/


























































































































































































































































































































Автор: Чеботин В.Н.
Теги: химия физика физическая химия физика твердого тела химия твердого тела хими
Год: 1982




Текст
В. И. Чеботин
Физическая
химия
твердого
тела
МОСКВА, «ХИМИЯ», 1982
УДК 541.1 :548.3
Чеботин В. Н.
Физическая химия твердого тела. — М.: Химия,
1982. — 320 с, ил.
Дано систематическое изложение наиболее важных разделов
современной физической химии твердого тела — теории разупорядоченности в
кристаллах с различными типами химической связи, теории явлений переноса,
определяющих протекание химических реакций в твердых телах,
адсорбционных явлений и кинетики роста твердых фаз.
В книге используется единый математический аппарат, основанный на
методах статистической термодинамики, для описания как статических, так
и кинетических характеристик твердых тел с различными типами
химической связи: металлов, валентных полупроводников и ионных кристаллов.
Предназначена для научных работников, инженеров, аспирантов и сту«
дентов, специализирующихся в области химии и физики твердого тела.
320 с, 12 табл., 67 рис., список литературы 106 ссылок.
Рецензенты: ТРЕТЬЯКОВ Ю. Д. докт. хим. наук проф.,
ЖУКОВСКИЙ В. Н. докт. хим. наук проф., рекомендована
Ученым советом Института электрохимии Уральского научного
центра АН СССР.
Редактор В. И. КОЗЛОВА
Художник А. Я. МИХАЙЛОВ
Художественный редактор Н. В. НОСОВ
Технический редактор В. М. СКИТИНА
Корректор В. М. БЕЛЯЕВА
ИБ № 1387
Сдано в наб. 20.05.82. Подп. к печ. 5.11,82. T-14668. Формат бумаги 60X90Vie.
Бумага тип. № 2. Гарн. литературная. Печать высокая. Усл. печ. л. 20,0.
Усл. кр.-отт. 20,0. Уч.-изд. л. 20,21. Тираж 5500 экз. Заказ № 321. Цена 3 р. 10 к.
Изд. № 2399.
Ордена «Знак Почета» издательство «Химия». 107076, Москва, Стромынка, д. 13.
Московская типография № 11 Союзполиграфпрома при Государственном
комитете СССР по делам издательств, полиграфии и книжной торговли.
Москва, 113105, Нагатинская ул., д. 1.
1805000000-001
4 050(01)-82 !'82
© Издательство «Химия», 1982 г
Содержание
Предисловие . ...... 5
Основные обозначения о
Глава 1.
КЛАССИФИКАЦИЯ И ОСНОВНЫЕ ФИЗИКО-ХИМИЧЕСКИЕ
свойства твердых тел и
1.1. Кристаллические и аморфные твердые тела 11
1.2. Металлы .... .12
1.3. Неметаллические твердые тела 14
1.4. Диффузия и ионная проводимость. Атомные дефекты ... 20
1.5. Эффективные заряды дефектов 26
1.6. Электронная проводимость неметаллических кристаллов.
Электронные дефекты 28
1.7. Энергетические состояния электронов в нестехиометрических
кристаллах 41
1.8. Протяженные дефекты 44
1.9. Твердые тела со структурной разупорядоченностью .... 50
1.10. Структура и транспортные процессы в стеклах 54
Глава 2.
СТАТИСТИЧЕСКАЯ ТЕРМОДИНАМИКА ДЕФЕКТНЫХ
КРИСТАЛЛОВ 57
2.1. Модельные представления 57
2.2. Химические потенциалы точечных дефектов 59
2.3. Закон действия масс для квазихимических реакций между
дефектами 65
2.4. Химические потенциалы атомов и ионов . . .69
Глава 3.
РАЗУПОРЯДОЧЕННОСТЬ МЕТАЛЛОВ И
ИНТЕРМЕТАЛЛИЧЕСКИХ СОЕДИНЕНИИ 71
о'о* ^>азУП0РяД°ченн°сть чистых металлов . ... 72
ч ?" ^азУпоРяД°че,нность интерметаллических соединений .... 76
«э.З. Концентрации дефектов в нестехиометрических
интерметаллических соединениях 79
^•4. Упорядочение твердых растворов замещения 87
«э.э. Фазы внедрения 97
Глава 4.
РАЗУПОРЯДОЧЕННОСТЬ ПОЛУПРОВОДНИКОВ .... 101
А' Собственные полупроводники ... . . . . 102
?•*• Примесные полупроводники 104
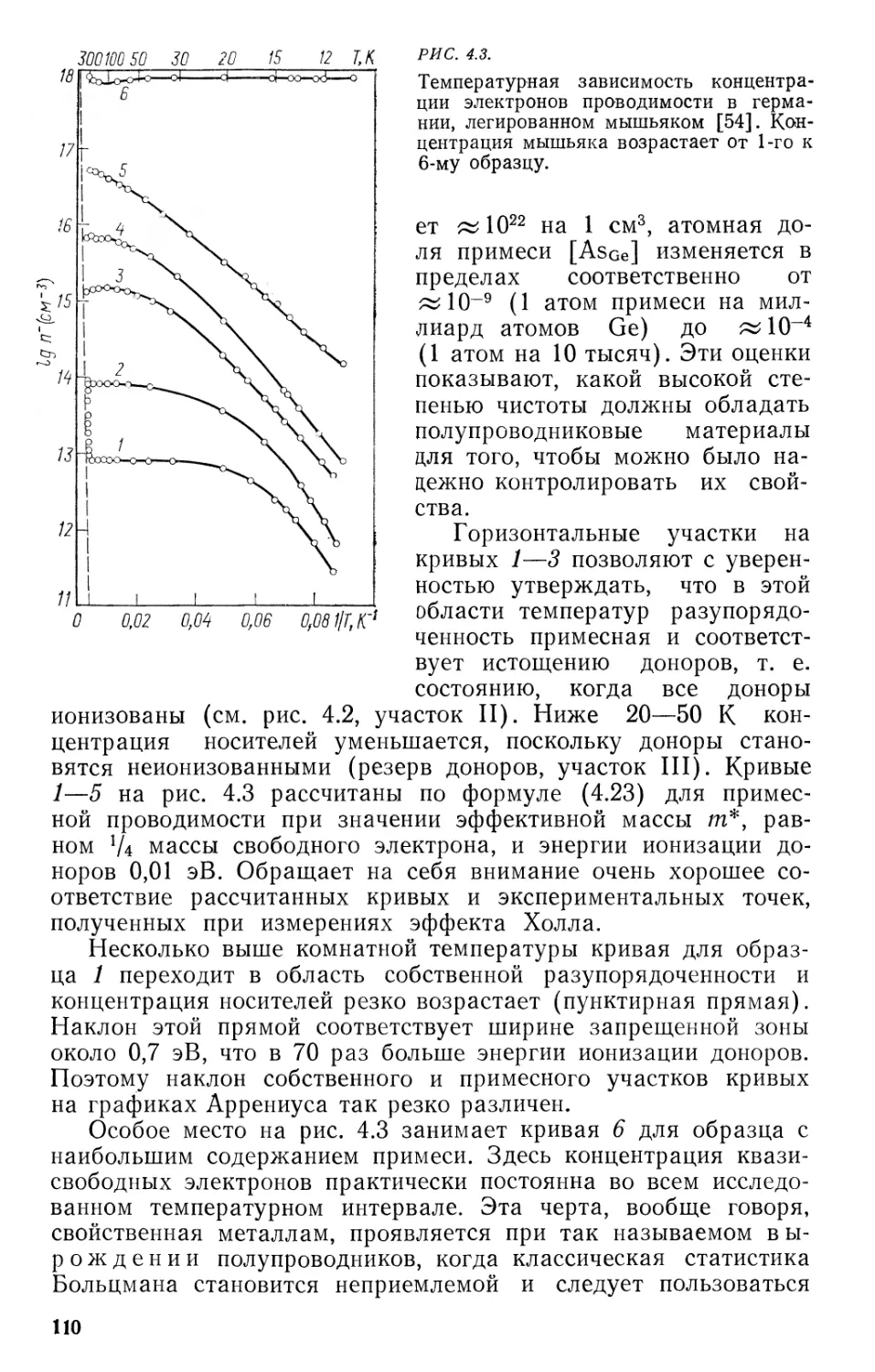
4«о. Собственно-дефектные полупроводники 111
• Пестехиометрические полупроводниковые соединения . . . . 115
Глава 5.
РАЗУПОРЯДОЧЕННОСТЬ ИОННЫХ КРИСТАЛЛОВ ... 129
5*2' Собственная ионная разупорядоченность . 131
„ ' • Афимеоная ионная разупорядоченность .136
5.3. Ассоциация дефектов в примесных ионных кристаллах . . 141
5.4. Разупорядоченность чистых нестехиометричеоких ионных
кристаллов
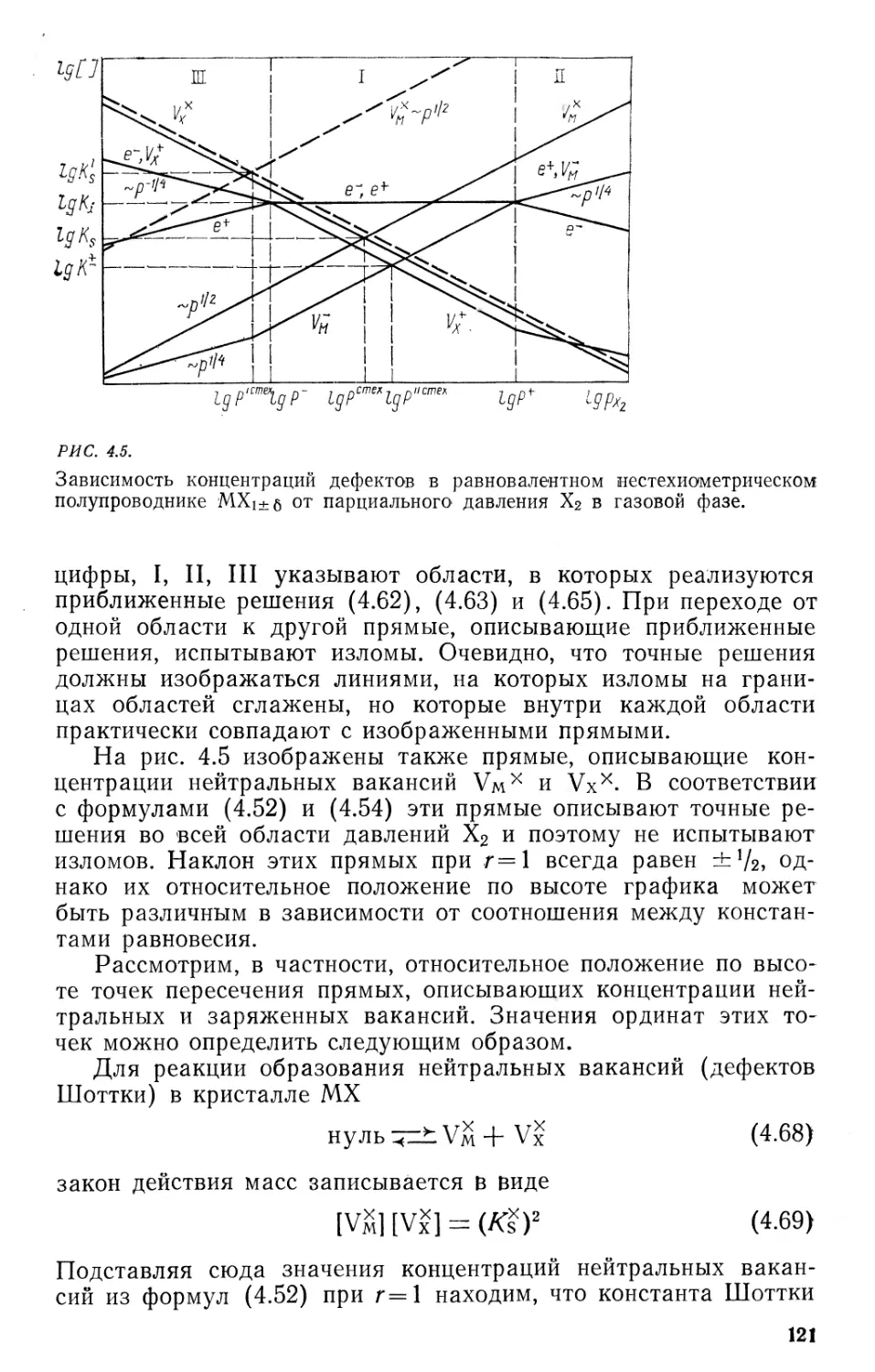
5.5. Разупорядоченность примесных нестехиометрических кристаллов
Глава 6.
ЯВЛЕНИЯ ПЕРЕНОСА В ТВЕРДЫХ ТЕЛАХ 168
6.1. Феноменологические уравнения 168
6.2. Статистический вывод основного уравнения переноса . . . 173
6.3. Подвижность дефектов. Ионная проводимость 179
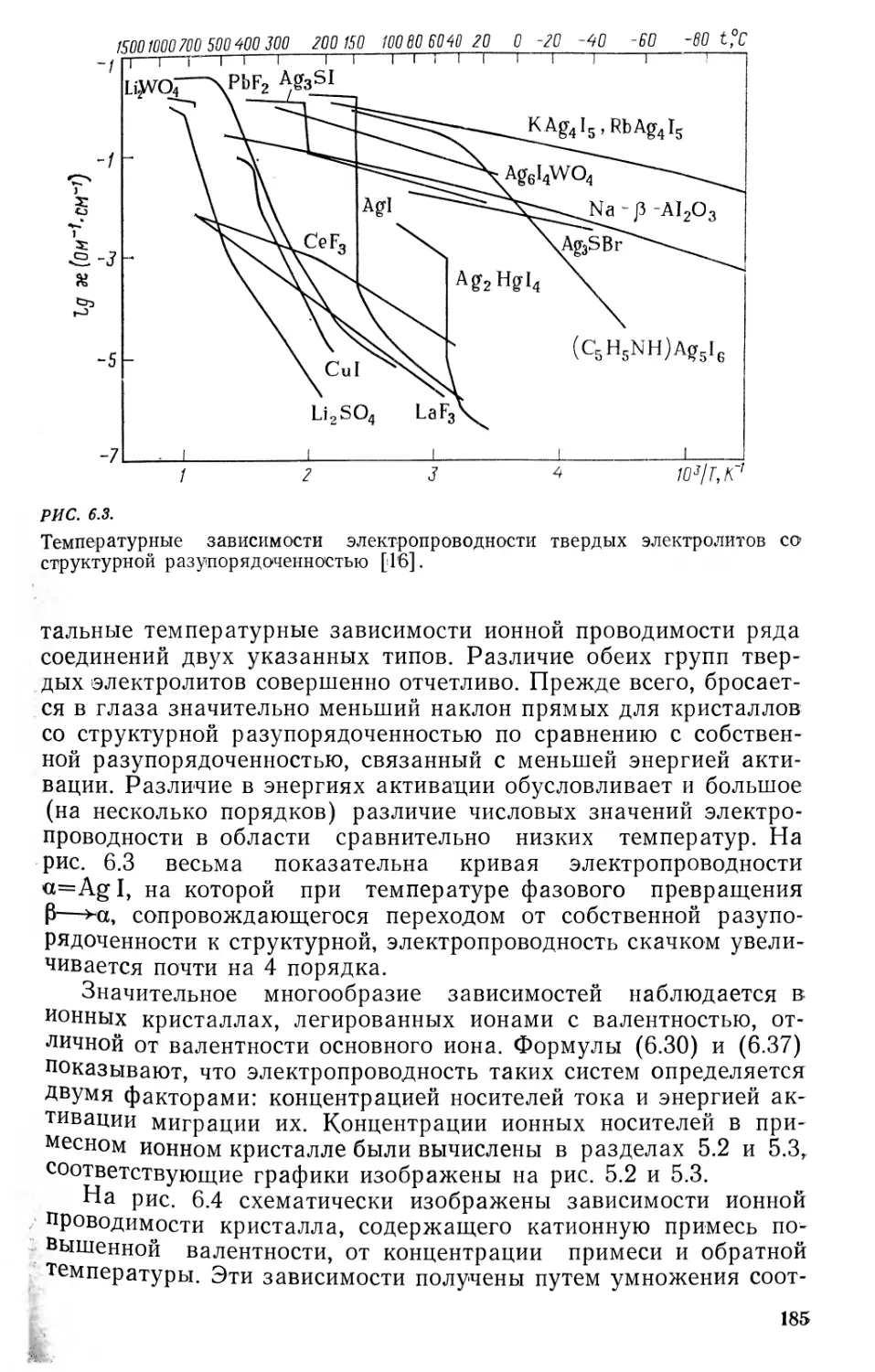
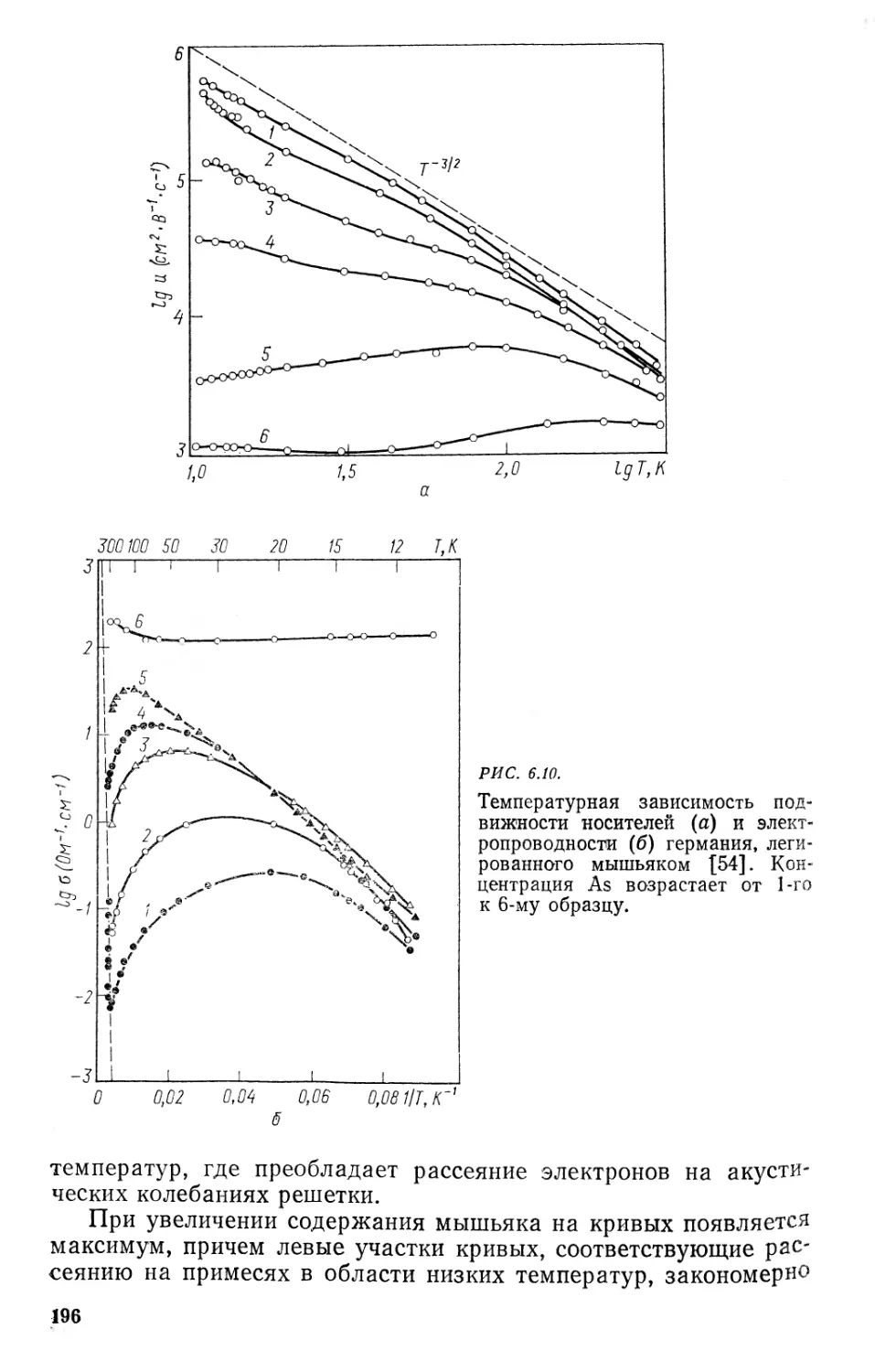
6.4. Подвижность электронов и дырок. Электронная проводимость . 189
6.5. Числа переноса ионов 206
6.6. Диффузия при хаотических блужданиях . .... 212
6.7. Корреляционный эффект при самодиффузии . . ... 221
6.8. Корреляционные эффекты при гетеродиффузии . . . . 229
6.9. Основное уравнение переноса при коррелированной диффузии . 233
6.10. Химическая диффузия 238
6.11. Взаимная диффузия в сплавах замещения 244
6Л<2. Сопряженная диффузия заряженных частиц 249
6.13. Взаимная диффузия в твердых электролитах 253
6.14. Термоэлектрический эффект в твердых электролитах .... 256
6.И5. Термоэлектрический эффект в невырожденных электронных
проводниках 262
Глава 7.
КИНЕТИКА РОСТА ТВЕРДЫХ ФАЗ 268
7.1. Феноменологические уравнения .... 268
7.2. Рост интерметаллических фаз при диффузионном контроле . . 271
' 7.3. Окисление . металлов при диффузионном контроле .... 274
7.4. Поверхностная разупорядоченность кристаллических фаз . . 281
7.5. Переход частиц через поверхность раздела твердых фаз . . 284
7.6. Адсорбция газо)в на твердых телах 289
7.7. Рост интерметаллических фаз при замедленном переходе через
межфазные границы 293
7.8. Окисление металлов при замедленных межфазных процессах . 296
Приложение. Некоторые сведения из химической термодинамики 305
Литература ,.**.. 319
Предисловие
Большие и все возрастающие потребности новых областей
техники стимулируют бурное развитие сравнительно молодой
области современного естествознания — химии твердого тела,
основными задачами которой являются синтез твердых веществ,
изучение их различных физико-химических свойств, реакций с
их участием и в конечном итоге создание материалов с заранее
заданными свойствами. Успешное решение этих задач
немыслимо без глубокого развития физической химии твердого тела,
которая, являясь составной частью химии твердого тела, призвана
изучать физические основы механизмов химических процессов
в твердых телах. Совершенно очевидно, что область ее
применения столь же необъятна, как и материаловедения в целом, и
включает в себя такие направления, как вопросы структуры,
химической связи, транспортных процессов и химического
взаимодействия, в частности химических реакций в
кристаллических и аморфных твердых телах.
Перечисленные направления сравнительно долгое время
развивались в значительной мере изолированно друг от друга,
однако в настоящее время становится все более очевидной
органическая связь между, казалось бы, совершенно различными
явлениями. Поэтому физическая химия твердого тела как
единая область науки формируется именно сейчас. Процесс этот
пока далек от завершения и, вероятно, потребует еще одного-
двух десятилетий. Тем не менее в настоящее время имеются
важные предпосылки для объединения различных
существующих подходов к решению многих задач в единую теорию,
которая в рамках единого формализма описывала бы достаточно
широкий круг явлений. При этом желательно, чтобы такая
теория опиралась на достаточно простые физические принципы и
использовала по возможности простой математический аппарат.
Данная книга является одной из первых попыток решения
этой задачи; ее автор стремился к охвату возможно более
широкого круга фундаментальных физико-химических явлений в
кристаллах в рамках единого и вместе с тем достаточно
простого формализма.
Излагаемая здесь теория опирается на представления о ра-
3Упорядоченности кристаллических тел, приводящей к
нарушению строгой периодичности их кристаллической структуры.
Основы таких представлений, заложенные в 20—30-х годах в
классических работах советского физика Я. И. Френкеля и
немецких физикохимиков К. Вагнера и В. Шоттки, явились рево-
люционным шагом вперед по сравнению с
кристаллографическими концепциями, господствовавшими в физике твердого тела
на рубеже XIX и XX столетий. В настоящее время ни у кого
нет сомнений в том, что идеально упорядоченный кристалл не
способен к какому-либо транспорту вещества в его объеме и
является, по существу, мертвым объектом в отношении
процессов, представляющих основное содержание химии твердого
тела. Действительно, протекание большинства твердофазных
химических реакций так или иначе связано с (переносом вещества
и электричества в твердых телах, поэтому разупорядоченность
кристаллической решетки является одним из важнейших
факторов, определяющих реакционную способность твердых тел.
За последние десятилетия было вскрыто большое
многообразие дефектной структуры реальных кристаллов, особенно
твердых растворов и сильно нестехиометрических соединений.
Тем не менее в современной теории разупорядоченности и
транспорта в твердых телах доминирующая роль остается за
концепцией точечных дефектов (или простейших комплексов,
состоящих из точечных дефектов и обладающих некоторыми
целочисленными эффективными зарядами). Важнейшим
достоинством такой концепции является то, что она позволяет
сформулировать в рамках статистической термодинамики
достаточно простой математический аппарат, в значительной мере
напоминающий хорошо разработанный аппарат классической
физической химии растворов. Несмотря на ограниченность
такого подхода, он дает возможность построения логически
стройной картины, правильно отражающей подавляющее
большинство закономерностей, связанных с разупорядоченностью
твердых тел.
В имеющейся литературе существует также значительный
разрыв между описанием явлений электронного переноса в
твердых телах (главным образом, в металлах и валентных
полупроводниках) и различными диффузионными процессами.
Вместе с тем статистико-термодинамическая теория
разупорядоченности позволяет рассмотреть проблему транспорта в
твердых телах в рамках единого формализма и тем самым
значительно уменьшить указанный разрыв. При этом оказывается
возможным связать воедино большое число самых различных
явлений, обусловленных переносом вещества и электричества в
твердых телах, в том числе и кинетику гетерогенных процессов,
сопровождающих твердофазные химические реакции.
Изложенные соображения в основном и определили
содержание данной книги; практическси все рассмотренные здесь
явления представлены в свете статистико-термодинамическои
теории разупорядоченности и переноса в твердых телах.
Разумеется, избранный ракурс не в состоянии полностью
осветить всю область физической химии твердого тела. При
этом из поля зрения выпадает, прежде всего, теория
химической связи, рассматриваемая специальной научной дисципли-
ной квантовой химией твердого тела. В книге обойден вопрос
о катализе, механизм которого тесно связан с особенностями
электронного строения поверхности твердых тел. Далее,
остаются неохваченными вопросы фазовых равновесий, а также
механизм и кинетика целого ряда твердофазных химических
реакций, таких, как термическое разложение твердых тел, их
горение, цепные реакции и т. д., протекание которых связано с
факторами, не учитываемыми в рамках изложенной здесь
теории. Изложение этих и некоторых других опущенных здесь
вопросов неизбежно привело бы к нарушению целостности
содержания книги, а также к излишнему дублированию учебных
пособий и монографий, достаточно подробно освещающих
указанные разделы физической химии твердого тела.
Книга рассчитана на читателей, знакомых с курсами общей
физики и неорганической химии в объеме обычных вузовских
программ. Дополнительные сведения по химической
термодинамике, используемые в книге, но не входящие в указанные
курсы, кратко изложены в Приложении.
Автор признателен коллегам по Институту электрохимии
Уральского научного центра АН СССР за большую помощь при
оформлении рукописи. Он будет рад получить критические
замечания читателей о содержании книги и указания на
имеющиеся в ней недостатки.
В. Чеботин
Основные обозначения
А — акцептор
А — компонент неионного соединения или твердого раствора
Ав — атом А в узле подрешетки В
АО, Ai — атом А в междуузлии
АП — вакансия в узле А
[A] —доля мест, занимаемых атомами А в соответствующей подре-
шетке
ак — активность я-компонеита
В —компонент неионного соединения или твердого раствора
Ва — атом В в узле подрешетки А
Bi — атом В в междуузлии
[B] —доля мест, занимаемых атомами В в соответствующей подре-
шетке
С — константа равновесия реакции взаимодействия кристалла в
газовой фазой
ск — атомная доля я-частиц
D —донор
DK — коэффициент хаотической диффузии /с-частиц
DK* — коэффициент диффузии радиоактивного изотопа я-частиц
DK — индивидуальный коэффициент химической диффузии я-частиц
Dab — коэффициент взаимной (сопряженной) диффузии частиц А и В
Е — энергия диссоциации комплекса
Ес — энергия дна зоны проводимости
Ен — энергия потолка валентной зоны
AEi —энергия ионизации /-дефекта
е — заряд электрона (абсолютное значение)
F — примесный атом
F ф (А) — атом F в узле А
F — изохорно-изотермический потенциал
/к — корреляционный множитель диффузии /с-частиц
G — изобарно-изотермический потенциал
Я — энтальпия
iK —плотность электрического тока, переносимого /с-частицами
Jк —плотность потока /с-частиц
К — константы равновесия реакций образования дефектов: Kaf —
антифренкелевских, Kf — Френкеля, Ks — Шоттки
Ki — константа равновесия реакции собственной ионизации
кристалла
Ki — константа линейного закона (роста продукта реакции
Кр — константа параболического закона роста продукта реакции
к — сорт частиц
k — постоянная Больцмана
L —толщина фазы
8
M — металлический компонент кристалла
Mj £+ междуузельный катион с эффективным зарядом £+
т — показатель степени в зависимости концентрации дефектов от
парциального давления неметалла в газовой фазе
т*—эффективная масса электрона в зоне проводимости или дырки
в валентной зоне
yyL — число узлов катионной подрешетки в единице объема
Ni — число междуузлий в единице объема
N* — эффективное число состояний в единице объема зоны
проводимости (валентной зоны)
л- — число электронов проводимости в единице объема
п+ — число электронных дырок в единице объема
РХ2 — парциальное давление неметаллического компонента кристалла
в газовой фазе
QK* — теплота переноса /с-частиц
qK — заряд я-частицы (алгебраическое значение)
г — стехиометричеекий коэффициент в формуле соединения АВГ
или МХг
Го — кратчайшее расстояние между соседними атомами в кристалле
S — энтропия
Т — абсолютная температура
t — время
tK —• число переноса /с-частиц
U — внутренняя энергия
Uк — энергия активации перескока /с-частиц
ик — подвижность я-частиц
V — объем
'а , Vbx —нейтральные вакансии в подрешетках А и В
Vm^~~— вакансия в катионной подрешетке с эффективным зарядом £~
Vxs —вакансия в анионной подрешетке с эффективным зарядом С+
vK — скорость движения /с-частиц
W — энергия реакции
№l —энергия решетки
w — энергия образования дефектов, отнесенная к одному
точечному дефекту
X — неметаллический компонент кристалла
Xi^~~— междуузельный анион с эффективным зарядом £~
х — координата
zK — валентность ^-компонента кристалла (абсолютное значение)
% & — коэффициенты переноса заряда
Тк — константа равновесия реакции ионизации /с-дефекта
Ук — коэффициент активности /с-компонента
£к , С,е~ — эффективные заряды /с-дефектов
V) — перенапряжение
В степень заполнения поверхности при адсорбции
v — коэффициент термо-э. д. с.
^ удельная ионная проводимость кристалла
Кк — парциальная удельная ионная проводимость /с-компонента
\хк — химический потенциал к-компонента, отнесенной к одной
частице
\iK — электрохимический потенциал ^компонента, отнесенный к
одной частице
V/c — частота колебаний к-частиц
5л —координационное число я-атомов
G — удельная электронная проводимость кристалла
ф —электростатический потенциал
% — множитель, учитывающий изменение частот колебаний атомою
при образовании дефектов
Q — термодинамическая вероятность
О) — частота прыжков атомов при диффузии
V — оператор градиента
Глава 1
Классификация и основные физико-
химические свойства твердых тел
1.1. Кристаллические и аморфные
твердые тела
Основным признаком твердых тел, отличающим их от
жидкостей и газов, является малая текучесть, позволяющая им
сохранять свою форму при воздействии внешних сил. В
кристаллических твердых телах малая текучесть непосредственно связана
со строго упорядоченным расположением атомов в узлах
кристаллической решетки, характеризующимся высокой
устойчивостью. В аморфных твердых телах более или менее упорядо-
ченно взаимное расположение только ближайших соседних
атомов, а дальнейший порядок в их расположении отсутствует,
так что по структуре аморфные тела больше приближаются к
жидкостям, нежели к кристаллам. С этим связаны особенности
механических свойств аморфных тел, в частности, плавный
переход от жидкого к твердому состоянию, не позволяющий
провести четкую границу между обоими состояниями. Хорошо
известным примером являются стекла, которые принято относить
к твердым телам при температурах, при которых их
коэффициент вязкости превышает 10й—1014 (Н-с)/м2 (1012—1015 Пз).
Несмотря на принципиальное отличие структуры,
кристаллические и аморфные тела можно противопоставлять друг
другу в основном лишь при сравнении их механических свойств.
Для физической же химии твердого тела наибольшее значение
имеют транспортные свойства, связанные с явлениями переноса
вещества и электричества и определяющими подвижность
частиц во внешних полях — химическом, электрическом, тепловом
и др. Такие свойства в значительной мере общи для
кристаллических и аморфных тел и определяются, главным образом,
характером химической связи, который в свою очередь непо-
редственно связан со строением электронных оболочек атомов,
образующих твердое тело. Поэтому тип химической связи обыч-
те к?1адУт в осн°ву физико-химической классификации твердых
ве я Деление твердых тел по характеру химической связи
сьма условно, тем более, что зачастую в одном и том же ве-
4ecJB°e ПР°ЯВЛЯЮТСЯ связи разных типов, учет характера хими-
все °И СВЯЗИ п03в°ляет в самом первом приближении разбить
разлМ1ШгообРазие твеРдых тел на Два больших класса, резко
ичающихся по своим транспортным свойствам: твердые те-
ла с металлической связью, осуществляемой свободными
электронами, и неметаллические твердые тела, валентные
электроны которых локализованы.
В дальнейшем, где это особо не оговаривается, мы будем
рассматривать кристаллические твердые тела, обладающие
регулярной структурой.
1.2. Металлы
Металлическая связь в твердых телах осуществляется
свободными или коллективизированными валентными электронами,
принадлежащими всей кристаллической решетке в целом.
Природа металлической связи раскрывается квантовой теорией
твердого тела, изложению которой посвящено большое число
изданий [3—7].
Хотя строгая квантовая теория металлов весьма сложна,
качественно правильное представление об их основных
физико-химических свойствах вытекает из простой и наглядной модели,
согласно которой металл представляется как геометрически
упорядоченный остов, состоящий из положительно заряженных
металлических ионов, погруженный в отрицательно заряженную
электронную жидкость. Очень высокая (^1022 см-3)
концентрация подвижных электронов обусловливает специфические
свойства металлов, такие, как высокая электропроводность и
теплопроводность, а также принципиальные отклонения от
классического закона Дюлонга — Пти для теплоемкости твердых
тел. Для наших целей нет необходимости подробно вдаваться
в квантовую теорию твердого тела, а достаточно отметить
основные черты энергетического спектра электронов. Пользуясь
представлениями зонной модели твердых тел, ероследим
происхождение свободных электронов в металлических кристаллах
по следующей простой схеме.
Хорошо известно, что в свободных атомах электроны могут
иметь не любые значения энергии, а только вполне
определенные дискретные значения, отделенные друг от друга широкими
запретными интервалами. Энергетические уровни электронов в
свободных атомах обычно изображают схематически в виде
ряда горизонтальных черточек, положение которых соответствует
энергии данного состояния (см. правую часть рис. 1.1). При
образовании твердых тел энергетический спектр электронов
претерпевает существенные изменения из-за квантовомеханическо-
го взаимодействия электронов. Согласно принципу запрета
Паули, лежащему в основе квантовой теории атома, в одном
квантовом состоянии, определяемом соответствующим набором
квантовых чисел, не может находиться больше двух электронов
(с противоположными спинами). При образовании твердого
тела путем сближения отдельных атомов, по мере усиления
взаимодействия между ними, действие принципа Паули начинает
распространяться на весь макроскопический объем кристалла,
in
РИС. t.l. Е
Расщепление уровней и образование
энергетических зон при сближении
атомов на примере натрия.
который при этом нужно рассматривать как одну гигантскую
молекулу, состоящую из огромного числа атомов. Запрет
Паули выполняется в твердом теле за счет того, что при
взаимодействии атомов друг с другом энергетические уровни, бывшие
одинаковыми у разделенных атомов, начинают смещаться на
величину тем большую, чем сильнее взаимодействие атомов,
т. е. чем теснее они сближены. В результате из N одинаковых
уровней у далеко разделенных атомов в твердом теле
возникает энергетическая зона, состоящая из N близко расположенных,
но тем не менее различных уровней. Образование
энергетических зон при сближении атомов показано на рис. 1.1 для
натрия. Здесь по оси абсцисс отложено расстояние между
соседними атомами; г0 — соответствующее равновесное расстояние в
твердом теле.
На рис. 1.2 показана общая схема энергетических зон
твердого тела. Зоны, образованные разрешенными уровнями,
называются разрешенными зонами, а зоны, лежащие между
ними и соответствующие запрещенным значениям энергии —
запрещенными зонами.
При перемещении электрона от одного атома к другому он
должен изменить свою энергию на величину разности энергий
соответствующих уровней. Ширина как разрешенных, так и
запрещенных зон бывает обычно порядка одного или нескольких
электронвольт; так как в образце твердого тела с объемом
1 см3 содержится порядка 1022 атомов, то, следовательно,
расстояние между соседними уровнями в одной и той же зоне
составляет ^Ю22 эВ. Энергия же, которую электрон набирает на
расстоянии свободного пробега при движении в электрическом
поле, бывает обычно тгорядка Ю-8—10~4 эВ; средняя энергия
тепловых- колебаний атомов, равная 3 kT, при комнатной
температуре составляет около 0,08 эВ. Эти цифры показывают, что
Как энергии внешнего электрического поля, так и энергии
тепловых колебаний атомов более чем достаточно для перевода
лектрона с одного энергетического уровня на другой в преде-
ах одной и той же зоны (но не из одной зоны в другую).
днако чт°бы движение электронов по кристаллу действие
имеЬН° пРоисх°Дило> необходимо, чтобы хотя бы в одной зоне
nvc ИСЬ ОДновРеменно уровни, заполненные электронами, и
У тые уровни, т. е. чтобы хотя бы одна из разрешенных зон
13
РИС. 1.2.
Схема энергетических зон твердого тела.
была заполнена частично. Типичным
примером веществ с частично
заполненной зоной являются щелочные
металлы. Так, в атоме натрия целиком
заполнена первая квантовая оболочка
Is, вторая оболочка 2s и третья 2р,
содержащие соответственно 2, 2 и
6 электронов. Внешний,
одиннадцатый валентный электрон находится на
уровне 3s, который может вместить
два электрона с противоположными спинами. Таким образом,
уровень 3s в атоме натрия заполнен только наполовину. При
образовании твердого тела возникающая из этих уровней зона
3s, соответствующая валентным электронам, также
оказывается заполненной ровно наполовину, и валентные электроны
коллективизируются, свободно перемещаясь по кристаллу.
Несколько сложнее обстоит дело со щелочноземельными
элементами, занимающими место во второй группе
Периодической системы. Эти элементы имеют на s-уровне по два
электрона и, казалось бы, в твердом состоянии должны иметь целиком
заполненные s-зоны и пустые зоны более высоких энергий,
исключающие перемещение электронов. На самом деле они
обладают ярко выраженными металлическими свойствами.
Объяснение этого кроется в следующем. Уровни s и р лежат
довольно близко друг от друга, так что при образовании твердого
тела зоны s и р перекрываются между собой. Это можно видеть
на рис. 1.1 (зоны 3s и Зр). Таким образом, объединенная s—р-
зона в щелочноземельных металлах, содержащая по 2
электрона на каждый атом, также оказывается заполненной частично,
что и обеспечивает металлические свойства этих элементов.
1.3. Неметаллические твердые тела
В кристаллах с неметаллическими типами химической связи в
их основном состоянии внешние, валентные электроны целиком
заполняют соответствующую зону. Поэтому в отличие от
металлов в этих веществах валентные электроны локализованы
на соответствующих атомах и не могут перемещаться по
кристаллу. Различают в основном следующие типы кристаллов с
неметаллическим характером связи.
Ионные кристаллы. Простейшим типом неметаллической
химической связи обладают ионные кристаллы, являющиеся
соединениями электроположительных элементов (металлов) с
электроотрицательными элементами (неметаллами). Как из-
14
тно металлы способны сравнительно легко отдавать один
В<ли несколько валентных электронов, превращаясь в положи-
и н0 заряженные ионы (катионы), а неметаллы —
присоединять лишние электроны с образованием отрицательно
заряженных ионов (анионов). В большинстве случаев внешние
электронные оболочки как катионов, так и анионов имеют
структуру подобную структуре электронных оболочек атомов
благородных газов, в частности, обладают сферической симметрией.
Особенности энергетического спектра электронов в ионных
кристаллах удобно проследить на наиболее типичном
примере кристалле NaCl. При образовании этого соединения
валентный З^-электрон атома натрия переходит к атому хлора, а
оставшиеся внутренние электроны с более низкой энергией
полностью заполняют свои оболочки. У атома хлора на внешней
Зр-оболочке содержится только 5 электронов вместо 6
возможных, так что З^-электрон, перешедший от атома натрия,
заполняет эту оболочку до конца. Таким образом, в NaCl не
содержится ни одной частично заполненной зоны (рис. 1.3).
Разность энергий между последней заполненной зоной,
соответствующей валентным Зр-электронам хлора (так называемой в а-
лентной зоной), и первой пустой зоной (зоной
проводимости) в NaCl составляет около 8 эВ и слишком велика для
того, чтобы электроны из валентной зоны могли перейти в
зону проводимости в результате теплового возбуждения.
Поэтому при обычных условиях электроны в NaCl локализованы на
соответствующих ионах.
Локализованный характер распределения электронной
плотности широко распространен и в кристаллах, содержащих ионы
с частично заполненными внутренними электронными
оболочками, например в соединениях переходных элементов. На
первый взгляд такие кристаллы должны обладать металлическими
свойствами. Однако они обычно не наблюдаются. Это связано
с тем, что электронные уровни частично заполненных оболочек
расщепляются в электрическом поле окружающих анионов на
ряд подуровней, образующих в кристалле неперекрывающиеся
подзоны, причем каждая подзона в обычных условиях
заполнена целиком.
Описанное строение электронных оболочек в ионных
кристаллах позволяет успешно использовать классическую
упрощенную модель, согласно которой образующие кристалл ионы
представляются заряженными твердыми шариками, причем заряд
онов полагается равным валентности соответствующего эле-
ента. В рамках этой модели связь в ионных кристаллах ОбуС-
^С 1.з.
ЙтллГ^сТ энеРгетических зои в
1^) | 3от
прододимости
Запрещенная зона
Валентная
>(С1),
зона
15
ловлена электростатическим притяжением разноименно
заряженных ионов. Чтобы притяжение преобладало над
отталкиванием между одноименно заряженными ионами, кратчайшие
расстояния между последними должны превышать кратчайшие
расстояния между разноименными ионами. Поэтому в ионных
кристаллах ближайшими соседями катионов являются анионы,
и наоборот.
Валентные кристаллы. В отличие от ионных кристаллов
валентные кристаллы (или кристаллы с ковалентной связью)
образуются из одного или нескольких химических элементов,
занимающих в Периодической системе промежуточное положение
между металлами и неметаллами, например С (алмаз), Si, Ge,
SiC, InSb, PbSe.
Как и в двухатомных молекулах газов (Н2, 02, HC1 и т. д.),
в валентных кристаллах каждый валентный электрон
принадлежит двум соседним атомам. Так, в кристалле алмаза
каждый из четырех валентных электронов обобществлен данным
атомом и одним из четырех его ближайших соседей, так что в
отличие от ионной связи ковалентная связь имеет резко
выраженную направленность. Как и в ионных кристаллах, в
кристаллах с ковалентной связью общее число валентных
электронов равно числу разрешенных состояний в соответствующей
зоне, так что все разрешенные энергетические уровни в
валентной зоне заняты. Поэтому и в кристаллах с ковалентной связью
валентные электроны пространственно локализованы и не
могут перемещаться по кристаллу под действием внешнего поля.
Различная природа ионной и валентной связей должна
проявляться в различных характерах распределения плотности
электронов в кристаллах. Действительно, в ионном кристалле
каждый электрон локализован на одном определенном ионе,
поэтому плотность электронов в промежутках между ионами
должна быть равна нулю. В валентных же кристаллах
плотность электронов в промежутках между атомами должна быть
сравнительно большая. Однако, как показывают исследования
электронной плотности, такого резкого различия в реальных
кристаллах не наблюдается; даже в галогенидах щелочных
металлов, обладающих наиболее ярко выраженными
свойствами ионных кристаллов, плотность электронов в промежутках
между ионами заметно отличается от нуля. Это означает, что
в природе вообще не существует кристаллов с идеально ионной
связью. В реальных ионных кристаллах валентные электроны,
в основном тяготея к соответствующему аниону, тем не менее
в течение заметной доли времени находятся в непосредственной
близости к соседнему катиону, осуществляя тем самым кова-
лентный характер некоторой доли сил связи между ионами.
Очевидно, что доля ковалентной связи тем более, чем в
большей степени металл и неметалл проявляют промежуточные
аморфные свойства. С другой стороны, идеально ковалентная
обнаруживается лишь в кристаллах простых веществ, со-
СВЯЗших из тождественных атомов (алмаз); в химических со-
СТ°нениях, атомы которых хоть сколько-нибудь различаются
€ДИктооотрицательностью, валентные электронные облака
смекаются в направлении от менее электроотрицательных атомов
Щ более электроотрицательным, придавая тем самым
химической связи частично ионный характер.
Существует несколько способов оценки доли ионной связи в
реальных кристаллах [8]'. Чаще всего реальное распределение
электронной плотности в твердых телах характеризуют
эмпирической величиной — так называемым эффективным
зарядом иона, определяемым из измерений каких-либо
физических характеристик кристалла (электрических, магнитных,
оптических и др.). Числовое значение эффективного заряда
подбирается таким образом, чтобы путем подстановки его в
формулы классической физики ионных кристаллов получить
экспериментальное значение измеряемой физической величины.
Лри таком определении эффективных зарядов совершенно
естественно, что их значения, найденные с помощью измерений
различных физических характеристик, должны различаться.
Однако эти различия сравнительно невелики, поэтому можно
считать, что экспериментальные значения эффективных
зарядов близки к значениям «истинного» заряда ионов, т. е. к
величине локализованного из них электрического заряда.
В табл. 1.1 приведены отношения эффективных зарядов
ионов г* к номинальным значениям валентностей г,
предусматриваемым классической моделью, для кристаллов бинарных
соединений. Эти отношения убедительно показывают
ограниченность классической ионной модели твердого тела. Эффективные
заряды ионов близки к номинальным только для галогенидов
щелочных и частично щелочноземельных металлов. Для
оксидов заряд иона кислорода близок к —1, а для халькогенидов
и прочих соединений заряды анионов по абсолютной величине
существенно меньше единицы.
Решение квантовомеханического уравнения Шредингера
побывает, что присоединение двух электронов к атому
кислорода энергетически невыгодно [12]. Поэтому существует мнение,
в пп!^0Г0заРяДНЬ1е отрицательные ионы вообще не существуют
тела6М Н6 менее значительная часть физики и химии твердого
зарядь007'306113 На конЦепЦии ионной модели, согласно которой
При эт И°нов Равны валентностям соответствующих элементов.
во> что°М Может показаться парадоксальным то обстоятельсг-
в общемИСП°ЛЬЗОВанИе заведомо огрубленной модели приводит,
Ся в том К пРавильным результатам. Причина этого заключает-
Жит неко ЧТ° ^ольшинство формул классической теории содер-
Раются Т0Рые ЭМ(пирические параметры; их значения подби-
теопии п ~ КИМ °браз°м, чтобы обеспечить наилучшее согласие
ft MC ЭКспеРИментальными данными.
Таблица 1.1. Отношение эффективных зарядов ионов к номинальным
в бинарных кристаллах [9—//]
Кристалл
LiF
LiCl
LiBr
Li I
NaF
NaCl
NaBr
Nal
KF
KC1
KBr
KI
RbF
RbCl
RbBr
Rbl
CsF
CsCl
CsBr
Csl
CuCl
CuBr
Cul
AgF
AgCl
AgBr
z* /z
0,83 1
0,72
0,68
0,55
0,94 I
0,74
0,71
0,70
I 0,96
0,79
0,77
0,70
0,93
0,87
0,82
0,81
0,88
0,87
0,81
0,74
0,98 (66)
0,96 (62)
0,91 (60)
0,89
0,69
0,67
Кристалл
Agl
T1C1
TIBr
Til
MgF2
CaF2
SrF2
BaF2
ZnF2
CdF2
EuF2
PbF2
MnF2
FeF2
1 CoF2
NiF2
BeO
MgO
MgS
CaO
CaS
SrO
SrS
BaO
BaS
ZnO
z*/z
0,61
0,88
0,84
0,83
0,76
0,82
0,85
0,87
0,77
0,80
0,84
0,81
0,79
0,68
0,73
0,76
0,55
0,59
0,62
0,60
0,52
0,61
0,53
0,74
0,55
0,53
Кристалл
ZnS
ZnSe
ZnTe
CdO
CdS
CdSe
CdTe
HgO
HgS
HgTe
PbO
PbS
PbSe
PbTe
IFeO
CoO
NiO
BN
BP
A1N
A1P
AlAs
AlSb
GaN
GaP
GaAs
Z*/Z
0,43
0,34
0,27
0,59
0,43
0,41 !
0,38 1
0,57
0,28
0,26
0,60
0,37
0,35
0,28
0,46
0,44
0,41
0,38
0,25
0,40
0,24
0,21
0,16
0,41
0,19
0,14
Кристалл
GaN
GaP
GaAs
GaSb
InP
InAs
InSb
A1F3
GaF3
InF3
ScF3
YF3
LaF3
jCeF3
PrF3
|NdF3
|ErF3
A1203
Y203
La203
! Pr203
Nd203
Sm203
тю2
Th02
uo2
z*/z
0,41
0,19
0,14
0,10
0,22
0,19
0,11
0,60
0,60
0,61
0,76
0,68
0,76
0,75
0,71
0,82
0,76
0,51
0,62
0,60
0,52
0,59
0,60
0,74
0,60
1 0,58
Наглядным примером условности ионной модели может
служить система кристаллохимических радиусов ионов в твердых
телах. Радиусы ионов определяются, как правило, из условия
аддитивности их в кристаллах соединений: гАЕ = гА-{-Гв, где Гдв—
расстояние между центрами ионов в соединении. Вообще
говоря, это соотношение определяет не сами радиусы, а разность
радиусов ионов В и С в соединениях АВ и АС: гАЕ—гАС = гв—Гс-
Поэтому любая шкала ионных или атомных радиусов,
построенная по принципу аддитивности, определяет их с точностью
до некоторой достаточно произвольной постоянной (радиус
иона — эталона); аддитивность сохраняется, если радиусы всех
катионов увеличить, а радиусы всех анионов уменьшить на
произвольную постоянную. Поэтому существует многообразие
эмпирических шкал ионных радиусов, отличающихся радиусом
эталонного иона [13]. Более того, для описания
кристаллохимии соединений с промежуточным характером связи с равным
успехом можно пользоваться как ионной, так и атомной
моделью строения кристалла и соответственно использовать как
ионные, так и атомные радиусы, поскольку правило
аддитивности выполняется в обоих случаях.
18
Молекулярные кристаллы. К этому классу относятся затвер-
шие благородные газы, а также твердые тела, образован-
Д6 насыщенными молекулами, в частности, органических ве-
ные г 141. Связь в молекулярных кристаллах обусловлена так
щес а,|змьши ван-дер-ваальсовыми силами. Природу этих сил
на е всего понять, рассматривая атом в модели Бора. Сово-
Пупность положительного ядра и отрицательных электронов в
каждый момент времени можно рассматривать как
электрический диполь, непрерывно изменяющий свою ориентацию в
результате движения электронов вокруг ядра. Такой диполь в
каждый момент времени индуцирует диполи на окружающих
атомах, в результате чего между ними возникают
электрические силы притяжения; несмотря на непрерывное изменение
ориентации диполей, среднее по времени значение этих сил
оказывается отличным от нуля.
Кристаллы с водородными связями. Атом водорода имеет
всего один электрон и при ионизации превращается в протон,
размеры которого ничтожно малы по сравнению с размерами
атомов других элементов. Такая особенность иона водорода
обусловливает ряд специфических свойств твердых тел с
водородными связями. Так, из-за малых размеров протон может
быть связан не более чем с двумя атомами. Водородная связь
играет большую роль в органических кристаллах.
Как в молекулярных, так и в кристаллах с водородными
связями в основном состоянии все разрешенные уровни энергии
заняты электронами, в этом отношении они не отличаются от
ионных и валентных кристаллов и при низких температурах
обычно являются изоляторами.
Описанная структура энергетического спектра электронов
в неметаллических кристаллах допускает некоторые
исключения. ^Наиболее известным из них является магнетит Fe304,
имеющий сложную кристаллическую структуру шпинели. В этом
соединении железо находится в двух валентных формах — в
виде двух- и трехзарядных ионов, так что его химическая
формула соответствует соединению оксидов FeO-Fe203.
В структуре шпинели ионы кислорода образуют плотней-
шую кубическую упаковку, а ионы железа занимают
неэквивалентные узлы. Из трех ионов железа, приходящихся на
каждую^ формульную единицу оксида Fe304, один трехзарядный ион
е размещается в тетраэдрическом узле, окруженном четырь-
я ионами кислорода, а два других Fe3+ и Fe2+ статистически
Р определены по октаэдрическим узлам, окруженным шестью
отлиМИ КИслоР°Да- Так как двух- и трехзарядные ионы железа
ра чаются только зарядом, это соответствует хаотическому
иона^6^^6™10 ^ электРонов> формально принадлежащих
СКа м ^е2+, по 2 N эквивалентным узлам. Поэтому энергетиче-
полне3°Н^' С00тветствУюЩая этим электронам, оказывается за-
сителе^011 только наполовину, а концентрация электронных но-
и тока в магнетите — сравнимой с таковой для металлов;
'9
перенос осуществляется перескоками электронов с ионов Fe2+
на ионы Fe3+ (подробнее см. в разделе 6.4).
В дальнейшем мы не будем рассматривать такие
соединения со шпинельным типом электропроводности, а ограничимся
«нормальными» неметаллическими кристаллами, основное
состояние которых отвечает целиком заполненной валентной зоне
и пустой зоне проводимости.
1.4. Диффузия и ионная проводимость.
Атомные дефекты
Изучение диффузионных процессов является одним из
краеугольных камней химии твердого тела. В отличие от газов и
жидкостей в твердых телах отсутствует механическое
перемешивание, поэтому при большинстве химических превращений в
твердых телах диффузия является единственным фактором,
обеспечивающим контакт между пространственно
разделенными участниками реакции.
Характерной особенностью диффузии в твердых телах
является сильная зависимость ее скорости от температуры.
Поэтому диффузионные процессы, незаметные при низких
температурах, при достаточно высоких проявляются во всех без
исключения твердых телах.
Большое значение для химии твердого тела имеет и другое,
родственное диффузии явление — ионная, или
электролитическая, проводимость, обнаруживаемая многими
твердыми телами при повышенных температурах. В таких твердых
телах, как и в традиционных электролитах — водных растворах,
прохождение электрического тока сопровождается
макроскопическим .переносом вещества. Поэтому в электрохимической
литературе твердые тела с преобладающей (по отношению к
электронной) ионной проводимостью называют твердыми
электролитами.
Круг веществ, которые можно отнести к твердым
электролитам в достаточно широком интервале температур, сравнительно
ограничен [15, 16]. Сюда относятся в основном ионные
кристаллы— галогениды и отчасти оксиды металлов с преобладающим"
ионным характером связи, некоторые сложные композиции на
их основе, а также кристаллические соли и стекла, содержащие
ионы щелочных металлов. В большинстве же твердых тел с
промежуточным характером химической связи ионная
проводимость или сравнима с электронной, или же вообще незаметна
на ее фоне. Тем не менее ионная проводимость в той или иной
мере присуща всем твердым телам с достаточно высокой долей
ионной связи [17—19]. Поскольку и диффузия, и ионная
проводимость в ионных кристаллах сводятся к перемещению одних
и тех же частиц — ионов, очевидно, что в основе обоих этих
явлений должен лежать единый механизм.
20
Многие проявления диффузии, например, такие, как
спекание твердых тел или окисление металлов, встречаются в
повседневной практике и известны с незапамятных времен. Поэтому
начало изучения диффузионных процессов в твердых талах
относится еще ко времени домикроскопической физики; более
ста лет насчитывают и исследования ионной проводимости. Тем
не менее представления о процессах переноса вещества в
твердых телах очень долго имели чисто феноменологический
характер, микроскопический же механизм этих процессов оставался
совершенно неясным даже в первой четверти текущего
столетия.
В начале этого века в физике твердого тела господствовали
кристаллографические концепции, согласно которым
кристаллические твердые тела составлены из регулярно и плотно
упакованных атомов или ионов, занимающих все разрешенные
позиции— узлы кристаллической решетки. Такое представление
не оставляло места сколько-нибудь плодотворным моделям
процессов переноса вещества в кристаллах. Действительно, в
целиком заполненной кристаллической решетке транспортные
процессы могут осуществляться только путем непосредственного
обмена местами соседних атомов. Такой механизм еще мог бы
как-то объяснить диффузию в твердых телах, но никак не
объясняет ионную проводимость. Действительно, обмен местами
одноименно заряженных ионов не приводит к перемещению
электрического заряда. Обмен же местами катиона и аниона
требует настолько больших затрат энергии (я^15 эВ), что
вероятность такого события ничтожно мала (при комнатной
температуре один раз за ^10200 лет, при температуре плавления —
один раз за ж 1050 лет).
Решающий шаг к пониманию механизмов транспортных
процессов в твердых телах был сделан в 1926 г. Френкелем,
показавшим, что в любом кристалле при конечных температурах
должны существовать локальные нарушения регулярной
кристаллической структуры — дефекты кристаллической
решетки.
Необходимость существования дефектов в кристаллах
вытекает из элементарных статистических соображений.
Действительно, при любой конечной температуре средняя энергия
тепловых колебаний атомов также конечна, но в результате
флуктуации всегда имеется некоторая вероятность того, что
отдельные атомы получают очень большую энергию,
достаточную для создания локальных нарушений регулярной
кристаллической структуры. Очевидно, чем выше температура, тем вы-
ще должна быть степень разупорядоченности решетки.
По Френкелю, разупорядоченность кристаллической
решетки достигается переходом определенного числа атомов из
регулярных узлов решетки в междуузлия, т. е. в такие позиции
между узлами, которые в идеальном кристалле не заняты. При
этом возникают два типа дефектов: междуузельные ато-
21
мы (ионы) и незанятые узлы кристаллической решетки,
получившие название вакансий. Оба типа дефектов, двигаясь по
кристаллу, дают вклад в диффузию, а в случае ионных
кристаллов—и в ионную проводимость. Движение но кристаллу
междуузельных атомов осуществляется путем их перескоков по
незанятым междуузлиям, движение вакансий — путем
последовательных перескоков в незанятый узел соседних атомов. При
таком перескоке вакансия переходит в соседний узел, который
был первоначально занят перескакивающим атомом, поэтому
направление ее движения противоположно направлению
перескоков атомов.
Внедрение атомов в междуузлия может происходить
сравнительно легко в кристаллах с достаточно просторной
упаковкой, при которой размеры междуузлий сравнимы с размерами
атомов, и затруднено в кристаллах с плотной упаковкой
атомов. Поэтому несколько позднее Шоттки предложил другую
модель разупорядоченности твердых тел, содержащую только
вакансии. По Шоттки, вакансии образуются при выходе
атомов из узлов в объеме кристалла на поверхность, в результате
которого на поверхности происходит достраивание
кристаллической решетки, а в объеме кристалла возникают вакансии.
В бинарных химических соединениях, в частности, в ионных
кристаллах такая модель предполагает существование
вакансий в подрешетках обоих компонентов в эквивалентных
количествах.
Большое влияние на транспортные свойства твердых тел
оказывают инородные примеси, растворенные в кристалле
основного вещества. В этом случае наряду с вакансиями и меж-
дуузельными атомами необходимо учитывать третий тип
атомных дефектов кристаллической решетки — дефектов
замещения. Этим термином обозначают узлы кристаллической
решетки, занятые атомами иного сорта, нежели атомы,
предусматриваемые идеальной кристаллографической структурой.
В кристаллах химических соединений с неионной связью,
например интерметаллических или валентных соединений,
дефекты замещения могут возникать и при отсутствии примеси,
когда атомы А частично размещаются в узлах подрешетки В и
наоборот. Такая разупорядоченность называется
антиструктурной. В бинарных ионных кристаллах антиструктурная
разупорядоченность не наблюдается, так как размещение
катионов в анионной подрешетке и наоборот потребует слишком
больших затрат энергии. Для кристаллов многокомпонентных
ионных соединений типа шпинелей, содержащих катионы двух
или более сортов, характерно разупорядочение катионов,
подобное антиструктурному. При этом катионы в идеальном
кристалле, занимающие неэквивалентные узлы, более или менее
хаотически распределяются по узлам обеих подрешеток. Такое
разупорядочение особенно важно для понимания свойств
магнитных материалов — ферритов.
22
Существует несколько различных способов описания
структуры дефектных кристаллов; мы остановимся на двух,
наиболее употребительных.
1 Наибольшее распространение получил способ,
предложенный Крёгером [20]'. По Крёгеру, любой дефектный кристалл
представляется как совокупность структурных
элементов, показывающих, какой атом или ион (или группа атомов,
например, N03, S04 и др.) находятся в данном узле или меж-
дуузлии. Незанятые узлы или междуузлия также считаются
самостоятельными структурными элементами.
Для обозначения структурных элементов обычно
используют химические символы, соответствующие данному
химическому элементу, например Na, A1, О и т. д. При рассмотрении
общего случая кристалла простого вещества, состоящего из
атомов одного сорта, мы будем обозначать эти атомы буквой А.
В общем случае формулу кристалла произвольного бинарного
химического соединения будем записывать в виде МХГ, где
буквой М обозначается более электроположительный компонент
(в ионном соединении — катион), а буквой X — более
электроотрицательный компонент (анион). Символ F используется для
обозначения чужеродных примесей. Вакансии или любые
незанятые позиции в кристаллической решетке обозначаются
буквой V.
Место расположения частицы указывают нижним индексом;
междуузлие помечается индексом i (от латинского слова inter-
stitium — промежуток). Так, атомным дефектам соответствуют
следующие структурные элементы: Va означает вакансию в
узле, который в идеальном кристалле должен быть занят атомом
А. (в подрешетке А); А* — междуузельный атом A; Fa — атом F
в узле подрешетки А. Такая система обозначений позволяет
•описывать и структурные элементы идеального кристалла: АА—
означает атом А в своем нормальном узле; Vi — незанятое
междуузлие.
Система структурных элементов выгодна своей
наглядностью, так как она описывает твердое тело как ансамбль частиц,
наиболее близко соответствующих общепринятым в химии
представлениям об атомном строении вещества, и в то же
время строго учитывает специфику твердых тел, связанную с их
кристаллической структурой.
Вместе с тем система структурных элементов неудобна для
статистико-термодинамического описания дефектных
кристаллов. Дело в том, что в кристаллах химических соединений
общие количества позиций в различных подрешетках жестко
связаны между собой определенными стехиометрическими
соотношениями. Это накладывает на концентрации различных
структурных элементов определенные условия связи, так что
структурные элементы нельзя рассматривать как компоненты
системы, концентрации которых можно варьировать независимо друг
0Т друга.
23
2. Указанного недостатка лишена другая система описания
дефектных кристаллов, применяемая в химии твердого тела —
система относительных составляющих единиц.
Согласно этой системе дефектный кристалл представляется
как раствор дефектов в идеальной кристаллической решетке
рассматриваемого вещества, причем под дефектом
подразумевается любое локальное отклонение реальной структуры от
идеальной [17, 21]. Так, вакансия означает недостачу атома в
соответствующем узле; дефект замещения — недостачу основного
атома и наличие вместо него чужеродного. Другими словами,
в системе относительных составляющих единиц дефект
эквивалентен разности между отвечающим ему структурным
элементом и тем, который должен быть расположен в данной
кристаллографической позиции идеального кристалла. При таком
определении дефектов их следует рассматривать как некие
квазичастицы, свойства которых определены по отношению к
фону идеальной кристаллической решетки. В отечественной
литературе для обозначения относительных составляющих единиц
преимущественно используется номенклатура Хауффе [21].
В этой системе основной символ указывает химический
элемент, ответственный за образование дефекта; сорт дефекта
помечается специальным значком □, О, •. Вакансия в подрешет-
ке А обозначается АП; атом (ион) А в междуузлии — АО; атом
(ион) F в узле подрешетки А—F#(A).
Все выше изложенное показывает, что в отличие от
традиционного химического подхода при обоих способах описания
дефектных кристаллов мы имеем дело не с системой истинных
атомов, ионов или молекул, а с системой некоторых
квазичастиц— структурных элементов или относительных составляющих
единиц. Тем не менее в химии твердого тела принято
изображать любые процессы с участием таких квазичастиц в виде
уравнений реакций, формально подобных обычным
химическим реакциям в растворах или газах. Специфика
твердофазных реакций часто подчеркивается тем, что такие реакции
называют квазихимическими, а сам метод — квазихи-
мическим методом. При таком методе согласно двум
описанным выше способам представления дефектных
кристаллов квазихимические реакции записываются двумя способами:
через структурные элементы и через относительные
составляющие единицы.
1. Первый способ записи наиболее близок обычному
химическому методу описания реакций. При этом учитываются все
структурные элементы, претерпевающие изменения в ходе
реакции, в том числе и отвечающие совершенному кристаллу —
нормально занятые узлы АА и незанятые междуузлия Vi.
Например, переход атома А из нормального узла в междуузлие
по модели Френкеля изображается реакцией
АА + V} - VA + A,
(1.1а)
Реакция
Мм + Хх -> Хм -f - Мх
(1.2а)
соответствует процессу антиструктурного разупорядочения.
Следует, правда, иметь в виду, что при некоторых реакциях
происходит увеличение объема кристалла за счет
достраивания решетки на поверхности. В этом случае структурные
элементы, отвечающие регулярной структуре, исчезают в объеме,
но восстанавливаются в поверхностном слое кристалла и
поэтому выпадают из уравнения реакции. Так, образование
вакансий по модели Шоттки при выходе атомов на поверхность
кристалла соответствует следующим реакциям:
для простого вещества А
Ад -> VA + Ад, или нуль -► VA (1.3а)
для бинарного соединения МХГ
Мм + гХх -> VM + /*VX + Мм + >"ХХ, или нуль -> VM + rVx
(1.4а)
«Нуль» в левой части этих уравнений означает, что
образование вакансий по модели Шоттки не связано с исчезновением
каких-либо исходных структурных элементов решетки и может
интерпретироваться как растворение вакуума в кристаллах.
2. При втором способе записи квазихимических реакций
учитываются взаимные превращения относительных
составляющих единиц — идеальной решетки и дефектов. При этом
идеальная решетка, вообще говоря, также рассматривается как
участник реакции, т. е. как самостоятельная составляющая
единица реального кристалла. В такой системе рассмотренные
выше процессы изобразятся следующими реакциями.
• При образовании дефектов Френкеля и антиструктурном
разупорядочении число узлов решетки не меняется и уравнения
реакций должны содержать только образующиеся дефекты:
нуль->АП+АО (1-16)
нуль->Х«(М) + М«(Х) (1.26)
В этом случае «нуль» означает отсутствие дефектов в
исходном состоянии кристалла.
При образовании дефектов Шоттки происходит увеличение
числа узлов решетки вследствие выхода атомов из объема на
Поверхность. Поэтому реакции образования вакансий по
модели Шоттки в кристаллах простого вещества А и бинарного
соединения MX,- запишутся соответственно в виде
нуль ->АП4-А (1.36)
нуль -> МП +тХП + МХГ (1.46)
ед6 И ^^г — означают атом простого вещества и формульную единицу со-
коиНеНИЯ В дополнитель'ных узлах, возникающих при реакциях на поверхности
оК
Уравнения реакций (1.1а) — (1.4а) и (1.16) — (1.46)
представляют собой разные формы записи одних и тех же
процессов, поэтому естественно, что при последовательном
рассмотрении они приводят к одинаковым результатам. Этот вопрос
будет более подробно рассмотрен во второй главе, где
используются оба подхода. В остальных главах, где это особо не
оговаривается, по указанным выше соображениям мы будем
отдавать предпочтение системе Крёгера и представлять
квазихимические реакции через структурные элементы.
1.5. Эффективные заряды дефектов
Атомные дефекты проявляют себя во многих
физико-химических процессах как электрически заряженные частицы. Так, в
кристалле германия, состоящем из электронейтральных атомов,
вакансии обнаруживают довольно высокую
электроотрицательность и могут захватывать электроны из валентной зоны,
превращаясь в отрицательно заряженные центры. Образование
такой отрицательно заряженной вакансии равносильно удалению
из кристалла нейтрального атома германия и замене его
электроном. Аналогично, междуузельные атомы металла в
валентном кристалле, например атомы лития в германии, могут
находиться в двух состояниях: в виде нейтральных атомов или
положительно заряженных ионов. Естественно, что такие
заряженные дефекты создают в окружающей области кристалла
электрическое поле, подобное полю заряженных ионов в
растворах электролитов.
Тенденция к проявлению электрических свойств дефектов
еще более ярко выражена в ионных кристаллах.
Проиллюстрируем это (рис. 1.4) на примере вакансий в узле I катионной
подрешетки ионного кристалла, предполагая, что заряд
катионов соответствует классической модели и равен -\~ге.
В идеальном кристалле, не содержащем дефектов, сила,
действующая на ион, помеченный цифрой II, со стороны
любого другого иона полностью уравновешивается силой
взаимодействия его с симметрично расположенным ионом, так что
равнодействующая всех сил, действующих на ион II, вследствие
полной симметрии кристалла равна нулю. Если же теперь уда-
, _ л- — л- — + лить ион *> то на ион ^ будет
+ , действовать нескомпенсирован-
. __ + г\ ^ _ ная сила электростатического
^ притяжения со стороны иона III,
JB Л ^ ^. так что ион II под действием
__ Q-h) -*{^) I I (з)^ + этой силы сместится направо от
+ ~ + Q +
9
РИС. 1.4.
N_ + — Н- — + Схема взаимодействия вакансии с
соседними ионами.
26
вакансии. Легко видеть, что в точности такая же сила как по
величине, так и по направлению действовала бы на ион II со
стороны узла I, если бы вместо удаления положительного иона
мы поместили в этот узел равный по величине отрицательный
заряд. Таким образом, вакансии в ионном кристалле следует
приписать эффективный электрический заряд, равный по
величине и противоположный по знаку заряду удаляемого иона.
Аналогичное рассмотрение дефекта замещения показывает,
что его взаимодействие с окружающими ионами
характеризуется эффективным зарядом, равным разности зарядов
примесного иона и основного иона, замещенного примесью. В случае
когда примесный и замещенный им основной ион имеют равные
заряды, эффективный заряд дефекта замещения равен нулю.
Аналогичная картина обнаруживается в кристаллах с
промежуточной ионно-ковалентной связью. Как указывалось в
разделе 1.3, истинное распределение электронной плотности в
таких кристаллах существенно отличается от предполагаемого
классической ионной моделью и в большинстве случаев
неизвестно. Поэтому определить строго истинные заряды дефектов
в реальных кристаллах невозможно. К счастью, это и не
нужно. Из схемы, приведенной на рис. 1.4, видно, что
электростатическое воздействие дефекта, находящегося в узле I,
определяется не истинным значением его заряда, а тем, насколько его
заряд отличается от заряда окружающих его ионов, состояние
которых отвечает таковому в идеальном кристалле. Поэтому в
рамках квазихимического метода зарядовое состояние атомного
дефекта можно однозначно определить, приписав ему некоторый
эффективный заряд, показывающий, на какую величину
его заряд отличается от заряда соответствующего структурного
элемента в идеальной решетке. Другими словами, эффективные
заряды дефектов определяются как разностные значения по
отношению к общему фону распределения плотности истинных
зарядов в решетке идеального кристалла, знать которое
совершенно не обязательно. Этот вывод находится в полном
соответствии с представлением дефектов как относительных
составляющих единиц кристалла, все свойства которых определяются
по отношению к фону идеальной кристаллической решетки.
Приведенное выше определение эффективных зарядов
дефектов равносильно следующему: эффективный заряд дефекта
равен изменению суммарного заряда кристалла при
образовании в нем одного дефекта данного сорта. Так как заряд
кристалла может изменяться только на величины, кратные заряду
электрона, из последнего определения эффективных зарядов
Дефектов вытекает, что они всегда кратны абсолютному
значению заряда электрона. В этом отношении они радикально от-
личаются от эффективных зарядов ионов, используемых в
теории химической связи, которые определяются истинным
распределением электронной плотности и поэтому могут принимать
яюбые как целочисленные, так и дробные значения.
27
Следуя принятой в химии традиции, будем помечать
эффективные заряды дефектов верхним индексом в единицах, равных
абсолютному значению заряда электрона с указанием
соответствующего знака*. Так, символы FA+ и F©(A) (соответственно
в системе структурных элементов и относительных
составляющих единиц) обозначают дефект замещения с положительным
эффективным зарядом, равным по абсолютному значению
заряду электрона; символы Vmz~ и MGz~ означают вакансию в
подрешетке М с отрицательным эффективным зарядом, в z раз
превышающим заряд электрона. Нулевой эффективный заряд
обозначается косым крестиком: Aix, AOx. Заметим также, что
согласно принятому определению эффективных зарядов
структурные элементы, отвечающие идеальному кристаллу, всегда
считаются электронейтральными независимо от характера
химической связи: Ммх, Vix .
В некоторых случаях необходимо указать истинный заряд
иона; в этих случаях мы будем пользоваться обычной
химической символикой, без нижнего индекса. Так, символ Fe2+
означает двухзарядный катион железа в обычном смысле.
1.6. Электронная проводимость
неметаллических кристаллов.
Электронные дефекты
Согласно сложившейся традиции твердые тела часто
подразделяют на три больших класса в зависимости от удельного
электросопротивления при комнатной температуре: проводники
(Ю-6—10~4 Ом-см), полупроводники (10~2—1012 Ом-см) и
изоляторы (10й—1020 Ом-см). При этом к проводникам относят
в основном металлические твердые тела, а к полупроводникам
и изоляторам — неметаллические как с ковалентной, так и с
ионной связью.
Однако, как показало развитие теории твердого тела,
деление на полупроводники и изоляторы чрезвычайно условно; в
зависимости от температуры и содержания примеси одни и те же
твердые тела могут быть как полупроводниками, так и
изоляторами. В то же время как в тех, так и в других характер разу-
порядоченности в значительной степени определяется типом
химической связи. Поэтому в дальнейшем мы будем
придерживаться более узкой классификации неметаллических твердых
тел и подразделять их, с одной стороны, на полупроводники и
изоляторы с преобладающей ковалентной связью, а с другой —
на соединения с преобладающей ионной связью.
Разумеется, такое деление также достаточно условно
благодаря существованию большого числа соединений с характером
* По Крёгеру, положительные эффективные заряды обозначают также
точками, а отрицательные — штрихами: Fa", Mi "-, Vm" и т. д. Однако такая
система обозначений неудобна в общем случае z-кратных эффективных
зарядов.
28
связи, промежуточным между ковалентным и ионным. Тем не
менее оба типа неметаллических твердых тел радикально
отличаются от металлов по строению их энергетического спектра
электронов.
Как было показано в разделе 1.3, в большинстве кристаллов
с неметаллическими типами связи, находящихся в их основном
квантовом состоянии, отвечающем строго регулярному
строению кристаллической решетки, все разрешенные уровни
энергии в валентной зоне заняты электронами, в то время как в
следующей за ней зоне проводимости все уровни свободны. Это
приводит к локализованному характеру распределения
электронов, исключающему возможность их перемещения по
кристаллу. Поэтому неметаллические кристаллы в основном квантовом
состоянии являются изоляторами.
Однако вследствие тепловых флуктуации или иных причин,
которые будут подробно рассмотрены ниже, во всех твердых
телах наряду с атомными дефектами имеются электронные
дефекты — нарушения регулярности строения электронных
оболочек атомов или ионов. Электронная разупорядоченность
приводит к тому, что при конечных температурах все реальные
твердые тела обладают большей или меньшей электронной
проводимостью.
Первый тип электронных дефектов в неметаллических
кристаллах возникает, если энергия электрона по каким-либо
причинам лежит внутри зоны проводимости, которая в идеальном
кристалле должна быть пустой (такое состояние электрона
аналогично возбужденному состоянию электронов в
изолированном атоме, когда его энергия соответствует следующему
после основного квантовому уровню). Поскольку большинство
разрешенных уровней в зоне проводимости остается свободным,
возбужденный электрон, изменяя свою энергию в пределах
зоны проводимости, может перемещаться по кристаллу,
принимая участие в электропроводности. Именно благодаря этому
обстоятельству низшая разрешенная зона, не занятая в
основном состоянии кристалла, и называется зоной
проводимости, а электроны, энергетические уровни которых лежат в
зоне проводимости,— электронами проводимости. Мы
будем обозначать их символом е~; знак минус указывает на их
отрицательный эффективный заряд.
Механизм движения электронов проводимости в кристаллах
с неметаллической связью в общем случае довольно сложен,
однако в предельных случаях можно пользоваться двумя
приближенными моделями, описанными ниже (см. раздел 6.4).
Первая модель применима к кристаллам с достаточно
широкой зоной проводимости, особенно к кристаллам с ковалент-
нои связью. В этом случае электрон можно описывать как
квазисвободную частицу, отличающуюся от свободного электрона
вакууме некоторой эффективной массой, отличной от массы
вободного электрона. При малых концентрациях электронов в
29
зоне проводимости их поведение аналогично поведению
идеального газа; отличие состоит в том, что в силу волновых свойств
квазисвободные электроны «размазаны» по соответствующим
атомным орбиталям. Последние образуют своеобразную
трехмерную сетку «туннелей», пронизывающих всю
кристаллическую решетку, причем движение квазисвободных электронов по
«туннелям» является безактивационным и не требует затрат
энергии. Такой механизм переноса часто называют
туннельным. В неметаллических кристаллах с широкой зоной, так же
как в металлах, подвижность электронов проводимости
уменьшается с температурой вследствие рассеяния на тепловых флук-
туациях, существующих при колебаниях атомов (фононах).
Вторая модель применима в основном к ионным кристаллам
с узкой зоной проводимости, в пределе даже превращающейся
в тонкий уровень, аналогичный уровням энергии в
изолированном атоме. Такая ситуация возможна в тех случаях, когда
электронные оболочки соседних ионов не перекрываются и тем
самым действие принципа Пули ослаблено. Вследствие слабого
перекрывания электронных оболочек электроны проводимости
локализованы на определенных ионах, даже несмотря на то,
что эквивалентные уровни соседних ионов свободны.
Природа такой автолокализации электронов проводимости
заключается в поляризации избыточным зарядом электрона
окружающей среды, в результате которой ионы несколько
смещаются из своих равновесных положений. В этом случае
избыточный электрон вместе с окружающим его полем поляризации
называется поляроном. При перескоке локализованного
электрона из одного узла в другой ему приходится
преодолевать силы связи с полем поляризации, т. е. затрачивать
некоторую энергию активации, в результате чего его подвижность
экспоненциально растет с температурой. Такой механизм
движения электронов обычно называют прыжковым.
При прыжковом механизме электрон проводимости
основное время локализован на одном ионе (или небольшой группе
ионов), обеспечивая ему отрицательный эффективный заряд,
В этом случае электрон можно рассматривать как дефект
замещения нормального иона того же химического элемента с
зарядом, на минус единицу отличающимся от нормального: Ад~.
При перескоке электрона эффективный заряд переносится на
соседний ион:
A^AIAM - AMAIA^
Второй тип электронных дефектов возникает, когда в
валентной зоне некоторая часть разрешенных уровней не занята
электронами. В этом случае становится возможным
перемещение по кристаллу валентных электронов по вакансионному
механизму: свободный уровень, отвечающий некоторому атому,
может быть занят электроном соседнего атома;
освободившийся при этом уровень соседнего атома занят электроном следу-
30
ющего соседа и т. д. Недостающий валентный электрон, энергия
которого соответствует энергии незанятого уровня в валентной
зоне, называют электронной дыркой; легко видеть, что
движение валентных электронов по кристаллу в случае почти
заполненной валентной зоны можно описывать как движение
дырки в противоположном направлении.
В приближении широкой валентной зоны электронные
дырки рассматриваются как квазисвободные частицы, аналогичные
электронам в зоне проводимости и отличающиеся от них
положительным зарядом, численно равным заряду электрона; мы
будем обозначать их символом е+.
В приближении узкой валентной зоны движение дырок
требует энергии активации и происходит по прыжковому
механизму:
AUUfAyA -> kUiktM
В дальнейшем, когда механизм движения электронов
проводимости и дырок можно не конкретизировать, для их
обозначения мы будем отдавать предпочтение символам зонной
теории е~ и е+.
Описанные механизмы электронной проводимости, вообще
говоря, присущи всем твердым телам с неметаллическими
типами связи, хотя явления, приводящие к появлению электронных
дефектов — электронов проводимости или электронных дырок—
могут быть достаточно разнообразными. Во всех случаях
электропроводность, обусловленную движением отрицательно
заряженных электронов проводимости, называют проводимостью
я-типа (от латинского negative — отрицательный);
электропроводность, обусловленную движением электронных дырок,
обладающих положительным эффективным зарядом —
проводимостью р-типа (от латинского positive — положительный).
Рассмотрим теперь основные механизмы явлений,
приводящих к возникновению электронных дефектов в различных типах
неметаллических твердых тел.
1. Собственные полупроводники. Простейшим механизмом
образования электронных дефектов в неметаллических твердых
телах является непосредственное возбуждение электрона из
состояния с энергией, соответствующей валентной зоне, в
состояние с энергией, соответствующей зоне проводимости; при этом
возникает пара дефектов: электрон проводимости и электронная
Дырка. В символах зонной теории этот процесс изображается
Уравнением квазихимической реакции
нуль -► е~~ + е+ (1.5а)
В приближении узких зон возникновение электрона
проводимости и дырки эквивалентно образованию в двух узлах
решетки дефектов замещения с отрицательным и положительным
эффективными зарядами:
2A£->AI+At (1.56)
31
Поэтому по аналогии с превращением двух свободных атомов
в отрицательный и положительный ионы реакцию образования
электрона проводимости и дырки часто называют собствен-
ной ионизацией кристалла. Термин «собственный» под^
черкивает, что этот процесс не связан с какими-либо
чужеродными включениями, а протекает в собственной решетке
кристалла. Электронная проводимость при собственной ионизации
обусловлена как электронами, так и дырками и называется
собственной проводимостью.
Для протекания элементарного акта собственной ионизации
кристалла необходимо сообщить атому, на котором
локализован валентный электрон, энергию, не меньшую, чем ширина
запрещенной зоны. Из всех энергетических факторов,
обеспечивающих передачу необходимой энергии, важнейшим является
тепловой. Поскольку распределение энергий тепловых флуктуации
носит статистический характер, очевидно, что собственная
ионизация в принципе может происходить при любой температуре,
отличной от абсолютного нуля, причем вероятность процесса
быстро растет с ростом температуры, а при данной
температуре определяется величиной минимальной энергии активации,
равной ширине запрещенной зоны.
Ширина запрещенной зоны в твердых телах является
индивидуальной характеристикой каждого вещества и изменяется в
весьма широких пределах, от сотых долей до нескольких элект-
ронвольт (табл. 1.2).
Особенно большие значения ширины запрещенной зоны
наблюдаются в ионных кристаллах — галогенидах и оксидах; эти
значения приблизительно на два порядка превышают значения
средней энергии тепловых колебаний при комнатной темпера™
туре, поэтому в таких кристаллах вероятность возбуждения
электронов из валентной зоны в зону проводимости ничтожно
мала вплоть до температуры плавления.
Иное поведение обнаруживают твердые тела с достаточно
узкой запрещенной зоной, по ширине сравнимой с энергией
тепловых колебаний атомов. В этих кристаллах, являющихся
изоляторами при низкой температуре, при повышении
температуры становится возможной собственная ионизация под
действием тепловых колебаний, что приводит к появлению в них
электронной проводимости. Такие вещества называются
собственными полупроводниками, поскольку по величине
электропроводности они занимают промежуточное положение
между проводниками (металлами) и изоляторами. В принципе
же полупроводники с точки зрения зонной теории не
отличаются от изоляторов, точнее, отличие носит лишь
количественный характер и заключается в меньшей ширине запрещенной
зоны.
