. Polymer Sym., 2, 45 (1966).
70. Lewis A. F., Gillham J. K., J. Appl. Polymer Sci., 6, 422 (1962).
71. Gillham J. K., Proc, of the Second Toronto Symp. on Thermal Anaiyil»]
McAdie H. G., ed.. Chemical Institute of Canada, Toronto, 1967, p 70,
72. Ashby G. E., J. Polymer Sci., 50, 99 (1961).
Глава 12
ПРИМЕНЕНИЕ ЦИФРОВЫХ
И АНАЛОГОВЫХ ВЫЧИСЛИТЕЛЬНЫХ МАШИН
В ТЕРМИЧЕСКОМ АНАЛИЗЕ
А. ВВЕДЕНИЕ
Несмотря на широкое использование цифровых и аналоговых
вычислительных машин в большинстве лабораторных химических
методов [53 ], применение их в термическом анализе довольно огра-
ничено. Согласно опубликованным данным, оно носит пассивный
характер и не предусматривает сколько-нибудь значительного конт-
роля эксперимента с помощью вычислительной техники. Активное
применение вычислительной техники, предусматривающее управ-
ление аппаратурой термического анализа, практически отсутствует.
В данной главе сделана попытка подытожить важнейшие при-
менения вычислительных машин в различных методах термическо-
го анализа. Ввиду трудностей поиска литературы по этому вопросу
автор не ставил своей целью дать исчерпывающее рассмотрение.
Однако можно считать, что наиболее важные исследования в эту
главу включены. Для удобства изложения материала глава рас-
членяется на разделы по применению вычислительной техники в
термогравиметрии, дифференциальном термическом анализе, диф-
ференциальной сканирующей калориметрии и других методах.
Б. ТЕРМОГРАВИМЕТРИЯ
Одним из первых применил цифровую вычислительную машину
для расчета термогравиметрических данных Соулен [1]. В связи с
большим объемом расчетов, требуемых для получения кинетических
констант по данным термогравиметрического анализа, была раз-
работана машинная программа для расчета температуры, массы
и скорости реакции на основе напряжения постоянного тока, по-
ступающего с термовесов. Была использована вычислительная ма-
шина фирмы «Ремингтон рэнд юнивак» с системой компиляции типа
Math-Matic, в которой для расчета 60 значений каждого параметра:
температуры, массы, суммарной потери массы и скорости реакции,
а также для хранения этих значений с целью последующего расчета
кинетических констант была использована 23-командная программа
на естественном английском языке. Вместо использования системы
регистрации данных цифровые значения снимались вручную с лен-
484
Глава 12
Применение вычислительных машин в термическом анализе 485
точного самописца с интервалом в 1 мин и использовались в качест
ве входных данных вычислительной машины. При этом было уста
новлено, что программа на английском языке недостаточно эффек-
тивна для программ этого типа и что при использовании машинного
языка можно разработать более эффективную программу.
Почти все другие случаи применения вычислительных машин и
термогравиметрии касаются расчетов, относящихся к кинетике ре
акций. Шемпф и др. [2] разработали программу POLY 2 для опре
деления предэкспоненциального множителя в уравнении Аррени-
уса и энергии активации. Эта программа, предназначенная для
оценки массы w и температуры образца Т в функции времени /,
может применяться только для расчета реакций первого порядка,
хотя при небольшой модификации ее можно использовать и дли
других реакций. Выборка значений массы производилась методом
наименьших квадратов применительно к полиномиальным выра-
жениям с помощью уравнения
w = 2С<г+1) *<0’
1=0
(12.1)
где п — требуемый порядок полинома; С — коэффициент полинома;
/ — время. По кривой зависимости w от t, полученной таким обра-
зом (с применением дополнительной подпрограммы FREEB па
языке ФОРТРАН), рассчитывалась константа скорости реакции
для любой точки ТГ-кривой с использованием уравнения
п—1
2 с(,+„ <м
i=0
dwjdt
w
(12-2)
где п — требуемый порядок полинома. Анализ значений lg k в за
висимости от 1000/Т выполнялся методом наименьших квадратов
для следующего полинома первого порядка:
lg k = lg А — Еа/2,3037? (1000/Т). (12.3)
_ Полная программа представлена структурной схемой па
фиг. 12.1. Точность машинной аппроксимации ТГ-кривой составля
ла 0,2 мг, тогда как предел точности для считывания значений
веса был равен 0,1 мг.
Сестак и др. [3] разработали две программы для алгоритми п«
ции расчетов кинетических данных на основе ТГ-кривых. Они ш
пользовали следующее фундаментальное уравнение:
^ = ехр(=|)(1-а)«, (12.4)
Фиг. 12.1. Структурная схема программы расчета на ЭВМ [2].
‘где а — степень разложения и п — порядок реакции. Кинетические
[параметры Е, Z и п оценивались путем использования двух диффе-
ренциальных методов. В первом методе применялась аппроксима-
ция ТГ-кривой полиномом /-го порядка с помощью метода наимень-
486
Глава 12
ших квадратов
а = Ао + AiX + • • • -j- AjXJ, (12.1i)
где / (~13) и А — константы, полученные при аппроксимации эк«
периментальных данных с помощью метода наименьших квадратоп
Второй метод состоял в попытке использования простейших ено
собов получения производной наблюдаемой ТГ-кривой путем чш
ленного дифференцирования с использованием первых трех членом
ряда
= Г V -------------------+ i ~ , |
\ / / L ~ t)V
+ 4оу7_2 — Wj_3)-------1----]/QwMaKC (12.0)
где w — потери массы и Q — постоянный временной интервал счн«
тывания. Эта программа была написана на языке АЛГОЛ 60. 14
зультаты, полученные при машинной обработке данных ТГ-крнншм
с использованием различных программ, и результаты ручного счет
приведены в табл. 12.1. Наблюдаемые расхождения отнесены дй
счет различий требований к входным данным. Также подробно пред
ставлена структурная схема второй программы.
Таблица 12 I
Сравнение результатов ручного и машинного расчетов [3]
Использованные экспе- риментальные данные Результаты ручного расчета Машинная программ*
производная [4] интеграл [5] частотный множитель [6]
1
СаС2О4 -> СаСО8 + +СО (г.) (из работы [4]) Е = 74 ккал 67+15 74,1+3,5 74 72,1 5Н.П7
л = 0,7 0,6 1 1 1 o,r>ui
Галлагер с сотрудниками описали несколько систем сбора Г1
данных путем записи на магнитную ленту или перфоленту. Струн»
турная схема их первой системы [7] приведена на фиг. 12.2. II
этой системе выходные данные с термовесов Кана (модель 1)
и хромель-алюмелевой термопары преобразуются в цифровую <|к>|>
му и записываются на перфоленту для последующей машинной
обработки. Период синхронизации счетчика устанавливался рпп
ным 1 с для считывания в канале термопары и 99 с для канала мне
сы. Время коммутации было почти мгновенным, и данные записи
вались на перфоленту при работе счетчика в таком режиме, чю
его мертвое время было пренебрежимо малым. Эффективное исполь»
зование усреднения результатов каждого считывания в предела»
Применение вычислительных машин в термическом анализе
487
указанных интервалов времени приводит к уменьшению шума, что
важно для расчета производной по времени.
Цифровые данные были перенесены с перфоленты на карты и
были получены таблицы зависимости э.д.с. от температуры для
Фиг. 12.2. Структурная схема цифровой термогравиметрической системы
Галлагера и Чери [7].
компенсированной термопары с использованием аппроксимации
методом наименьших квадратов применительно к уравнению
Т (°C) = 22,2877 4-25,7003 (мВ)2 4-0,0017 (мВ)3. (12.7)
£Это уравнение обеспечивало точность ±1° С в интервале тем-
ператур 200—1000° С. Была разработана программа расчета сред-
ней температуры для каждой пары последовательных значений
считанной температуры и увязки этой температуры со средним
значениями считанной массы, причем считывание массы произво-
дилось в интервале между считываниями температуры. Затем ЭВМ
модели 600 фирмы «Дженерал электрик» табулировала и представ-
ляла в графической форме потерю массы в процентах и скорость
потери массы в единицах мг/мин в зависимости от температуры. Ско-
рость потери массы была определена по разности значений массы
при последовательных отсчетах (с интервалом 100 с) и последую-
щей корректировке для получения значений в единицах мг/мин.
Дальнейшей корректировки или подгонки разностных данных не
требовалось.
488
Глава 12
При изотермических измерениях с применением термовепш
Кана (модель RG) была использована система сбора данных,
представленная на фиг. 12.3 [11, 12]. Эта система принимала до
четырех аналоговых входных сигналов, два из которых соответ
вовали массе и температуре. С помощью преобразователя напряжс
ние преобразовывалось в частоту. Результаты расчетов с четырех
Фиг, !2.3. Система сбора данных Галлагера и Джонсона [11, 12],
каналов подавались на четыре шкалы с определенным временным
интервалом. Блоком управления служил блок интерфейса накопи*
теля на магнитной ленте. При работе в'автоматическом режиме дан
ные повторно обрабатывались с заданным временным интервалом
и записывались на магнитную ленту вместе с кодами идентификации
канала. В целях идентификации или контроля можно было ис-
пользовать пятый канал для введения шестизначных чисел. Обри*
ботка данных состояла в передаче их с ленты на дисковый накопи-
тель ЭВМ модели 635 фирмы «Ханиуэлл» в блоки, соответствующие
каждому каналу. Затем при последовательной обработке данных,
как это было описано ранее 112], с помощью ЭВМ были получены
графики с каждого канала в функции времени.
Галлагер и др. [8—10, 13] также описали модификацию терме»*
весов фирмы «Перкин-Элмер» для получения данных в цифровой
форме. В этом приборе платиновая обмотка нагревателя печи слу-
жит одновременно термочувствительным элементом. Она образует
Применение вычислительных машин в термическом анализе
489
одно плечо мостовой схемы; на другое плечо поступает выходное
напряжение программирующего потенциометра. То же самое на-
пряжение используется для питания цифрового устройства регули-
рования температуры и непосредственно связывается с температу-
рой с помощью магнитных термогравиметрических калибровочных
эталонов (по точке Кюри).
о ,1 10,1 10,2 20,2 20,3 30,3 30.4 40,4 40,5 с
Фиг. 12.4. Временная последовательность, используемая в термовесах с
цифровым вычислительным устройством (8].
Оба входные напряжения преобразовывались в частоту и рас-
считывались для заданного времени в последовательности, показан-
ной на фиг. 12.4. Сигнал температуры регистрировался в течение
0,1 с, и затем выполнялось автоматическое переключение на сигнал
массы, который регистрировался в течение 10 с. Выходные данные
постоянно наносились на перфоленту для ввода в ЭВМ.
Таблица 12.2
Кинетические уравнения, использованные при машинном анализе [9]
1. Степенной закон
2. Геометрия усадки
3. Двумерная регулируемая диффузия
4. Уравнение Ерофеева
Б. Объемная регулируемая диффузия
6. Уравнение Джандера
7. Уравнение Праута—Томпкинса -
8. Второй порядок
9. Экспоненциальная зависимость
л — 4 * д, 2,1,2
1 — (1 — а)1/"; п = 2, 3
(1 — а) In (1 —a) -f- а
[—1п(1 — а)]1/п; п=1, -|-,2, 3, 4
3 2/3
(’-“Г
п-а-»)1/3 4 * 6 7 8 9!2
а
1П 7--
1 — а
1
1
In а
Первая стадия машинной обработки данных, записанных на
перфоленте, состояла в перенесении их на перфокарты и в использо-
вании последних для трех ступеней обработки [9]. Первая ступень
состояла в получении графической зависимости массы от времени
(фиг. 12.5). Вторая — в использовании начальных и конечных зна-
чений массы для каждого интервала с целью определения величины
490
Глава 12
Фиг. 12.5. Кривые зависимости массы от времени, по данным Галлагерп и
Джонсона [9].
