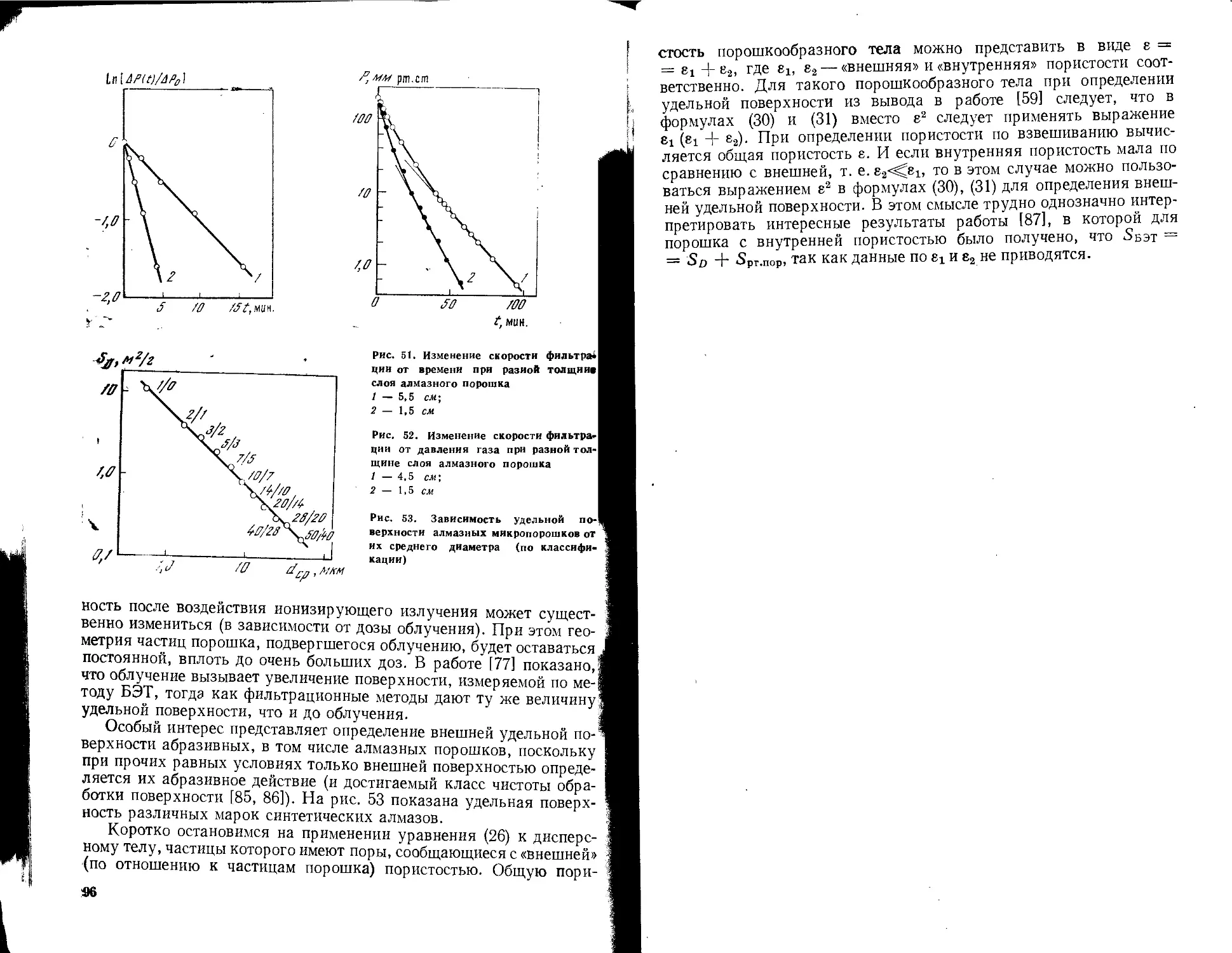
/
























































Автор: Дерягин Б.В. Федосеев Д.В.
Теги: неорганическая химия химия химическая кинетика физика твердого тела издательство наука
Год: 1977




Текст
АКАДЕМИЯ НАУК СССР
Ордена Трудового Красного Знамени
Институт физической химии
Б. В. ДЕРЯГИН, Д. В. ФЕДОСЕЕВ
РОСТ АЛМАЗА
И ГРАФИТА
ИЗ ГАЗОВОЙ ФАЗЫ
Издательство «Наука»
Москва 1977
ПРЕДИСЛОВИЕ
Ф "до™ e’eV Л Тм изигаз0В0Й *аз«- Дерягин Б. В.,
ед о се ев Д. В. М., «Наука», 1977, стр. 116.
Монография посвящена исследованию влияния поверхностных
сил на образование и рост новой фазы - алмаза и графита -
из метана при пониженном давлении. Предложена новая теория
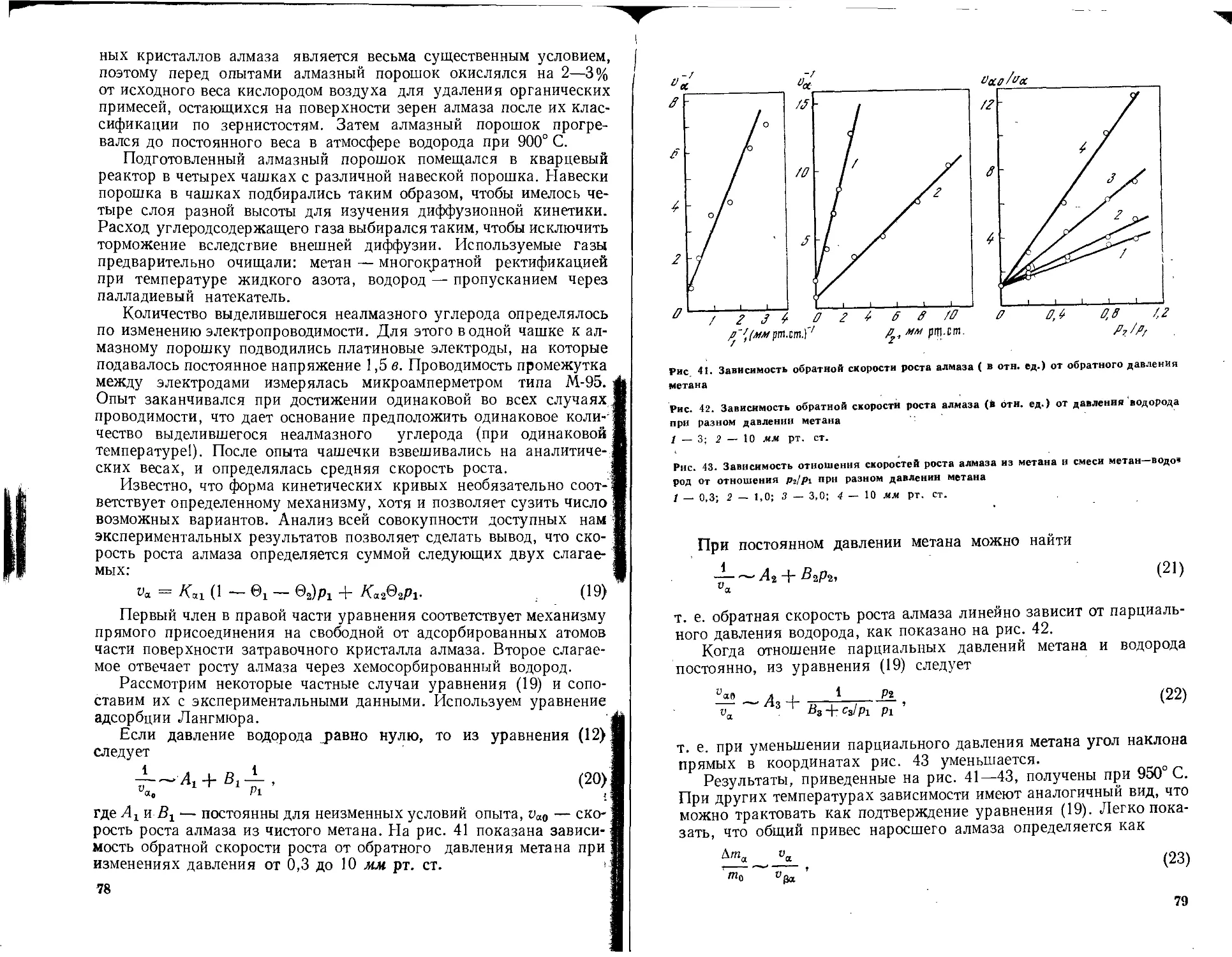
роста алмаза н графита, исследована кинетика роста, описаны
нитевые и изометрические кристаллы алмаза и графита. Пред-
ложена оригинальная методика определения удельной поверх-
ности порошков. р
Книга предназначена для специалистов в области физической
химии, физики твердого тела и химической кинетики.
Таблиц 5. Иллюстраций 61. Библ. 103 назв.
, 20503-556
055(02j-77“ 99-76
основу настоящей монографии легли исследования по синтезу
раза в области его термодинамической метастабильности, выпол-
щые в Отделе поверхностных явлений Института физической
чии АН СССР. Сама возможность роста алмаза при атмосферном
1еньшем давлении не только не была очевидной, но казалась мало-
юятной. Хотя еще в 1911 г. и были описаны первые эксперименты
наращиванию алмаза из углеводородов, практическая реализа-
я этой идеи вызывала большие сомнения в связи с тем, что рост
1фита, как термодинамически устойчивой фазы, затруднял до-
1ТОЧНО четкую идентификацию получаемых результатов. Поэто-
исследования по росту алмаза из газовой фазы, начатые в ИФХ
1 СССР в 1956 г., сначала преследовали цель надежного доказа-
тьства роста алмаза. Последующие работы были направлены на
учение кинетических характеристик процесса синтеза, морфоло-
и и свойств вновь синтезированной фазы.
В книге использованы уже опубликованные работы авторов
нографии, а также различные публикации (журнальные статьи
патенты) как отечественных, так и зарубежных ученых. Следует
и этом отметить, что число патентов значительно превосходит ко-
чество других публикаций. Это обстоятельство несомненно
|язано с практическим значением исследований в данной об-
1СТИ.
Теоретическое значение физико-химического синтеза алмаза из га-
вой фазы обусловлено не столько самими результатами, сколько
м, что в этих исследованиях наиболее тесно связаны теория поверх-
•стных явлений, термодинамика и физико-химическая кинетика,
оэтому мы вправе говорить о физико-химическом синтезе алмаза,
менно ориентирующее влияние поверхностных сил на образование и
)ст ровой фазь1~спосо'бствует'~!р6сту алмаза на поверхности алмаз-
ях затравочных кристаллов. Термодинамика дает указание на
пгнципиальнукГ'возможность этого процесса, хотя оговаривает
о значительно меньшую вероятность, нежели образование ста-
(льной фазы углерода — графита. Кинетика открывает путиповы-
ения вероятности..роста _алмаза из газовой фазы и у пр авл ения
1. Именно кинетические соображения указывают на возможность
Кта алмаза, да неалмазных“~~поверхндстях~ чтои_было доказано
Хпериментально— Исследования процесса зародышеобразования
роста новой фазы в сочетании с поверхностными явлениями по-
© Издательство «Наука», 1978 I)
3
зволили свести основные закономерности роста алмаза и гра
к обычной химической кинетике гетерогенных реакций.
Книга содержит четыре главы. В главе I излагаются осно
закономерности теории нуклеации в гомогенных и гетероге;
системах. Методической основой этой главы является исполь
ние большого ансамбля Гиббса для процесса зародышеобра;
ния. В этой же главе рассматривается влияние поверхностных
на образование и свойства тонких слоев. Глава II посвящена
зико-химической теории роста графита и ее экспериментал
проверке. Значение этой главы состоит в том, ---------
ОСНОВНЫЕ ЗАКОНОМЕРНОСТИ
НУКЛЕАЦИИ
..................... ptl Jt/inputicl
стабильной фазы. Глава III посвящена, в основном, кинетикеL его
та алмаза на высокодисперсных алмазных подложках. В глав!
рассматривается рост алмазных пленок, нитевидных и изоме’
ных кристаллов.
Авторы надеются, что изложенные
„__я.
кинетикой, изучением поверхностных явлений,
и физикой твердого тела.
3|ии 1лавы состоит в том, что изучение м
необ?п?имппграфита В условиях’ близких к условиям роста ал1НТИпуЮщее влияние поверхностных сил на образование и рост
стабипЗ^^яизв1сканитя1твозможноста Регулирования роста < ф£ы наиболее ярко проявляется при росте алмаза в усло-
стабильнои фазы. Глава Ш rwna„.P„, п „„„„„------------Iй Ф метастабильности [1, 2]. Исследования в области поверх-
в»ных сил [3] позволили заложить теоретическую^основу реали-
:i Щ самой возможности роста алмаза на алмазной подложке из
д *п, - вой фазы Эта возможность осуществляется благодаря изб
В П кТ выРажают глубокую благодарность С. П. Внук лЬНОму воздействию кристаллической решетки алмаза на ее
ской‘ Н РЛ п™ ' В' Лаврентьеву, Б. В. Спицыну, И. Г. Bapi адйвание, а не только на образование критического зародыша
’ /Л?0ЛЯНСК0И’ К- с- Успенской,, А. Е. Городецком ;ИЛьной фазы — графита — и его дальнейший рост. Более то
дру трудникам участникам большой и трудоемкой раб< воздействие алмазной решетки может вызывать образование и
лован^ХпТ?™’ ЧТ° изложенные в книге Результаты ис<г других модификаций углерода: карбинов, кубического графи-
кинетикпйУДы1 ° езны Ученым> занимающимся физико-химичес [ других политипов углерода. При этом надо учитывать также и
кинет™ —-........... _ . кристаллохш е^^ские факторы, воздействующие на систему. Скорее всего,
нно благодаря воздействию кинетических факторов возможен
г алмаза на неалмазных подложках.
Несмотря на то, что основным методом роста алмаза из газовой
я является наращивание из индивидуальных углеводородов,
эзя отрицать влияние промежуточных радикалов, образующихся
гстеме Химическая кристаллизация уже предполагает наличие
авновесности (хотя, возможно, и стациоиаРност^„^““;
1Я неравновесность особенно проявляется в методе химических
нспортных реакций [4].
В данной книге не рассматриваются общие вопросы кристал-
ации, которым посвящен ряд содержательных работ 16 У],
злагаются результаты исследований по общей теории нуклеа-
' :: гомогенной и гетерогенной. При этом используется новый под-
, предложенный одним из авторов [10, 11] и связанный с приме-
ием большого ансамбля Гиббса. Большое внимание уделено так-
ориен!ирующему влиянию поверхностных сил на образование
>ост новой фазы.
Теория гомогенной нуклеации
(иная с М. Фольмера и других ученых — Р. Беккера и И. Де-
та, Я. Б. Зельдовича, Я-И. Френкеля, Р. А. Каишева—до
тоящего времени теория нуклеации во всех работах строит-
на основе расчета цепочки событий, ведущих к образованию
ойчивого зародыша новой фазы, исходя из состояния метаста-
5
бильной фазы и проходя через все стадии докритических заро-
дышей, включая самые ранние. Авторы настоящей работы находят
эти представления принципиально неудовлетворительными, считая
невозможным в ряде случаев рассмотрение процесса зародыше-
образования по этому прямому пути. Действительно, начальная
стадия формирования ядра конденсации включает образование
димеров, тримеров и n-меров. Строгий расчет этой стадии требует
такой информации о свойствах n-меров, которой мы не распола-
гаем и вряд ли скоро сможем располагать, особенно для много-
атомных молекул.
Даже если бы эта информация имелась в достаточном объеме,
ее использование для точного расчета начальной стадии образова-
ния критических зародышей натолкнулось бы на такие расчетные
трудности, что вряд ли можно было бы надеяться получить анали-
тические формулы общего значения. Именно эти причины в зна-
чительной степени породили такое положение, когда в литературе
приводятся для вероятности образования устойчивых капель при
конденсации паров формулы, в которых встречаются десятки раз-
личных выражений для предэкспоненциального множителя. Не-
которые из них различаются на 15 десятичных порядков.
Еще хуже обстоит дело с расчетом образования зародышей при
кавитации и гомогенном вскипании жидкостей. По существу в этом
случае начальная стадия должна быть охарактеризована как со-
вершенно предположительная. Ниоткуда не следует, что полости
в самом начале сферичны, как это обычно принимается. Более
правдоподобно, что они трещинообразны. Когда берут за основу мо-
дель сферической полости, сталкиваются с дополнительной прин-
ципиальной (хотя обычно и не оговариваемой) трудностью, от-
сутствующей в теории конденсации пара. В последней начальная
стадия есть образование реальных димеров из вполне реальных и
определенных микрообъектов — молекул. В теории кавитации и
вскипании источником пузырьков служат межмолекулярные про-
межутки, флуктуативно вырастающие в зародышевые сферы. Та-
ким образом, исходный для формирования последних «материал»
имеет совершенно неопределенную форму и чисто условное число
и размер. Поэтому, в частности, недостающий линейный размер в
формуле для вероятности кавитации Зельдович [12] предлагает
приравнять либо размеру молекул, либо размеру критического
пузырька. Вследствие этого следует считать выведенную формулу
полуэмпирической, содержащей по сути в качестве неопределенного
множителя функцию безразмерного отношения двух линейных раз-
меров.
Существует, однако, возможность построения теории нуклеации
по менее прямому пути без рассмотрения микроскопической ста-
дии этого процесса с помощью общего статистического подхода.
Ограничимся изложением расчетов по этому методу для процес-
са гомогенной конденсации пара. Сконструируем большой ансамбль
Гиббса, в котором системой (подсистемой) с переменным числом
6
молекул служит объем V, выделенный внутри объема умеренно пере-
сыщенного пара W с жесткими адиабатическими стенками. Примем,
что
if < V < W, (1)
где vc — объем критической зародышевой капли. Выделение объе-
ма V произведем с помощью несмачиваемой пористой перегородки *,
не пропускающей околокритические и закритические зародыши.
Объем IF — V играет роль термостата и резервуара молекул,
задающего температуру и химический потенциал до тех пор, пока
в результате нуклеации в нем не образуется капля, которая сни-
мает имеющееся пересыщение, после чего наступает состояние,
близкое к двухфазному равновесию капля —пар. Поэтому будем
рассматривать только те состояния объема IF — V, которые близ-
ки к состоянию метастабильного равновесия пересыщенного пара,
в которых отсутствуют околокритические и закритические заро-
дышевые капли. При умеренном пересыщении такие состояния
объема IF — V могут быть весьма длительными, и практически
они не отличимы от состояний истинного термодинамического рав-
новесия.
В этом случае вероятность различных состояний резервуара
IF — F, принадлежащих к метастабильной «однофазной» области,
удовлетворяет классическому уравнению
ш~Сехр (Е', X')] ,
где 3 — энтропия резервуара, рассматриваемая как функция энер-
гии Е' и числа частиц N1, а С — множитель, постоянный для дан-
ного резервуара, независимо от того, в каком состоянии находится
объем V. Одновременно вероятность w пропорциональна вероятно-
сти тех состояний объема F, характеризуемых числом молекул N
и энергией n-квантового состояния Е, которые сосуществуют с дан-
ным состоянием резервуара. Поэтому можно в дальнейшем вероят-
ность w считать пропорциональной вероятности различных состоя-
ний объема F. Как будет показано, коэффициент пропорциональ-
ности С в дальнейших расчетах вероятности нуклеации сокращается
и необходимость заботиться о нормировке отпадает.
Объем V может находиться в одном из следующих классов
(подансамблей) состояний.
1. Заключенный в объеме пар находится в состоянии, близ-
ком к метастабильному равновесию, и зародыши околокритических
и закритических размеров отсутствуют.
2. В объеме V имеется околокритический зародыш (число моле-
кул 2V2). Состояние объема, помимо общего числа молекул N,
Ее лиофобность предполагается столь большой, а поверхность — столь малой,
что конденсацией на ней можно пренебречь.
7
более детально может характеризоваться числом молекул пара
Ni и числом молекул в капле Х2.
3. В объеме V образуется закритический зародыш, что с вероят-
ностью, близкой к достоверности, приведет к заполнению всего
объема V жидкостью. Не будучи в состоянии просочиться через по-
ры перегородки, она образует в них выпуклые мениски, которые на-
ходятся в равновесии с пересыщенным паром термостата.
В таком состоянии система объема W может пребывать относи-
тельно длительное время, колеблясь вокруг положения двухфазного
равновесия, прежде чем она вернется в результате «большой» и
потому крайне «редкой» флуктуации в один из классов состояний —
1 или 2. Класс состояний 3 в дальнейшем нас интересовать не бу-
дет, так как мы должны определить только вероятность перехода
из класса 1 через класс 2 в класс 3, но не «устойчивость» последнего.
Однако существование класса 3 необходимо для того, чтобы скон-
струированный ансамбль был статистически равновесным.
Итак, ограничимся рассмотрением только состояний объема V
классов 1 и 2. Если отношение V/vc не слишком велико, то наиболее
часто будут реализоваться состояния класса 1, отвечающие мета-
стабильному равновесию и, следовательно, относительному мини-
муму свободной энергии. По сравнению с ними состояния класса 2
как близкие к максимуму свободной энергии будут реализоваться
сравнительно редко, с относительно малой вероятностью. Эта ве-
роятность тем меньше, чем меньше объем V и чем больше критический
зародыш и, следовательно, работа его образования. Вероятность
одновременного присутствия нескольких околокритических заро-
дышей можно при этом считать пренебрежимо малой.
Необходимо, однако, внести ясность в определение нижней и
верхней границ размера околокритических зародышей, характери-
зуемых числом молекул N2. Для дальнейших расчетов методом
перевала необходимо, чтобы существовал интервал размеров заро-
дыша, характеризуемых числами N2 и Х2, настолько широкий,
что в силу соблюдения условий
(3)
кинетика нуклеации будет контролироваться стадией прохожде-
ния зародышем этого интервала. С другой стороны, этот интервал
должен быть достаточно узок, чтобы исключить возможность со-
существования в объеме V двух или более зародышей с размерами,
лежащими внутри этого интервала. Оба требования можно сов-
местно удовлетворить, если N достаточно велико. Соответствен-
ные значения и А'', будем считать нижней и верхней границами
размеров околокритических зародышей.
В силу соблюдения второго условия функция ф (Х2) распреде-
ления состояний сконструированного нами ансамбля Гиббса по
размерам околокритического зародыша в объеме V имеет одно-
значный смысл во всем интервале N™ — Nl2 и пропорциональна
8
распределению околокритических зародышей по размерам Nt.
Функция ф(Х2) ПРИ Х2 = Х2 имеет глубокий минимум и при боль-
ших Х2 возрастает, стремясь к очень высокому максимуму, отве-
чающему двухфазному равновесию капля—пар в объеме W — V.
Рассмотрим теперь стационарное распределение вероятности
различных состояний объема V, когда статистически равновесное
распределение ансамбля нарушается устранением состояний клас-
са 3 по мере их возникновений и заменой состояниями класса 1.
Вероятность нуклеации v найдем из формулы
v = Q/gi, (4)
где Q (при выбранном нами статистическом подходе) — поток со-
стояний объема V. Для стационарного состояния Q постоянно для
всех состояний ансамбля, распределение которых не зависит от вре-
мени. В области состояния класса 2 этот поток совпадает с потоком
зародышей в пространстве их размеров Х2. Величина g± в форму-
ле (4) — это суммарная вероятность состояний ансамбля, относя-
щихся к классу 1, т. е. относящихся к метастабильному состоянию
исходного пересыщенного пара в отсутствие околокритических И
закритических зародышей.
При этом, конечно, в пересыщенном паре могут присутствовать
микрозародыши типа димер—тример (при Х2 < X™), но их при-
сутствие автоматически учитывается в дальнейших расчетах glt
поскольку в конечные формулы входят термодинамические пара-
метры пересыщенного пара — его химический потенциал и производ-
ные последнего, зависящие от наличия флуктуационных микроза-
родышей, образующихся тем более интенсивно, чем больше пере-
сыщение пара.
Таким образом, и поток (вероятности) состояний и вероятность
состояния ансамбля класса 1 вблизи метастабильного равновесия
нами вычисляются на основе одного и того же большого ансамбля
Гиббса. При этом устраняется необходимость рассмотрения и
расчета начальной, микроскопической стадии кинетики образова-
ния критического зародыша, что является основной трудностью
теории нуклеации, особенно в конденсированных средах (кавита-
ция, кипение).
Ограничиваясь случаями умеренных пересыщений и, следова-
тельно, высокого потенциального барьера, можно рассчитать Q
методом перевала, когда поток состояний совпадает с потоком заро-
дышей. С этой целью используем, следуя Я. Б. Зельдовичу [12],
уравнение Крамерса для случая стационарного процесса нуклеа-
ции:
= (5)
ах2 [ * ам2 \ <р / J v
где п(Х2) — распределение, отвечающее стационарному процессу,
D — обобщенный коэффициент диффузии состояний объема V
в прбстранстве чисел молекул Х2. Интегрирование следует вести
9
при граничных условиях, аналогичных общепринятым в теории
нуклеации *:
п/ф = 1 при Х2 = NT, (б)
и/ф = 0 при Х2 = N2. (6')
Интегрируя уравнение (5), получим
___ 1 »____ Г dN2
~ V ’ ~i D<f (Л'г)
Полные энергия и число частиц теплового резервуара не зави-
сят от энергии и числа частиц объема V. В результате вероятность
различных квантовых состояний объема равна
= Cexp|-^-S [Е'— ДЕ. Х'ДХ]| , (8)
где Е' и N2 —средние значения энергии и числа частиц резервуара
в состоянии метастабильного равновесия с объемом V, содержащим
критический зародыш.
Для состояний объема V класса 2
ДХ - (Х2 - NT) + (Х2 - NT) AXj + ДХ2, (9)
ДЕ = ЕП^2-Ё^, (10)
где Nx и N2 — числа молекул в объеме V, входящих в состав пара
и, соответственно, капли; EnNi^ — энергия «-квантового состоя-
ния при соответствующих числах молекул, индекс «О» указывает
на принадлежность к состоянию лабильного равновесия с резер-
вуаром, черта над Ед означает усреднение по всем квантовым состоя-
ниям (при Xi = Хо> N2 = N). Разлагая S в уравнении (8) в ряд
по степеням ДЕ и NN, получим
2-Т f 1- О / С \ Г/\1 - Г Р ДО l^"nVl,V2 1
wnN,Nt = Сехр х S (£ , X exp ДХ---------------------------------g-------).
(И)
Заменяя &T на Fc 4- TSCO и суммируя по и, получим
> , WnVt^A еХР{---------в-------/ ’
п 4
Ас = С ехр 15 (£', X') + So (Ёс0, N{, X')]J .
(12)
(12')
* Условие (6) есть условие «сшивания» области состояний класса 1, мало воз.
мущенных процессом нуклеации, и области состояний класса 2, контроли.
рующей кинетику процесса. Условие (6) отражает условие изъятия из объе.
ма 1У капель, достигших размеров v^>vc-
16
Для FN1N1 положим
FniN2 = FNi + Fn2 + 6 In (1 /Ni), (13)
где F.v, — свободная энергия пара; FN2 — свободная энергия кап-
ли в координатах ее центра тяжести; третий член в правой части
уравнения (13) учитывает, по аналогии с теорией растворов, под-
вижность капли в объеме V (энтропию смешения).
Следует заметить, что энергетические спектры и числа молекул
(Л\ + Х2) для состояний объема V, входящих в класс 1, и состоя-
ний, близких к критическому, отнюдь не перекрываются и четко
разделены щелями, ширина которых пропорциональна размерам
критического зародыша. Чтобы различать эти состояния, нет необ-
ходимости прибегать к условному различию между «уже» двухфаз-
ными и «еще» однофазными состояниями объема V, а достаточно
руководствоваться тем, к каким областям энергии Ео и чисел
частиц N2 принадлежит то или иное состояние. Поэтому ничто не
мешает рассматривать метастабильные и лабильные (околокрити-
ческие) состояния объема как части единого большого ансамбля
Гиббса независимо от того, относится состояние к классу 1 или 2.
Однако использование для расчета скорости нуклеации метода пере-
вала уравнения Крамерса возможно (и необходимо) только для со-
стояний класса 2, так как требуется определение функции <p(N2):
Ф (А'Ч) = ЕЕ WnNtNf
Ni п
Для состояний класса 1 функция <p(N2) теряет смысл, и в то же вре-
мя соответствующие состояния исчерпывающе характеризуются
числами Nun.
Для определения <р(Х2) разложим экспоненту в правой части,
уравнения (12) вблизи состояния лабильного равновесия в ряд
до квадратичных по ANX и ДХ2 членов [учитывая уравнение (9)1:
У, иш, = Ас ехр 4 [peAN - AN -
п
z dNy * dN2 J
Член, содержащий AN1AN2, опущен, ибо при последующем сум-
мировании по ДХх он дает малый вклад.
Вычисляя производные, получим вместо уравнения (14) следую-
щее:
У]wnNtNt = A exp -2Q [ — (-^) — 1 (ДлМ2 —
п V L ' 1' J 1
(d2F \ 1
(15>
11
Членом с (JV2) 2 в дальнейшем пренебрежем. Суммируя по АЛ7Ъ
лолучим
Ф (^2) = ф (Л^2 + A/V2) = wnNiNt =
ДЛЧ п
= АСЕ«р{-4-(М(ЛМх
дач
xexp{-Tsw<M'!)1}- <1б>
Полагая *
FNt = N$(T) + es, ' (17)
где s — поверхность, а а — поверхностное натяжение капли (сле-
довательно, s ж (ТУг)*’)» получим
О'|=Ч’<7'> + т^- <18>
р ^Nt\C 2_ SSC . g,
\ dN% J 9 {Ncy • k '
Подставляя уравнение (19) в (16), находим
<Р(ЛГ2) = ф(Г8 + ДЛГ2) = Ас £ехр {- (^(ДЛ^)2} х
ДУ,
<20)
Таким образом, ф(1У2) имеет в функции (&N2lN2) минимум, весьма
глубокий и узкий, если поверхностная энергия критической капли
os0 велика по сравнению с ©. Подставляя уравнение (20) в (7), по-
лучим
АСГ)С )\ехр [—)° (Д^) ]
<ss
90(Лф2
dAN2
и, наконец,
<21>
2 ’ дли
Здесь Dc — «обобщенный коэффициент диффузии» зародыша вбли-
зи лабильного равновесия.