2. Примесные полупроводники. Наличие электронов в зоне
проводимости или дырок в валентной зоне может быть
обусловлено примесью, имеющейся в полупроводнике или изоляторе.
32
Т блииа 1 2. Ширина запрещенной зоны в неметаллических твердых телах
при комнатной температуре (в эВ)
Кристалл
В
Графит
Алмаз
Si
Ge
a-Sn
Р
Те
AlAs
AlSb
GaP
GaAs
GaSb
Ee-Eh
1,5
<0,1
5,3
1,12
0,75
0,08
0,33
0,35
2,2
1,6
2,25
1,4
0,78
Кристалл
InP
InAs
InSb
ZnS
ZnSe
ZnTe
CdS
CdSe
CdTe
PdS
PdSe
PdTe
NaCl
Ec~Eh
1,25
0,33
0,18
3,7
2,8
2,2
2,42
1,74
1,45
0,3
0,27
0,3
8
Кристалл
CuBr
AgCl
AgBr
Agl
RbAg4I5
Ag2S
Ag2Te
CaO
(X-AI2O3
P-AI2O3
ZnO
Zr02+CaO
Ec~Eh
2,9
3
2,5
2
3,0
0,9
0,17
7 J
8
6
3,2
5
Если примесь размещается в узлах кристаллической
решетки по способу замещения, ее влияние на энергетический спектр
электронов оказывается существенно различным в зависимости
от числа валентных электронов в примесном атоме.
Атомы изовалентной примеси, имеющие то же число
валентных электронов, что и замещаемые ими атомы
полупроводника, не вносят непосредственных искажений в строение
энергетического спектра электронов в кристалле. Их
возможное влияние носит косвенный характер и сводится в основном
к некоторому изменению энергетических параметров кристалла
(ширина зон, подвижности электронных дефектов и др.) без
изменения качественной картины.
Радикальные изменения энергетического спектра электронов
вызываются гетеровалентными примесями, атомы
которых по числу валентных электронов отличаются от замещаемых
ими атомов полупроводника. Для определенности рассмотрим
влияние гетеровалентных примесей в типичном
полупроводнике с ковалентными связями — германии, имеющем структуру
типа алмаза.
Атом германия, занимающего место в четвертой группе
Периодической системы, имеет во внешней электронной оболочке
етыре валентных электрона. Каждый из этих электронов при-
егс?^Жит Jie только данному атому, но и одному из четырех
лен оЖа^ших соседей, что обеспечивает насыщенность кова-
Ма НОи связи. По своем} влиянию на зонную структуру гер-
я гетеровалентные примеси делятся на две группы,
решет РИМесные атомы, располагающиеся в кристаллической
ных э^ П° СПос°бу замещения, имеют большее число валент-
нения Л??ТР°Н0В> чем замещаемые или атомы основного соеди-
пятой* г ^ИмеР0м может служить фосфор, занимающий место в
РУппе Периодической системы и имеющий, следователь-
3—321
33
У^ттургтр^-т-гтг^гр РИС. 1.5.
Х/ х зош > ^ Схема энергетических уровней электронов, свя-
.- у прооошмости ■ - занных с донорами и акцепторами.
донорные уробни Но, пять валентных электронов. При за-
акцепторные уровни мещении атома германия атомом фос-»
1 , фора происходит следующее.
th --~~-^-~^-г~~г-г-^~ Четыре электрона атома фосфора рас-
Г- . далентная, ', пределяются между четырьмя ближай-
;'-„ //'.. 3Лна ■ ,- V . шими атомами германия, а пятый элект-
c^-^c-s+^'' / /s / рон оказывается лишним в системе ва^
лентной связи германия. Согласно
зонной модели в кристалле германия каждому атому соответствует
четыре разрешенных энергетических уровня в валентной зоне,
поэтому пятый электрон примесного атома фосфора, в
соответствии с принципом запрета Паули, может находиться только в
следующем квантовом состоянии с более высокой энергией. Для
невзаимодействующих дефектов это состояние соответствует
зоне проводимости, поэтому избыточный электрон должен
давать вклад в проводимость я-типа.
Однако такое состояние избыточного электрона не является
энергетически самым выгодным; наинизшее энергетическое
состояние достигается, когда избыточный электрон
захватывается электростатическим полем положительно заряженного
ионного остатка фосфора, имеющего четыре электрона в валентной
зоне. Понижение энергии избыточного электрона в поле
ионного остатка примеси приводит к тому, что его энергетический
уровень оказывается лежащим в запрещенной зоне, ниже дна
зоны проводимости Ес на некоторую величину AED, равную
энергии его связи с ионным остатком (рис. 1.5).
Наглядное представление об электронной структуре
примесного центра дает атомная модель Бора, согласно которой
электроны в атоме движутся по стационарным орбитам. В этой
модели основному состоянию примесного атома фосфора
в кристалле германия соответствует движение четырех
валентных электронов по внутренним орбитам, а пятого — по
некоторой внешней орбите большего радиуса. Поэтому очевидно, чю
энергия связи пятого электрона должна быть гораздо меньше,
чем четырех остальных (AED значительно меньше ширины
запрещенной зоны), и он легко может быть оторван от
примесного центра и переведен в квазисвободное состояние с
энергией, лежащей в зоне проводимости. Этот процесс аналогичен
процессу ионизации свободного атома при отрыве от него
валентного электрона, поэтому его удобно изобразить в виде
квазихимической реакции
Pge-РЙе + е- (1-6)
где Рх — электронейтральный примесный атом фосфора; Р+ — ионизованные
примесный центр (ионный остаток); е- — электрон проводимости.
к Примесные атомы имеют меньшее число валентных элек-
ов чем замещаемые ими атомы основного соединения. По
Т^°ошению к германию таким свойством обладает, например,
бпо занимающий место в третьей группе Периодической систе-
и имеющий в валентной оболочке три электрона. Поскольку
мы образования насыщенной ковалентнои связи в кристалле
дл манИЯ нужно по четыре электрона от каждого атома,
примесному атому бора для этого не хватает одного электрона, и один
из принадлежащих ему разрешенных уровней энергии
оказывается свободным. Этот уровень может быть заполнен, если при
тепловых флуктуациях атом бора захватит недостающий
электрон у одного из соседних атомов германия. В результате
примесный атом бора превращается в отрицательно заряженный
ион, а в валентной зоне кристалла, образуемой уровнями
валентных электронов германия, возникает электронная дырка:
В&-> В5"е + £+ (1.7)
Захват примесным атомом валентного электрона и
последующее удаление образовавшейся дырки на бесконечно
большое расстояние от отрицательного иона примеси (переход ее
в квазисвободное состояние) требуют преодоления кулоновских
сил притяжения. Поэтому энергетический уровень захваченного
электрона оказывается расположенным в запрещенной зоне,
выше потолка валентной зоны Ей на величину Л£д, равную
энергии реакции ионизации примеси (1.7) (см. рис. 1.5).
Таким образом, на примере германия мы приходим к
следующему выводу. Примесные атомы, имеющие большее число
валентных электронов, чем замещаемые ими атомы основного
вещества, могут служить поставщиками электронов в зону
проводимости полупроводника или изолятора. На этом основании
такие примесные атомы получили название доноров (от
латинского слова donator — жертвователь). Напротив, атомы
примеси, имеющие меньшее число валентных электронов, чем
атомы основного вещества, могут захватывать электроны из
валентной зоны полупроводника или изолятора с образованием в
алентной зоне дырок. Такие примеси получили название а к-
Д е п т о р о в (латинское acceptor — получатель).
Лето убедиться в том, что правило это применимо и к крис-
в *^лам с ионным характером химической связи. Рассмотрим
Вечачестве примера оксид магния MgO. В чистом кристалле, от-
ных Ю1цем ТОЧному стехиометрическому составу, число валент-
в ва ЭлектР?нов в точности равно числу разрешенных уровней
Св лентн°й зоне. При этом значение истинного заряда ионов и
Вен Нная с ним доля ионной связи не играют роли; сущест-
атомо ЧТ° ПрИ обРазовании кристалла MgO из нейтральных
бой пи Магния и кислорода каждый атом магния вносит с со-
^a валентных электрона.
пУ зам РаствоРении примесного иновалентного металла по ти-
еЩения в подрешетке магния число валентных электро-
3*
нов, вносимых в кристалл каждым примесным атомом, будет
отличаться от двух. Так, примесь трехвалентного алюминия, за-
мещающая магний в кристалле MgO, вносит с собой три" ва
лентных электрона на каждый атом, из которых один
оказывается избыточным, поэтому соответствующий ему уровень
энергии в основном квантовом состоянии лежит в запрещенной зоне.
При возбуждении этот электрон может перейти в зону прово
димости, превратившись в квазисвободную частицу (в случае
узкой зоны — в полярой), причем дефект замещения магния
алюминием приобретает положительный эффективный заряд:
А1Й* -> Al^g + е~ (1.8)
Напротив, при растворении в кристалле MgO металличес
кой примеси с одним валентным электроном, например Li, его
валентный электрон заполняет только один энергетический
уровень в валентной зоне кислорода, а второй остается
свободным. Если в кристалле отсутствуют какие-либо дополнительные
атомные дефекты, которые могли бы 'привести к заполнению
незанятого уровня, последний на общем фоне представляет со-
бой электронную дырку в валентной зоне. Ион же лития,
отдавший в валентную зону кислорода один электрон, находится в
виде однократно заряженного положительного иона и на фоне
двукратно заряженных ионов магния проявляет себя как
дефект замещения Li~Mg с отрицательным эффективным зарядом.
Однако такое состояние не является энергетически самым
выгодным; под действием кулоновеких сил в основном
состоянии дырка может быть локализована на одном из ближайших
к примеси ионов кислорода или же «размазана» по нескольким
ионам кислорода. В любом случае примесный центр в его
основном квантовом состоянии можно рассматривать как
квазичастицу с нулевым эффективным зарядом; возбуждение такого
дефекта происходит при отрыве дырки от примесного центра
и переходе ее в квазисвободное (или поляронное) состояние:
LiMg-> LiMg + e^ (1.9)
Таким образом, в рассмотренном примере примесь
алюминия, имеющего в нейтральной форме больше валентных
электронов, чем замещаемый им магний, обладает свойствами
донора; примесь лития, имеющего меньше валентных электронов,
чем магний,—свойствами акцептора в соответствии со
сформулированным выше правилом. Оба рассмотренных примера
валентного кристалла Ge и ионного кристалла MgO показывают,
что, несмотря на существенно различный характер
распределения электронной плотности, свойства донорных и акцепторные
примесей в них вполне аналогичны. Реакции ионизации
примесных центров (1.6) — (1.9) в обоих типах кристаллов выражу
ются сходными уравнениями и могут быть записаны в
обобщенной форме
Dx -> D+ + e" (1.Ю)
36
пля донорной примеси и в форме
Д Ах->А~ + е+ (1.11)
пяя акцепторной примеси.
Качественная схема энергетических уровней доноров и ак-
odob, показанная на рис. 1.5, также оказывается одинако-
ц„ „L вСех неметаллических кристаллов как для полупровод-
НОИ ДЛЯ D
ков так и для диэлектриков, независимо от характера их хи-
Н^ческой связи; при этом каждому типу примеси
приписывается свой энергетический уровень в запрещенной зоне. Правда,
следует иметь в виду, что энергетические состояния электронов,
локализованных на примесных атомах, соответствуют узким
уровням только при достаточно малых концентрациях
примесей когда их взаимодействие не существенно. При больших
концентрациях взаимодействие примесных атомов приводит к
расщеплению уровней в самостоятельную зону, лежащую
внутри запрещенной зоны основного вещества, а в некоторых
случаях даже сливающуюся с зоной проводимости.
Количество электронных дефектов в полупроводнике,
локализованных на примесных атомах или же находящихся в
квазисвободном состоянии, при фиксированной температуре
сильно зависит от энергии ионизации примесей \Е, описываемой
реакциями (1.10) и (1.11). Поскольку эта величина строго
индивидуальна для различных веществ, изучение энергетического
спектра является первоочередной задачей физики
полупроводников.
Характерную величину энергии ионизации примесного до-
норного атома в полупроводнике легко оценить с помощью
простого расчета, используя атомную модель Бора. Согласно этой
модели локализованный на примеси электрон движется вокруг
положительного иона по круговой орбите некоторого радиуса
R, так что кулоновская сила притяжения e2/sR2
уравновешивается центробежной силой
e2jzR2^ т*ъ2]П (1.12)
^_е~7заРяД электрона; 8 — диэлектрическая проницаемость кристалла;
эффективная масса электрона в зоне проводимости; v — линейная
скорость электрона на орбите.
v. Другой стороны, условие квантования в модели Бора
выражается равенством
(jpdq
nh
число- h1 V—ИмпУльс электрона; q — его координата; п — главное квантовое
ш, л —постоянная Планка.
0pg НтегРирование по замкнутому контуру в случае круговой
§pdq = m*v2nR = nh (1.13)
37
Исключая из уравнений (1.12) и (1.13) скорость электрона v,
получаем для радиуса орбиты:
R=JL_(JLb\2 (1Л4)
Энергия связи электрона с ионом примеси равна полной
энергии электрона с обратным знаком:
А г-, с2 m*v2 e2
eR 2 2eR
Учитывая (1.14), получаем:
Эти формулы в равной мере применимы и к акцепторным
примесям, находящимся в электронейтральной форме; в этом
случае по боровской орбите движется электронная дырка.
В кристалле германия эффективная масса электрона
проводимости составляет 12% от массы свободного электрона; 8=15,8
[2]. При этих значениях и при значении я=1, отвечающем
основному состоянию примесного атома, формулы (1.14) и (1.15)
дают R==7 нм, AED = 0,0065 эВ. Экспериментальные значения
энергий ионизации примесей в германии лишь в 1,5—2 раза
превышают расчетное, что, безусловно, является хорошим
результатом для такой грубой оценки.
Оба полученных результата интересны следующим.
Во-первых, радиус боровской орбиты избыточного электрона
примеси в германии огромен по сравнению с межатомным
расстоянием (0,244 нм); электрон «размазан» по большому числу
атомов германия, расположенных на расстоянии 7 нм от
примесного центра. Поэтому примесный центр с локализованным
на нем электроном не является атомом в строгом смысле
слова и должен рассматриваться как некая квазичастица,
физические свойства которой лишь формально уподобляются
свойствам атома.
Во-вторых, энергии ионизации примесей в кристалле
германия почти на два порядка меньше энергии собственной
ионизации германия (ширины запрещенной зоны). Поэтому тепловые
колебания, недостаточные для собственной ионизации герма-
ния, легко ионизуют примесные атомы. Так, уже при комнатной
температуре средняя энертия тепловых колебаний в 6—8 раз
превышает энергию ионизации примесей, поэтому
подавляющая часть избыточных электронов или дырок находится в
квазисвободном состоянии, обеспечивая тем самым довольно
высокую проводимость п- или р-гипа в зависимости от типа
примеси.
3. Нестехиометрические ионные соединения. Согласно зако
ну кратных отношений в формуле бинарного соединения MX г
стехиометрический коэффициент г должен быть в точности ра-
38
цению валентностей обоих компонентов: г = гм/гх. Од-
вен отН еодых телах закон кратных отношений выполняется
нако в ^дИЖеНно. Согласно современным представлениям
ляшь пр д0Г0 точного стехиометрического состава существу-
вблизн ^ ая конечная область составов твердого тела МХГ±6
€Т неко ^ом0генн0сти), в которой соединение содержит избы-
(область ^^ другого компонента, сохраняя при этом свою ос-
Т0Кную кристаллографическую структуру и обнаруживая
Н° ое изменение физико-химических свойств по составу
rm^-241 Отклонения от точного стехиометрического состава
ктерны и для кристаллов многокомпонентных — тройных и
более сложных соединений [25—27].
Ширина области гомогенности по ту и другую сторону от
стехиометрического состава для каждого конкретного
соединения определяется многими факторами, в первую очередь
такими, как характер химической связи и кристаллографические
особенности, и может варьировать в довольно широких
пределах. В неметаллических твердых телах имеется тенденция к
расширению области гомогенности по мере уменьшения доли
ионной связи и увеличения доли ковалентной. Так, если в
соединениях, наиболее полно отвечающих модели ионного кристалла —
галогенидах щелочных металлов, область гомогенности
настолько узка, что не фиксируется методами химического
анализа, то в оксидах, и особенно в халькогенидах; ее ширина
может достигать многих атомных процентов.
Отклонения состава твердых тел от стехиометрического
сильно влияют на их транспортные свойства, и в первую
очередь на электропроводность. Зависимость электропроводности
от степени нестехиометричности наиболее сильно проявляется
в твердых телах, характер связи в которых близок к ионному,
таких, как галогениды и оксиды металлов.
Важной особенностью любой однородной среды, содержащей
подвижные заряженные частицы, является ее способность
сохранять макроскопическую электронейтральность. Эта
способность непосредственно вытекает из одного из
фундаментальных уравнений электростатики — уравнения Пуассона,
связывающего вторую пространственную производную
электростатического потенциала Аф (Д — оператор Лапласа) с плотностью
объемного электрического заряда р:
Дср= -—р (1.16)
8
е е — диэлектрическая проницаемость среды.
скопиГЛаСН? ЭТ0МУ Уравнению накопление в какой-либо макро-
тРичеЧеСК°^ °^ласти электропроводной среды объемного элек-
электг)К0Г0 заРяда пРивоДит к возникновению неоднородного
н*ою ^ческого п°ля (Дф^О), сильно повышающего внутрен-
нергию системы. Заряженные частицы, двигаясь в элект-
39
рическом поле, перераспределяют объемный заряд до тех пор,
пока потенциал не станет одинаковым по всему объему. При
этом в любой макроскопической области Аф = 0; в соответствии
с уравнением (1.16) такое состояние достигается только при
р=0, т. е. при соблюдении электронейтральности любой
макроскопической области однородной проводящей среды.
Следует оговориться, что условие электронейтральности
может не выполняться при резких нарушениях однородности
среды, например, на поверхности твердого тела, а в случае
поликристаллических образцов — и на внутренних поверхностях,
т. е. границах зерен и пор. Это приводит к возникновению
вблизи поверхности двойного электрического слоя, сильно
влияющего на поверхностные свойства твердых тел [16]:. Однако
строение поверхности не оказывает сколько-нибудь существенного
влияния на объемные свойства, поэтому при рассмотрении
объемных свойств твердых тел соблюдение условия
электронейтральности следует считать обязательным.
Для нестехиометрических ионных кристаллов учет условия
электронейтральности сразу приводит к важному результату.
Поскольку в ионных кристаллах оба компонента находятся в
виде заряженных ионов, любое отклонение состава кристалла
от стехиометрического должно сопровождаться образованием
электронных дефектов, компенсирующих заряд ионов,
избыточных по сравнению со стехиометрическим составом. Так, при
избыточном содержании металла в ионном соединении МХг+б
положительный заряд избыточных катионов должен быть
скомпенсирован соответствующим числом избыточных электронов.
Как было показано в разделе 1.3, при образовании ионного
кристалла валентные электроны металла переходят на
электронные оболочки неметалла, образуя вместе с его валентными
электронами заполненную валентную зону. Очевидно, что
общее число валентных электронов в кристалле равно числу
разрешенных уровней в валентной зоне только при строго стехио-
метрическом составе. При избыточном содержании металла его
внешние электроны уже не могут разместиться в валентной
зоне и вынуждены частично занимать более высокие
энергетические уровни в зоне проводимости или же какие-либо
локальные уровни донорного типа в запрещенной зоне.
При недостаточном содержании металла по сравнению ее
стехиометрическим составом электронейтральность
обеспечивается недостатком соответствующего числа электронов.
Энергетические уровни недостающих электронов могут быть
расположены либо в валентной зоне, что соответствует квазисвободным
электронным дыркам, либо в запрещенной зоне. В последнем
случае они соответствуют локализованным состояниям дырок и
аналогичны локальным энергетическим уровням акцепторного
типа в примесных полупроводниках.
Природа локальных уровней в нестехиометрических
соединениях будет подробно рассмотрена ниже; здесь следует отме-
40
тить, что возбуждение избыточных электронов в зону
проводимости с локальных уровней донорного типа и захват
электронов из валентной зоны локальными уровнями акцепторного типа
в нестехиометрических соединениях подобны описанному для
примесных полупроводников. Более того, и сами электронные
дефекты в нестехиометрических ионных кристаллах и в
примесных полупроводниках имеют в значительной степени общую
природу: в тех и других электронные дефекты возникают из-за
несоответствия числа электронов числу разрешенных уровней в
валентной зоне. Поэтому, несмотря на большую ширину
запрещенной зоны, нестехиометрические ионные кристаллы
обнаруживают ряд свойств, типичных для примесных
полупроводников; в частности, они обладают значительной электронной
проводимостью я-типа при избытке металлического компонента и
р-типа при избытке неметаллического компонента.
1.7. Энергетические состояния электронов
в нестехиометрических кристаллах
Важнейшей особенностью кристаллов нестехиометрических
соединений, отличающей их, например, от собственных
полупроводников, является довольно высокая концентрация атомных
дефектов даже при отсутствии чужеродных примесей. Так,
избыток металлического компонента М в нестехиометрическом
кристалле МХг_б может реализоваться либо за счет
соответствующего числа междуузельных катионов, либо за счет вакансий
в анионной подрешетке. Напротив, при избытке
неметаллического компонента X кристалл МХг+б должен содержать
соответствующее число либо междуузельных анионов, либо
вакансий в катионной подрешетке.
Все атомные дефекты, взаимодействуя с электронами
проводимости или дырками, в зависимости от их химической
природы могут проявлять донорные или акцепторные свойства,
аналогичные свойствам примесных атомов. Так, если внешняя
электронная оболочка междуузельных атомов заполнена
меньше чем наполовину, как в случае электроположительных
элементов, атом склонен терять эти электроны, передавая их в зо-
НУ проводимости кристалла и превращаясь в положительный
ион. При этом атом действует как донор. В противном случае
Междуузельные атомы (предпочтительнее превращаются в
отрицательные ионы, играя роль акцепторов.
Аналогичные превращения могут испытывать и вакансии,
захватывая своим полем электроны; последние при этом могут
ьггь локализованы на одном из атомов, ближайших к вакан-
и, или же «размазаны» по целой группе атомов, аналогично
роению атома фосфора в кристалле германия, рассмотренно-
У в предыдущем разделе. Если все валентные уровни атомов,
еДних с вакансией, заняты, вакансия может захватить элект-
41
рон только с набором квантовых чисел, отвечающим зоне
проводимости; в этом случае энергия электрона соответствует до-
норному уровню в запрещенной зоне (см. рис. 1.5). При
термическом возбуждении электрон снова переходит в зону
проводимости и вакансия выполняет функцию донора. Если же на
соседних с вакансией атомах имеется свободный уровень, он
может быть заполнен при захвате полем вакансии валентного
электрона, и вакансия играет роль акцептора. Захват
вакансией валентного электрона эквивалентен переходу в валентную
зону дырки, первоначально локализованной на соседних к
вакансии атомах.
В валентных кристаллах вакансии могут быть как
донорами, так и акцепторами. В ионных кристаллах анионные
вакансии являются, как правило, донорами, а катионные —
акцепторами. Сущность происходящих при этом электронных переходов
удобно проследить на простейшем примере образования
центров окраски в кристаллах галогенидов щелочных металлов.
Кристаллы галогенидов щелочных металлов, в обычных
условиях бесцветные, приобретают интенсивную окраску, если их
прогреть в парах соответствующего щелочного металла; это
явление носит название аддитивного окрашивания. Так,
кристалл NaCl, прогретый в парах натрия, окрашивается в
желтый цвет, КО, прогретый в парах калия,— в красный цвет.
Очевидно, что при этом возникают какие-то дефекты в
структуре кристаллов, ответственные за их окраску. Эти дефекты
получили название jF-центров (от немецкого слова Farbenzent-
гит — центр окраски).
Зная в общих чертах свойства кристаллов галогенидов
щелочных металлов, нетрудно установить природу /^центров. При
прогревании в парах металла кристалл получает избыток
металлических ионов по сравнению со стехиометрическим
составом и одновременно равное число избыточных электронов.
Избыточные ионы металла могут разместиться в кристалле двумя
способами: или в узлах соответствующей им подрешетки на
поверхности кристалла, притянув к себе равное число анионов
из глубины кристалла, так что в глубине образуются анионные
вакансии, или же внедряясь в междуузлия. В кристаллах
галогенидов щелочных металлов первый способ энергетически
оказывается значительно более предпочтительным, поэтому можно
утверждать, что аддитивно окрашенные кристаллы этих
соединений содержат в равных количествах анионные вакансии Vx+
и избыточные электроны е~.
Изолированные анионные вакансии имеют положительный
эффективный заряд и поэтому могут притягивать электроны
проводимости, образуя при этом электрически нейтральные
комплексы. Такой комплекс, называемый /^-центром, можно
представить в виде некоторого квазиатома, изображенного
схематически на рис. 1.6, а. Окружности изображают боровские
орбиты локализованного электрона; основному энергетическо-
42
. ^- 4- - + ~ + - + _ + _ +
+ / \ / \
/ /
Vx+
+ -I + - 1+ T m
^ *l ~
/
^ 4. — Ф »* *b ~* + -* + ~- .j. -. +
м?ИС I*.
Модель F-центра (а) и У-центра (б) в кристаллах галогенидов щелочных
металлов.
му состоянию отвечает внутренняя орбита с наименьшим
радиусом. При тепловом, оптическом или другом возбуждении
электрон 'переходит на более удаленную от вакансии орбиту; при
обратном переходе происходит излучение кванта света, что и
проявляется в окраске кристалла. При отрыве и удалении
электрона от вакансии он переходит в зону проводимости;
соответствующая квазихимическая реакция описывает ионизацию
f-центра, аналогичную ионизации примесного донора:
Fx-+Vt + e~ (1.17)
При нагревании кристалла в парах галогена возникает
противоположная картина. В этом случае избыток неметалла
обеспечивается вакансиями в катионной подрешетке, а компенсация
заряда осуществляется электронными дырками. Захват дырок
катионными вакансиями приводит к образованию атомоподоб-
ных центров, в которых дырка движется вокруг вакансии по
одной из стационарных орбит (рис. 1.6,6). Такие центры
называются F-центрами. Оптические свойства 1/-центров
аналогичны свойствам F-центров; различие состоит в том, что полоса
поглощения У-центров лежит в инфракрасной области.
Ионизация 1/-центров, приводящая к высвобождению дырок,
протекает согласно уравнению
Vx->Vu + e+ (1.18)
аналогичному уравнению реакции ионизации примесных
акцепторов.
В кристаллах, состоящих из ионов многовалентных
элементов, возможны более сложные процессы. Так, при валентности
металла z\ катионная вакансия, в чистом виде имеющая
эффективный заряд —z\, может захватить дырку, образовав при этом
Центр, подобный У-центру, но обладающий в отличие от
последнего эффективным зарядом —(z\—1). Далее, этот центр может
43
захватить вторую дырку, изме"ив s**6™^ к^ионн^ва-
црятт (7л 9) и так далее. В оощем ^^у
чения ^i z;, и id*. jx ок после чего ее эффективный
кансия может захватить z\ дыри^, и^ ч'Ч'
мояд будет равен нулю. Аналогичный захват электронов
проводимости анионными вакансиями приводит к тому, что их
эффективные заряды могут иметь любое целочисленное
положительное значение в пределах от -\-z2 до нуля (где г2 —
валентность неметалла).
Примесные нестехиометрические соединения имеют, как
правило, несколько локальных донорных или акцепторных уровней
в запрещенной зоне, из которых одни связаны с собственными
атомными дефектами — междуузельными атомами или
вакансиями, а другие — с примесными центрами, рассмотренными в
предыдущем разделе.
Описанное изменение эффективных зарядов дефектов при
захвате электронов или дырок может иметь место и в стехио-
метрических кристаллах, содержащих достаточное число
электронных дефектов, в частности, в валентных полупроводниках.
Так, в кристалле германия вакансии обладают акцепторными
СВОЙСТВаМИ И МОГуТ НаХОДИТЬСЯ В ДВуХ СОСТОЯНИЯХ VXGe И V"Ge-
1.8. Протяженные дефекты
До сих пор мы рассматривали атомные дефекты, связанные с
неправильным расположением отдельных атомов, каждый из
которых проявляет себя как некоторая изолированная
квазичастица. Такие дефекты обычно называют точечными
(нульмерными). Вместе с тем в реальных твердых телах всегда существует
большое число разнообразных нарушений идеальной
кристаллической структуры, одновременно охватывающих значительные
группы атомов и проявляющих себя как протяженные (одно-,
двух- и трехмерные) дефекты. Детальное описание
протяженных дефектов не является целью данной книги, так как это
потребовало бы существенно иного подхода и иного
формализма, не меняя при этом принципиального содержания
излагаемой здесь теории. Поэтому мы ограничимся лишь краткой
характеристикой простейших протяженных дефектов,
рассматривая их по степени усложнения пространственной структуры.
Одномерные (линейные) дефекты описываются линиями,
протяженность которых велика по сравнению с межатомными
расстояниями и во многих случаях достигает макроскопической
протяженности кристаллов. Такими дефектами являются
дислокации, особенно подробно изученные для
монокристаллических простых веществ -— металлов и атомных
полупроводников [2]. Схематическое изображение простейших видов
дислокаций— краевой и винтовой — дано на рис. 1.7 и 1.8. Как
видно из рис. 1.7, в случае краевой дислокации нарушение
правильной периодичности кристаллической решетки достигается
наличием неполной атомной плоскости, пронизывающей лишь
44
РИС. 1.7.
Краевая дислокация.
часть кристалла; край этой «лишней» плоскости и представляет
собой дислокационную линию.
Образование винтовой дислокации (см. рис. 1.8) можно
представить следующим образом. Сделаем в кристалле
мысленный разрез в вертикальной плоскости, заштрихованный на
рис. 1.8, и сдвинем левую область вниз на одно межатомное
расстояние. Дислокационная линия при этом будет направлена
по вертикали, как показано стрелкой. При такой конфигурации
атомные плоскости образуют вдоль дислокационной линии
своего рода «винтовую лестницу», чем и обусловлено название
такой дислокации.
Дислокации присутствуют практически во всех кристаллах.
Их концентрация сильно зависит от способа получения и
обработки материала и может изменяться в очень широких
пределах от 102—103 см~2 в наиболее совершенных кристаллах
германия и кремния до 1011—1012 см~2 в сильно деформированных
металлических кристаллах. И только путем создания
специальных условий удается выращивать тонкие нити («усы»)
кристаллов, не содержащие дислокаций.
Дислокации играют чрезвычайно важную роль в
механических свойствах твердых тел, прежде всего таких, как
ползучесть и прочность. Так, согласно теории упругости кристаллы
с идеальной структурой должны бы выдерживать относительную
упругую деформацию сдвига, достигающую примерно 50%.
Между тем реальные кристаллы подвергаются пластическому
течению уже при деформациях ^10~4—10~3. Это объясняется
45
РИС. 1-8-
Винтовая дислокация.
весьма высокой подвижностью
дислокаций под влиянием
механического напряжения,
приводящего к сравнительно
легкой сдвиговой деформации
кристаллов. Механизм такого
процесса наглядно
изображается рис. 1.9. Отсюда
становится очевидной одна из важнейших задач современного
материаловедения — получение бездислокационных материалов,
которые обладали бы повышенной прочностью.
Винтовые дислокации играют важную роль в кинетике роста
кристаллов. Так, при выращивании кристаллов в условиях
малого (»1%) пересыщения наблюдаемая скорость роста
значительно превышает теоретическую, рассчитанную для
идеального кристалла. Известен случай [2], когда такое превышение
достигало Ю1300 раз! Такую величину расхождения между
теорией и экспериментом следует считать своего рода рекордом,
не имеющим себе равных в истории физикохимии.
Механизм роста кристалла вдоль линии винтовой
дислокации легко понять на основании рис. 1.8. Очевидно, что
осаждаемым атомам легче всего занять места на поверхности
кристалла рядом с заштрихованной плоскостью, в результате эта
плоскость сместится влево. Когда ее поворот вокруг линии
дислокации достигнет 360°, на поверхности кристалла вырастет
новый монослой атомов. Такой механизм обычно реализуется
при достаточно низких температурах, когда даже относительно
небольшие различия в энергии активации осаждения играют
важную роль. При высоких температурах, имеющих особо
важное значение для химии твердого тела, роль дислокаций в
кинетике роста становится менее существенной.
Для проблемы точечных дефектов, представляющей
основное содержание данной книги, дислокации важны как источ-
• ••••••
• Ф Ф Ф f Ф Ф
4 Ф Ф Ф I Ф f
нТО
• • • • •
Ф Ф 4 1 Ф
Ф Ф Ф Ф Ф
Ф Ф Ф Ф Ф
Ф Ф Ф Ф Ф
• Ф Ф Ф Ф Ф
Ф Ф Ф Ф Ф Ф
Ф Ф Ф Ф Ф Ф
Ф Ф Ф i ф Ф
(г (Г в
РИС. 1.9.
Возникновение и передвижение краевой дислокации под влиянием
механического напряжения.
46
ет
it
iu i
лХач-1
mn
uttiTT
tttU-J-
шлм
ffittt
iffiit
tffi^t
inTT
П
4
1 I
-Д-4
1 1
Ti
1 {
Hfet-a
*#£. t.to.
ида зерен с малым углом разориентировки.
или сток вакансий и междуузельных
^томов. Действительно, при образовании
а акансий высвободившиеся атомы не
обязательно должны выходить на
поверхность кристалла; при наличии краевых
дислокаций они могут оседать на линии
дислокации, достраивая неполную
атомную плоскость. Напротив, при разборке
этой плоскости высвободившиеся атомы
будут либо аннигилировать с
вакансиями в объеме кристалла, либо внедряться
в междуузлия. Очевидно, что такие
процессы должны играть важную роль в ки- ■"•р1"
нетике образования точечных дефектов,
однако они не в состоянии изменить равновесные значения их
концентраций, поскольку последние определяются объемными
энергетическими характеристиками кристаллов.
Двухмерные (поверхностные) дефекты более разнообразны,
нежели одномерные. Среди них наиболее важное значение
имеют границы зерен в поликристаллических образцах,
представляющие собой поверхности с самой различной
конфигурацией. Достаточно простые модели таких поверхностей раздела
можно представить лишь в случаях, когда атомные плоскости в
соседних кристаллитах разориентированы на достаточно малый
угол 6 (рис. 1.10). При этом атомные плоскости,
оканчивающиеся на поверхности раздела, образуют двухмерную сетку краевых
дислокаций, для которых остается в силе все сказанное выше.
При больших углах разориентировки картина становится
гораздо более сложной. В этом случае с определенным успехом
можно рассматривать межкристаллитную границу как
некоторый слой с аморфной структурой. В ряде случаев как
отдельные дислокации, так и межкристаллитные границы могут
давать заметный, а иногда и решающий вклад в диффузионные
процессы в реальных твердых телах [28, 29].
Трехмерные (объемные) дефекты представлены, главным
образом, порами и различного рода инородными включениями.
Поверхность пор в принципе обладает теми же свойствами, что
и плоская поверхность; в частности, она служит источником или
стоком вакансий и междуузельных атомов [30].
Наряду с перечисленными типами протяженных дефектов
особое место занимают различные образования, возникающие
при ассоциации точечных дефектов. Дело в том, что точечные
Дефекты можно рассматривать как невзаимодействующие
частицы лишь в области весьма малых концентраций, не
превышающих 0,1% (ат.).
47
В большинстве чистых простых веществ и бинарных
соединений, не содержащих посторонних примесей, реально
достижимые концентрации дефектов не превышают указанного
значения.
Однако существует ряд соединений, концентрация дефектов
в которых значительно выше. Наиболее известным из них яв~
ляется вюстит FeO, который вообще не существует в стехиомет-
рическом состоянии, а всегда содержит значительное
избыточное количество кислорода. Так, при 1000 °С состав вюстита
соответствует формуле Fe0,88O, что достигается наличием 12(/0
(ат.) вакансий в подрешетке железа. Высокие отклонения от
стехиометрии наблюдаются у сульфидов железа и меди, Fe^ssS
и Cui,73S. Напротив, оксиды тугоплавких металлов Ti02, V205,
Nb205, M0O3, W03 и некоторые другие обнаруживают большой
дефицит кислорода, что указывает на высокие концентрации
вакансий кислорода.
Поскольку дефекты в указанных оксидах электрически
заряжены, естественно, что при высоких концентрациях сильное
электростатическое взаимодействие должно приводить к их
упорядоченному расположению.
В этом отношении исключение составляют монооксиды
титана и ванадия TiO и VO [31]. В этих необычных соединениях
обе подрешетки — катионная и анионная — содержат
одновременно очень высокие концентрации вакансий [в сумме около
30% (ат.) даже при стехиометрическом составе].. Причина та»
кой аномалии заключается в специфической электронной
структуре этих материалов. Ионы титана и ванадия в
кристаллической решетке этих оксидов имеют сильно перекрывающиеся
d-орбитали, благодаря чему они связаны между собой
значительной металлической связью. Поэтому обсуждаемые
соединения часто называют полуметаллами. Металлическая связь
приводит к сильному экранированию эффективных зарядов
вакансий и, следовательно, к ослаблению их электростатического
взаимодействия. Поэтому вакансии в TiO и VO в значительной
мере проявляют себя как слабо взаимодействующие точечные
дефекты, хаотически распределенные по узлам обеих подрешс-
ток.
Взаимодействие дефектов при высоких концентрациях может
приводить по крайней мере к двум различным эффектам [32,
-33]. Первый из них заключается в ассоциации атомных
дефектов в различного рода комплексы. Простейшие из них, парные,
будут рассмотрены ниже (см. раздел 5.3) для примесных
ионных кристаллов. Более сложные комплексы, содержащие более
двух атомных дефектов, получили название кластеров.
Типичным примером системы с ярко выраженным
образованием кластеров является вюстит FeO. Вследствие значительной
нестехиометричности в сторону избыточного содержания
кислорода согласно модели точечных дефектов в катионной
подрешетке этого соединения должны содержаться эквивалентные
48
p0C * " /cp3+.2VFe) в решетке вюстита.
Кластер (^е
вакансий и ионов Fe3+ с повышен-
тст положительным зарядом (электрон-
ВЬ1Ш дырки в вюстите не являются квази-
НЫ^5одными частицами, а локализованы на
СВ°ионах). На самом деле оказалось, что
^вюстите содержатся также междуузель-
В те ионы железа, а концентрация вакансий
Ндвое выше, чем это вытекает из величины отклонения от сте-
хиометрического состава.
Механизм такого явления состоит в следующем. Вследствие
электростатического взаимодействия катионная вакансия и ион
Fe3+ притягиваются друг к другу, стремясь занять соседние
узлы в решетке FeO, обладающие октаэдрической координацией
по кислороду (рис. 1.11). Однако энергетически более выгодной
оказывается конфигурация, при которой ион Fe3i~ переходит в
междуузлие с тетраэдрической координацией, в результате чего
появляется вторая вакансия в октаэдрическом узле, причем
расположение иона Fe3+ по отношению к обеим вакансиям
симметрично. Таким образом, образовавшийся кластер содержит
четыре иона кислорода, занимающие вершины тетраэдра, два
иона Fe2+ и две вакансии в октаэдрических позициях и один ион
рез+ в ценТре тетраэдра. Такая конфигурация является ничем
иным, как элементом шпинельной структуры магнетита Fe304,
и ее можно рассматривать как субмикродомен фазы магнетита
в фазе вюстита. При высоких температурах описанные
кластеры слабо взаимодействуют друг с другом и распределены по
решетке хаотически. Однако при понижении температуры они
притягиваются, образуя кластеры более высоких порядков. При
достаточно высокой концентрации такие мультикластеры
проявляют себя как сверхструктура и могут обнаруживаться рент-
геноструктурным анализом.
Описанная структура кластеров в решетке FeO объясняет
С0кУЮ степень нестехиометричности, достигаемую в этом ок-
ока6' ^е**СТВИтельно> отклонение от стехиометрии в этом случае
и Ь1вается равносильным растворению в вюстите магнетита
Вто степенное преобразование структуры первого в структуру
сти 05°' ЧТ° Д0пУскает довольно широкую область гомогенно-
оразующихся промежуточных фаз.
°кситт °** ТИП пРотяженных дефектов образуется в высших
обнаруХ пеРех°Дных металлов Ti02, V205, Nb205, M0O3, W03,
с°ставЖИВаЮЩИх большие отклонения от стехиометрического
Может9 В СТ0Р0нУ дефицита кислорода (например, рутил Ti02
МоделиВ°Сстанавливаться вплоть Д° состава TiOijs). Согласно
точечных дефектов, они должны были бы содержать
4^321
49
0- Катион
О-Анион
РИС. U2.
Образование структуры сдвига в оксиде типа Re03.
высокие концентрации вакансий кислорода. На самом деле
дефицит кислорода приводит к частичной перестройке решетки
этих соединений в плоскостях, получивших название
плоскостей кристаллографического сдвига.
Проиллюстрируем этот эффект на примере триоксидов
металлов со структурой Re03. Эта решетка состоит из кислород^
ных октаэдров, соединенных вершинами; катионы размещены
в центрах октаэдров. При частичной потере кислорода в не»
стехиометрическом оксиде возникает соответствующее
количество кислородных вакансий, которые упорядочиваются в
некоторых плоскостях; одна из возможных конфигураций показана
на рис. 1.12. Затем части кристалла, разделенные этими
плоскостями, сдвигаются относительно друг друга так, чтобы
вакансии аннигилировали. Это достигается тем, что кислородные
октаэдры соприкасаются не только вершинами, но и ребрами.
За счет такого уплотнения структуры вдоль плоскости сдвига
возникает слой с повышенным отношением металл : кислород.
Поскольку плоскости сдвига могут располагаться на
различных расстояниях друг от друга, при таком типе разупорядо-
чения можно реализовать целый набор составов оксида,
описываемых формулами Ren03n-i и Re7703n-2, где п — ряд
натуральных чисел. Такой ряд соединений фиксированного состава
формально аналогичен гомологическому ряду углеводородов
CnH2n-{-2 и CnH2n+i и поэтому также получил название
гомологического ряда. Аналогично, в структуре рутила Т\Ог
существует гомологический ряд оксидов Tin02n-\ и т. д.
50
льных кристаллах, как правило, существует широкий
Расстояний между соседними плоскостями сдвига, что
набор Р т каЖущееся непрерывное изменение состава окси-
°беиГсЬазовой диаграмме.
^а о думеется, образование как кластеров, так и структур сдви-
отменяет существования в этих кристаллах точечных де-
Га Нов поскольку оно вытекает из общих принципов статисти-
^еКкой'термодинамики. Образование протяженных дефектов
Не<шь снимает избыток точечных дефектов, приводящий к их
лИ н0Му взаимодействию; остающиеся в малых концентрациях
Очечные дефекты должны вести себя в принципе так же, как
« в обычных, слабо разупорядоченных кристаллах.
1.9. Твердые тела со структурной
резупорядоченностью
До сих пор мы описывали состояния твердых тел с
неупорядоченным расположением атомов с помощью представлений о
точечных дефектах как локальных нарушениях регулярной
кристаллической структуры. Такие представления совершенно
естественны для кристаллов, строение которых сравнительно
мало отличается от регулярного и в которых концентрации
дефектов невелики по сравнению с концентрацией узлов решетки.
Понятие точечного дефекта в некоторой степени утрачивает
смысл в концентрированных твердых растворах, содержащих
статистически распределенные атомы разных сортов в
сравнимых концентрациях. Однако и здесь представления о точечных
дефектах часто оправданы, поскольку выбор базисной
регулярной структуры, относительно которой определяются точечные
дефекты, в большинстве случаев не представляет затруднений.
Существует, однако, особый класс соединений, по
характеру связи близких к ионным, в которых степень разупорядочен-
ности одного из компонентов (чаще всего катионов) настолько
велика, что выбор базисной упорядоченной структуры сильно
затруднен или вообще невозможен. В соединениях этого типа
неупорядоченное распределение катионов, как правило, связа-
Но с кристаллографическими особенностями их структуры и
поэтому называется структурной раз упорядочены о-
стью [16]|. Структурную разупорядоченность ионных кристал-
л°в не следует смешивать с антиструктурной разупорядоченно-
етью^ интерметаллических или валентных соединений,
заключающейся в неправильном размещении атомов, а именно атомов
** в Узлах В и наоборот. В случае антиструктурной разупорядо-
нности базисная регулярная структура кристалла четко
определена, и неправильно расположенные атомы можно рассмат-
п«Вать как точечные дефекты. В случае же структурной разупо-
Доченности понятие точечного дефекта лишено смысла.
Da UJ,e^[ отличительной чертой твердых тел со структурной
упорядоченностью является большое число разрешенных
51
кристаллографических позиций, в которых могут располагаться
катионы, существенно превышающее число самих катионов.
В этом отношении строение катионной подрешетки твердых
тел со структурной разупорядоченностью напоминает
электронное строение металлов, в которых число разрешенных состояний
электронов существенно превышает число самих электронов.
Простейший пример структурно-разупорядоченного
кристалла представляет а-модификация иодида серебра, существующая
при температурах выше 146 °С. В структуре a-Agl ионы иода
образуют объемно-центрированную кубическую подрешетку.
Между ними находится большое число кристаллографических
пустот, имеющих сравнимые размеры: на каждый ион серебра
приходится 3 позиции, находящихся между 2 анионами
(двукратная координация), 6 позиций с четырехкратной
координацией и 12 позиций с трехкратной. Как показывают вычисления,
энергии ионов серебра в разных позициях различаются
незначительно, на величины zzkT, поэтому катионы статистически
распределены по всем этим позициям, образуя некоторое подо»
бие катионной жидкости, в которую погружена жесткая
анионная подрешетка. Разумеется, такое представление слишком
упрощено; в отличие от электронов в металлах катионы в
ионных кристаллах не являются свободными частицами, и их
перескоки из одной позиции в другую требуют некоторой энергии
активации. Однако разрешенные позиции в a-Agl настолько
тесно прилегают друг к другу, что образуют своеобразные
кристаллографические туннели, по которым катионы могут
двигаться с минимальными затруднениями. Это обеспечивает очень
высокую ионную проводимость a-Agl по ионам серебра, при
температуре фазового превращения почти на 4 порядка
превышающую проводимость низкопроводящей ^-модификации и срав
нимую с проводимостью ионных расплавов или растворов
электролитов. Особенности структуры a-Agl обусловливают и
другое его важное свойство как твердого электролита —
униполярный характер проводимости, в которой участвуют
практически только ионы серебра.
Большой практический интерес представляет синтез
соединений, которые сохраняли бы структурную разупорядоченность
и связанные с ней электролитические свойства при комнатной
температуре. Иодид серебра оказался подходящим базисным
соединением для этой цели; на его основе синтезировано
несколько десятков твердых электролитов, удовлетворяющих
поставленному требованию. Это достигается добавлением к иоди-
ду серебра каких-либо ионов, стабилизирующих кубическую
структуру и препятствующих ее превращению при низких
температурах в гексагональную, плотноупакованную по анионам.
Среди полученных таким образом твердых электролитов
наилучшими характеристиками обладают твердые электролиты
KAg4Is и RbAgJs, в которых 20% ионов серебра замещено
ионами калия или рубидия, и AgeUWO^ в котором 20% ионов иода
52
но ионами W042~. Проводимость этих соединений также
^^^влена практически исключительно ионами серебра.
'^rf°no6nbiun свойствами обладают и высокотемпературные
лификации халькогенидов серебра и меди.
а"МТак, Фазы a'A^2S и a-Ag2Se, существующие при температу-
соответственно выше 178 и 130°С, изоморфны и a-Agl. Oc-
РаХц йХ структуры является жесткий объемно-центрированный
ноВкаС из анионов S2- или Se2~, а два катиона Ag+ статисти-
К ски распределены по тем же 21 кристаллографическим пози-
4%яи чт0 и в a~AgI- Поэтому механизм ионной проводимости
ц эТих соединениях аналогичен описанному выше для a-Agl.
Однако в отличие от a-Agl халькогениды серебра и меди
обладают наряду с ионной также значительной электронной
составляющей проводимости, обусловленной отклонениями их
состава от стехиометрического; при достаточно больших
отклонениях от стехиометрического состава электронная
проводимость достигает весьма высоких значений и становится
преобладающей.
Благодаря такой ососбенности халькогенидов серебра и
меди путем стабилизации их структуры различными добавками
удалось получить ряд материалов, обладающих высокими
значениями ионной и электронной составляющих проводимости
при комнатной и более низких температурах. Так, композиции
0,69 Ag2S+0,285 Agi,7Te + 0,025 Ag4P207 и 0,925 Ag2Se+
+■0,075 Ag3P04 при комнатной температуре имеют наряду с
ионной проводимостью ^0,25 Ом-1-см-1 электронную
проводимость «102—103 Ом-1-см-1, что делает их удобными
материалами для электродов гальванических ячеек с твердыми
электролитами.
Особое место среди структурно-разупорядоченных
соединений занимает бэта-глинозем р-А1203. Этим термином
обозначают семейство полиалюминатов одновалентных металлов,
отличительной особенностью которых является возможность
получать при низких температурах высокую униполярную
проводимость по целому ряду однозарядных катионов.
Собственно Р-глинозем является полиалюминатом натрия. Его состав
отвечает эмпирической формуле Na20-/zAl203, в которой п
может изменяться в пределах от 5,33 до 8,5. Обрабатывая р-гли-
нозем в различных расплавленных солях, удается полностью
заместить в нем ионы натрия другими одновалентными катио-
^а^и ~~~ Щелочных металлов, серебра, аммония и др., а при
обработке в водороде — протонами. Высокая подвижность этих
атионов обусловлена особенностью их размещения в структуре
то u инозем имеет сложную слоистую структуру, основой ко-
Р°и являются двухмерные шпинельные блоки, упакованные
д ами алюминия и кислорода. Блоки отделены друг от друга
гЗЫйгаточно широкими щелями, в которых располагаются свя-
а*ощие ионы кислорода и ионы натрия или замещающего его
53
металла. В отличие от блоков щели упакованы неплотно, так
что общее число позиций, в которых могут размещаться
одновалентные катионы, значительно превышает число последних.
Кроме того, взаимное расположение разрешенных позиций
таково, что перескоки катионов из одной позиции в другую
требуют незначительной энергии активации, сравнимой с
энергией тепловых колебаний. Так как катионы могут двигаться лишь
в плоскости щелей, монокристаллические образцы р-А1203
обладают резко выраженной анизотропией электропроводности.
Специфическая природа структурной разупорядоченности
сильно упрощает теорию транспортных процессов в твердых
телах, где такая разупорядоченность наблюдается.
Действительно, если в большинстве кристаллических тел основной
задачей теории транспортных процессов является вычисление
концентраций дефектов решетки, ответственных за перенос
вещества и зависящих4от многих факторов, то в структурно-раз-
упорядоченных кристаллах соотношением между числом
занятых и свободных позиций задается кристаллографической
■структурой и поэтому является индивидуальной
характеристикой вещества.
1.10. Структура и транспортные
процессы в стеклах
Стекла относятся к числу наиболее важных технических
материалов с аморфной структурой, поэтому их основные свойства
изучены довольно подробно [34]. Транспортные процессы в
стеклах, находящихся в твердом состоянии, подчиняются, в
общем, тем же закономерностям, что и в неметаллических
кристаллических телах. Так, при низких температурах они обычно
являются изоляторами. При повышенных температурах многие
стекла, особенно содержащие щелочные металлы, являются
ионными проводниками; в других случаях, например, в халько-
генидных стеклах, наблюдается электронная проводимость
полупроводникового типа. Хотя зонная теория твердых тел
описывает лишь кристаллы и неприменима к аморфным телам, тем
не менее принято считать, что энергетический спектр электронов
в стеклах в основном подобен полупроводниковому, в
частности, содержит локальные уровни донорного или акцепторного
типа в запрещенной зоне, связанные с какими-то структурными
особенностями, аналогичными дефектами решетки в
кристаллах.
Стекла не имеют единой структуры. Каждый
стеклообразный материал содержит множество различных структурных
элементов; какие из них преобладают, зависит от многих
факторов, главным образом, от состава и от скорости охлаждения
из расплава в области стеклообразования. При этом
важнейшей особенностью стекол, как и всех аморфных твердых тел,
является отсутствие дальнего порядка в расположении атомов.
54
a 6 в
•-A o-O ©-Na
щс. Lis.
Двухмерные модели структуры кристаллического (а) и стеклообразного (б)
жсида А203 и натриевого стекла (в) [34].
Гем не менее рентгеновские исследования показывают, что
элижний лорядок (число ближайших соседей и расстояние до
еШх) в стеклах выражен вполне отчетливо.
Простейшая модель строения двухкомпонентного стекла —
истого оксида AOr (B203, Si02, Ge02, Р205) была предложена
Захариазеном. Он обратил внимание на важность направлен-
шх ковалентных связей в стеклообразной жидкости и описал
:труктуру стекла как трехмерную сетку, не имеющую
периодичности, но имеющую энергию связи, сравнимую с
кристаллической. Он предложил несколько эмпирических правил стеклооб-
разования, из которых важнейшие: а — атом кислорода связан
не более чем с двумя атомами А; б — кислородные
многогранники имеют общие вершины и образуют трехмерную сетку.
Так, в стеклообразном диоксиде кремния сетка построена из
кислородных тетраэдров, окружающих атомы кремния.
Тетраэдры имеют общие вершины, так что каждый атом кислорода
связан с двумя атомами кремния (м ости ков ый кислород).
Если в кристаллической Si02 ориентация двух тетраэдров с
общими вершинами одинакова, то в стекле их взаимная
ориентация может изменяться в довольно широких пределах.
Указанные особенности строения стекла наглядно
иллюстрирует двухмерная модель структуры гипотетического оксида
^03 (рис. 1.13,а, б). Видно, что в сетке имеются пустоты
различных размеров.
:, При добавлении в стекло оксида второго компонента воз-
РЖны два основных способа размещения его атомов. Если вто-
Р°и компонент близок по своим химическим свойствам к
компоненту А (например, G, В, Р в случае силикатных стекол), он
вещает А в сетке. Такие катионы называются сеткообраз-
Ва тел я ми. Напротив, при значительном различии их хи-
55
мических свойств (щелочные и щелочноземельные металлы) до-
бавляемые катионы размещаются в межсеточных пустотах; они
называются модификаторами. Поскольку их внедрение
в пустоты происходит в процессе образования стекла, размеры
полостей определяются размерами добавляемых ионов.
На рис. 1.13, в изображена двухмерная модель строения
натриевого стекла. Избыточный кислород, вводимый в стекло при
добавлении Na20, занимает места в сетке, в результате чего
часть кислорода оказывается связанной только с одним атомом
А. Такой кислород, называемый немостиков ым, может быть
представлен в виде субионов О". Ионы Na+ расположены в
полостях сетки в непосредственной близости к субионам немости-
KOBOiro кислорода и связаны с ним кулоновскими силами.
Перенос ионов в щелочных стеклах можно объяснить,
предположив, что около каждого немостикового кислорода имеется
несколько энергетически эквивалентных позиций, доступных
для размещения щелочного катиона и отделенных друг от
друга энергетическими барьерами. Такая модель допускает два
типа ионных переходов.
Первый заключается в повороте диполя 0~—Na+ путем
перескока катиона из одной -позиции в другую вокруг данного
немостикового кислорода. Такие переходы дают определенный
вклад в диэлектрические потери в переменном электрическом
поле, однако не приводят к поступательному движению ионов
в постоянном поле.
Второй тип движения ионов протекает в два этапа. Сначала
катион покидает свою нормальную позицию около
немостикового кислорода и занимает одну из разрешенных позиций
около второго немостикового кислорода. В результате этого
возникает пара «дефектов»: первый немостиковый кислород не
имеет в ближайшем окружении ни одного катиона, а второй
имеет два катиона. Дальнейшая миграция катионов может быть
обеспечена как заполнением «вакансии» около первого немо^
стикового кислорода путем переходов соседних катионов, так
и перескоками «избыточного» катиона от второго
немостикового кислорода по разрешенным позициям.
В этой схеме отчетливо видна аналогия с переносом тока
дефектами Френкеля в кристаллах; роль междуузельных
катионов здесь играют избыточные катионы возле немостикового
кислорода. Первый тип описанного механизма соответствует
образованию «дефектов», а второй — их миграции.
Продолжая эту аналогию, Хаггинс разработал метод опи
сания стеклообразных оксидов с помощью математического ап
парата статистической термодинамики дефектных кристаллов
(см. гл. 2). Он предложил рассматривать аморфное тело ка-
совокупность некоторых элементарных структурных единиц -
структонов. Каждый структон представляет собой атом ;
его ближайшим окружением, характеризующимся координат*
онным числом и кратностью связей. Это позволяет различать
56
мальные» структоны, отвечающие основному типу ближне-
*#DPооЯдка в стекле, и «дефектные» структоны, связанные с его
jp° Чтением. Так, в описанной модели щелочного стекла не-
^а тиковый кислород, имеющий в ближайшем окружении один
ы°с1очНой катион, представляет собой нормальный структон с
№е ВЬ1М эффективным зарядом. Совокупность таких нормаль-
ЯУЛ структонов аналогична идеальной решетке кристалла. Не-
Н^стиковые субионы кислорода, не содержащие в первой
координационной сфере ни одного катиона или же содержащие
ва катиона, являются дефектными структонами с
эффективными зарядами — еи+е соответственно.
Наряду с сеточной моделью Захариазена часто
используется другая, кристаллитная модель строения стекол. Она
предполагает, что некоторые группировки атомов объединены
в более упорядоченные состояния, чем обычная сетка. Эти
области подобны слабо разупорядоченным кристаллам,
разделенным зонами с меньшим порядком Механизм ионного
транспорта в рамках этой модели предполагается в значительной
мере аналогичным механизму транспорта в кристаллах,
поэтому часто говорят 'просто о вакансионном, или междуузельном,
механизме переноса вещества в стеклах. Впрочем, сеточную и
кристаллитную модели не следует рассматривать как
взаимоисключающие. Скорее всего, они являются предельными
приближениями, предполагающими различные степени
упорядочения: если по сеточной модели упорядочение ограничено
расстояниями 0,5—0,6 нм, то по кристаллитной—1,0—1,2 нм.
Существующая неопределенность в моделях строения
аморфных тел не позволяет сформулировать и надежные
механизмы переноса в этих системах. Однако в рамках различных
моделей почти всегда можно подобрать те или иные схемы,
сводящие процессы переноса в аморфных телах к миграции
некоторых локальных образований, аналогичных точечным
дефектам в кристаллах. Поэтому почти все существующие
теории ионного транспорта в аморфных телах в значительной
мере сводятся к теории транспорта в кристаллах с точечными
дефектами.
Глава 2
Статистическая термодинамика
Дефектных кристаллов
■"•■■ Модельные представления
тп\ГМеНеНИе теРм°Динамики к твердым телам часто бывает за-
ПочтНеН° СИльньш взаимодействием атомов в твердом теле,
еко и не поддающимся более или менее точному математиче-
Дов ^ 0ПИсанию. Поэтому термодинамические функции кристал-
в отличие от термодинамических функций газов или вод-
57
ных растворов удается определить лишь в сравнительно
немногих случаях при использовании предельно упрощенных
модельных представлений.
Наибольшее распространение получила модель,
соответствующая приближению идеальных растворов: кристалл
считается однородной средой, в которой по узлам кристаллической
решетки статистически распределены точечные дефекты;
концентрации дефектов настолько малы, что их взаимодействием
между собой можно пренебречь.
В большинстве случаев применение такой модели
оправдывает себя вплоть до концентраций дефектов «0,1% (мол.);
так как собственная разупорядоченность в чистых кристаллах
в большинстве случаев не превышает этой величины, теория
дает для них правильные результаты. Однако в некоторых
сильно нестехиометрических соединениях, а также в твердых
растворах концентрации дефектов могут достигать больших
значений, вплоть до десятков мольных процентов. В этих случаях
взаимодействие дефектов настолько существенно, что
приближение точечных дефектов становится недостаточным. Этот
вопрос был подробно обсужден в предыдущей главе (см. раздел
1.8), где были рассмотрены протяженные дефекты — кластеры
и структуры сдвига, образующиеся в результате
взаимодействия дефектов. В данной главе протяженные дефекты не
рассматриваются, а обсуждаются лишь точечные дефекты,
существующие в любом кристалле независимо от наличия в нем
любых протяженных дефектов — дислокаций, кластеров или
плоскостей сдвига.
В дальнейшем мы не будем накладывать особых
ограничений на концентрации точечных дефектов, допуская некоторое
взаимодействие между ними. В соответствии с этим энергия
любого дефекта, вообще говоря, предполагается зависящей от
концентраций всех имеющихся дефектов. Однако
взаимодействие предполагается достаточно слабым для того, чтобы не
повлиять на хаотичность распределения дефектов по узлам или
междуузлиям, и это распределение считается совершенно
беспорядочным.
Колебания атомов будем описывать с помощью модели
Эйнштейна, согласно которой каждый атом имеет три степени
свободы и совершает гармонические колебания с определенной
частотой v, причем колебания всех атомов независимы.
Образование точечного дефекта изменяет силы связи соседних
атомов и, следовательно, частоту их колебаний.
При вычислении термодинамических функций дефектного
кристалла очень важное значение имеет вопрос о выборе его
компонентов. Здесь мы используем три различных подхода,
когда в качестве компонентов кристалла выбираются
относительные составляющие единицы (см. разделы 2.2 и 2.3),
структурные элементы (см. раздел 2.3) и атомы или ионы (см. раздел
2.4).
-58
2.2. Химические потенциалы
точечных дефектов
В формализме относительных составляющих единиц дефектный
кристалл представляется как раствор дефектов в идеальной
кристаллической решетке, причем и решетку, и дефекты
каждого сорта можно рассматривать как независимые компоненты
системы. Поэтому точечным дефектам каждого сорта к можно
приписать определенное значение химического потенциала:
H = dG\dNK (2.1)
равное изменению изобарного потенциала кристалла при
увеличении в нем числа дефектов данного сорта на единицу (см.
Приложение). Таким образом, для вычисления химических
потенциалов всех дефектов необходимо выразить изобарный
потенциал кристалла как функцию числа дефектов каждого сорта.
Изобарный потенциал любой системы состоит из трех
слагаемых:
G= U~TS±pV (2.2)
где U — энергия; Т — абсолютная температура; S — энтропия; р— давление;
V — объем системы.
Для конденсированных систем, в том числе твердых тел, при
обычных давлениях последним слагаемым в (2.2) можно
пренебречь и считать изобарный потенциал практически равным
изохорному потенциалу F:
G^F^U—TS (2.3)
(такое приближение равносильно также замене энтальпии #=
= U-\-pV на энергию V).
В данном разделе мы будем представлять все дефекты в
■формализме относительных составляющих единиц, однако для
простоты записи будем обозначать их символами Крегера,
общепринятыми для структурных элементов. В дальнейшем
символика Хауффе будет использоваться лишь для записи квази-
;химических реакций в тех случаях, когда нужно четко
подчеркнуть различие в формализмах относительных составляющих
единиц и структурных элементов.
Рассмотрим сначала кристалл простого вещества А,
содержащий следующие дефекты: вакансии Va^, междуузельные
атомы AiS± и примесные атомы F, образующие дефекты
замещения Fas± в узлах решетки А. Эффективные заряды дефек-
т°в ±t,e считаем произвольными.
Изобарный потенциал бездефектного кристалла в рамках
"Модели Эйнштейна записывается в виде
G° = — NL WL -f 3№Tln (toJkT) (2.4)
где AfL — число узлов решетки кристалла; WL — энергия связи атомов в
идеальном кристалле (энергия решетки), отнесенная к одному атому; k —
постоянная Больцмана; Т — абсолютная температура; h — постоянная Планка.
59
Первое слагаемое в формуле (2.4) описывает
потенциальную энергию идеального кристалла без учета энергии
колебаний; второе — изобарный потенциал системы 3NL
гармонических осцилляторов, соответствующих тепловым колебаниям Л\
атомов.
Изменение изобарного потенциала кристалла при
образовании в нем дефектов согласно формуле (2.3) обусловлено
изменением энергии AU и изменением энтропии —TAS. В
приближении слабо взаимодействующих дефектов изменение энергии
при их образовании является аддитивной функцией числа
дефектов каждого сорта:
Ш = Е mWvk + NIWaT + Л^Й (2.5)
е
где до*>-—-энергия образования дефекта; нижние индексы относятся
соответственно к дефектам Va, Ai и Fa; суммирование проводится по всем
возможным значениям эффективных зарядов дефектов.
Энтропия кристалла при образовании дефектов резко
возрастает главным образом из-за того, что дефекты могут
статистически распределяться по большому числу узлов и
междуузлий. Эта конфигурационная составляющая энтропии может
быть вычислена по формуле Больцмана [см. Приложение,
формула (П.4)], в которой термодинамическая вероятность
нахождения системы в данном состоянии, характеризующемся
данными концентрациями дефектов, равна числу всех различимых
перестановок дефектов по узлам и междуузлиям. Это дает для
конфигурационной энтропии узлов:
Д5Г* = kIn п ,+ п g+ *L* v l+ v T¥ (2.6)
и для конфигурационной энтропии междуузлий:
Д5Г* = Ш -гг-^г N''' t+ (2.7)
тде Ni — число междуузлий в кристалле, произведение П берется по всем
значениям эффективных зарядов дефектов.
Выполняя логарифмирование и пользуясь формулой
Стерлинга для факториалов больших чисел
\nN\ = NlnN— TV (2.8)
справедливой при больших N, получаем для конфигурационной
составляющей изобарного потенциала A GKOH* = —Т (SAKOH*+
_|_5iKOH*):
АОконФ _ kT[_NL ln Nl + E N*± ln Nz± + £ N$± ln N<± +
е е
+ (NL-ZN&-I>N&)ln(NL--ZN$i — ZN&) —
ее ее
-NtInЛ/i + ЕTVlf In Nit + (N{-ZNft) In (N{ - STVlf)] (2.9)
e e e
*60
Определим теперь изменение колебательной составляющей
изобарного потенциала кристалла, происходящее при
образовании дефектов. Во-первых, образование NVa вакансий
приводит к исчезновению 3NVA одномерных энштейновских
осцилляторов, так что изобарный потенциал изменяется на величину
HG^l = —3ZN$tkT\n(folkT) (2.10)
с
Во-вторых, атомы, окружающие вакансию, будут колебаться в
направлении к ней с меньшей частотой vva, чем частота
колебаний в идеальном кристалле, поскольку удаление атома
приводит к уменьшению квазиупругой постоянной сил связи
соседних атомов. Будем считать, что частота колебаний атомов в
направлении, перпендикулярном направлению к вакансии,
остается прежней, тогда изменение изобарного потенциала
составит
AG^2= EASA^nn(vVA/v) (2.11)
где Са — координационное число атома А (число ближайших соседей).
При внедрении атома в междуузлие появляется три
дополнительных осциллятора с частотой v'; кроме того, соседние
атомы изменяют частоту колебаний по направлению к междуузель-
ному атому до некоторого значения vai>v. Это приводит к
изменению изобарного потенциала на величину
ДОа1Л = SMf kT(3\n (fo'kT) + Mn(vA1/v) (2.12)
Наконец, при замене основного атома А примесным атомом
F число осцилляторов не меняется, а изменение изобарного
потенциала связано только с изменением частоты колебаний
соседних атомов:
ДОрал = $A£JV™ kT\n (vFA/v) (2.13)
Таким образом, полный изобарный потенциал дефектного
кристалла определяется суммой выражений (2.4), (2.5) и
(2.9) — (2.13). Дифференцируя эти выражения по числу
дефектов каждого сорта, «получаем для их химических потенциалов*:
^ = ^ + ^j~31n^ + ^Aln-^+ln^] (2.14)
vfi = ^AT + kTkln^- + g, In ^- + In 1^Ц (2.15)
_ \ kT " [Vjx] I
* Все энергетические характеристики атомов и дефектов, в том числе и
химические потенциалы, мы относим к одной частице. Для перехода к
мольным величинам в последующих уравнениях достаточно умножить энергии wK
й \1°к на число Авогадро, а постоянную Больцмана k заменить на газовую
постоянную R.
61
V& = w& +кт[ *А1п^А + In i^-U (2.16)
Здесь с помощью квадратных скобок обозначены доли мест
(узлов или междуузлий), занятых дефектами
соответствующего сорта:
\^] = NhlNL][\r] = NkflNl; [FF] = N&INL) (2.17a)
а также доли нормально занятых узлов и незанятых
междуузлий:
]А£] = 1 - £ [VX*] - £ [F5f]; [V,x] = 1 - Е [А^] (2.176)
Заметим, что при малых концентрациях дефектов число
узлов практически равно общему числу атомов и доли мест
дефектов, расположенных в узлах решетки, практически
совпадают с их атомными долями (отношение числа дефектов к
числу атомов). На этом основании вместо долей мест часто говорят
об атомных долях дефектов. Однако доля мест не равна
атомной доле для междуузельных атомов, если общее число
междуузлий не равно числу узлов и, следовательно, общему числу
атомов.
Доли 'мест, занимаемых дефектами, являются наиболее
удобными единицами измерения концентраций дефектов при
описании квазихимических реакций в твердых телах. Однако в
некоторых случаях, например при записи условия
электронейтральности, удобнее оперировать с числом частиц в единице
объема. Поэтому в дальнейшем мы будем использовать оба
способа выражения концентраций: символ [к] будет
обозначать концентрацию дефектов сорта к, выраженную в долях
мест; NK — их число в единице объема.
Формулы (2.14) — (2.16) выведены для кристалла простого
вещества. В кристаллах химических соединений атомы
размещаются в узлах нескольких подрешеток. При слабом
взаимодействии между дефектами каждая подрешетка дает
независимый от других вклад в изобарный потенциал и вычисление
химических потенциалов дефектов производится независимо для
каждой подрешетки совершенно аналогично описанному для
простого вещества. Поэтому химические потенциалы атомных
дефектов в кристаллах как простых веществ, так и
химических соединений определяются одними и теми же формулами
(2.14) —(2.16).
Рассмотрим теперь электронные дефекты в
неметаллических кристаллах — электроны проводимости и дырки. Как уже
отмечалось, механизм их движения по кристаллу в разных
случаях может описываться двумя приближениями.
В приближении узких энергетических зон, соответствующем
прыжковому механизму миграции, электронные дефекты рас-
62
сматриваются как локализованные частицы, причем электрон
проводимости эквивалентен точечному дефекту замещения
нормального атома отрицательным ионом АА", а дырка —
положительным ионом Ад+. В этом случае их химические потенциалы
определяются непосредственно формулой (2.16), которая
может быть переписана в виде
\ v [A><] /
(2.18)
Здесь энергии образования электронных дефектов записаны в
обозначениях зонной теории: энергии электронов в зоне
проводимости Ес я в валентной зоне Eh.
В приближении широких энергетических зон электроны
проводимости и дырки рассматриваются как квазисвободные
частицы и не являются точечными дефектами, а «размазаны» по
решетке кристалла, причем их энергии распределены по
уровням зон в широком интервале. В этом случае химический
потенциал электронов (уровня Ферми) вычисляют методами
квантовой статистики. В наиболее распространенном случае
достаточно высоких температур и малых концентраций
квазисвободных электронов и дырок (при отсутствии вырождения)
их химические потенциалы определяются выражениями
*7 = Ee + kT\n(n-\N*)
р+ = ~Eh + kT\n{n+jN*)
где п~ и п+ — числа электронов и дырок в единице объема соответственно,
параметр
TV* = 2 {2ъщ*кТ)т!/Г3 (2.20)
носит название эффективного числа состояний в зоне
проводимости или в валентной зоне, отнесенного к единице объема; т* —
эффективная масса электрона или дырки в соответствующей зоне. Роль эффективных
энергий электрона и дырки в данном случае играют граничные энергии зон—-
энергия дна зоны проводимости Ес и энергия потолка валентной зоны Ей,
взятая с обратным знаком.
Для удобства сравнения формул (2.19) с предыдущими
формулами для точечных дефектов перейдем от абсолютных
концентраций электронов и дырок пт к относительным
концентрациям
\e*\ = n*INh (2.17b)
аналогичным долям мест точечных дефектов (NL — число
узлов в единице объема). Тогда формулы (2.19) запишутся в виде
?7 = ЕС + kTln (ЛГ£/Л*) + kTln [e~] 2])
|х+ = - Eh + kTln (М. }N*) + kTln [e+]
63
Сравнение выражений (2.18) и (2.21) показывает, что оба
приближения дают для химических потенциалов электронных
дефектов качественно одинаковый результат —
логарифмическую зависимость от концентраций. Отличие состоит в том, что
в приближении широких зон (2.21) отсутствует вибрационный
член, обусловленный изменением частот колебаний соседних
атомов, что вполне естественно при нелокализованном
характере распределения зарядов электронных дефектов. Вместо него
в формулах (2.21) содержится член, определяемый
эффективной плотностью состояний yV* и отражающий волновые
свойства электронов.
Полученные выражения для химических потенциалов
дефектов можно значительно упростить, если концентрации дефектов
достаточно малы (используемое приближение слабо
взаимодействующих дефектов лучше всего оправдано именно при
малых концентрациях). В этом случае доли нормально занятых
узлов [АА] и незанятых междуузлий [Vi] можно принять
равными единице, тогда выражения для химических потенциалов
как атомных дефектов (2.14) — (2.16), так и электронных
дефектов (2.18) и (2.21) представятся одной общей формулой для
дефектов сорта /с:
^к = }:к + кТЩк\ (2.22)
где [к] — доля позиций, занятых дефектом сорта к.
Стандартные значения химических потенциалов, отвечающие
условию .-[/e]i=l, равны
iK=wK-kT\nlK(T) (2.23)
Через %к (Т) обозначены следующие величины:
для атомных дефектов
(2.24)
для точечных электронных дефектов (в приближении
широких зон)
Хе = (Ч\)А (2.25а)
для квазисвободных дефектов (в приближении широких
зон)
Xe = N*INL (2.256)
Таким образом, стандартные значения химических
потенциалов дефектов ц°к зависят только от температуры и не зависят
от концентраций дефектов. Значения параметров %к (Т) при
заданной температуре определяются в случае точечных
дефектов спектром колебаний кристалла, а в случае квазисвободных
электронов и дырок — их эффективной массой, содержащейся
64
в выражении для N*. Формулы (2.25, а, б) соответствуют двум
различным моделям электронных дефектов и являются
предельными; в реальных кристаллах %е имеет некоторые
промежуточные значения.
В формализме относительных составляющих единиц
дефектный кристалл представляется раствором дефектов в решетке
идеального кристалла, поэтому последняя является
самостоятельным компонентом системы, аналогичным растворителю в
случае жидкого раствора. Химический потенциал решетки
определится как производная от изобарного потенциала по числу
узлов l/Vl. При пренебрежении малыми членами,
соответствующими малым концентрациям дефектов, дифференцирование
дает:
^ = ^ = _ NL — kTlnxL(T) (2.26)
XL(T)=(kT/fa)3 (2.27)
Полученную формулу можно рассматривать как частный
случай формулы (2.22), соответствующей «концентрации
решетки» равной единице. Поэтому формула (2.22) в формализме
квазихимического метода описывает все компоненты дефектного
кристалла при малых концентрациях дефектов.
2.3. Закон действия масс
для квазихимических реакций
между дефектами
В предыдущем разделе химические потенциалы дефектов были
вычислены в формализме относительных составляющих единиц.
В данном разделе мы рассмотрим применимость закона
действия масс к квазихимическим реакциям с участием дефектов в
рамках обоих формализмов — как относительных составляющих
единиц, так и структурных элементов.
Относительные составляющие единицы. Результаты,
полученные в предыдущем разделе, имеют далеко идущие
последствия. Формула (2,22) есть не что иное, как выражение для
химического потенциала /с-компонента идеальной системы [см.
Приложение, формулу (П.38)]. Следовательно, при малых
концентрациях дефектов, при которых справедлива формула
.. (2.22), дефектный кристалл представляет собой идеальную
термодинамическую систему, аналогичную разбавленным
жидким растворам, и при его описании можно пользоваться
традиционным математическим аппаратом физической химии
идеальных растворов.
В частности, к любой равновесной квазихимической
реакции между дефектами, описывающейся уравнением (П.23),
Можно применять закон действия масс в форме (П.40):
П[/1РуП[/р'=/Г (2.28)
J i
5—321 65
где роль мольных долей участников реакции играют доли мест,
занимаемых дефектами, определяемые отношениями (2.17)!
Индекс i относится к исходным реагирующим дефектам, индекс
/ — к продуктам реакции; а* и (3j— соответствующие стехиомет-
рические коэффициенты. Выражение для константы равнове^
сия К можно определить, подставив соотношение (2.23) в
(П.41):
K=Il$m.taiexp[—W{T)lkT] (2.29)
J i
где введено обозначение
W(Т) = Ер.*®, - ЕaiWi (2.30)
Из последнего определения видно, что W(T) есть изменение
энергии системы при протекании в ней единицы реакции или,
короче, энергия реакции без учета энергии колебаний*
Последняя наряду с энтропией колебаний входит в множители
%к{Т), определяемые спектром колебаний дефектного
кристалла.
Энергия реакции, вообще говоря, зависит от температуры
[35, 36]. Одной из причин такой зависимости может быть,
например, тепловое расширение кристалла, приводящее к
уменьшению энергии отталкивания атомов, обусловленного
перекрыванием их электронных оболочек. В первом приближении эту
зависимость можно учесть, разложив энергию в ряд и ограни™
чившись членом, линейным <по температуре:
W(T)= W(0)+ — T (2.31)
dT
Тогда формула (2.29) для константы равновесия запишется в
виде
/Г=Х(Г)ехр(— WjkT) (2.32)
где предэкспоненциальный множитель
1(Т) = Ш*;тГехр{-±^ (2.33)
является некоторой степенной функцией температуры, а
эффективная энергия реакции определяется при 7=0 К и,
следовательно, от температуры не зависит.
Заметим теперь, что уравнение закона действия масс (2.28)
содержит только концентрации относительных составляющих
единиц — дефектов и узлов кристаллической решетки. Как
отмечалось выше, это уравнение справедливо в ограниченной
области малых концентраций, когда химические потенциалы Де"
фектов описываются выражением (2.22), типичным для
идеальных растворов. При более высоких концентрациях для
химических потенциалов дефектов следует пользоваться болев
сложными уравнениями (2.14) — (2.16). Эти уравнения наряДУ
66
нцентрациями дефектов содержат концентрации «нормаль-
° *х>> структурных элементов АА и Vi, характерных для
бездефектного кристалла. Поэтому в области, где эти концентрации
оеменны, уравнения закона действия масс должны также со-
ожать эти переменные и, следовательно, иметь более
сложный вид, нежели в приближении идеальных растворов. В этом
н учае целесообразно вообще отказаться от формализма
относительных составляющих единиц и вывести уравнение закона
действия масс в формализме структурных элементов.
Структурные элементы. Уравнение закона действия масс
Ш.40), выведенное в Приложении, получается
непосредственно из выражения (П.38) для химических потенциалов
компонентов идеального раствора. В формализме структурных
элементов такая процедура, вообще говоря, становится
неправомочной. Дело в том, что в случае кристаллов химических
соединений между концентрациями структурных элементов,
принадлежащих различным подрешеткам, существуют
определенные стехиометрические соотношения. Например, для
бинарного кристалла МХГ с дефектами Шоттки отношение полной
концентрации узлов анионной подрешетки [Xx] + [Vx] и
концентрации узлов катионной подрешетки [Mm]+[Vm]
постоянно и равно стехиометрическому коэффициенту г=гм/^х- Это
приводит к тому, что структурные элементы в различных под-
рещетках нельзя добавлять независимо друг от друга;
изменение числа структурных элементов в одной подрешетке
неизбежно приводит к изменению их числа в другой. Таким образом,
в отличие от формализма относительных составляющих единиц,
структурные элементы, вообще говоря, нельзя рассматривать
как независимые компоненты системы и, следовательно, им
нельзя приписать определенные значения химических
потенциалов [37]. Тем не менее закон действия масс легко
сформулировать в общем виде и в рамках формализма структурных
элементов, если оперировать с изменениями термодинамических
функций всего кристалла при протекании в нем единицы ква-
зихймической реакции.
Пусть взаимные превращения структурных элементов в
дефектном кристалле описываются резиновой квазихимической
реакцией (П.23)
Еа^^Ер^В, (2.34)
i ]
вето И ^7 — исх°Дные и конечные структурные элементы; аг- и Pj—-соот-
ствующие стехиометрические коэффициенты.
Представим изменение изобарного потенциала кристалла,
Рванное протеканием единицы реакции, в виде
/Ю = W - kT^ (Т) — ГА5К0НФ (2.35)
Уа Ичина W описывает изменение потенциальной энергии крис-
Ла при протекании единицы реакции; второе слагаемое со-
У 67
1
ответствует изменению изобарного потенциала осцилляторов.
Оба эти слагаемые были подробно рассмотрены выше в рамках
формализма относительных составляющих единиц; здесь они
имеют тот же самый смысл. Через структурные элементы
несколько по-иному запишется лишь третье слагаемое в правой
части (2.35), соответствующее изменению конфигурационной
энтропии кристалла при протекании в нем единицы реакции
(2.34).
Запишем конфигурационную энтропию каждой из подреше^
ток дефектного кристалла (в том числе и междуузлий) через
концентрации всех содержащихся в ней структурных элементов:
5конФ = k ln j (2.36)
Используя формулу Стирлинга, перепишем это выражение в
виде
5конФ -k{^Nl)\ni:Nl — k^NiInNt (2.37)
i i i
Отсюда получается очень простое выражение для
парциальной конфигурационной энтропии i-ro структурного элемента:
dS"°**ldNt = - k ln (Л/;/Е Nt) = —k ln [I] (2.38)
i
где [i] — доля мест, занимаемых i-ы структурным элементом в
соответствующей подрешетке.
Последнее выражение позволяет легко вычислить изменение
конфигурационной энтропии 'при протекании единицы реакции
(2.34):
д£ксшф лсконф
i l dNt ^ jVj dNj
= k f E a7 In [/] - E py ln [/]) (2.39)
Подставляя полученный результат в (2.35) и учитывая, что при
равновесии AG должно быть равно нулю, получаем после
потенцирования уравнение закона действия масс:
П [jf) П [*р = X (Т) ехр (- WjkT) (2.40)
j i
С учетом соотношения (2.32) для константы равновесия
видим, что полученное уравнение в точности совпадает с
уравнением закона действия масс (2.28), найденным ранее в рамка^
формализма относительных составляющих единиц. Однако,
несмотря на формальное тождество этих уравнений, они
имеют существенное отличие: уравнение (2.40) содержит
концентрации всех структурных элементов, участвующих в реакции -^
не только дефектных, но и «нормальных», отвечающих
идеальному кристаллу, например [Адк ] и [Vi+]. Поэтому запись за-
действия масс через структурные элементы является более
^Г^ей нежели запись через относительные составляющие еди-
ь1 'И только в предельном случае малых концентраций
деликтов, когда концентрации «нормальных» структурных эле-
нтов' можно полагать равными единице, оба уравнения
совпадают.
2 4. Химические потенциалы атомов
и ионов
Во многих физико-химических задачах бывает удобнее
пользоваться не квазихимическим, а обычным химическим методом
описания дефектных кристаллов. В этом случае оперируют с
химическими потенциалами не дефектов, а реальных
составляющих кристалла — атомов или ионов, т. е. атомы или ионы
рассматриваются как компоненты системы.
Химические потенциалы атомов или ионов в дефектном
кристалле легко получить из выражений для химических
потенциалов дефектов. Рассмотрим, например, ион
электроположительного компонента М кристалла МХГ с произвольным
целочисленным зарядом lm+ (в случае незаряженного атома £м = 0; по
классической ионной модели £м равно валентности этого
компонента Zm). По определению, химический потенциал
рассматриваемого иона равен изменению изобарного потенциала
кристалла при добавлении в него одного иона. Ион может быть
введен в кристалл двумя способами: или внедрен в междууз-
лие, или размещен в узле, заполнив существовавшую там
вакансию. При первом процессе изменение изобарного
потенциала соответствует появлению междуузельного дефекта и
равно его химическому потенциалу, при втором процессе —
исчезновению вакансии и равно ее химическому потенциалу со
знаком минус. Таким образом, химический потенциал иона М^ +
равен
р:м+ = ^м+ = _^м- (2>41)
М Mi i VM v f
Согласно условию сохранения заряда в этой формуле
эффективные заряды дефектов — междуузельного иона и вакансии по
абсолютному значению равны заряду добавляемого иона £.
Аналогичное соотношение можно записать и для
электроотрицательного компонента кристалла Х&~~х:
££+ пРимесном кристалле добавление одного примесного иона
щ I в°зможно при заполнении вакансии в соответствую-
и подрешетке Vm^m~h образовании на ее месте дефек-
замещения FMA?;± с эффективным зарядом A£=£f—£m.