а, т. е. прореагировавшей части образца. По расчетным значениям
а для каждой точки ЭВМ строила кривые в соответствии с 18 урин-
нениями, приведенными в табл. 12.2. Соответствующие уравнении
определялись путем визуальной проверки выходных машинных
кривых на их линейность. Набор таких кривых зависимости
— 1п(1—а) от времени показан на фиг. 12.6. Выбор уравнения заии-
сел от требуемой точности аппроксимации, определяемой в свою
очередь стандартным отклонением при расчете значений k на третьей
ступени обработки. Третья ступень состояла в выборе наиболее под-
ходящего кинетического уравнения или уравнений, построении
кривых наилучших значений lg£ в зависимости от обратной абсо-
лютной температуры и аппроксимации методом наименьших кппд
ратов для лучшей подгонки к прямой. Полученные значения эиср
гии активации Е и предэкспоненциальных членов печатались вмес-
те с графиком.
Вахуска и Воборил [20] разработали для ЭВМ фирмы GIER про-
грамму VACHVO II [211, с помощью которой по эксперименталь-
Применение вычислительных машин в термическом анализе
491
ным данным рассчитывались в цифровой форме производные по
времени первого, а также второго порядка как для массы, так и для
температуры образца. Новым вариантом этой программы является
программа VYRVACHVON [22], в которой некоторая полиноми-
альная функция проходит через экспериментальные точки и ее ход
Фиг. 12.6. Кривые зависимости —In (1—а) от времени в случае дегидрата-
ции водного нитрата двухвалентного марганца [9].
определяется методом наименьших квадратов. Затем машина рас-
считывает «скорректированные» входные данные по заданной выра-
женной функции и, используя их, выполняет дифференцирование
в числовой форме. Обе программы написаны на языке GIER —
АЛГОЛ.'
" Хотя подробные данные о программах и методиках отсутству-
ют, известно, что ряд исследователй использовали ЦВМ для ана-
лиза кинетических данных, полученных с помощью термограви-
метрии [14—19]. В одном из этих исследований [19] использовался
программируемый калькулятор модели 9100 А фирмы «Хьюлетт-
Паккард».
Хьюз и Харт [231 разработали программу аналогового модели-
рования BASE, которая была использована для прогнозирования
ТГ-кривой. Расчет включал построение ТГ-кривой по уравнению
г,
f(a)= А уеХр(=^)л, (12.8)
т,
где /(а) — некоторый закон скорости разложения а твердого ве-
щества; А — предэкспоненциальный множитель; а = dT/dt — ско-
492
Глава 12
рость нагревания; Е — энергия активации. С целью составлении
исправляющих тестов программы, они приняли
y==exp(^r)=eZ’ (12Л,)
где z = —E/RT и dz = (E/RT2)dT. Тогда
dy = ezdz, (12.10)
dy = ег dT = 2,4ez dT, (12.11)
» RTz RT2 v /
ИЛИ
y=2,4exp/'—'i —. (12.12)
\ RT j RT2 v '
Так как интеграл производной у равен у и поскольку выри-
жение 2,4ехр(—E/RT)E/RT2 идентично у, при использовании вы-
ражения yEJRT2, где у = ехр(—Е/RT), генерируем функции
f(y) (т. е. 2,4r/£ZR7’2), интеграл которой равен у. На фиг. 12.7 этот
Фиг. 12.7. Диаграмма исправляющих тестов [23J.
процесс представлен диаграммой исправляющих тестов программы.
Описанные расчеты были применены к реакции дегидратации
СаСгОа-НгО — системы, которая хорошо изучена рядом исследо-
вателей. На фиг. 12.8 приводятся расчетные и экспериментальны
кривые, полученные при использовании данных Фримена и Кэрро-
ла [4]. Интересно отметить, что кривая, рассчитанная методами
с использованием одновременно таблиц Акахира и эксперименталь-
ных данных, хорошо согласуется с кривой, полученной машинным
расчетом.
Разница между результатами для двух одинаковых образцов,
испытанных в разных лабораториях предположительно в одинако-
вых условиях, привлекла внимание к влиянию скорости и режима
нагревания, а также пульсаций температуры после нагревания па
кривые потерь массы. Поскольку эти проблемы не могли быть ре
Применение вычислительных машин в термическом анализе
493
шены непосредственно на подручном экспериментальном оборудо-
вании, Гейл и Эггер [24] использовали аналоговое устройство для
исследования роли указанных переменных. Расчеты были выпол-
нены на аналоговой вычислительной машине, в которой кривые
скорости нагревания программировались как соответствующие
дифференциальные уравнения, и интеграл этих уравнений по темпе-
ратуре использовался как вводный член в уравнении Аррениуса.
Фиг. 12.8. Расчетные и экспериментальные ТГ-кривые для CaCsO4- НгО [23].
----— расчет; ---- эксперимент.
При интегрировании последнего были получены соответствующие
кривые потерь массы. Кроме того, проводилась специальная обра-
ботка данных для оценки влияния постоянных тепловых помех и
пульсаций в запрограммированном уровне температур. Следует от-
метить, что, когда изменение скорости является экспоненциальной,
а не линейной функцией температуры, симметричные пульсации
в итоге не дают взаимной компенсации помех. Аналоговая вычис-
лительная машина выдает график, представляющий собой доста-
точно точную картину рассмотренных выше процессов.
494
Глава 12
В. ДИФФЕРЕНЦИАЛЬНЫЙ ТЕРМИЧЕСКИЙ АНАЛИЗ (ДТА)
И ДИФФЕРЕНЦИАЛЬНАЯ СКАНИРУЮЩАЯ
КАЛОРИМЕТРИЯ (ДСК)
Почти всегда'использование вычислительных машин в ДТА было
связано с расчетами кинетики реакций. Эти расчеты позволяли дос-
таточно точно воспроизводить кривую ДТА химической реакции
Фиг. 12.9. Влияние энергии активации на кривую ДТА [25].
8 = 18,е; 6' = 0,0594; п = 1.
путем подбора кинетического уравнения. Одним из первых приме-
нили этот метод Рид и др. [251, которые произвели оценку и срав-
нение количественных расчетов кинетики реакций, выполненных
Борхардтом и Даниельсом [26], а также Киссинжером [27]. Кри
вая ДТА была переведена в цифровой код с помощью уравнений
типа
_ е; = Сф"е~Е/6, (12.13)
d6r
где е — энергия активации; 0/ dty/d6r— порядок реакции,
ф = N/No (число молей); £ = ссА(Ао /К)”-1; 9/ — безразмер
ная скорость нагревания. При использовании конечных разностей
уравнение будет иметь следующий вид:
р(6г + й)+^г Iя у
L 2 J
X exp [=^- (6, + h) +6(6Г)] ,
(12.14)
Применение вычислительных машин в термическом анализе
495
где h — размер шага А0Г. На фиг. 12.9 приведена типичная, за-
кодированная для расчета на ЭВМ кривая, которая отражает влия-
ние энергии активации s на кривую ДТА. Изменены как форма,
так и расположение кривых, а зависимость получается обратной
зависимости, наблюдаемой при изменениях частотного фактора.
Сквара и Сатава [28], пользуясь алгоритмом, разработанным
на языке АЛГОЛ 60 для ЭВМ модели 4130 «Эллиот» фирмы NCR,
вычислили по кривым ДТА прореагировавшую долю образца (сте-
пень разложения а). С помощью этого алгоритма вычисляются а и
lg g(cc) и строится зависимость последнего от температуры. При срав-
нении результатов ДТА, полученных на ЭВМ, с эксперименталь-
ными данными для нескольких реакций диссоциации получено хо-
рошее соответствие, что указывает на возможность применения дан-
ного метода расчета.
Вильбурн и др. [29, 30] исследовали возможность использова-
ния моделирующей системы для улучшения характеристик уста-
новки ДТА и изучения термических эффектов, отраженных на кри-
вой ДТА. Для определения связи градиентов температуры в образ-
цах и количества выделившегося или поглощенного тепла в соот-
ветствии с принятым уравнением был использован метод конечных
разностей. На ЭВМ модели 1909 фирмы ICT был произведен рас-
чет влияния этих физических параметров на форму и величину
пика температуры типичной кривой ДТА.
Горник [54] описал применение расчетов на ЭВМ при исследо-
ваниях методом ДТА кинетики кристаллизации полимеров. Ра-
счеты температуры полимерного образца при охлаждении произ-
водились на ЭВМ модели 5500 фирмы «Бэрроуз». Мори и др. [55]
использовал ЭВМ модели ИЗО фирмы IBM для построения эталон-
ных кривых давления в зависимости от температуры типа in р =
— f (1/7}, в которых давление пара определялось по кривым ДТА и
ДСК, а теплота испарения подсчитывалась по методу Хаггенмахера,
модифицированному Фиштайном.
Дэвид и др. [31 ] использовали для измерений температур фазовых
переходов цифровой датчик температуры в сочетании с аналоговым
самописцем, разрешающая способность которого составляла 0,05°С
при скорости нагревания 10 град/мин.
Амштутц [32] описал систему сбора данных фирмы «Меттлер»,
которая может обрабатывать восьмиразрядные числа любого форма-
та или уровня напряжения На фиг. 12.10 приведена ее структур-
ная схема. В расширенном варианте эта система имеет цифровой
и аналоговый многоканальные переключатели, клавишные пульты
управления и блоки переключателей ручного ввода данных, реле
времени и программирующие устройства. Применительно к ДТА
данная система производит ряд операций, включающих запись не-
обработанных данных на перфоленту или магнитную ленту в авто-
номном режиме и обработку данных от простых расчетов площади
496
Глава 12
пика с помощью программируемых настольных калькуляторов до
более сложных численных расчетов теплоты и кинетики реакций,
также анализа чистоты в оперативном режиме.
Одно из основных применений вычислительных машин в диф-
ференциальной сканирующей калориметрии (ДСК) заключается
1тА1о| >1тА2о| ~~ ~ - — и—>ГСТ2О| >1СТ15 k-—>j СТЮ | t-U А
—НТА321 |ТАЗО| „ ^tJtA3i| |gaio| |СТ14] | СТ191 [стю| Система передачи данных
модель 2000 фирмы «Меттлер» фирмы «Меттлер»
Фиг. 12.10. Система передачи данных фирмы «Меттлер», объединенная с си
стемой расчета кривых ДТА фирмы «Меттлер» (модель ДТА 2000) [32].
ТАЮ — блок ДТА; ТА20 — усилитель ДТА; TA30 — селектор программы; ТА31 — усилитель
мощности; ТА32 — регулятор температуры; GA10 — самописец; СТЮ —блок передачи данных;
СТ14 — клавишный пульт с цифровой индикацией; СТЮ — многоканальный переключатель
панели управления; СТ 16 — цифровая индикация; СТЮ —реле времени; СТ20 — цифровой
вольтметр; А — ленточный перфоратор.
в определении чистоты органических и неорганических соедине-
ний. Обзор методов точного определения чистоты с помощью ра-
счетов на ЭВМ сделан Джоем и др. [33]. Одна из первых про-
грамм для определения чистоты по данным ДСК составлена Дрис-
коллом и др. 134]. Для этой программы требуются следующие вход-
ные данные: масса образца,-молекулярный вес, константы измери-
тельных приборов, исходная температура в точке, в которой кривая
все еще совпадает с базовой линией, а также координаты различных
точек кривой. Одно измерение должно быть произведено на пике
кривой плавления, при этом нет необходимости соблюдать одина-
ковые интервалы между измеряемыми точками кривых. Информа-
ционная емкость программы — 99 пар считанных данных.
Для расчета АН/ по этой программе производится деление кри-
вой на 99 равных температурных интервалов и последующее интег-
рирование. Для каждого температурного интервала производится
коррекция температуры и площади под базовой линией, затем опре-
деляются «парциальные» площади. Далее программа предусмат-
ривает последовательную 0,5%-ную коррекцию площади для каж-
дой «парциальной» площади и всей площади в целом и расчет I/F.
