/






































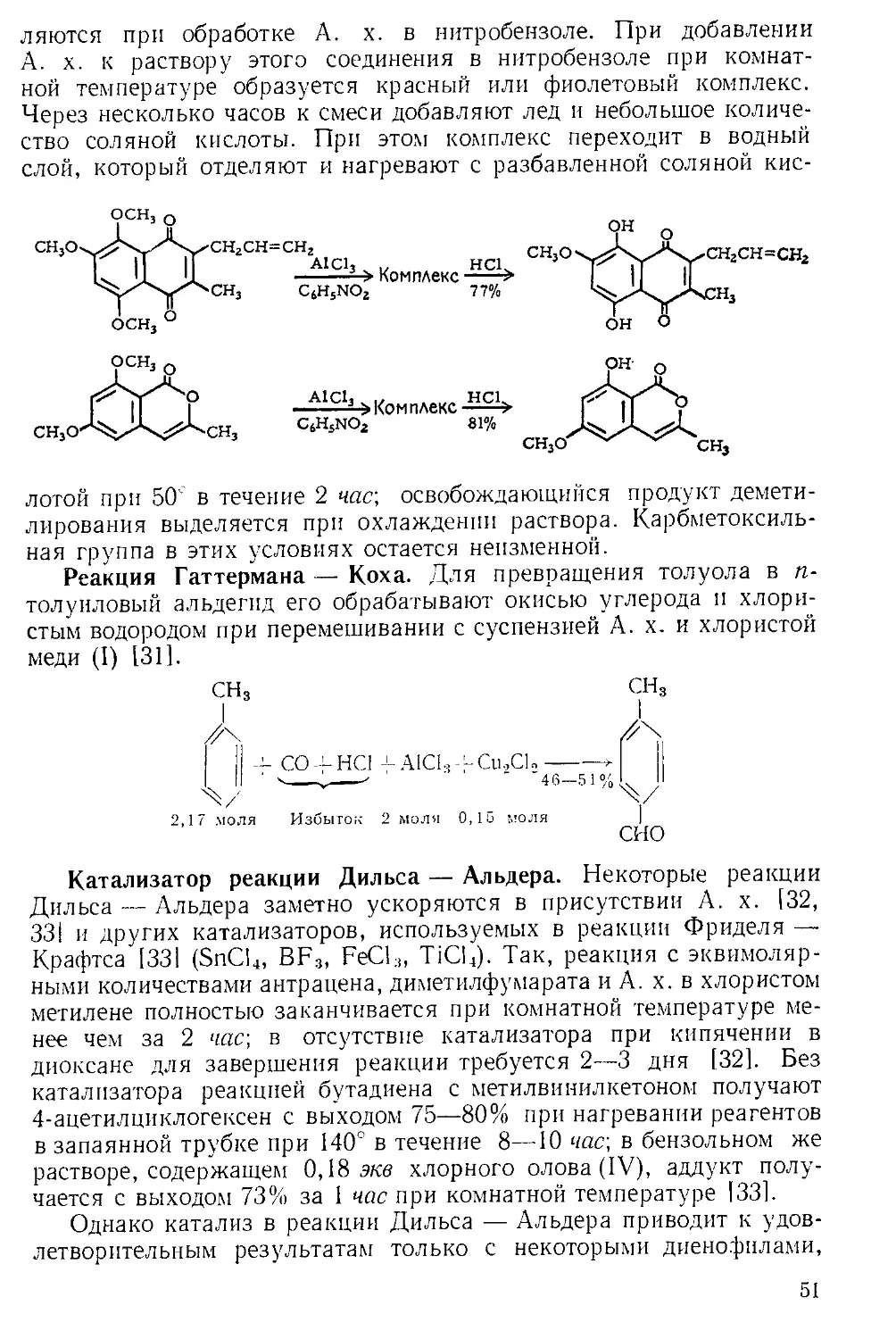

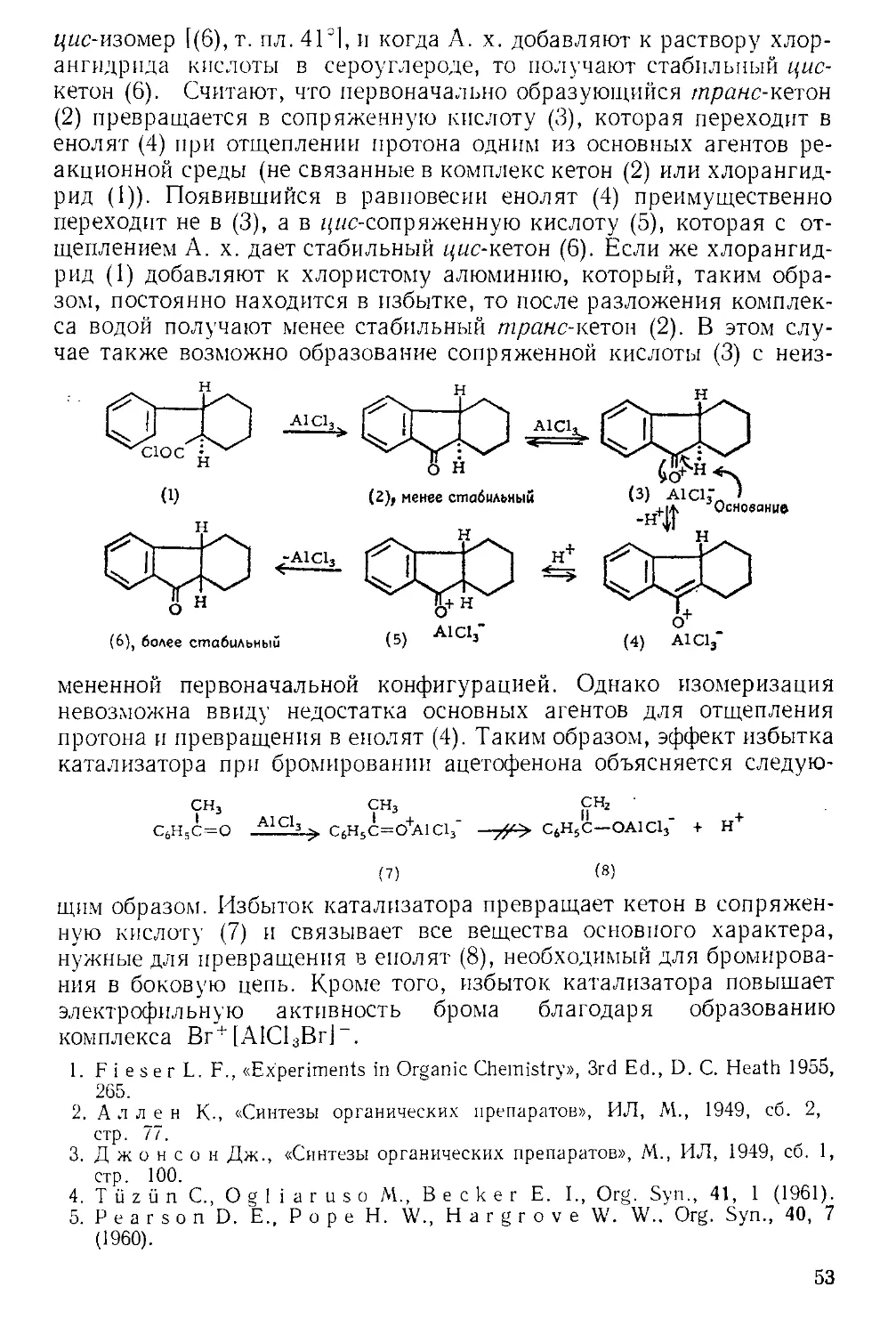



























































































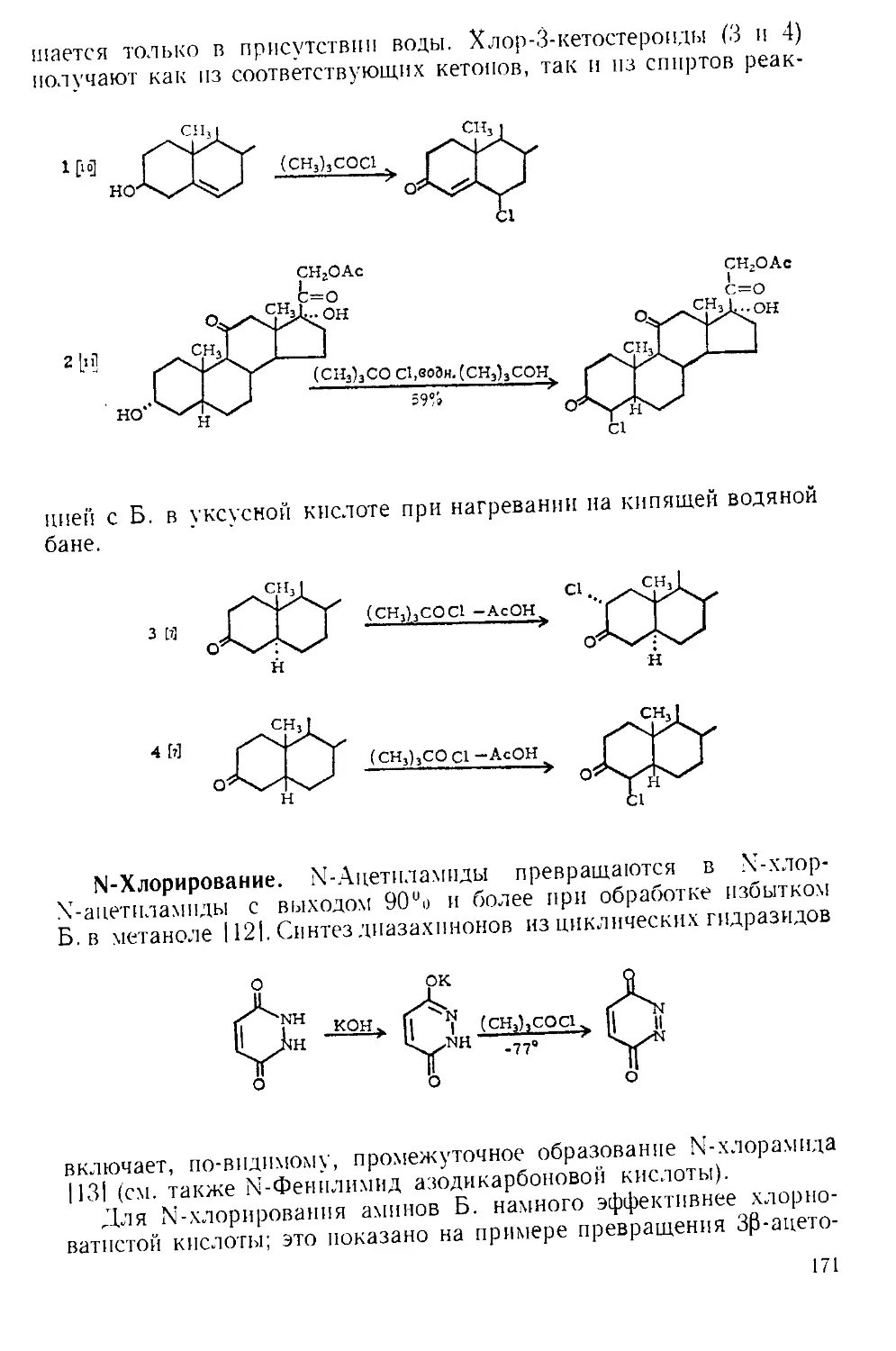
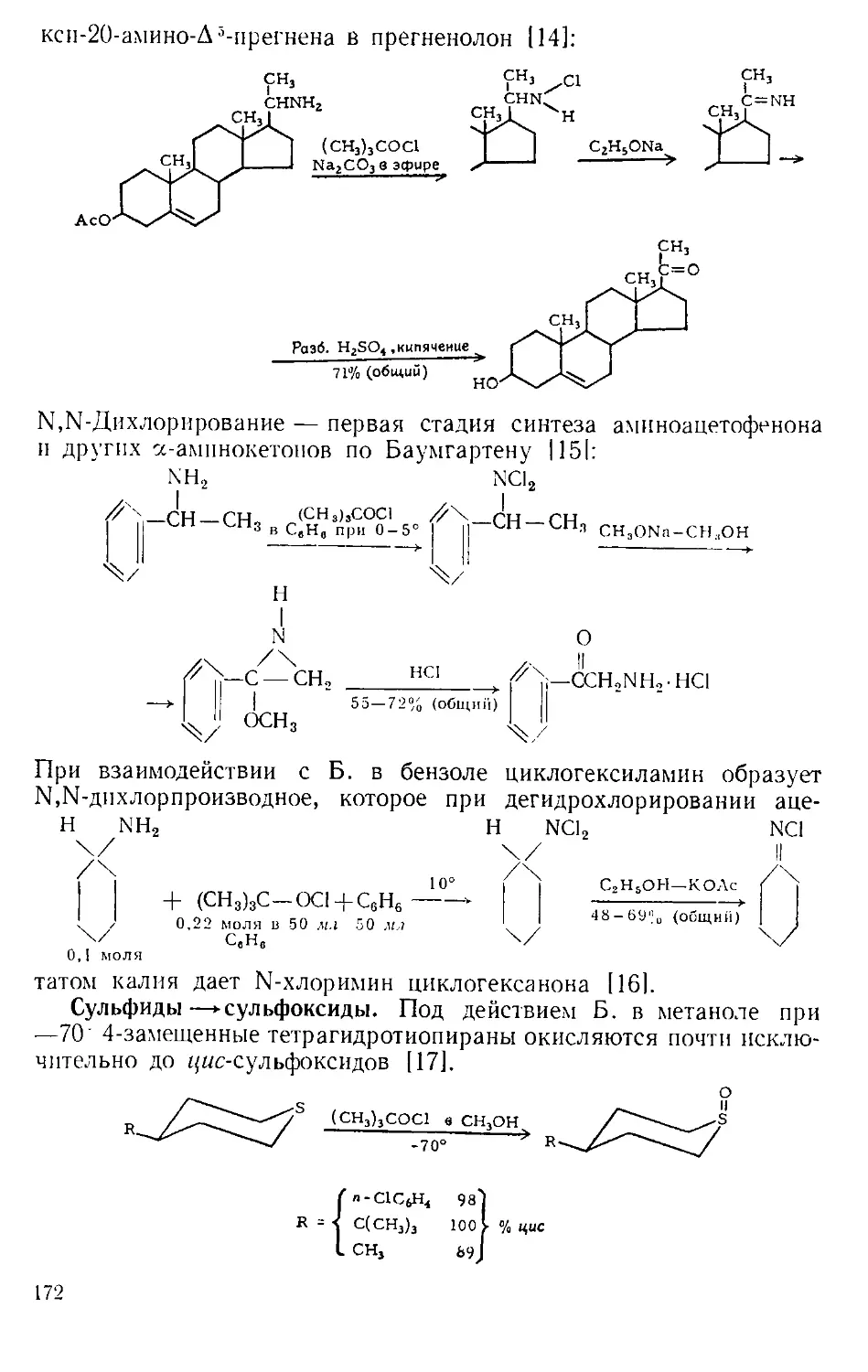
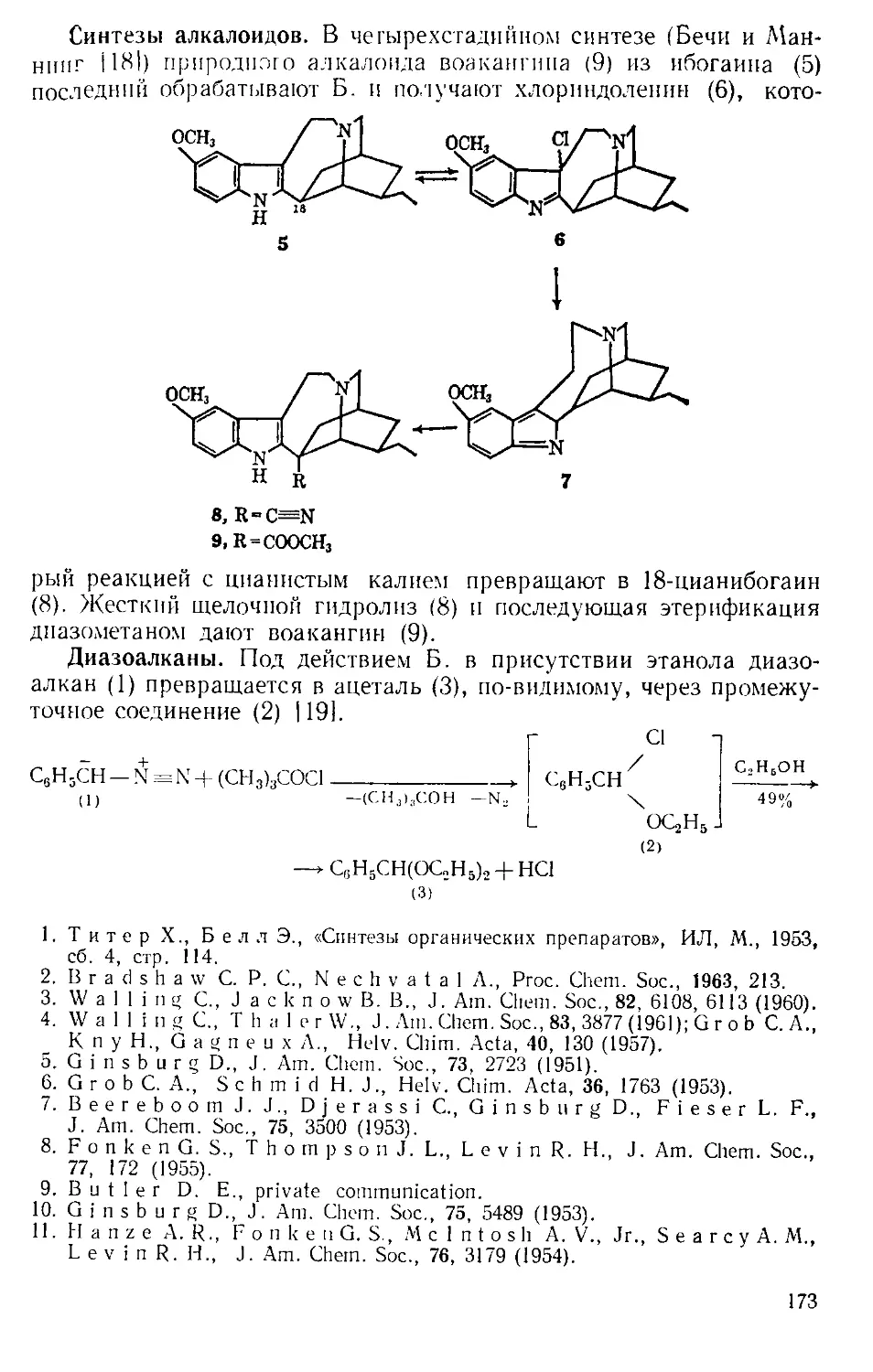
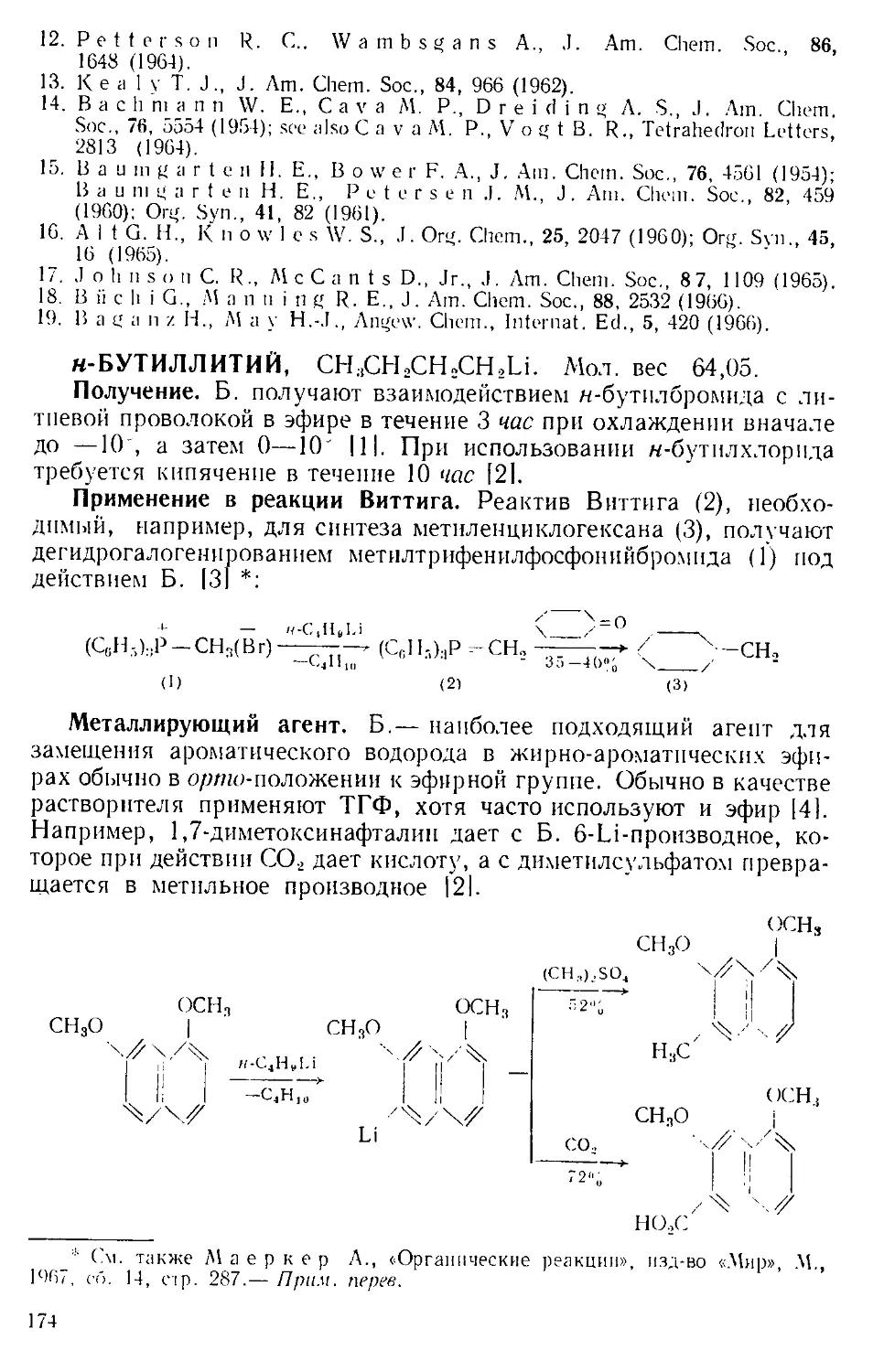
























































































































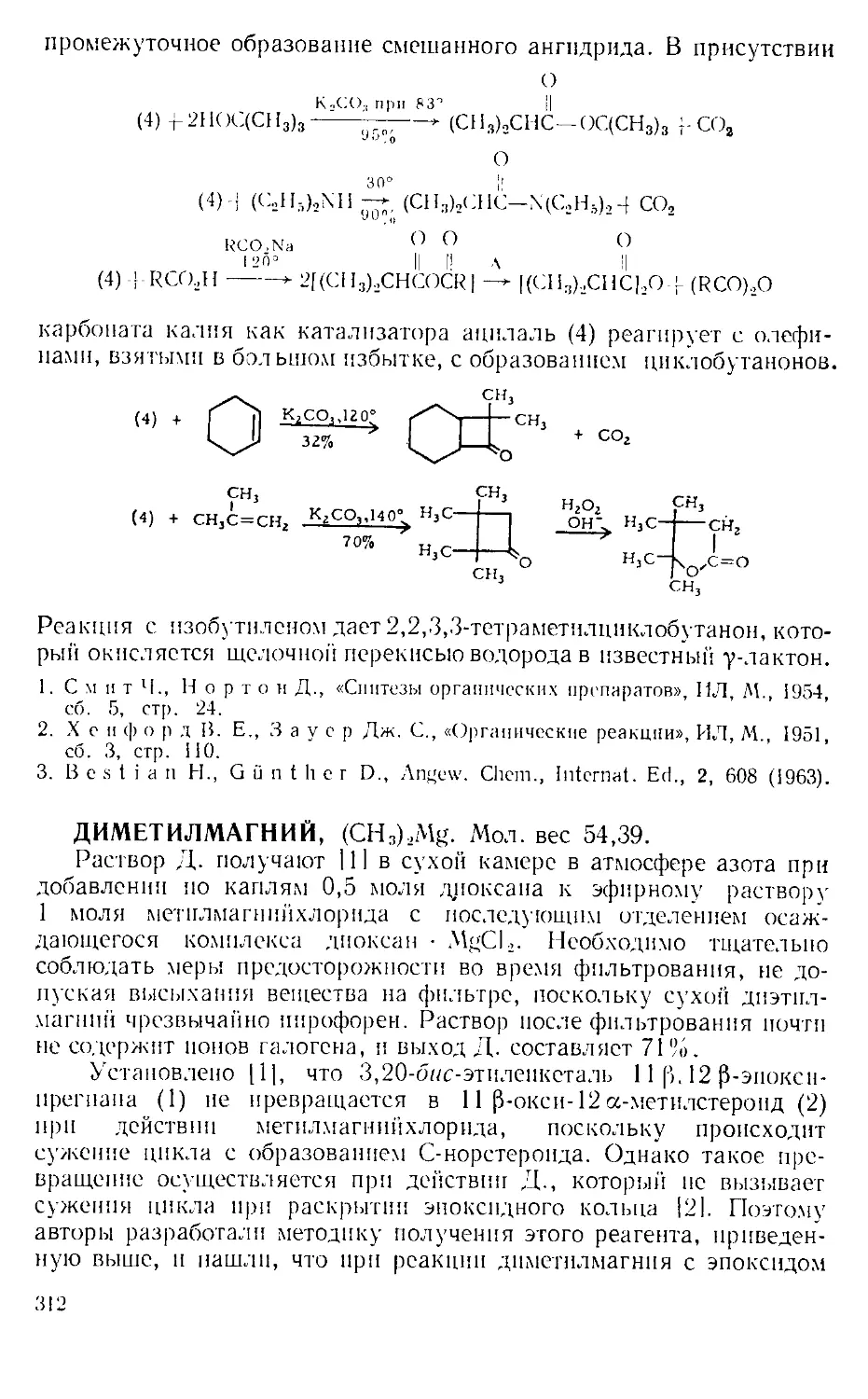


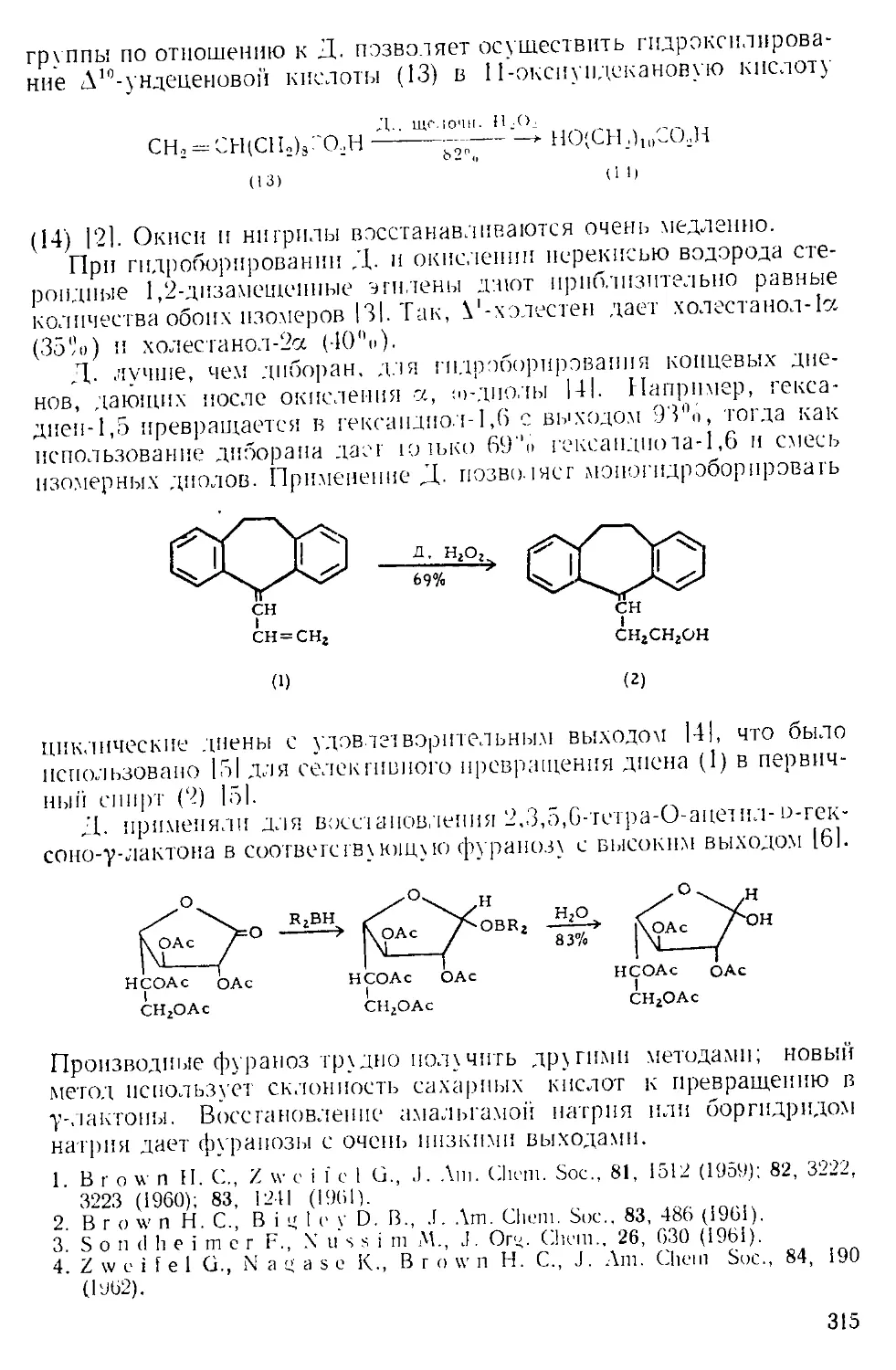


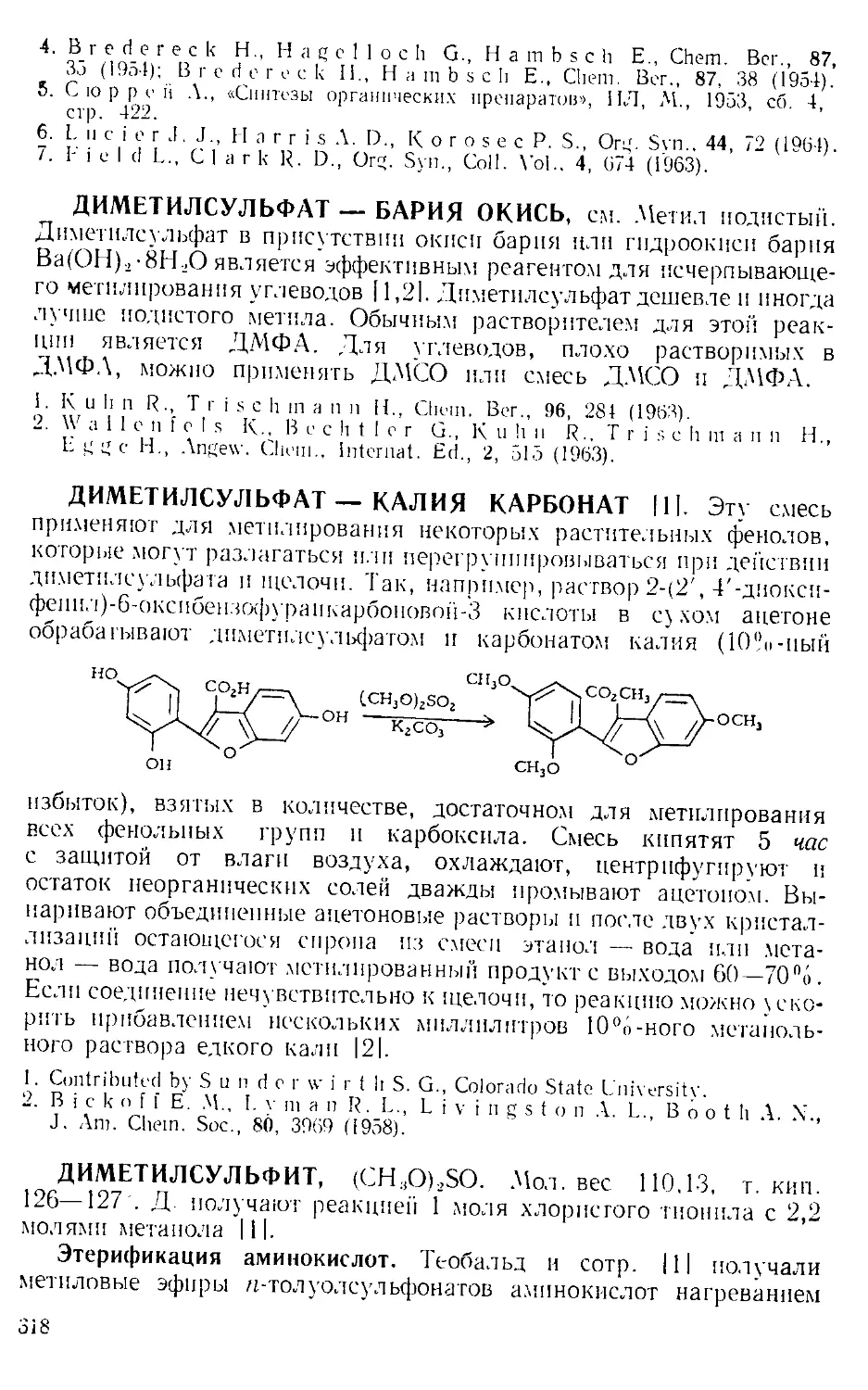


























































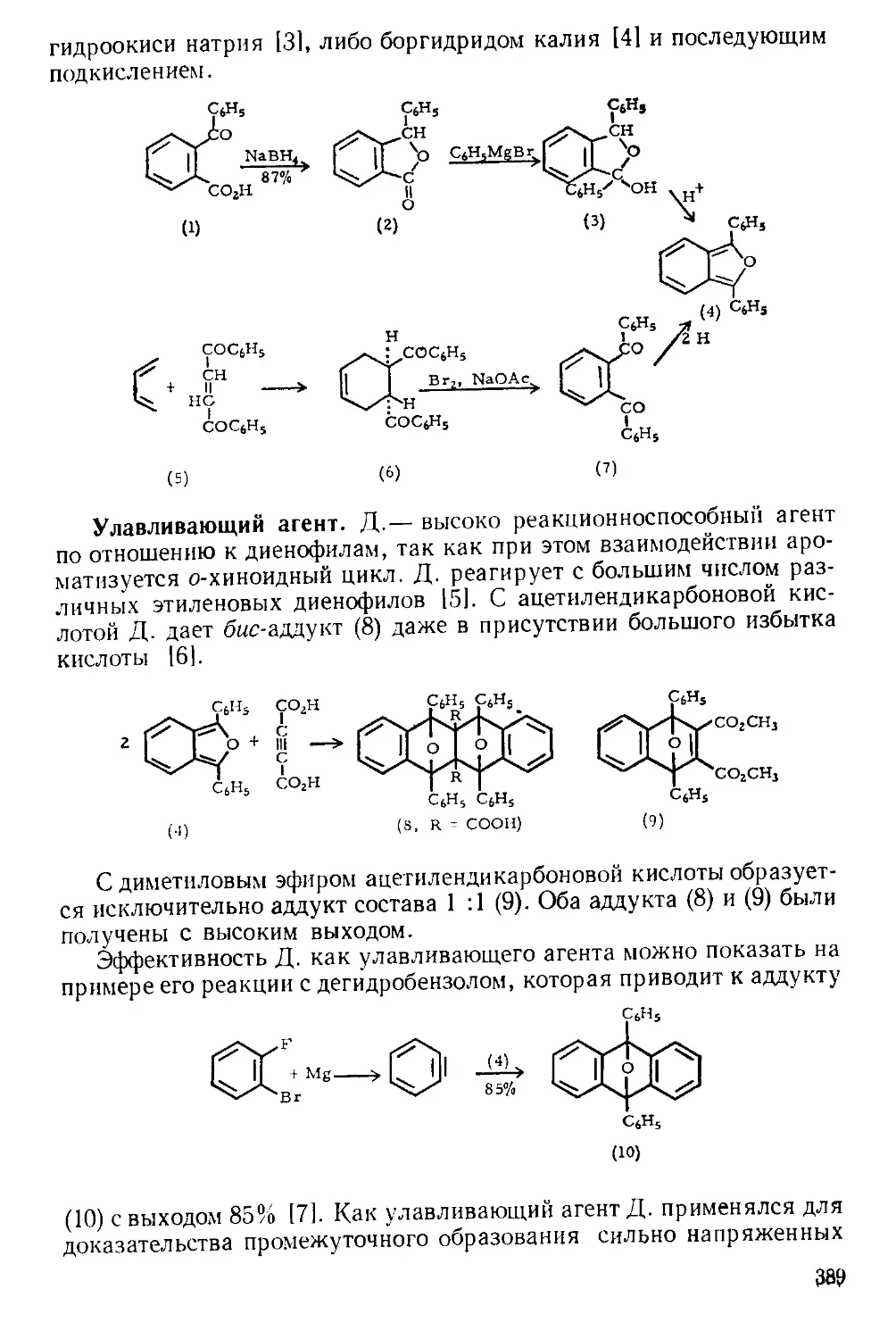






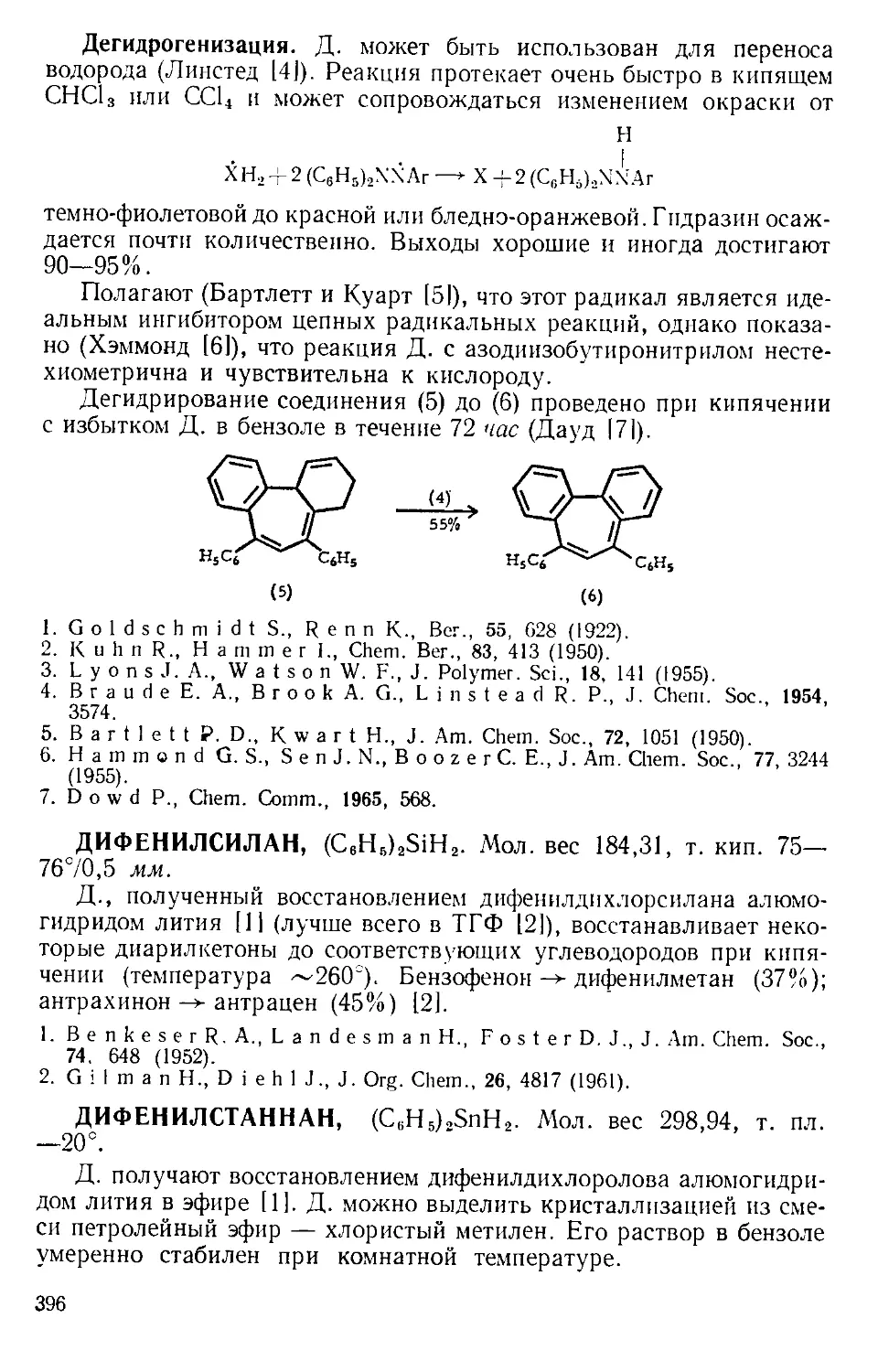
















































Текст
ИЗДАТЕЛЬСТВО
« М И Р »
Reagents for
Organic Synthesis
LOUIS F. F I ESE R
Sheldon Emery Professor of Organic Chemistry
Harvard University
MARY F I E S E R
Research Fellow in Chemistry
Harvard University
JOHN WILEY AND SONS, INC.
NEW YORK — LONDON — SYDNEY
I У t> s
Л. ФИЗ ЕР, М. ФИЗ ЕР
Реагенты
для органического
синтеза
TOMI
(А — Е)
ПЕРЕВОД С АНГЛИЙСКОГО
доктора хим. наук Н. С. ЗЕФИРОВА,
канд. хим. наук В. С. ПЕТРОСЯНА,
доктора хим. наук А. Ф. ПЛАТЭ
и канд. хим. наук С. С. ЧУРАНОВА
ПОД РЕДАКЦИЕЙ
академика -И. ’Л. КНУНЯНЦА
и докюра хнм. неук Р.’ Г/ КОСТЯНОВСКОГО
МОСКВА 1970
ИЗДАТЕЛЬСТВО «МИР»
УДК 661.7/54-41
Редакция литературы по химии
Инд. 2-5-3
70
ПРЕДИСЛОВИЕ
Книга Л. Фпзера и М. Физер, вышедшая в 1967 г., оказалась
удобным источником информации непосредственно в процессе вы-
полнения эксперимента, пособником быстродействия в научном ис-
следовании и потому превратилась в необходимое руководство хи-
мических лабораторий. Не отходя от лабораторного стола, здесь
можно найти наплучшие способы и средства осуществления как об-
щеизвестных, так и совершенно новых синтетических методов. Чи-
татель наверняка с удивлением узнает, что давно знакомый ему
реагент является далеко не самым лучшим для интересующего его
синтеза.
Кроме характеристики 1120 реагентов в книге почти всегда дают-
ся методы их получения и примеры использования с указанием за-
грузки, условий реакции, способа выделения и выхода продукта.
В большинстве случаев рассмотренные методики можно воспроиз-
вести непосредственно по приведенному краткому описанию или
схеме. Отсылка к проверенным источникам позволяет, если это не-
обходимо, быстро найти подробную методику.
Многие из методов и реагентов, рассмотренных авторами, мож-
но, конечно, найти в других пособиях по органической химии (на-
пример, 17% ссылок, как указывают авторы, приходится на сборни-
ки «Синтезы органических препаратов»). Но неизмеримое преиму-
щество этой книги в ее малом объеме, которого удалось достичь
благодаря тщательному отбору наиболее употребпмых, эффективных
и доступных реагентов; благодаря описанию наиболее принципи-
альных и рабочих характеристик; благодаря иллюстрациям наиболее
яркими и поучительными примерами с анализом старых недоразу-
мений н ошибок. •
Отбор реагентов и примеры их использования несут, естествен-
но, отпечаток индивидуальности авторов, интересы которых сосре-
доточены в области химии природных соединений, в частности сте-
роидов. Но это даже обогащает книгу, так как именно в этой, бурно
развивающейся области разработаны многие новые общие методы
синтеза.
Возможность быстро найти в этой книге и метод и реагент спо-
собствует немедленной реализации возникшей идеи и в высокой сте-
пени устраняет поводы для откладывания эксперимента. А должно
быть ясно каждому, что идея, не проверенная сразу, чаще всего ос-
тается не проверенной никогда.
5
Под впечатлением от этой книги при редактировании раздела
«Медный порошок» одним из нас был разработан эффективный
метод металлирования этиленимина. Этот реагент, перегоняющий-
ся без изменения над натрием, легко с ним реагирует в присут-
ствии каталитических количеств меди. Через образующийся эти-
ленамид натрия удается, например, получить такое интересное
соединение, как этиленимпноформ (i>N).,CH |Р. Г. К ост Янов-
ский, Ю. И. Эльнатанов, X. Хафизов, Изв. АН СССР
(сер. хим.), № 8, 1920 (1970)|.
Первая проба составления такого справочника сделана Физером
в книге, которая у нас была переведена и вошла в сборник «Сов-
ременные методы эксперимента в органической химии» (Госхимиздат,
1960 г., стр. 344—421, раздел «Реагенты»). Многие химики, по-ви-
днмому, давно уже убедились в его ценности.
Книга претерпела некоторую перестройку, связанную с перево-
дом названий (номенклатуры). Предметный указатель построен по
принципу, принятому в «Большой советской энциклопедии». Наз-
вания соединений, например всех производных кислот, следуют за
наименованием кислоты, т. е. основного вещества. При этом чита-
тель, конечно, найдет и вошедшие в обиход тривиальные названия
реагентов. Некоторые очень емкие английские термины оставлены
при переводе без изменения. Например, вместо очень длинного «уг-
леводороды, содержащие двойную связь в конце цепи», оставлено
«терминальные олефины». Или вместо «эффект содействия или уско-
рения реакции соседними функциональными группами» — «анхимер-
ный эффект» и т. д.
Перевод этой книги издается в четырех томах. В 1969 г.
Физеры выпустили дополнительный том (538 стр.), включающий
ряд новых методов и описание 616 реагентов для их выполнения.
Этот дополнительный материал выйдет в русском переводе пятым
томом настоящего издания в 1971 году.
И. Кнунинц
Р. Костяновский
ОСНОВНЫЕ СОКРАЩЕНИЯ
Ас — ацетил
АсОН — уксусная кислота
ВиОН —бутанол
Bz — бензоил
СЬ — карбобензокси
ЕЮН —этанол
МеОН — метанол
Ms —мезил, СН35О.,—
Ph —фенил
Phth —фталоил
РгОН —пропиловый спирт
Ру —пиридин
Ts —тозил, n-CH3CeH4SO.,—
TsCl —тозил хлор ид
TsOH — и-толуолсульфокислота
NBA — N-бромацетамид
NBC — N-бромсукцинимид
Глим — 1,2-диметоксиэтан
Диглим —диметиловый эфир диэтиленгликоля
ДМСО — ди.метилсульфоксид
ДМФА —диметплформа.мид
ДНФ —2,4-дпнитрофторбензол
ТГФ —тетрагидрофуран
Триглим —диметиловый эфир триэтиленгликоля
Тритил —(СВН5)3С—
ЭДТК — этилендиаминтетрауксусная кислота
ОСНОВНЫЕ СОКРАЩЕНИЯ
Ас — ацетил
АсОН — уксусная кислота
ВиОН — бутанол
Bz — бензоил
СЬ — карбобензокси
ЕЮН —этанол
МеОН — метанол
Ms —мезил, CH3S0.2—
Ph —фенил
Phth —фталоил
РгОН —пропиловый спирт
Ру —пиридин
Ts —тозил, n-CH3CeH4S0.2—
TsCl —тозилхлорид
TsOH —и-толуолсульфокислота
NBA —N-бромацетамид
NBC — N-бромсукцпнпмпд
Гли.м — 1,2-диметокспэтан
Диглим —диметиловый эфир диэтиленгликоля
ДМСО — диметилсульфоксид
ДМФА —диметилформамид
ДНФ — 2,4-дпнитрофторбензол
ТГФ —тетрагидрофуран
Триглим —диметиловый эфир триэтилеигликоля
Тритил — (CBH5)SC—
ЭДТК —этилендиаминтетра)ксусная кислота
A .
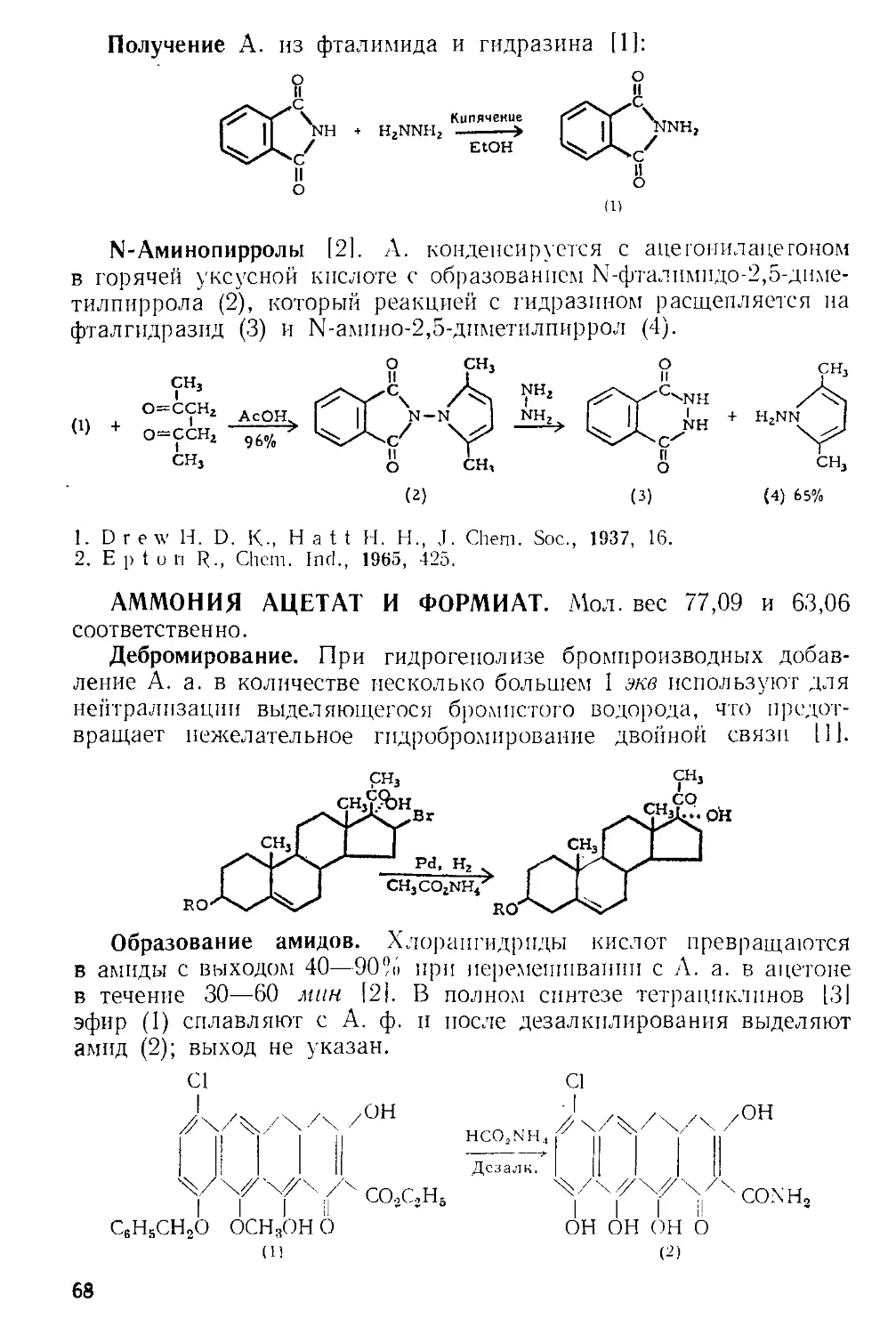
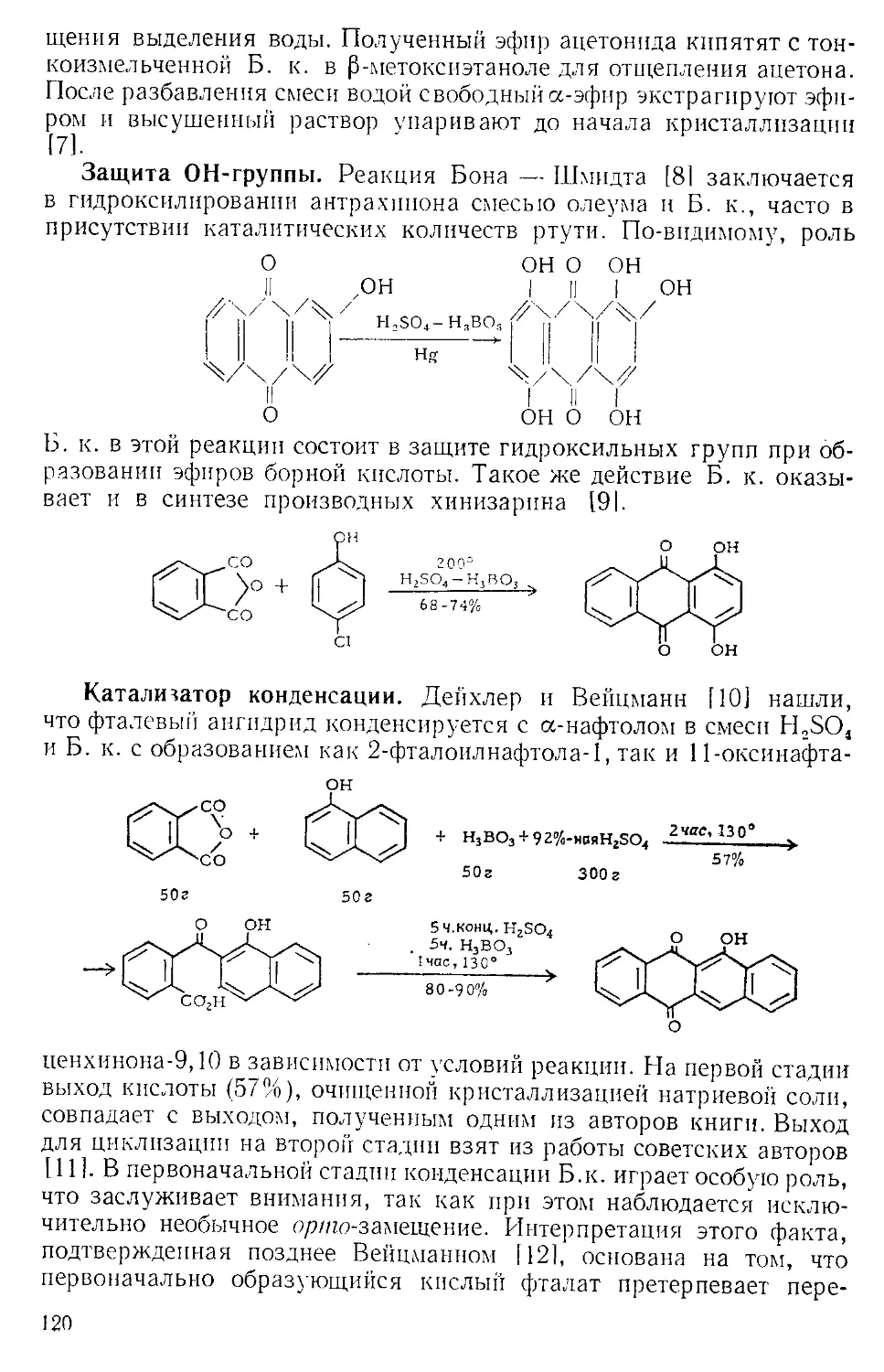
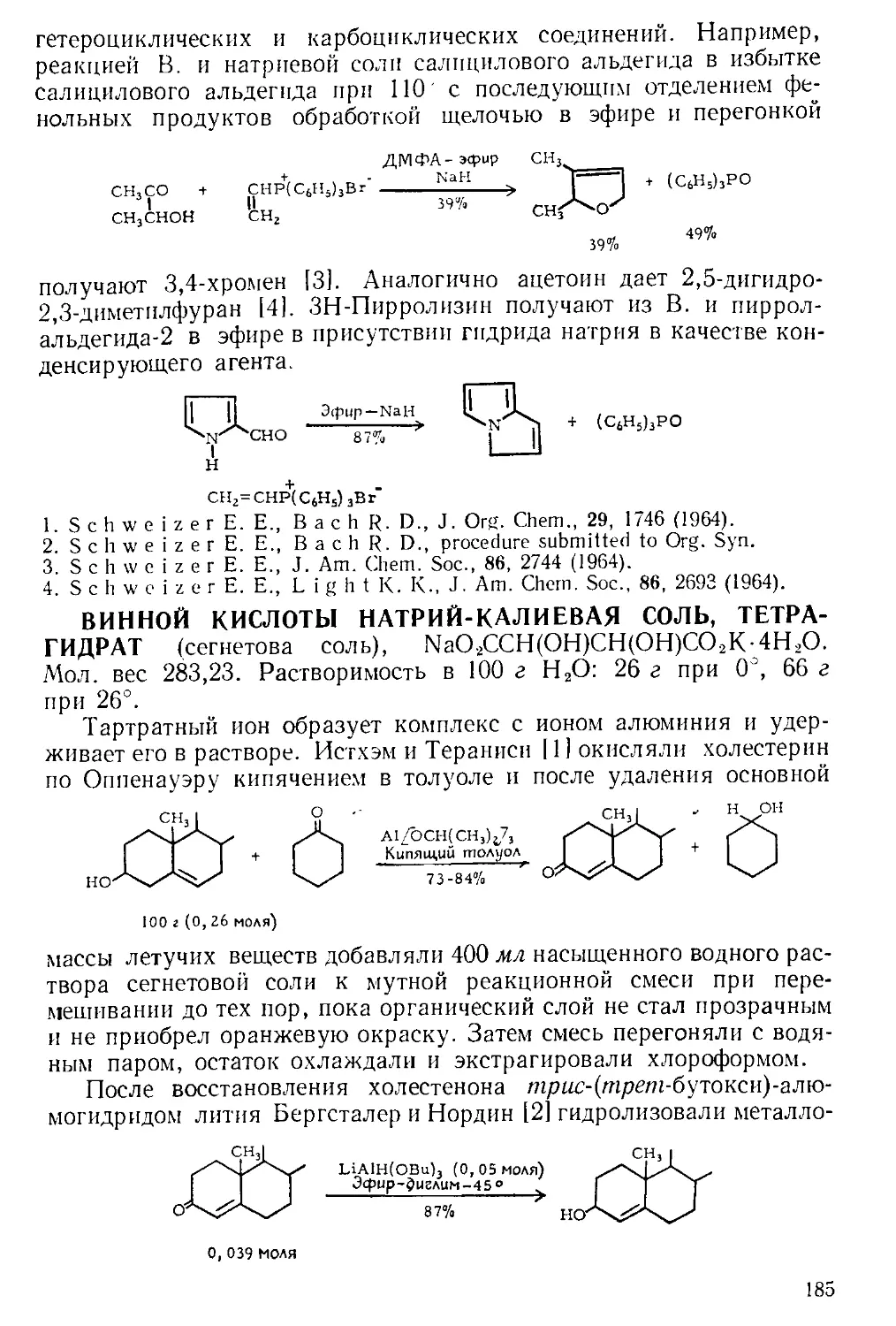
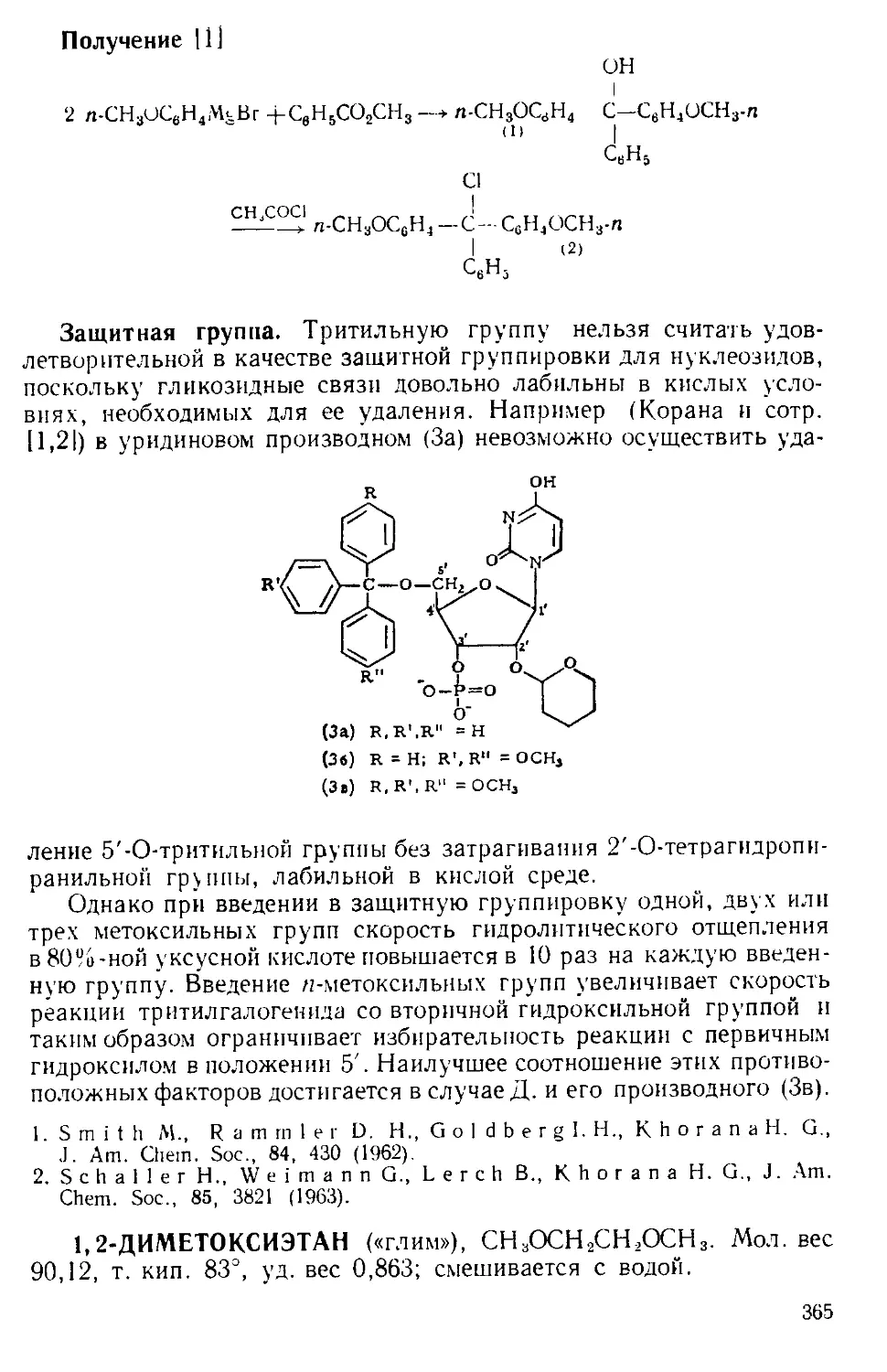
1-АДАМАНТИЛХЛ0РФ0РМИАТ (2). Мол. вес 214,69, i. ил. 46—47°.
А. получают из 1-оксиадамантана и фосгена в пиридине 111.
СЩ6-Ру
4«
до
+ СОС12 -----2---->
100%
ОСОС1
Пептидные синтезы. А. успешно использован для защиты амино-
группы в аминокислотах; объемная 1-адамантилоксикарбонильная
группа удаляется при сольволизе трнфторуксусной кислотой.
А. использовали также в синтезе пептидов методом смешанных ангид-
ридов; однако при этом наблюдалась значительная рацемизация.
1. Haas W. L., Krumkalns Е. V.,Gerzon К., J. Am. Chcni. Soc., 88,
1988 (1966).
АДАМСА КАТАЛИЗАТОР, см. Платиновые, катализаторы.
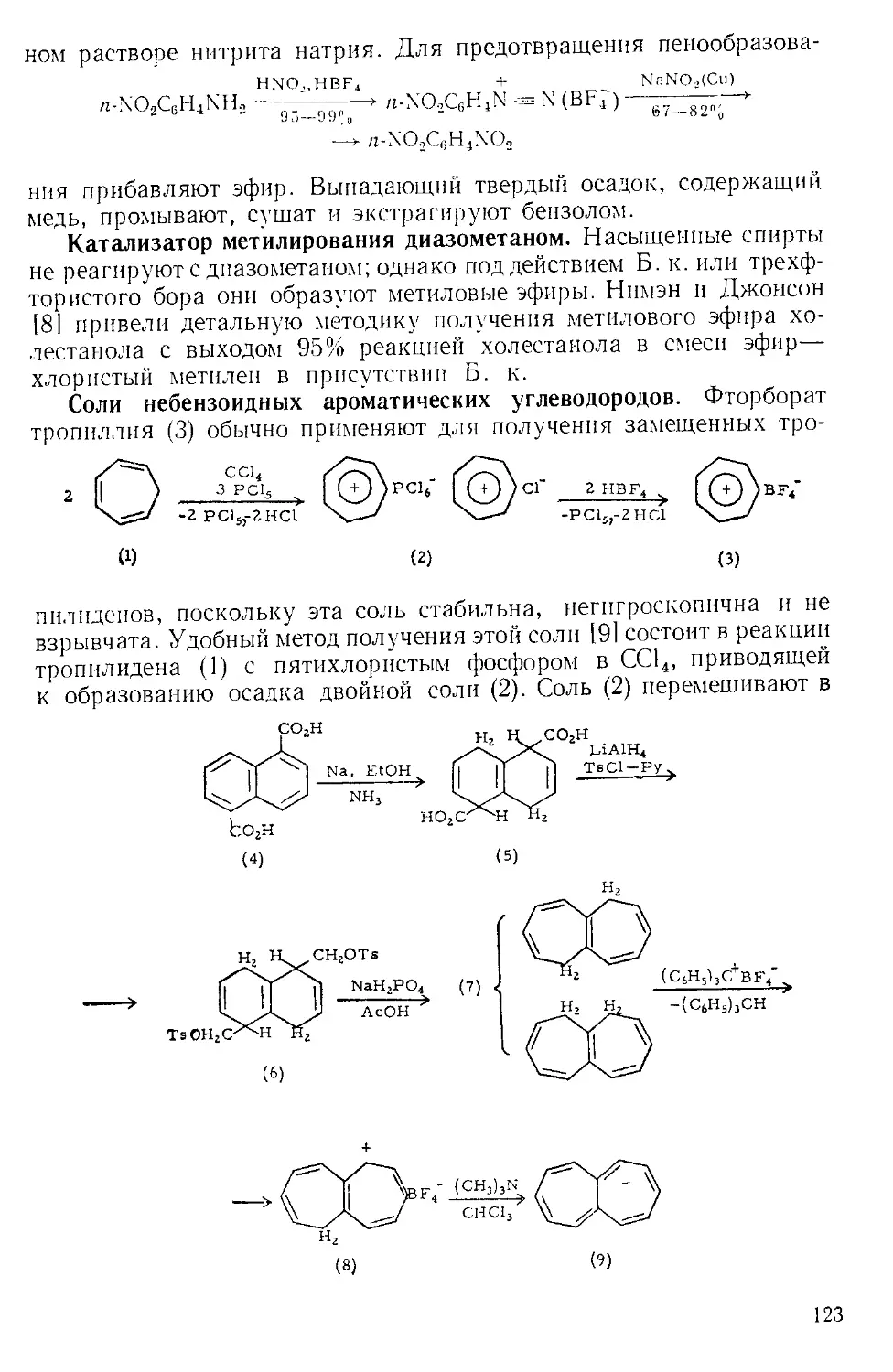
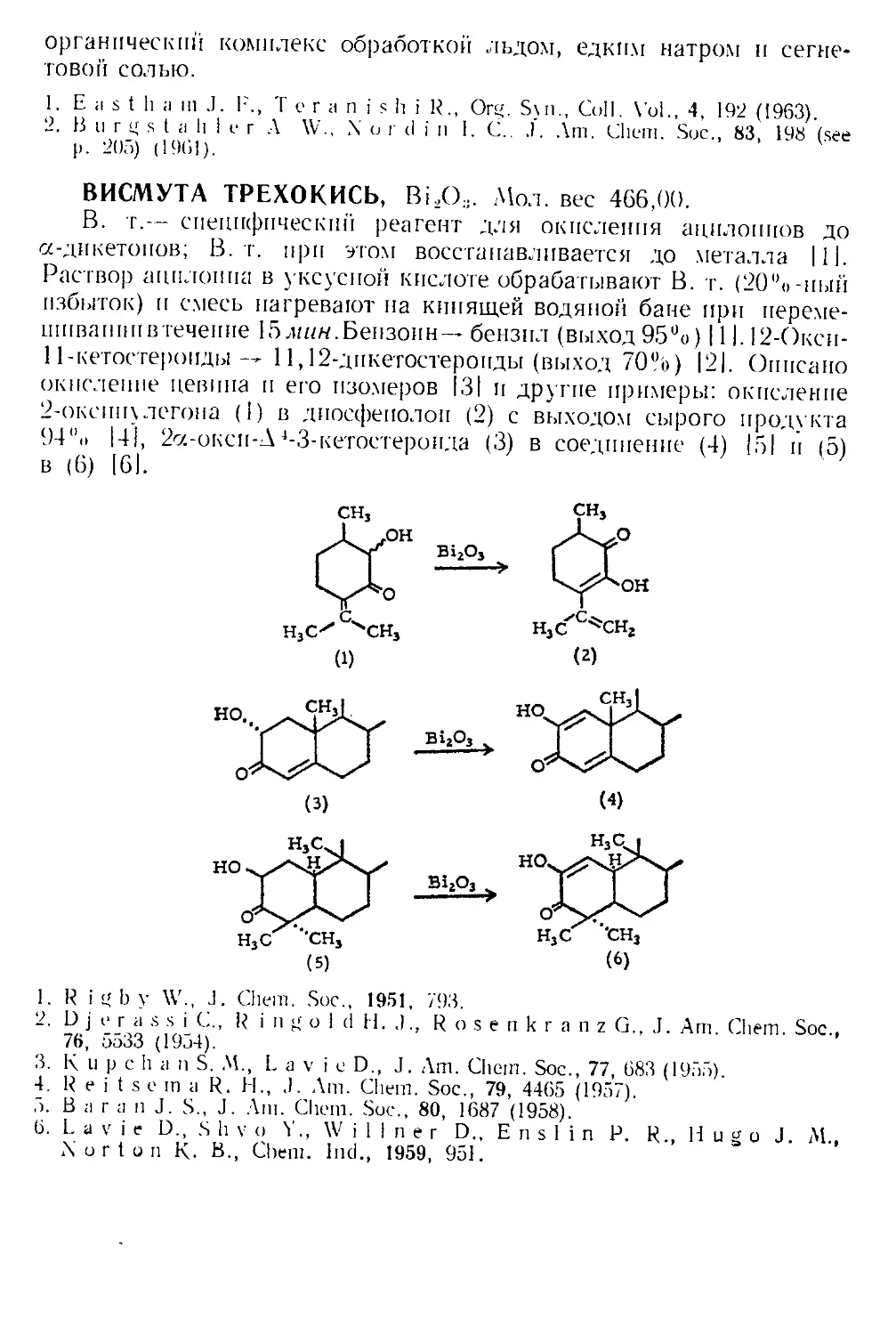
АДИПИНОВАЯ КИСЛОТА, НО,С(СН2)4СО2Н. Мол. вес 146,14,
т. пл. 151—153°, pKaj 4,43, рКа2 5,52; в 100 г воды при 15° раство-
ряется 1,4 г.
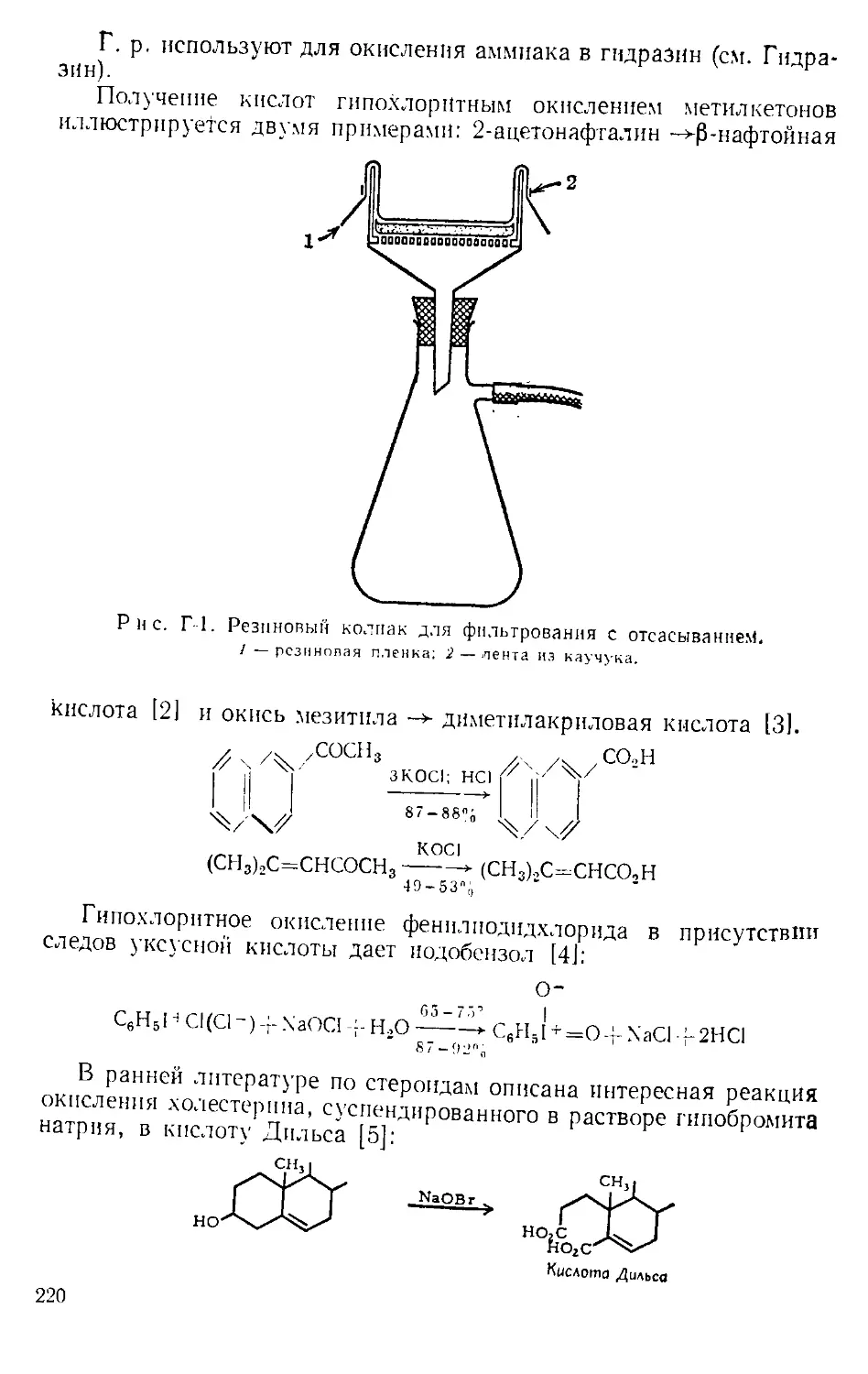
Катализатор для детализации. В обычном методе детализации
А 4-3-кетостероид (1) кипятят с этиленгликолем в бензоле в присут-
ствии каталитических количеств п-толуолсульфокислоты с водоот-
делителем до тех пор, пока не соберется теоретическое количество
воды. В этих условиях двойная связь обычно мигрирует в 5,6-поло-
жение (2). Бернштейн и сотр. 111 показали, что при замене /г-толуол-
сульфокпслоты более слабой адипиновой кислотой образование
А4-кеталя (3) происходит без миграции двойной связи. Катализ ща-
велевой кислотой, которая по своей силе является промежуточной
9
между адипиновой и /i-толуолсульфокпслотой, приводит к смеси
А4- и Д5-кеталей. В настоящее время известно, что кеталь (3) можно
получить также при использовании строго определенных количеств
п-толуолсульфокислоты (см. п-Толуолсульфокпслота).
1. Brown J. J., Lenhar d R. И., В e r n s t e i n S., J. Am. Chem. Soe.,
86, 2183 (1964).
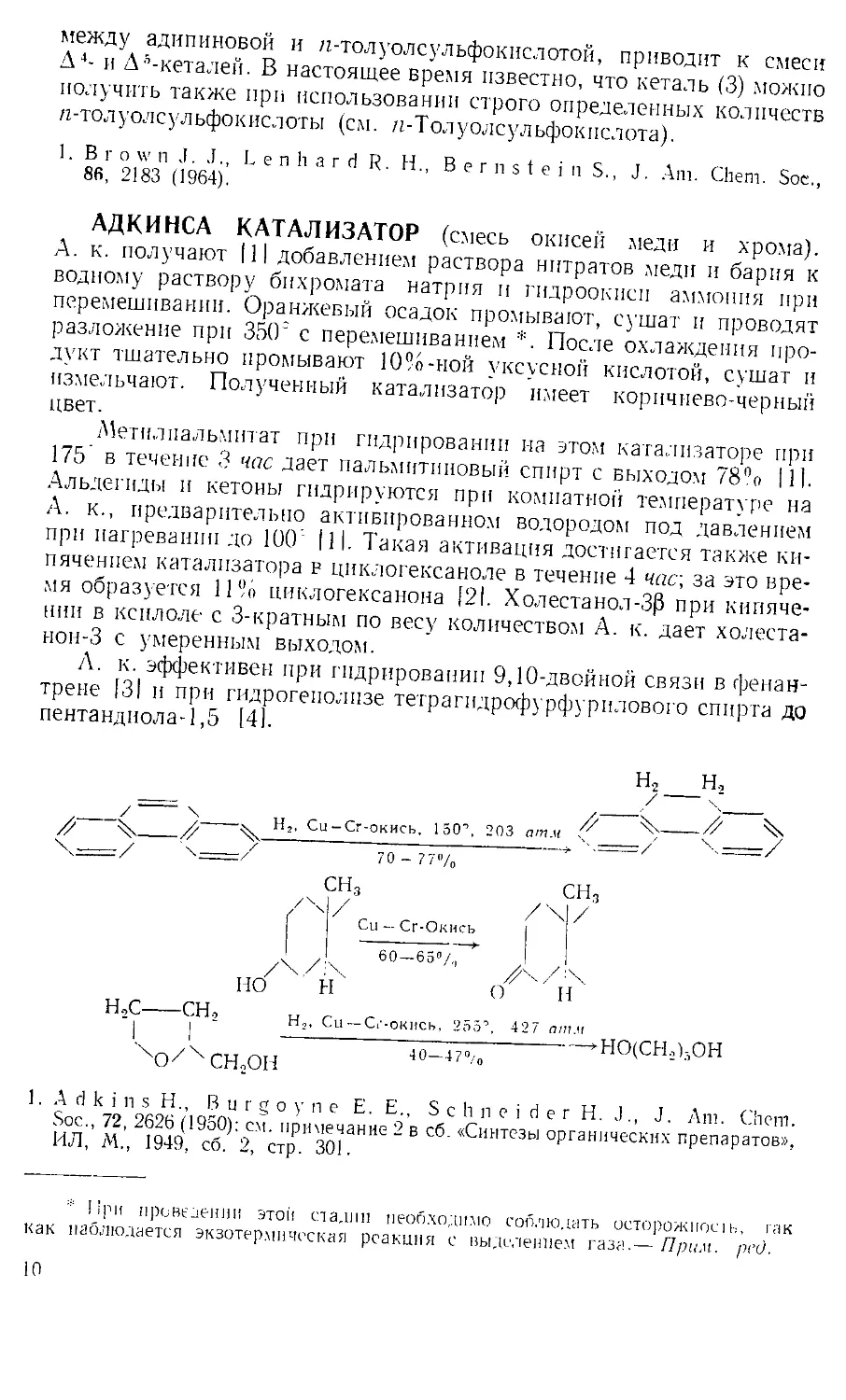
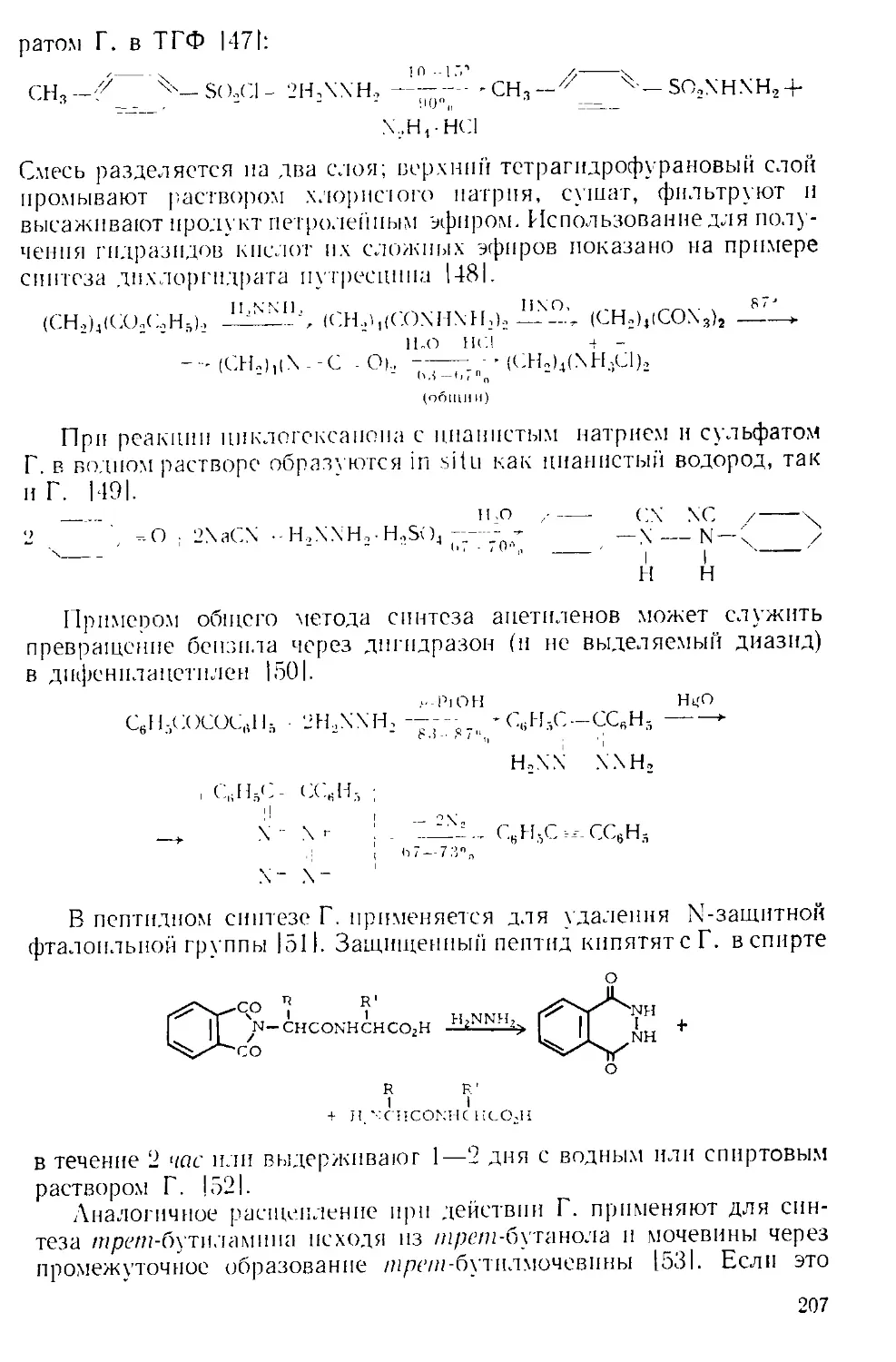
АДКИНСА КАТАЛИЗАТОР (смесь окисей меди и хрома).
А. к. получают 111 добавлением раствора нитратов меди и бария к
водному раствору бихромата натрия и гидроокиси аммония при
перемешивании. Оранжевый осадок промывают, сушат и проводят
разложение при 350' с перемешиванием *. После охлаждения про-
дукт тщательно промывают 10%-ной уксусной кислотой, сушат и
измельчают. Полученный катализатор имеет коричнево-черный
цвет.
Метилпальмитат при гидрировании на этом катализаторе при
175' в течение 3 час дает пальмитиновый спирт с выходом 78% 111.
Альдегиды и кетоны гидрируются при комнатной температуре на
А. к., предварительно активированном водородом под давлением
при нагревании до 100 |1|. Такая активация достигается также ки-
пячением катализатора в циклогексаноле в течение 4 час, за это вре-
мя образуется 11% циклогексанона [21. Холестанол-ЗР при кипяче-
нии в ксилоле с 3-кратным по весу количеством А. к. дает холеста-
нои-3 с умеренным выходом.
А. к. эффективен при гидрировании 9,10-двойной связи в фенан-
трене [31 и при гидрогенолизе тетрагидрофурфурилового спирта до
пентандиола-1,5 [4].
у уп-2 Н3, Си —Сг-окнсъ, 255э,
хо/ 4 сн2он 4°-'47”'"
427
-------->НО(СН.,)5ОН
1. A d k i n s Н., В и г д о у п е Е. Е., S с h n е i d е г Н. ,1., J. Am. Chern.
Soc., 72, 2626 (1950): см. примечание 2 в сб. «Синтезы органических препаратов»,
ИЛ, М., 1949, сб. 2, стр. 301.
* При проведении этой стадии необходимо соблюдать оеторожнооь, гак
как наблюдается экзотермическая реакция е выделением газа.— Прим. ра).
10
2. Nes W. R„ J. Org. Chem., 23, 899 (1958).
.3. P h i 1 1 i p s D. D., Org. Svn., Coll. Vol., 4, 313 (1963).
4. К a у (j) м а н Д., P и в У., «Синтезы органических препаратов», ИЛ, М.,
1953. сб. 4 стр. 414.
п-АЗОБЕНЗОЛ КАРБОНОВОЙ КИСЛОТЫ ГИДРАЗИД,
Мол. вес 240,26, т. пл. 209°.
А. к. г. получают из соответствующего этилового эфира и гид-
разина. Этот реагент с карбонильными соединениями образует
высокоплавкие гидразоны оранжевого цвета |1|. 3'- и 4'-нитропро-
изводные А. к. г. дают ярко окрашенные гидразоны с еще более вы-
соким» температурами плавления [21.
1. W е s t р 11 а 1 U., F е i е г Н., L й <1 е г i 1 z О., From m е 1., Biochem.
Z., 326, 139 (1954/55).
2. Z а у е И S. 41. A. D., F a k h г I. М. I., Ann.. 662, 165 (1963).
АЗОДИИЗОБУТИРОНИТРИЛ,
CN CN
1 I
(СН3)гС—N = N —С(СН3)2.
Мол. вес 136,20, т. пл. 102J.
А. очищают перекристаллизацией из метанола и высушивают в
вакууме над фосфорным ангидридом 111. А. разлагается в мягких
условиях (40') с образованием цианпропильных радикалов и иоэто.
му используется для инициирования радикальных реакций |21_
С.Х CN CN
I I
СН3-С—\ = Х—С-СН3 —« 2СН3—С-4-\„
! I I
сн;! СН;, сн.,
Штокманн предложил простой стереоспецифичный .метод син-
теза экзо-норбориилметилкетона путем радикального присоединения
ацетальдегида к норборнену в присутствии А. как инициатора.
При инициировании реакции перекисными радикалами выход резко
131.
CN CN Автоклав
сн3спо + (ch3)2c-n=n-c(ch3)2 40
8 0%
132 г О, 01 моля
<1 G. 8., М a h о n е v L. R., N and) U. S., J. Am. Chem. Soc.,
87, 740 (1963).
.2. У о 1 л и и г Ч., .X о й з е р Э., «Органические реакции», изд-во «Мир», М.,
1966, сб. 13, стр. 103.
3. Stockman n Н., J. Org. Chem., 29 , 245 (1964).
ухудшается
0,5 моля
1. Hammon
11
АЗОДИКАРБОНОВОЙ КИСЛОТЫ ДИАМИД,
О о
II I1
H2NC—N = N —CNH2.
Мол. вес 116,08.
Получение II) см. Диимид.
1. Т h i е 1 е J., Ann., 271, 129 (1892).
АЗОДИ КАРБОНОВОЙ КИСЛОТЫ ДИ-/п/^от-БУТ ИЛОВЫЙ
О о
II II
ЭФИР, (СНз)зСОС—N=N—СОС(СН3)з. Мол. вес 230,26, т. пл.
90—92 s.
Получение |1]. Раствор азида mpem-бутилового эфира азоди-
карбоиовой кислоты (1) и трет-бутилового эфира гидразппкар-
боновой кислоты (2) в пиридине выдерживают при комнатной
температуре в течение недели, затем разбавляют водой. Ди-шреш-
-бутиловый эфир гидразоднкарбоновой кислоты (3) выпадает
И + - Pv(—HN.)
(СН3)зСОС— N = N = N + H2NNHCO2C(CH3)3 —----1
<1) (2) 77—80%
О н н о
— (сн3)зСос-х-.\’-сос(снг)3
(3) 88-90%
О о
II II
—> (СН3)3СОС—N = N—СОС(СН3)з
(4)
в виде белоснежного мелкокристаллического порошка; его отде-
ляют и окисляют N-бромсукцинимпдом всмеси пиридин •—хлори-
стый метилен до А. к. д. э., который после кристаллизации из
лигроина получают в виде больших лпмониожелтых кристаллов.
Диенофил в реакции Дильса — Альдера [21. Это соединение
менее реакционноспособно, чем соответствующий диэтиловый эфир.
1. Car р in о L. A., Crowley Р. ,1., Org. Syn., 44, 18 (1964).
2. С а г р i п о L. A., Terr v Р. Н., Crowley Р. J., J. Org. Chem., 26,
4336 (1961).
АЗОДИ КАРБОНОВОЙ КИСЛОТЫ ДИКАЛИЕВАЯ СОЛЬ,
KO.,CN-NCO2K. Мол. вес 168,24.
Эту соль получают [1] гидролизом амида азодпкарбоновой кисло-
ты (1). При ее подкислении в присутствии азобензола образуется
лабильный промежуточный диимид (3), который гладко восстапав-
12
о о
|| || кон н +
HoNCX - Х’СХ'Н,------КО,СХ -= NCO.K — HN = NH
(1) (2) (3)
н и
лпвает азобензол в гидразобензол [21. С небольшим избытком реаген-
та олеиновая кислота восстанавливается в стеариновую (выход 51 %).
1. Thiele .1,, Ann., 271, 127 (1892).
2. Т a in е I е n Е. Е., van, D е w е у R. S., Т immons R. J., J. Chem.
Soc., 83, 3725 (1961).
АЗОДИ КАРБОНОВОЙ КИСЛОТЫ ДИЭТИЛОВЫЙ ЭФИР,
С2Н5О,С—N = N—СО,С2Н.-„ Мол. вес 174,16, т. кип. 93—9575 мм,
т. заст. 67
Получение. В двух описанных методиках синтеза [1, 21, почти
одинаковых на первой стадии (выход около 83°6), окисление прово-
дят хлорноватистой (выход 8296) [11 или дымящей азотной кислотой
а) НОС1
H.NNH, с) HNO,
С1СО,СоН, 7:1 С,Н5О2С— NH —NH—СО,С,Н5-------------->
N.!2CO,
—>• С2Н5О2С—N = N — СО„С.,Н -
(выход 75/6) [21. Второй метод безопаснее и дает более чистый и
стойкий продукт. Препарат, полученный при окислении гипохлори-
том, всегда содержит около 1% хлора и может взорваться при пере-
гонке (сообщение Б. Мак-Кузика).
Реакции Дильса — Альдера. С некоторыми сопряженными дие-
нами этот диенофил реагирует по схеме 1,4-присоединения (см. ни-
же, пример 1 131; см. также Антрацен-9,10-дшшин). Пример 2
иллюстрирует две возможности другого направления реакции: ос-
/ СО„С.2Н5
N
N
ЧСО.,С„Н5
(а)
новиои продукт образуется через циклическое переходное состояние
(нерадикально) (б) со сдвигом двойной связи, а побочный продукт —
в результате радикального аллильного присоединения (в) [3, 4].
Представляет интерес поведение циклических сопряженных диенов:
циклопептаднен [5| дает 1,4-аддукт (г), циклогептатрнен реагирует
по схеме аллильного присоединения без сдвига связей (в) [61 и 1,3-
циклогексадиен — по циклическому механизму (нерадикалыюму) со
сдвигом двойной связи (б) 13, 4]. Однако при облучении смеси
13
Главный продукт
Побочный продукт
А. к. д. э. и 1,3-циклогексадиена подучается обычный аддукт ре
акции Дильса — Альдера с выходом 87?6 17). При взаимодействи.
А. к. д. э. со стероидными Л г,-7-диенами наблюдается нуклеофильное
присоединение по С. с отщеплением водорода у С9 [81. С диметиловым
эфиром аиетилепдикарбоновой кислоты А. к. д. э. реагирует мед-
леннее, но дает также необычные продукты [9].
Синтезы а-кетоальдегидов. Необычная реакция А. к. д. э. с
диазокетонами открыла путь к синтезу а-кетоальдегидов, выде-
ляемых в виде их фенилозазонов [101.
о
я-о„хсвн/сн - \С.ЩО.СХ=ХСО..С.Н5 -—4-
(1) (2> 79%
о
хтг ы ген w/rn с н ' COHSXHN-H.„ АсОН
—► /г-О.,\тС6Н4ССН = .\.\(СО.,СлН5).,---— >.
(3) - - - 75%
М\’НС6Н,
II
—> П-ОЛУСРДССН = nnhc3h5
(4)
14
Реагенты (1) и (2) осторожно кипятят в толуоле в течение 15 мин,
растворитель удаляют в вакууме и продукт (3) перекристаллизо-
вывают из этанола (т. пл. 107 ). Гидролизом (3) в смеси уксусная
кислота — этанол в присутствии фенилгпдразина получают феиил-
озазоп (4).
Дегидрирование. Показано, что А. к. д. э. вызывает фотохими-
ческое дегидрирование изопропилового спирта до ппнакопа I 111
и циклогексанона до циклогексанона 1121. Этот же реагент осуще-
ствляет нефотохпмпческое окисление спиртов, меркаптанов, анили-
нов и гидразобензолов с образованием диэтилового эфира гндразодн-
карболовой кислоты 1131.
1. Р э б д ж о н Н., «Синтезы органических препаратов», ИЛ, М., 1953, со. 4,
стр, 575. Полученный этим методом препарат рекомендуется несколько раз
промыть холодным 3°6-ным раствором соды и перегонять небольшими пор-
циями непосредственно перед употреблением |R о d g m а п A., J. Отд. Chem.,
17, 1666 (1952)].
2. К а и е г J. С.. Org, Svn., Coll. Vo!., 4 , 411 (1963).'
3. G i 1 1 i s В. T„ BecliP. E., J. Ore. Chem., 27, 1947 (1962); 28, 3177 (1963).
4. Franzes B., Surr i dge J. H., .1. Org. Chem., 27, 1951 (1962); F r a n-
z. u s B.. ibid., 28, 2954 (1963); Thaler W. A., Franzus B., ibid. 29,
2226 (1964).
5. DielsO., Blom J. H„ Koll W., Ann., 443, 242 (1925): G a s s-
m a n P. G.. Mansfield К. T., procedure submitted to Org. Syn.
6. C i ii H a m on J. M., W eissK., J. Org. Chem., 26, 2644 (1961).
7. A s k a n i R., Chem. Ber., 98, 2551 (1965).
8. v a n d e r G e n A., LakemanJ., Gras Al. A. M. P., H u i s m a n H. O.,
Tetrahedron, 20, 2521 (1964).
9. van der Gen A., LakemanJ., Pandit H. K., Huisma n H O.,
Tetrahedron, 21, 3641 (1965).
10. 1-' a Ii r E., S c h e c k e n b a c 11 F., Ann., 655, 86 (1962).
11. Sell \v e ii c k G. O., For in aneck H., Angew. Chem., 70, 505 (1958).
12. Co о k s о n R. C., S t e \ e n s I. D. R., WattC. T., Chem. Comm., 1965,
259.
13. Voncda F., S u z u k i K., 1\ i t t a Y., J. Ain. Chem. Soc., 88, 2328
(1966).
АЗОТ 111. Cледы кислорода в продажном азоте можно удалить
пропусканием его через раствор Фпзера, однако при этом газ ув-
лажняется и становится непригодным для использования в качестве
инертной среды, например в реакциях Гриньяра и Впттига. Эффек-
тивная очистка азота от следов кислорода и влаги достигается про-
пусканием его через смесь, полученную добавлением 0,5 г LiA!H4
к раствору 10 г беизпипаколина в 50 мл пиридина. Кетон быстро рас-
щепляется [21 с образованием кроваво-красного раствора трпфеппл-
метилыгого аниона, который наряду с гидридом металла реагирует
со следами кислорода и влаги. В качестве источника карбаниона
О
(С6Н,5)лССС6Н5 —-f---> (С.Н-,);;С- Li + 4- НОСН.,С6Н5
См. «Синтезы супmni'iecKrix препаратов», ИЛ, AL, 1960, со. 10, стр. 83.—
Прим, персе.
можно использовать также трифеиилметаи |3|. Эффективен только
красный раствор. Следы пиридина, увлекаемые током азота, можно
удалить пропусканием газа через промывную склянку с коиц. сер-
ной кислотой. Однако пиридин часто используется как раствори-
тель для реакций карбанионов |4l, и в этом случае его присутствие
в азоте, естественно, не мешает.
1. Contributed byLansbur у I3. Т., State University of New York at Buffalo.
2. Lansbury P. T., J. Am. Chem. Soc., 83, 429 (1961).
3. Lansbury P. T., T h e d f о r d R., J. Ory. Chem., 27. 2383 (1962).
4. A v г а ш о f f M., S [) r i n z a k Y., J. Am. Chem. Soc., 82, 4953 (I960).
АЗОТА ОКИСЛЫ (в основном N.,O4 и немного N.,O;i и NO2).
А. о. получают (Докс 111) из мышьяковистого ангидрида и конц.
азотной кислоты в довольно сложном генераторе; сухой газ погло-
щают диэтилмалонатом (400 г) при охлаждении льдом до привеса
около 200 г. Темно-зеленую жидкость выдерживают несколько ча-
сов на льду п затем в течение двух дней при комнатной температуре
(красные пары). Затем газ п воду удаляют в вакууме водоструйного
насоса и диэтиловый эфир мезоксалевой кислоты перегоняют при
50 мм и затем (ярко-желтая жидкость) при 103—108715 мм.
К ..О,
СН2(СО,С,Н-,)., ' О-.-...-. С(СО,С.,Н5),
А. о. получают также (Брук 121) осторожным нагреванием сме-
си 83 мл дымящей (98%-ной) азотной кислоты, 33 мл конц. серной
кислоты и 100 г мышьяковистого ангидрида. Красные пары жидко-
сти, отгоняющейся в интервале 20—303, конденсируют при охлаж-
дении смесью льда с солью. Л. о. хранят в склянке со стеклянной
пробкой при 0° и отбирают пипеткой. А. о. применяют для окисле-
ния гидрохинонов в хиноны.Например, суспензию 20 а топкоизмель-
ченного 2,3-дихлор-5,6-дициангидрохииона в 300 мл СС14 сильно
ОН О
Cl. J ZCN Cl JI ZCN
III > II I
Clz |/XCN С1/ у III * * * * * * XCN
ОН о
перемешивают при комнатной температуре и в течение 5 мин об-
рабатывают 6 мл реагента. Перемешивание продолжают еще 5 мин,
хинон отфильтровывают п кристаллизуют' из смеси хлороформ —
бензол. С таким же успехом этот метод используют для окисления
2,3-дициаи-, 5-хлор-2,3-дициап-, тетрахлор- в 2,5-диметилгпдрохи-
ноиов; тетрабром- п тетрахлоркатехинов и 3,3',5,5'-тетрабром-4,4'-
диокепдифенила. Для окисления незамещенного гидрохинона реа-
гент не пригоден.
16
1. Dox A. \V., Ora. Syn., Coll. Vol., 1, 266 (1941).
2. Brook A. G., J. Chcni. Soc., 1952, 5040.
АЗОТА ПЯТИОКИСЬ (азотный ангидрид), N2O5. Мол. вес 108,02,
т. пл. 30', т. кии. 47'.
А. л.— летучее нестабильное белое твердое вещество. Его полу-
чают дегидратацией безводной азотной кислоты пятиокисыо фосфора
с возгонкой в токе кислорода, содержащего некоторое количество
озона, и хранят при —78“ 11]. А. п. получают также из азотной ки-
слоты и ангидрида трифторуксусной кислоты [21.
А. и. реагирует с пропиленом в хлористом метилене ври —25“
с образованием нитрата 1-нитропропаиола-2 и смеси нитроолефинов,
ОХО,
сн,сн = сн., -—2м сн3снсн.,\'о., o,nch.,ch =- сн„
27% ’ ' “ 16»%
в которой преобладает 3-нитропропен. С трпнс-стпльбеном А. п.дает
с высоким выходом смесь нитронитратов, однако с цис-стильбеном
их выход очень мал 131. В обоих случаях преобладает продукт цис-
присоединения, образующийся, по-видимому, через циклическое
переходное состояние |1].
Вторичные алифатические амины под действием А. и. превра-
щаются в нитрамины с очень хорошим выходом |4]; реагент в СС14
прибавляют к раствору избытка амина в СС14 при —25“.
I(CH3)2CH1.,NH-VN2O-,-->.[(CH3),CH].,NNO., + [(CH3).,CH],NH,NO3
91»/о
При нитровании дифенилкарбоновой-2 кислоты кони, азотной
кислотой при комнатной температуре получается смесь 2'-нитроди-
феннлкарбоиовой-2 кислоты (2) и 4'-витродпфенилкарбоновоп-2
кислоты (3) в соотношении 79 : 21. Нитрование А. п. в СС1} при (Г
(30 мин) дает с хорошим выходом единственный продукт — кислоту
(2) 151.
Цезар и Гольдфранк |6| описали установку для получения
А. в. (40—60 г в день), необходимого для синтеза эфиров азотной
кислоты по схеме
ROH - Х2О5 — ROXO, НОХО,
Раствор А. п. в хлороформе применяют для исчерпывающего
нитрования крахмала. Эта реакция ускоряется в присутствии фто-
рида натрия, который осаждает образующуюся азотную кислоту
в виде комплекса NaF-HONO.,.
Уолфром и Розенталь 171 применили этот метод для получения
пентанитратов d-глюкон- и о-галактонампдов.
1. StevcnsT. Е., Е ш raons ДА'. D., J. Am. Chem. Soc., 79, 6008 (1957).
2, Robson J. H., J. Am. Chem. Soc., 77, 107 (1955).
3. Stevens T. E., J. Org. Chem., 24, 1136 (1959).
4. E m mens W. D., Pagano A. S., Stevens T. E., J. Org. Chem., 23,
311 (1958).
5. Hey О. H„ LeonardJ.A., R e e s C. W., J. Chem. Soc., 1962, 4579.
6. CaesarG. V., G о 1 d f r a n k M., J. Am. Chem. Soc., 68, 372 (1946): G r u-
enhut S., Gol (Hran k Al., C u s h i n g Al. L., Caesar G. V.,
Inorg. Syn., 3, 78 (1950).
7. AV olfrom M. L., R osenthal A. J. Am. Chem. Soc., 75, 3662 (1953)
АЗОТА ЧЕТЫРЕХОКИСЬ, N2O4. ЛАол. вес 92,02, т. пл. —9,3’,
т. кип. 21,3е.
А. ч. очищают окислением при 0’ током кислорода до изменения
синего цвета на красно-коричневый; после перегонки над Р.,0.-, силь-
но охлажденная А. ч. затвердевает в почти бесцветные кристаллы 11 ].
Нитрование олефинов [2|. Реакцию А.ч. с олефинами обычно
проводят в присутствии кислорода (чтобы предотвратить образование
N.,O3) с применением в качестве растворителей простых или слож-
ных эфиров и получают виц-динптроалканы, нитронитриты или ни-
тронитраты 131. Холестерилацетат нитруют смесью газообразной
6, 07, 6, 4О.«х
NH3 9 5%
NO2
59 мк
А. ч. (из баллона при комнатной температуре) и кислорода в эфире
при 0: до появления окраски выходящего газа (около 1 час) 14].
Основной продукт реакции — 6₽-нптро-5а-нитрат — выделяют кри-
сталлизацией и превращают в 6-нптрохолестерилацетат реакцией
с аммиаком в эфире. В ИК- спектрах этих соединений имеются
18
полосы поглощения, характерные для трех типов групп: нитрат
6,07 мк; нитроалкан 6,40 мк\ нигроолефин 6,59 мк.
При нитровании олефинов по Сейферту |51, например цмс-цик-
лооктена, обрабатывают А. ч. как обычно в присутствии кислорода
в эфире. Затем без разделения смесь аддуктов обрабатывают 3 мо-
N2O< в эфире
Аддукт
3 (c2H,)3N?
9
лями триэтиламииа в течение 30 мин и с высоким выходом получают
сырой 1-нитроциклооктен-1. 1-Нитрооктадецен-1 из октадецена-1
этим методом получен с выходом 80%. В силу высокой основности
триэтиламин имеет явное преимущество перед обычно используемым
в этих реакциях аммиаком.
Нитрование ацетиленов. В этой реакции А.ч. приводит обычно
к образованию сложной смеси нестабильных соединений. Так, в про-
дуктах нитрования гексина-3 Фримэн и Эммонс |6| идентифициро-
вали пять веществ. Соединения с концевой этинильной группой дают
смесь особенно неустойчивых продуктов. Реакцией дифенилацетиле-
n..o4 С»Н-,—С—NO., С,Н5—С—NO.,
C2H5CsCC2H5— - II ' II ’ +
с2н,—с—no2 o.,n—с—с2н5
4,5% 31%
О NO., О О
II I II Д
4-С,Н5С-С-С2Н5 + С,Н5С-СС„Н5 + С.,Н5СО2Н
I
NO., 16% 6%
8°/0
на с А. ч. Кэмпбелл и сотр. [71 получили три соединения, относи-
тельное содержание которых варьирует в зависимости от условий
реакции. Обычно основными продуктами являются цис- и транс-
1,2-динитростпльбены; в небольших количествах образуется также
сильно окрашенный 5-нптро-2-фенилизатоген:
С6Н5 С6Н5
Х’О2 с6н5
с==с
I I
с6н5 no2
Реакция Понзио. По реакции Понзио бензальдоксим под дейст-
вием А. ч. в эфире превращается в фемилдинптрометан 181. В улуч-
С6Н-,СН = NOH C6H,CH(NO,),
3° /о
шенной методике [9] при осторожном добавлении эфирного раствора
19
оксима к кипящему раствору А. я. в эфире удается избежать выде-
ления дыма.