* Можно продолжить расчеты и для любого другого вида зависимости FN от
12
Для вычисления gi — «статистического веса метастабильного
состояния» — будем исходить из той же формулы (8), применяя
ее теперь к состояниям класса 1. Исключая состояния 2 и 3, имеем
ДУ = У —Уо, (22)
ДЕ = E„v - Ео, (23)
где /Vo — среднее число молекул в объеме V; Ео — среднее значение
энергии. Разлагая wn в ряд, получим
= С ехр [- 4 S (Ёо, tfo)] exp [-§• Д У - . (24)
Заменяя Е через Fo 4- ES0 и суммируя по п, находим
У, = Л ехр ДА/' —- -°-] , (25)
где "
А0 = С ехр {- 4 [S (£", АГ) + So (Ео, А^о)]} • (26)
Штрихи указывают на то, что резервуар тепла находится в равно-
весии с объемом V, находящемся в метастабильном состоянии.
Разлагая разность FN — Fo в ряд по Д-V до квадратичного чле-
на и суммируя по N, получим
=Е, Е=Е л«ехр [- 4 (ДМ • (27)
п п ДМ
Подставляя в уравнение (4) значения© из (21) и gi из (27), найдем
AC[F
До3 /л (Ntf
v =
—Еехр [~ 4§ (ж)с < дМ
gs ДАЧ________________
0 Еехр i ("Sr) (ДМ
дм
(28)
Сумма, стоящая в числителе формулы (28), отличается от суммы,
стоящей в знаменателе, только тем, что в первой молекулы пара
занимают объем V за вычетом объема критического зародыша Vе.
Ввиду того, что vc V, этим различием можно пренебречь, и
обе суммы сократить. Раскрывая значения А' и Ао, имеем
V! = 1/4- ехр {4 [S (Ее, Г) + So (Ро, < М) -
— S(E', A")-S0(E, Д\))]}. (29)
Выражение в квадратных скобках в экспоненте равно приросту
энтропии всего объема W7, возникающему при образовании крити-
ческого зародыша, по сравнению с состояниями класса 1 метаста-
1S
сильного равновесия. Ввиду постоянства энергии и объема системы
это изменение энтропии равно взятому с обратным знаком приросту
термодинамического потенциала системы, деленному на абсолют-
ную температуру, изменением которой при нуклеации мы прене-
брегаем ввиду больших размеров системы. Так как объем V ограни-
чен перегородкой, не пропускающей критических зародышей, то
в выражении для термодинамического потенциала системы после
нуклеации необходимо сохранить член^Т In (l/NJ, где — число
молекул в объеме V. В результате получим
vx = —1 /Л-^- ехр (— , (30)
3 Ул Я' у 9 6 / 4 '
где kg* — работа образования критического зародыша. Ввиду
того, что Ag* стоит в показателе, важно вычислить его значение с
достаточной точностью.
С целью вычисления значения Dc подставим для него выражение,
аналогичное использованному Эйнштейном в теории броуновского
движения:
в cW2
Др dt
(31)
где dNjdt — квазиравновесная скорость фазового перехода, за-
висящая от разности химических потенциалов капли и пара Ар.
Величину
со =
1 dN2
Др dt
(32)
можно вычислить методами кинетической теории газов, если из-
вестен коэффициент конденсации а, и в паре отсутствуют поли-
мерные ассоциаты.
В более общем случае вычисление со и D представляет собой осо-
бую задачу, относящуюся собственно не к теории нуклеации, а к
теории фазового перехода пар—жидкость. Поэтому она здесь не
рассматривается. При достаточно малом содержании в паре поли-
мерных ассоциатов D легко вычисляется, и мы получим
О = (33)
где рх — плотность Пара; v — средняя скорость молекул; т —
их масса. Подставляя уравнение (33) в уравнение (30), имеем
v,/£ехр(--£) (34)
(где рх — плотность критического зародыша) или для единицы
объема
Полученная формула при а = 1 совпадает, включая предэкспо-
ненциальный множитель, с формулой, выведенной существенно
иным методом Френкеля [131, что можно рассматривать как под-
тверждение непротиворечивости предложенного нами общего мето-
да. Различие с формулой Френкеля может проявиться при уточне-
нии значений D и U.
При выводе вероятности нуклеации мы дополнили гиббсовское
выражение для работы образования критического зародыша членом
&Т1п (l/A^, учитывающим энтропию смешения. В результате это-
го получена необходимая зависимость vx от Nx и, следовательно,
от объема пара. Что касается влияния трансляционных степеней
свободы зародыша, то оно с ростом критических размеров последней
стремится к нулю и не учитывается, поскольку наша цель — най-
ти асимптотическое выражение для скорости нуклеации. При же-
лании соответствующая поправка может быть внесена [15].
2. Влияние поверхностных сил
на образование и свойства новой фазы
Поверхность, на которой происходит образование новой фазы,
не является инертной по отношению к этому процессу, а, напро-
тив, оказывает на него существенное влияние. В зависимости от
характера связей, образующих подложку, и связей в конденсируе-
мой среде влияние подложки может быть различным.
Рассмотрим грань ионного кристалла, например, поваренной
соли. Вблизи одних граней ионы разного знака расположены стро-
го в одной плоскости. Вблизи граней с другими индексами — в со-
седних параллельных плоскостях (плоскости совершенной спай-
ности гипса или слюды). Во втором случае соответствующая плос-
кость раскола может разделить кристалл на две части, несущие
одинаковые по величине заряды противоположного знака. При взаи-
мном раздвижении обеих частей в образующейся между ними щели
возникает мощное электрическое поле с напряженностью порядка
миллионов и десятка миллионов вольт на 1 см. При определенных
условиях (в хорошем вакууме) эта щель может достигнуть ширины
порядка десятков микрон, прежде чем произойдет взаимная ней-
трализация противоположных зарядов за счет «пробоя вакуума».
Измерить на опыте достигаемую к моменту разряда разность
потенциалов, равную произведению напряженности электрического
поля на разрядный промежуток, помогло сделанное В. В. Кара-
севым, Н. А. Кротовой и Б. В. Дерягиным [16] наблюдение эмис-
сии электронов свежеобразованными поверхностями. Часть элек-
тронов, вырываемых мощным электрическим полем с отрицательно за-
ряженной поверхности образующейся щели, бомбардируют противо-
положную, положительно заряженную поверхность щели, другая
часть электронов, вылетая под более косым углом, может вырваться
из щели наружу.
15
Эти электроны будут разогнаны разностью потенциалов, равной
половине разности потенциалов в щели ЛК Эта разность потен-
циалов Д У/2 связана со скоростью электронов v по формуле
е ДУ и2
т 2 ~2~ ’
где е/т — отношение заряда к массе электрона. Скорость элек-
тронов измерялась по их отклонению в магнитном поле известной
напряженности. Все это проводилось в камере, откачанной до вы-
сокого вакуума. Найденные значения ДУ варьировали в зависимо-
сти от рода кристалла от десятков до сотен киловольт. Аморфные
тела эмиссии электронов не обнаружили, что и понятно.
Заметим, что специальный интерес представляет изучение этого
явления при нарушении контакта двух неодинаковых тел, напри-
мер при отдирании полимерной пленки от стеклянной пластинки.
Зная одновременно работу, затраченную на отслаивание, в основ-
ном идущую на преодоление притяжения противоположно заряжен-
ных поверхностей до наступления разряда, и максимальную ско-
рость эмитируемых электронов, можно было определить плотность
зарядов (на единицу площади) образуемых поверхностей; по суще-
ству плотность зарядов обкладок того двойного электрического слоя,
который, как предположил еще Г. Гельмгольц, образуется при
контакте разнородных тел. Измерения дали значения 103—105 абс.
электрост. ед. заряда на 1 см2. Плотность зарядов на свеже-
образованных гранях кристаллов может существенно превосходить
эти значения.
Каков бы ни был заряд граней кристалла в момент их образо-
вания, он быстро исчезает, особенно во влажном воздухе, за счет
стекания зарядов вдоль поверхности. Вместе с этими зарядами ис-
чезает и мощное поле, образуемое их совокупным действием,—
поле, которое, как и поле равномерно заряженных пластин конден-
сатора, убывает с расстоянием весьма медленно. Остаются только
электрические поля разноименных ионов, у которых концы силовых
линий локализованы на поверхности грани на близком расстоянии
друг от друга. Помимо этого поле поверхностных сил образуется
молекулярными силами (исходящими из поверхностных атомов
и ионов), природа которых связана со строением электронных обо-
лочек атомов.
Наконец, атомы могут взаимодействовать за счет сил валент-
ной связи. Примером такой химической связи служит валентная
связь двух атомов углерода, возникающая при их сближении.
Именно эта связь и определяет кристаллическую форму, прочность
и другие свойства алмаза.
Общее для всех этих сил, образующих суммарное поле поверх-
ностных сил,— их периодическое, как бы волнообразное, распре-
деление над гранью кристалла, обусловленное периодическим рас-
положением атомов. Под влиянием такого характера поля новые
молекулы или атомы, попадая на поверхность грани, стремятся
1в
занять такие положения, которые подчиняются той же периодич-
ности, с которой расположены атомы на грани и под ней,— перио-
дичности, обусловленной действием тех же самых сил.
Иными словами, поверхностные силы стремятся обеспечить про-
должение свойственной кристаллу «кирпичной кладки» и за счет
нового строительного материала, состоящего из тех же атомов.
Впрочем, кирпичная кладка кристалла действием его поверхно-
стных сил может быть навязана и атомам другого рода, но близких
размеров (радиусов), способных образовывать кристаллы аналогич-
ного строения и формы (почти изоморфные данному кристаллу).
Такое навязывание своей структуры инородному наслоению назы-
вается эпитаксией, наслоение из тех же атомов — автоэпитаксией.
При определенных условиях за счет эпитаксии может сформиро-
ваться метастабильная кристаллическая модификация, т. е. менее
устойчивая, чем обычная, в которую она иногда самопроизвольно
переходит и которая в отсутствие инородной затравки, как правило,
не образуется.
Здесь необходимо различать два случая. В одних случаях эпи-
таксия способна образовать инородный кристалл любых размеров
(очевидно, на более поздней стадии действует уже автоэпитаксия
новообразованной кристаллической формы). Во втором случае эпи-
таксия имеет как бы ограниченный радиус действия и способна на-
вязывать заданную структуру только слоям ограниченной толщи-
ны. Так, при кристаллизации на каменной соли паров сульфида
кадмия метастабильная форма последней наблюдается только при
толщине слоя менее 200А. Очевидно, на большем расстоянии от
«подложки» сказывается относительно малая устойчивость мета-
стабильной модификации. Радиус эпитаксиального действия по-
верхностных сил также конечен, если между гранью затравочного
кристалла и кристаллизующимся веществом имеется инородная
аморфная подложка. Согласно работам Р. Брадлея, Д. Ротена,
Г. И. Дистлера, эпитаксия способна влиять на расположение ато-
мов кристаллизующегося вещества через прослойки порядка 1000—
1500 А. Механизм такого явления (по выражению Г. И. Дистлера,
передачи структурной информации через бесструктурную прослой-
ку) неясен.
В случае жидких прослоек подвижность молекул позволяет
предположить, что под влиянием подложки тонкие граничные слои
жидкости способны приобретать определенную ориентированную
структуру, аналогично тому, как это происходит с жидкими кри-
сталлами. В отличие от последних толщина образующейся гранич-
ной фазы варьирует от сотых до десятых долей микрона, т. е. до-
вольно мала. Впрочем, удивляться надо скорее тому, что ориенти-
рующее или эпитаксиальное действие подложки не ограничивается
только одним-двумя молекулярными слоями, а простирается на
сотни слоев.
Ввиду того, что представление о способности жидкостей приоб-
ретать особую структуру и свойства в таких слоях является в нау-
ке существенно новым, следует кратко изложить имеющиеся дока-
зательства. Б. В. Дерягиным и Р. Грин-Келли [17] было обнаруже-
но, что тонкие слои воды толщиной в сотни ангстрем, внедряющие-
ся в пластинчатые кристаллы натриймонтмориллонита (глинистого
минерала) при погружении его в воду и вызывающие набухание
кристаллов, обладают двойным преломлением. Известно, что двой-
ное преломление, отличающее кристаллы от жидкостей, объясняет-
ся тем, что свойства кристаллов, в том числе оптические, неодина-
ковы по различным направлениям. Ориентация молекул воды под
влиянием граничащих с ними кристаллических слоев может также
вызывать подобную анизотропию свойств. Влияние особой струк-
туры тонких слоев на расклинивающее давление смачивающих пле-
нок было показано Н. В. Чураевым [18].
Особенно интересно, что в ряде случаев подобные ориентиро-
ванные слои с особыми свойствами отделены от остального объема
жидкости с нормальными свойствами резкой границей раздела. Это
было доказано Б. В. Дерягиным и Ю. И. Поповским [191 измерени-
ем удельной теплоемкости слоев нитробензола на стекле при по-
степенно возрастающей толщине. До определенной толщины тепло-
емкость сохраняла постоянное значение, на 20% меньшее теплоем-
кости объемного нитробензола. Толщина слоя с пониженной тепло-
емкостью уменьшается с повышением температуры от 0,1 мкм при
15° С до 0,05 мкм при 66° С.
Существование резкого скачка свойств на определенном рас-
стоянии от подложки было еще ранее обнаружено В. В. Карасе-
вым и Б. В. Дерягиным [20] при измерении зависимости вязкости
некоторых органических жидкостей от расстояния до твердой стен-
ки. Все это дает право называть такие слои особой, граничной фа-
зой, поскольку наличие резкой границы раздела есть основное оп-
ределение фазы. Различие с обычными фазами заключается в том,
что толщина граничной фазы — вполне определенная для данной
температуры величина.
Замечательно, что бензол, не имеющий, в отличие от нитробензо-
ла, полярных групп в молекуле, образует граничные фазы на стек-
ле только тогда, если последнее покрыто мономолекулярным ад-
сорбированным слоем нитробензола. Можно полагать, что ориен-
тированный на поверхности стекла монослой нитробензола вызы-
вает своего рода эпитаксиальное действие, распространяющееся
в бензоле от слоя к слою и ориентирующее несколько десятков
монослоев последнего. Отсюда видно, что состояние поверхности,
ее чистота могут играть решающее значение для процесса эпитак-
сиального наращивания. В дальнейшем нас будут в основном ин-
тересовать процессы, обусловленные автоэпитаксией, в условиях,
когда затравочный кристалл является метастабильной модифика-
цией.Процесс наращивания алмаза на алмазные затравочные кристал-
лы назван физико-химическим синтезом, поскольку он основывает-
ся на явлениях, изучением которых занимается физическая химия
поверхностных явлений.
18
Физико-химический синтез алмаза проводится из углеродсо-
держащих сред: углеродсодержащего газа (метан, ацетилен, окись
углерода и др.) и из растворов углерода в расплавленных ме-
таллах. При синтезе алмаза из газа на поверхности затравочного
кристалла алмаза происходят гетерогенные реакции разложения
вида
СН4~* С + 2Н2, С2Н2—• 2С +Н2.
алмаз алмаз
Углерод высаживается на алмазном затравочном кристалле
также из пересыщенного раствора углерода в расплавленном ме-
талле.
Процессы химической кристаллизации вообще, а тем более ме-
тастабильных фаз, разработаны еще далеко не полно. Поэтому
изложим качественные предпосылки, лежащие в основе эпитакси-
ального синтеза алмаза.
Возможность такого процесса может быть обоснована двояко:
во-первых, исходя из молекулярного механизма роста кристаллов;
во-вторых, исходя из общей теории образования новой фазы (нук-
леации). Если имеется грань кристалла, вблизи которой концент-
рация атомов углерода превышает соответствующую равновесную,
то избыток атомов углерода будет выделяться на грани. При этом
они будут находиться под влиянием силового поля кристалличе-
ской подложки (затравки), стремящейся продолжить ту «кирпич-
ную кладку», которая привела в свое время к образованию самой
кристаллической подложки.
Однако если пересыщение углерода по отношению к алмазу
слишком велико, и процесс выделения атомов углерода идет слиш-
ком быстро, могут создаться условия, при которых сделается
возможным образование и рост зародышей графита как термоди-
намически более выгодной модификации, имеющей меньшую энер-
гию, что перевесит влияние поверхностных сил, т. е. влияние авто-
эпитаксии. Вначале образование графита и рост алмаза будут идти
параллельно на разных участках поверхности, но в дальнейшем—
если пересыщение углерода достаточно велико — графит покроет
всю поверхность кристалла алмаза и его рост прекратится.
Условия, при которых этого можно избежать, выводятся из
общей теории зарождения и роста кристаллов Гиббса—Фольме-
ра—Каишева. По этой теории следует, что вероятность появления
устойчивого, так называемого критического, зародыша новой моди-
фикации (в нашем случае графита), способного к уже беспрепятст-
венному дальнейшему росту, тем меньше, чем больше работа его
образования. Эта работа пропорциональна поверхностной энергии
трехмерного зародыша «критических» размеров, которые тем боль-
ше, чем меньше пересыщение атомов углерода по отношению к их
равновесию с графитом. Поэтому выгодно брать малое пересыще-
ние, превышающее, однако, минимально необходимое, для того
чтобы мог происходить рост алмаза.
19
Существует фундаментальный факт, затрудняющий рост графи-
та на поверхности затравочного кристалла алмаза. В то время как
рост алмаза происходит за счет достройки уже имеющейся крис-
таллической решетки, для образования кристалликов графита
атомы углерода должны расположиться в ином порядке, не соглас-
ном с расположением атомов подложки. Речь должна идти о рожде-
нии новой кристаллической формы, а такие роды всегда трудны.
Трудности свойственны рождению любой новой фазы, как, напри-
мер, капли из пара, пузырька из жидкости, кристалла из газа или
жидкости.
Причины затруднений впервые вскрыл Гиббс — создатель клас-
сической термодинамики гетерогенных систем. Энергия таких си-
стем, включающих внешнюю среду и кристалл, равна сумме объем-
ной энергии, пропорциональной объему кристалла, и поверхност-
ной энергии, пропорциональной поверхности. Процесс может идти
беспрепятственно, если он сопровождается уменьшением энергии,
подобно тому, как тела стремятся падать вниз, так как при этом
уменьшается их потенциальная энергия. Точнее, здесь надо гово-
рить о «свободной энергии», известной из курса термодинамики.
Рост кристаллов идет за счет того, что при избытке в окружающей
его среде строительного материала энергия системы уменьшается
пропорционально пересыщению и приросту объема образующегося
нового кристалла. Увеличение размера кристаллов играет тем
меньшую роль, чем больше отношение поверхности к объему, а оно
велико только у микроскопических, «зародышевых» кристалликов.
Однако у очень малых кристалликов определенного, так назы-
ваемого критического, размера рост поверхностной энергии, соп-
ровождающий увеличение размеров, может как раз уравновесить
уменьшение объемной энергии. При больших размерах перевеши-
вает влияние объемной энергии, и кристалл может безостановочно
расти. Для зародышей меньше критического, наоборот, перевеши-
вает влияние поверхностной энергии, и они будут уменьшаться
в размерах. Поэтому для «рождения» жизнеспособного нового кри-
сталлика необходимо, чтобы он перешел через размеры критиче-
ского зародыша, что кажется противоречащим законам термоди-
намики.
Противоречие объясняется тем, что изменение размеров очень
маленьких кристалликов (и вообще зародышей новой фазы) не пол-
ностью подчинено термодинамике. Здесь вмешивается явление
флуктуаций, которое происходит от хаотичного, беспорядочного
характера движения атомов и молекул. Наиболее наглядным и об-
щеизвестным примером флуктуаций является броуновское движе-
ние взвешенных в газе или жидкости частиц.
В случае малого зародышевого кристалла в одни моменты число
атомов, присоединяющихся к нему, перевешивает число атомов,
отрывающихся от него, в другие моменты баланс отрицателен. При
очень счастливом стечении обстоятельств зародыш может достичь
критического размера и даже перевалить через него, несмотря на то,
20
что при этом свободная энергия системы увеличится. Если система
изолирована и ее полная энергия постоянна, увеличение свободной
энергии системы возможно только за счет уменьшения энтропии,
служащей, как известно, мерой молекулярного беспорядка. Мы
имеем здесь пример того, как хаотичное движение атомов и моле-
кул, приводящее, как правило, к росту беспорядка, может — имен-
но в силу своего неупорядоченного характера — приводить иногда
к небольшому уменьшению энтропии и, следовательно, к увеличе-
нию порядка, т. е. беспорядок рождает порядок.
Применение теории флуктуаций, созданной М. Смолуховским,
к возникновению из пересыщенной среды закритических, жизне-
способных зародышей позволило М. Фольмеру, Р. Беккеру и
И. Дерингу, И. Странскому, Р. А. Каишеву, Я. И. Френкелю,
Я. Б. Зельдовичу и другим ученым создать теорию образования
новой фазы, в частности кристаллической. Было показано, что
вероятность появления новой фазы, а следовательно и число возни-
кающих в единицу времени в пересыщенной среде жизнеспособных
зародышей, резко уменьшается по мере увеличения той свободной
энергии, которая должна быть затрачена на создание критического
зародыша. Еще В. Гиббс показал, что эта энергия, или, иначе, ра-
бота образования критического зародыша, равна одной трети его
свободной поверхностной энергии. Чем больше пересыщение, тем
меньше размеры критического зародыша и тем меньше работа его
образования. Поэтому с ростом пересыщения растет и число обра-
зующихся в единицу времени жизнеспособных зародышей. Это
число растет также с увеличением подвижности атомов, т. е. с
повышением температуры. Поэтому, например, алмаз при темпе-
ратурах 1500—2000° С способен быстро переходить в графит, а
при комнатной температуре сохраняться в течение геологических
периодов.
Таким образом, скорость образования и дальнейшего роста но-
вой фазы зависит от температуры, пересыщения и наличия кристал-
лической подложки.
Эта скорость также рассматривается в теории роста кристаллов
Фольмера—Каишева—Странского. При этом, несмотря на при-
сутствие уже образовавшегося кристалла (затравки), на определен-
ных стадиях роста граней в начале наращивания нового слоя «кир-
пичной кладки» скорость роста может лимитироваться необходимо-
стью затраты работы образования двухмерного (плоского), толщиной
в один атом, критического зародыша. При очень малом пересыщении
атомами углерода вероятность образования таких зародышей так
мала, что рост кристалла идет с «запинками», подобно остановкам
плохо смазанных поверхностей при скольжении. Однако на опре-
деленных участках поверхности кристалла рост может идти не-
прерывно, «без запинок», ввиду того, что либо работа образования
зародыша нового слоя кирпичной кладки равна нулю [например,
в случае алмаза на гранях с индексами (100)], либо (как в случае
винтовой дислокации на поверхности кристалла) никаких двухмер-
21
них зародышей не требуется, и процесс роста идет безостановоч-
но и равномерно, подобно подъему по винтовой лестнице без пло-
щадок.
Термодинамические расчеты показывают, что равновесное дав-
ление углеродсодержащего газа, пара или раствора углерода над
алмазом приблизительно вдвое больше, чем над графитом. Поэтому
все же имеется опасность появления неалмазного углерода, бло-
кирующего поверхность. Основная задача физико-химического
синтеза алмаза заключается именно в предотвращении влияния
выделяющейся стабильной фазы графита на синтез алмаза. Это
достигается различными способами. Можно периодически преры-
вать синтез и удалять графит с затравочных кристаллов. Можно
стараться подобрать такие условия синтеза, (когда образующийся
графит будет заращиваться, «замуровываться» алмазом. И, наконец,
можно пытаться создать такой режим синтеза, когда зародыши
графита не будут достигать критических размеров. Последний
путь, несомненно, предпочтительнее, так как он открывает возмож-
ность осуществления непрерывного процесса.
Возможность выделения двух конкурирующих фаз — алмаза
и графита — существенно усложняет процесс кристаллизации. В ки-
нетике гетерогенных химических реакций широко используются
понятия и определения, заимствованные из учения о гомогенных
химических реакциях. Во многих случаях это вполне оправданно,
например, при каталитических реакциях. Во многих же гетероген-
ных процессах, например, процессах роста и травления кристал-
лов, происходит обмен веществом между газовой и твердой фазами,
что приводит к ряду принципиальных особенностей гетерогенных
реакций, идущих с образованием новой фазы. Эти особенности
позволяют выделить физико-химический синтез веществ в качестве
отдельного направления химического (неорганического и органи-
ческого) синтеза, подобно тому как в настоящее время из общих ме-
тодов анализа выделился физико-химический анализ.
3. Теория гетерогенной нуклеации
Рассмотренный выше подход к образованию новой фазы был при-
менен к гетерогенной нуклеации [14]. Основная формула, описываю-
щая нуклеацию, согласно работе [15], может быть представлена
в виде
V, = exp (- l^kT), (36)
у л
где N[ — число молекул в объеме V; Ag* —• работа образования
критического зародыша; р,2 — химический потенциал конденси-
рованной фазы; N2 — число частиц в зародыше; индекс «с» отно-
сится к состоянию лабильного равновесия. Коэффициент диффузии
22
1У определяется как .
Dc = kTs-л, (37)
где x = 5 — поверхность критического зародыша; Vo —
в данном случае число частиц, присоединяющихся к зародышу
на единицу поверхности в единицу времени; Др — разность хими-
ческих потенциалов твердой и газовой фаз.
Рассмотрим процесс гетерогенной конденсации, ограничиваясь
плоским двухмерным зародышем, имеющим форму диска толщиной
в один атом. Пусть также установление адсорбционного равновесия
не является самым медленным процессом.
Примем, что поверхность твердого тела, на котором происходит
нуклеация, находится в равновесии с газовой средой. Выделим на
поверхности площадь 5 = п.г2с, содержащую N[ адсорби-
рованных частиц в двухмерном газе. Здесь гс — радиус критического
зародыша. Для дискообразного зародыша при конденсации на ана-
логичную подложку легко показать, что
(др/дМг)с = — х/4 057(Л/г)2, (38)
где 8е — боковая поверхность критического зародыша. Практиче-
ски все частицы будут присоединяться к зародышу благодаря поверх-
ностной диффузии. Тогда, согласно работе [5],
|Z0 = щпйехр(— gJkT)
и
х = (пд/(о/£7")ехр (— gJkT).
Здесь ns — поверхностная концентрация адсорбированных частиц;
а — средняя длина их пробегов при поверхностной диффузии;
gx — энергия активации поверхностной диффузии;
и = (&Т/К) [ 1 — exp (— hv/kT)],
где h — постоянная Планка; v — частота колебаний атомов на
поверхности. Подставляя значение х в уравнение (37), получим
Dc = nsscaa exp (— gJkT). (39)
Учитывая, что Nx ~ J — at0 rtsS~ ' ° — 2 /2л N^h или j ад т тГ 10 - 2 Р2 V ties и вводя обозначение 10 = v^s, получим rhkT resCXP( gltrg)’ (4°)
где m —- масса одной частицы (атома или молекулы); р2 — плот-
23
ность конденсированной фазы. В случае конденсации из пара
”s = ----—и—ехр (~) , (41)
(2лт/гТ) |ги F \ kT > v ’
где g2 — теплота адсорбции; Р — давление пара; а — коэффициент
конденсации. Подставляя значение для tis в уравнение (40), полу-
чим
I — —— 1/" ° pz exD Г — gi — Ag* ~1 /42}
0 8л;сорайГ V rchkT Г еХр [ kT J'
Поверхностная концентрация адсорбированных частиц в двух-
мерном газе пропорциональна степени заполнения поверхности 0:
ns = ®rts0,
где ns0 — плотность, соответствующая монослою. Пусть процесс
образования новой фазы идет на поверхности благодаря химичес-
кой реакции разложения
ВА—»В + А ,
газ газ
и выделяющиеся при разложении атомы вещества В наращиваются
на подложке из того же материала. Обозначим парциальные дав-
ления pi для ВА и р2 для А и рассмотрим процесс при постоянном
давлении.
Константа равновесия позволяет вычислить равновесные пар-
циальные давления над веществом В. Используя изотерму адсорб-
ции Лангмюра, в случае равновесия получим плотность адсорби-
рованных частиц
ns - nsovie - i+Pie/bl + p2e/bi ns0.
Если же Pl > рц, то плотность адсорбированных частиц и соответ-
ствующее пересыщение взаимосвязаны уравнением
»s = et = pL 1 + р^А + р2 А
«so ~ Pie 1 + Pi/6i + Pz/bz
В этом случае работа образования критического зародыша (диско-
образной формы) [51 будет равна
._________________ne2/i2__________
® — Г 1 -I- Pie/bi + Р2А 1 ‘
kT | In P1/Ple + In -f+л/И+’рЛ 1
Здесь е—удельная краевая энергия. Если получается
выражение
— kT In (pt/ple)
Именно второй член в квадратных скобках знаменателя уравнения
(43) учитывает влияние поверхности на пересыщение относительно
24
(43)
Л82Л2
(44)
образования критического зародыша на данной поверхности. Сход-
ство уравнения (44) с уравнением для работы образования крити-
ческого зародыша из пара чисто формальное. В случае конден-
сации из пара пересыщение падает при увеличении температуры
подложки и увеличивается при повышении давления пара (пропор-
ционально плотности потока падающих частиц). При химической
кристаллизации могут быть разные случаи. Например, при разло-
жении метана на графите пересыщение уменьшается при увеличении
давления и увеличивается при повышении температуры. В случае
ацетилена пересыщение не зависит от давления и уменьшается при
повышении температуры.
Действительно, рассмотрим реакцию разложения метана
CH4^C-f-2H2. ___
Константа равновесия К(Т) позволяет определить парциальные
давления этих компонентов (другими пренебрежем):
pielple — К (Т),
причем Pi + Р2 = Ро = const. В большой области температур и дав-
лений
Р1е Pie ~ pot
поэтому приближенно
Р*/р1е = К(Т).
Если же тем или иным способом над затравкой создать давление
радикала СН, то пересыщение будет увеличиваться при повышении
давления. Для более полного анализа следует рассмотреть зависи-
мость (43).
Зависимость постоянной адсорбции от температуры можно пред-
ставить как = 610 exp(g2/kT). Подставляя это выражение в урав-
нение (40), получим
J _ am m f б____________________Р?_______ Г 2g2 - gi — Ag* 1
~ 26*0 р2 |/ rJikT (1 4- Pxlbl+ Рг1Ъ^ Р L kT J ‘
(45)
Если процесс роста является безактивационным и лимитирует-
ся стадией образования критических зародышей, то скорость роста
Rx (массовая или линейная) будет пропорциональна 0®, т. е.
D _ с р<_______PYn Г 2ga ~ gi — Ag* 1 /
- C1 (1 + Pxfbx + Pilbtf eXP [ kT J • ( '
Это выражение показывает, что кажущийся порядок химической
реакции кристаллизации может быть любым — от нулевого до
второго. Если кристаллизация из газовой фазы проводится в слое
25
порошка, то по глубине слоя «порядок» реакции также может ме-
няться, и реакция всегда проникает на бесконечную глубину.