Для обеспечения минимального стандартного отклонения точек
от расчетной кривой применяется анализ методом регрессии с ис-
пользованием метода наименьших квадратов. При дальнейших
расчетах используются «наилучшие значения». ЭВМ выдает следую-
щие величины: AHf, То, Тт, содержание примесей (мол.%), пре-
Применение вычислительных машин в термическом анализе
497
делы используемых значений 1/F, процент коррекции и криоскопи-
ческую константу. Кроме того, рассчитывается молярный процент
примеси, откорректированный в предположении образования твер-
дого раствора. В этой работе рассматривается также «линеариза-
ция» кривой зависимости Ts от 1/F [35]. Джой и др. [33] трансли-
ровали программу Дрисколла с языка ФОРТРАН на язык основ-
ной программы, которая может работать при использовании уст-
ройства ввода — вывода ЭВМ, действующего в режиме разделения
времени. Другие программы определения чистоты по данным ДСК
были разработаны Бэрроллом и Диллером [361, а также Скоттом
и Греем [37 ].
Эллерстайн [38], используя программу Quiktran фирмы IBM,
производил расчеты кривых ДСК по уравнению
=--------- ГАШ1 + Пг (12.15)
Alg4 2,303/? L AigA- J
где I — ордината смещения между базовой линией и кривой, aAr
соответствует площади кривой при температуре Т. Графическая
зависимость левой части уравнения от выражения в скобках
имеет вид кривой с наклоном £/2,303/?. Точка пересечения этой
кривой с осью ординат определяет порядок реакции и. Затем ре-
зультаты расчета по этой программе вводятся в стандартную библио-
теку программ IBM Quiktran (FITLIN), которая дает «наилучшее»
линейное приближение для расчетных точек.
Грей [39] разработал программу, с помощью которой на осно-
вании кривой ДСК и положения базовой линии определяется ход
изотермы, производится интегрирование накапливаемой (кумуля-
тивной) и полной площади (кал/г), производится коррекция темпе-
|ратуры с учетом теплового запаздывания, табулируются и откла-
дываются по оси ординат удельная теплоемкость и кумулятивная
площадь в единицах теплосодержания (энтальпии). С помощью
системы анализа результатов термического анализа типа AD S VI
фирмы «Перкин-Элмер» аналоговые данные с измерительных при-
боров установки ДСК переносятся в цифровой форме на бумажную
ленту. Каждые 2 с, или каждые 0,133°, данные представляются
в цифровой форме. Затем автоматический графопостроитель вы-
числительной машины строит кривую ДСК, а также накапливаемую
площадь пика в единицах удельного теплосодержания (кал/г).
Кросли и др. [40 ] разработали метод машинной обработки дан-
ных для определения площади изотермической кривой вместо при-
менения планиметра. Обработка данных разделяется на две стадии:
а) решения относительно не зависящей от механизма реакции доли
а реагирующего вещества, сохранившейся к моменту времени /,
и различных функций а и б) решения относительно зависящих от
механизма реакции констант скорости. Для первой фазы расчета
была разработана программа DATAR, состоявшая в следующем:
17—230
498
Глава 12
непосредственно в вычислительную машину последовательно вво
дилнсь точки, соответствующие необработанным данным и расио
ложенные с равным интервалом вдоль оси времени кривой Д( К
Можно вводить до 1000 точек, но обычно для расчета с удовлетвори
тельной точностью достаточно 40—50 точек. Величина а в момен i
времени t рассчитывалась с помощью уравнения
t
J (ORD) dt
^макс
a(t) = — (12.16)
J (ORD) dt
^макс
Для нахождения интегралов использовалась методика Симпго
на, модифицированная для случая нечетного числа интервалон
С помощью этой программы рассчитывались и выдавались в напева
тайном виде следующие величины: время в секундах и минутах, а,
1ла, 1/а2, (1—а)аи lgl00[(l—а)/а.]. На второй стадии расчета для
определения истинных констант скоростей kt и k2 использовала» т.
программа PARACT.
Программы для расчета кинетики реакции по данным ДСК были
также разработаны Кауфманом и Бичем [41 ], а также Роджерсом
и Смитом [42] Хойвел и Линд [43] использовали вычислительную
машину для определения поправок к данным ДСК, учитывающих
тепловое запаздывание и изменения теплоемкости. Сондак 1111
получил простое уравнение для линеаризации результатов опре-
деления чистоты методом ДСК-
Г. ДРУГИЕ МЕТОДЫ ТЕРМИЧЕСКОГО АНАЛИЗА
Анализ изопериболических калориметрических данных требу er
длительных графических построений и громоздких расчетов дли
получения откорректированных значений изменений сопротивле-
ния в ходе реакций и калибровочных экспериментов. Протекающая
в таких экспериментах реакция графически выражается двумя пр я
мыми линиями: скоростью начального периода (СНП) и скоростью
конечного периода (СКП), соединенными между собой кривой, дли
которой аналитическое выражение неизвестно. Гейер и Бартел |'15|
разработали программу расчетов, связанных с этими эксперпмеп
тами.
Фридман и др. [46—48] разработали цифровой преобразователь
для записи на перфоленту результатов анализа выделившегося i ;i hi
(АВГ) и масс-спектрометрического анализа (МС). На фиг. 12.11
приведена использованная ими система сбора данных. Она основа-
на на применении высокостабильного программируемого источнике
питания, который подает стробирующий импульс в интервалы между
Применение вычислительных машин в термическом анализе
499
Данные, нанесенные
на перфорированную ленту
Фиг. 12.11. Система сбора данных для анализа выделившегося газа и масс-
спектрометрического анализа по Фридману [47].
выбранными дискретными пиками массы ионов. На практике ана-
логовые стробирующие напряжения задавались приблизительно для
20 пиковых значений в интервале значений т/е от 1 до 203, что кон-
тролировалось с помощью цифрового вольтметра. Результаты были
затем проанализированы с помощью программы для режима раз-
деления времени, основанной на методе наименьших квадратов,
с использованием алгебраического уравнения, включавшего пять
констант. Если все расчетные точки находятся в пределах значений,
эквивалентных наблюдаемой задержке 10 нс, то приближение яв-
ляется приемлемым, и путем интерполяции и экстраполяции полу-
17*
500
Глава 12
чают машинную распечатку для всех значений аналогового стропи
рующего напряжения в зависимости от массового числа. Результгны
эксперимента, полученные в виде записи на перфоленте, счигы
ваются оптическим считывающим устройством и сохраняются к
запоминающем устройстве малой вычислительной машины. Затем
большая вычислительная машина сортирует данные по массовым
Фиг. 12.12. Вычислительная система для анализа методами ТГ и МС по ГпЛ
сону [49].
числам и с помощью графопостроителя строит график. После этой»
в результаты, нанесенные на график, вводятся поправки с учетом
фонового уровня помех и чувствительности измерительных приборов,
производится редакционная корректировка и данные приводятся
к 1 мг массы исходного образца.*
Гибсон [49] описал установку для одновременного исследоии
ния методами ТГ и МС, в которой использовалась вычислительная
машина модели PDP 8/L, связанная с масс-спектрометром для обра
ботки данных в оперативном режиме.
На фиг. 12.12 приведена структурная схема этой системы. Вы
ходной аналоговый сигнал масс-спектрометра интегрируется, пре
образуется в цифровую форму 12-битовым преобразователем и па
писывается на магнитную ленту. Возможны два раздельных режп
ма работы: регистрация данных и контроль спектра. Они различи
ются только способом сбора информации. Во время работы мжч
спектрометра графопостроитель выдает кривую зависимости выспи
бождения газа в реальном масштабе времени, которая на самом деле
представляет собой просто характеристику ионного тока электрон
ного умножителя для каждого регистрируемого спектра. Другие
программы включают следующие операции:
1. Построение картины процесса высвобождения частиц виде*
Применение вычислительных машин в термическом анализе
501
лившегося газа путем нормирования наибольшего пикового значе-
ния относительно 100 и идентификация получающегося в процессе
вычислений спектра масс.
2. Печатание или построение графиков спектров для всех полу-
ченных спектров масс.
3. Построение графиков зависимости пика отдельной массы
(например, mle = 18) от температуры.
4. Получение графиков спектра или машинной распечатки с
положительной идентификацией шкал масс и с численным обозна-
чением ионной интенсивности.
5. Получение графиков спектра или машинной распечатки с вы-
читанием фонового спектра для исключения влияния примесей или
фона.
Сакамото и др. [56] описали кинетические расчеты для поли-
метилметакрилата с использованием масс-спектрометрического тер-
мического анализа. С использованием значения энергии активации,
рассчитанного по экспериментальным данным, вычислительная
машина строила графические зависимости логарифма приведенной
скорости dddQ от логарифма приведенного времени 6. Затем эта
кривая сравнивалась с расчетными теоретическими кривыми для
различных механизмов реакции. Расчетная кривая, которая лучше
всего согласовывалась с экспериментальной, являлась кривой ре-
акции первого порядка. Пфейл [51 ] рассмотрел применение циф-
ровых вычислительных машин для статистического анализа ТГ-
измерений. При термическом анализе биологических систем также
весьма необходимы и полезны графические зависимости, получаемые
с помощью вычислительных машин.
Следует отметить, что вычислительные машины с системами сбора
данных, предназначенные для работы с установками термического
анализа, выпускаются фирмами «Перкин-Элмер» [37], «Дюпон» и
«Меттлер инструмент» [32]. Система сбора данных термического
анализатора описана также Йи и др. [52].
ЛИТЕРАТУРА
1. Soulen J. R., Anal. Chem., 34, 136 (1962).
2. Schempf J. M., Freeburg F. E., Roger D. J., Angeloni F. M., Anal. Chem.,
38, 520 (1966).
3. Sestak J., Brown A., Rihak V., Berggren G., Thermal Analysis, Schwen-
ker R. F., Garn P. D., eds., Vol. 2, Academic Press, N. Y., 1969, p. 1035,
1085.
4. Freeman E. S., Carroll B., J. Phys. Chem., 62, 394 (1958).
5. Coates A. W., Redfern J. P„ Nature, 201, 88 (1964).
6. Satava V., Sestak J., Silikaty, 8, 143 (1964).
7. Gallagher P. K., Schrey F., Thermal Analysis, Schwenker R. F., Garn P. D.,
eds., Vol. 2, Academic Press, N. Y., 1969, p. 929.
8. Gallagher P. K., Schrey F., Thermochim. Acta, 1, 465 (1970).
9. Gallagher P. K., Johnson D. W., Thermochim. Acta, 2, 413 (1971).
10. Gallagher P. K., Johnson D. W., Thermochim. Acta, 3, 303 (1972).
502
Глава 12
11. Gallagher Р. К., Johnson D. W., Thermochim. Acta, 5, 455 (1973).
12. Johnson D. W., Gallagher P. K., J. Phys. Chem., 75, 1179 (1971).
13. Gallagher P. K-, Schrey F., Prescott B., Thermochim. Acta, 2, 405 (1971)
14. Ozawa T., J. Thermal Anal., 2, 301 (1970).
15. Zsako J., J. Phys. Chem., 72, 2406 (1968).
16. Zsako J., Varhelyi C., Agosescu M., Studia Babes-Bolyai Univ., 2, 33 (1970)
17. Zsako J., Varhelyi C., Agosescu M., Studia Babes-Bolyai Univ., 2, 113 (1970)
18. Sharp J. H., Wentworth S. A., Anal. Chem., 41, 2060 (1969).
19. Reich L., Gregory W., Stivala S. S., Thermochim. Acta, 4, 493 (1972).
20. Vachuska J., Voboril M., Thermochim. Acta, 2, 379 (1971).
21. Vachuska J., Rykalova N., GIER Computer Library, NRI, Czech. Acad
Sci., № 1064.
22. Rykalova N., Vachuska J., GIER Computer Library, NRI, Czech. Acad
Sci., № 1399.
23. Hughes M. A., Hart R., ICTA III, Davos, Switzerland, Aug. 23—28, 19/1,
paper 1-21.
