/





































































































































































































































































































































































































































































































































Автор: Рао Ч. Н. Р. Гопалакришнан Дж.
Теги: химия материаловедение химия твердого тела
ISBN: 5-02-029203-6
Год: 1990




Текст
С. N. R. RAO. FRS
J. GOPALAKRISHNAN
Solid State and Structural Chemistry Unit. Indian Institute of Science.
Bangalore. India
New directions
in solid state chemistry
Structure, synthesis, properties, reactivity
and materials design
The right of the
University of Cambridge
to print and sell
all manner of books
was granted by
Henry VIII in 1534
The University has printed
end published continuously
since 1584.
CAMBRIDGE UNIVERSITY PRESS
Cambridge
London New York New Rochelle
Melbourne Sydney
Ч. Н. Р. РАО
Дж. ГОПАЛАКРИШНАН
НОВЫЕ НАПРАВЛЕНИЯ
В ХИМИИ
ТВЕРДОГО ТЕЛА
Структура, синтез, свойства,
реакционная способность
и дизайн материалов
Перевели с английского кандидаты химических наук
В. Е. Федорову 3. М. Логвиненко.
П. П. Самойлов, В. А. Логвиненко
Под редакцией академика Ф. А. Кузнецова
НОВОСИБИРСК
«НАУК А»
СИБИРСКОЕ ОТДЕЛЕНИЕ
1990
УДК 541.1
Новые направления в химии твердого тела: Структура, синтез,
свойства, реакционная способность и дизайн материалов: Пер. с
англ/Рао Ч. Н. Р., Гопалакришнан Дж.— Новосибирск: Наука.
Сиб. отд-ние, 1990.— 520 с.
ISBN 5-02-029203-6.
Монография посвящена актуальным проблемам современной химии
твердого тела — новым аспектам строения твердых веществ различного типа,
наиболее распространенным методам их идентификации и характеризации, синтеза
и взаимосвязи между строением и свойствами, фазовым переходам и
реакционной способности, роли дефектов и нестехиометрии. Описаны достижения мировой
науки в исследовании наиболее интересных и практически важных материалов.
В русское издание книги включена глава, рассматривающая новые материалы —
высокотемпературные сверхпроводники.
Книга заинтересует специалистов в области химии и физики твердого
тела и материаловедения.
Табл. 35. Ил. 220. Библиогр.: 1073 назв.
Перевели с английского кандидаты химических наук В. Е. Федоров,
3. М. Логвиненко, П. П. Самойлов, В. А. Логвиненко
С. N. R. Rao, J. Gopalakrishnan. New directions in solid state chemistry:
Structure, synthesis, properties, reactivity and materials design
Cambridge University Press
Cambridge, 1986
Утверждено к печати
Институтом неорганической химии
СО АН СССР
1708000000—121 ^^ ... тт . ., „
Р 042(02)-90—КБ-17—89—1990 © Cambridge University Press, 1986
ISBN 5—02—029203—6 © Перевод на русский язык,
предисловие. Издательство «Наука», 1990
ОГЛАВЛЕНИЕ
От редактора перевода S
Предисловие к русскому издапию 11
Предисловие 12
1. Структура твердых тел: старые и новые аспекты 13
1.1. Введение —
1.2. Описание кристаллов —
1.3. Связи в кристаллах 15
1.4. Неорганические структуры 26
1.5. Силикаты и алюмосиликаты 42
1.6. Несвязывающис взаимодействия в ионных кристаллах 48
1.7. Новые аспекты рассмотрения структур неорганических
твердых тел 50
1.8. Политипизм 56
1.9. Органические кристаллические структуры 59
1.10. Соединения включения и клатраты 63
1.11. Некристаллические, или аморфные, твердые тела 66
1.12. Квазикристаллы 71
1.13. Модели и графическое изображение —
1.14. Солитоны 72
2. Новые и улучшенные методы характеризации 73
2.1. Введение —
2.2. Структурная характеризация 74
2.2.1. Рентгеновская дифракция 76
2.2.2. Электронная дифракция 77
2.2.3. Нейтронная дифракция и родственные методы 79
2.2.4. Электронная микроскопия 82
2.2.5. Рентгеновская абсорбционная спектроскопия (EXAFS,
XANES) 90
2.2.6. Спектроскопия ядерного магнитного резонанса с вращением
под магическим углом 100
2.2.7. Электронная спектроскопия 104
2.2.8. Другие спектроскопические методы 110
2.2.9. Заключительные замечания 111
2.3. Характеризация состава и чистоты 112
3. Стратегия синтеза 115
3.1. Введение —
3.2. Получение кристаллических материалов 119
3.2.1. Керамические методы —
3.2.2. Химические методы 121
3.2.3. Методы высокого давления 127
3.2.4. Дуговые методы 133
3.2.5. Настыльное плавление 135
3.2.6. Химическое осаждение из газовой фазы —
3.2.7. Синтез органических твердых веществ 137
6 Оглавление
3.3. Микрокристаллические частицы и кластеры (предельное состояние
твердого тела) 139
3.4. Аморфные материалы 141
3.5. Рост кристаллов 143
4. Фазовые переходы . • • 151
4.1. Введение —
4.2. Термодинамика —
4.3. Мягкие моды 156
4.4. Центральные пики 158
4.5. Критические явления 159
4.6. Структурные изменения при фазовых переходах 160
4.7. Механизмы фазовых переходов 161
4.8. Органические твердые тела 165
4.9. Несоразмерные фазы 166
4.10. Кооперативный эффект Яна — Теллера 175
4.11. Переходы спинового состояния 179
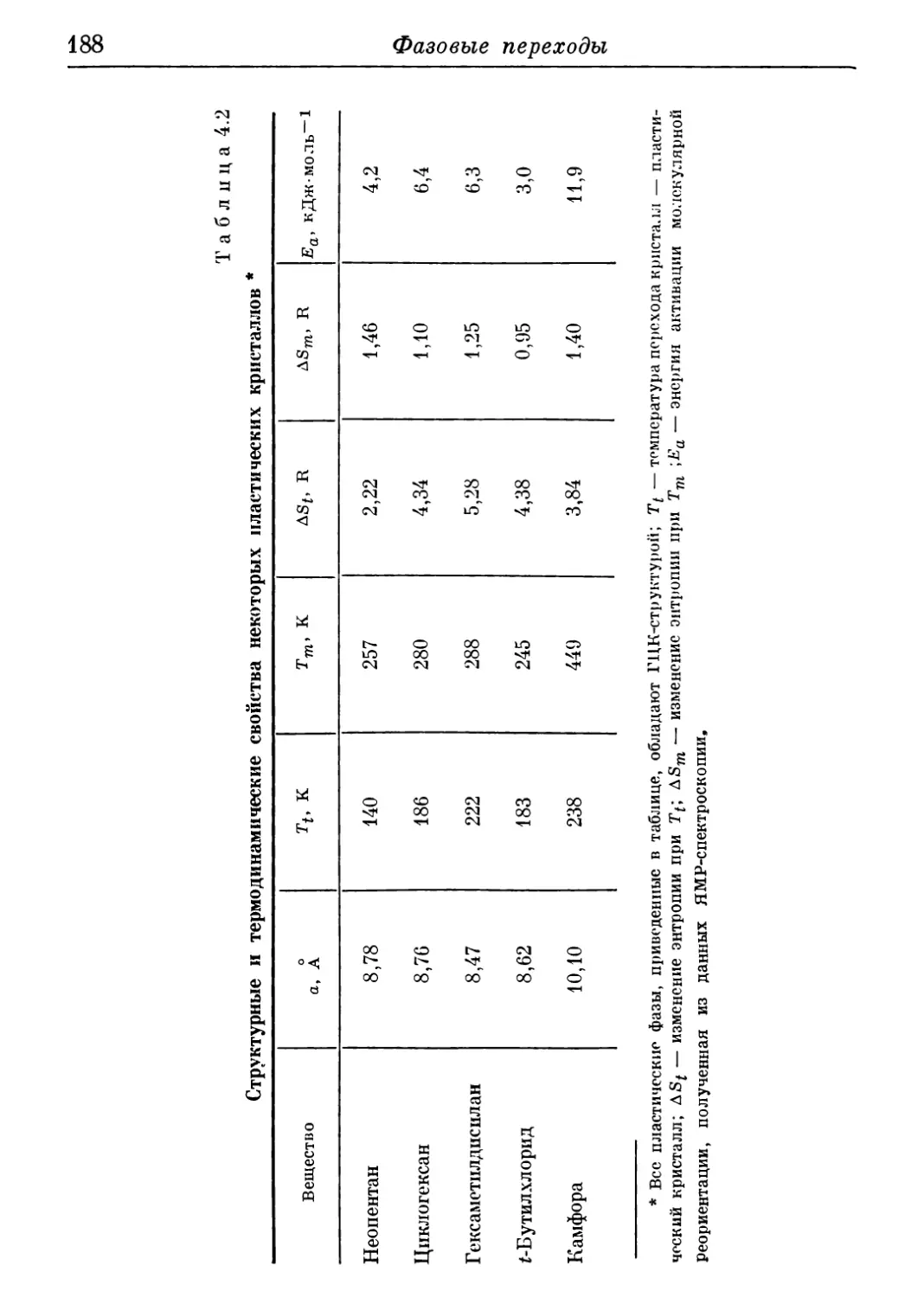
4.12. Пластическое кристаллическое состояние 187
4.13. Жидкокристаллическое состояние 193
4.14. Некристаллическое состояние и фазовые переходы в стеклах 197
4.15. Методы Монте-Карло и молекулярной динамики 201
4.16. Приложения фазовых переходов 203
5. Новый взгляд на старую проблему: дефекты и нестехиометричность 205
5.1. Введение —
5.2. Точечные дефекты 208
5.2.1. Равновесие точечных дефектов 212
5.2.2. Параэлектрические и молекулярные примеси в ионных
твердых телах 218
5.2.3. Центры окраски 219
5.3. Дислокации 220
5.4. Планарные дефекты 223
5.5. Упорядоченные точечные дефекты и сверхструктуры 226
5.5.1. Допированные галогениды щелочных металлов —
5.5.2. Халькогениды и карбиды металлов 227
5.5.3. Оксиды металлов со структурой каменной соли 229
5.5.4. Флюоритоподобные твердые тела 233
5.6. Кристаллографический сдвиг j . . . 235
5.7. Блочные структуры 239
5.8. Бесконечно адаптивные структуры 242
5.9. Прорастание кристаллов 243
5.10. Оксиды со структурой дефектных перовскитов: пример
исследования 247
6. Взаимосвязь структура — свойство 258
6.1. Введение —
6.2. Электроны в твердых телах 259
6.2.1. Зонная модель —
6.2.2. Модель локализованных электронов 263
6.2.3. Модель приближения химической связи 264
6.3. Свойства 266
6.3.1. Магнитные свойства 267
6.3.2. Электрические свойства 278
6.3.3. Сверхпроводимость 284
6.3.4. Диэлектрические и оптические свойства 286
6.4. Несколько примеров 291
6.4.1. Оксиды металлов 292
Оглавление 7
6.4.2. Сульфиды металлов 304
6.4.3. Фториды металлов 312
6.5. Переходы металл — неметалл 320
6.6. Металлические кластеры 331
6.7. Соединения со смешанной валентностью 332
6.8. Низкоразмерные твердые тела 341
6.9. Ферроики 349
6.10. Жидкие кристаллы 362
7. Моделирование твердых тел для особых целей:
аспекты дизайна материалов 366
7.1. Введение —
7.2. Твердые ионные проводники 368
7.3. Фотоэлектрохимия 375
7.4. Магнитные материалы 383
7.5. Материалы для хранения водорода 388
7.6. Аморфные материалы 393
7.7. Органические материалы 397
7.8. Пленки Ленгмюра — Блоджетт 401
7.9. Жидкие кристаллы —
7.10. Нелинейные оптические материалы 404
7.11. Люминесцентные неорганические материалы 405
7.12. Лазерные материалы 406
8. Реакционная способность твердых тел 407
8.1. Введение —
8.2. Природа твердофазных реакций 408
8.3. Реакции, включающие только одну твердую фазу 409
8.4. Реакции твердое — газ 412
8.5. Реакции твердое — твердое 417
8.6. Реакции твердое — жидкость 420
8.7. Химия интеркаляции 421
8.8. Реакции органических твердых тел 435
8.9. Гетерогенный катализ 445
9. Химия высокотемпературных сверхпроводников 457
9.1. Введение —
9.2. Купраты со структурой K2NiF4 458
9.3. Дефектные перовскиты в системе La—Ва—Си—О 460
9.4. YBa2Cu307 и родственные 123-оксиды 461
9.5. Система LnQ Ba0^„Cu-C) АА 466
о— X о+х w 14+0*
9.6. Висмутовые и таллиевые купраты 467
9.7. Природа кислорода и меди в сверхпроводящих купратах 468
9.8. Приготовление и родственные аспекты 472
9.9. Заключительные замечания 473
Дополнение переводчика 474
Список литературы 476
Предметный указатель 507
Формульный указатель 516
ОТ РЕДАКТОРА ПЕРЕВОДА
Многие черты научно-технического прогресса определяются
уровнем изученности и технологической освоенности новых
твердотельных материалов. На использовании комплекса свойств открытых
и освоенных в последние десятилетия материалов основаны ведущие
отрасли современной техники: электроника и связь, энергетика,
наземный, воздушный и космический транспорт, машиностроение.
Все это было бы невозможным без большого набора
полупроводников, оптических и магнитных материалов, пьезо- и сегнетоэлектри-
ков, новых композитных, керамических и полимерных материалов.
Открытие и освоение этих передовых материалов обязано
достижениям в областях науки, изучающей твердые тела. В свою очередь,
достижения в упомянутых областях человеческой деятельности,
определенно связанные с использованием свойств твердых тел,
являются мощным стимулом развития самой науки о твердом теле
и твердотельного материаловедения. Одним из значительных
проявлений взаимосвязи науки и технического прогресса было
проведение Международным союзом по теоретической и прикладной химии
(ИЮПАК) в 1987 г. Всемирной конференции по передовым
материалам — основе инноваций в энергетике, транспорте и связи (17—
22 мая, Токио). Мы сочли полезным напомнить читателю об этом
собрании не только потому, что в опубликованных трудах
конференции содержится много материала по тематике монографии
Ч. Н. Р. Рао и Дж. Гопалакришнана, но и в связи с тем, что
Ч. Н. Р. Рао, в то время президент ИЮПАК, был одним из главных
организаторов и активным участником этого важного для
твердотельного материаловедения научного собрания.
Предлагаемая читателю книга представляет, на наш взгляд,
значительное явление в развитии науки о твердом теле. Авторы
делают акцент на химической проблематике этой области науки.
В настоящее время перед химией твердого тела стоит задача
перейти от рассмотрения отдельных сторон строения, способов
синтеза и изучения реакционной способности твердых тел к построению
содержательных зависимостей набора свойств от особенностей
строения твердых тел, к разработке научно обоснованных способов
получения материалов с заданным набором свойств из числа известных
или «конструирования» не существовавших ранее материалов для
реализации необходимой функции. Такая возможность обусловлена
От редактора перевода
9
зрелостью лежащих в основе химии твердого тела разделов науки:
кристаллохимии, теории электронного строения химических
соединений, учения о дефектах в кристаллических и некристаллических
твердых телах, о механизмах и динамике процессов в твердых телах,
как термически активированных, так и происходящих при
радиационном или механическом воздействии, накопленной информации
о взаимосвязи деталей строения со свойствами твердых тел.
Авторы поставили задачу взаимосвязанно рассмотреть
соображения, развиваемые в упомянутых выше разделах науки о твердом
теле. Нельзя сказать, что такая цель ставится впервые. Можно
упомянуть, например, замечательные монографии К. Хауффе «Реакции
в твердых телах и на их поверхности», Б. Ф. Ормонта «Введение в
физическую химию и кристаллохимию полупроводников», Н. Б. Хен-
нея «Полупроводники», Ф. Крегера «Химия несовершенных
кристаллов». Ч. Н. Р. Рао и Дж. Гопалакришнан в настоящей монографии,
несомненно, сделали следующий шаг по пути обобщений.
Характерной особенностью книги, что еще раз подчеркивает
ее значимость, является универсальность. Она будет с интересом
воспринята читателями с разным уровнем подготовки. Студенты и
начинающие ученые найдут в ней краткое изложение основных идей
науки о твердом теле и убедительные аргументы в пользу особого
внимания к химическим аспектам этой науки, специалисты — много
примеров углубленного рассмотрения важнейших проблем химии
твердого тела. В книге достаточно материала и для тех читателей,
которых интересует область научных симпатий авторов. Выделить
область научных интересов авторов можно не только по ссылкам на
первоисточники, но и по детальности рассмотрения отдельных
вопросов. В ряде случаев (например, полупроводники, лазерные
материалы) описание схематично. Но включение такого материала
оправдано желанием дать общую картину.
Еще одна важная особенность книги — направленность всего
содержания на дизайн материалов с различными функциональными
свойствами. Непосредственно этой проблеме посвящены главы 6 и 7.
При изложении материала в разделах, касающихся общих
вопросов — электронного строения, кристаллохимии, дефектов в твердых
телах, способов экспериментального исследования материалов,—
авторы расставляют акценты таким образом, что подводят читателя
к идеям о целенаправленности выбора материала на основе
рассмотрения строения составляющего его вещества на атомном уровне.
Ценной в монографии является попытка показать пределы
применимости и ограниченность простых моделей, используемых,
например, в кристаллохимии или в химии дефектов. Рассматривая
проблему точечных дефектов, авторы показывают, что
количественные модели Вагнера — Шоттки, сыгравшие очень важную роль в
развитии общих представлений о процессах в твердых телах и с
успехом примененные (например, Ф. Крегером) для интерпретации
электрических свойств некоторых классов соединений, в общем случае,
10
От редактора перевода
при значительной концентрации дефектов, при их упорядочении,
оказываются неточными. Авторы не дают в монографии
альтернативного формализма, но качественное рассмотрение должно
заставить исследователей задуматься.
Следует специально сказать о гл. 9, посвященной химии
высокотемпературных сверхпроводников (ВТСП). В этой чрезвычайно
интересной и бурно развивающейся области авторы работают очень
активно и находятся на передовом рубеже исследования новых ВТСП-
материалов. Профессор Ч. Н. Р. Рао и его школа всемирно известны
своими пионерскими многолетними работами в области синтеза и
изучения широкого спектра неорганических материалов с
металлическими свойствами, в том числе и сверхпроводящими. Открытие
ВТСП-купратов и быстрое понимание их строения в существенной
мере подготовлено в том числе и работами по химии твердого тела
материалов с перовскитоподобной структурой, проведенными
Ч. Н. Р. Рао и его сотрудниками.
В период подготовки книги к русскому изданию профессор
Рао дважды переписывал эту главу. Затем мы с авторами решили,
что угнаться за развитием проблемы сейчас невозможно.
Представленный материал соответствует уровню исследований на середину
1988 г. Подобные сведения о собственных результатах автора можно
найти во многих десятках публикаций, подготовленных им с
сотрудниками со времени открытия ВТСП. По нашему убеждению,
это открытие могло исходить из его лаборатории. От этого
коллектива с большой степенью вероятности можно ожидать очередных
сюрпризов, которые, несомненно, появятся еще в области новых
ВТСП-материалов. Нужно отметить, что, несмотря на изложенные
обстоятельства, глава имеет не только исторический интерес —
основные идеи в кристаллохимии и химии твердого тела
ВТСП-материалов, содержащиеся в книге, достаточно полно отражают
современные представления.
Авторы имеют тесную профессиональную связь с советскими
учеными, однако, к сожалению, они не избежали обычного для
зарубежных публикаций недостатка — незнания советской научной
литературы. Учитывая характер книги (авторы не претендуют на
полноту охвата мировой литературы), мы с переводчиками решили
не давать дополнительного списка литературы.
Перевод монографии выполнен В. Е. Федоровым (главы 1, 9),
П. П. Самойловым (главы 2, 4), 3. М. Логвиненко (главы 3, 5—7),
В. А. Логвиненко (гл. 8).
Апрель 1989 г. Ф. А. Кузнецов
ПРЕДИСЛОВИЕ К РУССКОМУ ИЗДАНИЮ
Перевод нашей книги на русский язык — для нас большая
честь. Это особенно важно из-за огромного интереса советских
ученых к химии твердого тела, а также из-за того, что в Советском
Союзе у нас много замечательных друзей и коллег. Между Индией и
Советским Союзом существует традиционное сотрудничество в
области химии твердого тела. При содействии наших академий наук
мы организуем совместные симпозиумы по материаловедению и
химии твердого тела, и они всегда весьма успешны. Неорганические
материалы — важная область и в долгосрочных программах
сотрудничества в химии и технологии. Мы надеемся, что наши
совместные работы будут расширяться. Перевод этой книги мы
рассматриваем как еще один знак дружбы и сотрудничества между учеными
Индии и Советского Союза.
Для русского издания мы добавили главу по химии
высокотемпературных сверхпроводящих оксидов. Эта область возникла в
1986 г. уже после первого издания книги. Мы считаем, что с этим
дополнением книга действительно сможет представлять «Новые
направления в химии твердого тела», как утверждает ее название.
Мы искренне признательны нашим друзьям в Советском Союзе
за перевод этой книги. Мы особенно благодарны академику
Ф. А. Кузнецову и его коллегам в Институте неорганической химии
СО АН СССР (г. Новосибирск) за их помощь и поддержку.
Ч. Н. Р. Рао
Дж. Гопалакришнан
ПРЕДИСЛОВИЕ
Несмотря на то что наука о твердом теле является сферой
интенсивной исследовательской деятельности физиков и
материаловедов, вклад химиков в создание новейших сложных материалов
поистине уникален. Химия твердого тела в основном занимается
развитием новых методов синтеза, путей идентификации и характе-
ризации материалов, описанием их структуры и, сверх того, новыми
стратегиями получения материалов с желаемыми и
контролируемыми свойствами: электронными, магнитными, диэлектрическими,
оптическими, адсорбционными или каталитическими. Отрадно, что эта
новая независимая область химической науки получает все
большее признание.
Мы попытались в краткой форме представить
кульминационные моменты развития современной химии твердого тела и наметить
ее новые направления. При этом мы не описываем различные
принципы, свойства и методики, которые в конце концов стали
символом этой научной дисциплины,— наша задача показать ее
привлекательность.
Мы позаботились и о включении некоторых обобщающих
сведений в каждый раздел, чтобы дать возможность студентам и
начинающим исследователям извлечь пользу из книги. Монография не
разделена на главы традиционным образом (с обсуждением
кристаллохимии, свойств твердых веществ, реакционной способности
и т. д.)— проблема освещается так, чтобы отразить современный путь
развития предмета. Из-за этого размеры глав неизбежно стали
несколько неравномерными. Эта книга может быть использована не
только как дополнение, но и как основа хорошо спланированного
курса химии твердого тела. Авторы будут более чем вознаграждены,
если студенты, преподаватели и специалисты в области химии
твердого тела найдут книгу интересной.
Мы привлекаем важный материал из современной литературы,
включая некоторые самые последние ссылки, но, касаясь некоторых
новых концепций, вынуждены быть лаконичными из-за
ограниченного объема книги. Возможно, что какие-то ссылки не включены
из-за недооценки работы или по недосмотру, и мы бы хотели, чтобы
читатель нас извинил за это.
Большая часть книги запланирована и написана в то время,
когда один из авторов (Ч. Н. Р. Рао) был приглашен в
Кембриджский университет (за счет фонда Джавахарлала Неру). Он
благодарен профессору Дж. М. Томасу, члену Королевского общества,
и другим сотрудникам кафедры физической химии и членам
Королевского колледжа в Кембридже за доброту и гостеприимство.
Авторы признательны профессору Роберту Кану, а также
Попечительскому совету университета Н ью-Дел и за ценные советы
и поддержку этой работы.
Бангалор, июнь 1985 г. Ч. Н. Р. Рао, Дж. Гопалакришнан
1. СТРУКТУРА ТВЕРДЫХ ТЕЛ:
СТАРЫЕ И НОВЫЕ АСПЕКТЫ
1.1. ВВЕДЕНИЕ
Химия твердого тела имеет дело с различными типами
твердых тел, неорганических и органических, кристаллических и
некристаллических. Глубокие знания структуры твердых тел, так же
как и природы связи, важны для понимания этой науки, так как
свойства твердых тел в существенной степени определяются их
структурой. Кристаллохимия неорганических твердых тел широко
представлена в литературе (см., например, [Adams, 1974; Rao, 1974;
Wells, 1984]), но недавно предприняты попытки рассмотреть новые
пути образования неорганических структур и понять их
стабильность. В этой главе мы кратко рассмотрим основные факты
неорганической кристаллохимии после обсуждения некоторых
фундаментальных данных, касающихся кристаллов и различных типов связи
в них. Перед заключительным рассмотрением моделей,
используемых для понимания структур некристаллических или аморфных
твердых тел, мы обсудим также политипизм, структуру органических
кристаллов и родственные темы.
1.2. ОПИСАНИЕ КРИСТАЛЛОВ
Правильный кристалл состоит из бесконечного множества
ячеек, простирающихся в трех измерениях. Поскольку природа
ячейки не влияет на трансляционную периодичность, периодичность
обычно представляется замещением повторяющейся ячейки точкой,
получающаяся совокупность таких точек в пространстве
называется решеткой. В пространственной решетке трансляционные
векторы а, Ъ и с в трех кристаллографических направлениях определяют
параллелепипед, который называется примитивной ячейкой.
Примитивная ячейка или подходящая комбинация из них, выбранная
как повторяющаяся ячейка решетки, называется элементарной
ячейкой. Для определения кристаллографической элементарной
ячейки в трех измерениях необходимо выбрать три трансляционных
вектора и углы между осями ос, Р и у. Мы можем определить семь
кристаллических систем, основанных на шести решеточных
параметрах (табл. 1.1). Имеются 14 независимых способов расположения
точек в пространстве (в трех измерениях), приводящих к 14 решет-
кам Браве (рис. 1.1), перечисленных в табл. 1.1. В двух измерениях
возможны пять плоских решеток, в то время как в одном измерении
14 Структура твердых тел: старые и новые аспекты
Таблица 1.1
Кристаллические системы и решетки Браве
Система
Кубическая
Тетрагональная
Ромбическая
Ромбоэдрическая
Гексагональная
Моноклинная
Триклинная
Характеристика
элементарной ячейки
а = Ъ = с
а = р = у = 90°
а = Ь Ф с
а = р = Y = 90°
а ф Ъ фс
а = р = у = 90°
а = Ь = с
а = р = у ф 90°
а = Ь Ф с
а = р =905; y = 120c
афЪфс
а = у = 90° Ф Р
аф Ь Ф с
аф$Фу
1
1
}|
Характеристическая
симметрия
Четыре оси вращения 3-го
порядка
Одна ось вращения 4-го
порядка или одна
инверсионная ось 4-го порядка
Три взаимно
перпендикулярные оси вращения 2-го
порядка или одна ось
вращения 2-го порядка,
пересекающаяся с двумя зеркальными
плоскостями
Одна ось вращения 3-го
порядка
Одна ось вращения 6-го
порядка
Одна ось вращения 2-го
порядка или одна зеркальная
плоскость
Нет
Решетка
Браве
Р, I, F
Р, I
F
R(P)
P
P
С
P
Примечание. Решетки: Р — примитивная, F — гранецентрированная, I —
объемно-центрированная, С — базоцентрированная.
существует только один способ расположения точек — в линейную
решетку.
Найдено, что из десяти основных элементов симметрии (пять
собственных осей вращения 1, 2, 3, 4, 6 и пять инверсионных, или
несобственных, осей, 1-центр инверсии i, 2-зеркальная плоскость
т, 3, 4 и 6) кристаллы обладают одним или более. Набор элементов
симметрии, пересекающихся в общей точке внутри кристалла,
называется точечной группой. Десять основных элементов симметрии
вместе с их 22 возможными комбинациями составляют 32 класса
кристаллов. Имеется два дополнительных элемента симметрии,
винтовая ось и плоскость скольжения, которые возникают в кристаллах,
но не имеют аналога в молекулярной симметрии. Комбинация этих
элементов, включающих трансляцию, с точечной группой симметрии
называется пространственной группой. Для триклинной системы,
например, имеется только две возможные пространственные
группы Р1 и Р1, тогда как для моноклинной системы существуют
тринадцать возможных пространственных групп, возникающих из двух
возможных решеток Браве Р и С и трех точечных групп 2, т и 2/т.
Всего имеется 230 возможных пространственных групп.
Связи в кристаллах
15
Рис. 1.1. Четырнадцать решеток Бра-
ве. Цифры 2, 3 и т. д. обозначают оси
симметрии, а буквы Р, F, I и С — тип
решетки.
*$
Гт?1
Ш \& иьУ
231
Кубическая
ЛА
\Л*
4t 4P
Тетрагональ ногя
*&Л
U*^
FP1
222F 2221 222Р
Ромбическая
Цветная симметрия. Обсуждая
симметрию точечных и
пространственных групп, мы обозначали
каждую точку в решетке набором
пространственных координат (я, z/,
Z) И Изучали деЙСТВИе раЗЛИЧНЫХ Гексагональная
операций симметрии на точки.
Шубников предположил, что в
дополнение к трем пространственным
координатам к каждой точке может
быть добавлена четвертая
координата s. Эта координата s
одновременно допускает одно из двух
возможных значений; s можно приписать
спину частицы, в которой двумя
разрешенными значениями
являются спин, направленный вверх, и
спин, направленный вниз. В
абстрактных терминах они могут
соответствовать двум цветам — черному и белому. Новая
операция симметрии, названная операцией антисимметрии Л, вводится
для того, чтобы обратить значение s от черного к белому или
от спина, направленного вверх, к спину, направленному вниз.
Если эту операцию рассматривать в сочетании со всеми другими
операциями симметрии точечных и пространственных групп,
общее число возможных кристаллографических точечных и
пространственных групп возрастает соответственно до 122 и 1651. Эти
увеличенные числа групп часто относят к магнитным или
цветным группам и используют при описании симметрии магнитоупо-
рядоченных кристаллических твердых тел [Cracknell, 1975].
Моноклинная
Триклинноя
1.3. СВЯЗИ В КРИСТАЛЛАХ
Классификация кристаллов, основанная на типах связи,
является полезной в понимании соотношений структура — свойства
в твердых телах. В зависимости от типа связи легко определяются
пять типов твердых тел: ионные, ковалентные, металлические,
молекулярные (ван-дер-ваальсовы) и соединения с водородными
связями (табл. 1.2). Однако в реальных ситуациях твердые тела могут
иметь более чем один тип связи.
Таблица 1.2
Типы твердых тел
Тип
Присутствующие единицы
I
Характеристика
Примеры
Приблизительная
энергия когезии,
кДж-моль-1
Ионный
Ковалснтнып
М етал л ический
Вап-дер-ваальсовый
(молекулярный)
С водородной связью
Положительные и
отрицательные ионы
Атомы (связанные друг с
другом)
Положительные ионы,
уложенные в скопление
электронного «газа»
Молекулы или атомы
Молекулы,
удерживающиеся вместе с помощью
водородных связей
Хрупкие, диэлектрические и
довольно высокоплавкие
Твер,дые, высокоплавкие
и непроводящие (когда
чистые)
Высокая проводимость
Мягкие, низконлавкнс,
летучие и диэлектрические
Иизкоплавкис диэлектрики
NaCl
LiF
Алмаз
SiC
Na
Fe
Аргон
CH4
HoO (лед)
HF
795
1010
715
1010
НО
395
7,6
10
50
30
Связи в кристаллах
17
Ионные кристаллы. Ионные кристаллы образуются между
высокоэлектроположительными и высокоэлектроотрицательными
элементами, когда имеет место передача электрона от первых к последнимг
в результате чего противоположно заряженные ионы получают
электронную конфигурацию завершенных оболочек. Рентгеноди-
фракционные исследования ионных кристаллов обнаруживают
существенно сферическое распределение заряда вокруг ионов. Гало-
гениды щелочных и щелочноземельных металлов являются
типичными примерами ионных кристаллов. Сферическое распределение
заряда вокруг ионов в этих твердых телах приводит к симметричным
структурам с ионами одного заряда, окружающими ионы
противоположного заряда, и ионами с таким же зарядом, удаленными друг
от друга так далеко, как только это возможно.
Энергия когезии ионных кристаллов обусловлена главным
образом электростатическим взаимодействием и может быть рассчитана
на основе модели точечных зарядов. Следуя Борну, энергия когезии
([/) кристалла, содержащего противоположно заряженные ионы с
зарядами Zt и Z2, описывается как сумма двух термов, один из
которых обусловлен притяжением, а другой — отталкиванием:
U = у- + Дехр (--£). (1.1)
Здесь константа Маделунга А показывает, что мы рассматриваем
кристалл, а не только пару ионов, и является характеристикой
расположения ионов в кристаллах (а именно кристаллической
структуры); В — константа отталкивания; р — экспонента отталкивания и
R — расстояние между двумя противоположно заряженными
ионами. Терм в уравнении (1.1), отвечающий за отталкивание, объясняет
стабильность ионных кристаллов без коллапса, поскольку ионы с
завершенными электронными оболочками сопротивляются
перекрыванию их электронных оболочек с таковыми соседних ионов.
Константы В и р являются соответственно мерой силы и области
действия отталкивающего взаимодействия.
Для одно-одновалентного кристалла, как NaCl (Z7 = Z2 = 1),
уравнение (1.1) становится
£/= _ Л£1 +Яesp(--£). (1.2)
В может быть оценена исходя из того, что потенциальная энергия
U является минимальной при равновесном расстоянии Re между
парой противоположно заряженных ионов:
f(\U\ n Ае2 В ( Яе\ ,4 ov
2 Ч. II. Р. Рао, Дж. Гопалакришнан
18 Структура твердых тел: старые и новые аспекты
Таблица 1.3
Параметры энергии когезии некоторых галогенидов щелочных металлов со
структурой NaCl
Кристалл
NaCl
NaBr
KG1
KB r
RbGl
ИЬВг
о
Re, A
2,820
2,989
3,147
3,298
3,291
3,445
В, 10 8 эрг
1,05
1,33
2,05
2,30
3,19
3,03
о
P, А
0,321
0,328
0,326
0,336
0,323
0,338
U, кДжмоль"1
795
757
724
695
695
673
Соответственно
"-НЁ ('-*)■ <">
Уравнение (1.4) является выражением для энергии решетки ионного
кристалла, подобного NaCl, и впервые получено Борном и Майером.
Уравнение может быть использовано прямо для расчета энергии
когезии ионных кристаллов при условии, что мы знаем Лир.
В ранней работе по определению когезии ионных твердых
веществ было предположено, что энергия отталкивания меняется
обратно пропорционально расстоянию между ионами (B/Rn). Оба
параметра п и р получаются из сжимаемости твердых тел, причем
сжимаемость при О К равна
Л0 V dV /V=Ve
где Ve — объем кристалла, соответствующий Re. Значения В и р
для различных галогенидов щелочных металлов даны в табл. 1.3.
Pauling [1960] предложил следующие значения п для различных
электронных конфигураций с завершенными оболочками: Не — 5;
Ne — 7; Аг — 9; Кг — 10; Хе — 12. Значение п для галогенида
щелочного металла получается как среднее для двух ионов.
Например, п для NaCl в соответствии с этим методом равно 8.
Константа Маделунга А является функцией кристаллической
структуры и поэтому может быть вычислена из геометрии ионов в
кристалле. Так, константа Маделунга для структуры NaCl может
быть описана в виде ряда
а-1 12_ _8___6
1 У 2 + У3 2 + ' * ' '
который сходится к значению 1,74 756. Этот ряд, однако, является
лишь условно сходящимся, потому что суммирование может быть
Связи в кристаллах
19
остановлено в любой точке. Значение, полученное для конечного
ряда, является характеристикой конечного кристалла.
Значение А, приведенное выше для структуры NaCl, получена
для почти бесконечного кристалла. Энергии когезии галогенидов
щелочных металлов, рассчитанные с использованием уравнения (1.4)т
даны в табл. 1.3. Эти значения относятся к статическому кристаллу
и не содержат вклада ван-дер-ваальсовых сил и коррекцию для
энергии нулевой точки. Однако термы малы и вносят небольшую долю
в общую энергию решетки. Модифицированное выражение для
энергии решетки твердого тела типа MX, включающее все четыре термау
имеет вид
u^-^: + BM-T-)-{^ + ^) + ihv— (1-6>
где термы в правой части представляют собственно энергию Маде-
лунга, энергию отталкивания, вклад сил Ван-дер-Ваальса и
коррекцию нулевой точки, причем vmax является наивысшей частотой
решеточной колебательной моды. В терме Ван-дер-Ваальса члены i?7e
и RJ8 представляют соответственно диполь-дипольные и диполь-
квадрупольные взаимодействия. Энергии когезии ионных твердых
тел широко рассматривались в литературе [Tosi, 19641.
Экспериментальные энергии решетки ионных кристаллов получены из
термодинамических данных с использованием цикла Борна — Габера.
Соответствие между экспериментальными и теоретическими
значениями энергий решеток хорошее, что говорит в пользу ионной
модели для галогенидов щелочных металлов и подобных твердых тел.
Однако ионная модель является плохим приближением для
кристаллов, содержащих большие анионы и маленькие катионы, когда ко-
валентный вклад в связь становится существенным. Кроме того,
расчеты энергии когезии не могут быть использованы a priori для
предсказания структуры ионного соединения по той причине, что
метод использует экспериментальные межатомные расстояния в
сочетании с формальными ионными зарядами. Мы обсудим некоторые
ограничения ионной модели позднее, когда будем рассматривать
структуры неорганических кристаллов.
Электростатическая (маделунговская) часть энергии решетки
(MAPLE) служит для определения маделунговских потенциалов ионов
в кристаллах [Норре, 1975]. MAPLE ионного твердого тела
рассматривается как сумма вкладов катионов и анионов; тогда константа
Маделунга А кристалла будет суммой парциальных констант Маде-
лунга катионной и анионной подрешеток. Таким образом,
Л (NaCl) = ^c(Na+) + Aa{C\~)
1,74756 ... = 1x0,87378 ... +1x0,87378 ...
4(CaF2) = ^c(Ca2+) + 2^a(F-)
2*
20 Структура твердых тел: старые и новые аспекты
5,03878 ... = 3,27612 ... +2x0,88133 ...,
где Ас и Аа — парциальные константы Маделунга катионной и
анионной подрешеток соответственно. Маделунговские потенциалы
являются почти независимыми от кристаллической структуры, и
значения MAPLE для различных полиморфных модификаций данного
состава отличаются только приблизительно на 1 %. Расчеты,
основанные на ионной модели, в действительности предсказывают
неправильные структуры в некоторых случаях: например, LiF был
предсказан со структурой вюртцита вместо структуры
каменной соли.
Ковалентпые кристаллы. Когда атомы в кристалле имеют
близкие электроотрицательности, образование связи происходит через
обобществление валентных электронов, причем каждый атом
вкладывает в связь один электрон. Сила притяжения между двумя
атомами с одинаковой электроотрицательностью вызвана
перекрыванием атомных орбиталей, в результате чего общая энергия
понижается. Возникающая в результате такого взаимодействия кова-
лентная связь является классической двухэлектронной связью.
Типичные ковалентные твердые тела образованы элементами TV
группы Периодической таблицы, такими как углерод, кремний и
германий. Эти вещества кристаллизуются в структуре алмаза, где каждый
атом связан с четырьмя другими посредством ковалентных связей.
Атомы в ковалентных твердых телах стремятся достичь электронной
конфигурации завершенной оболочки путем обобществления
электронов с соседями. Например, в алмазе атомы углерода (с 2$22/?2-ва-
лентной электронной конфигурацией) образуют четыре
эквивалентные тетраэдрически расположенные £/?3-гибридизованные орбитали,
каждая заполнена одним электроном. Эти гибридные орбитали
перекрываются с подобными орбиталями соседних атомов углерода,
образуя четыре ковалентных связи; валентная оболочка вокруг каждого
атома, таким образом, является заполненной восьмью электронами.
В отличие от ионной, ковалентная связь имеет отчетливый
направленный характер.
Большое число бинарных соединений АВ, образованных
элементами IIIА и VA или ПА и VIA групп (так называемые III—V и
II—VI соединения), также кристаллизуются в алмазоподобных
структурах. Среди I—VII соединений в этой структуре
кристаллизуются галогениды меди (I) и Agl. В отличие от алмаза, связи в
таких бинарных соединениях не являются совершенно ковалентными
вследствие разницы в электроотрицательности между
составляющими атомами. Это может быть понято в терминах частичного ионного
характера или ионности связей в этих кристаллах.
Pauling [1960] выразил ионность ковалентной связи А—В в
терминах разницы между электроотрицательностями двух атомов:
/, = 1 - ехр [-(ХА - Хв)2/4]. (1.7)
Связи в кристаллах
21
Таблица 1.4
Параметры ионности Филлипса и Полинга для некоторых кристаллов А В
Кристалл*
Si(D)
BN(Z)
GaAs(Z)
ZnO(\V)
CuBr(Z, W)
MgS(W, R)
NaCl(R)
Rbl(R)
Eh, эВ
4,77
13,10
4,32
7,33
4,10
3,71
3,10
1,60
Ev эВ
0
7,71
2,90
9,30
6,90
7,10
11,8
7,1
/| (ФИЛЛИПС)
0
0,256
0,310
0,616
0.735
0,786
0,935
0,951
1l (Полинг)
0
0,42
0,26
0,86
0,80
0,67; 0,78
0,94
0,92
* Кристаллические структуры, данные в круглых скобках: D — алмаз, Z —
цинковая обманка, W — вюртцит, R — каменная соль.
Здесь ft представляет частичный ионный характер связи, в то
время как ХА и Хв — полинговские электроотрицательности элементов
А и В. Видно, что fi = 0, когда ХА = Хв, и ft = 1, когда ХА —
— Хв^> 1. В кристаллах это выражение модифицируется, для того
чтобы учесть тот факт, что атом образует более чем одну связь и
число образующихся связей не всегда равно его формальной
валентности. Для кристаллов ANB8~N выражение для ионности
читается как
/.с = 1 _ (NiM) + (7V/./M), (1.8)
где М — число ближайших соседей. Полинговская шкала ионности
объясняет структуры многих бинарных соединений. Mooser, Pearson
[1959] аналогично показали, что диаграмма зависимости среднего
главного квантового числа от разницы в электроотрицательности
четко отделяет различные координационные структуры бинарных
соединений. Phillips [1973a] предложил новую шкалу ионности для
кристаллов типа АВ, основанную на допущении, что среднее
значение зонной щели Eg этих кристаллов состоит из обоих вкладов, как
ковалентных, так и ионных, в соответствии с выражением Eg2 =
= Е h2 + Et2, где Е h и Et являются гомополярной и ионной частями.
Параметр ионности определяется выражением
/*С = |- (I-9)
Этот параметр получается отнесением статической диэлектрической
константы к Eg и принятием Е h в таких кристаллах
пропорциональным а2'5, где а — постоянная решетки. Параметры Филлипса для
некоторых кристаллов представлены в табл. 1.4.
22 Структура твердых тел: старые и новые аспекты
Филлипс показал, что все кристаллы с ftc ниже критического
значения 0,785 обладают тетраэдрической структурой алмаза (или
вюртцита); когда ftc > 0,785, оказывается предпочтительной
шестерная координация (структура каменной соли). Полинговская шкала
ионности также дает такие структурные предсказания, но шкала
Филлипса более универсальна. Соответственно MgS (/fc = 0,786)
показывает пограничное поведение. Энергии когезии тетраэдрически
координированных полупроводников были рассчитаны с
использованием параметров ионности. Следует отметить, что борновская модель
ионных твердых тел не применима для таких частично ионных
веществ.
Приближение, основанное на орбитальных радиусах атомов*
эффективно объясняет структуры 565 твердых веществ типа АВ
[Zvmger, 1981]. Орбитальные радиусы, выведенные из жесткоостов-
ных псевдопотенциалов, обеспечивают величину эффективного
размера атомных остовов, окруженных валентными электронами.
Линейные комбинации орбитальных радиусов, которые соответствуют
филлипсовским структурным индексам Е h и Еь используются как
координаты в построении структурных карт для твердых тел АВ.
Металлические кристаллы. Металлы существенно отличаются
от неметаллических твердых тел как по структуре, так и по
физическим свойствам. Они обычно кристаллизуются в плотноупаковап-
ных структурах с большими координационными числами (12 или 8).
Характерно, что металлы демонстрируют высокую тепло- и
электропроводность, указывая на присутствие электронов, которые
являются относительно свободными для движения через кристалл.
Понятно, что невозможно объяснить эти черты в терминах
локализованных связей ковалентного типа между ближайшими соседями.
Pauling [1960] описал металл как структуру, в которой одноэлект-
ронные связи и связи с участием электронных пар резонируют
между разными парами атомов. Отмечено [Brewer, 1967, 1984] значение
корреляции Энгеля (соотносящей структуры с электронными
конфигурациями) в понимании структуры металлов и сплавов. Согласно
этим работам, важность кислотно-основных взаимодействий в
металлических системах состоит в том, что электроны не являются
свободными для заполнения всех частей физического пространства,
они в основном ограничены орбитальными объемами. Поэтому
электроны в металлических системах занимают разрешенное пространство
и взаимодействуют с двумя или более ядрами.
Теория металлического состояния, предложенная Друдэ в
начале этого столетия, объясняет многие характерные черты
металлов. В этой модели, которая называется теорией свободных
электронов, все атомы в металлическом кристалле сообща принимают
участие в связывании, каждый атом поставляет определенное число
(валентных) электронов на связь. Эти «свободные электроны»
принадлежат кристаллу как целому. Кристалл рассматривается как
Связи в кристаллах
23
объединенный электростатическим взаимодействием между ионными
остовами и свободными электронами. Внутри кристалла свободные
электроны движутся под действием постоянного потенциала, в то
время как на границах имеется большая разница потенциалов,
которая предотвращает вырывание электронов. Свободные электроны
внутри потенциальной ямы, однако, не подчиняются статистике
Максвелла — Больцмана; эти электроны не дают существенного
вклада ни в удельную теплоемкость, ни в парамагнетизм металлов.
Зоммерфельд модифицировал теорию Друдэ введением законов
квантовой механики. Согласно квантовой механике электроны
ассоциируются с волновым характером, длина волны К определяется
выражением А, = hip, где р — момент, равный ти. Удобно ввести
параметр А, который называется волновым вектором, для
определения свободных электронов в металлах; величина волнового
вектора равна
7 2я 2кпги /Л im
k = — —к— <1Л°)
Энергия свободного электрона в металле является существенно
кинетической энергией, обусловленной его движением:
Е = 1 ти2 = 4^ = ^-. (1.11)
Поэтому соотношение между Е и к должно быть параболическим.
Вскоре было понято, что параболический закон свободных
электронов не описывает реальной ситуации. Электроны в кристаллах
испытывают влияние периодического потенциала, обусловленного
присутствием ядер, а не постоянного потенциала. В настоящее время
состояние электронов в твердых телах рассматривают в виде
картины энергетических зон, которые отделены друг от друга
запрещенными щелями (см. также гл. 6). Образование энергетических зон,
разделенных запрещенными щелями, можно понять исходя из
«приближения сильной связи». Приближение является существенно
таким же, как и в теории молекулярных орбиталей, используемой для
дискретных молекул. В металлах высшая занятая энергетическая
зона является частично заполненной, поэтому электроны легко
возбуждаются на незанятые уровни внутри зоны при действии
электрической или тепловой энергии, вызывая высокую проводимость.
Кристаллы с ван-дер-ваальсовыми связями (молекулярные
кристаллы). Инертные газы, подобно аргону и криптону, также, как и
молекулы, которые не обладают постоянным дипольным моментом (С02,
метан, бензол), конденсируются с образованием кристаллов с
маленькими энергиями когезии. Тот факт, что атомы и молекулы, в
которых все химические связывающие силы, очевидно, являются
насыщенными, могут быть сконденсированы в кристаллическое
состояние, показывает, что кроме обычных сил ионного, ковалентного или
металлического типа имеются определенные слабые остаточные си-
24 Структура твердых тел: старые и новые аспекты
лы притяжения. Об относительной слабости этих сил
свидетельствуют низкие точки плавления, маленькие энергии когезии и низкие
теплоты плавления таких твердых тел. Происхождение этих сил
обусловлено мгновенными флюктуациями в распределении
электронов, возникающими из наведенного диполь-дипольного
взаимодействия между атомами или молекулами. Если распределение заряда,
скажем, в атоме инертного газа будет жестким, флюктуации
зарядовой плотности не будет и, следовательно, не произойдет
взаимодействия. Однако даже зарядовое облако атома инертного газа до
некоторой степени деформируется, увеличивая мгновенное несферическое
распределение, которое индуцирует дипольный момент на соседних
атомах. Именно этот наведенный момент и является причиной
притягивающего взаимодействия между атомами. Энергия наведенного
диполь-диполыюго взаимодействия меняется как R'6. Такие силы
притяжения называются ван-дер-ваальсовыми, или
дисперсионными, силами. При очень коротких расстояниях сближения
появляются силы отталкивания вследствие перекрывания зарядовых облаков
разных атомов. Поэтому общий ван-дер-ваальсовый потенциал
состоит из двух термов — дисперсионного терма и терма
отталкивания — и определяется выражением
t/y=-4- + 4>- (1-12)
/?6 Л12 7
Уравнение (1.12) представляет хорошо известный потенциал Лен-
нарда — Джонса.
В последние годы оказалось возможным оценить энергию
когезии, характеристики упаковок и другие свойства молекулярных
кристаллов количественно на основе несвязывающих
взаимодействий между атомами [Kitaigorodsky, 1973]. Аналитические
выражения имеют форму
А •
Uij = — -£- + B{j exp (— Cyry),
ij ij
где константы А, В, С или п получаются подгонкой уравнения к
экспериментальным данным для таких кристаллических структур,
как углеводороды. Эти параметры были оценены [Williams, 1967]
путем тщательного подбора большого числа наблюдаемых
уравнений для алифатических и ароматических углеводородов. Уравнение
(1.13) принимает в расчет только дисперсионные и отталкивающие
взаимодействия, электростатические взаимодействия в
молекулярных кристаллах несущественны. Кулоновские взаимодействия могут
(1.13)
Связи в кристаллах
25
быть включены как действие точечных зарядов qe на атомы
[Williams, Starr, 1977] следующим образом:
о
ии - - ПГ + в^ ехР (- с^) + Ц^' (1Л4)
Таким образом, энергия решетки кристалла известной структуры
(с определенными атомными позициями) рассчитывается с учетом
всех возможных расстояний между парами атомов в различных
молекулах. Метод атом-атомных потенциалов был применен для
исследования явлений, принадлежащих как к статическим, так и к
динамическим решеткам, и этот объект был рассмотрен Kitaigorodsky
[1973] и совсем недавно — Ramdas, Thomas [1980]. Типичными
проблемами, которые исследовались этим методом, являются дефекты и
сдвиги плоскостей, фазовые переходы и молекулярные вращения
в кристаллах.
Кристаллы с водородными связями. Имеется много примеров,
когда водородные связи, образующиеся между двумя
электроотрицательными атомами, подобно кислороду или фтору, определяют
стабильность и свойства кристаллов. Когда атом водорода
связывается с сильно электроотрицательным атомом, электронная пара,
образующая связь, является настолько неравномерно смещенной,
что атом водорода можно рассматривать как положительный заряд,
пригодный для притяжения отрицательного конца атома с непо-
деленной парой электронов в другой молекуле. Атом водорода в
кристаллах с водородными связями обычно располагается
асимметрично между двумя такими электроотрицательными атомами. По
сравнению с ковалентными и ионными, водородные связи являются
слабыми, имеющими энергии в области 12—40 кДж-моль-1.
Водородная связь является причиной когезии в кристаллах таких веществ,
как лед и белки.
Лед является прототипом кристалла с водородными связями.
Даже в жидком состоянии вода имеет исключительные физические
свойства благодаря водородным связям. В кристаллическом
состоянии структура Н20 является аномальной. В то время как многие
такие гидриды кристаллизуются в структурах с плотной упаковкой,
точно так же, как молекулярные кристаллы, лед имеет гексагональ-
о
ную структуру с довольно коротким (2,76 А) межмолекулярным
расстоянием О ... О. Каждый атом кислорода в структуре тетра-
эдрически окружен четырьмя другими атомами кислорода.
Если пренебречь присутствием атомов водорода, то структура
льда будет точно подобна структуре вюртцита, причем атомы
кислорода воды находятся в позициях цинка и серы. Водородные
связи играют большую роль в определении свойств кристаллов кислых
солей, подобно КН2Р04.
26 Структура твердых тел: старые и новые аспекты
1.4. НЕОРГАНИЧЕСКИЕ СТРУКТУРЫ
Структуры металлов, как и многих немолекулярных твердых
тел с ненаправленными связями, легко описываются
геометрическими терминами в предположении, что атомы представляют собой
жесткие сферы, которые упаковываются как можно плотнее, чтобы
сэкономить пространство и минимизировать энергию. Обе плотпейшие
упаковки кристаллов как кубической (АВСАВС), так и
гексагональной (АВАВ) симметрии (КПУ и ГПУ соответственно) имеют
эффективность заполнения пространства 74,1 %. Обе эти плотноупакован-
ные структуры обладают двумя типами пустот (или междоузлий):
тетраэдрическими с координационным числом, равным 4, и окта-
эдрическими с координационным числом, равным 6. В ансамбле из
N сфер одинакового размера будет 2N тетраэдрических и N окта-
эдрических пустот. Объемно-центрированная кубическая упаковка
(ОЦК) сфер приводит к степени заполнения пространства,
равной 68,1 %.
Металлические элементы. Большинство металлов
кристаллизуются в одном или нескольких типах трех кристаллических
структур КПУ (А1), ОЦК (А2) и ГПУ (A3), показанных на рис. 1.2.
Многие из них являются полиморфными и показывают переход из одной
структуры в другую с изменением температуры или давления:
Т1(ГПУ)_21!!Д.ОЦК
Fp(OTTK) 1182К, КПУ 167бК,ОПК
Сз(ОЦК) 41кбар,КПУ
Высокотемпературные модификации обычно имеют меньшие
координационные числа, как и модификации низкого давления.
Структуры некоторых металлов, таких как Zn, Cd и Hg, не
соответствуют этой картине. Кристаллическая решетка ртути имеет
ромбоэдрическую структуру, в которой каждый атом является шести-
координированным и следует правилу (8 — 7V), указывающему на
наличие ковалентной компоненты в связи. Цинк и кадмий
кристаллизуются в искаженной ГПУ-структуре с большим расстоянием
между индивидуальными слоями, чем в идеальном случае
(отношение с/а > 1,633), указывающим на то, что связь между атомами
внутри слоя сильнее, чем связь между слоями. Атомные радиусы
для металлических элементов в случае координационного числа,
равного 12, были определены Гольдшмидтом, и соотношение между
длинами связей и природой связывания в металлах обсуждалось
Pauling [1960].
Неметаллические и полу металлические элементы. Среди
неметаллических элементов инертные газы кристаллизуются в плотно-
упакованных структурах (подобных металлам), причем силы Ван-
Неорганические структуры
27
Рис. 1.2. Структуры металлов: А1(а); А2(б); (уО^СГ^уО ryO fVQ
А3(в), показана также проекция гексагональ- YI ^т"^ I SrT Ч7 Т
ной ячейки на плоскость ху.
о
о
о
о
о^^&° (j>xb—^cS
о
0=^=9
дер-Ваальса являются ответственными
за когезию. Неметаллические простые
вещества элементов V группы (N, Р,
As, Sb), VI группы (О, S, Se, Те) и
VII группы (галогены)
кристаллизуются в молекулярных формах, в
которых внутримолекулярные связи
имеют ковалентный характер, в то время
как межмолекулярные связи
являются связями ван-дер-ваальсового типа.
Среди этих веществ азот и кислород
образуют молекулярные твердые тела, в
составленные кратно связанными
двухатомными молекулами. В случае других элементов атом из
N-vl группы Периодической таблицы имеет (8 — N) ближайших
соседей, так как в валентной оболочке имеется N электронов,
которые могут образовать (8 — N) ковалентных связей. Хлор,
бром и иод кристаллизуются в ромбической структуре, в
которой двухатомные молекулы расположены в виде труб вдоль оси с.
Среди халькогенов сера, селен и теллур образуют кластеры,
включающие складчатые кольца (Se, S8 и т. д.) или зигзагообразные цепи
с двумя соседями. Кольца конечного размера или бесконечные цепи
связываются друг с другом ван-дер-ваальсовыми силами. Элементы
V группы, кроме азота, кристаллизуются в структурах, содержащих
три близких соседа. Фосфор существует в нескольких полиморфных
модификациях; белый фосфор содержит нестабильные блоки Р4,
а форма красного фосфора (фосфор Гитторфа) кристаллизуется в
трубчатой структуре, в которой каждый атом Р окружен тремя
другими атомами пирамидально. Мышьяк, сурьма и висмут
кристаллизуются в изоморфных слоистых структурах, где в слое каждый атом
связан ковалентными связями с тремя другими, образуя
гофрированный лист; вся структура, получающаяся путем суперпозиции
таких слоев, дает ромбоэдрическое окружение. Кроме имеющихся
трех соседей в их собственном слое, каждый атом имеет три соседа в
смежных слоях на больших расстояниях. Разница в расстояниях
между двумя группами соседей уменьшается при переходе от
мышьяка к висмуту, указывая на приближение к металлической связи в
висмуте.
Элементы IV группы (С, Si, Ge, Sn) образуют четыре ковалент-
ные связи ($р3-гибридизация), давая начало структурам с полностью
ковалентными связями. Алмаз, кремний, германий и серое олово
принадлежат к этому типу, в котором каждый атом связан с четырь-
28 Структура твердых тел; старые и новые аспекты
^
--J^d-^^p-^^^ Рис. 1.3. Структуры алмаза (а)
и графита (б).
! ! ! | ' ! 1 ' '
мя другими и кристалл как
fy—jcS^—jc^—г1 целое является гигантской
j j| j ] i ] ]|| молекулой. Элементарная
--^ЦпоР--^ ячейка алмаза (рис. 1.3) со-
d^Se^ZK^fs* стоит из ГЦУ-мотивов
углерода, в которых дополни-
а тельные атомы углерода
находятся в чередующихся те-
траэдрических позициях (А4). Графит кристаллизуется в
гексагональной слоистой структуре (см. рис. 1.3), в которой каждый
атом углерода связан ковалентно с тремя другими атомами (яр2-
гибридизация). Смежные слои взаимно смещены друг
относительно друга, так что гексагональные кольца из атомов углерода в
данном слое не лежат точно один над другим; если два таких слоя
обозначить буквами А и В, то кристаллическая структура
гексагонального графита состоит из последовательности АВАВ; в более
редкой ромбоэдрической модификации графита последовательность
слоев имеет вид АВСАВС. В структуре графита расстояние С—С
о о
равно 1,42 А внутри слоя и 3,35 А между слоями. Для других
элементов группы, кроме углерода, структура графита не найдена.
Олово обладает диморфизмом, а-форма (серое олово) (стабильная
ниже 291 К) имеет структуру алмаза, а |3-форма (белое олово) —
тетрагональную структуру. Свинец кристаллизуется в кубической
плотноупакованной структуре, типичной для металлов.
Неорганические соединения. Немолекулярные неорганические
твердые тела, содержащие два элемента, один металлический, а
другой неметаллический, образуют бинарные соединения составов АВ,
А2В3, АВ2, АВ3 и т. д. Связь в таких соединениях является ионной,
если разница в электроотрицательности между А и В достаточно
большая. Кристаллические структуры таких соединений в основном
определяются природой связи и относительными размерами А и В.
Если связь имеет преимущественно ионный характер, как во
фторидах и оксидах, структура может быть описана в геометрических
терминах на основе плотной упаковки ионов. Так как в большинстве
таких соединений анионы значительно больше по размеру, чем
катионы, структуры могут быть представлены в виде плотнейшей
упаковки анионов (ГПУ или КПУ), междоузлия (пустоты) которых
заняты катионами. Когда катионы больше, чем анионы, плотнейшая
упаковка будет из катионов, пустоты в которой заняты анионами.
Кристаллические структуры бинарных ионных соединений поэтому
определяются относительными размерами пустот и ионов,
образующих плотнейшую упаковку. Можно показать, что если радиус
большей среды, образующей плотнейшую упаковку, равен /?, то в окта-
Неорганические структуры
29
Таблица 1.5
Отношения радиусов для различных катион-
ных геометрий в ионных соединениях
Отношение гс/га
0-0,155
0,155-0,225
0,225-0,414
0,414-0,732
0,732-1,000
>1,000
Координационное число (КЧ)
и геометрия
2; Линейная
3; Треугольная
4; Тетраэдрическая
6; Октаэдрическая
8; Кубическая
12
эдрической пустоте такой упаковки свободно разместится меньшая
сфера с радиусом г = 0,414 Я, ав тетраэдрической пустоте — сфера
с еще меньшим радиусом, равным г = 0,225 R. Если мы представим
большие сферы анионами, а меньшие — катионами, размеры пустот
определят предельное отношение радиусов rjra для октаэдрической
и тетраэдрической координации катионов анионами. В табл. 1.5
даны области отношений радиусов для различной координационной
геометрии в бинарных ионных соединениях.
Ионная модель, согласно табл. 1.5, предсказывает переходы
в твердых телах типа АВ от структур с координацией 8 : 8 к
координации 6 : 6 и от 6 : 6 к 4 : 4 при определенных значениях отношения
радиусов. Действительные структуры галогенидов щелочных
металлов, однако, отличаются от предсказанных, как показано на
рис. 1.4. Такие неудачи обусловлены ограничениями простой ионной
модели [O'Keeffe, 1977]. Ионная модель предполагает, что анионы
существенно больше по размеру, чем катионы. Радиусы оксидного pi
о
фторидного ионов (1,40 и 1,33 А соответственно) [Shannon, Prewitt,
1969] действительно больше, чем радиусы большинства катионов.
Ионные радиусы, приведенные в литературе, получены разделением
длин связей на основе известных свойств свободных ионов и поэтому
соответствуют свободным ионам, а не ионам в кристаллах. Анионы
в кристаллах подвергаются действию положительного потенциала
Маделунга, и этот эффект приводит к сжатию зарядового облака;
аналогично катионы подвергаются действию отрицательного
потенциала, который имеет противоположный эффект. Когда эти эффекты
принимаются во внимание, разница между радиусами аниона и
катиона оказывается не столь существенной. Поэтому более важным
кажется отношение длин связей к силе связей, чем ионные радиусы;
чтобы исключить неопределенность и иметь удовлетворительную
процедуру разделения длин связей на катионную и анионную части,
таблицы ионных радиусов было бы лучше рассматривать как
таблицы длин связей с определенной рациональной силой связи.
Несмотря на то что экспериментальная электронная плотность имеет
минимум в точке между катионом и анионом, это неглубокий минимум.
30 Структура твердых тел: старые и новые аспекты
LL* Na* K+Rtf Cs+
г.о-i
US
!
I
0.4
r~i
Br# /
cr« /
A '
1
L-/ 6
F /•
/
1/ /
/ /
/
/
/
/
1
т1-
•
•
/
•/
/
1
I
•
•
/
/4
/
/
/
•
5
г
• '■ 1
/
• .
• о
• •
• a
об
me
Рис. 1.4. Сравнение реальных структур
галогенидов щелочных металлов с
предсказаниями ионной модели. Рисунок
разделен на три области линиями,
соответствующими отношению г J r = 0,414
и 0,732. Области соответствуют
структурам с КЧ 4,6(а, б) и 8(6, в). Видно,
что предсказания ионной модели не
полностью соответствуют реальным
структурам галогенидов щелочных
металлов [Phillips, 1973b].
0,6 1fl 1,4
Радиус катооное, А
Если мы используем точку
минимума для определения
радиусов катионов и анионов, мы
придем к зарядам на ионах, которые
существенно меньше, чем
формальные заряды. Если разделение
сделать с сохранением суммарных
зарядов на ионах, радиусы
анионов окажутся меньше, а радиусы
катионов больше, чем принятые
значения.
Таблицы ионных радиусов, имеющиеся в литературе [Shannon,
Prewitt, 1969; Shannon, 1976], получены из данных по длинам
связей в оксидах и фторидах, поскольку они принадлежат к наиболее
ионным твердым телам. Однако эти значения не являются обычно
применимыми; в случае таких твердых веществ, как нитриды и
сульфиды, экспериментальные длины связей корректно не
воспроизводятся. Недавно был предложен новый набор радиусов, применимый
к сульфидам [Shannon, 1981]. Значения ионных радиусов,
полученных из оксидов и фторидов, бесполезно использовать даже для
оксофторидов. Например, наблюдаемые длины связей La—О и La—F
о
в LaOF равны 2,42 и 2,60 А соответственно, в то время как
соответствующие значения, рассчитанные с использованием радиусов Шэн-
о
нон — Прювитта,— 2,54 и 2,47 А. Согласно полинговской
концепции электростатической силы связи и правилу электростатической
валентности, сумма сил связей у аниона должна быть равна валентности
аниона, и это будет предсказывать силу связи La—F, равной 1/4,
и силу связи La—О, равной 1/2 [O'Keeffe, 1981]. Это подразумевает
ситуацию, где La координирован двенадцатью ионами фтора или
шестью ионами кислорода. Когда мы используем радиусы лантана,
соответствующие таким координациям, длины связей в LaOF
предсказываются достаточно разумно. Такие рассмотрения
обнаруживают, что ионный радиус является запутанной концепцией,
исключающей количественный расчет, что затрудняет использование
этого параметра.
Неорганические структуры
31
Наоборот, полинговская концепция электростатической силы
связи и последующие развития соотношений сила связи — длина
связи (см., например, [Brown, 1981]), кажется, будут более
применимы.
В ионной модели подрешетка анионов рассматривается в
приближении плотнейшей упаковки равновеликих сфер. Однако в
реальных кристаллах плотности анионных упаковок намного меньше,
чем идеальное значение, равное 0,74. Например, в а-А1203,
обладающем ГПУ-упаковкой анионов, плотность анионной упаковки
составляет только 0,595. Хотя анионы топологически уложены так,
будто они имеют плотнейшую упаковку, в действительности они не
находятся в контакте друг с другом. В работах O'Keeffe [1977] и
O'Keeffe, Hyde [1981] использован специальный термин «эвтаксия»
для описания такой ситуации. Согласно этим работам, структуры
ионных кристаллов лучше описываются как структуры с
максимальным объемом (минимум электростатической энергии) для
фиксированных расстояний катион — анион. Эта идея
проиллюстрирована с использованием нескольких обычных ионных структур. В
качестве примера мы приведем ZnO (структура вюртцита). Структура
вюртцита гексагональная, пр. гр. Р()3тс, атомы Zn находятся в
позициях 2(a): 0,0, иг\ 1/3, 2/3, 1/2 + щ; атомы кислорода 2(a): 0,0,
w2 и т. д. Два параметра у = с/а и и = их — и2 полностью
определяют структуру. Четыре тетраэдрических расстояния Zn—О равны,
когда у2 = 4/(12и — 3), и поэтому объем V = YTlP(u — 1/4)/2и3,
где I — расстояние катион — анион, V, полученное из этого
уравнения, является максимальным, когда и = 3/8 и у = 1,633. Для
ZnO и = 0,383 и у = 1,6022, а для ВеО и = 0,378 и у = 1,602.
Для нескольких других ионных твердых тел с этой структурой и
и 7 близки к идеальным значениям, предсказанным из соображений
максимального объема.
Координационное число катиона определяется отношением гс'гаУ
которое подразумевает, что чем больше радиус катиона, тем больше
его координационное число. Можно привести несколько примеров,
которые показывают, что это допущение неправильное. В MgAl204
(шпинель) большие катионы Mg2+ имеют тетраэдрическую
координацию, а меньшие А13+ — октаэдрическую координацию.
Совсем недавно обсуждались соотношения связевая
валентность — длина связи, а также их применимость к твердым телам
(см., например, [Ziolkowski, 1985; Savborg, 1985; Domenges et a].,
1985]). Эти соотношения были также распространены на равносвязе-
вую энтальпию и энергию связи. Концепция связевой валентности,.
вероятно, в будущем будет использоваться более обычно.
Структуры АВ. В соединения типа АВ входят каменная соль
(Bl), CsCl (В2), цинковая обманка (ВЗ), вюртцит (В4) и NiAs (B8).
Эти структуры представлены на рис. 1.5. В первых четырех
структурах катионные и анионные подрешетки совершенно эквивалент-
32 Структура твердых тел: старые и новые аспекты
Рис. 1.5. Структуры соединений АВ.
а — каменная соль; б — CsCl; в — цинковая
обманка; г — вюртцит; д — NiAs.
ные и координационная геометрия
вокруг катиона и аниона
одинаковая. Структуру каменной соли
(NaCl) демонстрирует большое число
соединений типа АВ. Эта структура
(см. рис. 1.5) может быть
представлена двумя взаимно проникающими
подрешетками типа ГЦК,
состоящими из ионов Na+ и С1~,
расположенными так, чтобы создать вокруг
• Металл каждого иона октаэдрическую коор-
о Неметалл динацию другими ионами
(координация 6 : 6); структура содержит
четыре формульные единицы NaCl в
д элементарной ячейке. Структуру
можно также рассматривать как
результат кубической плотной упаковки ионов хлора (а*С1_ =
= 1,81AJ с ионами натрия (rNa+ = 0,95 А], занимающими окта-
эдрические междоузлия. Структура такжэ может быть
представлена катион-центрированными октаэдрами NaClc>,
простирающимися в трех измерениях с обобществлением всех своих ребер.
Типичными примерами соединений АВ, кристаллизующихся в этой
структуре, являются: галогениды щелочных металлов, за
исключением CsCl, CsBr и Csl; NH4C1, NH4Br и NH4I выше 457, 411 и 255 К;
оксиды и халькогениды щелочно-земельных металлов, за
исключением солей бериллия и MgTe; оксиды двухвалентных 3^-металловт
за исключением СиО; оксиды и халькогениды двухвалентных
лантаноидов и актиноидов.
Хотя обычно считается, что эту структуру принимают ионные
соединения, некоторые менее ионные материалы, подобно нитридам,
фосфидам и арсенидам трехвалентных лантаноидов так же, как карбиды
и нитриды элементов титановой и ванадиевой группы,
кристаллизуются в этой структуре. Несколько создинений типа III—V, таких как
GaP и InP, и соединений типа II—VI, подобно CdS и CdSe,
подвергаются переходам в эту структуру при высоких давлениях. Кроме
вышеприведенных примеров бинарных соединений в структурах,
родственных NaCl, кристаллизуются также несколько тройных
соединений, содержащих два различных иона металла. Например, в
соединениях типа АА'В2 два имеющихся различных катиона могут
быть распределены статистически, как в у~1^АЮ2, a-NaT102
и NaLaS2, или упорядочены в чередующихся плоскостях (111),
простирающихся вдоль телесной диагонали, как в LiNi02 и AgBiS2;
Неорганические структуры
33
возможны также другие типы упорядочения катионов.
Структура NaCl найдена в таких соединениях, как TiO, VO pi
NbO, обладающих высоким процентом катионных и анионных
вакансий. Тройные оксиды типа Mg6Mn4+08 кристаллизуются в этой
же структуре, где 1/8 катионных позиций остаются вакантными.
В структуре каменной соли кристаллизуются такие твердые
растворы, как Li^.^Mg^Cl (0<[ я <! 1); стехиометрический MgCl2 на
самом деле можно рассматривать как дефектную структуру каменной
соли с 50 % упорядоченных катионных вакансий.
Структуру CsCi (см. рис. 1.5), состоящую из взаимно
проникающих примитивных кубических подрешеток из ионов Cs+ и С1~
с координацией 8 : 8, демонстрируют многие твердые тела: CsCl,
CsBr и Csl; NH4C1, NH4Br и NH4I в низкотемпературных
модификациях; галогениды таллия; сплавы типа р-латуни, CuZn, AuZn; CuCN,
CuSH и T1CN в высокотемпературной форме и галогениды щелочных
металлов (за исключением солей лития) при высоких давлениях.
Структура цинковой обманки (ZnS) близка родственной
структуре алмаза (см. рис. 1.5). Структура цинковой обманки с
координацией 4 : 4 получается, если в структуре алмаза одну половину
атомов углерода, образующих ГЦК-подрешетку, заменить атомами
цинка, а другую половину — атомами серы. Типичными примерами
соединений АВ, кристаллизующихся в структуре цинковой обманки,
являются: галогениды меди (I); y-hgl; халькогениды бериллия (за
исключением ВеО); халькогениды цинка, кадмия и ртути (за
исключением оксидов); много соединений типа III—V, таких как BN, В1\
BAs, A1P, AlAs, AlSb, GaP, GaAs, GaSb и SiC. Известны
производные структуры от структуры цинковой обманки следующих типов:
халькопирит CuFjS2 с упорядочением одно- и трехвалентных кати< -
нов в позициях цинка; структуры со статистическим распределением
различных катионов, как это найдено в CuSi2P3 и Cu2GeS3; Cu3SbS4
(фаматинит), получающийся при замещении в халькопирите
позиций, занимаемых Fe3+, ионами Си+ и Sb5+. Структура цинковой
обманки может также содержать катионные и анионные вакансии:
например, Ga2S3 кристаллизуется в этой структуре, где 1/3
катионных позиций вакантны. Аналогичным образом Ag2HgI4 имеет
структуру» производную от структуры цинковой обманки, с 1/4
вакантных катионных позиций.
Структура вюртцита является близкородственной структуре
цинковой обманки, она имеет такую же 4 : 4 тетраэдрическую
координацию, возникающую из гексагональной плотной упаковки
анионов, в которой половина тетраэдрических позиций занята катионами.
Примерами соединений АВ. кристаллизующихся в этой структуре,
являются CuCl, CuBr, Cul, P-AgI, BeO, ZnO, ZnS, MnS, MnSe, MnTe,
A1N, GaN, InN, SiC и NH4F. Структура вюртцита является, в
известном смысле, промежуточной между структурами NaCl и
ZnS.
3 Ч. Н. Р. Рао, Дж. Гопалакришнан
34 Структура твердых тел: старые и новые аспекты
Рис. 1.6. Структуры соединений АВ.
а — ТП; б — красный РЬО; в — HgS; г —
PtS.
В гексагональной структуре
NiAs атомы никеля октаэдрически
координированы атомами
мышьяка, а атомы мышьяка
окружены шестью атомами никеля,
лежащими в вершинах тригональной
призмы (см. рис. 1.5). Кроме того,
каждый атом никеля имеет в
качестве ближайших соседей вдоль
оси с два других атома никеля.
Структура может быть
представлена гексагональной плотной
упаковкой атомов мышьяка, в которой все октаэдрические позиции
заняты атомами металла; октаэдры NiAs6 обобществляют грани вдоль
оси с, приводя атомы металла к сближению. По сравнению со
структурой NaCl, структура NiAs имеет меньшую константу Маде-
лунга (1,748 и 1,693 соответственно). В соединениях АВ со
структурой NiAs связь А—В является достаточно ковалентной и имеется
связь металл — металл между атомами А. Соединения со
структурой NiAs в основном являются металлами и характеризуются
широким разнообразием составов. Типичными примерами
соединений с этой структурой являются сульфиды, селениды, теллуриды,
фосфиды, арсениды, антимониды и висмутиды переходных металлов.
Среди менее обычных структур АВ можно упомянуть
следующие: РЬО, SnS, TIL HgS и PtS (рис. 1.6). РЬО, SnS и ТП
иллюстрируют эффект неподеленной пары электронов катиона на структуру.
Хотя неподеленную пару формально можно рассматривать как пару
электронов, занимающую 5s- или бз-орбиталь, оказывается, что
имеется выраженный направленный характер, связанный с этими
электронами, что и влияет на структуру. РЬО существует в двух
модификациях; красная разновидность (свинцовый глет) имеет
тетрагональную слоистую структуру, в которой атомы свинца занимают
вершины тетрагональной пирамиды, а атомы кислорода имеют тет-
раэдрическую координацию из атомов металла. Каждый атом
свинца несет неподеленную пару, которая направлена между двумя
атомами металлов в смежных слоях. Сине-черная модификация SnO
изоморфна красной модификации РЬО. Желтая модификация РЬО
(массикот) имеет ромбическую цепочечную структуру, состоящую
из зигзагообразных цепей — РЬ—О — РЬ —, параллельных оси b. HgS
(киноварь) состоит из спиральных цепей, закрученных вдоль оси с,
в которых каждый атом имеет линейную координацию из двух
атомов серы. Появление линейной координации в этой и других d10-
системах, по-видимому, связано с образованием сильно направлен-
Неорганические структуры
35
Рис. 1-7. Структуры соединений АВ2.
о — флюорит; б — рутил; в — кристобалит; г —
куприт.
ш'-+-
• Металл
О неметалл
PtS вследствие ян-теллеров-
ных линейных sp- или sd-гибридных
орбиталей вследствие близких
значений энергий орбиталей ns, np и
(п - l)d. PtS, PtO, PdO и PdS
кристаллизуются в тетрагональных
структурах, в которых атом металла
имеет плоскоквадратную (dsp2)
координацию, а анионы — тетраэдри-
ческую координацию. СиО
кристаллизуется в искаженной структуре
ского искажения.
Структуры АВ2. Фториды и оксиды типа АВ2, которые
являются отчетливо ионными соединениями, кристаллизуются в
структурах, определяемых размерными соображениями. Как и в структурах
АВ, именно координационная топология анионов вокруг катиона и
определяет структурную упаковку. Координация может быть 8,6
или 4-кратная, соответствующие координационные числа аниона 4,3
или 2. Таким образом, мы имеем для ионных соединений АВ2
следующие структуры (рис. 1.7): флюорит (8 : 4), рутил (6 : 3) и
кремнезем (4:2).
Флюорит (С1) состоит из кубической плотноупакованной под-
решетки катионов, в которой все тетраэдрические позиции заняты
анионами (см. рис. 1.7). Имеется очевидное родство со структурой
цинковой обманки. Анионы тетраэдрически координированы
катионами; катионы находятся в кубической координации. Эта структура
также имеет близкое родство со структурой CsCl, где анионы
образуют примитивную кубическую структуру, в которой
чередующиеся кубы заняты катионами. Структура флюорита обычно
встречается в соединениях с отношением радиусов, большим чем 0,732:
фториды больших двухвалентных катионов, таких как Са, Sr, Ba, Cd, Pb,
Hg; тройные фториды одно- и трехвалентных катионов
приблизительно равных размеров, как, например, NaYF4, KLaF4, Na2ThF6,
K2UF6; диоксиды больших четырехвалентных катионов, таких как
Zr, Hf, Ce, Pr, Tb, Po, Th, Pa, U, Pu и Am; оксофториды
трехвалентных лантаноидов; ряд интерметаллических соединений и фаз
внедрения, таких как AuAl2, SiMg2, GeMg2, CoSi2, Be2C, а также
дигидриды многих лантаноидов. Структура флюорита также
легко приспосабливается к нестехиометрии, включая как дефицит,
так и избыток анионов. Несколько нестехиометрических оксидов
и оксофторидов лантаноидов и актиноидов кристаллизуются
в структурах, родственных структуре флюорита. Антифлюоритная
структура, которую принимают многие оксиды, сульфиды, селе-
3*
36 Структура твердых тел: старые и новые аспекты
ниды и теллуриды щелочных металлов (например, Li20, Na2S
и т. п.), является производной от структуры флюорита, она
получается путем перемены местами анионов и катионов
(координация 4 : 8).
Структура рутила (Ti02) (C4) является тетрагональной,
стабильной для ионных соединений с отношением rjra < 0,73. Она состоит
из бесконечных фрагментов, образованных октаэдрами Ti06 и
связанных через противоположные ребра вдоль оси с (см. рис. 1.7).
В этой структуре кристаллизуются фториды двухвалентных
металлов (Mg, Cr, Mn, Fe, Co, Ni, Cu, Zn) и оксиды четырехвалентных
металлов (Ti, V, Сг, Mn, Nb, Та, Mo, W, Ru, Os, Ir, Sn, Pb, Те).
Как и в структуре флюорита, в этой структуре можно заменить
стехиометрически эквивалентные количества соответствующих
катионов и получить тройные соединения; так AlSb04, GaSb04,
CrNb04, FeNb04, RhV04 и другие имеют структуру рутила. Эта
структура является также основой для фаз Магнели с общей
формулой Мп0271-1, найденных в восстановленных оксидах титана
и ванадия.
Si02 встречается в следующих трех формах:
Низкотемпературный кварц (С8) Тридимит (СЮ) Кристобалит (С9)
(ниже 1143 К) (1143-1743 К) (выше 1743 К)
Различные формы кремнезема обычно состоят из тетраэдров
Si04, все вершины которых поделены. Идеальная структура кристо-
балита (см. рис. 1.7) может быть описана таким образом: атомы
кремния занимают позиции атомов углерода в структуре алмаза, а атомы
кислорода внедрены посередине между каждой парой атомов
кремния. В получившейся структуре кремний имеет тетраэдрическую,
а кислород — линейную координацию (4 : 2). BeF2 также имеет
эту структуру. В тридимите мы тоже имеем координацию 4 : 2, но
он является родственным вюртциту в точности таким же образом,
как и кристобалит родствен цинковой обманке. В кварце
тетраэдры Si04 располагаются в виде гексагональных спиралей; в
зависимости от направления спирали кварц демонстрирует /- или d-опти-
ческую активность.
Структуру куприта (Cu20) составляет объемно-центрированная
кубическая упаковка из атомов кислорода; атомы меди занимают
центры четырех из восьми кубиков, на которые может быть
разделена ОЦК-ячейка (см. рис. 1.7). В этой структуре атомы меди
имеют линейную, а атомы кислорода тетраэдрическую координацию
(2 : 4). Уникальность этой структуры среди других неорганических
материалов заключается в том, что она состоит из идентичных
Неорганические структуры
37
Рис. 1.8. Структуры соединений АВ2.
а — пирит; б — Gdl2; в — MoS2.
взаимно проникающих каркасов,
не связанных непосредственно
друг с другом.
В соединениях АВ2, где В —
галоген (иной, чем фтор) или халь-
коген (S, Se, Те,) связь А—В
является существенно ковалентной,
и вследствие этого встречаются
различные типы структур.
Некоторые важные структуры
показаны на рис. 1.8.
Структура пирита (FeS2)(C2)
состоит из молекулярных ионов
S2~ (см. рис. 1.8). Структура
имеет близкое родство со структурой
NaCl, из которой она может быть получена путем замещения атомов
Na на атомы Fe, а С1 на S|_, при этом центр иона S2~ занимает
позицию хлора. Каждый атом железа имеет октаэдрическую
координацию, но каждый атом серы окружен тетраэдрически (один атом S и
три атома Fe). В этой структуре кристаллизуются несколько халь-
когенидов переходных металлов.
Структуры Cdl2 и CdCl2 (С6 и С19) являются
близкородственными. В структуре Cdl2 атомы иода образуют гексагональную плот-
ноупакованную слоистую последовательность, в которой атомы
кадмия занимают поочередно межслоевые октаэдрические позиции
(см. рис. 1.8). Последовательность слоев имеет вид АсВ АсВ...,
где А и В обозначают слои из атомов иода, а с обозначает атомы
кадмия. CdCl2 имеет также подобную слоистую структуру, но здесь
анионные слои имеют кубическую плотную упаковку вместо
гексагональной плотной упаковки; катионы занимают чередующиеся
межслоевые октаэдрические позиции, как и в Cdl2. В этих слоистых
структурах со слабой (ван-дер-ваальсовой) межслоевой связью
кристаллизуются несколько дигалогенидов и дихалькогенидов металлов.
Более ионные из этих соединений обычно предпочитают структуру
CdCI2 (константы Маделунга для структур CdCl2 и Cdl2 равны
соответственно 4,489 и 4,383). MgCl2, MnCl2, FeCl2, NiCl2, NbS2 и TaS2
являются такими примерами твердых тел, которые обладают
структурой CdCl2. Дииодиды двухвалентных металлов и дихалькогениды
четырехвалентных металлов кристаллизуются в основном в
структуре Cdl2.
Структура молибденита (MoS2) (C7) является слоистой,
близкородственной CdCl2 и Cdl2. Каждый слой MoS2 состоит из листа, пред-
• Металл
О Неметалл
38 Структура твердых тел: старые и новые аспекты
ставленного атомами металла, который заключен, как в сэндвиче,
между двумя листами атомов неметалла (см. рис. 1.8). Эти слои
упакованы один над другим вдоль гексагональной оси с, причем
последовательность листов, составленных из атомов, имеет вид АвА
ВаВ..., где большими буквами обозначены анионы, а маленькими
буквами — катионы. В этой структуре молибден имеет тригонально-
призматическую координацию из анионов. Полагают, что эта
координация образуется за счет с^р-гибридизации орбиталей металла.
Примерами соединений, кристаллизующихся в этой структуре,
являются MoS2, MoSe2, MoTe2, WS2, WSe2, a-NbSe2 и a-TaSe2.
Структура Hgl2 (C13). также является слоистой структурой,
включающей тетраэдрическую координацию атомов ртути и
ГПУ-упаковку атомов иода.
Структуры А2В3. Кристаллические структуры, принимаемые
соединениями А2В3, разбиваются на две главные категории в
зависимости от преимущественного характера связи А—В: ионного или
ковалентного. Ионные соединения А2В3, типичные для полуторных
оксидов d-переходных металлов и лантаноидов, кристаллизуются
главным образом в двух структурах: А1203 (корунд) и С-редкс
земельный оксид. Структура корунда (тип D5,), типичная для Ti203,
V203. Сг203 и a-Fe203, состоит из гексагональной плотноупакован-
ной подрешетки анионов, в которой катионы занимают две трети
октаэдрических пустот; катионы имеют октаэдрическую
координацию, а атомы кислорода — координацию, близкую к тетраэдри-
ческой (6 : 4). Структура ильменита является производной от
структуры корунда и получается путем замены катионов в чередующихся
слоях атомами Fe и Ti. Полуторные оксиды железа и алюминия
также существуют в метастабильной у-форме, которая имеет ка-
тионодефицитную кубическую шпинельную структуру. Структура
С-редкоземельного оксида, обыкновенно характерная как для
полуторных оксидов лантаноидов и актиноидов, так и для Мп203,
является производной от структуры флюорита; она получается при
удалении 1/4 анионов, так что образуется шестикратная
координация атомов металла, но координационная геометрия не является
точно октаэдрической. Оксиды редкоземельных элементов также
кристаллизуются в двух других структурах — А (гексагональная)
и В (моноклинная) формы, в которых лантаноид-ион имеет
семерную координацию. Среди полуторных оксидов Bi203 является
уникальным, он кристаллизуется по крайней мере в четырех
полиморфных модификациях: а (моноклинная), (3 (тетрагональная), у (ОЦК)
и б (ГЦК), из которых 5-фаза, имеющая дефектную структуру
флюорита, демонстрирует высокую ионную проводимость.
Среди ковалентных структур А2В3, структура Bi2Te3 (тип СЗЗ)
состоит из плотноупакованных атомов уалькогеиа, в которых
висмут находится в октаэдрических позициях. Кроме того, Sb2S3 и
Bi2S3 кристаллизуются в цепочечной структуре.
Неорганические структуры
39
Рис. 1.9. а — структура псровскита. Без
большого атома А в центре куба структура
становится структурой кубического Rc03; б — структура
K2NiF4, состоящая из слоев каменной соли (KF)
и перовскита (KNiF3). Октаэдры NiF6 сочленены
экваториальными вершинами, ограничивая тем
самым взаимодействие Ni—F—Ni в плоскости ху.
а
Структуры АВ3. Важной
структурой АВ3, которая является базовой для
структур нескольких оксидов, фторидов и
оксофторидов металлов, является
структура Re03 (тип D09). Это кубическая
структура (рис. 1.9, а), составленная из
соединенных общими вершинами
октаэдров Re06 с линейными связями Re —
—О—Re. W03 обладает искаженной
структурой Re03, многие сложные оксиды
ниобия содержат блоки типа Re03,
простирающиеся вдоль главной оси
тетрагональной структуры. Другими соединениями,
кристаллизующимися в структуре типа
Re03, являются NbF3, TaF3, *TiOF4 и
MoOF2. Многие трифториды переходных
металлов также состоят из сетки
соединенных через вершины октаэдров AF6, но
углы А—F—А не являются линейными.
В идеальной структуре Re03 анионы
занимают 3/4 позиций упаковки типа КПУ.
Когда анионы имеют упаковку типа ГПУ, угол становится
равным 132°. Типичными примерами последних являются PdF3 и RhF3.
Другие фториды переходных металлов AF3 (A—Ti, V, Cr, Fe, Co,
Мо и Ru) обладают структурами, промежуточными между
структурами Re03 и PdF3, где угол A—F—А около 150°. Структуры Sc(OH)3
и 1п(ОН)3 родственны Re03. Пниктиды некоторых переходных
металлов АВ3 (А—Со, Ni, Rh, Pd, Ir; В—Р, As, Sb) кристаллизуются
в структуре скуттерудита (CoAs3), которая соотносится простым
образом со структурой Re03; если атомы неметалла структуры
Re03 сместить из центра ребер так, чтобы образовывались плоско-
квадратные группы В4, полученная таким образом структура и
будет структурой скуттерудита. Известны также антиструктуры Re03:
например, Cu3N. Li3N, который показывает суперионную
проводимость по литию [Huggins, Rabenau, 1978], принимает
гексагональную структуру, состоящую из чередующихся слоев Li и Li2N.
Вопреки общему мнению о том, что незначительная статистическая
заселенность ионов проводимости является необходимым условием
40 Структура твердых тел: старые и новые аспекты
для суперионной проводимости, в Li3N позиции ионов лития,
ответственных за ионную проводимость, почти полностью заполнены.
Трифториды лантаноидов и актиноидов предпочитают другую
структуру — так называемую структуру тисонита (D06). В этой
структуре атом металла имеет необычную одиннадцатикратную
(5+6)-координацию. Ковалентные галогениды АВ3
кристаллизуются в слоистых структурах (D05), которые типичны для СгС13 к Bil3;
эти структуры соответственно являются родственными структурам
CdCl2 и Cdl2.
Перовскиты. Перовскиты с общей формулой АВХ3 можно
рассматривать как производные от структуры Re03 (см. рис. 1.9, а).
Каркас ВХ3 в перовските подобен таковому в структуре Re03,
состоящей из соединенных через общие вершины октаэдров ВХ6.
Большой катион А занимает объемно-центрированную 12-координи-
рованную позицию. В идеальной кубической перовскитовои
структуре, где атомы точно касаются друг друга, расстояние В—X
равно а/2, а расстояние А—X равно у 2(а/2), где а — длина кубической
элементарной ячейки, и имеется следующее соотношение между
радиусами ионов: (i?A + Ях) = ]^2(RB + i?x)- Гольдшмидт нашел,
что структура перовскита в соединениях АВХ3 все еще
сохраняется, даже если это отношение выполняется не точно, а определяется
фактором толерантности t, равным
t = ,_ А х . (1.15)
У2(ЯВ + ЯХ)
Для идеальной перовскитовои структуры фактор t равен
единице. Однако перовскитовая структура найдена также и для более
низких значений фактора (~ 0,75 < t ^ 1,0). В таких случаях
структура искажается до тетрагональной, ромбоэдрической или
ромбической симметрии. Это искажение происходит из-за малого
размера иона А, который служит причиной такого наклонения
октаэдров ВХ6, чтобы оптимизировать связи А—X. Katz, Ward [1964]
дали альтернативное описание перовскитовои структуры в терминах
плотной упаковки ионов А и О. В этой модели перовскитовои
структуры АВ03 плотноупакованные слои А03 уложены один на другой,
а катионы В занимают октаэдрические пустоты, окруженные
атомами кислорода.
В перовскитоподобных структурах кристаллизуется большое
число перовскитовых оксидов и фторидов. Важнейшие сегнетоэлект-
рическке оксиды — BaTi03, NaNb03, KNb03, NaTa03 и КТа03 —
имеют искаженную перовскитовую структуру при обычных
температурах и переходят в кубическую при высоких температурах.
Перовскитовая структура может допускать наличие вакансий в
позициях А или X, приводящих к нестехиометрическим соединениям
Ао.^ВХд и ABX3_X. Типичными примерами являются вольфрамовые
бронзы A^W03 и браунмиллерит CaFe02)5. Перовскитоподобные
Неорганические структуры
41
оксиды показывают также нестехиометрию с избытком анионов, как
в случае LaMn03+3C, где кажущийся избыток анионов, вероятно,
вызван вакансиями La.
Ряд сложных оксидов с общей формулой А2В04
кристаллизуется в структуре K2NiF4, которая является близкородственной пе-
ровскитовой. Тетрагональную структуру K2NiF4 (рис. 1.9, б)
можно рассматривать как состоящую из перовскитовых слоев толщиной
в одну элементарную ячейку, которые упакованы один над другим
вдоль оси с. Смежные слои смещены друг относительно друга на 1/2
1/2 1/2, так что ось с тетрагональной структуры примерно в три раза
больше ребра ячейки кубического перовскита. Структура является
двумерной в том смысле, что октаэдры NiF6 связаны вершинами
только через экваториальные анионы. Оксиды со структурой K2NiF4
интенсивно исследовались Ganguly, Rao [1984], их «двумерные»
магнитные и электрические свойства довольно интересные. Факторы
толерантности для структуры K2NiF4 могут быть выражены так же,
как и для перовскитов.
Оксидные бронзы являются тройными соединениями с общей
формулой AiXByOz, где В — переходный металл, ByOz — его оксид
высшей валентности, а А — обычно электроположительный металл,
подобный щелочным металлам, и 0<.г^ 1. Оксидные бронзы
можно рассматривать как раствор металла А в матрице оксида-хозяина
Ву02. В этом классе твердых веществ хорошо известны
вольфрамовые бронзы АдЛУ03. Они проявляют интересные свойства: яркий
цвет, металлический блеск и высокую электрическую проводимость,
подобную металлам. Кроме вольфрама оксидные бронзы также
получены для таких металлов, как V, Nb, Mo и Re. Менее
электроположительные металлы, такие как лантаниды, Си, Zn, Cd, In, Tl, Sn и
Pb, могут быть также включены в бронзы, как металлы А. Со
структурной точки зрения кубические вольфрамовые бронзы являются
простейшими, их можно рассматривать как перовскиты, в которых
позиции А заполнены неполностью. В зависимости от природы иона
А и значения х вольфрамовые бронзы могут иметь гексагональную,
тетрагональную или кубическую структуру.
Шпинели. Некоторые тройные оксиды с составом АВ204 имеют
структуру минерала шпинели MgAl204; ее элементарная ячейка
содержит восемь формульных единиц (Mg8Alle032). Структура
(рис. 1.10) состоит из кубической плотноупакованной подрешетки
оксидных ионов, в которой ионы магния занимают 1/8 тетраэдри-
ческих (А) позиций, а ионы алюминия — половину октаэдрических
(В) позиций. Такое распределение ионов, представленное формулой
(A)t[B2]0O4, является так называемой «нормальной» шпинельной
структурой, прототипом которой служит MgAl204. Важная
вариация —«обращенная» шпине льная структура (B)t[AB]0O4; в этом
случае половина ионов В находится в тетраэдрических позициях,
а другая половина вместе с ионами А занимает октаэдрические
позиции. Известно также промежуточное между этими двумя экстре-
42 Структура твердых тел: старые и новые аспекты
• А
.В
о X
Рис. 1.10. Структура шпинели.
Характеристические октанты для А и В,
содержащие тетраэдрические и октаэдрические
позиции катионов в структуре, показаны
слева. ГЦК-решстки ионов в Л-нозицип
в элементарной ячейке шпинели показаны
справа.
мальными случаями распределение
катионов — полное разупорядочение
катионов по всем имеющимся 24 ка-
оксидных шпинелей наиболее распро-
: 3 шпинели А2+В2+04, где A—Mg, Мгь
тиоиным позициям. Среди
странены так называемые 2
Fe, Co, Ni, Си и Zn, а В—V, Сг, Mn, Fe, Co, Rh и т. п. Из шии-
нельных ферритов AFe204, которые имеют обращенную структуру,
состоят технически важные ферримагнитные материалы. Магнетит
Fe304 имеет обращенную шпинельную структуру с распределением
Fe?+[Fe2+Fe3+]0O4.
Пирохлоры. В этой структуре часто кристаллизуются тройные
оксиды состава А2В207. Ее можно рассматривать как производную
от структуры флюорита, из которой систематически удалена 1/8
часть анионов, так чтобы вокруг ионов В образовалась почти окта-
эдрическая координация; катионы А сохраняют кубическую
координацию, как в флюорите. Анионы имеют два типа координационного
окружения. Шесть анионов имеют тетраэдрическую координацию
типа (2А + 2В), тогда как седьмой анион имеет тетраэдрическое
окружение типа (А + ЗВ). Хорошо известными примерами этой
структуры являются соединения 3 : 4 Ln2B207, где Ln — ион
лантаноидов, а В — четырехвалентные ионы Ti, Zr, Ru и Sn. Среди
2:5 — пирохлоров Cd2Nb207 является важным сегнетоэлектри-
ческим материалом. Структура пирохлора встречается также в
некоторых аинонодефицитных соединениях, подобных РЬ2В207_л. (х <
< 1,0), где В — Ru, Re или Ir, так же, как в ТШЬ03 и Т1Та03.
Мы не обсуждали структуры интерметаллических соединений.
Существует обзор [Girgis, 1983] по систематике структур
интерметаллических соединений с классификацией их на валентные,
электронные и соединения, основанные на размерных факторах.
1.5. СИЛИКАТЫ И АЛЮМОСИЛИКАТЫ
Основным строительным элементом всех силикатов является
тетраэдр Si04. Низкое координационное ичсло и направленные
свойства связей Si—О, благодаря частичной ковалентности, делают
силикатные структуры менее плотноупакованными, чем другие
оксидные структуры. Сложность силикатных структур обусловлена
озможностью связывания тетраэдров Si04 многообразными спосо-
Силикаты и алюмосиликаты
43
Рис. 1.11. Анионы в силикатных
структурах.
а - Si04~; >5-Si207~; *-Si3Og~; 2-SieoJg~
a - цепь (siOg-) n; e — слой (si^-) n
бамрт, а также возможностью
замещения Si4+ другими ионами,
подобно А13+. В зависимости от числа
вершин (0,1, 2, 3 или 4) тетраэдра Si04,
поделенных с другими тетраэдрами,
получаются силикаты различного
типа (островные, одиночные или
двойные цепи, кольца, слои или
трехмерные сетки). Когда два тетраэдра
соединяются вместе общей
вершиной, ребром или гранью,
стабильность результирующего элемента уменьшается в отношении
1,0 : 0,58 : 0,33 вследствие электростатического отталкивания
катионов. В силикатной структуре обобществляются только
вершины. В этих силикатах в качестве противоионов выступают
различные катионы, главным образом щелочных, щелочноземельных или
переходных металлов. Ион А13+ с отношением его радиуса к
радиусу оксидного иона, равным 0,36, может находиться в четверной или
шестерной координации; замещение Si4+ катионами А13+ в
силикатах создает избыточный отрицательный заряд на анионном
каркасе, который компенсируется при введении дополнительного
положительного заряда. Эти факторы ответственны за бесчисленные
силикатные структуры (рис. 1.11).
В ортосиликатах тетраэдры SiOt не соединены общими
вершинами. Изолированные тетраэдры соединены катионами, такими как
Mg2+, Fe2+ или А13+ (которые занимают октаэдрические позиции),
или Zr4+ (который имеет восьмерную координацию). Например,
форстерит (Mg2Si04) можно рассматривать как упаковку тетраэдров
Si04 и октаэдров MgOe. Форстерит и файялит (Fe2Si04) образуют
серии непрерывных твердых растворов, называемых оливинами.
В гранатах возможно широкое замещение ионов А2+— Mg, Fe, Mn
или Са (КЧ-8) и B3+-Cr, A1 или Fe (КЧ-6). Отношение Si : О
может быть и более 4, если катион требует кислорода для образования
комплексного иона, как (А120)4+ в кианите (Al2Si05) и его
полиморфных модификациях — силлиманите и андалузите. Островные
структуры также получаются, когда два тетраэдра Si04 соединяются
через общий атом кислорода, образуя фрагмент (Si207)6", как в пи-
росиликатах.
Когда два атома кислорода каждого тетраэдра Si04 поделены
с другими, образуются как кольцевые анионы, так и линейные це-
44 Структура твердых тел: старые и новые аспекты
почечные анионы. Три тетраэдра Si04 могут образовывать
кольцевой анион (Si30())6~7 как в бенитоите (BaTiSi3Oq); шесть тетраэдров
Si04 могут образовывать гексагональный кольцевой анион
(Si6018)12~, как в берилле (Be3Al2Si6018). Анионы (Si03~)n с
линейными одиночными цепочками найдены в энстатите (MgSi03) и диоп-
сиде (MgCaSi206); эти соединения называются пироксенами. Когда
чередующиеся тетраэдры Si04 соединены двумя и тремя общими
вершинами, в образующихся анионах (Si4Oii~)n, представляющих собой
двойные цепочки, которые имеются в амфиболах, оказываются
поделенными в среднем 2,5 атомов кислорода. Типичным амфиболом
является тремолит Ca2Mg5 (Si4011)(OH)2. Амфиболы отличаются от
пироксенов наличием в первых из них ионов ОН" и F". Амфиболы
и пироксены демонстрируют направленные характеристики своих
свойств, как в асбестах, вследствие наличия цепочечных
силикатных анионов. Хотя термин «асбесты» первоначально был
использован для обозначения волокнистых амфиболов, таких как тремолит,
содержащий двойные цепи Si4On, коммерческие асбесты в основном
состоят из хризотила Mg2Si205(OH)4, который является слоистым
силикатом.
Когда общими становятся три вершины каждого тетраэдра
Si04, образуется двумерная слоистая структура с формулой
(Si205~)n- К такому семейству филлосиликатов, содержащих
непрерывные тетраэдрические слои состава Т205 (Т—Si, Al, Fe и т. п.),
относятся глинистые минералы; тетраэдрические слои связаны в
элементарной структуре с октаэдрическими слоями и с группами
координированных (или индивидуальных) катионов [Brindley,
Brown, 1980]. Ячейка, образованная путем связывания одного
октаэдрического слоя с одним тетраэдрическим слоем, обозначается как
слоистая 1 : 1, а открытая поверхность октаэдрического слоя
состоит из ОН-групп; подобное связывание может происходить на
другой стороне октаэдрического слоя, образуя слой 2:1; причем обе
поверхности такого слоя состоят из гексагональной сетки базальных
атомов кислорода (рис. 1.12). Имеются структуры, в которых слои
не являются нейтральными и баланс зарядов поддерживается
катионами. Глинистые минералы могут быть классифицированы по типу
слоя (1 : 1 или 2 : 1), заряда на слоях и природы материалов,
находящихся между слоями. На рис. 1.12 показаны структуры типичных
групп. Дальнейшее подразделение возможно на основе природы
октаэдрических слоев (ди- или триоктаэдрических) и
последовательности в упаковке.
Каолинит получается при упаковке (Si2052")n и А12(ОН)Г с
образованием Al2Si205(OH)4; здесь две трети октаэдрических пустот в
плотноупакованном слое из ионов ОН" заняты ионами А13+, что дает
диоктаэдрическую ячейку А12(ОН)4. Пирофиллит [Al2Si4O10(OH)2]2
образуется при укладке типа сэндвич слоя гиббеита А1(ОН)3 между
Силикаты и алюмосиликаты
45
i I Слой
2 1 Слой
Тетраэдре/ чес кий
Окт агэ др и ческе/й
Temp аэдрический
с tin fi = «L^L^ 9jl ~ 9',4 A
7,1-7,3 A V^V*rt I
О О О С С О О
I
9,6-10,1 А
!
i
Серпентин-каолин Тальк - пирофилл ит Слюда (ж * I
(X * (I)
Хрупкая слюда (ж * 2)
'?0 '"О ОСС
О О О О О О С)
14,4- 15,бА
I
14,0-14,4 А
Смектит(* * 0,25-0,6)
и вермикулит (ж * 0.6-0,9)
Хлорит
(ос -переменная)
4 Тетраэдрическии катион
о Октаэдрический катио/-/
(П> Межслоевой катион
о Обменньш катион
О Кислород
О Ти дроке иль нал группа
% Молекула водь/
© Кислород + гидроксил
(в проекции)
Рас. 1.12. Слои 1 : 1 и 2 : 1 в глинистых минералах и структуры типичных
представителей [Brown, 1984].
двумя силикатными слоями. Если диоктаэдрический гиббеит
заменить триоктаэдрическим бруситом Mg(0H)2, получается тальк
[Mg3Si4Oi0(OH)2]2. Если часть ионов Si4+ замещается ионами А13+,
а для поддержания зарядовой нейтральности вводятся ионы
щелочных металлов, мы получим слюды, типичным примером которых
служит мусковит [KAl3Si3O10(OH)2]2. Структуры типичных глинистых
минералов показаны на рис. 1.12.
Если поделенными являются все четыре вершины тетраэдров
Si04, получаются трехмерные сетки. Типичными примерами
являются различные формы кремнезема (кварц, тридимит и кристобалит),
рассмотренные ранее. Когда часть Si4+ замещается ионами А13+,
а баланс заряда достигается размещением больших катионов в
междоузельных позициях, получаются полевые шпаты. Такими
примерами служат альбит NaAlSi308 и ортоклаз KAlSi308. Если
большие каналы с относительно открытым каркасом заняты Na+,
К+ или Са2+, то получаются цеолиты и лазуриты. Необходимо
сделать краткое введение в цеолитные структуры ввиду их большой
важности как сорбентов и катализаторов.
46 Структура твердых тел: старые и новые аспекты
Рас. 7.75. а — содалитовая
клетка или р-клстка; б — цеолит Л;
в — содалит; г — фоязит
(цеолит X и Y).
Рис. 1.14. а — ячейка пентасила
(строительный блок ZSM-5 и ZSM-11); б —
цепочка из пентасильных фрагментов; в —
цепи связываются по очереди, образуя
о
каркас с отверствиями в 5,5 А (тстраэд-
рические позиции показаны кружочками).
Цеолиты — микропористые алюмосиликаты с общей формулой
Mx/n[(Al02)x(Si02)y]-m]i20 и могут рассматриваться как открытые
структуры кремнезема, в которых часть х!(х -\- у) тетраэдрических
позиций замещена алюминием. Получающийся отрицательный заряд
алюмосиликатного каркаса нейтрализуется пригодными для обмена
катионами М с валентностью п. Пустое пространство, которое может
быть больше 50 % объема, занято т молекулами воды па
элементарную ячейку. Строительным блоком цеолитов является усеченный
октаэдр (кубооктаэдр), показанный на рис. 1.13, а; это так
называемая р-клетка, или содалитовая клетка. Тетраэдрические атомы,
отмеченные на рис. 1.13 знаком о, находятся в углах многоугольника,
а атомы кислорода (на рисунке не показаны) — примерно в середине
между ними. В зависимости от структуры пористость цеолитов
состоит из одно-, двух- или трехмерных сеток соединяющихся каналов
и полостей молекулярных размеров. Соответственно цеолиты А
образуются путем связывания содалитовых клеток посредством двойных
четырехчленных колец, тогда как содалит образуется путем прямого
объединения по граням четырехчленных колец в соседних содалито-
Силикаты и алюмосиликаты
М
вых клетках; фоязит (цеолит X и Y) образуется путем связывания
содалитовых клеток через двойные шестичлеиные кольца
(см. рис. 1.13). В литературе описано тридцать семь цеолитовых
минералов и большое число синтетических цеолитов [Meier, Olson,
1978 J. Прогресс в химии цеолитов обусловлен работами Ваггег [1978],
который предложил общий термин «поротектосиликат» для описания
кремпийсодержащих кристаллических материалов, включая как
цеолиты, так и твердые тела, которые похожи на них кристаллической
структурой, но не обладают ионообменными свойствами.
Стехиометрия элементарной ячейки природного фоязита
описывается составом Na5J(AlO2)56(SiO2)13(5]250H2O, а стехиометрия Lin-
de А (искусственного) — {Na12[(A102)12(Si02)12]27H20}8. В цеоли-
товом каркасе позиции Si и А1 могут занимать такие элементы, как
Be, Mg, В, Ga и Р. Используя газообразный SiCl4, цеолиты можно
полностью деалюминировать с получением цеолитовых кремнеземов;
«ультрастабилизация» цеолитов является эффективной при тепловой
обработке замещенных 1\тН4+-цеолитов, где NH4+ замещены ионами
Na+. Особенно примечательным является новый класс
высококремнеземистых цеолитов с оптимальным диаметром поры 5,5 А; одним из
таких цеолитов с фантастическими каталитическими свойствами
(например, для процесса превращения метанол — бензин) является
цеолит марки ZSM-5. Он имеет формулу Hx[(A102)x(Si02)9G_x.] •
• 16Н20, и его строительным блоком является ячейка пеитасила;
ячейки пеитасила образуют цепи, которые, поочередно связываясь,
образуют трехмерный каркас с 10-членными круглыми отверстиями
диаметром 5,5 А, как показано на рис. 1.14. Упорядочение атомов
Si, A1 внутри тетраэдрического каркаса трудно определить
общепринятыми структурными методами, но можно воспользоваться правилом
Лоуэиштейна о том, что два атома А1 не могут быть смежными на
тетраэдрических позициях.
Шабазитная группа цеолитов, которая включает офретит, эрис-
нит и левин, представлялась вначале состоящей из неродственных
членов с необычным составом, однако, если признать, что тетраэдри-
ческие позиции могут быть заняты тетраэдрами А104 или Si04, легко
можно увидеть систематику в упаковочной последовательности в
этой семье, такую как в политипах (см. разд. 1.8). Например,
офретит, эрионит и левин с составами (Na2, Ca...)2Al4Si14036, (Na2,
Ca...)4)5Al9Si27072 и Ca9Al18Si36O108 соответственно (и повторяющие-
о
ся упаковочные расстояния 7,6; 15,1 и 23 А) имеют упаковочные
последовательности ААВ, ААВААС и ААВССАВВС.
48 Структура твердых тел: старые и новые аспекты
1.6. НЕСВЯЗЫВАЮЩИЕ ВЗАИМОДЕЙСТВИЯ
В ИОННЫХ КРИСТАЛЛАХ
Значения ионных радиусов, используемые в настоящее
время, основаны на представлении анион-анионного контакта. Однако,
как отмечено ранее, в реальных кристаллах не существует контакта
анион — анион (eutaxy), а возможность контакта катион — катион
в кристаллах до недавнего времени игнорировалась.
При рассмотрении кристаллических структур Si02 и других
твердых тел со структурой типа В9 показано [O'Keeffe, Hyde, 1981,
1982] существование для катионов несвязывающего радиуса,
который лимитирует катион-катионное приближение в кристаллах. Идея
несвязывающих радиусов для атомов независимо была предложена
Bartell [1968] для объяснения геометрии некоторых молекул;
например, несвязывающее расстояние Si ... Si в дисилильных эфире, амине
о
и метане является постоянным (~ 3,0G А) независимо от мостикового
атома. Брэг ранее отмечал постоянство расстояний Si ... Si в
отдельной мостиковой цепи силикатов, но значение этого факта не было
осмыслено. Теперь можно признать, что отличительной чертой
силикатных структур с простой связью является большой угол (~ 145J)
при мостиковом атоме кислорода, связывающем два тетраэдра Si04.
В высоко- и низкотемпературной формах кристобалита угол Si —
—О—Si около 145°, хотя в принципе угол может меняться от 180
до 109,5°. Первоначальная интерпретация больших углов Si—О —
—Si обращалась к dn — рл-связыванию между кремнием и
кислородом, однако тот факт, что BeF2 также приспосабливает кристобалит-
ную структуру с подобным углом, исключает это объяснение.
Реальная причина, по-видимому, состоит в несвязывающем
взаимодействии между атомами кремния; Si—О—Si и другие подобные простые
Т—X—Т мостиковые углы (Т — тетраэдрически координированный
катион, а X — анион) в «тетраэдрических» структурах определяются
несвязывающими контактами Si ... Si (T ... Т). Из исследования
141 силикатной структуры найдено [O'Keeffe, Hyde, 1979], что кон-
о
тактные расстояния Si ... Si находятся вблизи 3,06 А в большинстве
из них, что дает величину несвязывающего радиуса для кремния
о
1,53 А. Положение с несвязывающими радиусами катионов в
кристаллах становится более сложным, так как подобные несвязываю-
щие контакты, вероятно, существуют в структурах, содержащих
угловые элементы других типов, такие как Р—О—Р, Ge—О — Ge
и А1—О—Si. В смешанных случаях несвязывающие расстояния
являются суммой индивидуальных несвязывающих радиусов.
Возникает естественный вопрос, имеется ли какая-нибудь
корреляция между углами (9) Si—О—Si (Т—X—М) и длинами (I)
связей Si—О (Т—X)? Геометрически расстояние d Si ... Si (T...T) и I
связаны выражением I — (d/2) cosec (9/2). Корреляция между I и 9
Несвязывающие взаимодействия в ионных кристаллах 49
должна существовать, когда катионы находятся в контакте (d
является постоянной). Когда Э велик, контакта Si ... Si не существует
и поэтому здесь корреляции между d и I не должно быть. Если 0 <С
<С 145°, то между d и I корреляция должна существовать, указываяг
что «тетраэдрические» структуры определяются взаимодействиями
типа Т—X и Т ... Т, а не X ... X.
Несвязывающие радиусы дают нам возможность рационально
объяснить те аспекты кристаллохимии, которые не могут быть
поняты в терминах ионной модели. Например, трудно объяснить, почему
кремний является четырехкоординированным в большинстве
оксидов. Такая координация является маловероятной из-за отношения
радиусов классической ионной модели; Si4+ слишком мал, чтобы
разместить вокруг него четыре атома кислорода. В некоторых
случаях (например, в SiP207) кремний известен как шестикоордипиро-
ванный; кремний является октаэдрически координированным во
фторидах, хотя ионные радиусы О2- и F~ подобны. Причиной,
вероятно, служит то, что если координационное число кремния больше
четырех, координационное число кислорода должно быть более двух;
трехкоординационный кислород обычно требует угла Si—О—Si 120°.
о
Угол Э ^ 120° с нормальной длиной связи Si —О 1,6 А требует рас-
о
стояния d(Si...Si) ^ 2,77 А, которое намного меньше нормального
о
d(Si ... Si) 3,06 А. В SiP207, однако, присутствие высоковалентного
фосфора увеличивает отношение анион/катион, и, таким образом,
кислород остается двухкоординированным.
Поэтому структуры этого типа, по-видимому, определяются
катион-катионным несвязывающим отталкиванием, а не отношением
радиусов катиона к аниону или анион-анионным отталкиванием.
Кроме того, центрированные анионом полиэдры могут быть более
важными для решения стабильности фаз, чем обычные полиэдры,
центрированные катионом. Таким образом, различные формы Si02
переходят в стишовит (тип рутила) при высоком давлении не
потому, что тетраэдры Si04 превращаются в октаэдры SiOe при высоких
давлениях, а потому, что давление является необходимым для
превращения OSi2 в ячейки OSi3.
Обсуждалось несколько применений несвязывающих радиусов
в кристаллохимии [O'Keeffe, Hyde, 1978, 1979]. Возможно, что в
определенных структурах отношение несвязывающего радиуса
к длине связи Т—X Rll является более важным, чем отношение
ионных радиусов гс/гс. В табл. 1.6 даны значения R и I в «тетраэдриче-
ских» кислородных соединениях вместе с отношениями Rll.
В одноугловых ситуациях критические отношения для анионной
координации следующие: Rll ^ sin 18072 = 1,0 для двойной;
Rll < sin 18073 = 0,866 дляотройной; Rll < sin 109°2872 = 0,816
для четверной; Rll ^ sin 9072 = 0,707 для шестерной и Rll ^
^ 1/К 3 = 0,577 для восьмерной. Если отношение Rll в твердом теле
4 Ч. Н. Р. Рао, Дж. Гопалакришнан
50 Структура твердых тел: старые и новые аспекты
Таблица 1.6
Иесвлзывающий радиус R и длина связи с
кислородом I в тетраэдрических соединениях
и их отношение [O'Keeffe, Hyde 1981]
Атом
Li
Be
В
О
Na
Ms
Al
Si
P
S
7n
Ge
о
R, A
1,50
1,35
1,26
1,12
1,68
1,66
1,62
1,53
1,46
1,45
1,65
1,58
/, A
1,97
1,65
1,49
..._
2,37
1,95
1,77
1,64
1,55
1,50
1,98
1,77
R/l
0,76
0,82
0,85
—
0,71
0,85
0,92
0,93
0,94
0,97
0,83
0,89
неблагоприятно, становятся конкурентоспособными альтернативные
структуры. В алмазе RII равно 0,812, и поэтому он не является
стабильной модификацией; трехкоординированпый графит более
стабилен. Другой способ приспособления неблагоприятных отношений
Ril состоит в замене одноуглового соединения двуугловой связью
между полиэдрами (общее поделенное ребро центрированных
катионом полиэдров вместо поделенной вершины).
1.7. НОВЫЕ АСПЕКТЫ РАССМОТРЕНИЯ СТРУКТУР
НЕОРГАНИЧЕСКИХ ТВЕРДЫХ ТЕЛ
Описание кристаллических структур твердых тел с целью
выявления взаимосвязей различных семейств, является важным
аспектом кристаллохимии. Общепринятое описание основано на
связывании центрированных катионом координационных полиэдров
через вершины, ребра и/или грани; структура Re03 описывается
октаэдрами Re06, связанными через вершины; структура NaCl —
октаэдрами NaCle со всеми обобществленными ребрами и т. д. Такой
подход недавно был распространен [Simon, 1981, 1982] на описание
структур соединений с конденсированными металлическими
кластерами. В качестве фундаментальных структурных блоков, из которых
можно построить структуры более сложных металлокластерных
твердых тел, служат два важных октаэдрических металлических
кластера МвХ8 и МвХ12 (рис. 1.15). Конденсация кластеров МвХ8
Новые аспекты
51
Рис. 1.15. Стр>ктуры кластеров: Рис. 1.16. Структуры Ti5Te4 (a);
МвХ8(а); МвХ12(б). Gd2Cl3 (б); KMo3S3 (в), образованные
конденсацией октаэдрических
металлических кластеров [Simon, 1981].
через противоположные вершины октаэдров М6 в результате
приводит к одномерной колончатой структуре, типичным примером
которой является Ti5Te4 (рис. 1.16, а). Аналогично связывание М6-окта-
эдров кластеров М6Х8 через т/ж//с-ребра приводит к цепочной
структуре Gd2Cl3 (рис. 1.16, б); связывание М6-октаэдров блоков М6Х8
противоположными гранями приводит к одномерным фазам Шевреля
KMo3S3 и TlFe3Te3 (см. рис. 1.16, в). Кластерное соединение
NaMo406 построено из кластеров Мо6012 путем связывания Мо6-
октаэдров через mpa/fc-ребра. Структура состоит из цепей Мо406,
простирающихся параллельно тетрагональной оси с, а атомы
щелочного металла находятся между цепями (рис. 1.17). Структура NbO
сама по себе может быть рассмотрена, как построенная из
кластеров Nb6012 трехмерносвязаиных через вершины октаэдрических
кластеров Nb6—Nb6/2012/4.
Хотя приближение связанных координационных полиэдров
является полезным в кристаллохимии, оно имеет определенные
ограничения, которые создают трудность для распространения его на
менее правильные и более сложные структуры. В последние годы
появилось несколько альтернативных подходов к описанию
кристаллических структур немолекулярных твердых тел. Одним из них
является рассмотрение сложных структур как построенных из
простых блоков, которые связаны общими кристаллографическими
операциями, такими как трансляция, вращение и отражение, так же
как и прорастанием [Hyde et al., 1974; Hyde, 1979; Andersson, 1983].
Когда эти операции повторяются с регулярными интервалами, из
простых прототипных структур образуются сверхструктуры, а когда
операции осуществляются нерегулярно — это обеспечивает способ
описания «дефектных» структур. Используя операцию трансляции,
можно показать, что структуры кристаллографического сдвига
(например, W20O58) будут производными от исходного Re03 и рутило-
вых структур [Magneli, 1978], и мы обсудим это далее в гл. 5.
Операция трансляции может быть использована для получения структур
сплавов, например, структур СиА12 и Zr4Al3 из структуры (3—W
4*
52 Структура твердых тел: старые и новые аспекты
Рис. 1.17. Структура NaMo406 [McCarley, 1982].
(или Cr3Si) [Donxing et al., 1983]. На рис. 1.18 мы видим
структуру Cr3Si, состоящую из рядов атомов Сг с атомами Si в междоузлиях
(основной ячейкой структуры является тетраэдрическая звезда,
образованная пятью неправильными тетраэдрами), и структуру Zr4Al3.
Структуры боридов переходных металлов также связаны
трансляционной операцией.
Используя операцию вращения, из каркаса Re03 можно
получить структуры как тетрагональной вольфрамовой бронзы, так и
Mo5Oj,, причем операция превращает восемь квадратных туннелей
вокруг каждой колонны в четыре пентагональных и четыре триго-
нальных туннеля [Hyde et al., 1974] (рис. 1.19). Структура W5Si3
связывается с Cr3Si при помощи такого вращения [Andersson, 1983].
Показано [Hyde et al., 1980; Parthe, 1981], как операция отражения
{двойникования) применима к описанию структур большого числа
твердых тел, таких как металлические сплавы, карбиды, бориды,
оксиды, сульфиды металлов и силикаты. В двойниковых кристаллах
плоскость двойникования зеркальная, она связывает
индивидуальные двойники. Операция отражения, являющаяся результатом
двойникования, может быть легко понята в случае мотивов плотноуиако-
ванных атомов. Например, отражающее двойникование в мотиве ГПУ
атомов приводит к новому типу междоузлий (тригональные призмы)
в плоскости двойникования (1122), как показано на рис. 1.20; если
Новые аспекты
53
■mwwm
W0bhm
.г*
Рис. 1.18. а — структура Cr3Si; томные Рис. 1.19. Превращение каркаса Rc03
и открытые кружочки — Сг в позици- (слева) в каркас ТТВ (справа) опера-
ях 1/2 и 0 соответственно; двойные цией упорядоченного поворота [Hyde,
кружки — два Сг (1/4 и 3/4). Si — О 1979].
или 1/2, где тетраэдрические звезды
имеют общие вершины; б — структура
Zr4Al3, спроектированная на плоскость
1110]; открытые и темные кружки — эти позиции занимают
различаемы в позициях 0 и 1/2; двойные круж- ные атомы, плоскость
двойники (алюминий)-атомы в позициях 4/4 кования становится составной,
и 3/4. Атомы алюминия располагаются « « т
также в общих вершинах тетраэдриче- в неи встречаются две двоини-
ских звезд. Оставшиеся атомы — Zr ковые индивидуальности. По-
[Andersson, 1978]. вторяя операции двойникова-
ния, можно генерировать
новые семейства сверхструктур. Например, Fe3C (цементит)
является структурой, основанной на двойниковапии ГПУ;
структуры Fe5C2, Pd5B2 и несколько сплавов редких земель родственны
Fe3C. Такие двойниковые структуры наблюдаются в LuFe03,
дефицитных (по А-позициям) перовскитах, СаТа206 и LaNb3On.
Концепция двойникования может быть распространена до «многократного
двойникования»1; такой силикат, как паулингит, как было
показано, получен действием операции «многократного двойникования» на
жисмондин [Anderson, Faith, 1983].
Другими идеями, полезными для описания сложных
кристаллических структур, являются плоские сетки и упаковка стержней.
Метод плоских сеток, детально исследованный O'Keeffe, Hyde
[1980], заключается в рассмотрении сложных кристаллических
структур как упаковок двумерных плоских сеток атомов. Сетки
не нуждаются в плотной (eutactic) упаковке. В приведенной выше
работе систематически были получены все двумерные периодические
сетки, даны примеры их распространения в соединениях
установленной структуры, обсуждены переходы между сетками и приведены
примеры. Одной из простейших плоских сеток является сетка
типа З6; такое обозначение указывает, что в любой точке сетки
сходятся шесть треугольников. Другой родственной сеткой является так
называемая сетка Кагомэ 3.6.3.6 (рис. 1.21). Сетка З6 может быть
трансформирована в сетку 3.6.3.6 путем вращения треугольных
Многокомпонентные двойники*
ГА Структура твердых тел: старые и новые аспекты
Рис. 1.20. Фрагмент ГПУ-атомов, спроектированный на
плоскость (1100), показывающий отражение двошгикова-
ния на плоскость (1122) [Hyde et al., 1980].
Рис. 1.21. а — сетка З6; б — сетка Кагомэ 3.6.3.6 [O'Keef-
fe, Hyde, 1980].
групп атомов. Сетке Кагомэ соответствуют анионные слои {111}
структуры Re03. В этой структуре упаковка сеток такая же, как
в кубической плотной упаковке ABC.... Такая последовательность
сеток Кагомэ может быть трансформирована в гексагональную
плотную упаковку АВАВ... или примитивную гексагональную
упаковку AAA, или ВВВ, или в смесь двух типов путем простого
преобразования сетки 3.6.3.6 в сетку 3<J. Такое превращение К11У -«->- ГПУ,
которое сохраняет вершинное связывание октаэдров, будет точно
соответствовать превращению структуры ReO:j в структуру PdF3,
как впервые было отмечено Jack, Guttmann [1951]. Это превращение
элегантно описывается как поворот чередующихся октаэдров на
+ 30° вокруг тригоналыюй оси (рис. 1.22). Топотактическое
превращение гексагонального LiNb03 в кубический HNb03 [Rice, Jackel,
1982] означает, что механизм на самом деле является
действительным в реальных химических системах.
Некоторые кристаллические структуры можно рассматривать
как структуры, построенные из плотноупакованных цилиндрических
стержней полиэдров или нитей атомов, как показано на рис. 1.23
[O'Keeffe, Andersson, 1977]. Гексагональная сотообразная упаковка
цилиндрических стержней соответствует упаковке с плотностью
0,9069. В квадратной упаковке стержней, дающей тетрагональную
ячейку, плотность упаковки равна 0,7854; пример такой упаковки
найден в структуре СиС12-2Н20, которая состоит из нитей атомов
меди вдоль [001 ] с приблизительно квадратными группами,
расположенными перпендикулярно к нитям. Такую же плотность
упаковки, как и в случае прямоугольной упаковки, дает двухслойная
упаковка стержней, в которой стержни не параллельны (рис. 1.23,
в); примером структуры, соответствующей такой упаковке, является
PtO. Упаковка стержней, показанная на рис. 1.23, г и обладающая
примитивной кубической симметрией, имеет плотность 0,589; этому
типу упаковки соответствуют структуры |3— W (A15) и Cr3Si.
Для изучения электронной структуры твердых тел, включая
халькогениды молибдена и твердые тела со структурой ThCr2Si2,
были успешно использованы полуэмпирические расчеты по методу-
Новые аспекты
55
Рас. 1.22. Превращение октаэдриче-
ского каркаса Ио03 (слела) в каркас
PdF3 (справа). Стрелками показаны
мысленные вращения октаэдров
[О'КесПе, Hyde, 1980].
Рис. 1.23. Стержневые упаковки.
а — гексагональная (сотообразнан); б — тетрагональная; в — тетрагональная слоистая; г —
примитивная кубическая стержневая lO'Keeffe, Andersson, 1977].
молекулярных орбиталей в рамках расширенного метода. Хюккеля
(РМХ) IWhangbo et al., 1980; Hughbanks, Hoffmann, 1983;
Hoffmann, Zheng, 1985J. Идеи молекулярного РМХ были применены
к простым неорганическим твердым телам для объяснения
структурных взаимосвязей [Burdetl et al., 1981, 1982J. В качестве
представительного фрагмента структуры твердого тела в расчетах РМХ
рассмотрена типичная ячейка IBurdett, 1980, 1982]. С использованием
складчатого кольца А3Х3Нб в качестве фрагмента предсказаны
структуры нескольких тетраэдрически координированных твердых
тел АХ с контактами XX, АХ и АА в системах, содержащих 7, 8 и
9 электронов на ячейку АХ. Предсказания подтвердились в
структурах ковеллина (Си^-часть) и 8-электронных вюртцитов и
сфалеритов, таких как ZnS, A1N и LiGa02. Последовательность заселен-
ностей связывающих орбиталей, рассчитанная для фрагмента, имеет
такой же порядок, как и длины связей в этих твердых телах.
Складчатый лист энергетически предпочтительнее плоского аналога,
поскольку различие электроотрицательностей между А и X в кольце
А3Х3 возрастает.
По аналогии с молекулярными системами искажения структур
АХ и АХ2 объяснены как отклик на присутствие избыточных
электронов на разрыхляющих орбиталях [Burdett et al., 1981, 1982].
Например, симметричная структура каменной соли, стабильная для
8 электронов на ячейку АХ, искажается до менее симметричных
структур из-за разрыва связи, когда добавляется больше
электронов; в GeP (9 электронов) у каждого атома разрывается одна связь,
а в GeS и GeTe (10 электронов) — три взаимно перпендикулярные
связи вокруг каждого атома. Аналогично структура красного РЬО
рассматривается как искаженная версия CsGl, поскольку на свинце
находится неподеленная пара электронов. Структуры твердых тел
АХ2, СаС2 ->- FeS2 (пирит и марказит) -> CaF2 (флюорит), ТЮ2 (ру-
56 Структура твердых тел: старые и новые аспекты
тил) —>- РЬС]2 -> XeF2 могут быть поняты как реакция на добавление
избыточных электронов в симметричную структуру СаС2, которая
имеет 10 электронов на ячейку СаС2. Фрагменты Х2 при
дополнительных электронах сначала наклоняются, образуя структуры
пирита и марказита. С 16 электронами на ячейку АХ2 связь Х2
разрывается, формируя структуры, содержащие изолированные атомы X
(флюорит и рутил). В конечном итоге при наличии 22 электронов
образуется структура молекулярного XeF2 путем разрыва шести из
восьми связей АХ флюоритовой структуры. Расчеты по методу РМХ
также дают диаграмму, подобную той, которая получается при
построении зависимости главного квантового числа от разницы элекгро-
отрицательности, показывая возможность существования структур
твердых тел типа AmXn с КЧ ~ 4, 6 и 8 [Burdett, Rosenthal, 1980].
Координационное число определяется как несвязывающими
отталкиваниями X — X, так и стабилизирующими взаимодействиями.
Метод МО (ковалентный анализ), кажется, будет альтернативным по
отношению к ионной модели. Burdett, Lee [1985 J
продемонстрировали зависимость структур твердых тел от числа валентных
электронов, для этого был использован метод моментов.
1.8. ПОЛИТИПИЗМ
Некоторые твердые тела кристаллизуются в более чем одной
модификациях, химически и кристаллографически подобных, но
отличающихся одним из размеров элементарной ячейки (обычно
параметром с). Это явление, которое можно рассматривать как
полиморфизм в одном измерении, называется политипизмом [Verma,
Trigunayat, 1974; Rao, Rao, 1978; Krishna, 1983]. Политипизм
обычно демонстрируют твердые тела, имеющие плотноупакованные и
слоистые структуры, в которых первичная координация вокруг
атома осуществляется более чем одним способом, как в кубической, в
противовес гексагональной плотной упаковке. Поэтому политипы
вещества имеют одинаковое первичное координационное окружение.
Политипы составлены структурными ячейками, идентичными в двух
измерениях (слоях), эти ячейки упакованы друг над другом. Из-за
структурной идентичности составляющих слоев политипы данного
твердого тела имеют одинаковые размеры элементарной ячейки в
плоскости слоя, но различные в направлении, перпендикулярном
слоям. Другое следствие идентичности слоев заключается в том, что
меняющиеся размеры элементарных ячеек всех политипов
отдельного вещества кратны основному повторяющемуся расстоянию,
которое соответствует толщине одного слоя.
Хотя политипизм структурно подобен полиморфизму, с точки
зрения термодинамики это различные явления. В то время как
полиморфные изменения во многих случаях аналогичны изменениям при
Политипизм
57
переходе от твердого состояния к жидкому или от жидкого к
газообразному, описываемому правилом фаз Гиббса и уравнением Клау-
зиуса — Клапейрона (см. гл. 4), политипизм не поддается такому
термодинамическому описанию. Различные политипы вещества
обычно образуются при одинаковых термодинамических условиях, часто
сосуществуют в одном синтезе. Известны политипы, имеющие
гигантские размеры элементарной ячейки (несколько тысяч ангстрем)
вдоль одной оси, где дальний порядок распространяется на большие
расстояния.
Политипизм был открыт Баумхауером в 1912 г. на примере SiC.
SiC существует в двух полиморфных формах: а — с гексагональной
и р — с кубической структурой. Форма а имеет огромное число
политипов (около 130). Гексагональный параметр с в этих политипах
о о
варьирует от 5 А в двухслойном (2Н) до ~ 15 А в наиболее общем
о
шестислойном (6Н) и до ~ 12000 А в 4()80-слойпом политипе. В
настоящее время известно, что политипизм проявляется в различных
неорганических твердых телах, таких как ZnS, Cdl2, РЫ2, слоистых
дихалькогеиидах переходных металлов, слоистых силикатах и
некоторых перовскитах АВХ3. В Cdl2 каждый сэндвич I—Gd—I имеет
о
толщину 3,42 А; два таких сэндвича составляют простейший (2Н)
о
политип. Параметр с в политипах Cdl2 меняется от 6,84 А в случае
2Н до ~ 410 А в случае 120 R. РЫ2, кристализующийся в той же
структуре, что и Cdl2, существует в нескольких политипах
(около 40). Слоистые дихалькогениды переходных металлов МХ2 (М —
— Nb, Та, Мо; X—S, Se) кристаллизуются в гексагональной или
ромбоэдрической структуре, содержащей сэндвичи X—М—X.
Координация атомов М или октаэдрическая (АвС), как в структуре Cdl2,
или тригоналыю-призматическая (АвА), как в структуре MoS2.
Сэндвичи X—М—X, упаковываясь различными способами, дают
политипы с чисто октаэдрической (IT), чисто тригонально-призматической
(2Н, 3R, 4НС) или смешанной координацией (4Н,}, 6R); некоторые
из них показаны на рис. 1.24. Октаэдрическая форма (IT) обычно
стабильна при высоких температурах и может быть сохранена при
комнатной температуре путем закалки от температуры выше 1100 К
в атмосфере халькогена.
Политипизм в слоистых силикатах хорошо описан. Среди слюд
известно более чем 19 политипов. Например, повторяющаяся ячейка
в мусковитовой слюде [KAl2(OH)2(Si3A])O10] состоит из листа октаэд-
рически координированных ионов алюминия, сэндвичеобразно
заключенного между двумя идентичными листами, образованными из
тетраэдров (Si, A1)04, причем большие ионы К+ локализованы в
межслоевых позициях. Поверхностные атомы кислорода тетраэдров
в индивидуальных слоях обладают псевдогексагональной
симметрией, которая позволяет индивидуальным составным слоям быть
58 Структура твердых тел: старые и новые аспекты
6^-6
АСЬА АСоВАЬСВсА АСоСВсВАЬА CqCBqBC CoCAcAC
Рис. 1.24. Политипы дихалькогснидов
переходных металлов (сечения 1120); большие кружки —
халькоген; малые кружки — металл.
ориентированными одним из шести возможных способов. Тот факт,
что все ориентации являются эквивалентными, по-видимому,
ответствен за проявление политипизма в слюдах.
Известно, что политипизм демонстрируют некоторые оксиды
АВ03, где А — большой катион, подобный Ва, а В — маленький
катион d-переходной серии. Эти оксиды состоят из плотноупаковаи-
ных упорядоченных слоев А03, которые упаковываются друг над
другом, а катионы В занимают все межслоевые кислородные
октаэдры. Упаковка слоя А03 в структуру может быть кубической (К)
или гексагональной (Г) в зависимости от двух смежных слоев,
которые образуют в середине последовательность ABA или ABC. Если
упаковка исключительно кубическая, октаэдры с катионом В делят
только вершины в трех измерениях, образуя перовскитовую (ЗС)
структуру (рис. 1.25). Если упаковка полностью гексагональная,
октаэдры с катионом В делят противоположные грани, образуя цепи
вдоль оси с, как в BaNi03 (2H). Между двумя крайними случаями
может быть несколько политипных структур, состоящих из
смешанных, кубических и гексагональных упаковок слоев А03; например,
политипы 6Н и 4Н имеют последовательность упаковки ККГ ККГ
и КГКГ соответственно. Типичными оксидами АВ03,
показывающими политипизм, являются ВаСгОд, ВаМп03 и BaRu03.
Одной из интересных особенностей политипов является наличие
различных упаковочных последовательностей, особенно в политипах
с большими периодами. Такое сосуществование различных
политипов, казалось бы, противоречит правилу фаз Гиббса. Разница в
Органические кристаллические структуры 59
Рис. 1.25. Соединение октаэдров В06
в псровскитовые политипы АВ03.
а — ЗС; б — 2Н; в — 6Н; г — 4Н; д — 9R.
энергиях между политипными
формами твердых тел мала, и пе- а б
реходы между политипами
следовало бы поэтому связать с
небольшими изменениями в
энтальпии в отличие от внутриполитип-
ных переходов. Несмотря на
широкие исследования политипов
различных твердых веществ, это
явление до сих пор остается
далеким от понимания. Для толкования политипизма были развиты
некоторые объяснения IRao, Rao, 1978; Krishna, 1983]. Основной
вопрсс, однако, остается: какие силы ответственны за дальний
порядок на больших расстояниях, которые вызывают в политипах
специфические упаковочные последовательности и гигантские
элементарные ячейки?
Была найдена удобная аналогия между одномерными изингов-
скими цепочками спинов 1/2 и политипами. На основании такой
аналогии объяснено образование различного рода иолитипов [Ra-
mascsha, Rao, 1977; Uppal et al., 1980]. Для объяснения
существования большого числа политипов данного вещества с успехом была
использована [Ramasesha, 1984] модель конкурирующих
взаимодействий, или анизотропная модель Изинга второй
координационной сферы, впервые описанная в [Elliott, 19(51]. Исследование
[Ramasesha, Rao, 1977], где использован термин «взаимодействие
бесконечного порядка» (помимо термина ближнего порядка), наводит
на мысль, что за дальний порядок в политипах могут быть
ответственны упругие силы.
1.9. ОРГАНИЧЕСКИЕ КРИСТАЛЛИЧЕСКИЕ СТРУКТУРЫ
Органические твердые тела в последние 10—15 лет
привлекают большое внимание, особенно в связи с их широкими
технологическими возможностями. Важными характеристиками твердых тел
являются сверхпроводимость (квазиодномерные материалы), фотопро-
водящие свойства и фотохимические превращения в твердом
состоянии. В органических твердых телах, образованных неполярными
молекулами, когезия в твердом состоянии в основном обусловлена
силами Ван-дер-Ваальса. Ввиду относительно слабых когезионных
сил органические кристаллы как класс являются мягкими и
низкоплавкими. Неполярные алифатические углеводороды имеют
склонность кристаллизоваться в приблизительно плотноупакованных
LJ
60 Структура твердых тел: старые и новые аспекты
структурах, вследствие ненаправленного характера ван-дер-вааль-
совых сил. Например, метай при температуре выше 22 К
кристаллизуется в кубической плотноупакованной структуре, в которой
молекулы показывают значительное вращение. Межмолекулярное
о
расстояние С—С, равное 4,1 А, подобно ван-дер-ваальсовым связям,
о о
присутствующим в криптоне (3,82 А) и ксеноне (4,0 А). Такие плот-
ноупакованные структуры не найдены в молекулярных кристаллах
полярных молекул.
В веществах с неполярными ароматическими молекулами, таких
как бензол, нафталин и антрацен, когезия почти полностью
обусловлена ван-дер-ваальсовым взаимодействием, однако энергии когезии
существенно больше, чем энергии в алифатических неполярных
молекулах. Повышенная энергия сцепления обусловливается более
высокой поляризуемостью л-облаков. Расположение молекул в этих
ароматических твердых телах диктуется плоской геометрией
молекул. Например, бензол кристаллизуется в ромбической структуре,
в которой позиции вершин и центров граней элементарной ячейки
заняты молекулами. Нафталин и антрацен кристаллизуются в
моноклинных структурах, в которых молекулы располагаются так, что
их длинная ось лежит почти параллельно направлению Z.
Для обоих кристаллов параметры а и Ь элементарных ячеек
одинаковые, но в антрацене ось с настолько длиннее, насколько это
необходимо для приспособления большой молекулы антрацена. Во
всех этих трех ароматических кристаллах несвязывающее расстоя-
о
ние С—С равно ~ 3,7 A. Robertson [1951] кристаллические
ароматические твердые тела разделил на три категории. В типе А,
примерами которого являются бензол, нафталин и антрацен, соседние
молекулы перекрываются только слегка. В кристаллах типа В
имеется значительное перекрывание между соседними молекулами.
Тип В далее подразделяется на Вх и В2; в типе Bt перекрывающиеся
молекулы укладываются в бесконечные стопки вдоль одной из
кристаллографических осей, как, например, 9-замещенные антрацены.
В категории В2 перекрывающиеся молекулы существуют парами,
причем каждая пара занимает точку решетки, как в кристаллах
пирена.
Хотя долгое время признавалось, что кристаллические
структуры неполярных молекул являются более или менее искаженными
вариантами плотных упаковок, лишь недавно было предложено
систематическое формулирование принципов органических
кристаллических структур IKitaigorodsky, 1973]. Основная идея Kitaigo-
rodsky проста и происходит из факта, что органические молекулы
не точно сферические по своей форме и отсюда их упаковка в
кристаллах не может быть идеальной ГПУ и КПУ. Если бы мы приняли
приближенно органическую молекулу как сферу, то определенные
части молекулы выдавались бы из воображаемой сферы в некоторых
Органические кристаллические структуры 61
Рис. 1.26. Структуры кристаллов иода (а) и бензола (б).
направлениях. Kitaigorodsky отметил, что выступы одной молекулы
будут укладываться в углубления другой (принцип ключ —замок).
Хотя принцип формируется просто, он имеет далеко идущие
следствия, относящиеся к кристаллографии органических твердых тел.
Применение этого принципа, например, показывает, что гз 230
пространственных групп только 13 — наиболее возможные кандидаты
для молекулярных кристаллов, составленных молекулами
неправильной формы. Принцип иллюстрируется структурами галогенов
и бензола (рис. 1.26). При низких температурах хлор, бром и иод
имеют одинаковую ромбическую структуру. Структура состоит из
цепей двухатомных молекул, связанных двойной винтовой осью,
образующих слои. Эти слои упакованы друг над другом, так что
выступы одного слоя подгоняются под углубления другого.
Аналогично в ромбической структуре бензола молекулы в плоскости ху
упакованы вдоль направления Z таким образом, что они не подходят
друг к другу в соседних слоях. Приближение Kitaigorodsky было
полезным при расчете энергии решетки и молекулярных конформа-
ций, как отмечено ранее (см. разд. 1.3). Этим методом были
вычислены константы элементарной ячейки кристаллов бензола с ошибкой
3 % [Mason, Rae, 1968]. Вычисленная энергия решетки
52,3 кДж-моль-1 при 270 К удовлетворительно сравнима с
энтальпией сублимации.
Упаковка молекул во многих органических кристаллах
определяется межмолекулярными водородными связями, причем молекулы
располагаются в кристаллах так, чтобы облегчить образование
водородной связи. Например, метанол кристаллизуется в ромбической
62 Структура твердых тел: старые и новые аспекты
Ось с
Рис. 1.27. Структуры комплексов с переносом
заряда (КПЗ).
а — TMPD — TCNB; б — М2Р — TCNQ.
структуре, содержащей протяженные цепи (вдоль оси с) молекул
СН3ОН, связанных водородными связями, причем каждый кислород
вовлечен в две водородные связи. Эти цепи соединены поперечно
силами Ван-дер-Ваальса. Другим типичным примером органического
кристалла, в котором когезия в основном обеспечивается
водородными связями, является щавелевая кислота. В а- и р-формах
индивидуальные молекулы имеют плоскую трбше-конфигурацию и
образуют протяженные цепи.
Комплексы с переносом заряда (КПЗ). Этот класс органических
твердых тел, образованных двумя различными молекулами, одна
из которых действует как донор (D), а другая — как акцептор (А)
электронов, показывает интересные электронные свойства в твердом
состоянии ISoos, Klein, 1976]. Молекулы D и А в этих твердых
телах упакованы в бесконечные стопки; упаковка может быть
смешанной... DAD А..., как в КПЗ между тетраметил-р-фенилендиамином
(TMPD) и тетрацианобензолом (TCNB), или образованной
отдельными правильными стопками ...DDDD... и ...АААА..., как,
например, в комплексе 5,10-дигидро-5,10-диметилфеназин(М2Р)-
тетрациано-р-хинодиметана (TCNQ) (рис. 1.27). Эти примеры КПЗ
относятся к категории комплексов 1 : 1, в которых происходит
полный перенос электрона от D к А, с образованием ион-радикальных
солей. Имеется несколько комплексов, в которых отношение донора
к акцептору разное — 1 : 2, 2 : 3 и т. д. Из них во многих
взаимодействие с переносом заряда слабое, образующее только
нейтральные молекулярные комплексы. Семейство комплексов тетратиофуль-
вален (TTF)—TCNQ в последние годы привлекает интерес химиков
и физиков [Torrance, 1979] из-за их необычно высокой металлопо-
добной проводимости вплоть до GO К. Структура этого комплекса
состоит из отдельных однородных стопок доноров и акцепторов, и
энергия когезии твердого тела имеет значительный вклад кулоновско-
Соединения включения и клатраты 63
го взаимодействия, которое зависит от степени переноса заряда.
Соль с переносом заряда, образованная между гексаметилентетрасэ-
ленафульвалепом (HMTSF) и TCNQ, показывает металлическую
проводимость вплоть до очень низких температур (структуры
молекул D и А см. ниже).
NC CN
TCNQ
LX.J
000 COCO
1.10. СОЕДИНЕНИЯ ВКЛЮЧЕНИЯ И КЛАТРАТЫ
Имеются твердые соединения со структурой хозяина,
которая размещает определенный тип гостей [Atwood et al., 1984].
Хозяином обычно служит кристалл с большими открытыми
плоскостями, внутри которых могут быть размещены гости. В качестве гестей
могут быть атомы, ионы или молекулы. В зависимости от того,
находятся ли гости в туннелях, межслоевом пространстве или в клетках
хозяина, соединения включения могут быть классифицированы как
одно-, дву- или трехмерные. Типичными примерами соединений
включения являются газовые гидраты, клатраты fi-гидрохинона и
соединения включения мочевины и тиомочевины. Когда вода
затвердевает в присутствии газов, подобно галогенам и благородным
газам, летучий компонент захватывается в клеткоподобной структуре,
образуемой молекулами воды. Такие газовые гидраты бывают двух
типов I и П. Типичным примером газового гидрата I типа является
С12'7-т-Н20, впервые выделенный сэром Хэмфри Деви. Это — к у-
о
бическая структура (а ~ 12 А), состоящая из сшитой водородными
связями сетки, образованной 40 молекулами воды, содержит две
маленькие и шесть больших полостей. Большие полости заполнены
полностью, а маленькие — заполнены частично молекулами хлора.
64 Структура твердых тел: старые и новые аспекты
Рас. 1.28. Пентагонододекаэдр и сечение слоя
связанного пентагонододскаэдра додскасила — ЗС; показана
также элементарная ячейка [Gies et al., 1982].
В структуре типа II кубическая элементарная ячейка имеет размеры
о
~ 17 А со 136 молекулами воды и 16 маленькими и 8 большими
полостями. В гидрохиноновых клатратах молекулы, подобно N2, HC1
и НСООН, захватываются в доступные пустоты каркаса хозяина,
связанного водородными связями; молекулы гостей в стуктуре
относительно свободно вращаются. В соединениях включения на основе
мочевины и тиомочевины молекулы гостей размещаются в каналах
хозяйской структуры. Соединения включения мочевины с
молекулами-гостями, такими как тг-углеводороды С10—С50, спирты, кислоты
и сложные эфиры с нормальной неразветвленной цепью, кристал-
о
лизуются в гексагональной структуре с а ~ 8,2 и с ~ 11 А. На
элементарную ячейку приходится шесть молекул мочевины,
образующих взаимопроникающую винтовую спираль. Каждый кислород
связан водородными связями с четырьмя атомами азота, а каждый
азот — с двумя атомами кислорода. Структура обеспечивает круг-
о
лые каналы с диаметром ~ 5 А, параллельные оси с, которые
заняты молекулами гостей. Интересно, что мочевина сама по себе
кристаллизуется в другой (тетрагональной) структуре и переходит в
гексагональную структуру, когда происходит включение. Кроме того,
когда молекулы гостей удаляются, то гексагональная структура
разрушается. Соединения включения тиомочевины аналогичны, но
больший диаметр канала позволяет размещать молекулы большего
диаметра, такие как разветвленные цепочечные углеводороды.
Уже давно известно, что подобную тенденцию к образованию
тетраэдрически связанных каркасных структур проявляют Si02 и
Н20 [Kamb, 1965]. Минерал меланофлогиш изоструктурен
кубическому газовому гидрату I, в то время как синтетический клашрасил-
додекасил-ЗС изоструктурен кубическому газовому гидрату II IGies
et al., 1981, 1982]. На рис. 1.28 показано сечение додекасила — ЗС.
Кроме молекулярных клатратов, в которых связывание между
хозяином и гостем слабое (ван-дер-ваальсового типа), известны неко-
Соединения включения и клатраты
65
торые металлические клатраты, в которых взаимодействие хозяин —
гость сильнее. К этой категории принадлежат клатратные соли
окиси серебра [Ag708]+X~(X — N03, C104, BF4). Эти соединения,
приготовленные электролитическим окислением кислых растворов
солей серебра, являются черными кристаллами с металлическим
блеском; они кристаллизуются в довольно сложной клетчатой структуре,
состоящей из полиэдров Ag608, каждый полиэдр размещает один
ион X". Четыре таких полиэдра совмещены в одной элементарной
ячейке с одним атомом серебра в промежуточном положении. По
существу, в структуре имеются два типа серебра со средней
степенью окисления 17/7. Смешанное состояние окисления серебра
вместе с уникальной структурой делает эти вещества
металлическими и обладающими парамагнетизмом Паули.
Другим классом трехмерных соединений включения являются
бориды металлов, состоящие из кластеров бора; типичные примеры
этого класса-соединений — гексабориды щелочноземельных
металлов и лантаноидов с формулой МВ6. Структура гексаборидов
аналогична CsCl, кластеры Вв занимают вершины куба, а
большой атом М располагается в объемно-центрированной позиции.
Физические свойства гексаборидов можно понять на основе модели
связывания [Longuet-Higgins, Roberts, 1968]. В кристалле МВб
на элементарную ячейку приходятся шесть атомов бора, которые
требуют 20 электронов (6 — для образования ковалентных связей
в кластерах В6 и 14 — для связей между кластерами В6) для
заполнения валентной зоны полностью; из этих электронов 18
доставляются 6 атомами бора, а 2 дополнительных электрона должны прийти
от катионов. Гексабориды двухвалентных металлов, подобно СаВ6
(представляемые как Са2+В2~), являются диэлектриками или
полупроводниками, в то время как гексабориды трехвалентных
металлов, таких как лантаноиды, являются металлическими, поскольку
электрон от катиона занимает низшую несвязывающую
металлическую зону.
Важным классом соединений включенияг демонстрирующих
замечательные физические свойства, являются тройные халькогениды
молибдена (фазы Шевреля) с формулой Мл:Мо6Х8(М—РЬ, РЗЭ и т. д.;
X—S, Se, Те и х ~ 1) (для подробного обзора см., например [Yvon,
1979]). Структура (рис. 1.29) ромбоэдрическая, состоит из восьми
блоков Мо6Х8, которые окружены пустотами. Наибольшая пустота
имеет форму куба и находится в центре, образованном восьмью
атомами халькогена, принадлежащими различным блокам Мо6Х8.
Меньшие пустоты находятся вблизи середины ромбоэдрических векторов
элементарной ячейки. Большие атомы М, такие как РЬ, занимают
кубическую пустоту, тогда как меньшие атомы, подобно Си,
занимают другие пустоты. Интересные электронные свойства фаз Шевреля,
которые включают высокополевую, высокотемпературную
сверхпроводимость и сосуществование сверхпроводимости и магнитного
5 Ч. Н. Р. Рао, Дж. Гопалакришнан
06 Структура твердых тел: старые и новые аспекты
о 6
Рис. 1.29. Структура фаз Шсвреля МлМо„Хя.
о — строительный блок МовХ8; б — упаковка восьми блоков Мо«Хн в
ромбоэдрическую структуру. Ьубическан полость в центре запита оолъшпм
атолом М LYvon, 197 9].
порядка, обусловлены передачей электронов от атомов гостей М
к хозяину MoGX8.
В гл. 8, в которой рассматривается химия интеркаляции, мы
обсудим другие типы соединений включения, образованные
слоистыми графитом и TaS2, и другие ряды хозяев.
1.11. НЕКРИСТАЛЛИЧЕСКИЕ, ИЛИ АМОРФНЫЕ,
ТВЕРДЫЕ ТЕЛА
Хотя большая часть химии твердого тела имеет дело с
кристаллическими материалами, в последние десять — пятнадцать лет
сильно везрес интерес к некристаллическим (или аморфным)
твердым телам, вероят! о, из-за универсальности, с которой почти все
материалы могут быть переведены в аморфное состояние, а также
и из-за различных применений этих твердых тел. В то время как
термин «аморфное, или некристаллическое, твердое тело» является
сбщим термином, термин «стекло» применяется только по отношению
к аморфным твердым телам, полученным при медленном или
быстром охлаждении расплавов. Сегодня мы имеем металлические,
органические и полимерные стекла. Стекла характеризуются особым
переходом — так называемым стеклованием, которое мы обсудим
в гл. 4.
При температуре стеклования стекло плавится или застывает
в виде переохлажденной жидкости [Zallen, 1983; Elliott et al., 198(5].
В крг.сталлехимии существование дальнего порядка составляет
основу всякого обсуждения; симметрия кристаллов, зоны Бриллю-
эпа и связанные с этим аспекты кристаллов являются первостепенно
Некристаллические, или аморфные, твердые тела 67
важными в химии твердого тела из-за наличия дальнего порядка.
В некристаллическом состоянии нет дальнего порядка, но ближней
порядок сильно заметен. Хотя отсутствие дальнего порядка
затрудняет получение точной структурной информации из дифракции и
других экспериментов, мы можем многое сказать о структуре
некристаллических твердых тел. Некоторые методы, такие как EXAFS
и ЯМР под магическим углом, способны давать точную информацию
о структурных чертах этих твердых тел (см. гл. 2), но многое в
нашем понимании этих твердых тел проясняется при структурном
моделировании аморфного состояния.
Одной из ранних моделей для описания аморфного состояния
была модель непрерывной беспорядочной сетки для ковалентных
неорганических стекол [Zachariasen, 1932]. Сейчас мы различаем
три типа непрерывных беспорядочных моделей:
— непрерывная беспорядочная сетка (применима к ковалентным
стеклам);
— беспорядочная плотная упаковка (применима к
металлическим стеклам);
— модель беспорядочной спирали (полимерные стекла). Все эти
модели включают описание аморфного состояния в терминах
статистических распределений. Эти модели широко обсуждались в
современной литературе.
В модели непрерывной беспорядочной сетки (НБС) для
неорганических ковалентных стекол сделано предположение, что структура
образуется из полиэдров, таких как Si04 (в случае Si02), связанных
таким образом, что дальний порядок нарушен вследствие
разрешенной свободы вращения соседних полиэдров вокруг связующего атома
(угол Т—О—Т может принимать различные значения). Мы хотели
бы отметить, что такая структура не является истинно
беспорядочной в статистическом смысле, как подразумевается термином
беспорядочной сетки. Благодаря наличию полиэдров, в стеклах
существует хорошо установленный локальный порядок, и если полиэдры
связываются вместе, образуя, скажем, кольца, в таком случае
распределение двугранных углов для ближайших ячеек не является
одинаковым (т. е. беспорядочным), а вместо этого оказываются
предпочтительными определенные двугранные углы. На рис. 1.50 мы
показываем типичную беспорядочную сетку стекла А2В3. Модель
НБС была использована как для халькогенидов IV — V — VI, так
и для элементарных стекол Si и Ge. НБС стекол может быть
топологически одно-, двух- или трехмерной, как это подтверждается
примерами Si, As2Se3 и Si02 соответственно.
Продемонстрировано [Polk, 1971], что можно построить
протяженную непрерывную беспорядочную сетку (содержащую 440 атомов),
основанную на инвариантном координационном числе, равном
четырем, для тетраэдрически связанных аморфных полупроводников,
таких как Si и Ge. Даже когда все длины связей близки к равновес-
5*
68 Структура твердых тел: старые и новые аспекты
Рис. 1.30. Разупорядочснная каркасная
модель Захариазена для стекла А2В3.
ному значению, не производится
серьезного искажения длин связей,
если разрешено умеренное
растягивание в углах. Существенно, что в
такой непрерывной сетке имелись
5-, 6- и 7-членные кольца, хотя в
кристаллических полупроводниках
встречаются только 6-членные
кольца; кроме того, плотность
получающейся некристаллической
структуры в результате отличается от ал-
мазоподобпой структуры примерно
на 1 %. Отсюда следует вывод, что
в простых ковалентно связанных
стеклах (Si, Si02 и т. п.) ближний
порядок родственен такому же порядку, который имеется в
соответствующих кристаллах. Даже в ионном стекле, образованном
BeF2, координация 4 : 2 сохраняется.
Помимо ближнего локального порядка, характеризующегося
в ковалентных стеклах полиэдрическими элементами, существенный
интерес представляет промежуточный порядок. Возрастает
количество данных в пользу существования в структуре некоторых
ковалентных стекол (например, сплавы Ge—Se) довольно хорошо
определенных кластеров атомов, содержащих 10 или более атомов; на их
присутствие указывают данные как колебательной спектроскопии,
так и дифракционных методов.
Стеклообразные металлы и сплавы структурно могут быть
рассмотрены в терминах беспорядочной упаковки жестких сфер (БУЖС).
Понятие плотной беспорядочной упаковки фигурировало в
классической работе Bernal [1965] по структуре моноатомных жидкостей.
Он использовал шариковые подшипники, упакованные в резиновые
камеры, перемешанные и помещенные в черную краску, причем
позиции анализировались вручную и визуально. Позднее варианты
такого подхода были связаны с компьютерным моделированием
(рис. 1.31). Все эти подходы показали, что можно генерировать
беспорядочно упакованную модель, которая является достаточно
плотной, чтобы воспроизводить эксперимент (63,7 % занятости наличного
пространства по сравнению с 74,05 % в кубической или
гексагональной плотной упаковке сфер). Такая модель не включает в себя
кристаллических областей при условии, что сферы не ограничены
правильной поверхностью. Хорошо известно, что в плотнейшей
упаковке сфер (как в кристаллических металлах, таких как Ag, Au и
Pt) имеются дыры тетраэдрической и октаэдрической формы. Bernal
Некристаллические, или аморфные, твердые тела 69
Рис. 1.31. Кластер, получаемый самопроизвольно при
охлаждении капли Леннарда — Джонса из 129 атомов,
моделированный методом молекулярной динамики.
Жирными линиями показаны элементы пснтагональной
симметрии [Ноаге, Barker, 1976].
отметил, что в плотной беспорядочной упаковке модели жестких
сфер имеются дыры пяти типов; в дополнение к тетраэдрическим и
октаэдрическим дырам имеются три другие. Эти дыры являются
полиэдрами с равными треугольными гранями (т. е. дельтаэдриче-
скими объектами), которые достаточно малы, чтобы вмещать другую
сФеРУ такого же размера. Они называются каноническими дырами
Бернала. Большое число структур, обладающих почти идентичными
расстояниями между ближайшими соседями, может быть образовано
путем совместной упаковки пяти дельтаэдров. Сконструирована
[Finney, 1970] плотная, беспорядочно упакованная структура,
содержащая 7994 «атома» с эффективностью упаковки 44 %. Функция
(общего) радиального распределения для этой структуры хорошо
сравнима с таковой, определенной экспериментально для аморфного
^7вРг4> хотя соответствие для индивидуальных частных парных
корреляционных функций не является удовлетворительным.
Модель БУЖС хорошо объясняет структуру аморфных
металлических сплавов, таких как Zr—Си. Однако она неудовлетворительна
Для так называемых металлостеклооб разных сплавов, образованных
из примерно 80 % переходного металла и 20 % металлоида (В, Si,
70 Структура твердых тел: старые и новые аспекты
Рит. п.), причем это является эвтектической композицией. В этих
материалах, вероятно, существует химическое упорядочение и, как
предполагается, присутствуют кристаллические мотивы, такие как
тригонально-призматичекие блоки. Предполагается [Gaskel], 1979],
что структура стеклообразных сплавов может также содержать такие
хорошо упорядоченные локальные блоки. Таким образом
достигается отсутствие соседства металлоид — металлоид (что согласуется
с дифракционным доказательством), которое не является случаем,
обсуждаемым моделью БУЖС, где предполагается, что маленькие
атомы металлоидов, такие как Р, находятся в канонических
полиэдрических дырах в структуре и, таким образом, ничто не мешает им
быть ближайшими соседями. Степень химического упорядочения в
аморфных материалах представляет большой интерес. Методы, при
помощи которых аморфные материалы могут приспосабливаться
к большой нестехиометрии, также достойны внимания. Поскольку
не существует хорошо определенной архитектурной ячейки в
аморфном состоянии, возможна непрерывная вариация в составе. Это
обусловливает особую ситуацию, не реализующуюся в
кристаллических твердых телах, при которой существует локальное насыщение
валентных потребностей в ковалентно связанных системах или
создаются структурные группировки в ионных системах, что приводит
к электрической нейтральности без необходимости в
компенсирующих дефектах. Модель беспорядочной плотной упаковки может быть
использована для ионных стекол в ограниченной степени.