69
Поэтому химический потенциал примесного иона равен
Jf+= - аСм~ i „дс± (2.43)
rF lVM lFM
На основании соотношений (2.41) — (2.43) химические
потенциалы ионов в дефектном кристалле могут быть выражены
через концентрации различных атомных дефектов с
подходящими значениями эффективных зарядов. Так, используя
формулу (2.14), выразим химические потенциалы основных
компонентов кристалла через концентрации вакансий:
^ - - «&Г + kA 31n Tr + ?м In ^ + Ш -Тр-1 (2-44)
* м
м vm I kT
Jx =_Цх+ + ^(з1п_Л;-+Нх1п-^+1п-4^-| (2.45)
х vx ( kT vvx [Vх] J
Для примесного компонента F^ с учетом (2.14) и (2.16)
получаем:
(4+ = <+ _ «&г + кт (зш ±г + ем in т. + in ■ [^±] ]
(2.46)
Этим выражениям равноценны выражения, в которых в
качестве характеристических дефектов выбраны междуузельные
ионы:
г + г + f ■ Ь' vMI [М^м+ ] 1
^ =<f+ ^(31nTF + ^ln-f+ ln-yvfTj (2-47)
ft = *xf + Аг{з1п ~ + 6, in -^ + In J^J. j (2.48)
[M = ^Cp+ + kT\3\n — + ?! ln--^ + In —Цт^} (2.49)
rF Fi [ kT v [ViXl ' V
В последней формуле [FM~ ] означает концентрацию между-
узельных примесных ионов в твердом растворе внедрения.
Формулы (2.41) — (2.46) весьма похожи друг на друга и
могут быть представлены в виде единого выражения для ионов
А ±:
tf=(tf)° + kT\n-^- (2.50)
[Va ]
70
в качестве характеристических дефектов выбраны вакан-
:ЗД^С^ gqr . электронейтральным атомам (£ = 0) соответствуют
CJiH ояженные вакансии Vax . Стандартное значение химическо-
гГпотенДиала равно
(tfy - -«& + *г(31п -j^ + SAln -JL.) (2.51)
Аналогично, формулы (2.47) — (2.49) можно представить в
вИде единого выражения:
& = (Ф° + kTln( [A^]/[Vf ]) (2.52)
Здесь в качестве характеристических дефектов выбраны меж-
дуузельные атомы или ионы А^. При малых концентрациях
междуузельных атомов или ионов доля незанятых междуузлии
fVi] в этой формуле может быть принята равной единице.
Стандартное значение химического потенциала определяется
выражением
Параметры —wVA^ и Wm^ означают работу,
совершаемую ;при 'переносе иона А^ из вакуума в узел решетки или в
междуузлие.
Формулы (2.50) и (2.52) существенно отличаются от
выражения для химических потенциалов компонентов идеальной
системы [см. Приложение, формулу (П.38)]. Поэтому при
выборе атомов или ионов в качестве компонентов дефектный
кристалл следует рассматривать как систему, сильно отличающуюся
от идеальной даже при малых концентрациях дефектов;
активности ионов согласно формуле (П.42) определяются
концентрациями дефектов и равны
а^ = [АА]/[Ус/]
(2.54)
4х = [Ада]
Глава 3
Разупорядоченность металлов
и интерметаллических соединений
м^РактеР и концентрация дефектов в твердых телах зависят от
ми°ЖеСТВа ФактоРов> связанных как с окружающими условия-
» так и с предысторией образцов, т. е. способом и условиями
факпРИгот°вления и хранения [38, 39]. Одним из важнейших
торов, определяющим дефектную структуру твердых тел,
71
является тепловой режим их обработки. При достаточно
длительной выдержке образцов при повышенной температуре
(отжиге) обычно достигается равновесие квазихимических
реакций между дефектами, так что концентрации всех возможных
дефектов принимают определенные равновесные значения,
связанные между собой законом действия масс. Эти концентрации
можно частично сохранить и при низких температурах путем
быстрого охлаждения (закалки) образцов; при этом
предполагается, что реакции между дефектами при низких
температурах замедляются настолько, что новое равновесие не
устанавливается [20].
Вычисление равновесных концентраций дефектов является
важнейшей и вместе с тем одной из наиболее простых задач
теории разупорядоченности твердых тел. Решению этой задачи
для разных типов твердых тел посвящены настоящая и две
последующие главы.
В настоящей главе будут рассмотрены атомные дефекты
кристаллической решетки простых металлов, а также
интерметаллических или других соединений металлов, имеющих частич-^
но заполненную зону проводимости и обладающих
металлическим характером электропроводности. Общей отличительной
чертой таких кристаллов является наличие большого числа
коллективизированных электронов, ведущих себя как
свободные частицы, что значительно упрощает рассмотрение реакций
между атомными дефектами.
Вообще говоря, атомным дефектам в решетках металлов,
так же как и в неметаллических кристаллах, должны
соответствовать определенные локальные уровни электронов в
запрещенной зоне, поэтому атомные дефекты в металлах могут
захватывать электроны или дырки и иметь различные
эффективные заряды. Однако коллективизированные электроны
вследствие очень высокой их концентрации сильно экранируют
заряды дефектов, так что электростатическое поле, создаваемое
каждым дефектом, практически полностью гасится на
расстояниях порядка межатомного. Таким образом, благодаря
экранирующему действию коллективизированных электронов атомньк
дефекты в металлах можно рассматривать как нейтральные и
полагать их эффективные заряды равными нулю.
3.1. Разупорядоченность чистых металлов
Рассмотрим основные квазихимические реакции, приводящие к
образованию незаряженных дефектов в решетке чистого
металла М — вакансий Умх и междуузельных атомов Mix, считая эти
реакции равновесными.
А. Образование вакансии обеспечивается выходом атома М
из узла на поверхность кристалла путем достраивания
кристаллической решетки.
72
п формализме относительных составляющих единиц (обоз-
ения Хауффе) соответствующая равновесная квазихимичес-
Йая реакция запишется в виде
0^z!:MGx + M (3.1)
«нуль» означает отсутствие дефектов; МПх —вакансия; М-—атом в но
Г<ом поверхностном узле кристалла.
Чтобы записать ту же реакцию в формализме структурных
элементов, учтем, что при переходе атомов из объема на
поверхность кристалла, атомы, находившиеся ранее на
поверхности, становятся атомами в объеме, так что число узлов в
объеме увеличивается, а на поверхности не изменяется. Поэтому в
формализме структурных элементов (обозначения Крегера)
реакция образования вакансий в кристалле простого вещества
запишется в виде
Мм ^z£ Vm + Мм , или нуль z^± Vm (3.2)
Применяя к этой реакции закон действия масс в форме (2.40),
получаем очень простое соотношение
[V&\ = Kv (3.3)
где константа равновесия равна
*V=( —1 ехР1~77 (3'4)
vvm
а энергия реакции
Ww = w$M— Wh (3.5)
складывается из двух величин: энергии шхум, затрачиваемой
при удалении атома М из узла в объеме кристалла в вакуум, и
энергии кристаллической решетки WL с обратным знаком,
выделяющейся при переносе атома из вакуума в поверхностный
Узел (энергия WL в случае простых веществ называется также
теплотой сублимации кристалла).
Соотношение (3.3) показывает, что концентрация вакансий
в чистом металле зависит только от температуры и при данной
температуре есть величина постоянная для каждого
конкретного вещества. Из вывода формулы (3.5) видно, что она остается
справедливой и для металлов, содержащих некоторую примесь,
еели вьшолняются два условия: наличие примеси не
сказывается на энергии реакции \WL и не приводит к значительному
отклонению [Ммх) от единицы. Поскольку оба условия
выполнятся при малых содержаниях примеси, можно заключить, что
р°РмУла (3.3) справедлива для разбавленных твердых раство-
Ва ддСОХРаняЮ1ВДх кристаллическую структуру чистого вещест-
73
Б. Внедрение атома М из узла на поверхности в междуузлис
в объеме кристалла, сопровождающееся уменьшением числа
узлов:
М^МОх, или Мм+УГ^ггМГ (3.6,
При малой концентрации дефектов [Ммх] [Vix]«l и закон
действия масс записывается в форме
[Mix] = /Cmi (3.7)
с константой равновесия
Km - (v/vM1)61 ехр(— WmjkT) (3.8j
Выражение для энергии реакции
rMi=^Mi+ WL (3 9)
содержит теплоту сублимации WL и энергию внедрения атома
из вакуума в междуузлие wxm-
Формулы (3.3) и (3.7) для равновесных концентраций
вакансий и междуузельных атомов аналогичны и отличаются
энергиями реакций, поэтому в кристаллах должны преобладать
те дефекты, для которых энергия реакции образования меньше.
Энергию выхода атомов на поверхность Wv,
сопровождающегося образованием вакансий, можно грубо оценить из
следующих соображений. В объеме кристалла координационное
число атомов в среднем вдвое выше, чем на поверхности.
Поэтому энергия связи объемных атомов вдвое превышает
энергию их связи на поверхности, т. е. равна удвоенной теплоте
сублимации WL. При удалении атома из узла жесткой решетки
в объеме кристалла в вакуум должна затрачиваться энергия
^xvm, равная энергии связи, поэтому энергия рассматриваемой
реакции в жесткой решетке должна быть равна Wv = 2WL—
—Wl = Wl. Однако при удалении атома из узла прежнее
положение атомов не сохраняется; атомы, окружающие вакансию,
несколько смещаются из своих узлов в направлении к вакансии,
что приводит к некоторому уменьшению энергии дефектного
кристалла. В результате такой деформации кристалла происхо-
хит компенсация некоторой доли энергии шхум, затраченной на
удаление атома из узла, и wxym оказывается меньше 2Wu %
суммарная энергия реакции Wy— меньше Wl.
Соотношение между энергией выхода атомов на
поверхность >Wy и теплотой сублимации WL хорошо иллюстрирует
табл. 3.1, из которой следует, что для металлов значения Wl
лежат в пределах 20—50% от значений теплоты сублимации и
в основном составляют несколько десятых долей электрон-
вольта.
При высоких температурах энергия тепловых колебаний И
сравнима с \Wy, поэтому концентрация вакансий, возникающие
в результате собственного разупорядочения, достигает ощути-
74
,m 3 1 Энергия образования вакансии Wv и теплота
кристаллах простых веществ (в эВ) [20]
Кристалл
А 1
Л1
Ag
Ди
Си
К
Li
Na
Pb
v\
W
С (алмаз)
С (графит)
Ge
Si
Те
wv
0,75—0,79
1,01-1,10
0,82—0,98
1,0 —1,05
0,39
0,40
0,39
0,59
1,2 —1,4
3,14
4,16
3,3
2,07—2,18
2,32
0,8
WL
3,34
2,9
3,6
3,46
0,83
1,41
1,0
2,0
5,78
8,3
^7,4
7,4
3,9
4,7
1,25—1,68
сублимации Wl
wY/wL
0,23
0,36
0,25
0,30
0,47
0,28
0,39
0,30
0,22
0,38
«0,56
0,45
0,56
0,49
0,55
мых значений. Так, вблизи температуры плавления доля
вакантных узлов в металлах составляет обычно ^10~3.
Большинство простых металлов кристаллизуется в плотно-
упакованные решетки, в которых размеры междуузлий
значительно меньше размеров атомов. Поэтому внедрение атомов в
междуузлия для них требует больших затрат энергии. Кроме
foro, энергия реакции перехода атомов в междуузлия с
поверхности кристалла Wmi содержит дополнительное слагаемое —
теплоту сублимации WL. В результате значения энергии
реакции Wmi оказываются слишком большими по сравнению с
энергией тепловых колебаний, и концентрация междуузельных
атомов в большинстве металлов ничтожно мала даже при самых
высоких температурах. Так, если в меди при 1000 DC доля
вакантных узлов составляет ^10~4, то доля междуузельных
атомов я^10~39. Поэтому собственные атомы в междуузлиях
металлических кристаллов не играют сколько-нибудь
существенной роли, и в решетках чистых металлов вакансии можно
считать единственным типом собственных атомных дефектов.
*. Формулы (3.3) —(3.5) позволяют определить химический
РРтенциал металла в состоянии слабой разупорядоченности.
Подставляя их в выражение (2.44) при £м = 0 и [Мм]«1,
получаем;
РМ = - ИГь+ЗЛЛп^
(ЗЛО)
Т £Ь
и е. выражение для химического потенциала jj°m бездефектно-
^ кристалла в модели Эйнштейна [ср. формула (2.4)]. Таким
Разом, при малых концентрациях дефектов в разупорядочен-
Вен кРисталле химический потенциал металла постоянен и ра-
его химическому потенциалу в совершенном кристалле.
75
3.2. Разупорядоченность
интерметаллических соединений
В кристаллах химических соединений атомы располагаются в
узлах двух или более подрешеток. Отношение чисел узлов
подрешеток строго фиксировано, поэтому в кристаллах
химических соединений могут возникать лишь определенные
комбинации дефектов нескольких сортов, а именно такие, при
которых отношение чисел узлов не изменяется в ходе реакции.
Рассмотрим бинарное интерметаллическое соединением АВГ
с металлическим характером энергетического спектра
электронов. В таком соединении могут существовать следующие
нейтральные атомные дефекты: междуузельные атомы обоих
сортов, вакансии и неправильно занятые узлы в обеих подрешетках.
Для сохранения баланса чисел узлов и атомов эти дефекты
должны образовывать парные комбинации. Наиболее важны
следующие три такие комбинации.
А. Дефекты Шоттки — вакансии в подрешетках обоих
компонентов, возникающие согласно реакции
нуль^±:Апх + гВПх + АВ/. (З.Па^
(в системе относительных составляющих единиц) или
нуль^±У^ + гУх (3.116)
(в системе структурных элементов). Применяя к этой реакции
закон действия масс, получаем уравнение
[Vx][VBx] = tfs+r (3.12)
где константа равновесия равна
^s+r = (v/vvA)5A(v/vVB)rlBexp(— ^) (3.13)
энергия реакции
Ws = w$A + rw$B— WL (3.14)
содержит энергию решетки WL с обратным знаком, отнесенную
к формульной единице МХГ.
Б. Дефекты Френкеля — междуузельные атомы одного из
компонентов и вакансии в подрешетке этого же компонента:
нуль^±АОх+АПх, или А2 + Vix :jz>: Aix + Va (3.15)
При малых концентрациях дефектов [АХА] »[Vix]»l, и закон
действия масс выражается уравнением
[А,х][У£] = /Г,?м (ЗЛО)
с константой равновесия
ATfa = Wv')3 (v/vAl )*• (v/vVA )'Aexp ( ~ x?F) <ЗЛ7)
76
и энергией реакции
W¥A = щ& + w^A (3.18)
Гоотношения для междуузельных атомов В и вакансий в под-
шетке В совершенно аналогичны.
^& в Антиструктурные дефекты — атомы А в подрешетке В и
атомы В в подрешетке А:
нуль ч^А#х (В) + В#х (А), или А£ + Ввх^± Ах + Вх
(3.19)
где А#Х(В) и В#Х(А) — дефекты замещения атомов В атомами А и
наоборот; Авх и ВаХ — структурные элементы, соответствующие этим дефектам.
При малых концентрациях дефектов [АХА] ^[Вхв]^ 1 и
закон действия масс выражается уравнением
[k£][B%] = Ki (3.20)
с константой равновесия
Wa_\ (3.21)
kT
Kl-(4\AfA(^ABfB™p{-
и энергией реакции
Wa=w$b + w$a (3.22)
В приведенных здесь формулах энергии реакций,
обозначенные W, относятся к разному числу возникающих дефектов,
если гф\. Для дальнейшего удобно вести энергии разупорядо-
чения, отнесенные к одному точечному дефекту:
ws=Wsj(\+r); wF=WFj2; wA=WJ2 (3.23)
тогда после извлечения корней из выражений (3.13), (3.17) и
(3.21) константы равновесия трех рассмотренных реакций
запишутся в единой форме
/C=X(v)exp(— w\kT) (3.24)
где множители %(v) определяются спектром колебаний и в
рассматриваемом приближении не зависят от температуры.
Кроме описанных трех основных комбинаций дефектов в
интерметаллических соединениях иногда встречается комбинация,
состоящая из вакансий в одной подрешетке и антиструктурных
Дефектов в другой. Примером может служить NiAl, в котором
такие дефекты возникают согласно реакции [40]:
нУль:^ш#х(А1) + 2№Пх + №А1, или Ni^^tNi^+2Vi?i
(3.25)
Теоретически возможны еще два типа разупорядоченности:
Томы обоих сортов в междуузлиях и комбинация из между-
77
узельных атомов одного из компонентов и антиструктурных
дефектов в его подрешетке. Однако вследствие высокой энергии
внедрения в междуузлия эти виды разупорядоченности не
наблюдались ни в одном из известных соединений.
Полученные здесь уравнения закона действия масс
существенно отличаются от выражений, приведенных в разделе (3.1)
для кристалла простого вещества. Если в простых веществах
концентрации дефектов при фиксированной температуре
постоянны, то в бинарных соединениях постоянно произведение
концентраций дефектов обоих сортов. Эти соотношения аналогичны
уравнению для «произведения растворимости», связывающего
концентрации противоположно заряженных ионов в
насыщенных водных растворах слаборастворимых солей; согласно этим
соотношениям увеличение концентрации одних дефектов ведет
к уменьшению концентрации других.
В соединениях, имеющих строго стехиометрический состав
АВГ (г — целое число или простая дробь), концентрации
атомных дефектов связаны простыми соотношениями, вытекающими
непосредственно из уравнения реакции их образования.
А. В случае разупорядоченности Шоттки из уравнения
реакции (3.11) вытекает, что на одну вакансию Vха приходится
г вакансий Vх в, т. е.
N$B = rN^A
Подставляя в это равенство очевидные соотношения
A^A = [V£bVLA; A/& = [V£]M.B; NL = rNLA
{где NLA и NLb — объемные концентрации узлов в подрешетках
А и В), получаем для равновесных долей вакантных узлов в
стехиометрическом кристалле:
[V*] = [VBX]
Подставляя последнее равенство в уравнение закона
действия масс (3.12) и извлекая из него корень степени Г+г,
находим:
[VX] = [V?]=/Ts (3.26а)
Отсюда вытекает, что константа Шоттки Ks имеет простой
смысл: это есть равновесная концентрация вакансий в
кристалле стехиометрического соединения, разупорядоченного, по
Шоттки, аналогично тому, как Kv есть равновесная концентрация
вакансий в кристалле простого вещества (см. раздел 3.1).
Для кристаллов стехиометрических соединений, имеющих
френкелевскую или антиструктурную разупорядоченность,
равновесные концентрации дефектов определяются аналогично.
Б. В случае разупорядоченности , Френкеля объемные
концентрации дефектов равны
Nva — Nm или NxB-Nti
78
Выражая объемные концентрации через- доли мест с
помощью соотношений
^vxa = [V^]^la; WU = [A,x]7Vi; Л#в = [VBXJ М,в;
^ш = [В1х]ЛА,
(где Ni — объемная концентрация междуузлий), представим эти
равенства в виде
[v£] = £[An; r[vBx] = ^[Bix]
где g — число междуузлий, приходящихся на одну формульную единицу АВГ«
Подставляя полученные соотношения в уравнение закона
действия масс (3.16) и извлекая из него квадратный корень,
находим равновесные относительные концентрации дефектов
Френкеля в стехиометрическом кристалле:
[vX] = ff[A,x]=/7^FA (326б)
№] = (Цг)[В?]=-/ТГгКп
В случае антиструктурной разупорядоченности
Лва = Л£в или [ВХ] = г[А£]
Подставляя последнее равенство в (3.20) и извлекая
квадратный корень, получаем для равновесных концентраций:
[В%]=г[А$]=уг7КА (3.26в)
Формулы (3.26а) и (3.266) показывают, что константы Kfa>
Kfb и Ка с точностью до постоянных множителей порядка
единицы равны равновесным концентрациям френкелевских или
антиструктурных дефектов в стехиометрическом кристалле.
Антиструктурная разупорядоченность твердых тел относится
к числу наиболее распространенных типов. Она типична для
соединений, в которых размеры и электроотрицательности
составляющих их атомов близки; это в особенности относится к
интерметаллическим соединениям, таким, как CuZn, CuPt,
CuPd, AgMg, AuZn, AuCu, AuCu3, PtCu3, MnNi3, FeAl, Fe3Al,
ReNi3, FeCo и jr. д. [41, 42]. Более того, в таких соединениях
при повышенных температурах разупорядочение становится
настолько сильным, что атомы разных сортов статистически
распределяются по узлам решетки, а кристалл приобретает черты
неупорядоченного сплава. Этот вопрос будет подробно
рассмотрен ниже, в разделе 3.4.
3.3. Концентрации дефектов в нестехиометрических
интерметаллических соединениях
В кристаллах химических соединений, состав которых
отличается от стехиометрического, равенства (3.26) нарушаются,
поскольку избыточное содержание одного из компонентов реали-
79
зуется за счет преобладания одного типа атомных дефектов,
Так, при разупорядоченности Шоттки избытку компонента А
соответствует преобладание вакансий в подрешетке В и
наоборот. Поэтому концентрации дефектов в нестехиометрических
кристаллах являются функциями состава.
Вычислим равновесные концентрации нейтральных атомных
дефектов в нестехиометрическом кристалле АВ14_6 в
зависимости от степени отклонения от стехиометрического состава б.
Здесь мы полагаем для простоты, что число узлов в обеих под-
решетках одинаково и стехиометрический коэффициент г равен
единице. В таком кристалле концентрации атомов связаны
соотношением
[В]-(1+8) [А] (3.27)
Рассмотрим отдельно три основных типа атомной
разупорядоченности.
А. Дефекты Шоттки. Концентрации атомов связаны с
концентрациями вакансий соотношениями [А] = 1—[VXA]; [B] = l—
—(Vxb]. Подставляя эти равенства в (3.27), находим:
[Vg]=[VX]-8 + 8[VX]
При малых концентрациях дефектов последним слагаемым в
этом уравнении можно пренебречь, и оно запишется в форме
[VX] = [V£] + 8 (3.28)
Полученное уравнение следует решать совместно с уравнением
закона действия масс для дефектов Шоттки (3.12), которое
при /1= 1 имеет вид
[V^][VBX]-/Cs2 (3.29)
Решение системы уравнений (3.28) и (3.29) приводит к
формулам
[VX1 - (8/2) [1 ± К1 + №/8)2] (3 30)
tV^] = (8/2)[-l±TAl + (2/Cs/8)2]
Знак перед корнем выбирается так, чтобы концентрации
вакансий были положительными. При этом условии положительным
значениям б соответствует плюс, отрицательным — минус.
Рассмотрим два предельных случая.
I. Малые отклонения от стехиометрического состава,
|'6|<2/Cs. В этом случае в формулах (3.30) можно пренебречь
единицами по сравнению с большим отношением 2/Cs/6 и
решения принимают упрощенный вид
[V£]«[Vg]«ffs (З.ЗП
II. Большие отклонения от стехиометрического состава,
|6|>2/CS. В этом случае значение корня в (3.30) можно раз-
80
г"—' ^~
^ч
\р* fl
^v В
L Al V
^^S^.
>^2i^
/
№f
У
у
/ X ^
У ь1^
J^T^^ I I 1
НО -А
о
8 б/К
РИС. 3.1.
Зависимость концентрации нейтральных атомных дефектов в нестехиометри-
ческом бинарном соединении ABj+6 от степени отклонения от стехиометриче-
ского состава б.
ложить в ряд по малому параметру 2/Cs/|6|. Ограничиваясь
членом ряда, линейным по этому параметру, получаем:
(3.32)
яри 8>0 [V£]^8; [VBx]^/Cs2/8
при8<0 [VU«tff/|»|; JV§]« |8|
Точные зависимости концентраций вакансий от б,
определяемые формулами (3.30), изображены на рис. 3.1.
Полученные решения для концентраций вакансий позволяют
вычислить химические потенциалы компонентов
рассматриваемого бинарного соединения АВ14-б в зависимости от степени
нестехиометричности. Искомые формулы получаются путем
подстановки приближенных решений (3.31) и (3.32) в формулу
(2.43) для химических потенциалов атомов в зависимости от
концентраций вакансий, которая при £м'=£х = 0 и [АА]~[Вв]~
^ 1 имеет вид
После
РА = Ра - АЛп [VX1
ЦВ=|4 —A74n[V£]
(3.33а)
(3.336)
е подстановки концентраций вакансий получаем для сте-
хиометрического состава 16
= 0:
отклонениях от стехиометрического состава,
Pa=(0) = jx1 — kTlnKs] v-B(0) = VB — kTlnKs (3.34)
При
больших
(3.35)
<0 VA^VA + kTi— 21n/Cs + ln | 8 | ); \*.в~\ъ-кТ1п\Ъ\
1 81
h\ м-м'(о)
X/ кт
1 \ !5
г-*-^ \\
1 -5 -6 ~ty-2jO}
^^ Г0,5
\ /'У
I s ~f>5
^/* -м
2/
/
//
J >■
V\\/ ! I
\
/Г4
1
0/К\
РИС. 3.2,
Зависимость химических потенциалов
компонентов бинарного соединения
№i+6 от степени отклонения от сте-
хиометрического состава б:
1 и 2 — соответственно А и В в случае
разупорядоченности Шоттки или Френкеля;
3 и 4 — соответственно А и В в случае
антиструктурной разупорядоченности.
Полученные результаты удобно представить в виде
разностей:
8> О 1М - ра(0) = - kТЫ [b/Ks )
Рв — Рв(0)=:кТ\п(ЪК$) (3.36)
8<0 pa—Ра(0) = ЛПЩ8//Г8|
№-М0) = —ЛГ1п \b/Ks\
описывающих отклонения химических потенциалов
кристалла от их значений при стехиометрическом составе, которые
рассматриваются как стандартные. Зависимости (3.36)
изображены на рис. 3.2 пунктирными линиями, касающимися кривых
/ и 2; сплошные линии 1 и 2 изображают точную зависимость
химических потенциалов Л и Б от б, определяемую
формулами (3.33).
Б. Дефекты Френкеля. Предположим для простоты, что
число междуузлий равно числу узлов каждой из подрешеток.
Тогда уравнения, определяющие полные концентрации атомов в
кристалле, запишутся в виде [А] = 1—[Vxa]+[Aix]; [В]=1, ее-
ли разупорядочен компонент А, и [А]=1; [В] = 1 — [Vxb] +
+ [Bjx], если разупорядочен компонент В. Подставляя эти
соотношения в (3.27) и полагая приближенно 1/(1+6) = 1—б,
получаем вместе с законом действия масс системы уравнений:
[Vfl - [Aix] + 8
[A,x][VU = tf?A
(3.37)
при разупорядоченности А и
[b.xi;
[Vg] + §
[В,Х][У£]=/С™
(3.38)
82
при разупорядоченности В.
Эти системы совершенно аналогичны системе уравнений
(3.28) и (3.29), поэтому их решения можно получить из
решения (3.30) заменой [Vмв] на [Aix] и [VхА] на [Bix], а
константы Шоттки К$ — соответственно на Kfa и Kfb. На графике
рис. 3.1 кривые для междуузельных атомов совпадают с
кривыми для вакансий при разупорядоченности Шоттки.
I. -Б области малых отклонений от стехиометрического
состава, |6|<c2/Cf, решения аппроксимируются формулами
[V*] *[**]«** (339)
[Bix] « [V£] « Kfb
II. При больших отклонениях от стехиометрического
состава, |6|^>2/Cf, приближенное решение имеет вид
8>0 [V2]«8; [A,xJ«K?a/8 (ЗЩ
S<0 [Vft«/&/|8| ; [А,х]^ |8|
при разупорядоченности компонента А и вид
8>о [вГ]-8; [vg]«/c|B/8 (341)
8<0 |В,х]«/Г|в/|8| ; [Vb]- I 8 |
при разупорядоченности В.
При френкелевской разупорядоченности компонента А его
химический потенциал определяется аналогично описанному
для кристалла с дефектами Шоттки с помощью формулы
(3.33а), поскольку концентрация вакансий Vax известна из
решений (3.39) и (3.40). Однако в этом случае концентрация
вакансий в почти упорядоченной предрешетке В требует
дополнительного определения. Хотя в этом случае доминирующими
являются дефекты Френкеля, образование вакансий Vbx все
равно протекает по механизму Шоттки. Поэтому в кристалле,
находящемся при равновесных условиях, концентрации
вакансий в обеих подрешетках по-прежнему связаны между собой
уравнением закона действия масс (3.29) для реакции Шоттки.
Поэтому на основании формулы (3.336) для химического
потенциала компонента В получаем:
Ив = р°в+ kT(—21n Ks + In [Va\ (3.42)
Подставляя в формулу (3.33а) и (3.42) значения [Vax] из
приближенных решений (3.39) и (3.40), находим для
химических потенциалов компонентов при стехиометрическом составе,
|*|-0:
Ра(0) = l^A— kTlnK?A /343ч
^в (0) = цв + kT (In Kfa - 21n Ks )
6*
83
и при больших отклонениях от стехиометрического состава,
16|»2KFA:
8>0 на~на — kTln*
Ив « Р-в + А?Г(— 21n /Ts + In 8) 44)
8<0 ^а«^ + АГ(-21п/Гра+1п |8 | )
^B~^B + ^r(~21n/Cs+21n^FA — In | 8 | )
При френкелевской разупорядоченности компонента В
решение проводится аналогично; в этом случае
На = На + kT{— 21n /Cs + In [Vх] ) (3.45)
и подстановка [VBX] из решений (3.39) и (3.41) в формулы
(3.45) и (3.336) дает для стехиометрического состава:
На (0) - на + k T (- 21n Ks + In /Tfb) (3 46)
Ив (0) = Ив — кТ1пКгв
и для больших отклонений от стехиометрического состава,
|6|>2/Cfb:
8>0 иа^^ + ^^(~21пЛгз + 21п/Срв —1п8)
Ив ~ Ив + kT(— 21п/Сгв + in 8)
8<0 Ha«^a + ^(—21n/Ts + ln | 8 ' )
Нв ~ Нв — kTln | 8 [
(3.47)
Вычитание формул (3.43) из (3.44) и формул (3.46) из
(3.47) дает для отклонений химических потенциалов
компонентов от их значений при стехиометрическом составе \х—]л(0)
выражения, совершенно аналогичные (3.36) и отличающиеся от
них только тем, что вместо константы Шоттки они содержат
константы доминирующей разупорядоченности Kfa или Kfb.
Графически зависимости химических потенциалов от б при
обоих типах разупорядоченности Френкеля изображаются теми
же кривыми / и 2, что и при разупорядоченности Шоттки (см.
рис. 3.2).
В. Антиструктурные дефекты. В этом случае уравнения для
полных концентраций атомов записываются в виде [А] =
= [Аах] + [Авх]; [В]=[Вв*|+[ВАх]/Гак как концентрации
нормально занятых узлов равны [Аах] = 1—[Вах]; [Ввх]=1—(АвхЬ
это дает [А]=1—[ВАХ] +[ABXJ; {B]=l—[Авх]+
[ВАХ].Подставляя полученные соотношения в (3.27) и пренебрегая малым
слагаемым б([Авх]—[ВАХ]), находим:
[ВХ]=[А£] + 8/2 (3.48)
84
Это уравнение вместе с законом действия масс (3.20) образует
систему, аналогичную системе (3.28) — (3.29) для дефектов
Шоттки и получающуюся из последней при замене [Vax], [VbxJ
и Ks соответственно на [ВАХ], [Авх] и КА, а б на 6/2.
I. При малых отклонениях от стехиометрического состава,
6<С4/СА, решение аппроксимируется формулой
[Ва1^[А£]^/<а (3.49)
П. При больших отклонениях от стехиометрического состава,
|6|^>4/Са, приближенные решения имеют вид
&>0 [В*] «8/2; [Авх]-2/ф
8<0 [В£]«2*а/|8| ; [Ag] « | 8| /2
Кривые для зависимости концентраций антиструктурных
дефектов от б идут более полого, чем для дефектов Шоттки или
Френкеля; наклон асимптот, изображенных пунктирными
прямыми на рис. 3.1, для антиструктурных дефектов вдвое меньше,
чем для дефектов Шоттки и Френкеля. Такое различие
обусловлено следующим. В случае дефектов Френкеля и Шоттки все
избыточное содержание одного из компонентов реализуется
через дефекты, так что при больших отклонениях от
стехиометрии концентрации доминирующих атомных дефектов
приблизительно равны |б|. В случае же антиструктурных дефектов
половина избыточного компонента размещается в узлах
собственной подрешетки и только оставшаяся половина расходуется на
образование дефектов. Поэтому при больших |б| концентрации
доминирующих дефектов приблизительно равны |б/2|.
Для вычисления химических потенциалов компонентов
кристалла с антиструктурной разупорядоченностью необходимо
определить равновесные концентрации вакансий, не
являющихся доминирующими дефектами. Для этого запишем реакции,
при которых наряду с антиструктурными дефектами
возникают вакансии:
Aa^A£+2V£
В£ ^zh Ъ1 + 2VBX
Уравнения закона действия масс для них при малых
концентрациях дефектов имеют вид
[ASJ [Vх]2 = СА
[B2][V£]a=Cg
(3.51)
(3.52)
или
ln[VX]zz:^lnCA-^ln[Ag]
(3.53)
ln[VBx]=-~lnCB-^ln[B^]
85
Подставляя полученные равенства в формулы (3.33), получаем
для химических потенциалов:
РА = ^ + (-у-) (_31п СА + In [Ag])
о fkT\ « (3*54)
N = N^ l~\ (-31n CB + In [Bfl)
откуда после подстановки концентраций антиструктурных
дефектов из решений (3.49) и (3.50) следует для стехиометричес-
кого состава (6 = 0):
МО) = ^ +(-у-) (-31п Са+ЫКа)
)ьт\ (3-55)
^в (0) = |хв + Ш- ) (_31n CB + In Ka)
и для больших отклонений от стехиометрического состава,
|6|>4#А:
8 > 0 |1А = ^ + (£772) (—31п С а + 21п /Га — In 3/2)
рв = jxb + (А7-/2) (—31п Св + In 8/2) 56)
8<0 ца => pl + (ЛГ/2) (-31п СА + In | 8/2 | )
Рв = ^в + (^П2)(^31пСв + 21п/Га-1п | 3/2 | )
Если выбрать в качестве стандартного состояния стехиомет-
рический состав, то при больших значениях |б| химические
потенциалы компонентов определяются как разности выражений
(3.56) и (3.55):
8 > 0 на - ра (0) = - (£772) In (8/2/Га)
|1В-Рв(0) = (ЛГ/2)1п(8/2/Са) (3 57)
3 < 0 iM - Ра (0) = (£772) In (8/2/Са )
Рв - Рв (0) = — (kTj2) In (8/2/Га )
Эти зависимости изображены на рис. 3.2 пунктирными
кривыми, прилегающими к кривым 3 и 4\ сплошные кривые 3 и 4
изображают точную зависимость химических потенциалов
соответственно А и В от б. Эти кривые идут значительно более
полого, чем кривые 1 и 2 для кристаллов с дефектами Шоттки
или Френкеля, поэтому те же отклонения химических
потенциалов от стандартных значений достигаются при более высоких
концентрациях антиструктурных дефектов, нежели дефектов
Шоттки и Френкеля.
86
3.4. Упорядочение твердых растворов
замещения
Изложенная выше теория описывает строение чистых металлов
и интерметаллических соединений при малой степени разупо-
рядоченности, когда отклонения от упорядоченной структуры
можно представлять как точечные дефекты. Очевидно, что
такое представление весьма ограничено. Оно явно не применимо
к твердым растворам с широкой областью гомогенности, в
которых концентрации обоих компонентов могут изменяться в
пределах до десятков атомных процентов. Кроме того, сплавы,
состав которых близок к стехиометрическому и которые при
низких температурах рассматриваются как интерметаллические
соединения, при достаточно высоких температурах ведут себя
как твердые растворы. Это связано с тем, что при высоких
температурах атомы различных сортов статистически
распределяются по узлам решетки. При этом понятие точечных
дефектов теряет смысл, и для таких сильно разупорядоченных систем
необходимо специальное описание [41—43].
В настоящем разделе за исходное состояние сплава
принимается состояние полной разупорядоченности; в
противоположность изложенному в предыдущих разделах, рассматриваются
процессы, приводящие к упорядочению такого сплава. Для
определенности мы будем рассматривать бинарный сплав А—В,
представляющий собой твердый раствор замещения, в котором
атомы А и В могут замещать друг друга в узлах одной и той
же подрешетки.
Дальний порядок. Для многих сплавов, разупорядоченных
при высоких температурах, при охлаждении на
рентгенограммах обнаруживаются сверхструктурные линии, что однозначно
указывает на изменение симметрии их кристаллической
решетки. При этом существует некоторая критическая температура,
ниже которой такие линии имеются и выше которой они
полностью отсутствуют. Исследования термодинамических
функций показывают, что при критической температуре сплав
испытывает фазовый переход II рода, характеризующийся плавным
изменением теплосодержания и пиком на кривой температурной
зависимости теплоемкости. Оба эти эффекта связаны с
переходом сплава из неупорядоченного состояния в упорядоченное; в
последнем атомы каждого сорта располагаются
преимущественно в узлах определенной подрешетки, в результате чего
симметрия кристалла понижается. При этом порядок определяется
распределением атомов по всем, в том числе и удаленным,
узлам в кристалле и поэтому называется дальним
порядком.
Рассмотрим твердый раствор замещения А—В с
произвольными концентрациями компонентов. Для простоты
ограничимся случаем, когда число узлов в обеих подрешетках А и В
одинаково и равно NL, причем узлы первой подрешетки окружены
87
только узлами второй подрешетки и наоборот. Этим условиям
удовлетворяют (3-латунь Си—Zn и родственные ей сплавы,
кристаллизующиеся в объемноцентрированную кубическую
структуру типа CsCl (координационное число g = 8), а также сплавы
со структурой типа NaCl (g = 6) и сплавы, структура которых
представлена плоскими квадратными сетками (g = 4).
Заметим, что при больших концентрациях компонентов
сплава, особенно при высокой степени его разупорядоченности,
понятие дефекта в известной мере утрачивает смысл. Поэтому
формализм относительных составляющих единиц в этом случае
неприменим, и сильно разупорядоченный сплав следует
описывать с помощью формализма структурных элементов. Тогда
основная квазихимическая реакция обмена атомами между
обеими подрешетками запишется в виде
А£ + Ввх:^2:А£ + В£ (3.58)
Применение к этой реакции закона действия масс дает
уравнение
wnwa -№ti-m (3.59)
[А*] [В*] V kT
Здесь, как и в формализме относительных составляющих
единиц, квадратные скобки означают долю мест в
соответствующей подрешетке, т. е. отношение числа структурных элементов
к числу узлов подрешетки. Через AU обозначено изменение
энергии сплава при протекании единицы реакции (3.58). При
высоких концентрациях компонентов сплава AU зависит от его
состава, поэтому экспонента в уравнении (3.59) не является
константой.
Для дальнейшего необходимо ввести некоторый параметр,
который характеризовал бы степень упорядоченности сплава.
Заметим прежде всего что в случае больших концентраций
компонентов твердого раствора замещения при описании их
распределения можно пренебречь относительно малой
концентрацией вакансий и полагать
[АЯ] + [ВЯ = [А£] + [Вё] = 1 (3.60)
Далее, при отсутствии вакансий в неупорядоченном сплаве
доли узлов, занятых атомами А и В, равны их атомным долям:
с = Na =Na> с = Nb =Nb (361)
А NA+NB 27VL ' B NA+NB 2NL
Поэтому в качестве характеристики степени упорядоченности
сплава целесообразно выбрать какой-либо параметр,
содержащий разность между истинной долей узлов в подрешетке,
занятых определенными атомами, и соответствующей атомной до-
88
лей. Выберем в качестве такой характеристики величину <ц:
Ч = 2([А£]-сА) = 2([В£]-св) (3.62)
Она называется степенью дальнего порядка.
Учитывая соотношения
[Аах] + [Авх]--^-=2,а
(3.63)
непосредственно вытекающие из (3.61), и равенство (3.60),
легко выразить концентрации структурных элементов через
атомные доли и степень дальнего порядка:
[АХ] = сА+ч/2; [Ag]=cA-i)/2
[В$] = св-ф, [Ввх] = св + ^/2
(3.64)
Согласно определению (3.62) для неупорядоченных сплавов
любого состава гг| = 0. При наибольшем упорядочении, когда
[ААХ] максимально, т] достигает своего максимального
значения. В случае стехиометрического сплава АВ (са=1/2)
максимальное значение г] достигается при полном упорядочении
([ААХ]=1); из формулы (3. 62) следует, что это значение
равно г)макс = 1. В сплавах, состав которых отличается от
стехиометрического, нельзя получить полное упорядочение,
отвечающее идеальной периодичности кристаллической решетки.
Поэтому в нестехиометрических сплавах т]Макс, соответствующее
наибольшему упорядочению, всегда меньше единицы.
Действительно, в сплавах, в которых cA^V2, при наибольшем
упорядочении все атомы А располагаются в узлах А и [Авх]=0; из
(3.64) следует, что в этом случае
Чмакс = 2^а (ПРИ са <х/г) (3.65а)
В противоположном случае, при СА^7г, при наибольшем
упорядочении все узлы А заведомо заняты атомами А, т. е.
[ААХ] = 1, и из (3.62) следует:
W = 2 (1 - сА) = 2св (при сА > i/a) (3.656)
Зависимости (3.65) изображены графически на рис. 3.3.
Вычислим теперь энергию частично упорядоченного сплава.
Поскольку кулоновское взаимодействие атомов в металлах
экранировано электронной жидкостью, взаимодействием
удаленных атомов можно пренебречь и ограничиться учетом
взаимодействия с ближайшими соседями. В таком приближении
энергия сплава А—В запишется в виде
U = "АА^АА + "ВВ^ВВ + nABU AB С3'66)
89
РИС. 3.3.
Зависимость максимальной степени
дальнего порядка от концентрации в бинарном
сплаве с одинаковыми числами узлов в
обеих подрешетках.
где tiij — число пар атомов (i, /), находящихся на кратчайшем расстоянии
друг от друга (число связей, иц — энергия парного взаимодействия атомов.
Если координационное число первой координационной сферы
равно g, общее число атомов, являющихся ближайшими
соседями атомов А, составит INA. Вычитая из этого значения число
пар А—В пав, получим число атомов А, являющихся
ближайшими соседями атомов А. При этом каждый атом А учитывается
дважды, поэтому для нахождения числа связей А—А
указанную разность нужно разделить на 2:
nAA^±(WA — пАВ) (3.67а)
Аналогично
2
пт = ^-(Шв~пАВ) (3.676)
Подставляя соотношения (3.67) в (3.66), находим:
U*=±[l (NA uAA + NB uBB) + nABQ) (3.68)
Величина Q — энергия смешения, равная
Q = 2uAB-uAA — uBB (3.69)
Физический смысл этой величины легко установить путем
следующих соображений. Заменим в чистом металле А один атом
атомом В, а в чистом металле В — атомом А. При замене
атома А на В изменение энергии составит ^(иАв—Uaa), при
замене атома В на А — 1(иАв—ивв). Работа суммарного процесса
равна
*(2иАВ-иАА-иАВ):=5(г
Таким образом, энергия смешения представляет собой
работу обмена местами атомов А и В в чистых металлах, деленную
на координационное число.
Энергия смешения играет большую роль в теории
упорядочения твердых растворов. Из формулы (3.68) видно, что для
сплавов, образование которых сопровождается выделением
тепла (Q<0), энергетически выгодно увеличение числа
разноименных пар пАв, т. е. возникновение упорядоченной структуры
90
с чередованием атомов разных сортов. Такие сплавы
называются упорядочивающимися. Напротив, для сплавов,
образование которых сопровождается поглощением тепла (Q>0),
энергетически выгодно уменьшение яАв, а следовательно,
увеличение Паа и 'Явв, т. е. образование таких областей, в которых
атомы окружены преимущественно атомами того же сорта.
Такие сплавы называются распадающимися. При
достаточно низких температурах такие сплавы распадаются на части,
состоящие из чистых компонентов или из твердых растворов, по
составу более близких к чистым компонентам, нежели
исходный твердый раствор. По этой причине энергию смещения Q
иногда называют энергией упорядочения, если она
отрицательна, или энергией распада, если она положительна.
Для вычисления энергии реакции (3.58) определим
изменение числа связей между разноименными атомами пАв.
Очевидно, что среднее число атомов В, являющихся
ближайшими соседями узла А, равно |[Ввх]. Поэтому и при
протекании единицы реакции (3.58) происходит уменьшение пАв на
£[Ввх] за счет исчезновения структурного элемента ААХ и
аналогично на £[ААХ] за счет исчезновения [Ввх]. При
образовании новых структурных элементов Авх и Вах величина пАв
увеличивается на |([Вах]+[АВХ]).
Таким образом, общее изменение числа разноименных пар
при протекании реакции (3.58) составляет
Ляав = £ ([Ag] + [ВХ] - [АХ] - [Вх] ) (3.70)
или при учете соотношений (3.64)
Дгс = — 2fy (3.71)
Подставляя последнее равенство в (3.68), получаем для энергии
реакции (3.58):
Д£/= —5tjQ (3.72)
При учете (3.64) и (3.72) уравнение закона действия масс
(3.59) превращается в трансцендентное уравнение
относительно степени дальнего порядка:
(2сА+У1)(2св + У1) V\kT 7 1
Найдем критическую температуру Т=Ткр, при которой ц
обращается в нуль. Для этого разложим экспоненту в ряд по
малому параметру lQr)/kT, ограничившись двумя членами ряда.
Выполняя затем элементарные преобразования с точностью до
членов, линейных по х\, находим:
KI кр
91
0,2 0,4 0,6 TlTKp(0,5)
п
1,0
0,8
0,6
Ofi
0,2
-
—
-
L
—v.».
\ *
\*
M, I I J
3 0,2 0,4 0,6 0,6 TjTKp
РИС. 3.4.
Температурная зависимость степени дальнего порядка в бинарном сплаве с
одинаковыми числами узлов обеих подрешеток. Числа на кривых указывают
атомные доли компонентов [42].
РИС. 3.5.
Температурная зависимость степени дальнего порядка в Р-латуни [42].
Отсюда для критической температуры получаем значение
TKV = -cAcBtQ\k (3J4)
Знак (—) здесь является математическим выражением
сформулированного выше правила, что упорядочение сплавов
возможно лишь при отрицательных значениях энергии смешения
(Гкр>0 при Q<0).
Формула (3.74) показывает, что степень дальнего порядка
обращается в нуль при конечной температуре, выше которой
дальний порядок отсутствует и аг) = 0. Как уже указывалось в
начале данной главы, исчезновение дальнего порядка при 7кр
является фазовым переходом II рода, поэтому 7КР часто
называют тем пер ату р ой перехода.
С помощью (3.74) параметр gQ можно выразить через Гкр
и произведение сАсв. Подставляя полученное выражение в
уравнение (3.73), запишем его в виде
(2сА^)(2св-7])
(2cA+rl)(2cB-\-rl)
ехр
'кр
сАсвТ
(3.75)
Это уравнение легко решить численно относительно г\ в
зависимости от относительной температуры Т/Ткр при
фиксированных значениях атомных долей сА и св.
Рассчитанные таким образом кривые изображены на рис. 3.4;
температура отнесена к значению температуры перехода сте-
хиометрического сплава (сА=св=1/2). Как видно из
приведенных графиков, вблизи критической температуры кривые идут
довольно круто, и обращение в нуль при 7КР этими графиками
иллюстрируется очень наглядно.
На рис. 3.5 изображена теоретическая кривая для р-латуни
стехиометрического состава CuZn, рассчитанная по уравнению
92
(3.75) при значении Гкр = 742 К, в сравнении с
экспериментальными точками. Несмотря на некоторое количественное
расхождение, расчетная кривая качественно правильно описывает
экспериментальные данные. Это говорит о том, что
квазихимический метод в ряде случаев может успешно применяться для
описания не только твердых тел с точечными дефектами, но и
сильно разупорядоченных систем.
Ближний порядок. Как следует из изложенного выше, при
температурах выше критической дальний порядок в сплаве
исчезает, так что распределение атомов по подрешеткам
становится равномерным. Твердые растворы, для которых ц = 0,
обычно называют неупорядоченными. Это, однако, еще
не означает, что атомы в неупорядоченном твердом растворе
распределены совершенно хаотически. Для полного описания
структуры твердого раствора наряду со степенью дальнего
порядка необходимо еще знать, насколько полно атомы данного
•сорта окружены (в среднем по кристаллу) атомами
какого-либо определенного сорта. В этом случае порядок определяется
характером окружения атома его ближайшими соседями и
потому называется ближним порядком. Установление
ближнего порядка приводит к тому, что размещение атома
определенного сорта в каком-либо узле влияет на вероятность
размещения атомов в соседних узлах. В результате даже в
неупорядоченном сплаве при отсутствии в нем дальнего порядка
распределение атомов отличается от хаотического и характеризуется
подчас сильно выраженным ближним порядком. В этом
отношении можно провести аналогию между твердыми растворами, с
одной стороны, и растворами электролитов или ионными
расплавами, с другой.
Как известно, отличительной чертой любой жидкости
является полное отсутствие дальнего порядка. В то же время в
концентрированных растворах электролитов, а тем более в ионных
расплавах положительные ионы окружены в первой
координационной сфере преимущественно отрицательными ионами и
наоборот, поэтому ближний порядок в них выражен достаточно
ярко.
Здесь мы будем для простоты считать все узлы
эквивалентными. Этому условию удовлетворяют рассмотренные выше
бинарные твердые растворы при отсутствии в них дальнего
порядка.
Характер и степень ближнего порядка можно полностью
описать, если определить число связей пАВ между
разноименными атомами, когда они находятся на кратчайшем расстоянии
друг от друга. Тогда числа связей между одноименными
атомами пАА и пАв будут однозначно определяться с помощью
формул (3.67).
Как уже отмечалось выше, в неупорядоченном твердом
растворе средняя вероятность занятия узлов атомами данного
сорта равна их атомной доле ci. Поэтому при хаотическом рас-
93
пределении атомов в твердом растворе вероятность обнаружить
в соседних узлах два атома А равна /?аа=£2а. В случае
частичного ближнего упорядочения, обусловленного взаимодействием
ближайших соседей, это выражение следует умножить на боль-
цмановский фактор, учитывающий энергию взаимодействия
пары:
Рак = gc\ exp (— UAAJkT) (3.76а)
где g — некоторый нормировочный множитель.
Аналогично
Рав =Рва = Sca св ехР (—иАв1кТ) (3-766)
Рвв = gel exp (—um\kT) (3.76в)
Примем, что вероятность того или иного способа размещения
пары в соседних узлах пропорциональна числу
соответствующих пар:
пав Рав + Рва . яав Рав + Рва
^АА РАА пВВ РвВ
(3.77)
Перемножая левые и правые части двух последних уравнений,
получаем:
_^£_ = 4exp(-Q/£7) (3.78)
ЛАА ПВВ
где Q — энергия смешения, определяемая формулой (3.69).
Уравнение (3.78) часто называют условием
квазихимического равновесия. Такое название обусловлено тем, что это
уравнение формально совпадает с выражением закона действия
масс для квазихимической реакции разрыва связей А—А и
В—В и образования вместо них двух связей А—В:
(А- А) + (В — В):^±2(А —В) (3.79)
Будем характеризовать ближний порядок в сплаве параметром
x = nABlWL (3.80)
Так как общее число связей между ближайшими атомами в
рассматриваемом кристалле равно £A/l, параметр х есть
относительная доля связей между разноименными атомами.
С помощью соотношений (3.61) и (3.67) числа различных
пар атомов выразятся через этот параметр и атомные доли
следующим образом:
пАВ = WLx; пАА = WL (сА — х]2)\ пш - Wl (св — х\2)
(3.81)
и уравнение квазихимического равновесия (3.78) превращается
в уравнение относительно одной неизвестной величины х:
(2Сд-,П2св-,)-еХР(-^Г) С3'82)
Рассмотрим сначала наиболее простой случай, когда сплав
имеет стехиометрический состав АВ (сА=св=1/2). В этом
случае уравнение (3.82) упрощается:
exp(-Qj2kT) (3.83)
1-х
и его решение имеет вид
х = [1 + exp (QI2kT)]"1 (3.84)
Подставляя полученное выражение в (3.81), находим числа
различных пар атомов:
»аа - "вв = Щг [1 + exp (-Q/2UT) J-1
Если Q = 0 или 7=оо, то согласно формулам (3.85) пАв =
= lNL/2; иаа = явв==§#1У4, что отвечает хаотическому
распределению атомов. Если Q отрицательно, то при понижении
температуры пАв стремится к предельному значению gJVL, равному
полному числу связей, а пАа и пВв уменьшаются в пределе до
нуля. Это означает предельно высокую степень ближнего
порядка, когда все атомы имеют в ближайшем окружении
исключительно атомы другого сорта. Напротив, при положительном
Q и достаточно низких температурах пАв мало, а пАА и пВв
имеют значения, близкие к половине числа всех связей |М/2.
Это означает, что атомы каждого сорта окружены такими же
атомами; в этом случае сплав распадается на части, состоящие
из почти чистых компонентов А и В.
Для характеристики ближнего порядка в стехиометрических
сплавах АВ удобно пользоваться специальным параметром,
равным разности между долей разноименных пар и долей
одноименных:
WL
(3.86)
Он может быть назван степенью ближнего порядка.
Подставляя сюда значения чисел пар из (3.85), получаем очень
простую формулу
a = th(™Q/4£7) (3.87)
При Q = 0 во всем интервале температур а = 0, что отвечает
хаотическому распределению атомов. При Q¥=0 график темпера-
95
РИС, 3.6.
Температурная зависимость степени
ближнего порядка о* в стехиометрических
сплавах АВ:
/ — упорядочивающиеся сплавы; 2 —
распадающиеся сплавы.
турной зависимости <т изображен на рис. 3.6. Он состоит из
двух кривых: кривая / соответствует упорядочивающимся
сплавам (Q<0), кривая 2 — распадающимся сплавам (Q>0). При
Г=0 степень ближнего порядка достигает предельных значений,
равных +1 для упорядочивающихся сплавов и —1 для
распадающихся. При повышении температуры а асимптотически
приближается к оси абсцисс.
Для нестехиометрических сплавов решение уравнения (3.82)
более громоздко, однако основные закономерности легко
проследить в предельных случаях. Перепишем уравнение (3.82) в
более компактном виде
{2сАх~1 - 1) (2свх-г—1) = s (3.88)
где использовано обозначение
£ = ехр(<2/£Г) (3.89)
Уравнение (3.88)—квадратное относительно неизвестной г-1,
поэтому оно имеет точное решение:
х = 4сА св [l ± /l+4cAcB(t-i)]~l (3.90)
Это решение упрощается в следующих предельных случаях.
I. Малая энергия смешения (высокие температуры): |Q|<C
<^kT. В этом случае экспонента е приблизительно равна 1 и из
(3.90) следует хж2сАсв- При этом из двух возможных знаков
перед радикалом в (3.90) мы выбрали ('+), так как в
противном случае х обращается в бесконечность при Q = 0, что
противоречит физическим условиям. С помощью формул (3.81) теперь
легко определить числа различных пар ближайших атомов:
*ab = 2Wl'a<V. nAA = WL(?A; »bb = Wi/b (3-91)
Как и следовало ожидать, полученные значения отвечают
беспорядочному распределению атомов.
II. Большая отрицательная энергия смешения (низкие
температуры): —Q^>kT. В этом случае приближенные решения
вытекают непосредственно из уравнения (3.82). Действительно, при
большом отрицательном значении QJkT экспоненту в (3.82)
б
1,0
0,5
0
0,5
1,0
-\7 J
| Т-— 1 1 1
-2—J—4Ж
96
можно положить равной бесконечности, а это отвечает двум
корням квадратного уравнения (3.82): х(1) = 2сА и х^ = 2св. Для
числа разноименных связей получаем, таким образом, два
приближенных решения:
Первое решение имеет смысл при Na<Nb. Действительно, в
этом случае при высокой степени ближнего порядка все
атомы А окружены атомами В, т. е. все имеющиеся %NA связей
атомов А с окружающими соседями являются разноименными.
Напротив, при Na>Nb все атомы В окружены атомами А, и
следует использовать второе решение. Таким образом, в случае
больших отрицательных значений энергии смешения
приближенное решение имеет вид
»ab«Wa (NA<NB)
«ab«UVb (^а>^в)
III. Большая положительная энергия смешения (низкие
температуры) : Q>&7\ В этом случае экспонента е в решении (3.90)
велика по сравнению с единицей, и оно может быть
представлено в упрощенном виде:
х = 2 /сАсвехр (—Qj2kT) « 1 (3.93)
При понижении температуры параметр х, а следовательно, и
пАв монотонно уменьшаются; напротив, Паа и ^вв
асимптотически приближаются к предельным значениям,
соответствующим полным числам связей атомов А и В:
лаа«^а/2; *BB«eWB/2 (3.94)
В этом случае почти все атомы А окружены атомами А и
почти все В окружены атомами В, что соответствует распаду
твердого раствора на почти чистые компоненты.
Резюмируя изложенное, можно заключить, что ближний
порядок в твердых растворах отсутствует только при энергии
смешения, равной нулю, или при бесконечно высоких
температурах. При Q^O степень ближнего порядка имеет конечное
значение при любой конечной температуре. В этом отношении
ближний порядок существенно отличается от дальнего порядка,
который полностью исчезает при температуре перехода.
3.5. Фазы внедрения
В противоположность твердым растворам замещения,
образуемым атомами с близкими значениями радиусов, твердые
растворы внедрения состоят из атомов, радиусы которых сильно
различаются: г/гм<0,59. Фазами внедрения называют
7—321
97
упорядоченные твердые растворы неметаллов в переходных
металлах, в которых атомы неметалла занимают междуузель-
ные позиции в решетке металла-растворителя. К фазам
внедрения относят нестехиометрические гидриды (М—Н), нитриды
(М—N), карбиды (М—С) и некоторые низшие оксиды
(например, TiO, NbO) переходных металлов [44, 45]. Напомним, что
переходными называются металлы, в изолированных
атомах которых при наличии s-электронов в электронной оболочке
с главным квантовым числом п предпоследняя (п=1)-оболочка
не заполнена до конца: отсутствует часть ее ^-электронов, а у
элементов второго и третьего больших периодов отсутствует
также и часть f-электронов.
Отличительной чертой переходных металлов в
конденсированном состоянии является наличие объединенной s—d-зоны
с большим числом электронов связи, что приводит к большим
значениям энергий связи, температур плавления и модулей
упругости этих металлов. Благодаря указанной особенности
электронного строения переходных металлов фазы внедрения, как
правило, обладают металлическими свойствами; их
электропроводность близка к электропроводности металлов, которая, как
и в металлах, уменьшается с температурой. Хотя строгая
теория электронного строения фаз внедрения до сих пор не
разработана, принято считать, что атомы металлоидов отдают часть
своих валентных электронов в 5—d-зону проводимости
металлов, причем концентрация электронов проводимости зависит от
состава фазы.
Переходные металлы кристаллизуются в три плотноупако-
ванные структуры: гранецентрированную кубическую (ГЦК),
компактную гексагональную (КГ) и объемноцентрированную
кубическую (ОЦК). В этих структурах имеется по два типа
междуузлий, в которых могут размещаться внедренные атомы
металлоидов: позиции с октаэдрической координацией
(координационное число £ = 6) и с тетраэдрической (g = 4).
В ГЦК и КГ решетках число октаэдрических междуузлий
равно числу атомов металла, а число тетраэдрических — вдвое
больше. При этом размеры междуузельных пустот таковы, что
в октаэдрические междуузлия могут внедриться без
деформации решетки атомы, радиус которых гх не превышает 0,41 от
радиуса атомов металла гм. Для менее просторных
тетраэдрических позиций это отношение не должно превышать 0,225.
В случае более крупных атомов металлоида их внедрение
приводит к увеличению межатомных расстояний.
В ОЦК решетке на один атом приходится 3 октаэдрических
междуузлия и 6 тетраэдрических. При этом для них
наблюдается противоположная картина: в тетраэдрические позиции
могут внедряться без деформации решетки более крупные
атомы (гх/гм^0,291), чем в октаэдрические (гх/гм^0,154).
Реальное распределение внедренных атомов по различным
позициям определяется двумя факторами: размерами пустот и
т
их координационным числом, поскольку с увеличением
последнего энергия притяжения между атомами разных сортов
возрастает по абсолютному значению. В ГЦК и КГ решетках оба
фактора приводят к предпочтительному размещению
внедренных атомов в октаэдрических позициях. В ОЦК же решетке
первый фактор отдает предпочтение тетра-позициям, в то
время как второй — окта-позициям, поэтому в структуре ОЦК
встречается размещение атомов в обоих типах междуузлий.
Характерной чертой фаз внедрения являются очень широкие
области гомогенности, зачастую достигающие десятков атомных
процентов. Поэтому понятие точечных дефектов для таких
систем, вообще говоря, лишено смысла. Так, в системе Pd—Н
отношение числа атомов водорода к числу атомов палладия
может непрерывно изменяться от нуля до значения, равного
единице и соответствующего стехиометрическому составу PdH,
причем во всей области составов структура металлической подре-
шетки отвечает ГЦК структуре чистого палладия. При стехио-
метрическом составе PdH все октаэдрические позиции заняты
водородом и взаимное расположение атомов Pd и Н отвечает
чередованию ионов в структуре NaCl. Подобное строение
характерно и для всех остальных фаз внедрения, однако для их
подавляющего большинства имеют место либо ограниченные
области гомогенности, разделенные на диаграмме состояний
двухфазными областями, либо полиморфные превращения ка-
тионной подрешетки, происходящие при увеличении
концентрации металлоида (так называемый концентрационный
полиморфизм). Эти превращения чаще всего протекают в
последовательности ОЦК—ИКГ—НГЦК.
Описанные особенности строения фаз внедрения зачастую
делают неоднозначным выбор базисного вещества — твердого
раствора внедрения. Действительно, при малых концентрациях
внедренных атомов базисным веществом целесообразно
называть чистый металл, а металлоид рассматривать как примесь,
размещенную в междуузлиях. Типичным примером такой
системы является аустенит — твердый раствор углерода в 7~железе>
в котором атомы углерода занимают окта-позиции ГЦК
подрешетки железа. При описании таких систем можно с успехом
пользоваться аппаратом статистической термодинамики (гл. 2),
оперирующим с междуузельными атомами, в частности,
определять химический потенциал металлоида по (2.45):
рх=?°+кТ1п^ (3.95)
[Vix]
где доля незанятых междуузлий [Vi] близка к единице.
Напротив, при высоких концентрациях металлоида, близких
к предельно допустимым, совокупность междуузельных позиций
можно рассматривать как самостоятельную подрешетку,
принадлежащую металлоиду, в которой основная часть позиций
занята, а незанятые позиции обладают свойствами точечных
дефектов — вакансий. В качестве примеров таких систем можно
привести ГЦК фазу Zr—С, в которой отношение углерода к
цирконию меняется от 0,56 до значений, близких к единице, а
также гексагональную фазу Nb—<N с отношением азота к
ниобию, изменяющимся в интервале от 0,695 до 1. Очевидно, что
для подобных систем целесообразно выбирать в качестве
базисного вещества эквиатомное бинарное соединение MX при строго
стехиометрическом составе, а при отклонениях от последнего
представлять фазу внедрения как нестехиометрическое
соединение MXi-б. Химический потенциал металлоида в таких
системах будет выражен через концентрацию вакансий согласно
формуле (2.43):
рх = рх + АПп^ (3,96)
При достаточно высоких температурах фазы внедрения
представляют собой неупорядоченные твердые растворы, в
которых внедренные атомы хаотически распределены по
разрешенным позициям. Однако следует помнить, что в таких
твердых растворах, как и в растворах замещения, может
существовать заметный ближний порядок в расположении занятых и
свободных междуузельных позиций. При понижении
температуры до некоторого критического значения занятые и свободные
междуузельные позиции упорядочиваются, что приводит к
возникновению подчас сложных сверхструктур, обнаруживающих
себя дополнительными линиями на рентгенограммах. Причиной
упорядочения является, прежде всего, отталкивание
электронных оболочек атомов металлоида, препятствующее их
размещению в соседних позициях. В целом явления упорядочения в
фазах внедрения вполне аналогичны описанным в предыдущем
разделе для твердых растворов замещения, поэтому нет
необходимости рассматривать их более подробно. Заметим только,
что, как и в растворах замещения, в данном случае степень
дальнего порядка может быть равна единице только для сте-
хиометрических составов, а для нестехиометрических систем
она всегда меньше единицы.
В некоторых системах область гомогенности фаз внедрения
может простираться до значения отношения неметалла к
металлу, большего единицы, что соответствует сверхстехиометричес-
ким составам MXi+б. В таких фазах сверхстехиометрическое
содержание металлоида может реализоваться либо путем
образования равного количества вакансий в катионной подрешет-
ке (например, в TiOi+б, Zri+б), либо путем дополнительного
размещения атомов металлоида в виде парных гантелей Х2 в
основных позициях (например, UCi+e), либо размещения их в
нормально не занятых позициях, которые играют роль междууз-
лий (например, путем занятия окта-позиций в фазах YH2+ б>
100
PuH2+6, в которых атомы водорода обычно занимают тетра-по-
зиции).
В первом случае разупорядоченность охватывает
металлическую подрешетку, в которой при фиксированном составе
содержится постоянное число вакансий. В остальных случаях ка-
тионная подрешетка в высокой степени упорядочена. Однако
это не означает идеального порядка, поскольку, как и в
кристаллах простых веществ или интерметаллических соединений,
в катионной подрешетке фаз внедрения неизбежно
существование термически активированных вакансий. Их концентрация
подчиняется тем же законам статистической термодинамики,
что и для рассмотренных выше веществ. Так, для субстехиомет-
рической фазы MXi-e произведение концентраций вакансий в
металлической и металлоидной подрешетках удовлетворяет
уравнению закона действия масс для реакции Шоттки (3.12):
[VU][V£] = /Ti (3.97)
Поскольку при фиксированном значении б [Vxx] = const,
концентрация металлических вакансий экспоненциально зависит
от температуры:
[V&]~exp(— WsjkT) (3.98)
Наряду с катионными вакансиями фазы внедрения могут
содержать также точечные антиструктурные дефекты,
аналогичные рассмотренным в разделе 3.3 для нестехиометрических
интерметаллических соединений, однако их роль для фаз
внедрения, в общем, незначительна.
Глава 4
Разупорядоченность полупроводников
В настоящей главе рассматриваются кристаллы
полупроводников или изоляторов — простых веществ или химических
соединений с неметаллическим характером энергетического спектра
электронов, в основном состоянии которых валентная зона
полностью занята, а зона проводимости пуста. Будем
рассматривать главным образом кристаллы с преобладающей ковалент-
ной связью, в которых атомные дефекты в их основном
состоянии электронейтральны [46—48].
Образование нейтральных атомных дефектов в
неметаллических кристаллах, в том числе и в полупроводниках,
происходит благодаря тем же квазихимическим реакциям, что и в
металлах или интерметаллических соединениях. Поэтому все
соотношения, выведенные в предыдущей главе для дефектов в
металлических кристаллах, в полной мере применимы к незаря-
101
женным атомным дефектам в полупроводниках [49]. В
настоящей главе основное внимание уделяется вычислению
равновесных концентраций электронных дефектов — электронов
проводимости и дырок.
Как и в предыдущей главе, здесь теория строится на
использовании квазихимического метода, в частности, уравнений
закона действия масс. Между тем уже отмечалось (см. гл. 2,
раздел 2.2), что формулы (2.19), описывающие химические
потенциалы электронных дефектов в приближении широких зон и
являющиеся теоретической основой для использования закона
действия масс, справедливы лишь в области достаточно
высоких температур и малых концентраций электронных дефектов,
когда можно пренебрегать их взаимодействием. В противном
случае благодаря запрету Паули возникает квантовомеханичес-
кое вырождение электронов и правильные результаты могут
быть получены только в рамках квантовой статистики Ферми—
Дирака [50—53].
В настоящей и в следующей главах мы будем
ограничиваться указанной областью высоких температур и достаточно
малых концентраций, с тем чтобы получить наиболее простые
соотношения. Поэтому следует иметь в виду, что все изложенное
в настоящей главе относится к невырожденным
полупроводникам.
4.1. Собственные полупроводники
Как было описано в гл. 1 (см. раздел 1.6), в собственных
полупроводниках доминирующей реакцией разупорядочения
является собственная ионизация, в результате которой
образуются электроны проводимости и дырки:
нуль :?г± £~ + <?+ (4.1)
Применение закона действия масс в форме (2.28) дает
уравнение, определяющее произведение концентраций электронных
дефектов
[е-][е+) = К? (4.2)
с константой равновесия
/С,2 = ХГ К ехр (- ^^ ) (4.3)
где энергия реакции Ес — Eh равна ширине запрещенной зоны; множител!
%е были определены в гл. 2 [см. формулы (2.25)] в рамках двух моделей;
перескакивающих электронов, соответствующих приближению узких
энергетических зон, и квазисвободных электронов, соответствующих приближению
широких зон.
В полупроводниках с преобладающей ковалентной связью,
рассматриваемых в настоящей главе, наиболее точные
результаты дает приближение широких зон, в котором электроны про-
102
водимости и дырки описываются как квазисвободные частицы;
в этом случае множители %е определяются отношением
ге = n*jnl
Величина
А* = 2 (2тс/гг*£Г)3/2/Г3 (4.4)
вычисляемая в рамках квантовой статистической теории,
имеет смысл эффективной плотности состояний в зоне
проводимости или в валентной зоне и определяется значением
эффективной массы электрона или дырки в соответствующей зоне (h —
постоянная Планка). Величина Л/l, как и в предыдущих главах,
обозначает плотность узлов решетки. В таком приближении
формула (4.3) для константы собственной ионизации
принимает вид
^ = yV*/7VLexp(-^=^) (4.5)
Уравнение (4.2) аналогично уравнениям (3.12), (3.16) и
(3.20), полученным в предыдущей главе для реакции
собственного атомного разупорядочения, и является основным
соотношением, определяющим концентрации электронных дефектов в
собственных полупроводниках.
Если собственный полупроводник не содержит других
заряженных дефектов, кроме электронов проводимости и дырок,
то их концентрации связаны между собой условием
электронейтральности кристалла [е~] = [е+]. Подстановка его в уравнение
(4.2) дает для относительных концентраций электронов и
дырок:
[е-] = \е+\ = Кх (4.6)
или при учете (4.5) для абсолютных концентраций (в единице
объема):
«- = п+ = Л*ехр(--^=^) (4.7)
Из соотношения (4.6) вытекает физический смысл
константы собственной ионизации Кс это есть равновесная
концентрация электронных дефектов в собственном полупроводнике,
аналогично тому как константа Шоттки есть равновесная
концентрация дефектов Шоттки в кристалле с атомной разупорядочен-
ностью. Формула (4.7) показывает, что концентрация
электронных дефектов экспоненциально растет с температурой
аналогично концентрациям собственных атомных дефектов,
рассмотренных в предыдущей главе.
Электропроводность собственного полупроводника
аддитивно слагается из вкладов электронов проводимости и дырок.
Поскольку для каждого типа носителей их вклад в электропровод-
103
ность связан с их концентрацией известным соотношением (см.
гл. 6, раздел 6.1)
°к=\Як\ иЛ (4.8)
[где qK — заряд носителей; ик — их подвижность (скорость
движения в электрическом поле с напряженностью, равной
единице)], из формулы (4.7) следует, что электропроводность
собственного полупроводника быстро растет при увеличении
температуры:
асо6 = eN* (гГ + и+) ехр (- Щ^) (4.9)
4.2. Примесные полупроводники
Как было описано в разделе 1.6, в примесных
полупроводниках доминирующими реакциями, приводящими к
возникновению квазисвободных электронов и дырок, являются реакции
ионизации примесных доноров
Dxt^D+ + ^ (4.10а)
и примесных акцепторов
Ax^irA~ + £+ (4.106)
Уравнения закона действия масс в применении к этим
реакциям записывается в виде
[D+][<n=rD[Dx] (4Л1а
[А~][е+]=:ГА[АХ] (4.116)
с константами равновесия
TD = (N*/NL)exp (-AED;kT) (4.12a)
ГА = (N*/NL) exp {-AEk\kT) (4.126)
и энергиями ионизации доноров и акцепторов AED и АЕА.
Рассмотрим для определенности полупроводник,
содержащий примесные донорные центры D. Будем считать, что донор-
ные уровни лежат значительно выше середины запрещенной
зоны, так что энергия ионизации донорных центров AED
значительно меньше половины ширины запрещенной зоны 1/2(Ес—
Eh), как это действительно имеет место в случае германия
(см. гл. 1, раздел 1.6). Пусть некоторая часть доноров [Dx]
находится в нейтральной форме, а другая часть—в ионизованной;
суммарная концентрация доноров
[D] = [DX] + [D+] (4.13)
предполагается фиксированной и известной. Концентрации
заряженных дефектов в таком полупроводнике связаны условием
электронейтральности
[e~] = [D+] + [e+] (4.14)
104
Уравнения (4.11а), (4.13) и (4.14) совместно с уравнением
собственной ионизации
[е-][е+] = К? (4.15)
образуют полную систему относительно неизвестных
концентраций [е~~\ [е4-], [D+] и [D*]. Однако в общем случае ее
решение не выражается через аналитические функции, поэтому мы
найдем ее приближенные решения путем следующих
упрощений.
Заметим прежде всего, что из уравнений (4.11а) и (4.13)
после исключения из них [Dx] вытекает соотношение
&1= Г° (4.16)
показывающее, что доля ионизованных доноров монотонно
растет при уменьшении концентрации электронов проводимости и
приблизительно равна единице при [e~]<crD. Это позволяет
упростить систему уравнений в области сравнительно малых
концентраций донорной примеси.
Действительно, очевидно, что в предельном случае исчезаю-
ще малых концентраций примеси [D]—Я), искомое решение
системы уравнений должно переходить в найденное в
предыдущем разделе решение (4.6) для чистого кристалла, согласно
которому концентрации электронов проводимости и дырок равны
константе собственной ионизации Ки С другой стороны,
сравнение формул (4.5) и (4.12) показывает, что прИ(Д£,в<1/2 (Ес—
Ен) константа ионизации доноров Td значительно больше
константы собственной ионизации Ки Следовательно, для
рассматриваемого полупроводника должна существовать некоторая
область сравнительно небольших концентраций доноров примеси,
в которой концентрация электронов проводимости [е~]
сравнима по величине с константой К\ и в то же время значительно
меньше Гб. Поэтому для указанной области в знаменателе
уравнения (4.16) можно пренебречь слагаемым [е~] по
сравнению с TD, после чего это уравнение принимает вид [D+]«[D],
показывающий, что практически все доноры находятся в
ионизованном состоянии.
Подставляя найденное значение [D+], а также значение
концентрации дырок [e+] = Ki2/[e~], вытекающее из (4.12), в
уравнение (4.14), получаем квадратное уравнение относительно
концентрации электронов проводимости:
[e-]*=[D)[e-] + Ki (4-17)
Его решение имеет вид
[е~\ = y [D] {1 + уТ+ТЩоЩ (4-18)
Полученное решение допускает дальнейшие упрощения в
двух следующих предельных случаях.
105
I. [,D]^C2/Ci. При таком соотношении обеими единицами в
фигурных скобках (4.18) можно пренебречь по сравнению с
большим значением дроби и для концентрации электронных
носителей тока получаем приближенное соотношение
[е-]«[е+]^/С, (4.19)
отвечающее рассмотренной в предыдущем разделе собственной
разупорядоченности полупроводника. Из приведенного выше
неравенства видно, что решение (4.19) и, следовательно,
собственная разупорядоченность реализуются не только в чистых
полупроводниках, для которых [D]«0, но и в примесных
полупроводниках при достаточно высоких температурах, при которых
константа собственной разупорядоченности К[ достигает
значений, высоких по сравнению с концентрацией примеси.
Физически это означает, что при высоких температурах энергия
тепловых флуктуации возрастает настолько, что процесс
собственной ионизации становится преобладающим по сравнению с
процессом ионизации примесных доноров.
П. В противоположном случае, при [D]>>2/Ci, в формуле
(4.18) под радикалом можно пренебречь малой дробью
(2/Ci/[D])2 по сравнению с единицей и решение принимает
упрощенную форму
[<T]«[D+]«[D] (4.20)
показывающую, что практически все электроны проводимости
обязаны своим существованием донорным центрам. Из условия
существования этого решения [D]^$>2Ki видно, что оно
реализуется при достаточно высоких концентрациях доноров или при
достаточно низких температурах, при которых энергия
тепловых флуктуации настолько мала, что процесс собственной
ионизации становится незаметным на фоне процесса ионизации
примесей.
Проведенный анализ показывает, что любой полупроводник,
содержащий фиксированную концентрацию примеси, может
иметь в зависимости от температуры как примесную, так и
собственную разупорядоченность. Переход от примесной
разупорядоченности к собственной происходит при некоторой
критической температуре Гкр, отвечающей условию [D] =2/G(TKp).
Подставляя сюда значение К\ из формулы (4.5), находим:
т — Ec — Eh С4 2П
'КР— 2k\n(N*jND) к '
Из этого соотношения следует, что с увеличением концентрации
примеси логарифм в знаменателе уменьшается и значение
критической температуры растет. Поэтому в сильно легированных
полупроводниках переход к собственной проводимости может
быть достигнут только при достаточно высоких температурах.
До сих пор мы предполагали, что концентрация электронов
106
проводимости остается малой по сравнению с константой
ионизации доноров Гб. Из решения (4.20) видно, что это условие
выполняется при ограниченных концентрациях примеси: [D]<C
<СГб. Поэтому при больших концентрациях примеси, сравнимых
с rD и тем более превосходящих ее, полученное выше решение
(4.18) утрачивает силу, и для этой области концентраций
необходимо использовать иное приближение.
Уравнение закона действия масс для собственной ионизации
(4.15) показывает, что с ростом концентрации электронов
проводимости концентрация дырок [е+] монотонно уменьшается.
Уже в области концентрации примеси, при которых
реализуется упрощенное решение (4.20), концентрация дырок, равная
/C2i/[D], уменьшается до значений, гораздо меньших константы
К\ и тем более малых по сравнению с концентрациями
доминирующих дефектов [£~]~[D+]. Поэтому при нахождении
решения системы уравнений в области высоких концентраций
примеси можно пренебречь концентрацией дырок в условии
электронейтральности (4.14), которое при учете соотношения (4.16)
для [D+] принимает вид квадратного уравнения
[08 + rD[*-] = rD[D] (4.22)
Решение последнего определяется выражением
[е-\ = (Г„/2) {-1 + jA+(4[D]/rD)} (4.23)
Асимптотическое поведение полученной зависимости [е~] от
[D] определяется соотношениями между [D] и Td.
В области умеренных концентраций примеси и достаточно
высоких температур, при которых 4 [D]<Xd, значение
квадратного корня в формуле (4.23) можно разложить в ряд по
малому параметру 4 [DJ/Гб, ограничившись двумя членами ряда.
После этого решение (4.23) принимает упрощенную форму
le~] ~[D+]^[D], совпадающую с найденным выше решением
(4.20), отвечающим предельному случаю И. Поэтому нужно
проанализировать противоположный предельный случай.
III. 4 [D]>rD (очень высокие концентрации примеси или
низкие температуры). В этом случае единицами в фигурных
скобках (4.22) можно пренебречь и решение принимает вид для
относительных концентраций заряженных дефектов:
[<r-]^[D+]«([D]rD)2 (4.24)
или, при учете выражения (4.12а) для Гб, для абсолютных
концентраций:
пГ^М&ж (N*ND )2 exp (~AEDl2kT) (4.25)
Из решения (4.24) видно, что вследствие принятого
неравенства Td<C:4[D] в области III (рис. 4.1) концентрация элект-
107
РИС. 4.1.
Зависимость концентрации электронов проводимости и дырок от содержания
донорной и акцепторной примесей:
I — область собственной разупорядоченности; II и III — области примесной разупорядо-
ченности при истощении доноров или акцепторов (II) и при резерве доноров или
акцепторов (III).
ронов проводимости, а следовательно, и приблизительно равная
ей концентрация ионизованных доноров мала по сравнению с
суммарной концентрацией доноров: [D+]<C[D]. Это означает,
что основная часть доноров находится в нейтральной форме,
т. е. в полупроводнике имеется значительный резерв неиони-
зованных доноров. При фиксированной концентрации
примеси такая ситуация реализуется в области достаточно низких
температур; с повышением температуры, в соответствии с
формулой (4.25), доля ионизованных доноров растет, а резерв не-
ионизованных донорных центров постепенно истощается.
Поэтому область II, в которой реализуется решение (4.20) с
преобладающей долей ионизованных центров, называют областью и с-
тощения доноров.
Полученные зависимости концентрации электронов
проводимости и обратной ей концентрации дырок от концентрации
доноров схематически изображены в правой части рис. 4.1;
римские цифры указывают номер приближенного решения,
соответствующего данному участку кривой.
Рассчитанные в данной главе концентрации электронов
проводимости в зависимости от температуры, как и другие
экспоненциальные функции обратной температуры, удобно
изображать в координатах Аррениуса: логарифм
концентрации— обратная температура 1/7. Преимущества таких
координат становятся очевидными, если прологарифмировать
формулу (4.7) для собственной разупорядоченности и (4.25)—для
примесной:
Щ п7оь = In TV* - ^^ jr (4.26)
In Яп-рим zz: — (In N* + In ND) - -^ -y (4.27)
Если пренебречь сравнительно слабой (степенной)
зависимостью плотности состояний N* от температуры, то соотношения
108
РИС. 4.2. 5
Температурная зависимость концентрации электронов rS
проводимости или дырок в примесном
полупроводнике.
(4.26) и (4.27) являются уравнениями
прямых линий в координатах \пп~—1/7,
причем тангенс угла наклона этих прямых
равен половине энергии соответственно
собственной или примесной ионизации, деленной
на постоянную Больцмана. Полученные
зависимости изображены схематически на 11ТкР ~ Iff
рис. 4.2.
Мы вычислили равновесные концентрации электронов
проводимости и дырок в полупроводнике, содержащем примесные
доноры. Для полупроводника, содержащего акцепторную
примесь, задача решается совершенно аналогично: вместо
уравнений (4.13) и (4.14) для него следует записать
[А] = [АХ] + [А"] (4.28)
[е+] = [А"] + [е~] (4.29)
и решать эти уравнения совместно с (4.116) и (4.15). Решения
системы можно непосредственно получить из приведенных выше
для случая донорной примеси путем замены [е~] и [D+]
соответственно на [е+] и [А""]. Графики для концентрации дырок и
электронов проводимости для этого случая приведены в левой
части рис. 4.1. Температурная зависимость концентрации дырок
в этом случае аналогична рассмотренной выше для электронов
проводимости (см. рис. 4.2).
Решения, полученные в данном разделе, имеют чрезвычайно
важное значение для физикохимии полупроводников. Они
показывают, что в области примесной разупорядоченности
концентрации электронов проводимости или дырок, а следовательно,
величина и характер электропроводности полупроводников
могут регулироваться содержанием донорной или акцепторной
примеси. Легирование полупроводников необходимым
количеством подходящих примесей позволяет создавать нужную
концентрацию носителей тока и поэтому является одним из
важнейших путей получения материалов с заданными
физико-химическими свойствами.
Это хорошо иллюстрирует рис. 4.3, на котором показана
температурная зависимость экспериментальных значений
концентрации квазисвободных электронов в монокристаллических
образцах германия n-типа, легированных мышьяком.
Концентрация мышьяка в этих образцах изменяется в широких
пределах: от 8-Ю12 атомов на 1 см3 (кривая 1) до ^1018 (кривая6).
В то же время его атомная доля во всех образцах остается
очень малой: так как концентрация атомов германия составля-
109
300 WO 50 30
П IK
0,02 0f04 0,06 0fi81lrtICs
РИС. 4.3.
Температурная зависимость
концентрации электронов проводимости в
германии, легированном мышьяком [54].
Концентрация мышьяка возрастает от 1-го к
6-му образцу.
ет ^1022 на 1 см3, атомная
доля примеси [Asce] изменяется в
пределах соответственно от
^10~9 (1 атом примеси на
миллиард атомов Ge) до « Ю-4
(1 атом на 10 тысяч). Эти оценки
показывают, какой высокой
степенью чистоты должны обладать
полупроводниковые материалы
для того, чтобы можно было
надежно контролировать их
свойства.
Горизонтальные участки на
кривых 1—3 позволяют с
уверенностью утверждать, что в этой
области температур разупорядо-
ченность примесная и
соответствует истощению доноров, т. е.
состоянию, когда все доноры
ионизованы (см. рис. 4.2, участок II). Ниже 20—50 К
концентрация носителей уменьшается, поскольку доноры
становятся неионизованными (резерв доноров, участок III). Кривые
1—5 на рис. 4.3 рассчитаны по формуле (4.23) для
примесной проводимости при значении эффективной массы т*,
равном lU массы свободного электрона, и энергии ионизации
доноров 0,01 эВ. Обращает на себя внимание очень хорошее
соответствие рассчитанных кривых и экспериментальных точек,
полученных при измерениях эффекта Холла.
Несколько выше комнатной температуры кривая для
образца 1 переходит в область собственной разупорядоченности и
концентрация носителей резко возрастает (пунктирная прямая).
Наклон этой прямой соответствует ширине запрещенной зоны
около 0,7 эВ, что в 70 раз больше энергии ионизации доноров.
Поэтому наклон собственного и примесного участков кривых
на графиках Аррениуса так резко различен.
Особое место на рис. 4.3 занимает кривая 6 для образца с
наибольшим содержанием примеси. Здесь концентрация
квазисвободных электронов практически постоянна во всем
исследованном температурном интервале. Эта черта, вообще говоря,
свойственная металлам, проявляется при так называемом в ы-
рождении полупроводников, когда классическая статистика
Больцмана становится неприемлемой и следует пользоваться
ПО
квантовой статистикой. Вырождение полупроводников
проявляется при достаточно низких температурах и высоких
концентрациях примеси.