24. Gayle J. B., Egger С. T„ Anal. Chem., 44, 421 (1972).
25. Reed R. L., Weber L., Gottfried B. S., I & EC Fundamentals, 4, 38 (1965)
26. Borchardt H. J., Daniels F., J. Am. Chem. Soc., 79, 41 (1957).
27. Kissinger H. E., Anal. Chem., 29, 1702 (1957).
28. Skvara F., Satava V., J. Thermal Anal., 2, 325 (1970).
29. Wilburn F. W., Hesford J. R., Flower J. R., Anal. Chem., 40, 777 (1968)
30. Melling R., Wilburn F. W., McIntosh R. M., Anal. Chem., 41, 1275 (1969)
31. David D. J., Ninke D. A., Duncan B., Am. Lab., Jan., 31 (1971).
32. Amstutz D., ICTA III, Davos, Switzerland, Aug. 23—28, 1971, paper 1 39
33. Joy E. F., Bonn J. D., Barnard A. J., Thermochim. Acta, 2, 57 (1971)
34. Driscoll G. L., Duling I. N., Magnotta F., Analytical Calorimetry, Poi
ter R. S., Johnson J. F., eds., Vol. 1, Plenum Press, N. Y., 1968, p. 271
35. Driscoll G. L., Duling I. N., Magnotta F., Sun Oil Quart., 3, 24 (1969)
36. Barrail E. M., Diller R. D., Thermochim. Acta, 1, 509 (1970).
37. Scott L. R., Gray A. P., Perkin-Elmer Instrument News, 19, №3, 1 (1969)
38. Ellerstein S. M., Analytical Calorimetry, Porter R. S., Johnson J. F., ed ,
Vol. 1, Plenum Press, N. Y., 1968, p. 279.
39. Gray A. P., Perkin-Elmer Instrument News, 20, 8 (1969).
40. Crossley R. W., Dorko E. A., Diggs R. L., Analytical Calorimetry, Poi
ter R. S., Johnson J. F., eds., Vol. 2, Plenum Press, N. Y., 1970, p. 429
41. Kauffman G. B., Beech G., Thermochim. Acta, 1, 99 (1970).
42. Rogers R. N., Smith L. C., Thermochim. Acta, 1, 1 (1970).
43. Heuvel H. M., Lind К. C. J. B., Anal. Chem., 42, 1044 (1970).
44. Sondack D. L., Anal. Chem., 44, 888 (1972).
45. Gayer К. H., Bartel J., Thermochim. Acta, 3, 337 (1972).
46. Friedman H. L., J. Macromol. Sci., Al, 57 (1967).
47. Friedman H. L., Griffith G. A., Goldstein H. W., Thermal Analysis,
Schwenker R. F., Garn P. D., eds., Vol. 1, Academic Press, N. J., 1969,
p. 405.
48. Friedman H. L., Thermochim. Acta, 1, 199 (1970).
49. Gibson E. K-, Thermochim. Acta, 5, 243 (1973).
50. Ingraham T. R., Proc, of the Second Toronto Symp. on Thermal Analysis,
McAdie H. G., ed., Chemical Institute of Canada, Toronto, 1967, p 21,
51. Pfeil R. W., Proc, of the Third Toronto Symp. on Thermal Analysis,
McAdie H. G., ed., Chemical Institute of Canada, Toronto, 1969, p. 187.
52. Yui H., Kato R., Okamoto H., Maezono A., 7th Japanese CaloriiiicllV
Conference, Nagoya, Japan, Nov. 25—26, 1971.
53. Perone S. P., Anal. Chem., 43, 1288 (1971).
54. Gornick F., J. Polymer Sci., Part C, 25, 131 (1968).
55. Morie G. P., Powers T. A., Glover C. A., Thermochim. Acta, 3, 259 (1972),
56. Sakamoto R., Ozawa T., Kanazaski M., Thermochim. Acta, 3 , 291 (1972),
Глава 13
ТЕРМИНОЛОГИЯ В ТЕРМИЧЕСКОМ АНАЛИЗЕ
А. ВВЕДЕНИЕ
В 1967 г. Макади [1] опубликовал рекомендации комитета
по стандартизации при Международной конфедерации по термиче-
скому анализу (МКТА) относительно представления эксперимен-
тальных данных ДТА и ТГ. К каждой кривой ДТА или ТГ в публи-
кациях должна прилагаться следующая информация:
1. Характеристика всех веществ (образец, эталон, растворитель)
путем указания их названия, эмпирических формул или эквивалент-
ных данных по химическому составу.
2. Данные о способе получения исследуемых веществ, их преды-
стории, условий предварительной обработки, степени химической
чистоты.
3. Средняя скорость изменения температуры по линейному за-
кону в исследуемом интервале температур.
4. Характеристика атмосферы, окружающей образец (давление,
состав, степень чистоты), с указанием типа атмосферы: статическая,
самогенерируемая или динамическая (с потоком газа, пропускае-
мого через образец или обтекающего его). Если образец находится
в атмосферных условиях, следует указать давление и влажность.
Если давление отличается от атмосферного, следует подробно ука-
зать метод его измерения.
5. Размеры, форма и материал держателя образца, а если обра-
зец нагружен, то и способ его нагружения.
6. Нанесение по оси абсцисс шкалы времени или температуры
в определенном масштабе с увеличением слева направо.
7. Указание методов идентификации промежуточных и конеч-
ных продуктов реакций.
8. Точное воспроизведение всех записей самописца.
9. По возможности расшифровка и подтверждение дополнитель-
ными экспериментальными данными каждого термического эффекта.
При опубликовании ТГ-данных необходимы следующие допол-
нительные сведения:
10. Характеристика термовесов, включающая указание место-
нахождения термопары.
Б04
Глава 13
11. Вес образца, нанесение шкалы веса по оси ординат. Потери
веса следует наносить сверху вниз. Отклонения от этого принципа
следует особо оговаривать. При желании можно использовать оси
ординат с дополнительными шкалами, такими, как степень разло-
жения или молекулярный состав.
12. Если применяется метод термогравиметрии по производной,
то необходимо указать метод получения производной и соответствен-
но обозначить единицы измерений по оси ординат.
При опубликовании кривых ДТА требуются следующие допол-
нительные данные:
10. Вес образца, а для раствора,— условия его разбавления
(концентрация, растворитель).
11. Характеристика прибора, включающая данные о форме и
материале термопар и расположении дифференциальной и измери-
тельной термопар.
12. Шкала ординат должна указывать отклонение АТСС при
данной температуре. Рекомендуется при нанесении на график от-
клонения температуры образца от температуры эталона отклады
вать вверх положительные разности температуры, вниз — отри-
цательные. Отступления от этого принципа следует особо огова
ривать.
В 1969 г. председатель комитета по терминологии при МКТ А
Маккензи [21 опубликовал первый сборник, содержащий перечень
терминов, рекомендуемых для применения в термическом анализе.
Этих рекомендаций следует придерживаться во всех публикациях
по термическому анализу на английском языке.
Б. ОБЩИЕ РЕКОМЕНДАЦИИ
1. Следует принять термин термический анализ (thermal analy-
sis), а не «термография», поскольку последний термин имеет по крап
ней мере еще два других значения (и одно из них медицинское).
Прилагательное должно при этом быть: «термоаналитическпп»
(сравните, например, «физическая химия» и «физико-химический»);
термин «термоанализ» не рекомендуется к употреблению (на той же
логической основе).
2. Термин дифференциальный (differential) используется как
прилагательное, образованное от существительного (difference
разность). Термин по производной (derivative) следует использован»
для первой производной (математической) параметра любой тер
мической кривой.
3. Термина «анализ» следует по возможности избегать, так как
рассматриваемые методы не включают анализ в том смысле, в каком
его употребляют химики. Однако, термин дифференциальный тер
мический анализ столь широко распространен, что изменять его не-
целесообразно.
Терминология в термическом анализе
505
4. Термин кривая предпочтительнее термина «термограмма»
по следующим причинам:
а. Термин «термограмма» применяется для обозначения резуль-
татов, получаемых в медицинской термографии.
б. В применении к определенным видам кривых (например,
термогравиметрическим кривым) термин «термограмма» не соответ-
ствует определению, которое дается в словарях.
в. Применяемые иногда для ясности термины «дифференциаль-
ная термограмма», «термогравиметрическая термограмма» и т. п.
являются не только громоздкими, но и запутанными.
5. В комплексном исследовании ^термин совмещенный (simul-
taneous) следует применять при одновременном использовании
двух или более методов исследования одного и того же образца.
Тогда термин комбинированный (combined) будет означать исполь-
зование отдельных образцов для каждого экспериментального ме-
тода.
6. Термин термическое разложение (thermal decomposition) и
другие аналогичные термины будут рассмотрены комитетом позже.
В. ТЕРМИНОЛОГИЯ
В табл. 13.1 приведены принятые наименования и сокращения,
а также термины, которые по различным причинам отклонены. Ко-
митет согласен с предложением, внесенным во время обсуждения
сборника по терминологии 12 ] и заключающимся в том, что некото-
рые сокращения следует рекомендовать для международного исполь-
зования независимо от языка.
Комитет не высказал мнения по терминологии в смежных мето-
дах (таких, как термометрическая титрометрия или калориметрия).
Таблица 13.1
Рекомендуемая терминология
Рекомендуемый термин
Принятое
сокращение1)
Нерекомендуемый термин
1. Общее понятие
Термический анализ
2. Методы, связанные с из-
менением веса
а. Статические:
Определение изменения
веса при постоянном дав-
лении
Изотермическое определе-
ние изменения веса
б. Динамические:
Термог рави метр и я
ТГ (TG)
1 :
Термография
Термоанализ
Изотермический термог рави-
метрический анализ
Термогравиметрический ана-
лиз
Продолжение
Рекомендуемый термин Принятое сокращение1) Нерекомендуемый термин
Термогравиметрия по про- ТГП (DTG) Динамический термограви- метрический анализ Дифференциальная термогра-
изводной 3. Методы, связанные с из- менением энергии Кривые нагревания2) Кривые скорости нагрева- ния2) Обратные кривые скорости нагревания2) Дифференциальный тер- мический анализ ДТА (DTA) виметрия Дифференциальный термо- гравиметрический анализ Термогравиметрический ана- лиз по производной Термический анализ Термический анализ по про- изводной Динамическая дифференци- альная калориметрия
Дифференциальный тер- мический анализ по про- изводной Дифференциальная ска- нирующая калориметрия 4. Методы, связанные с вы- делением летучих веществ Обнаружение выделен- ного газа ДСК (DSC) ОВГ (EGD) Обнаружение истекающего газа
Анализ выделенного газа3) АВГ (EGA) Анализ истекающего газа
5 Методы, связанные с из- менением размеров Дилатометрия Дилатометрия по произ- водной Дифференциальная дила- тометрия 6. Совмещенные методы Совмещенный метод ТГ и ДТА и т. д. Термический анализ при ис- парении ДАТА (Дифференциальный и термогравиметрический анализ) Дериватография Дериватографический анализ
*) Сокращения следует записывать прописными буквами» расположенными слитно. Во из-
бежание недоразумений ие следует использовать обширные сокращения.
2) В режиме охлаждения эти кривые будут называться: кривые охлаждении, кривые ско-
рости охлаждения и обратные кривые скорости охлаждении.
8) Метод анализа должен быть четко указан. Следует избегать таких сокращений, как
МТА (масс-спектрометрический термический анализ) и МДТА (масс-спектрометрический и
дифференциальный термический анализ).
Терминология в термическом анализе
507
Рассмотрение методов термического анализа, не получивших пока
широкого применения, отложено.
Г. ОПРЕДЕЛЕНИЯ И УСЛОВНЫЕ ОБОЗНАЧЕНИЯ
1. ОБЩЕЕ ПОНЯТИЕ
Термический анализ (Thermal analysis). Общий термин, охваты-
вающий ряд смежных методов, в которых измеряется зависимость
параметров какого-либо физического свойства вещества от темпера-
туры,
2. МЕТОДЫ, СВЯЗАННЫЕ С ИЗМЕНЕНИЕМ ВЕСА
а. Статические методы
Определение изменения веса при постоянном давлении (Isobaric
weight-change determination). Метод, позволяющий регистрировать
равновесное значение веса вещества в зависимости от температуры Т
при постоянном парциальном давлении летучего компонента или
компонентов.
Запись представляет собой изобару изменения веса. Обычно
по оси ординат откладывают сверху вниз вес, а по оси абсцисс —
слева направо температуру Т.
Изотермическое определение изменения веса (Isothermal w’eight-
change determination). Метод, позволяющий регистрировать вес
вещества в зависимости от времени t при постоянной температуре.
Запись представляет собой изотерму изменения веса. Обычно
по оси ординат откладывают сверху вниз вес, а по оси абсцисс —
слева направо время.