Полимерные твердые тела, такие как полистирол, очень часто
являются некристаллическими. Для описания таких твердых тел
наиболее подходит модель беспорядочной спирали. Во многих
полимерах присутствуют как кристаллические, так и аморфные области;
в таких материалах хорошо определенные спиральные области
уложены в беспорядочно скрученную матрицу.
Характеристикой аморфного состояния являются флюктуации
как состава, так и плотности и сопутствующие им флюктуации в
природе связывания. Большинство многокомпонентных оксидных
стекол состоят из заряженных ионносвязанных атомов кислорода,
а также и двухкоординированных ковалентно связанных атомов
кислорода, их относительные пропорции существенно различны.
В системе As2Se3—As/Teg присутствуют и ионные, и ковалентные
связи между подобными группами атомов. Сетки, подобные
Ge^Se^^, могут содержать жесткие и подвижные области,
собранные из мсстиковых фрагментов Sen.
Химические реакции и структурные перегруппировки в
аморфных твердых телах являются характеристическими, регулируемыми
химией этих материалов. Например, явление фотохромизма
(коммерчески используемое свойство) включает перенос заряда и реиоииза-
цию частичек галогенида серебра часто в присутствии
сенсибилизирующих ионов, таких как Си+. Координация кислорода германием
Квазикристаллы
71
изменяется как функция модифицирующего оксида в германиевом
стекле от четырех до шести и снова до четырех в областях, богатых
модифицирующим оксидом. В боратных стеклах непрерывная
вариация концентрации тетраэдрических и плоских (3-координированных)
атомов бора достигается модифицирующими оксидами. Известно,
что как сверх координированные, так и низкокоординированные
заряженные халькогепные (или пиикогенные) дефектные центры
присутствуют во всех халькогенидных (и пниктидпых) стеклах,
они играют критическую роль в транспортных и родственных
свойствах. Эти заряженые пары происходят из предпочтительной реакции
связей, которые первоначально претерпевают гомополярное
расщепление.
1.12. КВАЗИКРИСТАЛЛЫ
После недавнего открытия [Shechtman et al., 1984],
которое показало, что быстро закаленный сплав А1 — 14 % Мп
является квазикристаллическим с икосаэдрической симметрией, были
предприняты интенсивные попытки в исследовании таких сплавов [Мае-
kay, Kramer, 1985; Chattopadhyay et al., 1985] с целью выяснения,
будет ли в кристаллографии концептуальная революция.
Предполагается [Pauline, 1985], что икосаэдрическая симметрия является
результатом многократного двойникования кубических кристаллов,
однако многие характерные черты таких уникальных сплавов,
особенно их электронные дифракционные картины, все еще не полностью
объяснимы.
1.13. МОДЕЛИ И ГРАФИЧЕСКОЕ ИЗОБРАЖЕНИЕ
Важность моделей в визуализации, понимании и
иллюстрации структур твердых тел нельзя переоценить. Помимо хорошо
известных моделей заполнения пространства шарами и стержнями,
часто строят модели, специально пригодные для специфической
структурной проблемы. Особенно полезны структурные модели,
состоящие из атомов (разного цвета), от которых отходят тонкие
гибкие трубочки под разными углами связи, образующие многовершин-
ники (тетраэдрические, октаэдрические и т. д.). Это не только
полезная, но и необходимая процедура при построении моделей в
некоторых областях химии твердого тела. Например, оксидные слои,
построенные из картонных октаэдров, более эффективны для
визуализации сдвинутых и блочных структур. Клетки и каналы в
цеолитах и глинах наиболее удобно исследовать, используя стереомоде-
ли типа Дрейдинга. Непорядочные сетки в аморфных материалах
также лучше всего изучаются с помощью моделей.
72 Структура твердых тел: старые и новые аспекты
В последние годы стала привычной компьютерная графика
с цветными дисплеями, используемая не только для лучшей
визуализации сложных структур, но и для изучения необычных
структурных характеристик, дефектов и превращений, а также и
реакций. На рис. 1.32 показан кластер Naj[+ внутри содалитовой клетки
цеолита Y (компьютерная графика); кластер хорошо
приспосабливается внутри полости, ограниченной ван-дер-ваальсовой
поверхностью (сеткой) каркасных атомов. Замечательная способность
компьютерной графики, несомненно, будет использоваться и в
дальнейшем, а доступность недорогих вычислительных машин ускорит
процесс.
1.14. СОЛИТОНЫ
В последние несколько лет многие явления, такие как
дислокации, электронные структуры полиацетиленов и других твердых
тел, джозефсоновские контакты, динамика спинов и волны
зарядовой плотности в низкоразмерных твердых телах, ионная
проводимость и фазовые переходы, объясняются с помощью концепции
солитонов. Солитоны являются точными аналитическими решениями
нелинейных волновых уравнений, соответствующих колоколообраз-
Рис. 1.32. Кластер Na43+ в содалитовой ячейке цеолита Y. Справа
показаны К43+ и Rb43+ [S. Ramdas, Y. M. Thomas, Кембриджский
университет].
Введение
73
ным или ступенькообразным изменениям в переменной [Ogurtani,
1983]. Они могут двигаться через материал с постоянной амплитудой
и скоростью или оставаться стационарными; когда два солитона
сталкиваются, они не видоизменяются. Солитонная концепция,
вероятно, будет широко применяться в химии твердого тела для
объяснения разнообразных явлений, включая образование прорастаний
и других необычных структур.
2. НОВЫЕ И УЛУЧШЕННЫЕ МЕТОДЫ ХАРАКТЕРИЗАЦИИ
2.1. ВВЕДЕНИЕ
Характеризация — неотъемлемая часть всех исследований
в химии твердого тела и материаловедении. Ее различными
аспектами являются: 1) химический состав и химическая гомогенность
образца; 2) примеси, которые могут изменять свойства; 3) структура
образца, проявляющаяся в его кристалличности или иным способом,
сингония, элементарная ячейка и, где возможно (или необходимо),
атомные координаты, связь и ультраструктура; 4) природа и
концентрация несовершенств (дефектов) влияющих на свойства.
Несмотря на то что одному исследователю не по силам обеспечить
полную характеризацию данного твердого тела за любое данное время,
все же без хотя бы минимального уровня характеризации никакое
исследование не может быть начато. Границы «характеризации»
столь безбрежны, что почти все аспекты химии твердого тела могут
быть включены в ее сферу. В соответствии с рекомендациями Мате-
риаловедческого консультативного совета США «характеризация
описывает те особенности состава и структуры материала .(включая
дефекты), которые являются важными для каждого отдельного
приготовления, изучения свойств или использования и достаточны для
воспроизводства материала». Предмет характеризации дост аточно
рассматривался в литературе [Meinke, 1973; Newnham, Roy 1973;
Honig, Rao, 1981], и здесь мы изложим подробно главные ее
особенности. Достижения последних нескольких лет в методах
характеризации, особенно в решении структур, поистине велики и открыли
новые перспективы в химии твердого тела.
С начала нашего века рептгенодифракционные методы играют
главную роль в идентификации и характеризации тверд ых тел.
Наши идеи о природе связи и рабочие критерии отличия ближнего
и дальнего порядка кристаллов от аморфных веществ во многом
проистекают из ренттеновской дифракции* Несмотря на то что дифрак-
тометрия остается наиболее полезным инструментом для получения
структурной информации, усредненной по большому числу элемен-
74 Новые и улучшенные методы характеризации
тарных ячеек, она имеет некоторые ограничения. Следовательно,
необходимы новые, более мощные методы, и некоторые из них
сейчас доступны. Несомненно, будут продолжены усилия по разработке
более новых и лучших методов для характеризации твердых тел.
2.2. СТРУКТУРНАЯ ХАРАКТЕРИЗАЦИЯ
Различные доступные методы характеризации структуры
могут быть разбиты на следующие группы: оптические,
дифракционные, электронно-микроскопические, спектроскопические и другие
методы, использующие различные физические свойства как
инструменты характеризации (табл. 2.1). Оптические методы включают
исследование вещества под оптическим (поляризационным)
микроскопом, измерение показателей преломления, определение
топографии поверхности. Такие исследования обеспечивают ценную
информацию о морфологии, симметрии кристалла, физических дефектах,
магнитных и электрических доменах и т. д. Детальное определение
оптических индикатрис и показателей преломления может
обеспечить информацию о том, являются ли кристаллы оптически
изотропными (кубические или аморфные твердые тела), одноосными
(тетрагональная, гексагональная или ромбоэдрическая сингония) или дву-
осиыми (ромбическая, моноклинная и триклипная сингония).
Оптическое изучение травленых поверхностей обеспечивает информацию
о внутренних поверхностях и линейных дефектах. Выходящие
дислокации показывают ямки травления на поверхности, и таким образом
обеспечивается стандартный метод определения плотности
дислокаций в металлах. Симметрии ямок травления дают дополнительную
информацию о симметрии кристалла и его ориентации, поскольку
они могут быть расклассифицировании по десяти двумерным
точечным группам. Сегнетоэлектрические домены травятся с различными
скоростями в зависимости от ориентации вектора поляризации по
отношению к поверхности. Ферромагнитные домены могут быть
визуализированы декорированием поверхности. Информация о форме
частиц и распределении их размера в поликристаллах также может
быть получена из оптических исследований. Спектроскопические
методы обеспечивают различного рода информацию о твердых телах:
локальная симметрия, точечные дефекты, химическое окружение
атомов, связь, фазовые переходы.
Полное описание структуры твердого тела требует определения
сингонии, пространственной группы, размеров элементарной ячейки,
атомных координат и, наконец, действительного распределения
электронной плотности вокруг атомов. Чтобы обеспечить такую
информацию, используют дифракцию рентгеновских лучей, электронов
или нейтронов. Условия дифракции описываются хорошо известным
законом Брэгга: rik — 2d sin 6, где X — длина волны падающего
Структурная характеризация
75
Таблица 2.1
Структурная характеризация твердых тел
Детали структуры
Экспериментальные методы
1. а). Морфология,
идентификация фаз, природа
аморфного и кристаллическою
состояния, сингония,
пространственная группа,
положения атомов
б). Ультраструктура
2. Симметрия положения
3. Совершенство крисгаллов
4. Амплитуда тепловых колебаний
5. Фазовые переходы
6. Зонная и электронная
структура
7. Дефекты и упорядочение
8. Магнитное упорядочение и
спиновая конфигурация
9. Химическое окружение ядра
10. Степени окисления металлов
Рентгеновская дифракция, оптическая
микроскопия, электронная дифракция,
электронная микроскопия, нейтронная
дифракция, EXAFS, XANES
Электронная микроскопия высокого
разрешения (HREM)
Спектроскопические методы (ИК, Раман,
УФ и видимая область, ЯМР, ЭПР)
Микроскопия и рентгеновская топография
Рентгеновская дифракция, мессбауровская
спектроскопия
Дифференциальный термический анализ
(ДТА) или дифференциальная
сканирующая калориметрия (ДСК), рассеяние света
и различные спектроскопические методы,
дифракционные методы (особенно
рентгеновская дифракция), измерение
термического расширения и других свойств,
изменяющихся при переходе
Электронная спектроскопия *, электронная
плотность рентгеновской дифракцией и
спиновая плотность нейтронной
дифракцией
Термогравиметрия, трансмиссионная
электронная микроскопия, нейтронная
дифракция, ЭПР и оптическая спектроскопия
Магнитная восприимчивость, нейтронная
дифракция, мессбауровская спектроскопия
Различные спектроскопические метолы,
особенно ЯМР высокого разрешения
твердого тела, мессбауровская
спектроскопия, EXAFS, XANES. Электронная
спектроскопия *
Редокс-титрование, XPS, EELS, AES и
рентгеновская спектроскопия
* Включая рентгеновскую и УФ-фотоэлектронную спектроскопию (XPS и UPS),
Оже-электронную спектроскопию (AES) и спектроскопию потерь электронной энергии (EELS),
излучения, d — межплоскостное расстояние в кристалле и 9 —
брэгговский угол. Соотношение между дифракционной картиной и
структурой кристалла устанавливается в терминах обратной
решетки. Среди других дифракционных методов рентгеновская
дифракция — обычно используемый метод как для рутинной характериза-
Щяя, так и для детальных структурных исследований, но специали-
76 Новые и улучшенные методы характеризации
сты в химии твердого тела все чаще применяют электронную
и нейтронную дифракцию для получения информации, которой не
дает рентгеновская дифракция.
2.2.1. Рентгеновская дифракция
Имеются три стандартных метода рентгеновской дифракции:
а) метод Лауэ, использующий неподвижный монокристалл и
полихроматическое излучение; б) метод вращающегося кристалла,
применяющий монокристалл, вращающийся в луче
монохроматического рентгеновского излучения; в) порошковый метод, где образец
поликристаллов вращается в луче монохроматического
рентгеновского излучения. Метод Л а уз может быть применен для определения
симметрии кристалла и его ориентации, но не для определения
структуры. Метод вращающегося кристалла может быть использован
для определения монокристалличности и параметров элементарной
ячейки образца. Главной трудностью в методе вращающегося
кристалла является перекрывание рефлексов с различными значениями
hkl. Эта проблема преодолена в методе Вейзенберга движением
регистрирующей пленки параллельно оси вращения во время
вращения кристалла. Кроме того, экран с узкой щелью между кристаллом
и пленкой делает возможной регистрацию на движущейся пленке
отражений только от одного слоя линий (рефлексы, для которых
один из индексов является константой). Движение пленки позволяет
избежать перекрывания рефлексов. Улучшенный вариант этого
метода (прецессия с помощью ретиграфа) позволяет регистрировать
слои обратной решетки в неискаженной форме. Дифрактограммы
порошкообразных образцов дают возможность легко получать
информацию о кристаллической структуре. Дифрактометры обычно
используют для идентификации кристаллических структур, анализа
и т. д.; для точного определения параметров элементарной ячейки
часто используют фокусирующую камеру Гинье.
Для получения детальной структуры существенно знание
интенсивности дифракционных линий, поскольку эти интенсивности
связаны со структурными факторами. Автоматический монокристалльный
рентгеновский дифрактометр со структурным программным
обеспечением делает решение структурных проблем рутинным.
Доступность синхротронного рентгеновского излучения с непрерывно
варьируемой длиной волны превращает рентгеновскую дифракцию в еще
более мощный структурный инструмент для изучения твердых тел.
Специалисты в области химии твердого тела широко используют
для обработки профилей дифракционных линий метод Ритвелъда
[Rietveld, 1969; Manohar, 1983]. Пакет структурных программ для
определения этим методом неизвестных структур сейчас стал коммер-
ьески доступным (см. разд. 2.2.3).
Структурная характеризация 11
Кроме структуры, рентгеновская дифракция дает множество
другой ценной информации. Например, порошковая
рентгенография, особенно с прецизионными дифрактометрами, широко
используется для идентификации фаз, количественного анализа смеси фаз,
анализа размера частиц, характеристики физических несовершенств
(причем две последние характеристики получают из анализа ушире-
ния линю.) и изучения реакций in silu. В случае аморфных твердых
тел из данных рентгеновской дифракции получают функции
радиального распределения, которые дают число атомов в единице объема
на любом расстоянии г от заданного атома. Функция радиального
распределения (ФРР) аморфных твердых тел, полученная
преобразованием Фурье интенсивности рассеяния, обнаруживает
накладывающиеся на фоновую кривую пики на расстояниях,
соответствующих среднему межатомному расстоянию в первой координационной
сфере, второй координационной сфере и т. д. Хотя различные
координационные сферы могут вносить вклад в высоколежащие пики,
площадь под первым (и вторым) пиком прямо дает координационное
число. Структурная интерпретация ФРР аморфных материалов
затруднена потому, что ФРР является средней величиной, описываю
щей пространственно-усредненное структурное окружение около
данного атома; имеется также и химическое усреднение по всем
типам атомов в образце. Трудности, с которыми сталкиваются при
описании жидкостей, те же, что и в случае аморфных твердых тел.
Сама по себе ФРР не очень чувствительна к вариациям в модели.
Исследование температурной зависимости рентгеновской
дифракции кристаллических веществ полезно при изучении фазовых
переходов, термического расширения и амплитуд термических
колебаний атомов в твердых телах. Аналогично дифракционное
исследование при высоких давлениях служит для исследования фазовых
переходов, индуцируемых давлением.
2.2.2. Электронная дифракция
Электронные лучи отличаются от рентгеновских в двух
отношениях, делающих электронную дифракций) ценным методом
структурного изучения твердых тел. Во-первых, малой длиной
волны электронных лучей и, во-вторых, зарядом, который они
несут. Меньшая длина волны приводит к меньшим брэгговским
углам в дифракции электронов. Радиус сферы Эвальда 1/Х,
следовательно, для электронной дифракции много больше, чем для
рентгеновской дифракции. Это позволяет непосредственно регистрировать
обширные части обратной решетки для маленького неподвижного
кристалла. Благодаря заряду взаимодействие электронов с атомами
примерно в тысячу раз сильнее, чем рентгеновских лучей, и это
делает возможным почти мгновенную регистрацию дифракции
электронов, однако сильное взаимодействие накладывает строгие уело-
78 Новые и улучшенные методы характеризации
Рис. 2.1. Элсктронограммы.
а — La2Ni40; б — La4LiCoOg, показывающие
сверхрешеточные рефлексы В; в 6 — полоски
видны из-за некоторого разунорндочения
[Ganguly, Rao, 1984]; в — злектронограмма
квазикристаллического сплава А1 — 14 % Мп.
вия на толщину образца. Элек-
тронограммы легко получаются на
коммерческих электронных
микроскопах. Использование
дифракции электронов может быть
выгодным при исследовании
упорядочения дефектов, сворхструк-
тур, образцов мелких частиц
и т.д. Поэтому упорядочение ка-
тионных и анионных вакансий
(15 % каждых) в TiO или
анионных: вакансий в CaFe02>5 и
СаМпО.?,5 лучше исследуется
электронной дифракцией; такое
упорядочение вызывает
сверхструктурные пятна на дифрактограм-
мах. Например, La2Ni04
обнаруживает сверхструктурные пятна,
соответствующие а у 2-типу
элементарной ячейки (рисе 2.1), не
определяемой рентгеновской
дифракцией; показано, что в
LaJLiCoOg, Li+- и Со3+-ионы
упорядочены в двух измерениях;
рисунок 2.1 также показывает
характеристическую симметрию
пятого порядка в квазикристалле.
Электрсны с энергиями в области
10—200 эВ имеют длину волны
порядка межатомных расстояний
в кристаллах. Эти электроны
имеют очень маленькую
проникающую способность. Дифракция
низкоэнергетических электронов
(LEED) дает дифрактограммы,
характеризующие атомные окружения в поверхностных слоях.
Такие дифракционные карт* ны могут быть интерпретированы в
терминах двумерных решеток [Somorjai, 1981].
Структурная характеризация
79
2.2.3. Нейтронная дифракция и родственные методы
Тепловые нейтроны со скоростью около 4000 м/с, имеющие
о
длину волны ^ 1,0 А, используются в экспериментах по дифракции
нейтронов. В то время как рентгеновские лучи первично
рассеиваются внеядерными электронами в атоме, нейтроны рассеиваются в
основном атомными ядрами. Поскольку амплитуды нейтронного
рассеяния не показывают плавной зависимости от атомного номера,
нейтронная дифракция особенно полезна для определения позиций
легких атомов (особенно водорода) в кристаллах. Дополнительное
рассеяние нейтронов может возникать из-за их магнитного момента
(благодаря взаимодействию магнитного момента нейтрона с
постоянными магнитными моментами атомов или ионов в кристалле). В
отсутствие внешнего магнитного поля магнитные моменты атомов в
парамагнитном кристалле распределены случайным образом, так что
магнитное рассеяние нейтронов таким кристаллом также случайно.
Оно вносит вклад только в диффузный фон дифракционной картины.
В магнитоупорядоченных материалах (таких как ферро- и
антиферромагнетики), однако, магнитные моменты упорядочены. Дифракция
нейтронов является экспериментальным методом, посредством
которого могут быть определены различные магнитные структуры. В
дополнение к этим двум упругим — рассеивающим эффектам нейтроны
могут также подвергаться неупругому рассеянию кристаллами. Оно
включает энергию обмена между решеткой и нейтронами.
Неупругое нейтронное рассеяние кристаллами используется в изучении
квантованных колебательных мод (фононов) и динамики в твердых
телах [Lovesey, Springer, 1977; Satyamurthy et al., 1981].
Поскольку нейтронное излучение слабее по интенсивности, чем
рентгеновское, нейтронная дифракция требует монокристаллы
больших размеров (10—100 мм3 по объему по сравнению с кристаллами
около 0,1 мм3, используемыми для рентгеновской дифракции).
Однако возможно получение полезной структурной информации
анализом профилей нейтронной дифракции для поликристаллических
материалов [Cheetham, Taylor, 1977].
В методе полнопрофильного анализа, развитом Ри^вельдом
[1969], структурные параметры подгоняются к полному профилю
линий порошкограммы вместо традиционного приближения
подгонки интенсивностей индивидуальных пиков (рис. 2.2).
Предполагается, что дифракционная картина является суммой ряда брегговских
рефлексов с гауссовой формой линий. Метод включает расчет
функции интенсивности Yt при каждом значении брэгговского угла 6 и
ее сравнение с соответствующими наблюдаемыми значениями.
Функция Уг, которая является мерой вклада интенсивности в профиль
в точке 20, может быть записана как Yt = Wh kF^ где Wifk задает
вклад интенсивности брэгговского пика в точке 2ЭЛ к профилю в
точке 20^, a Fk—структурный фактор, соответствующий Zc-му брэг-
80 Новые и улучшенные методы характеризации
too
28, град
Рис. 2.2. Полнопрофильный анализ нейтронной дифрактограммы Nb02.
Показаны разностный профиль и положение рефлексов [Cheetham,
Rao, 1976].
говскому пику. Полнопрофильный анализ в особенности применим
в нейтронной дифракции, потому что пики точно описываются
функцией Гаусса. Структуры большого ряда оксидов, гидридов, цеолитов
ICheetham, 1986] и др. были уточнены с использованием
порошковых нейтронных дифрактограмм, и часто достигается точность
около 10 % (R = 0,1), что близко к точности, полученной в
рентгенографических работах на монокристаллах. Сейчас доступны
некоторые модификации оригинальной ритвельдовской программы, которые
учитывают анизотропные температурные факторы и константы
молекулярной гибкости. Доступность источников интенсивных
импульсных осколочных нейтронов делает этот метод еще более эффективным
[Lovesey, Sterling, 1984].
Изучение нейтронной дифракции на монокристаллах
обеспечивает богатую структурную информацию. Метод широко используется
для локализации легких атомов типа водорода, для определения
зарядового распределения и для изучения поверхности.
Упорядочение спинов в магнитных твердых телах также легко исследуется
этим методом. Из факторов магнитных*-форм может быть получена
информация о ковалентности. До настоящего времени большинство
работ по исследованию поверхности сделано на кристаллах
ориентированного графита. Нейтронная дифракция используется для
исследования аморфных твердых тел, таких как стекла, жидкости, а
также спиновые стекла. Парциальные структурные факторы получены
в этих работах методом изотопного замещения. Рентгеновское
излучение (из синхротрона) также может обеспечить такую информацию,
если использовать для этих целей аномальное рассеяние вблизи
края поглощения»
Структурная характеризация
81
Нейтронное рассеяние обепечивает ценную информацию о
динамике процессов, протекающих в твердом теле. Так, квазиупругое
рассеяние дает нам информацию по молекулярному движению,
диффузии водорода, туннелирующим состояниям и критическому
рассеянию (центральная мода) так же, как и о спиновой диффузии.
Очень высокое разрешение получено в геометрии обратного
рассеяния, и это делает возможным разделение упругой и квазиупругой
компонент. Преимущество нейтронного рассеяния над рассеянием
света в том, что оно дает S(Q, со), тогда как рассеяние света дает
5(0, со); функция рассеяния S(Q, со) описывает динамические
свойства исследуемой системы, где Q — изменение волнового вектора
нейтрона и ho — изменение энергии в процессе рассеяния. Плотность
колебательных состояний легко получается с некогерентным
неупругим рассеянием. Эта информация подобна таковой, полученной
ИК- и Раман-спектроскопией, но здесь нет правил отбора; для этих
исследований могут использоваться порошки. Изучение
когерентного неупругого рассеяния (с монокристаллами) наиболее полезно для
исследования фононов и мягких мод, так же как и магнонов. С
новыми источниками нейтронов возможно энергетическое разрешение
в несколько микроэлектрон-вольт, а временное разрешение достигает
10-е _ Ю-» с.
Можно привести много интересных примеров использования
нейтронов для изучения проблем, связанных с поверхностью
твердых тел. Различные материалы исследованы нейтронными методами
для получения (ядерной или магнитной) структурной информации.
Мы приведем несколько примеров из недавних литературных
данных.
Нейтронная дифракция и рассеяние использованы для
исследования структуры и колебательных мод, а также туннельных
переходов метана, адсорбированного на графите [Bomchil et al., 1980].
Исследован туннелирующий водород в графите, интеркалированном
щелочными металлами [Beanfils et al., 1981]. Изучение неупругого
нейтронного рассеяния in situ [Vasudevan et al., 1982] открыло
существование двух состояний, в которых водород может
сорбироваться на MoS2. Изучалась структура NHn, интеркалированного в
TaS2, и его трансляционная диффузия; причем эти результаты
использовались вместе с данными ЯМР; показано, что молекулы
аммиака выстроены таким образом, что неподеленная пара азота
примерно параллельна слою. Было также исследовано движение
молекул воды в аллюмосиликатных минералах [Olejnik et al., 1970].
Метод нейтронного спинового эха особенно полезен для изучения
магнитных фазовых переходов.
6 Ч. Н. Р. Рао, Дж. Гопаланришнан
82 Новые и улучшенные методы характеризации
2.2.4. Электронная микроскопия
Электронная микроскопия, очевидно, становится наиболее
многосторонним инструментом для изучения ультраструктуры
материалов, идентификации новых и известных фаз и одновременно для
получения информации о составе. Обычно электронную дифракцию
и изображение используют вместе с трансмиссионной электронной
микроскопией (ТЕМ). Изображения отдельных атомов могут быть
получены современными инструментами. Новые достижения в
приборостроении, в частности сканирующих инструментов (которые
регистрируют сигналы благодаря обратному рассеянию электронов
или за счет излученных вторичных электронов), дают возможность
определять исключительно маленькие количества материалов, а
также составлять карту топографии поверхности широкого ряда
материалов (минералов, металлов, катализаторов, полимеров и т. д.).
Эти возможности еще больше увеличиваются в сканирующих
трансмиссионных электронных микроскопах (STEM). На рис. 2.3
схематически показаны различные типы взаимодействий электронного
луча с образцами твердых тел.
Разрешающая способность микроскопа связана, кроме всего
прочего, с длиной волны используемого излучения. Для обычного
белого света с эффективной длиной волны 550 нм разрешающая
способность около 200 нм. Из-за этого, по-видимому, нет пути
улучшения разрешающей способности оптического микроскопа. Хотя
существует электромагнитное излучение с длиной волны много короче
длины волны видимого света, например рентгеновское излучение,
нет подходящих линз для фокусировки таких лучей. С осознанием,
что движущиеся электроны обладают маленькой длиной волны и
с открытием, что электронные пучки могут фокусироваться, как
магнитными, так и электростатическими полями, примерно в 1930 г.
появились микроскопы с много большей разрешающей способностью,
в которых использовались электронные пучки. Поскольку длина
волны электронных пучков примерно в 105 раз меньше таковой для
видимого излучения, электронный микроскоп способен изобразить
атомные структуры при условии, что длина волны — единственный
фактор, определяющий разрешающую способность.
Механизм формирования изображения в ТЕМ включает как
упругое, так и неупругое рассеяние. Контраст изображения в
электронном микроскопе является результатом амплитудного (фазы
прошедшего и дифрагированного пучка не рекомбинируют) и фазового
контраста (прошедший и дифрагированный пучки рекомбинируют,
сохраняя свои амплитуды и фазы). Для кристаллических образцов
имеется дополнительный контраст, вызванный сильным выделенным
брэгговским рассеянием в определенных направлениях. Это
называется дифракционным контрастом, который является частным
случаем амплитудного контраста. Схематическая диаграмма, которая
Структурная характеризация
83
ПадаА
ЛУ
2
7 к
Ч
оиций
'Ч
1 ofaQjeu К
А
5
Hcomtu
ЛУ
юненнь/и
V
Сканирующая
у и аналитическая тс
\ электронная 1
микроскопия *£
Трансмиссионная jf
электронная ^tl
>микроскопия ^^^Г
и анализ энергий >
Ш S
ffir 0L
7Т BF
^П0 lmt
r—L» IL
\ lm2
X/Im
-^ Экран
\ll
DPX/
Рис. 2.3. Различные типы
взаимодействий электронов с твердым телом.
1 — рентгеновский или оптический фотон;
2 — обратно-рассеянные электроны; 3 —
вторичные электроны; 4 — когерентное
упругое рассеяние; 5 — неупругое
рассеяние; 6 — некогерентное упругое
рассеяние.
Рис. 2.4. Принципы действия
трансмиссионного электронного микроскопа.
S — образец; OL—линза объектива; BF —
обратная фокальная плоскость; lmt—
промежуточное изображение; IL — промежуточная
линза; 1т2— второе промежуточное
изображение; PL — проекционная линза; FIM —
конечное изображение; DP — дифракто-
грамма,
иллюстрирует способ формирования изображения, показана на
рис. 2.4 вместе с дифракционным способом. Щелчком выключателя,
который активирует промежуточные линзы, можно получить
изображение или дифрактограмму той же самой области образца на
экране или фотопластинке. Соответствует ли экспериментальное
изображение действительной структуре, решают различные
факторы, как то: толщина образца, угловое распределение
дифрагированных лучей, прошедших через апертуру объектива, степень
расфокусировки (используемая для получения требуемого контраста) и т. д.
Минимальное расстояние разрешения задается как d ~ 0,6 5]/4Х3/4,
где S — коэффициент сферической аберрации объектива, X — длина
волны электронов. Важно помнить, что в получении изображения
электронный микроскоп выполняет преобразование Фурье: кристалл
в реальном пространстве трансформируется в дифрактограмму в
обратном пространстве, а дифрактограмма трансформируется в
изображение в реальном пространстве.
Чтобы проиллюстрировать возможности метода, на рис. 2.5
показано изображение решетки материала типа ZSM-5.
ТЕМ обычно дает проекционное изображение, где одновременно
•се уровни тонких образцов находятся в фокусе, затрудняя тем
«амым интерпретацию особенностей поверхности образцов. Недавно
84 Новые и улучшенные методы характеризации
Рас. 2.5. Изображение ZSM-5-силиката в электронном микроскопе
высокого разрешения. Изображение, смоделирэванное компьютером, показано
как вставка. Даны соответствующая элекгронограмма и рисунок струкгу
ры. Заметим, что как большие (~ 6,5 А диамэтром), так и малые каналы
обнаруживаются на изображении. Зигзагообразный путь линий
инверсионной симметрии, соединяющих межканальные щели, указан полосами.
появились некоторые важные усовершенствования, позволяющие
избежать этих затруднений. Посредством необходимых модификаций
электронный микроскоп может быть превращен в настоящую
лабораторию как низке-, так и высокотемпературную, работающую
в различных атмосферах и т. д. Высоковольтные микроскопы,
работающие при миллионе вольт или более, часто используются для
изучения с очень высоким разрешением в различных реальных
условиях. Как это будет показано ниже, кембриджский электронный
микроскоп высокого разрешения, работающий при 500 кВ,
фактически дает почти атомное разрешение в благоприятных случаях.
В сканирующем электронном микроскопе (SEM) тонко
сфокусированный электронный пучок движется от одной точки образца
к другой, образуя растровую картину, точно такую, как в
телевизионном изображении. Интенсивности рассеянных или вторичных
э-лектро:шв непрерывно измеряются и демонстрируются на телемони-
Структурная характеризация
85
Рис. 2.6. STEM-изооражонио 1 мае. % Pd на древесной угле:
средний размер около 100 А [Тгеасу et al., 1978].
торе, образуя растровое изображение. При использовании
современных источников высокоинтенсивных электронов (LaB6) легко дости-
о
гается поверхностное разрешение 20—50 А. Разрешение
ограничивается проникновением электронов под поверхность, а также чистотой
поверхности. Поверхностная сканирующая электронная
микроскопия дает топографические изображения, которые легко
интерпретируются. Магнитные домены также могут быть исследованы в методе
отражения с подходящим модифицированным сканирующим
микроскопом.
В сканирующей трансмиссионной электронной микроскопии
комбинируется прохождение электронов через образец со
сканирующими и аналитическими возможностями. Информация об образце
включает анализ возмущений, испытываемых электронами в их
столкновениях с атомами-молекулами образца. В этом методе сканированное
о
изображение может быть получено с 5 А электронным лучом и
разрешение изображения подобно получаемому с трансмиссионными
электронными микроскопами высокого разрешения. Кроме того, несколько
сканированных сигналов собираются одновременно, позволяя тем
самым улучшить изображение. Эта техника используется для
изучения широкого многообразия минералов и материалов, особенно
катализаторов. На рис. 2.6 показана микрофотография частиц ката-
86 Новые и улучшенные методы характеризации
■■■«■■«■■I
Е —
в
Рас. 2.7. HREM-изображеггао (зарегистрировано при 200 кВ) образца
номинального состава Bi0, 2W03. Показаны также электронограмма и
профиль линии рентгеновской эмиссии. Полосы обусловлены атомами Bi
в гексагональных туннелях [Ramanan et al., 1984].
Стру? туркая характеризация
87
лизатора, демонстрирующая мощь этой техники. Таким способом
можно видеть частицы порядка 10 А.
Аналитическая электронная микроскопия является сегодня
наиболее тонким доступным инструментом для микроструктурного
анализа. В этом методе мы можем получить как структуру высокого
разрешения, так и элементный состав образца. Вероятно это лучший
метод получения локального элементного состава малых областей
гетерогенных твердых тел. Когда высокоэнергетичный электронный
луч падает на образец, мы получаем упруго- и неупругорассеянные
электроны, характеристическое рентгеновское излучение, обратно-
рассеянные электроны и вторичные (излученные) электрсны
(см. рис. 2.3). Все они могут быть собраны одновременно в
микроскопе с маленькой площади образца, для того чтобы провести
анализы структуры и состава. Упругорассеянные электроны дают
изображения и дифрактограммы. Информацию о составе дают электроны
с потерей энергии и спектр рентгеновского излучения. Данные по
обоим спектрам обычно собираются в микрокомпьютер,
обрабатываются и выводятся на дисплей. В современных приборах, чтобы
сохранить источник высокоинтенсивных электронов и
минимизировать загрязнение, рабочий вакуум достигает 10~9 торр. На рис. 2.7
показан пример анализа рентгеновской эмиссии на электронном
микроскопе в случае кристалла номинального состава Bi0 2W03
действительный состав был найден рентгеновским эмиссионным
анализом (Bi007WO3); он был также подтвержден рентгеновской
фотоэлектронной спектроскопией. Присутствие атомов Bi в полосах
гексагональной вольфрамовой бронзы могло быть установлено
только с помощью электронной микроскопии в сочетании с
рентгеновским эмиссионным анализом. Еще один пример
электронно-микроскопического анализа дает лизардит, являющийся серпентином, у
которого несогласованность между слоями октаэдров Mg(0, OH)6 и
тетраэдрами Si04 снята замещением алюминия; то что это так, могло
быть установлено рентгеновским эмиссионным анализом. Другие
примеры рентгеновского эмиссионного анализа в электронном
микроскопе описаны Jefferson [1982]. Спектроскопия потерь
электронной энергии (EELS) может быть использована для анализа, а также
для получения информации о состояниях окисления металлов и
электронной структуре оксидов и других материалов.
Использование EELS в электронном микроскопе для качественного и
количественного анализа ультрамикроколичеств недавно рассмотрено в
обзорах Carpenter [1981], Egerton [1982]. L — края поглощения
переходных металлов и К — край поглощения кислорода в оксидах
переходных металлов (рис. 2.8) открывают важную структурную
информацию [Rao et al., 1984]. Плазмонные полосы металлов в EELS
также имеют диагностическое значение [Sparrow et al., 1983].
Современный аналитический микроскоп легко производит изо-
о
сражения с поточечным разрешением от 3 до 4,5 А (с увеличен и-
88 Новые и улучшенные методы характеризации
К край
поглощения кислорода
МпО
К край
поглощения марганца
МпО
Рас. 2.8. Мп(Ь)-край и О(К)-
край в EELS оксидов
марганца, измеренных в электронном
микроскопе [Rao et al., 1984].
610 650
ЛЕ/эВ
ем в области 300 000—
800 000); специальные
микроскопы высокого
разрешения (200 кВ) могут
даже дать разрешение
о
2,5А. В оптимальных
условиях мы можем
получать дифрактограммы от
о
кристаллов менее 5 А
в диаметре, в других
случаях от кристаллов
диаметром 100 Ли более.
Для получения лучших
аналитических
результатов толщина кристаллов
должна быть
достаточно мала (предпочтительна
о
толщина 500 А и меньше).
Когда образец находится
в свободном состоянии или скружен слабо рассеивающей матрицей,
этой методикой можно легко обнаружить 10~18 г материала; при
оптимальных условиях эта величина даже на несколько порядков
меньше. Когда присутствует матрица, чувствительность зависит от
атомного номера элементов, составляющих эту матрицу.
Наиболее важный аспект электронной микроскопии в химии
твердого тела связан с ее способностью проливать свет на проблемы,,
которые находятся за пределами возможностей рентгеновской или
нейтронной кристаллографии. Изображения в электроном
микроскопе высокого разрешения (HREM) показывают локальную
структуру кристаллов с удивительной детальностью; на рис. 2.9
приведено изображение Bi01WO3, полученное на кембриджском
университетском 500 кВ микроскопе, чтобы показать улучшенное
разрешение по сравнению с изображением, полученным на 200 кВ
микроскопе (см. рис. 2.7). На изображении легко видеть
ультрамикроструктуры несовершенств. Дифракция отдельных участков и их
изображение часто могут дать сведения об атомном окружении в
областях, содержащих только несколько атомов. Поскольку
рассеивающие факторы для электронов в тысячу раз больше таковых
для рентгеновского излучения, структурная информация может быть
Структурная характеризация
89
Рас. 2.9. HREM-изображение (зарегистрированное при 500 кВ) образца
номинального состава В10,г\\Ю3. Показан и рисунок структуры. Можно
видеть атомы Bi в гексагональных туннелях [Ramanan et al., 1984].
получена с очень маленькими образцами (~ 5 А диаметром). HREM
также очень полезен для идентификации новых фаз, которые часто
могут встречаться как включения в цепочки или слои, а также как
прорастания в других фазах (рис. 2.10).
Иногда экспериментальное изображение получают для тонких
образцов (< 100 А), а его интерпретация контролируется
использованием рассчитанного изображения, полученного с учетом
многократного рассеяния электронов, характеристик линз и толщины
образца. Обычно применяется так называемый многослойный метод
IGooelman, Moodie, 1974] для моделирований изображения, которое
рассчитывают для ряда значений толщины кристалла и
расфокусировки.
Приложения электронной микроскопии для исследования
различных типов явлений, структур, дефектов и реакций, имеющие
значения в химии твердого тела, за последние годы компетентно
90 Новые и улучшенные методы характеризации
Рис. 2.10. Изображение решетки оксида семейства (Bi202V2+-
'(Ал_ Вл03/г+ )2~ с номинальным /г 8, показывающее
существование оксидов с и --- 4 II 5 [Hutchison et al., 1977].
рассмотрены несколько раз [Allpress, Sanders, 1973; Anderson, 1978;
Kihlborg, 1979; Beer et al., 1981; Thomas, 1982], из них некоторые
мы будем сбсуждать в соответствующих разделах.
2.2.5. Рентгеновская абсорбционная спектроскопия
(EXAFS, XANES)
Традиционно измерения края рентгеновского поглощения
использовали для определения состояния окисления металлов в
сложных материалах. Расширенная топкая структура
рентгеновского поглощения (EXAFS) обеспечивает такую структурную
информацию, как длины связей и координационные числа даже для
порошкообразных образцов, кристаллических или аморфных, причем тонкая
структура является результатом главным образом ближнего порядка
вокруг поглощающего атома. Этот метод также полезен для изучения
поверхности твердого тела (SEXAFS). Наблюдение тонкой
структуры вдали от К-края поглощения материалов датируется началом
20-х годов, когда ее называли крониговской структурой, но только
с 19G5 г. в этой области были получены заметные успехи. Именно
чувствительность EXAFS к локальной структуре делает его
уникальным структурным методом [Stern et al., 1978; Sandstrom, Lytic,
Структурная характеризация
91
5 1
I
* 0,90 А
С 1
^ J
? 1
I 0,70 J
<5
^
^ J
i 1
х п чп • L
•»i UyOU *■
^ 0
XL
is
1 Iм
Ip, 10,02-
\Л-о,ог
j
1 1 r-
200
ftlX
'w
v_
1 1
< A Jo
V/W')
1 '
400
Е-Е0,эВ
Рас. 2.ii. Cu EXAFS-спектры
катализатора Gu75 — Ni25/Al203.
Вставка показывает нормированныэ
данные для биметаллического
катализатора в сравнении с Си/А1203
(штриховая линия) [по G. Sankar, S. Vasude-
van, С. N. R. Rao].
Рис. 2.12. Схематическое представление
EXAFS-процесса. Атом (черный
кружок) поглощает рентгеновские лучи,
испуская фотоэлектронную волну,
которая претерпевает обратное рассеяние
ближайшими атомами (заштрихованные
кружки). Сплошная окружность
означает исходящую электронную волну,
а прерывистая — рассеянную
электронную волну. Когда волны
перекрываются, имеет место интерференция.
1979; Тео, 1980; Parthasarathy et al., 1982]. Край поглощения сам
по сэбе представляет собой характеристическую особенность
элемента, и EXAFS является следствием локальной дифракции низко-
энергетичных электронов. В известном смысле EXAFS можно
рассматривать как LEED-эксперимент, «проводимый таким образом,
что и электронная пушка и фазово-чувствительный детектор
расположен внутри образца» [Sandstrom, Lytic, 1979]. Очевидно, что
изучение EXAFS различных элементов в данном материале
приводит к полному определению локальной структуры. Это равносильно
знанию парциальных структурных факторов (ближнего порядка) для
каждого элемента.
Ранее отмечалось, что EXAFS является результатом двух
фундаментальных процессов: К-(или L-) поглощения рентгеновского
фотона, что является фотоэлектронным эффектом, и эффективной
дифракции электрона, излученного таким образом. В случае одного
изолированного поглощающего атома (поглотителя) можно видеть
только характеристичное увеличение в поглощении, и(г=]п/0//), при
энергии, соответствующей краю, и затем экспоненциальный спад.
Когда поглотитель окружен другими атомами, ц, проявляет волнооб-
92 Новые и улучшенные методы зсарактеризации
разные изменения иногда вплсть до 2000 эВ от края. Изменения на
расстоянии 30 эВ до края составляют EXAFS. В качестве примера
на рис. 2.11 показан EXAFS биметаллического катализатора.
Фотоэлектроны излучаются поглощающим атомом вслед за
поглощением рентгеновских фотонов. Расходящаяся (сферическая)
фотоэлектронная волна, излучаемая поглотителем, отражается
окружающими «рассеивателями». Фаза отраженной волны зависит от
длины волны фотоэлектрона, атомного номера рассеивателя и
расстояния между поглощающим и рассеивающим атомами. Отраженная
(фазово-сдвинутая) волна подвергается самоинтерференции с
расходящейся из положения излучения волной. То, что эта
самоинтерференция ответственна за EXAFS, можно видеть из последующего
рассмотрения. Вероятность W рентгеновского поглощения (т. е.
фотоэмиссии) определяется как
W = 2n2e2((oc2m)-'\Mfs\ 2р(£у), (2.1)
где MfS = </|p: e|s>, e — заряд электрона, 2ясо — частота
рентгеновского фотона, с — скорость света, т — масса электрона, р —
вектор его момента, г — вектор электрического поля излучения и
p(Ef) — плотность конечных состояний. \s} — является волновой
функцией электронов К-ячейки, |/> — волновая функция того
конечного состояния электрона, в которое он переходит. Правила
отбора требуют, чтобы |/> имела р-характер.
Для Е ^ 30 эВ конечное состояние в действительности является
континуальным. Из уравнения (2.1) очевидно, что единственный
терм, который может вызвать тонкую структуру, это \Mis\, так как
другой терм является монотонной функцией. |$>-состояние в (Mf3}
соответствует очень низкой потенциальной энергии и, следовательно,
остается неизменяемым; таким образом, только </| -состояние может
вызвать волнообразные изменения. Конечное состояние
фотоэлектрона, которое соответствует сферической волне,
распространяющейся от поглотителя, показано схематически на рис. 2.12 [Wong, 19801
вместе с рассеянной волной, которую мы можем называть обычно
фазово-сдвинутой по отношению к расходящейся волне. Отраженная
волна дополнительно сдвигается по фазе на поглощающем атоме,
и интерференция между этой волной и расходящейся на К-ячейке
(которая составляет порядка десятой или пятнадцатой части
атомного радиуса) вызывает изменение в </| .
Штерн и сотрудники рассмотрели действие такой
самоинтерференции на конечное состояние, принимая во внимание относящиеся
к делу фазовые сдвиги, и показали, что EXAFS, %^k), может быть
записано как
X (к) = 2 А5 (к) sin [2kRj + £; (к)], (2.2)
где к — |к|, суммирование ведется по всем /-координационным
ячейкам и Rj — расстояние от поглотителя до у-й ячейки, тогда как
Структурная характеризация
93
£7-(к) есть общий фазовый сдвиг, вызываемый как у'-й ячейкой (т];-(к)),
так и потенциалом поглощающего иона (б(к)). £Дк) может быть
аппроксимирована полиномом второго порядка
£,(k) = U + Ък + Ък\ (2.3)
Aj(k) представляет общую амплитуду рассеивания у-й ячейкой и
может быть записана ISandstrom, Lytle, 1979]
А'(k)=5тFi (k) exp - (2°^2) • {2Л)
Nj и Gj в уравнении (2.4) представляют соответственно число атомов
в у-й ячейке и серднеквадратичпое отклонение межатомных
расстояний от Rj, которое является результатом эффектов как
статического, так и динамического (термического) разупорядочения.
Амплитуда рассеивания Fj(k) задается
Fj(k) = fj(n, к) exp (-2/ВД, (2.5)
где /;(я, к) — амплитуда обратного рассеяния (которая
эквивалентна фактору обратного рассеяния электрона) и Я — средняя длина
свободного пробега фотоэлектрона. Уравнение (2.2) объясняет,
почему одноатомные газы не проявляют EXAFS, и очень просто
показывает, что колебания поглощения определяются членами уравнения
под знаком синуса.
В то же время очевидно, что фурье-преобразование уравнения
{2.2) будет давать информацию о всех /-ячейках в прямом
пространстве, которые вносят вклад в EXAFS. i?y-, получаемые таким образом,
однако, ограничены £1? /с-зависнмой частью £7(к). Поскольку
интенсивность расходящейся сферической волны уменьшается очень
быстро с увеличением Л, отдаленные атомы вносят очень малый вклад
в тонкую структуру. Эффекты многократного рассеяния также
относительно мало важны, и они в действительности не учтены при
выводе уравнения (2.2). EXAFS не должен содержать информацию о
«затененных» или «заслоненных» атомах, но имеются исключения из
этого правила. Существуют и другие теоретические приближения,
использующие подобные эффекты для объяснения EXAFS.
Так как фурье-преобразование уравнения (2.2) дает только
функцию радиального распределения около поглотителя, заметим,
что информация, полученная из EXAFS, ограничена усредненным,
одномерным представлением структуры. Более того, чтобы
преобразование было свободно от пульсаций, данные должны базироваться
на протяжении по меньшей мере 500 эВ от края поглощения. Другие
ограничения метода Фурье, такие как ошибка обрыва ряда,
являются дефектами, характерными и для других похожих методов, хотя
для минимизации этих эффектов может быть введена функция
«окна». Наиболее важным параметром, включаемым в преобразование,
является функция общего фазового сдвига ^(к).
94 Новые и улучшенные методы характеризации
Для вычисления £;-(к) были развиты некоторые методы расчета
ab initio и ее значения табулированы [Тео, Lee, 1979], но точность
их ограничена из-за многих приближений, используемых в таких
расчетах. Часто поправки фазового сдвига устанавливаются
экспериментально с использованием структурной информации,
полученной другими методами на подобных соединениях. EXAFS-данные
обрабатываются для получения радиальных структурных функций
(РСФ). Первые, не EXAFS-компоненты вычитаются из этих данных.
Докраевое поглощение учитывается с использованием полиномов
Викторина (International Tables for Crystallography, 1969) в форме
АХ3 + В№. Монотонное уменьшение поглощения вдали от края,
называемое фотоэлектронным спадом, |я0, вычитается после
аппроксимации или в виде полинома второй степени или сплайн-функции
lEccles 1978]. Стандартизованная %(к) выражается как
Х(к) = (|х - |г0)/|х0. (2.6)
Фотоэлектронный волновой вектор к рассчитывается с помощью
соотношения %2к2 = 2т(Е — Е0), где Е — энергия рентгеновского
фотона, Е0 — энергия, от которой она отсчитана, и т — масса
электрона. %(к) умножается на кп(п = 2 или 3), чтобы увеличить слабый
EXAFS при больших к [Lytle et al., 1975]; кп%(к) являются фурье-
преобразованиыми, дающими РСФ 0 (/?). В модельных соединениях
первый пик на Rx представляет собой расстояние до ближайшей
соседней ячейки и может быть сравнен с известным расстоянием /?1в
Тогда можно определить а* как (R\ — /?г), которая представляет
собой экспериментально определенную фазовую коррекцию. В
принципе 2а* должно быть равно теоретически установленной /с-зависи-
мой части £j-(k), т. е. £г, если идентичность окружения рассеивателя
предположена корректно. Нужно подчеркнуть, что, если
идентичность рассеивателей не установлена (т. е. в некоторых ковалентно
связанных и неупорядоченных системах), рекомендуется
использование а* (а не £х). Кроме того, /с-зависимость £;(к) вводит внутренние
ограничения на ее количественную точность.
Вторая стадия улучшения EXAFS-данных достигается через
фурье-фильтрацию [Eccles, 1978]. Здесь отдельный пик на 0{R),
соответствующий i-m рассеивающей ячейке, трансформируется
обратно в /с-пространство и преобразованная функция
приспосабливается к параметризованному выражению для EXAFS lEccles, 1978;
Cramer, 1978]
X'(k) =кПсг*М-сг*) sin{C4 + {Сь + 2Ri) k + с^у (2.7)
%'(к) имеет шесть подгоночных параметров (CS) и состоит из
фазового и амплитудного термов (см. уравнение (2.3)). Rt может быть
обработан как подгоночный параметр. Здесь надо отметить, что £;(к)
Структурная характеризация
95
является свойством только пары поглотитель — рассеиватель и не
зависит от особенностей химической связи; £у(к), следовательно,
переносимо от соединения к соединению. В методе фурье-фильтра,
а* для пары поглотитель — £-я ячейка задается а* — Rt — IVt,
где Rt — расстояние на исправленной кривой до г-й ячейки и k't —
соответствующий пик на РСФ. Используя этот метод, можно
избежать двусмысленности благодаря вкладам от различных ячеек в
оцениваемые фазовые сдвиги. Параметр беспорядка af
(представленный С2) есть мера «размаха» межатомных расстояний в /-й ячейке
ILytle el a]., 1975]. В данной ячейке / а, составляется из вкладов от
статического разупорядочепия astat и колебательного или
динамического разупорядочения оу[Ь (связанного с фактором Дебая — Уол-
лера) и может быть записано о) = aftat + °~vib- Исследованием
температурной зависимости ст7- могут быть получены оценки
индивидуальных вкладов. Сравнение EXAFS-выражепия и выражения для
интенсивностей рентгеновской дифракции (XRD) предполагает, что
EXAFS более чувствителен [ехр(—З^ь/с2)] к разупорядочению,
чем XRD. Расстояния, рассчитанные из EXAFS, могут быть точны
до =t:0,02 А, но такая точность критически зависит от качества
вводимых данных. Nj, которое представляет координационное число,
оценивается сравнением С1 в известной и неизвестной ситуациях.
Его точность обычно мала, и интерпретации, основанные только на
таких значениях TV,, должны делаться с осторожностью.
EXAFS-экспериментальное устройство имеет три главных
компонента: источник рентгеновского излучения, монохроматор (и
коллиматор) и детектор. Для EXAFS широко используется синхротроп-
ное излучение, но, если эта аппаратура недоступна, подходящим
источником может быть вращающийся анод.
EXAFS как мощный метод для изучения ближнего порядка
(локальной структуры) эффективно используется для исследования
биологических объектов, аморфных материалов, катализаторов и
различных типов материалов, важных в химии твердого тела.
Имеются компетентные обзоры по рассматриваемой теме [Тео, 1980;
Parthasarathy et al., 1982]. Мы приведем несколько примеров,
относящихся к аморфным материалам, катализаторам, ионным
проводникам и интеркалатам. Исследования по суперионной фазе Agl
(высокотемпературная а-фаза, рис. 2.13) и фазе p-Agl
(диэлектрическая фаза) с использованием Agfe EXAFS показали, что ионы
Ag занимают искаженные тетрагональные позиции в
объемно-центрированной кубической решетке иодид-ионов. Кроме того, ионы
серебра смещены в направлении наиболее вероятного пути перескока
ионов [Воусе et al., 1977]. Сравнением с теоретически рассчитанным
EXAFS получены также свидетельства прыжково-диффузиопиой
модели проводимости. Подобное исследование RbAg4I5 IStuties et al.,
1979] указало, что Ag+ занимает тетраэдрические пустоты в иодид-
96 Новые и улучшенные методы характеризации
Рис. 2.13. I — функция радиального
распределения иодид-ионов около каждого атома Ag: в 0-
фазе Agl при: а — 293 К и б — 371 К; и в а-
фазе при: в — 471 К; г — 575 К. Каждая
функция нормирована на содержание четырех ионов
иода в первом пике. Стрелки на в и г указывают
на вклад иона серебра, занимающего центр
грани между смежными тетраэдрами. II — функция
радиального распределения иодид-ионов около
каждого атома Си в Cul: в у-фазе при: а — 295 К;
б — 573 К; в — 623 К; и в а-фазе при: г —
743 К. Стрелки указывают обнаружение ионов
Си в центре грани тетраэдра в мостиковом
положении между тетраэдром и октаэдром. III —
схематическое двумерное представление
разрешенной (заштрихованной) и исключенной
областей центров катионов в модели исключенного
объема для: а — диэлектрической и б —
проводящей фаз. Центры анионов обозначены
маленькими кружочками. Видна связь между
разрешенными областями в суперионной фазе [Воусе,
Hayes, 1979].
ной решетке и что существуют корреляции Ag+ — Ag+ в
суперионной фазе. В Cul, однако, EXAFS показывает, что Си+-ион находится
как в тетраэдрической, так и в октаэдрической позициях [Воусе,
Hayes, 1979]. Оказывается, что наиболее предпочтительный путь
движения Си+ — вдоль направления [111]. Анализ поддерживает
модель исключенного объема для Си—I парной корреляционной
функции. Рис. 2.13 дает схематичную диаграмму модели
исключенного объема; показана также форма первого пика на кривой
радиального распределения при различных температурах для Agl и Cul.
В последние годы появились работы по изучению интеркалатов.
В исследовании EXAFS Вг2 в графите найдено, что, хотя интеркалат
остается молекулярной структурой, расстояние Вг—Вг
увеличивается до такой степени, что сочетается с периодичностью решетки
[Heald, Stern, 1978]. В серии псевдостехиометрических соединений
щелочной металл — графит (КСП, п — 2, 3, 4) результаты EXAFS
наводят на мысль, что металл в этих соединениях можно предста-
Структурная характеризация
97
вить как неупорядоченный двумерный решеточный газ [Caswell et
al., 1980]. Изучены интеркалаты щелочных металлов в NbS2 и
найдено увеличение расстояния Nb—Nb [Bourdillon et al., 1980].
Изучение EXAFS стекла As203 показывает, что беспорядок
возрастает с увеличением расстояния от As, тем самым отвергая
существования А8406-единиц [Pettifer, McMillan, 1977]. Результаты
EXAFS показывают, что в стекле As2Te3 имеется более высокая
степень ближнего порядка по сравнению с кристаллом как результат
увеличения ковалентности [Pettifer et al., 1977]. Для стекол
системы As2Se3—As2Te3 расстояния до соседей из второй координационной
сферы изменяются вследствие увеличения ионности для
промежуточных составов [Parthasarathy et al., 1982]. В пленках Si—H
обнаруживается мышьяк в качестве замещающей примеси [Knights et al.,
1977]. EXAFS стекла ZnCl2 в процессе перехода в стеклообразное
состояние показал, что нет увеличения беспорядка при Tg, как в
случае стекла Ge02 [Wong, Lytle, 1980]. В аморфном GaAs не
найдено свидетельств химического разупорядочения [Theye et al., 1980];
в GaP, однако, оказывается, что часть связей вокруг Ga становится
типа Ga—Ga. Ряд металлических стекол был исследован методом
EXAFS [Wong, 1980]. Этот метод также использовался для
изучения ионных стекол. EXAFS-изучение Nd3+ в стекле BeF2, например,
обнаружило, что в стекле не только другое координационное число
Nd3+ (семь), но также имеется уменьшение расстояния Nd—F по
сравнению с кристаллическим NdF3 [Rao et al., 1983]. EXAFS
стекла NdF3—BeF2 и кристалла NdF3 показан на рис. 2.14.
EXAFS очень полезен при изучении катализаторов, особенно
для исследования природы металлических кластеров на поверхности
нанесенных металлических катализаторов [Sinfelt et al., 1984]. Уже
исследовано множество систем, и еще имеется заметное поле
деятельности для исследований в этой области. Так как EXAFS дает
расстояния и координационные числа и является селективным в
отношении поглотителя, можно каждый раз изучать один тип металла
(см. рис. 2.11). Так, EXAFS-исследование сульфидированных
катализаторов Со—Мо—А1203 показывает, что Со(Н)- и Мо(1У)-ионы
находятся в октаэдрической координации с расстояниями металл —
сера, не отличающимися от таковых в CoS и MoS2 [Sankar at al.,
1984]. До завершения обсуждения EXAFS нужно сказать, что, если
проблема выбрана неправильно, результаты могут оказаться
малоценными, так как информация о первой координационной сфере
(или основной длине связи) не может в целом быть неожиданной.
Структура рентгеновского поглощения вблизи края (XANES)
является полезной в определении координационного числа и
степени окисления металлических ионов [Sankar et al., 1983]. На
рис. 2.15 и 2.16 показаны XANES в некоторых соединениях,
как и в катализаторах. Переходы 15 — 3d кобальта и Is — id
молибдена чувствительны к координации, причем становятся более
7 Ч. Н. Р. Рао, Дж. Гопалакришнан
Новые и улучшенные методы характеризации
0,22
~т i i i г~
3,0 5У2 , 7У4 9,6
к,А"'
~Ot2 ' i i i II ' О L
0 2 4 6 в 1012 О
к,А"
т i г~
г 4 .в a
R.a
И 1 1 1 Г"
3,0 S,27 J,S3 9.8
к,А-1
Рис. 2.14. NdLjjEXAFS стекла NdF3— BeF2 (вверху) п
кристаллического NdF3 (внизу).
а — нормированные EXAFS-спектры; б — фурье-преобразование; в —
обратное преобразование (линия) и смоделированный EXAFS (точки) в области
0,2—3,2 А° [Rao, К. J. et al., 1983].
интенсивными, когда координация тетраэдрическая. Соответственно
из рис. 2.15 видно, что кобальт в предшественнике — Со/Мо/А1203 —
катализаторе тетраэдрически координирован, тогда как в сульфиди-
рованной форме он координирован октаэдрически [Sankar et al.,
1984]. Достойно внимания то, что данные XANES на рис. 2.15 п
2.16, полученные с использованием обычного источника
рентгеновского излучения, идентичны результатам, полученным для синхро-
тронного источника.
Оказывается, что химические сдвиги К- и L-краев поглощения
металлов обычно больше, чем химические сдвиги энергий связи,
найденные из рентгеновских фотоэлектронных спектров.
Следовательно, химические сдвиги рентгеновских краев поглощения полезны
в фиксации степени окисления металлов; химический сдвиг заметно
увеличивается с окислительным числом [Sarode et al., 1979].
Полезной является корреляция между химическими сдвигами и
эффективным зарядом на атоме металла. Химические сдвиги АЕ, оказывается,
следуют уравнению
АЕ = aq + bq2, (2.8)
где q — эффективный заряд атома [Sarode et al., 1979]. Уравнение
(2.8) подтверждено в некоторых сериях соединений переходных
Структурная характеризация
99
!
I
S
*
Со/Мо/А1г03 Супьфидироеанный [6.' 12)
Co.MoS2C4 6)
СоО
СоИо04
СоА1г04
Со/Мо/А1г03(* -12)
-Ю О 10 20 30 40 50
(Е-Е)0, эВ
Рис. 2.15. XANES типичных соединений
кобальта и кобальтовых катализаторов [Sankar
et al., 1984].
MoS4
03(6Ч2)
Ct/ль фидированный
20000 20020
Энергия, эВ
20040 Ю9вО 20000 20020 20040
Энергия, эВ
7*
Рис. 2.16. XANES типичных соединений молибдена и молибденовых
катализаторов [Sankar et al., 1984].
100 Новые и улучшенные методы характеризации
металлов [Manthiram et al., 1980]. Как показано позже [Sankar
et al., 1985], оказывается, что особенности в XANES следуют
уравнению (2.8). Химические сдвиги могут быть непосредственно
использованы для установления состояния окисления или числа d-элект-
ронов в системе. Измерения края поглощения (и EXAFS)
действительно были использованы для исследования состояния марганца
в диспергированных катализаторах на основе Mn02 [Brown et al.,
1984]. Недавно было определено число незанятых d-электронных
состояний в платиновом катализаторе, используя интенсивность L2-
и Lg-краев поглощения [Mansour et al., 1984]; в равной степени это
могло быть выполнено и спектроскопией потерь электронной
энергии (EELS). EELS, проведенная в электронном микроскопе, может
непосредственно давать информацию об интенсивностях L2 и L3,
которые связаны с незанятыми d-состояниями металла [Rao et al.,
1984]; кроме обеспечения пространственного решения EELS может
дать одновременно химический анализ и структуру (через
дифракцию электронов) мельчайшего образца (10~12—12~18 г).
2.2.6. Спектроскопия ядерного магнитного резонанса
с вращением под магическим углом
ЯМР-спектроскопия является, вероятно, наиболее широко
используемым химиками физическим методом; она обеспечивает
мощные средства исследования структуры и динамики. До настоящего
времени почти все работы были сделаны на соединениях в жидком
или твердом состоянии, причем спектры в твердом состоянии обычно
широкие и лишены особенностей благодаря дипольным
взаимодействиям. Даже широкие полосы полезны для исследования структуры
твердых тел (например, гипса) методом ЯМР «широких линий».
Измерение времен релаксации обеспечивает ценную информацию
о диффузии, реориентации и подобных аспектах динамики.
Использование ЯМР-спектроскопии как рутинного метода для структурных
исследований твердых тел, однако, до недавнего времени было за
пределами экспериментальной возможности. Успехи в технике и
приборостроении сделали возможным получение химической
информации из ЯМР-спектров порошков, и этот метод, несомненно, будет
использоваться в самой полной степени в ближайшем будущем. Уже
сейчас имеются значительные успехи в применении твердотельного
ЯМР в исследовании силикатов, в особенности цеолитов [Fyve
et al., 1983; Lippmaa et al., 1980; Magi et al., 1984].
Дипольные взаимодействия, ответственные за уширения ЯМР-
сигналов, могут быть в принципе элиминированы в случае
распространенных ядер, таких как протон, но маленькая область химсдви-
гов протона уменьшает пользу этого приема. Спектры хорошего
разрешения легко получаются в случае небольшой концентрации ядер.
Структурная характеризация
101
а Цеолит МаУ иА1
27А
\23,5 МГц
/04,2 МГц
200 /00 0 -100
и.д. От А1(Н20)^
9 Pb(N03)2 207РЪ
83,4 МГц
-Я50 -£00 -650
м д. от РЪ(СЮ4)2
5 Стекло 7070 11В
/2<?,4МГи,
50 О -50
м.д. от BF3- 0Et2
2 15NhJ5N0
JJLu
X3Z
40,5 МГц,
nh;
200
О ~200
t* g от NO3
-4O0
Рис. 2.17. ЯМР-спектры с вращением под магическим углом.
а — 27А1-спектр цеолита NaY при 23,5 МГц и 104,2 МГц; заметим,
что высокополевой спектр лучшего качества; б — "В-спектр стекла
Corning 7070 при 128,4 МГц; в — 207РЬ-спектр в Pb(N03)2 при 83,4 МГц
(широкая линия вверху'по лучена без вращения); г — 15К-спектр 15NH4
"NO3 при 40,5 МГц. Показан также увеличенный спектр NO* [Fyfe
et al.t 1983].
Магнитное взаимодействие для таких ядер задается
Яобщ = Я(зееман) + Я"*,-(диполь) +
+ Я,;(диполь) + ЯДаниз),
(2.9)
где дипольное взаимодействие Htj — взаимодействие между ядром i
и большим количеством ядер у (например, 13С и ХН). Терм Htj мал
из-за низкой концентрации ядер i (например, 13С). Дипольные
взаимодействия Ни можно удалить приложением сильного
«распаривающего» поля на резонансной частоте. Другой вклад в уширение
появляется из анизотропии химсдвига (Ht аниз). Хаотичные
движения молекул в жидкой фазе усредняют химическое экранирование
ядер в результате различных ориентации по отношению к
магнитному полю. В твердом состоянии, однако, получается широкая
характеристика химического окружения. Анизотропия химсдвига может
быть понята из уравнения
ЯДаниз) = (3cos20 —1) (другие члены) +
+ (3/2 sin29)a /50,
(2.10)
где 0 — угол между направлением оси прецессии и направлением
магнитного поля, В0 — магнитное поле, a — изотропное химическое
102 Новые и улучшенные методы характеризации
150 ioo д 1~~-?Л7
Октаэдрическая
ТетраэЗрическая
150 100 50 О -50 -ЮО
Октаэдрическоя
Тетраэдричесхая рис. 2.18. ЯМР-спектр 27А1 с вращением
под магическим углом при 104,22 МГц.
а — цеолит NaY; б — v-оксид алюминия; в —
берилл [Fyfe et al., 1983].
экранирование и / — спиновой
угловой момент. Когда 6 = 54°44',
(3 cos26 —1) становится равным
нулю и мы получаем изотропный хим-
сдвиг. Это значение 8 называется
магическим углом, и вращение
образца около оси, наклоненной под
этим углом к магнитному полю,
элиминирует Ht (аниз) при условии, что
частота прецессии близка к размаху
частоты сигнала. Если частота
прецессии меньше, можно получить
центральную линию с изотропным
химическим сдвигом, с несколькими
боковыми полосами.
Мы видим, как вращение под
магическим углом и распаривание
высокочастотным полем делают
разрешение подобным таковому для
жидкой фазы. Интенсивность
сигналов от ядер с меньшей
концентрацией увеличивается посредством
использования кросс-поляризованных
последовательностей импульсов.
Превосходные спектры разнообразных
ядер в твердом теле получены с
применением методов вращения под
магическим углом и
кросс-поляризации. Нужно заметить, что не существенно, действительно ли
концентрация интересующих ядер всегда мала,— главным критерием
является то, что второй член в уравнении (2.9) был мал. Поэтому
получены также спектры ядер со 100 %-й концентрацией
(например, 31Р и 27А1). В твердых телах, не содержащих ядер, подобных
протону (например, в силикатах), ни высокочастотное
распаривание, ни кросс-поляризация не пригодны; только вращение под
магическим углом может дать нербходимые спектры высокого
разрешения. ЯМР высокого разрешения в твердых телах достаточно
широко рассмотрен в литературе [Richards, Packer, 1981; Andrew,
1981; Fyfe et al., 1983].
На рис. 2.17 показаны ЯМР-спектры с вращением под
магическим углом в твердых телах для различных ядер; некоторыми дру-
50
-75
Мд от Al(H20)V
Структурная характеризация
103
I 1 1 1 1 1 1 1 1 I 1 ■ 1 1
~80 -90 -700 -770 -7 ZO -вО ~90 ~700 ПО
1 О J
-80 -90 -WO -HO -120 -8O -90 -WO -HO
Рис. 2.19. ЯМР-спектры 29Si в цеолитах гс вращением под
магическим углом при 79,8 МГц.
а — гмелинит; б — аналцит; в — морденит; г — Z-цеолит [Fyfe et£al.f
1983].
гими возможными ядрами являются 170, 23Na, 31Р, 57Fe, 113Cd и
119Sn. На рис. 2.18 приведены ЯМР-спектры с вращением под
магическим углом материалов, содержащих алюминий, чтобы показать,
как легко различаются октаэдрическая и тетраэдрическая
координации А1. На рис. 2.19 показаны спектры 29Si нескольких цеолитов,
чтобы подчеркнуть, как атомы кремния, по-разному
координированные атомами алюминия, проявляют широко различные значения
химсдвигов. Числа над пиками означают п в Si (nA\) с пятью
возможными значениями (от 0 до 4). Si—А1-упорядочение в цеолитах
не может быть установлено методами рентгеновской дифракции или
другими подобными методами; только благодаря использованию
ЯМР высокого разрешения мы добились знания существенных
моментов для этого важного класса материалов. Распределение А1
и Si по позициям в слоистых глинах также исследовано ЯМР-спект-
роскопией [Lipsicas et al., 1984]. ЯМР-спектры 27А1 алюмосиликат-
ных гелей показали, что атомы алюминия являются гексакоордини-
рованными в гелях, богатых алюминием, тогда как в гелях, богатых
кремнием, они тетракоординированы [Thomas et al., 1983]. ЯМР-
спектры под магическим углом 31Р в твердом РС15 показывают два
сигнала, обусловленные частицами РС14+ и РС1в~. ЯМР 19F
графитовых интеркалатов вскрыл природу частиц AsF^. [Moran et al., 1984].
ЯМР-излучение силикатов показало, что химсдвиг Si связан с углом
Si—О—Si [Smith, Blackwell, 1983]. Из наблюдаемой формы линии
недавно [Dupree, Pettifer, 1984] была проверена функция
распределения угла Si—О—Si в стеклообразном Si02, которая первоначально
104 Новые и улучшенные методы характеризации
была установлена из диффузного рентгеновского рассеяния [Moz-
zi, Warren, 1969]. Выведено полуэмпирическое соотношение между
химсдвигом кремния и несвязывающим расстоянием Si—T (Т—Si,
А1) в цеолитах и других силикатах [Ramdas, Klinowski, 1984];
имеется также соотношение между химсдвигом и расстоянием Si—О
[Grimmer, Redaglia, 1984]. Большая разница времен релаксации
в аморфной и кристаллической форме дает возможность изучить
эволюцию кристалличности силикатного стекла [Rao et al., 1985].
Распределение углов Si—О—Si в стекле может быть также
определено анализом профиля линий ЯМР [Selvaraj et al., 1985].
2.2.7. Электронная спектроскопия
Методы электронной спектроскопии стали основными для
исследования электронной структуры твердых тел и поверхности
[Rao, 1985; Mason et al., 1986]. Большинство из них включают
анализ кинетической энергии излученных или рассеянных электронов.
Среди наиболее важных методов электронной спектроскопии для
изучения твердых тел можно назвать фотоэлектронную
спектроскопию, использующую или рентгеновское (XPS), или УФ-изл учение
(UVPS), Оже-спектроскопию (AES) и спектроскопию потерь
электронной энергии (EELS). Все эти методы чувствительны к состоянию
о
поверхности и зондируют ~ 25 А твердого тела или меньше.
Следовательно, чистота поверхности и ультравысокий вакуум (~ 10~10—
10"11 торр) являются необходимыми условиями для изучения
твердых тел этими методами. Когда вместе с этими методами
используется дифракция низкоэнергетических электронов (LEED),
получается двумерная структура поверхности твердого тела; LEED также
обеспечивает информацию по реконструкции поверхности, когда
какие-либо атомы или молекулы адсорбированы на поверхностях.
Различные методы электронной спектроскопии широко рассмотрены
в литературе [Fiermans et al., 1978; Rao, Hedge, 1981; Rao, Sarma,
1982]. Мы коротко опишем принципы методов и укажем несколько
важных применений.
В фотоэлектронной спектроскопии монохроматический
фотонный пучок ударяется о твердое тело, и энергия получающихся при
этом фотоэлектронов анализируется. Энергия связи электронов Ев
получается из уравнения энергетического баланса
hv = EB + Eh + Ф, (2.11)
где Ek — кинетическая энергия электрона, Ф— рабочая функция
твердого тела. В ультрафиолетовой фотоэлектронной спектроскопии
обычно используются излучения Hel (21, 22 эВ) и Hell (40,8 эВ).
В XPS обычно используют излучение А1Ка (1486,6 эВ) и MgKa
(1253,6 эВ).
Структурная характеризация
105
В Оже-спектроскопии падающий электронный пучок вызывает
ионизацию остовного электрона, и эта остовная уровневая вакансия
заполняется за счет безызлучательного электронного перехода с
высокоэнергетического уровня с одновременным излучением Оже-
электрона с более высокого уровня. Таким образом, энергия Оже-
перехода KL1L23 в случае атома с эффективным атомным номером
Z задается уравнением
Якцц, (Z) = Ек (Z) - Еч (Z) - ЕЧз (Z + А) - 0. (2.12)
Здесь конечное состояние остается с двумя остовными вакансиями
по отношению к нейтральному атому и уровень L23 будет иметь
эффективный атомный номер на А выше, чем однократно
ионизованный атом. Энергия Оже-перехода не зависит от энергии первичных
электронов. Оже-спектроскопия особенно полезна в определении
степеней окисления элементов и характеризации элементов на
поверхности твердого тела в присутствии или в отсутствие адсорбции
газов. Сканирующая Оже-микроспектроскопия дополняет
сканирующую электронную микроскопию для картирования элементов в
материалах.
В спектроскопии потерь электронной энергии измеряют потерю
в энергии падающих электронов, вызванную взаимодействием с
веществом. В зависимости от энергии первичного пучка можно
получить информацию о колебательных и электронных переходах,
включая адсорбированные частицы; также может быть получена ценная
информация об электронных состояниях поверхности адсорбента
(субстрата). Информация по колебательным спектрам
адсорбированных молекул, полученная из EELS, подобна таковой, полученной
из отражательно-абсорбционной инфракрасной спектроскопии.
Для колебательной EELS энергия первичных электронов порядка
4 эВ, тогда как для изучения электронных возбуждений
используется энергия порядка 100—200 эВ.
Фотоэлектронная спектроскопия обеспечивает ценную
информацию, как о валентных зонах, так и об остовных уровнях твердых тел.
Появление синхротронного излучения делает фотоэлектронную
спектроскопию более мощным методом, так как широкая область энергий
излучения обеспечивает большую глубину проникновения. Можно
различить электронные состояния поверхности и объема твердого
тела, используя надлежащее излучение. EELS предоставляет
возможность изучения как колебательных, так и электронных
возбуждений в поверхностных частицах (как адсорбата, так и адсорбента)
в зависимости от энергии первичного электронного пучка. Низко-
энергетичная EELS во многом подобна ИК- или Раман-спектрам
и соответствует колебательному спектру твердого тела и адсорбата.
Особенно полезна EELS в изучении взаимодействий газ — твердое
тело применительно к катализу. Недавние работы по поверхности
ясно выявили необходимость использования нескольких методов
106 Новые и улучшенные методы характеризации
"1 I I I I I | I I 1 1 1 Т 1 1 1 1 1 1 1 1 1 1 1 1 | 1 1
О 5 10 О 5 Ю
Энергия связи, эВ Энергия связиу эВ
Рис. 2.20. UVPS (Hell-спектры) в области валентной зоны, показывающие
эффекты конечного состояния.
а — Sm; б — ТЬ. Валентная зона Sm показывает сосуществование как двух-, так и
трехвалентных частиц из-ва валентной нестабильности (смешанная валвнтнозть) [Rao, Sarma,
1982].
для получения полезной и стоящей информации. Так, чтобы изучать
поверхностное окисление металлов, можно использовать не только
UVPX, XPS, AES и LEED, но и другие методы, такие как
электронно- или фотостимулированная десорбция. Аналогично, чтобы
исследовать взаимодействие такого газа, как СО, с металлом,
применяют EELS и спектры тепловой десорбции помимо XPS, UVPS, AES
и LEED.
Фотоэлектронная спектроскопия широко используется для
исследования валентных связей металлов, сплавов и других
неорганических твердых тел. Помимо обеспечения экспериментальных
данных о плотности состояний UVPS и XPS показывают эффекты
конечных состояний при переходе в области валентных зон как для
переходных, так и редкоземельных металлов и их соединений.
Структура, обусловленная эффектами конечного состояния, полезна в
идентификации смешанных валентных и спиновых состояний
(рис. 2.20 и 2.21). Спектры остовных уровней (в XPS) оксидов
металлов и других соединений показывают характеристику энергии
связи металла в определенном окисленном состоянии. XPS-энергия
связи увеличивается с повышением степени окисления металла, как
и химсдвиги рентгеновских (К или L) краев поглощения; эти две
величины пропорциональны одна другой. Характеристичные спин-
Структурная характеризация
107
Энергия связи, эВ 4
Рис. 2.21. а — UVPS (Hell-спектры) FeO, Fe304 и Fe203, показывающие
структуры конечных Зй-состояний [Alvarado et al., 1979]; б — теоретический
спектр Fe304, рассчитанный в терминах конечных состояний Fe2+ и Fe+3
[Alvarado et al., 1976].
орбитальные расщепления (пр) и других уровней найдены в XPS
соединений редкоземельных и переходных металлов. ns-Орбитали
обнаруживают расщепление благодаря обменному взаимодействию
(с d-или /-орбиталями), причем это взаимодействие становится
максимальным, когда d- или /-орбиталь заполнена наполовину. Другими
электронными взаимодействиями, обнаруживающими себя в
спектрах остовных уровней, являются конфигурационные взаимодействия,
включающие 3$-состояние переходных металлов, Ы —
^-взаимодействия в соединениях редкоземельных элементов и взаимодействия
типа переноса заряда (металл — лиганд). Благодаря
взаимодействиям типа переноса заряда вблизи остовных уровней металла (а иногда
и остовных уровней лиганда) обнаруживаются сателлиты и их
интенсивность критично зависит от перекрывания орбиталей металла
и лиганда. Сателлиты обнаружены как при более высоких, так и при
более низких энергиях относительно главного пика остовного
уровня. На рис. 2.22 показаны такие типичные сателлиты.
Спектры остовных уровней полезны в изучении смешанной
валентности (валентной нестабильности или межконфигурационных
флуктуации) в редкоземельных системах (например, SmS, Се),
которая появляется, когда Еехс = Еп — {Еп_г + Ее) « 0, где (Еп —
108 Новые и улучшенные методы характеризации
Си О
i i i i i i i i ' ' ' i ' ' ' ■ i '
О 5 Ю 15
Энергия связи, эВ
■ i ■ '
20
Рис. 2.22. XPS СиО и ШО в 2р-
области металлов, показывающие
присутствие сателлитов [Sarma et
al., 1983].
— Еп_х) — разница в
энергии 4/п и 4/71"1 состояний, а
Ее — энергия промотирован-
ного электрона. Временная
шкала этих флуктуации
меньше, чем 10"11 с, что су-
25 щественно короче, чем
шкала в мессбауровских
исследованиях, но больше шкалы в XPS (<C 10"16 с). Поэтому XPS
показывает присутствие различных частиц, не обнаруживаемых мес-
сбауровской спектроскопией и другими методами [Rao et al.,
1979; Rao, Sarma, 1980]. Соответственно мессбауровская
спектроскопия не показывает Fe3+- и Ге2+-состояний в Fe304 при
комнатной температуре, но фотоэлектронная спектроскопия
обнаруживает их (см., например, рис. 2.21). Фотоэлектронной
спектроскопией можно легко исследовать переходы металл-диэлектрик в
твердых телах, так как металлические фазы показывают резкий обрыв
в интенсивности на уровне Ферми. Изменение спинового состояния
иона металла обычно сопровождается изменениями в валентной
связи и в остовных уровнях. XPS используется для анализов состава
(при изучении поперечных
разрезов). XPS с угловым
разрешением (вместе с рентгеновской
фотоэлектронной дифракцией)
также дает ценную
структурную информацию и элементный
анализ [Evans et al., 1979].
Оже-электронная спект роско-
пия широко используется для
анализов состава, особенно для
микроанализа поверхности.
Химсдвиги Оже-сигналов, так
же как и отношение интенсив-
ностей Оже-линий лиганда
(скажем, кислорода) и металла,
использованы для исследования
поверхностного окисления ме-
1000 \900 800
\КинетическаЯ{
энергия, эВ
"45^5 "23
Рис. 2.23. Оже-спектры соединений
меди и ниобия.
А — Cu(L,VV); В — Cu(L,M23V); С—
Cu(LaM23M2a) [Yashonath et al., 1983],
200 180 160 140
Кинетическая энергия, эВ
Структурная характеризация
109
/iF«0
TF«7°3
-tr^t
• f«t)
20
-F.,0,
,F«»
•FtO /
Рис. 2.24. Соотношение между числом валентных
электронов N и интенсивностями линий в Оже-спектре
железа и его оксидов: Z — степень окисления; а —
L3M46M46/L3M23M46; б — ЬзМ4бМ4б/Ь3М2зМ2о; в —
ЬзМ2зМ45/Ь3М2зМ2з [Rao, Sarma, 1982].
таллов. Показано, что отношения интенсивностей Оже-линий
металла связаны с числом валентных электронов (рис. 2.23) и,
значит, могут быть использованы для изучения поверхностного
окисления или для определения степени окисления металла. На
рис. 2.24 показано, как отношения интенсивностей Оже-линий для
железа и его оксидов связаны с числом валентных электронов
[Raoet al., 1980]. Кроме того, подобные соотношения имеют силу и
для соединений второго и третьего ряда переходных металлов
[Yashonath et al., 1983]. В дополнение к изменениям во
внутриатомных Оже-процессах оксиды металлов и подобные системы также
обнаруживают межатомные Оже-переходы, которые могут иметь
диагностическое значение.
EELS адсорбированных частиц в значительной степени
проясняет механизмы каталитических реакций. Например, СО,
адсорбированный на металлах, дает характеристичные частоты валентных
колебаний, благодаря мостиковым и концевым частицам; кроме того,
при более низких частотах видны валентные колебания металл —
углерод. Адсорбция этилена и ацетилена на металлах
сопровождается перегибридизацией, как свидетельствуют С — Н- и С — С-
валентные колебания, обнаруживаемые в области, обычной для
насыщенных углеводородов. Изучения адсорбированных частиц
методом EELS дополняются колебательными спектрами
металл-кластерных соединений, содержащих подобные молекулярные
фрагменты. Другими используемыми для характеризации поверхности
методами являются масс-спектроскопия вторичных ионов,
спектроскопия ионного рассеяния и метод термической десорбции, причем
последний дает десорбционное поведение адсорбированных частиц
как функцию температуры. Большинство экспериментов используют
110 Новые и улучшенные методы характеризации
больше чем один метод для исследования поверхностей, и
коммерческие спектрометры предлагают различные комбинации методов,
такие как UVPS - XPS, AES - SIMS — XPS, XPS - EELS —
LEED. Эффективность различных методов электронной спектроско*
пии и родственных методов была рассмотрена [Riviere, 1982]. Brems-
strahlung-изохроматичная спектроскопия получает использование
для исследования незанятых уровней [Laubschat et al., 1984].
2.2.8. Другие спектроскопические методы
Абсорбционная и флюоресцентная спектроскопия в УФ и
видимой области, ИК- и Раман-спектроскопия так же, как и
различные резонансные спектроскопии, широко используются в
характеризации твердых тел. Спектроскопия в УФ и видимой области зондирует
электронные переходы определенных атомов или ионов в твердом
теле, обеспечивая информацию о степенях окисления ионов, их
локальной симметрии и дефектных центрах. Этот метод особецно
подходит, когда поглощающий ион присутствует в малой
концентрации в непоглощающей матрице. Типичным примером является Сг3+
в оксидных решетках, этот ион в матрице А1203 (рубин) дает
интенсивное красное окрашивание, в Be3Al2(Si04)3 (изумруд) —
прекрасный зеленый цвет и в LiNb03 — тусклый зеленый цвет. Ион
занимает октаэдрические позиции во всех трех случаях, и различия в
оптическом поглощении возникают из-за неодинаковых
кристаллических полей. Оптическое поглощение и люминесценция в
широкозонных полупроводниках, таких как GaAs, полезны для
характеризации оптической зонной щели, экситонов, центров
рекомбинации и т. д. ИК- и Раман-спектроскопия обеспечивают существенную
информацию по колебаниям атомов и молекул в твердых телах.
Полный анализ колебательного спектра твердого тела в терминах
нормальных колебаний и правил отбора может дополнять структурные
определения дифракционными методами. Кроме того, ИК- и Раман-
спектроскопия могут быть использованы для изучения фазовых
переходов (поведения мягкой моды, если таковая имеется), явлений
порядок — беспорядок, адсорбированных частиц (методом SERS,
IRAS и подобными специальными методами) и для идентификации
фаз и смеси. Романовская микроскопия, кроме того, может быть
полезна в материаловедении, в особенности при изучении стекол.
Электронный спиновый резонанс (ЭПР) обнаруживает
присутствие неспаренных электронов, локализованных на парамагнитных
ионах, или парамагнитные дефекты в твердом теле. ЭПР главным
образом применим к изоляторам с низкими диэлектрическими
потерями. Из наблюдаемых расщеплений резонанса можно получить
информацию о кристаллическом поле и спин-орбитальном
взаимодействии парамагнитного иона. С хорошим разрешением можно видеть
сверхтонкое и даже суперсверхтонкое расщепление резонанса.
Структурная характеризация
111
Сверхтонкое расщепление возникает из взаимодействия между
спином электрона и ядерным спином, тогда как суперсверхтонкое
расщепление является следствием взаимодействия спина электрона с
ядерными спинами ближайших атомов. ЭПР-исследования обычно
проводятся на монокристаллах, содержащих парамагнитные ионы
в маленькой концентрации. Такие ионы, как Мп2+ и Gd3+, имеющие
S-основные состояния, являются полезным зондом для изучения
различных типов структурных изменений (анти- и сегнетолектриче-
ских или переходов Яна — Теллера) в твердом теле. Органические
спиновые зонды могут быть использованы для исследования стекол,
пластических кристаллов и других конденсированных систем,
составленных органическими молекулами.
2.2.9. Заключительные замечания
Мы кратко рассмотрели некоторые важные достижения в
методах структурной характеризации. Имеется много других методов,
таких как мессбауровская спектроскопия, аннигиляция позитронов
и резерфордовское обратное рассеяние, которые имеют широкие
применения. Мессбауровская спектроскопия особенно полезна для
исследования различных степеней окисления, спиновых состояний
и координации ионов металлов, фазовых переходов, магнитного
упорядочения, факторов Дебая — Уоллера и т. д. Применения этого
многостороннего метода в химии твердого тела широко рассмотрены
в литературе.
Важно отметить, что сочетание методов часто необходимо для
понимания явления в целом или решения структурных проблем в
химии твердого тела. Все средства химии твердого тела, обычно
используемые для исследования кристаллических твердых тел,
являются также полезными для исследования аморфных твердых тел,
хотя некоторые методы особенно подходящи и более эффективны для
изучения аморфного состояния. Так, рентгеновское рассеяние дает
полезные функции радиального распределения аморфных
материалов, но EXAFS дает длины связей и координационные числа; ЯМР-
спектроскопия, рассеяние света и нейтронов являются другими
важными методами для изучения таких твердых тел. Комппгоновское
рассеяние [Williams, Thomas, 1983] также может обеспечить
информацию о природе ближнего порядка; этот метод показывает,
например, что аморфный углерод более близок графиту.
Разнообразие методов используется для изучения глин, силикатов и
катализаторов, чтобы понять связь между структурной химией и
реакционной способностью (см., например, [Thomas, 1984]).
Несомненно, в дальнейшем будут развиты новые методы и
улучшены старые. Таким многообещающим методом для поверхностной
топографии на атомном уровне является сканирующая туннельная
микроскопия [Binning et al., 1982]. Этот метод использует только
112 Новые и улучшенные методы характеризации
связанные частицы, не требует линз и не является разрушающим.
Туннельные эффекты связаны как со структурой, так и с химией
периодических и непериодических поверхностей [Binning, Rohrer,
1984].
2.3. ХАРАКТЕРИЗАЦИЯ СОСТАВА И ЧИСТОТЫ
Химическая характеризация твердых тел обычно включает
характеризацию основной фазы, которая состоит в определении
стехиометрии, гомогенности и степени окисления, если
присутствует многовалентный элемент, и характеризацию сопутствующих фаз
и примесей. Наиболее важные доступные методы для этих целей
вместе с их точностью, чувствительностью и областью применения
даны в табл. 2.2.
Одна из главных проблем в приготовлении твердых тел —
необходимость выяснить, является ли продукт, полученный
твердофазной реакцией, одной фазой или смесью фаз. Когда продукт
однофазный, его легко охарактеризовать химическим и рентгеновским
дифракционным методами. Твердофазные реакции часто дают
поликристаллические смеси фаз. Полный химический анализ и рентгено-
фазовый анализ порошков сами по себе малополезны в
идентификации фаз. Исследование под поляризационным микроскопом могло
бы обнаружить присутствие смеси фаз. Выращивание
монокристаллов является альтернативой для целей полной характеризации, но
не всегда возможно вырастить монокристаллы всех фаз смеси.
Рентгеновский микроанализ в электронном микроскопе (аналитическая
электронная микроскопия, рассмотренная в разд. 2.2) наиболее
полезен для характеризации поликристаллических смесей,
полученных по многокомпонентной твердофазной реакции [Cheetham,
Skarnulis, 1981; Jefferson, 1982]. Этот метод обеспечивает средства
идентификации различных присутствующих фаз и определения
состава индивидуальных кристаллитов в смеси поликристаллов. Он
подобен электронному микроанализу, за исключением того, что в нем
используются тонкие кристаллы. В пределах тонкого кристалла
отношение концентраций двух элементов прямо пропорционально
отношению интенсивностей характеристического рентгеновского
излучения от этих элементов
^JLy = kXyiJIy) (2.1о)
где Сх — Су — концентрация элементов х и у в кристалле, а 1Х
и 1У — характеристичные интенсивности рентгеновского излучения
этих двух элементов. Это приближение, известное как метод
отношения, особенно применимо для характеризации, использующей
трансмиссионный электронный микроскоп (включая STEM).
Единственное, что необходимо, это использовать в качестве стандартов
хорошо охарактеризованные соединения, содержащие элементы
Характеризация состава и чистоты
113
Таблица 2.2
Характеризация состава твердых тел
Метод
1. а) Гравиметрия
б) Титриметрия
2. Электрохимические
методы:
а) кулонометрия
б) полярография
3. Тонкослойная
хроматография
4. Оптическая
спектроскопия:
а) спектрофотометрия
б) эмиссионная
спектроскопия
в) пламенная атомно-
абсорбционная,
атомно-флюорес-
центпая
спектроскопия
5. Масс-спектрометрия
6. Нейтронно-активаци-
онный анализ
7. Рентгеновская
флюоресценция
8. Электронно-зондовый
микроанализ и
рентгеновский эмиссионный
анализ в электронном
микроскопе
9. Спектроскопия потерь
электронной энергии в
электронном
микроскопе
Применение
Содержание основной
и сопутствующей
фаз
То же и примеси
Содержание основной
фазы
Содержание основной
и сопутствующей
фаз, примеси
Сопутствующая фазы
и примеси
Примеси
»
»
»
»
Содержание основной
и сопутствующих
фаз
Гомогенность
основной и
сопутствующих фаз
Чувствительность
1—100 мг
1—10 мг
10"2 М в растворе
В растворе
(10—100).10"e
0,001-Ю"6
10—1000 мкг
<0,005.10"6в
растворе
(0,1—100).10"e
(ОД—10).10"в в
растворе
(0,002-1,0). Ю-6
в растворе
(0,001—0,1). 10"в
(0,001-0,01)-Ю-6
(20—200).10"в
ОД -10~в с
предварительным
концентрированием
0:01—ОД % в
пятне диаметром
1—5 мкм
Определяемый
минимум — 10 "18—
10-2о г
Точность, %
0,003-0,01
од
0,01
0,001-0,005
ОД—2
5-10
5-50
5—10
5—10
0,5—5
5-10
5-20
2—10
ОД—0,5
2-10
0,5
Минимально
определяемая
доля
0,1-3
хку, для определения константы кху; при помощи этой величины
можно определять отношение концентраций CJCy в неизвестном
соединении. Этот метод применим к множеству твердотельных систем,
таких как оксидные твердые растворы, смешанные сульфиды
металлов, тройные оксиды и сплавы. Ограничением этого метода является
8 Ч. II. Р. Рао, Дж. Гопалакришнан
114 Новые и улучшенные методы характеризации
то, что его обычно нельзя использовать для определения элементов
легче натрия; однако, используя герметичные детекторы, обходят
эту проблему, и можно, например, определить кислород в оксидах.
Спектроскопию потерь электронной энергии в электронном
микроскопе можно использовать для проведения количественного и
качественного анализа твердых тел, и этот метод является в анализе
состава «абсолютным» в отличие от рентгеновского эмиссионного
анализа, где требуется эталон [Egerton, 1982]. Эффективность EELS
как количественного аналитического метода (особенно, когда это
проводится в электронном микроскопе), однако, не является
общепринятой.
Химическая чистота является важным критерием в химии
твердого тела, и для достижения этого прилагаются большие усилия,
как по распространению стандартных методов очистки, так и по
развитию новых. Однако должно быть ясно, что нет простого набора
методов, подходящих для решения всех проблем очистки. Точный
выбор требуемого метода будет зависеть от химии материала и
конкретной примеси, от которой необходимо избавиться. По природе
методы очистки могут быть физическими или химическими.
Физические методы включают сублимацию, отгонку летучих примесей,
перекристаллизацию из расплава, жидкостную экстракцию и
хроматографию, тогда как химическими методами являются ионный
обмен, электролиз жидкостей или самого твердого тела и очистка
при помощи химических реакций. Наиболее важным методом из
физических методов очистки твердых тел является зонная плавка;
метод основан на факте, что растворимость примеси в твердой и
жидкой фазе разная. В зависимости от степени чистоты для определения
количества примеси используется соответствующий метод (см.
табл. 2.2). Например, электросопротивление может быть мерой
концентрации примеси в проводящих материалах.
Элементарным методом, полезным в определении стехиометрии
и степени окисления, является окислительно-восстановительное
титрование. Например, иодометрическое титрование может быть
использовано для определения Ni3+ в La2 Ni04 или Мп4+ в LaMn03. Харак-
теризация нестехиометрических составов часто включает
определение стехиометрии такими методами, как термо- и гравиметрия с
последующим определением структуры электронной микроскопией,
рентгеновской дифракцией и подобными методами, чтобы найти,
являются ли дефекты (ответственные за дефицит или избыток
анионов) упорядоченными. Измерение ЭДС с использованием ячеек с
твердотельными электролитами и электропроводности при
различных парциальных давлениях кислорода или любых других
газообразных частиц, участвующих в равновесии дефектообразования
(см. гл. 5 и 6), обычно также применяется для характеризации
нестехиометрических соединений. Характеризации нестехиометрических
оксидов рассмотрена в литературе [S^rensen, 1981].
3. СТРАТЕГИЯ СИНТЕЗА
3.1. ВВЕДЕНИЕ
Во всех исследованиях веществ в твердом состоянии
решающим фактором является возможность получения чистых, хорошо
окристаллизованных образцов. Следовательно, знание различных
экспериментальных методов, пригодных для синтеза твердых
веществ, является важной и неотъемлемой частью химии твердого
тела [Hagenmtiller, 1972; Honig, Rao, 1981]. Анализ развития науки
о твердом состоянии показывает, что во многих случаях именно
синтез нового соединения является толчком к новым направлениям
в научном исследовании (табл. 3.1 и 3.2). Для многих специалистов
синтез твердых веществ может означать синтез монокристаллов из
элементов или простых соединений (например, Si, Ge, III—V-полу-
проводники, галогениды щелочных металлов и т. д.) с целью
исследования специфических свойств или для технологических
приложений. Однако синтез твердых веществ является более общей
деятельностью, результаты которой особенно зависят от химиков.
Существует ряд подходов к синтезу твердых веществ и к росту кристаллов.
Прогресс в технологии дал возможность исследователям,
работающим в области химии твердого тела, использовать широкий ряд
Таблица 3.1
Некоторые ранние синтезы твердых тел, которые привели к важным
результатам
Прототип
InP
Zr02(CaO)
Натриевый р-оксид
алюминия
V3Si
ВаТЮ3
LiNbOg
BaFe12019
LnNi5 (Ln —
редкоземельный элемент)
Аморфный Si
Са5(Р04)3Х : Sb3+, Mn2+
ZnS/CdS
Первое сообщение
Thiel [1910]
Ruff [1929]
Stillwell [1926]
Wallbaum [1939]
Tammann [1925]
Sue [1937]
Adelskold, Schreweli-
us [1938]
Klemm [1943]
Konig [1944]
McKeagetal. [1949]
Kroger [1940]
Последующее развитие
HI—V полупроводники
Твердый электролит,
кислородный сенсор
Твердый электролит, Na—S-ба-
тарея
Высокотемпературные
сверхпроводники А15
Сегнетоэлектрики,
керамические конденсаторы
Нелинейная оптика
Ферриты, устройства памяти
Сильные магниты, материалы
для хранения водорода
Солнечные элементы
Фосфор для флюоресцентного
источника
Фосфор для катодно-лучевой
труоки
116
Стратегия синтеза
Таблица 3.2
Некоторые недавно синтезированные материалы технологического назначения
Сиалон (Si, A1)3(0, N)4
Smo,4Y2,eGai,2Fe3,8°i2
MJMOeSeg и родственные фазы Шевре-
ля
LnRh4B4 (Ln — редкоземелышый
элемент)
Алюмосиликаты (включая ZSM-5)
РЬ2Ки2_хРЬ*+07_у
Полиацетилен (п- и р-тяп
допирования)
Диацетиленовые полимеры
Органические жидкие кристаллы
Высокотемпературные керамики
Пузырьковые устройства памяти
Высокополевые сверхпроводники
Сосуществование сверхпроводимости и
магнетизма
Катализ (например, метанол-бензин)
Электрокатализатор (для использования
в кислородных электродах)
Твердотельные батареи
Материалы для нелинейной оптики
(лучше, чем LiI03)
Дисплейные устройства
условий в препаративных целях. Таким образом, общими приемами
при синтезе стали сверхбыстрое закаливание веществ от очень
высоких температур, нагревание интенсивными лазерными лучами,
плавление при нагревании электронным пучком или настыльным методом
и использование высоких давлений. Если требуются
некристаллические твердые вещества, для их синтеза применяются методы
напыления, закаливания и др. При решении проблемы получения
веществ, которые требуют высоких температур, часто с успехом
используются простые химические методы.
Можно выделить четыре категории синтеза твердых веществ:
1) получение ряда соединений для исследования специфических
свойств: например, синтез перовскитов — для изучения
электрических или синтез шпинельных ферритов — для изучения магнитных
свойств; 2) получение неизвестных членов структурно родственного
класса твердых веществ, для того чтобы расширить (или
экстраполировать) взаимосвязь структура — свойство, как, например,
синтез слоистых халькогенидов и их интеркалатов, или новых
производных TTF-TCNQ — с целью изучения их сверхпроводимости;
3) синтез нового класса соединений (например, сиалоны (Si, А1).{
(О, N)4 или допированные полиацетилены); 4) получение известных
веществ с заданными характеристиками (кристалличность, форма,
чистота и т. д.), как в случае кристаллов Si, III—V-соединений и
галогенидов щелочных металлов. За синтезы, относящиеся к третьей
категории, несут ответственность химики.
В основе создания новых материалов, обладающих желаемыми
свойствами, лежит понимание кристаллохимии. Только тогда, когда
у нас есть правильное описание структуры (применительно к инте-
Введение
117
Т а б л и ц а 3.3
Некоторые гомологические ряды оксидов
Ti„02n_1
Mon°3n-l
w„o3n_2
рг„02„_2
Bi(W03)n
В'^П0ЗП+3
LanNin°3n-i
Ч.СОп08»-1
(Bi202) [An_1Bn03n+1]
^п+тРгРгп+1
AnBn° зп+2
4<тг<9 или 16<м<36
1 8<rc<12
n = 20, 24, 25 и т. д.
n = 4, 7, 9, 10, 11, 12
/t = 6, 8, 15 и т. д.
и = 1,2, 3
я = 2и выше
/г = 2 и выше
А — Ва, Bi и т. д.
В - Ti, Nb, W, Fe, Cr
n = 1-8
A — Sr, В — Ti
A — La; В — Ni
n = 1, 2, 3 и т. д.
A — Na, Ca
В — Nb
n * = 4, 5, 6, 7
* В этой системе между п = 4 и 4,5 известно большое число когерентно сросшихся
фаз с большими периодами.
ресующим нас свойствам), мы можем приступить к попыткам
синтеза. Как отмечалось в гл. 1, даже для одних только оксидов
металлов мы имеем несколько структур: перовскиты, шпинели,
бронзы и пирохлоры; в процессе характеризации и структурного
толкования оксидов идентифицированы новые семейства. В
соответствии с этим несколько членов из гомологических рядов оксидов
(табл. 3.3) идентифицированы скорее с помощью рентгеноструктурно-
го анализа или электронной микроскопии, чем химическим анализом,
причем их точный состав часто устанавливался при определении
структуры. Препаративная химия твердого тела становится
наиболее продуктивной тогда, когда есть тесная взаимосвязь между
синтезом, характеризацией (включая определение структуры) и
изучением свойств. Далее мы будем обсуждать методы получения твердых
веществ в различных агрегатных состояниях, начиная с аморфных
и микрокристаллических, через поликристаллические порошки и
кончая монокристаллами.
Полезно установить разницу между получением новых твердых
веществ и получением твердых веществ новыми методами.
Получение нового твердого вещества не обязательно подразумевает исполь-
118
Стратегия синтеза
зование нового метода синтеза. Можно привести ряд примеров,
когда получение новых твердых веществ с новыми структурами
и свойствами достигалось рутинными методами. Из таких
соединений типичными являются Na3Zr2PSi2012(NASICON), Na1+3CAln017+3C/a
(fj-форма), BaFe12019 и фазы Шевреля MMoeS8 (М — Си, РЬ и т. д.).
Все эти вещества получены обычным методом — взаимодействием
легкодоступных составных частей при высоких температурах
(керамический метод). При работе с сульфидами реакция проводится
в запаянных ампулах (по понятным причинам), но в других случаях
даже эта предосторожность не обязательна. Важность таких
синтезов заключается не в методе, а в подборе правильных составных
частей в правильной пропорции с учетом как химии, так и структуры
и свойств, требуемых для новой фазы. Это иллюстрируется синтезом
Na3Zr2PSi2012, твердого натриевого ионного проводника, где состав
выбирался с учетом предпочтительной координации включенных
атомов, устойчивости их состояний окисления, природы
образующегося каркаса и с учетом того, возможна ли в каркасе трехмерная
подвижность ионов натрия [Goodenough et al., 1976]. Другой
пример запланированного новаторского синтеза — получение
синтетического костного материала для протезирования [Roy, 1977]. Здесь
проблема заключается не только в синтезе химического соединения,
составляющего основу кости человека, а именно гидроксиапатита
кальция, но и в получении его 100%-й связности и пористости
натуральной кости. Этот синтез можно было бы выполнить двумя
путями: некоторые морские кораллы (СаС03) обладают такой же
пористостью и связностью, как человеческая кость, а арагонитная форма
СаС03 топотаксиально может быть превращена в гидроксиапатит
гидротермальным способом. Таким образом, реакция морских
кораллов с фосфорной кислотой в гидротермальных условиях дает
синтетический костный материал.
Синтез новых твердых веществ не всегда идет по плану,
препаративная химия твердого состояния внесла свой вклад в курьезность
открытий новых материалов. Открытие NaMo4Oe, прототипа
соединений с кластерной цепочкой, является примером такого вклада
[Torardi, McCarley, 1979]. Это произошло в результате совершенно
простого эксперимента, ставившего своей целью получение
NaZn2Mo30Q, натриевого аналога Li-содержащего кластерного
соединения LiZn2Mo308. С этой целью смесь Na2Mo04, ZnO, Мо02 и Мо
запаивалась в молибденовой трубке и нагревалась при 1370 К.
Вместо ожидаемого NaZn2Mo308 получились блестящие иглы
NaMo4Oe. Оказалось также, что кристаллическая структура
NaMo4Oe относится к самым уникальным оксидным структурам,
состоящим из бесконечных цепочек октаэдрических кластеров Мовэ
сочлененных противоположными ребрами (см. рис. 1.17).
Получение кристаллических материалов 119
3.2. ПОЛУЧЕНИЕ КРИСТАЛЛИЧЕСКИХ МАТЕРИАЛОВ
3.2.1. Керамические методы
Наиболее общий метод получения твердых веществ —
взаимодействие исходных компонентов при нагревании. Если все
компоненты твердые, то метод называется керамическим. Если один
из компонентов летучий или чувствителен к воздуху, реакция
выполняется в запаянных эвакуированных ампулах. Этим методом
получены тройные оксиды, сульфиды, фосфиды и т. д. Знание
фазовой диаграммы вообще полезно для выбора желаемого состава и
условий синтеза. Необходима некоторая предосторожность при
выборе материала контейнера; контейнеры из платины, Si02 и А1203
используются для синтеза оксидов металлов, тогда как графитовые
контейнеры подходят для сульфидов и других халькогенидов,
а также для пниктидов.
Керамические синтезы часто требуют относительно высоких
температур (вплоть до 2300 К), которые вообще можно достичь в печах
сопротивления. Дуговые и настыльные методы могут давать
температуры вплоть до 3300 К, тогда как высокомощные С02-лазеры —
до 4300 К.
Керамические методы обладают несколькими недостатками.
1. Если во время реакции расплав не образуется, вся реакция
должна идти в твердой фазе, сначала на границе фаз в точках контакта
между компонентами, а затем посредством диффузии составных
частей через полученную фазу. В процессе реакции пути диффузии
становятся длиннее и длиннее, а скорость реакции все меньше и
меньше; реакцию можно несколько ускорить, чередуя нагревание с
измельчением. 2. Нет никакого способа управления процессом
реакции; только методом проб и ошибок можно определить подходящие
условия, обеспечивающие полноту реакции. Из-за этого реакция
часто заканчивается образованием смесей реагентов и продуктов
реакции. Выделение требуемого продукта из таких смесей очень
затруднительно, если не невозможно. 3. Часто трудно получить
гомогенный по составу продукт, даже если реакция идет почти
до конца.
Несмотря на эти ограничения, керамические методы широко
используются в твердофазных синтезах. В качестве примера
успешного использования этого метода следует упомянуть синтез моно-
халькогенидов редкоземельных металлов, таких как SmS и SmSe.
Метод [Jayaraman et al., 1975] состоит в нагревании элементов,
сначала при пониженных температурах (870—1170 К) в
эвакуированных кварцевых ампулах; содержимое ампул затем
гомогенизируется, запаивается в танталовые ампулы и нагревается при
со 2300 К пропусканием через ампулу большого тока. Богатые
металлом галогениды лантаноидов и других ранних переходных элемен-
120
Стратегия синтеза
тов синтезированы с использованием танталовых контейнеров [Сог-
bett, 1980, 1981]. Несколько галогенидов и халькогенидов,
содержащих конденсированные металлические кластеры, также получены
керамическим методом [Simon, 1981]. Богатые металлом субоксиды
щелочных металлов получены по реакции металла с требуемыми
количествами кислорода до полного поглощения газа (р < 10~5 торр)
[Simon, 1975]; RbeO, Rb902, Csn03, Cs30, Cs40 и Cs70 являются
примерами таких фаз. Чрезмерная чувствительность к воздуху и
низкие температуры разложения — плавления делают очень
затруднительными синтез и исследование этих фаз. Эти оксиды,
следовательно, изучаются in situ. Для преодоления некоторых ограничений
керамического метода использовались различные его модификации.
Одна из них — уменьшение пути диффузии. В поликристаллической
смеси реагентов каждая частица имеет размер около 10 мкм, что
определяет расстояния диффузии со 10 000 элементарных ячеек.
При использовании методов сушки вымораживанием и распылением,
методов соосаждения, образования золя — геля и других подобных
методов можно уменьшить размер частицы до нескольких сот
ангстрем (см. разд. 3.3) и таким образом осуществить более тесное
смешение реагентов. В методе сушки распылением соответствующие
компоненты, растворенные в растворителе, распыляются в виде мелких
капелек в горячую камеру. Растворитель полностью испаряется,
остается однородная смесь реагентов, нагревая которую, при
повышенных температурах можно получить желаемый продукт. В методе
сушки вымораживанием реагенты в обычном растворителе
замораживаются в жидком азоте, а растворитель удаляется при низких
давлениях. В методе соосаждения катионы соответствующих металлов
соосаждаются из обычной среды, как правило, в виде гидроксидов,
карбонатов, оксалатов, формиатов или цитратов, которые далее
нагреваются при соответствующих температурах для получения
конечного продукта. Эти методы использовались для получения
поликристаллических образцов некоторых оксидов, таких как
ферриты, перовскиты и (J-оксиды алюминия. Золь — гель-процесс
[Johnson, 1981] включает в себя образование концентрированного золя
из оксидов или гидроксидов и превращение его в полутвердый гель
при удалении растворителя; удаление растворителя осуществляют
пропусканием мелких капелек золя через колонку с алифатическим
спиртом (например, 2-этил-1-гексанол). Для получения конечного
продукта дегидратированный гель нагревается при соответствующей
температуре. Этим методом получено несколько сложных оксидных
материалов, таких как NASICON, PbTi^^Zr^Og, твердые растворы
Th02 — U02 и модификации Zr2(P04)2S04 [Alamo, Roy, 1984];
золь — гель-метод становится также важным при синтезе
некристаллических твердых веществ [Scholze, 1984].
Получение кристаллических материалов 121
3.2.2. Химические методы
Усиленные попытки преодолеть ограничения керамического
метода привели к альтернативным путям твердофазного синтеза.
Новые методы, которые основаны на знании структурной химии
и реакционной способности твердых тел, не только обеспечили
синтез известных соединений высокой степени чистоты и гомогенности
при более низких температурах, чем в обычных методах, но и
привели к получению новых фаз. Из этих альтернативных методов
значительными оказались три следующих метода: метод твердофазного
предшественника и методы, основанные на топохимических редокс-
реакциях и топохимических ионообменных реакциях. Акцент во
всех этих трех методах делается на синтезе при низких
температурах, с тем чтобы получаемые продукты были в тонкоизмельченном
состоянии с большой поверхностью, что очень важно для катализа
и других областей применения. Более важно, что синтез при
температурах, значительно более низких, чем температуры спекания,
сохраняет существенные признаки исходной структуры с
минимальной структурной перестройкой (топохимические методы). Синтезы
топохимическими методами часто дают метастабильные фазы,
которые нельзя получить обычными методами.
Идеальное условие выполнения твердофазной реакции с целью
быстрого получения гомогенного продукта при более низкой
температуре состоит в том, чтобы обеспечить гомогенное смешение
реагентов на атомном уровне. Керамический метод и его модификации
этого, однако, не дают. Единственный путь — получение
индивидуальной фазы (химического соединения), в которой реагенты
находятся в требуемой стехиометрии. Такая твердая фаза, называемая
предшественником, при нагревании дает продукт с желаемой
стехиометрией и гомогенностью. Метод предшественника использовался
для синтеза тройных оксидов [Wold, 1980]; ферриты шпинельного
типа, MFe204(M — Mg, Mn, Co, Ni), получены термическим
разложением ацетатных предшественников общей формулы
М3Рев«(СНзСОО)17ОзОН-12С5Н5К. Преимущество этого метода состоит
в том, что предшественник можно получить в кристаллическом виде,
с высокой чистотой и с желаемым отношением M/Fe. Более того,
продукты являются гомогенными и образуются при довольно низких
температурах. Другими примерами синтеза оксидов металлов по
методу предшественника являются синтезы хромитов, МСг204, путем
разложения бихроматов, (NH4)2M(Cr04)2-6H20; синтезы BaTi03 и
LiCr02 путем разложения Ba[TiO(C204)2] и Li[Cr(C204)2(H20)2] и
синтез LaFe03 путем разложения La[Fe(CN)6]. Еще один пример
синтеза с предшественником — синтез керамических оксидов
гидролизом алкоксидов металлов.
Не всегда возможно найти подходящее индивидуальное
соединение-предшественник для синтеза всех необходимых фаз, так как
122
Стратегия синтеза
стехиометрия предшественника может не соответствовать
стехиометрии продукта. Недавно описан метод предшественника на основе
твердого раствора, который сохраняет все достоинства
предшественника — индивидуального соединения [Horowitz, Longo, 1978; Lon-
go, Horowitz, 1981; Vidyasagar et al., 1984, 1985]. Стратегия здесь
заключается в том, чтобы использовать изоструктурные соединения
с общим анионом так, чтобы они образовывали непрерывные ряды
твердых растворов. Этот метод использован для синтеза нескольких
тройных оксидов в системе Са—Мп—О, таких как Са2Мп308т
СаМп306, СаМп408 и СаМп7012, методом термического разложения
карбонатных твердых растворов Са^^Мп^СОд со структурой
кальцита. Ca^^Fe^COg применен для получения необычных ферритов;
CagFe^JVin^Og с тремя типами полиэдров из переходного металла
и кислорода получен именно таким способом; метод распространен
на предшественники — твердые растворы на основе гидроксидов,
оксалатов и нитратов. Соединения, подобные LaNi03, и четверные
оксиды получены с использованием гидроксидов со структурой
La(OH)3; ВаРЬ03, Ва2РЬ04 и BaSrPb04 — с использованием
нитратных предшественников.
Предшественники — твердые растворы — имеют несколько
преимуществ. 1. Реагирующие катионы смешиваются однородно, тем
самым исключаются диффузионные проблемы (диффузионный интер-
о
вал в этом методе составляет 10 А) и негомогенность состава
конечного продукта. 2. Конечный продукт образуется при значительно
более низкой температуре, чем при обычном керамическом синтезе;
это дает возможность проверки субсолидусных областей фазовой
диаграммы, что иначе было бы невозможно. Продукты, полученные
при более низких температурах, будут иметь большую удельную
поверхность, что является важным требованием в каталитических
синтезах. Метод предшественника — твердого раствора —
использован для низкотемпературного синтеза Мо — W-сплавов [Cheet-
ham, 1980] путем восстановления водородом (NH4)e[Mo7_:3CW:x.024].
Метод разложения из газовой фазы предшественников при
относительно низких температурах также используется для получения
твердых веществ. Так, SiC образуется при разложении CH3SiH3
или (CH3)2SiCl2, тогда как Si4N4 получается при взаимодействии
SiCl4 (или SiH4) и NH3; порошки сиалона — по реакции метакаоли-
на с газообразным аммиаком; сульфиды переходных металлов — по
реакции предшественников-оксидов, -сульфатов и -хлоридов с H2S
илиС82; пирохлоры типа Pb2 [Ru2_3cPbx+]07-y HBi2[Ru2_3cBix+]07_y—
низкотемпературным методом с использованием сильно щелочной
среды [Horowitz et al., 1981]. Высокотемпературная фаза (^> 1300 К)
Zr02 получается гидролизом циркониевых солей при комнатной
температуре, кубическая б-форма Bi203 может быть
стабилизирована при комнатной температуре введением таких добавок, как Y203
Получение кристаллических материалов 123
и W03, 7'^е2^з получается дегидратацией y-FeOOH органическими
основаниями [Desiraju, Rao, 1982].
Метод электролиза расплавленных солей используется для
синтеза таких оксидов, как голубые молибденовые бронзы.
Оксиды переходных металлов и халькогениды, МХП (М-металл),
обладающие слоистыми или цепочечными структурами, можно ин-
теркалировать при комнатной температуре литием или другими
щелочными металлами, получая при этом восстановленные фазы
АЛМХП (А — Li, Na, К); образование таких фаз впервые
обнаружено в 1959 г. [Rudorff, 1959]. Этот процесс отличается несколькими
особенностями, которые представляют интерес как с синтетической
точки зрения, так и с целью использования фаз в качестве катодов
в батареях [Murphy, Christian, 1979]. 1) Реакция интеркаляции
обратима и может выполняться химически или электрохимически;
2) реакция по своей природе является топохимической, с
минимальной структурной перестройкой «хозяина» — МХП; 3) как катионы
А, так и электроны, перенесенные в структуру «хозяина», обладают
значительной подвижностью в фазах АХМХП, делая их смешанными
ионно-электронными проводниками, что является основой их
успешного применения в качестве катодных материалов в твердотельных
батареях. Наилучшим из известных примеров интеркаляции
щелочного металла является интеркаляция лития в TiS2 с образованием
LixTiS2 (0 < ж< 1,0) [Whittingham, 1978; Whittingham, Chianel-
li, 1980]. Интеркаляцию можно проводить двумя способами. В
первом, химическом способе, интеркаляция лития в TiS2 проводится
с помощью раствора и-бутиллития в таком углеводороде, как ге-
ксан:
xC4H9Li + TiS2^LixTiS2 + -J C8H18.
Второй способ получения литиевых интеркалатов —
электрохимическое восстановление TiS2. Поликристаллический TiS2 в виде
электрода погружается в полярный органический растворитель
(например, диоксолан), в котором растворен хлорат (VII) лития; анодом
служит пластинка из LiAl. При замыкании двух электродов ионы
лития интеркалируют TiS2, при этом компенсирующие электроны
проходят по внешней: цепи. Электрохимическая интеркаляция
особенно благоприятна тем, что скорость реакции можно
контролировать приложением внешнего напряжения к ячейке; когда
напряжение превышает значение, соответствующее изменению свободной
энергии реакции интеркаляции AG, идет обратная реакция,
а именно реакция деинтеркаляции.
Интеркаляция лития проведена в большое количество структур-
хозяев МХП. Деинтеркаляция лития была проведена как
электрохимически, так и химически. Деинтеркаляция лития из 1лМХл
с использованием мягких окислителей, таких как I2/CH3CN и
124
Стратегия синтеза
Вг2/СНС13, предлагает низкотемпературный путь синтеза таких фаз,
как Ыд-МХтг и МХП, которые невозможно получить другим способом
(Lia.VS2 и LixV02); аналогично можно получить Mo6S8 взаимодействием
СиЛ.Мо688 с кислотой [Chevrel et al., 1974]. В табл. 3.4 приведены
характерные примеры интеркаляции и деинтеркаляции лития; из
таблицы видно, что интеркаляция лития наблюдается в широком
ряду оксидов и сульфидов, имеющих одно-, дву- и трехмерные
структуры. В большинстве случаев структурные признаки хозяина
в целом сохраняются; в некоторых случаях есть специфические
структурные изменения, которые могут рассматриваться в рамках
топохимического механизма, как, например, в LiRe03 и Li2Re03
[Cava et a]., 1982]. В LiRe03 и Li2Re03 хозяин Re03 претерпевает
значительное изменение при внедрении лития без разрыва связей;
каждая из 12-координированных полостей в каркасе Re03
превращается в две октаэдрические полости, которые заполняются литием.
Внедрение лития в плотноупакованные оксиды, такие как Fe203,
Fe304, Mn304 и Ti02, приводит к интересным структурным
изменениям. В случае Fe203 внедрение лития изменяет анионный
структурный мотив от гексагональной до плотной кубической упаковки.
Внедрение лития в Мп304 подавляет ян-теллеровское кооперативное
искажение в оксиде. Интеркалированный литием анатаз, Li0>r,TiO2>
превращается при ~ 770 К в шпинель LiTi204, которая имеет
сверхпроводящие свойства; деинтеркаляция лития из Li2FeS2
электрохимическими методами или иодом обратимо ведет к LiFeS2, который
имеет структуру LiTiS2.
Интеркаляция натрия и калия отличается от интеркаляции
лития; в слоистом AYMXn литий координирован всегда октаэдрически,
тогда как натрий и калий занимают октаэдрические или тригональ-
но-призматические позиции; октаэдрическая координация
предпочтительна для больших значений х и низших формальных степеней
окисления М; для меньших значений х и больших степеней
окисления М координация натрия и калия — тригонально-призматиче-
ская; цезий, интеркалированный в МХП, всегда имеет тригонально-
призматическую координацию. Интеркаляция натрия и калия в
слоистые оксиды и сульфиды МХ2 приводит к структурным
превращениям, включающим изменение в последовательности упаковки
анионного слоя.
Ряд неорганических твердых веществ, имеющих слоистую или
трехмерную сетчатую структуру, является твердыми ионными (кати-
онными) проводниками. Натриевые Р- и р "-оксиды алюминия —
типичные примеры таких веществ. Ионы натрия в этих твердых
веществах быстро движутся в слоях, что дает образование ряда
пустых позиций и легких маршрутов в движении иона. Коэффициенты
диффузии обычно порядка 10~7 см2-с"1. Твердые ионные
проводники, такие как р-оксиды алюминия, являются хорошими ионообмен-
никами. Обмен может проходить легко при комнатной температуре
Получение кристаллических материалов 125
Таблица 3.4
Интеркадяция и деинтеркаляция лития в матрицы состава МХП
Матрица
TiS2
vs2
NbS2(3R)
TiS3
MoS3
M02 (рутил)
Ti02 (анатаз)
Со02
vo2
V02(B)
Fe203
Fe304
Mn304
M0O3
v2o5
Re03
Примечания
Li:)CTiS2 : 0<^1. Фаза гомогенна по всему
составу
Li^VSa : 0<я<;1. Фазы получаются при де-
интеркачяции лития из LiVS2 с
использованием I2/CH,CN. Три различные фазовые
области: 0,25<д<0,33; 0,48<я<0,62 и
0,85<д^1, кроме VS2
Li05NbS2HLi070NbS2
Li2TiS3 и Li2+xTiS3 (0<*<1)
LixMoS3(0<:r<4)
Li^MOa (x^\) (M -Mo, Ru, Os, Ir)
LixTi02 (0<z<0,7)
LiQ 5Ti02 необратихмо превращается в
шпинель LiTi204 при 500 °C
Li^GoOcj (0<;г<1). Фазы, полученные при
электрохимической деинтеркаляции
LiCo02
LixV02 (0<а:<1). Фазы, полученные при
химической деинтеркаляции LiV02 с
использованием Вг2/СНС13
LixV02 (0<я<2/3). Химическая интерка-
ляция с использованием /г-бутиллития
Li3CFe203 (0<д;<2). При интеркаляции
анионный мотив превращается из ГПУ в
КПУ
LiJCFe304 (0<я<2). Подмотив Fe204 шпи-
нельной структуры остается без изменения
LixMn304 (0<я<1, 2). Внедрение лития
подавляет тетрагональное искажение Мп304
LixMo03 (0<*<1,55)
Ь^У205 (0<х<1,1). Интеркадяция лития с
использованием Lil
Li^ReOa (0<a:<2). Три фазы:
0<*<0,35; х= 1; 1,8<дг<2 ,
Литературный
источник
Whittingham
[1978]
Murphy et al.
[1977]
Whittingham
[1978]
Whittingham
[1978]
Jacobson et al.
[1979]
Murphy et al.
[1978]
Murphy et al.
[1982]
Mizushima et al.
[1980]
Vidyasagar, Gopa-
lakrishnan
[1982]
Murphy, Christian
[1979]
Thackeray et al.
[1982]
Thackeray et al.
[1982]
Thackeray et al.
[1983]
Dickens, Pye
[1982]
Dickens et al.
[1979]
Murphy et al.
[1981]
Cava et al. [1982]
126
Стратегия синтеза
как в водной среде, так и в среде расплавленной соли. Так,
натриевый р-оксид алюминия обменивается ионами гидроксония и
аммония и другими одновалентными и двухвалентными катионами, что
приводит к образованию различных Р-оксидов алюминия [Farring-
ton, Briant, 1978; Tofield, 1982]. Как известно, ионный обмен в
неорганических твердых веществах — общее явление, не ограниченное
одними твердыми ионными проводниками. Изучены кинетические и
термодинамические аспекты ионного обмена в неорганических
твердых веществах [England et al., 1983]. Результаты показывают,
что ионный обмен — довольно широко распространенное явление,
имеющее место даже при таких малых коэффициентах диффузии,
как 10"12см2-с-1, и при температурах, намного ниже температур
спекания. Ионный обмен можно проводить со значительной
скоростью даже в стехиометрических твердых веществах; вакансии для
подвижных ионов (обусловленные нестехиометричностью или
допированием) не требуются. Поскольку обмен идет топохимически,
удается получать метастабильные фазы, что невозможно при
высокотемпературных реакциях.
Показано, что большое количество оксидов металлов, имеющих
слоистые, туннельные или плотноупакованные структуры, могут
обменивать ионы в водных растворах и в среде расплавленной соли
с образованием новых фаз [England et al., 1983]. Типичными
примерами таких реакций являются следующие:
LiNO,
a-NaCr02 -+ a-LiCr02,
^300° С; 24ч £
AgNO,
КАЮ2 *-+> p-AgA102,
a-LiFe02^-^CuFe02.
Структура каркаса при обмене в основном сохраняется за
исключением небольших изменений, связанных со структурными
особенностями входящего иона. Так, когда a-LiFe02 превращается в CuFe02
путем обмена с расплавленным CuCl, структура изменяется от а-
NaCr02 до структуры делафоссита, чтобы обеспечить линейную
анионную координацию Си+. Аналогично, когда КАЮ2 превращается
в p-AgA102 путем ионного обмена, здесь имеет место изменение
структуры от кристаболитного типа до обычного упорядоченного
вюртцитного типа; изменение, вероятно, идет так, чтобы обеспечить
тетраэдрическую координацию Ag+. Есть сообщение об интересной
реакции ионного обмена — о превращении LiNb03 и LiTa03 в
HNb03 и НТа03 соответственно (путем обработки горячим водным
раствором кислоты) [Rice, Jacket, 1982]. Обмен лития на протоны
сопровождается топотактическим превращением ромбоэдрической
структуры LiNbOg в кубическую перовскитовую структуру HNb03.
Предлагаемый механизм превращения состоит в обратимом процессе
превращения кубического Re03 в ромбоэдрические LiRe03 и
Li2Re03 [Cava et al., 1982], включающем скручивание октаэдров
Получение кристаллических материалов 127
вдоль кубического направления [111], с тем чтобы превратить 12-
координированные туннельные пустоты перовскита в две 6-коорди-
нированные позиции ромбоэдрической структуры. Существует
другое интересное структурное изменение, сопровождаемое ионным
обменом [Delmas et al., 1982]. В Na0t7CoO2, в котором
последовательность анионных слоев АВВАА, ионы кобальта находятся в
чередующихся межслоевых октаэдрических позициях, а ионы натрия —
в тригональной призматической координации между блоками Со02.
Когда это вещество обменивается ионом с LiCl, получается мета-
стабильная форма LiCo02 с последовательностью слоев АВСВА;
около 520 К фаза необратимо превращается в устойчивый LiCo02
(АВСАВС).
Большое число неорганических твердых веществ обменивается
протонами с образованием новых фаз, некоторые из них обладают
высокой протонной проводимостью; типичные примеры таких
соединений — HTaW06-H20 [Groult et al., 1982], пирохлоры НМ03-
• xR20 (M — Sb, Nb, Та) IChowdhry et al., 1982] и слоистая фаза
HTiNb05 [Rebbah et al., 1982]. Ионный обмен наблюдался также
в сульфидах металлов. Например, KFeS2 подвергается топохими-
ческому обмену калия в водных растворах на катионы
щелочноземельных металлов с образованием новых фаз, в которых
сохраняются тетраэдрические цепочки [FeS4/2]oo [Boiler, 1978].
3.2.3. Методы высокого давления
Применение высокого давления для твердофазных синтезов
приобрело особое значение после синтеза алмаза в 1955 г. Ранние
исследования в этой области касались фазовых диаграмм давление —
температура для элементов Si, Ge и Bi. С развитием технологии
высокого давления после 60-х годов стало доступным коммерческое
оборудование, позволяющее использовать одновременно как
высокие давления, так и высокие температуры. Существуют обзоры, в
которых изложены экспериментальные аспекты, а также успехи
исследований в этой области [Goodenough et al., 1972; Joubert,
Chenavas, 1975; Pistorius, 1976].
Экспериментальное оборудование, требуемое для синтеза под
высоким давлением, можно разделить на две основные категории
в зависимости от применяемых давлений. Для давления 1—10 кбар
часто используется гидротермальный метод; в этом методе реакция
проводится либо в открытой, либо в закрытой системе. В открытой
системе твердое вещество находится в прямом контакте с
реагирующими газами (F2, 02 или N2), которые также служат усилителями
давления. В этом типе синтезов обычно используется контейнер
из золота. Этот метод использован для синтеза таких соединений
переходных металлов, как Rh02, Pt02 и Na2NiF6, в которых
переходный металл находится в высокой степени окисления.
Гидротермальный синтез при высоком давлении в условиях закрытой системы
128
Стратегия синтеза
также используется для получения оксидов высоковалентных
металлов; здесь к реагентам добавляется внутренний окислитель,
подобно КСЮ3, который при разложении в условиях реакции
дает необходимое давление кислорода. Например, оксидные пиро-
хлоры палладия (IV) и платины (IV), Ln2M207, получены этим
методом (970 К, 3 кбар). Гидротермальным способом получены также
(H30)Zr2(P04)3 [Subramanian et al., 1984] и семейство
керамик с нулевым термическим расширением (Са0 5Ti2P3012)
[Roy et al.,1984].
Цеолиты обычно получаются при гидротермальных условиях
в присутствии щелочи [Ваггег, 1981]. Щелочь, источники кремния
и алюминия смешиваются в соответствующих соотношениях и
нагреваются (часто ниже 370 К). Вся реакционная смесь представляет
собой водный гель, состоящий из щелочи (гидроксид щелочного или
щелочноземельного металла, органические четвертичные
основания и т. д.), свежеосажденного А1(ОН)3 и золя диоксида кремния
или щелочи, растворимого алюмината и золя диоксида кремния.
В щелочной среде алюминий находится в виде анионов А1(ОН)4";
ионы ОН" действуют как катализатор процесса минерализации,
тогда как катионы, присутствующие в реакционной смеси, определяют
разновидность образуемого цеолита. Накоторые цеолиты, кроме
воды, капсулируют также некоторые неорганические соли. Вопрос
образования-цеолитов в присутствии органических оснований
находится в развитии, этот путь перспективен для синтеза богатых
кремнием цеолитов. Силикалит с тетраэдрическим каркасом,
включающим трехмерную систему каналов (шириной в 10 колец, достаточной,
чтобы адсорбировать молекулы до 0,6 нм в диаметре), получен по
реакции гидроксида тетрапропиламмония (ТПА) и реакционноспо-
собной формы диоксида кремния между 370 и 470 К. Кристаллы
предшественника имеют состав (ТПА)2-0,48 Si02-H20, органический
катион удаляется в процессе химической реакции или термического
разложения с целью получения микроскопического силикалита,
который можно рассматривать как новую полиморфную разновидность
диоксида кремния [Flanigen et al., 1978]. Клатрасил (кремнеземный
аналог газового гидрата), 1Н-додекасил, получается из водного
раствора тетраметоксисилана и N(CH3)4OH; после добавления амино-
адамантана раствор обрабатывается гидротермально в атмосфере
азота в течение 4 дней при 470 К [Groenen et al., 1983].
Для твердофазных синтезов вообще используются давления в
области 10—150 кбар; такие давления достигаются с помощью трех
различных видов аппаратуры. 1. В аппаратуре поршень — цилиндр
(рис. 3.1), состоящей из камеры из карбида вольфрама и поршневого
агрегата, образец находится в подходящей металлической капсуле,
окруженной датчиком давления (пирофиллит). Давление
генерируется движением поршня через диафрагму в цилиндре. В
конструкцию можно ввести внутреннюю микропечь, сделанную из графита
Получение кристаллических материалов 129
Рис. 3.1. Аппаратура поршень —
цилиндр.
Поршень
Пир осриллит
Образец,
или молибдена. С помощью этой
конструкции в объеме 0,1 см3
легко достигаются давления вплоть
до 50 кбар и температуры вплоть
до 1800 К. 2. В аппаратуре
типа наковальни (рис. 3.2), впервые
сконструированной Бриджменом,
образец подвергается действию
давления просто путем сдавливания его между двумя
противоположными наковальнями. Массивная опора для наковален обеспечивается
при помощи фланцев. Хотя при этом можно достичь давлений
около 200 кбар и температур вплоть до 1300 К, метод не популярен
в твердофазных синтезах, так как можно работать лишь с
миллиграммовыми количествами. Развитие принципа противоположных
наковален — конструкция четырехгранной наковальни, где четыре
наковальни на массивной опоре, расположенные тетраэдрически,
движутся по направлению к центру, где в пирофиллитовой среде разме-
v...^ ) Металлической
прокладка
Образец у*~ щ^^^— Торец наковальни
(©Г
I I Накоеальмя \
\^^ J Бриджменс
-Щ&
Рис. 3.2. Устройства по типу наковальни,
о — противолежащая; б — тетраэдрическая; в — кубическая.
9 Ч. II. Р. Рао, Дж. Гопалакришнан
130
Стратегия синтеза
Присоединение
к водяной рибашке
Водяная рубашка
Карбид вольфрама
\ Стальные ограничительные
кольца
Рис. 3.3. «Бэлт»-аппаратура.
щен образец вместе с
нагревающим устройством. Мно-
гонаковальная конструкция
расширена до кубической
геометрии, где шесть
наковален действуют на грани
пирофиллитового кубика,
помещенного в центр. 3.
«Бэлт»-аппаратура (рис. 3.3)
дает, наверное, наиболее
удачное сочетание высокого
давления и высокой
температуры в твердофазном
синтезе. Эта аппаратура, которая
используется в США для
синтеза алмазов, в известной
степени является сочетанием
конструкций поршень — цилиндр и противоположные
наковальни; массивной опорой снабжена как наковальня, так и цилиндр.
Аппаратура состоит из двух конусообразных поршней, сделанных
из карбида вольфрама, которые вталкиваются навстречу друг
другу через камеру определенной формы. Камера и поршни
поддерживаются в горизонтальном положении с помощью нескольких
стальных колец, причем это обычно позволяет достичь довольно
высоких давлений (~ 150 кбар) и высоких температур (~ 2300 К).
В «бэлт»-аппаратуре образец находится в капсуле из
благородного металла (для халькогенидов используется контейнер из BN
и MgO), которая вставляется в графитовую гильзу с пирофиллитом,
гильза служит в качестве внутреннего нагревательного устройства.
В обычном опыте с высоким давлением образец загружается,
поднимают давление до желаемого значения и затем повышается
температура. После выдерживания под давлением примерно 30 мин
образец быстро охлаждается (400 К-с-1), находясь все еще под
давлением. Давление снимается после того, как образец охладится до
комнатной температуры.
Исследование под высоким давлением требует подходящей
калибровки и измерения давлений. Калибровка выполняется с
помощью стандартных веществ, для которых известно, что они
подвергаются структурным превращениям при определенных давлениях.
Твердые вещества обычно при высоких давлениях подвергаются
переходам в более плотно упакованные структуры с увеличением
координационного числа. Например, при действии давления Si и Ge
превращаются из четырех- в шестикоординированную структуру
(белого олова); железо переходит из восьми- в двенадцатикоордини-
Получение кристаллических материалов 131
рованную структуру, тогда как NaCl переходит в структуру CsCl.
Простое правило, что высокие давления приводят к понижению
объема и повышению координационного числа, основывается на модели
жесткой сферы, которая предполагает постоянный радиус и
сжимаемость атомов в различных структурах. Единственное общее правило,
которое мы можем определить, состоит в том, что переходы под
давлением идут в направлении большей эффективности упаковки;
менее эффективно упакованные структуры могут с большей
вероятностью подвергаться структурным изменениям под действием
давления. Фазовые превращения твердых веществ под давлением
могут либо обладать, либо не обладать способностью к закаливанию.
Многие формы твердых веществ, существующие при высоком
давлении, могут сохраняться при обычных условиях в виде метастабиль-
ных фаз; типичный пример — алмаз, способный к закаливанию.
Вообще превращения, принадлежащие к реконструктивному типу и
включающие разрыв и образование связей, могут быть закалены;
превращения, включающие лишь слабое движение атомов, закалить
трудно, именно это наблюдается при моноклинно-тетрагональном
превращении Zr02, а также при тетрагонально (тип красного РЬО)-
вюртцитном переходе SnO. Класс перовскитоподобных политипов
состава АВХ3 (X — F, О) представляет собой большой класс
веществ, имеющих закаливаемые фазы высокого давления [Goodenough
et al., 1972]. В этих случаях давление уменьшает фактор
толерантности t и, следовательно, происходит стабилизация кубической
плотной упаковки слоев АХ3 (октаэдры из В-ионов с общими
вершинами) в противоположность гексагональной плотной упаковке
(октаэдры из В-ионов с общими гранями). В гидроксидах переходных
металлов МООН при высоких давлениях структура боемита (при
нормальном давлении) переходит в структуру диаспора и структуру
типа InOOH.
Методы высокого давления использованы для синтеза новых
твердых веществ, которые нельзя получить другими методами.
Вообще образование нового соединения из его компонентов требует,
чтобы новый состав имел более низкую свободную энергию, чем
сумма свободных энергий компонентов. Давление может
способствовать понижению свободной энергии различными путями [Goodenough
et al., 1972]. 1. Давление может делокализоватьвнешниеd-элект-
роны в соединениях переходных металлов путем увеличения степени
взаимодействия между d-электронами на соседних катионах, тем
самым понижая свободную энергию. Типичные примеры — синтез
перовскитов АСг03 (А — Са, Sr, Pb) и Сг02. 2. Давление может
стабилизировать более высокие валентные состояния переходных
металлов, таким образом стимулируя образование новой фазы. Например,
в системе Са — Fe — О при обычных давлениях устойчив только
CaFe02,5 (браунмиллерит). При высоких давлениях кислорода
железо окисляется до состояния 4+ и, следовательно, образуется CaFe03
9*
132
Стратегия синтеза
со структурой перовскита. 3. Давление может подавлять сегнето-
электрическое смещение катионов, а это стимулирует синтез новых
фаз. Например, синтез бронз А^МоОд требует заселения пустых
d-орбиталей молибдена; при обычных давлениях Мо03
стабилизируется сегнетоэлектрическим искажением октаэдров Мо06 вплоть до
точки плавления. 4. Давление может изменять позиционно
предпочтительные значения энергий катионов, облегчая тем самым
образование новых фаз. Например, невозможно синтезировать ильмени-
ты состава А2+Мп4+03 (А — Mg, Co, Zn) из-за сильного предпочтения
двухвалентными катионами тетраэдрических позиций; при
атмосферном давлении получается смесь А[АМп]04 (шпинель) и Мп02
(рутил) вместо одной фазы АМп03. Однако при высоких давлениях
последняя фаза имеет корундовый тип структуры, в которой оба иона
А и М находятся в октаэдрической координации. 5. Давление может
понижать 6.92-поляризацию в оксидах, содержащих изоэлектронные
катионы Tl+, Pb2+, Bi3+. Например, перовскитоподобный PbSn03
нальзя получить при атмосферном давлении, так как смесь РЬО +
+ Sn02 более устойчива, чем перовскит.
Значительный интерес представляет вопрос стабилизации
необычных состояний окисления и спиновых состояний переходных
металлов (а именно La2Pd207). Такую стабилизацию можно
объяснить, используя корреляции структурных факторов с электронной
конфигурацией. Шестикоординированное высокоспиновое железо
(IV) стабилизируется в фазе La1,5Sr0,5-Li0,5Fe0,5O4, которая имеет
структуру K2NiF4 [Demazeau et al., 1982a]. Удлиненные октаэдры
Fe06 и наличие ионных связей Li — О являются результатом того,
что структура K2NiF4 благоприятствует высокоспиновому состоянию
Fe(IV). Ионы Li и Fe в этом оксиде упорядочены в плоскости ab,
что подтверждается пятнами сверхъячейки на картине электронной
дифракции. Такой оксид получается в условиях окисления. CaFe03
и SrFe03, синтезированные под давлением кислорода, также
содержат октаэдрический Fe(IV); тогда как Fe(IV) в SrFe03 находится
в высокоспиновом состоянии с ^-электроном в узкой зоне а* вплоть
до 4 К, Fe(IV) в CaFe03 диспропорционирует на Fe(III) и Fe(V)
ниже 290 К [Takano, Takeda, 1983]. La2LiFe06, полученный под
высоким давлением кислорода, имеет структуру перовскита с
железом в пятивалентном состоянии [Demazeau et al., 1982a].
Никель в степени окисления 3+ существует в перовските
LaNi03, который можно синтезировать при атмосферном давлении,
однако другие редкоземельные никелаты могут быть получены при
высоких давлениях кислорода. La2Li0,5Ni0,5O4 и MLaNi04 (М — Sr
или Ва) со структурой K2NiF4 являются другими примерами
оксидов с Ni(III) [Demazeau et al., 1982b]. MNiOg.^ (M — Ва или
Sr), полученный при высоком давлении, также содержит Ni(IV)
[Takeda et al., 1976]. В La2Li0,5Co0,5O4 имеется доказательство
превращения низкоспинового СоШ1) в промежуточное, а также в высо-
Получение кристаллических материалов 133
коспииовое состояния. Ионы Li и Со упорядочиваются в плоскости
аЪ этого оксида со структурой K2NiF4; сильно вытянутые октаэдры
Со06, по-видимому, стабилизируют промежуточное спиновое
состояние [Mohanram et al., 1983]. Сообщается, что ионы Со в
промежуточном спиновом состоянии образуются также в Sr4TaCo08 и Sr4NbCo08.
При высоком давлении кислорода получены оксиды со структурами
перовскита и K2NiF4 для трехвалентной меди [Demazeau et al.,
1982b]. Для получения Cs2NiF6 и других фторидов использовалось
высокое давление F2 [Hagenmuller, 1985].
Как отмечалось ранее, твердофазные реакции при обычных
давлениях обычно являются медленными, даже когда продукт
термодинамически устойчив. Давление имеет заметное влияние на кинетику
реакции, значительно уменьшая время реакции и в то же самое
время приводя к более гомогенным и кристаллическим продуктам.
Например, LnFe03, LnRh03 и LnNi03 (Ln-редкоземельный
элемент) в условиях высокого давления и высокой температуры
получаются, по существу, за часы, тогда как при обычных давлениях
реакции требуют несколько дней (LnFe03 и LnRh03) или не идут
вообще (LnNi03). В нескольких рядах соединений (АХ)(АВХ3)
при атмосферных давлениях образуются конечные члены АВХ3 и
А2ВХ4, имеющие структуры соответственно перовскита и K2NiF4,
а не промежуточные фазы, такие как А3В2Х7 и А4В3Х10. Давление
облегчает синтез: например, Sr3Ru207 и Sr4Ru3O10 образуются в
течение 15 мин при 20 кбар и 1300 К. TaS3, NbSe3 и подобные твердые
вещества получаются в течение 30 мин при 2 ГПа и 970 К.
3.2.4. Дуговые методы
Имеются обзоры по использованию электрической дуги для
получения веществ, а также для роста кристаллов тугоплавких
веществ [Loehmann et al., 1969; Muller-Buschbaum, 1981 ]. По существу,
дуга для синтетических целей получается при прохождении сильного
тока от вольфрамового катода к тигельному аноду, который является
контейнером для синтезируемого вещества (рис. 3.4). Конец катода
заточивается таким образом, чтобы поддерживать высокую плотность
тока. Типичными условиями работы являются токи порядка 70 А
при напряжении 15 В. Дуга поддерживается в инертной (Не, Аг,
N2) или восстановительной (Н2) атмосферах. Даже следы кислорода
действуют на вольфрамовый электрод, и, следовательно, перед
пропусканием в дуговую камеру газы освобождаются от кислорода
(с помощью геттера из нагретой титановой губки). Дуга может
поддерживаться в атмосфере кислорода, если вместо вольфрамовых
электродов использовать графитовые. Тигель (анод) изготавливается
из цилиндрического медного блока и охлаждается водой во время
процесса.
134
Стратегия синтеза
■* » Jl **'$""
.- Ар>»оча>-:ь;й ев^д
гонооь/й &ь/вод
Ввод -fen=-'
аргона ^
Кольцо
Ввод водь
Рукоятка
Ручка
Вводи вывод воды
Кольцо
Водоохла^даемь /й
■подъемный стержегь
- Веод аргона
Тиаель из графить
высокой плотности
Винтовой привод
р?од'.' вывод для водь/
Рис. 3.4. Дуговая (а) и трсхдуговая (б) печи постоянного тока.
При синтезе исходные вещества помещаются в медный тигель,
дуга зажигается путем контакта катода с анодом. Ток медленно
увеличивают и одновременно отводят катод так, чтобы поддерживать
дугу. Затем дугу располагают так, чтобы она как бы обмывала
образец в тигле. Ток увеличивают до тех пор, пока реагенты не
расплавятся. Когда дуга выключается, продукт затвердевает в форме
королька. Из-за огромного градиента температуры между расплавом
и водоохлаждаемым тиглем обычно тонкий твердый слой образца
отделяет расплав от медного пода; образец образует как бы свой
собственный тигель, и поэтому нет загрязнения образца медью.
Загрязнения образца за счет испарения вольфрамового катода
можно избежать, используя водоохлаждаемые катоды. Дуговой метод
успешно применяется при синтезе различных оксидов Ti, V и Nb.
Получен ряд низковалентных оксидов редкоземельных элементов
LnOi,5.3с методом дугового плавления Ln203 с металлическим Ln.
Более изощренный вариант дугового метода — трехдуговои
метод (см. рис. 3.4), который развит [Fan, Reed, 1972] для роста
монокристаллических тугоплавких твердых веществ. Главное отличие
между однодуговои и трехдуговои печами состоит в использовании
трех симметрично расположенных торированных вольфрамовых
катодов и графитового тигля в качестве анода вместе с
водоохлаждаемым вытянутым стержнем. Что касается роста кристаллов с аомощью
трехдуговои печи, то здесь поликристаллический образец
загружается в тигель, все три дуги зажигаются одновременно, и образец
доводится точно до плавления. Вытянутый стержень затем
опускается вниз до тех пор, пока кончик только коснется расплава. При
медленном вытягивании стержня вверх между застывшим твердым
Получение кристаллических материалов
135
веществом и расплавом в тигле образуется «шейка». Это
эквивалентно использованию зародышевого монокристалла в обычном методе
вытягивания. Осторожное движение вытянутого стержня при
зажженных дугах приводит к образованию бульки
монокристалла. Этот метод использован для роста монокристаллов Ti306,
Ti203, NbO.
Дальнейшая модификация дугового метода — дуговой
транспортный метод (метод переноса в дуге), где вещество, монокристалл
которого должен выращиваться, переносится электрической дугой.
Вертикальная дуга между катодом (заполненным расплавом) и
анодом с зародышевым кристаллом поддерживается на постоянном
уровне с помощью постепенного подъема анода. Расплавленное вещество
переносится дугой от катода к аноду, где оно растет на зародышевом
кристалле. Этим методом с применением Ni-электродов получены
кристаллы NiO [Honig, Rao, 1981].
3.2.5. Настыльное плавление
Этот метод пригоден не только для получения оксидов
металлов, но также и для роста кристаллов этих оксидов. Метод состоит
в обработке вещества радиочастотным электромагнитным полем
(200 кГц — 4 МГц, 20—50 кВт) [Harrison et al., 1980]. Вещество
помещается в контейнер, состоящий из набора охлаждаемых водой
холодных «пальцев», расположенных на водоохлаждаемом
основании (все делается из меди), причем пространство между «пальцами»
достаточно большое, чтобы позволить электромагнитному полю
проникать внутрь, но достаточно малое, чтобы избежать утечки
расплава. Процесс бестигельный, тонкий твердый настыль отделяет расплав
от водоохлаждаемого контейнера. Этим методом можно выращивать
монокристаллы очень больших размеров, а масса исходных веществ
может быть вплоть до 1 кг; температуры в этом методе доходят
вплоть до 3600 К, что позволяет выращивать кристаллы веществ,
подобных Th02 и стабилизированному Zr02. Стехиометрия оксида
контролируется легко путем использования подходящего газа
(смесей СО/С02, воздуха или кислорода). Настыльным методом
выращены большие кристаллы СоО, МпО и Fe304, метод в будущем
несомненно, найдет много приложений. В СоО и МпО ионы
трехвалентных металлов исключаются нагреванием кристаллов в подходящей
смеси СО/С02. Настыльным методом недавно выращены кристаллы
La2Ni04 и Nd2Ni04.
3.2.6. Химическое осаждение из газовой фазы
Осаждение твердого вещества на подложке посредством
химической реакции в парах называется химическим осаждением из
газовой фазы. Процесс включает в себя: 1) образование реагентов
136
Стратегия синтеза
в газообразном состоянии; 2) транспорт газа в зону осаждения;
3) осаждение твердого вещества из газовой фазы. Этот метод стал
важным методом твердофазных синтезов и находит применение в
синтезе широкого спектра твердых веществ, начиная с аморфных
и поликристаллических покрытий до эпитаксиально выращенных
монокристальных пленок, используемых в приборах [Tietjen, 1973].
Как синтетический метод химического осаждения из газовой фазы
имеет несколько преимуществ. Он, по существу, позволяет
получать материал почти любой геометрии. Поскольку образование
продукта не ограничивается кинетическими факторами и диффузией,
можно синтезировать твердые вещества при относительно низких
температурах; легко контролируются также гомогенность и
стехиометрия. Дополнительное преимущество состоит в том, что во время
синтеза достигается избирательное допирование в контролируемой
концентрации.
Химическое осаждение из газовой фазы широко применяется
для получения кремния и III—V-полупроводников, используемых
в приборах. Существуют два различных процесса получения
кремния методом химического осаждения из газовой фазы: 1)
восстановление водородом SiCl4 и 2) пиролиз SiII4. Осаждение осуществляется
либо гомоэпитаксиально на кремнии, либо на изолирующей
подложке, такой как сапфир. Несколько методов развито для синтеза
III—V-соединений. Например, GaAs получается методом
химического осаждения из газовой фазы с использованием AsCl3 и паров Ga
в атмосфере водорода; здесь AsCl3 действует не только как источник
As, но и как транспортный агент для Ga. Другой метод синтеза этого
класса соединений основан на использовании реакции между
летучим гидридом элемента V группы с элементом III группы в
присутствии газообразного НС1:
AsH3 + Ga + НС1 -> GaAs -|- 3/2H2 + HC1.
В третьем методе используются гидриды элементов V группы вместе
с летучими металлоорганическими соединениями элементов III
группы, такими как триметилгаллий и триметилалюминий.
Методами химического осаждения из газовой фазы получаются
также такие диэлектрики, как Si02, Si3N4 и A1203. Si3N4,
высокотемпературная керамика, образуется по реакции NH3 с SiH4 или
SiCl4. Реакция с SiH4 имеет то преимущество, что она идет при более
низких температурах. Химическое осаждение из газовой фазы
особенно пригодно для синтеза сверхпроводящих и магнитных
материалов. Один такой материал, Nb3Si, является важным
сверхпроводником типа А15. Получение этой фазы по реакции элементов при
пониженных температурах дает лишь несверхпроводящую фазу со
структурой Ti3P. Желаемая фаза со структурой А15 получается только
при осаждении из газовой фазы ниже 1120 К. Гранаты с редкоземель-
Получение кристаллических материалов
137
ными элементами, пригодные как магнитные материалы с
пузырьковыми доменами, получены в виде монокристаллов осаждением из
летучих галогенидов металлов.
3.2.7. Синтез органических твердых веществ
Синтез — это главное занятие химиков-органиков, но мы
не можем здесь рассматривать этот вопрос в различных аспектах.
Однако синтез органических твердых веществ со свойствами,
представляющими интерес для ученых, работающих в области твердого
тела, включает некоторые заслуживающие внимания способы, и
поэтому несколько таких типичных систем мы обсудим. TTF — TCNQ
и аналогичные донорно-акцепторные комплексы, а также
полиацетилен и другие проводящие органические полимеры изучаются той
областью науки о твердом теле, в которой сегодня ведутся активные
исследования [Hatfield, 1979]. Синтетические методы получения
TTF, TCNQ и родственных донорных и акцепторных молекул
описаны в литературе [Wheland, Martin, 1975; Narita, Pittman, 1976].
Соли с переносом заряда обычно получаются смешением горячих
ацетонитрильных растворов донора и акцептора. Для химии
твердого тела важен не синтез этих органических соединений как таковой,
а подбор таких доноров и акцепторов, чтобы твердые вещества с
переносом заряда имели желаемую структуру и свойства. Общая
концепция, основанная на молекулярной- упаковке, стехиометрии и
степени переноса заряда, развита для дизайна донорно-акцепторных
комплексов, проявляющих металлические свойства [Torrance, 1979].
Так, соли с переносом заряда 1:1, обладающие одинаковой
изолированной упаковкой и неполным переносом заряда, проявляют
металлические свойства. Сделаны попытки соединить эти три фактора
молекулярных свойств доноров и акцепторов. Оказывается, что
изолированная упаковка благоприятна тогда, когда перекрывание
донор — акцептор очень слабое. Степень переноса заряда
определяется главным образом по энергии ионизации донора (потенциал
восстановления катиона). График зависимости удельной проводимости
при комнатной температуре от потенциала восстановления катиона
для донорно-акцепторных твердых веществ семейства TCNQ
показывает, что металлический характер наблюдается только в узкой
области потенциала восстановления, близкой к 0. Когда потенциал
имеет большую отрицательную величину, происходит полный
перенос заряда, что приводит к ионным солям, а когда потенциал
положительный, получаются нейтральные комплексы [Torrance, 1979].
Факторы, которые определяют стехиометрию донорно-акцепторных
комплексов, точно не известны. В семействе солей TTF-галогенид
наблюдаемая нестехиометрия объяснена в терминах энергии Маде^
лунга, показывающей, что потенциал ионизации донора и сродство
138
Стратегия синтеза
к электрону акцептора являются важными факторами,
определяющими стехиометрию.
Сделаны также попытки создать одномерный органический
сверхпроводник, основанный на донорно-акцепторном взаимодействии
[Bechgaard, Jerome, 1982]. Для этого надо избежать пайерлсовского
искажения, которое приводит к потере металлических свойств
(см. разд. 6.8). Это достигнуто при получении комплексов с
переносом заряда для селеновых аналогов TTF, таких как TMTSF (тетра-
метилтетраселенафульвален). Вещества этого класса типа TMTSF2X
(X — С104, Re04, PF6 и AsF6) действительно проявляют
сверхпроводимость, причем перхлорат имеет сверхпроводящий переход при
1,2 К и атмосферном давлении, а другие при высоких давлениях.
Полиацетилен (СН)^ является прототипом проводящих
органических полимеров [Etemad et al., 1982], удельная
проводимость которых может изменяться допированием на 12 порядков,
от проводимости диэлектрика (< 10~9 Ом-1-см-1), через
полупроводник к металлу (> 103 Ом-1-см-1). Полиацетилен можно
получить, вводя газообразный ацетилен в стеклянный реактор,
внутренняя стенка которого увлажнена метилбензольным раствором
катализатора Циглера (С2Н5)3А1 и (ra-C4H90)4Ti [Shirakawa, Ikeda,
1971; Shirakawa et al., 1973]. На всех поверхностях, которые
увлажнены раствором катализатора, растет прочная пленка
полиацетилена. Полимеризация при комнатной температуре дает смесь 80 %
цис- и 20 % /npa/^-изомеров. При температуре около 350 К
получается почти 100 % цис-(СН)х; при 420 К (растворитель — декан) —
100 % пграис-формы. Цис-пгранс-изомершация идет при ~ 470 К.
Образованные пленки состоят из случайно[ориентированных волокон,
причем каждое волокно в диаметре составляет несколько сот
ангстрем. Удельная проводимость (СН)^ при комнатной температуре
вависит от содержания цис- и транс-форм, причем она изменяется
в пределах между значениями 10~5 Ом-1-см-1 для т/жмс-полимера
(Eg ~ 0,8 эВ) и Ю-9 Ом^-см-1 для uuc-nommepa (Eg ~ 1,0 эВ).
В результате широкого перекрывания сопряженных я-электронов
(СН)Х отличается от обычных органических полупроводников,
состоящих из слабовзаимодействующих молекул (например,
антрацен), или от насыщенных полимеров, не имеющих я-электронов
(например, полиэтилен).
Подобно неорганическим полупроводникам, полиацетилен
можно допировать различными донорами и акцепторами, чтобы
получить полупроводники п- или р-типов. Допирование до высоких
уровней (выше ~ 1 %) приводит к переходу полупроводник — металл
с образованием нового класса металлов с широким интервалом
электроотрицательности [MacDiarmid, Heeger, 1979; Mammone,
MacDiarmid, 1984]. Допирование можно осуществлять химически или
электрохимически. Химическое допирование можно проводить,
подвергая пленку (СН)Х действию известного давления газа летучего
Микрокристаллические частицы и кластеры 139
компонента, которым допируют, например, I2, AsF5 и т. д., до тех
пор пока получится желаемое значение удельной проводимости.
Удаление паров допирующей добавки, по существу, «замораживает»
удельную проводимость на этой стадии. Допирование можно также
проводить обработкой полимерной пленки раствором допирующего
вещества в соответствующем растворителе (например, 12 в пентане,
нафталид натрия в ТГФ) или обработкой расплавленным натрием
или калием. Полиацетилен может действовать как источник
электронов или как приемник электронов, в зависимости от того,
окисляется он (допированный р-тип) или восстанавливается (допирован-
ный га-тип). (СН)Х начинает терять электроны при величине
приложенного потенциала +3,10 В или приобретать электроны при
-|-1,75 В относительно электрода Li/Li+. Электрохимическое
допирование можно выполнять, используя в качестве анода и катода
ленты из (СН)Х. Например, когда электроды, погруженные в раствор
LiC104 в пропиленкарбонате, соединяются с источником
постоянного тока (4 В), на аноде идет окисление (СН)Х с образованием
[(СН)^+(С104~)ДТ, а на катоде — восстановление с образованием
[Liy+(CH)v~]iX. Возможность обратимых
окислительно-восстановительных реакций полиацетилеиа в электрохимической ячейке
обеспечивает основу для создания перезаряжаемых аккумуляторов,
легких по весу и с высокой плотностью энергии [Nigrey et al., 1981].
Получен ряд полигетероциклов. Например, окислительная
электрохимическая полимеризация пиррола дает полипиррол,
неизменно содержащий допирующие анионы электролита. Полипир-
рольная пленка толщиной ~ 10 мкм, содержащая AsF6, получается
при электролизе смеси 0,2 М пиррола и 0,004 М раствора тетра-
тг-бутиламмоний-гексафторарсената в диметилсульфате в течение
20 мин, с использованием In — Sn-оксидного анода и платинового
катода при плотности тока 2 мА-см~2. Полимер растет на
поверхности In — Sn-оксида [Hotta et al., 1983]. Синтезированные таким
образом допированные полимеры можно превращать в нейтральные
полимеры обращением полярности сразу после электрохимической
полимеризации. Аналогичными методами получаются политиенилен
и поли-(З-метилтиеиилен), а также металлоорганические полимеры,
обладающие высокой удельной электропроводностью [Sheats et al.,
1984].
3.3. МИКРОКРИСТАЛЛИЧЕСКИЕ ЧАСТИЦЫ И КЛАСТЕРЫ
(ПРЕДЕЛЬНОЕ СОСТОЯНИЕ ТВЕРДОГО ТЕЛА)
Существует особая область малых агрегатов или кластеров,
которые попадают между атомным (или молекулярным) доменом и
конденсированным веществом. Эти малые частицы и кластеры
обладают уникальными свойствами и имеют некоторые технологические
140
Стратегия синтеза
приложения. Образование этих частиц включает разные типы
фазовых изменений: газ — твердое, жидкость — твердое, твердое —
твердое или газ — жидкость — твердое, определяемые нуклеацией;
здесь необходимо контролировать размер растущих зародышей
[Multani, 1981].
Микрокластеры металлов вплоть до 500 атомных массовых
единиц получены испарением-конденсацией в инертном газе; пары
металла истекают из горячей ячейки через узкое отверстие. Средний
размер частиц, получаемых этим методом, варьирует в пределах
о
5—200 А, причем микрокластеры содержат от 1 до 500 атомов
(например, Sr^—Sb500; Bix—Bi280). Для определения размеров таких
кластеров использована масс-спектроскопия. Описана аппаратура
о
для синтеза частиц диаметром 30—60 A [Grandquist, Buhrman,
1976]. Метод дуговой плазмы использован для получения
микрокластеров керамических материалов, таких как карбид вольфрама
о о
(50—80 А) и карбид кремния (100—200 А). Двухфазные металло-
керамические композиты А1203, Si02 или Zr02 с Си, Pt, Sn, или Ni
получены золь-гель-процессом [Roy, Roy, 1984]. Ксерогель состоит
из микрокристаллической или некристаллической керамической
матрицы, в которой металлический компонент диспергирован в виде
маленьких островков.