Выражение (46) можно представить в виде
Rl = Сх (1 + Pjh + Р2/62)2 ехр (-~rt) ’
где Е = 2^2 — g± — Ag* — кажущаяся энергия активации про-
цесса. Поскольку А^* ~ ИТ, Е будет убывать при росте темпе-
ратуры. В общем случае зависимость кажущейся энергии активации
от температуры более сложная, нежели Е constx -(- (const2/7,)>
и эта зависимость тем сильнее, чем меньше пересыщение. Если же
I &g* | <<| 2g., —gi I, то величина Е практически постоянная.
Рассмотренный выше механизм справедлив лишь при условии
существования разреженного двухмерного газа, когда степень
заполнения поверхности значительно меньше единицы.
Рост несовершенных кристаллов может происходить без обра-
зования критических зародышей. Без образования критических
зародышей может также происходить, например, рост грани (100)
кристалла алмаза. В этом случае
т = Г+7Л‘+рА <47>
при условии, если присоединение новых атомов к растущей ступе-
ни является безактивационным процессом. В противном случае
в кажущуюся энергию активации должен входить еще один член
g3, учитывающий работу на разрыв связей в молекуле химически
реагирующего соединения:
Е = 2^ — gi — g3 — Ag*. j
Основным препятствием на пути эпитаксиального синтеза ал-
маза является не то обстоятельство, что алмаз представляет собой
метастабильную форму углерода, а тот факт, что при этом может
образовываться стабильная форма углерода — графит. Атомы угле-
рода обнаруживают свою принадлежность к той или иной модифи-
кации углерода лишь в кристаллической решетке. Поэтому даже
в идеально чистых условиях исходную углеродсодержащую среду
можно рассматривать как источник примесей. Выделяясь на за-
травочном кристалле, неалмазный углерод ухудшает эпитаксию,
блокирует поверхность и, в конечном счете, прекращает рост алма-
за. Поэтому при эпитаксиальном наращивании алмаза его рост
следует рассматривать не изолированно, а в сопоставлении с со-
ответствующими процессами выделения графита.
Сопоставим скорости образования двухмерных зародышей ал-
маза и графита на грани (111) затравочного кристалла алмаза при
различных пересыщениях, следуя работам [21, 221. Грань (111)
кристалла алмаза состоит из двух слоев атомов и представляет собой
гексагональную гофрированную сетку, проекция элементарного
ребра которой на плоскость (111) имеет длину 1,44 А, т. е. довольно
26
мало отличается от длины связи С—С в базисной плоскости графита
(1.42 А). Поэтому естественно предположить, что образующийся
на грани (111) алмаза зародыш графита будет ориентирован таким
образом, как если бы он возникал на базисной плоскости графита.
Обозначим частоту (скорость) нуклеации, т. е. число критиче-
ских зародышей, образующихся на единицу поверхности в единицу
времени, через 1.
В общем случае работу образования AG двухмерного зародыша
с периметром I и высотой h можно записать [5] в виде
AG = /е 4- yPh&Gv + уР (ас_х — щ_а -4- стс_а). (48)
Здесь уР— площадь поверхности зародыша; ас_х, ox-v,
<тс_а — удельные свободные межфазные энергии на границах
зародыш—кристалл (с—х), кристалл—среда (х—v) и зародыш—
среда (с—v) соответственно; е — удельная краевая свободная
энергия;
AGa = — kT/Q In (р/ре), (49)
где р — парциальное давление углеродсодержащего газа; ре —
равновесное давление этого газа; й — объем, приходящийся на
один атом в кристаллическом состоянии; Т — абсолютная темпе-
ратура.
Принимая во внимание тот факт, что расстояние между соседни-
ми слоями графита (3,35 А) значительно превосходит длину связи
С—С в кристалле алмаза (1,55 А), можно ожидать, что энергия
связи поверхности кристалла алмаза с графитовым зародышем
будет равна энергии связи между слоями графита, т. е.
= $x~v ^c-v> (50)
В этом случае последний член правой части уравнения (48) ис-
чезает. Найдя максимум AG относительно /, получим уравнения для
работы образования зародыша критического размера AG* и соот-
ветствующего ему периметра I*:
&G* — — e2/4y/zAGa, (51)
/* = —е/2у h\Gv = 2AG*/e. (52)
В соответствии с теорией Фольмера, скорость образования за-
родышей надкритического размера равна
I = I* а ехр (— &G*lkT). (53)
Величина а зависит от характеристик адатомов на поверхности.
Мы считаем, что она остается одной и той же как для зародыша
графита, так и для зародыша алмаза. Это предположение равно-
сильно утверждению о существовании на поверхности кристалла
своего рода двухмерного газа, в котором атомы, идущие на построй-
ку зародышей алмаза и графита, совершенно неразличимы. Такое
допущение в общем случае не представляется очевидным.
' 27
Обозначая далее индексами аир величины, относящиеся соот-
ветственно к алмазу и графиту, для отношения скоростей образо-
вания зародышей получаем:
т— = —~—— ехр
70 8яАО0
AG Al
ц"3
ag*
(54)
Удельная краевая свободная энергия для кристалла графита
легко вычисляется обычным способом [231:
е3 = <p3/2d.
Здесь фр — энергия связи ближайших соседей; d — постоянная
решетки базисной плоскости графита.
Равновесная форма зародыша графита, найденная по теореме
Вульфа,— правильный шестиугольник и, следовательно,
В случае кристалла с алмазной решеткой неприменим метод
определения удельной краевой энергии по отрыву участка поверх-
ностной сетки, имеющего форму параллелограмма, поскольку об-
разующиеся при таком отрыве края неравноценны в смысле проч-
ности их связи с кристаллом. Вероятно для того, чтобы обойти труд-
ности при определении удельной краевой энергии, равновесную
форму зародыша обычно находят из рассмотрения последователь-
ности присоединения частиц к плотноупакованной грани (111)
[24]. Нами удельная краевая свободная энергия для грани (111)
кристалла алмаза была найдена исходя из физического смысла
свободной краевой энергии как избытка свободной энергии атомов
края по отношению к свободной энергии внутренних атомов по-
верхности. При этом принималось во внимание различие в геомет-
рическом положении атомов верхнего и нижнего слоев. В резуль-
тате были получены два значения удельной краевой свободной
энергии
еа = фа/З^ И Еа =-2 <pa/3d
соответственно для ступеней с нормалью к направлениям [211]
и [211]. Равновесная форма зародыша — правильный треугольник
со сторонами [НО], [011], [101] va = 1/12 удельная краевая
энергия —
ба = Фа/З^. (55)
Теперь для отношения работ образования критических зароды-
шей имеем выражение
AG* _ 2,5 In (р/реа)
AG* 1п(р/реЭ) ' '
28
Термодинамические расчеты показывают, что в интервале тем-
ператур от 1000 до 2000° К (т. е. практически до температуры спон-
танной графитации алмаза)
реа ~ 2ре?. (57)
Вводя обозначения In (plpe$l\n 2) = х, получаем вместо (54)
/« 0,8х Г 0,9(Фа/И7Ч1-0,бх)-
/0 ~ х-1 ехр| х(х-1)
(58)
Максимум этого выражения соответствует х — 2,7. При х ->
-»I IcJIp —> 0; при х —> оо /а//е 0,8. При х = 1,67 показатель.
Рис. 1. Зависимость отношения
скоростей зародышеобразования
алмаза и графита от пересыщения
экспоненты обращается в ноль и = 2. График функции (58)
приведен на рис. 1. Легко видеть, что наиболее благоприятной об-
ластью для эпитаксиального синтеза алмаза является область пе-
ресыщений, несколько превышающих значение plpe$ = 21-7, При
дальнейшем росте пересыщения роль поверхностных сил умень-
шается.
Попытаемся вычислить максимальный прирост затравочных
кристаллов в течение одного опыта. Обозначим через Va и ско-
рости образования однослойных пленок алмаза и графита на по-
верхности алмаза, через Sum соответственно полную поверхность,
и массу затравочных кристаллов, и пусть р — поверхностная плот-
ность образующихся пленок (одна и та же для алмаза и графита).
Тогда можно составить уравнения
dmldt = pI4Sa, (59),
dSJdt = — SaV^, (60)
где t — время, a Sa — чистая поверхность подложки. Из уравне-
ний (59) и (60) легко получить для абсолютного прироста алмаза за
время t
t
Ьта = ^pVaSadt = pS[l — ехр(— /Гр)] VjV^ (61)
/ о'
2S
или в относительных единицах
Дта __ pVxS [1 - exp (- /у0)] 1
т ~ V^m
Из уравнения (62) следует, что относительный прирост алмаза!
не может превышать некоторого значения
(Д/Ма/ш)тах — (63)
При больших пересыщениях можно предположить, что Va/73 =
=/а//р. Поскольку в этой области Za/Zp —> 1, то для порошка
с удельной поверхностью 5УД = 20 мг1г максимально возможный
привес составит (Ama/rn)max = 0,014, или Г,4%.
Следует иметь в виду, что приведенный расчет относится лишь 1
к зародышеобразованию на грани (111). Грани (НО) и (001) могут!
расти за счет образования нульмерных и одномерных зародышей. I
В тех случаях, когда число этих граней в затравочных кристаллах |
достаточно велико, прирост может быть несколько выше. 1
Полученный результат, несмотря на грубость сделанных допу-1
щений, удовлетворительно согласуется с экспериментальными дан-1
лыми, которые будут изложены в главе III. I
2. РОСТ ГРАФИТА ИЗ ГАЗОВОЙ ФАЗЫ
1. Физико-химическая теория роста графита
Рост графита из газообразных углеводородов изучался многими ис-
следователями. Обзор этих работ можно найти в монографиях
[25, 26]. Нет недостатка также в различных теориях, описывающих
этот практически важный процесс. Нами предложено рассматри-
вать образование и рост графита при термическом разложении
углеводородов с позиций теории нуклеации и поверхностных явле-
ний [27, 28]. Действительно, известно, что при росте графита по-
верхность частиц сажи ограняется базисными плоскостями графита
[25]. При кристаллизации из газовой фазы рост поверхностей низ-
ких индексов контролируется двухмерным зарождением моносло-
ев [5]. Поэтому примем, что самой медленной стадией является
образование двухмерных критических зародышей графита.
Основные предпосылки теории следующие: однородная поверх-
ность, адсорбция не является самой медленной стадией, справед-
ливость применения изотермы адсорбции типа уравнения Ланг-
мюра. Тогда рост новой фазы должен лимитироваться скоростью’
образования двухмерных зародышей, являющихся источниками
ступеней роста, движущихся до встречи с другой аналогичной
ступенью.
Такой подход к данному вопросу переводит химическую гете-
рогенную реакцию разложения метана в область физической хи-
мии, рассматривающей рост графита как образование зародышей
новой фазы из двухмерного адсорбированного газа. Тогда энергия
активации и порядок реакции становятся некоторыми фиктивными
величинами, зависящими от конечного и начального состояний;
системы, поскольку не учитываются промежуточные продукты
и состояния. Поэтому предлагаемый подход к росту графита из
углеводородов газовой фазы может быть назван физико-химиче-
ским.
Двухмерные критические зародыши графита могут образовы-
ваться как из двухмерного адсорбированного газа, так и в резуль-
тате взаимодействия падающих на поверхность молекул с докри-
тическим зародышем. В предположении независимости этих двух
процессов суммарная скорость нуклеации будет равна
v = t>! + v2; — б? ; v2 ~ (1)
3i
где 0Х — степень заполнения поверхности метаном; Д — поток
молекул метана из газовой фазы.
Скорость гетерогенного зародышеобразования может быть пред-
ставлена [5] на основе теории Фольмера — Баккера—Деринга—
Зельдовича как
I = Z мп*, (Г)
где Z — неравновесный фактор (фактор Зельдовича); со — частота,
•с которой отдельные атомы присоединяются к критическому за-
родышу;
п* = ns ехр (— Ag*ikT),
где ns — поверхностная концентрация адсорбированных частицу
Ag* — работа образования критического зародыша. Величина ns
связана со степенью заполнения поверхности соотношением
Ms -—
где п0 — поверхностная концентрация адсорбированных молекул,
образующих монослой. С учетом сделанных выше ограничений
2
(2)
где
Здесь р1е — равновесное давление газа; а — длина диффузион-
ного скачка; со — эйнштейновская частота; h — характерная дли-
на элементарной ячейки (/г3 — объем, приходящийся на один атом);
а0 — коэффициент аккомодации; т — масса молекулы; gx — энер-
гия активации поверхностной диффузии; q — теплота адсорбции;
Ь10 — константа, слабо зависящая от температуры в соотношении
для коэффициента адсорбции = Ь10 ехр (—qlkT).
Введем кажущуюся энергию активации
= — 2q + gt + &g*.
Тогда для скорости роста новой фазы, которая лимитируется ско-
ростью зародышеобразования, получим
п2
P1 = К1а "Ым ехр Ei/Rr>- (3)
Рост графита, описываемый формулой (3), вызывается образованием
критических зародышей вследствие поверхностной диффузии.
В случае бинарной газовой смеси, например метана и водорода,
вместо уравнения (3) легко получить
Р?
= К™ (1 + P1/bi + р2/&2)2 ехр E1/RT)’ . (4)
где ръ — парциальное давление водорода.
Если критические зародыши образуются прямым присоедине-
нием к докритическому зародышу молекул из газовой фазы, то
скорость роста
р?
Vi = г+^+Жехр (“ E2,RT)' (5)
где Е2 = — q + \g*.
В предположении независимости обоих процессов скорость рос-
та графита из чистого углеводорода (метана) будет равна
2 , 2
v = К1° (П7^ехР (- Ei/Rr> + ехр (~ EtlRT}-
(6)
Два следствия могут быть получены из уравнения (6). Во-пер-
вых, порядок реакции принципиально является переменным, изме-
няясь от второго к первому. Действительно, при 1
v (Кх + K2)pt
При 1
v аа. К2Ь2рг.
Здесь
Д1 = Kw ехр (—Ei/RT); К2 = ехр (—E2IRT).
Во-вторых, энергия активации является также переменной и за-
висит как от температуры, так и от парциального состава. Дей-
ствительно, работа образования дискообразного критического за-
родыша может быть записана [5] в виде
~ kT In (0t/0le)
в предположении, что энергия связи подложки с зародышем равна
энергии связи между слоями кристаллического вещества. Здесь е —
удельная краевая свободная энергия, не зависящая от температу-
ры, 01е — степень заполнения поверхности при равновесии. Для
двухкомпонентной смеси (метан и водород)
0! __ Р1 1 + Pje/bl + P2e/b2
®le — Pie 1 + Р1/61 + Рз/^2 ’ ' '
Для работы образования критического зародыша получим
а ♦ /Л\
= Г ^ + Р1е/Ь1 + Р2е/Ь2] ( )
kT [Ш (Р1/Р1е) + In i+pi/bl + ptibT\
Из уравнения (9) следует, что при определении пересыщения необ-
ходимо учитывать не только термодинамические свойства вещества
(удельную энтальпию, энтропию), но и характеристику поверхно-
2 Б. В, Дерягин, Д. В. Федосеев
33
ста. При этом следует учитывать также изменение равновесного
парциального давления реагирующего газа при изменении давления
и температуры. При росте графита из углеводородов разбавление
системы водородом всегда приводит к уменьшению пересыщения и,
следовательно, к увеличению работы образования критического
зародыша, что, в свою очередь, вызывает увеличение кажущейся
энергии активации. Однако влияние разбавления водородом угле-
водородов может по-разному сказываться на росте алмаза и гра-
фита, что зависит от характера поверхности.
Из выражения (6) следует, что скорость роста графита из двух-
мерного газа возрастает не беспредельно, а до определенного значе-
ния, равного K-Jh, тогда как скорость роста при прямом присоеди-
нении может возрастать безгранично (конечно, в отсутствие диф-
фузионного торможения).
В зависимости от величин констант, входящих в выражение (6),—
bY, b2, /<2, для разных газов можно ожидать различного соот-
ношения между скоростями и Несомненно также, что это со-
отношение определяет качество получающихся осадков углерода.
Когда скорость зародышеобразования мала, можно ожидать полу-
чения графита с хорошей структурой, тогда как при больших ско-
ростях роста ориентация плоскостей будет нарушаться. Когда
vi v2> базисные плоскости графита прорастают на большее
расстояние, и новый слой атомов углерода, образовавшийся на
них, наследует структуру предыдущих. Напротив, при vx
v2, любое нарушение структуры будет жизнеспособным, посколь-
ку оно может присоединять к себе новые атомы. Возможно, при рос-
те графитовых усов реализуется именно этот случай. Следует
учитывать, что скорость роста графита вдоль базисных плоскостей
во много раз превышает скорость роста в направлении, перпенди-
кулярном к ним. Графитовые усы (по крайней мере, высокопрочные)
имеют преимущественную ориентацию базисных плоскостей гра-
фита вдоль направления роста.
Рост графита по механизму прямого удара наблюдается при
выполнении условия
1 + Рх/^1 >
Чем меньше коэффициент адсорбции, тем при меньших давлениях
можно ожидать протекания реакции по первому порядку. Общий
вид зависимости скорости роста от давления реагирующего газа
приведен на рис. 2.
Сказанное выше относится к установившемуся процессу роста,
когда поверхность покрыта несколькими монослоями графита.
Разница в скоростях роста на различных поверхностях связана
не только с различной адсорбционной способностью, но и с разли-
чиями в величине поверхностной диффузии и работе образования
критического зародыша. Кроме того, для многих материалов, рас-
творяющих углерод или образующих карбиды, следует учитывать,
«сток» атомов углерода в глубь материала. Величина этого стока
34
будет уменьшаться со временем и лишь по истечении некоторого
времени начнут образовываться островки графита.
В случае углеродных материалов очевидно наименьшая скорость
роста будет именно на базисных плоскостях графита. Действитель-
но, известно, что на поверхности алмаза или окисленной предва-
рительно сажи скорость роста графита в начальный период много
больше установившейся. При достаточно высоких температурах
й малых линейных скоростях роста графита материал подложки
на скорость нуклеации. Однако при достаточно больших слоях на-
росшего графита (или алмаза) это воздействие будет ничтожным.
Переход от первого механизма роста :;>> v2) ко второму <<
ц2) для различных газов происходит при разных давлениях и
температурах. Этот переход в общем случае связан с концентра-
дией углеводорода, например, в смеси с водородом. При очень ма-
лых степенях заполнения более вероятен механизм роста прямым
ударом. При любом возможны условия, при которых присоеди-
нение прямым ударом к докритическому зародышу будет более ве-
роятным, чем присоединение из двухмерного адсорбированного
газа. Разумеется, состав адсорбированного двухмерного газа не мо-
жет быть тождествен газовой среде. В двухмерном газе возможны
самые различные наборы радикалов и углеводородов. В разной сте-
пени они будут способствовать росту новой фазы. Так, например,
подбором соответствующих условий можно наращивать алмаз
из различных углеводородов газовой фазы, стимулируя их пре-
вращения в необходимые радикалы или соединения в адсорбиро-
ванном состоянии. При этом не исключается возможность подавле-
ния роста той или иной формы углерода.
В области роста по механизму прямого удара на скорость про-
цесса возможно влияние температуры падающих на поверхность мо-
лекул углеводородов, поскольку, например, холодные молекулы
менее склонны взаимодействовать с подложкой. Активируя газо-
вую среду термически или электрически (в разряде), можно добить-
ся значительного изменения скорости роста.
2*
35
2. Экспериментальная проверка предложенной теории
роста графита
Несмотря на большое число работ по изучению роста графита из
различных углеводородов, провести последовательную проверку
сделанных выше расчетов затруднительно, поскольку эксперимен-
ты разных исследователей проводились часто в несопоставимых
условиях на различных подложках. Кроме того, в оригинальных
публикациях зачастую опускаются некоторые подробности экспе-
римента, совершенно необходимые при сопоставлении результатов
разных авторов. Поэтому в ИФХ АН СССР были поставлены работы
по изучению кинетики роста графита из метана при давлениях ниже
атмосферного и температурах до 1100° С. Эти же условия характер-
ны для эпитаксиального роста алмаза.
Эксперименты проводились на двух установках. Первая, в ос-
нове которой лежат кварцевые пружинные весы Мак-Бэна, исполь-
зовалась также при росте алмаза из газовой фазы и будет описана
в главе III. Общий вид и схема второй установки приведены на
рис. 3. Кварцевый реактор 1 в виде цилиндрической трубы длиной
— 600 мм и внутренним диаметром 60 мм нагревается печью со-
противления, питаемой от трансформатора со стабилизатором.
В реактор помещается кварцевая чашка 2 с известной навеской
сажи. Контроль температуры осуществляется с помощью потенцио-
метра 8 и хромель-алюмелевой термопары, помещенной в кварце-
вый чехол и вставленной в чашку с сажей. Вакуум создавался
с помощью вакуумного блока 7. Давление в системе измерялось,
по ртутному манометру 5 с помощью катетометра 6. Метан наби-
рался в баллон 4 и очищался от примесей многократным заморажи-
ванием в ловушке 3 при температуре кипения жидкого азота и
размор ажива нием.
Первая серия экспериментов осуществлялась при постоянном
давлении метана, постоянной температуре и переменном давлении
водорода. Метан конденсировался в ловушке при температуре ки-
пения жидкого азота. Давление насыщенных паров метана при этой
температуре 10 мм рт.ст. Реактор откачивался до давления 10“4 мм
рт. ст. Предварительно на саже осаждалось такое количество угле-
рода, чтобы все частицы были покрыты несколькими атомными
слоями. Без такой операции трудно получить воспроизводимые
результаты.
После нагрева до рабочей температуры метан напускался в ре-
актор и измерялось суммарное давление метана pt и выделившегося
при разложении метана водорода р2 в зависимости от времени, т. е.
Pi + Ръ — /(0- Тогда dfidt есть скорость выделения водорода.
Зная объем вакуумной системы и dfidt, можно определить скорость,
роста графита. Все другие возможные углеводороды, способные об-
разовываться при крекинге метана, имеют при температуре жидкого
азота значительно меньшее давление насыщенных паров, поэтому
их вклад в величину f(f) был ничтожным. После опыта чашки с на-
36
Рис. 3. Общий вид (а) и схема ус-
тановки ((F) для исследования роста
графита из метана
веской взвешивались. Количество выделившегося углерода, сле-
довательно, можно было измерять по количеству выделившегося во-
дорода, по израсходованному метану и взвешиванием. Во всех слу-
чаях отклонение в определенном количестве углерода не превышало
3—5% от веса вновь образовавшейся фазы.
Вторая серия экспериментов проводилась при постоянной тем-
пературе и переменном давлении метана и водорода. В реактор
напускался метан до определенного давления и измерялось изме-
нение общего давления в системе. Пересчет на образовавшийся
графит аналогичен предыдущему.
В предварительной серии опытов была измерена удельная по-
верхность сажи в зависимости от привеса новой фазы. В случае
равномерного покрытия сажевых частиц слоем углерода нетрудно
получить
4=(1 + тг)1'3’ (’°)
где So — удельная поверхность исходной навески т0; S — удель-
ная поверхность навески после наращивания Лт графита. На
рис. 4 приведено рассчитанное значение отношения So/S по форму-
ле (10) и измеренное по методу БЭТ. Отметим, что именно подобные
результаты легли в основу предложенного П. А. Теснером [29]
кинетического метода определения удельной поверхности. Варьи-
рованием исходной навески были определены границы кинетической
области процесса, в которой и проводились все эксперименты.
37
Из формулы (4) следует, что при постоянном давлении метана
Vvolv = 1 + Лр2, (11)
где п0 — скорость роста графита при нулевом давлении водорода;
А — ——61—
1^1 + Р1)
На рис. 5 приведена полученная экспериментально зависимость
'|Ли0/и от парциального давления водорода при 900° С. Как видно,
экспериментальные значения хорошо укладываются на прямую,
следующую из формулы (11). Аналогичная зависимость получена
при температурах от 800 до 1050° С; с изменением температуры ме-
няется лишь наклон прямых в координатах4рис. 5.
При постоянном давлении водорода из формулы (4) следует
1 =ЙК-. (12)
У» рх
где
В = ехр(^) = ехр(^Г)
На рис. 6 приведена зависимость 1/]/"v от обратного давления ме-
тана при постоянном парциальном давлении водорода 12 мм рт. ст.
и температуре 900° С. При других температурах получены анало-
гичные зависимости. При больших (свыше 20 мм рт. ст.) давлениях
метана наблюдается отклонение от указанной закономерности. Это
объясняется тем, что при повышении давления увеличивается ско-
рость реакции по механизму прямого удара.
На рис. 7 приведены данные в координатах Аррениуса, при
постоянном давлении метана (10 мм рт. ст.) и переменном давлении
водорода. При увеличении давления водорода наклон прямых уве-
личивается. На рис. 8 дана зависимость кажущейся энергии акти-
вации, найденной на рис. 7, от парциального давления водорода.
Из рисунка видно, что кажущаяся энергия активации изменяется
от 76 ккал!моль для чистого метана (Юлы/рт. ст.) до 98 ккал!моль
для давления водорода 10 мм рт. ст. (и том же парциальном
давлении метана). Величина 76 ккал/моль хорошо согласуется с оп-
ределенной в работе [30] энергией активации разложения метана,
равной 78 ккал/моль, рассчитанной экстраполяцией к нулевому дав-
лению метана. При разбавлении водородом энергия активации все
более приближается к энергии активации гомогенной реакции рас-
пада метана, равной 108 ккал/моль [31].
Хотя выше говорилось о разложении метана, нельзя исключать
разложения на поверхности сажи других соединений, образующих-
ся из метана в газовой фазе. Если навеску сажи взять меньше не-
которой критической, то начинает протекать реакция в объеме и
стенка реактора покрывается пироуглеродом. В этом случае, ра-
зумеется, нет соответствия между количеством графита, высадив-
шегося на саже, и рассчитанным по изменению общего давления
38
Рис. 4. Изменение удельной поверхности сажи при наращивании графита
Рис. 5. Зависимость скорости роста графита от давления водорода при постоянном пари
циальном давлении метана
Рис. 6. Зависимость скорости pocia
графита от давления метана при по-
стоянном парциальном давлении водо-
рода
Рис. 7. Зависимость скорости роста
графита от температуры
Рис. 8. Зависимость кажущейся энер-
гии активации роста графита от давле-
ния водорода
Z, /М27/7//%7/7^
в системе. Однако при навесках больше некоторой критической, весь
углерод выделяется только на саже, и стенка реактора остается
совершенно чистой. Это явление можно, вероятно, объяснить сле-
дующим образом.
Гетерогенная реакция разложения метана идет на всех поверх-
ностях. Так, в работе [30] показано, что при низких давлениях
метана (< 20 мм рт. ст.) скорости роста пироуглерода на саже и
на кварцевом волокне не различаются. Нами не было отмечено су-
щественного различия в скоростях роста пироуглерода (не алма-
за!) на алмазных порошках и саже. При развитой поверхности
затравочных кристаллов (например, частиц сажи) основная часть
углерода осаждается на ней. В наших экспериментах [27] общая
поверхность навески сажи превышала поверхность реактора в не-
сколько десятков раз. В то же время в объеме протекает гомогенная
реакция разложения метана, например, по механизму, предложен-
ному в работе [31], с образованием промежуточных соединений и
радикалов. Они также присутствуют на поверхности сажи [32],
которая для них является стоком. Если эта поверхность мала, то
происходит накопление промежуточных продуктов в газовой фазе
выше некоторой критической концентрации, и тогда развивается
быстрая реакция в объеме.
Таким образом, период индукции реакции гомогенного разло-
жения метана можно регулировать с помощью сажи с той или иной
общей поверхностью. Однако эта поверхность должна быть доста-
точно доступной, поэтому толщина слоя не может превышать
глубину эффективного проникновения реакции. Так, при 1000° С
глубина эффективного проникновения реакции в саже с удельной
поверхностью 10 зФ/г равна 1 мм.
Эксперименты по росту графита, описанные выше, позволяют
установить следующие значения констант, входящих в уравнение
роста графита из метано-водородных смесей в установившемся ре-
жиме:
bi = 3-104 exp (—25000/7?Г) мм рт. ст.,
Ь2 = 8-109 ехр (—58000/7??) мм рт. ст.,
Ki = 2,9-10'6 ехр (—28000/7??) ~ г/см2-сек-мм рт. ст.,
= 3-10-2 ехр (—58000/7??) ~г!см2-сек-мм рт. ст.
Эти значения позволяют сопоставить результаты расчета с извест-
ными экспериментальными данными. На рис. 9 приведены экспе-
риментальные данные, заимствованные из работы [30] для роста
графита из метано-водородных смесей при 900° С при постоянном
давлении метана (50 мм рт. ст.) и переменном давлении водорода,
и данные, полученные нами при постоянном давлении метана, рав-
ном 10 мм рт. ст.
На рис. 10 представлена зависимость скорости роста графита
в широком диапазоне давлений метана (от 10-1 до 104 мм рт. ст.)
40
Рис. 9. Зависимость скорости роста гра-
фита от парциального давления водорода
1 — [301; 2 — результаты авторов. Сплош-
ные кривые — расчетные
Рис. 10. Зависимость скорости роста гра-
фита от давления метана
/ — [30]; 2 — [33]; 3 — [341; 4 — [35];
5 — результаты авторов. / — 800; // —
900; III — 1000° С. Сплошные кривые —
расчетные
Рис. 11. Зависимость константы скорости
реакции от температуры (см. уравнение
(6))
Точки — эксперимент [26]; кривая — рас-
чет
для температур. 800, 900 и 1000° С по данным работ [30, 33—38]
и авторов.
При больших давлениях метана уравнение (6) вырождается в
v = Крг,
(13)
где /С = K2bi — константа скорости образования пироуглерода
с энергией активации 83 ккал!моль. Величина этой константы сопо-
ставлялась с экспериментальными данными (рис. 11), приводимыми
в монографии [26, стр. 95]. Отклонения от расчетной прямой при
больших температурах могут быть вызваны различными причинами:
диффузионным торможением (внутренним и внешним), реакциями
в объеме, недостаточно точным определением температуры и дав-
ления газовой смеси.
Найденные значения констант адсорбции могут характеризовать
лишь хемосорбцию. Поскольку
Ei = —2q + gi + Ag = 28 ккал/моль,
E2 = —<7 + Ag = 58 ккал!моль,
легко найти Ag = 83 ккал/моль и gr = —5 ккал/моль. Расчет по
макроскопическим (объемным) константам даст для работы образо-
вания критического зародыша величины от 50 до 80 ккал/моль
в зависимости от пересыщения. Значение Ag = 83 ккал/моль равно
энергии активации реакции по формуле (13), т. е. при больших
степенях заполнения поверхности энергия активации процесса
близка к работе образования критического зародыша.