Современная теория не дает полного описания
вырожденных полупроводников из-за сложного характера
взаимодействия дефектов. Однако качественно явление вырождения можно
проиллюстрировать по такой схеме. При повышении
концентрации примесей в полупроводнике их взаимодействие
усиливается настолько, что волновые функции электронных дефектов
начинают перекрываться. При этом вследствие запрета Паули
локальные энергетические уровни электронов примесей
расщепляются в самостоятельную зону, которая в пределе может слиться
с зоной проводимости. Естественно, что при этом энергия
ионизации примесей упадет до нуля, а все избыточные электроны
будут коллективизированы кристаллом подобно металлу. По
этой причине сильно вырожденные полупроводники иногда
называют полуметаллами.
4.3. Собственно-дефектные полупроводники
Независимо от характера электронной разупорядоченности в
любом полупроводниковом кристалле с неионной химической
связью всегда имеется собственная атомная разупорядочен-
ность, включающая один или несколько сортов нейтральных
атомных дефектов; их равновесные концентрации определяются
соотношениями, выведенными в гл. 3. Однако общая картина
атомной разупорядоченности в полупроводниках обычно
значительно сложнее, чем в металлических кристаллах, поскольку
атомные дефекты в результате взаимодействия с электронами
проводимости или дырками могут переходить в заряженное
состояние. При значительных концентрациях заряженных
атомных дефектов их следует учитывать в условии
электронейтральности и, таким образом, нужно рассматривать
комбинированную атомно-электронную собственную разупорядоченность.
Рассмотрим валентный кристалл простого вещества А,
содержащий наряду с электронами проводимости и дырками
вакансии. Для определенности предположим, что вакансии
обладают свойствами доноров, т. е. могут переходить в
ионизованное состояние согласно равновесной реакции
VX-TzhVt + e- (4.30)
Уравнение закона действия масс для этой реакции имеет вид,
аналогичный уравнению (4.10а) для реакции ионизации
примесных доноров:
[V£]K] = rv[V£] (4.31)
Нейтральные вакансии в кристалле простого вещества
образуются в результате выхода атомов из узлов на поверхность
согласно реакции
нуль^=^АПх + А, или нульт—*• Уд (4.32)
ill
Их концентрация была вычислена в гл. 3; согласно формуле
(3.5) она равна константе равновесия К v. Поэтому уравнение
(4.31) можно переписать в виде
[Vt][e~\ = (Kt)2 (4.33)
где Kv+= (TvKv) '2 имеет смысл константы рав«овесия реакции образования
пары заряженных дефектов — вакансии и электрона проводимости.
Уравнение этой реакции легко получить, складывая
почленно уравнения реакций (4.30) и (4.32):
нуль z^hVt + e- (4.34)
Уравнение (4.33) следует решать совместно с уравнением
закона действия масс для собственной ионизации (4.2) и
условием электронейтральности
[е~] = [Vt] + [e+] (4.35)
Умножая обе части последнего равенства на [е-] и используя
(4.2) и (4.33), получаем для концентрации электронов
проводимости:
[е~] = /Kf+(Kt)2 (4.36)
Отсюда видно, что упрощенные формы решения указанной
системы уравнений получаются в следующих предельных случаях.
I. Ki^>K+Y- В этом случае получаем для концентраций
электронных дефектов:
[£Г]«[е+]«Я1 (4.37)
решение, типичное для собственных полупроводников.
Концентрация заряженных вакансий при этом равна
[Vj]«(/^")2//r,«/ri (4.38)
и мала по сравнению с [е~] и [е+].
II. /u<CK+v. В этом случае решение описывается
приближенной формулой
[0«[Vi]«/^ (4.39)
из которой виден смысл константы /C+v: это есть равновесная
концентрация нового типа собственной разупорядоченности,
включающего в себя электроны проводимости и заряженные
вакансии. Концентрация электронных дырок в этом случае
равна
[e+] = K?IKf<Kf (4.40)
и мала по сравнению с концентрациями доминирующих
дефектов [е~] и [V+д].
В рассматриваемом случае проводимость обусловлена
электронами и кристалл является полупроводником п-типа.
112
III. Если вакансии обладают акцепторными свойствами,
возникает противоположная картина: вакансии частично
приобретают отрицательный эффективный заряд, и их образование
происходит согласно уравнению реакции
нуль-^±:У1 + £+ (4.41)
которому соответствует закон действия масс
№][е+] = (К7)2 (4.42)
Условие электронейтральности в этом случае записывается в
виде
l^] = [VI] + [e-] (4.43)
и при /Ci<C"V упрощенное решение описывается формулой
[е+] « [VJ] « Kv (4.44)
В этом случае концентрация электронов проводимости равна
[e~]=Ki2JK7<K7 (4.45)
и мала по сравнению с концентрацией дырок. Таким образом,
проводимость обусловлена дырками, и кристалл является
полупроводником р-типа.
В рассмотренных предельных случаях II и III электроны
проводимости и дырки образуются в результате реакции
ионизации собственных атомных дефектов — вакансий. Поэтому
кристаллы, в которых такой механизм является доминирующим,
называют собственно-дефектными полупроводниками.
Полученные здесь решения иллюстрируются рис. 4.4, на
котором изображены концентрации дефектов в кристалле чистого
теллура в зависимости от обратной температуры. При темпера-
ряс. 4.4.
W3JT, К'1
Температурная зависимость концентраций собственных дефектов в
кристаллическом теллуре [20].
8—321
113
турах ниже 200 °С этот кристалл ведет себя как типичный
собственный полупроводник, в котором преобладающими
дефектами являются электроны проводимости и дырки, присутствующие
примерно в равных концентрациях. Однако вследствие большей
подвижности электронов проводимости по сравнению с
подвижностью дырок в этой области температур электропроводность
теллура обусловлена в основном электронами.
Вакансии в кристалле теллура обладают акцепторными
свойствами, поэтому наряду с квазисвободными электронами и
дырками в нем имеются отрицательно заряженные
(ионизованные) вакансии. В рассматриваемой области низких температур
концентрация ионизованных вакансий мала по сравнению с
концентрациями электронов и дырок, однако с повышением
температуры она растет быстрее, чем концентрации электронов и
дырок, и при температуре я^200°С достигает сравнимого с
ними значения. При температурах, превышающих 200 °С,
доминирующими дефектами являются дырки и отрицательно
заряженные вакансии, имеющие приблизительно равные концентрации.
В этой области температур концентрация электронов
проводимости значительно меньше концентрации дырок и кристалл
является собственно-дефектным полупроводником р-типа. (Смена
знака носителей в кристалле теллура обнаруживается
измерениями эффекта Холла.) Переход от собственной проводимости
к собственно-дефектной проявляется и в изменении наклона
прямолинейных участков графиков, изображающих
концентрации доминирующих дефектов: в собственной области он равен
половине ширины запрещенной зоны, а в собственно-дефектной
области — половине энергии реакции (4.41), деленной на
постоянную Больцмана.
Собственно-дефектными полупроводниками могут быть и
кристаллы химических соединений. Так, в бинарном соединении
MX, обладающем атомной разупорядоченностью Шоттки,
электроны проводимости могут возникать за счет ионизации
вакансий более электроотрицательного компонента X, играющих роль
доноров. Образование доминирующих заряженных дефектов —
электронов и ионизованных вакансий X в этом случае
соответствует квазихимической реакции
нуль^=> V& + Vt + е~ (4.46)
Аналогично, дырки возникают при ионизации вакансий
электроположительного компонента М; образование доминирующих
заряженных дефектов происходит при реакции
нуль ^=> Vx + Vm + e+ (4.47)
Однако в случае бинарных соединений количественные
соотношения между концентрациями дефектов гораздо более сложны,
чем в случае простых веществ, вследствие отклонений их
состава от стехиометрического, подробно рассматриваемых в
следующем разделе.
114
4.4. Нестехиометрические полупроводниковые
соединения
Для кристаллов неметаллических соединений, обладающих
преимущественно ковалентной связью, характерны в основном те
же типы атомной разупорядоченности, что и для
интерметаллических соединений, а именно дефекты Шоттки, Френкеля и
антиструктурные дефекты. Однако здесь картина значительно
усложняется из-за взаимодействия атомных дефектов с
квазисвободными электронами и дырками, в результате которого
атомные дефекты могут находиться как в нейтральной, так и в
заряженной форме. Поэтому при вычислении равновесных
концентраций дефектов в полупроводниковых соединениях
необходимо учитывать все квазихимические реакции, протекающие с
участием как нейтральных, так и заряженных дефектов, в том
числе квазисвободных электронов и дырок.
В равновесных условиях степень отклонения от стехиомет-
рического состава регулируется путем обмена атомами
кристалла с окружающей средой, чаще всего газовой. В дальнейшем
мы будем предполагать, что кристалл находится в
термодинамическом равновесии с окружающей газовой фазой, которая
содержит электроотрицательный компонент в виде
двухатомных молекул Х2 (например, в случае оксида — молекулярный
кислород 02) при контролируемом парциальном давлении Х2.
В этом случае отклонения от стехиометрического состава
обеспечиваются либо поглощением избыточного компонента X из
газовой фазы при высоких парциальных давлениях Х2, либо
удалением некоторого количества X из кристалла в газовую
фазу при низких парциальных давлениях Х2. При равновесии
точному стехиометрическому составу при фиксированной
температуре соответствует строго определенное значение
парциального давления Х2Рстех. Обмен кристалла с газовой фазой
компонентом X можно описать квазихимическими реакциями,
в которых наряду с дефектами кристаллической решетки
участвуют молекулы Х2 газовой фазы, причем при равновесии
кристалла с газовой фазой к этим реакциям можно применять
закон действия масс.
В настоящем разделе мы рассмотрим бинарное нестехиомет-
рическое соединение МХг+б, обладающее преимущественно
ковалентной связью и являющееся при стехиометрическом
составе (6 = 0) собственным полупроводником. Это означает, что при
стехиометрическом составе доминирующими заряженными
дефектами являются электроны проводимости и дырки,
возникающие в результате собственной ионизации кристалла (4.1), и
условие электронейтральности выражается приближенным
равенством
[е-]^[е+]^К.х (4.48)
8*
115
(приближенный характер равенства обусловлен наличием
некоторого количества заряженных атомных дефектов, которое в
случае собственного полупроводника считается пренебрежимо
малым). Степень отклонения от стехиометрического состава б
будем считать малой по сравнению с единицей.
Атомная разупорядоченность Шоттки. Предположим
сначала, что доминирующими атомными дефектами являются
вакансии в подрешетках М и X (дефекты Шоттки). Тогда изменение
состава кристалла по сравнению со стехиометрическим может
происходить путем удаления атома X в газовую фазу согласно
реакции
нуль^ХПх + уХ2, или Xx^V£ + yX2 (4.49)
при которой в подрешетке X возникает незаряженная вакансия,
или же путем поглощения атомов X из газовой фазы:
уХ2:^:МХг + МПх, или -^-Х2+±гХ$ + Х& (4.50)
Во втором случае происходит достраивание решетки на
поверхности кристалла; атом М, необходимый для образования на
поверхности новой формульной единицы МХГ (при б<^1),
переходит на поверхность из узла в объеме кристалла, и в этом узле
возникает незаряженная вакансия Vxm-
При не слишком низких температурах газовую фазу можно
считать идеальной системой и химический потенциал
молекулярного газа Х2 описывается формулой (см. П. 35).
Па-=^ + кТ1пРх2 (4-51>
где рх2 — парциальное давление Х2 в газовой фазе.
Поэтому к реакциям с участием молекул газовой фазы
можно применять закон действия масс в форме (2.28) или (2.40) с
тем отличием, что вместо доли [Х2] в него следует подставлять
парциальное давление рх„.
Для реакции (4.49) при [Ххх]^1 закон действия масс
запишется в виде уравнения
1
[Хх]/?Хо = Сух
или
[Щ^С^р1 (4.52а)
где константа равновесия определяется выражением
Cvxx^/-(v)exp(-^i^ii2) (4.53a>
где множитель %(v) определяется спектром колебаний кристалла.
116
В уравнении реакции (4.50) при [Ххх]^1 закон действия
масс записывается в виде
г
[V^] = C^m/?x2 (4.526)
с константой равновесия
CVM = Z (v) exp I — 2-1 (4.536)
Формулы (4.52) выражают важный результат, что
равновесные концентрации нейтральных атомных дефектов в несте-
хиометрическом кристалле зависят только от температуры и
парциального давления Х2 в газовой фазе и не зависят от
концентраций других дефектов. Этого нельзя сказать о
заряженных дефектах, концентрации которых взаимосвязаны.
Действительно, если вакансии электроотрицательного компонента X
обладают свойствами доноров, а вакансии электроположительного
компонента М — свойствами акцепторов, то их ионизация
описывается реакциями
Vx*--^+.- (454)
Применение закона действия масс к этим реакциям дает
уравнения, связывающие концентрации противоположно
заряженных дефектов:
[Vj][£T] = rVx[v]n (455)
[VM][e+j=rVM[VU]
где константы раеновесия Г определяются энергиями ионизации вакансий ш
описываются формулами (4.12).
Подставляя в уравнения (4.55) значения концентраций
нейтральных вакансий из (4.52), представим их в несколько ином
виде
—1
[«пл=с^; (45б)
[Vm] [e+] = СумРх,
с константами
Сух = ГухСух*, Сум = Гум Сум
Легко видеть, что соотношения (4.56) являются уравнениями
закона действия масс для квазихимических реакций:
(4.57>
(-^-JX2^:a-X£ + Vm + ^
117
согласно которым взаимодействие кристалла с газовой фазой
приводит к образованию заряженных вакансий, заряд которых
компенсируется квазисвободными электронами и дырками.
Уравнения (4.56) совместно с уравнением закона действия
масс для собственной ионизации полупроводника (4.2)
[e-][e+] = tf
и условием электронейтральности
п~ + МГм = я+ + Л/ух
или в атомных долях
[e"] + [VM] = [e+]+r[Vj] (4.58)
образуют полную систему относительно неизвестных
концентраций [е-], [е+], [V~m] и [V+x]. Решение этой системы уравнений
описывается формулами
[Vm]^PK]; [V$] = *[e+]
где введены обозначения
(4.59)
а = Ctx КГ2Рхй112; р ='(ЯмКГ2Ръ (4.60)
Из последних определений видно, что параметры а и §
зависят от температуры и давления Х2 в газовой фазе и могут
изменяться в очень широких пределах. Поэтому решения (4.59)
для концентраций заряженных дефектов можно упростить,
рассматривая различные предельные случаи.
I. Область промежуточных давлений Х2. Выше
мы предположили, что при стехиометрическом составе кристалл
является собственным полупроводником с приблизительно
равными концентрациями электронов проводимости и дырок и с
пренебрежимо малыми концентрациями заряженных вакансий.
Из решений (4.59) видно, что условия [У"м]<С(У~] и [V+x]<C[e+]
выполняются при значениях а и §, одновременно малых по
сравнению с единицей, т. е. при одновременном выполнении
условий
Pi » С^хКГ2; р1 « Кх (С7м Г1
При фиксированной температуре оба эти условия выполняются,
когда рх2 лежит в интервале
р- = (Сух)2 Kt « Рх « К (Сум ) г ^ Р+ (4.61)
118
следовательно, во всем этом интервале давлений кристалл
является собственным полупроводником. Концентрации дефектов
при а, §<1 описываются упрощенными формулами (4.59)
[е~] « [е+] « Кх
Г
[Мт]~ Сум/CrVl (4.62)
—1
показывающими, что концентрации доминирующих дефектов
е~ и е+ во всем рассматриваемом интервале давлений остаются
приблизительно постоянными, в то время как малые
концентрации ионизованных вакансий меняются при изменении рх2> На
границах интервала давлений (4.61) Р~ и Р+ условие малости
концентраций ионизованных вакансий нарушается. На верхней
границе интервала Р+ параметр |3 становится равным единице
и концентрация V~m сравнивается с концентрацией электронов
проводимости; на нижней границе Р~, напротив, а=1 и [V+x]
сравнивается с [е+].
П. Область больших давлений Х2. Если давление
Х2 в газовой фазе значительно превышает критические
значения Р~ и Р+, то параметры аир удовлетворяют неравенствам
а<С1; §^>1 и решения (4.59) упрощаются до вида
[6+]^[Vm]«(C7m)2Px42
-Л -г
[€-]*> Ki(C™)2 Pi (4.63)
[V$]^Ctx(CvM)2K-2pi 2
Согласно этим соотношениям доминирующими дефектами
являются дырки и отрицательно заряженные вакансии М,
присутствующие приблизительно в одинаковых концентрациях.
Концентрации электронов проводимости и положительных вакансий X
малы по сравнению с концентрациями дырок и отрицательных
вакансий М. Электронная проводимость обусловлена
преимущественно дырками и относится к р-типу.
Примечательно, что характер доминирующих дефектов в
рассматриваемом предельном случае такой же, как и в случае
собственно-дефектного кристалла простого вещества с
акцепторными вакансиями. Однако природа этих дефектов несколько
иная: вакансии М и дырки хотя и являются собственными
дефектами, в данном случае их доминирующая роль обусловлена
избыточным содержанием X по сравнению со стехиометричес-
119
ким составом. Образование доминирующих дефектов в
рассматриваемом случае обеспечивается реакцией
у Х2 ^ rX£ + Vm + e+ (4.64)
III. Область малых давлений Х2. Если давление Х2
в газовой фазе значительно меньше критических значений Р~
и Р+, то параметры аир удовлетворяют неравенствам а>>1;
|3<С1, и решения (4.59) принимают упрощенный вид
[e-}^r[Vi}^(rCyX)2pl
[er\ = KhrC^fpl (4.65)
_1_ r_ 1_
[Ум] = С^м(гС^х)2КГ2р1 4
Здесь картина противоположная описанной выше для
области больших рх2- Доминирующими дефектами являются
заряженные ионные вакансии и электроны проводимости,
образующиеся при реакции
X$+*:-jX2 + Vi + e- (4.66)
Электронная проводимость обусловлена преимущественно
электронами и относится к п-тииу. Концентрации дырок и
отрицательных вакансий V~~m малы по сравнению с концентрациями
электронов проводимости и положительных вакансий V+x.
Полученные приближенные решения показывают, что
концентрация всех рассмотренных дефектов в нестехиометрическом
полупроводнике являются степенными функциями давления Х2.
Поэтому при фиксированной температуре их удобно
изобразить графически в координатах логарифм концентраций —
логарифм давления Х2. В таких координатах все приведенные
приближенные решения изображаются прямыми линиями с
тангенсом угла наклона, равным показателю степени при рх2- Так,
для области I уравнения прямых имеют вид
l*[^ = lg[*+] = lgtfi
lg[VS] = const + (-~)lg/>x2 (4-67)
lg[V£] = const _^-i-jlgpXi
На рис. 4.5 изображена зависимость концентраций дефектов
от рх2 в логарифмических координатах для равновалентного не-
стехиометрического кристалла MXi+6 (2m = ;zx; r=l). Римские
12 0
1дР'сш1дР~ lgPcm*lgP,lcme* lgPf lgpXz
РИС. 4.5.
Зависимость концентраций дефектов в равновалентном нестехиометрическом
полупроводнике MXi±g от парциального давления Х2 в газовой фазе.
цифры, I, II, III указывают области, в которых реализуются
приближенные решения (4.62), (4.63) и (4.65). При переходе от
одной области к другой прямые, описывающие приближенные
решения, испытывают изломы. Очевидно, что точные решения
должны изображаться линиями, на которых изломы на
границах областей сглажены, но которые внутри каждой области
практически совпадают с изображенными прямыми.
На рис. 4.5 изображены также прямые, описывающие
концентрации нейтральных вакансий VMX и Vxx. В соответствии
с формулами (4.52) и (4.54) эти прямые описывают точные
решения во всей области давлений Х2 и поэтому не испытывают
изломов. Наклон этих прямых при г=\ всегда равен ±!/2,
однако их относительное положение по высоте графика может
быть различным в зависимости от соотношения между
константами равновесия.
Рассмотрим, в частности, относительное положение по
высоте точек пересечения прямых, описывающих концентрации
нейтральных и заряженных вакансий. Значения ординат этих
точек можно определить следующим образом.
Для реакции образования нейтральных вакансий (дефектов
Шоттки) в кристалле MX
нуль^УЙ+ Vx (4.68)
закон действия масс записывается в виде
IVjfcl [V£] = (АГГ)2 (4.69)
Подставляя сюда значения концентраций нейтральных
вакансий из формул (4.52) при г=\ находим, что константа Шоттки
121
в этом уравнении равна
j_
КЧ = (С^мС^х)2 (4.70)
В точке пересечения прямых для нейтральных вакансий
имеем:
[VM] = [V?]-/Ts (4.71)
Образование ионизованных вакансий (заряженных дефектов
Шоттки) в кристалле MX может происходить при реакции
нуль^ГУм + vt (4.72)
для которой закон действия масс записывается в виде
[Vm] [Vt] = (Ki)2 (4.73)
Константу равновесия этой реакции найдем, подставляя в
последнее уравнение концентрации ионизованных вакансий из
решений (4.62) для области I при г=1:
j_
/fs±=(CvMC^x)2/?r1 (4.74)
В точке пересечения прямых для заряженных вакансий имеем:
[Vm] = [Vt] = Ki (4.75)
На рис. 4.5 прямые, описывающие концентрации
нейтральных вакансий, лежат выше соответствующих прямых для
заряженных вакансий, т. е. вакансии находятся преимущественно в
незаряженном состоянии. Соответственно этому точка
пересечения прямых для нейтральных вакансий лежит выше точки
пересечения прямых для заряженных вакансий и Ksx>Ks±.
Такая ситуация характерна для соединений с ковалентной связью,
в которых атомы М и X мало отличаются по
электроотрицательности. В соединениях же, компоненты которых заметно
отличаются по величине электроотрицательностей и в которых,
следовательно, значительна доля ионной связи, значения
константы Ks± могут превышать значения /CsK, тогда вакансии
будут находиться преимущественно в ионизованном состоянии.
В этом случае прямые для нейтральных вакансий на рис. 4.5
будут лежать ниже прямых для заряженных вакансий. Однако
концентрации нейтральных вакансий не входят в систему
уравнений для заряженных дефектов, поэтому все исследованные
решения для заряженных дефектов е~~, е+, Vm" и Vx4"
сохраняют силу и в этом случае.
Если вакансии находятся преимущественно в нейтральной
форме, точка пересечения прямых, описывающих концентрации
нейтральных вакансий, лежит при давлении рстех^ приблизи-
122
тельно соответствующем стехиометрическому составу
кристалла. Действительно, при стехиометрическом составе должно
выполняться равенство
[VU] + [Vm] = [V£] + [Vt] (4.76)
или, при пренебрежении концентрациями заряженных
вакансий, [VM*]^[Vxx].
На рис. 4.5 точка пересечения сплошных прямых [VMK
и [Vxx] соответствует давлению Рстех, лежащему внутри
интервала Р~—Р+, в котором преобладающими дефектами являются
электроны и дырки. Это соответствует предположению,
принятому при постановке рассматриваемой задачи о том, что при
стехиометрическом составе кристалл является собственным
полупроводником. Однако такое предположение не ограничивает
применимости полученных приближенных решений, поскольку
при их нахождении использовались лишь ограничения,
накладываемые на параметры а и р. На рис. 4.5 возможный ход
зависимостей для концентраций нейтральных вакансий изображен
также пунктирными прямыми. Точка их пересечения,
отвечающая стехиометрическому составу, находится при давлении Рстех,,
лежащем вне интервала собственной электронной разупорядо-
ченности Р~—Р+, а именно в области II, в которой
доминирующими дефектами являются электроны проводимости и
положительные вакансии Vx+. Поэтому такой кристалл при
стехиометрическом составе является собственно-дефектным
полупроводником я-типа, а переход к собственной проводимости
происходит лишь при избыточном содержании X. В остальном же
поведение такого кристалла не отличается от ранее
рассмотренного.
Если Ks±>Ksx и вакансии находятся преимущественно в
заряженном состоянии, из соотношения (4.76) следует, что
стехиометрическому составу отвечает условие [Vm~]~[Vx+].
Поэтому стехиометрическому составу соответствует
приблизительно абсцисса точки пересечения прямых для заряженных
вакансий Р"стех# в этом случае Р"стех всегда лежит внутри
интервала Р~—Р+ и стехиометрический кристалл всегда является
собственным полупроводником.
Важным условием, ограничивающим применимость
полученных приближенных решений, является условие существования
собственной электронной разупорядоченности в области I,
согласно которому параметры а и ,§ должны быть одновременно
малы по сравнению с единицей, а давление Х2 должно
удовлетворять требованию (4.61): Р-<СРх2<^+. Очевидно, это
возможно, только если Р-<СР+. Используя значения Р" и Р+ из
(4.61) при г=\ видим, что условие Р~<^Р± равносильно
условию
CvmC$x « Kt (4.77)
ш
или, при учете (4.74), условию
Ki <C Я", (4.78)
Смысл этого неравенства очевиден: образование ионизованных
дефектов Шоттки в полупроводнике должно протекать менее
интенсивно, чем собственная ионизация кристалла, приводящая
к образованию электронов проводимости и дырок.
Если условие (4.78) не выполняется, а выполняется
обратное соотношение
/Cs±»^i (4.79).
существует область давлений Х2, в которой аир одновременно
велики по сравнению с единицей. В этом случае собственная
ионизация кристалла не является определяющей, а
доминирующими дефектами в области промежуточных давлений Х2
являются ионизованные вакансии Vm~ и Vx+. Такое поведение
характерно для ионных кристаллов с достаточно широкой
запрещенной зоной, которые, хотя и могут обнаруживать
значительную электронную проводимость, не являются
полупроводниками в строгом смысле этого слова. Теория разупорядоченности
ионных кристаллов будет изложена в следующей главе.
Другие типы атомной разупорядоченности. Мы подробно
рассмотрели нестехиометрический полупроводник МХг+б, в
котором преобладающими атомными дефектами являются
вакансии в подрешетках обоих компонентов (дефекты Шоттки).
Задачу нетрудно обобщить и на случай произвольной атомной
разупорядоченности, включающей дефекты Френкеля по
компоненту М или X или антиструктурные дефекты.
Для этого выпишем квазихимические реакции образования
незаряженных атомных дефектов при обмене кристалла
атомами X с газовой фазой. Внедрение атомов М в междуузлия
может происходить при переходе X в газовую фазу с низким
давлением Х2 согласно реакции
МХГ^± МОх + «- Х2, или М£ + гХх + Vix jzt
^±Mfx+ — X2 (4.80a)
при которой на поверхности кристалла разрушается
формульная единица МХГ. Напротив, внедрение в междуузлие атома X
происходит при поглощении избыточного X из газовой фазы с
высоким рх2:
~ Х2 ^ ХОх, или -j Х2 + V,x 5=t X,x (4.806)
124
Образование антиструктурных дефектов также происходит
либо при потере X в газовую фазу с низким давлением Х2:
МХ,«М#Х(Х) + -^Х2, или МЙ + (г + 1)Хх^
^Мх +^^Х2 (4.80в)
либо при поглощении X из газовой фазы с высоким рх2:
Г±1х2^=±МХг + ХФх(М)} или L±Lx2^±rX$ + X&
(4.80г)
При этих реакциях происходит либо разрушение, либо
образование новой формульной единицы МХГ на поверхности
кристалла.
Применяя закон действия масс к реакциям (4.80), получаем
зависимость равновесных концентраций незаряженных
дефектов от давления Х2 в газовой фазе:
—г
]MiX] = С^р
х2
1
1 J Fx> (4.81)
[MxJ —СмхрХ2
[Amj = СХмРх2
Для дальнейшего необходимо предположить, какими
свойствами— донорными или акцепторными — обладают
рассматриваемые атомные дефекты. Поскольку М обозначает
электроположительный элемент, а X — электроотрицательный,
естественно предположить, что междуузельные атомы М обладают
свойствами доноров, а междуузельные атомы X — акцепторов.
Антиструктурные дефекты ведут себя иначе. Если атом
электроположительного элемента М имеет меньшее число валентных
электронов, чем замещаемый им электроотрицательный атом X,
то дефект Мх обладает свойствами акцептора (см. раздел 1.6),
напротив, дефект Хм обладает свойствами донора. Таким
образом, реакции ионизации рассматриваемых атомных дефектов
протекают согласно уравнениям
MiX^=^Mib + ^~
Xх +± ХГ + е+ (4 82)
Мх:^Мх+£Ф
Хм"^1±:Хм-{-£
125
Записывая уравнения закона действия масс для этих реакций
и подставляя в них концентрации нейтральных дефектов из
(4.81), получаем:
—г
[Mt][e-] = aiPl
j_
[ХГ] [е+] = Cxipy
Ха (4.83)
[Мх][е+] = Сйх^+1)/2
[Xt)[e-] = C^p^+1)l2
Уравнения (4.83) следует решать совместно с
аналогичными уравнениями для вакансий (4.56), уравнением собственной
ионизации (4.2) и обобщенным условием электронейтральности:
п~ + WM + Nxi + Шх = п+ + N<tx + N^ + Л#м
Перейдем в последнем уравнении от абсолютных концентраций
дефектов к относительным, учитывая, что в кристалле МХГ,
содержащем в единице объема NL узлов М, имеется rNL узлов X
и gNL междуузлий (г и g— целые числа или простые дроби).
Тогда условие электронейтральности запишется в виде
[e"l+[vsi + e:[xn + r[Mx]-
= [*+] + г [Vt\ + g [Mt\ + [Xjfc] (4.84)
Здесь мы не будем искать точное решение системы уравнений,
а воспользуемся приближенным методом решения, обычно
используемым в задачах такого типа. Обратим внимание на то,
что в каждом из приближенных решений, найденных выше для
атомной разупорядоченности Шоттки, упрощенное условие
электронейтральности содержит в левой и правой части по
одному члену, а именно концентрации положительных или
отрицательных доминирующих дефектов: [е"-]«[е+], [е+]~[\Л\Г~] и
[e-]^r[Vx+] соответственно для областей давления I, II, III.
Поскольку в рассматриваемом общем случае все уравнения
аналогичны уравнениям для разупорядоченности Шоттки, можно
полагать, что каждое приближенное решение обобщенной
системы уравнений также будет соответствовать упрощенному
условию электронейтральности (4.84), содержащему в обеих частях
по одному члену. Перебирая таким образом все возможные
комбинации противоположно заряженных дефектов, получаем
следующие приближенные решения для доминирующих
дефектов.
I. Собственная электронная разупорядоченность
[ё~\ « [е+] « Кх (4.85)
126
преобладает в промежуточной области давлений Р~"<СРх2<СР+
при любом типе атомной разупорядоченности, если реакция
собственной ионизации протекает более интенсивно, чем
образование парных комбинаций из заряженных атомных дефектов.
Условия существования такой области давлений находятся
аналогично изложенному выше для разупорядоченности Шоттки и
сводятся к тому, что константа реакции собственной ионизации
/Ci должна значительно превышать константы реакций
образования парных комбинаций заряженных атомных дефектов
/C+fm, К^х и Л^а, аналогичные константе /^s для заряженных
дефектов Шоттки (4.74).
Преобладание заряженных атомных дефектов при
упрощенных условиях электронейтральности типа [V~M]~g[Mi+] и т.п.
требует выполнения противоположного условия: /G-C/Pfm,
/Pfx, Д^а. Последнее отношение характерно в основном для
ионных кристаллов с широкой запрещенной зоной, поэтому
такие решения здесь можно не рассматривать.
И. В области больших парциальных давлений Х2, рх2^Р~~,
Р+, концентрации доминирующих заряженных дефектов
описываются следующими приближенными решениями (для полноты
картины здесь приводятся и решения для разупорядоченности
Шоттки).
A. При разупорядоченности Шоттки и (Б) при
разупорядоченности Френкеля в подрешетке М доминируют одни и те же
дефекты:
1 г
H^[Vm]^(C™)2< (4.86)
B. При разупорядоченности Френкеля в подрешетке X
доминируют дырки и отрицательные междуузельные ионы:
[e+}^g[XT]~{gCu)2 pi (4.87)
Г. При антиструктурной разупорядоченности избыточное
содержание X, напротив, связано с электронами проводимости:
[е-]^[Х&]~(С£т)р(^т (4.88)
III. В области низких парциальных давлений, рх2<^Р~, Р+,
концентрации доминирующих заряженных дефектов
описываются следующими решениями.
А. При разупорядоченности Шоттки:
L zi
[e-]~r[V$]~(r&x)2 Pi (4.89)
Б. При разупорядоченности Френкеля в подрешетке М:
1
[е~) « g [Mt] « (gCtu )l РхГ (4-90)
127
В. При разупорядоченности Френкеля в подрешетке X
решение совпадает с (4.89).
Г. При антиструктурной разупорядоченности:
[е+] ж г [Ш] « (гСйх)2 р^г+т (4.91)
Все полученные приближенные решения для концентраций
доминирующих электронных носителей — электронов
проводимости и дырок можно представить в виде общей формулы
JL
" = Л(7)К>Р(-^) (4.92)
где Л(Т)—некоторая степенная функция температуры, включающая
различные мультипликативные комбинации предэкспоненциальных множителей
констант равновесия %(Т)\ е — эффективная энергия активации процесса
образования электронных носителей, включающая различные аддитивные
комбинации энергий квазихимических реакций; m — параметр, описывающий
зависимость концентраций доминирующих электронных носителей и,
следовательно, электронной проводимости от давления Х2, имеющий различные значения
при различных упрощенных формах условия электронейтральности.
Значения параметра т, отвечающие полученным
приближенным решениям, сведены в табл. 4.1.
Если изобразить полученные решения графически в
координатах логарифм концентраций — логарифм рХз, аналогично
изображенному на рис. 4.5, то тангенс угла наклона прямых на
каждом участке будет равен 1/т.
Из полученных решений видно, что электропроводность не-
стехиометрического полупроводника минимальна в собственной
области I, лежащей при промежуточных давлениях Х2. При
всех типах атомной разупорядоченности электропроводность
Таблица 4.1. Значения параметра m в формуле (4.92)
для концентраций электронных носителей в пестехиометрическом
полупроводнике МХг±б
Тип собственной атомной
разупорядоченности
Шоттки
Френкеля по М
Френкеля по X
Антиструктурная
Решение
I
II А
III A
I
II А
III Б
I
II В
III A
I
II Г
III Г
Условие
электронейтральности
\е~
>+;
'е-'
е~
е+[
[е~[
[е-'
>-:
е~'
е+"
'е-'
] = [е+]
= [vM-l
-[е+]
= [VM-]
=g[M«+]
= [f]
=g[x,-]
=r[Vx+]
= [Хм+]
=г[Мх-]
m
oo
4/r
—4
oo
4/r
-4/r
OO
4
—4
4 (r+1)
-4/(r+l)
128
растет при увеличении рх2 в области высоких давлений Х2 (при
избытке X) и при уменьшении Рх2в области низких давлений
Х2 (при недостатке X по сравнению со стехиометрическим
составом). Различное влияние разных типов атомной разупорядо-
ченности проявляется в том, что при разупорядоченности Шот-
тки и Френкеля избыточному содержанию X при высоких рх%
соответствует проводимость р-типа, а при антиструктурной раз-
упорядоченности проводимость n-типа. При низких давлениях
рх2 недостатку X соответствует обратный тип проводимости:
n-тип при разупорядоченности Шоттки и Френкеля и р-тип при
антиструктурной разупорядоченности.
Глава 5
Разупорядоченность ионных кристаллов
Как было описано в гл. 1 (см. раздел 1.3), чисто ионной связи
в природе не существует; все реальные ионные кристаллы
обладают той или иной долей ковалентной связи. Поэтому термин
«ионный кристалл» обычно используют для обозначения
соединений с преобладающей ионной связью.
В рамках квазихимического метода описания дефектных
кристаллов, используемого в данной книге, основные черты
дефектной структуры кристаллов определяются целочисленными
значениями эффективных зарядов дефектов, которые могут
существенно отличаться от зарядов ионов, а истинные заряды
ионов не играют существенной роли. Поэтому излагаемая здесь
теория в значительной степени применима и к соединениям с
промежуточным характером связи, а в некоторых случаях даже
к кристаллам, связь в которых ближе к ковалентной, нежели
к ионной. В этих случаях критерием применимости теории
являются не заряды ионов, а эффективные заряды доминирую*
щих атомных дефектов, определяемые энергетическим
спектром электронов в кристалле.
Рассмотрим в качестве примера 1,1-валентный кристалл МХ?
содержащий равные концентрации вакансий в катионной и ани*
онной подрешетках. В соответствии с изложенным в разделе 1.6
катионные вакансии в таком кристалле являются акцепторами
и могут находиться в двух состояниях — нейтральном Vm x И
отрицательно заряженном Vm~; анионные вакансии являются
донорами и могут находиться в нейтральном состоянии Vxx
или положительно заряженном Vx+. Переход вакансий из
нейтрального состояния в заряженное соответствует
квазихимической реакции
VU+Vx^Vm+v£ (5.1)
9—321
129
У////зот///// У////зотУ/// рис-*■'■
// прободимости ууу '// проводимости у у Положение донорных уровней анион-
'/'////. '////у/А '/'/// '/ ' ' ' '' ных вакансий в валентных (а) и ион-
. . , ных \(б) кристаллах.
Vm Vx+
и заключается в переходе
зависит от относительного положения донорных и акцепторных
уровнен в запрещенной зоне. При этом возможны две
ситуации, изображенные схематически на рис. 5.1.
Если донорные уровни анионных вакансий лежат ниже
акцепторных уровней катионных вакансий (рис. 5.1,а), то в
основном состоянии кристалла электроны локализованы на
анионных вакансиях, обеспечивая их электронейтральность. В свою
очередь катионные вакансии лишены электронов и также
находятся в нейтральном состоянии. Такая ситуация типична для
валентных соединений с малой долей ионной связи.
Если, напротив, донорные уровни анионных вакансий лежат
выше акцепторных уровней катионных вакансий (рис. 5.1,6),
то в основном состоянии кристалла электроны локализованы на
катионных вакансиях, обеспечивая их отрицательный
эффективный заряд, а лишенные электронов анионные вакансии имеют
положительный эффективный заряд. Такая ситуация типична
не только для кристаллов с характером связи,
приближающимся к ионному, но и для соединений с промежуточным
характером связи. Поэтому дефектную структуру таких соединений
можно описывать в рамках ионной модели, согласно которой
основное состояние соответствует заряженным вакансиям.
Аналогичная картина обнаруживается и в соединениях
многовалентных элементов. Например, в оксидах металлов
эффективные заряды ионов кислорода близки к —1 (см. табл. 1.1),
несмотря на то, что валентность кислорода равна 2. Таким
образом, по характеру химической связи оксиды металлов
относятся к соединениям, промежуточным между ионными и
валентными кристаллами. Тем не менее экспериментальные данные
показывают, что в большинстве оксидов металлов, таких, как
MgO, NiO, Zr02 и др., вблизи стехиометрического состава
эффективный заряд вакансий кислорода равен двум и
соответствует классической ионной модели, согласно которой кислород
представляется двухзарядным ионом. Причина такого сильного
несоответствия эффективных зарядов ионов и их вакансий в
настоящее время неясна. Однако для теории разупорядоченно-
сти это обстоятельство оказывается чрезвычайно удобным.
Благодаря ему при описании дефектной структуры оксидов ме-
130
таллов можно не обращать внимания на характер их
химической связи, а рассматривать эти соединения как типичные
ионные кристаллы, удовлетворяющие классической ионной
модели [35, 36].
В дальнейшем при рассмотрении ионной разупорядоченности
кристаллов предполагается, что основное энергетическое
состояние ионных дефектов отвечает их полной ионизации, при
которой эффективные заряды дефектов по абсолютному значению
равны валентностям соответствующих компонентов кристалла.
5.1. Собственная ионная разупорядоченность
Природа собственных ионных дефектов в ионных кристаллах в
принципе не отличается от природы атомных дефектов в
интерметаллических или полупроводниковых соединениях.
Основными точечными дефектами в ионных кристаллах являются
ионные вакансии и междуузельные ионы, отличающиеся от
соответствующих атомных дефектов в неионных кристаллах лишь тем,
что в основном энергетическом состоянии они заряжены.
В стехиометрических ионных кристаллах при отсутствии
электронной разупорядоченности ионные дефекты всегда обра-^
зуются в определенных комбинациях. Как и в кристаллах
неионных химических соединений, эти комбинации должны быть
такими, чтобы в ходе реакции сохранялось постоянным
отношение чисел узлов разных подрешеток. Кроме того, на реакции
образования заряженных дефектов накладывается
дополнительное условие: сумма эффективных зарядов всех возникающих
дефектов должна равняться нулю, с тем чтобы в ходе реакции
не нарушалась электронейтральность кристалла. В бинарном
ионном соединении МХГ, содержащем полностью ионизованные
вакансии или междуузельные ионы, обоим указанным
условиям удовлетворяют следующие комбинации ионных дефектов.
А. Дефекты Шоттки — полностью ионизованные катионные
или анионные вакансии, образующиеся согласно реакции
нуль:^МП*м~ + гХП*х+ + MX,, или
нуль +± V*M~ + rV*x+ (5.2)
Условие электронейтральности этой реакции выполняется,
поскольку коэффициент г в бинарном ионном кристалле равен
отношению валентностей компонентов: г = ^м/^х.
Б. Дефекты Френкеля — полностью ионизованные
междуузельные катионы и катионные вакансии, образующиеся
при реакции
нуль^МО*м+ + МП*м~, или
Мй + Vix ц=£ М*м+ + V^M~ (5.3)
9*
131
В. Противоположные дефекты — полностью ионизованные
междуузельные анионы и анионные вакансии, образующиеся
при реакции
нуль^ХО*х"~Н-ХП*х+, или
Xxx + VfeX^+V^+ (5.4)
называют антифренкелевскими дефектами.
Как уже указывалось в гл. 1, в кристаллах бинарных
ионных соединений антиструктурные дефекты, типичные для
интерметаллических соединений, не встречаются, так как размещение
катионов в анионных узлах и наоборот требует слишком
больших затрат энергии.
Применение закона действия масс к реакциям образования
собственных ионных дефектов (5.2) — (5.4) дает следующие
уравнения, связывающие их концентрации.
A. Для дефектов Шоттки:
[V^J[V^+r = ^+r (5.5)
Б. Для дефектов Френкеля:
KM+][v*M"]-/:l (5.6)
B. Для антифренкелевских дефектов:
[X*x~][V*x+] = /C|f (5.7)
Константы равновесия этих реакций определяются теми же
формулами, что и для кристаллов интерметаллических
соединений:
Kl+r(^VM)iK(4\xfxexp(- WslkT) (5.8)
^ = (v//)8(v/vM1)5'(v/vVM fMexp(-WP/kT) (5.9)
/Caf - (ФУ (v/vxl )*' (v/vvx )4xp (- WAP/kT) (5.10)
Однако в отличие от интерметаллических соединений в случае
ионных кристаллов энергии описываемых реакций включают
энергии образования полностью ионизованных дефектов:
Ws = w**~ + rw£ -Wt (5.11)
Wp-w2k+ + wZ[A~ (5.12)
Mi VM v '
WAF = wZyr + wZx+ (5.13)
XI ' VX v '
132
В кристаллах чистых стехиометрических соединений, в
которых разупорядоченность представлена одной из трех
описанных комбинаций полностью, ионизованных дефектов, их
концентрации связаны между собой условием электронейтральности.
А. Для дефектов Шоттки оно записывается через
абсолютные объемные концентрации вакансий в виде
zMNZ^ = zxN*x+ (5.14)
м VM x VX v '
Подставляя в это уравнение очевидные соотношения
г _ г _ z + г.
NJt = [Vmm]Mj*; N* =[УПМ.х;М.х = (гм/гх)М
VM ~~ L М J^Lm' VX ~~ l X ' LA' LA VM
LM
(где NLm и iVLx — объемные концентрации узлов катионной и
анионной подрешеток), получаем другую форму условия
электронейтральности, связывающего относительные концентрации
вакансий:
L M J L x J
Подставляя это равенство в уравнение закона действия масс
(5.5) и извлекая из него корень степени 1+г, находим
равновесные концентрации дефектов Шоттки в чистом стехиометри-
ческом ионном кристалле:
[V^M"]=[V/+]=^s (5.15)
Аналогично вычисляются и равновесные концентрации френ-
келевских и антифренкелевских дефектов.
Б. Для дефектов Френкеля условие электронейтральности
записывается в виде
j/jvT _ д*м+ (5.16)
VM Mi v '
или при учете соотношений
Л/ум = [Vm] jVLm ; NMi = [Mi] N{; N> = gNw
в виде
Подставляя полученное равенство в (5.6) и извлекая
квадратный корень, получаем для равновесных относительных
концентраций дефектов Френкеля:
[V/~] - g [М*м+] = ygKr (5.17)
где g — число междуузлий, приходящихся на одну формульную единицу МХг.
133
В. Для антифренкелевских дефектов условие
электронейтральности имеет вид
или
r[VZx*+] = g[X**-]
Подстановка в уравнение (5.7) дает для равновесных
относительных концентраций:
[V*x+] = (g/r) [Х*х~ ] = VJjrKAF (5.19)
Формулы (5.17) и (5.19) определяют смысл констант Kf
и Kaf" с точностью до постоянных множителей порядка
единицы они равны равновесным концентрациям френкелевских или
антифренкелевских дефектов в чистом стехиометрическом
кристалле.
Какие из рассмотренных дефектов являются
доминирующими, зависит от энергий их образования. На основании формул
(5.11) — (5.13) следует ожидать, что в плотноупакованных
решетках ионных кристаллов образование дефектов Шоттки
должно быть энергетически более выгодным по сравнению с
образованием дефектов Френкеля и антифренкелевских, поскольку в
случае дефектов Шоттки значительная часть энергии,
затрачиваемой на образование вакансий, компенсируется
отрицательным членом (—Wl) при достраивании решетки на поверхности
кристалла. Это хорошо иллюстрирует табл. 5.1, в которой для
галогенидов щелочных металлов и хлорида серебра приведены
вычисленные значения энергий образования вакансий,
необходимых для удаления иона из узла в вакуум, энергии решетки
Wl, равной работе по разделению кристалла на изолированные
ионы, и результирующие значения энергии дефектов Шоттки
Ws, отнесенные к паре ионов.
Из таблицы видно, что энергия решетки компенсирует
основную часть работы, затрачиваемой на образование пары
вакансий, так что для энергии образования дефектов Шоттки
получаются значения ^2 эВ.
Таблица 5.1. Составляющие энергии образования дефектов Шоттки
в галогенидах щелочных металлов и AgCl (в эВ) [55]
Кристалл
LiF
NaCl
NaBr
KCl
^"VM
6,67
5,03
4,71
4,77
^+VX
6,76
5,11
4,83
4,68
^L
11,09
7,95
7,54
7,20
ws
2,34
2,20
2,01
2,25
Кристалл
KBr
RbBr
AgCl
^"VM
4,53
4,46
5,58
^+VX
4,49
4,36
5,31
WL
6,88
6,72
8,92
ws
2,14
2,10
1,98
134
Таблица 5.2, Энергии образования дефектов Шоттки Ws, Френкеля Wf
и антифренкелевских дефектов Waf в ионных кристаллах (в эВ) [56—61]
Кристалл
NaF
NaCl
NaBr
Nal
KF
KC1
KBr
KI
RbF
RbCl
RbBr
Rbl
CsF
CsCl
CsBr
Csl
T1C1
TIBr
Til
AgCl
AgBr
Agl
CuCl
CaF2
SrF2
BaF2
MgO
CaO
SrO
Ce02
Th02
uo2
xvs
2,49
2,26
2,11
1,77
1,92
2,20
2,13
2,00
1,69
2,05
2,03
1,94
1,8-2,1
1,2-1,8
1,0-1,6
1,1 — 1.0
0,79
0,69
0,64
1,98
—
—
2,27
7,0—8,6
6,9-8,1
6,3-6,9
4—9
5,5-8
5-7
2,0
1,9
—7
wF
3,53
2,88
2,56
2,00
4,27
3,46
3,16
2,73
4,70
3,71
3,35
2,70
—
—
_
—
1,55
0,86
0,21
—
—
0,6—0,8
1,05
6,0
—
—
—
—
—
1 4,2
5,2
| ~10
^AF
3,39
4,60
4,84
5,14
2,57
3,73
4,17
4,26
2,35
3,51
3,68
3,83
—
—
__
—
—
—
—
—
—
—
—
2,6—2,7
2,2—2,4
1,6—2,0
—
—
—
20
16
^9
w
w эксп
2,0—2,4
1,68-1.74
—
2,64
2,2-2,6
2,0—2,5
1,6—2,2
_
2,0—2,1
1,98
1,8—2,1
—
1,1—1,8
2,0
1,9—2,1
0,8—1,3
—
—
1,1—1,7
0,9—1,3
0,6—1,0
—
2,2—3,1
1,7—2,3
1,6—1,9
4,8—5,2
—
—
2,2*
—
——
* Отнесено к трем точечным дефектам.
В кристаллах галогенидов щелочных металлов энергии
образования френкелевских и антифренкелевских дефектов
значительно выше, чем дефектов Шоттки (табл. 5.2), поэтому
разупорядоченность Шоттки для этих соединений при стехио-
метрическом составе является доминирующей.
Хотя во многих кристаллах тип доминирующих дефектов
неизвестен, принято считать, что разупорядоченность Шоттки
является наиболее типичной, а френкелевские и антифренке-
левские дефекты являются скорее исключением, чем правилом.
Разупорядоченность Френкеля доминирует в кристаллах
галогенидов серебра и меди, в которых энергии образования
дефектов Френкеля ниже, чем дефектов Шоттки. Это вызвано тем,
что в кристаллах этих соединений значительная доля энергии
135
связи обусловлена ван-дер-ваальсовыми силами притяжения,
сильно снижающими энергию внедрения катионов Ag+ и Си+
в междуузлия.
Антифренкелевская разупорядоченность наблюдается в
кристаллах галогенидов щелочноземельных металлов, обладающих
структурой типа флюорита CaF2. Это связано со специфическим
строением решетки флюорита, в которой имеются достаточно
просторные междуузлия, по размерам сравнимые с размерами
ионов, благодаря чему внедрение анионов в междуузлия
сопряжено со сравнительно низкими затратами энергии.
В связи с тем что дефекты Шоттки являются наиболее
распространенным типом собственной разупорядоченности ионных
кристаллов, в дальнейшем изложении предпочтение будет
отдаваться этому типу дефектов.
5.2. Примесная ионная разупорядоченность
Наличие эффективных электрических зарядов у точечных
дефектов в ионных кристаллах накладывает жесткое условие
связи между их концентрациями, благодаря которому
обеспечивается электронейтральность кристалла в целом. Поэтому наличие
заряженных примесных центров в ионных кристаллах может
приводить к радикальному изменению их дефектной структуры
в зависимости от значений эффективных зарядов этих центров.
Характер примесной разупорядоченности ионных
кристаллов, так же как и собственной, тесно связан с особенностями
энергетического спектра электронов, прежде всего с большой
шириной запрещенной зоны. Если в полупроводниках наиболее
энергетически выгодным способом компенсации избыточного
заряда примеси является образование дополнительных электронов
в зоне проводимости или дырок в валентной зоне, то в ионных
кристаллах разупорядочение электронов энергетически
невыгодно по сравнению с образованием ионных дефектов.
Действительно, при большой ширине замещенной зоны
переход электрона в зону проводимости требует его размещения
на весьма высоком энергетическом уровне, а образование дырки
в валентной зоне — удаления электрона с весьма низкого
уровня. Оба эти процесса сопряжены, таким образом, со
сравнительно большими затратами энергии. Поэтому в ионных кристаллах
наиболее энергетически выгодным способом компенсации
избыточного заряда примеси является увеличение концентрации
одного из двух типов собственных ионных дефектов,
доминирующих в чистом кристалле, а именно того, эффективный заряд
которого противоположен эффективному заряду примесного
центра.
В предыдущей главе при описании зарядовых состояний
примесных центров в полупроводниках мы подразделяли их на
донорные и акцепторные. Напомним, что свойствами доноров
обладают примеси, имеющие большее число валентных электро-
136
нов, чем замещаемые ими атомы, а свойствами акцепторов —
примеси с меньшим числом валентных электронов. Для
удобства мы сохраним эту терминологию и в случае ионных
кристаллов. Поскольку в дальнейшем будет рассматриваться только
один сорт примесных центров — ионы постороннего металла,
замещающие основные катионы, донорными центрами будут
являться примеси металлов с большей валентностью, чем
основной металл; акцепторными центрами будут примеси металлов с
меньшей валентностью, чем основной. Примерами донорных
центров в ионных кристаллах могут служить примеси металлов
II группы Периодической системы, замещающие в ионном
соединении металл I группы, или примеси металлов III группы,
замещающие металл II группы, и т. д. Примеси металлов I
группы, замещающие металл II группы и т. д., относятся к
акцепторному типу.
В ионных кристаллах основному состоянию доноров отвечает
положительный эффективный заряд, а акцепторов —
отрицательный. Так, при добавлении донорной примеси в ионный
кристалл с дефектами Шоттки компенсация положительного
заряда примеси осуществляется катионными вакансиями. Так как
энергетический уровень примесного донора обычно лежит
вблизи дна запрещенной зоны, а уровень катионной вакансии в
ионных кристаллах расположен гораздо ниже и зачастую близок к
потолку валентной зоны (см. рис. 5.1,6), избыточный валентный
электрон донора в его основном состоянии захватывается
катионной вакансией, обеспечивая ей отрицательный эффективный
заряд. Поэтому в основном состоянии такой примесный
кристалл в отличие от полупроводника не содержит электронов в
зоне проводимости.
Рассмотрим в общем случае бинарный ионный кристалл
МХГ, содержащий в узлах катионной подрешетки примесные
катионы FzF+ с валентностью zF. При полной ионизации примесных
центров эффективный заряд дефектов замещения основных
катионов примесными FMA2± равен Az = zF—zM и может быть как
положительным (при £F>Zm), так и отрицательным (при
£f<Zm). Поэтому условие электронейтральности примесного
ионного кристалла в общем случае произвольной ионной раз-
упорядоченности запишется в виде
zYNZ^ — zMJV*M~ + zM7V*M+ - zyN*~~ + ДгЛ£* d = 0 (5.20)
x VX M VM M Mi ^X XI F
или в атомных долях дефектов
[vz/]-[vy] + g-[M^+]
где коэффициент
Hz
137
(5.21)
^- — 1 (5.22)
2м
означает относительный избыточный заряд примесного центра.
При фиксированном значении концентрации примеси
[FMAz±] уравнения закона действия масс (5.5) — (5.7) совместно
с условием электронейтральности (5.21) образуют полную
систему уравнений относительно неизвестных концентраций
дефектов [VM*M_], [Vx2X+], [Mi*M+]H [Xi*x-]. Решение этой
системы может быть легко найдено для любых кристаллов с
определенным типом собственной ионной разупорядоченности.
Проиллюстрируем это на примере бинарного кристалла с
равными валентностями катионов и анионов (zm=Zx = z; г=1),
обладающего в чистом состоянии разупорядоченностью Шоттки.
В этом случае уравнения (5.5) и (5.21) упрощаются:
[^-КП + Т^Г]^ (5-23)
и после исключения [Vxz_] сводятся к квадратному уравнению
относительно [Vmz~"]
Ю2 - Т [^г±1 К~] - *s = 0 (5.24)
решение которого имеет вид
К1 = (Т/2) [FTl {l ± /1 + (2^/т^мг±])21 (5-25)
Знак перед радикалом выбирается так, чтобы [Умг~] было
положительным.
Полученное решение допускает упрощения в следующих
предельных случаях.
I. Малая концентрация примеси или высокие температуры:
\у\ [;FmA2±]<C2/Cs. В этом случае обеими единицами в фигурных
скобках (5.25) можно пренебречь и решение принимает вид
[VzM-]^[Vf]«/Ts (5.26)
приблизительно отвечающий собственной разупорядоченности
Шоттки в чистом кристалле.
П. Высокая концентрация примеси донорного типа
(дефектов замещения с положительным эффективным зарядом:
FmA2±=Dma*+; Az, y>0)« При выполнении условия у[^мЛ2Г+]>
>2/(s формула (5.25) упрощается до вида
[VZM-] = T[<+] (5.27а)
а из первого уравнения системы (5.23) следует, что
III. Высокая концентрация примеси акцепторного типа
(дефектов замещения с отрицательным эффективным зарядом:
138
FMA2:±=AMA2;~; Az, *y<0). В этом случае перед знаком радикала
в (5.25) следует брать знак (—), а значение корня можно
разложить в ряд по малому аргументу (2Ks/y[FMlz~])2. Органичи-
ваясь линейным членом разложения, получаем:
[Vm~1«-</tHm"]«Ks (5.28а)
а при учете первого уравнения системы (5.23):
К+]«-Т[АГ1 (5-28б>
При Az=y = 0 рассмотренная система уравнений вырождается
в систему, отвечающую собственной разупорядоченности и
исследованную в предыдущем разделе. Поэтому случай равнова-
лентной примеси, zF=zM, не приводит к новым решениям;
влияние равновалентной примеси сводится лишь к искажению
различных энергетических параметров и в рамках излагаемой здесь
теории игнорируется.
Приведенные формулы для упрощенных решений
показывают, что при любой фиксированной концентрации примеси
ионный кристалл может иметь либо собственную, либо примесную
разупорядоченность в различных температурных интервалах.
Примесная разупорядоченность, определяемая предельными
решениями (5.27), (5,28), реализуется при достаточно низких
температурах, при которых процесс собственного разупорядоче-
ния не вносит существенного вклада (константа Шоттки Ks
мала). Напротив, при высоких температурах (больших
значениях Ks) собственное разупорядочение является доминирующим и
практически подавляет влияние примеси. В этом отношении
поведение ионных кристаллов аналогично поведению
полупроводников, подробно рассмотренному в предыдущей главе.
Критическая температура Гкр, при которой примесная
разупорядоченность переходит в собственную, определяется условием
1т|[Рм±] = 2/Г5(Гкр) (5.29)
и может быть определена уравнением, аналогичным (4.21) для
полупроводников. Качественное отличие от полупроводников
состоит в том, что энергия реакции собственного разупорядочения
полупроводников, определяемая шириной запрещенной зоны, в
данном случае заменяется константой равновесия реакции
образования пары собственных дефектов Шоттки.
Мы рассмотрели подробно решение системы уравнений для
разновалентного кристалла с дефектами Шоттки. Для
кристаллов с различными валентностями катионов и анионов система
сводится к уравнениям, более сложным, нежели квадратное, и в
общем случае не имеет аналитического решения. Однако
асимптотическое поведение этого решения легко получить из общей
системы уравнений (5.5) —(5.7) и (5.21) путем упрощения усло-
139
вия электронейтральности (5.21). В предельном случае высоких
концентраций примеси можно считать, что избыточный заряд
примесных центров (эффективный заряд дефектов замещения
Дг±) целиком компенсируется эффективным зарядом
собственных дефектов (вакансий или междуузельных ионов
соответствующего знака). Поэтому, оставляя в уравнении (5.21) по паре
компенсирующих друг друга слагаемых, для кристаллов с
различными типами собственной ионной разупорядоченности
получаем следующие приближенные решения.
А. Разупорядоченность Шоттки при произвольных
валентностях компонентов zM, zx, zF. В случае примеси донорного типа
(Дг, y>0) уравнение (5.21) принимает простой вид
^мМ~]*Т[В*г±] (5.30)
совпадающий с (5.27а) для кристалла с равновалентными
компонентами. В случае примеси акцепторного типа (Дг, Y<0)
уравнение (5.21) принимает форму
[v"Mx+]«-T[A^-] (5.31)
совпадающую с (5.286).
Б. При преобладании френкелевских или антифренкелевских
дефектов эффективные заряды собственных дефектов совпадают
по абсолютному значению, благодаря чему в уравнениях (5.6)
и (5.7) не содержится отношения валентностей r = zM/£x.
Благодаря этому решения системы уравнений оказываются
совершенно аналогичными рассмотренным выше для равновалентного>
кристалла с дефектами Шоттки. Они могут быть получены из
решений (5.25) — (5.28) непосредственной заменой [Vm2-]—»--
—>[XizX~], [Vxz+]—^[Mi2M+] с точностью до коэффициентов,
g", г и у, значения которых «1.
Изложенную теорию- легко применить и к случаю, когда
ионный кристалл легируется по анионной подрешетке. Для этого во
всех приведенных уравнениях достаточно поменять местами
катионы и анионы, сменив при этом знаки у показателей
эффективных зарядов дефектов.
На основании изложенного можно сделать важный в
практическом отношении вывод: легирование ионных кристаллов ино-
валентными примесями позволяет контролировать концентрации
дефектов (вакансий или междуузельных ионов), ответственных
за транспортные процессы, и таким образом регулировать в.
весьма широком диапазоне совокупность их физико-химических
свойств. В этом отношении ситуация аналогична рассмотренной
в предыдущей главе для полупроводников; выводы теории
указывают пути сознательного воздействия на материалы с целью-,
придания им нужных физико-химических свойств
140
5.3. Ассоциация дефектов
в примесных ионных кристаллах
Решения, полученные в предыдущем разделе, показывают, что
при легировании ионных кристаллов иновалентной примесью
можно достичь очень высоких концентраций противоположно
заряженных дефектов. Так, диоксид циркония, широко
используемый в качестве твердого электролита, допускает растворение
в катионной подрешетке ионов иттрия от 14 до 67 атомных
процентов. Поекольку иттрий входит в решетку в виде трехзарядных
ионов, на фоне четырехзарядных ионов циркония он образует
дефекты замещения с эффективным зарядом —1. Компенсация
этого заряда осуществляется анионными вакансиями с
эффективным зарядом + 2. Поэтому условие электронейтральности
твердого раствора Zr02-rY203 имеет вид NY~=2Nv02+, или, в
атомных долях, i[YZr~]=4[Vo2+] (коэффициент 2 заменен на 4, так
как концентрации узлов анионной подрешетки вдвое выше, чем
катионной: Nlo = 2NLzt). Таким образом, в области
существования твердых растворов атомная доля вакансий кислорода
изменяется в пределах от 3,5 до 17% (ат). Аналогичной
особенностью обладает и диоксид циркония, содержащий примесь
двухвалентного кальция; его отличие заключается в том, что
дефекты замещения CaZr2~ имеют эффективный заряд —2.
В области столь высоких концентраций заряженных дефектов
становится важным их кулоновское взаимодействие [36],
которое тем сильнее, чем меньше среднее расстояние между
частицами, т. е. чем выше концентрация примеси. Благодаря силам
кулоновского притяжения противоположно заряженные
дефекты— центр замещения и вакансия — стремятся расположиться
как можно ближе друг к другу. Поэтому при достаточно низких
температурах наиболее устойчивы будут конфигурации, при
которых примесный ион и вакансия находятся в соседних узлах,
образуя своего рода квазимолекулу, которую принято называть
комплексом. Для отрыва вакансии от примесного центра и
удаления их на большое расстояние, другими словами, для
диссоциации комплекса, необходимо затратить энергию, равную
энергии их взаимного притяжения.
При повышенных температурах процесс диссоциации
комплексов может осуществляться за счет энергии тепловых
флуктуации. В этом отношении процесс диссоциации комплексов в
ионных кристаллах аналогичен процессу электролитической
диссоциации солей, кислот или оснований, хорошо известному из
электрохимии растворов электролитов.
Рассмотрим подробнее случай ионного кристалла,
содержащего в узлах катионной подрешетки примесь повышенной
валентности, образующей положительно заряженные дефекты
замещения донорного типа 1>мЛг+, заряд которых компенсируется
катионными вакансиями с отрицательным эффективным
зарядом VM2M~. При этом будем для простоты считать эффективные
141
заряды обоих типов доминирующих дефектов равными по
абсолютному значению Az=zM=z; у=1. Примером такой системы
может служить твердый раствор iNaCl + MgCb с
доминирующими дефектами Mg+Na и V~Na-
Обратимая реакция ассоциации — диссоциации комплекса
(DmVm)x описывается квазихимическим уравнением
(Dm • VM]X -^± D^+ + Vz~ (5.32)
здесь косой крестик у символа комплекса указывает на его
электрическую нейтральность. Применим к этой реакции
уравнение закона действия масс в форме (2.28):
[Dm+]|V^]=^[(DmVm)x] (5.33)
где /<а — константа равновесия реакции ассоциации — диссоциации.
Согласно формуле (2.32) величина Ка связана с энергией
диссоциации комплекса Е соотношением
Кл = *(Т)ехр(-Е1кТ) (5.34)
Если исключить из рассмотрения область крайне малых
концентраций примеси, отвечающую собственной разупорядоченно-
сти [в случае кристалла с дефектами Шоттки — решение (5.26)],
и ограничиться областью примесной разупорядоченности, в
которой концентрации анионных вакансий или междуузельных
катионов подавлены высокой концентрацией дефектов
замещения, то для решения задачи необходимо составить систему трех
уравнений относительно неизвестных концентраций свободных
дефектов [JDmz+] и [Vm*-] и комплексов [(DmVm) хЬ Одним из
уравнений такой системы является уравнение (5.33), два других
вытекают из уравнения электронейтральности:
Рм+1 = № (5-35)
и из выражения для полной концентрации примеси:
P^] + [(DmVm)x] = [D] (5.36)
в котором полная концентрация примеси [D] считается
фиксированной и известной.
Подставляя в уравнение (5.33) значения [Омг+] и
i[(DmVm)x] из (5.35) и (5.36), получаем квадратное уравнение
относительно концентрации свободных вакансий:
KU2 + Ка [Vf] - Ка [D] = 0 (5.37)
решение которого имеет вид
ГО = КЬ = (*а/2) {- 1 + 1Л + 4Р]/*.} (5-38)
Найденное решение допускает упрощения в следующих
предельных случаях.
142
При достаточно малых значениях концентрации примеси или
высоких температурах, 4[Ю]<СКа, формула (5.38) дает после
разложения квадратного корня в ряд:
КЛ = Р^«Р] (5.39)
Полученное приближенное решение совпадает с найденным в
предыдущем разделе решением (5.27а), отвечающем
предельному случаю II. Из уравнения (5.33) находим, что концентрация
комплексов равна
[(DMVM)x] = [D]2/^a (5.40)
и в силу принятого неравенства мала по сравнению с общей
концентрацией примеси [D].
Таким образом, при сравнительно низких концентрациях
примеси основная ее часть находится в виде свободных дефектов
замещения [Dm2+], а концентрация нейтральных комплексов
пренебрежимо мала. Другими словами, рассмотренный случай
отвечает полной диссоциации комплексов. Здесь, однако,
следует помнить о том, что концентрация примеси должна
оставаться большой по сравнению с константой собственной разупо-
рядоченности Ks, Kf или Kaf, в противном случае влияние
примеси вообще пропадает и в кристалле будет доминировать
собственная разупорядоченность (предельный случай I в
предыдущем разделе). Поэтому более строгое условие, при котором в
примесном кристалле реализуется решение (5.39), следует
записывать в виде
tfs«[D]«/fa (5.41)
Такому условию всегда можно удовлетворить, поскольку
вследствие сравнительной малости энергии диссоциации Ka^>Ks.
Противоположное условие согласно принятой в предыдущем
разделе нумерации нужно отнести к следующему предельному
случаю.
IV. Очень высокие концентрации примеси или низкие
температуры, 4[D]>/Ca. При этом формула (5.38) упрощается до
вида
JL -L
№ = [DJ+] « ([D] /Q 2 = (X [D])2 exp (-E\2kT) (5.42)
В соответствии с принятым неравенством, указанные
концентрации свободных дефектов малы по сравнению с общей
концентрацией примеси. Другими словами, примесь находится
преимущественно в ассоциированном состоянии, т. е. имеется
значительный резерв комплексов, аналогичный резерву нейтральных
доноров в полупроводнике (предельный случай III в
разделе 4.2).
Мы подробно проанализировали решение системы уравнений
для равных абсолютных значений эффективных зарядов доми-
143
нирующих дефектов — примесных центров донорного типа и ка-
тионных вакансий. Легко видеть, что полученные соотношения
остаются справедливыми и для примесных центров акцепторного
типа и анионных вакансий, если их эффективные заряды
по-прежнему равны по абсолютной величине. В этом случае
решения могут быть получены из приведенных выше путем
прямой замены донорной примеси на акцепторную и катионной
вакансии на анионную. Примером такой системы может
служить твердый раствор Zr02 + CaO, в котором акцепторными
центрами являются дефекты замещения Cazr2-, а их заряд
компенсируется вакансиями кислорода Vo2+. В таком твердом
растворе при сравнительно небольших концентрациях кальция и
высоких температурах реализуется решение (5.286), отвечающее
предельному случаю II (истощение комплексов, [CaZr2~] « [Са];
У=—V2):
2[V2+]^[Ca] (5.43)
В области высоких концентраций кальция и низких
температур реализуется пятый предельный случай, аналогичный IV и
отвечающий резерву нейтральных комплексов. По аналогии с
решением (5.42) можно записать:
V. 4[Са]»/С; [CaZrV0 ]х « [Са]
г_ (5-44)
[V20+] - [Са£г] «(х [Са])2 ехр (- E/2kT)
Характерной особенностью всех рассмотренных случаев
является то, что в условие электронейтральности входили только
концентрации свободных дефектов, не ассоциированных в
комплексы, комплексы же являлись электрически нейтральными
квазичастицами. Однако в некоторых примесных системах
эффективные заряды дефектов замещения и компенсирующих их
вакансий не совпадают по абсолютному значению. В этом случае
простейшие парные комплексы, включающие один ион примеси
и одну вакансию, обладают отличным от нуля эффективным
зарядом, поэтому их концентрация входит в уравнение
электронейтральности кристалла. Это приводит к решениям, отличным
от рассмотренных выше.
Проиллюстрируем это на примере твердого раствора
Zr02-f-Y203, в котором при отсутствии комплексообразования
(предельный случай III) доминирующими дефектами являются
неассоциированные дефекты замещения Y~~zr и вакансии
кислорода, причем условие электронейтральности имеет упрощенную
форму:
4[V20+]^[Y-]«[Y] (5.45)
При ассоциации указанных дефектов они могут образовывать
парные комплексы (YZrVo)+ с эффективным зарядом +1 или
144
тройные комплексы (2YzrVo) x с нулевым эффективным
зарядом. Свободные вакансии кислорода в этом случае возникают
при диссоциации парных комплексов:
(YZrV0)+ ^zh Y" + V2+ (5.46)
которой отвечает уравнение закона действия масс:
[Y-][V20+]=A-a[(YZfVo)+] (5.47)
Решение нового, шестого типа получается в предельном
случае сильной ассоциации дефектов, когда концентрация
свободных вакансий становится пренебрежимо малой, а условие
электронейтральности принимает упрощенную форму:
VI. [(YzrVo)+]«[Y-] (5.48)
При учете последнего равенства из уравнения (5.47) получаем:
[ V20+] ~ Kt = г exp {-ElkT) (5.49)
т. е. концентрация свободных вакансий кислорода не зависит от
содержания примеси иттрия.
Полученные зависимости концентраций свободных вакансий
от концентрации примесей донорного и акцепторного типа
схематически изображены на рис. 5.2 для ионного кристалла,
обладающего в чистом состоянии дефектами Шоттки; римскими
цифрами обозначены области существования соответствующих
предельных решений.
Обращает на себя внимание подобие приведенных кривых
кривой, изображенной на рис. 4.1 для концентрации электронов
проводимости в примесном полупроводнике. Это подобие не
является случайностью и обусловлено тем, что как в
полупроводниках, так и в ионных кристаллах возбуждение
собственных дефектов (электронных или ионных) происходит в резуль-
■РИС. 5.2.
Зависимость концентрации вакансий в ионном кристалле с дефектами Шоттки
от концентрации примесей донорного или акцепторного типа:
I — область собственной разупорядоченности; II и III — области примесной разупорядо-
ченности при отсутствии ассоциации дефектов; IV—VI — области примесной
разупорядоченности при резерве комплексов.
10-321
145
.
V
Ws\
wrjk
1
1
Jn,nr
\1
X
t\
\
\
\
\
I
1
1
'F,TZ
^\
|SEN
I
^/f 1
к |
■■» >
РЯС. 5.3.
Температурная зависимость концентрации
доминирующих вакансий в ионном кристалле с дефектами
Шоттки.
гате тепловых флуктуации и, таким
образом, имеет сходную физическую природу.
Аналогия полученных решений с
решениями для полупроводников становится еще
более наглядной при сравнении
температурит- ной зависимости концентрации
доминирующих вакансий в примесном ионном
кристалле, изображенной на рис. 5.3, с температурной зависимостью
концентраций электронов проводимости в полупроводнике (см.
рис. 4.2). Видно, что энергия собственного разупорядочения в
ионном кристалле (энергия образования дефектов Шоттки Ws)
играет ту же роль, что и энергия собственной ионизации
(ширина запрещенной зоны Ес—Ей) в полупроводнике, а энергия
диссоциации комплекса аналогична энергии ионизации доноров
или акцепторов.
5.4. Разупорядоченность чистых
нестехиометрических ионных кристаллов
В начале данной главы отмечалось, что в кристаллах с
преобладающей ионной связью основное зарядовое состояние атомных
дефектов отвечает классической ионной модели, так что
абсолютные значения их эффективных зарядов совпадают со
значениями валентностей соответствующих элементов. Это
обстоятельство учитывалось и при дальнейшем описании как
собственной, так и примесной разупорядоченности ионных
кристаллов, состав которых определяется точными стехиометрическими
отношениями.
Однако, как было подробно описано в гл. 1 (см. раздел 1.7),
в нестехиометрических кристаллах разупорядочение электронов
приводит к существованию ионных дефектов с различными
степенями ионизации, так что их эффективные заряды могут
принимать любые целочисленные значения от нуля до основного,
равного валентности соответствующего элемента.
Таким образом, рассматривая в данном разделе чистое не-
стехиометрическое ионное соединение МХг±а, будем
предполагать существование в нем следующих ионных дефектов: катион-
ных вакансий Vm^~~ (£=0,1, ...,2м), анионных вакансий Vx^+
(£ = 0,1, ...,zx), междуузельных катионов Mi^+ (£=0,1, ...,Zm) и
междуузельных анионов Х£~ (£=0,1,..., zx).
Образование этих дефектов может протекать как путем
собственного разупорядочения, рассмотренного в разделе 5.1, так
и при обмене кристалла атомами неметалла X с окружающей
146
газовой фазой, аналогичном описанному в разделе 4.4 для не-
стехиометрических полупроводниковых соединений. Запишем
основные квазихимические реакции, осуществляющие такой
обмен.
A. Образование на поверхности кристалла новой формульной
единицы МХГ, при котором катион переходит из узла в объеме
кристалла, а г анионов возникают путем адсорбции и
ионизации г/2 молекул Х2 из газовой фазы:
^Х2:^МХг + МПс~ + Се+, или -~-X2^i± rX^+VT+C^
(5.50а)
Эффективный заряд возникающей при этом катионной вакансии
в объеме кристалла компенсируется £ электронными дырками в
валентной зоне, так что кристалл приобретает электронную
проводимость р-типа.
Б. Переход аниона из узла в объеме кристалла в газовую
фазу:
нуль +± — Х2 + ХПС+ + С*", или Х£ q=± - X,+Vx+ +r,e~
(5.506)
Эффективный заряд возникающей при этом анионной вакансии
компенсируется соответствующим количеством электронов,
появляющихся в зоне проводимости.
B. Разрушение на поверхности кристалла формульной
единицы МХГ, при котором катион переходит в междуузлие в
объеме, а анионы превращаются в г/2 молекул Х2 в газовой фазе:
МХГ ^1— Х2 + MQC+ + С£~, или Мм + гХ$ +
+ Vх ^±~Х2 + М?+ + 1е~ (5.50в)
Эффективный заряд междуузельного катиона компенсируется
электронами проводимости.
Г. Переход неметалла X из газовой фазы в междуузлие в
объеме кристалла:
— Х2 +т XOc~ + te+, или — Х2 + Vix ^ Х:"+ Св+ (5.50г)
Компенсация заряда междуузельных анионов осуществляется
электронными дырками.
Здесь, как и в случае нестехиометрических
полупроводниковых соединений, предполагается, что кристалл находится в
термодинамическом равновесии с окружающей его газовой фазой,
содержащей неметалл Х2. Поэтому реакции (5.50) могут
протекать обратимо. Кроме того, здесь, как и в предыдущей главе,
мы рассматриваем только невырожденный электронный газ, для
10*
147
которого оправдано использование уравнений закона действия
масс в их обычной форме.
Применяя закон действия масс к уравнениям перечисленных
реакций, получаем уравнения для концентрации всех
рассматриваемых дефектов:
[v£
[^
-}[е+
}[е-
[МП [е~
к
}[е+
f =
]' =
■]С =
? =
Сух
^Xi
Pi
2
Px2
—г
pI
1
pI
К этим уравнениям следует также добавить уравнение закона
действия масс (4.2) для реакции собственной ионизации
кристалла, связывающее концентрации электронных дефектов:
[е-] [е+] = К? (5.52)
а также условие электронейтральности:
гм гх
=г 2с i vx+i + * 2; [м>+] +[е+] (5-53)
С=1 С=1
которое является обобщением уравнения (4.84) для случая
произвольных эффективных зарядов ионных дефектов. Здесь, как
и ранее, коэффициент g равен отношению числа междуузлий к
числу узлов катионной подрешетки, а коэффициент r=zM/zx —
отношению чисел узлов анионной и катионной подрешеток.
Уравнения (5.51) — (5.53) образуют полную систему
относительно неизвестных концентраций [W~~], [Vxs+], fMiS+],
[Xi^~" ], [e~] и [е+], где степени ионизации £ имеют, вообще
говоря, произвольные целочисленные значения от нуля до
валентности соответствующего элемента. Однако при дальнейшем
решении этой системы уравнений целесообразно исключить из нее
концентрации незаряженных атомных дефектов (£ = 0),
поскольку они не входят в условие электронейтральности (5.53).
Решения же для концентраций незаряженных атомных дефектов
получаются непосредственно из уравнений (5.51). Действительно,
при £ = 0 в этих уравнениях концентрации [е~] и [е+] исчезают
148
и, например, для концентраций нейтральных вакансий получаем
решения
г_ _-1
Ю = с£„/£; [Vyx] = c*xPl
совпадающие с формулами (4.52), полученными для нестехио-
метрического полупроводника.
Таким образом, в дальнейшем мы будем интересоваться
только концентрациями заряженных ионных дефектов, для
которых степени ионизации £ принимают целочисленные
значения от единицы до валентности соответствующего элемента.
Для решения такой системы уравнений поступим так же,
как в случае нестехиометрического полупроводника (см.
раздел 4.4): разобьем весь интервал изменения парциального
давления неметалла в газовой фазе рХ2 на области, в которых
условие электронейтральности имеет настолько упрощенный вид, что
в левой и правой его части остается лишь по одной неизвестной
переменной — концентрации тех или иных доминирующих
дефектов с противоположными эффективными зарядами, и таким
образом переберем все сочетания противоположно заряженных
дефектов.
В рассматриваемом случае все приближенные решения
разбиваются на три группы.
I. Первую группу составляют решения, отвечающие области
промежуточных давлений неметалла, при
которых концентрации электронов проводимости и дырок
пренебрежимо малы по сравнению с концентрациями ионных дефектов.
Очевидно, что именно решения этой группы должны описывать
собственную разупорядоченность ионных кристаллов вблизи
стехиометрического состава, где электронная
разупорядоченность отсутствует.
Условию малости концентраций электронных дефектов
удовлетворяют три сочетания противоположно заряженных
дефектов в условии электронейтральности:
a. :i[v^-]«^[v^]
Б. :ЛУ^]^^[М^[ (5.54)
В. C^[X^]^C2r[V^+]
описывающие соответственно обобщенные типы разупорядочен-
ности Шоттки, Френкеля и антифренкелевскую, включающие
ионные дефекты с произвольными степенями ионизации.
Четвертая форма упрощенного условия электронейтральности
Ct [XjH « С2 [МГ+] (5.55)
соответствует гипотетическому антишотткиевскому типу разупо-
рядоченности и, как отмечалось в гл. 3, в твердых телах не
наблюдается.
149
Концентрации доминирующих дефектов (5.54) легко
определить с помощью их подстановки в уравнения (5.51). В общем
случае они зависят от парциального давления неметалла в
газовой фазе (см. табл. 5.3). Характер этой зависимости
определяется соотношением между эффективными зарядами
компенсирующих друг друга дефектов. Однако, как уже указывалось
в начале главы, в ионных кристаллах при небольших
отклонениях от стехиометрического состава эффективные заряды
дефектов практически всегда совпадают по абсолютному значению
с валентностями соответствующих элементов.
Таким образом, вблизи стехиометрического состава для
чистых ионных кристаллов практическое значение имеют лишь
сочетания из четырех типов полностью ионизованных дефектов:
[Vm2M~], [VxzX+], [MizM+] и lXizX-], т. е. обычные дефекты Шот-
тки, Френкеля и антифренкелевские дефекты, отвечающие
классической ионной модели. Соответствующие решения
[Vm4~]«[VZxx+]«/Cs
L (5.56)
[vifWfM^]^2^
[V"XX+] « (glr) [Х*~] « (glrfKAF
подробно проанализированы в разделе 5.1 при описании
собственной разупорядоченности стехиометрических ионных
кристаллов и в данном случае отличаются от них только приближенным
характером соотношений (5.56), вытекающим из
приближенности условий электронейтральности (5.54).
Из общестатистических соображений ясно, что даже при
строго стехиометрическом составе кристалла концентрации
электронных дефектов отличны от нуля. Количественно это
выражается уравнением (5.52), согласно которому произведение
концентраций электронов проводимости и дырок равно «квадрату
константы собственной ионизации /G2. Концентрации обоих
типов электронных дефектов равны друг другу при некотором
определенном составе, близком к стехиометричеокому, и сильно
изменяются при смещении в обе стороны от стехиометрического
состава. Если парциальное давление неметалла в газовой фазе
Рх2 значительно превышает величину Рстех, отвечающую стехио-
метрическому составу, доминирующими реакциями,
определяющими характер электронной разупорядоченности, являются
реакции поглощения сверхстехиометрического неметалла (5.50а)
или (5.50г), приводящие <к появлению дырок в валентной зоне
кристалла. Напротив, при давлениях, значительно меньше
Рстех, доминирующими являются реакции (5.506) и (5.50в),
приводящие к потере неметалла и появлению электронов в зоне
проводимости.
150
Зависимость концентраций электронов проводимости и дырок
от парциального давления неметалла в газовой фазе в общем
случае рассматриваемой группы решений легко найти из
уравнений (5.51) и (5.52) при учете соотношений (5.54).
A. Для кристалла с преобладающими дефектами Шоттки
произвольной степени ионизации путем деления друг на друга
первых двух уравнений системы (5.51) можно исключить
концентрации обоих сортов вакансий:
к+]:,КГС!-С,й (5.57)
отсюда с помощью соотношения (5.52) получаем:
[е+]^А+р™'^ (5.58а)
В этих формулах значения эффективных зарядов £i и £2
относятся соответственно к катионным и анионным вакансиям:
£i = 1,...,zm; £2 = 1,..., Zx; величины С, А- и Л+ — некоторые
экспоненциальные функции температуры, представляющие собой
определенные комбинации констант равновесия.
Аналогичным образом получаем решения для концентраций
электронных дефектов и в случае кристаллов с другими типами
ионной разупорядоченности при произвольных степенях
ионизации ионных дефектов.
Б. Дефекты Френкеля:
Ci[v^-]«:2£[M^]
(где Ci, С2= 1,.. •, zM)
[е+] « A Vx?'+r2)
B. Антифренкелевские дефекты:
(где ;,, С2 = 1, • • •, гх)
(5.586)
(5.58в)
Полученные решения показывают, что при любом типе
доминирующей ионной разупорядоченности концентрация
электронов проводимости уменьшается, а дырок — растет с ростом
парциального давления неметалла в газовой фазе. В наиболее
важных случаях, когда доминируют полностью ионизованные дефек-
151
ты, из всех приведенных решений вытекает одинаковая
зависимость концентрации электронных дефектов от давления
неметалла:
г J I ±ll2zx
[п- }~Рх.2
Впрочем, эта зависимость получается непосредственно из
любого уравнения системы (5.51), если заменить в нем
эффективный заряд ионного дефекта на значение валентности
соответствующего элемента и учесть, что согласно решениям (5.56)
концентрации полностью ионизованных дефектов не зависят от рХ2.
Приведенные решения первой группы справедливы в
ограниченной области парциальных давлений неметалла, в которой
концентрации электронных дефектов остаются малыми по
сравнению с постоянной концентрацией доминирующих ионных
дефектов. При некотором значении давления р+^>Рстех
концентрация дырок сравнивается с концентрацией ионных дефектов и
приведенные решения утрачивают силу. То же происходит при
некотором значении давления Р~<С^стех, при котором
концентрация электронов проводимости достигает значения, равного
концентрации ионных дефектов. Таким образом, при давлениях,
более высоких, чем Р+, и более низких, чем Р~, реализуются
решения, относящиеся к другим группам.
П. Область высоких давлений неметалла. К этой
группе относятся решения, отвечающие комбинированной ион-
но-электронной разупорядоченности, когда доминирующими
являются ионные дефекты с отрицательным эффективным зарядом,
а компенсирующими их заряд дефектами-—электронные дырки.
Условие неэлектронейтральности в этом случае имеет две
упрощенные формы, подстановка которых в уравнения (5.51)
позволяет непосредственно рассчитать концентрации доминирующих
дефектов.