Микрокристаллы, полученные из относительно больших частиц
(все еще субмикронного порядка), используются в тех приложениях,
которые связаны с диффузией в твердом теле (или реакцией) и
спеканием. Они синтезируются любым из следующих методов:
распыление — высушивание, замораживание — высушивание, золь-
гель, пирогель (высокотемпературное распыление) или жидкое вы-
о
сушивание. Так, для синтеза а- и Y-Fe203 (150—200 А) использован
метод замораживания — высушивания, тогда как для получения
Y3Fe5012 и PbZr0)52Ti0t48O3(PZT) использован метод золь—гель. Во
всех этих методах работа начинается с водными растворами или
смешанными золями соответствующей стехиометрии; растворы
распыляются, перенос тепла или массы достигается в миллисекунды.
Температура для золь-гель-процесса около 300 К (как для 2-этил-
гексанола, так и для жидкого высушивания в ацетоновой
бане), 78 К — для процесса замораживание — высушивание, около
600 К — для распыления — высушивания, около 1200 К — для
пирогельного метода. Эти синтезы обеспечивают гомогенность на
молекулярном уровне, малый размер зерен, низкую пористость,
большую площадь поверхности и теоретическую плотность
спеченных компактных материалов.
Трансмиссионная и сканирующая электронная микроскопия
используется для прямого изучения микрокластеров, тогда как
распределение по размерам (или среднему диаметру) обеспечивается
методом седиментации и др. Средний диаметр частицы определяется
Аморфные материалы
141
по величине поверхности методом BET (Brunauer — Emmett —
Teller) и методом уширения рентгеновской линии.
Микрокристаллы проявляют свойства, заметно отличные от
свойств массивных образцов. Найдено, что частичное изменение
параметра кристаллической решетки увеличивается с уменьшением
размера частиц в Fe203. Магнитные сверхтонкие поля в
микрокристаллической фазе a-Fe203 и Fe304 ниже таких же полей в
массивных кристаллических фазах. Тетрагональность (т. е. отклонение
аксиального отношения от единицы) сегнетоэлектрического ВаТЮ3
уменьшается с уменьшением размера частицы; в PZT с
уменьшением размера частицы низкочастотная диэлектрическая постоянная
уменьшается, а температура Кюри растет. Малый размер частиц
в микрокристаллах, по-видимому, не может обеспечивать
низкочастотные колебания решетки.
3.4. АМОРФНЫЕ МАТЕРИАЛЫ
Только в последние годы пришло понимание того, что
большинство материалов могут быть переведены в аморфное состояние.
Большинство материалов при достаточно быстром (и достаточно
глубоком) охлаждении из жидкого состояния можно получить в
виде стекол. Существуют различные методы получения стекол [Аг-
gell, 1981; Elliott et al., 1985]: охлаждение переохлажденных жидких
фаз, осаждение из газовой фазы, разупорядочение ударом, разупо-
рядочение облучением, десольватация и гелеобразование (рис. 3.5).
Все эти методы, за исключением первого, дают вещества, которые
нельзя легко охарактеризовать, особенно в отношении их энтропии
(по сравнению с соответствующими кристаллами).
Получено большое число стекол, обладающих различными
типами связей. Универсальность стеклообразования иллюстрирует
табл. 3.5. Чтобы получить стекла из металлических материалов,
скорость охлаждения должна быть очень большой, необходимый
перепад температуры составляет около 1000 7мс (~ 106 К-с-1.) Для
этой цели используется метод
«формования из расплава», в котором
металлическое стекло, закручиваясь
медным ротором, отскакивает от него в ^^
виде узких лент при скорости, пре- /
вышающей 1 км/мин. Большинство Г| J
других стекол, получаемых закалива- $г§[
нием расплавов, требует меньших ско- l&d
Рис* 3.5. Различные пути получения
аморфных твердых тел.
142
Стратегия синтеза
Таблица 3.3
Типичные стеклообразные аморфные твердые тела
Вещество
Si02
Ge02
As2S3
Se
Поливинилацотилсн
BcF2
75 % AgI-25 % Ag2Se04
Auo,8Sio,iGoo.i
Пронилонкарбоиат
Изонентан
1I20
C2H5OH
Fe8uB20
Тип связи
Ковалентная
»
»
Ковалентная (полимер)
»
Ионная
»
Металлы ческая
Ван-дер-Ваал ьсова
»
Водородная
»
Металлическая
Температура
стеклования, К
1450
820
470
320
310
570
75
295
160
65
139(?)
90
Кристаллизуется
ростей; Si02 требует скорости охлаждения всего лишь ОД К-с-1,
это достигается свободным охлаждением расплава. Для получения
металлических стекол использованы также методы закалки со
скоростями охлаждения 105—108 К-с"1. Особый интерес представляет
метод лазерного стеклования, в котором поверхность
кристаллического материала становится стеклообразной при действии на нее
движущегося сфокусированного лазерного луча, так что само
твердое вещество действует как поглотитель тепла; скорость
закаливания в этом методе 10й К-с"1.
Для получения аморфных веществ используется метод
осаждения из газовой фазы. В этом методе атомы, молекулы или ионы
вещества (в разбавленной газовой фазе) осаждаются на охлаждаемую
подложку. Большинство аморфных материалов, осаждаемых из
газовой фазы, при нагревании кристаллизуется, но некоторые из них
претерпевают переход второго рода (близкий к переходу в
стеклообразное состояние), который препятствует кристаллизации.
Такие переходы претерпевают аморфный лед и аморфный твердый
метанол. Структурные характеристики аморфных твердых тел,
полученных осаждением из газовой фазы, сравнимы с характеристиками
стекол тех же самых веществ, полученных методом закаливания
расплава.
Разупорядочение кристаллических материалов при действии
на них ударом (например, как в случае кварца и других минералов)
или большими дозами радиации (нейтронами или альфа-частицами)
также приводит к аморфному состоянию. Метамиктовая форма [Pabst,
Рост кристаллов
143
1952], полученная облучением, неустойчива по сравнению со
стеклообразной формой. Такие вещества, как ацетат магния, переходят
в стеклообразное состояние в процессе десольватации. В методе
гелеобразования материал сначала получается в виде геля при
гидролизе органических производных (например, этил силикат):
гель затем высушивается и переходит в стеклообразное состояние.
Аморфные твердые вещества можно также получить
твердофазными реакциями исходя из кристаллических твердых веществ.
Например, найдено, что поглощение водорода кристаллическим
Zr3Rh превращает его в гидрированный аморфный материал [Yeh
et' al., 1983].
3.5. РОСТ КРИСТАЛЛОВ
Кристаллы важны как для фундаментальных исследований
твердых веществ, так и для производства приборов. Идеальные
требования — большой размер, высокая чистота и максимальное
совершенство (отсутствие дефектов). Может быть, во время роста
необходимо также включать селективные допирующие примеси, для
того чтобы достичь требуемых электронных свойств. Существует
ряд методов роста кристаллов (табл. 3.6); в литературе этот вонрсс
обсужден широко [Laudise, 1970; Banks, Wold, 1974; Mroczkowski,
1980; Honig, Rao, 1981].
Наиболее общие методы выращивания кристаллов включают
затвердевание из расплава в случае однокомпонентных систем или
кристаллизацию из раствора. Некоторые методы роста кристаллов
из расплава схематично представлены на рис. 3.6. В методе Чохраль-
ского, обычно известном как метод вытягивания, материал плавится
в индукционных печах или в печах сопротивления в подходящем
тигле, не взаимодействующем с веществом. Температура расплава
подбирается так, чтобы она была несколько выше точки плавления,
а затравочные кристаллы погружаются в расплав. После
достижения термического равновесия затравка медленно вытаскивается из
расплава. По мере вытягивания затравки идет непрерывный рост
на границе раздела. Диаметр растущего кристалла может
контролироваться подбором скорости вытягивания, скорости понижения
уровня расплава и потоками тепла в систему и из системы. Метод
имеет то преимущество, что граница раздела при росте кристалла
не приходит в контакт со стенками тигля и, следовательно,
исключается образование нежелательных зародышей. Метод использован
для роста кремния, германия, III—V-полупроводников,
керамических оксидов типа А1203, таких редкоземельных перовскитов, как
LnA103, LnFe03, гранатов, шеелитов и т. д. Оксидные материалы
могут выращиваться на воздухе, тогда как другие требуют
закрытых систем и контроля атмосферы. В трехугловом методе (описанпом
144
Стратегия синтеза
Таблица 3.6
Методы роста кристаллов
I. Рост кристаллов в однокомпонентных системах
а). Твердое — твердое
Отжиг напряжений, расстекловывание или полиморфный фазовый переход
б). Жидкость — твердое
1. Направленное затвердевание (Бриджмена — Стокбаргера)
2. Охлаждение с затравкой (Киропулоса)
3. Вытягивание (Чохральского, а также трехдуговой)
4. Зонный метод (горизонтальная зона, вертикальная зона)
5. Плавление в пламени (Вернейля)
6. Медленное охлаждение в настыл ьной плавильной камере
в). Газ — твердое
Сублимания — конденсация или напыление
II. Рост кристаллов, включающий более одного компонента
а). Твердое—твердое
Осаждение из твердого раствора
б). Жидкость — твердое
1. Рост кристаллов из раствора (выпаривание, медленное охлаждение и
температурный перепад)
2. Водный растворитель; органические растворители; растворители —
расплавы; гидротермальный
3. Рост кристаллов при реакции:
химическая реакция; электрохимическая реакция
4. Рост кристаллов из расплава; например, конгруэнтное плавление
интерметаллических соединений
в). Газ — твердое
1. Рост кристаллов по обратимой реакции (метод газотранспортных
химических реакций (ГХР)
2. Рост кристаллов по необратимой реакции (эпитаксиальный процесс)
в разд. 3.2.4) кристаллы вытягиваются из расплава. Метод
Киропулоса подобен методу Чохральского, но вместо вытягивания
затравочного кристалла из расплава фронт кристаллизации движется
в расплав. В методе Бриджмена — Стокбаргеравлоль расплава
устанавливается резкий градиент температуры, что приводит к зароды-
шеобразеванию в более холодной области. Коническая геометрия
дна тигля ограничивает количество образуемых зародышей.
Другой метод выращивания кристаллов из расплава — метод
плавающей зоны, в котором часть исходного материала,
поддерживаемая вертикально в виде стержня, плавится при соответствующей
температуре. Когда расплавленная зона движется вдоль стержня,
происходит постепенное плавление образца на одном конце зоны
и рост кристалла — на другом. Если на одном конце поместить
затравку, весь стержень можно превратить в монокристалл. Метод
имеет то преимущество, что нет никакого загрязнения материалом
тигля. Метод похож на очистку способом зонной плавки и обычно
используется для роста монокристаллов кремния. В методе плавления
в пламени (Вернейля) порошок образца подается непосредственно
в кислородно-водородное пламя и расплав капается на затравку.
Рост кристаллов
145
^ZBoda
74
Газ
*г*
Затравка
Расплавленная соль
Горячая зона
Экраны
Перегородка
Охлаждающая
вода
Холодная зона
Подвод анергии
Вакуум
Сито
Питателе
Газ
°л\ /
~^\/уПл
Расту -
сций крис
талл
,Г1
Загрузка
бункер
Пламя
— Газ
- Затравка
I Подставка из оксида
алюминия
Рас. 3.6. Методы роста кристаллов из расплава.
а — Чохральского; б — Киропулоса; в — Бриджмена — Стокбаргера; г — Вернейля.
Поскольку кристаллизация происходит на верхушке, растущий
зародыш медленно опускается, способствуя тем самым росту больших
кристаллов. Этот метод использован для роста таких тугоплавких
оксидов, как рубин и сапфир. Разновидностью метода плавления
в пламени является метод плазменной горелки, в котором порошок
пропускается через горячую плазму, получаемую и
поддерживаемую высокочастотным током. Выяснилось, что для роста больших
кристаллов нескольких оксидов становится пригодным настыльный
метод (см. разд. 3.2.5).
Часто рост кристаллов из расплава кроме основных
компонентов включает один или более таких компонентов, как примеси,
специально введенные добавки и т. д. В этих случаях важно знать
распределение второго компонента между растущим кристаллом и
расплавом. Это распределение происходит согласно фазовой диаграмме,
устанавливающей связь равновесных растворимостей второго
компонента (примеси) в жидкой и твердой фазах.
10 ч. Н. Р. Рао, Дж. Гопалакришнап
146
Стратегия синтеза
Известен ряд методов роста кристаллов, использующих
растворимость в соответствующем растворителе. Кристаллизация
требует пересыщения, которое может достигаться разницей
температур между зонами растворения и роста, испарением растворителя
или химической реакцией. В методах растворения трудно избежать
загрязнения продукта другими компонентами в растворе или в
плавне. Тем не менее метод флюса используется для роста
кристаллов Ge02, Si02, BaTi03, KTa03, а-А1203, GdA103 и т. д. Роль
растворителя заключается в том, чтобы понизить температуру
плавления растворенного вещества, кристалл которого выращивается.
Хотя для роста кристалла требуется пересыщение, экспериментально
найдено, что скорость роста из раствора значительно больше
ожидаемой, причем значительный рост идет даже при низких степенях
пересыщения (I %). Это расхождение было объяснено Франком
и Кабрерой в рамках влияния дислокаций на рост кристалла. Рост
совершенного кристалла требует образования зародышей нового
слоя на совершенной поверхности после завершения предыдущего
слоя, в то время как при наличии винтовой дислокации рост
кристалла не требует образования зародышей нового слоя.
Дислокация дает ступенчатую поверхность, на которой рост может
происходить даже при низких степенях пересыщения. Фигуры спиралей
роста, получающиеся в результате винтовых дислокаций,
наблюдались на ряде кристаллов, выращенных из раствора и газовой
фазы, что подтверждает модель Франка.
Среди методов роста из раствора кристаллизация из водного
раствора хорошо известна. Материалы с низкой растворимостью
в воде переводятся в раствор с помощью комплексующих агентов
(минерализаторов). Например, селен можно вырастить из водных
сульфидных растворов, используя реакцию
2Se + S2- ±*Se2S2-.
Важным методом роста кристаллов с низкой растворимостью в воде
является гидротермальный метод, рассмотренный в разд. 3.2.3-
Гидротермальный синтез обычно выполняется в автоклаве (рис. 3.7)-
Автоклав состоит из нижней питающей и верхней ростовой частей*
разделенных перегородкой. Растворенное вещество помещается в
питающую часть, и несколько затравочных кристаллов
суспендируются в ростовую часть. Сосуд заполняется водой до определенного
объема, если необходимо, добавляются минерализаторы, затем
сосуд закрывается и поднимается температура, при этом
обеспечивается температурный градиент. Поскольку для большинства веществ
растворимость с увеличением температуры увеличивается, питающая
часть поддерживается при более высокой температуре, чем ростовая.
Растворитель насыщается в растворяющей зоне, движется путем
конвекции в более холодную ростовую зону, где он пересыщается
и осаждает растворенное вещество на затравочном кристалле. Когда
раствор достигает питающей части, он становится недостаточно на-
Рост кристаллов
147
Плунт ернь /и
хвостовик
Съемный винт
Опорная шайба
Основная мусрта
Корпус
Запорное кольцо
Плунжер
Затравки
Перегородка
Питатель
Рис. 3.8. Метод геля для роста
нерастворимых в воде ионных твердых тел.
Рис. 3.7. Аппаратура для
гидротермального синтеза.
сыщенным, таким образом дальше растворяя вещество и продолжая
цикл. Этим методом выращиваются большие кристаллы кварца.
Если растворенное вещество имеет обратный ход растворимости,
как в случае А1Р04, температурный градиент между ростовой
и питающей частями должен быть обратным. В гидротермальном
синтезе важным фактором является степень заполнения автоклава
растворителем; она должна быть более 32 %, для того чтобы
обеспечить высокую плотность растворителя. Обычно гидротермальные
эксперименты выполняются в щелочной среде, причем ион ОН"
действует как комплексующий агент. Гидротермальный синтез
использовался для стабилизации необычных степеней окисления
в соединениях переходных металлов и для синтеза
низкотемпературных и метастабильных фаз [Rabenau, 1985].
Кристаллы нерастворимых ионных твердых веществ, таких как
СаС03 и BaS04, нельзя вырастить обычными методами из раствора
или даже методом контролируемой химической реакции из-за
мгновенного осаждения продукта при смешивании реагентов. Для роста
кристаллов таких веществ пригоден метод геля, этот простой метод
зависит от контролируемой диффузии реагентов через гель,
{/-образная трубка заполняется силикагелем, полученным подкислением
раствора метасиликата натрия (рис. 3.8). Реагенты добавляются
с двух сторон {/-образной трубки; они медленно диффундируют
навстречу друг другу. В отдельных местах, где концентрация
превышает растворимость продукта, образуются зародыши и дальше
они растут в большие кристаллы. Этот метод применялся для роста
кристаллов тартрата кальция, вольфрамата кальция и иодида
свинца, и даже в процессе роста избирательно вводились примесные
ионы (Mn2+, Cu2+, Cr3f и т. д.) [Henisch et al.: 1965].
10*
148
Стратегия синтеза
Такие твердые вещества, как KF, РЬО, PbF2 и В203, в
расплавленном состоянии являются мощными растворителями (флюсами)
для многих неорганических веществ и, следовательно, могут
использоваться в качестве среды для роста кристаллов. Обычный метод
состоит в растворении вещества в подходящей комбинации флюсов,
содержащейся в платиновом тигле, при одновременном поддержании
температуры несколько выше точки насыщения. После охлаждения
тигля с программированной скоростью плавень выливается или
обрабатывается минеральной кислотой, оставляя после себя
кристаллы. Типичными примерами твердых веществ, выращенных по
методу флюса с использованием плавней РЬО — PbF2, являются
железо-иттриевый гранат и его изоморфные разновидности.
Последняя разработка, касающаяся роста кристаллов из расплавленных
сред, заключается в использовании расплавленных металлов в
качестве растворителей, из которых вырастают кристаллы
интерметаллических соединений. Кристаллы редкоземельных боридов, LnB4
и LnB6, выращены из жидкого алюминия, а редкоземельно-родиевых
станнидов — из расплавленного олова [Fisk, 1972; Espinosa,
1980].
Для получения и роста кристаллов применяют электролиз
солевых растворов в расплавах [Wold, Bellavance, 1972; Banks, Wold,
1974; Feigelson, 1980]. Электролитический метод обычно включает
в себя восстановление катиона и отложение продукта, содержащего
восстановленный катион, на катоде. Andrieux с сотрудниками
использовали этот метод для синтеза разнообразных соединений
переходных металлов. Типичные примеры роста кристаллов таким
методом следующие: 1) рост ванадиевых шпинелей, MV204 (M—Mn, Fe,
Со, Zn и Mg), из расплавов Na2B407 и NaF с использованием
углеродных тиглей и углеродного катода (железного тигля и железного
катода — для соединения с железом); 2) синтез NaM02 (M—Fe,
Со, Ni) путем электролиза расплава NaOH, содержащегося в
алюминиевых тиглях, с электродами из указанных металлов; 3)
синтез вольфрамовых бронз со щелочными металлами AxW03 путем
электролиза вольфраматов щелочных металлов с использованием
платиновых электродов. Хотя большинство ранних исследований
по электролизу расплавленных солей носили эмпирический
характер и были направлены на получение больших монокристаллов
путем подбора состава расплава и плотности тока, последние
исследования (например, [Whittingham, Huggins, 1972]) по получению
вольфрамовых бронз со щелочными металлами показали, что если
в процессе электролиза поддерживать по всей ячейке постоянную
разность потенциала, а не плотность тока, то можно вырастить
кристаллы определенного состава. Метод электролиза использован
для получения и роста кристаллов широкого ряда твердых веществ,
таких как бориды, карбиды, силициды, фосфиды, арсениды и
сульфиды.
Рост кристаллов
149
Устройство экспериментальной ячейки для электролитического
роста может быть чрезвычайно простым или сложным в зависимости
от изучаемой системы. Например, электролитический рост щелочных
вольфрамовых бронз требует набора тиглей Гуча, маленького,
вставленного в большой, причем внутренний тигель служит анодным,
а внешний — катодным пространством; используются электроды
из платины или золота. Инертная атмосфера совершенно не
обязательна, так как кислород воздуха не влияет на соотношение ток —
потенциал. В противоположность этому электролитическая ячейка,
которую использовали [Didchenko, Litz, 1962] для синтеза CeS и ThS,
разработана очень тщательно: она состоит из графитовых и алун-
довых тиглей (служащих электродными пространствами) вместе с
молибденовыми электродами и включает обеспечение инертной
атмосферой.
Рост кристаллов по методу газотранспортных химических
реакций (ГХР) можно разделить на две категории, в зависимости от
того, какой характер носит изменение газ — кристалл: физический
или химический. Если состав пара и кристалла одинаков, процесс —
физический; примерами таких процессов являются сублимация —
конденсация и распыление. Процесс считается химическим, если
во время роста идет химическая реакция; в этом случае состав
твердого вещества отличается от состава пара. Использование в качестве
препаративного метода химического осаждения из газовой фазы
обсуждалось в разд. 3.2.6 этой главы. Теперь мы лишь кратко
обсудим родственный метод ГХР, который используется как для
получения новых твердых фаз, так и для выращивания их в виде
кристаллов. Этот метод широко использован [Schafer, 1972; Rosenber-
ger, 1981]. В методе ГХР конденсированная фаза реагирует с газом
с образованием летучих продуктов. Между реагентами и продуктами
существует равновесие:
*А(тв) + /сВ(г) + ... **/С(г) + ... .
Этот метод использует температурную зависимость
представленного выше гетерогенного равновесия для транспорта твердого
вещества А через газовую фазу посредством газообразного интерме-
диата С. То, что процесс включает настоящий транспорт, а не просто
испарение и конденсацию, видно из того факта, что твердое вещество
А не обладает заметным давлением пара при температуре
эксперимента; более того, транспорт А не наблюдается без транспортного
агента В.
Химический транспорт обычно выполняется путем поддержания
разности температур между участком загрузки и ростовым участком
в закрытой системе (рис. 3.9). Прямая реакция идет на участке
загрузки, с образованием газообразных продуктов, а обратная —
на ростовом участке, с выделением кристаллов. Температуры 7\
и Т2, подобранные соответственно для ростового участка и участка
загрузки, зависят от того, какая реакция — эндо- или экзотерми-
150
Стратегия синтеза
Т= 1270К
тугого к
Зона загрузки PHCl =10 мм пРа3°°К 3она Роста
Рел04^ 3HCl-2FeCl3+FeCl2+4H20
Рис. 3.9. Газовый химический
транспорт в закрытой системе.
ческая. Для
эндотермических реакций необходимо,
чтобы Т2 была больше 7\;
обратная зависимость —
для экзотермических
реакций. Факторами,
определяющими рост
кристаллов по этому методу,
являются выбор химической реакции для транспорта (равновесие
химического транспорта из газовой фазы не должно лежать на
экстремуме, чтобы не затруднять обратимость процесса), значения
Тг и Т2 и концентрация используемого транспортного агента.
Примеры, приведенные в табл. 3.7, ясно говорят о том, что этот метод
может использоваться для получения или роста кристаллов почти
любого типа твердых веществ при условии, если можно найти
подходящий транспортный агент, который дает летучие продукты.
Металлы переносятся с использованием летучих галогенидов.
Метод ГХР действительно является общераспространенным и
используется для роста таких оксидных фаз, как V8015, свободный
от загрязнения соседствующими фазами Магнели V70]3 и V9017.
Обычно используемыми транспортными агентами являются 12, ТеС1А
и С12. Если оксид металла легколетучий (например, Sn02 —►- SnO j-
-j- l/202), образующиеся пары могут вновь соединиться с
образованием монокристаллов в более холодной зоне.
Молекулярно-лучевая опилгаксия является важным методом
получения полупроводников (соединений III —V). Изощренность
современной препаративной науки о веществах в твердом состоянии
видна на примере этого метода, который использует взаимодействие
многочисленных молекулярных пучков с монокристаллической
подложкой.
Таблица 3.7
Некоторые кристаллы, выращенные по методу ГХР
Исходный материал
Si02
Fe304
Cr203
МО + Fe203
(M-Mg, Co, Ni)
Nb + Nb205
NbSe2
Конечный продукт
(кристаллы)
Si02
Fe304
Cr203
MFe204
NbO
NbSe2
Транспортный агент
HF
HC1
Cl2 + 03
HC1
CI,
h
Температура, К
1 470 ->- 770
1270 -+- 1070
1070 -+ 870
—
—
1100-> 1050
Введение
151
Если поликристаллический образец нагревать в течение
достаточно длительного времени ниже его точки плавления, наблюдается
увеличение среднего размера кристаллов. Движущей силой этого
изменения является уменьшение поверхности кристалла. Число
полученных кристаллов зависит от числа активных зародышей,
способных расти за счет поликристаллической матрицы. Металлы
обычно выращивают в виде кристаллов в твердом состоянии
способом так называемого «отжига напряжения», который состоит в
рекристаллизации отжигом со снятием напряжения. Рост в твердом
состоянии особенно пригоден для твердых веществ с инконгруэнт-
ным плавлением и полиморфным превращением ниже точки
плавления.
4. ФАЗОВЫЕ ПЕРЕХОДЫ
4.1. ВВЕДЕНИЕ
Многие твердые вещества претерпевают превращения из
одной кристаллической структуры в другую по мере изменения
температуры или давления, и это явление обычно определяют как
полиморфизм. В то время как полиморфизм относят к фазовым
переходам, включающим изменения в атомных конфигурациях в
кристаллах, имеются также переходы с изменением электронных или
спиновых конфигураций. Явление фазовых переходов представляет не
только большой академический интерес, но имеет и важное значение
в технологии. Фазовые переходы проявляются большим
разнообразием систем (табл. 4.1), от простых металлов и сплавов до сложных
неорганических и органических материалов. В последние два
десятилетия в связи с обнаружением новых типов фазовых переходов и
появлением новых подходов к объяснению явления представления о
предмете исследования необычайно расширились. Традиционно эта
тема жизненно важна в металлургии [Porter, Easterling, 1981], но
многие ее аспекты имеют очень большое значение в химии твердого
тела [Rao, 1984]. Различные аспекты фазовых переходов, такие
как критические явления, мягкие моды, механизмы и изменения в
свойствах при фазовых переходах рассмотрены с единых позиций
[Rao, Rao, 1978]. В этой главе мы будем касаться некоторых
основных моментов темы и исследуем некоторые классы фазовых
переходов.
4.2. ТЕРМОДИНАМИКА
В течение фазового перехода при равновесной температуре
свободная энергия твердого тела остается непрерывной, а такие
термодинамические величины, как энтропия, объем, теплоемкость, пре-
Таблица 4.1
Типичные обратимые фазовые переходы
1.
I. Переходы при нагревании *
Кварц а—Р
CsCl CsCl (8 : 8)-NaCl (6 : 6)
Agl Гексагональный — кубический
NH4C1 CsCl (8 : 8)-NaCl (6 : 6)
NaN02 Ромбический — ромбический
RbN03 Тригональный — CsCl
CsCl — Гексагональный
Гексагональный — NaCl
II. Переходы под давлением
КС1 NaCl (6 : 6)-CsCl (8 : 8)
RbCl NaCl (6 : 6)-CsCl (8 : 8)
Si02 Кварц — коэзит
Коэзит — стишовит
ZnO Вюртцит — NaCl
CdTiOg Ильменит — перовскпт
CdS Вюртцит — NaCl
FeCr204 Шпинель — тип Cr3S4
2
tvk
848
752
427
469
439
439
461
551
Pv кбар
19,6
5,7
18,8
93,1
88,6
40,4
17,4
36,0
3
AVt, см3-моль-1
1,33
10,3
-2,2
7Д
1,66
6,0
3,12
3,13
AFj, см3-моль-1
-4,11
-6,95
-2,0
-6,6
-2,55
-2,90
-7,2
-6,5
4
AHfJ кДж-моль"1
0,36
2,42
6,15
4,47
1,05
3,97
2,72
2,72
AHt, кДж-моль-1
8,03
3,39
2,93
57,27
19,23
15,88
-10,45
-30,51
* Все, за исключением переходов в кварце и NaNOj, показывают заметный гистерезис. ДУ^ ДЯ^ — изменение объема и
энтальпии, связанное с переходом при температуре Т^.
Термодинамика 153
терпевают дискретные изменения. В зависимости от того, какая
производная свободной энергии Гиббса проявляет дискретное
изменение при переходе, фазовые переходы классифицируются как
переходы первого рода, второго рода и т. д. В фазовом переходе первого
рода обнаруживают такие изменения первые производные свободной
энергии Гиббса (S и V):
(w)P = -s'' [Щт = у- ^
Для фазовых переходов второго рода такие изменения обнаруживают
вторые производные свободной энергии:
{&)r-b{£)r~r*(™)-v- (4-2>
Здесь Ср, а, Р — соответственно теплоемкость, коэффициент
объемного термического расширения и сжимаемость. Переходы первого
рода, включающие дискретные изменения в энтропии и объеме,
изображены на рис. 4.1. На этом рисунке кривые Gn Gu представляют
изменения свободных энергий фаз I и II соответственно, тогда как
77n Hu и VIt Vu — изменения энтальпии и объема. Мы видим, что
кривые свободных энергий пересекаются (AG = 0) строго при
температуре фазового перехода Tt или при давлении фазового
перехода Pt.
Для перехода второго рода кривые G(P, T) не пересекаются,
и мы видим непрерывные изменения энтальпии или объема в течение
перехода. В таких переходах обычно обнаруживает изменения
теплоемкость. Переходы второго рода почти всегда ассоциируются с
какими-либо процессами разупорядочения (рис. 4.2). Параметр
дальнего порядка изменяется с температурой в переходе второго
рода, причем его максимальное значение — единица, когда имеется
совершенный порядок, и нуль, когда имеется полный хаос.
Существует несколько переходов, когда теплоемкость стремится к
бесконечности при температуре перехода (приводя к ^-образной зависимости
Ср от температуры), и такие переходы относят к Х-переходам.
Фазовые переходы первого рода связаны с значительным гистерезисом,
и эффекты гистерезиса часто используют для характеристики «рода»
перехода. Величина термического гистерезиса в первую очередь
зависит от изменения объема, сопровождающего переход, но она
подвержена влиянию и других факторов [Rao, Rao, 1966; Nata-
rajan et al., 1969].
Уравнение Клаузиуса — Клапейрона описывает термодинамику
фазового перехода первого рода:
(4.3)
154
Фазовые переходы
I
Тс Температура
Рис. 4.1. Изменение: а — свободной энергии и эн- Рис. 4.2. Изменение па-
тальпии с температурой и б — свободной энергии раметра порядка при фа-
и объема с давлением при фазовом переходе первого зовом переходе второго
рода. рода (а) и при фазовом
переходе первого ро-
да (б).
В случае перехода второго рода AF, AS равны нулю и уравнение
(4.3) будет давать неопределенность 0/0. Мы можем, однако, вывести
аналог уравнения Клаузиуса — Клапейрона для таких переходов:
(4.4)
dr
VTAa
В табл. 4.1 приведены термодинамические данные для фазовых
переходов нескольких типичных веществ.
Каждый фазовый переход связан с изменением в симметрии и
в порядке. Чтобы облегчить количественный анализ, Ландау ввел
концепцию параметра порядка £ и выразил свободную энергию
Ф как
Ф(Р, Г, S) = Ф0(Л Т) + аЪ + Ы? + с? + d£* + ..., (4.5)
где Ф0(Р, Т), а, Ъ, с, d — константы. Здесь £ = 1 в полностью
упорядоченной фазе (при низких температурах), £ = 0 в полностью
разупорядоченной фазе после перехода. Так как изменение знака
£ не должно менять значение Ф, коэффициенты нечетных степеней
£ должны быть равны нулю. Уравнение (4.5) тогда становится
Ф(Р, 7\ Q = Ф0(/>, Т) + bt? + dt? + ... . (4.6)
Равновесное значение параметра дальнего порядка £ получается
из условий
(^)Р.т = ^ + 2^>=°'
Из этих соотношений мы получаем решения £ = 0 и £2 = —b/2d.
Так как £ = 0 в разупорядоченном состоянии, Ь > 0 при темпера-
Термодинамика
155
турах выше температуры перехода; тогда находим, что Ъ < О для
упорядоченной фазы. Таким образом, Ь меняет знак при фазовом
переходе второго рода. Предполагая, что Ь линейно изменяется с
температурой, Ъ(Р, Т) = В(Т — Тс), где Тс — температура
перехода или критическая температура, мы получаем
£2 = -ЫЫ = -В(Т - Tc)/2d. (4.7)
Можно легко показать, что изменения в теплоемкости при переходе
задаются B2Tc/2d. Таким образом, видно, что теория Ландау дает
основу для переходов второго рода. Очень важно отметить что
среднее значение параметра порядка (£) стремится к нулю выше Тс
и является конечной величиной ниже Тс для перехода второго рода;
для перехода первого рода изменение в (О дискретно (см. рис. 4.2).
Переходы второго и более высоких родов часто относят к
непрерывным переходам. Идентификация параметра порядка легка для
некоторых переходов и непроста для других. Например, в железе
параметр порядка, связанный с переходом ферромагнетик —
парамагнетик, есть намагниченность; в сегнетоэлектриках, таких как
ВаТЮ3, параметром порядка является поляризация. Для фазового
перехода SrTi03 при 108 К угол поворота кислородного октаэдра
(рис. 4.3) является параметром порядка.
Другой важный вклад Ландау относится к изменениям
симметрии, сопровождающим фазовые переходы. Для переходов второго
рода, или структурных переходов, симметрия кристалла изменяется
дискретно, вызывая появление (или исчезновение) определенных
элементов симметрии, в отличие
от фазовых переходов первого
рода, где нет связи между симме-
триями низко- и
высокотемпературной фаз. Если р(х, I/, z)
описывает вероятность
распределения атомных позиций в
кристалле, то р будет отражать группу
симметрии кристалла. Это
означает, что для Т > Тс р должно
быть согласовано с группой
симметрии G высокотемпературной
(высокосимметричной) фазы. Для
Т <С Тс р должно быть
согласовано с группой симметрии G0
низкотемпературной (низкосимметрич-
Рис. 4.3. Изменения кислородного ок-
таэдра в SrTi03 при переходе при 108 К. h
156
Фазовые переходы
ной) фазы. Можно записать
P = Po + Sp = G0 + 2cM(I\),
(4.8)
где WtK — базисные функции, преобразующиеся в соответствии с
неприводимым представлением I\; G0 и р0 соответствуют симметрии
G0, ар — симметрии G; бр имеет ту же симметрию, что и р.
Уравнение (4.6) фактически может быть записано как
Ф = Ф0 (Р, Т) + Ъ? + С4 2 Vafi (Vi),
(4.9)
где £2 = ^С\,С\ = Yi£ и 2т? = 1- /а является инвариантом
четвертого порядка, построенным из yt. Переход от
высокосимметричной G к низкосимметричной G0 сопровождается соответственно
появлением параметра порядка ( £), когда Ъ меняет знак.
4.3. МЯГКИЕ МОДЫ
Тот факт, что параметр порядка исчезает выше Гс, не
означает, что Природа не подозревает о происходящем вдали Тс.
Действительно, это обнаруживается во многих случаях в поведении
определенных колебательных частот (мод). Еще в 1940 г. Раман и Не-
дунгади открыли, что ос — (3-переход в кварце сопровождается
уменьшением частоты полносимметричной оптической моды по мере
приближения к температуре фазового перехода снизу. Исторически
это первое наблюдение мягкой моды. По существу, мягкая мода
является коллективным возбуждением, частота которого аномально
^/
^о
^/
^f^4
Тс
V 1
1
1
з/
а
Т
2^
6
3
I
I
Рис. 4.4. Изменение мягкомодовой ча- Рис. 4.5. Изменение с температурой
стоты, со2, с температурой при фазовых мягкомодовой частоты в SrTi03 [Shi-
переходах второго рода (а) и фазовых ^ d
переходах первого рода (б). Числа у rano, Yamada, 1969].
кривых представляют кратность
вырождения.
Мягкие моды
157
Рис. 4.6. Различные виды мягких
мод, наблюдаемых при фазовых
переходах. Смягчение может
наблюдаться в любом месте зоны Бриллюэ-
на [Venkataraman, 1979].
уменьшается по мере
приближения к точке фазового
перехода. На рис. 4.4 показана
температурная зависимость
частоты мягкой моды. В то
время как при переходе второго
рода частота мягкой моды
достигает нуля при Гс, при
переходе первого рода изменение
фазы осуществляется до того,
как частота моды может
достичь нуля.
В 1950 г. Кохран и Андерсон независимо предсказали мягкие
моды, отметив, что фазовые переходы в некоторых сегнетоэлектри-
ках могут быть результатом динамической нестабильности решетки.
Действительно, Борн и Хуанг ранее показали, что кристаллическая
решетка становится нестабильной, если одна из ее частот
нормальных колебаний становится чисто мнимой. Каули в 1962 г., изучая
рассеяние нейтронов, обнаружил, что одна из оптических мод SrTi03
смягчается. Рис. 4.5 показывает температурную зависимость одной
из колебательных мод SrTi03, связанной с угловыми осцилляциями
кислородных октаэдров. В последние годы мягкие моды обнаружены
в различных системах, включая сверхпроводники и органические
кристаллы [Blinc, Zeks, 1974; Rao, Rao, 1978; Scott, 1974]. Фоно-
ны центра зоны и фононы границ зоны, также и акустические фоно-
ны могут обнаруживать поведение мягкой моды (рис. 4.6).
Связь между мягкими модами и параметрами порядка в фазовых
переходах можно понять следующим образом. Параметр порядка £
не является статической величиной, хотя <£> служит таковой;
£ может флуктуировать локально, и эти флуктуации производятся
подходящей суперпозицией атомных смещений, связанных с
колебаниями мягкой моды. Соответственно находим, что квадрат частоты
колебания со2 = В(Т — Тс) точно, как в уравнении (4.7). Не
каждый фазовый переход связан с мягкой модой. Независимо от того,
есть мягкая вода или нет, флуктуационный спектр вблизи Тс
обнаруживает характерное поведение. Флуктуации являются, таким
образом, ключевой особенностью, связанной с фазовым переходом.
Недавно изучено поведение мягкой моды под давлением [Samara,
1984].
т, > т2 > т3
158
Фазовые переходы
4.4. ЦЕНТРАЛЬНЫЕ ПИКИ
Одной из наиболее впечатляющих особенностей спектра
рассеяния нейтронов или света около фазового перехода является
появление пика упругого или квазиупругого рассеяния с почти
нулевым сдвигом частоты, причем последняя совершенно не связана с
квазигармоническим мягкомодовым описанием динамики [Fleury,
Lyons, 1983]. Первые наблюдения дивергенции интенсивности
рассеянного света осуществили Яковлев с сотрудниками [Yakovlev
et al., 1956], которые наблюдали это явление при а — (3-переходе
кварца. Интенсивность рассеянного света резко увеличивается,
иногда почти в 10 000 раз вблизи Гг, и максимальное значение
ширины линии довольно мало (~ 1 см-1). На рис. 4.7 с целью
иллюстрации показаны типичные центральные пики.
Одно время существовало мнение, что центральные пики
появляются благодаря примесям, дефектам, другим подобным внешним
или внутренним факторам. Предложен ряд моделей и механизмов,
основанных на флуктуациях энтропии, флуктуациях плотности фо-
нонов, диэлектрической релаксации, молекулярной реориентации,
солитонах, фазонах (флуктуация в фазе волны вдоль к-вектора),
динамическом и статическом кластерировании и т. д. Сейчас
признано, что одиночные центральные пики при температуре вблизи Тс
VH.gii 11001
_0 5 0 0 5
Энергия, мэВ
20 0 20
Сдвиг частоты,
ГГы,
Рис. 4.7. Центральные пики.
спектр рассеянных нейтронов KMnF3 (Г
90 К)
[Shapiro et al., 19741; б — квазиупругое рассеяние света
TbV04 (T ~ 32 К) [Harley et al., 1980].
Критические явления
159
могут быть вызваны динамическими или статическими явлениями.
Измерения формы и ширины линии позволяют выяснить механизмы,
но нет одной причины, которой можно объяснить появление
центральных пиков во всех системах.
4.5. КРИТИЧЕСКИЕ ЯВЛЕНИЯ
Многие физические свойства отклоняются от нормы вблизи
Тс (например, показывают большие значения по мере приближения
к Тс с любой стороны). Интересно, что такие дивергенции подобных
свойств в различных фазовых переходах почти идентичны, как это
показано на рис. 4.8, на котором представлены типичные
зависимости Ср от температуры. Эти дивергенции могут быть определены
количественно в терминах так называемых критических экспонент.
Экспоненты критической точки задаются так:
*-Ч-т£гЧ (4Л0)
где 8 = (Т — Тс)/Тс и /(e)—функция, экспонентой которой
является X. Наиболее важными экспонентами являются те, которые
связаны с удельной теплоемкостью (а), параметром порядка (|3),
магнитной восприимчивостью (у) и длиной корреляции (v). Длина
корреляции относится к области, в которой индивидуальные составные
части целого (такие, как атомы, атомные моменты и т. д.) скоррели-
рованы. Оказывается, что индивидуальные экспоненты для
различных переходов достаточно близки (например, (3 « 0,33). Более инте-
т Ш\ КН2Р04 VI 1 Fe
О. ' 1 1 1 Г" С_> 1—,—|—г-1—»—
« 120 130 1020 10601100
т,к т,к
Рис. 4.8. Сравнение температурных
зависимостей теплоемкости для двух
фазовых переходов второго рода:
KH2P04 (a), Fe (б).
Рис. 4.9. Некоторые наблюдаемые
переходы в п — d-плоскости. Три цезиевых
соли являются одномерными, тогда как
полимеры структурно трехмерны, но не
имеют магнитного упорядочения [Rao,
1984].
Ферромагнитный EuS
jH CsCuCU K2NiF4 АнтиферромагнитньЛ
RbMnFj
2 \ CsNLF3 Co CI 2 Сверхтекучий Не
с
/J CsCoCl3 K2CoF4 Жидкости
q\ Полимеры
160
Фазовые переходы
ресно, что соотношения типа а + 2(5 -f- у = 2 остаются в силе для
переходов независимо от детальной природы систем.
Такая универсальность в экспонентах критической точки
интриговала ученых длительное время, но сейчас понята в свете
концепций Каданова о масштабной инвариантности [Kadanoff, 1966],
связываемой с флуктуациями вблизи Тс. Каданов использовал
«самоподобие» среди флуктуации вблизи Тс и установил, что гиббсовский
потенциал должен быть однородной функцией вблизи Тс. В 1972 г.
был применен метод ренорм-групп для расчета точного значения
экспонент. Все фазовые переходы могут быть охарактеризованы в
терминах размерности системы d и размерности параметра порядка
п (рис. 4.9). Уилсон и Фишер трактовали размерность систем как
непрерывно изменяющийся параметр е = (4 — d), где е
непрерывно изменяется. Подробнее критические явления описаны Domb
[1985]. Реиорм-группа применяется во многих случаях: трикрити-
ческие точки, переходы в двумерных системах, обобщение модели
Изинга для более, чем двух компонентов, во фрактальной
концепции [Mandelbrot, 1983].
4.6. СТРУКТУРНЫЕ ИЗМЕНЕНИЯ
ПРИ ФАЗОВЫХ ПЕРЕХОДАХ
На основе нашего знания кристаллохимии мы можем
предсказать природу структурных изменений при фазовых переходах в
простых ионных кристаллах. Для таких прогнозов воспользуемся
фактом, что при термических переходах высокотемпературная
форма имеет симметрию выше, чем низкотемпературная. При переходах
под давлением фаза высокого давления будет более плотно
упакована (большее координационное число) и будет иметь меньший объем.
Так, под давлением твердые тела со структурой типа NaCl обычно
переходят в твердые тела со структурой типа CsCl. Нагревание
твердых тел со структурой типа CsCl обычно приводит к твердым телам со
структурой типа NaCl. В твердых телах типа АВ2 мы могли бы
ожидать структуру искаженного рутила или некоторую другую
низкосимметричную структуру, переходящую в структуру рутила или
флюорита при высоких температурах. Можно полагать, что
искаженный перовскит переходит в кубическую структуру при высоких
температурах. Относительную стабильность кристаллических структур
может объяснить борновская модель ионных кристаллов с
соответствующими параметрами отталкивания и Ван-дер-Ваальса; в
частично ковалентных твердых телах параметр ионности можно
использовать для предсказания предпочтительной кристаллической
структуры (см. гл. 1, разд. 1.3).
Существует классификация [Buerger, 1951] фазовых переходов
на основе структурных изменений, затрагивающих первую или
следующие координационные сферы.
Структурные изменения при фазовых переходах 161
1. Переходы, затрагивающие
первую координационную сферу
(например, CsCl — NaCI,
арагонит — кальцит):
а) реконструктивные
(медленные);
б) дилатационные (быстрые).
2. Переходы, затрагивающие
вторую или последующие
координационные сферы (ВаТЮ8, а - Рис 410 Дилатационный механизм по-
р-кварц): рехода от структуры CsCl в структуру
а) реконструктивные (медлен- NaCI [Buerger, 1951].
ные); б) диспласивные (быстрые).
Реконструктивные переходы обычно требуют разрушения и
образования связей, но они же могут совершаться по простому дила-
тационному механизму. Бюргер предлагал такой механизм для
перехода из структуры CsCl в структуру NaCI (рис. 4.10). Известно, что
такие деформационные взаимосвязи существуют между различными
структурами |Rao, Rao, 1978] и они важны для понимания
механизмов фазовых переходов. При диспласивных переходах в структуре
координационных полиэдров происходят лишь небольшие изменения.
Структурные переходы также классифицируются на ферродис-
торсионные (без изменения числа формульных единиц в
элементарной ячейке) и антиферродисторсионные (с изменением числа
формульных единиц в элементарной ячейке). Ферродисторсионпые
переходы могут быть или диспласивными (например, ВаТЮ3) или типа
порядок — беспорядок (например, NH4C1).
Антиферродисторсионные переходы также могут относиться к этим двум типам (например,
SrTi03 и NH4Br соответственно). Ферродисторсионные
диспласивные переходы проявляются только у сегнетоэлектриков, но
антиферродисторсионные диспласивные переходы обнаруживаются и у
сегнетоэлектриков, и у антисегиетоэлектриков. В таких материалах,
как КН2Р04, имеется сильное смешение переменных типа порядок —
беспорядок и диспласивного типа. Переход Snl4 под давлением
уникален в том отношении, что фаза высокого давления аморфна [Su-
gai, 1985]. Является ли аморфное состояние результатом разрушения
топологии упаковки тетраэдров под давлением?
4.7. МЕХАНИЗМЫ ФАЗОВЫХ ПЕРЕХОДОВ
Фазовые переходы в твердых телах также плодотворно
классифицируются на основе механизма. Важными, обычно
встречающимися типами переходов являются: 1) переходы зародышеобразова-
ния и роста; 2) переходы порядок — беспорядок и 3) мартепситиые
переходы.
И Ч. Н. Р. Рао, Дж. Гопалакришнан
162
Фазовые переходы
Ъ \ тг
wvtj
Рис. 4.11, Изменение барьера за-
родышеобразовання AG ядер с
радиусом г при различных
температурах.
2 5 Ю 20 50 100 200 300400
t, MUH
Рис. 4.12. Зависимости Avrami для
переходов серого олова.
1 — 298 К; 2 — 300,5 К; 3 — 303 К; 4 —
305,5 К; 5 — 308 К*
В переходах типа зародышеобразования и роста зародыши
повой фазы, имеющие критический размер, сначала должны
образоваться в исходной фазе. Изменение свободной энергии AG вследствие
образования сферических ядер задается уравнением
AG = —4/3nr3AG_ + 4яг2?,
(4.11)
в котором первый член в правой части представляет уменьшение в
объемной свободной энергии, а второй — увеличение поверхностной
свободной энергии. Когда радиус сферического зародыша г мал,
второй член будет доминировать, и зародыши, следовательно,
будут термодинамически нестабильны. Однако когда зародыши
достигнут критического радиуса rc, AG начнет уменьшаться, и
дальнейший рост зародыша будет термодинамически предпочтительным.
На рис. 4.11 показано изменение AG с изменением г при различных
температурах. Видно, что при температуре термодинамического
перехода, Г0, AG всегда положительно и никакого
зародышеобразования не может происходить. Всегда, для того чтобы произошел
фазовый переход, необходимо небольшое переохлаждение либо
небольшой перегрев. Принимая (dAG/dr) равным нулю, мы можем
получить гс и AGC из уравнения (4.11):
rc = 27/AGr,
Gc = 16nY3/3AGD2.
Можно показать, что скорость зародышеобразования задается
R = А ехр [-(AGC + Ea)/RT], (4.12)
где А — предэкспоненциальный множитель, а Еа — энергия
активации перескока атомов через границу раздела объем — зародыш. Как
Механизмы фазовых переходов
163
только зародышеобразование произошло, фазовый переход до
своего завершения протекает путем роста (или размножения) ядер
критического размера. Степень роста будет также связана со свободной
энергией активации.
Для анализа кинетических данных по переходам
зародышеобразование — рост часто используются эмпирические уравнения. Одним
из обычных уравнений является уравнение Аврами:
х = 1 — ехр (—ktn), (4.13)
где х — объемная доля перешедшей фазы, a t — время. Уравнение
(4.13) адекватно описывает кинетику переходов, причем значение
п меняется в зависимости от природы перехода (всегда между 1 и 5).
На рис. 4.12 показаны графики Аврами для перехода серого олова.
Значение п для перехода белое — серое олово равно 3, тогда как для
перехода серое — белое находится между 1,5 и 2. Значения кип
в уравнении (4.13) получаются откладыванием lglg [1/(1 —х)] от
lg t для получения прямых линий. Переход анатаз — рутил для
TiO.: является другим примером перехода типа
зародышеобразование — рост.
Переходы порядок — беспорядок обычно связаны: 1) с
позиционным разупорядочением, 2) ориентационным разупорядочением или
3) разупорядочением состояний электронных (или ядерных) спинов.
Конфигурационная энтропия разупорядочения задается уравнением
AS = R In (QXI/Qx), (4.14)
где QIX и Qx — общее число конфигураций (или ориентации) в
получающейся и исходной фазе соответственно. Два известных
примера позиционного разупорядочения — переходы в (5-латуни и Agl.
Agl переходит из гексагональной фазы в кубическую, причем
последняя обнаруживает высокую ионную проводимость вследствие
разупорядочения Ag+-noHOB. Увеличение энтропии в этом переходе
(16,74 Дж-моль^-К-1) даже больше, чем ожидаемое увеличение в
конфигурационной энтропии (R In 12/2=14,86 Дж-моль^-К-1),
наводит на мысль, что ионы Ag+ в высокотемпературной кубической
фазе используют много больше позиций, чем доступные 12 тетра-
эдрических позиций.
Имеется много примеров ориентационного разупорядочения в
неорганических твердых телах. Большинство твердых веществ,
содержащих оксоанионы или другие ди- или полиатомные ионы,
проявляют ориентационные переходы порядок — беспорядок
[Parsonage, Staveley, 1978]. Так, при 83 К у KCN имеется переход из
фазы III в фазу II, AS этого перехода около R In 2; это указывает на
то, что CN~-noHbi получают две возможные ориентации. При II—I-
переходе в KCN при 168 К AS = R In 4, указывая, что в кубиче-
11*
164
Фазовые переходы
Рис. 4.13. Представление
пластинки мартенсита (б), образованной
из прямоугольного блока
исходного кристалла (а). Заметим, что
плоские поверхности A2B2C2D2 и
Ax'Bx'C/Dx' остаются
неизменными в течение фазового перехода.
ской фазе I CN-иоиы ориентированы в любом из восьми возможных
направлений. Аналогично при III—Н-переходе NH4C1 при 243 К
AS = 0,7 R, показывая, что в разупорядоченной фазе II х\Н4+-
ионы используют две возможные ориентации (и что нет
свободного вращения).
Хотя первоначально мартенситные фазовые переходы были
открыты в сталях, сейчас считается, что они обеспечивают механизм
переходов во множестве неорганических (и, возможно, даже
органических) твердых веществ. Мартеиситный переход является
структурным изменением, вызванным атомными смещениями (а не
диффузией), соответствующими однородной деформации, приводящей к
инвариантной плоской деформации, посредством которой исходная и
образующаяся фазы связаны замещенным решеточным соответствием,
иррациональной плоскостью габитуса и точным орпентационным
соотношением (рис. 4.13). Это систематическое изменение в
положениях атомов на макроскопические расстояния привело к
метафорическому определению переходов как «военных» [Christian, 1905].
Мартенситные переходы часто осуществляются с высокими
скоростями (порядка скорости звука). Интересно, что переход CsCl — NaCl,
для которого Бюргер бесхитростно предполагал дилатационный
механизм (см. рис. 4.10), является мертенситным переходом с ориента-
ционными связями между двумя фазами. Действительный
деформационный механизм незначительно отличается от механизма на
рис. 4.10. Другими примерами твердых тел, испытывающих
мартенситные фазовые переходы, являются Zc02) КТао.^^о.збОз И
сверхпроводящие А — 15-соединения, такие как V3Si.
Фазовые переходы, обсуждаемые до сих пор, протекают без
изменения состава. Спинодалъное разложение, часто наблюдаемое в
бинарных твердых растворах металлов и в стеклах, напротив,
является результатом термодинамической нестабильности, вызванной
их составом [Calm, 1968]. Особым признаком этого типа
твердофазных переходов является отсутствие какого-либо барьера зароды-
шеобразования. Имеется класс переходов, называемых эвтектоид-
ным разложением, в которых одна фаза разлагается на две фазы
различных составов, причем их морфология состоит из
параллельных слоев или стержней одной фазы в матрице другой.
У}р-44^
D2D,
В,
А4Кд
D,
Органические твердые тела
165
4.8. ОРГАНИЧЕСКИЕ ТВЕРДЫЕ ТЕЛА
Хотя время от времени сообщается о фазовых переходах в
органических кристаллах, изучение их механизмов не проведено так
широко, как в случае неорганических твердых тел. До недавнего
времени многие полагали, что фазы, участвующие в фазовых
переходах органических кристаллов, структурно не связаны, а эти
переходы рассматривались как в чем-то подобные плавлению. Однако
в последние несколько лет появилось несколько исследований
органических кристаллов, в которых изучались детали механизма
переходов. В 1,8-дихлоро-10-метилантрацене под давлением
исследованы [Jones et al., 1975] фазовые переходы, происходящие по
бездиффузионному диспласивному механизму (в чем-то подобному мар-
тенситному переходу) с определенной ориентационной связью. По-
видимому, иррациональная плоскость габитуса может быть
составлена из плотноупакованных плоскостей и свойства межфазной
границы могут быть сформулированы в терминах скольжения дислокаций.
Обратимый топотактический фазовый переход перхлората 5-метил-
1-тио-5-азониациклооктан-1-оксида объяснен [Parkinson et al., 1970]
в рамках периодических скользящих отдельных дислокаций;
раньше думали, что этот переход включает кооперативную инверсию
и вращение половины молекулярных катионов [Paul, Go, 1969].
у-Фаза р-дихлорбензола характеризуется необычайно высокими
внутримолекулярными частотами колебаний, и а — 7~пеРех°Д
показывает атермальное зародышеобразование [Ganguly et al., 1979];
а — |5-переход связан с разупорядочением. Бензотиофен
обнаруживает переход порядок — беспорядок [Rao et al., 1982]. Переход
желтой формы 2-(4'-метоксифенил)-1,4-бепзохинона в красную
представляет собой зародышеобразование и миграцию хорошо
определенных границ; молекулы в двух фазах являются диастереомерами
[Desiraju et al., 1977]. Фазовый переход паратерфенила,
включающий вращательное разупорядочение, объяснен определяющим
парным взаимодействием между несвязанными атомами [Ramdas,
Thomas, 1976].
Широко представлены в литературе фазовые переходы в
кристаллах с водородной связью, таких как сегнетоэлектрические
гидрофосфаты и сегнетова соль. Недавно методами колебательной
спектроскопии исследованы фазовые переходы алкановых дикислот [Rao
et al., 1982]. Переход в малоновой кислоте особенно интересен.
При обычных температурах элементарная ячейка малоновой
кислоты содержит два циклических димерных кольца, ортогональных друг
ДРУГУ; при температуре выше ее фазового перехода при 360 К два
водородно-связанных кольца становятся подобными, как об этом
свидетельствуют ИК- и Раман-спектры [de Villepin et al., 1984].
В среднем в высокотемпературной фазе водородные связи слабее,
чем в низкотемпературной. В полностью дейтерированной кислоте
166
Фазовые переходы
фазовый переход происходит при более высокой температуре (360 К)
и колебательные полосы показывают положительный изотопный
эффект. По-видимому, этот переход управляется колебательными и
торсионными, проявляющими тенденцию к смягчению модами водо-
родно-связанных колец (около 90 и 50 см-1 соответственно при
температуре ниже температуры фазового перехода). Слоистые
соединения типа (RNH3)2MC14 (M—Mn, Fe, Cd, Си) обнаруживают
несколько фазовых переходов, связанных с водородными связями
RNH3-rpynn, которые приводят к упорядоченным
низкосимметричным структурам при низких температурах. В метиламмониевых
соединениях, например, ионы (CH3NH3) + достигают С^-симметрии
только в высокотемпературной фазе; ИК-спектры ясно показывают
природу переходов через изменения в колебательных частотах алкил-
аммонийных групп [Rao et al., 1981].
4.9. НЕСОРАЗМЕРНЫЕ ФАЗЫ
Твердые тела обычно кристаллизуются в правильные струк-
хуры с периодической упаковкой атомов. В некоторых кристаллах
взаимодействие между электронами и ионными остовами или
наличие двух видов переходов различной симметрии делает правильное
расположение атомов нестабильным по отношению к малым
искажениям. Тогда стабильным должно быть такое состояние, в котором
плотность заряда или положения атомов имеют длиннопериоди-
ческие модуляции [Riste, 1977; Bak, 1982]. Период модуляций
может быть соразмерен и несоразмерен с решеткой, лежащей в основе,
причем несоразмерная фаза не показывает истинной периодичности,
но имеет два независимых периода. Вообще говоря, несоразмерные
фазы имеют место в системах с конкурирующей периодичностью,
когда одним является период, лежащий в основе решетки, а другим
может быть период еще одной атомной решетки со своей
периодичностью, приводящей к несоответствию при совместной упаковке
двух решеток. Такое несоответствие ответственно за несоразмерные
двумерные структуры, найденные во многих слоистых твердых
телах, таких как интеркаляционные соединения графита [Makovicky,
Hyde, 1981]. Кроме фаз с такой составной несоразмерностью, как
в проводниках (таких как TaS2), так и в диэлектриках (таких как
NaN02, K2Se04) встречаются фазы в которых несоразмерность
является результатом периодического искажения правильной
решетки (диспласивная несоразмерность). Несоразмерные фазы
встречаются в оксидах и сульфидах металлов и в других материалах, в
которых точечные дефекты (вакансии) самоупорядочиваются, вызывая
сверхструктуры (см. гл. 5, разд. 5.5). Давно известны
несоразмерные магнитные структуры, к этой категории принадлежат и
известные спиральные магнетики [Dzyaloshinskii, 1964]. Термин модули-
Несоразмерные фазы
167
е/а0
Рис. 4.14. Одномерная модель, по- Рис. 4.15. Образование ВЗП в одномерном
называющая взаимодействие меж- металлс Синусоидально модулированная
ду рядом атомов (кружки) и ие- "LKl<XJLJL J
риодическим потенциалом (вол- электронная плотность показана наверху,
нообразная линия) которая при- представляют ионы,
водит к соразмерной (а), несораз- lull4il " ^ "
мерной (б) и хаотичной
структуре (в) [Вак, 1982].
ровапные структуры используется для описания несоразмерных
возмущений (т. е. таких, в которых отношение вынужденной
периодичности к периодичности элементарной ячейки иррационально).
Расширенное определение модулированной структуры может быть
использовано для описания любого периодического или почти
периодического возмущения кристаллической структуры, с
расстоянием повторяемости, заметно большим, чем размеры элементарной
ячейки; такое определение будет также включать многообразие сверх-
структур [Cowley et al., 1979].
Образование несоразмерных фаз может быть визуализировано
рассмотрением одномерной упаковки атомов (рис. 4.14). Ряд
атомов, обладающий периодом а, взаимодействует с потенциалом
периода Ь. Мы можем различить три возможных случая: 1) когда
взаимодействие достаточно сильно, так что решетка согласуется с
основным потенциалом и период решетки а является простой
рациональной частью периода потенциала 6; 2) когда а и Ъ не связаны простым
рациональным отношением, структура становится несоразмерной;
3) третьей возможностью является хаотическая структура, в
которой атомы распределены случайным образом по минимумам
потенциала. Несоразмерная волна составляется амплитудной волной
(амплитудой) и фазовой волной (фазон). Присутствие высших
гармоник несоразмерной модуляции приводит к так называемой
дьявольской лестнице.
В проводящих твердых телах плотность электронов
проводимости пространственно модулирована, образуя волны зарядовой
плотности (ВЗП). Периодическое искажение, сопровождающее ВЗП
(благодаря взаимодействию электронов проводимости и решетки),
ответственно за несоразмерную фазу [Overhauser, 1962; Di Salvo,
Rice, 1973; Riste, 1977]. Существование ВЗП и периодического
искажения может быть понято в рамках модели, предложенной Пайерлсом
и Фрёлихом для одномерных металлов. Рассмотрим ряд однородно
168
Фазовые переходы
размещенных цепочек ионов (расстояние равно а), связанный с
электронами проводимости с энергией Е(к) и волновым вектором к.
При О К все состояния заполнены вплоть до уровня Ферми Е? =
= Е(к$). Если электронная плотность синусоидально
модулирована, как на рис. 4.15, то
р(х) = p0(s) [1 + Pi cos(Qx + Ф)], (4.15)
где pj — амплитуда зарядовой модуляции и Q — ее волновой
вектор; фазовый фактор Ф описывает положение ВЗП, относящейся
к ионам основной решетки. Смещение волны на расстояние и
эквивалентно изменению в фазе от Ф до Ф -f Qu, и зарядовая модуляция
вызывает модуляцию позиций ионов по уравнению
Un - U0 sin (nQa + Ф), (4.16)
где п — целое число, определяющее положение ионов.
Периодическое искажение решетки с волновым вектором Q открывает на Ер
щель в энергетическом спектре. Состояния ниже £Р, которые
опущены вниз по энергии, занимаются, тогда как состояния, которые
повышены,— освобождаются. Следовательно, общая энергия
понижается. Если выигрыш в энергии больше, чем потенциальная
энергия, требуемая на искажение, ВЗП будет проявляться спонтанно.
При О К такое искажение будет всегда иметь место в одномерной
системе вне зависимости, насколько слабы электрон-решеточные
взаимодействия. По мере повышения температуры электроны будут
термически возбуждаться через щель, уменьшая тем самым выигрыш
в энергии. Наконец, при температуре перехода Tt щель исчезает.
Выше температуры перехода остаются активными процессы, которые
вызывают кристаллическое и зарядовое искажение. Не приводя к
стабильному искажению решетки, они вызывают аномалию в
поведении продольных акустических фононов, называемую гигантской
аномалией Кона: эта аномалия проявляется в энергии фононов, которая
уменьшается, когда волновое число увеличивается, переходя через
2A*F. Согласно этой простой модели энергия фононов при 2AF
уменьшается до нуля, когда температура приближается к Tt. Ниже Тх
этот фонон заморожен в статическое искажение.
Кроме электрон-фононного взаимодействия в возможности
образования ВЗП важна форма поверхности Ферми. Поверхности фер-
ми-систем, содержащих линейные или плоские группировки атомов,
отражают их одно- или двумерный характер. Поскольку поверхности
Ферми связывают множество состояний с одним и тем же волновым
вектором Q, периодическое искажение, имеющее волновой вектор
Q, будет давать щели в этой же степени связанных вектором Q.
Энергия, выигранная созданием таких щелей около поверхности
Ферми, может перекрыть затраты потенциальной энергии на
искажение решетки, тем самым делая это искажение стабильным.
Несоразмерные фазы
169
Дихалькогениды переходных металлов Va группы дают
классические примеры металлических проводников, проявляющих ВЗП-
индуцируемые периодические искажения решетки [Wilson et al.,
1975; Williams, 1976]. Эти вещества кристаллизуются в нескольких
политипных структурах, IT (СсЩ-тип), 2Н (MoS2-Tnn) и 4Н, в
которых переходный металл имеет октаэдрическую, тригонально-приз-
матическую и оба типа координации соответственно. Искаженные
низкотемпературные фазы показывают сателлитные
(сверхструктурные) рефлексы на электронограммах (рис. 4.16), которые отделены
от брегговских рефлексов на mQ, где т — целое число.
Местонахождение сателлитных пятен помогает определить период ВЗП. Многие
из этих дихалькогенидов проявляют два перехода, один при Г1С
и другой при Тс. При Tjc сверхрешеточные рефлексы несоразмерны
с основной гексагональной решеткой, причем волновой вектор гхка-
жения Q равен (а*/п) -\- б. Часть б становится равной нулю при
^с с Q> точно равным а*/3. Ниже Тс фаза является соразмерной,
обладая элементарной ячейкой, которая в п раз больше основной
ячейки. В температурном интервале Тс <С Т < Г1С несоразмерная
структура существует с б(=^= 0), которая уменьшается с
температурой и резко падает до нуля при Тс. Для несоразмерной фазы не
может быть определена элементарная ячейка; нет такой элементарной
ячейки, которая могла бы содержать точный период как волновой,
так и основной кристаллической структуры. Наблюдаются
сопровождающие этот переход аномалии в электросопротивлении этих
халькогенидов (рис. 4.17).
К2 [Pt(CN)4 ]Вг0>3 • .гН20(КСР) является примером одномерного
соединения (см. гл. 6, обсуждение низкоразмерных твердых тел),
состоящего из параллельных стопок Pt(CN)4, образующих линейные
цепи атомов платины. Внутри каждой цепочки атомы металла раз-
о
мещены равномерно с расстоянием Pt—Pt, равным 2,88 А (немного
о
большим, чем это расстояние в металлической платине — 2,77 А),
как это показано на рис. 4.18, Стопки отделены друг от друга на
о
9,87 А. В то время как в диэлектрическом K2[Pt(CN)4]-xH20
высшая занятая зона (содержащая (й22)-орбитали платины) полностью
заполнена, в частично окисленном КСР, 0,ЗВг удаляют
эквивалентное количество электронов платины, делая с£22-зону заполненной на
85 %. КСР подвергается широкому переходу металл — диэлектрик
около 200 К и является хорошим диэлектриком при низких
температурах. Дифрактограммы КСР при комнатной температуре показы-
ьают диффузные сателлитные линии вместе с брегговскими
рефлексами [Comes et al., 1973]. Диффузные линии показывают смещения
атомов параллельно цепи с волновым вектором Q = 0,ЗС*, где
С* является постоянной обратной решетки вдоль направления цепи.
При понижении температуры до 77 К диффузные сателлиты
становятся острее. Исследования рассеяния нейтронов [Carneiro et al.,
170
Фазовые переходы
Рис. 4.16. Электронные дифрактограммы IT — TaS2 и 4Нь — TaS2.
I — IT — TaSe2 при 500 К; II — 4Hb— TaS2 при 300 К, показывающая
сверхструктуру с 13а0; III — 4Hb— TaS2 при 320 К; IV — 4НЬ— TaS2 при 350 К; V—4Hb—TaS2
при 460 К (заметим, что картинка похожа на I); VI—4Hb+— TaS2, вновь
охлажденный до 300 К, 13а0— сверхструктура появляется вновь (по: [Wilson et alM 1975])»
Несоразмерные фазы
171
ш
L- CN
Рис. 4.18. Схематическая структура
K2[Pt(CN4)]Br0, 33H20 (KCP).
Схематически показана упаковка
[Pt(CN4]-rpynn в цепи.
О ТОО
I I I I I I I > л
2О0 ЗОО 400 500 6бС
Рис. 4.17. Электросопротивление
параллельно слоям дихалькогенидов
переходных металлов [Wilson et al.,
1975].
1976] установили существование 2/ср аномалии Кона в КСР.
Низкотемпературные результаты показывают трехмерное упорядочение
решеточного искажения в соседних цепочках, но поперечная длина
корреляции не очень велика. Соли тетратиафульвалена (TTF) имеют
кристаллическую структуру, составленную из двух несоразмерных
подрешеток [Johnson, Watson, 1976].
Экзотическим, анизотропно проводящим твердым телом,
имеющим несоразмерные фазы, является Hg3_xAsF6, состоящая из
цепочек атомов ртути и решетки «хозяина» AsF6 [Pouget et al., 1978;
Emery, Axe, 1978]. Атомы Hg хорошо упорядочены вдоль
направления цепи, и внутрицепочечное расстояние Hg—Hg несоразмерно с
параметром решетки «хозяина» вдоль этой цепочки. При комнатной
температуре между соседними цепочками ртути нет никакого
фазового упорядочения, но при охлаждении оно постепенно появляется.
В положении ртутных цепочек имеется периодичность,
обусловленная правильной сеткой открытых каналов в подрешетке «хозяина»
AsFe. При 120 К конкурирующее ортогональное взаимодействие
между цепочками приводит к трехмерному упорядочению и фазово-
172
Фазовые переходы
му переходу. Трехмерной низкотемпературной фазе сопутствует
несоразмерная модуляция цепочек Hg, указывающая на
взаимодействие с решеткой «хозяина». Hgg.^AsFe действительно любопытное
твердое тело, в котором, грубо говоря, одномерные колонки атомов
Hg не соответствуют целому числу ячеек AsF6. Кроме анизотропии
электрических и оптических свойств, это вещество является
сверхпроводником при низких температурах.
Как отмечалось ранее, многие диэлектрики обнаруживают
несоразмерные фазы, причем при понижении температуры обычно
имеют место переходы в последовательности: обычная —
несоразмерная — соразмерная фазы. Переход между обычной и
несоразмерной фазами обозначается как несоразмерный. Например,
K2Pb[Cu(N02)6] показывает несоразмерный переход при 281 К (Т1С)
и переход в соразмерную фазу при 273 К (Тс). Соответствующие
переходы в K2Se04 — при 129,5 К и 93 К, причем последний связан
с сегнетоэлектричеством.
Несколько галоидных производных тетраалкиламмония типа
lN(CH3)4l2MCl4, где М—Cd, Zn и т. д., также показывают такие
несоразмерные переходы. Предложена модель [Heine, McConnell, 1981,
1984], объясняющая несоразмерные переходы в диэлектриках.
Согласно этой модели, если имеются две различные моды перехода 4я
и Ф (с различными симметриями) из высокотемпературной
неискаженной (разупорядоченной) фазы, они не могут присутствовать в
однородной соразмерной фазе. Они, однако, могут
взаимодействовать в модулированной (несоразмерной) фазе, чтобы понизить
свободную энергию, если их симметрия удовлетворяет определенному
связывающему соотношению. Одна из мод доминирует при низких
температурах, приводя к соразмерной фазе. В этих работах
использована теория фазовых мягкомодовых переходов Ландау,
распространяющая свободную энергию на высокотемпературную сторону и
ожидающая несоразмерную нестабильность при Г1С. В NaN02
группы N02 упорядочены с q = 0 ниже Тс (все выстроены в одном
направлении вдоль оси Ь) с сопровождающим смещением ионов Na+.
В несоразмерной фазе группы N02~ выстроены определенно вдоль
оси Ъ с синусоидальной вероятностью Р и вдоль Ъ с вероятностью
1 — Р, где Р = 1/2 + ^ cos qx и q меняется от 0,1 до 0,12 а*.
Другим переходом является локальный сдвиг элементарной ячейки,
описываемый как 8 = Ф sin qx. В ян-теллеровской системе
K2Pb[Cu(N02)6] действующая пара мод является эквивалентной
типам искажения октаэдрического комплекса [McConnell, Heine,
1982] х2 - у2 и 3z2 - г2.
Рассмотрим коротко фазовые переходы K2Pb[Cu(N02)6],
который при комнатной температуре имеет кубическую (F3m) структуру
[Joesten et al., 1977]. Это вещество подвергается фазовым переходам
при 281 К и 273 К. Промежуточная несоразмерная фаза является
тетрагональной, тогда как низкотемпературная соразмерная фаза
Несоразмерные фазы
173
имеет симметрию ниже ромбической со сверхструктурными
рефлексами при (h -f 1/2, к -\- 1/2, I -\- 1/2) в кубических индексах.
Дифракционные данные согласуются с антиферродисторсиониой
структурой, состоящей из двух различных подрешеток, в одной подрешет-
ке ось а находится в направлении тетрагонального растяжения
октаэдра Cii(N02)6, тогда как в другой направлением растяжения
становится ось Ь. Несоразмерная фаза более интересна.
Значительной особенностью является то, что она показывает сателлитные
рефлексы при (h =±= 0,425, к =*- 0,425,0) в кубических индексах,
указывая, что она обладает несоразмерной структурой. Предложенная
модель для статической структуры этой фазы включает локальное
ян-теллеровское искажение, которое модулировано вдоль [НО] с
\к0\ = 0,425. Структура, основанная на этой модели, показана на
рис. 4.19. В рамках псевдоспиновой модели соразмерная фаза
характеризуется как наклоненная спиновая структура, тогда как
несоразмерная фаза описывается как «винтовая» спиновая структура,
в которой ориентация псевдоспина изменяется в плоскости,
описывая перемещение «винтового» типа. В общем фазовые переходы в
K2Pb[Cu(N02)e] характеризуются нормальной ->- несоразмерной ->■
-> соразмерной структурами, которые сопровождаются
структурными изменениями из кубической в псевдотетрагональную и затем в
псевдомоноклинную фазу. Изменения в электронном состоянии ян-
теллеровского иона Си2+ характеризуются упорядочениями спинов
типа пара -> винт —>■ наклон. Появление несоразмерной фазы
является следствием распространения локальной ян-теллеровской
моды с несоразмерным волновым вектором к.
Для исследования несоразмерных фаз эффективно используется
рассеяние света и нейтронов [Ахе, 1980; Klein, 1983]. Мягкие моды,
связанные с несоразмерными переходами, показывают интересные
особенности. Например, в K2Se04 смягчение имеет место при
некоторых нечетных значениях q, как показано на рис. 4.20. На рис. 4.21
показана часть обратной решетки высокотемпературной
(нормальной) ромбической фазы; соответствующая решетка сегнетоэлектри-
ческой (соразмерной) фазы также показана на этом рисунке.
Из рис. 4.20 видно, что минимум мягкой ветви изменяется с
температурой, хотя он близок к q = (1/3, 0, 0). Смещение б от этой точки
(см. рис. 4.21) уменьшается с температурой. В несоразмерной фазе
сателлитные брегговские рефлексы развиваются в область обратного
пространства и дают меру б вплоть до Тс (93 К), при Тс б разрывно
становится равной нулю, приводя к переходу между несоразмерной
и соразмерной фазами. Предложен [Iizumi et al., 1977] терм
взаимодействия типа СL4r3(g) + 4f3(—q)]P, объясняющий фазовые переходы.
Для изучения фазовых переходов в K2Se04 методом ЭПР также
используется Se04~, получаемый 7~°блучением. ЯМР-спектроско-
пия используется для исследования переходов в таких твердых
телах, как Rb2ZnCl4.
174
Фазовые переходы
Z = О
Рис. 4.20. Дисперсия мягкой моды
в K2Se04 llizumi et al., 1977].
9*" т v
г
Рис. 4.21. Часть (010) плоскости в
обратной решетке K2Se04.
Слева — решетка высокотемпературной
фазы и справа — решетка сегнетоэластич-
ной фазы. В высокотемпературной фазе
смягчение происходит в точке, смещенной
на б от q = (1/3, 0, 0), показанной
крестиком. В несоразмерной фазе сателлитные
рефлексы обнаруживются при X.
Рис. 4.19. Модель структур
несоразмерной (а) и соразмерной (б) фаз
K2Pb[Cu(N02>>6. Стрелками показаны
смещения атомов при ян-теллеровских
активных фононах.
а — фононная мода имеет волновой вектор
к = (0,425, 0,425, 0) и б —волновой вектор Рис 4.22. Криптон, адсорбирован-
фононной моды к = (1/2;. 1/2, 1/2) [Yamada, ruL' *"' „¥¥/„п г' А " F y
1977].
ный на графите.
а — соразмерная /З-структура. Атомы
криптона занимают 1/3 сотовой ячейки;
б — несоразмерная структура [Вак, 1982].
Имеется обзор [Makovicky, Hyde, 1981] несоразмерных
структур в интеркаляционных соединениях графита, соединениях типа
бруцита, сульфидах и подобных слоистых системах. Простой дву-
Кооперативный эффект Яна — Теллера 175
мерной несоразмерной системой является графит с монослоями
адсорбированных газов. При низких плотностях и высоких температурах
эти монослои образуют двумерную жидкую фазу. При низких
плотностях и низких температурах имеет место соразмерная фаза
(рис. 4.22). Некоторые аспекты несоразмерных структур еще
должны быть исследованы (например, эффекты давления на
несоразмерные переходы). Неясно, почему эти несоразмерные переходы обычно
наблюдаются в области лабораторных температур.
4.10. КООПЕРАТИВНЫЙ ЭФФЕКТ ЯНА — ТЕЛЛЕРА
Согласно теореме Яна — Теллера, если основное состояние
иона в кристалле орбитально вырождено, даже в отсутствие других
возмущений, кристалл будет искажаться до кристалла более низкой
симметрии, чтобы снять это вырождение. Имеется некоторое
количество твердых тел, проявляющих тетрагональное искажение из-за
присутствия ян-теллеровских ионов, таких как Си2+ или Мп3+.
Искажение сохраняет центр тяжести ^-уровня катиона, и,
следовательно, ион может достичь одинаковой стабилизации через
искажение до тетрагональной симметрии с с/а > 1 или < 1. Если имеется
взаимодействие между электронными состояниями и
колебательными модами, мы имеем ситуацию, называемую динамическим эффектом
Яна — Теллера. Рассмотрим кратко гамильтониан ян-теллеровской
системы. Ян-теллеровский вклад в гамильтониан записывается как
AQS2, где Q — ядерная координата, S — электронный оператор,
а А — мера силы взаимодействия. Если включить упругую
энергию 1/2 mcx)2Q2, потенциальная энергия обнаружит два минимума с
ЕЗТ = А212ты2 при Q = —Q0, где Q02 = А/тех)2. Имеются четыре
возможных значения Q для данного значения энергии, и значения
Q так же, как и область энергий, изменяются, если изменяется
значение А. Если feo) <C £jt> тогда при низких температурах кТ <С
<С -EjTi и система будет находиться на дне потенциальной ямы,
и искажение будет заметным (как в статическом эффекте Яна —
Теллера). При высоких Г, когда кТ ^ EJT, отмечаются термически
индуцируемые флуктуации из одной потенциальной ямы в другую
(динамический случай). Если 7ш ^ EjT, даже при самых низких
температурах, то никакое искажение не будет стабилизироваться.
Когда электронные состояния взаимодействуют с дважды
вырожденными решеточными модами, энергия должна зависеть от двух
линейно независимых нормальных мод в виде (Qx2 + Q22)1/2 и не должно
быть минимума на поверхности потенциальной энергии.
Гамильтониан взаимодействия ян-теллеровского иона с локальным
искажением записывается в виде Н =2iVQ™01r, где локальное искажение
фгш преобразуется подобно m-й компоненте неприводимого
представления Г пространственной группы симметрии, сила связи за-
176
Фазовые переходы
дается V, Огт — оператор электронной энергии; Qrm выражается
в виде частот нормальных колебаний кристалла.
Так как механизм эффекта Яна — Теллера включает
взаимодействие между лигандными смещениями (и, следовательно, ян-тел-
леровскими ионами), благодаря упругим свойствам решетки, целый
кристалл может стать нестабильным по отношению к искажениям,
если концентрация ян-теллеровских ионов велика. Фазовые
переходы, вызванные такими взаимодействиями, могут привести к
параллельной или другой ориентации всех искажений. Кооперативный
эффект Яна — Теллера является таким фазовым переходом,
порожденным взаимодействием между электронными состояниями одного
из ионов в кристалле и фонолами. Этот переход может быть и
первого, и второго рода, и в любом случае наблюдается понижающее
симметрию искажение и расщепление электронных уровней. Несколько
лет назад обсуждался кооперативный эффект Яна — Теллера во
многих шшшельных соединениях [Englman, 1972]. Сегодня наше
понимание кооперативного эффекта Яна — Теллера, однако, многим
обязано исследованиям этого эффекта в редкоземельных арсенатах и
вападатах со структурой циркона. Эти вещества, будучи оптически
прозрачными, могут быть изучены спектроскопически, по переходам
между энергетическими уровнями, включенными в фазовый переход.
Измерения теплоемкости, упругих констант и других свойств по-
новому осветили это явление [Gehring, Gehring, 1975]. Мода
упругого возбуждения (мода деформации) является мягкой модой во
многих кооперативных ян-теллеровских переходах второго рода.
Большинство экспериментальных результатов по
кооперативному эффекту Яна — Теллера может быть объяснено на основе теории
молекулярного поля. Это возможно потому, что взаимодействие
между электронной деформацией и упругой деформацией является в
известной степени далыюдействующим. Используя простую теорию
молекулярного поля, можно получить выражения для параметра
порядка, поперечной восприимчивости, виброниых состояний,
удельной теплоемкости и упругих констант. Детальное обсуждение этой
теории и ее применения можно найти в имеющемся превосходном
обзоре [Gehring, Gehring, 1975]. На рис. 4.23 представлены
возможные ситуации разной степени сложности, которые могут возникнуть
в ян-теллеровских системах.
Как отмечалось ранее, редкоземельные цирконы являются
идеальными случаями для изучения кооперативного эффекта Яна —
Теллера. В TmV04 при высоких температурах самое нижнее
электронное состояние Тт3+-иона является дублетом Е. Псевдоэффект
Яна — Теллера возникает тогда, когда наблюдается случайное
вырожденное или почти вырожденное состояние. DyV04 является
таким примером, где два крамерсовских дублета разделены на 9 см-1,
другим примером является TbV04. Обсуждались и довольно
необычные особенности низколежащих электронных состояний редко-
Кооперативный эффект Яна — Теллера 177
Рис. 4.23. Различные степени сложности, которые могут возникнуть в ян-
теллеровских системах [Gehring, Gehring, 1975]. К. II.— кристаллическое
поле. Простые кружки означают фазовые переходы первого рода,
двойные — переходы второго рода. Кружки с прерывистой линией указывают
на переход, который является переходом первого рода из-за
существования ангармоничных сил.
земельных цирконов [Pytte, Stevens, 1971; Elliott et al., 1972]*
Фононные моды и упругие константы этих материалов также
обсуждались в обзоре [Gehring, Gehring, 1975]. На рис. 4.24 показана
температурная зависимость расщепления фононной моды Eg и
температурная зависимость параметров ячейки TbV04. В кубических
шпинелях АВ204, где А и В — ионы переходных металлов, эффект
Яна — Теллера может быть вызван ионами А и В или обоими
одновременно. Самое низкое электронное состояние может быть
дублетом (£-шпинель) или триплетом (Г-шпинель). Так как электроны,
ответственные за эффект Яна — Теллера, имеются вне этих ионов,
они сильно взаимодействуют с решеткой, вызывая структурные
фазовые переходы при довольно высоких температурах. Это
контрастировало бы с поведением редкоземельных цирконов, которые
показывают переходы при очень низких температурах. Редкоземельные
пниктиды (структуры NaCl) обнаруживают фазовые переходы
первого рода, включающие магнитное упорядочение и структурные
искажения; в этих системах имеется связь магнитных и ян-теллеровских
взаимодействий.
В уже упомянутом выше обзоре [Gehring, Gehring, 1975]
имеется сводка по ряду материалов, проявляющих кооперативный эффект
Яна — Теллера, вместе с литературными ссылками. Мы коротко
рассмотрим совершенно очаровательные переходы в РгАЮ3. Последний
*2 Ч. Н. Р. Рао, Дж. Гопалакришнан
178
Фазовые переходы
I Г0,2-\
fro,r
(12 00)
Ромбическая
(О 72 О)
\
\ Тетрагональная
/
У
—\—
10
—I—
20
1—
30
т,к
б
40—300
Рис. 4.24. а — температурная зависимость Eg фононной моды TbV04 в рама-
новском рассеянии [Elliot et al., 1972]; б —температурная зависимость
параметров решетки TbV04 [Will et al., 1972].
претерпевает три фазовых перехода, начиная с высокотемпературной
кубической перовскитовой структуры через тригональную и
ромбическую фазы к тетрагональной при ~ О К. Эти переходы можно
понять в терминах вращений октаэдров АЮ6, взаимодействующих
с электронными уровнями иона Рг3+ через электрон-фононные
взаимодействия [Harley et al., 1973]. Переход из ромбической структуры
в моноклинную (второго рода) при 151 К управляется
взаимодействием низколежащих экситонов с фононами, фазовый переход
первого рода из тригональной модификации в ромбическую при 205 К
появляется тоже аналогично благодаря электрон-фонному
взаимодействию [Birgeneau et al., 1974]: Для таких систем параметры
порядка могут быть получены из расщепления электронных уровней,
макроскопической деформации и соответствующих внутренних
смещений, и все эти параметры
порядка должны в принципе
показывать одинаковую тем-
"ti~* d • г\ пературную зависимость. Все
W
I
I
*095-
I
Ч
\
^
О 0,4
\
0%6
Т/Тс
V
1,0
Рис. 4.25. Сравнение различных
параметров порядка для перехода
второго рода при 151 К, в РгА103
[Sturge et al., 1975]. Сплошная
линия —кривая для параметра
порядка по данным ЭПР внутренних
смещений cos2 2<p. Темные
кружки — параметр порядка по
данным оптического поглощения.
Квадраты — макроскопическая
деформация по данным упругого
рассеяния нейтронов.
Переходы спинового состояния
179
три выше упомянутые параметра порядка получены для перехода
второго рода в РгАЮ3 при 151 К; расщепления электронных
состояний из оптического поглощения, флюоресценции и
электронной Раман-спектроскопии, внутренние смещения — ЭПР-спектро-
скопией, макроскопические деформации — рассеянием нейтронов.
Эти три параметра порядка были сравнены, и найдено довольна
хорошее соответствие (рис. 4.25). Параметры порядка также
следуют зависимости (Гс— Г)1/2 в пределах 0,2° от Тс [Sturge et aJ.r
1975].
4.11. ПЕРЕХОДЫ СПИНОВОГО СОСТОЯНИЯ
Ионы переходных металлов, имеющие электронную
конфигурацию d4, d5, d6 или d7, могут существовать в низко- или
высокоспиновом основном состояниях в зависимости от силы октаэдри-
ческого поля лигандов. Когда сила поля лигандов близка к обменной
энергии, соответствующей переходу между термами основного
состояния [Tanabe, Sugano, 1954], может иметь место фазовый переход
между двумя спиновыми состояниями (константа скорости 106 с-1
или больше). Переходы между спиновыми состояниями
сопровождаются изменениями магнитной восприимчивости и других свойств.
Переход из низкоспинового состояния в высокоспиновое у
комплексов железа и кобальта в последние годы широко
исследуется с применением изучения магнитной восприимчивости,
теплоемкости и спектроскопических методов. Особенно эффективна в
исследовании переходов в комплексах Fe2+ мессбауэровская
спектроскопия, поскольку низко- и высокоспиновое состояния связаны с
заметно различными изомерными сдвигами. Имеются обзоры по
переходам между спиновыми состояниями в комплексах [Gutlich, 1981a;
Konig et al., 1985]; рассматриваемая в них тема особенно уместна
в обсуждении свойств цитохромов. В некоторых комплексах переход
оказывается растянутым, тогда как в других он резкий. Во многих
комплексах резкий переход сопровождается структурными
изменениями и изменением энтальпии (рис. 4.26), и было предложено
несколько гипотез и моделей для объяснения различных особенностей
этих переходов. Переходы между спиновыми состояниями
наблюдаются не только в комплексах переходных металлов. Они найдены
в разнообразных твердых телах, таких как оксиды и сульфиды
металлов, а также в других системах. Однако те, кто работает с
комплексами переходных металлов, по-видимому, не знают о
наблюдениях на системах оксидов металлов, сделанных специалистами в
области химии твердого тела (см., например, [Ramasesha et al., 1979])r
и все же переходы в этих двух типах систем имеют много сходства.
Недавно эта проблема была обсуждена с единых позиций [Rao,
1985а], при этом были рассмотрены различные модели переходов
12*
180
Фазовые переходы
WO г
Рис. 4.26. Температурная зависимость.
а— относительная дифракционная
интенсивность высокоспиновых (бГ2) и низкоспиновых
(%) фаз [Fe (4,7-(CH3)oPhen)2(NCS2)] по
рентгеновским данным; 6 — высокоспиновая
(?г 6Г2) часть по данным мессбауэровских
спектров. В а по ординате отложена
относительная дифракционная интенсивность линий
при 6(5Г2)= 4,92° и в(1А1) = 4,95° (СиКа-
излучение) LKonig et al., 1979].
между спиновыми состояниями.
Мы здесь кратко остановимся на
некоторых важных особенностях
этих переходов, чтобы
проиллюстрировать, как
экспериментально и теоретически исследуются
такие фазовые переходы.
Скачкообразные переходы
между спиновыми состояниями в
комплексах, по-видимому, обычно
являются фазовыми переходами
первого рода с заметным температурным гистерезисом (см. рис. 4.26).
Структурные изменения в этих переходах также сопровождаются
скачками в рентгеновских факторах Дебая — Уоллера. Точная
природа структурного изменения (симметрия, размеры ячейки и т. д.)
в большинстве комплексов, обнаруживающих переходы между
спиновыми состояниями, не исследована. Можно было бы ожидать
изменения в размерах элементарной ячейки из-за разных размеров
высоко- и низкоспиновых ионов, но изменение симметрии должно
быть не обязательно. Аномальное изменение в теплоемкости найдено
в таких комплексах, как [Fe(Phen)2(NCS)2], которые подвергаются
резким изменениям в заселенности спиновых состояний [Sorai, Seki,
1974]. Термодинамически некоторые растянутые переходы (см.,
например: [Konig et al., 1983]) могут быть второго рода. На графиках
зависимости магнитного момента или восприимчивости от
температуры для некоторых комплексов обнаруживается плато. Например,
в комплексах Fe(III) вместо изменения иэфф между 5,9 ив (6АХ) и
2,0 ив (2Т2) можно видеть плато в широкой области температур при
промежуточном значении (1Эфф. Измельчение делает переход
растянутым, оставляющим больше низкоспиновых молекул при высокой
температуре; это может быть вызвано тем, что измельчение дает
те же эффекты, что и приложение высокого давления.
Спиновые переходы чувствительны к допированию ионами
других металлов. В таких комплексах, как [FercZn1_A.(2-pic)3]Cl2 X
X С2Н5ОН, природа спинового перехода зависит от х, указывая на
важность кооперативного взаимодействия между электронным
состоянием иона металла и решеткой [Gutlich, 1981b]. Более
интересно, что в некоторых случаях заметно влияние допирования на
Переходы спинового состояния
181
1400\
1200Л
100О\
Г * 800-
н
600Л
400-
200 \
200 400 600 800 1000
т, к
200
400
Т.К
600 800
Рис. 4.27. Вклад кобальта в
обратную магнитную восприимчивость
редкоземельных кобальтатов. Кривая
для LaCo03 показана на вставке [Ма-
dhusudan et al., 1980].
температуру перехода. Здесь
снова для понимания
наблюдения нужно помнить об эффекте
давления допируемым ионам
металла. В случае соединения
[Fe(3-OCH3-Sal Een)2PF6
допирование Сг3+ имеет только
небольшой эффект на
температуру перехода, но замещение Со3 +
резко увеличивает температуру
перехода IHaddad et al., 1981].
Это происходит, вероятно, по-
о
тому, что радиус Со3+ (0,53А)
значительно меньше радиуса
Fe3+ и можно было бы ожидать положительного эффекта
давления. Такие эффекты давления на фазовые переходы допиро-
ванных ионов металлов в действительности хорошо известны
[Rao, Rao, 1978]. Например, замещение Gd3+ вместо Sm в SmS
делает его металлическим, твердый раствор при охлаждении взрыво-
образно переходит в структуру с низкой плотностью. Замещение
Сг3+ вместо V3+ в V203 имеет отрицательный эффект давления на
переход металл — диэлектрик в V203. Частичное замещение Fe2 +
на Мп2 + в [Fe(Phen)2(NCS)2] круто понижает температуру перехода.
Мп2+ имеет много больший радиус (0,82 А), чем Fe2+, и должен
давать .отрицательный эффект давления; в действительности большая
концентрация Мп2+ делает переход растянутым. С02+ с радиусом
о
0,72 А имеет менее заметный эффект на переход; Zn2+, радиус
которого такой же, как у Fe2+, совсем не влияет. Остаточный
парамагнетизм при низких температурах в [Fe,_xMx(Phen)2(NCS)2] также
может быть интерпретирован в рамках относительных размеров ионов
М2+, причем больший размер иона увеличивает парамагнетизм [Giit-
lich, 1981b]. Такой эффект давления допированных ионов металлов
найден также при замещении на Мп2 + железа в FeS2. Замещение
железа на марганец стабилизирует высокоспиновое состояние Fe2+;
обычно железо в этом сульфиде находится в низкоспиновом
состоянии. Приложение давления к Fe^ JMn^Sg приводит к переходу
высокоспиновое — низкоспиновое состояние.
Спиновые переходы в оксидах металлов, вообще говоря,
ограничены примерами систем, в которых имеются Со3+-ионы. Широко
182 Фазовые переходы
изучались в этом отношении редкоземельные кобальтаты с общей
формулой LnCo03(Ln—La, Y или лантаноиды) [Raccah, Goodenough,
1967; Bhide et ah, 1972; Madhusudan et al., 1980]. В этих оксидах
Co3+ находится в иизкоспиновой ^-конфигурации ПРИ низких
температурах и переходит в высокоспиновое состояние (ttge2g) с
увеличением температуры (вплоть до 300—400 К). При высоких
температурах поведение зависит от редкоземельного иона; кобальтаты
легких редких земель (La, Nd и т. д.) обнаруживают свидетельства
переноса заряда между низко- и высокоспиновыми состояниями, тогда
как для более тяжелых (Но, Ег) редкоземельных кобальтатов такого
не наблюдается. Мы здесь главным образом рассмотрим эффект
спиновых переходов, а не переноса заряда. Спиновый переход в LaCo03
совершенно уникален в том отношении, что два спиновых состояния
упорядочивают сами себя, вызывая изменения в симметрии от R3c
до R3. Об упорядоченности спиновых состояний свидетельствуют
плато на зависимости обратной магнитной восприимчивости от
температуры (рис. 4.27), причем такое поведение отмечено ранее для
MnAsx.^P^.. Кобальтаты легких редких земель обнаруживают
температурную аномалию благодаря спиновым переходам со значением
Д# около 400 Дж/моль или больше; при температуре перехода
изменяются факторы Дебая — Уоллера и Лэмба — Мессбауэра и
отмечаются структурные особенности. Интересно, что при переходных
температурах отношение заселенностей высоко- и низкоспинового
состояний обычно равно единице. Графики в координатах обратная
магнитная восприимчивость — температура для NdCo03, PrCo03 и
других кобальтатов имеют максимум (см. рис. 4.27) и не имеют
плато, как в случае LaCo03; постепенное изменение магнитных моментов
в этих кобальтатах похоже на таковое в комплексах, обсужденных
выше. Изучение твердых растворов La1_xNd:x;Co03 показало переход
от поведения, подобного NdCo03 (максимум на графике %-1 от Г),
к поведению, подобному LaCo03 (плато на графике х-1 от Т). Это
наблюдение позволяет полагать, что мы имеем дело с двумя
крайностями в поведении этих систем. Кроме того, температура спинового
перехода увеличивается при переходе по ряду редкоземельных
элементов от La и далее. Это может быть обусловлено эффектом
поляризации редкоземельного иона на расщепление кристаллического
поля, причем последнее увеличивается по редкоземельному ряду,
сопровождаясь уменьшением объема элементарной ячейки. я*-Орби-
таль также должна значительно стабилизироваться по мере
движения по ряду, внося вклад в увеличение расщепления
кристаллического поля. Нужно отметить, однако, что разница в энергиях
низко- и высокоспиновых состояний в кобальтатах составляет 100—
500 см"1.
Сообщалось [Chenavas, Joubert, 1971] о синтезе Со203 под
высоким давлением. Фаза высокого давления, по-видимому, состоит
Переходы спинового состояния
183
только из низкоспиновых ионов Со3+, которые при нагревании
переходят в высокоспиновое состояние с положительным изменением
объема в 6, 7 %. Эти результаты свидетельствуют о переходе
первого рода, но нет полной определенности, что приготовлена
действительно чистая фаза Со203.
Переходы между спиновыми состояниями замечены в
квазидвумерных оксидах со структурой K2NiF4. La4LiCo08 — один из таких
оксидов [Mohanram et al., 1983]. Его считали первым примером,
имеющим максимум на графике обратная восприимчивость —
температура, обусловленный переходом ионов Со3 + из низкоспинового
состояния в высокоспиновое. Имеется интерпретация [Demazeau
et al., 1980] данных по восприимчивости в терминах существования
переходов низкоспиновое — промежуточное состояние и
низкоспиновое — высокоспиновое состояние. Как показано электронной
дифракцией, в этом оксиде ионы Li+ и Со3+ двумерно упорядочены
(в плоскостях ah) [Mohanram et al., 1983] (см. рис. 2.1). Sr4TaCo08
и Sr4NbCo08, которые имеют структуру K2NiF4, показывают
поведение восприимчивости, напоминающее плато на графике %-1 — Т
для LaCo03. Ионы Со3+, по-видимому, переходят в этих оксидах из
низкоспинового в промежуточное спиновое состояние (t\geg).
Сообщалось [Demazeau et al., 1982], что Ni3+ в LaSrNi04
находится в низкоспиновом ^е^-состоянии, но в LaBaNi04 — в высоко-
спиновом t\ge\. Изучение магнитной восприимчивости LaSr1_:cBa;3CNi04
показало, что ^-электроны в этой системе всегда находятся в
протяженных состояниях, формирующих ах2_2 -зону; с увеличением
х ширина зоны уменьшается, что сопровождается увеличением
объема элементарной ячейки. Поскольку с увеличением х только
небольшая часть ионов Ni3+ может быть в высокоспиновом состоянии,
перехода не наблюдается [Mohanram et al., 1983].
Первая модель переходов между спиновыми состояниями [Ches-
nut, 1964] дала феноменологическое описание систем, содержащих
триплетные экситоны, таких как ионные соли TGNQ-радикалов,
в терминах относительно высокой концентрации экситонов. При
рассмотрении свободной энергии как квадратичной функции
концентрации экситонов показано существование фазовых переходов в
таких системах. Позднее [Konig, Kremer, 1971] проведен
полуэмпирический анализ изменений заселенности спиновых состояний
в предположении тетрагонального искажения и температурно-зави-
симой разницы в энергиях этих состояний. На основе рассмотрения,
подобного теории регулярных растворов Гуггенгейма, дано
объяснение изменений, индуцированных давлением, в высокоспиновых
комплексах Fe (II) [Slichter, Drickamer, 1972]. Основываясь на
подобных рассмотрениях, сделана попытка объяснить спиновые
переходы первого рода [Sorai, Seki, 1974]. Свободная энергия Гиббса
при некоторой температуре может быть записана как
184
Фазовые переходы
G = xGHS + (1 - *)GLS + NkT{x In x +
+ (1 -s)ln(l -x)}, (4.17)
где Ghs " Gls — свободные энергии Гиббса высокотемпературной
(высокоспиновой) и низкотемпературной (низкоспиновой) фаз
соответственно. Равновесная мольная доля высокоспинового состояния
х задается уравнением
х = 1/{1 + exp (AG/NkT)}, (4.18)
где AG(=Ghs—6xs) включает колебательный терм. Показано [Sorai,
Seki, 1974], что спиновый переход является кооперативным,
протекающим через взаимодействия между электронным состоянием
иона металла и фононной системой; сила взаимодействия будет
определять форму кривой конверсии спиновых состояний, причем
слабое взаимодействие приводит к растянутой конверсии.
Имеется также модель [Spierin<< et al., 1982], где
высокоспиновое и низкоспиновое состояния комплекса рассматриваются как
жесткие сферы объема Fhs и Fls соответственно, и кристалл
считается изотропной упругой средой, характеризуемой объемным
модулем упругости и константой Пуассона. Комплекс рассматривается
как неупругое включение, внедренное в сферу объемом Vc.
Уменьшение упругой энергии несжимаемой сферы в расширяющемся
кристалле приводит к отклонению от больцмановского распределения
высокоспиновых комплексов. На этой модели основано объяснение
эффектов давления и температуры на спиновые переходы в
комплексах Fe (II) [Usha et al., 1985].
Имеется и микроскопическая модель [Kambara, 1979], в
которой взаимодействие между с?-электронами и решеткой задается по
определению. В предположении ян-теллеровского взаимодействия
между rf-электронами и локальным внутримолекулярным
искажением гамильтониан системы можно записать как
N
Н= 2 Ы + 1/2 NMa2Q\ (4.19)
i=l
где hj является гамильтонианом для Fe2+ иона в г-й молекуле и
Q — смещение шести лигандов в нормальной координате с
симметрией Eg точечной группы О/,, М — эффективная масса, со —
частота моды. Гамильтониан ht записывается как
h = К + V0 + Vso + (dV/dQ)oQ + 1/2 (d*V/dQ*)oQ2, (4.20)
где h0 — гамильтониан свободного иона, V0 — октаэдрическое
поле лигандов, V — поле лигандов, обусловленное искаженной
молекулой, Vso — спин-орбитальное взаимодействие.
Переходы спинового состояния
185
Свободная энергия на ион представляется как
F = - кТ 2 ei exp (- Ei/kT), (4.21)
г = 1
и равновесные значения (? находятся минимизацией F
относительно Q. Члены, содержащие Q или Q1, появляются в собственных
значениях. Разница в энергиях между bT2g и А ^-состояниями (500—
1000 см-1) в неискаженной фазе задается как Е0 — 2{)D<j — ЪВ —
— 8С, где В и С — параметры Рака. Параметр ян-теллеровского
взаимодействия а, появляющийся в выражении для Q, находится из
соотношений EJT = 1/2 а2, где EJT = 100—1000 см-1 и а,
следовательно,— между 0 и 50 см"1/2. В случае линейного взаимодействия
(последний член в уравнении 4.20 равен нулю) в широкой области
значений а имеет место переход первого рода с Q в качестве
параметра порядка. Если вводится взаимодействие второго порядка, могут
иметь место три возможных типа перехода с (> > 0 +± (? = 0, Q >
;>0:+±(?<;0 и Q < 0 +± Q == 0. Температурное изменение \1Эфф,
рассчитываемое из уравнения (4.22), находится в хорошем согласии
для скачкообразных спиновых переходов, когда используются
приемлемые значения Е0 и а:
h2» = ^(Xu + 2xi). (4.22)
где х является магнитной восприимчивостью; выражение для %
включает значения Et, которые, в свою очередь, определяются Е0,
a, Q и константами, которые связывают силовые постоянные и
частоты колебаний.
Позднее [Kambara, 1981] была предложена учитывающая
молекулярные взаимодействия трактовка внутримолекулярных
искажений как динамических переменных. Изучался также эффект
взаимодействия между ионОхМ переходного металла и деформацией решетки;
в таком случае решетка должна управлять спиновым переходом,
сопровождающим кооперативное внутримолекулярное искажение даже
в отсутствие сильного взаимодействия. Когда нет искажения,
решеточная деформация с температурой изменяется непрерывно, вместе
со спиновым состоянием. В узкой области силы взаимодействия
имеют место резкие изменения в деформации решетки, вызываемые
кооперативным внутримолекулярным искажением. Результаты
рассмотрения межмолекулярных взаимодействий подобны результатам
для статического рассмотрения [Kambara, 1979], причем
единственная разница — в значениях различных параметров. Показано
[Sasaki, Kambara, 1982], что кооперативное молекулярное искажение,
связанное со спиновыми переходами, может индуцироваться
магнитным полем. Небольшой сдвиг в температуре перехода
186
Фазовые переходы
[Fe(Phen)2(NCS)2], по-видимому, имеет место в поле 5,5 Т [Qi et al.,
1983]. Этот аспект заслуживает дальнейшего исследования.
Предложена [Ban, Sivardiere, 1972] модель для спиновых
переходов, сснованная на известной модели синглет-триплетных
переходов в ион-радикальных солях [Chesnut, 1964], которая включает в
гамильтониан смещение «дыхательной» моды Q и константы
упругости:
Я = NW2 + 2 (А - VQ) щ. (4.23)
г
Здесь (Д—VQ) представляет разницу в энергиях низко- и
высокоспиновых состояний и nt = 0 и 1 для низко- и высокоспинового
состояний соответственно. Эта модель предсказывает переход первого
рода из низкоспинового состояния в высокоспиновое с увеличением
температуры для 0,43 < (У2/4£Д) < 1. Расширение этой модели с
включением обменных взаимодействий между высокоспиновыми
состояниями приводит к предсказанию теплоты намагничивания [Bari,
Sivardiere, 1972]; однако магнитное упорядочение не наблюдалось
ни в каких системах, имеющих спиновые переходы. Имеется двух-
подрешеточная модель [Bari, 1972], предложенная для объяснения
структурного перехода, сопровождающего переход между
спиновыми состояниями в LaCo03. В этой модели гамильтониан
записывается в виде
Я = NW2 + 2 (А - VQ) щ + 2 (А + VQ) щ, (4.24)
гА гВ
где смещение подрешетки А противоположно смещению подрешет-
ки В. Эта модель показывает переход первого или второго рода из-за
упорядочения с последующим спиновым переходом первого или
второго рода. Экспериментально спиновый переход наблюдается до
перехода упорядочения. Эта работа, однако, указывает на
важность упругих свойств твердого тела и их связи с решеточным
искажением.
Рассмотренные модели являются статичными, но могут быть
решены с включением динамики решетки. При такой постановке
проблемы было найдено [Ramasesha et al., 1979], что динамическая
модель не показывает фазового перехода; это верно также и для двух-
подрешеточной модели. По-видимому, динамика решетки размывает
любой фазовый переход, являющийся результатом статического pi
линейного взаимодействия смещений спиновых состояний.
Динамическая двухподрешеточная модель, в которой высоко- и
низкоспиновое состояния смещены ионно-клеточной модой (с ионом
переходного металла, двигающимся относительно центра октаэдрической
клетки), однако, показывает ненулевую заселенность
высокоспинового состояния при низких температурах и плато на кривой
восприимчивости. Взаимодействие в таком рассмотрении квадратично по
Пластическое кристаллическое состояние 187
отношению к смещению, и гамильтониан записывается как
Я = l(PQ2/2M) + \.12М<ьЧ)г]1 + Aez +
+ li2aQ2M®2ox, (4.25)
где со — частота колебаний и а — спиновая матрица.
Взаимодействие спиновых состояний по кубу смещений подрешеток дает в
зависимости от параметров взаимодействия переходы первого и второго
рода, но заселенность высокоспинового состояния при низких
температурах всегда нулевая [Ramasesha et al., 1979]. Можно
объяснить различные особенности систем с переходами между спиновыми
состояниями включением взаимодействия спиновых состояний в
смещения дыхательной и ионно-клеточной моды, причем первая является
линейной, а вторая — квадратичной. Ионно-клеточная мода будет
смешивать спиновые состояния.
Предыдущее обсуждение должно было показать, как химически
интересное явление спинового перехода становится очень сложным
для понимания в твердом теле. Нужно отметить, что несмотря на
очень широкие исследования имеется еще много аспектов переходов
между спиновыми состояниями (критическое поведение, мягкая
мода), которые необходимо исследовать.
4.12. ПЛАСТИЧЕСКОЕ КРИСТАЛЛИЧЕСКОЕ СОСТОЯНИЕ
Обычно кристаллы обладают как трансляционным, так и
ориентационным порядком. Кристаллы некоторых веществ (особенно
веществ, образованных сферическими или шаровидными
молекулами) проявляют ориентационное разупорядочение, тогда как
трансляционный дальний порядок сохраняется. Такое ориентационно
разупорядоченное кристаллическое состояние обозначают как
пластическое кристаллическое состояние. Переход из кристаллического
в пластическое кристаллическое состояние демонстрируется
молекулярными кристаллами, так как молекулы приобретают при высоких
температурах ориентационные степени свободы. Характерной
термодинамической особенностью пластических кристаллов, которая
привела к признанию их существования Тиммермансом, является то, что
изменение энтропии при переходе кристалл — пластический
кристалл значительно больше, чем изменение энтропии, сопровождающее
плавление пластического кристалла (табл. 4.2). В дополнение к
низкой энтропии плавления пластические кристаллы плавятся
обычно при относительно высокой температуре: например, пластические
кристаллы неопентана плавятся при 257 К, тогда как н-пентан,
который не образует пластического состояния, плавится при 132 К.
В некоторых случаях пластические кристаллы сублимируются без
плавления; камфора, гексафторид серы и гексахлоран являются ти-
пичными примерами.
Таблица 4.2
Структурные и термодинамические свойства некоторых пластических кристаллов *
Вещество
Неопентан
Циклогексан
Г^ксаметилдисплан
f-Бутилхлорид
Камфора
о
а, А
8,78
8,70
8,47
8,62
10,10
Tt, К
140
186
222
183
238
тт> К
257
280
288
245
449
ASt, R
2,22
4,34
5,28
4,38
3,84
ASm, R
1,46
1,10
1,25
0,95
1,40
Еа, кДж-моль-1
4,2
6,4
6,3
3,0
11,9
* Все пластические фазы, приведенные в таблице, обладают ГЦК-структурой; Tt — температура перехода кристалл —
пластический кристалл; AS^ — изменение энтропии при Tft ASm ~ изменение энтропии при Тт \Еа — энергии активации молекулярной
Реориентации, полученная из данных ЯМР-спектроскопии,
Пластическое кристаллическое состояние 189
Из-за ориеитационной свободы пластические кристаллы обычно
кристаллизуются в кубических структурах (см. табл. 4.2). Важно,
что пластические кристаллы принимают кубические структуры,
даже когда симметрия молекулы несовместима с такой симметрией
кристалла. Например, Z-бутилхлорид в пластическом
кристаллическом состоянии имеет ГЦК-структуру даже несмотря на то, что
у изолированной молекулы есть ось вращения третьего порядка,
которая несовместима с кубической структурой. Такие кажущиеся
противоречия между симметрией решетки и симметрией молекулы
дают указания вращательного разупорядочения в пластическом
кристаллическом состоянии. Нужно, однако, заметить, что
молекулярное вращение в пластических кристаллах редко является
свободным; часто оказывается, что имеется конфигурация с более чем
одним минимумом потенциальной энергии. Это позволяет молекулам
быстро переходить из одной ориентации в другую, причем
различные ориентации в пластическом кристалле хаотичны.
Как и предполагает название, пластические кристаллы обычно
мягкие, часто деформирующиеся под собственным весом. Давление,
требуемое для получения тока пластического кристалла, как,
например, для экструзии через маленькое отверстие, обычно
значительно меньше (в 2—14 раз), чем давление, требуемое для
экструзии истинного кристалла того же самого вещества. ^-Бутиловый
спирт, триметилуксусная кислота и d-камфора — это обычные
лабораторные примеры пластических кристаллов. Тема пластических
кристаллов довольно широко рассматривалась в литературе [Aston,
1963; Sherwood, 1979], и мы ограничим наше обсуждение природой
ориентационного движения [Rao, 1985b].
Существование высокой степени ориеитационной свободы
является характерной особенностью пластического кристаллического
состояния. Мы можем представить три типа вращательных
движений в кристаллах; свободное вращение, вращательная диффузия
и прыжковая реориентация. Свободное вращение возможно, когда
взаимодействия слабые, и эта ситуация не применима к
пластическим кристаллам. В классической вращательной диффузии
(предложенной Дебаем для объяснения диэлектрической релаксации в
жидкости) можно ждать, что ориентационное движение молекул следует
уравнению диффузии, описывающему взаимосвязи эйнштейновского
типа. Этот тип диффузии неизвестен в приложении к пластическим
кристаллам. Что более уместно рассмотреть в случае пластических
кристаллов — это столкновительно затрудненное молекулярное
вращение.
Модель вращательной диффузии была обобщена включением
молекулярных реориентаций с углами довольно большой величины
[Gordon, 196(5]. Согласно этой модели предполагается, что
молекулы свободно вращаются в интервалах между столкновениями.
Обсуждались два предельных случая, которые включают ступени
190
Фазовые переходы
угловой диффузии (произвольного размера). Эти случаи известны
как /- и М-диффузионные модели. В первой из них направление,
так же как и величина вектора углового момента молекулы,
благодаря столкновениям следует распределению Больцмана. В М-диф-
фузионной модели ориентация вектора углового момента хаотична,
но его величина неизменна. В приложении этой модели к линейным
молекулам получающееся общее уравнение диффузии следует
классической механике для малых времен. При больших временах
частота вращения в Л/-диффузионной модели является неизменной
в последовательных ступенях, тогда как она подчиняется больц-
мановскому распределению в /-диффузионной модели.
В другой предложенной модели [Hill, 1963; Wyllie, 1971]
молекулы, захваченные в потенциальную яму, обнаруживают
либрации (возмущенные угловые осцилляции). Флуктуации в ориентации
соседних молекул приводят к изменению формы потенциальной
ямы и, следовательно, вызывают угловое движение молекул с
большими амплитудами. Возможно, что диффузия потенциальной ямы
на сферической поверхности приводит к реориентационному
движению молекул. Если потенциальная поверхность связана с
пространственной группой кристалла, молекулярная ориентация будет
ассоциироваться с демпфированными либрациями [Guthrie,
McCullough, 1961]. Вращения пространственной группы кристалла
будут приводить к разрешенным потенциальным ямам.
Не существует одной модели, способной точно описать
молекулярные реориентации в пластических кристаллах. Были
рассмотрены модели, которые включают особенности различных моделей,
описанных выше. Например, диффузионное движение, прерываемое
ориентационными прыжками, может рассматриваться ответственным
за молекулярную реориентацию. Эта модель по некоторой степени
имела успех в случае циклогексана и неопентана [Lechner, 1972;
Dr Graaf, Sciesinski, 1970]. До конца еще не ясно, является ли
реориентационное движение кооперативным. Имеются,
по-видимому, некоторые свидетельства взаимосвязи между реориентационным
движением и движением соседних молекул. Кажется, что есть
простор для сравнительных экспериментальных исследований,
использующих дополнительные методы, чувствительные к автокорреляции
и к мономолекулярной корреляции.
Природа вращательного движения, ответственного за ориента-
ционное разупорядочение в пластических кристаллах, еще не
полностью понята, и используется ряд экспериментальных методов для
исследования этой интересной проблемы. Здесь может быть
взаимодействие между вращательным и трансляционным движением,
причем простейшей формой последнего является самодиффузия.
Коэффициент диффузии D задается уравнением Эйнштейна
Z> = ^>, (4.26)
Пластическое кристаллическое состояние 191
где (г2 > — среднеквадратичное расстояние прыжка их — среднее
время между последовательными прыжками молекулы в позиции
решетки. Коэффициент диффузии зависит от величины скорости
молекулы, а также и от постоянства этой скоростг. И метод
радиоактивной метки, и методы ЯМР использованы для измерения
коэффициентов самодиффузии в пластических кристаллах. Во многих
случаях эти два метода дают подобные результаты, так как они
относятся к подобным процессам во временном масштабе
корреляционной функции скорости. Коэффициенты диффузии аномально
высоки (в сравнении с непластическими органическими
кристаллами), причем самые мягкие кристаллы имеют самые высокие
коэффициенты диффузии.
ЯМР-спектроскопия дает времена спин-решеточной (7\) и спин-
спиновой релаксации (^о). Сделав необходимые предположения
относительно магнитных взаимодействий, ответственных за
релаксационные процессы, можно связать эти времена релаксации с
молекулярными движениями. Так как ядерная спиновая релаксация
является результатом всех процессов, которые вызывают
флуктуацию магнитного поля на ядре, корреляционная функция будет в
общем соответствовать более чем одному виду движения, включая
все возможные взаимодействия. Уравнения для времен релаксации
обычно имеют вид
1 сх
~Т~ = 7Т~~! 2~~2\"> (4-^')
1 (1 + 0) X )
где константа пропорциональности С, так же как и т, определяется
природой ядерных и других взаимодействий. Если релаксационный
процесс осуществляется главным образом благодаря дипольным
взаимодействиям, ЯМР-спектроскопия дает информацию о
вращательной диффузии. Если доминирует ядерная квадрупольная
релаксация, мы получаем информацию о молекулярной реориентации.
В табл. 4.2 приведены энергии активации для молекулярных
реориентации, полученные из ЯМР-спектроскопии.
Для анализа молекулярного движения в пластических
кристаллах наиболее ценны эксперименты по рассеянию тепловых или
низкоэнергетичных нейтронов. Эти эксперименты измеряют
изменения в центрах масс молекул. Коэффициенты диффузии,
полученные из нейтронных экспериментов, очень отличаются от таковых,
полученных из экспериментов с радиоактивной меткой, так как
нейтронное рассеяние определяется главным образом вращательной
диффузией. Функция рассеяния имеет вид
S(Q,co) = j JG(R, f)exp(/Qr — uoOdQdco, (4.28)
где G(R, t) является вращательной корреляционной функцией для
рассеивающего центра, Q — вектор рассеяния, со — угловая часто-
192
Фазовые переходы
та и R — вектор равновесного положения. Неупругая часть
рассеяния дает информацию о вращательном движении, тогда как упругая
часть связана с трансляционным движением. Если реориентацион-
ное движениа диффузионно, мы можем использовать уравнение
диффузии, подобное упомянутому ранее, причем ширина
квазиупругого пика связана с вращательной диффузией. Часто применяются
другие функции, чтобы увидеть, происходит ли вращательное
движение через прыжки. Хорошим примером изучения рассеяния
нейтронов пластическими кристаллами является работа по ^-бутил-
цианиду [Frost et al., 1980].
Одним из наиболее прямых методов исследования реориента-
ционного движения молекул является спектроскопия в далекой
ИК-области или диэлектрическое поглощение. В отсутствие
колебательной релаксации времена релаксации, полученные из И К- и
диэлектрических методов, эквивалентны. В обоих этих методах мы
получаем корреляционную функцию, (u(£0), n{t0 -f- t)), Для
движения вокруг выделенной молекулярной оси относительно главных
осей вращения и направление колебания. Рэлеевское рассеяние,
так же как и рамановское, наиболее полезно в изучении
молекулярных ориентационных процессов. С помощью измерения
изотропного и анизотропного рамановского рассеяния ориентационные
процессы могу г быть отделены от других механизмов, которые
вызывают уширение линии. Анализ формы инфракрасных и рамановских
полос обеспечивает информацию о кратко- и долгосрочном
поведении корреляционной функции.
Времена корреляции и параметры энергии активации,
полученные разными методами, могут как согласовываться, так и не
согласовываться друг с другом. Сравнение этих данных дает возможность
проверить применимость используемой модели и изучить, отражает
ли измерение любой отдельный основной молекулярный процесс
или корректен ли используемый метод анализа. Чтобы правильно
охарактеризовать вращательное движение в пластических
кристаллах, может быть, действительно необходимо сравнить времена
корреляции, полученные несколькими методами. Так, значения из
ЯМР-спектроскопии и рэлеевского рассеяния дают нам возможность
отличить коррелированные и некоррелированные вращения.
Молекулярное разупорядочение не отражается в данных ЯМР, с этой
целью необходимы дифракционные исследования.
Ранее уже отмечалась необходимость дальнейших
экспериментальных и теоретических усилий для понимания молекулярной
реориентации в пластических кристаллах. Неясно, является ли
скоррелированным или нескоррелированным молекулярное
движение в большинстве пластических кристаллов. Поскольку
стабильность пластических кристаллов критически зависит от величины и
формы межмолекулярного потенциала, интересно исследовать,
могут ли быть использованы для описания пластического кристалла
Жидкокристаллическое состояние
193
феноменологические атом-атомные потенциалы. Было проведено
[Bounds et al., 1980] изучение молекулярной динамики
пластического состояния метана, а также изучение методом Монте-Карло
его пластического и стеклообразного состояния, полученного
закалкой пластического состояния. Методами молекулярной динамики
изучен и CF4 [Nose, Klein, 1983a; см. также Rao, 1985b].
4.13. ЖИДКОКРИСТАЛЛИЧЕСКОЕ СОСТОЯНИЕ
Жидкокристаллическое состояние является другим
промежуточным состоянием материи, через которое проходят некоторые
молекулярные кристаллы при их нагревании, до того, как стать
изотропной жидкостью. При плавлении термин «мезофаза» часто
используется для описания такого состояния материи. Мезофазы
имеют характеристики как жидкости, так и кристалла. В жидких
кристаллах нет трансляционного упорядочения, но есть ориента-
ционное. Природа мезофазы зависит от формы молекул.
Сферические или шаровидные молекулы легко приобретают вращательные
степени свободы и образуют пластические кристаллы. Стержнеоб-
разные молекулы более легко приобретают трансляционную, чем
вращательную, свободу, что и приводит к жидким кристаллам.
Жидкие кристаллы стали объектом широкого изучения, и в этом
разделе мы перечислим только различные типы переходов,
обнаруживаемых жидкими кристаллами. Увеличивается поток сообщений
о новых разновидностях жидких кристаллов и их переходах, и на
эту тему есть много хороших обзорных работ [Gray, Winsor, 1974;
Chandrasekhar, 1977; Chandrasekhar, 1983; Frank, 1983; Hilsum,
Raynes, 1985].
Вообще жидкие кристаллы можно разделить на лиотропные
и термотропные. Лиотропные жидкие кристаллы образуются ам-
фифильными веществами в присутствии растворителей.
Большинство современных интересных жидкокристаллических систем
являются термотропными по природе, у которых
жидкокристаллическое состояние является результатом изменения температуры.
Термотропные жидкие кристаллы могут быть образованы как
мономерными, так и полимерными молекулами. Нематические, холестери-
ческие и смектические жидкие кристаллы — это три класса жидких
кристаллов, образованных стержнеобразными молекулами. Так как
холестерические кристаллы — это всего лишь нематические
кристаллы, образованные хиральными молекулами, современная
классификация признает только два различных класса —
нематические и смектические жидкие кристаллы. В нематической фазе
молекулы расположены почти параллельно друг другу в направлении
длинной оси (рис. 4.28, а), но на больших расстояниях нет
позиционной корреляции направления длинной оси. Холестерические
13 ч. Н. Р. Рао, Дж. Гопалакришнан
194
Фазовые переходы
Рис. 4.28. Схематическое изобра-
//\\ч»//| [/>z-r-i/~" жение жидких кристаллов: не-
Ix'/'A (' ъ '///'.h/.j/m матического (а), хирального не-
\м |/// /^xVJvV^^ /;\: i, / / /, |/ /,* // матического (б) и А-смектическо-
/|4,/ ///
1 г--
А.^
.ля
р
^7-s
71
-А
||^Л
^7-п
/1\/J||,/|\1/ ГО (в).
'//'Iх// \/'AZAV'/s* //4///"i'/n'
фазы являются спонтанно
0 5- в скрученными нематическими
фазами, причем скручивание
вызвано оптической активностью молекул (рис. 4.28, б). Ряд
производных холестерола обнаруживают эту фазу, откуда и их
название. Смектические фазы являются, в фределиановском смысле,
слоистыми фазами с отсутствием позиционной корреляции на
больших расстояниях в индивидуальном слое (рис. 4.28, в).
Смектические фазы в дополнение к ориентационному упорядочению,
как в нематических фазах, обладают некоторым позиционным
упорядочением; центры молекул находятся в среднем в
эквидистантных плоскостях.
Слоистая структура смектических фаз подтверждается на
рентгеновских дифрактограммах, на которых наблюдаются острые
рефлексы на малых брегговских углах, соответствующих толщине слоя.
Известно много структурных вариантов (полиморфных
модификаций) смектических фаз в зависимости от способа расположения
молекулы в смектическом слое, угла наклона молекул по отношению
к плоскости слоя и степени корреляции структуры от слоя к слою.
Признаны две основные смектические фазы: без упорядоченных
слоев и с упорядоченными слоями. В первой группе распределение
центров молекул в слое соответствует распределению в двумерной
жидкости. Во второй группе слои построены правильно, так что
положения центров молекул лежат в двумерной решетке.
Дальнейшие подразделения на SA, SB, SG, Sp и SL первой группы и Sb, Se,
Sq и Sh второй группы делаются для указания наклона молекул и
упаковки внутри слоев. Смектические фазы, показанные на рис. 4.28.в,
соответствуют S^.
Локальное направление упорядочения молекул в жидких
кристаллах описывается единичным вектором л, называемым
директором, который дает направление предпочтительной оси в каждой
точке образца. Концепция директора очень ценна в нематических
кристаллах. Степень порядка в нематической фазе описывается
параметром порядка £, определяемым как £ = -у <3 cos2 9 — 1), где 9 —
угол между длинной осью молекулы и п, а угловые скобки
означают статистическое среднее. £ = 1 соответствует полному
ориентационному упорядочению, а £ = 0 получается при полном хаосе.
В нематических кристаллах £ имеет промежуточное значение,
зависящее от температуры. В хиральных нематические кристаллах (холе-
стерические фазы) в дополнение к ориентационному упорядочению де-
Жидкокристаллическое состояние 195
тального порядка существует пространственная вариация (твист)
директора, приводящая к геликоидальной структуре. Твист
характеризуется высотой, значение которой может быть порядка нескольких
сотен нанометров. Сравнивая нематические и хиральные нематические
фазы, нужно отметить следующее:
1) никогда не наблюдается переход от хиральной нематической к
нематической фазе и наоборот;
2) и нематическая, и хиральная нематическая фазы неограниченно
растворимы друг в друге — добавление небольшого количества
оптически активного вещества (вне зависимости является ли оно
мезогеном) переводит нематическую фазу в хиральную немати-
ческую с большой высотой;
3) смесь двух веществ противоположной хиральности может дать
нематическую фазу, а при определенных составах высота
становится бесконечной;
4) дифрактограммы хиральной нематической и нематической фаз
подобны, показывая отсутствие позиционного дальнего порядка.
Термодинамика фазовых переходов в системах с жидкими
кристаллами широко исследована. Изменения энтальпии, связанные с
переходом кристалл — жидкий кристалл (нематическая фаза),
A/7cN- много больше теплоты перехода, A#Nb из
жидкокристаллической фазы в изотропную жидкость. А#ш — обычно порядка
2 % от A/?cn- По-видимому, большинство фазовых переходов с
участием жидких кристаллов термодинамически являются
переходами первого рода, за исключением, возможно, межсмектических
переходов. То, что переход нематический жидкий кристалл —
изотропная жидкость должен быть первого рода, показано на основе
симметрийных аргументов [Gennes, 1971]. В духе теории фазовых
переходов Ландау в окрестности фазового перехода свободная
энергия может быть разложена в степенной ряд от параметра порядка
G = G0(p, Т) + Ctf + С£> + С& + .... (4.29)
Уравнение (4.29) подобно выражению, используемому для фазового
перехода второго рода, за исключением наличия кубического члена.
Фазовый переход второго рода имеет место тогда, когда С2 проходит
через нуль при Т = Г*< Гш, где Tni — температура перехода
нематический кристалл — изотропная жидкость. Условиями того,
чтобы переход второго рода был исчерпан переходом первого рода
при Гш> Т*, являются минимум свободной энергии при
ненулевом значении параметра порядка £ши равенство свободных энергий
нематической фазы и изотропной жидкости при этом значении £nl
Первое условие выражается dG/dt, и второе условие уравнением
G(£ni) c G(£ = 0) в следующих уравнениях, пренебрегающих
членами высоких порядков:
2С2 + 3C8£ni + 4C&i = 0, (4.30)
13*
196
Фазовые переходы
Т а б л и ц а 4.3
Некоторые примеры органических молекул, обнаруживающих
жидкокристаллическое состояние *
р-Азоксианизол C^£N^£l
'3
-о-
сн,о —<Г )>—м"
'з
р-Гептил-р - цианобифенил С 301'5»к N^£I
и-р-этоксибензилиден-р'-(р-метилбутил) анилин C-^i N* ^^1
/-а
W-Q-'
>,/?' -Динонилаз
/?, /?' -Динонилазобензол С 511£ S03-^£S А327£ I
В А
C9i
р.р' -Дигептилоксиазоксибензол c5BiKSc3-^KN5H]<I
^-ОЧ*""0^
\
* С — кристаллическая фаза; N — нематическая фаза; N* — хирально-нематиче-
ская фаза; S^, SB, Sq — смектические фазы; I — изотропная жидкость; цифры —
температуры перехода по Кельвину.
С2 + С£т + C4Cni = 0. (4.31)
Эти два условия выполняются одновременно, когда £ni = — С3/
/2С4. Но в нематическом упорядочении, в отличие от других
переходов порядок — беспорядок, положительные и отрицательные
значения параметра порядка соответствуют различному физическому
упорядочению молекул. Следовательно, в выражении для
свободной энергии кубической и более высокого порядка члены не могут
быгь тождественны нулю и должен быть фазовый переход
первого рода.
Характерные примеры органических молекул, которые
обнаруживают жидкокристаллическое состояние, ириведены в табл. 4.3.
Некристаллическое состояние и фазовые переходи в стеклах 197
Видно, что все эти молекулы вытянутые, стержнеподобные,
содержащие кольца и двойные связи, причем последние предотвращают
принятие молекулами нелинейных конформаций. В течение
последних лет осуществляется значительная работа, чтобы объяснить
взаимосвязь между молекулярной структурой и типом,
проявляемого жидкокристаллического состояния, особенно для понимания
склонности к смектическому и нематическому состоянию [Gray,
1983]. Мы рассмотрим некоторые аспекты этой проблемы в разд. 6.10
В последние годы показано, что дискообразные молекулы тоже
образуют жидкие кристаллы [Chandrasekhar, 19831. Типичными
примерами являются гексазамещенные эфиры бензола (I) и
некоторые эфиры порфирина (II). В жидкокристаллическом состоянии
дископодобные молекулы стыкуются апериодически в колонки
(подобно жидкости), а различные колонки упаковываются в двумерную
сетку (подобно кристаллам). Фазы имеют трансляционную
периодичность в двух измерениях, но подобно жидкости разупорядочены
в третьем. Кроме колончатой фазы (D), дископодобные молекулы
обнаруживают и нематическую фазу (Nd), сообщалось и о фазовых
переходах между ними.
Недавно было показано, что определенные спиропираны
обнаруживают некоторые признаки жидких кристаллов, но имеют
структуры, отличные от структуры общепринятых жидких кристаллов;
эти твердые тела называются квазижидкими кристаллами [Shvart-
sman, Krongauz, 1984].
4.14. НЕКРИСТАЛЛИЧЕСКОЕ СОСТОЯНИЕ
И ФАЗОВЫЕ ПЕРЕХОДЫ В СТЕКЛАХ
Ранее (см. разд. 3.4) отмечалось, что все материалы могут
быть превращены в аморфные. Химики часто не признают, что
большое количество разнообразных материалов — катализаторы, ката-
лизаторные носители, ксерографические фоторецепторы, оптические
волокна, солнечные батареи большой площади и многие
бионеорганические вещества — являются некристаллическими [Elliot et al.,
19861. Некристаллические вещества не обладают дальним порядком,
198
Фазовые переходы
Рис. 4.29. Изменения в объеме или
энтропии при охлаждении жидкости. При
Т, может происходить кристаллизация
или, если жидкость переохлаждается
ниже 7\, образуется стекло.
Температура, соответствующая изменению в
наклоне кривых зависимости v (или S)
от Т, определяется как температура
стеклования Tg. Значение Tg изменяется
со скоростью охлаждения /?(/?!> Л2).
TgTg Tf и их структуры родственны струк-
т ^ турам замороженных жидкостей.
Большинство некристаллических
(или аморфных) материалов становятся кристаллическими при
соответствующей тепловой обработке. Например, обычные стекла при
некоторой температуре расстекловываются; металлические стекла
кристаллизуются при нагревании (700 К или около).
Кристаллизация никоим образом не является общей особенностью стекол,
как это видно на примере плавленного кварца. Стеклообразный
углерод является материалом, используемым в
высокотемпературных приложениях. Кристаллизация стеклообразного В203
возможна только путем различных ухищрений.
В то время как кристаллизация является важным фазовым
переходом некристаллических материалов, явление перехода в
стеклообразное состояние и из него характерно только для стекол, а не
для всех аморфных материалов. При температуре стеклования Tg
переохлажденная жидкость отклоняется от равновесного поведения
(рис. 4.29), и истинное значение Tg само по себе зависит от скорости
охлаждения. Tg обычно составляет около 0,67Г/, где Tf—
температура плавления. Стеклование как переход имеет некоторые
признаки, по-видимому, подобные признакам фазового перехода второго
рода. Например, Ср и другие вторые производные свободной
энергии Гиббса резко увеличиваются при Tg. Согласно рис. 4.29
энтропия жидкости должна была бы стать равной энтропии кристалла
заметно выше 0 при условии отсутствия мешающих переходов. Эта
проблема обходится постулированием существования перехода при
температуре Т0 (термодинамическая температура перехода), которая
ограничивает уменьшение энтропии. Чтобы избежать такой
парадоксальной ситуации, как отсутствие перехода при Т < Г0,
предполагают, что стекло вынуждено кристаллизоваться при Т "> Т0.
Интересно, что значения Г0, установленные из калориметрии
(график Ср от lgT), близки к значению, полученному из уравнения
(4.32), которое описывает температурное изменение вязкости и
других динамических свойств:
t
>
(П
Cmewq^^S
Ri —^^7"
к ^1
Кристалл i .
11
&
У = Т0ехр Ш(Т - Г0)].
(4.32)
Некристаллическое состояние и фазовые переходи в стеклах 199
Идеальное стекло определяется как стекло, в котором T'g
приближается к Т0. Неидеальность стекла будет тогда измеряться
энтропией, замороженной при Tg (равна нулю при Tg = TQ). До сих
пор существование термодинамического перехода при Т0 не
наблюдалось, так как ниже Tg времена релаксации слишком велики.
Экспериментальная Tg сама определяется, когда время релаксации
во временной шкале эксперимента становится постоянным.
Ситуация при Т < Т0 в отсутствие перехода при Т0 относится к так
называемому парадоксу Каузмана [Kauzman, 1948]; предполагается, что
стекла будут кристаллизоваться при температурах выше Г0.
Разнообразные релаксационные, спектроскопические и другие
методы были использованы для исследования поведения различных
свойств стекол в окрестности Tg, но природа этого перехода до
конца не понята [Wong, Angell, 1976; Parthasarathy et al., 1983]. Среди
различных моделей, предложенных для этого перехода, заслуживают
внимания модели свободного объема [Cohen, Grest, 1981] и
конфигурационной энтропии [Gibbs, 1963]. В модели свободного объема
динамические свойства связываются со свободным объемом, тогда
как в модели конфигурационной энтропии они связаны с
конфигурационной энтропией по уравнению W(Т) = А ехр [B/TSC], где
А и В — константы. Согласно последней модели решение парадокса
Каузмана требует перехода при температуре Г0, где
конфигурационная энтропия исчезает. Различные модели переходов в
стеклообразное состояние недавно рассмотрены в обзоре [Parthasarathy
et al., 1983].
Энтропия, замороженная ниже перехода в стеклообразное
состояние, имеет довольно большую конфигурационную компоненту.
Роль коллективной энтропии рассматривалась, и предполагается,
что коллективная энтропия стремится к нулю при Tg, когда
перестают существовать свойства, подобные свойствам жидкости [Kan-
no, 1983]. Это должно требовать, чтобы переход в стеклообразное
состояние был нечувствителен к предыстории охлаждения, но это
противоречит эксперименту. Компьютерные эксперименты [Ноаге,
Barker, 1977] так же, как и электронно-микроскопические
исследования, указывают на существование упорядоченных областей или
кластеров (с некристаллическими мотивами), распределенных в
ткани материала низкой плотности. Когда растут число и размеры
кластеров, отмечается замораживание в стекло, что приводит к
элиминации конфигурационной энтропии; ткань материала в
межкластерных областях должна сохраняться, претерпевая
конфигурационные изменения, тем самым отвечая за существенное значение
конфигурационной энтропии. Была развита кластерная модель
переходов в стеклообразное состояние, в которой в качестве параметра
порядка используется относительный размер кластера [Rao, Rao,
1982]. Эта модель предсказывает, что переход в стеклообразное
состояние, по существу, второго рода, в отличие от модели свободного
объема, которая предсказывает переход первого рода.
200
Фазовые переходы
Термин переход в стеклообразное состояние обычно
используется в связи с позиционно разупорядоченными метариалами, но
этот переход найден и в твердых телах, которые характеризуются
другими степенями свободы. Например, ориентационное разупоря-
дочение в пластических кристаллах может быть закалено, давая
стеклокристаллы, которые обнаруживают стеклоподобные переходы
[Suga, Seki, 1980; Parthasarathy et al., 1984]. Разупорядочение в
дипольных взаимодействиях может быть заморожено, давая диполъ-
ные стекла (подобно спиновым стеклам), типичными примерами
чего являются КВг, допированный ионами CN", или КТаОя, допи-
рованный Li+ [Hochli et al., 1979; Bhattacharya et al., 1982].
Замороженные жидкие кристаллы также обнаруживают
стеклоподобные переходы [Johari, Goodby, 1982; Johari, 1982]. Таким образом,
стеклообразное состояние, включает разупорядочение дальнего
порядка разных типов, тогда как стеклование само по себе
проявляется, когда пересекаются временные шкалы эксперимента и
релаксации.
Моделирование локальной структуры жидкости имеет свое
начало в классической работе Бернала, который для этой цели
использовал механические ансамбли. В некоторых работах указывалось
на важность не заполняющих пространство симметрии (таких как
икосоэдрическая и пентагональная) в упорядочении агрегатов в
аморфное состояние. Роль таких агрегатов или кластеров в
стекловании уже отмечалась. Работы по компьютерному моделированию
[Angell, et al., 1981] дали полезную информацию о прототипе
перехода в стеклообразное состояние в простых жидкостях, хотя
использование высокой скорости закалки ограничивает применимость
этих результатов. Высокие фиктивные температуры (температура,
при которой пересекаются зависимости V ст Т для жидкости и
стекла) смоделированных стекол предполагают высокие скорости
диффузии. И жесткие, и мягкие сферы могут компактироваться в
аморфные ансамбли, но в этих случаях отсутствует характерный разрыв
в Ср. Ансамбль жестких сфер показывает, что диффузия не будет
изменяться линейно со свободным объемом при низкой температуре
и в уравнении
D = Aexv[-B/{V - V)]. (4.33)
V0D представляет собой, по-видимому, берналовский предел
плотности случайной упаковки жестких сфер. Тем не менее
компьютерное моделирование яснс показывает существование увеличения Сл,
которое является отличительным признаком перехода в
стеклообразное состояние в эксперименте. Работы по моделированию
молекулярной динамики, однако, дают ценное представление о ионном
движении в стеклах [Angell, 1981].
Вода, наиболее обычное вещество в природе, в твердом
состоянии может быть аморфной. Аморфная твердая вода может быть по-
Методы Монте-Карло и молекулярной динамики 201
лучена осаждением из разбавленной газовой фазы на холодную
подложку [Sceats, Rice, 1982]. Получающийся материал выглядит
как стекло и является рентгеноаморфным. В таких осадках
обнаруживаются фазовые переходы, характерные для стекла [McMillan,
Los, 1965], и Тб, предсказанная из экстраполяции данных по
бинарным солевым растворам (139 К), считается совпадающей с Tg,
полученной из тепловых измерений [Angell, Sare, 1970]. Измерение
теплоемкости аморфной твердой воды, проведенное в
адиабатическом калориметре, показало, что теплоемкость при фазовом
переходе, как и ожидалось, увеличивается [Sugisaki et a]., 1968].
Экстраполированная Tg, однако, не считается совместимой с
теплоемкостью жидкой воды. Структура аморфной твердой воды,
приготовленной осаждением из газовой фазы, не отличается от структуры,
ожидаемой для основного состояния низкотемпературной жидкой
воды. Ультрабыстрое закаливание жидкой воды дает аморфные
образцы, подобные образцам, приготовленным из газовой фазы, кроме
того, при таком охлаждении нет кристаллизации [Brugeller, Mayer,
1980; Dubocket et al., 1982]. Кристаллизация аморфной твердой
воды происходит около 162 К. Довольно загадочным является
недавнее сообщение по поводу аморфной твердой воды [McFarlane,
Angell, 1984] о том, что на ДСК кривой нет никаких проявлений
переходов, характерных для стекол в области температур,
предсказанной экстраполяцией данных для бинарных солевых растворов.
По-видимому, вода в аморфном твердом состоянии так же трудпа
для понимания, как и жидкая вода.
4.15. МЕТОДЫ МОНТЕ-КАРЛО И МОЛЕКУЛЯРНОЙ ДИНАМИКИ
Методы Монте-Карло и молекулярной динамики являются
двумя важными способами изучения текучих сред и твердых тел
[Klein, 1985]. В этих методах рассчитываются свойства конечной
системы частиц, взаимодействующих через известный межчастичный
потенциал. Принципом метода Монте-Карло является осуществление
стохастического усреднения свойств методом Метрополиса [Metro
polis et al., 1953]. Большая часть расчетов по Монте-Карло
проведена для ячеек постоянного объема и гиббсовского канонического, или
NVT-ансамбля. Однако стало множиться число работ [Wood, 1968;
McDonald, 1972] по расчетам изотермически-изобарических
ансамблей, в которых объем ячейки меняется в процессе моделирования,
но фиксируется ее форма. В расчетах молекулярной динамики
ньютоновские уравнения движения решаются численно для системы
частиц [Alder, Wainright, 1957]. Так как полная энергия системы
сохраняется, ансамбль, порожденный моделированием, соответствует
микроканоническому, или NVE-ансамблю. Основным
преимуществом метода Монте-Карло является то, что
изотермически-изобарические расчеты ближе к экспериментальным условиям фиксирован-
202
Фазовые переходы
ных температуры и давления. Динамические свойства легко
получаются из молекулярной динамики.
Метод молекулярной динамики при постоянной энергии и метод
Монте-Карло для канонического ансамбля не могут быть
использованы для изучения фазовых переходов, происходящих в результате
изменения температуры или давления, так как форму ячейки в
процессе моделирования изменить нельзя. Известна работа
[Andersen, 1980], в которой метод молекулярной динамики расширен до
учета изменений в размерах ячейки. Имеется и модификация этого
метода, учитывающая изменения в форме и размере ячейки
[Parrinello, Rahman, 1980]. Это дает возможность изучать фазовые
переходы методами молекулярной динамики [Nose, Klein, 1983b].
Например, был исследован р — а-переход в Agl, где катионная
подрешетка высокотемпературной фазы плавится, приводя к высокой
ионной проводимости [Parrinello et al., 1983]. Метод Монте-Карло
в расчетах канонического или изотермически-изобарического
ансамбля не позволяет варьирования формы ячейки моделирования.
Метод Монте-Карло был обобщен, чтобы получить возможность
изучения фазовых переходов и подобных явлений [Yoshanath, Rao,
1985а, b], учитывая изменение формы ячейки моделирования. В
молекулярных системах это делается включением ориентационных
координат через эйлеровские углы, что дает возможность
исследовать полиморфные или структурные фазовые переходы в твердых
телах. При моделировании твердого тетрахлорметана найдено, что
ячейка вращается в пространстве. Вращение моделируемой ячейки
возможно в исследованиях Монте-Карло переменной формы (как и
методом молекулярной динамики переменной формы).
Существующими экспериментальными и теоретическими
методами трудно исследовать соотношение между структурой твердого
тела и межчастичным потенциалом взаимодействия. Было проведено
исследование [Yashonath, Rao, 1985b] изменений в
кристаллической структуре одноатомного твердого тела при изменении
потенциала взаимодействия. При леннард-джонсовском потенциале твердое
тело имело гранецентрированную кубическую структуру, а когда
потенциал взаимодействия изменялся до цезиевого, структура
твердого тела переходила в объемно-центрирсванную кубическую.
Трансформация была обратной, когда потенциал взаимодействия
изменялся вновь до леннард-джонсовского, точно так же, как в случае
подобного исследования методом молекулярной динамики [Parrinello,
Rahman, 1980].
Недавно для изучения перехода из кристаллической фазы в
фазу пластического кристалла стал использоваться новый метод
молекулярной динамики, в котором форма ячейки при
моделировании может непрерывно изменяться. Фазовые переходы в СС14
изучены с использованием как методов молекулярной динамики, так и
метода Монте-Карло [McDonald et al., 1982; Yashonath, Rao,
Приложения фазовых переходов
203
1985а]. Фаза высокого давления СС14 (III) переходит в фазу
пластического кристалла примерно при той же температуре, которая
найдена в эксперименте. Переход упорядоченной моноклинной фазы в
фазу пластического кристалла при атмосферном давлении в CF4
также был изучен методом Монте-Карло, и результаты в общем
согласуются как с результатами эксперимента, так и с
результатами, полученными методом молекулярной динамики [Nose, Klein,
1983а]. Методом Монте-Карло исследованы и ориентационные стекла,
получаемые закаливанием пластических кристаллов [Yashonath,
Rao, 1983]. Успешно изучено методом Монте-Карло и явление стек-
лообразования в изопентане [Yashonath, Rao, 1985c]. В этой работе
предсказываются известные особенности перехода в стеклообразное
состояние и ясно показано, насколько сильно отличаются упаковка,
координационное число и корреляционные функции в
стеклообразном состоянии по сравнению с жидким состоянием; показано также,
как влияет на структуру стекла отжиг. В дальнейшем методы
компьютерного моделирования будут использоваться для предсказания
фазовых переходов и структуры твердых тел, включая жидкие
кристаллы .
4.16. ПРИЛОЖЕНИЯ ФАЗОВЫХ ПЕРЕХОДОВ
Изменения свойств при фазовом переходе часто вызывают
технологический интерес, и найдены некоторые приложения
материалов с фазовыми переходами. Приложения фазовых переходов
можно классифицировать по использованию:
1) образования и (или) движения подвижных границ между
двумя или более термодинамическими состояниями,
сосуществующими ниже критической температуры Гс;
2) изменений в физических свойствах по мере приближения
температуры к Iс\
3) изменений в свойствах при Гс;
4) метастабильных фаз, получаемых при контролируемой
кинетике зародышеобразования или диффузии, требуемой для перехода
в стабильные фазы.
Для первой группы (при Т < Гс), в которой используются
изменения суммарных свойств материала контролем зарождения и
движения границ доменов, мы имеем большие классы материалов,
подобных сегнетоэлектрикам, сегнетоэластикам, ферромагнетикам,
сверхпроводникам или жидким кристаллам. В ферромагнетиках
магнитное упорядочение дальнего порядка ниже температуры Кюри
индуцирует спонтанную намагниченность Ms, приводящую к
большому количеству магнитных доменов, в каждом из которых вектор
Мs ориентирован в направлении, отличном от направления в
смежных доменах (см. разд. 6.З.1.). Смежные домены разделены домен-
204
Фазовые переходы
ными стенками, на которых вектор Ms меняет свою ориентацию.
Внешнее магнитное поле изменяет размеры доменов, увеличивая
домены благоприятной ориентации за счет других доменов. Ниже
Тс сегнетоэлектрики характеризуются различными ориентацион-
ными состояниями, но кооперативное кристаллографическое
искажение индуцирует спонтанную поляризацию. Границы доменов в сег-
нетоэлектриках могут управляться внешним электрическим полем.
В сегнетоэластиках ниже Тс кооперативное искажение кристалла
индуцирует спонтанную деформацию. Приложенное внешнее
давление управляет границами доменов сегнетоэластика. Магнитное (или
электрическое) и упругое упорядочение дальнего порядка
сосуществуют во многих материалах, так что спонтанная намагниченность
(или поляризация) управляется приложенным давлением или
спонтанная деформация — приложенным магнитным или электрическим
полем. Сосуществование магнитного и электрического дальнего
порядков известно для веществ, подобных BiFe03, которое является
антиферромагнетиком — сегнетоэлектриком. Исследованы и
использованы магнитно-электрические явления [Freeman, Schmid, 1975].
Оптические свойства сегнетоэлектриков, сегноэластиков и
ферромагнетиков находят много применений, причем типичным примером
является сегнетоэлектрик — сегнетоэластика Gd(Mo04)3 с ТС
ниже 433 К, который прозрачен в видимой области.
Жидкие кристаллы нашли широкое применение в оптических
дисплейных устройствах, а также в определении примесей и
однородности температуры. Эти свойства связаны с ориентационным
упорядочением молекул в температурной области между Тс и точкой
плавления. Многообещающими являются возможные применения
сегнетоэлектрических жидких кристаллов. Сверхпроводники (II
рода) могут быть использованы для создания сильных магнитных
полей при низкой энергии; способность сверхпроводников I рода
захватывать магнитный поток в домены несверхпроводящего
материала также может иметь приложения.
Два важных свойства, которые изменяются около Тс, — это
смягчение колебательной оптической моды до диспласивного
перехода и температурная зависимость спонтанной намагниченности в
ферромагнетиках ниже Гс. Эти свойства используются в
диэлектрических и пиромагнитных детекторах соответственно. Инваровые
(сплав железа с никелем) сплавы имеют параметры решетки ниже
ферромагнитной температуры Кюри, по существу, температурно-
независимые, так как индуцируемые магнитным полем изменения
решетки налагаются в этой системе на обычное термическое
расширение. В металлическом Bi2Ru207_x (структура дефектного пиро-
хлора) сопротивление почти не зависит от температуры в области
(150 < Т <С 500К). По-видимому, это вызвано электронным
взаимодействием с мягкими колебательными модами выше Тс (150 К),
где происходят фазовые изменения. Электрон-фонное взаимодейст-
Введение
205
вие с мягкими модами в материалах будет, вероятно, иметь
некоторые важные применения. Устройства на использовании мягкомодо-
вых аномалий обсуждались в литературе [Fleury, 1973].
Что касается свойств при Гс, мы можем представить
использование скрытой теплоты фазового перехода первого рода для
накопления энергии и регулирования температуры. Магнитные переходы
первого рода могут быть использованы для переключателей.
Переходы полупроводник — металл могут использоваться в
выключателях, делителях напряжения или оптических переключателях.
Нет необходимости подчеркивать важность метастабильных фаз,
продолжающих существовать при обычных температурах и
давлениях. Контроль эвтектоидного разложения аустенита, метастабильной
ОЦК-фазы железо — углерод ниже 996 К очень важен в индустрии
стали. Аустенит делается закалкой ГЦК-фазы. Когда содержание
углерода больше 0,22 мае. %, кубический аустенит переходит в
тетрагональный мартенсит по бездиффузионному механизму. Много
фаз высокого давления может быть сохранено при атмосферном
давлении. Примерами таких фаз являются алмаз и нитрид бора.
Некоторые метастабильные фазы могут быть приготовлены
низкотемпературными химическими методами (см. гл. 3). Кубический
NaSb03, проявляющий быструю проводимость ионов Na+, считается
метастабильной фазой, приготовленной ионным обменом из KSb03.
Хорошо известные примеры метастабильных материалов — стекла и
другие аморфные материалы, которые имеют широкие приложения.
5. НОВЫЙ ВЗГЛЯД НА СТАРУЮ ПРОБЛЕМУ:
ДЕФЕКТЫ И НЕСТЕХИОМЕТРИЧНОСТЬ
5.1. ВВЕДЕНИЕ
То, что соединения имеют определенный состав,
принимается как истина; но все же существует несколько неорганических
твердых веществ с широкой областью составов или без простого
соответствия между составом и детальной структурой (или
химической индивидуальностью). С 20-х годов известно, что стехиометриче-
ский FeOli00 не попадает в ряд устойчивости оксидов железа (II)
(FeOb05—FeObl5). Точечные дефекты в кристаллах, такие как
вакансии и внедрения, впервые описанные Шоттки, Френкелем и Вагнером,
объясняют транспортные свойства ионных твердых веществ, но
существуют серьезные препятствия к применению идеологии точечных
дефектов к твердым веществам, обладающим широкой областью
стехиометричности или к тем твердым веществам, в которых
наблюдаются упорядочение дефектов или протяженные дефекты (такие
206
Новый взгляд на старую проблему
(?) С-} (?) (^ (?) f^i (?) (^) f?\ (~*\ Рис. 5.1. Точечные дефекты в ионных
w \ZJ vl/ \_J vl/ \^J KL) \^J \U \^J твердых телах: дефект Шоттки, ва-
С^) О (~*) О С-*) 0 С~} (?)-Г-) (?) кансионная пара, дефект Френкеля и
^^ ^.^ ^-^ ^-^ ^-^ ^jfi*^ алиовалентная примесь (определение по-
о О п О о О о (j о О нятий -см-разд- 5-2)-
000oDQgO0
О I I О Qy О Q (D\^) П О как ПЛ0СК0СТИ
кристаллографией ООоПОрК^Пр) ческого сдвига). Хотя нет какого-
^ ^ ^ ^ ^ ^ ^ либо четко очерченного перехода
между режимом точечных
дефектов и режимом высокоупорядоченных структурных искажений в
нестехиометрических твердых веществах, мы можем определенно
сказать, что модель точечных дефектов в самом деле имеет силу
лишь тогда, когда концентрация дефектов (или отклонение от
стехиометрии) крайне мала. Только в таких разбавленных в
отношении точечных дефектов системах можно удовлетворительно
связать электронные свойства и нестехиометричность с
концентрацией точечных дефектов.
Несколько общих соображений относительно беспорядка и не-
стехиометричности в кристаллах были бы здесь уместны. Твердые
вещества редко достигают состояния совершенного порядка, даже
когда они охлаждаются почти до абсолютного нуля. При обычных
температурах кристаллические твердые вещества обычно
отклоняются от совершенной упорядоченности и содержат несколько типов
искажений, которые в действительности отвечают за многие важные
для твердого состояния явления, такие как диффузия,
электропроводность, пластичность и т. д. Типы беспорядка в твердых веществах
представлены следующими категориями дефектов: точечными,
линейными, планарными и объемными. Точечные дефекты возникают
при отсутствии атомов (или ионов) в узлах решетки, при наличии
атомов в промежуточных и в ошибочных позициях (невозможных
в ионных твердых веществах) и при наличии чужеродных атомов
(рис. 5.1.). Наличие точечных дефектов приводит к поляризации
окружения в кристаллической структуре, вызывая небольшие
смещения соседних атомов или ионов. Например, катионная вакансия
в ионном твердом теле будет иметь отрицательный эффективный
заряд и вызывать смещения соседних ионов. Энергия образования
точечного дефекта зависит главным образом от расположения атомов
в их непосредственном соседстве, поскольку релаксационные
эффекты с расстоянием быстро уменьшаются. Линейные дефекты в
кристаллах — это дислокации, представляющие собой ряд атомов, которые
не обладают правильной координацией. Планарные дефекты —
пограничные дефекты: дефекты границы между малыми кристаллитами,
дефекты упаковки, плоскости кристаллографического сдвига, сдвоенные
границы двойников и антифазные границы. Сегрегация точечных
дефектов может вызывать объемные (трехмерные) дефекты.
Введение
207
Соединения с переменной стехиометрией относят к бертолли-
дам, а соединения с почти постоянным составом называют далътони-
дами, что отражает спор, который произошел между Дальтоном и
Бертолле свыше ста лет тому назад. Используется также термин
«нестехиометрическое соединение», хотя не всегда с тем же самым
значением. Нестехиометрическое соединение можно определить (в
рамках современных критериев) как кристаллическое соединение в
равновесии с его окружением, которое ведет себя как
термодинамически дивариантная система; хотя свойства кристалла (например,
размеры ячейки) могут изменяться с изменением состава, симметрия
остается той же самой по всему ряду устойчивости состава
[Anderson, 1984]. Разумно напомнить, что стехиометрия кристалла
однозначно определяется составом повторяющейся элементарной ячейки.
Формула элементарной ячейки может быть сложной, по-видимому,
с иррациональными отношениями составляющих ее атомов, как
в NbO2t4906, Nb02t4681, Nb02t4167, Pr0lt833 и РгОЬ714 (на самом деле
эти оксиды являются кристаллографически хорошо определенными
соединениями соответственно Nb530132, Nb470116, Nb12029, Рг12022 и
Рг7012); такие фазы являются структурно родственными и
объясняют природу нестехиометрических соединений. В нестехиометриче-
ских соединениях среднее число атомов на элементарную ячейку
не эквивалентно числу позиций, и в одной из подрешеток (анионной
или катионной) имеется их дефицит или избыток.
Нестехиометрические соединения — соединения со смешанной
валентностью и с нецелочисленными отношениями электрон/атом.
Электронные свойства этих соединений решающим образом зависят
от природы и от степени стехиометричности. Электронная
проводимость во многих таких соединениях обеспечивается прыжковым
механизмом между катионами различных валентностей (например,
Рг3+ и Рг4+ в Рг12022). Нестехиометричность с широкой областью
составов более обычна для оксидов, сульфидов и родственных
веществ, в которых связь не полностью ионная. В ионных
нестехиометрических соединениях могут происходить структурные
перегруппировки в результате кулоновских взаимодействий между
дефектами и наличия в соединениях катионов переменной валентности. В
металлических системах, однако, делокализованные электроны могут
экранировать центры локального дефицита или избытка электронов
вследствие взаимодействий дальнего порядка [Anderson, 1984]; такие
системы могут образовывать сверхструктуры, хотя вероятность
структурной перегруппировки значительно уменьшается. Гетероион-
ные твердые растворы являются модельными системами по
сравнению с настоящими нестехиометрическими соединениями. Такие
твердые растворы образуются при замещении ионом-гостем определенной
валентности другого иона определенной валентности в твердой
матрице (например, CaF2—YF3, CdCl2—NaCl), при замещении ионом
определенной валентности иона переменной валентности матрицы
208 Новый взгляд на старую проблему
(например, Ni^^ Li^O) или при замещении ионом-гостем
переменной валентности иона матрицы определенной валентности
(например, валентность иона Мп в оксидных матрицах может быть 2+, 3 +
или 4+). В отличие от многих нестехиометрических соединений,
в которых и структурные, и компенсирующие электронные дефекты
(заряды) участвуют в процессе упорядочения, в гетероионных
твердых растворах структурные (и электронные) дефекты становятся
локализованными, приводя в последнем случае к расширению ряда
нестехиометрических составов.
Ранее упоминалось, что дефекты упорядочивают сами себя,
образуя сверхструктуры; точечные дефекты, такие как вакансии,
элиминируются формированием планарных дефектов (плоскостей сдвига)
или структурными сингулярностями. Сейчас в литературе избыток
информации по реальной структуре дефектных твердых веществ,
особенно для случаев с иррациональными отношениями катион/анион
[Anderson, Tilley, 1974; Tilley, 1980], но редко бывает необходимо
обращаться к точечным дефектам. Фактически многое из того, что
есть в литературе последних лет по химии дефектных твердых
веществ, не имеет отношения к режиму точечных дефектов; вместо
этого химики рассматривают главным образом вопросы
сверхструктур, сдвигов, блочных структур и прорастаний. Поведение дефектов
в твердых веществах на самом деле изменяется от режима точечных
дефектов, контролируемого энтропией, с одной стороны, до систем,
контролируемых энтальпией, таких как плоскости
кристаллографического сдвига, — с другой. При этом сверхрешеточное
упорядочение и дефектные комплексы занимают промежуточное положение.
Эти аспекты химии твердого тела принадлежат поистине к
магистральному направлению неорганической и структурной химии.
5.2. ТОЧЕЧНЫЕ ДЕФЕКТЫ
В ионных твердых веществах обычные точечные дефекты —
это пары по Шоттки (пары катионных и анионных вакансий) и
дефекты по Френкелю (катионы или анионы плюс вакансия). При
большой концентрации пар по Шоттки измеренная пикнометриче-
ская плотность твердого вещества значительно ниже плотности,
рассчитанной из рентгеновских данных (например, VO). Галогени-
ды щелочных металлов — хорошие примеры твердых веществ с
дефектами по Шоттки; в AgCl и AgBr мы имеем кат ионные дефекты
по Френкелю, тогда как в CaF2 и SrF2 — анионные дефекты по
Френкелю» Замещение катиона-матрицы по Френкелю ионом другой
валентности приводит к целому ряду дефектных ситуаций. Если в
NaCl вводится небольшая концентрация Sr2+ или Cd2+, то образуются
катионные вакансионные пары из М2+. Число пар по Шоттки или
дефектов по Френкелю, п, в таком ионном твердом веществе, как
Точечные дефекты
209
NaCl, оценивается с помощью статистической термодинамической
модели. В случае пар по Шоттки п задается уравнением
п = N exp (St/2k) exp (— ErflkT) = N (-^rj* exp (- Ef/2kT), (5.1)
где N — общее число узлов решетки, v и v' — частоты колебаний
совершенных и дефектных кристаллов соответственно, St — тепловая
энтропия, Ef — энергия образования пары по Шоттки.
В металлических системах существуют некоторые необычные
типы дефектов, заслуживающие внимания. Найдено [Taylor,
Doyle, 1972], что в сплавах NiAl атомы алюминия в области, богатой
алюминием, не замещаются на Ni-подрешетку — вместо этого
имеются вакансии в позициях Ni. Например, при 55 ат. % А1 18 %
никелевых позиций вакантны, тогда как позиции А1 заполнены. Такие
вакансии, определяемые составом, считаются конституционными
вакансиями. Как впоследствии было показано, и другие сплавы
имеют такие вакансии, типичными из них являются NiGa и CoGa.
Другой, скорее, странный аспект дефектов — это образование
решеток пустот, когда такие металлы, как Мо, облучаются
нейтронами или более тяжелыми частицами [Gleiter, 1983]. Решетки
пустот возникают из скопления вакансий, и они родственны
сверхрешеткам. Как правило, соседние пустоты в Мо разделены
о
на 200 А. Предложено объяснение стабильности решеток пустот
на основании континуальной теории упругости [Stoneham, 1971;
Tewary, Bullough, 1972].