В заключении раздела следует отметить, что при росте графита
в слое дисперсного порошка (для высокопористого катализатора)
проявляется эффективная глубина торможения реакции ее продук-
тами (водородом). Эта величина отлична от эффективной глубины
проникновения реакции, связанной с расходованием реагирующего
газа при диффузионной кинетике, но внешне описывается теми же
зависимостями.
3. Рост графита в иеизотермичных условиях
Эксперименты по росту углерода в неизотермичных условиях были
проведены на установке, общий вид которой показан на рис. 12.
Эта же установка использовалась для получения свободных угле-
родных пленок — графитовой фольги. Основной частью установки
является шаровой реактор, в котором на охлаждаемых токовводах
закрепляется пластинка из тугоплавкого металла: вольфрама, тан-
тала, молибдена. Верхняя часть реактора съемная. Вакуумирова-
ние обеспечивается вакуумным постом до давления 10-4 мм рт. ст.
Контроль температуры осуществляется оптическим микропиромет-
ром. Форма металлической подложки выбиралась такой, чтобы
имелся участок постоянной температуры площадью ~1 см2. Во
всех опытах использовался ацетилен. Во время эксперимента раз-
лагалось не более 5% исходного газа. Согласно работе [261 обра-
зующийся при этом водород не вносит искажений в кинетику роста
графита.
Известно [261, что при температурах выше 1000° С реакция рос-
та графита из ацетилена хорошо описывается уравнением первого
порядка по исходному газу с энергией активации 28 ккал!моль
в изотермичных условиях. В наших экспериментах температура
подложки изменялась от 1300 до 1650° С, тогда как газовая среда
имела приблизительно комнатную температуру, и стенки реактора
не нагревались выше 40° С.
На рис. 13 приведены рассчитанные константы скорости реакции
в неизотермичных (кривая 7) и изотермичных [26] (кривая 2) ус-
ловиях. Из рисунка видно, что энергия активации изменяется от
Рис. 12. Установка для получения гра-
фитовой фольги
Рис. 13. Зависимость константы скорости
реакции роста графита из ацетилена при
неизотермичиых (/) и изотермичных (2)
условиях процесса
80 до 40 ккал!моль при повышении температуры до 1650° С, прибли-
жаясь к константе скорости реакции в изотермичных условиях.
Полученные графитовые пленки исследовались морфологически
и методом электронной дифракции.
Морфологически пленки, полученные при низких температурах,
представляют собой пластинки с гладкой поверхностью, на которых
(при 1300° С) изредка встречаются отдельные полусферические
образования. При этом даже на относительно больших толщинах
эти пленки копируют полосатую структуру подложки (рис. 14, а).
Повышение температуры увеличивает число сферических образен
ваний, которые при 1500° С . принимают форму гексагонов (см.
43
Рис. 14. Морфология поверхности графитовой фольги при разных температурах (х 100)
а — 1300; б — 1500; в — 1650° С
рис. 14, б). При температуре выше 1600° С вся поверхность пленки
покрыта полусферическими выступами различных размеров и
форм (см. рис. 14, в).
Структурные исследования показали, что повышение темпера-
туры изменяет как размеры кристаллитов, образующих пленку,
так и текстуру. Если при 1300° С размер кристаллитов составляет
400 А, то повышение температуры до 1650° С уменьшает их размер
до 100 А. При температурах 1300—1400° С наблюдаются отдельные
монокристальные включения размером до 10 мкм, т. е. соизмери-
мых с толщиной пленки. Пленки, полученные при низких темпе-
ратурах, имеют текстуру с углом 20—30°, тогда как при высоких
температурах текстура отсутствует полностью и кристаллиты
расположены хаотично относительно друг друга. Эти условия,
возможно, соответствуют переходу к реакции прямым ударом. На
рис. 15 приведена одна из электронограмм от поверхности графито-
вой фольги.
Принципиально возможно получение монокристальной графи-
товой фольги. Очевидно, этого можно добиться подбором условий
синтеза, тщательной очисткой газа и термостатированием всего
реактора.
Несмотря на то что графитовая фольга имеет значительную проч-
ность на разрыв (до 50 кГ/мм2), при резком охлаждении в области
градиента температур она легко разрывается, как показано на
рис. 16. Вероятно, это вызывается накоплением термонапряжений
в пленке, которая разрывается лишь при определенной толщине.
4. Нитевидные кристаллы графита
Нитевидные кристаллы иногда называют материалом будущего,
имея в виду их уникальные свойства: высокую, близкую к теорети-
ческой, прочность, высокий модуль Юнга и т. д. Эти свойства ните-
44
Рис. 15. Электронограмма от [поверхно-
сти графитовой фольги
Рис. 16. Разрыв графитовой фольги в об-
ласти градиента температуры
видных кристаллов обусловлены их строением. В настоящее время
получены и изучены нитевидные кристаллы многих веществ [36],
и исследования в этой области интенсивно продолжаются.
Впервые нитевидные кристаллы графита получил Бэкон [37],
используя электрическую дугу. При пиролизе различных газов их
наблюдали также авторы работ [38, 39].
Нами нитевидные кристаллы графита были получены при ис-
пользовании лучистого нагрева различного типа подложек. В ка-
честве углеродсодержащих газов применялись углеводороды.
Наилучшие результаты получены с ацетиленом [40].
Источником излучения служили установка радиационного на-
грева УРАН-1 и газоразрядный лазер ЛГ-25. Методика работы
45
с установкой УРАН-1 описана в главе IV. Здесь остановимся на опи-
сании установки с лазерным нагревом.
Схема установки для роста нитевидных кристаллов графита на
основе инфракрасного лазера ЛГ-25 с длиной волны 10,6 мкм
представлена на рис. 17. Установка состоит из блока питания 7,
лазера 2, реактора 4, укрепленного на трехкоординатном столике,
и вакуумной системы. Входные и выходные отверстия в реакторе
изготовлены из хлористого натрия. Прошедший пучок излучения
улавливается ловушкой 6. После вакуумирования реактор напол-
няется исследуемым газом, который разлагается только на подлож-
ке 5, оставаясь при этом холодным. Линза из хлористого натрия 3
позволяла фокусировать световой пучок до размера 200 мкм, что
обеспечивало получение на графите температур, до 3000° С. Исполь-
зование лазерного излучения с длиной волны 10,6 мкм (инфракрас-
ная область) имеет то преимущество перед нагревом с помощью
мощной ксеноновой лампы, что исключает постороннюю засветку и
позволяет проводить непрерывное пирометрирование образца.
Нитевидные кристаллы росли в стационарном режиме, а также
при импульсном периодическом режиме, впервые примененном
нами для наращивания алмазных пленок и выращивания нитевид-
ных кристаллов алмаза. Периодическое импульсное пересыщение
создавалось перекрытием светового пучка. При использовании
установки радиационного нагрева УРАН-1 импульсы создавались
при вращении диска с прорезями.
В зависимости от давления ацетилена и температуры могут
быть получены различные нитевидные кристаллы графита. Однако
температура подложки не должна быть ниже 900° С. На рост гра-
фитовых усов влияет как природа и состояние подложки (обычно
металлы: вольфрам, тантал, титан, рений и др.), так и условия обте-
кания ее потоком газа вследствие естественной конвекции. На ме-
таллических подложках, неоднократно использованных в опытах,
нитевидные кристаллы растут реже, нежели на свежих. Особенно
часто растут такие кристаллы на срезах металла, а также на не-
однородностях поверхности. Если на поверхность металла нанести
перед опытом царапину, то вдоль нее вырастут нитевидные кристал-
лы, как бы декорируя эту царапину. Когда на поверхности молибде-
на был осажден вольфрам с различной ориентацией, то наибольшее
число нитевидных кристаллов графита выросло на поверхности
с ориентацией <100>.
Для исследования в электронном микроскопе графитовые усы
выращивались на молибденовых шайбочках, которые затем вкла-
дывались непосредственно bl объектодержатель электронного мик-
роскопа *.
На рис. 18 приведены фотографии нитевидных кристаллов гра-
фита. Они могут быть как прямыми, так и изогнутыми. Интересно
* Электронномикроскопические исследования проведены А. Е. Городецким и
А. П. Захаровым.
46
Рис. 17. Схема установки для роста нитевидных кристаллов графита
Рис. 18. Нитевидные кристаллы графита (х 10 000)
а — прямые; б — изогнутые
Рис. 19. Полые кристаллы графита (х 10 000)
а — снимок в светлом поле; б — снимок в темном поле
отметить, что при определенных условиях получаются графитовые
усы с периодической структурой, полые внутри, как показано на
рис. 19. Очевидно, их структура связана со способом получения.
Электронная микродифракция показала, что, несмотря на ис-
пользование металлической подложки, в составе усов не обнаруже-
ны примеси металлов или соответствующих им карбидов. Дифрак-
ционные исследования показали, что, хотя имеется значительная
разориентировка кристаллитов, базисные плоскости графита вы-
тянуты вдоль направления роста графитовых усов, что объясняет
их высокую прочность на разрыв.
Значительный интерес представляют наращивание нитевидных
кристаллов (усов или вискеров) на поверхности непрерывных угле-
родных волокон. Этот процесс предназначен для улучшения адге-
зии углеродных непрерывных волокон в композиционных материа-
лах [41] и носит название «вискеризация»*. Основная трудность
вискеризации заключается в необходимости роста именно нитевид-
ных кристаллов, а не покрытии волокна слоем пироуглерода. Оп-
тические методы нагрева в связи с этим обладают определенными
преимуществами. На рис. 20 и 21 показаны вискеризованные волок-
на. Диаметр исходного волокна равен 6 мкм. Дифракция электро-
нов позволила установить, что, наряду с графитом, на углеродных
волокнах выделяется карбин [42], а также неидентифицированные
углеродные фазы. В табл. 1 приведены результаты расчета некото-
рых электронограмм.
Таблица 1
Расчет алектрочограммы углеродных усов (d, А)
Эксперимент Идентификация электроно- граммы Эксперимент Идентификация электроно- граммы
графит карбин графит карбин
4,55 4,46 2,47 2,46
3,90 — — 2,13 2,12 —
3,40 3,38 — 1,58 1,541 1,62
2,83 — 2,81 1,47 — 1,48
2,62 — 2,56 1,23 1,23 , —
Прочность на разрыв нитевидных кристаллов графита опреде-
лялась на сконструированной простой установке, позволяющей по-
степенно повышать приложенную нагрузку. Нитевидные кристал-
лы крепились цеокриновым клеем к более толстым волокнам, кото-
рые в свою очередь приклеивались к кварцевым нитям. Результаты
измерения прочности графитовых усов приведены на рис. 22. Как
* Редакторы перевода [41] предлагают термин «ощетинивание».
48
Рис. 20. Вискеризованное углеродное волокно (X 20 000)
Рис. 21. Вискеризованное углеродное волокно (X 5000)
Синмок в растровом микроскопе
видно из рисунка, характер зависимости прочности графитовых
усов на разрыв аналогичен всем нитевидным кристаллам [36].
Как уже указывалось, при росте графитовых усов необходимо'
подавить образование сплошного слоя графита. Принципиально
этого можно добиться, создав отдельные активные места на поверх-
ности и запассивировав ее остальную часть. Именно поэтому гра-
фитовые усы успешно растут под каплями металлов, хорошо смачи-
вающих графит. Активировать участок можно также, если нагреть
его до более высокой температуры, чем окружающую поверхность,
и обеспечить при этом большой поток углеродных атомов к нему.
49
Таким образом, для роста нитевидных кристаллов необходимо опре-
деленное сочетание кинетических процессов и состояния поверх-
ности.
Для приведенного выше способа получения графитовых усов
характерно большое различие в температурах подложки и углерод-
содержащего газа. Поэтому рассмотрим влияние неизотермичности
условий и неоднородности поверхности. Согласно изложенным
в разделе 4 соображениям, рост графита описывается уравнением
V = К10? + КА/, (14)
где — степень заполнения поверхности; / = б/1р1/(2лт/гТ)1^ —
лоток углеродсодержащего газа; /G и /(г — константы.
Рис. 22. Зависимость прочности на
разрыв нитевидных кристаллов гра-
фита от диаметра
Пусть температура подложки равна 7\, а газа — Т2. Аналогич-
но выводу уравнения изотермы адсорбции Лангмюра приравняем
в стационарном состоянии число десорбирующихся молекул числу
молекул, адсорбирующихся в единицу времени. Легко получить
@1 = Р1 [ZViexp(-g/^;H2^^Ta)^ + = , (j 5)
где q — теплота адсорбции; ах — коэффициент конденсации, зави-
сящий от температур газа и подложки. Если принять, что коэффи-
циент конденсации не зависит от температуры, то степень заполне-
ния в неизотермичных условиях тем меньше, чем ниже температура
газа (при постоянстве температуры подложки). Чем меньше коэф-
фициент конденсации, тем меньше степень заполнения поверхности.
Таким образом, неизотермичность условий будет сказываться на
обоих членах уравнения (14).
При неоднородной поверхности на степень заполнения влияют
как различие в величинах коэффициента конденсации, так и раз-
личие в теплотах адсорбции. Поэтому в уравнении адсорбции типа
уравнения Лангмюра для разных участков величины b неодинаковы.
Поверхность реальных тел неоднородна, поэтому различные
его участки имеют разные значения константы адсорбции. Рассмот-
рим неоднородную поверхность, состоящую из активных участков
50
с величиной bit разделенных участками с величиной Ь, причем bi
В этом случае частота образования критических зародышей
на активных участках Ц будет много больше частоты образования
критических зародышей на остальной поверхности.
При более полном рассмотрении необходимо учесть различие
в работах образования критического зародыша на разных участках
поверхности. Возможно, что присоединение в базисной плоскости
графита может идти без образования критического зародыша, тогда
как на базисной плоскости графита его образование необходимо.
Вероятно именно этим объясняется наблюдаемый в работе [35]
результат, когда скорость роста вдоль и перпендикулярно базис-
ным плоскостям графита различалась в 103 раз.
3. КИНЕТИКА
ФИЗИКО-ХИМИЧЕСКОГО СИНТЕЗА
АЛМАЗА
I. Из истории синтеза алмаза
История изучения алмаза, по-видимому, начинается с И. Ньютона,
который в 1675 г., основываясь на измёрениях преломления света,
алмазом и горючими жидкостями, сделал вывод, что алмаз должен
гореть. Этот вывод впервые экспериментально подтвердил Р. Бойль,
установивший, что алмаз изменяется от действия огня. В 1694 г.
во Флорентийской Академии были проведены публичные опыты по
сжиганию алмаза в фокусе большого увеличительного стекла.
В 1774 г. Лавуазье, исследуя продукты сгорания алмаза, пока-
зал, что известковая вода мутнеет при пропускании через нее га-!
зов сгорания — признак образования углекислого газа. В 1797 г
английский химик С. Теннант сжег алмаз в закрытом золотом со
суде, наполненном кислородом, и установил, что образуется угле
кислый газ, количество углерода в котором в точности соответст-
вовало весу сгоревшего алмаза. В 1799 г. во Франции Г. Гитон
и Ф. Клуе пришли к выводу, что алмаз — чистый углерод. Этот
вывод они подтвердили, получив сильным нагреванием чистого
железа с алмазом превосходную сталь. В том же году Гитон уста-
новил, что графит также является углеродом. i
В 1774 г. известным минералогом того времени А. М. Карамы- I
шевым в Петербургском Горном училище также были проведены
опыты по сжиганию алмазов. Уже в XIX в. процесс горения алмаза ,
исследовали Г. Дэви и М. Фарадей. В этот же период начались
опыты с целью получения искусственного алмаза.
В 1880 г. английский ученый Дж. Хэнней опубликовал сооб-
щение о получении искусственных алмазов. Он нагревал до красно-
го каления заклепанные трубы типа орудийных стволов, в которые
была помещена смесь углеводородов, растительного масла и метал- «
лического лития. Двенадцать из полученных им кристалликов хра-
нятся в Британском музее.
В 1893 г. французский химик А. Муассан провел исследования,
которые заключались в следующем. Насыщенное углеродом желе-
зо при температуре до 3000° С выливалось в ледяную воду. В ре-
зультате образования застывшей корки внутри охлаждающейся
массы получалось высокое давление. Растворив в кислотах остыв-
ший слиток, Муассан обнаружил несколько крупинок, не взаимо-
действующих с кислотами и царапающих рубин. Их плотность была
между 3 и 3,5 г/см\ и при сгорании они образовывали газ.
52
В этом же году профессор минералогии Петербургской медицин-
ской академии К- Д. Хрущев, независимо от А. Муассана, полу-
чил при кристаллизации углерода в расплавленном серебре про-
зрачные и темные кристаллики. Они царапали самый твердый после
алмаза минерал — корунд и сгорали, образуя углекислый газ.
Все приведенное выше относится к попыткам синтезировать ал-
маз из растворов углерода. Вероятно, первая попытка исполь-
зовать углеродсодержащие газы была предпринята в 1911 г.
В. Боултоном, который пытался наращивать монокристаллы ал-
маза из светильного газа [43].
Уже в начале XX в. появились сомнения: действительно ли
Б опытах Муассана, Хрущева и других были получены алмазы?
В 1943 г. английские физики Ф. Банистер и К. Лонсдейл с
. помощью рентгеновских лучей произвели проверку всех сохранив-
шихся образцов. Они обнаружили, что только алмазы, полученные
Хеннеем, обладают решеткой алмаза и, следовательно, являются
подлинными. В то же время все попытки воспроизвести результаты
Хеннея не увенчались успехом.
Попытки теоретически определить условия для перевода графи-
та в алмаз предпринимались многократно. Однако наиболее ус-
пешно с этой задачей удалось справиться О. И. Лейпунскому
[44], который показал, что все поиски синтетических алмазов про-
изводились в тех условиях, при которых графит является термоди-
намически более устойчивой формой углерода, чем алмаза. Для
того чтобы можно было перевести графит в алмаз, необходимо, во-
первых, сжать его давлением до 60 000 или более атмосфер, во-вто-
, рых, нагреть графит до высокой температуры (~ 2000° С). В этих
условиях, по крайней мере, часть графита перейдет в алмаз.
Для реализации идеи получения синтетических алмазов иссле-
дователям пришлось преодолеть значительные технические трудно-
сти, связанные с получением в заданном объеме одновременно вы-
соких давлений и температур. Это потребовало использования осо-
бых конструкционных материалов и многочисленных инженерных
усовершенствований. Впервые искусственные «рукотворные» ал-
мазы были получены в США в 1955 г. группой ученых: Ф. Банди,
Г. Холл, Г. Стронг и Р. Венторф. Затем синтетические алмазы были
получены в Швеции, Великобритании и других странах.
В Советском Союзе искусственные алмазы были получены кол-
.лективом сотрудников Академии наук СССР под руководством
академика Л. Ф. Верещагина. Синтезированные в Институте высо-
ких давлений АН СССР поликристаллические материалы баллас
и карбонадо во многих отношениях не уступают природным. Зна-
чительную роль в разработке методов синтеза алмазов и в разви-
тии промышленности синтетических алмазов сыграл коллектив
Института сверхтвердых материалов Академии наук Украинской
ССР, руководимый В. И. Бакулем.
Одинаковый состав графита и алмаза привел к естественным по-
пыткам превратить графит в алмаз применением высоких давлений,
53
которые, конечно, должны способствовать более плотному располо-
жению атомов, имеющемуся у алмаза. Для того, однако, чтобы пе-
регруппировка атомов из решетки графита в решетку алмаза могла
совершиться, необходима достаточная подвижность атомов. Это
достигается подъемом температуры. В настоящее время для превра-
щения графита в алмаз применяют давления порядка 100 000 атм
и температуры порядка 1600—2000° С. Впрочем, даже при этих
температурах перестройка решетки идет настолько медленно, что-
для ее осуществления добавляют растворители углерода — метал-
лы группы железа.
Из термодинамики находят, отвлекаясь от вопроса о скорости
перехода, весьма точные указания относительно температур и дав-
лений, при каких графит способен переходить в алмаз, при каких —
не способен. При каждой фиксированной температуре при давлении
выше равновесного графит неизбежно перейдет в алмаз, тогда как
при давлении ниже равновесного алмаз является «метастабильной»
модификацией углерода, и переход графита в алмаз невозможен-
Положение, однако, меняется, если поставить вопрос о возмож-
ности роста уже имеющегося кристалла алмаза. Рост алмаза мо-
жет происходить при давлениях и температурах, соответствующих
области его метастабильности, если тем или иным способом создать
вокруг него среду, содержащую атомы углерода в достаточной
концентрации. Рост алмаза в этом случае можно объяснить тем, что
концентрация атомов углерода, принесенных к поверхности, ока-
зывается выше, чем концентрация атомов, с которой алмаз может
находиться при определенной температуре в равновесии, т. е. выше
концентрации насыщения.Такие условия можно создать, если,
поместить алмаз в среду атомов углерода, например испаряемых,
нагретым до высокой температуры графитом, или в раствор углеро-
да в расплавленных металлах.
Этого условия — условия создания вокруг алмаза пересыщенной
концентрации атомов углерода — можно добиться и иначе, не при-
бегая к графиту как источнику атомов углерода, а используя какой-
либо углеродсодержащий газ. Если его молекулы способны разла-
гаться при температуре алмаза, выделяя атомы углерода, то при
достаточной плотности газа концентрация образующихся атомов,
углерода может превысить концентрацию насыщения. При этих,
условиях кристалл алмаза может расти. Этот способ по сравнению
со способом, в котором источником атомов углерода служит графит,
выгоднее в том отношении, что не требует поддержания зоны с тем-
пературой, фактически намного выше температуры алмаза, так как
пересыщение атомов углерода достигается за счет концентрации
углеродсодержащего газа и скорости теплового распада его моле-
кул.
Основанный на этой идее метод укрупнения алмазных кристал-
лов был предложен Б. В. Дерягиным и Б. В. Спицыным в 1956 г.
и независимо описан в патенте США В. Эверсолом с приоритетом,
от 1958 г.
54
В первом способе рост граней алмаза осуществлялся в атмосфе-
ре четырехиодистого углерода при 1000° С при его распаде по
реакции
С + 2J2.
алмаз
В. Эверсол [45] заявил процесс наращивания затравочных кри-
сталлов алмаза в потоке окиси углерода или углеродсодержащих
газов, способных при диссоциации образовывать метильные радика-
лы. В патенте указан интервал температур 600—1600° С; в то же
время рекомендованный оптимальный интервал — от 900 до 1100° С.
Парциальное давление углеродсодержащих газов, включающих
метильные группы, по патенту должно быть меньше 75 мм рт. ст.,
причем наилучшие результаты были получены в пределах
0,1 — 1 мм рт. ст. При использовании окиси углерода или смеси оки-
слов углерода давление может достигать 2500 атм. Предпочтитель-
ными являются давления около 100 атм.
В связи с тем, что в ходе опытов, наряду с алмазом, на затравоч-
ных кристаллах откладывается и неалмазный углерод, процесс
синтеза носил периодический характер: после периода наращива-
ния всегда следовал период очистки, стравливания неалмазного
углерода и новый период наращивания и т. д. Наращивание про-
должалось 3—4 часа, очистка проводилась в водороде по реакции
С +2Н2^СН4
графит
при давлении 10—50 атм и температуре 1000° С в течение 14 час.
Этот способ очистки основан на разной кинетической активности
алмаза и графита к взаимодействию с водородом.
В. Эверсол использовал в основном порошок с размером частиц
.до 1 мкм и наращивал до 60% новой фазы. Дифракцией электронов
было доказано, что структура наращенной новой фазы соответст-
вует алмазу. Плотность алмаза наращенного на 21%, определенная
методом флотации, оказалась равной 3,48 г!смя, тогда как исходные
.кристаллы, очищенные прокаливанием в водороде, имели плот-
ность 3,49 г!смя, т. е. практически ту же. Содержание углерода
в наращенном алмазе составляло 99,4 + 0,40% против 99,6 +
дЬ 0,40% в исходном алмазе.
Наибольшее увеличение веса исходного порошка было 59,50%.
В этом случае проведено 85 последовательных циклов наращивания
<по 1,8 часа) и очистки (по 4,5 часа). Обработка результатов экс-
периментов Эверсола показывает, что чем меньше продолжитель-
ность цикла наращивания, тем выше его скорость. Изменение
времени наращивания от 4 до 16 час. не увеличивало общего при-
веса алмаза, но средняя скорость наращивания, количество нара-
щенного нового алмаза в единицу времени, естественно, уменьша-
лось, что указывало на то, что поверхность растущего алмаза по
истечении определенного времени полностью блокируется графитом.
55
В 1964 г. в США Г. Гибшманом был подан патент на способ вы-
ращивания алмаза на алмазных затравках из газа, содержащего
в основном окись углерода, при температуре 600—1100°С и дав-
лении до 1200 атм. Необходимым условием по патенту является
присутствие по меньшей мере одного из благородных металлов.
По своему содержанию патент Гибшмана является во многом
повторением патента Эверсола, но существенно отличается тем, что
применение катализаторов позволило значительно увеличить ско-
рость выделения углерода на затравочных кристаллах алмаза,,
в качестве которых использовался порошок микронных размеров
[46].
В 1968 г. Дж. Ангус, Г. Уилл и В. Станко [47] опубликовали
статью, посвященную воспроизведению и проверке патента Эвер-
сола. Наращенные алмазные порошки были исследованы посред-
ством измерения плотности, спектрального, рентгеновского, элек-
тронографического анализов, исследований микроволнового погло-
щения. Все эти методы современного анализа показали полную
идентичность наращенного и исходного алмазов. В 1971 г. на Меж-
дународном конгрессе по синтетическим алмазам в Киеве Дж. Ан-
гус докладывал о наращивании полупроводникового алмаза на
затравочных алмазных порошках. Полупроводниковый алмаз по-
лучался за счет легирования бором алмаза, растущего из метана
[48].
Западногерманская фирма «Сименс и Гальске» получила во Фран-
ции патент на «Метод получения алмазоподобного углерода при
низких давлениях» 149]. Способ заключается во введении в горя-
чую реакционную зону соединений углерода, имеющих структуру,
полностью или частично соответствующую строению алмазной
решетки. В качестве исходного продукта используется, в частно-
сти, циклопентан. Его кольцо с пятью атомами углерода составле-
но почти без напряжений. Кроме циклопентана и циклогексана, при-
годны другие соединения углерода, разлагающиеся при высоких
температурах на составляющие, в которых цепочки атомов углеро-
да соответствуют решетке алмаза. Для ввода исходного вещества
в реакционную зону использовался газ-носитель: инертные газы,
водород или газообразные при комнатной температуре углеводоро-
ды. Для облегчения образования зародышей алмазоподобной
формы углерода в зону реакции добавляют «катализаторы»—
вещества, способные в условиях реакции образовывать карби-
ды кубической структуры. Это — кремний, титан, элементы
V группы периодической таблицы (преимущественно ниобий
и тантал), а также сплав вольфрама и кобальта. Катализатор
вводят в зону реакции либо в виде подложки для получения кри-
сталлических слоев, либо в виде металлсодержащих летучих соеди-
нений для получения порошкообразного углерода, имеющего ал-
мазоподобную структуру. Предпочтительная температура реакции
800—1200° С. Для активации реакционной смеси применяют раз-
личные формы электрического разряда.
56
В другой заявке этой же фирмы [50] патентуются всевозможные
способы синтеза алмаза при помощи химических транспортных
реакций. Для образования алмаза необходимо не слишком большое
удаление от равновесной концентрации углеродсодержащих газов
над алмазом во избежание паразитного выделения неалмазного
углерода. Отметим, что химические транспортные реакции в настоя-
щее время широко применяются для получения чистых и сверхчи-
стых веществ. Общая теория химических транспортных реакций на
основе термодинамики неравновесных процессов была дана в рабо-
те [51].
В патенте Г. Бринкмана и соавторов [52] заявлен метод эпитакси-
ального наращивания алмаза из растворов углерода в металлах,
причем источником углерода служит графит, находящийся при более
высокой температуре, чем затравочный кристалл алмаза. Метод
может быть реализован в двух видах: контурном и бункерном.
При контурном методе жидкий металл постоянно циркулирует по
замкнутому контуру и атомы углерода переносятся от графита
к алмазу. Скорость циркуляции выбирается в зависимости от раз-
ности температур источника углерода и алмаза. В бункерном методе
жидкий металл насыщается углеродом от стенок графитового тигля,
затем температура понижается на величину, определяемую задан-
ным пересыщением по отношению к алмазу, и затравочный кристалл
вводится в расплав. Углерод выделяется на затравке преимущест-
венно в форме алмаза. Когда пересыщение снимается, цикл повто-
ряется снова. Этот способ не представляется перспективным не
только из-за его цикличности и инертности системы, но и по другой
причине. Действительно, выше указывалось, что равновесная кон-
центрация над алмазом вдвое превышает соответствующую концен-
трацию над графитом. Значит, еще во время охлаждения расплава
графит выделяется на стенках камеры (выполненных из графита),
и к тому времени, когда в расплав вводится затравочный кристалл
алмаза, пересыщение в значительной степени оказывается снятым.
В патенте тех же авторов [53] заявляется способ получения алма-
за из атомов углерода, испускаемых источником углерода при вы-
сокой температуре (до 4000° С). Сообщений о подобных методах
синтеза из паров углерода известно довольно много.
Проблемы синтеза алмаза тесно связаны с пониманием процесса
его образования в природе. Сейчас определенной точки зрения на
этот вопрос нет, хотя предположений имеется достаточно 154].
Интересную идею о влиянии кавитации на образование и
рост алмазов развивает Э. М. Галимов. Вопросы образования ал-
мазов в метеоритах рассмотрены в книге Г. П. Вдовыкина [55].
Однако природа настолько щедра на различные условия, что впол-
не возможно, чтобы все предложенные методы' реализовывались
либо одновременно, либо поочередно. Исключение, вероятно, пред-
ставляет рост алмаза из продуктов разряда между графитовыми
электродами при низких давлениях благородных газов и последующей
конденсации этих продуктов на охлажденной до 80° К подложке [56].
57
2. Кинетика роста алмаза на алмазных порошках
Исследование кинетики роста алмаза проводилось на высоко-
дисперсных классифицированных алмазных порошках [57]. Вы-
сокоразвитая поверхность последних делала возможным исполь-
зование обычной методики, основанной на применении весов
Мак-Бена. Использовались природные и синтетические алмазные
порошки. Их удельная поверхность определялась методами БЭТ
и фильтрации разреженного газа.
Основная трудность в исследовании кинетики роста алмаза
из газовой фазы заключается в параллельном выделении неалмаз-
ного углерода, что затрудняет трактовку результатов эксперимен-
тов. Поэтому в кинетических исследованиях приходится исполь-
зовать лишь часть кривой вес—время.
Наращивание высокодисперсных алмазных порошков имеет и
самостоятельное практическое значение, поскольку они широко
используются в качестве абразивов для финишных и суперфиниш-
ных операций при обработке различных поверхностей [58].