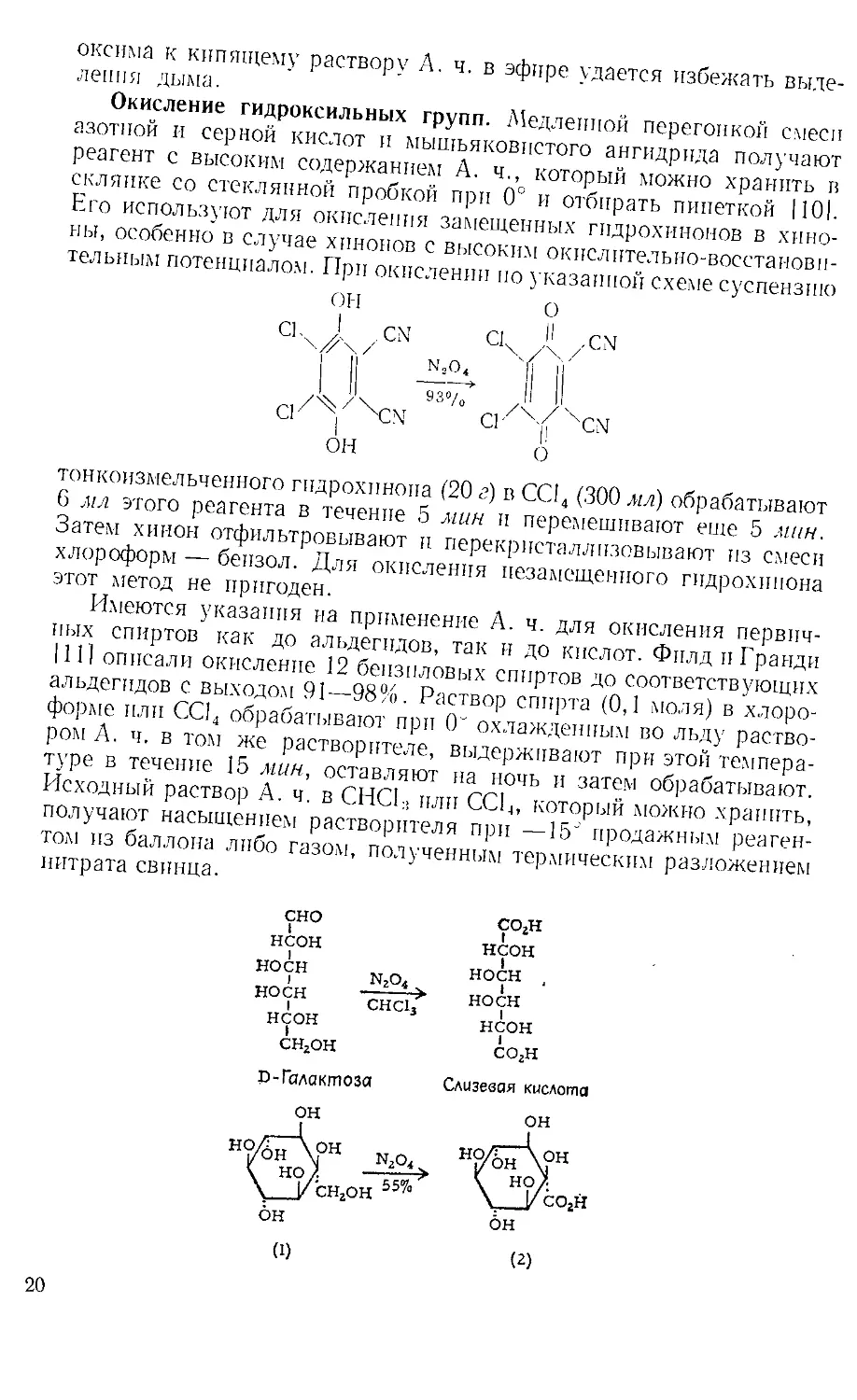
Окисление гидроксильных групп. ^Медленной перегонкой смеси
азотной и серной кислот и мышьяковистого ангидрида получают
реагент с высоким содержанием А. ч., который можно хранить в
склянке со стеклянной пробкой при 0° и отбирать пипеткой 1101.
Его используют для окисления замещенных гидрохинонов в хино-
ны, особенно в случае хинонов с высоким окислительно-восстанови-
тельным потенциалом. При окислении ио указанной схеме суспензию
он о
С1х/[ . CN С1х /CN
Г Г ~ 1 и'
A A 93»/0 II Д
СЕ у С'\ СР А/ су
ОН о
тонкоизмельченного гидрохинона (20 г) в CCI4 (300 мл) обрабатывают
6 мл этого реагента в течение 5 мин и перемешивают еще 5 мин.
Затем хинон отфильтровывают и перекристаллизовывают из смеси
хлороформ — бензол. Для окисления незамещенного гидрохинона
этот метод не пригоден.
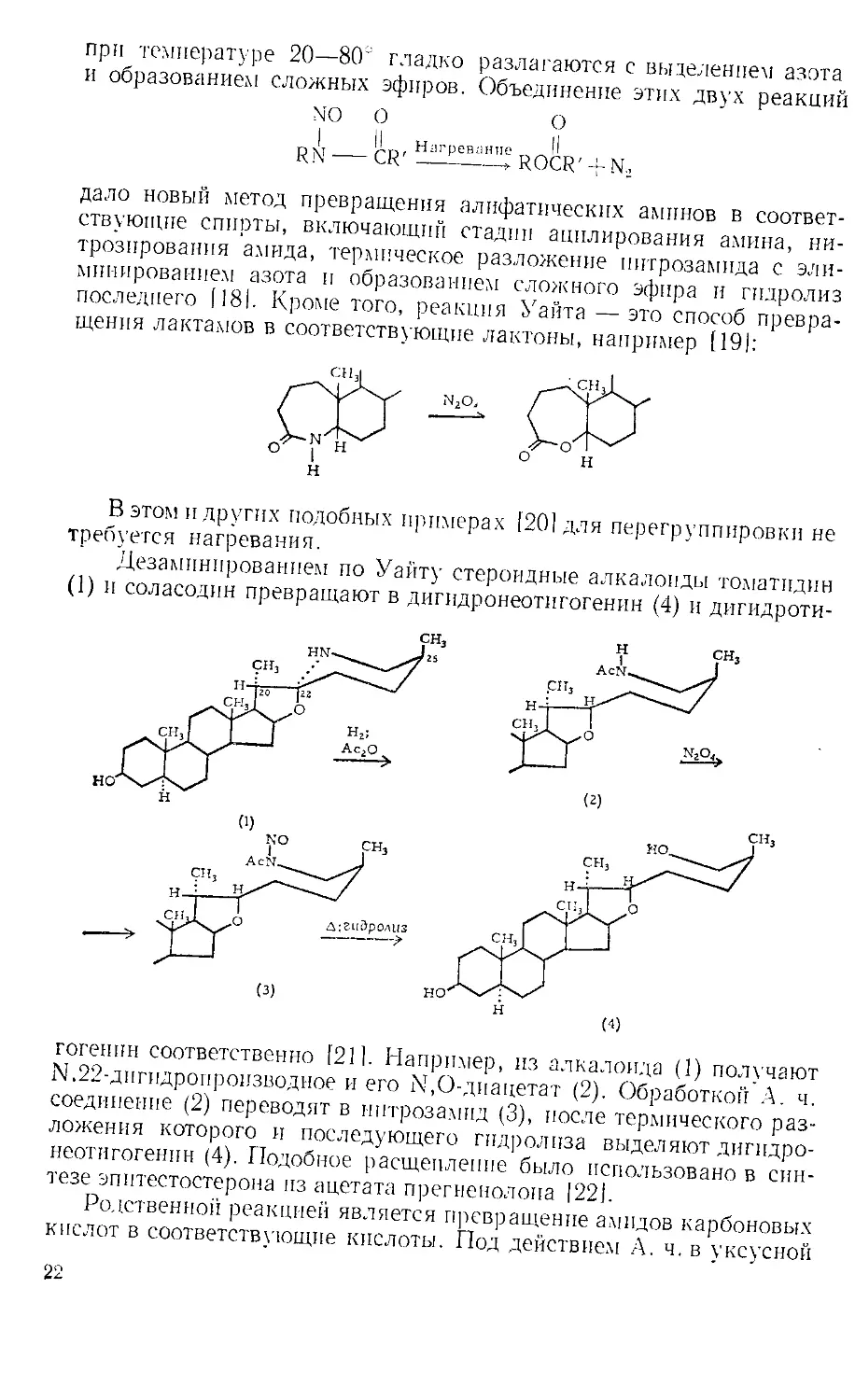
Имеются указания на применение А. ч. для окисления первич-
ных спиртов как до альдегидов, так и до кислот. Филд и Гранди
1111 описали окисление 12 бензиловых спиртов до соответствующих
альдегидов с выходом 91—98%. Раствор спирта (0,1 моля) в хлоро-
форме или СС14 обрабатывают при 0“ охлажденным во льду раство-
ром А. ч. в том же растворителе, выдерживают при этой темпера-
туре в течение 15 мин, оставляют на ночь и затем обрабатывают.
Исходный раствор А. ч. в СНСГ. пли СС14, который можно хранить,
получают насыщением растворителя при —15J продажным реаген-
том из баллона либо газом, полученным термическим разложением
нитрата свинца.
сно со,н
I I
неон неон
носн .. „ носн ,
I МгО4 !
НОСН "ТЩДА носн
I Cllclj ]
неон неон
I I
снгон СО2Н
B-Галактоза Слизевая кислота
он
он
(I)
он
он
(2)
20
Маурер и Дрефаль [121 описали применение А. ч. для окисления
D-галактозы в слизевую кислоту, а Постерпак л Реймонд 1131 —
для окисления двух производных инозита до соответствующих пно-
зпткарбоновых кислот. Например, для окисления ио приведенной
схеме к 468 мг тонкоизмельченного инозита (1) при охлаждении
льдом добавляли 10 ,ю А. ч. и смесь выдерживали 6 дней при 0J.
Лаигенбек и Рихтер 1141 изучили окисление ряда первичных
спиртов и диолов и обнаружили, что, за исключением производных
бензилового спирта, всегда получаются моно- или дпкарбоповые
кислоты. Кинетическое исследование показало, что альдегиды пе
являются промежуточными соединениями в этой реакции.
Скрайбнер [151 нашел, что фторированные первичные спирты
тина (1) не изменяются иод действием А. ч., однако их можно окис-
лить с хорошим выходом двуокисью азота (NO,), которую получают
пропусканием окиси азота и воздуха через трубку из стекла пирекс,
заполненную битым кварцем, при температуре 400 . При этом из
II(CF,)„C11,OI1 -°; Н(СГ,)„('.П(С)Н),
(I) zi--=6, 8, 10 (2)
011
\о. I
HCF,CF,CHCF.,CF,H______________> 1-ICF,CF„CCF,CF,H
| Превращение 57° 0 |
ОН он
(3) (4)
первичных спиртов (1) образуются гидраты альдегидов (2), а из
вторичных (3)- -гидраты фторкетонов (4); однако соединение (4)
легче получит!, окислением хромовой кислотой.
Окисление амидов. При исследовании реакции А. ч. с амидами
Уайт 11(51 готовил растворы реагента в СС.1., пли уксусной кислоте и
определял его концентрацию йодометрическим титрованием (в ат-
мосфере N,). Эта реакция является общим методом получения N-ал-
кил-1\1-питрозампдов из моноалкплампдов, наиршмер превращение
(D—(2):
И О NO О
CH3CH(CH.i)CH.,N—CQH, —iAlPAl'l; OH,CH(CII,)CH,N — ССсН-,
(1) S7" „ (О
Для связывания образующейся азотной кислоты реакцию проводят
в присутствии избытка ацетата натрия, иначе в результате обратной
реакции денптрозпроваппя равновесный выход соединения (2) сос-
тавляет только 60%. Уайт [17] отметил, что Ы-алкил-Ы-нитрозамиды
Н О NO О
II! I Г
R N — CR' Щ N ,О_| дщ R N-CR' -J- Н NO3
HNOa-J- NaOAc —»• NaNO-Щ- ИОАс
21
при температуре 20—80 гладко разлагаются с выделением азота
и образованием сложных эфиров. Объединение этих двух реакций
NO О О
I Н Н лгоевлнпр Н
RN----CR' --ре-Г^ ROCR' + N.,
дало новый метод превращения алифатических аминов в соответ-
ствующие спирты, включающий стадии ацилирования амина, ни-
трознроваиия амида, термическое разложение нитрозамида с эли-
минированием азота п образованием сложного эфира и гидролиз
последнего [181. Кроме того, реакция Уайта — это способ превра-
щения лактамов в соответствующие лактоны, например [19):
В этом и других подобных примерах [20! для перегруппировки не
требуется нагревания.
Дезаминированием по Уайту стероидные алкалоиды томатпдин
(1) и соласодин превращают в дигидронеотигогенин (4) и дигидроти-
гогеннн соответственно [211. Например, из алкалоида (1) получают
М.22-дигидропроизводное и его 1\,О-диацетат (2). Обработкой А. ч.
соединение (2) переводят в нитрозамид (3), после термического раз-
ложения которого и последующего гидролиза выделяют дигпдро-
неотигогенпн (4). Подобное расщепление было использовано в син-
тезе эпитестостерона из ацетата прегненолона [221.
Родственной реакцией является превращение амидов карбоновых
кислот в соответствующие кислоты. Под действием А. ч. в уксусной
кислоте при 0е из амида ацетилальдоновой кислоты получают с хо-
рошим выходом ацетилальдоновую кислоту 1231. Еще один пример
приведен на следующей схеме 1241:
СОХ'Н, СО,Н
I I
СН, СН,
CHjOCH СНчОСН
I 2_'Л 1
СИ, сн,
I I
сн, сн.
I !
CONH, СО,Н
Окисление фосфор- и серусодержащих соединений. Фосфиты
окисляются /X. ч. в хлористом метилене при тем пературе сухого льда
1251. Л. ч. окисляет триалкил- и триарилфосфины до соответствую-
щих фосфиноксидов, а диалкилсульфиды — до сульфоксидов (даль-
(RO)3P ——(ro);,P+-0-
75-85»,„
нейшего окисления до сульфона не происходит).
Синтез солей диазония. Скрайбнер [261 показал, что при добав-
лении А. ч. к эфирному раствору бензилиденанилина осаждается
CoH5N = СНС6Н5 + N,O4 C6H5N N(NO3 ) + О = СНС6Н5
У и : о
безводный чрезвычайно взрывчатый нитрат фенилдиазония.
Изомеризация циклических сульфоксидов. А. ч. ускоряет изо-
меризацию 4-замещенных окисей тетрагпдротиоппрана (Джонсон
и А1ак-Кантс [27|). Обработка как цис- (1), так и транс-нзожра (2)
этим реагентом приводит к равновесной смеси с указанным на схеме
соотношением изомеров:
о
и
(ch3)3c-J> (сн3)3с^А''
W (2) 19%
Отсюда следует, что для шестичленных сульфоксидов более стабиль-
ным является изомер с аксиальным положением сульфоксидной груп-
пировки, чем с экваториальным.
Реакция с гексахлорциклопентадиеном. Нагреванием этого диена
с А. ч. в автоклаве при 60 в течение 4 час « чают тетрахлоршиоюпентендпон-1,2 |28| — с высоким выходом полу- - желтое кристаллическое
23
соединение, т. пл. 44—45,5°:
С1 С1
1. Review: S h е с h t е г 11., Rcc. Chem. Piogr., 25, 55 (1964).
2. CoxJ.R.. Jr., W e stiic i m c r F. 14., J, Am. Chem. Soc., 80, 5141 (1958).
3. L e v у S c a i f e C. W. et al., J. Cliein. Soc., 1946, 1093, 1096, 1100; 1948
52; 1949, 2627.
4. Л nagnostopoiilos С. E., F ieser L. F., J. Am. Chem. Soc,, 76
532 (1954).
5. Seifert \V. K„ J. Org. Chem., 28, 125 (1963).
6. Free m a n J . P., E m m о n s W. D., J. Am. Chem. Soc., 79, 1712 (1957).
7. Campbell K. I\:., Shavel J., Jr., C a m p b e 1 1 13. 1\., J. Am. Chem.
Soc., 75, 2400 (1953).
8. Ponzi о G., J. prakt. Chem.. 73, 494 (1906).
9. Fieser L. F., I) о e r i n g W. E., von, J. Am. Chem. Soc., 68, 2252 (1946).
10. 13 г о о к A. G., J. Chem. Soc., 1952, 5040.
11. F i e 1 d В. O., Gr un <1 v J ., J . Chem. Soe., 1955, 1110.
12. M a u r e r K„ Dr el a h’l G„ 13er., 75, 1489 (1942).
13. Post e r n a к Th., R e у m о n <1 D., Helv. Chitn. Acta, 36, 1370 (1953).
14. L a n g e n b e с к \V., Richter M., Chem. 13er., 89, 202 (1956).
15. Scribner R. M., J. Org. Chem., 29, 279 (1964).
16. Whit e E. 11., J. Am. Chem. Soc., 77, 6008 (1955).
17. Wh i t e E. H., J. Am. Chem. Soc., 77, 6011, 6014 (1955).
18. F u j i i T., Tash iro M.. Ohara К., К u m a i M., Chem. Pharm.
Bull., 8, 266 (1960).
19. S ten n s <> n R„ J. Org. Cliein.. 28, 188 (1963).
20, В 1 a <1 <> n P„ Л1 с M e e к i n W„ J. Chem. Soc., 1961, 3504.
21. Sato Y., Lath a m II. G., Jr., J. Org. Chem., 22, 981 (1957).
22. A 1 v a r e z F., Steroids, 2, 393 (1963).
23. II u r d C. D., SowdcnJ. C., J. Am. Chem. Soc., 60, 235 (1938).
24. 13 r e n neis e n К., T a m m Ch., R cichstein T., Helv. Cliim. Acta 39,
1233 (1956).
25. A d d i s о n С. C., S h с 1 d о n J . C., J. Chem. Soc., 1955, 2/05.
26. Scribner R. M., J. Org. Chem., 29, 3429 (1964).
27. J о h n s о n C. R., M с C a n t s D., Jr., J. Am. Chem. Soc., 86, 2935 (1964);
87, 1109 (1965).
28. Scr i b n e r R. ЛЕ, J. Org. Chem., 30, 3657 (1965).
АЗОТА ЧЕТЫРЕХОКИСЬ — БОРА ТРИФТОРИД, N2OrBF3.
Реагент получают пропусканием газообразного трпфторпда бора
в раствор Л. ч. в нитрометане при 0 . Это аморфное твердое белое
вещество не растворяется в растворителях, с которыми не реагирует
111. При перемешивании сто с бензолом в течение недели образуется
нитробензол с выходом 39%. В отличие от нитрования азотной кис-
лотой этот реагент дает неудовлетворительные выходы и несколько
иное соотношение изомеров (преобладает о/л/ю-замещеиие) [21.
1. 13 а с h m a n G. В., F е и е г И., В 1 и е s t е 1 п 13. R., Vogt С. AL, J. Ат.
Chem. Soc., 77, 6188 (1955),
2. В а с h т an G. В., Vogt С, ЛЕ, J. Am. Chem. Soc., 80, 2987 (1958).
24
АЗОТИСТОВОДОРОДНАЯ КИСЛОТА, Н X X X . Мол. вес
43,03, т. кип. 37”.
Внимание: А. к. очень ядовита, работать под тягой!
Получение. Раствор А. к. в бензоле или хлороформе |1, 2J
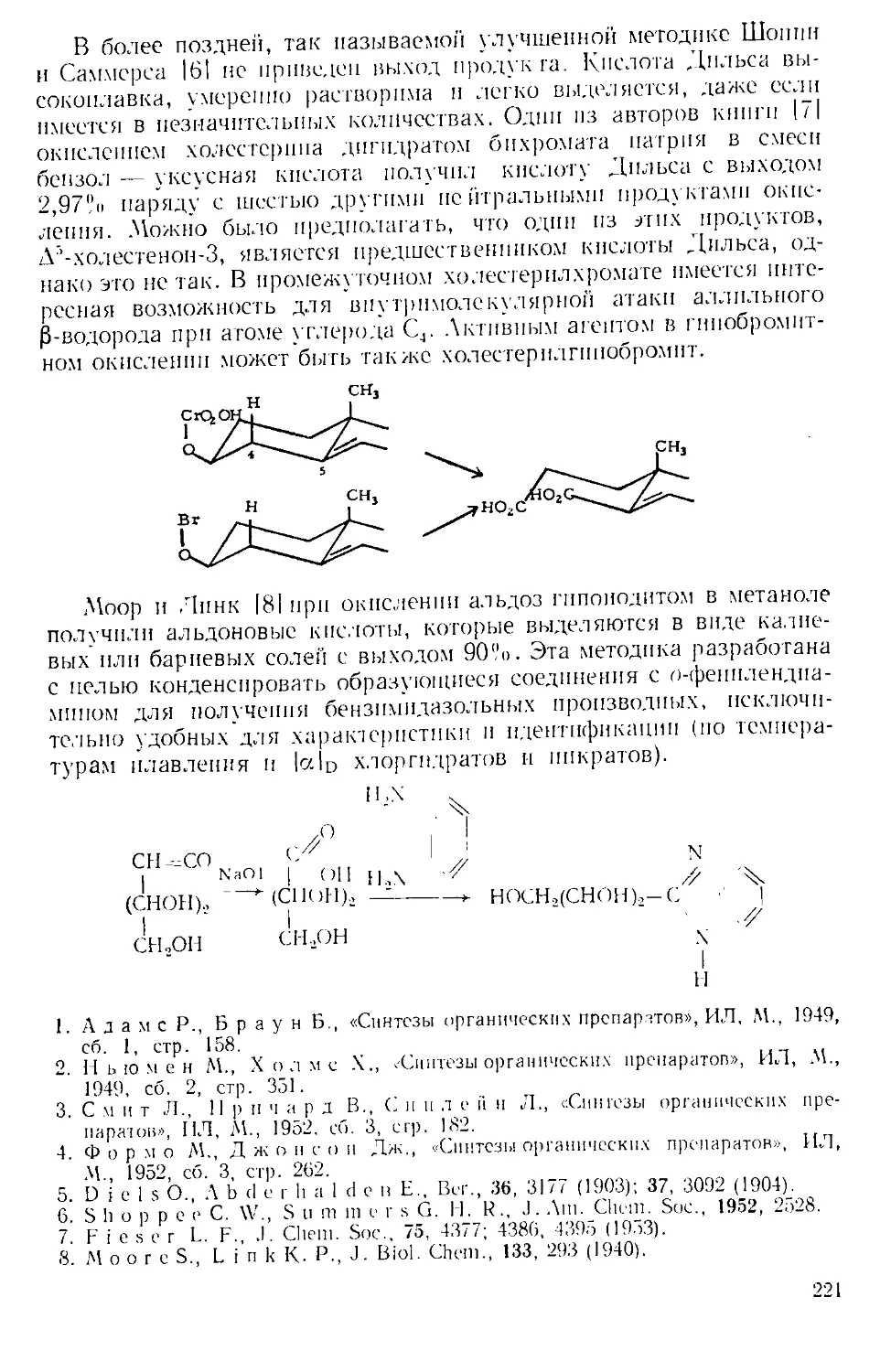
получают в приборе, состоящем из трехгорлой колбы, снабжепиоп
мощной мешалкой, капельной воронкой, термометром^ и газоот-
водной трубкой. К пасте из 65 г (1 эка) азида натрия п 65 мл теплой
воды добавляют 400 мл бензола или хлороформа, смесь охлаждают
до 0 и по каплям прибавляют 0,5 :-жв конц. серной кислоты, поддер-
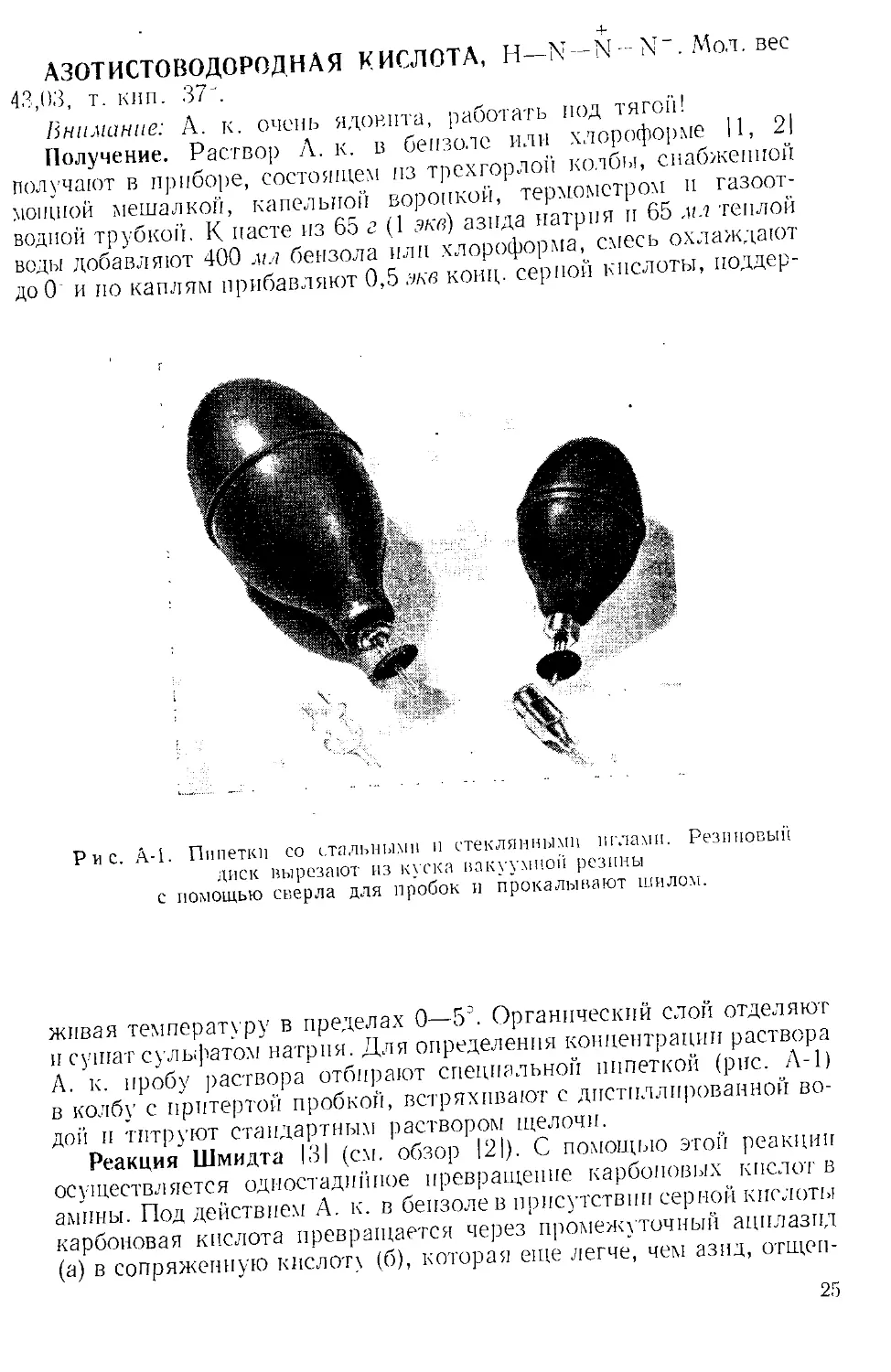
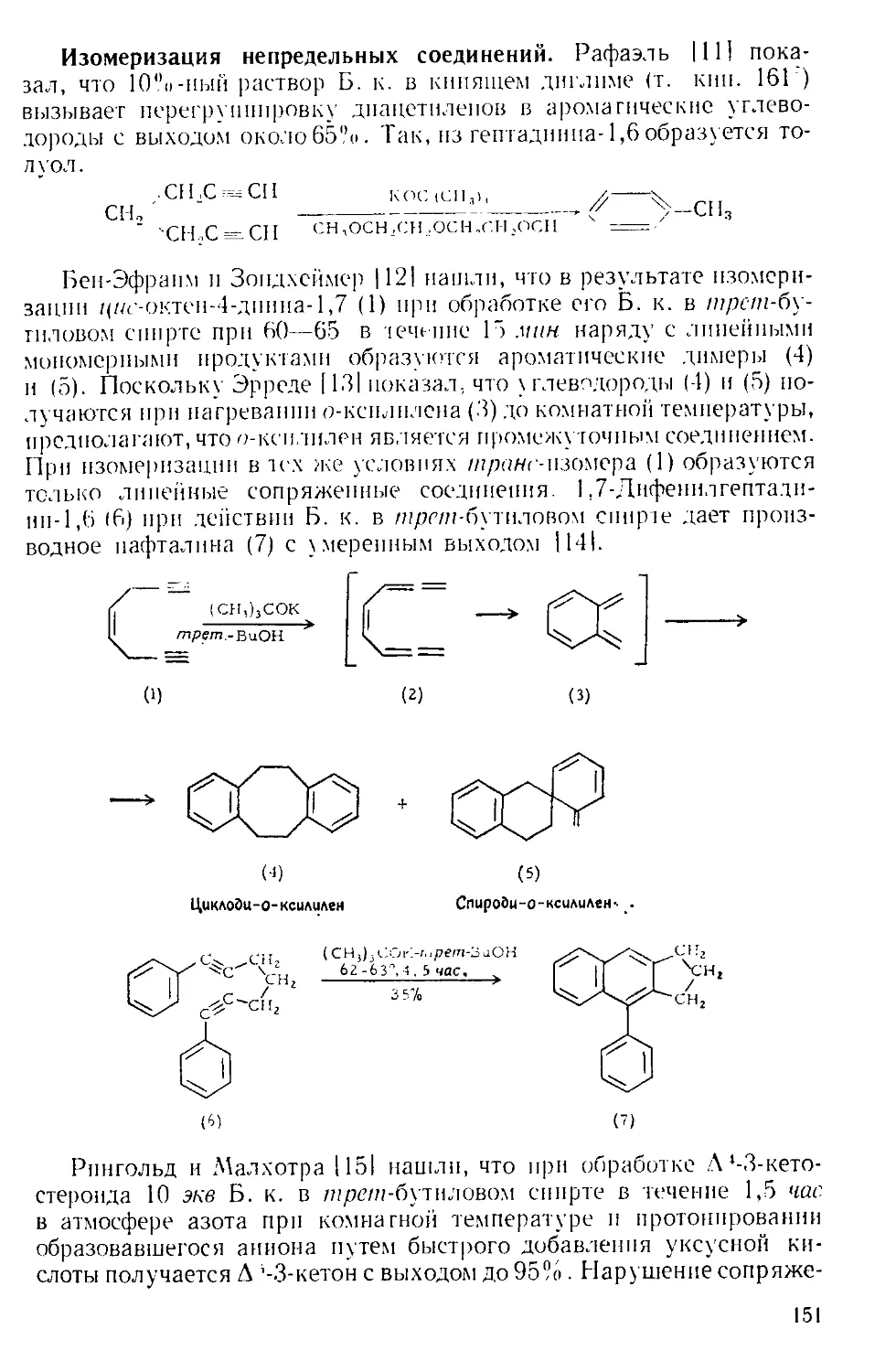
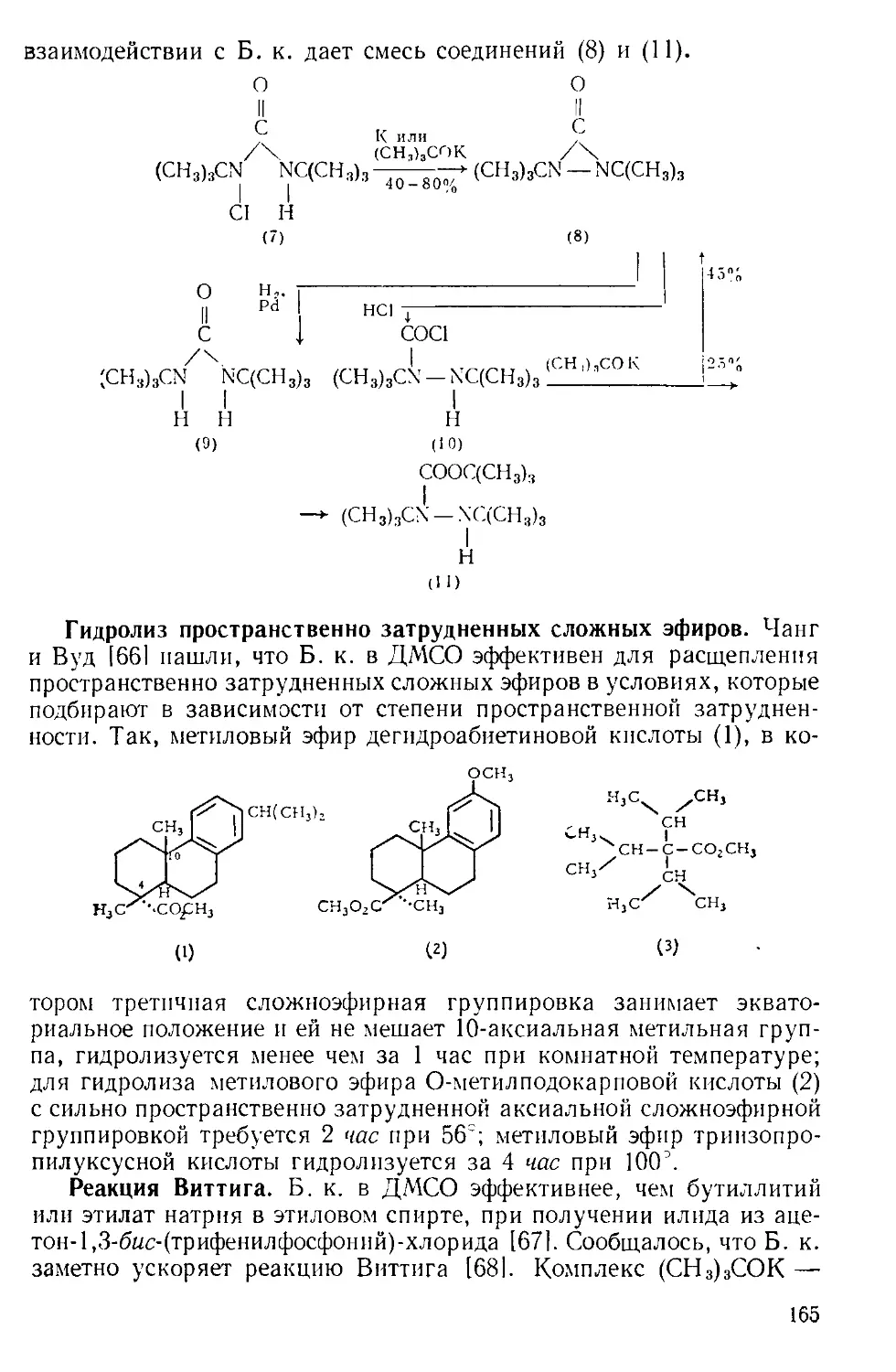
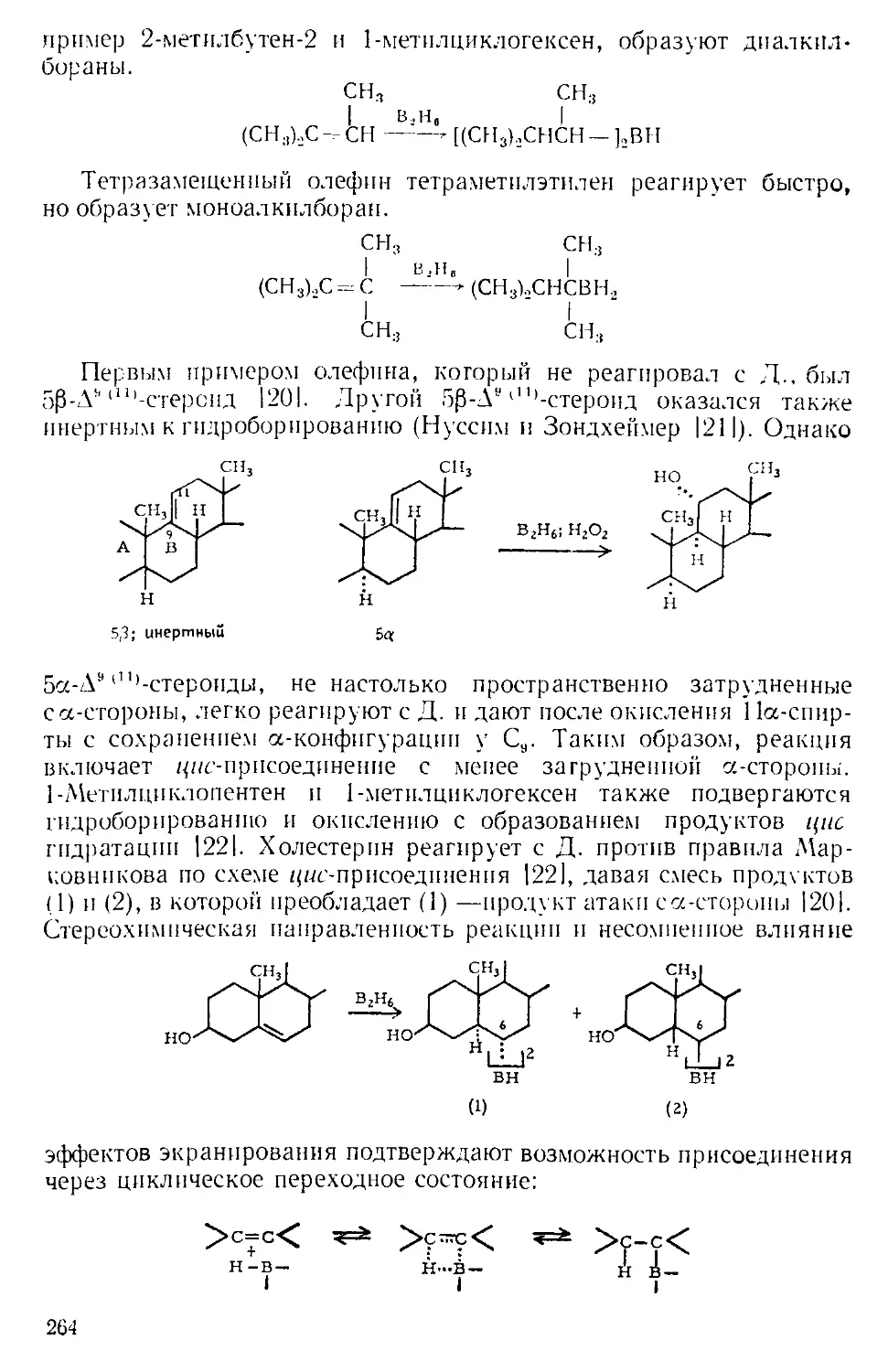
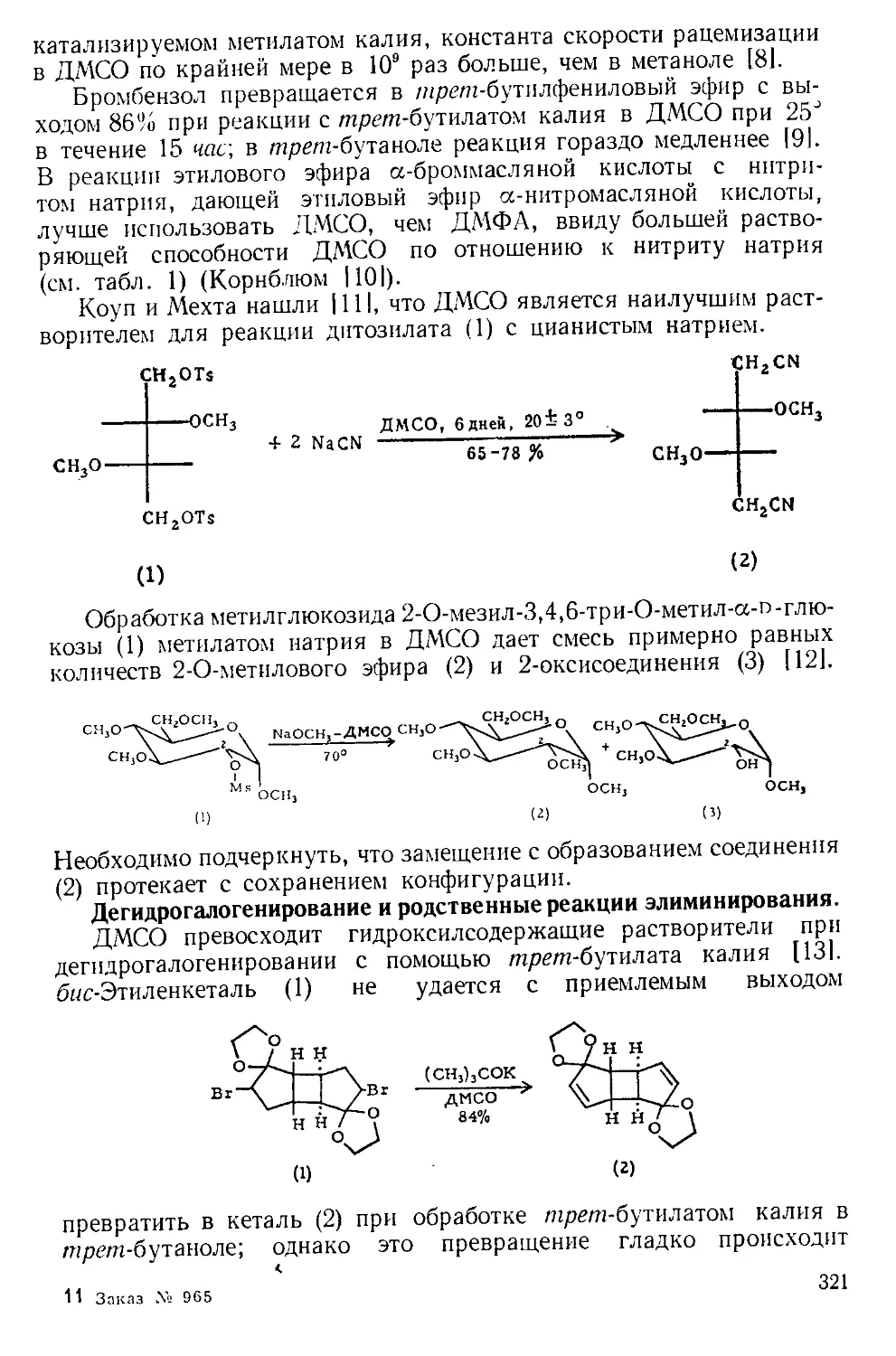
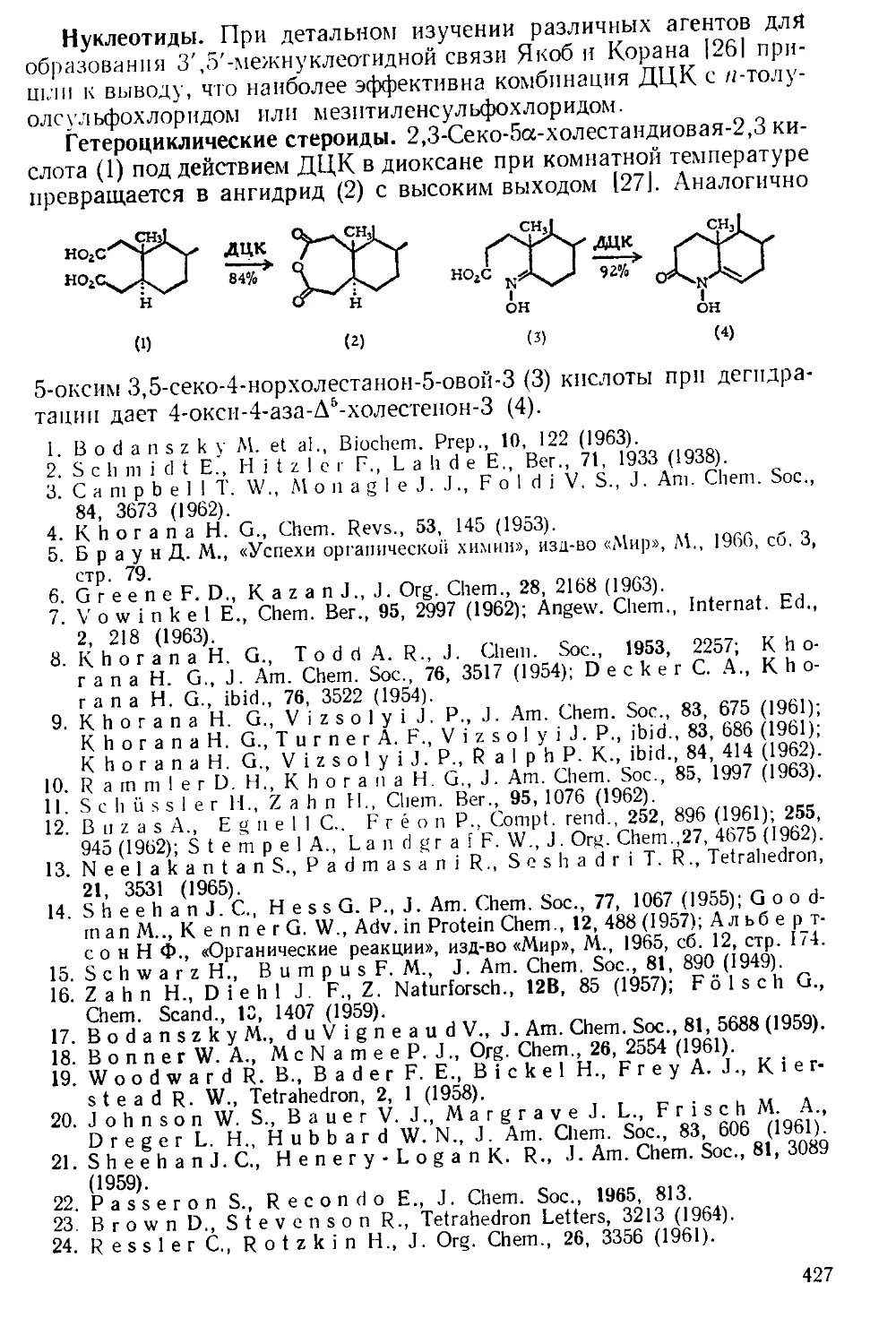
Рис. А-1. Пипетки со стальными п стеклянными иглами. Резиновый
диск вырезают из куска вакуумной резины
с помощью сверла для пробок и прокалывают шилом.
живая температуру в пределах 0—5Э. Органический слой отделяют
и сушат сульфатом натрия. Для определения концентрации раствора
А. к. пробу раствора отбирают специальной пипеткой (рис. А-1)
в колбу с притертой пробкой, встряхивают с дистиллированной во-
дой и титруют стандартным раствором щелочи.
Реакция Шмидта 131 (см. обзор |2|). С помощью этой реакции
осуществляется одностадийное превращение карбоновых кислот в
амины. Под действием А. к. в бензоле в присутствии серной кислоты
карбоновая кислота превращается через промежуточный ацплазпд
(а) в сопряженную кислоту (б), которая еще легче, чем азид, отщеп-
25
ляет азот с перегруппировкой в изоцианат (г). Эта одностадийная
О О н
h .. - - Н+ i1 | л-
RCO,Н - НХЭ _ RC -N = X = X — RC-N -X X —>
(а) (б)
О н
III — н + но
RC— N +-----> О = С — X — R —- RXH, - СО,
(в) ' (Г) (д>
реакция дает высокие выходы и препаративно достаточно проста.
Например, раствор стеариновой кислоты (15 а) в бензоле (500 мл)
обрабатывают кони, серной кислотой (30 мл) и перемешивают при
40’, добавляя 5%-ный раствор А. к. в бензоле (1,2 же) [4]. По окон-
чании выделения газа кислотный слой выливают в воду для осажде-
ния сульфата гептадециламина, который отделяют и кристаллизуют
СН3(СН„)1еСО„Н - и CH3(CH,)leXH,-- х,-усо,
96%
из этанола.
Бензальдегид в условиях реакции Шмидта образует бензонит-
рил с выходом 70% [2|; в этом случае перегруппировка С — N не
имеет места. В случае кетонов, в том числе и а, p-ненасыщенных, на-
он н
нха, н+ । ! ж _ч,
С6Н5СН = О-------СеШС------X-XsN —4
Н
ОН Н
j I _____и л +
—> С6Н5С-Х+ ——> с6н6с х
I
II
блюдается перегруппировка с образованием амидов. Ацетофенон
дает ацетанилид с выходом 77%, причем, как и в бекмановской
перегруппировке ацетофеноноксима, мигрирует фенильная группа
О ОН H
СН3СС6Н5 —4. НД СН3С - у . ( . \ —%
I
С6н5
он н он о н
I I —н+ I HI
—>СН,С—Х+ —сн3с=хсвн5—>сн3с— хсвн5
I
QH5
[2]. Кроме того, превращение ацетофенона в ацетанилид осуществ-
ляют продуванием сухого А. к. и воздуха в смесь кетона, бензола и
конц. серной кислоты. В условиях реакции Шмидта циклогексанон
26
превращается с расширением цикла в
е-капролактам [2|.
Реакция с хинонами. Добавление водного раствора азида натрия
к раствору а-нафтохпнона в уксусной кислоте сопровождается быст-
рым выделением азота л осаждением оранжево-красных игл 2-амп-
нонафтохинона-1,4 с выходом 87% 15). Реакция интерпретируется
как 1,4-прнсоединенпс А. к. с образованием азидонафтогндрохн-
нона (2) и последующее диспропорционирование с переносом гид-
рохиноновых водородов к азидной группе. Подобно этому Р-нафто-
хинон и его 3-бромпроизводное с А. к. дают соответствующие 4-амп-
нохиноны-1,2. Бензохинон-1,4 реагирует с А. к. с образованием
только аддукта — азидогидрохинона 161. Окислительно-восстанови-
тельный потенциал производных бензола ниже, чем нафталина, и,
очевидно, недостаточен для того, чтобы осуществился переход типа
(2)(3). Фепантренхипон, в котором невозможно подобное 1,4-прн-
соедпненпе, при взаимодействии с А. к. в присутствии серной кис-
лоты дает фепантридон |7|.
н
Мизитп и сотр. 181 нашли, что с некоторыми алкилхпнонамн в
конц. серной кислоте при 0: А. к. реагирует совершенно иначе с
образованием 2,5 /7-2,5-азепиндионов типа (5). К раствору хинона
(1) в конц. серной кислоте при 0' добавляют порциями 1 же азида
натрия. По окончании выделения газа смесь выливают в лед с водой
и осадок перекристаллизовывают из водного этанола. Выходы сос-
тавляют 75—85%. ЯМР-спектр амида (5) показывает, что NH-npo-
тон имеет непосредственное спин-спиновое взаимодействие с впнпль-
ным протоном, что свидетельствует о соседстве этих групп. В этой
реакции типа реакции Шмидта атакуется наименее пространствен-
но затрудненная карбонильная группа, а мигрирует группа, об-
27
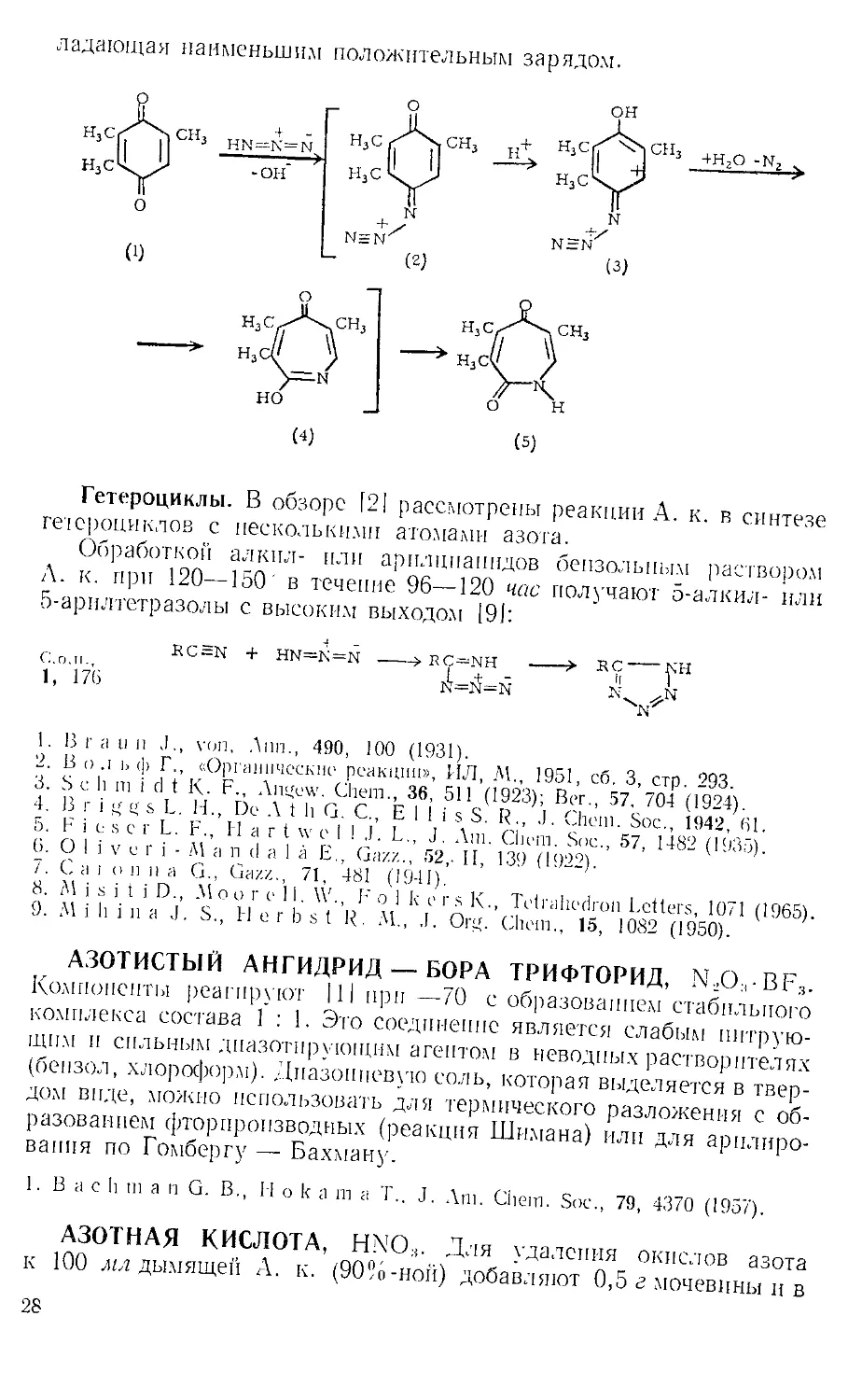
ладающая наименьшим положительным зарядом.
(4) (5)
Гетероциклы. В обзоре [21 рассмотрены реакции А. к. в синтезе
гетероциклов с несколькими атомами азота.
Обработкой алкил- или арилциаиидов бензольным раствором
Л. к. при 120—150 в течение 96—120 час получают 5-алкил- или
5-арилтетразолы с высоким выходом [91:
С. о. I (.,
1, 17(5
RC=N + N
НС—NH
1. Braun J., ven. Ann., 490, 100 (1931).
2. В o.i ь ф Г., «Органические реакции», ИЛ, М., 1951, сб. 3, стр. 293.
3. S с h m i (1 t К. F., Aneew. Chem., 36, 511 (1923); Вег., 57, 704 (1924).
4. В г i ц ц s L. Н., De A t h G. С., Ellis S. R., .1. Ghent. Soc., 1942, 61.
5. F i e s с г L. F., H a r 1 w a I I J. L., J. Am. Chem. Soc., 57, 1482 (1935).
6. Oliveri- M a n <1 a 1 a E., Gazz., 52,- II, 139 (1922).
7. C a i о n n a G., Gazz., 71, 481 (1941).
8. M i s i i i D., M our ell. \V., F о 1 k e r s 1\., Tetrahedron betters, 1071 (1965).
9. M i h i n a J. S., 11 e r b st R. M., J. 0гц. Chem., 15, 1082 (1950).
АЗОТИСТЫЙ АНГИДРИД — БОРА ТРИФТОРИД, NAO, BF3.
Компоненты реагируют Шири —/0 с образованием стабильного
комплекса состава 1:1. Это соединение является слабым нитрую-
щим и сильным диазотирующим агентом в неводных растворителях
(бензол, хлороформ). Диазоппевую соль, которая выделяется в твер-
дом виде, можно использовать для термического разложения с об-
разованием фторпроизводных (реакция Шимана) или для арнлиро-
ванпя по Гомбергу — Бахману.
1. Bachina n G. В., II о k a m а Т.. J. Am. Chem. Soc., 79, 4370 (1957).
АЗОТНАЯ КИСЛОТА, Н\’О:(. Для удаления окислов азота
к 100 ли дымящей А. к. (90%-нон) добавляют 0,5 г мочевины и в
28
течение 20 мин пропускают воздух; при этом жидкость обесцвечи-
вается 111.
Безводную А. к. (уд. вес 1,53) получают перегонкой дымящей
азотной кислоты из смеси с равным объемом кони, серной кислоты
121.
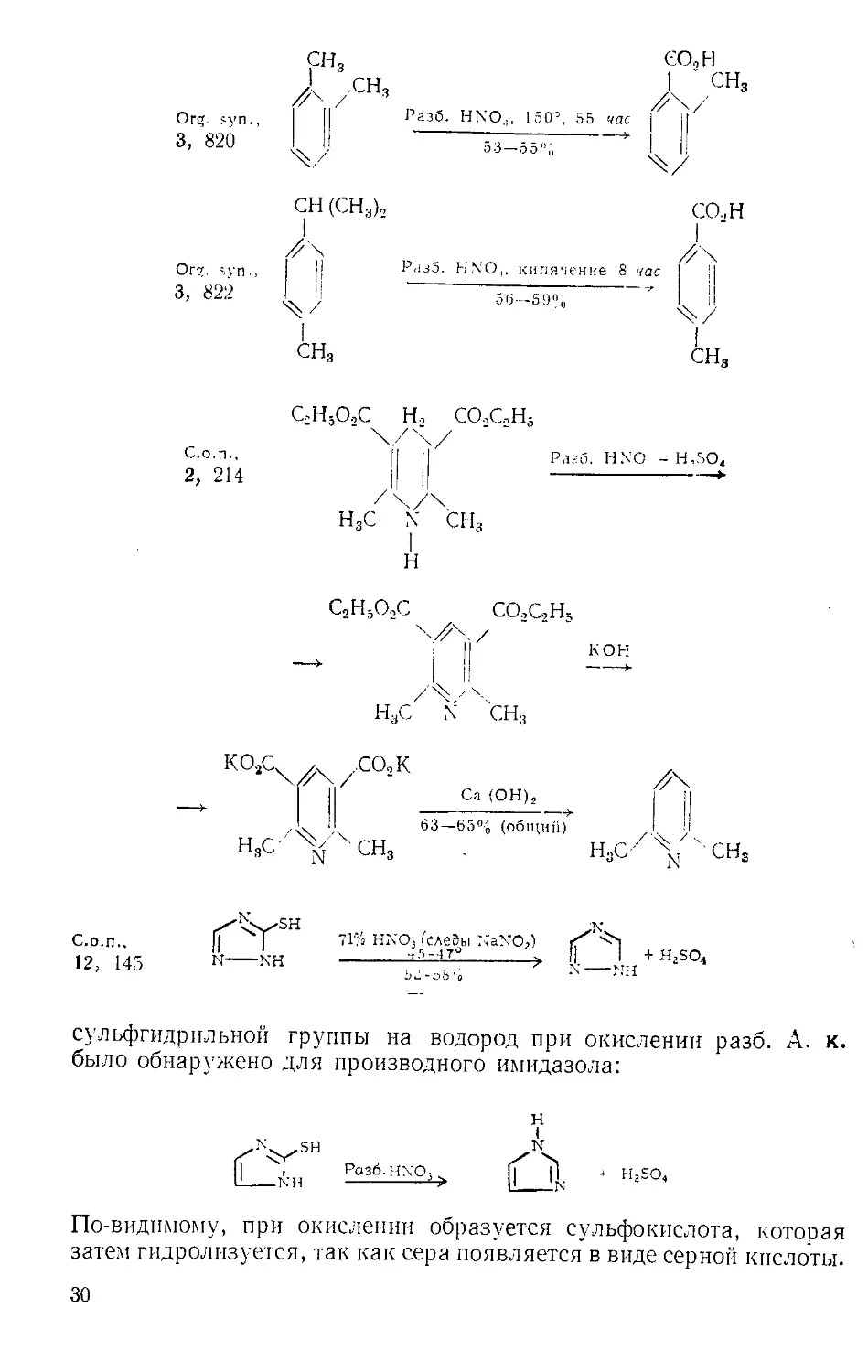
Окисление. Наряду с нитрованием А. к. применяют в основном
в качестве окислительного агента. Типы окислительных реакций,
их условия и выходы иллюстрируются примерами из сборников
«Синтезы органических препаратов» (С.о.и.) с указанием номера то-
ма и страниц. На приведенных схемах 71%-ная HNO;j соответствует
конц. А. к. (уд. вес 1,42). Последняя реакция, окисление З-.меркап-
то-1,2,4-триазола в 1,2,4-трпазол, требует пояснений. Замещение
I; 176
71%-ная HNO,, 25—30®
С1СН2СНрСН2ОН-----------:------> С1СН2СН„СО2Н
2 " * 78— 79% 2 - 2
Г1Л38 Q|£±12^ochch2ch2ch2chph
\/ '
о
— Н()оССН,СН,СН.,СО.,Н
С.о н.,
1, 288
71'io Н.\’О3,кипячение
Org. syn .
3, 791
сосн3
I
/л
у
сн3
Разб. HNO., кипячение,
затем КМп( )4—NaOH
СО2Н
29
Org. syn
3, 822
C.o.n..
2, 214
Org. syn.,
3, 820
CH(CH3)„ CO,H
I
//\
I |l РазЗ. HNO,. кипячение 8 час
55-59% "
I I
CH3 CH3
C;H5O,C H, CO2C2H5
H3c X CH3
I
H
C,H5O.,C CO,C,H4
KO3C4A/.CO2K
-> I II —I II
zk ‘I 63—65% (общи/i) I
H3C '%/XCH3 . H3C' % ' CHS
12, 145 N-------N-H ----------> y.-_Jn! + H‘SO*
сульфгидрильной группы на водород при окислении разб. А. к.
было обнаружено для производного имидазола:
Роз6.Н\О3
н
t
N
сч
* H2SO4
По-видимому, при окислении образуется сульфокислота, которая
затем гидролизуется, так как сера появляется в виде серной кислоты.
30
Ансел/i [4] показал, что некоторые 1,4-бензохиноны, особенно
три- п тетразамещенные, можно получить с высоким выходом окис-
лением гидрохинонов конц. А. к. в эфире при низкой температуре.
Раствор или суспензию 1 г гидрохинона в эфире перемешивают при
температуре от 0 до —20', добавляют конц. А. к. (около 10 мл) до
образования отдельного слоя или пока не перестанет углубляться
окраска эфирного слоя. После отделения от слоя А. к. эфирный раст-
вор обрабатывают твердым бикарбонатом натрия до прекращения
выделения газа и упаривают досуха.
он О
I СНз /\/СНз
| Н\О,-эфир (-1Ш) || ||
НзС/'У ЧСМ ° НзС/4!/ XN
ОН О
И) (2)
Удобная методика окисления, не включенная в вышеприведен-
ную таблицу, состоит в следующем 151. Смесь бензоина, 20 мл ук-
сусной кислоты и 10 мл 71 “о-ной А. к. нагревают на кипящей водя-
ной бане 2 час, охлаждают под краном и обрабатывают 75 мл воды.
Выход бензила (т. пл. 90—92') составляет 93—98%; после одной
кристаллизации из метанола получают чистый продукт с т. пл. 95—
96э.
Борел и Дойел [61 окислили З-нитро-4-нитрозотолуол до 3,4-ди-
нитротолуола с 96%-ным выходом (сырой продукт) добавлением
10 г вещества к 50 а дымящей А. к. при температуре 25“.
1. F г е е /и a nJ. Р., S h е р а г d 1. G., Org. Syn., 43. 81 (1963).
2. 21 и а и )', «Синтезы органических препаратов», ИЛ, М., 1952, сб. 3, стр. 411.
3. M а г с k w a I d \У„ Вег., 25, 2354 (1892).
4. Ansell М. F., X ash В. W., W i 1 s о n D. A., J. Chem. Soc. 1963, 3028.
5. Org. Expt., 215.
6. Borel E., Deuel H., Helv. Chim. Acta., 36, 806 (1953).
АКРИЛОНИТРИЛ, CHS=CHCN. .Мол. вес 53,06, т. кип. 78,5,
уд. вес 0,81.
Очистка [11: А. тщательно промывают 10%-ной H.,SO4, 10%-ным
Na2CO:J, насыщенным раствором Na.2SO4, сушат хлористым каль-
цием; фракционируют обычным образом и затем перегоняют в ва-
кууме.
А. применяют для цианэтилирования спиртов, меркаптанов, аль-
дегидов, кетонов, малонового эфира, нитрометана, бромоформа
и т. д. [2] *.
I. Bev ington J. С., Eaves D. Е., Trans. Faraday Soc., 55, 1777 (1957).
2. В г и s о n Н. A., Org. Reactions, 5, 79 (1949).
* См. также Терентьев А. П. и К о с т А. Н., «Реакции и методы ис-
следования органических соединений», Госхимиздат, М.— Л., 1952, том 2, стр.
47.— Прим, перев.
31
р-АЛАНИН, H.2NCH,CH,CO2H. Мол. вес 89,10.
А. п другие аминокислоты (см. Глицин) являются эффективными
катализаторами конденсации по Кневенагелю [1].
СН:)
(’-Аланин |
С„Н5СОСН3 + сн„со,с,н5-------> С,Н5С = ссо„с„н,
| - ' ° s 1-87..-,% - | - - °
С\' с\г
Каталитические количества А. значительно улучшают выход
при конденсации дифенилциклопропенона с дпнитрплом малоновой
кислоты и этиловым эфиром циануксусной кислоты |2|.
В присутствии А. цнклогександион-1,4 конденсируется с дпни-
трило.м малоновой кислоты практически количественно |3]. Реаген-
о
II
I I _____ rt 4 87’, несколько часов
+ 2СН, (СМ)2 + ₽-Алгнта + Н,0 —------------>
I J “ 94%
\/ 119 г 1г 200 лы
II
О
100 г
С (CN),
II
~0
"н
С (CN),
ты сплавляют при нагревании на кипящей водяной бане, добавляют
водный раствор А. и продолжают нагревание. Образующийся те-
траиптрпл выпадает в виде кристаллов; их промывают водой и эфи-
ром и получают чистый продукт с т. пл. 216—217 \ При проведении
этой реакции в бензоле с применением в качестве катализатора смеси
уксусной кислоты и ацетата аммония выход снижается до 76,5%.
1. П р а у т Ф., Хартман Р., Хуанг Е., «Синтезы органических препара-
тов», ИЛ, М., 1956, со. 7, стр. 10; Е g a w a Y., Suzuki М., О k u d а Т.,
Chem. Pharm. Bull., 11, 589 (1963).
2. Л и drea d е s. S., J. Am. Chem. Soc., 87, 3941 (1965).
3. Л.с к e г D. S., II e r t 1 e г W. R., J. Am. Chem. Soc., 84, 3370 (1962).
АЛКИЛАМИНБОРАНЫ, R3_„NH„-BH3.
Нот и Бейер HI описали получение этих реагентов и их приме-
нение для восстановления альдегидов, кетонов и хлорангидридов
32
кислот до соответствующих спиртов. Восстановительная способ-
ность значительно варьирует в зависимости от структуры реагента.
Этиламннборан C2H5NH2-ВН3 восстанавливает 3 моля «-бензохи-
нона, тогда как метиламинборан, диметиламинборан (CH3),NH-BH3
и mpe/71-бутиламинборан (CH3)3CNH2 • ВН3 восстанавливают только
2 моля этого соединения.
1. Noth Н„ Веуег Н„ Chem. Вег., 93, 928, 939, 1078 (1960).
АЛКИЛСУЛЬФ АТЫ. Окрашенные препараты диметил- и ди-
этилсульфата очищают медленной перегонкой в вакууме водоструй-
ного насоса (присутствующая ROSO2OH разлагается на H2SO4 и
R2SO4). Высшие алкилсульфаты удобно получать через соответ-
ствующие сульфиты [1].
1. Сутер К-, Г е р х а р т X., «Синтезы органических препаратов», ИЛ,
М„ 1949, сб. 2, стр. 135.
АЛКИЛЦИАНИДЫ [II. н-Пропил- и н-бутилцианиды получают
при кипячении и эффективном перемешивании 0,5 моля соответ-
ствующего первичного алкилбромида, 0,6 моля цианистого натрия
и 150 мл этиленгликоля [2]. Температура паров, вначале близкая к
температуре кипения галогенида, быстро повышается до постоянного
значения по мере расхода галогенида. Лучшим растворителем для
превращения хлоридов (от н-бутил-до н-октил-) в цианиды являет-
ся полиэтиленгликоль-300 [3]. Время реакции варьирует от 15 мин
до нескольких часов. Нитрилы выделяют перегонкой с выходом
90—95%.
Для превращения вторичных алкилхлоридов в соответствующие
цианиды лучшим растворителем является диметилсульфоксид [4].
Хлорид медленно добавляют к нагретой смеси цианистого натрия
и диметплсульфоксида, после чего нагревание продолжают еще
I—3 час. Реакционную смесь выливают в воду и продукт экстраги-
руют эфиром или хлороформом. После высушивания, удаления раст-
ворителя и перегонки алкилцианиды получают с выходом около
70%.
1. Contributed by Susi Р. V., American Cyanamid Co.
2. LewisR.N., S u s i P. V., J. Am. Chem. Soc., 74, 840 (1952).
3. Brandstrom A., Chem. Scand., 10, 1197 (1956).
4. S m i 1 e у R. A., Arnold C., J. Org. Chem., 25, 257 (1960).
АЛКИЛЫ ИОДИСТЫЕ. Йодистый метил и иодистый этил на
свету быстро разлагаются с выделением иода, поэтому долго хранив-
шиеся и потемневшие препараты не пригодны для использования.
Такие образцы должны быть очищены встряхиванием с несколькими
порциями разбавленного раствора тиосульфата натрия или бисуль-
фита натрия до обесцвечивания, промыванием водой, сушкой над
хлористым кальцием и перегонкой. Бесцветные перегнанные пре-
параты небходимо хранить в темных склянках, защищая от сол-
2 Заказ № 965 33 '
нечных лучей. В качестве предохранительного средства можно при-
бавить несколько капель чистой ртути, что позволяет хранить по-
дпетые алкилы неопределенно долго без их окрашивания, если пре-
парат защищен от длительного действия света. Продолжительное
освещение может представлять некоторую опасность, поскольку в
результате фотохимической реакции могут образоваться заметные ко-
личества токсичного метплмеркурподида. Небольшие количества
ртути можно также применять для удаления слабой розовой или
красной окраски у образца иодпда, в котором разложение только
началось.
АЛЛЕН, СН2=С=СН2. /Мол. вес 40,06, т. кип. — 32л
Получение. Большинство синтетических методов приводит к
трудноразделимым смесям аллена и метилацетилена; препарат, не
содержащий метилацетилена, можно получить дехлорированием
2,3-дихлорпропена цинковой пылью [11:
Кнпячепие
CHo = C-CHoCl+Zn —95%-ный ЕЮН — Н.,0----->СНо-=С = СН2
. I 1 2 3 /и - 80% - Л
С1
2,34 моля
4,6 г-атом
400 мл S0 мл
Синтезы производных циклобутана. Ангидрид 3-метиленцикло-
бутандикарбоновой-1,2-кислоты (3) получен [21 реакцией А. с мале-
иновым ангидридом в 645 мл бензола в присутствии 0,25 г гидро-
хинона. Реагенты загружают в охлажденный автоклав (—70е), за-
^со
HCSCCH2CH X
Н2С—снч /
со
(1) 2, 5 моля (2) 5,1 моля
(3) 22-26%
(4) 7-9%
тем перемешивают 8—10 час при 200—210°. Обычная обработка с
последующим тщательным фракционированием дает соединения (3)
и (4) с выходами, указанными на схеме.
Превращение в производное циклооктана. В присутствии ката-
лизатора б«с-(трифенилфосфит)-никельдикарбонила А. превращает-
ся в тетрамер, идентифицированный как 1,3,5,7-тетраметиленцикло-
октан [31.
Н2С,
1. С г i р р s Н. N., К i е f е г Е. F., Org. Syn., 42, 12 (1962).
2. S t е v е n s о n H. В., С г i p p s H. N., Wi I I jams J. K-, Org. Svn.,
43, 27 (1963).
3. BensonR. E., LindseyR. V., Jr., J. Am. Chem. Soc., 81, 4247 (1959).
34
АЛЛИЛЛИТИЙ. А. впервые был получен следующим образом
[11. Реакцией аллилмагнийхлорида с трифенилхлорстаннаном (1)
в эфире, ТГФ или бензоле получают аллилтрифенилстаннан (2),
обработка которого раствором фениллития дает суспензию тетра-
фенилстаннана в растворе А.Непосредственно эту смесь используют,
например, для реакции А. с карбонильными соединениями. А. полу-
СН. = СНСН..С1 -I- (CGH5)3SnCI -у Mg — rc6H5)3SnCH„CH = CH.,
(1) (2), т. пл. 7-1°
I C6HaLi
4'
CH., = CHCHoLi + (CeH5)4Sn
чают также переметаллированием при взаимодействии фениллития
с тетрааллилстаннаном [2]:
4C6H5Li-j-(CH2 = CHCH2)4Sn — 4CH2 = CHCH2Li4-(C6H5)1Sn
А. взаимодействует с различными органическими и металлоорга-
ническими соединениями с образованием аллильных производных.
Новый удобный метод синтеза А. состоит в металлировании ал-
лилфенилового эфира литием в ТГФ [3].
ТГФ. —15°
CH2 = CHCH,OC6H5-;-2Li-----> CH., = CHCH,Li-yCeH5OLi
‘ ь а 1 62-66% ‘ "
1. S е у f е г t h D., WeinerM. A., J. Org. Chem., 24, 1395 (1959).
2. S c v f e r t h D„ Weiner M. A., J. Org. Chem., 26, 4797 (1961).
3. Eisch J. J„ Jacobs A. M., J. Org. Chem., 28, 2145 (1963).
АЛЮМИНИЯ АМАЛЬГАМА, из Al (26,98) и HgCl2 (271,52).
Получение, a) [1]. Алюминиевые стружки, не содержащие
масла, обрабатывают разбавленным раствором едкого натра до силь-
ного выделения водорода, отделяют декантацией, споласкивают во-
дой один раз так, чтобы осталось некоторое количество щелочи, и
затем дважды обрабатывают раствором 0,5%-ной сулемы в течение
1—2 мин. Блестящий амальгамированный металл быстро промы-
вают водой, затем спиртом и эфиром и сразу используют в реак-
ции. А. а. быстро реагирует с водой с выделением водорода в коли-
честве, эквивалентном содержанию алюминия, и может быть исполь-
зована для сушки эфира или спирта.
б) Алюминиевую фольгу чистят наждачной бумагой и амальга-
мируют небольшим количеством сулемы (Ньюмен [2]).
Восстановление. А. а.— нейтральный реагент и поэтому исполь-
зуется для восстановления веществ, чувствительных к щелочам.
Под действием А. а. в эфирном растворе щавелевоуксусный эфир
восстанавливают до эфира яблочной кислоты с выходом 70—80%,
СО2С,Н5 со2с,н5
СО WMBpe СНОИ
| 70-80% |
СН2СО2С2Н5 СН,СО2С.,Н5
2*
35
тогда как с амальгамой натрия выход составляет всего 50% [11.
Нитробензол восстанавливается А. а. при нагревании в этаноле до
анилина, а при более низкой температуре в эфире — до фенилгид-
роксиламина [11. Жирноароматические кетоны восстанавливаются
А. а. до пинаконов (выходы 30—60%) [2|; с алифатическими кето-
нами выходы незначительные. К раствору ацетофенона в смеси бен-
СН3 СН3
I I
2СвН5С = О + А1 + HgCl2+ Абс. С2Н5ОН + QH6 —С,Н5СОН
31,8 г 8 г 0,5 г 130 мл 130.4,1 54-59% ।
С„Н5СОН
I
сн3
ХИзойеры)'
зола с этанолом добавляют сулему и очищенную наждачной бумагой
алюминиевую фольгу толщиной 0,4 мм, нарезанную квадратами
(2,5 см). При нагревании смеси начинается бурное взаимодействие,
которое затем продолжается самопроизвольно и затихает. Смесь
кипятят с обратным холодильником до исчезновения алюминия
(2 час), охлаждают, подкисляют разб. соляной кислотой, продукт
экстрагируют бензолом, перегоняют и перекристаллизовывают.
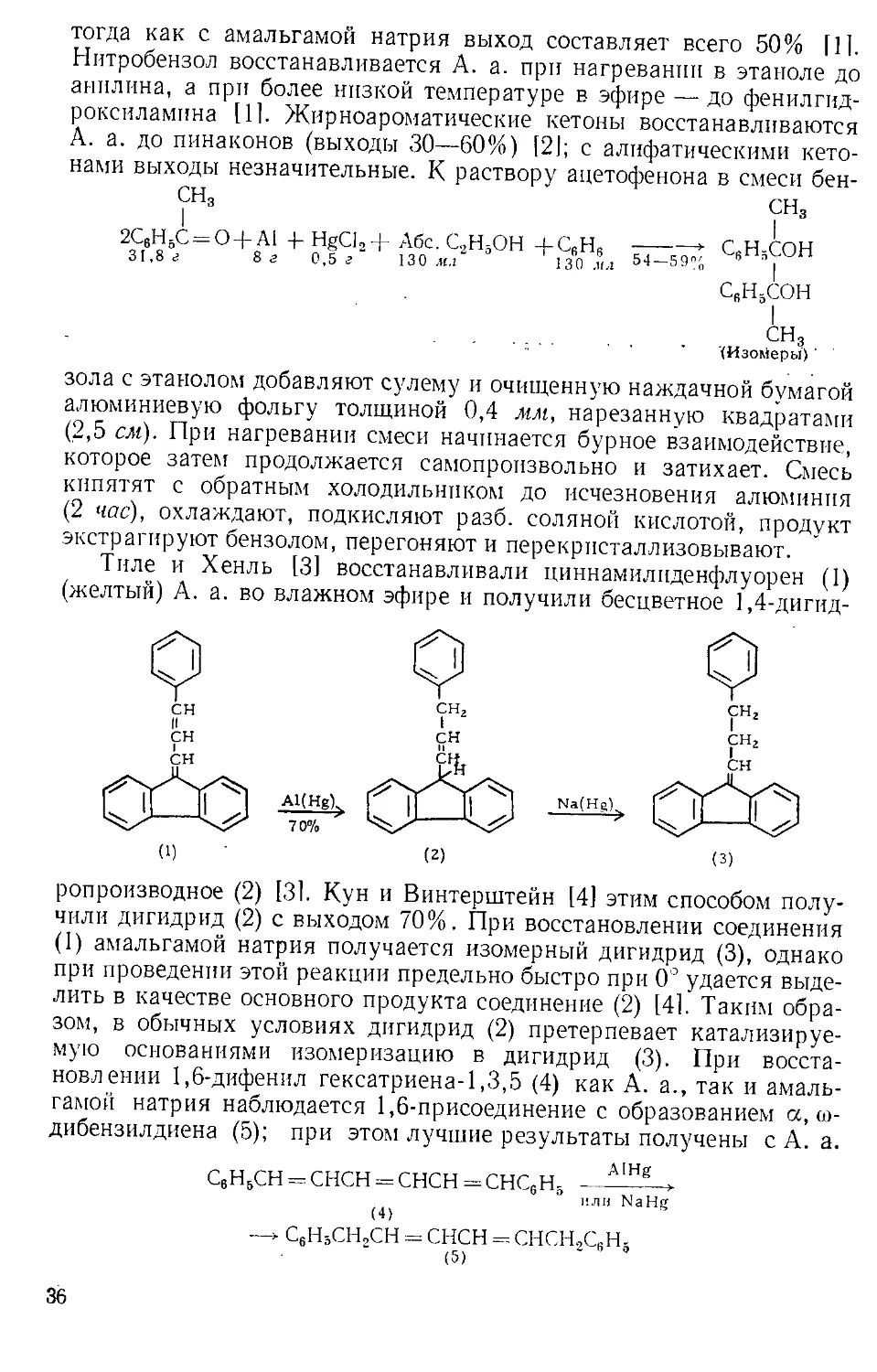
Тиле и Хенль [3] восстанавливали циннамилнденфлуорен (1)
(желтый) А. а. во влажном эфире и получили бесцветное 1,4-дигид-
Na(Hg)^
(3)
ропроизводное (2) [31. Кун и Винтерштейн [4] этим способом полу-
чили дигидрид (2) с выходом 70%. При восстановлении соединения
(1) амальгамой натрия получается изомерный дигидрид (3), однако
при проведении этой реакции предельно быстро при 0° удается выде-
лить в качестве основного продукта соединение (2) [41. Таким обра-
зом, в обычных условиях дигидрид (2) претерпевает катализируе-
мую основаниями изомеризацию в дигидрид (3). При восста-
новлении 1,6-дифенил гексатриена-1,3,5 (4) как А. а., так и амаль-
гамой натрия наблюдается 1,6-присоединение с образованием а, а>-
дибензилдиена (5); при этом лучшие результаты получены с А. а.
AIHg
СвН5СН = СНСН = СНСН=СНС6Н5-------
или NaHg
(4)
—> с6н5сн,сн = снсн=снсн„свн5
(5)
36
Кун и Фишер |5| нашли, что тетрафеннл кумулены (6) и (8)
восстанавливаются А. а. в тетрагидрофуране, содержащем 4% воды,
до днгидропроизводных (7) н (9).
AIHg
(С6Н6),С=С=С = С(С,Н5), (С6Н5)2С = С = СНСН(С6Н6)2
(6) " (7)
А1НО
(СвН5)2С=С = С = С=С=С(СвН5)2 -> (С6Н3)2С = С==СНСН==С = С(СвН5)2
7 5 %
18) О)
О восстановлении р-кетосульфоксидов в метилкетоны (RCOCH2-
• SOCH3B RCOCH3) см. в описании реагентов — производных ди-
метилсульфоксида: Метилсульфнннлметилид натрия.
Десульфуризация. При исследовании строения антибиотика
глиотоксина (1) Джонсон и сотр. [61 нашли эффективны!! способ
его десульфуризации до дестпоглиотоксина (2) под действием А. а.
в нейтральном спиртовом растворе: при этом никель Ренея дает
продукт дальнейшего восстановления.
1. W i s 1 i с е п u s Н.. Kaufmann L.. Вег., 28, 1323 (1895).
2. X е w m а п М. S., .1. Огц. Chem.. 26, 582 (1961).
3. Thiel е .1., Н е п 1 е F., Ann., 347, 307 (1906).
4. Kuhn R., Wintersto i п A., Helv. Claim. Acta-, 11, 123 (1928).
5. К в h n R., F ischer 11., Cheni. Ber., 94, 3060 (1961).
6. Dutch e r J. D., J о h n s о n J. R., Bruce W. I-'.. J. Am. Chem. Soc., 67,
1736 (1915); Johnson J. R., В u c h a n a n J. B., J. Am. Chem. Soc., 75,
2103 (1953).
АЛЮМИНИЯ БРОМИД, А1Вг3. Мол. вес 266,73, т. пл. 97,5°,
т. кип. 268’, получение [11.
Бромалкилирование. Реакцией адамантана (1) с этиленом в при-
сутствии А. б. при температуре от —30 до —20' в гексане получают
1-0-бромэтиладамантан (2) с выходом 57% (Штеттер и Гебель [2|):
37
Выход (кристаллизующегося продукта) повышается до 75%, если
реакцию проводить при охлаждении смесью сухого льда с ацетоном
и вводить рассчитанное количество этилена [31. Однако результаты
зависят от качества продажного препарата, и лучшими образцами
следует считать те, которые хранятся в запаянных ампулах.
Расщепление эфирной связи. А. б.— наиболее подходящий реа-
гент из многих испытанных для деметилирования 2,6-диметокси-
4-метнлбензальдегпда |4|. Раствор А. б. в сероуглероде добавляют
при перемешивании к альдегндоэфиру в том же растворителе. После
перемешивания в течение 1 час комплекс, выделяющийся в виде крас-
СНО
CH3oxJx ZOCH3
I II +А1Вт3
Д/ 2 2 г в 230 .«.1
| CS2
СН3
5 г в 250 M.i CS2
Перемешивав не,
затем Н2О
сно
ной смолы, кристаллизуют из воды и получают альдегидофенол удов-
летворительной степени чистоты.
Поданным Прея [51 , пиридиниевая соль А. б. (2 Ру: 1 А1Вг3) —
прекрасное средство для расщепления простых фениловых эфиров;
только дифениловый эфир устойчив к действию этого реагента.
К раствору 33 г пиридина в 50 мл бензола при перемешивании до-
бавляют раствор 50 г А. б. в 300 мл бензола. Выпавшую соль очи-
щают переосаждением из пиридина эфиром. Для деметилирования
смесь фенилового эфира с 3 частями этой соли (1:3) нагревают до
220J.
Бромирование [6]. Бромдихлорметан (т. кип. 89—90") получают
с хорошим выходом при бромировании хлороформа А. б. и броми-
стым водородом.
1. Н и х о л ь с о и Д., Винтер П., Ф и н б е р г X., «Неорганические син-
тезы», ИЛ, М., 1952, сб. III, стр. 33.
2. S t е t t е г Н„ G о е Ь е I Р., Chem. Вег., 95, 1040 (1962).
3. N a z е г М. -Z., unpublished.
4. Adams R., Mathieu J., J. Am. Chem. Soc., 70, 2120 (1948).
5. Prey V., Ber., 75, 537 (1942).
6. L a k e D. E., A s a d о r i a n А. А., пат. США 2553518 (1951).
АЛЮМИНИЯ ГИДРИД, А1Н3. Мол. вес 30,01. См. также
LiAlH,—А1С13.
А. г. получают из алюмогидрида лития и хлористого алюминия
3LiA!H44-A!Cl3—* 4А1Н3-]-3LiCl
в эфирном растворе 111. По восстановительному действию А. г. по-
добен ЫА1Н4 [11, но более специфичен при восстановлении а, 0-не-
предельных альдегидов в аллиловые спирты (Йоргенсон |2|). Так,
если ЫЛ1Н4 склонен к восстановлению кратных связей в аллиловых
38
или пропаргиловых спиртах, то при использовании А. г. не тре-
Л1Н,
С„Н,СН-= СНСНО----> С„ИзСН^СНСН2()Н
буется вмешательства для предотвращения такого процесса.
Охлажденный раствор LiAIH4 в эфире обрабатывают некоторым
избытком (от стехиометрического) хлористого алюминия и переме-
шивают при комнатной температуре до растворения хлористого алю-
миния и полного выпадения хлористого лития. После добавления
эфирного раствора субстрата смесь выдерживают при комнатной
температуре в течение от 30 мин до 5 час. Присутствие больших
количеств LiAlHj в растворе не изменяет ход восстановления А. г.
Несмотря па все предосторожности, метиловый эфир о-кумаровой
кислоты не удается восстановить LiAlHj до о-оксикоричного спирта,
который при восстановлении А. г. получают с выходом 75%.
Браун и Юун |3] получили относительно устойчивый раствор
А. г. добавлением 0,5 моля 100%-ной серной кислоты к 1 молю
LiAlH4 в ТГФ:
ТГФ
2LiAlH4-|-H2SO4 --> Li,SO4-i-2AlH3+2H,
При этом наблюдается выделение водорода и количественное осаж-
дение сульфата лития. А. г. в противоположность LiAlH4 избира-
тельно восстанавливает карбонильную группу диенонов.
1. Wiberg Е., Angew. Chem., 63, 485 (1951).
2. Jorgenson М. J., Tetrahedron Letters, 559 (1962).
3. Brown H. C., Y о о n N. M., J. Am. Chem. Soc., 88, 1464 (1966).
АЛЮМИНИЯ ИОДИД, АН,. Мол. вес 407,72.
Л. и., получаемый в растворе реакцией амальгамированного
алюминия с подом в эфире, применяют для превращения холестери-
на в холестсрплиодид с хорошим выходом |1]. Порошкообразный
алюминий быстро встряхивают с 1%-ным раствором хлорной
ртути (II) в эфире, образующийся серый порошок промывают декан-
тацией и постепенно обрабатывают эфирным раствором иода. Смесь
кипятят до полного растворения, прибавляют холестерин и раствор
кипятят 8 час. В этих же условиях холестанол-3₽ не изменяется.
В более поздней работе (21, в которой не упоминается вышеука-
занная статья, было установлено, что А. и. настолько чувствителен
к воздуху и влаге, что для его получения требуется специальная ап-
паратура. Как катализатор реакции Фриделя — Крафтса этот ре-
агент менее эффективен, чем хлористый или бромистый алюминий.
1. Broome J., В г о w п В. R., S u m mers G. Н. R., J. Chem. Soc., 1957,
2071.
2. Kline E. R, Campbell B. N„ Spaeth E. C„ J. Org. Chem., 24, 1781
(1959).
АЛЮМИНИЯ ОКИСЬ ДЛЯ ДЕГИДРАТАЦИИ СПИРТОВ.
А. о., полученная гидролизом изопропилата алюминия, довольно
39
кислый агент, под действием которого образующиеся олефины пре.-
терпевают изомеризацию [1], которая предупреждается обработкой
катализатора аммиаком или применением А. о., полученной из алю-
мината натрия.
Эффективную дегидратацию терпеновых и сесквитерпеновых
спиртов проводят на нейтральной А. о. (первой степени активности),
к которой добавляют 1—2% пиридина во избежание перегруппиро-
вок, наблюдающихся при использовании кислых реагентов
(Рудлоф |2]). Этим методом из аллильного лактоноспирта (1) (полу-
ченного из сантонина) синтезирован диеновый лактон (2) с выходом,
достаточным для последующего фотохимического синтеза дигидро-
костунолида (3) (Кори и Хартманн [31).
Катализатор для дегидратации готовят также (Штолль и сотр.
[4]) увлажнением смеси 90 г А. о., 60 г инфузорной земли и 20 г из-
мельченной пробки и нагреванием этой смеси до красного каления.
Через трубку, наполненную катализатором и нагретую до 380—
400°, пропускают пары циклопентанола (50 г) в токе азота. Цикло-
пентен отделяют, сушат и перегоняют; выход 81%.
Катал,, 390°
1. Р i п е s Н„ Р i 1 1 a i С. N., J. Am. Chem. Soc., 83 , 3270 (1961); Schap-
p e 1 i F. G., Pines H., J. Org. Chem., 31, 1735 (1966).
2. Rudloff E., von, Can. J. Chem., 39, 1860 (1961).
3. Corey E.J., H artmann A. G., J. Am. Chem. Soc., 87, 5736 (1965).
4. S t о 1 1 A., L i n d e n m a n n A., J ucker E., Helv. Chim. Acta, 36, 268
(1953).
АЛЮМИНИЯ ОКИСЬ ДЛЯ ХРОМАТОГРАФИИ. Образец А. о.,
эквивалентный А. о., «промытой кислотой», получают перемешива-
нием обычной А. о. с этилацетатом; смесь выдерживают в течение
1—2 дней, фильтруют и сушат при 80°. А. о., промытая 5—10%-ной
40
уксусной кислотой, является подходящим адсорбентом для соеди-
нений, чувствительных к основаниям (енолацетаты, а, Р-непредель-
ные ацетаты) [1]. Для предупреждения изомеризации при хромато-
графии Р,у-непредельных кетонов А. о. промывают теплой водной
щелочью, теплой уксусной кислотой и водой (до нейтральной реак-
ции), а затем реактивируют нагреванием при 200° в течение 30 час
12].
1. Farrar К. R., Hamlet J. С., Henbest Н. В., Jones Е. R. Н.,
J. Chem. Soc., 1952, 2657; Clayton R. В., Cr awsha w A., Hen-
best H. B., Jones E. R. H., Lovell B. J., Wood G. W., J. Chem.
Soc., 1953, 2009.
2. S h о p p e e C. W., Summers G. H. R., J. Chem. Soc., 1950, 689.
АЛЮМИНИЯ ОКИСЬ ДЛЯ ЭТЕРИФИКАЦИИ [1]. Следующий
простой и быстрый метод удобен при получении от 20 мг до 20 г
сложного эфира. К бензольному раствору спирта, например эрго-
стерина, добавляют небольшой избыток бензоил хлорида или п-фе-
нилазобензоилхлорнда и 2 же пиридина. По окончании реакции
смесь пропускают через небольшую колонку с окисью алюминия
(второй степени активности) и после упаривания бензола добавляют
толуол для азеотропной отгонки пиридина на кипящей водяной бане.
1. Contributed by MolauG. Е. Dow Chemical Co.
АЛЮМИНИЯ ОКИСЬ, ОКРАШЕННАЯ МОРИНОМ [1].
Раствор 300 мг морина (3,5,7,2',4'-пентаоксифлавон) в 500 мл
метанола добавляют при перемешивании к суспензии 500 г А. о. в
500 мл метанола. После обесцвечивания раствора А. о. собирают и
сушат 2 час при 150 . Получается лимонно-желтый препарат, имею-
щий вторую степень активности [2]. Колонка с этим адсорбентом
флуоресцирует в УФ-свете (приблизительно 360 ммк), и бесцветные
вещества, которые поглощают в ближнем УФ, наблюдаются на ко-
лонке в виде темных полос на светящемся фоне. Этот препарат при-
меняют для разделения природных полиненасыщенных диолов 131.
I. Brock mann Н., V о 1 р е г s F., Chem. Вег., 80, 77 (1947).
2. Brockmann Н., Schodder Н., Вег., 74, 73 (1941).
3. Anet Е. F. L. J. et al., J. Chem. Soc., 1953, 309.
АЛЮМИНИЯ ХЛОРИД, A1C13. Мол. вес 133,35. (См. также
Натрия алюмотетрахлорид.)
Во многих методиках не указано, необходимо ли использовать
свежевозогнанный А. х. или можно брать продажный тонкоизмель-
41
ченный с избытком, учитывая присутствие в реагенте инертных при-
месей.
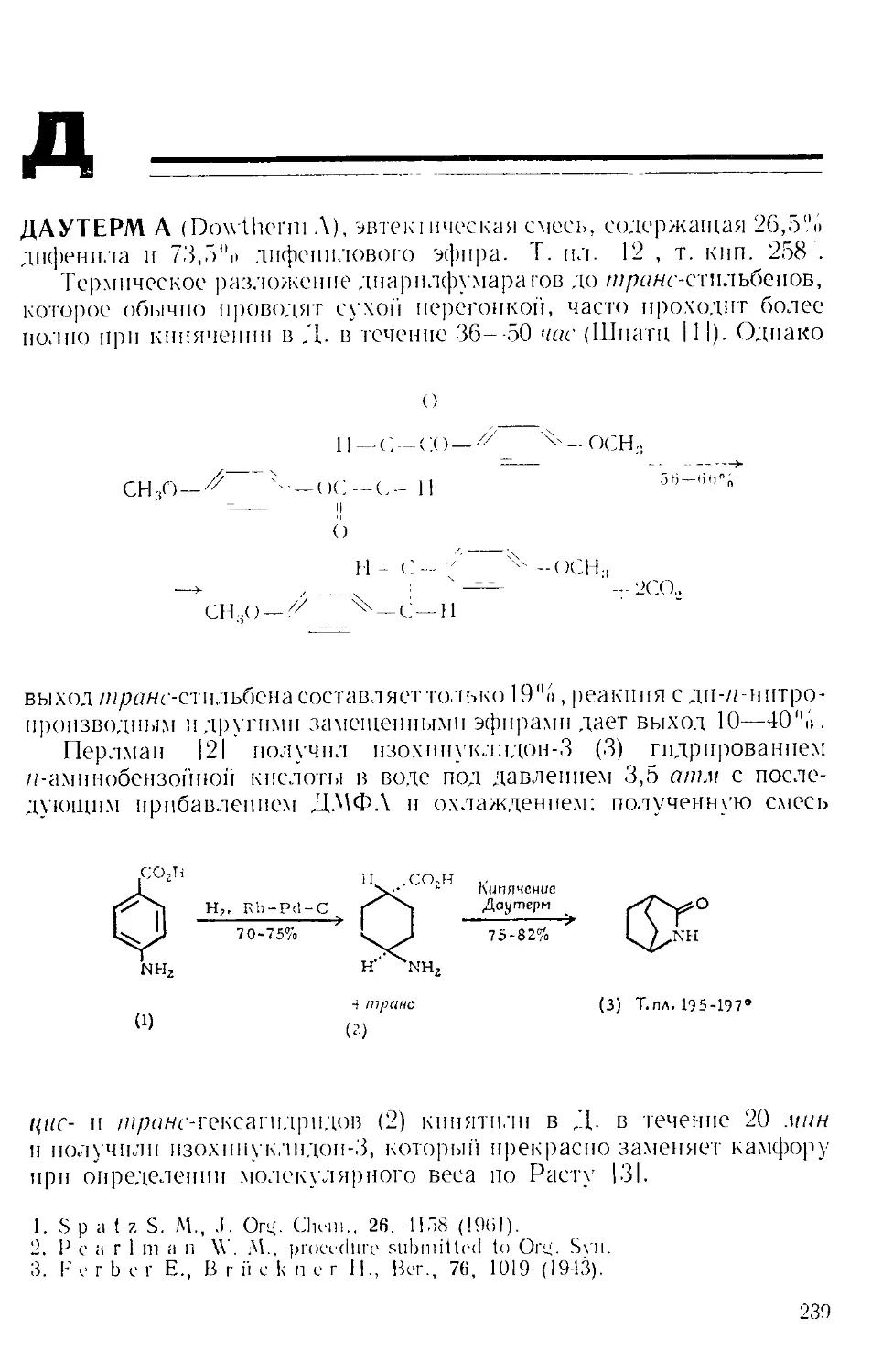
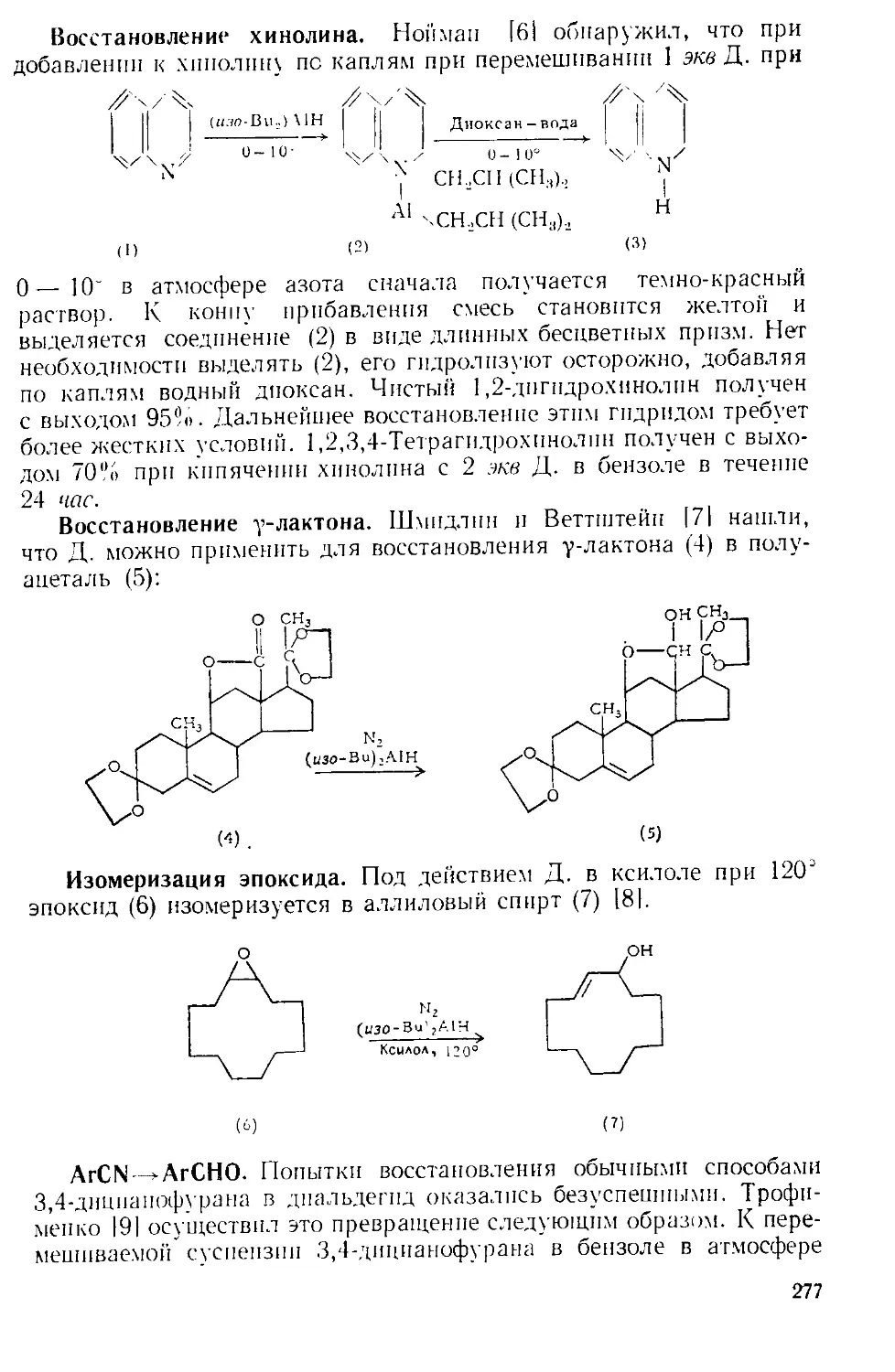
Техника эксперимента. Во многих реакциях А. х. добавляют
к раствору небольшими порциями, избегая контакта с влагой возду-
ха. Эту операцию удобно проводить следующим образом. А. х. взве-
шивают в колбе Эрленменера и соединяют ее отрезком широкой труб-
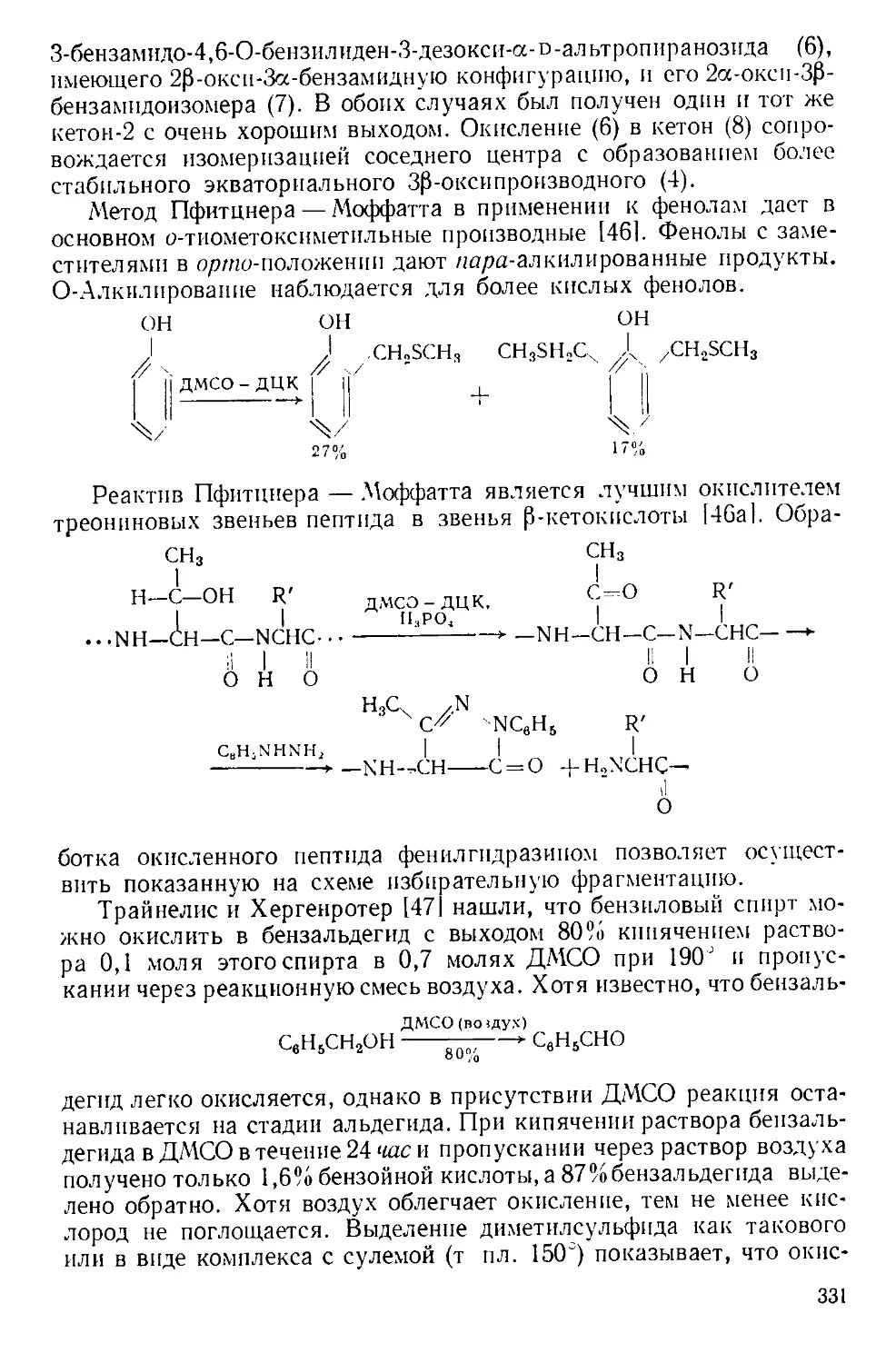
ки с горлом реакционной колбы, как показано на рис. А-2 111.
Рис. А-2. Способ добавления твер-
дого реагента.
Рис. А-3. Ловушки для по-
глощения газов.
Для постепенного добавления реагента колбу поднимают, по-
стукивая по ней пальцами.
В большинстве реакций, катализируемых А. х., выделяется га-
зообразный хлористый водород. Поэтому всегда, за исключением
работы с микроколичествами, желательно соединить верх обратного
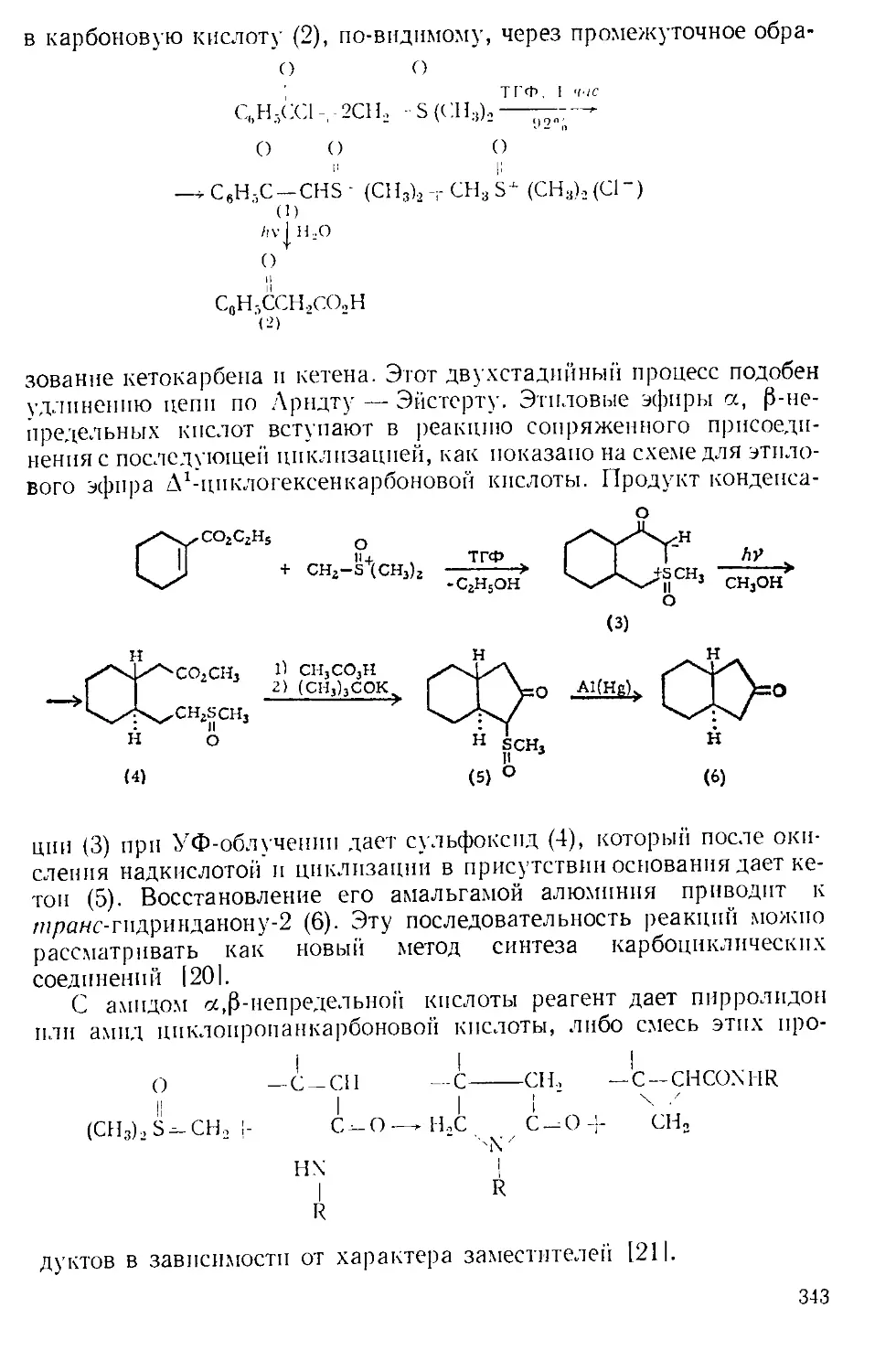
холодильника с какой-либо из газовых ловушек, изображенных
на рис. А-3 12,31.