Если в ионном твердом веществе типа MX, состоящем из
одновалентных ионов, присутствуют примеси двухвалентных катионов
(например, Cd2+, Sr2+), то при низких температурах отрицательно
заряженные катионные вакансии (создаваемые двухвалентными
ионами) соединяются с примесными ионами. Аналогично отрицательно
заряженные катионные и анионные вакансии стремятся образовать
нейтральные пары. Такие нейтральные вакансионные пары важны
в диффузионных процессах, но они не участвуют в
электропроводности. Энергия взаимодействия вакансионных пар или пар
примесь — вакансия уменьшается с увеличением расстояния между
двумя противоположно заряженными узлами.
Для исследования вакансионных пар алиовалентная примесь —
катион и других точечных дефектов в ионных твердых веществах
использован ряд методов. Наиболее ценными из них являются
измерения диэлектрической релаксации, оптической абсорбции, метод
эмиссионной спектроскопии и измерения ионных термотоков;
исследования методов ЭПР Мп2+ в NaCl показали наличие пар примесь —
вакансия по меньшей мере пяти различных симметрии. Методы,
которые дали богатую информацию по энергиям миграции, образования
и другим видам энергий дефектов в ионных твердых веществах, —
14 ч. Н. Р. Рао, Дж. Гопалакришнан
210
Новый взгляд на старую проблему
ЮОО/1уК
Рис. 5.2. Удельная ионная проводимость
«чистого» NaCl как функция температуры.
Собственная проводимость на стадиях I и II;
стадия III соответствует проводимости кати-
онных вакансий, являющихся результатом
примесей. Стадия IV — соединение вакансий
с образованием нейтральных пар.
это методы по измерению диффузии и
удельной электропроводности.
Электропроводность в ионных твердых
веществах осуществляется движением
ионов через вакансии, или междо-
узельные ионы. В случае движения
через вакансии электропроводность о
задается уравнением
о = о0ехр-[(^-+Ет)1кт], (5.2)
где Е1 и Еш — энергии образования вакансий (пар по Шот-
тки) и ионной миграции (обычно катионной миграции)
соответственно. Если кристалл допируется алиовалентными примесями,
концентрация вакансий намного больше при низких температурах,
чем собственное равновесное значение, а наклон кривой зависимости
In а от Т'1 непосредственно дает значение Ет/к; наклон при более
высоких температурах будет давать (Ef/2-\-Em). Продемонстрируем
различные области на графике зависимости электропроводности
типичного ионного твердого вещества (рис. 5.2.). Данные по
диффузии с использованием меченых атомов несут дополнительную
информацию по энергиям дефектов.
Прямое наблюдение точечных дефектов в металлах возможно
с применением полевой ионной микроскопии. Примесные точечные
дефекты можно успешно исследовать методом электронной
микроскопии в сочетании с электронной диффракцией и электронной
спектроскопией. Сделана попытка прямого наблюдения допирующей
примеси во флюоритах методом EXAFS в сочетании с компьютерным
моделированием [Catlow et al., 1984].
Энергии, связанные с образованием, взаимодействием и
миграцией дефектов, в большом числе твердых тел определены
экспериментально [Corish et al., 1977; Wollenberger, 1983]. В твердых инертных
газах энергии образования вакансий порядка 0,1 эВ; в металлах —
около 1 эВ (0,73; 1,03 и 1,49 эВ для А1, Си и Pt соответственно).
Энергия образования вакансии в этих твердых веществах
увеличивается с температурой плавления Тт. В металлах энергия миграции
Ет обычно порядка 7-10~4 Гт, за исключением натрия. Кроме того,
Точечные дефекты
211
значения Ет в металлах близки к значениям Е . Например, Ет для
А1, Си и Pt равны соответственно 0,65; 0,94 и 1,42 эВ. Обнаружено,
что энергии связи дивакансий в золоте и серебре около 0,2 эВ.
Энергии образования пар по Шоттки в галогенидах Na и К в пределах
2,0—2,5 эВ, тогда как энергия миграции катиона по вакансионному
механизму — в пределах 0,6—0,9 эВ. В AgCl и AgBr энергия
образования дефекта по Френкелю — около 1,3 эВ, энергия миграции по
Френкелю для катиона (Ag+) составляет 0,1 эВ или меньше; ионы
Ag + движутся без какого-либо барьера, вероятно, по междоузельно-
му механизму, включающему смещение иона Ag + из позиции (000)
в позицию (111) (с участием междоузельного (111) иона). В CaF2
и SrF2 энергия образования анионных дефектов по Френкелю —
порядка 2,5 эВ, а энергия анионной миграции составляет 1 эВ.
Экспериментальные энергии связи двухвалентных катионов и катион-
ных вакансий в NaCl и КС1 составляют около 0,5 эВ. Энергии
взаимодействия противоположно заряженных вакансий с образованием
пар тоже имеют такой порядок. Следует отметить, что образование
дефектов — всегда эндотермический процесс. Энергии образования
вакансий в ионных твердых телах, например, составляют обычно 2 эВ
или более. Собственная концентрация дефектов в этих твердых телах,
следовательно, чрезвычайно мала даже при высоких температурах.
Равновесная концентрация дефектов в бинарных твердых телах в
пределах 0,8Гт составляет ~ 10~5.
Энергии образования дефектов и другие энергетические параметры
дефектов оценены теоретически. В случае металлов энергия
образования вакансии должна быть равна (Es—Er), где Es — энергия
удаления атома с поверхности, а Ег — энергия релаксации.
Вакансия в твердом ионном теле несет на себе эффективный заряд
противоположного знака, и энергия поляризации, следовательно,
становится значительной. Впервые сделана успешная оценка
энергий образования вакансий в ионных твердых телах [Mott,
Littleton, 1938], при этом была оценена энергия поляризации после
релаксации решетки. В таких расчетах энергии дефектов смещения
и поляризация оцениваются с использованием специфических
ионных взаимодействий в ближайшем окружении дефекта с
одновременным рассмотрением остальной части решетки как диэлектрического
континуума. Таким расчетам обычно удовлетворяют хорошо
известные потенциалы Борна для ионных твердых тел. В литературе есть
компетентный обзор по теоретическим исследованиям точечных
дефектов [Stoneham, 1975; Corish et al., 1977]. В связи с доступностью
быстродействующих компьютеров в расчеты энергий дефектов
введены некоторые усовершенствования. Например, большое число
ионов включается в дефектную область, а потенциалы подбираются так,
чтобы соответствовать диэлектрическим и упругим свойствам (см.,
например, [Uppal et al., 1978]). Чтобы учесть изменение
взаимодействий ближнего порядка, обусловленных поляризацией при примене-
14*
212
Новый взгляд на старую проблему
нии оболочечной модели, вводятся также адекватные поправки.
Эти новые расчетные методы наиболее успешны в понимании
упорядочения дефектов, протяженных дефектов и нестехиометричности
[Catlow, Mackrodt, 1982; Catlow, James, 1980]; в этой же главе мы
рассмотрим некоторые результаты. Кроме энергий дефектов,
теоретически оценена также энтропия образования дефектов [Gillan,
Jacobs, 1983].
5.2.1. Равновесие точечных дефектов
Значение взаимодействий между точечными дефектами даже
при их довольно низких концентрациях признано несколько лет
тому назад. Хотя для того чтобы можно было полностью описать
свойства твердых тел, принимаются во внимание действительная
дефектная структура и модификации ближнего порядка
целесообразно все процессы, включенные в междоузельные дефектные
равновесия в кристалле (с низкой концентрацией дефектов), а также его
равновесие с внешним окружением представлять наборсм связанных
квазихимических реакций. Эти равновесные реакции в таком случае
регулируются законом действия масс. Свободную энергию и
константы равновесия для каждого процесса можно получить, если мы
знаем энтальпии и энтропии реакций из теории или эксперимента.
Такое приближение используется для исследования набора
равновесий дефектов [Kroger, 1974]. Чтобы написать уравнение реакций
в обычном виде, мы будем использовать систему обозначений по
Крёгеру и Либовицу [Libowitz, 1973]. В этой схеме АА означает
атом А в позиции А; МА означает атом М в позиции А. В твердом
MX Vm и Vx означают вакансию в позиции атома металла М и
вакансию в позиции аниона X соответственно. Символ I или i
используется для обозначения междоузлия. Поскольку концентрации
дефектов обычно низкие, мы будем использовать концентрации вместо
активностей. Исследованы использование активностей и
применимость метода Дебая — Хюккеля к дефектным ионным твердым телам.
Ks = [VmHVxI, (5.3)
где Ks — константа равновесия. Поскольку MX находится в
равновесии с внешней фазой (газ Х2), равновесие дефектов подчиняется
уравнениям (5.4) и (5.5):
Хх - l/2X2(i?) + Vx,
#vx = Рх722 [VX]/[XX] = рЦ221ух]/(1 - [VxD. (5.4)
гДе />х2 — парциальное давление Х2. Если Х2 влияет на
концентрацию вакансии М, [Хх] равно единице:
l/2X,(g) ^ Хх + VM + aV„
#vm = [VM] [VrlVPx! • (5.5)
Точечные дефекты
213
Кроме того, нейтральные дефекты могут подвергаться ионизации
Vx ^ Vx++£" и Vm ^ V^[ + h+, где ё~ и h+ — электроны в зоне
проводимости и дырки в валентной зоне соответственно. Константы
равновесия для приведенных выше реакций можно представить как
*Vx-[v£][*-]/[Vx] <5-6)
и
4m=[V5][^]/[Vm]. (5.7)
Всякий раз, когда в твердом теле посредством ионизации
генерируются электроны и дырки, устанавливается другое равновесие: О =^=
^ е~ +fe+.
Константа, соответствующая собственному равновесию,
Kt = [е-№\ = [п][р], (5.8)
где [п] и [р] — концентрации электронов и дырок соответственно.
Электронные дефекты, такие как электроны и дырки, в твердом теле
появляются в состоянии теплового равновесия, а их равновесные
концентрации подчиняются также закону действия масс.
Величина Kt определяется энергией ширины запрещенной зоны.
Электронейтральность требует, чтобы
Ы + IVS1 = lp] + [V£l. (5.9)
Таким образом, у нас есть семь неизвестных—[Vm], [Vxl, IVmI»
[Vxs [/г], [р] и рх2 и семь уравнений — (5.3) — (5.9). Все эти
уравнения, кроме (5.9), можно выразить в виде линейных уравнений в
логарифмическом (концентрационном) виде:
}nKs = ln [VM] + ln [Vx], (5.3)'
In Kyx = 1/2 In Px2 + ln [Vx], (5.4)'
In #VM = In [ VM] - 1/2 In p^ (5.5)'
In К\щ = In [n] + In [Vi] - In [Vx], (5.6)'
Ih^Vm = In [p] + In [VM] - In [VM], (5.7)'
In Kt = In [n] -fin [p]. (5.8)'
В приведенных выше уравнениях введены необходимые упрощения,
такие как Vx -С 1, [Vi] « [Хх] = 1 и т. д. Хотя в принципе можно
получить решения этих уравнений, когда известны различные
значения К, вид уравнений (5.9) делает это очень затруднительным. В
соответствии с приближением по Брауэру для больших отрицательных
отклонений от стехиометрии [Vm~] и [р] становятся незначитель-
214
Новый взгляд на старую проблему
ньтми:
м = [vii, (зло)
In [п] = ln IVtl
Используя соотношение (5.10), можно выразить все концентрации
через In px • Мы можем теперь ввести величину 7?, используя
уравнение (5.5)':
Л = ^умрк/22 = [Ум]. (5.11)
Таким образом, из уравнения (5.3)' мы имеем fVxl = KJR, а из
уравнения (5.6)' [п] = [v£] = #Vx [Vx]1/2 = K\X{KS;R?12.
Поскольку константы равновесия по Шоттки для ионизованных
дефектов известны, выгодно выразить Ks в терминах К\ с помощью
соотношения
Ks = К\К\/КумКух- (5.12)
Теперь без труда видно, что если графически представить
зависимость In (концентрации) любого из дефектов как функцию In R.
наклон будет равен ±1/2 или ±1.
При больших положительных отклонениях от стехиометрии мы
можем использовать приближение
In [VmI =ln [pi (5.13)
и соотношения между R и концентрациями различных дефектов
получаются таким же образом, как приведено выше. Вблизи стехио-
метрического состава концентрация дефектов зависит от
собственных констант равновесия дефектов Ks и Kt. Для материалов с
большой зонной щелью, таких как галогениды щелочных металлов Ks ^>
^>Кг, и поэтому уравнение (5.9) ведет к соотношению
In [VmI -In [Vil. (5.14)
С помощью этого ссотношения мы можем составить ряд границ для
областей, соответствующих большому отрицательному и большому
положительному отклонению от стехиометрии. Мы обозначаем эти
области как область I (большое отрицательное отклонение),
область II (близкое к стехиометрическому составу) и область III
(большое положительное отклонение). Согласно соотношению (5.14),
значение R, соответствующее пограничному значению /?ItII,
определяется соотношением
\RK\K\jwfKi) = \K\Ki/K\wR)
или
Л1>п = KifKU- (5.15)
Точечные дефекты
215
Таблица 5.1
Равновесия дефектов в кристалле MX, имеющем дефекты Шоттки *
Дефек т
Область I
Область II
Ks » К{
К{ » Ks
| Область III
Р
К*г
1/2
AVM
\ К )
Д-1/2
1/2
Я1/2
R
K\i{\ \ 1/2
AsAVM \ R
к,
К\К, \1/2
KYMR j
,1/2
R = KVMP*
(*l)1/2*i
(*i)1/2
R
(*:)
1/2
ВД
^VX^VM^
№)m
(*i)1/2
л:?/2л:!
^1'МЯ
л:.
Км*)1/2
(*V*)1/2
д
(ПмД)1/2
(*W*)1/2
Согласно аналогичным соображениям
(ПмЯ)1/2 = К\/{К\иЯ)т
или
Лц.ш = К1/К\м- (5.16)
Ширину области IT можно найти по следующему уравнению:
In Дц,1п - In Д1§11 = ЫК8ЧК{). (5.17)
Используя соотношения (5.14) — (5.16) вместе с уравнениями
(5.3)' — (5.8)', мы можем определить концентрации различных
дефектов в области II по обеим сторонам стехиометрического состава.
Стехиометрия точно определяется условием
[VM] + [VmI = [Vx] + [Vil.
Поскольку tfs*> Kt в области II, [V5l«[V£], a [VM1 и [Vx]
ничтожно малы. Следовательно, [п] » [p]. и R0, соответствующее
стехиометрическому составу, равно (КЛ1)//£ум- Кроме того, как
(VmI, так и [Vxl совершенно нечувствительны к R или к большим
216 Новый взгляд на старую проблему
Рис. 5.3. Графики зависимости концентраций
дефектов в твердом теле MX от парциального
давления X2(tf*> К-).
Рис. 5.4. Относительная парциальная
мольная свободная энергия кислорода AGo2 "для
Fe О как функция состава (lg x) [S«J>ren-
sen, 1981].
-370-]
-330
\-290A
PCD
<]
-250-
7,5
т~Т"
1>0
-1д>
0,5
изменениям в рх2- Концентрации различных дефектов в трех
областях приведены в табл. 5.1. Зависимость In Ц)]от1п R схематично
представлена на рис. 5.3, где [D] означает концентрацию дефектов.
Ситуация с полупроводниками точно противоположна
ситуации для диэлектриков с широкой щелью, рассмотренных ранее.
Условие, которое остается в силе для полупроводников, это — Kt^>
*^>KS{, и, следуя вышеизложенной методологии, можно найти
выражения для концентраций различных дефектов.
Анализ равновесий дефектов в КВг является типичным
примером такого анализа [Kroger, 197Л]. Поскольку К(и К8{ равны
соответственно ЗхЮ"35 и 8х10~14, область II на рис. 5.3 расширяется
примерно на 43 порядка (см. уравнение (5.7)) и, следовательно,
концентрация дефектов не оказывает влияния на парциальное давление
Вг2, которое пропорционально R2 (см. уравнение 5.5)). Отклонение
от стехиометрии б, определяемое по разности 2 [Vm] —2 I^xL
также мало (~ 1СГ3) по всей этой области.
В оксидах металлов природу дефектов и нестехиометричности
контролирует парциальное давление кислорода. Дефекты
ответственны за нестехиометричность, а соответствующее окисление или
восстановление катионов, можно описать в рамках квазихимических
реакций дефектов. Рассмотрим пример монооксидов переходных
металлов, М1_зсО(М—Mn, Fe, Co, Ni), которые проявляют
металл-дефицитную нестехиометричность. Для случая образования в М1вЖ0
Точечные дефекты
217
вакансий металла можно написать следующие уравнения:
1/20, ^ VM + 00,
VM + e-^ VM" + fc+t
2ММ + 2fe+ ^ 2ММ+.
Полная реакция
1/2 02 *± V2M" + 2ММ + 00. (5.18)
Здесь Ум"означает дважды ионизованную вакансию металла, Мм+—
ассоциацию М2+ и h+ (которую можно рассматривать как ион М3+),
a h+ — дырку в валентной зоне. Используя закон действия масс,
вводя условие нейтральности, [Л+] = 2[Ум2~]» и выражая вакансии
катиона в М^^О через х, константу равновесия для (5.18) можно
записать как
К ,+ = Ь*рБ™. (5.19)
Vm 2
Состав оксида, следовательно, зависит от парциального давления
кислорода, что задано
х ос рЦ*. (5.20)
Если дефекты представляют собой однозарядные вакансии Vm"» to
хосрЦ*. (5.21)
Термодинамику и нестехиометричность оксидов удобно обсуждать
в терминах относительной парциальной мольной свободной энергии
ДСо2 [SySrensen, 1981]:
ДСо2 = fXo2 - \i°o2 = RT In Po2, (5.22)
где |Xq2 — химический потенциал кислорода в твердом теле; Цо2 —
химический потенциал кислорода в его стандартном состоянии
Ро2 — равновесное парциальное давление кислорода в атмосфере,
существующей вокруг нестехиометрического оксида.
Для М^дО соотношение между Л£о2 и составом х можно
представить в общем виде
ДСо2 = nRT In х. (5.23)
Зависимость AG0 от In x должна быть линейной, причем наклон
определяет значение /г, которое дает информацию о типах включен-
ных дефектов. Для двухзарядных вакансий металлов Vm" n = 6;
218
Новый взгляд на старую проблему
для Vm~ п = 4 и т. д. Изотермы AGq2 — Inx можно построить по
данным равновесных термогравиметрических или электрохимических
измерений. Типичный график AGq2 — lg# для системы Fe^^O
показан на рис. 5.4, на котором даны значения п, соответствующие
различным областям. Мы видим, что п = 6 относится к х ^ 0,09,
хотя и п = 5, соответствующее парам (VmVm)4-, также тесно
примыкает к этой области; значения п = 3 и 4, наблюдаемые для х<^
< 0,09, можно объяснить в рамках дефектных комплексов [4Vm~—-
— Fef J5- и [16 Vm" — 5 Fe3+] 17~ соответственно. Существует
структурное доказательство наличия обоих дефектных комплексов в Fe^^O.
Комплекс 16 : 5 соответствует элементу структуры Fe304. Однако
знаменательно, что термодинамические данные не подтверждают
существования в Fe1-3C0 комплекса Коха — Коуэна [9Vm,4Vm~ —
— 4Fe? J5 (см. разд. 5.5.3). Кроме того, термодинамические данные
с очевидностью свидетельствуют о наличии комплекса Коха —
Коуэна в системе Mn^^O IS^rensen, 1981]. Эти результаты подчеркивают
важность одновременных термодинамических и структурных
исследований для характеристики дефектных структур оксидов.
5.2.2. Параэлектрические и молекулярные примеси
в ионных твердых телах
В твердые тела, такие как NaCl и КС1, можно вводить
биполярные ионы, например CN" и ОН". Некоторые ионы-добавки
малого размера, как Си4" и Li+, в решетке хозяина типа КС1
стабилизируются в нецентральных позициях, (несколько удалены от позиций
решетки), образуя диполи. Эти диполи присутствуют в
кристаллическом поле и способны к поляризации и ориентации во внешнем поле,
отсюда их название — параэлектрические примеси. В галогениды
щелочных металлов можно вводить также молекулярные ионы,
подобно S2". Se2", N2~ и 02". Их оптические спектры и релаксационное
поведение имеют диагностическую ценность при изучении решеток
хозяина. Эти примеси характеризуются электрическим дипольным
вектором и упругим дипольным тензором. В последние годы
проведены исследования по ,ципольным моментам и направлениям
ориентации большого числа параэлектрических примесей. Реориентаци-
онные перемещения могут быть классическими или включать кван-
тово-механическое туннелирование.
Найдено, что такие ионы, как S2", Se2" и SSe", в большинстве
гологенидов щелочных металлов располагаются вдоль направлений
(110), тогда как в Nal, KBr и KI связь S—S группы S2~
ориентирована вдоль направления <100>. В случае О^ р-орбитали
параллельны направлению (100) в натриевых солях и параллельны
направлению (110) в рубидиевых и калиевых солях. Описаны об-
Точечные дефекты
219
ширньте спектроскопические исследования по молекулярным ионам
N02", N03", СЮ4- и МпОЦ-. Имеется несколько обзоров по таким
допированньтм примесью твердым телам [Bridges, 1975; Grimes,
1976; Corishet al., 1977].
5.2.3. Центры окраски
При обсуждении равновесия дефектов мы обсуждали
нейтральные дефекты, такие как Vx; Vx соответствует анионной вакансии,
которая захватывает электрон. Такие вакансии приводят к
интересному оптическому явлению. Дефекты, связанные с электронами
или дырками, вызывают окрашивание и известны как центры
окраски. Название «центры окраски» охватывает и такие примесные
центры, как Т1+, которые отвечают за абсорбцию и люминесценцию
в видимой области. Электроны, захваченные анионными
вакансиями, называются F-центрами. Абсорбционные максимумы F-центров
в LiCl, NaCl, KC1 и RbCl находятся соответственно при 385, 465,
563 и 624 нм (окраска между желтой и голубой). Соответствующие
эмиссионные полосы показывают большие стоксовские сдвиги. Силы
осциллятора абсорбционной полосы, а также эмиссии F-центра очень
высокие. Время жизни излучения ~ 10~6 с, что почти на два
порядка выше, чем при оптических переходах. Спектральные
характеристики F-центра можно понять при рассмотрении его как частицы
в ящике или как водородно-подобного атома. Если размер ящика
принимается таким, как расстояние до ближайших соседей, то
модель частицы в ящике хорошо работает, объясняя зависимость
энергии F-полосы от расстояния до ближайшего соседа.
Кроме F-центров в литературе описаны многие другие центры
окраски [Fowler, 1968, 1975; Stoneham, 1975]. F"-ueHTp создается
захватом анионной вакансией двух электронов. В щелочноземельном
оксиде единственный электрон в анионной вакансии дает
заряженный дефект, соответствующий Р+-центру. Два смежных F-центра
образуют М-центр; для галогенидов щелочных металлов три
смежных центра в равностороннем треугольнике на плоскости (111)
обозначаются совместно как Fg-центр. Ук-центр в галогенидах щелочных
металлов представляет собой дырку, захваченную двумя смежными
галогенид-ионами в направлении (110). Три смежных центра плюс
электрон составляют R-центр. Исследованы абсорбционные и
эмиссионные спектры многих таких центров окраски, они часто проявляют
нуль-фононную эмиссию. В щелочноземельных оксидах образуются
как синглетные, так и триплетные F-центры, поскольку в центре
имеются два электрона. Для исследования электронов,
захваченных в центрах окраски, успешно применяется электронная спин-
резонансная спектроскопия (ЭПР). Например, Ук-центр в КС1
(самозахваченная дырка между двумя хлоридными ионами,
соответствующая иону С12~) обнаруживает семь линий (2x3+1) в ЭПР-спект-
ре, находящихся на равном расстоянии, что обусловлено сверхтон-
220
Новый взгляд на старую проблему
ким расщеплением, поскольку каждый атом хлора имеет ядерный
магнитный момент 3/2. F-центры дают скорее широкие ЭПР-сигналыг
соответствующие огибающей линии многих сверхтонких
расщеплений; чтобы обнаружить сверхтонкое взаимодействие, используется
метод ENDOR.
5.3. ДИСЛОКАЦИИ
Дислокации являются линейными дефектами, они были
впервые привлечены для объяснения механических свойств твердых
тел, особенно сил сдвига. Дислокации играют важную роль во
множестве явлений твердого состояния, начиная с фазовых переходов
и кончая химическими реакциями; этот вопрос широко исследован
и представлен в литературе [Fine, 1973; Nembach, 1974]. В
последние годы признано влияние дислокаций на превращения и свойства
органических твердых веществ [Thomas, Williams, 1971; Jones,
Thomas, 1979].
Дислокация обозначает границу между смещенной и
несмещенной частями кристалла; простейший вид дислокации — краевая
дислокация, включающая дополнительный слой атомов в кристалле.
Атомы в такой краевой дислокации не полностью координированы.
Две части кристалла сдвинуты одна относительно другой на одно
атомное расстояние. Винтовая дислокация превращает последующие
плоскости атомов в поверхность спирали. На рис. 5.5 представлена
картина краевой дислокации в Ba2Bi4Ti5018, полученная методом
высокоразрешающей электронной микроскопии (HREM).
Дислокации в органическом твердом веществе показаны на рис. 5.6.
Наиболее важной характеристикой дислокации является связанное с нею
смещение скольжения, или вектор Бюргерса, Ь, который легко
определяется по контуру вокруг дислокации, называемому контуром
Бюргерса (рис. 5.7). Краевая дислокация перпендикулярна
вектору Ь, а винтовая дислокация параллельна ему. Деформация из-за
атомных смещений вблизи дислокации высокая. Вдали от ядра
дислокации смещения быстро уменьшаются, что видно из рис. 5.5.
Деформационная энергия дислокации, пропорциональная |Ь|2,
значительно больше вблизи краевой дислокации, чем вблизи винтовой.
Вообще дислокации не бывают ни чисто краевого, ни чисто
винтового типов, а проявляют смешанный характер. Такая смешанная
дислокация может быть подразделена на краевую и винтовую.
Если вектор Бюргерса соответствует трансляционному вектору
решетки, дислокация называется совершенной дислокацией; в
противном случае она называется несовершенной, или частичной,
дислокацией. Совершенные дислокации включаются в скольжение (где
нет никакого изменения в кристаллической структуре), а
несовершенные дислокации — в фазовые переходы (например, мартенситные
переходы, описанные в гл. 4). Совершенные дислокации расщепля-
Дислокации
221
Рис. 5.5. Картина краевой дислокации в Ba2Bi4Ti5018, полученная методом
HREM. Отметим большое значение b=5 mm [Hutchison et al., 1977].
ются на частичные дислокации с векторами Бюргерса Ьх и Ь2
(каждый <Ь) в нескольких случаях; этот процесс уменьшает
деформационную энергию, а также приводит к структурным изменениям
(например, введение дефектов упаковки). Необходимым условием
для такого расщепления является \Ъ±\2 + |Ь2|2< |Ь12.
Краевая дислокация ограничена в движении по своей плоскости
скольжения (консервативное движение), а скольжение из-за
движения дислокации также ограничено плоскостью скольжения.
Движение винтовой дислокации может охватывать плоскость, где она
начинается или движется к какой-либо другой плоскости, параллельной
линии дислокации (поперечный сдвиг). Если бы краевая дислокация
двигалась перпендикулярно плоскости скольжения, она требовала
бы, чтобы ряд атомов на краю дополнительной полуплоскости был
удален или же чтобы то же самое число вакансий мигрировало в те
же позиции; такой диффузный процесс называется подъем
дислокации. Движение дислокации обычно обозначается плоскостью сколь-
жения и направлением скольжения (вектор Бюргерса). Например, в
антрацене плоскость скольжения и направление скольжения
следующие: (010) (001 >; (100) (010). а в нафталине они такие: (001)
(010). Взаимодействие дислокаций (принадлежащих к различным
плоскостям скольжения) во время их движения ведет к порогам и
уступам или даже к вакансиям. Когда винтовая дислокация изги-
222
Новый взгляд на старую проблему
Рис. 5.7. Цикл Бюргерса для краевой (а)
и винтовой дислокаций (б). При движении
на определенное число размеров решетки
направо и вниз, а затем на соответствующее
число налево и вниз мы получаем
законченный путь в нижней части а; в верхней
части, содержащей краевую дислокацию,
цикл не замыкается, что показано
вектором Ь. В a b перпендикулярен линии
дислокации, тогда как в б — параллелен.
Рис. 5.6. Картина сетки дислокаций в кристалле антрацена, полученная методом
HREM (данные любезно предоставлены Г. М. Паркинсоном и Дж. М. Томасом,
Кембриджский университет).
бается и имеет краевой компонент, вакансии мигрируют к
изогнутому концу дислокаций, вызывая спиральную дислокацию. Когда идет
расщепление дислокации на плоскости (111) по выражению
(1/2)а<011> = (1/3)а(111) + (1/6)а(211),
(5.24)
та часть, которая имеет вектор Бюргерса 1/За (111), называется
сидячей дислокацией, или частичной дислокацией Франка. Другая
часть (1/6а(211 )) называется скользящей дислокацией, или частичной
дислокацией Шокли.
Дислокации легко размножаются в процессе пластической
деформации, несколько механизмов этого исследованы электронной
микроскопией. Дислокации разрушаются в процессе регенерации и
рекристаллизации во время отжига после пластической деформации.
Поскольку дислокации вызывают в металлах и других твердых
телах напряжения низкой текучести, упрочнение твердого тела
сопровождается или уничтожением дислокаций, или иммобилизацией их.
Дислокации обычно исследуются электронной микроскопией.
Для изучения дислокаций часто используются также декорирование
(например, дислокаций в кремнии медью) и химическое травление в
сочетании с оптической микроскопией. Плотность дислокаций (число
Планарные дефекты
223
дислокационных линий, проходящих через единицу поверхности в
кристалле) зависит от метода получения и термомеханической
истории образца. В то время как усы чистого металла растут почти без
дислокаций, тщательно выращенные монокристаллы обычно имеют
около 102 дислокаций на квадратный сантиметр; отожженные
поликристаллические металлы имеют плотность около 10 ~8 дислокаций
на квадратный сантиметр. В органических твердых веществах и
молекулярных кристаллах плотности дислокаций обычно низкие
(~ 104 см-2) в кристаллах, выращенных из газовой фазы, а в
кристаллах, выращенных из расплавов, эти значения высокие
(~ 106 см~2). Энергии дислокаций в органических твердых веществах
высокие, поскольку у них параметры решетки по величине на
порядок больше, чем в неорганических твердых веществах. Однако в
органических твердых веществах энергии образования точечных
дефектов намного ниже (~ 1 эВ). Если плотность дислокаций около
1010 см-2, число атомов в высокомодифицированной центральной
области дислокаций будет около 1018 см-3. Эти атомы находятся в
переполненных, разреженных или искаженных позициях. Средняя
избыточная энергия атомов, находящихся в таких позициях, может
быть порядка 20—40 кДж-моль-1. Эти высокие энергии изменяют
ход химических реакций в органических твердых веществах или
влияют на него. Кинетические барьеры, изменяя скорости реакций
(везде, где реакции контролируются кинетически), понижаются.
Реакции далее облегчаются, так как часть энтальпии реакции
обеспечивается дислокациями. Каталитические примеси могут мигрировать
предпочтительно к дислокациям, тем самым значительно увеличивая
скорости реакций на таких участках. Молекулы в ядрах дислокаций
(особенно винтовой дислокации) ориентированы отлично от молекул
в свободной от дислокаций области. Это очень часто вызывает
необычные стереохимические ориентационные упорядочения,
необходимые для прохождения реакции.
5.4. ПЛАНАРНЫЕ ДЕФЕКТЫ
Поверхность кристалла, или межфазная граница,
представляет собой планарный двумерный дефект. Есть значительная
разница в окружении атомов, ионов или молекул на поверхности и в
объеме кристалла (где они находятся в состоянии «совершенного»
порядка). Изменение в окружении может приводить к уменьшению числа
взаимодействующих соседей или к искажению полиэдров вблизи
атомов поверхности. Свободная энергия поверхностных атомов
выше, чем атомов в объеме кристалла. Поверхностная энергия
кристаллических твердых веществ обычно порядка 0,5—2 Дж-м-2. В
поликристаллических материалах имеются межзеренные границы между
различными зернами. Когда в материале присутствует более чем
224
Новый взгляд на старую проблему
одна фаза, неизбежно имеются поверхности раздела с
значительными межфазными энергиями.
Поверхность раздела между двумя фазами может быть
когерентной, некогерентной или полукогерентной. Если есть совершенное
соответствие в межатомных расстояниях между соприкасающимися
плоскостями, считается, что поверхность раздела когерентная.
Эти поверхности раздела нельзя наблюдать раздельно,
следовательно, они обладают очень низкими межфазными энергиями
(^ 0,015 Дж-м~2). Хорошие примеры когерентных поверхностей
раздела найдены в суперсплавах на никелевой основе, где плоскости
(100) Ni3Al образуют когерентные поверхности раздела с гранями
(100) гранецентрированной кубической (ГЦК) матрицы. Фаза
MgFe204, выделившаяся в MgO, образует когерентные поверхности
раздела. Причина здесь в том, что как в MgO, так и в MgFe204 атомы
кислорода кубически плотно упакованы и, следовательно,
выделившаяся фаза и матрица непрерывны. В случае кобальта, где
существуют структуры ГЦК и гексагональная плотнейшая упаковка
(ГПУ), когерентная поверхность раздела соответствует плоскости
(0001) гексагональной фазы и плоскости (111) кубической фазы.
Когда разница в межатомных расстояниях достаточно большая,
образуются полукогерентные поверхности раздела. Поверхности
раздела, связанные с эпитаксиальным ростом, обычно являются
полукогерентными и содержат мотивы дислокаций. Малоугловые границы
зерен также составляют полукогерентные поверхности раздела.
Энергия таких границ, которые также состоят из дислокаций, зависит
от относительных ориентации двух зерен. Граница с малым углом
наклона состоит из совокупности периодически расположенных
краевых дислокаций, как показано на рис. 5.8.
Малоугловая твист-граница получается из совокупности
винтовых дислокаций. Энергии границ между зернами обычно низкие,
и считается, что множество дислокаций, составляющих малоугловые
границы, образуются при отжиге. Есть особые ориентации, для
которых энергии межзеренных границ минимальны, а дислокации
такие, что векторы Бюргерса этих дислокаций ассимилируют
атомное несоответствие вдоль границы. Сетки таких неподходящих
дислокаций, называемых дислокациями van der Merwe, легко
идентифицируются трансмиссионной электронной микроскопией.
Упаковки ГЦК (ABC ABC.) и ГПУ (АВАВ...) являются двумя
обычными мотивами в твердых телах (см. разд. 1.4). Можно
выделить две ситуации, соответствующие образованию дефектов
упаковки: а) переход частичной дислокации типа Шокли (ABC ABC ABC ->■
->- ABC AB ABC ABC) и б) смещение неполной атомной плоскости,
образующее краевую дислокацию (част. Франка). Энергии дефектов
упаковки изменяются широко от одного твердого тела к другому.
В а-А1203 скольжение при 1200 К включает три различные
области дефектов упаковки, так как вектор Бюргерса расщепляется на
Планарные дефекты
225
Рис. 5.8. Дислокации на Рис. 5.9. Коинсидентная граница (001) в цеолите L.
границе с малым углом. Верхняя часть кристалла поворачивается по
отношению к нижней части на 32,2°, образуя тем самым
коинсидентную границу [Terasaki et al., 1984].
четыре части; область между двумя любыми частями
соответствует дефектам упаковки.
Слой, имеющий зеркальную плоскость симметрии (по
отношению к слоям по o6eiM сторонам), может быть образован переходом
частичных дислокаций Шокли, такой слой известен как граница
двойников. Двойникование (отражение) является важной
кристаллографической сперацией, кзторая связывает сложные структуры
с простыми (см. разд. 1.G). Двойникование наблюдается во многих
минералах, оно отвечает за совместное выделение некоторых из них
(кальцит, полевые шпаты). Мы можем также иметь планарный
дефект, включающий вращение одной части кристалла на конкретной
плоскости по отношению к той, у которой имеется ряд позиций в
решетке на поверхности раздела, общих со структурами обеих
сторон. Такой планарный дефект, коинсидентная граница, показан на
рис. 5.9. Антифазные домены составляют другой тип двумерного
дефекта; при переходе через антифазную границу заполнение подре-
шетки сменяется. Например, если АВ — упорядоченный сплав из
атомов А и В, занимающих соответственно подрешетки а и |3 по
одну сторону антифазной границы, по другую сторону А и В занимают
соответственно подрешетки (3 и а. Доменная граница в
ферромагнетиках, сегнетоэлектриках и родственных материалах похожа на
аптифазную границу, только спины или электрические диполи по
разным сторонам границы поляризуются в различных, но
эквивалентных низкоэнергетических направлениях.
Планарные дефекты соответствуют более высокоэнергетическому
состоянию по сравнению с объемом твердого тела, и, следовательно,
15 ч. II. Р. Рао, Дж. Гопалакришнан
226
Новый взгляд на старую проблему
такие примеси, как различные алиовалентные ионы или растворенные
атомы, притягиваются поверхностями раздела или границами зерен
способом, похожим на тот, который превалирует в дислокациях.
Например, ионы Са2+ мигрируют к границам зерен в
поликристаллическом сапфире. В металле с гранецентрированной кубической
решеткой атомная сегрегация имеет место в области ленточного
дефекта структуры с гексагональной плотыейшей упаковкой. Такие
атомные миграции ведут к понижению энергий.
5.5. УПОРЯДОЧЕННЫЕ ТОЧЕЧНЫЕ ДЕФЕКТЫ
И СВЕРХСТРУКТУРЫ
Сверхрешеточное упорядочение вакантных позиций или меж-
доузельных атомов в результате взаимодействий между ними все
больше подтверждается экспериментальными данными.
Сверхрешеточное упорядочение точечных дефектов найдено в галогенидах,
оксидах, сульфидах, карбидах металлов и в других системах; по
вопросу о взаимосвязи между таким упорядочением и нестехиомет-
ричностью написаны обширные обзоры [Anderson, 1974, 1984;
Anderson, Tilley, 1974]. Сверхрешеточное упорядочение точечных
дефектов найдено также в сплавах и в некоторых интерметаллических
соединениях [Gleiter, 1983]. Мы рассмотрим особенности некоторых
типичных систем, чтобы проиллюстрировать это явление, которое
сводит к минимуму значение изолированных точечных дефектов во
многих химически интересных твердых телах.
5.5.1. Допированные галогениды щелочных металлов
Растворимость галогенидов двухвалентных катионов в
галогенидах щелочных металлов сильно зависит от температуры.
Например, растворимость CdCl2 и МпС12 в NaCl составляет примерно
Зх10~2 мол. % при 650 К и примерно 2х10~5 мол. % при 450 К.
Благодаря поверхностной диффузии в таких допированных
кристаллах, выделение допентов может идти при низких температурах;
образование димеров и тримеров пар примесь — вакансия, как
предполагается, объясняет скорости таких процессов. Suzuki [1961]
охарактеризовал семейство галогенидов типа МХ2 • 6АХ, в которых
двухвалентные катионы и катионные вакансии выстраиваются в
сверхструктуру 2x2x2 в структуре галогенида щелочного металла
(АХ). Слои структуры а на рис. 5.10 чередуются со слоями
структуры каменной соли б так, что вакантные позиции и
компенсирующие примеси двухвалентного катиона занимают позиции второй
координационной сферы, а не ближайшие соседние позиции. Лег-
Упорядоченные точечные дефекты и сверхструктуры 227
Рис. 5.10. Сверхструктуры в фазах Сузуки.
а — слой сверхструктуры, где П — катионная вакансия,
а <8> — примесь двухвалентного катиона М; б — слой
каменной соли.
кость образования фаз Сузуки является
результатом их топологической совместимости
со структурой каменной соли. Исследования с компьютерным
моделированием [Corish, Quigley, 1982] показывают, что
образованные частицы зависят от относительных размеров катионов до-
пента и матрицы.
5.5.2. Халькогениды и карбиды металлов
Халькогениды Зс^-переходных металлов (за исключением Мп)
имеют широкие области стехиометричности и непрерывные ряды фаз
с общими структурными типами, родственными структурам Cdl2
и NiAs. В промежуточной области составов (между МХ2 и MX)
найдено сверхструктурное упорядочение, причем занятые и пустые
октаэдрические позиции имеют двумерный порядок в гексагональном
мотиве позиций. Многие сверхструктурные фазы претерпевают
превращения порядок — беспорядок. Не во всех халькогенидных
системах наблюдается непрерывная область составов. Так,
Cr0)67S(Cr2S3) — высший состав в системе Сг—S, тогда как в теллу-
ридной системе такой состав — МТе2. Система Ni2-a.Te2 существует
между Nilt84Te2 и NiTe2; когда х < 0,4, структура является
дефектной структурой типа NiAs, а при х > 0,4 структура типа Gdl2. Это
справедливо для материалов, отожженных при низких
температурах; при высоких температурах, однако, вообще нет дальнего
порядка.
В системе Ti—S идентифицирован ряд фаз между TiS и TiS2,
ксе они имеют широкие области стехиометричности. Например, для
TiS и TiS2 это соответственно составы Ti0>94S—Ti1>o3S и Ti0i'62S—
—Tio,55S- В этих сульфидах иррациональная часть позиций может
быть занята в металл-дефицитных слоях и упорядочение
сверхрешетки будет неполным. Например, в Ti5S8 идеальная
последовательность слоев была бы ...STi1)0STi0)?STi1)0STi0>2... . Хотя в таких
сульфидах не наблюдается хорошо определенной сверхструктуры,
существует ближний порядок с высокой степенью упорядочения, что
доказывается данными по диффузному рассеянию между брегговскими
рефлексами от основной гексагональной подъячейки.
Исследованы размытость полос и несоразмерность сателлит-
ных максимумов для составов TiSb5—TiS1)73 и предложена модель
с двумя типами упорядочения ближнего порядка, где взаимодействие
между атомами Ti препятствует заселению ближайших соседних
позиций, но позволяет заселение второй или третьей координацион-
15*
228
Новый взгляд на старую проблему
Рис. 5.11. Упорядоченная сверхструктура
вакантных позиций в катионных слоях халько-
генидов со структурой NiAs. Часть занятых
позиций: а — 1; б — 3/4; в — 1/2; г — 1/4.
• d • d • • о • п • пых сфер [Moret et al., 1977]. Идентифи-
• d • d ddod цированы микродомены с двумя типами
• о • □ • d • а • □ локального порядка.
• о • а о а а а уз системах Сг^.Х^ —S, Se, Те) су-
• d • а • • d • d • ществует ряд сверхструктуриых фаз с
в г узкими областями стехиометричности.
Халькогениды находятся внутри области
составов 0 ^ х ^ 0,33, причем типичные из них — Cr7S8, Cr3S4 и
Cr5S8 с последовательностью слоев ...SCrli0SCrOf75S...,
...SCr110SCr0i5S... и ...SCr, 0SCr0 25S... соответственно; в Cr2S3
последовательность — ...SCr1>0SCr0)33S... . Во всех этих сульфидах
найдено строгое чередование заполненных и дефектных слоев с
хорошим дальним порядком, лишь Cr7S8 проявляет некоторое разу-
порядочение пустых позиций. На рис. 5.11 показаны типичные
упорядоченные сверхструктурные упаковки вакантных позиций.
Подобные упорядоченные фазы известны в системах V1-a. и MXVS2 (M—
—Fe, Co, Ni). Известно, что в сульфидах железа происходит
двумерное сверхструктурное упорядочение. Так, в Fe7S8(4C)
упорядоченная структура определяется строгим чередованием заполненных
и дефектных металлических слоев .. .SFe, i0SFe0t 75SFeli0SFe0.75S...
с пустыми октаэдрическими позициями в незаполненных
металлических слоях IBertant, 1950]. Более сложные последовательности
найдены в Fe9Sl0 (5C) и FenS12 (ОС); здесь символ пС означает число
слоев атомов железа в повторяющейся последовательности. Во всех
этих сульфидах вблизи 370 К вакантные позиции в слое железа
становятся статистически распределенными [Nakazawa, Morimoto,
1971]. В сульфидах железа предложен общий базис для
упорядочения структур и для иррациональных сверхрешеточных кратиостей,
приводящий к регулярной (но необязательно соразмерной)
модуляции [Kato, Kitamura, 1981]; следствием этого типа упорядочения
является распределение вакантных позиций по спирали с
переменным шагом.
Моносульфиды d1- и с?2-переходных металлов со структурой
каменной соли обладают катионодефицитной нестехиометричностыо.
Например, в системе Zr—S известны составы между Zr0i63S и ZrS;
структурные исследования Zr077S показывают, что
сверхструктурное упорядочение не согласуется с простым типом распределения
катионных вакансий [Conrad, Franzen, 1970]. Моноклинная структура
имеет 32 позиции для 23 катионов лишь с одной полностью
заполненной двукратной позицией и другими эквивалентными позициями
с различными степенями частичной занятости. Кроме того, в любом
Упорядоченные точечные дефекты и сверхструктуры 229
данном слое (111) кубической подъячейки (гексагональный слой
катионных позиций) заполнение позиции модулируется в каждом
ряду. В системе Sc—S и Sc2S3 дает сверхструктуру (2х /2x3 /2)
катионных вакансий в матричной структуре каменной соли,
причем вакантные катионные места во вторых соседних позициях
выстраиваются по спиралям вдоль оси Ь [Dismukes, White, 1964].
Сульфид скандия испаряется конгруэнтно выше 1970 К вблизи состава
Sc0)8S. Этот сульфид полностью разупорядочивается при высоких
температурах, но обнаруживает сверхструктурное упорядочение при
отжиге (при 970 К), с вакантными позициями, упорядоченными в
чередующиеся слои (111) катионных позиций. Ниже 570 К
наблюдается второй тип сверхструктурного упорядочения (включающий
упорядочение вакантных позиций внутри дефектного катионного
слоя); эта сверхструктура несоразмерна [Franzen et al., 1983].
Упорядоченные сверхструктуры Zr^^S, Sc^^S и Lu^^S исследованы в
рамках теории Ландау [Franzen, Folmer, 1984].
Неметаллические позиции в карбидах переходных металлов
заполняются лишь частично, карбиды вообще проявляют нестехиомет-
ричность. Сверхструктуры, обусловленные упорядочением
вакантных позиций, найдены в карбидах ванадия; V6C5 и V8C7 являются
двумя такими фазами. Эти фазы при высоких температурах
подвергаются переходам порядок — беспорядок и, как ни странно, дают
ряды стехиометричных составов [Emmons, Williams, 1983]. Фазы
с составом между V6C5 и V8C7 имеют два фазовых перехода и
обладают значительной степенью ближнего порядка. Ближний порядок
может сохраниться даже выше температуры перехода порядок —
беспорядок. Сверхструктурное упорядочение отмечено также в
нескольких других карбидах (например, Nb6C5).
5.5.3. Оксиды металлов со структурой каменной соли
TiO и VO — два важных оксида, которые заслуживают
особого внимания из-за большой доли в них дефектов (вплоть до 20 %
вакансий). В обоих оксидах есть фазовый переход между низко- и
высокотемпературной формами. Высокотемпературная форма TiO
с усредненной структурой каменной соли охватывает широкую
область составов между TiO065 и Ti0125 при 1770 К;
низкотемпературная форма (моноклинная) с узкой областью составов образуется
ниже 1270 К iWatanabe et al., 1970]. Параметр решетки элементар-
о
пой ячейки типа NaCl — 4,182 А. Сверхструктура, соответствующая
моноклинной элементарной ячейке с а' = 5,855 А = у 2д, Ъ' =
- 9,340 А = /5а, с' = 4,142 А « а и у = 107°32' « cos^(l//10),
которая показана на рис. 5.12, а, может быть преобразована в иска-
о
жениую кубическую структуру с а = 4,200, b = 4,43, с = 4,142 А
230
Новый взгляд на старую проблему
Рис. 5.12. а — упорядоченные
дефекты в моноклинном TiO«
• (Ti5/605/6); б —
упорядоченные дефекты в нестехиометри-
ческом (ромбическом) TiO^;
в — когерентное прорастание
а и б вдоль плоскостей (120)
структуры каменной соли.
Линиями указаны границы
элементарных ячеек
сверхструктур [Anderson, 1984].
t T' о о -jvo цУП
и у = 89,9°; эта структура, как оказывается, подходит для оксидов
TiO0t9—ТЮ1Л. При большем отклонении от стехиометричности
сверхструктура соответствует ромбическому искажению, как
показано на рис. 5.12, б. Орторомбические и моноклинные элементы
могут прорастать в исходную структуру каменной соли, как показано
на рис. 5.12, в. Дифрактограмма для разупорядоченной формы ТЮ
показывает диффузное рассеяние, что наводит на мысль о наличии
некоторого ближнего порядка в распределении вакансий [Terauchi,
Cohen, 1979]. Показано, что не может быть совершенного дальнего
порядка в низкотемпературной форме, по крайней мере для
температур отжига, близких к фазовому переходу ITerauchi et al., 1978].
VO также обладает высокой долей вакансий, точно так же как
ТЮ, но упорядочение дефектов при низкой температуре, вероятно,
совершенно отлично от такого же упорядочения в
низкотемпературном ТЮ. Следовательно, VO реально не смешивается с ТЮ [Loeh-
man et al., 1968]. Сверхструктура в VO образуется только в области,
богатой кислородом, где кислородная подрешетка почти заполнена,
а некоторое количество атомов ванадия занимают тетраэдрические
позиции; типичные исследованные составы — V52064 и V244O320. Ди~
фракционные исследования показывают присутствие ванадия в тет-
раэдрических позициях. Каждый тетраэдрический катион в качестве
ближайших соседей имеет четыре вакантных октаэдрических
позиции (рис. 5.13), а образованный таким образом^ кластер, будучи
топологически похожим на структуру каменной соли, приводит
к сверхструктуре 2/2x2)^2 X 2. В V52064 все позиции решетки
(отличные от позиций кластеров дефектов) заняты так, как в
структуре каменной соли, но в VOb30 некоторое количество
октаэдрических позиций частично занято; в сверхрешетке V52064 все вакантные
октаэдрические позиции кластерированы и совсем нет свободных
вакансий [Andersson, Gjonnes, 1970; Morinaga, Cohen, 1979].
Образцы VO^, закаленные после высоких температур, на картине
электронной диффракции имеют диффузное рентгеновское рассеяние и
сателлиты [Morinaga, Cohen, 1979; Rao et al., 1976], что
обусловлено ближним порядком, включающим, вероятно, тетраэдрические
атомы ванадия. Заслуживает внимания то, что сверхструктура фазы
Упорядоченные точечные дефекты и сверхструктуры 231
^>
^9
Рис. 5.13. 4 : 1-кластер одного тетраэд- Рис. 5.14. Кластер (дефектов) Ко-
рического катиона и четырех вакантных ха — Коуэна в Fe О (вюстит).
октаэдрических позиций в одном октан- f/j _ теТраэдрические атомы Fe; о - ани-
те структуры каменной соли. оны; □ — октаэдрические вакансии Fe.
VOb30 является результатом упорядочения кластеров с характерной
структурой, образованной скорее путем локальной перегруппировки
матричной структуры, чем путем упорядочения простых дефектов
[Anderson, 1984]. Такие кластеры ответственны за нестехиометрич-
ность в монооксидах переходных металлов (после ванадия), таких
как Fe^a-O, и в нестехиометрических соединениях, относящихся
к флюоритной структуре. Другой момент, который следует отметить,
состоит в том, что поскольку ТЮ и VO обладают металлическими
свойствами, делокализованные электроны могут экранировать
взаимодействие дальнего порядка и могут существовать изолированные
точечные дефекты. Однако в более ионных оксидах, таких как
Fej.^O, кулоновские эффекты будут сильны; окружение точечных
дефектов в значительной степени модифицируется, что
благоприятствует образованию хорошо определенных кластерированных
вакансий.
Вюстит всегда катионнодефицитен и имеет состав Fe085O—
Fe0)95O. Реакция образования вюстита записывается следующим
образом:
2 Fe2+ + 1/2 02 -> 2 ¥еЦТ + VFe,0KT + О2
(5.25)
Поскольку вюстит имеет структуру NaCl, по-видимому, Fe3+ и вновь
созданные вакансии остаются в октаэдрических позициях. По
данным нейтронной дифракции сделан вывод, что значительная часть
ионов Fe3+ занимает тетраэдрические позиции по реакции [Roth,
1960]
-Г £<экт т Vj > г еТ(>Тр
+ Уре.окт-
232
Новый взгляд па старую проблему
Поэтому предложена [Roth, 1960] модель комплексов (VFe—Fe^ip—
— Yfe), в которой Fe3+ окружен кислородом тетраэдрически. Однако
на основании данных рентгеновских кристаллографических
исследований закаленного Fe0f9O высказано предположение, что комплекс
дефектов столь же велик, как элементарная ячейка FeO, состоящая
из четырех тетраэдрических ионов Fe3+ в чередующихся тетраэдри-
ческих позициях (рис. 5.14) из 13 вакансий на позициях Fe2+ [Koch,
Cohen, 1969]. Такой кластер Коха — Коуэна соответствует
химической формуле FeO, но если рассматривать общий заряд
комплекса, включающий эффективные заряды на вакансиях, то он (кластер)
является отрицательно заряженной элементарной ячейкой с апти-
сфалеритной структурой. Поскольку упаковка иона кислорода
остается ненарушенной, эти кластеры дефектов могут существовать с
когерентными поверхностями раздела с матрицей. Отношение
вакансий для тетраэдрических ионов Fe3+ в кластере Рота — 2/1 = 2,
тогда как в кластере Коха — Коуэна — 13/4 = 3,25. На основании
экспериментов по нейтронной дифракции выявлено, что в
температурном интервале 1073—1473 К и в нестехиометрической области
составов Feo90—Fe095O отношение октаэдрических вакансий к тет-
раэдрическому Fe3+ было почти 3,25, а не 2 [Cheetham et al., 1971, а].
Обнаружены кластеры, где это отношение составляет 2,5 [Gavar-
ri, 1978].
Теоретическими расчетами [Catlow, Mackrodt, 1982] показано,
что такие кластеры образуются с общим понижением свободной
энергии. Энергии связи в кластерах дефектов изменяются между
1,98 эВ (кластер 4 : 1) и 2,52 эВ (кластер 8 : 3), для кластера 13 : 4
Коха — Коуэна это значение составляет 2,1 эВ. Кластер 16 : 5,
который является элементом шпинельной структуры Fe304, также
совершенно устойчив (энергия связи 2,38 эВ). Это говорит о том,
что он может быть промежуточным веществом при диспропорциони-
ровании вюстита. Исходя из совершенной структуры каменной соли,
окисление по всей нестехиометрической области может быть надежно
представлено следующими стадиями [Anderson, 1984]:
изолированные вакансии ->- дипольные ассоциаты ->- кластеры 4:1-*-
кластеры 6:2, 8:3, 13:4 и аналогичные сложные кластеры дефектов ->
—>- кластеры 16 : 5 с общими углами -> Fe304 (обратная шпинель).
Такие кластеры могут существовать также в системе Мп^^О;
рассчитанные энергии связи в кластерах (см. [Catlow et al. ]) ниже в
Мп1_д:0, чем в Fe1_xO. Свободные точечные дефекты, если вообще
они есть, могут существовать в Мп^^О, а не в Fet_xO. Строение
вюстита при высоких температурах еще не выяснено, эта проблема
исследована при изучении несоразмерных сателлитных пиков на
дифрактограммах [Bauer et al., 1980]. Оказывается, что нет
никаких простых кластерных частиц, диспергированных в матрице
структуры каменной соли, которые могли бы объяснить структуру при
высоких температурах. Для описания структуры, по-видимому, луч-
Упорядоченные точечные дефекты и сверхструктуры 233
ше подходит функция, которая может дать вероятность нахождения
Fe?ip и VFe окт вместе со смещением смежных атомов.
Несоизмеримые модуляции приводят к иррациональной сверхструктуре,
причем отношение VFCt0KT/Fe?t*p также, как и периодичность
модуляции, усредняется по всему кристаллу.
5.5.4. Флюоритоподобные твердые тела
Флюоритный структурный тип объединяет множество
производных ст флюорита структур с избытком или дефицитом анионов.
Катиоиная подрешетка остается, по существу, совершенной.
Элементарная ячейка соединения МХ2, имеющего структуру флюорита,
соответствует гранецентрированной кубической плотной упаковке
ионов М2+, причем все восемь тетраэдрических позиций заполняются
ионами X". Междоузельная позиция с высокой симметрией
находится в центре. Теперь признано, что в этих структурах беспорядок
возникает главным образом или от анионных вакансий, или от меж-
доузельных анионов. Широко исследованными среди них являются
диоксиды, либо анионодефицитные M02_x, либо анионоизбыточные
М02+х. Оказывается, что природа комплексов дефектов в этих
флюоритах, по существу, не зависит от изучаемой химической системы.
В соединениях M02_x кластеры дефектов соответствуют прочиосвя-
занным вакансиям вдоль телесной диагонали (111), как показано
на рис. 5.15; центральный катион является шестикоордипированным
и окружен шестью семикоординированными катионами IThornber,
Bevan, 1970]. Этот кластер дефектов часто называют кластером
Бевана. Пс-видимому, несколько оксидов cocTaiBa M7012 (например,
Рг7012) и промежуточных фаз в системе Y203—YOF, а также Y203,
стабилизированный Zr02, содержат кластеры Бевана. Поскольку
геометрия этих кластеров допускает четыре различные
эквивалентные ориентации, сна легко
приспосабливается к флюоритной
структуре и отвечает за
широкий ряд устойчивости так
называемых а-фаз (МОЬ71—М02).
Таким образом линейные ряды
кластеров вдоль (111)
исходной структуры флюорита
образуют мотив в
гомологическом ряду Ргп02п_2, а он
прорастает с увеличением доли
флюоритной структуры в
высших оксидах.
. о Катион
Рис. 5.15. Кластер Бевана в оксидах ш Пустая анионная
МО со структурой флюорита. позиция
234
Новый взгляд на старую проблему
1/2а„
Рис. 5.16. Кластеры Уиллиса
В U02+*-
О — кислород; • — атомы урана
в структуре U02. Кластер дефектов
2:2:2 состоит из двух междо-
узельных атомов кислорода (•)
и двух обычных атомов кислорода,
смещенных от их идеальных
позиций (П) к новым междоузельным
позициям Ш). Кластер дефектов
2:1:2 должен состоять из одного
междоузельного атома кислорода
и двух обычных атомов кислорода,
смещенных от их идеальных
позиций [Anderson, 1972b].
Оказывается, что
оксиды состава М02+г,
содержащие избыток
кислорода, размещают избыток
О2- в
высокосимметричных междоузельных
позициях, если степень не-
стехиометричности
низкая. Но когда иестехиометричность высокая, наблюдаются
значительные искажения, а междоузельные ионы кислорода более не
занимают позиции 1/2 1/2 1/2 во флюоритиой структуре. Так, по
данным нейтронной дифракции на U409 установлено, что
дополнительные ионы кислорода в элементарной ячейке U02 в значительной
степени смещены от позиции 1/2 1/2 1/2 в направлении (110); более
того, два флюоритных кислорода, ближайших к междоузельному
кислороду, также смещены в направлении (111), образуя две
вакансии. Кластер из двух вакансий одного междоузлия одного вида и
двух междоузлий другого вида известен как кластер 2:1:2
Уиллиса [Willis, 1904]. Более общий кластер 2:2:2 включает другое
междоузлие в обычно незанятой октаэдрической флюоритной
позиции, так что имеются две вакансии, два междоузлия, каждое двух
видов. Считается также, что расширенные кластеры Уиллиса,
такие как 3:4:2 (что означает число вакансий : междоузлия I
типа : междоузлия II типа) существуют в нестехиометрических
материалах типа М02+х. Различные типы кластеров Уиллиса в U02+x,
вместе со схемой флюоритной сверхъячейки показаны на рис. 5.16.
Аналогичные кластеры дефектов найдены в твердых растворах
редкоземельный фторид — щелочноземельный фторид, таких как
система CaF2—YF3 ICheetham et al., 1971b]. Поскольку катионы в
гранецентрированной кубической упаковке не меняются,
кластеры дефектов срастаются с исходной структурой когерентно. Gett-
mann, Greis [1978] обнаружили сверхструктурные фазы состава
Ca10Y5F35, Ca9Y5F33 и Ca7+yY6_j,F31_y, принадлежащие к
гомологическим рядам MnF2n+5 с п = 15, 14 и 13 соответственно. В
системе YbF2—YbF3 идентифицированы [Greis, 1977] сверхструктуриые
Кристаллографический сдвиг
235
фазы Yb3F7, Yb14F33 и Yb13F32_v. В минерале Ca14Y5F43, который
является членом этого гомологического ряда, анионы
систематически смещены, образуя новый тип кластера М1бХ37, а катионы
близки к позициям флюоритной структуры [Bevan et al., 1982];
кластер М1вХ37 мог бы дать альтернативную интерпретацию
флюоритов с избытком аниона, но не объясняет известные
структурные особенности и спектроскопическую информацию [Laval, Frit,
1983]. Расчеты энергии дефектов также не говорят в пользу этого
кластера [Catlow et al., 1983]. По-видимому, природа кластеров
дефектов в анионоизбыточпых флюоритах не выяснена полностью.
В таких системах, как CaO—Zr02 и Y203—Zr02, наблюдаемое
поведение проводимости не поддерживает модель простой
изолированной вакансии. Согласно рентгеновским данным, а также данным
по нейтронной и электронной дифракции, имеются эффекты сильного
локального упорядочения, но природа ближнего порядка не ясна.
Было сделано также предположение о возможном образовании
микродоменов упорядоченных сверхструктур. В анионоизбыточпых
структурах типа YF3—YOF и Zr02—Nb205 избыток междоузельных
анионов начинает скапливаться на определенных
кристаллографических плоскостях, причем граница движется наружу до тех пор,
пока эта особенность не распространяется по всему поперечному
сечению кристалла.
5.6. КРИСТАЛЛОГРАФИЧЕСКИЙ СДВИГ
В предыдущем разделе мы рассмотрели, как дефекты в не-
стехиометрических соединениях ассимилируются в решетке хозяина
как структурные элементы, так что не являются больше точечными
дефектами в обычном смысле и более не дают никакого вклада в
диффузию и проводимость. Важным способом приспособления анионо-
дефицитной нестехиометричиости в определенных оксидах
переходных металлов является исключение точечных дефектов так
называемым кристаллографическим сдвигом, обозначаемым cs [Anderson,
1972а; Anderson, Tilley, 1974]. Термин «кристаллографический
сдвиг» впервые был применен Уодсли для объяснения структур
гомологических рядов промежуточных оксидов W, Mo, Ti и V,
обнаруженных Магнели с сотрудниками. Поскольку точечные дефекты
исключаются посредством cs, процесс по отношению к позициям
решетки является «не сохраняющим»; как следствие cs —
уменьшается число анионных позиций по сравнению с исходной структурой.
Принцип cs лучше всего понятен на примере структуры Re03.
Структура Re03 построена из октаэдров Re06 обобщением всех их вершин.
Проекция структуры Re03 вдоль одной из ее кристаллографических
осей показана на рис. 5.17, а. Когда в структуру вводится дефицит
анионов, анионные вакансии исключаются путем сдвига одной части
236
Новый взгляд на старую проблему
Рис. 5.17. а — структура Re03, имеющая
кислородные вакансии, которые исключаются при
кристаллографическом сдвиге; б —
структура после кристаллографического сдвига.
структуры относительно другой вдоль
определенного кристаллографического
направления. При
кристаллографическом сдвиге (120), показанном па
рис. 5.17, плоскость позиций кислорода
в исходной структуре исключается, приводя к группам октаэдров,
которые имеют общие грани в плоскости кристаллографического сдвига.
Плоскость, в которой октаэдры соединяются по-разному, составляет
плоскость сдвига (рис. 5.17, в). Направление и величина смещения
исходной структуры вдоль плоскости сдвига определяются вектором
сдвига. Таким образом, плоскость сдвига, показанная на рис. 5.17,
обозначается как 1/2 (110) (120). Только если вектор сдвига имеет
компоненту, перпендикулярную плоскости сдвига, возможно
исключение анионных позиций. Наличие такого планарного дефекта в
кристалле дает хорошую четкость изображения на микроснимках,
полученных методом высокоразрешающей электронной микроскопии
(HREM), так что при изучении структур с кристаллографическим
сдвигом этот метод становится бесценным. На рис. 5.18 для
наглядности показана картина кристаллографического сдвига (120) в
кристалле W03_x, полученная методом HREM. Кристаллографический
сдвиг дает способ согласования анионодефицитной нестехиометрич-
ности без введения точечных дефектов и без изменения координации
катионов; анионная координация, однако, в плоскости
кристаллографического сдвига изменяется. В структуре Re03
кристаллографический сдвиг может быть не только по плоскостям (120), а также
и по плоскостям (Т30), (Г40) и т. д., а также по плоскостям (001),
хотя кристаллографические сдвиги по плоскостям (120), (130)
являются более обычными. В структуре рутила кристаллографический
сдвиг вообще имеет место по плоскостям (121) и (132), включающим
октаэдры с общими гранями.
Изолированная плоскость кристаллографического сдвига или
случайный набор плоскостей кристаллографического сдвига,
известные как дефект Уодсли, также приводят к нестехиометричности.
Регулярно повторяющиеся плоскости кристаллографического сдвига
приводят к гомологическим рядам стехиометричных промежуточных
фаз. Появление в кристалле таких эквидистантных плоскостей
кристаллографического сдвига указывает на кооперативный механизм.
Формула фазы с кристаллографическим сдвигом зависит от индекса
плоскости кристаллографического сдвига, так же как и от ширины
пласта исходной фазы между плоскостями кристаллографического
Кристаллографический сдвиг
237
Рис. 5.18. Картина решетки с кристаллографическим сдвигом (120)
в кристалле WO по данным HREM [Iijima, 1975]. Видно, что
расстояние между октаэдрами W06 составляет 3,8 Л.
сдвига. Если МОа — формула исходной граничной фазы, формула
гомологического ряда оксидов, получающихся в результате
кристаллографического сдвига, может быть представлена в виде МпОап_т,
где п — ширина (число октаэдров) исходной структуры между
плоскостями кристаллографических сдвигов, am — число исключенных
анионных позиций. Для семейства фаз Re03 с кристаллографическим
сдвигом мы можем показать, что в плоскости кристаллографического
сдвига (120) теряется один кислород на каждую группу из четырех
октаэдров с общими ребрами; в плоскости кристаллографического
сдвига (130) теряются два кислорода на каждую группу из шести
октаэдров с общими ребрами; в общем виде (т — 1) атомов
кислорода теряется в плоскости кристаллографического сдвига (1т0).
Если бы плоскости {1яг0} образовывали упорядоченную упаковку
МОз-х, общая формула гомологического ряда была бы Mn03n_(m_j).
Wn03n_j и Wn03n_2 представляют два гомологических ряда оксидов,
полученных из исходной структуры Re03 на основе
кристаллографических сдвигов (120) и (130) соответственно. Типичные
гомологические ряды оксидов с кристаллографическим сдвигом приведены
в табл. 5.2.
238
Новый взгляд на старую проблему
Таблица 5.2
Типичные гомологические ряды оксидов с кристаллографическим сдвигом *
Исходная
структура
wo3
моо3
Ti02
Плоскости сдвига
{120} Re03
{130} Re03
{120} Re03
{311}Мо03
{121} рутил
{132} рутил
Формула
' wno3n_1
wno3n_2
(п = 20, 24, 25, 40)
Mon°3n-l
(п = 8, 9)
Моп°зп-2
(п = 18)
Tin02n-i
(4 < п < 10)
Ъп02п-1
(16<м<37)
Приблизительная область
составов
wo3-wo2]93
wo2i93-wo2,87
—
ТЮ1,75-ТЮ1,90
Ti01)9o-Ti0li9375
* Кристаллографический сдвиг с одним набором непересекающихся плоскостей сдвига.
Фазы с кристаллографическим сдвигом (табл. 5.2) можно
рассматривать как трансляционные модуляции исходной структуры,
причем границы трансляции являются плоскостями
кристаллографического сдвига (см. разд. 1.0). Хотя в литературе описан ряд
твердых тел с плоскостями кристаллографического сдвига, лишь
недавно появилось понимание механизма образования и
упорядочения плоскостей кристаллографического сдвига [ТШеу, 1980; CatJow,
James, 1980]. По-видимому, упругая деформация играет решающую
роль в упорядочении плоскостей сдвига, и как континуальные, так
и атомистические теории ценны для понимания этой интересной
проблемы. Очевидно, для стабилизации этих структур необходимой
является экстенсивная релаксация вблизи плоскостей сдвига.
Интересная особенность, которую следует отметить в связи
с кристаллографическим сдвигом, состоит в том, что даже очень
слабо восстановленный рутил (Ti01)997) имеет плоскости
кристаллографического сдвига. В медленно охлаждаемых образцах Ti02_x. с 0,0 <
< х ^ 0,01 пары плоскостей кристаллографического сдвига
последовательно выделяются и разделяются [Blanchin et al., 1984]. Когда
0 < х ^ 0,0035, наблюдаются новые дефекты-пластиночки {100}
вместе с плоскостями кристаллографического сдвига; это можно
понять в рамках междоузельных катионных дефектов IBursill et al.,
1984]. В W03_x., кроме плоскостей кристаллографического сдвига,
наблюдаются гексагональные туннели и пентагональные бипирами-
дальные колонки. Проанализированы внешняя и/или внутренняя
Блочные структуры
239
природа обширных дефектов в ТЮ2_Х и W03_x вычерчиванием
контуров Бюргерса непосредственно на электронограммах высокого
разрешения [Bursill, Smith, 1984]. Это исследование имеет значенье
при определении природы малых дефектов в этих оксидах.
Цитируемые авторы получили также электронограммы высокого разрешения,
показывающие частичную границу дефектов из анионных вакансий.
5.7. БЛОЧНЫЕ СТРУКТУРЫ
Для описания переменного отношения металл : кислород в
бинарных и некоторых тройных оксидах ниобия принят новый
структурный принцип. Структуры этих оксидов можно получить из
Re03 двумя наборами почти ортогональных кристаллографических
сдвигов IWadsley, Anderson, 1970; Anderson, Tilley, 1974]. Два
набора повторяющихся плоскостей сдвига, которые проходят через
весь кристалл, сращивают структуру Re03 в прямоугольные блоки
(или колонки) октаэдров тХп в поперечном сечении и бесконечной
длины в одном направлении (направление Ь). Блоки структуры Re03
имеют общие ребра с идентичными или похожими блоками. Атомы
металла в получающейся структуре сохраняют октаэдрическую
координацию, но они находятся на двух уровнях (z = 0 и z = 1/2),
причем смещаются на 1/2 диагонали октаэдра в соседних блоках.
Кислородная подрешетка получающейся структуры та же самая,
что и в Re03, но координационное число кислорода разное: 1 —
в необобществленных вершинах блока; 2 — с внутренней стороны
блока; 3 — на ребрах и 4 — в позициях общих вершин. Блоки могут
связываться между собой тремя различными путями: 1) вершины
могут быть раздельными, создавая тетраэдрические позиции для
катионов; 2) ребра узловых октаэдров могут сочленяться на том же
самом уровне или 3) блоки могут образовывать ленты
перекрыванием общих ребер между блоками на том же самом уровне.
Соединения типа 2 и 3 через вершины связывают блоки на любом одном
уровне в группы из р блоков (р = 1, 2 или оо). Данный набор
блоков можно представить символом (тХп)р. Если р = 1 или 2, на
свободных вершинах создаются тетраэдрические катионные позиции.
Состав блока (тХп)р дается формулой Mmn7,+ 103mnp.p(mfл)+4- Для
структуры, включающей два или более отдельных блоков типа
г, /, ..-, химическая формула была бы
Тот факт, что геометрические параметры m, n и р могут изменяться,
делает возможным чрезвычайно малые вариации в отношении
металл : кислород, которые приспособлены перестройкой блочной
структуры, сохраняющей совершенный порядок. Более интересно
2
240
Новый взгляд на старую проблему
Рас. 5.19. Схема образования блочной структуры Н —Nb205 из структуры
Ro03 посредством кристаллографического сдвига по плоскостям (209) и
(601). Блоки (3x4)!+ (Зх5)а в структуре II —Nb205 показаны на схеме
справа [Anderson, Tilley, 1974].
то, что можно иметь различные блочные структуры для одного и
того же самого состава. Nb2Or>, например, существует в
четырнадцати различных полиморфных модификациях, десять из которых,
невидимому, обладают блочными структурами. Структура II—Nb205
наиболее устойчивой высокотемпературной полиморфной
модификации получается из структуры Re03 путем повторяющегося
кристаллографического сдвига в двух почти ортогональных направлениях,
как показано на рис. 5.19; плоскости кристаллографического едни-
га, используемые здесь,— (209) и (001). Повторяющаяся блочная
структура обозначается (ЗхА)1 -\- (Зх5)оо. Изображение решетки
Н —Nb205 показано на рис. 5.20. Между Nb02>4167 и NbO2>f000
идентифицирован ряд блочных структур, состоящих из различных типов
блоков: например, (3x4)^— в Nb12029, (3x4)^ -f (3x3)j— в Nb22054
и Nb470116 и (Зх4)2—в Nb25062. Кроме бинарных оксидов ниобия,
блочные структуры также хорошо охарактеризованы в системах
Ti02—Nb205 и W03—Nb205 в области составов M02654— М02>;ш.
В литературе есть публикации с описанием блочных структур
IWadsley, Andersson, 1970; McConnell et al., 197G], они исследованы
методом HREM [Gruelm, Mertin, 1980].
Следует отметить, что в блочных структурах возможны дефекты,
приводящие к нестехиометричности. В фазах с блочной структурой
известны как дефекты У од ели, так и точечные дефекты. Нестехиомет-
ричность вводится впедрением рядов или колонок блоков различного
размера, которые геометрически совместимы с матричной
структурой. Так, в ряду Nb02t48—NbO250 структурные единицы ячейки
Nb25062 (3x4)^ срастаются со структурой (3 Х4)х + (3 х5)оо Н —
—Nb205. Более интересно, что точечные дефекты привлекаются для
объяснения нестехиометричности в определенных блочных структу-
Блочные структуры
241
pax. Германий-пиобиевый оксид, GeO2-0Nb2O5, является типичным
примером этого. Фаза, несомненно, изоструктуриа с PNb9025,
блочная структура — (3x3)!. Состав, однако, не совместим с этой
структурой, если нет кислородных вакансий или междоузсльных катионов
или и того, и другого. Определение кристаллической структуры
показало, что позиции кислорода заняты, все октаэдрические кати-
онные позиции внутри блоков также полностью заняты. Избыточные
♦ #♦*♦*# : ;■;
;♦♦♦.♦'♦♦
за ♦♦♦♦■; '•• •" : 1 !,5^
.. *&i-,X у,.•.■:■■•>:•■ . . . ■ ••.■■.;Й5"
Рис. 5.20. а — картина решетки Н — Nb205,
имеющей два блока различной величины [Allpress,
Sanders, 1973]; б — идеализированная
структура.
"Ь Ч. Н. Р. Рао, Дж. Гопалакришнан
242
Новый взгляд на старую проблему
катионы располагаются в каналах, образованных углами колонок*
Внутри этих каналов доступны как тетра-, так и октаэдрические
позиции, они лишь частично заняты. Относительное количество
катионов в тетра- и октаэдрических позициях определяет
стехиометрию. Внутри любого одного канала тетра- и октаэдрические катионы
могут быть упорядочены, но корреляция между упорядочением в
соседних каналах незначительная. Поскольку нет никакого
доказательства сверхрешеточного упорядочения в германий-ниобиевом
оксиде, статистическая модель с симметрией 14/тп, как и для PNb9025l
оказывается подходящей. Раман-спектр Ge02-9Nb205 согласуется
с моделью случайного заполнения [McConnell et al., 1976].
5.8. БЕСКОНЕЧНО АДАПТИВНЫЕ СТРУКТУРЫ
В гомологическом ряду Ti^O^.! кристаллографический
сдвиг имеет место в плоскостях (121) для п <i 10 и (132) для п > 10
(см. табл. 5.2). Отличительной структурной особенностью составов,
для которых п = 10—14, является то, что плоскость (121)
непрерывно проходит от (121) до (132) через все возможные ориентации, такие
как (253), (374), (495) и т. д. Как следствие этого — почти любой
состав в ряду TiOl900—ТЮ1>937 может оставаться структурно
упорядоченным. Такие непрерывные ряды упорядоченных структур,
очевидно, могут иметь иррациональные плоскости
кристаллографического сдвига. Такие структуры называются бесконечно адаптивными
структурами, эту концепцию первым ввел Anderson [1973]. В
указанном ряду составов плоскость кристаллографического сдвига
меняется непрерывно, причем она поворачивается в своей собственной
зоне, от одного устойчивого направления к другому.
Пример бесконечно адаптивных структур, в которых нет
кристаллографического сдвига, найден в L—Та205 и его твердых
растворах с W03 вплоть до состава HTa2054W03. Структура L—Та20Г)
родственна структуре U308 и состоит из Пентагона л ыгых бипи-
рамнд с общими ребрами и искаженных октаэдров.
Добавление W03 к L—Та205 приводит к уникальным сверхструктурам
для каждого состава [Roth, Stephenson, 1970]. Получающиеся
гомологические ряды соответствуют M2mO(16m_2)/3(Ml0O26, M22058
и т. д.). Бесконечные ряды структур могут быть построены путем
упорядоченной упаковки этих элементов в различных соотношениях.
Любой состав в этой системе между М02625 и М02|б36 можно создать,
используя а единиц М]0О2б и Ъ единиц М22058, чтобы путем
соответствующего подбора а и b получить формулу (М]0а+226О26а+586), которая
соответствовала бы любому составу ряда. Другая система с
бесконечно адаптивными структурами — Ba;)(Fe2S4)q; несколько таких
систем найдено в последние годы [Rao, 1985]. Отмечено, что упругая
Прорастание кристаллов
243
энергия, связанная с вынужденным когерентным совмещением двух
типов плоскостей решетки, дает дальнодействующее отталкивающее
взаимодействие, необходимое для образования этих длиннопериоди-
ческих структур [Kittel, 1978]. Используя теорему Saint — Venant
по теории упругости, среднюю упругую энергию на единицу
площади плоскости при образовании состава АНВ (когерентным
совмещением плоскостей А и В) задаем формулой
v - титг (*•! + ■»-■£ и [^-J ~w • (5'26)
где Ля = аА — ав = (еА — £в)#> аА и ав — постоянные решетки
чистых А и В, е — деформация, К — объемный модуль упругости,
d — межплоскостное расстояние, ев — принимается равным — NeA.
5.9. ПРОРАСТАНИЕ КРИСТАЛЛОВ
Эпитаксия и политипия — примеры прорастания по
границам раздела твердое — твердое. Эпитаксия представляет большой
технологический интерес, поскольку она имеет дело со способами
нанесения тонких, монокристальных пленок (полупроводниковых
материалов) на подходящую подложку с целью использования их
в интегральных схемах, приборах ночного видения и т. д. Плоскость
двойникования и граница срастания (коинсидентная граница)
являются планарными дефектами (см. разд. 5.4) или поверхностями
раздела, которые представляют собой кинетически стабилизированные
неравновесные дефекты, они не приводят к изменениям в
стехиометрии материала; аналогично и политипия также не приводит к
изменениям в составе. Известны также многие другие, не приводящие
к изменениям прорастания [Rao, Thomas, 1985], причем необычным
примером является энантиомерное прорастание в гексагелиценах.
Другой интересный пример — прорастание цеолита ZSM-5 и ZSM-11.
Встречается прорастание амфиболов с тройными, четверными и более
широкими лентами связанных пироксеновых цепей. В большинстве
таких систем прорастание непериодическое или разупорядоченное.
Непериодическое прорастание с изменениями состава найдено
во множестве систем. Все случаи эпитаксии фактически являются
примерами поверхностей раздела, включающих изменения в составе.
Изолированные плоскости кристаллографического сдвига —
хорошие примеры таких межфазных границ твердое — твердое.
Нефритовый жадеит (амфибол) дает тройные цепи различного состава.
Другие системы, проявляющие такое непериодическое прорастание:
р- и Р"-оксиды алюминия, соседние члены оксидов с формулой
Bi2An_1B„03n+3, как и Vn02n_i. На рис. 5.21 показано изображение
решетки Sr3Ti207 в области А, а также слои различных членов
семейства 8гл+1Т1д03п+1 с п = 3, 4, 5, 7, 8. Недавно рассмотрены
16*
244
Новый взгляд на старую проблему
Рис. 5.21. Картина решетки Sr3Ti207,
показывающая непериодическое прорастание
других членов семейства Sr Ti. 03, ,
71+1 71 71+1
[Tillcy, 1977].
А — п = 1; В — п = 3; С — п = 4; D —
и — 5; Е — п = 7; F — п = 8.
непериодическое и периодическое
прорастания в общем контексте
химии межфазных границ I Rao, Thomas, 1985 |. В принципе
прорастание можно было бы рассматривать как класс модулированных
структур, где граница прорастания является периодическим
возмущением.
Некоторые непериодические прорастания имеют определенную
степень порядка, и часто бывает трудно решить, сколько
повторений отдельных последовательностей составляют периодическое
прорастание. Однако имеется много систем, где периодическое
прорастание имеет место на относительно больших расстояниях |Rao,
1985], приводя к гомологическим рядам структур с большими
параметрами ячейки (табл. 5.3).
Примером периодического прорастания является семейство
гексагональных бариевых ферритов, M?)Yr/, образованных срастанием
BaFe12019 (M) и Ba2Me2Fe12022 (Y) с Me—Zn, Ni или Mg [Anderson,
Hutchison, 1975]. На рис. 5.22 мы демонстрируем электронограмму
типичного феррита. Эти бариевые ферриты, которые можно
рассматривать как сросшиеся перовскитовые и шпинельные слои, имеют
широкий ряд параметров элементарных ячеек; например, в MY
о о
с —- 26 А, а в M8Y27 с = 1455 А. Сложность этих прорастаний
иллюстрируется фазой M12Y47 состава Ba10(.Ni94Fe708O12(.8, которая со-
Таблица 5.3
Типичные гомологические ряды, получающиеся в результате периодического
прорастания
Ba2n+pMe2nFei2(n+p)022M + i9p(Me-Zn' Ni и т- Л-)*; * = 1-47; р = 2-12
Bi4Am+n-2Bm+n°3(m + n)+e(A-BaHJIHBi'B-Ti'Nb'Cl,HT-A-); m, n = 1, 2, 3,4
AxW03 (A — щелочной или щелочноземельный металл, Bi и т. д.) (0,0 < х < 0,1)
AxP4O8(WO3)2m;/» = 4-10
(Na, Ca)nNbn03n+2; n = 4-4,5 **
(A3NbeSi402e)n(A3Nb8_xMx021)***(A—Ваили8г,М-Т1, Ni, Zn и т. д.); п = 1-15.
* Отношение кислород : металл в области 1,38—1,43 согласуется с изменением в
последовательности упаковки.
** В этих перовскитах ABO _j_x с избытком кислорода х изменяется между 0,444 и
0,50, но нет никакого признака точечных дефектов.
*** В этих системах с отсутствием точечных дефектов отношение кислород : металл
изменяется между 1,9 и 2,0.,
Прорастание кристаллов
245
Рас. 5.22. Картина прорастания рошотки MYMYe в барий-феррито-
вой системе [Anderson, Hutchison, 1975].
держлт 12 М в примитивной ячейке. M^Y^-срастания не могут
образовываться пр i замене ионов Fe3+ на ионы А13+, что говорит о
важности магнитных взаимодействий в стабилизации длиннопериодич-
ных структур. Интересный класс периодических прорастаний
образуется, когда пласты W03 срастаются с полосками гексагональной ок-
сидновольфрамовой бронзы (НТВ); эти проросшие оксидновольфрамовые
бронзы (ITB) имеют сложную упорядоченную последовательность
с большой периодичностью на довольно больших расстояниях
[Kihlborg, 1978]. На рис. 2.7 и 2.9 мы приводили изображение
срастания оксидповольфрамовых бронз с висмутом, где пласты W03
срастаются с полоской гексагональной оксидновольфрамовой
бронзы шириной в один туннель. На рис. 5.23 показана решетка высоко-
упорядоченной проросшей оксидновольфрамовой бронзы Cs^WOJTB.
Образование сросшихся бронз, возможно, связано с условиями роста
и является хорошим примером модели отторжения примеси
[Anderson et al., 1972]. По-видимому, прорастания с большим периодом для
силикониобатов бария [Studer, Raveau, 1978] и (Na, Ca)nNbn03n+2
[Portier el al., 1975] похожи на непрерывные серии упорядоченных
структур или на бесконечно адаптивные структуры [Anderson,
1973]. Упорядоченные прорастания, образованные семейством
оксидов с общей формулой (Bi202)2+(Am_1Bm03m+1)2_, широко
исследованы [Gopalakrishnan et al., 1984; Jefferson et al., 1984]. Эта система
проявляет заметный порядок даже в отсутствие магнитных или ку-
246
Новый взгляд па старую проблему
Рис. 5.23. Картина решетки Cs^WOg [Kihlborg, 1978]. Фаза проросшей
вольфрамовой оксидной бронзы имеет полосы гексагональной
вольфрамовой оксидной бронзы (шириной в два туннеля), разделенные семью
октаэдрами VVOe.
лоповских упорядочивающих взаимодействий и образует
гомологические ряды Bi4Am+n_213mf n03(mf „)+ ({. Приведем полученное с высоким
разрешением изображение решетки Bi9Ti6Cr027, образованного
прорастанием Bi4Ti3012(m = 3) и Bi5Ti3CrOI5 (n ------ 4), вместе с
компьютерным изображением и структурной моделью (рис. Г).24), чтобы про-
-" •*. J* ■*- *,
Рис. 5.24. Картина решетки Bi0TieCr027, полученная методом HREM
(500 кВ) [Gopalakrischnan et al., 19S4].
Оксиды со структурой дефектных перовскитов 247
иллюстрировать упорядоченную группировку слоев с т — 3 и п =
= 4. Эти системы представляют собой случаи, когда, по-видимому,
за периодическое прорастание ответственны только упругие силы.
В различных исследованных системах с прорастанием
(см. табл. 5.3) нет никаких доказательств наличия точечных
дефектов. Однако первопричина периодичности дальнего порядка в
сложных системах с упорядоченным прорастанием носит загадочный
характер. Значение упругих сил при образовании политипов,
структур со сдвигом и бесконечно адаптивных структур было отмечено
ранее. Видимо, упругие силы отвечают и за периодическое
прорастание IRao, 1985]. Рабочий критерий образования периодических
прорастаний можно получить, выражая энергию упругой
деформации через изменение объема, сопровождающее образование
прорастаний [Gopalakrichnan et al., 1984]. Исследована природа атомных
смещений, возникших в результате упругой деформации путем
сравнения полученных изображений высокого разрешения с
рассчитанными по различным моделям |Jefferson et al., 1984]. Такое
уточнение структуры дает новую информацию о поверхности раздела
твердое — твердое. Для понимания проросших структур могли бы быть
использованы волны массовой плотности (солитоны).
5.10. ОКСИДЫ СО СТРУКТУРОЙ
ДЕФЕКТНЫХ ПЕРОВСКИТОВ:
ПРИМЕР ИССЛЕДОВАНИЯ
Перовскиты представляют собой важный класс
неорганических твердых тел, и было бы полезно рассмотреть все разнообразие
дефектных структур в этом семействе оксидов. Нестехиометричность
в перовскитовых оксидах может получаться в результате дефицита
катиона (в позиции А или В), дефицита или избытка кислорода.
Некоторые сросшиеся структуры, образованные перовскитоподобны-
ми оксидами, и родственные структуры упоминались в предыдущем
разделе, а здесь мы будем рассматривать главным образом
упорядочение дефектов и сверхструктуры в этих оксидах.
Вакансии в позиции А. Поскольку группа В03 образует в перов-
€Китовой структуре устойчивую сетку, большие катионы А в
позициях с КЧ-12 могут отсутствовать или частично, или полностью.
Вольфрамовые бронзы Ax.W03, которые представляют собой важный
класс нестехиометрических оксидов, могут рассматриваться как
перовскиты с дефицитом в позиции A [Ekstrom, Tilley, 1980]. Из
трех известных структур вольфрамовых бронз: гексагональной,
тетрагональной и кубической, только кубическая структура
относится к перовскитовой структуре с вакансиями в позиции А.
Кубические бронзы принято считать перовскитовыми вольфрамовыми
бронзами (РТВ). Можно различить два типа РТВ-бронз: с малыми значе-
248
Новый взгляд на старую проблему
ниями х(х <С 1,0) и с большими — х ^ 1,0. РТВ-бронзы с большим
значением х обладают характеристиками истинных бронз (например,
металлические свойства и химическая инертность). Важным
вопросом, касающимся нестехиометричности этих фаз, является вопрос
о том, упорядочиваются ли атомы и вакансии в позиции А.
Проведенное ранее методом нейтронной дифракции исследование
Na0>75WO3 [Atoji, Rundle, I960] показало, что ионы натрия в
удвоенной перовскитовой ячейке упорядочены по шести позициям А из
восьми. Однако природа упорядочения вакансий в позициях А в
металлических РТВ-бропзах не ясна. Случайные изолированные или
слабовзаимодействующие дефекты могут наблюдаться в
металлических системах, в отличие от неметаллических аналогов.
Известно, что, кроме перовскитовых вольфрамовых бронз, пе-
ровскитовые оксиды с дефектами в позициях А образуются, когда
В—Ti, Nb, Та и т. д. Такие соединения проявляют металлические
свойства и имеют перокскитовые структуры, если атом В находится
в низкой степени окисления. Такие составы, как А0 f)Nb03 (А—Ва,
РЬ и т. д.), где ниобии находится в высшей степени окисления,
имеют иеперовскитовые каркасные структуры. Интересным примером
перовскита с дефектами к позициях А является Cu0t5TaO3, который
кристаллизуется в псевдокубической перовскитовой структуре.
Элементарная ячейка Сп0>5ТаО3 — ромбическая с а --- 7,523, Ь --- 7,525
о
и с -■- 7,520 А; па ячейку приходится восемь формульных единиц.
Атомы тантала образуют каркас из Та03, как в кубическом перов-
ските, тогда как атомы меди упорядочиваются в позициях A [Longo,
Sleight, 1975; Vincent et al., 1978]. Кроме перовскитовых
тетрагональных и гексагональных вольфрамовых бронз, заслуживает
внимания новый вид вольфрамовых бронз (ITB), образованных
срастанием пластов W03 (шириной в п октаэдров) с полосами
гексагональной бронзы (НТВ) шириной от одного до трех туннелей [Kihlborg,
1978]. Прорастание в ITB-бропзах бывает периодическим и
непериодическим. Полоски НТВ-бронзы (см. рис. 5.23) имеют ширину
в два туннеля, это — наиболее устойчивая конфигурация проросших
вольфрамовых бронз со щелочным металлом. Недавно проведено
исследование проросших висмут-вольфрамовых бронз, в которых
полески НТВ-бронзы всегда имеют ширину в один туннель I Rama-
nan et al., 1984]. Интересно, что в системе Bi^WOg при х ^ 0,02
образуется фаза РТВ-бронзы, при этом ITB-бронзы получаются
только при х > 0,02 (см. рис. 2.27 и 2.29). W03 и Re03 можно
рассматривать как предельные случаи перовскитов с вакансиями в
позиции А. Оба оксида обладают каркасом из октаэдров, соединенных
вершинами, но в отличие от Re03, W03 никогда не бывает
кубическим. При повышении температуры он подвергается нескольким
полиморфным превращениям, начиная с низкотемпературной три-
клинной структуры до более симметричных форм.
Оксиды со структурой дефектных перовскитов 249
Вакансии в позиции В в перовскитовых оксидах энергетически
являются неблагоприятными из-за большого формального заряда и
малого размера катионов в позиции В. Если в позициях В
существуют вакансии, то должны быть другие компенсирующие факторы,
такие как ковалентность связи В—О и взаимодействие В — В. В то
время как ковалентность связи В — О должна бы увеличиваться
с увеличением заряда и уменьшением размера катиона В,
взаимодействие В—В должно быть более благоприятным при гексагональной
упаковке слоев А03, чем при кубической. Таким образом, следует
ожидать, что вакансии в позиции В будут наблюдаться более часто
в перовскитовых оксидах с высокозарядными катионами В, имеющих
структуры гексагональных политипов. В самом деле, перовскиты
с вакансиями в позиции В с гексагональным или смешанным
гексагонально-кубическим типом упаковки А03, являются намного более
обычными, чем с исключительно кубической упаковкой слоев А03.
Составлена сводка кубических перовскитов, имеющих вакансии в
позиции В [Rauser, Kemmler-Sack, 1980]. Некоторыми примерами
перовскитов с упорядоченными вакансиями в позиции В являются
следующие: Ba2Sm2/3UOe (5), Ba2Ce^+Sb5+06 (7), Ba2Ce4+Sb*/+506 (9),
Ba2Sm3+U5/jj06 (11) (цифры в скобках означают число катионов В
па вакансию). Упорядочение этих вакансий проявляется
образованием сверхструктур.
Известно много хорошо охарактеризованных гексагональных
перовскитов с вакансиями в позиции В, причем вакансии в этих
оксидах упорядочиваются между гексагональными слоями, в
которых октаэдры ВОв сочленяются гранями. Таким образом, Ва5Та4015
имеет пятислойную последовательность (ггккк) [Shannon, Katz,
1970], где октаэдрическая позиция между г — г-слоями вакантна.
Такая упаковка приводит к изолированным кластерам из четырех
октаэдров, каждый с двумя общими противоположными вершинами
[Та4015]10". Похожая структура Ba3Re209 [Calvo et al., 1978]
состоит из последовательности слоев (ггк)3, где октаэдры Re06
соединены вершинами, вакансии в этом случае находятся в октаэдриче-
ских позициях между г — г-слоями. Некоторым исключением в этой
общей тенденции являются Ba3W209 и Ва3Те209 (см., например
[Jacobson et al., 1981]). Оба эти соединения имеют только
гексагональную последовательность слоев Ва03. В Ba3W209 2/3 октаэдри-
ческих позиций в каждом слое заполнены вольфрамом, тогда как
в Ва3Те209 октаэдрические позиции в двух смежных слоях
полностью заняты, причем каждый третий слой является полностью
вакантным. Оба эти упорядочения приводят к группам [В209]6~
(В—W, Те), состоящим из двух октаэдров В06, соединенных общей
гранью (биоктаэдры с общей гранью). В обеих структурах атомы В
в значительной степени замещаются вакансиями, что приводит к
минимизации отталкивания В—В.
250 Новый взгляд на старую проблему
Оксидные перовскиты, содержащие упорядоченные вакансии в
позиции В, проявляют новые люминесцентные свойства. Например,
Ba3W209 ниже 150 К характеризуется эффективной голубой (460 нм)
фотолюминесценцией, в отличие от BaW04 и Ba3W06, которые
состоят соответственно из изолированных тетраэдров W04 и
октаэдров W06 [Blasse, Dirksen, 1981]. Высказано предположение, что за
эффективную люминесценцию ответственно именно кластерирование
октаэдров. Интересно, что перовскиты с упорядоченными
вакансиями в позиции В имеют эмиссию разного цвета, когда к ним
добавляются различные редкоземельные активаторы. Например, когда 12-
слойный (ггкк)3-перовскит Ba^^Sr^LaScWaDO^ допируется
одновременно Еи3+ и ТЬ3+, получается зеленая тербиевая эмиссия при
547 нм (Tb3+ : bDA->7Fb) и красная европиевая при 615 нм (Еи3+ :
: bD0 ->■ 7F2). Первое получается, когда фосфор возбуждается в зоне
переноса заряда в WOe при 300 нм, а последнее — при возбуждении
в результате переноса заряда от кислорода к Еи3+ при 340 нм.
Аналогично Sr3La2W2nO]2, допированный Еи3+ и Тт3+, имеет красную
и голубую эмиссию при подходящем возбуждении, а такое же
допирование Ег3+ и Тт3+ дает зеленую и голубую эмиссию [Brown, Kem-
mler-Sack, 1983]. Семейство оксидов висмута с общей формулой
BimBm_103m(B—Ti, Nb, W), впервые описанное в 1950 г. [Aurivil-
lius, 1950], также можно отнести к перовскитам с дефицитом
катионов В. Типичными членами этого семейства являются Bi2W06,
Bi3TiNb09 и Bi4Ti3012.
Анионодефицитные перовскиты и структуры с упорядоченными
вакансиями. Нестехиометричность, обусловленная анионными
вакансиями в оксидах с перовскитовой структурой, является более
обычной, чем нестехиометричность, включающая вакансии в катионной
подрешетке. Нестехиометричность, связанная с анионными
вакансиями, известна для структур с каркасами как АВ03, так и В03.
Типичным примером последней структуры является нестехиометрич-
ный триоксид вольфрама WO^^. Анионодефицитная
нестехиометричность в АВОд.д. вообще не обеспечивается механизмом
кристаллографического сдвига. Упорядочение дефектов в оксидах AB03_x
основано на консервативном механизме, в том смысле, что вакансии
ассимилируются в структуре, приводя к большим сверхъячейкам
основной перовскитовой структуры. Однако тип образуемой
сверхструктуры зависит от катиона В.
Одними из наиболее охарактеризованных перовскитовых оксидов
с упорядочением анионных вакансий являются оксиды Ca2Fe205
и Ca2FeA105 со структурой браунмиллерита [Grenier et al., 1981].
Эти составы можно было бы рассматривать как анионодефицитные
перовскиты с 1/6 вакантных анионных позиций. Ромбическая
элементарная ячейка структуры браунмиллерита (а = 5,425, Ъ = 5,598
о
и с = 14.768 А для Ca2Fe205) получается в результате вакансионного
упорядочения; эта ячейка соотносится с кубическим перовскитом так,
Оксиды со структурой дефектных перовскитов 251
v v v
D Q Q Q D D
а а а а о а Г1101 с
[11 (Я
Рис. 5.25. а — вакансионное
упорядочение в плоскости ab Ca2Fc205 со
структурой браунмиллерита; б —
чередующаяся последовательность ок-
таэдрических и тетраэдрических
слоев в направлении с. • — железо;
• — кислород; а — кислородные
вакансии.
I *' п * о V a
с, * g.
c0-2gc
e
Рис. 5.26. a — вакансионное
упорядочение в плоскости ab в Ga2Mn205.
Вакансионное упорядочение
параллельно направлению с в Са2Мп205 и
Са<>Со205 показано вбив соответст-
• — кислород; а —
кислородные вакансии.
а2ъи2
венно.
что а а* /2ас, Ъ ~ V^2ac, с ~ 4ас. Кислородные вакансии
упорядочиваются в чередующихся плоскостях (001) В02 структуры
кубического перовскита так, что чередующиеся ряды кислорода 1110]
пропускаются (рис. 5.25). Таким образом, последовательность слоев
вдоль оси с: CaO—Fe02—CaO—FeO.... Малое смещение атомов
железа в слоях FeO приводит к тетрагональной координации железа в
этом слое, что, в свою очередь, ведет к образованию чередующихся
слоев из октаэдров Fe06 и тетраэдров Fe04 (OTOT'...) в
направлении с (см. рис. 5.25). Есть сообщение о новом оксиде Ca2FeCo05,
имеющем структуру браунмиллерита, в которой Fe3+ и Со3 + занимают
соответственно тетра- и октаэдрические позиции I Vidyasagar et al.,
1984].
Есть сообщения о Са2Мп20Г) и Са2Со205, которые имеют составы,
похожие на браупмиллерит IPoeppelmeier et al., 1982; Vidyasagar
et al., 1984], но их структуры имеют различные типы упорядочения
вакансий. Са2Мп205 имеет ромбическую структуру с а = 5,432,
252
Новый взгляд на старую проблему
Ъ = 10,242 и с = 3,742 А, причем связь с кубической перовскитовой
структурой такова: а и с ~ ас. Кислородные
вакансии в Са2Мп205 упорядочены в каждсй плоскости (001) В02
кубического персвскита так, что 1/2 атсмов кислорода в
чередующихся рядах Ц10] удалена (pre. 5.26). Ссстав плоскости теперь
становится МпОЬ5. Если эти плоскости собрать в стопку в
последовательности СаО—Мп01>5—СаО—Мп01>5..., то получится структура
Са2Мп205. Важной особенностью этой структуры, которая отличает
ее от структуры браунмиллерита, является то, что марганец в ней
находится в квадратно-пирамидальной координации. Зй4-Конфигура-
ция Мп3+ благоприятствует скорее квадратно-пирамидальной
координации, чем тетраэдрической, определяя тем самым упорядочение
анионных вакансий в Са2Мп205. Недавно охарактеризованы члены
серий Ca2Fe2_3.Mn3.O5 с тремя различными типами координации
вокруг иона переходного металла (октаэдрическая, тетраэдрическая и
квадратно-пирамидальная) IVidyasagar et a]., 1986]. Са2Со205 также
кристаллизуется в ромбической структуре с а = 11, 12, Ъ = 10,74
о
и с — 7,48 А. Сравнение этих параметров с такими же параметрами
Са2Мп205 обнаруживает удвоение осей a m с в Са2Со205.
Упорядочение вакансий в Са2Со205 похоже на такое упорядочение в Са2Мп20Г),
но удвоение оси с, вероятно, обусловлено упорядочением,
схематично показанным на рис. 5.26. Установлено, что Sr2Mn205 имеет
то же самое упорядочение дефектов, что и в Ca2Mn205 [Caignaert et
el., 1985]. Однако Sr2Co205 при низких температурах
кристаллизуется в анионодефицитиой 2Н-перовскитовой структуре, а выше
1170 К превращается в структуру браунмиллерита [Grenier et al.,
1979]. SrCo03_3; (0 < x < 0,5), полученный под высоким давлением
кислорода (50—2600 бар), принимает кубическую перовскитовую
структуру.
SrTi02t5, SrV02)5 и KTi02 5 представляют собой несколько
другой тип анионовакантных перовскитов состава AB025. KTi02>5
проявляет небольшое сходство с перовскитами, причем каждый атом
Ti имеет тригонально-бипирамидальную кислородную координацию.
Рентгенографические исследования SrTi02t5 и SrV02>5 обнаружили
кубические перовскитовые элементарные ячейки с а с^. 3,9 А, что
означает, что анионные вакансии расположены случайным образом.
Нет никаких данных о сверхрешеточном упорядочении в SrTi02>5
[Tofield, 1978; см. также [Alario-Franco, Regi, 1977]. Заслуживает
внимания тот факт, что как SrTi03_x, так и SrV03_x. имеют
металлические свойства, следовательно, d-электроны, связанные с
катионами В, являются делокализованными.
Во многих перовскитах состава АВ03_х нестехиометричность
охватывает область составов 0 <! х ^ 0,5; до сих пор мы обсуждали
только структуры конечных членов с х = 0,5. Интересно
посмотреть, какие структуры преобладают на промежуточных составах.
Оксиды со структурой дефектных перовскитов 253
Показано, что SrFeOg.^ имеет кубическую структуру при 0 < х <
< 0,12, тетрагонально-искаженную перовскитовую структуру —
при 0,15 < х <С 0,28 и смесь фаз со структурой перовскита и браун-
миллерита — при 0,28 < х < 0,5 [MacChesney et al., 1965].
В SrFe02t75 обнаружена, по данным электронной дифракции,
сверхъячейка, связанная с перовскитом так, что а с^2у2ас, Ъ ~ 2ас
и с с^2^2ас [Tofield et al., 1975]. Упорядочение вакансий,
предполагаемое в этой фазе, похоже на упорядочение в браунмиллерите;
каждый второй атом кислорода в чередующихся слоях [110 ]с
теряется, переводя половину атомов железа в пятикоординированное
состояние, причем другая половина сохраняет октаэдрическую
координацию, как в перовските. В Sr^Nb^^FeO^-y обнаружены
сверхструктуры, включающие случайное удвоение одной из осей перов-
скитовой ячейки (в направлениях а, Ъ или с) [Alario-Franco et al.,
1982]; считается, что это обусловлено существованием
микродоменной текстуры, где домены прорастают в трех измерениях случайным
образом.
Детально исследована анионодефицитная система CaMn03_.x в
области составов 0 < х ^ 0,5 [Reller et al., 1984].
Идентифицировано пять различных составов с упорядоченными вакансиями
кислорода: CaMn02,5, CaMn02)556, CaMn02,667, CaMn02i75 и СаМпО2,80;
во всех этих составах сохраняются структурные особенности
исходного перовскита. Структура СаМп02>5, имеющая
квадратно-пирамидальную координацию Мпзь, обсуждалась ранее. Для других
составов отношение квадратных пирамид к октаэдрам уменьшается
от СаМп02>5 к СаМпО3,0- Эти структуры можно представить в виде
сверхрешеточных повторов исходного неискаженного перовскита,
но с поворотом сверхструктурной ячейки на угол R в плоскости
(001). Сверхрешетка в СаМп028 составляет но повернута
на 26,5°; эта структура дает тетрагональную элементарную ячейку,
о о
а = Ь = 8,34 А, с — 7,46 А. Чтобы проверить эту структуру,
смоделированы изображения решетки и сравнены с данными HREM;
полученное соответствие говорит в пользу приведенной структурной
модели. Структуру СаМп0275 можно представить как 1^2x2^27?
45° или 1/"2х4)/"2Д 45°, тогда как структуру СаМп02,в67 — ]^2х
ХЗк 27? 45°. Наблюдались четыре упорядоченные структуры
СаМп02>5, но наиболее обычная структура -/2Х2/2Д 45° [Роер-
pelmeier et al., 1982]. Рассмотрена связь между дефектными
структурами фаз системы СаМп03_^ и их каталитической активностью
[Reller et al., 1984].
Вакансионное упорядочение в аниоиодефицитном LaNi03
исследовано несколько лет тому назад [Gai, Rao, 1975a].
Термогравиметрия на воздухе и в кислороде с очевидностью показала
образование серии фаз с общей формулой La^Ni^C^n-i (я = 7, 9, 13 и
254 Новый взгляд на старую проблему
30). Исследование анионодефицитных образцов методом
электронной дифракции привело к предположению об образовании
сверхструктур, обусловленном упорядочением вакансий. Методом
электронной дифракции идентифицирована ромбическая или объемно-
центрированная тетрагональная ячейка с параметрами а ~ с ~
~ 2 у2ас. Методом электронной дифракции исследовано вакансион-
ное упорядочение в La2Ni205 и La2Co205 [Vidyasagar et al., 1985].
Тогда как La2Co205 принадлежит к структурному типу браунмил-
лерита, La2Ni205 имеет новую вакансионно-упорядоченную
тетрагональную сверхструктуру перовскитового типа с параметрами а с^
~ с ~ 2ас, содержащую Ni2+ как в октаэдрической, так и в
плоскоквадратной координации. Имеются указания (HREM) о наличии
плоскостей кристаллографического сдвига в слабо восстановленном
LaNi03.
Методом термогравиметрии исследована устойчивость перовски-
тов LaB03 (В—V, Cr, Mn, Fe, Co или Ni) в восстановительной
атмосфере при 1270 К [Nakamura et al., 1979]. Результаты показывают,
что устойчивость зависит от атома В, изменяясь в ряду LaV03 ~
~ LaCr03 > LaFe03 > LaMn03 > LaCo03 > LaNi03. В
экспериментальных условиях, использованных в этом исследовании,
оксиды типа ЬаВО^д. не получаются.
Исследована система Ca^La^^FeOg,;, (составы между Ca2Fe205
и LaFe03) и CaTij.^Fe^Og^y (составы между Ca2Fe205 и CaTi0?)
с тем, чтобы попытаться охарактеризовать анионовакансионное
упорядочение в промежуточных составах между браунмиллеритом
и перовскитом [Grenier et al., 1981 ]. Эта работа обнаружила
существование гомологических рядов фаз с общей формулой АпВп03п_1
(A—La, Ca; В—Ti, Fe). Структура этих фаз состоит из
чередующихся в направлении с последовательности (п — 1) перовскитоподобных
слоев В06 (октаэдрические слои) и слоев тетраэдров М04 (тетраэд-
рические слои). При п = 2 получается браунмиллеритная фаза,
в которой последовательность октаэдрических и тетраэдрических
слоев — ...ОТОТ' ОТОТ..., Ти Г означают две различные
ориентации тетраэдров в структуре (см. рис. 5.25). При п = оо
тетраэдрических слоев не будет вообще, а последовательность будет такой,
как в перовските — ...ОООО... . Другими членами этой серии
являются Ca2LaFe308, Ca3Fe2Ti08 {n = 3) и Ca2Fe2Ti2Ou (n = 4).
Последовательность слоев в фазах с п — 3 и п = 4 соответственно
...ООТ ООТ... и ...ОООТ ОООТ... . Интересным членом этой серии
является Ca4YFe5013, который соответствует составу с п = 2,5 в
:>том ряду. Структура этого оксида, определенная HREM (1 мэВ)
[Bondo et al., 1981], состоит из регулярного прорастания элементов
серии AnBn03n_1 со значениями гс=2итг = 3ис
последовательностью слоев полиэдров — ...ОТООТОТООТ... . Другие известные
фазы с упорядоченным прорастанием — Ca7Fe6Ti018 (n = 2,33) и
Ca5Fe4Ti013 (n = 2,5).
Оксиды со структурой дефектных перовскитов 255
Недавние исследования [Alario-Franco et al., 1983; Calbet et al.,
1983] показали, что нестехиометрическое поведение в таких Fe-
содержащих системах является очень сложным, причем тип
упорядочения дальнего порядка (или отсутствие его) зависит от
условий получения образца. Промежуточные составы в системе
Сад-Ьа^д-РеОз.у (2/3<д;< 1) проявляют два различных типа
поведения. Когда их получают в инертной атмосфере при
относительно низких температурах (1370 К), они имеют разупорядочен-
ную структуру прорастания, включающую в себя фрагменты
Ca2LaFe308 (п = 3) и Ca2Fe205. Когда образцы получаются при
1670 К на воздухе, они обогащаются кислородом и имеют
совершенно отличное от первых поведение. Дифрактограмма порошков
последних образцов дает обманчиво простую кубическую перовскито-
о
вую структуру с а ~ 3,85 А. Данные электронной дифракции
и HREM говорят о том, что эта структура более сложная, состоящая
из микродоменов браунмиллерита или Ga2LaFe308, прорастающих
в трех измерениях с образованием структуры, которая в среднем
является перовскитоподобной кубической структурой. Размер ин-
о
дивидуальных доменов порядка ~ 106А3, с увеличением содержания
кислорода он уменьшается. Примечательно, что даже при самых
высоких температурах синтеза система не проявляет классического
нестехиометрического поведения с случайным распределением
точечных дефектов; точечные дефекты всегда структурно упорядочены,
образуя микродоменную текстуру.
Анионодефицитные гексагональные перовскиты проявляют
интересное структурное поведение, включающее политипизм.
ВаМОз-я (М—Mn, Fe, Со и т. д.) — прототип таких систем. Несте-
хиометричность в этих системах — результат подходящих
соотношений при кубической и гексагональной сборке слоев А03_л. HREM
дает прямое подтверждение слоистого типа структуры политипов
ВаМ03 С слоями Ba03 [Gai, Rao, 1975b].
Получены данные по системе ВаМп03_х, которые отчетливо
демонстрируют сложность структуры этой системы [Negas, Roih,
1971 ]. С уменьшением содержания кислорода в структуре
увеличивается кубический тип стыковки слоев. Структурные изменения,
сопровождающие нестехиометричность кислорода, однако, не
являются истинными политипическими превращениями. Система
BaFeOg.x (Mori, 1970; Zanne, Gleitzer, 1971] является такой же
сложной и проявляет различное структурное поведение: BaFe02>5
(триклинная структура браунмиллеритного типа), BaFe02e2_2>64
(ромбоэдрическая), BaFe02 67 (триклинная), BaFe02,75_2,81
(тетрагональная) и BaFe02G9_2>95 (6Н-гексагональная). Фаза 12Н также
описана, ее состав точно не известен. Структурное исследование
6Н BaFe0279 [Jacobson, 1976] показывает, что вакансии
кислорода распределены не поровну между кубическими и гексагональны-
256
Новый взгляд па старую проблему
ми слоями, которые имеют последовательность (ккг)2.
Гексагональные слои имеют состав Ва02,5, а кубические — Ва02>835.
По-видимому, упорядочение вакансий кислорода в слоях Ва02>5 подобно
упорядочению в слоях Sr02)5 в SrFe02 5, где один ряд кислорода
[110] из каждых четырех удаляется, превращая часть октаэдров
Fe06 с общей гранью в тетраэдры FeO { с общими вершинами.
Структуру 6Н BaFe02i79 следует противопоставить структуре
аналогичной фазы в системе с марганцем. Например, в 4Н Ba05Sr0>5MnO285
все вакансии находятся в гексагональных слоях, превращая пары
октаэдров с общими гранями в исходной 4Н-структуре в триго-
нальные бипирамиды с общими ребрами [Hutchison, Jacobson, 1977].
Различие в структурном поведении между системами с железом и
марганцем приписывается различию в координационных
тенденциях Fe3+ и Мп3+: высокоспиновый Mn3+(d4) предпочитает триго-
нально-бипирамидальное или квадратно-пирамидальное
окружение, тогда как высокоспиновое Fe3+(d5) принимает как октаэдри-
ческую, так и тетраэдрическую координацию.
Интересны структуры членов системы ВаСо03_т. В этой
системе при 1178 К найдены следующие фазы: 2Н BaCoO3>0_2>S5,
7Н ВаСоО2,57Г,_2>520, 12Н ВаСо02)4П_2)43, 15Н ВаСоО2>23_2>10 И орто-
ромбический ВаСоО2>07 [Zanne et al., 1972]. Из всех различных фаз
в этой системе надежно установлена фаза 12Н. Структура 12Н
ВаСо02>б определена методами HREM и порошковой нейтронной
дифракции [Jacobson, Hutchison, 1980]. Последовательность слоев
в одной фазе — (кккггг)2. Кислородные вакансии в этой структуре
являются неслучайными, они получаются в результате замещения
1/6 слоев Ва03 на слои Ва02. Как следствие — 1/3 атомов кобальта
тетраэдрически координируется кислородом. Тетраэдры Со04
соединяются вершинами в ряды из четырех октаэдров с общими гранями,
которые содержат оставшийся кобальт. Предполагая, что атомы
кобальта в тетраэдрических позициях имеют степень окисления 4+,
а в октаэдрических — 3+, приходим к идеальному составу фазы
12Н — BaCo02j67. Замещение слоев Ва03 слоями Ва02, ведущее к
тетраэдрической координации атомов металла В, является новым
путем аккомодации дефицита кислорода в перовскитах АВ03,
который может быть распространен также и на другие системы.
Интересный случай |Er-Rakho et al., 1981] перовскита AB03-.x
с высоким дефицитом кислорода (х > 0,5). где металл В находится
в трех различных координационных окружениях, представляет
собой La3Ba3Cu60H_j,. Фазы с переменным значением ц имеют
тетрагональную ячейку, связанную со структурой перовскита, так что
ат ^ у 2ас и ст^ Заг, где ас — параметр кубического перовскита.
Определена детальная структура Ьа3Ва3СиГ)014Л. Кислородные
вакансии упорядочиваются в слоях АО структуры перовскита,
последовательность слоев в упорядоченной структуре вдоль [001] —
Си204 — А2Оп — Cu204 — А2П2 — Си204 — А2Оп... Этот тип упо-
Оксиды со структурой дефектных перовскитов 257
рядочения анионных вакансий приводит к трем различным
координационным окружениям для меди: плоскоквадратное, квадратно-
пирамидальное и искаженное октаэдрическое. Предполагается, что
в этой структуре ионы Си3+ занимают октаэдрические позиции.
А нионоизбыточная нестехиометричность в оксидах со
структурой перовскита — не такое общее явление, как анионодефицитная
нестехиометричность, так как, вероятно, введение междоузельного
кислорода в перовскитовые структуры энергетически
неблагоприятно. Имеется несколько систем, которые проявляют несомненную
избыточную по кислороду нестехиометричность: LaMn03+5C,
BdLl_xLdLxTi03+x/2 и EuTiOo+x. Методом нейтронной дифракции
исследована [Tofield, Scott, 1974] кислородная нестехиометричность
в LaMnOg+я- В этой работе исследователи предполагают три
возможные модели для расположения избыточного кислорода в перов-
скитовой структуре: 1) междоузельный кислород в позициях (-тгОО I или
/ 1 1 1 \
ITT 4~ J кубической перовскитовой структуры; 2) катионные вакансии
в положениях А и/или В, сохраняющие совершенную кислородную
подрешетку и 3) образование новых кислородных фаз. Данные по
дифракции LaMn03>l2 показывают, что избыток кислорода
распределяется по вакансиям в положениях А и В с частичным удалением La
(в виде La«,Os), состав перовскита при этом — Ьа0,д4По,обМп0>98По,о20з-
Там, где есть большой избыток кислорода, образуются новые
фазы, обладающие перовскитоподобными структурами. Один из
путей расположения избыточного кислорода с одновременным
сохранением особенностей перовскитовой структуры заключается в том,
чтобы проводить срез кубического перовскита параллельно (110),
с образованием слоев состава (Ап_1Вп0371+2)оо, и располагать их
один над другим, добавляя между ними слои из атомов А. Этот
процесс приводит к гомологической серии с общей формулой AnBn03n+2.
Важными представителями этого семейства с п = 4 являются
Ca2Nb207 и La2Ti207.
Нестехиометричность в перовскитоподобных оксидах. Первым
семейством оксидов, которое мы здесь рассматриваем, будут оксиды
со структурой K2NiF4. Эти оксиды с общей формулой A2BQ4
содержат чередующиеся персвскитовые слей АВ03 и слои каменной
соли АО. По структурной химии этих оксидов недавно опубликован
обзор [Ganguly, Rao, 1984]. Избыточная по кислороду
нестехиометричность найдена в La2Ni04, кислород-дефицитная
нестехиометричность — в Са2Мл04, который может быть восстановлен топо-
тактически до Ca2Mn03>6 [Poeppelmeier et al., 1982]. Размеры
элементарной ячейки — а = 5,30 Ь = 10,05 и с = 12,24 А, при этом
связь с тетрагональной структурой K2NiF4 такова: а~ у Дт, Ь сы.
и с сы ст» Анионные вакансии упорядочиваются вдоль
17 ч. Н. Р. Рао, Дж. Гопалакришнан
258
Взаимосвязь структура — свойство
чередующихся рядов [110]с, как в СаМп02>5, с образованием слоев
из квадратных пирамид (Мп05). Охарактеризован оксид Ca2Fe03,5
с а = 14,79, Ь = 13,71 и с = 12,19 A [Vidyasagar et al., 1984].
Структура этого оксида отличается от структуры Ga2Mn03t5.
Вероятно, упорядочение анионных вакансий в Ca2FeO;j)5 похоже на
упорядочение в структуре браунмиллерита, состоящей из
чередующихся рядов тетраэдров и октаэдров в перовскитоподобных слоях
структуры K2NiF4.
Оксидные пирохлоры А2В2ОвО' могут иметь вакансии в
положениях А и О' с сбразованием фаз А2В2ОвП(АВ03) и AnB2OeD
(АВ2О0); небольшое число соединений А2В2Обпри условии, что А и В
являются высокополяризуемыми, но не слишком
электроположительными ионами, принимает структуру пирохлора, а не перовскита
[Subramanian et al., 1983]. Типичными из этих оксидов являются
Т12В2Ов (В—Nb, Та, U) и A2Sb206. В системе A—Sb—О (А—
— Na, К, Ag) известна серия нестехиометрических соединений
ASbwOz, максимальное отношение А : Sb зависит от А. Некоторые
из этих соединений являются суперионными проводниками.
6. ВЗАИМОСВЯЗЬ СТРУКТУРА - СВОЙСТВО
6.1. ВВЕДЕНИЕ
Установление взаимосвязи между свойствами веществ и их
структурой является главной задачей современной химии, а также
первостепенной заботой химиков, занимающихся твердым
состоянием. С точки зрения химии твердого тела важны следующие
свойства: электронные, магнитные, диэлектрические и оптические. Мы
кратко рассмотрим эти свойства, затем остановимся на основных
моментах химии твердого тела некоторых интересных классов материалов.
Важным классом материалов является класс ферроиков,
обладающих несколькими ориентационными состояниями, которые могут
переключаться из одного в другое при приложении соответствующей
силы; сегнетоэлектрические материалы образуют подгруппу этого
класса материалов. Другие обсуждаемые классы материалов — это
соединения смешанной валентности, низкоразмерные твердые тела
и жидкие кристаллы, которые приобрели большое значение в
последние годы. Мы также уделим внимание различным типам
переходов металл — неметалл; при обсуждении этой темы кратко
рассмотрим вопрос, что делает металл металлом.
Электроны в твердых телах
259
6.2. ЭЛЕКТРОНЫ В ТВЕРДЫХ ТЕЛАХ
Чтобы скоррелировать структуру и физические свойства
твердых: тел, важно иметь описание валентных электронов, которые
связывают атомы в твердом теле. Имеются две предельные модели
описания внешних электронов атома в твердых телах: зонная теория
и теория локализованных электронов, или теория поля лигандов.
Когда есть заметное перекрывание между орбиталями соседних
атомов, применима зонная теория Блоха и Вильсона. Теория
предполагает, что в твердом теле валентные электроны распределяются
одинаково между всеми одинаковыми атомами. В этой модели
энергия U, требуемая для переноса валентного электрона с одной
позиции на занятую орбиталь другой эквивалентной позиции, мала по
сравнению с шириной зоны, W(U <^W)\ в предельном случае
U = 0. Теория локализованных электронов применима тогда, когда
межатомные взаимодействия слабые и электроны прочно связаны
с атомным остовом. Локализованные электроны характеризуются
большим значением U (порядка потенциала ионизации атома минус
сродство к электрону в нулевом приближении) и малым значением
W(U ^> W). Когда U ~ W, мы имеем третью возможность сильно
коррелированных электронов в твердых телах. Внешние s- и р-элект-
роны, которые слабо связаны с атомным остовом и сильно
взаимодействуют с соседними атомами, хорошо описываются зонной
моделью. 4/-Электроны редкоземельных элементов, которые
экранированы от соседних атомов 5.925/?6-электронами, в твердых телах всегда
локализованы и хорошо описываются теорией поля лигандов.
Следует подчеркнуть, что 4/-электроны локализованы даже в
редкоземельных металлах, это говорит о том, что электропроводность
не всегда является свидетельством делокализации электронов.
Внешние d-электроны в соединениях переходных металлов
промежуточные; в неорганических комплексах переходных металлов d-электро-
ны локализованы, причем они могут быть описаны теорией поля
лигандов. В оксидах переходных металлов и родственных им
материалах мы сталкиваемся со свойствами как локализованных, так и
делокализованных электронов. В переходных металлах d-электроны
делокализованы, находясь в узких d-зонах, которые перекрываются
с широкими ,9 — р-зонами.
6.2.1. Зонная модель
Зонная теория — теория одноэлектронной независимой
частицы; эта теория предполагает, что электроны распределяются
среди ряда доступных стационарных состояний согласно статистике
Ферми — Дирака. Эти состояния задаются решениями уравнения
Шрёдингера
H¥nft=E¥n,, (6.1)
17*
260
Взаимосвязь структура — свойство
где гамильтониан Н включает не только член кинетической энергии
(р2/2т)1 но также потенциал внутрикристаллического поля V{x)y
который отвечает за взаимодействие электрона с другими частицами
в кристаллической решетке. V{x) имеет трансляционную симметрию
кристаллической структуры, а именно V{x + а) = V{x), где а —
постоянная решетка. Одномерные волновые функции (функции
Блоха), следовательно, имеют вид ехр(£, /с, x)unh{x), где unh является
периодичной, с той же периодичностью, что и потенциал внутри
кристаллического поля:
">п h(x + а) = unh(x). (6.2)
Волновая функция модулируется unk(x).
Каждая волновая функция характеризуется целым числом,
индексом зоны гс, и волновым вектором к. В отличие от простой модели
свободного электрона, в которой квадратичная зависимость энергии
от волнового числа к непрерывна, зависимость энергии от А: в зонной
модели квантована, имея разрывы на значениях к = п/а, 2п/а,
Зп/а и т. д. Зависимость энергии Епк от Ьк для различных значений
п описывает электронную зонную структуру кристаллического
твердого тела. Разрывы в энергии, которые вызывают разрешенные
(энергетические зоны) и запрещенные (зонные щели) области
энергии, вытекают из периодического потенциала: при значениях к =
= ±nnla электронная волна отражается по Брэггу, поскольку
к = ±nnla становится эквивалентной условию Брэгга, 2asin0 =
= тгЯ, так как к = 2яД, a sin0 = 1. Так, например, при к = п/а
решениями уравнения (6.1) является expU(jt/a);r]±exp[—i{nla)x],
что соответствует двум различным разрешенным энергиям,
приводящим к энергетической щели. При значениях /с, далеких от ±nnlay
энергия обладает квадратичной зависимостью от /с, как для
свободного электрона.
Применение статистики Ферми — Дирака показывает, что при
0 К все состояния, лежащие по энергии ниже энергий Ферми, EF,
заполняются электронами, тогда как значения выше EF являются
незаполненными. В металле EF лежит внутри зоны (наивысшая
занятая зона частично заполнена). В диэлектрике или
полупроводнике EF лежит между зонами в энергетической щели, разделяющей
самую высокую заполненную зону {валентная зона) и самую
низкую пустую зону {зона проводимости). Таким образом, зонная
теория Блоха — Вильсона классифицирует кристаллические твердые
тела на металлы и диэлектрики в соответствии с наличием или
отсутствием в их электронной структуре частично заполненных зон.
Схемы энергетических зон для различных типов твердых тел
показаны на рис. 6.1; примеры электронных материалов даны в табл. 6.1.
Представленная выше простая картина твердых тел не всегда
справедлива; существует еще класс кристаллических твердых тел,
известных как моттовские диэлектрики, электронные свойства кото-
Электроны в твердых телах 261
Рис. 6.1. Схематическое
изображение зонных структур твердых тел.
а _ диэлектрик (hT <c Eg)\ б —
собственный полупроводник (hT ~ Eg)'%
«иг — несобственные
полупроводники. Показаны соответственно донорные
ж акцепторные уровни в
полупроводниках п- и р-типов. д —
полупроводник; е — металл; ж — полуметалл;
верхушка валентной зоны лежит выше
нижнего уровня зоны проводимости.
рых полностью противоречат
элементарной зонной теории.
Типичные примеры моттов-
ских диэлектриков — МпО,
СоО и NiO, имеющие
структуру каменной соли. Здесь
единственными состояниями
в области Ферми-уровня должны быть Зй-состояния. d-Орбитали
катиона в структуре каменной соли должны расщепляться окта-
эдрическим кристаллическим полем анионов на подуровни t2g и eg.
В монооксидах переходных металлов TiO—NiO (?d2 — 3d8) d-уров-
ни должны быть частично заполнены и, следовательно, простая
зонная теория предсказывает им металлические свойства. Предсказание
справедливо для TiO и в некоторой степени для VO. Но стехио-
метрические МпО, СоО и NiO — хорошие диэлектрики с
антиферромагнитным упорядочением. Диэлектрическую природу FeO можно
понять, если мы предположим, что подуровень t2g полностью
заполнен для конфигурации 3d6 (если Fe2+ находится в низкоспиновом
состоянии), но диэлектрическую природу МпО, СоО и NiO нельзя
понять в рамках простой зонной теории. Аналогичные проблемы
возникают с оксидами переходных металлов, имеющими другие
структуры. Анализ симметрии показал [Kleiner, 1967], что полуторные
оксиды со структурой корунда и с 1, 3 и 5й-электронами на катион
в принципе могут быть диэлектриками: электрические свойства
Ti203(3d]), Cr203(3d3) и Fe203(3d5) действительно подтверждают это
предсказание; Ti203, однако, имеет металлическую проводимость
выше ~ 400 К. Согласно этой модели было бы наиболее вероятно
также ожидать, что V203(3d2), Mn203(3d4) и Co203(3de), если бы они
существовали в структуре корунда, были металлическими. В
действительности только V203 имеет структуру корунда выше ~ 150 К,
и эта фаза является металлической.
Поведение некоторых оксидов при 0 К как диэлектриков можно
объяснить, если прибегнуть к концепции антиферромагнитного
упорядочения; магнитный порядок ведет к удвоению размера
примитивной ячейки и, следовательно, к обменному расщеплению всех зон.
Это может иметь силу для NiO и МпО, но не для СоО; чтобы объяс-
Зона проеодимости
Е-Ес'—i ' I [е'ес'л'-Д'-4е.е^ '
Е«*-г
Е-Е,
kTj Е, иП.
E-EvH/iv/Д ^23e-Ev
E-EAi-i-
Валентная зона
l_U
Е-Е,
VW
E=EV
Е-Ес
262
Взаимосвязь структура — свойство
Таблица 6.1
Различные типы материалов для микроэлектроники
Металлические материалы:
Зй-соедиыения
4й-соединения
5с£-соединения
другие
Полупроводники или
диэлектрики:
Зс^-соединения
Ad- и ^-соединения
другие
Материалы с переходом
неметалл — металл *:
Зс^-соединения
^-соединения
4/-соеДинения
другие **
Материалы со сверхпроводящим
переходом
ТЮ, Сг02, TiS, CoS2, CuSa
NbO, Mo02, Ru02
Re03, A^WOg
Допированный полиацетилен, TTF—TCNQ
MnO, CoO, NiO, Fe203, Cr203, MnS, MnS2, FeS2
Mo03, W03, Nb205, Rha03
Органические кристаллы, например антрацен
V203, V02, V306, V407, Ve013, Ti203, Ti306,
NiS, CrS, FeS, NiS2
Nb02
EuO, SmS, SmTe
Допированные полупроводники (например,
P : Si), расширяемые металлы (например, Hg,
Cs при Т > 2200 К), пленки Хе—Na;
ТЮ, NbO, LiTi204, BaPb0 8BiQ g03, NbS2,
Pb0 9MoeS8, LuRh4B4
* В большинстве случаев это — термические переходы, за исключением
^-соединений (где переход идет под давлением).
** Допированные полупроводники, расширяемые металлы, растворы металл —
аммиак и пленки инертный газ — металл (где переход идет в результате изменения
концентрации или плотности донора).
нить поведение последнего оксида как диэлектрика, мы должны
предположить, что кристаллографическое искажение с понижением
симметрии ниже TN приводит к энергетической щели. Однако поведение
при более высоких температурах этим не объясняется, поскольку
предсказывается, что соединения при температурах выше TN
проявляют металлическую проводимость (МпО, СоО и NiO являются
полупроводниками при всех температурах). Элементарная зонная
теория не объясняет свойства этих оксидов даже качественно.
Причина этого заключается в том, что не учитывается электронная
корреляция, являющаяся результатом большого значения U и малого —
W [Hubbard, 1963]. Зонная теория, модифицированная с учетом
электронных корреляций и электрон-фононных взаимодействий,
должна предсказывать низкотемпературное диэлектрическое
состояние некоторых из этих оксидов. Электронная структура моттов-
ских диэлектриков рассчитана в рамках приближения Хартри —
Фока [Brandow, 1977]. В литературе есть обзор по расчетам энергий
зон методом Хартри — Фока, Ха-методами и согласно теории
функциональной плотности [Koelling, 1981]; что касается
полуэмпирических методов (см. [Messmer, 1977]).
Электроны в твердых телах
263
6.2.2. Модель локализованных электронов
Модель локализованных электронов, или теория поля ли-
гандов,— по существу, то же, что и теория Гайтлера — Лондона
для молекулы водорода. Модель предполагает, что кристалл состоит
из ансамбля независимых ионов, фиксированных в решетке на своих
позициях, и что перекрывание атомных орбиталей незначительное»
Когда межатомные взаимодействия слабые, внутриатомный обмен
(расщепление, согласно правилу Хунда) и электрон-фононные
взаимодействия благоприятствуют локализованному поведению
электронов. Это увеличивает время релаксации носителя заряда от 1СГ16 в
в обычном металле до ~ 10~12 с, время такого порядка требуется
при колебании решетки в полярном кристалле.
Локализованные d- или /-электроны сохраняют свой
одноатомный характер, за исключением того, что состояния, возникающие
от различных dn- или /"-электронов, расщепляются
кристаллическим полем и спин-орбитально взаимодействуют. Расщепления муль-
типлета, обусловленные спин-орбитальным взаимодействием,
больше, чем расщепления кристаллическим полем 4/п-уровней; для 3dn-
уровней — обратная зависимость. Различие в энергии
вырожденных уровней dn(fn) и dn+1(/n+1) соответствует свободной атомной
энергии С/, уменьшенной в результате межатомного взаимодействия
в твердом теле.
Модель локализованных электронов имеет то преимущество*
что она может предсказывать диэлектрическое основное состояние
твердых тел. При повышенных температурах, однако, электрон-
фононные и электрон-электронные взаимодействия становятся
существенными, особенно когда зоны узкие (например, d-зоны).
Выполнено много работ по изучению влияния электрон-фононных
взаимодействий на транспортные свойства оксидных материалов. Сила
электрон-фононного взаимодействия (полярона) определяет
эффективную массу электронов. Если взаимодействие достаточно большое»
носители заряда движутся вместе со связанной с ними поляризацией
(малый полярон), подвижность должна быть, следовательно, малой
и термически активизируемой. Вопрос переноса заряда
рассматривается в рамках классической теории диффузии. В присутствии
электрического поля диффузия электронов предпочтительно идет через
кристалл, образуя суммарный ток. Такие нескоррелированные
прыжки (хоппинг) поляронов встречаются в соединениях, имеющих один
катион в разных валентных состояниях (например, Fe304, Pr7012t
NaaV205). Если электрон-решеточное взаимодействие мало,
перемещение электрона подвергается слабому влиянию, и мы,
следовательно, имеем большие поляроны.
264
Взаимосвязь структура — свойство
6.2.3. Модель приближения химической связи
Для объяснения электронных свойств оксидов переходных
металлов и родственных им материалов найдено полуэмпирическое
приближение, базирующееся на кристаллохимии [Goodenough,
1971, 1974]. С помощью эмпирических критериев перекрывания
орбиталей катион — катион и катион — анион Goodenough
попытался объяснить природу с?-электронов в соединениях переходных
металлов и дать унифицированное описание магнитных и
электрических свойств большого числа оксидов и сульфидов переходных
металлов и родственных им твердых тел. Это, хотя и качестведное,
приближение обращено к химической интуиции и облегчает
понимание условий, которые приводят к локализованным и делокализован-
ным rf-электронам. Концептуальные фазовые диаграммы строятся
в рамках энергии переноса Ъц, которая определяется по уравнению
Ъи = WiHVj) cz euWj), (6.3)
где Н — член, отвечающий за взаимодействие, а егу- — одноэлект-
ронная энергия; Ъц определяет энергию взаимодействия между
локализованными орбиталями Wt и Ч^ на соседних подобных атомах i
и у; она пропорциональна интегралу перекрывания (Ч^-Ч^-). Хотя
Ъц нельзя точно измерить или рассчитать, можно предсказать
изменение этой величины в ряду изоструктурных твердых тел, поскольку
реально оценить относительные величины интеграла перекрывания
и орбитальных энергий.
В твердых телах, где взаимодействие катион — катион
значительное, btj можно связать с Д"1, где Д — расстояние катион —
катион. В тех случаях, когда взаимодействие катион — анион —
катион существенно, Ъц связывается с параметром ковалентного
смешения катион-анионных орбиталей, А. Для октаэдрически
координированных катионов, как в каменной соли и в структурах типа
перовскита, упомянутыми параметрами смешения являются Аа и
Ая в следующих «молекулярных» волновых функциях:
Чге - Ne(fe - АаФ0), (6.4)
Т^ЛГ*^-^), (6.5)
где Ne и Nt — постоянные нормировки; /е и ft — rf-орбитали
катиона с симметрией eg и t2g соответственно, Ф — spa- и /?л-орбитали
аниона соответствующей симметрии. В общем случае Аа > Ая, а Ъц ~
~ А2. При малых значениях Ъ внешние d-электроны локализуются,
а при больших — делокализуются. В ряду изоструктурных
твердых тел имеется критическое значение энергии переноса Ьс, которое
разделяет два состояния — делокализованных и локализованных
электронов. Поскольку Ъ связано с Д и А, выражения для
критических значений Дс и Ас даются согласно положению металла в Пери-
Электроны в твердых телах
265
одической системе, главному квантовому числу d орбитали, степени
окисления и общему значению спина катиона, а также
электроотрицательности аниона. Рассчитанные значения Rc в оксидах
структурного типа каменной соли, рутила и корунда действительно отделяют
вещества, показывающие локализованное электронное поведение,
от веществ, проявляющих коллективные электронные свойства.
Определены также критические энергии переноса Ът, Ъб и fecs,
характеризующие изменение различных физических свойств. В случае,
когда Ъ > Ът ~ bg, спонтанный магнетизм исчезает и появляется
металлическая проводимость. Другими важными электронными
свойствами, которые могут быть связаны с 6, являются: температура
антиферромагнитного упорядочения TN ~ b2/U для Ъ < Ьс и
ширина d-зоны W = 2zb, где z — число ближайших соседей.
Типичная концептуальная (Т — 6)-фазовая диаграмма для
случая одного d-электрона на взаимодействующую орбиталь (nt =: 1)
доказана на рис. 6.2. Физический смысл этой фазовой диаграммы
следующий. Магнитное взаимодействие в системе с щ = 1 —
антиферромагнитное. Поскольку TN пропорционально fe2, TN
увеличивается вплоть до критического значения Ьс. Для случая, когда Ъ <
< Ьс, локализованные электроны проявляют выше TN
парамагнетизм Кюри — Вейса. Когда Ъ > Ьс, состояния подобны зонным, при
bm исчезает спонтанный магнетизм (TN = 0 при Ъ = bm) и
предсказывается переход первого рода полупроводник — металл. Когда
Ь > bm, d-электроны действительно коллективизированные, они
имеют зависимые от поверхности Ферми электронные свойства и
парамагнетизм Паули. Для b > bcs при низких температурах сверх-
проводящее состояние может быть
4J стабилизировано. Wilson [1972]
считает, что отношение WIU
эквивалентно bij, и использует концеп-
Энершия переноса Ъ —•*
ГЧ-1
Рис* 6.2. Концептуальная фазовая
диаграмма Т — Ь для nt= 1 [Go-
odenough, 1974].
Рис. 6.3. Схематическая диаграмма
энергетических уровней Re03 [Go-
odenough, 1971].
266
Взаимосвязь структура — свойство
туалъные фазовые диаграммы Goedenough для понимания моттов-
ских диэлектриков (см. разд. 6.5) в соединениях переходных
металлов.
Чтобы применить концептуальные фазовые диаграммы для
интерпретации электронных свойств изоструктурного ряда соединений
переходных металлов, Goodenough строит одноэлектронные
диаграммы энергетических уровней, предполагая наиболее вероятной
гибридизацию катионных и анионных орбиталей. Эти диаграммы
похожи на диаграммы одноэлектронных молекулярно-орбитальных
уровней энергии изолированных молекул [Cotton, 1971 ], за
исключением того, что дискретные энергетические уровни становятся
зонными состояниями, там, где это необходимо, в зависимости от катион-
катионных и катион-анион-катионных взаимодействий в твердых
телах. Типичная диаграмма одноэлектронных уровней энергии
Re03 приведена на рис. 6.3. Мы видим большое сходство этой
диаграммы с диаграммой такого изолированного октаэдрического
комплексного иона, как Ti(H20)e+. В диаграмме Goodenough для Re03
заполненные связующие состояния (в основном, анионные 2s-, 2p-
уровни) и пустые разрыхляющие состояния (главным образом, кати-
ониые 6s-, бр-уровни) разделены большой щелью (~ 5 эВ),
получающейся в результате различия в электроотрицательности
составляющих атомов; 5с£-состояния катиона, которые являются
разрыхляющими по отношению к 2s- и 2р-состояниям аниона, располагаются
в щели. Электронные свойства материала определяются природой
d-состояний, локализованных или делокализованных. В Re03 обе
орбитали, t2g и eg* являются обобществленными из-за больших
величин %п и Яа; ^-уровень частично занят, что делает материал
металлическим. Подобная диаграмма применима к перовскитам АВ03;
локализованы или обобществлены состояния t2g и eg в перовските,
зависит от величины ковалентного смешения (Хя и Х^) орбиталей В
и О (см. разд. 6.4.1). Подобные диаграммы одноэлектронных
энергетических уровней могут быть построены для различных рядов
твердых тел, имеющих структуру каменной соли, рутила и пирита.
В разд. 6.4 мы будем обсуждать использование некоторых из этих
диаграмм.
6.3. СВОЙСТВА
Физические свойства твердых тел проявляются как отклик
на внешнее воздействие (физические силы). Такими важными
воздействиями являются механическое напряжение, электрическое и
магнитное поле и температура. Давление вызывает деформацию,
магнитное поле — намагничивание, а электрическое поле —
электрический ток. Физические свойства можно подразделить на четыре
категории: равновесные, свойства стационарного состояния, гисте-
Свойства
267
резисные и необратимые [Koerber, 1962; Newnham, 1975].
Равновесные свойства измеряются тогда, когда система находится в
тепловом равновесии с окружающей средой. При измерении свойств
стационарного состояния система не находится в тепловом
равновесии с окружающей средой, но она не изменяется во времени.
Примером такого свойства является удельное электрическое
сопротивление, которое связывает градиент потенциала с потоком заряда*
Гистерезисные свойства — это такие свойства, для которых трудно
определить функциональную связь между экстенсивными и
интенсивными переменными (связь между двумя переменными необратима,
она зависит от того, увеличивается или уменьшается интенсивная
переменная), типичным примером могла бы быть зависимость
намагниченности от магнитного поля для ферромагнитного материала.
Исследование необратимых свойств оставляет перманентное
изменение в материале. Например, твердость по Моосу определяется
царапанием. Равновесные свойства и свойства стационарного состояния
кристаллов выражаются математически с помощью тензоров [Nye,
1957]. Далее мы приводим краткое описание магнитных,
электрических, диэлектрических и оптических свойств твердых тел.
6.3.1. Магнитные свойства
Когда вещество помещается в магнитное поле Я, в нем
развивается определенная намагниченность М (магнитный момент на
единицу объема), согласно соотношению М = %Н, где % —
магнитная восприимчивость. Магнитная индукция определяется согласно
уравнению
В = Н + АпМ = [х#, (6.6)
где |i — магнитная проницаемость; мы видим, что \i = 1 + 4л%•
Вещества с малой отрицательной магнитной восприимчивостью
называются диамагнетиками, тогда как вещества с положительной
восприимчивостью известны как парамагнетики. Упорядочение
атомных магнитных моментов в твердых телах приводит к другим типам
магнитного поведения, которые будут обсуждаться далее.
Магнитные свойства обусловлены орбитальным и спиновым
движениями электронов в атомах. Соотношение между магнитным
моментом [А и угловым моментом J электрона с зарядом е и массой т
можно записать
|li = -(e/2m)J. (6.7)
Угловой момент квантуется в единицах fe/2-я, а самое низшее, не
равное 0, значение \i называется магнетоном Бора р,
р = eh/Anm. (6.8)
Его значение составляет 9,2732-1024 Дж-Г"1 (система СИ) или
9,2732-Ю"21 эрг-Э-1 (система CGS).
268
Взаимосвязь структура — свойство
Орбитальное движение электрона около ядра в атоме приводит
к магнитному моменту jjl, который связывается с величиной
орбитального углового момента L выражением
^=^ = PtL(L + 1)]1/a- <6-9>
Аналогично спиновый магнитный момент задается соотношением
^ = ^7 = 2HS(S + l)]1/a, (6.10)
где S — величина спинового углового момента, L и S —
соответственно орбитальные и спиновые квантовые числа. Эффективный
магнитный момент [х связан с величиной общего углового момента
/ уравнением
g-Фактор (Ландэ) является мерой относительного вклада спинового
и орбитального углового моментов в эффективный магнитный
момент, g = 0, когда 5=0; g = 2, когда L = 0. Уравнение 6.11
дает магнитный момент отдельного атома. Атом с угловым моментом
/ в магнитном поле имеет 2/ -j- 1 уровней энергии с энергиями
Еж = -tfMjH, (6.12)
где Мj принимает целочисленные значения в интервале от +/ до
—/. Для совокупности N не взаимодействующих атомов
заселенность различных уровней энергии подчиняется распределению Боль-
цмана. Относительная заселенность данного подуровня
p(EM)=::v(-EM/kT). (6.13)
2exp(-£M/W)
Общий магнитный момент может суммироваться, если известна
заселенность различных подуровней. Если расстояние между мульти-
плетными уровнями велико по сравнению с расщеплением уровней
с данным /, средняя намагниченность может быть выражена как
M = Ng$JBj(Y), (6.14)
где Bj(Y) известна как функция Бриллюэна, а Y = g$H/kT.
Bj{Y)==2J + i_coih
J +
4)rJ-^rcoth(y/2)- (6Л5>
Для парамагнитных веществ применимо условие У<С1, причем
в этом случае
5j(F) = (J + l)F/3. (6.16)
Молярная восприимчивость тогда будет
Хм = § с* NtfH (J + 1)/3*Г - f • (6-17)
Свойства
269
Это закон Кюри, в котором константа Кюри
С = Ng^4 (J + l)/3ft = PlttW2/3kT, (6.18)
где Peff = ^[J(J + 1)11/2. Уравнение (6.17) выведено в
предположении, что каждый парамагнитный атом является независимым.
В твердых телах во внешнем поле часто происходит взаимодействие
между атомными моментами. Поэтому парамагнитная
восприимчивость чаще описывается законом Кюри — Вейса (6.19), чем законом
Кюри:
Хм = f=V <6-19>
где 0 — константа Вейса. При выводе уравнения (6.17) сделано и
другое предположение: все атомы имеют одно и то же значение J.
В большинстве случаев это предположение обоснованно, так как
возбужденные J-состояния имеют энергии много больше, чем кТ.
Эффект примешивания более высоких энергетических состояний
проявляется в добавлении небольшого температурно-независимого ван-
флековского парамагнетизма к намного большей кюри-вейсовской
компоненте.
Редкоземельные металлы и их соли при обычных температурах
являются типичными парамагнетиками, подчиняющимися закону
Кюри — Вейса. Экспериментальные значения Peff обычно хорошо
согласуются с величинами, рассчитанными из значений J для
основного состояния, причем это находится в соответствии с
локализованной природой 4/-электронов. Sm и Ей проявляют расхождения с
теорией, которые объясняются наличием более высоких мультиплетов
с энергиями, сравнимыми с kT. Eu3+ заслуживает особого внимания;
основное состояние (7F0) с / = 0 для этого иона должно
предопределять нулевое значение Peff- Наблюдаемый ненулевой момент,
следовательно, обусловлен заселенностью более высоких
мультиплетов.
Парамагнетизм переходных металлов и их солей отличается от
парамагнетизма резкоземельных элементов. Это происходит из-за
участия с?-электронов в образовании связей в кристалле (см.
разд. 6.2.3). В соединениях первого ряда переходных металлов,
где катионам приписывается определенная электронная
конфигурация, соответствие между рассчитанным и экспериментальным
значениями Peff плохое. Если, однако, предположить, что орбитальный
момент не дает вклада в парамагнетизм и что момент полностью
обусловлен спином, то во многих случаях получается хорошее
соответствие между экспериментом и теорией. Причина такой потери
(тушения) орбитального момента лежит в неоднородном
кристаллическом поле среды, которое сильно взаимодействует с d-орбиталями
катиона.
270
Взаимосвязь структура — свойство
Делокализованные электроны простых металлов (Li, Na и т. д.)
имеют слабый температурно-независимый парамагнетизм
(парамагнетизм Паули), источник которого можно понять в рамках теории
свободного электрона [Kittel, 1976]. Большинство электронов в зоне
проводимости не отзываются на приложенное поле, так как
большинство состояний полностью заняты. Только электроны вблизи
Ферми-уровня EF с энергией, примерно равной кТ, вносят вклад
в восприимчивость. Эта часть приблизительно равна T/TF, где
TF = EF/k, Поскольку TF ^> T почти для всех доступных
температур, восприимчивость мала и задается уравнением
% ~ N$VkTF. (6.20)
Магнетизм в твердых телах более сложный, чем в изолированных
атомах, из-за возможности взаимодействия (спаривания) между
атомными моментами. Спаривание между моментами, которые
ответственны за кооперативный магнетизм твердых тел, имеет свое начало
в принципе Паули: электроны с параллельными спинами стремятся
удалиться друг от друга. Пара электронов с одинаковыми спинами,
каждый из которых локализован на одном атоме, по энергии ниже,
чем пара электронов с противоположными спинами, на величину
так называемой энергии внутриатомного обмена. Следовательно,
имеется статистическая корреляция для электронов с одинаковым
спином, окруженных пустотой, обусловленной локальным
обеднением. Это явление называется обменным взаимодействием, в нем
можно различить два класса. Прямой обмен происходит между
моментами на атомах, которые достаточно близки, чтобы иметь
значительное перекрывание своих волновых функций. Обменное
спаривание сильное, но уменьшается быстро с увеличением межатомного
расстояния. Кроме того, косвенный обмен спаривает моменты в
области относительно больших расстояний. Он может действовать
через посредничество немагнитного иона (суперобмен) или через
делокализованные электроны (RKKY) (Ruderman, Kittel, Kasuya,
Yosida). Суперобмен имеет место в диэлектриках, тогда как RKKY-
взаимодействие делокализованных электронов является важным в
металлах.
Обменная энергия Нех атомов i и/ в твердом теле, разделенных
расстоянием rtj и имеющих спины S( и Sj соответственно, может быть
выражена как
#ех = -2/Ы^. S;, (6'21>
где / называется обменным параметром. Для прямого
внутриатомного обмена как между двумя электронами, принадлежащими
одному и тому же атому, / положительно и приводит к правилу
Хунда. Для прямого межатомного обмена / может быть
положительно или отрицательно. Если / положительно, благоприятна парал-
Свойства
271
лельная ориентация спинов. Для отрицательного /
антипараллельная ориентация спинов приводит к низкой энергии. Для непрямого
обмена / может быть положительным или отрицательным
(суперобмен) или осциллирующим (RKKY).
В диэлектриках суперобмен описывает взаимодействие между
локализованными моментами иснов, которые слишком разделены,
чтобы взаимодействовать по прямому обмену. Он действует через
посредничество немагнитного иона. Суперобмен является
результатом того, что локализованные электронные состояния, описываемые
формальной валентностью, стабилизируются примесью
возбужденных состояний, включающей электронный перенос между катионом
и анионом. Типичный пример этого — линейное взаимодействие
катион — анион — катион в оксидах со структурой каменной соли,
где антипараллельная ориентация спинов на соседних катионах
благоприятна при ковалентном смешении р-орбитали аниона с d-
орбиталями каждого катиона. Возможен ряд суперобменных
взаимодействий в зависимости от структуры твердого тела и электронной
конфигурации катионов. Две важные формы суперобмена —
корреляционный суперобмен и делокализационный суперобмен. Делока-
лизационный суперобмен включает переход от одного катиона к
другому, благодаря катион-катионному или катион-анион-катионному
взаимодействию. Корреляционный суперобмен ограничивается лишь
катион-анион-катионным взаимодействием, поскольку он требует
спиновой корреляции между взаимодействующими катионами.
Описаны правила для определения знака и величины суперобмена
[Anderson, 1959; Kanamori, 1959; Goodenough, 1963]. Например,
линейное взаимодействие катион — анион — катион между
наполовину заполненными орбиталями является антиферромагнитным.
Взаимодействие катион — анион — катион иод углом 90° между
наполовину заполненными орбиталями является ферромагнитным
при условии, что орбитали связаны с ортогональными анионными
орбиталями. Суперобмен, включающий а-связи, сильнее, чем
суперобмен для д-связей. Так, температура Нееля TN (см. с. 274) для
LaCr03 меньше, чем для LaFe03. В монооксидах Зй-переходных
металлов TN увеличивается в ряду MnO < FeO < СоО < NiO, так
как в этом порядке увеличивается (i-взаимодействие. Среди катионов
одной и той же электронной конфигурации суперобмен сильнее
у катиона с высокой валентностью (например, Fe3+ > Мп2+).
Взаимодействие по RKKY обеспечивает механизм для
спаривания локализованных магнитных моментов в делокализованных
электронных системах. Это является уникальным, так как /
осциллирует от положительного к отрицательному, по мере того, как
расстояние между магнитными ионами изменяется. Этот механизм
можно понять следующим образом: магнитный ион индуцирует
осциллирующую поляризацию спина для электрона проводимости по
соседству с ним. С расстоянием от локального спина осциллирующая
272
Взаимосвязь структура — свойство
«
3
X
§
2
■3
Л,
2
3
ь
ф
к
!з
о
а
а
*
«
2
X
аль
ф
1:1
S
2
(0
я
ь
ф
X
маг
СО
а
со
с
*
2
ю
со
ч
О
2
со
X
*-
ф
я
&
2
a
а
-8-
1
со
а
со
с
а
ф
с
>>
и
2
СО
S
^
ф
ж
ь
СО
2
Миктомагнетизм
кластерное стекло
Идеальное
спиновое стекло
?
СО
55
f-
ф
ж
&
2
К
а
ф
о
si
со
2 £
X Ф
х х
Ф k
ж 2
о 2
5 s
£ о*
2 а
А&
Гелимагнетиэм
(асперомагнетизм
кристаллов)
Рис. 6.4. Магнитное поведение твердых тел. Показано 14 типов магнитного
поведения [Hurd, 1982].
поляризация спина затухает довольно медленно (пропорционально
г"3). Эту модулированную спиновую поляризацию делокализован-
ных электронов чувствуют моменты других магнитных ионов внутри
ряда, что приводит к осциллирующему непрямому обмену (рис. 6.5).
Взаимодействие по RKKY ответственно среди других свойств за
магнитное упорядочение в редкоземельных металлах и за ряд
явлений, включая ядерный магнитный резонанс в металлах. В разупоря-
доченных металлических системах, где расстояние между
магнитными ионами случайное, взаимодействие по RKKY ведет к
положительному и отрицательному обмену. Это ведет к так называемой
фрустрации. Фрустрированная система имеет не единственную
микроскопическую структуру в основном состоянии; в системе
существует бесконечный ряд эквивалентных состояний. Концепция
фрустрации имеет более широкое распространение в других
разделах науки, где она называется «структурной неустойчивостью».
Свойства
273
Теперь мы кратко опишем различные типы магнетизма в
твердых телах. Магнетизм в твердых телах может быть кооперативным
или некооперативным. В кооперативном типе магнетизма важным
является взаимное взаимодействие между моментами, тогда как
в некооперативном типе индивидуальные моменты ведут себя
независимо друг от друга. В твердых телах есть пять основных типов
магнетизма (см. рис. 6.4). Диамагнетизм, который является
свойством замкнутых электронных оболочек, представляет собой
некооперативный магнетизм, характеризуемый слабой отрицательной
температурно-независимой удельной магнитной восприимчивостью.
Идеальный парамагнетизм представляет собой другой
некооперативный магнетизм, который возникает из атомных моментов,
идентичных и локализованных в изотропном окружении, достаточно
отделенных один от другого. Температурно-зависимая восприимчивость
следует закону Кюри (уравнение (6.17)). Идеальный парамагнетизм
является скорее исключением, чем правилом, так как
восприимчивость реальных систем следует закону Кюри — Вейса. Поведение,
согласно закону Кюри — Вейса, предполагает кооперативный
магнетизм, который может проявлять себя, если тепловая энергия
моментов в парамагнетике достаточно снижена; при температурах
Т < Т0 существует упорядоченное магнитное состояние.
Ферромагнетизм является кооперативным явлением, в котором
имеется коллинеарный дальний порядок всех моментов в твердом
теле (/ > 0). Ферромагнитное твердое тело спонтанно
намагничивается даже в отсутствие поля. Чтобы максимизировать свою магнето-
статическую энергию, кристаллический ферромагнетик делится на
домены, которые спонтанно намагничиваются почти до насыщения,
но момент каждого домена ориентируется так, чтобы получить
общий нулевой момент. Внешнее поле изменяет размер доменов,
увеличивая домены благоприятной ориентации за счет других
доменов. Таким образом, при ферромагнетизме внешнее поле является
именно тем фактором, который необходим для того, чтобы на
макроскопическом уровне сделать очевидным упорядочение,
существующее на микроскопическом уровне. Обычно намагничивание резко
возрастает в более низких полях, когда домены с более
благоприятными ориентациями расширяются за счет других и насыщаются,
когда достигается максимум в доменной ориентации. С увеличением
температуры тепловая энергия увеличивается, становится сравнимой
с обменной энергией и в конечном счете превышает ее. Спонтанная
намагниченность, таким образом, с температурой уменьшается до
исчезновения при температуре Кюри Тс. Выше Тс идеальный
ферромагнетик становится парамагнетиком, подчиняясь закону Кюри —
Вейса. Известны как кристаллические, так и аморфные
ферромагнетики; типичными примерами последних являются различные
составы Fe, Co. Ni с В (металлические стекла) [Cahn,
1980].
18 Ч. Н. Р. Рао, Дж. Гопалакришнан
274 Взаимосвязь структура — свойство
Антиферромагнетизм, подобно ферромагнетизму,
характеризуется упорядочением дальнего порядка идентичных спонтанных
моментов. Но поскольку обменный параметр J отрицательный,
моменты соседних атомов точно противоположны; общая спонтанная
намагниченность вообще не наблюдается. Антиферромагнетик ниже
температуры Нееля TN (температура упорядочения) состоит из
двух идентичных взаимопроникающих подрешеток, в которых
спины одной подрешетки противоположны спинам другой подрешетки.
Простой антиферромагнетизм может существовать только в
кристаллических твердых телах, поскольку аморфную систему нельзя
разделить на две идентичные подрешетки. Большинство
антиферромагнетиков — диэлектрики (например, MnO, NiO, MnS и т. д.), но
антиферромагнетизм известен также среди металлов и сплавов
(например, %Сг, a-Mn, Pt3Fe). Антиферромагнетики с делокализован-
ными d-электронами не следуют закону Кюри — Вейса выше TN.
Ферримагнетизм требует две или более магнитные частицы,
которые химически различны. Они занимают два типа позиций
решетки в ферримагнетиках, образуя две подрешетки А и В, как
в шпинелях. Моменты ионов в каждой подрешетке взаимодействуют
ферромагнитно, но взаимодействие между моментами А и В
антиферромагнитное. Поскольку полные моменты А и В различны,
имеется результирующая спонтанная намагниченность. Температурная
зависимость ферримагнетизма похожа на температурную
зависимость ферромагнетизма, за исключением того, что спонтанная
намагниченность с ростом температуры уменьшается быстрее. В
парамагнитном состоянии имеется отклонение от закона Кюри — Вейса,
особенно вблизи Тс. Ферримагнетизм может также существовать
в аморфпом состоянии, но подрешетки А и В являются хаотичными.
Примерами аморфных ферримагнетиков являются LnFe2 (Ln — Gd
или Tb), где взаимодействия Fe — Fe и Ln — Ln являются
ферромагнитными, но взаимодействие Fe — Ln является
антиферромагнитным.
Магнитный порядок в ферро-, антиферро- и ферримагнетиках
включает простую коллинеарную группировку атомных моментов,
параллельных или антипараллельных. Это общая закономерность,
но бывают особые более общие случаи, когда возможны неколлине-
арные группировки. Магнитное состояние, в котором
локализованные моменты в кристалле упорядочены, но ниже некоторой
температуры упорядочения находятся в различных ориентациях, таких, что
некоторые из них более вероятны, чем другие, называется гелимагне-
тпизмом (рис. 6.4). MnAu3 является таким примером, в котором
моменты показывают геликоидальную структуру. В тетрагональной
структуре магнитные моменты лежат в плоскости аЬ,
перпендикулярной направлению с, но их направление вращается от одной плоскости
к другой примерно на 50°. Резкоземельные металлы проявляют
более сложные формы гелимагнетизма, где моменты вращаются скорее
Свойства
275
Рис. 6.5. Взаимодействие между
магнитными примесями по RKKY-механиз-
му. Взаимодействие между магнитной Дг)
примесью в начале координат 0 и на |
расстоянии г = ОА —
антиферромагнитное; взаимодействие со второй примесью 0
на расстоянии г = ОВ —
ферромагнитное. Видно, что взаимодействие носит
колебательный характер, оно медленно
затухает по зависимости 1/г3.
J
вдоль поверхности конуса, чем в плоскости. Некоторые твердые
вещества, состоящие из двух или более наклоненных под углом
антиферромагнитных решеток, все-таки сохраняют общую
намагниченность. Они называются слабыми ферромагнетиками или
наклоненными антиферр^магнетиками. Слабый ферромагнетизм может
возникать из различий в анизотропии отдельного иона или из
взаимодействия Дзялошинского — Мориа или из того и другого. NiF2
с рутильным структурным типом является примером слабого
ферромагнетизма, вызванного анизотропией отдельного иона.
Взаимодействие Дзялошинского — Мориа, которое проявляется в
асимметричной группировке анионных моментов, возникает в результате
сильного спин-орбитального взаимодействия аниона, которое нарушает
сверхобменное взаимодействие в определенных системах.
Типичными примерами слабых ферромагнетиков, являющихся результатом
такого взаимодействия, являются a-Fe203, CoC03 и CrFv
Признан ряд других типов магнитного порядка, причем многие
из них относятся к аморфным и разупорядоченным твердым телам
(см. IHnrd. 1982]). Эти новые типы магнитного порядка включают
мета-, сиеро-, асперо и сперимагнетизм, спиновые стекла и микто-
магнетизм (см. рис. 6.4). В аморфных твердых телах, где
кристаллическое поле изменяется от точки к точке, уравнение (6.21)
преобразуется, чтобы включить локальную анизотропию
tf=-SDi(S*)?-2/(r«)SrSjt (6.22)
г ij
где D — сила аксиального кристаллического поля, Sz — проекция
общего спина иона на локальное выделенное направление z.
Уравнение (6.22) выражает энергию взаимодействия, получающуюся как
из локальной анизотропии, так и из обменных эффектов. В аморфном
твердом теле D фиксированно и положительно, а выделенные оси
zt распределены хаотично.
В метамагнетизме наблюдается наведенный полем магнитный
переход из состояния с низкой намагниченностью в состояние с
относительно высокой намагниченностью [Stryiewski, Giordano, 1977].