Изучение эпитаксиального наращивания алмазных порошков:
проводилось на различных установках, схема одной из которых
показана на рис. 23. Основная часть установки — кварцевый реак-
тор 1 и пружинные кварцевые весы 2. В реактор вводилась на тон-
кой кварцевой нити кварцевая чашка с навеской алмазного порош-
ка, изменение массы которого фиксировалось при помощи кате-
тометра 3.
Реактор нагревался печью омического нагрева 4. Температура
реактора, поддерживаемая с точностью+10°, измерялась хромель-
алюмелевой термопарой 5, помещенной в «палец» реактора. Ваку-
ум в системе создавался ртутным диффузионным и форвакуумным
насосами. Давление в реакционной зоне, поддерживаемое с точ-
ностью +0,01 мм рт. ст., измерялось термопарной лампой 6 и
манометром Мак-Леода 7.
Для наращивания алмаза использовали метан. Перед опытом
часть его из баллона 8 отбиралась в охлаждаемую жидким азотом
ловушку 9, откуда поток метана через систему кранов поступал
в реактор. Это имело то преимущество, что давление метана в ло-
вушке оставалось постоянным и равным 10 мм рт. ст. В качестве
затравки использовались алмазные порошки марок AM 1/0 (при-
родный) и АСМ 1/0 (синтетический) с известной удельной поверх-
ностью и размерами частиц до 1 мкм.
В результате экспериментов не обнаружено различия между
удельными скоростями наращивания на синтетическом и природ-
ном алмазах в сопоставимых условиях.
На рис. 24 показано увеличение начальной массы навески
30 мг при наращивании из метана, давлении 0,1 мм рт. ст. и тем-
пературе 1050° С. Из рисунка видно, что с течением времени ско-
рость роста уменьшается, что вызвано блокированием поверхности
58
алмаза неалмазным углеродом *, который, как было обнаружено
экспериментально, растет медленнее, чем алмаз. По окончании
опыта графит удаляется. После очистки от графита зафиксировано
увеличение массы исходной затравки на 2,7%. Идентичность на-
росшего алмаза исходному доказана различными способами (рент-
геновской и электронной дифракциями, измерением плотности и
другими методами).
Предварительными экспериментами была установлена кинети-
ческая область процесса эпитаксиального синтеза. Для определе-
ния уравнения кинетики проведены эксперименты в интервале тем-
ператур 850—930° С при давлении 0,13—0,14 мм рт. ст. метана.
В диапазоне давлений 10“2—0,5 мм рт. ст. порядок реакции ока-
зался близок к первому. В результате этих экспериментов получе-
но кинетическое уравнение
va = 3,5-1011 ехр(—80000/7? 7')с0 г/см2-сек, (1)
где с0 — концентрация метана, г/см3. Это уравнение (несомненно
приближенное) положено в основу всего дальнейшего анализа.
Перевод процесса в диффузионную область дает значительное уве-
личение скорости роста, что имеет не только научный интерес.
В то же время удовлетворительное совпадение расчетов для диффу-
зионной области с экспериментальными результатами явится про-
веркой основного уравнения (1).
При наращивании микронного порошка в изучаемой области
давлений внутри пор имеет место режим кнудсеновской диффузии.
В этом случае эффективный коэффициент диффузии может быть
записан [59] как
п, _ 24 / 2 \1,2 (RT)'i2
тде Т — абсолютная температура; So — удельная поверхность;
7? — газовая постоянная; М — молекулярная масса; б — «пори-
стость», равная отношению объема пор ко всему объему тела. Если
р — плотность алмаза, ad — насыпная масса алмазного порошка,
до
(2)
6 = 1 — d/p.
Так как для алмаза р = 3,5 г!см3 и для микронных алмазных по-
рошков d = 1,25 г/см3, то
D'm 5-Ю3^^1'
оо
Анализ процесса наращивания в слое порошка удобно вести,
заменяя коэффициент диффузии эффективным коэффициентом диф-
(2')
Выделяющийся при эпитаксиальном синтезе черный неалмазный углерод
будем называть графитом.
59
Рис. 23. Схема установки для исследования кинетики роста алмаза
фузии, а константу скорости гетерогенной реакции — эквивалент-
ной ей константой гомогенной реакции в слое порошка: К' =
= KS (S — поверхность, приходящаяся на единицу объема по-
рошка). Такой метод, предложенный Я- Б. Зельдовичем, широко
применяется в процессах гетерогенного катализа в слоях высоко-
дисперсных катализаторов [60]. В данной работе стефановский
поток в слое порошка не учитывается, поскольку число Кнудсена
Рис. 24. Изменение веса затравочного алмазного порошка при наращивании алмаза
из метана
Восходящая часть кривой соответствует наращиванию, нисходящая — очистке от гра-
фита
Рис. 25. Зависимость эффективной глубины проникновения реакции синтеза алмаза
от температуры для порошка марки АСМ 1/0
60
значительно больше единицы, т. е. молекулы газа практически не-
будут сталкиваться друг с другом.
Рассмотрим уравнение диффузии для реакции первого порядка-
р., d2c ,,,
D^ = Kc
при граничных условиях
с = с0 при х = 0, dc]dx = 0 при х = I,
т. е. решается одномерная задача (не учитываются краевые эффек-
ты). Решение легко получить в виде
с ch(//L — х/£)
~о ch (//£) ’ '
где L = (D’/КУ!г — эффективная глубина проникновения реак-
ции в слой пористого материала. Количество образовавшегося
алмаза равно количеству вещества, продиффундировавшего через?
внешнюю поверхность слоя:
dmldt = (K'D'pC() th (Z/L). (4>
Отметим, что в случае синтеза алмаза из газа принципиально-
не может быть стационарного решения, т. е. в крайнем случае мож-
но использовать квазистационарное рассмотрение в отличие от
процессов катализа и кристаллизации стабильной фазы. Причина —
параллельное выделение неалмазного углерода, блокирующего по-
верхность. Это приводит к тому, что, поскольку удельная скорость,
кристаллизации алмаза из газа выше, чем скорость образования
графита, эффективная глубина проникновения реакции возрастает
со временем, захватывая новые и новые слои алмазного порошка.
Кроме того, выделяющийся при реакции разложения метана водо-
род тормозит выделение неалмазного углерода в глубинных слоях
порошка, и вполне возможно, что неалмазный углерод локализует-
ся вблизи поверхности слоя.
Уравнение (4) можно привести к виду
va = va0 = l/L = vaof [—), (5>
где ua0 — удельная скорость роста, определяемая уравнением (1).
Используя уравнения (1) и (2'), можно вычислить эффективную
глубину проникновения реакции для данного порошка при различ-
ных температурах (рис. 25).
При проведении процесса в кинетической области сомножитель
f(l/L) равен единице и меньше ее в диффузионной области. При
I L удельная скорость роста уменьшается обратно пропорцио-
нально толщине слоя. Вероятно, значительная часть порошка не
принимает участия в синтезе, а является простым балластом.
На рис. 26 приведены результаты экспериментов в координа-
тах Аррениуса и расчетная кривая по формуле (5), описывающей
6£
процесс в кинетической переходной и диффузионной областях син-
теза. Скорости роста алмаза определялись из начального участка
кривой вес—время. Результаты расчета удовлетворительно сов-
падают с экспериментальными данными.
На переход из кинетической области в диффузионную влияет
не только повышение температуры, но и толщина слоя порошка,
его дисперсность и разбавление продуктом реакции разложения
метана — водородом. На рис. 27 приведена зависимость безраз-
мерной скорости роста алмаза (отношение роста при данной толщине
слоя к скорости роста при толщине слоя 0,1 мм) от толщины слоя
при различных температурах. Из рисунка видно, что чем выше
температура, тем больше отклонение экспериментальных значений
от расчета. Тем не менее, учитывая приближенный характер рас-
чета, совпадение следует признать удовлетворительным. Основной
причиной расхождения между расчетными и экспериментальными
значениями является то обстоятельство, что алмазный порошок
комкуется, при этом комки могут быть различного размера вплоть
до 1 мм. При проведении процесса синтеза в кинетической области,
когда глубина эффективного проникновения реакции много боль-
ше размера комков f(l/L) = 1, это явление не сказывается на ре-
зультатах вычислений. Если же размеры комков становятся срав-
нимы или больше эффективной глубины реакции, имеет место рас-
хождение между экспериментальными результатами и расчетом.
Кроме того, строго говорить о толщине слоя, когда некоторые ком-
ки больше его средней толщины, нельзя. Вторая причина отклоне-
ний — влияние краевых эффектов, поскольку уравнение (1) полу-
чено для бесконечно протяженного слоя. Действительно, если тол-
щина слоя порошка 2 мм, а радиус кварцевой чашечки, в которой
ведется эпитаксиальное наращивание алмаза, равен 6 мм, то крае-
вые эффекты могут внести существенный вклад.
Отношение толщины слоя порошка к эффективной глубине про-
никновения реакции при постоянной температуре можно записать
как
/ const
~Т ~ d(l — dip) ’
где d — насыпная плотность порошка; р — плотность алмаза. Это
отношение имеет минимум при d/p = 0,5.
Эксперименты проводились при отношении d/p = 0,36. Дей-
ствительно, при d —> 0 отношение 1/L -» оо. При d —> р, т. е. при
насыпной плотности, стремящейся к плотности вещества, также
UL —» оо, так как в этом случае эффективная глубина проникнове-
ния реакции стремится к нулю. В связи с этим могут приобрести
большое значение такие способы оптимизации процесса синтеза
алмаза, как создание виброкипящего слоя, и другие.
Уравнение (5) также удовлетворительно описывает скорость
роста алмазного порошка различной степени дисперсности.
В табл. 2 приведены экспериментальные результаты наращивания.
62
Рис. 26. Зависимость логарифма удель-
ной скорости роста алмаза из метана
от температуры
Сплошная прямая — расчет по [урав-
нению (1); пунктирная — расчет по
уравнению (5)
Рис. 27. Зависимость относительной
удельной скорости наращивания алмаза
от толщины слоя при различных тем-
пературах
Точки — эксперимент; сплошные ли-
нии — расчет
синтетических алмазных порошков при давлении метана 0,13 мм
рт. ст. и температуре 1000° С. Навеска (~0,1 г) помещалась в оди-
наковые кварцевые чашечки. Скорость роста определялась по
начальному участку кривой вес—время. Все полученные данные
отнесены к средней скорости роста порошка АСМ 1/0.
Удельная поверхность S всех алмазных порошков определялась
методом фильтрации разреженного газа через слой порошка
в стационарном режиме. В качестве рабочего газа использовали
гелий, имеющий наибольшую длину пробега среди других газов,
что позволило провести измерение удельной поверхности более-
крупных порошков. Результаты определения удельной поверхности
адсорбционным способом хорошо согласуются с приведенными выше
данными. Это связано с отсутствием в микрокристаллах алмаза
внутренней пористости.
Существенно искажает результаты графит, сопровождающий
выделение алмаза. Процесс образования графита на углеродной
поверхности широко исследуется, но еще нет четкой картины гете-
рогенной реакции разложения углеводородов. Тем более это от-
носится к росту алмаза и графита на поверхности затравочного-
кристалла алмаза. В главе I показано, что при низких давлениях
метана скорость роста графита пропорциональна степени заполне-
ния поверхности не в первой степени, а в большей. В то же время
63
1 а о л и ц a &
Влияние дисперсности алмазного порошка АСМ на массовую скорость
роста алмаза
Марка порошка АСМ 1 ъ V-10’, %/мин V » Марка порошка АСМ т о СО v -10*, %/мин V »
экспери- мент расчет экспери- мент расчет
1/0 0,1 8 1 1 10/7 2 10 0,125 0,23
0,3/0 0,3 12 1,5 1,2 20/14 1 0,5 0,08 0,12
5/3 4 4 0,5 0,46
* Безразмерная скорость роста алмаза.
Pi
(6)
.для роста алмаза может быть принята мономолекулярная реакция
разложения, т. е. скорость роста алмаза будет пропорциональна
заполнению поверхности метаном, согласно изотерме Лангмюра,
равной
Pi + + (bt/b?) р2 ’
где pi, р2 — парциальные давления метана и. водорода в объеме;
bi, b2 — отношения скоростей адсорбции и десорбции метана и
водорода соответственно. Для скорости роста алмаза получим
_ Д_________________
Va &i i + Р1/&1 + Р2/62 ‘
Р1
(7)
Из этого выражения следует, что зависимость величины обратной
удельной скорости роста алмаза от парциального давления водорода
в смеси должна быть линейной при постоянном парциальном давле-
нии метана. На рис. 28 приведена такая зависимость, полученная
при давлении метана 0,12 мм рт. ст. и температуре 1020° С.
Влияние водорода на процесс синтеза алмаза двоякое. Разбав-
ление водородом уменьшает скорость роста алмаза, но в то же вре-
мя водород еще более сильно тормозит образование сажи. Поэтому,
разбавляя исходный метан водородом, можно значительно дольше
вести процесс наращивания алмаза без блокировки поверхности
последнего неалмазным углеродом.
Такого же эффекта удается добиться, ограничивая расход мета-
на. Тогда в виде алмаза выделяется до половины углерода, содер-
жащегося в прошедшем через реактор газе. Образующийся при
этом водород существенно тормозит выделение графита. В общем
случае для более полного анализа процесса следует рассматривать
и внешнее диффузионное торможение.
64
Рис. 28. Зависимость величины обратной скорости роста ялмаза от давления водорода
в смеси при постоянном парциальном давлении метана
Рис. 29. Зависимость безразмерной скорости роста алмаза от расхода метана при раз-
личных давлениях
7 — 0,14; 2 — 0,37 мм рт. ст.
При проведении процесса во внешней диффузионной области,
согласно [60], скорость наращивания алмаза будет определяться
уравнением
dma (Nu Ь/27?) соу7
dt — (Nu DI2g)c0-\-va ’
где D — истинный коэффициент диффузии метана; R — радиус
чашки с затравочным алмазным порошком, обтекаемой с торца
потоком метана; Nu — диффузионный критерий Нуссельта; va —
скорость роста алмаза при достаточно большом расходе, когда
внешнее диффузионное торможение отсутствует. Действительно,
предыдущую формулу можно представить как
eta [14- 1 1
_ - ця[1 + nC()Nu/^ I •
Гидродинамика этого процесса сложна, поэтому ограничимся его
приближенным .рассмотрением. Обозначим
dmjdt
V ~ (dmjdt)g^x ’
где g — расход метана. Обычно реализуется следующая зависи-
мость между критериями Нуссельта и Рейнольдса: Nu ж a Re‘/!!.
Поскольку в рассматриваемом случае Re ~g, Nu можно
записать
G-const (t4v)2-
Экспериментальные данные приведены на рис. 29.
х/43 Б. В. Дерягин, Д. В. Федосеев
85
Учитывать влияние расхода метана необходимо при измерении
зависимости скорости реакции от давления. Кроме того, условия
кнудсеновского течения выполняются лишь в слое алмазного за-
травочного порошка, но не выполняются ни в самом цилиндриче-
ском кварцевом реакторе (в котором ведется эпитаксиальный син-
тез), ни в кварцевой чашке (на дно которой насыпан затравочный
алмазный порошок). Поэтому следует учитывать влияние стефанов-
ского потока продуктов реакции (водорода).
При синтезе алмаза из газа обязательно образуется неалмаз-
ный углерод (графит). Блокируя поверхность затравочного кристал-
ла алмаза, графит уменьшает суммарную скорость роста алмаза.
Обозначим через р скорость зарастания поверхности затравочного
кристалла алмаза графитом. Тогда, при равнодоступной поверхно-
сти всех частиц алмазного порошка (так же как и монокристалла),
поверхность роста алмаза убывает со скоростью
dSJdx — —psa . .
и зависит от времени как
Sa = S ехр (—0т), (8)
где S — полная исходная поверхность затравки. Скорость роста
алмаза поэтому также падает:
dmeJd-i — Sva ехр (—Рт).
Здесь va — удельная скорость роста алмаза; та — привес алмаза
(т. е. количества алмаза, наросшего на затравке). Для графита
соответствующее уравнение имеет вид
dm^dt = Syp [1 — ехр(—0т)].
Привес алмаза ко времени т составит
tyS
Wa = —[1 — ехр(—Рт)], (9)
тогда как количество выделившегося графита
= Sv^t----[ I ~ ехр (—0т)], - (10)
где у3 — удельная скорость роста графита на графите. ..
При т —> оо массовая скорость роста алмаза dmjdt —> 0, и ко-
личество наращенного алмаза не превосходит величины zzzamax =
= t'aS/p. В то же время массовая скорость роста графита
dm.fjdt -» Sv$, и количество выделившегося графита неуклонно
растет.
Приведенный выше приближенный анализ процессов совместной
кристаллизации алмаза и графита (сделанный в предположении
их независимости друг от друга) показывает, насколько важен выбор
66
условий проведения эпитаксиального синтеза. Действительно, ана-
лиз скоростей образования критических зародышей алмаза и гра-
фита, сделанный в работе [21], показывает, что возможны условия,
когда величина Р становится очень малой. Тогда максимальное
количество вновь образовавшегося алмаза может быть весьма зна-
чительным. Вполне вероятен такой процесс, когда образующийся
неалмазный углерод (графит) заращивается («замуровывается»)
алмазом и процесс эпитаксиального синтеза будет идти безостано-
вочно. В то же время величины иа, иэ, р связаны между собой и
изменять их независимо друг от друга нельзя.
Суммарный привес за время т определится как
т = та 4- = -|-[1 — ехр (—рт)] (оа — ор) 4-SypT. . - (11)
В экспериментах по эпитаксиальному синтезу алмаза на затра-
вочных алмазных порошках реализовывался случай щ О v$.
Определим характерное время процесса т' как момент времени,
соответствующий пересечению касательных к кривой т(т) в точках
т = 0 и г = оо. Собственно наклон этих касательных определяет
скорости роста алмаза и графита в отсутствие выделения конкури-
рующей фазы. Точка их пересечения дает искомое значение харак-
терного времени т' = 1/р.
Поскольку р есть скорость заращивания графитом поверхности
затравочного кристалла, а экспериментально показано, что чем
выше удельная скорость роста алмаза, тем больше и удельная
скорость роста графита, то характерное время должно быть обра-
тно пропорционально скорости нуклеации. На рис. 30 приведена
зависимость характерного времени наращивания от обратного зна-
чения скорости роста алмаза на порошке природного алмаза раз-
мером частиц до 1 мкм (порошок AM 1/0) при давлении 0,12 мм рт. ст.
Эти результаты получены на установке и по методике, описанным
выше. При повышении температуры скорость роста алмаза увели-
чивается, но характерное время процесса уменьшается.
Когда эффективная толщина выделившегося слоя графита со-
ставляет в среднем один атомный слой, практически полностью
подавляется рост алмаза из газа. Следует тем или иным способом
удалить неалмазный углерод, а затем продолжить наращивание
алмаза в следующем цикле. Был предложен и разработан метод
контроля, основанный на большом различии в электропроводности
алмаза и выделяющегося неалмазного углерода.
На рис. 31 показан реактор 1, в который через шлиф введена
кварцевая чашка с алмазным порошком 2 на специальной подвеске 3,
Контакты, изготовленные из графита или платины, прижимаются
к слою порошка пружиной (4 — электроды, 5 — печь сопротивле-
ния). Экспериментально установлено, что проводимость алмазного
порошка сильно изменяется при незначительном выделении графи-
та. Это обстоятельство можно объяснить тем, что графит высажи-
вается на поверхность затравочных кристаллов алмаза в виде тон-
3” Б. В. Дерягин, Д. В. Федосеев 57
//ify
Рис. 30. Зависимость характерного времени синтеза алмаза от величины обратной ско-
рости роста (в отн. ед.)
Рис. 31. Реактор с подвеской для нзмерения^проводимости алмазного порошка в процессе
синтеза
рис. 32. Изменение проводимости слоя алмазного затравочного порошка в процессе син-
теза
/ — выход на режим; 2 — наращивание; 3 — остывание; 4 — напуск воздуха; 5 — окис~
леиие
кой проводящей пленки, которая к тому же улучшает контакт
между зернами порошка. На рис. 32 приведена зависимость про-
водимости от времени, полученная при наращивании алмаза из
метана при давлении 0,15 мм рт. ст. и температуре 1000° С. В этом
опыте получено (после удаления неалмазного углерода) 1,5% нового
алмаза на порошке марки АСМ 1/0.
Проведенные измерения проводимости обнаружили некоторые
полупроводниковые свойства эпитаксиально наращенных порош-
ков: наличие значительного температурного коэффициента прово-
димости, способности к накоплению заряда и перезарядке. Вероятно
здесь имеется уникальная система с чисто поверхностной прово-
димостью, состоящая из атомов одного и того же вещества — угле-
68
рода. Подобные эффекты в некристаллических системах (стеклах)
привлекают интерес исследователей. Возможно, мы имеем право
говорить о «структурном легировании», поскольку атомы углерода,
образующие структуру алмаза, как бы легируют атомы углерода,
находящиеся в решетке алмаза.
Исследования, описанные в этом разделе, позволили перейти
к наращиванию больших количеств алмазных порошков, что дало
возможность испытать их в инструменте.
Найденное выше уравнение кинетики синтеза алмаза из метана
и рассчитанная с его помощью диффузионная кинетика наращива-
ния алмазных порошков характеризуют (приближенно) брутто-
процесс. Нет сомнения, что как и при получении пирографита,
реакция проходит ряд промежуточных стадий, связанных с образо-
ванием новых соединений и радикалов. Согласно схеме Касселя [61],
в газовой фазе пиролиз метана идет через этилен и ацетилен. При
синтезе алмаза такое положение может также иметь место. Нами
было указано на возможность роста алмаза из углеводородов с крат-
ными связями, хотя в одном из первых патентов по синтезу алмаза
из газовой фазы такая возможность отрицалась [451.
Недавно стали известны патенты США, в которых указан рост
алмаза из этилена, ацетилена и их смесей [62]. Учитывая практиче-
скую ценность этих результатов, представляется интересным ис- t
следовать кинетику роста алмаза из этих газов. Кроме того, непо-
средственное изучение элементарных процессов в гетерогенных
реакциях представляет определенные трудности, особенно при сов-
местной кристаллизации двух фаз — алмаза и графита. Поэтому
независимое исследование возможных промежуточных реакций
может служить источником информации о всем процессе в целом.
Эксперименты проводились на установке и по методике, описан-
ным выше. В качестве подложки использовался синтетический ал-
мазный порошок марки АСМ 1/0 с удельной поверхностью 10 л?/г.
Изучалась только начальная стадия роста алмаза в пределах ли-
нейной части зависимости вес—время, когда количество выделив-
шегося неалмазного углерода было незначительно и его влиянием
можно пренебречь.
На рис. 33 показана зависимость скорости роста алмаза от дав-
ления ацетилена и этилена при 830° С. Аналогично метану, реакция
образования алмаза из этих углеводородов подчиняется уравнению
первого порядка. Сопоставление с известными скоростями роста
для метана показывает, что при постоянном давлении скорость
роста алмаза увеличивается в следующем ряду: метан, этилен,
ацетилен.
Изучение кинетики синтеза при температурах 780—950° С
(результаты приведены в координатах Аррениуса на рис. 34) при
давлении 0,06 мм рт. ст. позволило установить энергию активации:
для роста алмаза из ацетилена 37 ккал/моль, для роста алмаза из
этилена 80 ккал/моль. Для роста алмаза из метана на синтетическом
алмазном порошке энергия активации равна 70 ккал/моль. Отличие
3**
69
Тис. 33. Зависимость удельной скорости роста алмаза от давления ацетилена (/) н эти-
лена (2)
Рис. 34. Зависимость логарифма Удельной скорости роста алмаза из ацетилена (/) и эти?
лена (2) от обратной температуры
в 10 ккал/моль от результатов, полученных на природных порош-
ках, вызывается, вероятно, примесями металлов, используемых
при синтезе алмаза в области высоких давлений. Все значения
энергий активации определялись с точностью +3 ккал/моль.
Если для роста алмаза из метана и этилена энергии активации
совпадают, то для роста из ацетилена энергия активации реакции
много меньше. Это нельзя объяснить диффузионным торможением
в слое исходного затравочного порошка, поскольку эксперимен-
тально было показано, что диффузионное торможение проявляется
при температурах более высоких чем 950° С. Поэтому можно пред-
положить, что стадия разложения ацетилена не является лимити-
рующей.
Существенно разным является влияние водорода на рост алмаза
из ацетилена и этилена. Экспериментальные данные для смеси
углеводорода (парциальное давление 0,06 мм рт. ст.) и водорода
(парциальное давление 0,12 мм рт. ст.) приведены на рис. 35. Эти
эксперименты показали, что энергия активации роста алмаза из
смеси ацетилен—водород возрастает ккал/моль, тогда как для
смеси этилен—водород — уменьшается до 56 ккал/моль.
Сопоставление относительных скоростей роста алмаза в зависи-
мости от разбавления водородом при парциальном давлении угле-
водорода 0,06 мм рт. ст. и температуре 950° С показывает (рис. 36),
•что наибольшее влияние разбавление водорода оказывает на эти-
лен и наименьшее — на метан. Однако нельзя утверждать, что
.полученные зависимости являются одинаковыми для всех условий.
В зависимости от разбавления водородом энергия активации
70
Рис. 35. Зависимость логарифма удельной скорости роста алмаза нз смесей ацетилен—
Водород (7) н этилен—водород (2) от обратной температуры
Рис. 36. Зависимость относительной скорости роста алмаза из различных углеводородов
от парциального давления водорода
1 — ацетилен; 2 — этилен; <3 — метай
роста графита из метана может меняться, увеличиваясь от 75 до
.103 ккал!моль (см. главу II). В этом случае можно предположить,
что только при больших разбавлениях водородом процесс идет не
по схеме Касселя, а лимитируется отрывом одного атома водорода
и образованием метильного остатка [311. Действительно, известно
163, 64], что энергия отрыва первого атома водорода от метана равна
101,4 ккал, тогда как энергия разрыва связи С—Н в радикале СН2
составляет 89,9 ккал. Поэтому можно ожидать, что при росте алмаза
могут иметь место различные механизмы роста в зависимости от
разбавления системы водородом. На это указывает также то обстоя-
тельство, что разбавление метана водородом значительно слабее
уменьшает скорость роста алмаза, нежели скорость роста графита.
На различие между ростом алмаза и графита указывает также
тот факт, что энергия активации роста алмаза из ацетилена значи-
тельно (на 9 ккал/моль) превышает энергию активации роста графита
из ацетилена [26]. Поскольку разбавление водородом увеличивает
энергию активации роста алмаза, можно ожидать, что его рост идет
через хемосорбированный на затравочной поверхности водород,
что позволяет объяснить меньшее водородное торможение.
3. Циклический метод наращивания
алмазных порошков
Наращивание алмазного порошка алмазом происходит до тех пор,
пока его поверхность не покроется слоем графита. Затем графит
необходимо удалить, чтобы подготовить затравочные кристаллы
71
ДБ,мг
Рис. 37. Пять последовательных цик*
лов наращивание—очистка
Рис. 38» Установка для наращивания
алмазных порошков
алмаза к новому наращиванию. Эверсол [45] использовал для этой
цели травление графита в водороде при давлении 50 атм и темпе-
ратуре 1000 С в течение нескольких часов. Однако такой способ
очистки существенно осложняет аппаратуру для синтеза.
Нами был предложен ^осуществлен циклический метод наращи-
вания алмазных порошков, в котором графит удалялся окислением
кислорода [65]. При этом используется различная кинетическая
актавность алмаза и графита к окислению [66]. При температуре
до 700 С на воздухе скорость окисления графита выше, чем скорость
окисления алмаза. Поэтому можно удалить неалмазный углерод
и получить практически чистую алмазную поверхность. Разумеется,
что при этом удалится и часть наращенного алмаза. При проведении
наращивания с весовым контролем (как описано в предыдущем раз-
деле) об удалении графита можно судить по изменению скорости
газификации. При контроле синтеза по электросопротивлению
наращиваемого порошка об удалении графита судят по возраста-
нию сопротивления. После проведения очистки цикл наращивания
повторяется.
На рис. 37 приведены пять последовательных циклов наращи-
вание очистка синтетического алмазного порошка марки АСМ 1/0,
и получено 9,5% нового алмаза. На рис. 38 приведена фотография
установки для наращивания до 10 карат исходного алмазного по-
рошка. Эта установка позволила нарастить значительное количество
алмаза и провеет его всестороннее исследование [2]. Электроне-
графическое исследование показало, что наращенный алмаз не
отличается от природного. Определение элементного состава про-
водилось сжиганием навески алмаза в токе кислорода. Было уста-
новлено, что если в исходном природном порошке среднее содержа-
ние углерода 93,10%, то в наращенном на 27% оно равно 93,95%.
Это увеличение процентного содержания углерода связано с тем,
что в процессе эпитаксиального синтеза наращивается чистый
углерод, в то время как при синтезе методом высоких давлений
алмаз включает в себя большое количество примесей.
Определение удельного веса проводилось по методу градиент-
ной трубки. Для приготовления среды требуемого высокого удель-
ного веса использовалась жидкость Клеричи (удельный вес
4,3 г/см3). Разбавляя ее водой при специальном перемешивании,
Можно иметь в вертикальном сосуде определенный градиент плот-
ности. Для получения реперных точек использовались образцы
с известным удельным весом. Положение крупинок алмазного по-
рошка позволяло легко и быстро определять их удельный вес,
который составил для исходного порошка 3,46 г/см3, а для нара-
щенного на 27% —3,48 г/см3.
Исследование, проведенное методом микродифракции электро-
нов, показало, что наращенный углерод имеет структуру алмаза.
Аналогичные результаты были получены при изучении рент-
геновской дифракции алмазных порошков по методу Дебая. Так,
наращенный на 50% порошок синтетического алмаза марки АСМ 1/0
исследовался на рентгеновской установке УРС-60. Было установ-
лено, что он имеет параметры решетки те же, что и исходный алмаз-
ный порошок, т. е. 3,5656 + 3-10-1 А. Примеси других веществ
не были обнаружены, хотя метод позволяет уверенно определять
компоненты смеси, если их количество не менее 2—3 вес.%.
Испытания исходного и наращенных алмазных порошков (в виде
свободного абразива и паст) показали, что наращенный порошок
дает повышенную «чистоту» (гладкость) поверхности кремния после
обработки. Так, исходный порошок давал класс чистоты 13а, тогда
как наращенный — 14а.
Проведенные эксперименты показали, что толщина слоя затра-
вочного порошка существенно влияет на производительность эпи-
таксиального синтеза. При большой толщине средняя скорость
роста алмаза существенно уменьшается вследствие диффузионного
торможения. Поэтому была поставлена работа по интенсификации
роста алмаза [67].
Размеры реакторов для эпитаксиального синтеза алмаза су-
щественно зависят от слоя затравочного алмазного порошка: чем
тоньше слой, тем более компактным может быть изготовлен реактор.