б. Динамические методы
Термогравиметрия (ТГ) [Thermogravimetry (TG)]. Метод, поз-
воляющий регистрировать вес вещества в зависимости от темпера-
туры или времени при нагревании или охлаждении в заданной среде
с регулируемой скоростью.
Запись представляет собой термогравиметрическую (ТГ) кри-
вую. Вес следует откладывать по оси ординат сверху вниз, а вре-
мя t или температуру Т — по оси абсцисс слева направо.
Термогравиметрия по производной (ТГП) [Derivative thermo-
gravimetry (DTG)J. Метод, позволяющий получить первую произ-
водную термогравиметрической кривой по времени или по темпера-
туре.
Запись представляет собой производную термогравиметрической
кривой (ТГП). Производную следует откладывать по оси ординат
а время t или температуру Т — по оси абсцисс слева направо.
508
Глава 13
Д. МЕТОДЫ, СВЯЗАННЫЕ С ИЗМЕНЕНИЕМ ЭНЕРГИИ
Кривые нагревания (Heating curves). Эти кривые представляют
собой запись температуры вещества, помещенного в среду, на-
греваемую с регулируемой скоростью, в зависимости от времени.
Температуру Т следует откладывать по оси ординат снизу вверх,
а время t — по оси абсцисс слева направо.
Кривые скорости нагревания (Heating-rate curves). Эти кривые
представляют собой запись первой производной кривой нагревания
по времени dT/dt в зависимости от времени или температуры.
Производную dTIdt следует откладывать по оси ординат, а вре-
мя t или температуру Т — по оси абсцисс слева направо.
Обратные кривые скорости нагревания. (Inverse heating-rate
curves). Эти кривые представляют собой запись первой производ-
ной кривой нагревания по температуре dtldT в зависимости от вре-
мени или от температуры.
Функцию dtldT следует откладывать по оси ординат, а время t
или температуру Т — по оси абсцисс слева направо.
Дифференциальный термический анализ (ДТА) [Differential
thermal analysis (DTA) ]. Метод, позволяющий регистрировать
разность температур исследуемого вещества и эталона в функции
времени или температуры. Исследуемый образец и эталон находятся
в идентичных температурных условиях в среде, нагреваемой или
охлаждаемой с регулируемой скоростью.
Запись представляет собой кривую ДТА. Разность температур
АТ следует откладывать по оси ординат снизу вверх (для эндотер
мических реакций —- сверху вниз), а время t или температуру Т
по оси абсцисс слева направо.
Дифференциальный термический анализ по производной. (Deri
vative differential thermal analysis). Метод, позволяющий регистр и
ровать первую производную кривой дифференциального термиче
ского анализа в зависимости от времени или температуры.
Запись представляет собой производную кривой дифференциала
него термического анализа (ДТА). Производную следует отклады-
вать по оси ординат, а время t или температуру Т— по оси абсцисс
слева направо.
Дифференциальная сканирующая калориметрия (ДСК) [Differen-
tial scanning calorimetry (DSC) ]. Метод, позволяющий регистриро-
вать энергию, необходимую для выравнивания температур иссле-
дуемого вещества и эталона в зависимости от времени или темпера-
туры. Исследуемый образец и эталон помещаются в идентичные
температурные условия, в среду, нагреваемую или охлаждаемую
с регулируемой скоростью.
Запись: представляет собой кривую ДСК, где по оси ординат
откладывается количество тепла в единицу времени, а по оси абс-
цисс,— t или Т.
Терминология в термическом анализе
509
Е. МЕТОДЫ, СВЯЗАННЫЕ ^ВЫДЕЛЕНИЕМ ЛЕТУЧИХ ВЕЩЕСТВ
Обнаружение выделенного газа (ОВГ) [Evolved gas detection
(EGD) J. Этот термин относится к любому методу обнаружения не-
зависимо от того, образуется или не образуется летучий продукт
в процессе термического анализа.
Анализ выделенного газа (АВГ) [Evolved gas analysis (EGA)].
Метод, позволяющий определить природу и оценить количество
летучего продукта (или продуктов), образовавшегося в ходе терми-
ческого анализа.
Ж. МЕТОДЫ, СВЯЗАННЫЕ С ИЗМЕНЕНИЕМ РАЗМЕРОВ
Дилатометрия (Dilatometry). Метод, в котором определяются
изменения размера (или размеров) образца в зависимости от тем-
пературы. Графически зависимость выражается дилатометрической
кривой.
Дифференциальная дилатометрия', дилатометрия по производ-
ной (Differential dilatometry; derivative dilatometry). При исполь-
зовании этих определений следует учесть различие в терминах,
приведенных в разд. Б.2 настоящей главы.
3. КОМПЛЕКСНЫЕ МЕТОДЫ
Этот термин относится к одновременному использованию ДТА,
ТГ и других методов. Маккензи и др. [31 возражают против исполь-
зования термина «термогигрометрический анализ» (ТГА), который
применяли Стилл и Клали [4 ]. Они считают, что этот метод являет-
ся просто частным случаем метода «обнаружения выделенного газа»
(ОВГ), для которого уже есть международное обозначение. Комитет
по терминологии при МКТА считает совершенно неоправданным вве-
дение сокращений, являющихся вариантами уже принятых тер-
минов, и настоятельно просит всех научных работников вводить
и публиковать новые сложные термины и сокращения только после
очень серьезного обсуждения.
В 1972 г. во втором сборнике комитета по терминологии Маккен-
зи и др . [5 ] опубликовали более определенные, чем ранее, рекомен-
дации.
И. ДТА
Образец (sample) — исследуемое вещество независимо от того,
является оно чистым или находится в смеси с растворителем (на-
полнителем).
Эталон (reference material) — вещество, обычно термически
неактивное в представляющем интерес интервале температур.
510
Глава 13
Образцы (specimens) •— образец и эталон.
Держатель образца (sample holder) — контейнер, в котором
находится образец, или устройство, его удерживающее.
Держатель эталона (reference holder) — контейнер, в котором
находится эталон, или устройство, его удерживающее.
Узел держателя образца (specimen holder assembly) — узел,
в котором помещаются образец и эталон в целом. Если нагревающее
или охлаждающее устройство находится в одном узле с контейнером
или держателем образца и эталона, то оно должно рассматриваться
как часть этого узла.
Блок (block) — узел держателя образца, в котором корпус
сравнительно большой массы находится в контакте с держателями
образца и эталона.
Дифференциальная термопара (differential thermocouple), или
АТ-термопара (AT thermocouple) — система термопар, используе-
мая для измерения разности температур. Если используется другой
термочувствительный элемент, то его следует соответственно назы-
вать.
к. тг
Термовесы (thermobalance) — прибор для непрерывного взвеши-
вания образца при его нагревании или охлаждении.
Образец (sample) — исследуемое вещество независимо от того,
является оно чистым или находится в смеси с растворителем (на-
полнителем). В ТГ обычно используются чистые образцы без раст-
ворителя или наполнителя, а при одновременном применении ТГ
и ДТА могут быть использованы образцы в виде смесей.
Держатель образца (sample holder) — см. предыдущий р аздел.
л. ДТА И ТГ
Температурная термопара (temperature thermocouple), или Т-
термопара (Т thermocouple), — система термопар, используемая
для измерения температуры. Положение термопары относительно
образца всегда должно быть определено. При использовании дру-
гого термочувствительного элемента следует указывать его назва-
ние.
Скорость нагревания (heating rate) — скорость возрастания
температуры, обычно измеряемая в единицах град/мин (по шкале
Цельсия или Кельвина). Соответственно скорость охлаждения
(cooling rate) — скорость понижения температуры. Скорость на-
гревания или охлаждения, как правило, постоянна (температура
зависит от времени по линейному закону).
При одновременном использовании методов ДТА и ТГ применя-
ются те же определения, что и при раздельном использовании этих
методов.
Терминология в термическом анализе
511
Наряду с рассмотренными ранее [1,2] прн использовании мето-
дов ДТА и ТГ рекомендуется применять следующие определения.
М. ДТА
При использовании метода ДТА следует помнить, что, хотя по
оси ординат обычно откладывается АТ, выходной сигнал с АТ-
термопары в большинстве случаев изменяется с температурой, и
регистрируемый результат измерения — это на самом деле выход-
ная э.д.с., т. е. Е [переводной коэффициент b в уравнении АТ =
ЬЕ не является величиной постоянной, так как Ь = f(T)]. Ана-
логичная ситуация имеет место и при использовании других термо-
чувствительных элементов.
Все приведенные ниже определения относятся к одному пику,
подобному показанному на фиг. 13.1. Кривые, имеющие перегибы
или больше одного максимума или минимума, могут рассматривать-
ся как результат наложения одиночных пиков.
Базовая линия (base line) соответствует тем участкам кривой
ДТА (АВ и DE), для которых АТ «0.
Пик (peak) (BCD) — часть кривой ДТА, которая сначала от-
клоняется от базовой линии, а затем возвращается к ней.
Эндотермический пик (endothermic peak), или эндотерма (endo-
therm), — пик, образующийся в том случае, когда температура
образца падает ниже температуры эталона (т. е. АТ — отрицатель-
ная величина).
Экзотермический пик (exothermic peak), или экзотерма (exo-
therm), — пик, образующийся в том случае, когда температура об-
разца становится выше температуры эталона (т. е. АТ — положи-
тельная величина).
Ширина пика (peak width) (B'D') —• временной или темпера-
турный интервал между точками начала отклонения кривой ДТА
от базовой линии и возврата к ней.
Фиг. 13.1. Схематическая кривая ДТА [5].
512
Глава 13
Высота пика (peak height) (CF) — расстояние по перпендикуля-
ру к оси абсцисс между интерполированной базовой линией и вер-
шиной пика (точка С).
Площадь пика (peak area) (BCDB) — площадь, ограниченная
пиком кривой ДТА и интерполированной базовой линией.
Экстраполированная точка начала процесса (extrapolated onset)
(G) — точка пересечения касательной, проведенной в точке наи-
большего наклона восходящей ветви пика (ВС), с экстраполиро-
ванной базовой линией (BG).
н. тг
Все определения относятся к одностадийному процессу, показан-
ному на фиг. 13.2. Многоступенчатый процесс можно рассматри-
вать как последовательность одностадийных процессов.
Плато (plateau) (А В)— часть ТГ-кривой, по существу, с по-
стоянным весом.
Начальная температура (initial temperature) Tt (точка В) —
температура (по Цельсию или Кельвину), при которой интеграль-
ное изменение веса достигает предела чувствительности термовесов.
Конечная температура (final temperature) Tf (точка С) — тем-
пература (по Цельсию или Кельвину), при которой интегральное
изменение веса достигает максимума.
Температурный интервал реакции (reaction interval) — разность
начальной Tt и конечной температур.
Недавно Макади [6] опубликовал рекомендации МКТ А от-
носительно представления экспериментальных данных, касающих-
ся обнаружения и анализа выделенного газа (ОВГ и АВГ). К каж-
дой кривой должна прилагаться следующая информация:
1. Характеристика всех веществ (образец, эталон, раствори-
тель) путем указания их названия, эмпирических формул или экви-
валентных данных по химическому составу.
Терминология в термическом анализе
513
2. Данные о способе получения исследуемых веществ, их преды-
стории, условий предварительной обработки, степени химической
чистоты.
3. Четкое описание температурных условий во время опыта.
4. Средняя скорость изменения температуры по линейному
закону в исследуемом интервале температур. При нелинейном из-
менении температуры должна быть подробно описана программа
изменения температуры.
5. Размеры, форма и материал держателя образца, а если обра-
зец нагружен, то и способ его нагружения.
6. Нанесение по оси абсцисс шкалы температуры или времени
в определенном масштабе с увеличением слева направо.
7. Нанесение по оси ординат любой измеряемой величины. Обыч-
но снизу вверх откладывается увеличение концентрации выделив-
шегося газа. При измерениях плотности выделившегося газа снизу
вверх откладывается увеличение плотности газа. Случаи нанесения
по оси ординат других величин следует оговаривать особо.
8. Указание методов идентификации промежуточных и конеч-
ных продуктов реакций.