A. Для кристаллов, обладающих при стехиометрическом
составе дефектами Шоттки, и (Б) для кристаллов, обладающих
дефектами Френкеля, в области высоких давлений неметалла
реализуется одно и то же приближенное решение (при
£=1, ...,2м):
С [V£] - [е' ] « (С^м )1/(:+1) pi ' " (5.59а, б)
B. Для кристаллов, обладающих при стехиометрическом
составе антифренкелевскими дефектами, реализуется решение
('при £=l,...,zx):
Cg[X;n^k+]«(C^)1,(c+1)px2i (5.59b)
III. Область низких давлений неметалла. К этой
группе относятся решения, при которых доминирующими явля-
152
ются ионные дефекты с положительным эффективным зарядом и
электроны проводимости.
A. Для кристаллов, обладающих при стехиометрическом
составе дефектами Шоттки, упрощенное решение в области низких
давлений имеет вид (при £=1, ...,zx):
О [Vcx+] - [е~] - (С* )1/(:+1) р^_ (5.60а)
Б. Для кристаллов, имеющих при стехиометрическом
составе дефекты Френкеля, реализуется упрощенное решение (при
£=1,...,2м):
-гС+1>
С£ [М;+] « [е'\ - (С£ )1/(с+1) Pi (5.606)
B. Для кристаллов с преобладающими антифренкелевскими
дефектами решение совпадает с (5.60а).
Все решения для концентраций электронов проводимости и
дырок, определяемые формулами (5.58) — (5.60), можно
представить одной общей формулой (в абсолютных (концентрациях):
1
n=A(T)plexp(-slkT) (5.61)
совпадающей с формулой (4.92) для нестехиометрических
полупроводников. Здесь А (Т)—некоторая сравнительно слабая
степенная функция температуры; е — эффективная энергия
активации процесса, приводящая к образованию электронных
дефектов. Показатель степени при парциальном давлении рХ2
всегда положителен для дырок и отрицателен для электронов
проводимости.
Значения характеристического параметра m определяются
валентностями и эффективными зарядами доминирующих
ионных дефектов; они сведены в табл. 5.3.
Подвижность электронных носителей тока — электронов
проводимости и дырок обычно на несколько порядков выше
подвижности ионных дефектов. Поэтому в области малых
отклонений от стехиометрического состава, где реализуются решения
группы I, электронная составляющая проводимости, вообще
говоря, сравнима с ионной. Так, галогениды щелочных металлов,
серебра и меди в воздухе или в вакууме являются чисто
ионными проводниками; в парах соответствующего металла или в
атмосферах, содержащих галоген, их электронная проводимость
имеет приблизительно тот же порядок, что и ионная. Чистые
оксиды при составах, близких к стехиометрическому, в
большинстве случаев являются смешанными ионно-электронными
проводниками.
Напротив, при больших отклонения от стехиометрического
состава, где реализуются решения II и III групп, концентрации
доминирующих ионных и электронных носителей одинаковы,
153
Таблица 5.3. Значения параметра m в формуле (5.61)
для концентраций электронных дефектов
в чистом нестехиометрическом ионном соединении МХг±б
Тип собственной ра-
зупорядоченности
при стехиометриче-
ском составе
Шоттки
Френкеля
Антифренкелевская
Решение
IA
НА
ША
1Б
ПБ
ШБ
IB
ПВ ,
ШВ
Условие электронейтральности
Si[^MSn = lAVxli+]
То же при gj = гм, £2 = гх
ЦУ^~] - [е*]
MVxl+\ = [е-]
Ci[HmCH = ШЩ1г+}
То же при £х = £2 = гм
№*П = [«-■]
ШМр-] = Щ
U[Xill+\ = ZAVxU+]
То же при £х = £2 = гх
ШХр-] = Р]
tr[VxZ+] = [е-]
m
±~ 1 -\-г
±2гх
—(£+1)
-2(2 +1)
£i + S2
±2гх
2
2
±(Ei + Es)
±2гх
2(С+1)
-2(£+1)
поэтому в силу большей подвижности последних нестехиометри-
ческие соединения в этой области составов являются
практически чисто электронными проводниками п- или р-типа.
В данном разделе мы опустили еще одно упрощенное
решение:
[е-] - [*+] - AT,
(5.62)
которое следовало бы выделить в четвертую отдельную группу.
Однако это решение типично для собственных полупроводников
и было подробно проанализировано в предыдущей главе. Для
ионных кристаллов, таких, как галогениды и оксиды металлов,
в которых ширина запрещенной зоны очень велика (^5—
10 эВ), прямое возбуждение электронных дефектов путем
собственной ионизации не играет сколько-нибудь существенной
роли, и решение (5.62) в них не реализуется.
Проиллюстрируем полученные решения на примерах трех
типов соединений с определенными отношениями валентностей
катионов и анионов.
1. Одно-одновалентное соединение MX (гм = гх=1;
г=1) с дефектами Шоттки. К этому типу соединений относят-
154
ся галогениды щелочных металлов; их ионная разупорядочен-
ность при стехиометрическом составе была обсуждена в
разделе 5.1. При Zm=Zx = 1 ионизированные вакансии могут
находиться в единственном зарядовом состоянии, £i = £2 = l, поэтому для
области I, отвечающей промежуточным значениям рх2, решения
(5.56а) и (5.58а) записываются в форме
(5.63)
[V-] « [V+] - Ks
1
1
[e-]^A-pXi* ; [<Г] =
j
*a+pI
Для областей II и III, отвечающих соответственно высоким и
низким значениям рх2, из решений (5.59а) и (5.60а) получаем:
II. [V-]^[e+)~(C-M)2 pi (5.64)
III. [V+]«[e_]«(C+ )2 pi (5.65)
Если прологарифмировать соотношения (5.63) — (5.65), то
получаются уравнения, связывающие логарифмы концентраций
дефектов с логарифмом парциального давления неметалла:
I. igra«ig[v+]«ig*s
lg [e~] = const — -~ lg pXa (5.66)
lg [е+] = const + ~~lSPx,
И- lg TO «lg[e+]« const + -flg/>x, (5.67)
III. lg[V+] «lg[e-]* const—i-lgpXi (5.68)
Геометрически они представляют собой уравнения прямых
линий в координатах логарифм концентрации — lgpx2 с
различными угловыми коэффициентами. Соответствующие графики
схематически изображены на рис. 5.4.
Интересно сравнить этот рисунок с рис. 4.5, на котором
изображены аналогичные графики для нестехиометрического
полупроводника. При сравнении бросается в глаза аналогичный ход
прямых для доминирующих заряженных дефектов в каждой из
различных областей давлений I—III. Существенное отличие
обоих типов нестехиометрических соединений наблюдается только
в области I, прилегающей к стехиометрическому составу. Здесь
оба соединения имеют различный тип доминирующих
собственных дефектов: полупроводник — электронные дефекты, ионный
кристалл — ионные. В областях же II и III при больших откло-
155
hgp- lgPcme* l3P+ lgp/2
РИС. 5.4.
Зависимость концентраций дефектов в 1,1-валентном нестехиометрическом
ионном кристалле MXi±6 с дефектами Шоттки от парциального давления Хг
в газовой фазе.
нениях от стехиометрического состава собственная разупорядо-
ченность утрачивает доминирующую роль по сравнению с разу-
порядоченностью, вызванной нестехиометричностью и, тип
преобладающих дефектов в обоих соединениях совпадает.
Отличие в ходе прямых для недоминирующих дефектов,
наблюдаемое на обоих рисунках, связано с различными
соотношениями констант равновесия К\ и константы Шоттки для
заряженных вакансий: если для полупроводника /Cs+<CKb то для
ионного кристалла Ks^Ki-
2. Соединение М^хХ с одновалентными катионами
(2м=1; r=l/zx) и дефектами Френкеля. В этом случае
доминирующие ионные дефекты также имеют только одно
зарядовое состояние £i = g2 = 1 - Поэтому решения (5.566) и (5.586)
для области промежуточных давлений Х2 записываются в форме
i- [v-]«g-[M+]«g-2/cF
_r ^ (5.69)
le-\~A-pl; [e+]~A+pl
Решения для областей больших и малых давлений получаем из
(5.596) и (5.606):
II. [\f-]~[e+)^(C-M)2 pi (5.70)
Ш.£[М+]«К-]«(С+)2 pi (5.71)
При 2Х=1; r=l (галогениды) полученные решения
совпадают с решениями (5.63) — (5.65) для 1,1-валентного кристалла с
дефектами Шоттки, поэтому графики зависимости концентраций
156
дефектов от рХ2 могут быть получены из графиков рис. 5.4
прямой заменой [Vx+] на g[Mi+].
При zx = 2; г = г/2 (оксиды, халькогениды) все показатели
степеней при рХ2 вдвое меньше, чем для 1,1-валентного кристалла.
Поэтому в данном случае вдвое меньше наклон всех прямых на
рис. 5.4. В частности, концентрации дырок и электронов в
области I пропорциональны рХ2±1/4> а в областях II и III
пропорциональны рх2±1/8. Тем не менее качественный ход графиков при
zx=2 остается таким же, как и при zx = l.
3. Оксид двухвалентного металла МО (2m=2x = 2;
/-=1) с дефектами Шоттки. В этом случае решение носит
более сложный характер, чем в двух предыдущих случаях,
поскольку ионные дефекты могут находиться в двух зарядовых
состояниях, £=1; 2. Однако, как отмечалось выше, вблизи сте-
хиометрического состава наиболее вероятным является
полностью ионизованное состояние вакансий £ = 2, которое мы будем
считать основным. Тогда для области промежуточных давлений
кислорода (решение I) из формул (5.56а) и (5.58а) получаем:
I- Ю«[У20+]«/С5
Mi~ L v О
—1
(5.72)
4 • r^+i ~ д+ «4
[e-]^A-pl; [e*\~A+pl
Границы области I определяются следующими условиями.
При определенном значении давления кислорода Р+
концентрация дырок возрастает до значения, равного удвоенной
концентрации полностью ионизованных катионных вакансий. Поэтому
при дальнейшем увеличении ро2 в области II условие
электронейтральности переходит в упрощенную форму: 2[Vm2~] ~ [е+],
и из соотношения (5.59а) при £=2 для этой области получаем:
НА. [e+]^2[V2-]«(C^)3p06s (5.73)
Аналогично, граница между областями I и II (малые
давления кислорода) определяется значением Р~, при котором
концентрация электронов проводимости становится равной
удвоенной концентрации полностью ионизованных анионных вакансий.
При дальнейшем уменьшении рог решение определяется
формулой (5.60а) при, £=2:
ША. [е~] « 2 [V20+] « (C2v+0 )3 pi (5.74)
В области существования решений (5.73) и (5.74)
по-прежнему концентрации однократно ионизованных вакансий [Vm"~]
и [Vo+] считаются пренебрежимо малыми. Однако это условие
выполняется только на ограниченных участках областей II и III,
непосредственно прилегающих к области I, т9 е. при не очень
157
больших отклонениях от стехиометрического состава. Чтобы
убедиться в этом, запишем реакции вторичной ионизации
вакансий:
,+ (5-75)
Уравнения закона действия масс для них имеют вид
K+lK] = rvo[V+]
[v2M-][e+] = rVM[v-]
2+1Г._1 ^ rir+1 (5.76)
откуда легко определить отношения концентраций однократно и
двукратно ионизованных вакансий:
[Vm] [e+] [V+] [е-]
[V^f] rVM [V^] rvo
(5.77)
Из последних равенств вытекает, что указанные отношения
растут по мере роста электронных дефектов, доминирующих в
соответствующей области давлений кислорода: дырок' в
области II и электронов в области III. Поэтому при очень больших
отклонениях от стехиометрического состава, т. е. при [е+]^Тум
в области II и при [е~~]Гуо в области III преобладающими
ионными дефектами становятся однократно ионизованные вакансии,
и решения получаются из (5.59а) и (5.60а) при £=1:
ПБ. [e+]«[V-]«:(C-M)2p04, (5.78)
2. zl
ШБ. [e-]*[Vj]-(C+0)2^043 (5.79]
При этом согласно соотношениям (5.77) концентрации
полностью ионизованных вакансий оказываются не зависящими от
давления кислорода: в области ПБ [Vm~2]~Tvm, а в области
ШБ [Vo2+]^rVo.
Полученные решения изображены графически на рис. 5.5.
В более сложных соединениях, когда валентности металла и
неметалла различны, катионные и анионные вакансии имеют в
основном состоянии разные эффективные заряды. Поэтому для
них графики, подобные изображенным на рис. 5.4 и 5.5, могут
иметь существенно несимметричный ход, в частности,
характеризоваться различными угловыми коэффициентами в областях с
недостатком и избытком неметалла. Значения показателя
степени при давлении неметалла в формуле (5.61) могут быть
легко определены из табл. 5.3 при всех возможных значениях
эффективных зарядов £.
158
РИС. 5.5.
Зависимость концентраций дефектов в нестехиометрическом оксиде
двухвалентного металла MOi±6 с дефектами Шоттки от парциального давления
кислорода в газовой фазе.
5.5. Разупорядоченность примесных
нестехиометрических кристаллов
В гл. 1 (см. раздел 1.6) было показано, что изменение
зарядовых состояний примесных центров может быть описано при
использовании эффективных зарядов с помощью уравнений
квазихимических реакций, одинаковых для кристаллов как с
валентной связью (полупроводников), так и с ионной. Поэтому в
данном разделе мы исследуем роль примесей в общем случае
нестехиометрического бинарного соединения МХг±б с
произвольным (но не металлическим) типом связи, включающем в
себя как частные случаи полупроводники и ионные кристаллы.
Как и в разделах 4.2 и 5.2, будем считать, что примесные ионы
размещаются в узлах катионнои подрешетки с образованием
дефектов замещения донорного или акцепторного типа.
Образование электронных дефектов в зоне проводимости или
в валентной зоне примесного кристалла может происходить при
ионизации нейтральных донорных или акцепторных примесных
центров согласно реакциям
DX^D+ + 6T
дх * + (5-8°)
Применяя к указанным реакциям закон действия масс,
получаем уравнения
[^][D"-] = rD[Dx]
(5.81)
[£+][А-] = ГА[Ах]
, Аналогичные уравнения можно записать и для реакций
последующей ионизации примесных, центров, в ходе которых од-
159
нозарядные доноры или акцепторы превращаются в двухзаряд-
ные и т. д. Однако, как будет видно из дальнейшего изложения,
при упрощенном решении выписывать эти уравнения не
обязательно.
Поскольку в общем случае эффективные заряды ионных
дефектов могут принимать произвольные целочисленные
значения от нуля до валентности соответствующего элемента,
условие электронейтральности примесного кристалла запишется в
виде
С=1 С=1 С=1
= г ^ С [ V=+] + g 2 С [МГ+] + 2 С [Dc-3 + [в'] (5.83)
где максимальные значения эффективных зарядов донорных и
акцепторных центров равны абсолютному значению разности
валентностей примесного и основного металлов:
\lzD\=zD — zM; \&гА\=гм — гА
Концентрации донорных и акцепторных центров с
различными степенями ионизации связаны с суммарными концентрациями
примеси [D] и [А] очевидными соотношениями
I д* I i д*i
р] = 2 Рс+]; [А] = 2 [АС~] <5-84)
Уравнения (5.81) — (5.84) следует включить в систему
уравнений (5.51), (5.52), сформулированных в предыдущем разделе
для чистого нестехиометрического кристалла. Решение
полученной обобщенной системы проводится аналогично изложенному
выше.
Совокупность упрощенных решений обобщенной системы
уравнений будет содержать и все решения, найденные в
предыдущем разделе для чистого кристалла. Прежде всего, это
очевидно для исчезающе малых концентраций примеси. Так, если
концентрация примеси мала по сравнению с одной из констант
собственного разупорядочения ионных кристаллов /Cs, Kf или
Kaf (обладающих соответственно дефектами Шоттки, Френкеля
или антифренкелевскими дефектами), влияние примеси
несущественно и концентрации доминирующих дефектов во всем
интервале давлений неметалла определяются решениями I—III,
полученными в предыдущем разделе. Аналогично, в случае
полупроводника реализуются решения I—III, приведенные в
разделе 4.4, если концентрация примеси мала по сравнению с
константой собственной ионизации Ки
160
Решения II и III групп, найденные в разделах 4.4 и 5.4, могут
реализоваться и при значительных концентрациях примесей,
если отклонения от стехиометрического состава достаточно
велики. В этих случаях концентрации заряженных дефектов,
обусловленных нестехиометричностью соединения, значительно
превышают концентрацию примеси, и ее влияние на дефектную
структуру кристалла становится несущественным.
Таким образом, в дополнение к найденным ранее решениям
I—III групп для чистых кристаллов нужно рассмотреть все
упрощенные формы условия электронейтральности, содержащие в
его левой или правой части концентрации заряженных донорных
или акцепторных примесных центров. Соответствующие им
приближенные решения разбиваются на три дополнительные
группы.
IV. Четвертую группу составляют решения, отвечающие
упрощенным формам условия электронейтральности: [D+]«
~ [£~] и [А~] »[е+]. В этих случаях собственные ионные
дефекты не играют заметной роли и кристалл ведет себя как обычный
примесный полупроводник, подробно рассмотренный в разделе
4.2. В случае донорной примеси согласно формулам (4.20) и
(4.24) приближенные решения для примесного полупроводника
описываются следующими формулами.
A. В области резерва нейтральных доноров:
1
[e-]«[D+]«([D]rD)2«[D] (5.85а)
Б. В области истощения доноров:
[е~] « [D+] * [D] (5.856)
В случае акцепторной примеси приближенные решения
имеют аналогичный вид.
B. В области резерва нейтральных акцепторов:
[е+] « [А"] « ([А] ГА)2 « [А] (5.85в)
Г. В области истощения акцепторов:
[е+] « [А"] « [А] (5.85г)
Области давления неметалла, в которых реализуются
решения (5.85), называют областями контролируемых
электронных дефектов. Такой термин оправдан тем, что
концентрации электронов проводимости и дырок в этих областях
не зависят от рХ2 и целиком контролируются содержанием ге-
теровалентной примеси в кристалле. Тот результат, что
концентрации доминирующих дефектов согласно полученным решениям
не зависят от давления неметалла в газовой фазе, связан с
незначительной ролью собственных ионных дефектов — вакансий
11—321
161
Таблица 5.4. Значения параметров \s\ и \т\ в формулах (5.91)—(5.93)
для концентраций дефектов в примесном нестехиометрическом
соединении МХГ± б
Решение
IVA
IVB
IVB
ivr
VA
VB
VB
vr
VIA
VIB
VIB
vir
VIA'
VIB'
VIB'
vir
Условие электронейтральности
j [D+] = [el {
j [A-] = [e4 |
[D+]=UVMH 1
[D+] = lg [xf~] J
[A-] = ^[VXE+] 1
[A-]=^[Mi£+] j
b[Ab-] = C,g[Mf.+] j kl J
Лг[0Дг+]=гм VMZM~] )
r -,M = Дг [D]
AZ[DAz+]=2xg[XiZx-] j
Дг[ААг-1=г„[Ухгх+1 )
Условие
нормировки примесей
ID] = [DX]
[D] = [D+]
[A] = [AX]
[A] = [A"]
[D] = [Dx]
[A] = [AX]
[D] = [Db+]
[A] = [Ab-j
[D] = [DAz+]
[A] = [АДг~]
|s|
2 )
1
2
1
£+1
?2
c2
E)
Um J
|m|
1 oo
|2(£+l)/r
\2(C+1)
(2(C+1)
\2(C+l)/r
f 2?2/r
\2E,
( 2?2
I 2£2/r
2гх
2zx
или междуузельных ионов, на концентрации которых прежде
всего и сказывается изменение рХ2-
Из-за малости концентраций собственных ионных дефектов,
а также их низкой подвижности по сравнению с подвижностью
электронов в области контролируемых электронных дефектов
кристалл является практически чисто электронным
проводником п- или р-типа в зависимости от сорта примеси.
V. Пятая группа включает решения, присущие только несте-
хиометрическим соединениям. В этих случаях положительный
эффективный заряд донорных центров компенсируется
отрицательным эффективным зарядом собственных ионных дефектов
или, напротив, отрицательный заряд акцепторных центров —
положительным зарядом ионных дефектов. Соответствующие
упрощенные формы условия электронейтральности приведены в
табл. 5.4.
Решения этой группы предполагают, что основная часть
донорных или акцепторных центров находится в нейтральной
форме и только незначительная их доля ионизована. Используя
162
терминологию физики полупроводников, здесь можно говорить о
значительном резерве нейтральных доноров или акцепторов.
Поскольку в рассматриваемом случае примесные центры
находятся в основном в нейтральной форме, из всех заряженных
состояний необходимо учесть только однозарядные, а
состояниями с высшими степенями ионизации можно пренебречь.
Рассмотрим наиболее подробно решение, отвечающее одной
из упрощенных форм условия электронейтральности,
принадлежащих рассматриваемой группе:
A. [D+]«C[VCM-1 (5.86)
а при условии нормировки:
[D] « [Dx] (5.87)
Подстановка этих соотношений в уравнение (5.81а) дает для
концентрации электронов проводимости:
K1 = rD[DK[V^l (5.88)
Подставляя сюда концентрацию вакансий из уравнения (5.51а)
и учитывая (5.52), находим:
[е J — (AdIL>J/^Cvm) Aj /?Хз
ь+i __ /rrc~ /Г rniW+1> /r2/(c+1V/2(c+1) (5.89)
[e J — (u>VM/i D [u\ A} /?Х2
Наконец, для концентраций доминирующих заряженных ионных
дефектов из (5.51) получаем:
Р+] - с [vjn - (rD [D] f^ (cc- )^кгш^1) р\ {Ш)
(5.90)
Остальные решения данной группы находятся аналогично
изложенному. Мы не будем выписывать их в деталях, а
ограничимся записью обобщенных формул. Из решения (5.89) видно,
что абсолютные концентрации электронов проводимости и
дырок можно выразить общей формулой [ср. формулу (5.61)
предыдущего раздела]
J. _L
п = А (Т) [F]s р£ ехр (~фТ) (5.91)
где [F] —полная атомная доля донорной или акцепторной примеси.
В этой формуле знак показателя степени 5 при
концентрации примеси определяется следующим правилом: донорные
примеси (с большим числом валентных электронов) увеличивают
концентрацию электронов проводимости и уменьшают
концентрацию дырок; акцепторные примеси (с меньшим числом
валентных электронов) вызывают противоположный эффект. Знак
И* 163
цоказателя степени т при давлении неметалла определяется
тем же правилом, что и в чистом соединении: плюс для дырок и
минус для электронов.
Концентрации доминирующих ионных дефектов в решениях
данной группы зависят от различных параметров: от содержания
примеси, температуры и давления неметалла в газовой фазе.
При донорной добавке (решения VA и VB) концентрации кати-
онных вакансий или междуузельных анионов определяются
формулой (5.90) или в общем виде
i-JL _L
N*~ = A(T) [D] * pm ехр(- -J^ (5.92)
т. е. растут с ростом содержания донорной примеси и давления
неметалла. Аналогично, при акцепторной добавке концентрации
анионных вакансий или междуузельных катионов растут с
ростом содержания акцепторной примеси и убывают с ростом
давления неметалла:
7Vc+ = A(r)[A] * p *ехр(-^) (5.93)
Значения параметров s и т приведены в табл. 5.4.
VI. В решениях шестой группы условие
электронейтральности принимает те же упрощенные формы, что и в предыдущей
группе, однако здесь основная часть примеси ионизована
(истощение нейтральных доноров или акцепторов).
Соответственно и концентрации преобладающих ионных дефектов в
данном случае не зависят от температуры и давления неметалла и
определяются суммарным содержанием примеси. Однако
концентрации электронных дефектов зависят от всех внешних
условий: содержания примеси, температуры и давления неметалла.
Соответствующие формулы для концентраций электронных
дефектов получаются непосредственно из уравнений (5.51)
путем замены в них концентраций доминирующих вакансий или
междуузельных ионов на равные им концентрации примесей.
Легко видеть, что в общем случае для концентраций электронов
проводимости и дырок для решений рассматриваемой группы
остается справедливой формула (5.91), причем знаки
показателей степеней s и т определяются теми же правилами, что и в
решениях предыдущей группы. Поскольку концентрации
электронных дефектов в решениях данной группы малы по сравнению
с концентрациями ионных дефектов, а подвижность их
значительно выше, доля электронной проводимости здесь может быть
любой и, вообще говоря, сравнима с ионной.
В противоположность решениям IV группы, решения V и VI
групп можно отнести к области контролируемых ионных
дефектов. Электронная составляющая проводимости при
благоприятных условиях в этом случае может быть подавлена
164
соответствующей добавкой донорной или акцепторной примеси,
а также выбором подходящего содержания неметалла в газовой
фазе.
Для ионных кристаллов особый интерес представляют
решения VI группы (VIA" — VIP в табл. 5.4) при полной ионизации
основных дефектов. Действительно, условия
электронейтральности в этих случаях соответствуют примесной ионной разупорядо-
ченности, подробно рассмотренной в разделе 5.2, и определяют
концентрации доминирующих ионных дефектов практически во
всех примесных кристаллах с преобладающей ионной
проводимостью.
Зависимость концентраций электронов и дырок от давления
неметалла в этих решениях определяется тем же самым
значением m = 2zx, что и в случае чистых соединений с полностью
ионизованными дефектами. Такое совпадение не случайно: из
уравнений (5.51) видно, что т становится равным 2гх в любом
случае, когда концентрация полностью ионизованных дефектов
не зависит от давления неметалла.
Последнее обстоятельство можно отнести и к твердым
электролитам со структурной разупорядоченностью, если состав их
не слишком отличается от стехиометрического. Действительно,
при структурной разупорядоченности разрешенные позиции для
катионов можно рассматривать в качестве вакансий, причем их
концентрация определяется строением кристаллической
решетки и не зависит от внешних факторов, и из (5.51а)
непосредственно вытекает <[е+] ~рУ*гХ. Разумеется, при структурной раз-
упорядоченности пользоваться соотношениями закона действия
масс следует с осторожностью, ибо они могут быть значительно
искажены коллективным характером взаимодействия ионов в
таких твердых телах.
Подводя итоги изложенному в двух последних разделах, а
также в разделе 4.4, можно сделать следующие выводы.
1. Ионные точечные дефекты в кристаллах, не содержащих
гетеровалентных примесей, являются преобладающими лишь
при составах, близких к стехиометрическому. Их концентрация
при этом экспоненциально растет с температурой и не зависит
от давления неметалла в газовой фазе. В кристаллах,
содержащих гетеровалентную примесь, ионные дефекты доминируют в
определенной области давлений неметалла, в которой составы
обоих соединений в твердом растворе также близки к стехиомет-
рическим. При этом концентрация основных ионных дефектов
определяется содержанием примеси. При больших отклонениях
от стехиометрического состава как полупроводники, так и
ионные кристаллы имеют комбинированную ионно-электронную раз-
упорядоченность, обеспечивающую преобладающую
электронную проводимость.
2. В некоторых случаях концентрации электронных дефектов
в нестехиометрических кристаллах описываются формулами
физики полупроводников — собственных или примесных (реше-
165
ния I, табл. 4.1 и IV, табл. 5.4). Это возможно в тех случаях,
когда энергия образования ионных дефектов превышает энергию
образования электронных дефектов. Однако такие решения
больше характерны для твердых тел с ковалентными связями и
не типичны для ионных кристаллов.
3. Во всех остальных случаях, за исключением указанных
решений, концентрации электронов проводимости и дырок в не-
стехиометрических соединениях существенно зависят от
парциального давления его неметаллического компонента в газовой
фазе. При этом концентрация дырок, как правило, растет с
ростом давления неметалла, а концентрация электронов
уменьшается.
4. Что касается температурной зависимости концентраций
дефектов, то в случае нестехиометрических соединений она
определяется температурной зависимостью целого ряда констант
равновесия квазихимических реакций между дефектами и не
имеет такого наглядного физического смысла, как в случае
собственных или примесных полупроводников. В конечном итоге
она определяется энергетическим эффектом весьма сложной
квазихимической реакции, приводящей к возникновению
дефектов данного сорта.
Экспериментальный материал в целом неплохо подтверждает
результаты изложенной теории. Здесь мы ограничимся
четырьмя иллюстрациями, показывающими наиболее простые
зависимости.
На рис. 5.6 приведены экспериментальне результаты (точки)
по плотности твердых растворов Zr02 — Y203 в зависимости от
состава. Эти результаты сравниваются со значениями, рассчи-
1др0г(По)
-10 -9 -6
I I I I I I U -12\ 1 L_J
10 15 20 25 30 35 ' _/5 -Щ -13
[Y203 7,% (мол.) ц р0г (щ)
РИС. 5.6.
Зависимость плотности твердых растворов Zr02 — Y203 от состава [62].
Прямая 1 рассчитана по междуузельной, прямая 2 — по вакансионной модели.
РИС. 5.7.
Зависимость концентрации катионных вакансий в нестехиометрическом
FeOi+6 от давления кислорода [63].
166
3fl igpo2M
ig[vv]
-2lgPojna)
2,0 -1,5
1др0г(бср)
РИС. 5.8.
Зависимость концентрации катионных вакансий в нестехиометрическом
Cu20i+6 °т давления кислорода. Экспериментальные точки — данные разных
авторов [64, 65].
РИС. 5.9.
Зависимость концентрации катионных вакансий в нестехиометрическом
V203+6 0T давления кислорода [Щ при температурах (в °С):
J — 1400; 2 — 1500; 3 — 1600; 4 — 1700.
тайными на основании двух моделей дефектной структуры
твердых растворов. Согласно первой модели (прямая ))
компенсация отрицательного эффективного заряда примесных центров Y~Zr
осуществляется междуузельными компонентами Zri4+, согласно
второй модели (прямая 2) компенсация осуществляется
кислородными вакансиями Vo2+. Как видно, имеется очень хорошее
соответствие экспериментальных значений плотности с
расчетными по вакансионной модели.
На рис. 5.7 изображены зависимости атомной доли
катионных вакансий в нестехиометрическом вюстите от парциального
давления кислорода в газовой фазе. Прямые линии имеют
наклон, равный 7б, что соответствует зависимости [VFe] ~Ро21/б-
Такая зависимость вытекает из решения (5.73), согласно
которому катионные вакансии имеют эффективный заряд +2.
Следует заметить, что такая зависимость для оксидов является
довольно распространенной.
На рис. 5.8 и 5.9 показаны зависимости от давления
кислорода концентрации вакансий в оксидах СщО и V2O3. Наклоны
прямых соответствуют зависимостям [Vcu] ~po21/4; [Vv] ~Po28/4-
Такие зависимости легко объясняются при предположении, что
в обоих соединениях преобладают электронейтральные
вакансии; они непосредственно вытекают из уравнений закона
действия масс для квазихимических реакций образования
нейтральных вакансий:
Си
2
4-о2^ьзо*
2V>
(5.94)
(5.95)
167
Глава 6
Явления переноса в твердых телах
Явления переноса в твердых телах, вообще говоря,
исключительно многообразны как по природе переносимой материи
(вещество, электричество, теплота), так и по природе вызывающих
перенос полей (химическое, электрическое, магнитное,
температурное). В данной главе рассмотрена лишь сравнительно
небольшая часть основных явлений переноса вещества и электричества:
электропроводность, диффузия и термоэлектрический эффект,
занимающие наиболее важное место в физической химии
твердого тела.
В дальнейшем изложении также приняты следующие
ограничения: поля считаются слабыми и однородными, а твердые
тела — изотропными. В этом случае все рассмотренные явления
переноса сводятся к одномерным задачам о движении частиц
вдоль оси х, имеющей направление градиента соответствующего
потенциала.
Для обозначения градиентов в целях сокращения записи мы
будем пользоваться символом теории поля «набла» V, имея в
виду, что в одномерном случае градиент некоторой величины Ф
равен ее производной по координате х:
vO == grad<£ = d<$)\dx
6.1. Феноменологические уравнения
Плотность тока, протекающего через твердое тело в
электрическом поле, согласно эмпирическому закону Ома
/= —ху? (6.1)
пропорциональна напряженности поля (—Vxp). Коэффициент
пропорциональности х, называемый удельной
электропроводностью, является специфической характеристикой
вещества и зависит от ряда внешних факторов, таких как
температура, давление и др.
Если в переносе тока принимают участие частицы разных
сортов (ионы или электроны), то общий ток складывается
аддитивно из вкладов каждого сорта носителей:
1 = ЪК = Ъя^к (6.2)
к к
где qK=±zKe — заряд частицы сорта к; JK — плотность потока я-частиц
(число я-частиц, проходящих за единицу времени через единичное сечение).
Парциальные плотности тока iK также подчиняются закону
Ома:
Ik=-kkV? (6.3)
168
где Кк — парциальные удельные электропроводности,
обусловленные частицами сорта к.
Доля /с-частиц в суммарной удельной электропроводности
обозначается как число переноса /с-частиц:
tK = KKfc " = 2X (6.4)
к
Парциальные удельные электропроводности легко связать с
концентрациями /с-частиц. Действительно, если частицы
движутся со средними скоростями vK, то плотность их потока будет
равна
Л=ЛГЛ (6-5)
где NK— число /с-частиц в единице объема.
Среднюю скорость миграции частиц в электрическом поле
можно в первом приближении положить пропорциональной
напряженности поля:
ък •- + ик v ср (6.6)
Коэффициент пропорциональности ик численно равен средней
скорости миграции частиц в электрическом поле, напряженность
которого равна единице. Он называется подвижностью
носителей сорта /с. Двойной знак здесь учитывает
противоположные направления положительно заряженных (верхний знак) и
отрицательно заряженных носителей. Подставляя (6.6) в (6.5)
и переходя от потоков частиц к 'плотностям электрического
тока, получаем:
'* = TgKNKuK^^ (6.7)
Сравнение этого выражения в законом Ома (6.3) показывает,
что
*к = I Чк I NJ*k = ztfiNjlK (6.8)
Наряду с подвижностью частиц в электрическом поле,
определяемой эмпирическим уравнением (6.6), часто используют
несколько иную величину:
и£= ±uKjqK^uK/zKe (6.9)
Чтобы выяснить ее физический смысл, подставим ее вместо ик
в уравнение (6.6):
^ = -?Xv? (6.Ю)
Произведение —*7*V*p есть сила F, действующая на заряженную
частицу в электрическом поле, поэтому уравнение (6.10) можно
переписать в виде
vk = ufkF (6.11)
169
Отсюда следует, что uKF численно равна средней скорости,
приобретаемой частицей под действием силы, равной единице.
Подвижности и/ является более универсальной характеристикой,
нежели ик\ она определяет скорость частицы в любом поле,
которое может действовать на частицу с некоторой силой F.
Кроме того, в разделе 6.4 будет показано, что подвижность uKF
не зависит от заряда частицы. Поэтому она может относиться
как к заряженным частицам (ионам и электронам), так и к
нейтральным атомам. Ее обычно используют в теории диффузии
(см. раздел 6.5), в то время как подвижность ик используется
преимущественно в теории электропроводности (см. разделы 6.3,
6.4).
Движение частиц в концентрационных полях описывается
двумя уравнениями диффузии, называемыми также законами
Фика.
Первый закон Фика выражает обычно наблюдаемую
на опыте пропорциональность между плотностью частиц
сорта к в направлении оси х и х-составляющей градиента
концентрации этих частиц:
Jk = DkVNk (6.12)
Коэффициент пропорциональности DK называется
коэффициентом диффузии /с-частиц.
Второй закон Фика, описывающий зависимость
концентраций от времени, непосредственно вытекает из первого при
учете закона сохранения вещества. Выделим в диффузионной
среде два единичных сечения, перпендикулярных направлению
диффузии х и находящихся на расстоянии dx друг от друга.
Если число частиц в объеме между выделенными сечениями
меняется со временем, то очевидно, что увеличение их числа за
единицу времени равно разности потоков, входящих и
выходящих из объема:
J к (х) - J к (х -±-dx)=- ^JKdx
С другой стороны, так как величина выделенного объема
численно равна dx, это увеличение равно (dNjdt)dx. Сравнивая оба
выражения и используя (6.12), получаем выражение второго
закона Фика:
—" =v(D„vAy (6.13)
dt
Второй закон Фика часто записывают в форме
^ = DK—K = DKv2NK (6.14)
dt к а*2 к v K v '
считая коэффициенты диффузии не зависящими от
концентрации и, следовательно, от координаты х.
Часто удобнее выражать диффузионные потоки частиц через
градиенты не концентраций, а их химических потенциалов. Соот-
170
ветствующее уравнение было предложено Вагнером,
рассмотревшим движение заряженных частиц в ионном кристалле,
находящемся одновременно в химическом (концентрационном) и
электрическом полях [21]. В основу вывода Вагнер положил
следующие допущения.
1. Частицы каждого данного сорта (катионы, анионы,
электроны) движутся независимо от того, движутся или находятся в
покое частицы других сортов.
2. Имеются две движущие силы для заряженных частиц
каждого сорта: диффузионная, обусловленная градиентом их
химического потенциала, и электрическая, обусловленная
электрическим полем, — причем действие каждой из этих движущих сил
не зависит от действия другой движущей силы.
Эти предположения являются предположениями
термодинамики необратимых процессов; они ниоткуда не следуют и
являются, по существу, постулатами, правильность которых
проверяется правильностью всех результатов теории в совокупности.
В соответствии с этими предположениями поток заряженных
частиц сорта к можно записать в виде
JK = J?* + Jl* (6.15)
где /кдиф — вклад в обший поток диффузионных движущих сил; /«эл — вклад
электрических сил.
Последнюю величину легко определить из закона Ома; так
как каждая /с-частица переносит заряд qKi то плотность тока
(6.3) соответствует потоку частиц
Лл= ^-vcp (6.16)
Як
В соответствии с предположением (2) вклад диффузионных
сил /кдиф в общий поток не зависит от /кэл и, следовательно, от
имеющейся напряженности электрического поля —V<p. Поэтому
для определения /Ииф удобно поступить следующим образом.
Положим напряженность поля равной такой величине —V<ppaBH,
чтобы электрические силы полностью уравновешивали
диффузионные, так чтобы общий поток /с-частиц был равен нулю:
удиф+(7эл)Равн_„ Q (617)
Но из термодинамики известно, что в этом случае по всему
кристаллу должен быть постоянным электрохимический потенциал
/с-частиц, т. е.
vi-Ww + ^V?paBH = 0 (6.18)
Отсюда следует, что равновесное значение напряженности
электрического поля определяется величиной градиента химического
171
потенциала /с-частиц («напряженностью химического п о-
л я»):
равн 1 /г» in\
V? = VP-« (6.19)
Як
Подставляя полученное равенство в (6.16) и (6.17), находим:
■C*=-^WK (6.20)
Як
Для суммарного потока /с-частиц получаем уравнение
Вагнера:
J к = — ^г (Vft* + ?*V?) (6.21)
или, поскольку выражение в скобках есть градиент
электрохимического потенциала /с-частиц:
JK=—4w* (6-22)
Як
Смысл полученной формулы достаточно прост. Аналогично
законам Ома и Фика, утверждающим, что поток /с-частиц
пропорционален напряженности электрического поля или же
градиенту концентрации этих частиц, уравнение Вагнера (6.22)
показывает, что в электрохимическом поле (т. е. при наличии
градиентов электрического и химического потенциалов) поток
заряженных /с-частиц пропорционален «напряженности
электрохимического поля» (—VjxK).
Уравнение (6.21) обобщается и на более сложный случай,
когда к химической и электрической движущим силам
добавляется тепловая, если кристалл находится в температурном
поле. Наиболее строгий вывод такого обобщенного уравнения
предполагает термодинамика необратимых процессов [67].
В этом случае при независимом движении частиц разных
сортов для потока частиц сорта /с в химическом, электрическом и
температурном полях следует записать
я1
(6.23)
Это уравнение может быть названо основным
уравнением переноса заряженных частиц.
Следует отметить, что уравнение (6.23), как и все
предыдущие, применимо лишь в области слабых полей. Они выражают
общее правило, что в слабых полях поток частиц
пропорционален движущей силе соответствующего поля. В электрическом
поле движущей силой является кулоновская сила (—0cV(p); в
общем случае эффективная движущая сила равна выражению в
квадратных скобках (6.23), взятому с обратным знаком.
172
Величина ик* носит название энергии переноса /с-частиц*.
Уравнения Ома, Фика, Вагнера и основное уравнение
переноса носят феноменологический характер и описывают потоки
вещества и электричества независимо от конкретного механизма
движения носителей. Поэтому содержащиеся в них параметры
должны либо определяться экспериментально, либо
вычисляться в рамках микроскопической теории, связанной с выбором
конкретных механизмов переноса.
6.2. Статистический вывод основного
уравнения переноса
Согласно основному (положению молекулярно-кинетической
теории в любом агрегатном состоянии, в том числе и в твердых
телах, атомы находятся в непрерывном тепловом движении.
Специфической чертой твердых тел является то, что атомы
основное время колеблются около равновесных положений в
узлах кристаллической решетки и лишь изредка в результате
тепловых флуктуации перескакивают в соседние устойчивые
позиции, совершая таким образом хаотическое блуждание по
кристаллу. В однородном изотропном кристалле при отсутствии
движущих сил прыжки атомов во всех направлениях происходят с
одинаковой вероятностью, поэтому какого-либо направленного
движения их не существует. Направленный поток атомов
возникает, если вероятности прыжков в каком-либо определенном
направлении становятся преобладающими; это происходит либо
при наличии градиентов концентраций атомов различных
сортов, либо под действием движущих сил, облегчающих прыжки
в данном направлении.
Для определенности рассмотрим сначала вакансионный
механизм переноса в смешанном ионном кристалле, содержащем
* В общем случае термодинамика необратимых процессов определяет
потоки вещества и энергии выражениями
Jк — Z LtfXi + LKUXU ; Ju — £ LuiXt + LUUXU
i I
где обобщееные силы равны
При
независимом движении частиц разных сортов LKi=LiK=0 при 1^Фк;
^KK=KKfqK2. Согласно соотношению взаимности Онзагера £иг=£гы=Хг^г*/<7/2.
Ь частном случае изотермической системы Хи=0, и потоки связаны
соотношением
Отсюда следует, что ui* есть энергия, переносимая через кристалл единичным
потоком частиц сорта i при изотермических условиях.
173
РИС. 6.1.
Зависимость потенциальной энергии ионов от координаты х при отсутствии
внешних полей (а) и во внешнем поле (б).
переменную концентрацию ионов сорта /с. Пусть концентрация,
электростатический потенциал и температура изменяются в
направлении оси х. Выделим в кристалле две соседние атомные
плоскости, перпендикулярные оси х и находящиеся друг от
друга на кратчайшем расстоянии г0, и рассмотрим конфигурацию,
изображенную на рис. 6.1, а, при которой ион сорта /с и
вакансия находятся в соседних узлах, соответственно в плоскостях I
и II.
Обозначим через to*4" частоту, с которой выделенный ион
может перескочить из плоскости I в один из ближайших узлов в
плоскости II при условии, что этот узел вакантен. Тогда число
/с-ионов, перескакивающих из плоскости I в плоскость II за
единицу времени через единичное сечение, будет равно
произведению частоты (о*4-, вероятности того, что узел в плоскости II
вакантен, [Vk]" = Nk"/Nl, и числа /с-ионов на единице площади в
плоскости irV Аналогичным образом число прыжков /с-ионов в
обратном направлении определим как произведение ^"[V^T/'.
Поверхностная концентрация /с-ионов Тк простым образом
выражается через локальное значение их объемной концентрации
N'K. Поскольку расстояние между соседними атомными
плоскостями равно Го, в кубе с единичным ребром содержится 1/г0
плоскостей и N'K ионов /с. Следовательно, их число на единице
поверхности одной плоскости равно T/K = foN/K. Плотность
потока /с-ионов запишется как разность чисел их прыжков слева
направо и справа налево:
JK = r„ (TV/ [VK]"«»+ - N'K[VKY О (6.24)
Рассмотрим подробнее частоты прыжков со«+ и сок~. При
отсутствии полей в однородном кристалле они могут быть записаны
в виде произведения
<% = а>7= vK exp (—bKlkT) (6.25)
где v,c — частота попыток перескоков; экспоненциальная функция определяет
больцмановскую вероятность того, что ион успешно преодолеет потенциаль-
174
яый барьер при элементарном акте перескока, высота которого называется
энергией активации перескока.
Хотя частота попыток перескоков vK точно не определена, ее
можно оценить как дебаевскую частоту колебаний ионов vd,
умноженную на число узлов /с-подрешетки в плоскости II,
находящихся на ближайшем расстоянии от узла I (например, в
структуре NaCl это число равно 4 при расстоянии между
соседними катионными узлами а = У2г0).
В присутствии .полей энергетическая картина кристалла
искажается, так что иону I при перескоке необходимо затратить
энергию UK + 8UK (рис. 6.1,6). Предположим, что необходимая
для перескока энергия набирается в трех точках: в исходном
узле I, в седловой точке (посредине между соседними
устойчивыми позициями) и в конечном положении П. Действительно,
при перескоке иона критическим состоянием является такое,
когда он находится в седловой точке между двумя вакансиями в
узлах I и П. При этом решетка сильно деформирована:
ближайшие соседи седловой точки должны раздвинуться настолько,
чтобы предоставить перескакивающему иону туннель
достаточной ширины; смещены и соседи обеих вакансий. Избыточная
энергия UK+[bUK обеспечивается тепловыми флуктуациями и
может набираться по частям в разных точках траектории: в
исходной, седловой и конечной, соответственно долям а, р и
'у(а-Ь|3+7=:1)> зависящим от механизма передачи энергии в
кристалле.
Обозначим температуру в плоскости I, в седловой точке и в
плоскости II соответственно через Т—6Г, Т и Г+б7\ Тогда
частота прыжков &к+ запишется через произведение вероятностей
накопления энергии в соответствующих точках:
ioj = V|f exp ^-^ — exp — v h
X
Частота прыжков в обратном направлении <о«г записывается
совершенно аналогично и может быть получена из (6.26)
прямой заменой 8UK на —8UK и 67 на —6 Г.
Считая все поля достаточно слабыми, разложим
экспоненциальные функции в (6.26) в ряды по малым параметрам 8Т/Т
и 8uK/kT и ограничимся линейными членами. Так, первую
экспоненту в (6.26) преобразуем следующим образом:
еХР[ k(T-bT) \~е + dU W +
+ ^дт—(-ьт)-е \l--pf ьтг) (6-^7)
Как видно из рис. 6.1,6, энергия активации при прыжках в обо
их направлениях отличается на величину, равную разности энер
175
гий перескакивающего иона в узлах II и I:
2bU = (w"K + ял») - {w'K + дк9')
где wK — потенциальная энергия взаимодействия иона с окружающей средой
в отсутствие электрического поля, которая, вообще говоря, зависит от
состава кристалла и, следовательно, от координаты х; qK — заряд иона.
В рассматриваемом случае слабых полей
IUK = Ц- {xwK + <7куср)
87"=: ^хТ
2 (6.28)
yv; = 7V/(l+r0vlnK-])
[V,]" = [VKr(l+roVln[VK])
Подставляя соотношения (6.26) — (6.28) в (6.24) и
ограничиваясь членами, линейными по градиентам, получаем:
(6.29)
Здесь через хк обозначена величина
~2
Kr№KNKK [WK] ; п
kT
exp
[~Ti) <M°)
Концентрация /с-ионов в узлах /с-подрешетки помечена двойным
индексом: NKK (одиночный индекс /с будет в дальнейшем
использоваться для обозначения полной концентрации /с-компонента в
кристалле NK).
Если в формуле (6.29) положить все градиенты, кроме Vcp,
равными нулю, то получающееся выражение совпадает с
законом Ома. Отсюда вытекает, что кк есть не что иное, как
парциальная удельная электропроводность частиц сорта к.
Три первых слагаемых в уравнении (6.29) можно выразить
через градиент химического потенциала /с-частиц, деленного на
температуру. Действительно, согласно формулам (2.50) и (2.51)
имеем:
^ = -^r+k (31n — + iKln ) + k\n-^- (6.31)
T T \ kT ^ K ^K } [V«] V
откуда
A {**) = ^ - ^- ^ + *Y in K] - k? In [ VJ (6.32)
Подставляя производные правой части в (6.29), находим:
К = - ■* [7-v (f) + ЯКУ9 + "« + 3"-+<—?>"« vr] (6.33)
176
Сравнивая полученное уравнение с основным уравнением
переноса заряженных частиц (6.23), видим, что они будут
совпадать, если положить
uK = wK + 3kT + (а _ Т) UK (6.34)
В этой сумме wK = —Wvk — энергия покоящейся /с-частицы;
3kT — средняя энергия ее колебаний. Следовательно, (а—y)UK
есть энергия поступательного движения частицы, или, в
терминологии термодинамики необратимых процессов, теплота
переноса частиц сорта /с*. Такое название обусловлено тем, что
указанная энергия равна количеству избыточной теплоты,
переносимой /с-частицей, движущейся через кристалл при отсутствии
в нем градиента температур.
Таким образом, при вакансионном механизме миграции ионов
теплота переноса /с-частиц
Q* =(a-i)UK (6.35)
равна разности энергий, поставляемых элементарному акту
перескока кристаллической решеткой в исходной и конечной
устойчивых позициях перескакивающего иона. Так как а и у не
превышают единицы, теплота переноса не может быть больше
высоты потенциального барьера между устойчивыми положениями
иона UK.
Полученный результат вполне понятен. Действительно, при
локальном искажении решетки, связанном с переходом иона из
узла I в седловую точку, в узлах I и II определенные
/количества теплоты, соответственно aUK и yUK, переходят в
потенциальную энергию деформации решетки. На втором этапе
элементарного акта, когда ион переходит из седловой точки в узел II,
процесс превращения энергии идет в обратную сторону с тем отли*
чием от первоначального, что теперь узлы I и II меняются
местами. Поэтому на втором этапе элементарного акта в узле II
потенциальная энергия uUKf а в узле I yUK превращаются в
теплоту. В результате этого элементарный акт перескока иона
сопровождается переносом количества тепла (а—y)UK из узла I
в узел II. Теплота же $UK, затраченная в седловой точке, по
причинам симметрии выделяется в той же точке и через
кристалл не переносится.
Мы вычислили потоки ионов при вакансионном механизме
переноса. Для междуузельного механизма переноса задача ре-
Многие авторы называют теплотой переноса величину ик*. В этих
случаях для величины QK* целесообразно использовать термин Пригожина
«приведенная теплота переноса». При любой терминологии эти величины
связаны соотношением
uK = hK+ Ql
где hK парциальная удельная энтальпия /с-частиц, в случае твердых тел
практически равная их парциальной удельной энергии ик.
12—321
177
шается совершенно аналогично. Легко убедиться в том, что для
междуузельного механизма вместо (6.29) получается
аналогичное уравнение
К = - ^ (?да* + kT^ln Ы-ьъ in[v,]+
Гк 1
+ ^V? + (a~;)t/KV^ (6.36)
с ионной проводимостью к-частиц, определяемой выражением
Vo^k, [V,] / UK\ /йо7ч
**=—«=—ехрг#) (6-37)
где шК1 и NKi—энергия и концентрация тс-ионов в междуузлиях; [к{\ и
[Vi] —доли междуузлий, занятых /с-ионами и незанятых соответственно.
На основании формул (2.52) и (2.53) имеем:
i^ = J^+*f31n^rE|ln-^U*ln-i^ (6.38)
Дифференцируя последнее выражение и подставляя
производные правой части в (6.36), получаем уравнение переноса, в
точности совпадающее с уравнением (6.33), полученным выше
для вакансионного механизма переноса ионов.
Очевидно, что приведенный анализ справедлив и для
электронных проводников при прыжковом механизме миграции
электронов и дырок. В этом случае проводимость р-типа аналогична
вакансионному механизму ионной проводимости, а /г-типа —
междуузельному механизму.
При туннельном механизме миграции электронов и дырок
основное уравнение переноса (6.23) также остается справедливым,
однако в нем значение парциальной электронной проводимости
уже не определяется формулами (6.30) и (6.37) (см. раздел 6.4).
Изложенный вывод основного уравнения переноса позволяет
глубже понять физический смысл эффективных движущих сил
миграции частиц в твердых телах. Из рассмотренных
градиентов потенциалов полей только напряженность
электростатического поля создает кулоновскую силу —<7>cV(p, непосредственно
действующую на заряженную частицу. Действие градиента
химического потенциала имеет статистический характер и
проявляется путем увеличения вероятностей прыжков атомов в одном
направлении за счет градиента концентрации как самих атомов,
так и вакансий. При этом поток частиц направлен в сторону
уменьшения их концентрации и увеличения концентрации
вакансий. Температурное поле оказывает аналогичное действие,
увеличивая вероятность прыжков в более горячей области
кристалла за счет более интенсивных флуктуации тепловых колебаний
кристаллической решетки. Соответствующий поток частиц
направлен в сторону понижения температуры.
178
6.3. Подвижность дефектов.
Ионная проводимость
формулы (6.30) и (6.37) определяют парциальную удельную
электропроводность, обусловленную движением ионов сорта к
по вакансиям в соответствующей подрешетке и по междуузли-
ям. Смысл этих выражений становится более ясным, если
перейти к подвижности носителей тока с помощью
феноменологического соотношения (6.8):
•A = LzeNu (6.39)
где ze — абсолютная величина эффективного заряда носителей; N — их
объемная концентрация; и — подвижность носителей, определяемая как средняя
скорость их движения в электрическом поле единичной напряженности.
Однако в рассматриваемой задаче выбор конкретных типов
носителей не однозначен. С одной стороны, можно и в
дальнейшем сохранять в центре внимания микроскопическую картину
перескоков точечных дефектов, рассматривая их в качестве
носителей тока. Тогда в формуле (6.39) под N следует понимать
концентрации вакансий и междуузельных ионов. Определенные
таким образом подвижности точечных дефектов, ответственных
за перенос тока, называют микроскопическими [68].
Сравнивая (6.39) и (6.30), находим для подвижности
вакансий в /с-подрешетке:
uv = ^^ [кк]ехр (--¥*-) (6.40)
vk kT L ,tJ r\ kT ,
При сравнении (6.39) с (6.37) получаем аналогичную
зависимость для подвижности междуузельных ионов сорта к:
kX ~~ kT
V,]exp(- gr) (6.41)
Концентрации ,[кк] и ;[Vi] во многих случаях можно полагать
равными единице. Так, если индекс к относится к основному
иону, при слабой разупорядоченности в формуле (6.40) доля
узлов, занятых ионами к, »[Як]«1. Аналогично, в чистых
кристаллах или в твердых растворах замещения при слабой
разупорядоченности доля незанятых междуузлий в (6.41) [Vi]«l.
В этих случаях уравнения (6.40) и (6.41) формально совпадают.
Полученные зависимости показывают, что подвижность
точечных дефектов — носителей тока определяется больцманов-
ской вероятностью перескоков дефектов в соседние устойчивые
положения. Это связано с тем, что перескоки ионов
представляют собой активационный процесс; энергию U, которую нужно
затратить при перескоке часто называют энергией
активации миграции иона.
Иная ^картина возникает при другом возможном выборе
носителей. Если в определенной степени абстрагироваться от
конкретного микроскопического механизма перескока дефектов
и снова перейти к макроскопическому рассмотрению переноса,
12* т
то в качестве носителей тока можно рассматривать всю
совокупность /с-ионов. Тогда в формуле (6.39) N будут означать
полную концентрацию ионов сорта к. Определенные таким
образом подвижности иногда называют макроскопическими
[68].
Так как при малых концентрациях междуузельных ионов
сорта к практически все /с-ионы расположены в узлах, можно
положить NKK = NKi и для вакансионного механизма переноса из
(6.30) получаем:
«^^^^[VJexpf--^ <6-42)
kT L "J г \ kT
Аналогично для междуузельного механизма переноса при
гул ~ \
W*gK М. (_Щ (6.43)
kT [к] \ kT )
где gK=Ni/NLK — отношение числа междуузлий к числу узлов я-подрешетки
Выражения (6.42) и (6.43) определяют уже с р е д н ю ю
подвижность /с-ионов, которая в случае слабой разупорядоченности
может быть значительно меньше подвижности точечных
дефектов благодаря малым множителям [VK] и {ki\. Этот результат
совершенно естествен: при слабой разупорядоченности
подвижными, или активными, являются лишь ионы, находящиеся в
соседних с вакансией узлах или в междуузлиях; основная же
доля ионов, расположенная в узлах решетки и не имеющая
вакансий в соседних узлах, неподвижна. Поэтому усредненная
макроскопическая подвижность ионов гораздо меньше
микроскопической подвижности активных ионов (или дефектов).
Формула (6.42) справедлива и для твердых электролитов со
структурной разупорядоченностью, если ионы в них
статистически распределены по большому числу доступных позиций. Более
того, при сильной разупорядоченности само понятие дефекта
теряет смысл, поэтому единственно правильным будет выбор в
качестве носителей тока ионов и, следовательно, определение
их подвижности по (6.42). Если число избыточных позиций в
g раз превышает число ионов, то при статистическом
распределении доля незанятых позиций равна
[V] = 5/(5+ 1) (6.44)
и при достаточно больших значениях | приближается к
единице. В этом случае формула (6.42) для структурно-разупорядо-
ченных ионов принимает простой вид
Таким образом, подвижность структурно-разупорядоченных
ионов определяется тем же выражением, что и
микроскопическая подвижность точечных дефектов в случае слабой
разупорядоченности (6.40), (6.41), и значительно выше средней макро-
180
скопической подвижности ионов в почти упорядоченных
кристаллах (6.42), (6.43). Это обстоятельство является основным
фактором, обеспечивающим в области умеренных и низких температур
значительно более высокую проводимость структурно-разупоря-
доченных фаз по сравнению с проводимостью кристаллов с
собственной разупорядоченностью типа Френкеля или Шоттки.
Различие в природе ионной проводимости твердых
электролитов со структурной разупорядоченностью и собственными
точечными дефектами проявляется даже не столько в ее
числовых значениях, сколько в физическом смысле параметров,
определяющих ее температурную зависимость. Действительно, для:
структурно-разупорядоченных фаз при [V]«1 формула (6.30)
имеет вид
сто vr02<7W / U
y. у = ——— ехр '
(-£) <™>
kT
где N — концентрация структурно-разупорядоченных ионов.
В этой зависимости эффективная энергия активации
(энергия, стоящая в числителе больцмановской экспоненты) равна
энергии активации миграции носителей U.
В случае собственной разупорядоченности типа Френкеля,,
Шоттки или антифренкелевских дефектов концентрации
вакансий или междуузельных ионов того или другого знака
определяются константами соответствующих реакций собственного раз-
упорядочения Kf, Ks или /Caf. На основании результатов
вычислений, проведенных в разделе 5.1, они могут быть представлены
единой формулой
[VJ, [/4] = X(v)exp(--Jr-) (6.47)
Величина w — энергия разупорядочения, отнесенная к одному
точечному дефекту:
ws = TT7' ™f=^f/2; ^af=^af/2 (6-48)
[ср. с формулами (3.23) и (3.24) для интерметаллических
соединений] При учете (6.47) формула (6,30) для кристаллов с
собственной разупорядоченностью принимает вид
\ = Тт еХР(--^-) (6'49)
отличающийся от (6.46) дополнительным множителем %(v) и
слагаемым w в показателе экспоненты. Эти параметры
описывают среднюю свободную энергию образования одного точечного
дефекта; их происхождение отвечает больцмановской
вероятности термического возбуждения носителей. Таким образом, в
случае собственной ионной проводимости кристалла, обусловленной
181
точечными дефектами, ее эффективная энергия активации
состоит из двух слагаемых: энергии активации образования
дефектов w и энергии активации миграции их по решетке U.
Следует отметить, что в формулах (6.46) и (6.49) энергии
U и ш, вообще говоря, зависят от температуры: U = U(T);
w = w(T). Причиной такой зависимости может быть, например,
тепловое расширение кристалла. Действительно, при тепловом
расширении вследствие увеличения межатомных расстояний
силы взаимодействия между ионами ослабевают, так что
образование дефектов и их миграция требуют уже меньших затрат
энергии. Это обстоятельство легко учесть, если предположить,
что энергии образования и миграции дефектов зависят от
межатомного расстояния г0(Т) при данной температуре и что эта
зависимость может быть описана формулой, аналогичной
формуле Борна — Ланде для энергии отталкивания ионов в
решетках ионных кристаллов:
U = bros (6.50)
где bus — константы.
Для w формула аналогична. Тогда, ограничиваясь линейным
членом в разложении энергии по разности г0(Т)—г0(0), можем
записать:
U(T) = /7(0) + d^-{r0(T) - r0(0) ] (6.51)
U(T)= U{0) --L[j(0)ar0T (6.52)
'о
где а — коэффициент линейного термического расширения.
Выражение для w(T) аналогично. В результате в формулах
(6.46) и (6.49) появляется дополнительный множитель перед
экспонентой, а в показателе экспоненты остается значение
энергии активации при Г = 0:
."- = *?е,р(-£) (ИЗ)
,r«=£|!exp(_Ji±Ji) (6.54)
где для предэкспоненциальных множителей введены
обозначения
£стр __ vrfqW ( asU
2 г 1 (6.55)
В« = 1
В формулах (6.53) и (6.54) в отличие от (6.46) и (6.49) энергии
U и ш относятся к Г = 0; £/=(7(0); w=w(0).
k >
■exp
!
as (W + UK)
k \
182
Анализ экспериментальных данных показывает, что в
большинстве случаев значения предэкспоненциальных множителей
в формулах типа (6.53) и (6.54) скоррелированы со значениями
энергии активации так, что приближенно выполняется линейная
зависимость
\пВ = а + Ы) (6.56)
Это эмпирическое правило получило название
компенсационного эффекта. Для его объяснения предлагалось довольно
много различных механизмов; тем не менее строгой теории
компенсационного эффекта до сих пор не создано. Изложенная
здесь упрощенная теория показывает, что одной из возможных
причин компенсационного эффекта может быть температурная
зависимость энергии активации, обусловленная тепловым
расширением кристалла. Чтобы убедиться в этом, достаточно
прологарифмировать формулы (6.55), в результате чего получается
непосредственно уравнение компенсационного эффекта (6.56).
В ионных кристаллах с собственными точечными дефектами
носителями тока являются дефекты двух типов с
противоположными зарядами. В случаях френкелевских или антифренкелев-
ских дефектов различие в их подвижностях представляет,
скорее всего, теоретический интерес, ибо макроскопически здесь
наблюдается униполярная проводимость либо по катионам, либо
по анионам.
Таблица 6.1. Энергии активации миграции дефектов
в ионных кристаллах (в эВ) [16]
Кристалл
LiF
LiCl
LiBr
Lil
NaCl
NaBr
KC1
KBr
Кристалл
AgCl
AgBr
P-AgI
Катионная
вакансия
0,65—0,75
0,40
0,39
0,38—0,43
0,65—0,85
0,80
0,59—0,78
0,66—0,73
Междуузель-
ньш катион
0,055—0,16
0,15
0,65 (|| с);\
0,29 (1 с)
Анионная
вакансия
0,86
0,95-1,11
0,87—1,20
Катионная
вакансия
0,27—0,35
0,34
0,50 (И с);
0,30 (± с)
Кристалл
KI
RbCl
RbBr
Rbl
CsCl
CsBr
Csl
T1C1
Кристалл
CaF2
SrF2
BaF2
CdF2
SrCl2
Катионная
вакансия
0,65—1,21
0,54—0,99
0,80
0,60—0,91
0,60—0,79
0,58
0,58—0,67
0,5
Междуузель-
ный анион
1,02—1,64
0,94
0,62—0,85
Анионная
вакансия
1,36
1,45
1,6
0,35
0,27
0,3
0,2
Анионная
вакансия
0,51—0,56
0,94—1,0
0,42—0,85
0,39—0,44
0,34—0,46
18£
Более сложный случай представляют ионные кристаллы с
дефектами Шоттки, где при равной концентрации вакансий
обоих знаков характер преобладающего переноса определяется
отношением подвижностей обоих сортов вакансий :[18, 36]. Как
видно из формулы (6.40), основным фактором, определяющим
величину подвижности дефектов, является энергия активации
миграции U. В большинстве кристаллов с дефектами Шоттки
характер электропроводности и, в частности, число переноса
ионов определяется этим параметром.
В табл. 6.1 представлены экспериментальные значения
энергии активации миграции основных дефектов для наиболее
хорошо изученных ионных кристаллов с дефектами Шоттки,
Френкеля и антифренкелевскими.
Несмотря на естественный разброс числовых значений U,
полученных разными авторами, таблица показывает совершенно
определенную закономерность. Щелочные галогениды с
катионами меньшего радиуса, чем у анионов, обнаруживают заметно
меньшую энергию активации миграции катионов, нежели
анионов. Поэтому все они, как правило, при умеренных
температурах являются преимущественно катионными проводниками.
Заметная анионная составляющая электропроводности появляется
лишь в области температуры плавления.
Напротив, в галогенидах цезия и таллия, где радиусы
катионов близки к радиусам анионов, энергии активации анионных
вакансий ниже, чем катионных, и ток в этих соединениях
переносится преимущественно анионами.
Различие между электропроводностью кристаллов с
собственными точечными дефектами и со структурной разупорядочен-
ностью отчетливо проявляется при сравнении рис. 6.2 и 6.3, на
которых представлены в координатах Аррениуса эксперимен-
12001000 800 ВОР 500 400 300 200 100 t,°C
РИС. 6.2.
Температурные зависимости электропроводности ионных кристаллов с
собственной разупорядоченностью [16].
184
РИС. 6.3.
Температурные зависимости электропроводности твердых электролитов со
структурной разупорядоченностью [16].
тальные температурные зависимости ионной проводимости ряда
соединений двух указанных типов. Различие обеих групп
твердых электролитов совершенно отчетливо. Прежде всего,
бросается в глаза значительно меньший наклон прямых для кристаллов
со структурной разупорядоченностью по сравнению с
собственной разупорядоченностью, связанный с меньшей энергией
активации. Различие в энергиях активации обусловливает и большое
(на несколько порядков) различие числовых значений
электропроводности в области сравнительно низких температур. На
рис. 6.3 весьма показательна кривая электропроводности
a=AgI, на которой при температуре фазового превращения
Р—их, сопровождающегося переходом от собственной разупо-
рядоченности к структурной, электропроводность скачком
увеличивается почти на 4 порядка.
Значительное многообразие зависимостей наблюдается в
ионных кристаллах, легированных ионами с валентностью,
отличной от валентности основного иона. Формулы (6.30) и (6.37)
показывают, что электропроводность таких систем определяется
Двумя факторами: концентрацией носителей тока и энергией
активации миграции их. Концентрации ионных носителей в
примесном ионном кристалле были вычислены в разделах 5.2 и 5.3Г
соответствующие графики изображены на рис. 5.2 и 5.3.
На рис. 6.4 схематически изображены зависимости ионной
проводимости кристалла, содержащего катионную примесь по-
: Вышенной валентности, от концентрации примеси и обратной
температуры. Эти зависимости получены путем умножения соот-
185
эе.
Т=const
1 !/П
2L '
-^/ 1
/
/
/
/
/ ^
1^ш
1
1
1
1
1
#7
РЯС. 6.4.
Зависимость ионной проводимости кристалла от концентрации примеси (а) и
температуры (б):
I — область собственной проводимости; II — область примесной проводимости, где
комплексы отсутствуют; III — область примесной проводимости, где почти все вакансии
связаны в комплексы.
ветствующих решений разделов 5.2 и 5.3 для концентраций но»
сителей на больцмановский фактор ехр(—U/kT)\ обратно
пропорциональной зависимостью х от Т пренебрежимо по
сравнению с экспоненциальной. Обращает на себя внимание симбат-
яый ход кривых для концентраций доминирующих носителей (см.
рис. 5.2 и 5.3) и для ионной проводимости (см. рис. 6.4,
соответственно а и б). В частности, тангенс угла наклона прямых
участков на рис. 6.4, б отличается от такового на рис. 5.3 на
постоянную величину U/k, связанную с затратами энергии при
элементарном акте миграции носителя.
Таким образом, изложенная упрощенная теория дает
достаточно простые выражения для ионной проводимости кристаллов
с различными типами разупорядоченности. Однако для
примесных кристаллов экспериментальные кривые обнаруживают под-
час весьма причудливый ход, не укладывающийся в рамки изло-
женной упрощенной теории. Зависимость электропроводности от
температуры и содержания примесей удается достаточно надеж-
но интерпретировать лишь для тщательно приготовленных об-
разцов, состав которых точно известен. На рис. 6.5 показан
пример двух образцов NaCl, механизм электропроводности которых
расшифрован достаточно надежным образом. Для «чистого»
образца (с малыми контролируемыми примесями) при высоких
температурах имеется самый крутой участок (I, кривая /)■
Здесь электропроводность определяется формулой (6.49) с
энергией активации, равной Ws + UNa. В области более низких тем^
ператур (участок II) происходит переход от собственной ионной
проводимости к примесной, когда концентрация носителей
определяется лишь содержанием легирующей добавки и не зави-
186
сит от температуры. В этом случае электропроводность
описывается формулой (6.30), в которой концентрация катионных
вакансий с точностью до постоянного множителя порядка
единицы равна концентрации примеси. Наклон графика логарифм
электропроводнвсти — обратная температура на этом участке
определяется лишь энергией активации миграции катионных
вакансий Un&-
При еще более низких температурах, на участке III,
наблюдается несколько более крутой ход прямой, связанный с
ассоциацией дефектов в парные комплексы типа примесный ион —
вакансия (см. раздел 5.3). Согласно формуле (5.42) в этом
случае концентрация свободных катионных вакансий
пропорциональна ехр(—E/2kT)y где Е — энергия диссоциации
комплекса. Поэтому эффективная энергия активации
электропроводности, вычисляемая из наклона соответствующего участка кривой,
равна Unz + E/2.
При легировании кристалла двухвалентным металлом его
электропроводность сильно возрастает (кривая 2). При этом
собственная разупорядоченность не достигается и при наиболее
высоких температурах участок II имеет наклон,
соответствующий энергии активации миграции носителей. В области более
низких температур ход кривой 2 повторяет ход кривой 1 для
«чистого» образца, так что участок III также связан с
ассоциацией дефектов в парные комплексы. Новый изгиб кривой Арре-
ниуса при переходе от участка III к IV, скорее всего, связаьг-с
700500 300 200 100 "t,°C 700 570 470 370 t,°C
РИС. 6.5.
Температурная зависимость электропроводности NaCl:
1 — чистого; 2 — легированного Мп [0,0325% (мол.)] [69].
РИС. 6.6.
Температурная зависимость электропроводности LiF — чистого (/) и
легированного Mg в концентрациях [в % (мол.)]:
2 — 0,006; 3 — 0,013; 4 — 0,024; 5 — 0,043; 6 — 0,077; 7 — 0,12 [70].
187
РИС. 6.7.
Зависимость ионной проводимости твердых
растворов СеОг — LaOi,5 от концентрации
LaOb5 [71]. Числа на
кривых—'температура в °С.
распадом твердого раствора, при
котором к энергии активации
миграции Unsl добавляется
соответствующее слагаемое.
Второй типичный график
температурной зависимости электропро»
водности примесных кристаллов
представлен на рис. 6.6 для LiF,
легированного MgF2. Здесь отчетливо
виден изгиб кривой, связанный с
переходом от примесной к
собственной электропроводности
кристалла. На вставке этого рисунка
О 20 40 60 80 показана концентрационная зависи-
[LaOlf$]fi(M0/i) мость электропроводности
образцов при 470 °С, близкая к зависимости ^^[Mg]1^,
предсказываемой формулой (5.42).
Полученные здесь зависимости, как правило, не выполняются
в кристаллах, содержащих иновалентную примесь при больших
концентрациях, превышающих «1%. В качестве иллюстрации
на рис. 6.7 изображены изотермы ионной составляющей
электропроводности твердых растворов Се02 — LaOi,5, в которых
ионный ток переносится вакансиями кислорода. На этих кривых
отчетливо видны два участка: восходящий (при малых
концентрациях La), качественно соответствующий упрощенной теории,
и нисходящий (при больших концентрациях La); ход
последнего резко противоречит результатам теории.
Причина такого расхождения кроется в сильном
взаимодействии дефектов при их высокой концентрации, которое приводит
не только к образованию двойных и тройных комплексов
примесь— вакансия (см. раздел 5.3), но и к значительному
увеличению энергии активации миграции вакансий U и, следовательно,
к сильному снижению электропроводности в области высоких
концентраций примеси.
Здесь можно провести некоторую аналогию с
металлическими сплавами, в которых взаимодействие различных атомов
приводит к установлению в них ближнего порядка,
уменьшающегося с повышением температуры. Как и в сплавах, в смешанных
ионных кристаллах взаимодействие дефектов наиболее ярко
проявляется при пониженных температурах. При высоких
температурах взаимодействие дефектов менее заметно на фоне
интенсивного теплового движения и восходящий участок кривых
на рис. 6.7, соответствующий простой теории, становится более
188
протяженным. Это приводит к тому, что при повышении
температуры максимум на изотермах рис. 6.7 смещается в сторону
больших концентраций примеси. Если бы в кристалле было
возможно достижение бесконечно высокой температуры,
максимум должен был бы вообще исчезнуть, и тогда кривые
полностью соответствовали бы изложенной здесь теории.
6.4. Подвижность электронов и дырок.
Электронная проводимость
При описании электронной проводимости в твердых телах мы
будем исходить из феноменологического соотношения (6.8),
связывающего парциальную удельную электропроводность /с-частиц
с их подвижностью ик, определяемой как средняя скорость
движения частиц в электрическом поле, напряженность которого
равна единице. В общем случае в твердых телах электронная
проводимость обусловлена движением электронов проводимости
и электронных дырок, поэтому формулу (6.8) следует
записывать в виде
о = е(п~и~ + п+и ') (6.57)
Здесь вместо ке мы используем специальное обозначение для
удельной электронной проводимости сг, чтобы подчеркнуть ее
отличие от ионной проводимости х. Как и в предыдущих главах,
л означает число электронов проводимости и дырок в единице
объема; верхние индексы (—) и ( + ) указывают знак носителя
и относятся соответственно к электронам проводимости и
дыркам.
Концентрации электронных носителей в зависимости от
температуры и состава кристаллов были вычислены в гл. 4 для
полупроводников и в гл. 5 для нестехиометрических ионных
кристаллов. Поэтому в данном разделе задача описания
электронной проводимости твердых тел сводится к определению
подвижности электронных носителей.
В отличие от ионных проводников, для которых, как
отмечалось в предыдущем разделе, выбор носителей бывает двояким,
для электронных проводников носители принято выбирать
вполне однозначно: это электроны в зоне проводимости для
проводников n-типа и электронные дырки для проводников р-типа.
Поэтому для них подвижность всегда относится к указанным
частицам.
Подвижность квазисвободных электронов и дырок.
Рассмотрим сначала кристаллы с широкими зоной проводимости и
валентной зоной, в которых движение электронов проводимости и
Дырок происходит по туннельному механизму и не связано с
затратами энергии активации. В зонной теории в приближении
широких зон электроны проводимости и дырки описываются как
189
квазисвободные частицы, движение которых подчиняется
формально тем же законам, что и свободных классических частиц;
отличие состоит в том, что эффективная масса квазисвободных
электронных носителей т* отличается от истинной массы
электрона. Благодаря своим волновым свойствам квазисвободные
электроны и дырки при движении в идеальной кристаллической
структуре вообще не должны испытывать сопротивления и в
электрическом поле должны разгоняться до бесконечно высокой
скорости. Однако в реальных кристаллах происходит
торможение (рассеяние) электронов как на локальных нарушениях
периодичности кристаллической решетки (примеси и другие
дефекты), так и на тепловых колебаниях атомов, которые также
можно рассматривать как нарушения периодичности решетки.
Рассмотрим поведение квазисвободного электрона (дырки) в
кристалле, помещенном в электрическом поле. Если в отсутствие
поля электроны двигались хаотически со средней скоростью vo,
то под действием поля они приобретают некоторую
дополнительную скорость Av в направлении градиента электрического
потенциала (дырки — в противоположном направлении).
Обозначим через т среднее время между двумя соударениями
электрона с колеблющимися атомами (время свободного пробега
электронов). Так как сила, действующая на электрон в
электрическом поле, равна eV<p, под ее действием электрон движется
с ускорением (efm*)d<$\dx. Следовательно, скорость миграции
электронов в направлении поля между столкновениями
составляет
Л^^гУР (6.58)
а их подвижность равна
а = ezj2m* (6.59)
Выделим в кристалле элемент объема в форме
прямоугольного параллелепипеда, одно ребро которого имеет некоторую
длину К и расположено параллельно направлению движения
электрона, а два других ребра, перпендикулярных направлению
движения электрона, имеют длину, равную единице. Далее,
обозначим через 5 эффективную площадь поперечного сечения
атома, на которой рассеивается электрон (эффективное
сечение рассеяния). Поскольку объем параллелепипеда
численно равен Я, он содержит XNL атомов, и суммарная площад!
сечения рассеяния всех атомов, заключенных в
параллелепипеде, составит kNLS. Теперь выберем значение % таким, чтобы
kNLS = l/2. Это означает, что суммарная площадь сечения
заключенных в параллелепипеде атомов, на которых рассеивается
электрон, составляет 7г от общей площади грани,
перпендикулярной его движению. Следовательно, вероятности электрона
столкнуться с атомами внутри параллелепипеда и вне его
одинаковы и равны 1/2, а характеристическая длина К означает
190
среднее расстояние, проходимое электроном между
столкновениями (длина свободного пробега).
Таким образом, мы видим, что длина свободного пробега
электрона связана с эффективным сечением рассеяния
соотношением
Х= lj(2NLS) (6.60)
С ДРУГ°Й стороны, значение Я может быть выражено через
среднюю скорость электрона. В слабых полях добавочная скорость
направленной миграции под действием поля Av мала по
сравнению со средней скоростью их хаотических блужданий v0
Поэтому последнюю можно приравнять к истинной средней скорости
движения электронов и положить
k = v0>z (6.61)
Сравнивая (6.60) и (6.61), находим:
т=1/(2М^0) (6.62)
и на основании (6.59):
U = 4m*NLSvn (6-63)
Для неионных кристаллов (металлов и валентных
полупроводников) в области не очень низких температур (^102—103 К)
доминирующим механизмом рассеяния квазисвободных
электронов является рассеяние на тепловых колебаниях решетки
(акустических фононах). В этом случае эффективное сечение
рассеяния представляет собой площадь сечения, занятую в
среднем колеблющимся атомом. В свою очередь, мерой этой
площади является среднее по времени значение квадрата смещения
атома из его положения равновесия:
(Ar)2 = (Ajk)2 + (Ay)2 + (Az)2 (6.64)
Поэтому эффективное сечение рассеяния должно быть
пропорционально среднему значению квадрата смещения:
5 = S°(Ar)2 (6.65)
Согласно модели Эйнштейна колеблющийся атом эквивалентен
трем гармоническим осцилляторам, направления колебаний
которых взаимно перпендикулярны. Поэтому потенциальная
энергия колебаний согласно классической механике определяется
выражением
UU0T (АГ) =z 2кЧЪ2 (ДГ)2 (6.66)
где М — масса атома.
^ другой стороны, согласно молекулярно-кинетической
теории средняя потенциальная энергия колеблющегося атома равна
191
половине его полной энергии и составляет
Тпот о
и1ЮТ = ^kT (6.67)
Сравнивая (6.66) и (6.67), видим, что
3k Т
(ДГ)2=_^1_ (6.68)
Подставляя (6.68) в (6.65) и (6.63), получаем для не очень
низких температур:
м = зЩ>07 (аб9)
Рассмотрим теперь кристаллы с различными типами
химической связи.
Металлы. В кристаллах с металлическим характером
химической связи число электронов в зоне проводимости сравнимо с
числом разрешенных квантовых уравнений. Важным следствием
этого является сильное квантовомеханическое вырождение
электронной жидкости, связанное с особенностями квантовой
статистики Ферми — Дирака. Согласно этой статистике
подавляющая часть энергетических уровней, расположенных в зоне
проводимости металла ниже значения химического потенциала
электронов (уровня Ферми), занята электронами, в то время
как уровни, лежащие выше уровня Ферми, в основном
свободны. Поэтому в металлах оказываются практически
невозможными переходы между далеко расположенными уровнями,
требующие больших затрат энергии, и в переносе участвуют
только электроны, энергия которых незначительно (на величины
ttkT) отличается от энергии Ферми. В свою очередь, значение
последней определяется природой металла и практически не
зависит от температуры. В результате этого оказываются
практически не зависящими от температуры средняя кинетическая
энергия электронов проводимости в металле и средняя скорость
их блужданий, связанная с кинетической энергией классической
формулой
UK™ = m*v02l2 (6.70)
В этом случае согласно формуле (6.69) подвижность
электронов в металлах обратно пропорциональна температуре:
и = ЫТ (6.71)
Такая зависимость вытекает из (6.57) для удельной
электропроводности металлов:
a^BjT (6.72)
Значения констант Ъ и В определяются природой металла. При
комнатной температуре подвижность электронов в металлах
192
РИС. 6.8.
Температурная зависимость удельного
электросопротивления натрия [72].
обычно составляет ^102—104 см2/
/(В-с), а удельная
электропроводность ^Ю4—106 Ом-1-см-1.
Типичный пример зависимости
удельного электросопротивления от
температуры изображен на рис. 6.8
для металлического натрия. При
температурах, достаточно удаленных от
абсолютного нуля и от точки
плавления, удельное сопротивление р=1/а линейно растет с
температурой в соответствии с теоретической зависимостью (6.72).
Однако эта зависимость нарушается при очень низких
температурах, при которых энергия колебаний атомов уже не равна ЪкТ,
а убывает при уменьшении температуры по законам квантовой
статистики. Линейная зависимость р от Т также нарушается в
области предельно высоких температур, близких к точке
плавления. Это явление связано с быстрым возрастанием при
высоких температурах концентраций вакансий, которые приводят к
дополнительному рассеянию электронов и к появлению
избыточного сопротивления металла. Измерения избыточного
сопротивления иногда используют для оценки концентраций вакансий в
металлах.
Валентные полупроводники. При достаточно высоких
температурах и малых концентрациях примеси электронный газ в
полупроводниках в отличие от металлов не вырожден и
подчиняется классической статистике Больцмана, согласно которой
кинетическая энергия электрона в среднем равна
m*v02j2 = —kT
(6.73)
Определяя из этого соотношения среднюю скорость электронов
и подставляя ее в (6.69), получаем для их подвижности
-3
где значение коэффициента
•■ЬТ'
екШч*
(3k) m
(6.74)
(6.75)
NLS°
определяется природой полупроводника.
В сложных кристаллических решетках наряду с
акустическими колебаниями существуют оптические, которые также могут
13—321
193
участвовать в рассеянии электронов. Согласно квантовой теории
рассеяния число таких столкновений электрона
пропорционально эффективному числу оптических фононов:
[ехр(Ьопт1/гТ)-1]~1
При сравнительно низких температурах единицей в квадратных
скобках можно пренебречь, откуда для подвижности вытекает
зависимость
u^exp(bonTlkT) (6.76)
более сильная, чем степенная зависимость (6.74) для рассеяния
на акустических фононах. Поэтому, если рассеянием на
оптических фононах нельзя полностью пренебречь, в
высокотемпературной области может наблюдаться зависимость
и==ЬТ~р (6.77)
где значение показателя степени р больше 3/2 и может
достигать 3.
При низких температурах (порядка единиц — десятков
градусов Кельвина), когда тепловые колебания не оказывают
существенного сопротивления движению электронов, в
полупроводниках наблюдается другой механизм рассеяния носителей, а
именно на ионизованных примесях, В отличие от нейтральных
примесных центров примесные ионы создают вокруг себя
достаточно сильное электрическое поле, искривляющее траектории
движения электронов. Задача о таком виде рассеяния
аналогична задаче о рассеянии а-частиц на атомах решетки, решенной
Резерфордом. Так как при понижении температуры скорость
движения электронов уменьшается, время, проводимое им в
окрестностях примесного иона, возрастает и соответственно
усиливается рассеяние и уменьшается подвижность. Теория такого
рассеяния дает зависимость
Л
u = bTf[F] (6.78)
где [sF] — концентрация примесных ионов.
Здесь любопытна следующая особенность. В примесных
полупроводниках концентрация примесных ионов равна
концентрации электронных носителей: [D+] = [e~]; [A~] = [e+]. Поэтому
формула (6.57) с учетом (6.76) дает для электропроводности
полупроводника при рассеянии на примесных ионах значение,
не зависящее от концентрации последних.
При наличии двух или более механизмов рассеяния
вероятности столкновений, обусловленных каждым механизмом,
суммируются. Так как вероятности столкновений обратно
пропорциональны соответствующим длинам свободного пробега, а
следовательно, подвижностям при каждом механизме рассеяния,
194
рИС. 6.9.
Температурная зависимость
подвижности электронов
проводимости и дырок в валентных
полупроводниках при рассеянии на
акустических фононах и
ионизованной примеси; кривая 2
соответствует большей концентрации
примеси.
для итоговой подвижности можно записать:
к
//
А /
—-А <-
II
U
В этом случае для атомных полупроводников
3 -3
1
[Р]Г
ЦТ
(6.79)
(6.80)
Эта зависимость изображена схематически на рис. 6.9 в
логарифмических координатах, в которых оба слагаемых формулы
(6.80) изображаются прямыми линиями с наклоном ±3А.
С увеличением концентрации примесей левый участок графика,
связанный с рассеянием на ионах примеси, смещается вниз, а
правый участок, связанный с рассеянием на тепловых
колебаниях, остается неизменным. Поэтому максимум кривой на
рис. 6.9 смещается вправо, в сторону более высоких температур.
Для большинства полупроводниковых материалов максимум
подвижности находится в области очень низких температур
(20—80 К).