В зависимости от условий синтеза, в основном от температуры,
реакция может не проникать на всю глубину слоя, и тогда часть
затравочного порошка не будет принимать участия в процессе,
а ее присутствие будет снижать значение удельной производитель-
ности.
73
Добиться относительной равномерности наращивания по всей
глубине слоя можно по-разному. Во-первых, понизить температуру
и перевести процесс из диффузионного режима в кинетический.
Однако в этом случае необходимо рассыпать порошок тонким сло-
ем, а скорость роста алмаза сильно уменьшить. Например, пусть
эффективная глубина проникновения реакции равна 0,1 мм. Тогда
10 карат алмазного порошка потребуется рассыпать на площади
в 160 см~, причем за один цикл будет наращиваться не более 0,1 %
алмаза. Во-вторых, можно повысить температуру и увеличить
толщину слоя алмазного порошка, проводя таким образом процесс
в диффузионном режиме, и осуществить тем или иным способом
непрерывное перемешивание. Второй путь является, несомненно,
предпочтительным.
Кристаллизацию алмаза и графита нельзя рассматривать как
независимые процессы, поскольку само явление образования новой
фазы происходит на одной и той же поверхности и из одного и того
же газа. Однако вследствие различных скоростей реакции эффектив-
ные глубины проникновения реакции в глубь дисперсного тела
для алмаза и графита различны:
U = /БтИ и
Здесь D' — эффективный коэффициент диффузии в слое порошка,
а величина Д' является некоторой фиктивной константой скорости
гомогенной реакции, связанной с константой скорости гетерогенной
реакции соотношением К' = KS0, где So — удельная поверхность
порошка. Этот метод, предложенный Я- Б. Зельдовичем, оказался
очень перспективным и сводится к рассмотрению некоторой фик-
тивной гомогенной реакции в дисперсном теле вместо соответствую-
щей гомогенной реакции.
Естественно, что величина эффективного коэффициента диффузии
характеризует проникновение газа, например метана, в глубь
порошка, поэтому она одинакова при росте и алмаза, и графита.
Поскольку было установлено, что скорость роста алмаза превышает
скорость роста графита, то L0 в случае раздельного осаж-
дения. При одновременном образовании двух фаз эффективная глу-
бина проникновения реакции одна, потому что она связана с рас-
ходом реагирующего газа. Однако эта величина не остается по-
стоянной и меняется со временем вследствие постепенного закрытия
поверхности графитом. Если принять, что образование графита
и алмаза происходит одновременно, то суммарная константа ско-
рости реакции может быть записана в виде
К' = к'л (1 - 6) -Ф Д0,
где 6 — степень покрытия поверхности графитом, равная отноше-
нию поверхности, занятой графитом, к общей поверхности. Тогда
эффективная глубина проникновения реакции равна
74
D'
L У <(l-5) + Kp
и меняется от = }^£>7(К'а + К'з) в начальный момент времени
(6 = 0) до (6 = 1)ч
При равнодоступной поверхности всего затравочного кристалла
поверхность алмаза Sa, не занятая графитом, будет убывать в ки-
нетической области со скоростью
dSa
dt
где Р — скорость зарастания поверхности графитом. Если же тол-
щина слоя порошка I много больше эффективной глубины проник-
новения реакции и осуществляется непрерывное перемешивание, то
dS _ _ Q'С
где Р' = Р/, а для f можно принять
f-ur-
При I <С L величина f = 1, а при возрастании толщины слоя
f — 1/L. В диффузионной области количество алмаза, наращенное
к моменту времени / на единицу внешней поверхности слоя порошка,
будет равно
Ма = -^[1-ехр(- p7)J va.
Максимальное количество алмаза будет равно
ДЛ _ ldS<>oao
max — 5-- •
Основные экспериментальные исследования проводились в ре-
акторе с виброкипящим слоем. Реактор выполнялся из кварца
в виде тора, имеющего два ввода—для входа и выхода газа, и
нагревался с помощью специальной ленты, намотанной непосред-
ственно на тор. Реактор укреплялся на вибростолике, который
сообщал алмазному порошку не только непрерывное перемешива-
ние, но и движение по тору. Контроль за синтезом осуществлялся
с помощью электросопротивления.
В одной из серий экспериментов в реактор первоначально было
загружено 1415 мг порошка АСМ 1/0 (синтетический алмазный
порошок с размером частиц до 1 мкм). Синтез проводился при тем-
пературе 1030° С и давлении метана 0,6 мм рт. ст. Очистка от гра-
фита осуществлялась кислородом воздуха.
На рис. 39 показано наращивание в виброкипящем слое порошка
АСМ 5/3 (размер частиц 3—5 мкм). При виброперемешивании были
75
рнс. 39. Последовательное наращивание
алмазного порошка в установке с вибро*
кипящим слоем
Рис. 40. Влияние температуры на рост
алмаза
1 — общий привес; 2 — привес чистого
алмаза
подтверждены сделанные ранее выводы об оптимальной температуре
наращивания алмазных порошков. На рис. 40 приведены данные
о зависимости скорости наращивания от температуры для алмаза
АСМ 1/0. Видно, что оптимальная температура синтеза находится
в интервале 1040—1060° С.
4. Механизм роста алмаза
Кинетика гетерогенных химических реакций, протекающих с обра-
зованием новой фазы, зависит как от реакционной способности га-
зовых молекул, так и от характера поверхности. Влияние поверх-
ности двоякое. Во-первых, она влияет на кинетику благодаря своим
молекулярным и валентным силам. Формально это сводится к из-
менению работы образования критического зародыша, входящей
экспоненциально в константу скорости реакции. Во-вторых, по-
верхность определяет частоту столкновений в двухмерном адсорби-
рованном слое или частоту столкновений падающих молекул (или
атомов) с докритическими зародышами. Опять же формально, это
влияние поверхности выражается через адсорбционные характери-
стики системы поверхность—газ (адсорбент—адсорбат).
Разумеется, это справедливо в предположении, что адсорбция
не является самой медленной стадией процесса. Неявно также пред-
полагается, что хемосорбированные молекулы и радикалы доста-
точно подвижны, чтобы во время роста кристалла мигрировать по
поверхности. На такое допущение можно пойти с достаточной уве-
ренностью, поскольку миграция поверхностных атомов доказана
вполне убедительно (см., например, [68]).
В зависимости от реакционной способности подложки и газовых
молекул, характера связей в кристалле подложки и новой фазе,
условий синтеза можно предположить различные механизмы роста.
Рассмотрим их иа примере роста алмаза из метана. Водород —
76
единственный газообразный продукт реакции. Пусть р± и р2 — дав-
ление метана и водорода в газовой фазе, ©! и @2 — доли поверхно-
сти, занятые метаном и водородом, соответственно.
1. Если реакция роста алмаза идет на части поверхности, сво-
бодной от адсорбированных молекул, то кинетика роста алмаза
определится уравнением
= Кг (1 - ©! - 02)h, (12)
где /1 = /э1/(2л mkiy!’ — поток метана из газовой фазы на поверх-
ность растущего кристалла.
2. Алмаз может расти прямым присоединением к подложке
через хемосорбированный водород; тогда
= Ki®2,jl- (13)
3. Рост новой фазы, благодаря зарождению двухмерных за-
родышей из двухмерного адсорбированного газа, будет определяться
уравнением (см. раздел 5)
= К3®1 (14)
4. Если процесс роста алмаза определяется встраиванием угле-
рода из адсорбированного газа непосредственно в решетку, то
= Кг® г- (15)
5. Когда скорость гетерогенной реакции определяется реакцией
между адсорбированными метаном и водородом, имеем
v5 = К,® г®,. (16)
В принципе скорость роста алмаза должна определяться супер-
позицией уравнений (12)—(16). В предположении независимости
всех шести процессов
(17)
1=1
В действительности лишь один или несколько механизмов могут
вносить определяющий вклад в общую скорость гетерогенной ре-
акции. Ниже мы покажем, что алмаз растет по механизмам 1 и 2
в предположении справедливости уравнения адсорбции типа Ланг-
мюра, согласно которому для бинарной смеси
0 ____ ___Pi/H____ (Д) _____Рг/^г___ /1 о\
1 1 + Pi/^i + РаЛ’а 2 Г + Pi/^i + Рг/^2
Естественно, что вид принятого уравнения адсорбции скажется
на характере получающихся результатов.
Эксперименты проводились на установке, описанной в главе II
и в работе [27]. Использовался синтетический алмазный порошок
АСМ 1/0 с удельной поверхностью 10 м2/г, определенной методом
фильтрации разреженного газа [77]. Чистота поверхности затравоч-
77
ных кристаллов алмаза является весьма существенным условием,
поэтому перед опытами алмазный порошок окислялся на 2—3%
от исходного веса кислородом воздуха для удаления органических
примесей, остающихся на поверхности зерен алмаза после их клас-
сификации по зернистостям. Затем алмазный порошок прогре-
вался до постоянного веса в атмосфере водорода при 900° С.
Подготовленный алмазный порошок помещался в кварцевый
реактор в четырех чашках с различной навеской порошка. Навески
порошка в чашках подбирались таким образом, чтобы имелось че-
тыре слоя разной высоты для изучения диффузионной кинетики.
Расход углеродсодержащего газа выбирался таким, чтобы исключить
торможение вследствие внешней диффузии. Используемые газы
предварительно очищали: метан — многократной ректификацией
при температуре жидкого азота, водород — пропусканием через
палладиевый натекатель.
Количество выделившегося неалмазного углерода определялось
по изменению электропроводимости. Для этого в одной чашке к ал-
мазному порошку подводились платиновые электроды, на которые
подавалось постоянное напряжение 1,5 в. Проводимость промежутка
между электродами измерялась микроамперметром типа М-95.
Опыт заканчивался при достижении одинаковой во всех случаях
проводимости, что дает основание предположить одинаковое коли-'
чество выделившегося неалмазного углерода (при одинаковой
температуре!). После опыта чашечки взвешивались на аналитиче-
ских весах, и определялась средняя скорость роста.
Известно, что форма кинетических кривых необязательно соот-
ветствует определенному механизму, хотя и позволяет сузить число
возможных вариантов. Анализ всей совокупности доступных нам
экспериментальных результатов позволяет сделать вывод, что ско-
рость роста алмаза определяется суммой следующих двух слагае-
мых:
— Aai (1 - ®1 - ®a)Pi 4~ (19)
Первый член в правой части уравнения соответствует механизму
прямого присоединения на свободной от адсорбированных атомов
части поверхности затравочного кристалла алмаза. Второе слагае-
мое отвечает росту алмаза через хемосорбированный водород.
Рассмотрим некоторые частные случаи уравнения (19) и сопо-
ставим их с экспериментальными данными. Используем уравнение
адсорбции Лангмюра.
Если давление водорода равно нулю, то из уравнения (12)
следует
----'Ai+Bj— , (20)-
Ч Pl i;
где Ах и В± •— постоянны для неизменных условий опыта, уа0 — ско-
рость роста алмаза из чистого метана. На рис. 41 показана зависи-;
мость обратной скорости роста от обратного давления метана при
изменениях давления от 0,3 до 10 л£и рт. ст. >'
78
Рис, 41. Зависимость обратной скорости роста алмаза ( в отн. ед.) от обратного давления
метана
Рис. 42. Зависимость обратной скорости роста алмаза (в оти. ед.) от давления водорода
при разном давлении метана
1 — 3; 2 — 10 мм рт. ст.
Рис. 43. Зависимость отношения скоростей роста алмаза из метана и смеси метан—водо«
род от отношения Рг'рг при разном давлении метана
1 — 0,3; 2 — 1,0; 3 — 3,0; 4 — 10 мм рт. ст.
При постоянном давлении метана можно найти
— — Аг + В2рг,
(21)
т. е. обратная скорость роста алмаза линейно зависит от парциаль-
ного давления водорода, как показано на рис. 42.
Когда отношение парциальных давлений метана и водорода
постоянно, из уравнения (19) следует
___ Д I_________1_________Р2
3 + Сз/ Р1 Р1
(22)
т. е. при уменьшении парциального давления метана угол наклона
прямых в координатах рис. 43 уменьшается.
Результаты, приведенные на рис. 41—43, получены при 950° С.
При других температурах зависимости имеют аналогичный вид, что
можно трактовать как подтверждение уравнения (19). Легко пока-
зать, что общий привес наросшего алмаза определяется как
Awg_____v_x_
тй '
(23)
79
Рис. 44. Зависимость привеса алмаза
от обратного давления метана
Рис. 45. Зависимость проводимости алмазного порошка от времени синтеза прн 950° С
/ — исходный; 2 — облученный у-лучами; 3 — облученный электронами
где т0 — исходная навеска; Дща — количество наращенного ал-
маза; Vjja — скорость роста графита на алмазе.
Подставляя в уравнение (23) скорости роста алмаза, определяе-
мые уравнением (19), и графита (см. глава II) в виде ~
получим
—-~а4 + В4 —
«о 41 4 Pl
(24)
На рис. 44 приведена зависимость общего привеса от обратного-
давления метана в смеси метан—водород при их отношении 2:1.
Совпадение с ожидаемым, согласно уравнению (24), результатом 1
можно расценивать как подтверждение предложенного механизма.
Все приведенное выше относится к чистой поверхности идеаль-
ного кристалла алмаза. В действительности, изучая кинетику роста
алмазных порошков, следует иметь в виду, что вклад ребер и вер-
шин отдельных кристаллитов в общую поверхностную энергию*
может быть весьма значительным, особенно у высокодисперсных
порошков. Наличие различных дефектов на поверхности реального
кристалла должно сказаться на кинетике нуклеации. В связи с этим
рассмотрим влияние дефектов, вызванных облучением, на рост ал-
маза и графита из метана, следуя работе [69].
Облучение быстрыми электронами проводилось на электронном
ускорителе на 1,5 ТИж Навеска алмазного порошка порядка 2—3 г
помещалась в медную ячейку, интенсивно охлаждаемую водой.
Температура алмаза во время облучения не превышала 20° С. Ве-
личина интенсивности электронного тока устанавливалась предва-
рительно при помощи коллектора Фарадея с входным отверстием,
равным окну ячейки. При измерениях соблюдалось равенство-
расстояний от входного окна электронного ускорителя до ячейки.
80
с алмазами и до входного окна коллектора. Постоянство условий
облучения и необходимая величина потока электронов контролиро-
валась по значению тока в камере-мониторе. Энергия электронов
составляла 1 Мэв. Электронный поток был равен 12 эв! см2. Доза,
определенная по интенсивности электронного тока, равна 3,6-
• 1022 эв!см2. Облучение у-лучами источника 60Со производилось
в течение 10 суток при комнатной температуре. Мощность у-лучей
5600 рад!сек, доза 4,8-109 рад.
Затравочный алмазный порошок марки АСМ 10/7 (синтетический
алмазный порошок с размерами частиц от 7 до 10 мкм) помещался
в кварцевую чашку и взвешивался. Затем в чашку вставляли
графитовые электроды и на специальной кварцевой подвеске по-
мещали внутрь вертикального реактора, который вакуумировался.
Одновременно с повышением температуры через реактор пропускали
водород. По достижении температуры синтеза через реактор уста-
навливался поток метано-водородной смеси при соотношении 1 : 2.
Во время опыта измерялась проводимость затравочного порошка.
По окончании опыта порошок охлаждался в водороде, затем в ре-
актор напускался воздух и чашка с порошком взвешивалась.
По привесу можно судить об общем количестве выделившихся ал-
маза и графита, а по проводимости — об относительном количестве
графита.
Эксперименты были проведены при 950 и 1000° С и общем давле-
нии метано-водородной смеси 0,35 мм рт. ст. Навеска во всех слу-
чаях 400 мг (2 карата). Результаты экспериментов приведены в
табл. 3.
Характер изменения относительной проводимости при 950° С
при наращивании на исходном и облученном порошках приведен
Таблица 3
Влияние облучения на рост алмаза и графита
Затравка Привес, мг Проводимость, отн. ед.
Температура 1000° С (1,5 часа)
Исходная 0,92 6,6-102
Облученная электронами 0,68 103
То же 0,84 1,1-10»
Температура 950° С (2 часа)
Исходная 0,76 6,3-10
Облученная электронами 0,76 6,3-10
Облученная у-лучами ........ 0,48 3,6-102
То же 0,49 2,8-102
81
на рис. 45. При 1000° С различие в проводимости значительно
меньше. При эпитаксиальном синтезе имеется определенная скорость
(3 (в см2!см^-сек или тг-1)заращивания алмаза графитом. Чем мень-
ше р, тем больше нарастает алмаза. Поверхность, на которой может
расти алмаз, вследствие блокировки графитом, убывает по экспо-
ненциальному закону = S ехр (—р/). Максимальный привес
алмаза не может превысить величину naS/p и он тем меньше, чем
больше величина р. Скорость роста алмаза, как было показано выше,
значительно больше скорости роста графита. Поэтому при интер-
валах времени, сравнимых с 1/р, наибольший вклад в привес вносит
выделившийся алмаз. При наращивании облученного алмаза вели-
чина р будет больше, поскольку дефекты, вызванные воздействием
излучения на вещество, вносят изменения в силовое поле поверх-
ности кристалла и делают более вероятным образование на них
зародышей стабильной фазы — графита.
Можно предположить, что на начальной стадии процесса имеет
место декорирование дефектов поверхности графитом, подобно
автодекорированию поверхности алмаза алмазом [70] или декори-
рованию вольфрамом при осаждении последнего из газовой фа-
зы [71]. Действительно, из данных табл. 3 видно, что при нара-
щивании на облученном порошке по сравнению с исходным при
950° С проводимость вырастает до значительно большей величины,
тогда как общий привес новой фазы уменьшается. В то же время
при 1000° С различие заметно меньше, что, вероятно, связано с от-
жигом дефектов при повышенной температуре. Так, при 950° С
относительная проводимость облученного порошка в среднем в 5 раз
больше, чем у исходного, тогда как при 1000° С это отношение равно
всего 1,6.
Из рис. 45 следует, что облучение влияет в основном на началь-
ный участок кривых проводимость—время. Плато от начала экспе-
римента до 20 мин. вызвано тем, что за это время еще не успевают
образоваться мостики между отдельными участками графита на
поверхности затравочных кристаллов. Увеличение наклона кривой
проводимость—время свидетельствует о том, что облучение при-
водит к возрастанию скорости образования неалмазного углерода.
Если бы облучение вызвало лишь графитацию части поверхности,
указанная кривая просто сдвигалась бы влево. |
В этих экспериментах не было обнаружено различия при облу-
чении алмаза быстрыми электронами и у-лучами.
Согласно предложенному механизму роста алмаза, продукт ре-
акции (водород) должен оказывать на эпитаксиальный рост
алмаза специфическое действие. Известно, что в обычной химиче-
ской кинетике конечный продукт реакции смещает равновесие
реакции в сторону исходного вещества. При синтезе алмаза дело
обстоит совершенно по-другому. Из (19) можно показать, что
возможны условия, при которых рост алмаза в присутствии
водорода ускоряется, причем это не связано с уменьшением скорости
роста графита.
82
Незнание значений констант скоростей реакций, входящих
в уравнение (19), делает такой расчет более чем ориентировочным.
Поэтому ограничимся тем, что приведем некоторые эксперимен-
тальные данные, полученные В. П. Варниным. Им исследовалось
наращивание порошка АСМ 10/7 в метано-водородной среде. Кон-
троль за процессом осуществлялся с помощью регистрирования
изменения электропроводимости. После опыта порошок взвеши-
вался. Исходная навеска была равна 400 мг. Все приведенные
в табл. 4 результаты получены при 1000° С. Внутреннее и внешнее
диффузионное торможение отсутствовало.
Таблица 4
Влияние водорода на рост алмаза
Ноыер опыта Давление, мм рт* ст. Продолжи- тельность опыта, мин. ПривеС, мг П роводимост ь, отн. ед.
метана водорода
1 0,30 0 60 0,80 1,7-10*
2 0,15 0,15 80 1,2 6,6-10*
3 0,06 0,27 108 ' 0,81 3,0-10*
4 0,03 0,27 153 0,72 . 4,5-103 .
5 0,03 0 45 0,12 4,8-10?
Из таблицы следует, что разбавление водородом либо незначи-
тельно уменьшает скорость роста алмаза, либо даже увеличивает ее
(см. опыты 4 и 5). Действительно, уменьшение парциального дав-
ления метана в 10 раз при одновременном сильном разбавлении
продукта реакции уменьшает скорость роста алмаза всего в 2 раза.
В то же время для чистого метана сохраняется приблизительно
первый порядок скорости реакции. Предложенный механизм и
подтверждающие его экспериментальные данные позволяют рас-
считывать на создание таких условий синтеза, при которых растет
лишь один алмаз без графита, либо образующийся графит зара-
щивается растущим алмазом. Реализация одного из этих методой
описана в главе IV (см. стр. 98).
5. Синтез алмаза из смесей углеводородов
(неаддитивность скоростей реакций)
При кристаллизации из газовой фазы по механизму прямого удара
всегда можно ожидать аддитивности скоростей реакции при кри-
сталлизации из смесей двух газов. Действительно, в этом случае
скорость реакции прямо пропорциональна потоку молекул на по-
верхность затравочного кристалла, который, в свою очередь, прямо
пропорционален парциальному давлению. Изменением коэффици-
83
ента диффузии можно пренебречь, если процесс проводится в за-
ведомо кинетической области.
Аддитивность скоростей реакций для смесей метан—ацетилен и
этан—этилен наблюдалась для роста графита [26, 27]. Однако,
согласно развиваемым выше представлениям, и для роста графита,
и для роста алмаза из газовой фазы можно ожидать, что в опреде-
ленных условиях сумма скоростей отдельных реакций не будет
равна суммарной скорости роста твердой фазы из смеси газов.
Согласно предложенному в предыдущем разделе механизму
роста алмаза из газовой фазы рост алмаза может идти на поверх-
ности, свободной от адсорбированных молекул углеводорода,
а также через хемосорбированный водород. Рассмотрим синтез
алмаза из смеси двух углеводородов, ограничиваясь случаем отсут-
ствия водорода в системе *. Если ру и р2 — давления углеродсо-
держащих газов, a и 02 соответствующие степени заполнения
поверхности, то скорости роста алмаза равны
Vi = Ki (1 — ©l)pl, V2 = K2 (1 — ©2)p2-
Скорость роста из смеси будет определяться также свободной
поверхностью затравочных кристаллов алмаза
«3 = (KlPl + К2Р2) (1 — ©з),
где 03 — степень заполнения поверхности молекулами двух газов.
При использовании изотермы адсорбции типа изотермы Лангмюра
скорость роста равна
_ KiPi К2р2
8 1 Pi/^i + Рг!Ь2
Так как
A'iPi KiPi
1 + Pi/^i + Р2/&2 1 + Pi/^i
(25)
и
А~2р2 КгР2
1 P1/61+Рз/^а 1 + рфг
ТО V3 < Vi + v2.
Случай, когда v3 = vr + v2, возможен при условии p^bi 1 и
р21Ь2<^ 1> т. е- пРи высоких температурах или при очень низких
давлениях (относительно Ьх или Ь2).
Для проверки изложенного были проведены эксперименты по
росту алмаза из смесей метан—ацетилен и этилен—ацетилен по мето-
дике, описанной выше. Использовался синтетический алмазный
порошок марки АСМ 1/0 с удельной поверхностью 10 м2/г. Газы
предварительно смешивались в баллоне и выдерживались в течение
12 час. для выравнивания концентраций. Давление в реакторе
* Практически уже образующийся при разложении углеводород может оказы-
вать влияние на синтез.
84
Рис. 46. Зависимость логарифма удельной скорости роста алмаза из различных углеводо-
родов от обратной температуры
1 — метан; 2 — ацетилен; з — смесь метан—ацетилен (2 : 1); 4 — аддитивная скорость
роста алмаза
Рис. 47. Зависимость логарифма удельной скорости роста алмаза из различных углеводо-
родов от обратной температуры
1 — этилен; 2 — ацетилен; 3 — смесь этилен—ацетилен (1 : 1); 4 — аддитивная ско-
рость роста алмаза
во всех случаях было равным 0,06 мм рт. ст. Выше 950° С экспери-
менты не проводились, чтобы избежать попадания в диффузионную
область.
Из рис. 46 видно, что действительная скорость роста алмаза
из смеси метан—ацетилен меньше аддитивной скорости, что и сле-
дует из формулы (25). Различие в скоростях больше при низких
температурах и уменьшается при увеличении температуры.
Рис. 48. Зависимость энергии активации роста алмаза от состава смеси метан—ацетилен
Рис. 49. Зависимость энергии активации' роста алмаза от состава смеси этилен—ацетилен
4 Б. В. Дерягин, Д. В. Федосеев 85
Аналогичный результат получен для смеси метан—ацетилен
(1 : 4), только при этом различие в скоростях реакции меньше:
аддитивная скорость превышает действительную на 20% при тем-
пературе 850° С.
Из рис. 47 следует, что аддитивная скорость превышает экспе-
риментально найденную на 30% при температуре 850° С. При повы-
шении температуры различие в скоростях уменьшается.
Энергия активации роста алмаза из смесей является переменной.
Изменение энергии активации роста алмаза из смеси метан—ацети-
лен показана на рис. 48. На рис. 49 приведена зависимость энергии
активации синтеза алмаза из смеси этилен—ацетилен. В зависимости
от состава смеси энергии активации определялись с точностью
±3 ккал/моль. Нетрудно убедиться, что такбго характера зависимо-
сти должны следовать из формулы (25). Действительно, следующую
из нее константу реакции (например, первого порядка) можно пред-
ставить в виде
Кз = Кг
или
1 Д-Д-р2/62
Поэтому энергия активации будет зависеть от состава смеси и не
может стать меньше, чем энергия активации для роста алмаза из
ацетилена, или превысить энергию активации для роста алмаза
из метана или этилена. Следует отметить, что это положение спра-
ведливо лишь при рассматриваемом механизме роста и справед-
ливости уравнения адсорбции типа изотермы Лангмюра. При дру-
гих механизмах роста новой фазы возможно значительное увели-
чение энергии активации.
Найденная неаддитивность скоростей реакции представляет
практический интерес для избирательного роста твердой фазы из
газовой. Вероятно при этом могут открыться возможности управ-
ления ростом алмаза и графита при совместной кристаллизации.
Кроме того, легко видеть, что над алмазом смесь газов может обо-
гащаться одним из компонентов.
1 + (K2p2/KiPi)
1 + Pilb2
1 + (К1Р1/К2Р2)
6. Фракционирование стабильных изотопов углерода
Алмаз, как и все другие формы углерода, характеризуется опреде-
ленным соотношением стабильных изотопов углерода — 12С и
13С [73]. Поэтому представляло интересным исследовать изотопный
состав алмаза эпитаксиального синтеза. Такая работа была постав-
лена и выполнена авторами данной монографии совместно с Э. М. Га-
лимовым и В. С. Прохоровым [74].
Не затрагивая геохимической стороны вопроса, укажем, что изо-
топный состав алмазов имеет две особенности по сравнению с изо-
топным составом других углеродсодержащих минералов. Во-пер-
вых, все алмазы имеют приблизительно один и тот же изотопный
86
состав. Во-вторых, отклонения от некоторой средней величины
относительного содержания изотопов незначительны.
Этими двумя особенностями отличаются бесцветные прозрачные
алмазы. Алмазы типа карбонадо обогащены легким изотопом угле-
рода. В работе [75] указывается, что цветные алмазы Якутского
месторождения имеют различный изотопный состав. Более того,
разные части одного и того же окрашенного кристалла отличаются
в процентном содержании стабильных изотопов. Авторы связывают
эти различия с условиями кристаллизации.
Синтез алмаза осуществлялся из метана при температуре 1000—
1050° С и давлении метана 0,2—0,5 мм рт. ст. Перед опытом метан
очищался ректификацией при температуре жидкого азота. В каче-
стве подложки исследовались синтетические и природные алмазные
порошки марок АСМ 1/0 и AM 1/0 с размером частиц до 1 мкм.
Удельная поверхность алмазных порошков составляла около
10 м2!г. Алмаз наращивался циклическим способом, после этого
в течение определенного времени синтез прекращался и удалялся
образовавшийся графит. После очистки цикл наращивания повто-
рялся. Процесс синтеза и очистки контролировался либо весовым
методом, либо по изменению проводимости наращиваемого порошка.
Изотопный состав углерода исследовался на масс-спектрометре
МИ-1305 с конструктивными изменениями, обеспечивающими пре-
цизионный изотопный анализ. В процессе подготовки весь углерод
алмаза и графита переводился в двуокись углерода в установке и
по методике, описанным ранее [76]. Погрешность, оцениваемая на
уровне доверительной вероятности 0,95, составляла ±0,05%.
Погрешность по воспроизводимости была равна ±0,02%. Резуль-
таты экспериментов обрабатывались по формуле
г (“C/i-C), 1
613C4WcV~ Т^-
Индекс «а» относится к алмазу, «0»—к стандарту. Отношение
13С/12С в стандарте равно 1123,72-10-5. Предварительно была опре-
делена величина 6/13С в исходном метане, которая оказалась рав-
ной —4,62%. Контрольные опыты показали, что пропускание ме-
тана через ловушку с жидким азотом не вносит изменений ' в его
изотопный состав.
Изотопный эффект при кристаллизации из газовой фазы удобно
характеризовать по отношению к исходному газу — метану, введя
определение
д = [^с±С)7-Т00%’
где индекс «1» относится к исходному метану. Величина А может быть
представлена как
613Са — б12^
Л = 1 — 613СХ ’
4*
87
но, учитывая, что 613Ci<C 1, получим
Д = 613Q _ S13Q.
Если при синтезе новая фаза обогащается тяжелым изотопом угле!
рода (по сравнению с исходным метаном), то Д > 0. Когда нара!
щенный углерод обогащен легким изотопом, то Д < 0. Если же,
наращенная фаза наследует изотопный состав исходного газа, то
Д = 0.
В табл. 5 приведены обработанные результаты измерений изо-
топного состава для алмаза. Из таблицы видно, что наращенный
алмаз «тяжелее», чем исходный газ, т. е. обогащен изотопом 13С
по сравнению с исходным метаном. В то же время при наращивании
графита эффект фракционирования имеет'другой знак — нара-
щенный графит обогащен изотопом 12С по сравнению с исходным
метаном.
Результаты исследования изотопного состава графита приведены
также в табл. 5. Если бы не было фракционирования при синтезе
графита, то наращенная на 25% сажа должна иметь 613С =—2,27%.
Для проверки влияния характера подложки на процесс фракцио-
нирования поставлен специальный опыт по наращиванию графита
на алмазных затравочных кристаллах. Было наращено 20% гра-
фита, анализ которого дал 613С = —6,6%, т. е. кристаллическая
решетка подложки не оказывает в этом случае влияния на эффект
фракционирования.