9. Точное воспроизведение всех записей самописца.
10. Характеристика атмосферы, окружающей образец (давле-
ние, состав, степень чистоты) с указанием типа атмосферы: само-
генерируемая или динамическая (с потоком газа, пропускаемого
через образец или обтекающего его). Расход газа, общий объем,
конструкция и температура всей системы между образцом и детек-
тором.
11. Характеристика используемых установок (тип и промыш-
ленная марка) с подробным указанием расположения термопар и
взаимного расположения систем нагревания образца и измерения
количества выделенного газа.
12. Если при использовании метода АВГ абсолютные парамет-
ры не измеряются, следует точно знать соотношение между вели-
чиной сигнала с детектора и концентрацией измеряемого компонен-
та. Например, устанавливается зависимость сигнала при иониза-
ции в пламени от числа атомов углерода в молекуле и характера
межатомных связей или от концентрации исследуемого вещества.
ЛИТЕРАТУРА
1. McAdie Н. G„ Anal. Chem., 39, 543 (1967).
2. Mackenzie R. C., Taianta, 16, 1227 (1969).
3. Mackenzie R. C., Keattch C. J., Hodgson A. A., Redfern J. P., Chem. Ind.,
272 (1970).
4. Still J. E„ Cluley H. J., Chem. Ind., 1777 (1969).
5. Mackenzie R. C., Keattch C. J., Dollimore D., Forrester J. A., Hodgson A. A.
Redfern J. P., Thermochim. Acta, 5, 71 (1972).
6. McAdie H. G., Anal. Chem., 44, 640 (1972).
ПРИНЯТЫЕ СОКРАЩЕНИЯ
НАИМЕНОВАНИЙ МЕТОДОВ
АВГ — анализ выделенного газа
ВТСО — высокотемпературная спектроскопия отражения
ГХ — газовая хроматография
ДИП — детектирование по ионизации в пламени
ДСК — дифференциальная сканирующая калориметрия
ДСО — динамическая спектроскопия отражения
ДТА — дифференциальный термический анализ
ДТМА— дифференциальный термомеханический анализ
КККО— анализ по крутильным колебаниям композитного образин
МС — масс-спектрометрия
ОВГ — обнаружение выделенного газа
ПТГТ — производная термогазотитрометрической кривой
ТАЧ — термический анализ по выделенным частицам
ТБ Г — термобарогравиметрия
ТГ — термогравиметрия
ТГП — термогравиметрия по производной
ТГТ — термогазотитрометрия
ТД — термолюминесценция
ТМА — термомеханический анализ
ТМАГ — термомагнитный анализ
ТСХ — тонкослойная хроматография
ФТА — фототермический анализ
ЭЛТА — электротермический анализ
ЭП — электропроводность
ЭТА — эманационный термический анализ
ПРЕДМЕТНЫЙ УКАЗАТЕЛЬ
Автоматический гравиметрический
анализ 110
Адиабатический калориметр 417
Алюминия окись, прокаливание 116
Аминокислоты 302
Аммония нитрат 37
— — ДТА-кривые 289
— — фотометрические кривые 402
— перхлорат, ДТА-кривые 284
— — ОВГ — ДТА-кривые 337
— фосфомолибдат, ТГ-кривые 127
Амперометрический термический ана-
лиз 454
Анализ выделенного газа 509
--------определение 330
Аналитические осадки, сушка ИЗ
Аналоговая вычислительная маши-
на, расчеты 493
Анисальдазин, ДТА-кривые 276
Анкерит 401
Антрацен 140
«Аполлон-15», образцы лунного
грунта 364
Асфальты, исследования методом
ДСК 280
Ацетанилид, дилатометрическая
кривая 448
— ДТА-кривая 279
Базовая линия 511
— — участок до начала перехода
158
— — — после завершения пере-
хода 158
Бактериальные декстраны 303
Бальзамической пихты древесина
303
Бензойная кислота 140, 162, 182
— — ДТА-кривая 268, 272
Беизотрифторид, кривая плавле-
ния 415
Биологические объекты, исследова-
ния методом ДТА 302
Боксит 149
Брушита и монетита смеси ТГ-кри-
вая 120
1
н- Бутан, ДТА-кривая 272
Бэра — Ламберта закон 371
Вант-Гоффа уравнение 164, 417,
434
Весы для взвешивания группы об-
разцов 104
— Кана 76, 101, 486, 488
— кварцевые 101
Взрывчатые вещества 141
----исследования методом ДТА
279
— — органические, исследования
методом ДТА 275
Вибрин 135 (смола), ДТА-кривая 310
ВТСО-кривые 385
Высокотемпературная спектроскопия
отражения 373
— ячейка ДТА фирмы «Дюпон» 249
Высокотемпературное сканирующее
микроотражепие 404
Газовая хроматография 343
Галлоизит 186
Гексоген, ТГ- и ДТА-кривые 142
Гетит 295, 296
Гибсит 295, 296
Гипс, ДТА-кривые 299
Гипс обожженный (штукатурный) 299
Глины, исследование методом ДТА
294
Глиняная посуда, определение вре-
мени изготовления методом
термолюминесценции 472
Горизонтальные плато на ТГ-кри-
вой 21
Дакрон 310
Держатель образца 510
— — блок 172
----для ДТА
— — — микрообразцов 215
— — — текстильных волокон 218
— — запаянный в трубку 218
516
Предметный указатель
Держатель образца изолированный
— — изохорический 214
— — конструкции 46
— — нагреваемый 375
— — постоянная времени 170
— — фирмы «Дюпон» 192
— — чашка 172
— эталона 510
Дериватограф 91, 96
Дериватография и другие методы
447
Детектор ионизации в пламени 355
Дилатометрия 410, 442, 509
— по производной 509
— применение для исследования
полимеров 449
Дильса — Альдера диеновый синтез
278
Динамическая газовая атмосфера 165
— спектроскопия отражения 373
Дифенил 315
Дифференциальная наблюдаемая
температура разложения 130
— сканирующая калориметрия, оп-
ределение 204, 508
— термометрия 145
Дифференциальный термомеханичес-
кий анализ 443
ДСК, определение примесей 434
— теория 207
ДСК-ячейка 255
ДСО — ОВГ, совмещенный метод 377
ДТА, Бойерсма теория 151
— Волда теория 151
— высокие скорости нагревания 296
— Грея теория 155
— держатель образца высокого дав-
ления 235
— — — постоянного давления 235
— использование в промышленнос-
ти 264
— историческая справка 148
— каталоги кривых 265
— Керра и Кульпа теория 150
— кинетика реакций 198
— кривые 263
— — влияние атмосферы печи 163
— — — держателя образца 168
— — — инертного наполнителя 186
— — — массы образца 183
— — — размера и упаковки частиц
образца 183
— — — скорости нагревания 159
— — общий вид 158
— метод Борхардта и Даниельса 201
— — Киссинжера 200
ДТА, метод Маррея и Уайта 199
— — Мейкока 204
— — Пилояна 200
----- Рейха 204
ДТА — ОВГ — МС, совмещенный
метод 362
— определение 145, 508
— основные методические парами!
ры 188
— пики кривых, природа 262
— по производной, определение 508
— приборы 231
— — автоматизация 241
— — вакуумный 243
— — высокого давления 235
— — для изучения кинетики ре.тц
цнй в растворах 245
— — — микроанализа 240
— — — работы при 'высоких тем
пературах 237
— ----------- низких температурах
244
— — калориметрическая ячейка
251
— — с высокочастотным нагревом
252
— — — держателями образцов и
виде герметичных трубок 232
— — — регулируемыми атмосфе-
рой и давлением 247
— — схема 213
— — термисторного типа 247
— применение для изучения ор
ганических соединений 267
— Пэкора теория 154
— расчеты Рида 200
— Спейла теория 150
— стандартные эталонные материи
лы 222, 291
— теория 150
Жидкие кристаллы 273
Известняк, исследование методом
ДТА 393
Изменение веса при постоянном дан
лении 507
Изопериболический калориметр 4'18
Изотермическое определение пзме
нения веса 507
Интегральная наблюдаемая темпер»
тура разложения 130
Интервал реакции температурный
15, 513
Инфракрасная спектроскопия 349
Иодид ртути 375
Предметный указатель
517
Калибровка по температуре 85, 122
Калибровочный коэффициент 190
Калия метилсульфат 340
— фталат кислый, ТГ-кривая 125
— хлорат 281
— этилсульфат 340
Калориметр фирмы «Дельтатерм»
256
Калориметрические методы 410
Кальве калориметр 257
Калькулятор фирмы «Хьюлетт—
Паккард» 491
Кальцит 36, 120, 301
Кальция силиката гидрат 121
Камера сканирования в ПК-области
спектра 400
Каолин 30, 184, 185
Каолинит 173, 301
Каолинит — гекторит, смесь, ТГ-
кривые 120
Катализатора активность 469
Кварц 173
— ДТА-кривые 300
Кинетика реакций, метод Ахара и
др. 61
— — — Бройдо 62
— — — Вахуски и Воборила 60
— — — Горовица и Метцгера 57
— — — Дойля 57
— — — Дэйва и Чопра 60
— — — Ингрэхема и Марье 59
-------Коутса и Редферна 57
— — — Магнусона 61
— — — Ньюкирка 56
— — — Фармера 61
— — — Фримена и Кэррола 56
— — определение механизма по ки-
нетическим данным 66
— — сравнение различных методов
62
Кипения температуры определение
270
Киссинжера уравнение 160
Клапейрона уравнение 163
Кнудсена эффузионный метод 140
— ячейка 360
Количественный дифференциальный
термический анализ 189
Кольцевая термопара 215
Коменовая кислота 269
Комплексное исследование 505
Комплексы переходных металлов,
термохимия 291
Конечная температура 512
Кофлера нагреваемый столик 399
Крахмал, ДТА-кривые 304
Кривая зависимости температуры от
времени 411
Кривые нагревания 149, 508
— охлаждения 149
— с постоянными значениями Л 374
— свечения 467
— скорости нагревания 508
Криоскопические методы 409
Критическая температура, опреде-
ление методом ДТА 322
— — Кюри точка 85
— — определение методом ДТА 324
Кубелка — Мунка теория 371
р-Лактоглобулин 302
Легированный фторид лития 467
Лексан, ТГ-кривая 132
Лигнит, ДТА-кривая 166
Линейный дифференциальный транс-
форматор 444
Лунного грунта образцы, ТГ —
МС-кривые 365
Лунные породы, кривые свечения 409
Лэнгмюра уравнение 138
Магнезит 301
Магнитная восприимчивость 473
Магния и кальция карбонатов смесь,
ТГ-кривая 125
Майлар, ТГ-кривая 132
Максимальный градиент темпера-
туры 30
Марлекс-50 162
«Маскирующее» воздействие 187
Масс-спектрометрия 358
Машинное масло 280
Международная конфедерация по
термическому анализу 13
Меконовая кислота 269
Метод микроскопических измерений
интенсивности деполяризован-
ного света 401
Мнкродетекторная система «Карл»
(модель 1000) 377
Микроскопия расплава 398
Многотарельчатый держатель об-
разца 29
Моносалицилальдоксин цинка 20
Монурон 140
МС — АВГ, совмещенный метод 359
Наблюдаемая температура разло-
жения 130
— — соответствующая минимуму
пика кривой 158
— — — началу отклонения кривой
158
518
Предметный указатель
Наблюдаемая температура соответст-
вующая окончанию отклонения
кривой 158
Нагреваемый столик фирмы «Мет-
тлер» 400
Нагревательные элементы печей 223
Найлон 315
Найлон-6, ДТА-кривая 307
Найлон-66 310 312
— ДТА-кривая 307
— ориентированное волокно, ис-
следование методом ТМА 451
— ТГ-кривая 132
Нафталин — бензойная кислота,
фазовая диаграмма смеси 320
— определение содержания приме-
сей 428
Начальная температура 512
Неопрен 310, 312
Неорганические вещества, иссле-
дование методом ДТА 282
Непрерывного титрования метод 347
Ниагарские известняковые отло-
жения, кривые свечения 469
Никель в катализаторах, определе-
ние содержания 288
Ниобат калия 453
Нитроглицерин, кривые ОВГ —
ДТА 337
Нитрония перхлорат, разложение
368
пара-Нитрофенилгидразин, ДТА-
кривая 278
Нитроцеллюлоза 302
Ныотоиа закон 156
Обнаружение выделенного газа 509
— — — калибровка по темпера-
туре 342
— — — определение 330
Образцы 510
Обратные кривые скорости нагре-
вания 508
ОВГ и хроматография 346
Огнеупорная глина 149
N-окиси Ы-арил-Ы'-тозилоксиди-
имида 277
Окислы германия и алюминия, кри-
вые свечения 469
Оксалаты редких металлов, разло-
жение 282
— смеси кальция, стронция и ба-
рия, ТГ-кривые 123
Оксилюминесценция 480
Оксина фосфомолибдат 128
Органические кислоты, ДТА-кри-
вые 268
— соединения, определение дав-
ления ТГ-методом 138
— — производные, ДТА-кривые
277
— — углерода, определение 357
Орлон 310
Относительная жесткость 478
Отражательная способность образца,
определение методом микроско
пии 403
Ошибки в термогравиметрии, вы-
талкивающая сила воздуха 38
— — — измерение температуры 41
— — — источники 37
— — — конвективные потоки 40
Параметр механического затухания
колебаний 478
Пенополистирол 364
н-Пентан, чистота 432
Пентаэритрит 302
Пентаэритрита тетранитрат 277, 302
Переходы второго рода, обнаружи
ваемые методом ДТА 261
Пик 511
— высота 511
— максимальная температура 261
— минимальная температура 261
— площадь 511
— температуры, измерения 179
— ширина 511
М-пикрнл-Ы-метилнитрамин 277
Пириты, определение 120
Пиролиза кривая 14
Пиросинтез 25?