В зависимости от природы полупроводника, содержания
примеси и температуры подвижность носителей в полупроводниках
может изменяться в широком диапазоне — от ^ 1 до
^106 см2/(В-с).
На рис. 6.10, а изображена в логарифмических координатах
температурная зависимость подвижности квазисвободных
электронов в германии, легированном мышьяком, для той же серии
образцов, для которой в гл. 4 обсуждалась температурная
зависимость концентрации электронов (см. рис. 4.3). На
рис. 6.10, а обращает на себя внимание, во-первых, довольно
широкий диапазон изменения подвижности (3 порядка
величины), во-вторых, различный характер кривых при малых и
больших содержаниях примеси.
При наименьшем содержании мышьяка (атомная доля 10~9
для образца 1) подвижность монотонно убывает с ростом
температуры. При этом кривая 1 идет почти параллельно
пунктирной прямой, тангенс угла наклона который равен —3/2 и
соответствует теоретической зависимости (6.80) в области высоких
13* 195
300100 50 30 20 15 12 J, К
РИС. 6.10.
Температурная зависимость
подвижности носителей (а) и
электропроводности (б) германия,
легированного мышьяком 154].
Концентрация As возрастает от 1-го
к 6-му образцу.
температур, где преобладает рассеяние электронов на
акустических колебаниях решетки.
При увеличении содержания мышьяка на кривых появляется
максимум, причем левые участки кривых, соответствующие
рассеянию на примесях в области низких температур, закономерно
196
смещаются вниз, в качественном соответствии с выводами тео-
^И На рис. 6.10,6 изображен график Аррениуса для
электропроводности тех же образцов. Как и на рис. 4,3 для концентрации
носителей, пунктирная прямая относится к области собственной
электропроводности.
За исключением кривой 69 отвечающей вырожденному
состоянию, кривые для примесной электропроводности имеют
четкие максимумы. Нисходящие участки кривых в области
промежуточных температур (от 20—80 К до комнатной) соответствуют
горизонтальным участкам на рис. 4.3 для концентрации
носителей в области истощения доноров; здесь уменьшение
электропроводности с ростом температуры обусловлено уменьшением
подвижности носителей при их постоянной концентрации.
Ионные кристаллы. Как уже указывалось в разделе 1.6,
механизм движения электронов в ионных кристаллах зачастую
отличается от такового в валентных полупроводниках и
осуществляется перескоками их из устойчивого состояния на одном ионе
в аналогичное состояние на другом. В отличие от туннельного
механизма движения квазисвободных электронов в валентных
полупроводниках такой механизм движения локализованных
электронов называют прыжковым.
Различное поведение электронов и дырок в кристаллах с
ковалентной и ионной химической связью обусловлено
различным характером взаимодействия избыточного электрона с
окружающей средой кристалла. В любом веществе избыточный
электрон, находящийся в зоне проводимости, или дырка в
валентной зоне поляризуют окружающую среду. В телах,
состоящих из нейтральных атомов, таких, как валентные
полупроводники, поляризация сводится к образованию электрических
диполей на каждом из окружающих атомов благодаря смещению
их внешних электронных оболочек относительно положительного
ионного остатка. Такой вид поляризации соответствует
высокочастотной (оптической) диэлектрической постоянной и
характеризуется очень малым временем релаксации, при котором
поляризационное искажение среды успевает следовать за
вызвавшим его избыточным электроном при движении последнего по
кристаллу. Поэтому энергетическое состояние кристалла не
изменяется при переходе электрона от одного узла к соседнему,
и движение электронных носителей по кристаллу не требует
затрат энергии, т. е. электроны проводимости и дырки
являются квазисвободными частицами.
Иная картина наблюдается в ионных кристаллах. В них
заряженные ионы, окружающие избыточный электрон, под
действием его электрического поля целиком смещаются из своих
положений равновесия; такое явление называется
поляризацией смещения и дает определенный вклад в низкочастотную
(статическую) диэлектрическую постоянную среды. В отличие
от высокочастотной поляризации электронных оболочек поля-
197
ризация смещения характеризуется сравнительно малым
временем релаксации и при переходе электрона от одного узла к
другому не успевает следовать за ним. Поэтому перескок электрона
в соседний узел сопровождается исчезновением поля
поляризации вокруг исходного узла и возникновением его вокруг
конечного. Такой акт в общем случае может быть сопряжен с
разрывом связи электрона с полем поляризации и, следовательно, с
затратами энергии. Поэтому избыточные электроны и дырки в
ионных кристаллах, вообще говоря, уже не являются
квазисвободными частицами. Вместе с сопровождающим их полем
поляризации они образуют особые квазичастицы, получившие
название поляронов.
Принято различать поляроны большого радиуса и поляроны
малого радиуса. К первому типу относят поляроны, у которых
эффективный радиус поляризованной области гораздо больше
постоянной решетки; ко второму типу — поляроны, у которых
эффективный радиус сравним с постоянной решетки или
меньше ее.
Для поляронов большого радиуса квантовая теория
строится в континуальном приближении, когда область
поляризации рассматривается как сплошная среда с определенными
значениями высоко- и низкочастотной диэлектрических
постоянных. В такой теории, используя разумные приближения,
удается свести задачу о движении полярона к формализму зонной
теории и таким образом описать полярон большого радиуса
снова как квазисвободную частицу. Однако его эффективная
масса т** оказывается больше, чем соответствующее значение
т* в жесткой решетке, когда поляризация смещения
отсутствует. Это увеличение эффективной массы полярона тем сильнее,
чем больше так называемая константа связи а,
характеризующая поляризуемость кристалла благодаря смещению
ионов. Так, в предельных случаях слабой и сильной связи
эффективная масса полярона большого радиуса удовлетворяет
соотношениям [73]
/га** «(l + — \m* (а<!)
^ 6 ; (6.81)
m**^0,U2a*m* (а > 1)
Так как в зонной теории эффективная масса носителей обратно
пропорциональна ширине соответствующей зоны, соотношения
(6.81) предсказывают уменьшение ширины зон за счет
поляризации смещения, причем в случае сильной связи (а^>1) —очень
резкое.
В достаточно чистых ионных кристаллах при низких
температурах доминирует рассеяние поляронов на акустических
колебаниях решетки, аналогичное описанному выше для
квазисвободных электронов. Вклад столкновений с оптическими фоно-
нами при низких температурах также аналогичен случаю
квазисвободных электронов и описывается зависимостью (6.76), так
1ЯЯ
что подвижность поляронов в низкотемпературной области
убывает с ростом температуры. При высоких температурах
kT^hvonr> поляроны большого радиуса рассеиваются
преимущественно на оптических фононах; в этой области температурная
зависимость их подвижности довольно слабая и в первом
приближении может быть аппроксимирована степенной функцией
и = ЬТр (6.82)
с показателем степени — Уг^Р^1^.
Наиболее интересные результаты получаются для поляронов
малого радиуса, теория которых строится с учетом
дискретного строения кристалла. Такое состояние может возникать при
наличии избыточного электрона в узкой зоне проводимости или
дырки в узкой валентной зоне ионного кристалла и при
достаточно высокой поляризуемости смещения ионов. В этом случае
при абсолютном нуле температур зависимость эффективной
массы полярона от константы связи оказывается еще более
сильной, чем в случае поляронов большого радиуса:
/га** ~ exp (const • а) (6.83)
соответственно (6.83) уменьшается и ширина зоны,
образованной энергетическими уровнями поляронов.
Энергетические зоны поляронов малого радиуса, весьма
узкие уже при 7 = 0, при повышении температур еще более
сужаются, а эффективная масса поляронов еще более возрастает,
причем в области высоких температур, kT^hv0117, — по
экспоненциальному закону:
/га** = яг* exp (const • Т) (6.84)
Увеличение эффективной массы поляронов приводит к
уменьшению ускорения, испытываемого ими в электрическое поле за
период между столкновениями, а следовательно, и к
уменьшению подвижности. Поэтому в предельном случае сильной связи
поляроны малого радиуса правильнее рассматривать не как
квазисвободные частицы, а как некие квазичастицы,
локализованные на одном-двух определенных ионах. Так, в LiF при
температуре 110 К подвижность дырок настолько мала
(1,7-10—14 см2-В-1-с-"1), что дырка совершает всего один
прыжок в минуту, что на 15 порядков ниже частоты колебаний
атомов. Этот результат нельзя объяснить в рамках зонной
теории. Более того, в предельном случае сильной связи полярон-
ные зоны становятся настолько узкими, что зонная модель
утрачивает смысл, так как неопределенность энергии, вытекающая
из общего квантовомеханического соотношения
неопределенностей и связанная с волновыми свойствами электронов,
становится сравнимой с шириной поляронной зоны. В этом случае в
более строгой, нежели зонная, теории стационарные состояния
поляронов малого радиуса следует описывать локализованными
199
функциями, соответствующими электрону, расположенному в
узле решетки.
Локализованное состояние избыточного электрона или дырки
в описанной ситуации не связано с какими-либо посторонними
факторами, а является следствием особенностей взаимодействия
электронов с собственной решеткой кристалла. Поэтому его
часто называют автолокализацией. Физически состояние
автолокализации обусловлено достаточно большим значением
энергии взаимодействия избыточного электрона с
индуцированным им полем поляризации, поскольку для перескока
электрона в соседний узел необходимо преодолеть это взаимодействие и,
следовательно, затратить определенную энергию активации.
В сильно полярных кристаллах, таких как галогениды и оксиды
металлов, эта энергия активации достигает десятых долей элект-
ронвольта. Поэтому перескоки избыточного электрона (дырки)
в соседний узел могут происходить благодаря тепловым флук-
туациям, причем тем чаще, чем выше температура.
Таким образом, полярон малого радиуса основное время
жизни проводит в автолокализованном состоянии, когда
избыточный электрон или дырка вращаются вокруг определенных
ионов и только изредка перескакивают к соседним, в конечном
итоге хаотически блуждая по кристаллу. В этом отношении
картина прыжкового механизма миграции электронных
носителей в ионных кристаллах аналогична механизму ионной
проводимости, рассмотренному в предыдущем разделе, причем
движение электронов проводимости аналогично движению между-
узельных ионов, а дырок — движению вакансий.
В начале настоящего раздела мы условились относить
подвижность к избыточным электронам проводимости и дыркам,
поэтому соответствующие формулы можно непосредственно
получить из (6.40) и (6.41). При сравнительно малых
концентрациях носителей, когда их число гораздо меньше числа уровней
в зонах, доли занятых уровней в валентной зоне и свободных
уровней в зоне проводимости, соответствующие символам [Vi]
и [кк] в формулах (6.41) и (6.40), можно положить равными
единице. Тогда для подвижности обоих типов электронных
носителей при прыжковом механизме получаем единое выражение:
Отсюда на основании (6.57) находим удельную электронную
проводимость:
0==:TF-[n"exp(~I7-) + "+exp(-T7)] <6-86)
где п~ я п+ — числа поляронов п- и р-типа в единице объема.
Прыжковый механизм электропроводности играет важную
роль в оксидных соединениях переходных металлов со структур
200
ой шпинели, в которых катионы находятся в двух валентных
состояниях (см. раздел 1.3). Так, в кристалле магнетита Fe304
перенос электронов осуществляется перескоками электронов
между ионами Fe2+ и Fe3+, причем, несмотря на постоянные
значения концентраций обоих валентных состояний железа,
электропроводность магнетита при высоких температурах
экспоненциально растет с температурой с энергией активации около
О 1 эВ. По-видимому, здесь также можно применить модель
поляронов малого радиуса, в которой энергия активации
перескока электрона U связана с поляризацией смещения ионов
кислорода в неэквивалентных полях ионов Fe2+ и Fe3+.
Отмеченная выше аналогия в поведении ионных дефектов и
поляронов малого радиуса при прыжковом механизме миграции
отнюдь не случайна, несмотря на различную природу носителей
тока. Эта аналогия при высоких температурах обусловлена
чертой, общей для обоих типов проводников, — термической
активацией носителей тока, подчиняющейся статистике Больцмана.
Однако при низких температурах поляроны малого радиуса
проявляют черту, совершенно не свойственную ионным
дефектам и отражающую волновые свойства электронов —
способность их туннелировать сквозь решетку без затрат энергии
активации. Такая черта, как было описано выше, совершенно
естественна в приближении широких зон, когда поляроны
обладают свойствами квазисвободных частиц. Между тем
оказывается, что и в случае узких зон может существовать такая
область промежуточных температур, в которой зонная теория уже
не применима, но наряду с прыжковым механизмом существует
конечная вероятность электрона протуннелировать сквозь
потенциальный барьер, разделяющий локализованные состояния.
В этом случае общая подвижность полярона определяется
суммой вкладов обоих механизмов:
и = и1ук+ипрыж (6.87)
Если при туннельном механизме доминирует рассеяние на
оптических фононах, подвижность поляронов обратно
пропорциональна эффективному числу фононов:
«тун~ [ехр(/Ь0ПТ/£Г)— 1] (6.88)
и при kT<^hvonT экспоненциально уменьшается с ростом
температуры. В этом случае эффективная энергия активации
подвижности, определяемая как
U* = dlna (6.89)
d(ljkT)
отрицательна и равна —hvonT. Если же при туннельном
механизме доминирует рассеяние на акустических фононах,
подвижность поляронов убывает с температурой ~T~S^ в соответствии
с формулой (6.74).
201
РИС. 6.11.
Температурная зависимость
подвижности поляронов малого радиуса в
ионных кристаллах при рассеянии на
оптических фононах.
//Г
График температурной зависимости подвижности поляронов,.
соответствующий формуле (6.88) при рассеянии на оптических
фононах, схематически изображен на рис. 6.11 в координатах
Аррениуса. Здесь обращает на себя внимание ход кривой,
противоположный представленному на рис. 6.8; в отличие от
квазисвободных электронов в атомных полупроводниках
подвижность поляронов малого радиуса имеет минимум в области
промежуточных температур. Пунктирный участок кривой
изображает переход к рассеянию туннелирующих поляронов на
заряженных точечных дефектах решетки, играющих в ионных
кристаллах ту же роль, что и примесные ионы в валентных
полупроводниках.
Анализ показывает, что температурная зависимость
подвижности электронных носителей тока в ионных кристаллах может
быть весьма сложной, особенно при наложении различных
механизмов рассеяния. Это приводит к значительным трудностям
при интерпретации экспериментальных данных по
температурной зависимости электропроводности нестехиометрических
ионных кристаллов. Положение осложняется еще и тем, что
концентрации как ионных, так и электронных дефектов в таких
системах описываются простыми выражениями (5.61) и (5.91)
только в предельных случаях, когда можно ограничиваться
учетом двух типов доминирующих дефектов. В промежуточных же
случаях интерпретация экспериментальных результатов часто
оказывается чрезвычайно затруднительной. Поэтому
электропроводность нестехиометрических кристаллов обычно описывают
полуфеноменологическими выражениями, аналогичными
формулам (5.61) и (5.91) для концентрации носителей:
Л
а = Л/4г9ехр(-г/£Г) (6.90)
для чистых кристаллов и
а = A [F]s /?xmexp(—ejkT) (6.91)
для примесных. При такой записи температурная зависимость
множителя А аппроксимируется экспоненциальной, а
соответствующий показатель экспоненты включается в эффективную
энергию активации е. Затем для исследуемого интервала
температур, потенциальных давлений Х2 в газовой фазе и концент-
\ V
1 \к
/ Ы>
/ ~ К
опт
N
\
\ |
\
202
РИС. 6.12.
Температурная зависимость
электропроводности оксидов металлов
в атмосфере воздуха.
150012001000 800 600 500 400i"C
раций примеси в образце ^>
определяют эмпирические S
значения параметров т и s ^
и путем их сравнения с тео- §
ретическими значениями ?
(см. табл. 5.3 и 5.4) подби- ^
рают модель дефектной ^
структуры, наиболее близко
соответствующую
экспериментальным данным.
Несмотря на свой
полуэмпирический характер,
формулы (6.90) и (6.91) в
целом неплохо описывают
температурную зависимость
электропроводности нестехиометрических кристаллов,
наблюдаемую на эксперименте. Это хорошо иллюстрирует рис. 6.12, где
в координатах Аррениуса изображены экспериментальные
температурные зависимости электропроводности наиболее
подробно изученных оксидов металлов в атмосфере воздуха (ро2 =
= 0,21 бар), построенные по данным различных авторов [24, 74].
Действительно, здесь в большинстве случаев опытные данные
описываются прямыми линиями. Ломанные линии,
наблюдающиеся для некоторых оксидов, чаще всего свидетельствуют об
изменении типа преобладающих дефектов при изменении
температуры в достаточно широком интервале.
Несколько сложнее обстоит дело с зависимостью
электропроводности тех же оксидов от давления кислорода, изображенной
в логарифмических координатах на рис. 6.13. Здесь
прямолинейные участки кривых, отвечающие формуле (6.90) при
постоянных значениях параметра ту наблюдаются лишь в
ограниченном интервале давлений; в целом для кривых характерно
значительное изменение наклона в исследованном интервале lgPo2.
Из проводников n-типа, характеризующихся уменьшением
логарифма электропроводности с ростом lg/?02, исключение
составляют Ti02 и Се02, для которых значение параметра m
лежит в сравнительно узких пределах от —4 до —5,5. Из
проводников р-типа, характеризующихся увеличением логарифма
электропроводности с ростом р<э2, таким исключением,
по-видимому, является СоО, для которого параметр т изменяется от
+ 6 до +4.
Ряд оксидов, таких как MnO, A1203, Sc203 и СаО,
обнаруживает амфотерные свойства: характер изменения их электро-
203
проводности соответствует я-типу при достаточно низких
давлениях кислорода и р-типу при достаточно высоких давлениях.
При некоторых промежуточных давлениях, отвечающих стехио-
метрическому составу этих оксидов, их электропроводность про-
ходит через минимум.
Для оксидов Y203, Zr02, НЮ2, Th02, U02 и Ta205 в широкой
области давлений наблюдаются постоянные значения
электропроводности. Исследование чисел переноса ионов и электронов
по методу, описанному в следующем разделе, показывает, что в
области постоянных значений электропроводность таких
оксидов является в основном ионной, а при переходе к наклонным
участкам — смешанной ионно-злектронной.
В областях смешанной ионно-электронной проводимости ее
зависимость от давления неметалла легко найти, исходя из
следующих соображений.
Очевидно, что доля ионной проводимости нестехиометриче™
ского кристалла максимальна при минимальной концентрации
электронных носителей, т. е. вблизи стехиаметрического состава.
Согласно решениям I группы гл. 5, в области, непосредственно
прилегающей к стехиометрическому составу, доминирующими
дефектами чаще всего являются полностью ионизованные
вакансии или междуузельные ионы, причем их концентрации в
этой области практически не зависят от давлений неметалла.
Концентрации же электронных носителей в этой области
изменяются пропорционально рх2±1/22х. Поэтому, полагая, что
подвижности как ионных, так и электронных носителей не зависят
i —J . J i I I ! I I
~:6 44 42 40 -8 -5 -4 -2 0
L9PoJfop)
РИС. 6.13.
Зависимость электропроводности оксидов металлов от парциального давлении
кислорода при температурах:
800 °С (пунктирные линии); 1000 °С (сплошные линии); 1500 °С (штрихпунктирные линии).
204
О -5 -Ю -151др0г№)
-] Г
РИС 6.14.
Ионная и электронные составляющие
проводимости твердого электролита
0§2 ZrO2+0,10 YOb5+0,08 MgO в
зависимости от парциального
давления кислорода при 1000°С
(сплошные прямые) и 800 °С (штрихпунк-
тирные прямые) [75].
-5 -10 -15 -20 -25
1др0г(бар)
от /?х2, для суммарной электропроводности нестехиометрическо-
го ионного кристалла вблизи минимума можно записать:
X + О = А ~\- А р.
+ А+р1
2*Х
(6.92)
Точно такая же зависимость получается и для примесных
ионных кристаллов, если в соответствующей области давлений
неметалла доминирующими собственными дефектами
являются полностью ионизованные вакансии или междуузельные
ионы, концентрации которых контролируются примесью и не
зависят от рх2 (см. табл. 5.4, четыре последние строки).
Типичный график зависимости ионной и электронных составляющих
электропроводности приведен на рис. 6.14 для твердого
электролита на основе диоксида циркония, где наклон прямых для
электронных составляющих соответствует зависимостям а^
~Р±1/2о2-
При анализе экспериментальных графиков типа
изображенных на рис. 6.13 легко допустить ошибку в определении
параметра т, если полностью игнорировать ионную проводимость
даже при составах, казалось бы, далеких от
стехиометрического. Действительно, из формулы (6.92) видно, что по мере
Удаления от стехиометрического состава тангенс угла наклона
касательной к кривой lg (>с+б)—lgpx2 изменяется от нуля,
отвечающего минимуму кривой, до значений ±112%х,
отвечающих практически чисто электронной проводимости р- или я-ти-
па.^ При относительно больших значениях ионной
составляющей минимум кривой может быть очень пологим и ошибка в
определении параметра т по наклону ограниченного участка
кривой может быть значительной. В качестве примера такие
графики приведены на рис. 6.15, а для оксида лантана;
пунктирные касательные к восходящим участкам кривых имеют
наклон, соответствующий значению га = 6. Между тем
практически горизонтальный ход левых участков кривых позволяет
предположить, что исследованный интервал давлений
кислорода соответствует окрестностям стехиометрического состава и
205
igpojna) ° 5 >° PSt№
РИС. 6.15.
Зависимость электропроводности La203 от давления кислорода в координатах
lg(>c+o)—lgpo2 (a) [76] и (>с+а)—ро2х/4(б).
РИС. 6.16.
Температурная зависимость коэффициентов самодиффузии металлов и
элементарных полупроводников [79].
общая электропроводность складывается из ионной и
электронной р-типа:
х+а^х + Л+т^ (6.93)
Последняя зависимость представляет собой уравнение
семейства прямых линий в координатах (и+б)—Ро21/4- Такие
координаты использованы на рис. 6.15,6, на котором
экспериментальные точки удовлетворительно укладываются на прямые.
Отрезки, отсекаемые прямыми на оси ординат, характеризуют
не зависящую от ро ионную составляющую
электропроводности. Аналогичное построение целесообразно проводить и при
электронной проводимости я-типа; в этом случае прямые
должны получаться в координатах (х+6)—Рх2~1/2гх-
6.5. Числа переноса ионов
При любом исследовании транспортных свойств или
реакционной способности твердых тел прежде всего встает вопрос о
том, является ли данное вещество ионным или электронным
проводником или же обладает обоими типами
электропроводности. Количественной характеристикой природы
электропроводности являются числа переноса ионов и электронов, опреде-
206
яемые согласно (6.4) как доли обоих типов
электропроводности:
t^-j-; te = ~- (6-94)
Здесь общая ионная проводимость складывается из
парциальных проводимостей всех сортов ионов, а электронная
проводимость о — из вкладов электронов и дырок.
К настоящему времени разработано довольно большое
число методов определения чисел переноса. Наиболее прямыми из
них являются различные модификации метода Тубандта,
сводящиеся к измерению количества вещества, выделившихся на
электродах при электролизе исследуемых образцов, и
сопоставлению результатов измерений с количеством пропущенного
электричества. Недостатком метода Тубандта являются
значительные трудности его экспериментального оформления
(спекание смежных образцов, прорастание дендритов и т. д.).
В экспериментальном отношении более просты методы,
основанные на использовании блокирующих электродов или
ионных фильтров, позволяющих измерять раздельно электронный
и ионный токи. Так, блокирующие электроды могут быть
выполнены из любого индифферентного металла (обычно
платина); при малых напряжениях они пропускают только
электронный ток и позволяют определить электронную проводимость.
Ионными фильтрами служат таблетки твердых электролитов,
проводящих ток по соответствующим ионам, но не
пропускающих электроны. Эти методы особенно эффективны при
измерении малой электронной проводимости на фоне большой ионной
и наоборот. Однако и они не свободны от побочных явлений,
сопутствующих пропусканию постоянного тока и затрудняющих
интерпретацию результатов измерений.
Указанных недостатков лишены косвенные методы
определения чисел переноса, основанные на измерении
электродвижущих сил различных гальванических ячеек, в которых в
качестве электролитов используются образцы исследуемых твердых
тел. Обычно э. д. с. измеряется компенсационным методом,
когда ток через ячейку не проходит и, следовательно, состояние
образцов наиболее близко к равновесному. Несомненными
достоинствами методов, основанных на измерении э.д. с,
являются высокая точность и воспроизводимость измерений и
простота экспериментального оформления.
Мы остановимся на наиболее простом методе э. д. с,
предложенном Вагнером и в настоящее время являющимся самым
распространенным. Рассмотрим сначала простейший случай
бинарного соединения МХГ, выполняющего функции твердого
электролита в гальванической ячейке типа
(I) Pt, М(ам)|МХг|М(ам), Pt (II) (6.95а)
207
или типа
(I) Pt, Х2 (рх2) | MX, | Х2 (/?х2), Pt (ID (6.956)
В ячейке (6.95а) Pt (или любой другой инертный материал)
служит токоподводами; электроды могут быть выполнены из
сплавов, содержащих металлический компонент исследуемого
соединения в различных концентрациях (а'м и а"м — активно^
сти М в сплавах; если один из электродов выполнен из
чистого металла М, его активность принимается за единицу).
В ячейке (6.956) электроды из индифферентного материала
(Pt) находятся в атмосферах с различным парциальным
давлением неметаллического компонента Х2.
Будем считать, что ток через образец МХГ переносится
благодаря раздельному движению ионов Мгм+, Xzx~ и электронов
е- (безразлично, по электронному или дырочному механизму).
Тогда поток частиц каждого /с-сорта из трех перечисленных
будет определяться уравнением Вагнера (6.21):
Jk = *_ (Wk + qjiV^ (6.96)
(для электронов ке = о). Если электрический ток через ячейку
отсутствует, потоки ионов и электронов связаны между собой:
Х?Л=0 (6-97)
К
Подставляя сюда потоки из (6.96) и разрешая полученное
уравнение относительно градиента электростатического
потенциала, находим:
W=-I-^VPv (6.98)
Суммирование проводится по заряженным частицам: катионам,
анионам и электронам (tK — число переноса /с-частиц).
Поскольку катион Мгм+ есть атом Мх, лишенный 2м
электронов, а анион Xzx~ — атом Xх с Zx лишними электронами,
химические потенциалы ионов можно выразить через
химические потенциалы соответствующих нейтральных атомов и
электронов:
М ' х —
1 М • М М v e
(6.99)
Кроме того, воспользуемся соотношением Гиббса
в форме
х dx м dx
— Дюгема
(6.100)
208
аведливой при не очень больших отклонениях от стехиомет-
°йческого состава, когда отклонение Nm/Nx приблизительно
оавно zxJzm- Подставляя (6.99) и (6.100) в (6.98) и выполняя
суммирование, получаем:
v^^^W-+±W~ = ^W^+±w7 (6.10!)
пе и = хКк+°) —суммарное число переноса катионов и анионов.
Электродвижущая сила ячейки складывается из скачков
потенциала на границах раздела электролит — электрод и
падения потенциала по толщине электролита L:
L
£=(ср0_?') + (?"_/) + fv?d* (6.102)
6
где индексы 0 и L относятся к поверхностям электролита, граничащим с
электродами I и II: штрихи указывают номер токоподвода.
Для определения скачков потенциала на межфазных
границах запишем условие равновесия на границах относительно
4 электронов [см. Приложение, формулу (П.47)]:
e(f — ?') = ^ — Pe\ e{f — yL) = Ve — \4 (6.103)
Если токоподводы выполнены из одного материала,
химический потенциал электронов в них одинаков, \х'е = уь'е, и
подстановка (6.101) и (6.103) в (6.102) приводит к формулам
Вагнера для э.д. с. ячеек (6.95а) и (6.956) соответственно:
Щ
^0) (6.104)
Е = — Г ttdv^
zYe J
х (0)
Здесь пределами интегрирования являются значения
химических потенциалов атомов М и X в приэлектродных слоях
электролита.
Вывод формул Вагнера (6.104) легко обобщить на
многокомпонентный ионный кристалл произвольного состава. При
этом безразлично, является ли он твердым раствором или
химическим соединением.
Для определенности рассмотрим кристалл, состоящий из
нескольких сортов катионов Mz*+ и общего аниона Xх х-.
Тогда выражение (6.98) для градиента электростатического
потенциала распишется в виде
*<? = -!-*-v#+
14—321
р— 1 С * л..х
(6.105)
209
Здесь индекс к нумерует только катионы; \х+к и \х~х означают
химические потенциалы катионов с зарядом zKe и анионов с
зарядом —zxe.
Перейдем с помощью соотношений (6.99) от химических
потенциалов ионов к химическим потенциалам атомов: \ххк для
металла сорта к, jlixx для неметалла. Тогда, учитывая
очевидное равенство
*1=1*к+*х (6-Ю6)
К
после элементарных преобразований получаем значения э.д. е.:
а) для ячейки, на электродах которой фиксированы
активности некоторого произвольного металла М из набора /с-сортоь
1 Щ (L) (L)
Е=-Т7 ^м + -V f ^мх,-1тг1 'Ах (6-107«)
м (0) м (0) к к (0)
б) для ячейки с фиксированными давлениями Х2
х (0) к к (0)
Здесь символом \хкх обозначена комбинация
P,x=tf+-7j-Px (6-108)
очевидно, это есть химический потенциал бинарного соединения
/с-металла с неметаллом, отнесенный к формульной единице
MKXZ /гх, если это соединение рассматривать как компонент
сложной системы.
Если катионный состав электролита постоянен по всей
толщине, интегралы по химическим потенциалам бинарных
соединений dfiMX и d\iKx B (6.107) обращаются в нуль, и мы снова
получаем для э.д.с. формулы Вагнера (6.104).
При выводе формул (6.104) мы предполагали, что
электрический ток через ячейку не протекает. Однако следует иметь
в виду, что даже при разомкнутых токоподводах парциальные
токи ионов и электронов, вообще говоря, отличны от нуля.
При этом катионы и анионы движутся навстречу друг другу,
в стороны понижения своего химического потенциала; заряды
же, переносимые ионами от одного электрода к другому,
стекают обратно благодаря электронной проводимости
электролита. Поэтому истинное равновесие в ячейке возможно только
при полном отсутствии электронной проводимости, ti=l.
Однако в большинстве случаев условия таковы, что
лимитирующей стадией процесса переноса вещества через ячейку
является его миграция через твердый электролит (при
достаточной толщине последнего), а переход частиц через межфазные
границы протекает без существенных затруднений. В этих слу-
2 10
состояния на границах близки к равновесным и химиче-
4&ие потенциалы М и X в приэлектродных слоях электролита
СК(Ьормулах (6.104) можно заменить на соответствующие
значения в электродах ц'м и ji"x. Тогда, используя общее
термодинамическое соотношение для металла
jj.m = const + &Г1пам (6.109a)
й полагая газ Х2 идеальным
Рх = const + —- In p (6.1096)
можем переписать (6.104) в виде
kl
Е =— [tdlna
М
kT "
Z: = f ttd\npXo
*M* n
2zve
(6.110)
При чисто ионной проводимости электролита, когда ti=l
во всем интервале изменения химических потенциалов,
формулы Вагнера (6.110) переходят в формулы Нернста:
£о^_^_1па«
гме
ме ям
(ели)
Е° = Ш— In -^~
2г*е Р\
Значения э.д. с, удовлетворяющие уравнениям Нернста
(6.111), называются термодинамическими. Смысл этого
термина заключается в том, что при отсутствии электронной
проводимости разомкнутые ячейки находятся в равновесных
условиях, и формулы (6.111) в отличие от формул Вагнера
могут быть выведены чисто термодинамическим методом.
Естественно, что и результат при этом выражается только через
термодинамические переменные — активности или парциальные
Давления компонентов.
Формулы Вагнера (6.110) обычно записывают в более
компактном виде. Для этого воспользуемся известной теоремой
0 среднем значении подынтегральной функции, согласно кото-
Р°й для непрерывной функции f(x) справедливо равенство
b _
f/(*) dx = (a- b)f(x) (6.112)
a
^Десь f(x) есть значение f(x) при некотором фиксированном
значении аргумента х, заключенном в интервале а^.х^Ь. Бо-
ее того, если функция f(x) в данном интервале монотонна, ее
14*
* 211
значение f(x) в точке х=х заключено в промежутке между
крайними значениями f(a) и f(b) на концах интервала (а—Ъ)
В нашем случае интервал изменения аж или pXg всегда
можно выбрать достаточно узким, чтобы в нем функция /;(ам)
или ti(px2) была монотонной. Тогда на основании (6.112) и
при учете (6.111) уравнения (6.110) записываются в единой
форме
E=t^ (6.113)
Здесь U есть значение числа переноса ионов, отвечающее
некоторым средним значениям активности М или давления Х2:
причем это значение U заключено в интервале между
крайними значениями числа переноса ионов в приэлектродных слоях
электролита: и(а'ж) и ti(a"u) или соответственно ti{p'x2) и
ti(p"x2)> Таким образом, метод Вагнера позволяет определять
средние числа переноса ионов по измеренным значениям э. д. с.
соответствующих ячеек и их рассчитанным термодинамическим
значениям:
tt = ElE^ (6.114,
Однако при этом существует некоторая неопределенность в
том, каким именно условиям следует приписывать найденное
таким образом значение iu Однако очевидно, что эта
неопределенность может быть сведена до минимума, если выбирать
значения а'ж и а"ж или р'х2 и р"х2 достаточно близкими друг
к другу. Такое сближение пределов интегрирования ограничено
только точностью измерений; его можно проводить до тех пор,
пока измеряемые значения э. д. с. не уменьшатся до величин,
сравнимых с погрешностью эксперимента. Для окисных систем
удовлетворительные по точности результаты получаются, если
один электрод ячейки (6.956) находится в воздухе, а второй —-
в кислороде при атмосферном давлении.
6.6. Диффузия при хаотических блужданиях
В твердых телах наблюдается множество диффузионных
процессов, различающихся как механизмом диффузионного
перемещения частиц, так и их феноменологическим проявлением.
Основные из них — следующие: диффузия при хаотических
блужданиях атомов, диффузия меченых атомов (самодиффузия
и гетеродиффузия), химическая диффузия, взаимная диффузия,
заряженных частиц [21, 28, 36, 49, 77, 78].
В данном разделе рассматривается простейший из указан-
ных процессов — диффузия при хаотических блужданиях
атомов в кристаллах.
Вследствие тепловых флуктуации в разупорядоченных
твердых телах при любой конечной температуре атомы,
перескакивая из одного устойчивого положения в другое, хаотически
212
блуждают по кристаллу. Такие блуждания происходят и в со-
еошенно однородных образцах, при отсутствии каких-либо по-
!!ей в частности при отсутствии градиентов концентраций. Это
приводит к непрерывному взаимному перемешиванию атомов,
которое мы будем называть хаотической диффузией.
Хаотическим блужданиям в равной мере могут быть
подвержены как основные атомы твердого тела, так и примесные.
Поэтому теория хаотических блужданий описывает в рамках
одного и того же формализма диффузию как основных атомов (с а-
модиффузию), так и чужеродных примесей (гетеродиф-
фузию).
Как будет показано в дальнейшем, скорость хаотической
диффузии самым тесным образом связана со скоростями
различных диффузионных процессов, протекающих в твердых
телах при наличии полей: химического, электрического и
температурного. Поэтому количественное описание явления хаотической
диффузии представляет собой одну из важнейших задач всей
теории диффузионных процессов в твердых телах.
В настоящее время основным методом экспериментального
исследования диффузии в твердых телах является
использование меченых атомов: в диффузионную среду вносится
радиоактивный изотоп интересующего нас химического элемента и по
скорости выравнивания его концентрации определяется его
коэффициент диффузии. В частности, опыт можно поставить так,
чтобы суммарная концентрация обычного и радиоактивного
изотопов одного и того же элемента была постоянной по всему
образцу, тогда диффузионный процесс сведется к встречной
миграции обоих изотопов. Для не слишком легких элементов, когда
массы обоих изотопов отличаются незначительно, разные
изотопы одного и того же элемента химически не различимы.
Поэтому процесс встречной диффузии обоих изотопов можно в первом
приближении отождествить с процессом хаотического
перемешивания одинаковых атомов, а измеряемый коэффициент
диффузии радиоизотопа считать равным его коэффициенту
хаотической диффузии.
При постоянных концентрациях всех химических элементов,
входящих в состав образца, его дефектная структура не
зависит от содержания радиоизотопа. В частности, концентрация
вакансий постоянна во всей диффузионной зоне. Постоянна и
суммарная концентрация /с-атомов (ионов) в междуузлиях, а
концентрации обоих изотопов в междуузлиях в любом сечении
образца пропорциональны соответствующим полным
концентрациям изотопов в кристалле. На этом основании можно
записать:
VA^ = 0; vM, = 0; -L vNKl = -±- VNK (6.115)
0гда при обоих механизмах диффузии — вакансионном и меж-
АУузельном — уравнения (6.29) и (6.36) превращаются в одно
213
и то же выражение для потока /с-ионов обычного изотопа:
кТк
JK = 5-\Л^ (6.116)
Для потока радиоактивного изотопа уравнение аналогично.
Полученное соотношение представляет собой уравнение
первого закона Фика (6.12) для процесса диффузионного
перемешивания изотопов ^-элемента. Следовательно, величина,
определяемая выражением перед градиентом концентрации
DK = ^-4 (6.117)
имеет смысл коэффициента диффузии ионов сорта к. По
причинам, которые будут подробно рассмотрены в следующем
разделе, здесь необходимо подчеркнуть, что вывод уравнения (6.117)
основан на предположении о хаотических блужданиях
ионов в кристалле. Поэтому коэффициент DK называется
коэффициентом хаотической диффузии частиц
сорта к.
Соотношение (6.117), связывающее коэффициент
хаотической диффузии с парциальной удельной электропроводностью,
называется соотношением Нернста — Эйнштейна. Оно
справедливо для любых заряженных частиц, в том числе для
электронов и дырок, при условии, что диффузия и перенос тока
осуществляются одними и теми же дефектами.
Соотношению (6.117) можно придать несколько иной вид,
если перейти от парциальной ионной проводимости %к к
подвижности /с-носителей в электрическом поле ик, определяемой
формулой (6.6) как скорость частицы в поле единичной
напряженности. Подставляя равенство (6.8) в (6.117), получаем вторую
форму соотношения Нернста — Эйнштейна для заряженных
частиц:
Ож = —и„ (6.118)
гке
Если проследить за выкладками, выполненными в разделе
6.2 и в данном разделе, нетрудно убедиться в том, что все
полученные уравнения остаются формально справедливыми и для
нейтральных атомов в случае металлических сплавов или
валентных кристаллов, если для них положить заряд qK равным
нулю. Однако в этом случае в основном уравнении переноса
(6.33) и в соотношении Нернста — Эйнштейна (6.117)
содержится неопределенность xK/q2K = 0/0. Ее, однако, легко исключить,
если воспользоваться определением подвижности атома и¥к
(см. раздел 6.1) как скорости, приобретаемой /с-частицей под
действием единичной силы. Из соотношений (6.8) и (6.9)
следуем
ет связь между парциальной ионной проводимостью и
подвижностью и¥к:
*« = ?>Х (6.119)
Подставляя это равенство в (6.117), получаем третью форму
соотношения Нернста—Эйнштейна:
DK= kTuFK (6.120)
которая в отличие от (6.117) и (6.118) применима как к ионам
и электронам, так и к нейтральным атомам.
Мы определили коэффициент хаотической диффузии как
величину, характеризующую способность атомов к взаимному
перемешиванию при отсутствии химического поля (градиента
концентраций химических элементов). Однако формула (6.120)
показывает, что коэффициент хаотической диффузии является
более фундаментальной характеристикой вещества, отражающей
способность составляющих его частиц перемещаться под
действием любого поля, если последнее создает определенную силу,
действующую на частицы.
Заменим в основном уравнении переноса (6.23) ионную
проводимость на коэффициент хаотической диффузии с помощью
соотношения Нернста — Эйнштейна (6.117):
г _ NKDK
kT
dx\ T K dx T dx
(6.121)
В таком виде основное уравнение переноса не содержит
указанной выше неопределенности и применимо к частицам с
любыми зарядами — как к ионам, электронам и дыркам, так и к
нейтральным атомам.
Как и в случае ионной проводимости, при описании
диффузии в твердых телах существует* неоднозначность выбора
носителей. С одной стороны, при слабой разупорядоченности можно
считать носителями точечные дефекты — вакансии и между -
узельные атомы (ионы). В этом случае их коэффициенты
диффузии получаются при непосредственной подстановке формул
(6.40) и (6.41) для подвижности дефектов в соотношение
Нернста— Эйнштейна (6.118):
Dv« = Vo2 К! ехр(- UJkT) (6.122)
^ = Vo2[Vi]exp(-^/^r) (6.123)
Их иногда называют микроскопическими
коэффициентами диффузии.
С другой стороны, носителями можно считать все атомы
(ионы), абстрагируясь от микроскопической дефектной
структуры кристалла. В этом случае в соотношение Нернста —
Эйнштейна (6.118) следует подставлять формулы (6.42) и (6.43);
соответствующие коэффициенты диффузии называют макро-
21S
скопическими. Для вакансионного и междуузельного
механизмов миграции получаются несколько различающиеся
выражения макроскопического коэффициента хаотической диффузии
л:-атомов:
DK = >W [VJ exp (-UjkT) (6.124)
Дк = Уо2 e"™lKl] exp (-UjkT) (6.125)
[K\
где gK — отношение числа междуузлий к числу узлов /с-подрешетки.
Формулы (6.122) — (6.125) допускают некоторое упрощение.
Рассмотрим отдельно три основных случая.
A. В а к а нсионн ы й механизм диффузии. Вакансионный
механизм миграции /с-частиц предполагает достаточно высокую
концентрацию вакансий в /с-подрешетке по сравнению с
концентрацией /с-частиц в междуузлиях. Поэтому последнюю можно
считать пренебрежимо малой, а концентрацию /с-частиц в узлах
полагать равной их полной концентрации в данном сечении
кристалла: [кк] = [/с]. Тогда из уравнений (6.122) и (6.124)
получаем:
DvK = ^r04K]exp(-UKjkT) (6.126)
Ас = VV [V,] exp (-UK\kT) (6.127)
DJDvk = [Vk]'[/c] (6-128)
Б. Междуузельный механизм диффузии в чистых
кристаллах и твердых растворах замещения. В подавляющем
большинстве указанных систем концентрация междуузельных
атомов или ионов имеет значение, малое по сравнению с
единицей. Поэтому для большинства таких систем можно поло-
жить долю незанятых междуузлий [Vi] равной единице. Тогда
уравнения (6.123) и (6.125) принимают вид
DKi = >y02 exp (- UjkT) (6.129)
DK - Vefe, -^f exp (-UjkT) (6.130)
AJAd = £,[*.]/[*] (6.131)
B. Фазы внедрения. В этих системах, подробно
рассмотренных в разделе 3.5, концентрация внедренных атомов
металлоида Х{, вообще говоря, сравнима с общей концентрацией
междуузлий. Поэтому для фаз внедрения в общем случае
понятие точечного дефекта лишено смысла, и коэффициент
диффузии внедренных атомов следует определять формулой (6.125) в
макроскопическом приближении. Если атомы X целиком
размещаются в междуузлиях, совокупность междуузлий можно
рассматривать как самостоятельную подрешетку, причем [X] —
= g[Xi]. В этом случае формулы (6.123) — (6.125) принимают
одинаковый вид:
Dx = DXi - vxr02 [V,] ехр(- UxjkT) (6.132)
216
При малых концентрациях внедренных атомов [Vi]^l, и
(6 132) еще более упрощается:
Dx = DXi - vxr02exp(—£Ух/&7) (6Л33)
Напротив, при больших концентрациях внедренных атомов,
приближающихся к предельно допустимым, доля занятых междууз-
лий [XK]'Ss[Xi] близка к единице, а незанятые междуузлия
играют роль точечных дефектов (вакансий). В этом случае из
(6.122) и (6.132) получаем:
^vx = \r02exp(—UxjkT) (6Л34)
дх= vxro2[Vi]exp(— Ux kT) (6.135)
Dx\Dw* = [Vx] (6.136)
Формула (6.132) была получена в предположении, что
внедренные атомы металлоида диффундируют путем
непосредственных перескоков из одной октаэдрической позиции в другую
(с прохождением, например, тетраэдрической позиции). Однако
в фазах внедрения с плотнейшей упаковкой металлических
атомов такой механизм диффузии не является единственно
возможным. Например, в карбидах переходных металлов размер
промежутка между атомами металла, через которые должен
протиснуться атом углерода, примерно вдвое меньше размера
последнего, поэтому прямой перескок требует значительной
энергии активации. Миграция внедренных атомов значительно
облегчается при наличии вакансий в металлической подрешет-
ке; в этом случае переход атома металлоида может
осуществляться двумя последовательными прыжками из междуузлия в
металлический узел и их узла в соседнее междуузлие.
Вероятность такого перехода должна быть пропорциональна
вероятности того, что расположенные рядом металлический узел и меж-
Дуузлие одновременно вакантны. Поэтому в выражении (6.132)
наряду с [Vi] появляется дополнительный множитель — доля
вакантных узлов металлической подрешетки:
Dx =vxr02[VM][Vi]exp(-t/x/^r) (6.137)
В фазах внедрения, отвечающих формуле MXi_6,
произведение концентраций вакансий металла и металлоида согласно
Уравнению (3.97) равно константе Шоттки:
[V£] [Vxl = X(v) exp(-|f) (6.138)
и Формула (6.137) принимает вид
Dx = vxr02x(v)exp (- -V"1) (6-139>
Проведенный здесь анализ показывает большое
многообразие выражений для коэффициента хаотической диффузии, зна-
217
чительно отличающихся друг от друга. Это обстоятельство
иногда приводит к недоразумениям, связанным с
неоднозначностью выбора носителей, описанной выше. Во избежание
подобных недоразумений необходимо всегда четко определять,
что выбрано в качестве носителей — дефекты или атомы и,
следовательно, каков смысл коэффициентов диффузии —
микроскопический или макроскопический.
Однако в большинстве случаев микроскопические
коэффициенты диффузии дефектов представляют лишь теоретический
интерес; их использование ограничивается в основном
теоретическими задачами, в которых по тем или иным причинам
необходимо вычислить поток точечных дефектов. На основе же
экспериментального исследования диффузионных процессов, как
правило, рассчитываются значения коэффициентов диффузии
реальных атомов или ионов. Поэтому в подавляющем
большинстве случаев при описании диффузии в твердых телах
используются макроскопические коэффициенты диффузии атомов или
ионов DK.
В случае ионных кристаллов выбор в качестве носителей
ионов имеет еще одно преимущество: при таком выборе NK в
соотношении Нернста — Эйнштейна (6.117) имеет фиксированное
значение и коэффициент пропорциональности перед %к при
заданной температуре известен. А так как DK и кк поддаются
измерению, соотношение (6.117) может быть проверено
экспериментально.
По изложенным соображениям при выборе носителей мы
будем отдавать предпочтение атомам или ионам. Поэтому в
дальнейшем изложении во всех случаях, когда это специально не
оговаривается, коэффициенты диффузии будут относиться к
атомам или ионам, но не к дефектам, т. е. будут иметь
макроскопический смысл.
Во всех приведенных формулах для коэффициентов
хаотической диффузии энергия активации подвижности носителей U,
вообще говоря, зависит от температуры. Это было подробно
обсуждено в разделе 6.3, где было показано, что учет этой
зависимости с точностью до членов, линейных по температуре,
приводит к появлению перед больцмановской экспонентой
дополнительного множителя exp(asU/k), а энергия активации в
показателе экспоненты принимает значение U (0),
соответствующее нулевой абсолютной температуре. При учете этого
обстоятельства на основании полученных решений нетрудно
установить характер температурной зависимости макроскопических
коэффициентов диффузии. Как и в случае ионной проводимости,
здесь следует рассмотреть две предельных ситуации.
А. Фиксированные концентрации дефектов,
участвующих в переносе: [VK], [/Ci]= const. В неионных кристаллах —
интерметаллических и полупроводниковых соединениях, а также
в фазах внедрения такая ситуация типична при значительных
отклонениях от стехиометрического состава, если степень откло-
218
нения фиксирована составом образца и не зависит от
температуры. В ионных кристаллах концентрации вакансий или между-
узельных ионов постоянны в области контролируемых ионных
дефектов и соответственно примесной ионной проводимости
благодаря присутствию фиксированного количества гетерова-
лентной примеси. В этих случаях формулы (6.127), (6.130) и
(6.132) для макроскопических коэффициентов диффузии атомов
(ионов) сорта к имеют вид
DK = D°K exp {—U\kT) (6.140)
Здесь множитель D°K равен значению DK, экстраполированному
к бесконечно высокой температуре; для вакансионного и между-
узельного механизмов, а также для фаз внедрения при
фиксированной концентрации незанятых междуузлий и прямом переходе
внедренных атомов он определяется выражениями
соответственно:
D°K = vKr0HVK}exp(asUK:'k)
Dl = \r0*gK M exp (asUJk) (6.141)
[К \
Dl = vxr0*[Vl]exp(asUKlk)
Б. Зависящие от температуры концентрации
дефектов, участвующих в переносе. Такая ситуация типична для
кристаллов простых веществ, а также для стехиометрических
соединений в области собственной атомной (ионной) разупорядочен-
ности, когда концентрации дефектов определяются константами
равновесия реакций разупорядочения и экспоненциально растут
с температурой:
[VJ, Sp]- = Mv)exp(-*>JkT) (6.142)
В этом случае температурная зависимость макроскопических
коэффициентов диффузии определяется уравнением
DK = D°exp(-^±ik) (6.143)
где предэкспоненциальный множитель при всех механизмах
диффузии равен
^ = vK^X(v)expi^±^«) (6.144)
k
К этой же группе относятся решения для нестехиометриче-
ских соединений, находящихся в равновесии с окружающей их
газовой фазой. В этом случае концентрации дефектов
определяются выражением
1
[VK], [к1]=Ар™* exp(-eKlkT) (6.145)
219
и для коэффициента диффузии получаем:
Z)K = D°exp(-^J^) (6.146)
где предэкспоненциальный множитель зависит от давления
компонента Х2 в газовой фазе:
1
DK = AvKr02px^ exp — к к) (6.147)
В формулах (6.140) —(6.147) энергии UK, w и е имеют
постоянные значения, соответствующие Г = 0. Поэтому предельные
решения (6.140), (6.143) и (6.146) для макроскопических
коэффициентов диффузии содержат экспоненциальную зависимость
от температуры. Этот теоретический результат хорошо
подтверждается всей совокупностью экспериментальных данных,
часть которых для чистых металлов, элементарных
полупроводников и оксидов металлов изображена на рис. 6.16 и 6.17 в
координатах Аррениуса.
Как и в случае ионной проводимости, на рис. 6.16 и 6.17
бросается в глаза чрезвычайно широкий диапазон изменения
коэффициентов диффузии в различных твердых телах, даже
выше 500 °С простирающийся приблизительно на 10 порядков
величины. В этом отношении твердые тела радикально
отличаются, например, от жидкостей, где коэффициенты диффузии
имеют более или менее близкие значения, чаще всего лежащие в
интервале 10~6—10~5 см2/с.
Представляет интерес сделать некоторые числовые оценки,
характеризующие процесс диффузии в твердых телах. Как бу-
2000 1500 1200 1000 800 600 500 t,°C
РИС. 6.16.
Температурная зависимость коэффициентов самодиффузии металлов и
элементарных полупроводников [79].
220
2000 1500 1200 WOO 800 700 600 t,°C
Ofl 0,6 0,8 1,0 W3!T,K'!
РИС. 6.П.
Температурная зависимость коэффициентов самодиффузии металлов
(сплошные линии) и кислорода (пунктирные линии) в оксидах [79].
дет показано в следующем разделе, коэффициент хаотической
диффузии может быть выражен через частоту хаотических
прыжков соответствующего атома со простым соотношением
£> = а2со/6 (6.148)
где а — длина прыжка (расстояние между соседними устойчивыми
позициями) .
Выберем для него значение D^10~10 см2/с, типичное для
многих твердых тел при высоких температурах (см. рис. 6.16,
6.17). Так как длина прыжка в кристаллах составляет а^
^2-10"8 см, из формулы (6.148) находим соя^К)6 с-1. Для
сравнения заметим, что частота колебаний атомов v^lO13 с-1, что
на 7 порядков превышает значение со. Это означает, что атом
перескакивает в соседний узел только раз за ^10 млн.
колебаний, а остальное время колеблется около одного устойчивого
положения. Несмотря на такую малую вероятность прыжков, за
1 ч атом проходит при хаотическом блуждании по кристаллу
большое расстояние асо/ — около 1 м. В то же время наиболее
вероятным местонахождением его при отсутствии движущей
силы диффузии остается исходный узел, а среднеквадратичное
смещение от этого узла, определяемое при хаотическом
блуждании как
Г=У"^1а2 (6.149)
за 1 ч составляет всего 15 мкм.
6.7. Корреляционный эффект при самодиффузии
В предыдущем разделе мы определили коэффициент хаотической
диффузии как величину, характеризующую скорость взаимного
перемешивания одинаковых атомов при их хаотическом блуж-
221
РИС. 6.18.
Сравнение коэффициента хаотической
диффузии D серебра в AgCl, рассчитанного из
данных по ионной проводимости, с
коэффициентом диффузии радиоактивного серебра D*
[80].
дании по кристаллу в отсутствии
внешних движущих сил. Количественно эта
величина была определена с помощью
уравнения (6.116), выражающего
первый закон Фика для диффузии
основного изотопа в присутствии
радиоактивного изотопа того же элемента.
Тем самым мы отождествили
коэффициент хаотической диффузии данного
элемента с коэффициентом диффузии
его основного изотопа при случайных блужданиях по кристаллу.
Соотношение Нернста—Эйнштейна в форме (6.118)
показывает, что коэффициент хаотической диффузии определенных
атомов не зависит явно от их концентрации и определяется
специфической характеристикой вещества — подвижностью атомов
под действием приложенной силы. Поскольку радиоактивный и
основной изотопы одного и того же элемента различаются
только массой, для не слишком легких элементов подвижности
обоих изотопов практически не различаются. Следовательно,
согласно изложенной выше теории коэффициенты диффузии обоих
изотопов должны быть одинаковыми и равны коэффициенту
хаотической диффузии, удовлетворяющему соотношениям
Нернста — Эйнштейна.
Однако прецизионные измерения, выполненные на ряде
ионных кристаллов, показали, что измеряемый коэффициент
диффузии радиоактивных изотопов не совпадает точно с
коэффициентом хаотической диффузии, вычисленным из значений
ионной проводимости с помощью соотношения (6.117). Типичный
пример показан на рис. 6.18 для кристалла хлорида серебра, где
кривая для коэффициента диффузии радиоактивного серебра
£>* лежит заметно ниже кривой, рассчитанной по ионной
проводимости. Это обстоятельство учитывают введением в
соотношение Нернста—Эйнштейна для коэффициента диффузии
радиоизотопов специального множителя fK, называемого
корреляционным (от латинского correlatio — соотношение,
взаимозависимость) :
°«=-Мгх«=/л (6Л50)
Природа корреляционного множителя сводится к
следующему. При измерениях ионной проводимости ток определяется
400300 200 100 30 t?C
Ifi 1,8 2,2 2,ь J,0WJ/LK'}
222
прыжками обоих изотопов, так что вероятность каждого
последующего прыжка не зависит от того, какой изотоп
перескакивал в предыдущем акте. При измерениях же диффузии имеют
значение лишь прыжки радиоизотопов, что приводит к
корреляции между его последовательными прыжками. Действительно,
согласно рассмотренной нами схеме вакансионного механизма
диффузии вероятность перескока, радиоизотопа из узла 1 в
вакансию II пропорциональна вероятности нахождения последней
в узле II (см. рис. 6.1). Перед первым прыжком эта вероятность
равна доле вакансий в силу беспорядочного распределения
ионов, предполагаемого при выводе формулы (6.116). Однако
уже после первого прыжка картина становится
несимметричной: вакансия находится слева от радиоизотопа (в узле I,
радиоизотоп— в узле II) с вероятностью, равной единице. Справа
же от радиоизотопа вероятность нахождения вакансий
по-прежнему равна средней доле вакансий и при обычных
экспериментальных условиях чрезвычайно мала. Поэтому вероятность
последующего прыжка радиоизотопа в вакансию справа
значительно выше, чем слева, и суммарный поток радиоизотопов
слева направо должен быть меньше вычисленного по формуле
(6.116). Таким образом, при вакансионном механизме диффузии
корреляционный множитель fK в соотношении (6.150) меньше
единицы.
Формально это означает, что вследствие корреляционного
эффекта значительная часть последовательных прыжков
меченого атома происходит в противоположных направлениях и не
приводит к его диффузионному перемещению. Другими словами,
коррелированные прыжки не дают вклада в диффузию, а
эффективная частота прыжков, приводящих к диффузионному
перемещению меченого атома, меньше истинной на множитель fK.
В строгой статистической теории коэффициент диффузии
меченого атома в отсутствие внешних полей определяется
выражением
£>* = Z2/6 (6.151)
где L2 — среднее значение квадрата вектора смещения атома от исходного
узла за единицу времени.
Если обозначить через at единичный вектор смещения
атома при t-прыжке, суммарное смещение за единицу времени
после со прыжков запишется в векторной форме как сумма
где а — абсолютное значение единичного смещения; 6г/ — угол между
направлениями векторов I- и /-прыжков.
Подставляя (6.152) в (6.151), находим:
D*=f^L (6Л53)
223
где величина
/=i+-^2cose<-; (6Л54)
учитывает взаимосвязь значений косинусов углов между
различными прыжками атомов и имеет смысл корреляционного
множителя.
Если корреляция отсутствует, все направления прыжков
равновероятны. Тогда для каждого значения cos 8г/ найдется
равновероятный ему прыжок в противоположном направлении
со значением cosQiK = —cos 6*/. В результате сумма средних
значений косинусов в (6.154) будет равна нулю, а
корреляционный множитель /=1, что соответствует формуле (6.148) для
коэффициента хаотической диффузии /с-частиц.
Убедимся теперь в том, что формула (6.148) эквивалентна
выведенным в предыдущем разделе, например (6.124) для ва-
кансионного механизма диффузии при отсутствии корреляции
между прыжками. Поскольку со есть число прыжков
выделенного атома в единицу времени, произведение а2со представляет
собой сумму квадратов смещений, набираемых атомом за со
прыжков. При хаотическом блуждании все прыжки
равновероятны, поэтому со/2 прыжков сопровождается смещением атомов
вправо (Ах^О) и со/2 — влево (Ах^О; каждая группа
включает также половину прыжков в плоскости ух при Ах = 0).
Следовательно, величина а2со/2 представляет собой сумму квадратов
элементарных смещений щ, набираемых атомом при со/2
прыжках в направлении положительной оси х (Ах^О):
о/2 со/2
a*i»/2 = V а? = 2 I (А^)2 + (ДУ/)2 + ^zf] (6.155)
В силу хаотического характера прыжков в изотропной среде
(например, в кубической решетке) квадраты смещений атомов
вдоль осей координат равны друг другу, и из (6.148) и (6.155)
получаем:
со/2 -_
D = 2(A^=-i^p- (6.156)
Последняя формула позволяет глубже понять физический
смысл коэффициента хаотической диффузии, который
оказывается численно равным сумме квадратов элементарных
смещений атома в произвольно выбранном направлении (в данном
случае х^О) за единицу времени при хаотическом блуждании.
При этом формула (6.156) справедлива не только для
кристаллических тел, но и для аморфных, жидких, газообразных.
С другой стороны, при вакансионном механизме диффузии
согласно схеме, рассмотренной в разделе 6.2, число прыжков из
плоскости I в плоскость II равно v[V]exp(—U/kT), следователь-
224
но, при расстоянии между плоскостями г0 сумма квадратов
смещений в (6.156) равна
о)/2
2 (Д*/)2 = V [V] ехр (- U/kT) (6.157)
i = \
я для коэффициента хаотической диффузии мы снова получаем
формулу (6.124).
В случае коррелированных прыжков меченых атомов
определение коэффициентов корреляции сводится к вычислению
суммы в формуле (6.154). Такое вычисление, вообще говоря,
сопряжено со значительными трудностями, особенно для твердых
растворов. Простые и надежные результаты получаются лишь в
простейшем случае — диффузии собственных атомов в чистом
веществе (самодиффузии). В этом случае имеется только одна
частота прыжков со, и задача вычисления 2 cos 8;/ является
геометрической, поэтому корреляционные множители самодиффузии
вычисляются с высокой точностью. Оказывается, что они имеют
различные значения для различных структур и механизмов
переноса. Как и следовало ожидать, при вакансионном
механизме диффузии корреляционные множители всегда меньше
единицы; их значения для наиболее важных структур приведены
в табл. 6.2.
При междуузельном механизме диффузии картина несколько
сложнее, потому что в одной и той же структуре возможны
различные типы прыжков междуузельных ионов. Простейший
тип — прямой междуузельный механизм —
осуществляется непосредственными прыжками ионов из одного междууз-
лия в соседние. Так как подавляющее число междуузлий
свободно, при прямом междуузельном механизме все прыжки
равновероятны, т. е. корреляция отсутствует, и /= 1.
В кристаллах с достаточно плотной упаковкой атомов
прямой междуузельный механизм сопряжен со значительной
энергией активации, необходимой для протискивания междуузельного
иона между атомами в соседних узлах. В таких случаях
предпочтительнее непрямой междуузельный механизм
миграции, при котором междуузельный атом выталкивает атом из
Таблица 6.2. Значения корреляционного множителя
при диффузии по вакансиям [81]
Решетка
Типа алмаза
Простая кубическая
Гранецентрированная кубическая
Объемноцентрированная кубическая
Гексагональная плотноупакованная
Образец
С
CsCl
NaCl !
Na
Co
f
0,5000
0,6531
0,7815
0,7272
/x=/„=0,7812
/z=0,7815
15—321
225
Таблица 6.3. Значения корреляционного множителя
при диффузии по междуузлиям [81]
Решетка
Гранецентриро-
ванная
кубическая
Простая
кубическая
Типа флюорита
Образец
AgCl
CsCI
CaF2 j
1
Междуузельная
позиция
Центр малого куба
Центр грани куба
Середина ребра
куба
Центр куба
1
Тип прыжка
Прямой
Коллинеарный
Неколлинеарный
Прямой
Коллинеарный
Неколлинеарный
»
Прямой
Коллинеарный |
Неколлинеарный
Прямой
Коллинеарный
(Са)
Неколлинеарный
(Са)
> (F) j
cos e
f
— 1
1 0,3333
1
-3-0,7273
— -у-1,4464
— 1
1 0,3333
~2~ 0,6216
0 1
--£-1,8240
— 1
1 0
0 1
— 1
1 0,4
0 1
1
-д- (0,7391
ближайшего узла в следующее междуузлие, а сам занимает его
место. В этом случае последующие прыжки скоррелированы
довольно сильно, причем корреляционный множитель существенно
зависит от взаимной ориентации векторов междуузлие Г—>узел
и узел—^междуузлие II (cos 6 в табл. 6.3). Наименьшие
значения / соответствуют коллинеарным прыжкам, при которых оба
вектора направлены по одной линии. Согласно табл. 6.3 это
значение даже равно нулю при коллинеарных прыжках атомов
через центры граней в структуре типа CsCI; это обстоятельство
требует дополнительного пояснения.
Объемноцентрированная решетка типа CsCI состоит из двух
простых кубических подрешеток, вставленных друг в друга так,
что узлы одной подрешетки находятся в центрах кубов другой
(рис. 6.19). Междуузельные атомы могут размещаться в двух
позициях, различных по отношению к подрешетке данного
компонента: 1 — в центре грани куба элементарной ячейки; в 2 — в
середине ребра куба. Пусть после очередного прыжка
радиоизотоп занимает некоторый узел 3. Если диффузия
осуществляется прыжками по позициям 1, находящийся в ней атом
может совершить прыжок в одном из четырех направлений,
указанных стрелками, в том числе с вероятностью lU в один из уз-
226
РИС. 6.19.
Междуузельные позиции в структуре CsCl:
I — центр грани; 2 — середина ребра куба элементарной ячейки.
лов, вернув при этом радиоизотоп в исходное
междуузлие. Если же этого не произойдет (с
вероятностью %), радиоизотоп может
изменить свое положение только после некоторой
серии прыжков междуузельного атома, в
результате которой он снова окажется по
соседству с узлом 3. При этом междуузельный атом может
оказаться в одном из четырех междуузлии, соседних с узлом 3. Учет
вероятностей всех возможных путей при такой серии прыжков
приводит к значению корреляционного множителя /=Уз.
Ситуация существенно иная при диффузии по позициям 2—
серединам ребер куба элементарной ячейки. В этом случае
междуузельный атом имеет только два соседних узла и коллинеар-
ные перескоки возможны только вдоль одной линии ребра. Если
после предыдущего прыжка междуузельный атом находится
справа от радиоизотопа, он может вытолкнуть радиоизотоп
только влево, в исходное междуузлие (с вероятностью У2 в
ближайшем акте и с равной вероятностью — после серии прыжков).
Таким образом, в данном случае смещение радиоизотопа в
предыдущем прыжке междуузлие—^узел целиком гасится в
последующем противоположном прыжке. Взаимодействием же
радиоизотопа с другими междуузельными атомами можно
пренебречь из-за малости их концентрации, и мы получаем / = 0, т. е.
коллинеарные прыжки по серединам ребер практически не дают
вклада в диффузию радиоизотопов. В то же время такой
механизм является полноправным при ионной проводимости, когда
корреляция отсутствует.
При неколлинеарных прыжках, когда векторы
междуузлие—кузел и узел—^междуузлие не лежат на одной линии,
возможны, в свою очередь, три ситуации. Если угол 8 между
обоими векторами тупой, картина качественно похожа на описанную
для коллинеарных прыжков, и корреляционный множитель
меньше единицы. Если векторы перпендикулярны друг другу и
8 = 90°, последующие прыжки оказываются
некоррелированными и /= 1. Если же угол между направлениями векторов острый
и векторы, грубо говоря, направлены навстречу друг другу,
после прыжка радиоизотопа из междуузлия в узел выбитый им
атом оказывается в междуузлии, расположенном где-то сзади по
направлению движения изотопа. Поэтому существует
повышенная вероятность того, что при последующем акте радиоизотоп
будет выбит из узла примерно в том же направлении, что и при
первом акте. Таким образом, в этом случае корреляция между
прыжками ускоряет поступательное смещение радиоизотопов,
и корреляционный множитель оказывается больше единицы.
15*
227
0,8
ОМ
Ofi
Прямой междуузельный f = 1,000
Неколлинеарный f= 0,727
РИС. 6.20.
Сравнение теоретических и
экспериментальных значений корреляционного
множителя для диффузии серебра в
хлориде серебра при различных механизмах
перескоков [82].
Коллинеарный
f=0,333
300
350
400
t;c
При достаточно точных
экспериментальных данных
значения корреляционного множителя
можно использовать для
установления механизма переноса ионов.
Это хорошо иллюстрирует рис. 6.20 для диффузии серебра в
хлориде серебра, в котором ток переносится преимущественно
междуузельными ионами серебра. На этом графике
представлены значения отношения коэффициента диффузии
радиоизотопов серебра Z)*Ag к коэффициенту хаотической диффузии
серебра DAg, рассчитанному из данных по электропроводности AgCl
с помощью соотношения Нернста — Эйнштейна (6.59) (так
называемое отношение Хэвена). Пунктирные линии
рассчитаны с использованием теоретических значений
корреляционного множителя для трех разных типов перескоков междуузель-
ных ионов с учетом вклада вакансий серебра в диффузию и в
электропроводность. Положение экспериментальной кривой
обусловлено суперпозицией двух типов непрямых междуузель-
ных механизмов переноса серебра, при отношениях частот кол-
линеарных и неколлинеарных прыжков от 1,0 до 2,5 в
исследованном интервале температур. Аналогичная картина
наблюдается и в AgBr.
При сравнительно низких
температурах во многих системах
обнаруживается отношение Хэвена больше
единицы (рис. 6.21). Это объясняется
ассоциацией противоположно
заряженных катионных и анионных вакансий
в парные комплексы (VMVX), которые
принято называть бивакансиями.
При одинаковых валентностях
катионов и анионов эффективный заряд би-
вакансий равен нулю, поэтому они не
-*
-3
1
«N
5:
^-Ю
<=а
£ъ
»-о
-//
чр.
WO 600
А
-
-
1
1
500 400 t,°G
i i i j
л°
oft 1
V
" V
\ \
\\
, iV
W 1tZ Ifi 10% K"
РИС. 6.21.
Сравнение коэффициента самодиффузии D
яатрия в NaBr, рассчитанного из данных по
ионной проводимости, с коэффициентом
диффузии радиоактивного натрия D* [83].
228
рИС. 6.22.
Коэффициент диффузии
радиоактивного хлора в КС1,
легированном Sr, при 670 °С в зависимости
от содержания Sr:
/ — составляющая по одиночным
вакансиям; 2 — по бивакансиям; 3 —
суммарный [84].
-5 -5 -4 lg[Sr]
дают вклада в электропроводность, не участвуют в
диффузионных процессах. На рис. 6.22 приведена экспериментальная
изотерма коэффициента диффузии радиоизотопа хлора в КС'1,
легированном Sr (кривая 5), а также рассчитанные кривые для
его составляющих, обусловленных диффузией по одиночным
вакансиям / и бивакансиям 2.
На кривой / отчетливо различаются два участка:
горизонтальный, соответствующий собственной разупорядоченности
[Vk~] = [Vci+] =/Cs, и наклонный, где концентрация свободных
анионных вакансий уменьшается с ростом содержания
стронция в соответствии с решением (5.276) для примесной
разупорядоченности.
Диссоциация бивакансий протекает согласно реакции
(VKVCi)x^±VK+Vdi (6.158)
поэтому их концентрация связана с концентрациями свободных
вакансий уравнением закона действия масс:
[Vr] [V^i] = /Св | (VKVc. )x] (6.159)
аналогичным уравнению (5.33) для комплексов примесь —
вакансия. Видно, что в соответствии с соотношением Шоттки.
[Vk][VS]=/CI (6.160)
концентрация бивакансий не зависит от содержания стронция в
твердом растворе (горизонтальная прямая 2). Поэтому
суммарная кривая 3 на рис. 6.22 имеет S-образный вид.
Ассоциация дефектов имеет особо важное значение для
диффузии иновалентных примесей, для которых перенос в виде
комплексов зачастую дает решающий вклад. При этом
поступательное движение комплекса осуществляется путем
последовательных прыжков примеси в вакансию и поворотов диполей
примесь — вакансия вокруг примеси.
6.8. Корреляционные эффекты
при гетеродиффузии
Все изложенное в предыдущем разделе для самодиффузии в
принципе применимо и к диффузии в кристаллах чужеродной
Примеси (гетеродиффузии). Однако корреляционные эффекты
10
и
-— з
2
1 1
229
для примесей оказываются более сложными, чем для
радиоизотопов основного компонента, поскольку в этом случае
необходимо учитывать различие в частоте прыжков основных и
примесных атомов. Поэтому корреляционный множитель для
примесей в общем случае довольно сложным образом зависит от
частоты различных прыжков.
Тем не менее в наиболее важных случаях статистическая
теория гетеродиффузии приводит к достаточно простым
выражениям для корреляционного множителя. Так, при вакансион-
ном механизме диффузии в кубических твердых растворах
замещения корреляционный множитель компонента определяется
формулой Маннинга:
Л^г— (6-161)
где (х)к — частота обмена местами меченого /с-атома с вакансией; Q —
суммарная частота таких серий прыжков, при которых вакансия удаляется из
узла, занятого ею рядом с меченым атомом при предыдущем прыжке.
Частота Q характеризует вероятность удаления вакансии из
положения, обусловленного предыдущим прыжком меченого
атома, в любое другое (случайное) положение. Поэтому она
называется эффективной частотой удаления
вакансии.
В случае бинарного раствора, если индекс к относится к
примеси, присутствующей в малой концентрации, частота
удаления Q определяется частотой обмена местами вакансии и
атомов растворителя, а точнее, пропорциональна этой частоте
с коэффициентом пропорциональности, определенным для
каждой определенной кубической решетки. В этом случае согласно
формуле (6.161) при фиксированной частоте прыжков примеси
сок корреляционный множитель для примесных атомов тем
больше, чем выше частота прыжков атомов растворителя. При
этом возможны два предельных случая.
Если частота прыжков атомов растворителя гораздо выше
частоты прыжков примесных атомов, Q^>2cdk, на основании
(6.161) имеем /к^1. Физически это означает, что вакансия
успевает много раз обменяться местами с атомами
растворителя, пока примесный атом находится в определенном узле.
В этом случае примесный атом «не помнит» первоначального
положения вакансии, и его последовательные прыжки
оказываются некоррелированными.
В противоположном предельном случае, Q<C2coK, формула
(6.161) дает jPk^Q/2cdk. Так как со и Q — приблизительно
экспоненциальные функции температуры, в этом случае f также
приблизительно экспоненциально изменяется с температурой.
На основании (6.153) для коэффициента диффузии атома
примеси получаем:
D*K= — a2Q (6.162)
230
т. е. коэффициент диффузии примеси не зависит от частоты ее
прыжков, а определяется частотой прыжков атомов
растворителя. Этот результат связан с тем, что в этом предельном
случае примесный атом часто обменивается местами с вакансией,
фактически оставаясь привязанным к ней, так что fK^l.
Поступательное же движение комплекса примесь — вакансия
■обеспечивается сравнительно редкими прыжками в вакансию
атомов растворителя, причем такие прыжки являются
лимитирующей стадией диффузионного процесса. Вместе с тем
коэффициент диффузии примеси в этом случае может значительно
превышать коэффициент самодиффузии основных атомов,
поскольку из-за деформации решетки по соседству с примесью
энергия активации прыжков основных атомов может быть
существенно ниже, а частота прыжков выше, чем в чистом
кристалле.
Если концентрации компонентов в твердом растворе
сравнимы между собой, эффективная частота удаления вакансии
от меченого атома Q в формуле (6.161) оказывается зависящей
от концентрации. Следовательно, и корреляционные множители
компонентов концентрированных твердых растворов зависят от
состава последних. Строгие статистические выражения в этом
случае весьма сложны, однако при определенных упрощающих
предположениях легко получить уравнения, связывающие
корреляционные множители компонентов с экспериментально
измеренными величинами— коэффициентами диффузии меченых
атомов в твердом растворе заданного состава.
Рассмотрим неупорядоченный бинарный твердый
раствор А—В, образованный по типу замещения, и предположим,
что диффузия в нем протекает по вакансионному механизму.
Состав твердого раствора будем характеризовать атомными
долями компонентов
NA Nn
£д — .. .. ; св —
NA+NB B NA±NB
связанными между собой очевидным соотношением
*А + *В=1 (6Л63)
(Заметим, что при сравнительно малых концентрациях
вакансий, Nv<CNa-\-Nb, атомные доли сА и св практически
совпадают с долями узлов [А] и [В], занятых атомами А и В.)
Учтем, что частота прыжков атомов А в вакансию связана
с коэффициентом их диффузии £>*А уравнением (6.153):
- ™*А (6.164)
Аналогичное соотношение имеем и для атомов В.
231
Относительно частоты удаления Q предположим в первом
приближении, что она пропорциональна среднему значению
коэффициента диффузии меченых атомов А и В:
Q = b{ckD\ +cBD*B) (6.165)
где коэффициент пропорциональности Ь определяется только геометрией
решетки и не зависит ни от концентраций, ни от подвижностей атомов А и В.
Подставляя выражения (6.164) и (6.165) в уравнение Ман-
нинга (6.161) и разрешая его относительно корреляционного
множителя атомов А, получаема
/А = 1 ^ — (6.166)
ФЬ (cADA + cBDB )
и аналогичное выражение для /в.
Неизвестную константу Ъ легко определить из условия, что
в частном случае самодиффузии, когда А и В являются
изотопами одного элемента (D*a=D*b), /а должно быть равно
корреляционному множителю /0, подробно рассмотренному в
предыдущем разделе. Учитывая (6.163), находим:
ФЬ ~ [ /о
и для корреляционных множителей получаем выражения
<1-/о)Яа
/в=1-
А А "Г В В (6167)
(1 —/0) Z>S
CADl + CKD
А^А "t" CBX^B
Формулы (6.167) упрощаются в случае разбавленных
растворов, когда доля одного из компонентов, например ев, мала
по сравнению с единицей. В этом случае, полагая Са~1,
получаем:
/А~/о; 1-/в«(1-/о)(£>«/£>') (6.168)
Эти соотношения показывают, что в разбавленных твердых
растворах замещения корреляционный множитель атомов
растворителя имеет приблизительно такое же значение, как при
самодиффузии, а корреляционный множитель примеси может
изменяться в пределах от 0 до 1 в зависимости от соотношения
DbIDa, как уже было описано выше.
Из общих выражений (6.167) вытекает важное
соотношение
^а/а + св/в=/о (б-169)
232
показывающее, что, несмотря на существенную
концентрационную зависимость индивидуальных корреляционных множителей
}а и /в, их среднее значение
7 = сА/А+св/в (6.170)
сохраняется постоянным и равным корреляционному
множителю при самодиффузии. Это означает, что увеличение /А
приводит к уменьшению /в и наоборот. Другими словами, изменение
корреляционных множителей компонентов твердых растьоров,
а следовательно, и их потоков не является независимым, а
подчиняется строгой корреляции. Природа этого явления
вполне понятна: движение атомов одного сорта приводит к
локальному изменению концентрации вакансий вблизи движущегося
атома, а такое изменение вследствие корреляционного
эффекта, в свою очередь, влияет на поток атомов другого сорта.
Таким образом, мы видим, что в случае гетеродиффузии
корреляционный эффект приводит к нарушению справедливости
первого постулата Вагнера о независимом движении частиц
разных сортов (см. раздел 6.1). При гетеродиффузии
существует корреляция не только между последовательными прыж*
ками каждого атома, но и между потоками атомов разных
сортов, движущихся по узлам одной подрешетки. Поэтому
следующей задачей является уточнение основного уравнения
переноса, выведенного в разделе 6.2, путем отказа от I постулата
Вагнера и учета корреляционного эффекта.
6.9. Основное уравнение переноса
при коррелированной диффузии
При вакансионном механизме диффузии, рассмотрением
которого мы ограничимся в данном разделе, корреляционный
эффект будет обусловлен локальным изменением концентрации
вакансий в узлах, соседних с движущимся атомом. Поэтому
при выводе обобщенного уравнения переноса мы
воспользуемся той же схемой (см. рис. 6.1), что и в разделе 6.2. Тогда
поток частиц сорта к запишется в виде
JK = r0(N^pW — N^pK^) (6.171)
где рк" — вероятность того, что узел в плоскости II, ближайшей к меченому
атому к, вакантен; рк'— аналогичная вероятность для плоскости I.
Отличие уравнения (6.171) от (6.24) состоит в том, что
локальные вероятности р'к и р"к вследствие корреляционного
эффекта не совпадают с их средними значениями для плоскостей
I и II, равными долям вакантных узлов [VK]' и [Vk]". Таким
образом, учет корреляционного эффекта в уравнении переноса
сводится к учету указанного отличия рк от [VK].
233
Введем дополнительные переменные 8р'к и др"к,
определяющие отклонения локальных вероятностей нахождения
вакансий в соответствующих узлах от их средних значений:
/>* = [VJ'+ 8/>* ; pl=[VK]" + bpl (6.172)
Полагая эти отклонения относительно малыми, в линейном
приближении можно записать:
":=iv«i'(i+i^); p;=[vj"(hw) <бл7з)
Используя последние соотношения вместе с соотношениями
(6.25) — (6.28) и выполняя последующие преобразования
аналогично изложенному в разделе 6.2, получаем обобщенное
уравнение переноса:
[%\ а*« т \ кТ{<ьРн-ьРк) )
'.=--£Н£) + ™ + -г"-+
ro[VK]
(6.174)
Это уравнение отличается от (6.23) лишь последним
слагаемым, учитывающим корреляционный эффект.
Для дальнейшего целесообразно перейти от парциальной
электропроводности пк к коэффициенту хаотической диффузии
DK с помощью соотношения Нернста — Эйнштейна (6.117), а
также выделить отдельно часть потока, не связанную с
корреляционным эффектом:
JK = fK-NJ)Kbl^ (6.175)
Здесь J°K означает поток /с-частиц при отсутствии
корреляционного эффекта:
(6.176)
,о __ NKDK
•J к —
kT
Myj + ^ + ^V^
Такая форма записи двух последних уравнений позволяет
проводить дальнейший анализ как для заряженных ионов, так и
для нейтральных атомов (*7«; = 0).
Для определения второго слагаемого в уравнении (6.175)
будем рассуждать следующим образом. Локальные изменения
концентрации вакансий около меченого атома возникают,
только если этот атом движется относительно окружающей среды.
Если же все атомы движутся с одинаковой скоростью (как,
например, при электропроводности в электрическом поле при
равных зарядах носителей), вероятности нахождения вакансий
в любом узле остаются одинаковыми, и корреляционный эффект
не возникает. Далее, если меченый атом движется быстрее, чем
окружающая среда в целом, очевидно, что он должен остав-
234
лять за собой некий шлейф, характеризующийся повышенным
содержанием вакансий. В этом случае вероятность найти
вакансию сзади по направлению его движения выше, чем
спереди: др'к>6р"к. Противоположный случай, когда меченый атом
отстает от движения среды, аналогичен его движению в
направлении, противоположном движению среды, и рг к<р" к. На
этом основании можно полагать, что разность др'к—др"к
должна определяться ^относительной скоростью движения
выделенного атома vK—vK, где vK — средняя по всем сортам атомов
скорость движения среды.
Уравнение (6.176) для потока частиц в отсутствие
корреляционного эффекта выведено в линейном приближении по
движущим силам. Следовательно, и поправку на
корреляционный эффект нужно вычислять в линейном приближении по
движущим силам. А так как скорости движения частиц в этом
приближении пропорциональны движущим силам, становится
очевидным, что поправку на корреляционный эффект
достаточно определить с точностью до члена, линейного по переменной
(vK—vK). Итак, мы приходим к выводу, что указанную
поправку в рассматриваемом приближении следует записать
IlL^ = HK(vK-vK) (6.177)
При этом существенно, что в используемом приближении
коэффициент пропорциональности Нк не зависит от величины
движущей силы, так как в противном случае мы получили бы
приближение более высокого порядка, нежели линейное.
Относительная скорость движения /с-атомов связана с
потоками очевидным соотношением
ЕЛ-
(6.178)
1к
ЪЩ
где суммирование ведется по всем сортам атомов, располагающихся в узлах
я-подрешетки.
Таким образом, уравнение (6.175) для потока /с-частиц
может быть переписано в виде
/к = Л°-ЯА(Ук-СкХ/,) (6.179)
/
где сп=ИЛЫг—доля атомов к в узлах соответствующей подрешетки (при
i
малых концентрациях вакансий [V*] доля ск приблизительно равна доле
узлов [кк], занятых атомами к).
Для определения неизвестного параметра Нк
воспользуемся отмеченным выше условием, что Нк не зависит от
движущей силы (напряженности полей). Рассмотрим процесс само-
диффузии компонента /с, когда наряду с обычными атомами к
235
кристалл содержит малое количество радиоизотопа /с-элемен-
та и диффузионный процесс сводится к взаимному
перемешиванию обоих /с-изотопов при неподвижных атомах прочих
сортов (1фк). В этом случае в выражении (6.179) для потока
радиоизотопа последним слагаемым можно пренебречь
вследствие условия с*кжО и записать:
Л =Jll-HKDtfK (6.180)
(снежинка в верхнем индексе подчеркивает, что
соответствующий поток относится к /с-радиоизотопу).
С другой стороны, по определению корреляционного
множителя, при самодиффузии
Л =ЛЛ*° (6-181)
Разрешая уравнение (6.180) относительно /*к и сравнивая
результат с (6.181), находим:
tf*=T7T (6Л82)
Корреляционный множитель fK и коэффициент хаотической
диффузии DK имеют значения, одинаковые для обоих
изотопов /с-элемента, следовательно, рассчитанное таким образом
значение параметра Нк относится не только к радиоактивному,
но и к основному изотопу /с-элемента.
Таким образом, уравнение переноса (6.179), учитывающее
корреляционный эффект, принимает вид
А=/Л°Н1--ЛК1Л (6.183)
i
В частности, при отсутствии корреляции (fK=\) поток равен
Рк и определяется уравнением (6.176), выведенным в
разделе 6.2.
Если в узлах /с-подрешетки размещаются атомы т сортов
(я=1, 2, ..., т), уравнение (6.183) дает систему т линейных
алгебраических уравнений относительно неизвестных потоков
J\, /2, ..., Jm- Решение этой системы позволяет определить
указанные потоки как линейные комбинации некоррелированных
ПОТОКОВ /°ь /°2, ..-, 1т-
Если атомы сорта к являются примесью, присутствующей
в малых концентрациях, ск<£.\, в уравнении (6.183) можно
пренебречь последним слагаемым, и оно упрощается до вида
JK=h& (6.184)
В частности, при чисто диффузионных процессах, когда поток
примеси Рк определяется первым законом Фика, уравнение
(6.184) принимает вид
JK=- fKDKsNK =-Dl vNK (6.185)
236
Поэтому в случае малых концентраций примесей их
эффективный коэффициент диффузии совпадает с коэффициентом
диффузии соответствующих радиоактивных изотопов Z)*K.
Этого нельзя сказать об основных атомах, для которых уравнение
переноса (6.179) не сводится к такому простому виду, как
(6.184).