В настоящее время нельзя дать исчерпывающего объяснения на-
блюдаемым эффектам — это дело дальнейших исследований. По-
этому ограничимся общим анализом. Наблюдаемые эффекты не мо-
гут быть объяснены разной подвижностью метана 13СН4 и 12СН4
в газовой фазе. Известно, что при диффузии в высокопористой
среде должно наблюдаться обогащение газа легким изотопом.
Однако тот факт, что алмаз обогащается тяжелым изотопом,
Таблица 5
Фракционирование при росте алмаза и графита
Привес, % Величина &13С, % А, % Привес, % Вели
измерен- ная га Си О га и- д О m X га X £ S чага 1 измерен- । ная
Природный алмаз АМ-1 Синтетиче
А(
0 -0,86 — —
0 -0,87 — — 0 —3,07
13,3 —1,02 -2,20 +2,42 24 -3,0
60,0 —1,02 -1,73 +2,73
чина % А, % Привес, % Величина ЕТС, % А, %
для нара- щенного алмаза измерен- ная для нара- . щепного I алмаза
гский алмаз СМ-1 0 Са —2,31 жа
25,0 —3,17 —6,61 -2,15
—2,71 +1,75 44,6 107,5 -3,47 —3,94 -6,06 —5,46 -1,40 -0,99
88
а графит — легким, исключает такое объяснение. Предположение
о термодинамическом изотопном эффекте следует также исключить,
так как величина коэффициента разделения в системе метан—алмаз
при высоких температурах пренебрежимо мала. Так, уже при
325° С коэффициент разделения, следующий из термодинамического
изотопного эффекта, равен 1,006 [73] и продолжает уменьшаться
при повышении температуры, тогда как наблюдаемый на опыте
коэффициент разделения при 1000° С равен 1,025.
Можно предположить, что открыто новое явление, и его объяс-
нение вряд ли может быть тривиальным. Но ясно, что это явление
относится к той области физической химии, в которой изучаются
поверхностные явления и процессы зарождения новой фазы.
Эффект разделения стабильных изотопов углерода существенно
зависит от скорости кристаллизации. При очень малых скоростях,
как и при очень больших, эффект разделения не наблюдается.
Лишь при определенной скорости образования роста новой фазы,
при определенных пересыщениях и температуре, будет происходить
фракционирование.
Для объяснения разделения изотопов при физико-химическом
синтезе алмаза и графита было сделано нескол ько ориентировоч-
ных расчетов. В соответствии с общей концепцией, изложенной
в этой книге, рост графита рассматривался как процесс, опреде-
ляемый двухмерным зародышеобразованием. В такой постановке
удается связать разделение изотопов с процессами поверхностной
миграции атомов и молекул. Тогда, поскольку зародыши графита
служат как бы стоком для легкого изотопа углерода, они им обо-
гащаются.
Однако такой механизм не является единственно возможным.
Очень интересной представляется общая трактовка Э. М. Галимова.
Поверхность алмаза покрыта хемосорбированным слоем атомов
водорода. Поэтому одной из стадий присоединения атома углерода
к затравочному кристаллу алмаза является образование комплек-
сов СН4-Н. Адсорбированные молекулы метана будут претерпевать
термическую диссоциацию. Так как энергия связи 13С—Н выше,
чем энергия связи 12С—Н, то преимущественно образуются радикалы
12СН3 и комплексы 13СН4-Н (конечно, относительно к общей кон-
центрации соединений соответствующих изотопов).
На тот факт, что в наращивании графита преимущественно
участвуют радикалы СН3, указывают результаты экспериментов
по росту алмаза и графита из смеси метана и водорода. Разбавление
метана водородом препятствует диссоциации метана. Поэтому
скорость роста графита падает значительно сильнее, нежели алмаза.
В этих условиях коэффициент разделения может быть прибли-
женно приравнен отношению скоростей реакций изотопных
форм [73]:
а Кх SlS2 ( Ц'2 Н*
— ‘ Pi / ’
89
где Ki и /С2 — константы скоростей реакции изотопных форм;
Sj и S2, S* и S* — числа симметрии для изотопных форм соединений
и переходных комплексов соответственно; ьц и ti2 — приведенные
массы изотопных форм. Расчет показывает, что а (СН4-Н) = 1,025
и а (СН3) ~ 0,975, что находится в хорошем согласии с наблюдае-
мыми в эксперименте коэффициентами разделения стабильных
изотопов углерода.
7. Определение удельной поверхности порошков
Исследование синтеза алмаза в диффузионной области требует зна-
ния проницаемости слоев алмазных порошков. С этим связано само
определение внутренней диффузионной обла'сти. Это стимулировало
создание нового прибора для определения удельной поверхности
порошков методом фильтрации разреженного газа [77]. Поскольку
этот метод имеет общий интерес, остановимся на нем подробнее.
Под удельной поверхностью понимают поверхность порошка,
деленную на его массу.
В сорбционной технике наибольшее применение находят поро-
шки, частицы или гранулы которых имеют развитую внутреннюю
пористость. Поры между гранулами или транспортные поры облег-
чают или ускоряют приток внешней среды к частицам, а внутрен-
ние поры обеспечивают развитую поверхность с большой емкостью
поглощения тех или иных компонентов среды. Примерами таких
сорбентов служат силикагель, алюмогель и др. Очевидно, что
в этом случае для характеристики рабочих свойств порошка необхо-
димо раздельно характеризовать развитость транспортных пор и
пор внутри частиц.
В соответствии с этим принято различать внешнюю и внутреннюю
удельную поверхность частиц. Первая — Se — характеризует сред-
ний статистический размер (поперечник) частиц d и одновременно
(при компактной упаковке) средний просвет пор, согласно формулам
~i _ 4 . _ 4 е
а~ SeP ’ SeP 1-е ’
где р — плотность частиц; &е — «внешняя» пористость, приравнивае-
мая объемной доле транспортных пор порошка [59].
Внутренняя удельная поверхность S; характеризует поверхность
пор внутри частиц. Общая удельная поверхность очевидно равна
5 = 8е -[- S(.
Для многих материалов (абразивные и полировальные порошки,
наполнители, пигменты и красители, порошки для кристаллизации
капель в переохлажденных облаках и др.) интерес представляет
только внешняя удельная поверхность, т. к. рабочие функции
выполняет только внешняя поверхность.
В тех случаях, когда частицы этого класса имеют внутренние
поры, они могут только ухудшать их технологические свойства^
90 '
в подобных случаях совершенно неоправданно измерять с целью ха-
рактеристики полную удельную поверхность порошков этого рода.
Неоправданным является измерение удельной поверхности порош-
ков этого класса даже при отсутствии пор внутри частиц методами
типа адсорбционных, так как эти методы [78, 79], как правило, бо-
лее трудоемки и сложны. Их же основное преимущество — измерять
полную поверхность — в данном случае является бесполезным.
Гораздо рациональнее поэтому применять более простые фильтраци-
онные методы, которые несравненно удобнее. Во всех случаях, в том
числе для сорбентов, практически важно иметь оптимальную дис-
персность порошков или хотя бы знать средний размер их зерен.
Несмотря на очевидность и элементарность этих соображений, мы
считаем необходимым их привести, так как использование адсорбци-
онных методов измерения удельной поверхности по традиции весьма
распространено и в тех случаях, когда интерес представляет только
определение среднего размера частиц порошка.
Одна из причин преимущественного использования адсорбцион-
ных и других методов, измеряющих полную поверхность, состоит
в том, что фильтрационные методы часто применяют в таком виде,
который чреват серьезными ошибками. Методы же устранения та-
ких ошибок недостаточно разработаны и сформулированы.
Все фильтрационные методы основаны на определении сопро-
тивления течению жидкости или газа через слой порошка. Это сопро-
тивление растет с ростом удельной поверхности. Однако одновремен-
но оно зависит от пористости и структуры слоя частиц. Эта зависи-
мость затрудняет извлечение однозначной информации об удельной
поверхности на основании измерений сопротивления фильтрации.
В зависимости от соотношения средней длины свободного пробега
молекул газа X и характерного размера поровых пространств 7 раз-
личают два предельных режима течения газа: вязкий или пуазейлев-
ский, характеризующийся соотношением % /, и молекулярный
или кнудсеновский, характеризующийся соотношением X !>> I.
Соответственно существуют методы определения удельной поверх-
ности, основанные на вязком течении (см., например, [80]) и ме-
тоды, использующие свободно-молекулярный поток. Методы,'осно-
ванные на газопроницаемости, имеют то преимущество, что они
устраняют взаимодействие, могущее влиять на взаимное расположе-
ние частиц порошка или их размеры (при агрессивном воздействии).
Особо следует отметить необходимость осушения рабочего газа
перед измерением, так как капиллярная конденсация может су-
щественно изменить результаты измерения. Во всех методах га-
зопроницаемости точность и достоверность результатов зависит
от упаковки порошков, их агрегативности. Для методов, использу-
ющих вязкий поток, равномерная укладка более важна, чем в случае
свободно-молекулярного режима.
Для режима кнудсеновского течения (кнудсеновской диффузии
предложены как стационарные [59, 81], так и нестационарные мето-
91
ды [81]. Превышение значений удельной поверхности, найденной
нестационарным методом, над значением, найденным стационарным
методом, для того же образца порошка указывает на внутреннюю
пористость частиц, однако не позволяет ее количественно измерить.
В основу методов определения удельной поверхности дисперс-
ных тел при кнудсеновском режиме течения положен коэффициент
кнудсеновской диффузии D, теоретически найденный Б. В. Деряги-
ным [59] для модели пористого тела или порошка в виде шесоприка-
сающихся непористых шаров. Этот коэффициент равен
D = 1/---------, (26)
где е — пористость; S — удельная поверхность; р2 — насыпная
плотность порошкообразного тела; М — молекулярный: вес; R —
универсальная газовая постоянная; Т — температура.
Метод, основанный на уравнении (26), предполагает1 независи-
мость результатов измерения удельной поверхности от пористости
образца. Однако в реальных системах возможно влияние пористости
на величину удельной поверхности. Основное требование к образ-
цам — равномерность укладки и отсутствие агрегатов. В противном
случае, измерения будут давать удельную поверхность, характери-
зующую набор агрегатов из исходного порошка.
В тех случаях, когда пористость образца велика, т.. е. близка
к единице, может реализоваться, как было показано Б. В. Деряги-
ным и С. П. Бакановым [84], особый режим течения газа, промежу-
точный между кнудсеновским и вязкостным. Он возникает при таком
разрежении газа, когда длина пробега молекул % удовлетворяет од-
новременно двум неравенствам
Здесь d/(e — 1) равно среднему расстоянию между двумя соударе-
ниями о поверхность пор, проходимому при чисто кнудсеновском
режиме, когда длина пробега %m^>d/(e— 1), т. е. величина d/(e — 1)
имеет смысл «среднего просвета» (она при этом значительно больше
расстояния ближайших «соседей»). Этот режим был назван псевдо-
молекулярным. Для него коэффициент D отличается от данного
уравнением (26) дополнительным числовым коэффициентом 0,9.
Этот режим течения хорошо оправдывается при фильтрации через
тонковолокнистые высокопористые фильтры.
Такая модель пористых тел получила полезные применения и
при расчете других кинетических процессов, например, диффузи-
онного разделения изотопических смесей. Эта модель, несмотря на
заведомую идеализацию, дает более близкие к действительности ре-
зультаты расчетов, чем модель цилиндрических капилляров.
С учетом сказанного выше была разработана и изготовлена уста-
новка для определения удельной поверхности порошков!. Основное
отличие от известного прибора Дерягина [81] заключается! в том, что,
92
во-первых, с целью обеспечения наиболее оптимального и естествен-
ного положения частиц порошка в образце и сохранения формы
частиц порошка при проведении опыта, вместо прессования порошка
применялась виброукладка, и, во-вторых, измерения проводились
в квазистационарном режиме течения. При этом пористость образца
измерялась во время фильтрации газа. Изменение пористости
во время эксперимента (или в вакууме) преследовало цель освобо-
диться от адсорбированной воды и посторонних летучих примесей,
способствующих агрегированию порошка. Виброукладка позволяет
измерять удельную поверхность легко деформируемых или агрегиру-
ющих порошков (например, шариков полиэтилена).
Общий вид и принципиальная схема установки показаны на
рис. 50. Стеклянная кювета 1 с исследуемым порошком помещалась на
вибратор 2 и присоединялась к установке через шлифы и вакуумные
резиновые трубки 3. Вибратор 2 питался от ЛАТРа 4 и позволял
проводить виброукладку с частотой 50 гц и различной амплитудой
с одновременным перемешиванием частиц. В работах [82] показано,
что на виброукладку широкого класса порошковых материалов суще-
ственное влияние оказывает частота вибрации; влияние амплитуды
значительно слабее. Там же [82] показано, что плотность виброуло-
женного порошка значительно увеличивается при увеличении часто-
ты вибрации до 50 гц. Если же частота вибрации больше 50 гц, то
ее влияние незначительно. Таким образом достигается наиболее
оптимальная и естественная укладка частиц порошка, что и отвечает
«бесструктурной» модели дисперсного тела [59].
С целью сокращения продолжительности опыта и упрощения
прибора и работы с ним при определении коэффициента диффузии
вместо стационарного течения газа через слой порошка применялось
квазистационарное течение [83]. Под этим понимается течение, ско-
рость которого меняется со временем настолько медленно, что в
каждый момент сохраняется применимость уравнения (26). Если обо-
значить давление над слоем порошка в объеме V через Р, а давление
под слоем — через Ръ то число диффундирующих моделей газа
через слой порошка высотой h в кювете площадью $ в единицу вре-
мени t будет равно
:__L — — d — <£. (27)
RT dt — h >
Предполагая, что Рг Р (это обеспечивается вакуумным насо-
сом) и интегрируя уравнение (27), получим
1п[т?] = -°тт'- - <28>
Из уравнений (26) и (28) получим выражение для удельной поверхно-
сти (в см2/г)
е_ 24-| /~2~-|/^Г jr» S2 t
13 V л Y М V т In [Р (/)/Ро1 * V '
93
F
Рис. 50. Общий вид (а) и прин-
ципиальная схема прибора (б)
для определения удельной по-
верхности порошков
Если при комнатной температуре фильтруется воздух, то I
(30)
где пористость е — Г— Р1/Р2; Pi — плотность порошка в кювете пос-
ле виброукладки; р2 — плотность частиц порошка, г/см3; рх =
= tn/^h; т — навеска порошка, г; £ — площадь кюветы, см2;
h — высота слоя порошка, см; k — —4,30-104 f2/V; b~ln[P (/)/Р0].
34
При фильтрации газа из объема V в объем (в этом случае ваку-
умный насос используется только для предварительного вакуумиро-
вания объема Vj) вместо уравнения (30) получим
где
t,= -4,3-W (4- + х)'
&1 = 1п[АР(0/ДРо].
Для упрощения обработки результатов измерений шкала мано-
метра была проградуирована в величинах Ь< = 1п (РгЛР0).
Как видно из уравнения (28), в координатах Ьг- — t результат
эксперимента должен выражаться прямой линией. На рис. 51 при-
ведены результаты экспериментов по фильтрации из объема в вакуум
для различных толщин образцов алмазного порошка. Тангенс угла
наклона прямых на рис. 51 характеризует внешнюю удельную по-
верхность порошков. Хотя возможно осуществление непрерывной
автоматической записи, экспериментально удобнее снимать 4—5
значений через определенные промежутки времени.
При столь больших первоначальных давлениях фильтрующего-
ся газа, когда вначале не выполняется условие кнудсеновского те-
чения, по мере изменения давления изменяется и наклон кривой,
как показано на рис. 52.
Квазистационарным методом определялась удельная поверхность
различных порошков. Для сравнения удельная поверхность неко-
торых порошков определялась методом низкотемпературной ад-
сорбцией аргона на приборе «Areatron» и при стационарной фильтра-
ции разреженного газа. Для осуществления стационарного режима
фильтрации на том же приборе использовался метан и его свойст-
во иметь давление насыщенного пара 10 мм рт. ст. при температуре
кипения жидкого азота. В этом случае опыты проводились следую-
щим образом. Метан напускался в установку и измерялось его на-
чальное давление Ро. Затем один из баллонов погружался в сосуд
дьюара с жидким азотом и метан конденсировался в нем. После кон-
денсации метан фильтровался через порошок и измерялся перепад
давления на слое порошка. По окончании опыта метан разморажи-
вался и измерялось его давление. Зная время фильтрации, началь-
ное и конечное давления метана в известном объеме V, легко опреде-
лить X [77].
Отметим, что в настоящее время сложилось такое положение,
что удельная поверхность, измеренная методом БЭТ, без должных
оговорок часто принимается в качестве не вызывающего сомнений
эталона, хотя метод БЭТ обладает существенными ограничениями.
На адсорбционную способность поверхности твердого тела су-
щественнно влияет ее энергетическое состояние, наличие дефектов
^примесных атомов, смещенных атомов и других дефектов). Поверх-
95
фильтра*
толщин»
фильтра*!
зной тол-1
ьной по-'
рошков от
классифи-
ность после воздействия ионизирующего излучения может сущест- 1
венно измениться (в зависимости от дозы облучения). При этом гео- 1
метрия частиц порошка, подвергшегося облучению, будет оставаться J
постоянной, вплоть до очень больших доз. В работе [77] показано,
что облучение вызывает увеличение поверхности, измеряемой по ме-Я
тоду БЭТ, тогда как фильтрационные методы дают ту же величину И
удельной поверхности, что и до облучения. Л
Особый интерес представляет определение внешней удельной по-Ч
верхности абразивных, в том числе алмазных порошков, поскольку I
при прочих равных условиях только внешней поверхностью опреде- I
ляется их абразивное действие (и достигаемый класс чистоты обра- I
ботки поверхности [85, 86]). На рис. 53 показана удельная поверх- I
ность различных марок синтетических алмазов. |
Коротко остановимся на применении уравнения (26) к дисперс- |
ному телу, частицы которого имеют поры, сообщающиеся с «внешней» 1
(по отношению к частицам порошка) пористостью. Общую пори- 1
S6 J
стость порошкообразного тела можно представить в виде s =
= в! + в2, где Si, 82 — «внешняя» и «внутренняя» пористости соот-
ветственно. Для такого порошкообразного тела при определении
удельной поверхности из вывода в работе [59] следует, что в
формулах (30) и (31) вместо 8а следует применять выражение
Si (ei + е2). При определении пористости по взвешиванию вычис-
ляется общая пористость е. И если внутренняя пористость мала по
сравнению с внешней, т. е. 82<^8!, то в этом случае можно пользо-
ваться выражением е2 в формулах (30), (31) для определения внеш-
ней удельной поверхности. В этом смысле трудно однозначно интер-
претировать интересные результаты работы [87], в которой для
порошка с внутренней пористостью было получено, что Збэт —
= Sd + Зрт.пор, так как данные по 8Х и 82 не приводятся.
4. ЭПИТАКСИАЛЬНЫЕ АЛМАЗНЫЕ ПЛЕНКИ
И НИТЕВИДНЫЕ КРИСТАЛЛЫ
1. Синтез и свойства алмазных пленок
Эпитаксиальные алмазные пленки, очевидно, могут быть получены
лишь в условиях, когда скорость роста алмаза много больше ско-
рости роста графита. Это также возможно, если подобрать некото-
рый гипотетический травитель, который не действовал бы на алмаз,
но газифицировал зародыши графита. Все усилия по синтезу эпи-
таксиальных алмазных пленок направлены именно на подавление
роста графита при сохранении значительной скорости роста алмаза.
Первые попытки получения алмазных пленок были связаны с на-
ращиванием крупных монокристаллов алмаза (с линейным размером
3—4 мм) в тех же условиях, что и алмазных порошков. Небезынте-
ресно отметить, что при этом на алмазных монокристаллах получены
линейные скорости роста, значительно превышающие линейные ско-
рости роста частиц алмазных порошков.
Возникает естественно важный вопрос, почему нельзя вести нара-
щивание алмазных порошков столь же высокими линейными скоро-
стями. Одна из важнейших причин этого может быть разъяснена
на основе теории роста кристаллов. Для появления каждого нового
слоя атомов на грани алмаза большей частью необходимо, чтобы сна-
чала образовался двухмерный критический зародыш. Вероятность
его образования за единицу времени пропорциональна площади,
поэтому средний промежуток времени между образованием двух за-
родышей обратно пропорционален площади. Если за этот промежу-
ток времени вся грань успеет вымоститься монослоем атомов, то ли-
нейная скорость роста окажется равной частному от деления рас-
стояния соседних атомных слоев на интервал времени между образо-
ванием двух критических зародышей. Если бы эта закономерность
была безгранично применима, то линейная скорость роста, прямо
пропорциональная площади граней, была бы пропорциональна
квадрату линейных размеров кристалла. Однако этот закон справед-
лив с оговоркой.
Когда площадь кристаллической грани переходит известные гра-
ницы, то либо за время появления на ней второго критического за-
родыша она не успеет замоститься, либо это не сможет произойти
из-за того, что грань кристалла не является идеально гладко вымо-
щенной плоскостью и состоит из террас, разделенных ступеньками.
Для замощения каждой террасы именно на ней должен образоваться
критический зародыш.
98
Таким образом, предельная, максимальная линейная скорость
роста граней равна расстоянию между соседними слоями, деленному
на среднее время между появлением двух плоских критических за-
родышей на одной и той же террасе. Размеры террас не только раз-
личны у разных кристаллов и на разных гранях, но и могут менять-
ся, например уменьшаться по мере роста кристалла. В этом послед-
нем случае рост кристалла замедляется.
Иначе обстоит дело с блокированием граней алмаза выделяю-
щимся графитом, образующим на них трехмерные зародыши одина-
ково растущие как в толщину, так и в направлении граней. Образую-
щиеся трехмерные зародыши графита, в отличие от двухмерных за-
родышей алмаза, не способны замостить грань одноатомным слоем
графита, поэтому блокирование каждой грани происходит под сово-
купным действием кристаллических зародышей графита, последо-
вательно образующихся и растущих на ней бок б бок. Вследствие
этого время, по истечении которого процесс блокирования практиче-
ски прекращает или в определенной степени замедляет эпитаксиаль-
ный рост алмаза, не зависит от размера кристалла и не укорачивает-
ся с его увеличением. Отсюда понятно, почему в наших опытах с
большими кристаллами (3—4 мм) можно было получать большую
линейную скорость роста, чем для порошков.
На ранних стадиях кристаллизации из газа заметно проявляется
эффект декорирования, т. е. фиксации деталей строения поверхности
реального кристалла. На рис. 54 показано автодекорирование вы-
росшим алмазом поверхности затравочного кристалла алмаза на
начальной стадии кристаллизации. При дальнейшем росте появля-
ются отдельные островки новой фазы (рис. 55, а), которые затем,
сливаясь, образуют сплошной фронт кристаллизации (рис. 55, б).
Чтобы увеличить скорость роста алмаза, необходимо удалять
графит. Травителем для селективной газификации графита может
Рис, 54. Автодекорированне поверхности алмазного монокристалла на начальной стадии
кристаллизации (х 100 000)
99
Рдс. 55. Стадии последовательного роста эпитаксиальной алмазной пленки
л — начальная стадия; б — стадия образования сплошного слоя
служить водород. Как было показано в предыдущей главе, разбавле-
ние углеродсодержащего газа водородом сильно уменьшает скорость
роста графита, но почти не отражается на скорости роста алмаза.
Также известно, что водород хорошо травит графит при давлениях до
50 атм и температуре 1000° С [45]. Однако можно предположить, что
на графит действует именно атомарный водород. При повышении дав-
ления молекулярного водорода концентрация атомарного водорода
увеличивается пропорционально корню квадратному из общего дав-
ления. Поэтому для газификации используют высокие давления.
Однако можно каталитически [88] или с помощью электрического
разряда [89] создать сверхравновесную концентрацию атомарного
водорода. Так, в работе [48] для удаления графита с алмазных по-
рошков использовался атомарный водород, генерируемый нагретой
вольфрамовой нитью.
Соединение в одном реакторе двух процессов — роста алмаза
(и графита) из углеродсодержащего газа и каталитического получе-
ния атомарного водорода в непосредственной близости от затравоч-
ного монокристалла алмаза — позволило авторам совместно с
Н. Д. Полянской получить и исследовать структуру достаточно
толстых эпитаксиальных алмазных пленок.
Структурные исследования, проведенные с помощью дифрак-
ций медленных электронов под малыми углами, позволили создать
следующую картину роста. Первоначально растет слой совершенно-
го монокристалла (рис. 56, а), затем его структура ухудшается и
наряду с монокристаллическими участками растет поликристал-
лический алмаз (см. рис. 56, б). На следующей стадии растет как
алмаз, так и графит, причем алмаз сохраняет монокристальную
структуру, что видно по линиям Кикучи, которые часто бывают
двойными вследствие наследования двойниковой структуры кри-
сталла-затравки. Далее растет поликристаллический алмаз совмест-
но с графитом и наконец один графит (см. рис. 56, в). Ниже приведе-
ны результаты расчета и табличные значения межплоскостных рас-
стояний на стадии, соответствующей рис. 56, а:
<1ЭКСП, А 2,05 1,27 1,073 0,837 0,814 0,721 0,631 0,598 0,557
^тдбл> А 2,05 1,26 1,072 0,885 0,813 0,721 0,680 0,597 0,558
Некоторые свойства алмазных эпитаксиальных пленок изучены
в работах [90]. Микротвердость алмазных пленок измерялась
методом вдавливания трехгранной алмазной пирамиды на приборе
ПМТ-3. Глубина вдавливания составляла 1 мкм. Измеренная вели-
чина микротвердости равнялась 9500 + 400 кПмм\ что соответст-
вует данным для природного алмаза.
Элементный состав пленок по данным микрорентгеноспектраль-
ного анализа, проведенного на приборе MS-46 («Камека»), отвечает
содержанию углерода 99,5 ± 0,5%. Плотность пленок синтетиче-
ского алмаза, определенная по скорости флотации для трех образ-
цов, оказалась равной 3,29 ± 0,04 г!см3. Показатель преломления-
101
Рис. 56. Электронная дифракция от поверхности плеики при последовательных стадиях
наращивания алмазного монокристалла , л
а — алмазная пленка; б — алмазно-графнтоваЯ пленка; в — графитовая пленка
измеренный по полоске Бекке в иммерсионных средах сера—се-
лен, составил 2,38 ± 0,02 против 2,40 для природного алмаза.
Все эти результаты позволяют утверждать, что методом эпитак-
сиального синтеза возможно получение алмазных пленок, по свойст-
вам максимально близких к природным алмазам.
2. Импульсный способ наращизания алмаза
Импульсный способ наращивания алмаза [91] был предложен в
1967 г. Он преследовал две цели: избавиться от выделения графита
и сделать поверхность алмазного кристалла как можно более актив-
ной. Первая цель достигается созданием периодического импульсно-
го пересыщения, вторая — повышением температуры поверхности
затравки до 2000° К. При создании импульсов пересыщения и пауз
между ними образование и рост алмаза и графита будут протекать
различно. Во время импульса пересыщения образуются критические
зародыши алмаза и графита. Однако при этом зародыши алмаза про-
должают уже имеющуюся подложку (автоэпитаксия!) и будут двух-
мерными. Зародыши графита, выросшие на чужеродной подложке,
трехмерны уже в силу необходимости создания новой фазы. Работа
образования трехмерных зародышей всегда больше работы образо-
вания двухмерных; кроме того, надо учесть, что некоторые грани ал-
маза, например (100), могут расти без образования критического за-
родыша вообще. Поэтому число критических зародышей алмаза
много больше числа критических зародышей графита.
При росте алмаза и графита из метана размеры критическхх заро-
дышей алмаза и графита уменьшаются при повышении температуры.
Во время импульсов пересыщения при периодическом повышении
температуры затравочного кристалла минимальные размеры кри-
тических зародышей соответствуют максимальной температуре. Во
время пауз между импульсами, когда пересыщение по углероду па-
дает, критические размеры зародышей будут увеличиваться, и
часть возникших зародышей алмаза и графита перейдет в разряд
докритических, неустойчивых. Так как работа образования крити-
ческих зародышей графита на поверхности алмаза значительно выше
этой величины для зародышей алмаза, то даже при температуре им-
пульса число зародышей первого, успевших за время импульса пе-
ревалить через критические размеры, будет пренебрежимо мало, а
за время пауз зародыши графита, ставшие докритическими, газифи-
цируются и исчезнут.
С помощью импульсного способа кристаллизации наращены эпи-
таксиальные алмазные пленки, получены нитевидные и изометрич-
ные кристаллы алмаза.
Для создания импульсного пересыщения использована установ-
ка радиационного нагрева, основным элементом которой являлась
ксеноновая лампа сверхвысокого давления. Концентрирование све-
товой энергии осуществлялось одним или двумя эллиптическими
зеркалами-отражателями. Для прерывания лучистого потока исполь-
103
зовался диск с прорезями, который вращался электромотором.
В случае биэллиптической схемы диск перекрывал лучистый поток
в промежуточном фокусе. Изменяя скорость вращения диска, вели-
чину и количество прорезей, можно в широких пределах менять
продолжительности импульсов и пауз между ними.
Затравочный алмазный монокристалл крепился игольчатыми
рениевыми держателями и помещался внутрь шарового кварцевого
реактора, в который после вакуумной тренировки напускался угле-
родсодержащий газ. Оптическая система регулировалась таким об-
разом, чтобы кристалл находился в фокальном пятне установки, раз-
мер которого был больше кристалла. За поверхностью монокри-
сталла во время опыта можно было наблюдать в микроскоп. Его
средняя температура измерялась с помощью оптического пирометра
во время пауз освещения. В качестве углеродсодержащего газа в ос-
новном использовался метан, а также этан, гексан и октан. Затрав-
ками служили природные двойники алмаза, ограненные плоскостя-1
ми (111). j
После проведения наращивания поверхность кристалла рассмат-i
ривалась в микроскоп, фотографировалась, а сам кристалл взве-
шивался с точностью + (2 -г- 3)-10~6 г.
В результате экспериментов было установлено, что оптималь-
ная средняя температура кристалла составляет 900—1100° С, тогда
как в импульсе на поверхности кристалла температуру следовало
доводить до 2000° С и выше. Хотя, как известно, температура гра-
фитации алмаза составляет 1500—1700° С, перехода алмаза в гра-
фит не происходит, вероятно, вследствие малой продолжительности
импульса, во время которого не успевает образовываться критиче-
ский зародыш графита. Продолжительность импульса пересыщения
менялась от 5-Ю"4 до 10-1 сек, а продолжительность пауз между
ними от 5-Ю-2 до 5-Ю"1 сек. При больших частотах импульсов их
влияние уменьшается, очевидно, вследствие тепловой инерции за-
травочного кристалла алмаза, а при продолжительных импульсах
(более 2 сек.) происходит растрескивание и графитация алмаза. Пе-
ред опытом поверхность монокристалла алмаза делалась матовой
травлением на воздухе с целью локализации и усиления нагрева
именно самой поверхности. В процессе роста шероховатости по-
степенно сглаживаются, поэтому на определенном этапе синтеза
шероховатость приходится возобновлять.