Типичным метамагнетиком является антиферромагнетик ниже его
18*
276 Взаимосвязь структура — свойство
температуры Нееля, но с увеличением приложенного поля силы
кристаллической анизотропии преодолеваются, резко изменяя
магнитную структуру. Суперпарамагнетик — это ферромагнетик, у
которого размер частицы слишком мал, чтобы поддерживать
многодоменную структуру. Малый размер частиц устраняет
заторможенность на магнитных моментах, позволяя им флюктуировать так,
как в идеальном парамагнетике. При суперпарамагнетизме
гистерезиса зависимости намагниченности от поля не наблюдается (М от
Н — единственная кривая при данной температуре).
Суперпарамагнетизм отмечается тогда, когда тонкие ферромагнитные частицы
распределены в аморфных гелях или в немагнитной матрице. С перо-
магнетик — это такой магнетик, в котором локализованные моменты
данных частиц закреплены в случайных ориентациях, ни с общим
намагничиванием, ни с регулярным типом локального упорядочения,
независимо от ближайших соседей. Предполагается, что сперомагне-
тизм существует главным образом в системах, где Jtj беспорядочно
или отрицательно, но сильно фрустрировано.
Кристаллический сплав, содержащий магнитные атомы данного
вида, включенные в решетку немагнитного «хозяина», может
упорядочиваться сперомагнитно при охлаждении ниже критической
температуры. Это называется «спин-стекольным замораживанием»,
которое характеризуется пиком восприимчивости в низком поле при
температуре, обозначаемой Tsg- Хаотичность ориентации моментов
напоминает хаотичность атомных позиций в обычном стекле, а это —
спиновое стекло. Ориентации замороженных моментов
фиксируются неопределенно и не флюктуируют со временем, но выше Tsg
моменты флюктуируют с образованием парамагнитного состояния.
Существенным является то, что, когда Т —>• Ts&, различные спины
начинают взаимодействовать друг с другом по всей области дальнего
порядка и система стремится к конфигурации своего основного
состояния для специфического распределения спинов. Установлено
существование такого основного состояния для случайно
взаимодействующих спинов без какого-либо ближнего порядка [Edwards,
Anderson, 1975]. Типичными примерами спиновых стекол являются
железо или марганец, растворенные в меди или в золоте. Проблема
спинового стекла продолжает привлекать внимание [Ford, 1982;
Mydosh, 1982]. Спин-стекольное поведение обнаружено в LaNi03,
допированном Mn [Vasanthacharya et al., 1984], и в диэлектрических
системах, например, Eu^^Sr^S и Мп^^О (см., например, [Hauser,
Waszczak, 1984]).
Состояние спинового стекла существует в кристаллических
сплавах только в пределах ограниченной области концентрации
магнитной добавки. Концентрация должна быть достаточно высокой,
чтобы обеспечивать взаимные взаимодействия, но достаточно низкой,
чтобы избежать образования кластера. Для концентрации ниже
предела разбавления получается режим Кондо, а магнитные атомы
Свойства
277
Рис. 6.6. Изображение спинового 1/11*111 11 IIMTI *•••! tt I «It 1^11 •
стекла с примесями ~ 10 ат. %. IIIII Ц llil 11TZ111*1*11 • ПИ II1
В некоторых областях видны микто- •1«\1Пн|1111111««*•*!• !!• •!•!
магнетические кластеры [Mydosh, I ПЯ IIIIIIIII 1ГП\1 ПГШ^Г! I
19751. ••••••••••••••••••^•••••#*>ж« • •
11*11У1 * 111'YiYiW 111 tTt 11 1"У» 1
i i>: iy. iy.li 1 iZri i; г :у. :Т 1111111
• •••• •••• «ч\ • • • • • • • •-•■жъ • •••••••
экранированы от взаимодей- 1iym\ym11 iWl ЦП!!!!!!!!!! 1^11
ствия с другими магнитными IIIIIIItIIIIIIIlTlIII.il/IIIIII
атомами. Когда концентрация IГ11111 IT." 1}11•11111lllll 1/11 1}
магнитных атомов достаточно ШМПППППШЛГПП-кГПИ
велика, образуются кластеры. ГШПШГ^! I/I I ift ft I I^t 11111
Миктомагнетик похож на спи- Цу>^1! II11II III III1H1 111 11) 11
новое стекло, за исключением I^^MII>III^>IIIJlTlIITiTI-IIII
того, что локальные корреля- IIlMtHIIIIl/^I IIIIIIItit-MIi:
ции магнитных атомов из-за Yll III III ItlllHIIItl^ till IIIM
кластерирования являются
доминантными. Имеется прямой
обмен в кластерах, но моменты индивидуальных кластеров
косвенно спариваются путем взаимодействия по RKKY. Имеющее мик-
томагнитные кластеры 10%-е спиновое стекло схематично
представлено на рис. 6.6.
Проблема разбавленных магнитных примесей в системах с дело-
кализованпыми электронами вообще является особенно важной как
для магнетизма, так и для электропроводности. Взаимодействие
между изолированным атомом d- переходного металла и дел окал изо-
ванными электронами металла описывается s — d-смешением
[Bell, Caplin, 1975]. Из-за этого смешения делокализованный
5-электрон временно остается в d-состоянии перед тем. как туннелиро-
вать обратно в делокализованные состояния. Другими словами,
d-уровень гибридизуется с состояниями зоны проводимости, образуя
виртуальное связанное состояние (VBS). Концепция VBS способна
объяснить магнитные свойства примесей Зй-металлов в таких
металлических растворителях, как Al, Cu, Ag, Au и Zn. Таким образом,
VBS-модель объясняет, почему в некоторых растворителях примесь
переходного металла магнитная, а в других — нет. Например, Fe
магнитно в Au, Ag и Си и не магнитно в Zn и А1. VBS-примеси
бывают двух видов — локальные и зонные. Если смешение слабое,
доминируют локальные эффекты и магнетизм сохраняется. Сильное
смешение зонных состояний разрушает магнетизм. Сила смешения
приблизительно пропорциональна Ер металла-растворителя. Если
Е¥ высокая, как в А1(~ 12 эВ), все Зй-металлы в нем теряют свой
магнетизм. EF благородных металлов меньше (~ 4 эВ), и примеси
многих переходных металлов сохраняют в них свой магнетизм.
Наличие магнитной примеси в металлическом растворителе
спин-поляризует s-электроны. Это облако антиферромагнитной спин-
поляризации, резонирующее около магнитного иона, называется
278
Взаимосвязь структура — свойство
связью Кондо. Ниже характеристической температуры, называемой
температурой Кондо Тк, антипараллельное спиновое облако
нейтрализует момент иона и теоретически уменьшает его до нуля.
При абсолютном нуле и изолированной магнитной примеси
превалирует идеальная ситуация Кондо; в очень разбавленном магнитном
сплаве при низких температурах получается приблизительный
режим Кондо.
Кроме обсуждаемого упорядочения моментов в основном
состоянии, могут происходить различные магнитные возбуждения в
различных магнитных состояниях; это вопрос в литературе представлен
хорошо [Mattis, 1981]. Одним из элементарных возбуждений
является квантовая спиновая волна (магнон) в классическом
ферромагнетике. В основном состоянии ферромагнетика все спины
ориентированы параллельно. Мы можем образовать возбужденное состояние,
переворачивая один из спинов. Вместо этого мы можем позволить
всем спинам частичное обращение, в этом случае система спинов
принимает волноподобный характер. Элементарные возбуждения
спиновой системы, имеющей волноподобную форму, называются
магнонами. Магноны являются аналогами фононов, их можно
подобным образом рассматривать для вывода зависимостей магнонной
дисперсии. Магнонный спектр твердого тела можно определять по
неупругому нейтронному рассеянию.
6.3.2. Электрические свойства
Электропроводность при постоянном токе, теплопроводность,
эффекты Зеебека и Холла являются некоторыми общими электрон-
транспортными свойствами твердых тел, которые характеризуют
природу носителей заряда. По электрическим свойствам твердые
материалы подразделяются на металлы, полупроводники и
диэлектрики, в которых носители заряда движутся в зонных состояниях
(см. рис. 6.1); имеются другие полупроводники и диэлектрики, где
носители заряда локализованы и их движение включает
диффузионный процесс [Honig, 1981]. Мы кратко рассмотрим важные
соотношения, касающиеся интерпретации транспортных явлений в
твердых телах.
Выражение для электропроводности металла можно получить
в рамках теории свободного электрона. Когда прикладывается
электрическое поле Е, свободные носители в твердом теле ускоряются,
но ускорение разрушается из-за рассеяния колебаниями решетки
(фононами) и другими дефектами. Суммарный результат состоит в
том, что носители заряда достигают скорости дрейфа (vd) согласно
уравнению
<vd> = ^TL = -»dE, (6.23)
Свойства
279
где е — заряд электрона, т — его масса, т — время релаксации
(среднее время между фактами рассеяния) и ud — подвижность
дрейфа (скорость дрейфа на единицу электрического поля).
Плотность тока J можно выразить
J = ^<yd>=-^E. (6.24)
Уравнение (6.24) выражает закон Ома, где константа
пропорциональности между J и Е определяет электропроводность а (удельная
проводимость). Таким образом,
а = neh/m = пеЧ/mv, (6.25)
поскольку средняя длина свободного пробега I = vr, где v — общая
скорость (а не дрейфовая). Для металла i\ т и I в уравнении (6.25)
относятся к электронам с энергией EF, поскольку только такие
носители (вблизи Ер) могут рассеиваться. Таким образом, выражение
для электропроводности металла
/8я\1/з 27/
а = Ы пе1/т
или
ст=ЫЫ F v• (6-26)
где —3 < г < 3 является индексом рассеяния. Далее, согласно
уравнению (6.26), т представляет собой эффективную массу
носителей заряда. Температурная зависимость электропроводности в
металле возникает в результате изменения 1(и) с температурой;
I обратно пропорциональна числу фононов. Выше температуры Де-
бая плотность фононов изменяется как Т и, следовательно, аосГ"1,
тогда как ниже этой температуры плотность фононов изменяется
с Г3. Поскольку при низких температурах рассеяние слабое,
приводящее к эффекту памяти, а ос Г5.
В полупроводниках, где число носителей небольшое, их общее
количество соответствует приложенному полю. Средняя
кинетическая энергия -2mv2 может быть приравнена тг^» и> таким образом.
о = пеЧЦЗткТ)1'*. (6.27)
Для собственных полупроводников, в которых есть и электроны,
и дырки,
о = nte{un + ир), (6.28)
где щ = (NcNv)^2exp(-Eg/2kT). (6.29)
Здесь Nc и Nv являются эффективными плотностями состояний в
зоне проводимости и валентной зоне соответственно; и — подвиж-
280
Взаимосвязь структура — свойство
ности. Окончательное выражение для электропроводности
собственного полупроводника дается уравнением
о = 2eup{b + i)[2n(mnmp)V2kT/h2]V2exv(-Eg/2kT)4 (6.30)
где Ъ = ип/ир. Поскольку предэкспоненциальный множитель не
очень чувствителен к температуре, график зависимости In о от ЦТ
должен давать прямую линию с наклоном, пропорциональным Eg.
Для случая полупроводников, допированных донорами (гс-типа)
и акцепторами (р-типа), можно различить три области. В
низкотемпературной примесной области (kT <C Eg) электропроводность
дается уравнением
о= g^~Na) [le*(2nmnkTf*exV(-Ed/2kT)], (6.31)
где число доноров Nd больше числа акцепторов на единицу объема.
В уравнении (6.31) g — статистический весовой фактор; g = -~ для
электронов и 2 — для дырок. Таким образом, а со ехр(—Edl2kT),
поскольку Г1/4 в предэкспоненте имеет малое влияние. В
высокотемпературной (собственной) области уравнение (6.30) представляет
собой выражение для электропроводности. При промежуточных
температурах (истощенная область) примесная электропроводность
постепенно переходит в собственную, а температурная зависимость
электропроводности обусловлена, в основном, температурной
зависимостью подвижности, и ~ Т~п\ п~ у], поскольку число носителей
остается постоянным (все примесные уровни ионизированы).
Полупроводники, характеризуемые локализованными
носителями заряда {хопперы), отличаются от «делокализованных»
полупроводников, рассмотренных выше, тем, что подвижность в случае
хопперов активизируется, и = uQ(T)ex])(—EJkT), и выражение для
электропроводности
Nt
g [ Na
eu0(T) exp(- (Et + Eu)/kT), (6.32)
где gt и g — факторы вырождения, Nt — плотность локализованных
позиций, обеспечивающих носители заряда, Ех — энергия,
требуемая для ионизации носителей заряда, и Еи — энергия активации,
связанная с подвижностью.
Термоэлектрический эффект обусловлен градиентом в
электрохимическом потенциале, вызываемом температурным градиентом в
проводящем материале. Коэффициент Зеебека а — константа
пропорциональности между напряжением и температурным градиентом,
который вызывает это напряжение в отсутствие тока, он
определяется как (АУ/АГ), когда ДГ->0, где AV — термоэдс, вызываемая
Свойства
281
температурным градиентом А Г; он связан с энтропией на носитель
заряда (а = —S*le). Коэффициент Пельтье я — константа
пропорциональности между тепловым потоком, переносимым
электронами, и плотностью тока; а и я связаны соотношением а = л/Т.
Выражение для коэффициента Зеебека металлов:
_^Ite/3„,"-_±[£][£]. (6.33)
Таким, образом, металлы характеризуются малым |а|, который с
температурой увеличивается линейно.
Коэффициент Зеебека собственного полупроводника дается
уравнением
а
а=±7[— -Иг
(6.34)
где 5* — энтропия, связанная с переносом носителей заряда.
В таких ионных твердых телах, как оксиды металлов, S*/k <
<10мкВ/К. EF — энергия Ферми по отношению к
соответствующему краю зоны, а — положительно для дырочной проводимости
и отрицательно для электронной проводимости, поскольку
^ = In [с/(1 - с)] или In [-£ ], (6.35)
где (1 — с) (или N) представляет собой плотность незанятых
состояний в соответствующей зоне и с (или п)— плотность носителей
заряда в зоне. Для примесных полупроводников |а| обычно велико,
ет 0,1 до 1 мВ/К, и уменьшается с увеличением температуры.
Максимальное значение а наблюдается для малого сие изменением с от
0 до 1 изменяется знак а. Для локализованных примесных
полупроводников (малый хоппинг полярона) с обычно постоянно и,
следовательно, а является почти температурно-независимой.
Эффект Холла. Коэффициент Холла R для твердого тела с
единственным типом носителя заряда определяется
R = ± £, (6.36)
где Сг ~ 1(Зя/8 < Сг < 315я/512). Если R и о известны, то их
произведение является мерой дрейфовой подвижности
oR = neud(^) = Crud. (6.37)
Электрические транспортные свойства обеспечивают полезный
критерий для того, чтобы различить локализованные и
коллективизированные электроны в твердых телах. Таким образом,
температурная зависимость дрейфовой подвижности и для
коллективизированных электронов (Ь > Ьс) отличается от поведения для малых
282
Взаимосвязь структура — свойство
поляронов (Ь < Ьс). Для коллективизированных электронов и
изменяется как Г-3/2, и, когда зоны узкие, подвижность становится
термически активируемой; и со ex^(-EJkT), где Еа— энергия
активации для хоппинга. Для локализованных полупроводников,
имеющих прыжковую проводимость, подвижность небольшая
(<]0,1 см2/(В-с)), а в области зоны — большая (>1 см2/(В-с)).
Экспериментальное произведение коэффициента Холла и удельной
электропроводности дает меру дрейфовой подвижности. Квантовый
эффект Холла [Klitzing et al., 1980] найдет много приложений. Хол-
ловское сопротивление квантуется согласно соотношению R = h/ie2
(i — целое число).
Движение ионов через твердые тела приводит как к переносу
заряда, так и к массопереносу. В то время как перенос заряда
проявляет себя в виде ионной проводимости в присутствии
приложенного электрического поля, макроскопический массоперенос
(диффузия) имеет место при градиенте концентрации. Как ионная
проводимость, так и диффузия возникают в результате наличия в твердых
телах точечных дефектов (см. разд. 5.2). Для твердого тела,
проявляющего исключительно ионную проводимость, электропроводность
записывается как
а = 2 пгЯгеЩу (6.38)
i
где сумма берется по всем заряженным частицам i\ nt— означает
концентрацию £-го иона с зарядом qte и подвижностью ut. В галоге-
нидах щелочных металлов, где дефекты Шоттки приводят к
электропроводности, п обозначает концентрацию вакансий в катионной
и анионной подрешетках, пс и па. Для чистого кристалла пс = паг
но их подвижности не равны, причем ис намного больше, чем иа-
Выражение для электропроводности в твердом теле с дефектами
Шоттки, где доминирующий вклад в проводимость определяется
движением катионов, имеет вид
o=(^Jexp[-EA/kT]. (6.39)
Здесь сг0 = Ne2a2v/k для данного кристалла постоянная, ЕА = -~Е$-{-
+ Ет. N — число мест решетки одного вида на единицу объемау
а — длина прыжка, v — соответствующая частота колебания, Es —
энергия образования дефекта Шоттки, Ет — энергия активации,
требуемая для миграции иона.
Выражение для коэффициента диффузии:
D = va2 ехр [- ({ Es + Em) j кт\. (6.40)
Свойства
283
Сравнивая выражения для ионной проводимости и коэффициента
диффузии, мы видим, что
о Ne2 и е /а /л\
_=__ или ir = If. (6.41)
Уравнение (6.41) известно как соотношение Нэрнста — Эйнштейна,
первоначально выведенное для подвижности коллоидных частиц
в жидкости, но пригодное также для ионных твердых тел.
В ранее выполненном исследовании по ионной проводимости
данные представлены в виде выражения log а от Г"1, которое
показало две линейные области, соединенные «коленом», причем
положение «колена» зависит от чистоты образца. Но поскольку
подвижность ионов, обусловленная точечными дефектами, зависит не
только экспоненциально от Г-1, но также обратно пропорциональна Т
(уравнение (6.39)), более подходящей является зависимость log(aT)
от Г-1. Ранее, в гл. 5, мы показали типичный график для «чистого»
NaCl (см. рис. 5.2) и объяснили природу различных областей.
Энергии, связанные с различными процессами, можно легко оценить по
наклону линейных областей. Так, во внутренней области Ех п =
= Em«+ EJ2, Еш = Emc EIV = Етс + EJ2, где Е8 - энергия
образования пар Шоттки, Етс — энергия миграции катиона и Еа —
энергия ассоциации между примесью и противоположно
заряженной вакансией. Температурная зависимость коэффициента
диффузии катиона в галогенидах щелочных металлов имеет подобный
характер. Коэффициенты диффузии, определенные методом меченых
атомов, DT, и коэффициенты, полученные из электропроводности,
используя соотношение Нэрнста — Эйнштейна, Da, имеют
неуловимые различия, так как ионное движение в экспериментах с
мечеными атомами скоррелировано. Отношение Хавена, DT/D0,
пропорционально корреляционному множителю. Электрически
нейтральные дефекты вносят вклад в DT, а не в Da, и, следовательно,
отношение Хавена в присутствии нейтральных дефектов будет необычно
большим. Наличие электронной проводимости в твердом теле
приводит к необычно малым отношениям Хавена. В литературе широко
представлены данные по механизмам диффузии и ионной
проводимости в рамках точечных дефектов в ионных твердых телах [Corish
et al., 1977; Haven, 1978]. В некоторых твердых телах
проводимость одного из ионов необычно большая, сравнимая с
проводимостью водных электролитов; такая проводимость быстрых ионов
обсуждается в разд. 7.2. Ионный транспорт в стеклах
рассматривается в рамках теории слабого электролита [Ingram et al., 1980].
Эффект смеси щелочных металлов в ионных стеклах там, где два изо-
валентных иона замедляют друг друга, является удивительным.
284 Взаимосвязь структура — свойство
6.3.3. Сверхпроводимость
Когда электросопротивление материала исчезает ниже
критической температуры Гс>0К, мы называем это сверхпроводящим
переходом. Камерлинг Оннис впервые в 1911 г. наблюдал этот
переход в ртути. На рис. 6.7, а схематически показаны два возможных
типа перехода; резкий скачок в удельном сопротивлении — в
чистых гомогенных материалах и широкий переход — в негомогенных
материалах. Типичные удельные сопротивления материалов в
сверхпроводящем состоянии порядка 10~23 Ом-см, в металлах с обычной
проводимостью ~10-13 Ом-см. Температурный интервал АГС, в
пределах которого происходит переход между нормальным и
сверхпроводящим состояниями, может быть всего лишь 10"4 К или несколько
градусов в зависимости от материала. В сверхпроводящем состоянии
материалам свойствен удельный диамагнетизм, выталкивающий
любое магнитное поле ниже критического поля Нс (эффект Мейсснера—
Охзенфельда). Когда Н > Яс, сверхпроводимость разрушается и
материал переходит в нормальное состояние. Эффект Мейсснера —
Охзенфельда является фундаментальным свойством сверхпроводящего
состояния и часто используется как способ обнаружения
сверхпроводящих переходов. Энтропия сверхпроводящего состояния ниже
энтропии нормального состояния; разница в энтропии между
сверхпроводящим и нормальным состояниями материала вблизи О К
связана с электронной удельной теплоемкостью у: (S8 — Sn)t-*o = —У?»
Поведение у в сверхпроводящем состоянии отлично от его поведения
в нормальном состоянии; у в нормальном состоянии является
линейной функцией температуры, но в сверхпроводящем состоянии
ее температурная зависимость экспоненциальная. Сверхпроводящий
переход в нулевом магнитном поле является фазовым переходом
второго рода, поскольку наблюдается скачок в удельной
теплоемкости, а не изменение энтальпии.
Различаются два типа сверхпроводников, в зависимости от их
магнитного поведения. Материалы, в которых сверхпроводимость
при критическом поле Нс резко разрушается, известны как
сверхпроводники I рода (см. рис. 6.7, б). Нс— функция температуры.
Такое поведение имеют чистые материалы. В сверхпроводниках
f г^ *
ОТ Н Н И \А~
а б в
Рис. 6.7. Свойства сверхпроводников.
а — кривая удельное сопротивление — температура для
чистого (сплошная линия) и примесного (штриховая
линия) сверхпроводника; б — намагниченность как
функция внешнего поля для сверхпроводника I типа; в —
кривая намагниченности для сверхпроводника II типа.
Свойства
285
II рода магнитный поток начинает проходить сквозь материал
при поле Нс , которое ниже термодинамического критического
поля Нс. Затем с увеличением силы поля намагниченность постепенно
уменьшается до тех пор, пока при Н = НС2 сверхпроводящее
состояние полностью разрушается (см. рис. 6.7, в). Считается, что между
Нс и Нс материал находится в вихревом состоянии (стержнепо-
добные области с нормальной проводимостью внутри объема сверх -
проводящего вещества).
Возможность необычных технических приложений
сверхпроводимости обусловила ее широкое исследование; при этом число
сверхпроводящих материалов непрерывно растет. Среди них чистые
металлы, сплавы, интерметаллические соединения и
полупроводники. Среди важных структурных типов сверхпроводящих материалов
можно назвать следующие: фазы А15 (Р — W), каменная соль,
вольфрамовая бронза, перовскит, шпинель и фазы Шевреля;
соединения А15 (например, Nb3Ge, Nb3Sn) имеют Тс в области 20 К.
Известны тройные фазы, проявляющие сверхпроводимость [Subba
Rao, Balakrishnan, 1984]. Среди тройных сверхпроводников фазы
Шевреля являются необычными, так как они не только имеют самые
высокие критические магнитные поля *, но, что более интересно,
в некоторых из них сосуществуют магнитное упорядочение и
сверхпроводимость. Сверхпроводимость известна также для
стеклообразного состояния, типичные примеры — Be и Ga, температура
перехода которых в стеклообразном состоянии (8 К) выше, чем в
кристаллическом состоянии [Duwez, Johnson, 1978]. Некоторые
полупроводники становятся сверхпроводящими при высоких давлениях;
фазы Si и Ge при высоком давлении являются сверхпроводящими.
Хотя большинство немагнитных металлов проявляют
сверхпроводимость, существенно то, что щелочные металлы, а также такие высо-
копроводящие металлы, как Au, Ag, Си, не становятся
сверхпроводящими вплоть до очень низких температур. Некоторые
органические соли с переносом заряда типа бис(этилендитиоло)тетратиафуль-
вален трииодида (BEDT—TTF)2I3 проявляют сверхпроводимость
при обычных давлениях [Jerome, 1985]. Примечательно то, что это
вообще происходит в органических твердых телах при обычных
давлениях.
Неоднократно предпринимались попытки понять причину
сверхпроводимости и предсказать ее появление в материалах. Бардин,
Купер и Шриффер (БКШ) объяснили природу сверхпроводящего
состояния как результат образования связанных электронных пар
(куперовские пары), энергия которых меньше энергии свободного
электрона в обычном металле. Спаривание идет через взаимодейст-
1 Это было верным до открытия высокотемпературных сверхпроводников
на основе купратов (см. гл. 9). (Пер.)
286
Взаимосвязь структура — свойство
вие электрон — решетка таким образом, что электрон с волновым
вектором +к и спин, направленный вверх, спариваются с
электроном с волновым вектором —к и спином, направленным вниз. Теория
БКШ не отвечает на вопрос, какие вещества могут стать
сверхпроводящими, а какие — не могут. Высказано предположение
[Matthias, 1955], что появление сверхпроводимости сильно зависит от
кристаллической структуры и среднего числа валентных электронов
на атом. Например, в сплавах Nb3Ge и структурах ОЦК Тс
максимальна, когда концентрация электронов находится между 4—5
или 6—7. Сверхпроводимость объяснялась в рамках структуры и
химической связи [Krebs, 1975]. Согласно концепции Кребса
сверхпроводимость возможна только тогда, когда структура позволяет
образование протяженных (делокализованных) состояний, которые
по крайней мере в одном пространственном направлении не
пересекаются планарными или коническими узловыми поверхностями,
и если соответствующая энергетическая зона не полностью занята.
Несмотря на значительные успехи в этой области, невозможно
существенным образом поднять температуру сверхпроводящего
перехода (скорость ~37десять лет)2 [Mtiller, 1980]. По теории
сверхпроводимости имеется обзор x\llen, Mitrovic [1982], где обсуждаются
различные аспекты максимального значения Гс. По-видимому,
любой предел в Гс, определяемый мягкой модой, ошибочен; вероятно,
решающим фактором может быть решеточная нестабильность.
6.3.4. Диэлектрические и оптические свойства
Диэлектрическая постоянная среды является константой
пропорциональности в отношении между диэлектрическим
смещением и силой электрического поля. Поляризация среды
Р = гЛеЕ = 8,(ег- 1)Е, (6. 42)
где eD—диэлектрическая постоянная вакуума,
ег—диэлектрическая постоянная материала по отношению к вакууму и %е—
электрическая восприимчивость. Молярная поляризация в случае
твердого тела выражается уравнением Клаузиуса — Moccommu
ег + 2 р Зв,
(6.43)
V
где М — молекулярная масса, р — плотность, N0— число Аво-
гадро и а — поляризуемость.
Реориентация диполей в направлении поля характеризуется
временем релаксации т. Когда т мало по сравнению с частотой
приложенного напряжения, происходит мгновенная поляризация; когда
2 Как известно, ситуация резко изменилась в 1986 г., когда были открыты
новые высокотемпературные сверхпроводники (см. гл. 9). (Пер.)
Свойства
287
т большое, результирующая поляризация будет свободна от ориен-
тационного вклада. Дисперсия поляризации и, следовательно, гг
имеют место тогда, когда частота и т-1 близки друг к другу; ег в этой
области является сложной величиной, er = er — i'er. В области
дисперсии наблюдается отставание по фазе между приложенным
полем и мгновенной поляризацией; когда фазовый угол б мал,
tg б = ег/ег. Кроме того,
8г = еоо — ' 2°°2, (6.44)
1-|-(0Т
i-!Zi££L. (6.45)
1 +С0 Т
Здесь es и Goo — соответственно статическая (низкочастотная) и
оптическая (высокочастотная) диэлектрические константы.
Восприимчивость можно записать как
р Na/г
Х« = 1Х=ег~1= l-(AW3e,)- <6-4б>
Когда (Na/3ev)= 1, то и поляризация, и восприимчивость
стремятся к бесконечности. При критической температуре Тс разупорядо-
чивающий эффект температуры уравновешивается ориентирующим
эффектом внутреннего поля. При таких условиях %е определяется
по закону Кюри — Вейса:
371
Xe = 5-riv <6-47>
Здесь Тс— температура Кюри. Этот закон обычно записывается
в таком виде:
вг^еоо + у-^, (6.48)
где константа Кюри С = Np2/3evk. Он имеет значение при
обсуждении диэлектрических свойств сегнетоэлектриков и родственных
материалов.
Поведение диэлектриков в переменных электрических полях
можно рассматривать в рамках принудительной гармонической
осцилляции. Смещение тогда задается уравнением
еЕп exp (i сот/т)
со* — со + iya
где у — константа демпфирования, со0— нормальная частота
колебания, е и т — заряд и масса колеблющейся частицы. Комплексная
восприимчивость, % = %'— j%", может быть получена из этого
уравнения. Максимум в %" при частоте со означает, что материал погло-
288
Взаимосвязь структура — свойство
щает энергию поля (резонансное поглощение), а %' в этой области
изменяется с частотой быстро (аномальная дисперсия).
Диэлектрическая «катастрофа», которая имеет место тогда, когда отношение
между молярной рефракцией атомов в газообразном состоянии и
молярным объемом в конденсированном состоянии становится равным
единице, означает начало металлизации твердого тела [Edwards,
Sienko, 1983].
Эффектом электромагнитного излучения на вещество является
индуцирование диполя. В прозрачной диэлектрической среде
уменьшается лишь скорость электромагнитного излучения, которая
зависит от показателя преломления среды, определяемого плотностью.
Константа распространения электромагнитных волн определяется
по уравнению
Я = -^+-^, (6.50)
где пик — действительная и мнимая части показателя преломления.
Коэффициент поглощения равен 2к(о/с, в то время как коэффициент
отражения
R = (yi"~^a + f . (6.51)
("+1)2 + *2
Два показателя преломления связываются с функцией
диэлектрической проницаемости по уравнению
бг((о) = гг + is'r = [и (со) + i/c(co)]2, (6.52)
и мы, следовательно, имеем е/ = п2— к2 и ег = 2пк, которые
связываются друг с другом соотношениями Крамерса — Кронига
[Hodgson, 1971]. Если известно пк по всему широкому ряду частот,
можно определить п2— к2, и наоборот. Экспериментально измеряют R
и для понимания поведения системы используют соотношения
Крамерса — Кронига. На рис. 6.8 мы приводим спектр отражения
кристалла CdS и показатель преломления п, полученный из данных по
отражению с помощью анализа по Крамерсу — Кронигу [Balkan-
ski, 1971].
Отклик электронов в твердом теле на приложенное
электромагнитное поле истолковывается уравнением
©J + ю* + iy<*
где (ор — известная как плазменная частота, определяется как
[(4ппе2)/т]^2; со0 — истинная частота, т — масса электрона и -у —
константа демпфирования. Для свободных электронов, как в
металле, (о0= 0, а для диэлектриков со0 имеет конечное значение. Мнимая
Свойства
289
150200250300550
Волновое число, см"1
i ' i ■ |^|*Т
150 200 250 300 350
Волновое число, см"'
б
Рис. 6.8. Оптические свойства
CdS.
а — экспериментальный спектр
отражения монокристаллов CdS;
б — показатель преломления CdS,
полученный по данным а с
применением уравнения Крамерса —
Кронига.
и реальная части
диэлектрической постоянной при
плазмепной частоте
равны друг другу.
В непоглощающей области электромагнитного спектра
диэлектрическая постоянная прозрачного твердого тела изменяется как
функция частоты осциллирующего электрического поля.
Показатель преломления изменяется с частотой аналогично (он обычно
увеличивается с со). Такое дисперсионное поведение материалов имеет
значение при выборе материалов для призм и т. д. Твердые тела
поглощают электромагнитное излучение различными способами в
зависимости от природы связи.
Сильносвязанные электроны и ионы вызывают узкое
резонансное поглощение, электроны — в ультрафиолетовой области,
а ионы — в инфракрасной области. Резонансное поглощение в
инфракрасной области называется также поглощением излучения.
Поглощение излучения слабосвязанными электронами обычно
обусловлено межзонными переходами', такие полосы поглощения в
полупроводниках — широкие и без особенностей, но с острым краем
поглощения. Край поглощения в ZnS (Eg = 3,7 эВ)— при 0,34 мкм,
тогда как в InSb (Eg = 0,2 эВ)— при 6,2 мкм. Прямые межзонные
переходы имеют место, когда разница в энергиях зоны проводимости
и валентной зоны точно соответствует энергии перехода, hv =
- Ес(к) — Ev(k). Если при том же к нет минимума энергии в зоне
проводимости и максимума в валентной зоне, происходят непрямые
переходы.
Поглощение излучения имеет место в результате возбуждения
электронов примесей в твердых телах. Ионы переходных металлов
дают полосы поглощения, обусловленные d—d-переходами, которые
являются запрещенными переходами по Лапорту. Как донорные,
так и акцепторные примеси в полупроводниках дают полосы
поглощения в результате фотоионизации при энергиях ниже, чем энергии
щели. Экситоны (электрон-дырочные пары) в диэлектриках дают
характеристичные полосы, причем энергия зависит от того,
насколько сильна связь в электрон-дырочной паре.
Электроны в металлах и полупроводниках приводят к
поглощению свободного носителя, причем коэффициент поглощения
пропорционален квадрату случайной длины волны (следовательно, он
высок для большинства металлов в инфракрасной области). Отражаю-
19 ч. Н. Р. Рао, Дж. Гопалакришнан
290
Взаимосвязь структура — свойство
щая способность металлов связана с плазменной частотой сор
соотношением
Поскольку величина в круглых скобках для металлов очень малая,
отражающая способность вплоть до высоких частот почти полная.
Неупругое рассеяние излучения в твердых телах
устанавливается по Раман-эффекту, который включает зарождение или
аннигиляцию фононов или магнонов. Если участвует один фонон,
результат рассеяния рассматривается как Раман-эффект первого
порядка; в Раман-эффекте второго порядка участвуют два фонона.
Поляризуемость, связанная с фононной частотой, можно
представить в виде степенных рядов фононных амплитуд
а = а0+ ахи + «2^2+ аз^3+ ••• • (6.55)
В эффекте первого порядка аги всегда является конечной величиной;
эффект второго порядка возникает из-за появления члена а2и2.
Бриллюэновское рассеяние является специальным случаем раманов-
ского рассеяния, где участвующие фононы — из акустической
ветви.
Пара электрон — дырка, получаемая в фотоэкситонном
процессе, превращается в первоначальное состояние путем выделения
энергии в решетку через зарождение фононов или излучательную
рекомбинацию, приводящую к люминесценции. Такое снятие
возбуждений может быть индуцировано в местах примесей, называемых
активаторами (например, Т1+ в галогенидах щелочных металлов).
Процессы поглощения—эмиссии при фотолюминесценции не должны
идти на одних и тех же примесях. Например, в Са3(Р04)2, допиро-
ванном Се и Мп, поглощение идет на Се (сенсибилизатор), а
рекомбинация — на Мп (активатор). Этот процесс включает энергию
переноса между двумя центрами, и интенсивность люминесценции,
следовательно, зависит от среднего расстояния между примесными
центрами. Частота люминесценции зависит от иона активатора; так,
ZnS, допированный Ag+, Cu2+ и Мп2+, имеет эмиссионные полосы
соответственно в голубой, зеленой и желтой частях спектра. Пары
электрон — дырка могут вводиться в полупроводники при
наложении высоких напряжений, и излучательная рекомбинация
приводит к электролюминесценции. Для получения
электролюминесценции в красной части спектра в используемых обычно дисплеях
на светодиодах применяется GaP.
Лазерное излучение (распространение света посредством
стимулированной эмиссии излучения) в таких материалах, как рубин
(0,01 ат. % Сг3+ в А1203), можно понять согласно схеме на рис. 6.9.
Обращение заселенности достигается оптической накачкой энергии
на уровни 3 и 4, возбуждение с которых снимается через безызлу-
Несколько примеров
291
Рис. 6.9. Схематическое
изображение энергетических уровней
иона Сг3+ в рубине, используемом
в лазерном процессе.
§* hi)
% ^Накачка
<м
■ ■■и.Щ|У..Л1/««г
2(гЕ)
Эмиссия /5900 см
t(4A2)
чательныи процесс на
уровень 2. Лазерное излучение
происходит при переходе с
уровня 2 на уровень 1 (из-
о
лучение 6943А).
Полученный свет обладает особым свойством когерентности: он имеет ту же
фазу и направление распространения, как и падающий фотон.
Лазерное излучение получено в полупроводниковых диодах (сильно
допированных III—V-полупроводниках с р—гс-переходами) при
наложении смещающего напряжения.
До сих пор мы предполагали, что поляризация, получаемая
в твердом теле, пропорциональна электрическому полю (линейное
поведение). Полученные электрические поля в лазерных пучках
могут быть очень высокими, и, следовательно, становятся
значительными эффекты нелинейности. Поляризация тогда должна содержать
кратные частоты согласно уравнению
Р = e^i^o exp (hot) + ev%2E20 ex? (i2at) + е^з^о3 exp(i3o)*) + (6.56)
В случае линейного поведения вклад имеет только первый член.
Число фотонов с частотами 2со, Зсо ...будет малым. Генерация
гармоник при 2о) возможна лишь тогда, когда %2 = 0; это действительно
может быть в таких нецентросимметричных кристаллах, как КН2Р04
6.4. НЕСКОЛЬКО ПРИМЕРОВ
Связь между структурой и свойствами исследована в
большом многообразии твердых тел, таких как оксиды, халькогениды,
пниктиды и галогениды металлов. Кроме того, с целью изучения
модельных систем для проверки теоретических предсказаний
специалисты в области химии твердого тела получили новые
классы твердых тел, а также новые материалы на основе известных
типов твердых тел. В этом разделе мы выбрали три класса твердых
тел, а именно: оксиды, сульфиды и фториды металлов, чтобы
обсудить взаимосвязь структура — свойства. Свое внимание мы будем
особенно концентрировать на их электрических и магнитных
свойствах.
19*
292
Взаимосвязь структура — свойство
б/i.t. Оксиды металлов
Оксиды переходных и непереходных металлов удобно
обсуждать отдельно. Типичные оксиды непереходных металлов —
Na20, MgO, A1203 и Si02. Их электронная структура состоит из
заполненной валентной зоны (сформированной главным образом из
2р-уровней кислорода) и пустой зоны проводимости
(сформированной из внешних оболочек атомов металла), разделенных большой
щелью (~10 эВ). Следовательно, при обычных условиях они
являются диамагнитными диэлектриками. Поскольку собственная
энергия активации для электронной проводимости выше, чем энергияг
требуемая для получения и миграции точечных дефектов, ионная
проводимость в таких оксидах при умеренно высоких температурах
превалирует над электронной [Adler, 1975].
Оксиды переходных металлов входят в большую группу
твердых тел с разнообразными электрическими и магнитными
свойствами [Goodenough, 1971; Rao, Subba Rao, 1974]. Можно различить
два класса оксидов переходных металлов: оксиды, в которых ион
металла имеет ^-электронную конфигурацию, и оксиды, в которых
d-оболочка частично заполнена (табл. 6.2). Первый класс оксидов
имеет заполненную 2р-валентную зону кислорода и пустую d-зону
проводимости металла. Энергетические щели находятся в интервале
3—5 эВ; высокочистые материалы этого класса имеют собственную
электронную проводимость только при высоких температурах.
Оксиды металлов с с?°-катионами в октаэдрических позициях проявляют
спонтанные сегнетоэлектрические и антисегнетоэлектрические иска-
Таблица 6.2
Примеры различных типов оксидов переходных металлов
Оксиды <2°-металлов:
Sc203, ТЮ2, V206, Cr03, Zr02, Nba05, Мо03,
НЮ2, Ta206, W08
Оксиды ^-металлов:
TiO, NbO, Сг02, Re02, Ru02, Os02, Мо02,
Rh02, W02, Ir02, Re03,
Ti203, Ti306, Ti407, Ti609, V203, V305, V407,
V02, Nb02, Fe304
MnO, FeO, CoO, NiO, Cr203, Fe203, Mn304
Оксиды /п-металлов:
PrOa, Ln203 (Ln — редкоземельный элемент)
PrnOan-a>Tbn02n_2,EuO
Диамагнитные полупроводники или
диэлектрики без примесей; при
допировании или слабом
восстановлении проявляют
примесную проводимость /i-типа
Металлические и парамагнетики
Паули (Сг02 — ферромагнетик)
Проявляют температурно-индуци-
рованный переход неметалл —
металл
Моттовские диэлектрики
Диэлектрики или хоипинговые
полупроводники. Парамагнетизм,
характерный для
^-конфигурации. ЕиО проявляет переход
неметалл — металл
Несколько примеров
293
жения. Многие из них теряют кислород при высоких температурах,
становясь нестехиометрическими. Потеря кислорода или включение
в эти оксиды атомов электроположительного металла вводит
электроны в зону проводимости. Природа электронной проводимости
таких материалов зависит от силы электрон-фононного
взаимодействия и от ширины зоны проводимости, получаемой из d-состояний
металла. Когда взаимодействие велико, а зона узкая, образуются
небольшие поляроны, такие материалы проявляют прыжковую
проводимость, например Na^V^O^ Если зона проводимости широкая,
материал проявляет металлические свойства, например, NaxW03.
Оксиды переходных металлов, содержащих частично
заполненные d-состояния, могут обладать свойствами металлов или
полупроводников. Небольшая группа из них обнаруживает температур-
но-зависимые переходы неметалл — металл (см. табл. 6.2).
Магнитные свойства также совершенно разнообразны: от парамагнетизма
Кюри — Вейса, через спонтанный магнетизм, до парамагнетизма
Паули. В табл. 6.2 мы также приводим типичные оксиды
редкоземельных металлов, содержащих локализованные 4/п-электроны
[Eyring, 1974, 1979].
Оксиды металлов с ^-конфигурацией проявляют металлические
свойства, когда перекрывание между орбиталями валентных
оболочек атомов большое. Можно различить два типа металлического
поведения: одно обусловлено сильным катион-катионным
взаимодействием, возникающим в результате незначительного расстояния
катион — катион, а другое обусловлено сильным катиои-анион-
катионным взаимодействием, возникающим в результате большого
ковалентного смешения 2р-орбиталей кислорода с d-орбиталями
катиона. Некоторые изоструктурные серии оксидов переходных
металлов со структурами каменной соли, корунда, рутила, перовскита
и другими обнаруживают систематические изменения в электронных
свойствах, при этом по крайней мере один член серии проявляет
характеристичные свойства делокализованных электронов, а
другие — свойства локализованных электронов. В этих твердых телах
энергия Ферми, EF, которая совпадает с энергией множества dn-
уровней, лежит внутри энергетической щели между анионными
связующими и катионными разрыхляющими состояниями и
определяет электронные свойства. Мы будем обсуждать электронные
свойства нескольких типичных изоструктурных серий оксидов
переходных металлов в свете концепций, представленных в разд. 6.2.
Монооксиды Зй-переходных металлов от TiO до NiO имеют
структуру каменной соли и свойства, приведенные в табл. 6.3; тогда как
TiO и VO имеют свойства, характерные для делокализованных (или
почти делокализованных) d-электронов, МпО, FeO, СоО и NiO имеют
свойства локализованных электронов. Эти свойства можно
объяснить в рамках возможных катион-катионных и катион-анион-
катионных взаимодействий в структуре типа каменной соли
294
Взаимосвязь структура — свойство
Таблица 6.3
Свойства оксидов 3d-металлов со структурой каменной соли
Оксид
тю
VO
МпО
FeO
СоО
NiO
о
Я, А
2,94
2,89
3,14
3,03
3,01
2,95
о *
Яс, А
3,02
2,92
2,66
2,95
2,87
2,77
Свойства
Металл, парамагнетик Паули
Полуметалл, слабая температурная
зависимость х
Полупроводник, парамагнетик Кюри —
Вейса, TN = 122 К
Полупроводник, парамагнетик Кюри —
Вейса, TN = 198 К
Полупроводник, парамагнетик Кюри —
Вейса, TN --= 293 К
Полупроводник, парамагнетик Кюри —
Вейса, TN = 523 К
* Goodenough [1971].
(рис. 6.10, а). Прямое катион-катионное взаимодействие может идти
через перекрывание катионных ^-орбиталей по граневой
диагонали кубической структуры. Когда это взаимодействие сильное
(R <С Вс и Ъ > fcc), катионные ^g-орбитали превращаются в t2g*~
зону катионной подрешетки; если эта зона частично занята,
материал будет металлическим. Свойства TiO согласуются с этой
картиной (рис. 6.10, б). VO менее металличен, и его магнитная
восприимчивость при низких температурах становится зависимой от
температуры (рис. 6.11), так как больший заряд ядра сжимает
радиальную протяженность З^-орбиталей ванадия и, следовательно,
уменьшает перекрывание £2#-орбиталей. Увеличение заряда ядра
вдоль серии TiO—NiO приводит к понижению энергии множества
Зй-уровней по отношению к вершине анионных 2р-состояний. Это
имеет два последствия. 1. Радиальная протяженность З^-волновых
функций уменьшается, что приводит к уменьшению энергии катион-
катионного переноса Ьсс и обеспечивает локализацию £2#-электронов
для всех членов ряда после VO. 2. Энергия катион-анион-катион-
ного переноса Ьса, включающая ^-орбитали катионов,
увеличивается вдоль по ряду TiO—NiO, поскольку параметр ковалентного
смешения к между орбиталями катиона и аниона обратно
пропорционален разнице энергии между катионными d-состояииями и
анионными р-состояниями; эта разница в энергии уменьшается по ряду,
будучи минимальной для NiO. Структура каменной соли позволяет
катион-анион-катионное сверхобменное взаимодействие через eg-
орбитали атомов металла (см. рис. 6.10, а), которое объясняет
антиферромагнитное поведение МпО, FeO, СоО и NiO. Увеличение ТN
Несколько примеров
295
Рис. 6.10. а — орбитали в плоскости (100) структуры типа
каменной соли, показывающие катион-катион и катион-
анион-катионное взаимодействие; б — схематическая
диаграмма уровней энергии TiO.
в ряду объясняется в рамках увеличения энергии
катион-анионного переноса Ьс по ряду. Эти четыре оксида являются типичными
моттовскими диэлектриками, остающимися полупроводниками
вплоть до самых высоких температур (1400 К).
Ряд тройных оксидов LiM02 (M—V, Сг, Mn, Fe, Co и Ni)
кристаллизуется в упорядоченной структуре каменной соли. В
соединениях с М—V, Сг, Со и Ni ионы М3+ и Li+ упорядочиваются в
чередующихся катиониых плоскостях (111), вводя выделенную ось [111]
и ромбоэдрическую симметрию. В LiV02 расстояние V—V =
о о
= 2,84 А < i?c(V3+) = 2,95 А. Восприимчивость при высоких
температурах имеет характер типа Кюри — Вейса, но обнаруживает
резкий спад ниже 460 К, указывая, вероятно, на катион-катионное
связывание в плоскостях (111) [Bongers, 1975]. NaV02, который
изоструктурен с LiV02, не проявляет подобного магнитного
перехода, поскольку расстояние V—V в этом соединении больше, чем
i?c(V3+). Соединения АСг02(А—Li, Na, К), которые изоструктурны
с LiV02, имеют двумерный антиферромагнетизм [Le Flem et al.,
1977]. По-видимому, в LiCo02 и LiNi02 ионы трехвалентного
переходного металла находятся в низкоспиновом состоянии. Литий
может быть обратимо удален из некоторых оксидов химическим или
электрохимическим способами, что делает их полезными
электродными материалами в твердотельных батареях (см. гл. 3).
ЕиО является интересным оксидом в семействе структур типа
каменной соли [Honig, Van Zandt, 1975]. Он является
ферромагнитным (Тс = 69 К) диэлектриком, когда он чистый и стехиометрлч-
ный. В отличие от З^-оксидов сверхобменное взаимодействие в ЕиО
296 Взаимосвязь структура — свойство
1 i г 1 1 1 1 г—1
100 200 ЗОО
т,к
Рис. 6.12. Удельное
электросопротивление ЕиО, отожженного в
богатой европием (EuOl;)c) и кислородом
(Ей _ О) атмосфере [Honig, Van
Zandt, 1975].
является ферромагнитным, так как перенос заряда идет от 4/-орби-
тали к пустой 5й-орбитали. Электронную структуру можно
рассматривать как состоящую из локализованных 4/7-состояний Ей,
отделенных от пустых зон проводимости Eu, 5d (t2g) и 6s, энергией около
1,4 эВ. Ниже локализованных 4/7-состояний лежит заполненная
зона 2р-состояний кислорода. Стехиометрический ЕиО имеет при
300 кбар переход диэлектрик — металл. Механизм этого перехода
включает промотирование 4/7-электрона в 5^-зону с сопутствующей
флюктуацией валентного состояния европия. ЕиО имеет нестехио-
метричность с обеих сторон стехиометрического состава Еи^^О
и ЕиО^.. Транспортные свойства этих двух дефектных оксидов
различны. В Eu^^O дырки вводятся в 4/7-уровень, а проводимость
обусловлена небольшим прыжком полярона; не наблюдается
никакой аномалии в удельном сопротивлении при Тс. В ЕиО^^.
электроны вводятся в 5<2-зону. Образцы имеют резкий переход
полупроводник — металл при 50 К < Тс и выраженный максимум в удельном
сопротивлении выше Тс (рис. 6.12). Этот переход — удивительный,
так как он гигантский (удельное сопротивление в некоторых
образцах изменяется почти на 13 порядков), а металлическое состояние
наблюдается в области низких температур. Хотя электронные
свойства этого интересного оксида не совсем поняты, ясно, что
перерос. 6.11. Температурная
зависимость обратной мольной магнитной
восприимчивости VO . х изменяется
от 0,79 для верхней кривой до 1,32 —
для нижней, х = 1,02 и 0,99 для
кривых 5 и 6 снизу [Banus, Reed,
/Ю7П1
Несколько примеров
297
ход в EuOj.^ связан с резким изменением числа носителей.
Изменение, видимо, вызывается расщеплением зоны проводимости ниже
Тс. Стабилизированный зонный край пересекает донорный уровень,
связанный с анионными вакансиями, так что при низких
температурах донорные уровни отдают свои электроны в 5с?-зону. В другой
модели [Torrance et al., 1972] предполагается, что носители заряда,
захваченные вакансионными позициями, взаимодействуют с
магнитными моментами соседних ионов Еи2+ посредством обмена. В
низкотемпературном ферромагнитном состоянии все спины
выстраиваются в одном направлении, и взаимодействие не мешает прыжку
между позициями Ей. Волновая функция связывающего состояния
имеет тенденцию к увеличению и перекрывается в случае достаточно
большой концентрации дефектов так, что имеют место образование
зоны и металлическая проводимость. Когда температура
увеличивается, спины Ей постепенно разупорядочиваются и прыжок из
вакансии становится все менее вероятным. Электрон стремится быть
более локализованным, образуя так называемые магнитные поля-
роны. В магнитноразупорядоченном состоянии проводимость
осуществляется через прыжковый механизм (хоппинг).
Диоксиды некоторых переходных металлов кристаллизуются
в структуре рутила (см. рис. 1.7, б). Их электронные свойства
приведены в табл. 6.4. Структура рутила дает возможность
взаимодействию катион — анион — катион под углом 135° между
октаэдрами, сочлененными вершинами, и взаимодействию катион —
анион — катион под углом 90° между октаэдрами, сочлененными
ребрами. Кроме того, вдоль направления с возможно прямое катион-
катионное взаимодействие. Кроме октаэдрического
кристаллического поля катионов, тетрагональная структура дает аксиальное поле,
которое расщепляет трижды вырожденные г2£-орбитали на
невырожденную Ц- и дважды вырожденную ^-орбитали. Тогда как t^
может образовывать я*-зоны с ря-орбиталями аниона, Ц образует
d-зоны катионной подрешетки, если расстояние катион-катион вдоль
оси с R < Rc. Таким образом, структура рутила уникальна и дает
возможность материалу стать металлическим либо посредством ка-
тион-катионного и/или посредством катион-анион-катиониого
взаимодействия. На основании этих соображений рассмотрены
электронные свойства оксидов со структурой, подобной структуре
рутила [Rogers et al., 1969; Goodenough, 1971].
Некоторые диоксиды переходных металлов имеют искаженную
(моноклинную) структуру рутила, при этом расстояния металл —
металл вдоль цепочки чередуются, что указывает на образование
металл — металл-связей. Имеется прямая корреляция между
отношением с/а (рис. 6.13). и образованием металл — металл-связей в
семействе оксидов со структурой рутила. Отношение с/а для
конфигураций 3d1, 4d2, 5d2, Ad3 и 5d3 минимально, и соответствующие
диоксиды имеют моноклинное искажение, обусловленное образованием
298
Взаимосвязь структура — свойство
Таблица 6.4
Свойства диоксидов переходных металлов со структурой типа рутила
Оксид
Ti02
vo2
Сг02
P-Mn02
Мо02
wo2
Ru02***
о
R, А
2,96
2,88
(2,65; 3,12)**
2,92
2,87
2,52; 3,10
2,49; 3,08
3,14
о *
Нс, А
3,0
2,94
2,86
2,76
—
Свойства
Диамагнитный полупроводник, Eg~3 эВ
Переход полупроводник — металл при
Tt = 340 К
Ферромагнитный (Тс = 398 К) металл
Антиферромагнитный (TN = 94 К)
полупроводник. Аномалия в удельном
сопротивлении при TN
Диамагнитный металл. Структура,
искаженная до моноклинной симметрии в
результате образования связи М—М
Диамагнитный металл. Структура,
искаженная до моноклинной симметрии в
результате образования связи М—М
Паули-парамагнитный металл
* Goodenough, 1971.
** Моноклинная структура.
*** Os02f Rh02, Гг02 и Pt02 имеют похожие свойства.
металл — металл-связи. Среди диоксидов первого ряда переходных
металлов только Ti02 и V02 являются оксидами со структурой
рутила, межкатионное расстояние в них меньше Rc. Ti4+ вообще не
имеет d-электронов, и, следовательно, вещество является
диэлектриком. Ti02 легко восстанавливается, а образуемые таким образом
члены ряда Tin02n_1 имеют интересные электронные свойства [Rao,
Subba Rao, 1974; Honig, Van Zandt, 1975].
V02 при комнатной температуре моноклинный и переходит
в тетрагональную структуру при 340 К. Структурный переход
сопровождается переходом полупроводник — металл. В
тетрагональной форме, поскольку R < 7?с, £ц-орбитали перекрываются и дают
d-зону катионной подрешетки; эта зона перекрывается с я*-зоной,
образованной ^-орбиталями. Поскольку высшая зона частично
заполнена, терагональный V02 является металлическим (рис. 6.14, а).
В моноклинной форме катионы смещены с образованием пар
катион — катион вдоль оси с, причем расстояния, при которых идет
о
взаимодействие, чередуются между 2,65 и 3,12 А, вместо одного рас-
о
стояния 2,88 А в тетрагональной фазе. В этом моноклинном
искажении имеется также сегнетоэлектрическая компонента. При гомео-
полярном металл — металл-связывании искаженная структура
захватывает d-электроны V, а вещество является немагнитным и полу-
Несколько примеров
299
vo,
/ 2 i 4 £ в 7 6 9
Число d-электронов на катоон.
Рис. 6.13. Изменение отношения с/а
в оксидах типа рутила в зависимости
от d-электронной конфигурации
[Rogers et al., 1969].
\л-/з(а
*ц-А(1)
ftud)
tnd)
• tlx (f)
Рис. 6.14. Схематические диаграммы энергетических зон для рутилоподобных
оксидов.
а — тетрагональный Vo2; б — моноклинный Vo2, образующий металл-металл—связи t -
электронами; в — ферромагнитный Сг02, обладающий одновременным наличием
локализованных * -электронов и коллективизированных я*-электронов; г — МоО* и Wo2, с металл —
металл-связью и коллективизированными я*-электронами [Rogers et al., 1969; Goodenough,
проводниковым (рис. 6.14, б). В низкотемпературной фазе нет
магнитного упорядочения, а резкий спад в магнитной восприимчивости
при температуре перехода как раз является свидетельством
исчезновения ферми-поверхности. В разд. 6.5 мы обсуждаем переход
металл — диэлектрик в V02. В твердых растворах V02 с другими
оксидами со структурой типа рутила температура перехода
понижается замещающими примесями, которые расширяют решетку,
и повышается ионным замещением, которое сжимает решетку.
Эффект состоит не только в простом химическом моделировании
гидростатического давления; здесь важно другое: уменьшает или
увеличивает замещающий ион сегнетоэлектрическую компоненту
перехода.
Объяснение свойств Сг02, Мо02 и W02 не такое простое, как
в случае V02. Тетрагональный Сг02 с самым большим отношением
с/а (среди диоксидов Зс?-металлов) является ферромагнитным и
металлическим, тогда как и Мо02, и W02, оба — с искаженной
структурой, являются немагнитными и металлическими. Свойства
переходного металла с двумя d-электронами на атом решающим образом
зависят от того, R < Rc или R > Rc. В Сг02 один из d-электронов
находится в Ц, а другой — в t±; ^-электрон должен быть
локализован, поскольку R > Rc (большое отношение с/а). Но
^-электрон сам находится в я*-зоне, образованной посредством катион-
анион-катионпого взаимодействия. Внутриатомный обмен расщеп-
300
Взаимосвязь структура — свойства
ляет состояния на а- и р-спиновые компоненты (рис. 6.14, в). а-Спи-
новое состояние является единственным занятым состоянием, а
ферромагнитное катион-анион-катионное взаимодействие между
локализованными £ л-электронами и коллективизированными
^-электронами делает материал одновременно ферромагнитным и
металлическим.
В Мо02 и W02 R < Rc и один d-электрон на атом металла
включается в гомеополярную связь катион — катион посредством
£ц-орбиталей. Дополнительный d-электрон частично заполняет я*-
зону, делая оксиды металлическими (рис. 6.14, г). Эта упрощенная
картина, однако, не объясняет того факта, что порядок связи М—М
в этих оксидах больше единицы. По-видимому, истинная ситуация
является более сложной, включая одновременное участие t^-
электронов в образовании связи М—М, а также я-связи М—О.
В Мп02, с тремя d-электронами на катион, как £ц, так и я*-электро-
ны локализованы (R > Rc и Ьп<С &с), оксид имеет чистоспиновый
магнетизм и является полупроводником. В этом оксиде имеется
аномалия удельного сопротивления и удельной теплоемкости при
температуре магнитного упорядочения, TN= 94 К.
Полуторные оксиды первого ряда переходных металлов,
кристаллизующиеся в структуре корунда (Ti203, V203, Сг203 и а ==
= Fe203), имеют интересные свойства. Ti203 и V203 обнаруживают
переходы полупроводник — металл при температурах 410 К и 150 К
соответственно. В Ti203 переход широкий и в широком
температурном интервале. В V203 переход сопровождается
антиферромагнитным упорядочением. Сг203 и a-Fe203, оба — антиферромагнитные
диэлектрики. Эти свойства можно качественно разумно объяснить
в рамках катион-катионного и катион-анион-катионного
взаимодействий в структуре корунда [Goodenough,) 1971, 1974]. В этой
структуре важными являются два различных катион-анион-катион-
ных взаимодействия под углом (135 и 90°) и катион-катионные
взаимодействия (в базальной плоскости и вдоль гексагональной оси с).
Структура обеспечивает тригональную компоненту в октаэдрическое
кристаллическое поле, которое расщепляет ^-орбитали на alg
(направленную вдоль сн) и eg(n), направленную по базальной
плоскости. В Ti203 с одним d-электроном на атом и малым гексагональным
отношением с/а, оба расстояния катион — катион меньше, чем
критическое значение (Дсс < Rbb < Дс), a^-зона, следовательно,
заполнена и отделена от пустой ^(я)-зоны конечной щелью,
объясняющей полупроводниковое поведение при низких температурах. В V203
с двумя d-электронами на атом ванадия и большим отношением с/а
Rcc < Rbb< Rc, а ^(я)-зона является более устойчивой, чем а1ё-
зона. Металлическая природа ромбоэдрического V203 указывает
на то, что обе зоны до некоторой степени перекрываются (см.
разд. 6.5.).
В Сг203 и Fe203 alg- и ^(я)-орбитали заполнены наполовину.
В Сг203 ^-электроны локализованы, но a^-электроны, вероятно,
Несколько примеров
301
Рис. 6.15. Перовскитовая структура,
демонстрирующая возможность ка-
тион-анион-катионного
взаимодействия вдоль ребра куба.
являются промежуточными,
так как расстояние между
катионами в направлении с менее
Яс, что вызывает изменение
атомного момента с
изменением температуры. В Сг203, где
совсем нет ^(а)-электронов,
катион-аниои-катионное
взаимодействие слабее, чем в Fe203,
с двумя ^(а)-электронами.
В соответствии с этим TN в
Fe203 намного выше (953 К),
чем в Сг203 (307 К); Fe203 имеет слабый, паразитный
ферромагнетизм в интервале температур 253 < Т < 953 К. В этом
температурном интервале атомные моменты лежат почти в базаль-
ной плоскости. Антисимметричное спиновое спаривание
параллельно оси с, а анизотропный суперобмен ставит спины в базальной
плоскости под углом и создает суммарный момент. При Т < 260 К
спины располагаются параллельно оси с и паразитный ферромагнетизм
исчезает. При наложении сильного магнитного поля в направлении
с наблюдается спиновый переход первого рода в области 260 К
(Morin-nepexod).
Имеется ряд семейств изоструктурных тройных оксидов,
электрические и магнитные свойства которых изучены широко. В
ферромагнитных шпинелях (см. с. 41, 42, 274) сильное А—В-взаимодейст-
вие между катионами в тетра- и октаэдрических позициях
благоприятствует аитипараллельной ориентации спинов. Электрические
свойства оксидных шпинелей в значительной степени определяются
взаимодействием В-катионов. В гранатах (например, V3Fe5012)
антипараллельное спаривание магнитных моментов тетраэдриче-
ского и октаэдрического Fe3+ приводит к ферримагнетизму.
Структура перовскита идеально подходит для изучения катион-
анион-катионного взаимодействия (180°) катионов в октаэдрических
позициях (рис. 6.15); катион-катионное взаимодействие
незначительное из-за большого расстояния вдоль диагонали грани куба.
Оксиды со структурой перовскита имеют разнообразные
электронные свойства. Так, ВаТЮ3— сегнетоэлектрик,
SrRu03—ферромагнетик, LaFe03—слабый ферромагнетик, ВаРЬ^В^О'з—
сверхпроводник. Имеются обзоры по свойствам известных перовскитов
[Goodenough, Longo, 1970; Nomura, 1978]. Некоторые оксиды со
структурой перовскита имеют металлическую проводимость; типич-
302
Взаимосвязь структура — свойство
ными примерами таких соединений являются Re03, A^WOg, LaTi03,
AMo03(A—Ca, Sr, Ва), SrV03 и LaNi03. Металлическая
проводимость в оксидах со структурой перовскита обусловлена
исключительно сильным катион-анион-катионным взаимодействием.
На рис. 6.16 мы приводим важные оксиды со структурой
перовскита, содержащие атомы переходных металлов в В-позициях;
оксиды, имеющие одинаковую d-электронную конфигурацию, на рисунке
сгруппированы в колонки по Goodenough Ц971, 1974]. Колонки
составлены в порядке уменьшения энергии переноса b В-катион —
анион (В—О-ковалентность) сверху вниз. Параметры ковалентного
смешения Ха и Хп (и, следовательно, Ъа и Ьп) увеличиваются с
увеличением валентности В-катиона; для одинаковой валентности
смешение изменяется в таком ряду: Ъй > Ad > 3d. Влияние А-катионов
на В—О-ковалентность непрямое; кислотные А-катионы уменьшают
В—О-ковалентность, во всех соединениях Ха> Хп.
Штриховые линии на рис. 6.16, соответствующие Ъп— Ът (Ьт —
критическое значение для спонтанного магнетизма), Ъп=- Ъс и Ьа =
= Ьс, отделяют оксиды, характеризующиеся поведением
локализованных электронов, от оксидов со свойствами
коллективизированных электронов. Соединения в колонке 1 являются диэлектриками,
так как В-катионы имеют й°-электроныую конфигурацию.
Большинство соединений в колонке 2 (S ■■== 1/2) являются
металлическими и парамагнетиками Паули; линия bn= bm отделяет LaTi03
от GdTi03, так как GdTi03 является полупроводником с
температурой ферромагнитного перехода Тс= 21 К. АМо03 (A—Ca, Sr,
Ва) и SrCr03 в колонке 3 (S = 1) являются металлическими и
парамагнетиками Паули. Другие соединения в этой колонке являются
полупроводниками и антиферромагнетиками. Линия bn= bm
отделяет металлический и парамагнитный (парамагнетизм Паули) SrCr03
от антиферромагнетика-полуметалла СаСг03. Линия Ьп = ЪС
отделяет РЬСг03 от LaLaV03, так как последний проявляет
кристаллографический переход при Т < ТN, характерный для локализованных
электронов. По-видимому, область Ьт>Ъ^>Ъс должна быть очень
узкой, как обнаружено при исследовании электрических,
магнитных и других связанных с ними свойств. При исследовании этой
области важными являются эксперименты с применением давления.
Так, для СаСг03 dTNldP < 0, тогда как для YCr03 и СаМп03 dTNl
IdP > 0. Поскольку увеличение давления увеличивает Ьп (при
уменьшении размеров решетки), для Ъп < bc dTN!dP > 0 (поведение
по типу локализованных электронов) и для Ът > bn > bc dTNldP < О
(поведение по типу коллективизированных электронов).
Соединения в колонках 4—6 являются антиферромагнитными
диэлектриками. Поскольку внутриатомный обмен ~S(S + 1) уменьшает кова-
лентное смешение, естественно, что максимум на кривых bn— bc
и Ь0 = Ъс, соответствующий наименьшим значениям Ьл и 6С,
наблюдается в середине колонок с <S = 5/2. Редкоземельные ортоферриты
с S — 5/2 являются антиферромагнитными диэлектриками и прояв-
Спин В-катиона при локализованных d-зтктронйх
S=0
В8* W0
S=—
15 2
S=1
Ъ 2
S-2 S-
S-2
S=|- S-1
KxRe03
A+B5
л А2+В4+
A3+By
3 Re03
NaxW03
LaxW03 NaxReOs
KTaOj
KNb03
BaHf03
PbHf03
BaZr03
CaZr03
PbZrO,
BaTl03
SrTL03
CaTi03
EuTL03
PbTL03
LaY03
LaScOj
GdSc03
YScOj
KxMo03
NaxMo03
Sr»Nb03
SrV03
CaV05
BaMo05
StMo03 (SrTcOa)
CaMoO.
i;
SrCrOJ,
/CaCr03 taMn03
I
/ РЪСтО,
SrRu03
CaRu03
/ i ' ' \
LaTL03/ ,LaV03 UCr03JLaMn03 LaFeOj10-aCoQ3)j
/GdTl03 ._
/ , YV03 VCr03, YMn03 YFe03
/ Ъ^Ъяг-Ъс* Ъб>Ъс>Ък j Ьс>Ъб>Ъа1
Ъя~Ът b^sbc Ьв*Ъс
i
I 1
GdCr03|GdMn03GdFe03 GdCo03 1
I
1
\ SrFe03
\
\
v
\
\
\
\
\
\
\
\
S-тг S-0
SrlrO,
\ LaRh03
\ LaNl03 LaCo03
\ GdCoOj
\YNL03
Ъл*Ъс Ъп*Ъс Ьл«Ьт
Рис. 6.Iff. Перовскитоподо бные оксиды, содержащие ионы переходных металлов в различных спиновых
конфигурациях. Оксиды сгруппированы по энергии переноса b [Goodenough, 1971].
304
Взаимосвязь структура — свойство
ляют паразитный ферромагнетизм. Важными вкладами здесь
являются: а) спины Fe3+, наклоненные в общем направлении или из-за
кооперативного искажения кислородных октаэдров, или из-за
анизотропного суперобмена, и б) наклоненная антиферромагнитная
редкоземельная подрешетка как результат взаимодействия двух под-
решеток.
LaCoOg на рис. 6.16 показан дважды: в колонке с S = 2 и в
колонке с S = 0, в конце ее, так как Со3+ может находиться в низко-
или высокоспиновой конфигурации. Соединение имеет переход из
локализованного в коллективизированное электронное состояние
(см. разд. 6.5). В колонке 9 рис. 6.16 помещаются перовскиты,
содержащие й4-катионы. Из трех соединений в этой колонке SrRu03-
ферромагнитный металл (Тс = 160 К); CaRu03 —
антиферромагнетик (TN = 110 К) со слабым ферромагнетизмом. Поскольку оба
соединения имеют один и тот же мотив Ru03, изменение от ферромаг-
нитно-антиферромагнитного спаривания значительное. Хотя SrFe03
помещается в той же самой колонке на основании предположения,
что Fe4+: 3d4 находится в низкоспииовом состоянии, в недавней
работе [Takano, Takeda, 1983] показано, что Fe4+ в этом оксиде
находится в высокоспиновом состоянии вплоть до 4 К. С другой стороны,
CaFeOg проявляет диспропорционирование Fe3+^- Fe4+-> Fe5+ ниже
290 К. В предпоследней колонке, содержащей катионы с S — 1/2,
металлический LaNi03, обладающий парамагнетизмом Паули,
должен быть отделен от антиферромагнитных YNi03 и LuNi03,
указывая тем самым на то, что в LaNi03 ЪС> bm, а в YNi03 b0<C bm.
Аналогично LaCo03 должен быть отделен от LaRh03, так как последний
является полупроводником с узкой щелью, между заполненной
£2я(я*)-зоной и пустой ^(а*)-зоной.
6.4.2. Сульфиды металлов
Структуры и свойства халькогенидов переходных металлов
совершенно отличны от структур и свойств соответствующих оксидов
из-за большой ковалентности связей металл — халькоген. Кова-
лентность понижает формальный заряд на атоме металла и
благоприятствует образованию связей металл — металл. Факторы,
которые вносят вклад в различие между оксидами и сульфидами
металлов, следующие: а) большая поляризуемость анионов,
благоприятствующая образованию слоистых структур с ван-дер-ваальсовыми
связями между слоями, б) связь сера — сера, приводящая к
молекулярным анионам, и в) участие Зй-орбиталей аниона в образовании
связи, что стабилизирует тригонально-призматическую
координацию анионов. Электрические и магнитные свойства сульфидов
переходных металлов исследованы широко [Hulliger, 1968; Jellinek,
1972; Rao, Pisharody, 1976]. В то время как электрические свойства
сульфидов изменяются от полупроводниковых до металлических
Несколько примеров
305
(в некоторых случаях до сверхпроводящих), магнитные свойства
изменяются от слабого диамагнетизма (через локализованный
магнетизм) до температурно-независимого парамагнетизма Паули. Эти
изменения рассмотрены в общем виде на основании качественной
зонной модели [Jellinek, 1968]. Проведено сравнительное изучение
свойств фторидов, оксидов и сульфидов двухвалентных переходных
металлов в рамках концептуальных фазовых диаграмм (см.
разд. 6.2.3) [Goodenough, 1974].
Общие признаки моносульфидов и дисульфидов переходных
металлов можно разумно объяснить в рамках диаграмм одноэлек-
тронной энергии, похожих на такие диаграммы для оксидов; (п -\-
+ \)s- и (п + 1)р-орбитали переходных металлов взаимодействуют
с 3s- и Зр-орбиталями серы с образованием валентной зоны (в
основном благодаря сере) и зоны проводимости (в основном благодаря
металлу), причем энергетическая щель между ними составляет
несколько электрон-вольт. тгй-Орбитали переходного металла, которые
приходятся на щель и являются частично заполненными, определяют
электронные свойства. Они могут оставаться локализованными на
катионе или образовывать зонные состояния в зависимости от
кристаллической структуры и межъядерных расстояний. В некоторых
случаях d-зоны перекрываются либо с зоной проводимости, либо с
валентной зоной. d-Состояния обычно расщепляются на подуровни,
благодаря полю лигандов, искажению Яна — Теллера и
внутриатомному обменному взаимодействию. Кроме этого, металл — металл-
взаимодействия могут также расщеплять d-зоны на узкие подзоны.
йп-Множества в моносульфидах Зс£-металлов становятся более
устойчивыми с увеличением атомного номера до тех пор, пока
оболочка заполнится наполовину, как и в монооксидах. Большая ко-
валентность в моносульфидах, однако, ведет к меньшему значению
U и, следовательно, к меньшей энергии переноса для образования
делокализованных состояний. Меньшая энергия переноса означала
бы также большее критическое расстояние Пс для образования d-
зон катионной подрешетки через катион-катионное взаимодействие.
Такие d-зоны катионной подрешетки образуются в TiS и VS. Стехио-
метрический VS неустойчив при атмосферном давлении, и,
по-видимому, он диспропорционирует на V7S8 и V9S8. Магнитная
восприимчивость нестехиометрических фаз V^^S (рис. 6.17) обнаруживает
наличие как локализованных, так и делокализованных d-электро-
нов. В CrS и MnS d-электроны локализованы. Локализованные
моменты, найденные в CrS и MnS, а также искажение Яна — Теллера
в CrS обнаруживают, что 6а< Ьс. В CoS, NiS и CuS мы находим
определенное доказательство для делокализованных а*-электронов
благодаря тому, что Ьа > Ьс. В CuS значение U очень маленькое,
поскольку ba > bm, что приводит сР°-множества меди близко к
вершине валентной зоны, делая таким образом сомнительной формальную
валентность меди. Действительно, структура CuS (ковеллит) отли-
20 Ч. II. Р. Рао, Дж. Гопалакришнан
306
Взаимосвязь структура — свойство
,4000
>
ъ
с;
о
го
Z
1
зооо-
200СИ
7000-1
Рис. 6.17. Удельная магнитная
восприимчивость сульфидов ванадия как
функция температуры и состава [De
Vries, Haas, 1973].
чается от структуры других
моносульфидов, содержащих
два типа атомов меди и ионов
S2- и S2~. Электронные
свойства FeS (рис. 6.18)
обнаруживают, что Ъ0
авизированные d-электроны.
Ьс; в этом
сульфиде сосуществуют как де-
локализованные, так и коллек-
Высокотемпературная (NiAs)
структура FeS искажается при 413 К с образованием треугольных
металлических кластеров. Спонтанный магнетизм (TN = 600 К),
спиновый переход при 433 К и высокоспиновое состояние
железа указывают на то, что в FeS электроны с а-спином
локализованы, а электроны с (3-спином делокализованы. NiS
со структурой NiAs является метастабильным и при TN — 264 К
претерпевает фазовый переход первого рода без изменения
симметрии кристалла; изменение электрических свойств (см. рис. 6.18)
при TN указывает на то, что переход осуществляется от сильно
коррелированной антиферромагнитной фазы к слабо коррелированной
Паули-парамагнитной фазе и что Ь0 = Ьт. Фотоэмиссионные
исследования моносульфидов Зй-металлов в области валентной зоны
согласуются с их электронными свойствами, показывая четкий
металлический характер для TiS, VS и CuS и диэлектрический характер
для CrS и MnS [Gopalakrishnan et al., 1979].
Электронные свойства дисульфидов переходных металлов 4, 5
и 6-й групп, кристаллизующихся в структуре Cdl2 (IT), MoS2 (2Н,
3R) и подобных политипных структурах, разумно объясняются
с помощью подобных рассуждений. Ковалентыое взаимодействие
между четырехвалентными катионами и сульфидными ионами такое
сильное, что во всех образуются коллективизированные d-состояиия.
Их свойства решительным образом зависят от электронной
конфигурации и координации переходного металла. Так, можно ожидать,
что дисульфиды й°-катионов (Ti, Zr, Hf) являются диамагнитными
полупроводниками, дисульфиды ^-катионов (V, Nb, Та) —
металлами и дисульфиды й2-катиоиов (Mo, W) в тригонально-призматиче-
ской координации — полупроводниками (рис. 6.19). Электрические,
магнитные и оптические свойства слоистых дисульфидов [Hulliger,
1977; Subba Rao, Shafer, 1979] в общих чертах согласуются с этой
картиной, за исключением TiS2, который имеет металлический
характер. По-видимому, TiS2 приобретает металлический характер,
путем слабого перекрывания пустой Зй-зоны с верхушкой заполнен-
Несколько примеров
307
Рис. 6.18. Изменения
физических свойств NiS и FeS при
фазовом переходе.
а — удельная теплоемкость; б —
удельное сопротивление; в — тер-
моэдс; г — удельная магнитная
восприимчивость и д —
параметры решетки. [Coey et al., 1976].
ной валентной зоны (см.
рис. 6.19). Это
заключение подтверждается
фотоэмиссионными;
исследованиями [Williams, 1976],
которые показывают
наличие определенной
плотности состояний вблизи
EF в отличие от других
дисульфидов элементов IV
b группы. Первый пик на
спектре TiS2, несомненно,
является пиком d-типа и
показывает ожидаемое
увеличение в
интенсивности между Hel и Hell.
Результаты подтверждают
i 80-\
Х70\
\бо\
о" so]
10г-
а
"^ 40-
Л 20-
со
т о-
^ 4~
^ 2-
о
«4»
Q^SfiO-
Л 3,4.3-
1С
*
Ц
a
\6
в
И
и
350
400
т,к
FeS
450
наличие в этом соединении, в отличие от других дисульфидов
элементов IV b-группы, определенного перекрывания Ti (3d)/S(3p).
Предложены качественные схемы энергетических зон для ди-
халькогенидов элементов Vb и IVb-групп, содержащих переходные
металлы в тригонально-призматической координации [Wilson, Yof-
fe, 1969; Huisman et al., 1971]. Тригонально-призматическое поле
лигапдов расщепляет d-орбитали на три типа орбиталей: dz2 (более
низкая по энергии), dx2_y2, dxy и dxz, dyz, которые образуют узкие
зоны. d-Зоны могут перекрываться или могут не перекрываться с
валентной зоной или зоной проводимости. В одной модели [Huisman
et al.) имеется значительное перекрывание d-зон с зоной
проводимости и валентной зоной, тогда как в другой (Wilson — Yoffe) d 2-
'4s,4P(TL)
3d(r0E
N(E)
>4d(M)
зона не перекрывается с
валентной зоной. Обе модели
объясняют металлические и
полупроводниковые свой-
Рис. 6.19. Схематическое
изображение зонных структур.
а — TiS2 (IT); б — NbS, (2H); в —
MoS2.
20*
308
Взаимосвязь структура — свойство
ства соответственно d1- и d2- дихалькогенидов. Расчеты зонной
структуры и фотоэмиссионные исследования подтверждают многие
из признаков простых моделей. Дисульфиды и диселениды
переходных металлов Vb группы при низких температурах имеют вну-
триполитипные переходы благодаря образованию волн зарядовой
плотности (см. разд. 4.9). Эти переходы сопровождаются
аномалиями в магнитной восприимчивости, а также в
электросопротивлении.
Пиритные дисульфиды MS2 (M = Мп—Си), состоящие из ионов
S2~n двухвалентных катионов в октаэдрических позициях,
демонстрируют широкий набор электронных свойств [Bongers, 1975]. MnS2
является антиферромагнитным (TN = 78 К) диэлектриком; восири-
имчивость выше ТN характерна для высокоспинового Мп2+ (3d5).
FeS2 является немагнитным полупроводником, что говорит о
наличии низкоспинового Fe2+ (3de). CoS2 является ферромагнитным
металлом (Тс = 115 К); магнитное насыщение соответствует 0,8 не-
спаренных электронов на атом кобальта. NiS2 неожиданно является
антиферромагнитным полупроводником (TN ~ 50 К, 9 = —1800 К);
CuS2 — металлический и сверхпроводящий (Тс < 1,5 К).
Изменения в свойствах пиритов MS2 от локализованных d-электронов
в MnS2 до сильно скоррелированных делокализованных электронов
в CoS2 и NiS2 и до слабо скоррелированных делокализованных
электронов в CuS2 можно понять в рамках ожидаемого изменения ba ~
~ f-'g'kfj2 при переходе от одного соединения к другому. В MnS2 d-
электроны локализованы, как в МпО и MnS. В других пиритах
валентное смещение е^-орбиталей металла посредством а-связывания
достаточно сильное (ba > for), что дает не только узкую а*—зону
делокализованных d-электронов, но также и низкоспиновое
состояние для катионов. Так, FeS2 имеет заполненную t2g{en и 0])-зону,
которая отделена от ^(а*)-зоны некоторой зонной щелью ~0,9 эВ
(рис. 6.20). d-Зоны CoS2 и NiS2, по существу, похожи, за
исключением того, что а*-зона расщеплена корреляционным
взаимодействием. Заполненная на одну четверть, е#*-зона CoS2 является спин-
поляризованной, образуя а- и Р-спиновые компоненты. Если бы
две компоненты не перекрывались, CoS2 был бы ферромагнитным
полупроводником, а ферромагнитный момент соответствовал бы
одному электрону. Металлическая проводимость и уменьшенный
момент CoS2 указывают на малое перекрывание ос- и Р-спиновых
компонент. NiS2 с наполовину заполненной а*-зоной является моттов-
ским диэлектриком; е^-зона в результате отталкивания электронов
расщепляется на высоко- и низкоспиновые подзоны. В отличие от
NiS2, NiSe2 является металлом, а твердые растворы NiS2_xSex
[Bouchard et al., 1973; Wilson, 1985] обнаруживают переход
полупроводник — металл (рис. 6.21). При высоких давлениях (30—40 кбар)
NiS2 обнаруживает переход в металлическое состояние [Wilson,
1972]. В CuS2 ba > fem, a d-зона Си, вероятно, опускается ниже вер-
Несколько примеров
309
Fe
FeS,
Рис. 6.20. Схематическое изображение зонной структуры пирита
FeS2 [Goodenough, 1974].
шины анионной Зр-зоны, порождая дырки, что подтверждается р-
типом металлической проводимости. Рентгеновские фотоэлекарон-
ные спектры пиритных дисульфидов в остовной и валентной областях
согласуются с электронными структурами, обсуждаемыми здесь
[van der Heide et al., 1980].
Трисульфиды (и триселениды) Ti, Zr, Hf, Nb и Та
кристаллизуются в одномерных структурах, образуемых тригональными
призмами MSe, которые соединяются противоположными гранями.
Атомы металлов в этих сульфидах формально находятся в
четырехвалентном состоянии, а часть серы существует в виде молекулярных
ионов —M^S^S2-. TaS3 имеет переход металл-диэлектрик пай-
ерлсовского типа при низких температурах (см. разд. 4.9). NbS3
принимает пайерлсовскую искаженную структуру диэлектрика,
предполагающую возможность перехода при высоких температурах
в металлическую фазу, но он не переходит полностью в
неискаженную структуру. По структурным свойствам и структурным
переходам этих сульфидов написаны обзоры [Rouxel et al., 1982; Meers-
chaut, 1982 ]3.
3 Эти соединения подробно рассмотрены в монографии В. Е. Федорова
«Халькогениды переходных тугоплавких металлов. Квазиодномерные
соединения».— Новосибирск: Наука. Сиб. отд-ние, 1988.— 222 с. (Пер.)
310
Взаимосвязь структура — свойство
Рис. 6.21. Удельное электросопротивлениеt
демонстрирующее переход
полупроводник — металл N182-^56^. [Bouchard et al.r
1973].
Известно большое число
бинарных сульфидов, богатых металлом
(S/M <С 1,0), обладающих металл —
металл-связями [Franzen, 1978].
Типичные из них — V3S, V5S4, Zr2S,
Hf2S, Ta6S и Nb2lS8. Стехиометрия
таких твердых тел не может быть
понята в рамках традиционных
концепций валентности из-за наличия
обширного металл —
металл-связывания.
Тройные сульфиды, содержащие
два различных атома металла,
являются многочисленными. Оба атома
металла в таких твердых телах
могут быть переходными металлами,
или один металл является щелочным
или щелочноземельным, а другой — переходным металлом.
Примеры первой категории — тиошпинели и фазы MM2S4,
принадлежащие к Cr3S4-Tnny структур. По свойствам этих соединений
ранее опубликован соответствующий обзор [Goodenough, 1967].
Тройные сульфиды, содержащие щелочные металлы и переходные
металлы, которые имеют цепочечные и слоистые структуры,
рассмотрены недавно [Bronger, 1981 ]. Среди большого числа тройных
сульфидов заслуживают внимания BaVS3 и Ba]+;xFe2S4. BaVS3,
кристаллизующийся в цепочечной структуре (CsNiCl3), "ниже 240 К
обнаруживает структурные и электронные переходы, возникающие
в результате спаривания V4+ с З^-электронами в цепочке V—V
[Sayetat et al., 19821. Ba1+:rFe2S4 является единственной сульфидной
системой, имеющей бесконечно адаптивные цепочечные структуры.
Электрические и магнитные свойства имеют радикальные изменения
в связи с нестехиометричностью бария. Стехиометрический BaFe2S4,
похожий на KFeS2, является линейным цепочечным
антиферромагнетиком с полупроводниковым типом поведения. Другие члены
семейства являются сульфидами железа смешанной валентности,
имеющими парамагнетизм Кюри — Вейса, возникающий от
дополнительных электронов, связанных с Fe2+ [Swinnea, Steinfink, 1982].
Известны тройные сульфиды редкоземельных элементов LnMS3(Ln—La,
Nd, Gd; M—Ti, V, Сг), кристаллизующиеся в слоистой структуре,
состоящей из чередующихся слоев LnS и MS2. Соединения титана
и ванадия имеют свойства делокализоваппых d-электронов, тогда
Несколько примеров
311
как соединения хрома имеют локализованные d-электроны Сг3+
[Murugesan et al., 1981]. Изменение электронных свойств в этих
рядах при переходе от титана и ванадия к хрому напоминают перовски-
ты LaM03 и сульфиды NaMS2. Тройные халькогениды, которые
привлекают к себе значительное внимание из-за своих необычных
электронных свойств,— это фазы Шевреля, МлМо6Хя(Х—S, Se, Те).
Структура и электронные свойства этих твердых тел в литературе
обсуждены широко [Yvon, 1979; Maple, Fisher, 1982; Scholl-
horn, 1983 ]*.
Среди бинарных сульфидов редких земель [Holtzberg et al.,
1970] заслуживают внимания моносульфиды, кристаллизующиеся
в структуре каменной соли, и сульфиды Ln3S4, имеющие структуру
Th3P4. Моносульфиды, за исключением Sm, Eu и Yb, являются
металлическими; магнитные моменты свидетельствуют в пользу
трехвалентного состояния редкоземельных элементов. Металлическая
ирирода этих сульфидов возникает от одного из электронов
редкоземельного элемента, который становится делокализованным в 5d-
состоянии. Моносульфиды Sm, Eu и Yb являются диэлектриками,
а их магнитные свойства указывают на конфигурации
двухвалентных редкоземельных атомов /6, /7 и /14 соответственно.
Двухвалентные моносульфиды под давлением переходят в металлическое
состояние в результате валентной флюктуации в редкоземельном элементе
[Jayaraman et al., 1973, 1975]. Широко исследован переход в SmS.
Здесь изоструктурный переход в металлическую фазу идет под
давлением 6,5 кбар, он также может индуцироваться замещением Sm
таким трехвалентным редкоземельным элементом, как Gd3+.
Изменение до металлического состояния происходит в результате дело-
кализации 4/-электрона в 5^-зону (4/65с?° ->- Afd1). которая
сопровождается валентной флуктуацией самария от 2+ к 3+.
Ln3S4(Ln — La, Се, Рг, Nd, Sm, Eu, Gd), кристаллизующиеся
в структуре Th3P4, представляют другой изоструктурный ряд
сульфидов. Eu3S4 и Sm3S4 являются соединениями со смешанной
валентностью (промежуточной), содержащими трех- и двухвалентные
катионы в эквивалентных положениях. Оба сульфида имеют
прыжковую полупроводимость с энергией активации 0,1—0,2 эВ.
Магнитная восприимчивость этих твердых тел согласуется с наличием двух-
и трехвалентных ионов редкоземельных элементов. В Eu3S4
наблюдается внезапное изменение в удельном сопротивлении в области
185 К. В то время как есть указание на некоторое зарядовое
упорядочение в Eu3S4 при низких температурах, в Sm3S4 вообще нет
упорядочения вплоть до очень низких температур. Энергия
активации для прыжка электронов в En3S4 и Sm3S4 уменьшается с давле-
4 Этим фазам посвящена монография «Сверхпроводимость в тройных со-
единениях»/Под ред. Э. Фишера, М. Мейла: Пер. с англ.—М.: Мир, 1985.—
Т. 1,2. (Пер.)
312
Взаимосвязь структура — свойство
нием, dEJdP = 1,8±0,3 мэВ/кбар для Eu3S4 [Rohler, Kaindl, 1980].
Линейная экстраполяция изменения энергии активации с давлением
предполагает, что энергия активации должна стремиться к нулю
в области 110 кбар. а материал должен превращаться в гомогенное
соединение со смешанной валентностью и с металлической
проводимостью. Недавние исследования по нейтронной диффракции Eu3S4
[Wickelhaus et al., 1982] обнаружили небольшое ромбоэдрическое
искажение ниже температуры перехода, но оно слишком мало,
чтобы указывать на разделение редкоземельных ионов на Еи2+ и Еи3+.
Более вероятно, что состояния редкоземельного иона расщепляются
на две или более промежуточные валентности, причем каждая имеет
собственную энергию активации электропроводности. Смешанная
валентность Sm3S4 и Eu3S4 полностью еще не понята [Holtzberg,
1980; Wachter, 1980]. Другие соединения Ln3S4, содержащие
трехвалентные редкоземельные элементы, имеют металлический характер
поведения. Они проявляют металл-дефицитную нестехиометрич-
ность, причем предельным составом является Ln2S3. В ряду Ln3S4—
—Ln2S3 имеется непрерывное изменение характера поведения от
металлического до диэлектрического (структура остается той же
самой); некоторые члены обладают сверхпроводимостью [Ikeda et al.T
1980]. Исследована термоэлектрическая эффективность некоторых
из этих материалов [Taher, Gruber, 1981].
6.4.3. Фториды металлов
В отличие от оксидов и сульфидов металлов фториды
являются высокоионными твердыми телами, проявляющими свойства
локализованных электронов в силу высокой электроотрицательности
фтора. Ковалентность связи металл — фтор никогда не является
достаточно большой, чтобы приводить к делокализованным
электронам (исключение, вероятно составляет PdF3 при высоких
давлениях). Малая ковалентность уменьшает также катион-катионные
взаимодействия во фторидах. Тогда как электронные свойства
фторидов определяются природой связи металл — фтор,
кристаллическая структура определяется размером иона и энергией Мадедунга.
о
Радиус иона фтора (1,33 А) сравним с радиусом иона кислорода
о
(1,40 А), и мы, следовательно, находим структурные подобия между
несколькими семействами оксидов и фторидов металлов (табл. 6.5).
Близость размеров также дает возможность в некоторых случаях
замещать ион кислорода на ион фтора (например, V02_a;Fx
и MoOg^Fx). Поскольку фтор может образовывать ион только с
одним отрицательным зарядом, формальная валентность катиона во
фториде ниже, чем в изоструктурном оксиде, но исключительно
большая электроотрицательность фтора благоприятствует более высоким
степеням окисления в случае некоторых металлов. Электрические,
Несколько примеров
313
Таблица 6.5
Структурные аналогии между фторидами и оксидами
Формула
АХ
АХа
АХ3
АВХ3
АаВХ4
АВ2Х4
АзВ3СаХ1а
Структура
NaCl
Рутил
Re03
Перовскит
K2NiF4
Шпинель
Гранат
Примеры
Фториды и оксиды щелочных и
щелочноземельных металлов
Дифториды и диоксиды переходных
металлов
VF3 и Re03
KNiF3 и SrTi03
K2NiF4 и La2Ni04
Li2NiF4 и MgFe204
Na3Li3Fe2F12 и Y3Fe6012
магнитные и оптические свойства фторидов металлов совершенно
удивительные; в последние годы они усиленно исследовались [Tres-
saud, Dance, 1977, 1982; Portier, 1976; Hagenmuller, 1983a, 1985].
Электрические свойства оксидов охватывают свойства от хороших
металлов до хороших диэлектриков, а все фториды переходных
металлов являются хорошими диэлектриками.
Известны фториды, имеющие структуры вольфрамовых бронз
АЛМК3(М—Fe, V). В системе K^FeFg, например, структура
гексагональной вольфрамовой бронзы получается для составов 0,18 < х <
<; 0,26, тетрагональной бронзы — для 0,40 < х < 0,50 и
кубической перовскитовой бронзы — для 0,95 < х ^ 1,0. Подобные
структуры бронз известны в системе KXVF3. В отличие от оксидных бронз
фторидные бронзы являются антиферромагнитными (или ферри-
магнитными) диэлектриками.
Фториды металлов как ионные проводники лучше, чем оксиды
металлов [Portier et al., 1983]. Некоторые фториды металлов
являются твердыми ионными проводниками с удельной проводимостью,
сравнимой с ее значением для наилучших ионных проводников
(рис. 6.22) (см. также разд. 7.2); типичные примеры здесь — PbSnF4,
Pb!_xBixF2+x и NH4Sn2F5. Многие из фторидных ионных
проводников имеют флюоритоподобные структуры. Наличие катионов с не-
поделенной парой, а также стехиометрический избыток фторид-иона,
по-видимому, вообще благоприятствуют высокой ионной
проводимости, что иллюстрируется обычно системой Pbl_xBixF2+x
[Hagenmuller et al., 1981]. Избыточные ионы фтора, занимающие междо-
узельные позиции в структуре флюорита, смещают соседние ионы
фтора с их идеальных позиций. Получающийся беспорядок
увеличивает удельную проводимость для составов вплоть до х — 0,25. Для
х > 0,25 удельная проводимость уменьшается благодаря
увеличивающемуся порядку; при х = 0,5 образуется новая упорядоченная
фаза PbBiF5. Как правило, максимум в удельной проводимости и ми-
314
Взаимосвязь структура — свойство
юоо
Рис. 6.22. Удельная ионная проводимость фторид-ионных
проводников. Для сравнения приведены также данные для
типичных ионных проводников с Li+, Na+, Ag+ и О2*
[Hagenmuller, 1983a].
нимум в энергии активации соответствуют приблизительно середине
ряда твердых растворов. По-видимому, PbSnF4, который имеет
удельную проводимость 10"1 Ом"1-см"1 при 370 К с энергией
активации 0,14 эВ, является лучшим из известных проводников с
фторид-ионом.
Фторидные ионные проводники потенциально пригодны для
многих электрохимических устройств, которые включают батареи у
электрохромные дисплеи и газовые сенсоры. Для измерения
давления 02, а также давления газов-восстановителей, таких как Н2>
в качестве твердого электролита можно использовать PbSnF4 в
ячейках типа Ag|PbSnF4|Pt. Графит, интеркалированный фтором, может
использоваться в качестве электрода в батареях типа Li|LiC104
в пропиленкарбонате |CFa.. Фториды-диэлектрики в принципе могут
применяться в качестве конденсаторов, но они являются плохими
Несколько примеров
315
изоляторами; их удельная емкость вообще не очень высокая, но,
когда это и наблюдается, материал имеет большие диэлектрические
потери из-за высокой ионной проводимости.
Фториды переходных металлов представляют собой идеальные
модельные системы для проверки теоретических моделей магнетиков
(см. разд. 6.8). Поскольку они вообще являются хорошими
диэлектриками, их магнитные свойства можно изучать без помех в
электронной делокализадии. Кроме того, ионы переходных металлов
неизменно находятся в октаэдрическом окружении, а октаэдры MFe
могут соединяться друг с другом с образованием одно-, дву-, или
трехмерных (ID, 2D, 3D) фторидных структур или существуют как
независимые единицы. Примером последней структуры является
A3NiF6 (A — щелочной металл), содержащий изолированные
октаэдры NiF6, которые имеют переход низкоспиновое —
высокоспиновое состояние Ni3+.
Когда октаэдры MF6 сочленяются, магнитное взаимодействие
между локализованными спинами на соседних атомах описывается
гейзенберговским обменным членом Н^ = ^JijSiSj, где /и — об-
менный интеграл. Определяющий вклад в Jtj идет от суперобмена
(см. разд. 6.3.1). Когда взаимодействие идет через а-связывание,
J —-- —2b2/U, знак / можно предсказать исходя из правил Anderson,
Kanamori и Goodencugh. Если суперобмен идет между наполовину
заполненными орбиталями катиона, как в KNiF3, взаимодействие —
антиферромагнитное. Если электронный перенос идет с
полузаполненной орбитали катиона на незанятую орбиталь, взаимодействие
будет ферромагнитным, как для Ni2+ и Мп4+. Хотя в случае оксидов
и фторидов действуют одинаковые правила магнитных
взаимодействий, взаимодействие во фторидах намного слабее, что видно из
температур упорядочения. Так, ТN = 738 К для LaFe03, тогда как для
FeF3 с похожей антиферромагнитной структурой ТN =■■ 394 К. FeF3
и CoF3 имеют слабый ферромагнетизм ниже TN. Благодаря
прозрачности в видимой области, FeF3 находит применение в качестве маг-
нетооптического материала. Он может также использоваться как
магнитный «пузырьковый» материал в устройствах памяти вместо
LaFe03 (см. разд. 7.4).
Ферримагнитные фториды составляют особенно важный класс
магнитных материалов [Tressaud, Dance, 1977]. Ферримагнетизм
известен во множестве фторидов со структурами хиолита,
6Н—BaTi03, веберита и др. Эти структуры отличаются от структур
ферримагнитных оксидов. В случае оксидов ферримагнетизм —
общее явление для структур шпинели, граната и магнетоплюмбита,
где ферримагнетизм обусловлен заселением магнитными ионами
тетра-, окта- и додекаэдрических позиций. Во фторидах, однако,
катионы переходного металла неизменно занимают октаэдрические
позиции; ферримагнетизм является результатом способа сочленения
316 Взаимосвязь структура — свойство
Температура, К
6
Рис. 6.23. а — структура хиолита: б — температурная зависимость
намагниченности хиолитов Na3M5F14 [Tressaud, Dance, 1977].
октаэдров. Например, во фторидах с гексагональной структурой
BaTi03 одна из подрешеток состоит из октаэдров, соединенных
вершинами, и других октаэдров, соединенных гранями; одна треть ионов
металла находится в октаэдрах, соединенных вершинами, а две
трети — в октаэдрах, соединенных гранями. Обе подрешетки имеют
различные магнитные моменты, которые спариваются антиферромаг-
нитно, приводя к ферримагнетизму; CsFeF3 является типичным
материалом, обладающим этой структурой. Взаимодействие Fe—Fe
между октаэдрами с общими гранями является ферромагнитнымг
а такое взаимодействие между октаэдрами с общими вершинами —
антиферромагнитным; суммарный результат приводит к
ферримагнетизму с Тс = 60 К. Изоструктурный RbMnF3 является, однако,
антиферромагнитным, поскольку в этом соединении все
взаимодействия между соседними октаэдрами антиферромагнитны. Хио-
литы Na3M5Fe14(M—V, Cr, Fe, Co) — важный класс ферримагнит-
ных фторидов. Октаэдры MFe в этой структуре имеют общие
вершины, образуя слои M6F1!; одна треть октаэдров соединена четырьмя
вершинами, а две трети — двумя. Ионы натрия занимают вакантные
позиции в слоях и между слоями (рис. 6.23, а). Внутри каждого
слоя магнитные взаимодействия — антиферромагнитные
(суперобмен около 180°). Суммарные моменты каждого слоя располагаются
параллельно оси с, приводя к ферримагнетизму (рис. 6.23, б).
Несколько примеров
317
Другой интересный класс магнитных фторидов — вебериты
Na2M2+M3+F7, в которых цепочки сочлененных вершинами
октаэдров M2+Fe соединяются изолированными октаэдрами M3+F6.
Структура обеспечивает различные возможности магнитного поведения.
Если магнитны оба катиона, твердое тело вообще является ферримаг-
нитным из-за суперобмена М2+—F—М3+. Когда ионы М+3
немагнитны, как в Na2NiAlF7, структура имеет магнитно-изолированные
цепочки (M2+F5)?l, которые проявляют одномерное магнитное
поведение. Когда ионы М2+диамагнитны, изолированные октаэдры M3+Fe
приводят к парамагнитному поведению в этой структуре.
Ферромагнетизм, который требует непрямого или прямого
магнитного взаимодействия между неодинаково занятыми орбиталями
катиона, проявляют фториды типа M2+Mn4+F6(M—Ni, Zn, Cd),
кристаллизующиеся в упорядоченной структуре VF3. NiMnF6 имеет
самую высокую из известных до сих пор для ферромагнитных
фторидов Тс = 39 К. PdF3 — интересный фторид со структурой типа
упорядоченного NiMnFe (который соответствует Pd2+Pd4+F6). При
обычных давлениях он является ферромагнитным диэлектриком, но при
высоких давлениях, по-видимому, он подвергается валентному
переходу Pd2+ + Pd4+ ->- 2Pd3+, сопровождаемому переходом в
металлические состояние [Demazeau et al., 1980].
Ряд фторидов переходных металлов имеют низкоразмерный маг
нетизм [Tressaud, Dance, 1982]. Низкоразмерный магнетизм (см.
разд. 6.8) может возникать даже в кристаллографически
трехмерных структурах благодаря кооперативному эффекту Яна — Теллера
(см. разд. 4.10). Фториды, содержащие й4-(высокоспиновые), d7-
(низкоспиновые)- и ^9-катионы, имеют такое искажение октаэдров.
Различаются два возможных упорядочения [Reinen, 1979]: ферро-
дисторсионньте (все длинные связи металл — лиганд параллельны)
и антиферродисторсионные (длинные связи металл — лиганд
направлены поочередно перпендикулярно друг к другу). Магнитные
взаимодействия в упорядочениях ферро- и антиферродисторсиоиного типов
различны. В случае й4-катионов вытянутые октаэдры с антиферро-
дисторсионным упорядочением проявляют ферромагнетизм, тогда
как сжатые октаэдры с ферродисторсионным упорядочением —
антиферромагнитное взаимодействие. Перовскит KCrF3 является
антиферромагнитным твердым телом, принимающим антиферродистор-
сионную структуру. Магнитная структура здесь похожа на структуру
LaMn03 (А-тип). Сверхобменное взаимодействие d°x2 2—р —
^ведет к ферромагнитным слоям, которые антиферромагнитно
взаимодействуют через пустые dx2_ 2-орбитали. KCuF3 — одномерный анти-
ферромагиетик (тип А) со спинами, лежащими в плоскости ab. Здесь
магнитное поведение снова является следствием
антиферродисторсиоиного упорядочения искаженных октаэдров. Взаимодействие
между двумя наполовипу заполненными dx2_ 2-орбиталями идет
318
Взаимосвязь структура — свойство
Рис. 6.24. Орбитали в двух
полиморфных модификациях KCuF3.
Рис. 6.25. Данные по удельной
магнитной восприимчивости
монокристалла CsNiF3 [Tressaud, Dance,
1982].
Сплошная линия — теоретическая кривая
для J/k = 10,2 К.
вдоль оси с, тогда как межцепочечное взаимодействие идет через
взаимодействие орбиталей: заполненная d 2 — наполовину
заполненная dx2_ 2 в плоскости ab. Существуют две формы тетрагонального
KCuF3, одна — с симметрией 14/mcm, другая — с симметрией
P4/mbm; одномерный характер в последнем случае является более
выраженным, что можно видеть из расположения орбиталей
(рис. 6.24). CsNiF3 — единственный фторид, кристаллизующийся
в гексагональной перовскитоподобной 2Н-структуре, где существуют
бесконечные цепочки октаэдров NiF6 с общими гранями
параллельно оси с. Он имеет одномерный ферромагнетизм при высоких
температурах (70 < Г< 3000 К) и трехмерный антиферромагнетизм —
при низких температурах (рис. 6.25). Данные по нейтронной
дифракции и теплоемкости показывают, что трехмерный переход на^-
блюдается при 2,65 К. Трехмерная магнитная структура состоит
из ферромагнитных плоскостей, параллельных оси с, которые
взаимодействуют антиферромагнитно.
Среди фторидов со слоистыми структурами семейство K2NiF4
широко исследовано. K2NiF4 сам по себе является двумерным
гейзенберговским антиферромагнетиком (см. разд. 6.8) с ТN = 97 К
и J/k = —50 К. Изоструктурный K2CoF4 ведет себя как двумерная,
S = 1/2, изииговская система (TN = 107,8 К, J/k = —97 К)
(рис. 6.26). K2CuF4 и его рубидиевые и цезиевые аналоги,
кристаллизующиеся в ромбической искаженной структуре K2NiF4,
являются двумерными гейзенберговскими ферромагнетиками. Искажение
структуры и ферромагнитные свойства возникают в результате
упорядочения вытянутых осей октаэдров CuF6 поочередно, в
направлениях а и Ь [Friebel, Reinen, 1974]. Константа внутрислойного обмена
J/k = —11 К, значение для константы межслойного
взаимодействия —0,03 К.
Несколько примеров
319
0,001 0,01 0,1
(TN-T)/TN
Рис. 6.26. Приведенная намагниченность подрешетки K2GoF4 как функция
приведенной температуры. Сплошная линия — теоретический расчет для
двумерной изинговской модели [Tressaud, Dance, 1982].
Ионные фториды с большими оптическими щелями имеют
высокую прозрачность для электромагнитного излучения. MgF2,
например, прозрачен от ~106 см"1 (что соответствует пороговой
энергии для электронного перехода из валентной зоны в зону
проводимости) до ~103 см"1 (максимальная частота колебаний решетки).
Прозрачность фторидов металлов приводит к их использованию
в качестве окон и призм в оптических приборах (см. разд. 7.6). Это
свойство делает их привлекательными в качестве матричных
материалов для люминесцентных ионов; хорошим примером здесь
является эмиссия Еи2+ в некоторых фторидных матрицах. В оксидных
и хлоридных матрицах Еи2+ дает широкую эмиссию из состояний
с ^б^-конфигурацией, но во фторидных матрицах, где связь между
Еи2+ и матрицей слабая и, следовательно, более слабое поле лиганда,
эмиссия идет из 6Р7/2-состояния, которое по энергии лежит ниже,
чем соответствующие ^З^-конфигурации. В дотированных Еи2+
BaAlF5, BaSiF6 и BaY2F8 наблюдается монохроматическая
(27 800 см"1) эмиссия 6Р7/2 -> 857/2.
Трехвалентные редкоземельные элементы (за исключением Се3+
и Еи3+) для получения люминесценции не могут легко возбуждаться
ультрафиолетовым излучением, но многие из них имеют
возбужденные уровни близко к уровню 6Р7/2 Еи2+. Таким образом, можно
320 Взаимосвязь структура — свойство
наблюдать резонансный и электронно-колебательный перенос
энергии от возбужденного Еи2+ к ионам трехвалентных редкоземельных
элементов, если их ввести в подходящую матричную решетку.
BaY2F8, можно одновременно допировать двухвалентным Ей и
трехвалентными ТЬ, Но, или Ег, что приводит к сильной зеленой
эмиссии от трехвалентных ионов благодаря энергии переноса Еи2+;
в отсутствие Еи2+ эмиссии нет [Hagenmuller, 19836]. Фторидные
матрицы успешно используются также в фосфорах, которые
конвертируют инфракрасный свет в видимый. Этот процесс включает
перенос двух фотонов от сенсибилизатора к активатору, допирован-
ному в ту же самую решетку; Yb3+ и Ег3+ функционируют
соответственно как сенсибилизатор и активатор.
6.5. ПЕРЕХОДЫ МЕТАЛЛ — НЕМЕТАЛЛ
Переходы металл — неметалл в целом можно разбить на три
категории: 1) переходы в кристаллических твердых телах,
которые наблюдаются между делокализованными состояниями с
изменением в структуре; 2) моттовский переход между делокализован-
ным и локализованным состояниями и 3) андерсоновский переход
между делокализованным и локализованным состояниями,
характерный для некристаллических твердых тел. Первый класс
переходов описывается в рамках зонной теории электронов в твердых телах.
Зонная структура кристаллического твердого тела, состоящего из
атомов с четным числом электронов (заполняющих целое число зон),
может быть возмущена с переходом в такое положение, где пустая
и заполненная зоны пересекаются или перекрываются в результате
изменения давления или температуры, или при подходящем
допировании. Такие переходы металл — неметалл с пересечением или
перекрыванием зон обнаружены во многих материалах (например,
Yb, оксиды Ti и V), обычно они сопровождаются изменением в
кристаллической структуре, а в некоторых случаях также и в магнитном
упорядочении. Моттовский переход из металлического состояния
в неметаллическое происходит тогда, когда ширина зоны
уменьшается настолько, что становится меньше энергии
электрон-электронного отталкивания локализованных электронов,
индуцированного электронной корреляцией. В андерсоновском переходе
локализация происходит в результате беспорядка (как в аморфных
материалах), т. е. переход металл — неметалл идет тогда, когда ширина
зоны электрона становится меньше ширины распределения энергий
случайных положений. В этом разделе мы рассмотрим упрощенную
оценку различных переходов металл — неметалл с пояснительными
примерами; для более детального понимания читатель отсылается
к специализированным монографиям по этому вопросу [Mott, 1974;
Honig, Van Zandt, 1975; Elliott, 1984; Edwards, Rao, 1985; Rao,
Edwards, 1986].
Переходы металл — неметалл
321
Рис. 6.27. Температурная зависимость
удельного электросопротивления Ti203,
VO,
при переходе
диэлектрик.
металл —
ю'
ю6-
10s-
к ю3-
о
,; ю2-
% ю-
!'■
ю
3 4 5 6
ГООО/Т,К
Переходы между делокализо-
ванными состояниями. Некоторые
оксиды Ti и V имеют переход
металл — неметалл, обусловленный
изменениями в зонной структуре,
которые вызываются изменениями <$ ю-г\
в кристаллической структуре и
симметрии, а также в магнитном
упорядочении. Мы рассмотрим три
оксида — V203, V02 и Ti203,
которые являются типичными
примерами соединений с переходами
металл — неметалл, с различной степенью изменений в удельной
электропроводности (рис. 0.27) и с различной сложностью.
V203 подвергается переходу первого рода (моноклинный —
корунд), сопровождаемому скачком в электропроводности при 150 К
в 10 миллионов раз и магнитным переходом (антиферромагнетик —
парамагнетик). Отношение с/а и объем при переходе резко
изменяются. Высокотемпературная металлическая фаза имеет
значительное удельное сопротивление. При наложении давления V203
становится все более и более металлическим, это наводит на мысль о том,
что этот оксид находится вблизи критической области;
соответственно допирование хромом и титаном имеет заметное влияние на
переход, причем последний имеет эффект положительного давления,
а первый — отрицательного. V203 обнаруживает также переход
второго рода около 400 К с небольшой аномалией в удельной
проводимости (см. рис. 6.27).
V02 подвергается переходу первого рода (моноклинный —
рутил), сопровождаемому скачком в удельной проводимости при 340 К
в 10 тысяч раз. В низкотемпературной фазе магнитного
упорядочения вообще не наблюдается, материал остается полностью
парамагнитным. В чистом V02 имеется промежуточная моноклинная фаза с
небольшой температурной областью устойчивости, она
стабилизируется при допировании алюминием и хромом.
Ti203 подвергается переходу второго или более высокого рода
(без изменения симметрии кристалла) со 100-кратным скачком
удельной электропроводности, в области 410 К переход сопровождается
постепенным изменением отношения с/а и объема. Материал
остается парамагнитным. Замещение титана на ванадий вплоть до 10 %
делает систему металлической, причем отношение с/а твердого
21 Ч. Н. Р. Рао, Дж. Гопалатфишнан
322
Взаимосвязь структура — свойство
раствора соответствует отношению для высокотемпературной
металлической фазы Ti203.
Предложено несколько объяснений появления энергетической
щели при температуре, ведущей к переходу металл —- неметалл
[Van Zandt, Honig, 1974]. Постепенный переход в Ti203, связанный
с небольшим изменением проводимости, можно объяснить простой
моделью, включающей пересечение (или уширение) зон с
увеличением температуры. Модель, основанная на искажении кристалла
(при этом низкотемпературная низкосимметричная структура
связывается с энергетической щелью), может объяснить основные
особенности перехода в V02. Однако большой скачок в удельной
проводимости V203 трудно объяснить на основании модели искажения
кристалла, даже если принимать в расчет антиферромагнетизм
низкотемпературной фазы (удваивающий период и, следовательно,
открывающий щель). В литературе есть информация о моделях,
принимающих во внимание взаимодействия электрон — решетка, энергию
экситонов и другие факторы. В соответствии с одной из моделей
кривая плотности состояний для d-зоны V203 миеет ряд чередующихся
пиков и глубоких спадов [Kuwamoto et al., 1980]. Ферми-уровень
вблизи одного из минимумов может замещаться на зонную щель,
которая открывается при допировании хромом или алюминием.
Изменение в стехиометрии кислорода (или допирование титаном) смещает
ферми-уровень так, чтобы материал стал металлическим.
Кроме трех вышеупомянутых оксидов переходы металл —
неметалл проявляют также многие из фаз Магнели (Т1Л02п-1 и Vn02n_1)t
Ti305, Nb02 и несколько других оксидов. Переходы металл —
неметалл обнаружены также в NiS, CrS, FeS и других халькогенидах;
переход в NiS можно объяснить магнитным переходом
(антиферромагнетик — парамагнетик) (см. разд. 6.4.2).
Из-за эффектных переходов металл — неметалл особого
внимания заслуживает система (V1_JCM:c)203. По современному состоянию
вопроса, связанного с этими переходами, можно рекомендовать обзор
Honig [1982]. Переходы металл — неметалл в этой системе
схематично показаны на рис. 6.28, где римские цифры соответствуют
различным областям составов.
I. 0,0 < х <С 0,005 (М—Сг, А1). При низких температурах оксид
является антиферромагнитным диэлектриком и проявляет основной
переход металл — диэлектрик около 167 К. Выше 350 К удельное
сопротивление увеличивается в 3 раза и становится постоянным
выше 800 К.
П. 0,005 < х <С 0,018. Эти составы имеют три перехода. Первый —
из антиферромагнитного диэлектрика в металлическую фазу около
170 К, тогда как второй — из металлической фазы в парамагнитный
диэлектрик при Т = Т0 в области 190—385 К. Т0 с увеличением х
уменьшается, и в то же самое время увеличивается температура
перехода антиферромагнитный диэлектрик — металл. Таким образом,
Переходы металл — неметалл
323
Рис. 6.28. Переход металл —
неметалл в системах (VlxMx)203
(M-Cr, Al, Ti) и V2(l_y)03
[Honig, 1982]. Объяснение
различных кривых в тексте.
область устойчивости ме- ^
таллической фазы умень- ^
шается с увеличением
содержания хрома или
алюминия. Удельное со- м ^ t _
противление парамагнит- о .
ного диэлектрика посте- '
пенно уменьшается, уступая место второй металлической фазе.
III. 0,018 < х < 0,10. В этой области составов имеется
переход антиферромагиитиый диэлектрик — парамагнитный диэлектрик
в области 170 К, а вторая металлическая фаза получается выше
800 К; первая металлическая фаза отсутствует.
IV. Система (V^^Ti^O., похожа на I, за исключением того, что
температура перехода антиферромагнитный диэлектрик —
металлическая фаза понижается; для х ^ 0,055 прерывность перехода
металлическая фаза — антиферромагнитный диэлектрик исчезает.
V. V2(1_y)03. Температура перехода антиферромагнитный
диэлектрик — металлическая фаза уменьшается с увеличением у и для
у > 0,009 переход вообще не наблюдается.
VI. 0,055 < х < 0,1 для Ti или 0,009 < у < 0,035. Материал
обладает металлическими свойствами с высоким удельным
сопротивлением и, возможно, с антиферромагнитным упорядочением
около 10 К.
Все вышеупомянутые переходы сопровождаются изменениями
коэффициента Зеебека, структурных параметров, теплоемкости и
других характеристик. Система V203 объясняется в рамках
термодинамической модели, которая использует различные выражения
для свободной энергии электронов в делокализоваином и
локализованном режимах [llonig, Spalek, 198G].
Переходы локализованное электронное состояние — делокализо-
ванное электронное (зонное) состояние. Кобальтаты редкоземельных
элементов, LnCo03 (Ln—La или любой другой редкоземельный
элемент) около 1200 К имеют фазовые переходы первого рода,
которые, по-видимому, в основном определяются изменением
электронной энтропии [Raccah, Goodenough, 1967; Bhide et al., 1972; Jadhao
et al., 1976]. Кобальтаты становятся металлическими выше
перехода, но в удельной проводимости заметный скачок вообще не
наблюдается. Проводимость в системе увеличивается постепенно во всей
широкой области температур до тех пор, пока она становится
21*
324
Взаимосвязь структура — свойство
t 6004OO 200 ЮО
-J—I 1 1 ■
1 б" M (
Нд <
_ шр3.~0,2эВ _
Вь, сокоспиноеыи 00 ( tf
*29
Ы„Г7777?:
Т*0К
Тз>72/0К
Рис. £.50. Схематическая диаграмма
энергетической зоны в редкоземельных кобаль-
татах, объясняющая переход металл —
неметалл.
4М
5
О
1000/1,К 1
Рис. 6.29. Температурная
зависимость удельного
электросопротивления LaCo03. NdCo03 и GdCoOg.
У кривых указаны значения Е
[Rao, Rao, 1978].
J.5-
__gy86*io~z
0J69*Г0'}
0y56*10~z ,
з | -=^Ot33x70'3
0J1*10~Z
Тос,/з
7000/1, К■ —~
Рис. 6.31. Переход металл — неметалл в системе Ag 6Se [Shukla et al., 1981].
металлической (рис. 6.29). Температурное изменение электронных
(и спиновых) конфигураций кобальта исследовано методами мессба-
уэровской и рентгеновской фотоэлектронной спектроскопии. При
низких температурах ионы кобальта находятся в диамагнитном
низкоспиновом состоянии {tig), а с увеличением температуры переходят в
высокоспиновое состояние (ttgel), причем в спектре Мёссбауэра два
спиновых состояния отчетливо различаются. Электронный хоппинг
между двумя спиновыми состояниями приводит к состояниям с
переносом заряда (Со2+ -f- Co4+) и связанному с этим увеличению удельной
электропроводности. При дальнейшем увеличении температуры eg-
электроны стремятся образовать а*-зону; в соответствии с этим в
температурной области 700—1000 К смещение центра в спектрах
Мёссбауэра уменьшается из-за увеличивающегося перекрывания
орбитали катион — анион. При приближении к температуре пере-
Переходы металл — неметалл
325
хода первого рода (1200 К) спектры Мёссбауэра имеют единственную
резонансную линию (с химическим сдвигом, близким к нулю). Выше
температуры фазового перехода кобальтаты становятся
металлическими, благодаря изменению природы d-электронов от
локализованных к делокализованным (рис. 6.30). Поскольку при переходе
не отмечалось никакого изменения симметрии кристалла, считалось,
что суммарное изменение энтропии по характеру было электронным.
По-видимому, возможно увеличение симметрии LaCo03 при
температуре перехода (от ромбоэдрической до кубической). Однако
недавние исследования [Thornton et al., 1982] предполагают, что переход
eg —>■ о* происходит непрерывно, без связанного с ним структурного
изменения. Природа металлического состояния LaCo03 не вполне
понятна.
Смешанные проводники. Халькогениды серебра, Ag2+6X (X—S,
Se, Те) переходят в высокосимметричную фазу (около 410 К), где
ионы Ag + распределяются случайно, приводя к суперионной
проводимости. Эти материалы являются также хорошими электронными
проводниками (близкими к металлическим) и демонстрируют
интересное электронное поведение в зависимости как от температуры,
так и от состава [Shukla et al., 1981]. Так, в высокотемпературной
фазе Ag2+6Se имеет металлический характер поведения электронной
проводимости при высоких значениях б. С уменьшением б
электронная проводимость обнаруживает признаки интересного перехода
(рис. 6.31), причем ионная проводимость высокотемпературной
фазы, по существу, не зависит от б. Величина изменения электронной
проводимости при фазовом переходе также определяется
стехиометрией. В низкотемпературной фазе около 400 К материал
проводит как полуметалл, проводимость в значительной степени
уменьшается при низкой температуре, причем величина уменьшения
зависит от значения б.
Моттовский переход. Много лет тому назад [Wigner, 1938]
введено понятие электрон-электронных взаимодействий и высказано
предположение, что при низких плотностях свободный электронный
газ должен «кристаллизоваться» в непроводящее состояние. Mott
[1949] предположил, что диэлектрическое состояние может быть
получено в тех материалах, где зоны в области ферми-уровня узкие.
Если суммарное уменьшение кинетической энергии не перекрывает
суммарного увеличения потенциальной энергии, обусловленного
дополнительным кулоновским отталкиванием между электронами в
ионизированных состояниях частично заполненной зоны, то основное
состояние системы должно быть непроводящим. В последующих
работах Mott [1956, 1967] предположил, что если в вышеупомянутом
типе диэлектрика электрон удалять с одного атома и помещать на
другой, то свободная дырка и свободный электрон притягиваются
друг к другу посредством кулоновского взаимодействия и образуют
связанное состояние, или экситон, который не дает возможности ни
326 Взаимосвязь структура — свойство
электрону, ни дырке участвовать в электрической проводимости.
Однако если концентрация носителей высока, то электрон и дырка
будут притягиваться посредством экранируемого кулоновского
взаимодействия с константой экранирования. При более высоких
значениях константы экранирования взаимодействие становится очень
слабым и может произойти резкий переход из состояния с
отсутствием свободных носителей в состояние с их большим числом.
Согласно Мотту, этот переход должен наблюдаться при критическом
значении постоянной решетки и он необязательно означает фазовый
переход, поскольку это изменение должно быть резким только при
Т = О К. Такие переходы диэлектрик — металл называются мот-
товскими переходами.
Что делает твердые тела металлическими! Показано, что
энергетическая щель в твердом теле исчезает при некотором
критическом значении постоянной решетки, приводя к металлу [Hubbard,
1963]. Согласно Хаббарду, критерием перехода металл — неметалл
является W ^ U. Переход металл — неметалл происходит при
критическом значении WIU ~ 1, 2.
Согласно Мотту, переход металл — неметалл в твердом теле
может быть при наложении давления, когда параметры решетки
проходят через критическое значение. Поскольку константы решетки
реальных кристаллов могут изменяться только в ограниченном
интервале даже при наложении сверхдавлений, проблема моттовских
переходов лучше исследуется в допированных полупроводниках и
других химических системах. Мотт предсказал, что переход металл—
неметалл должен быть при критической концентрации носителей
заряда тгс, определяемой как nlaH с^ 0,25, где ан — боровский
радиус атома в неметаллической области. Моттовский критерий
использован для описания начала металлического характера поведения
в широком ряду систем [Edwards, Sienko, 1983]. Экспериментально
найдено, что начало металлического характера поведения в
большом числе систем описывается критерием п1/3ан. = 0,26=5=0,05, где
#н* теперь означает радиус реальной волновой функции для
изолированных частиц, полученный непосредственно из измерений в
области локализованного электрона.
Переход металл — неметалл стал злободневной темой со времен
сэра Хэмфри Дэви, когда были открыты натрий и калий; до тех пор
было известно, что только такие высокоплотные вещества, как
золото, серебро и медь, обладающие блеском и другими родственными
свойствами, являются металлическими. Множество материалов
имеют переход из неметаллического в металлическое состояние в
результате изменений в кристаллической структуре, составе,
температуре и давлении. Хотя большинство элементов по природе являются
металлическими, некоторые из элементов, обычно являющихся
неметаллами, становятся металлическими при наложении давления
или при плавлении; соответственно, кремний является металли-
Переходи металл — неметалл
327
ческим в жидком состоянии и неметаллическим в твердом состоянии.
Такие металлы, как Cs и Hg, становятся неметаллическими при
высоких температурах, когда они расширяются и делаются менее
плотными. Растворы щелочных металлов в жидком аммиаке становятся
металлическими при достаточно высокой концентрации щелочного
металла. Вольфрамовые бронзы (например, Na^W03) становятся
металлическими при критической концентрации электронов (Na). Есть
много других систем, которые проявляют концентрационно-зависи-
мые переходы металл — неметалл, типичные примеры таких систем:
тонкие пленки металл — благородный газ (например, Na : Аг), до-
пированные полупроводники (например, Р : Si), сплавы (например,
Си : Аи) и некоторые сложные оксиды металлов.
Поучительным критерием для понимания металлического
состояния является критерий, предложенный Герцфельдом в 1927 г.
[Herzfeld, 1927]. Согласно этому критерию материал является
металлическим при RIV ^ 1, а диэлектриком — при R/V < 1. Здесь
R означает молярную рефракцию атомных частиц в газообразном
состоянии, а V — молярный объем в конденсированном состоянии.
Этот критерий объясняет металлический характер элементов в
природе [Edwards, Sienko, 1983 J. Показано, что скрытая теплота
испарения является удовлетворительным критерием различия между
металлическими и неметаллическими элементами [Rao, Ganguly, 1986].
Андерсоновский переход. В разупорядоченных (или
некристаллических) материалах вектор обратной решетки становится
бессмысленным, а /с не является более хорошим квантовым числом.
Электронная структура этих материалов не может быть описана в
терминах зонной структуры (Е от к). В отличие от кристаллов, где
диэлектрикам соответствует заполненная зона, в разупорядоченных
материалах электронные состояния локализованы. Волновая
функция сконцентрирована около центра из нескольких атомов и имеет
незначительную амплитуду в любом другом месте твердого тела.
Амплитуда с увеличением расстояния R резко спадает (ехр — оси,
где а — величина, обратная длине локализации). Важным понятием,
введенным в связи с рассмотрением электронных состояний
разупорядоченных материалов, является понятие края подвижности [Mott,
Davis, 1979]. Согласно этой концепции существует критическая
энергия, ниже которой электроны пространственно локализуются в
области позиции единственного атома, а выше которой — они дело-
кализуются в пространстве (как в кристалле), хотя фазы волновых
функций от позиции к позиции могут быть хаотичными. На рис. 6.32
показаны результаты, где состояния на концах зоны являются
локализованными. Anderson [1958] в своей классической работе
показал, что локализация появляется как следствие беспорядка в
системе совершенно независимых электронов. Беспорядок в этой модели
представляется в виде хаоса энергий положений (рис. 6.33).
Критерий локализации состоит в том, что разница в энергии между
328 Взаимосвязь структура — свойство
Кристаллическое
твердое тело \
|1 I
Протяжен- Ь\ Нет
ные состоя-^}состоянии
^ия ъ'
% Протяжен
^нъ/е состоя]
ъния
Некристаллическое
{твердое ^ | тело
I
\/7ротяжен§ )£>Локализо-\
&} стояния
\ные состо&^ванные со-.
\ЯНиЯ оЫ^лчлл^у/я I
Рис. 6.33. а — андерсоновская
локализация, обусловленная беспорядком в
потенциалах положений. Для сравнения
приведены также потенциалы в
регулярной решетке (б). W — ширина од-
ноэлектронной зоны в отсутствие
случайного потенциала V0. Локализация
определяется отношением W/V.
Энергия электронов Е —
Рис. 6.32. Схематическая диаграмма плотности состояний для кристаллических
и аморфных^ пол у проводников в области высших занятых (EJ и низших пустых
(Е\ состояний.
локализованными состояниями больше, чем ширина зоны.
Андерсеновский переход — это переход локализация — делокализация в
результате движения ферми-уровня через край подвижности при
изменении в составе, температуре или каких-либо других переменных.
Электропроводность в таких разупорядоченных материалах идет
по процессу связанного с фонолами туннелирования, в котором
вероятность переноса между двумя положениями задается уравнением
р =-. v ехр (—2аД) exp (-WlkT), (6.57)
где первый экспоненциальный множитель дает вероятность
туннелирования между двумя .положениями с соответствующими
локализованными волновыми функциями, а второй экспоненциальный
множитель означает вероятность фоиона с энергией W (разница в энергии
между двумя положениями); v — фононная частота. При низких
температурах вероятность переноса электрона может быть
оптимальной при тунпелировании не в ближайшее положение, а в более
удаленное, которое имеет разницу в энергии W. Такая оптимизация
привела Мотта к предложению закона
о = о0ехр (—Л Г"1/*), (6.58)
где А = 2,'l[a3/kN(EF)]1l*, N(EF) означает плотность состояний на
уровне Ферми. Такое поведение обнаружено в аморфных
полупроводниках, а также в оксидах типа Laj.^Sr^CoOg. Исследована
картина хорошо определенного края подвижности электрической
проводимостью при высоких температурах в результате транспорта
электрона в зоны делокализованных состояний за край подвижности. Пред-
Переходы металл — неметалл
329
Рис. 6.35. Характеристика удельного
сопротивления Nd^xSr^CoC^ [Rao et
al., 1975].
Рис. 6.34. Удельная проводимость и диэлектрическая
восприимчивость кремния, допированного фосфором, при Т ~ Ю-2 К. Диэлектрическая
восприимчивость имеет дивергенцию при приближении к переходу со стороны
диэлектрика. Со стороны металла наблюдается резкий, но непрерывный порог
в о(п) [Hess et al., 1982].
ложена теория, основанная на подвижности полярона [Emin, 1977].
Исследована концепция края подвижности [Abrahams et al., 1979],
при этом использовались методы подобия (как при фазовых
переходах), с целью показать, что во время локализационного перехода
подвижность и проводимость изменяются непрерывно. Это
подтверждено исследованиями допированного фосфором кремния (рис. 6.34);
измерены значения удельной проводимости ниже минимальной
металлической проводимости (amin « 500 Ом-1-см-1 при 0 К),
предложенной Mott [1972, 1983] в качестве предельной проводимости разу-
порядоченного материала при приближении к локализационному
переходу (Е = Ес) со стороны металлического состояния. Резкое
расхождение в диэлектрической восприимчивости напоминает
критерий Герцфельда.
Известно, что состояние щелей в аморфных материалах приводит
к заряженным дефектам, при этом транспорт идет через хоппинг би-
поляронов. В халькогенидных стеклах биполяроны соответствуют
координированным центрам (над — Cf и под — Q~).
Переходы металл — неметалл в оксидах, контролируемые
составом. Имеется много систем, которые изменяют поведение от
неметаллического к металлическому в зависимости от состава. Например,
в системе (Vi-xTix)203, где х = 0,90 или = 0,10, материал имеет
металлический характер поведения при всех температурах; переход
неметалл — металл с изменением х — очень резкий. Здесь мы
коснемся тех оксидных систем, где переход металл — неметалл в
исходном материале отсутствует, но замещение одного катиона на другой
и 1—и 1 г
2 4 6"
п (ГО 7*ЫЪ)
330
Взаимосвязь структура — свойство
ОЛ
>-М
s
о
<
х=о,го
х*о,ю о
°°о0
Х=0У05
«о о0о
-2Н
-Jl x = opr
Х=0,0
юо гоо
т, к
«Я*?
Рис. 6.36. Характеристика удельного сопротивления LaNi1_JCMn:c03.
постепенно приводит оксид в металлическое состояние [Rao,
Edwards, 1985; Rao, Ganguly, 1985]. Ln^Sr^CoOg (Ln—La или
другой редкоземельный элемент) является хорошим примером такой
системы [Rao et al., 1975; Rao et al., 1977]. LnCo03 (x = 0) при
обычных температурах является диэлектриком, но с увеличением
х его удельное сопротивление постепенно уменьшается до тех пор,
пока при х = 0,4—0,5 оксид становится металлическим (рис. 6.35).
При каком-либо промежуточном значении х температурный
коэффициент удельного сопротивления изменяет знак, а значение
удельной проводимости при этом составе (102—108 Ом"1-см-1) близко к
«минимальной металлической проводимости», которую ввел Mott
[1972] в работе по разупорядоченным материалам. Интересно то, что
значение amin имеют те оксидные системы, в которых температурный
коэффициент удельного сопротивления изменяет знак.
Другой пример такого поведения — LaNi^^-M^C^ (M—-Mn, Сг
и т. д.), где температурный коэффициент удельного сопротивления
Металлические кластеры
331
меняет знак при критическом значении х (рис. 6.36); LaNi03 (x = 0)
по своему поведению является d-зонным металлом, его удельное
сопротивление увеличивается с увеличением х. La]_xSrTV03 также
обнаруживает такое поведение amin [Sayer et al., 1975].
Переходы металл — неметалл, контролируемые составом, имеют
место в бронзах AxW03; но А0>3МоО3 (А—К, Rb) обнаруживает
переход металл — неметалл, связанный с волнами зарядовой плотности,
неомическим транспортом и квазиодномерным характером [Schlen-
ker et al., 1985]. Похожий неомический транспорт обнаружен в
NbSe3 и TaS3.
6.6. МЕТАЛЛИЧЕСКИЕ КЛАСТЕРЫ
Как упоминалось в разд. 3.3, можно получить кластеры из
атомов металла различных размеров. Полностью подтверждено
существование кластеров из атомов щелочного металла в газовой фазе.
Такие кластеры имеют намного меньшую энергию ионизации, чем
отдельный атом, а также высокое сродство к электрону.
Следовательно, в металлическом кластере вероятность переноса электрона
значительно выше. Действительно, для цезия известно, что при
увеличении плотности цезия (от отдельных атомов в газе с низкой
плотностью до жидкости) образуются большие кластеры и перенос
заряда становится очень благоприятным; когда плотность приближается
к критическому значению, энергии кластерных зон перекрываются>
приводя к хорошей проводимости. При первом взгляде на такие
металлические кластеры всегда возникает вопрос, сколько атомов
металла должно быть в кластере, чтобы имитировать или отражать
свойства металла как такового I Edwards, Sienko, 1983]. Если
интересоваться таким транспортным свойством, как электропроводность,
то для того чтобы электроны стали делокализовапными, должна быть
полная делокализация электронной волновой функции; для этого
необходимо, чтобы в агрегате было большое число атомов. Однако
спиновая делокализация требует меньшего числа атомов, поскольку
она имеет место без образования свободных носителей. Ширина
сезоны или обрыв на Ферми-крае в фотоэлектронном спектре также
могут использоваться для того, чтобы отличать металлические
кластеры от компактного металла. Металлические частицы (подобно
частицам на поверхностях металлических катализаторов на подложках
(см. гл. 8)) представляют собой модель металлического агрегата,
находящегося где-то между металлическими кластерами с высокой
нуклеарностыо и компактными металлами.
Имеются семейства кластерных соединений (рис. 6.37),
содержащих металлические кластеры, окруженные лигандами [Lewis,
Green, 1982]. В соединениях с небольшими кластерами электроны
спариваются, но в больших кластерах электронные уровни становят-
332
Взаимосвязь структура — свойство
А Дг
Kh6(C0)16
0*6(С0)1в
C(CO)J2" [H2Rhe(C0)J:
Рис. 6.37. Типичные кластеры из атомов металла,
найденные в карбонитах переходных металлов.
Для ясности карбонильные группы не показаны.
Светлыми кружками обозначены атомы углерода
в кластерном соединении [Edwards, Sienko,
1983].
Кластеры: 1 — треугольный; 2 — тетраэдрический; 3—
октаэдрический; 4— шапочная тригональная бипирамида
(политетраэдрическая структура); 5 — четырехшапочный
октаэдрон (фрагмент структуры ГЦК); 6 — антикубок-
таэдрон (фрагмент структуры ГПУ); 7—фрагмент
структуры ОЦК; 8 — пентагональная структура; 9 — то же.
-,3- ся близкими, как в металлических
частицах. В таких кластерах следует ожидать
квантовый эффект размера. Обнаружен
существенный парамагнетизм кластера в
H2Os10(CO)24 ниже 70 К [Benfield et al.,
1982], что ожидается из приблизительно-
[Rh15C2(co)28j го диаметра осмиевой частицы в 10 А;
избыточный парамагнетизм в соединениях
осмия увеличивается с размером кластера
[Johnson et al., 1985].
Кластерные соединения металлов
моделируют поверхностные частицы,
получаемые взаимодействием молекул с
поверхностью металлов [Muetterties et al.,
1979]; это имеет значение в понимании
гетерогенного катализа. Создание селективных катализаторов для
химической индустрии, использующей в качестве сырья СО (и,
возможно, С02), привело к основным успехам в исследовании
металлических кластеров. Разработаны критерии различия между
кластерным и моноядерным катализом. Типичными катализаторами,
исследованными до сих пор, являются |Ir4(CO)12_JC(PPh3)x], где Ph —
фенил, а х = 1, 2 или 3.
[RbH(cov3
9
[*1э(со)2?]4
6.7. СОЕДИНЕНИЯ СО СМЕШАННОЙ ВАЛЕНТНОСТЬЮ
Считается, что химические соединения, содержащие элемент
(обычно металл) в двух различных формальных степенях окисления,
проявляют смешанную валентность. Химия смешанной валентности
стара, как сама химия; некоторыми хорошо известными
соединениями со смешанной валентностью являются берлинская лазурь
(Fe4[Fe(CN)e]3'14H20), магнетит (Fe304) и вольфрамовые и
молибденовые сини (гетерополисоединения). Химия смешанной
валентности включает большое разнообразие твердых тел с удивительными
Соединения со смешанной валентностью
333
Таблица 6.6
Типичные твердые тела со смешанной валентностью
Соединение
рь8о4
Sba04
Fe4 [Fe(CN)e]314HaO
Vn02n—1
LixNi^O
La1_JCSr:)CMn03
BaBi^P^O,
LiTi204
K2Pt(CN)4Br030.3H2O
Na^WO,
MxMo0S8
Ре484-ферридоксины
Классификация по
схеме Робина-Дэя
Класс I
»
Класс II
»
»
»
Класс III
»
ь
ь
»
*
Значение
Красный свинец
Минерал сервантит
Красящее вещество и пигмент
(берлинская лазурь)
Переходы полупроводник —
металл
Хоппинговый полупроводник
Ферромагнетизм
Сверхпроводимость
*
Молекулярный металл, пайерл-
совская неустойчивость
Бронзовый блеск и металл при
больших значениях х
Сверхпроводимость
Электронный перенос (энзимы)
свойствами (табл. 6.6), образуемых почти третьей частью элементов
Периодической таблицы; в последнее время отмечается рост интереса
к этим вопросам [Day, 1981]. Поскольку переменная валентность
является предпосылкой для смешанной валентности, она
действительно общее свойство соединений переходных металлов, Се, Ей и
ТЬ, а также некоторых постпереходных элементов с устойчивыми
ns2- и м^-электрониыми конфигурациями, таких как Ga, Sn, Sb, TI,
Pb, Bi. Большинство соединений со смешанной валентностью
содержат такие электроотрицательные анионы, как галогениды, оксиды,
сульфиды или молекулярные лиганды, содержащие
электроотрицательные атомы. Для того чтобы твердое тело назвать соединением со
смешанной валентностью, мы должны приписать элементу,
имеющему смешанную валентность, определенные степени окисления,
которые различаются на целые числа (одна или по большей мере две
единицы). Fe304, содержащий железо в степенях окисления 2 + и 3+,
является соединением со смешанной валентностью, тогда как сплав
Nb3Ge, где мы не можем определить степени окисления составных
частей, не относится к таким соединениям. Мы должны также
определить различие между соединениями со смешанной валентностью
типа Fe304 и системами с флуктуирующей валентностью, такими как
Се и SmS, где имеет место флуктуация электронной конфигурации
между 4/п и Afn-W [Falicov et al., 1981] (см. разд. 2.2.7).
Электронные свойства, связанные с /-электронами и привлекающие в послед-
334
Взаимосвязь структура — свойство
нее время большое внимание,— это такие свойства, которые
объясняются моделью тяжелого фермиона (например, CeCu2Si, UPt3), там,
где носители имеют большие эффективные массы [Stewart, 1984].
В некоторых соединениях со смешанной валентностью наличие
более одной степени окисления можно определить по формуле, как,
например, в РЬ304 и Vn02n_1, тогда как в других соединениях
формула дает, видимо, суммарную степень окисления, хотя состояние
интересующего нас элемента является, скорее всего, необычным.
Типичные примеры последнего случая — Sb204, BaBi03 и Pt(NH3)Cl3;
экспериментальные данные говорят о том, что в этих соединениях мы
имеем дело не с Sb (IV), Bi (IV), Pt (III), a c Sb (III, V), Bi (III, V)
и Pt (III, IV), и эти соединения в действительности следует
представлять как Sb3+Sb5+04, BaBiJ^BiJ^Oa и [Pt(NH3)4]2 + [PtCl6]2-.
Во всех таких системах рентгеновская фотоэлектронная
спектроскопия может легко устанавливать наличие смешанной валентности
[Rao et al., 1979].
Предложена [Robin, Day, 1967] классификация соединений со
смешанной валентностью на основе коэффициента валентной делока-
лизации а, величина которого зависит от разницы энергий между
двумя состояниями М1+М(в+1)+ и М(Ап+1)+Мв+, где А и В - два
различных положения. Когда АЕ большое, как в РЬ304, а мало; такие
соединения относятся к классу I. Если два положения подобны, но
кристаллографически различимы, считают, что соединения
принадлежат к классу II. В классе III а становится большим, а два
положения, занятые катионами со смешанной валентностью,—
идентичными. Свойства, связанные с различными классами, должны
быть различными (см. табл. 6.6). Например, в соединениях класса I
хоппинг электронов между положениями не благоприятен,
поскольку АЕ большое. В соединениях класса III электроны должны быть
делокализованы. Лиганды, которые соединяют катионы, играют
роль в межвалентном переносе: чем больше перекрывание металл —
лиганд, тем выше вероятность переноса электрона [Mayoh, Day,
1972]. Для того чтобы описать перенос электрона в соединениях со
смешанной валентностью как таковых, следует рассматривать
взаимодействие между электронными и колебательными движениями.
Экспериментально частота оптического межвалентного перехода дает
величину энергии, требуемой для термически активируемого
переноса электрона. Интенсивность оптического межвалентного перехода
дает информацию об а. Одним из наиболее характерных признаков
II класса соединений со смешанной валентностью является широкая
межвалентная полоса поглощения в видимой и инфракрасной
области. Для расчета контуров полос поглощения разработана модель
колебательного взаимодействия [Piepho et al., 1978]; хорошим
примером анализа таких профилей полос поглощения является недавнее
исследование (CHgNH^Sb^Sn^^Cle [Prassides, Day, 1984]. Если
электроны делокализованы не полностью и прыгают от положения к
Соединения со смешанной валентностью
335
положению, как в маргинальных полупроводниках, сила
взаимодействия между электронами и решеткой (поляронами) становится
важным фактором.
Смешанная валентность наблюдается в минералах (например,
Fe304), металл-цепочечных соединениях, димерах, олигомерах,
комплексах металлов и даже в органических и биологических системах
[Brown, 1980; Day, 1981]. Среди димерных и олигомерных
комплексов металлов со смешанной валентностью большое внимание к себе
привлек аминный комплекс Ru (II, III) с пиразином в качестве
мостика ^(NH^Ru— nQn—Ru(NH3\J5+ ' синтезированный в
1973 г. [Creutz, Taube, 1973]. Важным вопросом, касающимся этого
класса комплексов, является вопрос о том, принадлежат ли они к
классу II или III. Поскольку вокруг каждого иона металла лиган-
ды идентичны, этот комплекс, как на первый взгляд кажется, следует
рассматривать принадлежащим к классу III. Оптическое
поглощение дает интенсивную полосу в видимой области (550 нм), которая
является характеристичной для Ru (II). Это, несомненно,
подтверждает идею о том, что Ru (II) можно идентифицировать по временной
шкале оптического перехода (Ю-14 с). Однако межвалентная
полоса ~ 1550 нм не чувствительна к действию растворителей и слишком
узкая, чтобы относить комплекс к классу II. XPS имеет дублеты в
спектрах внутренних уровней Ru(3d) и (Зр). Это свидетельствует
о том, что отдельные степени окисления можно также различить по
этой временной шкале (10~6 с). Высказаны аргументы [Hush, 1975]
в пользу того, что образование при фотоионизации внутренней дырки
должно релаксировать систему в локализованное состояние, даже
если первоначально она была делокализована; следовательно,
по-видимому, фотоэлектронная спектроскопия внутренних оболочек не дает
способ определения степени делокализации в валентной оболочке.
Инфракрасная спектроскопия показала, что деформационное
колебание NH3 (800 см-1), валентное колебание Ru—NH3 (449 см-1)
и валентное колебание Ru-пиразин (316 см-1) в рассматриваемом
комплексе происходят при значениях, промежуточных между
соответствующими значениями для комплексов Ru(II) и Ru(III). Это
является доказательством того, что валентный электрон делокали-
зуется во временной шкале инфракрасной спектроскопии (10 ~12—
10~13 с). Результаты различных экспериментов по этому комплексу
несколько противоречивы из-за различной временной шкалы;
по-видимому, скорость переноса электрона составляет где-то 105—1012 с-1.
Смешанная валентность в соединениях типа Laj.^Sr^CoOg [Rao
et al., 1975, 1977] определяется как по скорости переноса
электрона, так и по составу; для MoFe204 и других твердых тел это
рассматривается как результат быстрого переноса электронов [Ramdani
et al., 1985].
336
Взаимосвязь структура — свойство
В металл-цепочечных соединениях мы можем различить два
типа систем со смешанной валентностью: в первом типе — цепочка
полностью состоит из атомов металла (класс III), а во втором —
атом металла и мостиковый лиганд чередуются (класс II). Красная
соль Вольфрама lPt(C2H5NH2}4] [Pt(C2H5NH2)4Cl2]-4H20 является
примером первой системы, состоящей из ионов Pt(II) и Pt(IV) с
мостиковыми ионами хлора. В соединениях типа КСР смешанная
валентность достигается частичным окислением ионов Pt до
усредненного состояния (2 + х) с х ж 0,3. Частичное окисление
сопровождается удалением некоторого количества катионов, как в
Kb75Pt(CN)4-l,5H20, или введением дополнительных анионов
K2Pt(CN)4Br0t3-3H2O. Соединением со смешанной валентностью
является даже цепочечное соединение ртути Hg3-aAsF6 (CM
разд. 4.9).
Fe304 имеет структуру обратной шпинели с ионами Fe2+ и
половиной ионов Fe3+, расположенными в октаэдрических позициях
(позиции В) кислородного каркаса, а оставшаяся половина ионов Fe3 +
располагается в тетраэдрических позициях (позиции А). При
температуре около 850 К он подвергается переходу ферримагнетик —
парамагнетик, а при температуре около Tv = 123 К — другому
переходу (Verwey-nepexod). Материал является полупроводником как
выше, так и ниже Verwey-перехода. Некоторые изменения
наблюдались также вблизи 200 К и 12 К, но эти изменения не очень
значительны. Свойства Fe304 в области Verwey-перехода и выше его
представляют большой интерес для исследователей; этому переходу был
посвящен целый выпуск «Philosophical Magazine» (B42, № 10, 1980).
Детальные структурные исследования методом нейтронной
дифракции и другими методами свидетельствуют в пользу того, что
зарядовое упорядочение Fe2+ и Fe3+ (и, следовательно, дальний порядок)
устанавливается ниже TY. Катионные цепочки а и Ь располагаются
вдоль направлений [110] и [НО] соответственно (на чередующихся
плоскостях (001)). В то время как вдоль одной цепочки а за тремя
ионами Fe2+ в непрерывном ряду располагается ион Fe3+, в смежной
цепочке а за тремя ионами Fe3+ следует один ион Fe2+. В цепочке
Ъ катионное упорядочение идет в паре ионов Fe3+, за которой
следует пара ионов Fe2+ и далее — поочередно. Последовательные
плоскости — Ъ — а — Ъ — располагаются перпендикулярно оси с, так
что ближайшие катионы в трех последовательных плоскостях
собираются в группы, образуя гексагональные кольца с чередующимися
ионами Fe2+ и Fe3+. Все катионы fo-цепочек входят в состав колец,
и только 1/4 катионов а-цепочки включаются в кольцо. Синхронное
смещение трех электронов к трем ближайшим позициям внутри
любого кольца приводит к обмену между Fe2+ и Fe3+. Ниже Tv
значительная часть гексагональных колец всегда существует в
«обращенной» зарядовой конфигурации, тем самым делая случайным
заряд вдоль fr-цепочек, но оставляя 3/4 катионов а-цепочки в упоря-
Соединения со смешанной валентностью
337
доченном состоянии. Это объясняет прежние экспериментальные
данные о том, что при низких температурах упорядочиваются именно
а-плоскостные катионы.
Существование сверхструктурных линий как раз выше Tv при
критическом нейтронном рассеянии и детальное исследование
упругого и неупругого нейтронного рассеяния говорят о существовании
мягкой моды с волновым вектором к = (00^), которая
«конденсируется» при к = (001/2). Если исходить из смещения одного иона
Fe2+ (или одного иона Fe3+) в плоскости а при z = 1/8 в
соответствующее положение в плоскости а при z = 9/8, приходим к
комплементарному заряду (а именно Fe3+ или Fe2+). Для того чтобы
дублировать ту ионную конфигурацию при z = 17/8, которая преобладает
при z = 1/8, должно быть увеличение вдвое размера элементарной
ячейки вдоль оси с. Формально это соответствует существованию
волны зарядовой плотности (с длиной волны К = 2 с), которая
сильно взаимодействует с соответствующей фононной модой с тем же
самым волновым вектором. При переходе упорядочение зарядов
(ведущее к образованию волн зарядовой плотности) происходит
одновременно с атомными смещениями, что обусловливает понижение
симметрии.
Хотя структурная характеристика Fe304 вблизи Verwey-nepe-
хода почти удовлетворительна, многие более тонкие детали до сих
пор еще не поняты IHonig, 1982, 1986], включая вопрос об истинной
структуре низкотемпературной фазы (ромбоэдрическая, ромбическая,
моноклинная или триклинная). Электрические свойства в области
Tv также не полностью охарактеризованы. Существует
неопределенность относительно природы изменения удельной проводимости с
температурой. Не ясно, действительно ли имеет место делокализо-
ванный характер носителей заряда, предполагаемый некоторыми
исследователями. По-видимому, большинство данных по
транспортным свойствам предполагают модель малого полярона. Удивительно,
что удельное сопротивление при Tv уменьшается в 100 раз вместе
с потерей дальнего порядка. Недавние исследования показали, что
Verwey-переход и связанные с ним изменения в удельной
проводимости и теплоемкости очень чувствительны к кислородной
стехиометрии [Honig, 1986].
Существует несколько интересных семейств неорганических
соединений со смешанной валентностью, которые мы здесь не
обсуждаем (см. [Yvon, 1979; McCarley, 1982]). Например, имеются
кластерные соединения, такие как фазы Шевреля, M^MoeXg (X—S, Se),
конденсированные цепочечные кластерные соединения, такие как
TlMo3Se3, Ti5Te4, NaMo4Oe и MxPt304. Галогениды TTF и
комплексы TTF—TCNQ (см. разд. 1.9) представляют собой молекулярные
системы со смешанной валентностью, в которых смешанная
валентность относится ко всей молекуле; заряд на TTF в таких соединениях
нецелочисленный. Структура TTF—Вг0>79 и подобных твердых тел
22 Ч п. Р. 1\.о. Дж. Гонаткп.'рппш ш
338 Взаимосвязь структура — свойство
ОЮО-ООнЫ
4
ОЧХХХ>Ч>
б ОЮОЮО-О»
ооооо
очхмх>-о
ооооо •
очхмхх>
ооооо _
ооооо
ооооо
• ооооо 1*
-в€ММ
ООООО £
3,57 А 4,54 А
ооооо
ооооо ооооо
ооочхх>
ф
ооооо-о^
Рис. 6.38. Кристаллическая структура TTF—Вг0,79.
Проекции: а — по оси а; б — по оси б [Torrance, Silverman, 1977].
состоит из стопок молекул TTF, параллельных оси с. Ионы Вг-
также сгруппированы в колонки параллельпо оси с. Однако траис-
о о
ляциопные расстояния катионных (3,57 А) и анионных (4,54 А)
колонок различны (рис. 6.38). Структура несоразмерна вдоль оси с,
так как периодичности решеток двух подъячеек не являются
простыми кратными числами (или долями) одной и другой. Важно отметить,
Соединения со смешанной валентностью 339
Рис. 6.39. Кристаллическая структура HMTTF — TGNQ.
о — проекция на плоскость, перпендикулярную оси упаковки; б — проекция на
плоскость, содержащую ось упаковки [Greene et al., 1976].
что периодичность подрешетки TTF не зависит от стехиометрии,
тогда как периодичность бромидной подрешетки— зависит.
Следовательно, перепое заряда в соли можно выразить как /= 3,57/с(Вг).
По-видимому, нестехиометрические составы в системах TTF-
22*
340
Взаимосвязь структура — свойство
галогенид стабилизируются в силу электростатических маделунгов-
ских факторов. Расчеты показывают, что энергия Маделуига
максимальна при содержании галогена 0,7—0,8. Вне этого состава
вдоль оси упаковки начинает доминировать отталкивание между
одинаковыми зарядами, уменьшая общую энергию связи.
Оптическое поглощение для TTF—Вг0,79 имеет новый пик в области
0,7 эВ. Этот пик, который не наблюдается в спектрах стехиометри-
ческих солей, приписывается переходу смешанно-валентного
переноса заряда (внутри упаковки) между нейтральным TTF и смежным
TTF+.
Структура HMTTF—TCNQ, типичного комплекса типа TTF-
TCNQ, показана на рис. 6.39 (HMTTF-гексаметилентетратиофульва-
лен). Выделенная упаковка в этой структуре является
характерным признаком высокопроводящей органической системы (с
переносом заряда) семейства TTF—TCNQ. В системе HMTTF—TCNQ рас-
о
стояние между донорными молекулами составляет 3,57 А, тогда как
о
между молекулами TCNQ вдоль стопок — 3,23 А. Чтобы анионные
и катиопные подрешетки были соразмерны друг с другом, молекулы
упаковываются в зигзагообразной конфигурации так, что нормали к
молекулярным плоскостям не параллельны оси упаковки, а
образуют с нею угол. Углы для донорных и акцепторных стопок
различны. В структуре HMTTF—TCNQ их значения составляют
соответственно 23,8 и 34,2°.
От других солей с такими катионами, как щелочные металлы и
тстраметиламмоний, семейство TTF—TCNQ отличает неполный
перенос заряда (/< 1). Члены семейства TTF—TCNQ отличаются
также от галогеиидов TTF; в последнем случае, где заряд на каждом
атоме галогена равен единице, частичный перенос заряда
(смешанная валентность) реализуется путем образования нестехиометри-
ческих соединений, тогда как в семействе TTF—TCNQ состав сте-
хиометричный (1:1), но смешанная валентность получается в
результате частичного переноса электрона.
Доказательство неполного переноса электрона (смешанной
валентности) получено в ряде физических исследований. Исследования
оптической абсорбции показали низкоэнергетический пик при 0,3 эВ,
которого нет в непроводящих солях, таких как K+(TCNQ)~. Более
того, абсорбция поляризуется параллельно оси упаковки.
Абсорбция, следовательно, обусловлена, несомненно, смешанно-валентным
электронным переходом внутри упаковки. TTF—TCNQ при 59 К
подвергается переходу из проводящего в непроводящее состояние.
Этот переход характеризуется едва различимой периодической
модуляцией решетки, обусловленной взаимодействием электронов
проводимости с решеткой (ВЗП)— волна зарядовой плотности. Это
проявляется в форме сателлитных рефлексов вокруг брэгговских пиков
на диффракционной картине. Так как структура модулируется сину-
Низкоразмерные твердые тела
341
соидально, брэгговские пики, обусловленные усредненной
структурой, по существу, остаются без изменения. Поскольку сателлитные
пики получаются в результате взаимодействия между электронами
проводимости и решеткой, их положения определяются степенью
переноса заряда /; для TTF—TCNQ по диффракционным сателлитам
получено значение / = 0,59. Сравнение переноса заряда в ряду
солей TCNQ с потенциалом восстановления катионов показывает,
что только те катионы, которые имеют потенциал восстановления
Ег = 0,0—0,5 В (относительно насыщенного коломельного
электрода), приводят к смешанно-валентному или неполному переносу
[Torrance, 1979]. Если потенциал слишком высокий (перилен, пирен,
антрацен и т. д.), переноса заряда не наблюдается вообще, а если
потенциал слишком низкий, перенос — полный.
Влияние переноса заряда / на удельную проводимость солей
TCNQ можно представить следующим образом: для того чтобы имела
место электропроводность, электроны должны двигаться от одного
TCNQ к другому. Если перенос заряда полный, процесс можно
представить так:
TCNQ" + TCNQ- ->- TCNQ0 + TCNQ2-,
т. е. он подразумевает образование дианиона. Понятно, что энергия
этого процесса должна запрещать перенос заряда, и,
следовательно, соли TCNQ с / = 1 являются диэлектриками. В солях со
смешанной валентностью электроны могут легко двигаться вдоль стопки
согласно процессу TCNQ" + TCNQ0 ->- TCNQ0 + TCNQ-, который
не требует образования дианионов. Однако эта картина только
качественная. Более корректное описание должно включать зонную
модель. Связь между смешанной валентностью и электрическими
свойствами видна в HMTTF—TCNQ и HMTTF—TCNQF4. Обе
системы изоструктурны, но HMTTF—TCNQ — металлическая, тогда как
HMTTF—TCNQF4 — полупроводник; проводимость последней
почти на семь порядков меньше первой. Это различие возникает из-за
того, что TCNQF4 — намного более сильный акцептор, чем TCNQ,
и следовательно, перенос заряда в HMTTF—TCNQF4 — полный.
6.8. НИЗКОРАЗМЕРНЫЕ ТВЕРДЫЕ ТЕЛА
Химиков в общем занимают трехмерные структуры, и
большая часть химии твердого тела имеет дело с трехмерными твердыми
телами. Однако растет интерес к низкоразмерным твердым телам,
которые имеют необычную анизотропию своих свойств. Известно,
что графит в двух измерениях — металлический, а в третьем —
полупроводник; поразительные различия в свойствах слюды
(чешуйки) и асбеста (волокна)— обычное явление. Платиновое цепочечное
соединение КСР, о котором шла речь ранее, только в направлении
342 Взаимосвязь структура — свойство
цепочек отражает видимый свет и проводит электричество как
металл. Если рассматривать кристалл КСР с поляроидной ориентацией
так, что электрический вектор света параллелен оси цепочки, этот
кристалл обладает сильной отражательной способностью и имеет
медно-красный цвет; если поляроид повернут по правому углу, то
кристалл имеет бледно-желтый цвет и прозрачен, как любой другой
ионный кристалл. Ситуация здесь похожа на случай с красной солью
Вольфрама. Большинство синтетических или молекулярных
металлов являются низкоразмерными твердыми телами: многие из этих
экзотических материалов, которые испытываются на
сверхпроводимость, также являются низкоразмерными [Keller, 1975, 1977;
Miller, Epstein, 1978; Hatfield, 1978; Alcacer, 1980; Miller, 1982J.
Низкоразмерные твердые тела удобно разбить на две категории:
цепочечные (преимущественно одномерные) и слоистые
(преимущественно двумерные). Примеры цепочечных соединений — КСР и
другие цепочечные платиновые соединения, полимерный (SN)^,
полиацетилен, Hg3_a;AsF6 с цепочками Hg, [(CH3)4N]-MnCl3, KCuF3 и
RbFeCl3. Примеры слоистых соединений — графитоподобные
системы, халькогениды Та и Nb, K2NiF4, (RNH3)2MC14 и СоС12 (R —
СН3 и т. д., М—Сг, Мп и т. д.). Мы кратко рассмотрим магнитные,
электрические и оптические свойства, а также фазовые переходы для
типичных представителей этого необычного класса материалов Шау,
1983; Subramanyam, Naik, 1985; см. также Phil. Trans. Roy. Soc.
London, 1985, A314I.
Для понимания магнитного поведения твердых тел необходимо
учитывать не только размерность решетки (1—3), но также и
размерность спина или параметр порядка (1 — 3), что приводит к девяти
возможным типам магнитных систем. Кроме того, параметр
взаимодействия / может быть положительным (ферромагнетик) или
отрицательным (антиферромагнетик), а это дает 18 различных типов
возможных систем (табл. 6.7). Рассмотрен магнетизм в различных
модельных системах [Jongh, Miedema, 1974]. Если имеют место
взаимодействия только ближнего порядка, одномерные магнитные
системы, в которых наблюдается магнитное взаимодействие только
в одном направлении, не обладают магнитным фазовым переходом
(одномерные изинговские системы). Если имеются отклонения от
идеального одномерного поведения, то при ненулевых температурах
возможно наблюдать фазовый переход в трехмерно-упорядоченную
структуру. Если / — величина внутрицепочечного взаимодействия,
а /' — величина межцепочечного взаимодействия, то отношение
/7/ определяет температуру, при которой имеет место фазовый
переход. Двумерная изинговская модель со спином 1/2 в квадратной
решетке проявляет фазовый переход в отсутствие внешнего
магнитного поля. Это точное решение впервые было получено Онзагером.
Двумерная гайзенберговская система не может иметь какого-либо
дальнего порядка, и в такой системе магнитная под решетка не об-
Низкоразмерные твердые тела
343
Таблица 6.7
Примеры низкоразмерных магнитных систем
Соединение
KCuF3
(CH3)4NMnCl3
CsCuGl3
CsCoCl3
RbFeCl3
CsNiF3
K2CuF4
K2NiF4
CoCla
CoBiy6H20
K2CoF4
Размерность
спина
3
3
3
1
2
2
3
3
2
2
1
решетки
2
2
2
2
2
s
1/2
5/2
1/2
1/2
1
1
1/2
1
1/2
1/2
1/2
Взаимодействие*
АМФ
АМФ
ФМ
АФМ
ФМ
ФМ
ФМ
АФМ
ФМ
АФМ
АФМ
**
Те , К
38 (243)
0,84 (55)
10,4
8
2,55
2,61
6,5
97,2
10
3,14
107
гв/е
0,20
0,011
—
0,08
—
0,12
0,28
0,36
0,8
0,6
0,55
* ФМ — ферромагнитное; АФМ — антиферромагнитное.
** В скобках указаны температуры, при которых удельная восприимчивость
максимальна.