Проиллюстрируем применение общего уравнения (6.183) на
примере системы, рассмотренной в предыдущем разделе —
бинарного раствора замещения А - В с произвольными
концентрациями компонентов. В этом случае (6.183) дает систему
двух уравнений:
Л = /а/а + (1 —/a)CA{Ja+Jb) /g jggv
Jb = /вЛ + {I - /в) cB(JA + JB)
Разрешая эту систему относительно потоков /А и /в с учетом
соотношений (6.163) и (6.169), получаем:
J A = /aJa Н 7 (/aJa + /bJb)
/п (6.187)
Jb^/bJb + т {/aJa + /b-/b)
/о
Потоки /°А и /°в, отвечающие отсутствию корреляции,
записываются для конкретных условий согласно уравнению
(6.176). Так, при взаимной диффузии компонентов А и В в
отсутствие электрического поля и в изотермических условиях
(V(p = 0, VT=0) имеем
п ^д^д iVRDR
Уа= ТГ~^^ /в — ЧЬ^в (6-188)
Подставляя эти выражения в (6.187) и используя соотношение
Гиббса — Дюгема [см. Приложение, формула (П.22)]:
ЛО^А+Л^В = 0 (6.189)
получаем для потока А
(1--/a)/r
Da + - ~^ (cADB + cBDA)
/о
^Y Wa (6.190)
и аналогичное выражение для потока В.
Для дальнейших преобразований полезно заметить, что из
формул (6.167) для корреляционных множителей вытекает
соотношение
_L =1А1 ill (6.191)
^а /вО-/а)
в справедливости которого легко убедиться прямой
подстановкой значений 1—fK из (6.167) и последующей заменой D*K на
fKDK. Если теперь исключить DB из уравнений (6.190) и (6.191),
то для потока атомов А и аналогично для потока В находим:
/о к (6.192)
, /а/в ^в^в
Полученные уравнения показывают, что при взаимной
диффузии в бинарной системе потоки обоих компонентов
благодаря корреляционному эффекту изменяются в /а/в//о раз по
сравнению с потоками, которые были бы в этой системе при
отсутствии корреляции. Если концентрация В мала, /а«/в (см.
предыдущий раздел), и указанный множитель приблизительно
равен корреляционному множителю примеси /в. В этом случае
поток примеси определяется выражением
Ув = ~Г^в (6-193а)
таким же, как и при диффузии радиоактивных изотопов, в то
время как поток атомов растворителя
fBNADA /B NAD\ q
Л= ^— wA=-j—;^-WA (6-1936)
отличается от соответствующего потока при самодиффузии в
/V/o раз. Этот дополнительный множитель может быть
существенно меньше единицы. Его происхождение связано с
нарушением первого постулата Вагнера и обусловлено корреляцией
между движением атомов обоих сортов при гетеродиффузии.
6.10. Химическая диффузия
В предыдущих разделах мы оперировали с понятиями само- и
гетеродиффузии; самодиффузией мы называли процесс
перемешивания атомов изотопов одного химического элемента в
чистом кристалле, а гетеродиффузией — процесс диффузии в
кристалле инородных примесей. Как будет видно из
дальнейшего изложения, гетеродиффузия является частным случаем
более широкого круга явлений — химической диффузии.
В самом широком смысле химическая диффузия определяется
как диффузионный процесс в химическом поле, когда
имеется отличный от нуля градиент химического потенциала
данного элемента. Процессы диффузии нескольких сортов
частиц в одной подрешетке (взаимная диффузия) будут
рассмотрены в следующих разделах; здесь мы ограничимся
рассмотрением простейшего случая химической диффузии, когда в
к-подрешетке движутся частицы единственного я-сорта.
Если /с-частицы — единственные, занимающие узлы я-под-
решетки, скорость движения любого меченого атома из их ан-
238
самбля равна средней скорости движения всех /с-частиц.
Поэтому поправка (6.177) к уравнению переноса на
корреляционные эффекты равна нулю, и в обозначениях предыдущего
раздела JK = J°K' [Это также видно непосредственно из (6.183) при
1=к и ск=1.]
Другими словами, при единственном сорте движущихся
частиц в данной подрешетке корреляционный эффект
отсутствует и поток частиц имеет некоррелированное значение Рк.
При отсутствии корреляции из уравнения (6.176) следует,
что поток частиц /с-сорта в химическом поле (при Vcp = 0, VT=
= 0) определяется простым выражением
JK=-^WK (6.194)
показывающим, что поток частиц пропорционален градиенту
химического потенциала. Заметим, что последнее уравнение
является более общим, нежели феноменологическое уравнение
первого закона Фика, согласно которому поток любых частиц
считается пропорциональным градиенту их концентрации.
Действительно, в некоторых случаях наблюдаются диффузионные
процессы при отсутствии градиентов концентраций. Например,
в образцах, подвергнутых неоднородным механическим
напряжениям, атомы движутся из области с большей энергией (а
следовательно, и с большим химическим потенциалом) в область
с меньшим химическим потенциалом, несмотря на отсутствие
градиента концентрации. Более того, в таких образцах может
даже наблюдаться поток атомов в направлении увеличения их
концентрации, что формально соответствует отрицательным
значениям коэффициента диффузии в уравнении первого закона
Фика. Такой диффузионный процесс называется восходящей
диффузией.
Запишем общее термодинамическое соотношение [см.
Приложение, формулу (П.42)]:
\>.K = \& + kTlnaK (6.195)
где ак — активность /с-частиц, зависящая от состава раствора; juk0 —
стандартное значение химического потенциала, отвечающее значению ак=\.
Далее, пренебрежем изменением мольного объема
кристалла, связанным с изменением его состава, и будем считать
общую концентрацию узлов постоянной. Тогда при
дифференцировании (6.195) получим:
w* = ^r7TTTvIn М = 17~71^Г^У« (6-196)
d In [к] NK d In [к]
где [к] — полиая концентрация /с-частиц в сечении х, отнесенная к
концентрации узлов соответствующей подрешетки.
239
Подставляя (6.196) в (6.194), получаем уравнение первого
закона Фика:
J« = -D*-§T7TvN* (6Л97)
d In [к]
в котором произведение
D -D dlnaK (6.198)
* ~ к d In [к]
имеет смысл индивидуального коэффициента
химической диффузии частиц сорта к.
Формулу (6.198) обычно записывают в несколько ином виде.
Воспользуемся понятием коэффициента активности
^'-компонента раствора, определяемого как отношение
активности ^-компонента к его концентрации:
Т, = ак/[«] (6.199)
Тогда получаем другую форму записи соотношения между
коэффициентами химической диффузии и хаотической диффузии:
А. = М1 + тЙг) (6-200)
Соотношения (6.198) и (6.200) не содержат величины заряда
/с-частицы. Поэтому они справедливы как для нейтральных
атомов, так и для ионов. Тем не менее для ионов полезно вывести
еще одно подобное соотношение, связывающее индивидуальный
коэффициент химической диффузии с соответствующей
парциальной ионной проводимостью. Оно получается непосредственно
из уравнения (6.20), описывающего диффузионный поток
заряженных частиц в химическом поле. Перепишем это уравнение
в виде
%,, d\xu
формально совпадающим с уравнением первого закона Фика
(6.12). Очевидно, что в данном случае выражение перед
градиентом концентрации есть индивидуальный коэффициент
химической диффузии ионов сорта к:
d\xK
Ч1 ~dNK
D«=-T--^7- (6-202)
Согласно формуле (6.198) индивидуальные коэффициенты
химической диффузии разных атомов в твердом растворе
существенно различны. В качестве иллюстрации найдем их
отношение для бинарного твердого раствора А—В, в котором NA-\-
+^в = const. Запишем для такой системы соотношение Гибб-
са — Дюгема [см. Приложение, формулу (П.22)]:
Nax^ + NbWb^0 (6-203)
240
Подставляя сюда значения градиентов >\1К из (6.196), находим
d In aA dlnaB
rflnlA]" ~ din [В] (6.204)
тогда из (6.198) следует
DaIDb^DaIDb (6.205)
Коэффициенты хаотической диффузии различных атомов или
ионов в твердых телах обычно различаются на несколько
порядков. Следовательно, так же сильно различаются и
индивидуальные коэффициенты химической диффузии.
Из формулы (6.200) видно, что индивидуальные
коэффициенты химической диффузии равны (по крайней мере, с
точностью до корреляционного множителя fK) соответствующим
коэффициентам хаотической диффузии в случаях, когда
коэффициенты активности не зависят от состава раствора, в частности
в случае идеальных растворов, для которых все ук равны
единице.
В гл. 2 было показано, что дефектные кристаллы, вообще
говоря, являются сильно неидеальными системами. Поэтому
производная в формуле (6.200) может принимать большие
значения, что приводит к сильному отличию DK от DK.
Физический смысл этого отличия сводится к отличию
условий, при которых протекают процессы
самодиффузии и химической диффузии. При самодиффузии образец
остается химически однородным и движущей силой является
только градиент концентрации изотопов соответствующего
элемента. При этом вероятности прыжков меченого атома в
противоположных направлениях отличаются друг от друга только в
результате корреляционного эффекта, причем это отличие
сравнительно невелико (/^1). При химической диффузии вступает
в действие дополнительный фактор — градиент концентрации
вакансий, который может приводить к резкому различию
вероятностей прыжков в противоположных направлениях. В
результате этого возникает дополнительный направленный поток
атомов в сторону увеличения концентрации вакансий, так что в
ряде случаев градиент концентрации вакансий играет роль
движущей силы диффузии, гораздо более мощной, нежели
градиент концентрации атомов.
Согласно формулам (2.54) активность атомов или ионов
связана с концентрациями дефектов соотношениями
a*=Kl/[VK] (6.206)
ак = [«.[/[V,] (6.207)
Это позволяет вычислить индивидуальные коэффициенты
химической диффузии для трех основных механизмов,
рассмотренных в разделе 6.6.
16—321
241
А. Вакансионный механизм диффузии. Как и в
разделе 6.6, положим [Як] = [я], тогда после дифференцирова
ния (6.206) и подстановки производной gin aK/dln[K] в форму
лу (6.198) для коэффициента химической диффузии /с-частиц по
лучаем:
/ М d[VK]\
^^гмж) (6-208)
Отсюда видно, что DK совпадает с коэффициентом хаотической
диффузии DK только в тех случаях, когда концентрация
вакансий в к-подрешетке постоянна. В общем же случае DK сильно
зависит от характера дефектной структуры кристалла.
В данном разделе мы ограничимся рассмотрением систем,
в которых /с-подрешетка содержит только один сорт /с-частиц,
причем их концентрация может быть переменной. В качестве
примеров можно привести чистые нестехиометрические
соединения с недостатком /с-компонента, имеющие вакансии в ^-под-
решетке, или твердые ионные растворы с примесными
катионами пониженной валентности: CaF2+KF, Zr02+Y203, Zr02+CaO
и др., в которых анионная подрешетка содержит атомы одного
сорта и вакансии. Во всех указанных системах можно положить
M + [V/c]=l. Тогда производная в (6.208) будет равна —1 и
Dk = Dk{\ + [k]\\Vk]) = Dk]Vk (6.209)
При малых концентрациях вакансий [VK]<Cl, i[/c]^l и при
учете (6.122) имеем
Dk^DVk^Dk (6.210)
Полученные соотношения показывают, что коэффициент
химической диффузии DK может превышать коэффициент
хаотической диффузии DK (а следовательно, и измеряемый
коэффициент самодиффузии D*K = fKDK) на несколько порядков величины.
Б. Междуузельный механизм диффузии в
чистых кристаллах и твердых растворах замещения.
Дифференцируя (6.207) и подставляя производную в (6.198), в общем
случае находим:
/ 1 d\KX\ I d[V{}\
л =Dk[k] T-f -тгт—TiTi-Tri1 (6.211)
к к L J \ [к\] d [к] [ Vx] d[K] ] v '
Если междуузлия содержат атомы только одного сорта /с,
[к\] + \Vi]= 1. Далее рассмотрим системы, в которых основная
подрешетка атомов к полностью упорядочена, а суммарная
концентрация /с-атомов изменяется за счет изменения их
концентрации в междуузлиях: dNK = dNKi или, в долях мест,
d[K]=gKd[fcl] = -gKd\Vl] (6.212)
Примерами таких систем могут служить кристаллы чистых
нестехиометрических соединений с избытком ^-компонента, ко-
242
торый реализуется путем размещения /с-частиц в междуузлиях,
или твердые ионные растворы замещения с примесными
катионами повышенной валентности: CaF2+YF3, La203+Ce02 и др.,
в которых основная анионная подрешетка полностью
укомплектована, а избыточные анионы находятся в междуузлиях.
При выполнении соотношений (6.212) формула (6.211)
упрощается:
DK [К]
При малых концентрациях междуузельных атомов [fti]<Cl,
[Vi]«[fc]«l, и на основании (6.123) имеем
DK ^ DKi « DKjgK [Kl] » DK (6.214)
Отсюда видно, что коэффициент химической диффузии /с-ато-
мов может на несколько порядков превышать их коэффициент
самодиффузии. В этом отношении рассматриваемые системы
аналогичны описанным выше системам с вакансионным
механизмом диффузии.
В. Фазы внедрения. В этом случае совокупность меж-
дуузлий удобно считать самостоятельной подрешеткой
металлоида, а диффузию атомов металлоида — происходящей по вакан-
сионному механизму. Тогда коэффициент химической диффузии
металлоида будет непосредственно определяться формулой
(6.209). При этом значения отношения DJDK могут изменяться
в широком диапазоне в зависимости от концентрации
внедренных атомов. В предельном случае малых концентраций [к]<с1,
[VK] «1 коэффициент химической диффузии металлоида
практически совпадает с коэффициентом его хаотической диффузии
DK. В противоположном случае, когда металлоидная
подрешетка укомплектована почти полностью, [к]^1, а доля незанятых
позиций мала, [VK]<Cl, выполняется приближенное
соотношение (6.210), согласно которому коэффициент химической
диффузии металлоида значительно превышает его коэффициент
самодиффузии.
Если теперь воспользоваться формулами (6.124) и (6.125)
для коэффициента хаотической диффузии, то из выражений
(6.209) и (6.213) следует, что во всех трех рассмотренных
случаях коэффициент химической диффузии имеет одно и то же
значение:
DK = ^exp(^UKjkT) (6.215)
Примечательно, что это выражение не содержит каких-либо
концентраций в отличие от выражений (6.122) — (6.125) для
коэффициентов хаотической диффузии атомов и дефектов.
Индивидуальная химическая диффузия какого-либо
компонента проявляется в чистом виде в ограниченном числе случаев.
Сюда относятся системы, в которых, во-первых, данная
подрешетка содержит диффундирующие атомы только одного сорта
16*
24а
и, во-вторых, диффузия осуществляется нейтральными атомами
и не связана с переносом электрических зарядов.
Наиболее наглядным примером индивидуальной диффузии
является миграция атомов металлоида в фазах внедрения, где
базисное вещество (металл или интерметаллическое
соединение) обычно упаковано в решетку с высокой степенью
упорядоченности, поэтому его участие в диффузии крайне
незначительно. Напротив, атомы металлоида в нестехиометрических
фазах внедрения образуют сильно разупорядоченную
подсистему, и следовательно, обладают высокой диффузионной
подвижностью. Поэтому при наличии градиента химического
потенциала внедренных атомов они создают значительные
диффузионные потоки, пронизывающие жесткий каркас подрешетки
базисного вещества.
Другой пример индивидуальной химической диффузии
представляют нестехиометрические соединения с металлическим или
ковалентным характером связи, в которых перенос вещества
осуществляется нейтральными атомами. В этих системах атомы
одного компонента обычно образуют высокоупорядоченную под-
решетку, в то время как атомы второго компонента либо
частично размещаются в междуузлиях (при избыточном
содержании этого компонента), либо не полностью занимают узлы
своей подрешетки, так что последняя содержит значительную
концентрацию вакансий (при недостаточном содержании второго
компонента). В обоих случаях атомы разупорядоченного
компонента при наличии градиента их химического потенциала
создают диффузионный поток относительно жесткой подрешетки
упорядоченного компонента. Как и в случае фаз внедрения, для
таких нестехиометрических соединений эффективный
коэффициент диффузии, определяемый из экспериментальных данных
согласно уравнениям Фика, представляет собой
индивидуальный коэффициент химической диффузии разупорядоченного
компонента.
Как будет показано ниже, в твердых растворах замещения,
содержащих в одной подрешетке атомы разных сортов, а
также во всех системах, где носителями являются заряженные
частицы, химическая диффузия обеспечивается одновременным
движением по крайней мере двух сортов частиц и поэтому
имеет более сложный характер.
6.11. Взаимная диффузия
в сплавах замещения
В данном разделе мы рассмотрим процесс химической
диффузии в бинарных твердых растворах замещения, в которых атомы
обоих компонентов размещаются в узлах общей подрешетки.
Сразу же заметим, что все изложенное в данном разделе
справедливо для металлических сплавов, а также для твердых рас-
244
творов с ковалентной связью, в которых диффузия
обеспечивается нейтральными атомами.
В бинарных растворах замещения градиент концентрации
одного из компонентов неизбежно связан с противоположно на-
правленным градиентом концентрации другого. Поэтому в
диффузионном процессе участвуют атомы обоих компонентов,
причем их потоки направлены навстречу друг другу: атомы А
диффундируют в область, более богатую атомами В, а атомы В —
в область, обогащенную атомами А. Такой тип встречной
химической диффузии атомов двух сортов называют взаимной
диффузией.
Итак, рассмотрим бинарный твердый раствор замещения,
образованный нейтральными атомами А и В. В этом случае при
взаимной диффузии атомы А и В движутся по узлам обшей под-
решетки, следовательно, потоки скоррелированы между собой и
определяются выражениями (6.192). Перепишем эти выражения
в виде
*=-т!ь(1+-^)л
/о V diniAj/ ^6216)
/л /r / d In 7R \
Уравнения (6.216) имеют вид первого закона Фика (6.215),
причем роль индивидуальных коэффициентов диффузии играют
выражения
(6.217)
£>в-
Сравнение с формулой (6.200) показывает, что корреляционный
эффект при взаимной диффузии в бинарном твердом растворе
замещения приводит к уменьшению индивидуальных
коэффициентов химической диффузии обоих компонентов на множитель
/ = /а/в//о. Согласно формулам (6.199) и (6.204) в бинарном
сплаве при А^а+^в^ const
d in Ta d In Тв
din [A] "" d\n [B]
и из выражения (6.217) следует
п в
(6.218)
(6.219)
Таким образом, мы видим, что и в случае коррелированной
взаимной диффузии индивидуальные коэффициенты
хаотической диффузии различных атомов пропопциональны их коэффи-
245
РИС. 6.23.
Схема опыта Смигельскаса и Киркендаля
[85].
^Молибденовые
про дол оч т.
Медь
циентам хаотической диффузии и, следовательно, могут
отличаться друг от друга на несколько порядков. Поэтому при
равных абсолютных значениях градиентов концентраций в
рассматриваемом твердом растворе потоки обоих компонентов через
кристаллическую решетку, вообще говоря, также различны. Это
должно приводить к деформации диффузионной зоны
кристалла. В области более богатой малоподвижным компонентом
благодаря некомпенсированному притоку высокоподвижного
компонента будут возникать новые узлы решетки, что приведет к
разбуханию этого участка образца. В области же, обогащенной
высокоподвижным компонентом, благодаря его
некомпенсированному оттоку число атомов будет уменьшаться, что может
привести к двум эффектам.
Во-первых, вакансии, возникающие вследствие оттока
высокоподвижных атомов, могут коагулировать с образованием
субмакроскопических или даже макроскопических пор.
Развитие такого рода диффузионной пористости получило название
эффекта Френкеля.
Во-вторых, образующиеся поры могут залечиваться
благодаря смещению соседних участков при пластической
деформации кристалла; в этом случае диффузионная пористость не
возникает, но в области кристалла, обогащенной высокоподвижным
компонентом, уменьшается общее число кристаллографических
узлов, что приводит к уменьшению объема этой области. Такое
явление называют эффектом Киркендаля.
Этот эффект был обнаружен в опыте Смигельскаса и
Киркендаля [85]. В этом опыте на брусок из а-латуни (0,7 Си+
+0,3 Zn) помещались тонкие молибденовые проволочки и
снаружи наносился слой чистой меди (рис. 6.23); образец
обжигался в течение 56 дней при температуре 785 °С. Во время отжига
через поверхность раздела латунь — медь, отмеченную
молибденовыми проволочками, происходила взаимная диффузия
цинка и меди. В результате ее после отжига толщина слоя латуни
L уменьшалась, что однозначно указывает на более высокую
диффузионную подвижность цинка по сравнению с медью.
Выведем теперь основные уравнения, описывающие
взаимную диффузию в бинарном сплаве замещения с учетом эффекта
Киркендаля. При этом для простоты пренебрежем изменением
мольного объема раствора, связанным с изменением его
состава, с тем чтобы число узлов в единице объема можно было счи-
nt-nnmuNk
246
тать постоянным по всему образцу. Кроме того, пренебрежем
концентрациями вакансий и междуузельных атомов по
сравнению с полными концентрациями NA и JVB. При этих условиях
7VA + 7VB = NL = const (6.220)
й градиенты концентраций обоих компонентов будут равны по
абсолютному значению:
V7VA= —V^Vb (6.221)
Уравнение первого закона Фика для химической диффузии:
(6.215) описывает потоки атомов, пронизывающих
кристаллическую решетку. Другими словами, оно определяет
диффузионные потоки в системе координат, жестко связанной с атомными
плоскостями в каждом данном сечении образца. Для
определенности назовем эту систему внутренней системой
координат.
Однако, как мы видели, при эффекте Киркендаля
вследствие пластической деформации кристалла атомные плоскости,,
расположенные в различных участках образца, могут
двигаться относительно друг друга. Поэтому для корректного
описания процесса взаимной диффузии необходимо выбрать единую
систему отсчета, не зависящую от киркендалевской скорости
движения атомных плоскостей.
Свяжем искомую систему координат с внешней поверхностью
образца, находящейся вне диффузионной зоны, и назовем ее
внешней системой координат. Далее, обозначим через v(x)
скорость движения атомной плоскости, имеющей во внешней:
системе координату х, относительно начала координат.
Очевидно, что при таком определении v(x) есть скорость движения
меток (молибденовых проволочек в опыте Смигельскаса и
Киркендаля) относительно внешней поверхности образца.
В выбранной таким образом внешней системе координат
потоки атомов А и В через сечение х будут определяться
уравнениями
JA (х) = — DA v^Va + NAv [х) _
Ув (х) = — £>в VM* + NBv (x)
Выделим в образце два произвольных сечения,
перпендикулярных оси х, с фиксированными координатами х\ и хч во
внешней системе отсчета. Так как объем, заключенный между
сечениями, при этом будет постоянным, в соответствии с принятым
условием (6.220) должно быть постоянным и общее число
атомов А и В между сечениями. А для этого необходимо, чтобы
сумма потоков обоих компонентов сплава через каждое из
сечений была одинаковой. Теперь положим #i = 0 (начало
внешней системы координат), Х2 = х (произвольное сечение), тогда
247
сформулированное условие запишется в виде
J к М + 7в (х) = > (0) + 7В (0) (6.223)
Но в начале внешней системы координат диффузия отсутствует
и 7а(0)+^в(0) =0. Следовательно, в используемой системе
отсчета для "произвольного сечения должно выполняться условие
7a(x)+Jb(x) = 0
Подставляя в последнее равенство выражения для потоков
(6.222) и учитывая (6.220), получаем уравнение относительно
неизвестной киркендалевской скорости движения меток v(x):
/5А VNa + /Зв vtVb -- /Vl v (х) =0 (6.224)
откуда при учете (6.221) следует
v (х) = (Da - DB ) у [А] = (DB ~ DA) у [В] (6.225)
Теперь подставим (6.225) в уравнения (6.222) для потоков
во внешней системе координат; после элементарных
преобразований получаем уравнения, формально совпадающие с
уравнением первого закона Фика:
Л = —DabV^a; Jb = — DabVNb (6.226)
где через DAB обозначена линейная комбинация
индивидуальных коэффициентов химической диффузии атомов А и В:
БАВ = [A] DB + [В] 5А (6.227)
При учете определения индивидуальных коэффициентов
химической диффузии (6.217), а также соотношения (6.218)
можно выразить £)Ав через коэффициенты хаотической диффузии
атомов А и В:
DAB = ^~ (l + 4щкг) < М Db + ^ Da ) <6228)
Из уравнений (6.226) видно, что величина £)Ав имеет смысл
эффективного коэффициента взаимной
диффузии в бинарном сплаве замещения, причем Dab имеет
значение, общее для обоих сортов диффундирующих атомов. При
этом формула (6.228) показывает, что Dab в общем случае
зависит от концентраций компонентов твердого раствора.
Однако такая зависимость пропадает в случае разбавленных
растворов. Так, при исчезающе малой концентрации В, [В]-—Ю,
коэффициент активности стремится к постоянному значению, т. е.
dlnyA/d\n[A\—>:0, а корреляционный множитель fAfb!fo—*/в.
При этом согласно (6.228) эффективный коэффициент
взаимной диффузии Dab приближается к значению D*B = fB^B
коэффициента диффузии меченых атомов примеси В.
248
6.12. Сопряженная диффузия
заряженных частиц
Выше мы видели, что в твердых растворах замещения, состоя»
щих из нейтральных атомов, эффективные потоки атомов
обоих сортов выравниваются благодаря киркендалевскому течению
кристаллической решетки относительно внешней системы
отсчета. В принципе эффект Киркендаля может иметь место и в
системах, содержащих заряженные частицы (полупроводники
и ионные кристаллы), если в диффузионном процессе
одновременно участвует не менее трех сортов заряженных частиц.
Однако ситуация существенно отличается от описанной
выше, если в диффузии участвует два сорта заряженных частин
(ионов и электронов). Такой процесс мы будем называть
сопряженной диффузией заряженных частиц. В этом
случае фактором, выравнивающим потоки частиц обоих сортов,
является внутреннее электрическое поле, возникающее внутри
образца вследствие разделения электрических зарядов
(поляризации) при диффузии заряженных частиц.
В данном разделе рассмотрим простейший случай
сопряженной диффузии, когда корреляционные эффекты, описанные
в разделах 6.8 и 6.9, отсутствуют. Заметим, что этому условию
удовлетворяет случай, когда частицы обоих сортов движутся
по узлам различных подрешеток либо когда частицами одного
сорта являются ионы, а другого сорта — электроны или дырки.
Наконец, сюда же относится диффузия по междуузлиям путем
прямых перескоков (прямой междуузельный механизм), при
которой корреляционные множители равны единице.
При отсутствии корреляции между последовательными
прыжками в изотермических условиях (VT=0) поток частиц,
/с-сорта определяется уравнением Вагнера (6.21):
JK = -~^(WK + q«w) (6.229)
Як
Учитывая связь индивидуального коэффициента химической
диффузии с парциальной электропроводностью (6.202),
перепишем уравнение Вагнера в виде
y, = _D,vAk--£-v? (6-23°)
При диффузии заряженных частиц в твердых телах
внутреннее электрическое поле создается, главным образом,
благодаря изменению зарядов в двойных электрических слоях на
межфазных поверхностях. В объеме же кристалла в
соответствии с уравнением Пуассона (1.16) должно выполняться условие
электронейтральности (см. раздел 1.6):
2^=° (6-231)
к
249
Толщина двойных слоев в различных твердых телах
колеблется обычно от одного или нескольких межатомных расстояний
до нескольких микрометров. В подавляющем большинстве
диффузионных систем толщина диффузионной зоны обычно
значительно превышает толщину двойных слоев, поэтому можно
считать, что условие электронейтральности должно выполняться
практически для всей диффузионной зоны.
В рассматриваемом случае диффузии двух сортов
заряженных частиц (к=1; 2) концентрации всех остальных заряженных
частиц (кФ1; 2) остаются постоянными, поэтому из (6.231)
следует:
?iVWt + ?2V*V2 = 0 (6.232)
Далее, будем считать, что диффузия протекает в
квазистационарном режиме. Это означает, что заряжение двойных
слоев происходит только в самый начальный момент
диффузионного процесса, а в дальнейшем количество электричества,
расходуемого в единицу времени на изменение заряда двойных
слоев (ток смещения), остается пренебрежимо малым. Это
допущение приводит к дополнительному условию отсутствия
суммарного электрического тока в диффузионной зоне:
ЯЛ+ ?2Л=0 (6.233)
Условие (6.233) играет ту же роль, что и условие
материального баланса (6.224) при взаимной диффузии в системах,
состоящих из нейтральных атомов, и устанавливает условие связи
между потоками обоих сортов частиц. Согласно этому условию
относительные направления движения частиц зависят от знаков
их зарядов: одноименно заряженные частицы движутся в
противоположных направлениях, а разноименные — в одном
направлении, а именно в сторону уменьшения их химических
потенциалов.
Подставляя в условие (6.233) выражение (6.230) для
потоков обоих сортов частиц, находим для квазистационарной
напряженности внутреннего электрического поля:
- vp=Trh; <й ~ й> v"i = -^h & - 5j ^ (6-234)
При учете последнего состояния уравнение (6.230) для потоков
частиц обоих сортов приводится к виду первого закона Фика:
Л = - /312vAV, J2 = - Dt2vN2 (6.235)
с эффективным коэффициентом сопряженной диффузии
заряженных частиц:
512 = ttD2 + t2Dt (6.236)
250
Здесь t\ и h — числа переноса подвижных частиц:
/1 = _2L—; *2 = _Л_ (6.237)
Xj -f- %2 ^1 + У-2
В реальных кристаллах обычно имеется один сорт частиц,
обеспечивающих основную долю их электропроводности
(основные носители), в то время как доля участия в
электропроводности других сортов частиц мала (неосновные
носители). Поэтому представляет интерес рассмотреть случай щ^$>
В этом случае формула (6.236) для эффективного
коэффициента сопряженной диффузии принимает вид
D12 = 52 + ^L (6.238)
Используя определение индивидуальных коэффициентов
химической диффузии (6.198) и выражая щ и щ через коэффициенты
хаотической диффузии D{ и D2 с помощью соотношения Нерн-
ста — Эйнштейна (6.117), представим (6.238) в виде
~ /din я* qJN2d\naA
0" = °Ашъ + шгш^г1 (6-239)
Полученное выражение показывает, что эффективный
коэффициент сопряженной диффузии пропорционален коэффициенту
хаотической диффузии неосновных носителей D%. Смысл этого
результата вполне понятен: он означает, что движение
неосновных носителей является лимитирующей стадией диффузионного
процесса в целом.
Величины термодинамического множителя в (6.239)
(выражение в скобках) определяется дефектной структурой
кристалла. Проиллюстрируем это на примере нестехиометрического
ионного кристалла, являющегося смешанным ионно-электрон-
ным проводником с преобладающей электронной проводимостью
(1—е~~ или е+, 2—Mz+ или Xz~). Процессы сопряженной
диффузии в таких системах часто являются определяющими для
протекания химических реакций в твердых телах, например при
окислении металлов, которое будет подробно рассмотрено в
следующей главе (раздел 7.3). При этом по тем или иным
причинам в нестехиометрическом ионном кристалле МХг±б
создается градиент степени отклонения от стехиометрического
состава б, что, в свою очередь, приводит к возникновению
градиентов химических потенциалов ионов и электронов и,
следовательно, их диффузии. Внутреннее электрическое поле
устанавливается таким, чтобы обеспечить равенство нулю суммарного
тока, переносимого через кристалл ионами и электронами.
251
В рассматриваемом случае формула (6.239) принимает вид
~ Id In aL z2N; d In a A
где индексы i и е относятся соответственно к ионам и
электронам.
Первое слагаемое в формуле (6.240) уже было вычислено
в разделе 6.10 для различных механизмов диффузии; так, для
вакансионного механизма согласно (6.209) оно определяется
концентрацией вакансий ионов i:
Ш£'=Л_ (6.241)
d\nNt [V*]
Для оценки второго слагаемого в формуле (6.240) можно
воспользоваться двумя предельными приближениями квантовой
статистики. Если электронный газ в кристалле сильно
вырожден, положение уровня Ферми, а значит, и активность
электронов не зависят от их концентрации, следовательно, dlnae/d\nNe
равно нулю. В случае же невырожденного электронного газа
можно пользоваться классической статистикой Больцмана,
согласно которой
1^ = const 4- kT\nne (6.242)
Отсюда имеем:
In ае = const + In ne (6.243)
и формула (6.240) принимает окончательный вид (при [i]^l):
1 ^2
Die = £,(_ + _,.) (6.244)
Согласно решениям гл. 5 (см. раздел 5.4) в чистых ионных
кристаллах концентрация электронных дефектов,
компенсирующих избыточный заряд ионов, определяется степенью
отклонения от стехиометрического состава и равна [e]=z6.
Концентрация же вакансий либо значительно превышает концентрацию
электронных дефектов (область малых отклонений от
стехиометрического состава), либо меньше ее на множитель, равный
эффективному заряду доминирующих вакансий (области
больших отклонений от стехиометрического состава), т. е. по
порядку величины [1/;]^?6. При этих условиях оценка коэффициента
сопряженной диффузии в нестехиометрическом ионном
кристалле МХг±б по формуле (6.244) дает:
Db^zDft (6.245)
Таким образом, мы видим, что при достаточно малых
значениях б эффективный коэффициент сопряженной диффузии
может значительно (на несколько порядков величины) превышать
значение коэффициента хаотической диффузии ионов Д.
252
6.13. Взаимная диффузия
в твердых электролитах
Рассмотрим теперь частный случай сопряженной диффузии
заряженных частиц — взаимную диффузию катионов двух
сортов М' и М" (/с=1; 2), размещающихся по способу замещения
в узлах катионной подрешетки ионного кристалла,
обладающего чисто ионной проводимостью (твердого электролита).
Для простоты будем считать анионы неподвижными, а также
пренебрежем зависимостью мольного объема твердого раствора
от его состава. При выполнении последнего условия можно
записать для катионной подрешетки
Nx+N2 + Nv = NL (6.246)
и считать число узлов в единице объема NL величиной
постоянной. Кроме того, пренебрежем концентрацией анионных
вакансий и учтем, что по условию электронейтральности суммарный
заряд катионов обоих сортов равен заряду анионов и при
неизменной концентрации последних равен заряду основных
катионов М' в чистом кристалле МХГ ([М7] = 1, [М"] =0):
ztNt + z2N2 = ziNL (6.247)
Из условий (6.246) и (6.247) вытекают важные соотношения
между переменными:
7Vv=f— -\)n2 (6.248)
ZidNt -f z2dN2 = 0 (6.249)
Найдем сначала упрощенное решение, пренебрегая
корреляционным эффектом и полагая корреляционные множители для
обоих катионов f{ и /2 равными единице. В этом случае
эффективный коэффициент взаимной диффузии катионов
определяется выведенной в предыдущем разделе формулой (6.236).
Выражая в ней индивидуальные коэффициенты диффузии и числа
переноса через парциальные электропроводности с помощью
соотношений (6.202) и (6.237), представим (6.236) в виде
~ xtx2 / 1 dixt 1 da* \
Термодинамический множитель, заключенный в скобки,
легко вычислить с помощью формулы
PK = ri + kT(inNK — lnTVv) (6.251)
выведенной в гл. 2 для химического потенциала я-ионов.
Выполняя дифференцирование, находим:
~ __ kT %г%2
ui2 —
£'J Xj + эй
1 1 dNv\ 1 / 1 1 dNy
+
zf \Nt Nv dNx J ' z2z \ N2 Ny dN2 }
(&252)
253
Дальнейшие преобразования целесообразно выполнить
раздельно для двух типов твердых растворов, в которых ток
переносится преимущественно катионами.
А. Равновалентные катионы, zx = z2 = z. В этом случае
члены с концентрацией вакансий в (6.252) взаимно
уничтожаются вследствие условия dNi = —dN2, и для коэффициента
взаимной диффузии получаем:
°"=1шкъ+ч (6-253)
Б. Равновалентные катионы, z2>z\. В этом случае
согласно (6.248) концентрация вакансий в катионной подрешет-
ке однозначно определяется содержанием примеси М", и
выражение (6.252) приводится к виду
А2 = -^^—2 (6-254)
Заметим, что последняя формула остается справедливой и в
случае zx — z2, переходя при этом в формулу (6.253).
Найдем теперь более строгое выражение для эффективного
коэффициента взаимной диффузии ионов с учетом
корреляционного эффекта.
В случае коррелированной диффузии частиц двух сортов в
твердом растворе замещения их потоки определяются
выделенными ранее уравнениями (6.187)
/о
j2 = /2/2о + С-^М (МЧМ°)
/о
(6.255)
где некоррелированные потоки /°i и /°2 определяются
уравнением Вагнера (6.229).
Подставляя эти выражения в условие отсутствия
суммарного тока (6.233), получаем уравнение для напряженности
внутреннего электрического поля
- V? = —, 1 Г (— ™ + — V^2 ) (6.256)
где через Si и s2 обозначены комбинации
= 1+-т- -
l+^[£iAiLzA)_/l(l_/2)
/oL ^2 J
(6.257)
Из этого определения видно, что параметры s{ и s2 задаются
значениями корреляционных множителей f\, f2 и f0; при fi =
=/2=1; 5i = s2=l. Подстановка выражений (6.229) для
некоррелированных потоков /°i и /°2 и (6.256) для напряженности
254
внутреннего электрического поля при учете соотношения (6.249)
после несколько длинных, но элементарных преобразований
дает:
1 /о е2 (/Л + s2^) [ г? dNx "*" z£ dN2 JV * 25g
/0 £2 (Si*i + 52x2) \ ^2 dNi z£ dN2 I
Полученные уравнения представляют собой уравнение
первого закона Фика с эффективным коэффициентом взаимной
диффузии:
~ /i/o XtX2 / 1 d\*.y 1 d[L2 \
°12 = "Ж si*i + s3*2 1^7 "^vT + T/" ЖГ j (6-259)
Последнее выражение отличается от найденного ранее при
пренебрежении корреляционным эффектом (6.250) множителем
~ /l/2 Х1 ■+" Хо
/=4^- „,;, (6.260)
/О 51Х1 + 52х2
Следовательно, / можно назвать эффективным корреляционным
множителем при взаимной диффузии ионов в твердом
электролите. Учитывая проведенные выше преобразования [уравнения
(6.251) — (6.254)], для эффективного коэффициента взаимной
диффузии ионов с учетом корреляционных эффектов получаем:
Аз=-^Г— (6-261)
Формуле (6.261) можно придать другой вид, заменив
парциальные электропроводности щ и х2 на коэффициенты
хаотической диффузии с помощью соотношения Нернста —
Эйнштейна (6.117):
~ fzxz2DxD2
°12 = ^[M'JDi + ^JVnA* (6.262)
Отсюда видно, что, как и в случае взаимной диффузии в
сплавах (см. раздел 6.11), в твердых электролитах коэффициент
взаимной диффузии довольно сильно зависит от концентраций
диффундирующих частиц.
В предельном случае разбавленного твердого раствора
C2~[M"]<Cl; Ci«[M']«l, имеем %2<С^ь и из (6.262) следует
DX2^^D2 (6.263)
т. е. эффективный коэффициент взаимной диффузии, как и в
случае металлических сплавов, определяется коэффициентом
диффузии неосновных носителей М". Кроме того, в этом случае
255
fv
»/o (см. раздел 6.8), a Si~l, и по формуле (6.260) получаем
'/2. Подстановка этого значения в (6.263) дает:
^2
D12
d;
(6.264)
В случае Z2=9Lzi дополнительный множитель z2/z\ обусловлен
химическим фактором, отсутствующим в металлических
сплавах, — условием связи (6.248) между концентрацией вакансий
и концентрацией гетеровалентной примеси.
6.14. Термоэлектрический эффект
в твердых электролитах
Если электрохимическая ячейка, включающая в себя твердый
электролит, находится в температурном поле, так что ее
электроды поддерживаются при разных температурах, в ней
возникают различные термоэлектрические эффекты, связанные с
диффузией ионов под влиянием градиента температуры. Из них
наибольшее значение для физической химии твердого тела
имеет эффект Зеебека, заключающийся в возникновении
электродвижущей силы ячейки в неизотермических условиях
(тер мо-э. д.с).
Природа термоэлектрического эффекта выражается
основным уравнением переноса (6.23):
л = -
й
и„
Tv[^) + qKV1+-fvT
(6.265)
согласно которому в кристалле, находящемся в температурном
поле, возникают конечные потоки всех подвижных частиц,
приводящие к разделению зарядов в твердом электролите и,
следовательно, к установлению конечной разности потенциалов на
электродах.
Напряженность возникающего при этом поля может быть
вычислена следующим образом. Ограничимся для простоты
наиболее распространенным случаем, когда в каждой из подреше-
ток кристалла твердого электролита движется не более одного
сорта ионов (например, катионы в катионной подрешетке и
анионы в анионной). При этом условии корреляционные эффекты
отсутствуют и при описании диффузионных процессов можно
пользоваться уравнением (6.265).
Представим это уравнение в виде
J к
фд. dNi
dT
дТ
dy
+ 4"~dT
(6.266)
полагая, что химический потенциал /с-ионов зависит от
концентраций Ni и температуры, которые рассматриваются как не-
256
зависимые переменные. Здесь индекс i относится не только к
ионам различных сортов, но и ко всем сортам дефектов,
определяющих значения \хк (например, вакансий). Далее,
воспользуемся общетермодинамическим соотношением Гиббса — Гельм-
гольца [см. Приложение, формула (П.20)]:
-k(f)=-% (a267)
(где hK — удельная энтальпия я-частиц), а также учтем, что
разность между энергией, переносимой движущимися
частицами, и их средней энтальпией в состоянии покоя
Q* =u*K-hK (6.268)
есть избыточная теплота, переносимая частицами через
кристалл (теплота переноса). Тогда формула (6.267)
приводится к виду
jK = -^(Y*«.J>!L + q *L + S<L)vt (6.269)
я1 \i dNi dT dT T I
При разомкнутой внешней цепи ток через электролит не
протекает, т. е. I,qKJK = 0, и для производной
(6.270)
(6.271)
находим:
где tK=KK/2nK-
0 = J2L
игом— jrr
dT
A — У tfC (У d^K
игом — £., L* ,..
к Чк \l dNl
- число переноса «-частиц.
dT
T
Величина #гом согласно определению (6.270) представляет
собой изменение электрического потенциала в объеме твердого
электролита, приходящееся на изменение температуры в 1 °С;
она носит название коэффициента гомогенной тер-
мо-э. д. с.
Если ячейка состоит из твердого электролита и двух
электродов, выполненных из одинакового материала и находящихся
при температурах Т и Т-\-АТ, то измеряемая разность
потенциалов на электродах равна
Д?=К(Г+ДЛ- V(T) + -4%-bT (6.272)
dT
где V(T) —контактная разность потенциалов между электродом и
электролитом при температуре Т.
Здесь пренебрежено эффектом, связанным с
металлическими токоподводами, поскольку вследствие квантовомеханическо-
го вырождения химический потенциал электронов в них очень
слабо зависит от температуры и вклад металлических токопод-
17—321
251
водов оказывается пренебрежимо малым по сравнению с
вкладом объема электролита и его границ с электродами.
Уравнение (6.272) используется для определения коэффициента тер-
мо э.д. с. (коэффициент Зеебека) #, которому придается смысл
коэффициента пропорциональности между величиной термо-
э. д. с. ячейки и разностью температур на электродах:
Д<р = »ДГ (6.273)
Согласно этому определению коэффициент Зеебека численно
равен величине термо-э. д. с, возникающей в ячейке с разностью
температур на электродах 1 °С. Положительному значению
коэффициента Зеебека соответствует более положительный
потенциал на более горячем электроде.
Из соотношения (6.272) видно, что общий коэффициент
Зеебека включает две составляющие: коэффициент гомогенной
термо-э. д. с. ,&roM = dq)/dT и коэффициент гетерогенной термо-э. д. с.
ь^= vvn-vm (6274)
Если разность температур на электродах достаточно мала
(в практических условиях не превышает нескольких градусов),
правую часть выражения (6.274) можно разложить по малому
параметру АТ/Т с точностью до линейного члена. В этом случае
получаем:
»гетеР = dVjdT (6.275)
Коэффициент гетерогенной термо-э. д. с. существенно зависит
от характера межфазного равновесия, устанавливающегося в
различных ячейках на границах электрод — электролит. Мы
найдем его для двух типов простейших и тем не менее важных
в практическом отношении ячеек.
т т+ат
1. Ячейка типа М|М+|М с металлическими
электродами и твердым электролитом со структурной разупорядо-
ченностью по катионам М+. Напомним, что к твердым
электролитам такого рода относятся многочисленные системы,
производные от a-Agl (см. гл. 1, раздел 1.8). Поскольку в таких
системах число переноса катионов с высокой степенью точности
равно единице, а концентрации ионов серебра и вакансий в
металлической подрешетке определяются структурными
факторами и не зависят от температуры, все производные по
концентрациям в (6.271) обращаются в нуль, и эта формула
упрощается до вида
*ro»=-QlleT (6.276)
Гетерогенную составляющую термо-э. д. с. определим из
условия, что на границах электролита с электродами существует
равновесие
Мх z<zt M+ + е" (6.277)
258
обеспечивающее выполнение равенства
^Й + t (6-278)
[см. Приложение, формула (П.24), в котором химические
потенциалы для заряженных частиц следует заменить на
электрохимические] . Подставляя сюда значения электрохимических
потенциалов согласно (П.44) и вводя контактную разность
потенциалов между электродом и электролитом V, из (6.278)
находим:
Теперь, дифференцируя полученное выражение по температуре,
в соответствии с (6.275) получаем для коэффициента
гетерогенной термо-э. д. с:
игетер —
(d&_ ^_Щ (6280)
\ dT dT dT J
Согласно формуле (2.50) химический потенциал катионов в
твердом электролите определяется выражением
Й=Й0 + №Ё[ (6.281)
LvmI
отсюда имеем:
= h k In -777-7- (6.282)
dT dT [VM]
В кристаллах со структурной разупорядоченностью катионов
концентрации катионов и вакантных позиций не зависят от
температуры, следовательно, второе слагаемое в (6.282) есть
величина постоянная, равная —k In g, где g есть отношение числа
вакантных позиций к числу занятых (для a = AgI £=20).
Химический потенциал атомов в слабо разупорядоченном
чистом металле согласно формуле (3.10) равен
ц* = _ WL + ЗА Лп — (6.283)
Отсюда следует, что его производная
dT dT
(6.284)
определяется только колебательной составляющей стандартного
значения (jlixm)°. Если частоты колебаний атомов в электроде
17*
259
о,э
0 J
0,5
0,7
0,5
-0,7
0,5
0,7
0,5
0,3
KAg4I5
RbAgr4l5
U^w-^ m^h
[(CH3)4N]2Ag13I15
РИС. 6.24.
Температурная зависимость
коэффициента Зеебека для ячеек
с твердыми электролитами,
структурно - разупорядоченны-
ми по серебру [86].
и ионов в электролите
близки друг другу, можно
приближенно положить
dT
d (fw
dT
(6.285)
0,9
0,7
2
1 U*
1 1
J 4
of-Agr2HgrI4 ( ,/3-Ag2HgI4|
Наконец, учтем, что в
металлах вследствие
сильного квантовомеханиче-
ского вырождения
электронной жидкости
значение химического
потенциала электронов
(положение уровня Ферми)
практически не зависит от
температуры. Тогда,
подставляя соотношения
(6.282) —(6.285) в (6.280),
находим для
коэффициента гетерогенной
составляющей термо-э. д. с:
а,
k\nl (6.286)
2 1 J
103lT,K" "гетер
Объединяя последнюю формулу с (6.276), получаем
коэффициент полной термо-э.д.с. (коэффициент Зеебека):
= — k In 5 — Ом1еТ
(6.287)
Для рассмотренных систем согласно полученному
результату коэффициент Зеебека отрицателен и линейно меняется с
обратной температурой 1/Т. Поэтому значения теплоты переноса
катионов Q*m можно определить из экспериментальных данных
зависимости Ф от 1/Т; такие данные изображены на рис. 6.24
для ячеек, включающих твердые электролиты на основе cc-Agl
со структурной разупорядоченностью по серебру.
В табл. 6.4 определенные таким образом значения теплоты
переноса ионов серебра Q*Ag сопоставлены со значениями
энергии активации электропроводности UAg.
Сравнение показывает, что в рассмотренных электролитах
теплота переноса в большинстве случаев близка к энергии
активации, тем не менее оставаясь несколько меньше ее. В свете
теории, изложенной в разделе 6.2, значение коэффициента а
260
Таблица 6.4. Теплота переноса ионов серебра и энергия активации
электропроводности в структурно-разупорядоченных твердых
электролитах [16]
Электролит
a-Agl
Ag2HgI4
KAg4I5
RbAg4I5
NH4Ag4I5
Ag2I4W04
[(CH3)4N]2Ag13I,5
Теплота переноса, эВ
0,052—0,144
0,40—0,63
0,078—0,092
0,078—0,093
0,058—0,093
0,14
0,09—0,115
Энергия
активации, эВ
0,051
0,69
0,095
0,098
0,095
0,16
0,17
в формулу (6.35) близко к единице. Это означает, что
перескакивающий ион набирает необходимый запас энергии в
исходной позиции и при очередном колебании раздвигает соседей,
протискиваясь между ними в следующую устойчивую позицию.
Т Т+АТ
2. Ячейка типа 02, Pt|02~|Pt, 02 с газовыми
(кислородными) электродами и твердым электролитом с анионной
проводимостью. К электролитам такого типа относятся твердые
оксидные электролиты с примесной разупорядоченностью типа
диоксида циркония, стабилизированного добавками оксидов
металлов с меньшей валентностью (Y203, СаО и др.). В системах
такого рода число переноса ионов кислорода равно единице,
причем концентрация вакансий кислорода в первом
приближении не зависит от температуры и определяется содержанием
стабилизирующей добавки. Поэтому в формуле (6.271) первое
слагаемое в скобках обращается в нуль, и для коэффициента
гомогенной составляющей термо-э.д. с. можно полагать при
qo = — 2e:
Ku^^z (6.288)
2еТ
Теплота переноса относится к ионам кислорода.
В случае кислородных электродов равновесие на трехфазных
границах электролит — платина — газ поддерживается за счет
реакции
Q2-^±lo2 + 2r (6.289)
Условие равновесия записывается в виде уравнения
l£- + 2eV = -L р0й + 2|Г- (6.290)
Как и в предыдущем случае, химический потенциал
электронов в металле (Pt) не зависит от температуры. Поэтому диф-
261
ференцирование (6.290) по температуре согласно определению
коэффициента гетерогенной термо-э. д. с. (6.275) дает:
<WP = -(-^^) (6.291)
гетер 2е \ 2 dT dT j v
Содержащуюся здесь производную химического потенциала
ионов кислорода по температуре можно вычислить по формулам
(2.50) и (2.51): ^
^ = -Що + ^1п^+кт{з\п^ + 4\п^) (6.292)
Для химического потенциала молекул 02 в газовой фазе
используем выражение статистической физики, более подробное,
нежели приведенное в Приложении (П.35):
Р0 = const + kT
t 7 i ^ t (2Л1) k
1П/7П In T— In -
^°2 2 2/z36B
(6.293)
где рог—парциальное давление кислорода в газовой фазе; М —масса
молекулы 02; h — постоянная Планка; 0Вр — характеристическая температура
вращательного движения молекул кислорода.
Выполняя дифференцирование (6.292) и (6.293) по
температуре, подставляя их выражения в (6.291) и складывая
результат с (6.288), находим для суммарного коэффициента Зее-
бека:
% = ±[А + — In T+ —\пРо 4- —?-) (6.294)
Здесь все величины, не зависящие от температуры и
парциального давления кислорода, объединены в константу Л,
являющуюся функцией состава электролита. Вследствие больших
отрицательных значений параметра А (^ — 10ч—15)
коэффициент Зеебека, как и в предыдущем случае, отрицателен.
Полученная формула позволяет определить теплоту
переноса ионов кислорода Q*o по экспериментальной зависимости #
от обратной температуры. Для твердых электролитов Zr02+CaO
такой анализ дает значение Q*o, составляющее около 40% от
энергии активации электропроводности. Это означает, что в
твердых электролитах такого типа элементарный акт перескока
носителя в соседнюю устойчивую позицию носит более сложный
характер, нежели в кристаллах со структурной разупорядочен-
ностью катионов; согласно теории, изложенной в разделе 6.2,
параметр а существенно меньше единицы, и значительную часть
энергии, необходимой для перескока носителя, берут на себя
соседи, расступающиеся и облегчающие ему прыжок в
результате тепловых флуктуации.
Формула (6.294) предсказывает также достаточно жесткую
зависимость коэффициента Зеебека в ячейках с газовыми элект-
262
lffPo, Ш
РИС. 6.25.
Зависимость коэффициента Зеебека для
ячеек с твердыми электролитами
0,88 ZrO2+0,12 СаО (1) и 0,91 Zr02+
+0,09 Y203 (2) от парциального
давления кислорода при 800°С [87].
родами от парциального
давления кислорода в газовой
фазе. Соответствующие
эксперименты (рис. 6.25) показали, что
зависимость (6.294) выполняется
с вполне удовлетворительной
точностью. На этих графиках
наклон прямых соответствует
значениям коэффициента пропорциональности между # и 1пр'о2,
равным 0,0219 мВ/°С для твердого электролита 0,88 Zr02-f
+0,12 СаО и 0,0220 мВ/°С для 0,91 ZrO2+0,09 Y203, в то время
как его теоретическое значение при 800°С составляет
0,0216 мВ/°С. Такое хорошее соответствие теоретических и
экспериментальных результатов можно рассматривать как еще
одно свидетельство в пользу оправданности классической ионной
модели при описании явлений переноса в твердых
электролитах, несмотря на то что эффективные заряды ионов в них
существенно отличаются от номинальных.
/ о
19Рог Ш
6.15. Термоэлектрический эффект
в невырожденных электронных
проводниках
В данном разделе мы рассмотрим применение теории термо-
э. д. с. к ячейкам, включающим невырожденные электронные
проводники п- или р-типа (валентные полупроводники и несте-
хиометрические ионные кристаллы).
В общем случае электронная проводимость таких
материалов обеспечивается как электронами (число переноса t~)y так
и дырками (число переноса t+). Для простоты мы пренебрегаем
возможной ионной составляющей электропроводности, полагая
*-+/+= 1. Тогда общая формула (6.271) для коэффициента
гомогенной термо-э.д. с. запишется в виде {к = е~\ е+)
игом —
дгГ dT
h
dT
дп
где \х~, п~ и Q*~ — соответственно химический потенциал, концентрация и
теплота переноса электронов проводимости; \х+, п+ и Q*+ — аналогичные
величины для дырок.
Будем считать электроды ячейки изготовленными из одного
произвольного металла (например, Pt). Далее, как и в предыду-
263
щем разделе, будем пренебрегать изменением потенциала в
металлических токоподводах вследствие сильного вырождения
электронной жидкости, а также считать одинаковыми
химические потенциалы электронов в обоих электродах,
поддерживаемых при различных температурах.
Поскольку равновесие между электронным проводником п
металлическим электродом поддерживается за счет обмена
между ними электронами, из условия равенства электрохимических
потенциалов электронов между обеими фазами получаем для
контактной разности потенциалов:
V= — №~(М)-1П (6.296)
е
где |л~ (М) и \х~ — значения химического потенциала электронов
соответственно в металле и в кристалле.
При указанных выше условиях отсюда находим:
& = d[± (6 297)
игетер е tff \\j.aui i
Теперь учтем, что химические потенциалы электронов и дырок
удовлетворяют условию ir~+jJt+=0, непосредственно
вытекающему из уравнения реакции собственной ионизации кристалла:
нуль^=±: е~ + е+
На этом основании формулу (6.297) можно переписать
следующим образом:
а t~ d\iT t+ d\tr __ Г~ d\±~~ /+ dp+ (aoqo.\
агетер— е dT e dT ~~ е dT'^ e dT lu-zyo*
Выполняя дифференцирование согласна формуле
d^ ф dn . д\±
Ит~~1^ "dT дТ
(6.299 >
и объединяя результат с (6.295), получаем для суммарного ко-
эффициента Зеебека:
0 = Г»" + <Н"0+ (6.300^
(6.30П
Здесь величины
е [ Т дТ )
ф+
1 /Q* +
е \ Т
дТ
имеют смысл парциальных коэффициентов термо-э. д. с,
обусловленных электронами проводимости и дырками. Согласно
формуле (6.300) в проводниках с проводимостью только я-типа
(t~=ly /+=0) '&=#-; в проводниках р-типа (/- = 0; /+=г
264
Более подробные выражения для парциальных
коэффициентов термо-э.д. с. можно найти из (6.301) для двух моделей
электронных дефектов в твердых телах (см. раздел 1.6).
А. Модель локализованных электронных дефектов
(поляронов). В этом случае химические потенциалы электронов
проводимости и дырок связаны с их концентрациями
соотношениями (2.18), которые можно записать в несколько упрощенном
виде
1 (6.302)
где х — колебательные составляющие, определяемые
выражениями
In Х- = In [А*] + 6М In -^-; In У+ <= In [ А*] + 6М In -^
(6.303)
При фиксированном составе кристалла %~ и %+ — величины
постоянные.
Подставляя производные (6.302) в (6.301), находим:
е\ kT [е ] } (6 304)
kT
Поскольку при локализованных электронных дефектах их
движение по кристаллу происходит по прыжковому механизму и
требует энергии активации, теплоты переноса Q*~ и Q*+ можно
интерпретировать таким же образом, как и для ионных
проводников (см. раздел 6.2). Поэтому в первом приближении Q*~
;и Q*+ являются параметрами, постоянными для данного
вещества.
Б. Модель квазисвободных электронных дефектов.
В этом случае, при отсутствии вырождения электронного газа,
химические потенциалы электронов проводимости и дырок
связаны с их концентрациями соотношениями (2.19)
[х =Ec + kT\n^
plr^—Eh + kTln^r
N*
77*
(6.305)
^де эффективное число состояний
3
N* = 2[ 2nm^kT) (6.306)
265
также зависит от температуры. Отсюда имеем:
k \
гг 3\
П iV*" 2 /
дТ ~ \ N* 2
При квазисвободных электронных носителях их движение
происходит по туннельному механизму, не связанному с
энергией активации. В этом случае теплота переноса определяется
как избыточная кинетическая энергия движущихся электронов
или дырок по сравнению со средней тепловой энергией 3/2&Т.
В случае невырожденного электронного газа статистическая
теория полупроводников дает для теплоты переноса электронов
(дырок) выражение
Q* = [r+±.yT (6.308)
где г — параметр, характеризующий зависимость длины свободного пробега
носителей от их энергии: %~иг\ значение этого параметра зависит от
механизма рассеяния электронов и обычно лежит в интервале от 0 до 2.
Подставляя (6.307) и (6.301), получаем для парциальных
коэффициентов термо-э.д. с. так называемые формулы Пиеа-
ренко:
k I .o,i #*'
— I г + 2 -f In—т-
2 ^ п ' (6.309)
)+=_А/г+2 + 1пД!
Формулы (6.304) и (6.309) заставляют обратить внимание
на следующее очень важное обстоятельство. В невырожденных
проводниках и ионных кристаллах концентрации электронных
дефектов обычно бывают очень малыми по сравнению с числом
допустимых состояний: [е]<С1 или n/N<^l. Поэтому
выражения в скобках (6.304) и (6.309) существенно положительны, и
коэффициенты термо-э. д. с. для проводников /г-типа всегда
положительны, а для проводников р-типа всегда отрицательны.
Это обстоятельство широко используется для определения
знака носителей в полупроводниках и нестехиометрических ионных
кристаллах, тем более что техника экспериментального
исследования эффекта Зеебека чрезвычайно проста.
Установленное правило знаков поддается достаточно простой
интерпретации в случаях, когда концентрация электронных
дефектов растет с повышением температуры. В проводниках п-ти-
па электроны проводимости диффундируют от более горячего
конца образца, где их концентрации выше, к более холодному-
В результате они переносят отрицательный заряд на более
холодный конец, а горячий конец теряет часть электронов и
заряжается положительно. Такое распределение зарядов соглас-
266
\
рИС. 6.26. $
Зависимость коэффициента Зеебе-
ка нестехиометрического
кристалла от давления неметалла в
газовой фазе.
D
но определению (6.270)
соответствует
положительному коэффициенту
гомогенной термо-э. д. с. В
проводниках р-типа распределение спт
зарядов обратное, что coot- ln p ' L9Pxz
ветствует отрицательному
коэффициенту гомогенной
термо-э. д. с. Учет гетерогенной составляющей термо-э. д. с.
значительно корректирует величину коэффициента термо-э. д. с,
однако не меняет ее знака.
В области собственной проводимости, где концентрации
электронов и дырок близки, происходит диффузия обоих сортов
носителей от горячего электрода к холодному, и оба эффекта
в значительной степени компенсируют друг друга. Так, если
для квазисвободных электронов и дырок пренебречь различием
их эффективных масс и подвижностей, при гг=п+ имеем #- =
= <&+; t- = t+=l/2, и согласно (6.300) ®=0.
Описанные закономерности хорошо прослеживаются на не-
стехиометрических кристаллах, в которых благодаря изменению
степени отклонения от стехиометрического состава удается в
весьма широком интервале варьировать концентрации
электронов проводимости и дырок. Согласно решениям гл. 5 в общем
случае концентрации электронов и дырок в нестехиометриче-
ском кристалле в зависимости от температуры и давлении
неметалла в газовой фазе описываются уравнением
^ = Ат(Г)ехр(-4г)/421,т (6.310)
где А^(Т)—сравнительно слабые (степенные) функции температуры,
которые в первом приближении можно считать постоянными.
Подставляя это выражение в (6.304) или (6.309), по
формуле (6.300) находим:
гДе а и а+ — некоторые положительные параметры.
График зависимости # от 1п/?Х2 изображен схематически на
Рис. 6.26 для нестехиометрического кристалла МХг±б. Левая
часть графика отвечает преобладающей электронной
проводимости n-типа, правая — проводимости р-типа (соответственно
267
при t~tt\ и /+«1). Наклон обеих ветвей равен k/em. Заметим,
что согласно решениям гл. 5 (см. разделы 5.4 и 5.5) вблизи
стехиометрического состава значение т одинаково для п- и р~
областей и равно У2 2Х, т. е. !/2 для галогенидов lU для
оксидов. Резкий спад кривой в центральной части графика
соответствует переходу от n-типа проводимости к р-типу; здесь
согласно изложенному выше кривая проходит через нуль при
п-&п+, т. е. приблизительно при стехиометрическом состава
(6 = 0).
Глава 7
Кинетика роста твердых фаз
Химические реакции, приводящие к образованию твердых фаз,
чрезвычайно многообразны. Они различаются как по
агрегатному состоянию реагирующих веществ (газы, жидкости, твер^
дые тела), так и по особенностям локализации реакций в прост-
ранстве (в отдельных точках, на линии, на поверхности, в
объеме). Поэтому описание кинетики таких реакций также
очень разнообразно и должно учитывать конкретные условия, в
которых протекает каждая реакция [21, 88—92].
В данной книге, ввиду ее ограниченного объема, не
представляется возможным дать даже общее описание реакций с
участием твердых тел, и мы остановимся лишь на двух типах
реакций, широко распространенных в природе и достаточно
подробно изученных в лабораторных условиях: реакциях
образования интерметаллических соединений и реакциях окисления
металлов.
Оба указанных типа реакций имеют общую особенность:
продукт реакции разделяет исходные реагирующие вещества,
так что дальнейшее протекание реакции связано с диффузией
реагентов через слой продукта. Поэтому кинетика таких
реакций в значительной мере определяется транспортными
свойствами их продуктов, подробно рассмотренными в предыдущей
главе.
7.1. Феноменологические уравнения
Рассмотрим некоторую реакцию
А + гВ + АВг (7.1)
при которой из исходных простых веществ А и В возникает их
химическое соединение АВГ, представляющее собой достаточно
плотный твердый слой, разделяющий фазы исходных реагентов
А и В. Реагентами А и В могут быть металлы, в этом случае
АВГ представляет собой интерметаллическое соединение эти*
268
АВГ В, М
РИС. 7.1.
Схема движения частиц при росте интерметаллической фазы (а) и при
окислении металла (б).
металлов, либо металл и металлоид, образующие соединение
АВг полупроводникового типа, либо металл и неметалл
(кислород, халькоген S, Se, Те, галоген F, C1 и т.д.).
Агрегатные состояния исходных реагентов в рамках
излагаемой здесь теории не имеют значения, более того, они могут
находиться и в связанном состоянии: например, металлы—в
виде твердых растворов или интерметаллических соединений,
отличных от продукта реакции АВГ; неметалл, например
кислород, может содержаться в газовой смеси СО-гС02, Н2+Н2О
и др. Для последующего изложения важно лишь, чтобы
химические потенциалы исходных реагентов на границе их с
продуктом реакции АВГ были постоянными и не изменялись со
временем в ходе реакции.
Общая схема таких реакционных систем изображена на
рис. 7.1: для реакции двух металлов с образованием интерме-
таллида АВГ (а); для реакции металла М и неметалла Хг,
присутствующего в газовой фазе (б). Координата х отсчитывается
в фазе продукта реакции от значения х=0, отвечающего
границе с А(М), до значения x = L, отвечающего границе с В(Х2).
В соответствии с приведенной схемой скорость процесса
образования новой фазы АВГ определяется суммарным потоком
веществ А и В через фазу АВГ. Этот поток будет вычислен
ниже; в данном разделе мы рассмотрим феноменологическую
картину процесса, не вдаваясь в его механизм.
Согласно общим принципам термодинамики необратимых
процессов скорость любого неравновесного процесса считается
прямо пропорциональной некоторой «движущей силе»
процесса и обратно пропорциональной некоторой величине, которую
можно назвать эффективным сопротивлением реакции R*.
Понятие «движущей силы» в термодинамике необратимых
процессов отличается от понятия силы в механике, и ее выбор,
вообще говоря, не однозначен. Однако чаще всего используется
наиболее удобный способ ее выбора: движущая сила процесса
считается равной работе, совершаемой системой при протекании
в ней единицы реакции. Так, при протекании электрического
269
тока через проводник с сопротивлением R и разностью
потенциалов Дф движущей силой является электрическая работа
дДф. Для того чтобы выполнялся закон Ома для участка цепи
/=Дф//?, при таком выборе эффективное сопротивление
реакции должно быть равно R* = qR.
При протекании химической реакции ее движущей силой
согласно термодинамике необратимых процессов является ра^
бота реакции; при Г, р=const она равна изменению изобарно-
изотермического потенциала системы при протекании единицы
реакции ДС В дальнейшем для краткости мы будем называть
эту величину изобарным потенциалом реакции.
Таким образом, для скорости реакции, характеризуемой
скоростью роста толщины продукта dL/dt, можно формально
записать
*L=-^ (7.2)
dt R* К
Здесь R* имеет смысл некоторого эффективного сопротивления
реакции; знак (—) указывает на то, что реакция протекает в
сторону уменьшения изобарного потенциала системы.
Предположим, что R* складывается из двух слагаемых:
объемной составляющей
RV=9L (7.3)
пропорциональной толщине слоя продукта L, и постоянною
контактного сопротивления г, не зависящего от толщины слоя
продукта L. Тогда формула (7.2) принимает вид
дифференциального уравнения
dL AG с? л\
или = (7.4)
dt pL -\-г
-?- dL2 + rdL = — bGdt (7.5)
2 '
Интегрирование этого уравнения в пределах от L = 0 до L и от
t—О до t приводит к формуле, связывающей толщину слоя про-
дукта реакции со временем:
JL + -±- = t (7.6)
Кр Ki
где введены обозначения
Л; = -^; /Г, = -^- (77)
Уравнение (7.6) имеет две упрощенные формы,
соответствующие двум предельным случаям кинетики роста твердых
фаз.
1. Малая толщина фазы, L—Ю. Этот случай отвечает
начальной стадии реакции, когда объемное сопротивления р^
в формуле (7.4) пренебрежимо мало по сравнению с контакт-
270
иъ\ы сопротивлением г. В этом случае в уравнении (7.6) можно
пренебречь первым слагаемым и записать его в упрощенном
вйдУ
L = Ktt (7.8)
согласно которому толщина слоя продукта реакции растет
линейно со временем. Такая зависимость часто наблюдается на
практике при малой толщине слоя продукта реакции, поэтому
ее называют линейным законом роста твердой фазы.
Коэффициент пропорциональности Ki называется
константой линейного закона роста фаз.
2. Большая толщина фазы, L—*оо. В этом случае объемное
сопротивление pL велико по сравнению с контактным
сопротивлением г; в уравнении (7.6) первое слагаемое значительно
больше второго, и она может быть записано в упрощенном
виде
L* = Kpt (7.9)
представляющем собой уравнение параболы в координатах
t—L. На этом основании соотношение (7.9) называют
параболическим законом роста твердой фазы, а
коэффициент КР — константой параболического закона. Это
уравнение было получено эмпирическим путем Тамманом; оно
часто довольно хорошо описывает кинетику роста толстых слоев
фаз как в случаях образования интерметаллических
соединений, так и в случаях окисления металлов. Уравнение (7.9)
часто называют уравнением Таммана.
Приведенные уравнения позволяют легко интерпретировать
наблюдаемые закономерности временной зависимости скорости
роста фаз, однако они не вскрывают физической сущности
эффективных констант линейного и параболического законов
Ki и КР. Установить ее можно при более строгом физическом
рассмотрении, излагаемом ниже.
7.2. Рост интерметаллических фаз
при диффузионном контроле
Рассмотрим более подробно реакцию (7.1) образования
интерметаллического соединения АВГ из металлов А и В на
достаточно поздней стадии, когда процесс контролируется диффузией
А и В через интерметаллид АВГ. Другими словами, будем
считать, что диффузия является единственной лимитирующей
стадией процесса, а переход атомов А и В из металлических фаз
в интерметаллид через разделяющие их поверхности
происходит без затруднений, так что пограничные слои интерметал-
лида (при х=0 и x = L) находятся в термодинамическом
равновесии с фазами А и В:
М°) = МА); М^) = МВ) ало)
271
При вакансионном механизме диффузии в случае бинарного
интерметаллического соединения атомы А и В являются един.
^твенными компонентами, диффундирующими по узлам
соответствующих подрешеток, поэтому корреляционные эффекты
можно не учитывать. Тогда диффузионные потоки атомов за"
пишутся с помощью уравнения (6.194):
MADA NbDb
Ja = W~ wa ' Jb =— kT ^b (7.11)
■де DA и DB — коэффициенты хаотической диффузии атомов А и В; Лгд п
Vb — их объемные концентрации.
Учтем, что скорость увеличения толщины слоя интерметал-
шда dL/dt численно равна скорости увеличения объема этого
:лоя, приходящегося на единицу сечения. Если в интерметал-
шде подвижны атомы обоих компонентов, то наращивание его
уюя происходит одновременно с двух сторон: на правой гра-
шце с металлом В — за счет притока слева атомов А в
количестве /а, а на левой границе с металлом А — за счет притока В
5 количестве —/в. Так как объем, приходящийся в интерметал-
шде на один атом А, равен l/NA (и аналогично для В), мы,
:аким образом, получаем:
(XL, J A J r>
(7.12.
1одставим в полученное соотношение уравнение (7.11) и по-
южим в линейном приближении
V^a = 7 (7ЛЗ}
LDL^±-Kpdt (7.14)
[ аналогично для у|ив. Это дает дифференциальное уравнение
_1_
2
штегрирование которого в пределах от /=0, L = 0 до t, L
фиводит к уравнению параболического закона Таммана:
L2 = Kpt (7.15I
}десь через константу КР обозначена величина
Кр = -^ {DA K(0)-,,A (L)] + DB[v.B(L) -txB(0)] } (7.16)
шределяемая перепадами химических потенциалов
компоненте на границах интерметаллической фазы.
Значения перепадов химических потенциалов А и В легко
южно связать с изобарным потенциалом AG реакции (7.1 Ь
;оторый, по определению химических потенциалов как парци-
[льных изобарных потенциалов, равен
ДО = !*АВ _цА(А)-п*в(В) (7.17)
72
Зд^сь первое слагаемое в правой части означает химический
потенциал соединения АВГ, а два последних — химические
потенциалы его компонентов в металлических фазах А и В.
В фазе чистого соединения АВГ его химический потенциал
постоянен по всей толщине. Поэтому, в силу свойства
аддитивности, для него можно записать:
1^авг = !^а (°) + П>-в (0) = !^А (L) + п*в (L) (7.18)
Подставляя эти соотношения в (7.17) и учитывая равенства
(7.Ю), получаем:
ДО = цА (L) - [*А (0) = г [цв (0) - ,,в (L)) (7.19)
В результате формула (7.16) для константы параболического
закона приобретает простой вид
2AG / Dp
Кп =
kT
-(da + -^) (7.20)
Сравнение этой формулы с феноменологическим выражением
(7.7) показывает, что роль удельного сопротивления реакции р
играет величина
Р = Б1Т^, <™>
обратно пропорциональная суммарному коэффициенту
хаотической диффузии компонентов интерметаллического соединения.
Формуле (7.16) для константы параболического закона
можно придать иной вид, выразив ее через разности концентраций
атомов А и В на границах интерметаллической фазы. Для
этого воспользуемся общетермодинамическим соотношением для
,/с-частиц:
РК = \Ы + kTlnaK (7.22)
связывающим их химический потенциал с активностью, тогда
Для приращения химического потенциала в первом
приближении можно записать:
^ ^kT dlnaK A In [к] (7.23)
r* din [к] L J v
Полагая при этом долю узлов, занятых в соответствующей под-
решетке, [к]«1 и учитывая определение коэффициентов
химической диффузии (6.198), получаем:
KP = 2(DA^[A} + DB^[B)) (7.24)
Здесь для краткости обозначены через А [А] и А [В]
абсолютные значения перепадов концентраций атомов А и В по
толщине интерметаллида:
А[А] = [А](0)-[А](1); Д[В] = [В](/:)-[В](0) (7.25)
18—321 273
Как и разности химических потенциалов, при
незаторможенном переходе атомов через межфазные границы перепады
концентраций определяются равновесными условиями на
границах фаз. Величины AG, А [А] и А [В] сравнительно слабо
зависят от температуры, поэтому температурная зависимость
константы параболического закона согласно формулам (7.20) и
(7.24) в основном определяется экспоненциальной температур,
ной зависимостью коэффициентов диффузии наиболее
подвижного компонента.
7.3. Окисление металлов
при диффузионном контроле
Проблема окисления металлов в газовых средах, содержащих
сильные окислители — кислород, халькогены и галогены,
представляет собой одну из важнейших задач физикохимии
твердого тела, поскольку она связана с практической задачей
повышения коррозионной устойчивости металлов, а следовательно, и
с основами современной металлургической технологии.
Достаточно корректная теория таких процессов была предложена
Вагнером в начале 30-х годов. В данном разделе мы изложим
основное содержание этой теории, которая в современной
литературе обычно именуется теорией окисления
металлов Вагнера. Под окислением здесь понимается в широком
смысле взаимодействие металлов не только с кислородом, но
и с халькогенами или галогенами, приводящее к образованию
окалины — бинарного соединения МХГ, химическая связь в
котором в значительной степени носит ионный характер.
Вместо термина «окисление» часто также используется термин
«о к а л и н о о б р а з о в а н и е» [21].
В основе теории Вагнера лежат два важных допущения.
1. Перенос вещества через слой продукта реакции
окисления — окалины — осуществляется путем независимого
движения заряженных частиц — ионов (катионов Mzm+ и анионов
XzX _) и электронов е~ (несущественно, по какому механизму,
п- или р-типа). Согласно схеме, изображенной на рис. 7.1» о,
при этом катионы и электроны движутся в положительном
направлении— от границы окалины с металлом (х=0) к
границе с неметаллом (x=L), анионы — в противоположном
направлении.
2. Процесс переноса через слой окалины является
лимитирующей стадией процесса окисления; переход ионов и
электронов через поверхности раздела фаз протекает без затруднении,
поэтому на межфазных границах существует
термодинамическое равновесие. Это допущение аналогично принятому нами в
предыдущем разделе при описании образования
интерметаллических соединений; его значение будет ясно из дальнейшего и-*"
ложения.
274
Вычисление скорости процесса окалинообразования в
принципе аналогично изложенному в предыдущем разделе для
янтерметаллических соединений. Отличие состоит в
необходимости учета внутреннего электрического поля, возникающего
при диффузии заряженных частиц. Для этого обратимся к
приему, уже использованному в разделе 6.5 при вычислении
электродвижущей силы ячейки с ионным кристаллом, поверхности
которого поддерживаются при различных химических
потенциалах неметаллического компонента х.
При реакции окисления металла суммарный электрический
ток, переносимый через окалину ионами и электронами, равен
нулю; поэтому согласно (6.98) при /c=Mzm+, Xzx~, е~ имеем:
V? =■
■VP4
м '
X X | Гр —
Wy + — We
zYe
(7.26)
Здесь заряды катионов и электронов выражены через их
валентности г\ верхние индексы у градиентов химических
потенциалов указывают заряд соответствующих частиц в
классической ионной модели; tK означает числа переноса к-частиц.
Подставляя (7.26) в общее уравнение переноса /с-частиц
Jk=—ixfa^ + ^v?)
"к
(7.27)
получаем для потоков ионов через окалину:
J™ -—— I vh-
'М
4^2
Zye-
M
'mV^
ZN& , гМ ^X
MvrM
V^XX +Z^eWe
(7.28)
Vt\
+ ^mV^m — ^xVP-j
zX-
ZXfeWe
CN[
Для уменьшения числа неизвестных в правых частях этих
Уравнений перейдем от химических потенциалов ионов к
химическим потенциалам нейтральных атомов Мх и Xх с
помощью очевидных соотношений
tb
= ^x+zx^
(7.29)
после чего уравнения (7.28) принимают вид
Ум =—
■'м
zLe1
(1-^m)VIV +
м ^х
V^x
18*
(7.30)
275
ZY~~
J =
x A*
■^-wi + O-yv^
Скорость роста слоя окалины определяется потоками
катионов и анионов согласно уравнению, аналогичному (7.12):
dL
NM Ny
dt ^м iVx
(7.31)
Подставим сюда выражения для потоков (7.30) и учтем, что при
не слишком больших отклонениях от стехиометрического
состава в бинарной окалине МХГ отношение концентраций ее ком»
понентов составляет:
NxINm = zJzx (7.32)
Кроме того, запишем соотношение Гиббса — Дюгема (П.22),
выбрав в качестве компонентов окалины атомы металла и
неметалла:
М^м + JWx = ° С7-33)
а также учтем очевидные соотношения между суммарной
ионной (катионной+анионной) проводимостью к, электронной
проводимостью or и их числами переноса:
х = *м + хх
*м+'х + <* = 1
После этого в результате элементарных алгебраических
преобразований получаем для скорости роста слоя окалины:
dL ха х /7 r>r" 1
7Г"гмгх^м(х+а)^Х С-»3'
Проинтегрируем левую и правую часть последнего уравнения
по координате в пределах толщины окалины:
\*Ldx=—± f - w*dx (7.36)
Далее, учтем, что скорость роста окалины dLjut не зависит от
координаты х и может быть вынесена за знак интеграла. Пр11
малых отклонениях от стехиометрического состава окалины т°
же самое можно сказать о концентрации металла Л/м- A ii°"
скольку градиент химического потенциала yjix в одномерном
276
случае равен dpix/dx, произведение yiixdx равно просто d\xx,
уравнение (7.36) принимает вид
*LL== l f -2-dv* (7.37 \
dt zN[*Xp2NM J % + * X *
^(0)
Здесь пределами интегрирования в правой части являются
значения химического потенциала нейтрального неметалла в
окалине на ее границах с металлом и газом (х = 0\ L), а ионная и
электронная составляющие электропроводности к и а
предполагаются однозначными функциями jj,xx.
В соответствии со вторым предположением теории Вагнера
о термодинамическом равновесии на межфазных границах,
указанном в начале данного раздела, будем считать, что значения
химических потенциалов компонентов окалины в плоскостях
х=0 и x=L равны их значениям в металле и в газе
соответственно. Тогда пределы интегрирования в правой части (7.37),
а следовательно, и значение самого интеграла не зависят от
времени. Поэтому можно ввести обозначение
Рх№)
КР= г Д„- f -^-*x (7-38)
ц*(0)
и считать эту величину постоянной. В этом случае
интегрирование уравнения (7.37) в пределах от t—0> L = 0 до /, L
приводит к параболическому закону окалинообразования:
L2 = Kpt (7.39)
с константой этого закона КР.
Аналогично соотношению (7.19), в случае окалины при
r=zM/zx при равновесии на межфазных поверхностях можно^
записать
^(0)-^(L)=i^AO (7.40)
где AG — изобарный потенциал реакции образования окалины
из металла и газообразного неметалла:
Мх + — Х2 = МХГ (7.41)
Далее, представим интеграл (7.38) с помощью теоремы о
среднем значении в виде
V$(L)
j
" ^ = -2-[l»J(i)^^(0)] (7.42)
27?
Здесь среднее значение отношения квЦк+о) лежит в
интервале между его предельными значениями, отвечающими
плоскостям х=0 и x = L. Теперь при учете (7.40) для константы
параболического закона (7.38) можно записать:
В большинстве случаев характер электропроводности
окалины определяется ее нестехиометричностью и окалина
является преимущественно электронным проводником п- или р-типа.
Поэтому, полагая а^>х, формулу (7.43) можно переписать в
несколько упрощенном виде
2AG -
^ = -^г-х (7-44)
гМ.е iVM
показывающем, что константа параболического закона окали-
нообразования пропорциональна некоторому среднему
значению ионной составляющей электропроводности окалины к.
Наконец, значение КР можно выразить через коэффициенты
диффузии катионов и анионов, заменив в формулу (7.44)
к через выражение
* = TF &N*P* + 4"А) (7-45)
вытекающее из соотношения Нернста — Эйнштейна и
учитывая, что Nx=(z^/zx)NiA. После такой замены получаем:
Kp=-—[Dft + ^Dxj (7.46,
Последняя формула совпадает с формулой (7.20),
полученной в предыдущем разделе для константы параболического
закона образования интерметаллида. Этот результат легко
понять, если вспомнить о принятом выше упрощении а^>к. Такое
допущение означает: электронная проводимость окалины по
сравнению с ионной настолько велика, что уничтожает
внутреннее электрическое поле в окалине и процессы переноса в
ней уподобляются таковым в металлических фазах.
В противоположном же предельном случае к^>о
формула (7.43) упрощается до вида
К, = -^%--. (7.47)
показывающего, что константа параболического закона окали-
нообразования пропорциональна некоторому среднему
значению электронной проводимости окалины. Физически это
связано с тем, что перенос электронов через окалину является
наиболее затрудненной стадией процесса окалинообразования **.
следовательно, определяет его скорость.
.278
РИС. 7.2.
Кинетика окисления никеля и никель-хромовых сплавов в атмосфере
кислорода при 1О0О°С и различном содержании хрома [числа на прямых в % ат.]
[21].
РИС. 7.3.
Кинетика окисления никеля при 800 °С в атмосферах 6% 02+п % S02-b
+ (94—п) % С02 i('числа на прямых указывают п) [93].
Полученные результаты показывают, что при
параболическом характере окисления металлов скорость процесса
контролируется транспортными свойствами окалины. Последние, в
свою очередь, определяются ее дефектной структурой.
Поэтому очевидно, что, меняя каким-либо образом концентрации
доминирующих дефектов в окалине, можно направленно
воздействовать на скорость окисления металла.
Согласно изложенному в разделе 5.5 наиболее эффективным
способом изменения дефектной структуры нестехиометрических
ионных соединений является легирование их примесями с
валентностью, отличной от валентности основных ионов.
Поэтому следует ожидать, что добавление гетеровалентной примеси
в окисляемый металл или в окисляющую газовую среду
должно приводить к изменению дефектной структуры окалины и,
следовательно, скорости окалинообразования.
Рис. 7.2—7.4 демонстрируют такое влияние на примере
окисления никеля в атмосфере кислорода, приводящего к
образованию фазы NiO.
Известно, что монооксид никеля iNiO в кислородсодержащих
атмосферах является преимущественно электронным
проводником р-типа. Поэтому при интерпретации экспериментальных
результатов можно полагать с высокой степенью точности
<у^>к и для константы параболического закона пользоваться
формулами (7.44) и (7.46). В свою очередь, ионная
проводимость в NiO обусловлена практически исключительно катиона-
ми (£>№»£>о), движущимися по вакансионному механизму.
279
Поэтому добавки к №0 катионов повышенной валентности
увеличивающих концентрацию катионных вакансий (см.
раздел 5.2), должны приводить к увеличению КР и, следовательно
к ускорению процесса окисления. Этот эффект отчетливо
проявляется при легировании никеля хромом, влияние которого на
кинетику окисления показано на рис. 7.2. Здесь по оси
ординат отложен квадрат приращения массы окалины,
приходящейся на сечение 1 см2. Прямолинейный ход графиков в
координатах Am2—t соответствует параболическому закону
окисления, а наклон прямых определяется значением константы КР.
Как видно, увеличение содержания хрома в окисляемых
сплавах приводит к монотонному возрастанию Кр.