Реакционная смесь часто сильно загустевает, поэтому ось ме-
шалки должна быть жестко соединена с мощным мотором; магнит-
ные мешалки неприменимы [4|. Пирсон [51 рекомендует применять
мешалку из тефлоновой пластинки, вырезанной в форме полумесяца
и прикрепленной па штифте к нижнему концу оси.
Для многих реакций Фриделя — Крафтса предпочитают нитро-
бензол, хороший растворитель для большинства органических со-
единений, образующий растворимый комплекс с А. х. Так, 532 г
А. х. добавляют порциями по 50 г при перемешивании и охлаждении
к 2,1 л нитробензола; реагент растворяется и остается в растворе
42
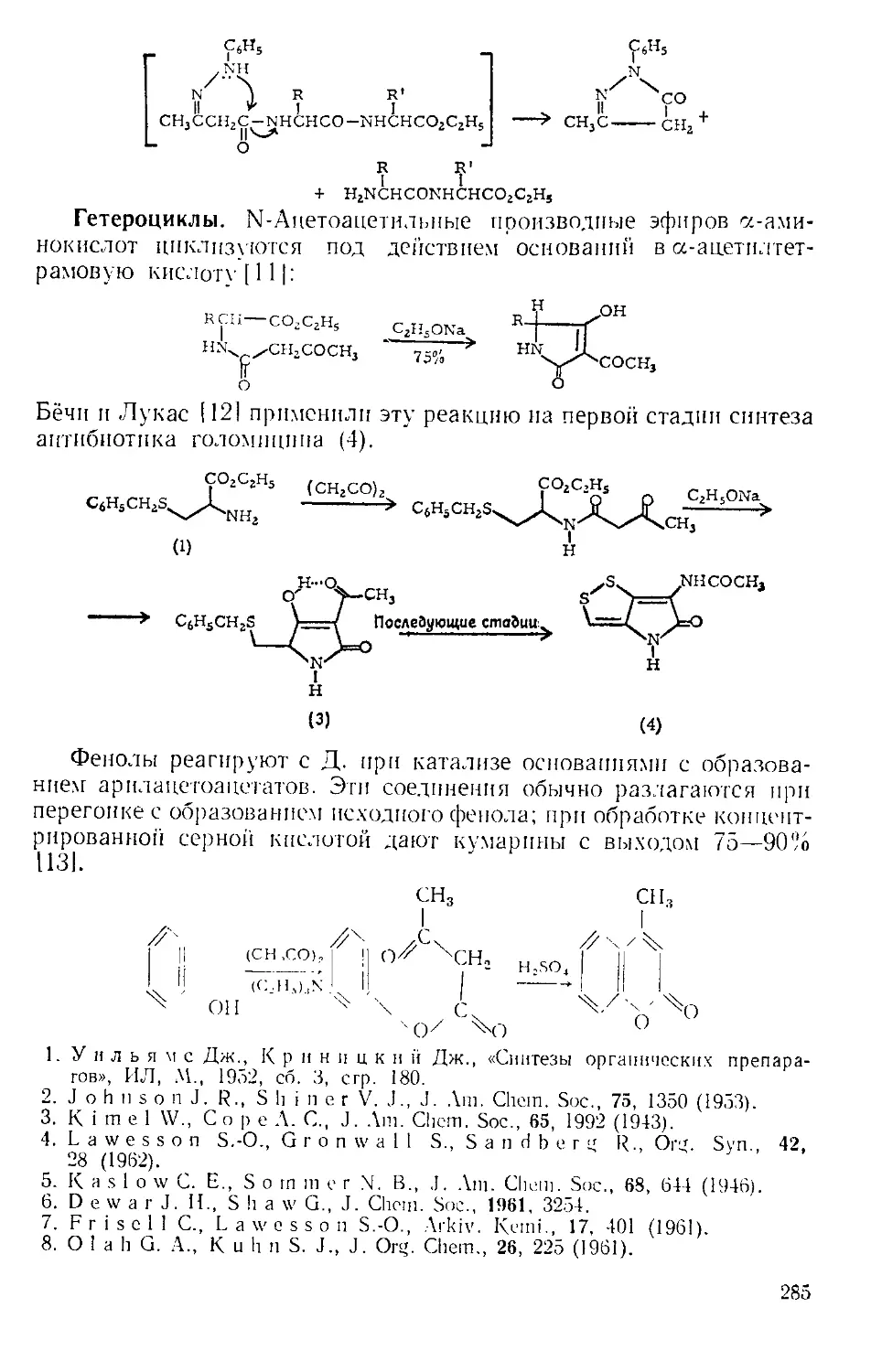
при комнатной температуре |61. По окончании реакции нитробензол
можно легко удалить с помощью прибора, изображенного на рис.
А-4 171. Небольшая круглодонная колба служит ловушкой, в кото-
рой пар барботирует через постоянно имеющееся небольшое количе-
ство жидкости. В таком приборе можно отгонять нитробензол с па-
ром со скоростью 400 г в час.
Рис. А-4. Прибор для перегонки с паром.
Реакции Фриделя — Крафтса. Синтез ди-шре/п-бутилбензола [8]
алкилированием бензола 2 же т^т-бутилхлорида наглядно иллю-
стрирует необходимость применения только каталитических коли-
честв А. х. и относительно легкое выделение из сложной смеси сим-
метричного продукта. Смесь mpem-бутилхлорида и бензола охлажда-
ют до 0—3° в колбе Эрленмейера на 125 мл с боковым отводом, ко-
торый соединяют с водоструйным насосом для поглощения хлори-
стого водорода, и при перемешивании добавляют четвертую часть
А. х., взвешенного в маленькой пробирке. Примерно через 2 мин
(индукционный период) при этой температуре начинается энергич-
ная реакция и оставшийся катализатор добавляют в три приема с
интервалом около 2 мин. По окончании реакции получающийся
43
продукт выпадает в виде белых кристаллов. Реакционную смесь
+ А1С13
1 г
---->
41%
оставляют на 5 мин без охлаждения, после чего обрабатывают водой
со льдом. После экстракции эфиром и перекристаллизации из 20 мл
метанола получают красивые иглы или пластинки чистого вещества
(8,4 г, 41%). Небольшую порцию продукта (0,8 г) можно выделить
из маточного раствора через соединение включения с тиомочевиной
(3,2 г).
Ацилирование по Фриделю — Крафтсу обычно резко тормозится
после введения одного заместителя ввиду его дезактивирующего дей-
ствия. Выходы при ацилировании, как правило, высокие, что можно
проиллюстрировать следующим примером 19]. При ацилировании
ангидридом требуется применение 2 же А. х. После разложения сме-
0,68 моля 4,35 моля
+ А1С13
1,5 моля
До 90°
96%
си льдом и соляной кислотой толуол отгоняют с паром, водный раст-
вор декантируют и сырую кислоту растворяют при нагревании на
кипящей бане (10 мин) в разбавленном растворе соды. Подкисление
фильтрованного раствора дает моногидрат кислоты, который сушат
до постоянного веса и получают чистую безводную о-толуилбензой-
ную кислоту.
Вышеуказанную процедуру выделения можно использовать и
для разделения изомеров, например при ацилировании аценафтена
янтарным ангидридом в нитробензоле при низкой температуре [10].
После отгонки растворителя с паром кислотные продукты растворя-
ют в горячем растворе соды. Раствор фильтруют и насыщают хло-
ристым натрием при кипячении. При этом избирательно кристалли-
зуется натриевая соль 3(3'-аценафтеноил)-пропионовой кислоты и,
44
таким образом, получается очень чистая 3-кислота. Для метиловых
эфиров соотношение растворимостей обратное, поэтому этерифика-
ция кислот в маточном растворе позволяет легко получить чистую
1-кислоту.
О, 65 МОЛЯ
снгсо
I 7D+AICI3 + C6H5NO2
СН2с6
О, 72МОЛЯ 1f46 моля 600 ЛЛ
В некоторых случаях один из изомеров можно выделить из сме-
си в виде комплекса с А. х. При получении 1-бензоилнафталина по
Перье [11] 14 г А. х. добавляют порциями при встряхивании к 14 г
хлористого бензоила и по окончании экзотермической реакции оса-
док полностью растворяют при осторожном нагревании на пламени
горелки. Образующийся желтый расплав комплекса при охлажде-
нии затвердевает в желтое или оранжевое кристаллическое веще-
ство, которое при перемешивании и слабом нагревании растворяют
в 80 мл сероуглерода. К охлажденному раствору добавляют порция-
ми 12,8 г нафталина. Взаимодействие сопровождается обильным вы-
делением хлористого водорода, но незначительным тепловым эф-
фектом. По окончании добавления смесь быстро нагревают для -за-
вершения реакции и тщательно охлаждают льдом. Комплекс А. х.
с I-бепзоилнафталином выделяется в виде темного масла, которое
кристаллизуется при растирании. Его отделяют, промывают и до-
бавляют порциями к разб. соляной кислоте для выделения 1-бен-
зоилнафталина; т. пл. 76°, выход 18,0 г (78%). Этот продукт достаточ-
но чист, так как образующийся в реакции 2-бензоилнафталин дает
с А. х. более растворимый в сероуглероде комплекс, который остает-
ся в маточном .растворе вместе с другими примесями. В реакции
Фриделя — Крафтса хлористого бензоила с фенантреном также по-
лучается смесь изомеров, однако 1-бензоилфенантрен легко выделя-
ется из смеси через комплекс Перье и получается в чистом виде
с выходом 8% 112].
При низкотемпературном ацетилировании антрацена в бензоле
образующийся красный комплекс отделяют на фильтровальной во-
СОСН3
I
z^Xz^X/^ Z^Az^x/X
I I || | + CH3COC1+ A1C13 - 5°до+10’ | | || |
1 I 1 1 1,68 моля 0,56 шля I I И I
A/'X/\7 57-60% 'Ч/'Ч/х/
0,28 моля в
320 лм CeHe
45
ронке с пористым стеклянным фильтром [13] и разлагают перемеши-
ванием с соляной кислотой и льдом, твердый осадок собирают и
длительно кипятят в 95%-ном спирте. Затем смесь быстро охлажда-
ют до комнатной температуры и отфильтровывают пушистый осадок,
который состоит главным образом из непрореагировавшего антра-
цена. Фильтрат вновь нагревают для растворения небольшого коли-
чества выпадающего осадка и оставляют медленно охлаждаться для
кристаллизации 9-ацетилантрацена. В этой методике не дано обос-
нования использованию пятикратного избытка хлористого ацетила.
Реакции типа Фриделя — Крафтса. Конденсацией бензола с
у-бутиролактоном получают с высоким выходом а-тетралон [14] в од-
600 г (4,5 моля)
1л 1,21 моля
А1С1 1<ипячение 16 час
91-96%
ну стадию вместо возможного сукциноилирования с последующим
восстановлением и циклизацией (см. также у-Бутиролактон). Ко-
личество А. х. берут на основании следующих данных:
Л1С13, г 400 500 600 700
Выход, % 68 86 94 95
Реакция Фриса иллюстрируется превращением диацетата гидро-
хинона в ацетил гидрохинон. По улучшенной методике [15] реакцию
проводят в две стадии. Диацетат гидрохинона измельчают (сито
ОСОСНз
I
А
| | + А1С13
3,0 моля
I
ОСОСНз
1,23 моля
1 15—120°
ОСОСНз
он
уч/сосн,
и
он
5% HCI в CHjOH | 75»;,
он
/СОСНз
ОН
20 меш), тщательно смешивают с А. х. и тремя порциями добавляют
при перемешивании в химический стакан, нагреваемый на масляной
бане. Через 20 мин смесь охлаждают, растирают в ступке и при пе-
ремешивании добавляют в воду со льдом. Полученный 2-ацетилгид-
46
рохннон, загрязненный моноацетатом ацетилгидрохинона и сле-
дами исходного вещества, промывают, сушат и перемешивают с
5%-ным раствором НС1 в метаноле. При выливании смеси в воду со
льдом ацетилгидрохипон выпадает в виде ярко-желтых кристаллов
с т. пл. 203—204 (чистое вещество плавится при 205—206:); гид-
рохинон остается в маточном растворе.
Реакция РС1;! с бензолом, приводящая к фенилдихлорфосфину
116], подобна алкилированию по Фриделю— Крафтсу. В этой ре-
| | + РС13 + А1С13
1,2 моля 0,4 моля
0,3 моля
/РСР-АЮз
Кипячение 3 час Н
“на । II
+ А1С13- РОС13
(3)
акции необходим по крайней мере 1 моль А1С13, поскольку он обра-
зует с конечным продуктом комплекс (1). При добавлении эквива-
лентного количества хлорокиси фосфора осаждается мелкозерни-
стый комплекс (3) и освобождается соединение (2), которое выделяют
экстракцией петролейным эфиром и перегонкой.
Карбоксилирование. Реакция мезитилена с оксалилхлоридом,
катализируемая К. х., приводит к триметилбензойной кислоте [171
Н3С СН:;
\/\/ ,л
I I] 10— КИПИЧС1
-bClCOCOCl + AlCl34-CS.,--------------
II 76%
Г
CH;i
1 моль 1,1 молл 1,1 моля 700 МЛ
со.,н
I “ СН3
I
сн3
и, по-видимому, включает стадии моноацилирования, гидролиза
и декарбонилирования. Однако последовательность двух последних
стадий не ясна. Диацилирование оксалилхлоридом достигается
47
при его взаимодействии с большим избытком М,М-диметиланилина[’4].
5°, затем кипячение
2C6H5N (СН3)2 + C1COCOCI -|- А1С13 -|- CS, -38_47~-”
1,5 моля 0,25 моля 1 моль 200 .мл ~ 0
О о
(CH3)2N —/ *>-С-С—N (СН3) 2
Реакции присоединения. При кипячении с А. х. наблюдается
присоединение хлороформа к тетрахлорэтилену с образованием
Кипячение 15 час
СС12 = СС12+ СНС13 + А1С13 ---------93О.----> СС13СС12СНС12
1 моль 2,5 моля 0,2 моля '°
«есгыьи-гептахлорпропана. После охлаждения смесь обрабатывают
льдом, органический слой отделяют, промывают, сушат и перего-
няют (т. кип. 110—1137Ю мм,т. пл. 29—30 ) [18]. Интересно отме-
тить, что тетрахлорэтилен не склонен к присоединению дихлоркар-
бена, получаемого из трихлорацетата натрия (см. Трихлоруксус-
ной кислоты натриевая соль).
. А. х. катализирует. присоединение бензола- к бензальацетофе-
нону с образованием р, ₽-дифенилпропиофенона ]191 и анилина к
бензонитрилу с образованием N-фенплбензамидина [201.
10°, затем 25°
сг,н5сн=снсосг>п5-сс6нв aici3 —
0.58 моля в 3.00 ли 1,7 л 1,2 моля /п
бензола
—> (СбН5)л СНСН2СОС6Н5 NH
30 мин при 200° ||
CfiH5C^N+C6H5NH2 + А1С13 ————>c6h5cnhc6h5
0.66 моля 0,67 моля 0,67 моля /0
Изокапроилхлорид присоединяется к ацетилену с образованием
Р-хлорвинилизоамилкетона. Четыреххлористый углерод насыщают
(СН3)2СНСН,СН2СОС1--НСе-СН -I- А1С13 + CCI.,______>
0,63 Моля Избыток 0,74 моля 200 г 56 64%
о
I!
—> (СН3)Д .НСНоСНХСН = СНС1
ацетиленом, прибавляют А. х., перемешивают и добавляют по кап-
лям хлорангидрид с одновременным пропусканием тока ацетилена
1211.
Циклодегидратация. В синтезе 4-метилкумарина [6] по методике,
которую предложили Сефна, Шах и Шах [22], смесь фенола, ацето-
уксусного эфира п нитробензола нагревают до 100° при перемеши-
вании, обрабатывают раствором А. х. в нитробензоле в течение
48
30—45 мин и нагревают 3 час при 130°.
СН3
СО СН3
2 моля 2 моля 300 л/л 4 моля в 2,1
нитробензола
Реакцией бензиловой кислоты с А. х. в бензоле получают флу-
оренкарбоновую-9 кислоту [231, при этом происходит циклодегид-
ратация с перегруппировкой.
Л,?, меля
Циклизация.- Под действием А. х. фенилацетилизотиоцианат
циклизуется в 2а-тиогомофталимид [241.
СН2СОС1 сн2
22%N2; А/Хсо+aici3+cs2 22222Х
61—79% | || | 0,22 моля 150 л 52~75%
W N
о
S
0,2 моля
II
S
Расщепление эфирной связи. А. х. является классическим реа-
гентом для деметилирования: АгОСН3+А1С13-*АгОА1С1.2+СН3С1.
Методика, описанная для деметилирования диметилового эфира
3-бромпирокатехина [251, применима также к 2,3-диметоксидифени-
Вг Вг
| ОСНз | ОН
/ A1CI, в С„Н„С1 //\/
I Л Кипячение 3.5 час / и7
I II 80% I I
\/\ ^/\
ОСНз ОН
лу 126]. В качестве растворителя можно использовать также бензол,
но при этом необходимо более длительное кипячение 127).
А1С1д в СвНя
Кипячение 6 час
CCLH
Разработан метод синтеза о-оксибензофенонов (Кулька ]281), в
котором защита пара-положен и я в метиловых эфирах фенолов
трстп-бутильной группой обусловливает о-бензоилирование. Под
действием А. х. /npem-бутильная группа отщепляется вместе с эфир-
ной.
А1С15 а СЬНЬ
65-70° 45WC
79%
Ланге 1291 осуществил гладкое деметилирование ванилина в про-
токатеховый альдегид путем добавления 4,4 же пиридина к суспен-
зии хлористого алюминия (1,1 экв) в растворе ванилина (1 экв) в
хлористом метилене и кипячения смеси в течение 24 час. Обычным
способом ванилин деметилируется неудовлетворительно, а данный
метод пригоден только для соединений, имеющих рядом с эфирной
группой свободную гидроксильную группу. Поэтому можно счи-
тать, что эта реакция включает промежуточное сольватное цикличе-
ское пятичленное соединение. Невозможность деметилирования
сно
а-Ванилин
о-ваиилина в этих условиях объясняется преимущественным ком-
плексообразованием атома алюминия с соседним карбонильным кис-
лородом, более основным, чем метоксильный атом кислорода.
Хардеггер исотр. (301 нашли, что алкоксильные группы в орто-
или п^н-положении к карбонильной группе селективно расщеп-
50
ляются при обработке А. х. в нитробензоле. При добавлении
А. х. к раствору этого соединения в нитробензоле при комнат-
ной температуре образуется красный или фиолетовый комплекс.
Через несколько часов к смеси добавляют лед и небольшое количе-
ство соляной кислоты. При этом комплекс переходит в водный
слой, который отделяют и нагревают с разбавленной соляной кис-
лотой при 50е в течение 2 час; освобождающийся продукт демети-
лирования выделяется при охлаждении раствора. Карбметоксиль-
ная группа в этих условиях остается неизменной.
Реакция Гаттермана — Коха. Для превращения толуола в п-
толуиловый альдегид его обрабатывают окисью углерода и хлори-
стым водородом при перемешивании с суспензией А. х. и хлористой
меди (I) [31].
СН3 СН3
I I
2,17 моля Избыток 2 моля 0,16 моля 1
сно
Катализатор реакции Дильса — Альдера. Некоторые реакции
Дильса — Альдера заметно ускоряются в присутствии А. х. [32,
331 и других катализаторов, используемых в реакции Фриделя —
Крафтса [331 (SnCI4, BF3, FeCl:i, TiCl4). Так, реакция с эквимоляр-
ными количествами антрацена, диметилфумарата и А. х. в хлористом
метилене полностью заканчивается при комнатной температуре ме-
нее чем за 2 час; в отсутствие катализатора при кипячении в
диоксане для завершения реакции требуется 2—3 дня [32]. Без
катализатора реакцией бутадиена с метилвинилкетоном получают
4-ацетилциклогексен с выходом 75—80% при нагревании реагентов
в запаянной трубке при 140° в течение 8—10 час; в бензольном же
растворе, содержащем 0,18 же хлорного олова (IV), аддукт полу-
чается с выходом 73% за 1 час при комнатной температуре [331.
Однако катализ в реакции Дильса — Альдера приводит к удов-
летворительным результатам только с некоторыми диенофилами,
51
такими, как два указанных выше, а также акролеин и акриловая
кислота. Другим ограничением является полимеризация некоторых
диенов в присутствии катализаторов реакции Фриделя — Крафтса.
Катализатор бромирования. А. х. в каталитических количествах
промотирует бромирование метильной группы ацетофенона с обра-
зованием бромацетофенона [34]. Эфир, и растворенный бромистый
СОСНз СОСН,Вг
А
I II + Вг, + А1С13 + Эфир--------||
0,42 моля 0,5 г 50 м.1 64 — 66%
0,42 моля
водород удаляют при пониженном давлении и оставшийся темно-
коричневый кристаллический осадок промывают встряхиванием
со смесью воды и эфира (1 : 1) до обесцвечивания. После перекри-
сталлизации из метанола получают бромацетофенон удовлетвори-
тельной чистоты.
Бромированием ацетофенона в присутствии большого избытка
А. х. (2,5же) без растворителя получают с хорошим выходом лг-бро.м-
ацетофенон (Пирсон 15, 351). К А. х. при перемешивании мощной
СОСНз СОСНз
I
| |U Вг2 Д- А1С13 2ААА%| ||
0,80 моля 1,65 моля '6 76%
0,67 моля Вг
мешалкой в течение 20—30 мин добавляют ацетофенон. Наблюдает-
ся разогревание, но температура не должна превышать 180А После
добавления трети необходимого количества ацетофенона смесь прев-
ращается в комок, однако к концу добавления она вновь расплавля-
ется и легко перемешивается. Затем в течение 40 мин прибавляют по
каплям бром и перемешивают еще 1 час при 80—85э до затвердева-
ния смеси. Продукт выделяют разложением комплекса льдом и со-
ляной кислотой. В дальнейшем «эффект избытка катализатора»
Пирсон [36] с успехом использовал для галогенирования в ядро
эфиров, хлорангидридов и нитрилов ароматических кислот. Напри-
мер, бромированием метилбензоата в виде комплекса с А. х. (2 же
А1С13) при 65° получают метиловый эфир л-бромбензойной кислоты
с выходом 86%.
Такое поразительное влияние избытка катализатора на направ-
ление бромирования можно понять на основании результатов иссле-
дования Хауза [37], полученных при изучении циклизации хлор-
ангидрида транофенилциклогексанкарбоновой-2 кислоты (1) в при-
сутствии А. х. с образованием цис- и /иранс-гексагидрофлуоренонов,
формулы (2) и (6). транс-Кетон [(2), т. пл. 92°] менее стабилен, чем
52
цис-изомер 1(6),т. пл. 4Г], и когда А. х. добавляют к раствору хлор-
ангидрида кислоты в сероуглероде, то получают стабильный цис-
кетон (6). Считают, что первоначально образующийся /иранс-кетон
(2) превращается в сопряженную кислоту (3), которая переходит в
енолят (4) при отщеплении протона одним из основных агентов ре-
акционной среды (не связанные в комплекс кетон (2) или хлорангид-
рид (1)). Появившийся в равновесии енолят (4) преимущественно
переходит не в (3), а в цис-сопряженную кислоту (5), которая с от-
щеплением А. х. дает стабильный цис-кетон (6). Если же хлорангид-
рид (1) добавляют к хлористому алюминию, который, таким обра-
зом, постоянно находится в избытке, то после разложения комплек-
са водой получают менее стабильный тдаис-кетон (2). В этом слу-
чае также возможно образование сопряженной кислоты (3) с неиз-
мененной первоначальной конфигурацией. Однако изомеризация
невозможна ввиду недостатка основных агентов для отщепления
протона и превращения в енолят (4). Таким образом, эффект избытка
катализатора при бромировании ацетофенона объясняется следую-
сн3 сн3 сн2
С6Н5С=О AIC‘3 > С6Н5С=О+А1С13‘ //У С4Н5С—OAICIj" + н+
(7) (8)
щим образом. Избыток катализатора превращает кетон в сопряжен-
ную кислоту (7) и связывает все вещества основного характера,
нужные для превращения в енолят (8), необходимый для бромирова-
ния в боковую цепь. Кроме того, избыток катализатора повышает
электрофильную активность брома благодаря образованию
комплекса Br+ [AlCl3Brj~.
1. F i е s е г L. F., «Experiments in Organic Chemistry», 3rd Ed., D. C. Heath 1955,
265.
2. Аллен К., «Синтезы органических препаратов», ИЛ, М., 1949, сб. 2,
стр. 77.
3. Джонсон Дж., «Синтезы органических препаратов», М., ИЛ, 1949, сб. 1,
стр. 100.
4. Tiiziin С., Ogliaruso М., Becker Е. I., Org. Syn., 41, 1 (1961).
5. Pearson D. E., Pope H. W., Hargrove W. W.. Org. Syn., 40, 7
(1960).
53
6. Вудрафф Э., «Синтезы органических препаратов», ИЛ, М., 1952, сб. 3,
стр. 305.
7. F i е s е г L. F., «Experiments in Organic Chemistry», 3rd Ed., D. C. Heath,
1955, 256 (см. «Синтезы органических препаратов», сб. 3, стр. 85, ИЛ, М.,
1952).
8. Org. Expts., 184.
9. Ф и з е р Л., «Синтезы органических препаратов», ИЛ, М., 1949, сб. 1, стр. 389.
10. Ф и з е р Л., «Синтезы органических препаратов», ИЛ, М., 1952, сб. 3,
стр. 84.
11. Perrier G., Bull. soc. chirn. France, [3|, 31, 859 (1904); F i e s e r L. F.,
«Experiments in Organic Chemistry», 2nd ed., 1941, 192.
12. В a c h m a n n W. E., J. Am. Chem. Soc., 57, 555 (1935).
13. Мер p и т т 4., мл., Б p a v н 4., «Синтезы органических препаратов», ИЛ,
М„ 1953, сб. 4, стр. 71.
14. О 1 s о n С. Е., В a d е г A. R., Org. Sym., Coll. Vol., 4, 8 (1963).
15. Reynolds D. D., Cathcart J. A., W i 1 1 i a m s J. L. R., J. Org.
Chem., 18, 1709 (1953).
16. Бюхнер В., Л о к x a p т Л., мл., «Синтезы органических препаратов»,
ИЛ, М., 1953, сб. 4, стр. 510.
17. S о к о 1 Р. Е., Org. Syn., 44, 69 (1964).
18. Ф а р л о у М., «Синтезы органических препаратов», ИЛ, М., 1949, сб. 2,
стр. 153.
19. Ill и л ь д н е к П., «Синтезы органических препаратов», ИЛ, М., 1949, сб. 2,
стр. 235.
20. Купер Ф., Партридж М., «Синтезы органических препаратов», ИЛ,
М., 1958, сб. 8, стр. 62.
21. Прайс Ч., П а п п а л а р д о Дж., «Синтезы органических препаратов»,
ИЛ, М.. 1953, сб. 4, стр. 529.
22. Set h n a S. М., Shah N. M., Shah R. C., J. Chem. Soc., 1938, 228.
23. Рихтер Г., «Синтезы органических препаратов», ИЛ, М., 1954, сб. 5,
стр. 86.
24. S m i t h P. A. S., Kan R. O., Org. Syn., 44, 91 (1964).
25. Mason H. S., J. Am. Chem. Soc., 69, 2241 (1947).
26. В г и c e J. M., S u t c 1 i f f e F. K-, J. Chem. Soc., 1955, 4435.
27. P e t t i t G. R., P i a t a k D. M., J. Org. Chem., 25, 721 (1960).
28. К u I k a M., J. Am. Chem. Soc., 76, 5469 (1954).
29. L a n g e R. G„ J. Org. Chem., 27, 2037 (1962).
30. H a r d e g g e r E., Widmer E., Steiner К., P f i f f n e r A., Helv.
Chim. Acta, 47, 2027, 2031 (1964).
31. Колеман Дж., Крэг Д., «Синтезы органических препаратов», ИЛ, М.,
1949, сб. 2, стр. 464.
32. Y a t е s Р., Е a t о n Р., J. Am. Chem. Soc., 82, 4436 (1960).
33. F г а у G. I., Robinson R., .1. Am. Chem. Soc., 83, 249 (1961).
34. Коупер P., Дэвидсон Л., «Синтезы органических препаратов», ИЛ,
М„ 1949, сб. 2. стр. 116.
35. PearsonD. Е., Р о р е Н. W., Hargrove W. W., Stamper W. Е.,
.1. Org. Chem., 23, 1412 (1958).
36. PearsonD. E., S t a m p e г W. E., S u t h e r s B. R., J. Org. Chem., 28,
3147 (1963).
3/. House H. O., Par a g a m i a n V., R о R. S., W 1 u k a D. J., J. Am.
Chem. Soc., 82, 1457 (1960).
АМИД КАЛИЯ, KNH,. Мол. вес 55,13.
Осторожно! А. к. огнеопасен, самовоспламеняется под действием
влаги. Остатки реагента следует уничтожать путем осторожной об-
работки этанолом или изопропанолом. Реакции с жидким аммиаком
необходимо проводить в вытяжном шкафу.
54
Получение, бензилирование нитрила. Методика получения
А. к. Ill, которую проф. Хаузер более подробно объяснил в своем
письме, состоит в следующем. Реакцию калия с жидким аммиаком
проводят в обычной трехгорлой колбе на 1 л, снабженной обратным
воздушным холодильником (без осушительной трубки), механиче-
ской мешалкой с шаровым затвором и капельной воронкой. Охлаж-
дения снаружи не требуется, поскольку испарение жидкого аммиа-
ка обеспечивает достаточное охлаждение для проведения реакции,
которую можно завершить за 1—3 час, в этом случае вполне приго-
ден воздушный холодильник. За 3 час объем реакционной смеси
уменьшается примерно на одну треть. В случае более длительного
времени реакции можно воспользоваться конденсатором со смесью
твердой углекислоты и ацетона. Атмосфера азота необходима толь-
ко при проведении реакции при температуре —78' для предохране-
ния реагента от влаги воздуха.
В колбу через трубку вводят из баллона 500 мл безводного жид-
кого аммиака и при перемешивании вносят 0,5 г калия, который
тщательно протирают фильтровальной бумагой после извлечения
из керосина. Как только жидкость окрасится в голубой цвет, до-
бавляют около 0,25 г гидрата азотнокислого окисного железа, а за-
тем продолжают прибавлять кусочки калия по 0,5 г (всего 9 г,
0,23 моля). Исчезновение голубой окраски примерно через 20 мин
указывает на полное превращение калия в А. к. В отличие от амида
натрия и амида лития, которые дают суспензию в жидком аммиаке,
А. к., по-видимому, в основном находится в растворе, хотя раствор
мутный п некоторые исследователи принимали его за суспензию.
Следующая стадия состоит в добавлении 0,23 моля дифенилаце-
тонитрила и перемешивании полученного зеленовато-бурого раст-
вора в течение 5 мин. Затем за 10 мин добавляют раствор 0,24 моля
хлористого бензила в 100 мл абсолютного эфира, при этом раствор
принимает оранжевую окраску. После перемешивания в течение
1 час аммиак испаряют на кипящей водяной бане п добавляют 300 мл
KNH-. — С,Н5СН.С1
(c6h5).,chcn —(cei-L,)„c-cN (к+)-------
NH, 115—99%
(С6Н5), ССН2СвН5
CN
эфира. После прибавления 300 мл воды в осадок выпадает а,а,Р-
трифеннлпропионптрпл. Эфир отгоняют и неочищенный светло-ко-
ричневый препарат отделяют и перекристаллизовывают из этанола;
две порции (т. пл. 126, 5—127,5 ) дают выход 95—9796.
А. к. получают также (Беннет и corp. |2|) на специальной уста-
новке, состоящей из двух колб на 5 л, причем каждая снабжена ме-
шалкой и конденсатором, охлаждаемым смесью твердой углекисло-
ты и изопропанола. Обеспечена продувка системы азотом и возмож-
ность переливания раствора из одной колбы в другую под давлени-
ем азота.
55
Дегидробромирование. Спирты и бромиды типа (1) склонны
к скелетной перегруппировке с превращением в олефины, причем
(особенно в присутствии кислых реагентов) преимущественно об-
разуются олефины типа (3) с внутренним положением двойной свя-
зи, а не терминальные типа (2). Хаузер и сотр. 131 нашли, что при
обработке соединения (1) менее чем 1 экв А. к. в жидком аммиаке
получается а-олефин (2), однако при избытке А. к. этот продукт
KNH.
С6Н5СНСН2Вг----->CeHsC=CH, CSH-,CH=CHCH2CH3
I ' I
сн2сн3 сн2сн3
(1) (2) (3)
l-Ji С0Н5ССН3
II
снсн3
(4)
изомеризуется в олефин с внутренним положением двойной связи
(4). Перегруппированный продукт (3) образуется в незначительных
количествах.
В модифицированном расщеплении по Гофману при отщеп-
лении НВг от триметилциклооктиламмонийбромида под действием
фениллития получается смесь олефинов, содержащая 80% цис-
циклооктена (Виттиг и Полстер [4]). Под действием А. к. в жидком
аммиаке преимущественно образуется трянс-циклооктен.
80% цис
85% транс
Перегруппировка Виттига: бензиловый эфиркарбинол. Виттиг
показал, что «дифенан» (1) перегруппировывается в 9-окси-9,10-
56
дигидрофенантрен (4) под действием фенилли-тия в течение 1 недели.
Хаузер и сотр. 15] нашли, что эту реакцию можно осуществить под
действием А. к. в жидком аммиаке за 1 час с выходом 90%.
Образование промежуточного дегидробензола. Общий принцип
синтеза, разработанного Беннетом 12], заключается в получении
промежуточного дегидробензола, в котором нуклеофильный центр
расположен так, что он может присоединяться внутримолекулярно
к тройной связи. Примером может служить синтез 3-ацетилоксин-
дола из о-хлорацетоацетанилида под действием А. к. в жидком амми-
аке. В другом синтезе о- и л/-галогензамещенные изомеры дали
один и тот же продукт.
।
н
1. Н auser С. R., Dunnavant W. R., Org. Syn., Coll. Vol., 4, 962 (1963).
2. H r u t f о r d B. F., Bunnett J. F., J. Am. Chem. Soc., 80, 2021 (1958);
Беннетт Дж. Ф., Хрутфорд Б. Ф., Уильямсон С. М.,
«Синтезы органических препаратов», изд-во «Мир», М., 1964, сб. 12, стр. 7.
3. Н a u s е г С. R., S k е 1 1 Р. S., В г i g h t R. D., R e n f г о w \V. B., J. Am.
Chem. Soc., 69, 589 (1947).
4. W i t t i g G., P о 1 s t e r R., Ann., 612, 102 (1958).
5. W e i n h e i m e r A. J., KantorS. W., H a u s e r C. R., J. Org. Chem.,
18, 801 (1953); H a u s e r C. R., KantorS. W., J. Am. Chem. Soc., 73, 1437
(1951); Hrutfor d B. F., Bunnett J. F., J. Am. Chem. Soc., 80, 2021
(1958).
АМИД ЛИТИЯ, LiNH2. Мол. вес 22,97. Хранят в узкогорлых
желтых склянках с завинчивающейся крышкой, которую тщатель-
но закрывают и заливают парафином. Если склянку открывать на
короткое время только при взятии навески А. л., то титр довольно
постоянен (первоначальный титр 98%) [1].
Получение и использование в альдольной конденсации сложных
эфиров с альдегидами и кетонами. Хаузер и Путербаух [2] добавляли
к 300 мл жидкого аммиака при перемешивании небольшую часть
1,1-граммовой навески литиевой дроби, а затем каталитическое ко-
личество нитрата трехвалентного железа. После исчезновения пер-
воначальной синей окраски небольшими порциями добавляли ос-
тавшуюся часть лития. Приблизительно через 20 мин образовав-
шуюся серую суспензию А. л. обрабатывали эфирным раствором
° 1) LiNHz, NH, СН3 О
II 2) С„Н6СОСНЭ, эфир I II Н.О
СН3СОС(СН3)3---------------> СеН5ССН2СОС(СН3)3
| ' ° %
OLi
ОН О
i II
—► СвН6С(СН3)СН2СОС(СН3)3
57
0,2 моля »1рт-бутилацетата, который добавляли по каплям. За-
тем жидкий аммиак отгоняли, прибавляли 1 же кетона и смесь ки-
пятили до завершения альдольной конденсации. Для конденсации
лучше использовать не амид натрия, а А. л. трет-Бутиловые слож-
ные эфиры также обладают некоторыми преимуществами, так как
они легко расщепляются при пиролизе или под действием кислот.
Дегидратацию до а, р-непасыщенных эфиров можно осуществить
тионилхлоридом пли пиридином. Хаузер и Линдсей [31 осущест-
вили альдольную конденсацию этилацетата в присутствии 2 эхе ос-
нования. Сиспдо |4| сообщил об алкилировании шреш-бутиловых
сложных эфиров.
А. л. используют вместо амида натрия для получения гетероцик-
лических аминов, обладающих антигистаминной активностью [51.
Например, смесь 2-аминопиридпна, бромгпдрата Р-дпметиламино-
этилбромида и 2 экв А. л. кипятили в течение 22 час в толуоле. Кей
/К Вг('.Н,СН2\(СН.|),.НВг Z\
ii [ 2IJNI-L>. толуол । н
II I 68% I II
'У/\ Д " \
N .NH, N ХСН2СН,\'(СН3)2
Н
[11 отметил, что с А. л. работать легче, чем с амидом натрия. Он
получил с очень хорошими выходами ряд гетероциклических вторич-
ных аминов указанного выше типа, которые можно превратить тем
же способом с высокими выходами в соответствующие третичные
амины.
1. К а у е 1. A., J. Am. Chem. Soc., 71, 2322 (1949); К а у е I. А., К о g a n I. С.,
ibid., 73, 5891 (1951); F er г а г о J. J., К а у е I. A., W е i s s U., J. Chem. Soc.,
1964, 2813.
2. H a u s e r C. R., P u t e r b a u g h W. H., J. Am. Chem. Soc., 75, 1068 (1953).
3. H a u s e r C. R., Lindsay.!. K., J. Am. Chem. Soc., 77, 1050 (1955); See
also Hauser C. R., C h a ni b e r s W. ,1., J. Org. Chem., 21, 1524 (1956).
4. S i s i d о К.. К a z. a m a X'., К о d a m a H., N ozaki H., .1. Am. Chem.
Soc., 81, 5817 (1959); SisidoK., Sei K.., N о z a k i H., ,1. Org. Chem., 27,
2681 (1962).
5. Hutt г e г С. P., Djerassi С., В e e a r s W. L., M a у e r R. L.,
Scholz C. R., J. Am. Chem. Soc., 68, 1999 (1946).
АМИД НАТРИЯ, NaNH,. Мол. вес 39,02, т. пл. 210=.
Продажный А. н. можно использовать только сразу же после
вскрытия склянки. При хранении он поглощает влагу, разлагается,
желтеет и становится взрывоопасным. Такой реактив уничтожают
обработкой твердым хлористым аммонием:
Ха\Н . \ Н:С: — > NaC!-i-2XH:l
Ввиду нестабильности реагента методики его применения в боль-
шинстве случаев включают описание способов получения.
Получение. Большая часть лабораторных методик основана на
открытии Ньюленда и сотр. [11, которые обнаружили, что реакция
58
натрия с жидким аммиаком заметно ускоряется черным катализа-
тором, который первоначально получали взаимодействием соли
окисного железа с окпслами натрия в жидком аммиаке. К жидкому
2 N а ~ 2 X Н з —, 2 X а \ И- J- Н,
аммиаку (500 мл) при перемешивании (—33 ) добавляют 0,3 г
Fe(NO3):i-6H.,O, а затем 1 г натрия. При пропускании в смесь воз-
духа первоначальная голубая окраска исчезает и выпадает черный
осадок катализатора. Затем добавляют еще натрий (25 а) отдельными
порциями в течение 20 мин.', голубая окраска исчезает и образуется
серая суспензия А. н. Растворимость полученного таким путем
Л. и. — 1 моль в 1 л жидкого аммиака при —33 .
Позднее было показано, что окисление воздухом начальной не-
большой порции натрия не обязательно, и поэтому просто ждут,
пока прекратится выделение водорода и выпадет черный катализа-
тор, и лишь после этого добавляют основное количество натрия.
Удовлетворительные методики, различающиеся лишь в деталях,
описали Хаузер |2, 3|, Джонс 14] и Кан 15]. Хаузер и некоторые
другие исследователи не применяют ни охлаждающей бани, ни об-
ратного холодильника, а просто по мере испарения добавляют но-
вые порции аммиака; испаряющийся аммиак служит защитой от
кислорода. Как Джонс, так и Кан использовали обратный холодиль-
ник, охлаждаемый сухим льдом, работали в атмосфере азота и при-
меняли ловушку для поглощения газов и барботер. Для получения
катализатора Кан использовал хлорное железо. Обычно лучше при-
менять мешалку Гершберга; мешалка с лопастями из тефлона не ре-
комендуется.
Дегидрогалогенирование (и дебромирование). Суспензию 2 мо-
лей А. и. в 1,5 л жидкого аммиака перемешивают при —33 и по
каплям добавляют 1 моль Р-бромстирола (Ньюленд п сотр. |1|). По
окончании бурной реакции смесь перемешивают еще 10 мин, а за-
С6Н,СН СНВг ;- 2.\а\тН, — СЙН-,С $ СХ’а ——, С6Н5С СН
73%
1 моль 2 моля
тем обрабатывают 600 мл воды. Фенплацетилен образуется с хоро-
шим выходом; часть исходного вещества была выделена обратно.
По такой же методике днбромстнрол в результате дегидробромпро-
вания дает фенплацетилен с выходом 64%. Хотя в некоторых усло-
виях ацетиленовые соединения с внутренним положением тройной
связи могут путем перегруппировки превратиться в аллены и в тер-
минальные ацетилены, сотрудники Университета Нотр-Дам нашли,
что Р-нонин не изомеризуется под влиянием Л. н. в течение 16 час
при —34'.
Для получения цпклогексилпропина (2) Леспио и Бургель 16]
растирали в ступке взятый в избытке продажный А. н. с 200 мл. ми-
нерального масла, перемешивали суспензию ири нагревании на ба-
59
не при 160—165° и по каплям добавляли соответствующий бромистый
винил (1) в течение 1,5 час.
160—165° НС1
CeHnCH,C^CH„ + 2NaNH,---------> С6НПСН„С = СХа---->
“1 о 1 ” 66%
| 3,1 моля и
Вг
(1) 1 моль
—>СвН11СН2С^СН
(2)
Изучая превращение олеиновой кислоты в стеароловую через
стадию дибромида, Кан и сотр. [5] нашли, что при использовании
для дегидробромирования А. н. выход в два раза выше, чем со спир-
товым раствором едкого кали. Суспензию А. н. в жидком аммиаке,
полученную из 1,87 г-атом натрия, перемешивали (конденсатор
с сухим льдом и ацетоном) и медленно из капельной воронки
добавляли эфирный раствор дибромида олеиновой кислоты. По-
СН3(СН2)7СН = СН(СН2)7СО2Н +
0,35 моля Эфир
3NaNH,; НС1
СН3(СН2)7СНСН(СН2)7СО2Н-> - гоп, ; „—г<Н3(СН2)7С^С(СН2)7СО2Н
। । 02— О2 о (ООЩИИ)
Вг Вг
еле непрерывного перемешивания в течение 6 час постепенно добав-
ляли 60 г твердого хлористого аммония для разложения избытка
А. н. После испарения аммиака оставшуюся твердую соль нагрева-
ли с водой в атмосфере азота и подкисляли; после двух кристалли-
заций из петролейного эфира получили чистую стеароловую кис-
лоту (т. пл. 46—46,5°).
Аналогичная методика использована и для превращения А10-
ундециленовой кислоты в А10-ундециновую кислоту [7].
Джонс и сотрудники описали два случая дегидрогалогенирова-
ния, сопровождающегося расщеплением простой эфирной связи.
Один из них — получение этоксиацетилена из диэтилхлорацеталя
[4]. Суспензию А. н. в жидком аммиаке перемешивали от руки в кол-
бе, снабженной холодильником типа «холодный палец» с сухим
льдом, и в течение 15—20 мин добавляли диэтилхлорацеталь. Через
Жидкий NH.3
С1СН2СН(ОС2Н5), 4 3NaNH,
0,50 моля 1.65 г-атом Na
NaCl-H.O
КаС^СОС,Н5--------^НС = СОС„Н6
57—60%
15 мин испаряли еще некоторое количество аммиака в токе азота и
колбу с очень пирофорным натриевым производным охлаждали
до —70° в бане с сухим льдом и трихлорэтиленом. Затем в один при-
ем добавляли насыщенный раствор хлористого натрия, предвари-
тельно охлажденный до —20', и после этого отгоняли легко ки-
60
пящий продукт, собирая его в приемник, охлажденный до —70°.
Дистиллят нейтрализовали водным раствором мононатриевой соли
фосфорной кислоты, водный слой замораживали, охлаждая его сухим
льдом, верхний слой отделяли, сушили и перегоняли. Фракциониро-
ванием получили чистый этоксиацетилен, т. кип. 49—51°/749 мм.
Вторая реакция, сопровождающаяся расщеплением простой эфир-
ной связи, приводит к образованию пентин-4-ола-1 из тетрагид-
рофурфурилхлорида 181.
сн3—сн, сн,—сн2
I I 3,5 моля NaNH, I I 3,3 моля NH.CI
СН2 СНСН,С1-------------->СН, C = CNa------------►
\/ ' NH„ I 75-85%
О ОК’а
1 моль
—НОСН,СН,СН,С сн
Для получения бутин-2-ола-1 (4) Уайтинг исотр. 19] приготовля-
ли раствор А. н. в колбе дыоаровского типа емкостью 4 я, снабжен-
ной механической мешалкой с пластмассовой лопастью (3 л NH3,
1,5 г гидрата азотнокислого окисного железа, 2,8 г-атом натрия)
и добавляли 1,26 моля 3-хлорбутен-2-ола-1 (2) в течение 30 мин.
Смесь перемешивали в течение ночи, затем добавляли в несколько
приемов 2,8 моля твердого хлористого аммония со скоростью, поз-
воляющей контролировать экзотермическую реакцию. Смесь пере-
носили в ведро из нержавеющей стали и оставляли на ночь в вы-
10%Na,CO., NaNH,
СН3С = СНСН,С1--------CH3C = CHCH2OH —-------------——
' I “ 63% 3 | 2 Жидкий NH
О С1
(1) (2)
—> СН3С = CCHoONa ---Д СН3С ССН,ОН
(3) (4)
тяжном шкафу для испарения аммиака. Продукт экстрагировали
эфиром и перегоняли при 55с/8 мм.
Дегидрогалогенирование под действием А. н. является также
удобным способом замыкания азиридинового кольца, как это ил-
люстрирует получение N-этилалленимина (1-этил-2-метиленазири-
дина) [10].
NaNH,-NH3
сн,=с—сн, nc,h5 ————> сн, = с — снг
I | 48—55%
Вг Н N
I
С,н5
RC=CNa и его производные. Тетроловую кислоту синтезируют
превращением пропина в его натриевое производное с последующим
61
карбонилированием и подкислением 111],
NaN Н. СО.
СН3С ss СН-----СН3С ss CNa-----> СН3С = ССО2Ха
но
30-39",;
СН3Са=ССО.,Н
Для получения гексина-1 Кэмпбелл и Кэмпбелл [12] пропускали
ацетилен в 3 л жидкого аммиака и постепенно добавляли 4 г-апюм
натрия с такой скоростью, чтобы голубое окрашивание не распро-
странялось на весь раствор (для этого каждый кусочек натрия по-
мещали на железный рыболовный крючок и с помощью гибкой же-
лезной проволоки опускали в аммиак или вынимали оттуда). Ток
ацетилена прекращали и по каплям добавляли «-бутилбромид
(4 моля). В конце прибавляли по каплям 500 мл водного раствора
аммиака и приливали дистиллированную воду. Обычной обработкой
получали чистый гексин-1, т. кип. 71—72°.
Жидкий NH., «-С,Н9Вг
НС СН + Na----------НС = CNa w-C4H9C = СН
—H,
70-7 7».,
Образование реакционноспособных анионов. В случае типичного
алкилирования кетонов Вандерверф и Леммерман [131 получали
А. н. в атмосфере азота в виде серой суспензии (1,5 л аммиака, сле-
ды азотнокислого окисного железа и 2,05 г-апюм натрия), прили-
вали 1,2 л эфира с такой скоростью, какую допускало испарение
аммиака, нагревали смесь для удаления аммиака, прибавляли ци-
моля
О
клогексанон и перемешивали и нагревали смесь при температуре ки-
пения в течение 3 час. Смесь охлаждали, медленно добавляли эфир-
ный раствор бромистого аллила (экзотермическая реакция) и для
завершения реакции кипятили 3 час.
Другие примеры приведены кратко в виде уравнений (2—5).
При использовании для получения 2-метилбутин-З-ола-З (2) А. н.
смачивали гептаном, растирали в тонкий порошок и оставляли для
испарения растворителя. В суспензию А. н. в эфире при охлажде-
нии до —10 и перемешивании пропускали ацетилен для образо-
вания аддукта и вытеснения аммиака. В примере (3) (Хаузер)
62
А. и. получали из жидкого аммиака и натрия в присутствии желе-
за в качестве катализатора; аммиак заменяли на эфир до при-
ливания эфирного раствора метилизобутилкетона. После образо-
вания натриевого производного добавляли этиловый эфир пзова-
лериановой кислоты в эфире, чтобы избежать сложноэфирной кон-
денсации. Диизовалероилметан выделили в чистом виде через его
медную соль. В примере (4) (Хаузер) фенилуксусную кислоту при-
бавляли непосредственно к суспензии А. н. в жидком аммиаке и
хлористый бензил добавляли к образовавшейся темно-зеленой сус-
пензии. По завершении алкилирования аммиак заменяли на эфир.
СН;1 СНДХ'а)
I | нс = сн
2[ 14р СН3С=О —— СН3С - О--------
— IX Fl it
СН3 СН3
I н+ |
—> CH5С-СsСН ------> СНрС — стон
। 40—46% ।
ONa ОН
NaNH- _ +
3[ 15]) СН3СОСН2СН(СН3).,----> CH2COCH.,CH(CH3)»(Na) —*
— N Н
О о
(СН.)..СНСН.СО.,СЛЦ |1_ || +
---(СН3)2СНСН2ССНССН,СН(СН3)2(Х’а) —>
О О
—~f (СН3)2СНСН2ССН2ССН2СН(СН3)2
э о I и 0
2NaNH. +
4[3]) С„Н5СН2СО,Н-----> CGH5CHCO2(Na), —*
CjHsCH.CI н +
ДН-ДНСО.А’а---------- СсН-,СНСО.,Н
- 80-84% ”|
с6н-,сн, с0н5сн2
«тоццо
Другие примеры алкилирования фенилуксусной кислоты и ее эфи-
ров описали Хаузер и сотр. [171. N-Метилирование индола [при-
мер (5)1 осуществляют в основном так, как описано в примере (4).
Примером использования А. н. для образования иона, который
вступает в реакцию с кетоном, может служить синтез нитрила
а, p-дифенилкоричной кислоты [181.
NaKH.. _ + (СГ,Н-).,С = О
CeH5CH2CN------>СсН5СНС\’(Ха) (С6Н5)2С = ССПН5
CN
63
Синтез 2-метилиндола по Верлею [19] включает циклизацию
промежуточного аниона. Выход указан в соответствии с данными
Аллена и Ван Аллана [20].
с2н5он-н2о_
80-83% :
При детальном исследовании образования дикарбанионов из
Р-дикарбонильных соединений Харрис и Харрис [21] нашли, что
А. н. является лучшим реагентом, чем амид калия, димсилнатрий
и гидрид натрия.
NH7 _ _ со2
RCOCH2COCH3--^ RCOCHCOCHj —- RCOCH2COCH2CO,H
Другие реакции. Конденсацию по Дарзану [22], например
конденсацию ацетофенона с этилхлорацетатом с образованием фе-
нилметилглицидного эфира, Аллен и Ван Аллан [23] провели пу-
тем добавления тонкоизмельченного А. н. к раствору реагентов в бен-
золе.
СН3 СН3
I NaNH2 |
С6н5с = О 4- С1СН»СО»С2Н5---- С0Н5 с-—:СНСО,С„Н5
О а ) -.23 62-64% “ а \ / - - а
о
Бразен и Хаузер [24] описали методику восстановительной пере-
группировки йодистого бензилтриметиламмония (1) в третичный
амин (2).
СН2Й(СН3)з(1) CH2N(CH3)2
А А/сн<
I II SI II
А/ А/
(1) (2)
Беннет и сотр. [25] применили А. н. для конденсации натриевой
соли нафталин-0-сульфокислоты с пиперидином и получили N-p-наф-
тилпиперидин.
Пархам и Уинберг [26] описали методику проведения реакции
диэтилбромацеталя (1) с сульфидом натрия и серой с целью полу-
64
чения соединения, средний состав которого соответствовал тетра-
сульфиду (2). Смесь этого соединения (1,38 моля) и эфира переме-
шивали при температуре от —35 до —45', добавляли жидкий ам-
миак, а затем по частям 8,7 г-апюм натрия. Прибавление последних
нескольких граммов натрия вызывает темно-синее окрашивание
BrCH,CH(OC.,H5), ; у 2S
(!) 3 моля 2,2.’ моля •!,"> г-атам 83 —42%
NaNH.,-NH,
—, (C„H5O),CHCH.,SSSSCH.,CH(OC,H,,),-:--i
2(C„H5O).,CHCH,SNa —2(C.,H5O)„CHCH.,SH
(3) 7 0 -83% >13 (2) (-1)
раствора. Через 30 мин непрерывно прибавляли твердый хлорис-
тый аммоний до исчезновения синей окраски. Аммиак удаляли, на-
триевую соль (3) растворяли в воде и добавляли соляную кислоту до
pH 8,0—8,5 (pH-метр). Чтобы избежать гидролиза нестойкой к дей-
ствию кислот апетальной группировки, устанавливали pH 8,0—8,5
и выделяли свободный дпэтилацеталь меркаптоу ксусиого альдегида
(4), пропуская в раствор двуокись углерода. Затем препарат
экстрагировали эфиром и перегоняли.
Синтез циклопропена. Фишер и Эппльквист [27] действовали
на металлилхлорид (1) А. и. в кипящем ТГФ и получили 1-метил-
циклопропен (2) с умеренным выходом; по-видимому, реакция
включает стадии образования и циклизации винилкарбена. Клосс
и Кранц [281 показали, что аналогичным образом можно получить
и циклопропен (3) из хлористого аллила:
СН3 СНо — ССН3'
I Na\'H.-Tr4>
СН2^С—СН,С1---------:---% :СН J
(1)
(2)
—> сн ссн3
50% ч z 3
СНо
Выход составляет лишь 10%, однако этот метод значительно
проще всех ранее описанных. Полученный таким образом циклопро-
пен вступает в реакцию с циклопентадиеном при 0° с образованием
по реакции Дильса — Альдера аддукта (4) с выходом 10%.
сн2=сн
СНгС1
NaNH;^ сн =сн
10% хс/
----*
0°
(3) (4)
1 V a u g h n Т. Н., Vogt R. R., N i euwl and J. A., J. Am. Chem. Soc.,
’ 56, 21'20 (1934).
2. Хаузе p 4. P., .4 даме Дж., Левин P., «Синтезы органических пре-
паратов», ИЛ, М., 1953, сб. 4, стр. 160.
3 Заказ № 965
65
3. Хаузер Ч. Р., Д ю и и а вант X’. Р., «Синтезы органических препа-
ратов», изд-во «Мир», М., 1964, сб. 12, стр. 56.
4. Д ж о п с Э., Э г л н н т о н Дж., У а й т и п г ДБ, Ш о у Б., «Синтезы
органических препаратов», ИЛ, М., 1956, сб. 6, стр. 105.
5. К а н Н., Д э з и р е д ж Ф., Б р о у и Дж., «Синтезы органических пре-
паратов», ИЛ, АБ, 1959, сб. 9, стр. 60.
6. Л е с и и о Р., Б у р г е л ь АБ, «Синтезы органических препаратов», ИЛ,
ДБ, 1949, сб. 1, стр. 511.
7. Каи И. \., «Синтезы органических препаратов», ИЛ, АБ. 1953, сб. 4,
стр. 494.
8. Д ж о и с 9 г л и н т о и Дж., У а й т и и г М.; «Синтезы органических
препаратов», ИЛ, ДБ, 1954, сб. 5, стр. 54.
9. А ш \ о р з е П., А1 э и с ф п л ь д Дж., У а й т и н г АБ, «Синтезы орга-
нических препаратов», ИЛ, АБ, 1956, сб. 7, стр. 22.
10. В о t t 1 и i А. Т., Oise n R. E., Org. Syn., 44, 53 (1964).
11. Kail e r J. С., В г о w и АБ, Org. Syn., 42, 97 (1962).
12. К м и б e л л К. И., К э м n б e л л Б. К., «Синтезы органических препа-
ратов», ИЛ, АБ, 1953, сб. 4, стр. 111.
13. В а п д е и в е р ф К., Л емче р м а н Л., «Синтезы органических препа-
ратов», ИЛ, АБ, 1953, сб. 4, стр. 18.
14. К о ф ф м а п Д. Д., «Синтезы органических препаратов», ИЛ, АБ, 1952,
сб. 3, стр. 198.
15. X а у з е р Ч., А д а м с Дж., Л е в и и Р., «Синтезы органических препа-
ратов», ИЛ, АБ, 1953, сб. 4, стр. 160.
16 Поттс К. Т., Сакстон Дж. Э., «Синтезы органических препаратов»,
нзд-во «А1ир», АБ. 1964, сб. 12, стр. 91.
17. Н а и s е г С. R., С h a in Ь е г s W. .]., J. Am. Cliem. Soc., 78, 4942 (1956);
Al e v er R. B., Hau s с r C. R., J. Org. Chem., 26, 3696 (1961); К e n v-
o n W. G„ Al с у e г R. В.. H a u s e r C. R„ J. Org. Chem., 28, 3108 (1963);
К а н s e г W. АС. К e и у о n W. G., H a u s e г C. R., procedure submitted to
Org. Syn.
18. В а в з о и e к С., С м о л и и Э., «Синтезы органических препаратов», ИЛ,
АБ, 195.3, сб. 4, стр. 356.
19. V с г 1 еу АБ A., Bull. Soc. chim. France, 35, 1039 (1924).
20. А л л e и 4., В a u А л л а и Дж., «Синтезы органических препаратов»,
И,4, АБ, 1952, сб. 3, стр. 302.
21. Harris Т. АБ, Н а г г i s С. АБ, J. Org. Chem., 31, 1032 (1966).
22. Darzeiis G.. Compt. rend., 139, 1215 (1904).
23. А л л e и 4., В а н А л л а н Дж., «Синтезы органических препаратов»,
11Л, АС, 1952, сб. 3, стр. 516.
24. Б р а з е п А’., X а у з е р Ч., «Синтезы органических препаратов», ИЛ, АБ,
1956, сб. 6, стр. 44.
25. Б с и пета Дж. Ф., Бро а ер то н Т., У Илья угон С., «Синтезы орга-
нических препаратов», изд-во «Мир», М., 1964, сб. 12, сгр. 100.
26. П а р х а м А’., А' и и б е pi г X., (Синтезы органических препаратов», ИЛ,
АБ, 1956, сб. 7, стр. 30.
27. F 1 s h е г F., А р р 1 е q u i s t D. E., J. Org. Chem., 30, 2089 (1965).
28. С 1 о s s G. L., К r a n t z K. D., .1. Org. Chem., 31, 638 (1966).
н-АМИЛНИТРИТ, C-H„ONTO. Мол. вес 117,15, т. кип. 104°
уд. вес 0,853.
Встречающиеся в литературе ссылки на «амилнитрит» или «пен-
тплпптрпт» относятся, вероятно, к изоамилнитрпту, более доступ-
ному продажному реагенту.
п-АМИНОАЗОБЕНЗОЛ, CGH5N = NC6H4NH2-n. Мол. вес 197,23,
т. пл. 128°.
66
Двухосновные кислоты типа янтарной пли глутаровой, получае-
мые при окислении природного сырья, часто трудно выделить или
очистить из-за неблагоприятного соотношения растворимости или
присутствия примесей, препятствующих кристаллизации. В этом
случае используют следующий прием. Загрязненную кислоту прев-
ращают в соответствующий ангидрид (Ас.,0 или АсС1), затем реак-
цией с п-ампноазобензолом в хлороформе — в мопоанилпд (2), ко-
торый с хлористым ацетилом циклизуют в имид (3) И).
СО
(СН,),,' С6Н5Х ХСеН.ХВСО'СН.фСО.Н -
\ / (• п
(1)
п = 2 и л и 3
СО
/СИ.,),,
' со
01
Такие имиды легко разделяются хроматографией на окиси
алюминия, так как они хорошо заметны в виде темпо-желтых полос.
По ПК- и УФ-спектрам легко осуществить контроль разделения и
идентификашпо продуктов.
1. Н с н b е s t Н. В., Owen Т. С., J. Chem. Soc., 1955, 2968.
1-АМИНОБЕНЗОТРИАЗОЛ,
т. пл. 82".
Мол.
вес 134,14,
О получении А. см. Гпдрокспламин-О-сульфокпслота; о получе-
нии из А. дегидробензола см. Свинца тетраацетат.
2-АМИНО-2-МЕТИЛПРОПАНОЛ-1, (CH,.)2C(NH,)CH2OH. Мол.
вес 89,14, т. пл. 25—29", эквив. нейтр. 88,5—99,0.
По данным Харриса и Сэндерсона 111, А.— исключительно спе-
цифичный реагент для осаждения левопимаровой кислоты из кани-
фоли. Полученную соль разлагают борной или фосфорной кислота-
ми. Этот метод подробно описан Ллойдом и Хедриком 121.
1. Н а г г i s G. С., Sanderson Т. F., J. Ain. Chem. Soc., 70, 334 (1948); see
also R о e b I i c h V. M., Baldwin D. E., O’C о n n о r R. Law-
rence R. V., ibid., 77, 6311 (1955).
2. Lloyd W. D., HedrickG. W., Org. Syn., 45, 65 (1965).
N-АМИНОФТАЛИМИД (1). Мол. вес 162,15, т. пл. 200—205°
(с разл.).
3* 67
Получение А. из фталимида и гидразина [1]:
N-Аминопирролы [2]. А. конденсируется с ацетонил ацетоном
в горячей уксусной кислоте с образованием Кт-фталнмидо-2,5-диме-
тилпиррола (2), который реакцией с гидразином расщепляется на
фталгидразид (3) и Ы-амино-2,5-диметилпиррол (4).
1. Drew Н. D. к., Hatt Н. Н., J. Chem. Soc., 1937, 16.
2. Epton R., Chem. Ind., 1965, 425.
АММОНИЯ АЦЕТАТ И ФОРМИАТ. Мол. вес 77,09 и 63,06
соответственно.
Дебромирование. При гидрогенолизе бромпроизводных добав-
ление А. а. в количестве несколько большем 1 же используют для
нейтрализации выделяющегося бромистого водорода, что предот-
вращает нежелательное гидробромирование двойной связи [1].
Образование амидов. Хлорапгидрпды кислот превращаются
в амиды с выходом 40—90?» при перемешивании с А. а. в ацетоне
в течение 30—60 мин [2|. В полном синтезе тетрациклинов [31
эфир (1) сплавляют с А. ф. и после дезалкилирования выделяют
амид (2); выход не указан.
С1
A/WX/x /0Н
I II I I II
| | | ii ои.\нг
ОН ОН он о
(2)
68
Аминирование. Взяв за основу работу, относящуюся к родст-
венным соединениям [41, Кертин исотр. 151 при сплавлеппн 2-фепил-
индапдиоиа-1,3 с А. а. в атмосфере азота получили З-ампно-2-фе-
35, 5г
нилинденон. Если плав красного цвета охладить до 80—90° и раз-
бавить водой, то амин получается в кристаллическом виде с высо-
кой степенью чистоты.
Каталитическая конденсация. А. а. рекомендован в качестве
катализатора для конденсации кетонов с циануксусной кислотой и
ее этиловым эфиром [61.
(А г,\:
.-—ч ! ch,co.nh4 ,---------. /
/ ! -О -1-СНХОоН—::—*/ х=с
\_____/ “ ' .
СО2Н
СН3 С\' СН3
| | сн.со.кн, | / "
С0Н5С О -1- СН,СО2С2 н5 С6Н5С -С
ХСО2С2Н5
1. R i n R о 1 d И. J., Loken В., Rosenkranz Q., Sondheimer F.,
J. Am. Chem. Soc., 78, 816 (1956); Loken B., KaufmannS., Rosen-
kranz G., Sond 11 e i m e r F., ibid., 78, 1738 (1956).
2. Finan P. A., F о I h c r g i 1 1 G. A., J. Chem. Soc., 1962, 2824.
3. В о о t ii c J. H., К e ii d e A. S., F i e I <1 s T. L., W i 1 k i ti s о n R. G.,
J. Am. Chem. Soc., 81, 1006 (193!)).
4. H о r t о n R. L., M u r d о c k 1\. C., J. О1Д. Clieim, 25, 938 (I960).
5. C u r t i n D. Y., К a m p ni e i e r J. A., F a r in e г Л1. L., J. Am. Chem. Soc.,
87, 874 (1965).
6. Co p с A. C. et al., J. Ain. Chem. Soc., 63, 3452 (1941); «Синтезы органических
препаратов», ИЛ, М., 1953, сб. 4, стр. 562.
АММОНИЯ МЕТАВАНАДАТ, NH4VO;i. Мол. вес 116,99.
А. м. как катализатор [11 при окислении гидрохинона перхлора-
том лучше, чем обычно используемая пятиокись ванадия (Биллман
и сотр. [2]).
ОН О
I II
I I +^'аС1О3 + МН4УОз-|-2%-наЯН,8О41°-^[| [I
[ 0,56 моля 1,4 г 1 .1 93—9 3",! [I
Y "ii
он о
Окисление начинается сразу после добавления А. м. к остальным
реагентам при перемешивании и температуре 40'; необходимо ох-
69
лаждение, чтобы избежать повышения температуры. Окисление за-
капчивается менее чем за 30 мин, т. е. в восемь раз быстрее, чем
обычно.
1. В i 1 1 m а п J. Н., WolnakB., BarnesD. К., J. Лт. Chem. Soc., 66,
652 (1944).
2. У н дера v д X., мл., У о л ш В., «Синтезы органических препаратов», ИЛ,
М., 1949, сб. 2, стр. 545.
АММОНИЯ НИТРАТ, NH4NO:). Мол. вес 80,05.
Л. н. применяют при получении диэтилацеталя акролеина в ка-
честве катализатора конденсации акролеина с ортомуравьиным
эфиром 111:
сн2снсно нс(ос,н3)3 -у х н j хсу
0,79 миля 0.97 -io i'.i 3 в 30 ' 0
абс. EtOI-I
— СН2 -СНСН(ОС2Н-,)2 нсо.,с.,н3
1. F 1 s с h е г Н. О. L., В а с г Е., Helv. Chim Acta, 18, 514 (1935); ВаиАл-
л а и Дж., «Синтезы органических препаратов», ИЛ, М., 1953, сб. 4, стр. 70.
АНИЗОЛ, СГ,Н3ОСН:|. Мол. вес 108,13, т. кип. 154 , уд. весО, 990.
По данным японских авторов 111, в реакциях гидроборирования
А. является удовлетворительным заменителем диглима (т. кип.
161). Однако в отличие от последнего А. не растворяется в воде.
1. К u g 1 t а Н., Takeda М., Chem. Pharm. Bull., 12, 1166 (1964).
АНИЛИН, CbH-,NH2. Мол. вес 93,12, т. кип. 1843, уд. вес 1,02,
рКЬ 9,30.
Потемневший при хранении А. обесцвечивают перегонкой над
небольшим количеством цинковой пыли.
АНТРАЦЕН-9, 10-ДИИМИН,
Мол. вес 208,26, т. пл. около 100э.
А. получают гидролизом и декарбоксилированием аддукта ан-
трацена с азодпкарбоновым эфиром. А. разлагается в кипящем
спирте на антрацен и диимпд 111. Взятый в 2—8-кратном избытке,
А. восстанавливает азобензол, а также реакционноспособные олефи-
ны и ацетилены.
1. С о г с у Е. J., Mock W. L., J. Am. Chem. Soc., 84, 685 (1962).
АРИЛДИАЗОНИЯ ГЕКСАФТОРФОСФАТЫ, ArXWNPF^
Превращение арнламинов в арилфториды по Шиману значитель-
но улучшается при использовании в качестве промежуточных со-
единений А. г. вместо тетрафторборатов арплдиазония [1|. А. г. ме-
нее растворимы, чем соответствующие тетрафторбораты, поэтому
70
получаются с большим выходом и легко очищаются промыванием.
Для получения 2-бромфторбензола |21 из о-бромаиилина хлоргидрат
Вг Вг Вг
J ХН, J XX J. F
/Л- / // - // -
I Г HC1-HNO.; НРГ„ ; Г1Г._ 165" I 1|
,j --------с----------------------! I;
Д, 'i%V
амина диазотируют обычным образом, добавляют 65/о-пую гекса-
фторфосфорную кислоту, А. г. отделяют п сушат. Для проведения
разложения А. г. помещают в колбу Эрлеимейера, соединенную ко-
роткой резиновой трубкой с горлом трехгорлой колбы, содержащей
минеральное масло и снабженной обратным холодильником и термо-
метром. Масло нагревают до 165—170 п А. г. добавляют порциями.
После охлаждения к реакционной смеси добавляют 10%-ный вод-
ный раствор карбоната натрия и продукт перегоняют с паром. Об-
щий выход 73—75%, тогда как по методике Шпмаиа он равен толь-
ко 37%. Общие выходы о-фторбепзойной кислоты в таком синтезе
и по Шпмапу составляют 61 и 9% соответственно.
Новый способ дезаминирования А. г. состоит в их спонтанном эк-
зотермическом разложении в тетраметилмочевпне 131:
о О
’ CH F
+ (CH.j.NCNtCH,). , , , хСП2г
ArX^XPF?-----------------ArH-J-N., : PFS--(C.H3).,XC\4
Эффект этого органического растворителя обусловлен, вероятно,
тем, что он смешивается с водой, тогда как другие гидрофобные
растворители не действуют. Наилучшие выходы (75—85%) получа-
ются для соединений, в которых ароматическое, кольцо содержит
электроноакцепторпые заместители (NO.,, СО,Н, Вг). Для анилина,
о- и н-толуидинов и о-апизидина выходы составляют 25—30%.
Сухую соль диазония добавляют к тетраметилмочевпне порциями
при комнатной температуре так, чтобы температура смеси не пре-
вышала 65'.
1. Rutherford К. G., Redmond W., R i g a m о n t i J., .1. Org. Chem.,
26, 5149 (1961).
2. R u t h e r f о r d К. G., Redmond W., Org. Syn., 43, 12 (1963).
3. RutherfordK. G., Redmond W., J. Org. Chem., 28, 568 (196.3).
АРИЛДИАЗОНИЯ ТЕТРАГАЛОГЕНБОРАТЫ, ArN=NBX4.
По Шимапу |1), ариламин диазотируют в соляной кислоте
и суспензию пли раствор хлористого арилдиазопия обрабатывают
раствором тетрафторборной кислоты, полученным 12—4] растворе-
нием борной кислоты в продажной 50—60%, -пой фтористоводород-
ной кислоте при 20—25' в колбе, внутри парафинированной или
покрытой слоем меди, свинца или серебра. А. т. отделяют, сушат
71
и пиролизуют. Например:
-1. IIBFj а- Нагревание
Ц21) Сг,н5х - XCI — с6н,л\bf; т' c„h5f-; n..-1-bf,
.11 .) / и()
_|. ИВГ4 а-
2|3J) n-C2H5O.,C(:0Hp\r - \С1 —_ ' /Z-CJ-I5()2CC6H4N — NBF“ —>
Нагрева юге
—у я-С.ДГ,О/:СиН4Г
Л. т. получают также [5] из амина и тетрафторбората нитрозония,
синтезированного по реакции (3).
3) 2H + BF" --> 2NO+BF; г Н,0
4) ArXH.,-’ \0‘BF; - , Лг\; BF7 ' И.,(4
z - 4 2 4 1
Тетрахлорбораты и тетрабромбораты арплдиазонпев получают
следующим путем |6|. Раствор амина в хлороформе добавляют к
раствору хлористого (или бромистого) пптрозила (5) в смеси хлоро-
форма п петролейного эфира при —18' и отделяют белый осадок со-
ли арилдпазоппя. К суспензии этой соли в смеси хлороформ — пе-
тролейпый эфир при перемешивании и температуре —18 добавляют
треххлористый бор (6) н отфильтровывают выпадающий белый оса-
док продукта. Эти соли при нагревании часто разлагаются со взры-
5) Ar NIL ; NOC1 ArX'+ci- ' Н,0
6) Аг\г'С1--|-ВС13--> АгХц ВО;
вом; однако можно провести контролируемое разложение с исполь-
зованием инертного разбавителя (лигроин, т. кип. НО—115).
В процессе получения п-динитробензола из н-питроанплпна 141,
сходном с реакцией Шпмана, разложение проводят не пиролизом,
а непосредственно в водном растворе. Кроме того, фторбораты арил-
дпазопиев, содержащие питрогруппу, гладко разлагаются в хлорбен-
золе 17|:
U. нвг4 +
7) п-О2ХСсН4Х ---- ХС1--> n-O.,NC0H4N = NBF;
Води. NaNOj(Cii)
---------------> /!-O.>NCgH4NO.>
07 —«ОО/-------“ b 4
Бергсталер и сотр. 181 диазотируют ограниченно растворимый
хлоргидрат ЗД-ди-шреш-бутнлаиилина при 0—5 , после фильтрова-
ния обрабатывают реакционную смесь раствором борфторпда
натрия и получают А. т. Эта соль подвергается медленному сольво-
лизу в смеси уксусной кислоты и уксусного ангидрида с образова-
72
нием соответствующего ацетата.
\--\Cl~ X . XBF7 ОЛс
I I I
”'>» // ч
I и N.1BF- / и ЛсОН-Лс.Л) -z ,)
I ----. I ----------Г \ 1|
\ Н-С(СН3)3 ;-С(СН3)3 Ц । —С(СН3)з
Г I I'
С(СН3)3 С(СН3)3 С(СН3)3
Реакцией А. т. с карбонилом никеля в спирте или уксусной ки-
слоте получают арилкарбоновые кислоты с выходом 2—76?6 191.
Дезаминирование амина можно осуществить превращением в
А. т. с последующим восстановлением цинком в спирте 1101 или,
более эффективно, боргидридом натрия (8) [111. Простейшая мето-
N а В Н t
8) ArN + BF;->ArH.
дика состоит в добавлении боргидрпда натрия порциями к охлажден-
ному раствору (или суспензии) А. т. в метаноле с последующим вы-
ливанием реакционной смеси в лед с соляной кислотой. По другой
методике холодный раствор боргидрпда натрия в ДМФА добавляют
к холодному раствору А. т. в том же растворителе. Выход 50—75?6.
1. Роэ Л., «Органические реакции», ИЛ, М., 1951, сб. 5, стр. 155.
2. Ф л о о д Д., «Синтезы органических препаратов», ИЛ, М., 1949, сб. 2, стр. 537.
3. Ш и м а и Лж., В и и к с л ь м го л лер В., «Синтезы органических препара-
тов», ИЛ, 51., 1949, сб. 2, стр. 534.
4. Старки Е., «Синтезы органических препаратов», ИЛ, М., 1949, сб. 2,
стр. 227.
5. W a n n a g a t U., Н о li 1 s t с i n G., Chem. Вег., 88, 1839 (1955).
6. Olah G. A.. T о 1 g у e s i \V. S., J. Org. Chem., 26, 2053 (1961).
7. V a 1 k a n a s G., H о p f f H., ,J. Chem. Soc., 1963, 1925.
8. В u г g s t a li 1 e r A. W., A b cl e I - R a h in a n M. O., Chien P.-L.
Tetrahedron Letters, 6! (1964).
9. Cl a rk J. C„ Coo ks о n R. C., ,1. Chem Soc., 1962, 686.
10. Roc A., Gralia in J. R., .1. Am. Chem. Soc., 74, 6297 (1952).
11. Hendr ickson J. B., J. Am. Chem. Soc., 83, 1251 (1961).
АЦЕТАЛЬДОКСИМ. CH3CH-NOH. Мол. вес 59,07, т. пл. 473.
А. реагируете солями диазонпя с образованием оксимов, которые
при кислотном гидролизе дают арилметилкетоны 111.
СН3 СН;!
I н + I
АгХ.,С1 : С1ЦСН - ХОН ... Arc -ХОН — ArC=-0
1. Beech W. 1С, J. Chem. Soc., 1954, 1297.
АЦЕТАМИД, CH.,CONH2. .Мол. вес 59,07, т. пл. 82°.
А. применяют при бромировании соединений, изменяющихся под
действием выделяющегося бромистого водорода, с которым А. об-
разует устойчивый комплекс CH:1CONH., • НВг, осаждающийся из
растворителей, обычно используемых для бромирования [11.
J. Z с i 1 е К., М е у е г II., Chem. Вег., 82, 275 (1949).
7.3
гг-АЦЕТ АМИДОБЕНЗОЛСУЛЬФОХЛОРИД,
о Н
СНаС - — SO ,С1.
Мол. вес 233,68, т. пл. 149.
А. получают из ацетанилида и хлорсульфоновой кислоты 111
и используют как катализатор для бекмановскои перегруппировки
оксидов в пиридине |2|.
1. См а й л с С., Стмоар т Дж., «Синтезы органических препаратов», ИЛ,
51., 19-19, со. I, ci]). 46(1.
2. К а и 1 in а п 11 St., J. Am. Cliein. Soc., 73, 1779 (1951); И eusser И. ct al.,
Helv. Cliim. Acta, 38. 1399 (1955); R о s e n k r a n z G., 51 ancera O.,S о n d-
h c i in e r F., Djcr assi C., J. О1Д. Chem., 21, 520 (1956).
АЦЕТИЛ АЦЕТОН (2,4-пентандион), Cl I3COCH2COCH3. Мол.
вес 100,11, т. кип. 134—136 , уд. вес 0,975.
Получение 111,
(СНэС0).,0 rCH.COCH3-J^'’ , СН3СОСН,СОСН3
Расщепление —N-производных. При расщеплении фенилгидра-
зонов п семпкарбазопов А. более аффективен, чем пировиноградная
кислота |2|. При взаимодействии А. с фенилгидразоиамп образуется
3,5-диметпл-М-фенплпиразол и освобождается карбонильное со-
единение:
Пептидные синтезы |3|. А реагирует с аминокислотами в ме-
танольном растворе едкого кали с образованием еиамииа, стабили-
зированного водородной связью. Эти производные применяют в син-
тезах пептидов карбодппмпдным методом или методом иианметило-
вых эфиров. Защитная группа снимается 2 и. соляной пли уксусной
кислотой.
74
Получение тетраацетилэтана [41.
WiOH (води.)
сн.(он
СНУСОСН2СОСН3 —7777^“-' (снисоснсоснз)^а +
О О
в эфире СН3С— СН—С —СНд
"4 1-5У«/СН3С-СН-С-СН3
о о
Синтезы гетероциклов. Ниже приведены примеры синтезов
2,4-дпметил-3-ацетил-5-карбэтокснп11ррола (1) [5] и 3,5-диметилпн-
разола (2) |6|.
NOH NH,
сн3сосн2со2с2н5 ДДщ» сп3соссо2с,н; ?-> сн3соснсо2с2нг —>
АсОН АсОН
СН3СО____^СН3
СН3СОСП;СОСН3? >---
55-60% (общий) н3сАССсО2С2Н5
(1)
СН3СОСН2СОСН3
1. Денун К., мл., «'Синтезы органических препаратов», ИД, М., 1952, сб. 3,
стр. 92.
2. R i е d W., Л1 ii li I е G., Ann., 656, 119 (1962).
3. D a n е Е., Drees F., Konrad Р., Dockner Т., Angew. Cliem., In-
ternal. Ed., 1, 658 (1962).
4. Ч a p л и з P. Дж., «Синтезы органических препаратов», ИЛ, Л1., 1961, сб. 11,
стр. 52.
5. Фишер Г., «Синтезы органических препаратов», ИЛ. М., 1952, сб. 3, стр. 269.
6. У а й л и Р., ХекенерП, «Синтезы органических препаратов», ИЛ, Л1.,
1953, сб. 4, стр. 189.
АЦЕТИЛГЕКСАФТОРАНТИМОНАТ, CH3CO+SbFp [1]. Мол. вес
278,80, т. пл. 173—175-.
Получение [21. Эту исключительно устойчивую ацилиевую
соль получают в условиях, исключающих наличие влаги, лучше в
замкнутой системе. Холодный раствор ацетилфторпда во фреоне
113 (1,1,2-трпфтортрпхлорэтан) обрабатывают фреоновым раство-
ром эквивалентного количества свежеперегнашюй пятпфторпстой
сурьмы п после перемешивания в течение часа собирают белый кри-
сталлический осадок, промывают его холодным фреоном ИЗ и вы-
75
сушивают в вакууме.
СН.,СОЕ '-C1CFCC1F., -' - SbFr, CH.,CO+SbF7
0,2 моля 70 м.1 0.2 моля
Ацетилирование. Л. (чистый плп в нитрометане) ацетилирует
с хорошим выходом ароматические соединения (1). Л.— эффектив-
ный реагент для О-ацетплированпя спиртов (2), S-ацетплпрования
меркаптанов и ГхДщетилнроваппя незамещенных и N-моноалкилами-
ДОВ.
О
1) СсН5СНДС1) ;-СНзСО SbF- «-CHsCCsH4CH:t(Cl).( HF T-SbF5
CH .NO.,
2) СН;((СНг)вСН2ОН -I CH3CO !-SbF" -p“; CH3(CH2)6CH2OCOC.H;t (- HF SbF5
1. Contributed by Olah G.. Case Institute of Technology.
2. О 1 a h G. A., К u h n S. JT о 1 у v e s i W. S., Baker E. 13., J. Am. Chem.
Soc., 84, 273.3 (1962).
АЦЕТИЛГИПОБРОМИТ, СН:)СО2Вг. Мол. вес 138,96.
Этот (или эквивалентный ему) реагент получают добавлением
N-бромацетамида к раствору ацетата лития в уксусной кислоте в
присутствии олефина. Например, в случае (1) наблюдается транс-
диакспалыюе присоединение А. с образованием ацетата бро.мгидрина
(2) с 74%-ным выходом 111. В этих условиях не затрагиваются груп-
пы, чувствительные к кислотам, а введенная ацетоксигруппа устой-
чива к окислению. Обработка (2) основанием дает Р-эпокспд (3), а
действие Zn — Си в этаноле приводит к регенерации олефина (1).
Другой метод получения A. J2] состоит в обработке при переме-
шивании суспензии ацетата серебра в четыреххлорпстом угле-
роде раствором брома в том же растворителе. Прозрачный раствор
А. добавляют при 0 к раствору олефинового субстрата в четырех-
76
хлористом углероде (например, Холестерил ацетата (4)]. Эта реакция
менее стереоспецпфпчна, чем в первом случае, но все же основной
ее продукт — /ирпнс-диаксиальпый ацетат бромгидрина (5), который
превращается в р-окпсь (6). В противоположность взаимодействию
A<J(’’’-соединения (1) с Л., полученным первым методом, метиловый
эфир За-ацетокси-А"11” -холеповой кислоты не реагируете раствором
А., приготовленным вторым способом.
1. Ro b i n s о n С. Н., Finkonor L., К i г t 1 е у М., G о и 1 d D., О 1 1-
v г t о Е. Р., J. Am. Chem. Soc., 81, 2195 (1959).
2. 1. е v i n c S. G„ W a 1 I M. E., J. Am. Chem. Soc., 81, 282G (1959).
АЦЕТИЛЕН, IIC СН. Мол. вес 26,04, т. кип. —83°.
Очистку Л., хранящегося в баллоне в ацетоновом раст-
воре, раньше проводили пропусканием газа через воду
для поглощения ацетона п затем через конц. серную
кислоту [11. Позднее в большинстве случаев А. пропускали через
охлажденную (—80') ловушку, снабженную предохранительным
ртутным затвором, а затем через пустую склянку, конц. серную кис-
лоту и через склянку с натронной известью [2,3]. По другой схеме А.
пропускают через колонку с окисью алюминия (10 меш) и затем че-
рез серную кислоту [3].
Ниже приведены некоторые характерные примеры синтезов с А.:
СН,
1 [1]) 2СиН5СН3-|-НС^СН—сн-у—СН3
О\’а О\’а
NaNH.. I НС=СН |
2 [4]) СН3СОСН3-' СН,С--> СН3С—С s= СН —>
!! I
СИ, сн3
он
н + I
—> СН3С — С^СН (40 — 46%, общий)
I
СН3
о
Л NaO /CsCH НОХ С = СН
। । /А / А
N а, N Н з \ / fl Н + ||
3 [5]) HCs-CH---HC = CNa— >
Ag.O
4 121) C1CH,CO2H-|-HCsCH--------—* C1CH,CO,CH — CH.,
I и . 2 I 42—49%
Na(NHs) H-C4IIsBr
5 [6]) HC ® CH —-- - НС = CNa уууцу w-C4H9C CH
AICI3
6 [7]) (CH,),CHCH.,CH,COC1 HC < i!----------+
— (CH3).,CHCH2CH.,COCH -- CHC1
|3-Хлорв11нш1нзоа.милкетон
77
- ГЧЬ ПС Ci! C ИГ ГН n С.ДЦСН.СНСНО
I [3]) HO=-:Cli----.HC -sCMgBr---------------- CcH5CH•= CHCHC r-CH
I
OH
1. Рейхерт Дж.. Ньюленд Дж., (-Синтезы органических препаратов»,
ИЛ, 51., 1919, co. 1, стр. 202.
2. У а ii л и Р._ «Синтезы органических препаратов», ИЛ, М., 1953, сб. 4, стр. 126.
3. С к а т т е б о ,т Л.. Д ж о н с Э. Р. X., У a ii т и н г 31. Ч., «Синтезы органи-
ческих препаратов», Г1Л, 31., 1961. co. II, стр. 69.
4. К о ф ф м а и Д., (Синтезы органических препаратов», ИЛ, 31., 1952, сб. 3,
стр. 198.
5. С а у и д е р с Дж., "Синтезы органических препаратов», ИЛ, 31., 1953, сб. 4,
стр. 600.
6. К '-> м и б с л л К., Кэмпбелл Б., «Синтезы органических препаратов»,
ПЛ, 31., 1953, сб. 4, стр. 111.
7. П р а й с Ч., Паппа л а р д о Дж., «Синтезы органических препаратов»,
ИЛ, 31., 1953, сб. 4, стр. 529.
АЦЕТИЛЕНДИКАРБОНОВАЯ КИСЛОТА, НО;!СС=ССО2Н,
т. пл. 176 .
А. к. получают из а, Р-дибромянтарной кислоты при действии
метанольного раствора едкого кали, калиевую соль Л. к. выделяют,
подкисляют и экстрагируют эфиром [II. А. к. применяется как
диенофил (см. также Ацетилендикарбоновой кислоты дпметнловый
эфир).
1. А б б о т т Т., А [> и о л и д Р., Томпсон Р., «Синтезы органических пре-
паратов», ИЛ, 31. 1919, сб. 2, стр. 71.
АЦЕТИЛЕНДИКАРБОНОВОЙ КИСЛОТЫ ДИМЕТИЛОВЫЙ
ЭФИР, CH;iO2CC^CCO2CH;). Мол. вес 110,11, т. кип. 96—98", 8 леи,
уд. вес 1,10; обладает сильным лакримогенным и кожно-нарывным
действием.
А. к. д. э. получают из калиевой соли ацетилендикарбоновой
кислоты 111.
А. к. д. э. широко используется как диенофил в реакции Диль-
са — Альдера (см. Растворители для реакции Дильса — Альдера).
Реакцией А. к. д. э. с гситафульвеном (1) получают бесцветный
аддукт (2), который легко окисляется кислородом воздуха в темно-
синий азулен (.3) 12J. Менее активные диенофилы с (1) не реагируют.
Новый эффективный синтез триметилового эфира геми.меллптовой
кислоты осуществляют пиролизом смеси А. к. д. э. и метилового эфи-
78
ра а-пиронкарбоновоп-6 кислоты (Бенкерт и сотр. 131). При сильном
нагревании, необходимом для реакции присоединения, образующий-
ся аддукт теряет лактонный мостик с отщеплением СО.,
со2сн3
со2сн3
с
HI
с
I
со3сн3
210-215°
70%
СО2СН3
СО2СН3
со,с.н3
+ со2
I. Хзнтресс Э., Лессли Т., Борнштей и Дж., (.Синтезы органиче-
ских препаратов'). ИЛ, М., 1953, сб. 4, стр. 186.
2. Doer inp W., von, \V i ! e у D. W., Tetralieriron, 11, 183 (1960).
3. W ciiker t E., Johnston D. B. R., Dave 1\. G., J. Org. Chem., 29,
2534 (1964).
АЦЕТИЛЕНДИКАРБОНОВОЙ КИСЛОТЫ ДИЭТИЛОВЫЙ
ЭФИР, H.-,C2O2,CCe=CCO2C2H-„ Мол. вес 170,16, т. кип. 96—98 8мм,
получение 111.
1. И tin tress Е. Н., Lesslie Т. Е., Born stein J., Org. Svn., Coll.
Vol., 4, 330 (1963).
АЦЕТИЛЕНИД ЛИТИЯ, LifeCH. Мол. вес 31,97.
Методики получения амида натрия в жидком аммиаке и его ре-
акции с ацетиленом с образованием ацетиленида натрия применимы
также для получения амида лития и А. л. III. Для превращения ке-
NHgFMT) НСыСН
Li---------- LiN’H.,--—' LiC^CH
-Н, - -Ml,,
тонов в этинилкарбинолы обычно используют ацетиленид натрия (ре-
акция Нефа), хотя часто применение А. л. дает определенные пре-
имущества, особенно в случае а,|3-ненасыщенных кетонов. Например,
Р-ионон (1) реагирует с А. л. с образованием карбинола (2) с 79%-ной
конверсией и выходом 95% 12]. С ацетиленидом натрия в аналогич-
ных условиях (3 час при —34') конверсия составляет 2796, а выход
74%. А2,4’“-Октатрпеналь (3) дает с А. л. этииилкарбпнол (4) с вы-
ходом 72%, тогда как с ацетиленидом натрия выход более чем в три
79
раза ниже [31.
LiC — CH(NH-)
СН3СН = СНСН - СНСН --= снсно-—— -
(3)
—СН3СН -= СНСН СНСН снснс = сн
I
он
(4)
Если кетон нестоек к действию жидкого аммиака, А. л. можно
получить в жидком аммиаке обычным способом и осадить эфиром.
После удаления аммиака к суспензии А. л. в эфире добавляют
кетон. Таким методом очень нестойкий 2-метил-3-бутокси-Д2-цик-
логексенои-1 (5) превращают в этин ил карбинол (6), который
перегруппировывается с отщеплением бутанола и дает (7) [4].
хОВи
« ОВи
С СН
(7)
1. R а р h а е 1 R. A., «Acetylenic Compounds in Organic Syntheses», Academic
Press, 1955, p. 193—194.
2. Oroshnik W., M e b a n e A. D., J. Am. Chem. Soc., 71, 2062 (1949).
3. Raphael R. A., loc. cit. [1], p. 197.
4. E s c h c n m о s e r A., Schreiber J., J ulia S. A., Helv. Chim. Acta,
36, 482 (1953).
АЦЕТИЛЕНИД ЛИТИЯ — ЭТИЛЕНДИАМИН,
LiC^CH • H2NCH2CH2NH2(LiC=CH ЭДА).
Эта стабилизированная форма ацетиленида лития[1] не требует
применения жидкого аммиака. Ее можно использовать в виде сухого
порошка, растворимого в таких растворителях, как диглим, диок-
сан, ДМФА и ДМСО.
Растворимость при 25° (г/100 г): в гексане 0, в бензоле 0,008,
в ксилоле 0,04, в диэтиловом эфире 0,05, в ТГФ 0,4, в дпоксане
0,6, в «-пропиламине 17,0, в н-бутиламине 12,1, в дн-«-бутилампне
0,03.
Реакция с галогенидами. Типичная реакция замещения галогена
проведена с 1-фтор-5-хлорпентаном в ДМСО (Паттисон и Дир [2]);
7-фторгептии-1 получается с очень высоким выходом.
F(C1I.,)5C1 -1-LiC СН .ЭДА —F(CH.,)5C = CH
'.'2\
Присоединение к кетонам. Хаффмен и Арапакос 131, используя
ацетиленид натрия в жидком аммиаке или ацетилен и 2-мсгилбути-
лат калия, превратили кетон (1) в этнннлкарбинол (2), но с очень
80
низким выходом, тогда как 1лС=СНЭДА реагирует гладко и с
высоким выходом.
1. BeumelO.F., Jr., Н а г г i s R. F., J. Org. Chem., 28, 2775 (1963); 29, 1872
(1964).
2. P a t t i s о n F. L. M., D e a r R. E. A., Can. J. Chem., 41, 2600 (1963).
3. Huffmann J. W., ArapakosP. G., J. Org. Chem., 30, 1604 (1965).
АЦЕТИЛЕНИД НАТРИЯ, CH^CNa. Мол. вес 48,03.
Реагент с целью применения его для синтеза 1-этпнилцнклогек-
санола получают, пропуская ацетилен в 1 л жидкого аммиака при
перемешивании и медленном добавлении 23 г натрия [II. Смесь об-
___. /____. /С-.^СН н+ ._____х СщСН
\=O4-NaC = CH—< X ____________>< ><
---/ \/ -ONa 65-75% 4--------/ ХОН
рабатывают циклогексаноном (1 моль) в течение 1 час и оставляют
для испарения аммиака. Твердый остаток разлагают льдом и во-
дой, смесь подкисляют и продукт реакции экстрагируют эфиром.
1. Саундерс Дж., «Синтезы органических препаратов», ИЛ, М., 1953, сб. 4,
стр. 600.
N-АЦЕТИЛ ИМИДАЗОЛ (1). Мол. вес 110,12, т. пл. 101,5—102,5.
А. получают растворением имидазола в небольшом избытке ук-
сусного ангидрида с последующей отгонкой в вакууме образовавшей-
ся уксусной кислоты и избытка уксусного ангидрида (выход коли-
чественный) [1]. При кипячении А. с эквимолярным количеством
пиррола (90 мин) происходит переацетилпрование с образованием
N-ацетилпиррола (выходы около 90%) [21, который трудно получить
обычными методами. Прямое ацетилирование пиррола дает смесь
2- и 3-ацетплпирролов примерно в равных количествах.
NCOCH3
(1)
1. ReddyJ.S.,MandellL., GoldsteinJ.H.,J. Chem. Soc., 1963, 1414.
2. Reddy J. S., Chem. Ind., 1965, 1426.
81
S-АЦЕТИЛМЕРКАПТОЯНТАРНОЙ КИСЛОТЫ АНГИДРИД (1),
Мол. вес 174,18, т. пл. 77:
А. к. а. получают присоединением тиоуксусной кислоты к малеи-
новому ангидриду; выход 83% 111.
Метод введения меркаптогрупп в белки (а также полиспирты,
например декстрин или поливиниловый спирт) состоит в добавлении
твердого А. к. а. к раствору белка при pH 7 в атмосфере азота 121.
Продукт гидролиза ангидрида удаляют на анионообменнике, соли —
на смешанном ионообмешшке пли диализом и ацилированный
протеин (2) выделяют лиофилизацией. Связь ацетил —S легко рас-
CIKCOSCHCO ГГКГ,|ГП и
. . рн7 CHsCOSCHCO.Jl pH 11,3
Му Н2Х-Протеин—> | -----'
СНоСОХН-протеин
(1) (2)
— HSCHCO,Xa
I
CHiCOXH-протеин
(3)
щепляется в разбавленном растворе едкого натра при pH 11,5 (3).
1. Brow п R., Jones W. Е., Pinder A. R., J. Chem. Soc., 1951, 2123.
2. Klotz 1. .'ll., H e i n e у R. E., J. Am. Chem. Soc., 81. 3802 (1959).
АЦЕТИЛ НИТРАТ, CH:iCO2.\’O2. Мол. вес 105,05. А. потенци-
ально взрывчат.
А. получают добавлением 4,5 г 70%-ной азотной кислоты к
35 м.1 уксусного ангидрида при 25—30е. А. реагирует с простыми ал-
кенами, давая смеси 0-нитроацетатов, нитроолефинов и р-нитронп-
тратов 111, например:
ОАс
AcONO- I
1) (СН3),С = СН2------ (СН3),ССН.,ХО,+
И",
СН3 0X0,
I I
+ сн2 = ССН,ХО, (СН3)2ССН,ХО,
3% Г1,,
ХО,
X//4-NO,
30% зо%
Стирол, стильбены и 1,1-диарилалкены дают нитроацетаты с вы-
ходом 40—70'%.
1. Во г И w ell F. G., G а г b i s с h Е. W., Jr., J. Am. Churn. Soc., 82, 3588
(1960); J. Org. Chem.. 27, 2322, 3049 (1962); ibid., 28, 1765 (1963).
82
а-АЦЕТИЛ-р-ЭТОКСИ^-КАРБЭТОКСИАКРИЛАМИД,
О COCH.)
II I
НХ-С-С = СНОС.2Н5
I
СО,С,Н3
Мол. вес 229,23, т. пл. 90;.
Этот реагент применяют для определения N-концевого остатка
пептида или белка [11.
1. Dewar J. Н., S h a w G., J. Chem. Soc., 1961, 3254.
а-АЦЕТОКСИАКРИЛОНИТРИЛ,
ОСОСНз
СН2 = С—CN
Мол. вес 110,10, т. кип. 173°/772 мм.
А. получают присоединением цианистого водорода к кетену;
лучший выход можно получить в присутствии мягких основных ка-
тализаторов, таких, как ацетат калия 11].
ОН ОСОСН3
HCN I СН.,=С. = О I
сн2 = С = О > СН, - CCN —=-> СН2 = С—CN
А. используют как диенофил; гидролиз аддукта приводит к ке-
тону, как показано на примере синтеза дегндроноркамфоры [2].
C-CN
।
ОСОСНз
62
ососн,
ОН"
CN 82%
Другой пример — синтез 7-нзопропилиденбнцикло-12,2,1 ]-геп-
тен-5-она-2 [3|:
1. D е a k i n S.. Wilsmore N. Т. М., J. Chem. Soc., 97, 1971 (1910): .1 о h n-
s t о n F., N e w t о n L. W., пат. CHIA 2395930 (1946) [C. A., 40, 4078 (1946)];
Hagemeyer H. J., Ir.. Ind. Eng. Chem., 41, /65 (1949).
2. Bartlett P. D., TateB. E,, J. Am. Chem. Soc., 78, 2473 (1956).
3. D e P u у С. H., StoryP. R, J. Am. Chem. Soc., 82, 627 (1960).
1-АЦЕТОКСИБУТАДИЕН. Мол. вес 112,16, т. кип. 42—43", 16 льи
51—52с/30 мм.
Получение. А.— енолацетат кротонового альдегида — можно по-
лучить кипячением этого альдегида с уксусным ангидридом и аце-
83
татом натрия [1]. Эта равновесная реакция дает низкий выход А.
СНО СНОСОСНз
| Кипячение 4 час I!
CH + (CH3CO)2O + CHsCO2Na ------—------- СН
|| 440 ? 2и0 г ° |
сн сн
I II
СН3 СН2
24 0 г
([2], использована такая же методика, выход 35%). Непрореагиро-
вавший кротоновый альдегид (сильный лакриматор) удаляют в ви-
де бисульфитпого производного. Лучший способ получения А., опи-
санный Хагемайером и Халлом 131,— это реакция кротонового аль-
дегида с изопропенилацетатом в присутствии каталитических ко-
личеств н-толуолсульфокислоты и ацетата меди (роль последнего
не выяснена). Кротоновый альдегид добавляют к реакционной сме-
си в течение 2—3 час с одновременным удалением ацетона для
смещения равновесия. Выход дважды перегнанного А. 90%.
ОСОСНз
СН3СН-СНСНО СН.,~Н’СН3-| n-CH3C6H4SO3H -I Cu(OAc).,
10.10 г '> кг 20 г аг 90%
- - CI J., - СНСН -- СНОСОСНз |-СН3СОСН3
Реакция Дильса — Альдера. А. использован Хиллом и сотр.
|4| в одностадийном синтезе предшественника лнкорина.
36час при 110°
+ с6н5сн3 ~ ’
90 мл
Н2С=СНСН=СНОДс +
42,42
Хилл и Карлсон [5] осуществили прямой синтез ароматического
цикла реакцией А. с производным ацетилена. А. нормально при-
ОАс
I
I +
'Ч
ОН ГН ОАс
со.,сн3
I
с
с
I
со.,сн3
84
Соединяется к хинонам, однако в уксусной кислоте при нагревании
на кипящей водяной бане аддукт частично ароматизуется [61.
о
1. \V i с h t е г 1 е О., Н u d 1 1 с k v Лк, Coll. Czech., 12, 564 (1947).
2. В lane Р. Y., Helv. Chim. Acta, 44. 1 (1961).
3. H a g e m e у e r H. .1. Jr., Hull D. C., Ind. Eng. Chem., 41, 2920 (1949).
4. H i 1 1 R. K., J о u 1 e J. A., Loeffler L. J., j. Am. Chem. Soc., 84, 4951
(1962).
5. li i 1 1 R. K-, Carlson R. M., J. Org. Chem., 30, 2414 (1965).
6. F 1 a i g \V„ Aim., 568, 1 (1950).
2- и 3-АЦЕТОКСИПИРИДИНЫ, C-,HjNOCOCH,, 111. Мол. вес
137,14.
Получение. 2-Изомер (т. кип. 110—112710 дви) получают дейст-
вием хлористого ацетила на натриевую соль 2-оксипирпдина; 3-изо-
мер (т. кип. 92 79 мм) — из 3-оксипиридина и уксусного ангидрида.
Ацетилирование. Эти реагенты применяют для ацетилирования
спиртов, фенолов, аминов, а также реакционноспособных аромати-
ческих соединений по Фриделю — Крафтсу. 2-Лцетоксипиридин
более активен, чем 3-изомер.
1. Ueno Y., Т а к а у а Т., Imoto Е., Bull. Chem. Soc. Japan, 37, 864 (1964).
N-АЦЕТОКСИФТАЛ ИМИД (2). А. получают реакцией N-ок-
епфталимида натрия с хлористым ацетилом |1]. А.— специфический
реагент |1, 2] для N-ацетилирования мурамовой кислоты (1), кото-
рая с уксусным ангидридом в пиридине дает продукты внутримоле-
(1) 358 лг (law)
25°
70%
НО’
СН3СН-О-
СО2Н
(2) 2 же
МОАс + (C2H5)3N + СН3ОН —>
СН,ОН
NH
Ас ОН
1 же
(3)
85
кулярной циклизации (лактамы). Осава и Джинлоз 121 обрабаты-
вают раствор (I) в метаноле при О' 2 же А. (2) п 1 же триэтиламииа
и смесь выдерживают при комнатной температуре в течение 20 час.
После выпаривания смеси остаток экстрагируют водой, раствор
фильтруют, доводят до pH 3,5 амберлитом 1R 120 и экстрагируют
этилацетатом. Сиропообразный продукт самопроизвольно кристал-
лизуется; перекристаллизацией из смеси метанол — этилацетат полу-
чают чистый (3) с 70||(1-иым выходом. Реакцией с 1 же X. получается
смесь соединений (I) и (3) III.
1. Carroll Р. 31., Nature, 197, (594 (1963).
2. О s a w а Т., ,1 е a n 1 о 7 R. W., J. Orc. Chem., ЗС. 448 (1965).
Зр-АЦЕТОКСИ-А5-ЭТИЕНОВАЯ КИСЛОТА,
Мол. вес 360,48, т. пл. 238°.
А. получают окислением ацетата прегненолона гипобромптом [1].
Соответствующий хлорангидрид применяют для расщепления на
антиподы 1а-оксидии.11клоиентад11ена 121, щ/с-щ/с-декалола-! [31
и /ирц«с-3-/п/7е/и-бутилцпклогексанола |4| (в этом случае общепри-
нятый метод расщепления через соли кислого фталата и алкалоидов
к успеху не привел).
1. Staunton J., Eisenbraun Е. ,1., Org. Syn., 42, 4 (1962).
2. W о о d ward R. В., Kat 7. T. .1., Tetrahedron, 5, 70 (1959).
3. D j erassiC., Staunton J., J. Am. Chem. Soc., 83, 736 (1961).
4. D j e г a s s i C., W a r a w a E. J., Wolff R. E., Eisenbraun E. J.,
J. Org. Chem., 25, 917 (1960).
АЦЕТОНДИКАРБОНОВАЯ КИСЛОТА, HO2CCH2COCH2CO2H.
Мол. вес 146,10, т. пл. 138' (с разд.).
А. к., получаемая с высоким выходом при действии олеума на
лимонную кислоту при 0—30' |1| нестабильна; ее необходимо пере-
СН.,СО2Н СН,СО,Н
I H..SO, I
НОССОоН----------------- C---0-LC0-H,0
I - 50-90% (сырой) | '
СН.,С0.,Н сн,со.,н
кристаллизовать из этплаиетата пли превратить этерификацией по
Фишеру |2| в диэтиловый эфир (т. кип. 145—148 /17 дни).
Классический синтез тропинона по Робинсону состоит в конден-
сации янтарного диальдегида и метиламина с А. к. |3|. Позднее
Шёпф [41 проводил реакцию в буферном растворе при pH 5 и ком-
натной температуре. В этих условиях, подобных физиологическим,
86
промежуточно образующаяся дикислота спонтанно декарбоксили-
руется до тропинопа (выход более 90%). (См. также Глутаровый
диальдегид.)
СНД.О.Л
СН,—СНО I
| " -СО
СН,-СНО ' ' I
сн,со,н
C11, — сн — снсо,н
i I
мен., со
! I
СН, — сн - снсо,н
сн - сн,
I I
хсн,со
сн,
сн, — сн — сн.
Грони нон
1. Адамс Р., Ч a ii л ь с Г., Р а с с г. с й л е р К., «Синтезы органических
препаратов», ИЛ, М., 1949, сб. 1, стр. 70.
2. А даме Р., Ч а й л ь с Г., «Синтезы органических препаратов», ИЛ, М.,
1949, сб. 1, стр. 536.
3. Robinson R., .1. C.licin. Soc., 111. 762 (1917).
4. S c h 6 p f C., L c li m a n n G., Лип., 518, 1 (1935).
АЦЕТОНЦИАНГИДРИН, (CH:!),C(OH)CN. Мол. вес 85,10, т. кип.
80' 15 лич.
Получение, а) А. иолечагог добавлением 40%-иой серной кислоты
к водному раствору ацетона и цианистого натрия при 10—20 11].
б) Реакцией цианистого калия с бисульфитным производным ацето-
на получается менее чистый продукт, который необходимо исполь-
зовать сразу |2|.
Применение для перециангидринирования. 17-Моноциангидрин
3,17-дикето-А4-стероида получен с высоким выходом путем обмена
цианистым водородом при реакции с А. 131.
Применение для присоединения цианистого водорода. А. дает
аддукты HCN с бензальацетофеноном л другими а,|3-непредельными
кетонами в присутствии 5—10%-ного водного раствора карбона-
(СН C(O1I)CN
с6н-,сн -= снсос6н5-----------г с6н-,сн — сн,сосвн5
CN
та натрия как катализатора |4]. Сопряженные стероидные
кетоны присоединяют HCN обычно с образованием смесей п с пло-
хим выходом. Гладкое присоединение HCN к Д'-ба-холестенону-З
проводят кипячением (3,5 w) раствора кетона нА. в смеси
ТГФ — метанол с небольшим количеством водного раствора карбоната
87
натрия (Жюлиа и сотр. [5]). После упаривания в вакууме хромато-
графией отделяют 1,2 г исходного вещества и получают 1а-циан-
5а-холестанон-3 с хорошим выходом.
1. К о к с Р., Стормонт Р., «Синтезы органических препаратов», ИЛ, М.,
1949, сб. 2, стр. 566.
2. Вагнер Э., Байзер М., «Синтезы органических препаратов», ИЛ, М.,
1952, сб. 3, стр. 193.
3. Е г с о 1 i A., R u g g i е г i Р., de, J. Am. Chem. Soc., 75, 650 (1953).
4. В e t t s В. E., Davey W., J. Chem. Soc., 1958, 4193.
5. J u 1 i a S., L i n ar es H., S i m on P., Bull. soc. chim. France, 1963, 2471.
ONOa
I
АЦЕТОНЦИАНГИДРИНА НИТРАТ, (CH3)2CCN . Мол. вес
130,11, т. кип. 65—66710 мм.