ладает спонтанной намагниченностью. Однако нет никакой
термодинамической аргументации, которая исключает возможность
отклонения удельной восприимчивости, хотя намагниченность в
нулевом поле может исчезать. Температура, при которой наблюдается
отклонение в удельной восприимчивости при отсутствии отклонения
от идеального поведения, называется температурой Стэнли —
Каплана Г§к- ^sk является нижним пределом для температуры
перехода Гс, имеющего место в силу отклонений от идеального
двумерного поведения. Только трехмерная магнитная система
обнаруживает дальний порядок, независимый от размерности спина.
KCuF3 и K2NiF4 являются хорошо известными примерами
соответственно одномерных и двумерных магнитных систем. KCuF3 имеет
тетрагональное искажение перовскитовой структуры. Расположение
орбиталей, показанное на рис. 6.24, таково, что имеется сильное
перекрывание вдоль оси с и почти нулевое перекрывание вдоль
плоскости аЪ\ J4J в KCuF3 составляет 2,7х10~2, данные по нейтронной
дифракции свидетельствуют об антиферромагнитном упорядочении
при 39,5 К, тогда как магнитная восприимчивость имеет максимум в
области 275 К. Данные по восприимчивости хорошо согласуются с
данными, рассчитанными для одномерной гайзенберговской системы
(рис. 6.40, а). В K2NiF4 взаимодействие между ионами Ni2+ внутри
слоя антиферромагнитное и есть еще небольшое взаимодействие
перпендикулярно слою. Магнитная восприимчивость K2NiF4 имеет
широкий максимум около 230 К (рис. 6.40, б), типичный для двумерных
344
Взаимосвязь структура — свойство
0.06
1 кс°F)
1 1 1 1 1
100 300
т,к
1
500
О»
Z
* 0,02
Рис. 6.40. а — график зависимости % от Т для KCuF3
(кружки) в сравнении с теоретическим графиком
(сплошная линия) для одномерной гейзенберговской
модели; б — график зависимости % от Т для K2NiF4
(кружки) и теоретический вариант (сплошная линия)
для двумерной гейзенберговской модели.
магнитных систем; d%/dT имеет максимум при 100,5 К, тогда как
данные по нейтронной дифракции говорят о том, что при 97,1 К
имеет место трехмерное упорядочение. Последние данные
свидетельствуют также о том, что корреляции ближнего порядка
сохраняются вплоть до 200 К. Таким образом, широкий максимум в
восприимчивости двумерной системы следует связывать с
взаимодействием ближнего порядка. Данные по удельной теплоемкости имеют
небольшую аномалию при 98,7 К и широкий максимум в области
200 К, который подтверждает, что выше Тс существует значительное
взаимодействие ближнего порядка.
K2GoF4 — двумерный изинговский антиферромагнетик с S =
= 1/2. Изинговская анизотропия является результатом
орбитального вырождения основного состояния Со2+ и искажения, требуемого
для снятия вырождения. Удельная восприимчивость этого
соединения (см. рис. 6.26) хорошо сопоставима с теоретической кривой
квадратичной изинговской решетки. Тс совпадает с температурой, при
которой (1%/(1Г максимальна. Кривая удельной теплоемкости имеет
логарифмическую особенность при Гс, подтверждая двумерное изин-
говское поведение.
Спиновую и решеточную размерность системы лучше всего
можно определить при изучении термодинамических свойств системы
вблизи температуры перехода. При отсутствии этих данных важной
величиной при определении размерности системы является
отношение Гс/9, где Тс — истинная критическая температура, 9 —
критическая температура, предсказанная теорией среднего поля.
В табл. 6.7 приведены соотношения Тс и 0 для различных
размерностей решетки и спина, а также для различных значений спина.
Исследования магнитных свойств низкоразмерных твердых тел
активно продолжаются, и недавно обнаружены многие интересные
Низкоразмерные твердые тела 345
системы. V305 имеет переход металл — неметалл при 425 К и
магнитный переход при более низких температурах [Jhans, Honig,
1981]. Максимум в магнитной восприимчивости находится при 12 К,
но действительный переход, найденный из данных по теплоемкости,
находится при 76 К. Переход при 76 К в V305 отражает
перестройку упорядочения из трехмерного в одномерное цепочечное
[Honig, 1982].
(Об электронных свойствах двумерных систем см. [Ando et al.,
1982].) La2Ni04 в области 500 К подвергается переходу металл —
неметалл в плоскости ab [Rao et al., 1984]; стехиометрические
образцы этого оксида, по-видимому, также подвергаются
антиферромагнитному упорядочению. О переходе из гайзенберговского в изин-
говский 2с£-типы сообщалось для нескольких твердых тел семейства
K2NiF4. Недавно опубликован обзор по антиферромагнитным
(Са2Мп04) и ферромагнитным (La05Srb5CoO4) оксидам со структурой
K2NiF4 [Ganguly, Rao, 1984]. В Ln2Cu04 ионы Cu2 + не вносят вклад
в удельную восприимчивость. Соединения с формулой А2СгХ4
являются прозрачными ферромагнетиками [Day, 1979]; детальные
структурные, магнетооптические и другие подобные исследования
этих интересных галогенидов продолжаются (см., например: [Janke
et al., 1983; Fyne et al., 1984]). (RNH3)2MX4 (R-CH3 и т. д.; M-Cr,
Mn и т. д.) проявляют ферромагнетизм (М—Сг) и
антиферромагнетизм (М—Мп), а их видимые спектры поглощения связываются с
магнитным упорядочением [Day, 1984]. FeCl2 со слоистой
структурой является метамагнетиком; под влиянием магнитного поля
антиферромагнитная фаза превращается в ферромагнитную фазу, что
сопровождается увеличением непрозрачности (из-за сосуществования
двух магнитных фаз с различными показателями преломления [Rob-
bins, Day, 1973]). В случае низкоразмерной системы конкурирующие
взаимодействия между упорядоченными цепочками или слоями
спинов приводят к наклоненным или несоразмерным структурам. NiBr2
и Nij_3.M3.Br2 (M—Fe, Mn) проявляют переход в гелимагнитную фазу
[Moore, Day, 1985].
Слоистые материалы со структурой K2NiF4 имеют интересные
фазовые переходы. Например, соединения типа (RNH3)2MX4
подвергаются нескольким фазовым переходам, которые обусловлены
различиями в способах связи NH3+-rpynn с галогенидными ионами
слоев, причем органическая цепочка R при образовании водородной
связи изгибается [Rao et al., 1981]. Эти соединения
рассматриваются как неорганические-органические молекулярные композиты [Day,
1985]. Получены также соли с диацетиленовыми катионами, которые
могут подвергаться полимеризации [Ledsham, Day, 1981 ].
Слоистые халькогениды, такие как TaS2, могут интеркалировать
такие большие молекулы, как амид стеариновой кислоты. Химию
интеркаляции этих и других слоистых материалов мы будем
обсуждать в гл. 8. TaS2 является металлическим и сверхпроводящим
346 Взаимосвязь структура — свойство
о ю 20 зо
1000/J, К"1
Удельная прово-
КСР как фуык-
температуры.
(Тс = 0,8 К), при включении пиридина сверхпроводящий переход
увеличивается до 3,5 К; межслоевое расстояние увеличивается от 3
о
до 6 А. Октадециламин увеличивает межслоевое пространство до
о
50 А, Тс возрастает до 3 К (см. разд. 3.2.2 и 8.7).
Красная соль вольфрама [Pt(C2H5NH2)4] [Pt(C2H5NH2)4Cl2] .
• 4Н20 (рис. 6.41) имеет межвалентную полосу поглощения в
видимой области, обусловленную переходом Pt (II) (dzz) ->■ Pt (IV) (dA,
возможно, с участием орбиталей мостиковых галогенидных ионов.
Резонансный Раман-спектр имеет большое число линий
симметричного валентного колебания X—Pt—X, обусловленное смещением
возбужденных состояний электронов вдоль этой колебательной
координаты [Clark, 1977]. В тетрацианоплатинитах энергия пика
изменяется обратно пропорционально кубу расстояния Pt—Pt,
показывая тем самым, что все диполи перехода располагаются вдоль
платиновой цепочки [Day, 1975].
Согласно ранее выполненной работе [Krogmann, 1969] большой
интерес проявлен к электрическим свойствам K2Pt(CN)4Br0>30 •
• ЗН20(КСР) и подобных соединений. Цель этих исследований
получить высокотемпературный сверхпроводник, как предсказывал
Little [1964]; другой аспект интереса — иайерлсовская
неустойчивость одномерных металлических проводников (по отношению к
искажению) [Peierls, 1955]. КСР является металлическим в области
Рис. 6.41. Структура красной
соли Вольфрама.
X
о
3
h
Ю
°\
ю?\
юА
Ю'6\
юА
Рис. 6.
димость
ци
Низкоразмерные твердые тела
347
комнатной температуры, при понижении температуры его удельная
проводимость начинает падать, и в конечном счете она имеет
температурную зависимость полупроводника (рис. 6.42). Несколько
других анионо- и катиоподефицитных платиновых комплексов
исследованы с точки зрения их отношения к переходам металл — неметалл
в результате паиерлсовского искажения; этот переход подвержен
сильному влиянию лигандов, анионов и катионов [Underhill,
1985].
Полимер (SN)^ является не только металлом, но при 0,26 К
он становится сверхпроводником [Hatfield, 1978]. Другое
квазиодномерное соединение, проявляющее сверхпроводимость, это
Hg3-jcAsFe (рис. 6.43), рассмотренное ранее в разд. 4.9 и 6.7.
Получены другие ртутные цепочечные соединения с золотистым блеском,
имеющие общую формулу Hga-^MFg (M—Sb, Nb, Та, As),
содержащие непересекающиеся цепочки из атомов ртути, параллельные осям
а и Ъ [Gillespie et al., 1985]; эти соединения превращаются в стехио-
метрический Hg3MF6 с серебристым блеском и слоистой структурой
из плотноупакованных атомов ртути. Измерены электрические
свойства этих соединений. В то время как анизотропная проводимость
является результатом того, что ферми-поверхность состоит из ряда
цилиндров (с осями вдоль оси с), сверхпроводящие свойства являют-
348
Взаимосвязь структура — свойство
и—i—i—г-^—I—I—I—г
4 2 0 2 4
% добавки
Ь
Зона
проводимости
rw»»rr> Полент -
пая зопо
zzzV(п)
Рис. 6.45. а — влияние допирования на удельную проводимость
(сплошная линия) и термоэдс (штриховая линия) полиацетилена [Etemad
et al., 1982]; б — солитоны в mpawc-полиацетилене: I —нейтральные,
II — положительные, III — отрицательные. Стрелкой указана граница
между двумя симметричными конфигурациями. А — акцептор; Д — донор
[Subramanyam, Naik, 1985].
ся результатом того, что ртуть поймана в ловушки в отдельных
областях [Datars et al., 1985].
Поскольку большой пик в проводимости в области 60 К
обнаружили сначала в TTF—TCNQ [Coleman et al., 1973] (рис. 6.44),
многие исследования выполнены на родственных системах с TTF
[Subramanyam, 1981; Soos, Klein, 1976]. Некоторые аспекты
относительно проводимости в этих системах упомянуты в предыдущем
разделе. Обнаружено, что (TMTSF)2CI04 и родственные соединения
(TMTSF-тетраметилтетраселенафульвален) проявляют
сверхпроводимость при низких температурах [Jerome, 1985].
Полиацетилен (CH)X стал, вероятно, самой большой новинкой
в этой области [MacDiarmid, Heeger, 1980; Chien, 1984].
Полиацетилен допируется химически или электрохимически с целью
получения материалов р- или я-типа. Проводимость можно поднять до
металлического состояния, добавляя /2, Li, AgCI04 и другие вещества
[Etemad et al., 1982; MacDiarmid et al., 1985]. В то время как
полиацетилен сам по себе имеет проводимость 10~9 Ом-1-см-1, [СН-
(AsF^yl^ и (СН—Jy)x имеют проводимости соответственно 10~4—
5х103 и 10"4—50 Ом-1-см-1 (рис. 6.45). Содержание цис-транс-
полимера во время синтеза можно контролировать.
н н н н
W W
/ W \
\
Цис
ннн
i с i
н
н н
Транс
Ферроики
349
Считается, что солитоны являются важными дефектными
состояниями в этих сопряженных полимерах (см. рис. 6.45). Недавние
исследования, однако, показали, что корреляционная энергия является
более важным фактором в получении энергетической щели в (СН)Х
[Soos, Ramasesha, 1983]. Другие полимеры, родственные
полиацетилену,— политиофен, полипиррол, полифениленсульфид и полипара-
фенилен (см. разд. 3.2.7). За последние пять лет проведены обширные
измерения на допировапных полиацетиленах, и, по-видимому, эти
материалы, в отличие от других проводящих полимеров, таких как
(SN)X, имеют хорошие технологические возможности.
Существует много других низкоразмерных твердых тел, которые
не рассматриваются здесь; один из них — полидиацетилен [Bloor,
1985]. Порфириновые молекулярные металлы проявляют
анизотропную проводимость, где зарядоносители принадлежат как металлу,
так и лиганду [Hoffman, Ibers, 1983]. Проводимость в некоторых
системах достигает значений больше, чем 103 Ом-1-см-1, и не
опускается до значения проводимости диэлектрика даже при низких
температурах. Здесь снова комплекс металл — лиганд является
смешанно-валентным (частично окисленным), с неполным переносом
заряда, а дырки, которые получаются при окислении иодом,
по-видимому, прыгают между лигаидом и металлом. Химическая
податливость порфириновых металлов действительно может представлять
определенные преимущества для возможных применений этих
материалов и достойна дальнейшего исследования.
6.9. ФЕРРОИКИ 5
Важной характеристикой ферромагнитных материалов
является гистерезисная петля в данных по зависимости между
намагниченностью и магнитным полем. Электрический аналог
ферромагнетика — ферроэлектрик (сегнетоэлектрик), который имеет ги-
стерезисную петлю в данных по зависимости между поляризацией и
электрическим полем и проявляет спонтанную поляризацию в
отсутствие внешнего электрического поля. Ферроэлектрические (сег-
нетоэлектрические) материалы обладают постоянными дипольными
моментами, причем диполи возникают из-за отсутствия центра
симметрии. Если энергетические соображения благоприятствуют анти-
параллелыюй ориентации постоянных диполей на соседних
плоскостях вместо параллельной, такой кристалл называется антифер-
роэлектриком (аптисегнетоэлектриком). Как ферроэлсктрики, так и
5 В данном разделе сохранена авторская номенклатура ферромагнитных
материалов как более универсальная. {Пер.)
350
Взаимосвязь структура — свойство
антиферроэлектрики демонстрируют аномалию в диэлектрической
постоянной при критической температуре, но последние не имеют
гистерезисной петли в зависимости поляризации от электрического
поля. В этом разделе мы будем обсуждать поведение общего класса
материалов, называемых ферроиками [Newnham, Cross, 1981],
которые включают в себя ферроэлектрики и антиферроэлектрики.
Ферроик можно определить как материал, обладающий двумя
или более ориентационными состояниями, или доменами, которые
могут переключаться из одного состояния в другое при приложении
одной или более соответствующих сил [Newnham, 1974]. В ферро-
электрике ориентационное состояние спонтанной электрической
поляризации может изменяться при приложении электрического поля.
В ферромагнетике ориентационное состояние намагниченности в
доменах может переключаться при приложении магнитного поля;
в ферроэласпгике (сегнетоэластике) направление спонтанной
деформации в домене может переключаться при приложении
механического напряжения. Такие переходы описаны как ферроикные переходы.
Во всех этих случаях границы доменов при приложении силы
движутся таким образом, чтобы завершить изменения в ориентации.
ВаТЮ3 — типичный ферроэлектрик, Сг02 — ферромагнетик, тогда
как CaAl2Si2Os — ферроэластик. Свойства, которые меняют
направление в вышеуказанных примерах, а именно: электрическая
поляризация, магнитная поляризация и упругая деформация — являются
главными величинами в том смысле, что они непосредственно
определяют свободную энергию системы. Ферроики, управляемые
изменением этих свойств, называются поэтому первичными ферроиками.
Во вторичных ферроиках эти свойства наблюдают как
наведенные. Ориентационные состояния различаются по производным
величинам, которые характеризуют наведенные эффекты. Так,
наведенная электрическая поляризация характеризуется диэлектрической
восприимчивостью kif, наведенная магнитная поляризация —
магнитной восприимчивостью ifj и наведенные деформации —
коэффициентом упругой деформации Cijhl. Ориентационные состояния во
вторичных ферроиках, следовательно, отличаются по значениям
ktj, %ij и Cijhi\ эти значения являются тензорными величинами, ранг
тензора равен числу подстрочных знаков. Такие наведенные
эффекты, как поляризация или намагниченность, могут получаться в
результате комбинированных эффектов, таких как наведенная
напряжением поляризация {пьезоэлектрический эффект), наведенная
напряжением намагниченность (пьезомагнитный эффект) или
комбинированный эффект двух типов полей, такой как эластоэлектрический
или эластомагнитный. Изменение свойств с направлением можно
наблюдать по соответствующим производным величинам. В табл. 6.8
мы приводим различные типы ферроикных эффектов. В основу
классификационной схемы разделения ферроиков на первичные,
вторичные и т. д. положено термодинамическое начало.
Ферроики
351
^^■
Ферроэлектри-
ки
Ферромагнетики
Ферроэластики
Ферробиэлект-
рики
Ферробимагне-
тики
Ферробиэлас-
тики
Ферроэласто-
электрики
Ферромагнето-
эластики
Ферромагнето-
электрики |
Та
Первичные и вторичные ферроики
Свойство
Изменяющее поле
Первичные ферроики
| Спонтанная поляризация
Спонтанная
намагниченность
Спонтанная деформация
Электрическое
Магнитное
Механическая сила
Вторичные ферроики
Удельная
диэлектрическая восприимчивость
Удельная магнитная
восприимчивость
Эластик
Пьезоэлектрические
коэффициенты
Пьезомагнитные
коэффициенты
Магнетоэлектрические
коэффициенты
Электрическое
Магнитное
Механическая сила
Электрическое поле и
механическая сила
Магнитное поле и
механическая сила
Магнитное и
электрическое
блица 6.8
Пример
ВаТЮ3
СгОа
CaAl2Sia08
SrTi03
NiO
а-Кварц
NH4C1
FeC03
Cr203
Свободную энергию Гиббса для кристаллического твердого тела
в дифференциальной форме можно записать так:
AG = -SdJ + PtdEt + MtdHi + гийоф (6.59)
где малое изменение свободной энергии моля вещества (dG)
представляет собой сумму теплового члена (SdT) и членов, получающихся в
результате действия различных сил. Здесь Pt и Et означают
соответственно электрическую поляризацию и электрическое поле; Mi и
Ht — магнитную поляризацию (намагниченность) и магнитное поле;
е^- и atj — упругую деформацию и соответствующее механическое
напряжение; i и / изменяются от 1 до 3. Если рассматривать ферро-
икные явления в изотермических условиях, то первый член в
уравнении (6.59) будет равен 0. Учитывая и спонтанный, и
индуцированный эффекты, разницу в свободной энергии Гиббса для двух ориента-
ционных состояний записываем так:
AG = АР\Е{ + ДМ-Я* + Аеуау + \ [А/с^ВД- + Дху# i#j +
+ ЬСцыацаы] + 2 [AaijEiffj + AdijkEiGjk + bQijhHiOjh]. (6.60)
Здесь А — разница в соответствующих значениях между ориента-
ционными состояниями I и II для спонтанных величин (s).
Следовательно, Ae'ij = e\j (II) — E|j (I) и т. д.— разница es для ^'-компонентов
352
Взаимосвязь структура — свойство
в состояниях II и I. В отсутствие внешнего поля AG = 0 и ориента-
ционные состояния энергетически вырождены.
Из уравнения (6.60) очевидны несколько возможных ферроикных
явлений в зависимости от доминантности отдельных членов. В
материале, который обладает большим значением спонтанной
поляризации, другие члены становятся несущественными и свободная
энергия в электрическом поле подчиняется выражению
AG = АР'\Ег = АР*Е, (6.61)
где Е — электрическое поле в выбранном направлении (скажем, z).
Аналогичные выражения можно написать для первичных ферройков,
включающих спонтанную намагниченность и спонтанное напряжение,
когда они являются доминантными величинами и взаимодействуют
с соответствующими внешними полями. Когда APjs = \Mts =
= Дег/ = 0, величины, выражающие значения AG, определяются
членами в двух квадратных скобках в уравнении (6.60). Первый ряд
величин — kkij, hytj и ^ijhl. Когда kktj Ф 0, а Да^- и t±dijh имеют
малый или незначительный вклад, как в ферроэлектриках,
свободная энергия определяется выражением
AG ж i MtijEiEj ж /ikE2. (6.62)
Доминантность других членов приводит к аналогичным выражениям
для других вторичных ферроиков.
Ферроэлектрический переход из одного ориентационного
состояния в другое (наблюдаемый по гистерезисной петле) электрически
является переходом первого рода. Порядок перехода между
доменными состояниями в ферроиках является простой суммой экспонент
полевых членов в выражении свободной энергии. Ферробиэласти-
ческие, ферробимагнитные, ферроэластоэлектрические, ферромагне-
тоэлектрические и другие подобные переходы являются переходами
второго рода. Здесь мы должны заметить, что коэффициенты вариации
сами по себе являются взаимозависимыми. Например, спонтанная
поляризация связана с полем, которое, в свою очередь, определяет
напряжение через сильное взаимодействие с решеткой, увеличивая
тем самым вклад электрострикционпых или пьезоэлектрических
коэффициентов. Следовательно, где бы ни существовала
спонтанная поляризация, рядом с нею должно быть спонтанное
напряжение, и наоборот. Когда бы спонтанная поляризация и спонтанная
деформация ни продуцировали ориентационные состояния,
совершенно независимые друг от друга ферроэластические и ферроэлект-
рические свойства могут реализоваться полностью, и такие
материалы называются полностью ферроэлектрическими — полностью фер-
роэластическими материалами. Другие взаимосвязи аналогичной
природы можно видеть из взаимозависимости различных величин
уравнения (6.60).
Ферроики
353
Рис. 6.46. Диаграмма,
иллюстрирующая несколько типов
параметров порядка,
приводящих к собственным и
несобственным ферроикам [Newnham,
Cross, 1981].
При высоких
температурах ферроэлектриче-
ские материалы переходят
в параэлектрическое
состояние (где диполи
ориентированы случайно),
ферромагнитные — в
парамагнитные, а ферроэла-
стические — в нормальное
состояние без двойников.
Переходы
характеризуются параметрами порядка
[Rao, Rao, 1978]. Эти параметры порядка являются
характеристичными свойствами, параметризованными таким образом,
что результирующее значение равно 1 для ферроикного
состояния при температуре ниже температуры перехода и равно О
для неферроикной фазы за пределами температуры перехода.
Поляризация, намагниченность и напряжение являются
собственными параметрами порядка для ферроэлектрических,
ферромагнитных и ферроэластических переходов соответственно. Всегда,
когда переходы подчиняются ожидаемым вариациям этих параметров,
они называются собственными ферроиками. Как мы отмечали
ранее, природа ферроикиых явлений такова, что параметр порядка,
который определяет переход, часто может отличаться от
«собственного» параметра порядка. Ферроики, в которых параметр порядка
не является «собственным» свойством, называются несобственными
ферроиками. Например, в молибдате тербия параметр порядка
представляет собой конденсированную оптическую моду [Dorner et al.,
1972]. Оптическая мода вызывает спонтанное напряжение, которое,
в свою очередь, вызывает спонтанную поляризацию путем
пьезоэлектрического взаимодействия. Следовательно, молибдат тербия
является собственным ферроэлектриком и несобственным ферро-
эластиком.
На рис. 6.46 изображены взаимосвязи собственных и
несобственных первичных ферроиков в виде гексагона [Newnham, Cross, 1981].
Параметр порядка собственных ферроиков находится по диагоналям
шестигранника, а его стороны представляют несобственные
ферроики. Они указывают на то, что источник ферроикных явлений —
кросс-взаимодействие. Несобственный первичный ферроик в этой
23 ч. Н. Р. Рао, Дж. Гопалакришнан
jlJE Магнетснрерро -
-^т эластичность
354
Взаимосвязь структура — свойство
классификации отличается от собственного вторичного ферроика
префиксом «ферро» только перед частью слова (относящейся к
первичному ферроику), а не перед всем словом. Так, термин «магнетофер-
роэлектрик» (магнетосегнетоэлектрик), например, Сг2Ве04)означает,
что материал является несобственным ферроиком, тогда как ферро-
магнетоэлектрик (например, Сг203) должен означать, что материал
является вторичным ферроиком.
Ферроэлектрики. Среди 32 классов кристаллов 11 обладают
центром симметрии, являются центросимметричными и, следовательно,
не обладают полярными свойствами. Из 21 нецентросимметричных
классов 20 проявляют электрическую полярность при наложении
нагрузок и называются пьезоэлектриками; один из
нецентросимметричных классов (кубический 432) имеет другие элементы
симметрии, сочетание которых исключает пьезоэлектрический характер.
Пьезоэлектрические кристаллы подчиняются линейной зависимости
Pt = gtjFj, между поляризацией Р и силой F, где gtj—
пьезоэлектрический коэффициент. Наоборот, пьезоэлектрический эффект
приводит к механической деформации или напряжению под влиянием
электрического поля. Десять из 20 пьезоэлектрических классов
обладают единственной полярной осью в непроводящих кристаллах,
изменение в поляризации может наблюдаться по изменению температуры;
такие кристаллы относятся к пироэлектрическим кристаллам. Если
полярность пироэлектрического кристалла в электрическом поле
может быть обращена путем наложения на электрическое поле, такие
кристаллы мы называем ферроэлектриками. Следовательно, для того
чтобы установить пьезоэлектрическую или пироэлектрическую
природу твердого тела, достаточно знать класс кристалла, но для фер-
роэлектриков необходимым условием является обратимая
поляризация. Хотя все ферроэлектрические материалы являются также
пьезоэлектриками, обратное — неверно; например, кварц — пьезоэлект-
рик, но не ферроэлектрик.
Одной из важных характеристик ферроэлектриков является то,
что диэлектрическая постоянная подчиняется закону Кюри — Вейса
(уравнение (6.48)), похожему на уравнение, связывающее
магнитную восприимчивость с температурой в ферромагнитных материалах.
Для иллюстрации этого поведения на рис. 6.47 показано изменение
диэлектрической постоянной с температурой для монокристалла
ВаТЮ3. Выше 393 К BaTiO з становится параэлектрическим (диполи
разупорядочиваются). Поликристаллические образцы при
температуре перехода имеют менее заметные изменения.
Титанат бария, который кристаллизуется в перовскитовой
структуре, при температуре выше 393 К имеет кубическую
симметрию с ионами Ва2+ в центре и октаэдрами Ti06 по вершинам. При
393 К структура превращается в тетрагональную, при 278 К —
в ромбическую и при 183 К — в ромбоэдрическую (см. рис. 6.47).
По сравнению с кубической в тетрагональной фазе идет удлинение
Ферроики
355
вдоль одного из ребер (направление [100]), в ромбической фазе —
удлинение вдоль одной из граневых диагоналей (направление [110])
и в ромбоэдрической — вдоль одной из телесных диагоналей
(направление [111 ]). При охлаждении кубической фазы кристалла (в
которой Ti4+ находится в центре октаэдра) ион Ti4+ движется
последовательно по этим трем направлениям. Кроме диэлектрической по-
стоянной и поляризации, теплоемкость и другие свойства также
обнаруживают аномальные изменения при этих трех фазовых
переходах BaTi03 (см. рис. 6.47).
Поляризация ферроэлектрического материала в электрическом
поле изменяется нелинейно. Зависимость Р—Е характеризуется
гистерезисной петлей, а наличие гистерезисной петли является
лучшим доказательством существования ферроэлектричества в
веществе. Причина образования гистерезисной петли заключается в
перегруппировке доменов под влиянием приложенного электрического
поля. Обычно домены распределяются случайно, что в итоге дает
нулевую поляризацию. Под действием электрического поля или
механического напряжения растут благоприятно ориентированные
домены за счет менее благоприятно ориентированных до тех пор, пока не
получится одна доменная конфигурация. В ферроэлектрической
фазе, по сравнению с параэлектрической, доменная структура сама по
себе родственна кристаллической. Так, в тетрагональной фазе
BaTiOg смежные домены могут иметь свои полярные оси под углом
90° и 180° (рис. 6.48).
Многие ферроэлектрические материалы проявляют смягчение
определенных колебательных мод. Мягкая мода ферроэлектрических
материалов детально исследована с помощью Раман-спектроскопии
и нейтронного рассеяния [Blinc, Zeks, 1974].
Ферроэлектричество свойственно большому числу материалов
[Jona, Shirane, 1962; Subbarao, 1974]. К ним относятся оксиды с
перовскитовой структурой (например, ВаТЮ3), твердые тела с
водородной связью (например, КН2Р04), соединения со структурами
типа вольфрамовых бронз, пирохлоры, простые соли (например,
(NH4)2S04, NaN02, KN03), квасцы, органические соединения
(например, мочевина, глицин-сульфат) и такие простые бинарные
соединения, как НС1, FeS, GeTe и V3Si. Ферроэлектрики с водородной
связью, подобно КН2Р04, демонстрируют изотопные эффекты по
дейтерию (Тс увеличивается при замещении водорода на дейтерий),
показывая тем самым важную роль водородной связи. Недавно
найдено, что даже аморфные образцы таких материалов, как LiNb03,
проявляют подобные аномалии [Varma et al., 1985].
Антиферроэлектрические материалы при Тс имеют
сверхструктуру в антиполярной фазе, а также аномалию в диэлектрической
постоянной. При Гс, когда антиферроэлектрическая фаза переходит в
параэлектрическую, наблюдаются также изменения других физи-
23»
356 Взаимосвязь структура — свойство
о
их
1,86А
2,00 А
о Q&Q—о°
2.17А
6 01
1
Ромбическая
Тетрагональная
^ ^Ромбоэдрическая
&
М
Злектроды
Кубиче
Ромбическая Тетрагональная
■Ч
Понижение температуры
^ 0,20-
6000]
2000
I
«о
Ромбоид- Ромби- Тетра- Куби-
\рическая ческая гональ-чес-
нся пая
-80 О 80
Температура
-до о во
Температура,
760
о
С
Рис. 6.47. Искажение октаэдра TiOe в тетрагональном ВаТЮ3 (вверху) и
возможные ориентации полярной оси, когда электрическое поле прикладывается вдоль
псевдокубического (001) направления BaTi03 (в середине). Полярные оси
показаны стрелками внутри каждого куба. Фазовые переходы в ВаТЮ3,
сопровождаемые изменениями в диэлектрической постоянной (а); спонтанной поляризации
(б); теплоемкости (в) и размерах решетки (г) (внизу).
Ферроики
357
Рис. 6.48. Доменные стенки в тетрагональном BaTi03.
АА'— девяносто градусная стенка; ВВ'— стовосьмидесятиградусная
ческих свойств. Однако антиферроэлектрические материалы не
имеют гистерезисной петли в зависимости Р—Е. Поскольку разница
в энергиях между антиферроэлектрическими и ферроэлектрическими
состояниями довольно мала, приложение большого электрического
поля, механического напряжения или изменение состава могут
приводить к тому, что антиферроэлектрические материалы становятся
ферроэлектрическими. PbZr03 с перовскитовой структурой —
типичный антиферроэлектрик. Другие примеры — NaNb03, BiFe03,
PbCo0>5W0>5O3, NH4H2P04 и Cu(HCOO)2.4H20.
В последние годы появились сообщения о большом числе
материалов, проявляющих, кроме ферроэлектричества, другие
интересные свойства (возможно, технологического значения). Вот
типичные из них:
ферроэлектрик — ферроэластик, Gd2(Mo04)3, KNb03,
ферроэлектрик — антиферромагнетик, YMn03, HoMn03,
ферроэлектрик — ферромагнетик, Fe3B70,3Cl, Bi9Ti3Fe5027,
антиферроэлектрик — антиферромагнетик, BiFe03, Cu(HCOO)2 x
X 4Н20,
ферроэлектрик — полупроводник, FeS, восстановленный SrTi03,
YMn03,
ферроэлектрик — сверхпроводник, SrTi03, GeTe, V3Si.
Первичные ферроики. Как упоминалось ранее, ориентационные
состояния в ферроэлектриках соответствуют направлениям
поляризации в доменах. Мы можем классифицировать ферроэлектрические
материалы по величине константы Кюри С (уравнение (6.48)).
Большие значения С (~ 105 К)— в оксидных ферроэлектриках, особенно
таких, которые содержат ионы Ti4+, Nb5+ и W7G+, и таких, которые
содержат одиночные пары ионов Pb2+, Bi3+ и Sb3+. Благодаря свсей
электронной структуре, эти ионы способствуют искажениям,
приводящим к спонтанно поляризуемым структурам. Вторая категория,
где С ~ 103 К, включает в себя в основном ферроэлектрики типа
порядок — беспорядок, где наблюдается ротационное упорядочение
биполярных ионов, как в NaN02, или позиционное упорядочение
протонов, как в КН2Р04. Третья категория с очень низким
значением С (~ 1 —10 К) включает такие несобственные ферроэлектрики,
как Gd2(Mo04)3, где размерность атомных смещений увеличивается,
по мере того как уменьшается значение С В то время как в титана-
тах смещения — одномерные, приводящие к сильным дипольным
моментам, в гидрофосфатах и NaN02 диполи упорядочиваются в
плоскостях (двумерные смещения), уменьшая общий эффект
поляризации. В таких несобственных ферроэлектриках, как молибдаты, смеще-
358
Взаимосвязь структура — свойство
ния трехмерные. Попутно можно заметить, что ферроэлектрики в
направлении спонтанной поляризации способны переключаться из
одного состояния в другое с помощью коэрцитивного поля, которое
не превышает диэлектрическую пробойную силу.
Переходы ферроэлектрик — параэлектрик можно понять на
основе теории Ландау — Девоншира, использующей поляризацию
как параметр порядка IRao, Rao, 1978]. Упорядоченная ферроэлект-
рическая фаза имеет более низкую симметрию, относящуюся к одной
из подгрупп высокосимметричной разупорядоченной параэлектри-
ческой фазы. Однако точная структура, в которую переходит пара-
электрическая фаза, определяется на энергетической основе.
Доменные структуры в ферроиках подвергаются влиянию
приложенных полей. В ферроэластиках доменные стенки (удвоенные
слои) могут двигаться при приложении силы. Спонтанные
напряжения, связанные с ферроэластиками, имеют величины порядка
нескольких долей процента, а коэрцитивные силы не более 104—
105 Н-см~2. Класс ферромагнетиков по определению, данному выше,
включает в себя как ферромагнетики (например, Сг02, 7-Fe203), так
и ферримагнетики (например, магнетит), поскольку оба они обладают
спонтанной намагниченностью. Магнитные домены, такие как в
гематите, можно наблюдать с помощью эффекта Фарадея, а движения
доменных стенок — в приложенном магнитном поле.
Вторичные ферроики. SrTi03 (один из первых ферроэлектриков)
и NaNb03 (антиферроэлектрик) могут рассматриваться как ферро-
биэлектрики (сегнетобиэлектрики). Диэлектрическая анизотропия
в антиферроэлектрических доменах может приводить к высоким
вначениям наведенной поляризации, которая ориентационно
различна в различных доменах. Таким образом, в них происходит
перегруппировка доменов под действием приложенного поля. Кварц —
классический пример ферробиэластичности. Двойниковая
структура а-кварца управляется правилами двойниковой морфологии.
Однако, когда прикладывается внешняя сила, наведенные напряжения
различаются в разных двойниках; как следствие в силовом поле
дифференциал наведенных напряжений приводит к движению
двойниковых стенок. Такое наведенное движение двойников Дофина (180°)
наблюдали в кварце [Thomas, Wooster, 1951; Aizu, 1970]. Оксид
никеля (NiO), хорошо известный антиферромагнитный материал,
также является ферробимагнетиком и ферроэластиком.
Считается, что хлорид аммония — ферроэластоэлектрик. В
низкотемпературной (~ 247 К) упорядоченной фазе типа CsGl, NH4C1
имеет домены, для которых значения dijk (эластоэлектрический или
пьезоэлектрический коэффициент) различны, а когда одновременно
прикладываются и электрическое поле, и механическая сила,
происходит ориентационное переключение.
Ферромагнетоэластичностъ проявляют FeC03 (сидерит) и CoF2.
По-видимому, потенциальных кандидатов в ферробиэлектрики, фер-
Ферроики
359
робимагнетики и ферробиэластики вообще можно ожидать среди
таких материалов, которые обычно являются соответственно анти-
ферроэлектриками, антиферромагнетиками и антиферроэластиками
(с внутренне компенсируемыми деформациями и обладающие
двойниками под углом 180°). Антиферроикные состояния предполагают
большие восприимчивости, которые, как можно ожидать, приводят
к вышеупомянутым наведенным эффектам.
Несобственные ферроики. Несобственные ферроики получаются
в результате явлений кросс-взаимодействия. Типичные примеры
несобственных ферроиков — электроферромагнитный
никель-иод-борацит Ni3B7013I [Ascher et al., 1966] и эластоферромагнитный литий —
аммоний тартрат ISawada et al., 1977]; Cr2Be04 — магнетоферро-
электрик [Newnham et al., 1978]. При охлаждении кубический
борацит подвергается превращению, в котором ионы Ni2+ дают
антиферромагнитное упорядочение. Эта фаза, которая является
антиферромагнетиком — пьезоэлектриком, при дальнейшем охлаждении
превращается в ромбическую фазу (при 64 К), которая полярна и
обнаруживает спонтанную поляризацию. Вследствие эффекта
поляризации происходит разрушение нейтральной спиновой ориентации
и образование слабоферромагнитного состояния. Поскольку
Ni3B7013I — ферромагнитный, из-за своего ферроэлектричества он
считается электроферромагнетиком — несобственным ферроэлект-
риком, параметром порядка которого является электрическая
поляризация. При магнитном переходе тетрагональный (парамагнитный)
Сг2Ве04 превращается в ромбическую антиферромагнитную фазу.
Антиферромагнитное взаимодействие вызывает слабые смещения и
перегруппировки в позициях ионов Сг34" таким образом, что
получаются полярные оси, приводящие к спонтанной поляризации.
Таким образом, Сг2Ве04 — ферроэлектрик, в котором движущая сила
возникает в результате магнитных взаимодействий, и он,
следовательно, относится к магнетоферроэлектрикам.
Релаксорные ферроэлектрики. Эти материалы имеют диффузные
переходы, растянутые по большому температурному интервалу,
которые связаны с большими изменениями диэлектрической
постоянной [Setter, Cross, 1981]. Величины пиков диэлектрических
постоянных уменьшаются с увеличением частоты, пики смещаются к более
высоким температурам, свидетельствуя о признаках
релаксационного механизма. Из оптических и электрических данных понятно, что
за это явление ответственны микродомены, возникающие в
результате флуктуации состава. Практическое значение этих релаксорных
ферроэлектриков объясняется тем, что во многих из них большие
удельные проницаемости связаны с очень низкими коэффициентами
электрострикции — свойство, желательное во многих технических
приложениях, как, например, в манометрах.
Электрострищия, в отличие от обратного пьезоэлектрического
эффекта,— это явление, в котором деформация и электрическое
360
Взаимосвязь структура — свойство
поле, приводящее к деформации, связаны соотношением е^- =
= Mijh Eh2, где Mijk — коэффициенты электрострикции. Известно,
что несколько релаксорных ферроэлектриков обладают низкими
коэффициентами термического расширения; хорошими примерами
таких материалов являются Pb(Mg1/3Nb2/3)03, 0,9Pb(Mg1/3Nb2/3)O3,
0,1РЬТЮ3 и Pb(Zn1/3Nb2/3)03 [Uchino'et al., 1981].
Электрооптические материалы. Зависимость показателя
преломления от электрического поля, или решеточной поляризации,
определяется как электрооптический эффект:
А (1/и2)у = 2 rijthEk + S кгШЕкЕг + .. ., (6.63)
k h,l
Д(1/»а)у = 2/ъ-,Л + %giitkiPhPi+ • • •• (6.64)
h h,l
В уравнении (6.03) первый член справа описывает линейный
электрооптический эффект, или эффект Покеля, а второй — квадратичный
электрооптический эффект, или эффект Керра. В уравнении (6.64)
ftj,k и SijM называются коэффициентами оптической
поляризации. Можно ожидать, что большие значения этих коэффициентов
связаны с большими линейными восприимчивостями, так как г
ос (D — 1)/, и ферроэлектрики являются очевидными кандидатами;
феррсэлектрические электрооптические кристаллы могут быть цент-
ричными, как в LiNb03, LiTa03 и Ba2NaNbr,Or), или ацентричными,
как в KH2P04(KDP), NH4H2P04(ADP) и Cs2H2As04.
Электрооптические композиты имеют несколько преимуществ по сравнению с
мопокристальными материалами, так как их легко производить, они
дешевле и лучше контролируются на ориентацию оптической оси.
Кроме того, в композитах можно изменять эффективное двойное
лучепреломление с помощью ферроэлектрической поляризации и
готовить их специально с помощью соответствующего ионного
замещения. Электрооптическая керамика — почти прозрачный ферроэлект-
рический материал; наиболее известный пример такой керамики —
Pb(Zr1_0rTix)O3 (или PZT). Состав (отношение Zr/Ti) можно
варьировать, а РЬ частично замещать на другие ионы (La, Bi и т. д.). PZT —
керамика, подвергнутая горячему прессованию, обнаруживает
электрически контролируемое рассеяние света (например, PBiZT) и
двойное лучепреломление (например, PLZT и PBiZT).
Композитные ферроики. С композитами из ферроика и другого
материала в настоящее время выполняются оригинальные
эксперименты [Newnham, Cross, 1981; Lynn et al., 1981; Rittenmeyer et al.,
1982; Safari et al., 1982]. Например, в пьезоэлектрически подобном
PZT коэффициент пьезоэлектрического напряжения g можно
определить для данного направления (скажем, z = 33); таким образом,
g33 — d33/(e0k33), где d и к — пьезоэлектрический коэффициент и
диэлектрическая восприимчивость соответственно. Существует
ситуация (например, в гидрофонах), где требуется, чтобы g33 было вы-
Ферроики
361
Рис. 6.49. Десять типов связности для двухфазного твердого тела.
Каждая фаза внутри себя имеет нуль-, одно-, рву- или трехмерную свяекость. Например,
в композите 3-1 затемненная фаза является трехмегнесвязанной, а незатемненная — одно-
мерносвязанной. Стрелками указаны связанные направления. Для типов связности 3-3 и 3-2
даны два варианта, так как на бумаге трурно различить два гзэимолгокикающих каркаса.
Варианты получаются путем поворота против чесоюй cii<j*h на 90° вокруг оси Z.
сокое. Далее, когда прикладывается гидростатическое давление,
уравнение для g видоизменяется: g = d/s0k, a d = d33 -f- 2d31
(k имеет аналогичное выражение); d при гидростатическом давлении
фактически равно 0, поскольку d33 = —2d31. Стратегия ферроик-
ных композитов состоит в создании композита керамика — полимер
таким способом, чтобы исключить d3l и уменьшить к. Уменьшение
к очевидно, поскольку введение полимерной фазы-наполнителя
уменьшает объемную фракцию пьезоэлектрика с высокой
диэлектрической постоянной. Метод, при котором достигается уменьшение (или
исключение) d3l, состоит в следующем [Safari et al., 1982]:
керамика подвергается действию давления только в направлении <33>f
при этом менее сжимаемая фаза (по сравнению с полимером)
поднимает общее давление в направлении (33) выше собственного
значения. В направлениях (11) и (22) давление, обусловленное
непосредственно керамикой, не обнаруживается вообще, поскольку по-
362 Взаимосвязь структура — свойство
лимер поглощает все напряжение. Кроме того, k == fk33, где / —
объемная фракция керамики. Значение g должно быть равно
^зз/(8о/&зз)» оно намного выше, чем в том случае, когда используется
кубическая разновидность пьезоэлектрика.
В вышеуказанном примере PZT соединяется внутри себя в одном
направлении, тогда как полимерная фаза соединяется между собой
во всех трех измерениях (связность 1—3). В двухфазных
композитах керамика — полимер можно наблюдать другие типы связностей
(2—3 или 3—3), как это видно из рис. 6.49. В двухфазном композите
со связностью 2—3 мы в принципе можем использовать два
различных свойства керамики в двух различных направлениях или одно
и то же свойство — дополнительным способом. Композиты 3—3
получаются так называемым методом выплавления воска.
Естественная модель связностей (3—3)— коралл. Кораллы сначала
заполняются расплавленным воском, а затем карбонатная структура
(коралл) вымывается кислотой. Затем вместо кораллового карбоната в
образовавшиеся пустоты вводят PZT. При отжиге выгорает воск,
а отожженный продукт заполняется полимером.
6.10. ЖИДКИЕ КРИСТАЛЛЫ
В разд. 4.13 мы кратко рассмотрели природу и структуру
жидких кристаллов. В последнее десятилетие мы стали свидетелями
большой волны интереса к жидким кристаллам из-за их применения
в дисплейных устройствах (устройства, которые превращают
электрический сигнал в визуальную информацию). Создание устройств
на жидких кристаллах основано на связи между молекулярной
структурой и фазовым поведением (относительно склонности к смек-
тическому — нематическому состоянию, T^j и т. д.), а также
физическими свойствами жидких кристаллов.
Важно понять взаимосвязь между молекулярной структурой
и типом проявляемого жидкокристаллического состояния, особенно
для того, чтобы объяснить склонность к смектическому и
нематическому состояниям жидкокристаллических систем [Gray, 1983]. Не
простой вопрос, будут молекулы упаковываться своими длинными осями
параллельно или так, что их концы будут располагаться в
плоскости (смектическое состояние). Имеются данные, показывающие, что
вообще удлинение алкильной цепи терминального га-алкил- или
n-алкоксизаместителя в гомологических рядах мезогенов (молекул,
способных к образованию жидких кристаллов) способствует
смектическому поведению из-за взаимной близости соседних алкильных
цепочек. Например, в ряду
сн3(сн2)по -hQ>~Q>-co2h
Жидкие кристаллы
363
нет смектической фазы вплоть до п = 4. Для п^Ъ появляется смек-
тическая фаза (Sc), область устойчивости которой увеличивается
с увеличением п. Вопрос предсказания типа смектической фазы,
которую может принимать мезоген, наиболее трудный. Это, по
существу, равносильно выяснению деталей кристаллической
структуры на основе молекулярной структуры. Имеются некоторые
корреляции между молекулярной структурой и тенденцией к образованию
смектической фазы (скрученная разупорядоченная слоистая
структура). Терминальные алкоксигруппы в ароматических мезогенах
благоприятствуют образованию смектических фаз Sc, тогда как их
алкильные аналоги часто приводят к фазам SA, в которых молекулы
не скручены. Тенденция молекул к повороту относительно плоскости
слоя и, следовательно, к предпочтительному образованию
смектической фазы S с приписывается существованию терминальных боковых
диполей, связанных с алкоксизаместителями.
Более точные структурные корреляции установлены в немати-
чески-холестерических системах. Мы приводим их в таком
суммарном виде согласно [Gray, 1983].
1) Добавление колец или звеньев с кратной связью, увеличивающее
жесткий остов молекулы, повышает температуру перехода нема-
тический кристалл — изотропная жидкость Гш.
/Р^\ /^\ 317М32С/С
С6Н13—\Q/— CO-0-^Q)— CN-,C— N—I
3S4K 502%6K
C—^N—-I
2) В гомологических рядах, содержащих гс-алкильные или гс-алко-
ксильные терминальные цепочки, Т^\ уменьшается с
увеличением длины цепочки; кроме того, регулярное чередование Гш для
фаз с нечетным и четным числом га-ал кил ьных групп, отмеченное
в начале ряда, с увеличением длины алкильной цепи имеет
тенденцию к исчезновению.
3) Влияние терминальной группы на Гш следует в ряду:
Ph > NHCOCHg > CN > OGH3 > N02 > CI > Br > N(GH8)a >
> CH3 > J > H.
4) Влияние на TNI связующих групп в остовной структуре можно
показать в общем виде
364
Взаимосвязь структура — свойство
7TNi имеет максимальное значение, если X — бензольное кольцо
(фениленовая группа), а минимальное — если X представляет
собой прямую связь между двумя бензольными кольцами; Tni
для других связующих групп находится между этими крайними
значениями.
5) Замещение в боковых положениях остовной структуры обычно
уменьшает Тт- Например, в ряду карбоксильных кислот
R0-4O>-<O>-c^H
TNi изменяется с X в следующем ряду: Н > F > СН3 « С1 >
> Вг > I « N02. Видно, что изменение TNi коррелирует с
размером X безотносительно к постоянному диполю С — X.
6) Разветвление цепи имеет огромный подавляющий эффект на T^i
особенно в положении 1; эффект уменьшается, если заместитель
движется к свободному концу цепочки, как показано в
следующем ряду:
NC -ЧП>— CH-N-ЧО)—CH=CH-C0-0R
R TNI(K)
-CH2CH2CH2CH2CH3 409,5
—CHCH2CH2CH2CH3 <293
СН3
-СН2СНСН2СН2СН3 385
СН3
-СН2СН2СНСН2СН3 376
сн3
-СН2СН2СН2СНСН3 392,5
СН3
Корреляции между молекулярной структурой и жикдокристал-
лическим состоянием успешно использованы для получения 4-ал-
кил- и ^-алкокси-^'-цианобифенилов, которые являются основой
для электрооптических дисплейных устройств. Доводы в пользу
создания этих нематогенов следующие. В семействе нематогенов с
общей формулой
-о-»-о-'
для максимальной стабильности требуется, чтобы два кольца
соединялись непосредственно (А — простая связь), но вследствие этого
понижается T^i. Этому можно противодействовать, используя для
Y группу CN, которая высоко расположена в ряду нематическои
Жидкие кристалла
365
эффективности. Использование алкильной или алкоксильной групп
для X способствует нематическому упорядочению и сохраняет
низкой температуру плавления. Соединения, составленные таким
образом, являются хорошими нематогенами, дающими устойчивые
бесцветные нематические фазы при комнатной температуре; типичный
пример —
295к 308к
С —N —1
Бензольное кольцо в нематогенах можно замещать на такие али-
циклические кольца, как циклогексан, бицикло(2.2.2)октан и кубан.
Это привело к открытию циклогексановых и бициклооктановых
аналогов цианобифенилов. Замещение бензольного кольца на
циклогексан или бициклооктан дает эффект увеличения T^i'.
С5Н„—ф—@—CN
_^ TWI(K)
Бензол зов
Циклоеексан 328
Бициклооктан 373
Наоборот замещение бензольного, циклогексанового или бицикло-
октанового кольца на кубановое дает заметный эффект подавления
Tni. Этот эффект объясняется в терминах гибкости кольца и
коллинеарности 1,4-связей. Бициклооктановое кольцо, будучи более
гибким, принимает участие в эффективной упаковке молекул,
образуя устойчивое нематическое состояние. Высокая потенциальная
энергия деформации кубанового кольца (650,8 кДж-моль"1) делает
его жестким и наименее подвижным и, следовательно, менее всего
благоприятствует нематическому состоянию.
Что касается технических целей, то используемые молекулы
должны быть устойчивыми к нагреванию и освещению.
Жидкокристаллические фазы должны быть устойчивыми в широком интервале
температур, близких к комнатной. Этого можно достичь, используя
смеси мезогенов. Наиболее успешной оказалась смесь
цианобифенилов, одного — с алкильпым заместителем (СбНп), а другого — с ал-
коксильным (С8Н170).
Физические свойства жидких кристаллов, как правило,
анизотропны (см., например, [de Jeu, 1980]). Анизотропные свойства,
которые имеют отношение к дисплейным устройствам,— это
показатель преломления, диэлектрическая проницаемость и ориентации
онная упругость [Raynes, 1983]. Нематический жидкий кристалл
имеет два главных показателя преломления, щ\ и тг±, измеряемые
соответственно параллельно и перпендикулярно оси нематического
366 Моделирование твердых тел для особых целей
кристалла. Двойное лучепреломление, An = щ — пх,
положительно, обычно около 0,25. Анизотропия диэлектрической
проницаемости, которая определяется как Де = 8ц — ех, является движущей
силой для большинства электрооптических эффектов в жидких
кристаллах. Электрический вклад в свободную энергию содержит член,
который зависит от угла между направлением п и электрическим
полем Е, и задается уравнением
^Е^убоДе^.Е)2. (6.65)
В отсутствие других помех свободная энергия минимизируется
вращением оси. Здесь существует две возможности: когда ец > ех
(положительная анизотропия), ось ориентируется параллельно
полю, а когда ех> 8ц ( отрицательная анизотропия), ориентация оси —
перпендикулярна полю.
Что касается нематического жидкого кристалла, здесь
предпочтительной ориентацией является та, в которой ось повсюду
параллельна. Другие ориентации имеют распределение свободной
энергии, которое зависит от констант упругости, Kit. Константы ориента-
ционной упругости К1г, К22 и К33 определяют соответственно
деформации скоса, скручивания и изгиба. Значения констант упругости в
жидких кристаллах ~ 10"11 Н, так что разница в свободной
энергии между различными ориентациями порядка 5-Ю"4 Дж-м"2, т. е.
по величине такая же, как поверхностная энергия. Следовательно,
тонкий слой жидкого кристалла, расположенного в виде сэндвича
между двумя поверхностями, принимает ориентацию, задаваемую
поверхностями. Этот факт лежит в основе электрооптических
эффектов в жидких кристаллах. Дисплейные устройства на жидких
кристаллах обсуждаются в гл. 7.
7. МОДЕЛИРОВАНИЕ ТВЕРДЫХ ТЕЛ
ДЛЯ ОСОБЫХ ЦЕЛЕЙ:
АСПЕКТЫ ДИЗАЙНА МАТЕРИАЛОВ
7.1. ВВЕДЕНИЕ
В предыдущей главе мы обсуждали некоторые интересные
и полезные свойства твердых тел. Необходимо отметить, что
разработка материалов с желаемыми свойствами так же представляет
собой неотъемлемую часть современной химии твердого тела. Чтобы
отдать должное какому-либо одному классу материалов, или како-
Введение
367
му-либо одному типу их применения, нам следовало бы широко
ваняться вопросами их создания, в том числе и технологическими
деталями. Поскольку это выходит за рамки монографии, мы кратко
рассмотрим несколько отдельных случаев использования
материалов.
Сегодня в технологии большое место занимает получение
керамик с высокими эксплуатационными свойствами [Bowen, 1980; Katz,
1980; Sanders, 1984]. Термин «керамика» сам по себе относится к
неорганическому, неметаллическому материалу, который
обрабатывается или затвердевает при высоких температурах. Некоторые
керамические материалы (например, кубический BN, SiC, Si3N4) —
твердые, устойчивые к окислению и легкие; они часто получаются
из доступного сырья и менее дорогостоящие, чем металлы.
Недостаток керамик в том, что они, как правило, хрупкие и их трудно
получать в виде больших сложных форм. Однородные субмикронные
порошки, требуемые для изготовления высокотехнологичных
керамик, получаются посредством золь — гель-процесса или из газовой
фазы. Нити, сделанные из Si3N4, SiC или углерода, становятся
жесткими при осаждении на них металлов или кремния, а литий-
алюмосиликатные стеклообразные керамики упрочняются с
помощью нитей SiC. Керамические материалы используются в
тепловых двигателях (например, Si3N4), в качестве формовочных
материалов, термисторов, подложек для электронной аппаратуры
(например, силикат бария), в магнитной аппаратуре и в других
приложениях. Материалы, используемые в термисторах, могут иметь как
отрицательные (NiFe204 и MgFe204 — для контроля и измерения
температуры), так и положительные температурные коэффициенты
сопротивления (MgAl204 и Zn2Ti04 — для нагревательных
элементов и переключателей); твердые ферриты (BaFe12019) и мягкие
ферриты (MnFe2Oj) находят широкое применение в устройствах
памяти, в магнитных лентах и в сердечниках трансформаторов.
Многие керамики являются проводниками ионов и находят
применение в батареях. Интересно, что легкие покрытия на космических
кораблях сделаны из композитных керамик (волокна из Si02 и
алюмоборосиликатные). Преобладающая часть материалов,
которыми занимаются химики, работающие в области твердого тела, это
керамики, и здесь важно, что большее внимание уделяется
керамикам с высокими эксплуатационными качествами. Эту главу мы
посвящаем обсуждению некоторых керамических материалов.
Другие важные аспекты, которые мы рассмотрим, это —
оптические материалы, органические материалы, полимеры,
фотоэлектрохимические материалы и материалы для хранения водорода.
Важное значение приобретают органические материалы, в связи с
развивающейся областью молекулярной электроники.
368 Моделирование твердых тел для особых целей
7.2. ТВЕРДЫЕ ИОННЫЕ ПРОВОДНИКИ
Существует большое (и возрастающее) семейство ионных
твердых тел, в которых определенные ионы проявляют необычайно
высокую подвижность. Эти материалы стали известны как твердые
ионные проводники (ТИП). В некоторых случаях быстрый ионный
транспорт сопровождается также заметной электронной
проводимостью. В науке и технологии твердые ионные проводники
представляют огромный интерес с точки зрения их потенциального
использования в качестве электродов или электролитных материалов в
устройствах электрохимического превращения энергии [Hagenmuller,
van Gool, 1978; Chandra, 1981; Goodenough, 1984].
Чтобы твердое вещество проявляло проводимость твердых
ионных проводников, оно должно удовлетворять следующим критериям:
иметь высокую концентрацию носителей заряда, высокую
концентрацию вакансий или междоузельных позиций и низкую энергию
активации для прыжка ионов. При этом существенным условием
является наличие набора энергетически эквивалентных позиций,
частично занятых подвижными ионами и удовлетворяющих условию
с(1 — с) Ф О, где с — доля занятых позиций. Чтобы предсказать
структуру, необходимую для удовлетворения первых двух
критериев, можно прибегнуть к кристаллохимическим данным, но
предсказать энергию активации не так легко. Проводимость, по
существу, определяется выражением
а = {yNqVkT)c{l - c)l2v0exp{-AHJkT), (7.1)
где A#m — энтальпия миграции подвижного иона, N — плотность
энергетически эквивалентных состояний; I — длина прыжка, с —
концентрация занятых позиций, v0 — частота прыжка; у --=
= -тг zexp(ASm/A;), где z —число ближайших соседних позиций.
Проводимость твердых ионных проводников не является
открытием последних лет; еще в 1914 г. ее наблюдали в некоторых
соединениях серебра [Tubandt, Lorenz, 1914]. Авторы, например,
нашли, что удельная проводимость Agl почти перед точкой плавления
фактически примерно на 20 % больше удельной проводимости
расплавленного AgT. Твердые вещества, в которых установлена ионная
проводимость ТИП, представлены как несколькими стехиометри-
ческими [Agl, RbAg4I5, AgSl], так и нестехиометрическими
соединениями, такими как fi-оксид алюминия Na1+a.Aln017+3C/2,
стабилизированный кальцием диоксид циркония 2т1_хСах02^х и Li^TiSg,
причем последнее соединение обладает как ионной, так и
электронной проводимостью. Ионная проводимость ТИП обнаружена в ряде
стекол, содержащих Li+ или Ag* [Rao, 1981].
Проводимость по типу твердых ионных проводников может
наблюдаться в одном, двух или трех измерениях в зависимости
Твердые ионные проводники
369
от кристаллической структуры фазы. Одномерная ионная
проводимость найдена, например, в LiAlSi04 ф-эвкриптит), который имеет
гексагональную структуру с ионами Li+ в каналах, параллельных
оси с. Двумерная проводимость наблюдается в соединениях,
кристаллизующихся в слоистых структурах; р-оксиды алюминия и LixTiS2—
типичные примеры таких соединений. Трехмерная проводимость
обнаружена в Li3N, а также в сложных структурах, состоящих из
цепочек полиэдров; представителем таких соединений является
Na3Zr2PSi20]2 (NASICON, натриевый суперионный проводник).
Известны также и литиевые аналоги (LISTCON), которые являются
твердыми растворами Li2ZnGe04 и Li4Ge04 [Bruce, West, 1982;
Mazumdar et al., 1984]. На рис. 7.1 показана удельная проводимость
ряда твердых ионных проводников как в кристаллическом, так
и в стеклообразном состоянии.
Подрешетка проводящего иона в твердых ионных проводниках
обычно считается «расплавленной». Модель расплавленной подре-
шетки для ионной проводимости в твердом теле впервые предложена
в 1936 г. на основании структурных и термодинамических данных
по Agl [Strock, 1936]. В большинстве твердых ионных проводников
энтропия фазового перехода в проводящее состояние (ТИП) больше
энтропии плавления. Например, в Agl энтропия перехода |$- в
ос-форму (ТИП) при 420 К составляет 14,7 Дж-град-1-моль""1,
тогда как энтропия плавления Agl при 861 К составляет лишь
11 Дж-град"1-моль-1.
Структурные исследования твердых тел, обладающих ионной
проводимостью (ТИП), показывают, что проводящие ионы
распределяются статистически по большому числу мест и имеют большие
амплитуды колебания. В a-Agl два иона серебра на
элементарную ячейку распределяются в значительной степени среди 12й-тетра-
эдрических позиций (пр. группа Im3m) (рис. 7.2), при этом
энергия активации для миграции ионов Ag+ едва ли составляет 0,05 эВ.
Карты электронной плотности показывают, что небольшое
количество электронной плотности приходится также на 24/г-позиции,
которые связывают мостиками позиции \2d. Характер электропереноса
в твердых ионных проводниках можно интерпретировать в модели
точечных дефектов, как для обычных ионных проводников, но
ионная проводимость нескольких твердых ионных проводников имеет
отрицательное отклонение от закона Аррениуса при высоких
температурах. Высказано предположение, что данные по проводимости
твердых ионных проводников могут быть описаны уравнением типа
a = ЪТ — а, применимым к жидкостям fO'Keeffe, 1973].
Что касается проводимости ионов Na+ и других ионов в твердых
ионных проводниках, то она установлена среди оксидов с
каркасными (скелетными) структурами [Goodenough et al., 1976]. В этих
структурах оксидов-хозяев одно-, дву- и трехмерное пространства
взаимосвязаны большим числом узких каналов. В то время как
24 Ч. ТГ. Р. Рио, Дж. Гопал:\1ф1П11н:ш
370 Моделирование твердых тел для особых целей
Температура, °С
900 ТОО 500 400 JOO 200 ЮО
' ' ' «—I 1 1 ■ ' '—
25
L_
I <^дШ£2эВ)
Кристаллический проводник
Аморфный проводник
К К-
<-тв 1Т„
У ^^М*(О.043В)(Тв1°'4"""'На")
Рис. 7.1. Ионная проводимость кристаллических и аморфных твердых ионных
проводников [Tuller et al., 1980]. (SG — суперионный проводник.)
Твердые ионные проводники 371
Рис. 7.2. Структура a-Agl с междоузельными катионными позициями: большие
заштрихованные круги — иодид-ионы; квадраты — октаэдрические бЬ-позиции;
сплошные кружки — тетраэдрические 12й-позиции; открытые кружки — 24/&-
позиции.
структуры вольфрамовой бронзы и Р-оксида алюминия содержат
одно- и двумерные междоузельные пространства, гексагональный
каркас NaZr2(P04)3 имеет трехмерное междоузельное пространство
(рис. 7.3), где вдоль оси с располагаются (в порядке Z — р — Z —
М — Z) вакантные тригонально-призматические места р,
октаэдрические места Zr4+, Z, и октаэдрические места для ионов Na+, M.
Для каждого заполненного места М имеется три пустых места М0,
образующих ГПУ-слои, перпендикулярные оси с. В NaZr2(P04)3
положения М заполнены, а М0 — пустые; в Na4Zr2(Si04)3, у
которого каркас такой же, как у NaZr2(P04)3, оба положения, М и М0,
заполнены. Обе фазы являются плохими проводниками ионов Na+,
но твердые растворы Na1+:cZr2P3_:!CSix012 относятся к проводникам
с энергией активации ~ 0,2 эВ для х ж 0,2 (NASICON); изучены
пути диффузии и одночастичные потенциалы иона Na+ в NASICON'e
[Kohler, Schulg, 1985]. Найдено, что твердые ионные проводники
типа LISICON имеют отрицательные объемы активации при
высоких давлениях [Bose et al., 1984].
Значительное внимание привлекла к себе натриевая форма р-
оксида алюминия, после того как было обнаружено, что ее можно
использовать в качестве твердого электролита в Na/S-батареях
[Weber, Kummer, 1967]. Новизна этого материала определяется
тем, что, хотя его температура плавления равна ~ 2200 К,
некоторое количество тока он может пропускать уже при 570 К. Ключ
к быстрому транспорту Na+ в этом твердом теле лежит в его
кристаллической структуре (рис. 7.4) и в пути транспорта с низкой
потенциальной энергией, который эта структура создает для миграции
Na+. Вообще р-оксиды алюминия, М1+зсА1п017+:х:/2 (М — Na+, K+,
24*
372 Моделирование твердых тел для особых целей
Зеркальная
плоскость
Зеркальная
плоскость
Рис. 7.4. Структура натриевого Р-оксида
алюминия. Показана только полопина
элементарной ячейки. Большие
заштрихованные кружки — натрий, открытые
кружки — кислород, малые сплошные
кружки — алюминий.
Ag+ и т. д.) являются слоистыми не-
стехиометрическими соединениями.
Для натриевой формы р-оксида
алюминия х = 0,20—0,30;
гексагональная структура этого твердого
вещества состоит из двух плотноупако-
ванных шпинелеподобных блоков толщиной в четыре атома
кислорода вдоль оси с; два блока разделяются зеркальной
плоскостью, в которой плотность атомов кислорода составляет 1/4 от
значения плотности в шпинельных блоках. Зеркальные плоскости
содержат подвижные ионы натрия; транспорт Na+ в этой структуре,
по существу, является двумерным. Нестехиометричность в этом
веществе возникает из возможности размещать в проводящих
плоскостях различные варьируемые количества ионов кислорода и натрия.
Измерена [Whittingham, Huggins, 1971] ионная проводимость
натриевой формы р-оксида алюминия, ее значение сопоставлено с
полученным ранее коэффициентом диффузии [Yao, Kumraer, 1967]:
DfcM^c-1) = 2,4.10-4ехр(-0Д65/А:Г) и аТ^К-Ом^-см"-1) = 2,37-
•К)3 ехр(—0,164//с7т). Отношение Хэвена (см. разд. 6.3.2) составляет
0.610, что находится в хорошем соответствии со значением 0,600,
рассчитанным в предположении междоузельного механизма для
миграции натрия, р"-Оксид алюминия, представляющий собой
стабилизированную магнием фазу Na1+acMg3CA]11_0CO17, является лучшим
ионным проводником, чем р-оксид алюминия [Farrington, Briant,
1979]. Проводимость и энергия активации этих оксидов алюминия
изменяется со стехиометрией и расположением дефектов; дефектные
структуры Р-оксидов алюминия исследованы методом электронной
микроскопии [Ganapathi et al., 1985].
Дифториды со структурой флюорита, такие как PbF2, обладают
ионной проводимостью ТИП, обусловленной легким транспортом
иона F" (см. разд. 6.4.3). К числу интересных соединений с ионной
проводимостью Li+ относится Li3N [Rabenau, 1978], причем
проводимость Li+ в нем имеет двумерный характер. По-видимому, за про-
димость в этом нитриде отвечают кооперативные базальные
возбуждения, включающие поворот шести ионов Li+ на 30° около общего
иона N3" по направлению к граневым позициям (позиции
посредине между ионами N3~ в слое Li2N). Во флюоритной структуре, по-
видимому, происходит поворот на 45° отдельного куба из ионов F";
сплав Цинтла LiAl также является проводником иона 1л+.
Значительным открытием недавних лет является открытие —
стекол с ионной проводимостью ТИП [Tuller et al., 1980]. Agl яв-
Твердые ионные проводники
373
ляется компонентом многих стеклообразных твердых ионных
проводников, содержащих Ag+; литиевые стекла с ионной
проводимостью ТИП, как правило, образугот сетчатые структуры. Примеры
стеклообразных твердых ионных проводников следующие: Li20 —
В203 (42,5-57,5), Li20 - SiO, (33.3-66,7), Agl - Ag2Mo04 (75-
25) и ZrF4 — BaF2 — ThF4 (58,7—31,3—10); в скобках указаны
составы. Кроме легкости изготовления стеклообразные ионные
проводники обладают преимуществами варьирования свойств через
контроль состава. Механизм ионной проводимости в стеклообразных
твердых ионных проводниках более сложен, чем в кристаллических.
До сих пор мы рассматривали модель твердых ионных
проводников, которые используются главным образом в качестве твердых
электролитов; другой класс соединений, применяющихся в качестве
электродных материалов в твердотельных батареях, представляет
собой смешанные электронно-ионные проводники (с высокой ионной
проводимостью). Проводимость здесь возникает в результате
обратимого электрохимического внедрения проводящих частиц. Для
успешного применения такого материала в высокоэнергетических
батареях нужно, чтобы степень внедрения была большой, а материал
должен претерпевать многократные циклы внедрение — удаление.
Исследован ряд систем с оксидами и сульфидами переходных
металлов в качестве твердых электродов [Murphy, Christian, 1979].
Твердые ионные проводники как электюохимические сенсоры,
электролиты и электроды с успехом используются в батареях, а
также в твердотельных дисплеях [Farrington, Briant, 1979; Ingram,
Vincent, 19841. Если твердый ионный проводник, содержащий
подвижные ионы М, разделяет два состава с различной активностью М,
то в нем устанавливается потенциал, что можно связать с различием
в химической активности М. Если задать одну активность, можно
определить неизвестную вторую. Этот принцип лежит в основе ряда
ион-селективных электродов; LaF3, допированный 5 % EuF2,
используется для контроля концентрации фторид-иона в питьевой
воде. Аналогично двуокись циркония, стабилизированная кальцием,
используется в ячейках типа Pt(TB), 02(r)]Zr02 — СаО|02(г), Pt(TB)
(срави.) для контроля концентрации кислорода.
Большинство успешных приложений твердых ионных
проводников — в твердотельных батареях. Можно различить два вида
батарей: 1) маленькие первичные ячейки, в которых главным
является время жизни и отсутствие саморазряда, и 2) перезаряжаемые
вторичные батареи, в которых главным критерием является
высокая плотность энергии. Батарея первого типа, которая
используется как регулятор сердечной деятельности, основана на литий-
иодидном твердом электролите. Здесь требованием является низкая
мощность и непрерывное функционирование в течение длительного
времени (~ 15 лет). Типичная ячейка в этом случае состоит из
литиевого анода, литий-иодидного электролита и комплекса поли-2-ви-
374 Моделирование твердых тел для особых целей
нилпиридин-12 в качестве катода. Батареи с высокой плотностью
энергии важны с точки зрения альтернативных источников энергии.
Важной вехой в развитии батарей с высокой плотностью энергии
является Na/S-батарея, в которой в качестве твердого электролита
используется р-оксид алюминия, а в качестве электродов — натрий
и сера [Tofield et al., 1978]. Напряжение в ячейке (2,08 В)
устанавливается в результате химической реакции между натрием и серой
с образованием полисульфида натрия. Теоретическая плотность
энергии Na/S-батареи довольно высокая (~ 750 Вт-ч-кг-1) по сравнению
с плотностью энергии 170 Вт-ч-кг-1) — для обычной кислотно-
свинцовой батареи. Основной недостаток этих батарей состоит в
использовании расплавленных натрия и серы, которые на воздухе
и во влажных условиях опасны. Что касается тока, то здесь интерес
представляют батареи литий — сульфид металла, которые дают
возможность использовать органические электролиты, а в качестве
электродов — твердые растворы (которые являются смешанными
электронно-ионными проводниками). Получили развитие работы в
области литий-титановых батарей (Li/TiS2), использующих
органический электролит для переноса лития; батарея имеет высокую
плотность энергии (480 Вт-ч-кг-1) и дополнительное преимущество
работы в условиях комнатной температуры [Whittingham, 19761.
В качестве электродов в батареях исследован широкий ряд
смешанных проводников, включая фторированный графит, р-фер-
риты щелочных металлов (изоструктурные с р-оксидами алюминия),
нестехиометрические сульфиды серебра, вольфрамовые и ванадиевые
бропзы и такие сплавы, как Li^Al и Li^Si. Кристаллографически
выстроенный Ni/Zr02 — композит, полученный восстановлением
быстро отвердевшей эвтектики NiO/Zr02 [Revcolevschi, Dhalenne,
1985], может найти применение как электродный материал.
Развиваются также работы по полностью полимерно-твердотельным
батареям, основанным на использовании системы с допированным
полиацетиленом; (CHNa^l иодид натрия — оксид полиэтилена |(GHIz)a..
Одно из самых ранних применений твердых ионных
проводников — использование стабилизированного диоксида циркония в
качестве электролита в высокотемпературных топливных элементах.
Топливный элемент представляет собой электрохимическое
устройство, которое превращает химическую энергию реакции сгорания
топлива в электрическую энергию: Н2(г) + 1/202(г) —►- Н20(г).
Газообразное топливо (например, Н2 или СО) циркулирует по
внутренней стенке электролитной трубки, а кислород или воздух —
по внешней. Температура работы элемента ~ 1200 К. Во время
генерации энергии кислород принимает электроны на воздушном
тэектроде, образуя ионы О2", которые проходят по электролитной
рлубке и реагируют на топливном электроде с Н2 или СО, образуя
Н20 или С02. Электроны, получающиеся в окислительном процессе»
проходят по внешней цепи назад к катоду.
Фотоэлектрохимия
375
Комбинация топливного элемента, где сжигается топливо, и
водной электролизной ячейки, где получается водород,— новый
вариант чистого энергетического цикла. Главное ограничение
топливного элемента — высокая рабочая температура (> 1000 К). Ключ
к решению проблемы в целом лежит в нахождении подходящего
электролитного материала, который может функционировать при
довольно низких температурах (400—600 К). Поскольку почти
невозможно найти низкотемпературный оксидный ионный проводник,
предпринимаются попытки в направлении развития твердых
протонных проводников; сообщается, что HNb03 [Sen, Sen, 1983] и
уранилфосфат [Skou et al., 1983] являются хорошими протонными
проводниками.
Метанол — удобное жидкое топливо, которое можно или
смешивать с бензином, или непосредственно сжигать в двигателе.
Утилизацию метанола можно рассматривать для автомобильного
транспорта, она должна быть дешевой, надежной; поэтому развиваются
работы по долгоживущим топливным элементам метанол — воздух.
Прежде чем реализовать производство элементов, необходимо
разрешить две принципиальные проблемы, связанные с материалами:
дешевый протонный электролит, устойчивый при ~ 570 К и
каталитический анод для превращения метанола и воды по реакции
СН3ОН + Н20 -* С02 + бе" + 6Н+.
Проекты топливного элемента должны составляться с учетом
решения этих проблем.
Твердые ионные проводники используются для развития элект-
рохромных дисплеев. W03 — бледно-желтый порошок, который
становится темно-голубым при поглощении небольшого количества Na
с образованием NaxW03. Поскольку обмен идет обратимо и очень
быстро, вещество используется в дисплеях.
7.3. ФОТОЭЛЕКТРОХИМИЯ
Поскольку эпоха недорогих природных топлив кончается,
заметно усиливается интерес к созданию альтернативных
источников энергии. Среди возрожденных источников энергии солнечная
энергия не только наиболее распространенный, но и чистый
источник. Ее превращению в полезные формы энергии, однако, мешают
такие проблемы, как низкая плотность потока и прерывистость
освещения. Даже наиболее эффективные устройства по
преобразованию солнечной энергии (а именно фотогальванические кремниевые
солнечные элементы, эффективность ~ 15 %) совершенно не
экономичны для широкомасштабного использования из-за стоимости
производства и сопутствующей стоимости инкапсуляции, стоимости
монтажа и стоимости земли. В связи с этим большое внимание
привлекает к себе превращение солнечной энергии в химическую (т. е. На
376 Моделирование твердых тел для особых целей
из воды) или электрическую с использованием полупроводниковых
электродов в фотоэлектрохимической ячейке. Этот вопрос
превосходно рассмотрен в недавних публикациях [Nozik, 1978, 1980;
Bard, 1980; Heller, 1981; Aruchamy et al., 1982; Kutal, 1983;
Wrighton, 1983].
Фотоэлектрохимический эффект включает в себя получение
напряжения и электрического тока при падении света на
полупроводниковый электрод, погруженный в раствор электролита и
соединенный с электродом сравнения [Becquerel, 1839]. Работа с германиевыми
электродами показала, что эффект Беккереля обусловлен
образованием системы полупроводник — электролит [Brattain, Garret,
1955]. Идея использования освещенного полупроводникового
электрода в электролитической ячейке с целью получения химических
продуктов или электрической энергии возникла из этой работы,
но возможность такого использования не была продемонстрирована
до тех пор, пока при работе с фотоанодом из тг-типа Ti02 не показали,
что окисления воды до кислорода можно достичь при значительно
более отрицательном потенциале, чем стандартный редокс-гютен-
циал пары Н20/02 [Fujishima, Honda, 1972].
Для понимания фотоэлектрохимии с применением
полупроводниковых электродов необходимо знание энергетики систем
полупроводник — электролит. Когда полупроводник погружается в
электролит, равновесие уровней Ферми (химических потенциалов)
устанавливается путем переноса большинства носителей заряда. Это дает
пространственно-заряженный слой (так называемый истощенный
слой) в полупроводнике. В результате зона проводимости и
валентная зона искажаются так, что устанавливается потенциальный
барьер, который предотвращает дальнейший перенос электронов
(рис. 7.5). В электролите также существует заряженный слой
(слой Гелъмголъца), он состоит из заряженных ионов электролита,
адсорбированного на поверхности твердого электрода.
Когда система полупроводник — электролит подвергается
действию света, фотоны с энергией, большей, чем энергия щели (Eg)y
поглощаются в истощенном слое, образуя пары электрон — дырка,
которые разделяются под действием электрического поля
пространственно-заряженной области. Индуцированный светом процесс и
последующее разделение пар электрон — дырка стремятся вернуть
ферми-уровень в первоначальное состояние, до того, как была
образована система полупроводник — электролит. Для того чтобы
пошла редокс-реакция, в электролит вводится минимальное
количество фотогенерируемых носителей. Для полупроводников тг-типа
в электролит вводятся дырки, которые могут привести к анодному
окислению, а для полупроводников р-типа в электролит
вводятся электроны для того, чтобы пошла реакция катодного
восстановления. В обоих случаях большинство фотогене-.
Фотоэлектрохимия
377
Го Зона ,
LD проводимостц\
Е. ^
'ЛВалет
'ЛВалентная<А
яоиа 'Л
#.
зона 'Л
V/////////A
Электрод
(полупроводник
п-типа)
О
-Е* (Редоке -
уровень)
Раствор
Поле
Истсщённь/й
Слой Гелынгольца
Электрод
(полу про -
водник п-типа)
б
Раствор
Рис. 7.5. Схематическое изображение электролитного соединения
полупроводник гс-типа — электролитный раствор, показывающее образование истощенного
слоя, изгиба зоны и гельмгольцевского слоя: перед погружением (а) и после
погружения (б) в раствор.
рируемых носителей уходят с полупроводника через омический
контакт и вводятся на электроде сравнения для того, чтобы
пошла реакция окисления — восстановления, обратная реакции
на полупроводниковом фотоэлектроде.
Следует различать два типа проводников полупроводник —
электролит в зависимости от природы
окислительно-восстановительной реакции. Если реакция на аноде отличается от реакции на
катоде, в электролите происходит чисто химическое изменение и ячейка
называется фотоэлектролизной; здесь падающая оптическая
энергия превращается в свободную энергию химической реакции в
ячейке. Если окисление на аноде просто обращается на катоде, то в
электролите вообще нет химического изменения. В этом случае
ячейка ведет себя как электрохимический фото гальванический эле-
мент, где падающая оптическая энергия превращается в
электрическую [Gerischer, 1975, 1979].
Фотоэлектролизные ячейки. Фотоэлектролизную ячейку можно
сделать двумя способами. В первом способе используются
полупроводниковый электрод и металлический электрод сравнения
(ячейка Шогптки); полупроводник может быть п-или jo-типа. В
другом варианте оба электрода являются полупроводниками, один —
п-, другой — jo-типа. Фотоэлектролиз воды с использованием
полупроводникового анода >?-типа (SrTi03) показан на рис. 7.6. Ячейка
может работать для получения Ы2 и 02 из Ы20 без дополнительного
ввода энергии, кроме световой энергии, с целью возбуждения
электронов в SrTi03. К сожалению, поскольку энергетическая щель
большая (~ И,2 эВ), в этой ячейке малая часть (< 5 %) спектра
солнечной энергии может быть превращена в химическую энергию
(лишь эта часть спектра солнечной энергии больше энергии щели).
378 Моделирование твердых тел для особых целей
Нагрузка
R/0 0/R^
n-5rTL0.
Водный электролипА Pt Катод
F Полупроводник
п-типа Электролит Металл
Рис. 7.7. Схематическое
изображение фотогальванической
ячейки с жидкостным
соединением и с использованием
полупроводника га-типа. R/0 —
редокс-пара в электролите.
Рис. 7.6. Фотоэлектролив воды
с использованием SrTi03 в
качестве анода [Wrighton, 1983].
Для эффективного превращения солнечной энергии необходимо,
чтобы щели в полупроводниковых электродах были бы порядка 1,3=Ь
±0,3 эВ.
Фотоэлектролизные ячейки р — га-типа можно сделать,
используя тот же самый полупроводниковый материал, один — допиро-
ванный, по р-типу, другой — по га-типу или с двумя^ различными
(га- и р-) полупроводниками. Фотоэлектролизная ячейка, которая
сделана с использованием поликристаллического га- и р-допирован-
ного Fe203, представляет собой ячейку первого типа [Leygraf et al.,
1982]. Ячейка состоит из таблеток Fea08, допированного Mg +
и Si4+, соединенных друг с другом одноименными полюсами.
Суспендированные в водных растворах NaOH или Na2S04 и освещенные
видимым светом диоды выделяют Н2 и 02. В р — га-ячеике
освещаются оба электрода и два фотона поглощаются с образованием пары
электрон — дырка. Пара состоит из меньшей части дырок
га-электрода и меньшей части электронов р-электрода. Важное преимущество
р — га-ячейки состоит в том, что для реакции в данной ячейке можно
использовать материалы с меньшей щелью; поскольку фототок с
уменьшением щели увеличивается, увеличивается и эффективность.
Возросший потенциал пары электрон — дырка в р — га-ячеике
может также элиминировать смещающий потенциал, обычно требуемый
в ячейках типа Шоттки. Как было показано, это действительно
наблюдалось в ячейке ra-Ti02/p-GaP, в которой фотоэлектролиз воды
можно было бы провести с нулевым смещающим напряжением при
искусственном дневном свете [Nozik, 1976]. Такие ячейки можно
было также использовать для проведения других реакций, в резуль
тате которых получаются химически полезные продукты. laKtL.U2
можно восстановить до формальдегида и метанола с помощью элект-
Фотоэлектрохимия
379
родов из p-GaP, а хлорид можно окислить до С12 с использованием
/1-ТЮ2.
Фото электролиз воды является наиболее притягательной
реакцией для превращения солнечной энергии, но эффективно выполнить
ее невозможно [Harris, Wilson, 1978]. Недавняя работа [Gant-
ron et al., 1980; Lemasson et al., 1982] показывает, что увеличить
эффективность анода из п-ТЮ2 можно, либо делая его кислород-
дефицитным, либо сплавляя его с V02. Эффективность ~ 21 %
зарегистрирована в ячейке с электродами из Т\02_х (х ~ 7-10"4)
и излучении 335 нм. Использование твердых растворов Ti^^-V^C^
более эффективно, так как в этом случае можно уменьшить щель до
~ 2,0 эВ. Фотокатоды, такие как р-InP или jo-GaAs (Eg «1,4 эВ),
в действительности являются более устойчивыми, чем га-тип
фотоанодов в гальванических элементах. Показано, что поверхностную
рекомбинацию электронов и дырок можно уменьшить химической
модификацией поверхности электрода [Heller, 1981]. Так,
адсорбция ионов Ru3+ на поверхности электродов заметно увеличивает
эффективность.
Электрохимические фотогальванические ячейки. В
электрохимической фотогальванической ячейке идут две, обратные друг другу,
электродные реакции: R -f- h+ -> 0+ (анод) и 0+ -+- е~ ->- R (катод),
где R и 0+ относятся соответственно к восстановленному и
окисленному состояниям. В электролите при этом не происходит никаких
изменений. Падающая оптическая энергия увеличивает свободную
энергию электронов в полупроводниковом электроде, которая
должна использоваться как электрическая энергия во время работы
ячейки.
Электрохимические фотогальванические ячейки (с жидкостным
соединением), по существу, похожи на фотоэлектролизные ячейки.
Схема фотогалъванической ячейки с жидкостным соединением и с
использованием полупроводникового электрода тг-типа показана на
рис. 7.7. Когда полупроводниковый электрод помещается в
электролит и подвергается действию положительного смещающего
напряжения, область вблизи поверхности полупроводника становится
истощенной по электронам. Когда в этом истощенном слое
поглощается свет с достаточной энергией, электрическое поле способно
отделить фотогенерируемый электрон (в зоне проводимости) от
дырки (оставленной в валентной зоне). Направление поля такое, чтобы
вести дырку к поверхности, где она превращается в
восстановленную форму R окислительно-восстановительной пары О/R в растворе.
Электрон перемещается по внешней цепи, выполняя работу eV,
восстанавливая 0+ на металлическом электроде. Таким образом,
в ячейке не происходит никакого химического изменения, а идет
превращение световой энергии hv в электрическую работу eV.
Напряжение разорванной цепи такой ячейки Voc, которое является
максимальным напряжением при нулевом токе, соответствует раз-
380 Моделирование твердых тел для особых целей
Таблица 7.1
Типичные примеры электрохимических фото-
гальванических ячеек на полупроводниках,
работающих в водной среде
Полупроводниковый
электрод
rc-CdS
n-CdSe
n-CdTe
rc-GaAs
n-Si
Доминирующая '
редокс-пара
s2-/sr
s2-/sr
Te^/Te2,-
Se2-/Se2"
Fe(cp)*/Fe(cp)+
Максимальная
экспериментальная
эффективность, %
1-2
7-8
10
9-10
2
* ср — (я — СаН6)
нице между потенциалом края зоны проводимости полупроводника
и окислительно-восстановительным потенциалом О/R в растворе.
Максимальный ток получается, когда два электрода закорочены.
Между этими двумя крайними случаями есть область, где можно
получить полезную работу.
Хотя принципы этой ячейки известны давно, практические
устройства для превращения солнечной энергии реализуются с трудом
из-за следующих проблем. 1. Фотокоррозия полупроводниковых
электродов; ее можно избежать большой скоростью окисления R,
которая сводит к минимуму концентрацию дырок на поверхности
полупроводника. 2. Улавливание дырок на поверхностных
состояниях вблизи зоны проводимости, которая в конечном счете
разрушается захватом электрона из зоны проводимости. 3. Сильная хемо-
сорбция R и О на полупроводнике неблагоприятно влияет на
кинетику электродных реакций и уменьшает общую эффективность.
Делаются некоторые усилия к тому, чтобы преодолеть эти ограничения.
Фотокоррозию можно предотвратить путем добавления к
электролиту окислительно-восстановительной пары, потенциал которой
более благоприятен, чем потенциал разложения, так что
предпочтительно идет окислительно-восстановительная реакция. Когда в
качестве анода в водных электролитах используется rc-GdS,
электрод фотокорродируется, поскольку легко идет реакция CdS +
+ 2/i+ -> S + Cd2+. При добавлении к электролиту NaOH и
полисульфида натрия [Ellis et al., 1976] фотокоррозия
предотвращается. Пара S2_/Sn~ преимущественно очищает систему от
фотодырок. На аноде сульфид окисляется до полисульфида (свободной
серы), а свободная сера снова восстанавливается на неосвещенном
Фотоэлектрохимия
381
катоде. Аналогично аноды из rc-Si стабилизируются использованием
невоБиого электролита, содержащего пару Fe(cp)2+/Fe(cp)2 [Legg
et al., 1S77; Chao et al., 1983]. К сожалению, подобный метод
стабилизации нельзя применить к фотоэлектролизным ячейкам.
В табл. 7.1 приведены некоторые примеры электродных материалов
и окислительно-восстановительных пар, используемых в
фотогальванических ячейках с жидкостным соединением.
Фотогальваиические ячейки с жидкостным соединением по
сравнению с твердотельными фотогальваническими ячейками обладают
важными преимуществами: 1) потенциальные барьеры (искажение
зоны) легко устанавливаются простым погружением электрода в
электролит; 2) могут использоваться поликристаллические
полупроводниковые пленки без резкого уменьшения эффективности и
3) искажение зоны можно изменять подбором
окислительно-восстановительного потенциала электролита. Однако последний фактор
лимитируется устойчивостью электрода. Если потенциал
электролита слишком положительный, в случае /г-электродов (или слишком
отрицательный — для /^-электродов), более благоприятной может
стать реакция разложения электрода, чем реакция в ячейке.
Недавние сообщения показывают, что фотогальванические ячейки с
жидкостным соединением очень скоро смогут конкурировать с
твердотельными солнечными элементами. При использовании
tt-GaAs^aPjc в ацетонрттриле и окислительно-восстановительной пары
Fe(cp)2/Fe(cp)2+ достигнута эффективность 13 % при естественном
освещении, причем это самая высокая эффективность, известная
для фотогальванической ячейки с жидкостным соединением [Gro-
net, Lewis, 1982].
Фотохимическое превращение солнечной энергии в гомогенной/мик-
рогетерогепнсй средах. Химическое превращение солнечной
энергии можно достичь проведением фотохимических реакций в
гомогенном или коллоидном растворах. Этот процесс, по существу,
имитирует фотосинтез углеводов в природе, т. е. химическая реакция
проводится так, что фотопродукты имеют более высокую свободную
энергию, чем реагенты. При обращении реакции можно извлечь
дополнительную энергию. В этом смысле заманчивой является реакция
фотохимического расложения воды:
Н20 (ж) !% Н2 (г) + 1/202 (г); AG°98 = 238 кДж -моль-1.
Реакция требует сенсибилизатора, так как сама вода не поглощает
свет выше 200 им. В качестве сенсибилизаторов этой реакции успеш-
по используются комплексы переходных металлов, обладающие
свойствами фотоокисления — восстановления. Однако главная
проблема здесь состоит в том, что большинство комплексов
переходных металлов включают одноэлектропный перенос на
поглощенный фотон, тогда как реакция разложения воды — процесс
382 Моделирование твердых тел для особых целей
многоэлектронный:
Н20 + 2е- = 20Н- + Н2,
2Н20 = 4Н+ + 02 + 4ег.
Следовательно, фотосенсибилизируемое разложение воды требует
наличия катализатора — аккумулятора зарядов, который является
посредником в многоэлектронных процессах. По существу, такой
катализатор аккумулирует необходимое число электронов для
передачи реагентам.
Приблизительная схема разложения воды, основанная на
химии фотоокисления — восстановления, приведена на рис. 7.8 [Ки-
tal, 1983]. Поглощение света инициирует реакцию переноса
электрона между донором D и акцептором А, приводя к образованию
энергетически богатых D+ и А". Восстанавливающая способность
А" относится к восстановлению Н20 подходящим катализатором,
тогда как катализируемое окисление воды идет с помощью D+.
Ru(bipy)g+ является примером фотосенсибилизатора, который
выполняет роль D+ и А", когда возбуждается поглощением с переносом
заряда при 450 им. Возбужденное состояние бипиридильного
комплекса *1\и(Ыру)з+, который имеет энергию на 2,1 эВ выше энергии
основного состояния, может функционировать как восстановитель
и как окислитель; восстановительное тушение *11и(Ыру)з+ дает
мощный восстановитель Ru(bipy)3+, а окислительное тушение —
сильный окислитель Ии(Ыру)з+.
Есть сообщение об успешном применении системы разложения
воды с использованием солнечной энергии (рис. 7.9) [Gratzel et al.,
1981]. Комплекс Ru(II) используется как сенсибилизатор, N-метил-
фиолетовый — как электронное реле, a Ti02 (частицы), допирован-
ный платиной и диоксидом рутения,— как бифункциональный
катализатор. При фотовозбуждении рутениевый комплекс возбуждается
и окислительно гасится метилфиолетовым. Последний вводит
электрон в зону проводимости платины, где он используется для
выделения водорода из воды. Сенсибилизатор уступает дырку Ru02,
которая используется для выделения кислорода. Коллоидный Ti02
играет в процессе очень важную роль; кислород, генерируемый на
его поверхности, восстанавливается до 02" и соединяется с
коллоидной частицей, таким образом обеспечивая удобный способ
разделения выделяемых водорода и кислорода. Водород выделяется
при освещении системы; кислород, накопленный на Ti02, можно
удалить в темноте добавлением фосфата натрия. Gratzel с
сотрудниками разработали другую интересную систему для расщепления
H2S. Система использует коллоидный CdS, допированный
сверхтонким Ru02 (0,1—0,5 %). Когда частицы катализатора
диспергируются в водном растворе H2S и освещаются видимым светом,
образуются водород и элементарная сера. Для обсуждения вопросов
Магнитные материалы
383
D-A/
Основное
состояние
Ruftlpy^MV2— MV^RuCblpy)^
TL02/Ru02
(-450 А)
Рис. 7.9. Схематическое изображение
разложения воды, инициируемое
видимым светом, с использованием Ru02 и
Pt в качестве редокс-катализаторов, со-
осажденных на коллоидном Ti02 [Grat-
zel, 1981].
Рис. 7.8. Фоторазложение воды, основанное на бимолекулярной фоторедокс-
реакции между донором электрона D и акцептором A. Gat. 1 и Gat. 2 —
катализаторы — переносчики заряда для восстановления и окисления воды
соответственно.
фотохимического разложения воды в коллоидных и
микрогетерогенных системах рекомендуется статья Kiwi et al., [1982].
Фотохимическое разложение воды на ее составные части
обеспечило значительный прогресс в двух направлениях:
фотоэлектрохимические ячейки с одним или двумя полупроводниковыми
электродами и гомогенные фотохимические реакции в растворах. Видимое
различие между двумя подходами исчезает при использовании
коллоидных металлов и суспензий полупроводников в растворе и при
использовании фотохимических диодов, которые состоят из
непосредственно контактирующих частиц двух электродов,
представляющих собой единое целое. Оказывается, что этот «беспроволочный»
фотоэлектролиз мог бы быть основным решением
крупномасштабного преобразования солнечной энергии. Однако исследования в этой
области мучительны из-за проблем невоспроизводимости (см.,
например, [Yesodharan, Gratzel, 1983]).
7.4. МАГНИТНЫЕ МАТЕРИАЛЫ
Использование ферритовых шпинелей в различных
прикладных целях хорошо известно (например, телефоны, компьютеры,
микроволновая техника). В последние годы получено много
магнитных сплавов [Chin, 1980; White, 1985]. Делаются попытки
создать молекулярные полиметаллические системы с
ферромагнитным упорядочением [Kahn, 1985]. В качестве моделей органических
ферромагнетиков исследованы высокоспиновые поликарбоны [Iwa-
raura, 1986].
384 Моделирование твердых тел для особых целей
Интерметаллические соединения на основе РЗЭ и Со состава
LnCo5 и Ln2Co17 представляют собой постоянные магнетики с
высокими эксплуатационными качествами [Nesbitt, Wernick, 1973;
Wallace, 1973; Menth et al., 1978]. Они характеризуются очень
высокими значениями собственной коэрцитивности (вплоть до 3-Ю6 А/м),
чрезвычайно высокой магнетокристаллической анизотропией и
максимальной энергией (ВН) вплоть до 240 кДж-м~3. SmCo5 является
наиболее выдающимся членом семейства LnCo5, обладающим самым
большим значением магнетокристаллической анизотропии (17 МДж-
• м~3) и большой коэрцитивной силой (1600 кА-м-1). Фазы Ln2Co17
имеют более высокие значения намагниченности насыщения, чем
LnCo5, и, следовательно, более высокие значения произведения ВН
[Wallace, Narasimhan, 1980]. LnCo5 кристаллизуется в
гексагональной структуре СаСи5, тогда как структуру Ln2Go17 можно
рассматривать как полученную из структуры LnCo5 упорядоченным
замещением 1/3 атомов Ln па пары из атомов Со. Магнетизм
соединений Ln — Со возникает в результате межатомного обмена
между спинами двух нодретпеток и спии-орбиталыюго взаимодействия
в атоме редкоземельного элемента. В более легких редкоземельных
элементах спины Ln и Со располагаются параллельно и, таким
образом, намагниченность насыщения большая. В интерметаллических
соединениях с более тяжелыми редкоземельными элементами спины
антипараллельпы, что ведет к низким значениям намагниченности
насыщения. Первопричина магнетокристаллической анизотропии
лежит в делокализованных электронах подрешетки кобальта и в
кристаллическом поле редкоземельных атомов.
С коммерческой точки зрения моиофаза SraCo5 и быстрозака-
лениая фаза Sm2(Co, Cu)17 являются важными магнитными
сплавами. Чтобы достичь высокой коэрцитивности, тонкие порошки SmCo5
(1—10 мкм) разравниваются, уплотняются в сильном магнитном поле
и отжигаются. Механизм коэрцитивности включает доменную
нуклеацию или пограничный пиининг на границах зерен. Таким
образом, высокая коэрцитивиость требует минимизации
существования доменных стенок, а это достигается, если сплав находится
в виде тонких частиц. Замещение Sm на Се или Nb понижает
намагниченность насыщения и магнетокристаллическую анизотропию,
а также Т'с. Замещение на Gd уменьшает температурный
коэффициент остаточной намагниченности. Частичное замещение Со на Си
в SmCo5 может увеличить коэрцитивиость при соответствующей
тепловой обработке. В случае замещенных магнетиков с медью для
проявления ими магнитной жесткости не требуется, чтобы они были
размолоты до тонких частичек. Электронная микроскопия показала,
что низкотемпературная тепловая обработка приводит к гомогенному
выделению второй фазы, вероятно, типа Sm2Co17, которая
когерентна со структурой SraCo5. Замещение кобальта на железо,
цирконий и гафний дает возможность оптимизировать намагниченность
Магнитные материалы
385
насыщения, коэрцитивность и произведение (ВН). Необычно
большие значения (ВН) и коэрцитивности сплавов делают их пригодными
в устройствах, где требуются малый размер и высокие
эксплуатационные качества [Chin, 1980]. Так, магниты для электронных
ручных часов, компасов, для магнитных стыковок и волноводов
делаются из сплавов Sm — Со; малые размеры постоянно-токовых
и синхронных двигателей также требуют использования этих
сплавов. Вообще редкоземельно-кобальтовые постоянные магниты
полезны в ряде мелкомасштабных специализированных приложений.
Некоторые сплавы в системах Fe — Ln — B(Ln — La, Tb или
Nd), о которых сообщалось недавно, обладают полезными
магнитными свойствами, которые делают их конкурентными SmCo5 в
устройствах с постоянным магнитом. Например, аморфные ленты
состава (Fe0i82B0tl8)0t90Tb005La0t05, полученные вытягиванием нити из
расплава, обнаруживают большой гистерезис и высокую коэрцитивность
при отжиге ~ 920 К; отжиг дает микрокристаллы нескольких фаз,
которые включают Fe3B, Ln6Fe23 и Ln3Fe21B[Das, Koon, 1983].
Самый мощный постоянный магнит из когда-либо известных
открыт за последние годы. Это Nd2Fe14B из системы Fe — Nd — В;
такой состав имеет необычную тетрагональную структуру с высокой
намагниченностью насыщения и коэрцитивностью. Структура этого
вещества определена, и материал уже получают промышленным
способом; сейчас предпринимаются значительные исследования с
различных точек зрения на этот экстраординарный магнитный материал
[Robinson, 1984; Herbst et al., 1985; Koon et al., 1985]. Последние
данные по этому вопросу можно найти в «Трудах 8-го
международного семинара по редкоземельным магнитам и их применению»,
который проходил в Дэйтоне в мае 1985 г., а также в «Трудах рабочего
совещания по постоянным магнитам Nd — Fe», опубликованных
Комиссией европейских сообществ (см. также [White, 1985]).
Большим достижением стало создание нового типа
твердотельного запоминающего устройства, называемого устройством на основе
«магнитного пузырька», для использования в компьютерах и других
системах хранения данных, основанных на перемещении магнитных
доменов через кристалл [Nielsen, 1976, 1979; Giess, 1980; Blunt,
1983]. Образование магнитных пузырьков схематично показано на
рис. 7.10. В тонкой пленке магнитного материала с соответствующей
намагниченностью и осью одноосной магнитной анизотропии,
перпендикулярной плоскости пленки, магнитные домены, образуемые
в пленке, будут состоять из змеевидного набора ленточных доменов,
по намагниченности направленных вверх и вниз, как показано на
рис. 7.10, а. При наложении магнитного (смещающего) поля
перпендикулярно плоскости пленки один ряд доменов будет расти за счет
другого (рис. 7.10, б). С увеличением приложенного поля домены
сжимаются и становятся цилиндрическими (рис. 7.10, в).
Цилиндрические домены называются «магнитными пузырьками», так как их
25 Ч. Н. Р. Рао, Дж. Гопалакришнан
386 Моделирование твердых тел для особых целей
а Без поля
I М(*2*итное поле
б Приложено маленькое
, ^ поле
\Mas"ugtHoe поле
в Приложено поле для
стабилизации пузырей
Рис. 7.10. Образование магнитных пузырьков
(схематично) [Blunt, 1983].
поведение описывается уравнениями,
похожими на уравнения, применяемые к
случаю мыльных пузырьков. В оптически
прозрачных материалах, таких как гранаты
и ферриты, пузырьки можно наблюдать
непосредственно, используя эффект Фара-
дея. Пузырьки движутся в направлении
областей более низкого смещающего поля,
и это используется в устройствах памяти.
Сохраняемая информация в форме
пузырьковых доменов движется через
неподвижную среду; в магнитофоне сохраняемая
информация в форме неподвижных доменов
находится на движущейся ленте. В
настоящее время делаются пузырьковые
устройства памяти емкостью в несколько мегабайт.
Ортоферриты, гексаферриты, металлические сплавы, такие как
GdCo6, все могут поддерживать пузырьки, но только гранаты
Ln3Fe5012 (Ln — редкоземельный элемент) обеспечивают требуемую
эластичность. В этих кубических материалах редкоземельный ион
находится в додекаэдрической позиции, а ионы Fe3+, распределенные
между октаэдрическими (2) и тетраэдрическими (3) позициями,
спариваются антипараллельно. В иттриево-железистом гранате
Y3Fe5012 намагниченность обусловлена избытком тетраэдрических
ионов Fe3+, приводя к намагниченности 1760 G, которая слишком
велика, чтобы поддерживать пузырьки диаметром 1—8 мкм.
Намагниченность контролируется химическими приемами; типичные составы,
используемые для пузырьков различного диаметра, следующие:
Sm0t4Y2ieGali2Fe3,8O12 (8 мкм)), Sm0)]8Ln0il9Ylt73Ca0j9Ge0)9Fe4)1O12
(6 мкм) и Sm0>51Lu0i42Ylt21Ca0>8eGe0>70Si0)1GFe4>14O12 (2—3 мкм).
Жидкофазная эпитаксия используется для роста тонких гранатовых
пленок на подложке (обычно немагнитный Gd3Ca5012); для этой цели
применяют метод травления из раствора гранатных оксидов,
растворенных в расплаве РЬО — В203.
Жидкие магниты (феррофлюиды) проявляют интересные
свойства, имеющие значение для технологии [Rosensweig, 1982; Рор-
plewell, 1984]. Поскольку кооперативный магнетизм является
свойством твердого состояния, магнитный флюид нельзя получить при
простом плавлении твердого вещества. Однако феррофлюиды
получают путем диспергирования тонких магнитных частичек в жидкую
среду и предохранения их от агломерации. Есть две силы, которые
стремились бы агломерировать магнитные частички суспензии: это
короткодействующие ван-дер-ваальсовы силы и дальнодеиствующие
Магнитные материалы
387
магнитные силы. Когда частички маленькие (~ 100 А в диаметре),
оказывается, что магнитные силы незначительны. Притяжение,
обусловленное ван-дер-ваальсовыми силами, предотвращается
покрытием магнитных частичек монослоем поверхностно-активного
вещества. Примером феррофлюида является диспергированный в масле
Fe304, к которому добавляется олеиновая кислота в качестве
поверхностно-активного вещества.
В присутствии магнитного поля на данный объем феррофлюида
действует сила. Так, в магнитном поле поток феррофлюида может
изменять направление, а поверхность феррофлюида — искажаться.
Частицы в феррофлюиде меньше, чем отдельный домен, и,
следовательно, не существует никаких доменных стенок. Но
намагниченность не одинакова по всей частице из-за поверхностной
анизотропии. Феррофлюиды проявляют новые свойства, которые включают
подъем немагнитных и магнитных объектов, и характеристичные
нестабильности, такие как нестабильность на границе между фер-
рофлюидом и обычной (прозрачной) жидкостью в присутствии
магнитного поля перпендикулярно межфазной границе. Нестабильность
на границе приобретает форму лабиринтного типа, которая похожа
на «волнистные» домены запоминающих устройств с магнитными
пузырьками.
Для феррофлюидов обычная форма уравнения Бернулли,
которое связывает давление несжимающейся жидкости с кинетической
и потенциальной энергией, должна быть модифицирована с тем,
чтобы включить магнитное давление, введенное приложенным
магнитным полем. Модифицированное уравнение имеет вид:
я
Р + 1/2ри2 + pgh — u.0 J MdH = constant, (7.2)
о
где Р — давление, и — линейная скорость потока, pgh —
гравитационная потенциальная энергия. Последний член описывает
магнитное давление на феррофлюиде, обусловленное внешним
магнитным полем. Уравнение (7.2) описывает поведение магнитных
жидкостей во внешних магнитных полях. Например, при постоянных
давлении и гравитационной энергии феррофлюид претерпевает
увеличение скорости при протекании через магнитное поле. В
результате должно уменьшаться поперечное сечение горизонтального
потока феррофлюида. Аналогично подъем магнитных и немагнитных
объектов в феррофлюиде можно понять в рамках модифицированного
уравнения Бернулли.
Феррофлюиды находят несколько технических приложений,
которые включают: сальники для вращающихся валов, дэмпферные
жидкости, акселерометры, оптические переключатели и затворы и
магнитные метки. Самое раннее применение — в конструкции
износостойких сальников для вращающихся валов. Подобное уплотне-
5*
388 Моделирование твердых тел для особых целей
ние используется в компьютерных дисководах. С использованием
феррофлюидов сконструированы различные многоступенчатые
уплотнения высокого давления. Когда сальник герметизируется,
каждая ступень создает кратковременную течь в следующую камеру.
Общее давление представляет собой сумму разностей в давлении по
всем ступеням. Сальники, которые работают при ~ 5000 об./м
и дифференциальном давлении ~ 470 кПа, обычные. В
громкоговорителях феррофлюид, окружающий звуковую катушку, ослабляет
нежелательный звуковой резонанс и рассеивает тепло, улучшая
качество звука. Феррофлюидный акселерометр использует магнит,
всплыьающий в феррофлюиде, в качестве детектора. Поток ферро-
флюида можно отклонять в магнитном поле, придавая ему характер
письма. Развиваются новые методы печати, основанные на ферро-
флюидных чернилах; этот метод пригоден в невидимой печати, где
буквы можно читать с помощью магнитного чтеца. Феррофлюиды
находят также приложение в медицине. Аневризмы, которые могут
привести к разрыву артерий головного мозга, можно лечить без
хирургического вмешательства, используя феррофлюид.
Феррофлюид поддерживается магнитным полем так, чтобы изолировать
разрыв от потока крови, позволяя тем самым разрыву
естественно рубцеваться. Важное свойство феррофлюида, которое
может найти применение в будущем,— это температурная
зависимость намагниченности, особенно вблизи точки Кюри, где
изменение большое.
7.5. МАТЕРИАЛЫ ДЛЯ ХРАНЕНИЯ ВОДОРОДА
Водород является одной из наиболее заманчивых форм
нетрадиционных энергоемких материалов, он может быть получен
несколькими способами: например, фотоэлектролизом воды с помощью
солнечной энергии (см. разд. 7.3). Большая доля интереса к
водороду как к топливу, к так называемой водородной экономике, исходит
из того, что для получения водорода в больших количествах методом
электролиза воды можно использовать дешевую и обильную
электрическую энергию (в результате ядерного синтеза или в солнечном
фотогальваническом элементе). Использование водорода в качестве
топлива ставит проблемы хранения. Водород можно хранить в виде
газа, жидкости или в твердом виде. Технология хранения
газообразного и жидкого водорода хорошо известна, но низкая температура
кипения (20,4 К) и низкая плотность (0,071 кг/л) затрудняют
хранение водорода в жидком виде. Хранение водорода в виде газа при
высоких давлениях нельзя усовершенствовать, не выходя за рамки
обычно используемых цистерн под давлением 150 атм. Более того,
всегда есть угроза образования взрывоопасной смеси водорода с
воздухом (синдром Hindenburg). Хранение водорода в форме твер-
Материалы для хранения водорода
389
Рис. 7.11. Изотермы давление — состав гидридов
LaNi6 и FeTi.
Температура, К : 1 — 343; 2 — 313; 3 — 303; 4 — 273;
5 — 413; 6 — 393; 7 — 373; 5 — 333; 9 — 313; 10 — 293
[Cohen, Wernick, 1981].
дого соединения (гидрида) —
альтернативная возможность, интересная по различным
причинам; он безопасен, компактность
такая же высокая, как для жидкой формы;
при хранении нет никаких потерь, хранение
обратимо. Кроме того, для хранения
водорода в виде гидрида не требуется никаких
энергетических затрат. Фактически можно
использовать энергию, выделяемую во
время реакции гидрирования. Хранение
водорода в твердом виде широко освещено в
литературе (см., например, [Reilly, Sandrock,
1980; Cohen, Wernick, 1981; Buschow, van
Mai, 1982; Angus, 1981; Ivey, Northwood,
1983]).
Идеальные требования к металлу, используемому для хранения
водорода, следующие: малая атомная масса, быстрая кинетика
сорбции водорода и способность к обратимому хранению больших
количеств водорода с равновесным парциальным давлением ~ 1 атм в
пределах комнатной температуры. Процесс сорбции водорода
представляется в виде изотерм давление — состав (рис. 7.11).
Первоначальная сорбция приводит к резкому увеличению давления
водорода. При некотором определенном давлении начинается реакция
гидрирования, и материал адсорбирует большие количества
водорода при почти постоянном давлении. В этой области сосуществуют
гидрид металла (р-фаза) и металл, насыщенный водородом (а-фаза).
Когда вся а-фаза превратится в Р-фазу, дальнейшее увеличение
давления приводит к небольшому изменению в отношении
водород/металл. Давление, соответствующее равновесию а + Н2 =^ р,
называется давлением плато. Когда давление понижается ниже давления
плато, реакция обращается и материал освобождает водород.
Сорбция не является строго обратимым процессом, в некоторых
случаях она сопровождается гистерезисом. Реакция гидрирования —
экзотермична, а обратная реакция эндотермична.
Хотя многие металлы образуют гидриды, их термодинамика
и кинетика неблагоприятны, чтобы удовлетворять требованиям
идеального материала для хранения водорода. Что касается металлов,
то здесь есть два обычных типа поведения. Металлы, подобно Pd,
V, Nb и Та, сорбируют водород обратимо, но отношение
водород/металл мало; такие электроположительные металлы, как щелочно-
ОА 0,8 ff2 1,6 2,0
390 Моделирование твердых тел для особых целей
Таблица 7.2
Интерметаллические соединения, пригодные для
хранения водорода
Соединение
LaNi6
SmCo5
CeNi3
FeTi
ZrMn2
Zr2Cu
X*
5-6
2,5
3
1-2
3,6
1,2
Давление на
плато, атм
2,9 (313 К)
3,3 (293 К)
0,09 (323 К)
5 (303 К)
0,01 (293 К)
0,20 (1073 К)
Теплота реакции.
кДж-моль- i-Hg
—31,0
-32,7
—
-28,0
-53,2
—120
* х означает число атомов водорода на
формульную единицу интерметаллического соединения.
земельные, редкоземельные, Ti и Zr, сорбируют большие количества
водорода с образованием гидридов, но реакция почти необратима,
что делает эти вещества непригодными для хранения водорода.
Сочетание этих двух классов металлов в виде интерметаллических фаз
оказалось, однако, очень успешным. В табл. 7.2 приведены
некоторые важные материалы для хранения водорода вместе с
характеристиками реакций. Интерметаллические фазы, пригодные для
хранения водорода, имеют стехиометрию АВ, А2В, АВ2 и АВ5, где А —
Ti, Са, La, Се, Sm, Mg или Zr, а В — Fe, Ni, Mn, Си или Al. Среди
них LaNi5 и FeTi — два кандидата, которые сейчас серьезно
рассматриваются как материалы для хранения водорода.
Прогнозировать подходящие интерметаллические соединения
не простое дело. Во-первых, не все комбинации двух металлов
действительно образуют интерметаллические фазы во всех случаях;
например, не известно ни одной интерметаллической фазы между
редкоземельными элементами и Nb, Mo, Та или W. Предложена модель
для предсказания того, будет или не будет давать
интерметаллическое соединение комбинация двух металлов [Miedema, 1973]. Эта
модель, однако, не предсказывает состав или кристаллическую
структуру интерметаллических фаз. Здесь необходима фазовая
диаграмма. Интерметаллические соединения обычно получаются путем
плавления исходных веществ в оксидном тигле из А1203, MgO или
Th02 в индукционной печи. Плавка в электромагнитном поле или
дуговая плавка также используются для того, чтобы избежать
загрязнения в результате реакции с материалом тигля. Если давление
пара одного из компонентов высокое при повышенных температурах
(Mg, Zn или Cd), то прибегают к методу синтеза в запаянных
ампулах, используя трубки из Та или Мо.
LaNi6 и FeTi, обладая способностью хранить относительно
большие количества водорода, кроме того, при подходящих дав-
Материалы для хранения водорода
391
Рис. 7.12. Зависимость равновесного
давления водорода от объема
элементарной ячейки различных соединений
типа LnNi5(Ln — редкоземельный
элемент). Открытые кружки — LnCo6
сплошные кружки — LnNi6; открытые
ники - LaCo6_5:cNi6a
how, van Mai, 1982].
треугольники — ^aCo Ni [Busc-
6
и
2
0
-2
-4
L
\Gd
S*
L_
CLY
N<A
°
wPT
1
3d
0s*
\.Lo
Nd\,
Pr
i i
\x=o,6
xo,t
0,2 fi\
i l
sALo
i J
6?
Ы. 66
Объём ячейки, A
86
fO
лениях и температурах быстро
сорбируют и десорбируют
водород (см. рис. 7.11). Теплота
образования гидрида составляет ~
~ 30 кДж-моль_1-Н2. Эта
теплота должна выделяться при
поглощении водорода и поглощаться
при освобождении водорода из
сплава. Термодинамическая
устойчивость интерметаллической фазы
по отношению к ее гидриду определяет пригодность материала для
хранения водорода. Обнаружено, что в ряду изоструктурных
интерметаллических фаз АВП последовательность в энтальпии
образования гидрида обратна последовательности в энтальпии
образования интерметаллического соединения [Miedema et al., 1977]. Это
приводит к правилу обратной стабильности, т. е. для менее
устойчивого исходного интерметаллического соединения — более
устойчивый гидрид, и наоборот, когда гидрид устойчив, его равновесное
(плато) давление по водороду низкое. Таким образом, поскольку
LaNi5 имеет AHf° — 60 кДж-моль-1, a LaCu5 — 101 кДж-моль"1,
никелевое соединение является лучшим материалом для хранения
водорода. Существует линейная связь между логарифмом
давления плато и объемом элементарной ячейки фаз типа LaNi5
(рис. 7.12), что является большой помощью в подборе
подходящих гидридов.
Сплавы типа LaNi5 кристаллизуются в гексагональной
структуре СаСи5, которая обеспечивает ряд междоузельных позиций
(девять) для сорбции водорода. Состав нормального гидрида LaNi5 —
LaNi5H6, но при высоких давлениях получен гидрид состава
LaNi6H8>36, где все междоузельные позиции, по-видимому, заняты.
Сорбция — десорбция водорода в LaNi5 — это топотактическая
реакция, которая идет без коренного изменения структуры (при
образовании LaNi5H6 пространственная группа изменяется от P6/mm
до Р31т, а решетка расширяется примерно на 25 %). FeTi
(структура CsCl) образует два гидрида, FeTiHi и FeTiH2; структура гидридов
отличается от структуры исходной интерметаллической фазы.
Важной особенностью LaNi6 и его аналогов является то, что в позициях
392 Моделирование твердых тел для особых целей
Ni они могут замещаться другими Зй-металлами или металлами
групп ША и IVA, а в позициях лантана — редкоземельными
элементами или кальцием. Замещение сильно изменяет равновесное
давление гидрида. Например, LaNi4Al имеет давление на плато
0,002 атм, тогда как GdNi5 имеет давление 150 атм при комнатной
температуре; LaNi5 имеет давление 2,9 атм. Таким образом, при
подходящем замещении можно специально получить
интерметаллические фазы со специфическими рабочими характеристиками
материала для хранения водорода [van Mai et al., 1974; Pourarian,
Wallace, 1982].
Сорбция водорода интерметаллической фазой требует
предварительной «активации». LaNi5 можно активировать просто действием
на него водорода при давлении порядка нескольких атмосфер. При
активации образец трескается и сильно раскалывается. После
нескольких циклов сорбции — десорбции водорода материал
становится мелким порошком (10—100 мкм). Более затруднительно
активировать FeTi, и для начальной сорбции требуются давления
значительно выше атмосферного, а также высокие температуры (400—
—600 К). Микроскопические процессы, которые идут при активации
и деактивации, до последнего времени еще не были известны.
Спектры Мессбауэра, магнитные и XPS-измерения [Shenoy et al., 1980;
Stucki, Schlapbach, 1980] обнаружили, что активация приводит к
образованию на поверхности микрокристаллов Зй-металлов,
которые, по-видимому, способствуют диссоциации молекул Н2,
облегчая тем самым быструю сорбцию.
Хранение водорода в виде интерметаллических гидридов имеет
несколько технических приложений (см. [J. Less Comm. Met. V.
73 и 74]), которые включают в себя производство сверхчистого
водорода, отделение дейтерия от водорода, использование водородного
топлива из гидрида в двигателях внутреннего сгорания и
использование интерметаллических фаз в качестве электродов в протонных
батареях или в сочетании с топливными элементами [Cohen, Wer-
nick, 1981]. Среди различных приложений заслуживает внимания
гидрид FeTi как источник топлива для автомобилей, химический
тепловой насос HYCSOS (система превращения и хранения водорода)
и использование металлических гидридов для выравнивания
нагрузки на электростанциях. Однако интерметаллические гидриды,
применяемые в автомобилях, должны конкурировать с Na—S-бата-
реями, используемыми в аналогичных целях. Химический тепловой
насос HYCSOS использует два таких различных материала для
хранения водорода, как LaNi5 и CaNi5, которые требуют разных
температур для получения одинакового давления плато [Sheft et al.,
1980]. Для разложения гидрида металла (А) с более высокой
свободной энергией диссоциации используется незначительная тепловая
энергия. Высвобождаемый водород сорбируется при промежуточной
температуре и хранится в виде второго гидрида (В), который имеет
Аморфные материалы
393
меньшую свободную энергию диссоциации. При действии теплового
насоса внешнее тепло используется для разложения второго
гидрида и ресорбции водорода при той же промежуточной температуре,
что и для первого гидрида. Теплота сорбции первого гидрида теперь
может обогревать пространство. Если теплоту сорбции при
промежуточной температуре выбрасывать наружу, то цикл с тепловым
насосом можно использовать для охлаждения пространства. Этот
подход можно применить для создания рефрижератора на основе
сорбции водорода, в котором единственный источник энергии —
нагрев при ~ 370 К, так что он идеально подходит для
кондиционирования воздуха с использованием солнечной энергии. Применение
гидридов для выравнивания нагрузки на электростанциях
основывается ка идее, что водород, полученный электролитически в менее
напряженное время, можно сохранять в виде гидрида (гидрид FeTi),
а потом, в часы пик, превращать в электричество, используя
топливные элементы [Reilly, 1978].
Существует несколько проблем в использовании
интерметаллических фаз для хранения водорода. Первая состоит в том, что даже
малые количества таких газов, как СО и пары Н20, могут понижать
емкость LaNi5 и FeTi по водороду. Поскольку интерметаллические
гидриды метастабильны по отношению к другим устойчивым фазам
(например, LaNi5Hr метастабилен по отношению к LaH2 и Ni),
происходит внутренняя деградация материала при повторном цикле.
Кроме того, выделение тепла и поглощение, объемные изменения,
связанные с сорбцией водорода, и тонкая природа частиц
материала, используемого для хранения водорода, создают проблемы по
эффективному использованию интерметаллических фаз.
7.6. АМОРФНЫЕ МАТЕРИАЛЫ
Множество материалов, имеющих насущное значение в
технологии, являются аморфными или квазикристаллическими. Кроме
оконных стекол, посуды и т. д., аморфные вещества находят ряд
других применений. Обнадеживающий фотогальванический
материал для превращения солнечной энергии в электрическую —
аморфный кремний. Свойства магнитных, диэлектрических и других
материалов в аморфном состоянии также сулят несколько приложений
[Zallen, 1983; Elliott, 1984]. Легкость производства стеклообразных
материалов делает их привлекательными для использования во
множестве случаев. У нас действительно есть стекла, прозрачные в
любой желаемой области электромагнитного спектра (300 нм —
300 см-1), полупроводниковые стекла, стекла, которые ведут себя
как ферроэлектрики (сегнетоэлектрики), и стекла с
контролируемым магнитным поведением. Ферроэлектрические (сегнетоэлектри-
ческие) стекла и стекла с контролируемой анизотропией свойств
представляют собой достойную для исследования область. Открыты
394 Моделирование твердых тел для особых целей
Таблица 7.3
Технологические приложения аморфных материалов
Природа аморфного
твердого тела
Полупроводники
Металлические
стекла
Халькогенидные
стекла
Стекла на основе
кремнезема
Проводящие стекла
с быстрой
ионной
проводимостью |
Пример
(а) Кремний (из силана)
HSi0,9H0,l
(б) Te0,8Ge0,2
(а) Стекла, содержащие
большие количества
Fe или Со (например,
*Ч7Р0,2С0Д)
(б) TiCu
As2Se8
SiOa с 10 % Ge02
NASIGLAS,
Nai+*Zr2-*/3SV
.p о
^3-^12-2x73
Приложение
Фотогальванические солнечные
элементы
Компьютерные элементы памяти на
основе перехода аморфное
состояние — кристаллическое
состояние (под действием
электрического поля)
Ферромагнитные ленты с низкими
потерями для использования в
сердечниках трансформаторов
Хранение водорода
Ксерография на основе фотопро-
водящего свойства
Оптические волокна
Твердые электролиты в батареях
и топливных элементах
некоторые сверхпроводниковые стекла (например, Pb0(9Cu0tl и
La0f8Auo,2)- Многие металлические стекла являются
ферромагнетиками. Наблюдение ферромагнетизма и сверхпроводимости в
стеклообразных системах показывает, что дальний порядок не является
обязательным условием для этих важных свойств. Одно из наиболее
привлекательных применений стекол — замена медных телефонных
проводов на оптические световоды. В табл. 7.3 показано
использование аморфных веществ в передовых технологиях. Кроме того,
стекла с быстрой ионной проводимостью могут стать ценными для
батарей и топливных элементов.
Интересны оптические свойства аморфных твердых веществ.
Эти вещества оптически изотропны. Более того, в спектрах
аморфных твердых веществ даже при низких температурах отсутствуют
те характерные признаки, которые есть в спектрах кристаллических
веществ. Однако в целом электронные спектры аморфных твердых
веществ (полосы с широким максимумом) похожи на спектры
кристаллов, что является отражением значения ближнего порядка в
определении этих характеристик. Края поглощения аморфных
материалов нечеткие, а на графике зависимости коэффициента
поглощения от энергии фотона есть экспоненциальный хвост, связанный
с внутренним беспорядком (рис. 7.13).
Аморфные материалы
395
2,2 2fl 2,6 2,8 $0 J,2
Эмереия фотона\ эВ
Рис. 7.13. Сравнение краев
оптического поглощения
аморфного и кристаллического As2S3
[Zallen et al., 1971].
40A
зоА
20Л
Кршмний
L- Кристалла-
r ческий
Аморфный
bo, эВ
Рис. 7.14. Мнимая часть диэлектрической
постоянной е2 как функция энергии фотона,
hv для аморфного и кристаллического
кремния {Pierce, Spicer, 1972]. Поскольку е2
пропорциональна коэффициенту оптического
поглощения а, е2— безразмерная
характеристика оптического поглощения.
Различие между аморфными и кристаллическими твердыми
веществами более четко обнаруживается при фононных, чем при
электронных, возбуждениях. Вклады всей плотности состояний фонона
проявляются в Раман-спектрах и в инфракрасных спектрах
аморфных твердых веществ. Все моды в природных аморфных
полупроводниках активны в инфракрасной области.
На рис. 7.14 мы приводим мнимую часть диэлектрической
постоянной кристаллического и аморфного кремния, полученную
анализом по Крамерсу-Кронигу ультрафиолетовых спектров отражения.
Оба имеют полосу поглощения, обусловленную переходом
связывающая орбиталь —>■ разрыхляющая орбиталь, но спектр поглощения
аморфного кремния гладкий, тогда как соответствующий спектр
кристаллического образца имеет структуру. Это различие в полосах
поглощения имеет значение в использовании их как материалов для
солнечных элементов. Аморфный кремний — лучший кандидат для
получения энергии в крупномасштабных солнечных элементах
(кристаллический кремний можно использовать только тогда, когда не
важны расходы, например, в качестве космического зонда),
поскольку его коэффициент поглощения намного больше в области
1—3 эВ. Оптические зонные щели для кристаллического и
аморфного кремния примерно одинаковы (~ 1 эВ), но нет никаких ограничи-
396 Моделирование твердых тел для особых целей
Рис. 7.15. Схематический спектр потерь
оптического волокна.
тельных правил отбора для того,
чтобы запрещать поглощение
света аморфным кремнием сразу
выше 1 эВ.
Другим важным
приложением аморфных твердых веществ в
оптике является использование
стекол на основе Si02 в
световодах [MacChesney, 1981; Goodman,
1983]. Это определяется высокой прозрачностью материала в
инфракрасной области ниже зонной щели. Распространение света по
волокну идет по закону полного внутреннего отражения. Для
случая передачи света на большие расстояния волокно должно иметь
очень малую потерю света. Два возможных источника потери —
рассеяние (из-за флюктуации плотности, согласно закону Рэлея)
и поглощение (обусловленное электронными и колебательными
возбуждениями). Зависимость общей потери от длины волны
выглядит в виде F-образной кривой (рис. 7.15). Рэлеевское
рассеяние сводится к минимуму с помощью длинноволнового излучения,
и участок малых потерь затем определяется пересечением рэлеевской
кривой с мультифононным краем поглощения (1,6 мкм в Si02 с
теоретической потерей 0,2 дБ-км-1). Фактическая потеря в
волокнах Si02 намного больше (~ 10 дБ-км-1) из-за поглощения ОН-
группами.
Потери можно уменьшить, применяя волокна из материалов,
для которых мультифононный край лежит в области более длинных
волн, чем у Si02. Это можно сделать, используя материалы с более
тяжелыми атомами или с более слабой силовой постоянной.
Испытаны галогенидные стекла (основанные на ZrF4 вместе с BaF2, LaF3
и т. д.) и стекла из Ge- или Sb-замещенной Si02; стекла Ge02—
— Sb203 теряют 5 дБ-км-1. Трудность здесь заключается в получении
очень чистых волокон, которые не кристаллизуются.
Халькогенидиые стекла, такие как WSe2, при облучении
светом демонстрируют необычные структурные изменения [Chopra et
al., 1981; Elliott, 1983; Owen et al., 1985]. Природа и причина
фотоструктурных изменений еще полностью не ясны, но фотоиндуцируе-
мые изменения сопровождаются изменениями в оптических и других
свойствах. Фотоструктурные изменения можно снять отжигом при
Tg. Эти эффекты имеют значение для некоторых технологических
приложений, в том числе оптическое хранение информации и
литография.
УФ- межзонные
переходы
ИК- многскро -
Органические материалы
397
7.7. ОРГАНИЧЕСКИЕ МАТЕРИАЛЫ
Органические твердые вещества имеют множество
интересных электронных и оптических свойств, использующихся в технике.
Низкоразмерные органические твердые вещества с высокой
удельной электропроводностью, а также органические жидкие кристаллы
обсуждались в гл. 6. Поистине плодотворной стала область
молекулярной электроники, основанной на органических твердых
веществах [Munn, 1984]. Органические твердые вещества обнаруживают
фотопроводимость и фотогальванические эффекты, а также
накопление и сбрасывание заряда. По электронным процессам в
органических кристаллах написан прекрасный авторитетный обзор [Pope,
Swenberg, 1982]. Здесь мы кратко укажем на некоторые важные
приложения органических материалов, обращая особое внимание на
современные полимерные материалы.
Наука и технология полимеров дала миру несколько
выдающихся современных материалов. Из нового поколения полимерных
материалов кевлар (сополимер р-фенилен диамин-терефталевой
кислоты) является поистине удивительным материалом в силу своей
высокой прочности, высокого модуля упругости и низкой плотности,
что делает его пригодным для различных приложений, к которым
относятся автомобильные шины, кабели для буровых площадок
в открытом море и пуленепробиваемые жилеты. Ароматические
полиамиды типа кевлара являются высококристалличными
полимерами с жесткими цепями; такие полимеры образуют кристаллы с
удлиненными цепями. Известны также совершенно аморфные
полиамиды; примером такого полиамида является полиамид,
образованный из терефталевой кислоты и смешанных тетраметил-гексаметилен-
диаминов, он имеет коммерческое название «Trogamid T». Этот
материал прозрачный и стекловидный и лучше сохраняет
механические свойства при повышенных температурах, чем многие
кристаллические полиамиды. Изоляционные полимеры находят применение
в качестве технических пластмасс, часто по своим механическим и
структурным функциям заменяющих металлы. Они заменяют
металлы, так как их можно легко изготовить в виде сложных форм,
обладают лучшей устойчивостью к коррозии и более легкие по весу.
Одна из самых ранних технических пластмасс, которая до сих пор
еще используется, это найлои-66, полученный из гексаметилендиами-
на и адипиновой кислоты. Полимеры, содержащие ароматические
кольца, такие как полифениленовые эфиры и полифениленовые
сульфиды, обладают большей структурной жесткостью, термической и
окислительной стабильностью и высокими температурами
размягчения [Anderson et al., 1980]. Ароматические полимерные композиты,
состоящие из полиэфирных эфир-кетонов (РЕЕК) и углеродных
волокон, разработанные недавно в Англии, обладают удивительным
сочетанием свойств: легкий вес, высокотемпературные эксплуатационные
398 Моделирование твердых тел для особых целей
качества и огнестойкость — все это делает их полезными
структурными материалами.
Введение атомов металла в полимерную структуру (металло-
органические полимеры) увеличивает ее жесткость и стабильность,
и, если атомы металла выстроены цепочкой, возможна высокая
электропроводность. Недавно получены проводящие металлооргани-
ческие полимеры: из полимерных металл-фталоцианинов в
сильнокислых средах можно получить нити; свежеприготовленные нити
могут быть допированы до проводимости 1—2 Ом-см-1. Атомы
металла в полимерах могут приводить к интересным оптическим
свойствам. Комплексы Ей и ТЬ с 4-метил-4'-винил-бипиридином при сопо-
лимеризации с метил-метакрилатом проявляют эффективную
флюоресценцию в области 500—600 нм при возбуждении излучением
335 нм. Новое применение металлоорганических полимеров состоит
в возможности использования их в качестве тепловых экранов для
мишеней в экспериментах по лазерному плавлению [Sheats
et al., 1984].
Чтобы получить ряд композитных многополимерных систем,
можно сочетать различные полимеры. Сочетания можно делать в
слоистых пластинках и пленках, гомогенных и гетерогенных смесях,
взаимопроникающих полимерных сетках, двухкомпонентных
волокнах и т. д. Свойства композитных полимеров примерно аддитивные,
но синергетические взаимодействия могут стимулировать свойства
и эксплуатационные качества, превышающие соответствующие
свойства индивидуальных компонентов. Многослойные пленки из
чередующихся слоев двух прозрачных полимеров с различными
показателями преломления могут демонстрировать интересные оптические
эффекты, подобно радужной окраске бабочек и рыб. Получены
полимерные «сплавы», обладающие лучшим сочетанием свойств, чем
у исходных компонентов. Полистирол и полипропиленоксид
смешиваются во всех соотношениях, а смеси имеют более высокие пределы
прочности при растяжении, чем их компоненты; рассмотрены
многокомпонентные полимерные системы [Alfrey, Jr., Schrenk, 1980].
Свойства некоторых органических твердых тел с высокой
удельной проводимостью металлического характера (102—103 Ом-1-см-1
при комнатной температуре) обсуждались ранее. Допированные
полиацетилены (например, галогенами, AsF5, хлорной кислотой и
другими допентами, дающими материалы n-типа) могли бы стать
пригодными в фотогальванических приборах. Обратимое
электрохимическое допирование полиацетилена делает возможным производство
перезаряжаемых батарей [Maclnnes et al., 1981]. Устойчивый
проводящий полимер, который можно легко получать, был бы основным
толчком в технологии; хотя эта цель еще должна быть реализована,
прогресс в этом отношении наблюдается.
Фотопроводящие полимеры, такие как поли(Г^-винилкарбазол),
испольвуются в ксерографии или электрофотографии, так как фото-
Органические материалы
399
генерированные носители зарядов могут перемещаться через
полимерную пленку с относительной легкостью, прежде чем станут
неподвижными или поглощенными [Roberts, 1980]. Некоторые
изоляционные полимеры, такие как поли(1,1-дифтороэтилен),
удовлетворяют требованиям электретных материалов (электрический аналог
постоянного магнита); они реагируютт на приложенные
электрические поля ограниченным движением заряда; при снятии поля заряд
обратно не проникает.
Фотохромные материалы обеспечивают способы хранения
информации на молекулярном уровне. Главным требованием здесь
является молекулярное превращение, связанное с изменением цвета,
которое является истинно обратимым. Пример такой реакции —
димеризация диена [Guru Row et al., 1983]:
/R a P C6H*
c6hs-ch=ch-ch=c ^ )=\b\==/
CN — CN \ \
C6H5 R
(R- C00CH5)
Реакция, к сожалению, не является полностью обратимой. В
приведенной ниже реакции привлекает чередующееся фотохимическое
замыкание и размыкание кольца [Darcy et al., 1981]:
X-0,S или NCH3
R-H,CH3 или С6Н5
сн3о
Если в случае фотохромных твердых веществ, чтобы вызвать
изменение цвета и обнаружить это, пользуются светом, то в случае элект-
рохромных твердых тел для этих целей применяют электрическое
поле. Некоторые семихиноны, окраска которых чрезвычайно
изменчива по действиям электрического поля, потенциально пригодны в
ячейках памяти.
Молекулы в кристаллах или диспергированные в решетках
хозяина часто бывают с целым рядом окружений, и это приводит к
уширению электронного спектра поглощения. Так, неоднородно
уширенная полоса поглощения (огибающая переходов) может
рассматриваться как суперпозиция нескольких различимых состояний.
Узколинейный лазер может насыщать один из переходов (под
огибающей), и соответствующие молекулы больше не будут участвовать в
400 Моделирование твердых тел для особых целей
Рис. 7.16. а — уширение
электронной полосы поглощения
молекулы, обусловленное
неоднородным окружением; б —
иллюстрация индуцированной лазером
фотохимической дырки, выжженной
на линии 0—0А, несвязанного
основного порфирина в п-октане при
2К; в — спектр возбуждения
линий 0—0 перехода S1—S{i
свободного основания в n-гексане,
показывающий разницу в частоте
(~100 см-1) между двумя тауто-
мерными формами (1) и (2)
свободного основания в одном типе
позиции. Облучение на линии Aj
превращает ее в А2 и наоборот;
г — дырка, выжженная на линии
At при 4,2 К [Williams, 1983].
процессе поглощения. Это явление называется выжигание дырки,
поскольку выглядит так, как будто дырку выжигают в спектре
поглощения (рис. 7.16). Если молекулы, возбужденные лазером,
подвергаются фотохимическому изменению, дырка, выжигаемая в полосе
поглощения, будет сопровождаться поглощением фотопродукта на
другой частоте. Это явление может быть полезным при оптическом
хранении информации [Castro, 1983]. Некоторыми из возможных
систем, пригодных для процессов с выжиганием дырки, являются:
свободный основный порфирин, фталоцианин, s-тетразин и хлорин,
а кроме того центры окраски в галогенидах (и азидах) щелочных
металлов и бинарные полупроводники; в последнем случае
граничные экситонные (нуль-фононные) переходы включаются в выжигание
дырки. Основные требования к материалам с выжиганием дырки
следующие: высокий квантовый выход, отсутствие фотоообесцвечи-
вания, нечувствительность к циклированию и фотохимическая
обратимость.
Электропроводность многих органических твердых веществ
заметно подвергается влиянию адсорбции газов. Это свойство можно
использовать для создания газовых сенсоров. Например, пленки
из фталоцианина можно использовать в качестве N02 — сенсоров
с чувствительностью 1 м. д. [Wright et al., 1983]. Полидиацетилены
обладают сильно анизотропными структурными и оптическими
свойствами; этот материал проявляет металлоподобную отражательную
способность, когда свет поляризован параллельно направлению
цепи, и голубое отражение, когда свет поляризован
перпендикулярно цепи. Некоторые из полидиацетиленов проявляют эффективную
генерацию второй гармоники [Gartio, Singer, 1982].
Жидкие кристаллы
401
7.8. ПЛЕНКИ ЛЕНГМЮРА — БЛОДЖЕТТ
Тонкие пленки материалов играют важную роль в
современной электронике. Пленки Ленгмюра — Блоджетт [Mort, 1980]
получаются из поверхностно-активных молекул, которые адсорбируют
на поверхности водного раствора. Поверхность осторожно сжимается
для получения плотноупако-
ванного монослоя, который
можно покрыть подходящими
субстратами путем погружения при
постоянном поверхностном
давлении. Полученные таким
образом слои имеют заданную
толщину, а повторное
погружение приводит к многослойно-
сти. Например, диацетилены и
другие полимеризующиеся
компоненты включаются в пленки
Ленгмюра — Блоджетт,
позволяя пленке селективно полиме-
ризоваться через маски, а
остатку растворяться (рис. 7.17).
Такая пленка дает резислг для
микроэлектронной схемы.
Характерно, что полидиацетиле-
новые пленки используются в
качестве волноводных структур
и в литографии, с ультрафиолетовым, рентгеновским и электрон-
ным излучением (с разрешением 1 мкм) на окисленных
кремниевых облатках. Магнитные пленки можно получать аналогично;
пленки стеарата Мп (II) исследованы с целью изучения двумерного
магнетизма [Pomerantz et a]., 1978]. Пленки Ленгмюра —
Блоджетт могут также найти применение в интегральной оптике и в
качестве газовых сенсоров.
7.9. ЖИДКИЕ КРИСТАЛЛЫ
Мы уже обсуждали (см. разд. 6.10) взаимосвязь структура —
свойство в жидких кристаллах. Дисплеи на жидких кристаллах
сейчас обычно используются в часах, калькуляторах, фотоаппаратах,
в оборудовании высшего качества, электронных играх и т. д.;
предсказывается, что они найдут применение в карманных компьютерах,
микропроцессорных портативных системах, портативных
телевизорах и телексных системах, принимающих данные от большого
компьютера. Дисплеи на жидких кристаллах основываются на
электрооптических свойствах ориентированных нематических жидких крис-
20 ч. И. Р. Рао, Дж. Гомалакршин.ш
Рис. 7.17. Пленка Ленгмюра —
Блоджетт, образованная полидиаце-
тиленом.
а — схематическая диаграмма впадины
Ленгмюра и б — структура типичной по-
л иди ацетиленовой пленки [Williams,
1983].
402 Моделирование твердых тел для особых целей
тал лов и их способности к переключению ориентации. Реориента-
ция директора (достигаемая приложением напряжения) приводит
к изменению оптических свойств. Есть три пути, в которых можно
сделать видимым изменение в ориентации жидкого кристалла.
Изменение показателя преломления можно наблюдать с помощью двух
поляризаторов (поляризатора и анализатора света, проходящего
через жидкий кристалл). Изменение в анизотропии поглощения
света, связанное с реориентацией директора, можно наблюдать с
помощью поляризатора или непосредственно. Реориентацию директора
можно наблюдать по индуцированному поглощению, получаемому
при добавлении молекул плеохроичного красителя [Raynes, 1983].
Среди различных дисплеев на жидких кристаллах наиболее
широко используются три: 1) дисплей на основе эффекта
динамического рассеяния света, 2) дисплей на основе «твист>кэффекта и 3)
дисплей на основе эффекта «гость — хозяин» [Shanks, 1982].
Дисплеи на основе эффекта динамического рассеяния света,
которые были первыми дисплеями на жидких кристаллах,
используют нематический жидкий кристалл с отрицательной
диэлектрической анизотропией (Де = ец — ех <! 0), допированный
электролитом для увеличения электропроводности. Дисплей в выключенном
состоянии прозрачный и изменяет цвет на молочно-белый при
напряжении >8 В. Изменение обусловлено турбулентным, вихреподоб-
ным потоком в жидком кристалле в результате анизотропии
электропроводности. Это вызывает пространственное изменение в
эффективном показателе преломления и, следовательно, рассеяние
падающего света.
Большинство коммерческих дисплеев на жидких кристаллах
основаны на электрооптическом «твист»-эффекте. В них
используется нематический жидкий кристалл, имеющий положительную
диэлектрическую анизотропию, дисплей работает на чистой
диэлектрической реориентации; они, таким образом, являются дисплеями на
основе истинного полевого эффекта, не требующими никакого
потока заряда в жидкий кристалл. Основная структура и действие
ячейки дисплея на основе «твист»-эффекта показаны на рис. 7.18. Тонкий
слой (6—15 мкм) жидкого кристалла помещается между двумя
стеклянными панелями, которые на внутренних поверхностях имеют
прозрачное электропроводящее покрытие (сделанное из оксида
индия — олова), куда накладывается электрическое поле поперек
жидкого кристалла. Поверхностный выстроенный слой, состоящий из
микробороздок (например, полированный полиамид), наносится на
проводящий слой. Направления бороздок на внутренних
поверхностях ортогональны друг к другу. Это приводит к повороту
директора нематического жидкого кристалла на 90° при движении от
одной граничной поверхности к другой (см. рис. 7.18). Падающий
поляризованный свет в результате «твист»-эффекта при
прохождении через ячейку направляется и передается, т. е. ячейка является
Жидкие кристаллы
403
Рис. 7.18. Ячейка на основе «твист»-эффекта
в отсутствии поля (слева) и в поле (справа)
[Raynes, 1983].
волноводом. Mauguin, который открыл
это замечательное свойство несколько
лет тому назад, показал, что
волновод является эффективным, только
если dAn > 2А,, где d — толщина слоя Я Ц=\
э- \ I! \
Поляризатор
^> Электрод ^
жидкого кристалла,
An
анизотро-
I
Электрод
Светлый
Поляризатор Тёмный
пия показателя преломления и X —
длина волны света. При переменном
напряжении, несколько большем, чем
пороговое значение, директор
ориентируется параллельно приложенному
полю, свойство волновода теряется, и поляризованный свет не
передается. Когда директор перестраивается вдоль электрического
поля, эффективное An опускается ниже уровня, требуемого
для волновода. Дисплеи на основе «твист»-эффекта становятся
популярными у потребителя и у специалистов по электронике, так как
они обладают несколькими преимуществами, по сравнению с
обычными дисплеями: 1) хорошее изображение достигается при низких
значениях напряжения (~ 2 В) с малым расходом энергии
(~ 1 мК'Вт-см-2); 2) приборы можно делать в компактной
плоскопанельной геометрии и 3) приборы обладают хорошей четкостью
изображения в широкой области освещения. Ячейку можно
использовать в методе пропускания или отражения. Интересная вариация
«твист»-эффекта — получение электрооптического цветового
переключателя с большой площадью, если сделать один из
поляризаторов окрашенно-дихроичным.
Третий важный тип дисплейного устройства на нематических
жидких кристаллах — устройство на основе эффекта «гость
—хозяин» [Taylor, White, 1974; Scheffer, 1983]. Жидкокристаллический
материал состоит из нематического жидкого кристалла с
положительной диэлектрической анизотропией, допированного хиральной
добавкой (холестерический жидкий кристалл) и плеохроичным
красителем. Смесь ведет себя подобно холестерическому жидкому
кристаллу, имеющему спиралевидный шаг, который зависит от
концентрации хиральной добавки. Молекулы плеохроичного красителя
выстраиваются в жидком кристалле так, что их длинные оси
параллельны директору (взаимодействие «гость — хозяин»). Краситель
поглощает свет, вектор Е которого направлен вдоль длинной оси,
и дисплей имеет цвет красителя. Когда прикладываемое напряжение
больше порогового значения, спиральная структура разрушается,
директор перегруппировывается параллельно приложенному полю.
Молекулы красителя тоже перестраиваются так, чтобы быть парал-
404 Моделирование твердых тел для особых целей
лельно директору. Свет теперь идет через ячейку без поглощения.
Антрахиноновые и тетразиновые красители, которые сильно дихро-
ичны и фотохимически устойчивы, используются в дисплейных
устройствах на основе эффекта «гость — хозяин».
Можно с уверенностью предсказать, что применение жидких
кристаллов в будущем будет распространяться на все более
усложненные области электроники. В особенности притягательна
возможность применения ферроэлектрических жидких кристаллов
(например, быстрые затворы, сложные мультиплексные дисплеи).
Единственный жидкий кристалл, который может проявлять ферроэлектри-
ческое свойство, это хиральныи смектическии кристалл, но, как
оказывается, не известно ни одного подходящего соединения,
проявляющего такое поведение.
7.10. НЕЛИНЕЙНЫЕ ОПТИЧЕСКИЕ МАТЕРИАЛЫ
Нелинейные оптические свойства второго порядка, такие
как генерация второй гармоники и линейный электрооптический
эффект, обусловлены нелинейной зависимостью между
поляризацией и приложенным электрическим полем. Генерация второй
гармоники включает в себя удвоение частоты света в материале;
линейный электрооптический эффект — изменение показателя
преломления материала в приложенном поле (см. разд. 6.3 и 6.9). Среди
электрооптических устройств — оптические переключатели и
модуляторы. Чтобы произошла генерация второй гармоники, кристалл
должен быть нецентросимметричным; для максимума генерации
второй гармоники кристаллы должны обладать такими направлениями
распространения волн, где двойное лучепреломление кристалла
аннулирует естественную дисперсию, приводя к одинаковым
показателям преломления на основной и второй гармониках (фазовое
совпадение). Мерой нелинейности материала являются б Миттера и
коэффициент оптической поляризации /, оба они связаны с нелинейным
откликом. На рис. 7.19 мы приводим значения б и / в
неорганических и органических материалах [Garito, Singer, 1982], для того
чтобы показать, что органические и полимерные материалы (MNA,
т — NA, MAP) обладают значениями б и /, большими по порядку
величины.
2-Метил-4-нитроанилин (MNA) имеет значения б и /, которые
в 50 раз больше, чем у KDP. Источником этих больших значений
б и / является природа возбужденных состояний я-электронных
систем. Поляризация я-облака в органических материалах (что
противоположно смещению или перегруппировке координат ядер в
неорганических материалах) приводит к большим нелинейным
эффектам, что делает их хорошими кандидатами для высокочастотных
приложений. Можно считать, что в органических твердых телах
Люминесцентные неорганические материалы 405
Рис. 7.19. Сравнение
нелинейных оптических характеристик
6 и / для органических и
неорганических твердых тел. 6
измеряется при 1,06 м; / — при
0,633 мкм, за исключением
GaAs, где / измеряется при
0,9 мкм [Garito, Singer, 1982].
восприимчивость
кристалла — аддитивное свойство
микроскопической
восприимчивости второго
порядка молекул р. Большое
значение |3 для р-нитро-
анилина получается в
результате наличия
высокоасимметричных , скорре-
лированных по заряду
возбужденных состояний
л-электронной структуры.
Оптические
нелинейные монокристаллические
полимеры можно получить
полимеризацией двузамещенных диацетиленовых мономеров в
твердом состоянии. Полимеризация инициируется термически
или облучением рентгеновскими лучами, электронами или УФ-све-
том. Заместители R и R' в диацетиленовой системе R—С=С—С=
=С—R', выбираются так, чтобы они были оптически нелинейными,
ацентричными органическим заместителями. Кроме того, анизотропия
в результате упорядочения полимерных цепочек ведет к большому
оптическому двойному лучепреломлению, что важно для фазового
совпадения. Одним из примеров таких полимерных цепочечных
структур является 1, 6-бис(2, 4-динитрофенокси)2, 4-гексадиен,
который показывает увеличение генерации второй гармоники с
увеличением полимеризации; интенсивность второй гармоники
диацетиленовых полимеров намного больше, чем у LiI03; исследуется
несколько новых полимерных структур [Williams, 1983]. Кроме
монокристалл ическйх полимеров, полимерные тонкие пленки
получаются по методу Ленгмюра — Блоджетт. Тонкие пленки предлагают
вниманию возможность ряда новых приложений, включая волновод-
ную структуру, литографию и т. д.
7.11. ЛЮМИНЕСЦЕНТНЫЕ НЕОРГАНИЧЕСКИЕ МАТЕРИАЛЫ
Люминесцентные неорганические материалы (фосфоры)
широко использовались в течение нескольких десятилетий, причем
материалы, родственные ZnS, открыты в середине XIX столетия. Фос-
Вторал еармс.чика Линейная
электрооптика
: 1000 1000 А
MNA
m~NA;MAP ?
РОМ
ос-кварц
GaAs
MNA
Органические
твердые тела
100 /00±m-NA
А-10
LLNbOj f
LLI(vf
KDpJ Диэлектрика и
pjc4- полупроводники
70±
6{Ю2н2/С)
f(lO~
UNb03
LlI03
KDP
CdS
ADP;GaAs
м2/С)
406 Моделирование твердых тел для особых целей
форы продолжают оставаться важным материалом; делаются
попытки улучшить эффективность и цвет существующих фосфоров, а также
получить новые [Hill, 1983; Hagenmuller, 1983]. Малые количества
примесей заметно влияют на люминесцентные характеристики
фосфоров; например, 1—2-Ю"6 % Си сдерживают люминесценцию ZnS.
В производстве флюоресцентных ламп широко используются
галофосфаты щелочноземельных металлов. Роль фосфора состоит в
том, чтобы поглощать излучение 254 нм ртутной лампы и испускать
свет при подходящей длине волны в видимой области. Примесные
атомы действуют как поглощающие и испускающие центры. Такие
хорошие люминесцентные характеристики имеет 3Ca3(P04)2-CaF2,
допированный сурьмой и марганцем; энергия, поглощаемая сурьмой,
делится между центрами эмиссии Sb и Мп. Для улучшения
эксплуатационных качеств фосфора Са замещают на Sr, Ва или Cd, a F —
на С1. В последние годы с применением редкоземельных добавок
получены фосфоры, излучающие в узких спектральных полосах.
Еи2+ в барий-магниевом алюминате дает голубую эмиссию, тогда
как Еи3+ в Y203 — красную. Се3+ и ТЬ3+ использованы в решетках
хозяина для получения зеленой эмиссии. Оказывается, что Еи2+,
добавленный в BaAlF5, BaSiF6 и BaY2F8, дает монохроматическую
эмиссию 6Р7/2 — 8^7/2- Найдено, что Na3Ce0>66Tb0t35(PO4)2 является
очень эффективным фосфором, поскольку эмиссия Се3+ очень хорошо
перекрывает возбуждение Tb3+(Z)4 — 7Fb).
В различных ситуациях требуются фосфоры для катодно-луче-
вых трубок с быстрой, а также устойчивой люминесценцией. Для
этой цели используются фосфоры семейства ZnS—CdS с Ag, Си и т. д.
в качестве добавок. Другие используемые решетки хозяина — CaSr
Zn2Si04, Y202S и Zn3(P04)2; типичные добавки — Мп, Се, Ей и ТЬ.
Для рассмотрения вопросов люминесценции в неорганических
твердых веществах рекомендуется книга Di Bartolo [1978].
7.12. ЛАЗЕРНЫЕ МАТЕРИАЛЫ
Типичные лазерные материалы, в которых лазерными
центрами являются примесные ионы в твердой матрице, приведены в
табл. 7.4 вместе с полупроводниковыми лазерами; в таблице также
дана длина волны эмиссии. GaAs, InP и другие аналогичные
полупроводники с теми характеристиками, которые необходимы для их
применения в приборах, удобно синтезировать методом химического
осаждения из газовой фазы, используя металлоорганические
соединения [Moss, 1983]. Среди других сфер применения лазерных
материалов — лазерные окна и интерференционные покрытия [Newnamr
1982]. Получены поликристаллические окна с повышенными
механическими свойствами из LiF, NaCl, KC1 и CaF2; быстрое
закаливание увеличивает порог повреждения лазера в 4,6 раза. Развиваются
Введение
407
Таблица 7.4
Типичные твердотельные лазеры
Лазерный материал
А1а03 (Сг»+)
CaF2 (Nd3+)
GaF2 (Sm3+)
CaF2 (Ho3+)
CaW04 (Nd3+)
GaAs
GaP
GaAe^i-*
InP
InAs
PbS
PbSe
SnxPb1_xTe
Эмиссия, мкм
0,6943
1,046
0,7085
2,09
1,06
0,84
0,72
0,64-0,86
0,91
1 3,10
4,27
8,53
9,5—28
Примечание. «Лазерные» ионы указаны
в скобках.
работы по использованию фторида гафния и фторцирконатов в
качестве новых материалов для инфракрасных стекол. Найдено, что
LiYF4 является хорошей лазерной матрицей в ультрафиолетовой
области или хорошим материалом для соответствующих окон.
Многие оптические элементы включают в себя
интерференционные покрытия, которые склонны к повреждению под действием
лазера. В целях увеличения порогов повреждения монослойные
пленки различных материалов получены высокочастотным
напылением или другими методами. Типичными используемыми здесь
материалами являются Ti02, Ti02 + Si02 и Si02 + MgF2. Исследуются
алмазоподобные углеродные пленки, получаемые ион-лучевым
осаждением. Предварительное облучение щелочногалогенидных стекол,
покрытых As2S3, NaF и т. д., также вызывает увеличение порога
повреждения.
8. РЕАКЦИОННАЯ СПОСОБНОСТЬ ТВЕРДЫХ ТЕЛ
8.1. ВВЕДЕНИЕ
Изучение реакций с участием твердых тел — важный аспект
химии твердого состояния для понимания влияния структуры и
дефектов на химическую реакционную способность твердых тел.
408 Реакционная способность твердых тел
Необходимо установить факторы, определяющие реакционную
способность твердого тела, чтобы иметь возможность синтезировать
новые твердые материалы с нужными структурой и свойствами.
Твердофазные реакции коренным образом отличаются от
реакций в гомогенной жидкой или газовой средах; в то время как
реакции в жидком или газообразном состоянии зависят в основном от
истинной реакционной способности и концентрации участвующих
химических форм, твердофазные реакции в большей степени зависят
от расположения химических составляющих в кристаллах. Эти
составляющие части зафиксированы в определенных позициях
кристалла, что вводит новую размерность в реакционной способности
твердого состояния, не существующую в других состояниях
материи. То есть химическая реакционная способность более часто
определяется кристаллической структурой и структурой дефектов
твердых тел, чем истинной химической реакционной способностью
составляющих элементов. Это свойство твердофазных реакций
обнаруживается в топохимических и топотактических реакциях.
Большинство фотохимических превращений твердых органических
соединений контролируется химией кристаллов. В этой главе мы
детально обсудим органические твердофазные реакции, а также
привлечем внимание к реакциям интеркалирования твердых тел,
приобретающим важное значение. Мы коротко обсудим и некоторые
стороны катализа, чтобы отметить роль химии твердого состояния
в этой решающей отрасли химической технологии.
8.2. ПРИРОДА ТВЕРДОФАЗНЫХ РЕАКЦИЙ
Удобно классифицировать твердофазные реакции [Gomes,
Dekeyser, 1976] по следующим категориям:
I. Твердое -^продукты (реакции разложения и полимеризации);
II. Твердое + газ -> продукты (например, окисление);
III. Твердое + твердое -> продукты (образование сложных
оксидов из простых);
IV. Твердое + жидкость -> продукты (интеркаляция);
V. Реакции на твердых поверхностях.
Твердофазные реакции могут включать одну или более таких
элементарных (скорость определяющих) стадий: I—адсорбция (и десорбция)
газовых частиц на твердой поверхности; II—химическая реакция на
атомном уровне; III—образование зародышей новой фазы и
IV—явления переноса в твердом теле. Кроме того, существуют и внешние
факторы, такие как температура, окружающая атмосфера,
облучение и т. д., влияющие на реакционную способность. В литературе
[Наппау, 1976] рассмотрены различные типы реакций
неорганических твердых веществ и факторы, влияющие на реакционную
способность твердых тел. Фазовые переходы также демонстрируют при-
Реакции, включающие только одну твердую фазу 409
знаки, подобные признакам твердофазных реакций, но они не могут
рассматриваться как реакции в химическом смысле. Действительно,
при исследовании полиморфного превращения предполагается
отсутствие химического изменения. Несколько факторов влияют на
твердофазные реакции, в особенности на реакции твердое + твердое
[Gomes, Dekeyser, 1976; Boldyrev, 1979, 1981]. Из них наиболее
важны размер частиц, газовая атмосфера, примеси. В гетерогенном
катализе хорошо известен рост реакционной способности при
уменьшении размера зерен; малый размер зерен также благоприятствует
прохождению реакций твердое + твердое. Газовая атмосфера может
существенно влиять на реакционную способность, если газ —
компонент, способный к обмену между твердыми фазами; образование
шпинели ZnFe204 облегчается в присутствии воздуха (но отнюдь
не азота). Найдено, что добавление примесей влияет на скорости
реакции. Прохождению реакции ZnO + CuS04->ZnS04 + CuO
содействует допирование оксида цинка оксидом лития. Поскольку реакция
Ge + 2Мо03 -*- Ge02 + 2Mo02 включает перенос электрона от Ge
к Мо03, более реакционноспособным является Ge дг-типа. Для
некоторых реакций благоприятно прохождение при температуре фазового
перехода для одного из реагентов (эффект Хедвала); образование
СоА1204 идет легче при у ->- а переходе А1203. Факторы, влияющие
на общую скорость процесса, локализация реакции в особой области
(и в нужном направлении) и реакционный механизм в твердофазных
реакциях были проанализированы [Boldyrev, 1971, 1981]. Метод
приготовления (т. е. предыстория) сказывается на топохимической
реакционной способности; влияние механической обработки твердых
тел заключается во введении различных типов дефектов, которые,
в свою очередь, воздействуют на общую скорость процесса.
Контроль реакционной способности твердого состояния не только во
времени, но и в пространстве — это основная цель.
8.3. РЕАКЦИИ,
ВКЛЮЧАЮЩИЕ ТОЛЬКО ОДНУ ТВЕРДУЮ ФАЗУ
К этой категории реакций принадлежат разложение
неорганических твердых веществ и димеризация, а также полимеризация
органических молекул в твердом состоянии при термическом или
фотохимическом воздействиях. Среди них реакции органических
твердых веществ включают различные структурные принципы и
поэтому будут рассмотрены в разд. 8.8.
Разложение солей, таких как карбонаты, оксалаты, азиды
металлов и перхлорат аммония, исследуется довольно долго, и
основные механизмы установлены относительно надежно. Из
кинетических исследований следует, что критической ступенью в реакциях
разложения является зародышеобразование. Кинетические данные,
410 Реакционная способность твердых тел
Рис. 8.1. Типичная кривая разложения
твердого вещества, .представленная в форме
зависимости степени разложения (а) от времени (/)•
представленные в виде зависимости доли
разложившегося твердого вещества от
времени, являются типичными S-образны-
ми кривыми (рис. 8.1), состоящими по
крайней мере из трех разных периодов:
Время, t индукционного (А), ускорения (В) и
затухания (С). Эти кинетические кривые
интерпретируются в терминах механизма, включающего
инициирование реакции на особых местах твердого вещества с
образованием зародышей (нуклеация в области А) и последующим их
ростом. Рост протекает как пограничная реакция на межфазной
границе раздела продукт — реагент. Поскольку реакция
продолжается, межфазная граница непрерывно распространяется, заставляя
реакцию ускоряться вплоть до точки перегиба В. За пределами этой
точки растущие зародыши, по-видимому, перекрываются, оставляя
непрореагировавшими изолированные куски материала. Снижение
скорости реакции в периоде С, вероятно, обусловлено уменьшением
межфазной поверхности, когда реакция близится к завершению.
На основании этой общей модели выведены различные кинетические
уравнения для количественного описания кинетики разложения
твердого тела. Точные механизмы, предполагаемые для образования
и роста зародышей, приводят к уравнениям, отличающимся друг
от друга. В уравнении А ерами — Ерофеева
-In (1 - а) = (kt)n, (8.1)
где а — степень прохождения реакции, к — константа скоростиг
предполагается, что зародышеобразование происходит по случайному
закону и изолированные зародыши растут в трехмерном
пространстве. Уравнение Праута — Томпкинса
In [a/(l - а)] =Ы (8.2)
предполагает, что рост происходит по цепному механизму с
разветвлением. Какое-либо единственное кинетическое уравнение обычно
не подходит для описания данных полного протекания реакции
разложения. В области затухания С реакция часто следует или
мономолекулярному закону (8.3), или закону сокращающегося куба (8.4):
In (I _ a)-i = м, (8.3)
[1 - (1 - a)i/3] = kt. (8.4)
Образование и рост зародышей наблюдались под микроскопом
для нескольких реакций разложения и дегидратации в
монокристаллах.