Аналогичный эффект вызывает добавление в атмосферу
окисления диоксида серы S02 [рис. 7.3, числа на прямых
указывают содержание S02 в атмосфере в % (об.)]. В этом
случае дефектная структура окалины может быть более сложной,
чем при легировании гетеровалентными катионами. Однако
подробные исследования показали, что при внедрении серы в
решетку монооксида никеля примесные центры служат
ловушками для электронных дырок, уменьшая их концентрацию. При
этом они, по крайней мере частично, приобретают
положительный эффективный заряд по отношению к фону кислородной
подрешетки:
S0 + в -> S0
Здесь нет необходимости подробно исследовать систему
квазихимических уравнений, описывающих дефектную структуру
оксида, легированного серой. Достаточно заметить, что наличие
примесных центров с положительным эффективным зарядом
вследствие условия электронейтральности неизбежно должно
приводить к увеличению концентраций всех точечных
дефектов, обладающих отрицательными эффективными зарядами, в
том числе катионных вакансий Vni2~ и Vni", а следовательно,
и к ускорению диффузии никеля через окалину. Действительно,
рис. 7.3 показывает сильное увеличение скорости окисления
никеля в присутствии S02 в атмосфере.
Противоположный эффект наблюдается при добавлении в
атмосферу окисления паров Li20. Соответствующие графики
приведены на рис. 7.4. Прямая 1 соответствует окислению
никелевой жести в чистом кислороде; в опыте 2 никелевая жесть
предварительно выдерживалась 5 ч при 1000 °С в атмосфера-
паров Li20, содержащей примесь кислорода, и затем
окислялась в атмосфере кислорода, содержащей примесь паров Li20-
Как видно из рис. 7.4, при таких условиях добавка лития
приводит к сильному снижению константы параболического
закона. Причина этого эффекта очевидна: ионы лития,
занимающие узлы в никелевой подрешетке окалины NiO, представляют
собой центры LiNf~ с отрицательным эффективным зарядом, по-
280
РЯС 7.4. ^ j
Кинетика окисления никеля при 1000 °С в ^
чистом кислороде (У) и в кислороде с при- V
месью паров Li20 (2) [21]. % £
этому их добавка приводит к умень- Jjj
щению концентрации отрицательно
заряженных катионных вакансий и,
следовательно, к снижению
скорости диффузии никеля. 0 2о чо 60 во t,4
Теория роста твердых фаз,
изложенная в двух последних разделах, основывалась на
предположении о равновесных условиях на межфазных поверхностях.
Как уже отмечалось, такие условия возникают при
незаторможенном протекании реакций на межфазных поверхностях,
когда диффузия через слой продукта реакции является
единственной лимитирующей стадией. Очевидно, что такие условия могут
реализоваться лишь при значительной толщине продукта,
когда его сопротивление диффузии достаточно велико.
Напротив, на начальных этапах процесса, когда толщина
продукта достаточно мала и мало его сопротивление
диффузии, становятся существенными затруднения при протекании
гетерогенных реакций на границах фаз. В этом случае условия
на межфазных поверхностях могут существенно отличаться от
равновесных, и изложенная выше теория должна быть
соответствующим образом модифицирована с учетом кинетики
перехода частиц из одной фазы в другую.
7.4. Поверхностная разупорядоченность
кристаллических фаз
Основы современных представлений о поверхностной разупо-
рядоченности твердых тел были заложены Френкелем, который
ввел понятие естественной шероховатости
кристаллических граней, возникающей в результате тепловых
флуктуации на поверхностях кристаллов. По Френкелю, идеально глад-
Кая поверхность граней кристалла может существовать только'
> при абсолютном нуле температуры. При конечных температу-
I pax тепловые флуктуации приводят к нарушению идеальной
-структуры не только объема кристалла, но и его поверхности.
В этом смысле природа поверхностной разупорядоченности
Твердых тел та же, что и природа собственных точечных
дефектов, подробно рассмотренная в предыдущих главах. Однако
явления, связанные с поверхностными дефектами, более
сложны, чем в объеме кристаллов. Здесь мы ограничимся
рассмотрением только трех простейших типов поверхностных дефек»
тов, играющих большую роль в протекании реакций с участием
твердых тел.
281
РИС. 7.5.
Простейшие типы поверхностной разупоря-
дочевности кристаллов:
/ — изломы ступени роста; 2 — вакансии в
поверхностном монослое; 3 — ад-атомы.
1. Ступени роста — края
атомных плоскостей, не полностью
покрывающих грани кристалла. Та-
кой термин обусловлен тем, что
рост или испарение кристалла
происходят путем достраивания или разборки именно таких
неполных плоскостей. По энергетическим соображениям очевидно,
что присоединение или отрыв атомов происходят
преимущественно в местах излома ступеней роста (рис. 7.5, позиции /).
2. Вакансии в поверхностном слое атомов (рис. 7.5,
позиции 2).
3. Одиночные атомы (ионы) 3, расположенные на
поверхности атомно-гладкой плоскости. Такие атомы, как
собственные, так и примесные, могут быть связаны с атомами
поверхностного слоя специфическими силами адсорбции и получили
название ад-атомов (ад-ионов).
Вакансии и ад-атомы играют большую роль в реакциях с
участием твердых тел. Так, при росте кристалла поступающие
из другой фазы атомы чаще всего осаждаются на его
поверхности в виде ад-атомов и затем диффундируют по поверхности к
местам роста — изломам ступеней, местам выхода на
поверхность дислокаций и т.д. При растворении или иной разборке
кристалла убыль вещества также может происходить путем
перехода в смежную фазу ад-атомов, которые образуются при
отрыве атомов от мест излома ступеней или иных мест
разборки и удаляются от этих мест в процессе поверхностной
диффузии. Аналогично, рост или разборка кристалла могут
осуществляться путем заполнения или соответственно образования
вакансий в поверхностном монослое; при этом вакансии
диффундируют от мест их зарождения или стока.
При равновесных условиях концентрации вакансий и ад-
атомов имеют определенные значения при каждой
определенной температуре; эти значения поддерживаются равновесными
реакциями их образования в местах зарождения или стока,
например в местах излома ступеней роста. Более того, даже при
отсутствии ступеней роста или других мест зарождения
дефектов возможно образование пар дефектов на атомно-гладкой
поверхности, когда какой-либо атом из поверхностного слоя
переходит в ад-состояние и затем путем поверхностной диффузии
удаляется от образовавшейся при этом поверхностной
вакансии. Этот процесс можно уподобить образованию дефектов
Френкеля в объеме кристалла, при этом ад-атом выступает
аналогом междуузельного атома. Описанная реакция может
282
быть изображена квазихимическим уравнением
АА-УА + Аад (7.48)
Применяя к ней закон действия масс, получаем уравнение
[Уа][Кл] = К* (7.49)
аналогичное уравнению образования дефектов Френкеля
Константа К может быть названа константой собственно
поверхностной разупорядоченности; она содержит в показателе
экспоненты энергию реакции (7.48):
/T2 = X(v)exp(—И^/АТ) (7.50)
Таким образом, очевидно, что в рамках изложенной простой
модели разупорядоченность поверхности кристаллов можно
описывать с помощью того же математического аппарата, что
и разупорядоченность объема. При этом существуют те же
ограничения в применимости аппарата, что и для объема
кристалла. Важнейшим из них является предположение о
малости концентраций поверхностных дефектов по сравнению с
концентрацией поверхностных кристаллографических узлов.
Разумеется, реальная ситуация гораздо сложнее описанной,
прежде всего, потому, что числа заполнения узлов на
поверхности реального кристалла могут значительно отличаться от
нуля или единицы. Это связано с тем, что атомы в
поверхностных слоях кристалла связаны друг с другом более слабыми
силами, нежели в объеме, хотя бы из-за меньшего числа
ближайших соседей. Поэтому образование дефектов кристаллической
решетки, например вакансий, на поверхности кристалла требует
меньших затрат энергии, чем в объеме, и концентрация
поверхностных дефектов при данной температуре должна
значительно превышать концентрацию объемных дефектов.
Тем не менее при описании поверхностного слоя кристалла
мы можем в первом приближении воспользоваться аппаратом
статистикотермодинамической теории, изложенной в гл. 2.
Тогда с помощью уравнений (2.50) — (2.53) можно определить
химический потенциал атомов (ионов) А, расположенных в
некоторых позициях сорта к на поверхности кристалла:
^^ак-*а>) + ^1п-[М (7.51)
где wAfc и [Ак]—энергия и концентрация атомов А в /с-позициях; [VK] —
концентрация незанятых к -позиций; член %a* обусловлен колебаниями
решетки; эффективные заряды атомов и вакансий здесь для простоты опущены.
Из изложенного в гл. 2, ясно, что уравнение (7.51) можно
^пользовать и при сильной поверхностной разупорядоченности,
к°гда [Ак] и [VK] существенно отличаются от нуля или
единицы. При этом, однако, следует помнить, что параметры
Wa* и %Ак могут заметно зависеть от концентраций [Ак] и
283
[Ук]. Эта зависимость пропадает только при слабой разупоря-
доченности, когда ад-атомы и вакансии можно рассматривать
как точечные дефекты.
7.5. Переход частиц через поверхность
раздела твердых фаз
В настоящем разделе дан вывод кинетического уравнения,
описывающего поток частиц через поверхность раздела двух
твердых фаз при неравновесных условиях, когда электрохимические
потенциалы этих частиц в обеих фазах имеют различные
значения. При этом мы будем рассматривать картину перескоков
частиц через потенциальный барьер, аналогичную рассмотрен»
ной в разделе 6.2 при выводе уравнения для потока частиц в
объеме твердой фазы. Основное отличие в данном случае со-
стоит в том, что разности концентраций и энергий частиц в
начальной и исходной позициях отличаются настолько, что
разложение в ряд, использованное в разделе 6.2, здесь
неприменимо и уравнение в общем виде будет нелинейным.
Пусть частицы (атомы, ионы, электроны) перескакивают
из некоторых устойчивых позиций I в твердой фазе I в устой»
чивые позиции II в фазе II, преодолевая при этом
потенциальный барьер высотой U. Профиль потенциальной энергии на
пути прыжка частицы изображен на рис. 7.6. В отличие от
рис. 6.1, демонстрирующего перескок частицы внутри одной
фазы, в рассматриваемом случае профиль энергетического
барьера может быть существенно несимметричным. Пусть
расстояние от вершины барьера до позиции I относится к расстоянию
от вершины до позиции II как а/Р (рис. 7.6), причем
а + Р = 1 (7.52)
Параметры аир, являющиеся индивидуальной
характеристикой контактирующих фаз, носят название
коэффициентов переноса заряда; смысл этого термина буд«:т ясен
из дальнейшего изложения.
Обозначим через Аф разность потенциалов между
позициями I и II, тогда согласно рис. 7.6 изменение энергии частииы
в позиции I по сравнению с таковым в седловой точке за счет
электрического поля
составит ш7)Д>ф, а в позиции П
—(Р^Аф. Если энергия
частицы в позициях I, II и в
седловой точке без учета
электростатической состав-
-'zii-P рис-7-6-
Схема энергетического барьера на
поверхности раздела твердых тел-
1 ^
ctqAq)
с(
«■■■■* - tffg
/з Т
284
ляющей равна соответственно w , w и ws, то вероятность
успешного перескока частицы слева направо при одном
колебании составит
/ wr -f aqAy — W
exp\ If
Чтобы получить число частиц, перескакивающих слева
направо, это значение нужно умножить на частоту колебаний v',
число частиц данного сорта в позициях I на единице
площади Г' и на вероятность [V]" того, что позиция II вакантна.
Записывая аналогичным образом число частиц,
перескакивающих справа налево, для общего потока частиц (числа
перескоков I—Я1 за единицу времени через единичное сечение)
получаем:
J=exp(-Tf)(v'I'[V]'exp ш
-/T^vrexp-^^^} (7.53)
Введем новую переменную — отклонение контактной
разности потенциалов в обеих фазах Д<р от ее равновесного
значения Дф0:
т] = Дер - Аср0 (7.54)
Эта величина широко используется в электрохимии; она носит
название перенапряжение перехода. Аналогичным
образом определим отклонения энергии частицы в позициях I и
II от соответствующих равновесных значений:
t№=*w — w0 (7.55)
и подставим соотношения (7.54) и (7.55) в уравнение (7.53):
J= exp^—— JJvT'[Vrexp —
— VT"[V]'exp — J (7.56)
Равновесное значение контактной разности потенциалов Аф0
можно исключить, учитывая, что при равновесии (Дш, г) = 0)
поток частиц / равен нулю. Отсюда находим:
ехр-^ = ; ^ехр——— (7.57)
Подстановка этой формулы в (7.56) дает после
элементарных преобразований:
У = У°/ { [АГ [V]Q Cxp Aw' + *W {АГ [V]° cxp Lw" - № j
VW[Vr kT [A]o[Vr kT )
(7.58)
285
В последнем уравнении отношение поверхностной
концентрации частиц к объемной концентрации Г/Г0 заменено на
отношение соответствующих долевых концентраций [А]/[А]0. Через
/v обозначена величина
_ [УПУ]"
/ v — 'г ~ (7.59)
[V]0[VJo
определяемая отношениями фактических концентраций
вакантных позиций I и II к их равновесным значениям. Величина /°
равна
jo = (/To [V]o )a (vTo [V]o f exp (-U\kT) (7.60)
Величина U означает высоту потенциального барьера в седло-
вой точке (см. рис. 7.6):
U = wso — w0 = wso — ($w'o + awl) (7.61)
Физический смысл величины /° легко видеть из уравнения
(7.58): при равновесии оба члена в фигурных скобках, а также
множитель fv обращаются в единицу и уравнение принимает
вид / = /°—/°. Следовательно, /° есть плотность потока частиц,
переходящих при равновесии через поверхность раздела фаз
слева направо и справа налево, другими словами, плотность
потока обмена частиц между обеими фазами при
равновесии фаз.
Уравнение (7.58) можно записать более компактно, если
воспользоваться определением химического потенциала частиц
(7.51). Действительно, из (7.51) вытекает, что отклонение
химического потенциала частиц от его равновесного значения
Л|! = |1—|хо равно
др = д^ + kT In [A] fV]o (7.62)
[A]o[V]
Потенцируя это равенство и подставляя результат в (7.58),
получаем уравнение замедленного перехода частиц
через поверхности раздела твердых фаз:
J = Jo/v(exp VJJ23 _ ехр *Е--М ) (7.63)
где Д|л' и А\х" — локальные значения отклонения химического потенциала
частиц от его равновесных значений в позициях I и П.
Наряду с макроскопическими переменными \х и т], не
зависящими от выбора микроскопической модели процесса,
уравнение (7.63) содержит множитель fv, определяемый формулой
(7.59). По своему происхождению этот множитель имеет
некоторую аналогию с корреляционным множителем диффузии в
кристаллических телах, подробно обсужденным в гл. 6. В обоих
случаях значение такого множителя определяется
отклонениями концентраций вакансий от некоторых стандартных значе-
286
ний: при диффузии — от среднестатистического, при переходе
через границу фаз — от равновесного. Однако в отличие от
корреляционного множителя диффузии, однозначно
определяемого характером диффузионного процесса и геометрией
кристаллической решетки, множитель /у зависит от степени
отклонения реальных условий на границе раздела фаз от
равновесных. Поскольку степень неравновесности процесса прямо
связана с потоками различных частиц через поверхность
раздела фаз, следует считать, что /у является функцией
плотностей потока всех частиц, участвующих в гетерогенной
реакции.
Это обстоятельство значительно усложняет задачу и в
общем случае требует ее решения на микроскопическом уровне,
т. е. с помощью уравнений, в которых неизвестными
переменными являются концентрации атомов и поверхностных дефектов.
Решить эту задачу в общем виде в настоящее время не
представляется возможным, поскольку структура поверхности
твердых тел чрезвычайно чувствительна не только к
температуре, но и к условиям приготовления образцов (температура,
давление прессования, временной режим обработки и др.).
Поэтому описание реальных кристаллов неизбежно связано с
использованием упрощенных модельных представлений о
строении поверхности реагирующих твердых тел.
Согласно изложенному в предыдущем разделе можно
выделить две предельные модели процесса, вытекающие из
упрощенной модели строения поверхности.
1. Переход через поверхность раздела фаз осуществляется
частицами, находящимися в плотноупакованных поверхностных
слоях, в которых доля незанятых позиций [V] мала по
сравнению с единицей. В этом случае реальные концентрации
вакансий могут значительно отличаться от равновесных и
множитель /у может быть существенно отличным от единицы. Для
описания такой ситуации необходимо записывать кинетические
уравнения в микроскопических переменных, например в форме
уравнения (7.58), где основной переменной будет являться доля
вакантных позиций [V].
2. Через поверхность раздела фаз переходят
преимущественно частицы, находящиеся в ад-состоянии. Согласно
описанной выше модели доля ад-позиций, занятых такими атомами,
считается малой, а доля вакантных позиций [V] — близкой к
единице. Поэтому в формуле (7.59) можно полагать {V]«
^ [V]0~l, а следовательно, и fv&l.
В реальных системах перенос через границу раздела фаз
осуществляется в основном частицами, локализованными на
некоторых активных центрах, которые занимают, в общем,
сравнительно небольшую долю поверхности раздела фаз.
Согласно современным представлениям физикохимии поверхности
твердых тел такими центрами могут служить ступени роста (в
первую очередь, их изломы), точки выхода на поверхность
287
дислокаций, линии выхода на поверхность границ зерен в
поликристаллах и т. п. В большинстве случаев при указанных
механизмах локальные значения концентрации вакантных позиций
при гетерогенных реакциях изменяются сравнительно
незначительно. Например, при росте атомного слоя, изображенного в
левой части рис. 7.5, ступень роста (край слоя) продвигается
вправо; при этом одновременно продвигаются вправо и
вакантные позиции, разрешенные для соответствующих атомов (узлы
решетки, непосредствено прилегающие справа к ступени),
причем число разрешенных вакантных позиций не меняется и
приблизительно остается равным равновесному.
В общем случае, при сравнительно сильной разупорядочен-
ности поверхности, относительные изменения концентраций
вакансий по сравнению с их равновесными значениями в
начальной и исходной позициях происходят в различных
направлениях. Так, если имеется направленный поток частиц из фазы I
в фазу II, можно полагать, что в позициях I истинная
концентрация вакансий будет ниже, а в позициях II — выше
равновесной. Согласно формуле (7.59) указанные эффекты частично
компенсируют друг друга, так что множитель /V в первом
приближении можно считать постоянным и приблизительно
равным единице.
При таком упрощении уравнение замедленного перехода
частиц через поверхность раздела твердых фаз (7.63)
принимает довольно простой вид
j = jo (exp W + W— ехр V'^j (7 64)
связывающий поток частиц с сугубо макроскопическими
переменными (I и т]. Поэтому уравнение (7.64) в дальнейшем
можно не связывать с микроскопическими моделями строения
поверхности твердых тел и использовать его в рамках
феноменологического описания кинетики гетерогенных реакций между
твердыми телами.
Остается еще отметить, что при малых отклонениях от
равновесных условий, А|х, qr\<^.kT, уравнение замедленного
перехода (7.64) может быть линеаризовано путем разложения экспо-
ненты в ряд по малым параметрам A\ifkT и qv\[kT с точностью
до линейных членов:
/=-£.(Д,1'_Дц" + ет) (7.65)
hi
Уравнение (7.65) представляет собой уравнение
термодинамики необратимых процессов, описывающее потоки частиц при
малых отклонениях от равновесия. При этом роль движущей
силы процесса перехода частиц через межфазную поверхность
играет разность их электрохимических потенциалов в
начальной и конечной позициях I и II:
Д^ = Др/ — Др/' + qi\ (7-66)
288
:0тсюда видно, что уравнение (7.64) является более общим,
нежели уравнения термодинамики необратимых процессов,
применение которых оправдано лишь в случае малых движущих
сил.
7.6. Адсорбция газов на твердых телах
Если твердое тело находится в газовой среде, на его
поверхности всегда содержится то или иное количество
адсорбированных молекул или атомов веществ, содержащихся в газовой
фазе. Адсорбция существенно изменяет свойства поверхности
твердого тела и поэтому играет важную роль в протекании
различных гетерогенных процессов [94—98].
Принято различать два типа адсорбции. К первому типу
относится такая, при которой адсорбированные молекулы
связаны с поверхностью относительно слабыми ван-дер-ваальсовы-
ми силами молекулярного взаимодействия (см. гл. 1,
раздел 1.3). Такая адсорбция называется физической. Она
проявляется в основном при низких температурах, при которых
энергия теплового движения недостаточна для разрыва слабых
молекулярных сил.
При высоких температурах преобладает второй тип
адсорбции — химическая, или хемосорбция. В этом случае
атомы адсорбированного вещества связаны с поверхностью
твердого тела специфическими химическими силами со
значительной энергией связи. Поверхность твердого тела в этом случае
представляет собой новую фазу, свойства которой могут
существенно отличаться от свойств объема твердого тела.
Поскольку образование химических связей обусловлено
короткодействующими силами, обычно предполагается, что хемосорбция
протекает лишь в одном монослое. На физическую же
адсорбцию это ограничение не распространяется; физическая
адсорбция может приводить к осаждению молекул поверх хемосор-
бированного слоя.
При физической адсорбции равновесие устанавливается
очень быстро, поэтому ее влияние на кинетику гетерогенных
реакций не существенно. Напротив, хемосорбция связана с
преодолением значительного потенциального барьера и поэтому
протекает сравнительно медленно; довольно часто она
является самой медленной стадией химического процесса и,
следовательно, определяет его скорость.
Сформулируем основные уравнения для скорости адсорбции
и равновесных концентраций адсорбированного газа. При этом
примем следующие упрощающие предположения.
*:■ 1. Поверхность твердого тела однородна в том смысле, что
вероятность адсорбции или десорбции одинакова для всех
Участков поверхности.
г 2. Элементарный акт адсорбции происходит при соударении
^олекулы газа с поверхностью твердого тела. Поэтому число
19—321 289
молекул, адсорбирующихся за единицу времени на единице
поверхности, пропорционально числу соударений молекул газа
с плоской поверхностью, равному согласно кинетической
теории газов
_ !_
п&л = (2«МкТ) 2р (7.67)
где М—- масса адсорбирующейся молекулы; р — парциальное давление
рассматриваемых молекул в газовой фазе.
3. Радиус действия адсорбционных сил очень мал, так что
адсорбироваться могут только те молекулы газа, которые
соударяются с чистой поверхностью твердого тела. Те же
молекулы, которые соударяются с уже адсорбированными
молекулами, упруго отражаются ими и возвращаются обратно в
газовую фазу. Поэтому для плотности потока адсорбции (числа
молекул газа, адсорбирующихся за единицу времени на
единице поверхности) можно записать:
Лд = (1-в)Яая (7.68)
где G — доля поверхности, занятой адсорбированным веществом.
4. Адсорбция протекает только в одном монослое, поэтому
поверхностная концентрация адсорбированного вещества равна
Г = 6ГЬ (7.69)
де Гь — эффективное число центров адсорбции на единице поверхности; в
случае атомно гладкой поверхности Гь имеет тот же порядок величины, что
и число узлов решетки.
При указанных предположениях скорость адсорбции не
зависит от ее механизма и определяется только свойствами газа
[см. уравнения (7.67) и (7.68)]. Обратный же процесс сильно
зависит от того, в каком виде находится адсорбированное
вещество — в виде молекул (02, СО, СОг, Н2, Н20) или в виде
атомов (О, S, CI, Br и т.д.). Поэтому рассмотрим два
отдельных случая.
1. Молекулы адсорбируются в том же виде, в котором они
находятся в газовой фазе (СО, С02, НгО). В этом случае поток
десорбции должен быть пропорционален концентрации
адсорбированного вещества, т. е. степени заполнения поверхности 6:
Лес = вЯдес (7.70)
где /гдес — плотность потока десорбции с единицы занятой поверхности.
Обозначим через Q энергию, необходимую для удаления
с поверхности адсорбированной молекулы (энергию де-
сорбции). Тогда в соответствии со статистикой Больцмана
для плотности потока десорбции можно записать:
Удес = 6vrL exp (—QlkT) (7.71)
где v — частота колебаний атомов в поверхностном слое (частота «попыток
десорбции»).
290
Таким образом, для плотности суммарного потока
адсорбции-десорбции / = /ад—/дес получаем:
у==_ ^.„evIYexp^-^J
(7.72)
(2™ШГ)
Отсюда легко найти равновесное значение Во при заданных
температуре и давлении, если учесть, что при равновесии / = 0.
При этом получаем уравнение
1 —(
Кр
(7.73)
имеющее смысл уравнения закона действия масс для
адсорбции с константой равновесия:
К--
1
■ехр
kT
(7.74)
(2izMkT) vrL
Разрешая уравнение (7.73) относительно степени
заполнения поверхности, находим:
fi _ _._КР _
\ + Кр
(7.75)
Это соотношение носит название изотермы адсорбции
Ленгмюра. График функции 60 изображен на рис. 7.7
(кривая 1) в зависимости от приведенного давления Кр. При
низких давлениях Во растет приблизительно линейно с Кр, а при
высоких плавно приближается к предельному значению 60=1.
2. Молекулы двухатомного газа Х2 при адсорбции
диссоциируют на атомы и адсорбция протекает в виде атомов X
(диссоциативная адсорбция). В этом случае плотность
потока адсорбции определяется тем же выражением (7.68), что
и при молекулярной адсорбции; десорбция же сопровождается
рекомбинацией адсорбированных атомов в молекулы, поэтому
плотность потока десорбции пропорциональна вероятности
одновременного нахождения двух адсорбированных атомов в
соседних позициях и,
следовательно, квадрату степени
заполнения.
Лес — аб2 ехр (— QjkT) (7.76)
РИС. 7.7.
Изотермы молекулярной (/) и
диссоциативной (2) адсорбции газов на
однородной повепхности твердого
тела.
19*
с некоторым коэффициентом пропорциональности а. Таким
образом, при диссоциативной адсорбции кинетическое уравнение
для потока молекул Х2 имеет вид
«-** -ав-expf-^ (7.77)
У =
kT
(2nMkT)
Отсюда для равновесия при / = 0 получаем уравнение закона
действия масс:
■Кр
(7.78)
с константой равновесия
К =
1
Q
(7.79)
(2nMkT) a
Решение уравнения (7.78) имеет вид
КР_
во= 2
1 +
V кр J
(7.80)
Соответствующая кривая 2 изображена на рис. 7.7 в
зависимости от приведенного давления Кр. В целом эта кривая
подобна изотерме Ленгмюра 1 и отличается от нее несколько
более крутым ходом.
Если степень заполнения поверхности 6 мала по
сравнению с единицей, в правых частях кинетических
уравнений (7.72) и (7.77) можно выразить 6 через химический
потенциал адсорбированного вещества и таким образом
выразить потоки адсорбции через макроскопические величины р и
\i. Для этого подставим формулу (7.51) для химического
потенциала адсорбированного вещества в виде
p = p° + kTln
1—(
(7.81)
Полагая 6<Cl, имеем для отклонения химического потенциала
от его равновесного значения:
А[г = АГ1п(6/е0) (7.82)
где 6о — равновесное значение 6.
Теперь преобразуем кинетические уравнения следующим
образом.
1. Молекулярная адсорбция. Представим (7.72)
при 8<1 в виде
у_ Р, а ..т_ „™/ Q\ 6
60vrL ехр (--£-) —
1 ° *Ч kT) e0
(7.83)
(2яЛГЛ7)'
292
Из условия равенства нулю потока при равновесии получаем
равенство
J» = Е~г = 80vrL exp (- ~jL)j (7.84)
(2nMkT)2
При этом /° равно плотности потоков адсорбции и десорбции и
может быть названо плотностью потока обмена между газовой
и твердой фазами; согласно равенству (7.84) /°
пропорционально парциальному давлению адсорбирующегося вещества в
газовой фазе. Учитывая (7.82) и (7.84), уравнение (7.83)
теперь можно представить в простом виде
j=J0[\— ехр (Дц kT) ] (7.85)
2. Диссоциативная адсорбция. В этом случае из
(7.77) имеем:
•/x3 = y°x2[l-(W2] (7.86)
где плотность потока обмена равна
4 = V—j- = a\* ехр (- -£-} (7.87)
{2nMkT)2
Используя выражение (7.82) для химического потенциала
адсорбированных атомов X, из (7.86) получаем:
Ух, = Д[1 -ехр(2Д(лх/£Г)] (7.88)
Поскольку удвоенный химический потенциал атомов 2A\ix
равен химическому потенциалу ^х2 адсорбированного вещества в
молекулярной форме Хг, (7.88) можно переписать в виде
Ух3-Д[1-ехр(Д^Г)] (7.89)
Последнее уравнение совпадает с уравнением
молекулярной адсорбции (7.85). Этот результат очень важен при
описании адсорбции двухатомных газов, поскольку при этом в
большинстве случаев точно не известно, в какой форме протекает
адсорбция: в форме молекул Х2 или атомов X. Согласно
полученному результату при сравнительно малых степенях
заполнения 6<С1 механизмом адсорбции можно не интересоваться и
пользоваться формальным уравнением (7.89).
7.7. Рост интерметаллических фаз
при замедленном переходе
через межфазные границы
Вернемся теперь к процессу роста интерметаллической фазы
АВГ из чистых металлов А и В (см. рис. 7.1,а). Однако здесь
мы рассмотрим предельный случай, противоположный рас-
293
смотренному в разделе 7.2, где считалось, что рост фазы
целиком определяется скоростью диффузии через интерметаллид.
Теперь мы будем считать, что диффузия протекает практически
без затруднений, а скорость всего процесса лимитируется
скоростью гетерогенных процессов — переходом атомов А и В из
чистых металлических фаз в интерметаллическое
соединение АВг.
Далее, будем считать, что химические потенциалы атомов
А и В в чистых фазах А и В на границах этих фаз с
интерметаллическим соединением имеют равновесные значения, а в
поверхностных слоях интерметаллической фазы отличаются от
равновесных значений на величины
ЬгАщгГА{0)-гА(к); Дцв = ,хв(/.)-цв(В) (7.90)
Тогда плотности потоков атомов А и В из металлических фаз
в интерметаллид будут определяться уравнением замедленного
перехода (7.64), в котором следует положить Дц/ = 0, Д(х//=А|1,
<7=0:
JA = /A(l-exp-^-)
v ; (7.91)
J в ~- Jb
(1 -ехР-тг)
С другой стороны, потоки атомов через границы фаз
непосредственно связаны со скоростью роста интерметаллической
фазы [см. уравнение (7.12)]. Приращение интерметаллида за
счет потока атомов А через границу А—АВГ равно
dL\dt = Jk\Nk (7.92а)
а за счет потока В через границу В—АВГ
dLldt=JBINB (7.926)
Подставляя выражения для потоков (7.91) в (7.92), получаем:
dLjdt = Kt (7.93)
где введено обозначение
Смысл этого обозначения будет выяснен позднее.
Уравнение (7.94) связывает между собой значения
отклонений химических потенциалов А и В в интерметаллиде от их
значений при равновесии с фазами А и В соответственно Д|м
и Д|1В. Второе уравнение, необходимое для их определения,
найдем из следующих соображений. Перепишем соотношения
(7.14) и (7.16) в виде
dL 1 Г
DA г /пч /гм , °В
foA (0) _ рА (I) ] + -JL [цв (Ц - ^в (0) ] (7.95)
294
и заметим, что условие незаторможенности диффузии в
интерметаллической фазе формально выражается в предельном
переходе DAjL—^оо или D^IL—^оо. Это означает, что диффузия
протекает без затруднений либо в случае очень высокой
подвижности атомов (Z)A—^оо или Db—*°°), либо в случае очень
тонкого слоя интерметаллида (L—>-0). Однако в обоих случаях
вследствие заторможенности межфазных процессов скорость
роста интерметаллической фазы dL/dt должна оставаться
конечной величиной. Следовательно, при рассматриваемых
условиях разности химических потенциалов в квадратных скобках
(7.95) должны быть близкими к нулю, так что
PA(0)«|iA(L); vB(0)^vB(L) (7.96)
По физическому смыслу это означает настолько быстрое
протекание диффузионного процесса, что в любой момент времени
химические потенциалы успевают выравняться по всей
толщине интерметаллической фазы, а значительные перепады AjiA
и Д|1в их значений целиком локализованы на границах
раздела фаз.
Принимая упрощенные условия (7.96), из (7.17) находим
для изобарного потенциала реакции:
AG-A^ + rA^ (7.97)
Уравнения (7.94) и (7.97) образуют вместе полную систему
относительно двух неизвестных К\хА и Д|хв- Эта система имеет
единственное решение, однозначно определяющее значения
перепадов химических потенциалов на границах фаз Дц,А и
Д^в- Здесь нет необходимости решать эти уравнения в общем
виде, тем более что одно из них — (7.94) — трансцендентное.
Для нас существенно то обстоятельство, что в этой системе
уравнений все коэффициенты перед неизвестными переменными
не зависят от времени. Поэтому не должны зависеть от времени
и значения Д^А и Д|1в, удовлетворяющие системе уравнений, а
следовательно, и величина Кь определяемая уравнением (7.94).
Поэтому интегрирование уравнения (7.93) в пределах от t=0>
L = 0 до t, L приводит к простому соотношению
L = Kxt (7.98)
представляющему собой математическое выражение
линейного закона роста интерметаллической фазы. Отсюда
становится ясным и смысл введенного выше обозначения К\\ эта
величина представляет собой константу линейного закона роста
фазы.
Согласно уравнению (7.94) она определяется довольно
сложными выражениями и в общем случае зависит как от
токов обмена /°, так и от отклонений химических потенциалов
от равновесных значений Д^х обоих компонентов. Однако эти
выражения значительно упрощаются в предельном случае, до-
295
пускающем аналитическое решение системы уравнений (7.94) и
(7.97). Предположим, что через одну из межфазных
поверхностей, например В—АВГ, атомы переходят без затруднений,
так что переход атомов А через границу А—АВГ является
единственной лимитирующей стадией процесса. Формально это
условие означает очень высокое значение потока обмена
атомов В на границе В—АВГ, т.е. /°в—^°°. В этом случае при
любых конечных значениях Ki разность во вторых скобках
выражения (7.94) должна быть очень малой, так что
ехр (Дц,в/£Г) ~1, или A|iB~0. Тогда из (7.97) вытекает
AG^A^a, т.е. все изменение изобарного потенциала реакции
оказывается сосредоточенным на границе фаз А—АВГ.
Для константы линейного закона в рассматриваемом
предельном случае получаем:
*'=4(i-expf) (7-99)
Изменение изобарного потенциала образования
интерметаллического соединения AG, как и любой спонтанно протекающей
реакции, отрицательно. Кроме того, при не слишком высоких
температурах и при достаточно высоком химическом сродстве
веществ А и В AG по абсолютному значению может быть
большим по сравнению с kT. Тогда экспонентой в (7.99) можно
пренебречь по сравнению с единицей, и формула (7.'99)
принимает очень простой вид
K^JIjNa (7.100)
согласно которому величина Ki целиком определяется
плотностью тока обмена наиболее медленной стадии процесса /V
В этом случае согласно определению (7.60) константа
линейного закона Ki является экспоненциально возрастающей
функцией температуры.
7.8. Окисление металлов при замедленных
межфазных процессах
Кинетика роста окалины на металле при диффузионном
контроле была рассмотрена в разделе 7.3. Здесь мы рассмотрим
противоположные предельные случаи, когда процесс окалино-
образования лимитируется скоростью процессов на
поверхностях раздела фаз: либо скоростью перехода металла через
границу М—МХГ, либо скоростью адсорбции неметалла на
границе Х2—МХГ (см. рис. 7.1, б). При этом, как и в случае
образования интерметаллических соединений, будем считать
диффузионный процесс в слое продукта реакции — окалины —
незаторможенным. Тогда на основе соображений, изложенных в
предыдущем разделе, по аналогии с формулой (7.97) можно за-
296
писать для изобарного потенциала реакции окалинообразова-
ния:
AG = pb* + rAfx*
(где косые крестики в верхнем индексе означают
электронейтральность выбранных компонентов окалины). Поскольку
A|ixx = V2A^x2 и г = 2м/2Х, это соотношение можно представить
в виде
AG==A^m + -~-ДРх, (7.Ю1)
При незаторможенной диффузии скорость роста окалины
следует связывать с потоками частиц через межфазные границы
с помощью кинетических уравнений, аналогичных (7.92):
либо с потоком катионов М2м+ через границу металл —
окалина
J м
— =-£- (7.102а)
dt NM v '
либо с потоком адсорбции молекул неметалла Хг на
границе окалина — газ
dL 2JY
=—Ь. (7.1026)
dt Nx v '
Здесь Nm и N.x — объемные концентрации катионов и анионов
в окалине; предполагается, что отклонения от стехиометриче-
ского состава окалины MX Zl/Z сравнительно невелики, такчто
JVM и Nx постоянны по всей ее толщине.
Рассмотрим подробнее уравнение замедленного перехода
(7.64), определяющее плотность потока катионов и электронов
через границу металл — окалина. Как и при описании роста
интерметаллида, предположим, что химические потенциалы
катионов и электронов в слое металла, граничащем с окалиной,
имеют равновесные значения, поскольку состав металлической
фазы в ходе реакции не изменяется. Тогда в уравнении (7.64)
как для катионов, так и для электронов следует принять
Л}г/ = 0, а Л|л"=Д|1 равным разности
Д[х = ^(МХГ)~^(М) (7.103)
где вследствие быстрой диффузии значения химических
потенциалов всех частиц в окалине |л,(МХг) считаются
приблизительно постоянными по всей ее толщине; \i(M) —значение
химического потенциала соответствующих частиц в окалине при ее
равновесии с металлом. Таким образом, плотности потоков
297
катионов и электронов через границу металл — окалина равны
/мм = ./Цехр -^±- ехр — J (7.104)
J p J p
Ь\*-е + N
kl l kT
exp l ^.j — ехр
(7.105)
Поскольку электроны в твердых телах обладают гораздо
более высокой подвижностью, чем ионы, их переход через
межфазную границу можно считать незаторможенным. Формально
это условие означает /°е=оо; следовательно, при конечных
плотностях потока электронов J-e выражение в квадратных
скобках (7.105) должно быть равно нулю. Это приводит к
простому соотношению между перенапряжением перехода и
отклонением химического потенциала электронов в окалине от
равновесного значения (7.103):
£7J = —ДрГ (7.106)
Поскольку электронная проводимость окалины обусловлена
ее нестехиометричностью, химический потенциал электронов
однозначно определяется составом и, следовательно,
однозначно связан с химическим потенциалом металла. Эту связь легко
установить, если учесть, что при равновесии ионного
кристалла с газовой фазой согласно результатам гл. 5 [см. уравнение
(5.61)] концентрация электронных дефектов — электронов
проводимости или дырок — связана с давлением неметалла Хг в
газовой фазе уравнением
[ет] = Арт11т (7.107)
где параметр т определяется значениями эффективных зарядов
доминирующих ионных дефектов (см. табл. 5.3). В
рассматриваемом случае реакционная система находится в существенно
неравновесных условиях благодаря замедленному протеканию
межфазных процессов. Тем не менее есть все основания пола-
гать, что в каждом элементе объема окалины существует
локальное равновесие между точечными дефектами — как
ионными, так и электронными. В этом случае каждому малому
элементу объема окалины можно формально приписать некоторое
определенное значение давления р(МХг) неметалла Хг,
отвечающее локальному равновесию дефектов. (В случае пористой
окалины это значение соответствует реальному давлению Хг в
поре.) Тогда концентрации электронных носителей также будут
определяться уравнением (7.107), несмотря на отсутствие
равновесия на границах фаз; однако вместо реального давления в
газовой фазе в этом уравнении содержится формальное
давление в окалине р(МХг).
298
В случае невырожденного электронного газа согласно
статистике Больцмана химический потенциал электронов
определяется формулой
р7 = Ре±ЬТ1п[ет] (7.108)
Следовательно, для его отклонения от равновесного значения с
учетом (7.107) можно записать:
А[х7 =—— A Inp (МХГ) (7.109)
m
С другой стороны, химический потенциал нейтрального
металла [Iх м в окалине также легко связать с давлением р(МХг).
Для этого воспользуемся соотношением Гиббса — Дюгема
(П.22) в форме
^М - чу %
гх zzx
а также уравнением для химического потенциала
молекулярного неметалла в окалине
VXu = \fXt + kTliip(MXr) (7.111)
Интегрируя уравнение (7.110), при учете (7.111) находим:
дР$ = -^д1п/>(МХг) (7.П2)
Сравнение последнего выражения с (7.109) приводит к
искомому соотношению
AfcT=—AfU (7Л13)
гмяг
Наконец, учтем очевидное соотношение между
химическими потенциалами катионов и нейтральных атомов металла:
*мМ++*м*7 = *Й (7-114)
Теперь, подставляя соотношения (7.106), (7.113) и (7.114) в
уравнения (7.102а) и (7.104), получаем для скорости роста
окалины:
^^^exp^-^^-J^l-exp^J (7.115a)
С другой стороны, скорость роста окалины связана с
плотностью потока адсорбции на границе окалина — газ
уравнением (7.1026). Подставляя в него (7.89), имеем:
dL 2,/X» Л А'ахЛ /7 11^
= ■——2-( 1 — ехр (/.1150)
299
Приравнивая правые части (7.115а) и (7.1156), получаем урав
нение, образующее совместно с (7.101) полную систему урав-
нений относительно двух неизвестных Д|лмх и iA\xx2. Поскольку
все коэффициенты перед неизвестными не зависят от времени,
единственное решение системы определяет значения Д|лмх и
Д|лХ2, от времени также не зависящие. Поэтому правые части
уравнений (7.115) остаются постоянными во времени и могут
быть названы константой линейного закона ока-
линообразования:
1 nm \ mkT j \ v kT }
= «7l1-exp^) (7Л161
Такое название вытекает из результата интегрирования урав-
нений (7.115) в пределах от / = 0, L = 0 до t, L:
L*eKtt (7.117)
представляющего собой выражение линейного закона роста
фазы.
Как и в случае образования интерметаллида, система (7.101)
и (7.116) в общем виде не решается аналитически, поскольку
уравнение (7.116) трансцендентно. Однако аналитические
решения легко находятся в двух предельных случаях.
1. Переход катионов из металла в окалину является
единственной лимитирующей стадией процесса окалинообразования;
адсорбция неметалла, как и диффузия в окалине, протекает без
затруднений. В этом случае ток обмена адсорбции-десорбции
/°х2 можно считать бесконечно большим; тогда согласно (7.116)
при конечных значениях константы Ki должно выполняться
равенство
ехр-^ = 1 или А[хХз = 0 (7.118)
и из (7.101) следует
Д^-ДО (7.119)
Это означает, что в случаях, когда скорость процесса определя
ется переходом катионов через границу металл — окалина, все
изменения изобарного потенциала реакции \G сосредоточены
на указанной границе.
Учтем теперь, что AG — отрицательная величина, по
абсолютному значению в большинстве случаев значительно
превосходящая kT, поэтому экспонентой exp(A\iMxIkT) в (7.116)
300
можно пренебречь по сравнению с единицей, и для константы
линейного закона получаем:
2. Единственной лимитирующей стадией процесса является
адсорбция неметалла на поверхности окалины; переход
металла через межфазную границу не заторможен. В этом случае,
полагая /м° = оо, из (7.116) и (7.101) находим:
Д^=0; A^zzz-^AG (7.121)
Kl = 2JxJNx (7.122)
Формула (7.122) аналогична упрощенной формуле (7.100),
найденной в предыдущем разделе для процесса роста
интерметаллического соединения. Эта аналогия вполне естественна,
поскольку обе формулы соответствуют процессам,
лимитируемым потоком нейтральных атомов или молекул. Формула же
(7.120) более сложна и содержит экспоненциальную
зависимость Ki от изобарного потенциала образования фазы AG. Это
обстоятельство обусловлено перенапряжением перехода,
существующим на границе металла с окалиной и значительно
упрощающим протекание наиболее медленной стадии процесса —
перехода катионов из металла в окалину.
Согласно формулам (7.120) и (7.122), а также
определениям потоков обмена (7.60), (7.84) и (7.87) в обоих
рассмотренных выше предельных случаях константа линейного закона
окалинообразования является экспоненциально возрастающей
функцией температуры.
В ряде случаев при изменении температуры опыта
наблюдается переход параболического и линейного законов друг в
друга. Типичным примером является окисление тория в
атмосфере кислорода. Соответствующие экспериментальные
зависимости прироста массы окалины Th02 от времени приведены
на рис. 7.8. Как видно из рис. 7.8, а, при сравнительно низких
температурах 250—350 °С прирост массы окалины
пропорционален корню квадратному из времени, что соответствует
параболическому закону (7.39). При более высоких температурах
375—400 °С параболический закон сменяется линейным
(рис. 7.8,6). Причиной этого является более быстрый рост с
температурой коэффициента диффузии кислорода, чем потока
обмена на межфазной границе. В результате диффузионные
затруднения, определяющие параболическую кинетику при
пониженных температурах, в области высоких температур
становятся несущественными по сравнению с затрудненностью
межфазных процессов, что и приводит к линейной кинетике
окисления.
301
О 2 Ч 6 t'lfauH 0 8 16 24 Ь,мин
а б
РИС. 7.8.
Кинетика окисления тория при различных температурах [99]:
а — параболическая; б — линейная зависимость.
В данной главе мы рассмотрели кинетику роста твердых
фаз в простейших реакционных системах, включающих лишь
один продукт реакции — бинарное интерметаллическое (или
валентное) соединение АВГ или бинарную окалину МХГ, и
вывели уравнения двух фундаментальных законов их роста —
параболического и линейного. При выводе уравнений этих
законов нам пришлось принять целый ряд упрощающих
предположений. Уже из обилия этих предположений видно, что
изложенная здесь теория сильно ограничена и может описывать
лишь ограниченный круг реальных систем, более или менее
отвечающих принятым упрощениям.
Условия, отвечающие изложенным выше модельным
представлениям, чаще всего удается обеспечить только при
постановке специальных экспериментов при строго контролируемом
режиме протекания реакций (температура, состав исходных
реагирующих веществ, геометрические факторы и т.д.). В
реальных же условиях, например при технологических процессах в
условиях промышленного производства материалов, ситуация,
как правило, оказывается гораздо более сложной.
Наблюдаемые при этом закономерности (если таковые вообще удается
установить) чаще всего не укладываются в рамки ни
параболического, ни линейного закона. Это связано прежде всего с тем,
что при технологических процессах их кинетика определяется
множеством факторов, не учитываемых изложенной выше
упрощенной теорией. К таким факторам относится образование
многофазного продукта реакции (несколько слоев фаз АВГ или
МХГ с различными значениями стехиометрических
коэффициентов г, например FeO, Fe304, Fe203, либо многофазные слои,
состоящие из крупинок фаз различного состава). Положение еще
более усугубляется, если в ходе реакции возникают пористые
слои окалины, а тем более растрескивающиеся в ходе реакции.
302
В этих случаях перенос вещества через продукт реакции
зависит от множества факторов, не столько физико-химических,
сколько механико-динамических, и не может быть описан в
рамках теории, опирающейся на представления о точечных
дефектах кристаллов.
Более того, даже в условиях образования компактного слоя
продукта твердофазной реакции принятые выше предположения
могут не отвечать реальным ситуациям. Так, в двух последних
разделах мы предполагали, что химический потенциал частиц,
переходящих через границу двух твердых фаз, в их исходных
позициях остается постоянным и равным его равновесному
значению в соответствующей фазе (А, В или М соответственно в
разделах 7.7 и 7.8). Однако при достаточно высокой скорости
процесса это условие может не выполняться. Частицы,
переходящие в соседнюю фазу, скорее всего сосредоточены на
некоторых активных центрах, концентрация которых может быть
значительно ниже общей концентрации поверхностных узлов.
Поэтому при достаточно высокой скорости реакции вполне
правдоподобна ситуация, при которой активные позиции
обеднены участвующими в реакции атомами. В этом случае в
кинетическом уравнении замедленного перехода (7.63) наряду
с переменной A\i", учитывающей изменение химического
потенциала частиц в приконтактном слое продукта реакции,
необходимо учитывать и его изменение Д[/ в приконтактном слое
исходных реагентов А, В или М. Это приводит к тому, что в
обсужденных выше системах двух уравнений (7.94) и (7.97) или
(7.101) и (7.116) появляется третье неизвестное А\х'.
Соответствующее третье уравнение системы может быть
сформулировано только на основе определенных модельных представлений
о переходе атомов А, В или ионов М2м+ из их решеточного
состояния в активное состояние, из которого они переходят в
фазу продукта реакции. Это приведет к тому, что все
переменные Ар, окажутся зависящими от времени и согласно
уравнениям (7.94) и (7.116) величина Кг окажется также зависящей
от времени. Следовательно, при этих условиях линейный закон
роста продукта реакции будет нарушен, и тем в большей
степени, чем больше химические потенциалы исходных реагентов
в их активном состоянии на границе с продуктом реакции
отличаются от их значений в объеме соответствующей фазы.
На практике наряду с параболической и линейной
кинетикой наблюдаются и совершенно иные закономерности роста
фаз — кубическая, логарифмическая, обратно логарифмическая,
асимптотическая и т.д. Каждая из них требует для своего
объяснения многих дополнительных предположений о механизме,
определяющем кинетику реакции. Наблюдаемые
закономерности удается более или менее убедительно объяснить на основе
изложенной здесь теории, но, как правило, при дополнительных
предположениях помимо принятых здесь.
303
Ситуация становится еще более запутанной, если в ходе
твердофазной реакции изменяется площадь поверхности, на
которой протекает реакция. Так, топохимические
реакции начинаются в одной или нескольких активных точках
поверхности раздела фаз и постепенно распространяются на
значительную долю поверхности.
Очень важное влияние на скорость твердофазных
гетерогенных реакций оказывают поверхностно-активные вещества,
обладающие каталитическими свойствами [106]. Большое
значение имеет способ приготовления образцов и вся их
предыстория. Поэтому описание кинетики химических реакций с
участием твердых тел в настоящее время никак не может быть
уложено в рамки какой-либо единой общей теории, и в данной
книге автор ограничился изложением теории, базирующейся
на предельно упрощенных модельных представлениях о
дефектной структуре и транспортных свойствах кристаллических
твердых тел.
ПРИЛОЖЕНИЕ
Некоторые сведения из химической
термодинамики
Здесь мы приводим некоторые важные соотношения химической
термодинамики, используемые в основном тексте книги.
Подробное изложение химической термодинамики можно найти в
книгах [101—105].
Основные термодинамические функции. Если
термодинамической системе сообщается извне бесконечно малое количество
теплоты dQ, то изменение энтропии системы составляет
dS>dQjT (П.1)
где Т — абсолютная температура; знак = соответствует обратимым
процессам; знак >—необратимым процессам (второе начало термодинамики).
В соответствии с таким определением энтропия является
мерой необратимости процесса; при самопроизвольных
процессах, протекающих в изолированной системе, энтропия может
только возрастать, достигая максимального значения при
установлении термодинамического равновесия. Так как при
необратимых процессах изолированная система, как правило,
переходит из менее вероятного состояния в более вероятное, то
энтропия должна быть связана с вероятностью Q данного состояния
системы:
5 = 5(9)
Явный вид этой зависимости может быть установлен
следующим образом. Энтропия системы есть аддитивная
величина, т. е. равна сумме энтропии отдельных частей системы, если
процессы в отдельных частях протекают независимо друг от
друга. [Для обратимых процессов это свойство энтропии
вытекает непосредственно из равенства (П.1), поскольку вся
теплота dQ, полученная системой, аддитивно складывается изтеп-
лот, полученных независимыми частями.] Таким образом,
энтропия системы, состоящей из двух независимых подсистем с
энтропиями S\ и S2, равна S = 5i+52 или
5(9) = 5(91) + 5(92) (П.2)
Но вероятность Q нахождения системы в данном состоянии
равна произведению соответствующих вероятностей Q\ и Q2
для обеих подсистем, так что равенство (П.2) можно записать
в виде
5 (2122) = S(21) + 5 (22) (П.З)
50—321
305
Функция, удовлетворяющая такому свойству
S=klnQ (П.4)
(формула Больцмана). Действительно, дифференцируя
(П.З) по Qi и Q2, получаем:
Sf(Q1Q2)Ql=Sf(Q2) '
откуда следует
S'(Qi)Q1 = S'(Qs)Q2 или S' (Щ 2 = const
Интегрирование последнего равенства дает S = constln Os
т.е. (П.4).
В отличие от теории вероятностей, где все вероятности
нормируются на единицу, в термодинамике используют
ненормированные вероятности и термодинамической
вероятностью называют число способов, с помощью которых можно
осуществить данное макросостояние системы. При таком
задании Q константа k в формуле (П.4), называемая
постоянной Больцмана, оказывается равной газовой постоянной,/?,
отнесенной к одной частице: k=\R/NA, где NA — число Авогадро*
Рассмотрим теперь причины, приводящие к изменению
энергии системы. В соответствии с законом сохранения энергии
таких причин может быть три: а) теплота dQ, сообщаемая
системе извне; б) работа dA, совершаемая системой над внешними
телами; в) переход некоторого числа частиц dNi в систему из
окружающей среды.
Таким образом, изменение энергии системы определяется
выражением
dU^dQ ~dA + % — dNt (П.6)
/ l
(первое начало термодинамики). Здесь производные
ia = dU/dNi берутся при dQ = dA==0 и носят название
химических потенциалов /-частиц.
Предположим, что работа может совершаться только
силами давления: dA = pdV; тогда объединение первого начала (П.6)
и второго начала (П.1) приводит к основному
уравнению термодинамики:
dU< TdS-pdV + ^VidNt (П.7)
i
Введем следующие функции:
H=U + pV (П.8)
и зохор н о-и зо тер м и чески й, или, сокращенно, изо-
хорный потенциал
f=U—TS (П.9)
306
и изобарно-изотермический, или, сокращенно
изобарный потенциал
G=U—TS + pV (П.10)
Дифференцируя эти определения функций, находим:
dH = dU + pdV + Vdp
dF-dU—TdS — SdT
dG = dU— TdS — SdT + pdV + Vdp
или, при учете (П.7):
dH < TdS + Vdp + 2 M^V* (П. 11)
i
dF^-SdT- pdV+^^dNt (П.12)
dGK — SdT+ Vdp + ^tdNt (П.13)
i
Уравнения (П.7) и (П.11) — (П.13) показывают направления
самопроизвольных процессов в системах с постоянным числом
частиц. Действительно, при Ni = const эти уравнения
принимают вид
<Ш<0 (5, V
d#<0 (5, р
dF^O (Г, V
rfG<0 (Т, р
Это означает, что в системе с постоянным числом частиц могут
протекать лишь такие необратимые процессы, которые
приводят к уменьшению соответствующих потенциалов £/, Я, F или
G, или же обратимые процессы, при которых эти потенциалы
не изменяются.
Если число частиц в системе может изменяться, то
направления самопроизвольных процессов можно проследить на
следующем примере. Приведем в контакт друг с другом две
системы, содержащие частицы одного сорта, но с различными
химическими потенциалами; для определенности будем считать,
что химический потенциал в первой фазе \л' выше, чем во
второй \i". Пусть при постоянных температуре и давлении из
первой фазы во вторую перешло dN частиц. Тогда в соответствии
с (П.13) изменение изобарного потенциала первой фазы
составляет dG'^.—\\'dN, а второй фазы dG"^.-\-\k"dN, так что
изменение изобарного потенциала объединенной системы,
включающей обе фазы, равно
dG = dG' + dG" < (р/' — fV) dN
const)
: COnSt) (ПИ)
: Const)
= const)
20*
307
и отрицательно вследствие |ЛЛ> |х,/г. Но число частиц в
объединенной системе осталось неизменным, так что к ней можно
применять критерий (П.14), который в случае dG<0 означает
необратимость и, следовательно, самопроизвольность процесса.
Таким образом, при наличии систем с различными химическими
потенциалами вещество переходит самопроизвольно из фазы с
большим химическим потенциалом в фазу с меньшим. Термин
«химический потенциал» введен именно в связи с этим
обстоятельством по аналогии с электрическими явлениями, где
положительный заряд переходит из областей с большим
электростатическим потенциалом в области с меньшим потенциалом..
Аналогично тому, как электрическое поле является движущей
силой для зарядов, гравитационное поле — для масс, темпера»
турное поле — для теплоты, «химическое поле» является
движущей силой для данного вещества.
Выведем теперь некоторые свойства химических потенциалов,
предполагая, что в системе могут протекать лишь обратимые
процессы. В этом случае в уравнениях (П.7) и (П.11) — (П.13)
остается знак равенства, и для химического потенциала i-ча-
стиц следует:
dU
/ дН
/, ~~ V dNt
dNi Js, v, Nj \ dhTi ]s,p, a7j
(-¥-) =(^A (П.15)
Таким образом, химический потенциал можно определять как
производную по числу частиц от любой из характеристических
функций U, Я, F и G, однако при этом он будет выражен
через различные переменные.
Большая часть физических и химических экспериментов
проводится при постоянных температуре и давлении, поэтому из
введенных характеристических функций наиболее удобно
пользоваться изобарным потенциалом, а химический потенциал
определять с помощью последнего из равенств (П.15):
Ъ=(—\ (П.16)
1 К dNt )т. р, Nj
Более наглядное представление о химическом потенциале
можно получить с помощью следующего рассуждения.
Возьмем некоторую термодинамическую однофазную систему с мае-
сой М. Увеличим при постоянных температуре и давлении
число частиц каждого сорта так, чтобы состав системы не
изменился, т. е. чтобы относительное увеличение числа частиц
dNi/Ni было одинаковым для всех сортов частиц и равно
относительному увеличению массы системы dMjM. Тогда изменение
308
изобарного потенциала системы в соответствии с уравнением
(П.13) составит
dG =2^= d~- 2^ (П.17))
i i
С другой стороны, новая часть системы dM имеет тот же состав.,
что и старая часть М, и физически неотличима от нее. Но
изобарный потенциал подобно другим экстенсивным свойствам
У, S, U, Я, F является аддитивной величиной, поэтому его
приращение должно быть пропорционально приращению массы
системы:
dG __ dM
G " М
Сравнивая с (П.17), видим, что
0 = 2^ (П18>
Таким образом, оказывается, что химический потенциал t'-ча-
стиц есть изобарный потенциал, приходящийся на одну
частицу i-сорта*. Другими словами, химический потенциал
совпадает с парциальным изобарным потенциалом i-частиц:
Ь= gi = Ui— Tst + pvt (П. 19)
где gi, иь Si и vt — парциальные величины: изобарный потенциал, внутренняя
энергия, энтропия и объем, приходящиеся на одну частицу t-сорта.
Для системы, состоящей из нескольких различных фаз,
результат будет тот же, если относительные увеличения масс всех
фаз одинаковы.
Проведенное рассуждение справедливо лишь для
изобарного потенциала; для других характеристических функций U, Н
и F приращения не равны 2^/rfiV/, поскольку приращения dV
i
и dS в формулах (П.7), (П.11) и (П.12) отличны от нуля.
Поэтому парциальные энергия, энтальпия и изохорный потенциал
не совпадают с химическим потенциалом.
Из определения термодинамических функций вытекают два
важных соотношения, широко использующихся в прикладных
задачах физической химии. Прежде всего, из сотношения (П.13)
для обратимых процессов следует
А° ~ с
д'т - *
Поскольку
G = H- TS
* Химический потенциал часто определяют как производную от G по
числу молей г-вещеетва, т. е. как изобарный потенциал, приходящийся на
моль i-частиц. Очевидно, что при таком определении химический потенциал
в Na раз больше используемого нами.
3095
это дает:
H=G-Г—
дТ
Отсюда, при учете очевидного соотношения
дТ [т)~ Т дТ Р
получаем
д ( G\_ H
дТ\Т) 72
Последнее равенство носит название соотношения Гиб-
бса — Гель м гольца. Дифференцируя его по Ni, приходим
к более употребительной форме соотношения Гиббса — Гельм-
гольца:
—; (П.20)
дТ \Т )
связывающей химический потенциал и удельную энтальпию
/-частиц.
Следующее соотношение получается из формулы (П. 18),
дифференцирование которой дает:
i i
Сравнение с (П.13) показывает, что
SdT —Vdp + ^ N^ = О (П.21)
i
Полученное уравнение называется соотношением
Гиббса — Д ю г е м а. Обычно оно используется в форме
2л/^ = 0 (Г, р = const) (П.22)
Условия термодинамического равновесия. Если в системе
установилось термодинамическое равновесие, то малые
отклонения от равновесного состояния, вызванные флуктуациями,
безусловно обратимы. Поэтому для рассмотрения малых
отклонений от равновесия можно пользоваться всеми приведенными
выше уравнениями, заменяя знаки ^ или ^ знаком равенства.
Так, при постоянном числе частиц в системе условия равновесия
следуют непосредственно из (П. 14) и выражают условия
минимума соответствующих потенциалов.
Рассмотрим требования, предъявляемые к химическим
потенциалам частиц, для двух типов равновесия.
А. Система состоит из нескольких фаз; частицы могут
переходить из одной фазы в другую. Рассматривая переход dNi
310
частиц из одной фазы в любую другую при постоянных Г, /?,
получаем для изменения изобарного потенциала
dG=-\>.l'dNl + v.t"dNi
(штрихи нумеруют фазы). Но для малых отклонений от рав*
новесия dG = 0) следовательно, \Х1=\\" для любой пары фаз,,
т. е. при равновесии фаз химические потенциалы компонентов
одинаковы во всех фазах.
Б. В системе может протекать химическая реакция, так что
концентрации химических веществ могут изменяться за счет
их взаимного превращения. Запишем уравнение реакции в
общем виде:
atAt + a2A2 + ...+± № + Р2В2 + . . .
ИЛИ
2«а^2ра (п-23>
i J
Здесь А/ — исходные реагирующие вещества; В7 — продукты
реакции; коэффициенты iCt,, р7- (так называемые стехиомет-
рические коэффициенты) показывают, в каком
отношении вещества А/ и В7 реагируют друг с другом. Будем
называть единицей реакции процесс превращения а\
частиц Аь (Х2 частиц А2 и т.д. в pi частиц Вь Рг частиц В2 и т.д.
Так как стехиометрические коэффициенты — числа порядка
единицы, а число частиц в системе очень велико, то очевидно,
что протекание единицы реакции есть чрезвычайно малое
отклонение от равновесного состояния, и изменение изобарного
потенциала при постоянных температуре и давлении должно
быть равно нулю. С другой стороны, при протекании единицы
реакции при постоянных Тир изобарный потенциал
уменьшается на величину aii^ за счет исчезновения ai частиц Аь на
величину сс2|1а2 за счет исчезновения а2 частиц Аг и т. д. и
возрастает на величину р1|лв1+|РгМ'В2 +• • • за счет возникновения
Pi частиц Вь р2 частиц В2 и т.д. Таким образом, имеем:
Это условие также справедливо, если вещества А/ и В7
находятся в разных фазах, поскольку химические потенциалы во
всех фазах при равновесии одинаковы.
Идеальные системы. Методы химической термодинамики
оказываются особенно плодотворными в тех случаях, когда
удается найти функциональную зависимость, связывающую
химические потенциалы и концентрации частиц. Это можно легко
сделать для таких простейших систем, как идеальные газы
или бесконечно разбавленные растворы.
Вычислим сначала энтропию идеального газа. Если в
объеме V содержится N молекул газа, то уравнение состояния
311
идеального газа запишется в виде
pV^NkT (П.25)
Изменение энтропии системы при обратимых процессах можно
вычислить с помощью основного уравнения термодинамики
(П.7). Предположим, что обратимый процесс протекает при
постоянных температуре и числе молекул. Так как в идеальном
газе молекулы не взаимодействуют друг с другом, то его
энергия U не зависит от объема или давления газа, а является
функцией только числа молекул и температуры. Поэтому при
Г, iV = const изменение энергии dU=Q, и уравнение (П.7) для
такого процесса запишется в виде
TdS^pdV (П.26)
Подставляя сюда значение р из (7.25), получаем:
dS^kN —V~
V
или, после интегрирования:
S = kN\nV+ty(T, Л) (П.27)
Здесь функция г|)(Г, N) играет роль константы интегрирования
по V; она не зависит от переменных р и V и зависит от величин,
которые поддерживались постоянными при интегрировании, т. е.
Тик. Чтобы определить характер зависимости ф от числа
молекул N, воспользуемся свойством аддитивности энтропии.
Увеличим число молекул в системе в п раз, оставив прежними
температуру и давление; тогда объем газа также увеличится в
п раз. Так как новая часть системы, состоящая из вновь
введенных частиц, по своим свойствам не отличается от старой,
то энтропия системы должна также возрасти в п раз. Таким
юбразом, для энтропии можно записать с помощью (П.27):
nS = nkNlnnV+ty(T, nN)
Сравнивая полученное равенство с (П.27), видим, что
Щ(Г, N) = nkNln я + Ф (Г, nN)
Это свойство г|з(Г, N) должно сохраняться при любом п.
Возьмем n=\/N, тогда последнее равенство принимает вид
ф(Г, N) = — kN\nN+Nty(T, 1)
Здесь функция г|)(Г, l)=i|)(r) зависит уже только от
температуры и не зависит от числа частиц. Подставляя полученное
выражение в (П.27), получаем для энтропии идеального газа:
S^N
Л1пХ + ф(7)'
(П.28)
Теперь можно вычислить энтропию смеси, состоящей из
нескольких различных идеальных газов. Для этого рассмотрим
J12
5; = TV,
/г In
сначала систему отдельных газов, разделенных
непроницаемыми перегородками. Если объем каждой отдельной фазы Vu a
число молекул каждого газа Ni, то по формуле (П.28)
энтропия каждого газа
У~Г+ЫТ)] (П.29)
Уберем теперь перегородки, разделяющие газы, и предоставим
им возможность свободно смешиваться при постоянной
температуре. Из формулы (П.29) следует, что энтропия каждого
газа увеличится при необратимом расширении на величину
Д5. = М>1п — (П.ЗО)
где V — объем газовой смеси после смешения газов.
Таким образом, энтропия газовой смеси определится как
сумма выражений (П.29) и (П.ЗО) для всех компонентов:
5 = 2лф1п-^- + 'МЛ
I
1*1
(П.31)
т. е. энтропия газовой смеси равна сумме энтропии
компонентов при условии, что каждый газ занимает весь объем смеси.
Отсюда для изобарного потенциала следует
G = U-%At\ kTln-^- + T^(T)}+ pV (П.32)
Так как смесь идеальных газов — тоже идеальный газ, то
PV=^NfiT
Подставим это равенство в предыдущее выражение и учтем,,
что величина
-RNt =Л (П.ЗЗ)
2,4
i
для идеального газа есть парциальное давление /-компонента
в смеси pt, а внутренняя энергия смеси аддитивно
складывается из парциальных энергий частиц:
i
Тогда выражение (П.32) приобретает вид
G = 2 Ni [Щ + kTlnpt - kTln kT— Щь(Т) + kT\ (П.34)
313
Сравнивая полученную формулу с (П.19), видим, что
химический потенциал /-компонента идеальной газовой смеси зависит
от его парциального давления следующим образом:
Ъ = Ъ0СП+ЬТ1пР1 (П.35)
где введено обозначение
v,°(T) = ui — kT(lnkT-l)—Tb(T) (П.36)
Из (П.35) видно, что рц° есть химический потенциал идеального
газа при р=1; эта величина зависит только от температуры
и не зависит от состава газовой смеси.
Если ввести в рассмотрение мольную долю /-компонента
;в смеси, определяемую как
то равенство (П.ЗЗ) может быть переписано в виде
Тогда из (П.35) следует другая форма записи химического
потенциала:
Ъ^^ + кТХъЩ (П.38)
Здесь величина
^° = щ -кТ(\пкТ-1)—Ц(Т) + кТ\пр (П.39)
в отличие от (П.36) определяет химический потенциал
/-компонента в чистой фазе (при [/]=1) и зависит не только от
температуры, но и от общего давления р.
По аналогии с идеальными газами, идеальной систе-
м о й называют любую систему, химические потенциалы
компонентов которой определяются формулой (П.38).
Эта формула справедлива для бесконечно разбавленных
жидких растворов (практически для растворов с достаточно
малыми концентрациями растворенных веществ).
Действительно, для разбавленных растворов имеет место закон Рауля,
согласно которому равновесное давление пара /-компонента
над раствором пропорционально мольной доле этого
компонента в растворе:
pfap) = const [/](pacTB)
При равновесии химические потенциалы компонентов в
растворе и в газовой фазе должны быть одинаковыми. Подставляя
выражение закона Рауля в формулу (П.35) и включая
АПп const в |д/, получаем для химического потенциала
/-компонента в разбавленном растворе формулу (П.38).
Применим полученную зависимость химических потенциалов
от концентраций для описания равновесия в системах (в газо-
.314
вых смесях или в идеальных растворах), где возможны
химические реакции. Подставляя (П.38) в условие (П.24), получаем:
l J i J
или, после деления на kT и потенцирования:
Пи}?]ПЩ~а1=К (П.40)
/ i
где введено обозначение
К=- ехр [— L ^ J (П.41)
Индекс i нумерует исходные реагирующие вещества, а индекс
/ — продукты реакции. Произведения П и суммы 2 берутся
по всем сортам реагирующих веществ.
Уравнение (П.40), связывающее мольные доли реагирующих
веществ при равновесии, носит название закона действия
масс. Величина /С, зависящая только от температуры и
давления, остается постоянной, если Г, р = const, и называется ко н-
стантой равновесия.
Закон действия масс в форме (П.40) не применим к
неидеальным системам, например к концентрированным
растворам, поскольку для них зависимости химических потенциалов
от концентраций более сложны по сравнению с зависимостью
(П.38). Для таких систем наряду с концентрациями
компонентов вводят дополнительную характеристику — активность
i-компонента щ, связанную с химическим потенциалом
соотношением
V4 = ^0 + kT\nal (П.42)
Это уравнение для активности имеет точно такой же вид, как
и (П.38) для мольной доли, поэтому для неидеальных систем
выполняется закон действия масс в форме
Ua\!na7ai = K (П.43)
получающейся из (П.40) простой заменой мольных долей
компонентов на их активности. Для идеальных систем активность
совпадает с мольной долей; для неидеальных систем
отношение активности к мольной доле (коэффициент
активности) может служить мерой неидеальности системы: чем
больше коэффициент активности какого-либо компонента
отличается от единицы, тем в большей степени не идеальна система.
Основной причиной неидеального поведения систем
является взаимодействие между частицами. Так, для идеального
газа значения энергии щ, отнесенные к одной i-частице, при
315
взаимодействии частиц между собой будут зависеть от объема,
приходящегося на одну частицу, т. е. от давления, так что
простая зависимость (П.35) будет искажена. Поэтому
коэффициент активности в значительной мере характеризует
взаимодействие частиц в системе.
Термодинамика заряженных частиц. Если частицы
обладают электрическим зарядом qi, то наряду с обычной энергией V
необходимо учитывать добавочную энергию системы 2JV;<7Kp,
обусловленную взаимодействием частиц с внешним
электрическим полем (ф — электростатический потенциал).
Эту добавочную энергию, разумеется, следует ввести и в
определения энтальпии и изохорного и изобарного
потенциалов (П.8) — (П. 10), что приведет к появлению добавочного
слагаемого q^ в выражениях для химического потенциала.
Таким образом, для заряженных частиц, находящихся во
внешнем поле, вместо химического потенциала щ следует
рассматривать обобщенный химический потенциал, так называемый
электрохимический потенциал:
\>ч = Уч + ЯЯ (П-44)
Так, для идеальных растворов вместо (П.38) нужно
использовать выражение
V4 = b°+bTln[i] + qff (П.45)
Для электрохимических потенциалов заряженных частиц
остаются в силе все закономерности, изложенные выше для
химических потенциалов нейтральных частиц. Так, условие
равновесия различных фаз, содержащих общие компоненты, для
заряженных частиц означает:
?/=?/" (П.46)
для любой пары фаз, или
ь"-ь' = д№ — 9") (П.47)
Последнее равенство широко используется для вычисления
контактных разностей потенциалов, возникающих на границах
различных фаз.
Условие (П.46) можно применять не только к различным
.фазам, но и к разным участкам одной фазы, находящейся во
внешнем поле. В этом случае его удобнее записывать в форме
Wi = 0 (П.48)
или
т =—яя<р (П-49)
В некоторых случаях учет энергии взаимодействия частиц
с электрическим полем не приводит к новым результатам.
316
В качестве примера рассмотрим условие (П.24) равновесия
химической реакции. Для заряженных частиц это условие
следует обобщить до вида
- 2 «| (ft + q#) + 2 h &J + Я ft) = ° (п-5°)
* у
Если все реагирующие вещества находятся в одной фазе, то
электрический потенциал меняется с расстоянием плавно и
сохраняет постоянное значение в любой точке, где происходит
элементарный акт химического взаимодействия. Поэтому
равенство (П.50) можно переписать в виде
—2 ал + 2 h^j + <p (- 2 a# i + 2 Ру?у) = °
Выражение, стоящее в скобках, есть изменение заряда системы
при протекании единичной реакции и при равновесии равно
нулю. Таким образом, мы приходим снова к условию (П.24);
это означает, что внешнее поле не влияет на химическое
равновесие в однородных системах, и при рассмотрении
равновесных концентраций в однородных системах можно пользоваться
законом действия масс в форме (П.40), не учитывающей заряд
частиц.
Этот вывод, конечно, не относится к таким системам, в
которых различные реагирующие вещества находятся в разных
фазах; в этом случае разность потенциалов на границе фаз
может иметь существенное значение.
Литература
1. Жданов Г. С. Физика твердого тела. М., Изд-во МГУ, 1961, 501 с.
2. Киттель Ч. Введение в физику твердого тела. Пер. с англ. М., Наука,
1978. 791 с.
3. Горбачев В. В., Спицына Л. Г. Физика полупроводников и металлов. М.,
Металлургия, 1976. Э68 с.
4. Барнард А. Теоретические основы неорганической химии. Пер. с англ.
М.', Мир, 1968. 362 с.
5. Левин А. А. Введение в квантовую химию твердого тела. М., Химия,
1974. 237 с.
6. Давыдов А. С. Теория твердого тела. М., Наука, 1976. 640 с.
7. Займан Дж. Принципы теории твердого тела. Пер. с англ. М., Мир,
1974. 472 с.
8. Бацанов С. С. Электроотрицательностъ элементов и химическая связь.
Новосибирск, Изд-во СО АН GOCP, 1962. 196 с.
9. Бацанов С. С — ЖНХ, 1975, т. 20, с. 25)95.
10. Бацанов С. С, Дулепов Е. В. — ЖСХ, 1973', т. 14, с. 541.
11. Tateno /.—J. Phys. Chem. Solids, 1970, v. 31, p. 1641.
12. Левин А. А., Сыркин Я. /С, Дяткина М. Е. — Усп. хим., 1969, т. 38.
13. Лебедев В. И. Ионно-атомные радиусы и их значение для геохимии и
химии. Л., Изд-во ЛГУ, 1969. 156 с.
14. Алесковский В. Б. Химия твердых веществ. М., Высшая школа, 1978.
15. Укше Е. А., Букун Н. Г. Твердые электролиты. М., Наука, 1977.
16. Чеботин В. #., Перфильев М. В. Электрохимия твердых электролитов.
М., Химия, 1978. 312 с.
17. Чеботин В. Я. Явления переноса в ионных кристаллах. Свердловск,
Изд-во УрГУ, 1968. 181 с.
18. Шурин А. Я. Химия несовершенных ионных кристаллов. Л., Изд-во ЛГУ,
1975. 270 с.
19. Физика электролитов. Пер. с англ., М., Мир, 1978. 555 с.
20. Крегер Ф. Химия несовершенных кристаллов. Пер. с англ. М., Мир,
1969. 654 с.
21. Хауффе К. Реакция в твердых телах и на их поверхности. Пер. с нем.
М, Издатинлит, Ч. 1, 1962. 415) с. Ч. 2, 1963, 275 с.
22. Третьяков Ю. Д. Химия нестехиометрических окислов. М., Изд-во МГУ,
1974. 364 с.
23. Мень Л. Я., Воробьев Ю. Я., Чуфаров Г. И., Физико-химические
свойства нестехиометрических окислов. Л., Химия, 1973. 224 с.
24. Кофстад Я. Отклонение от стехиометрии, диффузия и электропроводность
в простых окислах металлов. Пер. с англ. М., Мир, 1975. 396 с.
25. Нестехиометрические соединения. Пер. с англ. М-., Химия, 1971. 607 с.
26. Коллонг Р. Нестехиометрия. Пер. с франц. М., Мир, 1974. 288 с.
27. Третьяков Ю. Д. Твердофазные реакции. М., Химия, 1978. 360 с.
28. Бокштейн Б. С., Бокштейн С. 3., Жуховицкий А. А. Термодинамика и
кинетика диффузии в твердых телах. М., Металлургия, 1974. 280 с.
■29. Гегузин #. Е. Диффузионная зона. М., Наука, 1979. 343 с.
30. Гегузин Я. Е. Макроскопические дефекты в металлах. М., Металлургиз-
дат, 1962. 252 с.
31. Banus M. D., Reed Т. В. — In: The Chemistry of Extended Defects in Non-
Metallic Solids. Eyring L., O'Keeffe M., Eds. North-Holland Publ. Co.,
Amsterdam, 1970, p. 488.
32. Мень А. Я., Воробьев Ю. П., Чуфаров Г. И. Кристаллохимия и
термодинамика взаимодействующих дефектов. Итоги науки и техники, серия
«Химия твердого тела». Т. 1. М., ВИНИТИ, 1973. 88 с.
318
33. Mrowes S. — Rev. Int. Hautes Temp. Refract., 1977, v. 14, p. 225.
34. Хьюз К-, Изард Д. О. Перенос ионов в стеклах. В [9], с. 371—422.
35. Мотт Н., Герни Р. Электронные процессы в ионных кристаллах. Пер. с
англ. М., Издатинлит, I960. 304 с.
36. Лидьярд А. Ионная проводимость кристаллов. Пер. с англ. М.,
Издатинлит, 1962. 222 с.
37. Kroger Г. A., Stieltjes Г. Н., Vink Н. /. — Philips Res. Repts., 1959, v. 14.
38. Дамаск А., Дине Дж. Тачечные дефекты в металлах. Пер. с англ. М.,
Мир, 1966. 291 с.
39. Точечные дефекты в твердых телах. Пер. с англ. М., Мир, 1979. 379 с.
40. Neel L. — Compt. rend., 1950, v. 230, p. 190.
41. Кривоглаз M. А., Смирнов А. А. Теория упорядочивающихся сплавов.
М., Физматгиз, 1958. 388 с.
42. Смирнов А. А. Молекулярно-кинетическая теория металлов. М., Наука,
1966. 488 с.
43. Кожеуров В. А. Статистическая термодинамика. М., Металлургия, 1975.
44. Андриевский Р. А., Уманский Я. С. Фазы внедрения. М., Наука, 1977.
45. Смирнов А. А. Теория сплавов внедрения. М., Наука, 1979. 365 с.
46. У гай #. А. Введение в химию полупроводников. М., Высшая школа, 1975.
47. Сюше Ж. Я. Физическая химия полупроводников. Пер. с англ. М.,
Металлургия, 1969. 224 с.
48. Ормонт Б. Ф. Введение в физическую химию и кристаллохимию
полупроводников. М., Высшая школа, 1973. 656 с.
49. Болтакс Б. И. Диффузия и точечные дефекты в полупроводниках. Л.,
Наука, 1972. 384 с.
50. Шалабутов Ю. К Введение в физику полупроводников. Л., Наука, 1969.
51. Ансельм А. И. Введение в теорию полупроводников. М.— Л., Физматгиз,
1962. 418 с.
52. Стильбанс Л. С. Физика полупроводников. М., Сов. радио, 1967. 451 с.
53. Бонч-Бруевич В. Л., Калашников С. Г. Физика полупроводников. М.,
Наука, 1977. 672 с.
54. Debye P. P., Conwell Е. М. — Phys. Rev., 1954, v. 93, p. 693.
55. Kurosawa Г.— J. Phys. Soc. Japan, 1959, v. 14, p. 741.
56. Schulze P. D., Hardy J. R. — Phys. Rev., 1972, v. B5, p. 3270; v. B6.
57. Boswarva I. M. — Phil. Mag., 1967, v. 16, p. 827.
58. Shukla A. K., Ramdas S.t Rao C. N. R.— J. Phys. Chem. Solids, 1973, v. 34.
59. Catlow C. R. A., Norgett M. J. — J. Phys., C: Solid State Phys., 1973, v. 6.
60. Tharmalingam /C. —Phil. Mag., 1971, v. 23, p. 199.
61. Чеботин В. Я.— Труды Ин-та электрохимии УФ АН СССР, 1965, вып. 7.
62. Горелов В. П., Палъгуев С. Ф.— Неорг. мат., 1977, т. 13, с. 181.
63. Hauffe К, Pfeiffer H. — Z. Metallkunde, 1953, Bd. 44, S. 27.
64. Wagner С, Hammen H. — Z. phys. Chem., 1938, Bd. B40, S. 197.
65. O'Keefe M., Moore W. /. — J. Chem. Phys., 1962, v. 36, p. 3009.
66. Wakihara M., Katsura Г. —Met. Trans., 1970, v. 1, p. 363.
67. Хаазе Р. Термодинамика необратимых процессов. Пер. с нем. М., Мир,
1967, 544 с.
68. Фриауф Р. Дж. Основы теории процессов ионного переноса. В [19],
с. 165—217.
69. Kirk D. L., Pratt P. L. — Proc. Brit. Ceram. Soc, 1967, v. 9, p. 215.
70. Bar sis E.t Taylor A. — Proc. Brit. Ceram. Soc, 1967, v. 9, p. 203.
?1. Неуймин А. Д., Палъгуев С. Ф. — Труды Ин-та электрохимии УФ АН
СССР, 1961, вып. 2, с. 185; 1962, вып. 3, с 133.
72. Mac Donald D. К. С. — J. Chem. Phys., 1953, v. 21, p. 2097.
73. Аппелъ Дж., Фирсов Ю. В. Поляроны. М., Наука, 1975. 423 с.
74. Duquesnoy А. — Rev. Hautes Temp. Refractaires, 1965, v. 2, p. 201.
75. Hartung R. — Z. phys. Chem., 1973, Bd. 254, S. 393.
76. Rudolph J. — Z. Naturforsch., 1959, Bd. 14a, S. 727.
77. Шьюмон П. Диффузия в твердых телах. Пер. с англ. М., Металлургия,
1966. 195 с.
78. Маннинг Дж. Кинетика диффузии атомов в кристаллах. Пер. с англ. М.,
Мир, 1971. 278 с.
319
79. Askill J. Tracer Diffusion Data for Metals, Alloys and Simple Oxides.
IFI Plenum Data Corporation. N. Y., 1970, p. 107.
80. Compton W. D. — Phys. Rev., 1956, v. 101, p. 1209.
81. Compaan K, Haven У. — Trans. Faraday Soc, 1956, v. 52, p 786; 1958,
v. 54, p. 1498.
82. Weber M. D., Friauf R. /. — J. Phys. Chem. Solids, 1969, v. 30, p. 407.
83. Mapother D.t Crooks H. N.t Maurer R. /. — J. Chem. Phys., 1950, v. 18,
p. 1231.
84. Fuller R. G., Marguardt С L.f Reilly M. N.f Wells J. C, Jr. — Phys. Rev.,
1968, v. 176, p. 1036.
85. Smigelskas A., Kirkendall E. — Trans. AIME, 1947, v. 171, p. 130.
86. Magistris A., Pezzati E., Simistri C. — Z. Naturforsch., 1972, Bd. 27a,
S. 1379.
87. Fischer W. — Z. Naturforsch., 1967, Bd. 22a, S. 1575.
88. Хешей Н. Химия твердого тела. Пер. с англ. М., Мир, 1971. 223 с.
89. Мейер К. Физико-химическая кристаллография. Пер. с нем. М.,
Металлургия, 1972. 480 с.
90. Барре П. Кинетика гетерогенных процессов. Пер. с франц. М., Мир, 1976.
91. Кофстад П. Высокотемпературное окисление металлов. Пер. с англ. М.,
Мир, 1969. 392 с.
92. Кубашевский О., Гопкинс Б. Окисление металлов и сплавов. Пер. с англ.
М., Металлургия, 1965. 428 с.
93. Конев В. Н., Сунцов Н. В. Структура и свойства твердых тел. Ученые
зап. Уральского ун-та. Свердловск, 1969, № 96, серия физ., вып. 6, с. 18.
94. Трепнел Б. М. Хемосорбция. Пер. с англ. М., Издатинлит, 1958. 327 с.
95. Гохштейн А. Я. Поверхностное натяжение твердых тел и адсорбция. М.,
Наука, 1976. 400 с.
96. Межфазовая граница газ — твердое тело. Пер. с англ. М., Мир, 1970.
97. Волькенштейн Ф. Ф. Физикохимия поверхности полупроводников. М.,
Наука, 1973. 400 с.
98. Моррисон С. Химическая физика поверхности твердого тела. Пер. с англ.
М,, Мир, 1980. 488 с.
99. Levesque P., Cubicciotti D. — J. Arn. Ceram., Soc, 1951, v. 73, p. 2028.
100. Розовский А. Я. Кинетика топохимических реакций. М., Химия, 1974.
101. Акопян А. А. Химическая термодинамика. М., Высшая школа, 1963. 527 с.
102. Кричевский И. Р. Понятие и основы термодинамики. М., Химия, 1970.
103. Эверет Д. Введение в химическую термодинамику. Пер. с англ. М.,
Издатинлит, 1963. 300 с.
104. Полторак О. М. Лекции по химической термодинамике. М., Высшая
школа, 1971. 256 с.
105. Мюнстер А. Химическая термодинамика. Пер. с нем. М., Мир, 1971.
106. Полторак О. М. Лекции по теории гетерогенного катализа. М.} Изд-во
МГУ, 1968. 155 с.
Василий Николаевич
Чеботин
Физическая
химия
твердого
тела