А. н. получают нитрованием ацетонциангидрина смесью дымя-
щей азотной кислоты и уксусного ангидрида [1]. Внимание-. А. н.—
умеренно взрывчат. Его применяют для превращения первичных
и вторичных аминов в нитрамины [2]. Эта реакция интересна тем,
ono2
. ।
\ (CH.,)2CCN
NH----------->
О N-NO2 + O N — С(СН3),
X_____/ \____/ I
(1) (2) CN
что нитрование проводится в нейтральной или щелочной среде.
Например, N-нитроморфолин [(1), т. пл. 543] получают с выходом
57—64% реакцией морфолина с 2 же А. н. [11; к реакционной смеси
добавляют соляную кислоту, чтобы растворить непрореагировавший
морфолин и побочный продукт (2), а соединение (1) извлекают хло-
ристым метиленом. При нитровании других аминов, особенно в боль-
ших масштабах, в качестве растворителя для лучшего контроля
температуры можно применить ацетонитрил.
А. н. используют для нитрования соединений, содержащих ак-
тивную метиленовую группу, способную к образованию натриевых
производных; эта реакция лежит в основе общего метода синтеза
68
а-нитроэфпров [3].
ONO,.
NaCH (CO.,C.,H5), !СН±УЛ СН (СО.,С.,Н5).,-Г (CH..).,CONaCN
I
\’О2
1. Freeman J. Р., Shepard 1. G,, Огр. Syn., 43, 83 (1963).
2. Е ш m о n s W. D., Free m a n .1. Р., ,1. Am. Chem. Soc., 77, 4.387 (1955).
3. Е т т о n s \\'. I)., Free т а п .1. Р., .1. Лт. Chem. Soc., 77, 4391 (1955).
АЦЕТОУКСУСНОЙ КИСЛОТЫ трет-БУГИ,НОВЫЙ ЭФИР,
СН;!СОСН2СО2С(СНМол. вес 158,19, т. кип. 85720 мм.
А. к. б. э. получают реакцией дпкетена с треш-бутанолом в при-
сутствии этилата натрия 111 и используют в новом синтезе ацилои-
нов 121. Енолят Л. к. б. э. алкилируется с образованием соединения
(2), в сс-положение которого вводится бепзоплоксигруппа действи-
ем перекиси бензоила на енолят (3) с образованием соединения (4).
11 ОС (СН;/-С3Н,,Вг.
NaOBt NaOl't
СН^С-------О-------------‘.Н. с -он ---------------->•
I ! |
СН2- С -о СН.,С().,С (СНз)з
Дикетен (I)
О
I! | Nall, бензол Н1,т.»|евое (С.НЩС),)., 0»
> CH-.C— (.НСОоС (СН3)3 ~ производное *
(2) ‘ (.3) Ь//«
О С5Нп-н О
II I TsOl-I' ОН- II
СН3С- ССО..С (СН3)3_'77..'._+ СН3С - СНС,Н1Гн + (СН3)2С = СН2 -р со2
I ' 51% I
ососвн5 он
(4) Окт.чнон-2-ол-З
При нагревании эфира (4) с каталитическим количеством/7-толу-
олсульфокислоты происходит расщепление сложноэфнрной трст-
бутильиой группы п декарбоксилирование; последующее омыление
в мягких условиях дает ацилоин (чувствителен к щелочам).
Общий метод синтеза а,|3-непредельных кетонов типа (2) |3|
включает конденсацию по Кневенагелю А. к. б. э. с альдегидом
и нагревание продукта (1) с каталитическим количеством п-толуол-
() О
II Пиперидин lj TsOU. 1">0°
CH3CCH,CO,C (СН3)3-|-СН3СНО------------> СНзСССОХ (СН3)3-----------------
ЬА-О ц
сн
I
сн3
(11
о
I!
—* СН3ССН -= СНСН3 У- (СН3).2С СН., I со2
(2)
89
сульфокислоты, которое сопровождается отщеплением СО., и изо-
бутилена и образованием непредельного кетона (2).
1. Lawesson S.-О., Gronwall S., San cl berg R., Org. Svn., 42, 28
(1962).
2. L a \v esso n S.-О., G r 6 n w a 1 1 S., Chem. Scand., 14, 1445 (1960).
3. L a w esso n S.-О., Larsen E. H., S n n d s t г о m G., J a k о b s e n H. J.,
Chem. Scand., 17, 2216 (1963).
АЦЕТОУКСУСНОЙ КИСЛОТЫ ФТОРАНГИДРИД,
СНзСОСНХХЖ Мол. вес 104,08, т. кип. 132—134-.
А. к. ф. получают реакцией дикетена с безводным фтористым
водородом 111:
HF
СН, = С —CH,____. CH3COCH,COF
| I ' 60".,
O-C-=O
Реагент можно хранить при О' в течение недели, но при комнатной
температуре происходит медленное разложение с образованием де-
гпдрацетовой кислоты. А. к. ф. непригоден для ацилирования по
Фриделю — Крафтсу, но используется для ацетоацетилирования
спиртов и аминов.
CH3COCH,COF -1°” , CH,COCH,CO,R -L HF
as-8 6%
CH3COCH.,COF CH ,COCH.,COXHR —- RNH3F
"6 — 87%,
1. OlahG. А., К ti li n S. J., J. Org. Chem., 26, 225 (1961).
С) О
АЦИЛОВ ПЕРЕКИСИ, RCO,CR. Получение [1]: (a) RCOCI +
-HNaXC; (6) RCOCH-H2O.,+NaOH.
А. и. применяют для алкилирования 2-оксинафтохинона-1,4 12].
Эту реакцию проводят в уксусной кислоте при 90—95J. Побочными
продуктами являются RCO,H, RH, RR, ROCOCH3, ROH, RCO,R.
О О
М- 1916 Н
Недостатки двух способов получения А. и., основанных на ре-
акции Шоттен — Баумана, заключаются в образовании в щелочной
среде стойких эмульсий и снижении выхода в результате гидролиза
хлорангпдрпдов. Зпльберт и Сверл [31 разработали улучшенный
метод, включающий ацилирование 50—65%-ной перекиси водоро-
да хлорангидридамп кислот в смеси эфир — пиридин; чистые А. и.
получают с почти количественным выходом. Например, к раствору
90
хлорангидрида миристиновой кислоты в эфире прибавляют при О3
2/v-(;H3(CH;p.2('()Cl , бО'^НоО^ -Пиридин - |CH3(CIi.>)|2CO.;|2
0,1 МОЛЯ В 1 7; > At.l <).7о моля 0,12 моля
перекись водорода п затем при перемешивании и охлаждении по
каплям добавляют пиридин. А. и. осаждается по мере образования.
После прибавления реагентов ледяную баню убирают, продолжают
перемешивать в течение 1 час, добавляют эфир до получения гомоген-
ного раствора, промывают разбавленной! соляной кислотой, 5%-ным
бикарбонатом калия, водой п сушат сульфатом натрия. Содержание
А. и. определяют йодометрическим титрованием. По этой улучшен-
ной методике получены очень хорошие результаты со следующими
хлорангидридами [41:
С Нг) СОС1 (СНг) ,lCOC1
- (« = 5, 6, 7, 8) <л = Н г' А
1. Fieser L. F„ Leffler М. et al., J. Am. Chem. Soc.. 70. 3178 (1948).
2. FieserL.F., L e f f 1 e r M. et al., J. Ain. Chem. Soc., 70, 3174—3215 (1948).
3. S i 1 b e r t L. S., S w e r n D., J. Ain. Chem. Soc., 81, 2364 (1959).
4. F i e s e r L. F., Nazer Al. Z., Schir m e r J., unpublished results.
АЦИЛЫ ФТОРИСТЫЕ [1|. А. ф. получают обработкой 2 молей
ангидрида соответствующей кислоты 45 а (2,25 1моля)жпдкого безвод-
ного фтористого водорода при температуре —10° и перемешивании
магнитной мешалкой, сердечник которой закрыт тефлоном [2|.
Реакционную смесь выдерживают 1 час при —10', затем 2 час без
охлаждения, обрабатывают безводным фторидом натрия (15 г) для
удаления избытка фтористого водорода, фильтруют и перегоняют.
,О
RC.^'
' О у- HF — RCOF — RCO,H
R4>
Операции с безводным фтористым водородом необходимо проводить
в аппаратуре из плавленого кварца пли полимерного материала
(полиэтилена, полипропилена, тефлона, кель-F). Хлорангидриды
кислот превращают в соответствующие А. ф. или по вышеописанной
RCOCI ; HF — - RCOF y F1C1
методике, пли обработкой 2 молен хлорангидрида кислоты безвод-
ным газообразным фтористым водородом, пропуская его со скоростью
около 1 г/мин в течение 1 час при температуре от —5 до 0; и переме-
91
шнванип. Этим методом получены А. ф. из 21 кислоты с выходом
78-93%.
Формилфторид получают реакцией смешанного ангидрида ук-
сусной п муравьиной кислот с фтористым водородом при атмосфер-
ном давлении с непрерывным удалением образующегося формпл-
фторида (т. кип. —29 ) 121. Формилфторид используют для введения
zO
CH.;C/Z
о HF ——* HCOF + CH3CO,H
HQ. '
Ч)
альдегидной группы по Фриделю — Крафтсу и формилирования по
Шоттеи — Бауману.
Применение безводного фтористого водорода для получения А. ф.
впервые было предложено Колсоном [31 и Фреденхагеном [41 и
развито в препаративный метод в работе Ола и Куна |2|. Методика
получения бензоилфторида приведена в сб. «Синтезы органических
препаратов» 151.
1. Contributed by Olah G. A., К u h n S. J. (formerly of the Dow Chemical
Co., Framingham, .Mass.)
2. О 1 a h G. A., К u h n S. .1., J. Ani. Chem. Soc., 82, 2380 (1960); J. Org. Chem.,
26, 237 (1961).
3. Co 1 s о n A., Bull. soc. chim. France, [3[, 17, 55 (1897); Ann. chim., [7], 12,
255 (1897).
4. F r e tl e n h a g e п K-, Z. physik. Chem., Л164, 189 (1933); Fredenha-
gen K., Cadenbach G., ibid., A164, 201 (1933).
5. О 1 a h G. A., К u 11 n S., Org. Syn., 45, 3 (1965).
БАРИЯ ПЕРМАНГАНАТ. Мол. вес 375,22, растворимость 205,8
а/100 г Н2О при 0J. Получение 111. Окисление: —SH -—SO3H |2|.
1. Vanino L., «Handbuch (кт Praparativen Chemie», 3 rd cd., 1925, 479.
2. H о f m a n n K., Bridgwater A., Axe! г о d Л. E., J. Am. Chem.
Soc., 71, 1253 (1949).
БЕНЗАЛЬДЕГИД, С„Н,-,СНО. Мол. вес 106,12, т. кип. 179°
(57—59 /8 лш), уд. вес 1,046.
Препарат обычно вполне пригоден при первом открывании склян-
ки. Однако при дальнейшем использовании, если препарат дает
положительную пробу на бензойную кислоту с бикарбонатом натрия,
его промывают для удаления кислоты 5—10%-ным раствором би-
карбоната натрия, сушат над безводным карбонатом натрия и пере-
гоняют в вакууме в присутствии небольшого количества цинковой
пыли, защищая от действия воздуха.
БЕНЗИЛАМИН, C6H5CH2NH2. Мол. вес 107,15, т. кип. 184,5°
(70—7Г/10 мм), уд. вес 0,982, смешивается с водой.
а,[3-Эпоксикислоты. аф-Эпоксикислоты, получаемые окисле-
нием аф-непредельных кислот перекисью водорода в присутствии
вольфрамата натрия, с Б. дают исключительно а-беизилампноф-
оксикислоты, гидрогенолизом которых в присутствии катали-
о
СН-; Н п.о. СН3 /ч /Н едцен.кчь
>С = С< ------->- /С----------------------->
W СО,Н Н' ccui
СН2С6Н5
I
ОН NH ОН NH.,
СН3 | I ,Н н., Pd СН3 I I /н
-> )C-CZ ---------------> /С~с<
Н-' CO,H Hz ZCO.,H
затора (ЗО?6 Pd на угле) получают незамещенные окспамино-
кислоты |1|. Эта реакция стереоспецпфична: шракс-кпслоты дают
эритро', а цне-кпелоты — шрео-изомеры. Например, из транс-
кротоновой кислоты получен о,ь-треонпн.
Восстановительное аминирование. Обработка 3-кетостероидов
аммиаком и водородом в присутствии Pd — С дает лишь низкий
выход 30-аминостероидов. В этой реакции лучше использовать
Б., подвергая гидрогенолизу промежуточно образующийся вторич-
ный амин [2]. Реакция специфична для З-кетогрупны стероидов;
93
карбонильная группа в положениях 17 и 20 не затрагивается. На-
пример, гидрированием прогестерона в присутствии Б. и Pd — С
получают 30-амнно-5Р-прегнанон-2О с выходом 71 ?6.
1116; L i w s с h i t z V., R a b i ti s о h n Y., Haber A., ibid., 1962, 3589.
2. Sch in i t t .1. et al., Bull. soc. chilli France, 1962, 1846, 1855.
БЕНЗИЛАТ НАТРИЯ, C,fH3CH2ONa. Мол. вес 130,13.
Бензилат натрия в бензиловом спирте отщепляет мезильную груп-
пу в З'-мезилате (1) с сохранением окисной связи и без инверсии,
с6н5снгон
(I) 0,15 г
90-95°
+ C6H5CH2ONa с- >
9 ОД
(2)
давая 1-(2',5'-оксп-Р-о-лнксофураноз11л)-урац11Л (2) * [II. Реакция
такого типа без обращения получила название S\ 25-реакцпи. Раст-
вор Б. н. получают из бензилового спирта и раствора метилата на-
трия в метаноле.
1. С о d i и с t о п J. F., D о с г г 1. L., F о х J. J., J. Org. Cheni., 30, 476 (1965).
БЕНЗИЛЛИТИЙ, C„H5CH2Li. Мол. вес 98,07.
Б., один из наиболее употребительных реагентов для металлиро-
вания 111, получают с высоким выходом расщеплением бензилме-
тплового эфира в ТГФ при температурах от —5 до —15" 12]. Б. легко
образуется в ТГФ, но более стабилен в эфире, поэтому раствор этого
* Название, приведенное в цитированной ссылке, «1-(2',5'-э(1окси-[}-0-.тпксо-
фура иозплРурацил», по мнению авторов, неудачно. Термин еяюксид» лучше
«править для обозначения 1,2-окнсн, получаемой в результате присоединения
кислорода к двум углеродным атомам двойной связи (от epi — на).
94
реагента с концентрацией до 1,2 н. (темно-коричневый) получают
постепенным добавлением раствора бензилметилового эфира в ди-
этиловом эфире к суспензии лития в ТГФ (выход 83%) [31.
По Сейферту |4| Б. можно получить реакцией переметаллиро-
вания метиллитпя с трибензилхлорстаннаиом, который легко об-
(C6H3CH.,).,SnCl -J- 4CH-L1 3CRH,CH,Li (CH..)4Sn LiCI
89%
разуется реакцией бензплхлорпда с металлическим оловом в без-
водной среде |51. Б. получается с высоким выходом, а образующий-
ся при этом тетраметилстаинан (т. кип. 78') легко удаляется с раст-
ворителем.
1. Gilman Н„ McNinc h Н. A., J. Org. Chem., 27, 1889 (19621.
2. G i 1 m a n H., McNinc h H. A., J. Org. Chem., 26, 3723 (1961).
3. Gilman H., Schwebke G. L., J. Org. Chem., 27, 4259 (19,62).
4. S e у f e r t h D., Suzuki R., Mur phy C. J., S a b e t C. R., J. Organo-
metallic Chem., 1964, 431.
5. S i s i d о К., Takeda Y., К i n u g a \v a Z., J. Am. Chem. Soc., 83, 538
(1961).
БЕНЗИЛТРИМЕТИЛАММОНИЙИОДИД, C„H3CHA’(CH3) J.
Мол. вес 277,15, т. пл. 181—182\
В новом синтезе пурина |1| конденсацией 4-амино-1,3-диметил-
5-нитрозоурацила (1) с Б. как четвертичной солью основания Ман-
ниха получают 8-фенилтеофиллин (4) с выходом 35%. Реакция вклю-
чает конденсацию нитрозогруппы (1) с активной метиленовой груп-
пой Б., внутримолекулярное присоединение о-аминогруппы к анилу
и последующее расщепление по Гофману.
1. Т а у 1 о г Е. С., G а г с i а Е. Е., J. Am. Chem. Soc., 86, 4720 (1964).
БЕНЗИЛТРИМЕТИЛАММОНИЙМЕЗИТОАТ, СвН3СН2(СН3)3.
МО,ССвН2(СНз)з. Мол. вес 281,43.
Дегидрогалогенирование. По данным предварительного сооб-
щения [1], Б. применяется для дегидрогалогенирования а-галоген-
95
кетонов в мягких условиях. При обработке Б. в ацетоне 4|3-бромко-
простанон дает с хорошим выходом холестенон.
Вг
1. J ohnson W.S., KeanaJ.F. W., Tetrahedron Letters, 195 (1963), note 3.
БЕНЗИЛТРИМЕТИЛАММОНИЙХЛОРИД, C(iH5CH2(CH:t);,NCl.
Мол. вес 185,70.
Б.— катализатор синтеза глицидиловых эфиров, получаемых
взаимодействием натриевых солей жирных кислот с эпихлоргид-
рином [1|. Другие четвертичные аммонийгалогениды менее эффек-
тивны, по-видимому, из-за меньшей растворимости в эпихлоргид-
рине.
н-С,1Н.,чСО.А’а + С1СН.,СН - СН,----> н-СиН,,СО,СН,СН — СН,
, - 84%, неочищенный 1 -“ ,
О О /
1. Maerker G., Carmichael J. F., Port W., J. Org. Chem., 26, 2631
(1961).
БЕНЗИЛТРИМЕТИЛАММОНИЙЦИАНИД, C„H5CH2N(CH,)3CN.
Мол. вес 176,26.
Водный раствор Б. получают растворением 0,12 моля хлори-
стого беизилтриметиламмония и 0,12 моля цианистого натрия в
40—50 мл воды [II. Добавление алкилгалогенида к этому раствору
при перемешивании и охлаждении сопровождается частичным прев-
ращением его в нитрил и образованием галогенида бензилтриметил-
аммония, который действует как эмульгатор. После нагревания
эмульсии на кипящей водяной бане в течение 1—6 час нитрилы обыч-
но получаются с хорошим выходом.
RX РС6Н,,СН,К(СН3)3С.\Т —* RC.N-LC„H5CH,N(CH3)3X
1. S u g i m о t о N., F u j i t a T., S h i g e m a t s u N., A v a cl a A., Chem.
Pharm. Bull., 10, 427 (1962).
О-БЕНЗИЛФОСФОРИСТЫЙ-О.О-ДИФЕНИЛФОСФОРНЫЙ
АНГИДРИД;
о н
I
(СвН3О),Р—О—Р —ОСН,С6Н5. Мол. вес 404,28.
6
Получение [1|. При каталитическом действии 2.6-лутиднна Б. а.
реагирует со свободной гидроксильной группой сахара (1) с образо-
ванием бензилфосф’ат.а (2), который хлорируется N-хлорсукцнн-
96
имидом в (3). Гидролиз (3) дает кислый бензилфосфат (4), а бензиль-
ная группа удаляется гидрированием [2].
H CI
— CHOH 1 1 —CHOP—OCH.,C6H5 —CHOP—OCH2CeH5 1 ' ' 1 i!
— CH —сн о —сн о
1 OAc (1) j 1 OAc OAc (2) (3) OH OH 1 1 —CHOP —OCB->CcH5 —CHOP —OH । ,, - 6 pt । —CH 0 -- —CH 0 OAc OAc (4) (5)
1. ('.о г b v К. S., Kenner G. W., Т о d d A. R., J. Chem. Soc., 1952, 3669.
2. В г о ’Л и D. Al., FasmanG. D., М a g г a t h D. I., Todd A. R., J. Chem.
Soc., 1954, 1448.
БЕНЗИЛХЛОРМЕТИЛОВЫЙ ЭФИР, C6H5CH2OCH2C1. Мол. вес
156,61, т. кип. 105—109','П мм.
Получение [1]. Смесь бензилового спирта и водного раствора
формальдегида насыщают хлористым водородом при перемешивании
и охлаждении льдом.
Оксиметилирование кетонов [2]. Кетон превращают в енолят
обработкой гидридом натрия и затем алкилируют Б. э. Наилучший
растворитель — диоксан, так как в нем О-алкилирование минималь-
но. Приведенные примеры иллюстрируют высокую степень стерео-
сн3
1) NaH
2) RQCH2C1
55%
С6Н5СН2ОСН
СО2С2Н5
сн3
со2с2н5
NaH
КОСН2С1
——> X X >
с6н5сн?осн2 'сн3
специфичности этой реакции. Бензильную группу удаляют гидроге-
нолизом на подходящей стадии синтеза.
1. Н i 1 1 A. J., Reach DeW. Т., J. Am. Chem. Soc., 48, 257 (1926).
2. Graha m C. L., At c Q u i 1 1 i n F. J., J. Chem. Soc., 1963, 4634; McQuil-
I i n F. J., S i m p so n P. L., ibid., 1963, 4726; Gr a h a mC. L., McQ u i 1-
I i n F. J., ibid., 1964, 4521.
4 Заказ № 965 97
о о
БЕНЗОИЛА ПЕРЕКИСЬ, с0Н5СО2СС6Н5. Мол. вес 242,22,
т. пл. 107". Продажный препарат очищают псреосаждением из хо-
лодного хлороформа метанолом. Анализ 11J.
Катализатор полимеризации. Так как Б. и. при умеренном на-
гревании разлагается с образованием радикалов |2|, то ее можно
применять в качестве катализатора радикальной полимеризации.
Присоединение НВг против правила Марковникова. В присутст-
вии Б. и. бромистый водород присоединяется к несимметричным оле-
финам против правила Марковникова (Караш п Майо [31).
(СНз)2С-СН, + НВг (СН3)2СНСН,Вг
Хлорирование алканов. Караш [4] разработал метод низкотем-
пературного радикального хлорирования алканов хлористым суль-
фурилом, катализируемого Б. п.
RH-I SO.C1,^^£± RC1---HC1+SO.,
40 — 80°
Синтез ацилоинов. Согласно новому методу, который разрабо-
тали Лавессон и Грёнваль [4а], /7?рет-бутиловый эфир ацетоуксус-
ной кислоты, полученный из дикетена и /7гре/;г-бутанола, алкилиру-
ют и вводят бензоилоксигруппу в a-положение реакцией натриевого
производного с Б. п. При нагревании эфира до 160" в присут-
ствии каталитических количеств н-толуолсульфокпслоты происходит
отщепление /zzpe/п-бутпльной группы и декарбоксилирование. Омы-
СН2 — С О НОС(СН3)3, C.H-ONa
СН.2 — С—О 85-95%
СН2 = С—ОН
I
СН2СО2С(СН3)3
Дикетен
н-С5НпВг, C2H5ONa
67%
О СзН^-w
р I
СН3С—СНСО2С(СН3)3
NaH, С„Н»
Натриевое
(СсН5СО2)2(0°)
производное —-------------->
83%
О C5HL1-«
II | TsOH, он-
СН 3С—ССО,С(СН3)3———>
| 0 1 /о
ососвн5
р
СН3ССНС5Н11-«-|_
I
он
Октанон-2-ол-З
+ (СН3)2С=СН2 + СО2
ление бензоатной группы в мягких условиях дает ацилоин, неустой-
чивый в щелочной среде.
Бромирование N-бромсукцинимидом (NBC). Аллильное броми-
рование гептена-2 NBC проводят в атмосфере азота в присутствии
следов Б. п.; поскольку углеводород взят в избытке, выход приве-
ден в расчете на NBC [51. В случае бензильного бромирования 3-ме-
98
тилтиофена реакцию проводят без азота с избытком тиофена, при-
чем при увеличении количества катализатора время реакции со-
кращается [61.
>.U,< : I .< il.clM, н СНГ И, ‘ NBC + С113С11,СН,С11С11 = СНСН,
О, НМОЛЯ в’ЗОЛЛ ССК 0,27 моля 0,2 2 58 ’ ” ''”.|ГЛ ВГ
С-читая на NDC
£j]C\ Ж
2 моля
2,24 моля в
7 00 зм с6Н6
И—ПСЩЗ
(СбН^СОО)..
4 г
Кипячение20 мин
71-79%,
считая на N6C
Радикальное присоединение энантового альдегида к диэтиловому
эфиру малеиновой кислоты [71. Для соблюдения точного темпера-
турного режима реакцию проводят в колбе с рубашкой, заполнен-
ной кипящим трихлорэтиленом. Альдегид берут в большом из-
бытке; Б. п. прибавляют в два приема.
СН3(СН2).-,СНО+ НС—СО,С2Н5
2 МОЛЯ -1- (С|(Н-,СО.,).
НС —СО.2С,Н5 ' 1 2
1 моль
18—24 час,
8 1 — 85°
И-75%,
считая на
сложный эфир
о
и
СН3(СН2)5С—СНСО2С2Н5
СН2СО,С2Н5
Реакция с енаминами. Реакция Б. п. с енаминами дает косвен-
ный способ введения бензоилоксигруппы в соединение тппа(1) [81,
содержащее активную метиленовую группу.
(С0Н5СО2)2, н +
78% ~*
Катализатор бензильного бромирования, см. 1,3-Дибром-5,5-ди-
метилгидантоин.
Бензоилоксилирование малоновых эфиров. Б. п. реагирует с нат-
риевым производным диэтилового эфира этилмалоновой кислоты с
образованием диэтилового эфира О-бензоилэтилтартроновой кислоты
4*
99
[9]. Эта реакция является общей для 0-дикарбонильиых соединений.
NaH, С6Н, — (С,Н,СО,),
С2Н5СН(СО2С,Н8)2--------> С2Н5С(СО,С,Н,),--------->
-И2 Na +
с,н5
-> “ \С(СО,С2Н6)2
CeHjCO7
II
о
1. Б р а у н Г., «Синтезы органических препаратов», ИЛ, М., 1949, сб. 1. стр. 337.
2. W а 1 1 i n g С., Free Radicals in Solution, Wiley, 1957, 63 ff., 474 ff.
3. К h a r a s c h M. S., Mayo F. R., J. Am. Chem. Soc., 55, 2468 (1933).
4. К h a r a s c h M. S., Br own H. C., J. Am. Chem. Soc., 61, 2142 (1939).
4a. L a w e s s о n S.-О., Gronwal 1 S., Chem. Scand., 14, 1445 (1960).
5. G r e e n w о о d F. L., К e 1 1 e r t M. D., Sedlak J., Org. Svn., Coll.
Vol., 4, 108 (1963).
6. Кампень Э., T у л л a p Б., «Синтезы органических препаратов», ИЛ,
М., 1954, сб. 5, стр. 16.
7. Р a t г i с k Т. М., Jr., Е г i с к s о n F. В., Org. Syn., Coll. Vol., 4, 430 (1963).
8. Lawesson S.-О., Jakobsen H. J., Larsen E. H., Chem. Scand.,
17, 1188 (1963).
9. Lawesson S.-О., Busch T., Berglund C.. Chem. Scand., 15, 260
(1961); L a r s e n E. H., Lawesson S.-О., Org. Syn., 45, 37 (1965).
О
БЕНЗОИЛИЗОТИОЦИАНАТ C6H5CNT = С = S. Мол. вес 163,19.
Б. получают in situ реакцией хлористого бензоила с тиоцианатом
аммония (мол. вес 76,11) [1]. Б. реагирует с первичными и вторич-
ными аминами с образованием производных бензоилтиомочевины,
которые легко гидролизуются щелочами, давая монозамещенныетио-
мочевины:
о он nhc6h5 он_ nhc6h5
II CBH,NH, И I I —'—* I
C6H6CN = C=S ° _> C6H5C—-N—C = S H2N—C = S
1. DouglassLB., DainsF.B.,J. Am. Chem. Soc., 56 1408 (1934).
БЕНЗОИЛ ХЛОРИСТЫЙ, C6H5COC1. Мол. вес 140,57, т. кип.
197°, уд. вес 1,21.
Технический Б. х. очищают промыванием его бензольного раст-
вора водным раствором бикарбоната натрия, затем сушат и пере-
гоняют [1].
Хлорангидриды алифатических кислот (С,— Cfi) получают с вы-
ходом 80—90% отгонкой летучего RCOC1 из смеси кислоты с избыт-
ком Б. х. [2|, например, акрилоилхлорид (70%) [3], пропионил-
хлорид (80%) [4], изовалероилхлорид 15].
1. ОквудТ., Венсгербер К., «Синтезы органических препаратов», ИЛ,
М., 1952, сб. 3, стр. 488.
2. BrownH. С., J. Am. Chem. Soc., 60, 1325 (1938).
3. S t е ш р е 1 G. Н., Jr., et al., J. Am. Chem. Soc., 72, 2299 (1950).
100
4. F о г г e s t J. et al., J. Chem. Soc., 1946, 454.
5. F i n a n P. A., FotheryillG. A., J. Chem. Soc., 1963, 2723.
БЕНЗОЙНАЯ КИСЛОТА, С„Н3СО2Н. Мол. вес 122,12, т. пл.
122’, т. кип. 249°, растворимость в 100 г воды 0,21 г при 17,5°;
2,2 г при 75°, рКа 4,17.
Рюхард и сотр. [11 нашли, что кислоты, в особенности Б. к.,
катализируют реакцию Виттита карбэтоксиметилентрифенилфосфо-
рана с кетонами в бензольном растворе. Этот эффект менее заметен
с повышением температуры (при 40 слабее, чем при 23) и в хлоро-
форме (по сравнению с бензолом) 12]. Его объясняют образованием
с«н,со,н
(CeH5)3P = СНСО2С2Н5 + R,C = О ——RM = СНСО,С2Н5-I-(С„Н5)3РО
особых водородных связей в переходном состоянии. Каталитиче-
ский эффект не влияет на соотношение цис- и транс-изомеров в
продуктах [31.
1. R й с h а г d t С., Е i с h 1 е г S., PanseP., Angew. Chem., Internal. Ed.,
2, 619 (1963).
2. F 1 i s z a r S., H u dson R. F., S a 1 v a d о r i G., Helv. Chim. Acta, 47,
159 (1964).
3. HouseH.O., JonesV.K., F r a n k G. A., J. Org. Chem., 29, 3327 (1964).
БЕНЗОЙНОЙ КИСЛОТЫ НАТРИЕВАЯ СОЛЬ, C6H5CO2Na.
Мол. вес 144,11. Растворимость в 100 г Н2О: 62,5 г при 25°, 76,9 г
при 100°.
Эта соль в кипящем диметилформамиде является одним из наибо-
лее сильных нуклеофильных реагентов [11. В сахаре (1) Б. к. н. с.
замещает вторичную тозилатную группу у С4 с обращением конфи-
гурации (замещение Sn 2) без анхимерного эффекта соседней группы.
В 2',3',5'-тримезилоксиуридине (3) под действием Б. к. н. с. в кипя-
101
щем диметилформамиде замещаются все три мезильные группы, при-
чем образуются небольшие количества (4) и (5) 121. Б. к. н. с. замеща-
ет мезильную группу в (6) с образованием единственного продукта
(7) [31.
1. R е i s t Е. J., Goodman!.., Baker В. R., J. Am. Chem. Soc., 80, 5775
(1958); Reist E. J., Spencer R. R., Baker B. R., J. Ore. Chem., 24,
1618 (1959).
2. С о cl i n g t о n J. F., FecherR., Fox J. J., J. Am. Chem. Soc., 82, 2794
(1960).
3. Ryan K- J-, Arzoumanian H., A c t о n E. M., Goodman L., J.
Ain. Chem. Soc., 86, 2497 (1964).
БЕНЗОЙНОЙ КИСЛОТЫ СЕРЕБРЯНАЯ СОЛЬ, CeH5CO2Ag.
Мол. вес 228,99.
Для получения соли [1J смешивают растворы эквивалентных ко-
личеств азотнокислого серебра и бензоата натрия, осадок отфиль-
тровывают, промывают и сушат в вакуумном сушильном шкафу.
Перегруппировка Вольфа. При перегруппировке диазокетонов
по Вольфу под действием окиси серебра и метанола часто получают-
ся невоспроизводимые результаты. Ньюмен и Бил [1] разработали
методику проведения этой реакции в гомогенном растворе и в мяг-
ких условиях. К раствору диазоацетофенона в метаноле прибавляют
при комнатной температуре несколько капель фильтрованного раст-
вора 0,004 моля Б. к. с. с. в 9,1 мл триэтиламина; смесь чернеет и
О
CeH5CCH = X = X- СН8ОН + CeHsCO2Ag СвН5СН2СО,СН8-- X, Ag
Ь О %
0,003.3 моля в 53 лм 0,016 моля
53 ли СН3ОН в (CaH5).,N
начинает выделяться азот. По мере ослабления выделения газа
добавляют еще Б. к. с. с. (всего около 0,5 же) до завершения реак-
ции; выделяется 92% от теоретического количества азота. Экспе-
102
риментально подтвержден следующий механизм реакции:
О о
1! - - И- -F - +
RCC = X = X (С.,Н5)3Х щд RCC = X = X + (С,Н5)3ХН
I
н
о о
II — + — Ag + I + _ —N.
RCC = X = X---* RCC=\ = X----> RC = C = O
~A« .
О о
II + - II + —
RC = C = O+RCC = N = X -> RCH —С —O-г RCC = X = XT
I CH.,OH
H RCHXOoCH;,
Необходимость наличия а-водородного атома подтверждается тем,
что C.,H3COC(CH3)=N=N в реакцию не вступает. Для реакции не-
обходимо каталитическое количество окислителя и основание, ко-
торым служит триэтиламин, использующийся как растворитель для
соли серебра. В случае диазокетона, который при комнатной тем-
пературе вступает в реакцию с трудом, реакцию проводят в трет-
бутиловом спирте в качестве растворителя, ибо тогда температуру
можно повысить даже до кипения; при нагревании раствора Б. к. с. с.
с триэтиламином соль восстанавливается до серебра с потерей ката-
литической активности.
Реакция высоко стереоспецифична и не приводит к изменению
конфигурации. С другими катализаторами на 20—30% проходит
рацемизация [2]. Другие примеры см. в работе [3].
1. NewmanM. S., Beal III Р. F., J. Am. Chem. Soc., 72, 5163 (1950).
2. WibergK.B.,HuttonT. W., J. Am. Chem. Soc., 78, 1640 (1956).
3. Klei n J., Bergmann E. D., J. Org. Chem., 22, 1019 (1957).
БЕНЗОЙНЫЙ АНГИДРИД, (СвН5СО)2О. Мол. вес 226,23, т. пл.
42—433.
Декарбоксилирование а-кетокислот. Фенилглиоксиловая кис-
лота гладко декарбоксилируется при кипячении с Б. а. и пиридином
в бензоле [11.
О
II Кипячение 13 час
С6Н.-ССО;Н4-(СвН5СО).,ОА- Пиридин -фС6Н6--------------► СбН5СНО + СО2
13,ммолей 27 .ммолей 140 .кмолей 50 мл 75% 88%
Можно использовать и другие ангидриды, однако предпочитают
Б. а., что позволяет избежать конденсации типа реакции Перкина.
Механизм этой реакции предположителен.
1. С о h е n Т., Song I. Н., J. Am. Chem. Soc., 87, 3780 (1965).
103
о-БЕНЗОЛДИАЗОНИЙКАРБОКСИЛАТ (предшественник дегид-
робензола). Мол. вес 148,12.
II
О
Б. можно получить диазотированием антраниловой кислоты [1],
но его приходится хранить при 0°, и он иногда взрывается. Поэтому
реагент удобнее получать in situ апротонным диазотированием ан-
траниловой кислоты в присутствии акцептора (антрацена) |2].
Триптицен можно получить добавлением (по каплям) раствора ан-
траниловой кислоты в ТГФ к кипящему раствору амилнитрнта и
антрацена в метплен.хлориде в течение 4 час; выход 51%. По упро-
щенной методике (для студентов) [31 кипятят раствор антрацена и
изоамилнитрита в более высококппящем 1,2-диметоксиэтане, до-
бавляя в течение 40 мин антраниловую кислоту; выделение чистого
триптицена происходит без осложнений.
1. Stiles М., Miller R. G., Burckhardt U., J. Am. Chem. Soc., 85,
1792 (1963); M i 1 1 e r R. G„ S t i 1 e s M„ ibid., 85, 1798 (1963).
2. F r i e d m a n L., LogulloF. M., .1. Am. Chem. Soc., 85, 1549 (1963); L o-
g u 1 1 о F. M., Friedman L., procedure submitted to Org. Syn.
3. Org. Expts., Chapt. 62.
БЕНЗОЛСУЛЬФОКИСЛОТЫ АНГИДРИД, (CeHsSO2)2O. Мол.
вес 298,33, т. пл. 90=.
Б. а. применяют в синтезе сульфонов по Фриделю — Крафтсу
и для получения бензолсульфонатов из фенолов [1].
1. Fi el d L., J. Am. Chem. Soc., 74, 394 (1952).
БЕНЗОЛСУЛЬФОНАТ НАТРИЯ, C6HsSO3Na. Мол. вес 180,17.
4-NaNH..
0,4 моля
94«/,
Кипячение и пере-
мешмвание 12 час । к
Беннет и corp. [1] получили с высоким выходом N-феннлпиперидин
продолжительным кипячением Б. н. и амида натрия в избытке пи-
перидина.
1. Bunnett J. F., В г о t h е г t о n Т. К., Williamson S. М., Org.
Syn., 40, 74 (1960).
104
БЕНЗОЛСУЛЬФОНИЛИЗОЦИАНАТ, C6H5SO,N=C=O. Мол.
вес 183,19, т. кип. 69—7270,15 лш.
Получение. Б. получают реакцией бензолсульфохлорида с циа-
натом серебра [11 или из бензолсульфамида и фосгена [21.
Применение для характеристики пространственно затруднен-
ных спиртов и фенолов [31. Пространственно затрудненные гидро-
ксилсодержащие соединения под действием Б. легко превращаются
в уретаны — производные, удобные для характеристики соединений
этого типа. Например, при взаимодействии Б. с /пре/и-бутанолом в
толуоле (0,01 М раствор) при 0° уже через 1 мин обнаруживаются
(титрованием) лишь следы изоцианата. Фенилизоцианат при такой
СН3 СН3 О Н
I с н сн i i! I
СН3-С—OHy-C6H5SO„N = C = o771777_-. СН3С—ОС—NSOXeH-,
I ' I
сн3 сн3
Т. пл. 128°
же концентрации mpem-бутанола в толуоле при 100° за 19 дней рас-
ходуется только на 5996. Б. быстро реагирует также с 2,6-дпметилфе-
нолом, 2,6-диметоксифенолом, пентахлорфеиолом и даже с 2,6-ди-
пгрещ-бутилфенолом.
1. В i 1 1 е t е г О. С., Вег., 37, 690 (1904).
2. К г z i k а 1 1 а Н„ пат. ФРГ 817602; С. А., 47, 2206 (1953).
3. McFarland J. W., Howard J. В., J. Org. Chem., 30, 957 (1965).
БЕНЗОЛСУЛЬФОХЛОРИД, CsH,SO,Cl. Мол. вес 176,63, т. пл.
15°, т. кип. 251,5° (113—115710 мм), уд. вес 1,384.
Б. получают реакцией бензолсульфоната натрия с РС15 или с
РОС13 П1, а также хлорсульфнрованием бензола [21.
Бензолсульфамиды. В пробе Хинсберга первичные и вторичные
амины различают по образованию бензолсульфамидов соответствен-
но растворимых и нерастворимых в щелочи [31.
Прием Хинсберга использован при получении N-н-бутнлпирро-
лидина (3) из ди-н-бутиламина (1) по Гофману-Леффлеру [4] для от-
деления продукта реакции (3) от исходного (1) с применением бен-
золсульфохлорида. Третичный амин (3) отделяли перегонкой с па-
ром от нелетучего бензолсульфамида, получающегося из (1).
r~| СГНСП'щ
(«-Bu)2NH— | ' 'XN-bu-«^2i.
Cl
(1) (2)
CH,CH..
| " >N—CH,CH.,CH..CH3
CH.CHZ
(3)
Эфиры бензолсульфокислоты. Склонность группы OSO2C6H-,
к эффективному замещению иллюстрируется превращением пента-
105
эритрита в соответствующий тетрабромид [5] через промежуточный
С (СН,ОН)4 ~ 4CeH5SO,Cl -- Пиридин
0,96 моля 4,24 моля 650 м.1
-+C(CH2OSO.2CcH5h NaBr l5’s ?10ЯЯ) в 140-1,5 0^ c(CH_Brh
тетраэфир, гладко образующийся в пиридине. Флороглюцин под
действием Б. в пиридине дает смесь моно-, ди- и триэфиров (Кэм-
порис 161). Бензолсульфонирование этого и других многоатомных
фенолов проводили в водном растворе в присутствии гидроокиси
кальция, добавляя ее порциями к реакционной смеси до постоян-
ной щелочной реакции. Выход дибензолсульфоната пирокатехина
96%; выход триэфира из флороглюцина не указан.
Удобный метод циклизации 1,4- и 1,5-гликолей состоит в кипя-
чении их с третичным амином при постепенном добавлении Б. [7].
сн3снснгснгснсн3.с‘Н53ОгС1>Г сн3снсн2сн3снсн3
он ОН ОН OSO2C6H5
64%
Другие реакции. Б. применяется также для получения метил-
фенилсульфона 181 и тиофенола [91.
Na.SO,
C6H-,SOX1 NaH(;o^ C,;H-,SO.-,.\a ~Д£Н1°>У°- C6H5SO.,CH,
66(общий) 6 ° - 3
C6H5SO2C1 — H,SO4 (КОНЦ.)4- Zn (пыль) --- Лед CfiH5SH
6 0 0 <3 2,4кг 1,2 кг 7,2 кг
1. Адамс P., Марвел Ч., «Синтезы органических препаратов», ИЛ, М.,
1949, сб. 1, стр. 467.
2. Кларк Г., Бабкок Г., М э р р е ii Т., «Синтезы органических препара-
тов», ИЛ, М., 1949, сб. 1, стр. 469.
3. Org. Expts., 126.
4. К о л ь м э н Дж., Н и к о л ь с Г., Мартенс Т., «Си: тезы органических
препаратов», ИЛ, М., 1949, сб. 3, стр. 129.
5. Герцог Г., «Синтезы органических препаратов», ИЛ, М., 1953, сб. 4,
стр. 448.
6. Kampouris Е. М., J. Chem. Soc., 1965, 2651.
7. Reynolds D. D., Kenyon W. O., J. Am. Chem. Soc., 72, 1593 (1950).
8. Фил ьд Л., К л a p к P., «Синтезы органических препаратов», ИЛ, ЛЕ, 1960,
сб. 10, стр. 45.
9. Адамс Р., Марвел Ч., «Синтезы органических препаратов», ИЛ, М.,
1949, сб. 1, стр. 381.
бис-(БЕНЗОНИТРИЛ) ПАЛЛАДИЙ(П)ХЛОРИД,
(СкН5СЫ)2Рс1С1.,. Мол. вес 383,85.
Б.— стабильное желто-оранжевое соединение, растворимое в
бензоле, хлороформе и ацетоне. Его получают с хорошим выходом
растворением хлористого палладия в бензонитриле при нагревании
106
на кипящей водяной бане; продукт осаждается при охлаждении [1].
PdCI2 —2CBH-,CN (с«нг.слЪр<«'Д..
Бензонитрильный лиганд вытесняется олефином с образованием
очень нестабильных координационных соединений состава
(С„Н.2„РЙС!;).2.
Поданным Бломквиста и Мэйтлиса 121, в присутствии каталити-
ческих количеств Б. в бензоле, хлороформе или ацетоне дпфеннл-
ацетилен тримеризуется в гексафенилбензол с выходом 80—85%.
1. К h а г a s с li М. S., S е у 1 е г R. С., Л1 а у о F. R., J. Am. Chem. Soc., 60,
882 (1938).
2. В I о т q nisi Л. Т„ М a i t 1 i s P. M., J. Am. Chem. Soc., 84, 2329 (1962).
1,4-БЕНЗОХИНОН (я-хпноп). Мол. вес 108,09, т. пл. ИЗ'.
Темный технический продукт очищают (до ярко-желтого) воз-
гонкой в вакууме пли кристаллизацией из тетрахлорэтилена.
Б. используют для дегидрирования а-гпдроксиламинонитрилов
в а-оксиминонитрилы [11. Хинон добавляют небольшими порциями
при нагревании и встряхивании к раствору исходного вещества в
бензоле и смесь кипятят 3—5 час. Выход составляет 41—61%.
О ОН
il I
RCHCN /Т RCCN
NHOH I) ф NOH ф
!! I
О ОН
Б. служит акцептором водорода при окислении по Оппенауэру
Д5-3₽-оксистероидов в Д4“-диеноны-3. В сообщении об открытии
этой реакции Веттштейн 12] подробно описал окисление прегненоло-
на в 6-дегидропрогестерои, но не указал выхода. При обсуждении
механизма окисления по Веттштейну — Оппенауэру Манделл 13]
рассмотрел литературу и сообщил, что выход «до 50%».
107
1. Kissinger!.. W., U n g n a d e H. E., J. Org. Chem., 25, 1471 (1960).
2. W etts t e i n A., Helv. Chim. Acta, 23, 388 (1940).
3. Mandell L., J. Am. Chem. Soc., 78, 3199 (1965).
1,2,3-БЕНЗТИАДИАЗОЛ-1,1-ДИОКСИД
Мол. вес 168,18.
г
Синтез [1] этого предшественника дегидробензола очень трудо-
емок и требует предосторожностей. Б. взрывает при 60° и медленно
разлагается при температуре -~10; с образованием дегидробензола,
азота и сернистого газа.
1. Wittig G., Hoff matin R. W., Chem. Ber., 95, 2718 (1962).
БЕРЧА ВОССТАНОВЛЕНИЕ, Li(Na), NH3, ROH. Обзор [11.
Восстановлением производных 3-метилового эфира эстрадиола (1)
с последующим гидролизом эфира енола (2) успешно получают цен-
ные для гормональной терапии 19-норстероиды (3). Автор этого ме-
тода А. Дж. Берч (1949 г.) применял натрий в жидком аммиаке и
этанол в качестве донора протонов. По данным Уайлдса и Нельсона
[21 (1953 г.), выходы улучшаются при замене натрия литием, при
этом восстановление осуществляется даже в тех случаях, когда
натрий не эффективен. По Уайлдсу — Нельсону, в качестве сораст-
ворителя применяют эфир, спирт добавляют в конце реакции.
Разложение реакционной смеси водой проводят после испарения
аммиака. Эта реакция стала ключевой стадией в промышленном
синтезе двух 19-норстероидов, перспективных для клинического
применения. Обширные исследования ее эффективности в больших
масштабах [31 по предварительным результатам подтвердили преиму-
щество методики Уайлдса — Нельсона (Li). Тщательная проверка
показала, однако, что эта методика не дает воспроизводимых резуль-
татов и степень восстановления от опыта к опыту изменяется без
видимой причины. Подбором реагентов при восстановлении стан-
дартного субстрата литием в перегнанном жидком аммиаке было най-
дено, что mpem-бутанол является наиболее подходящим и как донор
протонов и как сорастворитель. По новой методике получены вы-
сокие (87%) и воспроизводимые выходы. Однако если аммиак на-
бирают непосредственно из баллона, без перегонки, то литий рас-
ходуется за 30 мин вместо обычных 3—5 час, раствор совсем не си-
неет, неочищенный продукт реакции содержит до 23% исходного
вещества, а из водного маточного раствора после выделения продук-
108
та реакции осаждается гидроокись железа. Опыт испорчен из-за
примесей коллоидных соединений железа в продажном аммиаке!
Изучение действия небольших количеств солей железа на реак-
цию металлов со спиртами в жидком аммиаке показало, что содержа-
ние 0,5-10“6 вес. ч. железа приводит к возрастанию скорости реакции
для лития в 2 раза, а для натрия в 50 раз. Поэтому при восстановле-
нии натрием и трет-бутанолом по Берчу высокий выход достигает-
ся в свободном от примесей железа жидком аммиаке. Очевидно, пре-
имущество лития перед натрием только в том, что реакция натрия со
спиртом катализируется железом гораздо в большей степени. В дей-
ствительности же натрий так же эффективен, как и литий, а в не-
которых случаях даже имеет определенное преимущество. Только
в случае 5-метокситетралина литий дает более высокий выход (62%),
чем натрий (45%). Это соединение восстанавливается особенно мед-
ленно, и поэтому большая скорость восстановления литием приобре-
тает решающее значение. Ботнер-Бай [4] нашел, что бензол восста-
навливается литием в 60 раз быстрее, чем натрием.
По-видимому (Ботнер-Бай, 1959), восстановление бензольного
цикла по Берчу состоит в присоединении одного электрона с обра-
зованием ион-радикала (2), который может протонироваться (3)
и затем присоединять второй электрон (4) и протон (5). Ввиду не-
достаточной кислотности аммиака для такого протонирования не-
обходим спирт.
Восстановление по Уайлдсу — Нельсону а-нафтола в 5,8-дигидро-
1-нафтол — первая стадия в синтезе пг-тетрагидронафтола-1 (об-
щий выход 84—88%) |5|.
ОН
I
/'Уч Li, МЦ
। сан.-,он
Восстановление по Берчу бензойной кислоты (Na, NHa, С2Н5ОН)
дает 1,4-дигидробепзойную кислоту с выходом 89—95% 16,7].
Другие подобные примеры восстановления рассмотрены в работе [7].
Фрайдом и Абрахамом 181 установлена важная роль природы со-
растворителя, применяемого для повышения растворимости при
восстановлении стероидных кетонов литием в жидком аммиаке в
присутствии донора протонов. В случае эфира или 1,2-диметоксиэта-
на наблюдается побочное пинаколиновое восстановление (с выходом
109
до 30%), которое подавляется ТГФ, диоксаном и в отсутствие сора-
створителя. Диглим дает промежуточные результаты.
1. В i г с h A. J., Quart. Rev., 4, 69 (1950); В i г с h A. J., S m i t h Н., ibid., 12,
17 (1958); W a t t G. W., Chem. Revs., 46, 317 (1950).
2. W 1 1 ds A. L., Nelson N. A., J. Am. Chem. Soc., 75, 5366 (1953).
3. Dryden H. L., Webber G. M., Burtner R. R., Celia J. A., J.
Org. Chem., 26, 3237 (1961).
4. К r a p c h о A. P., В о t h n e r-By A. A., .1. Am. Chem. Soc., 81, 3658 (1959);
82, 751 (1960).
5. GutscheC. D., Peter H. H., Org. Syn., Coll. Vol., 4, 887 (1963).
6. KuehneM. E., L a m b e r t B. F., J. Am. Chem. Soc., 81, 4278 (1959).
7. KuehneM. E., Lambert B. F., Org. Syn., 43, 22 (1963).
8. Fried J., Abraham N. A., Tetrahedron Letters, 1879 (1964).
БОРА ТРИБРОМИД, BBr3. Мол. вес 250,57, т. кип. 90,6°.
Б. т. получают из трехфтористого бора и бромистого алюминия
[1]. Для расщепления простых эфиров Б. т. добавляют к 3 же эфи-
ра при охлаждении льдом; смесь кипятят и после удаления алкил-
бромида эфир ортоборной кислоты гидролизуют 10%-ной щелочью.
Фениловые эфиры дают фенол и алкилбромид |2]. Фенилметиловые
Зм-Ви — О — Ви-«+ ВВг3 ту^->(и-ВиО)3В л- Зн-ВиВг
(н-ВиО)3В 4- Н2О , ' Зя-ВиОН -у Н3ВО3
эфиры деметилируют с высоким выходом, смешивая реагенты
ОСН3 ОВ"’ он
| Вг | Вг | Вг
в хлористом метилене или н-пентане при —80°, после чего смеси
дают нагреться до комнатной температуры (например, вератровая
кислота (90%); 3,4,3',4'-тетраметоксидифенил (85%) [3]).
Штеттер и Вульф [4] нашли, что Б. т. оказывает заметное ката-
литическое действие при бромировании адамантана и сообщили об
образовании в этих условиях 1,3-дибромадамантана. Однако Баух-
маи (51 установил, что в действительности в этих условиях получает-
ся 1-бромадамантан, а дибромид удается получить с выходом 74%
при добавлении к Б. т. следов бромистого алюминия. Б. т. селектив-
но расщепляет 5-метоксигруппу в 4-окси-5,6,7-триметокси-2-карб-
этоксихинолине и в его 3-карбэтоксиизомере [6]. Такая избира-
тельность объясняется образованием внутримолекулярной водород-
ной связи в аддукте.
НО
1. GambleE. L., GilmontP., S t i f f J. F., J. Am. Chem. Soc., 62, 1257
(1940).
2. В e n t о n F. L., D i 1 1 о n T. E., J. Am. Chem. Soc., 64, 1128 (1942).
3. McOml eJ. F. W., Watts M. L., Chem. Ind., 1963, 1658.
4. S t e t t c r H„ W u 1 f f C„ Chem. Ber., 93, 1366 (1960).
5. В a u g h m a n G. L., J. Ogr. Chem., 29, 238 (1964).
6. Schafer W., Franck B., Chem. Ber., 99, 160 (1966).
БОРА ТРИФТОРИД, BF3. Мол. вес 67,82, т. кип. —101°.
Б. т. хранится в баллонах и вводится в реакционную смесь бар-
ботированием.
Реакция Фриделя — Крафтса. Безводный Б. т.— умеренно ак-
тивный катализатор для алкилирования по Фриделю — Крафтсу.
Некоторые примеры приведены на следующей схеме [1]:
СН(СНз)., СН(СН3),
I I
//\ //'^
I |1-/г- или пзо-С3Н7ОН BFj | |1 + | ||
24% I
сн (СНз)з
13-14%
// , НГ)_/ \. вт., (0,7экч> ( 5б°/0 Цпклогексилбепзола
\ ___/ г \____/ 'i'1’ \ 27% п-Дпцнклогексилбензола
По данным Ола [2] Б. т. катализирует алкилирование аромати-
ческих соединений алкилфторидами, что иллюстрируется реакцией
бензола с изопропилфторидом:
СН (СН3)2
I
//' . //\
I 5Д-. |
Другие тригалогениды бора также обладают каталитическими
свойствами: В1з>ВВг:!>ВС1з>ВРз. Однако самые высокие выходы
обычно получают с Б. т. Так, выход кумола составляет с ВС13 63%,
с ВВ1’з 72?о, с BI:J 60% . Алкилхлориды, -бромиды или -иодиды в этих
условиях, как правило, не алкилируют. Смешанные же галоген-
производные, содержащие фтор, реагируют с отщеплением HF, на-
пример:
СН„СН2Вг
I
// \ //\
| || + FCH.,CH,Br BFa’-4 "С_ДР"_£°°_> | ||
V 9‘,;’ V
СН3СНСН.,С1
I
/л. // \
I || + FCH.,CH2CH2C1 BFj' 1 "а9с-”ри.-1-10°-, | ||
%/ 90'° %/
111
Во втором примере алкилирование сопровождается изомеризацией
атакующей группы.
Перегруппировка эпоксидов. Следующий интересный пример
описан Хаузом [3]. Бензольный раствор глицидного эфира (1) на-
сыщают Б. т. и оставляют на 30 мин при комнатной температуре;
обработка реакционной смеси дает а-кетоэфпр (2) с высоким выходом.
BF,
С6Н-,СН — CHCOoQH-, С6Н-СН„ССО„С„Н3
\0/ ° !!
и О
(1) (2)
Алкилирование и ацилирование. Алкилирование ацетоуксусного
эфира изопропанолом отличается тем, что Б. т. сначала очищают,
пропуская через насыщенный раствор борной кислоты в конц.
H2SO4, и затем барботируют в смесь реагентов при 0—7 [4]:
СН (СН3),
в г, I
СН3СОСН2СО2С2Н34ХСН3).,СНОН удц-фл СН3СОСНСО,С2Н-,
Неочищенный Б. т. используется в синтезе ацетилацетона; выход
указан для препарата, который очищают через ацетплацетонат ме-
ди, затем перегоняют [5].
СН3СОСН3 + (СН3СО)2О - СН3СОСН„СОСН3
Бекмановская перегруппировка альдоксимов и кетоксимов [6].
Нитрилы—»амиды [7]. Раствор нитрила в водной уксусной
кислоте насыщают газообразным BF3, затем температуру повышают
до 115—135° и выделяют амид с очень хорошим выходом.
1. McKennaJ. F., Sowa F. J., J. Am. Chem. Soc., 59, 470 (1937).
2. О 1 a h G. A., К u h n S. J., О 1 a h J. A., J. Chem. Soc., 1957, 2174;
О 1 a h G. A., К u h n S. J., J. Org. Chem., 29, 2317 (1964).
3. H о u s e H. O., Blaker J. W., M a d d e n D. A., J. Am. Chem. Soc., 80,
6386 (1958).
4. A d a m s J. T., L e v i n e R., H a u s e r C. R., Org. Syn., Coll. Vol., 3, 405
(1955).
5, Денун К., мл., «Синтезы органических препаратов», ИЛ, М., 1952, сб. 3,
стр. 92.
6. И а п s с г С. R., Н о f f с n b е г g D. S., J. Org. Chem., 20, 1482, 1496 (1955).
7. Hauser С. R., H о f f e n b e r g D. S., J. Org. Chem., 20, 1448 (1955).
БОРА ТРИФТОРИД--МЕТАНОЛ (51% BF3; 2 экв, 4,4 мл).
Ароматические кислоты этерпфиппруют в присутствии этого
реагента в избытке метанола с хорошим выходом [II.
1. Н а 1 1 a s G., J. Chem. Soc., 1965, 5770.
БОРА ТРИФТОРИДА — УКСУСНОЙ КИСЛОТЫ КОМПЛЕК-
СЫ. BF3-CH3COOH (твердый), BF3-2CH3COOH (жидкий). Жидкий
комплекс содержит 36% (по весу) уксусной кислоты,
112
Твердый комплекс (гигроскопичный!) получают насыщением
раствора уксусной кислоты в дихлорэтане трехфтористым бором с
последующим фильтрованием и промыванием осадка растворителем;
жидкий комплекс остается в растворе [11. Твердый комплекс при-
меняют для ацетилирования кетонов, например циклогексанона
О О
R сосн,
' \ / \/
I BF.,-CH.,COOH |
У ) 86% ф \
111. При перемешивании и охлаждении льдом в уксусную кислоту
пропускают BF3 до превращения реакционной смеси в твердую по-
рошкообразную массу (поглощается около 80 мол. %). К получен-
ному комплексу при охлаждении и перемешивании добавляют по
каплям циклогексанон; смесь оставляют при комнатной темпера-
туре па 4 час и выделяют продукт реакции.
Жидкий комплекс с 2 молями уксусной кислоты используют в из-
бытке уксусного ангидрида для С- и N-ацетилирования фенилаце-
тамида с образованием продукта (3) [2], который можно получить
с„н-сн2с\ BFa~2Ac2^с„н5снхо\нл —cgh,chconhcoch3
59%из(1) I
53% из (2) СОСН3
(I) (2) (3)
непосредственно из фенилацетонитрила (1), так как под действием
этого комплекса последний превращается в фенилацетамид (2).
1. М а п у i k R. М., F г о s t i с к F. С., Jr., Sanderson J. JHau-
ser C. R., J. Am. Chem. Soc., 75, 5030 (1953).
2. W о i f e J. F., Eby C. J., H a u s e r C. RJ. Orc. Chem., 30, 55 (1965).
БОРА ТРИФТОРИДА ЭФИРАТ, BF3 (C2H,)2O. Мол. вес 141,94,
т. кип. 126°, уд. вес 1,154.
Очистка. Продажный препарат быстро темнеет вследствие окис-
ления воздухом, однако при перегонке жидкость обесцвечивается.
Цвейфель и Браун [1] рекомендуют вначале добавить эфир (10 мл
на 500 мл эфирата) и перегонять в полностью стеклянном приборе
при 46 /10 мм над 2 г гидрида калышя для удаления летучих кислот
и уменьшения толчков при перегонке.
Дегидратация. Под действием Б. т. э. в уксусной кислоте (4 час
при 25') 11₽-оксистероиды гладко дегидратируются до А9 '“’-енов [21.
113
Межмолекулярная дегидратация иллюстрируется следующими
примерами. Б. т. э.— прекрасный катализатор конденсации кетонов
с димеркаптоэтаном 13] и тиолами [4|. При добавлении 1 мл Б. т. э.
к горячему раствору 0,6 г холестанона-3 в 15 мл уксусной кислоты
сразу начинается выделение игл этилентиокеталя. Еще более эф-
фективно действие Б. т. э. на суспензию или раствор кетона вдимер-
Холестанон-З
ВГ3-(С2Н5)2О
HSCH2CH2SH, СН3СО2Щ
92% *
сн3
каптоэтане, имеющем высокую растворяющую способность. Напри-
мер, этим способом холестандион-3,6 превращают в 3,6-бш?-этилеп-
тиокеталь с выходом 94% .тогда как реакция в уксусной кислоте при
комнатной температуре дает смесь 3-моио- и 3,6-бис-этилентиокеталей.
Б. т. э.— эффективный катализатор конденсации аллиловых
спиртов с 2-метилнафтогидрохиноном-1,4 (в диоксане при 50') [5]
и кетонов с аминами (с образованием азометинов) [61.
он
N-C6H4Cl-a
Реакция Тиле. Как катализатор превращения а-нафтохипона
О ОСОСНз
II I
О ОСОСНз
по Тиле Б. т. э. эффективнее серной кислоты; выходы соответственно
81 и 74,5% |7|. Однако в реакциях пространственно затрудненных
кетонов Б. т. э. уступает серной кислоте. Например, в присутствии
114
Б. т. э. уксусный ангидрид гладко присоединяется к 2,5-диметил-
бензохинону-1,4, но не реагирует с 2,6-диметилбензохиноном-1,4
181, вступающим в реакцию Тиле в присутствии H2SO4 [91.
Перегруппировка эпоксидов. Окись транс-стильбена изомери-
зуется в дифенплацетальдегпд 1101 при обработке Б. т. э. в бензоль-
ном растворе (перемешивание, комнатная температура). Смесь вы-
держивают 1 мин и разлагают водой. Органический слой промывают
/И 2.г'°. 1 мин
,СГ—^С/ -1- BF3• (С,НА,О----> (С6Н5).2СНСНО
ьи чс„н5 “ ' 74-«2%
0,2 моля в 450 ли 0,1 моля
водой, сушат и после удаления растворителя продукт перегоняют
при 0,6 мм. Изомеризация окиси изофорона ПИ в альдегид прово-
дится аналогично, но перед разложением смесь выдерживают 30 мин.
вг,-(сгнщо 25°-30-Л!ЦН>
0,16 моля
0,2 5 моля 0400 ЛМ
СЬН6
После промывания водой органический слой для отщепления фор-
мальдегида встряхивают 1—2 мин со щелочью и получают 2,4,4-три-
метилциклопентанон.
Кратко сообщалось 1121, что под действием Б. т. э. в бензоле а-
п [3-эпоксипроизводные тестостерона перегруппировываются в 4-ок-
си-А4-3-кетоны. Коллинз (Австралия) [131 более подробно иссле-
довал эту реакцию в ряду холестана и показал, что а- и 0-эпоксиды
ВГ3-(С2Н,)гО
С6Н6,23 час
<CtH6,18,5 час
(1) (2) (3)
дают окси-Д 4-холестенон-3 и еще одно соединение, идентифициро-
ванное как 5|}-Л-норхолестанон-3, образующееся, по-видимому,
из промежуточного 0-кетоальдегпда в результате деформилирова-
ния при хроматографии на нейтральной окиси алюминия.
1. Ц в е й ф е л ь Ж., Броун Г., «Органические реакции», изд-во «Мир»,
М., 1966, сб. 13, стр. 7—65.
2. И е у m а и п Н., F i е s е г L. F., J. Am. Chem. Soc., 73, 5252 (1951); Clin-
ton R. О. et al., ibid., 79, 6475 (1957).
3. F i e s e r L. F., J. Am. Chem. Soc., 76, 1945 (1954).
4. Schoberl A., W i e h 1 e r G., Angew. Chem., 66, 273 (1954).
5. Org. Expts., Chapt. 49.4.
6. T а у 1 о г M. Е., F 1 е t с h е г Т. L., J. Org. Chem., 26, 940 (1961).
7. F i e s e r L. F., J. Am. Chem. Soc., 70, 3165 (1948).
8. F i e s e r L. F., A r d а о M. I., J. Am. Chem. Soc., 78, 774 (1956).
115
9. E r d t m a n H., Svensk Kem. Tids., 44, 135 (1932); C. A. 26, 4803 (1932).
10. Reif D. J., House H. O., Org. Syn., Coll. Vol., 4, 375 (1963).
11. Ryerson G. D., WassonR. L., H о u s e H. O., Org. Syn., Coll. Vol.,
4, 975 (1963).
12. C a m e r i n о В., P a t e 1 1 i В., Vercellone A., J. Am. Chem., 78,
3540 (1956).
13. С о 1 1 i n s D. J., J. Clieni. Soc., 1959, 3919.
БОРА ТРИФТОРИДА ЭФИРАТ — УКСУСНЫЙ АНГИДРИД.
Есефи и Мазур |11 нашли, что смесь эфирата трехфтористого бора с
бромистым литием в уксусном ангидриде расщепляет алифатиче-
ские эфиры при комнатной температуре с образованием Соответ-
H. /ОСН3 Н ОАс
/\ X /X
| I ВГЩС.НЩО-LiBr I |
1/1-----------аСо-----N +1 \
7 частей 1 часть
ствующпх ацетоксисоединенпй [1]. Однако, поданным работы [2],
такое расщепление осуществляется только под действием смеси эфи-
рата трехфтористого бора и уксусного ангидрида, а галогенид лития
замедляет эту реакцию, ие подавляя элиминирования с образова-
нием олефина. Авторы получили холестерилацетат из холестерилме-
тилового эфира (гомоаллпльного) с 93°о-иым выходом (15 час при
О'). Расщепление соответствующего аллильного метилового эфира
(метиловый эфир А кхолестепола-3|3) завершается за 3 мин при
—18 (90%-ный выход); в немного более жестких условиях главным
продуктом является А3,° -диен. Расщепление насыщенных эфиров в
значительной степени сопровождается реакцией элиминирования.
1. Y о ussefyeh R. D., Mazur Y., Tetrahedron Letters, 1287 (1962).
2. а г а у a n а и С. R., Iyer К. N., J. Org. Chem., 30, 1734 (1965).
БОРА ТРИХЛОРИД, ВС13. Мол. вес 117,19, т. кип. 13°. Обзор
III. 13. т. очищают через комплекс с нитробензолом |2|.
Б. т. применяют для расщепления метиловых эфиров углеводов
|3]. Суспензию метилового эфира в хлористом метилене охлаждают
до —70', перегоняют в нее треххлористый бор и выдерживают смесь
при —70' в течение 1 час, периодически перемешивая.
Хотя продукты присоединения Б. т. к диметиловому и диэтило-
вому эфирам стабильны при комнатной температуре, этот реагент
расщепляет тетрагпдрофуран 14], а также реагирует с диметиловым
эфиром этиленгликоля даже при 0J с образованием хлористого ме-
тила и, возможно, соответствующих моно- и дихлоралкоксибора-
пов 15].
ЗСН3 (ОСН.2СН.2)2ОСН3
вег, I СН3(ОСН2СН2)2ОВС12 + СН3С1
1 [СН3(ОСН2СН2)2О]2ВС1 + 2СН3С1
116
Б. т. реагирует с ацеталями или со смесью альдегида и спирта с
образованием 1-хлоралкиловых эфиров (выходы 40—85%) [6], ко-
торые представляют особый интерес, так как при дегидрохлориро-
вании под действием триметиламина образуют виниловые эфиры.
RCH2CHO + ВС1 з + R 'ОН RCH2CHC1OR' -I- НОВС12
I (CH3)3N а. _
RCH = CHOR' + (CH3)3NHC1
Б. т. используют для деметилирования и декетализации произ-
водных инозита без отщепления тозильной группы [7]. Квебрахит
(1), или 2-О-метил-(—)-инозит, превращают в 3,4;5,6-ди-О-циклогек-
силиденовое производное (2) и тозилируют по 1-гидроксильной
группе. Реакция протекает медленно из-за аксиального положения
TsCl-Ру (3 Дня) ?
82%
ВС13(43моля),СН2С12
-80°,15 час____а
90%
(4)
этого гидроксила. Обработкой (3) смесью ВС13—СН2С13 при —80°
проводят деметилирование и декетализацию с образованием 1-О-то-
зилат-(—)-инозита (4).
Применение Б. т. для получения нового класса гетероцикличе-
ских соединений иллюстрируется синтезом 10-метил-10,9-бораза-
рофенантрена [8].
NH
ВСН3
1. Gerrard W., L а р р е г t М. F., Chem. Revs., 58, 1081 (1958).
2. В г о \v n Н. С., Н о 1 in е s R. R., J. Am. Chem. Soc., 78, 2173 (1956).
3. А 1 1 е п S., В о п п е г Т. G., BourneE.J., S a v i 1 1 е N. М., Chem. Ind.,
117
1958, 630; В о n ncr T. G., Bourne E. J., M c Na 1 1 у S., J. Chem. Soc.,
1960, 2929; F о s t e г А. В., H о r t о n D., SalimN., Stacey M., W e b-
b e r J. M., ibid., 1960, 2587.
4. Edwards J. D., Gerrard W., Lappert M. F., J. Chem. Soc., 1955,
1470.
5. В г о w n H. C., Tierney P. A., J. Am. Chem. Soc., 80, 1552 (1958).
6. В 1 а с к D. K-, LandorS. R., J. Chem. Soc., 1965, 5225.
7. GeroS. D., Tetrahedron Letters, 591, 1966.
8. D e \v a r M. J. S., Dewar R. В. K., G a i b e 1 Z. L. F., Or^. Syn., 46, 65
(1966).
БОРНАЯ КИСЛОТА, H3BO3. Мол. вес 61,84.
Дегидратация. Первичные, а также вторичные и третичные
спирты дегидратируются с хорошим выходом при нагревании до
350' с эквимолекулярным количеством Б. к. 111. Б. к. вызывает де-
гидратацию спирта (1) в триен (2); в этом случае обычные методы не
эффективны или дают (2) с очень низким выходом [2]. Однако в сис-
н3во,?
61%
темах, склонных к карбкатионным перегруппировкам, при дегид-
ратации Б. к. в основном образуются продукты перегруппировки,
например в случае 3,3-диметилбутанола-2 [31. Пиролиз ацетата сое-
СН3СН3 СН3СН3 СН3
СН3СНС — СН3 —2'5 СН3С = ССН3 + СН, = С—СН 4- СН2 = СНССНз
I Vu I I
ОН ^Нз СН3 сн3
(1) (2) 73,7% (3) 25,9% (4) 0.3%,
динения (1) дает исключительно нормальный продукт (4).
Цветные реакции. Димрот |41 разработал цветную реакцию
с использованием раствора смешанного ангидрида борной и уксус-
ной кислот, полученного растворением небольшого количества Б. к.
в уксусном ангидриде. 2-Оксиантрахинон и полиоксиантрахиноны,
содержащие Р- (но ней-!) гидроксильную группу, дают с этим раст-
вором (как и с чистым уксусным ангидридом) нормальные ацетаты
(г)
118
обычного цвета. Однако 1-оксиантрахинон образует интенсивно
окрашенный, легко идентифицируемый по яркой окраске бороацетат-
ный комплекс (1), который был выделен и проанализирован.
1,5-Дпоксиантрахпнон дает бис-производное (2). С помощью этой
пробы Димрот установил, что нафтазарин имеет структуру (3) и не
является, как это предполагали ранее, низшим «бензологом» ализари-
на. Фнзер 15] показал применимость этой цветной реакции к про-
изводным фенантренхинона и успешно использовал для установле-
ния строения шести продуктов дисульфирования фенантрена.
Найдено |6|, что лимонный сок, высушенный в присутствии Б. к.,
приобретает ярко-желтую окраску. Вещество, вызывающее появле-
ние окраски, идентифицировано как кверцетин или его З-ь -рамно-
зид-кверцитрии. Их можно определить в количествах 0,002 мг и
0,004 мг при обработке смесью (1 : 1) ацетона, насыщенного Б. к.,
и 10%-ного раствора лимонной кислоты в ацетоне. Эта проба поло-
жительна для флавонов, содержащих ауксохромы (ОН, ОСН3) в
Кверцетин (положительный тест) Физетин (отрицательный тест)
положении 5; для физетина, например, эта проба отрицательна.
Флаваноны, такие, как 2,3-дигидрофлавоны, не дают окраски.
Расщепление ацетонидов. Общий метод синтеза a-эфиров гли-
церина 17] заключается в кипячении смеси глицерина, ацетона, хло-
роформа и следов /1-толуолсульфокислоты с водоотделителем, пока
не соберется теоретическое количество воды. Затем добавляют кис-
лоту, например стеариновую, и смесь повторно кипятят до прекра-
сней
I Кипячение 2 час
СНОН + (СН з),С =о+СНС1 з J- TsOH--——--------
I 15.hu 20 мл 0,3 г -
СН2ОН
10 г
СН,ОН —СН —О ч /С 1 /с\ СН2—о/ хс О il СН2О.'(СН2)16СН3 1 Н сно х сн3 — СНгО7 ^СНз СНя(СНо)10СО.>Н, кипячение 2 час Н3 : : - J -н2о Н3 О СН2ОС(СН2)!3СН3 3во3, сн.,осн,сн2он | *СН0Н о □ /о | СН2ОН
119
щения выделения воды. Полученный эфир ацетонида кипятят с тон-
коизмельченной Б. к. в (3-метоксиэтаноле для отщепления ацетона.
После разбавления смеси водой свободныйа-эфир экстрагируют эфи-
ром и высушенный раствор упаривают до начала кристаллизации
[7].
Защита ОН-группы. Реакция Бона — Шмидта [8] заключается
в гидроксилировании антрахинона смесью олеума и Б. к., часто в
присутствии каталитических количеств ртути. По-видимому, роль
Б. к. в этой реакции состоит в защите гидроксильных групп при об-
разовании эфиров борной кислоты. Такое же действие Б. к. оказы-
вает и в синтезе производных хинизарина [91.
Катализатор конденсации. Дейхлер и Вейцманн [10] нашли,
что фталевый ангидрид конденсируется с а-нафтолом в смеси H2SO4
и Б. к. с образованием как 2-фталоилнафтола-1, так и 11-оксинафта-
ценхинона-9,10 в зависимости от условий! реакции. На первой стадии
выход кислоты (57%), очищенной кристаллизацией натриевой соли,
совпадает с выходом, полученным одним из авторов книги. Выход
для циклизации на второй стадии взят из работы советских авторов
[11]. В первоначальной стадии конденсации Б.к. играет особую роль,
что заслуживает внимания, так как при этом наблюдается исклю-
чительно необычное орто-замещение. Интерпретация этого факта,
подтвержденная позднее Вейцманном [121, основана на том, что
первоначально образующийся кислый фталат претерпевает пере-
120
группировку Фриса в о-оксикарбонильное соединение, стабилизи-
рованное образованием хелатного комплекса с Б. к.
Комплексы с углеводами. Бозекен [13] установил конфигурацию
аномеров сахаров, изучая их влияние на электропроводность
растворов Б. к.
Синтезы 2-моноглицеридов [141. Б. к. рекомендуют для уда-
ления 1,3-бензилиденовой группировки из 1,3-бензилиден-2-ацил-
глицеринов нагреванием в триэтилборате в присутствии тонкоиз-
мельченной Б. к. до полного превращения в диборат с последующим
гидролизом водой, как это показано на примере 2-моноолеата гли-
церина:
н н о СН2О
|| II | \ н3во3. (ЕЮ)3в
СН3(СН2)7С = С(СНо)7СОСН нссвн5------Д—-----»-
I / 10
СН2О
И Н “о СН2ОВ(ОН)2
II II I Н.,0
-+СН3(СН2)7С==С(СН,)7СОСН —•_>
I
СН,ОВ(ОН),
НН о СН2ОН
I I II I
—> СН3(СН»),С = С(СН»)7СОСН
I
СН..ОН
1. Brandenberg W., G а 1 a t A., J. Am. Chem. Soc., 72, 3275 (1950).
2. Campbell J. R. B., Islam A. M., Raphael R. A., J. Chem. Soc.,
1956, 4096.
3. C h a p m a n O. L., В о r d e n G. W., J. Org. Chem., 26, 4193 (1961).
4. D jmr ot h O., Faust T., Ber., 54, 3020 (1921); D imrot h O., Ann.,
446, 97 (1926); Dimroth O., Ruck F., ibid., 446, 123 (1926); D i m-
r о t h 0., R о о s H., ibid., 456, 177 (1927).
5. F i e s e r L. F., J. Am. Chem. Soc., 51, 2471 (1929).
6. WilsonC. W., J. Am. Chem. Soc., 61, 2303 (1939); other examples: W о 1 f-
r о m M. L. et al., J. Org. Chem., 29, 689 (1964).
7. Hartman L.. Chem. Ind., 1960, 711.
8. Bohn R., герм. пат. 46654 (1889); Schmidt R. E., герм. пат. 60855
(1891); Phillips AL, Chem. Revs., 6, 168 (1929).
9. Б и г e л о в, Рейнольдс, «Синтезы органических препаратов», ИЛ,
М., 1949, сб. 1, стр. 458.
10. D е i с h 1 е г Chr., Weizmann Ch., Вег., 36, 547 (1903).
11. П о с т о в с к и й И. Я., Голдырев Л. Н., ЖОХ, 11, 429 (1941); герм,
пат. 134985.
12. Weizmann Ch., HaskelbergL., В е г 1 i n Т., J. Chem. Soc., 1939,
398.
13. В о е s е k е n J., Adv. Carbohyd. Chem. 4, 189 (1949).
121
14. М а г t 1 п J. В., J. Am. Chem. Soc., 75, 5482 (1953); see also С о о к P. F. Е.,
S li о \v 1 е г Л. J., J. Chem. Soc., 1965 , 4594.
БОРФТОРИСТОВОДОРОДНАЯ КИСЛОТА, HBF4. Мол. вес 87,83.
Получают медленным добавлением борной кислоты к фтористоводо-
родной кислоте в медном, свинцовом, серебряном или покрытом
слоем парафина реакторе, при постоянном перемешивании [1—31.
Уайтинг и сотр. [4—6| готовили 20—30%-ные растворы реаген-
та путем абсорбции газообразных фтористого водорода и трехфто-
ристого бора в сульфолане (тетраметпленсульфон) и хранили их
в бутылях из тефлона. Растворы разбавляли в стеклянной посу-
де. Из одиннадцати кислот Льюиса, испытанных по их способности
изомеризовать А100 -окталин и щ/лэнс-А^окталин в равновесную
смесь Аа-окталпна, А1(й| -окталина и остальных четырех изомеров
в отношении 90 : 9 : 1, Б. к. в смеси бензол — сульфолан оказалась
наилучшей [41.
Раствор фенилгидроксиламина в Б. к. превращается в катион
С„Н-,\'Н ' , который реагирует с ароматическими соединениями, об-
разуя с низким выходом смесь продуктов ампиофенилирования [5].
C„H,NH+ 10% Ю"„ 28%,
Се1% (%Н.,\Н'СвН5 4-Н.ЛСД14С,;Н, 2-Н2\С6Н4С3Н5
(ОиЩПИ,) - . __
Процентное распределение
Раствор 85%-ной перекиси водорода в реагенте содержит ион Н3О2,
являющийся сильным, ио не селективным окислителем [6|.
Реакция Шимана и родственные реакции. Прибавление охлаж-
денного льдом раствора Б. к. при энергичном перемешивании к раст-
вору хлористого феиилдиазония при 0э приводит к образованию
желтоватого кристаллического осадка фторбората феиилдиазония,
который фильтруют, промывают ледяной водой, мета-
4- — HNOo -- — HBF4 + Пиролиз
C6H5NH3C1----—*-C3H5\T N(C1)------- С6Н5 N N (BF?) ——.-щ*
16 молей
- • <11Г,1‘ \ 2 111
полом и эфиром и сушат [21. Твердый фторборат помещают в колбу
объемом 12 л с мощным широким обратным холодильником, соеди-
ненным последовательно с тремя колбами Эрленмейера емкостью по
2 л, охлаждаемыми смесью льда и соли. Отводную трубку из послед-
него приемника выводят в тягу, поскольку при реакции выделяется
трехфтористый бор, дымящий на воздухе. Твердое вещество осто-
рожно нагревают на открытом пламени горелки так, чтобы обеспе-
чить непрерывное разложение, затем удаляют небольшое количество
фенола. Так же получают 4,4'-дифтордифенил 171 и п-фторбензойную
кислоту.
Синтез п-динитробензола осуществляют по аналогичной реакции
[1 ]. В этом случае суспензию фторбората диазония вводе прибав-
ляют при перемешивании к порошку меди, суспендированному в вод-
122
ном растворе нитрита натрия. Для предотвращения пенообразова-
HNO.,HBF4 + NaNO.,(Cu)
/г-ХО,СвН4МБ------------> /i-XOXeHjN N (BFn —;---1----►
. l 4 _ 9i-_99«o _ b 1 \ i) 67—82%
-> /i-XO,C6H4XO,
ния прибавляют эфир. Выпадающий твердый осадок, содержащий
медь, промывают, сушат и экстрагируют бензолом.
Катализатор метилирования диазометаном. Насыщенные спирты
не реагируют с дпазометаном; однако под действием Б. к. или трехф-
тористого бора они образуют метиловые эфиры. Нпмэн и Джонсон
[81 привели детальную методику получения метилового эфира хо-
лестанола с выходом 95% реакцией холестанола в смеси эфир—
хлористый метилен в присутствии Б. к.
Соли небензоидных ароматических углеводородов. Фторборат
тропнллия (3) обычно применяют для получения замещенных тро-
СС14
-г рс15,-гнщ
г hbf4
-pci5,-zhci
(2) (3)
пнлиденов, поскольку эта соль стабильна, негигроскопнчна и не
взрывчата. Удобный метод получения этой соли [91 состоит в реакции
тропнлидена (1) с пятихлористым фосфором в СС14, приводящей
к образованию осадка двойной соли (2). Соль (2) перемешивают в
(СН3),К
СНС13
(9)
123
абсолютном этаноле при охлаждении льдом до образования красно-
ватого раствора и добавлением водной Б. к. осаждают белый фтор-
борат тропиллия (3).
Даубен и Бертелли [10] синтезировали гептален (9); на первой
стадии восстанавливали нафталин-1,5-дикарбоновую кислоту натри-
ем и этанолом в жидком аммиаке до тетрагидродикислоты (5), ко-
торую восстанавливали алюмогидридом лития до соответствующего
диола. Сольволитическая перегруппировка дитозилата (6) в уксус-
ной кислоте, содержащей мононатрийфосфат, дает смесь 1,6- и
1,10-дигидрогепталенов (7). Отщепление гидрид-иона под действием
фторбората трифенилметилкарбония в хлористом метилене дает
фторборат 1-гепталения (8) — ярко-желтые кубические кристаллы,
которые довольно стабильны на воздухе. При добавлении избытка
триметиламина к раствору соли (8) в хлороформе при 0° и удалении
фторбората триметиламмония образуется красный фильтрат, из ко-
торого после выпаривания и хроматографии выделяют гептален в
виде красновато-коричневой вязкой жидкости.
Хафнер и др. [11] описали получение фторборатов азуления и
их применение для синтеза различных производных. Например,
при обработке эфирного раствора 4,6,8-триметилазулена (1) эфирным
(1) (2)
раствором Б. к. фиолетовая окраска исчезает и осаждается бесцвет-
ный фторборат 4,6,8-триметилазуления (2). Под действием бензаль-
дегида бесцветная соль (2) постепенно растворяется и через нес-
колько часов выпадает окрашенный фторборат 1-бензилиден-4,6,8-
триметилазуления (3) с выходом 97%.
1. Старки Е., «Синтезы органических препаратов», ИЛ, М., 1949, сб. 2,
стр. 227.
2. Флуд Д.. «Синтезы органических препаратов», ИЛ, М., 1949, сб. 2, стр. 537.
3. Ш и м а и Дж., Винкел ьм ю л л е р В., «Синтезы органических препа-
ратов», ИЛ, М., 1949, сб. 2. стр. 244.
4. Р о w е 1 1 J. W., W h i t i n g M. C., Proc. Chem. Soc., 1960, 412.
5. P а г i s h J. H., Whiting M. C., J. Chem. Soc., 1964, 4713.
6. Alder R. W., Whiting M. C., J. Chem. Soc., 1964, 4707.
7. Шима н Дж., Винкел ьмюлл ер В., «Синтезы органических препа-
ратов», ИЛ, М., 1949, сб. 2, стр. 534.
8. N е е m а п М., J о h n s о п W. S., Org. Syn., 41, 9 (1961).
9. Con row К., Org. Syn., 43, 101 (1963).
10. DaubenH. J., Jr., В e r t e 11 i D. J., J. Am. Chem. Soc., 83, 4657, 4659 (1961).
11. Hafner K-, Pelstcr H., Schneider J., Ann.. 650, 62 (1961); Haf-
п e г К., P e 1 s t e г И., P a t z e 1 t H., ibid., 650, 80 (1961).
124
N-БРОМАЦЕТАМИД (NBA), CH3CONHBr. Мол. вес 137,98,
т. ил. 102—105°. Получение [11.
Окисление вторичных спиртов. По данным Галлахера и сотр.
121, NBA очень удобен для окисления За-оксиД2-П,12-прегнано-
на-20 в соответствующий дион-3,20. К раствору 600 мг стероида
в смеси 3 мл /npem-бутанола и 0,9 мл пиридина добавляют 4 капли
воды и 600 мг NBA (4 же) и оставляют на ночь при перемешивании;
выход 90% . При окислении этого соединения смесью хромовой и ук-
сусной кислот выход составляет 84%. Гершберг и сотр. [31 исполь-
зовали подобную методику для селективного окисления За, 11а-
диоксипрегнанона-20 в 11а-оксипрегнандпон-3,20 (выход 80%).
Джонс и Кохер [41 применили NBA для избирательного окисления
стероидных За, 20-диолов в З-кето-20-олы (выход 95%). Ранее счи-
талось, что NBA окисляет жирно-ароматические спирты только
с бензильным положением гидроксильной группы [5], что впослед-
ствии было признано ошибочным [6]. Ниже приведен один из пяти
изученных примеров:
СН3
1 25° (в темноте)
СвН5СН2СН2СНОН + СНзСОХНВг + Ру-Ь Ацетон + Н,0-—-------*
0,003 моля 0,0025 моля 0,003 25 мл 8 м.г
моля
сн3
I
-^С6Н5СН2СН2С = О
Реакция с олефинами. При взаимодействии с олефинами NBA
в отличие от N-бромсукцинимида склонен к образованию транс-
дибромидов; реакция присоединения катализируется НВг [7]. Как
источник НОВг NBA используют в виде раствора во влажном аце-
тоне [8, 91 или диоксане [10]. Как показано на схеме, реакцией
А11-стероидов с NBA в водном ацетоне [91 получают транс-диак-
сиальный продукт. Например, суспензию А9 ‘“’-стероида в смеси
диоксана и 0,46 н. хлорной кислоты обрабатывают твердым NBA
[10,111.
1. О л и в е т о Ю., Герольд К., «Синтезы органических препаратов»,
ИЛ, М., 1953, сб. 4, стр. 89.
2. К о е с h 1 i п В. А., К г i t с h е v s k v T. H., Gallagher T. F., J.
Biol. Chem., 184, 393 (1950).
3. О 1 1 v e t о E. P., Herzog H. L, Her sh berg E. B.. J. Am. Chem.
Soc., 75, 1505 (1953).
4. Jones R. E., К о c h e r F. W., J. Am. Chem. Soc., 76, 3682 (1954).
5. L e с о m t e J., Dufour C., Compt. rend., 234, 1887 (1952); Lecom-
pte J., Gault H„ ibid., 238, 2538 (1954).
125
6. Corral R. A., О r a z i О. O., Chem. Comm., 1965, 5.
7. Buckles R. E., J. Am. Chem. Soc., 71, 1157 (1949); В u с к 1 e s R. E.,
JohnsonR.C., Probst W. J., J. Org. Chem., 22, 55 (1957).
8. Reich H., R e i c h s t e i n T., Helv. Chim. Acta, 26, 562 (1943);
О 11 G. H„ R e i c h s t e i n T., ibid., 26, 1799 (1943).
9. Lardon A., R e i c h s t e i n T., Helv. Chim. Acta, 26, 747 (1943).
10. F r i e d J., SaboE. F, J. Am. Chem. Soc., 75, 2273 (1953); 76, 1455 (1954).
11. For another procedure see В e r n s t c i n S. et al., J. Am. Chem. Soc., 78,
5693 (1956).
N-БРОМАЦЕТАМИД— ПИРИДИН — SO2. Для дегидратации
110-ОКСИ-11а-метил-5Р-прегнандиона-3,20 121 применили метод, опи-
санный в патенте [11.
Раствор спирта в сухом пиридине обрабатывают 1 же NBA при ком-
натной температуре (15 мин), затем в охлажденную смесь пропу-
скают ток SO» до тех пор, пока появляющаяся вначале темно-крас-
ная окраска не изменится до слабо-желтой. Другие примеры опи-
саны в патенте 131.
1. Drake Н. A., F о n k е n А., Н о w а г d R. В., англ. паг. 790452.
2. Wechtcr W. .1., S 1 о m р G., J. Org. Chem., 27, 2549 (1962).
3. D г а к е Н. A., HowardR. В., Fonkcn А. Е., пат. США 3005834.
N-БРОМАЦЕТАМИД — ФТОРИСТЫЙ ВОДОРОД. NBA— ф. в. в
присутствии эфира или ТГФ, акцепторов протона, повышающих
ионизацию HF, используют как источник фтористого брома [1, 21.
CH3CONHBr-H2F2
Эфир
Например, действием NBA—ф. в. циклогексен превращают с хорошим
выходом в транс- 1-фтор-2-бромциклогексан [21. Реакцию можно
использовать для синтеза фторстероидов [31.
I. Robinson С. Н„ F i n с к с и о г L., О 1 1 v с t о Е. Р., Gould D., J.
Am. Chem. Soc., 81, 2191 (1959).
2. В о w е г s A., IbanejL. С., Denol Е., Becerra R., J. Am. Chem
Soc., 82, 4001 (1960).
3. Bowers A., J. Am. Chem. Soc., 81, 4107 (1959).
N-БРОМАЦЕТАМИД — ХЛОРИСТЫЙ ВОДОРОД. Смесь яв-
ляется источником элементов хлористого брома. Например, стирол
и NBA по отдельности медленно добавляют к охлажденному вод-
ному раствору соляной кислоты; продукт /пр««с-присоединения —
126
2-бром-1-хлор-1-фенилэтан (40%-ный выход) 11]. Хлорбромирование
холестерина проводят насыщением его хлороформенного раствора
хлористым водородом с последующим добавлением NBA в хлоро-
форме; 5а-бром-6|3-хлорхолес'ганол-ЗР получают с хорошим выходом
121. Другие примеры в области стероидов см. [3|.
1. В и с к 1 е s R. Е., LongJ. W., J. Am. Chem. Soc., 73, 998 (1951).
2. Z i с у 1 о г .1. В., Shabica A. C., J. Am. Chem. Soc., 74 , 4891 (1952).
3. Robinso и С. H., Fine к e n о r L., О 1 i v e t о E. P., G о u 1 d D., J.
Am. Chem. Soc., 81, 2191 (1959).
БРОМ — АЦЕТАТ СЕРЕБРА. Лемье и Фразер-Рейд [II разра-
ботали метод синтеза 2,5-ангпдросахаров реакцией триацетата 2-де-
зоксп-2-иод-р-г>-метплглюкоппранозида с большим избытком брома
и ацетата серебра. При этом получают с высоким выходом смесь
аномерных 1,3,4,6-тетра-О-ацетпл-2,5-ангидро-1 -метокси-п -манноз.
1. Lem ieux R. U., F г a s е г - R е i d В., Can. J. Chem., 42, 547 (1964).
0-БРОМИОДБЕНЗОЛ, BrCfiHJ-o. Мол. вес 282,92, т. пл. 7—9°,
т. кип. 257° (120—121/15 лмг).
Б. получают из о-бромапплииа реакцией Зандмейера и применяют
в качестве предшественника дегидробензола, например при его
тримеризации в трифенилен |1]. В этом случае литий, покрытый
Li- Эфир -C6H6(NZ).
10°, 1,5 час
HCl, NaNO2, KI
72-83%
парафиновым маслом, расплющивают молотком в тонкую фольгу,
отмывают сухим эфиром и нарезают в виде мелкой стружки так,
чтобы она сразу попадала в эфир в реакционной колбе. К смеси
127
при охлаждении и перемешивании добавляют по каплям раствор
Б. в эфире.
1. Heaney Н., М i 1 1 а г I. Т., Org. Syn., 40, 105 (1960).
БРОМИСТОВОДОРОДНАЯ КИСЛОТА, ПОСТОЯННО КИПЯ-
ЩАЯ (48%-ная НВг). Т. кип. 126°, уд. вес 1,49, 8,8 моль/л, 70,7 г/
100 мл. Окрашенную кислоту очищают перегонкой над хлористым
оловом.
ROH—> RBr. Первичные алкилбромиды лучше получать в при-
сутствии каталитических количеств серной кислоты [1].
Кипячение 5 — 6 час
(СН3)2СНСН2СН2ОН-|-48%-ная HBr + H2SO4------------”
10 молей 12,5 моля 100 г
—► (СН3)2СНСН2СН2Вг
Бромгидрат 2-бромэтиламина получают кипячением 2-этанола-
мина с Б. к., отгонкой большей части кислоты и осаждением бром-
НОСН2СН,\’Н2 + 48%-ная НВг-----> ВгСН2СН2\’Н3Вг
83%
164 моля 52 моля
гидрата ацетоном [2]. Пентаэритрит превращают в монобромид
кипячением в уксусной кислоте, добавляя 48%-ную Б. к. двумя
порциями за 8 час [3]. Обработка смеси включает отгонку, добавление
НВг —АсОН
НОСН2С(СН2ОН)3 —%———” ВгСН2С(СН2ОН)з
7 о — 8 5 %
спирта и азеотропную отгонку с бензолом, кипячение с эфиром и,
наконец, экстракцию эфиром в аппарате Сокслета. Р-Бромпропио-
новую кислоту получают заменой гидроксильной группы и гидро-
лизом нитрильной группы [4].
Кипячение 2 час
HOCH,CH2CN 4- 48%-наяНВг----------------ВгСН2СН2СО2Н
2 2 I /и 82 — 83% 2 2 2
4,5 моля 13,6 моля
Гидролиз нитрилов. Аминонитрил (3), полученный из 3 молей
ацетона, кипятят в течение 2 час с 1 кг 48%-ной Б. к. и 600 мл воды,
затем отгоняют большую часть кислоты, остаток растворяют в ме-
таноле и обрабатывают пиридином для осаждения аминокислоты
(4) [5].
NaCN-NH,Cl NH, НВг; Ру
(СН3)2СО— ---—> (CH3)2CCN---> (CH3),CCN ——(СНз)2ССО,Н
। । 30 33% I
(1) 3 моля он nh2 (общнй) nh2 (2) (3) (4)
128
Расщепление окисей и простых эфиров. Под действием Б. к.
эпоксидный цикл окиси этилена раскрывается в очень мягких
10 =
СН2 —СН2Н-46Оо-ная НВг----------> ВгСН.,СН2ОН
\ / 87—92% - 2
3 моля 4,50 моля
Кипячение 2 час
~48%-ная НВг-рH2SO4-------——--------> Br(CH2)5Br
I I OU — OZ zq
о7
0,25 моля 1,5 моля 74 г
условиях (10=, 46%-пая кислота) [6], тогда как для расщепления
тетрагидроппрана требуется 48%-ная Б. к., добавление серной ки-
слоты п кипячение |7|.
Деметилирование а-метокспфеназпна проводят еще более кон-
центрированной Б. к. 181; однако выход при этом заметно ниже,
чем при деметилировании гваякола 48%-ной Б. к. [91.
В последнем случае смесь перегоняют на колонке с автомати-
ческим сепаратором для возвращения гваякола в реактор, а образую-
щийся метилбромид поглощают метанолом, охлаждаемым льдом.
Когда температура в головке колонки указывает, что весь гваякол
прореагировал, смесь охлаждают и экстрагируют толуолом. Эк-
стракт упаривают для удаления воды и после охлаждения получают
бесцветный кристаллический пирокатехин.
8-Метокспхпнолпн деметилируют кипячением с 48%-ной Б. к.
почти с количественным выходом [10]. 6-Метокси-1 -нафтойная ки-
слота деметилируется с выходом 90% при кипячении с 48%-пой
Б. к. в уксусной кислоте в течение 5 час [И].
Дегидратация. Медленной перегонкой пинакона в присутствии
Б. к. как катализатора получают 2,3-дпметилбутадиен 112]. Однако
5 Заказ № 965
Г29
его можно получить с более высоким выходом (79—86%) перегонкой
ОН ОН
| | Пепегонка
СН3С — ССН., : 48%-ная НВг----:-------СН.,^С — С-=СН.,
I I | ।
СН3СН3 СН3СН3
3 МОЛЯ 10 л/л
пинакона в вакууме водоструйного насоса через трубку, заполнен-
ную окисью алюминия и нагретую до 420—470' [131.
Восстановительное расщепление сульфамидов.
(1) 0, 13 моля
+ 48%-ная НВт + СН3СН2СО2Н +
2 7 0 л/л 45 лм
Кипячение 2 час (N-?
________________—
NaQH^
63-71%
Изояндолпн (3) получают из его тозилата (1) кипячением смеси
сульфамида, Б. к., пропионовой кислоты и фенола в токе азота
[14, 15]. Фенол предотвращает бромирование образующегося амина
[161.
1. К а м м О., МарвелС., (Синтезы органических препаратов», ИЛ, М., 1949,
сб. 1, стр. 108.
2. К о р т п з Ф., (.Синтезы органических препаратов», ИЛ, АТ, 1949, сб. 2,
стр. 126.
3. Wawzonek S., Al a t а г A., Issiclorides С. Н., Org. Syn., Coll.
Vol., 4, 681 (1963).
4. Кендалл E., M а к - К e н з и, «Синтезы органических препаратов»,
ИЛ, М., 1949, сб. 1, стр. 133.
5. Кларк X., Бин X., «Синтезы органических препаратов», ИЛ, М., 1949,
сб. 2, стр. 34.
6. Тэйер Ф., М а р в е л, X а й е р с, «Синтезы органических препаратов»,
ИЛ, АТ, 1949, сб. 1, стр. 528.
7. Андрюс Д., «Синтезы органических препаратов», ИЛ, АТ, 1952, сб. 3,
стр. 112.
8. С ю р р е й А., «Синтезы органических препаратов» ИЛ, АТ, 1953, сб. 4,
стр. 422.
9. Кларк X., Тэйлор Е., «Синтезы органических препаратов», ИЛ, М.,
1949, сб. 1, стр. 347.
10. К i п g F. Е., S h е г г е d J. A., J. Chem. Soc., 1942, 415.
11. Long L., Jr., Burger A., J. Org. Chern., 6, 852 (1941).
12. А л л еиЧ„ Бе л л А., «Синтезы органических препаратов», ИЛ, АТ, 1952,
сб. 3, стр. 187.
13. Ныото н Л., Кобурн Э., (Синтезы органических препаратов», ИЛ, АТ,
1952, сб. 3, стр. 189.
14. BornsteinJ., L о s h u a S. С., В о i s s е 1 1 е А. Р., J. Org. Chem., 22,
1255 (1957).
130
15. В о r n s t e i n J., S li i е I d s J. Е., В о i s s о 1 1 е Л. Р., procedure submitted
by Org. Syn.
16. Searles S., X u kina S., C h e m Revs., 59, 107/ (1959).
БРОМИСТЫЙ ВОДОРОД БЕЗВОДНЫЙ. Мол. вес 80,93, т. кип.
—66 .
Получение. Для получения Б. в. б. водород вводят в испари-
тель брома и смесь газов пропускают через трубку, заполненную
битым фарфором при температуре красного каления [1|. Конструк-
ция испарителя показана на рис. Б-1. Внутренняя
колба, в которую помещают бром, обогревается па-
рами кипящего бромистого этила (т. кип. 38,4 ).
Этот генератор дает 300 г Б. в. б. в час.
ROH(ROAc) —> RBr. Даурилбромид (додецил-
бромид) получают, пропуская Б. в. б. в спирт при
100 ; бромид отделяют от водного слоя бромисто-
водородной кислоты и промывают конц. серной
1113 г (100'1
«-СпНо-,ОН---—•—* Н.,Вг-Р Н.,0
кислотой [2]. Декаметилендибромид полхчают
аналогично из соответствующего диола 131.
В синтезе 2,3,4,6-тетра-О-ацетил-а-о-глюко-
пиранозилбромпда (а-ацетобромглюкозы) Б. в. б.
получают, как описано выше, или путем добавления
по каплям жидкого брома к кипящему тетралину [41.
Присоединение к метилакрилату [51. Для полу- Рис. Б-1.
чепия метилового эфира 0-бромпроиионовой кисло-
ты раствор метилакрилата в эфире при охлаждении льдом обра-
батывают Б. в. б., который получают первым способом и освобож-
дают от брома, пропуская через раствор фенола в четыреххлорис-
том углероде.
0’
( II. ;И'.ЗЫ Н. НВ.г -—> ВгСН,СН,СО.,СН3
з . S0-84% -
3 модя r 500 3 моля
эфп р ;i
Расщепление тритиловых эфиров [61. Для удаления тритильной
защитной группы раствор тритилового эфира (2) (6-О-тритил-1,2,3,4-
131
тетра-О-ацетил-р-г>-глюкоза) в уксусной кислоте обрабатывают на-
сыщенным раствором Б. в. б в уксусной кислоте и встряхивают
(') (2)
(3)
в течение 45 сек. Выделяющийся тритилбромид быстро отделяют
фильтрованием, фильтрат выливают в 1 л воды и смесь экстрагируют
хлороформом. Экстракт сушат п выпаривают; сиропообразный оста-
ток кристаллизуется при растирании в эфире.
1. Р у х о ф ф Дж., БерпеттР.,РпдЭ., «Синтезы органических препаратов»,
ИЛ, М., 1949, сб. 2, стр. 109.
2. Рид Э., Р у х о ф ф Дж., Бернетт Р., «Синтезы органических препара-
тов», ИЛ, М., 1949, сб. 2, стр. 112.
3. Мак-Юэн В., «Синтезы органических препаратов», ИЛ, М., 1952, сб. 3,
стр. 111.
4. Р с д с м а п К., Ниман К., «Синтезы органических препаратов», ИЛ, М.,
1952, сб. 3, с гр. 97.
5. Моз и н г о Р_, Паттерсон Л., «Синтезы органических препаратов»,
ИЛ, М., 1952, сб. 3, стр. 308.
6. Р е ii н о ,т ь д с Д., Эванс У., «Синтезы органических препаратов», ИЛ,
М., 1952, сб. 3, стр. 399.
БРОМИСТЫЙ ВОДОРОД — УКСУСНАЯ КИСЛОТА. Насыщен-
ный раствор бромистого водорода в уксусной кислоте применяют
для удаления 6-тритильной защитной группы при получении
1,2,3,4-тетра-О-ацетпл-О-Р-п-глюкопиранозы 11|.
D-Глюкоза 6-ТритилнроизооЭное АСг°>
Ру 35%
1. Рейнольдс Д., Э в а и с У., «Синтезы органических
М., 1952, сб. 3, стр. 399.
препаратов, ИЛ,
СН„СН,СО х
| ' ’ ;NBr
N-БРОМКАПРОЛАКТАМ, СН,СН,СН,/ . Мол. вес 192,07, т. пл.
64—66°.
Б. получают бромированием капролактама (выход 75%) и исполь-
зуют в качестве бронирующего агента, наряду с N-бромсукциними-
132
дом. При использовании Б. не требуется присутствия перекисного
катализатора или активации светом для инициирования реакции [1].
I. TaubB., Н i п о J. В., J. Org. Chem., 25, 263 (1960).
БРОММОЧЕВИНА, H,NCONHBr. Мол. вес 122,97.
Получение |1|. Смесь мочевины, карбоната кальция и воды
перемешивают при охлаждении льдом, добавляют по каплям бром,
0—5°
H,XCONH. + СаСО3 + Вг,-------> Н,ХСОХНВг
0.221 моля 0,126 моля 0,188
в 13,3 .ил воды моля
перемешивают в течение 2 час до прекращения выделения СО,. Про-
дукт выпадает в виде светло-желтых кристаллов. Б. в виде суспен-
зии применяют для окисления сахаров по описанной ниже ме-
тодике.
Окисление 0-защищенных альдоз [2|. Б. окисляет 3,5,6-три-
О-бензил-2-О-метил-о-глюкофуранозу в соответствующий лактон
с почти количественным выходом. Раствор 0,043 молей альдозы
с6н5сн2осиг
осн,.
HjNCONHBr
—-------->
9<%
в 180 мл метанола добавляют при 5—20° в течение 30 мин к суспен-
зии Б., полученной, как указано выше. Смесь разбавляют водой
и экстрагируют эфиром. Как окислитель этот реагент лучше N-бро-
мацетамида.
1. Kiss J., Chem. Ind., 1964, 32.
2. KissJ., Spiegelberg H., Helv. Chim. Acta, 47, 398 (1964).
БРОМНОВАТИСТАЯ КИСЛОТА ВОДНАЯ, см. N-Бромацетамид.
БРОМ — ОКИСЬ СЕРЕБРА. Реакция брома в присутствии
серебра (обычно окиси серебра) с третичными спиртами дает с хоро-
шим выходом продукты внутримолекулярного внедрения кислорода.
Под действием Б.— о. с. 1,3,3-триметилциклогексанол (1) превра-
щается в бициклический эфир (4) с 75%-ным выходом [1|. Предпола-
гается, что промежуточный продукт этой реакции — гипобромит
(2), разложение которого катализируется ионами серебра. Анало-
13:
гично 6а-метплхолестанол-6₽ дает 6,19-эндоксопроизводное:
1. SneenR. A., Mathen у X. Р., J. Am. Chem. Soc., 86, 3905 (1964).
0- БРОМПРОП ИОН И Л ИЗОЦИАНАТ, BrCH,CH2C-N = С = О.
Мол. вес. 178,00, т. кип. 68—70'710 льи. Б. стабилен в течение 4.не-
дель в темноте при —783.
Получение. Б. получают перегруппировкой N-бромсукцинимида
в кипящем хлороформе, содержащем немного хлористого аллила
и следы перекиси бензоила [11.
СН,С
СЬЬВг
CH,CZ
Идентификация. Б. взаимодействует со спиртами с образова-
нием твердых уретанов (BrCH2CH,CONHCOOR), которые можно
использовать для идентификации |2]. Реагент можно также исполь-
зовать для характеристики фенолов и ариламинов (но не алифати-
ческих аминов) [3].
1. J о h и s о n Н. W., Jr., В u b 1 i t z D. Е., J. Am. Chem. Soc., 80, 3150 (1958).
2. Johnson H.W., Jr., К г e у s s 1 er H.A., Needles H.L., J. Org.
Chem., 25, 279 (1960).
3. J о h и s о n H. W., Jr., Day R. J., T i n t i D. S., J. Org. Chem., 28, 1416
(1963).
0-БРОМПРОПИОНИТРИЛ, NCCH2CH2Br. Мол. вес 133,98, т.
кип. 64—66755 мм.
По данным Буцкуса 111, Б. очень удобен для цианэтилирования
аминов и спиртов в присутствии основания, но его преимущества
по сравнению с гораздо более дешевым акрилонитрилом не обосно-
ваны.
1. Б у ц к у с Р. Ф„ ЖОХ, 30, 1799 (1960).
СН,СО
N-БРОМСУКЦИНИМИД (NBC) CH.2COZ NBr 'Мол. вес 178,00,
т. пл. 1737
Обзоры [1—31. Получение [41. Продажный препарат, окрашен-
ный окклюдированным бромом, очищают перекристаллизацией из
воды (10 г на 1 г вещества) 15, 61; при этом удаляются неорганиче-
ские соли, в присутствии которых преобладает реакция присоедине-
ния по сравнению с аллильным замещением 161.
Аллильное бромирование. .Метод, описанный Вельюзом [7|,
используется в промышленном синтезе кортизона. Раствор 100 г
дифенилэтилена (1) в 1л СС14 обрабатывают 30 г NEC и кипятят на
свету в 15 мин до начала выделения бромистого водорода. Затем
смесь охлаждают, осадок образовавшегося сукцинимида отделяют
фильтрованием и раствор 22-бромпропзводного кипятят около 4 час
до прекращения выделения бромистого водорода, сопровождающе-
гося образованием диена (2). После удаления растворителя остаток
кристаллизуют из ацетона и получают 75,4 г (2). Из вещества, полу-
ченного из маточного раствора, после непродолжительного кипя-
чения с диметиланилпном, повторного ацетилирования и кристал-
лизации из ацетона получают дополнительно 6,4 г (2). Общий выход
(2) составляет 82%. Повторное аллильное бромирование дает (3)
(выход 72%), из которого кипячением с безводным ацетатом калия
в уксусной кислоте получают (4) с выходом 77,5% . Наконец, окисле-
ние хромовой кислотой в смеси уксусная кислота — дихлорэтан
приводит к отщеплению боковой цепи с образованием 21-ацетоксп-
20-кетона (5) с выходом 72%.