Давление метана варьировалось от нескольких долей мм рт. ст.
до 100 мм рт. ст. В диапазоне 20—100 мм рт. ст. изменение дав-
ления не оказывало существенного влияния на результаты синтеза.
(Можно отметить, что нитевидные кристаллы алмаза чаще образу-
ются при меньших давлениях — см. ниже.)
На рис. 57 приведен результат наращивания алмазного моно-
кристалла с исходным весом 9,55 мг. Привес пересчитывался на
толщину наращенного слоя. Метан в реакторе обновлялся каждые
4 часа; тогда же взвешивался кристалл. Условия эксперимента
были следующие: продолжительность импульса 2-Ю-2 сек., частота
104
следования импульсов 5 гц, средняя температура кристалла 950° С,
давление метана 25 мм рт. ст. Метан перед опытом очищался неодно-
кратным замораживанием при температуре жидкого азота и скачи-
ванием неконденсирующегося остатка.
Наращенные алмазные пленки исследовались методом дифрак-
ции быстрых электронов на электронографе ЭГ-100. Было обнару-
жено, что во многих случаях они имеют монокристальную струк-
туру, иногда с незначительными включениями графита. Кристалл
Рис. 57. Изменение веса моно-
кристалла алмаза и толщины
наращенной алмазной пленки
при импульсной кристаллизации
из газовой фазы
может наращиваться не по всей поверхности, а только на некото-
рых участках; при этом образующийся алмаз выделяется в виде от-
дельных микрокристалликов (10—30 мкм). Рассмотрение их с по-
мощью растрового микроскопа показывает, что это изометричные
монокристаллики алмаза, нередко с огранкой.
Авторами совместно с В. П. Варниным удалось наращивать ал-
маз с помощью импульсов пересыщения, создаваемых нагреваемой
электрическим током тонкой металлической пластинкой, которая
располагалась в непосредственной близости от кристалла. Однако
значительная тепловая инерционность такой системы является пре-
пятствием для осуществления импульсной кристаллизации.
Отметим, что с помощью импульсного способа удалось наращи-
вать алмаз под каплями расплавленных металлов. Преимущества
импульсного способа, способствующие росту метастабильной формы
углерода из газовой фазы, вероятно, не ограничиваются системой
алмаз—графит. Имеются основания ожидать, что применение пе-
риодического импульсного пересыщения может оказать существен-
ное влияние на кристаллизацию двух конкурирующих фаз вообще,
а также на получение метастабильных текстур при осаждении из га-
зовой фазы, расплавов и растворов.
3. Нитевидные и изометричные кристаллы алмаза
Нитевидные кристаллы (усы или вискеры) привлекают в настоя-
щее время к себе исследователей в связи с рядом их уникальных
свойств: совершенным монокристаллическим строением, способ-
ностью сохранять упругие свойства при высоких температурах,
способностью после деформации полностью восстанавливать свою
105
форму при последующем нагреве, исключительной прочностью на
разрыв, близкой к теоретической. Например, если прочность на
разрыв обычного железа равна 18—23 кПмм\ то усы из железа
диаметром 2 мкм и длиной ~2 мм имеют прочность на разрыв
1200—1300 кПмм\ Наибольшую прочность, достигнутую к настоя-
щему времени, показывают нитевидные кристаллы графита [36].
Нитевидные кристаллы до сих пор выращивались в области
термодинамической устойчивости. Значение открытия нитевидных
кристаллов алмаза двояко: во-первых, показана возможность выра-
щивания нитевидных кристаллов в метастабильных условиях;
во-вторых, выявлена принципиальная возможность роста алмаза
при низких давлениях из углеродсодержащих газов с очень значи-
тельными линейными скоростями до 0,25 мм! час, намного порядков
превосходящими, например, линейные скорости наращивания ал-
мазных порошков (несколько ангстрем или, в некоторых случаях,
десятки ангстрем в час) [92, 93].
Для проведения экспериментов была использована установка *
радиационного нагрева на основе ксеноновой лампы сверхвысокого
давления, разработанной под руководством В. П. Сасорова. Уста-
новка позволяла фокусировать излучение от лампы на поверхности
затравочного алмазного монокристалла, который крепился специ-
альными рениевыми игольчатыми держателями и помещался в сфе-
рический кварцевый реактор. Температура монокристалла измеря-
лась оптическим пирометром. За поверхностью затравочного мо-
нокристалла можно было наблюдать в микроскоп.
В этих экспериментах было установлено образование и последу-
ющий рост нитевидных кристаллов алмаза. Скорость их роста зави-
сит от температуры, давления и некоторых других параметров. По-
скольку плотность лучистого потока, применяемого для нагрева
кристалла, существенно неоднородна, в процессе роста нитевидных
кристаллов условия существенно меняются. Так, при образовании
нитевидного кристалла длиной 400 мкм облученность его основания
и вершины иногда различалась вдвое. Средняя скорость роста ните-
видных кристаллов составляла 10 мкм!час, но в отдельных случаях
достигала 50—250 мкм!час.
На рис. 58 приведена фотография нитевидного кристалла алмаза
длиной —0,4 мм, выросшего на грани октаэдра алмазного затравоч-
ного кристалла. На рис. 59 показан нитевидный кристалл алмаза,
выросший на боковой грани затравочного кристалла. Длина этого
вискера составляла —1 мм. На снимке виден рениевый держатель.
Специальные опыты показали, что при облучении потоком элект-
ронов спектр люминесценции нитевидных кристаллов аналогичен
спектру люминесценции подложки. Для более точной идентифика-
ции полученных образований после одного из опытов нитевидные
кристаллы были осторожно отделены от подложки, раздроблены и
* Установка радиационного нагрева была создана в Отделе поверхностных
явлений ИФХ АН СССР Б. В. Дерягиным, Г. Г. Лопатиной и Б. В. Спицы-
ным.
106
Рис. 59. Нитевидный кристалл алмаза длиной ~1 мм
помещены в объектодержатель электронного микроскопа. Резуль-
таты исследования дифракции электронов позволили определить
следующие межплоскостные расстояния: 2,06; 1,27; 1,068; 0,883;
0,827; 0,719; 0,684; 0,625 А. В то же время известно, что межплос-
костные расстояния в алмазе равны соответственно 2,05; 1,26;
1,072; 0,885; 0,813; 0,721; 0,680; 0,620 А.
Полученные электронограммы указывают на монокристалличе-
ский характер нитевидных кристаллов.
В некоторых опытах было отмечено, что на вершинах нитевидных
кристаллов алмаза имеются темные полусферы, возникшие, вероят-
но, в результате осаждения перед опытом металлических частиц
на поверхность алмаза — подложки. Поэтому были поставлены спе-
циальные опыты по росту алмазных усов под расплавленными кап-
лями металлов. Подобный метод кристаллизации называется ме-
тодом VLS («vapor—liquid—solid»— пар—жидкость—твердое тело).
107
Рнс. 60. Изометричиый кристалл алмаза диаметром 20 мм
Снимок сделан в сканирующем электронном микроскопе
В нашем случае углеродсодержащий газ разлагается на поверх-
ности расплавленного металла, углерод растворяется в металле,
диффундирует через него и высаживается на поверхности затравки.
Были получены нитевидные кристаллы алмаза с капельками за-
стывшего металла на вершине. Положительные результаты дали
металлы, хорошо растворяющие углерод и смачивающие алмаз:
никель, железо, марганец, тогда как в случае расплавленного
золота никаких образований получено не было [94]. Очевидно можно
считать, что одним (но не единственным) из возможных механизмов
роста алмазных нитевидных кристаллов является механизм по мето-
ду VLS.
Роль промежуточной металлической фазы для выращивания ал-
маза можно объяснить тем, что поверхностная энергия на границе
металл—графит намного больше, чем на границе графит—газ, в ре-
зультате чего резко увеличивается работа образования зародышей
графита, а его выделение подавляется.
Значительный интерес представляет факт . превращения ните-
видных кристаллов в изометричные [95]. На рис. 60 приведена фо-
тография изометричного кристалла алмаза диаметром 20 мкм, сде-
ланная с помощью сканирующего электронного микроскопа.
Было обнаружено, что первоначально растет нитевидный кри-
сталл, затем его рост останавливается, и он превращается в изо-
метричный. Подобный процесс, когда из вискера вырос огранен-
ный кристаллик средним размером 0,1 мм, наблюдался в микроскоп.
Этот кристаллик В. Г. Лютцау исследовал микрорентгенографиче-
ски по методу Лауэ (на прохождение) в трех местах: у его вершины,
108
в середине и у основания [95]. Использовалась рентгеновская остро
фокусная трубка БСВ-5 с молибденовым анодом (аппарат АРС-4).
Применялась рентгеновская микрокамера, которая позволяла лока-
лизовать площадь исследуемого участка на кристаллике до 12 мкм
в диаметре. Кассета с пленкой располагалась на расстоянии 5 мм
от объекта; оптическая система камеры позволяла устанавливать
участки кристаллика для исследований с точностью до нескольких
микрометров.
Рентгеновский пучок ориентировался в направлении, совпа-
дающим с осью [111]. Как следовало из рентгенограмм, все три иссле-
дуемых участка представляли собой монокристаллические обла-
сти, соответствующие ориентации в направлении первичного пучка
рентгеновских лучей. Это свидетельствует о том, что выросший из
газовой фазы кристалл, а следовательно, и образовавшийся в на-
чале вискер являются единым монокристаллом. Некоторая вытяну-
тость лауэ-пятен свидетельствует об искажении кристаллической
решетки (астеризме) исследованного кристаллика.
Известно, что алмаз может кристаллизоваться в природных ус-
ловиях в виде сферолитов [54]. О том, что при кристаллизации крем-
ния из газовой фазы получаются образования сферической формы,
также известно (см., например, [96]). Однако в рассматриваемом
случае получены изометричные кристаллы в заведомо метастабиль-
ных условиях.
Возможность роста нитевидных кристаллов алмаза под каплями
металлов по механизму VLS экспериментально доказана. В то же
время нельзя отрицать возможности роста по дислокационному ме-
ханизму. Так, число кристалликов, выросших на поверхности за-
травочного монокристалла, близко к числу дислокаций в природном
алмазе [97] и составляет приблизительно 104/слА Алмазная пленка
может наследовать дислокации подложки [98]. Кроме того, даже
в самых чистых условиях проведения экспериментов, выделяющий-
ся графит будет служить своего рода примесью, блокирующей рост
нитевидного кристалла алмаза. Влияние примеси на рост нитевид-
ных кристаллов рассмотрено в работах [99].
При описании изометричных кристаллов алмаза нельзя обойти
вниманием тот факт, что при наращивании высокодисперсных ал-
мазных порошков образуются трудноразрушаемые комочки. Даже
если перед экспериментом порошок тщательно растерт, после прове-
дения опыта комочки всегда возникают. Очевидно, здесь имеет место
спекание через выделившуюся новую фазу.
При описании роста графитовых нитевидных кристаллов уже
указывалось на специфическое воздействие лучистого нагрева на
кристаллизацию из газовой фазы. Теоретически этот вопрос рас-
смотрен в работе [100] в применении к кристаллизации кремния из
газовой фазы. Экспериментально было установлено, что скорость
роста кремния при лучистом нагреве превышает скорость роста
кремния в аналогичных условиях (давлении, температуре) при
использовании омического нагрева [101]. Это находит свое объяс-
109
Рис. 61. Покрытие из рутила на поверхности непрерывного волокна (X 200)
иение в электронной теории катализа и адсорбции Ф. Ф. Волькен-
Штейна [102], Действительно, воздействие облучения вызывает
увеличение плотности хемосорбированных атомов, что приводит
к увеличению скорости роста. При этом увеличивается лишь предъ-
экспоненциальный множитель, но энергия активации остается неиз-
менной, как и наблюдается экспериментально.
В заключение сделаем несколько замечаний об использовании
лучистого нагрева. В применении к гетерогенной кристаллизации
этот метод представляется весьма перспективным. Поэтому в ИФХ
АН СССР, в Отделе поверхностных явлений, были развиты различ-
ные методы лучистого нагрева для проведения исследований по фи-
зико-химической кристаллизации. Оптические печи на основе ксено-
новых ламп высокого давления описаны в работе [103]. Установка
на основе инфракрасного лазера описана в главе II.
Основными преимуществами лучистого нагрева являются сле-
дующие.
1. Возможность проведения экспериментов в контролируемой
газовой среде, когда нагревается локально лишь небольшой участок
затравки.
2. Практическая безынерциоиность нагрева.
3. Возможность концентрации очень высокой мощности на огра-
ниченном участке.
Действительно, возьмем случай, когда на непроводящее волокно,
например сапфировое, необходимо нанести покрытие из газовой
фазы при высокой температуре. Прежде всего возникает вопрос
о нагреве подложки в заданной среде. Омический и индукционный
нагревы волокна в этих условиях не подходят. Из традиционных
методов возможен лишь кондуктивный, когда вся среда, в котором
находится волокно, будет нагрета до температуры подложки. Одна-
110
ко при этом возникают два осложнения. Во-первых, хотя надо на-
греть только поверхность волокна, экспериментатор будет вынуж-
ден нагревать несравненно больший объем, и даже при использова-
нии сразу многих волокон неудобство не снимается. Во-вторых,
в газовой фазе при высоких температурах могут развиться гомо-
генные реакции, что повлияет на ее химический состав.
При использовании лучистого нагрева подобные осложнения не
возникают. При этом можно получать самые тугоплавкие покрытия
в установке, сделанной из стекла. В качестве иллюстрации на
рис. 61 приведена фотография покрытия из двуокиси титана (рути-
ла), нанесенного на плохопроводящую подложку. Отметим, что в за-
висимости от условий могут быть получены покрытия с различными
кристаллическими свойствами. Таким образом, в настоящее время
не только доказана сама возможность синтеза алмаза из газовой
фазы, но и исследованы многие закономерности его роста. Выявле-
ны основные области практического использования наращивания
затравочных алмазных кристаллов. На очередь встают новые вопро"
сы и из них самый главный: переход от алмазной подложки к дру-
гим, инородным, подложкам. В принципе рост алмаза из газовой
фазы на неалмазных подложках возможен, задача сводится к нахож-
дению оптимальных условий этого процесса. Теоретической основой
этого направления исследований служит теория образования и роста
новой фазы в сочетании с теориями кинетики адсорбции и химиче-
ской кинетики.
Непосредственно в изучении кристаллизации алмаза из газовой
фазы усилия исследователей будут направлены на разработку
более общей теории совместной кристаллизации алмаза и графита
с целью выбооа условий для предотвращения выделения последнего.
ЛИТЕРАТУРА
1. Б. В. Дерягин, Д. В. Федосеев.
Успехи химии, 39, 1161 (1970).
2. Б. В. Дерягин, В. Н. Бакуль,
Д. В. Федосеев. Ысник АН УССР,
№ 5, 80 (1971).
3. Сб. «Поверхностные силы в тон-
ких пленках и дисперсных си-
стемах». Под ред. Б. В. Деря-
гина. М., «Наука», 1972.
4. Б. В. Дерягин, Д. В. Федосеев,
Б. В. Спицын, О. В. Спирин.
В сб. «Физико-химические про-
блемы кристаллизации», вып. 2.®
Алма-Ата, Изд-во КазГУ, 1971,
стр. 24; Б. В. Дерягин, Д. В. Фе-
досеев, О. В. Спирин. ТЭХ, 6,
824 (1970).
5. Д. Хирс, Г. Паунд. Испарение^
и конденсация. М., «Металлур-
гия», 1966.
6. Р. Ф. Стрикленд-Констчбл. Ки-
нетика и механизм кристаллиза-«
ции. Л., «Недра», 1971.
7. А. А. Чернов. В сб. «Физико-хи-
мические проблемы кристаллиза-а
ции». Алма-Ата, Изд-во КазГУ,
1969, стр. 8.
8. Р. Лодиз, Р. Паркер. Рост мо-
нокристаллов. М., «Мир», 1974.
9. Л. С. Палатник, М. Д. Фукс,
В. М. Косевич. Механизм обра-
зования и субструктура конден-
сированных пленок. М., «Наука»,
1972.
10. Б. В. Дерягин. ТЭХ, 9, 471 (1973).
11. В. V- Deriaguin. J. Chem. Phys.,
61, 3664 (1974).
12. Д. Б. Зельдович. ЖЭТФ, 12,
525 (1942).
13. Д. И. Френкель. Кинетическая
теория жидкостей. М., Изд-во
АН СССР, 1945; ТЖТФ, 9, 192
(1939).
14. Д. В. Федосеев. ТЭХ, 10, 176
(1974).
15. В. V. Derjaguin. J. Colloid and
Interface Science, 38, 517 (1972).
16. В. В. Карасев, И. А. Кротова,
Б. В. Дерягин. ДАН СССР, 88,
777 (1953).
17. В. V. Derjaguin, R. Green-Kelly.
Trans. Faraday Soc., 60, 449
(1964).
18. H. В. Чураев. В сб. «Успехи
коллоидной химии», М., «Наука»,
1973, стр. 169; Н. В. Чураев.
В сб. «Поверхностные силы в тон-
ких пленках и устойчивость кол-
лоидов». М., «Наука», 1974,
стр. 90.
19. Б. В. Дерягин, Ю. М. Попов-
ский. ДАН СССР, 159, 89 (1964);
175, 385 (1967); 207, 1153 (1972).
20. В. В. Карасев, Б. В. Дерягин.
ЖФХ, 33, 100 (1959).
21. Д. В. Федосеев, В. П. Варнин,
Б. В. Дерягин. ДАН СССР,
193, 1290 (19701.
22. В. П. Варнин, Д. В. Федосеев,
Б. В. Дерягин. В сб. «Физико-
химические проблемы кристал-
лизации», вып. 2. Алма-Ата,
Изд-во КазГУ, 1971, стр. 18.
23. Б. Хонигман. Рост и форма
кристаллов. М., ИЛ, 1961. е
24. Л. А. Боровинский, Р. П. Во-*
ронцова. В сб. «Рост кристаллов».
М., «Наука», 1968.
25. Сб. «Химические и физические а
свойства углерода». Под ред.
Ф. Уокера. М., ИЛ, 1969.
26. П. А. Теснер. Образование угле-
рода из углеводородов газовой
фазы. М., «Химия», 1972.
27. Д. В. Федосеев, С. П. Внуков.
ДАН СССР, 209, 1162 (1973).
28. Д. В. Федосеев, С. П. Внуков,
В. П. Варнин. ДАН СССР, 215,
151 (1974).
29. П. А. Теснер, И. С. Рофалькес.
ДАН СССР, 80, 401 (1951).
30. П. А. Теснер, М. М. Полякова,
С. С. Михеева. ДАН СССР, 203,
402 (1972).
112
31. В. Н. Кондратьев. В сб. «Хи-д
мическая кинетика и цепные реак-
ции». М., «Наука», 1966, стр. 165.
32. П. С. Шантарович, Б. В. Пав-
лов. ЖФХ, 34, 960 (1960).
33. П. А. Теснер, М. М. Полякова,
С. С. Михеева. Труды ВНИИ
природных газов. Переработка
и использование природных га-
зов, вып. 40/48, 8 (1969).
34. Л. А. Анисонян, П. А. Теснер.
Труды ВНИИ природных газов.
Переработка и использование
природных газов, вып. 12/20,
71 (1960).
35. А. Е. Городецкий и др. ДАН
СССР, 203, 1336 (1972).
36. Г. В. Бережкова. Нитевидные
кристаллы. М., «Наука», 1969.1
37. R. Bacon. Bull. Amer. Phys. Soc.,
3, 108 (1958).
38. П. А. Теснер, А. И. Ечеистова.
ДАН СССР, 87, 1029 (1952).
39. Л. В. Радушкевич, В. М. Лукья-
нович. ЖФХ, 26, 88 (1952).
40. Б. В. Дерягин и др. В сб. «Гра-
фиты и их применение в про-
мышленности». Материалы се-
минара. М., «Знание», 1974,
стр. 30.
41. Монокристальные волокна и ар-
мированные ими материалы. Под *
ред. А. Т. Туманова. М., «Мир»,
1973.
42. В. И. Касаточкин и др. ДАН
СССР, 201, 1104 (1971).
43. V. Bolton. 7. Elektrochemie, 17,
971 (1911).
44. О. И. Лейпунский. Успехи хи-
мии, 8, 1519 (1939).
45. W. G. Eversole. Pat. USA 3030187,
3030188 (1962).
46. Н. G. Hybschman. Патент США,
3371996 ('1968).
47. G. С. Angus, Н. Will, Д’- Stanko.
J. Appl. Phys., 39, 2915 (1968).
48. Дж. Ангус, H. Гарднер, Д. По-
ферл, С. Шаухан, Т. Дабл, Пай
Сунг. В сб. «Синтетические алма-
зы в промышленности». Киев,
«Наукова думка», 1974, стр. 30.
49. Патент Франции 1366544 (1964).
50. Патент Франции 1367388 (1964).
51. В. V. Derjaguin, D. V. Fedoseev,
В. V. Spytsin. J. Crystal. Growth,
3, 4, 111 (1968).
52. G. A. Brinkman, C. Park, C. J.
Melchan. Патент США 3172539
(1964).
53. G. A. Brinkman, C. Park, C. J.
Melchan. Патент США 3175885
(1965).
54. В. Г. Васильев, В. В. Ковальский,
Н. В- Черский. Происхождение
алмазов. М., «Недра», 1968;
И. И. Шафрановский. Алмазы.
М.— Л., «Наука», 1964.
55. Г. П. Вдовыкин. Алмазы в метео-
ритах. М., «Наука», 1970.
56. В. М. Гольянов, А. П. Демидов.
Патент Франции 2157957 (1973).
57. Б. В. Дерягин, Д. В. Федосеев,
К- С. Успенская. ЖФХ, 47, 24
(1973); Д. В. Федосеев, Б. В. Де-
рягин, В. П. Варнин, К- С. Ус-
пенская. Там же, стр 28;
Д. В. Федосеев, В. П. Варнин,
Б. В. Дерягин. Там же, стр. 32.
58. В. И. Бакуль. В сб. «Синтети-Я
ческие алмазы», вып. 6(12). Киев,
«Наукова думка», 1970, стр. 3.
59. Б. В. Дерягин. ДАН СССР, 53,
627 (1946).
60. Д. А. Франк-Каменецкий. Диф-
фузия и теплопередача в хими-
ческой кинетике. М., «Наука»,
1967.
61. L. S. Kassel. J. Amer. Chem.
Soc., 70, 3949 (1932).
62. N. C- Gardner. Патенты США
3630678 (1971); 3661526 (1972)."
63. H. Н- Семенов. О некоторых про-
блемах химической кинетики и
реакционной способности. М.,
Изд-во АН СССР, 1958. .
64. Э. Г. Сабо. В сб. «Химическая
кинетика и цепные реакции».
Под ред. В. Н. Кондратьева. М.,
«Наука», 1966, стр. 47.
65. Патент Англии 1322242 (1972).—
66. К- С- Успенская и др. ТЭХ, 5,
133 (1969).
67, В. V. Derjaguin et al. Powder
Technol., 9, 15 (1974).
68. Сб. «Поверхностная диффузия и
растекание». Под ред. Я. Е. Ге-
гузина. М., «Наука», 1969.
69. Д. В. Федосеев, Г. Н. Пирогова,
В. П. Варнин. ДАН СССР, 214,
618 (1974).
70. Б. В. Дерягин и др. Физико-хи-
мический синтез алмаза из газа.
Киев, «Техтка», 1971.
71. Р. К- Чужко и др. В сб. «Физико-
химические проблемы кристал-
лизации», вып. 2. Алма-Ата,
Изд-во КазГУ, 1971, стр. 186.
72. Е. В. Денисович, П. А. Теснер.
ДАН СССР, 212, 660 (1973).
113
73. Э. М. Галимов. Геохимия ста-
бильных изотопов углерода. М.,
«Недра», 1968.
74. Д. В. Федосеев, Э. М. Галимов,
В. П. Варнин, В. С. Прохоров,
Б. В. Дерягин. Письма в ЖЭТФ,
14, 80 (1971); ДАН СССР, 201,
1149 (1971).
75. В. В. Ковальский, Э. М. Галимов,
В. С. Прохоров. ДАН СССР,
203, 440 (1972).
76. Э. М. Галимов, В. А. Гриненко,
В. И. Устинов. ПТЭ, № 3, 159,
(1965).
77. Б. В. Дерягин и др. Препринт
доклада на конференции «Син-
тетические алмазы — ключ к тех-
ническому прогрессу», Киев,
21—24 августа 1974 г.; Б. В. Де-
рягин, С. П. Внуков, Д. В. Фе-
досеев. В сб. «Расширенные тези-
сы докладов на 4-й Всесоюзной
конференции по теоретическим
вопросам адсорбции». М.,«Наука»,
вып. 1, 102 (1973).
78. С. Григ, К- Синг. Адсорбция,
удельная поверхность, пори-
стость. М., «Мир», 1970.
79. С. Orr, J. М. Dallavalle. Fine
particle measurement. New York,
Pergamon Press, 1959.
80. Г. С. Ходаков. Основные ме-
тоды дисперсионного анализа по-
рошков. М., Стройиздат, 1968;
В. В. Паничкина, И. В. Уварова.
Методы контроля дисперсности
и удельной поверхности метал-
лических порошков. Киев, «Нау-
кова думка», 1973.
81. Б. В. Дерягин, Н. Н. Захаваева,
М. В. Галаев, В. В. Филиппов-
ский. Определение удельной по-
верхности порошкообразных тел
по сопротивлению фильтрации
разреженного воздуха. М.,
Изд-во АН СССР, 1957.
82. И. Г. Шаталова, Н. С. Горбу-
нов, В. И. Лихтман. Физико-
химические основы вибрацион-
ного уплотнения порошковых ма-
териалов. М., «Наука», 1964; В. В.
Иващенко, И. П. Тартаковский,
Т. М. Голубев. Порошковая ме-
таллургия, № 8, 35 (1965); № 12,
28 (1970);
83. Д. П. Тимофеев. Кинетика адсор-
бции. М., Изд-во АН СССР, 1962.
84. Б. В. Дерягин, С. П. Баканов,
ДАН СССР, 115, 267 (1957);
ЖТФ, 27, 2056 (1957).
85. С. П. Внуков, Б. А. Аникин,
3. А. Рябикова. Синтетические \
алмазы, № 5, 36 (1971).
86. В. Н- Бакуль. Новые техничес-
кие условия на порошки из-
синтетических алмазов. Киев
УкрНИИНТИ, 1969.
87. J. F. Brock, С. Orr. Analyt.
Chem., 44, 1534 (1972).
88. Д. Элей. В сб. «Катализ. Вопросы
теории и методы исследования».
М., ИЛ, 1955, стр. 152.
89. В. И. Кондратьев. Кинетика хи-
мических газовых реакций. М.,
«Наука», 1968.
90. Б. В. Дерягин и др. В сб. «Физи-
ко-химические проблемы кри-ц
сталлизации», вып. 2. Алма-Ата/
Изд-во КазГУ, 1971, стр. 90;
Б. В. Дерягин и др. ДАН СССР,
213, 1059 (1973).
91. Б. В. Дерягин, Д. В. Федосеев.
ДАН СССР, 213, 1304 (1973).
92. Б. В. Дерягин и др. ДАН СССР,
181, 1094 (1968).
93. В. V. Derjaguin et al. J. Crys-
tal Growth, 2, 380 (1968).
94. Б. В. Дерягин и др. Кристалло-
графия, 14, 535 (1969).
95. Б. В. Дерягин, В. Г. Лютцау,
Д. В. Федосеев, В. А. Рябов.
ДАН СССР, 190, 86 (1970).
96. Е. И. Гиваргизов. В сб. «Рост
кристаллов», т. 8, ч. II. М.,
«Наука», 1968, стр. 250.
97. В. М. Зубков, А. С. Семенова-Тян-
Шанская, Н- И. Епишина. Ал-
мазы, вып. 1, 3 (1969).
98. А. В. Бочко, Б. В. Дерягин.
ДАН СССР, 181, 1378 (1968).
99. Д. В. Федосеев, Н. Д. Полянская.
В сб. «Физико-химические про-
блемы кристаллизации», вып. 2.
Алма-Ата,Изд-во КазГУ,1971,стр.
79; ДАН СССР, 198,1143(1971).
100. И. Г. Варшавская, С. В. Банце-
ков, Г. Г- Лопатина. В сб. «Фи-
зико-химические проблемы кри-
сталлизации», вып. 2. Алма-Ата,
Изд-во КазГУ, 1971, стр. 52.
101. Б. В. Дерягин и др. В сб. «Рост
кристаллов», т. 9. М., «Наука»,
1972, стр. 210.
102. Ф. Ф. Волькенштейн. Электрон-
ная теория катализа на полу-
проводниках. М., Физматгиз,
1960.
103. Г. Г. Лопатина и др. Оптические
печи. М., «Металлургия», 1969
ОГЛАВЛЕНИЕ
Предисловие............................... 3
1. ОСНОВНЫЕ ЗАКОНОМЕРНОСТИ НУКЛЕАЦИИ 5
1. Теория гомогенной нуклеации............ 5
2. Влияние поверхностных сил на образование и
свойства новой фазы...................... 15
3. Теория гетерогенной нуклеации......... 22
2. РОСТ ГРАФИТА ИЗ ГАЗОВОЙ ФАЗЫ.............. 31
1. Физико-химическая теория роста графита . . 31
2. Экспериментальная проверка предложенной
теории роста графита..................... 36
3. Рост графита в неизотермичных условиях ... 42
4. Нитевидные кристаллы графита............44
3. КИНЕТИКА
ФИЗИКО-ХИМИЧЕСКОГО 'СИНТЕЗА АЛМАЗА 52
1. Из истории синтеза алмаза.............. 52
2. Кинетика роста алмаза на алмазных порошках 58
3. Циклический метод наращивания алмазных
порошков............................... 71
4. Механизм роста алмаза.................. 76
5. Синтез алмаза из смесей углеводородов (не-
аддитивность скоростей реакций).......... 83
6. Фракционирование стабильных изотопов угле-
рода ................................... 86
7. Определение удельной поверхности порошков 90
4. ЭПИТАКСИАЛЬНЫЕ АЛМАЗНЫЕ ПЛЕНКИ
И НИТЕВИДНЫЕ КРИСТАЛЛЫ .................. 98
1. Синтез и свойства алмазных пленок...... 98
2. Импульсный способ наращивания алмаза . . 103
3. Нитевидные и изометричные кристаллы алмаза 105
Литература............................... 112
115