Плато 512
Подогреваемые кюветы в ИК-спек-
троскопии 397
Полиадипамиды 315
Поливинилбутираль пластифицнро
ванный, ТГ-кривая 134
Поливинилхлорид 318
— ТГ-кривая 133
Полиглюкозаны 305
Полимеры, анализ содержания доба-
вок 133
— исследование методом термогрв-
виметрии 129
Полиметиленсульфид 362
Полиметилметакрилат 362
— МС-анализ 501
Полиоксиметилен, ДТА-кривая 307
Полиолефины 314
Полипиромелл итимид, ТГ- кр и и а я
133
Полипропилен, ДТА-кривая 307
— оксилюминесценция 480
Предметный указатель
519
Полисебацамиды 315
Полистирол, ТГ-кривая 132
— исследование методом ТМА 449
Политетрафторэтилен 133
— ДТА-кривая 307
Полиэтилен 179, 314
— ДТА-кривая 307
— линейный, содержание 310
— ТГ-кривая 133
Почвы, ТГ-кривые 119
Прибор ДТА фирмы «Дельтатерм»,
модель III 378
— с поршневой системой 236
Приборы ДТА фирмы «Линзейс» 215
— ДСК фирмы «Перкин-Элмер» 252
— фирмы «Стоун» 215
Программа DATAR для ЭВМ 497
— IBM Quiktran 497
— PARACT 498
— POI.Y2 484
— VACHVO 490
— VYRVACHVOH 491
Процентное изменение массы 87
Прямая запись изменения массы 187
Радиационная дозиметрия 467
Ракетные топлива 279
Растворимость газов в твердых об-
разцах 37
Реакции с участием органических
соединений, исследование ме-
тодом ДТ А 279
Регистрирующие весы 73
Рекомендации комитета по стандар-
тизации 503
Родохрозит 301
Салициловая кислота 186
---ДТА-кривая 177
Самописцы 231
Сера, ДТА-кривая 288
Серебра иодид 286
— карбоната разложение 397
— нитрат в качестве эталона 194
— — ДТА-кривая 185
— хлорид-бромида твердые растворы
293
Сидерит 301
Система анализа результатов ADS
фирмы «Перкин-Элмер» 497
— сбора данных фирмы «Меттлер»
Скорость нагревания 510
Содержание влаги, определение
Сополимер этилена и винилацетата,
ТГ-кривая 135
Сополимеры, ТГ-кривая 135
Спейла уравнение 161
Спектрорефлектометр фирмы «Бе-
кман», модель ДК-2А 378
Спектроскопия диффузного отра-
жения, определение 371
Стандарты ICTA — NBS 221
Статическая газовая среда 165
Статические и динамические методы
426
Степень кристалличности 185, 305
— — полимера 315
Сульфид цинка, влияние платины
на процесс окисления 32
Сульфиды редкоземельных метал-
лов 137
Тальк 294
ТГ — ДТА — ОВГ — МС-метод 362
ТГ — ОВГ — МС-метод 362
Температура конечная 15
— наблюдаемая разложения 7
— плавления, определение 270
— начальная 15
— плавления, определение 270
— размягчения по Вика 447
— стеклования 261
Теплота испарения, определение ме-
тодом ДСК 321
— перехода 286
— реакции, расчет 196
— — точность и надежность изме-
рений 196
Термисторы 222
Термическая стабильность 15
Термические матричные реакции 392
Термический анализ, определение
9, 145, 410, 504
— — по выделенным частицам 351
— — — крутильным колебаниям
образца 477
— — размеров 443
— — реакции 346
— поляризационный анализ 402
Термическое разложение 505
Термоанализатор фирмы «Меттлер»
215
Термобарогравиметрический ана-
лиз 366
Термовесы автоматические 102
— визуальное наблюдение за об-
разцом 82
— влияние атмосферы печи 22
-----массы образца 34
— — размера частиц образца 35
— — скорости записи 21
— — — нагревания печи 17
— воздействующие факторы 17
520
Предметный указатель
— высокого давления фирмы «Дю-
пон» 96
— Гишара 90
— держатели образцов 77
— оборудование для уплотнения
образца 81
— определение 71
— — температуры 80
— печи 82
— постоянная времени прибора 87
— с газовым хроматографом 344
— системы записи 87
— со спиральной пружиной 93
— термоанализатор фирмы «Мет-
тлер» 97
— условия оптимальной чувстви-
тельности 32
— устройство для обеспечения ре-
гулируемой атмосферы 82
— — — регулирования темпера-
туры по заданной программе 83
— факторы, связанные с образцом
36
— фирмы «Дюпон» 94, 95
— — «Перкин-Элмер» 101
— Хонда 89
— Шевенара 91
Тер мо-Гр ав 93
Термогравиграмма 14
Термогравиметрическая кривая 14
Термогравиметрического анализа
кривая 14
Термогравиметрця 490
— динамическая 14
— и одновременное определение
плотности дыма 395
— историческая справка 15
— квазистатическая 14
— определение 14
— по производной 51, 507
— применение 108
— самогенерируемая атмосфера 45
— система сбора данных 486
— статическая 14
Термограмма 14, 505
Термодилатометрический анализ
442
Термолиза кривая 14
Термолюминесценция 463
— интенсивность 465
— теория 463
— установка для измерений мето-
дом ТЛ 466
Термометрические методы 410
Термомеханический анализ 442
— анализатор 446
Термопара, влияние сечения про-
водов 182
— дисковая 220
— калибровка 182
— несимметричное расположение
176
— потери за счет теплоотвода про-
водами 180, 182
— расположение 174
— тонкопленочная 220
— характеристики 219
Титана карбид 138
— тетрахлорид, определение чис-
тоты 433
Тонкослойная хроматография 353
Тремолит 190
Триамкинолондиацетат, ДТА-кри-
вая 269
1, 3, 5-Тринитрозо-1, 3, 5, 7-тетра-
азоциклооктан 277
Тринитротолуол, ОВГ-кривая 333
— ТСХ 353
Углеводороды, исследование мето-
дом ДТА 280
Уголь, теплотворная способность,
измерение методом ДТА 324
Указатель кривых ДТА SCIFAX
265
Универсальный прибор для исполь
зования при высоких темпера
турах 239
Усилитель слабых напряжений ио
стоянного тока 230
Установка для измерения теплового
расширения 445
— — определения температур за
твердевания 422
Устройство, регулирующее темпе-
ратуру по заданной программе,
229
Фарадеевская установка 475
Фармацевтические препараты, ДТЛ
кривые 269
пара-Фенацетин 140
Фенилдиазоний хлористый 203
Фотометр фирмы «Дюпон» 395
Фототермический анализ 393
Фотохимия 354
оршо-Фталевая кислота 277
Характеристическая цветовая кри-
вая 372
Хинолина фосфомолибдат 127
8-Хинолинол, ДТА-кривая 187
Холестерин 302
Холестерина эфиры 276
Предметный указатель
521
Холестеринпропионат 162
Хризотил 36
Целлюлоза 302
Целлюлозы триацетат, кривые
КККО 479
Циклогексан, кривые ДСК 437
Цифровой датчик температуры с
аналоговым самописцем 495
— преобразователь 498
Цифровые и аналоговые вычисли-
тельные машины 483
Четверные точки, определение ме-
тодом электропроводности 455
Чистые реактивы 138
Чувствительность термовесов 32
Щавелевой кислоты дигидрата ДТА-
кривая 268
Щелочные металлы, стеараты 266
ЭВМ, модель PDP 8/L 500
— фирмы «Бэрроуз» (модель 5500)
495
— ,— «Дженерал электрик» (мо-
дель 600) 487
— — «Ремингтон рэнд юнивак» 483
— — «Ханиуэлл» 488
---GIER 490
— — IBM (модель ИЗО) 495
— — ICT (модель 1909) 495
— — NCR (модель 4130 «Эллиот»)
495
Экзотермический пик 511
Экстраполированная точка начала
процесса 511
Электрическое сопротивление 452
Электропроводности метод 452
ЭЛТА и дилатометрия 458
ЭЛТА — ТГ — ТГП — ДТА-из-
мерения 456
Эмаль обмотки магнита 136
Эмационный термический анализ 458
--------- установки 459, 460
Эндотермический пик 511
ЭП—ДСК совмещенный метод 458
Эпоксидные смолы 308
Эталон 510
Эталоны для калибровки 194
Яичный альбумин 302
Ячейка ДСК фирмы «Дюпон» 254
Ag2[HgI4], термохромизм 390
ВаВг2-2Н2О 25
ВаС12-2Н2О 25
— дилатометрическая кривая 448
СаСОз, ТГ-кривые 24, 34, 36, 190
СаС2О4-Н2О 346
— дегидратация 31
— ТГ-кривая 18, 24, 26, 27, 35, 41,
114
СаНРО4-2Н2О 284
a-CaSO4-0,5 Н2О 30
CdO 185
СоС12-6Н2О, исследование методом
ДСО 387
[Co(N Нз)4(Н2О) (CN)Co(N Hs)4CN J
(С1О4)4 18
[Co(NH3)8H2O]X3 288
[Co(NH3)elCl3, ТГ —ТМАГ-кривая 475
Со(Пиридин)2С12, исследование ме-
тодом ВТСО 379
—------дилатометрии 447
[Со(Этилендиамин)2(Н2О)](ПОз)2, ре-
акции 393
[Cr(NH3)5-Н2О]Хз 288
CrVO4 136
1Сг(Этилендиамин)зС13, метод ДСО
392
Cu(NH3)4SO4-Н2О, дериватографи-
ческие и термо-газотитримет-
рические кривые 349
— ОВГ и МС-кривые 361
CuS — Cu2S, кривые пиросинтеза 298
СиЦЭтилендиамин) (H2O)2]SO4 382
CuSO4-5H2O, ДТА-кривые 288
— ДМО-кривые 406
— исследование методом ИК-спек-
троскопии 350
— ОВГ — ДТА-кривые 339
— СМО-кривые 405
— ТГ-кривые 19, 126, 29
;— электропроводности кривая 456
Fe3C, реакция с натрием 476
FeC2O4-2H2O 25
FeaOg, эманационная кривая 461
KASFe, дилатометрическая кривая
448
КВг, диски 397
KHSO4 19
KNO2 293
KNO3 285
KSCN 293
M2[HgI4], комплексы 375
MgCO3 30
— ДТА-кривая 295
MgSO4-7H2O, ДТА-кривая 155
Мп (СНзСО2)2-4Н2О, ТГ-кривая 49
Na2CO3, сушка 115
NaHCOs, 161, 293
Na4P2O7-ЮН2О, ТГ-кривая 118
Ni(NHs)eCl2, ТГ-кривая 50
NiS2 136
ОГЛАВЛЕНИЕ
Предисловие редакторов русского перевода......................... 5
Предисловие автора ко второму изданию............................ 6
Предисловие автора к первому изданию............................. 7
Глава 1. Общее введение.......................................... 9
Глава 2. Термогравиметрия....................................... 14
А. Введение................................................ 14
Б. Некоторые факторы, влияющие на характер термогравиметри-
ческих кривых............................................ 16
1. Факторы, связанные с измерительным прибором (термовеса-
ми) .................................................... 17
а. Скорость нагревания печи . .................... 17
б. Скорость записи................................... 21
в. Атмосфера печи...................................... 22
г. Форма держателя образца...................... .... 27
д. Условия оптимальной чувствительности................ 32
2. Характеристики образца................................ 34
а. Масса образца....................................... 34
б. Размер частиц образца............................. 35
в. Другие факторы, связанные с образцом................ 36
В. Источники ошибок в термогравиметрии...................... 37
1. Выталкивающая сила воздуха............................. 38
2. Конвективные потоки и турбулентность в печи............ 40
3. Измерение температуры.................................. 41
4. Другие ошибки.......................................... 44
Г. Термогравиметрия в собственной атмосфере.................. 45
Д. Термогравиметрия по производной (ТГП) . 51
Е. Кинетика реакций....................................... 54
1. Неизотермические методы .............................. 54
а. Метод Ньюкирка...................................... 56
б. Метод Фримена и Кэррола............................. 56
в. Метод Горовица и Метцгера........................... 57
г. Метод Коутса н Редферна............................ 57
д. Метод Дойля........................................ 57
е. Метод Ингрэхема и Марье........................... 59
ж. Метод Вахуски и Воборила . ......................... 60
Л з. Другие методы....................................... 60
2. Сравнение различных методов........................... 62
3. Определение механизма реакции по кинетическим данным,
полученным в неизотермических условиях.................... 66
Ли тература . . . ................... 68
Глава 3. Термовесы и вспомогательное оборудование к ним .... 71
А. Введение ................................................. 71
1. Регистрирующие весы................................... 73
а. Весы Кана........................................... 76
2. Держатели образцов.................................... 77
3. Печи и устройства для регулирования их температуры по за-
данной программе........................................ 82
Оглавление
Б23
4. Определение температуры и системы регистрации данных 83
Б. Типы термовесов............................................. 89
1. Введение................................................. 89
2. Термовесы Шевенара .................................... 91
3. Термовесы со спиральной пружиной....................... 93
4. Термовесы фирмы «Дюпон»................................ 94
5. Дериватограф...................................... ... 96
6. Термоанализатор фирмы «Меттлер»........................ 97
7. Другие типы термовесов *...............................101
8. Кварцевые весы........................................ 101
9. Автоматические термовесы...............................102
Литература ............................................. 105
Глава IV. Применение термо гравиметрии . . .....................108
А. Введение...................................................108
Б. Применение в аналитической химии..........................110
1. Автоматический гравиметрический анализ...................НО
В. Аналитические определения с помощью метода термограви-
метрии .......................................................113
1. Сушка осадков в весовом анализе................ ...... ИЗ
2. Сушка и разложение карбоната натрия.....................115