Аллильное бромирование холестерилбензоата в положение 7
является ключевой стадией в промышленном синтезе витамина П:!.
Согласно тщательно отработанной методике, обработка 20%-ным
избытком NEC проводится при кипячении в нетролейном эфире
на свету в течение 4 мин 18].
Исследования, проведенные в четырех лабораториях [91, показа-
ли глубокое сходство реакций NEC и брома. Поэтому цепной ради-
кальный механизм с участием атома брома является наиболее ве-
роятным для реакций NEC.
Окисление вторичных спиртов. В присутствии воды NEC яв-
ляется высоко избирательным окислителем. Например, окислением
135
Холестантриола-ЗР, 5а, 60 NBC в водном дйоксайе получают 6-кето-
холестандиол-Зр, 5а с выходом 93% [10].
Реагент избирательно атакует одну из
ксильных групп. Поэтому при ее окислении
трех вторичных гидро-
избытком NBC в водном
растворе бикарбоната натрия удается получить дезоксихолевую
кислоту — исходный продукт в синтезе кортизона 111].
Другие реакции. NBC применяли для получения НОВг в водном
1,2-диметоксиэтане [12], однако для этой цели более предпочтите-
лен N-бромацетампд.
Чистый NBC эффективно бромирует ароматические углеводороды
в боковую цепь, тогда как долго хранившийся реагент бромирует
ароматическое ядро 113]. Примеси, вызывающие этот эффект, мож-
но удалить откачиванием образца в вакуумэкспкаторе (0,5 мм)
над Р2О;> в течение 4 час.
Внимание! При работе с NEC возможен взрыв 114]. Баракат
и сотр. 1151 нашли, что при обработке NEC в водном растворе
а-амипокислоты (аланин) и двухосновные кислоты (щавелевая)
декарбоксилируются с выделением брома. Лак |16| показал, что
а-аминокислоты, пептиды и белки под действием NEC (2 экв) в вод-
ном растворе количественно декарбоксилируются. Эта реакция
заканчивается через 30 мин\ на аппарате Варбурга определяется
количественное выделение СО2. Единственный устойчивый продукт
136
этой реакции — альдегид.
NEC
СН3СНСО,Н-----> CH3CHO-HNH3 + COn
I
\н,
1. Waugh Т. D., «N-Bromosuccinimide, Its Reactions and Uses», Arapahoe
Chemicals, Inc., Boulder, Colo, 1951; H о r n e r L., W i k e 1 m a n n E. H.,
Newer Methods of Preparative Organic Chemistry, 3, 151 (1964).
2. Djerassi C., (allylic bromination), Chem. Revs., 43, 271 (1948).
3. F i 1 1 e r R. (oxidation and dehydrogenation), Chem. Revs., 63, 21 (1963).
4. C a m p a i g n e E., Tull ar B. F., Org. Syn., Coll. Vol., 4, 921 (1963).
5. DaubenH. J., McCoy L. L., J. Am. Chem. Soc., 81, 4863 (1959).
6. В a i 1 e у W. J., В e 1 1 о J., J. Org. Chem., 20, 693 (1955).
7. V e 1 1 u z L., Substances naturelies de synthese, 7, 31 (1953).
8. BernsteinS., В i n о v i L. J., Dortman L., Sax K. J., Subba-
row Y„ J. Org. Chem., 14, 433 (1949).
9. S k e 1 1 P. S. et al., J. Ain. Chem. Soc., 85, 2850 (1963); W a 1 1 i n g C. et al.,
ibid., 85, 3129, 3134 (1963); Russell G. A. et al., ibid., 85, 3136, 3139 (1963);
Pearson R. E., Martin J. C., ibid.. 85, .3142 (1963).
10. FieserL. F., RajagopalanS., J. Am. Chem. Soc., 71, 3938 (1949).
11. F i e s e r L. F., RajagopalanS., J. Am. Chem. Soc., 71, 3935 (1949).
12. T a m e 1 e n E. E., v a n, S t о г и i A., Hessler E. J., Schwartz M.,
J. Am. Chem. Soc., 85, 3295 (1963).
13. C h a p m a n N. B., W illiams J. F. A., J. Chem. Soc., 1952, 5044.
14. Martin R. H„ Nature, 168, 32 (1951).
15. В a r a k a t M. Z., E 1 - W a h a b M. F. A., E 1 - S a d r M. M., J. Am. Chem.
Soc., 77, 1670 (1955).
16. C h a p p e 1 1 e E. W., Luck J. M., J. Biol. Chem., 229, 171 (1957);K o-
n i g s b e r g N., Stevenson G., Luck J. M., ibid., 235, 1341 (1960).
NO2 Вг
4-БРОМ-2,5,7-ТРИНИТРОФЛУОРЕН,
Мол. вес 283,22, т. пл. 191 —192'.
2,4,7-Тринитрофлуорен образует два комплекса с 1-метоксибенз-
(cl-фенантреном: черный (1 : 1) с т. пл. 2123 и красно-коричневый
(1:1) с т. пл. 157~. Для установления строения подобных комплексов
методом рентгеноструктурного анализа Ньюмен и Блюм 111 синте-
зировали бромтринитрокетон и соответствующий иодтринптрокетон
(т. пл. 193—194 ). Оба соединения являются активными комплек-
сообразующими агентами; однако с 1-метоксибенз-[с]-фенантреном
получен комплекс только одного типа. Многие изученные соединения
образуют с этими реагентами комплексы в соотношении 1:1, за
исключением пренитола, который дает комплекс, содержащий 2 же
углеводорода на 1 же л-кислоты. Особенно интересно то, что бром-
и иодтринитрофлуореноны с бензолом образуют желтый и светло-
оранжевый комплексы, которые не изменяются в течение дня в ва-
кууме (1—2 мм pm. ан.) при комнатной температуре и отщепляют
бензол только при нагревании (не выше 100').
1. Newman М. S., В 1 u m J., J. Am. Chem. Soc., 86, 5600 (1964).
137
БРОМТРИФТОРМЕТАН, CF.,Br. Мол. вес 148,93, газ.
Б. реагирует с алкпллитием и алкил.магнийбромпдом подобно
дифтордпбромметану и приблизительно с таким же выходом 111.
3< ,11,1.: < i ,Br ( , 11,,< :i 1 ( НС И-f-.н..Вг 31.:Е
b J' I)
1. Franzen V., F i k e n t s c her L., Chem. Bor., 95, 1958 (1962).
БРОМТРИХЛОРМЕТАН, BrCCl,. Мол. вес 198,30, т. кип. 104°,
уд. вес 2,055. Очистка 111.
Б. используют как растворитель для реакции Хунсдикера 11],
в фотохимическом бромировании алкилбензолов 121
/IV
ВгСС13—-—> Вг -!- СС13
СС13 у ArCH, —* НСС! . ДгСН,
ArCH, -- ВгСС13 —* ArCl CBr-j- -СС13
и для образования дпхлоркарбеиа 13]:
ВгССЦ
H-BuLi
-BuBr
» :CC12
Циклогексен
91%
1. Bake r F. W., Holt z 11. D., Stock L. Al.. J. Ore. Chem., 28, 514 (1963).
2. H и v s e r E. S , .1. Am. Chem. Soc., 82, 391, 394 (I960).
3. Aliller W. T.. Jr., К i m C. S. Y., J. Am. Chem. Soc., 81, 5008 (1959);
С 1 о s s G. L., С 1 о s s L. E., ibid., 82, 5723 (1930).
п-БРОМФЕНАЦИЛБРОМИД, »-BrC6H4COCH2Br. Мол. вес
277,96, т. пл. 110с Получение |1|.
Карбоновые кислоты можно охарактеризовать в виде и-бром-
фенациловых эфиров 121. Спирты для идентификации превращают
в алкилксаитогеиаты калия и затем реакцией с Б. в ацетоне в хорошо
S S
C.S.,. КОИ ’ BiCH ,COC„H„Br
ROH-----:-----> ROCSK-------у-----> ROCSCH,COC6H4Br-/i
кристаллизующиеся //-бромфенацилалкилксантогенаты 13], которые
в аналитических целях удобнее, чем ксантогеиаты.
1. Л а н г л е 11, «Спигезы органических препаратов.-», ИЛ, М., 1949, сб. 1, стр. 138.
2. Wil cl F., Characterization of Organic Compoum's, Cambridge, 1947, 146, 147.
3. В e r g e r J., Uldall I., Chem. Scand., 18, 1353 (1964).
БРОМЦИАН, BrC—N. Мол. вес 105,93, т. ил. 52', т. кип. 61°.
Получение 11,2].
138
Б. чрезвычайно токсичен и представляет большую опасность
при неаккуратной работе. Все операции необходимо проводить под
тягой.
Реакция Брауна. При взаимодействии Б. с третичным амином
образуется дизамещенный цианамид п алкилбромид [3, 41:
R,NR + BrCN R2NC\T4-RBr
Ы.Ы-Диметил-а-нафтиламлн кипятят с Б. в течение 16 час, после
охлаждения к смеси добавляют эфир и отфильтровывают осадок
а-нафтилтриметиламмонийбромида; затем смесь экстрагируют раз-
бавленной соляной кислотой для удаления исходного амина и N-ме-
тил-а-нафтилцианамид перегоняют.
Эта реакция в частности использовалась при установлении строе-
ния алкалоидов. Например, при изучении лупинпна лупинан (1)
расщепляли под действием Б. в кипящем бензоле с образованием
BrCN >
90%
СН2СЦ2СН2СН2Вг
продукта (2), строение которого доказано превращением в хиноли-
новую кислоту (3) [61.
Расщепление тиоэфиров. Диалкплтиоэфиры расщепляются Б.
при несколько повышенной температуре (60—70~, в запаянной ампу-
ле) с образованием алкилтиоцпаната и алкилбромпда (Браун 161).
Три примера, приведенные на схеме, отчетливо показывают зави-
симость направления расщепления от величины группы:
CH3CH2SCH3
BrCN
CH3CH.,SCX + CH3Br
BrCN
CH3CH2CH,SCH,CH3--------- CH3CH,CH2SC.\' + CH3CH,Br
BrC.N
CH3CH2CH2CH2SCH2CH2CH3-------> CH3CH2CH2CH.,SCN +CH3CH2CH2Br
139
Эту реакцию Гросс и Виткоп использовали 17] для селективного
расщепления метиониновых пептидов по схеме
CH3S
СН2 BrCN
Н О СН, н2о
I I I ------
RCHN+—С — CHNHCOR' Вг
I
со,н
О СН, -CH3SCN
I! I
RCHNHC—CHNHCOR'
I
СО2Н
сн,
О'7 сн,
—> RCHNH,HBr-|-O = C —CHNHCOR'
I
со,н
Расщеплением гипофизарного гормона роста человека (HGH) под
действием Б. в 70%-ной муравьиной кислоте установлено положение
двух дисульфидных мостиков и подтверждена последовательность
188 аминокислот, выведенная из обычного ферментативного расщеп-
ления |7а|.
Реакция с 1,2-аминоспиртами. Ньюхолл и сотр. [81 восстанов-
лением оксимпнокетона (1) получали смесь цис- и троис-ампноспир-
тов (2) и (3), которую обрабатывали Б. Из фракции, имеющей основ-
ные свойства, выделен в кристаллическом виде /р/с-ампнооксазолин
(4), а из нейтральной фракции — транс-оксицианамид (5), который
140
под действием кислоты циклизовали в /?г^л«с-аминооксазолин (6).
Выходы низкие.
Ацилирование. При кипячении салицилата натрия с Б. в ук-
сусной кислоте наблюдается выделение СО2 и образуется ацетил-
салициловая кислота; эта реакция еще ждет своего объяснения[9].
1. Slotta К. Н„ Вег., 67, 1028 (1934).
2. X г, р т м а н В., Д р е д ж е р Е., «Синтезы органических препаратов»,
ИЛ, М., 1949, сб. 2, стр. 123.
3. В га tin, J., von, Вег., 40, 3914 (1907); 42, 2219 (1909); 44, 1252 (1911).
4. X е ii геман X. А., «Органические реакции», ИЛ, М., 1956, сб. 7, стр. 260.
5. Крессман Г., «Синтезы органических препаратов», ИЛ, М., 1953, сб. 4,
стр. 308.
6. Braun J., von, Engelbert z Р., Вег., 56, 1573 (1923).
7. Gross Е., W i t к о р В., J. Am. Chem. Soc., 83, 1510 (1961); J. Biol. Chem.,
237, 1856 (1962).
7a. L i C. H„ L i u W.-K., Dixon.l. S„ .1. Am. Chem. Soc., 88, 2050 (1966).
8. X ewha 1 1 \V. F., Poos G. I., R о s e n a u .1. D., Suh .1. T., J. Org.
Chem., 29. 1809 (1964).
9. T h v a g a r a j a n B. S., R a j a g о p a 1 a n I\., Tetrahedron Letters, 729
(1965).
р-БРОМЭТИЛМЕТИЛКЕТОНА ЭТИЛЕНКЕТАЛЬ (2). Мол. вес
195,07, т. кип. 76 /4 мм.
Б. э. получают реакцией соответствующего спирта с РВг:! 11]
или присоединением НВг к метилвинилкетону и обменной кетализа-
цией [2].
Для аннелирования по Робинсону. Сторк |3] использовал Б. э.
для синтеза (4) из (1); однако низкая реакционная способность и тен-
денция к отщеплению НВг препятствуют широкому применению Б. э.
1 W 1 1 1 1 m а п п L., S с h i n z Н., Helv. Chim. Acta, 32, 2151 (1949).
2. S t о г k G., В о г с h R., J. Am. Chem. Soc., 86, 935 (1964).
3. S t о r k G., «Third Internal. Svmp. on the Chem. of Nat. Prod.» Kvoto, 1964,
p. 134.
БУТАДИЕН, CH2--CHCH^CH2. Мол. вес 54,09, т. кип. —4,4°.
CI l3
I
HOCH
d-(—)-БУТАНДИОЛ-2,3, I . Мол. вес 90,42, aD—12,9°
I
CH3
чистый).
141
b, L-Камфора была расщеплена на антиподы газожидкостной
хроматографией диастереомерных кеталей, полученных с помощью
этого легкодоступного реагента (получают ферментативно) [1].
1. Casanova J., Corey Е. J., Chem. I ml., 1961, 16(54.
ь-(+)-БУТАНДИТИОЛ-2,3. Мол. вес 122,25.
Б. получают из d-(—)-б\'тандиола-2,3 (1) по следующей схеме
[И:
сн3
но-----н
н-----он
сн3
(1)
сн3 сн3
ТвС1-Р>; ТвО-------Н KSCN ч Н-----------SCN
* Н--------ОТв (СНэ)г5О NCS---------Н
СН3 СН3
(2) (3)
Н3С .СН3 снз
Ш-Р; 2-----( ЫА1Н4 Н-----------SH
s4 xs HS-----н
н СН3
о
(4) (5)
Для разделения рацематов соединений, содержащих карбонильную
группу (которая затем восстанавливается), конденсацией с опти-
чески активным дптиолом (5) получают смесь диастереомерных тио-
кеталей, разделение и десульфуризация которых приводят к опти-
чески активным дезоксосоединенпям.
1. Со г е у Е. М i t г a R. В., .1. Am. Cliem. Soc., 84, 2938 (1962).
лпре/га-БУТИЛА ГИДРОПЕРЕКИСЬ, (СН.,)3С—ООН. Мол. вес
90,12, т. пл. 3—4 , т. кип. 40 723 мм 11], 42718 л/л/ 12], 37—38716мм
[31, 35—36 /8,5 л/л/ [41. Получение 151. /Методика анализа 161.
Очистка. Б. г. очищают ректификацией [2,3] или с отделением
азеотропа до образования гомогенного раствора и последующей
перегонкой 11]. После нескольких вакуумных перегонок продажного
продукта (76—90%) удается получить препарат, содержащий более
99% Б. г. 13]. Продажный продукт очищают также ]4] экстракцией
холодным 15%-ным водным едким кали, последующей нейтрализа-
цией твердым хлористым аммонием и перегонкой в вакууме (98,4%).
Реакции с ненасыщенными соединениями. Б. г. гладко разла-
гается при 95—1 ОБ п гидроксилирует двойную связь даже а, 0-непре-
дельпых кетонов; реакция катализируется небольшими добавками
тетраокнси осмия и приводит к образованию /р/е-глпколей 17]. Под
действием Б. г. в бензольном растворе в присутствии тритона Б.
(гидрат окиси бензилтриметиламмония) как катализатора а,Р-не-
предельные кетоны превращаются в соответствующие эпоксиды с вы-
ходами 50—90°о [81. Отсутствие взаимодействия Б. г. с изофороном
и А4-3-кетостероидами указывает на определенные ограничения для
142
этой реакции, катализируемой основаниями. Р-Фенилглнцидный
альдегид гладко получен (Пайн 191) взаимодействием коричного аль-
дегида с Б. г. в метаноле в течение 5—6 час при постепенном добавле
нии к смеси разбавленного раствора едкого натра для иейтрализа-
pll fS.S, 33-ИГ; доЗав.т. NaOH (' Н3СН —СНСНО
СВН.-,СН=СНСНО + (СН3)3СО,Н ---------—------------► .хо/
0,3 моля 0,6 моля
в СИ,ОН
шш кислых побочных продуктов (pH около 8,5). Щелочная перекись
водорода дает плохие результаты.
Арилгалогенид —- фенол. Новый способ перехода от арилгало-
гснидов к фенолам состоит в обработке Б. г. 1 экв этилмапшйбромпда
с образованием МдВг-производного, к которому затем добавляют
1 экв фенилмагннйбромнда и после гидролиза реакционной смеси
с хорошим выходом получают фенол [101.
ОМуВг ОН
I
C,HsMgBr в- 0 Н.о | А
(сн3)3со2н —-г (CH3)sco2MgB —J
'Ч/ V
Декарбоксилирование. Виберг и сотр. [111 разработали инте-
ресный метод декарбоксилирования бипикло-|2,1,1 |-гексан-5-кар-
боиовой кислоты (1) с образованием незамещенного углеводорода
(4). Хлорангидрид (2) реакцией с Б. г. и пиридином в /z-цимоле при
перемешивании и охлаждении льдом превращают в эфир надкисло-
ты (3), который, не выделяя, получают в виде безводного свободного
от кислоты раствора в //-цимоле. При нагревании этого раствора вы-
о
2 А/С°гН soci2 ix^\\/cocl ноос(с.н31, z-^Kpooc(CH3)3
н ~95%~* Щ/Н Ру-л- цимол Щу Н
А <2)
НЗН'С<СсН3 115-130°
Н3с' \=/
45%,считая на(2)
деляется СО..; н-нимол используется как донор водорода при обра-
зовании углеводорода (4) и как акцептор ////щ///-бутоксильных радика-
лов с образованием (5). В случае 1-хлорзамешешюй кислоты (1)
общий выход в этой реакции составил только 26'16 *.
См. также ХСХ'-Карбопплдиимидазол.
* Эта реакция явилась к.иочепоп в синтезе губана из ксбанкарбоиовой
кислоты [Eaton Р., С о 1 е. Т., J. Am. Cliem. Soc., 86, 3157 (1964)].— Прим,
перев.
143
а-Окисление кетона. .Метиловый эфир 7-кетодегидроабиетиповой
кислоты (1) при нагревании в уксусной кислоте с Б. г. и следами
H2SO4 в течение 55 час при 50—55' дает ненасыщенный лактон
(2) с очень высоким выходом 112]:
1. В а г t 1 о t t Р. D., М inato Н., J. Am. Chem. Soc., 85, 1858 (1963).
2. S i 1 b е i t L. S., S w е г n D., J. Ain. Chem. Soc., 81, 2364 (1959).
3. Kochi .1. K„ Moca d 1 о P. E., J. Org. Chem., 30, 1134 (1965).
4. Bartlett P. D., Me В r i de J. M., J. Am. Chem. Soc., 87, 1727 (1965).
5. M i I и s A., S u r g с и о r D. M., J. Am. Chem. Soc., 68, 205 (1946); 'A' a 1-
I i ii g С., В u c k 1 e r S. A., J. Am. Chem. Soc., 77, 6032 (1955).
6. Wagner C. D., Smit h R. H., Peters E. D., Anal. Chem., 19, 976
(194-7).
7. Milas N. A., Sussman S., J. Am. Chem. Soc., 58, 1302 (1936); 59, 2345
(1937).
8. Y а и g N. F i n n c g a n R. A., J. Am. Chem. Soc., 80, 5845 (1958).
9. Pay n e G. 13.. .1. Org. Chem., 25, 275 (1960).
10. L a w e s s о n S.-О., Y a n g N. C., .1. Am. Chem Soc., 81, 4230 (1959).
11. Wiberg К. B., Ln wr v B. R., С о 1 b v T. H., J. Am. Chem. Soc., 83,
3998 (1961).
12. W e n k e r t E., Carney R. W. J., К a n e k о C., J. Am. Chem. Soc.,
83, 4440 (1961).
Н-БУТИЛАЗИД, h-C4H9N —N - N. Мол. вес 99,13, т. кип. 106,5°.
Б. получают 11 [добавлением раствора н-бутилбромида к суспензии
азида натрия в воде. Реагент расщепляет кетоны типа ArCOCH2R
в бензоле пли нитробензоле при 70—90 в присутствии каталити-
ческих количеств серной кислоты с образованием двух карбониль-
ных соединений [21. Ацетофенон дает бензальдегид (8О?6) и формаль-
дегид (83"<>); пропиофенон — бензальдегид и ацетальдегид. Реакция
интерпретируется следующим образом:
о он + ОН
1[+ I Bu\ = \ = N | -N,
С6Н5ССН3 —г С6Н.-,ССН3 > С6Н5ССН3
+ I + _ —н-’°
Bu\'+—X = N
но он
-г 9Н О I I
__,свщс=сн,<->сгн,-с-сн., с6н-,с-сн, —с6н5сно-сн.,о
I '-н<- | - РиКН,
BuX + ВиХ ВиХН
Она включает атаку азидом сопряженной кислоты кетона, выде-
144
ление азота и воды из катиона диазопия, гидратацию и расщеп-
ление.
1. 15 о \ е г .1. И., М о г g a n L. R., Jr., J. Am. Chem. Soc., 80, 2020 (1958).
2. В о у е г J. 11., Н a in с г J., J. Ain. Chem. Soc., 77, 951 (1955); В о у е г J. И.,
М о г g а п L. R., Jr., ibid., 81, 3369 (1959).
/ире/и-БУТИЛАМИН, (CH3)3CNH2. Мол. вес 73,14, т. кип. 45’,
уд. вес 0,698.
Б. используют вместо аммиака в окислительной конденсации
соединений с концевой ацетиленовой группой по Глазеру 111.
Этот пространственно затрудненный амин успешно использован для
дегидробромирования дибутокси-1-бром-3,3,3-трихлорпропилборана
121.
с13ссн.,снв(ос4н,,).,-у(сн3)3с\н, ci3cgh == снв(ос4н9).,
| о
Вг
Обычные реагенты в этом случае неприемлемы; с этилатом калия
и даже /прс/п-бутилатом натрия происходит только замещение атома
брома на алкокспльную группу, а продукты дегидробромирования
не обнаруживаются.
1 Cameron М. D., В е n nett G. Е., J. Org. Chem., 22, 557 (1957).
2 . Maltese n D. S., М a h R. W. И., J. Org. Chem., 28, 2174 (1963).
mpem-БУТИЛА ПЕРЕКИСЬ, (СН3)3СО2С(СН3)3. Мол. вес 146,22,
т. кип. 50—52290 мм. Продажный препарат очищают перегонкой
в вакууме II].
Разложение. Б. п. претерпевает гладкое термическое разложе-
ние с образованием /преш-бутоксильных радикалов, которые в за-
висимости от растворителя в той пли иной степени разлагаются
далее на метильные радикалы и ацетон |2|:
(СН3)3СО.,С(СН3)3 —* 2(СН3)3СО- — 2(СН3),С - О- - 2-СН3
Период полупревращения для первой стадии реакции, определяю-
щей скорость процесса, составляет примерно 20 час при 120 п 1 час
при 150 . Б. п. применяется для генерирования радикалов, в част-
ности для инициирования цепной полимеризации и других радикаль-
ных реакции в интервале температур 120—150'.
Метилирование ароматических соединений. Радикальное аро-
матическое замещение ввиду низкого выхода представляет скорее
теоретическое, нежели препаративное значение. Уотерс и сотр. [31
изучили метилирование моиозамещениых бензола метильными ра-
дикалами, полученными из Б. п. Смесь монометилпрованиых про-
(..Н,.\ 6 !1:;:,ш1. -СН3—CII3C„H4X ;- (CI 13):.СО1 1
изводных отделяли от исходного соединения с помощью ГЖХ и опре-
деляли соотношение трех образующихся изомеров по ПК-спектрам.
145
Например, смесь хлорбензола и Б. п. кипятят е одновременной от-
гонкой образующихся ацетона и треш-бутанола, поддерживая тем-
пературу около 130°. Соотношение изомеров полученных
лов показано на следующей схеме:
С1
хлортолуо-
С1
Cl
СН3
(-(СН3)3СО2С(СН3)3
100 ,ил
64
сн;
Распределение и номеров
Присоединение амидов к олефинам. Б. п. инициирует ради-
кальное присоединение формамида к гексену-1 с образованием ами-
да, имеющего неразветвленнын заместитель с выходом 5—10%
(Рихе [4]). При использовании циклической перекиси, полученной
сн,
сн3у ./ \ СН,
ЧС с/
СН-/| ро—
о-----------о
Н3С, /
>с
—oz |
о—
сн.
ч /СНз
с
1 ' СНз
-О
из окиси мезитила, полимеризация уменьшается и выход возрастает
до 35—40%. Метиловый эфир ундеценовой кислоты дает амид мети-
•R
СНз (СН,)3СН = СН, у HCONH, —уд СН3 (СН,)3СН.,СН,СО\Н,
•R
СН2=СН (СН,)8СО,СНз+ HCONH, - H,NCO (СН,)10СО2СН3
лового эфира декандикарбоиовой кислоты с выходом 40%.
Реакцией октена-1 с диметилформамидом в присутствии катали-
тических количеств ди-трт-бутилперекиси получена смесь N.N-ди-
метиламнда пеларгоновой кислоты 1(1), амидирование] и М-метил-N-
нонилформамида 1(2), ампноалкилпроваппе! в соотношении 60 : 40
(Фридман и Шехтер 151). В тех же условиях П-шрет-бутилформамид,
• R (! 8 7 '.с при 1 3 2
СН3 (СН,)5СН^С.Н, -У HCON (СН3)2 ---—-------”
СН3
—- СН3(СН,)3СН,СН,СО\(СН3), у- СНДС11,).-,СН,СН ,CH,NCI IO
<1> (0)
N-алкил- и .Ч,М-диалкилацстамиды реагируют с октеном-1, изби-
рательно образуя с хорошим выходом только продукт типа (1).
1. Beckwith A. L. J., W a t е г s W. АJ. Chem. Soc., 1956, 1108.
2. Уоллинг Ч., X о ii з с р Э., «Органические реакции», изд-во «Мир»,
1966, сб. 13, стр. 103.
146
3. Crowley В. R., Norman R. О. C., Waters W. A., J. Chem. Soc.,
1959, 1799.
4. R i e c h e A., Schmitz E., G r u n d e m a n n E., Angew. Chem., 73, 621
(1961).
5. Friedman L., Schechter H., Tetrahedron Letters, 238 (1961).
/ире/и-БУТИЛАТ АЛЮМИНИЯ, Al [OC(CH3)3]3. Мол. вес 246,32.
Получение [1|.
Окисление по Оппенауэру [2]. Окисление холестерина в холе-
стеной проводят [31 осторожным кипячением в течение 8 час смеси
продажного холестерина, Б. а., ацетона (акцептор водорода) и бен-
зола. Охлажденную смесь встряхивают с разб. серной кислотой,
продукт реакции выделяют из бензольного слоя и дважды кристал-
лизуют.
Катализатор конденсации. Б. а.— один из реагентов, катали-
зирующий самокондепсацпю ацетофенона в дипнон[4]. Реакционную
смесь в ксилоле перемешивают при нагревании с медленной отгон-
кой шдеш-бутаиола:
CIL, О
CeHsC = O ' СН3СС6Н..-А1[ОС(СН3)3]3л_свН4(СН3)2-^|^-
" “ ’ 0,55 моля 400 мл '°
1 моль
сн3 о
I н
----- С6Н5С = СНССаН5
1. Уэйн У., Адкинс Г., '.Синтезы органических препаратов», ПЛ, М.,
1952, сб. 3, стр. 119.
2. Джерасси К., «Органические реакции», ИЛ, М., 1953, сб. 6, стр. 235.
3. Описи a v э р Р., «Синтезы органических препаратов», ИЛ, М., 1952, сб. 3,
стр. 486.
4. У э й н У., А д кипе Г., (.Синтезы органических препаратов», ИЛ, М., 1952,
сб. 3, стр. 224.
/ире/и-БУТИЛАТ КАЛИЯ, (СН3),,СОК- Мол. вес 112,21. Белый
гигроскопичный порошок. Данные о растворимости Б. к. приведены
в таблице
147
Растворитель
Растворимость
(25 — 26-'). <1и0г
Гексан
Толуол
Эфир
т ре т - К у т и л о в ы й
спирт
Тетрагпдрофурап
0,27
2,27
4,34
17,80
25,00
Продажный Б. к. может содержать следы едкого кали или поташа,
но он вполне пригоден для большинства целей. Очистить Б.к. можно
возгонкой при 220 /1 мм, однако при этой температуре часть вещест-
ва теряется вследствие разложения.
Меры предосторожности. Б. к. весьма гигроскопичен, и рабо-
тать с ним лучше всего в сухой камере. Рекомендуется проводить
реакции в инертной атмосфере. Б. к. разъедает кожу и слизистые
оболочки. Он вступает в экзотермическую реакцию с кислородом,
поэтому длительное соприкосновение с кислородом или воздухом
при повышенной температуре небезопасно в пожарном отношении.
Температура воспламенения Б. к. 160—165 '.
Получение. Джонсон и сотр. Ill описали методику получения
раствора Б. к. в трет-6\тиловом спирте в атмосфере азота. Скат-
тебол и Соломон [2| получали раствор этим же методом и выпарива-
нием выделили твердый, не содержащий растворителя Б. к. (краткое
изложение метода см. Метиллнтий, алленовый синтез). Б. к. можно
также получить из калия, очищенного с помощью т/ж/«-амилового
спирта (см. Калий, методика Пирсона).
Б. к., вероятно, самое сильное основание из всех известных.
Как основание Б. к. сильнее этилата натрия, причем его применение
в отличие от последнего не вызывает осложнении, связанных с окис-
лительно-восстановительными побочными реакциями. В ряде слу-
чаев эго наилучшпй, а иногда и единственно приемлемый реагент.
Конденсации (см. также Калия фторид). Конденсацию бензо-
фенона с дпэтпловым эфиром янтарной кислоты по Штоббе осуще-
ствляют путем прибавления этих реагентов к свежеприготовленному
раствору Б. к. в шре/п-бутпловом спирте 11 ]. Аналогичным образом
СНоСОХоН-, ’’ °с (СЬ,3>-'(n'055 Mo-™’ сн,со,н
(с6н5)Д=о-|- ! ’ । -
СП.2СО2С2Н5 92-94% (С6Н5).2С = ССО2С2Н5
0,05 моля U,075 моля
(температура 10—15е) конденсируют циклогексанон с этиловым
Н8
эфиром хлоруксусной кислоты по Дарзану [31.
О О— СНСО.2С2Н5
/ КОС (СН3)3 (0, 153 моля) (
|J + C1CH2CO2C2H5~--------------------- IJ
0,148 моля 0,148 моля
Б. к. используют также в качестве катализатора основного типа
в видоизмененной конденсации по Дарзану, в которой кетон конден-
сируют не с галогензамещенным сложным эфиром, а са-галогеннпт-
рилом с целью получения глицидоиитрила 141. Этилат натрия в этой
реакции применять нельзя, так как он приводит к смеси эпоксини-
трила (I) и нмидоэфира (II):
NH
Конденсация динитрила (1) по Торпе была успешно осуществле-
на под действием Б. к. в/npem-бутиловом спирте |5|; провести цикли-
зацию с этилатом натрия пли натрием в диоксане не удалось. Леонард
и Шимельпфениг [61 осуществили циклизацию по Дикману cz,co-
диэфиров в присутствии Б. к. в кипящем ксилоле в атмосфере азота
при перемешивании скоростной мешалкой. Продукты реакции гид-
ролизовали и декарбоксилировали кипячением со спиртовым раст-
вором соляной кислоты. Монокетоны С|4 — С16 получены с выходами
24—48?»; дикетоны С13—С,о, С.,., и С.,4 — с несколько меньшими вы-
ходами. Метод оказался неприменимым для синтеза циклических
СО.,С.,Н5 кощснш zco , С°\
(СЩ)"< со2с,н,“------------<сн=)
СН2 со/
149
соединений Cs—Cl2. Леонард и сотр. [7] использовали Б. к. для цик-
лизации а-аминодиэфиров в циклические диаминодикетоны с числом
членов в кольце до 20.
Джонсон и сотр. |8] нашли, что Б. к.— наилучшее основание
для циклизации по Дикману диэфира (3) в эпиандростерон (4). В этом
случае как метилат натрия, так и гидрид натрия оказались неэф-
фективными.
Чтобы осуществить альдольную циклизацию метилкетона (5)
с образованием соединения (6), являющегося ключевым промежу-
точным соединением в полном синтезе цедрола, Сторк и Кларк [9]
применяли Б. к. в mpm-бутиловом спирте при комнатной темпера-
туре. Если реакцию проводить при температуре кипения, то полу-
чается нежелательный продукт (7). Конденсацию (5) в (6) не удалось
осуществить ни с mpm-бутилатом алюминия, ни с п-толуолсульфо-
кислотой.
О'
,.н
...сн3
сн3
КОС(СН3)3
(СН3)3СОН
25°
ОН сн3
т3с --------- .
(5) 20,1г
। КОС(СН3)3-(СН3)3СОН
Кипячение
(6)18, 6 г (сырой)
(7)
Ключевой стадией в новом синтезе циклических кетонов по ме-
тоду Хауза и Бабада [10] является циклизация соли фосфония типа
(8) в циклофосфоран (9), протекающая в присутствии Б. к. в трет-
бутпловом спирте. Считают, что эта реакция родственна конденсации
по Дикману. При // 2-—4 выходы составляют 52—84%.
о
+ в
(С6Н3)3Р^ сосдц
- сн2 сн2
1 kcHjJ
О’
(С6Н5)3Р
КОС(СН3)3 С 9нг
(сн3)3сон’ Исн9,(
о
н+ Л
н2о сн2 СН2
* l(CH2)J
(10)
(8)
150
Изомеризация непредельных соединений. Рафаэль ЦП пока-
зал, что 10%-ный раствор Б. к. в кипящем диглиме (т. кип. 161 )
вызывает перегруппировку дпацетилепов в ароматические углево-
дороды с выходом около 65%. Так, из гептадиипа-1,6образуется то-
Беи-Эфрапм и Зоидхсймер 1121 нашли, что в результате изомери-
зации /(//с-октен-4-дпина-1,7 (1) при обработке его Б. к. в трст-бу-
гиловом спирте при 60—65 в течение 15 мин наряду’ с линейными
мономерными продуктами образуются ароматические димеры (4)
и (5). Поскольку Эрреде 1131 показал, что углеводороды (4) и (5) по-
лучаются при нагревании о-ксплплена (3) до комнатной температуры,
предполагают, что о-ксилилен является промежуточным соединением.
При изомеризации в тех же условиях //гринс-нзомера (1) образуются
только линейные сопряженные соединения. 1,7-Дифенилгептади-
ип-1,6 (6) при действии Б. к. в /;;рс/?;-бутиловом спирте дает произ-
водное нафталина (7) с умеренным выходом 1141.
Цикло&ц-о- ксилилен
Спироби-о-ксилилен-- .
Рпнгольд и Малхотра 1151 нашли, что при обработке А'-З-кето-
стероида 10 же Б. к. в /прс/?(-бутиловом спирте в течение 1,5 час
в атмосфере азота при комнатной температуре и вротонпровании
образовавшегося аниона путем быстрого добавления уксусной ки-
слоты получается А '-З-кетон с выходом до 95%. Нарушение сопряже-
151
ния можно осуществить также в диглиме, однако в бензоле оно про-
исходит крайне медленно.
(СН,)3СОК
-(CHPjCOH
Крам 1161 наблюдал, что диметилсульфоксид (ДМСО) значитель-
но усиливает основность Б. к.; это привело к широкому использо-
ванию этой системы для карбанионных реакций. Гарднер [17],
например, нашел, что аллен (1) под действием Б. к. вД.МСО при 70’
в течение 1 час перегруппировывается в цис, ф/оциклононадиен (2).
Увеличение времени реакции до 3 час привело к частичному превраще-
нию диена-1,3 (2) в диен-1,4 (3) и дпен-1,5 (4). В изомере с сопряжен-
ными связями (2) отсутствует обычная резонансная стабилизация,
поскольку напряжения в кольце мешают расположению в плоскости.
Б. к. в сочетании с ДМСО вызывает перегруппировку цис,цис,цис-
цнклононатриена-1,4,7 (1) сначала в ф/с,ццс,/р/с-цпклононатриен-
1,3,6 (II) (10 мин при 18), а затем в /р/сдр/слр/с-циклононатриен-
1,3,5 (111) (1 час при 18 ) 1181. И, наконец, в результате валентной
изомеризации образуется бициклический диен (IV).
Шрисхейм (191 показал, что раствор Б. к. в ДМСО пригоден в
качестве гомогенной основной среды при изучении констант ско-
ростей, энергий активации и энтропий изомеризации олефинов.
Берч н corp. (201 нашли, что этот реагент можно применять при
изомеризации (2) первичного продукта, образующегося при вос-
становлении по Берчу метилового эфира эстрона (1) всопряжен-
152
ный диен (3).
Несопряженные триеновые сложные эфиры льняного масла
быстро изомеризуются в сопряженные эфиры при действии Б. к.
в ДМСО, ДЛ1ФА или тетраметилмочевпне (все они примерно одина-
ково эффективны 1211. В бутиловом спирте такая изомеризация пра-
ктически не происходит.
Катализатор алкилирования. При детальном изучении алкили-
рования ацетоуксусных эфиров Ренфроу и Ренфроу 1221 нашли, что
Б. к. является, как правило, наплучшнм основанием, особенно для
алкилирования а-замещенных сложных эфиров. В синтезе кортизо-
на по Саретту Б. к. —эффективное основание для двух последова-
тельных реакции алкилирования 14-кетона (1). При метилировании
(СН:)1, Б. к.) |23| образуется только один изомер, которому было
приписано строение экваториального 13а-метильного производ-
ного (2). Алкилирование (2) металлил иодидом (Б. к.) дает продукт,
у которого наиболее стабильная форма (3) та, в которой меньшая из
двух групп у С13 аксиальна (0) по отношению к 1 ip-гидроксильной
группе:
При синтезе ланостерина из холестерина Вудвард, Бартон и сотр.
124] нашли, что наиболее удовлетворительный метод введения 4-ге.м-
диметильной группировки — это метилирование А4- или А°-холе-
стенона-3 подпетым метилом и Б. к. в трет-б} типовом спирте. Этот
же метод был позднее использован для синтеза смоляных кислот;
сид
(сид,сок
(СН3)3СОН
153
в частности Айрлендом и Шпссом 1251, которые обратили внимание
на необходимость в некоторых случаях большого избытка основания
для обеспечения полной енолизацпи кетона.
Образование карбенов. В качестве примера образования дибром-
карбена из КОС (СН:;):| и СНВг:1 см. Метиллптий (Скаттебол [2|).
Хартцлер 1261 добавлял при перемешивании З-хлор-З-метилбу-
тин-1 к суспензии Б. к. (не содержащего спирта) в стироле и полу-
чил циклопропан-алленовую систему (3) с выходом 48%. Позднее
сн,
и
К0С(СН3)з СНС„Н5
(СН3).,С-С S сн----— (СН3)2С = С = С:---->
I — НС1
—* (СН3)2С = с = с - снсвн5
(3)
I КОС(СН,),
(СН3),С = С — СНС1
(1)
[271 он получил винилиденкарбен (2) иза-галогеналлена (4) и исполь-
зовал его во втором методе синтеза циклопропаналлена (3).
Дегидрогалогенирование. Браун |28| нашел, что в тех случаях,
когда отщепление НХ под действием этилата калия происходит
по правилу Зайцева с образованием преимущественно олефина
с внутренней двойной связью (1)->(2), можно добиться того, чтобы
главным продуктом реакции стал терминальный олефин (3); для
этого следует использовать Б. к. пли основания, еще более про-
странственно затрудненные, как это видно из данных, приведенных
в таблице.
Отщепление НХ от СН3СН2С(Вг) (СН3)2 (1)
Основание к-Олефнн (3), %
с»н5ок 29
mpt'm-C4H9OK 72
щрещ-С5НцОК 78
(С2Н5)3СОК 89
Попытки прямого дегидробромирования 16-бром-17-кетостеро-
идов, таких, как (6), приводили к неудовлетворительным резуль-
татам, по-видимому, вследствие значительной полимеризации реак-
ционноспособной циклопентеноновой системы. Хорошие результаты
дает защита кетогруппы, например, превращение ее в этилеикеталь
(7) и кипячение с обратным холодильником бромкеталя (7) с Б. к.
в ксилоле в течение 16 час [29|. После мягкого кислотного гидроли-
за получается метиловый эфир 15,16-дегпдроэстрона. Другой пример
154
использования той же последовательности реакций привел Зонд-
хеймер |301.
(?) (в) т
Мак-Элвен и Кунднгер 1311 использовали Б. к. в шреш-бутнло-
вом спирте для превращения бромацеталя в диэтилацеталь кетена.
16 О’
d г'П f'tJ /fxf' и \ (CHjIjCOK — (СНОСОВ --- ~ /гуг' и т
ВгСНоСН (0GH-,)n---------------------—► СН, = С (ОСД),
Г’7-7 3%
Кэзон 1321 нашел, что Б. к. наиболее подходящий реагент для
дегидробромнрования а-бромзамещенных кислот в А2-алкеновые
кислоты. Бромзамещенную кислоту прибавляют к раствору 3 же
(СН3),СОК (3 эк в)
СН;,(СН.,)8СН.,СНСО.,Н-----—---------’ СН3(СНг)яСН = СНСО.2Н
| 1 %
В г
Б. к. в ш/эем-бутпловом спирте и смесь кипятят 4 час. Едкое кали
в метаноле, этаноле или пропаноле дает смеси А2- и А3-изомеров.
Метод с применением Б. к. был использован для получения транс-
додецен-2-овой кислоты 1331 и а-метиленлауриновой кислоты 1341.
Дегидрогалогенирование с перегруппировкой. Эриксон и Во-
линский [351 показали, что реакцию бромметиленциклоалкана
с Б. к. можно использовать для получения сильно напряженных
циклоалкннов. Так, при нагревании ю-бромкамфена (1) в течение
4 часе Б. к. и 1.З-дифепнлнзобепзофураном (3) в качестве улавливаю-
щего реагента удалось выделить смесь эндо- н жзо-окисей (4) и (5)
с выходом 94%, что указывает на промежуточное образование
эндо-камфнна (2). При реакции Б. к. с бромметиленциклооктаном
получается сложная смесь продуктов (суммарный выход 65%).
Гарднер п сотр. 1361 показали, что галогенппклопропаны мож-
но легко дегидрогалогенировать Б. к. в ДМСО. Пользуясь этой си-
стемой, обычно получают продукт, который образуется из первона-
чально возникающего циклопропена в результате миграции двой-
ной связи в более устойчивое положение вне трехчленного кольца.
В некоторых случаях такая изомеризация сопровождается скелетной
перегруппировкой. Примеры:
Главный продукт Побочный продукт
Расщепление тозилатов и мезилатов. Б. к.— наиболее пригодное
основание для циклизации Ы,О-дитозил-3-аминоспиртов в азетиди-
ны (Воган 137)).
(сносок S (СН,),СОН
TsOCH2CH2CH2NH Ts Кипячение 16 vac
93% *
Чанг 1381 нашел, что мезилаты 3-оксистероидов претерпевают
стереоспецифическое расщепление с Б. к. в смеси бензола и ДМСО
при комнатной температуре до соответствующих З-оксипронзводных
156
без инверсии. В приведенных примерах выход олефинов 4 и 6%.
Соответствующие тозилаты в тех же условиях дают главным обра-
зом олефины.
СН3 СН3 СН., СН3
Г ' (СН,ЦСОК I7 (СН3)ЯСОК '
I ' 86% I J 87% I
MsO/X/A' НО ' С MsO 4 НО'
п г! г1 п
/СН..,
Ms = СНз—/ ^-so, -
XCH;J
Позднее Чанг и Вуд 1391 нашли, что при действии на 12а-мезил-
оксихолан (1) Б. к. в ДМСО при комнатной температуре в качестве
главного продукта получается Д''-холен (2) и в меньшем количест-
ве— холанол-12а (3). При кипячении мезилата (1) с Б. к. в tnpeni-
бутпловом спирте реакция не идет. Полезным применением этого
наблюдения оказалась реакция холсстерилтозилата в бензоле
с Б. к. и ДМСО при комнатной температуре, в результате которой
был получен Д:,’°-холестадпен с выходом 92'%. Снайдер н Сото (401
изучили действие Б. к. в ДМСО на сульфонаты первичных и
вторичных алифатических спиртов и конформационно гибких али-
циклических спиртов. Оказалось, что условия реакции значительно
более мягкие, чем в классических методах. Сульфонаты первич-
ных алифатических спиртов дают преимущественно трет- бутиловые
сложные эфиры. Эфиры циклических и вторичных ациклических
спиртов дают алкены с выходами около 80% и лишь незначитель-
ные количества простых эфиров.
(1) (2) 65% (3) 29%
Арнольд [411 обратил внимание на поразительное различие меж-
ду типичными первичными алкилбромидами и тозилатами при кипя-
чении их в течение длительного времени с Б. к. в /преш-бутиловом
спирте: тозилаты вступают в реакцию замещения и дают трст-
бутиловые простые эфиры, тогда как бромиды подвергаются реакции
отщепления и дают олефины (в небольших количествах могут обра-
зоваться и продукты других реакций). Вуд и Чанг (421 нашли,
что при применении ДМСО в качестве растворителя эти реакции
протекают за несколько минут; они отметили также различия в при-
роде реакций с тозилатами и бромидами стероидов.
157
Другой случай [З-отщепления с образованием олефина наблюдали
Шрисхейм и сотр. 1431 при разложении алифатических сульфокси-
дов и сульфонов в условиях основного катализа. С Б. к. в ДМСО
реакция легко протекает при 55 :
(СН3)2СН
^5=0
(CHj)jCH
(СН,)3СОК-ДМСО
___ц час> 5Л2___>
95%
СН3СН=СН2 + (CH3)2SOH
(СН3)2СОК-ДМСО
117 WC, 55°
80% ’
СН2=СНСН=СН2
Перегруппировки. Деринг и Урбан [44] кипятили раствор сухого
Б. к. и днбензила в бензоле в течение 2 час и выделили неочищенный
/пре/и-бутилбензилат с высоким выходом. При применении метила-
та натрия в тех же условиях выход метплбепзилата — продукта
бензиловой перегруппировки — составил только 18%.
О о ОН
(СИ,).,сок I
С6Н5С—ССвН5—(С6Н5)Х-СО.,С (СН5)з
•’0,0
В одной из стадий синтеза колхицина но Эшенмозеру 1451 —
реакции Дильса — Альдера между производным пирона (1) и хлор-
метнлмалепновым ангидридом — образуется аддукт (2), который в ре-
зультате ацидолиза и этерификации диазометаном был превращен
в хлорметиловый диэфир (3). Затем под действием Б. к. было осу-
ществлено расширение цикла с образованием соединения (4), в ко-
тором имеются два семнчленных кольца.
158
Йэтс и Андерсон |46| нашли, что Б. к. вызывает перегруппировку
4-бензонлоксиинклогексанона (5) до 2-бензоплциклопропанпро-
пионовой кислоты (10):
(9)
(10)
Сужение пятичленного кольца до четырехчленного наблюдается
редко, поскольку при этом возрастает напряжение. Однако Гера
|47| нашел, что D-норстеропды (2) можно получить в результате
перегруппировки типа пинаколиновой 16а-мезилата типа (1) под
действием Б. к. в /преш-бутиловом спирте.
(2) 16о- и 16)9- изомеры
Окисление по Оппенауэру. Поскольку при регулируемом окис-
лении хинина (1) хромовой кислотой хининон (2) получается лишь
с выходом 3%, Вудвард, Вандлер и Бручи [481 изучили метод
Оппенауэра. Попытки применить различные алкоголяты алюминия
в комбинации с разными кетонами в качестве акцепторов водорода
оказались совершенно безуспешными; однако удалось разработать
весьма удовлетворительную методику с использованием в качестве
основания сухого Б. к., бензола как растворителя и бензофенона как
окислителя. Смесь реагентов, взятых в указанных на схеме количест-
вах, в 500 мл бензола кипятили с обратным холодильником в ат-
159
мосфере азота в течение 18 час. Эту методику окисления использо-
вали Рапопорт 149] и Гэйтс 1501 для окисления неактивированных
спиртов ряда морфина, причем были получены очень хорошие
результаты. Согласно новой методике, в качестве акцептора водо-
рода используют флуоренон.
Автоокисление. Б. к.— прекрасный катализатор автоокисления
кетонов п сложных эфиров в а-гпдропсрекиси 1511. Так, если про-
дувать воздух через суспензию твердого алкоголята в растворе ме-
тплизопропилкегопа в диметиловом эфире этиленгликоля и трет-
бутнловом спирте при —8 , то образуется гидроперекись с выходом
80?о (определено титрованием), которую выделяют с выходом 40%.
О
О
СИ»
СНССН3
СН.,7
(СПИСОК (-8=)
40%
снх
сн:/
сссн.,
I
о»н
Было также показано 152], что 20-кетостероид при взбалтывании
с кислородом в присутствии Б. к. в /npem-бутиловом спирте окисляет-
ся до 17а-гидроперекиси с выходом 30—60%. Эти гидроперекиси
можно восстановить до 17а-спиртов цинком и уксусной кислотой
с высоким выходом.
В тех же условиях (Камерпно 1531) 3-кето-5Р-стеронды окисляют-
ся в 4-окси-Д4-3-кетоны (18—50 час при 25 ); 5а-изомеры окисляются
в 2-кетоны (енольные формы) и холестенон окисляется с низким вы-
ходом в диостерин-I. Поскольку в каждом из этих случаев атака
В форме енола
160
направлена на место енолизации, по-видимому, кислород атакует
енолят-апион.
При аналогичном автоокисленип лимонила (1) в сухом трет-
бутнловом спирте, содержащем Б. к., был получен диосфенол (2)
1541. Эта методика оказалась наиболее подходящей для синтеза
диосфеполов в ряду ланостерина.
Пайпс 1551 нашел, что Б. к. при температуре его разложения
(250—300 ) катализирует дегидрирование гпдроароматических угле-
водородов. Например, d-лимонен и Б. к. помешают в автоклав,
воздух вытесняют азотом и смесь нагревают до тех пор, пока давле-
ние не перестанет изменяться (8,5 час). Получают /г-цимол с хорошим
выходом, и образуется значительное количество водорода. Предпо-
лагают, что анион алкоголята в результате пиролиза дает ацетон
и метпл-анион и что последний катализирует перемещение двойной
СН3
I
I I (С1-13),СОК (0, 1 моля), 257±.‘°
сн3
/к
| || II., (0,38 моля)
I
' J 95 о,
I
с
H^C^-'CIL НзС/ЧСН3
0,5 моля
сн3 сн3
сн3-с-о- 2=-^ снз+с=о
I. I
СН3 СН3
связи и дегидрирование. С mpem-амилатом калия получены ана-
логичные результаты, тогда как в случае изопропилата калия выход
снизился до 3—4/6, а в случае метилата калия — до 1,5%.
Пиколины окисляются кислородом в системе (СН3)3СОК—ДМФ;
с //ф^-бутнловым спиртом в качестве растворителя реакция не
6 Заказ № 965
161
идет [561.
Автооквеление с использованием ДМСО в качестве растворителя
протекает следующим образом 1571.
(СПИСОК - ДМСО
(cjij.c сиен.с,.н- о.,—------------------(с6н-,).,со cji-со..н
II.. о
Расщепление кетонов. Сван [581 нашел, что нееиолизпрующийся
кетон, например бензофенон, гладко расщепляется при добавлении
3 же твердого Б. к. к эфирному раствору кетона, содержащему
1 моль воды, п кипячении смеси. При этом оказалось, что небольшая
нейтральная фракция состоит из чистого трпфенилкарбинола.
О о
(Си,),со к
( .Н:,<:с,.н,. • 11,0-—-------с ib.coii .-с,.ив
л'* /I)
Шрекер и Хартвэлл [591 показали, что эту реакцию можно исполь-
зовать для идентификации гексазамещенного бензофенона, получен-
ного из подофиллииа; в качестве растворителя они применяли диок-
сап, содержащий 1 же воды.
Гасман и Залар 1601 нашли, что кетоны гладко расщепляются
при взаимодействии с /иреш-бутплатом калия в ДМСО при комнат-
ной температуре; примером может служить расщепление бензпи-
наколпна и дегпдропоркамфоры. Во втором случае при кипячении
с Б. к. в шрт-бутпловом спирте выход Д3-цик.топеитепилуксусной
кислоты составил только 19%.
(С611.>,ССОС6Н5 ; КОС(СНз),
ДГ19О>^5<’> (с4н5)3сп + едцео.н
97% 10 0%
КОС(СИ3)з
ДМСО, 25°
30% *
Нитрование кетонов. Фойер и сотр. [611 тщательно изучили реа-
генты и условия, необходимые для эффективной реакции циклопента-
нопа с амилнптратом и основанием с целью получения 2,5-динптро-
162
цпклопентанона, который удобно выделить в виде негигроскопичной
дикалпевой соли (2).Наплучшие результаты получены при перемеши-
вании раствора возогнанного Б. к. в 95 м г ТГФ при —30 и одновре-
менном прибавлении по каплям сначала раствора цпклопентанона
о
Т 2 С5НпОЫОг (СН3),СОК(0.165моля^
7 2%
(1) 0, 05 моля
В г В г
KOBr ? O2NCCII2CH2CNO2
72%,исходя из(1) ‘ । I
(3)
в ТГФ, а затем раствора амплиитрата в ТГФ. Смеси дают нагреться
до 25', не прекращая перемешивания, зеленый осадок дикалпевой
соли 2,5-Д11нитрои11Клопеитанона (2) отфильтровывают, промывают
и высушивают. С целью очистки соль растворяют в воле и переосаж-
дают метанолом. Лучший выход (72%) получен с применением
65%-ного избытка Б. к.; при 10',’о-ном избытке основания выход
снижается до 48%. Выходы в пределах 4,5—37% получены со сле-
дующими основаниями: NaNH,, NaC (С6Н.,).(, NaCH.iNC(iH5,
NaOC (СН:1):), KCH:1NC(iH.-,, KNH.2. При введении двух нитрогрупп
в циклогексанон и высшие члены ряда также были получены дика-
лиевые соли, аналогичные (2), но они оказались очень гигроскопич-
ными, что затрудняет точное определение выхода. Однако все эти
соли расщепляются аналогично количественному расщеплению
(2) при действии водного раствора гипобромита калия при 0' с об-
разованием 1,1,4,4-тетрабром-1,4-дпиитробутана (3), выделенного
в виде осадка и очищенного перекристаллизацией из гексана. Вы-
ходы, определенные по продуктам расщепления, аналогичным (3),
оказались следующими: циклогексанон 53%, циклогептапон 54%,
ппклооктанон 35%.
Получение изонитрилов. Общий метод синтеза арилизоцианидов
(Угп 1621) иллюстрирует получение о-тол ил изонитрила. Суспензию
Б. к. в /?//7С/?;-бутнловом спирте получают в атмосфере азота при
перемешивании калия и спирта п нагревании сначала до расплавле-
ния металла, а затем до температ\ры кипения жидкости. Постепен-
но Б. к. выпадает в осадок и образует густую суспензию. Через
СН3 СП3
I .NHCHO । Х-..-С
/Л / //у
|| <!>(«-• 4 (СП.,,.,(.! >!< , ЗК<1 г-
I Ч ч J — / J и ( | I
'Ч/ %/
1 моль 0,6 моля 2,6 моля
у КРОз : -4 (СНз)зСОН
6
163
несколько минут после добавления N-о-толплформампда к горячей
суспензии образуется прозрачный раствор. Его охлаждают до 10—
20° н, поддерживая эту температуру, прибавляют в течение 30—
40 мин хлорокись фосфора. После выдерживания в течение 1 час
при 30—35° смесь обрабатывают и продукт реакции перегоняют.
Соединения с трехчленным циклом. Синтез первого достовер-
ного а-лактама осуществил Баумгартен 1631 путем медленного до-
бавления раствора Б. к. в толуоле к раствору амида (1) и трет-
бутнлгшюхлорита в толуоле при 5". Успех зависел от того, насколь-
ко удавалось поддерживать минимальную концентрацию трст-
бутнлового спирта, чтобы избежать расщепления реакционноспособ-
ного соединения (3) с образованием (4). Выход l-mpm-бутил-З-фе-
О
О
II (СП,).,СОК
C6H5CH.,C-NC(CH3)3------;------->
6 ° ) з/з (СН,),СОС1
I - IIC1
С6Н5СНгС—NC (СН3)3 : —-~-
н
С6Н5СН — с = о
О
(2)
N
/Ц/Д/ с„н,сн -- (ох'нс(сн3)3
С (СН3)3
(3)
(4)
нилазирндннона (3) (т. пл. 32—33°) 31 %. Шихан и Ленгьель [641
получили а-лактам (6) обработкой Б. к. раствора соединения (5) в
эфире в атмосфере азота при 25'. Высоко реакционноспособный ази-
ридинон (6) имеет т. пл. 22—24°. Грин и Стоуэлл [651 получили ди-
О
СН3 || (Снщс.ок СН3.
>C-CNHC(CH3)3------------» ----С = О
СН/ I СИ/ \ /
Вг 'У
С (СН3)3
(5) (б)
/7!ре/??-бутилд11аз11ридинон (8) при действии на М,ХГ-дн-/»рет-бутнл-
N-хлормочевину (7) калия в пентане или Б. к. в тре/п-бутиловом
спирте. Диазиридинон (8) (т. пл. 0—1“) обладает необычной терми-
ческой стабильностью, а также устойчив по отношению к нуклео-
фильным агентам. При гидрировании расщепляется связь N—N
и образуется соединение (9); хлористый водород вызывает расщепле-
ние связи С—N с образованием хлорангидрида (10), который при
164
взаимодействии с Б. к. дает смесь соединений (8) и (11).
О о
II II
у' К нли Я
/\ (СНОСОК /\
(CH3)3CN NC(CH3)3 (CH3)3CN — NC(CH3)3
I 1 4 и - e и %
CI H
(7) (8)
О H,. i-------------------------------------
II Pd HCH-----------------------------------
C I COCI
;сн3)зс/\с(сн3)з (СНз)зСХ- \'С(СН3)3(СН|)яС0К
I I I
H H H
(9) (10)
COOC(CH3)3
* (CH3)3CN — \C(CH3)3
I
H
(II)
Гидролиз пространственно затрудненных сложных эфиров. Чанг
и Вуд [661 нашли, что Б. к. в ДМСО эффективен для расщепления
пространственно затрудненных сложных эфиров в условиях, которые
подбирают в зависимости от степени пространственной затруднен-
ности. Так, метиловый эфир дегидроабиетиновой кислоты (1), в ко-
сн
I
с-согсн,
сн
сн3
(3)
/СН,
тором третичная сложноэфирная группировка занимает эквато-
риальное положение и ей не мешает 10-аксиальная метильная груп-
па, гидролизуется менее чем за 1 час при комнатной температуре;
для гидролиза метилового эфира О-метилподокарповой кислоты (2)
с сильно пространственно затрудненной аксиальной сложноэфирной
группировкой требуется 2 час при 56=; метиловый эфир триизопро-
пилуксусной кислоты гидролизуется за 4 час при 100’.
Реакция Виттига. Б. к. в ДМСО эффективнее, чем бутиллитий
или этилат натрия в этиловом спирте, при получении илида из аце-
тон-1,3-бис-(трифенилфосфоний)-хлорида [671. Сообщалось, что Б. к.
заметно ускоряет реакцию Виттига [681. Комплекс (СН3)3СОК —
165
—(CH.i)3COH (1 : 1) более эффективен, чем алкоголят, не содержа-
щий растворителя.
о
I < ДМСО
(СОН5)3Р —СН2ССН.,-Р(С6Н6)з + 2(СН3)3СОК-;-2п-С1СвН4СНО——<
— — 'Ь"о
С1 С1
о
II
—* н-С1СсН4СН = СНССН -«СНС„Н4С1-л 2(С5Н5)3РО
Реакция Вольфа — Кижнера. Крам и сотр. 1691 показали, что
восстановление можно проводить при комнатной температуре,
если в качестве растворителя брать ДМСО. Например, 1,96 г гид-
разона бензофенона прибавляют небольшими порциями в течение
8 час при перемешивании к 2 г Б. к. в 5 мл ДМСО. Выделяется азот
и раствор окрашивается в темно-красный цвет. Выход дифеиплме-
тана 90%. Грундон, Хенбест и Скотт 1701 показали, что гидразоны
кетонов превращаются в углеводороды при взаимодействии с Б. к.
в кипящем толуоле.
1. Джон с о н У., IIJ н с й дер У., «Синтезы органических препаратов». ПЛ,
М., 1953, сб. 4, стр. 279; Джонсон 5'. С., Доб Г. X., «Органические
реакции», ИЛ, М., 1953, сб. 6, стр. 1.
2. S k a t t с Ь и 1 L., Solomon S., procedure submitted to Or". Syn.
3. Hunt R. H., Chinn L. J., J о h nson W. S., Orc'. Svn., Coll. Vol.,
4 , 459 (19(53).
4. S t о г к G., W о r r a 1 1 W. S., P a p p a s J. J.. J. Am. Chem. Soc., 82, 4315
(19(50).
5. Bald w i n S., J. Or". Chem., 26, 3280 (1901).
6. LeonardX. J., SchimelpfenigC. W., Jr.. J. Orc. Chem., 23, 1708
(1958).
7. Leonard X. J., et al., J. Am. Chem. Soc., 74, 1704, 6251 (1952); 76, 3193
(1954); 77, 6234 (1955).
8. J о h n s о n W. S., Bannister В., P a p p о R., J. Am. Chem. Soc.,
78, 6331 (1956).
9. Stork G., С 1 a г к e F. H., Jr.. J. Am. Chem. Soc.. 83. 3114 (19(51).
10. H о u s e H. О., В a b a d H., J. Or". Chem., 28, 90 (1963).
11. Ej> I i nt on G., R a p h a e 1 R. A., W i 1 1 i s R. G., Z a b к i e \v i c z J. A.,
J. Chem. Soc., 1964, 2597.
12. В e n - E f r a i in D. A., S о n d h e i m e r F., Tetrahedron Letters, 313 (19(53).
13. E r r e d e L. A.. J. Am. Chem. Soc., 83, 949 (1961).
14. I wa i L, I de J., Chem. Pharm. Bull., 12, 1094 (1964).
15. R i n " о 1 d H. J., AlalhotraS. K., Tetrahedron Letters, 669 (1962); see
also Shapiro E. L. et al., Steroids, 3, 183 (1964).
16. C r a m D. J., R ickborn B., KnoxG. R., J. Am. Chem. Soc., 82, 6412
(1960).
17. D e v a p r a b h a к a r a D., C a r d e n a s C. G., G a r d п e r P. D., J. Am.
Chem. Soc., 85, 1553 (1963).
18. W a t t h e у J. W. H., W i n s t e i n S.. J. Am. Chem. Soc., 85, 3715 (19(53);
G 1 a s s D. S., W a t t h e у J. W. H., W i n s t e i n S. Tetrahedron Letters,
377 (1965).
19. S c h r i e s h e i m A., Muller R. J., Rowe C. A., Jr., J. Am. Chem.
Soc., 84, 3164 (1962), and eailier papers cited.
20. В i г c h A. J., G r a v e s J. M. H., SiddallJ.B., J. Chem. Soc., 1963,
4234.
166
21. U g е 1 s t a d J., J e n ssen В., Murk P. C., Chem. Scand., 16, 323 (1962).
22. Renfro w W. B., Renfrow A., J. Am. Chem. Soc., 68, 1801 (1946).
23. Lukes R. M., Poos G. L. В e у 1 e r R. E., Johns W. F., S a-
ret t L. H., J. Am. Chem. Soc., 75, 1707 (1953).
24. W о о <1 w a r <1 R. В., P a t c h e t t A. A., Barton D. H. R.,
I v e s D. A. .1., Kell у R. B., J. Chem. Soc., 1957, 1131.
25. Ireliin (1 R. E., S c h i e s s P. W.. J. Org. Chem., 28, 6 (1963).
26. Hartzler H. D., J. Am. Chem. Soc., 83, 4990 (1961).
27. Hart z I e r H. D., J. Org. Chem., 29, 1311 (1964).
28. Bro w n J.C., M о r i t a n i 1., J. Am. Chem. Soc., 75, 4112 (1953).
29. J о h n s о n W. S., J о h n s W. F., J. Am. Chem. Soc., 79, 2005 (1957).
30. S о n (I h e 1 m c r F., В u r s t e i n S., M e c h о u 1 a m R., ,1. Am. Chem.
Soc., 82, 3209 (I960).
31. M а к - Э л ь н e и С., К у н д и г е р Д., «Синтезы органических препара-
тов», ИЛ, AL, 1952, сб. 3, стр. 244; Применение см. G г е е w е R., S t г u v е А.,
Chem. Вег., 96, 2819 (1963).
32. С a s о п ,1., А 1 I i n g е г N. L., S и m г е I 1 G., a Org. Chem., 18, 850 (1953).
33. А л л е н Ч., К а л м М., «Синтезы органических препаратов», ИЯ, AL, 1959,
сб. 9, стр. 31.
31. А л л е и Ч., К а л м М., «Синтезы органических препаратов», ИЛ, AL. 1960,
сб. 10, стр. 37.
35. Erickson К. L., W о 1 1 п s к у J., J. Am. Chem. Soc., 87, 1142 (1965).
36. Osborn С. L., S h i е 1 d s Т. C., S h о u Ider s B. A., Krause J. F.,
С о r t e z H. V., G a r d n e r P. D., .1. Am. (Лет. Soc., 87, 3158 (1965).
37. Va ughan W. R., К lonowsk 1 R. S., M с E I h i nne у R. S., M i 1 1-
w a r d В. B., J. Org. Chem., 26, 138 (1961).
38. C h a n g F. C., Tetrahedron Letters, 305 (1964).
39. Chang F. C., Wood N. F., Steroids, 4, 55 (1964); see also В h a-
rucha K. R., Schrenk H. M., Expcrientia, 21. 278 (1965).
40. S n у d e r C. IL, S о t о A. R., J. Org. Chem., 29, 742 (1964).
41. V e e r a v a g u P., Л r n о 1 d R. T., Eigen in a n n E. W., J. Am. Chem.
Soc., 86, 3072 (1964).
42. Wood N. F., Chang F. C., J. Org. Chem., 30, 2054 (1965).
43. H о f m a n n J. E., W a I 1 a с e T. J., A r g a b r i g h t P. A., Schri-
e s h e i m A., Chem. Ind., 1963, 1243.
44. D о e r i n g W. E., von, U r b a n R. S., J. Am. Chem. Soc., 78, 5938 (1956).
45. S c h r e i b e r J., L e i in g r u b e r W., Pesaro M., Sch u de I P.,
T h r e 1 f a 1 I T., E s c h e n m о s e r A., Helv. Chim. Acta, 44, 540 (1961).
46. Y a t e s P.. Anderson C. D., J. Am. Chem. Soc., 85, 2937 (1963).
47. G h e r a E., Tetrahedron Letters, 4181 (1965).
48. W о о d w a r d R. B., W e n d 1 e r N. L., В r u t s c h у F. J., J. Am. Chem.
Soc., 67, 1425 (1945).
49. Rapoport H., Naumann R., Bissell E. R., Bonner R. AL, J.
Org. Chem., 15, 1103 (1950).
50. Gates M., T s c h u d i G., J. Am. Chem. Soc., 78, 1380 (1956).
51. Gers m a n n H. R., N i e u w e n h u i s H. J. W., В i с к e 1 A. F., Proc.
Chem. Soc., 279 (1962).
52. В a i 1 e у E. J., E 1 к s J., В a r t о n D. H. R., Proc. Chem. Soc., 214 (1960);
Bailey E. J., Barton D. H. R., E 1 к s J., T e m p 1 e t о n J. F., J.
Chem. Soc., 1962, 1578.
53. Camerino B., Patelli B., Sc laky R., Tetrahedron Letters, 554
(1961).
54. В a r t о n D. H. R., P r a d h a n S. K., S t e r n h e 1 1 S., Temple-
t о n J. F., J. Chem. Soc., 1961, 255.
55. Pines H., S c h a a p L., J. Am. Chem. Soc., 79, 2956 (1957).
56. Bartok W., R о s e n f e 1 d D. D., S c h r i e s h e i m FL, J. Org. Chem.,
28, 410 (1963).
57. В a r t о n D. H. R., J о n e s D. W., J. Chem. Soc., 1965, 3563.
167
58. S w a n G. A., J. Chem. Soc., 1948, 1408.
59. S c h г e с к e г Л. W., H a r t we 1 I J. L., J. Am. Chem. Soc., 75, 5924 (1953).
60. G.i ssm an P. G., Z al ar F. V., Tetrahedron Letters, 3031, 3251 (1964).
61. Feuer H., Shepherd J. W., S a v i d e s C., J. Am. Chem. Soc., 78,
4364 (1956).
62. У г и И., Мей p Р., «Синтезы органических препаратов», изд-во «Мир»,
М„ 1964, сб. 12, стр. 140.
63. Baumgarten Н. Е., J. Ain. Chem. Soc., 84, 49/5 (1962).
64. ShechanJ.C., Lengyel I., J. Am. Chem. Soc., 1356 (1964).
65. (ireeneF. D., Stowell J.C., J. Am. Chem. Soc., 86, 3569 (1964).
66. Chang F. C., Wood N. F., Tetrahedron Letters, 2969 (1964).
67. Denney D. B., SongJ., J. Org. Chem., 29, 495 (1964).
68. Schlosser M., C h r i s t in a n n K. F., Angew. Chem., Internal. Ed.,
3, 636 (1964).
69. Cram D. J., S a h у u n M. R. V., К n о x G. R., J. Am. Chem. Soc., 84,
1734 (1962).
70. G r u n d о п M. F., H e n b e s t H. B., S с о t t M. D., J. Chem. Soc., 1963,
1855; G r u n d о n M. F., S с о t t M. D., ibid., 1964, 5674.
/npe/n-БУТИЛГИПОБРОМИТ, (СН3)3СОВг. Мол. вес 153,03,
т. кип. 44—45°/85 мм.
Получение [1|. Водный раствор бромноватистой кислоты, получен-
ной из сульфита серебра и бромной воды, встряхивают с трет-бута-
нолом, продукт реакции экстрагируют трихлорфторметаном (фре-
он 11) п перегоняют; выход42%. Б.— красно-оранжевая жидкость,
по запаху напоминающая бром, устойчива при 0э в темноте в тече-
ние длительного времени, быстро разлагается при нагревании до
85° и при освещении, а также под действием бикарбоната натрия.
Б. с алканами и бромтрихлорметаном дает бромиды.
Реакцией Б. с олефинами и этанолом получают р-этоксибромиды
121:
\ = С// + С2Н5ОН + (СН3)3СОВг —> \-CZ + (СН3)3СОН
/ \ /1 I \
Вг ОС2Н5
СН2 = СНОС2Н5у- С2Н5ОН + (СН3)3СОВг —> ВгСН2СН(ОС,Н5)2
1. W а 1 1 i n g С., Padwa A., J. Org. Chem., 27, 2976 (1962).
2. G о n е s t e J.-M., Rergomard A., Bull. soc. chim. France, 1963, 470.
трет-БУТИЛГИПОИОДИТ, (CH3)3COI. Мол. вес 200,02.
Получение. Б. получают либо взаимодействием mpem-бутил-
гипохлорита с иодом в бензоле, либо обработкой mpem-бутилата
калия бензольным раствором иода [1]. Все операции проводят в тем-
ноте в атмосфере азота. Б. достаточно стабилен; его активность
немного уменьшается при освещении в течение 30 мин (25°).
6,19-Окиси стероидов. Ахтар и Бартон [1] нашли, что при ин-
тенсивном ультрафиолетовом облучении смеси Б. с 5а-бром-60-
оксистероидом получается 6,19-окись с выходом 60—80%, тогда
как под действием Б. в темноте зр-ацетоксихолестанол-60 окисляется
168
с высоким выходом до 6-кетона.
Декарбоксилирование [21. В бензольном растворе карбоновой
кислоты и /ире/н-бутилгипоиодпта, видимо, имеются равновесные
количества mpcm-бутанола и ацилгипоиодита, который на свету
(СН.,)3СО1 н RCOH ДД (СН3)3СОН + RCOI
II II
О О /IV
____RI-LCO«
разлагается до алкилиодида и СО2. Поэтому действие света на раст-
вор кислоты и Б.— удобный метод декарбоксилирования, сравнимый
с реакцией Хунсдикера. Например:
н СО2Н
+ (СН3)3СО1
/IV
С.н,
И I
+ СО2
90%
70%
Этим методом замещается карбоксильная группа при первичном,
вторичном и третичном атоме углерода. Адипиновая кислота, плохо
растворимая в бензоле, успешно декарбоксилируется в смеси бензо-
ла и сульфолана с образованием иодида (выход 62%).
N-Иодамиды и у-лактоны. Бартон и сотр. [31 нашли, что Б.—
эффективный реагент для получения N-иодамидов (бензамид, сукци-
нимид, амиды н-масляной и октадекановой кислот). При фотолизе
иодамиды, соответствующие структуре (2), легко превращаются
в у-лактоны (3).
(1) (А О)
1. Akhtar М., BartonD. И. R., J. Am. Chem. Soc., 86, 1528 (1964).
2. В а г t о n D. Н. R., F а г о Н. Р„ Serebryakov Е. Р., W о о 1-
s е у N. F., J. Chem. Soc., 1965, 2438.
3. В а г t о n D. Н. R., В eck w i t h A. L. J., Goosen Л,, J. Chem. Soc.,
1965, 181.
трет-БУТИЛГИПОХЛОРИТ, (CH3)3COC1. Мол. вес 108,57,
т. кип. 78°, уд. вес 0,91.
169
Б. получают 111 пропусканием хлора в щелочной раствор трет-
бутанола. Внимание: сообщалось о взрыве при получении Б. |2|.
Хлор следует пропускать с такой скоростью, чтобы температура
реакционной смеси не превышала 20 .
Галогенирование углеводородов. Уоллинг |3| нашел, что Б.
можно использовать для хлорирования толуола по радикально-
цепному' механизму при инициировании реакции светом пли ради-
калами. В присутствии, например, азоднпзобутнропптрила реакцией
Б. с толуолом при 40 получают бензнлхлорпд (84%), //фета-бутанол
(97?о) и по 1—3% хлортолуолов, хлористого метила и ацетона.
с.\ с.х С\'
I I I (С.Н.).СОС1 с,и,сн,
(СН..;).,С\ .. .\С(СН3).,-> (СН,),С • ------:-----+ (СН.,) .СО ----:—
' -Х_. - <с;Н.,).,С(С.\)С| ' -(СН.,),СОН
(СН.,)1со<:1
— С„Н-,СН.,--------------> С6Н-,СН.,С1
в •’ - -(СЛЬЬСО- 6 ’
Олефины под действием Б. дают с хорошим выходом аллилхло-
рпды |4|. Реакционная способность аллильной связи С—Н убывает
в ряду: третичная>вторичная>первпчная. При аллильном замеще-
нии не изменяется положение и цис- пли 777/?«яс-конфпгурацпя двой-
ной связи. Фенолы хлорируются обычно в «-положение |5|.
Окисление спиртов. Под действием Б. в СС14 или эфире в при-
сутствии пиридина инклогексанол окисляется в циклогексанон
и я-бутаиол— в масляный альдегид (Гроб и Шмид 161). З-Оксп-
стероиды окисляют в СО,, добавляя по каплям раствор Б. в ССГ
при комнатной температуре (Гинзбург и сотр. 171). Кетон можно
хлорировать непосредственно в процессе получения. Для этого
реакцию с Б. проводят в уксусной кислоте при 65 с последующим
нагреванием на кипящей водяной бане. Левин и сотр. |8| получали
З-кетостеропды также с высоким выходом окислением соответствую-
щих спиртов под действием Б. в сухом /77реш-бутаиоле.
Б. особенно успешно применяется при окислении смеси цис-
п 777/7йяс-2-фен11Лпиклогексаиолов 191. Раствор спирта в CCI, в при-
сутствии 1 же пиридина буквально титруют, добавляя Б. при темпс-
+ (снэ)3сон
ратуре от—5 до О'. Осадок хлоргидрата пиридина отделяют, рас-
твор промывают водой, сушат и перегоняют.
Хлорирование кетонов. Окислнтелыю-хлорпрующее действие
Б. иллюстрируется примерами превращения холестерина в 60-
хлор-А’-холестеиои-З (1) и реакцией окисления-хлорирования поли-
функционального стероида (2). В этом случае хлорирование завер-
яй
шается только в присутствии воды. Xлор-3-кетостероиды (3 и 4)
получают как из соответствующих кетонов, так и из спиртов реак-
цией с Б. в уксусной кислоте при нагревании на кипящей водяной
бане.
N-Хлорирование. N-Ацетилампды превращаются в Х’-хлор-
Х-анетиламнды с выходом 90% и более при обработке избытком
Б. в метаноле 112|. Синтез диазахпнонов из циклических гидразидов
включает, по-видимому, промежуточное образование N-хлорамида
|13| (см. также N-Фенили.мид азодикарбоновой кислоты).
Для N-хлорирования аминов Б. намного эффективнее хлорно-
ватистой кислоты; это показано на примере превращения 30-ацето-
171
кси-20-амино-Д 5-прегнена в прегненолон 114]:
Ы,Ы-Дихлорирование — первая стадия синтеза аминоацетофенона
и других а-ампнокетонов по Баумгартену 1151:
nh2 nci2
__ГЫ___сн (СН3)3СОС1 //\_____CJJ__СН
| || v.n3 в С1|Н() прн 0 5о । || ^п3 CH3ONa-CH„OH
Ч'/ н Х/
I
N О
С — СН2 HC1 CCH2NH2-HC1
— I II I 55-72% <«0 | ||
При взаимодействии с Б. в бензоле циклогексиламин образует
М.ЬБдпхлорпроизводное, которое при дегидрохлорировании аце-
0,1 моля
+ (СН3)3С—ОС1+С6Н6
0,22 моля 1з 50 мл 50 мл
СвНб
NC12
С2Н5ОН—КОАс
48-60% (общий)
татом калия дает N-хлоримин циклогексанона 116].
Сульфиды—► сульфоксиды. Под действием Б. в метаноле при
—70' 4-замещенные тетрагидротиопираны окисляются почти исклю-
чительно до qwc-сульфоксидов [17].
к
-70°
Q
(СН3)3СОС1 в СНдОН
(п-асдг,
R = ( С(СН3)3
I сн3
98‘
100 % цис
172
Синтезы алкалоидов. В чегырехстадийиом синтезе (Бечи и Ман-
нинг ]18|) природного алкалоида воакангииа (9) из ибогаина (5)
последний обрабатывают Б. и получают хлорнндоленин (6), кото-
8, R“C=N
9, R = COOCH3
рый реакцией с цианистым калием превращают в 18-цианибогаин
(8). Жесткий! щелочной гидролиз (8) и последующая этерификация
диазометаном дают воакангин (9).
Диазоалканы. Под действием Б. в присутствии этанола диазо-
алкан (1) превращается в ацеталь (3), по-видимому, через промежу-
точное соединение (2) 119].
CI
CeH5CH-NsN’ + (CH3)3COCl________________> C6H5CHZ
(1) -(СНДзСОН —N. X 49%
L ос2н5.
(2)
—> С6Н5СН(ОС2Н5)2 + НС1
(3)
1. Титер X., Б е л л Э., «Синтезы органических препаратов», ИЛ, М., 1953,
сб. 4, стр. 114.
2. Bradshaw С. Р. С., Nechvatal A., Proc. Chem. Soc., 1963, 213.
3. Walling C., JacknowB. В., J. Am. Chem. Soc., 82, 6108, 6113 (1960).
4. Walling C., Thaler W., J . Am. Chem. Soc., 83, 3877 (1961); Grob C. A.,
Kny H., G a g n e u x A., Helv. Chim. Acta, 40, 130 (1957).
5. G i n sb ur g D„ J. Am. Chem. Soc., 73, 2723 (1951).
6. G г о b C. A., Sch m i d H. J., Helv. Chim. Acta, 36, 1763 (1953).
7. Beereboom J. J., Djerassi C., Ginsburg D., Fieser L. F.,
J. Am. Chem. Soc., 75, 3500 (1953).
8. FonkenG. S., Thompson J. L., Levin R. H., J. Am. Chem. Soc.,
77, 172 (1955).
9. В u t 1 e r D. E., private communication.
10. Ginsburg D., J . Am. Chem. Soc., 75, 5489 (1953).
11. Hanze A. R., F о n k e n G. S., Mclntos h A. V., Jr., S e a г с у A. M.,
LevinR. H„ J. Am. Chem. Soc., 76, 3179 (1954).
173
12. Petterson R. C.. Wambsgans A., .1. Am. Chem. Soc., 86,
1648 (1964).
13. Real у T. J., J. Am. Chem. Soc., 84, 966 (1962).
14. Bachman n W. E., Cava M. P., Dre i d in» Л. S., J. Am. Chem.
Soc., 76, 5554 (1951); see alsoC a v a M. P., V о g t В. R., Tetrahedron Letters,
2813 (1964).
15. Baunif(arte и II. E., Bowe r F. A., J. Am. Chem. Soc., 76, 4561 (1954);
В a umijart e n H. E., Petersen .1. M., J. Am. Chem. Soc., 82, 459
(I960); 0гц. Syn., 41, 82 (1961).
16. A I t G. H., К n owle s W. S., J. 0гц. Chem., 25, 2047 (1960); Org. Svn., 45,
16 (1965).
17. Jo h n s о n C. R., M с C a n t s D., Jr., J. Am. Chem. Soc., 8 7, 1109 (1965).
18. В ii c h i G., M a n ning R. E., J. Am. Chem. Soc., 88, 2532 (1966).
19. В a g a n z H., M а у H.-.I., Angew. Chem., Internal. Ed., 5, 420 (1966).
w-БУТИЛЛИТИЙ, CH;tCH2CH»CH2Li. Мол. вес 64,05.
Получение. Б. получают взаимодействием н-бутилбромида с ли-
тиевой проволокой в эфире в течение 3 час при охлаждении вначале
до —10', а затем 0—10' 111. При использовании «-бутилхлорнда
требуется кипячение в течение 10 час 12].
Применение в реакции Виттига. Реактив Виттита (2), необхо-
димый, например, для синтеза метиленциклогексана (3), получают
дегидрогалогенированием метилтрифенилфосфонийбромида (1) под
действием Б. [3] *:
(CJlA^P-CH^Br)-^-^ (CJ1.-MP —сн,
(1) (2)
(3)
-сн.
Металлирующий агент. Б.— наиболее подходящий агент для
замещения ароматического водорода в жирно-ароматических эфи-
рах обычно в о/я/ю-положении к эфирной группе. Обычно в качестве
растворителя применяют ТГФ, хотя часто используют и эфир 141.
Например, 1,7-диметоксинафталин дает с Б. б-Бьпроизводное, ко-
торое при действии СО2 дает кислоту, а с диметилсульфатом превра-
щается в метильное производное |2|.
* См. также Маеркер
1967, со. 14, стр. 287.— Прим.
А., «Органические реакции», изд-во «.Мир», М.,
персе.
174
Получение карбенов. Б. и метиллитий применяются для полу-
чения карбенов 151. Например, реакцией Б. с бромтрихлорметаном
в эфире при -60' в присутствии циклогексена получают 7,7-дихлор-
норкаран (1). Из хлористого метилена при этом образуется хлор-
карбен (2), используемый в синтезе монохлорциклопропанов.
BuLi г- Циклогексен
D СВгС,а fLiCC1J-------------> :СС1г --
7,7-Дмхлорноркаран
(СН3)2
(СН,),
21 CHjCU
BuLi r~ «1
' У UCHC\J-----> :СНС1
—ВиН L
(СНз)?С^С(СН;)>
67%
/?;р«нс-Бутен-2 дает исключительно продукт щ/с-ирисосдинения (вы-
ход 40%). В реакциях моно- и дихлоркарбена выходы увеличивают-
ся с возрастанием нуклеофильности олефина. При получении феио-
ксикарбена из бензальхлорида Б. дает лучшие результаты по срав-
нению с /лреш-бутилатом калия [61.
Синтезы алленов. Превращение гещ-дигалогенциклопропанов
в аллены под действием магния 17| или Б. 181 является, по-видимому,
самым лучшим общим методом синтеза алленов. Например, децен-1
(1) превращают в ге.и-дпбромид (2), дегалогенирование которого
под действием Б. дает ундекадиен-1,2. В этом случае при исполь-
СНВг. - (СН ) ,СОК BuLi
СН3(СН2)7СН = СН2----" -' -» СН3(СН2)7СН—СН2 — -
Л) 3,’-“ \г/
/ \
Вг Вг
12)
—. СН3(СН,)7СН (. сн.
(3)
зованпи метпллптия выход на второй стадии повышается до 68°6.
Ди хлориды не изменяются при обработке метпллитием, но медленно
реагируют с бутпллитием.
1. Jones R. G., Gilman Н., Огц. Reactions, 6, 352 (1951).
2. Bar n е s R. А., В и s h W. М., .1. Am. Chem. Soc., 81, 4705 (1959).
3. Витти г Г., Ш с л л к о п ф IO., «Синтезы органических препаратов»,
изд-во «/Мир», 1964, сб. 12, стр. 89.
4. G i I m а n Н., Gray S., J. Огц. Chem., 23, 1476 (1958).
5. М i 1 1 е г W. Т., Jr., К i ni С. S. ХУ, J. Am. Chem. Soc., 81, 5008 (1959);
С 1 о s s G. L., С 1 о s s I.. E., ibid., 82, 5723 (1960).
6. S c h 6 1 1 k о p f U., Lerch A., P i t t e г о f f W., Tetrahedron Letters,
241 (1962).
7. Doerini! W. E., von. La F I a m in e P. A\., Tetrahedron, 2, 75 (1958).
8. Moore W. R., Ward H. R„ J. 0гц. Chem., 27, 4179 (1962).
/ирет-БУТИЛЛ ИТИЙ, (CH.)):1CLi. Мол. вес 64,05.
Получение этого реагента и его реакции с карбонильными сое-
динениями осуществляются достаточно гладко только при низкой
температуре (Бартлетт и Леффертс) [11. /лрстл-Бутилхлорпд добавля-
ло
ют при эффективном перемешивании к суспензии тонконзмельчен-
ного лития в эфире при температурах от—35 до—40'. По результатам
титрования выход Б. составляет 75%. Раствор Б. охлаждают до
температур от —60 до—70 , обрабатывают эфирным раствором
гексаметилацетона и выделяют с высоким выходом кристаллический
о
Эфир, |!
от—35 до—10" (CHt)yCCC(CH4),
(СН3)3ССГД Li----------* (CH,).,CLi------------->
' 1 75% ' от — 60 ;.о — 70"; 8 1%
С(СН3)..,
I
—> (СН3)3С— С—С(СН3)3
I
он
три-т/7ст-бутил карби нол.
Стайлсу и Майеру [2] по этой методике не удавалось получить
Б. из лития, не содержащего натрия, однако добавление 1—2%
натрия в расплав при получении тонкоизмельчепного лития при-
водило к успешным результатам. Подобные результаты были опи-
саны при получении других литиевых реагентов |3|. По данным
Кертина и Келя |4|, для активации лития можно также использо-
вать тонкоизмельченпую медную бронзу.
1. Bartlett Р. D., Lefferts Е. В., .1. Am. Che.п. Soc., 77, 2804 (1955).
2. Stiles М., Mayer R. P., .1. Am. Cliein. Soc., 81, 1501 note 38b (1959).
3. W r i g h t J. B., Outsell E.S., J. Am. Chem. Soc., 81, 5193, note 10 (1959);
KamienskiC. \V., EsmayD. L., J. Org. Chem., 25, 1807 (1960).
4. C u r t i n D. Y., К о e h 1 W. J ., Jr., J . Am. Chem. Soc., 84, 1967 (1962).
н-БУТИЛНИТРИТ, h-C4H(jONO. Мол. вес 103,12, т. кип. 77°
(24—273/43 мм).
Б. получают из н-BuOH, H.,SO4, Н.,О, NaNO., при О3 [11.
Азидный метод синтеза пептидов. Этот метод, широко исполь-
зуемый благодаря отсутствию рацемизации 121, осложняется тем,
что реакция гидразидов с азотистой кислотой наряду с азидами
дает соответствующие амиды. Их образование крайне незначитель-
но при диазотировании Б. или нитрознлхлоридом по Рудингеру
[3]. Для обеспечения максимального выхода необходимо немного
О OR'
о Н\О., । -I- — 14 МСЦСЛ СН
CbNHCHC—NHNH,---- CbNHCHC-X = X = N 21 _ .УСД
| ' | ~HN3
R R
О
—* CbNHCHC—NHCHCO,CH3
176
более 2 экв Б.; в качестве растворителя применяют ТГФ. При взаи-
модействии с нитрозилхлоридом в диоксане избытка реагента
не требуется.
Нитрозирование. Например, превращение хлорацетофенона в
«-хлоризонитрозоацетофенон [41:
ХОН
Кипячение II
С6Н5СОСН,С1 +я-С4Н9ОХО+ НС1(газ) 2_8|^ С6Н5СОСС1
0,1 моля в 100.4.1 0,1 моля °
эфира
1. Нойес В., «Синтезы органических препаратов», ИЛ, М., 1949, сб. 2, стр.
131.
2. S m а г t X'. A., Y о u п g G. Т., W i 1 I i a m s М. W., J. Chem. Soc., 1960,
3902; Wieland T., Detcrmann H., Angew. Chem., Internal. Ed.,
2, 358 (1963).
3. H о n z 1 .1., R и d i n » er J., Coll. Czech., 26, 2333 (1961).
4. Л e n ii и H., Хартунг В. «Синтезы органических препаратов», ИЛ, М.,
1952, сб. 3, стр. 469.
тре/п-БУТИЛНИТРИТ, (СНЕСОМО. Мол.вес 103,12,т. кип. 633.
Азидный метод синтеза пептидов. Б. и НС1 в диметилформамиде
используют (Гофманн и др. 111) в модифицированном Рудингером син-
тезе азидов (см. «-Бутилнитрит). По сравнению с обычным азидным
методом этот способ проще и дает более высокий выход.
1. Hofmann К. et al., J. Am. Chem. Soc., 87, 620 (1965).
О
Zttpe/tt-БУТИЛ-л-НИТРОФЕНИЛ КАРБОНАТ, zz-O.ACeHjOCOCiCH.^.
Мол. вес 239,22, т. пл. 78,5—79,5°.
Б. получают из /г-нитрофенплхлоркарбоната и /пре/п-бутанола;
применяют для синтеза /ирс/п-бутоксикарбониламинокислот [11:
О
I! N;i2CO3
п-О2ХС0Н4ОСОС(СН;,):,+ H2NCHRCO2H---->
о
II
—л (CH3),COCNHCHRCO.,Na-: п-О2ХС6Н4ОН
/?гре//г-Бутоксикарбонилы1ая защитная группа устойчива к гидри-
рованию и к действию натрия в жидком аммиаке.
1. А и d е г s о п G. W., М с G г е g о г А. С., J. Am. Chem. Soc., 79, 6180 (1957).
трет-БУТИЛ ХЛОРИСТЫЙ, (СНЭ):!СС1. Мол. вес 92,57, т. кип.
51°, уд. вес 0,85.
Получение. Смесь 50 мл /прг/ц-бутапола и 250 мл конц. соляной
кислоты сильно встряхивают в делительной воронке в течение
10 мин. Органический слой отделяют, сушат надСаСП и перегоняют;
выход 45—50 г.
177
трет- БУТ ИЛ ХРОМАТ, (СН.,),СОСгО2(ОН). Мол. вес 174,13.
В первом описании реагента приведен очень хороший выход
при окислении ацетата холестерина в 7-кетохолестерилацетат 111;
однако в последующем это не подтвердилось. В более удовлетвори-
тельной модифицированной методике Б. получают в СС14, содержа-
щем уксусную кислоту и уксусный ангидрид |2|. Некоторые авторы
считают, что и модифицированная методика дает низкие выходы
13, 41, хотя, по мнению других 15—7|, Б.— такой же или даже луч-
ший реагент, чем обычно применяемые (СгО3, реагенты Оппенауэра,
NBC). Применение Б. в пиридине не дает преимуществ перед исполь-
зованием хромового ангидрида в этом же растворителе 181.
1. Оррепаиег R. V., О Ь е г г а и с h Н., Analcs asoc. quim. argentina,
37, 246 (1949); С. A.. 44, 3871 (1950).
2. H с и s i e r K., Wettstci n A., Helv. Chim. Acta, 35, 284 (1952).
3. Rao P. \., К u r a t h P., ,1. Am. Chem. Soc,, 78 , 56(50 (195(5).
4. Bevier R. E., Obersier A. E., Hoffman F., Saretf L. H., J.
Am. Chem. Soc., 82, 170 (1960).
5. S a n n i e C., Panouse J.-J., VertalierS.. Bull. soc. chim. France, 22,
1039 (1955).
6. H a v n e s i\. B., Redraorc D., T i m m о n s C. .1., .1. Chem. Soc., 1963,
2420.
7. I\at z A., Helv. Chim. Acta, 40, 487 (1957).
8. Me n i n i E., N or у ni b er s k i J. К., Bioehcni. J., 84, 195 (1962).
2-н-БУТИЛЦИКЛОГЕКСИЛАТ НАТРИЯ
H CH2CH._>CH2CH3
>7 ONa
Б. н. получают при растворении 24 г натрия в 400 г 2-бутилцикло-
гексанола! 11. Б. и.- эффективный реагент для дегидрогалогенирова-
ния даже таких нереакционноспособных соединений, как борнпл-
хлорнд (1). 2,6-Дихлоркамфан (3) не дегпдрогалогенируется пи ди-
н3с сн,
\ ..сн,
н,с сн3
V .сн,
Н,С сн,
н3с сн,
178
этпламидом лития, ни трет-6\тплатом калия 12], однако при дей-
ствии Б. и. из (3) получают борпадиен (4) с высоким выходом |3|.
1. Hanack М., Н a h и 1 с RChem. Вег., 95, 191 (1962).
2. К w а г t 11., \ u 1 1 G , .1. Am. Chem. Soc., 78, 5943 (1956).
3. Hanack At, E g " e и s p e г h e r H.. H :i h n 1 e R., Atm., 652, 96 (1962).
I----o----1
у-БУТИРОЛАКТОН, CH.,CH.,CH.1CO. Мол. вес 86,90, т. кип.
206' (90—92717 мм), уд. вес 1,128.
О реакции с KCN см. Глутаровая кислота; о синтезе дицнкло-
гексилкетона см. Метилат натрия, катализатор конденсации.
Реакция Фриделя — Крафтса. Реакцией у-бутиролактона с бен-
золом можно получить один из двух образующихся продуктов
в зависимости от количества хлористого алюминия, взятого при рас-
чете на лактон; при молярном соотношении 1,25 у-фенилмасляную
кислоту получают с выходом 73% и только 11% а-тетралона; при
соотношении 2,5 выход кетона возрастает до 66% 111, а при соотно-
шении 3,7 выход а-тетралона почти количественный |2|.
Присоединение по концевой двойной связи. При действии сол-
нечного света или при освещении ртутной лампой у-бутнролактон
присоединяется к олефинам с концевой двойной связью (гептен-1,
октен-1, децен-1) с образованием 2-алкиллактона 131, побочный
продукт — 4-алкиллактон.
Ацетон,
соан. сеет
ьЗ’о
1. Truce W. Е., Olson С. Е., .1. Am. Chem. Soc., 74, 4721 (1952).
2. Olson С. Е., Bader A. R., Ощ. Syn., Coll. Vol., 4, 898 (1963).
3. Elad D., Youssef ye h R. D., Chem. Comm., 1965, 7.
В -
ВИЛЬСМЕЙЕРА РЕАГЕНТ. (См. Диметилформамид—хлор-
окись фосфора.)
ВИНИЛАЦЕТАТ, СН2-СНОСОСН3. Мол. вес 86,09, т. кип.
72—73, стабилизируется гидрохиноном.
Перевинилирование [1]. Смесь свежеперегнанного В. и лаури-
новой кислоты нагревают в атмосфере азота до растворения кислоты,
добавляют ацетат ртути (II), затем серную кислоту и смесь кипятят
в течение 3 час. Для нейтрализации кислоты добавляют тригидрат
СН3(СН.,)10СО.>Н4~СН3СО2СН =СН2 + Hg(OAc)., + 100%-ная H2SO4 —>
0.4 моля 2,4 моля 1,6 г 0,15 мл
Кипячение 3 час
-- _ > СНз(СН2)10СО2СН — сн2
ацетата натрия (0,83 г), затем отгоняют избыток В. и виниллаурат
дважды перегоняют, т. кип. 120—120,572 мм.
Синтез диенов через промежуточные циклобутаны [2]. В этом
синтезе стадию циклоприсоединения проводят под давлением.
В автоклав загружают свежеперегнанный В., 1 г гидрохинона, три
капли дипентена или терпинолена (для предотвращения полимери-
зации фторолефина), охлаждают смесью ацетон — сухой лед, ва-
куумируют и вводят из баллона трифторхлорэтилен.Смесь нагревают
CI Н
CFC1
II
cf2
СНОСОСНз
2 15°, 3 час F
27—37%”* F
J---!_ососн3
I I
F H
70075 — 10 лг.и
CFC1 = CHCH = CF,
при перемешивании до 215° и выдерживают при этой температуре
в течение 3 час. Получают черную вязкую реакционную смесь;
выход 2-хлор-2,3,3-трифторцпклобутилацетата низкий. Следующую
стадию отщепления уксусной кислоты с последующим разрывом
углеродной цепи выполняют при нагревании в электрической печи
в реакционной трубке из стекла викор.
Пептидный синтез. Виниловые эфиры аминокислот, получа-
емые переэтерификацией из В., используют в качестве актпвнро-
180
ванных эфиров в пептидном синтезе [3|, который наиболее успешно
проводится в циануксусном эфире. В этом растворителе подавляет-
ся образование окрашенных продуктов, обычно образующихся из
выделяющегося ацетальдегида. Рацемизация при этом, по-видимому,
незначительна.
I. С в е р н Д., Джордан Э., мл., «Синтезы органических препаратов», ИЛ,
М„ 1953, сб. 4, стр. 123.
2. Putnam R. Е, Anderson В. Т., Sharke у W. Н., Org. Svn., 43, 17
(1963).
3. Weygand F., Steglich W., Angew. Chem., 73, 757 (1961).
ВИНИЛ-(КАРБМЕТОКСИМЕТИЛ)-КЕТОН (метиловый эфир ак-
рилоилуксусной кислоты), СН,=СНСОСН2СО,СН3. Мол. вес
128,12, т. кип. 65 /19 мм; при хранении полимеризуется.
Получение. Метод, разработанный Назаровым и Завьяловым
11), был применен в крупном масштабе Бенкертом и сотр.
[21, а также Гогенлоэ-Орингеном 131, который получил карб-
этоксиметильное производное (выход на первой стадии 29%). По
методу, примененному группой Бенкерта, к суспензии 1,5 моля на-
триевого производного метилового эфира ацетоуксусной кислоты
в эфире медленно добавляют спиртовой раствор 1,6 моля 0-этокси-
пропиоиилхлорида. По окончании реакции хлористый натрий от-
деляют фильтрованием, а фильтрат насыщают аммиаком для осу-
ществления кетонного расщепления. При перегонке над п-толуол-
сульфокислотой в вакууме отщепляется этанол и образуется В.
С2Н5ОСН2СН,СОС! -{-CH3COCHCO,CH3(Na+) —>
1,6 моля
С„Н,,ОСН,СН„СО
'I
СН3СОСНСО.,(
TsOH, перегонка в вак. 12
---------—------------* СН,=СНССН,СО2СН3 -J- С,Н5ОН
1,5 моля
о
NH-. II
7/ДГ С2Н5ОСН2СН,ССН2СО,СН3 —>
о
0-130" [I
Синтез карбалкоксилированных циклических систем. Назаров
и Завьялов [1] использовали реагент для синтеза дикетоэфира (4)
из 2-метплциклогександиона-1,3 (1) и карбэтоксиметильного
реагента (2). Катализируемое основанием присоединение по Михаэлю
181
дает соединение (.3), альдолизаппя которого в (4) осуществляется при
перегонке в вакууме. Конденсацией по Маннпху из В., метиламина
и феннлацетальдегпда Гогенлоэ-Оринген [31 получил 4-пиперидон
(5) с умеренным выходом.
О о
СЩОХ СНАС П
|| 3<) — 4Q% | I
с,н5сн2сно- \н, с6н5сн, х'
СН:! сн3
Венкерт и corp. [21 описали два примера использования В. для
аннелпровання ио Робинсону с образованием продуктов, содержа-
щих карбметокспльпую группу. Первый пример — взаимодействие
с (6), приводящее к (7) — ключевая стадия в синтезе смолящих ки-
слот, а второй — превращение (8) в (9),— этап в решении проблемы
синтеза гибберелловой кислоты.
1. Назаров И. Н.. Завьялов С. И.. ЖОХ, 23, 1703 (1953).
2. W г и k е г t Е.. Л 1 о и s о Л.. 13 г с <1 е и |> е г ц .1. В.-son, К а п е к о С.,
Т а 11 а г а Л., J. Ani. Chem. Soc.. 86, 2038 (19(14).
3. 11 о И е п I о h е - О е h г i и 2 е п К., Monatsh., 93, 576 (1962).
ВИНИЛ-ф-КАРБМЕТОКСИЭТИЛ)-КЕТОН,
СН, СНСОСН,СН,СО,СН3. Мол. вес 142,15, т. кип. 103716 льи.
В. легко полимеризуется, поэтому его необходимо стабилизировать
гидрохиноном.
Получение 111.
о о
II A1C1., II
СН., - СН, л С|С(:Н,СН„СО,СН3-- С1СН,СН,('.СН,СН,СО,СН3 —>-
о
(С,НДЖ ;
—----> СН, --- СН - ССН,СН,СО,СН3
182
Синтезы ароматических соединений. Бухта [2| применил В.
для построения полициклических соединений, способных к аромати-
зации. Присоединением по .Михаэлю реагента (2) к 2-карбэтокси-
тетралону-1 (1) получают соединение (.3), подвергающееся внутри-
молекулярной конденсации Штоббе с образованием кислоты (4),
которую циклизуют в жидком HF и перегонкой продукта (5) над
цинковой пылью получают пирен (6). По этой схеме синтезирован
гептацнклическпп пероппреп 131.
1. В и с h t а Е., Bayer W., Heinz G., Xaturwiss., 45, 439 (1958); see also
Grundmail n G., Rus k e W., Chem. Ber., 86. 939 (1953); H а з a-
p о в И. 14., 3 а в ь я л о б С. И., С. Л., 47, 5364 (1953).
2. В и с li t а Е., В aver W., Xaturwiss., 45, 140 (1958).
3. В и с h t а Е., V i и с k е Р., Chem. Вег., 98, 208 (1965).
ВИНИЛЛИТИЙ, СН,- CHLi. Джусиге и Сейферт 111 пред-
ложили два способа получения В. в эфирном растворе. Фенплли-
iiiii в эфире реагирует с тетравпнплсвинцом, при этом осаждается
тетрафенилсвинец (т. пл. 230 ), а отфильтрованный раствор содер-
жит В. Второй способ включает обмен металлами в реакции лития
с тетравинилстаннаном; олово выделяется в виде черного осадка
и является единственным побочным продуктом. Полученный обоими
способами В. выделяется в виде исключительно пирофорного бе-
лого твердого вещества, которое при гидролизе дает этилен. Третий
1) 4<:„11.| ; РЬ(СН=-СН,), —, 4СН, -CHLi--РЬ(С6Н3),
2) 4Li ! Sn(CH -СН,), — , 1СН, -CHLi 8п
3) IG.H.Li Sn(CH _ - СН,), — 2СН,-CHLi । Su(C„l 1,),
способ получения В., предложенный Сейфертом и Вейнером [21,
состоит во взаимодействии фенпллптпя с тетравинилстаннаном.
Этот способ позволяет получить В. с хорошим выходом. Ниже прп-
183
ведены некоторые реакции В.:
н2о
СН, = CHLi + СО,--> СН, = СНСО,Н
37%
ОН
н2о I
CH, = CHLi + (CH3),C =О---» СН2 - СНС СН3
7 4% |
СН3
(w-C4H9)3SnCl + СН2 = CHLi_> (н-С4Н9)3 SnCH = СН., Д- LiCl
74%
Показано (Уэст и Глэйз 131), что В. можно получить непосредст-
венно из хлористого винила и мелкодисперсного лития, содержащего
около 2% натрия, при температуре 0—10°.
I. J uenge Е. С., S е у f е г t h D., J. Org. Chem., 26, 563 (1961).
2. Seyfertli D., W e i n e r M. A., J. Am. Chem. Soc., 83, 3583 (1961).
3. W e s t R., G 1 a z e W. H„ J. Org. Chem., 26, 2096 (1961).
ВИНИЛТРИФЕНИЛФОСФОНИЙБРОМИД,СН2=СНР(СсН5)3Вг-.
Мол. вес 269,24, т. пл. 188—190°; соль вызывает аллергическую
реакцию (чихание).
Получение [1, 21. Смесь p-бромфенетола, трифенилфосфина
и фенола (в качестве растворителя) перемешивают при 903 в течение
48 час, затем осаждают эфиром белый кристаллический фенокси-
этилтрифеиилфосфонпйбромид, при кипячении которого с этилаиета-
том отщепляется фенол и образуется В.
90е, 48 час
С„Н5ОСН,СН,Вг+ (СвН3)3Р + Фенол---> С6Н3ОСН2СН2Р+(СвН5)3Вг-—>
0,5 моля 0,5 моля 454 г
снчсо,с.н5
---- —------> СН2 = СНР + (СйН5)3Вг- + С6Н5ОН
75—85% (общин)
Присоединение по Михаэлю. В. вступает в реакции сопряженного
нуклеофильного присоединения с R(Ar)OH, R(Ar)SH, R,NH,
например [11:
C„H5SH4 CH„-=CHP + (C„H5)3(Br-)--> CcH5SCH,CH.,P+ (CcH5)3(Br-)
74%
Синтезы циклических соединений. Виггильиая группа реагента
виде двухуглеродного звена при построении
z чz СНО
t СНР + (СсН5)3Вг , 11 О', 24 час
^СН» L х. 82%^
0,5 моля 4 ОН
200 МА
—1| | |ч(С6Н5)3РО
о
57%
184
может
сно
/ ОМа +
0,5 моля
гетероциклических и карбоциклических соединений. Например,
реакцией В. и натриевой соли салицилового альдегида в избытке
салицилового альдегида при НО' с последующим отделением фе-
нольных продуктов обработкой щелочью в эфире и перегонкой
ДМ ФА - эфир
СН3СО + СНР+(С6Н5)3Вг’ ------->
I II 39%
CHjCHOH сн2
39%
+ (С6Н5)3РО
49%
получают 3,4-хромен 13). Аналогично ацетоин дает 2,5-дигидро-
2,3-диметилфуран [4). ЗН-Пирролизин получают из В. и пиррол-
альдегида-2 в эфире в присутствии гидрида натрия в качестве кон-
денсирующего агента.