Реакции, включающие только одну твердую фазу 411
Реакции термического разложения чувствительны к природе
предварительной обработки твердых тел. Возможно, самое раннее
наблюдение в этом отношении было сделано Майклом Фарадеем,
заметившим, что выветривание карбоната натрия стимулируется,
если кристалл поцарапать иглой. Современная интерпретация этого
наблюдения заключается в том, что дислокации, создающиеся
царапиной, обеспечивают центры для развития реакции. Повышенная
реакционная способность на дислокациях продемонстрирована на
нескольких реакциях разложения [Thomas, Williams, 1971; Paras-
nis, 1970]. Предварительное облучение твердых тел нейтронами,
протонами или ультрафиолетом, рентгеновскими или 7"лУчами
уменьшает индукционный период и увеличивает скорость
разложения без заметного влияния на энергию активации. Это наблюдение
приведено, чтобы показать, что предварительное облучение
увеличивает количество центров, способных к образованию зародышей,
сохраняя полностью тот же самый механизм. Хотя и установлено,
что в некоторых твердофазных процессах дислокации повышают
реакционную способность, пока нет ясного понимания их точной
роли. В общем повышенная реакционная способность на
дислокациях может возникать по двум причинам. Энергия ядра и энергия
упругой деформации, связанные с дислокациями, могут содействовать
процессу зародышеобразования, значительно снижая энергию
активации. Аномальная стехиометрия атомов или молекул на
дислокациях также может облегчать достижение переходного состояния,
реализация которого иным способом затруднена в идеальном
твердом теле. Кроме того, существуют и такие факторы, как ловушки
для электронов, дырок и экситонов вблизи дислокаций, также
могущие увеличивать реакционную способность термически или
фотохимически стимулируемого разложения твердых тел.
В тех случаях, когда реакционная способность твердого
вещества контролируется кристаллической структурой в большей мере,
чем химическими составляющими кристалла, реакцию считают
контролируемой топохимически. Природа продуктов реакции разложения
часто определяется топохимическими факторами, в особенности
когда реакция происходит внутри твердого вещества без выделения
новой фазы [Thomas, 1974; Manohar, 1974]. Топотактическая
реакция — это твердофазная реакция, в которой упаковка атомов в
кристалле реагента остается в значительной мере неизменной в течение
хода реакции, за исключением изменений в размерах в одном или
нескольких направлениях. Типичный пример топотактической
реакции — дегидратация Мо03-2Н20:
Мо03-2Н20 -> Мо03-Н20 + Н20 -> Мо03 + 2Н20. (8.5)
Две молекулы воды в Мо03-2Н20 структурно различны (рис. 8.2);
одна молекула, как и атомы кислорода, координирована
молибденом, образующим слои, другая молекула удерживается водородными
412 Реакционная способность твердых тел
связями между этими слоями. Обнаружено [Giinter, 1972] наличие
таких топотактических соотношений между дигидратом (D) и
моногидратом (М): (010)Ы|(010)М; [Г01Ы|[001]ми [101]D||[100]M; между
моногидратом (М) и безводным оксидом Мо03(А) соотношения
таковы: (010)м||(010)А; [Ю0]м||[001]а и [001]м||[100]а. Эти соотношения
указывают на то, что плоскость (010), по существу, остается
неизменной при прохождении реакции. Слоистая структура сохраняется
во всех трех фазах, причем дегидратация вызывает только сжатие
решетки по одному из кристаллографических направлений. Важный
факт, установленный недавно: желтый гидроксид Мо03-Н20 при
дегидратации в мягких условиях переходит в структуру, подобную
Re03; метастабильный оксид Мо03 (подобный Re03) при высоких
температурах превращается в слоистую структуру [Ganapathi et
al., 1986]. Недавно показано, что все соединения Moj.^W^Og-HgO
(включая х — 0) подвергаются топохимической дегидратации с
образованием оксидов со структурой W03 [Ganapathi et al., 1986].
Другая интересная реакция: разложение W03, Mo03, Ti02 и
т. д. с образованием фаз со смешанной валентностью:
МОп (тв) -+ МОп_х (тв) + 1 х 02, (8.6)
где М—W, Mo, Ti и т. д. В этих реакциях потеря кислорода из
начальной решетки не происходит случайным образом с созданием
изолированных кислородных вакансий; вместо этого потеря
кислорода сопровождается сжатием решетки в определенных
кристаллографических направлениях, создающим плоскости
кристаллографического сдвига (см. гл. 5). Разложение V206 с образованием V6013-
включает плоскость кристаллографического сдвига (130) cs, и два
окисла прорастают [Colpaert et al., 1973] точно так же, как фазы
Магнели Wn03n_1 и Wn03n_2, базирующиеся на плоскостях
кристаллографического сдвига (120) и (130), прорастают в восстановленном
W03 [Iguchi, Tilley, 1978].
8.4. РЕАКЦИИ ТВЕРДОЕ — ГАЗ
Видимо, наиболее широко изученной реакцией твердое
тело — газ оказалось потускнение металлов в присутствии таких
активных газов, как кислород и галогены [Smeltzer, Young, 1975;
Hauffe, 1976]. Потускнение1 это, по существу, окислительная
реакция, развивающаяся до появления таких твердых продуктов,
как оксиды или галогеииды металлов, образующие пленки на
поверхности металла. Склонность металлов к потускнению в
определенной атмосфере управляется и термодинамическими, и
кинетическими факторами. Термодинамическим критерием является то, что
* Используется также термин «окалинообразование».
Реакции твердое — газ
413
изменение свободной энергии в реакции
М (тв) + -у Хя (г) -^ МХП (тв) (8.7)
должно быть отрицательным. Этот критерий выполняется почти для
всех оксидов, халькогенидов и галогенидов металлов, за
исключением благородных металлов. Существуют некоторые металлы,
подобные алюминию, которые устойчивы к потускнению при действии
кислорода и серы, хотя свободные энергии образования оксида и
сульфида отрицательны. Для понимания таких эффектов мы должны
рассмотреть кинетику и механизм реакций твердое тело — газ.
Реакция металла с таким газообразным реагентом, как
кислород, включает, во-первых, реакцию на границе фаз с образованием
пленки продукта на поверхности металла. Дальнейшая реакция
зависит от природы пленки продукта. Если пленка пористая, она
не препятствует доступу реакционного газа к металлу, и,
следовательно, окисление происходит беспрепятственно: реакция идет по
линейному закону (постоянная во времени скорость), независимо
от толщины слоя продукта. Если образуется компактный непористый
продукт, реакционный газ не имеет прямого доступа к металлу;
дальнейшая реакция может протекать лишь за счет переноса по
крайней мере одного из реагентов сквозь продукт. Поэтому
механизм окисления металла в решающей степени зависит от того,
пористым или нет является слой продукта. Имеется полезный критерий
[Pilling, Bedworth, 1923], предполагающий, что пористый незащищаю-
щий слой продукта образуется, если отношение молярного объема
продукта к молярному объему реагирующего тела меньше единицы.
К этой категории относится окисление щелочных и
щелочноземельных металлов.
Высокотемпературные процессы окисления, образующие доволь-
о
но толстые (> 1000 А) слои продукта, демонстрируют
параболическую кинетику роста
^f = A; X* = kpt + C, (8.8)
где х — толщина слоя продукта в момент времени t и кр —
константа параболической скорости. Параболический закон скорости обычно
является признаком механизма, включающего диффузию реагентов
через слой продукта, определяющую скорость процесса. Если
окисление происходит при достаточно низких температурах, существует
начальная быстрая реакция, за которой следует возможное
прекращение реакции, с образованием тонкой пленки продуктов
о
(< 1000 А). В тонкой пленке продуктов не выполняется условие
электронейтральности, что связано с появлением объемных зарядов
[Cabrera, Mott, 1948], и поэтому диффузия сквозь слой продукта
под влиянием градиента химической концентрации не является ско-
414 Реакционная способность твердых тел
J- о
фффф
о о о о о
фффф
Рис. 8.2. Топотактическая дегидратация Мо03-2Н20.
Проекции: а — (Ш) МоО,-2Н20; б — (001) Мо08.НгО; « — (100)
MoOs. Открытые кружки обозначают межслоевую, черные
кружки — координированную воду [Cunter, 1972].
(ЮН
фффф
фффф
wm
рость-определяющей стадией. Рост тонкой
пленки продукта следует множеству кинетических
законов, таких как логарифмический, линейный,
кубический и закон четвертой степени.
Окисление переходных металлов при
высоких температурах, дающее толстые слои
продукта, следует кинетике параболического роста.
Реакция происходит за счет диффузии или
кислорода, или металла. Оксиды металлов групп IIIB —
IVB (за исключением монооксидов) растут за
счет избирательного переноса кислорода, в то
1 время как оксиды других переходных металлов
растут при преобладающем перемещении ионов металлов.
Параболический закон скорости для роста толстых слоев
продукта на металлах впервые описан Tammann [1920], а теоретическая
интерпретация в терминах противоположно направленной диффузии
реагентов через слой продукта введена позднее Wagner [1936, 1975].
Модель Вагнера можно описать качественно следующим образом:
когда металл подвергается воздействию кислорода при высоких
температурах, образуется компактный толстый слой продукта,
разделяющий металл и кислород (рис. 8.3). Лимитирующим скорость
процессом становится диффузия реагентов под влиянием
концентрационных градиентов в слое продукта. В принципе возможна диффу-
ция металла от I межфазной границы ко II и кислорода в обратном
направлении, но практически доминирует диффузия одного из
реагентов (или металла, или кислорода) в зависимости от дефектной
структуры данного оксида. Модель Вагнера предполагает, что
движущимися частицами являются ионы или электроны/дырки вместо
нейтральных атомов.
Имеется огромное количество
экспериментальных данных по
окислению металлов, которые в общих чертах
служат доказательством модели
Вагнера. Наиболее ранний пример —
изучение окисления меди [Wagner, Grune-
wald,1938]. Хотя металл образует два
оксида, Gu20 и CuO, продуктом
окисления является Cu20 при давлении,
Рис. 8.3. Схематическое изображение окисле- (X2=o2,Cl2.etc)
ния металла М газом X..
Реакции твердое — газ
415
близком к атмосферному, и высоких температурах. Cu20 — это
полупроводник р-типа, содержащий вакансии меди, и равновесие с
участием дефектов выглядит так:
02 (г) *± 20 + 4Vcu + 4A+. (8.9)
Считают, что окисление идет по реакции (8.9) на межфазной
поверхности II, за ним следует диффузия вакансий меди и дырок от
границы II к границе I; движение вакансий меди определяет скорость. По
закону действия масс мы получаем
[V^u] = [Л+] = const phf29 (8.10)
Предполагается, что электропроводность Cu20 меняется как
функция рог; экспериментально найдено, что она меняется как
функция ро7 и предполагается, что уравнение (8.10) приближенно
описывает действительную ситуацию с Cu20. Так как химический
потенциал кислорода ix2 задан выражением -тгНТ \пр0 , выраже-
ние для так называемой наблюдаемой константы скорости запишется
как
где а1 — проводимость Cu20 в равновесии с медыо и £Си — число
переноса для меди в Cu20. Экспериментальные и рассчитанные (по
параболическому закону) константы скорости окисления
удивительно хорошо согласуются. Окисление меди становится назависимым
от давления кислорода выше 0,1 атм. Это происходит из-за
образования слоя СиО на поверхности Cu20. Хотя стадией, определяющей
скорость, все еще остается диффузия вакансий меди через Cu20,
концентрация вакансий меди на межфазной границе Cu20/CuO
является фиксированной величиной и зависимой от давления
диссоциации СиО, а не от внешнего давления кислорода.
Окисление цинка в оксид цинка — еще один пример, в котором
кинетика интерпретируется в рамках модели Вагнера [Wagner,
Grunewarld, 1938]. Найдено, что при 670 К реакция не зависит
от давления кислорода между 0,02 и 1 атм. ZnO — это
полупроводник Ti-типа со сверхстехиометрическим избытком цинка,
расположенного в междоузлиях; равновесие с участием дефектов может быть
представлено в виде
ZnO = Zn?" + e+i02(r). (8.12)
Уравнение Вагнера упрощается до
*■ = 4 (ТГ-) **п0П (Р°г = c«nst)-1/9. (8.13)
416 Реакционная способность твердых тел
И ZnO, и СиО — существенно электронные проводники. Есть такие
реакции потускнения, продукты которых хорошие ионные
проводники (например, AgBr). В таких случаях потускнение металла
контролируется подвижностью электронов/дырок через продукт. Бромиро-
вание серебра с образованием AgBr в интервале 570—670 К —
пример такого типа процесса; у него следующие характеристики:
1) кинетика следует параболическому закону с константой
скорости/с, пропорциональной р1/2; 2) скорость уменьшается, если в
серебре есть небольшие примеси двухвалентных металлов, таких как
Cd, Zn или Pb, и 3) скорость увеличивается на два порядка, если
серебро и бромид серебра накоротко замкнуть внешним платиновым
проводом. Все эти наблюдения можно интерпретировать на основе
следующего механизма: AgBr обладает существенно ионной
проводимостью, возникающей из разупорядочения по Френкелю подре-
шетки серебра:
V* + AgAff = Agi+ + VXg. (8.14)
Параболический рост AgBr на Ag можно себе представить как
происходящий через атаку бромом на межфазной границе II с
образованием ионов Вг" и вакансий серебра; вакансии частично
ионизируются, создавая дырки.
1Вг2(г) = Вг + У1е + Л+. (8.15)
Концентрация дырок пропорциональна корню квадратному от
давления брома. Образование вакансий серебра на границе фаз II
приводит к градиенту концентрации Ag^ по толщине слоя продукта.
Реакция происходит за счет миграции Agf от межфазной границы I
к межфазной границе II и обратной миграции дырок. Константа
скорости бромирования серебра приближенно описывается как
A; = const[(p^)1/2-(p^)1/2], (8.16)
где рвт и Рвт —давления брома на межфазпых границах AgBr/Br2
и Ag/AgBr соответственно. Так как рвг «Срвт* наблюдаемая
пропорциональность константы скорости к корню квадратному из
величины давления брома согласуется с ранее приведенным выражением
для к. Влияние добавок двухвалентного металла проявляется через
VXg B уравнении (8.15). Рост скорости реакции Ag/AgBr, если Ag
и AgBr накоротко замкнуты внешним Pt-проводом, происходит,
поскольку контролирующая скорость подвижность дырок через
AgBr замещается более легким потоком электронов в
противоположном направлении через внешнюю цепь.
В противоположность параболическому росту продуктов в
реакциях металл — газ при высоких температурах низкотемпературное
потускнение металлов следует другим кинетическим законам. В не-
Реакции твердое — твердое
417
скольких случаях существует начальный период чрезвычайно
быстрого окисления, дающего тонкие пленки (< 1000 А), за которым
следует практически остановка реакции. Когда пленки тонки,
перенос реагентов происходит в градиенте электрохимического
потенциала, возникающего от объемно-заряженных слоев на поверхности.
Наличие электростатического поля в пленке во время окисления
доказано измерениями поверхностного потенциала; его влияние на
скорость окисления подтверждается воздействием внешней разности
потенциалов на растущую пленку, изменяющим скорость реакции.
Кроме того, иные явления, чем перенос, такие как зародышеобра-
зование и реакции на границе фаз, могут стать контролирующими
скорость; ранние стадии окисления могут быть осложнены также
образованием продуктов в форме усов, лопастей и пластинок.
Соответственно для описания окисления, приводящего к тонким
пленкам, оказались пригодными, кроме параболического, иные
кинетические законы: линейный, кубический и логарифмический.
Линейный закон скорости объясняется IFehlner, Mott, 1970]
существованием определяющей скорость поверхностной реакции между атомами
металла на поверхности и адсорбированным газом; кубический закон
скорости объясняется на основе поддерживаемого полем переноса
через слой продукта. Начальные стадии окисления, когда
доминируют хемосорбция газа и явления зародышеобразовапия, широко
изучены IBenard et al., 1959; Gwathmey, Lawless, 1960]. Важный
результат, выяснившийся в этих исследованиях,— то, что разные
грани кристалла проявляют разную реакционную способность.
8.5. РЕАКЦИИ ТВЕРДОЕ — ТВЕРДОЕ
Два твердых вещества могут реагировать исключительно в
пределах твердой фазы, образуя твердые продукты. Несколько лет
назад Хедвал (см. [Garner, 1955]) изучил реакции этого типа,
происходящие между некоторыми неорганическими твердыми веществами,
такими как оксиды, галогениды, карбонаты, сульфаты металлов
и т. д. Они принадлежат к двум классам: реакции присоединения
(например, ZnO + Fe203 ->■ ZnFe204) и реакции обмена (например,
ZnS + CdO ->■ CdS + ZnO). Общий механизм реакций твердое —
твердое состоит из начального образования одного или более
твердых продуктов, которые разделяют реагенты в пространственном
отношении. Последующая реакция требует массопереноса сквозь
слои продукта. Если, по крайней мере, один из реагентов
присутствует в форме монокристалла, а другой — в форме спрессованных
гранул, кинетика реакции подчиняется параболическому закону, как
и процесс потускнения металлов.
Реакции твердое — твердое, как правило, экзотермичны, а
движущей силой реакции является разность между свободными энер-
27 ч. Н. Р. Рао, Дж. Гопалакришнан
418 Реакционная способность твердых тел
гиями образования продуктов и реагентов. Понимание механизма
реакций твердое — твердое на количественном уровне возможно,
только если эти реакции изучаются при хорошо охарактеризованных
условиях, и число переменных сведено к минимуму. Это требует
монокристаллических реагентов и тщательного контроля
химического потенциала компонентов. Кроме того, полезно было бы знать
о равновесиях точечных дефектов в фазе продукта.
Реакции присоединения. Простейшими случаями реакций
присоединения являются реакции между ионными оксидами АО и В203
с образованием шпинелей АВ204. Поскольку продукт — тройной
оксид, экспериментальные условия определяются однозначно, если
кроме температуры и давления фиксирована и активность кислорода.
В литературе описано несколько тщательно выполненных
исследований этих реакций с монокристаллами ISchmalzried, 1971I.
Классический механизм, предложенный Вагнером (см. ISchmalzried, 1976]),
для этой реакции, включает встречную диффузию катионов сквозь
слой продукта. Поскольку реакция происходит между ионными
кристаллами, обоснованно ожидать, что эти потоки обусловлены
ионами; более того, потоки, обусловленные различно заряженными
ионами, должны быть связаны так, чтобы во время реакции
сохранить электронейтральность. Механизм встречной диффузии катионов
по Вагнеру при образовании шпинели показан на рис. 8.4; на этом
рисунке приведены и другие возможные механизмы.
Различные механизмы могут быть сгруппированы в два класса:
(I) механизмы, включающие перенос ионов только через слой
продукта, и (II) механизмы, в которых, кроме переноса катионов через
слои продукта, существует и перенос кислорода через газовую фазу.
Последний механизм возможен только в случаях, когда продукт
реакции проявляет электронную проводимость и включает ион
переходного металла, который может подвергаться окислению —
восстановлению.
При образовании оксидных шпинелей реакция на межфазной
границе АО/АВ204, по существу, заключается в гомогенной
перегруппировке катионов в окта- и тетраэдрических позициях
кубической плотноупакованной решетки кислорода, так как анионная под-
решетка, по существу, остается той же самой и в структуре типа
каменной соли (АО), и в структуре шпинели. Соответственно
найдено, что в реакции между монокристаллическим MgO и
поликристаллическим Сг203 продукт, растущий на границе раздела фаз MgO/
Cr203, также монокристалличен. Удивительно, что в аналогичной
реакции между поликристаллическим NiO и монокристаллическим
А1203 продукт, образованный на границе раздела фаз NiAl204/Al203,
также является монокристаллическим. На этой межфазной границе
реакция должна включать две ступени: изменение
последовательности анионных слоев от ГПУ в а — А1203 к КПУ, с последующим
перераспределением катионов. Приходится предположить, что пере-
Реакции твердое — твердое
419
Рис. 8.4. Механизмы реакции
образования шпинели АВ204 при
твердофазном взаимодействии между АО и В203.
стройка последовательности
упаковки анионов происходит
коррелированным движением,
включающим дислокации, а не
диффузионным движением.
Кинетика роста шпинели
обычно следует параболическому
закону скорости, если толщина слоя
продукта Ах превышает 1 мкм.
Можно понять такую
параболическую зависимость, если
предположить, что по ширине слоя
продукта существует градиент
активностей компонентов. Константа скорости для параболического закона
также рассчитывается, если известны коэффициенты самодиффузии
катионов через шпинель и свободная энергия Гиббса образования
продукта. Такой расчет предполагает поддержание определенных
условий реакции, но согласие с экспериментом достаточно хорошее.
Для получения дополнительной информации относительно
механизма образования шпинели используются инертные метки. Перед
началом реакции тонкая платиновая проволока помещается на
границе между двумя реагентами. Предполагается, что расположение
метки, после того как реакция в значительной степени прошла,
объясняет механизм диффузии. В то время как интерпретация
экспериментов с меткой проста в металлических системах и дает
желательную информацию, в ионных системах интерпретация более сложная,
так как диффузия затруднена в основном в катионной подрешетке
и неясно, с какой подрешеткои связываются метки. Использование
естественных меток, таких как поры в реагентах, служит
доказательством противодиффузии катионов в реакциях образования
оксидной шпинели. Трактовка кинетики реакций твердое — твердое
становится более сложной, если продукт частично растворим в
реагентах, а также если существует более чем один продукт.
Предшествующая трактовка предполагает, что цо крайней мере
один из реагентов — монокристалл, а геометрия реагент/продукт
хорошо охарактеризована. Однако многие технологически важные
реакции получения керамики проводятся обычно между
поликристаллическими порошками. В этих случаях кинетика зависит от
нескольких физических факторов: размера зерен, объемной
плотности, пористости и т. д. Яндер и Картер предложили модели для
реакций с порошками, сделав несколько упрощающих допущений
Uander, 1927; Carter, 1961].
2 7*
АО | АВ204 В203
?В ♦ ;А0 = к-
В,0, ♦ ЗА2* I
—-1 3AB2(V?BJ
, 2* 2-
A(WB3** ЗО2"
420 Реакционная способность твердых тел
Реакция обмена. Реакции обмена, или двойного разложения,
в которых два твердых вещества реагируют, образуя два разных
продукта, более сложны и осознаны менее полно. Предложены два
лимитирующих механизма [Jost, 1937; Wagner, 1938]. Оба
механизма допускают, что катионы более подвижны, чем анионы. В
первом из них предполагается, что реагенты АХ и BY разделены
продуктами ВХ и AY, образующими между ними прилежащие
когерентные слои. Для развития реакции требуется перенос ионов А и В
через слои продукта, что действительно и происходит в реакции
MS + М'О -> M'S + МО, где М—РЬ или Zn и М'— Cd. Вагнер
установил, что многие обменные реакции между твердыми веществами
довольно быстры и не могут быть объяснены с точки зрения
механизма Иоста, в котором необходима подвижность катионов сквозь
оба продукта. Вагнер предложил механизм с замкнутым ионным
циклом, в котором ионы должны двигаться через свои собственные
продукты. В этом механизме также не требуется, чтобы продукты
образовались в виде смежных слоев; вместо этого они образуют
мозаичную структуру, облегчающую одновременную диффузию катионов
через их продукты. Считается, что такие реакции обмена металлом,
как Си -!- AgCl -^ Ag + CuCl и 2Cu + Ag2S -> 2Ag + Cu2S,
подчиняются механизму Вагнера.
8.6. РЕАКЦИИ ТВЕРДОЕ — ЖИДКОСГЬ
Реакции твердое — жидкость более сложны, чем реакции
твердое — газ, и включают многообразие технически важных
процессов, таких как коррозия и электролитическое осаждение. Когда
твердое тело реагирует с жидкостью, продукты могут образовать
слой на твердой поверхности или же раствориться в жидкой фазе.
Там, где продукт образует слой, полностью покрывающий
поверхность, реакция аналогична реациям твердое — газ; если же
продукты реакции полностью или частично растворимы в жидкой фазе,
жидкость имеет доступ к реагирующему твердому телу, а химическая
реакция на межфазной поверхности становится поэтому важной в
определении кинетики.
Простейшей реакцией твердое — жидкость является растворе-
ние твердого тела в жидкости. Скорость, с которой твердое тело
растворяется в жидкости, зависит от того, какая именно
кристаллографическая плоскость (грань кристалла) подвергается
воздействию. Влияние кристаллографических плоскостей на растворение
понятно из наблюдения, что сферические монокристаллы при
растворении приобретают полиэдрические формы. При растворении в
кислотах оксида цинка плоскость, содержащая атомы кислорода
(0001), атакуется быстрее, чем плоскость, содержащая атомы
цинка (0001).
Химия интеркаляции
421
Подобно реакциям термического разложения, на растворение
твердых веществ существенно влияют дислокации. Ямки травления,
например, образуются в местах выхода дислокаций на поверхность
кристалла. Именно по этой причине травление — полезный способ
для того, чтобы сделать дислокации видимыми и даже определить
их плотность [Thomas et al., 1971]. Проведено измерение скорости
роста ямок травления на кристалле NiS04-6H20 для определения
снижения энергии активации зародышеобразования на
дислокациях. Показано также соответствие между ямками травления на одной
половине свежерасколотого кристалла антрацена и центрами
реакции фотодимеризации на другой половине того же самого кристалла
[Thomas, 1967].
В следующем разделе обсуждаются реакции между некоторыми
твердыми веществами со слоистой структурой (например, графит,
силикаты, дихалькогениды металлов, такие как TaS2) и основаниями
Льюиса, такими как аммиак и пиридин, с образованием соединений
включения.
8.7. ХИМИЯ ИНТЕРКАЛЯЦИИ
К интеркаляции относят твердофазную реакцию,
включающую обратимое внедрение частиц гостя G в структуру хозяина [Hs].
Хозяин обеспечивает взаимосвязанную систему доступных
незанятых мест П. Реакцию можно схематически представить так:
xG + D*[Hs] *± Gx.[Hs]. (8.17)
Считают, что эта реакция является топотактической, так как
при прохождении интеркаляции и деинтеркаляции матрица хозяина
сохраняет свою целостность в отношении структуры и состава.
Первая реакция интеркаляции (а именно интеркаляции графита
сульфатом) была описана Шафо в 1841 г., но лишь с 60-х годов к химии
интеркаляции проявили широкое внимание с точки зрения ее
важности в ряде технологически важных проблем, таких как обратимые
электроды для батарей с высокой плотностью энергии,
сверхпроводимость и катализ. Хотя традиционно интеркаляцию связывали со
слоистыми хозяевами, в настоящее время этот предмет включает
также и взаимодействия, в которых хозяин не обладает слоистой
структурой [Thomas, 1983; Schollhorn, 1984].
Кроме графита, в качестве вещества-хозяина использовалось
широкое разнообразие неорганических твердых тел: слоистые
оксиды переходных металлов, халькогениды, галогениды, оксогалоге-
ниды, слоистые силикаты, цеолиты и даже сплавы металлов.
Разновидности гостей простираются от простых атомных частиц, таких
как водород, щелочные металлы и галогены, к нейтральным
молекулам (Н20, NII3, м-алкил-амины с длинной цепью, амин-оксиды, гете-
28 Ч. Н. V. Рпо. Дж Голпттгпгтттт^н
422 Реакционная способность твердых тел
роциклы) и до сложной металлоорганики, такой как кобальтоцен,
хромоцен и дибензолхром. Эта проблема детально рассмотрена в
монографиях [Levy, 1979; Whittingham, Jacobson, 1982].
Графит. Графит, у которого размер ван-дер-ваальсовой щели
о
3,35 А,— уникальный материал для хозяина, так как он может быть
интеркалирован и донорами, и акцепторами электронов. Среди
наиболее изученных доноров электронов находятся щелочные
металлы. Тяжелые щелочные металлы К, Rb и Cs с энергиями ионизации
ниже, чем сродство к электрону графита (4,6 эВ), легко интеркали-
руются в графит. Внедрение щелочных металлов приводит к
расширению структуры хозяина вдоль оси с, причем удаление друг от
друга соседних плоскостей углерода зависит от размера атома щелочно-
о о
го металла; рост параметра достигает 0,38 А для Li; 1,15 А для Na;
о о о
2,0 А для К; 2,3 А для Rb и 2,60 А для Cs. Кроме геометрического
изменения, происходит перенос электрона от щелочного металла
к графитовой матрице, что доказывается переходом от
диамагнетизма к парамагнетизму Паули и значительным ростом проводимости
вдоль оси с. Некоторые другие физические свойства также
указывают на то, что графит восстанавливается при интеркаляции щелочных
металлов. Щелочные интеркалаты графита интенсивно окрашены и
поглощают водород, возможно, из-за образования гидрида;
например, золотистая фаза КС8 переходит в голубую KH0i67C8 в
присутствии водорода. Эта легкая реакция с водородом может быть
важной для катализа и, возможно, является причиной образования
NH3, если пропускать смесь Н2 и N2 через калиевый интеркалат
графита [Sudo et al., 1969].
Разнообразные электронные акцепторы, такие как HS04~, Вг2,
N03~, Сг02С12, Сг03, Мо03, и галогениды металлов, такие как MoF6,
FeCl3, FeCl2, были интеркалированы в графит. Графит в этих
соединениях является донором электронов. Природа электронного
переноса обнаруживается при измерении эффекта Холла, указывающего
на носители р-типа. Эти соли графита — хорошие проводники в
плоскости аЪ, с проводимостью, сравнимой с проводимостью
металлического алюминия.
Структурные исследования показывают, что частицы гостя, ин-
теркалированные в графит, хорошо упорядочены, что вызывает
появление ступенчатости. При максимальной концентрации монослой
частиц гостя внедрен между парой слоев графита (соединения первой
ступени); с уменьшением концентрации образуются интеркалаты
более высокой ступени. В соединении второй ступени гость внедрен
через два слоя. В соединении третьей ступени частицы гостя
внедрены через каждые три слоя и т. д. На рис. 8.5 схематически
показаны интеркаляционные соединения графита первой, второй и третьей
ступени. Одной из головоломных проблем является механизм
легкого взаимного превращения из одной ступени в следующую. Домен-
Химия интеркаляции
423
Рис. 8.5. Схематическое изображение
«ступенчатости» при образовании интер-
каляционных соединений графита.
Ступени: а — третья; б — вторая, в —
первая.
а б е
ная модель, основанная на силах упругости, предложенная для
объяснения ступенчатости [Daumas, Herold, 1969], получила прямое
подтверждение в электронно-микроскопических исследованиях
[Thomas et al., 1980]. Изучение интеркалатов FeCl3 и FeCl2 показало
сосуществующие ступени на микроструктурном уровне (рис. 8.6).
Найдено, что в присутствии жидкого HF графит подвергается
обратимой интеркаляции фтором. Материалы первой ступени имеют
состав C12(HF)Fn (1 < п <; 5), а предельная соль — бифторид второй
ступени C12HF2. Широкие исследования этих интеркалатов выявили,
что проводимость меняется от металлической до диэлектрической
[Mallouk et al., 1985].
Дихалъкогениды переходных металлов. Дихалькогениды
переходных металлов МХ2 групп IV, V и VI (М—Ti, Zr, Hf, V, Nb, Та,
Mo, W) обладают гексагональной или ромбоэдрической слоистыми
структурами. Блоки, из которых построена структура этих
соединений, это сэндвичи МХ2, в которых переходный металл М занимает
или тригоналыю-призматические, или октаэдрические позиции
между двумя слоями плотноупакованных атомов халькогена (X). Эти
X—М—Х-сэндвичи набраны стопкой вдоль гексагональной оси с.
Связи внутри групп МХ2 сильны (ионно-ковалентные), в то время
как связи между отдельными группами МХ2 слабы (ван-дер-вааль-
совы). Именно слабое межслоевое связывание и соответствующая
величина ван-дер-ваальсовой щели делают халькогениды
двумерными материалами, проявляющими интеркаляционные и другие
анизотропные физические свойства. Для халькогенидов МХ2
известны различные полиморфные модификации, в зависимости от того,
в октаэдрических или тригонально-призматических позициях
размещены атомы М и от характера размещения слоев МХ2 вдоль оси с.
Большое внимание уделено изучению интеркаляционных
соединений с дихалькогенидами [Whittingham, 1978; Subba Rao, Shafer,
1979]. Эти соединения могут быть разделены на три категории:
соединения с молекулами типа оснований Льюиса, такими как
аммиак, н-алкиламины, пиридины и т. д.; соединения с катионами
металлов или молекулярными катионами, такими как Li, Na, К
и др. или [(С5Нб)2Со]+; соединения, содержащие и катионы, и
нейтральные полярные (сольватированные) молекулы в
ван-дер-ваальсовой щели.
Известно множество органических интеркалатов дихалькогени-
дов [Jacobson, 1982]. Дисульфиды и диселениды образуют стабиль-
28*
•прайма
424 Реакционная способность твердых тел
Рис. 8.6. Электронно-микроскопическое изображение графитовых интеркалатов.
Вверху: фотография, показывающая сосуществование второй и третьей ступени интеркалата
графит — FeC]2. Внизу: фотография интеркалата графит — FeCl3, показывающая взаимное
проникновение областей с различными ступенями.
ные интеркалаты, в то время как дителлуриды не образуют. Интер-
каляция приводит к расширению решетки вдоль оси с, причем это
увеличение является характеристикой гостя. Стехиометрия и
устойчивость интеркалатов зависят от молекул и гостя, и хозяина, причем
наибольшая устойчивость наблюдается для 2HTaS2, 2HNbS2
и lTTiS2.
Интеркаляция органических соединений происходит при
прямой реакции халькогенидов непосредственно с чистой органической
жидкостью или в растворителе, таком как бензол или толуол. За
прохождением реакции следят, регистрируя расширение решетки,
с помощью рентгеновской дифракции. Поразительным примером
интеркаляцпи из этой группы является реакция н-алкиламинов
(CnH2n+1NH2), впервые описанная для сверхпроводника 2HTaS2 как
хозяина. Прямой реакцией была получена целая серия интеркалатов
от п = 1 до п = 18. При значениях п до 4 увеличение по оси с мало
о
и постоянно (~ 3,0 А), что, вероятно, указывает на расположение
углеводородных цепей параллельно слоям халькогенида. При
больших значениях п получается фаза определенной стехиометрии
A2/3TaS2, у которой параметр с растет линейно от количества
углеродных атомов в амине. В этих фазах углеводородная цепь
перпендикулярна (или почти перпендикулярна) слоям халькогенида. Сте-
Химия интеркаляции
425
Tos, Слои
[-—ToS, Слой
Рис. 8.7. Структуры интеркалатов TaS2— и-
ал кил амин.
а — двухслойная и б — монослойная конфигурации
интеркалированного амина.
хиометрия и расширение по оси с
указывают на двухслойное расположение
аминов (рис. 8.7). Для аминов с п —
=4—9 двухслойная структура
сохраняется, но молекулы наклоняются под
углом 56° к оси с. Существование
ступеней не всеобщее явление, хотя для
интеркаляции стеарин-амида в TaS2
были описаны соединения первой и второй ступени для составов 2 : 3
и 1 : 3 соответственно.
Дихалькогениды VI группы не образуют легко интеркалатов
с органическими молекулами. Но соединения м-алкиламмония
можно приготовить через ионообменные реакции гидратированного
натриевого соединения включения Na0a(H2O)0>6MoS2. Приготовленные
таким путем алкиламмониевые соединения проявляют расширение
решетки, зависящее от длины цепи алкила, подобно описанным
выше системам МХ2-м-алкиламин. Это приводит к фундаментальному
вопросу, не являются ли протонированные амины подлинными ин-
теркалированными частицами в обоих случаях, осложненных
восстановлением хозяина? Действительно, это составило основу повой
модели для реакций интеркаляции в МХ2 [Schollhorn, 1980].
Довольно хорошо изучена интеркаляция пиридина (Ру) и
замещенных пиридинов в 2HNbS2 и TaS2. Предельный состав близок
к (Py)o,5MS2 для халькогенидов Ti, Nb и Та, хотя известны и
меньшие включенные количества. При прямой реакции сухого пиридина
с 2HTaS2 при 450—475 К получаются две фазы со слабо различаю-
о
щимися параметрами с: 23,7 и 24,1 А. Электрохимическая
интеркаляция с катодом TaS2 и гидрохлоридом пиридиния в метаноле дает
о
фазу 23,7 А с выделением водорода. Эти пиридиновые интеркала-
ты — соединения первой ступени, где все ван-дер-ваальсовы щели
заняты пиридином. Соединения второй ступени можно получить,
если контролировать концентрацию пиридина. Аммиак образует
с TaS2 соединения и первой, и второй ступени. На изображении
о
решетки ясно видны повторяющиеся расстояния 6,12 и 18 А между
темными интерференционными полосами, отвечающими слоям TaS2,
и светлыми полосами, от пустых или заполненных органикой слоев.
Дефекты упаковки особенно преобладают в веществе второй ступени,
в котором отмечалось случайное чередование областей первой и
второй ступени [Moran et al., 1971].
426 Реакционная способность твердых тел
Стоит упомянуть о некоторых структурных аспектах
пиридиновых интеркалатов. При интеркаляции происходит изменение
последовательности упаковки 2HTaS2 от АсА/ВсВ к АсА/АЬА. Среди
трех возможных ориентации предпочтительна та, при которой
длинная ось CN параллельна слою дихалькогенида. По-видимому, и в
(Py)0j5NbS2, и в (Py)0f5TaS2 пиридин двумерно упорядочен, образуя
прямоугольную сверхрешетку 2а/3х13а (а— а для TaS2). Объем
сверхрешетки свидетельствует о том, что одна молекула пиридина
приходится на 2г единиц TaS2 (а не на 2TaS2). Это приводит к
идеальному составу (Py)0?46TaS2.
Природа взаимодействия гость — хозяин в органических интер-
калатах дихалькогенидов все еще является предметом дискуссии.
Обычно их считают соединениями с переносом заряда или донорно-
акцепторными соединениями, в которых заряд переносится от
органической молекулы к пустой (Ti, Zr, Hf) или наполовину
заполненной (Nb, Та) dz2 зоне халькогенида. Дихалькогениды VI группы
с заполненной низколежащей d-зоной не подвергаются прямой
интеркаляции. Данные многочисленных физических измерений
подтверждают перенос заряда. Градиент электрического поля на Nb,
наблюдающийся в спектре ЯМР Nb и (Py)05NbS2, указывает на
перенос ~ 0,2 электрона в зону проводимости ниобия. Сдвиг Is
уровня азота в рентгеноэлектронном спектре также качественно
указывает на перенос заряда к хозяину. Трудно мысленно
представить себе точную природу взаимодействия между неподеленными
парами а-электронов основания Льюиса (NH3 или Ру) и орбиталями
хозяина, поскольку ориентация молекул гостя не допускает прямого
перекрывания. Колебательный спектр, полученный методом
неупругого рассеяния нейтронов, также не согласуется с непосредственной
передачей неподеленной пары а-электронов. Для разрешения этого
противоречия Schollhorn [1980] предложил альтернативное описание
образования связи в интеркалатах пиридина и аммиака. В этой
модели образование связи рассматривается как электростатическое
взаимодействие между отрицательно заряженными слоями хозяина
и интеркалируемых катионов по аналогии с интеркалатами
щелочных металлов и металлоорганическими интеркалатами. Для случая
интеркалатов пиридин — TaS2 модель формируется как
2Pyridine ->- Bipyridine + 2Н+ + 2е~
.zPyridine + хИ+ -> #[РуН + ]
я[РуН + ] + (0,5 — *)Ру + хе~ + TaS2 ->
+ [PyH + UPy)0,5-*TaS2.
Ионная модель объясняет некоторые экспериментальные
наблюдения, такие как уменьшение общего объема, сдвиг энергии связи
ls-уровня азота, перенос заряда, доходящий до 0,25 е~ на TaS2, и
Химия интеркаляции
427
резкий перегиб кривой зависимости потенциала от заряда при
0,25 £~/TaS2 во время электрохимической интеркаляции. Ионная
модель также обеспечивает единое описание образования связи во
всех интеркалатах дихалькогенидов.
Удивительная группа интеркалатов в дихалькогенидах
металлов — это интеркалаты с металлоорганикой, такие как кобальтсцен
и хромоцен в TaS2 [Dines, 1975]. Все металлоорганические
соединения, которые были интеркалированы до сих пор, являются хорошими
восстановителями с низкими энергиями ионизации (менее 6,2 эВ).
Интеркаляция соответственно включает перенос электрона от гостя
к хозяину. Магнитные исследования дают прямое доказательство
переноса электрона; в то время как нейтральный кобальтоцен —
парамагнетик с одним неспаренным электроном, при интеркаляции
в TaS2 он теряет свой магнитный момент, что и указывает на
перенос электрона.
Открытие сверхпроводимости в молекулярных интеркалатах
слоистых дихалькогенидов привело к большой активности в этой
области, в основном для поиска того, можно ли достичь интеркаля-
цией высоких температур перехода в сверхпроводящее состояние.
Неудача в попытке обнаружить повышение Тс приводит к
убеждению, что сверхпроводимость, по существу, ограничена двумерными
металлическими слоями в МХ2. Два экспериментальных результата
подтверждают это и наводят на мысль, что между слоями отсутствует
межслоевое спаривание электронов посредством джозефсоновских
контактов (туннелирование сверхпроводящих пар). Первое указание на
это исходит из факта, что даже такое большое межслоевое расстоя-
о
ние, как GO А, в интеркалатах TaS2 не влияет на Тс. Второе
указание исходит из исследования изоструктурных интеркалатов TaS2
с кобальтоценом и хромоценом. В обоих случаях расширение
решетки идентично. Однако интеркаляция приводит к диамагнетизму в
случае кобальтоцена и парамагнетизму в случае хромоцена. Вопреки
присутствию локализованного магнитного момента в chromo-
cene)1/4TaS2 оба интеркалата обладают почти одинаковыми
сверхпроводящими свойствами, отчетливо показывая, что межслоевое
джозефсоновское взаимодействие не является необходимым
компонентом механизма сверхпроводимости.
Среди атомов металлов, которые интеркалируются в дихалько-
гениды МХ2, наибольшее внимание привлекают щелочные и
переходные металлы первого ряда. В противоположность графиту для
слоистых дихалькогенидов неизвестны интеркалаты с акцепторами
электронов. Интеркалаты с одновалентными катионами АМХ2 (А —
одновалентный катион, М — переходные металлы IV В или
V В групп и X—S или Se) важны как возможные электродные
материалы в твердотельных батареях. Обычно это проводники со
смешанной проводимостью, причем электронная часть связана со
слоями материала хозяина, а ионная — со слоями интеркалированных
428 Реакционная способность твердых тел
ионов. Так же как атомы М в МХ2 находятся в октаэдрических
(lTTiS2) или тригоналыю-призматических (2HNbS2, MoS2)
позициях, интеркалированные атомы А могут занимать октаэдрические или
тригонально-призматические позиции. Последняя координация
предпочтительнее для больших ионов щелочных металлов и при высокой
ионности связей MX. Обнаружено, что Си+ и Ag+ находятся в тет-
раэдрических позициях. Если А — большой ион щелочного металла
в тригонально-призматических позициях, в соединениях АМХ2
обнаруживаются ступени. Интеркалированные ионы А особым образом
упорядочиваются при достаточно низких температурах. В
зависимости от геометрии координации ионов А возможны три двумерные
подрешетки для АМХ2: треугольная решетка для октаэдрически
координированных ионов А, сотообразная решетка для ионов А
в тригонально-призматической координации и гофрированная
сотообразная для ионов А в тетраэдрнческих позициях. В Na^TiSa
найдены три различные сверхрешетки, происходящие из упорядочения
тригонально-призматически координированных ионов натрия: две из
них соответствуют гексагональным (2x2) и (]^3х У^З) решеткам,
а третья (2х]^3). Эти сверхрешетки наблюдаются при
определенных дробных составах (значениях х). Li^TiSg — единственная
наиболее важная система в этом семействе. Хотя в ранней работе она
была описана как однофазная система во всем интервале
стехиометрии (0 <[ # <[ 1) (химический потенциал довольно плавно меняется
в зависимости от х), недавние точные измерения величины
химического потенциала как функции состава обнаружили наличие ярко
выраженной структуры при определенных дробных составах,
указывающее на дальний порядок по Li+. Кроме того, на диффракцион-
ных картинах видны слабые, но резкие фигуры сверхструктуры.
Обе сверхструктуры, 2а0 и За0, идентифицированы анализом
диффузного рассеяния рентгеновских лучей. Прямое определение
энтропии интеркаляции дает дополнительное доказательство наличия
ill 1
упорядоченных структур при #==-т1>"'Т'Т и -т [Thompson, 1981].
Интеркаляция и катионных, и нейтральных (сольватированных)
молекулярных частиц в МХ2 особенно важна, если реакция
протекает в присутствии полярных растворителей, особенно при низких
температурах (рис. 8.8). Предложена упрощенная схема реакций,
объясняющая такой тип интеркаляции [Schollhorn, 1980]:
[Hs] ^ Gx+(S)w[Hs]x- =* Gt [Hs]*"
h3o+.h2oJ| (8.18)
Ях [Hs] S (H30+)x(H20)y [Hs]*"
—n2u
где Hs — хозяин, G — гость и S — растворитель.
Вышеприведенная схема должна иллюстрировать сложность топохимических реак-
Химия интеркаляции
429
х е"
X Ф
RED
ОХ
►н?о
-н20
I
.©^Ж
Рис. 8.8. Образование гидратированных фаз при топотактической
окислительно-восстановительной реакции интеркаляции в водных растворах
электролитов.
а — решетка хозяина МХ2; б — монослойный гидрат Ах (Н20)у [МХ2]Х ;
ный гидрат [Schollhorn, 1980]Г
в —двухслой-
ций интеркаляции в растворах, включающих и процессы
окисления — восстановления, сольватации и обмена. Важной стороной
сольватации катионов и связанной с этим полиэлектролитной
природой этих соединений включения является то, что интеркалаты
оснований Льюиса (L) могут рассматриваться как (LH+)xLj/[Hs]3C_,
где катион и сольватированные молекулы различаются только
дополнительным протоном, присоединенным к основанию.
Другие слоистые неорганические матрицы. Несколько в стороне
от слоистых дихалькогенидов переходных металлов существуют
такие многочисленные слоистые твердые соединения, как трихалько-
фосфиды металлов (например, MPS3), халькогениды металлов, халь-
кокарбиды и галогениды металлов, для которых также была изучена
интеркаляция. Трисульфид-фосфиды переходных металлов
[Johnson, 1982] кристаллизуются в слоистой структуре, подобной CdCl2.
При записи формулы в виде M2/3(P2)1/3S2 виДна связь со структурой
CdCl2: атомы серы образуют кубический мотив с плотной упаковкой.
Каждый второй слой октаэдрических позиций между слоями с
плотной упаковкой заполнен атомами переходного металла и парами
атомов Р2. В направлении, перпендикулярном слоям, структура
скрепляется ван-дер-ваальсовыми силами, и именно в ван-дер-вааль-
совы щели и происходит интеркаляция. Другой путь ощутить эти
твердые фазы — это мысленно представить их как соли металлов
с гексатиогиподифосфатным анионом P2Se~. Фазы MPS3 известны
для ряда таких двухвалентных металлов Mg, Mn, Ni, Zn и Cd. Среди
селенидов FePSe3, MgPSe3, MnPSe3 и др. найдены аналоги структуры
Cdl2. Точно так же, как и у дихалькогенидов переходных металлов,
известно три класса типов гостей, интеркалирующихся в MPS3 и
MPSe3; органические молекулы, щелочные металлы и металлоорга-
нические соединения. Среди органических молекул интеркаляция
н-алкиламина в MPS3 приводит к расширению решетки, что указы-
430 Реакционная способность твердых тел
вает на двухслойную конфигурацию с ориентацией цепи алкила
перпендикулярно слоям. В настоящее время детали структуры и
движущая сила этого вида интеркаляции не совсем понятны.
Щелочные металлы могут быть интеркалированы в MPS3 или
химическим, или электрохимическим методами. Химическая интер-
каляция происходит, если хозяин реагирует с таким металлооргани-
ческим соединением щелочного металла, как н-бутиллитий или наф-
талид натрия. Реакцию с н-бутиллитием можно изобразить
следующим образом:
;rC4H9Li + MPS3 -> LiJMPS3 +fC8H18.
NiPS3 дает состав с высоким содержанием лития: Li4i5NiPS3. Такое
большое содержание лития, обнаруженное в продуктах химической
интеркаляции, возможно, является результатом необратимой
деструкции решетки гостя с образованием смеси Li2S и аморфных
продуктов. Возможность электрохимической интеркаляции лития в
фосфидсульфиды MPS3 позволяет использовать их как катодные
материалы во вторичных батареях. Для NiPS3 электроинтеркаляция
дает две однофазных области Li^NiPSa, 0 <С х < 0,5 и 0,5 < х <
<С 1,5. Интеркаляция электрохимически обратима до тех пор, пока х
меньше 1,5. Если х больше, чем 1,5, происходит необратимая
реакция, приводящая к разрушению решетки хозяина.
Изучены оптические свойства MPS3 и Li^MPSg. Фазы гостя —
широкозонные полупроводники с шириной запрещенной зоны 1,3—
3,5 эВ. Электрохимически активными являются три соединения:
NiPS3, FePS3 и FePSe3, у которых наименьшая ширина запрещенной
зоны. При интеркаляции лития пропускание падает до тех пор, пока
вся область от 3300 до 500 нм не становится непрозрачной. Удельное
сопротивление на постоянном токе меняется параллельно
оптическим свойствам. Для NiPS3 интеркаляция лития уменьшает
удельное сопротивление приблизительно на восемь порядков. Результаты
наводят на мысль, что электрон от лития переходит в делокализо-
ванную зону проводимости, что приводит как к увеличенной
электронной проводимости, так и к поглощению свободных носителей
и увеличенному коэффициенту отражения в оптических спектрах.
Данные по магнитной восприимчивости и магнитному резонансу
на ядрах 7Li и 31Р для FePS3 и его литиевых интеркалатов
показывают, что железо остается в высокоспиновом двухвалентном
состоянии. В соединении LixNiPS3 электроны, добавленные интеркаля-
цией лития, видимо, переходят на 4^-орбиталь переходного металла
или на пустые высшие орбитали серы. Остается непонятным
исчезновение магнетизма в LixNiPS3 при х > 0,5.
Известно, что среди большого числа халькогенидов металлов
с общей формулой MXY только оксогалогениды (при М—Ti, Vt
Сг, Fe и А1) и тио- и селеногалогениды алюминия подвергаются
Химия интеркаляции
431
7,91 А
О С1 при г = О
О а л/м/z=i/2
12,85 А О 0 при Z = О
о F е при г - О
• Fe при z = V2
Pwc. 5.P. Схематическое представление интеркаляции кобальтоцена в FeOCl
[Halbert, 1982].
топохимической интеркаляции. Слоистые оксогалогениды металлов
кристаллизуются в четырех главных структурных типах: (I)
структура А10С1, в которой ион А13+ четырехкоординирован, (II)
структура FeOCl, в которой ион Fe3+ гексакоординирован, (III) структура
SmSI, в которой металл семикоординирован и (IV) структура PbFCl,
в которой свинец или восьми-, или девятикоординирован. Среди них
только структуры АЮС1 и FeOCl выполняют функции матрицы в
интеркаляции [Schafer-Stahl, Abele, 1980; Halbert, 1982].
Разновидности гостя, интеркалированные в хозяева-дихалькоге-
ниды,— это несольватированные ионы щелочных и переходных
металлов, сольватированные катионы металлов, катионные формы
органических и металлоорганических соединений. Для оксогалогени-
дов в качестве хозяев известны соответствия в каждом классе гостей,
хотя ряд хозяев ограничен оксогалогенидами переходных металлов,
имеющими способные к восстановлению катионы М3+. Интеркалаты
можно приготовить химическим или электрохимическим методами.
Об интеркаляции несольватированного Li+ сообщалось для FeOCl,
VOC1 или CrOCl. Соединения включения с сольватированными
щелочными металлами M3C+(H20)y(FeOCl)x~ получены прямым
восстановлением FeOCl с использованием водных растворов M4Fe(CN)6(M—
—Li, Na, К, Cs). В этих соединениях отмечается большое
расширение, перпендикулярное слоям. Сольватные молекулы воды могут
быть замещены молекулами ДМФ, ДМСО и т. д. Восстановление
FeOCl водным раствором FeS04 в присутствии хлоридов w-алкилам-
мония приводит к образованию серии соединений состава
(RNHg+^FeOCl)*-, в которых межслоевое пространство
увеличивается параллельно удлинению цепи алкила. Сообщалось и об
интеркаляции металлоценов в FeOCl, VOC1 и TiOCl (рис. 8.9). Более
легко восстанавливающийся хозяин FeOCl включает металлоцены
с более высокой энергией ионизации (например, ферроцен —
6,68 эВ), иллюстрируя соответствующий баланс восстановительных
свойств хозяина и гостя при образовании этих соединений.
432 Реакционная способность твердых тел
Диэлектрические листовые структуры. Несколько
неорганических твердых диэлектриков, обладающих листовыми структурами,
например силикаты, принадлежащие к семейству пирофиллита
[Thomas, 1982], и кислые фосфаты [Clearfield, 1981; Alberti, Constantino,
1982] некоторых четырехвалентных металлов образуют интеркалаты
со множеством донорных молекул; в этих случаях интеркаляция не
сопровождается редокс-процессом в отличие от халькогенидов и
оксогалогенидов переходных металлов, описанных в предыдущем
разделе.
Пирофиллиты являются типичными примерами листовых
силикатов, обладающих идеальной формулой Al2(Si4O10)(OH)2
(см. разд. 1.5). Их можно представить как продукты конденсации-
полимеризации листов А1(ОН)в (О) и тетраэдрических листов
Si203(OH)2 (T), причем порядок чередования слоев ТОТ. В этих
структурах возможны различные замещения Si4+ и А13+. Если
четверть Si4+ в пирофиллите замещена А13+ вместе с ионами К+ в
межслоевом пространстве для компенсации заряда, это приводит к
слюде мусковиту K[Al2(AlSi3O10)(OH)2]. В смектитах (монтмориллонит,
гекторит, вермикулит и т. д.) Li+, Mg2+ и Fe2+ (или Fe3+) замещают
А13+ в октаэдрических слоях, а А13+, в свою очередь, замещает Si4+
в тетраэдрических слоях. Общая формула монтмориллонитов
М;ЛА12_Х Mgx][Si4O10](OH)2, формула вермикулитов Mx(Mg)3X
X [(Al;x:Si4_JC)O10](OH)2, где М—одновалентный катион, способный к
обмену. Вот важные характеристики этих слоистых силикатов:
ионы М, занимающие межслоевое пространство между тетраэдрически-
ми слоями, являются способными к обмену, и основное количество
воды, присутствующее между слоями, может быть замещено (интер-
калировано) органическими молекулами. Смектиты и вермикулиты
обладают достаточно большими ионообменными емкостями, порядка
100 мг-экв на 100 г силиката. Важно, что органические молекулы,
интеркалированные в межслоевом пространстве этих силикатов,
претерпевают необычные химические реакции; чтобы реализовать
новые химические превращения органических молекул,
используется именно это свойство силикатов с листовой структурой.
Термическое разложение, димеризация, окисление — восстановление,
перенос водорода, лактонизация, образование простого и сложного эфи-
ров и конверсия первичных аминов во вторичные — вот некоторые
типы реакций, свободно протекающих в интеркалатах листовых
силикатов. Мы приведем несколько специфических примеров.
I. Конверсия бипротонированного 4—4'-диамино-гаранс-стиль-
бена в монтмориллоните в анилин при слабом нагреве интеркалата:
+ остаток пауглерожешюй глины.
Химия интеркаляции
433
II. Конверсия сложных эфиров в алкены и карбоновые кислоты
происходит гомогенно при 670 К. Но если реагент интеркалирован
в смектитах, реакция дает чистый продукт и идет при более низких
температурах. Например, карбоксилат метилциклогексана, интерка-
лированный в Al3f-замещенный монтмориллонит, на 98 %
превращается в циклогексен и уксусную кислоту при средних
температурах.
О1--
оси,—^-W Vch,cooh
III. Если 1, 1-дифенилэтилен нагревать с обратным
холодильником в присутствии Си2+-замещенного монтмориллонита, с хорошим
выходом получается 1-метил-1,3,3-трифенилиндан:
Ph2C =CPh2
но тот же самый 1,1-дифенилэтилен, реагируя в присутствии фтор-
гекторита, дает окисленные продукты — 1,1-дифенил этан и бензо-
фенон.
Важно провести различие между этими новыми реакциями,
идущими внутри индивидуальных листовых силикатов и включающими
интеркаляцию одного или более реагентов, и реакциями,
катализируемыми поверхностями глинистых минералов. В то время как
детали механизма реакций, включающих интеркаляцию в листовых
силикатах, не до конца понятны, в общем известно, что кислотные
центры по Бренстеду и Льюису, имеющиеся в листовых силикатах,
принимают участие в этих реакциях. Протонированные
промежуточные частицы, такие как карбониевые и оксониевые ионы, играют
решающую роль в таких реакциях.
Кислые соли четырехвалентных металлов с общей формулой
MIV(HX04)2-rcH20(MIV—Ti, Zr, Zn; X—P, As) — еще один важный
класс диэлектрических листовых материалов, образующих интер-
калаты. Эти соли можно получить в разных кристаллических
формах, обладающих волокнистой, слоистой или трехмерной
структурами. В свою очередь, слоистые кислые соли существуют в двух
различных модификациях: а и р. Структура а-формы типичной кислой
соли, Zr(HP04)2'H20, состоит из слоя атомов циркония,
расположенных, как в сэндвиче, между двумя листами фосфатных групп.
Три атома кислорода каждой Р04-группы так связаны с тремя
атомами циркония, что каждый атом циркония октаэдрически коорди-
434 Реакционная способность твердых тел
нирован шестью атомами кислорода шести разных фосфатных групп.
Четвертый атом кислорода каждой фосфатной группы несет
способный к замещению протон; он может полностью или частично
замещаться другими катионами. Таким образом составленные слои
упакованы друг над другом, образуя листовую структуру. Из-за
присутствия высокополярных групп -у Р—ОН эти материалы могут интер-
калировать ряд протонных акцепторов, таких как аммиак, амины и
спирты, благодаря их новым свойствам, таким как селективность по
определенным катионам, высокая устойчивость в сильнокислой и(или)
окислительной среде, высокая термическая стабильность и
устойчивость к облучению; кислые соли циркония фосфатного типа нашли
применение как неорганические ионообменники в особенности там,
где органические смолы не могут использоваться из-за их
нестойкости.
Каркасные матрицы. Сплавы редкоземельных и переходных
металлов, как LaNi5, туннельные структуры на основе W03 или
цеолитов — это те трехмерные матрицы, для которых интенсивно
исследуется интеркаляция. Обратимая интеркаляция атомов
водорода интерметаллидами с образованием таких тройных гидридов, как
LaNi5H6, TiFeH, ThFe3H5, привлекла внимание с точки зрения
потенциального использования этих твердых соединений для хранения
водорода (см. разд. 7.5). Включение доноров электронов (Li, H) в
трехмерные оксидные матрицы, такие как W03, V205 и т. д.,
технически важно как электродный процесс для твердотельных батарей
и электрохромных устройств.
Среди трехмерных каркасных матриц, образующих интеркалаты,
значительное внимание, из-за их технической важности, привлекли
цеолиты (см. разд. 1.5) [Derouane, 1982; Dyer, 1984]. Внутрикри-
сталлические полости в безводных цеолитах обеспечивают среду
с высокополярным окружением, которая может быть заполнена
полярными или ионными частицами с повышением энергии кристалла.
Обработка цеолитов (например, Na—Y-цеолита) парами натрия
приводит к образованию красного продукта, содержащего ионы Na^"1-,
интеркалированные в пустоты цеолита [Thomas, 1983].
Местонахождение этих кластеров внутри цеолита пока неизвестно, но они,
по-видимому, запрятаны в относительно недоступных пустотах
внутри цеолитов, и поэтому обычные растворители не проникают к ним.
Такая интеркаляция ионных частиц в цеолитах может
использоваться при переработке и хранении ядерных отходов.
Заключительные замечания. Короткий обзор интеркаляционной
химии, представленный здесь, показывает, что этот предмет
затрагивает замечательное разнообразие твердофазных реакций между дву-
или трехмерным материалом хозяина и атомами и молекулами гостя.
Не все материалы хозяина, проявляющие способность к интеркаля-
ции, затронуты в этом изложении. Например, мы не включили ин-
Реакции органических твердых тел
435
теркаляцию в Р-оксид алюминия и фазы Шевреля (см. разд. 1.10),
не обсуждались и реакции таких систем с линейными цепями, как
NbSe3 и AFeS2 с внедренными литием и аналогичными металлами.
Проблема интеркаляции привлекает широкое внимание в связи
с различными технически важными вопросами. Как уже
рассматривалось в гл. 7, ионный транспорт в натриевом ji-оксиде алюминия
привел к его использованию в качестве твердого электролита в
Na—S-батареях; обратимая интеркаляция лития в TiS2 нашла
применение в катодных процессах батарей с высокой плотностью энергии.
Водородные бронзы некоторых оксидов переходных металлов
полезны в системах безэмиссионных электрохромных дисплеев. Интерка-
латы графита важны для каталитических процессов; MoS2 и WS2,
интеркалированные Ni/Co,— основные катализаторы процессов гид-
родесульфуризации. Соединения включения TaS2 и фаз Шевреля
являются высокотемпературными сверхпроводниками.
8.8. РЕАКЦИИ ОРГАНИЧЕСКИХ ТВЕРДЫХ ТЕЛ
Химическая реакционная способность органических твердых
тел отличается от таковой неорганических и металлических твердых
тел по крайней мере в двух отношениях. В отличие от
металлических и неорганических кристаллов органические твердые тела
кристаллизуются в молекулярные структуры (см. гл. 1). Роль примесей
в реакционной способности также различна; точечные дефекты,
играющие важную роль в реакциях других классов твердых веществ,
не имеют такого значения в реакционной способности твердых
органических веществ, хотя и могут способствовать диффузии. Энергии,
требуемые для образования вакансий и междоузлий в органических
твердых телах, соизмеримы по величине с их энтальпией
сублимации, и поэтому концентрация точечных дефектов должна быть
ничтожной даже вблизи точки плавления. В реакционной способности
твердых органических тел более важную роль играют линейные и
планарные дефекты.
В их реакционной способности кристаллографический фактор
и особенности окружения более важны, чем истинная реакционная
способность молекулы. Давно признано, что реакционные
способности кристалла органического соединения и свободных молекул,
образующих этот кристалл, могут быть различны. Уже в 1889 г. Либер-
ман обнаружил, что трамс-коричная кислота димеризуется в твердой
фазе, хотя в растворе она всего лишь изомеризуется. За время
исследования реакций твердых органических тел в течение последних
двух десятилетий выяснились следующие факты [Schmidt, 1971;
Cohen, Green, 1973; Cohen, 1975; Morawetz, 1977; Thomas, 1974, 1979]:
(I) продукты реакций твердых органических веществ имеют меньшее
число разновидностей и часто отличаются от продуктов тех же
436 Реакционная способность твердых тел
реакций в гомогенной среде; (II) некоторые реакции протекают
гораздо быстрее в твердой фазе, чем в жидком состоянии, и (III) твердые
органические вещества постоянно кристаллизуются во многих
полиморфных разновидностях, и их реакционная способность
совершенно различна; если некоторые полиморфные модификации реагируют
с образованием определенных продуктов, другие не реагируют
вообще. На рис. 8.10 показаны поразительные примеры разницы в
реакционной способности между твердым состоянием и гомогенной
средой.
При отсутствии дефектов реакционная способность твердых
органических веществ в основном определяется молекулярной
упаковкой. Реакции, в которых кристалическая структура господствует
над собственной молекулярной реакционной способностью,
называются «топохимически контролируемыми» [Thomas, 1974].
Классическим примером топохимически контролируемой
органической реакции в твердой фазе является фотодимеризация транс-
коричных кислот, изученная Шмидтом с сотрудниками (см. [Gins-
burg, 1976]. Мономерные кислоты (например, о-этокси-гараис-корич-
ная кислота) кристаллизуются в трех полиморфных модификациях:
а, р и у. Во всех трех модификациях мономеры упакованы в
одномерные стопки, причем смежные стопки соединены водородными
связями. Три структуры отличаются друг от друга видом упаковки
и повторяющимся расстоянием между молекулами в стопке. а-Мо-
дификация, в которой молекулы размещены центросимметрично
о
с расстоянием между двойными связями около 4 А, дает центросим-
метричный димер (а-труксиновую кислоту), р-модификация, в
которой соседние молекулы трансляционно эквивалентны (зеркально
симметричное взаимоотношение), с расстоянием между двойными
о
связями в пределах 4 А, превращается в р-труксиновую кислоту.
В обоих случаях димеры уже заранее «предопределены» в
кристаллах мономеров, причем фотореакция всего лишь превращает
благоприятно ориентированные двойные связи в циклобутановые кольца
(рис. 8.11). у-Модификация, в которой молекулы не расположены
подходящим образом ни с точки зрения молекулярной ориентации,
ни с точки зрения необходимого расстояния между олефиновыми
двойными связями, устойчива к воздействию света. Как уже
отмечалось, если это соединение подвергать облучению в растворе или
расплаве, идет лишь только реакция транс-цис-изомершации без
димеризации.
Димеризация транс-коричных кислот — это далеко не
единственный пример топохимически контролируемых (2+2) реакций
фотосоединения дизамещенных этиленов в твердой фазе. Для
реакционной способности в фотореакциях требуется, чтобы двойные
связи в реагирующих кристаллах были параллельны и разделены
о
расстоянием ^4 А (см. далее); стереохимия фотопродуктов (про-
Реакции органических твердых тел
437
Рис. 8.10. Иллюстрация разницы в
реакционной способности органических
молекул в твердом состоянии и в
гомогенной среде [Thomas, 1979].
изводных циклобутана)
непосредственно отражает расположение
молекул в исходных кристаллах.
Другим важным примером (2+2)
фотоциклоприсоединения является
фотополимеризация 2, 5-дистирил-
пиразина (DSP) [Hasegawa etal.,
1968]. Молекулярная упаковка
в одной из модификаций DSP
(рис. 8.12) предпочтительна для
фотохимической полимеризации
через (2+2)-циклоприсоединение.
Эта реакция является топохимиче-
ски контролируемой, так как DSP,
выросший из паровой фазы и
кристаллизующийся в другой
структуре, не подвергается фотополимеризации. Кристаллографическое
исследование [Nakanishi, 1973, 1975] фотополимеризации DSP показало,
что и мономер, и полимер кристаллизуются в той же структуре
(пространственная группа РЬса). Полимер образует отдельную фазу
внутри кристалла мономера, обнаруживая трехмерные ориентацион-
ные взаимоотношения с исходной фазой.
Эта реакция использована [Nakanishi et al., 1973, 1975, 1980]
для получения широкого набора циклобутановых полимеров. Во
всех этих случаях реакция является топохимической в том смысле, что
продукт заранее предопределен в кристаллах мономера (с коллине-
арными реакционноспособными двойными связями на расстоянии
о
около 4 А), и фотополимеризация происходит посредством
последовательных реакций димеризации, образующих циклобутановые
кольца в качестве повторяющихся единиц. Термическая деполимеризация
также является топохимической реакцией. Электронная
микроскопия [Jones, Thomas, 1979] не дала доказательств гетерогенности
фотополимеризации DSP; нет указаний и на то, что дислокации
служат предпочтительными центрами инициации реакции. Л ахав
с сотрудниками использовал реакции такого типа для синтеза хираль-
ных полимеров [Addadi et al., 1980] (см. далее). (2+2)-Фотополиме-
ризация потенциально полезна в голографической интерферометрии
[Mizuno et al., 1977].
Важный аспект реакций органических веществ в твердом
состоянии касается такого обдуманного конструирования органических
29 ч. Н. Р. Рао, Дж. Гопалакришнан
438 Реакционная способность твердых тел
а»А Z " модификации
Рис. 8.11. Реакционная
способность в реакциях фотолиза
полиморфных модификаций
транс-формы коричной
кислоты.
ос
( ЦеНтросимметричнай упаковкаf
/3 ■* х
(
Зеркально-симметричная упаковка)
са„н
К —"■ Фотостабильна
(Упаковка, неблагоприятной
для димеризации)
молекул, чтобы они
упаковывались
специфическим способом,
обнаруживающим k предсказуемую
реакционную способность
в твердом состоянии
(техника организации
кристалла). Эта стратегия
включает выбор подходящих
систем и проведение
специфических замещений,
чтобы они кристаллизовались
в желаемые структуры.
Ключ к технике
организации кристалла лежит в
понимании природы
слабых межмолекулярных сил, контролирующих упаковку и структуру
твердых органических веществ. Например, известно, что
межмолекулярные несвязывающие взаимодействия, такие как С—О ... С1,
С=0 ... Ph или С1 ... С1, управляют упаковкой молекул
специфическим образом. Тот факт, что многие хлорзамещенные ароматические
о
соединения кристаллизуются с короткой (~ 4 А) осью элементарной
ячейки, используется в технике организации структур с
расстоянием между молекулами в ~ 4 А (ji-упаковка) (см., например, [Desi-
raju, 1984a]). Такие заместители, как гидрокси-, метил-, ацетокси- и
метоксигруппы, также исследовались с задачей выяснения
организации органических твердых тел. Оказывается, что метил- и хлорза-
местители способны к взаимной замене с образованием изоморфных
кристаллов [Jones et al., 1981]. По-видимому, ацетильные группы
предпочтительны для 0-у паковки, что отмечено для кумаринов [Gna-
naguru et al., 1985].
Интересный пример техники организации кристалла и топохи-
мической реакционной способности — производное 2-бензил-5-бен-
зилиден-циклопентанона (ВВСР), подвергающееся фотодимеризации
монокристалла в монокристалл [Jones et al., 1980]. Двойные связи
о
в кристалле мономера находятся на расстоянии, меньшем 4 А.
Существенно бездиффузионная природа реакции продемонстрирована
структурным анализом частично прореагировавших кристаллов
твердого раствора р-МеВВСР—р-С1ВВСР [Theocharis et al., 1984].
Реакции органических твердых тел
439
— вблизи у = о
- вблизи у = -
Рис. 8.12. Упаковка молекул дистирил-
пиразина в твердой фазе (проекция по
оси в), благоприятствующая
фотополимеризации [Jones, Thomas, 1979].
Расчеты с использованием атом-
атомных потенциалов помогли в
логическом обосновании
фотохимической активности производных
ВВСР [Thomas et al., 1985]. Это
приближение дало возможность
понять влияние заместителей на
способ молекулярной упаковки.
Кристаллическая структура а-бензили-
ден-у-бутилолактона была определена с помощью таких расчетов и
по наличию фотохимической активности [Kearsley, Desiraju, 1985].
Другим примером топохимической реакции с возможностью
обратимого фотохромизма является (2 + 2) фотодимеризация циннами-
лидин-цианоацетата [Guru Row et al., 1983].
Хотя обычно и считается, что надлежащим образом ориентиро-
u о
ванные двойные связи, находящиеся в пределах 4,2 А друг от друга,
очень важны для фотохимической активности, недавние
исследования [Gnanaguru et al., 1985] на замещенных кумаринах показали,
что фотодимеризация происходит даже в том случае, если активные
двойные связи ориентированы не параллельно и находятся друг от
о
друга на расстоянии, большем 4,2 А.
Полимеризация диацетилена (рис. 8.13), может быть, наиболее
ясный пример топохимического принципа. Показано [Wegner, 1971,
ХН
V
,сн7
\о
^
~\if
-НгС
^
#*'
n/L
г^
1,^
*H,<^L
^
г#°
,/CH^R
;/°H^R '
,/CH^R
hv
4¥V
с*
CH7
'\r
CH,
'\n
CH9
'\C
Рис. 8.13. Полимеризация диацетилена в твердой фазе.
29*
440 Реакционная способность твердых тел
1979], что мономеры диацетилена R—С==С—Cs=C—R в твердом
состоянии полимеризуются по реакции 1,4-присоединения у диаце-
тиленовой группы с образованием полимера, который может быть
изображен мезомерными структурами:
[ = (R)C-C=C-C(R) = ]n ~[-(R)C=C=C=C(R)-]n.
Бесцветные кристаллы мономера диацетилена можно
подвергнуть полимеризации при нагреве, облучении ультрафиолетом,
рентгеновскими или у-лучами с образованием интенсивно окрашенного
монокристаллического полиацетилена. Твердофазная реакция
превращает весь кристалл мономера в кристалл полимера без
разделения фаз; полимер образует с мономером твердый раствор во всей
области превращения мономер —►■ полимер. Например, если R—р-
толуолсульфонат, между мономером и полимером образуется
совершенный твердый раствор, и на скорости реакции проявляется яркий
автокаталитический эффект (при протекании реакции ее скорость
возрастает на два порядка). Этот автокатализ был отнесен к
влиянию напряжения на зарождение цепи и ее развитие [Baughman,
1978]. Недавние исследования указывают на важность локального
напряжения для отдельных ступеней, контролирующих развитие
цепи в полимеризации и диацетилена, и дистирилпиразииа [Nieder-
wald et al., 1983]. Интересно, что, если группу СН3 в р-толуолсуль-
фонате заменить на атэм хлора, диацетиленовый мономер не поли-
меризуется. Потерю химической активности относят за счет сильного
взаимодействия С1 и соседних фенильных колец, приводящего к
разделению потенциально активных атомов углерода.
До последнего времени механизм полимеризации диацетиленов
не был полностью установлен. Недавно показано [Gross, Sixl, 1982],
что реакция протекает по радикальному механизму, включающему
в качестве интермедиатов парамагнитные триплет (бирадикал)
и квинтет (диметилен). Один из важных выводов этого исследования:
оптическая полимеризация при низкой температуре дает гораздо
меньшую длину цепи (~10 ед.), чем термическая полимеризация
(~10 000 ед.). Вероятно, это связано с тем, что барьеры
потенциальной энергии, участвующие в обрыве цепи, могут быть преодолены
фотонами, но не термическим путем. Полидиацетилены имеют
несколько возможных приложений. Они обладают сильно анизотропными
оптическими свойствами; в соответствии с этим отражение света —
как от металла, если свет поляризован параллельно направлению
цепей, и голубое, если свет поляризован перпендикулярно цепям.
Степень полимеризации определяет цвет. Некоторые
полидиацетилены обнаруживают сильную генерацию второй гармоники.
Полидиацетилены могут быть внедрены в пленки Ленгмюра — Блоджетте
(см. разд. 7.8). Монокристаллы полидиацетилена, приготовленные
полимеризацией 1,6-ди (]Ч-карбозолил)2, 4-гексадина, демонстрируют
высокую степень внутреннего совершенства и обладают выдающи-
Реакции органических твердых тел 441
1984]. Полимеризация диаце-
Рис. 8.14. Упаковка в кристалле
несимметрично замещенных
диеновых молекул,
благоприятствующая фотополимеризации,
приводящей к асимметрическому
синтезу [Addadi et al., 1980].
мися механической
жесткостью и прочностью. Этот
материал имеет большие
возможности для
использования в качестве армирующих
волокон в композитах [Galiotis et al.
тиленов теоретически исследована на основе фрагментного
формализма [Burdett, 1980]. Результаты показывают, что реакция
разрешена фотохимически, но, кроме того, экспериментально она
проходит и термическим путем.
Систематические исследования топохимических реакций
органических твердых тел привели к возможности асимметрических
синтезов путем реакций в хиральных кристаллах. (В хиральном
кристалле элементы симметрии не соответствуют энантиомерам [Green
et al., 1979; Addadi et al., 19801.) Такой синтез, по существу,
является двухступенчатым: (I) синтез ахиральных молекул,
кристаллизующихся в хиральные структуры с подходящей упаковкой и
ориентацией активных групп и (II) проведение топохимической реакции так,
чтобы хиральность кристаллов была перенесена на продукты.
Первая ступень — неотъемлемая часть более общей проблемы
конструирования кристалла. Пример такой системы с почти достигнутой
количественной асимметрической индукцией,— семейство
несимметрично замещенных диенов:
CN
^C00R2
f^OOC
Q
Эта система была выбрана после тщательного рассмотрения
соотношения между молекулярной и кристаллической структурой:
известно, что симметрично дизамещенные производные бензола
кристаллизуются в структурах, которые подвергаются
фотополимеризации. Такие хиральные Г^-группы, как sec-бутил, индуцируют
кристаллизацию в хиральные пространственные группы: sec-бутиль-
ная группа также исключает упаковку, которая приводила бы
к реакции между эквивалентными двойными связями.
Неэквивалентность двойных связей обеспечена воздействием нитрила. Этиловый
эфир (R2 — этил, Rx — sec-бутил) диена кристаллизуется в
пространственной группе Р1, в которой молекулы несимметрично
замещенного диена упакованы таким образом (рис. 8.14), что две неэкви-
442 Реакционная способность твердых тел
Рис. 8.15. Реакция р— р'-диметил-
халькона в кристаллическом
состоянии, дающая неравные количества
энантиомеров дибромидов (вверху);
конформация р—р'-диметил хал ько-
на, дающая избыток энантиомера
дибромида (внизу) [Addadi et al.,
1980].
валентные двойные связи
параллельны друг другу на рас-
о
стоянии ^ 4 А вдоль
бесконечной стопки. Если это твердое
вещество облучать при 278 К
светом с длиной волны X >
>310 нм, то образуются диме-
ры, тримеры и олигомеры
(вплоть до молекулярной массы
10 000) с почти 100%-м
количественным выходом
энантиомеров.
Известно, что конформаци-
онно контролируемые реакции
газ —твердое с органическими твердыми соединениями приводят к
асимметрическим синтезам. Примером является присоединение брома
к нехиральным алкенам, кристаллизующимся в хиральных
пространственных группах [Addadi et al., 1980]. Предполагается, что
топохимически контролируемая реакция газ — твердое с
монокристаллом алкена даст энантиомерные дибромиды в неравных
количествах. Реакция брома с р, р'-диметил хал ьконом,
кристаллизующимся в пространственной группе Рг^^, действительно дает в
избытке один из энантиомеров (около 20 %) (рис. 8.15). Конформация
энантиомера р, р'-диметилхалькона, дающего избыток хирального
дибромида, показана на рис. 8.15, б [Rabinovich, Hope, 1975].
Установлено [Addadi et al., 1985], что хиральность может
быть индуцирована использованием селективных добавок,
модифицирующих морфологию кристалла. Так, если (R, S)-Tpeomm
кристаллизуется в присутствии примесей, легко получить избыток
энантиомера больше 95 %. Эта стратегия также дает средства для
определения абсолютной конфигурации молекул-добавок.
Одна из новых стратегий в органической химии твердого тела —
использование молекулярных комплексов внедрения типа хозяин —
гость некоторых стереоидов для проведения стереоспецифических
и зонноспецифических реакций [Addadi et al., 1980]. Деоксихолие-
вая (DCA) и апохолиевая кислоты (АРА) включают широкое
разнообразие молекул гостей, таких как углеводороды, спирты, эфиры
Реакции органических твердых тел
443
и кислоты. В этих соединениях включения только определенная
часть молекулы стероида взаимодействует с гостем таким образом,
что реакции являются селективными. Обычная структура комплекса
включения DCA — ромбическая (Р212121) и содержит каналы.
Размер и форма этих каналов согласованы с природой частиц гостя.
На рис. 8.16 показан типичный пример стереоспецифической реакции
с участием ди-£-бутил пероксомонокарбоната DCA. Фотолиз
комплекса дает два продукта; во время реакции один из перекисных
кислородных атомов эфира переходит в позицию 5 стероида. Топохи-
мическая природа реакции подтверждается таким наблюдением,
что эта реакция не происходит ни в растворе, ни в молекулярном
комплексе АРА, где иные размеры канала.
Анизотропную химическую активность органическим
кристаллам придает наличие полярной оси [Curtin, Paul, 1981; Desiraju,
1984b]. Обычно органические кристаллы принимают полярную (не-
центросимметричную) пространственную группу Р212121 и Р2Х.
В то время как хиральные кристаллы должны иметь полярные
направления, полярные кристаллы не нуждаются в хиральности.
Анизотропная реакционная способность видна, например, в реакции
аммиака с р-бромбензойным ангидридом, кристаллизующимся с
полярной осью; полярная ось и направляет реакцию.
р-Бромбензойный ангидрид является и хиральным, и
полярным. Хиральность кристаллического ангидрида использовалась
для разделения газообразного рацемического амина; хиральный
кристалл предпочтительно реагирует с одним из энантиомеров
амина. Таким образом, если кристаллы р-бромбензойного ангидрида
обрабатывать парами рацемического фенил этил амина,
получающийся амид содержит в избытке один из энантиомеров.
В топохимическом принципе предполагается, что
кристаллическая решетка обеспечивает колыбель для молекул,
препятствующую существенным изменениям в их конформации и
соответствующих положениях, но тем не менее позволяет молекуле нормально
вести себя в этой реакции. Недавние исследования [McBridge, 1983]
показали, что окружение в кристалле действует не только как
пассивная колыбель, но и обеспечивает заметное анизотропное
напряжение (эквивалентное десяткам тысяч атмосфер). Это напряжение
проявляет себя в необычной реакционной способности. Мы уже
ссылались на влияние напряжения, приводящего к
автокаталитическому эффекту в термической полимеризации диацетиленов. Прямое
доказательство наличия напряжения на молекулах в течение реакции
в твердом состоянии имеется в инфракрасных спектрах пероксида
ундеканоила. Хорошие спектры образцов, отожженных при высоких
температурах после фотолиза, имеют устойчивый сдвиг
асимметричного колебания в сторону низких частот. Сдвиг относят к
релаксации анизотропного локального напряжения величиной, возможно,
до ~40 кбар.
444 Реакционная способность твердых тел
со2н
ч-00-C-OO-H^t
Рис. 8.16. Реакция комплекса включения дезоксохолиевая
кислота (БСА)-ди-^-бутилпероксомонокарбонат, дающая стереоспеци-
фические продукты [Addadi et al., 1980].
Предшествующее обсуждение органических твердофазных
реакций может создать впечатление, что кристаллическая структура
является единственным фактором, контролирующим химическую
активность, а роль дефектов незначительна. Это не совсем верно,
поскольку существуют несколько реакций, в которых можно
натолкнуться на «неподходящие» продукты или «неправильную»
реакционную способность, причем ни то, ни другое не может быть объяснено
на основе структурных представлений [Thomas, 1979]. Так,
например, на основании структурного рассмотрения не следует ожидать
фотодимеризации антрацена или 1,8-дихлор-9-метилантрацена,
однако они димеризуются. Ожидают, что 9-цианоантрацен и 1,8-
дихлор-10-метилантрацен дадут ^мс-димеры, но они дают гпранс-
димеры. Возможно, что дефекты способствуют такой неожиданной
фотохимической активности этих твердых веществ. При учете роли
дефектов в реакционной способности следует различать случаи,
когда переход возбуждения в энергию является эффективным, а когда —
нет. В транс-коричных кислотах такой перенос неэффективен,
поэтому фотохимическая реакция происходит на центрах поглощения
фотонов. Структурные дефекты, следовательно, не важны. В
антрацене и его производных, где перенос энергии фотонов эффективен,
энергия возбуждения захватывается в дефектных позициях и
вызывает там реакцию.
В случае антрацена стабильная моноклинная фаза под влиянием
нагрузки переходит в триклинную фазу, в которой молекулы
ориентированы благоприятно для того, чтобы прошла димеризация. Хотя
триклинная фаза и не была выделена в качестве чистой фазы,
структура ее установлена при использовании низкотемпературной
электронной микроскопии и расчетов с помощью атом-атомных
потенциалов [Jones, Thomas, 1979]. В 1,8-дихлор-9-метилантрацене изо-
Гетерогенный катализ
445
[100] I
Рис. 8.17. I — проекция на плоскость а—с кристаллической
структуры 1, 8-дихлор-9-метилантрацена; II — расположение
1
молекул в этой же плоскости после трансляции (201) у [010],
переводящей соседние молекулы в геометрию, благоприятную
для образования mpawc-димера [Tnomas, 1974].
лированные дислокации с (201) -^ [010] трансляцией переводят
молекулы в необходимую форму (рис. 8.17), облегчая фотодимериза-
цию. Интересным примером является 1,5-дихлорантрацен. Вместо
ожидаемого 100%-го выхода димера «голова к голове»
фотохимическая реакция дает 80 % димера «голова к голове» и 20 % «голова
к хвосту». Для объяснения образования непредвиденного димера
«голова к хвосту» постулирован ориентационный дефект,
включающий поворот молекулы около одной из кристаллографических осей.
8.9. ГЕТЕРОГЕННЫЙ КАТАЛИЗ
Гетерогенный катализ — это область громадного
технологического значения, формирующая сущность нефтяной и химической
промышленности [Stinson, 1983]. Хотя в этой области и были
достигнуты впечатляющие успехи с точки зрения новых процессов и
новых катализаторов, мало известно о поверхностных реакциях на
молекулярном уровне. В последние годы стали доступны методы
исследования таких реакций; в характеризации катализаторов, кроме
методик по изучению поверхности, особенно полезны электронная
микроскопия и спектроскопия ЯМР [Thomas, Lambert, 1980; Rao,
1984; Rao, 1985] (см. также гл. 2). Информация, полученная этими
методами, имеет большое значение для гетерогенного катализа в по-
446 Реакционная способность твердых тел
нимании взаимоотношения между реакционной способностью
поверхности и ее структурой и составом. Вопреки успехам в методах харак-
теризации, мы все еще не способны поставлять «кулинарные рецепты»
синтеза катализаторов для специфических реакций; мы также не
можем полностью охарактеризовать поверхностные частицы,
участвующие в реакции. Отчасти эта проблема возникает потому, что в
большинстве своем катализаторы являются сложными,
поликристаллическими (или аморфными) твердыми веществами, в то время как
модельные исследования, как правило, проводятся на
идеализированных поверхностях.
Гетерогенный катализ — это огромная тема, и мы не собираемся
обсуждать ее здесь в деталях. Но, поскольку химия твердого тела
заметно содействует пониманию каталитических свойств твердых
веществ и созданию новых катализаторов, мы коротко и выборочно
обсудим несколько интересных в настоящее время гетерогенных
катализаторов одновременно с некоторыми основными идеями
гетерогенного катализа.
В качестве катализаторов используется множество твердых тел:
металлы, сплавы, глины, оксиды, сульфиды, нитриды, карбиды
металлов и т. д. Катализаторы могут быть однофазными веществами
или многофазными смесями; они могут быть кристаллическими,
микрокристаллическими или даже аморфными. Катализаторы могут
обладать диэлектрическими, полупроводниковыми или
металлическими свойствами. Несколько примеров гетерогенных
каталитических реакций приведено в табл. 8.1. Два аспекта катализа являются
важными: активность и селективность. Активность относится
к способности катализаторов ускорять химические реакции, так что
состояние равновесия быстро достигается. В некоторых случаях
степень ускорения может доходить до 1010 раз. Выразительная
иллюстрация каталитической активности — это реакция водорода и
кислорода в присутствии платины с образованием воды, идущая с
бурным взрывом; хотя реакция термодинамически разрешена, смесь
газов может храниться в отсутствие катализатора неопределенно
долгое время без взаимодействия. Селективность относится к
способности катализаторов направлять реакции в сторону получения
отдельных выбранных продуктов с исключением остальных. Примером
селективности является дегидроциклизация н-гептана в толуол.
Хотя м-гептан может подвергаться различным реакциям, таким как
гидрогенолиз, дегидрирование и изомеризация, платина селективно
катализирует реакцию дегидроциклизации. Другой пример — это
реакция пропилена с кислородом в присутствии молибдата висмута,
дающая акролеин; та же реакция в присутствии оксида висмута дает
бензол и гексадиен. Селективность в гетерогенном катализе была
предметом фарадеевской дискуссии химического Королевского
общества (Лондон, 1981). Стандартное определение катализатора:
вещество, которое ускоряет химическую реакцию, но само при этом
Гетерогенный катализ
447
Таблица 8.1
Некоторые примеры реакций, идущих при гетерогенном катализе, с указанием
катализаторов
Реакция
Крекинг углеводородов в присутствии
водорода
Гидродесульфуризация нефти
Получение метана из СО и Н2
Синтез аммиака
Синтез Фишера — Тропша
(гидрирование СО)
Окисление СО в автомобильных
отходящих газах
Полимеризация этилена и пропилена
Аммоокисление пропилена
Катализатор
Цеолиты
Кобальт и молибден на подложке из
оксида алюминия
Никель, нанесенный на оксид алюминия
Оксиды железа (II), алюминия и кальция
с оксидом калия в качестве промотора
Железо и кобальт, промотированные
К20, Th02
Платина или палладий на оксиде
алюминия
Галогениды триалкила алюминия и
титана (IV) (катализатор Циглера —
Натта)
Молибдаты висмута, антимонат уранила
не расходуется; тем не менее катализаторы, используемые в
коммерческих процессах, подвергаются изменениям и требуют
регенерации. Например, цеолиты, используемые как катализаторы при
крекинге нефти, имеют эффективное время жизни только несколько
секунд, но непрерывно регенерируются.
Гетерогенно катализируемая реакция включает несколько
стадий [Mady et al., 1976]: (I) массоперенос газообразных или жидких
реагентов к поверхности, (II) хемосорбция реагентов на
поверхности, (III) диффузия и химическая реакция на поверхности и (IV)
десорбция и диффузия продуктов от поверхности. Стадия III,
включающая образование поверхностных интермедиатов, является
ключевой. Образование поверхностных интермедиатов, которые в
конечном счете являются источником продуктов, впервые предположил
Себатье (см. [Burwell, 1973]) и убедительно доказали Сахтлер и Фа-
ренфорт [Sachtler, Fahrenfort, 1960] на реакции разложения
муравьиной кислоты, катализируемой различными переходными
металлами. Часто в гетерогенном катализе используется концепция так
называемых активных центров [Taylor, 1925]. Основная идея
состоит в том, что каталитическая активность твердого вещества
ограничена определенными специфическими центрами на поверхности.
Активные центры стали ассоциировать с поверхностными нерегуляр-
ностями или дефектами. В нескольких случаях было доказано
присутствие своеобразных атомных центров с низкой координацией или
с иной валентностью на тех поверхностях, которые активны в
химических реакциях. Таким образом, диссоциация водорода не требует
энергии активации, если происходит на ступеньках поверхности
448 Реакционная способность твердых тел
платины, однако определенная энергия активации необходима на
террасах той же поверхности [Salmeron et al., 1979]. На оксидных
поверхностях активными центрами могут быть точечные дефекты.
В катализаторах Циглера — Натта, применяемых в стереоспецифи-
ческой полимеризации пропилена, активными центрами являются те
позиции Ti3+ в a-TiCl3, которые имеют вакансии по хлору [Mady
et al., 1976]. Точно так же активными позициями в WS2,
используемого для гидрирования бензола, видимо, являются ионы W3+.
Одной из целей исследовательских работ в катализе является
установление факторов, способствующих каталитической активности
твердых тел. Среди разнообразных факторов обычно различают
геометрический и электронный. Согласно Баландину [Balandin,
1969], подчеркивающему роль геометрического фактора, активность
зависит от присутствия на твердой поверхности точно размещенных
рядов (мультиплетов) атомов, с которыми и взаимодействуют
молекулы реагента.
Доказательство важности геометрического фактора найдено,
хотя и в другом смысле, в исследованиях реакционной способности
поверхности платины [Somorajai, 1981]. Реакционной активности
способствовали поверхностные нерегулярности, такие как уступы
на гранях с высокими индексами монокристалла платины
(идентифицировано методом LEED). Для нескольких реакций с участием
углеводородов найдено, что поверхности с уступами и ступеньками
более активны, чем плоские грани (111). Реакции углеводородов
на поверхностях металлов бывают двух типов: или чувствительные
к изменению морфологии частиц катализатора, или нет. Образование
метана (ЗН2 + СО ->- СН4 + Н20) — пример
структурно-нечувствительной реакции, в то время как гидрогенолиз (С2Н6 + Н2 ->
->- 2СН4) — пример чувствительной реакции. Недавние работы
[Goodman, 1984] показали, что не только скорость реакции
образования метана независима от индекса Миллера (100) или (111)
монокристаллов никеля, но также что эти скорости одинаковы и для
монокристаллов, и для катализаторов с развитой поверхностью (Ni/Al203),
что указывает на нечувствительность реакции к поверхностной
структуре катализатора.
Электронный фактор в катализе относится к корреляции между
объемными электронными свойствами твердых веществ и их
каталитической активностью. Для катализаторов — переходных металлов
каталитическая активность коррелирует с процентом d-составляю-
щей в металлической связи [Pauling, 1949]. Обнаружена [Sinfelt,
1975] довольно хорошая корреляция между процентом d-составляю-
щей связи и активностью в гидрогенолизе для переходных металлов
второго и третьего ряда (рис. 8.18); однако наблюдается плохая
корреляция при сравнении активности элементов в различных рядах
переходных металлов (например, Pt и Ni). Сделан вывод, что, хотя
и существует некоторая корреляция активности в гидрогенолизе
Гетерогенный катализ
449
VTU Щ Ш2 Ш5 IB
Номер группы
Рис. 8.18. Корреляция между
каталитической активностью переходных
металлов в реакции гидрогенолиза
этана и процентом ^-составляющей
связи. Зачерненные кружки
обозначают активности, открытые кружки—
процент d-составляющей [Sinfelt,
1977].
4
~\ 1 1 1 r
B Y 1 УП УПТ1УШ?УПТл1В
Положение в Периодической
системе
Рис. 8.19. Каталитическая
активность сульфидов переходных
металлов при гидродесульфуризации ди-
бензотиофена [Whittingham, Ghianel-
ly, 1980].
с процентом d-составляющей связи, одного этого параметра
недостаточно для оценок.
Было много попыток соотнести объемные электронные свойства,
полупроводниковых оксидов с их каталитической активностью.
Электронная теория катализа оксидами металлов [Wolkenstein,
1960; Hauffe, 1966; Krylov, 1970] базируется на той идее, что хемо-
сорбция таких газов, как СО и N20, на полупроводниковых оксидах
связана с электронным переносом, приводящим к изменению
электрон-транспортных свойств твердого оксида. Например, при
окислении СО на ZnO обнаружена корреляция между концентрацией
носителей заряда и скоростью реакции [Chon, Prater, 1966].
Есть сообщение о следующих корреляциях каталитической
активности с разными объемными свойствами полупроводников: (I)
средняя ширина запрещенной зоны у полупроводников A(III)—B(V)
и А(П)—B(VI) и их активность в гидрировании изопропанола; (II)
энтальпия образования оксидов и их активность в окислении
пропилена и (III) количество d-электронов (и энергия стабилизации
450 Реакционная способность твердых тел
кристаллическим полем) у оксидов Sd-металлов и их активность при
разложении N20. Последняя корреляция [Dowden, 1972] особенно
важна, так как она обеспечивает связь между гетерогенным
катализом и химией координационных соединений переходных металлов.
Найдена корреляция каталитической активности сульфидов
переходных металлов в реакции гидродесульфуризации ароматических
соединений с положением переходного металла в Периодической
таблице [Whittingham, Chianelli, 1980] (рис. 8.19). Наиболее
активными являются сульфиды металлов, содержащие максимальное
количество d-электронов. При исследовании сульфидных катализаторов
гидродесульфуризации, содержащих Со, Мо и А1203, методами XPS
и рентгеновской абсорбционной спектроскопии показано, что на
поверхности существуют октаэдрически координированные Со (II)
и Мо (IV); поверхностные частицы, по-видимому, имеют избыток
серы, возможно, за счет дисульфидных мостиков [Sankar
et al., 1984].
Несмотря на различные попытки, не найдено никакой единой
универсальной корреляции между объемными свойствами и
каталитической активностью твердых тел. Теперь ясно к тому же, что
геометрический и электронный факторы не могут быть отделены друг
от друга и что для достижения успеха в понимании проблем
гетерогенного катализа каталитическая активность должна
рассматриваться одновременно с каталитической селективностью [Sachtler, 1981].
Некоторые важные катализаторы. Переходные металлы
используются как катализаторы разнообразных реакций:
гидрирования, гидрогенолиза и изомеризации углеводородов (металлы VIII
группы), окисления алкенов (Ag), синтеза аммиака (Fe, Ru, Os),
синтезов по Фишеру — Тропшу (Со, Fe) и т. д. [Sinfelt, 1975].
Платина является одним из самых разносторонних катализаторов,
применяемых и в окислительных, и в восстановительных условиях,
поскольку она катализирует дегидроциклизацию, изомеризацию и
гидрирование углеводородов, окисление NH3 и СО, а также пригодна
как электрокатализатор в электрохимических ячейках.
Металлические катализаторы, как правило, диспергируют в виде мелких
частиц на подложке — носителе, таких как Si02 и А1203. Такое
диспергирование обеспечивает доступность для реакции большой
поверхности катализатора. Вариация металлического катализатора
достигается распылением двух или более металлов на ту же самую
подложку (биметаллические катализаторы) [Sinfelt, 1975, 1977].
Типичными примерами биметаллических катализаторов являются Pt—Re
и Pt—Ir. распыленные на оксиде алюминия и применяемые как
катализаторы в реформинге углеводородов (реакция, используемая
для увеличения октанового числа бензина). Следует отличать
биметаллические катализаторы от более распространенных
катализаторов-сплавов, потому что первые состоят из кластеров только
немногих атомов обоих металлов, тонко диспергированных на носитель;
Гетерогенный катализ
451
Рис. 8.20. Активность сплавов Ni — Си
при дегидрогенизации циклогексана п ~
гидрогенолизе этана [Sinfelt, 1977].
юЧ
■10*
>ю4-
\юЛ-\
•с
4
Дегидроеенизаи, ия
\
х^Ъдрогенолиз этана
О 20 40 60
Медь, атомные %
-у—
80
100
более того, такие биметаллические
катализаторы содержат сочетания
металлов, которые обычно не
образуют сплавов в объеме.
Сплавы металлов уже давно
исследуются как катализаторы
для понимания электронного
фактора в катализе. Раньше думали,
что каталитические свойства
систематически зависят от состава
сплава. Однако это оказалось
совсем не так, поскольку было
найдено, что во многих случаях состав поверхности сплава отличен
от состава его объема. Важным аспектом катализа сплавами
является специфичность по отношению к некоторым реакциям. Так,
например, активность никеля в гидрогенолизе этана заметно
снижается при его сплавлении с медью, хотя по отношению к
дегидрированию циклогексана активность остается постоянной в широком
интервале объемного состава сплавов Ni—Си (рис. 8.20) [Sinfeltt
1977]. Разница объяснима с точки зрения состава поверхности
сплавов. Гидрогенолиз этана на никеле идет на несколько
порядков быстрее, чем на меди. Поскольку поверхность сплавов Ni—Си
обогащена медью по сравнению с объемом, снижение
каталитической активности при увеличении концентрации меди совершенно
понятно.
Аморфные (быстро закаленные) металлы и сплавы также
исследовались как катализаторы [Schlogl, 1985; Kisfalndi, et al., 1985].
Было найдено, что адсорбционные характеристики монооксида
углерода на металлических стеклах отличаются от таких характеристик
для кристаллических материалов. Найдено, например, что СО
быстро диссоциирует на поверхности металлического стекла Ni76B12Si2,
но всегда молекулярно сорбируется на металлическом никеле [РгаЬ-
hakaran, Rao, 1985].
Сегодня катализ металлокластерными соединениями — область
большого интереса [Lewis, Green, 1982; Haggin, 1982]. Кластеры
металлов (см. разд. 6.6), содержащие агрегаты атомов металла
окруженные лигандами, представляют собой модельные системы,
имитирующие поверхностные интермедиа™, образованные хемо-
сорбцией молекул на металлических поверхностях [Muettertieset al.,
1979]. Примером кластерного катализатора является СКч3(СО)12,
который оказался активным в гидрировании СО с получением алкенов
и алканов. Кластерные катализаторы на основе осмия проявляли
452 Реакционная способность твердых тел
заметную селективность при образовании С2, и распределение
продуктов было почти независимо от отношения Н2/СО в исходной
реакционной смеси. Кластеры металлов могут быть рассмотрены как
промежуточное вещество между моноядерными комплексами
металла и маленькими частицами металла, диспергированными на
инертных носителях.
Хотя обычно считается, что нет взаимодействия между
металлами и подложкой в катализаторах на носителях, появляется все
больше доказательств, что носитель во многих случаях может влиять
на каталитические реакции [Sleight, 1980]. Одним из таких
примеров является спилловер адсорбированных частиц с металла на
носитель. Водород, адсорбированный на металлах (например, Pt),
подвергается диссоциации и «перетекает» к носителю (например, А1203
или Si02), где реагирует с адсорбированными там органическими
молекулами [Sermon, Bond, 1974]. Более важно то, что
каталитические свойства металлов могут необыкновенно измениться при
взаимодействии с определенными носителями. Сильное взаимодействие
металл — носитель (SMSI) впервые замечено у металлов VIII
группы, нанесенных на Ti02 [Tauster et al., 1978]. Основные
особенности SMSI — это подавление адсорбции Н2 и СО и коренные
изменения в активности и селективности таких реакций, как
гидрирование СО. Так, в реакции образования метана катализатор
Pt/Ti02, восстановленный выше 750 К, в 10 раз активнее, чем
Pt/Al203, и в 100 раз активнее, чем Pt/Si02. Катализаторы SMSI
проявляют большую активность и селективность в синтезах
Фишера — Тропша (СО—Н2); вероятно, такое поведение SMSI-катали-
заторов обязано необычным сорбционным характеристикам СО и Н2.
Для понимания механизма SMSI были предприняты
значительные усилия. Экспериментальное исследование SMSI-катализаторов
обнаружило заметные морфологические изменения,
сопровождающиеся восстановлением оксида-носителя; в катализаторе Pt/Ti02
методом диффракции электронов было доказано образование Ti407.
Для объяснения эффекта SMSI предложена модель с переносом
заряда, основанная на расчетах молекулярных орбиталей методом Ха
[Horsley, 1979]. Взаимодействие, по-видимому, появляется за счет
переноса заряда от восстановленного катиона (Ti3+) к соседнему
атому металла. Исследования Pt/Ti02 и Pt/SrTi03 [Bahl et al., 1980]
с помощью XPS дают доказательства такого переноса заряда.
В обоих случаях химический сдвиг 4/ остовного уровня Pt
указывает на значительный перенос отрицательного заряда к атомам
платины. Недавние работы показали, что SMSI обусловлен диффузией
оксида титана к поверхности металла [Santoe et al., 1983; Jiang
et al., 1983]. Исследование SMSI в Ni/Ti02 методами AES и EELS
[Takatani, Chung, 1984] обнаружило, что восстановление водородом
при 700 К никелевой пленки, нанесенной на Ti02, приводит к
выделению TiOj,. (х ~ 1,0) на поверхность никеля; по-видимому, вое-
Гетерогенный катализ
453
становленный оксид титана быстро диффундирует сквозь никель.
Представляется, что и физическое заполнение поверхности никеля
ТЮд, и химическое взаимодействие никеля с восстановленным
оксидом титана на поверхности содействуют SMSI-поведению. О подобной
миграции оксида титана к металлической поверхности сообщалось
и для других переходных металлов [Ко, Gorte, 1984]. SMSI
относится не только к ТЮ2-носителям. О нем сообщалось и для
носителей из оксидов других переходных металлов, таких как Nb205
и Та205; по-видимому, существует корреляция между SMSI и
способностью к восстановлению оксидных носителей. Хотя пока можно
лишь ожидать полного понимания SMSI, нет сомнений, что SMSI-
катализаторы представляют собой одну из важных особенностей
гетерогенного катализа, имея дальним логическим следствием наше
понимание каталитического поведения переходных металлов.
Среди разных оксидных катализаторов значительное внимание
привлечено к молибдатам висмута, катализирующим селективное
окисление и аммоокисление пропилена с получением акролеина
и акрилонитрила [Grasselli, Burrington, 1981]:
СН3СН=СН2 + 02-> СН2-СНСНО + Н20,
СН3СН=СН2 + NH3 + | 02->CH2=CHCN + 3H20.
Обе реакции включают образование интермедиата аллила
(СН2—СН—СН2). Для проявления каталитической активности в этой
реакции требуется последовательное восстановление и окисление
катализатора. При использовании в качестве катализатора молиб-
датов висмута хемисорбированный углеводород окисляется
решеточным кислородом; активные центры воспроизводятся быстрой
диффузией решеточных кислородов из объема к поверхности
катализатора. Каталитический цикл завершается восстановлением
газообразного кислорода и включением его в решетку в форме анионов.
Эксперименты с 180 показали, что кислород в продуктах реакции
происходит непосредственно из катализатора; поскольку решеточный
кислород легкодоступен, скорость окисления пропилена не зависима
от парциального давления кислорода. Предположено [Sleight, 1977],
что восстановление происходит на молибденовых центрах, а реокис-
ление — на висмутовых. Вырождение (смешение) уровней Mo(4d)
и Bi(6p) в катализаторе позволяет электронам, добавившимся к
уровню Mo(4d) во время восстановления, стать доступными на
висмутовых центрах для реокисления. Другим требованием к селективности
является то, что структура должна быть приспособленной к легкому
удалению и присоединению кислорода.
Координационно-ненасыщенный молибден на поверхности, подвергающийся легкому
взаимному переходу между октаэдрической и тетраэдрической
координацией, и обеспечивает, по-видимому, селективность катализаторов
на основе молибдатов висмута [Grasselli et alM 1981]. Из трех молиб-
454 Реакционная способность твердых тел
датов висмута Bi2Mo3012 обладает дефектной шеелитовой структурой
(с тетраэдрически координированным молибденом), а 7"Г^2Мо06
кристаллизуется в слоистой структуре с октаэдрически
координированным молибденом. Структура Bi2Mo209 точно не известна, но,
возможно, относится к шеелитам. Селективность и активность
катализаторов по отношению к реакции окисления алкенов, по
существу, постоянна при их составах от Bi2MoOl2 до Bi2MoOe. Возможно,
что координация молибдена на поверхности катализатора иная,
чем в его объеме.
Цеолиты. Может быть, наиболее важными оксидными
катализаторами являются цеолиты, широко применяемые в нефтяной и
химической промышленности для каталитического крекинга и
гидрокрекинга, изомеризации алкенов и алканов и ароматического ал кил и-
рования [Rabo, 1976; Dwyer, 1984; 1. Наиболее замечательным
каталитическим свойством цеолитов является их селективность к форме;
например, начиная с таких разнообразных реагентов, как метанол,
этанол, гексадекан, глицериды, ненасыщенные длинноцепочечные
спирты и кислоты, некоторые цеолиты могут вырабатывать
относительно узкий спектр углеводородов промежуточного размера
(алифатические — с максимальным выходом С3 и С4; ароматические —
в интервале от Сб до С10), состав которых близок к составу бензина.
Цеолиты — это алюмосиликаты, содержащие каналы и клетки
молекулярных размеров (см. разд. 1.5). Мы обсудили структуру
цеолитов, полученных из содалитовых ячеек (см. рис. 1.13),
обладающую большими полостями (клетками), соединенными порами
разного размера. В семействе так называемого пентасила — цеолитов
с высоким содержанием оксида кремния (ZSM-5 и ZSM-11) (см.
рис. 1.14) структура пор является уникальной, состоящей из
пересекающихся каналов. Селективный к форме катализ цеолитами
зависит от структуры пор. Размер самых маленьких пор в цеолитах
о о
около 2,6 А, и самых больших около 7,4 А. Величина отверстий пор
определяется свободным отверстием кислородного кольца,
ограничивающего пору. Максимальные величины отверстий пор 2.6, 3.6,
о
4.2, 6.3 и 7,4 А для 4-, 6-, 8-, 10- и 12-членных колец соответственно.
Гидроксильные группы, связанные с атомами алюминия,
являются активными центрами в катализе цеолитами. Они легко
образуются деамминированием аммоний-замещенных цеолитов при
нагревании вблизи 670 К. Если цеолиты нагревать при более высоких
температурах (>800 К), кислотные центры по Бренстеду
превращаются в кислотные центры по Льюису за счет потери Н20.
Поскольку цеолиты легко синтезировать с широким интервалом отношения
Si/Al, можно контролируемым способом варьировать концентрацию
активных центров. В недавнем исследовании активных центров
в кислотных катализаторах на базе ZSM-5 было показано [Hagg
et al., 1984], что активность по отношению к крекингу л-гексана
Гетерогенный катализ
455
2 4 6 8 Ю
п в СпН2п«-2
1982], которое показало,
Рис. 8.21. Соотношение между диффузией
и селективным к форме катализом
цеолитами [Gorring, 1973].
а — коэффициенты диффузии н-ларафинов в
калиевой форме Т-цеолита; б — распределение
продуктов при крекинге н-трикозана на н-прионите.
линейно зависит от содержания
алюминия и при экстраполяции на
нулевое содержание алюминия
достигается пулевая активность.
Такая количественная корреляция
объяснима, только если все атомы
алюминия находятся в тетраэдри-
ческих позициях. Это заключение
согласуется с исследованием
методом ЯМР под магическим углом
высокого разрешения [Fyfe el al.
что весь алюминий, захваченный во время синтезов ZSM-5, даже на
самом низком уровне концентрации идет в тетраэдрические позиции.
Такие корреляции между деталью структуры и каталитической
активностью очень многозначительны.
Диффузия молекул реагента через внутрикристаллические поры
является важной предварительной необходимостью для адсорбции
и селективного к форме катализа [Rees, 1984]. В отличие от
классической и кнудсеновской диффузии в цеолитах мы встречаемся
с новым типом диффузионного явления, называемого
конфигурационной диффузией. Диффузия здесь контролируется соответствием
в размере, форме и конфигурации диффундирующих частиц
аналогичным параметрам внутри решетки хозяина. Конфигурационная
диффузия создает основу селективного к форме катализа цеолитами.
Соотношение между диффузией и селективностью к форме
иллюстрируется на рис. 8.21, где коэффициенты диффузии к-алканов С3—С14
при 610 К в К-форме Т-цеолита сравниваются с распределением
продуктов крекинга, полученных из /t-трикозана на Н-эрионите
при той же температуре. Эти два цеолита структурно сопоставимы.
Как очень маленькие (вплоть до С6), так и очень большие (н-С9 —
н-С12) молекулы диффундируют через цеолит быстрее всего, в то
время как молекулы промежуточного размера (к-гептан и н-октан),
о о
чья длина (11,56 и 12,82 А) близко соответствует длине (13 А)
клетки эрионита, захватываются и поэтому имеют низкую
диффузионную способность. Такой близкий параллелизм между распределением
продуктов крекинга к-трикозана и данными по диффузии указывает
на взаимоотношение между диффузией и селективностью к форме
[Derouane, 19821.
Селективный к форме катализ в цеолите требует, чтобы реагенты
диффундировали внутрь к активным центрам, локализованным
456 Реакционная способность твердых тел
Таблица 8.2
Проценты распределения продуктов при
конверсии спиртов * на цеолите ZSM-5 при 644 К
Продукт
Метан
Этан
Этилен
Пропан
Пропилен
i-Бутан
«-Бутан
Бутены
МТентан
и-Пентан
Пентены
С6+ (алифатич.)
Бензол
Толуол
Этиленбензол
Ксилолы
С9 (ароматич.)
С10 (ароматич.)
С11+ (ароматич.)
Метанол
i,o !
0,6
0,5
16,2
1,0
18,7
5,6
1,3
7,8
1,3
0,5
4,3
1,7
10,5
0,8
17,2
7,5
3,3
0,2
Реагент
f-Бутанол
од
0,7
0,5
18,8
1,1
18,4
8,7
0,7
6,2
1,4
0,2
7,6
3,3
11,6
1,3
12,4
6,1
0,4
0,6
i-Гептанол
0,0
0,3
<0,1
16,4
0,2
19,3
11,0
<0,1
8,7
1,5
0,1
3,0
3,4
14,3
1,2
11,6
5,3
2,9
0,6
* Но Chang, Silvrstri [1077].
во внутрикриоталлическом объеме (в порах), и чтобы продукты после
реакции диффундировали в противоположном направлении. На
активных центрах наличие высокого локального электрического поля
может направлять реакцию в соответствии со стерическими
требованиями с образованием специфических продуктов. Таким образом,
селективность к форме может достигаться в силу геометрических
факторов, кулоновского поля на активных центрах и/или разницы
в скоростях диффузии. Соответственно различают три вида
селективности к форме [Dwyer, 1984]. Если геометрические факторы
таковы, что только определенные молекулы реагента могут
диффундировать сквозь поры, достигается селективность по реагенту.
Если же геометрические ограничения относятся к диффузии
молекул продукта, селективность называется селективностью по
продукту. Если пространственная конфигурация вокруг активного
центра такова, что она позволяет образовать только один тип интер-
медиата, исключая все возможные другие, то достигается
селективность по переходному состоянию. Так, о-ксилол преимущественно
Введение
457
подвергается изомеризации, а не диспропорционированию в таких
цеолитах, как ZSM-5 и морденит, где полости чересчур малы для
размещения относительно большого диарильного интермедиата,
участвующего в реакции диспропорционирования:
Особенно замечательна селективность к форме H+ZSM-5.
Активные центры на внутренних стенках катализатора легко передают
протоны органическим молекулам реагента, с образованием карбо-
ниевых ионов, которые, в свою очередь, через потерю воды и ряд
ступеней образования С—С-связей образуют смесь углеводородов,
аналогичную бензину. Начальными продуктами могут быть метанол,
этанол, кукурузное масло или масло jojoba. Поразительна
селективность к ферме этого катализатора, как можно увидеть из
распределения продуктов, полученных при дегидратации трех разных
спиртов (табл. 8.2). Такое распределение продуктов можно понять
в рамках представлений о промежуточном размере пор у ZSM-5,
в которых могут размещаться как линейные алканы и изоалканы,
так и моноциклические ароматические углеводороды размером
меньше 1, 3, 5-триметилбензола.
9. ХИМИЯ ВЫСОКОТЕМПЕРАТУРНЫХ
СВЕРХПРОВОДНИКОВ
9.1. ВВЕДЕНИЕ
До недавнего времени бытовали неутешительные прогнозы,
что температура сверхпроводящего перехода, возможно, никогда
не поднимется выше 23 К (такую температуру перехода показывают
соединения А 15, такие как Nb3Ge), несмотря на огромные усилия
ученых в течение семи десятков лет. Однако ситуация в конце 1986 г.
разительно изменилась, когда Беднорц и Мюллер IBednorz, Mul-
ler, 1986] показали возможность сверхпроводимости около 30 К
в системе La—Ва—Си—О. После этого был достигнут большой
30 ч. Н. Р. Рао, Дж. Гопалакришнан
458 Химия высокотемпературных сверхпроводников
прогресс в исследовании оксидных сверхпроводников, из которых
многие показывают сверхпроводимость при температурах, заметно
выше температуры жидкого азота (Тс ж 80—120 К). Химия твердого
тела оксидов металлов приобрела важное значение из-за этих
высокотемпературных сверхпроводников. Сегодня уже становится
понятно, что в поЕСках стабильных материалов с высокими
температурами перехода специалисты в области химии твердого тела будут
играть решающую роль.
Сверхпроводимость в оксидах металлов известна давно. Когда
SrTiOg восстанавливают или допируют ниобием, он показывает
сверхпроводимость при 0,2 К. Оксидные бронзы типа К^ЛУОз, где
К находится в гексагональных туннелях, становятся
сверхпроводящими в области 1—4 К. BaPb^^Bi^Og со структурой перовскита
становится сверхпроводником при 0,05 <С х ^ 0,3 с Тс в области
9—13 К [Sleight et al., 1975]; здесь Bi имеет номинально смешанную
валентность (+3, +5). Шпинель Li1+:x.Ti2_x04 показывает
сверхпроводимость в такой же температурной области [Johnston et al.,
1973]. Все новые семейства оксидов, показывающих
высокотемпературную сверхпроводимость, являются сложными оксидами меди,
производными перовскитовой структуры с номинальной смешанной
валентностью Си. До настоящего времени в основном три класса
оксидов меди показывают высокие Тс: 1) оксиды со структурой
K2NiF4 типа La2_a:MJCCu04 (М — Sr, Ba); 2) дефектные перовскиты
типа LnBa2Cu307 (Ln — Y или РЗЭ) и 3) оксиды, родственные
оксидам семейства Ауривиллиуса, образованные Bi и Ti. В этом
разделе мы коротко обсудим химию высокотемпературных оксидных
сверхпроводников, выдвигая на первый план роль структуры,
стехиометрии и химии кислорода. Недавно опубликованы некоторые
обзоры по химии твердого тела оксидных проводников [Nelson
et al., 1987; Rao, 1988a, b; Williams et al., 1987], однако эта тема
развивается так быстро, что трудно поспевать за ежедневными
новыми результатами. Есть надежда, что специалисты в области химии
твердого тела откроют новые и более лучшие материалы (возможно,
без меди или даже без кислорода), демонстрирующие стабильную
высокотемпературную сверхпроводимость.
9.2. КУПРАТЫ СО СТРУКТУРОЙ K2NiF4
La2Cu04 и другие сверхпроводящие оксиды, производные
от него, такие как La^xSr^Ba^CuO^ обладают структурой K2NiF4,
где ионы переходного металла взаимодействуют только в
плоскости ab. Химия твердого тела оксидов со структурой K2NiF4 была
подробно обсуждена [Ganguly, Rao, 1984]. Эти квазидвумерные
оксиды меди являются производными от перовскитовой структуры,
Купраты со структурой K2NiF4 459
^la(Sr.Ba)
Рис. 9.1. Структура сверхпроводящего
La .Sr (Ba^CuC^ при комнатной температуре
2"Л Х [Wang et al., 1987].
содержащей группы Cu04, связанные
вершинами. В ромбическом La2Cu04
окружение иона меди — тетрагонально-удлиненное
вследствие искажения Яна—Теллера и
поэтому имеется четыре короткие связи
Си—О и две более длинные аксиальные
связи Си—О. La2Cu04 с избытком
кислорода или дефицитом La, содержащий Си
номинально в смешанном валентном
состоянии (2 +, 3+), показывает
сверхпроводимость в области 20—40 К [Beille et
al., 1987; Fine et al., 1987]. В отличие от
La2Cu04, который является ромбическим,
Pr2Cu04 и Nd2Cu04 — тетрагональные,
обладающие совершенно другими
структурами, содержащими плоские анионные
слои (Cu02)2~, крестообразно связанные
слоями (Nd202)2+; эти оксиды не
являются сверхпроводящими. La2Cu04, ромбический при комнатной
температуре, становится тетрагональным около 500 К.
Частичное замещение La в La2Cu04 на Ca, Sr или Ва
стабилизирует тетрагональную структуру при комнатной температуре, когда
х в La2_:x.Sr^(Baa., Cax)Cu04 больше, чем ~ 0,05. Эти вещества
содержат вытянутые октаэдры CuOe, подобно La2Cu04, и сохраняют
квазидвумерный характер. На рис. 9.1 показана структура
La2_;tSr:;c(BaJCu04. Короткое расстояние Си—О находится вблизи
о о
1,9 А, в то время как длинное (аксиальное) расстояние равно ~2,4А.
Тетрагональная структура оксидов La2_:cSr3C(Ba:c)Cu04 переходит
в ромбическую вблизи 180 К [Day et al., 1987] и становится
сверхпроводящей в области 20—40 К в зависимости от состава [Cava
et al., 1987a; Chu et al., 1987a; Mohan Ram et al., 1987a, Uchida
et al., 1987]. Сообщалось, что в этих оксидах эффект Мейсснера
достигает 70 %. Замещение стронция кальцием снижает Гс,
указывая, что факторы, управляющие Гс, являются более сложным, чем
только один размер щелочноземельного заместителя. Ромбическое
искажение в этих оксидах не показывает никакого резкого
изменения при Тс. Однако в любой данной серии La2_;3CM:)cCu04(M—Ca, Sr
или Ва) Тс является максимальной при особом значении х (например,
0,15 в случае Ва). Параметры сна также показывают некоторую
систематику в зависимости от х или Тс.
30*
460 Химия высокотемпературных сверхпроводников
Частичное замещение (<Ю %) La в La2_xSr:x.(Ba:c)Cu04 ионами
редкоземельных элементов, такими как Рг, Nd, Gd и Ей, может быть
проведено без потери сверхпроводимости, но Тс понижается.
Структурные черты этих оксидов должны быть почти такими же, как и
структурные характеристики La2_JCSra.(Ba:x)Cu04. Частичное
замещение Си никелем заметно снижает Гс, а при ~5 %-ном замещении
сверхпроводимость разрушается; замещение цинком имеет
подобный эффект, хотя размеры ионов Ni2+ и Zn2+ отличаются от ионов
Си2+ в разных направлениях [Mohan Ram et al., 1987; Tarascon
et al., 1987a]. Заслуживает внимания факт, что эти
сверхпроводящие оксиды являются фактически пограничными на переходе
металл — диэлектрик и их сопротивление при комнатной
температуре соответствует минимальной металлической проводимости по
Мотту.
9.3. ДЕФЕКТНЫЕ ПЕРОВСКИТЫ В СИСТЕМЕ La—Ва—Си—О
Поучительно исследовать структуры некоторых других
родственных перовскитам оксидов системы La—Ва—Си—О, имеющих
интересные особенности. Тетрагональный оксид La4BaCii5013+6 со
смешанной валентностью является дефектным перовскитом,
построенным из октаэдров СиОб и пирамид Cu05 [Michel et al., 1985].
В этом оксиде имеются кислородные вакансии вдоль [001]
кубической перовскитовой ячейки, и получающийся каркас образует
гексагональные туннели, где локализуются ионы La и Ва; расстоя-
о
ния Си—О находятся между 1,85 и 2,30 А, и оксид остается
металлическим от жидкого гелия до комнатной температуры. При
удалении некоторого количества кислорода из La4BaCu5013+6 мы можем
сделать его полупроводником; то же самое также достигается
варьированием отношения La/Ba. Эта оксидная система показывает
только такие контролируемые составом переходы металл— диэлектрик,
а не сверхпроводимость.
Другой интересный тетрагональный перовскит с дефицитом
кислорода в системе La—Ва—Си—О — La3Ba3Cu6014+6, который
является полупроводником [Er-Rakho et al., 1981].
Предполагалось, что структура этого оксида включает такую
последовательность: Cu204, AxOn, Cu204, А2П2» Cu204, А2Оп, где □ — вакансия
кислорода вместо последовательности АО, Cu02, вдоль оси а, как
в кубическом перовските; около 22% анионных позиций не заняты,
что разрешает интеркаляцию кислорода без изменения структуры.
Структура этого оксида была определена повторно [David et al.,
1987а], и мы рассмотрим этот аспект позднее. Можно приготовить
различные композиции с общей формулой Ln3_xBa3+:x:Cu6014+6
(Ln—La, Nd, Y и т. д.); структура этих оксидов обычно
тетрагональная. При х = 1 состав соответствует знаменитой в настоящее
время системе 123, LaBa2Cu307, которая является ромбической.
YBa2Cu307 и родственные 123-оксиды
461
9.4. YBa2Cu307 И РОДСТВЕННЫЕ 123-ОКСИДЫ
Сверхпроводимость выше температуры жидкого азота в
системе Y—Ва—Си—О впервые была описана для состава Yli2Ba0j8CuO4,
который был приготовлен по аналогии с оксидами типа
La2_a.Ba:)CCu04 [Wu et al., 1987]. Эта композиция была двухфазной,
состоящей из зеленого диэлектрика (Y2BaCu05) и черного
сверхпроводящего оксида (сейчас известного как YBa2Cu307). Система
Y—Ва—Си —О исследована [Rao et al., 1987] при получении
композиций типа Y3_3CBa3+:)CCue014+6 по аналогии с
кислород-дефицитным перовскитом La3_JCBa3+:!CCue014+6 7 описанным ранее. В некоторых
из этих композиций зеленая диэлектрическая фаза Y2BaCu05
присутствовала лишь в незначительной степени, и состав
сверхпроводящей фазы был получен как YBa2Cu307, основанный на
рентгенографических исследованиях порошков Yg.^Bag+^CueO^+e- В
фазовой диаграмме системы Y203—ВаО—СиО единственно реальным
высокотемпературным сверхпроводником, по-видимому, будет
только YBa2Cu307.
Структура сверхпроводящего YBa2Cu307 (Tc = 95=±5 К)
является интересной. Она родственна перовскитовой структуре, в
которой 2/3 ионов Си образуют плоские группы Cu04, происходящие
из квадратно-пирамидальной координации Си, в то время как
1/3 ионов Си образуют одномерные цепи сплющенных плоских
групп Cu04, не связанных в направлении a [David et al., 1987b].
Структура, данная на рис. 9.2, а, показывает соединенные
вершинами плоские группы Cu04, связанные в листы в плоскости ab,
а также цепи, параллельные оси Ъ. Из двух типов атомов Си один
окружен четырьмя атомами кислорода на расстояниях 1,929 и
о о
1,960 А; пятый атом кислорода расположен на расстоянии 2,30 А,
формируя квадратно-пирамидальную координацию для Си, и
образует складчатые листы Cu02. В другой группе атомы Си
окружены четырьмя атомами кислорода на расстояниях 1,942 и 1,845 А;
здесь атомы кислорода образуют почти квадраты, связанные
вершинами так, что в результате образуются цепи вдоль оси Ъ.
Расстояние 01—04 довольно короткое (~2,6 А), а атомы кислорода 01
имеют большие амплитуды колебаний. Цепи Си—О ответственны
за ромбическую структуру YBa2Cu307. В тетрагональном YBa2Cu3Oe
(6 = 1,0) цепи полностью исчезают, как показано на рис. 9.2, б,
а атомы кислорода занимают только 2/3 анионных узлов перовскита
и являются упорядоченными таким образом, что 1/3 атомов Си имеет
координационное число 2, в то время как 2/3 имеют
координационное число 5 [Bordet et al., 1987; Jorgensen et al., 1987]. Связь Си—04
о
в этом оксиде действительно очень короткая (~1,80 А). Наркс. 9.2, в
мы также видим тетрагональную фазу (0,3 ^ б ^ 1,0), где
кислородная неупорядоченность может приводить к разупорядоченнъш
462 Химия высокотемпературных сверхпроводников
%Ж}
Си 2
e+Y
Си 2
Атоны О 02, 03
ксолорода
07,04
YBa2Cu306
6
''.* 07,0-5" Разупорядочемнь/е
Рис. 9.2. Структуры YBa2Gu307_6.
о — ромбическая структура сверхпроводящей фазы с б = 0,0; б — тетрагональная
структура сверхпроводящей фазы с б = 1,0 без цепочек Си—О; в — разупорядоченная
структура тетрагональной фазы, содержащая октаэдры CuOe с малым заполнением позиций.
октаэдрам Cu06 [Jorgensen et al., 1987]. Кислородная
нестехиометрия в YBa2Cu307_6 может быть понята как в терминах беспорядка,
так и в терминах структурного искажения. Если позиции 01
(кислород вдоль цепочек) являются преимущественно заселенными по
отношению к позициям 05 (вдоль оси а), то структура будет
ромбической, а материалы сверхпроводящими. Если позиции 01 и 05
заселены одинаково, то структура будет тетрагональной.
Упорядочение кислорода в цепочках, по-видимому, критично для
высокотемпературной сверхпроводимости. Незначительное смещение атомов
кислорода 01 (возможно, 05) также следует учитывать. Закалка
YBa2Cii307 от высоких температур приводит к потере кислорода,
а связанный с этим переход из ромбической в тетрагональную форму
происходит вблизи б = 0,6 в области 620—700 К в зависимости от
давления кислорода. Тс оксида сильно чувствительна к значению б
(рис. 9.3, 9.4).
Все сверхпроводящие оксиды с общей формулой LnBa2Cu307
(Ln—Nd, Sm, Eu, Gd, Dy, Ho, Er, Tm, Yb, Lu) имеют ромбическую
структуру типа YBa2Cu307 и показывают ТС ~ 90 К [Mohan Ram
et al., 1987b; Murphy et al., 1987; Tarascon et al., 1987b]. Оксиды
этого состава, образуемые Се, Pr, Tb, не являются
сверхпроводящими и обладают другими структурами. По-видимому, в сверхпро-
YBa2Cu307 и родственные 123-оксиды 463
2У0Л
us Л
г,о
0.5
А
Л
Л
дддд ДДДДДА'
ддддддд s =0,24
О
•п
D
Д
Да
д
д
а
д
D
дао
д а
д о
Д D
-rttt ППгт.
,***
,^'
1СР'
сро 8=0,12
6 = 0,08
\-зо
V20
''*•• 6 = 0,37
100
200
300
Рис. 9.3. Поведение электросопротивления
YBa2Gu30 . с различными значениями 6 (данные
авторов).
водящих Ln(Y)Ba2Cu307 роль иона Ln(Y) состоит не только в
формировании структуры.
На рис. 9.5 показано изменение параметров элементарной
ячейки YBa2Cu307_6 в зависимости от б. Ясно видно, что структура
изменяется от ромбической к тетрагональной при б » 0,6. С
изменением б температура сверхпроводящего перехода Тс показывает
интересную вариацию (рис. 9.6). Тс почти постоянна вблизи 90 К
вплоть до б ^ 0,2 и падает к почти постоянному значению, равному
~ 60 К, в области б ^ 0,2—0,5. Это плато Тс вблизи 60 К также
отражается на различных электрических и магнитных свойствах
[Cava et al., 1987]. Имеются указания, что составы Об67 и Ов%5
могут обладать некоторыми уникальными структурными
особенностями, вероятно, обусловленными упорядочением вакансий. Те =
464 Химия высокотемпературных сверхпроводников
оч
сп -2-
-з-\
_| I
YBa2Cu307.x
х = 0,68
40 60
Температура 7 К
700
Рас. 9.4. Эффект Мейсс нера (при 50 G) для YBa2Cu30 . [Johnston
et a]., 1987].
= 90 К обнаруживается только тогда, когда цепочки Си—О
являются достаточно заселенными и упорядоченными.
Частичное замещение Си в YBa2Cu307 такими ионами, как Zn,
Fe и Ni, снижает Гс, обычно с превращением в оксид с дефицитом
кислорода или с тетрагональной структурой. Замещение Ва
лантаном имеет подобный эффект. Все описанные до сих пор
сверхпроводящие оксиды с Тс =■- 95=t5 К, родственные YBa2Cu307,
обладают ромбической структурой и цепочками Си—О. Возможно,
однако, что тетрагональная структура реализуется вследствие наличия
беспорядочных цепочек Си—О, или обусловлена отсутствием
дальнего порядка в расположении цепочек Си—О. В то время как
ромбическая структура может простираться вплоть до высоких значений б
(0,7 или более) в YBa2Cu307_6 [Cava et al., 1987b], значение Тс
в этих композициях всегда существенно ниже, чем 60 К, если б
превосходит 0,5.
Многочисленные измерения наводят на мысль, что при Тс в
YBa2Cu307 имеется фазовый переход. Это, вероятно, должен быть
изоструктурный переход, включающий только изменение в
ромбическом искажении. Нс1(0) и Нс2(0) для YBa2Cu307 равны, соответ-
о
ственно около 1 и 120 Тл с длиной когерентности 14 А и глубиной
о
магнитного проникновения 225 А. Эффект Мейсснера, определенный
для этого оксида, достигает ~ 70 %.
YBa2Cu307 и родственные 123-оксиды
465
sso-
•Ъ3,88
| л
К*84']
О—СЮо--„о
£Р
--о-
&
V-a-
О?
0,4 0,6
8
—I—
0,8
80Л
40Л
1,0
11,9-
•< 11,8-
Q--Q13
,-О-П*
.-о-сг
J>-
.чг-
^-0
Рас. Р.5. Изменение rc от б для
YBa2Gu304_6: по данным магнитной
восприимчивости (заштрихованная
область), по данным
электросопротивления (темные кружки).
0,2
0,4 06
08
W
Рис. 9.5. Изменение параметров элементарной ячейки YBa2Gu307_6 от 6, при
6 ~ 0,6 наблюдается изменение структуры из ромбической в тетрагональную.
Электронно-микроскопические исследования YBa2Cu307
показывают присутствие двойников. Двойники являются следствием
ромбической структуры, которая, в свою очередь, формируется в
результате образования цепочек Си—О вдоль оси fe, таким образом
устанавливаются соотношения между связью, кристаллической
структурой и микроструктурой. Считается, что двойники играют
критическую роль в определении как Тс, так и плотности
критического тока. Поэтому контроль за плотностью двойников требует
соответствующего контроля как кислородной стехиометрии и
упорядочения, так и кристаллической структуры. По-видимому,
двойники сами по себе являются результатом различных ориентации
фрагментов (СиО^ и на границах двойников происходит перемена
осей а и Ъ элементарной ячейки. В соответствии с нашей моделью
избыточный кислород должен присутствовать вдоль границы в таких
двойниках. Высокоразрешающие исследования показали, что не
наблюдается реального изменения в структуре или/и в стехиометрии
о
на протяжении 200 А. Странно, что мы не видим двойников
в Lali80Sr0i2CuO4, который является также ромбическим в
сверхпроводящем состоянии. Однако эта ромбическая структура
отличается от структуры оксидов 123, которые имеют цепи Си—О.
466 Химия высокотемпературных сверхпроводников
Сообщалось [Bhargava et al., 1987], что Тс в YBa2Cu307
существенно возрастает при циклировании образца вблизи 240 К; это
объяснялось изменением в упорядочении кислорода. Хотя вероятно,
что переход связан со смещениями 01 (цепочечного) или других
атомов кислорода, это также можно было бы представить
изменением в плотности двойников в образце. Измерения акустических
свойств и теплоемкости также показывают наличие такого
перехода. Изучение Раман-спектров обнаруживает непрерывный переход
вблизи 234 К из симметрии D2h к С2и. Найдено, что многие из
образцов оксидов, родственных YBa2Cu307 (или семейства 123),
показывают преходящие высокие Тс в области 200—250 К. К сожалению,
стабилизировать эти фазы не оказалось возможным.
9.5. СИСТЕМА Ln3-xBa3+xCueOi4+6
Семейство Ln3_3CBa3+:cCu6014+6(Ln—La, Y и т. п.) или
Ьп(Ва2_хЬпх)Си307+6 представляет собой класс оксидов, среди
которых важными членами являются хорошо известные
высокотемпературные сверхпроводники с общей формулой LnBa2Cu307.
Ранние сообщения о высокотемпературной сверхпроводимости в
La3_a.Ba34_xCue014+6 вызвали значительный интерес, поскольку эти
тетрагональные фазы не содержали одномерных цепей Си—О.
Однако недавние нейтронно-дифракционные исследования [David et al.,
1987а] показали, что La3Ba3Cue014+6 имеет структуру, подобную
структуре тетрагонального YBa2Cu307_6, с частичным замещением
La в позиции Ва. Считается, что члены системы La3_a.Ba3+:cCue014+6
являются разупорядоченными изоморфами ромбического
YBa2Cu307_6, содержащего цепочки Си—О [Serge et al., 1987].
В этих 336-оксидах обе позиции кислорода 01 и 05 заполнены
частично и температура сверхпроводящего перехода увеличивается
с ромбичностью. В большинстве случаев найдено, что члены системы
La3_xBa3+:cCu6014+6 обладают тетрагональной структурой с довольно
низкими температурами начала сверхпроводимости (~ 50 К);
ромбические композиции образовывались в результате определенных
условий, особенно когда нагрев при высоких температурах был
минимальным [Ganapathi et al., 1988]. Тетрагональная структура этих
оксидов основана на перовскитовой субъячейке с утроенной
периодичностью. Соответственно тетрагональные оксидные составы
показывают 90° микродомены. Ромбический LaBa2Cu307_6 демонстрирует
экстремальную чувствительность свойств по отношению к
кислородной стехиометрии; избыток кислорода обычно встречается в этом
123-оксиде, и структура таких соединений тетрагональная.
В системе La3_xBa3+xCu6014+6 La и Ва могут легко
обмениваться местами; то же самое справедливо для Nd и других более легких
Висмутовые и таллиевые купраты
467
лантаноидных ионов. Поэтому такие оксиды можно получить с La
или Nd в широкой области составов. Структура всех этих оксидов
тетрагональная. Когда редкоземельный ион маленький, как в
случае Gd или Y, обмен с Ва становится затруднительным. Оказалось
возможным приготовить несколько членов системы Y3_xBa3+:)CCu6014+a
путем разложения нитратов при относительно низких температурах
[Umarji et al., 1988]. Полученные таким образом оксиды являются
тетрагональными с низкими Тс; однако все эти соединения богаты
кислородом. Тетрагональный богатый кислородом YBa2Cu3074e
(Т £^ 50 К) также был получен низкотемпературным методом.
Изучение системы Ln3_3CBa3+JCCue014+6 ясно выявляет важность ромбич-
ности и кислородной стехиометрии для высокотемпературной
сверхпроводимости в этом семействе оксидов.
9.6. ВИСМУТОВЫЕ И ТАЛЛИЕВЫЕ КУПРАТЫ
Открыты два новых семейства сверхпроводящих купратов со
структурами, родственными оксидам семейства Ауривиллиуса с
Тс в области 100 К. Сначала были открыты сверхпроводящие
висмутовые купраты [Michel et al., 1987], Тс была в области 7—22 К
в оксидах типа Bi2Sr2Cu207+6. Затем найдена высокая ТС(~ 100 К)
в системе Bi—Ca—Sr—Си—О [Maeda et al., 1988]. Высокую
критическую температуру сверхпроводимости Тс(~ 110 К)
демонстрировали висмутовые купраты типа Bi2(Ca, Sr)3Cu208+6 [Chu
et al., 1988; Hazen et al., 1988; Rao et al., 1988; Tarascon et al.,
1988]. На рис. 9.7 мы сравниваем структуру Bi4Ti3012 со структурой
Bi2CaSr2Cu208. На рис. 9.8 показано изменение сопротивления и
динамической восприимчивости образца, чтобы продемонстрировать
поведение высокотемпературной сверхпроводимости. Висмутовые
купраты имеют меньшую кислородную стабильность и,
по-видимому, являются более стабильными. Точный состав материала с
Тс ~ 110 К более сложный, чем тот, который указан ранее.
Таллиевые купраты систем Т1—Ва—Си—О и Т1—Са—Ва —
—Си—О демонстрируют высокотемпературную сверхпроводимость с
началом перехода в области 100—120 К [Hazen et al., 1988; Gan-
guli et al., 1988; Sheng, Hermann, 1988]. На рис. 9.9 и 9.10 показано
поведение сопротивления и восприимчивости типичных
представителей системы Т1—Са—Ва—Си—О. Структура оксида 2122
аналогична таковой соответствующих висмутовых соединений
Bi2CaSr2Cu208+6.
Электронно-микроскопические исследования висмутовых и тал-
лиевых купратов показывают широкое присутствие дефектов и
дислокаций точно так же, как и в оксидах семейства Ауривиллиуса.
468 Химия высокотемпературных сверхпроводников
В120г
TL Перовскит
BLO
TL Перовскит
BLO
Т1 Перовскит
В1202
В12
(Са
Си-
(Са
Си
(Са
02
>Sr)0
Перовскит
,Sr)0
-Перовскит
,Sr)G
(Си02)
(Си02)
В1202
BL4TL3012
(с* J2,0A )
О
В1
TL
О
О
Bl2 CaSr2Cu20e
(с* 31 А)
Рис. 9.7. Структура ромбической фазы Ауривиллиуса Bi4Ti3012 и
подобной структуры ромбического Bi2GaSr2Gu208.
В этих системах также присутствуют прорастания и, может быть,
они ответственны за высокие TCJ найденные в некоторых из этих
образцов.
9.7. ПРИРОДА КИСЛОРОДА И МЕДИ
В СВЕРХПРОВОДЯЩИХ КУПРАТАХ
Ранее мы видели, какую критическую роль в
сверхпроводимости YBa2Cu307_6 играет кислород. Тщательное изучение с
использованием фотоэлектронной и Оже-спектроскопии более всего
прояснило этот аспект. Исследование рентгеновских
фотоэлектронных спектров при изменении температур на La18Sr0 2Gu04 и
YBa2Cu307 в областях O(ls) иСи(2р) [Sarma, Rao, 1987; Sarma et al.,
Природа кислорода и меди в сверхпроводящих купратах 469
Рис. 9.8. Данные по удельному сопротивлению и
динамической восприимчивости для образца Bi2(Ca,
Sr)3Cu2Og+6 [Tarascon et al., 1988].
1987] указывает на присутствие разновидностей молекулярных
кислородных частиц с высокой O(ls) энергией связи ~ 533 эВ, помимо
характерных пиков около 529 и 531 эВ, обусловленных
соответственно оксидным и 0"-ионами. Доля этой разновидности,
ответственной за полосу 533 эВ, по-видимому, возрастает с понижением
температуры. Эти кислородные частицы идентифицируются как
пероксидоподобные фрагменты. Спектр Си(2р) показывает
присутствие хорошо экранированного й10-состояния (Си1+) при 933 эВ
вместе с плохо экранированным ^-состоянием (Сп2+) при 942 эВ.
Доля состояния d10 возрастает с понижением температуры.
В обсуждаемых Си(2р) Оже-спектрах нет, однако, свидетельств
наличия Си3+. Оже-спектры показывают присутствие Си1+, доля
которого возрастает с понижением температуры. В висмутовых и
таллиевых купратах также найдены частицы типа Си1+ и 0~. Изме-
470 Химия высокотемпературных сверхпроводников
/.04
0J5-\
asoA
0,25А
• * охО
> oD
Лз
>D
XoD
Хо _
^S
I
/00
т,к
200
• 2122
о 2212
Х2213
0 2223
300
Рис. 9.9. Поведение удельного сопротивления фаз
2122, 2212, 2213 и 2223 системы Т1—Са—Ва—Си—О
(данные авторов).
рения края рентгеновского поглощения также на дают доказательств
для Си3+.
Было предположено, что в YBa2Cu307 и La2_:vBaJC(Sr:x.)Cu04
дырки присутствуют на атомах кислорода, а не меди.
Кислородные дырки (О") затем будут димеризоваться, образуя частицы перок-
сидного типа (022_). Поэтому реакция
202
•2СГ или ОГ + 2е,
возможно, будет важна для сверхпроводимости.
Вероятно, средний заряд на кислороде в этих оксидах близко
к —l,3=t0,l. Дырки в валентных зонах 0(2р) предпочтительны для
состояния Cu1+(d10) точно так же, как и дырки в валентных
зонах S(3p) или Se(4p) обычно предпочтительнее в халькогенидах
Си1+. Поскольку нет реальных доказательств для Си3+, а связи
Природа кислорода и меди в сверхпроводящих купратах 471
01
-^
i
*
<ъ
>4~
Я;
*Q
&**"
•
/
• D
•
а
• а
•
D 2/22
• 222 3
Г
Г'2
03
-76
I I I I I н
/Я0
200
J00
т. к
Рис. 0J0. Данные по динамической
восприимчивости фаз 1222 и 2223 системы Т1—Са—Ва—Си—О.
Си—О высококовалентны (1,8—1,95 А), мы можем заключить, что
вклад от состояния с дырками на Си, действительно,
незначительный.
Наличие кислородных дырок в форме О1" и 0\~, кажется также
будет связано со сверхпроводимостью в системе Ва (Pb, Bi)03.
Недавние исследования наводят на мысль, что дырки О1" присутствуют
во множестве оксидов металлов, которые, как подразумевалось,
содержат металлы в высоких состояниях окисления. Низкая
энергия переноса заряда между ионами Мп+ и О2", по-видимому,
приводит к частицам М(п~р)+ и О1" во многих оксидах, где свяьь металл—
—кислород ковалентная, типичными такими примерами будут
Ba2Cu205 и LiNi02. Если это так, то в свете этого наблюдения
необходимо переосмыслить физику и химию многих оксидов металлов.
472 Химия высокотемпературных сверхпроводников
9.8. ПРИГОТОВЛЕНИЕ И РОДСТВЕННЫЕ АСПЕКТЫ
Синтезы сверхпроводящих оксидов открывают широкие
возможности и дают пищу для сомнений IRao, 1988a]. Наиболее общим
методом использования является керамический, включающий
высокотемпературную реакцию соответствующей смеси оксидов и
карбонатов. Цитраты и оксалаты также использованы в случае
La2_:x.Ba:!C(SrA;)Cu04 и YBa2Cu307 без определенных преимуществ;
однако нитраты, кажется, имеют определенное преимущество. В
синтезах YBa2Cu307 и родственных оксидов удобно использовать Ва02
вместо ВаС03. Начиная с небольшого избытка CuO, переход
делается резче. Добавление BaF2, кажется, дает подобный эффект.
Эффект частичного фторирования еще должен быть тщательно
исследован, хотя имеются указания, что Тс существенно увеличиваются.
Метод предшественника был бы идельным альтернативным
путем для синтезов этих оксидов. К сожалению, не найдено простого
соединения-предшественника или предшественника-твердого
раствора. Однако смесь нитратов, кажется, пригодна в приготовлении
оксидных сверхпроводников, особенно тех, которые требуют
низких температур (например, Y3_A.(La3_x)Ba3KxCu6014). Осаждение
из высокощелочной среды не было успешным. Метод золь — гель
в традиционном смысле кажется трудным, потому что трудно
приготовить алкоксиды Y(La), Ва и Си в форме гомогенной, смеси.
Возможно, заслуживает рассмотрения вероятность использования
алкоксидов меди II (вместо алкоксидов меди I) для золь — гель-
пути. Гели можно приготовить, добавляя подходящие количества
нитрата иттрия и ацетата меди к известному количеству гидроксида
бария. Оксиды системы La—Ва—Си—О могут быть аналогично
приготовлены, исходя из нитрата лантана.
Нагревание под высоким давлением кислорода удобно для
получения стехиометрических образцов или образцов с избытком
кислорода. Так, сверхпроводящий La2Cu04+6 получен при обработке
под высоким давлением кислорода. Плазменное окисление
оказывается другим удобным способом приготовления таких материалов.
Плазменным окислением был приготовлен La2Cu04+6; YBa2Cu307_d
(б ^ 0,5—1,0) легко окисляется до фазы, близкой к стехиометрич-
ной, путем плазменного окисления. YBa2Cu307_6 приготавливали
при использовании различных обработок. Время отжига
(пропитывания) в кислороде варьируется довольно заметно, от нескольких
часов до нескольких дней. В общем, большая часть приготовления
дает образцы с Тс в области 90—95 К. Некоторые исследователи
сообщали более высокие значения Тс (>100 К) в образцах, различно
отожженных или пропитанных.
Висмутовые купраты приготовлены при нагревании смеси
оксидов/карбонатов. Кажется, что отжиг ниже точки плавления
является решающим для получения хороших сверхпроводящих
Заключительные замечания
473
свойств. Таллиевые купраты приготовлены при нагревании смеси
в течение минимального периода в предварительно нагретой печи.
Т1203 легко сублимирует и требует осторожности, поскольку
является ядовитым.
Кажется, что выдержка YBa2Cu307 и других оксидных
сверхпроводников в лабораторной атмосфере в течение длительного
периода приводит к деградации материалов. В случае YBa2Cu307
замечены образования гидроксидов и карбонатов составляющих
металлов и карбонизация поверхности. Висмутовые купраты,
вероятно, более стабильны, чем оксиды 123.
Образцы YBa2Cu307 и другие оксиды, приготовленные
низкотемпературными методами, в основном демонстрируют низкую Тс.
Отчасти проблема, возможно, связана с малым размером частиц,
который, в свою очередь, определяет природу межзеренных границ.
Монокристаллы La2_xBaJC(SrJL.)Cu04 и YBa2Cu307_6 выращены
некоторыми исследователями по методу флюса. Найдено, что более
легко растут кристаллы висмутовых купратов при использовании
метода флюса.
9.9. ЗАКЛЮЧИТЕЛЬНЫЕ ЗАМЕЧАНИЯ
Огромные возможности применений оксидов с высокой Тс
делают эту область исследований очень интересной. Ясно, что
высокотемпературные оксидные сверхпроводники являются самым
сенсационным открытием, после открытия транзистора.
Существует большой простор для получения надежных данных
по этим оксидным сверхпроводникам, и эта тема открывает
безграничную сферу исследований по химии твердого тела оксидов
металлов. Возможно, что стабильный при комнатной температуре
сверхпроводник находится сейчас где-нибудь в лаборатории химии
твердого тела. Необходимо исследовать многие как оксидные, так и
неоксидные системы, в особенности системы, содержащие такие
металлы, как V, Nb, Pb и Ru, а также и структуры прорастания.
Особенно радует, что даже в этот короткий период достигнуто
понимание роли структуры и стехиометрии в сверхпроводимости.
Так, цепи Си — О—Си ответственны за ромбическую структуру
YBa2Cu307, а ромбическая структура существенна для образования
Двойников, которые контролируют многие из свойств. Кроме того,
все исследованные оксиды с высокой Гс, являются перовскитами.
Купраты являются в основном сверхпроводниками II рода.
Наиболее важной характеристикой сверхпроводников,
необходимой для применений, является высокая плотность критического
тока (~ 105 А-см~2). Ожидалось, что проволоки, пленки и ленты
с такими характеристиками будут делаться из оксидных материалов.
Купратные сверхпроводники, по-видимому, вообще связаны с ма-
31 Ч. Н. Р. Рао, Дж. Гопалакришнан
474 Химия высокотемпературных сверхпроводников
77К
110
—г-
-2
-1
Магнитное поле. мТ
Рис. 9.11. Производные сигналов поглощения
Т12СаВа2Си2Оя+б при различных температурах
(9, 11 G/Hz; данные авторов).
лыми длинами когерентности. Важным свойством этих
гранулированных сверхпроводников является поглощение микроволн [Bhat
et al., 1987] (рис. 9.11). Это свойство может иметь применение
для обнаружения сверхпроводимости в упомянутых материалах.
ДОПОЛНЕНИЕ ПЕРЕВОДЧИКА
Как уже отмечалось редактором перевода академиком
Ф. А. Кузнецовым, угнаться за развитием химии
высокотемпературных сверхпроводников в настоящее время невозможно ввиду
чрезвычайно интенсивного развития исследований в этой области.
Любой обзор рискует устареть прежде чем будет опубликован.
Дополнение переводчика
475
Действительно, с момента написания профессором Рао главы
по высокотемпературным сверхпроводникам в литературе появились
данные о синтезе новых типов ВТСП-материалов, о которых не
упоминается в данной главе. Поэтому нам показалось уместным хотя бы
перечислить эти новые группы веществ.
Здесь следует сказать прежде всего о фазах YBa2Cu408 (Tc ~
~ 80 К) и Y2Ba4Cu70]4+;x. (ТГ ~ 40 К). Структуры этих фаз
являются производными от структуры YBa2Cu307. Решетка YBa2Cu4Og
во многом повторяет решетку базовой структуры YBa2Cu307 и
отличается от последней только наличием сдвоенных цепей из ромбов
Cu04, соединенных через общие вершины и ребра. Структуру
другой фазы Y2Ba4Cu7014+:x; можно представить как упорядоченное
чередование слоев структуры YBa2Cu307 (с одиночными цепочками
{Си03}оо) и YBa2Cu408 (со сдвоенными цепочками {Cu204}oo).
Другие родственные фазы найдены в таллиевых купратных
системах: здесь кроме фаз с двойными таллий-кислородными слоями,
принадлежащих к гомологическому ряду Tl2Ba2Can_1Cnn02rif4,
получены фазы с одиночными таллий-кислородными слоями, которые
описываются общей формулой ^В'л2С^п_^С\1п02п+215+х (п = 1—4).
Сверхпроводники этого типа характеризуются довольно высокими
критическими температурами: Тс ~ 90 К для /г = 2, Гс ~ 116 К
для п = 3 и Тс ~ 122 К для п ■— 4.
Синтезированы новые свинецсодержащие купраты, не
содержащие висмута или таллия, с общей формулой Pb^^R^Mt.^CiisOg+j,
(R = Y, La, Pr, Nd, Eu, Dy, Но и другие РЗЭ; М =■ Са, Sr) с Тс ~
~ 40—70 К. Структура этих фаз имеет много общих черт с другими
ВТСП-материалами.
И, наконец, следует упомянуть о синтезе фаз типа Nd^^Sr^.y-
• CeyCu04_6 (Тс ~ 23 К), кристаллизующихся в структуре Nd2Cu04.
Особенность данных сверхпроводников состоит в том, что они
обладают электронной проводимостью в отличие от всех других ВТСП-
материалов, характеризующихся дырочным типом проводимости.
В заключение отметим, что строение и физико-химические
свойства всех ВТСП-материалов рассмотрены в серии статей,
опубликованных в «Жури. ВХО им. Д. И. Менделеева», 1989, т. 34, вып. 4.
31*
СПИСОК ЛИТЕРАТУРЫ
ГЛАВА 1
Adams D. M. Inorganic Solids // An Introduction to Concepts in Solid State
Structural Chemistry.— London: Wiley, 1974.
Andersson S.// J. Solid State Chem.— 1978.— V. 23.—P. 191.
Andersson S./7 Angew. Chem. Int. Ed. (Engl.).—1983.—V. 22.— P. 69.
Andersson S., Faith L./7 J. Solid State Chem.—1983.—V. 46.—P. 265.
Atwood J. L., Davies J. E. D., MacNicol D. D. (Eds). Inclusion Compounds.—
London: Academic Press, 1984.— V. 1 and 2.
Barrer R. M. Zeolites and Clay Minerals as Sorbents and Molecular Sieves.—
New York: Academic Press, 1978.
Bartell L. S. /7 J. Chem. Ed.- 1968.— V. 45.— P. 754.
Bernal J. D. // Liquids: Structure, Properties, Solid Interactions/Ed. by
T. J. Hughel.— Amsterdam: Elsevier, 1965.
Brewer L. // Acta Met.— 1967.— V. 15.— P. 553.
Brewer L. // J. Chem. Ed.— 1984.— V. 61.— P. 101.
Brindley G. W., Brown G. (Eds). Crystal Structures of Clay Minerals and
their X-ray Diffraction.— London: Mineralogical Society, 1980.
Brown G.//Phil. Trans. Roy. Soc. London.—1984.—V. A311.—P. 221.
Brown I. D. // Structure and Bonding in Crystals/Ed. by M. O'Keeffe,
A. Navrotsky.— New York: Academic Press, 1981.
Burdett J. K. // J. Am. Chem. Soc— 1980.— V. 102.— P. 450.
Burdett J. K./7 Advances in Chemical Physics/Ed. by I. Prigogine, S. A.
Rice.— New York: Wiley-Interscience, 1982.
Burdett J. K. /7 Inor?. Chem.— 1985.— V. 24.— P. 2244.
Burdett J. K., Hughbanks T. // Inorg. Chem. —1985.—V. 24.— P. 1741.
Burdett J. K., Rosenthal G. L.// J. Solid State Chem.—1980.—V. 33.—
P. 173.
Burdett J. K., McLarnan T. J., Haaland P.//J. Chem. Phys.— 1981.—
V. 75.— P. 5774.
Burdett J. K., McLarnan T. J. // Inorg. Chem.— 1982.— V. 21.— P. 1119.
Burdett J. K., Lee S.//J. Solid State Chem.—1985.—V. 56.—P. 211.
Chattopadhyay K., Lele S., Ranganathan S., SubbannaG. N., Thangaraj N.//
Curr. Sci., (India).— 1985.—V. 54.— P. 895.
Cracknell A. P. Magnetism in Crystalline Materials.— Oxford: Pergamon
Press 1975.
Domenges В., McGuire N. K., O'Keeffe M. // J. Solid State Chem.— 1985.—
V. 56.— P. 94.
Douxing L., Stenberg L.t Andersson S.// J. Solid State Chem.— 1983.—
V. 48.— P. 368.
Elliott R. J.// Phys. Rev.—1961.—V. 124.—P. 346.
Elliott S. R., Rao С N. R., Thomas J. M. // Angew. Chem. Int.
Ed. (Engl.).—V. 25,- P. 31.
Finnev J. L. // Proc. Roy. Soc. London.— 1970.— V. A319.— P. 479.
Ganguly P., Rao С N. R. // J. Solid State Chem.— 1984.— V. 53.— P. 193.
Gaskell P. H.//J. Non-Cryst. Solids.—1979.—V. 32.— P. 207.
Gies H., Liebau F. // Acta Crystallogr.— 1981.— V. A37.— P. C187.
Gies H., Liebau F., Gerke H. // Angew. Chem.— 1982.— V. 94.— P. 214.
Глава 1
477
Girgis К. // Physical Metallurgy (3rd Edn)/Ed. by R. W. Cahn, P. Hassen.—
Amsterdam: North-Holland, 1983.
Hoare M. R., Barker J. // The Structure of Non-Crystalline Materials/Ed.
by P. H- Gaskel.— London: Taylor, Francis, 1976.
Hoffmann R., Zheng С // J. Phys. Chem.— 1985.— V. 89.— P. 4175.
Hoppe R. // Crystal Structure and Chemical Bonding in Inorganic
Chemistry/Ed. by С J. M. Rooymans, A. Rabenau.— Amsterdam: North-Holland,
1975.
Huggins R. A., Rabenau A. // Mater. Res. Bull.— 1978.— V. 13.— P. 1315.
Hughbanks Т., Hoffmann R.// J. Amer. Chem. Soc—1983.—V. 105.—
P. 1150.
Hyde B. G., Bagshaw A. N., Andersson S., O'Keeffe M. // Ann. Rev. Mater.
Sci.- 1974.—V. 4.- P. 43.
Hyde B. G. // Modulated Structures.— 1979, AIP Conference Proceedings/
Ed. by J. M. Cowley, J. B. Cohen, M. B. Salamon, B. J. Wuensch.— New York:
American Institute of Physics, 1979.
Hyde B. G., Andersson S., Bakker M., Plug C. M., O'Keeffe M. // Prog. Solid
State Chem.—1980.—V. 12.—P. 273.
Jack К. Н., Guttmann V. // Acta Crystallogr.— 1951.— V. 4.— P. 246.
Kamb B. // Science.— 1965.— V. 148.—P. 232.
Katz L., Ward R.//Inorg. Chem.—1964.—V. 3.— P. 205.
Kitaigorodsky A. I. Molecular Crystals and Molecules.— New York:
Academic Press, 1973.
Krishna P. (Ed.). Crystal Growth and Characterization of Polytype
Structures.— Oxford: Pergamon Press, 1983.
Longuet-HigginsH. C, Roberts M. de V.// Proc. Roy. Soc. London.—1968.—
V. A224.— P. 336.
McCarley R. E.//Phil. Trans. Roy. Soc. London.—1982.—V. A308.—
P. 141.
Mackay A. L., Kramer P. // Nature.— 1985.— V. 316.— P. 17.
Magneli A.// Pure, Applied Chem.—1978.—V. 54.—P. 1261.
Mason R., Rae A. I. M.//Proc. Roy. Soc. London.—1968.—V. A304.—
P. 501.
Meier W. M., Olson D. H. Atlas of Zeolite Structure Types.— Pittsburgh;
Pennsylvania: Int. Zeolite Assoc. Polycrystal Book Serv., 1978.
Mooser E., Pearson W. B. // Acta Crystallogr.— 1959.— V. 12.— P. 1015.
Ogurtani Т.О. // Ann. Rev. Mat. Sci.— 1983.— V. 13.— P. 67.
O'Keeffe M.// Acta Crystallogr.— 1977.— V. A33.—P. 924.
O'Keeffe M. // Structure and Bonding in' Crystals/Ed. by M. O'Keeffe,
A. Navrotsky.— New York: Academic Press, 1981.— V. 1.
O'Keeffe M., Andersson S. // Acta Crystallogr.— 1977.— V. A33.— P. 914.
O'Keeffe M., Hyde B. G. // Acta Crystallogr.— 1978.— V. B34.— P. 3519.
O'Keeffe M., Hyde B. G.//Trans. Amer. Crystallogr. Assoc—1979.—
V. 15.— P. 65.
O'Keeffe M., Hyde B. G. // Phil. Trans. Roy. Soc. London.— 1980.—
V. A295.— P. 553.
O'Keeffe M., Hyde B. G. // Structure and Bonding in Crystals/Ed. by M.
O'Keeffe, A. Navrotsky.—New York: Academic Press, 1981.—V. 1.
O'Keeffe M., Hyde B. G. // J. Solid State Chem.— 1982.— V. 44.— P. 24.
Parthe E. // Structure and Bonding in Crystals/Ed. by M. O'Keeffe, A.
Navrotsky.— New York: Academic Press, 1981.— V. 2.
Pauling L. The Nature of the Chemical Bond (3rd edn).— New York:
Cornell University Press, 1960.
Pauling L. // Nature.— 1985.— V. 317.— P. 512.
Phillips J. C. Bonds and Bands in Semiconductors.— New York: Academic
Press, 1973a.
Phillips J. С // Treatise on Solid State Chemistry/Ed. by N. B. Hannay.—
New York: Plenum Press, 1973b.—V. 1.
478
Список литературы
Polk D. E. // J. Non-Cryst. Solids.— 1971.— V. 5.— P. 365.
Ramasesha S. // Prajnana. —1984.— V. 23.— P. 745.
Ramasesha S., Rao С N. R. // Phil. Mag.— 1977.— V. 34.— P. 827.
Ramdas S., Thomas J. M. // Chemical Physics of Solids and their Surfaces.
Specialist Periodical Report/Ed. by M. W. Roberts, J. M. Thomas.— London:
The Chemical Society, 1980.—V. 7.
Rao С N. R. (Ed.) Solid State Chemistry.— New York: Marcel Dekker,
1974.
Rao C. N., R., Rao K. J. Phase Transition in Solids.— New York: McGraw-
Hill, 1978.
Rice С E., Jackel J. L. // J. Solid State Chem. —1982.— V. 41.— P. 308.
Robertson J. M. // Proc. Roy. Soc. London.— 1951.— V. A207.— P. 101.
Shannon R. D., PrewittC. T. // Acta Crystallogr.— 1969.— V. B25.— P. 925.
Shannon R. D. // Acta Crystallogr.— 1976.— V. A32.— P. 751.
Shannon R. D. // Structure and Bonding in Crystals/Ed. by M. O'Keeffe,
A. Navrotsky.— New York: Academic Press, 1981.— V. 2.
Shechtman D., Blech I., Gratias D., Cahn J. W. // Phys. Rev. Lett. —1984.—
V. 53.— P. 1951.
Simon A. // Angew. Chem. Int. Ed. (Engl.).— 1981.— V. 20.— P. 1.
Simon A. // Ann. Chim. Fr.— 1982.— T. 7.— P. 539.