3. Температура прокаливания окиси алюминия...............116
4. Сложные смеси гидратов..................................117
5. Анализ глин и почв......................................119
6. Определение содержания свободной извести и карбоната
кальция в гидратах силиката кальция .................... 121
7. Определение содержания магния, кальция, стронция и бария 122
8. Кислый фталат калия................................. 125
9. Фосфомолибдаты некоторых аминов.......................127
Г. Полимеры ............................................... 129
1. Введение..............................................129
2. Относительная термическая стабильность................132
3. Содержание добавок....................................133
4. Состав смесей полимеров и сополимеров.................134
5. Другие применения.....................................135
Д. Применение в неорганической химии....................... 136
Е. Определение давления пара и исследование возгонки .... 138
Ж» Другие применения . 140
Литература .............................................. 142
Глава 5. Дифференциальный термический анализ и дифференциальная
сканирующая калориметрия > . ...................145
А. Дифференциальный термический анализ.....................145
1. Введение..............................................145
2. Историческая справка..................................148
3. Теория................................................150
4. Факторы, влияющие на кривые ДТА.......................157
а. Скорость нагревания.................................159
б. Атмосфера печи .....................................163
в. Держатели образцов..................................168
г. Расположение термопары .............................174
д. Термопары ..........................................180
5. Характеристики образца .... 183
а. Масса образца.......................................183
б. Размер и упаковка частиц образца.................. 183
в. Влияние инертного наполнителя.......................186
524
Оглавление
6. Основные методические указания........................188
Б. Количественный дифференциальный термический анализ . . 189
1. Введение..............................................189
2. Калибровка............................................190
3. Эталоны для калибровки................................194
4. Расчет Д/7............................................196
5. Точность и надежность измерений АН.............196
В. Кинетика реакций ........................................198
Г. Дифференциальная сканирующая калориметрия................204
1. Введение..............................................204
2. Теория................................................207
Литература.............................................. 209
Глава 6. Приборы дифференциального термического анализа и диффе-
ренциальной сканирующей калориметрии.......................213
А. Дифференциальный термический анализ......................213
1. Введение..............................................213
2. Держатели образцов....................................213
3. Системы определения Д7’ и 7’.............214
4. Печи и устройства для регулирования их температуры по за-
данной программе........................................ 223
5. Усилитель слабых напряжений постоянного тока и регистри-
рующие системы............................................230
6. Приборы ДТА...........................................231
а. Введение............................................231
б. Держатели образцов в виде герметичных трубок . . . 232
в. Приборы ДТА высокого давления.......................235
г. Приборы ДТА для работы при высоких температурах . . 237
д. Приборы ДТА для микроанализа........................240
е. Автоматизация приборов ДТА..........................241
ж. Другие типы приборов ДТА............................243
Б. Дифференциальная сканирующая калориметрия................252
Литература ............................................. 257
Глава 7. Применение дифференциального термического анализа и диф-
ференциальной сканирующей калориметрии................261
А. Введение .-..............................................261
Б. Каталоги кривых ДТА.......................................265
В. Органические соединения..................................266
Г. Неорганические вещества...................................280
Д. Глины и минералы..........................................294
Е. Биологические объекты и природные органические материалы 302
Ж. Полимеры .................................................305
3. Другие применения . ......................................313
Литература .............................................. 325
Глава 8. Обнаружение и анализ выделенного газа................. 330
А. Введение.................................................330
Б. Обнаружение выделенного газа..............................330
В. Анализ выделенного газа (АВГ)............................342
1. Газовая хроматография..................................343
2. Титриметрические методы.............................. 347
3. Инфракрасная спектроскопия.............................349
4. Термический анализ по выделенным частицам (ТАЧ) . . 351
5. Тонкослойная хроматография (ТСХ)......................353
Оглавление 525
6. Метод ионизации в пламени.............................355
7. Масс-спектрометрия (МС) ..............................358
а. Совмещенный метод МС—АВГ.............................359
б. Совмещенный метод ОВГ—МС.............................361
в. Совмещенный метод ДТА—ОВГ—МС.........................362
г. Совмещенные методы ТГ—ОВГ—МС и ТГ—ДТА—ОВГ—
МС......................................................362
8. Термобарогравиметрия (ТБГ)............................366
Литература .............................................. 368
Глава 9. Спектроскопические, фотометрические и оптические методы
термического анализа............................................371
А. Высокотемпературная и динамическая спектроскопия отраже-
ния .........................................................371
1. Введение..............................................371
2. Высокотемпературная спектроскопия отражения .... 373
3. Аппаратура............................................375
4. Применение методов ВТСО и ДСО для исследования неорга-
нических соединений.......................................379
а. Переход октаэдрической структуры соединения Со(Пири-
дин)2С12 в тетраэдрическую..............................379
б. Соединение (Си(Этилендиамин) (H2O)2]SO4 382
в. Соединение CuSO4-5H2O................................384
г. Соединение СоС12-6Н2О................................387
д. Соединение №(Пиридин)4С1г...........................389
е. Термохромизм Ag2[HgIJ ...............................390
ж. Термические матричные реакции.......................392
Б. Фотометрические методы....................................393
В. Высокотемпературная инфракрасная спектроскопия .... 397
Г. Методы термической оптической микроскопии.................398
1. Микроскопия расплава..................................398
2. Интенсивность деполяризованного света и фотометрическая
термическая микроскопия...................................400
Литература .............................................. 406
Глава 10. Криоскопические методы определения чистоты веществ 409
А. Введение.................................................409
Б. Теория....................................................411
В. Экспериментальное оборудование...........................417
Г. Погрешности, недостатки и другие факторы, влияющие на ре-
зультаты измерений .......................................422
1. Недостатки динамического метода.......................423
а. Переход Твердая фаз а -> Жидкость....................423
б. Переходы в твердой фазе.............................424
в. Скорости диффузии...................................424
г. Влияние перемешивания................................424
д. Тепловые потоки.....................................424
е. Перепады температур при плавлении...................425
ж. Влияние контакта между поверхностями................425
• 2. Недостатки статического метода.........................425
а. Неопределенность в оценке концентрации примесей . . 425
б. Образование твердых растворов........................425
в. Применение теории твердых растворов..................426
3. Сравнение результатов, полученных статическим и динами-
ческим методами.........................................426
4. Рекомендации............................................427
526
Оглавление
Д. Практические примеры определения содержания примесей и 428
другие проблемы...........................................
Е. Определение примесей методом дифференциальной сканирую-
щей калориметрии............................................434
1. Введение..............................................434
2. Теория................................................434
3. Некоторые особенности использования дифференциального
сканирующего калориметра для определения чистоты ве-
щества ...................................... . ... . 435
4. Применение метода ДСК Для определения чистоты веществ 439
5. Оценка метода........................................ 439
Литература ...................... 440
Глава 11* Другие методы термического анализа »*. 442
А. Введение............ . .................................442
Б. Дилатометрия (термомеханический анализ)..................442
1. Введение..............................................442
2. Измерительная аппаратура..............................444
3. Применение............................................447
В. Электропроводность.......................................452
Г. Эманационный термический анализ..........................458
Д. Термолюминесценция . ..................................463
Е. Термомагнитный анализ....................................473
Ж. Термический анализ по крутильным колебаниям композитного
образца ..................................................477
3. Оксилюминесценция .......................................480
Литература . •• . ......................... 481
Глава 12. Применение цифровых и аналоговых вычислительных машин
в термическом анализе.....................................* . 483
А. Введение ...............................................483
Б. Термогравиметрия.........................................483
В. Дифференциальный термический анализ (ДТА) и дифференци-
альная сканирующая калориметрия (ДСК) -.....................494
Г. Другие методы термического анализа.......................498
Литература................................... ... 501
Глава 13. Терминология в термическом анализе...................503
А. Введение................................................503
Б. Общие рекомендации ......................................504
В. Терминология.............................................505
Г. Определение и условные обозначения ......................507
1. Общее понятие.........................................507
2. Методы, связанные с изменением веса...................507
а. Статические методы . ,.............................507
б. Динамические методы ...............................507
Д. Методы, связанные с изменением энергии...................508
Е. Методы, связанные с выделением летучих веществ , . . . 509
Ж- Методы, связанные с изменением размеров..................509
3. Комплексные методы.......................................509
И. ДТА.................................................... 509
К- ТГ.......................................................510
Л. ДТА и ТГ ................................................510
М. ДТА .....................................................511
Н. ТГ.......................................................512
Литература .... 513
Принятые сокращения наименований методов.......................514
Предметный указатель ....... 515
УВАЖАЕМЫЙ ЧИТАТЕЛЬ!
Ваши замечания о содержании книги, ее оформ-
лении, качестве перевода и другие просим присы-
лать по адресу: 129820, Москва, И-110, ГСП,
1-й Рижский пер., д. 2, издательство «Мир».