И П Эфир—NaH ,
%Асно ~ А |] + (С‘Н=Ьро
н
СН2=СНР(С6Н5)3Вг’
1. Schweizer Е. Е., Bach R. D., J. Org. Chem., 29, 1746 (1964).
2. Schweizer Е. E., Bach R. D., procedure submitted to Org. Syn.
3. S c h w e i z e r E. E., J. Am. Chem. Soc., 86, 2744 (1964).
4. S c h w e izer E. E., Light К. K., J. Am. Chem. Soc., 86, 2693 (1964).
ВИННОЙ КИСЛОТЫ НАТРИЙ-КАЛИЕВАЯ СОЛЬ, ТЕТРА-
ГИДРАТ (сегнетова соль), NaO2CCH(OH)CH(OH)CO2K-4H2O.
Мол. вес 283,23. Растворимость в 100 г Н2О: 26 г при 0°, 66 г
при 26°.
Тартратный ион образует комплекс с ионом алюминия и удер-
живает его в растворе. Истхэм и Тераниси HJ окисляли холестерин
по Оппенауэру кипячением в толуоле и после удаления основной
А1/ОСН(СН3)^73
Кипящий толуол
73-84%
Н ОН
100 г (0, 26 моля)
массы летучих веществ добавляли 400 мл насыщенного водного рас-
твора сегнетовой соли к мутной реакционной смеси при пере-
мешивании до тех пор, пока органический слой не стал прозрачным
и не приобрел оранжевую окраску. Затем смесь перегоняли с водя-
ным паром, остаток охлаждали и экстрагировали хлороформом.
После восстановления холестенона трис-(треш-бутокси)-алю-
могидридом лития Бергсталер и Нордин 12] гидролизовали металло-
0, 039 Моля
LiAlH(OBu)3 (0, 05 моля)
Эфир-$иглим-45°
87%
185
органический комплекс обработкой льдом, едким натром и сегне-
товоп солью.
1. Е a s t 11 a in J. I'., Т е г a n i s 11 i R., Org. Sin., Coll. Vol., 4, 192 (1963),
2. В ii г 4Д s t a li 1 e г A W., X о r d i n I. C.. .1. Am. Chem. Soc., 83, 198 (see
p. 205) (1961).
ВИСМУТА ТРЕХОКИСЬ, Bi2():;. Мол. вес 466,00.
В. т.— специфический реагент для окисления ацилоинов до
а-дикетоиов; В. т. при этом восстанавливается до металла |1|.
Раствор ацилоина в уксусной кислоте обрабатывают В. т. (20%-ный
избыток) и смесь нагревают на кипящей водяной бане при переме-
шивании влечение 15мин.Бензоин— бензил (выход 95%) 111.12-Окси-
11-кетостероиды — 11,12-дпкегостеропды (выход 70%) |2|. Описано
окисление цевина и его изомеров 131 и другие примеры: окисление
2-оксипхлегона (1) в диосфеполоп (2) с выходом сырого продукта
94"<> 141, 2'7,-оксп-А 4-3-кетостеропда (3) в соединение (4) 151 и (5)
в (6) |61.
1. R is» by W., J. Chem. Soc., 1951, 793.
2. D j er a s s i C., R i ngo I (I H. ,1., R о s e n k r a n z G., J. Am. Chem. Soc.,
76, 5533 (1954).
3. К u p c h a n S. At, L a v i e D., J. Am. Chem. Soc., 77, 683 (1955).
4. R e i t s e in a R. H., J. Am. Chem. Soc., 79, 4465 (1957).
5. В а г a n J. S., J. Am. Chem. Soc., 80, 1687 (1958).
6. L a v i e D., S h v о Y., Willner D., E n s 1 i n P. R., Hugo J. AL,
X or ton К. B-, Cbem. Ind., 1959, 951.
ГАЗЫ ИНЕРТНЫЕ.
Ннсгинын газ 1 Мол. Rec 1 Уд вес (воздух — I )
Гелин 4,003 0,137
Азот 28,016 0,967
Аргон 39,941 I ,38
Дзот, не содержащий кислорода (менее 20 частей на миллион),
применяется для создания сухой инертной атмосферы в камерах,
для проведения координационной полимеризации, для получения
реактивов Гриньяра. Сухая инертная атмосфера очень важна при
получении найлона из соли адипиновой кислоты и гексаметиленди-
амина.
Гелий широко используется в газовой хроматографии. Он часто
применяется для продувания систем, включающих ловушки с жид-
ким азотом, поскольку этот инертный газ при температуре жидкого
азота не конденсируется. Однако чаще для этой цели применяется
аргон.
Аргон обычно используется в тех случаях, когда азот нежелате-
лен из-за его реакционной способности, например в реакциях с при-
менением лития может образоваться нитрид лития. Аргон рекомен-
дуется применять при реакциях активных металлов, требующих
нагревания. Поскольку аргон тяжелее воздуха, он очень эффективен
для заполнения систем; при подаче в систему аргон заполняет ее
снизу, вытесняя воздух в верхнюю часть.
ГАЛОФОРМНАЯ ПРОБА 111. Эта проба, улучшенная Фуджи-
вароп |21, позволяет определять фтороформ, хлороформ, бромоформ и
йодоформ, а также предшественники галоформа или дигалокарбена.
Несколько капель испытуемого раствора добавляют к смеси 3 л/л
10%-ного едкого натра и 2 мл пиридина. Проба считается положи-
тельной, если при встряхивании при комнатной температуре в те-
чение 1—2 мин смесь приобретет розовый пли яркий красно-голу-
бой цвет. Если изменение окраски происходит не при комнатной
температуре, а при нагревании в течение 1 мин на кипящей водяной
бане, то это указывает на присутствие предшественника галоформа
или дихлоркарбеиа. Интенсивность окраски пропорциональна кон-
центрации галоформа пли вещества, легко образующего галоформ
в этих условиях.
187
Этот чувствительный тест позволяет использовать при галоформ-
ном расщеплении более доступный гипохлорит натрия вместо
гипоиодита натрия для определения метилкетонной или метилкар-
бинолыюй структуры. Г. п. применяется также в тех случаях, когда
определение галоформа по ИК-спектрам или ГЖ.Х не дает хороших
результатов, например при некоторых реакциях, где галоформ-
ный тип расщепления осуществляется в незначительной степени |3|.
1. Contributed by .1 о u 1 1 i ё М. М., University of Pennsylvania.
2. F и j i w а г а К., Sitz. Nat. Ges. Rostock, 6, 33 (1916); [C. A., 11, 3201 (1917)1.
3. Pierce Л. C., J о и 1 1 i ё M. M., J. Org. Chem., 28, 658 (1963).
ГАЛЬВИНОКСИЛ
С(СН3)з С(СН3)з
j_ __lx
О=< ; = СН->-()
i I
С(СН:1)3 С(СН3):)
Мол. вес 421,62, т. пл. 158~ (с разл.); темно-синий.
Г.— пространственно затрудненный, фенокспльный ра-
дикал; получается при окислении 3,3',5,5'-тетра-треш-бутил-4,4'-
диоксидифенилметана либо двуокисью свинца в эфире и изооктане
111, либо щелочным раствором феррицианида 12]. Г.— эффективный
акцептор радикалов; он количественно реагирует с радикалами за
счет неспаренного электрона на атоме углерода или кислорода [31.
В противоположность первоначальным данным, этот радикал не
стабилен по отношению к кислороду |4|.
1. Со р р i n g е г G. М., J. Am. Chem. Soc., 79, 501 (1957).
2. К. h а г a s с h М. S., .1 о s h i В. S., J. Org. Chem., 22, 1435 (1957).
3. Bartlett P. D., Riichardt C., J. Am. Chem. Soc., 82, 1756 (I960);
Bartlett P. D., F u n a h a s h i T., ibid., 84, 2596 (1962); Bart-
lett P. D., G о n t a r e v В. H., S a k u r a i H., ibid., 84, 3101 (1962);
Greene F. D., Adam W., С a n t r i 1 1 J. E., ibid., 83, 346! (1961).
4. G r e e n e F. D., Adam W., J. Org. Chem., 28, 3550 (1963).
ГЕКСАМЕТИЛБЕНЗОЛ, Ce(CH3)(i. Мол. вес 162,26, т. пл. 166°,
т. кип. 265
Получение. В специфической методике 111 получения Г. па ката-
лизаторе из активной окиси алюминия при 530' раствор 100 г фе-
нола в 1 л метанола вводят по каплям со скоростью ПО мл!час
в установку, в которой должна быть предусмотрена трубка для от-
вода газа под тягу. Твердый светло-желтый продукт собирают в при-
емник и переносят с метанолом на воронку для отсасывания. Кри-
сталлизация из спирта дает бесцветные кристаллы, т. пл. 165—166°,
выход 55—57%. Механизм реакции неизвестен.
Реакция. Об интересной окислительной перегруппировке см.
Трифторнадуксусная кислота.
1. Cullinane N. М., С h а г d S. J., D a w k i n s С. W. С., Org. Svn. Coll.
Vol., 4, 520 (1963).
188
ГЕКСАМЕТИЛДИСИЛАЗАН, (CH3)3SiNHSi(CH3)3. Мол. вес
161,40, т. кип. 125,5% Г. получают из триметилхлорсилана и аммиа-
ка с выходом 65% [11.
Пептидный синтез. Обзор [21. Г. реагирует с аминокислотами
(кипячение в присутствии нескольких капель серной кислоты) с об-
разованием триметилсилильного эфира N-триметилсилилампно-
кпслоты. При этом гидроксильная группа (серин) и меркаптогруппа
(цистеин) также сплилируются. Эти производные можно использо-
RCHCOJI ,-(CH3)3Si\HSi(CH3)3^ RCHCO2Si(CH3)3-(-NH3
I ' I
NH, NHSi(CH3)3
вать в пептидных синтезах методом смешанных ангидридов, п-нн-
трофениловых эфиров, а также с помощью хлорокиси фосфора или
имидазола. Интересно, что силильная группа удаляется просто при
обработке водой в обычных условиях. Триметилхлорсплан не при-
годен, так как аминокислоты являются амфотерными соединениями.
Производные спиртов. Желчные кислоты [31 и сахара [41 при
обработке Г. при комнатной температуре в пиридине превращаются
в О-триметилсилпльпые эфиры, которые более летучи, чем обычные
простые эфиры, и могут быть использованы для ГЖХ.
1. S а и с г R. О., J. Am. Cliein. Soc., 6fi, 1707 (1944).
2. В i г к о f е г Ritter A., Anijew. Chem., Internal. Ed., 4, 417 (1965).
3. M о k i t a M„ W c 1 I s W. W., Anal. Biochem., 5, 523 (1963).
4. S wed e v С. С., В en I I e у R., M a k i 1 a M., Wells W. W., .1. Am.
Chem. Soc\ 85, 2497 (1963).
ГЕКСАМЕТИЛЕНТЕТРАМИН (уротропин), (CH2)liN4. Мол. вес
140,19, белое твердое вещество.
Г. получают выпариванием смеси формалина и водного концент-
рированного раствора аммиака.
Г. применяется в медицине под названием уротропин в качестве
антисептика мочевых путей. Его действие обусловлено медленным
выделением формальдегида и аммиака; моча при этом приобретает
щелочную реакцию.
Гексоген. Дымящая азотная кислота разрушает мостиковую
систему молекулы Г. и нитрует периферические атомы азота. Обра-
зующийся гексоген — взрывчатое вещество с баллистической силой
150,2 (баллистическая сила тротила принята за 100). Гексоген
впервые применен в первой мировой войне и широко использовался
во время второй мировой войны. Выход гексогена повышается
вдвое при добавлении к реакционной смеси нитрата аммония и
189
уксусного ангидрида (Бахман и Шихан 111). При этом из 1 моля Г.
OjN
\ N-NOj + 3 СН2О + .NH,
N—f
O2l/
Гексоеен (гп.пл. 203’)
получают 2 моля (а не 1) гексогена:
QHpA'j : 4Н\О3--: 6(СН3СО)2О
—>2С;)Нг>\вО6л. 12СН3СО()Н
Гексоген
Три рассмотренные ниже именные реакции с применением Г.
широко используются в синтезах.
Реакция Делепина [21. Соединения с подвижным галогеном
реагируют с Г. в неводной среде с образованием иммонпевой соли,
которую гидролизуют раствором хлористого водорода в этаноле
и получают .хлоргпдрат соответствующего амина; формальдегид
удаляется в виде летучего диэтилформаля. Например, 2,3-дпбром-
проиен-1 (1), содержащий аллильный галоген, образует соль (2), из
которой при кислом гидролизе получают амин (3) 131. Амины полу-
Вг Вг
I | _
СН.,-ССН,Вг-гС6Нг\4 —-----— СН,^ССН.,\СвН,.,\3(Вг) —
(1) (2f
Вг
EtOH-HCl NaOH I
-----_о „-----> СН^-ССНАНо
(3)
чаются также из сравнительно активных бензил- и фенацилгалогени-
дов. Промежуточно образующиеся соли гидролизуют холодным
спиртовым раствором хлористого водорода до первичных аминов
типа ArCH.NH . и ArCOCKNH.. |4|.
О О
II (CH,),X4 [j с.н,он
АгССНоВг---------- АгССН,Вг (СН,)6\Т4-----
' -'6 4 НС1
о
II
ЛгССН2.\Н3+С|-
Реакция Соммле |5, 61. Эта реакция также включает взаимодейст-
вие галогенида (обычно бензильного тина) и Г. с образованием соот-
ветствующей соли, которую далее гидролизуют в некислой водной
среде (вода или водный спирт). При взаимодействии бензил.хлорида
с Г. (см. схему) в водном этаноле образующийся при гидролизе
бензиламин реагирует с продуктами, получающимися из Г., и дает
190
бензальдегид, который отгоняют с паром. Первые две стадии тож-
СН,С1
СН..\С6Н1.,\3
сно
ШОП !
дественны реакции Делеппна, и в присутствии соляной кислоты
реакция завершилась бы на стадии образования хлоргпдр.чта бен-
зиламина. Превращение бензиламина в бензальдегид по Соммле
и нейтральной среде было изучено .Апжиалом (1948 г.) 161. Бензил-
амин обратимо реагирует с формальдегидом, присутствующим в реак-
ционной смеси, с образованием метилеибензнламина (2), который при
отсутствии Г. диспроиорцпонпруется с бензиламином таким обра-
зом, что соединение (2) восстанавливается в метплбензилампп (4),
а соединение (1) окисляется в имин (3), который при гидролизе дает
с низким выходом бензальдегид. В условиях реакции Соммле (pH
CbHjCHA’Ho ; СаН.,СН,\ сн, —: с„нг>сн \н СВН5СН,ХНСН3
(1) (2) <3) (4)
I
I Н..О
!--' сйн-,сно ; \н3
С)
3,0—6,5) Г. взаимодействует как метиленовое производное аммиака
(СН, —NH), которое принимает водород от бензиламина и восста-
навливается в метиламин. Эта реакция ингибирует диспропорцио-
нирование, приводящее к метилбепзпламину (4), поэтому выход
бензальдегида возрастает. Процесс можно изобразить следующей
схемой:
('.„НДН.АН, 1 СН, -ХН |-Н,О—-СвН.,СН() !-хн3--сн;|хн,.
Этот окислительно-восстановительный процесс соответствует реак-
ции Канниццаро и, вероятно, включает аналогичную стадию пере-
носа гидрид-иона, причем в качестве акцептора выступает кислота,
сопряженная основанию Шиффа:
Кроме того, в этой реакции возможен 1,3-перенос протона в соедине-
нии (2) с последующим гидролизом:
п,о
СвН5СН.,Х - СН, - с„н,сн - хсн.,—- - CJH.CHO ' н,хсн3
(2)
В методике синтеза 2-тпофенальдегида 171 промежуточно обра-
зующуюся соль получают в кипящем хлороформе, после чего про-
191
водят перегонку с паром для осуществления заключительной стадии
и отделения продукта по мере его образования. В синтезе 3-тио-
фенальдегида [81 промежуточную соль также получают в хлоро-
c6h„n4 8 СНС1Д>
94-97%
Н2О,перегонка с паром! Г* !
*<S>CHO
форме, затем экстрагируют водой и после перегонки с паром полу-
чают смесь альдегида и метиламина. Продукт экстрагируют эфиром
и для удаления амина экстракт промывают кислотой.
СбН.2М4 в CHCL г Н20,перееонка спором
f СОЛЬ ".I < .—।
(не изолирована) 54-72%
а-Нафтойный альдегид получают по Соммле [9] с высоким выхо-
дом.
СН2С1 СНО
Реакцию проводят при кипячении в 50%-ной уксусной кислоте
(2 час) и четвертичную соль не выделяют. Затем прибавляют соля-
ную кислоту и кипятят еще 15 мин для гидролиза и разрушения ос-
нований Шиффа, которые могут загрязнять продукт.
Насыщенные алкилгалогениды образуют четвертичную соль
с высоким выходом; однако на второй стадии выходы низкие. Напри-
мер, для w-гептилиодида выход составляет 46% 1101. В ряду хлор-
метильных производных азотистых гетероциклов такая реакция
не имеет большого значения, так как эти соединения обычно недо-
ступны или неустойчивы. Однако в качестве исходных веществ для
синтеза соответствующих альдегидов можно использовать амино-
метильные производные, легко получающиеся при гидрировании
нитрилов. Это показано на примере 3-аминометилпиридина [11]:
СНА'Н, СНО
/W /Ч/
CeH12N4, АсОИ, НС1
Реакция Даффа 112]. Г. конденсируется с фенолами в орто-
положение с образованием основания Шиффа, которое при гидро-
лизе даето-оксиальдегид:
о-НОС6Н4СН = NCH,C0H4OH-o —
nh2
I
—> о-НОС5Н4СНО + СНоС0Н4ОН-о
192
Дафф нагревал глицерин с борной кислотой до 170? до полного удале-
ния воды, прибавлял 25 г фенола н 25 г Г. п нагревал при 170"
в течение 15 мин, после чего выделял о-окспальдегпд перегонкой,
с паром. Для нескольких фенолов выходы были в пределах 2—8,5 г.
1,3- Диметиловый эфир пирогаллола замещается в пара-положен не
с образованием сиреневого альдегида (1) с выходом 31—32?о 1131.
Бэкер 1141 применил для 3,4-дпметилфенола методику [ 15], вклю-
ОН
СН3О ( ОСН3
' .7
ii
i
СНО
О)
OII
( сно
Z, /
н3с
СН3
чающую нагревание фенола с Г. в уксусной кислоте в течение 5 час
на кипящей водяной бане, добавление кипящей воды и соляной
кислоты н отгонку альдегида (2) с паром; выход 32%.
1. В j с 1, m а п n W. Е., S li е е li а п .1. (2. .1. Am. Chem. Soc., 71, 1842 (1949).
2. Del ё р i и е М., Compt. rend., 120, 501 (1895).
3. В о t t i п i A. T„ D e v V., К 1 i n c k J., Огщ Syn., 43, 6 (1963).
4. Lon» L. M., Troulima n H. D., J. Am. Chem. Soc., 71, 2473 (1949).
5. S о in ni e 1 e t M., Conipt rend., 157, 852 (1919).
6. А и ж u а л С. Дж., «Органические реакции», ИЛ, М., 1956, сб. 8, стр. 263.
7. В ибер г К., «Синтезы органических препаратов». ИЛ, М., 1953, сб. 4,
стр. 474.
8. К а м п е н ь Э., Б у р ж у а Р., Ма к - Керт и У., «Синтезы органических
препаратов», ИЛ, М., 1954, сб. 5, стр. 66.
9. А н ж и а л С., Т е т а з Дж., В и л ь с о н Дж.. «Синтезы органических
препаратов», ИЛ, М., 1953, сб. 4, стр. 346.
10. А п )« у а 1 S. J., Р е п m a n D. R., W а г w i с k G. Р. ,1. (diem. Soc., 1953,
1737. '
И. А п t< у а 1 S. .1., В а г 1 i п G. В.. \V a i 1 е s Р. С., J. Chem. Soc., 1953, 1740.
12. D u f f J. C„ ,1. Chem. Soc., 1941, 547.
13. А л л e h 4., Л e 11 б н e p Г., «Синтезы органических препаратов», ИЛ, М.,
1953, сб. 4, стр. 441.
14. Baker W., М с О ni i е J. F. W., Mile s D., J. Chem. Soc., 1953, 820.
15. Duff H. C., Bills E. J., J. Chem. Soc., 1932, 1987; 1934, 1305.
ГЕКСАМЕТИЛТРИАМИД ФОСФОРИСТОЙ КИСЛОТЫ,
l(CH,).,N].,P. Мол. вес 163,19, т. кип. 162—164" (49-51712 лги),
1,4620.
Получение |1, 2] по методу Марка [31. Раствор 1 моля трех-
хлорпстого фосфора в 1,5 л эфира перемешивают в токе азота при
охлаждении до 2 (ледяная баня) и пропускают газообразный ди-
РС1;, 1-6(СИ;|),_,\И [(CH3),N]3P 43(CH3),NHX1
метиламин (т. кип. 7,8 ) с такой скоростью, чтобы температура не
превышала 15 (5—7 час). Белый, легко перемешиваемый осадок
хлоргидрата диметиламина, который образуется после выдержива-
7 Заказ № 965
193
нпя реакционной смеси в течение ночи при комнатной температуре
в атмосфере азота, отфильтровывают и промывают эфиром. Удаление
растворителя из прозрачного фильтрата на роторном испарителе
дает почти чистый Г. ф. к. Его можно очистить с небольшими поте-
рями обработкой активированным углем в пентане или перегонкой.
Г. ф. к. лучше хранить под азотом.
Эпоксидирование. Реакция Г. ф. к. с ароматическими альде-
гидами представляет простой одностадийный метод получения сим-
метричных и несимметричных эпоксидов |51. Этот реагент отщепляет
Cl CI о С1
2 СНО |(CH3)..XLP---------- z/ СН —СН —'z % J-
0,4 моля 0.232 моля "r>-f0''Q тр'лнс, 3.'- 4U,J„ цис
1 |(CH;,k\]3po
альдегидный атом кислорода с образованием гексаметилтриамидо-
фосфата. Раствор альдегида в бензоле перемешивают в колбе
с обратным холодильником и добавляют эфирный раствор триамида,
поддерживая температуру 24—36 . При охлаждении водопроводной
водой для добавления требуется 30—50 Прозрачный рашвор
нагревают при 50' в течение 15 мин, растворитель удаляют на ротор-
ном испарителе и остаток перемешивают с водой и пентаном. Пов-
торная экстракция пентаном дает белый кристаллический продукт,
являющийся смесью щи- и /иронс-эиокситов (по ЯМР-сиектру).
Алифатические и гетероциклические альдегиды дают главным
образом аддукты состава 1 : 1 например |4|:
О X(CH3i., О Х(СН.,).,
I ’ I
RCH с Р-Х(СН3)„ — RCH — Р-Х(СН3),
I I
Х(СН3|., Х(СН3),
Ньюмен и Блюм 161 показали, что Г. ф. к. может циклизовать
диальдегиды, такие, как (1), в ароматические эпоксиды типа (2):
1. Carmody D. R., Zletz А., пат. США 2898732.
2. BurgA. В., S 1 о t а Р. J., Jr., J. Am. Chem. Soc , 80, 1107 (1958).
3. Mark V., procedure submitted to Org. Svn.
4. Mark V., J. Am. Chem. Soc., 85, 1884 (1963).
5. Al ark V., Org. Syn., 46. 52 (1966).
6. Newman Al. S., В 1 u m S., J. Am. Chem. Soc., 86, 5598 (1964).
194
ГЕКСАМЕТИЛТРИ AM ИД ФОСФОРНОЙ КИСЛОТЫ (гекса-
метапол), |(('H;i)2N 1:>РО. Мол. вес 1/9,19, т. пл. 4 , т. кип. 232°,
диэлектрическая постоянная 30(25 ).
Результаты большого числа реакций магний- и натрийоргани-
ческих соединений, проведенных в Г. ф. к. и ТГФ, сравниваются
в работе Норма пта 111.
Френкель и corp. 121 показали, что натрий, калий и литий раст-
воряются в Г. ф. к. с образованием синего раствора концентрации
до 1 Л7. Раствор тончив is течение нескольких часов, после чего
его окраска изменяется на краситю. Этот апротонный растворитель
обладает высокой растворяющей способностью ио отношению к ме-
таллоорганическим соецшениям; в нем с высоким выходом можно
получать и реактивы Гриньяра и лптийоргаипческие соединения.
Г.ф. к. подобен аммиаку, ио работать с ним легче.
Шрисхейм п corp. |3| показали, что системы Г. ф.к. —
(С1ГФ.СОК и даже Г. ф. к. — КОН очень эффективны в автоокисле-
иии при комнатной температуре алкплбензо.тов с различными боко-
выми цепями до ароматических карбоновых кислот. Для очень
слабокислых углеводородов (толуол, ксилолы, этилбензол) выходы
составляют 10—50%. Г. ф. к. гораздо более активен в этой реакции,
чем тетраметплеисульфокспд, диметилсульфоп пли диметилсульфо-
ксид, что объясняется его устойчивостью к основаниям, высокой
ионизирующей способностью по отношению к углеводородам,
а также низкой температурой плавления и высокой температурой
кипения, что позволяет широко варьировать условия реакции.
Растворитель высушивают перегонкой в токе азота над цеолитом
(прокаленным в токе азота при 350 в течение 4 час) и хранят в су-
хой камере (Уоллес и Шрисхейм |4|). Найдено |4|, что как раство-
ритель для автоокислснпя тиолов и дисульфидов до сульфокислот
в условиях основного катализа (КОН пли NaOH) Г. ф. к. предпоч-
тительнее, чем ДМФА или тетраметилмочевина. Г. ф. к. также наи-
лучшпп реагент для катализируемого основаниями автоокисленпя
алкилпктфепов, толхола 151 и циклических кетонов (С.-,—С12) до соот-
ветствующих двухосновных кислот.
1. XormantH., Bull. soc. chim. France. 1963, 1888; 1965, 859; Cuvign у J.,
H о r in a n t 14., ibid., 1964, 2000; C u v i g и у T., Normant J., Xor-
mailt H., Compt. rend., 258, 3502 (1904).
2. Fraenkel (_i., Ellis S. H., 1) i s; D. T., J. Am. Chem. Soc., 87, 1406
(1965).
3. Hof m a n n J . E., S c !i r i e s h e i in A., R о s e n 1 e 1 d D. D., J. Am. Chem.
Soc.. 87, 2523 (1965).
4. Wallace T. J., S c h i i e s h e 1 m A., Tetrahedron, 21, 2271 (1965).
5. W a 1 I a c e T. J., В a r <> n F. A., .1. Org. Chem., 30. 3520 (1965).
6. W alia с e T, J., R о b iner H., Scbr ic s h e i m A., J. Org. Chem., 30,
3768 (1965).
7*
195
ГЕКСАФТ0РБУТИН-2, CF3C=CCF3. Мол. вес 162,04, т. кип.
от —25 до —24'.
Г. получен дехлорированием CF3CCI ~CCICF3 цинком [ 1 ]; однако
эго соединение удобнее получать реакцией ацетилендикарбоновой
кислоты с четырехфтористой серой в присутствии четырехфторнстого
титана 12]. Этот ацетилен — очень активный диенофил в реакции
Дильса — Альдера 131. Он реагирует с бутадиеном при комнатной
температуре с образованием замещенного циклогексадиена-1,4(1).
со2н
с SF4(TiF4)
С 80% *
СО2Н
СРз
с
III
с
I
CF3
Г. присоединяется к дуролу при 200? с образованием замещенного
бициклооктатриспа (2).
1. IIcnncA.L., Finnee а п W. Л., J. Am. Chem. Soc., 71, 298 (1949).
2. Putnam R. E., Harder R. J., Castle J. E., J. Am. Chem. Soc., 83
391 (1961).
3. KrespanC. G., McKusickB. C., CairnsT. L., J. Ain. Chem. Soc.,
83, 3428 (1961).
ГЕКСАФТОРФОСФОРНАЯ КИСЛОТА, HPF6, см. Арилдиазо-
ння гексафторфосфаты.
ГЕКСАХЛОРЦИКЛОПЕНТАДИЕН (1). Мол. вес 272,80, т. кип.
235—238=.
Г. реагирует с норборнадиеном (2) по схеме реакции Дильса —
Альдера с образованием аддукта (3), применяющегося в качестве
инсектицида и названного «альдрин» в честь К. Альдера. Этот реа-
гент является предшественником 5,5-дпметоксн-1,2,3,4-тетрахлор-
циклопентадиена (см).
(') (2)
ГЕКСАЭТИЛТРИАМИД ФОСФОРИСТОЙ КИСЛОТЫ,
l(C:H5)..NJ3P 11,21. Мол. вес 247,36, т. кип. 245 — 246 (120 —
122 /10 л/л;), /г*0 1,4750. (О получении и реакциях см. Гексаметил-
триамид фосфористой кислоты.)
1. М 1 с h а е I i s A., Ann., 326, 129 (1903).
2. S t u с b с С., L a n k e 1 m a H. P., J. Am. Chem. Soc., 78, 976 (1956).
196
ГИАМИН 1622 И ГИАМИН 3500. Эти четвертичные аммониевые
гермициды применяются для дезодорации лабораторий при раз-
брызгивании их разбавленных водных растворов:
СНз сн:1 сн3
СН3-С-СН,-С-^________
СН3 СН3 СН3
I намин 1Ь22. моногидрат. Мол. вес 466,09
сн3
С„Н,„ +, - \ + - СН., > (С1)
сн3
Гиамин 3300. Средний мол. вес 358
Эти соединения нетоксичны, но могут вызвать раздражение при
вдыхании из-за высокой поверхностной активности. 0,1 % -ный водный
раствор одного из этих соединений достаточен для уничтожения боль-
шинства запахов. Запах меркаптанов исчезает при действии более
концентрированного раствора. Для дезодорации пролитого меркап-
тана необходим 5%-ный раствор.
ГИДРАЗИН, HA’X'Hj. Мол. вес32,05, т. кип. 113,5\ уд. вес 1,01;
100%-иын гидрат гидразина H.NNH, Н,0 содержит 64% N,H4.
Исторические сведения. Описание методики получения сульфата
гидразйна приведено в качестве свидетельства упорства и мужества
химиков, основавших в 1921 г. «Organic Syntheses». Авторы этой
методики — Адамс, редактировавший первый том, и Браун. Проверял
методику Конант, редактор второго тома, где она была опублико-
вана, и Ханауэй. Основная реакция достаточно проста и заключает-
ся в окислении аммиака гипохлоритом с последующим выделением
продукта в виде сульфата:
2.ХН3 —-2Д Н2\\Н,3 • H,SO4
Готовя! раствор 300 г (7,5 моля) едкого натра в 1,5 л воды в колбе
емкостью 5 л, к нему прибавляют 1,5 ха льда и при охлаждении льдом
Пропускают ток хлора до привеса, соответствующего поглощению
3 молей. Раствор гипохлорита помещают в чашку для выпаривания
(36 см) п прибавляют 1,5 л водного аммиака, 900 мл воды и 375 мл
10%-його раствора желатины (в качестве «загустителя»); раствор
упаривают до одной трети первоначального объема. После охлажде-
ния «раствор фильтруют с отсасыванием сначала через вдвое сложен-
ное полотенце, а затем через один слой ткани, поверх которой поме-
щают обычную фильтровальную бумагу для удаления мелкодисперс-
ных твердых примесей, после чего раствор помещают в кристалли-
затор» ... (для превращения в сульфат). Сырой сульфат гидразина,
обычно получаемый с выходом 34—37%, после перекристаллизации
дает 19 г (12%) чистого препарата. В то время эта методика никому
197
не казалась трудной, на что указывает следующее оптимистическое
замечание: «Один человек может, выпаривая одновременно 6 чашек
гидразиновой смеси, провести за 9 час 20—25 опытов. Время для
выпаривания раствора, как указано в экспериментальной части,
при использовании четырехпламенной горелки Бунзена составляет
от 2 до 3 час».
Безводный гидразин. Безводный Г. можно получить при вы-
держивании 95%-ного Г. в течение ночи с 20 вес. % едкого кали.
Полученный гель фильтруют и перегоняют фильтрат без доступа
влаги |21.
Смесь 100%-ного гидрата гидразина с равным весовым количест-
вом гранулированного едкого натра рекомендуют кипятить в течение
2 час с последующей перегонкой в слабом токе азота, поступающем
через капилляр (Дэй и Уайтинг 12а I). Перегонка гидрата Г. на воз-
духе может привести к взрыву.
Реакции. См. также Азодикарбоновой кислоты диэтиловый
эфир; Диимид; Жирара реактив; Триэтиловый эфир аминотрикар-
боновой кислоты.
Производные гидразина. N.N'-Диметилгидразин можно по-
лучить метилированием М.М'-дпбензоильиого производного и
гидролизом [31.
о н н о
2С«Н,СОС| !, | | 'I (CH3O),SO2
HoNNHo------:---'CeH-,C—N—N—СС„Щ ----------2--►
66—'V 6 -1 6 5 36-93%
О СН3 СН3 О СНзСНз
Н I I ' |1 1-1CI | |
—> СВНЬС- N — N — СС„Н5 -уе-р-г HN —- NH
Тиле 141 описал метод получения монометилгидразнна, приведен-
ный в сб. «Синтезы органических препаратов» [51. Бензальазин,
получающийся в виде желтых игл при конденсации бензальдегида
H.NNH. (CHjO).SO.
2С,Н,СНО —:-----’ С()Н5СН X — N = СНС„Н,---—*
6 5 91—94% ’ ’ " ”
Беняальалив
OSO,OH н
зп.,о I
— СвН5СН -= X — \--.СНС6Н5 Г 2C(iH5CHC)^CH3XXH;412SO4-CH3OH
I •’ 1 • ’ %
сн3
с Г., в кипящем бензоле реагирует с диметилсульфатом с образо-
ванием четвертичной соли, которая выделяется в виде твердого
вещества. Вода разлагает эту соль на бензальдегид, сульфат метил-
гидразина и метанол. Бензальдегид и бензол отгоняют и после выпа-
ривания водного раствора получают сульфат метплгидразина.
Фенилмочевина дает с умеренным выходом 4-фенилсе.мнкарбазид
при нагревании на кипящей водяной бане с 42%-ным раствором Г.
в воде в течение 12 час |6|.
С6Н5ХНСОХН, -1 Н,\\Н, —-----С„Н-,ХНСОХ’НХН,
198
Восстановление по Вольфу — Кижнеру. Обзор литературы по
классической методике Вольфа— Кижнера опубликован Тоддом |7|.
Модифицированная методика Хуанг-Миплона |8| позволяет эф-
фективной с высоким выходом проводить эту реакцию в больших
масштабах при атмосферном давлении. Например, в случае 0-(//-фе-
ноксибепзоилНфонпоновой кислоты (исторические сведения см. |9|)
смесь 500 г (1,85 моля) кетокпслоты, 3.50 г КОН (3,2 же), 250 мл
85%-ного Г. (.3,6 же) и 2,5 л трпэтпленгликоля (или днэтилепглп-
коля) кипятят 1,5 час с обратным холодильником для получения
гидразона, после чего убирают обратный холодильник, и избыток
Г. и воду отгоняют, повышая температуру до начала разложения
гидразона (195—200). Смесь кипятят с обратным холодильником
еще 4 час, охлаждают, разбавляют 2,5 л
() 8Т"„ H.XXH., <3.6
КОН (3.2
ссшлссол
- о - '
.Мол ве< 2 70. 2/
воды и медленно выливают в 1,5 л 6 и. соляной кислоты. Выпавший
осадок светло-кремового цвета отделяют и сушат. Средний выход
препарата с т. пл. 64--6Б составляет 451 г (95%). Чистая у-(,ч-фе-
ноксифеинл)-.масляная кислота плавится при 71—72'.
Хуанг-Минлоп показал 110|, что стероидные кетогруипы в поло-
жениях С:|, С-, С, ., С17 и С.и довольно хорошо восстанавливаются
этим способом, ио Сц-кетогрупиа остается незатронутой. Два
11-кетостероида восстановили путем нагревания 0,5 -1 г препарата
со свежеполученным метилатом натрия в метаноле и безвод-
ным Г. в запаянной ампуле при 200' (Моффет н Хантер 1111). Бар-
тон показал [ 121, что можно восстановить обе карбонильные группы
в 7,11 -дпкетолапостанплацетате безводным Г. при повышенной
температуре. Раствор натрия в ди этилен гл и коле нагревают до 180',
перегоняют в этот раствор Г., который предварительно кипятят
в течение 3 час над едким натром. Перегонку Г. прекращают, когда
смесьпачнетспокойно кипеть при 180 . Раствор охлаждают, прибав-
ляют дикетон, кипятят в течение ночи, отгоняют Г., повышая темпе-
ратуру до 210' , и раствор кипятят при этой температуре еще сутки.
199
Нагата и Итадзаки [131 разработали методику восстановления
пространственно затрудненных 11-кетостероидов, не требующую
применения безводного Г. Кетон (1 моль) нагревают с 66 молями
гидрата Г., 8 молями дихлоргидрата Г. и 150 молями триэтиленглп-
коля при 130 в течение 2,5 час. Гранулированный КОН (22 моля)
прибавляют к реакционной смеси, температуру постепенно повы-
шают до 210° (отгонка Г. и воды) и выдерживают при этой темпера-
туре 2,5 час, выход 9О°6.
Одна стадия в синтезе морфина по Гэйтсу и Чуди [141 включает
удаление кетогруппы в промежуточном соединении, имеющем две
фенольные метоксильные группы. В обычных условиях восстанов-
ления по Хуанг-Минлону происходит интенсивное деметилирование,
и высокий выход продукта реакции достигается при восстановлении
в течение 1 час при 150—155’.
Для получения гидразонов Шёнберг [151 рекомендует кипятить
эквивалентные количества кетона и Г. в w-бутаноле (т. кип. 117,7 )
в течение 2 час. Применение этанола требует более длительного
кипячения, а использование этилен- или пропиленгликоля может
привести к побочным реакциям. Так, при использовании гликолей
для превращения флуоренона в соответствующий гидразон Бальтцли
и сотр. 1161 наблюдали образование значительного количества флу-
орена (необычно легкое восстановление по Вольфу — Кижнеру).
При кипячении реагентов в «-бутаноле в течение 4 час был получен
гидразон с выходом 67?». Однако наилучший метод синтеза гидра-
зонов включает реакцию кетона с N, N'-диметилгидразином (см.)
с последующей обменной реакцией с Г.
Температуру восстановления можно значительно снизить и
таким образом исключить некоторые побочные реакции, если при-
менять сильное основание — шре/«-бутилат калия в кипящем то-
луоле (Хенбест [171). В безводных условиях гидразон (1) дает
СН3 \\’Н2 сн:!
Сн3—С —ССН., —(СН,),сокшо^ сн (^„снхн.-у \
| - "----------/ 3 | - /--------------------------/
СН:! CH3Z сн3 СН/
(I) (2)
с высоким выходом продукт восстановления (2), тогда как в обыч-
ных условиях реакции Вольфа — Кижнера около 50% исходного
вещества подвергается 0-элиминированпю с образованием 3,3-
диметилбутена-1. Семикарбазон Д4-холестенона-3 при кипячении
81%
с mpem-бутилатом калия в толуоле до прекращения выделения газа
(газовая бюретка) реагирует очень медленно, но дает почти чистый
200
Д‘-холестен с высоким выходом. В случае гидразона восстановление
завершается за 6 час, но выход составляет только 65%.
Модифицированное восстановление по Хуанг-Миилону было
с успехом применено для превращения метилового эфира холевой
кислоты в лптохолевую кислоту без очистки промежуточных сое-
динении 1181. Экваториальную З-окспгруппу избирательно сукпино-
нлпровали, гидроксильные группы при С;1 и С12 окисляли и метило-
вый эфир За-сукцинокси-7,12-дикетохолановой кислоты нагревали с
85%-иым Г. и этиленгликолем при 100 в течение 1 час. Получаю-
щийся прозрачный раствор охлаждали и прибавляли порциями гра-
нулированный КОН при комнатной температуре для связывания
присутствующей воды. Смесь нагревали при 200 в течение 2 час,
охлаждали и отделяли выпавшую калиевую соль центрифугирова-
нием. Раствор этой соли в воде подкисляли и получали лптохолевую
кислоту с выходом 95% в расчете на метилхолат. Восстановление
дпкетона при обычных условиях реакции Хуанг-Минлона дает
очень загрязненный продукт с низким выходом, так как этот дике-
тон неустойчив в сильнощелочной среде.
О восстановлении по Вольфу — Кпжперу при комнатной тем-
пературе см. Дпметнлсульфоксид, эффекты растворителя.
Удобная модификация методики Хуанг-Мннлона предложена
Гарднером 1191 для последней стадии изящного синтеза пимелиновой
кислоты. Фурфурол в присутствии пиридина (следы пиперидина)
конденсируется с малоновой кислотой с образованием фурплакри-
ловой кислоты (2), которая при обработке этанолом и хлористым
водородом дает дпэтиловый эфир у-кетоппмелпновой кислоты (6).
Эта реакция, описанная в патентной литературе 1201, включает
(см. схему) 1,-^присоединение этанола с образованием соединения
(3), миграцию двойной связи с образованием соединения (4) и затем
соединения (5) и расщепление никла с образованием диэфира (6).
При восстановлении кетодиэфира (6) по методике Хуанг-Минлона
Гарднер столкнулся с затруднением, связанным с продолжительной
экстракцией эфиром кислой реакционной смеси, во время которой
201
растворители этиленгликоль и диэтиленгликоль частично переходят
сно
сн;(согн)
86%
НС1
EtOH
W (5)
СН2 СН2 (HOCH.С.Н
Н,О I I Н.\ХН„ КОН
—-> ЕЮС CCH.CH,COOEt---------------- НООС(СН.),СООН
75% || ||
О о
<6) (7;
в эфирный экстракт. Поэтому в качестве растворителя использовали
триэтаноламин.Смесь 53,8 г (0,234 моля) диэфира (6),230 мл 95%-ного
триэтаноламина, 0,91 моля 85по-ного КОН н 37 мл (0,65 моля)
85%-ного гидрата Г. кипятят 2,5час. Когда температура повысится
до 195' заменяют водяной обратный холодильник па воздушный
и кипятят еще 2 час. Охлажденною смесь обрабатывают 450 мл
кони, соляной кислоты и непрерывно экстрагируют эфиром (суспен-
дированные соли остаются в водной фазе н не мешают этой операции).
После однократной кристаллизации из конц. соляной кислоты
О О О
z С(СН2)8С ч J1. . о о
I I | | ±12.± \аО2С(СН2)5С(СН.2)8С(СНг)5СО2\а —>
\/ (3) (II
H2NNH.. (HOC.H.,CH,),N. кон
69 — 72». „ iij (li
HOsC(CH.,),0CO2H
202
(несколько раз отделяют кристаллы) получают нимелиновую кис-
лоте (т. пл. 103,5-105 ) практически с количественным выходом.
Триэтаноламин был также использован для синтеза эйкозанди-
карбоновой кислоты (5)(см. схему на стр. 202). Конденсация цикло-
гексанона с морфолином дает №-1-циклогексеп11лморфолнн (1) 1211,
ацилирование этого енамина хлораигидрпдом себацнповой кислоты
приводит к дикетону (2) |221. Кислотный гидролиз бмс-енамина (2) и
щелочное расщепление б//с-Р-дпкетона (3) дают соль дикетодикислоты
(4), которая восстанавливается в эйкозаидикарбоиовую кислоту (5).
Восстановление, см. обзор 123]. Хлоргидрат пиридоксина можно
восстановить с высоким выходом в 4-дезокснпиридокспн простым
кипячением этой соли с 95?о-ным Г. в течение 18 час [24]. Оксиме-
тильная группа при С.-, не затрагивается.
Г. без добавки и с добавкой щелочи десульфирует циклические
и ациклические этилентпокетали 1251. Субстрат (1 вес.ч.)в 8—20
об.ч. дпэтилен- или трпэтиленгликоля нагревают с 3 — 5 об.ч.
64?о-ного Г. («гидразин гидрат») и 1,25—2,5 вес. ч. КОН в прибо-
S— СН, 1 %-п XXII.. кон
( - носи/.н о, н..< н .он
S-СН., i,u
ре, имеющем приспособление для собирания выделяющегося газа
над водой, до начала выделения газа (90—135 ), после чего
постепенно повышают температуру до 155—190 , чтобы поддер-
живать выделение газа. Время реакции варьируется от 30 мин
до 3 час. Хотя добавка щелочи и не является необходимой, она зна-
чительно понижает температуру реакции,
Вартон 1261 предложил новую реакцию восстановления Г.<х,Р-
эпоксикетонов до аллиловых спиртов. Если суспензию стероида
(Ч
(1) быстро нагреть с избытком 64%-ного Г. на кипящей водяной
бане, а затем кипятить с обратным холодильником, то выделяется
азот и с хорошим выходом получается аллиловый спирт (2). Стеро-
идный а,р-эпоксикетон (3) реагирует аналогично, но с меньшим вы-
203
ходом [271.
Восстановление гидразином по Вартону было использовано
в двух лабораториях для синтеза 1-оксистероидов (1) 1281 и (1а)
[291. Эта реакция оказалась стереоспецифичной (Клайн и Охлофф
1301). Так, энантиомерные эпоксиды пиперитоиа (2) н (4) дают исклю-
чительно энантиомерные аллиловые спирты (3) и (5).
(2) (3)
2<х-Бромхолестанон-3 (6) восстанавливается Г. в А'-холестен
(9), вероятно, через промежуточное образование алкенплдпимнда
(8) 130 al. Раствор бромкетона в циклогексене прибавляют при пере-
мешивании к кипящей смеси гидрата Г., ацетата калия и циклогек-
сена. В качестве растворителя используют циклогексен, чтобы
предотвратить восстановление продукта реакции днпмидом, если
последний образуется.
Гидразин и металлический катализатор. В присутствии палладия
на древесном угле гидразин с высоким выходом восстанавливает
ароматические нптросоедннення до аминов (Пьетра 1311). Так, на-
гревают при перемешивании суспензию 30 г 2-нитрофлуорена
204
в 250 мл 95%-ного этанола до 50°, прибавляют 0,1 г 1О°о Pd/C (смо-
ченного спиртом) п затем по каплям в течение 30 мин добавляют
15 мл 64%-ного Г. |32|. Снова прибавляют 0,1 г увлажненного ката-
лизатора п смесь нагревают до слабого кипения. За 1 час нитрофлуо-
рен растворяется, н реакционная смесь становится почти бесцвет-
ной. После обработки реакционной смеси получают чистый амин
в виде бесцветных кристаллов.
Этот метод дает прекрасные результаты и применяется для
получения толуидинов, аминодифенилов, фенилеидиаминов, амино-
фенолов, /г-аминобензойноп кислоты 131,331, а также для восстанов-
ления полициклических ароматических нптросоедпненип 1341. При
восстановлении 1331 азокси- пли азобензола получается гидразобен-
зол с выходом 80—90%.
До опубликования работы Пьетра (1955 г.) Ферст [351 применил
никель Ренея в качестве катализатора восстановления ароматиче-
ских нитросоедипений гидразином. Тарбелл 1361 с успехом применил
этот метод для нитростприлтрополона, для которого другие восстано-
вители (гидроокисьжелеза (II), су.тьфпдаммония, гидросульфит нат-
рия, водород и платина в дноксаие)оказались неэффективными.Этот
метод был также использован в ряду флуорена для восстановления
пптрогрупп, не затрагивающего карбонильной или трифторацетил-
амнногруппы [371. Галогеннитробензолы гладко восстанавливаются
в галогенанилины [38]. о-Нитробепзонитрил дает с высоким выходом
амид антраниловой кислоты [391.
Стероидные азиридины синтезировали путем восстановления
мезилатов вицинальных азидоспиртов гидратом Г. и никелем Ренея
(Понсольд [401).
Подробный обзор данных по этой реакции приводит к выводу
(Ферст п сотр. 1411), что Ni, Pd, Pt и Ru одинаково пригодны и этот
метод восстановления часто лучше прямого гидрирования; кроме
того, он не требует специальной аппаратуры.
Было показано, (Хжельт |42|), что а, ^-ненасыщенные кислоты
можно восстанавливать Г. и никелем Ренея в щелочной среде.Так,
раствор шронс-коричной кислоты в небольшом избытке водного
едкого натра обрабатывают небольшим количеством никеля
Ренея и при взбалтывании и нагревании до 90' в течение 2 час
205
Прибавляют 2 моля Г.
N i
С„Н-,СН ( НСО.,Н 2Н.,\\Н.,—г->С,|Н-1СН.,СН.,С().,Н
S. > ()
Как было указано выше, галогенинтробензолы гладко восстанав-
ливаются Г. п никелем Ренея до галогенапплииов (.Чеггеттер н Браун
1381); однако с палладием в качестве катализатора наблюдается зна-
чительное дегалогенирование. Аналогичные результаты, полученные
Мосби 1431, стимулировали исследования 1441, которые показали,
что взаимодействие с H.,NNH2—Pci (в С.Н-.ОН или СН:|ОСН.,ОН)
является универсальным методом дегалогенирования ароматиче-
ских галогенпроизводных (присутствующие в соединении нптро-
гр у и пы восс та 11 а вл 11 ва юте я).
Синтез гетероциклов. 3,5-Дпметилппразол получают из ацетил-
ацетона и сульфата Г. в водном растворе щелочи (реакция с гидра-
том Г. иногда протекает очень бурно) 1451.
ОН—ССН3 боди. NaOH
сн3с о + h2nnh2' h2so4 ---------У >
ОН ‘ 73-77%
5-Ам11но-2,3-диг11дрофталаз1шдпо11-1,4 (3) (известный под назва-
нием люминола, поскольку окисление этого соединения сопровож-
дается сильной хемилюминесценцией) может быть получен в течение
25 мин следующим образом 146!. Смесь I г З-иптрофталевой кислоты
и 2 мл 8%-ного водного раствора Г. нагревают па открытом пламени
в пробирке (20'' 150 мм) до растворения твердого вещества и при-
бавляют 3 мл трнэтнлепглнколя. Раствор сильно кипятят, чтобы
отогнать избыток воды, и затем нагревают 2 мин при 215—220 для
проведения циклизации. Желтое нитросоеднненпе (2) выделяют при
охлаждении и восстанавливают гидросульфитом натрия в люминол
(3). Разбавленный раствор (3) в щелочи выливают в делительную
воронку п прибавляют водный раствор перекиси водорода и
феррицианида калия; эта смесь даег хемилюминесценцию, хорошо
заметную в темноте.
\О.,О хн,о
! ' 1 I
// - //''
ХН„ 215-22U- | I ХН Xa.s.o, I | ХН
I ’---------- ! — — । I I
хн. । i| \н ; и \н
Ж х / 'X .
хо.
.А. ш,н
!!
ХСО2Н
(1)
(2) (3)
О синтезе индазола см. Муравьиной кислоты этиловый эфир.
Другие реакции. Получение гидразидов из кислот можно про-
иллюстрировать на примере реакции н-толуоле\льфохлорнда с гид-
206
ратом Г. в ТГФ 1471:
СН.,-/ х'— SCKG1 - 2HAXH,----------СН,—z/ ч —SO„XHXH24-
—' SO",, XXX— ‘
Х.,Н,НС1
Смесь разделяется на два слоя; верхний тетрагидрофурановый слой
промывают раствором хлористого натрия, сушат, фильтруют и
высаживают продукт петролейпым эфиром. Использование для полу-
чения гидразидов кислот их сложных эфиров показано на примере
синтеза дихлоргидрата путресцина 1481.
(СН2)4(СО2(ДН5), ——(СН2),(СОХНХН,)2 —-% (СН2),(СО\3),
11Д-) HCI т -
— (СН.2)1(\--С - -—_пЧСН2)4(\Н?,С1)2
(общи и)
При реакции циклогексанона с цианистым натрием и сульфатом
Г. в водном растворе образуются in situ как цианистый водород, так
и Г. 1491.
---. Н.О /------С\ ХС /-----------\
2 (> 2\,Ч .\ H.WII H..S-. р--- - — v_____N_' )
х " (►7 • 7 0л - ' X /
X--- „ ---- ! !
Н Н
Примером общего метода синтеза ацетиленов может служить
превращение бензила через дшшдразон (и не выделяемый диазид)
в дифенилацетилен 1501.
.•-Pi ОН НцО
С6Н-,С()СОС,.Щ 2Н..ХХН,------ -С,;ЩС—СС„Н-,-----<-
- Ы- ,Ю"„ ' /
н,хх ххн,
С,;ЩС- < д ;
-- \ \ • ; - - 4 . с.н,'. ссщ.,
: ; <17-тл\
В пептидном синтезе Г. применяется для удаления N-защитной
фталоильной группы 1511. Защищенный пептид кипятят с Г. в спирте
в течение 2 час или выдерживают 1—‘2 дня с водным или спиртовым
раствором Г. 1521.
Аналогичное расщепление при действии Г. применяют для син-
теза итрс/п-бути.тамина исходя из /прста-бутанола и мочевины через
промежуточное образование /прс/и-бутилмочевины (531. Если это
207
соединение нагревать с фталевым ангидридом, то выделяется дву-
окись углерода и аммиак с образованием /ире/н-бутнлфталимида.
Это соединение расщепляют Г., добавляют соляную кислоту, от-
НО 240°
, , ,, H,SO, , „ , „J Н..,. Фталевый ангидрид
(СН3)3СОН + H3NCONH, НгД^<> (CH3)3CN CNH3 ----------------у—
31-33%
72-76%
+ NH3 + (СН3)3СЦ
H2NNHS; HC^ (СНз)зСКН3.НС1ЛХ
72-88% Ny
О
фнльтровывают фталгидразид и из фильтрата выделяют хлоргид-
рат /ире/и-бутпламина.
Цнклогександнон-1,3 (в енольной форме) реагирует с избытком
Г. в этаноле в присутствии воздуха с образованием нпридазпна (3),
который кристаллизуется в виде светло-желтых игл [54]. Если про-
водить реакцию в отсутствие кислорода, то (3) не образуется, а дан-
ные ЯМР указывают на промежуточное образование дпазпна (2).
Это соединение очень нестабильно, так как атомы водорода внутрен-
них метиленовых групп находятся на расстоянии не более 1,2А.
(2) (3)
Этим п объясняется легкость его окисления кислородом воздуха
в ароматический ииридазпп (3).
Реакция с бромацетофеноном 1551. Г. реагируете бромацетофе-
ноном (1) в этаноле при 60J с образованием моногндразона 2-фенил-
глиоксаля (2) по следующей схеме:
О О
II I
С.6Н5ССН2Вг ; ЗН.,ХХН2 > С6Н5ССН = ХХН2 ; Х2И5Вг ) 2Х’Н3
(I) (-’)
Выделение фенацплгпдразнна (3) при проведении реакции ниже 53
заставляет предположить следующий механизм:
(1) + 2H.NNH, -N2HjBr Свн5 о 4 (А7 ► 1 СН2Х? н ^6^5 с6н5 о HZ^XH
\н, [i нх н /NH2
(3) 1 н (4)
208
Соединение (2) при дегидрировании гидратом двуокиси марганца
дает диазометилкетон (5), а при обработке азотистой кислотой —
фен ил глиоксаль (6).
Расщепление — NCO2CH;i. Карбметоксильная группа при атоме
азота в гетероциклах отщепляется при кипячении с гидратом Г.
(Альдер и Никлас 1561). Эта реакция была применена 1571 для син-
теза 11,12-диазастероидов, как показано на схеме:
1. А д а м с Р., Браун Б. К., «Синтезы органических препаратов», ИЛ, М.,
1949, сб. 1, сгр. 158.
2. Taborsky R. G., J. Org. Chem., 26, 596 (1961).
2а. Day А. С., W h i t i п g At. C., procedure submitted to Org. Syn.
3. Хатт X., «Синтезы органических препаратов», ИЛ, А1., 1949, сб. 2, стр. 202.
4 Т li i е 1 е J., Ann., 376, 244 (1910).
5. X а т т X., «Синтезы органических препаратов», ИЛ, М., 1949, сб. 2, стр. 199.
6. У н л е р А. С., «Синтезы органических препаратов», ИЛ, М., 1949, сб. 1,
стр. 438.
7. Tod d D., Organic Reactions, 4, 378 (1948) *.
8. Huang - M i и 1 о n, J. Am. Chem. Soc., 68, 2487 (1946).
9. Fie s e r L. F., F i e s e r Al., «Topics in Organic Chemistry», 258, Heath
D. C„ 1963.
10. Hua n g -Minlo П, J. Am. Chetn. Soc., 71, 3301 (1949).
11. M о f f e t t R. B„ HunterJ. H., J. Am. Chem. Soc., 73, 1973 (1951).
12. Ba r to n D. H. R., 1 v e s D, A. J., T h о m a s B. R., J. Chem. Soc., 1955,
2056.
13. Nagata W., I t a z a k i H., Chem. Ind., 1964, 1194.
14. Gates Al., T s c h u d i G., .1. Ain, Chem. Soc., 78, 1380 (1956).
* См. также «Реакции и методы исследования органических соединений»,
Госхимиздат, М.— Л., 1951, том 1, стр. 7.— Прим, иерее.
209
15. S c h c n b е г g A., Fateen A. E. K.. Samtnour A. E. M. A., .1. Am.
Chem. Soc., 79, 6020 (1957).
16. Ba I Iz I у R„ M eli t a N. B., R u s s cl P. В., В г о о к s R. Е., G г i v-
s к у E. AL, S t e i n b e r g G. A., J. Org. Chem., 26, 3669 (1961).
17. Grundon M. F., Ilenbcst H. B., Scott M. D., J. Chem. Soc., 1963,
1855.
18. Sarel S., Yanuka 1’., .1. Org. Chem., 24, 2018 (1959).
19. G ardncr P. D., Rand L., H a v n e s G. R., J. Am. Chem. Soc., 78,
3425 (1956).
20. S i n g I c t о n F. G., пат. C111A 2436532 |C. A., 42. 5048 (1946)1.
21. 11 ii n i g S., L li с к e E., В г e п п i n g e r \V., Org. Syn., 41, 65 (1961).
22. И ii n i g S., L ii с к e E., В r v n n i n g e r W.. Org. Syn., 43, 34 (1963).
23. F u r s t A., Berio R. C., Hooto n S., Cheni. Revs., 65, 51 (1965).
24. T a b о r s к у R. G., J. Org. Chem., 26, 596 (1961).
25. Georgian V., HarrissonR., G u b i s c h .\., J. Am. Chem. Soc., 81,
5834 (1959).
26. W h a r t о n P. S., В о h 1 e n D. H., J. Org. Chem , 26, 3615 (1961); \V h a r-
t о и P. S.. ibid., 26, 4781 (1961).
27. hi ti a n g - Al i и 1 <> n, Chung- T u n g s h u n. Tetrahedron Letters, 1961,
666: Djerassi C.. Williams D FL. Berkoz B., .1. Org. Chem.,
27, 2205 (1962); Djerassi C.. von M u t z e n b e с к e r G . F a j к о s
.1., W i I I i a in s D. H., В u ci z i к i e w i e z H., .1. Ain. Chem. Soc., 87, 817
(1965): see also 43 e e n W. R., D о d s о n R. AL. J. Org. Chem., 29, 1142 (1964).
28. D j e r a s s i C., W i 1 1 i a m s D. I L, Berko /, B., .1 Org. Chem27, 2205
(1962).
29. ,\ а к a n о 1'., H a s c g a vv a AL. Bull. Chem. Pharin., 12, 9/1 (1964).
30. Klei n E., О h I <> f f G., Tetrahedron, 19, 1091 (1963).
30a. W hart о n P. S., D u n n у S.. 1\ r e b s L. S., .1. Org. Chem., 29, 958 (1964).
31. Pietra S.. Ann. Chim. (Route), 45. 850 (195.51.
32. Bavin P. AL G.. Org. Syn., 40, .5 (I960).
33. В a v i n P. AL G., Can. J. Chem., 36, 238 (1958).
34. D e w a r At. J . S., At о 1 e T., ,1. Cheni. Soc., 1956, 2.556.
35. Balco m D.. Furst A.. .1. Am. Chem. Soc., 75, 4334 (1953); Furst A.,
At о о r c R. Е., ibid., 79, 5492 (1957); At о о re R. E., Furst A.. J . Org.
Chem., 23, 1504 (1958).
36. Tarbell D. S., S m i t h R. F., Bockclh e i d e V., J. Am. Chem. Soc.,
76, 2470 (1954).
37. F I e t c li e r T. L., N amkung At. .1., .1. Org. Chem., 23, 680 (1958).
38. L e g g e t t е г В. E., Bro w n R. K., Can. J. Cheni., 38, 2363 (I960).
39. Butler K-, Part r i it g e Al. W., .1. Cheni. Soc., 1959, 2396.
40. P о n s о 1 d K., Chem. Ber., 97, 3524 (1964).
41. Furst A., Berio R. С., H no to n S.. Chem. Revs., 65, 51 (1965).
42. II j e I t e X. S., Cheni. Scand., 15, 1200 (19(d).
43. Alosb v \V. L., J. Org. Cheni.. 24. 421 (1959).
44. Mosby \V. L., Chem. Ind.. 1959, 1348.
45. У a ii л и P., X e к с н e p FL, «Синтезы органических препаратов», ИЛ, Al.,
1953, сб. 4, стр. 189.
46. Org. Expts., Chapt. 46; Редема н К. Т., Редеман К. Е., «Синтезы
органических препаратов», ИЛ, AL, 1953, сб. 4, стр. 40.
47. Erie d m a n L., L i t 1 e R. L.. R e i c h I e W. R.. Org. Syn., 40, 93 (1960),
48. Смит II., «Синтезы органических препаратов», ИЛ, Al.. 1958, сб. 8, стр. 12.
49. Овсрберджс р К., X у а н г П а о - г у и г, Б с р е н б a v м А!.. «Син-
тезы органических препаратов», 1451, AL, 1953, сб. 4, cip. 244.
50. С о р е. А. С., Smit h D. S., Cotter R. .1.. Org. Svn., Coll. Vol., 1, 377
(1963).
51. 1 n g H. R., Al a n s k e R. F., J. Chem. Soc., 1926, 2348.
52. Б у а с с о и P. А., «Успехи органической химии», изд-во «Мир», At., 1966,
сб. 3, стр. 158.
210
53. Смит Л., Эмерсон О., «Синтезы органических препаратов», М., ЙЛ,
сб. 4, стр. 105.
54. S t i 1 I е .1. К., Е г t z R., .1. Am. Chem. Soc., 86, 661 (1964).
55. Hauptmann S., Klue e M., S e i d i g K.-D., Wilde H., Angew.
Chem., Internal. Ed., 4, 688 (1965).
56. Aider K., Niklas H., Ann., 585, 97 (1954).
57. Willems A. G. M., v. EckR.R., Pandit U. K., HuismanH. O.,
Tetrahedron Letters, 1966, 81.
ГИДРАЗИНА АЦЕТАТ.
Пептидные синтезы. Г. а.— эффективный реагент для удаления
защитной N-фталоильной группы в нейтральной среде и поэтому
применяется в синтезе пептидов, неустойчивых в щелочной среде,
например а-.меланотроиина 11]. Это соединение было получено с вы-
О
V “ _____
Ac —|Se г — Туг — Met —Gin —His — Phe —Arg — Try — Gly —L-ya—Pro — Vai f-NHa
h2nnh2|ch3co2h
i___________L—_______________________________
Ac —|Ser — Tyr — Met — Gl'i — His — Phe—Arg — Try — Gly —Lys — Pro— Vai N H2
соким выходом при нагревании 440 лк? N-фталопльного производного
с 20 л/д 2 /И раствора Г. а. в метаноле при 50“ в течение 15 час.
I. S с h w у z с г R., С о s t о р а п ,т g i о t i s A., Helv. Chini. Acta, 46, 870 (1963).
ГИДРАЗИНА СУЛЬФАТ. H Л'Х’Н ,-H2SO,. Мол. вес 130,12.
Анализ пептидов. По методу Акабори для определения С-кон-
иевой аминокислоты пептид нагревают с безводным Г. при 100—
120 в течение 8 час\ при этом аминокислоты, находящиеся внутри
цепи, образуют гидразиды, а концевая аминокислота получается
в свободном виде. К сожалению, некоторые аминокислоты разла-
R R' R R'
i I HASH, I I
— \HCHC.ONHCHCO.,H--------- — NHCHCONHNH., - HACHCO..H
гаются в этих довольно жестких условиях. Брэдбери 111 показал,
чтос Г. с. эта реакция намного быстрее и ее можно провести при 60".
1. В; а <1 1> и г v J. IL. Bioclum. .1., 68, 475 (1958).
ГИДРОКСИЛАМИН-О-СУЛЬФОКИСЛОТА, NH3OSO7. Мол. вес
113,09, т. ил. 210“ (с разя.).
Г. с, гигроскопична, растворима на холоду в воде и метаноле,
медленно разлагается в воде при 25 .
Г. с. получают 111 добавлением сухого бис-(гидроксиламин)-суль-
фата к 30%-ному олеуму при перемешивании при О'. Продукт осаж-
дают эфиром (добавление ио каплям), промывают семь раз эфиром
211
для удаления серной кислоты и высушивают в вакууме. Чистота
(XH3OH).,SO;“ -|-2H„SO4SO3_____> 2XH3OSO7 -y3H.,SO4
продукта (92—98%) определяется реакцией с избытком иодид-иона
и титрованием выделяющегося иода раствором тиосульфата натрия.
Дезаминирование. Г. с. применяют для дезаминирования пер-
вичных аминов через метансульфамид или ароматические сульфа-
миды |2|. Например, N-бензилметаисульфамид обрабатывают
в водно-спиртовой щелочи 15—25 же реагента, неустойчивого в ще-
лочной среде.
+
NHaOSOj
C6H5CH.,XHSO.,CH3------CeH5CH.,XSO»CH3 —>
- - -H..SO, ь J -|
хн2
-------> СсН;СН.2Х = хн —> с6н5сн3
-CH3SO..H —N;
Промежуточное образование диимидов. Г. с. с циклогексаноном
в щелочной среде дает 1,1-диокспазоциклогексап (3) 131, который
быстро разлагается даже при комнатной температуре с образова-
нием циклогексанона, азота и гидразина. Промежуточное образова-
ние диимида при разложении следует из того, что в присутствии
хинона или азобензола наблюдается образование гидрохинона или
гидразобензола. Для получения диимида в щелочном растворе
можно использовать непосредственно Г. с. [41:
2XH3OSO3-J-4OH- -* H\^\H-' 2SO2~
Например, этилен, циклогексен, фумаровая и коричная кислоты
восстанавливаются при добавлении Г. с. к их водно-щелочным или
спиртово-щелочным растворам.
Аминирование. Г. с. применяют также для аминирования аминов
при получении несимметрично замещенных гидразинов 151. Напри-
мер, 1-аминопирпдииийиодид получают добавлением 0,3 моля
пиридина к свежеприготовленному раствору 0,1 моля Г. с. в холод-
ной воде. Смесь нагревают в течение 20 мин на кипящей водяной
____ NH3OSO3 __________ _ ,Н1 ____ J_
С % у----------- // %Х —ХН-------------- " %Х — NHoI-
4----/ K_>CO;t 4--------/' 63 — 72% ----
бане, охлаждают и обрабатывают при перемешивании 0,1 моля кар-
боната калия. Воду и избыток пиридина удаляют на роторном испа-
212
рителе в вакууме, а органическое вещество отделяют от сульфата
калия экстракцией этанолом. После добавления 0,1 моля подисто-
водородной кислоты и охлаждения до —20: Паминопиридпнийиодид
отделяют и перекристаллизовывают из абсолютного этанола. Этот
метод применим для первичных, вторичных и третичных аминов;
выходы колеблются от 30 до 80%.
Заслуживает внимания N-аминированпе бензотриазола (1)
в 1-аминобензотриазол (2) 161, соединение, особенно интересное как
предшественник дегидробензола (см. Свинца тетраацетат, различ-
ные реакции окисления).
I I
и nh2
(О <2>
Попытки использовать Г. с. для превращения карбоновых кис-
лот в амиды оказались безуспешными 171. В присутствии хлористого
алюминия (2 же) Г. с. аминирует ароматические углеводороды
с очень низкими выходами: бензол -«-анилин (28%), толуол—толуи-
дин (50?о), о-, л;-, ii- соответственно 51 : 13 : 36 [81.
Браун 191 показал, что с алкилборанами Г. с. легко образует
соответствующие амины, например:
СН3(СН2)3СН = СНг —/СН3(СН2)5СН2СН2;3В
64% (общий)
CH3(CH2)5CH2CH2NH2
Г. с. растворяется в диглиме и в этом растворителе превращает
органобораны, полученные как из пространственно незатруднен-
ных, так и затрудненных олефинов, в соответствующие амины,
например (Браун и сотр. 19а|):
Бензизоксазолы. Г. с. используют в обычном синтезе бенз-
изоксазолов из производных салицилового альдегида [101. Салицило-
вый альдегид в эфирном растворе обрабатывают избытком реагента
и безводным сульфатом натрия. Отделяющийся через несколько
минут оксимсульфонат натрия циклизуется под действием бикарбо-
213
ната натрйй.
и' ' NH,OSO7-Na.SO, и ^NOSOJ NaHCO3
I II 7,77; ” I I! '.-5% I !| N
ОН ОН 1Ха- 'о"
Диазиридины и диазирины. Г. с. и аммиак реагируют с цикло-
гексаноном с образованием 3,3-пентаметилендиазиридина, который
можно окислить окисью серебра в 3,3-пентаметилендиазирин 1111.
Этот жидкий продукт перегоняют, пользуясь защитным экраном,
и для предохранения от разложения хранят в эфирном растворе
в холодильнике.
----ч 0-10-
< ^=();-15н. води. H2\OSO3H------------►
4 00 ли 1 моль b 1 — /
Т. пл. 104-107
2-Оксициклогексанон с Г. с. и аммиаком дает диазиридин (выход
45%), окисление которого окисью серебра приводит к диазирину
(выход 70%) [121.
1. М a t s п g u m a H. J., Audriet h L. F., Inorg. Syn, 5, 122 (1957).
2. N i с k о n Л.., S i n z A., J. Ani. Chem. Soc., 82, 753 (I960); N i с k о n A.,
Hill A. S., ibid., 86, 1153 (1964).
3. Schmitz E., О h m e R., Angew. Chem., Internal. Ed., 2, 157 (1963); Chem.
Ber, 97, 2521 (1964).
4. S c h m i t z E., О h m e R., SchrammS., Angew. Chem., 73 , 807 (1961);
Appel R., Biichner W., ibid., 73, 807 (1961); Ann., 654, 1 (1962).
5. G о s I R., Meuwsen A., Org. Syn., 43, 1 (1963).
6. C ampbell C. D., R e e s C. \V., Chem. Comm., (1965) 192; see also
R ees C. W., Storr R. C„ ibid., 1965, 193.
7. BachmanG. B., Goldmacher J. E., J. Org. Chem., 29, 2576 (1964).
8. К о v a c i с P., Bennett R. P., J. Am. Chem. Soc., 83, 221 (1961); К o-
v a c i cP., Benue t t R. P., Foo t e J. L., ibid., 84, 759 (1962).
9. Bro w n H. С., Heydkamp W. R., Breuer E., Murph у \V. S.,
J. Am. Chem. Soc., 86, 3565 (1964).
9a. R a t h k e M. W., 1 non e X., Varma K. R., Brown H. C., J. Chem.
Am. Soc., 88, 2870 (1966).
10. К e m p D. S., WoodwardR. B., Tetrahedron, 21, 3019 (1965).
11. A b e n d г о t h H. J., Angew. Chem., 73, 69 (1961); Schmitz E.,
О h m e R., Schmid t R ,-D., Chem. Ber., 95, 2714 (1962); Schmitz E.,
О h m e R., Org. Syn., 45, 83 (1965).
12. SchniitzE., Stark A., Horig C., Chem. Ber., 98, 2509 (1965).
ГИДРОКСИЛАМИНА СУЛЬФАТ, HONH2 1 2H ,SO4. Мол. вес
82,07.
Реакция 1,3-дикарбоннльных соединений с гидроксиламином,
обычно используемым в виде хлоргидрата, широко применяется для
синтеза изоксазолов |1|. Реакцию обычно проводят в водном спирте,
214
иногда в присутствии основания.
H.NOH г-
с6н,сосн.,сосн, ——< С0Н5С—сн,сосн3
хон
-и.о
— - С„Н5С—сн
X С - сн3
о
При получении 3,5-диметилизоксазола вместо хлоргндрата пред-
почитают использовать более дешевый Г. с. |2|. Смесь ацетилаце-
тона, Г. с. и карбоната калия нагревают 1,5 час с обратным .холо-
дильником на кипящей водяной бане до отрицательной пробы
с FeCl3.
сн,ссн,ссн,
и и
о о
H,NOH
—---->
74-84%
li Т
N------О
1. Q ii i I i с о Л., in Weis s b е г с с r's Л. <Tlie Chemistry of Heterocyclic Com-
pounds», 17, 6 (1962).
2. Miller С. E., procedure submitted to 0гц. S\n.
ГИДРОКСИЛАМИНА ХЛОРГИДРАТ, HONH.HCI. Мол. вес
69,50, т. пл. 151 , в 100 г воды (17 ) растворяется 83,3 г Г. х.
Получение 111. Дипатриндисульфопаттидроксилампи (1), полу-
ченный пропусканием сернистого газа в водный раствор нитрита
и бисульфита натрия, реагирует с ацетоном с образованием оксима
ацетона (2), кислый гидролиз которого приводит к Г. х.
О = С(СН,Г
ХаХО.2 rXaHSO3-;-SO., —> HOX(SO:1Xa)2-«
<н
hoi - и .о
—<- НОХ -С(СНД;----НОХН, HCI , () = С(СН3),
( 1 1 17 ° п 121
Производные. Для получения калиевой соли бепзгпдроксамовой
кислоты |2| растворяют при кипячении 46,7 г (0,67 моля) Г. .х.
в 240 мл метанола и 1 моля КОН в 140 мл метанола и после охлаж-
дения до 30—40 второй раствор прибавляют к первому. Для пол-
ноты осаждения хлористого калия, к охлажденной льдом смеси при
С6Н-,СО.,С»Н5 ; НОХII., НС) -КОН-------
57-1,0%
о о
АлТН
— с„н,с-хнок--------- СДЦС-ХНОН
1'1
встряхивании добавляют 0,33 моля этплбепзоата. Смесь быстро
фильтруют и фильтрат выдерживают при комнатной температуре.
Кристаллы калиевой соли бензгидроксамовой кислоты начинают
выпадать через 20—180 мин и через 48 час соль отфильтровывают.
Смесь соли и эквивалентного количества разбавленной уксусной
кислоты перемешивают, hhiревакт ш; ,/рчзоьання прозрачного
раствора и оставляют для кристаллизации сначала при комнатной
температуре, а затем при 0 . Беизгидроксамовая кислота получается
в виде белых кристаллов. Перекристаллизация из этилацетата дает
чистый продукт, т. пл. 125—128'.
Для получения оксимочевины (т. пл. 137—141') |3| раствор
Г. х., уретана и едкого натра в воде оставляют при комнатной тем-
пературе на 3 дня, затем раствор осторожно нейтрализуют соляной
кислотой, экстрагируют эфиром и водный слой выпаривают досуха
при пониженном давлении. Остаток — смесь хлористого натрия
О Н
Х.зОН II |
Н.,ХШ,С.,Н5-ГН„\'ОННС1------> НАС-N—ОН
53-73%
и окенмочевпны — экстрагируют абсолютным этанолом, из которого
окси.мочевнна кристаллизуется.
Оксимы. По одной из методик 141 к смеси 0,55 моля бензофенона,
0,86 моля Г. х., 200 мл 95%-ного этанола и 40 мл воды добавляют
NaOH
(C„H5).,C-=O4-H,XOH HC1-----* (СвН5).,С=ХОН
>>8-'.Ы%
небольшими порциями 2,75 моля измельченного едкого натра. Смесь
кипятят 5 мин и выливают в разбавленную соляную кислоте'. Выпа-
дает достаточно чистый оксим, т. ил. 141 —142 .
Для получения оксима энантола |51 к суспензии 4 молей альде-
гида в растворе 5 молей Г. х. в 600 мл холодной воды при перемеши-
вании и охлаждении добавляют раствор 2,5 моля карбоната натрия
Ха. СО,
CH3(CH.,)5CHO-i НАОН-НС1 —:> СНз(СН.,)5СН — NOH
81 -‘33%
в 500 мл воды так, чтобы поддерживалась температура 453. Переме-
шивают 1 час, верхний маслянистый слой отделяют, промывают
дважды водой и перегоняют при 6 мм. Полученный по этой методике
оксим (т. пл. 44—46 ) можно сразу использовать для восстановления
в «-гептиламин действием натрия в абсолютном этаноле. Чистый
оксим плавится при 53—55 .
Нитрилы. Вератроиптрил получают [61 нагреванием смеси ок-
сима и уксусного ангидрида сначала осторожно, пока не закончится
бурное взаимодействие, а затем до кипения в течение 20 мин.
СН - ХОН С N
I I
// X n
I li —^1 II
1!—осн3 Н-осн3
Iz I'
ОСН3 ОСН3
216
После этого реакционную смесь выливают при перемешивании в хо-
лодную воду; вератрошприл выпадает в виде почти бесцветных
кристаллов с т. пл. 66—67".
В более простом способе обходятся без предварительного полу-
чения оксима и в качестве растворителя используют уксусную
кислоту 17]. Смесь 5 г анисового альдегида, 1,2 же Г. х. и 1,2 же
безводного ацетата натрия в 20 мл уксусной кислоты кипятят в тече-
ние ночи, отфильтровывают хлористый натрий, отгоняют раствори-
тель в вакууме, продукт реакции извлекают эфиром и перекристал-
лизовывают из петролейного эфира (т. пл. 59—60 ). Ванилин и пи-
перональ превращают по этой методике в нитрилы с выходом 80 и
50%. В случае пиридин-3-альдегида (выход 7?о) и н-масляного аль-
дегида получены неудовлетворительные результаты.
Превращение моногидрата D-глюкозы через оксим в пентааце-
тил-О-глюкопптрпл требует большой затраты времени; выход ко-
нечного продукта 50% |8|.
а-Кетокпслоты превращаются в нитрилы с одновременным де-
карбоксилированием при кипячении с 1 же Г. х. в водном растворе
О Г ХОН '
и ( н +
RCCO..HНА’ОН —* [RCCOJlJ —> RCX--CO., л-Н.,0
в атмосфере азота 191. Выходы достаточно хорошие, но некоторые
пространственно затрудненные кегокпслоты не реагируют.
Нуклеофильное замещение. Интересная реакция наблюдается
при растворении 20 г 1-нптронафталина и около 6 же Г. х. в 1,2 л
95%-ного этанола и перемешивании с нагреванием на водяной бане
при 50—60" и добавлением в течение 1 час раствора 100 г едкого
кали (около 10 же} в 630 мл метанола [101.
1,4-Присоединение-восстановление. Синтез P.L-0-амино-р-фе-
нплпропионовой кислоты (3) из коричной кислоты (1) включает
1,4-присоединение гидроксиламина к сопряженной системе с после-
217
дующим восстановлением гидроксиламипокислоты (2) гидроксил-
амином 1111. Горячий раствор 2 молей Г. х. в 100 мл воды добавляют
и .sou
СЦЦСН - СНСО..Н — -
id
СйИ5СНСН.,СО.,Н
I
хнон
<-')
1.NOH
-----* CJI5CHCH,CO.H
Л", |
хн.,
Ы)
к горячему раствору алкоголята, полученному из 2 г-атом натрия
и 1,6 л абсолютного этанола. После охлаждения смеси осадок хло-
ристого натрия отфильтровывают с отсасыванием. Затем прибав-
ляют 1 моль коричной кислоты и образующийся объемистый осадок
соединения (2) растворяют при кипячении; через 5—6 час начинается
осаждение аминокислоты (3). Спустя 9 час, когда осаждение закон-
чится, осадок отфильтровывают, промывают водой для удаления
хлористого натрия и абсолютным этанолом для удаления воды.
Восстановление. В реакции конденсации ацетиленов по Кадо —
Ходкевичх Г. х. применяется в качестве восстановителя соли
меди |12|.
Синтез изатина. Этот синтез, разработанный Запдмейерсм 1131,
был позднее модифицирован Марвелом и Хайерсом 114). Первая ста-
дия — конденсация анилина с хлоральгидратом и Г. х.:
II И
I I
с0н4—х —н CICCH(OH)., и.лон ис:-------► сльд-с-снохой
Cl Cl о
НОН НОН Л)
Кристаллический изонитрозоацетаннлид (1) осаждается с высоким
выходом при нагревании реагентов в водном растворе. Во второй
стадии соединение (1) медленно прибавляют к конц. серной кислоте
с перемешиванием и нагреванием до 60—70'; затем раствор выли-
вают на лед. Циклизацией и гидролизом соединение (1) с хорошим
выходом превращается в изатин (2).
Нг5О4 >
71-78%
+ nh4hso4
н
(2)
I. Се м о и В., «Синтезы органических препаратов», ИЛ., .4., 1949, сб., 1,
стр. 164.
2. Хаузер Ч., Ренфроу В. мл., «Синтезы органических препаратов»,
ИЛ.'М., 1949, сб. 2, стр. 8"’.
3. D е g h е n g h i R., Org. Syn., 40, 60 (I960).
4. Лахмап А., -Синтезы органических препаратов/, ИЛ. М.. 1949, сб. 2,
стр. 393.
5. Бусквет Е., «Синтезы органических препаратов», ИЛ, М., 1949, сб. 2,
стр. 394.
218
6. Б у к Дж., Л й д н В., «Синтезы органических препаратов», ИЯ, М., 1949,
сб. 2, с гр. 41.5.
7. Hunt J. II., Chem. Ind., 1961, 1873.
8. К ,т а р к \., II эд ж и С., «Сшисзы органических препаратов». М.. 1952,
сб. 3. cip. 372.
9. Л h ш а <1 Л.. Spencer I. I).. Can. J. (Ъеш.. 39. 1340 (1961).
10. Прайс Ч., В о о и г С и н г - т а \, <Сишезы opi апических препаратов»,
НЯ, ,М„ 1953, сб. 4. с гр. .376.
11. Ill гайгер Р.. (.Синтезы органических препараiон>. ИЯ, М., 1932, сб. 3,
стр. 61.
12. С 11 о г! k i е w i с z W., Ann. Chim . 113|. 2. 819 I1957).
13. S a n <i m e у e r T.. Helv. Chim. Acta. 2. 2 17, 2 (1919).
14. M a p в e л 4., ,\ a ii e p с Ж., «Синтезы opi пшческих нрепзратои», НЯ, M.,
1949, сб. 1, стр. 216.
ГИДРОПЕРЕКИСИ АЛКИЛОВ, ROOH, см. такжешрсш-Буш-
ла гидроперекись.
Получение, а. Обработка по Байеру и Виллшеру III АО" п-иой
перекиси водорода .шалкилсульфатом да* г низкие выходы загряз-
ненных и взрывоопасных метил-, этил-, н-иропи i- и /г-б\тп.п идро-
перекисей. По улучшенной методике |2|, применяя в качестве раст-
ворителя метанол, получают чистые н- и «шо/т-бу ги.пт1лроперек11сп
с выходом 20—40%.
б. Реакцией w-алкиловых эфиров метаису льфокислоты (СН:)SO.tR)
с перекисью водорода в метанольном растворе едкого кали удается
получить чистые «-алкилгидроперекпеи (от пропила до децила)
с выходом 70 —80°6 131 (для вторичных выход 20 —25%). По степени
устойчивости эти гидроперекиси не отличаются от ш/шш-бутплиной.
в. Чистые гидроперекиси алкилов можно получить медленным
добавлением алкилмагппйгалогенпдов к эфире, насыщенному кис-
лородом, при температуре —75 |4|.
1. Вас у е г А., V i 1 1 i у е г V., Вет., 33, 318, 34, 738 (1901).
2. L i и 3 s t г о in Е. G., .1. Am Chem. Soc.. 75. 5123 (1953).
3. W i I I i a in s H. R.. M о s h crll. S„ .1. Am. flu in. Soc.. 76. 2984. 2987 (1954).
4. Wallin ’ С., В u c k 1 с r S. A., .!. Am. (.licin. Soc., 75 4372 (1933).
ГИПОГАЛОГЕНИТОВ РАСТВОРЫ, см. также Натрия гппобро-
мит, Натрия гипохлорит.
Раствор гипохлорита натрия получают пропусканием хлора
в разбавленпу ю щелочь при 0 |1). Гппох юрит катышя выпускается
промышленностью в виде устойчивого, растворимого в воде
порошка. Для превращения в гипохлорит катня |2| раствор 250 г
гипохлорита калышя в 1 л теплой воды обрабатываю) теплым раст-
вором 175 г карбоната калия и 50 г едкого кали в 500 .я.? воды. После
тщательного встряхивания карбонат кальция отфильтровывают на
широкой воронке Бюхнера, промывают на фн.тигре перемешиванием
с 200 ты воды и отсасывают, насколько это возможно, с резиновым
колпаком (рис. Г-1). Фильтрат объемом около 1,5 л содержит приб-
лизительно 200 г (2,3 моля) КОС).
219
Г. р. используют для окисления аммиака в гидразин (см. Гидра-
зин).
Получение кислот гипохлорИтным окислением метилкетонов
иллюстрируется двумя примерами: 2-ацетонафталин —>|3-нафтойная
Рис. Г-1. Резиновый колпак для фильтрования с отсасыванием.
1 — резиновая пленка; 2 — лента из каучука.
кислота [2] и окись мезитила диметилакриловая кислота [3].
// X /СОСН8 . / СО2Н 3KOCI; HCI % |И у
1 ' 1 87-88% у J KOCI
(СН3)„С=СНСОСН3-------> (СН3).,С-СНСО,Н
49-53%,
Гипохлорнтное окисление феннлподидхлорида в присутствии
следов уксусной кислоты дает иодобензол [41:
О-
С6Н5М Cl(С1 -) у Х'аОСЛ Н,0 —X СвН,[ +=0 ф- NaCI г2HCI
87-92%
В ранней литературе по стероидам описана интересная реакция
окисления холестерина, суспендированного в растворе гипобромита
натрия, в кислоту Дильса [5]:
Кислота Дильса
220
В более поздней, так называемой улучшенном методике Шоппи
и Саммерса 161 нс приведен выход продукга. Кислота Дильса вы-
сокоплавка, умеренно растворима н легко выделяется, даже если
имеется в незначительных количествах. Одни из авторов книги |7|
окислением холестерина дигидратом бихромата натрия в смеси
бензол — уксусная кислота получил кислоту Дильса с выходом
2,97% наряду с шестью другими не игральными продуктами окис-
ления. Можно было предполагать, что один из этих продуктов,
А’-холестенон-З, является предшественником кислоты Дильса, од-
нако это не так. В промежуточном холестерилхромате имеется инте-
ресная возможность для внутримолекулярной атаки аллильного
Р-водорода при атоме углерода С4. Активным агентом в гипобромит-
ном окислении может быть также холестерилгипобромит.
Моор и .Чинк |8| при окислении альдоз гипоиодитом в метаноле
получили альдоновые кислоты, которые выделяются в виде калие-
вых или бариевых солей с выходом 90%. Эта методика разработана
с пелью конденсировать образующиеся соединения с о-фенплендпа-
мшюм для получения бензимпдазольных производных, исключи-
тельно удобных для характеристики и идентификации (по темпера-
турам плавления и IccId хлоргндратов и пикратов).
СН--.СО I у N
| I (’ll ИЛ A x
(СИОН)., (СПОН)., —--------------->. HOCH,(CH(')H),-C I
I I //
ОМОН CH.OH \
I
и
1. АдамсР., Б р а у н Б., «Синтезы органических препаратов», ИЛ, At., 1949,
сб. 1, стр. 158.
2. Ньюмен At., Холмс X,, «Синтезы органических препаратов», ИЛ, At.,
1949, сб. 2, стр. 351.
3. С м и т Л., Прпчард В., С и и л е 11 н Л., «Синтезы органических пре-
паратов», ИЛ, At., 1952. сб. 3, стр. 182.
4. Фор мо М., Д ж о и с о н Дж., «Синтезы органических препаратов», ИЛ,
М., 1952, сб. 3, стр. 262.
5. Diels О., Л b d е г li а 1 d е и Е., Вег., .36, 3177 (1903): 37, 3092 (1904).
6. S 11 о р р е е С. W., S и m in е г s G. И. R., J. Am. Cliein. Soc., 1952, 2528.
7. F i е s е г L. F., .1. Chem. Soc., 75, 4377; 4386, 4395 (195.3).
8. At о о г c S., Lin k К. P., J. Biol. Chem., 133, 293 (1940).
221
ГИППУРОВАЯ КИСЛОТА, CfiH-,CONHCH.,CO,H. Мол. вес
179,17, г. пл. 188 .
Получение 111. 1 моль .хлоруксусной кислоты прибавляют
к 3 .? конц. водного раствора аммиака (около 45 молей) и через
четыре дня раствор аммиака отгоняют. К остатку добавляют раствор
1,25 моля едкого натра н кипятят до исчезновения запаха аммиака.
Раствор фильтруют и при перемешивании, поддерживая охлажде-
нием температуру около 30 , обрабатывают одновременно раствором
едкого натра (2 моля) и хлористым бензоилом (1,1 моля) в течение
I час так, чтобы раствор постоянно оставался слабощелочным. После
этого реакционную смесь выливают в соляную кислоту и осадок
NH, N.iOll CJI.COCI
CICHXOjH -- H.,\CH2CO,H------* ИАСНХОЛа----------►
64-68%
С„НЬС(}КНСН2СО2Н
перекристаллизовывают из воды.
Синтез азлактонов. Применение Г. к. для азлактонного синтеза
аминокислот можно иллюстрировать методикой получения и.1.-фе-
нилаланина |2|. Смесь 0,5 моля бензальдегида, 0,5 моля Г. к. и
0,5 моля безводного ацетата натрия обрабатывают 153 г укосного
ангидрида и нагревают при перемешивания до разжижения почти
твердой массы и появления ярко-желтого окрашивания. Затем смесь
нагревают 2 час на кипящей водяной бане, слегка охлаждают, при-
бавляют этанол и оставляют до полного осаждения темно-желтых
КаОАс
С6Н5СНОД СН.,С().,Н.; 2(СН3СО).,О--> с,диен-С-С. -_о —►
I 62-64% | |
NHCOC6HS ,\ о
''с
I
с6н5
(1)
Р, (СН,СО)2О. Н1
---------------------------- СвН-,СНХНСО.,Н
64-67",,-------|
мь
<2)
кристаллов азлактона (1). К смеси азлактона (1), красного фосфора
и уксусного ангидрида при перемешивании добавляют 50%-иую
иодистоводородпую кислоту. Раствор кипятят 3—1 час, охлаждают
и после отделения фосфора фильтрованием выпаривают досуха при
пониженном давлении. Остаток растворяют в воде и раствор вновь
выпаривают досуха (для удаления HI). Полученный остаток раство-
ряют в воде, раствор несколько раз экстрагируют эфиром, освет-
ляют норитом, нагревают сначала осторожно для испарения эфира,
а затем до кипения и нейтрализм тот аммиаком по конго красном}7
(изоэлектрическая точка 5,48). Фенилаланин выделяется в виде бес-
цветных пластинок.
222
Ранее эта конденсация была использована только для аромати-
ческих альдегидов. Однако таким путем конденсируются и алифа-
тические альдегиды, если проводить реакцию в тетрагидрофуране
с использованием ацетата свинца вместо ацетата натрия |3|.
1. И п г е р с о :1 л Л., Б б к о к С., «Синтезы органических препаратов»,
ИД, М., 1949, сб. 2, стр. 158.
2. Джи л л е с п it X., С н а и д е р X., (Синтезы органических препаратов»,
ИЛ, АС, 1949, сб. 2, стр. 496.
3. В а 1 t a z z i Е., R о b i n s о п R., Chem. Ind., 1954, 191.
ГЛИОКСАЛЬ, ОСНСНО. Мол. вес 58,04, т. пл. 15', т. кип.
50,4 , уд. вес 1,14. Бисульфитное производное, ОСНСНО-2NaHSO4-
•Н,О, мол. вес 310,20.
Получение. Бисульфитное производное Г. получают кипячением
смеси паральдегида, двуокиси селена, диоксана и небольшого
количества 50%-нон уксусной кислоты в течение 6 час [11. Раствор
декантируют с осадка селена, удаляют дпоксан и паральдегид пере-
(СИ.,СНО);; 3H,Se();i—ЗОСНСНО-: 3Se 6Н.,()
гонкой с паром п обрабатывают небольшим избытком 25%-ного
раствора ацетата свинца для осаждения селенистой кислоты в виде
селенида свинца. Осадок отфильтровывают, раствор насыщают серо-
водородом, обрабатывают норитом и фильтруют горячим. Бесцвет-
ный фильтрат концентрируют при пониженном давлении и добав-
ляют раствор бисульфита натрия в 40%-ном этаноле. Выход бисуль-
фитного производного 62- 64%. Его кристаллизуют из водного
раствора при добавлении этанола в таком количестве, чтобы полу-
чился 40%-ный раствор.
Синтезы хиноксалинов. Конденсация бисульфитного произ-
водного Г. с о-фенилеидпампиом вводном растворе при 80’ приводит
с высоким выходом к хиноксалину, окислением которого перманга-
натом получают ппразип-2,3-дикарбоновую кислоту [2]. В отсут-
11,,X А /X ZCO2H
СН—О ХМ ISO I КМпО4 I ' 7
I л. -----А \ |-----> . ;
ствпе бисульфита натрия конденсация водного Г. с о-фенилендиа-
мином дает хиноксалин с выходом только около 30% и образованием
смолообразиых побочных продуктов.
Синтезы хинонов. В присутствии сильных щелочей Г. диспро-
порииоппруег в гликолевую кислоту:
о -СНСН о 1 Н,< > — 11(.)СН.,СО.,И
Однако найдено (Гомолка |3|), что при пропускании воздуха в вод-
ный раствор Г. в присутствии сильного буфера происходит конден-
сация с образованием гексаокспбеизола, который окисляется до
натриевой соли тетраокси.хпнона. По методике Фатнади и Загера
223
[41 600 г 30%-ного раствора Г. добавляют к водному раствор'"
сульфита натрия и бикарбоната натрия при 40—45 и в смесь про-
пускают сальный ток воздуха сначала в течение 1 час без нагрева-
ния, а затем 1 час при 80—90'. Уже через несколько минут начинают
выделяться зеленовато-черные кристаллы динатрпевой соли тетра-
окспхипона. Соль отфильтровываю!, промывают и нагревают
с 2 н. соляной кислотой; при охлаждении выпадают блестящие
черные кристаллы тетраоксихинона. Выход 11 —15 г (6,2—8,4°о).
Выход небольшой, однако исходное соединение доступно, а опера-
ции очень простые. Тетраоксихпнон при восстановлении хлористым
СНО 011 °
НО ) ОН НО / ,ОХа
СНО СНО N;1;sO1 " У о, %
СНО CIIONallCO., I ' j lx
НО % ОН XaOz ОН
ено ОН о
оловом дает гексаокспбензол, а при последующем окислении—роди-
зоновую кислоту и трихинонл.
Изонафтазарин можно синтезировать добавлением раствора
о-фталевого альдегида в диоксане к водному раствору бисульфит-
ного производного Г., цианистого калия и бикарбоната натрия и пе-
ремешиванием раствора в присутствии воздуха при 20' |5|. Раствор
О
/ч , сн0 /ч \ /он
с ;/ СНО КС\. \uHC0j. О.: НС! С ' \ '
I L "сно----------I
% z СНО (2NUHSO3) <)Н
6
быстро приобретает интенсивную фиолетовую окраску, при под-
кислении через 15 мин (продукт очень чувствителен к щелочи)
ос а ж даете я ярко-красный и зо и афта з а р и н.
1. Р о и з и о Л., Во Т., «Синтезы органических препаратов», ИЛ, М., 1952,
сб. 3, стр. 106.
2. Д ж о и с Р., М а к - Л а ф л и и К., «Синтезы органических препаратов»,
ИЛ, М.. 1953, сб. 4, стр. 426.
3. Н о m о I к а В., Вег., 54, 1393 (1921).
4. F a t i a d i A. .1., Sai>er W. F„ Ощ. Syn., 42, 90 (1962).
5. W e у й a n d F., Ber., 75, 625 (1942).
ГЛИОКСИЛОВОЙ КИСЛОТЫ м-БУТИЛОВОГО ЭФИРА n-TO-
ЛУОЛСУЛЬФИМИД, n-CH:1C,1H.iSO2N СНСО2С4Н9-н. Мол. вес
28.3,35, т. кип. 130—135''10-:| льи.
Получение. К кипящей смеси н-бутплглиоксилата [11 и N-суль-
финил-л-толуолсульфамида в бензоле медленно прибавляют раствор
224
хлористого алюминия в нитробензоле [21.
-so2
n-CH3CeH4SO,N—SO-:-O=CHCO2C4H8-h —>
G7%
— w-CH3C6H4SO2N = CHCO2C4H9-w
Диенофил [2|. Этот реагент образует аддукт с 2,3-диметилбу-
тадиеном (выход не указан) при кипячении в бензоле в течение
10 час. При омылении аддукта получают с низким выходом 4,5-ди-
метилпиридпн-2-карбоновую кислоту:
тт r ,SO2C6H4CH3-n СН3 z. SO2CeH4CH3-n
3 N Кипячение ц \г
I + II --------* I -*
НГ/'^ сн С6н„ Н I
4Мз<-; со.,с,н.ги н3с/Х/'со2с4н9-и
Н3С. /х
С2Н6ОН-КОН Н N
34% /[I I
Н3С' 7 'СО,Н
1. Wo I f F. J., W е i j 1 а г d J., Org. Syn., Coll. Vol., 4, 124 (1963).
2. Albrecht R., Kresze G., Chem. Ber., 98, 1431 (1965).
ГЛИЦИН, H2NCH2CO2H. Мол. вес 75,07,pK4 (CO2H) 2,4, рК2
9,8. Г. получают из метиленаминоацетонитрила [1] или из
хлоруксусной кислоты [2].
Г., как и другие аминокислоты и аминофенолы, является ката-
лизатором реакции Кневенагеля [3,4] (см. 0-Аланин). Так, бен-
зальмалононитрил получают с количественным выходом при
добавлении каталитических количеств Г. к 70%-ному водно-спир-
Н2К'СН2СО2Н
С6И-СНО-!-СН.ДСХ’),--------> C6H5CH=C(CN)2
100%
товому раствору бензальдегида, содержащему 1 же динитрила мало-
новой кислоты [4]. Г. катализирует альдольную конденсацию ди-
оксиацетона п гликолевого альдегида с образованием смеси, содер-
жащей небольшие количества кетопентоз [51.
1. А н с л о у В., К и н г Г., «Синтезы органических препаратов», ИЛ, М., 1949,
сб. 1, стр. 167.
2. О р т е н Дж., Хилл Р. М., «Синтезы органических препаратов», ИЛ, М.,
1949, сб. 1, стр. 168.
3. D a k i n Н. D., J. Biol Chem., 7, 49 (1909); Р г о u t F. S., J. Org. Chem., 18,
928 (1953).
4. Bastiis 1. B., Tetrahedron Letters, 955 (1963).
5. Gascoigne J. A., Overend W. G., Stacey M., Chem. Ind., 1959, 402.
ГЛУТАРОВАЯ КИСЛОТА, HO2CCH2CH2CH2CO2H. Мол. вес
132,11, т. пл. 98\ в 100 г воды (20") растворяется 63,9 г Г. к.; рКа4
4,33, рКа2 5,57.
8 Заказ А» 965 225
При получении Г. к. в первой 111 из четырех методик, описанных
в «Organic Syntheses», исходят из довольно труднодоступного 1,3-ди-
бромпропаиа, который превращают в динитрпл и далее подвергают
СХМС.Х' IIC1
BrCI I..C1I,CI I .Br -» \CCII,CII,C.II,C\------ HO.CCII ,CH,CII,CO.,H
кислотному гидролизу (4 час). Экстракция эфиром п кристаллиза-
ция из смеси эфир — бензол дает чистую Г. к. (т. пл. 97—98 )
с высоким выходом.
О получении Г. к. из формальдегида и малонового эфира см.
Формальдегид.
В более поздней методике [2| у-бутиролактоп нагревают при
190 - -195 в течение 2 час с цианистым калием, затем добавляют воду
для растворения калиевой соли цпанкислоты и теплый раствор
обрабатывают соляной кислотой, которую берут в количестве,
СИ..—СО
СП,
СН,СО,К СН,С(),Н сн,со,н
i сн, 1 Д' сн. 1 7 1- ।
CH,CN сн,со\н. сн,с.о,н
достаточном для выделения свободной кислоты из калиевой соли
и осуществления частичного гидролиза с образованием моноа.мпда
Г. к. После этого прибавляют конц. соляную кислоту и смесь кипя-
тят 1 час для завершения гидролиза. Раствор осветляют норитом,
фильтруют, фильтрат выпаривают досуха и продукт кристаллизуют
из хлороформа. Получают Г. к. с т. пл. 98—99J.
В четвертой методике |3| дигидроппран гидролизуют при кратко-
временном нагревании с 0,2 н. азотной кислотой на кипящей
СН, СН,
| . 1222112 (in сн, 222; где сн,
< I ~ I 11
О ' сн, сн-о Н(),С со,и
2
водяной бане с образованием б-оксивалерпапового альдегида. Полу-
ченный раствор добавляют к конц. азотной кислоте при охлаждении
до 10 и ниже и перемешивании в течение 3 час. Затем растворитель
удаляют перегонкой п остаток кристаллизуют из смеси эфир — бен-
зол. Получают сырую Г. к. с т. пл. 90'; выход 70—75°о.
1. At а р в е л, М а к - К о л ь м, (Синтезы органических препаратов», ИЛ, At.,
1949, сб. I, стр. 506. At а р в ел, Т у л е й, «Синтезы органических препара-
тов», ИЛ, At., 1949, сб. I, стр. 173.
2. Р а г i s G., В с г 1 i и g u е t L.., Gaudry R., Org. Svn., Coll. Vol., 4, 496
(1963).
3. И и гл и in Дж., мл., Да ii а н Дж., «Синтезы органических препаратов»,
ИЛ, At., 1953, сб. 4, стр. 138.
226
ГЛУТАРОВЫЙ ДИАЛЬДЕГИД, OCHCI 12СН,,СН;СНО. Мол.
вес 100,11, т. кип. 75—81 -15 д.и.
Г. д. получают перемешиванием смеси 120 г 2-этоксп-3,4-Д11ГИ-
дроннрана-2Н, 300 воды и 25 мл конц. соляной кислоты в тече-
ние 22 мин; за это время температура повышается до 38 и смесь
l’a.16. IK I
Г
ОС.,Н3 О^СН СН-О
становится прозрачной 111; через 1,5 час нейтрализуют бпкарбона-
том натрия, насыщают хлористым натрием п экстрагируют эфиром.
Перегонка дает 55 г Г. д.
Классический синтез псевдопельтьерпна Робинсона (1924 г.),
улучшенный в последующих работах Шёпфа (1935 г.), Циглера
(1950 г.) и Коупа (1951 г.) |2|, заключается в конденсации Г. д.
с метиламином и ацетоидпкарбоновой кислотой.
сно сн2со2н
+ HjNCHj + СО
„ сн2со,н
х—оно
-2 СО2?
58-68%
Псевдопельтьерин
Наплучшпе результаты достигаются, если Г. д. получают из
2-этокси-3,4-Д11П1Дро1шрапа-2Н и кони, соляной кислоты в дистил-
лированной воде, из которой удален кислород пропусканием тока
азота. Полученный бесцветный раствор Г. д. обрабатывают в атмо-
сфере азота водными растворами хлоргидрата метиламина,
анетондикарбоновой кислоты, Na>HPO4 • 12Н.0 и едкого натра.
При перемешивании раствора наблюдается выделение двуокиси
углерода и повышение pH от 2,5 до 4,5 в течение 24 час. Раствор
обрабатывают конц. соляной кислотой и нагревают на кипящей
водяной бане в течение часа для завершения декарбоксилирования.
После нейтрализации щелочью, многократной экстракции хлори-
стым метиленом и очистки получают бесцветный псевдопельтьерин
с т. пл. 63—64'.
I. I. о ii gley R. 1,, Jr., Е ш г г s о ii \V. S., .1. Ain. Cheni. Soc.. 72, 3079 (1950).
2. Рог the procedure cited and references to the earlier work see С о p e Л. C. D r y-
d c n H. L., Jr., И о w e 1 I С. E., Org. Syn., Coll. Vol., 4, 816 (1963).
ГРИНЬЯРА РЕАКТИВЫ.
Аппаратура. Для получения Г. р. очень удобен прибор Герш-
берга, показанный на рис. Г-2. Трехгорлую круглодонную колбу
8’ 227
на шлифах устанавливают на съемной паровой бане и снабжают об-
ратным змеевиковым холодильником, капельной воронкой с
трубкой для выравнивания давления и мешалкой Гершберга |1|
с ртутным затвором, сделанной из танталовой проволоки (рис. Г-3
и Г-4). Систему продувают сухим азотом, вводя его через верх холо-
Р и с. Г-2. Прибор для получения реактивов Гриньяра.
дильника и выпуская через капельную воронку. Когда воронка
закрыта, в приборе создается небольшое избыточное давление, на
которое указывает уровень ртути в предохранительном клапане.
Такое устройство [2| обеспечивает экономичный расход азота, а
также устраняет испарение растворителя с током инертного газа,
которое неизбежно при непрерывном продувании. Азот высокой
чистоты можно использовать непосредственно из баллона.
228
Атмосфера азота необходима прежде всего для предотвращения
разложения Г. р. при окислении воздухом по схеме [3]
2RMgX 4-0, —» 2R0MgX
а также для предохранения от влаги воздуха. В колбу помещают не-
обходимое количество магниевой стружки и пропускают азот, промы-
вая им также и пустую капельную воронку. После вытеснения
воздуха продолжают пропускать азот, осторожно подогревая колбу
открытым пламенем горелки, чтобы удалить влагу, адсорбирован-
ную на поверхности стекла и металла; азот предохраняет поверх-
ность нагретого металла от окисления. Затем колбу охлаждают,
ток азота уменьшают до едва ощутимого и добавляют небольшое
количество галогенида и эфира из капельной воронки. Перемеши-
вание начинают, когда имеется еще очень небольшое количество
жидкости; при этом кусочки магния разру-
шаются и этого часто оказывается достаточ-
ным для начала реакции. Если простое пере-
мешивание неэффективно, то в качестве од-
ного из приемов можно рекомендовать опус-
тить мешалку так, чтобы при движении она
придавливала кусочки металла к дну колбы и
до некоторой степени дробила их. Кроме того,
можно прибавить немного метилмагнийподи-
да или небольшой кристаллик иода. Согласно
работе |4], бром как инициатор эффективнее
иода. Катализатор Гпльмана [51 получают
Рис. Г-4. Ртут-
ный затвор для
мешалки.
взаимодействием магния и иода в смеси эфира с бензолом.
Анализ RMgX. Гильман [61 определял выход Г. р. разбавлением
до стандартного объема, добавлением аликвотной порции к избытку
229
0,2 н. кислоты и обратным титрованием 0,2 н. щелочью в присутствии
фенолфталеина. Ниже приведены средние данные для ряда опытов:
Исходное соединение КМцХ. %
Этилбромпд 93,1
«-Амилбро.мид 88,0
в/лор-Амилбромпд 66,8
//|р<’/л-Амилбромнд 2.3,7
«-Гексилбро.мид 92,0
«-Бхтплхлорид 91,2
«-Бутплиоднд 85,6
Бензилхлорпд 93.1
Бро.мбензол 94,7
Широко используется проба Гильмана [71 на присутствие Г. р.:
около 0,5 ли испытуемого эфирного раствора отбирают капиллярной
пипеткой и обрабатывают сначала 0,5 мл 1?о-ного раствора кетона
Мп.хлера в сухом бензоле, а затем медленно 1 мл воды. Наличие
в растворе Г. р. обнаруживается по характерному зеленовато-си-
нему окрашиванию, появляющемуся при добавлении нескольких
капель 0,2%-ного раствора пода в уксусной кислоте.
Газообразные галоидные алкилы. Для получения небольших
количеств метилмагнинбромида ампулу с бромистым метилом
(т. кип. 4,6 ) охлаждают льдом, вскрывают п резиновой трубкой
соединяют через хлоркальцпевую трубку с газоподводящей трубкой
реакционной колбы. Реагент вводят, нагревая ампулу рукой. Хло-
ристый метил (т. кип. —24) добавляют к смеси магния н эфира
непосредственно из баллона. Затруднения, встречающиеся иногда
при инициировании реакции, обычно можно преодолеть перемеши-
ванием и нагреванием реакционной смеси. Чтобы удержать реагент
в колбе, ее снабжают ртутным клапаном или резиновым баллоном
либо в боковой отвод вставляют конденсатор типа «охлаждающий
палец», наполненный сухим льдом. В последнем случае устраняется
опасность противодавления п обеспечивается высокая исходная
концентрация галоидного алкила, благоприятствующая началу
реакции. Из реакционноспособных галогенидов типа бромистого
аллила Г. р. получают в очень разбавленном растворе, чтобы свести
к минимуму реакцию сочетания.