Soos Z. G., Klein D. J. // Treatise on Solid State Chemistry/Ed. by
N. B. Hannay.— New York: Plenum Press, 1976.—V. 3.
Savborg O. // J. Solid State Chem.— 1985.— V. 57.— P. 154.
Torrance J. B. // Ace. Chem. Res.— 1979.— V. 12.— P. 79.
Tosi M. P. Solid State Phys./Ed. by F. Seitz, D. Turnbull.— New York:
Academic Press, 1964.— V. 16.
Uppal M. K., Ramasesha S., Rao C. N. R. // Acta Crystallogr.— 1980.—
V. A36.— P. 356.
Wells A. F. Structural Inorganic Chemistry (5th edn).— Oxford: Oxford
University Press, 1984.
Whangbo M.H., Foshee M. J., Hoffmann R. // Inorg. Chem.— 1980.—
V. 19.— P. 1725.
Williams D. E. // J. Chem. Phys.— 1967.— V. 47.— P. 4680.
Williams D. E., Starr R. // Comput. Chem.—1977.—V. 1.—P. 173.
Yvon K. // Current Topics in Materials Science/Ed. by E. Kaldis.—
Amsterdam: North-Holland, 1979.—V. 3.
Zallen R. The Physics of Amorphous Solids.— New York: John Wiley,
1983.
Ziolkowski J. // J. Solid State Chem.— 1985.— V. 57.— P. 269.
Zunger A. // Structure and Bonding in Crystals/Ed. by M. O'Keeffe, A. Nav-
rotsky.— New York: Academic Press, 1981.— V. 1.
ГЛАВА 2
AIP Conference Proceedings/Ed. by E. A. Stern.— New York: AIP, 1980.—
N64: Laboratory EXAFS Facilities.
Allpress J. G., Sanders J. V. // J. Appl. Crystallogr.— 1973.— V. 6.— P. 165.
Alvarado F., Erbudak M., Munz P. // Phys. Rev.—1976.—V. B14.—
P. 2740.
Anderson J. S. // Proc. Indian Acad. Sci.— 1978.— V. 87A.— P. 295.
Andrew E. R. // Int. Rev. Phys. Chem.— 1981.— V. 1.— P. 195.
Beanfils J. P., Crowley Т., Rayment Т., Thomas R. K., White J. W. // Molec.
Phys.— 1981.— V. 44.— P. 1257.
Beer M., Carpenter R. W., Eyring L., Lyman С E., Thomas J. M. // Chem.
Engg. News.— 1981.— V. 59.— P. 40. See also Jefferson D. A., Thomas J. M.t
Egerton R. F. // Chem. Brit.— 1981.— V. 17.— P. 514.
Глава 2
479
Binning G., Rohrer H. // Physica.— 1984.— V. 127B.— P. 37.
Binning G., Rohrer H., Gerber Ch., Weibel E. // Phys. Rev. Lett.— 1982.—
V. 49.— P. 57.
Bomchil G., Htiller A., Rayment Т., Roser S. J., Smalley M. V.,
Thomas R. K., White J. W. // Phil. Trans. Roy. Soc. London.— 1980.— V. B290.—
P. 537.
Bourdillon A. J., Pettifer R. F., Marseilha E. A. // J. Phys.—1980.—
V. G12.— P. 3889.
Boyce J. В., Hayes T. M., Stutius W., Mikkelsen Jr., J. C. // Phys. Rev.
Lett.— 1977.— V. 38.— P. 1362.
Boyce J. В., Hayes T. M. // Physics of Superionic Conductors/Ed. by M. B.
Saiamon.— Berlin: Springer-Verlag, 1979.
Brown N. M. D., McMonagle J. В., Greaves N. // J. Chem. Soc. Faraday, 1.—
1984.— V. 80.— P. 589.
Caswell N., Solin S. A., Hayes T. M., Hunter S. H.// Physica B+C.—
1980.— V. 99.— P. 463.
Cheetham A. K. // Proc. Ind. Nat. Sci. Acad.— 1986.—V. 52A.— P. 25.
Cheetham A. K., Rao С N. R. // Acta Grystallogr.— 1976.— V. B32.—
P. 1579.
Cheetham A. K., Taylor J. С // J. Solid State Chem.— 1977.— V. 21.—
P. 253.
Cheetham A. K., Skarnulis A. J. // Anal. Chem.— 1981.— V. 53.— P. 1060.
Cramer S. P. Stanford Synchrotron Res. Lab. Rep., 78/07.— Stanford, USA,
1978.
Dupree E., Pettifer R. F. // Nature.— 1984.— V. 308.— P. 523.
Eccles T. K. // PhD Thesis.—Stanford: Stanford University, USA, 1978.
Egerton R. F. // Phil. Trans. Roy. Soc. London.— 1982.—V. A305.—
P. 521.
Evans S., Adams J. M., Thomas J. M. // Phil. Trans. Roy. Soc. London.—
1979.— V. A292.— P. 563.
Fiermans L., Vennik J., Dekeyser W. (Eds). Electron and Ion Spectroscopy.—
New York: Plenum Press, 1978.
Fyfe С A., Thomas J. M., Klinowski J., Gobbi G. С // Angew. Chem. Int.
Edn (Engl.).—1983.—V. 22.-259.
Ganguly P., Rao С N. R. // J. Solid State Chem.— 1984.— V. 53.— P. 193.
Goodman P., Moodie A. F. // Acta Crystallogr.— 1974.— V. A33.— P. 701.
Grimmer A. R., Rdaeglia R. // Chem. Phys. Lett.— 1984.— V. 106.—
P. 262.
Heald S. M., Stern E. A. // Phys. Rev.— 1978.— V. B17.— P. 4069.
Honig J. M., Rao С N. R. (Eds). Preparation and Characterization of
Materials.— New York: Academic Press, 1981.
Hutchison J. L., Anderson J. S., Rao С N. R. // Proc. Roy. Soc. London.—
1977.- V. A355.- P. 301.
International Tables for Crystallography.— Birmingham, England: Ky-
noch Press, 1969.— V. 3.
Jefferson D. A. // Phil. Trans. Roy. Soc. London.—1982.—V. A305.—
P. 535.
Kihlborg L. // Direct Imaging of Atoms in Crystals and Molecules. Nobel
Sjinposium 47.— The Royal Swedish Academy of Sciences, 1979.
Knights J. C, Hayes T. M., Mikkelsen Jr., J. С // Phys. Rev. Lett — 1977.—
V. 39.— P. 712.
Laubschat C, Kaindl G., Sampathkumaran E. V., Schneider W. D. // Solid
State Commun.—1984.—V. 49.—P. 339.
Lippmaa E., Magi M., Samoson A., Engelhardt G., Grimmer A. R. // J. Amer.
Chem. Soc— 1980.— V. 102.—P. 4889.
Lipsicas M., Raythatha R. H., Pinnavaia T. J., Johnson I. D., Geise Jr.,
R. F., Costanzo P. M., Robert J. L. // Nature.— 1984. - V. 309.— P. 604.
Lovesey S. W., Springer T. (Eds). Dynamics of Solids and Liquids by Neut-
480
Список литературы
ron Scattering. Topics in Current Physics.— Berlin: Springer-Verlag, 1977.—
V. 3.
Lovesey S. W., Sterling G. С ,/ Physica.— 1984.— V. 1276.— P. 306.
Lytle F. W., Sayers D. E., Stern E. A. // Phys. Rev.— 1975.— V. Bll.—
P. 4825.
Magi M., Lippmaa E., Samoson A., EngelhardtG., Grimmer A. R. // J. Phys.
Chem.—1984.—V. 88.—P. 1518.
Manohar H. // Curr. Sci. (India).— 1983.—V. 52.- P. 39.
Mansour A. N., Cook Jr., J. W., Sayers D. E. // J. Phys. Chem.— 1984.—
V. 88.— P. 2330.
Manthiram A., Sarode P. R., Madhusudan W. H., Gopalakrishnan J., Rao
С N. R. // J. Phys. Chem.— 1980.— V. 84.— P. 2200.
Mason R., Rao C. N. R., Sheppard N., Roberts M. W. (Eds). Phil. Trans.
Roy. Soc— London, 1986.
Meinke W. W. // Treatise on Solid State Chemistry/Ed. by N. B. Hannay.—
New York: Plenum Press, 1973.—V. 1.
Moran M. J., Miller G. R., DeMarco R. A., Resing H. A. // J. Phys. Chem.—
1984.— V. 88.— P. 1580.
Mozzi R. L., Warren В. Е. // J. Appl. Crystallogr.— 1969.— V. 2.— P. 164.
Newnham R. E., Roy R. // Treatise on Solid State Chemistry/Ed. by N. B.
Hannay.— New York: Plenum Press, 1973.— V. 1.
Olejnik S., Stirling G. C, White J. W. // Special Disc. Faraday Soc —
1970.— V. 1.— P. 1.
Parthasarathy R., Sarode P. R., Rao K. J., Rao С N. R. /7 Proc. Ind. Nat.
Sci. Acad.—1982.-V. 48A.—P. 119.
Pettifer R. F., McMillan P. W. ,1 Phil. Mag.— 1977.—V. 35.— P. 871.
Pettifer R. F., McMillan P. W., Gurman S. J. // The Structure of
Non-Crystalline Materials/Ed. by P. H. Gaskel.— London: Taylor, Francis, 1977.
Ramanan A.,Gopalakrishnan J., Uppal M. K., Jefferson D. A., RaoC. N.R. /
Proc. Roy. Soc. London.— 1984.— V. A395.— P. 127.
Ramdas S., Klinowski J. // Nature.—1984.—V. 308.— P. 521.
Rao С N. R. // Proc. Ind. Nat. Sci. Acad.— 1985.— V. 51A.— P. 1.
Rao С N. R., Sarma D. D., Vasudevan S., Hegle M. S. /7 Proc. Roy. Soc.
London.— 1979.— V. A367.— P. 239.
Rao С N. R., Sarma D. D., Hegde M. S. // Proc. Roy. Soc. London,—
1980.— V. A370.— P. 269.
Rao С N. R., Sarma D. D. // Science and Technology of Rare Earth
Materials/Ed. by E. С Subbarao, W. E. Wallace.— New York: Academic Press, 1980.
Rao С N. R., Hegde M. S. // Preparation and Characterization of Materials/
Ed. by J. M. Honig, Rao С N. R.— New York: Academic Press, 1981.
Rao С N. R., Sarma D. D. // J. Solid State Chem.— 1982.— V. 45.— P. 14.
Rao С N. R., Sparrow T. G., Thomas J. M., Williams B. G. // J. Chem. Soc.
Chem. Commun.— 1984.— P. 1238.
Rao C. N. R., Thomas J. M., Selvaraj U., Klinowski J., Rao K. J.,
Ramdas S. // Angew. Chem. Int. Ed. (Engl.).— 1985.— V. 24.— P. 61.
Rao K. J., Wong J., Weber M. J. // J. Chem. Phys.— 1983.—V. 78.—
P. 6228.
Richards R., Packer K. J. (Eds). Phil. Trans. Roy. Soc. London, 1981.—
V. A229 P 475
Rietveld H. M. // J. Appl. Crystallogr.— 1969.—V. 2.— P. 65.
Riviere J. С // Phil. Trans. Roy. Soc. London.— 1982.— V. A305.— P. 545.
Sandstrom D. R., Lytle F. W. // Ann. Rev. Phys. Chem.— 1979.— V. 30.—
P. 215.
Sankar G., Sarode P. R., Rao С N. R. // Chem. Phys.— 1983.— V. 76.—
P. 435.
Sankar G., Sarode P. R., Srinivasan A., Rao С N. R., Vasudevan S.,
Thomas J. M./7 Proc. Indian Acad. Sci. (Chem. Sci.).—1984.—V. 93.—P. 321.
Глава 3
481
Sarma D. D., Kamath P. V., Rao С N. R. // Chem. Phys.— 1983.— V 73—
P. 71.
Sarode P. R., Ramasesha S., Madhusudan W. H., Rao С N. R. // J. Phys.—
1979.— V. G12.— P. 2439.
Satyamurlhy N. S., Dasannacharya B. A., Chakravarty R. // Preparation and
Characterization of Materials/Ed. by J. M. Honig, C. N. R. Rao.— Now York:
Academic Press, 1981.
Selvaraj U., Rao K. J., Rao С N. R., Klinowski J., Thomas J. M. //Chem.
Phys. Lett.- 1985.- V. 114.- P. 24.
Sinfelt J. H., ViaG. H., Lytle F. W. /7 Catalysis Reviews.— 1984.— V. 26.—
P. 81.
Smith J. V., Blackwell С S. // Nature.— 1983.— V. 303.— P. 223.
Somorjai G. A. Chemistry in Two Dimensions: Surfaces.— Ithaca: Cornell
University Press, 1981.
Sprensen О. Т. (Ed.). Nonstoichiometric Oxides.— New York: Academic
Press, 1981.
Sparrow T. G., Williams B. G., Thomas J. M., Jones W., Herley P. J.,
Jefferson D. A. // J. Chem. Soc. Chem. Comm.— 1983.— P. 1432.
Stern E. A., Rimildi S., Callen E., Heald S. M. // J. Mag., Mag. Mat.—
1978.- V. 7.—P. 188.
Stutius W., Boyee J. В., Mikkelsen Jr., J. С // Solid State Comm.— 1979.—
\m 3i p 539
Teo B. K. // Ace. Chem. Res.- 1980.- V. 13.— P. 412.
Teo B. K., Lee P. A. // J. Amer. Chem. Soc— 1979.— V. 101.— P. 2815.
Theye M. L., Launois H., Gheorghiu A. // J. Phys.— 1980.— V. C13.—
P. 6139.
Thomas J. M. // Ultramicroscopy.—1982.—V. 8.— P. 13.
Thomas J. M. II Phil. Trans. Roy. Soc. London.— 1984.—V. A311.—
P. 271.
Thomas J. M., Klinowski J., Wright P. A., Roy R. // Angew. Chem— 1983.—
V. 22.— P. 614.
Treacy M. M. J., Howie A., Wilson С J. // Phil. Mag — 1978.— V. A38.—
P. 569.
Vasudevan S., Hegde M. S., Rao С N. R. // J. Solid State Chem.— 1979.—
V. 29.— P. 253.
Vasudevan S., Thomas J. M.> Wright С J., Sampson С // J. Chem. Soc.
Chem. Comm.—1982.—P. 418.
Williams B. C, Thomas J. M. // Int. Rev. Phys. Chem.— 1983.— V. 3.—
P. 39.
Wong J. // Metallic Glasses. Topics in Applied Physics 46/Ed. by H. J. Giin-
therodt, H. Beck.— Berlin: Springer-Verlag, 1980.
Wong J., Lytle F. W. // J. Non-Cryst. Solids.— 1980.—V. 37.— P. 273.
Yashonath S., Sen P., Hegde M. S., Rao С N. R. // J. Chem. Soc.
Faraday I.— 1983.— V. 79.—P. 1229.
ГЛАВА 3
Alamo J., Roy R. //' J. Solid State Chem.— 1984.— V. 51.— P. 270.
Angell С. А. // Preparation and Characterization of Materials/Ed. by
J# M. Honig, С N. R. Rao.— Rew York: Academic Press, 1981.
Banks E., Wold A. // Solid State Chemistry/Ed. by C. N. R. Rao.— New
York: Marcel Dekker, 1974.
Barrer R. M. // Zeolites.— 1981.— V. 1.— P. 130.
Bechgaard K., Jerome D. // Scientific American.— 1982.— V. 247.— P. 50.
Boiler H. // Monatsh. Chem.— 1978.— B. 109.— S. 975.
Cava R. J., Santoro A., Murphy D. W., Zahurak S., Roth R. S. // J. Solid
482
Список литературы
State Ghem.— 1982.- V. 42.- P. 251.
Cheetham A. K. // Nature.— 1980.— V. 288.— P. 469.
Chevrel R., Sergent M., Prigent J. // Mater. Bes. Bull.— 1974.—V. 9.—
P. 1487.
Chowdhry U., Barkley J. R., English A. D., Sleight A. W. // Mater. Bes.
Bull.—1982.—V. 17.— P. 917.
Corbett J. D. // Solid State Chemistry: A Contemporary Overview/Ed. by
S. L. Holt, J. B. Milstein, M. Bobbins.— Washington, D. C: American Chemical
Society.
Corbett J. D. // Ace. Chem. Bes.— 1981.— V. 14.— P. 239.
Delmas C, Braconnier J. J., HagenmuUer P. // Mater. Bes. Bull.— 1982.—
V. 17.- P. 117.
DemazeauG., Buff at В., Pouchard M., HagenmuUer P.// J. Solid State
Chem.— 1982a.- V. 45.- P. 881.
DemazeauG., Buffat В., Pouchard M., HagenmuUer P. // Z. Anorg. Allgem.
Chem.—1982b.—V. 491.—P. 60.
DesirajuG. R., Rao M. // Mater. Res. Bull.— 1982.— V. 17.— P. 443.
Dickens P. G., French S. J., Hight А. Т., Pye M. F. // Mater. Res. Bull.—
1979.- V. 14.- P. 1295.
Dickens P. G., Pye M. F. // Intercalation Chemistry/Ed. by M. S. Whitting-
ham, A. J. Jacobson.— New York: Academic Press, 1979.
Didchenko R., Lifz L. M. // J. Electrochem. Soc— 1962.—V. 109.—
P. 247.
Elliott S. R., Rao С N. R., Thomas J. M. // Angew. Chem. Int. Ed.
(Engl.).— 1986.- V. 25.— P. 31.
England W. A., Goodenuogh J. В., Wiseman P. J. // J. Solid State Chem.—
1983.— V. 49.— P. 289.
Espinosa G. P. // Mater. Res. Bull.— 1980.— V. 15.— P. 791.
Etemad S., Heeger A. J., MacDiarmid A. G. // Ann. Bev. Phys. Chem.—
1932. v. 33. P. 443.
' Fan J. C. C, Reed T. B. // Mater. Res. Bull.— 1972.— V. 7.— P. 1403.
FarringtonG. C, Briant J. L. // Mater. Res. Bull.— 1978.— V. 13.— P. 763.
Feigelson R. S. // Solid State Chemistry: A Contemporary Overview/Ed.
by S. L. Holt, J. B. Milstein, M. Robbins.— Washington, D. C, 1980.
Flanigen E. M., Bennett J. M., Grose R. W., Cohen J. P., Patton R. L., Kir-
chner R. M., Smith J. V. // Nature.— 1978.— V. 271.— P. 512.
Fisk Z., Cooper A. S., Schmidt P. H., Castellano R. N. // Mater. Res. Bull.—
1972.— V. 7.— P. 285.
Goodenough J. В., Kafalas J. A., Longo J. M. // Preparative Methods in
Solid State Chemistry/Ed. by P. HagenmuUer.— New York: Academic Press, 1972.
Goodenough J. В., Hong H. Y., Kafalas J. A. // Mater. Bes. Bull.— 1976.—
V. 11.— P. 203.
GranqvistC.G., Buhrman R. A. // J. Appl. Phys.— 1976.— V. 47.— P. 2200.
Groult D., Pannetier J., Raveau B. // J. Solid State Chem.— 1982 —
V. 41.— P. 277.
Groenen E. J. J., Alma N. C. M., Bastein A. G. T. M., Hays G. R., Huis R.,
Kortbeek A. G. T. G. // J. Chem. Soc. Chem. Comm.— 1983.— P. 1360.
HagenmuUer P. (Ed.). Preparative Methods in Solid State Chemistry.— New
York: Academic Press, 1972.
HagenmuUer P. // Proc. Indian Acad. Sci. (Chem. Sci.).— 1983.— V. 92.—
P. 1.
HagenmuUer P. (Ed.). Solid Inorganic Fluorides.— New York: Academic
Press 1985.
Harrison H. R., Aragon R., Sandlxrg С J. // Mater. Res. Bull.— 1980 —
V. 15.— P. 571.
Hatfield W. E. (Ed.). Molecular Metals.— New York: Plenum Press, 1979.
Henisch H. K., Dennis J., Hanoka J. I. // J. Phys. Chem. Solids.— 1965 —
V. 26.— P. 493.
Глава 3
483
Honig J. M., Rao С. N. R. (Eds.). Praparation and Characterization of
Materials.— New York: Academic Press, 1981.
Horowitz H. S., Longo J. M. //Mater. Res. Bull.— 1978.— V. 13.— P. 1359.
Horowitz H. S., Longo J. M., Lewandowski J. T. // Mater. Res. Bull.—
1981.-V. 16.- P. 489.
Hotta S., Hosaka Т., Shimotsuma W. // Syn. Met.— 1983.— V. 6.— P. 319.
Jacobson A. J., Chianelli R. R., Rich S. M., Whittingham M. S. // Mater.
Res. Bull.— 1979.— V. 14.— P. 1437.
Jayaraman A., Dernier P. D., Longinotti L. D. // High. Temp. High. Pr.—
1975.- V. 7.- P. 1.
Johnson Jr. D. W. // Amer. Ceram. Soc. Bull.— 1981.— V. 60.— P. 221.
Joubert J. C, Chenavas J. // Treatise on Solid State Chemistry/Ed. by N. B.
Hannay.— New York: Plenum Press, 1975.— V. 5.
Laudise R. A. The Growth of Single Crystals.— New Jersey: Prentice-Hall,
1970.
Loehman R. E., Rao C. N. R., Honig J. M., Smith C. E. // J. Sci. Ind. Res.
(India).- 1969.- V. 28.- P. 13.
Longo J. M., Horowitz H. S. // Preparation and Characterization of
Materials/Ed. by J. M. Honig, С N. R. Rao.— New York: Academic Press, 1981.
MacDiarmid A. G., Heeger A. J. // Molecular Metals/Ed. by W. E. Hatfield.—
New York: Plenum Press, 1979.
Mammone R. J., MacDiarmid A. G. // Syn. Met — 1984.— V. 9.— P. 143.
Mizushima K., Jones P. C, Wisemann P. J., Goodenough J. B. // Mater.
Res. Bull.-1980.-V. 15.— P. 783.
Mohanram R. A., Singh K. K., Madhusudan W. H.,Ganguly P., RaoC. N. R.//
Mater. Res. Bull.—1983.—V. 18.—T. 703.
Mroczkowski S. // J. Chem. Ed.— 1980.— V. 57.— P. 537.
Multani M. // Preparation and Characterization of Materials/Ed. by J. M.
Honig, С N. R. Rao.— New York: Academic Press, 1981.
Muller-Buschbaum H. K. // Angew. Chem. Int. Edn. (Engl.).— 1981.—
V. 20.— P. 22.
Murphy D. W., Cros C, DiSalvo F. J., Waszczak J. V. // Inorg. Chem.—
1977.— V. 16.— P. 3027.
Murphy D. W., Di Salvo F. J., Carides J. N., Waszczak J. V. // Mater. Res.
Bull.— 1978.— V. 13.— P. 1395.
Murphy D. W., Christian P. A. // Science.— 1979.— V. 205.— P. 651.
Murphy D. W., Greenblatt M., Cava R. J., Zahurak S. M. // Solid State
Ionics.— 1981.— V. 5.— P. 327.
Murphy D. W., Greenblatt M., Zahurak S. M., Cava R. J., Waszczak J. V.,
Hutton R. S. // Rev. Chim. Miner.— 1982.—V. 19.— P. 441.
Narita M., Pittman С U. // Synthesis.— 1976.— P. 489.
Nigrey P. J., Mclnnes D., Nairns D. P., MacDiarmid A. C, Heeger A. J. //
J. Electrochem. Soc—1981.—V. 128.—P. 1651.
Pabst A. // Amer. Mineral.—1952.—V. 37.—P. 137.
Pistorius C. W. F. T. // Prog. Solid State Chem.— 1976.— V. 11.— P. 1.
Portier R., Carpy A., Fayard M., Galy J. // Phys. Stat. Solidi.— 1975.—
V. A30.— P. 683.
Rabenau A. // Angew. Chem. Int. Ed. (Engl.).— 1985.— V. 24.— P. 1026.
Rebbah H., Pannetier JM Raveau B. // J. Solid State Chem.— 1982.—
V. 41.— P. 57.
Rice С E., Jackel J. L. // J. Solid State Chem.— 1982.— V. 41.— P. 308.
Rosenberger F. // Preparation and Characterization of Materials/Ed. by
J. M, Honig, C. N. R. Rao.— New лгогк: Academic Press, 1981.
Roy R. // J. Amer. Caram. Soc— 1977.— V. 60.— P. 350.
Roy R., Agarwal D. K., Alamo J., Roy R. A. // Mater. Res. Bull.— 1984.—
V. 19.— P. 471.
Roy R. A., Roy R. // Mater. Res. Bull.— 1984.—V. 19.— P. 169.
Rudorff W. // Angew. Chem.—1959.—B. 71.—S. 127, 487.
484
Список литературы
Schafer H. // Solid State Chemistry. NBS Special Publication 364/Ed. by
R. S. Roth, S. J. Schneider Jr.— USA: National Bureau of Standards., 1972.
Scholze H. (Ed.). // Proc. Second Int. Workshop on Glasses and Glass
Ceramics from Gels. J. Non-Cryst. Solids.—1984.—V. 63.— P. 1—300.
Sheats J. E., Pittman Jr. С U., Carraher Jr., С. Е. // Chem. Brit.— 1984.—
V. 20.- P. 709.
Shirakawa H., Ikeda S. // Polym. J.— 1971.— V. 2.— P. 231.
Shirakawa H., По Т., Ikeda S. ,■/ Polym. J.— 1973.— V. 4- P. 460.
Simon A. // Crystal Structure and Chemical Bonding in Inorganic Chemistry/
Ed. by C. J. M. Rooymans, A. Rabonau.— Amsterdam: North-Holland, 1975.
Simon A. // Angew Chem.— 1981.— B. 20.— S. 1.
Subramanian M. A., Roberts B. D., Clearfield A. // Mater. Res. Bull.—
1984.— V. 19.— P. 1471.
Takano M., Takeda Y. // Bull. Inst. Chem. Res. (Kyoto University, Japan).—
1983.— V. 61.— P. 406.
Takeda Y., Kanamaru F., Shimada M.T Koizumi M. ,/ Acta Crystal logr.—
1976.— V. B32.— P. 2464.
Thackeray M. M., David W. I. F., Goodenough J. B. ,/ Mater. Res. Bull.—
1982.- V. 17.— P. 785.
Thackeray M. M., David W. I. F., Parks P. G., Goodenough J. B. // Mater.
Res. Bull.- 1983.- V. 18.- P. 461.
Tietjen J. J. // Ann. Rev. Mater. Sci.— 1973.— V. 3.— P. 317.
Tofield В. С // Intercalation Chemistry/Ed. by M. S. Whittingham,
A. J. Jacobson.— Now York: Academic Press, 1982.
Torardi C. C, McCarley R. E. // J. Amer. Chem. Soc— 1979.—V. 101.—
P. 3963.
Torrance J. B. // Molecular Methods/Ed. by W. E. Hatfield.— New York:
Plenum Press, 1979.
Vidyasagar K., Gopalakrishnan J. // J. Solid State Chem.— 1982.— V. 42.—
P. 217.
Vidyasagar K., Gopalakrishnan J., Rao C. N. R. // Inorg. Chem.— 1984.—
V. 23.— P. 1206. Also see: JCS Chem. Commun.— 1986.— P. 449.
Vidyasagar K., Gopalakrishnan J., Rao С N. R. // J. Solid State Chem.—
1985.— V. 58.- P. 29.
Wheland R. C, Martin E. L. // J. Org. Chem.— 1975.— V. 28.— P. 3101.
Whittingham M. S. // Prog. Solid State Chem.— 1978.—V. 12.— P. 41.
Whittingham M. S., Huggins R. A. // Solid State Chemistry. NBS Special
Publication 364/Ed. by R. S. Roth, S. J. Schneider Jr.— USA: National Bureau
of Standards 1972
Whittingham M. S., Chianelli R. R. // J. Chem. Ed.—1980.—V. 57.—
P 569.
Wold A. // J. Chem. Ed.— 1980.— V. 57.— P. 531.
Wold A., Bellavance D. // Preparative Methods in Solid State
Chemistry/Ed. by P. Hagenmuller.— New York: Academic Press, 1972.
Yeh X. L., Samwer K., Johnson W. L. // Appl. Phys. Lett.— 1983.— V. 42.—
P. 242.
ГЛАВА 4
Alder B. J., Wainright T. W. /; J. Chem. Phys.— 1957.— V. 27.— P. 1208.
Andersen H. С // J. Chem. Phys.— 1980.— V. 72.— P. 2384.
Anderson P. W. // Fizika Dielectrikov Ed. by G. I. Shanavi.— Moscow:
Acad. Nauk SSSR, 1959.
Angell C. A. // Preparation and Characterization of Materials/Ed. by
J. M. Honig, C. N. R. Rao.— New York: Academic Press, 1981.
Angell С A., Sare E. J. // J. Chem. Phys.— 1970.— V. 52.— P. 1058.
Глава 4
485
Angell С. A., Clarke J. H. R., Woodcock L. V. // Adv. Chem. Phys.— 1981.—
V. 48.— P. 397.
Aston J. G. // Physics and Chemistry of the Organic Solid State/Ed. by
D. Fox, M. Labes, A. Wessberger.— New York: Interscience, 1963.
Axe J. D. ,7 Phil. Trans. Roy. Soc London.— 1980.— V. B290.— P. 593.
Bak P. /7 Repts Prog. Phys.— 1982.— V. 45.— P. 587.
Bari R. A. // Proc. 7th Annual Conf. Magnetism and Magnetic
Materials.— Chicago: American Institute of Physics, 1972.
Bari R. A., Sivardiere J. // Phys. Rev.— 1972.— V. B.5— P. 4466.
Bhattacharya S., Nagel S. R., Fleishman L., Susman S. // Phys. Rev. Lett.—
1982.— V. 46.— P. 1267.
Bhide V. G., Rajoria D. S., Rao G. R., Rao C. N. R. // Phys. Rev.— 1972.—
V. B6.— P. 1021.
Birgeneau R. J., Kjems J. K., Shirane G., Van Uitert L. G. // Phys. Rev.—
1974.— V. BIO.— P. 2512.
Blinc R., Zeks B. Soft-Modes in Ferroelectrics and Antiferroelectrics.—
Amsterdam: North-Holland, 1974.
Bounds D. G., Klein M. L., PateyG. N. // J. Chem. Phvs.— 1980 — V. 72.—
P. 5348.
Brugeller P., Mayer E. // Nature.— 1980.— V. 288.— P. 569, 579.
Buerger M. J. // Phase Transformation in Solids/Ed. by R. Smoluchowski,
J. E. Mayer, W. A. Weyl.— New York: John Wiley, 1951.
Burgers W. G., Groen L. J. // Disc. Faraday Soc— 1957.— V. 23.— P. 183.
Bursil L. A., Thomas J. M., Rao K. J. // Nature.— 1981.— V. 289.— P. 157.
Cahn J. W. ,7 Trans AIME.- 1968.— V. 242.- P. 168.
Carneiro K., Shirane G., Werner S. A., Kaiser S. /7 Phys. Rev.— 1976.—
V. B13.- P. 4258.
Chandrasekhar S. Liquid Crystals.— Cambridge: Cambridge University
Press. 1977.
Chandrasekhar S. // Phil. Trans. Roy. Soc. London. —1983. —V. A 309.— P. 93.
Chenavas J., Joubert J. С // Solid State Comm.— 1971.— V. 9.— P. 1057.
Chesnut D. B. /7 J. Chem. Phys.— 1964.— V. 40.— P. 405.
Christian J. W. Iron and Steel Institute Special Report N 93.— 1965.
Cochran W. // Phys. Rev. Lett.— 1959.— V. 3.— P. 412.
Cohen M. H., Grest G. S. // Adv. Chem. Phys.— 1981.— V. 48.— P. 455.
Comes R., Lambert M., Launois H., Zeller H. R. // Phys. Rev.—1973.—
V. B8.— P. 571.
Cowley J. M., Cohen J. В., Salamon M. В., Wuensch B. J. (Eds). Modulated
Structures.— American Institue of Physics, 1979.
de Gennes P. G. /7 Mol. Cryst. Liq. Cryst.— 1971.— V. 12.— P. 193.
De Graaf L. A., Sciesinski J. // Physica.— 1970.— V. 48.— P. 79.
DemazeauG., Pouchard M., Thomas M., Columbert J. F., Grenier J. C, Four-
nes L., Soubeyroux J. L., Hagenmuller P. // Mater. Res. Bull.— 1980.— V. 15.—
P. 451.
DemazeauG., Marty J. L., Buffat В., Dance J. M., Pouchard M., Dordor P.,
Chevalier B. // Mater. Res. Bull.— 1982.— V. 17.— P. 37.
de Villepin J., Limage M. H., Novak A., Toupry N., Le Postollec M., Pou-
let H., Ganguly S., Rao С N. R. // J. Raman Spec— 1984.— V. 15 — P. 41.
Desiraju G. R., Paul I. C, Curtin D. Y. // J. Amer. Chem. Soc— 1977. —
V. 99.— P. 1594.
Di Salvo F. J. // Elrctron-Phonon Interactions and Phase Transitions/Ed.
by T. Riste.— New York: Plenum Press, 1977.— P. 107.
DiSalvo F. J., Rice Т. М. // Physics Today (April issue).— 1973.
Domb С ,7 Contemp. Phys.—1985.—V. 26.— P. 49.
Dubocket J., Lepault J., Freeman R. // J. Microse — 1982.— V. 128.— P. 219.
Dzyaloshinskii J. E. // Soviet Phys. JETP.— 1964.— V. 19.— P. 960.
Elliott R. J., Harley R. Т., Hayes W., Smith S. R. P. // Proc. Roy. Soc.
London.— 1972.— V. A328.—P. 217.
486
Список литературы
Elliott S. R., Rao С. N. R., Thomas J. M. // Angew. Ghem. Int. Ed. (Engl.).—
1986.— V. 25.— P. 31.
Emery V. J., Axe J. D. // Phys. Rev. Lett.— 1978.— V. 40.— P. 1507.
Englman R. The Jahn — Teller Effect in Molecules and Crystals.—
London: John Wiley, 1972.
Fleury P. A. // Phase Transitions/Ed. by H. Henisch, R. Roy., L. E. Cross.—
New York: Pergamon Press, 1973.
Fleury P. A., Lyons К. В. // Light Scattering near Phase Transition/Ed.
bv H. Z. Cummins, A. P. Levanyuk.— Amsterdam: North-Holland, 1983.
Frank С // Phil. Trans. Roy. Sos. London.—1983.—V. A309.— P. 71.
Freeman A. J., Schmid H. (Eds). Magnetoelectric Interaction Phenomena
in Crystals.— New York: Gordon, Breach, 1975.
Frost J. C, Leadbetter A. J., Richardson R. M. // Disc. Faraday Soc.—
1980.— V. 69.- P. 32.
Ganguly S., Fernandes J. R., Bahadur D., Rao C. N. R. // J. Chem. Soc.
Faraday 2.— 1979.—V. 75.—P. 923.
Gaskell P. H., Smith D. J., Catto С J. D., Cleaver J. R. A. // Nature.—
1979.- V. 281.— P. 465.
GehringG. A., Gehring K. A. // Repts. Prog. Phys — 1975 — V. 38.— P. 1.
Gibbs J. H. // Modern Aspects of the Vitreous State/Ed. by J. D.
Mackenzie.— London: Butterworths, 1963.
Goodenough J. B. // Phase Transitions/Ed. by H. Henisch, R. Roy, L. E.
Cross.— New York: Pergamon Press, 1973.
Gordon R. G. // J. Chem. Phys.— 1966.— V. 44.— P. 1830.
Gray G. W. Phil. Trans. Roy. Soc— Loudon.— 1983.— V. A309.— P. 77.
Gray G. W., Winsor P. A. (Eds). Liquid Crystals and Plastic Crystals.—
Chichester: Ellis Horwood, 1974.
Guthrie G. В., McCullough J. // Phys. Chem. Solids.—1961—V. 18.—
P. 53.
Giitlich P. // Structure and Bonding.— 1981a.—V. 44,— P. 83.
Giitlich P. // Mossbauer Spectroscopy and its Chemical Applications.
Advances in Chemistry Series 194.— American Chemical Society, 1981b.
Haddad M. S„ Lynch M. W., Federer W. D., Hendrickson D. N. // Inorg.
Chem.— 1981.—V. 20.—P. 131.
Harley R. Т., Hayes W., Perry A. M., Smith S. R. P. // J. Phys — 1973.—
V. Сб.— Р. 2382.
Harley R. Т., Lyons К. В., Fleury P. A. // J. Phys — 1980.— V. C13.—
P. L 447.
Heine V., McConnell J. D. C. // Phys, Rev. Lett.— 1981.— V. 46.— P. 1092.
Heine V., McConnell J. D. С // J. Phys.— 1984.— V. C17.— P. 1199.
Hill N. E. // Proc. Phys. Soc. London.— 1963.— V. 82.— P. 723.
Hilsum C, Raynes E. P. (Eds). // Phil. Trans. Roy. Soc— London.— 1983.—
V. A309.— P. 69.
Hoare M. R., Barker J. A. // The Structure of Non-Crystalline Materials/Ed.
by P. H. Gaskell.— London: Taylor, Francis, 1977.
Hochli U. Т., Weibel H. E., Boathner L. A. // J. Phys.— 1979.— V. C12.—
P. L563.
Iizumi M., Axe J. D., Shirane G., Simaoka K. // Phys. Rev.— 1977.—
V. B15.— P. 4392.
Joesten M. D., Takagi S., Lenhert P. G. // Inorg. Chem.— 1977.— V. 16.—
P. 2680.
Johari G. P. // J. Chem. Phys.— 1982.— V. 77.— P. 4619.
Johari G. P., Goodby J. W. // J. Chem. Phys.— 1982.— V. 77.— P. 565.
Johnson С. К., Watson С. R. // J. Chem. Phys.— 1976.— V. 64.— P. 2271.
Jones W., Thomas J. M., Williams J. O. // Phil. Mag.— 1975.— V. 32.—
P. 1.
Kadanoff L. P. // Physics.— 1966.—V. 2.— P. 263.
Kambara T. // J. Chem. Phys.—1979.—V. 70.—P. 4199.
Глава 4
487
Kambara Т. // J. Ghem. Phys.— 1981.— V. 74.— P. 4557.
Kanno H. // J. Non-Cryst. Solids.— 1983.—V. 37.—P. 203.
Kauzmann W. // Chem. Rev.— 1948.— V. 3.— P. 219.
Klein M. L. // Ann. Rev. Phys. Chem.— 1985.— V. 36.— P. 525.
Klein M. V. // Light Scattering near Phase Transitions/Ed. by H. Z.
Cummins, A. P. Levanyuk.— Amsterdam: North-Holland, 1983.
Konig E., Kremer S. // Theor. Chem. Acta.— 1971.— V. 20.— P. 143.
Konig E., RitterG., Irler W. // Chem. Phys. Lett.— 1979.— V. 66.— P. 336.
Konig E., RitterG., Kulshreshta S. K., Nelson S. M. // J. Am. Chem. Soc —
1983.—V. 105.- P. 1924.
Konig E., RitterG., Kulshreshta S. K. // Chem. Revs —1985.—V. 85.—
P. 219.
Lechner R. E. // Solid State Comm.— 1982.— V. 10.— P. 1247.
Madhusudan W. H., Jagannathan K., Ganguly P., Rao С N. R. // J. Chem.
Soc. Dalton.— 1980.— P. 1397.
Makovicky E., Hyde B. G. // Structure and Bonding.—1981.—V. 46.—
P. 101.
Mandelbrot В. В. The Fractal Geometry of Nature.— New York: Freeman
and Co., 1983.
McConnell J. D. C, Heine V. // J. Phys.— 1982.— V. C15.— P. 2387.
McDonald I. R. // Mol. Phys.—1972.—V. 23.—P. 41.
McDonald I. R., Bounds D. G., Klein M. L. // Mol. Phys — 1982 — V. 45.—
P. 521.
McFarlane D. R., Angell C. A. // J. Phys. Chem.— 1984.— V. 88.— P. 759.
McMillan J. A., Los S. C. // J. Chem. Phys.—1965.—V. 42.—P. 829.
Metropolis N. A., Rosenbluth A. W., Rosenbluth M. N., Teller A. H.,
Teller E. // J. Chem. Phvs.— 1953.— V. 21.— P. 1087.
Mohanram R. A., Singh K. K., Madhusudan W. H.t Ganguly P., Rao C. N. R.
// Mater. Res. Bull.—1983.—V. 18.—P. 703.
Natarajan M., Das A. R., Rao С N. R. // Trans. Faraday Soc— 1969.—
V. 65.— P. 3081.
Nose S., Klein M. L. // J. Chem. Phys. — 1983a.— V. 78.— P. 6928.
Nose S., Klein M. L. // Mol. Phys.— 1983b.— V. 50.— P. 1055.
Overhauser A. W. // Phys. Rev.— 1962.— V. 128.— P. 1437.
Parkinson G. M., Thomas J. M., Williams J. O., Goringe M. J.t Hobbs L. W. //
J. Chem. Soc. Perkin 2.— 1976.— P. 836.
Parrinello M., Rahman A. // Phys. Rev. Lett.— 1980.—V. 45.— P. 1196.
Parrinello M., Rahman A., Vashista P. // Phys. Rev. Lett.— 1983.— V. 50.—
P. 1073.
Parsonage N. G., Staveley L. A. K. Disorder in Crystals.— Oxford:
Clarendon Press, 1978.
Parthasarathy R., Rao K. J., Rao C. N. R. // Chem. Soc. Revs.— 1983.—
V. 12.- P. 361.
Parthasarathy R., Rao K. J., Rao С N. R. // J. Phys. Chem.—1984.—
V. 88.- P. 49.
Paul I. C, Go K. T. // J. Chem. Soc. В.— 1969.— P. 33.
Porter D. A., Easterling К. Е. Phase Transitions in Metals and Alloys.—
New York: Van Nostrand-Reinhold, 1981.
Pouget J. P., Shirane G., Hastings J. M., Heeger A. J., Miro N. D., Mac-
Diarmid A. G. // Phys. Rev.— 1978.— V. B18.— P. 3645.
Pytte E., Stevens K. W. H. // Phys. Rev. Lett.— 1971.— V. 27.— P. 862.
Qi Y., Muller E. W., Spiering H., Gutlich P. // Chem. Phys. Lett.— 1983.—
V. 101.- P. 503.
Raccah P. M., Goodenough J. B. // Phys. Rev.— 1967.— V. 155.— P. 932.
Ramasesha S., Ramakrishnan T. V., Rao C. N. R. // J. Phys.— 1979 —
V. C12.— P. 1307.
Ramdas S.T Thomas J. M. // J. Chem. Soc. Faraday 2.— 1976.— V. 72.—
P. 1251,
488
Список литературы
Rao С. N. R. // Асе. Chem. Res.— 1984.— V. 17.— P. 83.
Rao С. N. R. // Int. Rev. Phys. Chem.— 1985a.— V. 4.— P. 19.
Rao С N. R. // Proc. Ind. Acad. Sci. (Chem. Sci.).— 1985b.— V. 94.—
P. 181.
Rao С N. R., Rao K. J. Phase Transitions in Solids.— New York: McGraw-
Hill, 1978.
Rao С N. R.f Ganguly S., Swamy H. R., Oxton I. A. // J. Chem. Soc.
Faraday 2.- 1981.-V. 77.- P. 1825.
Rao С N. R., Ganguly S., Swamy H. R. // Croat. Chem. Acta.— 1982.—
V. 55.— P. 207.
Rao K. J., Rao С N. R. // J. Mat. Sci.— 1966.— V. 1.— P. 238.
Rao K. J., Rao С N. R. // Mater. Res. Bull.- 1982.- V. 13.- P. 1337.
Riste T. (Ed.). Electron-Phonon Interactions and Phase Transitions.— New
York: Plenum Press, 1977.
Samara G. A. // Solid State Phys.— 1984.— V. 38.— P. 1.
Sasaki N.. Kambara T. // J. Phys.— 1982.— V. C15.— P. 1035.
Sceats M. S., Rice S. A. // Water: A Comprehensive Treatise/Ed. by F.
Franks.— New York: Plenum Press, 1982.— V. 7.
Scott J. F. // Rev. Mod. Phys.— 1974.— V. 46.— P. 83.
Shapiro S. M., Axe J. D., ShiraneG., Riste T. // Anharmonic Lattices.
Structural Transitions and Melting/Ed. by T. Riste.— Leiden: Noordhoff, 1974.
Sherwood J. N. (Ed.). The Plastically Crystalline State.— New York: John
Wiley, 1979.
Shirane G., Yamada Y. // Phys. Rev.- 1969.- V. 177.- P. 858.
Shvartsman F., Krongauz V. // Nature.— 1984.— V. 309.— P. 608.
SlichterC. P., Drickamer H. G. // J. Chem. Phys.— 1972.— V. 56.— P. 2142.
Sorai M., Seki S. // J. Phys. Chem. Solids.— 1974.— V. 35.— P. 555.
Spiering H., Meissner E., Koppen H., Mtiller E. W., Gutlich P.// Chem.
Phys.— 1982.— V. 68.— P. 65.
Sturge M. D., Cohen E., Van Uitert L. G., Van Stapele R. P. // Phys. Rev.—
1975.—V. Bll.— P. 4768.
Suga H., Seki S. // Faraday Disc. Chem. Soc— 1980.— P. 69.
Sugai S. // J. Phys.— 1985.— V. C18.— P. 799.
Sugisaki M., Suga H., Seki S. // Bull. Chem. Soc. Japan.— 1968.— V. 41.—
P. 2591.
Tanabe Y., Sugano S. /7 J. Phys. Soc. Japan.— 1954.— V. 9.— P. 753, 766.
Usha S., Srinivasan R., Rao С N. R. // Chem. Phys.— 1985.— V. 100.—
P. 447.
Venkataraman G. // Bull. Mater. Sci.— 1979.— V. 1.— P. 129.
WHIG., Gobel H., Sampson С F., Forsyth J. B. // Phys. Lett.— 1972.—
V. 38A.— P. 207.
Williams P. M. // Crystallography and Crystal Chemistry of Materials with
Layered Structures/Ed. by F. Levy.— Dordrecht-Holland: Reidel, 1976.
Wilson J. A., Di Salvo F. J., Mahajan S. // Adv. Phys.— 1975.— V. 24.—
P. 117.
Wilson K. G., Fisher M. E. // Phys. Rev. Lett.- 1972.— V. 28.— P. 248.
Wong J., Angell С A. Glass Structure by Spectroscopy.— New York:
Marcel Dekker, 1976.
Wood W. W. // Physics of Simple Liquids/Ed. by H. N. V. Temperley,
J. N. Rowlinson, G. S. Rushbrooke — Amsterdam: North-Holland, 1968.
Wyllie G. // J. Phys.— 1971.— V. C4.— P. 564.
Yakovlev I. A., Velichkina T. S., Mikheeva L. F. // Sov. Phys. Doklady.—
1956.— V. 1.— P. 215.
Yamada Y. // Electron-Phonon Interactions and Phase Transitions/Ed. by
T Riste.— New York: Plenum Press, 1977.
Yashonath S., Rao С N. R. // Chem. Phys. Lett.— 1983.— V. 101.— P. 524.
Yashonath S., Rao С N. R. // Chem. Phys. Lett.— 1985a.— V. 119.— P. 22.
Глава 5
489
Yashonath S., Rao С. N. R. // Mol. Phys.— 1985b.— V. 54.— P. 245.
Yashonath S., Rao C. N. R. // Proc. Roy. Soc. London.— 1985c —
V. A400.— P. 61.
ГЛАВА 5
Alario-Franco M. A., Regi M. V. // Nature.— 1977.— V. 270.— P. 706.
Alario-Franco M. A., Joubert J. C, Levy J. P. // Mater. Res. Bull.— 1982.—
V. 17.— P. 733.
Alario-Franco M. A., Calbet J. M. G., Regi M. V.t Grenier J. C. // J. Solid
State Chem.—1983.—V. 49.— P. 219.
Allpress J. G., Sanders J. V. /7 J. Appl. Cryst.— 1973.— V. 6.— P. 165.
Anderson J. S. // Surface and Defect Properties of Solids/Ed. by M. W.
Roberts, J. M. Thomas.— London: The Chemical Society, 1972a.— V. 1.
Anderson J. S. // Solid State Chemistry: NBS Special Publication 364/Ed.
by R. S. Roth, S. J. Schneider.— Washington D. C: US Department of
Commerce, 1972b.
Anderson J. S. // J. Chem. Soc. Dalton Trans.— 1973.— P. 1107.
Anderson J. S. // Defect and transport in Oxides/Ed. by M. S. Seltzer, R. I.
Jaffe.— New York: Plenum Press, 1974.
Anderson J. S. /7 Proc. Indian Acad. Sci. (Chem. Sci.)- — 1984.— V. 93.—
P. 861.
Anderson J. S., Browne J. M., Hutchison J. L. // Nature.— 1972.— V. 237.—
P. 5351.
Anderson J. S., Tilley R. J. D. /7 Surface and Defect Properties of Solid/Ed.
by M. W. Roberts, J. M. Thomas — London: The Chemical Society, 1974.— V. 3.
Anderson J. S., Hutchison J. L. // Contemp. Phys.— 1975.— V. 16.— P. 443.
Andersson В., Gjonnes J. // Acta Chem. Scand.— 1970.— V. 24.— P. 2250.
Atoji M., Bundle R. E. /7 J. Chem. Phys.— I960.— V. 32.— P. 627.
Aurivillius B. /7 Ark. Kemi.— 1950.— V. 2.— P. 519.
Bando Y., Sekikawa Y., Nakamura H., Matsui Y. // Acta Crystallogr.—
1981.- V. A37.- P. 723.
Bauer E., Pianelli A., Aubry A., Jeannot F. // Mater. Res. Bull — 1980 —
V. 15.- P. 323.
Bertaut E. F. // Acta Crystallogr.— 1956.— V. 6.— P. 357.
Bevan D. J. M., Strahle J., GreisO.//J. Solid State Chem.—1982.—V. 44.—
P. 75.
Blanchin M. G., Bursill L. A., Smith D. J. // Proc. Roy. Soc. London.—
1984.-V. A391.- P. 351.
Blasse G., Dirksen G. J. // J. Solid State Chem.— 1981.— V. 36.— P. 124.
Bridges F. // CRC Crit. Rev. Solid State Science.— 1975.—V. 5.— P. 1.
Brown R., Kemmler-Sack S. // Naturwissenschaften.— 1983.— B. 70.—
S. 463.
Bursill L. A., Blanchin M. G., Smith D. J. // Proc. Roy. Soc. London.—
1984.- V. A391.- P. 373.
Bursill L. A., Smith D. J. // Nature.— 1984.— V. 309.— P. 319.
Caignaert V., Nguyen N., Hervieu M., Raveau B. // Mater. Res. Bull.—
1985.- V. 20.- P. 479.
Calbet J. M. G., Regi M. V., Alario-Franco M. A., Grenier J. С // Mater.
Res. Bull.- 1983.— V. 18.— P. 285.
Calvo C, Ng N. N., Chamberland B. L. // Inorg. Chem.— 1978.— V. 17.—
P. 699.
Catlow C. R. A., James R. // Chemical Physics of Solids and Surfaces/Sr.
Reporters M. W. Roberts, J. M. Thomas.— London: The Chemical Society, 1980.—
V. 8.
Catlow С R. A., Mackrodt W. С (Eds). Computer Simulation of Solids.
Lecture Notes in Physics.— Berlin: Springer-Verlag, 1982.
32 4. H. P. Pao, Дж. Гопалакришнан
490
Список литературы
Catlow С. R. A., Chadwick А. V., Corish J. // J. Solid State Chem.— 1983.—
V. 48.- P. 65.
Catlow C. R. A., Chadwick A. V., Greaves G. N., Moroney L. M. // Nature.—
1984.- V. 312.- P. 601.
Cheetham A. K., Fender B. E. F., Taylor R. I. ,7 J. Phys. C— 1971a.—
V. 4.— P. 2160.
Cheetham A. K., Fender B. E. F., Cooper M. J. 7 J. Phvs. C— 1971b.—
V. 4.- P. 3107.
Conrad B. R., Franzen H. F. // The Chemistry of Extended Defects in Non-
metallic Solids/Ed. by M. 0'Keeffc, L. Eyring.— Amsterdam:
North-Holland, 1970.
Corish J., Jacobs P. W. M., Radhakrishna S. // Surface and Defect
Properties of Solids/Sr. Reporters Roberts M. W., Thomas J. M..— London: The
Chemical Society, 1977.— V. 6.
Corish J., Quigley J. M. // Computer Simulation of Solids/Ed. by C. R. A.
Catlow, W. C. Mackrodt.— Berlin: Springer-Vcrlag, 1982.
Dismukes J. P., White J. G. // Inorg. Chem.— 1964.— V. 3.— P. 1220.
Drennan J., Tavares С P., Steele В. С. Н. // Mater. Res. Bull.— 1982.—
V. 17.- P. 621.
Ekstrom Т., Tilley R. J. D. // Chem. Scripta.— 1980.— V. 16.— P. 1.
Emmons G. H., Williams W. S. // J. Mater. Sci.— 1983.— V. 18.— P. 2589.
Ег-Rakho L., Michel C, Provost J., Raveau B. // J. Solid State Chem.—
1981.- V. 37.- P. 151.
Fine M. E. //' Treatise on Solid State Chemistry/Ed. by N. B. Hannay.— New
York: Plenum Press, 1973.— V. 1.
Fowler W. B. Physics of Colour Centres.— New York: Academic Press, 1968.
Fowler W. B. // Treatise on Solid State Chemistry/Ed. by N. B. Hannay.—
New York: Plenum Press, 1975.— V. 2.
Franzen H. F., Tuenge R. Т., Eyring L. // J. Solid State Chem.— 1983.—
V. 49.- P. 206.
Franzen H. F., Folmer J. С W. // J. Solid State Chem.— 1984 — V. 51.—
P. 396.
Gai P. L., Rao С N. R. // Z. Naturforsch.— 1975a.— В. 30А.— S. 1092.
Gai P. L., Rao С N. R. // Pramana.- 1975b.- V. 55.- P. 274.
Ganguly P., Rao С N. R. // J. Solid Slite Chem.— 1984.— V. 53.— P. 193.
Cavarri J. // Thesis. Univ. Paris VI.— 1978.
Gettmann W., Greis O. // J. Solid State Chem.— 1978.- V. 26.- P. 255.
Gillan M. J., Jacobs P. W. M. // Phys. Rev.— 1983.— V. B28.- P. 758.
Gleiter H. // Physical Metallurgy. 3rd edn. // Ed. by R. W. Cahn, P. Haa-
scn.— Amsterdam: North-Holland, 1983.
Gopalakrishnan J., Ramanan A., Rao С N. R., Jefferson D. A., Smith D. J. //
J. Solid State Chem.-1984.-V. 55.—P. 101.
Greis O. // Z. Anorg. Allgem. Chem.— 1977.— B. 430.— S. 175.
Grenier J. C, Ghodbane S., Demazeau G., Pouchard M., Hagenmuller P. //
Mater. Res. Bull.- 1979.— V. 14.- P. 831.
Grenier J. C, Pouchard M., Hagenmuller P. // Structure and Bonding.—
1981.— V. 47.— P. 1.
Grimes N. W. // Contemp. Phys.— 1976.— V. 17.— P. 71.
Gruehn R., Mertin M. // Angew. Chem. Int. Ed. (Engl.).— 1980.— V. 19.—
P. 505.
Hutchison J. L., Jacobson A. J. // J. Solid State Chem.— 1977.— V. 20.—
P. 417.
Hutchison J. L., Anderson J. S., Rao С N. R. // Proc. Roy. Soc. London —
1977 у А355 P. 301.
' Jijima S. // J. Solid State Chem.- 1975.- V. 14.— P. 52.
Jacobson A. J. // Acta Crystallogr.- 1976.- V. B32.- P. 1087.
Jacobson A. J., Hutchison J. L. // J. Solid State Chem.— 1980.— V. 35.—
P. 334.
Глава 5
491
Jacobson A. J., Scanlon J. С, Poeppelmeier К. R., Longo J. M., Cox D.
Б. //Mater. Res. Bull.- 1981.— V. 16.- P. 359.
Jefferson D. A., Uppal M. K., Rao С N. R., Smith D. J. // Mater. Res.
Bull.-1984.-V. 19.-P. 1403.
Jones W., Thomas J. M. // Prog. Solid State Chem.—1979.—V. 12.—
P. 101.
Kato K., Kitamura M. // Acta Crystallogr.— 1981.— V. A37.— P. 307.
Kihlborg L. // Chem. Scripta.— 1978.- V. 14.— P. 187.
Kittel С // Solid State Comm.- 1978.- V. 25.- P. 519.
Koch F., Cohen J. B. // Acta Crystallogr.— 1969.— V. B25.— P. 275.
Kroger F. A. The Chemistry of Imperfect Crystals.— Amsterdam: North-
Holland, 1974.- V. 2.
Laval P. L., Frit B. // J. Solid State Chem.— 1983.— V. 49.— P. 237.
LibowitzG. G. // Treatise on Solid State Chemistry/Ed. by N. B. Hannay. —
New York: Plenum Press, 1973.— V. 1.
LoehmanR., Rao С N. R., Honig J. M.//J. Phys. Chem.— 1968.— V. 73.—
P. 1781.
Longo J. M., Sleight A. W. // Mater. Res. Bull.— 1975.- V. 10.- P. 1273.
MacChesney J. В., Sherwood R. C, Potter J. F. // J. Chem. Phys.— 1965.—
V. 43.- P. 1907.
McConnell A., Anderson J. S., Rao С N. R. // Spectrochim. Acta.— 1976.—
V. 32A.— P. 1067.
Moret R., Huber M., Comes R. // J. Phys. Colloq.— 1977.— V. C7.— P. 202.
Mori S. // J. Phys. Soc. Japan.— 1970.— V. 28.- P. 44.
Morinaga M., Cohen J. B. // Acta Crystallogr.— 1979.— V. A35.—
P. 745, 975.
Mott N. F., Littleton M. J. // Trans. Faraday Soc— 1938.— V. 34.— P. 485.
Nakamura Т., Petzow G., Gauckler L. J. //Mater. Res. Bull —1979.—
V. 14.- P. 649.
Nakazawa H., Morimoto N. // Mater. Res. Bull.— 1971.— V. 6.—P. 345.
Negas Т., Roth R. S. // J. Solid Slate Chem.- 1971,- V. 3.- P. 323.
Nembach E. // Treatise on Solid State Chemistry/Ed. by N. B. Hannay.—
New York: Plenum Press, 1974.— V. 2.
Poeppelmeier K. R., Leonowicz M. E., Scanlon J. C, Longo J. M., Yelon
W. B. // J. Solid State Chem.— 1982.— V. 45.— P. 71.
Portier R., Carpy A., Fayard M., Galy J. // Phys. Stat. Solidi.—1975,—
V. A30.— P. 683.
Ramanan A., Gopalakrishnan J., Uppal M. K., Jefferson D. A., Rao C. N.
R. // Proc. Roy. Soc. London.- 1984.-V. A395.—P. 127.
Rao С N. R. // Chem. Scripta.— 1982.— V. 19.- P. 124.
Rao C. N. R. // Bull. Mat. Sci.— 1985.— V. 7.— P. 155.
Rao С N. R., Gai P. L., Ramasesha S.//Phil. Mag.—1976.—V. 33.—
P. 387.
Rao С N. R., Thomas J. M. // Ace. Chem. Res.— 1985.— V. 18.— P, 113.
Rauser G., Kemmler-Sack S. // J. Solid State Chem.—1980.—V. 33.—
P. 135.
Reller A., Thomas J. M., Jefferson D. A., Uppal M. K. // Proc. Roy. Soc.
London.— 1984.— V. A394.— P. 223.
Roth R. S., Stephenson N. С // The Chemistry of Extended Defects in Non-
metallic Solids/Ed. by L. Eyring, M. O' Keeffe M.— Amsterdam: North-Holland,
1970.
Roth W. L. // Acta Crystallogr.— I960.— V. 13.— P. 140.
Shannon J., Katz L. // Acta Crystallogr.— 1970— V. B26. — P. 102.
S0rensen О. Т. (Ed.). Nonstoichiomctric Oxides.— New York: Academic
Press 1981
Stoneliam A. M. // J. Phys.— 1971.— V. Fl.— P. 778.
Stoneham A. M. Theory of Defects in Solids.— Oxford: Clarendon Press,
1975.
32*
492
Список литературы
Studer F., Raveau В. // Phys. Stat. Solidi.— 1978.—V. A48.— P. 301.
Subramanian M. A., Aravamudan G., Subba Rao G. V. II Prog. Solid State
Chem.— 1983.— V. 15.— P. 55.
Suzuki K. // J. Phys. Soc. Japan.— 1961.— V. 16.— P. 67.
Taylor A., Doyle N. J. // J. Appl. Cryst.— 1972.- V. 5.— P. 201.
Terasaki 0., Thomas J. M., Ramdas S. // J. Chem. Soc. Chem. Comm.—
1984.- P. 216.
Terauehi If., Cohen J. В., Reed T. B. // Acta Crystallogr.— 1978.— V. A34 —
P. 556.
Terauehi H., Cohen J. B. 7 Acta Crystallogr.— 1979.— V. Л35.— P. 646.
Tewary U. K., Bullough R. // J. Phys.— 1972.— V. F2.— P. 269.
Thomas J. M., Williams J. 0. // Prog. Solid State Chem.— 1971.— V. 6.—
P. 271.
Thornber M. R., Bevan D. J. M. // J. Solid State Chem.— 1970.— V. 1.—
P. 536.
Tilley R. J. D. // J. Solid State Chem.- 1977.- V. 21.- P. 293.
Tilley R. J. D. // Chemical Physics of Solids and Surfaces/Sr. Reporters
M. W. Roberts, J. W. Thomas.— London: The Chemical Society, 1980.—V. 8.
Tofield В. С // Nature.- 1978.— V. 272.- P. 713.
Tofield B. C, Scott W. R. // J. Solid State Chem.— 1974.— V. 10.— P. 183.
Tofield B. C, Greaves C, Fender В. Е. F. // Mater. Res. Bull — 1975 —
V. 10.— P. 737.
Uppal M. K., Rao С N. R., Sangster M. J. L. II Phil. Mag.—1978.—
V. 38.- P. 341.
Vidyasagar K., Gopalakrishnan J., Rao C. N. R. // Inorg. Chem.— 1984 —
V. 23.— P. 1206.
Vidyasagar K., Reller A., Gopalakrishnan J., Rao С N. R. 111. Chem. Soc.
Chem. Comm.— 1985.— P. 7.
Vidyasagar K., Ganapathi L., Gopalakrishnan J., Rao С N. R. // J. Chem.
Soc. Chem. Comm.— 1986.— P. 449.
Vincent H., Bochu В., Aubert J. J., Joubert С. С, Marezio M. // J. Solid
State Chem.— 1978.— V. 24.— P. 245.
Wadsley A. D., Andersson S. // Perspectives in Structural Chemistry/Ed.
by J. D. Dunitz, J. A. Ibcrs.— New York: John Wiley, 1970.— V. 3.
Watanabe D., Terasaki O., Jostsons A., Castles J. R. // The Chemistry of
Extended Defects in Nonmetallic Solids/Ed. by L. Eyring, M. O' Keeffe. —
Amsterdam: North-Holland, 1970.
Willis В. Т. М. // J. Physique.— 1964.— V. 25.— P. 431.
Wollenberger H. J. // Physical Metallurgy, 3rd edn./Ed. by R. W. Cahn,
P. Haasen.— Amsterdam: North-Holland, 1983.
Zanne M., Gleitzer С // Bull. Soc. Chim. Fr.— 1971.— P. 1567.
Zanne M., Courtois A., Gleitzer С // Bull. Soc. Chim. Fr.— 1972.— P. 4470.
ГЛАВА 6
Abrahams E., Anderson P. W., Licciardello D. C, Ramakrishnan T. V. //
Phys. Rev. Lett.— 1979.— V. 42.— P. 673.
Adler D. // Treatise on Solid State Chemistry/Ed. by N. B. Hannay.— New
York: Plenum Press, 1975.— V. 2.
Aizu K. // Phys. Rev.— 1970.— V. B2.— P. 754.
Alcacer L. (Ed.). The Physics and Chemistry of Low Dimensional Solids.—
Dordrecht-Holland: Reidel, 1980.
Allen P. В., Mitrovic B. // Solid State Phys.— 1982.— V. 37.— P. 2.
Anderson P. W. // Phys. Rev. — *1958.— V. 109.— P, 1492.
Anderson P. W. // Phys. Rev.— 1959.— V. 115.— P. 2.
Ando Т., Fowler H. B.T Stern F. // Rev. Mod. Phys.— 1982.—V. 54.—
P. 437.
Глава 6
493
Ascher H., Reider H., Schmid H., Stossell H. // J. Appl. Phys.— 1966.—
V. 37.— P. 1404.
Balkanski N. (Ed.). Light Scattering in Solids.— Paris: Flammarion, 1971.
Banus M. D., Reed T. B. // The Chemistry of Extended Defects in Nonmetal-
Цс Solids/Ed. by L. Eyring, M. 0'Keeffe.—Amsterdam: North-Holland, 1970.
Bell A. E., Caplin A. D. // Contemp. Phys.— 1975.— V. 16.— P. 375.
Benfiel R. E., Edwards P. P., Stacy A. M. // J. Chem. Soc. Chem. Comm.—
1982.— P. 525.
Bhide V. G., Raioria D. S., Rao G. R., Rao С N. R. // Phys. Rev.— 1972.—
V. B6.— P. 1021.
Blinc R., Zeks B. Soft Modes in Ferroelectrics and Antiferroelectrics.—
Amsterdam: North-Holland, 1974.
Bloor D. // Phil. Trans. Roy. Soc. London.— 1985.—V. A314.— P. 67.
Bongers P. F. // Crystal Structure and Chemical Bonding in Inorganic
Chemistry/Ed. by C. J. M. Rooymans, A. Rabenau.— Amsterdam: North-Holland,
1975.
Bouchard R. J., Gillson J. L., Jarett H. S. // Mater. Res. Bull.— 1973.—
V. 8.— P. 489.
Brandow В. Н. // Adv. Phys.— 1977.— V. 26.— P. 651.
Bronger W. // Angew. Chem. Int. Edn (Engl.).—1981.—V. 20.—P. 52.
Brown D. B. (Ed.). Mixed-Valence Compounds.— Dordrecht-Holland:
Reidel, 1980.
Buttrey D. PhD Thesis.— Purdue University, USA, 1984.
Cahn R. W. // Contemp. Phys.— 1980.— V. 21.— P. 43.
Chien J. С W. Polyacetylene: Chemistry, Physics and Materials Science.—
New York: Academic Press, 1984.
Clark R. J. II. .// Ann. N. Y. Acad. Sci.- 1977.- V. 313.- P. 672.
Coey J. M. D., Roux-Buisson H., Brusetti R. // J. Physique. Colloq.— 1976.—
Т. С4.— P. 11.
Coleman L. В., Cohen M. J., Sandman D. J., Yamagishi F. G., Garito A. F.f
Heeger A. J. // Solid State Comm.— 1973.— V. 12.— P. 1125.
Corish J., Jacobs P. W. M., Radhakrishna S. // Surface and Defect
Properties of Solids/Sr. Reporters M. W. Roberts, J. M. Thomas.— London: The
Chemical Society, 1977.— V. 6.
Cotton F. A. Chemical Applications of Group Theory.— 2nd Edn.— New
Delhi: Wiley-Eastern, 1971.
Creutz C, Taube H. // J. Amer. Chem. Soc— 1973.— V. 95.— P. 1086.
Datars W. R., Razavi F. S., Gillespie R. J., Ummat P. // Phil. Trans. Roy.
Soc. London.—1985.—V. A314.—P. 115.
Datars W. R., Ummat P. K., Gillespie R. J. // Mater. Res. Bull.— 1985.—
V. 20.— P. 865.
Day P. // J. Amer. Chem. Soc— 1975.— V. 97.— P. 1588.
Day P. // Ace. Chem. Res.— 1979.— V. 12.— P. 237.
Day P. // Int. Rev. Phys. Chem.— 1981.— V. 1.— P. 149.
Day P. // Chem. Britain.— 1983.— V. 19.— P. 306.
Day P. // Phil. Trans. Roy. Soc. London.—1985.—V. A314.—P. 145.
du Jeu W. H. Physical Properties of Liquid Crystalline Materials.— New
York: Gordon, Breach, 1980.
de Jongh L. J., Miedema A. R. .// Adv. Phys.— 1974.— V. 23.— P. 1.
Demazeau G., Langlais F., Portier J., Tressaud A., Hagenmuller P. // High
Pressure Science and Technology.— 1980.— V. 1.— P. 579.
De Vries А. В., Haas С // J. Phys. Chem. Solids.— 1973.— V. 34.— P. 651.
Dorner В., Axe J. D.. Shirane G. // Phys. Rev.— 1972.— V. B6.— P. 1950.
Duwez P., Johnson W. L. // J. Less-Common Metals.—1978.—V. 62.—
P. 607.
Edwards P. P., Sienko M. J. // Int. Rev. Phys. Chem.— 1983.— V. 3.—
P. 83.
494 Список литературы
Edwards P. P., Rao С. N. R. (Eds). The Metallic and Nonmetallic States
of Matter.— London: Taylor, Francis, 1985.
Edwards S. F., Anderson P. W. // J. Phys.— 1975.— V. F5.— P. 965.
Elliott S. R. Physics of Amorphous Materials.— London: Longman, 1984.
Emin D. // Phil. Mag.— 1977.— V. 35.— P. 1189.
Etemad S., Heeger A. J., MacDiarmid A. G. // Ann. Rev. Phys. Chem.—
1982.— V. 33.— P. 443.
Eyring L. // Solid State Chemistry/Ed. by С N. R. Rao.— New York:
Marcel Dekker, 1974.
Eyring L. // Handbook on the Physics and Chemistry of Rare Earths/Ed.
by K. A. Gschneidner Jr., L. Eyring — Amsterdam: North-Holland, 1979.
Falicov L. M., Hanke W., Maple M. P. (Eds). Valence Fluctuation in
Solids.— Amsterdam: North-Holland, 1981.
Ford P. J. // Contemp. Phys.— 1982.— V. 23.— P. 141.
Franzen H. F. // Prog. Solid State Chem.— 1978.— V. 12.— P. 1.
Friebel C, Reinen D. // Z. Anorg. Allgem. Chem.— 1974.— B. 407.— S. 193.
Fyne P. J., Day P., Hutchings M. Т., Depinna S., Cavenett B. C, Pynn R. //
J. Phys.— 1984.— V. C17.— P. L245.
Ganguly P., Rao C. N. R. // J. Solid State Chem.—1984.—V. 53.—
P. 193.
Gillespie R. J., Brown I. D., Datars W. R., Morgan K. R., Tun Z., Urn-
mat P. K. // Phil. Trans. Roy. Soc. London.— 1985.— V. A314.— P. 105.
Goodenough J. B. Magnetism and the Chemical Bond.— New York: John
Wiley, 1963.
Goodenough J. B. // Colloquos Intcrnationaux du CNRS.— Paris, 1967.—
N 157. *
Goodenough J. В., Longo J. M. // Landolt-Bornstein Tabellen.— Berlin:
Springer-Verlag, 1970.— New Series, III/4a.
Goodenough J. B. // Prog. Solid State Chom.— 1971.— V. 5.—P. 149.
Goodenough J. B. // Solid State Chemistry/Ed. by G. N. R. Rao.— New
York: Marcel Dekker, 1974.
Gopalajtrishnan J., Murugesan Т., Hegde M. S., Rao C. N. R. // J. Phys.—
1979.- V. C12.- P. 5255.
Gray G. M. // Phil. Trans. Roy. Soc. London.— 1983.— V. A309.— P. 77.
Greene R. L., Mayerle J. J., Schumaker R., Castro G., Chaikin P. M.,
Etemad S., LaPlaca S. J. // Solid State Comm.— 1976.- V. 20.— P. 943.
Hagenmuller P. // Zeit. Chem.— 1983a.— B. 23.— S. 1.
Hagenmuller P. // Proc. Indian Acad. Sci. (Chem. Sci.).- 1983b.— V. 92.—
P. 1.
Hagenmuller P. (Ed.). Solid Inorganic Fluorides.— New York: Academic
Press, 1985.
Hagenmuller P., Reau J. M., Lucat C, Matar S., Villeneue G. // Solid State
Ionics — 1981.—V. 3/4.—P. 341.
Hatfield W. E. (Ed.). Molecular Metals.— New York: Plenum Press, 1978.
Hauser J. J., Waszczak J. V. // Phys. Rev.- 1984.- V. B30.- P. 5167.
Haven Y. // Solid Electrolytes/Ed. by P. Hagenmuller, W. Van Gool.—
New York: Academic Press, 1978.
Hess H. F., De Conde K., Rosenbaum T. F., Thomas G. A. // Phys. Rev.—
1982.— V. B25.— P. 5578.
Hodgson J. N. Optical Absorption and Dispersion in Solids.— London:
Chapman, Hall, 1971.
Hoffmann B. M., Ibers J. A. // Ace. Chem. Res.— 1983.— V. 16.— P. 15.
Holtzberg F. // Phil. Mag.—1980.-V. B42.-P. 491.
Holtzberg F., McGuire T. R., Methfessel S. Landolt-Bornstein Tabellen.—
Berlin: Springer-Verlag, 1970.— New Series. IH/4a.
Honig J. M. // Preparation and Characterization of Materials/Ed. by J. M.
Honig, C. N. R. Rao.— New York: Academic Press, 1981.
Honig J. M. // J. Solid State Chem.— 1982.— V. 45.— P. 1.
Глава 6
495
Honig J. M. // Proc. Ind. Acad. Sci. (Chcm. Sci.).— 1986.— V. 96.— P. 391;
Honig J. M., Spalek J. // Proc. Ind. Nat. Sci. Acad.— 1986.— V. 52A.— P. 32.
Honig J. M., Van Zandt L. L. // Ann. Rev. Mater. Sci.— 1975.— V. 5.—
p 225.
Hubbard J. // Proc. Roy. Soc. London.— 1963.— V. A276.— P. 238.
fluisman R., de Jong R., Haas C, Jellinek F. // J. Solid State Chcm.—
1971.— V. 3.— P. 56.
Hulliger F. // Structure and Bonding.— 1968.— V. 4.— P. 83.
Hulliger F. Structural Chemistry of Layer Type Phases.—
Dordrecht-Holland: Reidel, 1977.
Hurd С. М. // Contemp. Phys.— 1982.— V. 23.— P. 469.
Hush N. S. // Chem. Phys.- 1975.- V. 10.- P. 361.
Tkeda M., Gschneidner Jr., K. A., Beaudry B. J., Altzmany U. // Solid State
Comm.- 1980.— V. 36.- P. 657.
Ingram M. D., Moynihan С. Т.. Lesikar A. V. // J. Non-Cryst.
Solids.— 1980.- V. 38, 39.- P. 371.
Jadhao V. G., Singru R. M., Rao G. R., Bahadur D., Rao С N. R. // J. Phys.
Chem. Solids.—1976.—V. 37.—P. 113.
Janke E., Hutchings M. Т., Day P., Walker P. J. // J. Phys.- 1983.—
V. C16.— P. 5959.
Jayaraman A., Bucher E., Dernier P. D., Longinnotti L. D. // Phys. Rev.
Lett.-1973.-V. 31.- P. 700.
Jayaraman A., Dernier P. D., Longinnotti L. D. // High Temperatures —
High Pressures.—1975.-V. 71.
Jellinek F. Inorganic Sulphur Chemistry/Ed. by G. Nickless.— Amsterdam:
Elsevier, 1968.
Jellinek F. // MTP International Review of Science, Inorg. Chem.—
London: Butterworths, 1972.— V. 5.
Jerome D. // Phil. Trans. Roy. Soc. London.— 1985.— V. A314.— P. 69.
Jhans H., Honig J. M. // J. Solid State Chem.— 1981.— V. 38.— P. 112.
Johnson D. C, Benfield R. E., Edwards P. P., Nelson W. R., Vargas M. D. //
Nature.-1985.-V. 314.-P. 231.
Jona F., Shirane G. Ferroelectric Crystals.— Oxford: Pergamon Press, 1962.
Kanamori J. // J. Phys. Chem. Solids.—1959.—V. 10.—P. 87.
Keller H. J. (Ed.). Low Dimensional Cooperative Phenomena.— New York:
Plenum Press, 1975.
Keller H. J. (Ed.). Chemistry and Physics of One-dimensional Metals.—
New York: Plenum Press, 1977.
Kleiner W. H. // MIT Lincoln Laboratory Solid State Research.— 1967.—
Report N 3.
Klitzing K. V., Dorda G.. Pepper M. // Phys. Rev. Lett.— 1980.— V. 45.—
P. 494.
Kittel C. Introduction to Solid State Physics, 5th edn.— New Delhi: Wiley-
Eastern, 1976.
Koelling D. D. // Rep. Prog. Phys.— 1981.— V. 53.— P. 517.
Koerber G. G. Properties of Solids.— New Jersey: Prentice-Hall, Englewood-
Cliffs, 1962.
Krebs H. // Prog. Solid State Chem.— 1975.— V. 9.— P. 269.
Krogman K. // Angew. Chem. Int. Edn (Engl.).—1969.—V. 8.—P. 35.
Kuwamoto H., Honig J. M., Appel J. // Phys. Rev.— 1980.— V. B22.—
P. 2626.
Le FlemG., DelmasC, Menil F., Niel M., Cros C, FouassierC, PouchardM.//
J. Physique Colloq.—1977.—Т. С7.—P. 262.
Ledsham R. D., Day P. // J. Chem. Soc. Chem. Comm.— 1981.— P. 921.
Lewis J., Green M. L. H. // Phil. Trans. Roy. Soc. London.— 1982.—
V. A308.-P. 1.
Little W. A. // Phys. Rev.— 1964.— V. 134.— P. A1416.
496
Список литературы
Lynn S. Y., Newnham R. E., Klicker K. A., Rittenmyer K., Safari A., Schul-
ze W. A. // Ferroelectrics.— 1981.— V. 38.— P. 955.
MacDiarmid A. G., Mammone R. J., Kaner R. В., Porter S. J. // Phil. Trans.
Roy. Soc. London.—1985.—V. A314.—P. 3.
MacDiarmid A. G., Heeger A. J. // Syn. Met.— 1980.— V. 1.— P. 101.
Maple M. В., Fisher F. (Eds). Topics in Current Physic— Berlin: Springer-
Verlag, 1982.-V. 34.
Matthias В. Т. // Phys. Rev.— 1955.- V. 97.- P. 74.
Mattis D. C. The Theory of Magnetism.— Berlin: Springer-Verlag, 1981.—
V. 1.
Mayoh В., Day P. // J. Amer. Ghem. Soc— 1972.— V. 94.— P. 2885.
McCarley R. E. // Phil. Trans. Roy. Soc. London.—1982.—V. A308.—
P. 141.
Meerschaut A. // Ann. Chim. Fr.— 1982.— T. 7.— P. 131.
Messmer R. P. // Semi-empirical Methods of Electronic Structure
Calculation.— New York: Plenum Press, 1977.— Part B/Ed. by G. A. Segal.
Miller J. S. (Ed.). Extended Linear Chain Compounds.— New York:
Plenum Press, 1982.
Miller J. S., Epstein A. J. (Eds). // Ann. N. Y. Acad. Sci.— 1978.— V. 313.
Moore M. W., Day P. // J. Solid State Chem.— 1985.— V. 59.— P. 23.
Mott N. F. // Proc. Phys. Soc—1949.-V. 62A.- P. 416.
Mott N. F. // Canadian J. Phys.— 1956.— V. 34.— P. 1356.
Mott N. F. // Adv. Phys.- 1967.— V. 16.- P. 49.
Mott N. F. // Phil. Mag.— 1972.— V. 26 — P. 1015.
Mott N. F. Metal-Insulator Transitions.— London: Taylor, Francis, 1974.
Mott N. F. // Int. Rev. Phys. Chem.— 1983.— V. 4.— P. 1.
Mott N. F., Davis E. A. Electronic Processes in Non-crystalline Materials.—
Oxford: Clarendon Press, 1979.
Muller J. // Rep. Prog. Phys.— 1980.— V. 43.— P. 643.
Muetterties E. L., Rhodin T. N., Band E., Brucker С F., Pretzer W. R. //
Chem. Rev.—1979.—V. 79.—P. 91.
Murugesan Т., Ramesh S., Gopalakrishnan J., Rao C. N. R. // J. Solid State
Chem.—1981.—V. 38 — P. 165.
Mydosh J. A. // AIP conf. Proceedings.—1975.—V. 24.— P. 131.
Mydosh J. A. // Magnetism in Solids. Some Current Topics/Ed. by A. P.
Cracknell, R. A. Vaughan.— Edinburg University, SUSSP Publications, 1982.
Newnham R. E. // Amer. Mineralogist.—1974.—V. 57.— P. 906.
Newnham R. E. Structure-Property Relations.— New York:
Springer-Verlag, 1975.
Newnham R. E., Cross L. E. // Preparation and Characterization of
Materials/Ed. by J. M. Honig, C. N. R. Rao.— New York: Academic Press, 1981.
Newnham R. E., Kramer J. J., Schulze W. A., Cross L. E. // J. Appl. Phys.—
1978.- V. 49.- P. 6088.
Nomura S. Landolt-Bornstein Tabellen.— Berlin: Springer-Verlag, 1978.—
New Series. III/12a.
Nye J. F. Physical Properties of Crystals.— Oxford University Press, 1957.
Peierls R. E. Quantum Theory of Solids.—Oxford University Press, 1955.
Piepho S. В., KrauszE. R., Schatz P. N. // J. Amer. Chem. Soc— 1978.—
V. 100.- P. 2996.
Portier J. // Angew Chem. int. Edn (Engl.).— 1976.— V. 15.— P. 475.
Portier J., Reau J. M., Matar S., Soubeyroux J. L., Hagenmuller P. //
Solid State Lonics.— 1983.— V. 11.— P. 83.
Prassides K., Day P // J. Chem. Soc. Faraday II.— 1984.— V. 80 — P. 85.
Raccah P. M., Goodenough J. B. // Phys. Rev.— 1967,— V. 155.— P. 932.
Ramdani A., Gleitzer C, Gavoille C, Cheetham A. K., Goodenough J. B. //
J. Solid State Chem.- 1985.- V. 60.— P. 269.
Rao C. N. R., Subba Rao G. V. Transition Metal Oxides: Crystal Chemistry,
Глава 6
497
Phase Transition and Related Aspects.— Washington D. C: National Bureau of
Standards, 1974.— NSRDS — NBS monograph 49.
Rao С N. R., Bhide V. G., Mott N. F. // Phil. Mag.- 1975.— V. 32,—
p. 1277.
Rao С N. R., Pisharody K. P. R. // Prog. Solid State Chem.— 1976.—
V. 10.— P. 207.
Rao С N. R., Parkash P., Bahadur D., Ganguly P., Nagabhushana S. //
J. Solid State Chem.— 1977.— V. 22.— P. 353.
Rao C. N. R., Rao K. J. Phase Transition in Solids.— New York: McGraw-
НШ, 1978.
Rao С N. R., Sarma D. D., Vasudevan S., Hegde M. S. // Proc. Roy. Soc.
London.- 1979.- V. A367.- P. 239.
Rao С N. R., Ganguly S., Swamy H. R , Oxton I. A. // J. Chem. Soc.
Faraday.— 1981.— V. II.- P. 77, 1825.
Rao С N. R., Buttrey D., Harrison G., Ganguly P., Honig J. M. // J Solid
State Chem.— 1984.— V. 51.— P. 266.
Rao С N. R., Edwards P. P. // Proc. Indian Acad. Sci. (Chem. Sci.).—
1986.— V. 96.— P. 473.
Rao С N. R., Ganguly P. // Solid State Commun.— 1986.— V. 57.— P. 5.
Rao C. N. R., Ganguly P. // Localization* and Metal-Insulator Transitions/
Ed. by D. Alder, H. Frit/sche.— New York: Plenum Preps, 1985.
RaynesE. P. // Phil Trans. Roy Soc. London.— 1983.— V. A309.— P. 167.
Reinen D. // Structure and Bonding.— 1979.— V. 37.— P. 1.
Rittenmeyer K., Strout Т., Schulze W. A., Newnham R. E. // Ferroelect-
rics.- 1982.- V. 41.- P 189.
Robbins D. J. Day P. // Chem. Phys. Lett.— 1973.— V. 19.— P. 529.
Robin M. В., Day P. // Adv. Inorg. Chem. Radiochem.— 1967.— V. 10.—
P. 247.
Rogers D. В., Shannon R. D., Sleight A. W., Gillson J. L. // Inorg. Chem.—
1969.—V. 8.— P. 841.
Rohler J., Kaindl G. // Solid State Comm.— 1980.— V. 36.— P. 1055.
Rouxel J., Meerschaut A., Guemas L., Gressier P. // Ann. Chim. Fr.— 1982.—
T. 7.— P. 445.
Safari A., Halliyal A., Nawnham R. E., Lachman I. M. // Mater. Res.
Bull.— 1982.— V. 17.— P. 301.
Sawada A., Udagawa M., Nakamura T. // Phys. Rev. Lett.— 1977.—
y# 39 p 329.
Sayer M., Chen R., Fletcher R., Man Singh // J. Phys — 1975.— V. C8.—
P. 2059.
Sayetat F., Ghedira M., Chenavas J., Marezio M. // J. Phys.— 1982.—
V. C15.- P. 1627.
Schlenker C, Dumas J., Escribe-Filippini, Guyot H., Marcus J., Fourcan-
dot G. // Phil. Mag.— 1985.— V. B52.— P. 643.
Schollhorn R. // Inclusion Compounds/Ed. by J. L. Atwood, J. E. D. Da-
vies, D. D. MacNicol.— New York: Academic Press, 1983.
Schultz A. J., Williams J. M., Miro N. D., MacDiarmid A. G., Heeger A. J. //
Inorg. Chem.— V. 17.— P. 646.
Setter N., Cross L. E. // Ferroelectrics.— 1981.— V. 37.— P. 551.
Shukla A. K., Vasan H. N., Rao C. N. R. // Proc. Roy. Soc. London.—
1981.— V. A376.- P. 619.
Soos Z. G., Klein D. J. // Treatise on Solid State Chemistry/Ed. by
N. B. Hannay.— New York: Plenum Press, 1976.— V. 3.
Soos Z. G., Ramasesha S. // Phys. Rev. Lett.— 1983.— V. 51.— P. 2374.
Stewart G. R. // Rev. Mod. Phys.— 1984.— V. 56.— P. 755.
Stryjewskv E., Giordano N. // Adv. Phys.— 1977.— V. 26.— P. 487.
Subbarao E. С // Solid State Chemistry/Ed. by C. N. R. Rao.— New York:
Marcel Dekker, 1974.
498
Список литературы
Subba Rao G. V., Shafer M. W. // Intercalated Layered Materials/Ed. by
F. Levy.— Dordrect-Holland: Reidel, 1979.
Subba Rao G. V., Balakrishnan G. // Bull. Mater. Sci.—1984.—V. 6.—
P. 283.
Subramanyam S. V. // Preparation and Characterization of Materials/Ed. by
J. M. Honig, С N. R. Rao.— New York: Academic Press, 1981.
Subramanyam S. VM Naik H. // The Metallic and Nonmetallic States of
Matter/Ed. by P. P. Edwards, С N. R. Rao.— London: Taylor, Francis, 1985.
Swinnea J. S., Steinfink H. // J. Solid State Chem.— 1982.— V. 41.—
P. 114, 124.
Taher S. M., Gruber J. B. // Mater. Ros. Bull.— 1981.— V. 16.— P 1407.
Takano M., Takeda Y. // Bulletin of the Institute for Chemical Research.
Kyoto University, Japan.—1983.—V. 61.—P. 406.
Thomas L. A., Wooster W. A. // Proc. Roy. Soc. London.— 1951.—
V, A208.- P. 43.
Thornton G., Tofield B. C, Williams D. E. // Solid State Commun.— 1982.—
V. 44.- P. 1213.
Torrance J. B. // Ace. Chem. Res.— 1979.— V 12.— P. 79.
Torrance J. В., Shafer M. W., McGuire T. R. // Phys. Rev. Lett.— 1972.—
V. 29.— P. 1168.
Torrance J. В., Silverman B. D. // Phys. Rev.— 1977.— V. B15.— P. 788.
Tressaud A., Dance J. M. // Adv. Inorg. Chem. Radiochem./Ed. by
H. J. Emeleus, A. G. Sharpe,— New York: Academic Press, 1977.—V. 20.
Tressaud A., Dance J. M. // Structure and Bonding.— 1982.— V. 52.— P. 87.
Uchino K., Nomura S., Cross L. E., Newnhain R. E. // Ferroeletrics.—
1981.— V. 38.— P. 825.
Underhill A. E. // Phil. Trans. Roy. Soc. London.—1985.—V. A314.—
P. 125.
Van der Heide H., Hemmel R., Van Bruggen C. F., Haas C. // J. Solid State
Chem.-1980.—V. 33.-P. 17.
Van Zandt L. L., Honig J. M. // Ann. Rev. Mater. Sci.— 1974.— V. 4.—
P. 191.
Varma K. B. R., Harshvardhan K. S., Rao K. J., Rao C. N. R. // Mater.
Res. Bull.- 1985.— V. 20.— P. 315.
Wachter P. // Phil. Mag.— 1980.— V. B42.— P. 497.
Wickelhaus W., Simon A., Stevens K. W. H., Brown P. J., Ziebeck K. R. A.//
Phil. Mag.— 1982.- V. B46.— P. 115.
Wigner E. P. // Trans. Faraday Soc— 1938.— V. 34.— P. 678.
Williams P. M. Optical and Electrical Properties of Layered Materials/Ed.
by P. A. Lee.— Dordrecht-Holland: Reidel, 1976.
Wilson J. A. // The Metallic and Nonmetallic States of Matter/Ed. by
P. P. Edwards, С N. R. Rao.— London: Taylor, Francis, 1985.
Wilson J. A., Yoffe A. D. // Adv. Phys.— 1969.— V. 18.— P. 193.
Wilson J. A. // Adv. Phys.— 1972.— V. 21.— P. 143.
Yvon K. // Current Topics in Materials Science/Ed. by E. Kaldis.—
Amsterdam: North-Holland, 1979.— V. 3.
ГЛАВА 7
Alfrey Jr., Т., Schrenk W. J. // Science.— 1980.— V. 208.— P. 813.
Anderson B. C, Barton L. R., Collette J. W. // Science.— 1980.— V. 208.—
P. 807.
Angus H. С // Phys. Technol.- 1981.— V. 12.— P. 245.
Aruchamy A.. Aravamudan G., Subba Rao G. V. // Bull. Mater. Sci.—
1982.— V. 4.— P. 483.
Bard A. J. // Science.— 1980.— V. 207.— P. 139.
Глава 7
499
Becquerel E. // С. R. Acad. Sci.— 1839.— Т. 9.— P. 561.
Blunt R. // Chem. Britain.- 1983.- V. 19.— P. 736.
Bose D. N., Parthasarathy G., Mazumdar D., Gopal E. S. R. // Phys. Rev.
Lett.— 1984.— V. 53.- P. 1368.
Bowen K. // Mater. Sci. Engg.— 1980.— V. 44.— P. 1.
Brattain W. H., Garrett C. G // Bell. Syst. Tech. J.— 1955.— V. 34.—
P. 129.
Bruce P. G., West A. R. /7 J. Solid State Chem.— 1982.— V. 44.— P.
354.
Buschow K. H. J., van Mai H. H. // Intercalation Chemistry/Ed. by
M. S. Whittingham, A. J. Jacobson,— New York: Academic Press. 1982.
Castro G. // Photochemistry and Photobiology/Ed. by A. H. Zewail.— Har-
wood, 1983.
Chandra S. Superionic Solids: Principle and Applications.— Amsterdam:
North-Holland. 1981.
Chao S., Robbins J. L., Wrington M. S. // J. Amer. Chem. Soc— 1983.—
V. 105.— P. 181.
Chin G. Y. // Science.— 1980.— V. 208.— P. 888.
Chopra K. L., Harshvardhan K. S., Rajagopalan S., Malhotra L. K. //
Solid State Comm.— 1981.— V. 40.— P. 387.
Cohen R. L., Wernick J. H. // Science.— 1981.— V. 214.— P. 1081.
Darcy P. J., Heller H. G., Strydom P. J., Whittal J. // J. Chem. Soc. Per-
kin.- 1981.- V. 1.- P. 202.
Das B. N., Koon N. С // Met. Trans.— 1983.— V. 14A.— P. 953.
Di Bartolo (Ed.). Luminescence of Inorganic Solids.— New York: Plenum
Press, 1978.
Elliott S. R. // J. Non-Cryst. Solids.— 1983.— V. 59—60.— P. 899.
Elliott S. R. Physics of Amorphous Materials.— London: Longman, 1984.
Ellis А. В., Kaiser S. W., Wrighton M. S. // J. Amer. Chem. Soc— 1976.—
V. 98.- P. 1635, 6855.
Farrington G. C, Briant J. L. // Science.— 1979.— V. 204 — P. 1371.
Fujishima A., Honda K. // Nature.— 1972.— V. 238.— P. 37.
Ganapathi L., Subhanna G. N., Gopalakrishnan J., Rao С N. R. // J. Mater.
Sci.— 1985.— V. 20.- P. 1105.
Gantron J., Lemasson P., Marucco J. F. // Faraday Discussion of the
Chemical Society.— 1980.— N 70, 81.
Garito A. F., Singer K. D. // Laser Fokus.— 1982.— V. 18.— P. 59.
Gerischer H. // J. Electroanal. Chem.—1975.— V. 58.—P. 263.
Gerischer H. // Topics in Applied Physics/Ed. by В. О. Seraphin.— Berlin:
Springer-Verlag, 1979.— V. 31: Solar Energy Conversation.
Giess E. A. // Science.- 1980.- V. 208.- P. 938.
Goodenough J. B. // Proc. Roy. Soc. London.— 1984.— V. A393.— P. 215.
Goodenough J. В., Hong H. Y-P., Kafalas J. A. // Mater. Res. Bull.—
1976.— V. 11.- P. 203.
Goodman С H. L. // Chem. Britain.— 1983.— V. 19.— P. 745 and the re-
fercnces therein
Gratzel M.7/ Ace. Chem. Res.— 1981.— V. 14.— P. 376.
Gronet С M., Lewis N. S. // Nature,— 1982.— V. 300.— P. 733.
Guru Row T. N., Swamy H. R., Acharya K. R., Ramamurthy V., Venkate-
san K., Rao С N. R. // Tetrahedron Lett.— 1983.— V. 24.— P. 3263.
HagenmuPer P. // Proc. Indian Acad. Sci. (Chem. Sci.).— 1983.— V. 92.—
P. 1.
Hagenmuller P., van Gool W. (Eds). Solid Electrolytes.— New York:
Academic Press, 1978.
Harris L. A., Wilson R. H. // Ann. Rev. Mater. Sci.— 1978.— V. 8.— P. 99,
Heller A. // Ace. Chem. Res.— 1981.— V. 14.— P. 154.
Herbst J. F., Croat J. J., Pinkerton F. E., Yelon W. B. // J. Appl. Phys.—
1985.— V. 57.— P. 4086.
500
Список литературы
Hill С. G. А. // Chem. Britain.— 1983.— V. 19.— P. 723 and the references
therein.
Ingram M. D., Vincent C. A. // Chem. Britain.— 1984.— V. 20.— P. 235.
Ivey D. G., Norfchwood D. K. // J. Mater. Sci — 1983.— V. 18.— P. 321.
Iwamura H. // Pure and Appl. Chem.— 1986.— V. 58.— P. 187.
Kahn 0. // Angew. Chem. Int. Edn (Engl.).— 1985.— V. 24.— P. 834.
Katz R. N. // Science.- 1980.— V. 208.- P. 841.
Kiwi J., Kalyanasundaram K., Gratzel M. // Structure and Bonding.—
1982.— V. 49.— P. 39.
Kohler H., Schulg H. // Mater. Res. Bull,— 1985.- V. 20.- P. 1461.
Koon N. C, Das B. N., Rubinstein В., Tyson J. // J. Appl. Phys.— 1985.—
V. 57.- P. 4091.
Kutal C. // J. Chem. Educ— 1983.— V. 60.— P. 882.
Legg K. D., Ellis А. В., Bolts J. M., Wrighton M. S. // Proc. Natl. Acad.
Sci. USA.-1977.-V. 74.— P. 4116.
Lemasson P.. Etman M., Gantron J., Marucco J. F. // Proc. 4th Int. Conf.
on Photochemical Conversion and Storage of Solar Energy.— Jerusalem, 1982.
Leygraf C, Hendewerk M., Somorjai G. A. // Proc. Natl. Acad. Sci. USA.—
1982.- V. 79.- P. 5739.
MacChesney J. B. // Preparation and Characterization of Materials/Ed. by
J. M. Honig, С N. R. Rao.— New York: Academic Press, 1981.
Maclnnes Jr., D., Druy M. A., Nigrey P. J., Nairns D. P., MacDiarmid A. G.,
Heeger A. J. // J. Chem. Soc. Chem. Comm.— 1981.— P. 317.
Mazumdar D., Bose D. N., Mukherjee M. L. // Solid State Ionics.— 1984.
V. 14.— P. 143.
Menth A., Nagel H., Perkins R. S. // Ann. Rev. Mater. Sci.— 1978.— V. 8.—
P. 21.
Miedema A. R. // J. Less-Common Metals.— 1973.— V. 32.— P. 117.
Miedema A. R., Buschow K. H. J., van Mai H. H. // Electrochem. Soc. Conf.
Proc-1977.—V. 77—6.-P. 456.
Mort J. // Science.— 1980.— V. 208.— P. 819.
Moss R. H. // Chem. Britain.— 1983.— V. 19.— P. 733.
Munn R. W. // Chem. Britain.— 1984.— V. 20.— P. 518 and the
references therein.
Murphy D. W., Christian P. A. // Science.— 1979.— V. 205.— P. 651.
Nesbitt E. A., Wernick J. H. Rare Earth Permanent Magnets.— New
York: Academic Press, 1973.
Newnam В. Е. // Laser Focus.— 1982.— V. 18, N 2.— P. 53.
Nielsen J. W. // IEEE Transactions on Magnetics, MAG-12.— 1976.—
P. 327.
Nielsen J. W. // Ann. Rev. Mater. Sci.— 1979.— V. 9.— P. 87.
Nozik A. J. // Appl. Phys. Lett.— 1976.— V. 29.— P. 150. *
Nozik A. J. // Ann. Rev. Phys. Chem.— 1978.— V. 29.— P. 189.
Nozik A. J. // Faraday Discussions of the Chemical Society.— 1980.— N 70.
O'Keeffe M. // Fast Ion Transport in Solids/Ed. by W. van Gool.—
Amsterdam: North-Holland, 1973.
Owen A. E., Firth A. P., Even P. J. S. // Phil. Mag.- 1985.- V. B52.—
P. 347.
Pierce D. Т., Spicer W. E. // Phys. Rev- 1972.- V. B5.- P. 3017.
Pomerantz M., Dracol F. H., Segmuller A. // Phys. Rev. Lett.— 1978.—
V. 40.— P. 246.
Pope M., Swenberg С E. // Electronic Processes in Organic Crystals.—
Oxford: Clarendon Press, 1982.
Popplewell J. // Phys. Technol.— 1984.— V. 15.— P. 150.
Pourarian F., Wallace W. E. // J. Less-Common Metals.— 1982.— V. 87,—
P. 275.
Rabenau A. // Adv. Solid State Phys.- 1978.- V. 18.- P. 77.
Raynes E. P. // Phil. Trans. Roy. Soc. London.— 1983.— V. A309.— P. 167.
Глава 7
501
Rao К. J. // Preparation and Characterization of Materials/Ed. by J. M. Ho-
nig, С N. R. Rao.— New York: Academic Press, 1981.
Reilly J. J. // Hydrides for Energy Storage/Ed. by A. F. Andersen, A. J. Mee-
land.— Oxford: Pergamon Press, 1978.
Reilly J. J., Sandrock G. D. // Scientific American.— 1980.— V. 242.—
P. 98.
Revcolevschi A., Dhalenne G. // Nature.— 1985.— V. 316.— P. 335.
Roberts G. G. // Thin Solid Films.— 1980.— V. 68.— P. 135.
Robinson A. L. // Science.— 1984.— V. 223.— P. 920.
Rosensweig R. E. // Scientific American.— 1982.— V. 247,— P. 124.
Sanders H. J. // Chem. Engg. News.— 1984.— V. 62 (28).— P. 26.
Sen P. K., Sen S. // Solid State Ionics.— 1983.— V. 9, 10.— P. 1015.
Scheffer T. J. // Phil. Trans. Roy. Soc. London.— 1983.— V. A309.— P. 189.
Shanks I. A. / Contemp. Phys.- 1982.- V. 23.- P. 65.
Sheats J. E., Pittman С U., Carraher Jr., С. Е. // Chem. Britain.— 1984.—
V. 20.— P. 709.
Sheft I., Gruen D. M., Lamich G. J. // J. Less-Common Metals.— 1980.—
V. 74.— P. 401.
Shenoy G. K., Niarchos D., Viccaro P. J., Dunlap B. D., Aldred А. Т., Sand-
rock G. D. // J. Less-Common Metals.— 1980.— V. 73.— P. 171.
Skou E., Anderson I. G. K., Simonson K. E., Anderson E. K. // Solid State
Ionics.- 1983.- V. 9, 10.- P. 1041.
Strock L. W. // Z. Phys. Chem.— 1936.- В. В31.- S. 132.
Slucki F., Schlapback L. // J. Less-Common Metals.— 1980.— V. 74.—
P. 143.
Taylor G. N., White D. L. // J. Appl. Phys.— 1974.— V. 45.— P. 4718.
Tofield B. C, Deil R. M., Jensen J. /7 Nature.— 1978.— V. 276.— P. 217.
Tubandt C, Lorenz E. // Z. Phys. Chem.— 1914.— B. 87.— S. 513.
Tuller H. L., Button D. P., Uhlmann D. R. // J. Non-Cryst. Solids.—
1980.— V. 40.- P. 93.
van Mai H. H., Buschow K. H. J., Miedema A. R. // J. Less-Common
Metals.- 1974.— V. 35.- P. 65.
Wallace W. E. Rare Earth Intermetallics.— New York: Academic
Press. 1973.
Wallace W. E., Narasimhan K. S. V. L. // Science and Technology of Rare
Earth Materials/Ed. by E. С Subbarao, W. E. Wallace.— New York: Academic
Press, 1980.
Weber N., Kummer J. T. // Adv. Energy Conv. Eng. ASME Conf. Florida,
913, 1967.
White R. M. // Science.- 1985.— V. 229.— P. 11.
Whittingham M. S. // Science.—1976.—V. 192.—P. 1126.
Whittingham M. S., Huggins R. A. // J. Electrochem. Soc— 1971.—
V. 118.- P. 1.
Williams D. J. (Ed.). Nonlinear Optical Properties of Organic and
Polymeric Materials.— Washington: American Chemical Society, 1983.— ACS
Symposium Series 233.
Williams J. O. // Chem. Britain.— 1983.— V. 19.— P. 713.
Wright J. D., Chadwick A. V., Meadows В., Miasik J. J. // Mol. Cryst. Lig.
Cryst.-1983.-V. 93.-P. 315.
Wrighton M. S. // J. Chem. Educ— 1983.— V. 60.— P. 877.
Yao Y. Y., Kummer J. T. // J. Inorg. Nucl. Chem.— 1967.— V. 20.—
P. 2453.
Yesodharan E., Gratzel M. // Helv. Chim. Acta.— 1983.— V. 66.— P. 2145.
Zallen R. The Physics of Amorphous Solids.— New York: John Wiley, 1983.
Zallen R., Drews R. E., Emerald R. L., Slade M. L. // Phys. Rev. Lett.—
1971.— V. 26.- P. 1564.
502
Список литературы
ГЛАВА 8
Addadi L., Ariel S., Lahav M., Leiserowitz L., Papovitz-Biro R., Tang C. P. //
Chemical Physics of Solids and their Surfaces/Sr. Reporters M. W. Roberts,
J. M. Thomas.— London: The Royal Society of Chemistry, 1980.
Addadi L., Berkovitch-Yellin Z., Weissbuch I., Mil J. V., Shimon L. J. W.,
Lahav M., Leiserowitz L. // Angew. Chem. Int. Edn. (Engl.).— 1985.— V. 24.—
P. 466.
Alberti G., Constantino U. // Intercalation Chemistry/Ed. by M. S. Whit-
tingham, A. J. Jacobson.— New York: Academic Press, 1982.
Bahl M. K., Tsai S. C, Chung Y. W. // Phys. Rev.— 1980.— V. B21.—
P. 1344.
Balandin A. A. // Advances in Catalysis.— 1969.— V. 19.— P. 1.
Baughman R. H. // J. Chem. Phys.- 1978.- V. 68.— P. 3110.
Benard J., Gronlund F., Oudar J., Duret M. // Z. Electrochcm.— 1959.—
В 63 g 799
Boldyrev Y. V. // Ann. Rev. Mater. Sci.— 1979.— V. 9.— P. 455.
Boldyrev V. V. // Ann. Chim. Fr.— 1981.— T. 6.- P. 359.
Burdett J. K. // J. Amcr. Chem. Soc— 1980.— V. 102.- P. 5458.
Burwell Jr., R. L. // Catalysis-Progress in Research/Ed. by F. Basolo,
R. L. Burwell Jr.— New York: Plenum Press, 1973.
Cabrera N., Mott N. F. // Repts. Prog. Phys.— 1948.— V. 12.— P. 163.
Carter R. E. // J. Chem. Phys.— 1961.— V. 34.— P. 2010: V. 35.— P. 1137.
Chang С D., Silvestri A. J. // J. Catal.— 1977.- V. 47.— P. 249.
Chon If., Prater С D. // Disc. Faraday Soc— 1966.— V. 41.— P. 380.
Clearfield A. // Preparation and Characterization of Materials/Ed. by
J. M. Honig, C. N. R. Rao.— New York: Academic Press, 1981.
Cohen M. D., Green B. S. // Chem. Britain.— 1973.— V. 9.— P. 490.
Cohen M. D. // Angew. Chem. Int. Edn. (Engl.).— 1975.— V. 14.— P. 386.
Colpaert M. N., Clauws P., Fiermans L., Vernik J. // Surface Science.—
1973.— V. 36.— P. 513.
Curtin D. Y., Paul Т. С // Chem. Rev.— 1981.— V. 81.— P. 525.
Daumas N., Herold A. // С R. Acad. Sci. (Paris).— I960.— Т. C268.—
P. 373.
Derouane E. G. // Intercalation Chemistry/Ed. by M. S. Whittingham,
A. J. Jacobson.— New York: Academic Press, 1982.
Desiraju G. R. // Proc. Indian Acad. Sci. (Chem. Sci.).— 1984a.— V. 93.—
P. 407.
Desiraju G. R. // Endeavour.— 1984b.— V. 8.— P. 201.
Dines M. B. // Science.— 1975.— V. 188.— P. 210.
Dowden D. A. // Catal. Rev.— 1972.- V. 5.— P. 1.
Dwyer J. // Chemistry, Industry.— 1984.— 2 April Issue.— P. 258.
Dyer A. // Chemistry Industry.— 1984.— 2 April Issue.— P. 241.
Fehlner F. P., Mott N. F. // Oxidation of Metals.— 1970.— V. 2.— P. 59.
Fyfe С A., Gobbi G. C, Klinowski J., Thomas J. M., Ramdas S. // Nature.—
1982.— V. 296.— P. 530.
Galiotis C, Read R. Т., Yeung P. H. Y., Young R. J., Chalmers I. F.,
Bloor D. // J. Polymer Sci.- 1984.— V. 22.— P. 1589.
Ganapathi L., Ramanan A., Gopalakrishnan J., Rao C. N. R. // J. Chem.
Soc. Chem. Coram.- 1986.— P. 62.
Garner W. E. (Ed.). Chemistry of the Solid State.— London: Butterworths
1955.
Ginsburg D. (Ed.). Solid State Photochemistry.— Weinheim: Vcrlag Chemie,
1976.
Gnanaguru K., Ramasubbu N., Venkatesan K., Ramamurthy V. // J. Org.
Chem.— 1985.— V. 50.— P. 2337.
Gomes W. P., Dekeyser W. // Treatise on Solid State Chemistry/Ed. by
N. B. Hannay.— New York: Plenum Press, 1976.— V. 4.
Глава 8
503
Goodman D. W. // Ace. Chcm. Res.— 1984.— V. 17.— P. 194.
Gorring R. L. // J. Catal.— 1973.— V. 31.— P. 13.
Grasselli R. K., Burrington J. D. // Advances in Catalysis.— 1981.— V. 30.—
P. 133.
Grasselli R. K., Burrington J. D., Brazdil J. F. // Faraday Discussions of
the Chemical Society.— 1981.— V. 72.— P. 203.
Green B. S., Lahav M., Rabinovich D. // Ace. Chcm. Res.— 1979.— V. 12.—
P. 191.
Gross II., Sixl II. //Chcm. Phys. Lett.— 1982.— V. 91.— P. 262.
Giinter J. R. // J. Solid State Chem.- 1972.— V. 5.— P. 354.
Guru Row T. N., Swamy II. R., Acharya K. R., Ramamurthy V., Venkate-
san K., Rao C. N. R. // Tetrahedron Lett.— 1983.— V. 24.— P. 3263.
Gwathmey А. Т., Lawless K. R. // The Surface Chemistry of Metals and
Semiconductors/Ed. by H. С Gatos.— New York: Wiley, 1960.
Hagg W. 0., Lago R. M., Weisz P. B. // Nature.— 1984.— V. 309.— P. 589.
Haggin J. // Chcm. Engg. News.— 1982.— V. 60 (6).— P. 13.
Halbert T. R. // Intercalation Chcmislry/Ed. by M. S. Whittingham,
A. J. Jacobson.— New York: Academic Press, 1982.
Hannay N. B. (Ed.). Treatise on Solid State Chemistry.— New York:
Plenum Press, 1976.— V. 4.
Hasegawa M., Iguchi M., Nakanishi II. // J. Polymer. Sci.— 1968.— V. A-
1,6.— P. 1054.
Hauffe K. Rcactioncn in und an Festonstoffen, 2nd Edn.— Berlin: Springer-
Verlag, 1966.
Hauffe K. // Treatise on Solid State Chemistry/Ed. by N. B. Hannay.—
New York: Plenum Press, 1976.— V. 4.
Horsley J. A. // J. Amcr. Chem. Soc— 1979.— V. 101.— P. 2870.
Iguchi'E., Tilley R. J. D. // J. Solid State Chem.— 1978.— V. 24,— P. 121.
Jacobson A. J. // Intercalation Chemistry/Ed. M. S. Whittingham,
A. J. Jacobson.— New York: Academic Press, 1982.
Jander W. // Z. Anorg. Allgom. Chcm.—1927.—B. 163.—S. 1.
Jiang X. Z., Hayden T. F., Dumesic J. A. // J. Catal.— 1983.— V. 83.—
P. 168.
Johnson J. W. // Intercalation Chemistry/Ed. by M. S. Whittingham,
A. J. Jacobson.— New York: Academic Press, 1982.
Jones W., Thomas J. M. // Prog. Solid State Chem.— 1979.— V. 12.—
P. 101.
Jones W., Nakanishi H., Theocharis C. R., Thomas J. M. // J. Chem. Soc.
Chem. Comm.— 1980 — P. 610.
Jones W., Ramdas S., Theocharis С R., Thomas J. M , Thomas N. W. //
J. Phys. Chcm.- 1981.— V. 85.- P. 2594.
Jost W. Diffusion und Chemischc Reaction in Festenstoffen.— Dresden:
Steinkopf-Verlag, 1937.
Kearsley S. K. Desiraju G. R. // Proc. Roy. Soc. London.— 1985.—
V. A397.— P. 157
Ко С. S., Gorte R. J. // J. Catal.— 1984.— V. 90.- P. 59.
Krylov O. V. Catalysis by Nonmetals.— New York: Academic Press, 1970.
Levy F. (Ed.) Physics and Chemistry of Materials with Layered
Structures.— Dordrecht: Reidel, 1979.— V. 6.
Lewis J., Green M. L II. (Eds.). // Phil. Trans. Roy. Soc. London — 1982.—
V. A308 — P. 1.
Mady T. E., Yates J ., J. Т., Sandstrom D. R., Voorhoeve R. J. H. /
Treatise on Solid State Chemistry/Ed by N. B. Hannay.— New York: Plenum Press,
1976.— V. 6B.
Mallouk E., Hawkins B. L., Conrad M. P., Zilm К , Maciel G. E., Bart-
lett N. // Phil. Trans. Roy. Soc. London.— 1985.— V. A314.— P. 179.
Manohar H. // Solid State Chemistry7Ed. by C. N. R. Rao.— New York:
Marcel Dckker, 1974.
504
Список литературы
McBridge J. M. // Асе. Chem. Res.— 1983.— V. 16.— P. 304.
Mizuno Т., Hattori S., Tawata M. // J. Opt. Soc. Amer.— 1977.— V. 67.—
P. 1651.
Moran H. F., Ohstuki M., Hibino A., Hough С // Science.— 1971.— V. 174.—
P. 498.
Morawetz H. // Reactivity of Solids/Ed. by J. Wood 0. Lmquist, С Hel-
gesson, N.-G. Vannorberg.— New York: Plenum Press, 1977.
Muetterties E. L., Rhodin T. N., Band E., Brucker C. R., Pretzer W. R. //
Chem. Rev.—1979 —V 79.—P 91.
Nakanishi F., Tasai Т., Hasegawa M. // Polymer.— 1975.— V. 16.— P. 218.
Nakanishi II., Hasegawa M., Sasada Y. // J. Polymer Sci.— 1972.— V. 10.—
P. 1537.
Nakanishi H., Nakanishi F., Suzuki Y., Hasegawa M. // J. Polymer Sci.—
1973.— V. 11.- P. 2501.
Nakanishi Ho, Jones W., Thomas J. M., Hasegawa M., Rees W. L. // Proc.
Roy. Soc. London — 1980.— V. A369.— P. 307.
Niederwald H., Richter K.H., Guttler W., SchwoererM. // Mol. Cryst. Liq.
Cryst.—1983.-V. 93.—P. 247.
Parasnis A. S. // Modern Aspects of Solid State Chemistry/Ed. by С N.
R. Rao,— New York: Plenum Press, 1970.
Pauling L. // Proc. Roy. Soc. London.— 1949.— V. A196.— P 343.
Pilling N. В., Bedworth R. E. // Inst Metals.— 1923.— V. 29.— P. 529.
Prabhakaran K., Rao С N. R. // Surf Science.— 1985.— V. 163. —P. L 771.
Rabinovich D., Hope H. // Acta Crystallogr.— 1975.— V A31.— P. S12S.
Rabo J. A. (Ed.). Zeolite Chemistry and Catalysis.— Washington: Amer.,
Chem. Soc—ACS Monograph N. 171.
Rao C. Nc R. Solid State Chemistry: A Perspective.— Indian National
Science Academy, Golden Jubilee Publication, 1984.
Rao С N. R. // Proc. Ind. Nat. Sci. Acad.— 1985.— V. A51.- P. 1.
Rees L. V. С // Chemistry and Industry.— 1984.— 2 April Issue.— P. 252.
Sachtler W. M. II., Fahrenfort J. // Actes du DeuMcme Congres
International de Catalyse.—I960.—P. 831.
Sachtler W. M. II. // Faraday Discussions of the Chemical Society.— 1981.—
V. 72.- P. 7.
Sankar G., Sarode P. R., Srinivasan A., Rao C. N. R., Vasudevan S.,
Thomas J. M. // Prcc. Indian Acad. Sci (Chem. Sci.).— 1984.— V. 93.— P. 321.
Salmeron M., Gale R. J., Somorjai G. A. // J Chem. Phys.— 1979.—
V. 70.— P. 2807.
Santos J., Phillips J., Dumesic J. A. // J. Catal.— 1983.— V. 81.— P. 147.
Schafer-Slahl H., Abele R. // Angew. Chem. Int. Edn (Engl.).— 1980.—
V. 19.— P 477.
Schlogl II. //Proc. 5th Int. Conf. on Rapidly Quenched Metals.—
Amsterdam: North-Holland, 1985.
Schinalzricd II. // Solid State Reactions.— Weinheim: Verlag Chcmie, 1971.
SchmaVried II. // Treatise on Solid State Chemistry/Ed. byN. B. Hannay,—
New York: Plenum Press, 1976.— V 4.
Schmidt G. M. J. // Pure and Appl. Chem.— 1971.— V. 27.— P. 647.
SchohhornR. // Angew. Chem. Int. Edn (Engl.).— 1980.— V. 19.— P. 983.
Schollhorn R. // Pure and Appl Chem.— 1984.— V. 56.— P. 1739.
Sermon P. A., Bond G. С // Catal. Rev.— 1974.— V. 8.— P. 211.
Sinfelt J. A. V Prog. Solid State Chem.—1975.—V. 10.—P. 55.
Sinfelt J. A. // Science.— 1977.- V. 195.— P. 641.
Sleight A W. // Advanced Materials in Catalysis/Ed. by J. J. Burton,
R. L. Garten.— New York: Academic Press, 1977.
Sleight A. W. // Science.— 1980.— V. 208.— P. 895.
Smeltzer W. W., Young D. J. // Prog. Solid State Chem.— 1975.— V. 10.—
P. 17.
Somorjai G. A. Chemistry in Two Dimensions: Surfaces.— Ithaca: Cornell
University Press, 1981.
Глава 9
505
Stinson S. С. // Chem. Engg. News.— 1983.— V. 61(49).— P. 19.
Subba Rao G. V., Shafer M. W. // Physics and Chemistry of Materials with
Layered Structures/Ed. by F. Levy.— Dordrecht: Reidel, 1979.— v. 6.
Sudo M., Tchikawa M., Soma M., Ovisha Т., Tamaru K. // J. Phys. Chom.—
1969.— V. 73.— P. 1174.
Tammann G. // Z. Anorg. Allgem. Chem.— 1920.— B. Ill,— S. 78.
Takatani S., Chung Y. W. // J. Catalysis.- 1984.— V. 90.— P. 75.
Tauster S. J., Fung S. C, Garten R. L. // J. Amer. Chem. Soc— 1978.—
V. 100.— P. 170.
Tauster S. J., Fung S. C, Baker R. Т. K., Horsley J. A. // Science.— 1981.—
V. 211.— P. 1121.
Taylor H. S. // Proc. Roy. Soc. London.— 1925.— V. A108.— P. 105.
Theocharis C. R., Desiraju G. R., Jones W- // J. Amer. Chem. Soc.—
1984.— V. 106.— P. 3606.
Thomas J. M. // J. Chem Soc. Chem. Comm.— 1967.— P. 432.
Thomas J. M. // Phi). Trans. Roy. Soc. London.— 1974,— V. A277.—
P. 251.
Thomas J. M. // Pure and Appl. Chem.— 1979 — V. 51.— P. 1065.
Thomas J. M. // Intercalation Chemistry/Ed. by M. S. Whittingham,
A. J. Jacobson.— New York: Academic Press, 1982.
Thomas J. M. // Proceedings of the NATO DAVY ASL— Cambridge, UK7
1983.— Physics and Chemistry of Electrons and Ions in Condensed Matter.
Thomas J. M., Evans E. L., Clarke T. A. // J. Chem. Soc. A.— 1971 —
P. 2338.
Thomas J. M., Lambert R. M. (Eds). Characterization o\ Catalysis.—
Chichester: John Wiley, 1980.
Thomas J. M., Williams J. O. // Prog. Solid State Chem.— 1971.— V. 6.—
P. 119.
Thomas J. M., Millward G. R., Schlogel R. F., Boehm II. P. // Mater. Res.
Bull.— 1980.— V. 15.- P. 671.
Thomas N. W., Ramdas S., Thomas J. M. 7 Proc. Roy. Soc. London.—
1985.— V. A400.— P. 219.
Thompson A. H. // Physica (B + C) (Amsterdam).— 1981.— V. 105B —
P. 461.
Wagner С // Z. Phys. Chem.— 1936.— B. 32.— S. 447.
Wagner С // Z Anorg. Allgem. Chem — 1938.— B. 236.— S. 320.
Wagner С // Prog. Solid State Chem.— 1975.— V. 10.— P. 3.
Wagner С Grunewald K. // Z. Phvs. Chem.— 1938.— B. 40.— S. 455.
Wegner G. // Makromol. Chem.— 1971.— V. 154.— P. 35.
Wegner G. // Molecular Metals/Ed. by W. E. Hatfield.— New York:
Plenum Press, 1979.
Whittingham M. S. // Prog. Solid State Chem.— 197S.— V. 12.— P. 41.
Whittingham M. S., Chianelli R. R. // J. Chem. Educ— 1980.— V. 57.—
P. 569.
Whittingham M. S., Jacohson A. J. (Eds.). Intercalation Chemistry.—
New York: Academic Press, 1982.
Wolkenstein Th. // Advances in Catalysis.— 1960 — V. 12.— P. 189.
ГЛАВА 9
Bednorz J. G., Miiller K. A. // Z. Phys — 1986.— B. 64.— S. 187.
Beille J. et al. // С R. Acad. Sci. Paris.—1987.-T. 18.-P. 304.
Bhargava R. N. et al. // Phys. Rev. Lett.— 1987.— V. 59.— P. 1468
Bhat S. V., Ganguly P., Ramakrishnan T. V., Rao С N. R. // J. Phys. С
Solid State.— 1987.- V. 20.— P. L559.
Bordet P. et al. // Nature.— 1987.— V. 327.— P. 687.
Cava R. J. et al. // Phys. Rev Lett — 1987a.— V. 58.— P. 408.
H. P. Pao, Дж. Гопалакришнан
506
Список литературы
Cava R. J. et al. // Nature.— 1987b.- V. 329.— P 423.
Chu С W. et al. // Phys. Rev. Lett.— 1987a.— V. 58.— P. 405.
Chu С W. et al. // Phys. Rev. Lett.— 1988.— V. 60.— P. 941.
David W. I. F. et al. // Nature.— 1987a.— V. 328.— P. 328.
David W. I. F. et al. // Nature.— 1987b.— V. 327.— P. 310.
Day P. et al. // J. Phys. С Solid State.— 1987.— V. 20.— P. 1429.
Er-Rakho L., Michel C, Provost J., Raveau B. // J. Solid State Chem.—
1981.— V. 37.- P. 151.
Fine S. M., Greenblatt M., Simizu S., Friedberg S. A. // ACS Symposium
Series.— 1987.— Chapter 10.
Ganapathi L., Ganguli A. K., Mohan Ram R. A., Rao С N. R. // J. Solid
State Chem.— 1988.— V. 76.— P. 235.
Ganguli A. K.. Subbanna G. N., Umarji A. M., Bhat S. V., Rao С N. R. //
Pramana.— J. Phys.— 1988.— V. 30.— P. L483.
Ganguly P., Rao С N. R. // J. So4d State Chem.— 1984.— V. 53.— P. 193.
Hazen R. M. et al. // Phys. Rev. Lett.— 1988a.— V. 60.— P. 1174.
Hazen R. M. et al. // Phys. Rev. Lett.— 1988b.— V. 60.— P. 1657.
Johnston D. С et al. // Mat. Re?. Bull.— 1973.— V. 8.— P. 777.
Johnston D. С et al. // ACS Symposium Series 351.— 1987.— Chapter 14.
Jorgensen J. D. et al. // Phys. Rev.— 1987.— V. B35.— P. 7915.
Maeda H. et al. // Jpn. J. Appl Phys.— 1988.— V. 28.— P. L209.
Michel C, Er-Rakho L., Raveau B. // Mat. Res. Bull — 1985 — V. 20.—
P. 667.
Michel С et al. // Z. Physik.- 1987.— B. 68.- S. 421.
Mohan Ram R. A., Ganguly P., Rao С N. R. // Phase Transitions.— 1987a.—
V. 10.- P. 107.
Mohan Ram R. A., Vasantacharya N. Y., Rao С N. R. // J. solid State
Chem.—1987b.—V. 69.—P. 186.
Murphy D. W. et al. // Phys. Rev. Lett.— 1987.— V. 58.— P. 1888.
Nelson D. L., Whittingham M. S., George T. F. (Eds.). // ACS Symposium
Series 351.—1987.—Chemistry oi' High Temperature Superconductors.
Rao С N. R. // J. Solid State Chem.- 1988a.— V. 74.— P. 147.
Rao С N. R. Chemistry of Oxide Superconductors.— Oxford: Blackwell
Scientific Publications, 1988b.
Rao С N. R. et al. // Nature.— 1987.— V. 326.— P. 856.
Rao С N. R* et al. // Pramana.— J. Phys.— 1988.— V. 30.— P. L359.
Sarma D. D., Rao С N. R. // J. Phys. C. Solid State.— 1987.— V. 20.—
P. L659.
Sarma D. D.. Ganguly P., Sreedhar K., Rao C. N. R. // Phys. Rev.— 1987.—
V. B36.— P. 2371.
Serge С U. et al. // Nature.— 1987.— V. 329.— P. 227.
Sheng Z. Z., Hermann A. M. // Nature.— 1988.— V. 332.— P. 55, 138.
Sleight A. W. et al. // Solid State Commun.— 1975.— V. 17.— P. 27.
Tarascon J. M. et al. // ACS Symposium Series 351.— 1987a.— Chapter 20.
Tarascon J. M> et al. // Phys. Rev.— 1987b.— V. B36.— P. 226.
Tarascon J. M. et al.— 1988.— V. B38.— P. 2504.
Uchida S. et al. V Jpn. J. Appl. Phys.— 1987.— V. 26.— P. 4.
Umarji A. M., Somasundaram R., Rao С N. R. // Solid State Comm.-
1988.— V. 66.— P. 177.
Wang H. H. et al. // Inorg. Chem.- 1987.- V. 26.— P. 1190, 1474.
Williams J. et al. // Ace. Chem. Res.— 1987.
Wu M. K. et al. // Phys. Rev. Lett.— 1987.— V. 58.— P. 908.
ПРЕДМЕТНЫЙ УКАЗАТЕЛЬ
Азот 27
Алиовалентная примесь 206, 209
Алкановые дикислоты 165
Алмаз 16, 27, 28
Альбит 45
Алюмосиликаты 45, 103, 116
Аморфные материалы 393
приложения 394
Аморфные полупроводники 328
Аморфные твердые тела 66, 198
получение 141
Амфиболы 44
Амплитудой 167
Амплитуда тепловых колебаний
75
Андерсоновский переход 320, 327
Анизотропия химического сдвига 101
Аномалия Кона гигантская 168
Антифазная граница 225
Антиферродисторсионный переход
161
Антиферромагнетизм 272, 274
Антисегнстоэлсктрикп (антиферро-
электрнки) 161, 349, 359
Антрацен 60
Апохолиевая кислота 442
Аргон 16
Ароматические твердые тела 60
Асбесты 44
Асимметрический синтез 441
Асперомагнетизм 272, 275
Атомно-абсорбционая спектроскопия
НО
Атомные радиусы 26
Ауривиллпуса фазы 245, 458, 468
Аустенит 205
Бариевые ферриты 244
Батареи твердотельные 123, 373, 374
катоды 123
Na/S-батарея 374
Бензол 60
Бертоллиды 207
Бесконечно адаптивные структуры
242
Берилл 44
Биполяроны 329
Блочные структуры 239
Бориды 65
Боросиликат алюминия 367
Браунмиллерит 250
Bremsstrahlung-изохроматичная
спектроскопия НО
Бриллюэновское рассеяние 290
Бром 27
Бромнрованне серебра 416
f-Бутнлхлорид 188
Вакансионный механизм 211
Вакансионная пара 206
Валентная зона 260
Валентная флуктуация 107, 311
Ван-дер-Ваальсовы кристаллы 23,
59
Ван-флсковскпй парамагнетизм 269
Вектор Бюргерса 220
Взаимодействие Дзялошинского—
Морпа 275
Взаимодействие по RKKY 271, 272,
277
Винтовая ось 14
Виртуальное связанное состояние
277
Висмут 27
Вихревое состояние 285
Вода 25
аморфная твердая 201
разложение 381, 383
Волновой вектор 23
Волны зарядовой плотности 72, 167
Вольфрамовые бронзы 41, 52, 87, 88г
148, 245, 247, 285, 371
Вюртцит 31, 32, 33, 55, 149
Вюстит 231
Выжигание дырки 400
Газовые гидраты 63
Газотранспортные химические
реакции 149
Галогениды щелочных металлов 18,
29, 32
Галофосфаты щелочноземельных
металлов 406
Гексабориды 65
Гелимагнетизм 272, 274
Генерация второй гармоники 404
33*
508
Предметный указатель
Германий аморфный 67
Гетерогенный катализ 445
активность 446
геометрический фактор 448
селективность 446
цеолитами 454
электронный фактор 448
Гиббсит 44
Гидротермальный метод 127, 146
Гистерезис 153
Гистерсзисная петля 355
Глины 44
Гомологические ряды оксидов 117
Гранаты 43, 301, 386
Границы двойников 225
Графит 28
адсорбция Кг 174
интеркаляцпя 422
Дальтониды 207
Двойникованпе 52, 54
Дегидратация
Мо03-2Н20 411
Мо03Н20 412
Мо1-Л°з-Н20 412
Деинтеркаляцня 123
Деоксихолисвая кислота 442
Дефекты
Уодсли 236
упаковки 221, 224
Френкеля 206, 208
Шоттки 206, 208
энергия 210
Диамагнетизм 267, 272, 273
Диацетилсны 405, 440
полимеризация 400, 405
Дилатационный переход 161
Диоксид кремния 35, 36, 152
Диопсид 44
Дипольные стекла 200
Дислокации 220
van der Merwe 224
взаимодействие 221
винтовые 220
в органических кристаллах 220
краевые 220
смешанные 220
совершенные 220
спиральные 222
частичные 220
Франка 222, 224
Шокли 222, 225
энергия 223
Диспласивный переход 161
Дисплей на принципе гость —
хозяин 402, 403
динамического рассеяния 402
Диффузия 210
вращательная 189, 191
р-Дихлорбензол 165
Диэлектрическое поглощение 192
Диэлектрические свойства 286
Додекасил-Ш 128
Додекасил-ЗС 64
Допент 226, 398
Дрейфовая подвижность 281
Дуговая печь 134
Дуговые методы 133
транспортный (переноса в дуге)
трехдуговой 134
Дьявольская лестница 167
Жидкие кристаллы 116, 193, 362, 401
лиотронные 193
нсматические 193
приложения 401
свойства 362
смсктнчсскис 193
термотропные 193
Жидкокристаллическое состояние
193
Закон Брэгга 74
Кюри 269
Кюри — Всйса 269, 273, 287
Ома 279
Зародышеобразованис 409
Золь-гель процесс 120, 140
Зона проводимости 260
Зонная плавка 144
ZSM—5 47, 116, 243, 454
ZSM-11 243
Изопентановос стекло 203
Икосаэдричсская симметрия 71
Инваровые сплавы 204
Интеркаляцпя 123
A10G1 431
графита 422
дихалькогенидов 423
каркасных матриц 434
листовых структур 432
Li 123, 125
оксидов 123, 125
ступенчатость 422
сульфидов 123, 125
FeOCl 431
Интерметаллические гидриды 391
Иод 27, 61
Ионная диффузия 210, 282
Ионная модель 29
Ионная проводимость 210, 282
в стеклах 283
во фторидах 313
Ионные кристаллы 17
Ионные радиусы 29, 30
Ионные стекла 68, 70
Ионные твердые тела 16
Предметный указатель
509
с молекулярными примесями 218
с пара электрическими
примесями 218
с точечными дефектами 208
Ионный обмен 126
Ионный характер 20
Истощенный слой 280, 376
Кадмий 26
Каменная соль 32
Камера Гинье 76
Камфора 188
Каолинит 44
Каркасные структуры 369
Катализ 445
Катализаторы 91, 97, 99, 450
селективность к форме 454
SMSI 452
спилловер 452
Катодные материалы 123
Квазижидкие кристаллы 197
Квазикристаллнческпй сплав 78
Квазикристаллы 71
Квантовый эффект Холла 282
Кварц 36, 45, 152
ее—р-переход 152, 156, 161
мягкая мода
центральные пики 158
Кевлар 397
Керамический метод 118, 119
Кинетика разложения 410
Классы кристаллов 14
Кластеры (предельное состояние
твердого тела) 139
Клатрасил 64, 128
Клатраты 63
Ковалентные кристаллы 16, 20
Ковалентные стекла 67, 68
Коинспдентная граница 225
Комплексы
Коха — Коуэна 218
Creutz — Taube 335
с переносом заряда 62, 137
HMTSF—TGNQ 63
TMPD—TCNB 62
TMTSF 138
(TMTSF)2X 138, 348
TTF — галогениды 137, 337
TTF — TCNQ 62, 137, 337, 341,
347, 348
хозяин — гость 442
Композитные полимеры 398
Комптоновское рассеяние 111
Компьютерное моделирование 201
Компьютерная графика 72
Константа Кюри 269
Маделунга 17
Кооперативный эффект Яна — Тел-
лера 175, 317
Коричные кислоты 435, 436, 444
Коэффициент Зсебека 281
Псльтье 281
Край подвижности 327
Крамерсовский дублет 176
Красная соль Вольфрама 336, 346
Кремниевые солнечные элементы 375
Кремниевый фотоанод 380
Кремпий
аморфный 68, 395
получение 136
рост кристаллов 143
Кристаллические системы 13, 14
Кристаллографический сдвиг 51, 235
Кристаллохимия 13
Кристобалит 35, 36, 45
Критерий Герцфельда 327
Мотта 326
Хаббарда 326
Критическая температура 155
Критические экспоненты 159
Критические явления 159
Куперовские пары 285
Купраты сверхпроводящие
висмутовые 467—469, 472
таллпевые 467, 471, 473
типа La2Cu04 458—460
типа YBa2Cu307 461—466
Куприт 35, 36
Лазерное стеклование 142
Лазерные материалы 406
Лазеры 290
твердотельные 407
Лазурит 45
Левин 47
LISICON 369, 371
Люминесцентные материалы 405
Люминесценция 250, 290
Магнетизм Дзялошинского 166
Магнетит 42, 107
Магнетон Бора 267
Магнитная группа 15
Магнитные материалы 383
«Магнитные пузырьки» 385
Магнитные свойства 267, 272
высокотемпературных
сверхпроводников 464, 469, 471
оксидов 292
перовекптов 301
сульфидов 304
фторидов 315
Магноны 278
Малоугловые границы зерен 224
Мартенснтные переходы 164, 205
Масс-спектрометрия ИЗ
Масштабная инвариантность 160
Материалы для нелинейной оптики
116, 404
510
Предметный указатель
Материалы для хранения водорода
115, 388
Междоузельный механизм 211
Межзеренные границы 223
Межзонный переход 289
Мезофаза 193
Металлические кластеры 331
Металлические кристаллы 22
Металлические стекла 66, 69, 141,
273, 394, 451
Металлические элементы 26
Металоорганические полимеры 398
Метамагнстизм 272, 275
Метан 60
Метанол 61
2-Метил-4-ннтроанилин 404
Методы
Вейзенберга 76
вращающегося кристалла 76
дифракции порошка 76
Лауэ 76
молекулярной динамики 202
Монте-Карло 201
Рптвельда 76, 79
Методы роста кристаллов
Брпджмена — Стокбаргсра 144
газотранспортных химических
реакций 149
геля 147
гидротермальный 146
зонной плавки 144
Киропулоса 144
молекулярно-лучевой эпитак-
сни 150
«отжига напряжения» 151
плавающей зоны 144
плавления в пламени (Вернейля)
144
плазменной горелки 145
флюса 146, 148
Чохральского 143
электролитический 148
Методы синтеза
высокого давления 127
аппаратура типа наковальни
129
аппаратура типа поршень —
цилиндр 128, 129
«бэлт»-аппаратура 130
гидротермальный 127
деинтеркаляцни 123
дуговые 133
транспортный (переноса в дуге)
135
трехдуговой 134
закалки 142
интеркаляции 123
ионного обмена 126
керамическе 118, 119
золь-гель 120 (140)
соосаждения 120
сушки вымораживанием 120
(140)
сушки распылением 120 (140)
лазерного стеклования 142
настыльного плавления 135
органических твердых веществ
137
пирогеля 140
предшественника 121
разложения из газовой фазы 122
формования из расплава 141
химического осаждения из
газовой фазы 135, 149
электролиза расплавленных
солей 123
Методы характсризации 73
состава и чистоты 112
Микрокластеры 139
Микрокристаллические частицы 139
Миктомагнетизм 272, 275, 277
Минимальная металлическая
проводимость 329
Модель тяжелого фермиона 334
Модулированные структуры 167
Молекулярная динамика 201
Молекулярная электроника 397
Молекулярно-лучеЕая эпптаксия
150
Молекулярные твердые тела 16, 23
Молибдаты висмута 453
Morin-переход 301
Морфология 75
Моттовский диэлектрик 262, 266
Моттовский критерий 326
Моттовский переход 320, 325
Мусковит 45
Мышьяк 27
Мягкие моды 156, 174. 204
Направление скольжения 221
NASICON 118, 120, 369, 371
Настыльное плавление 135
Нафталин 60
Нейтронная дифракция 79, 80
Нейтронно-активационный анализ
113
Нейтронное рассеяние 79
пластичных кристаллов 191
Нейтронное спиновое эхо 81
Нейтроны импульсные 80
Некристаллические твердые тела 66,
197
получение 120, 141
Нелинейные оптические материалы
404
Неметаллические элементы 26
Неопентан 188
Предметный указатель
511
Несвязывающис взаимодействия 48
Несвязывающие радиусы 49, 50
Несоразмерные фазы 166
Нестехиометрические соединения
207
Нестохиомстричность 205
Низкоразмерные твердые тела 341
Нити 367
Нуклеация 410
Обменное взаимодействие 270
Обращенная шпинель 42
Оже-спектроскопия 104
Окалинообразованис 412
Окисление металлов
кинетика 413
меди 414
цинка 415
В-Оксиды алюминия 115, 118, 120,
124, 243, 368, 371, 372
Оксидные бронзы 41, 148, 245, 248
Оксиды
ниобия 240
переходных металлов 261, 292,
297, 300
редкоземельных (С-тнп) 38
Оливины 43
Операция вращения 51
отражения 51
трасляцни 51
Оптические волокна 396
Оптические свойства 286
Орбитальные радиусы 22
Органические твердые тела 59
реакции 435
синтез 137
фазовые переходы 165
Ориентационно разупорядоченные
фазы 187, 202
Ортоклаз 45
Ортофоррнты 386
Отжиг напряжения 151
Отношение радиусов 29
Хавсна 283
Офретит 47
Пайерлсовское искажение 167
Пара примесь — вакансия 209
Параболический закон скорости 413
Парадокс Каузмана 199
Парамагнетизм 267, 273
Ван-флсковский 269
Паули 270
Параметр
ионности 21
ковалентного смешения 264
порядка 154, 178, 195
собственный 353
Параэлектрические примеси 218
Парные свойства 357
Переходы
Verwey-переход 336
зародышеобразованне — рост 162
мартенситные фазовые 164, 205
металл — неметалл 320
неметалл — металл 262
под давлением 131, 152
порядок — беспорядок 163
Периодическое прорастание 244
Перовскитовые вольфрамовые
бронзы 247
Перовскиты 39, 40, 58, 247, 301
PZT 140, 141, 360
Пирит 37, 55
Пирогель 140
Пироксены 44
Пирофиллит 44, 129, 432
Пирохлоры 42, 258
дефектные 258
Пироэлектричество 354
Плавление в пламени 144
Плазменная горелка 145
Плазменная частота 288
Планарные дефекты 223
Пластическое кристаллическое
состояние 187, 202
Пленки
инертный газ — металл 262, 327
Ленгмюра — Блоджетт 401
Плоскость скольжения 14, 221
Плотная упаковка ионов 28
Плотнейшие упаковки 26, 28, 31
Пннктиды редкоземельных
металлов 177
Поглощение излучения 289
Поглощение свободного носителя
289
Полиацетнлсны 72, 116, 262, 348,
374, 398, 400
получение 138
Полиготероцпклы 139
Поликарбоны 383
Полимерные материалы 397
Полимерные стекла 66, 70
Полимеры 397
композитные 398
металлоорганичеекпе 398
«сплавы» 398
фотопроводящие 398
Полипиррол 139, 349
Полнтиснилсн 139
Политиофен 349
Политипнзм 56, 243
в перовскитах 58, 59
в SiC 57
в силикатах 57
в TaS2 57, 58
Полуметаллы 26, 261
512
Предметный указатель
Полупроводники 260, 279
аморфные 328
несобственные 261, 280
скомпенсированные 261
собственные 261, 279, 280, 281
п-типа 261, 280
р-типа 261, 280
Полупроводник — электролит 376
Полупроводниковые электроды 376,
378
Полярон 263
Порфирин 400
Потенциал Китайгородского 24, 60
Леннарда — Джонса 24, 202
Маделунга 19
Потускнение 412
Правило Лоуэнштейна 47
Предельное состояние твердого тела
139
Препаративные методы 115
Приближение сильной связи 23
Примитивная ячейка 13
Прорастание кристаллов 51, 73, 243
Проросшие бронзы 89, 245
Пространственная группа 14, 61
Пространственная решетка 13
Пространственно-заряженный слой
376
Протонные проводники 127, 372
Прыжки (хоппинг) 263, 281
«Пузырьковая память» 116
Пустоты 26, 29
Пьезомагнетизм 350
Пьезоэлектричество 350
Равновесие дефектов 212
в оксидах 216
Шоттки 209, 212
Разложение твердых веществ 409
Рамановская микроскопия НО
Рамановская спектроскопия 110, 192
Раман-эффект 290
Раствор металл — аммиак 262, 327
Растворение твердых тел 420
Расширяемые металлы 262, 327
Реакции
обмена 420
присоединения 418
твердое — жидкость 420
твердое — твердое 417
топотактические 408, 411
топохимическис 408, 411, 436
в органических твердых телах
436, 437
Реакционная способность твердых
тел 407
Режим Кондо 276
Резист 401
Реконструктивный переход 161
Релаксорные ферроэлектрики (сег-
нетоэлектрики) 359
Ренорм — группа 160
Рентгеновская абсорбционная
спектроскопия 90
фотоэлектронная спектроскопия
105
Рентгеновская дифракция 73, 76
аморфных твердых тел 77
профильный метод 76
Рентгеновская флюоресценция ИЗ
Рентгеновский эмиссионный анализ
86, 112
Решетки Браве 13, 14
Решетки пустот 209
Рост кристаллов 143
Ртуть 26
Рубин 110, 291
Рутил 35, 36, 55, 56
Сверхпроводимость 284
высокотемпературная 457—473
Сверхструктуры 226
в галогенидах щелочных
металлов 226
— карбидах 227
— оксидах 229
— оксидах со структурой ие-
ровскита 247
— твердых телах со структурой
типа NiAs 228
— флюорита 233
халькогенид 227
Световоды 396
Свстодиоды 290
Связность 361, 262
Связь Кондо 278
Сегнетоэлектрикп 40, 161, 349, 354,
357
релаксорные 359
Сетка Кагомэ 53
Сиалон 116
Силикалит 128
Силикаты 42
несвязывающие взаимодействия
48, 49
расстояние Si...Si 49
слоистые 44, 45, 57, 432
углы Si —О—Si 48, 49
Симметрия положения 75
Система
Fe—S 228
La—Ba—Си—О 460, 466
Sc—S 229
Ti-S 227
Zr-S 228
Сканирующая туннельная
микроскопия 111
Предметный указатель
513
Скуттерудит 39
Слой Гельмгольца 376
Слюда 45, 57
Смешанная валентность 108, 332
Совершенство кристаллов 75
Содалит 46
Содалитовая клетка 46
Соединения А-15 115, 136, 164, 285
Соединения включения 63, 443
мочевины 64
тиомочевины 64
Соединения
I—VII 20
II—VI 20
III—V 20, 136, 143
Соединения со смешанной
валентностью 332
Солитоны 72, 247
Солнечные элементы 395
Соосаждение 120
Соотношения Крамерса — Кронига
288
Соразмерные структуры 172, 173
Спектроскопия потерь электронной
энергии 87, 100, 105, ИЗ
адсорбированных молекул 105,
109
Спектр остовных уровней 106
Сперимагнстизм 272, 275
Сперомагнетизм 272, 275, 276
Спиновая волна 278
Спиновое стекло 272, 275, 276
Спиновые переходы 179
в комплексах 180
в оксидах 182
Спин-решеточная релаксация 191
Спин-спиновая релаксация 191
Спиральные магнетики 166
Сплавы Mo-W 122
Ni— Al 209
РЗЭ-Со 384
Стабилизированный диоксид
циркония 373
Статистика Ферми — Дирака 259,
260
Стеарат Мп(П) 401
Стекла 68, 141, 394, 395
дипольные 200
получение 141
методы
закалки J42
лазерного стеклования 142
формования из расплава 141
Стеклование 198
воды 201
изопентана 203
Стеклокристаллы 200
Стишовит 49
труктуры со сдвигом 51, 235
Структуры типа каменной соли 32,
50
Сульфиды
ванадия 305, 306
переходных металлов 262, 304
Суперобмен 271
Суперпарамагнетизм 272, 276
Сушка
вымораживанием 120, 140
распылением 120, 140
Сфалерит 31, 32, 33
Тальк 45
Твердые ионные проводники 368
аморфные 368
Твердотельные батареи 373
Твердые тела с водородной связью 25
Твердые электролиты 115
«Твист»-граница 224
«Твист»-эффект 402
Температура Кюри 273, 287
Нееля 274
Стэнли — Каплана 343
Теорема Saint-Venant 243
Теория БКШ 285
Блоха — Вильсона 259
Ландау 154
поля лигандов 259
свободных электронов Друдэ 22,
23
Термисторы 367
Термоэлектрический эффект 280
5-Тетразин 400
Точечная группа 14
Точечные дефекты 205, 208
миграция 209, 210
наблюдение 210
образование 208
в ионных твердых телах 208
в металлах 209, 211
в твердых инертных газах 210
равновесие 212
упорядочение 226
Трансмиссионная электронная
микроскопия 82
Тридимпт 36, 45
Тройные сверхпроводники 285
Упаковка стержней 53
Уравнение Аврами 163
Аврами — Ерофеева 410
Бернулли 387
Клаузиуса — Клапейрона 154
Клаузиуса — Моссотти 286
Крамерса — Кронига 288, 289
Нернста — Эйнштейна 283
Праута — Томпкпнса 410
Уранилфосфат 375
Условие Брэгга 260
УФ-сисктроскония 110
514
Предметный указатель
Фазовые переходы 152
в органических твердых телах 165
в пластичных кристаллах 187
второго рода 153,
гистерезис 153
компьютерное моделирование 202
механизмы 161
мягкие моды 156, 205
первого рода 153
под давлением 152
приложения 203
термодинамика 152
Фазон 167
Фазы несоразмерные 166
Фазы
Магнели 235, 322, 412
Шевреля 51, 65, 116, 118, 285,
311, 337, 435
Фактор толерантности 40
Фаматннпт 33
Ферримагнстизм 272, 274
Ферриты 42, 120, 150, 367
Ферродисторснонный переход 161
Ферроики 258, 349
вторичные 350, 358
композитные 360
несобственные 353, 359
первичные 350, 357
собственные 353
Ферромагнетизм 272, 273
Ферромагнетики 203, 383
молекулярные 383
органические 383
Феррофлюиды 386
Ферроэластнчность (сегнетоэластнч-
ность) 350, 358
Ферроэлектрпкп (сегнетоэлектрпкп)
40, 161, 349, 354, 357
релаксорные 359
Филлосиликаты 44
Флюорит 35, 55
Формование из расплава 141
Фосфор 27
Фосфоры 405
Фотоаноды 380
Фотогалъваннческис ячейки 379
на полупроводниковой основе 380
Фотоднмернзацпя 436
Фотокатоды 379
Фотокоррозия 380
Фотолюминесценция 290
Фотополимеризация
диацетнлена 439
днетирилпиразина 437
Фотопроводящне полимеры 398
Фоторазложение воды 381, 383
Фотоструктурный эффект 396
Фотохимическое превращение
солнечной энергии 381
Фотохромные материалы 399
Фотоэлектролиз 377
Фотоэлектрохимическая ячейка 376
Фотоэлектрохимия 375
Фоязит 47
Фрактали 160
Фрустрация 272
Фталоцианин 400
Фторидные ионные проводники 314,
372
Фториды металлов 312
Функция Блоха 260
Бриллюэна 268
Халькогенидные стекла 394, 396
Халькопирит 33
Хаотическая структура 167
Химические сдвиги
Оже-спектры 108
рентгеновские края поглощения
98, 106
Химическое осаждение из газовой
фазы 135, 149
Химия интеркаляции 421
Хиолит 315
Хлор 27
Хлорин 400
Хопперы 280
Хоппинг 263, 281, 282, 292, 297, 324,
329, 333, 334
Хранение водорода 115, 388
Хризотил 44
Цветная группа 15
Центральные пики 158
Центры окраски 219
Цеолиты 45, 46, 72, 103, 434
катализ 454
синтез 128
шабазитная группа 47
Цепочечные силикаты 43
Цианобифенилы 364, 365
Цикл Борна — Габера 19
Циклогексан 188
Цинк 26
Цинковая обманка 31, 32, 33
Шпинели 31, 41, 152, 274, 301
образование 418
эффект Яна — Теллера 177
Щавелевая кислота 62
Р-Эвкриптит 369
Эвтаксия 31, 48
Экситоны 289
Электретные материалы 399
Электрические свойства 278
аморфных твердых тел 329
оксидов 292
перовскитов 301
Предметный указатель
515
сульфидов 304
фторидов 312
Электролиз расплавленных солей
123
Электролитический метод 148
Электронная дифракция 77, 82
Электронная микроскопия 82
аналитическая 87
высокого разрешения 84, 88
сканирующая 84
сканирующая трансмиссионная
82, 85
трансмиссионная 82
Электронная спектроскопия 75, 104
Электронный спиновый резонанс 110
Электрон-фононное взаимодействие
263
Электрооптические материалы 360,
402
Электрооптнческнй эффект 404
«твист»-эффект 402
Электрострлкция 359
Электрохимические фотогальвани-
ческпе ячейки 379
Элементарная ячейка 13
Эмиссионная спектроскопия ИЗ
Энергия когезии
галогенидов щелочных металлов 18
ионных кристаллов 17, 18
молекулярных кристаллов 24
Энергия переноса 264
Энергия решетки
ионных твердых тел 19
Энергия Ферми 260
Энстатнт 44
Эпитакснальный рост 224
Эрионит 47
Эффект Беккереля 376
Мейсснера — Охзенфельда 284,
464
Хедвала 409
Холла 281
Яна — Теллера 175
Эффективная масса 263, 279
ЯМР-спектроскопня 100
алюмосиликатов 101
анизотропия хпмедвпга 101
пластических кристаллов 191
с вращением под магическим
углом 100, 102
Ян-Теллсровское взаимодействие 184
Ячейка на основе «твист-эффекта» 403
WtdtdtdtdWtdtdtdtdtd td td td td td to to td td td со td td >>>>>>>>>>>>>>>> > > A
^. и-. »-■• и-, н. ф и в ш ш р Р5э5эрйэ5э5эоэоэйэс?оэоэ u семисот H-'t-'StjqIIQuQCiqQPQCiqciqs ^ w
ЪГтк"&>&&*$ йнв vH *я3? 31 ?« 12it ЗГДО:© *go=* 11 "я2? ,и 3tt £
kQ
о tr1
l-H
JNJNJNjNtOCO^tO^i^cOH^^lNS^lNScotOCOlNSlNS^jNo^ to ^^051^0 tO^WOi CO W WOWmjs М М 3
СЛСЛСлОО^^^СПСЛ^СпО^О^^^ОСЯСЯЙСПпоО [СОО<1*ч<11^С0^01МЬЮ05С0н.|^ СЛ СП
^^^ч 4^-* ^OOOO-J^ C005«OM<ICnUlfl00*s05 ^^-" W 00^- СЛ СЛ СЛ- И-С5 4 *< S>
~ ^ ~ ^ ^^^T CO tO HH
OO ^ oo ^ ^40W 00 О 0*9
СЛ tO СЛ СП GO ~ & tO ^
-jo- en g:
н^ DO CO OO J~
£: -<i со ^
H
H-* CO ^
^ JO £H
nnnnnonnon о "Onnnnnnnnnnnnonnnnnnnnnnatdwwaaw и и
*"? ^ £* ^2.^ OOOO ^ 'o £ 4j О-О-О-РЬ О-П Р О5»5» Р BpjjjjjjBtBBjjpp ^ •— »-• — •— »— — *-• —
^ -* " " ® со .^| _«. ^ ± » ^^^^РиФ.Р.Ро1 сГ ПО ^ — — « Ч « ^ -
Я 2 ч О "Я? О? I он-р к- О со о-5
:-ч
3 ч
—3
g
р
о
&*
ъ
-J
Чо<
с
^tO^tX^OOoOh^ ОО оэ СО ^OOOO^coOOtO^toto-^tOtODO ^^^W tOrTTtOOOtO^COJNOO tO
t^^aicoocnoooto^ oo ^j^^tot^iss^^Oo^^oocncncntooooooocncoojcnS^o^cn^oociOo о
-^,н^00 JNQC Ю OiCn to Г.СО - - CO^-^^- CO rfs^cn" ^^ OO о ^ ^ СЯ О °^ X] 4N
COCO^^ fe 5 ^ 2 СПГ, JN
gcO 0°fe Я О СО ^
°o oog to со oo
to
CO
CO
Формульный указатель
517
CsCl
CsFeF3
CsNiFg
Cs2NiF6
Gs W03
X °
CuAl2
CuGN
CuFeS2
Cul
CuO
Cu20
CuS2
Cu3SbS4
CuSi2P3
Cu0f5TaO8
CuZn
DyV04
EuO
Eu3S4
Eu „Si- S
1-Х X
Fe
FeF3
Fe—La—В
Fe-Nd-B
FeO
Fe 0
Fe203
Fe304
FeOGl
FePSe3
FeS
FeS2
Fe—S
FeTi
FeTiH
FeTiH2
GaAs
GaP
Ga2So
Gd2Cl3
GdGo5
Gd2(Mo04)3
GdTi03
Ge02
GeS
Ge—Se
GeTe
Hg3 xAsF6
Hgl2
HgS
HNb03
HTa03
31, 32, 33
316
318
133
245
51
33
33
33, 96
415
35, 36, 415
308
33
33
248
33
176
295
311
276
159
315
385
385
107, 293
205, 232
38, 107, 140, 261,
300
42, 107, 141, 150,
333, 335, 387
431
429
306, 307
37, 308
228
434
391
391
97
97
33
51
386
204, 357
302
97, 142
55
68, 70
55
171, 342, 347
38
34
126, 375
126
кс8
KC1
KCN
K2CoF4
KCuF3
K2CuF4
K4Fe(CN)6
KH0,67G8
KH2P04
KM11F3
KM03S3
KNiF3
K2NiF4
K„Pb[Cu(N02)6]
K2[Pt(CN)4b
• Br0,3'^H2O
KSb63
K2Se04
KTa0,65Nb0,35O3
ктю2,5
La3-.xBa3+x-
•CU6014 + 6
La3Ba3Cu601Q _.
0 0 0 18—У
LaCoO,
LaoGu64
La^ M Gu04
2-х x *
ЬаоСо205
LaFc03
La4LiCo08
La2LiFeOe
La2Li0,5Ni0f5O4
LaMn03
LaMn03+JC
LaNi5
LaNi4Al
LaNi5He
LaN4-xMx°3
LaNi03
La2Ni205
La2Ni04
n n Зп-1
LaOF
Lai-.vSr»Co03
Lai.5Sr0,5Li0,5*
•Ре0,5О4
La, Sr V03
1-х x °
LaTi03
LiAlSi04
LiCo02
LiGr02
LiFoS?
LiGa02
Li4Ge04
Li3N
LiNbO^
422
18
163
319, 344
317, 343
318
431
422
159, 357
158
51
39
39, 41, 318, 343,
458, 459
172, 173
169
205
157, 166,
164
252
466
256
182, 323
458, 459,
458, 459
254
121, 315
78, 132,
132
132
317
41, 257
389, 390,
392
391
330
132, 133,
254
78, 135,
253
30
328, 335
132
331
302
369
127, 295
121, 126,
124
55
369
372
355
173
472
183
434
302
345
295
518
Формульный указатель
LiNi02
LiRe03
Li2Re03
Li0,6TiO2
Li—TiS2
Li^TiS2
LiV02
LiYF4
Li2ZnGe04
Ln2B207
Ln3-*Ba3+*
LnCo5
Ln2Co17
LnGo03
LnCrSg
Ln3Fe21B
LnNi6
LnNiOg
LnRhOg
Ln2S3
Ln3S4
LnTiS3
LnVS3
MgAl204
MgFe204
MgPSe3
MnFe264
MnO
Mn02
Mn203
MnPSe3
MnS2
Mo02
Mo03
Mo5014
MOn°3n-l
M°n°3n-2
Mo03.nH20
MoS2
MoftS8
NaGl
Na5Cr3F14
NaMo40G
NaN02
NaSb03
Na5V3F14
NaZr2(P04)3
Na3Zr2PSi2012
NbO
Nb02
Nb205
NbS2
NbS3
Nb21S8
NbSe2
295
124
124
124
374
123, 368
295
407
309
42
466, 467, 472
384
384
182
310
385
115
132, 133
133
312
311
310
310
31, 41, 367
367
429
367
261, 293
298, 300
261
429
308
298, 299
412
52
117, 238
238
411
37, 57, 58, 306,
307, 425
65
18, 19, 32
316
51, 52, 118
166, 172, 357
205
316
371
118
150
322
240
37, 57, 306, 307,
424
309
310
150
Nb3Si
Nb3Sn
[N(GH3)4]2MG14
NdCo03
NdF3
Nd2Fe14B
NH4Br
NH4C1
(NH3R)2MG14
NiAl
NiAs
NiBr2
NiFe204
NL Li О
1-Х* X
NiO
NiPS
NiS
NiSe2
NiS0 So
2-х* X
Ni.-«Te»
РЬ1-,ШЛ + *
PbCl2
PbF2
Pbl2
PbO
pbiRu1„acPbxo7.
PbSnF4
PbSn03
PbTi^^Zr^O,
PdF3
PdO
PdS
PrA103
PrCoOg
Pr7Ol2
Рг 0o 0
71 2П-2
Pt(NHg)2Cl3
PtS
RbAff4I5
RbCl
RbMnF3
RbN03"
Re03
(RNH3)2MG14
Ru(bipy)33+
Ru02
136
157
172
182
97
385
32, 33
32, 33, 358
166
209
31, 32, 34
345
367
208
261, 293
430
307
308
308
227
313
56
372
57
34
116, 122
313
132
120
39
35
35
177
182
233
117, 233
334
34, 35
95
152
316
152
39, 52, 265, 266,
302
342, 345
382
298, 382, 383
Sb204
Sb2S3
Sc-S
Sc,s3
Se
SiC
S13N4
Si02
Si
334
38
229
229
142
57, 122
136, 367
67, 68
67, 68
Формульный указатель
519
Sm
SmCo6
Sm2C017
SmS
SmSe
<SN),
S11I4
SnO
SnS
SrFeOg
SrFe03 _x
SrRu03
Sr3Ru207
SrTi03
SrTi02,5
Sr3Ti207
SrV02,5
SrVOg '
TaS2
TaS,
Ta6S
TaSe2
TbV04
ThGr2Si2
TiO
Ti02
Ti203
T13O5
Tin°2n-i
TiS2
Ti — S
2HTe4
Til
TmV04
U02+*
v6c5
106
384
384
119, 311
119
347
161
131
34
132
253
304
133
156, 157,
458
252
243
252
302
37, 57,
424
309
310
57
176
54
33, 229,
36, 298,
38, 300,
322
117, 238,
123, 306,
227
51
34
176
234
229
, 333
377,
58,
293,
376
321
■ 298,
, 307,
378,
166,
295
322
424
V8G7
VO
vox
vo2
V2Og
Vn°2*-1
VOCl
V, S
1-х
V3S
v»s4
V3Si
p - w
wo3
W03-*
wo2
Wn°3n-1
w„o,„ ,
n Зп.—2
W03-Ta205
XcF2
Xc-Na
Y3Fc5012
YBaaCu30_
* A 1-х
YNiOg
Y203 - YOF
Y203 - Zr02
ZnCl2
ZnFe204
ZnO
ZnS
Zn2Ti04
Zr4Al3
Zr02
ZrOo — CaO
ZrO; - Y203
Zr - S
229
33,
296
298,
38,
322
431
228
306
310
310
115,
51
39
236
298,
238,
117,
242
56
262
140,
461-
304
233
233,
97
409
31,
32,
367
51
164
368,
233
229
230, 293
321
321
164
299
412
238, 412
386
-466, 472
235
33, 415
57, 115, 405
373
Научное издание
Рао Чинтамани Нагеса Рамачандра
Гопалакришнан Джаннатха
НОВЫЕ НАПРАВЛЕНИЯ В ХИМИИ
ТВЕРДОГО ТЕЛА:
СТРУКТУРА, СИНТЕЗ, СВОЙСТВА,
РЕАКЦИОННАЯ СПОСОБНОСТЬ
И ДИЗАЙН МАТЕРИАЛОВ
Редактор издательства Т. П. Гришина
Художественный редактор С. J3. Марковская
Художник С. М. Кудрявцев
Технический редактор Г. Я. Драгун
Корректоры Е. Н. Зимина, Т. Ф. Погиблова
ИБ № 34604
Сдано в набор 04.09.80. Подписано к печати
13.08.90. Формат 60x90Vie. Бумага книжно-
журнальная. Обыкновенная гарнитура.
Высокая печать. Усл. печ. л. 32,5. Усл. кр.-
отт. 33,1- Уч.-изд. л. 35. Тираж 1000 экз.
Заказ № 787. Цена 7 р. 30 к.
Ордена Трудового Красного Знамени
издательство «Наука», Сибирское отделение.
630099 Новосибирск, ул. Советская, 18.
4-я типография издательства «Наука». 630077
Новосибирск, ул. Станиславского, 25.