Способ «вовлечения» (см. также Дибромэтап). Метод превра-
щения малоактивных алкплгалогенидов в их магниевые производ-
ные, разработанный Гриньяром [81, заключается в прибавлении
1 же бромистого этила к эфирному раствору инертного галогенида
и медленном добавлении этой смеси к такому количеству магния,
которое достаточно для взаимодействия с обоими галогенидами.
Вспомогательный галогенид очищает и активирует поверхность маг-
ния; кроме того, возможно, что он оказывает действие за счет обмен-
230
ной реакции. В представленном ниже перечне реакции Гриньяра
приведены результаты превращения броммезптплеиа в соответст-
вующую карбоновую кислоту в присутствии бромистого этила и
без него, нз которых видно, что способ «вовлечения» повышает
выход на 25"». В этом случае присутствие в растворе двух типов
Г. р. не создает трудностей, так как после карбоксилирования
С.,Н-.MgBr получается жидкая кислота, которая не мешает выделе-
нию твердой АгСО.Н. Однако часто присутствие второго Г. р.
нежелательно. В качестве «вовлекающего» реагента Пирсон |9|
предложил применять дпбромэтан, который реагирует с магнием,
образуя этилен и бромистый магний:
Br(.II.,CII.,Br Mg — MgBr., (.II., - CIL.
При этом очищается поверхность магния и он активируется для
реакции с инертным галогенидом без образования в смеси второго
Г. р. Эта методика почти всегда дает более высокие выходы по срав-
нению с использованием в качестве «вовлекающего» агента этпл-
бромнда.
Винилгалогениды. Из вииплгалогеипдов (даже иодидов) не
удается получить Г. р. обычным способом. Однако Норман 1101
нашел, что винилхлорид и випплбромпд легко образуют вииилмаг-
нпйгалогеипды в ТГФ пли в диэфпрах ди- и трпэтпленглнколя (см.
обзор 1101).
Сорастворители. Для некоторых реакций присоединения недо-
статочна температура кипения эфира. В этом случае к эфирному
раствору Г. р. добавляют растворитель с более высокой температу-
рой кипения (бензол, ксилол) и эфир отгоняют. Г. р. получают с вы-
соким выходом в лигроине, толуоле и ксилоле при добавлении 1 же
ТГФ на каждый эквивалент магния (Лейх 1111). Растворы Г.р.
в бензоле пли толуоле получают, применяя трнэгплампн в качестве
комплексообразующего агента (Эшби и Рид 1121). Раствор алкил-
rt-C4H„Cl-i- СвНй -У Mg -(-(СН-ДдХ- Wl' /oClH,,MgCI
0,5 экв 430 м.1 0,(> :жв 0,5 .же О3‘'|(
или арилгалогенида в бензоле прибавляют к магниевой стружке,
покрытой бензолом и содержащей триэтиламин. После добавления
примерно 30 л/л раствора смесь осторожно нагревают до начала
реакции. Оставшийся раствор добавляют в течение 2 час, поддержи-
вая температуру 40—50 . Полученный раствор, прозрачный и часто
бесцветный, фильтруют через воронку со стеклянным фильтром.
Выход определяют после удаления растворителя в вакууме, выде-
ляя реактив Гриньяра в виде твердого вещества, которое затем ана-
лизируют.
«Обратная реакция» Гриньяра. Г. р. получают в обычной
аппаратуре (см. рис. Г-2), после чего капельную воронку заменяют
сифоном, вставленным в пробку и доходящим до дна колбы, и раст-
вор Г. р. передавливают азотом в сухую капельную воронку,
также заполненную азотом. Воронку присоединяют к реакционной
231
колбе, содержащей раствор второго компонента. Для отделения
следов непрореагировавшего металла, который может помешать,
раствор Г. р. фильтруют через стеклянную вату, находящуюся
в нижней части трубки сифона.
Гидролиз. По окончании реакции MgX-производное разлагают
действием 25%-ной серной кислоты или постоянно кипящей соляной
кислоты, добавляя по каплям с такой скоростью, чтобы кипел эфир
(охлаждение). Можно также реакционную смесь вылить небольшими
порциями в смесь льда и разбавленной кислоты. Если продукт
реакции неустойчив в присутствии кислот, разложение можно про-
водить насыщенным водным раствором хлористого аммония при
25э. (Чтобы получить около 127 мл насыщенного раствора, 38 г
NH4C1 и 100 мл дистиллированной воды перемешивают до полного
растворения и охлаждают до 25е). Одна из методик рекомендует
прибавлять достаточно большой объем раствора, чтобы перевести
первоначально выпадающий осадок основных солей магния в раст-
вор (около 3 л на 1 моль магния) [131. Лучше добавить точное
количество раствора, достаточное для осаждения магниевых солей,
и отделить верхний слой почти безводного эфирного раствора про-
дукта реакции. Реакционную смесь охлаждают и при перемеши-
вании добавляют по каплям насыщенный раствор хлористого
аммония (25°) с такой скоростью, чтобы эфир равномерно кипел.
Обычно для четкого осаждения требуется 150—175 мл насыщенного
раствора на 1 моль магния, и это количество не следует превышать
[14]. Раствор, вначале мутный и непрозрачный, при добавлении
достаточного количества хлористого аммония внезапно становится
прозрачным, и белая соль осаждается в виде плотного слоя, который
может остановить мешалку. Смеси дают отстояться в течение не-
скольких минут, эфирный раствор декантируют, а осадок промы-
вают одной-двумя порциями эфира. Эфирный раствор не сушат и
сразу выделяют продукт реакции, отгоняя растворитель. Эта мето-
дика применима также к реакционным смесям, в которых до одной
трети всего количества растворителя составляет бензол. Если реак-
цию проводили в ТГФ, рекомендуется применять 100—120 мл раст-
вора хлористого аммония на 1 моль RMgX [15].
Примеры. Ниже приведены примеры реакций, описанных в сб.
«Синтезы органических препаратов» (С. о. п.) и «Organic Syntheses»
(О. S.), с указанием тома и страницы. Как правило, стадии получе-
ния Г. р. и гидролиза продукта присоединения не указаны, а общий
выход приведен для продукта реакции, имеющего удовлетворитель-
ную степень чистоты.
Углеводороды и образование связи С—С
С. о. п., 2, 408
СН3СН„СНоСНСН3_____СН3СН.,СН2СНСНз —
| (н-С4Н,)2О | 50-52%
Вг Т. кип. 143“ MgBr
СН3СН2СН2СН2СН3 (т. кип. 36°)
2.32
С. о. п., 1, 364
CeH,CH2MgCI
С. о. п., 2, 259
(C.H5O)2SO,
70—75%
СвН5СН2СН2СН3
MgBr сн,
хЛ/СНв 1 || (CHsO)2SO, Н3СХ 1 /СН3 1 II
1 II 52-60% I^J
сн3 СН3
С. о. п., 2, 30
C6H,CH,MgCl + 2n-CH3C6H4SO3C4H9-«---------
50— 39%
— С6Н5СН2С4Н9-н + н -С4Н9С1 + (TsO)2Mg
С. о. п., 1, 513
Вг Вг
I I
и мп ВгСН-С—СН2 _ __ <~т> г-г л'ГД
C6HltMgBr----------->- с6н11сн,с = сн2
60-64%
С. о. п., 4, 147
2СН2 = СНСН2С1 ———► СН2 = СНСН2 —СН2СН = СН2
55—65%
Спирты или продукты дегидратации
С. о. п., 1, 515
CeHuMgCl C«HUCH2OH
64—69%
С. о п., 1, 154
р
CH3CH2CH2CH2MgBr СН3СН2СН2СН,СН,СН2ОН
60—62%
С. о. п., 2, 193
2«-C4H9MgBr+HCO2CH,----------------------> (я-С4Н9)2СНОН
83—85%
С. о. п.,2, 321
(CH3)2CHMgBr + CH3CHO---------► (СН3)2СНСНСН3
53—54% |
ОН
O.S., 41, 49
СН2 = CHCH2MgBr SH,rC.HCt!2r СН, = СНСН2СНСН = сн2
57-59% |
он
С. о. п., 4, 419
CH3MgCl + СН3СН = СНСНО_______>. СН3СН = CHCHCHj
81-86% I
он
С. о. п.,4, 542
ОН
jM-ClCeHjMgBr-^^ л-С1С6Н4СНСН3
82—88%
233
С. о. п., 3, 424
(С3Н3)3СОН
2C„H-,MgBr4- С„Н-,СО,С„Н3------------>
89—43%,
С. о. и., 2, 490
3C,H5MgBr о=С|Ос-1112. (С„Н5).,СОН
32-88%
С. о. II., 1, 207
СНлСОлС Н% Перегонка
2C6H-,MgBr------:> (С6Н5).,ССН.~-------------> (С6Н-,).,С = СН2
| ' <>< — /()%
ОН
С. о. п., 3. 440
С. о. п., 11, 69
НС CMgBr - " 22. НС сснсн = СНС6Н3
38-34% |
он
С. о. п., 3, 164
Кислоты, сложные эфиры
С. о. п., 1, 410
(CH3)3CMgCl ———> (СН3)3ССО2Н
69 — 70%
С о. п., I, 267
СН3 СН3
I со. I
CH3CH,CHMSC1------------> СН3СН.,СНСО,Н
С. о. п., 2, .348
MgBr со.,н
231
О. S., 3, 553 (методом ..вовлечения")
СН.,
Л ..MgBr
/У со.
2Mg + C?H5Br —* I ------->
| J) 86-87%
н.,с 7 сн3
СН3
J. /СО.Н
~ Л'
H3C/'^/ ХСН3
С. о. п.,3, 555 (без ,,вовлечения”)
СН., СН:1
Н.,с/^/ N:H3 НзС/^/ 'СН3
СН3
2 .«).н
H/M^Z СН3
С. о. п., 2, 592
MgBr СО.,С,Н5
Г I
// \ О=С(ОС.,Н5)2 " "Ч
I !i i i H I
'Ч/ \// х/
C.o.n.,2, 211
П—ПСНз
C1CO2C;H; HjCl^ UcOjCjHj
57-58% |
H
Синтезы альдегидов и кетонов
C. o. n., 2, 295
H-C5H„MgBr "C(°S- ±^ w-C5HnCHO
45-30%
C. o. n., 4, 24
,.CHO
ArMgBr+HC(OC.,Ha)3
235
С. о. п„ 3, 321
О
C6H5MgBr -С-3°СН:^СН3ОСН2СС8Н5
71-78%
С. о. п., 4, 314
о
/r'U v ruru r'u м о С,1С1-- n С1ССН,СН2СО,СН3
(CH3).,CHCH„CH2MgBr------г R,Cd---------------->.
73—75%
О
II
—► (CH3)2CHCH.2CH2CCH.2CH„CO.2CH3
OS, 4, 601
Лактоны + R MgX
2CH3MgBr+CH3 (CH„)4CHCHoCH.CO-------► CH3 (CH2)4CHCH.,CH.,C (CH3),
i 7'1 5 Ob I I
1,4-Присоединение
O.S., 4, 93
CH3 CH3
I С„Н5СН2МяС1 I
СН3СН2С=ССО.,С,Н5 - QRC—• СН3СН2С----------СНСО2С,Н5
| --Уэ.о | |
CN С6Н5СН2 CN
O.S., 41, 60
СН3
I
«-C4H9MgBr+ СН3СН = снсо.,снсн,сн3 ——т—>
ЬЪ —
сн3
I КОН
—> сн3снсн2со2снсн2сн3 -—СН3СНСН,СО,Н
I 2 i 2 J 90—94% | - -
С4Нд-Я С4Нд-Я
Синтезы простых эфиров
O.S., 41, 91
О
И
CeH5MgBrJ С„Н-СО2С (СНЖ-^-^7* С6Н5ОС (СН3)3
236
O.S., 43, 55
f?
С6Н3СООС(СН3)3?
70-76%
OC(CH3)3
(TsOH) 155°
89-94%
+ (CH3)2C=CH2
•S^-MgBr
O.S., 4, 667
CH3I
------>
53-60%
Неуглеродные акцепторы
C.o.n., 1, 425
SbCl
3C6H5MgBr —(C6H-)3Sb
C.o.n,, 3, 386
CeH5MgBr + Se - -- -- CeH5SeH
•JI 1 I .1)
C,Q.n., 11, 13
(к-C H ,hSnCI2
CH., = CHMgBr ———— (H-C4He)2Sn (CH = CH^
'(ТГФ) /4—^1%
O.S., 4, 605
H
CeH5CH2MgCl + C6H5CH = NCH3 CeH5C-NCH,
o* j о 0 j
CH2C„H6
C.o.n., 8, 57
4C,H5MgBr+SnCI4 -777-57* (P>H5)4Sn
O.S., 4, 910
CeH5MgBr+(CeH5)sAsO (C6H5)4AsCl-HCl
Новые примеры [16, 17]
В 6?Hjone при 73г
CeH5.MgBr4-CJCH2CeH4F-o ------—7-------* CeH5CH2C3H4F-o
Ь O', о
° 9 /=N
CfcHjC-CI + 2 H-N --------------C6H5C-N I -------->
-c3h3n2h2ci \—I
-^-MeBr> c6hJC6h5 >
72%
237
1. Hers 11 berg E. B., Ind. Eng. Chem., 8, 313 (1936).
2. G i 1 m a n H., Hewlett A. P., Rec. trav., 48, 1124 (1929).
3. G i I m a n H., Wood A., .1. Am. Chem. Soc., 48, 806 (1929).
4. V an d e r Ker к G. J. M., L u i j t e n J. G. A., Org. Syn., Coll. Vol., 4.
882 (1963).
5. G i I in a n 11., К i r b у R H., Rec. trav., 54, 57/ (1935).
6. G i 1 in a n H., Zocllner E. A., D 1 с к e v .1. B., J. Am. Chem. Soc.,
51, 1576 (1929).
7. G i 1 m a n H., Schulze F.. J. Am. Cheni. Soc., 47, 2002 (1925)' G i I-
ni a n H., H e с к L. I.., ibid., 52, 4949 (1930).
8. Grig n a r d V., Conipt. rend., 198, 625 (1934); see also С 1 e m e n t H. ibid.,
198, 665 (1934); b r i о н E„ ibid., 198, 1244 (1934).
9. P e a r s о n I). E., С о w a n D., .1. Org. Chem., 24, 504 (1959).
10. X or m a n t H., Conipt. rend., 239, 1510 (1954); Bull. soc. chim. France, 1957,
728; «Успехи органической .химии», изд-во «Мир», ЛЕ. 1964, том 2, стр. 5.
11. Leigh Т., Chem. Ind.. 1965, 426.
12. Л s h b у Е. С., R е е d R., J. Org. Chem., 31, 971 (1966).
13. С к а т 1 е б ё л Л., Д ж о н с Э. Р. X., У a ii т и и г М. Ч., «Синтезы органи-
ческих препаратов», ИЛ, М., 1961, сб. 11, стр. 69.
14. Кобурн Э., «Синтезы органических препаратов», ИЛ, М., 1953, сб. 4,
ср. 419.
15. Сейферт Д., «Синтезы органических препаратов», ИЛ, М., 1961, сб. И,
с гр. 13.
16. V i и g i е I 1 о F. A., Q u о S.-G., S Ii c r i d a n .1., .1. Org. Chem., 26 3202
(1964).
17. S t a ;i b II. A., .1 о s t E., Ann., 665, 90 (1962); S t a a b H. A., Angew. Chem.,
Intermit. Ed., 1, 351 (1962).
ДАУТЕРМ А (Dowtherm Л), эвтекi ическая смесь, содержащая 26,5%
дифенила и 73,5",) дифенилового эфира. Т. ил. 12 , т. кип. 258 .
Термическое разложение диарплфумарагов до шдтшс-стпльбенов,
которое обычно проводят сухой перегонкой, часто проходит более
полно при кипячении в Д. в течение 36--50 час (Шпаги 111). Однако
о
11 _ с(;о— осн..
СН3О — ОС—(II
--- II
о
Н ( ''''--ОСН.,
, —... | -- 2СО..
сн.о—z/ с—и
выход щ/щнс-стильбенасоставляет только 19"6, реакция с дп-н-нитро-
производпым и другими замещенными эфирами дает выход 10—40% .
Перлман |2| получил нзо.хннуклндон-3 (3) гидрированием
н-ампнобеизойной кислоты в воде под давлением 3,5 атм с после-
дующим прибавлением ДМФА и охлаждением: полученную смесь
nh2
Н2, Rh-Ptl-C
70-75%
(1)
Кипячение
Даугперм
75-82%
цис- п щ/мнс-гексагидридов (2) кипятили в Д. в течение 20 мин
п получили изохпнуклндоп-З, который прекрасно заменяет камфору
при определении молекулярного веса но Расту 131.
1. Spat 7. S. М., .1. Ощ. Chem.. 26, Ш8 (19(H).
2. Р earl m a n W. М., procedure submitted to Огц. Sen.
3. F e r b e г E., В r ii c k n e г II., Ber., 76, 1019 (1943).
239
ДЕГИДРОАБИЕТИЛАМИН
(i)
Мол. вес 285,46.
Сьёберг и Сьёберг [1| показали, что Д. (1) дает хорошо кристал-
лизующиеся соли с различными пенициллинами. Они обнаружили
также, что этот оптически активный и относительно доступный
амин — превосходный реагент для расщепления кислот. Так, не-
однократные попытки расщепления (±)-у-фенилпропилянтарной
кислоты всеми обычными основаниями оказались тщетными; однако
расщепление удалось легко осуществить с помощью Д. (1). Другие
примеры расщепления см. в [21.
Для выделения (1) [1,31 раствор 2 кг смеси, содержащей 50% Д.
(1), 20% дегидропроизводного и 20% тетрагидропроизводного и
нейтральные вещества канифоли («амин D»), в 3,3 л толуола обраба-
тывают 460 г уксусной кислоты при 65—70 . Ацетат дегидроабиетил-
амина (т. пл. 141 —143 ) получается тремя порциями с общим выхо-
дом 960 г. Основание отделяется при нейтрализации теплого водного
раствора ацетата; его экстрагируют бензолом, экстракт сушат и
упаривают в вакууме. Получают вязкое, медленно кристаллизую-
щееся масло.
l.Sjoberg В., SjobergS., Arkiv. Kcmi., 22, 447 (1964).
2. G о t t s t е i n W. J., С h с и е у L. С., J. Org. Cliem., 30, 2072 (1965).
3. CheneyL. С., пат. США 2787637 (1957); [С. A., 51, 13, 926 (1957)|.
1,5-ДИАЗАБИЦИКЛО-|3,4,0]-HOHEH-5 (2). Мол. вес 124,19,
т. кип. 100—102 /12 мм, п™ 1,5190.
Получение [11:
(2)
Дегидрогалогенирование. Д.— прекрасный реагент для дегид-
рогалогенирования. Трусхейт и Эйтер [21 не смогли осуществить
дегидрохлорирование хлорэфира (1) обычными третичными аминами
(пиридин, хинолин, N.N-диметиланилин). Однако после перемеши-
вания (1) с бициклическим амидином (2) при 90° и последующей
240
обработки льдом и серной кислотой из нейтральной фракции им
удалось выделить смесь цис- и транс-метиловых эфиров тридецен-
10-ИН-12-ОВОЙ-1 кислоты (3) в соотношении 70 : 30 с выходом 85%.
НСЗССН2СН(СНг),СОгСН,
С1
(1) 109 г
(2) 117,4 г
90“
—*• HC=CCH=CH(CH2),CO2CHj
8 5 то
(3)
Фогель и сотр. [31 получили 2,7-диметилоксепин (7а) в виде ста-
бильной желтой жидкости (выход 60?«) при реакции дибромэпоксида
(5а) с трет-бутилатом калия в эфире. Вероятным промежуточным
(а) Н = CHj, (б) в = н
соединением является ненасыщенный эпоксид (6а). При попытке
синтеза незамещенного оксепина по этой методике получен только
фенол. Однако применение в качестве основания Д. позволяет
получить оксепин с хорошим выходом в виде желтого нестабильного
соединения (95%-ная чистота). При температуре выше 70° это сое-
динение (т. кип. 27°/14 мм) начинает перегруппировываться в фенол.
Оксепиновая структура (76) подтверждается легким присоединением
малеинового ангидрида уже при 20° и спектром ЯМР, в котором
имеются сигналы только в области, соответствующей олефиновым
протонам.
Изучение синтеза сопряженных енинов по реакции Виттига
подтвердило (Эйтер и Одигер [4!), что наиболее удобным методом
синтеза этих соединений является дегидробромирование 4-бром-
алкинов-1 под действием Д.:
C4HSCHO + ВгСН2С = СН С4Н5СНСН2С= СН «-----С4Н5СНО + BrAlj/^CHzCHCH
с4н5снсн2с=сн ---- ; > с4н.сн=снс=сн
I 54%
в с4н4 (30°)
Высокая эффективность этого реагента видна из приведенных
в таблице результатов получения ацетата витамина А при обработке
241
хлорида (8) различными основаниями при 100 в течение 15 мин |5|.
Экстинкция (для чистого
ацетата витамина А
г = 39 490)
Пиридин, ди меги лани лип,
N-метил.морфолии
1 -100
7 500
17 500
33 500
1. Rfppe Н. et ;il., Ann., 596, 210 (1955).
2. Т г u s с 11 с i t Е., Е i t с г К., Ann., 658, 65 (1962).
3. V о g е 1 Е., S с h u b а г t R., Boll \V. A., Angew. Chem., Internal. Ed., 3,
510 (1964).
4. E i t e г К., О e <1 i g e r IE, Ann.. 682, 62 (1965).
5. О e <1 i ger 11., К a b b e H.-.J., M 6 Iler F., E i t e r K., Cliein. Ber., 99,
2012 (1966).
ДИАЗОМЕТАН, CH., N -N«->CH2—N-N. Мол. вес 42,04. т. пл.
—145 , т. кип. —23 .
Д. токсичен и взрывоопасен, поэтому операции с большими
количествами следует проводить в хорошем вытяжном шкафу с за-
щитным экраном при тщательном соблюдении предосторожностей,
отмеченных ниже.
Определение. Д. получают и используют в эфирном растворе.
Содержание его определяют следующим образом. Аликвотную пор-
цию разбавляют абсолютным эфиром п обрабатывают избытком
0,2 и. раствора бензойной кислоты в абсолютном эфире до обесцвечи-
212
вания раствора. После этого добавляют воду и титруют избыток бен-
зойной кислоты 0,1 и. раствором едкого натра.
Получение. Впервые Д. получен Пех.маном 111 при действии
спиртового раствора едкого кали па иитрозометплуретан. Однако
обычно предпочитают другие способы, так как эта жидкость обла-
дает сильным кожионарывным действием. Из всех возможных мето-
дов здесь рассмотрены только пять наиболее удобных в порядке,
соответствующем их важности.
а) Из бис- (N-метил-N-нитрозамида) терефталевой кислоты
|2|. Это соединение можно храни ть неопределенно долго при комнат-
ной температуре в виде смеси с 30"» белого минерального масла.
Порнпю нитрозамида прибавляют в один прием к смеси эфира,
монометилового эфира дпэтилеиглпколя и 30"»-пого водного раст-
Х'О \о
СН,\С--Ч-С.\’С.Нз 2CII..X, 1-ХяОеС—— СО.,Ха
1| = • ' zG-Mj",; =
О о
вора едкого натра при О'; раствор Д. в эфире перегоняют с использо-
ванием эффективного холодильника в приемник, охлаждаемый
льдом. В 2 л этого дистнллата содержится около 0,8 моля Д.
б) Из метилнитрозамида и-толуолсульфокислоты. Мол. вес
214,25, т. пл. между 55 и 60 . Получение этого соединения см. 131.
Внимание! Имеется указание 141, что это соединение может раздра-
жать кожу.
О ХО
< S. X О).
= | Г1“„
о-
о
—> СН.. — ^'--S.-OEt- СП.,Х„
= ' I
O'-
Для получения эфирно-спиртового раствора Д. [51 в перегонную
колбу па 100 мл загружают раствор 5 г едкого кали в 8 мл воды.
Колбу снабжают холодильником с двумя последовательно соеди-
ненными охлаждаемыми льдом приемниками. Во второй приемник
помещают 25 мл эфира так, чтобы вводная трубка была погружена
в жидкость. При нагревании па водяной бане до 65' в колбу прибав-
ляют по каплям в течение 25 мин раствор 21,4 г (0,1 моля) нитроз-
амида в 130 мл эфира. После введения всего реагента в капельную
воронку помещают 20 мл эфира и добавляют его по каплям, про-
должая перегонку до тех пор, пока не будет отгоняться бесцветный
эфир. Во всем дистпллате содержится около 3 г Д.
Для получения эфирного раствора Д., не содержащего спирта
[5,61, в колбу помещают 35 мл моиометплового эфира диэтилепгли-
243
коля, 10 мл эфира и раствор 6 г едкого кали в 10 мл воды. При нагре-
вании па водяной бане до 70“ и периодическом встряхивании в колбу
прибавляют по каплям в течение 20 мин раствор 21,4 г нитрозамида
в 180 мл эфира. Дистиллат собирают, как указано выше; получают
около 3 г Д.
в) Из N-метил-N-нитрозо-N'-нитрогуанидина. Мол. вес 147,11,
т. пл. 118“. Получение 17].
К смеси 100 мл эфира и 30 мл 40%-ного водного раствора едкого
кали в перегонной колбе при 0; прибавляют порциями при встря-
хивании 10 г кристаллического реагента, после чего желтый раст-
вор Д. отгоняют из реакционной смеси [81.
NH
II кон
CH3N —С— NNO2 ——CH„N2
| | 73%
NO H
г) Из N-нитрозометилмочевины, CH3NCONH2. Мол. вес 103,09.
NO
Этот реагент легко доступен [9|, из него Д. получают по простой
методике (в) [10]. Однако этот реагент необходимо хранить в холо-
дильнике, а при температуре выше 20э его можно хранить лишь
несколько часов.
д) Из 6uc-(N-метил-N-нитрозамида) щавелевой кислоты (2).
Исходное соединение — N, N'-диметиламид щавелевой кислоты
(1) — получают с выходом 94?6 реакцией диамилоксалата с метил-
амином ] 11 ]. Нитрозирование N2O3 или N2O4 в СС14 дает нитрозамид
(2) в виде оранжевого твердого вещества (т. пл. 68J, мол. вес 174, 13)
CH3N—С —С—NCH3
I II II I
н О о н
(I)
2О3 или N2O4 в СС14
75% —”
CH3N—С —С—NCH3
I II II I
NO О О NO
(2)
[121. Это соединение стабильно, невзрывчато и его можно долго
хранить в темноте при комнатной температуре; оно не раздражает
кожу. Для получения эфирного раствора Д. 8,7 г нитрозамида (2)
в 250 мл эфира добавляют по каплям при перемешивании в течение
20 мин к раствору 3 г натрия в 100 мл бутанола при 60°. Желтый
дистиллат собирают в приемник, охлаждаемый до —30J.Титрование
бензойной кислотой показывает, что выход Д. составляет 3,2 г. Этот
реагент можно также использовать для получения Д. in situ или
в газообразном состоянии.
2C4H,ONa +
CH3N—С —С —NCH3--------> 2CH, = N = N+C4H9OC—COC4H9+2NaOH
I ii II I 76% ' II II
NO О О NO О О
244
Этерификация кислот. Широко используемая реакция Д. с кар-
боновыми кислотами была открыта Пехманом II], который нашел,
что Д. является эффективным агентом для метилирования фенолов,
минеральных кислот, синильной кислоты и фталимида. Д. метили-
рует также трополоны, алкил- [13] и арилсульфокислоты [14].
Индифферентность нейтральных спиртов по отношению к Д. застав-
ляет предположить, что катализ этерификации осуществляется за
счет протона кислых соединений.
О НО ОСН3 ОСН,
|! ш Е снг-\^\ и I
RC—ОН - * RC—ОН--------> RC — ОН---> RC-O
- N 2 - Н +
Метилирование спиртов. Хотя сами спирты инертны по отно-
шению к Д., они могут метилироваться Д. при катализе эфиратом
трехфтористого бора [15] или фторборной кислотой [161.Минераль-
ные кислоты нельзя использовать для катализа, поскольку они реа-
гируют с Д. Кислота Лыопса играет ту же роль, что и органические
кислоты. При исследовании строения D-глюкуронозида эст-
— +
BF, +- CH,-N = N
ROH ROBF3—:----------> ROCH3+BF3
I
H
риола Нимен [17] использовал как некаталитическое метилирова-
ние фенольного гидроксила, так и катализируемое BF3 метилирова-
ние спиртовых групп.
Хлористый алюминий также можно применять в качестве ката-
лизатора, причем выходы обычно выше, чем с трехфтористым бором
(Мюллер [18]).
Расширение цикла. По методике, предложенной Колером с сотр.
[19] для получения циклогептанона, к смеси циклогексанона, мета-
нола и каталитических количеств тонкоизмельченного карбоната
натрия медленно, при перемешивании прибавляют нитрозометил-
уретан (4,55 моля). Цпклогептанон получают с хорошим выходом;
основная примесь — окись метиленциклогексана. Эта методика
была использована для синтеза ряда циклических кетонов до С10.
Де Боер и Бэкер [20] получали Д. в присутствии циклогексанона
из нитрозометиламида /z-толуолсульфокислоты и едкого кали в вод-
ном этаноле, скорость реакции контролировали медленным добавле-
нием твердого нитрозамида к перемешиваемой смеси. Циклогептанон
выделяли в виде бисульфитного производного и таким образом очи-
245
щади от нримссн небольшого количества циклооктанона. Однако
выход продукта при этом составляет только 33—36%.
Образование из циклогексанона кетона (2) с расширением цикла
п эпоксида (3) показывает, что первая стадия реакции состоит в
присоединении но карбонильной группе:
-С о
I
—сн„
(О’
Эфнрат трехфторпстого бора заметно ускоряет образование кетона
и подавляет образование эпоксида 1211.
Эфнрат трехфторпстого бора и борфторпстоводородная кислота
используются также как катализаторы в синтезе гомологичных ке-
тонов из стероидных Д’-З-кетонов [221. Эта реакция катализируется
и хлористым алюминием |23|.
CH2N2(BF,)
40%
Холестенон А-Гомохолестенон
Леопард п corp. [24] нашли, что Д. реагирует с пммоний-
перхлоратамп с образованием очень реакционноспособных азирпдп-
ниевых солей, которые расщепляются водой или метанолом с расши-
рением цикла:
сю.
ch2n2
——%
8 7%
сю, сн3
СН3ОН
9 0%
Фпзер 1251 изучал действие избытка Д. на фталоил нафтол (I)
(l-окси-?, 12-дпгндронленденднон-7,12) и нашел, что продукт реак-
I и
246
mm имеет дооавочные метильную и метиленовую группы, однако
полная структура не была установлена. Спустя .32 года Кромби и
сотр. 1261, применяя 7-кратный избыток Д., повысили выход соеди-
нения до .30—40% и идентифицировали его как продукт расширения
цикла и метилирования (II). (.Метиловый эфир соединения 1 не
реагирует с Д.)
Реакция Арндта — Эйстерта. По этой реакции можно привести
некоторые новые данные, появившиеся после обзора 1271, опублико-
ванного в 1942 г. По улучшенной методике 1281 разложение диазо-
кетона проводится в высококипящсм спирте (бензиловый спирт,
О о
-C.II.C1 - \i!
RCC1 , 2С.Н.Л4-------» RCCH X X ~-
RCH- с о RCH..C(),.H
октанол-2) при 160—180'; применение катализатора в этой реакции
ие обязательно, но добавление коллидина улхчшает выход. 11сслс-
дованпя кислот с асимметрической мигрирующей группой пока-
зали, что миграция происходит с сохранением конфигурации, воз-
можно, через циклическое переходное состояние 1291.
Расширение цикла по Бюхнеру. Эта реакция нашла новое при-
менение в синтезе цпклогептатриеиа (тропплидепа) при взаимодей-
ствии Д. с бензолом в присутствии бромистой меди (см. .Медь бро-
мистая).
Получение йодистого иодметилцинка. Этот активный реагент
реакции Симмонса — Смита получают добавлением раствора Д.
к эфирному раствору подпетого цинка и олефина [301:
Zn 12 •!- СН.,\, - - - JCFLZnI -г X.,
Другие реакции. Реакции Д. с различными карбонильными
соединениями рассмотрены в обзоре Гутше 1311.
1. Р е с h m a n и Н., von, Вег. 27, 1888 (1894); 28, 855 (1895).
2. Moore .1. A., R eed D. Е„ Org. Syn., 41, l(i (1961).
3. ;i e Боер T., Бэкер X., «Синтезы органических препаратов», 1151, М.,
1958, сб. 6, стр. 67.
4. К и 1> i k D.. Stenberg V. I., Cliein. Ind., 1966, 248.
5. (I e В о e г T 11. .1.. Back е г Н. .1., Rec. trav., 73, 229 (1951).
(> . де Боер Т„ Бэкер X., «Синтезы органических препаратов», 11/1. М.,
1958, сб. 8. стр. 7.
7. М с К а у A. I-'., W г i g 11 t G. F„ .1. Am. Cliein. Soc., 69, 3028 (1947).
8. Me К а у A. F., J. Am. Cliein. Soc., 70, 19/4 (1948).
9. A p и д т Ф., «Синтезы органических препаратов», ИЛ. М., 1949, сб. 2, стр. 373.
10. А ]) и .1 т Ф.. «Синтезы органических препаратов». ИЛ, М., 1949. сб. 2. стр. 174.
11. Allenby О. С. W., W г i g h t G. F„ Can. .1. Res., 25B, 295 (1947).
12. R e I in I i n g e г U., Cheili. Ber., 94, 254/ (1961).
13. А г n <1 t I’., M а г t i и n s C... Ann., 499, 264 (1932).
14. Showcll .1. S., Russell .1. R., Swern D., ,1. Org. Cliein., 27, 28,53
(1962).
15. M ii Iler E., R и n d e 1 W., Angew. Cliein., 70. 105 (1958); M ii Iler E.,
Bauer M., R u n d c 1 W.. Z. Xaturlorsch., 14B, 209 (1959).
247
16. C a s e r i о М., Roberts J. D., N e e m a n M., Johnson W. S., J.
Am. Chem. Soc., 80, 2584 (1958); Tetrahedron, 6, 36 (1959); N e e m a n M.,
Johnson W. S., Org. Syn., 41, 9 (1961).
17. N e e m a n M., Hashimoto Y., J. Am. Chem. Soc., 84, 2972 (1962).
18. M ii I 1 e r E., Heischkeil R., Bauer M., Ann., 677, 55 (1964).
19. Koh ler E. P., T i s h 1 e r M., Potter H., Thompson H. T., J.
Am. Chem. Soc., 61, 1059 (1939); see also Cr a m D. J., H с 1 g c so n R. C., J.
Am. Chem. Soc., 88, 3515 (1966).
20. д e Боер T., Бэкер X., «Синтезы органических препаратов», ИЛ, М.,
1956, сб. 6, стр. 97.
21. Н о и s е Н. О., Grubbs Е. J., Ga nnon W. F., J. Am. Chem. Soc., 82,
4099 (1960).
22. Johnson W. S., N e e m a n M., В irke I an d S. P., Tetrahedron Let-
ters, № 5, 1 (1960); J о h n s о n W. S., N e e m a n M., В i r k e 1 a n d S. P.,
F e d о г и k N. A., J. Am. Chem. Soc., 84, 989 (1962).
23. M ii 1 1 e r E., Z e e h В., H e i sell ke i 1 R., F r i c k e F., S и h r H.,
Ann., 662, 38 (1963).
24. Leonard N. J., J a n n K., J. Am. Chem. Soc., 84, 4806 (1962); Leo-
na r d N. J., J a n n K., Paukstelis J. V., SteinhardtC. K., J.
Org. Chem., 28, 1499 (1963).
25. F i e s e r L. F„ J. Am. Chem. Soc., 54 , 4963 (1932).
26. Biffin M. E. C., Crombie L., E I v i d g e J. A., J. Chem. Soc., 1965,
7500.
27. Б a x w а н В., С т р у в е В., «Органические реакции», ИЛ, М., 1948, сб. 1,
стр. 53.
28. W i 1 d s A. L., М е a d е г A. L., J. Org. Chem., 13, 763 (1948).
29. W i b е г g К. В., Н u t t о n Т. W., J. Am. Chem. Soc., 78, 1640 (1956).
30. W i t t i g G., Schwartzenbach K-, Ann., 650, 1 (1961); Hoberg H.,
Ann., 656, 1, 15 (1962).
31. Г у т ш e К- Д-, «Органические реакции», ИЛ, M., 1956, сб. 8, стр. 469.
ДИАЗОМЕТАНДИСУЛЬФОНАТ КАЛИЯ (KO3S)2C=N=N.
Мол. вес 278,36.
Д. к. был получен Пехманом и Манком [1] при реакции водных
растворов цианида калия и бисульфита калия. Образующийся
умеренно растворимый аминометандисульфонат (1) при реакции с
нитритом калия дает Д. к. (2). Эта соль кристаллизуется из воды в
KCN 2^ K°3S\CHNH„ J22 c = N = N
KO3S z ' KO3SZ
(1) (2)
виде оранжево-желтых игл или призм и необычно устойчива по
сравнению с другими диазосоединениями. Описан улучшенный ва-
риант методики синтеза Д. к. [21.
Д. к. гладко реагирует с активированными олефинами, такими,
как акрилонитрил, метилвинилкетон и этилакрнлат, с образова-
нием Д1-пиразолинов (3), которые легко перегруппировываются
в Д3-пиразолины (4) (Коттенхан |3|). Д. к. не растворим в органиче-
ских растворителях, поэтому реакцию проводят в воде, а олефин
должен хотя бы частично растворяться в воде или водном этаноле.
При обработке Д2-пиразолиндисульфоната (4) основаниями происхо-
248
дит отщепление элементов KHSO3 и образуется пиразолмоносуль-
фонат (5); при обработке кислотой наблюдается гидролиз нитриль-
ной группы до карбоксильной.
+ - CHCN
(KO3S);.C=N=N + £
(2)
1. Реей шапп Н., von, Manck Ph., Ber., 28, 2374 (1895).
2. Bannard R. A. B., R о s s J. H., Canad. J. Chem., 32, 49 (1954).
3. Kottenhahn A. P., J. Org. Chem., 28, 3433 (1963).
ДИАЗОУКСУСНЫЙ ЭФИР, N—N=CHCO2C,H5. Мол.вес 114,10,
т. кип. 42710 мм, уд. вес 1,09.
Получение. Методика, описанная в сб. «Синтезы органических
препаратов» 111, основана на следующих реакциях:
1) 2СН, -=О + \аС\' + \Н4С1 —> CH.,^ NCH2CN + \'aCH 2Н,0
2) СН, ^\СН.гС\’+С.,Н3ОН -р2НС1 + 2Н,О —-
—> С1 \ Н:1СН,СО,С,Н5 + СН,0 + N Н4С1
3) С1\Н3СН,СО,С,Н54-\а\О, —► N .= СНСО2С>Н5-{-NaCl + 2Н,0
На первой стадии формальдегид (в виде формалина) превращают
в метиленаминоацетонитрил (61—71%) 111. Это соединение при ки-
пячении со смесью абсолютного этанола, насыщенного хлористым
водородом, и 95%-ного этанола, взятого в таком количестве, чтобы
имеющейся в нем воды было как раз достаточно для гидролиза,
превращается в хлоргидрат этилового эфира глицина |2|. Горячий
раствор фильтруют для отделения хлористого аммония; при охлаж-
дении выпадает хлоргидрат этилового эфира глицина в виде белых
игл (выход 87—90%). На заключительной стадии 131 к смеси хло-
ристого метилена и водного раствора полученного хлоргидрата при
—5° в атмосфере азота добавляютпри перемешивании предварительно
охлажденный льдом раствор нитрита натрия, а затем разбавленную
серную кислоту. По мере образования Д. э. экстрагируется хло-
ристым метиленом и таким образом предохраняется от действия
кислоты. Д. э. получают с выходом 79—88% в виде желтого
масла, обычно пригодного для использования без очистки. Такая
же методика, в которой для экстракции был использован эфир [4],
дает выход 90% (не проверено).
По новой методике [5] Д. э. получают из этилового эфира
N-ацетилглицина нитрозированием в СС14 и обработкой получив-
шегося этилового эфира Ы-нитрозо-Ы-ацетилглицина смесью окиси
249
п гидроокиси оария.
N..O, или К .О,. СС1,
сн3со\ нсн.,( х ——- ------------< сн;(со.хсн,со.,с2н5—>
j
ХО
В.-.О-Вп(ОН), - -
---------_> X - сНС0оС„1Ц
М"„------- - 5
Реакция Бюхнера. Одним из примеров расширения цикла по
Бюхнеру 161 является реакция Д. э. с бензолом. При повторном
исследовании этой реакции получены следующие результаты
(Грундман и Оттмаи [41).
Следы тяжелых металлов катализируют разложение дпазоэфира;
нагревание смеси Д. э. с 20 ч. бензола в стеклянной ампуле в авто-
клаве при 136—140 приводит к 7-карбэтоксиноркарадиену (1) со
значительно более высоким выходом. Эфир (1) превращается в амид
(2), который при щелочном гидролизе подвергается перегруппи-
ровке с образованием смеси четырех пиклогептатрпенкарбоповых
кислот. Их характеристики приведены па схеме.
По реакции Бюхнера синтезирован ветивазулен из замещенного
индана (I) (Пфау и Платнер |7|). При высокой температуре, которая
Вегпноал/лен
необходима для конденсации соединения I с Д. э., аддукт II частично
изомеризуется в соединение 111. Эта изомеризация осуществляется
250
полностью при омылении смеси. Заключительной стадией является
дегидрирование, сопровождающееся декарбоксилированием. Общий
метод синтеза трополонов можно иллюстрировать на примере полу-
чения стшштатовой кислоты |8]:
Присоединение к хинонам. Д. э. присоединяется по наиболее
реакционноспособной двойной связи 1 Д-иафгохипона без отщепле-
ния азота, вероятно, по схеме 1,3-дпполярпого присоединения, как
показано ниже 191:
47%
18%
Реакцию проводят в бензоле при нагревании. Выделение в качестве
продуктов реакции З-карбэтоксппафтиидазолхппопа-й.Э и иафто-
гпдрохппоиа показывает, что первоначальный аддукт подвергается
изомеризации и окислению.
Реакция с кетонами. Д. э. очень медленно реагирует с кетонами,
обычно с образованием эфиров енолов. Однако в присутствии трех-
фтористого бора кетоны реагируют при более низкой температуре
У'
- + От-40 до-55®
+ N=N=CHCO,C,H5 + BFjEt2O + EtOEt---------->
I---1 44%
0,3 моля 0,1 моля 0,3 моля
с выделением азота и в случае циклических кетонов с расширением
цикла. Образуются гомологичные Р-кетоэфиры с очень хорошим
выходом |10|. Для цпклопентанона наилучший выход получен при
251
трехкратном избытке смеси кетон —BF3 • О(С2Н5)2 (1 : 1) в эфире
при низкой температуре.
1. Адамс Р., Ланглей В., «Синтезы органических препаратов», ИЛ, М.,
1949, сб. 1, стр. 256.
2. Марвел К., «Синтезы органических препаратов», ИЛ, М., 1949, сб. 2,
стр. 577.
3. Сир л Н., «Синтезы органических препаратов», ИЛ, М., 1958, сб. 8, стр. 9.
4. GrundmannC., Ottmann G., Ann., 582, 163 (1953).
5. R e i m 1 i n g e r H., S k a t t c b б 1 L., Chem. Ber., 93, 2162 (1960).
6. В u c h п e r E. et al., Ber., 34, 982 (1901); 36, 3502, 3509 (1903); Ann., 377,259
(1910).
7. P f a u A. St., P 1 a t t n e r Pl. A., Helv. Chim. Acta, 22, 202 (1939).
8. JohnsR. B., Johnson A. W., Tiller M., J. Chem. Soc., 1954, 4605.
9. F i e s e r L. F., P e t e r s M. A., J. Am. Chem. Soc., 53, 4080 (1931).
10. T a i W. T„ W a r n h о f f E. W., Can. J. Chem., 42, 1333 (1964).
9-ДИАЗОФЛУОРЕН (1). Д. получают окислением гидразона
флуоренона [11 и применяют для отщепления кислорода от N-окисей
(I) i ЭКв
пиридина [21. При использовании 4 же Д. 2-ппколин и 2,6-лутндин
получены с выходом 45 и 62?о соответственно.
1. Н о н и ц с с к у К., С о ,ч о м о и и к а Е., «Синтезы органических препара-
тов», ИЛ, М., 1949, сб. 2, стр. 506.
2. S с h w е izer Е. Е., O’N е i 1 1 G. J., W е m р 1 о J. N., J. Org. Chem., 29,
1744 (1964)
ДИ-я-АМИЛАМИН, (С-.Нц^К’Н. Мол. вес 157,29, т. кип. ди-н-
амиламина 189:; т. кип. диизоамиламина 183’.
Абиетиновая кислота выделяется из кислотно-изомеризованной
экстракционной канифоли прибавлением продажного Д. к переме-
шиваемому раствору последней в ацетоне [1]. Выпавшую соль
(aD —18') кристаллизуют четыре раза из ацетона и очищенную соль
(aD —60 ) обрабатывают уксусной кислотой в водном этаноле. После
кристаллизации из разбавленного этанола получают абиетиновую
кислоту с выходом 40%.
1. Н а г г i s G. С., S a n d е г s о п Т. F., Org. Syn., Coll. Vol., 4, 1 (1963).
ДИАЦЕТИЛЕН, HC—:CC :_CH. »Мол. вес 50,06, т. пл. от —36 до
—35э, т. кип. 10,3",быстро полимеризуется при температуре около 0°.
Получение. Д. можно получить дегидрохлорированием легко
доступного 1,4-дихлорбутина-2 10%-ным NaOH [11. Если требуется
С1СН2С = ССН..С1 НС^СС = СН
моно- или динатриевое производное, например, для реакции с ал-
килгалогенпдом или кетоном, то их можно синтезировать непосред-
252
ственно из 1,4-дихлорбутина-2 при взаимодействии с 3 или 4 экв
амида натрия в жидком аммиаке [2,3].
3 NaNH,
НС = CC = CNa
R X или R2C=O
---------->
HC =CC =CR нлц
]HC = CC= CCR2
L OH
Cl CHjC=CCH2Cl
4 NaNH2
NaC = CC =CNa
R X или R 2C=O
RC — CC — CR или
RaCC = CC =CCR,
OH OH
Для получения небольших количеств Д. предложен новый метод,
включающий катализируемое основанием расщепление 2,7-диме-
тилоктадиин-3,5-диола-2,7 (синтезирован дегидроконденсацией
карбинола, полученного из ацетилена и ацетона).
СН3 СН3
| | 90°
СН3СС = СС Е= СССНзН- NaOH ——► НС СС СН + 2(СН3),С = О
I I 0,13 г 80% 83%
ОН он
0,1 моля в 100 мл
ксилола
Реакции. Реакции Д. (в виде натриевых производных) с кетонами
и алкилгалогенидами приведены выше. Другие реакции даны на сле-
дующей схеме:
1 [5]) HC = CCsCH4-CH3OH —> НС^ССН=СНОСН3
2 [6]) НС s СС S СН + СН, = CHCN НС ^СС CCH,CH,CN
3 [7]) НС = СС sCH + 2СН2О НОСН,С = СС =.= ссн,он
60-70°
4 [7]) R2NCH2OH + НССС == СН —► R2NCH2C -^СС = СН
•“ Г1 2 О
5 [7])R2NH + HC^CC = CH ^д_С°р-Г- Н2С = С —Се-СН
nr2
1. Johnson A. W., J. Chem. Soc., 1946, 1009.
2. А г m i t a g е J. В., J о n e s E. R. H., Whiting M. C., J. Chem. Soc.,
1951, 44; 1952, 1993; 1953, 3317.
3. Bohlmann F., Chem. Ber., 84, 545 (1951).
4. Tedeschi R. J., Brown A. E., J. Org. Chem., 29, 2051 (1964).
5. H e r b e r t z T., Chem. Ber., 85, 475 (1952); J о h n s о n A. W., J. Chem. Soc.,
1946, 1009; Fr an ke W. et al., Chem. Ber., 86, 793 (1953).
6. Franke W., Meister H., герм. пат. 828572.
7. Co pen haver J. W., В i g e 1 о w M. H., «Acetylene and Carbon Monoxide
Chemistry», 306—308, Reinhold, 1949.
253
/«;шн<?-/«/шнс-!,4-ДИАЦЕТ0КСИБУТАДИЕН (7). .Мол. вес
170.46, т. пл. 103—104 .
Получение. Реппе и coip. Ill впервые получили Д. по указанной
схеме из цпклооктатетраепа, по вначале приняли его за 1,2-дпаце-
токсп-А:'-цнклобуген. Криге и corp. |21 установили правильную
структуру, а Хилл и Карлсон |3| предложили удобный препаратив-
ный метод получения Д., в основе которого лежит синтез дпаиетата
(4) по Коупу реакцией цпклооктатетраепа с ацетатом ртути(П)
в уксусной кислоте с последующим пиролизом продукта присоеди-
нения. Так, 52 г цпклооктатетраепа добавляют при перемешивании
к суспензии 160 г ацетата рту гп в 400 ли уксусной кислоты. Через
КОД с
(6) (7а) (70)
10—15 мин выделившийся продукт присоединения отфильтровывают
ii разлагают при нагревании до 70—75 в течение 2 час. Теплый раст-
вор отделяют от образовавшейся ртути п выливают в воду.
Диацетат (4) затвердевает при охлаждении (83 г); к его раствору
в 250 мл бензола добавляют 54 г дпметплового эфира ацетнленднкар-
боновой кислоты и смесь кипя тя т 6 час. За тем раствор фильтруют и
перегоняют в вакууме. Из фракции с т. кип. 140—155 /20 .мм, содер-
жащей смесь дпметплфталата и диена (7), при охлаждении прием-
ника кристаллизуется (7). Кристаллы промывают пстролейиы.м эфи-
ром для удаления дпметплфталата и перекристаллизовывают из смеси
ацетон — петролейпый эфир. Получают бесцветные иглы Д., т. пл.
102—104 , выход 26—31 г (40 -48"<>, общий).
Реакция Дильса — Альдера. Удобным методом одностадийного
синтеза ароматических соединений является реакция Д. с диено-
филами в кипящем бензоле пли без растворителя при 100—110
(Хилл и Карлсон |3|). Примером может служить синтез а-нафтохи-
251
нона из хинона:
AcQH^
антрахинона)
Другой пример — реакция Д. с р-питростпролом, приводящая
к 2-пптродифенилу (20%). Этот новый метод синтеза несимметричных
дифенилов был использован Хиллом и Карлсоном 141 для получения
алкалоида исмпна по следующей схеме:
liaih4
Исмии
Синтез (/./-шпкимовой кислоты (5) был осуществлен двумя груп-
пами исследователей 15,6]; приведенная ниже схема взята из послед-
ней работы 161, в которой уточнена стереохимия промежуточных
соединений (I)—(3). Реакцией Д. с метилакрилатом получают аддукт
(1), который атакой с менее затрудненной тыльной стороны цикла
цпс-гидроксилируют в диол(2) и затем превращают в ацетонпд(З).
Под действием основания в различных условиях ие удается отще-
пить уксусную кислоту от соединения (3), однако превращение(З)
в эфир (4) осуществляется с высоким выходом при пиролизе. Это
показывает, что ацетокспгруппа была скорее в транс-, а не в цнс-
положенпи к соседней карбметоксильной группе 151. Удалением
ацетонидной защиты (60%-пая уксусная кислота) и омылением
получают (/./-шпкпмовую кислоту (5), которую удается разделить
255
на антиподы.
1. R е р р е W., S с h 1 i с h t i n g О., К 1 a g е г К-, Т о е р е 1 Т., Ann., 560, 1
(1948).
2. Criegee R., Horauf W., Schellenberg W. D., Chem. Ber., 86,
126 (1953).
3. H i 1 1 R. К., С a r 1 s о n R. M., Tetrahedron Letters, 1964, 1157; J. Org. Chem.,
30, 2414 (1965).
4. H i 1 1 R. К., С a r 1 s о n R. M„ J. Org. Chem., 30, 1571 (1965).
5. M с C r i n d 1 e R., Overton К. H., Raphael R. A., J. Chem. Soc.,
1960, 1560.
6. S m i s s m a n E. E., Suh J. T., Ox m an M., D an iel sR., J. Am. Chem.
Soc., 84, 1040 (1962).
ДИБЕНЗИЛКЕТОН, C«H5CH2COCH2C„H5. Мол. вес 210,26, т. пл.
32,5 — 33°. Продажный препарат (т. пл. 31—34') можно очистить
перекристаллизацией из абсолютного эфира при —70'.
Д. применяют для получения 2,6-диметил-3,5-дифенил-4Н-пи-
ранона-4 и тетрафенилциклопентадиенона.
О
II
ДИ БЕНЗИЛ ФОСФ АТ СЕРЕБРА, AgOP(OCH 2С0Н5)2. Мол. вес
385,11, т. пл. 230° (с разл.).
Саретт и сотр. [11 разработали улучшенные методики получения
этого реагента и его использования для синтеза кортизон-21-фосфа-
та. Зервас [2] описал другие примеры использования его в качестве
фосфорилирующего агента.
1. Cutler F. A., Jr., ConbereJ.P., LukesR.M., FisherJ.F.,Mer-
tel H. E., Hirschmann R., Ch emer da J. M., Sarett L. H.,
Pfister K., 3rd, J. Am. Chem. Soc., 80, 6300 (1958).
2. Z e r v a s L., Naturwiss., 27, 317 (1939).
256
ДИБЕНЗИЛФОСФИТ (дпбензплфосфонат) 111, (С,;Н.-,СН2О)2РОН.
Мол. вес 262,26, т. ил. 216,5 .
Д. получают реакцией треххлористого фосфора с бен-
зиловым спиртом в присутствии пиридина в бензоле |2|.
Применяют для получения днбепзплхлорфосфата (см.).
I. \'ew Nomimclature: IL'PAC, see .1. Chem. Soc., 1952, 5122.
2. F r e e d m a n О. M., Klass D. 1... Sei is m a n A. M., J. Am. Chem. Soc.,
76, 916 (1954).
О
ДИБЕНЗИЛХЛОРФОСФАТ, (СД1.-,СН2О).,РС1. Мол. вес 296,68.
Новая номенклатура 111. Обзор |2|. Получают .хлорированием дибен-
знлфосфита безводным N-хлорсукцннимпдом 131.
Д. применяют для фосфорилирования нуклеозидов |4|. Он реаги-
рует со свободной первичной пли вторичной гидроксильной группой
с образованием дибензилфосфатов. Гидрирование этого эфира или
обработка тиоцианатом калия в кипящем ацетонитриле приводит
к отщеплению обеих бензильных групп и образованию моноэфпра,
а иод действием хлористого лития отщепляется только одна бен-
зильная группа и образуется монобеизпловый диэфир 151.
О о
Р
RCH,OH У С1Р(ОСН.,С6Н5), —► RCH2OP(OCH.,C6H5)2
О Н2, Pt I I о
। или KCNS LiCI ,1 ..ОН
RCH„OP(OH)„ -------! 1------>RCH2OP'
ОСН.2СвН5
1. IL'PAC see J. Chem. Soc., 1952, 5122.
2. И р а у н Д. М., «Успехи органической химии», нзд-во «Мир», М., 1966, стр. 79.
3. Kenner G. W., Todd A. R., W eymoutb F. J.. J. Chem. Soc., 1952,
3675; Hall R. H., Khoratia H. G., ,1. Am. Chem. Soc., 76, 5056 (1954).
4. T о d d A. R., J. Chem. Soc., 1946, 647; К e n n e г G. W., Fortsch. Chemie
ory. Xaturstoffe, 8, 96 (1951).
5. Clark V. M., T о d <1 A. R., .1. Chem. Soc., 1950, 2030; Christie S. M. H.,
E 1 tn о r e D. T., Kenner G. W., T о d d .1. R., We v m о u t h F. J.,
ibid., 1953, 2947.
/п/ш«с-1,2-ДИБЕНЗОИЛЭТИЛЕН. Мол. вес236,27, т. пл. 109—
по-.
Получается из фумарплхлорида и бензола [1].
о
'I
НССОС1 н—с-ссвн6
b .50 —со- ;
CIOCCH + С,Н„ л- А1С13 ---------сон-с—с—н
1 моль 800.it; 2,6 моля ,s S3“'' ||
О
Диенофил. При кипячении Д. с 2,3-дифепилбутадиено.м в спирте
в течение 4 час получается аддукт с выходом 9О”о [21.
Превращение кислот в ангидриды. Д. в смеси с три-н-бутплфос-
фииом (или трпфенплфосфином) превращает карбоновые кислоты
9 Заказ № 965
257
в их ангидриды с выходом 50 -80°о [31. Отщепление элементов воды
происходит обычным путем -- третичный фосфин соединяется с ки-
слородом, а Д. присоединяет водород с образованием дпбензоилэта-
на. При добавлении к бензольному раствору Д. при комнатной тем-
пературе три-н-бутплфосфпна раствор разогревается и приобретает
красную окраск). Через 10 мин этот раствор добавляют к бензоль-
ному раствору 2 же пропионовой кислоты и смесь кипятят 2 час,
пропионовый ангидрид получают с выходом 78% вместе с дпбепзоил-
этаном (77%) и трп-«-бутилфосфиноксидом (84%). Предполагаемая
последовательность реакции приведена ниже.
О о °Р" (С|Н9):1
11 (С4Н„)3Р I
СвН6ССН = СНССвН5------"C6HS(:сн-сн = сс,.н5 —-
о-
ОР + (С4Н9)3
RCO.H |
-----*С0Н,С-=СНСН„ССвН5 -<
RCO-
i: О
о
0 9
_______RC0-"_______„ RC~ОРДСДЧЬ_________ R(-%o + о= Р(С4н9)3
~с,н5сосн,сн.сос„н5 _QCR_______________RC?
1. Л ю т ц Р., «Синтезы органических препаратов», ИЛ, М , 1952, сб. 3, стр. 1С9.
2. Allen С. F. Н., (} a t е s J. \V., Jr.. .1. Am. Chem. Soc.. 65, 1283 (1943).
3. К u w a j i rn a 1., M u k a i у a m a T., J. Org. Chem., 29, 1385 (1964).
ДИБОРАН, B2Htl. Мол. вес 27,69, т. пл. —165', т. кип. —92,5'.
Д. — самовоспламеняющийся газ; очень токсичен при вдыха-
нии., причем по запаху нельзя определить токсическую концентра-
цию, так как под действием Д. чувство обоняния притупляется.
Хотя чистый Д. самопроизвольно не воспламеняется на воздухе при
комнатной температуре, он обычно содержит продукты разложения,
которые могут вызвать самопроизвольное воспламенение. Примерно
1AI раствор ВН3 в ТГФ хранят при 0 — 5‘, образцы отбирают
шприцем.
Д. хранят также в баллонах из хромомолибденовой стали при
— 80= (минимум 100 г). Д. применяется как ракетное топливо.
Д. удобно получать по реакции (1)
3XaBH4-f-4BF:1—2B2HG % 3XaBF4 (1)
Раствор боргидрпда натрия в диглиме (диметиловый эфир ди-
этиленгликоля, т. кип. 16Г, смешивается с водой) прибавляют по
каплям при перемешивании к смеси эфирата трехфтористого бора
258
и диглима. Образующийся газ подается слабым током азота в реак-
ционною колбу, содержащую раствор олефина или другого реагента
обычно в тетрагпдрофуране [11. Схема прибора приведена в работе
Цвсйфеля и Брауна |2[ (стр. 32). Применяют 20%-ный избыток
борнщрида натрия и 50%-ный избыток эфирата трехфтористого
бора. Выводную трубку’ из реакционной колбы погружают в ло-
вушку с ацетоном, который реагирует с избытком Д. с образованием
дпизопропоксиборана [реакция (2)1, разлагающегося водой по урав-
нению (3).
4(СН;ЛСО 4- В.,Н, -> 2|(С1-13).,СНО].,ВН (2)
[(СН3).,СНО]2ВН -г ЗН.,0 —> 2(СН,).,СНОН + Н: - Н3ВО3 (3)
В.2Н„ -I- 6Н..О —> 6Н.. г 2Н;,ВО3 (4)
По другому способу |31 в колбу помещают хлористую ртуть и
диглим, пропускают при перемешивании ток азота и создают вакуум,
2.\аВНИ-НйХ1,—Д" 2Н£; !-2.\аС1Л-В,Н„ - Н ,.
1 г 1'3.6 1 8S"„
близкий к давлению паров растворителя. Выводную трубку соеди-
няют с серией последовательно соединенных ловушек, охлаждаемых
жидким азотом, и прибавляют из капельной воронки раствор бор-
гидрида натрия в диглиме. Сразу начинается экзотермическая реак-
ция, так что нагревания не требуется. В приборе поддерживают
вакуум до тех пор, пока весь Д. не соберется в ловушки. Вместо
хлористой ртути можно применять иод; однако в этом случае лучше
прибавлять иод к раствору боргидрида.
Восстановление. Реакция Д. с ацетоном 1см. уравнение (2)1
и другими простыми карбонильными соединениями с образованием
диалкоксиборанов 141, возможно, включает равновесие В..Н„^2ВН3,
и первую стадию можно описать следующим уравнением (5):
I. Н 1
R .С _ О---> [ДС-.сО- ►R..C —О > R с—о , .
, ; । ( ,5)
н—11*11, Ц...ЦЦ, II ВИ;
Моноалкоксиборан не выделен и, очевидно, реагирует со вторым
молем карбонильного соединения с образованием диалкокспборана.
Даже в случае ацетальдегида реакция завершается именно на этой
стадии, что, по-видимому, обусловлено пространственными препят-
ствиями. Реакции легкого образования и гидролиза [реакции (2)
и (3)1 диалкоксиборанов являются общим методом восстанов-
ления, который, однако, редко применяется из-за сравнительно
трудной доступности Д. Д. при комнатной температуре легко
восстанавливает альдегиды, кетоны, эпоксиды, карбоновые кислоты,
нитрилы, азосоедппенпя и амиды 151. Восстановление можно прово-
дить или пропусканием Д. в раствор органического соединения, или
прибавлением эфирата трехфторпстого бора к раствору боргидрида
натрия и восстанавливаемого соединения в ТГФ пли диглиме. Пред-
почитают ТГФ ввиду лучшей растворимости в нем Д.
9*
259
Сравнение относительной реакционной способности различных
групп по отношению к Д. дает следующий порядок активности [61:
—С( )2Н > — С - -= С— > R2C — О > RC ~ X >
°\
С — > —CO2R'
Не восстанавливаются следующие группы: — COCI,—NO,,—
SO2—, RX, АгХ, простые эфиры, фенолы. Следует специально ука-
зать на легкое восстановление с помощью Д. карбоксильной группы,
которая не восстанавливается NaBH.,. Порядок реакционной спо-
собности нитрилов и сложных эфиров по отношению к Д. изме-
няется на обратный при восстановлении LiBH4 171. Хлораигпдриды
кислот восстанавливаются боргидридом натрия 171, но не Д.
Существенная разница в реакционной способности труни 151
по отношению к Д. и боргидрнду натрия обусловлена тем, что
Na+BHf является основанием, реакции которого осуществляются
путем атаки попа BHf на центр с дефицитом электронов. Д., как
кислота Льюиса, преимущественно атакует группы с высокой элект-
ронной плотностью. Поэтому, применяя Д. или боргидрид натрия,
можно избирательно восстанавливать различные группы в при-
сутствии других. Например, карбоксильную i руппу можно изби-
рательно восстановить Д. в присутствии карбонильной или нитро-
группы. Преимущественное восстановление карбоксильной группы
антрахинона (I) приводит к образованию соединения (II) (выход
около 60"<>); хиноновый цикл затрагивается только при избытке
восстановителя 181. Лптрахппон можно восстановить в антрацен под
действием Д., получаемым in situ |8|. Раствор антрахинона (2 г)
в диглиме приобретает темно-красную окраску при добавлении
боргидрпда натрия: после обработки эфпратом трехфторпстого бора
и перемешивания в течение 2 час получают антрацен (1,2 е).
Восстановление легко доступных альдоксимов и кстокспмов под
действием Д. в ТГФ — простой и удобный метод получения моноза-
мещеииых гидроксил аминов |91:
- В -
р [j I | Води. Na ОН
RR'C ХОН _'-A PITCH —X—О-В—----------------- RR'CHXHOH
R—алкил или арил; R' -Н или алкил
260
Нитросоединеиня не изменяются под действием Д., однако соли
нитросоединенпй восстанавливаются до гидроксиламинов:
RC--- NO., Ме+ ВаН|,: Н+. ил" RCHNHOH
i I
R' R'
В обычных условиях лактоны восстанавливаются алюмогидри-
дом лития до диолов. .Метод восстановления 6-лактонов Д. в ТГФ
до циклических ацеталей был применен для синтеза некоторых
необычных оксастеропдов [101.
Пространственно незатрудненные олефины реагируют с Д. и
образуют триалкилбораны. Эти соединения, сравнительно устой-
чивые к действию водных кислот и оснований, подвергаются неожи-
данно легкому протолизу уксусной кислотой. Таким образом, гидро-
зон,соон
3RCH^CH2T 1/2В2Н6 -- (RCH,CH.,)3B----;-----« 3RCH,CH3 Г(СН3СО,)3В
борирование олефинов с последующим протолизом является мето-
дом восстановления двойных связей [111. В применении к олефинам
этот метод едва ли имеет преимущества по сравнению с гидрирова-
нием. Однако на этой основе был предложен метод гомогенного гид-
рирования, катализируемого алкплборанамп |12|. Циклогексен
или каприлен, содержащие 3,8% трп-н-бутилборана, количественно
гидрируются в течение 3 час при 220 и давлении водорода 70 атм.
Эта реакция пригодна для восстановления высокополимеров в раст-
н, к-сн=сн, зн.
BR3 —R,BH-------------«- R'CH2CH2BRj---R'CH,CH3
— R Н
воре.
Гидроборированием и протолизом можно восстановить алкины.
Так, гексин-3 под действием Д. образует ненасыщенное производное
С2Н5
I
С
О” С2Н,—С—Н о° с,н,—С—н
3 С Т-1/2В2Н6-------, : —- I!
| С.,Н-,—С—Bl/3 AlOH С,Н-,—с—н
С.,Н5
бора, которое при обработке безводной уксусной кислотой превра-
щается в чистый ф(с-гексен-3 131. Попытки прямого гидроборирова-
ния гексина-1 Д. в диглиме показали, что этот терминальный
261
ацетилен реагирует частично с присоединением 2 молей Д. Однако
гладкое восстановление до гексена-1 можно провести, применяя в
качестве восстановителя пространственно затрудненный бис-( 1,2-ди-
метплпропил-1)-боран (Браун использует для этого соединения три-
виальное название диизоамилборан). Присоединение атома бора к
концевому атому углерода доказано окислением перекисью водорода
и выделением w-капропового альдегида с выходом 88%.
СН3 СН., СН3
I 1 2В,Н„ I I С,Н,С i.H
2СН3С - - СНСНз-----* (СН;,СН —СН);ВН----------
ДсОН /гфН8СН--СН,
Н-С-С4Н9 /
'н— С — BR,
Н2°^с5нпсно
Представляет интерес синтез циклопропанов из аллилхлоридов
[141. ПрисоединениеД. к ал тилхлориду в эфире дает смесь моно-, ди-
и три-(у-хлорпропил)-боранов. Щелочным гидролизом смеси полу-
СН,
СН., -СНСН,С1 — НД Н„В(СН.,СН.,СН„С1)3_„ cfl—сн,
43%
(п = 0. 1. ?)
чают циклопропан. Этим способом были полечены метил-, «-пропил-,
фенил- и бензилциклопропаны с выходом 45—71%.
Д. восстанавливает с высоким выходом амиды до соответствую-
щих аминов (Браун и Гейлп 1151). Медленнее восстанавливаются
СОХ’(СНз), СН,Х(СНч),
I I
Кипячение I ч-с в ТГФ
амиды, не замещенные по азоту; реакция требует кипячения в тече-
ние 8 час. Еще медленнее и с худшим выходом эти амиды восста-
навливаются алюмогпдридом лития. Алифатические амиды восста-
навливаются Д. в амины с хорошим выходом (Паианастасну и Брю-
ии) 1161. Д. используется, в частности, для восстановления произ-
водных фторацетампда, так как L.i А1Н, или Li Al Н,—А1С13 вызывают
гидрогенолиз связи С—F.
в .н„
FCH„CO?<HR —FCHjCHAHR
Гидроборирование. Джонсон и Ван Кампен 1171 заметили, что
три-н-бутилборан гладко окисляется щелочной перекисью водорода
в эфир борной кислоты, который гидролизуется в щелочной среде
262
до w-бутанола и борной кислоты. Последующие исследования
Брауна и сотр. |2,18) показали, что окисление почти количественно
II.02 IIC.1
(СН3СН2СН2СН.,)3В (СН3СН2СН2СН.,О).,В-’ЗСН3С112СН.,С112ОН '-Н;!ВО3
проводится при 25 ц растворитель, применяемый для получения
алкилборана, не мешает окислению, и поэтому реакцию можно про-
водить без выделения борорганического соединения. В связи с этим
реакция гидроборирования получила очень широкое распростране-
ние. Присоединение Д. к двойной связи и последующее окисление
в спирт широко используются в качестве метода непрямой гпдра-
II II
Н —В — н он
I
танин. Для этого двухстадийного окислительного процесса исполь-
зуется термин гидроборирование, чтобы отличить его от процесса
восстановления, включающего присоединение по связи Н—В и
протолиз. Гидроборирование несимметричных олефинов в отличие
от гидратации их серной кислотой с последующим гидролизом осу-
ществляется против правила Марковнпкова. Терминальные оле-
в..н, н2о.
RCH = СНЙ----” (RCH2CH2)3B RCH2CH.,OH
фины дают почти исключительно соответствующие первичные
спирты, например, октен-1 дает смесь октанола-1 и октанола-2
в соотношении 9:1. Такое направление присоединения попятно:
ВН3 — кислота Лыоиса с дефицитом электронов, образующая связь
с наиболее гидрогенизированным атомом углерода. Гексен-2 дает
примерно равные количества двух возможных алкилборапов; однако
если реакционную смесь кипятить несколько часов в диглиме п за-
тем окислять, то получается с высоким выходом гексаиол-1 1181.
Таким образом, присоединение по связи Н -В обратимо, и наблю-
дается перенос бора от внутренних к терминальным атомам углерода.
При изомеризации олефина с двойной связью внутри цени, например
З-этилпентена-2, в термодинамически менее стабильный олефин
происходит присоединение ио связи Н—В н устанавливается равно-
весие. После добавления децена-1 как акцептора бора медленной
отгонкой получают более летучий З-этплпентен-1 1191.
Терминальные олефины и простые олефины, такие, как бутен-2,
обычно образуют триалкилбораиы. Трехзамещепные олефины, ria-
в.п.
сн3сн=снсн3-------- СН3СНСН„СН3
I
В I 3
.263
пример 2-метилбутен-2 и 1-метилциклогексен, образуют диалкил-
бораны.
сн3 сн3
I В..Н, |
(СН - СН------- [(СН3),СНСН — ]2ВН
Тетразамещенпый олефин тетраметилэтилен реагирует быстро,
но образует моноалкилборан.
СН3 СН:!
I В..Н, I
(СН3).,С = С-----(СН3).,СНСВН„
I I
СН:! сн3
Первым примером олефина, который не реагировал с Д., был
5₽-А” ‘“’-стероид 1201. Другой 5р-А9 ‘“’-стероид оказался также
инертным к гидроборированию (Нуссим и Зондхеймер |211). Однако
5с/.-А9 ‘“’-стероиды, не настолько пространственно затрудненные
са-стороны, легко реагируют с Д. и дают после окисления 1 la-спир-
ты с сохранением a-конфигурации у С9. Таким образом, реакция
включает /р/с-прпсоединение с менее затрудненной а-сторопы.
1-Метилциклопентен и 1-метилциклогексен также подвергаются
гидроборированию и окислению с образованием продуктов цис
гидратации |22|. Холестерин реагирует с Д. против правила Мар-
иовникова по схеме ц/л’-присоединения 1221, давая смесь продуктов
(1) и (2), в которой преобладает (1) —продукт атаки са-стороиы 1201.
Стереохимическая направленность реакции и несомненное влияние
эффектов экранирования подтверждают возможность присоединения
через циклическое переходное состояние:
с=с
+
н -в-
I
I
264
.Миграцию атома бора при повышенной температуре из внутреннего
в терминальное положение можно объяснить последовательным от-
щеплением и присоединением через циклические переходные
состояния.
Если гидроборирование олефина проводят для последующего
окисления образующегося вторичного спирта в кетон, то можно
обойтись без промежуточного окисления перекисью водорода, а ал-
килборан непосредственно окислять хромовой кислотой. Так, дез-
окси-Д5-стеропд превращают в смесь стереоизомерных алкилбора-
нов, подобных (1)и (2), окислением которых хромовой кислотой
получают 6-кето-5а-стероид с хорошим выходом (Паппо 1231); перво-
начально образующийся 6-кето-5|3-стероид при этом изомеризуется.
Браун 1241 предложил эффективную двухфазовую системе (хромовая
кислота — эфир) для окисления в кетоны как алкилборапов, так и
свободных спиртов.
Д., как чистый, так и в присутствии олефина, обычно получают
по уравнению (1), прибавляя раствор боргидрида натрия в диглиме
к раствору эфирата трехфтористого бора в том же растворителе.
В связи с тем что диглим трудно доступен, были разработаны альтер-
нативные методики |25, 261. Так, диэтиловый эфир — подходящий
растворитель при получении Д. из алюмогпдрида лития и эфирата
трехфторпстого бора. Например, раствор алюмогпдрида лития
в эфире добавляют в атмосфере азота при перемешивании к раствору
эфирата трехфторпстого бора и холестерина в эфире |26|. После
окисления перекисью водорода и ацетилирования получают дпаце-
тат холестандиола-ЗР,6а с выходом 70?п п диацетат копростандио-
ла-Зр, 6Р (выход 15—20%). Позднее Браун и сотр.(271 изучили боль-
шое число различных методик, однако ни одна из них не имеет
особых преимуществ.
Избирательное присоединение алкилборапов к олефинам по связи
Н—В расширило границы применения реакции гидроборирования.
Так, бис-( 1,2-диметилпропил-1)-боран (БМБ), получаемый по урав-
нению (1), даже с избытком несимметричных олефинов дает преиму-
щественно пространственно менее затрудненные продукты; такое
избирательное взаимодействие неосуществимо другими способами
1281. В отличие от Д. этот реагент не восстанавливает карбопиль-
СН3 СН3
I о- |
2СН3С = СНСН3 — 1/2B.,Hg--► [(СН3),СНСН|2ВН (I)
БМБ
БМБ. Н.О,
СН2==СН(СН;)вСО,Н-----ГД——* СН.,(СН.,)0СО2Н (2)
Я о п j
он
БМБ. Н,О,
СН,СНСН»СН.,С-= О----:—- — СН.,СНСН.,СН,СНО (3)
I I (>% I - -
265
ную группу, поэтому его можно применять для гидроборирования
незащищенных кислот (2). у-Дактоны реагируют только с 1 молем
реагента, даже если последний присутствует в избытке (3).
Гидроборированием экранированных оптически активных оле-
финов (например, терпенов или стероидов) легко получают диалкил-
бораны, которые можно использовать для превращения подходя-
щего олефина в оптически активный спирт: например щщ-бутена-2
в (—)-бутанол-2 1291.
Гидроборирование под действием Д. в диглиме сравнитель-
но мало зависит от структуры олефина [301. Реакционная способ-
ность наиболее активного из изученных олефинов—2-метилбутена-1
только в 20—30 раз выше наименее активных 2,4,4-триметил-
пентепа-2 и 2,3-диметилбутена-2, тогда как при гидроборировании
БМБ самый активный октеп-1 оказался в 10 000 более реакционно-
способным, чем наименее активный из изученных — циклогексен.
Это исследование пе могло быть распространено на еще более инерт-
ные олефины, например 2,4,4-триметнлнентен-2. Гексин-1 и гексин-3
оказались еще более реакционноспособными, чем самые активные
из изученных олефинов.
Как указывалось выше, пространственно затрудненный 2,3-ди-
метнлбхтен-2 (1) дает при реакции с Д. в диглиме (1—2 час) моноал-
СН3 СН3 СИ.., СПз
- i в нь I I
СИ. С .С —> СНз—С—с—вн,
I °’ 'll'
СПз Н СНз
(1) (2)
килборан — 2,3-дпметилбутпл-2-боран (2) («текенлборан»). Этот ре-
агент находит особое применение ввиду больших пространственных
требований для реакций с его участием [311*. Описаны характерные
свойства и применение диалкилборанов, полученных при гидро-
борировании циклогексена, 1-метилииклогексена и а-пинена [321.
При восстановлении боргпдрпдом натрия или боргпдридом лития
пространственно незатрудненных кетонов или кетонов, существую-
щих в подвижном конформационном равновесии, главным продук-
том является наиболее устойчивый спирт. Так, холестанон-3 дает
экваториальный 30-снпрт, который более стабилен, чем За-эппмер.
2-Метплииклопентаноп и 2-метплциклогексанон восстанавливаются
гидридами металлов с образованием более стабильных транс-ст\'й-
тов с выходом 69—75%. Д., хотя он и отличается по своей вос-
станавливающей способности от гидридов металлов, также восста-
навливает эти два циклических кетона в /прпнс-спирты с выходом
69 и 65% 1331. Однако выход z^wc-сппрта становится преобладающим,
* Особое значение этот реагент приобрел для гидроборирования цикличес-
ких диенов с образованием бициклических структур, содержащих бор 131а].—
Прим, перев.
266
если восстанавливать более пространственно затрудненным алкилбо-
раном. Так, zp/c-2-метплциклопентанол и цнс-2-метилцпклогексанол
образуются с выходом 78 и 77% при восстановлении бпс-(1,2-диме-
тилпропил-1 )-бораном; выход повышается до 91 и 92% соответ-
ственно при использовании более объемного диизопинокамфил-
борана, полученного гидроборированием а-ппнена.
Гидроборированием енолапетата (1) получен 3-метиловый эфир
Обработка енамина (1) Д. и уксусной кислотой в ТГФ дает кис-
лоту (2), при кипячении которой с уксусной кислотой в диглиме
получают циклогексен с общим выходом 95% [35].
Расщепление циклопропанов. В отсутствие растворителя цикло-
пропаны подвергаются восстановительному расщеплению Д. 1361.
Так, при нагревании норкарана с Д. при 100" в течение 1—2 час
получается алкилборан (2), идентифицированный после расщепления
перекисью водорода в циклогексплкарбипол (3). В тетрагидрофуране
реакция не имеет места.
Расщепление простых эфиров. Смесь Д. и галогена, особенно
иода, легко расщепляет простые эфиры с образованием боратов, ко-
6С6Н5ОСН«0-31, -1-В.,Н„ —> 2В (ОС6Н5)з J-6CH31 ; ЗН,
В(ОС0Н5)3 Д-ЗН.,0- , ЗСсН,ОН- н3во3
267
торые гидролизуются водой при комнатной температуре за не-
сколько минут (Лонг и Фригуард 1371). Реакция применима к сим-
метричным или несимметричным, ароматическим, алифатическим
и циклическим эфирам и может иметь препаративное значение, на-
пример расщепление тетрагидрофурана Д. и подом дает 4-иодбута-
нол с выходом 90%.
1. Brown Н. С., Tierney Р. А., .1. Am. Chem. Soc., 80, 1552 (1958).
2. Ц в е ii ф ель Ж., В р а у и Г., «Органические реакции», изд-во «Мир».
А1., 1960, стр. 7, see also Brown Н. С.. «Hydroboration», Benjamin
\V. A., Inc.. 1962; Tetrahedron, 12, 117 (1961).
3. Frecguard G. F., LongL. H., Chem. Ind., 1965, 471.
4. Brov. nll.C., S c li I e s i n g e r H. I., В u r g A. B., J. Am. Chem. Soc., 61,
673 (1939).
5. Brown II. C., S u b b a R а о В. C., J. Am. Chem. Soc., 82, 681 (1960).
6. В г о w n H. C., Rorytn у k W., J. Am. Chem. Soc., 82, 3866 (1960).
7. Bro w n II. (2., S u b b a R а о В. C., J. Org. Chem., 22, 1135 (1957).
8. В a p a t D. S., S u b b a R а о B.C., L n n i M. K., Venkataraman K.,
Tetrahedron Letters, № 5, 15, (I960).
9. F с uer IL, Vine e n t B. F., Jr., J. Am. Chem. Soc., 84, 3771 (1962); F e-
u e г IL, V i n c e u t B. F., Jr., В a r t I e t t R. S., J. Org. Chem., 30, 2877
(1965): F e u e r IL, Bartlett R. S., V i n c e n t B. F., Jr., Ander-
son R. S.. .1. Org. Chem., 30, 2880 (1965).
10. Pettit G. R„ Kasturi T. R„ Green B., Knight J. C., J. Org.
Chem., 26, 4773 (1961).
II. Bro w n II. Murr а у К., J. Am. Chem. Soc., 81, 4108 (1959).
12. I) e Witt E. J., Ramp F. L., T r a p a s s о L. E., J. Am. Chem. Soc.. 83,
4672 (1961).
13. В г о w nil. C., Z w e i f e 1 G., J. Am. Chem. Soc., 83. 3834 (1961).
14. II a w t h о r n e AL F., J. Am. Chem. Soc., 82, 1886 (1960).
15. В г о w n II. С., H e i m P., J. Am. Chem. Soc., 86 , 3566 (1964).
16. P a p a ii a s t a s s i о u Z, В., В r u n i R. J., J. Org. Chem., 29, 2870 (1964).
17. J о h n sonJ.R., A'anC a m pen AL G,, J. Am. Chem. Soc., 60, 121 (1938).
18. В г о w n H. C.. S u b b a R а о В. C., J. Am. Chem. So"., 81, 6423 (1959).
19. В r о w n II. С., В h a t t M. V., J. Am. Chem. Soc., 82, 2074 (I960).
20. W e ch ter W. J., Chem. Ind., 1959, 294.
21. X u s s i in AL, S о n d h e i m e r F., Chem. Ind., 1960, 400.
22. В г о w n H. C., Z w e i f e I G., J. Am. Chem. Soc., 83, 2544 (1961).
23. P a p p о R., J. Am. Chem. Soc., 81, 1010 (1959).
24. Bro w n H. C., Garg С. P., J. Am. Cheni. Soc., 83, 2951 (1961).
25. D n I о w R., C h r e tien-Bessiere Y., Compt. Rend., 248, 416 (1959).
26. W о 1 f e S., X ii s s i ni AL, Al a z u r Y., S о n d h e i nt e r F., J. Org. Chem.,
24, 1034 (1959).
27. В г о w и H. C., Al и r r a v K. J., Snover J. A., Z w e i f e I G., J. Am.
Chem. Soc., 82. 4233 (I960).
28. В г о w n 11. C., Bigley D. B., J. Am. Chem., Soc., 83, 486 (1961).
29. В г о w n H. C., Z w e i f e I G., J. Am. Chem. Soc., 83, 486 (1961).
30. В г о w nH. С., Moer ikofer A. W., J. Am. Chem. Soc., 85, 2063 (1963).
31. Z w e i f e ! G., Brow n H. C., J. Am. Chem. Soc., 85, 2066 (1963).
31a. В г о w n H., P f a t t e n be r ge rC.D., J, Am. Chem. Soc., 89, 5475 (1967).
32. Z w e i f e 1 G., A v v a n g а г X. R., В г о w n H. C., J. Am. Chem. Soc., 85,
2072 (1963).
33. В г о w n H. С., В i g 1 e у D. B., J. Am. Chem. Soc., 83, 3166 (1961).
34. A i v a r e z F. S., Arreguin AL, Chem. Ind., 1960, 720.
35. L e w i s J. \\L, Pearce A. A., Tetrahedron Letters. 2039 (1964).
36. R ic kborn B., W о о d S. E., Chem. Ind., 1966, 162.
37. 1. о n g L. H., Freeguard G, E., Xature, 207, 403 (1966).
268
1.3-ДИБРОМ-5,5-ДИМЕТИЛГИДАНТ0ИН(«дибромантпн»). Мол.
вес 285,95, т. пл. 195" (с разл.).
о
Д. подобно N-бромсукипнимиду применяется для аллиль-
ного бромирования, дегидрирования, замещения активного водорода
и окисления вторичных спиртов. Обзор |1|.
Попытка бромирования 2,4-дпбензопламино-6-метилхпназо-
лина в боковую цепь бромом в хлороформе привела к отщеплению
4-бензопльной группы. Реакция с применением N-бромсукцинпмида
+ Дибромантин
О, 8 2
(С Н С О О) кЦпящ,емСС14,1 чдс45>мин
79%
О, 15 2
28
в хлороформе или четыреххлористом углероде также дала отрица-
тельный результат 121. Однако желаемое 6-бромметильное производ-
ное было получено с высоким выходом бромированием Д. в кипящем
СС14 в присутствии перекиси бензоила при освещении лампой
(500 ет) с вольфрамовой нитью.
1. R е с (I R. A., Chem. Prods., 23, 299 (1960); [С. А. 54, 21059 (I960)].
2. Oak е s V., R у d о n Н. N., U n d h е i m К., J. Chem. Soc., 1962, 4678.
ДИБРОМДИФТОРМЕТАН, CF2Br,. Мол. вес 209,86, т. кип. 24,5=.
Д. реагирует с алкпллптнем или алкплмагнийгалогенпдом с об-
разованием олефина по следующей схеме 111:
3RCH,Li (MgBr) -- CF2Br2 1?CH "CHCH2R + RCH2Br-(-2LiF-|-
LiBr(MgBrF . MgBr.,)
Эта реакция является удобным методом синтеза олефинов с нечет-
ным числом атомов углерода и центральным положением двойной
269
связи. Постулирован следующий механизм образования карбена:
- 70°
C3H7CH,l.i J CF.Br.,->- C3H.CH2Br4-:CF.,-i-LiBr
СД1-СНЛ : -.CF.,—C3H7CH..CF - LiF
Li
C3H,CH.,CF~C3H7CH.,Li — C,H-CH.,-C —CH.C3H. Л'Д.
I ' ' -LiF
F
— C3H7CH--CHCH2C3H7
1. Franzen V., Fikentscher L., Chem. Ber., 95, 1958 (1962).
ДИБРОММАЛОНОВОЙ КИСЛОТЫ ДИАМИД, CBr»(CONH2)2.
Мол. вес 259,91, т. пл. 203J.
Получение. Д. к. д. получают с выходом 94% при реакции
амида малоновой кислоты с бромом в присутствии ацетата натрия
для связывания образующегося бромистого водорода II].
Для фосфорилирования. Оксисоединеиия фосфорилируются при
реакции с 1 молем Д. к. д. и 2 молями трнэтилфосфита [2|. Амид
прибавляют к смеси трнэтилфосфита и w-пропанола, большой избы-
ток которого служит растворителем. После выдерживания в течение
некоторого времени амид малоновой кислоты отделяют филырова-
О
2ROH -l СВг., (СОХН.Д, - 2 (С.,Н-,О),Р -s- 2ROP (ОС.,Н5)., - СН., (СОХH.,)., +
- 2С.,Н5Вг
нием. При взаимодействии диэтилфосфата, Д. к. д. и трнэтил-
фосфита в эфире при О’ образуется тетраэтилпирофосфат.
О
11
2(С2Н5О).,РОН - СВг.ДСОХН,),-'-2 (С.,Н5О)3Р --д*
О о
— 2 (С,Н;О)..Р —О —Р (ОС.НД, -СН, (COXH,),- 2С,Н5Вг
J. BackesJ. V„ West R. \V., Wllitelev M. A., .1. Chem. Soc., 119, 365
(1921).
2. Mukai у a rn a T., H a t a T., Tasaka K., J. Org. Chem., 28, 481 (1963).
ДИБРОМЭТАН, ВгСН,СН2Вг. Мол. вес 187,87, т. пл. 9—10=,
т. кип. 131,7, уд. вес 2,180.
Реагент для «вовлечения». Д., по-видимому, лучше, чем этил-
бромнд, способствует превращению нереакцпонноспособных гало-
генидов в реактивы Гриньяра 111. Одно из преимуществ состоит
в том, что Д. реагирует с магнием с образованием бромистого магния
и этилена, и, таким образом, в реакционной смеси отсутствует
второй реактив Гриньяра. При медленном добавлении к нереак-
270
шюнноспособному галогениду и магнию обеспечивается взаимо-
действие галогенида с чистой блестящей поверхностью магниевой
стружки. Таким образом превращают сс-хлорнафталин в ^-нафтой-
ную кислоту с выходом 56% 111. Другой пример — превращение
гексахлорбензола в пентахлорбензойную кислоту 12]. Смесь гекса -
Cl MgCl CO..H
Cl. A п Cl ЗМс - 2BrC.H .CH ,Br Cl //'. Cl 1 ,! Cl co, ~N|/C'
1 1 ci у Cl 0,5 моля в 1 .i Cl 5фпря -2Л1йВг,-CCH. = CH: Cl 'Cl 1 Cl 65% Cl Д ' Cl Cl
хлорбензола, магния и эфира кипятят при перемешивании и в тече-
ние 48 час из воронки Гсршберга добавляют раствор Д. в бензоле
(истребуется особого внимания, если платиновая проволока точно
пригнана). В темно-коричневую смесь пропускают ток сухой двуо-
киси углерода, прибавляют 10%-ную соляную кислоту, продукт
реакции очищают через аммонийную соль и перекристаллизовывают
из 50%-ного метанола.
1. Pea rso iiD. Е., С о w а п D., В е с k 1 е г J D.. J . Org. Chem., 24, 504 (1959).
2. Pears о и D. Е., С о w а п D., Org. S\n., 44. 78 (1964).
ДИ-н-БУТИ.Л АМИН, (CH:)CH;CH2CIL)Л'Н. .Дол. вес 129,24,
т. кии. 159,6 , уд. вес 0,768, умеренно растворим в воде, рКЬ 2,7.
Абиетиновую и родственные кислоты очищают в виде солей с Д.
и регенерируют действием уксусной кислоты 111.
1. В u г g s t а й 1 е г A. W.. W о г d с n L. RJ. Am. Chem. Soc., 86, 96 (1964).
ДИ-юрет-БУТИЛИМИНОКСИЛ (радикал), [(CI I3)3CJ2N—О.
Мол. вес 144,23, г. кип. 74—75 35льп.
Металлический натрий реагирует с /??/х7П-нитробутаном в 1,2-ди-
метоксиэтане (гли.ме) с образованием соли |(CH:!):iC|..NO_Na'",
которая при гидролизе ласт радикал Д. — красную жидкость, ус-
тойчивую по отношению к кислороду, воде и водной щелочи [1|.
Д. применяют для улавливания радикалов, в частности при
фотолизе благодаря слабому поглощению в ближнем ультрафио-
лете |2|.
1. Hoffmann \. К., Feld ги ап Л. ЛЕ, G е 1 b 1 u m Е., Hodg-
son W. G., J. Am. Chem. Soc., 86, 639 (1964); procedure submitted to Org. Syn.
2. N e I sen S. F., Bartlett P. D., J. Am. Chem. Soc., 88, 143 (1966).
271
ДИ-н-БУТИЛОЛОВОДИХЛОРИД, «-(C4H9),SnCl2. Мол. вес
303,83; белое кристаллическое твердое вещество, т. ил. 43', т. кип.
170 60 леи (100 2 лы?).
Реакция с винилмагнийбромидом 111. В колбу, снабженную кон-
денсатором, охлаждаемым смесью сухого льда и ацетона, мешалкой и
капельной воронкой, помещают магниевую стружку, ТГФ н при
перемешивании добавляют 5 л?,? вшшлбромпда. После того как реак-
ция начнется, добавляют еще ТГФ п остальной раствор вппилбро-
I I Ф I'. 1 I (/< - (.,1 Г,। ,8пС1,
СН., СНВг Mg------> сн„ -СН.МцВт------------:---►
Г 1-2 1 %
1,3 мо.чя 1,2 г-и'ип.м
(«-C4H9),Sn (СН - СН,),
мида в ТГФ с такой скоростью, чтобы обеспечить слабое кипение.
По окончании реакции раствор реактива Гриньяра охлаждают, за-
меняют конденсатор на обычный обратный холодильник и прибав-
ляют раствор Д. при перемешивании и слабом кипении. Смесь
кипятят 20 час, гидролизуют насыщенным раствором хлористого
аммония (100— 120 мл на 1 моль реактива Гриньяра), декантируют
органический слой, промывают осадок солей эфиром, выпаривают
растворители и перегоняют Д. при 60 /0,4 мм.
1. S е у f е г t h D., Огщ. Syn., Coll. Vol., 4, 258 (1963).
2,6-ДИ-т/?ет-БУТИЛПИРИДИН, C^H^N. Мол. вес 191,31,
рКа 3,58 111.
Д. образует соль с хлористым водородом, но не дает производных
с иодистым метилом или трехфторнстым бором.
1. Brow и Н. С., Капнет В., J. Am. Clicm. Soc., 75, 3865 (1953).
2,2-ДИ-н-БУТОКСИПРОПАН (ди-н-бутилкеталь ацетона),
(СН,),С(ОС4Н,,-«),. Мол. вес 188,30, т. кии. 88—90 20 мм, уд. вес
1,4105.
Д. получают иерекетализацпей из 2,2-диметоксппропана и
«-бутанола 111. После отгонки азеотропа бензол—метанол
смесь охлаждают, катализатор нейтрализуют метилатом натрия и
ьон
(CI 1,),С (ОСН.,), - 2н-С,НвОН-> (СН3)„С (ОС4Н9-н), 2СН3ОН
Бензол
п р о д од ж а ют пере го н к у.
1. L о г е t t е N. В., Howard W. L., Org. S\n., 42, 1 (1962).
ДИВИНИЛРТУТЬ, Hg(CH-CH,),. Мол. вес 254,70, т. кип.
48—50 . 14 мм.
Д. получают прибавлением раствора сулемы в ТГФ к раствору
винилмагпийбромида в том же растворителе 111. Внимание] Раст-
вор очень ядовит, при хранении необходимо соблюдать предельную
осторожность.
272
При нагревании Д. на кипящей водяной бане с алифатической
или ароматической карбоновой кислотой без растворителя быстро
образуются виниловый эфир, этилен и металлическая ртуть (1) |2|.
Если для замедления реакции использовать инертный растворитель,
то можно получить промежуточный продукт (2). Соединения этого
RCO.,11 Иц (СИ СИЛ, --- RC(\CII СН, 1 СН, СН, ' Hg (1)
RCO,11 Hg(CH СИ,), - - RCO.HgCII СП,- СП, -СИ, (2)
АгОН Ия (СИ СИ,), - л ЛгОСН СП, -СП, (П Н. (3)
ArSH Нк(С11 - СП,), - - Ai-SCH СИ,- СИ, СН, Н»
типа представляют собой кристаллические вещества, разлагающиеся
при нагревании с образованием винилового эфира и ртути. Анало-
гично реагирует дивипплртуть с фенолами (3) и тиофеполамп (4),
образуя виниловые и тповиипловые эфиры |2|.
1. Re у 11 olds G. Г., Dessy R. Е„ J а 1 1 6 И. И., J. Org. Cliein., 23, 1217
(1958).
2. Foster D. J., T <> b 1 e r E., J. Am. Cliein. Soc., 83, 851 (1961).
ДИГИДРОПИРАН, ' о ..Мол. вес 84,11, т. кип. 85'. Д. полу-
чают из тстрагидрофурфурилового спирта при дегидратации с пере-
группировкой над окисью алюминия II—31.
0-, N-, S-защитная группировка. Д. реагирует с первичными
и вторичными спиртами и фенолами в условиях мягкого кислотного
катализа с образованием тетрагпдроппраппловых эфиров, которые
стабильны по отношению к основаниям, реактивам Гриньяра,
алюмогпдрпду лития, уксусному ангидриду в ппрпдппе л к окисле-
нию |4|. После окончания синтетических операций с другой! частью
молекулы эти эфиры можно легко расщепить кислотами (кипячение
с уксусной кислотой дает соответствующий ацетат) 151.
273
Д. применяют для защиты карбоксильной 161 и сульфгидрильной
[7| групп, а также имидазольного водорода в пуринах |8|. В каждом
из этих случаев соединение, содержащее активный водород, регене-
рируют при гидролизе разбавленной минеральной кислотой.
Пропаргиловые спирты, даже третичные (1), реагируют с Д.
в присутствии /ьтолуолсульфокпслоты как катализатора с образова-
RR'COII
I
С СН
нием тетрагидропиранпловых производных (2), которые стабильны
по отношению к основаниям, по ле> ко гидролизуются водными раст-
ворами кислот или сульфата магния 191.
Д., по-видимому, лучший реагент для защиты 2'-гидрокспльпой
группы в 5'-нуклеотидиых мономерах, которые далее подвергали
полимеризации 1101. Теграгпдроипраниловые эфиры обычно очень
чувствительны к кислотам, поэтому их следует получать непосред-
ственно перед использованием, а хранить в сухом сосюянпи при
возможно более низкой температуре.
Найдено, что Д. исизбирагелыю реагирует с аксиальными и
экваториальными гидроксильными группами инозита |11|.
Реакции. Д. гидролизуется разбавленной соляной кислотой! до
5-оксивалерианового альдегида 1121. Он реагирует с дпхлоркарбе-
ном, полученным из этиловою эфира трихлоруксусной кислоты
и метилата натрия в пентане, с образованием 7,7-дихлор-2-оксанор-
карана |13|.
Разб. нет > HO(CHjiCIIO
74 >79%
CI3CCO2C2H5, ?;aOCHi
68
Реакция с амидами. Д. реагирует с алифатическими и арома-
тическими амидами в бензоле или ДМФА в присутствии хлористого
RCOXH.
водорода как катализатора с образованием N-(reipai идроипраипл-2)-
амидов 1141. Эти соединения стабильны ио отношению к щелочи, но
гидролизуются концентрированной фосфорной кислотой с образова-
нием амида и 5-окспвалерианового альдегида.
274
1. Paul R., Bull. soc. chim. France, 53. 1489 (1933).
2. Schn iepp L. E., Ge i ! er H. H.. J. Am. Chem. See., 68, 164o (1946).
3. С о ii e p P., А н д p ю с Д., «Синтезы органических препаратов», ИЛ, М.,
1952. сб. 3, стр. 176.
4. Paul R.. Bull. soc. chim. France, 1, 971 (1934); W о о <i s G. F.. Kra-
пнт D. X., J. Am. Chem. Soc., 69, 2246 (1947); P a r Ii a ni W. E., A n cl e r-
s о n E. L„ ibid., 70, 4187 (1948); О t t A. C„ Murray M. F„ P e d e r-
son R. L., ibid., 74, 1239 (1952); .1 ones R. G., Ma n n M. .1., ibid., 75,
4048 (1953).
5. D a u b e n \V. G., Bradlo vv H. L.. J. Am. Chem. Soc., 74 , 559 (1952).
6. Bowman R. E., F о r d h a m W. D., .1. Chem. Soc., 1952, 3945.
7. ParhamW. E., DeLaitschD. M., .1. Am. Chem. Soc., 76 , 4962 (1954),
8. Robins В. K., G о d e f г о i E. F., Taylor E. C., L e w is R. L.,
.1 ackso n A., J. Am. Chem. Soc., 83, 2574 (1961).
9. R о b e r t s о n D. \., J. Org. Chem., 25, 931 (1960).
10. S t г а и s D. B., F г e s с о .1. R., J. Am. Chem. Soc., 87, 1364 (1965).
11. A n i> v a I S. J., Gero S. D., J. Chem. Soc., 1965, 5255.
12. P a u 1 R., Bull. soc. chim. France, 1, 976 (1934); В у д с Дж. мл., «Синтезы
органических препаратов». ИЛ, М.. 1953, сб. 4, стр. 399.
13. Р а г h a m W. Е., S с li w е i z е г Е. Е., М i е г z w a S. A., Jr., Org. Syn., 41,
76 (1961).
14. S р е z i а 1 е А. J., R a t t s К. \V., M a г с о G. J., J. Org. Chem.. 26, 4311
(1961).
ГИДРОПИРИДИЛ)-АЛЮМИН AT, см. Лития
алюмогидрид — пиридин.
ДИГЛИМ (диметиловый эфир диэтиленгликоля),
СН:1ОСН;СН..ОСН.,СН.,ОСН3. Мол. вес 134,17, т. кип. 161 \ смеши-
вается с водой.
Очистка 111. 1 .? технического Д. выдерживают 12 час над не-
большими кусочками гидрида кальция (10 г) и декантируют в колбу
для перегонки. Добавляют алюмогидрид лития в таком количестве,
чтобы обеспечить избыток активного гидрида (см. очистку ТГФ),
перегоняют прп 62—63 /15 мм и храпят в атмосфере азота.
1. Contributed bv Z W » i f с 1 G., Ltiiversitv of California, Davis, California.
H
ДИИЗОБУТИЛ АЛЮМИН И Й ГИДРИД, (CH.)>CHCII,AiCU,CH (СН,),
Мол. вес 142,21, т. кип. 140 ' 4 льч.
Д.— жидкость, растворяющаяся легче, чем алюмогидрид лития.
В качестве растворителей пригодны эфир, бензол, толуол и цикло-
гексан. ТГФ образует с Д. комплекс и поэтом)’ не применяется
в больших концентрациях. Д. ппрофорен, поэтому все операции
с ним, как ц с другими лптийорганическими соединениями, следует
проводить в безкислородпоп атмосфере 111.
Получение |2|. Гейслер и Бруно |3| получили Д. кипячением
160 мл 25%-ного раствора трипзобутплалюмиппя в гептане в течение
2—3 час. Перегонка дает 27 мл бесцветного Д. с т. кип.
80—90 70,05 мм. Другая методика предложена Эйшем и Каска
|3а|.
Восстаиовление ацетиленов. Д. применяют для восстановления
дпзамещенных ацетиленов до олефинов, поскольку олефины с внут-
275
решит положением двойной связи реагируют сД. только очень мед-
ленно при 70—80 , тогда как дизамещениые ацетилены восстанав-
ливаются даже при более низкой температуре |4|. При этом полу-
чаются почти исключительно фщ-олефпны.
1(СН,).с,н<:н.,1„А1н
С,Н5С = сс,н5 ---------------—-----> С,Н,СН^СНС.,Н5
X’.,: 45
ю,% цис
Гейслер и Бруно 131 применили Д. в синтезе изомеров линолевой
кислоты для восстановления некоторых дппнов до фщ-диенов. На-
пример, к 1-хлор-Д'’^-гептадекаднину (1) в атмосфере азота при 0J
Н
10 7 I N.
СН3 (СН,)5С ССН..С С (СН,)5СН,С1 (СИ3),СНСН,А1СН,СН (СН3), —
(1) 0,0’»2 моля (2) 0,1." моля
Н II пн
—. Комплекс -- -’°-СН, (СН,), С-ССН, С — С (СН,),СН,С1 +
4 (CII,);,CH-; А1 (ОСН;))з
добавляют по каплям большой избыток Д. По окончании реакции
комплекс разлагают, прибавляя по каплям при охлаждении смесь
метанола и петролейногоэфира, и получают цис-цис-лиен (3), изобу-
тан и метилат алюминия. При обработке разбавленной серной
кислотой метилат растворяется и выделяется хлорид (3). Чистый
ф/с-ф/с-дпен (3) получен с выходом 82% после повторного восста-
новления, которое проводят, чтобы обеспечить полное превраще-
ние дннна (1).
Если прибавить по каплям 0,41 моля Д. к 0,2 моля цпклододе-
цнпа при осторожном нагревании, то можно осуществить стерео-
специфическое восстановление до ф/с-цпклододенеиа, который вы-
деляется после разложения метанолом (выделяется изобутан) |5|.
ДСЩ)гСНСН,7гА1Н „
;—=т-—> Комплекс
2 час при 45°
(сн3)3сн
ГЖХ-аиализ указывает на присутствие менее 0,4'% ш/лгнс-изомера.
Эйш п Каска |3а1 неожиданно обнаружили, что 1-фенилпропнн-1
в зависимости от условий реакции можно восстановить в цис- или
транс- 1-фенилпропены. При соотношении реагентов 1 : 1 полу-
чается исключительно ццс-изомер.
276
Восстановление хинолина. Нойман [61 обнаружил, что при
добавлении к хпполпн\ пс каплям при перемешивании I экв Д. при
(и:ю-Ви.Д \ 1Н
0- 10-
| j Диоксан-вода
\ х 0-10°
I CH.CII (CH:t),
А1 ЧСН..СН (СН3).,
(2)
0— 10 в атмосфере азота сначала получается темно-красный
раствор. К кончу прибавления смесь становится желтой и
выделяется соединение (2) в виде длинных бесцветных призм. Нет
необходимости выделять (2), его гидролизуют осторожно, добавляя
по каплям водный дпоксан. Чистый 1,2-дпгндрохннолнн получен
с выходом 95%. Дальнейшее восстановление этим гидридом требует
более жестких условий. 1,2,3,4-Тетрагидрохинолин получен с выхо-
дом 70% при кипячении хинолина с 2 же Д. в бензоле в течение
24 час.
Восстановление у-лактона. Шмидлин и Веттштейи [71 нашли,
что Д. можно применить для восстановления у-лактона (4) в полу-
ацеталь (5):
Изомеризация эпоксида. Под действием Д. в ксилоле при 120э
эпоксид (6) изомеризуется в аллиловый спирт (7) [8|.
Мг
(U3O-Bu'2AIH
Ксилол, 120<з
(Ч (?)
ArCN—*АгСНО. Попытки восстановления обычными способами
3,4-дициаиофурана в дпальдегпд оказались безуспешными. Трофи-
менко |9| осуществил это превращение следующим образом. К пере-
мешиваемой суспензии 3,4-дицианофурана в бензоле в атмосфере
277
азота добавляют Д. Смесь нагревают при температуре около 50э
до полного растворения. К вязкой реакционной смеси прибавляют
метанол для разложения комплекса. Обычная обработка смеси при-
водит к чистому диальдегиду с умеренным выходом. Тиофеновый
Дсн3)гснснД2л1н
0,114 моля
OI
СНО
0,051моля eloO мл С6Н6
аналог дает соответствующий диальдегид с выходом 23%. 2,3,4,5-
Тетрацпанотпофен не восстанавливается. Бензонитрил дает бензаль-
дегид с выходом 90% 110].
Другие реакции восстановления. Миллер и сотр. 1101 нашли,
что бензойная кислота и метилбензоат восстанавливаются до бен-
зилового спирта, а w-бутиловый эфир капроновой кислоты — в «-бута-
нол и «-гексанол. Захаркин и Хорлпна 1111 описали восстановление
алифатических и ароматических сложных эфиров в альдегиды,
ортоэфиров — в ацетали, ацеталей — в простые эфиры и бензил-
этилового эфира — в толуол.
Расщепление сс.р-непредельных эфиров 112].
цен ,),СНСН .J .А1Н
С.,Н3ОСН СНСН. - :----------> СН. =СНСН, [(CHt).,CHCH.,]..A10C.,H3
50-100 '
Восстановление а, р-непредельного у-лактона в фуран [13]:
1. «Handling and Properties of Triisobutylaluminurn», Hercules Powder Co.,
Wilmington, Del.
2. Z i e g 1 e г К. et al., Ann., 589, 91 (1954); англ. пат. 778098 (1957); [С. A., 51,
15081 (1957)]; Ann., 629, 1 (1960).
3. G e n s 1 e r \V. .1., BrunoJ. J., J. Org. Chem., 28, 1254 (1963).
3a. Ei sc Ii J. J., К a s k a W. C., J. Am. Chem. Soc., 88, 2213 (1966).
4. Wilk e G„ Al ii 1 I e r H„ Chem. Ber., 89, 444 (1956); Ann., 629, 224 (1960).
5. Z i e g e ti b e i n W., Schneider N. M., Chem. Ber., 98. 824 (1965).
6. X eumann W. P., Ann., 618, 80 (1958).
7. Schmidlin J., Wettstein A., Helv. Chim. Acta, 46, 2799 (1963).
8. К i r c h h о f W., Chem. Ber., 93, 2712 (1960).
9. T г о f i m e n k о S., J. Org. Chem., 29, 3046 (1964).
10. Miller A. E. G., В i s s J. \V., Schwartzman L. H., J. Org. Chem.,
24, 627 (1959).
11. 3 a x a p к и и Л. И., Хорлииа И. М., Изв. АН СССР, 1959, 2255; Tet-
rahedron Letters, 14, 619 (1962).
12. Р i и о Р., L о г е n z i G. Р., J. Org. Chem., 31, 329 (1966).
13. M i n a t о H., X a j a s a k i T., Chem. Ind,, 1965, 899; J. Chem. Soc.,
1966, 377,
278
ДИИЗОПИНОКАМФИЛ БОРАН. Д. получают из а-пннена [1].
Этот оптически активный и пространственно затрудненный реагент
применяют для превращения сшоолефинов и кетонов в оптически
активные спирты, а также для расщепления рацематов олефи-
нов [1|.
Д. реагирует с жесткими бициклическими и конформационно-
подвижными мопоцнклическпми кетонами в диглиме преимущест-
венно с образованием менее стабильного из двух возможных спиртов
[2|. Сравнительные результаты для четырех кетонов приведены на
схеме и в таблице (на схеме приведены менее стабильные эпимеры
спиртов).
н
Количество менее стабильного спирта, %
X: (см. схему) LiAlH4, ТГФ В.Щ, ТГФ T i Ф Дни ?оп инока.мфи л бо- ран, диглим
1 25 41 74 83
2 25 26 79 94
3 73 74 64 98
4 91 52 65 (медленно) 100 (медленно)
1. Brown 14. С., ZweifelG., .1. Am. Chem. Soc., 83, 486 (1960);
Brow n H. С., В i « b у D. B„ ibid., 83, 3166 (1961); Brown H. C„ A y-
yanfier N. R., Zweifel G.. ibid., 84, 4341, 4312 (1962); 86. 397, 1071
(1964).
2. Brown 14. С., V a r in a V., J. Am. Chem. Soc., 88, 2871 (1966).
ДИИЗОПРОПИЛНАТРИЙАМИД, (z/3o-C3H7)2NNa. Мол. вес
123,18.
279
Д. получают из фенилнатрия и диизопропиламина; он является
очень хорошим основанием для ацилирования 3- или 4-ппколина[1].
у\ СН, СН,СОС6Н5
I -ГС6Н,СО2С2Н5 --------Г—--* !!
1. R е у п о 1 d s S., Levine R., J. Am. Chem. Soc., 82, 472 (1960).
ДИИЗОПРОПИЛ ПЕРОКСИДИКАРБОНАТ,
О О
II I!
(СН3),СНОСО2СОСН(СН.,)3. Мол. вес 206,19, т. пл. 8—10°.
В присутствии хлористого алюминия как катализатора Д. можно
использовать для превращения толуола в смесь крезолов с выходом
около 5О'1о. При этом наблюдается следующее соотношение изоме-
ров: орто- 34%, мета- 11 % и пара- 55% 111.
Д.— эффективный катализатор полимеризации. При нагревании,
растирании пли ударе Д. взрывается, поэтому его обычно хранят
в твердом состоянии при охлаждении.
В присутствии хлорной меди толуол под действием Д. превра-
щается в смесь изомерных толилизопропилкарбонатов, в которой
преобладает изомер |2|.
о О
|i " CuCJ, (60 = )
(СН3),СНОСО2 СОСН (СН3).,л-СвН5СН,—
— (СН3).,СНОН -СО.,Д-СН,С6Н1ОСО.,СН (СН;!),
Д. используют в качестве инициатора радикальных реакций при
умеренных температурах, например для присоединения хлористого
ацетила к гептену-1 131.
СН3 (СН.,)4 СН = СН3 4- Реагент - СН3СОС1 "°~ ' °-^CH., (СН.,)-СОС1
3 с 2 г я С.Н,, 80 t " '' Ц.,0 I 100%
СН3 (СН.,)ТСО.,Н
1. К о v а с i с Р., М о г n е w е с k S. Т., J. Am. Chem. Soc., 87, 1566 (1965);
К о v а с i с Р., Kurz At. Е., ibid., 87, 4811 (1965).
2. К о v а с i с Р., Kurz М. Е., J. Am. Chem. Soc., 88, 2068 (1966).
3. Allen С., С a doganJ. 1. G., Н е у D. Н., J. Chem. Soc., 1965, 1918.
ДИИМИД, HN — NH. Этот реагент получают «в момент выде-
ления» (in situ) при окислении гидразина кислородом воздуха в при-
сутствии ионов меди 111; окисление также можно проводить пере-
кисью водорода 111, феррицианидом калия |2|, окисью ртути |2|. Д.
восстанавливает олефины, алкины и азосоедпненпя. Восстановление
относительно малореакционноспособной двойной связи холестерина
достигается с выходом всего 20%. Простая методика |31 восстановле-
280
пггя азобензола до гидразобензола включает прибавление 95%-ного
гидразина и раствора, содержащего следы сульфата'меди, к суспен-
зии азосоедпиеппя в этаноле при О'; затем прибавляют 30?<>-ную
перекись водорода по каплям при 0” до исчезновения окраски.
Дпкалиевую соль азодикарбоновой кислоты (2), получаемую
гидролизом азодиамида (1), также используют для получения
о о о о
НАС—X—X— СХН., КОС — Х-Х-СОК Н-Х=Х-Н
(II (2)
Д., хотя этот способ менее удобен, так как требует большого избытка
реагента |1,4|. Можно синтезировать Д. из гидразида и-толуолсуль-
фокислоты, по и в этом случае необходимо применять 100%-ный
избыток реагента 12,51. Олефины восстанавливаются кипячением
этого реагента и субстрата в диметпловом эфиреднэтпленглпколя.
Д. восстанавливает симметричные кратные связи (С—С, С^С,
N---N) более гладко, чем поляризованные связи (C=N, С-О,
С- N, О N —-О“,О- Sx- С)~) IGI. Восстановление осуществляется
стерсоспецифично как /рщ-прпсоедпнеиие 12,7|. Изучение восстанов-
ления олефинов Д. путем образования его в этанольном растворе
олефина и гидразина окислением кислородом воздуха при 45—55'
показало, что в тех случаях, когда пространственное экранирование
двойной связи в олпрпне резко выражено, восстановление подчи-
няется заметному стерическому контролю подхода реагента (steric
approach control); если объем группировок в олефине умеренный,
наблюдается резкое уменьшение пространственных затруднений 181.
Согласно предварительному сообщению 191, Д. можно получить
действием едкого патра при 0' на хлоргндрат хлорацетилгндразипа
(см.). Другим предшественником Д. является гидроксилампп-О-
сульфокпслота (см.). Азобензол был количественно восстановлен
в гидразобензол, аллиловый спирт — в пропанол-1 (70?о). В отсут-
С1СН.,СО\Н\Н2-НС1 г 2ХаОН - СН._, - С —О - Н.,О 4~ НХ — ХН 2\аС1
1---<Н:)СО.,Н
ствие субстрата Д. дпспропорцпонирует на азот и гидразин, который
при действии кетена дает ацетплгпдразпн.
цис,транс,/и/шнс-Циклододекатрпен-1 ,5,9 можно селективно
восстановить Д. до рпг’-цнклододеиена (гидразингидрат, сульфат
меди, воздух) 110J. Это указывает на значительную разницу в реак-
281
ционной способности цис- и транс-двойных связей.
Обзоры 1111. Хамерсма и Снайдер 1121 детально изучили влияние
условий на реакцию восстановления диимидом, который получали
из дикалиевой соли азодпкарбоновой кислоты. При этом было
найдено, что воздух не оказывает заметного влияния, а скорость
восстановления в различных растворителях уменьшается в следую-
щем порядке: пиридин > диоксан> дпметилсульфоксид > мета-
нол > этанол > «-бутанол. Вода является сильным ингибитором
реакции в апротонных растворителях, ио практически неоказывает
влияния в гидроксилсодержащих растворителях.
1. Со г е у Е. J., MockW. L., Р a s t о Г>. J., Tetrahedron Letters, 347 (1961).
2. HunigS., Muller H.-R., T h 1 e r W., Tetrahedron Letters, 353 (1961).
3. Org. Expts, Chapt. 38.
4. v anTamelen E. E., Dewey R. S., T i m mon s R. J., J. Am. Chem.
Soc., 83, 3725 (1961).
5. D e w e у R. S., v a n T amelen E. E., J. Am. Chem. Soc., 83, 3723 (13ol).
6. vanTamele n E. E., Dewev R. S., Lease M. F., Pirkle W. H,,
J. Am. Chem. Soc., 83, 4302 (1961)'
7. С о r e v E. J., P a s t о D. J., M о c k W. L., J. Am. Chem. Soc., 83, 2957
(1961). ’
8. v a n T a m e I e n E. E., TiramonsR. J., J. Am. Chem. Soc., 84, 1067
(1962).
9. Buvle R., van О v e r s t г a e t e n A., E 1 о у F., Chem. Ind., 1964,
839.
10. Ohno M., Okamoto M., Tetrahedron Letters, 2423 (1964).
11. M i 1 1 e г С. E., J. Chem. Ed., 42, 254 (1965); H ii n i g S., Mill lerH, R.,
T h i e r \V., Angew. Chem., Internal. Ed., 4, 271 (1965).
12. HamersmaJ. W., S n у d e r E. J.. .1. Org. Chem., 30 , 3985 (1965).
1.3-ДИИОД-5.5-ДИМЕТИЛГИДАНТОИН, щс J”3
Мол. вес 379,94. < J?
Д. получают с выходом 74% при добавлении по каплям раствора
хлористого пода в четырех хлористом углероде к охлажденному льдом
водному раствору о,5-диметилгидантоина и эквивалентного коли-
чества едкого натра 111. Осадок отделяют, промывают ледяной
водой, этил ацетатом и сушат в вакууме при G0J; препарат исполь-
зуют без дополнительной очистки. Д. иодирует (в ядро) реакционно-
способные ароматические соединения (анилин, фенол, но не нафта-
лин). Д. не участвует в гомолитических реакциях, таких, как иоди-
рование толуола в боковую цепь в присутствии перекиси бензоила.
Подобно N-иодсукцпнимиду Д. реагирует с енолацетатамп насыщен-
ных п ненасыщенных кетонов с образованием сх-иодкетонов.
282
1. О r a z i О. О., Corral R. Л., В e r t о г e 1 1 о Н. Е., J. Org. Chem., 30,
1101 (1965).
ДИКЕТЕН, СН2С = О Мол. вес 84,07, т. пл.—8', т. кип.
СН.,=С-0
67—69 /92 мм.
Д. токсичен и все операции с ним необходимо проводить под
тягой. Д. получают, пропуская газообразный кетен через систему
из трех промывалок, охлаждаемых смесью ацетон — сухой лед,
вторую и третью промывалки заполняют сухим ацетоном. Около
2 молей кетена пропускают через эту систему в течение 4 час; за это
время температуру постепенно повышают до комнатной. Фракцио-
нирование дает Д. с выходом 50—55% [11. Строение |2|.
Реакция с соединениями, содержащими активный водород.
Д. гидролизуется водой с образованием ацетоуксусной кислоты;
со спиртами он дает эфиры ацетоуксусной кислоты [31, что можно
проиллюстрировать на примере получения ///реш-бутилацетоацетата
[41. Аналогичная реакция с ароматическими аминами приводит
О
СН, = С — О NaOAr(80 t r-u /ru rn r/r-u ч
I -;-ноС(Сн3)3-------------► сн3ссн..со2с(сн3)3
СН.-СО 75-80«„
к ацетоацетанилидам CH3COCH2CONHAr [51. Уретаны присоеди-
няются к дпкетену аналогично, давая CH jCOCHoCONHCO.C.H-,
[61.
Дегидратация спиртов. Эфиры ацетоуксусной кислоты приме-
няются для дегидратации спиртов |7|. Так, 1-метплцпклогексанол
экзотермически реагирует с днкетеном, затем в присутствии катали-
о
II
тических количеств /нтолуолсульфокислоты при повышенной темпе-
ратуре отщепляет СО3 и ацетон, что приводит к образованию
1-метилцпклогексена 98%-ной степени чистоты. Детдратаипя дру-
гими методами дает препарат, загрязненный довольно значительным
количеством метиленцпклогексана.
283
Реакция с фтористым водородом. Д. реагирует с безводным фто-
ристым водородом с образованием фторангидрида ацетоуксусной
кислоты 181:
СН., — С—О HF
| |-------> CH3COCH.,COF
СН„ СО G5%
а-Оксикетоны. Новая модификация синтезов с ацетоуксусным
эфиром позволяет получать а-оксикетоны [91. трет-Бутил ацето-
ацетат (1), полученный с высоким выходом из /пре/7г-бутанола и Д.,
CII. --C----о HOC (СН,),, C,H,ONa СН, —С — ОН
I I------------------> I
СН.-С=О 8э-95% СН.,СО.,С (СН3)3
(1)
н-С,Н,,Вг: C.HSON?. ° C3HU-H Ха11 с.Н,
-------— * !i I --------->-
G' °----------------------------------------------СН3С—СНСО,С (СН3)3
(2)
о С5Ни-н
i j ри и эвидпис 83°' |
ОСОС6Н3
(3) (4)
о
СН3ССНС5Нп-н + (СН3),С = СН., + со,
I
ОН
(5)
алкилируют (2) и бензоилоксплпруют ва-положение через натриевое
производное (3) действием перекиси бензоила в бензоле. При нагре-
вании эфира (4) происходит отщепление mpe/n-бутильной группы и
декарбоксилирование. Пиролиз проводят при 160° в присутствии
каталитических количеств л-толуолсульфокислоты. Последующее
омыление в мягких условиях приводит к ацплошп (5), чувствитель-
ному к щелочам.
N-Защитная группа в синтезе пептидов 1101. Д. реагирует
с хлоргидратамн эфиров аминокислот в этаноле при 0— 5 в присут-
ствии 1 же этилата натрия или третичного амина с образованием
кристаллических и легко очищаемых N-ацетоацетнльных производ-
ных (1). Защитная группа при обработке фенплгпдразнно.м уда-
ляется в виде 1-фенил-3-метилпиразолона-5:
R R
+ I (СН2СО), |
1) (Cl) h3nchco,c,h5 ——-д-ch3coch2conhchco,c2h5
С ,НДОNа
R R'
I I Cr,H.-,NHXH,
2) CH3COCH,CONHCHCO-NHCHCO2C,H3------------
284
С6Н5
. NH
nZ’A R
II * I
CH,CCH,C-NHCHCO
3 II''-*
о
R*
-NHCHCO2C2H5
R
+ HjNCHCONHCHCO2CjHs
Гетероциклы. N-Апетоацетильные производные эфиров a-ами-
нокислот циклизуются под действием оснований в а-ацетилтет-
рамовую кислоту [ 111:
нсп—СО.СДЦ
HN. /СН.СОСНз
C2H5ONa,
75%
Бёчи и Лукас 112] применили эту реакцию на первой стадии синтеза
антибиотика голомицпиа (4).
Фенолы реагируют с Д. при катализе основаниями с образова-
нием а рил ацетоацетатов. Эти соединения обычно разлагаются при
перегонке с образованием исходного фенола; при обработке концент-
рированной серной кислотой дают кумарины с выходом 75—90%
113].
сн3 СНз
1. Уильямс Дж., К р и н и ц к и й Дж., «Синтезы органических препара-
тов», ИЛ, М., 1952, сб. 3, сгр. 180.
2. Johnson! R., S h i и е г V. J., J. Am. Chem. Soc., 75, 1350 (1953).
3. К i т е 1 W., Соре А. С., J. Am. Chem. Soc., 65, 1992 (1943).
4. Lawesson S.-О., G г о n w а 1 1 S., S an d Ьегц R., OiM- Syn., 42,
28 (1962).
5. К a s 1 о w С. E., S о rn m e r N. B., .1. Am. Chem. Soc., 68, 644 (1946).
6. D e w а г J. IL, S h a \v G., J. Chcin. Soc., 1961, 3254.
7. F г i s c 1 1 C., Lawesson S.-О., Arkiv. Keim'., 17, 401 (1961).
8. О 1 a h G. A., К uhnS. J., J. On’. Chem., 26, 225 (1961).
285
9. L a wesson S.-О., Gr on wa I I S., Chem. Scand., 14, 1445 (1960).
10. D’A n g e 1 1 F„ F 1 1 1 r a F., S с о f f о n e E„ Tetrahedron Letters, 605 (1965).
11. Lacey R. N., J. Chem. Soc., 1954, 850.
12. В ii c h i G., Lukas G., J. Am. Chem. Soc., 86, 5654 (1964).
13. Lacey R. X., J. Chem. Soc.. 1954, 854.
ДИ КЕТЕН — АЦЕТОН, АДДУКТ (1.1,5-Триметил-2,6-диокса-
О
ii
циклогексен-4-он-З), н3С О СН3 . Мол. вес 142,15, т. пл. 13°,
т. кип. 65—6770,2 мм.
Аддукт дикетен+ацетон получают реакцией дикетена с ацетоном
в присутствии /1-толуолсульфокислоты 111. Это жидкость с приятным
запахом, стабильная в отсутствие щелочи. Аддукт можно использо-
вать во многих реакциях вместо более летучего дпкетена.
1. С а г г о 1 1 М. F., В a d о г Л. R., J. Am. Chem. Soc., 75, 5400 (1953).
ДИКЕТЕН — АЦЕТОФЕНОН, АДДУКТ. Мол. вес 204,22, т. пл.
93,57 О получении и применении см. Дпкетен— ацетон, аддукт.
ДИКОБАЛЬТОКТАКАРБОНИЛ, Co,(CO)s. Мол. вес 342,12,
оранжево-красные кристаллы, т. пл. 51—52 .
Получение. Д. удобнее всего получать из солей дв} хвалентного
кобальта, обычно из карбоната 111, но также и из ацетата. Его полу-
чают в эфире [21 или в бензоле [31 при реакции кобальта Ренея
с окисью углерода при 150' под давлением 224 атм. Кроме того, Д.
можно получить из карбоната двухвалентного кобальта и газооб-
разной смеси Н, и СО (1 : 1) [41 пли разложением НСо(СО)4 [51:
2НСо (СО)4 — Со, (СО)а 4- Н,
Строение. Высоко полярная структура, изображенная на схеме,
не согласуется с низкой температурой плавления этого соединения,
+о="с ? с=о+
+ - I/ \ I - +
О=С-Со,--------------------------Со —С=О
U У tso+
о
а также с его растворимостью в углеводородах. Истинная структура
Д. пока неизвестна.
Оксо-реакция (оксо-синтез). Патент, зарегистрированный в США
в 193<3 г. (Otto Roelen) и опубликованный в 1943 г., содержит
описание реакции присоединения окиси углерода и водорода к оле-
финам в присутствии таких катализаторов, как кобальт, окись тория
или кизельгур. Впервые эта реакция была описана как «оксо-син-
СН;1
125-200 итл |
СН3СН2СН=СН, + СО-РН,----------> СН3 (СН.,)з сно рсн3сн,снсно
- 12а-1а0“ 70-80% ’20-30%
тез». Позднее было найдено (Адкинс и Крзек [2,31), что применение
286
твердых кобальтовых катализаторов, таких, как дикобальтпента-
карбонил, значительно улучшает результаты, и реакция стала
известна как реакция гидроформилирования. Обычно в результате
реакции получается смесь двух возможных альдегидов, однако пре-
обладает альдегид с неразветвлеипой цепью.
Орчин и сотр. |6| экспериментально показали, что в условиях
оксо-реакции Д. быстро превращается в гидрид тетракарбонила
кобальта НСо(СО)4, который и является истинным катализатором,
и что реакция протекает через
фину. Другие примеры этой
схеме |3|:
сн3соосн2сн=сн2 + со +
+ СО + Н;
присоединение этого гидрида к оле-
реакцни приведены на следующей
Н2 СоДсО)8> СН3СООСН2СН2СН2СНО
69%
Со2(СО)я^ РД
> I----1сно
НССО,С,Н5 „
П I | ТГ Со2(СО)8
С2Н3О2ССН + со + н2 —-----------------
:но
:нсо2с2н5
:н2СО2с2Н5
Удобный способ получения парафиновых углеводородов с прямой
цепью включает «обратный оксо-процесс»:
Ni
240 атл Г -СО,
и-С1,Н31СН.,СН.,С.Н.,ОН--> Ci-,H31CH.2CH2CHO —-
—
_ h-<'j.-,H3ICH — СН2 —L и-С|7Нзв
J 05ио
Адкинс п Крзек |2| отмечали, что некоторые а, ^-непредельные
карбонильные соединения быстрее подвергаются восстановлению,
чем гидроформилированию. 'Гак, кротоновый альдегид и акролеин
восстанавливаются до а/-,масляного и пропионового альдегидов соот-
ветственно; метилвинилкетон восстанавливается до метплэтилке-
тона. Орчин и сотр. |4| нашли, что карбонильная группа может
также восстанавливаться, если реакцию проводить при более
высокой температуре (180—185'). Применение оксо-реакции к спир-
там позволяет получать гомологичные спирты:
Со. (СО)8
(СН3)..ССН32Н., ' СО ——(СН3)„СНСН.гСН.,ОН у НоО
он
Жирноароматические углеводороды получаются с высокими
выходами. Например, ацетофенон восстанавливается до этилбензола
с выходом 64?о, флуоренон до флуорена — с выходом 95%.
Скорость гидроформилирования зависит от структуры олефина
и уменьшается в следующем ряду: нормальные терминальные оле-
фины, нормальные олефины с двойной связью в середине цепи, раз-
ветвленные олефины [7]. Циклогексен реагирует медленнее, чем
циклопентеи, циклогептен или циклооктен.
287
Родственные реакции. Если в реакции оксо-сиитсза применять
вместо водорода этанол, то обычно получаются сложные эфиры, хотя
могут образоваться и другие продукты |8|.
/Ч»О
j Р : сол-(сн;1),а)Н—' j сосн(сн3),
7 н 7
Реакция с аминами приводит к одно- или двузамещенным ами-
дам, как правило, с хорошим выходом:
RCH СН. СО——524’ RCH.,CH.,CONHR'-LRCHCO\HR'
В присутствии Д. при 130’ и давлении от 25 до 200 атм можно
осуществить присоединение цианистого водорода к олефину 191.
Наил\чшие результаты получены с терминальными олефинами и
сопряженными диенами, которые дают главным образом продукты
Си,. (СО),
СН3СН—СН., ; НСХ ----> СН.,СНСН3
сх
] ,4-присоединеиия. Хорошо реагируют также аддукты Дильса —
Альдера из циклопентадиена, которые дают смеси изомеров.
При нагревании анила бензальдегида с Д. в бензоле под давле-
нием с хорошим выходом получается 2-фенилфталимидин 1101.
230s
СН^ХС6Н5+СО С0, (С0’’
еСН<
I il /с«н*
'со
Эйзепман 111] нашел, что в присутствии Д. в условиях оксо-
реакции окись пропилена реагирует с окисью углерода и метанолом
с образованием метилового эфира Р-окспмасляпой кислоты с выходом
40%. Позднее он установил |12|, что главным продуктом является
130 , 245 атм. Ci>, (CO)S
сн3сн—сн., ; ацонн со-----------------------------2—->ch.:chch.,co.,chs
з - 1 •> - 40% •' 8
апетоп п что при проведении реакции в отсутствие окиси углерода
в спирте пли бензоле эпоксиды с хорошим выходом изомеризуются
в кетоны:
сн3сн2сн—сн2
Сог(СО)8 сн сн ссн
77% И
Сог(СО)а^
73%
288
Реакция ацетилена с окисью углерода. Установлено [131, что
в присутствии Д. ацетилен реагирует с окисью углерода в апротон-
ных растворителях (нитрометан, ацетонитрил, ацетон) при 90"’
и высоком давлении с образованием шранс-бифурандиона. При
4 СН = СН + 2 СО 900-1000^
65-70%
H2SO<
о
цис .
транс
действии горячей конц. серной кислоты это соединение количест-
венно изомеризуется в т^пс-пзомер. В большинстве растворителей
образуется транс-изомер; однако в тетраметилмочевине в качестве
единственного продукта получается цпс-изомер.
Взаимодействие с ацетиленами. Формула Д., приведенная выше,
содержит шесть поляризованных карбонильных групп, непосредст-
венно связанных с атомом кобальта, и две мостиковых карбэниль-
Рис. Д-1.
ных группы, каждая из которых связывает два атома кобальта.
Гринфильд и сотр. 1141 нашли, что Д. реагирует с дифенилацетиле-
ном в пентане с замещением двух мостиковых карбонильных групп
и образованием глубоко окрашенного соединения по уравнению:
CeH5C^CC6H5 I Со, (СО)8 —-0—* CeH5C,CeH5Co, (CO)e + 2СО
10 Заказ № 9G5
289
Выход указан для соединения, дважды перекри.'талп.пзованного
из метанола. Вещество образует темно-красные кристаллы с т. пл.
НО ; для него характерна высокая летучесть и хорошая раствори-
мость в органических растворителях. Рентгено-структурный анализ
показал 1151, что это вещество имеет строение, изображенное на
рис. Д-1. При более высокой температуре (200—280 ) или, лучше,
при кипячении в растворителе при температуре 65—НО ацетилены
тримеризуются с образованием производных бензола 116|. Эта реак-
ция тримеризацпп была использована для синтеза 1,2,4-трп-/прс/п-
бутплбепзола— первого представителя ор/но-замещенных производ-
ных бензола, имеющих в качестве заместителей /7гре/?/-бутильные
Со. (СО).
нс ссн„с н „он —-------->
' 14%
сн,сн.,он
! сн.,сн.,он
%.
1
СН„СН2ОН
группы [171. Если шрет-бутплацетилеи обрабатывать Д. при ком-
натной температуре, то образуется комплекс с выходом 30%. При
Со., (СО). Вг,
3 (СН3)з сс сн — -- Со2 (СО)4 [(СН3)3СС2Н]3
С (СНЛ)3
J. ^(СНз^+гсоВг. -нсо
4“
I
С (CHg)g
обработке этого комплекса бромом в СС14 при 0 выделяется СоВг,
н с выходом 70% получается 1,2,4-три-/?;рт-бутилбензол. Этот
комплекс разлагается с образованием ароматического углеводоро-
да также при нагревании до 150—170J (4 час, 56%).
Восстановление ароматических соединений. Некоторые полицик-
лические углеводороды претерпевают в условиях оксореакцпи ча-
стичное восстановление (Н2, СО, 150—200% 210—252 атм). Бензол,
нафталин и фенантрен устойчивы в этих условиях, но антрацен вос-
станавливается до 9,10-дигидроантрацена [181. Другие примеры:
I 51'.°
290
1 Wender I., Sternberg H. W., At e t 1 i n S., Orchin At., Inorg.
Svn., 5, 190 (1957).
2. A cl к i n s H., К r s e к G., J. Am. Chem. Soc., 70, 383 (1948).
3. A d к i n s H., К г s e к G., J. Am. Chem. Soc., 71. 3051 (1949).
4. W e n d e г I., L e v i n e R., Orchin M., J. Am. Chem. Soc., 72, 4375 (1950).
Ь. «Неорганические синтезы», ИЛ, M., 1951, сб. II, стр. 230.
6. Wender к, L е v i п е R., О г с h i n М., J. Am. Chem. Soc., 71, 4160 (1949).
7. Wender I., M e t 1 i n S., Ergun S., Sternberg H. W., Green-
field H„ J. Am. Chem. Soc., 78, 5401 (1956).
8. Natta G., Pino P., E г с о 1 i R., J. Am. Chem. Soc., 74, 4496 (1952).
9. A r t h u r P., Jr., England D. C., Pratt В. C., W h i t in a n G. M.,
J. Am. Chem. Soc., 76, 5364 (1954).
10. At ii r a h a ,s 11 i S., J. Am. Chem. Soc., 77. 6403 (1955).
11. E i s e n m a n n J. 1.., Y a m a r t i n о R. L., HowardJ.F., Jr., J. Org.
Chem., 26, 2102 (1961).
12. Eisenmann J. L., J. Org. Chem., 27, 2706 (1962).
13. S a u e r J. C., Cr a m er R. D., Engelhardt V. A., Ford T. A.,
Holmquist H. E., H о w к B. W., J. Am Chem. Soc., 81, 3677 (1959).
14. G r e e n f i e 1 d H., St e r n b ergH. W., F г i e d c 1 R. A., W о t i z J .H.,
Markby R„ W e n d e r 1., J. Am. Chem. Soc., 78, 120 (1956).
15. S I у W. G.. J. Am. Chem. Soc., 81, 18 (1959).
16. Hubei W., Hoogzand C., Chem. Ber., 93, 103 (1960).
17. Kr ii er к el1., H о ogz a n dC., H ii b e I W., Chem. Ber., 94, 2817 (1961).
18. F r i e d m a n S., At e t 1 i n S., S v e d i A., Wender I., J. Org. Chem., 24,
1287 (1959).
ДИЛЬСА —АЛЬДЕРА РЕАКЦИЯ, РАСТВОРИТЕЛИ
Введение. Ниже приведены данные, касающиеся выбора раст-
ворителя для реакции Дильса — Альдера. В качестве типичной
выбрана реакция между тетрафенилциклопентадиеноном и диметп-
ловым эфиром ацетплендикарбоновой кислоты [ 1). Исчезновение
интенсивной фиолетовой окраски диенона свидетельствует об окон-
чании реакции. Если эту реакцию проводить при кипячении в о-ди-
со.сн,
I '
с
III
с
I
со2сн3
хлорбензоле (т. кип. 179), то фиолетовый цвет исчезает уже за 5 мин
121. В этом случае реакция необратима, так как первоначально
образующийся аддукт самопроизвольно отщепляет окись углерода
с ароматизацией вновь образовавшегося цикла.
Предварительные опыты (комнатная температура). Пространст-
венно затрудненные кетоны не образуют оксимов при высоких тем-
пературах, однако они могут давать оксимы, если силыющелочной
раствор реагентов выдерживать при комнатной температуре от
одного до шести месяцев (Пирсон (31). Эти интересные данные навели
на мысль о возможности реакции Дильса — Альдера при комнатной
температуре. В первом опыте суспензию 2 г тонкоизмельченного
тетрафенплциклопентадпенона в 10 мл растворителя и 2 мл (3 экв)
дпметилового эфира ацетплендикарбоновой кислоты выдерживали
to*
291
при комнатной температуре, периодически перемешивая палочкой.
В у-дп.хлорбензоле через 17 дней выпадает белый аддукт и раствор
обесцвечивается. Перекристаллизация из приготовленного при 80’
раствора с добавлением 15 м.1 95%-ного этанола дает чистый аддукт
с выходом 80% ,. В хлористом метилене реакция заканчивается через
II дней с образованием светло-коричневого раствора. Выпаривание
раствора и кристаллизация из 10 мл о-дихлорбензола п 15 мл
95%-ного этанола дает чистый аддукт с выходом 83%. В обоих слу-
чаях продуктом реакции является дпметнлтетрафепилфталат; таким
образом, элиминирование окиси углерода из первоначального ад-
дукта протекает даже при 25’.
В уксусной кислоте реакция прекращалась приблизительно
через 2 месяца; анализ выделенного сероватого осадка показал, что
кристаллы аддукта содер/кат включения фиолетового дненового
компонента. Подобные результаты дали опыты в бензоле и ТГФ.
Очевидно, для того чтобы результаты были сравнимы, нужно пере-
мешивать суспензию.
Реакция при температуре кипения. При кипячении суспензия
достаточно хорошо перемешивается (табл. 1), но кипение может
Таблица 1
Реакция теграфенилциклопснтадиснона (2 г) с диметиловым эфиром
ацетилендикарбоновой кислоты (2 .цл) (3 экв) в кипящем растворителе
(10 мл) (выход аддукта 80—97%)
Растворитель 1 Температура ] кипения. СС । Время Д.ЧЯ завершения р 'акции Окончачельный цвет раствора
о-Дпхлорбензол 179 5 мин Желтый
1 страхлор этилен 120,8 15 » »
Уксусная кислота 118,2 15 » »
Метилциклогексан 100,9 -10 » »
Тиофен 1, 2-Диметоксиэган 8-1 2 час. Оранжевый
83 2 » Желтый
1(иклогексаи 80,8 2. 1 » Бссцие I ный
Бензол 80, Г1 2 » »
Метплэтплкеюн 79,6 3,2 » »
Сероуглерод -16.3 23 » Коричневый
Хлористый метилен -40,7 24 » Бесцветный
а Истинная температура жидкости 86 — 90,5'’.
Сопровождаться толчками. Более серьезным является то, что тем-
пература кипения чистого растворителя не обязательно совпадает
с температурой реакции. Так, циклогексан п бензол кипят примерно
при одинаковой температуре, но очень различаются по способности
растворять диенон: 10 мл раствора в циклогексане и бензоле,
насыщенные при 25°, содержат 22 и 493 мг дпенона соответственно.
292
Температура кипящей смеси в койне реакции превышает температуру
кипения чистого бензола приблизительно на 10', тогда как при
растворении в циклогексане температура жидкости близка
к температуре кипения циклогексана. Поэтому нельзя сравнивать
скорости реакций в растворителях с одинаковой температурой ки-
пения.
Даже если аддукт осаждается в виде бесцветных кристаллов,
неисчезающая окраска жидкости в конце реакции нежелательна.
Поэтому не следует пользоваться для реакций, проводимых при
температуре кипения, растворителями с т. кип. выше 81 (табл. 1),
а также сероуглеродом. Два серусодержащих растворителя дают
особенно интенсивную окраску.
Реакция при 78,8е. Реакцию проводили при температуре в жид-
кости 78,8', которую поддерживали с помощью перемешиваемого
чистого кипящего бензола.
Выбор молярного соотношения реагентов основывался отчасти
на результатах, полученных с применением малеинового ангидрида
как диенофила (табл. 2). Время, приведенное в табл. 2, соответст-
Таблица 2
Реакция тетрафенилциклопентадиенона (2 г) в тетрахлорэтилене
(10 л/л) при 78,8°
Диенофил Количество, же Время, час
Малеиновый ангидрид3 1,5 8,0
Малеиновый ангидрид 3 2,4
Димеги.товый эс[>и_р ацетплеидикар- боновой кислоты" 3 3,5
а Белый твердый осад-ж (2,90 г) содержит малеиновый ангидрид;
кристаллизация из 100 лз бензола дает 2,18 г (87°п) вещества в форме
призм с т пл. 223° (с разя.).
” Белые кристаллы собирают, промывают метанолом и получают
2,47 г (Ю0"(|) вещества с т. пл. 264 — 266-. Кристаллизация из 10 ,ил
о-дихлорбензола с добавлением 15 мл 95%-иого этанола дает 2,40 г (97%)
призм с т. пл. 264 — 266°.
вует иодному исчезновению фиолетовой или розовой окраски. Эта
бимолекулярная реакция существенно замедляется на заключитель-
ной стадии, но применение 3 экв диенофила уменьшает время реак-
ции. Поэтому именно такое соотношение было выбрано в опытах,
результаты которых приведены в табл.З, с учетом того, что малеино-
вый ангидрид — более реакционноспособный диенофил, чем димети-
ловый эфир ацетплендпкарбоповой кислоты.
В табл. 3 приведено время реакции при 78,8е для четырех типов
растворителей. Данные о температуре кипения и растворимости,
включенные в таблиц), в некоторых случаях важны для проведения
эксперимента пли выделения продукта реакции. Так, ТГФ обладает
293
Таблица 3
Реакция тетрафенилциклопентадиенона (2 г) с диметиловым эфиром
ацетилендикарбоновой кислоты (2 мл) (3 экв) в растворителе (10 мл) при
78,8° и перемешивании
- * S
л; Растворитель — - о
.Т 3 • *5
— г- - г
Апротонные растворители, нерастворимые в воде
1 9 Четыреххлористый углерод Бензол ссц CJJ., 76,8 80,1 4,0 5,0 164 493
3 Циклогексан СП.,(С1Б),СН„ 80,8 3,6 22
4 Дихлорэтан С1СН.,СН.,С1 8.3,8 4,0 400
5 Фторбензол СДБ.Р 84,8 3.9 350
6 Диметилкарбопат (СН3О).,С—О 90 3,0 65
7 Хлор мольный эфир CICOXoH, 92 4,4 386
8 Бромистый метилен СН.,Вг., 98,2 3,1 888
9 Три метилортофор ми ат НС(ОСН3)3 104 5,5 —
10 4 етрахлорэтилен CI.X----CC1., 120,8 3.5 130
11 Паральдегид (СН.,С1Ю)., 124 3.3 23
12 Днметилсульфит 126 0,0 102
13 Дибромэтаи ВгСН.,СН.,Вг 131,7 3.5 352
1 1 Уксусный ангидрид (СН.,СО).,() 139,6 4.8 40
15 Ортомуравьпиый эфир нс<Ьс.,нд., 146 5,3 —
16 си.клт-Тетрахлорэтаи С1.НССНСБ 146,3 3,9 760
17 Бромоформ СНВгз 149,6 2,8 876
18 Циклогексанон 1 1 СН.,(СНД4С=О 156 3,7 260
19 Ортоугольвый эфир Диметилоксалат С(ОС.,Н-,), 158 5,0 —
20 (СО.,СП.,)., 163 5,5 —
21 о-Дихлорбеизол о-С1Х„Н4 179 3,5 296
22 Диэтилоксалат (ССБС.Н-,)., 185,4 5,0 32
23 Иодбеизол с6н31 188,5 3,7 396
24 Диметилсульфат (CH3O).,SO„ 188,5 3,5 38
25 Нитробензол C(iH5NO„ 210 5.0 132
26 Диэт илсульфат (C.,H.-O)2S(0, 210 10,5 170
27 Г ексахлорбутадиен (CCI„- CCI)., 21 1 4,5 —
28 Тетраэтокспсилан (C.,1 l5O).Si 71.20.и.и 6,0 56
29 Дифениловый эфир (т. пл. С6Н5ОС6Н5 26,9) | Апротонные растворители, частично смешивс 257 кнциеся 3,5 с водой 352
30 Этилацетат CH3CO2C2H5(8,5% при 15°) 77,1 5,3 94
31 Нитрометан CH3NO.,(9,5% при 20°) 101,3 4,0 43
32 Хлорпикрин Апротонные раство CC13XO2 (около l°0 при 20') p и me ли, смешивающиес 112 я с водо 3,2 1
33 Тетрагидрофураи 1 1 CH.,CHoCH.,CH.,0 65,4 5,1 960
34 Ацетонитрил CH3CN 81 ,6 0,0 45
294
П родолжение
к- Растворитель Т. кип.а , С Время, час Раствори- мость и
35 1, 2-Диметоксиэтаи СН3ОСН.,СН,ОСН3 83 5,2 214
.36 Диоксин 1 1 осн.,сн,осн,сн. 101 ,3 4,За 386
37 Ди.метилформа.мид НСОХ(СН3)„ 153 3,2 125
38 Тетраметилмочевипа (СН1).АС(=О)Х(СН3)., 175,6 2,5а 100
39 Дпметилсульфоксид (CHj.,S+ - О- 189 3,0а 47
40 Т риглим СН3д(СН.,СН,О)3СН3 222 3,3 115
41 Сульфолан 1 "1 CH.,(CH.,)3SO2 130/6,5 Л/Л1 3,0а —
42 Этиленкарбонат, т. пл. 39_ ОСН,СН.2ОС=О 238 6,1 —
Протонные растворители, смешивающиеся с водой
43 44 Трифторуксусиая кислота /ирс/п-Бутапол CF3CO.,H (СН.,)3СОН 72,5 82,5 З.О1 5,7 30 130
45 Уксусная кислота (т. пл. 16,6°) СН3СО,Н 118 5,8 30
46 Этиленбромгпдрин ВгСН.,СН.2ОН 150 4,0 —
47 Фенол (т. пл. 42,5:) свн-,бн 181 5,2 —
48 Хлоруксусная кислота (т. пл. 63”) СН.>С1СО,Н 189,5 2,7 —
49 Днхлоруксусная кислота (т. пл. 10 ) СНС1.,СО.,Н 193,5 2, 1а 257
50 Трпхлоруксусная кислота |т. пл. 58-) СС13СО.,Н 196 1 2J —
51 Трнэтиленглвколь И(ОСН.,СН.,)3ОН 287 8,0 23
а См. Marsden С., Мапя S., Solvents Guide. Interscience (1963).
® По исчезновению окраски исходных веществ
в Растворимость диенона (.ие/10 .11.1) при 25'.
такой резко выраженной растворяющей способностью по отноше-
нию к днепону (960 л/г/10 мл при 25’),что реакцию можно проводить
при температуре на 13,4' выше температуры кипения ТГФ. Другие
растворители, хорошо растворяющие дпенои: бромистый метилен
(888 мг), бромоформ (876 л/г) и сшилг-тетрахлорэтан (760 л/г). Для
выделения аддукта обычно достаточно охладить суспензию, но, если
необходимо, можно разбавить смесь другим растворителем или
отогнать растворитель. Выделение этого аддукта с выходом, близ-
ким к количественному, почти всегда не вызывает затруднений.
295
Ни в одном из .32 апротонных растворителей, не растворимых пли
частично растворимых в воде, нежелательная окраска не появ-
ляется.
За исключением ,\л 26, для которого время реакции составляет
10,5 чае, реакция протекает в течение 2,8—6,1 час. Для десяти водо-
растворимых апротонных раствори гелей время реакции приблизи-
тельно одинаково (в среднем 3,9 час); в четырех из них протекает
быстрая реакция, но появляется дополнительная окраска.
Для растворителей № 6 и №42 наблюдаются аномалии: днметил-
карбоиат (№ 6), т. кип. 90 , очень мало растворим в воде; этплен-
карбоиат (Ао 42), т. ил. .39 и т. кип. 238 , хорошо растворим в воде.
Определение молекулярного веса этпленкарбоиата в феноле но упру-
гости паров подтверждает его мономерность (Форлендер |4|).
Наиболее сильными эффектами растворителя обладают водораст-
воримые протонные соединения. В трихлоруксусион кислоте,
которая представляет собой жидкость при температуре кипения
бензола, время реакции составляет всего 1,2 час, тогда как для
триэтиленгликоля (А" 51) оно равно 8 час. Дихлоруксусная, три-
хлоруксусиая и трифторуксусная кислоты ускоряют реакцию, но
п вызывают образование нежелательных окрашенных примесей
в реакционной смеси.
Влияние добавок. Особенно интересным эффектом растворителя
является отрицательный катализ, который можно видеть из данных
табл. 4, полученных для перемешиваемой смеси при 78,8 . При
переходе от абсолютного этанола к 95%-ному этанолу время реакции
увеличивается с 6,5 до 9,3 час, а при добавлении к спирту 10 об. %
серной кислоты — до 9,6 час. Время реакции в уксусной кислоте
(5,8 чае, табл. 3) увеличивается до 9,3 чае при добавлении 10?о воды;
Таблица 4
Влияние добавок на реакцию в условиях,
указанных в табл. 3
10 лм чистого растворителя или 10 лм растворителя < добавкой Время час Выход, о Пеоиетя- чески 2.4 7 г)
Абсолютный этанол 6,5
95%-ный этанол J 9.3 2,38
Абсолютный этанол и 1 мл концент- 9,6
рировашюй серной кислоты Уксусная кислота • 1 мл Ле.,О 5,0
Л ксхснля кислота 1 мл воды 9,3
Эфнрат трехфтористого бора 5,6 1,78
а Растворимость диенона при 2.->“: 10 .иг/10 ,ил.
& Растворимость дненона при 2Й1': 8 1 .иг.'10 .и.т
296
наоборот, прибавление уксусного ангидрида, устраняющего следы
влаги, приводит к ускорению реакции. Трехфторпстый бор нс ката-
лизирует эту реакцию.
Масла как растворители. Наиболее пригодны рафинированное
минеральное масло и силиконовое масло, представляющее собой
узкую фракцию полнметилсплоксана с концевыми блокирующими
метильными группами:
СН3 сн3 1 СН3
1 CH3SiO 1 — SiO 1 1 —SiCH3 п =10— 14
1 СН3 1 СН:1 1 п СН3
Таблица 5
Опыты при 78,8' в условиях, указанных в табл. 3
I’dCTROpif ТСЛЬ Время, час Раство- римое гь дкеиоиа, .ис/1 0 .4.1 Разбави- тель Выход, г (теорети- чески 2.47 <?) Т пл., °C
Силиконовое масло Минеральное масло 3,2 5,0 24,4 СН3()Н 2,47 2,22 263—265 257—260
Результаты, приведенные в табл. 5, показывают, что реакция про-
текает быстро и с высоким выходом. Эти растворители настолько
стабильны, что нежелательная окраска, вероятно, не будет возни-
кать даже при гораздо более высокой температуре. Растворитель,
содержащий избыток диенофила, можно регенерировать для повтор-
ного использования, например разбавлением петролейным эфиром,
удалением аддукта и выпариванием.
Реакция при высокой температуре. Реакцию тетрафенилцикло-
пентадиенопа с дпфенплацетиленом, протекающую с выделением
окиси углерода и образованием гексафенилбензола, можно провести
при сильном нагревании смеси компонентов (по 0,5 г каждого) в про-
бирке 151. Этот способ неудобен для получения больших количеств
вещества, так как продукт реакции имеет очень высокую темнера-
с6н5
с
in
с
С6Н5
+ со
туру плавления (454—456 ). Удобная методика (61 основана на при-
менении в качестве растворителя бензофенона (т. пл. 48;, т. кип.
305,4 ). Смесь 40 г этого кетона, 0,021 моля теграфенплцпклопента-
диеноиа и 0,043 моля днфенплацетплена нагревают на микрогорелке
в круглодонной колбе с воздушным обратным холодильником до
297
сильного кипения (температура жидкости 301—303°). Фиолетовая
окраска начинает бледнеть через 10—15 мин и через 25—30 мин
становится красно-коричневой. После того как окраска перестанет
светлеть (45 мин), убирают горелку и прибавляют 8 мл дифенилового
эфира,чтобы предотвратить
осаждение бензофенона.
Кристаллы вещества отде-
ляют, вновь растворяют
при нагревании п оставля-
ют кристаллизоваться. Пос-
ле отделения и промывания
Рис. Д-2. Вакух мирсвапие
и запаивание капилляров.
Рис. Д-З. Прибор для определения темпера-
туры плавления.
кристаллов бензолом до обесцвечивания, прод\ кт реакции получают
в виде бесцветных пластинок с т. пл. 454—456'; выход 9,4 г (85%).
Растворителем для перекристаллизации может служить дифенило-
вый эфир (7 мл/г).
Вещество окисляется на воздухе, поэтому температуру плав-
ления определяют в вакуумированном капилляре. Это можно сде-
298
дать с помощью приспособления, показанного на рис. Д-2, которое
состоит из стеклянной трубки, закрытой с одного конца резиновой
пробкой со сквозным отверстием. Это отверстие смазывают глицери-
ном, капилляр берут ближе к запаянному концу и вставляют
в отверстие почти на всю длину (пометки, сделанные восковым
карандашом на капиллярах для того, чтобы их не перепутать, после
этой операции необходимо восстановить). Соединяют открытый
конец стеклянной трубки с вакуумным насосом, делают пометки на
капилляре для обозначения места, где его следует запаять, прибли-
жают к этому месту пламя микрогорелки, устойчиво располагая
руки на столе, чтобы не сделать неосторожного движения при рас-
плавлении капилляра, вносят капилляр в пламя. Когда стенки
капилляра опадут и расплавятся, сразу вытягивают капилляр из
пламени. Если трубка и капилляр не образуют прямой линии,
то вновь расплавляют и выравнивают.
Определение температуры плавления удобно проводить в спе-
циальном приборе, изображенном па рис. Д-З. Вакуумированный
капилляр следует запаивать как можно ближе к образцу, чтобы из-
бежать возгонки, тогда повторные определения температуры плав-
ления можно проводить на том же образце.
Значение температуры плавления, равное 456 , является средним
из нескольких определений, среднее значение для температуры
затвердевания составляет 4546 Если количество дифеннлацетилепа
уменьшить до 1,2 же, то выход остается тем же самым, но темпе-
ратура плавления понижается до 450—452'.
Эту реакцию пытались проводить и при других температурах
с использованием следующих растворителей: стеариновая кислота
(340—365 ), дп-н-бутилфталат (320—325 ), фенплсалнцилат (290 ).
Однако первые два растворителя непригодны из-за побочных реак-
ций, па которые расходуется некоторое количество диснона, а
в третьем реакция протекает слишком медленно.
1. D i t I 11 е у \V„ Т li е w а 1 t I., Tros к е и О., Ber., 67. 1939 (1934).
2. Org. Expts., 305.
3. Pearson D. E., К e a t о n O. D., J. Org. Chem., 28, 1557 (1963).
4. Vor I ander D., Ann., 280, 187 (1894).
5. Org. Expts., 307.
6. F ieser L. F., procedure submitted to Org. Syn.
ДИМЕДОН (5,5-Диметплцнклогександиоп-1,3). Мол. вес 140,18,
т. пл. 149'.
Д. получают конденсацией .малонового эфира с окисью мезитила
с последующим гидролизом и декарбоксилированием [1]. Вещест-
во почти нацело енолнзовано.
О-Ма+ ОН
С2Н6О,С СО,С,Н5 С.,Н-О,С
СН, СН, —; Ц3С I I ОН~- Н1 Н..С ) I !-СО2
<СН’>*Ч/ко НзС/ Ао 0Т-85% Н3С -Ч
299
Реакция с альдегидами. Л- особенно широко применяется для
выделения и идентификации низших альдегидов 121. С газообразным
формальдегидом Д. без катализаторов образует метнлен-бис-диме-
доп— соединение с высокой температурой плавления (188), при
образовании которого происходит десятикратное увеличение массы
альдегида.
ОН ОН
L .СН2 J
н;!с [| Р | СНз
Н.(С ' " z сн:,
Т. пл. 1
N-Защитная группа для аминокислот 131. Д. легко реагирует
с эфирами аминокислот (но не со свободными аминокислотами) с об-
разованием енамина (1). Еиамиинаягруппировка отличается большой
R
ОН
I
R
I
\HCHCOJi
I
I I 2 стадии
(СН J ,^о "
(2)
I
NHCHCO..R’
I
, / Д
СНС13 I I 5 н HCI
—’ (CH3).J ' г юн’
- оЧ)
(I)
R R"
I I
\HCHCO\HCHCO.,R"'
I I Н\о3
(СН^' i
(3)
R R”
\CHCO.\’HCHCO.2R'"
АОН
' А
(CH3J. I
(4)
устойчивостью по отношению к кислотам и щелочам. Например,
кипячение вещества (1) со смесью разбавленной кислоты и спирта
в течение 2 час приводит к удалению сложноэфирноп группы с обра-
зованием N-дпмедоноампнокислоты (2). Эту кислоту можно приме-
нять для конденсации с образованием дипептида (3) (карбодиимид-
ный метод). /Димедоновую группировку удаляют в случае необхо-
димости в виде 2,2-дпбромдпмедона или, что более удобно, реакцией
с азотистой кислотой, которая приводит к темно-синему пзонптрозо-
300
соединению (4), легко гидролизующемуся разбавленной соляной
кислотой.
1. Ill [) ;i ii н е р Р., Тодд X., «Синтезы органических препаратов», ИЛ, ЛЕ,
1949. сб. 2, стр. 220.
2. И о г и i n р Е. С., II о г п i п g ЛЕ G., .1. Огу. Chem., 11, 95 (1946).
3. II а 1 р < г п В., .1 a m е s L. В., .Australian .1. Chem., 17, 1282 (1964); Н а 1-
р е г и В., ibid., 18, 417 (1965); Н а I р с г п В., Cross /\. I)., ('hem. Ind.,
1965, 1183.
/яр«о(ДИМЕТИЛАМИДО)-ФОСФИН, см. Гексаметилтриамид
фосфористой кислоты.
я-ДИМЕТИЛАМИНОБЕНЗАЛЬДЕГИД, п-(СН :1)2№СвН4СНО.Мол.
вес 149,19, т. пл. 74 , т. кии. 176 — 177717 мм. Не совсем чистое
вещество окрашено в лимонный цвет; на свету приобретает ярко-
розовую окраску.
Получение 111:
UNO.. СИ..О • C,H;N(CH3)..
((-1 IJ A( „II,-• : :.Н.. Л„Н1У - нсиП------'
- - h-(CH3).,\C,.II4X СИС(1Н4Х(СН3).,-л
—лh-(CH3).,\c(1HjX -ch.,-; /i-ohccjIjX
Цветная реакция на пирролы [21. Одну каплю раствора иссле-
дуемого соединения в воде, эфире пли щелочи смешивают на часовом
стекле с одной или двумя каплями 5%-пого раствора н-диметил-
амииобензальдегида. Если в растворе имеется пиррол или вещество,
способное к образованию пиррола, то появляется фиолетовая ок-
раска. Альдегид конденсируется с таутомерной формой пиррола
(3) с образованием соединения (4), которое при действии иона водо-
рода превращается в окрашенный хиноидный ион (5). Положитель-
ную пробу дают производные пиррола, имеющие одну СН-группу,
такие, как индол, скатол (0-метилипдол), триптофан. Д. конденсиру-
ется с первичными аминами, образуя основания Шиффа, имеющие
желтый цвет. Кроме того, желтые вещества получаются при кон-
денсации этого реагента с соединениями, содержащими группу
—NHCONH.J3]. Однако только пирролы дают фиолетовые красители.
301
Проба на гидразин. Д. конденсируется с гидразином, давая
продукт (6), образующий далее хиноидный нон (7), имеющий
окраску от желтой до оранжевой |4|.
2 (СН3).,Х —'У— СНО ~ > (СН3).Х -^'2 сн
н +
(СН3).Х — Д СН = X
(Ч)
(СНл).А’ = (222 =° сн~хн
~~~ >
(СН3).,Х %- сн =.\
О)
1. Адамс Р,, Колеман Г., «Синтезы органических препарате-^, ИЛ, М.,
1919, сб. 1, стр, 193,
2. F е. 1 д 1 F-. Spot Tests, 2, 198, Elsevier (1954).
3, S с h г е i b e r ,1., \V itkop B., .1. Am, Chem. Soc.. 86. 2441 (1964).
4. F e i д 1 F., Spot Tests, 2, 219, Elsevier (1951).
ДИМЕТИЛАМИНОБОРАН, (CHJ.NHBH,. Мол. вес 58,93, т. пл.
37 , т. разд. 150 . Получение [11.
Основания Шиффа, например полученный из анилина и бензаль-
дегида бензилиденанилин, быстро восстанавливаются Д. в виде
раствора или суспензии в уксусной кислоте |2]. В отличие от других
боргидрпдов Д. можно применять в кипящей уксусной кислоте
ЗСЬН-.СН^ ХС.Н, -у (CH3).\HBH3-i-3CH:iCO.,H —
— ЗС„Н.-,СН.,ХНС6Н,, -У (СН.,),ХН (СН;1СО..);!В
с небольшой потерей активности (рекомендуется 25%-ный избыток
этого реагента). Не затрагиваются следующие функциональные
группы: NO.. CI, ОН, ОСН., СО.Н, СО2С;Н-„ > > ХН-.
1. X о 1 h II., В с V с г 1!.. Cheni. Вег.. 93. 928 '19901.
2. В i 1 1 m a n J. Н.. М с D о w е 1 1 .1. W., .1. Они Chem., 26. 1437 (1901).
3-ДИМЕТИЛАМИНОПРОПИЛАМИН, (СНЯ) ДСН ,CH.CH,NH2.
Мол. вес 102,18, т. кип. 131 —134’.
Этот диамин применяют для удаления избытка хлорангидрида
кислоты в новом методе синтеза сложных эфиров из третичных и вто-
ричных стероидных спиртов |1|. Раствор спирта в эфире обраба-
тывают этилмагнийбромидом с образованием магнийбромалкого-
лята, после чего проводят реакцию с хлораигидридом кислоты. Для
С.,Н,.М«Вг R'COCI
ROH —-------ROMgBr--------- ROCOR' -7-CiMgBr
302
удаления избытка хлорапгидрпда, который гидролизуется разбав-
ленной щелочью довольно медленно, прибавляют избыток Д., и
образующийся амид, обладающий основными свойствами, удаляют
промыванием разбавленной кислотой.
«:и >,\сн сн сн лн.
R'COLl----;---1—2 : > (CI[.,),\CH;CH,CH..\HCOR'
1. Evans D. D., Evans D. E., Lewis G. S., Palmer P. J., W e y-
e 1 1 D. J., J. Chem. Soc.. 1963, 3578.
М-(3-ДИМЕТИЛАМИНОПРОПИЛ)-ХТ -ЭТИЛКАРБОДИИМИД,
ХЛОРГИДРАТ, (СН ,)гХСН2СН2СН.Х-С—\’С_.Н.-,-НС1. Мол. вес
175,69. Получение 111.
Пептидный синтез |2|. .Этот водорастворимый карбодиимид
применяют в синтезе циклического гексапептида на стадии конден-
сации двух трипептидов и для проведения циклизации.
I. S h е е 11 а и 4. С, G i a i е k s 11 а и к Р. А., В oshart G. L., J. Or,'. Chem.,
26. 2525 (1961).
2. К о р р 1 с К. D., X 1 1 е с к i D. Е.. J. Am. Chon. Soc.. 84, 4457 (1962).
ДИМЕТИЛАМИНОСУЛ ЬФОНИЛМЕТ ИЛ ИДЛИТИЙ,
(CHJ2\SO2CH2Li. Мол. вес 129,08.
Реагент получают действием н-бутиллптпя на Х,\т-диметпламид-
метапсульфокислогы в ТГФ 111. При взаимодействии с кетонами он
образует с высокими выходами замещенные fJ-оксисульфампды.
(С„Н,).,С- О--'- (СН3) ,\SO.,CH .Li_> (С6Н-,)..ССН ,SO.,\ (СН3)„
ОН
1. Corey Е. .1.. С h <1 \ к о v s к \ М, .1. Am. CIktii. Sue.. 87, 1345 (1965).
N, N-ДИМЕТИЛАНИЛИН. .Нол. вес 121,18, т. кип. 193', уд. вес
0,96, рКЬ 9,62.
Очистка. Технический продукт обычно содержит примесь моно-
метпланплина, который можно удалить добавлением небольшого
количества уксусного ангидрида и перегонкой. Вторичный амин
превращается при этом в малолетучее ацетильное производное, а
уксусная кислота связывается в виде соли. Повышение температуры
при добавлении уксусного ангидрида указывает на присутствие
вторичного (пли даже первичного) амина; при прибавлении
уксусного ангидрида к чистому третичному амину наблюдается
понижение температуры. После ацетилирования Д. можно отде-
лить также экстракцией минеральной кислотой.
Ацилирование третичных спиртов. Из трех почти одинаковых
методик, приведенных в сб. «Синтезы органических препаратов»
для получения /прс/и-бутиловых эфиров, здесь описана одна, даю-
щая наплучшпй выход: получение ди-/ирс/п-бутилового эфира
303
малоновой кислоты [1!. Смесь спирта и Д. охлаждают льдом и при
зю СО,С(СН3)3
СН,(С()С1)„Ш2(С11.,):1С.()Н I с„и5\ (С.Нз), ^г8-40: сн,.
0,2 моля в 1 моль (),(>3 моля СО,С (СН3)3
CHCI, (60 AI.I)
перемешивании прибавляют раствор дихлораигидрида малоновой
кислоты в хлороформе с такой скоростью, чтобы температура не
поднималась выше 30 .
Смесь хлористого ацетила и Д. применяли для ацетилирования
5а-оксистеропдов; 50-гпдроксилы1ая группа при этом не затраги-
вается 121. Например, кипячение смеси 30-ацетокси-5а-окси-
Д' •"-эргостадиеиа, хлористого ацетила и дпметилапплина в хлоро-
форме в течение 16 час, обработка смеси, хроматография и однократ-
ная кристаллизация приводят к почти чистому 3₽,5а-диацетату
с очень хорошим выходом 131.
Дегидрогалогенирование. Применение Д. для дегидрогалоге-
нирования можно проиллюстрировать получением циклогексадиепа-
1,3 14]. Циклогексен, взятый в избытке, хлорируют трет-бугмл-
гииохлоритом в присутствии перекиси бензоила при температуре
кипения циклогексена. Вторую стадию проводят при такой
температуре, чтобы диен отгонялся по мере образования.
Декарбоксилирование. Тарбелл и сотр. |5] декарбоксилировали
3,5-дихлор-4-оксибензойную кислоту при кратковременном нагре-
вании ее раствора в Д.
ОН ОН
С1\/ /СЛ С1 ч// /С1
I II , , , 190- 200'. 2 час | 11
| || H — | ||
, 4,8 моля ' -
I
со,н
1,2 моля
304
Реакция с тетрацианэтиленом. Мак-Кузик и Мельби |6| прибав-
ляли тетрацианэтилен небольшими порциями к перемешиваемому
раствору Д. в ДМФА, похтержпвая температуру 45—50 . п-Три-
цнаивпппл-1Х,ГХ-диметплапилпн выпадает в виде темно-синих кри-
сталлов, которые перекристаллизовывают из уксусной кислоты
(темно-красный раствор).
(CH3);N %+ (NC).,C-C(CX)2
0.20 моля 5- 58%
0,22 моля в
63 ли ДМФЛ
—. (СН3).,Х — С---С(CN),Д-НСХ
= I
сх
Элиминирование (CH3),NH. Элиминирование диметиламина из
основания Манниха (1), полученного пза-феноксиацетофенона, фор-
мальдегида и аммиака, происходит особенно гладко при кратко-
временном кипячении в Д. |7|.
Кипячение
4 с.н.мсн,,), !
С6Н-ПССНСН.,Х(СН3).,--------> С6Н5С-С^СН„ 4- (СН3)„ХН
I I
ос,,н5 ОС6Н-,
(1)
Другие реакции. См. п-Диметиламннобензальдегид, Оксалил-
хлорнд.
1. РахаЧ., «Синтезы органических препаратов», ИЛ, М., 1954, сб. 5, стр. 19.
2. Р 1 a t t п е г А., Р е t г z i I k a Th., Lang W., Helv. Chim. Acta, 27, 513
(1944).
3. В 1 a (1 о n P. et al., .1. Chem. Soc.. 1952, 4883.
4. G г о b С. А., К и у И., (i а к п e u x A., Helv. Chim. Acta, 40, 130 (1957).
5. T a p б e л л Д., В и л ь с о и Дж., Ф анта п II.. «Синтезы органических
препаратов», ИЛ, М., 1953, сб. 4, стр. 23(5.
6. М а к - К у з и к Б., М ел ьб и Л., «Синтезы органических препаратов»,
1961, сб. И, стр. 67.
7. \V г i я h t J. В., J. Or у. Chem., 25, 1867 (1960).
ДИМЕТИЛАЦЕТАМИД, CH:1CON(CH3),. Мол. вес 87,12, т. кип.
165 . Получение II].
Ацетилирование. В присутствии хлорокиси фосфора Д. аце-
тилирует бепзофураны 121 (ср. ДМФА — РОС13).
1,7 г
Декарбоксилирование (эффект растворителя). Казинп и
Гудмен [31 нашли, что 7-нитропндолкарбоновая-2 кислота
305
декарбоксилируется при нагревании в присутствии каталитических
количеств медпой соли этой же кислоты. Выход повышается, если
в качестве растворителя применять Д., а не обычно используемый
хинолин. Кроме того, в этом случае облегчается выделение продукта
реакции.
COTI
89'6
(R CO?) ,Cu - C!Ij COX( CH3)
Е., J. Am. Chem. Soc., 59, 401 (1937).
Р., Chem. Вег., 97, 616 (1964).
L., Can. J. Chem., 42, 1233 (1964).
ДИМЕТИЛАЦЕТАМИДА ДИЭТИЛАЦЕТАЛЬ,
(СН;(),\С(ОГ,НД
1. R u h n f f J. R,, R e i <i E.
2. Deor h a D. S., Gupta
ch.
Мол. вес 1(51,24.
Д. д. — жидкость, не перегоняющаяся в вакууме.
Получение 111. Метод получения Д. д. точно такой же, как
и диэтплацегаля ди.метилформамида; выход 64%.
Реакции с соединениями, имеющими активную метиленовую
или метильную группу 111. Д. д. может обратимо отщеплять этанол
с образованием 1-ди.метиламино-1-эгоксиэтплена (2):
сн3 сн.
(CH3),X-C(OC2H5)2 (CH3).,\-COC2H5-C2H5OH
Соединения (1), (2) или их смесь применяют для конденсации с аце-
тофеноном, метиловым эфиром циануксусной кислоты, динптрплом
малоновой кислоты, нитрометаном, циклопентадиеном и 2,4-динит-
ротолуолом. Эти экзотермические реакции проводят без раствори-
теля. Только в случае циклопентадиена необходимо нагревание (кн-
СН3 СН.,
О,\ ^-СН3 -- (СН:;),\С(ОС,Н5),
- CH -C.\(CH3)2
пячение в течение 30 мин).
Реакция с аллиловыми спиртами. Взаимодействие Д. д. |в фор-
ме (2)1 с аллиловым спиртом сопровождается аллильной пере-
группировкой и приводит к диметиламиду аллилуксусной кислоты
,.\(СН3),
СН, — С. —
ОСН2СН = СН,_
(3)
А(СН3),
—> СН, = СНСН,СН,СХ
СН, =.- С ' ’ -У НОСН.СН сн,
ОС,Н5
(1)
306
(4) |1|. Эшенмозер и сотр. [2] нашли, что эта реакция является сте-
реоспецифичной (швейцарские исследователи применяли смесь дпме-
тилацеталя диметилацетамида и его дезметанольного производного).
Так, например, аллиловый спирт (5) с 4р-гидроксилыюй группой
дает соединение (6), а 4а-изомер (7) дает 2-эпимер (8). Бензиловый
хг.-(снр2
сн2=с'
________ ОСН,
.В ксилоле при 14 0”
(Ч
(7)
/М(СН,)2
СН2=сТ_
юсщ
В ксилоле при-14О5
(8)
спирт при взаимодействии с Д. д. образует два продукта (разде-
лены хроматографией), указанные на следующей схеме |2|:
СОСИ,
,CHSOH сн СН3 (к
I I —!; + к
I 1> 20 час в кипящем 0 ।
X/ и-дях.торбензоле х/ ХСН СН
I I
СО.\'(СН3)., сохуснд,
.10°,, 2О‘„
1. Meer w е i n Н., Florian W., Schone X., S t о р р G., Ann., 641,
1 (1961).
2. W i с k А. Е., Felix D., Steen К., Е ь с h е nmoser A., Flelv. Chim.
Acta, 47, 2425 (1964).
2,3-ДИМЕТИЛБУТАДИЕН,СН2=С- -С-СЩ.
СН,сн,
'Мол. вес 82,14, т. кип. 70'.
Д. получают из пинакона а) медленной перегонкой смеси с не-
большим количеством 48%-ной бромистоводородиой кислоты (вы-
ход 55—60%) |1| и б) быстрым пропусканием через трубку, запол-
ненную окисью алюминия и нагретую до 450—470 |2|.
1. Аллен Ч., Белл А., «Сипплы органических препаратов». ИЛ, М., 1952,
сб. 3, стр. 167.
2. И ь ю т о и Л., К о б у р и Э. «Синтезы органических препаратов», ИЛ, М.,
1952, сб. 3, стр. 189.
2,3-ДИМЕТИЛБУТИЛ-2-БОРАН («тексплборан»). (См. также
Дпборан.) Получают реакцией 2,3-дпметплбутена-2 с дибораном
в диглиме или ТГФ [11. С ц//с-ациклическими олефинами Д. взаимо-
307
СН3СНз
В-.н. I I
(СН3),С-С(СН.,), —* НС—С-ВН,
11“
СНз СНз
действует с большей скоростью, чем с //г/мм-олефинами, и это раз-
личие в скоростях позволяет выделять ///ринл-олефин из смеси изо-
меров. Д. — удобный гидроборирующий агент для диенов (диборан
дает полимерные продукты):
RBH..; 11.0,
СН, - СНСН..СН - сн, —
/НОСН.,СН,СН,СНСНз
ОН
НОСН,СН,С11,,СН,СН,0Н 30'%,
70%
Д. реагирует с ацетиленом с образованием соответствующего винил-
борапа.
1. Z V. е i f е I G., Brown Н. С., .1. Am. Chem. Soc., 85, 20(56 (1963).
N.N'-ДИМЕТИЛГИДРАЗИН, (CH.,),NNH,. Мол. вес 60,08,
т. кип. 63J, уд. вес 0,784, рКЬ 6,8, смешивается с водой.
Синтезы нитрилов. Метод превращения альдегидов в нитрилы
включает получение Ы,Ы-диметилгидразона (1), реакцию его с иоди-
RCHO + HjNN(CH3)2 -» rch=nn(ch,)2 ch?i>
-що (1)
-->R-C=N^(CH,),l'----> R-C = N + (CH,),N + B+Hf
В—ЭН
(2) (3)
стым метилом (или метиловым эфиром /г-толуолсульфокпслоты)
с образованием 1\!,1\1,.\1-трпметилгидразоиневой соли (2) и Р-элими-
нироваипе под действием метанольного раствора едкого натра 111.
Этот способ с успехом использован для превращения 2-хлор-
1-формилциклогексена-1 (4) в нитрил (7) (Пакетт |2|)*. Гидразон (5)
получают кипячением реагентов в течение 16 час с водоотделителем
Дина — Старка, конструкция которого описана в |3|.
' Обзор родственных внутримолекулярных перегруппировок см.
/7/ш.и. перев.
[За).-
308
Получение гидразонов. Реакцией альдегида или кетона с гидра-
зином обычно не удается получить гидразон с хорошим выходом
вследствие быстрого образования азина. Эту трудность можно
обойти превращением карбонильного соединения в ЫД’-диметил-
гидразон с последующей обменной реакцией с гидразином 141 при
кипячении М.М-диметплгидразона е 2 -3-кратным избытком без-
водного гидразина
1. S m i t Ii R. F„ W a I k e г L. E„ .1. 0гц. Chem., 27, 4372 (1962).
2. Paquette L. A., procedure submitted to Org. Syn.
3. II а тельсон С... Готтфрид С., «Синтезы органических препаратов»,
ИЛ, М„ 1952, сб. 3, стр. 505.
За. II а у м о в 10. А., Г р а и д б е р г И. И., Уси. химии, 35, 21 (1966).
4. \ е w к о ni е G. R., Fishel I). L., J. Org. Chem., 31, 677 (I960).
N.N-ДИМЕТИЛГЛИЦИНГИДРАЗИД, ХЛОРГИДРАТ (реактив
D), (CH;!).,NCH.,CON11NII., HCI. Мол. вес 153,62, т. пл. 181 .
Это соединение аналогично реактивам Жирара Т и Р и получается
тем же путем. Д. х. применяется для выделения кетонов из смесей
за счет их превращения в производные, которые экстрагируются из
эфирного раствора кислотой (разбавленная НС1) 111. Очистка его
несколько проще, по сравнению с реактивами Жирара Т и Р.
I. V i s с о п t i п i М., М е i е г J., Helv. Chim. Acta, 33, 1773 (1950).
1,2-ДИМЕТИЛ-4,5-ДИ-(МЕРКАПТОМЕТИЛ)-БЕНЗОЛ (3). Мол.
вес 198,34, т. пл. 66—67', по запаху сходен с о-дихлорбензолом.
Получение. Модификация 111 более раннего метода включает
кипячение и перемешивание смеси о-ксилола, параформа и соляной
кислоты, после чего дихлорметильное производное (2) кипятят с тио-
мочевиной в изопропиловом спирте.
Н3с , Н3С, , . СН.,С1
. ,| Кипя ение 22 час । Д
। ;-у (сн.,о)„ ; Конц. нс1--> I
Л 11 ISO г 1230 л:.1 "'° К ' ,
Н3С' А/ Н3С СН2С1
2 12г (2)
(1)
S = C(NH.,)..: Л , CH.,SH
•• (С.НД..СНОИ | ||
so% -* I ф
4 CH..SH
< 3)
Идентификация карбонильных соединений. Д. легко конден-
сируется с альдегидами и кетонами в условиях кислого катализа
и применяется для получения характеристических производных 111,
тем более, что оно не имеет неприятного запаха, характерного для
1,2-димеркаптоэтана. Альдегиды и кетоны различаются по скорости
реакции с Д.
I. S h a s h a k I., В е г g m а n 11 Е. D., J. Chem. Soc., 1966, С, 1005.
309
ДИМЕТИЛ КЕТЕН, (СН ,)2С -С-О. Мол. вес 70,09, т. кип. 34°.
Д. быстро окисляется кислородом с образованием взрывчатой
перекиси; несколько капель раствора при испарении на воздухе
могут детонировать. Д. димеризуется при хранении.
Получение, а) Из бромангидрида а-бромизомасляной кислоты |1].
Бромангпдрид прибавляют по каплям к смеси цинковых стружек
и этилацетата, находящихся в колбе, наполненной азотом и эвакуи-
рованной до 300 л/л/. Д. отгоняют вместе с этилацетатом и собирают
в ловушке, охлаждаемой смесью сухого льда п ацетона. Титрование
1 Icpei-оика при
(СН3).,С—С —O-yZn (стружка)-уЗОСН3С02С,Н3 —> (СН3),С = С-=О
Вг Вг
0,48 моля 0.61 г-атп.м
аликвотной пробы при О’ 0,1 и.раствором едкого натра пофенолфта-
лепну указывает, что дистиллат является 9—10%-ным раствором
Д. в этилацетате.
б) Из димера. Димер диметилкетеиа — тетраметилпиклобутан-
дион-1,3 получают действием триэтиламина на хлорангидрид изо-
(СНЭ)2СНС=О +
С1
(c2h5)3n
о
Li)-» г (сн3)2с=с=о
8fe%
Т. пл. 116°
масляной кислоты [2]. Хепфорд и Зауер [2] описали устрой-
ство прибора для пиролиза, в котором можно осуществить
деполимеризацию сравнительно высокоплавкого димера Д. с обра-
зованием Д.
в) Из диметилмалоновой кислоты (21. Смесь этой кислоты, ук-
сусного ангидрида и следов серной кислоты встряхивают до раство-
рения и выдерживают 2 дня для образования ангидрида диметнл-
малоновой кислоты. После этого прибавляют небольшое количество
карбоната бария, уксусную кислоту и ангидрид отгоняют в вакууме
О
СН3 СОЛ Н3С с
3 - 23- 3 '• юн ( Н,
С —Ac2O(H2SO|) —- С О-—г—> ' )-С = С = О-СО3
СН3 '"'СО2Н Нзс/ 'С/ (общпн| СНз'
2,3 г (следы)
6
(или полимер)
при температуре ниже 60'. Сухой остаток ангидрида диметилмало-
новой кислоты разлагают, повышая температуру так, чтобы нача-
310
тось достаточно интенсивное образование двуокиси углерода и Д.
(около 100 ).
г) Из диметилкетенацилаля диметилмалоновой кислоты (4).
Бестиаи и Гюнтер [31 медленно нагревали до кипения при давлении
О
СНз. СО2Н 60-^% ,С го /СН,
с - (сн3со).,о с о—_;о=с=с;
СН3 СО,Н НзС чС/ СН3
о
(1)4 моля 1е молей (2) (3)
СН, /С-О. сн3
с' с-=с
СН3' чс —OZ СНз
о
(-1) т. пл. 80°
10 .ил? диметилмалоновую кислоту с избытком уксусного ангидрида
и отгоняли образующуюся уксусную кислоту иа эффективной ко-
лонке. Кристаллизацией белого остатка из петролейного эфира
получают чистый ацилаль с выходом 80','о. Соединение термически
устойчиво до 180J, но в присутствии карбоната калия быстро рас-
щепляется уже при 150' на Д. и двуокись углерода. Расплавленный
ацилаль (4) прибавляют по каплям к небольшому количеству кар-
боната калия, находящегося в колбе, нагретой до 150С Образую-
щуюся газообразную смесь Д. и двуокиси углерода можно непо-
средственно использовать для реакции или сконденсировать Д. для
О
,С-0 СН, СН,
' \ 3 К .со., |.;о ‘-пз
С С = С. -------;----*2 С=:С = ОТСО,
''С-О 'СН3 СНз/
о
отделения от СО...
Ацилаль (4) можно применять для генерации Д. in situ.
Ацилаль (4) устойчив в кипящем /пре/и-бутаноле, но в при-
сутствии карбоната калия как катализатора гладко расщепляется
с образованием /////^///-бутилового эфира изомасляной кислоты.
Ацилаль (4) реагирует без катализатора при комнатной темпера-
туре с аминами, давая амиды изомасляной кислоты. В присутствии
карбонатов щелочных металлов или солей карбоновых кислот аци-
лаль (4) превращает кислоты в их ангидриды, возможно, через
Н3С.
н3с
311
промежуточное образование смешанного ангидрида. В присутствии
О
К-СО., при II
(4) -р2Н()С(СН3)3 • ----> (СН3)2СНС—ОС(СН3)3 р СОа
о
зо° I:
(4) -| (С2Н5)2М1 (СН3),СНС—Х(С,Н5)24 со,
КСОЖМ
120° II |! л II
(4) J RCO,H------> 2[(CH3),CHCOCR| — |(CH3),CHC]2O г (RCO),O
карбоната кадия как катализатора ацилаль (4) реагирует с олефи-
нами, взятыми в большом избытке, с образованием никлобутанонов.
Реакция с изобутиленом дает 2,2,3,3-тетраметилциклобутанон, кото-
рый окисляется щелочной перекисью водорода в известный у-лактон.
1. Смит Ч., Нортон Д., «Синтезы органических препаратов», ИЛ, М., 1954,
сб. 5, стр. 24.
2. X е и ф о р д В. Е., 3 а у о р Дж. С., «Органические реакции», ИЛ, М., 1951,
сб. 3, стр. НО.
3. В е s t i а п Н., G fl п t 11 е г D., Angew. Chem., Internal. Ed., 2, 608 (1963).
ДИМЕТИЛМАГНИЙ, (CH3)2Mg. Мол. вес 54,39.
Раствор Д. получают 111 в сухой камере в атмосфере азота при
добавлении по каплям 0,5 моля диоксаиа к эфирному раствору
1 моля метилмагнийхлорида с последующим отделением осаж-
дающегося комплекса диоксан MgCI,. Необходимо тщательно
соблюдать меры предосторожности во время фильтрования, не до-
пуская высыхания вещества на фильтре, поскольку сухой дпэтил-
магний чрезвычайно иирофорен. Раствор после фильтрования почти
не содержит ионов галогена, п выход Д. составляет 71?о.
Установлено [1], что 3,20-бнс-этилепкеталь 11 р.12 Р-эпокси-
прегпапа (1) не превращается в 11 Р-окси-12 а-метилстероид (2)
при действии метилмагнийхлорида, поскольку происходит
сужение цикла с образованием С-норстероида. Однако такое пре-
вращение осуществляется при действии Д., который по вызывает
сужения цикла при раскрытии эпоксидного кольца 121. Поэтому
авторы разработали методику получения этого реагента, приведен-
ную выше, и нашли, что при реакции диметилмагння с эпоксидом
312
(1) образуется /и/м/щ-дпаксиальиый продукт (2), выделенный после
снятия кетальной защиты в виде дикетона (3) с выходом 41 "о. Для
проведения этой реакции отгоняют эфир от раствора Д., заменяя его
на диоксан, прибавляют стероид (1), кипятят 24 час и разлагают из-
быток реагента насыщенным раствором хлористого аммония (приме-
нение ацетона, этплацетата или сухого льда приводит к очень бур-
ной реакции).
1. Christensen В. G., Strachan R. G., Т г е n п е г \. R., А г i-
s о и В. И., Н irsc h m a n n R., C h enier <! a J. M., J. Am. Chem. Soc.,
82, 3995 (1960).
2. BartlettP. D., В e г г у C. At., J. Am. Chem. Soc., 56, 2683 (1934).
бис-(\,2-ДИМЕТИЛПРОПИЛ-1 )-БОРАН («диизоамилборан»).
l(CH;()2CHCH(CH;t)l2BH. Мол. вес 154,10, т. пл. 35—4()\
Д. получают при реакции диборана с З-метилбутеном-2;
даже при избытке олефина образуется только дналкилбо-
раи. Полутвердый продукт реакции используется без дополни-
тельной очистки [1]. По другой методике раствор диборана в ТГФ
СН;!
I
(С (СН.Д.СНСН
2(СН3).,СН.----СНСН3л 1/„В.,Н1!-^ ВН (Д)
(СН3).,СНСН
I
СН,
обрабатывают теоретическим количеством З-метплбутена-2 в ТГФ
и после удаления растворителя получают Д. в виде белых, неста-
бильных на воздухе кристаллов.
Д. подобно диборану можно применять как восстановитель, при-
чем часто существенные преимущества его применения обусловлены
стерпческимп эффектами. Д. обычно присоединяется к концевому
атому углерода алкенов-1. Так, гексен-1 превращается при гидро-
борировании Д. и перекисью водорода в гексанол-1 с выходом 99%,
тогда как использование дпборана дает первичный спирт только
с 94%-ным выходом и вторичный спирт с выходом 6%. Стерпческпй
контроль возможен также для несимметричных олефинов с двойной
связью внутри цепи. Например, гщс-4-метилпеитеи-2 (1) при
реакции с дибораном и окислении Н.,0.2 дает примерно равные коли-
313
чества обоих возможных спиртов. Реакция (1) с Д. протекает очень
д. но.
(СН3),СНСН = СНСН3 (СН3).,СНСН.,СНСН3-------- (СН3)3СНСНХНСНч
‘I I
BR, ОН
<11 (2) <31 97“„
медленно (около 12 час), но дает почти исключительно изомер (2),
в котором атом бора связан с менее затрудненным атомом углерода
двойной связи.
/{нс-Олефин реагируют с Д. намного быстрее, чем /и/д7нс-изомер,
который может быть выделен после селективного восстановления
более реакционноспособного цис-изомера.
Ацетилен с тропной связью внутри цепи, гексин-3 (4), легко
реагирует с дибораном с образованием ненасыщенного производ-
ного (5), которое при обработке уксусной кислотой дает цис-гек-
Н ВН3 Н Н
13,11., | | АсОН | I
С,Н5С С(',ц.------ С,Н.-,С-.-СС,Н.-,-- с,н:,с-=сс.,н3
(ii (."a iG>
сен-3. При гидроборировании дпборано.м гексина-1 (7) (ацетилена
с концевой тройной связью) он вступает в реакцию наполовину,
тогда как оставшаяся часть присоединяет два фрагмента Н—В.
Однако с более объемистым реагентом Д. реакция завершается на
стадии образования моноаддукта, который при обработке уксусной
кислотой дает гексен-1 с выходом 90%. При этом атом бора нахо-
А;ОНД«-С4НДН^СН,
/ (9)
д /
Н-СД{9С« .Н -* н-С,Н,СН =CHBRZ
(7) (8)
h'Oj\<-c4h9ch,cho
< 1'))
дится у концевого атома углерода, как в соединении (8), посколь-
ку окисление этого непредельного соединения перекисью водорода
дает альдегид ПО).
Д. восстанавливает только определенные функциональные груп-
пы. В ТГФ приО' он восстанавливает альдегиды и кетоны в спирты,
у-валеролактон (11)в у-оксивалериановый альдегид (12) и N.N-ди-
метиламиды в альдегиды. В этих условиях не восстанавливаются кар-
боновые и сульфокислоты, амиды, сложные эфиры, хлорангидриды,
ангидриды и сульфоны. Нереакиионноспособность карбоксильной
д.
сн.,спсн,сн.,с=о - СН3СНСН2СН.,СНО
i ’I 'G% I
1--О------1 он
(11) (12)
314
группы по отношению к Д. позволяет осуществить гидроксилирова-
ние Д10-ундеценовой кислоты (13) в 11-оксиупдекановую кислоту
Д.. щг. ю'ш. И • О
сн2-сн(С112)8:'О..н-----------------* но'СнЛоСол!
(13) (11)
(14) |21. Окиси и нитрилы восстанавливаются очень медленно.
При гидроборировании Д. и окислении перекисью водорода сте-
роидные 1,2-дизамещепные этилены дают приблизительно равные
количества обоих изомеров |3|. Так, Х'-хэлестен дает холестанол-кх.
(35/о) и холестанол-2а (40%).
Д. лучше, чем диборан, для гидроборирования концевых дне-
нов, дающих после окисления х, .л-диолы 141. Например, гекса-
диен-1,5 превращается в гексаидиот-1,0 с выходом 93%, тогда как
использование диборана дает ютъко 69"» гександпота-1,6 и смесь
изомерных диолов. Применение Д. позволяет мопогидроборпрэвагь
Д. НгО;
69%
циклические диены с удовлетворительным выходом 141, что было
использовано 151 для селективного превращения диена (1) в первич-
ны!! спирт (2) lol.
Д. применяли для восстановления 2,3,5,0-тетра-О-ацетил-о-гек-
соно-у-лактона в соответствующею фуранозе с высоким выходом [6].
СЩОАс
R.BH
I
СЩОАс
НСОАс ОАс
СЩОАс
Производные фураноз трудно получить другими методами; новый
метод использует склонность сахарных кислот к превращению в
у-лактоны. Восстановление амальгамой натрия или боргидридом
натрия дает фуранозы с очень низкими выходами.
1. Brown Н. с., Z w о i I с 1 G., J. Am. Chem. Soc., 81, 1512 (1959); 82, 3222,
3223 (1960); 83, 1241 (1961).
2. Brown Н.С., В i » 1 e у D. В., J. Am. Chem. Soc., 83, 486 (1961).
3. Sondheimer F., \ u s s i m Al., J. Or». Chem., 26, 630 (1961).
4. Zweilel G., N a H a s c K., Bro w n H. C., J. Am. Chem Soc., 84, 190
(l'J62).
315
5. Hof I s о in mer R. D., Taub D., Wendler \. L., .1. Orc. Chem 28
1751 (19(13)
6. 1< о Ii и P., Samarita n <> R. H,, Lerner I.. AL, J. Am. Chem. Soc., 86
1457 (1901); 84, 5475 (1965).
ДИМЕТИЛСУЛЬФАТ, (CH.,0)^0.., см. также Мст ил подпетый.
Мол. вес 126,14, т. кип. 188,5 , уд. вес 1,35.
0-Метилирование. Auei илгндрохинои превращается в диме-
тиловый эфир при последовательном добавлении Д. и водного
раствора едкого натра к раствору этого соединения в 95°о-ном
этаноле 111:
СОСИ;,
А/011
По '
СОСПз
J zocn3
с,н,он nz
МаОН -{ (CH3O)2SO2
СН:,О
n-Глюкоза дает '2,3,4,6-гетра-О-мстил-о-глюкозу |21 при взаи-
модействии с Д. в водном растворе едкого натра при 55 с после-
+ (CH3),SO4 + NaOH
55е,затем кислотн. сиОролиз
46-557®
Н
ОН
дующим нагреванием с 2 и. соляной кислотой в течение 1 час при
100 для гидролиза а- и р-метилглюкозидов.
г-Капролактам превращается в иминоэфир, О-метилкаиролак-
тим, при кипячении с Д. в бензоле и добавлении карбоната калия
для разложения первоначально образующейся соли 131.
6 молей
+ (СЩ0)2ЗО2 + Сб11 К.СО}>
61-687»
6 молей 2 л
По методу Бредерека 141 для исчерпывающего метилирования
углеводов частично метилированный препарат растворяют в эфире,
прибавляют натриевую проволоку и затем раствор Д. в эфире.
Реакция заканчивается за 1 час.
N-Метилирование. Последней стадией синтеза 151 пноцианина
(6) является N-метилированиеа-окснфсиазина (4). Эгу реакцию про-
водят путем нагревания 2 г окепфеназииа (4) с 10 мл Д. в течение
10 мин при 100' с последующим охлаждением и промыванием темно-
коричневого метиле ул ьфата (5) эфиром. Раствор соли в воде подще-
316
лачивают п экстрагируют хлороформом до тех пор, пока не пре-
кратится ослабление синего окрашивания.
Общий метод для получения несимметричных вторичных ами-
нов 161 включает алкилирование N-бензилпдеиампна, такого, как
(1), при смешивании его с Д. в бензоле с последующим нагреванием
(сначала осторожным, а потом при кипячении), после чего разла-
гают соль (2) действием основания.
я-С,Н,.\’П. (СИ.,О),SO.
С6Н-,СНО------:--> С„11.,С11 \С4Н9-н-----*
(D
+ Н.О; \dOlI
—► С6Н5СН ..- NC4119-h(C1I3OSO-н-С4Н9\НСНз(VСвН5СНОит.д.)
(2) I (общий) (J)
СНз
Получение сульфонов 17]. n-Толуолсульфохлорид восстанав-
Na.SO.,
NaHCO., |(.Н.|Г)).зО, \аН(.О,
//-CH3C6H4SO.,CI------- n-CH3CeH4SO2Na----------------*
//-CH3C6114SO.,CH3
ливают сульфитом натрия и бикарбонатом натрия до /г-толуол-
сульфнпата натрия, который выделяется в твердом виде. Смесь
этой солн, диметилсульфата, бикарбоната натрия и воды кипятят
20 час и экстрагируют бензолом метил-/?-голилсульфоп.
1. Вайас Г., Шах Н., «Синтезы органических препаратов», ПЛ, М., 1953,
еб. 4, стр. 401.
2. Вест Э., Хо.'Н.ден Р., «Синтезы органических препаратов», ИЛ, М.,
1952, сб, 3, стр. 407.
3. Бенсон Р., К э й р и с Т., «Синтезы органических препаратов», ИЛ, М.,
1953, со,, 4, с гр. 305.
317
4. Bredereck Н., Н a g с 1 1 о с h G., Н a m b s с h Е., Chem. Вег., 87,
35 (1951); В re derec к II., Hambscli Е., Chem. Вег., 87, 38 (1954).
5. С ю р р е ii Л., «Синтезы органических препаратов», ИЛ, М., 1953, сб. 4,
стр. 422.
6. 1. u с i е г .1. J., 11 а г г i s A. D., KorosecP. S., Org. Sen.. 44, 72 (1964).
7. I- i е I d L„ С 1 а г к R. D., Org. Syn., Coll. Vol.. 4, 674 (1'963).
ДИМЕТИЛСУЛЬФ AT — БАРИЯ ОКИСЬ, см. Метил подпетый.
Диметилсулыфат в присутствии окиси бария или гидроокиси бария
Ва(ОН) ,-8Н.,0 является эффективным реагентом для исчерпывающе-
го метилирования углеводов 11,21. Диметилсульфат дешевле и иногда
лучше подпетого метила. Обычным растворителем для этой реак-
ции является ДМФА. Для углеводов, плохо растворимых в
Д.МФЛ, можно применять ДМСО или смесь ДМСО и ДМФА.
1. Kuhn R., Trischina n n Н., Chem. Вег., 96, 281 (1963).
2. \V а 1 1 с n f е I s К., В е с h t 1 е г G., К u h и R., Т г i s с h in а n и Н.,
Е g g с Н., Angew. Chem., Internal. Ed., 2, 515 (1963).
ДИМЕТИЛСУЛЬФАТ — КАЛИЯ КАРБОНАТ |1]. Эту смесь
применяют для метилирования некоторых растительных фенолов,
которые могут разлагаться или перегруппировываться при действии
диметилсульфата и щелочи. Так, например, раствор 2-(2', 4'-дпоксп-
фе1111л)-6-оксибензофураикарбо1и)вой-3 кислоты в сухом ацетоне
обрабатывают диметилсульфатом и карбонатом калия (10%-пый
избыток), взятых в количестве, достаточном для метилирования
всех фенольных групп и карбоксила. Смесь кипятят 5 час
с защитой от влаги воздуха, охлаждают, центрифугируют и
остаток неорганических солей дважды промывают ацетоном. Вы-
паривают объединенные ацетоновые растворы и после двух кристал-
лизаций остающегося сиропа из смеси этанол — вода или мета-
нол — вода получают метилированный продукте выходом 60—70%.
Если соединение нечувствительно к щелочи, то реакцию можно уско-
рить прибавлением нескольких миллилитров 10%-ного метаноль-
ного раствора едкого кали |2|.
1. Contributed by S u п d e r w i r 1 It S. G., Colorado State University.
2. Bicknff E. M., 1. v m an R. L., Livingston A. L., Boot h A. N.,
J. Am. Chem. Soc., 80, 3969 (1958).
ДИМЕТИЛСУЛБФИТ, (CH3O)2SO. Мол. вес 110.13, т. кип.
126—127 . Д. получают реакцией 1 моля хлористого тиоиила с 2,2
молями метанола 111.
Этерификация аминокислот. Теобальд и сотр. Ill получали
метиловые эфиры н-толуолсульфонатов аминокислот нагреванием
318
смеси соответствующей аминокислоты, 1,1 же /i-толуолсульфокис-
лоты п большого избытка (5 —15 же) Д. В некоторых случаях выхо-
101У, 4.5 Ч.1С
С6Н5СН3СИСО.,Н--(CH3O).,SO--TsOH---------CJI-.CII.,C[[CO,C113 J-
I y-'"" I
XH2 + XH-(TsO~)
0.01 моля 0,05 моля 0.011 моля
Sn. СЦ..ОЯ
ды очень высокие; однако метилирование аланина, валина и лизина
протекает не полностью. Этот метод менее удовлетворителен, чем
этерификация тиошглхлоридом и метанолом (см. Тионнл хлори-
стый); однако н-толуолсульфонат ь-пролина очищается легче, чем
хлоргидрат.
[.Theobald J. М., \V i 1 i a m s M. W., Y о u п" G. T., J. Chem. Soc.,
1963, 1927.
O"
ДИМЕТИЛСУЛЬФОКСИД(ДМСО), Cli:)S Cfl3. Alon, вес 78,14,
т. пл. 18,5°, t. кип. 189' (86 /18 мм, 63/8 мм), уд. вес 1,1014, очень
гигроскопичен, смешивается с водой. Обзор 111.
Данные по химическим превращениям ДМСО сведены в раздел
«Реакции». Однако известно большое число случаев, когда никаких
изменений или по крайней мере никаких видимых изменений не
происходит, но благодаря специфической растворяющей способ-
ности ДМСО удается проводить такие реакции, которые в других
условиях не идут. Кроме того, эффекты ДМСО как растворителя
ускоряют целый ряд реакций, причем невозможно провести резкую
границу между растворяющей способностью и эффектами. Эффек-
ты ДМСО как растворителя объясняют резким увеличением реак-
ционной способности анионов по сравнению с таковой в гидроксил-
содержащих растворителях с близкой диэлектрической постоянной,
в которых реакционная способность анионов резко уменьшена за
счет образования водородных связей. Это же свойство может увели-
чивать и растворяющую способность ДМСО.
РАСТВОРЯЮЩАЯ СПОСОБНОСТЬ
Органические и неорганические соединения. ДМСО очень
хорошо растворяет не только многие органические соединения, но
также п некоторые неорганические соли (см. табл. 1).
Расщепление дигитонидов. Новый метод расщепления стероид-
ного дигитоиида, например комплекса дигитонииа с холестерином
(1 : 1), основан на высокой растворяющей способности ДМСО для
дигитонина и довольно умеренной для стероидного спирта, что при-
водит к полной диссоциации дигитонидов при нагревании на кипя-
319
Тиб.ища /
Растворимость солей (г/100 г ДМСО при 25 )
Хлорное железо (611,0) 30 Нитрат натрия 20
Ацетат ртути 100 , Нитрит натрия 1
Бихромаг натрия (2Н,О) 10 Хлористое о.юно (2[1,О) 40
Иодид натрия 30 > Хлористый цинк 30
щей водяной бане 12]. При охлаждении стероидный спирт осаждается
и его экстрагируют гексаном; дигитонин выделяют после отгонки
ДМСО в вакууме.
Дигитонин;Т.пл.235°с разл.; [а.]0-54,30(метанол)
Молекулярное вращение. ДМСО считается лучшим раствори-
телем, чем пиридин для исследования молекулярного вращения
лактонов |3|.
ЭФФЕКТЫ РАСТВОРИТЕЛЯ
Замещение. Первичные и вторичные алкилхлориды легко и эк-
зотермически реагируют с цианистым натрием в ДМСО, давая
с очень хорошим выходом соответствующие нитрилы [4,51. При
этом взаимодействие осуществляется быстрее и с лучшими выхо-
NaCN, ДМСО
(СН3)3ССН2С1 —-------► (CH3)3CCH2CN
дамп, чем в реакциях соответствующих бромидов или иодидов в вод-
ном этаноле |4|. Неопентил- и неофилгалогениды можно превратить
этим методом в нитрилы без перегруппировки [51. В ДМСО повы-
шаются скорости и других реакций бимолекулярного нуклеофиль-
ного замещения у атома углерода: замещение галогена азид-, тио-
цианат- п галогенид-ионамн 161, а также тозильной группы бромид-
иопом 171. ДМСО ускоряет также электрофильное замещение у атома
углерода. При дентерообмене в ( |-)-2-метил-3-фенилпропионитр11ле,
СН:( СН3
I снаон, ДМСО I
CelI5CH.,CCN------------> С8Н,СН.,СС.Х
,!,
(+) (±)
320
катализируемом метилатом калия, константа скорости рацемизации
в ДМСО по крайней мере в 109 раз больше, чем в метаноле [8].
Бромбензол превращается в mpem-бутилфениловый эфир с вы-
ходом 86% при реакции с mpem-бутилатом калия в ДМСО при 25°
в течение 15 час, в mpem-бутаноле реакция гораздо медленнее (91.
В реакции этилового эфира а-броммасляной кислоты с нитри-
том натрия, дающей этиловый эфир сс-нитромасляной кислоты,
лучше использовать ДМСО, чем ДМФА, ввиду большей раство-
ряющей способности ДМСО по отношению к нитриту натрия
(см. табл. 1) (Корнблюм ПО)).
Коуп и Мехта нашли ЦП, что ДМСО является наилучшим раст-
ворителем для реакции дитозилата (1) с цианистым натрием.
CH2OTs
CH2CN
ОСН3
сн3о
4- 2 NaCN
ДМСО, 6дней, 20±-3°
6S-78 %
СН3О
•ОСН3
CH2OTs ch2cn
(1) (2)
Обработка метилглюкозида 2-О-мезил-3,4,6-три-О-метил-а-о-глю-
козы (1) метилатом натрия в ДМСО дает смесь примерно равных
количеств 2-О-метилового эфира (2) и 2-оксисоединения (3) [12].
Необходимо подчеркнуть, что замещение с образованием соединения
(2) протекает с сохранением конфигурации.
Дегидрогалогенирование и родственные реакции элиминирования.
ДМСО превосходит гидроксилсодержащие растворители при
дегидрогалогенировании с помощью mpem-бутилата калия [131.
бнс-Этиленкеталь (1) не удается с приемлемым выходом
(сн3)3сок^
дмсо *
84%
превратить в кеталь (2) при обработке тргт-бутилатом калия в
/ггрет-бутаноле; однако это превращение гладко происходит
11 Заказ № 965 321
в ДМСО. ДМСО — превосходный растворитель для перевода арил-
сульфонатов вторичных спиртов в соответствующие олефины 1141.
В реакции а-бромкетостероидов с бикарбонатом натрия в ДМСО
при транс-диакспальной конфигурации атома галогена и соседнего
водорода преобладающим процессом является ///рп«с-элпмпнпрова-
иие 1151. Если атомы брома и соседнего водорода имеют /р/с-эква-
ториальио-аксиальную конфигурацию, то вместе с элиминированием
наблюдается конкурентная реакция гидролитического замещения
в соответствующий диосфенол.
При Р-элиминироваипп различных изопропильных производных
с образованием пропилена легкость протекания процесса умень-
шается в зависимости от отщепляемого заместителя X в следующем
порядке: Br>SO.,R>SOR>SCN>SR>SH>CN [16].
(СН,),СОК—ДМСО
СЩСНСНз----— ------* СН3СН -=СП2
Крам и сотр. |17, 181 описали методику превращения бромбензола
в фенил-///ре/7/-бутиловый эфир в течение 1 мин при реакции с трет-
бутилатом калия в ДМСО при 125—130 . Полагают, что промежу-
точным соединением в этой реакции является дегпдробензол. В этой
реакции использовали ДА1СО, высушенный реактивом Фишера, и
322
продажный /ирет-бутилат калия. Авторы считали выход хорошим
[17], однако он составляет всего 42—46%.
I
-I- (CH3)3ok-;-ch3s+ch31’- (сн3)3сон
125—130’
0,159 моля
0,67 моля
4 8 мл
ZOC(CU3)3
150 мл
(( н^сон | ।
Миграция двойной связи. Прайс и Снайдер П91 нашли, что
специфические эффекты ДМСО в значительной степени ускоряют
прототропную перегруппировку аллиловых эфиров в ццс-пропенило-
(СН,),СОК. ДМСО СНз OR
СН„ = СНСН.,OR---------------->- ;С = СУ
IV ЧН
вые эфиры в присутствии mpem-бутилата калия как катализатора.
На такой изомеризации основан новый способ защиты гидро-
ксильной группы углеводов (Каннигэм и др. [201). Для этого реак-
цией с бромистым аллилом и щелочью легко получают аллиловый
(с.н3),сок. дмсо RO. /СН:; н +
ROCH.,CH = CH,---~--------->- С С -------------ROH +
н хн
(1) (2)
+о=снсн„сн3
эфир (1), который с трудом расщепляется водными кислотами
или щелочами. Изомеризация по методу Прайса дает ццс-пропенн-
ловый эфир (2), который, будучи енольным эфиром, легко гидроли-
зуется разбавленными кислотами. Пропениловый эфир (2) можно
также расщепить окислением перманганатом или озонированием.
Смесь mpem-бутплата калия и ДМСО широко применяется для
изомеризации олефинов 1211, циклогексадиенов [221, циклооктади-
(сн3)1сок-дмсо^
Ivac при 70°
енов 1231, циклононадпенов [231 и циклононатриенов [231. Напри-
мер, цпклооктадиен-1,5 в мягких условиях быстро и почти количест-
венно превращается в цпклооктадиен-1,3.
Алкилирование и ацилирование кетонов и нитрилов. ДМСО
значительно увеличивает скорость алкилирования енолят-анионов
|24, 251. Динитрил малоновой кислоты и пентандиои-2,4 довольно
ДМСО-NaH
СН.,(СХ)., । 2CII31------> (СН3).,С(СХ)г
114
323
плохо диалкилируется обычными методами. Однако при диалкили-
ровании иодистым метилом в ДМСО в присутствии гидрида натрия
выход составляет 60—64% 1251. Те же реагенты позволяют осущест-
вить эффективное ацилирование метилкетонов метиловыми эфирами
кислот с образованием симметричных Р-дикетонов [26]. Наилучшие
ДМСО- Na и
RCO„CH3 -j- CH3COR------>- RCOCHoCOR ; СН3ОН
результаты получены при молярном соотношении NaH: сложный
эфир: кетон 2:2: 1. Раствор гидрида натрия в ДМСО эффективен
NaH-ДМСО
1удс при85°^
92%
(2)
ври конденсации Торпе 1,4-динитрила (1)-в 0-цианенамип (2), а
также для циклизации по Дикману [27].
Термическое разложение. По обычной методике Кижнера —
Вольфа гидразон и основание нагревают при температуре около
200°. Крам и др. [281 описали интересный метод применения раст-
вора основания трет-бутилата калия в ДМСО, который позволяет
провести реакцию при комнатной температуре [28|. Например, 1,96 г
(СН,).,СОК. дмсо
(C6H5)2C=NNHo-----------------------> (CeH5)XH.,-i-N2
бензофенонгидразона прибавляют в течение 8 час при перемешива-
нии к смеси 2 г возогнанного mpem-бутилата калия и 5 мл ДМСО.
Эта методика препаративно неудобна, однако она имеет большой те-
оретический интерес. Аналогично [26] элиминирование N-окисей
аминов по Коупу, которое обычно проводят путем пиролиза при
температуре 120—150°, протекает гладко при 25° в безводной смеси
ДМСО и ТГФ.
О
| ДМСО, 2 5°, ТГФ
С6Н5СН\ + (СН3),---------► С0Н6СН = СНо -j- (СИД,ХОН
I
СНз
Исчерпывающее метилирование полисахаридов. Полисахариды
эффективно метилируются в ДМСО диметилсульфатом в присутствии
окиси бария или (и) гидроокиси бария [29]. В этой реакции в каче-
стве основания можно использовать и едкий натр [301. Гранулиро-
ванный едкий натр и диметилсульфат добавляют в атмосфере азота
при перемешивании в течение 8 час к недеградированному крахмалу
в ДМСО. После перемешивания еще в течение 16 час, смесь нагре-
вают для разложения диметилсульфата, охлаждают, разбавляют и
324
нейтрализуют. Продукт реакции экстрагируют хлороформом и пе-
реосаждают из ацетона эфиром; выход 91 %, ОСН3=42,3%. В этол»
случае высокая растворяющая способность ДМСО несомненно спо-
собствует проявлению эффектов растворителя.
Гидролиз. Скорость щелочного гидролиза этилацетата в смеси
ДМСО — вода возрастает с увеличением концентрации ДМСО [311.
РЕАКЦИИ
Дегидратация. Дегидратацию вторичных бензиловых и третич-
ных бензиловых н алифатических спиртов можно проводить при
нагревании спирта с 4—8 молями ДМСО при 160—185° в течение
9—16 час (Трайнелис 132]). 1-Алкилциклоалканолы дегидратиру-
ются главным образом в эидоцпклические олефины, как показано на
следующей схеме:
95%
2-Метилгексанол-2 при такой дегидратации дает примерно равные
количества 1- и 2-олефпнов. Эти результаты указывают на катализ
по кислотному механизму; однако такому механизму противоречит
тот факт, что добавление анилина или натриевого алкоголята «-ок-
тилового спирта не влияет на выход олефинов и направление эли-
минирования. По-видимому это не чисто термическое элиминиро-
сн3 сн3 сн3
। ДМСО I I
СН3СН„СН,СН2ССН3 —.—> CH/TLCELCELC = СН2 4-СН3СНоСН2 = ссн3
| н'"“ 46 54
Qpj - — - — - - — — , '
Анализ методом ГЖХ
ванне, что видно из сравнения данных для ДМСО и других раствори-
телей, приведенных в табл. 2. Дегидратирующее действие раствори-
телей как с большей, так и с меньшей диэлектрической константой
в корне отличается от действия ДМСО при 160°. Не обнаружено
также корреляции между дегидратирующим действием и дипольным
моментом растворителя. ДМСО способен ускорять ионизацию спир-
тов с промежуточным образованием карбониевых ионов. Примерами
сн2—сн2
RC CR
И 11
О О
ДМСО^
19 0°
К = СН5 или С(,Н5
^25
Таблица 2
Влияние растворителей на 1-фенилпропанол-1 при 160J
Растворитель Диэлектри- ческая кон- станта, Е Ди ПОЛЬНЫН момент, |i Возвращен- ный спирт.
Ацетамид 59 3,7 100
Диметплсульфоксид 48,9 3,95 3
Нитробензол 36 3,98 82
Бензонитрил 25 3,91 85
Хинолин 9 2,25 93
Дпфеннлметан 2,7 2,95 91
синтеза с помощью дегидратации ДМСО являются превращения
ацетонплацетона в 2,5-днметилфуран и 1,4-дифенилбутандиона-1,4
в 2,5-дифенилфуран с выходом 66% и 60% соответственно.
Проведена идентификация некоторых продуктов разложения
ДМСО, образующихся при дегидратации спиртов в ДМСО, которая
протекает при повышенных температурах и в течение довольно
продолжительного времени.
Гиллис и Бек 133] нашли, что при нагревании 1,4-дпола (1) в
ДМСО вместо ожидаемого сопряженного диена получается цикли-
Р\^снгон
ДМСО, 13 уде при 156-166°^
98% ’
(2)
(1)
ческая окись (2). Вторичные и третичные 1,4-диолы реагируют с
ДМСО аналогично; такая дегидратация превосходит известные ме-
тоды синтеза тетрагидрофуранов. Для этой реакции постулировано
циклическое переходное состояние:
v
V
п- >4 -Н.
?+ —
S (сн3)г >С— C...O...S+(CH3)7~*
н
Конденсация Михаэля — Кневенагеля обработкой смеси 1, 2,
(3)
С° ДМСО-KF
53% '
снс6н5
сьн5
С6н5 с6н5
(5)
(<)
326
З-трпфенилцпклопентадпепа (3) п 1,2, З-трифенилпропенона (4) фто-
ридом калия в ДМСО дает ярко-желтый дигидрогексафенплпен-
тален (5) (Ле Гофф 1341).
Окисление. Корпблюм 135] нашел, что бромацетофепоп, а также
его «-галоген-, //-нитро- и л-фенплнронзводные можно окислить до
ДЛКЮО'З )
С6Н3С()СН2 Вг ———•> С6Н3СОСНО
i 1? 1>
глиоксалей растворением в ДМСО и выдерживанием при комнатной
температуре в течение 9 час. а-Бромдезоксибеизопп гладко окисля-
ется в бензил при 45 в течение 44 час. Для бепзилбромндов этот
ДМСО (4 3 )
СвН3СОСНВгС6Н3-----—----> С6Н3СОСОСсН-,
метод не дает удовлетворительных результатов. Однако последую-
щие исследования 1361 показали, что не только беизилгалогениды,
по даже насыщенные первичные хлориды, бромиды и подпды можно
превратить в альдегиды окислением соответствующих тозплатов.
Например, 1-иодгептап (7 г) прибавляют в темноте к раствору
тознлата серебра (11 а) в ацетонитриле при 0—5 и смесь остав-
ляют на ночь при комнатной температуре. Неочищенный тозплат,
ТЮАй ДМСО
СН:1(СН,)-СИ.,1 -—7* CH;,(CH,)5CH.,OTs-- СН;1(СН.,)3СН()
CH;1CN 70*',, (общий)
полученный добавлением воды и экстракцией эфиром, прибавляют к
раствору 150 мл ДМСО, нагретому до 150 (наблюдается небольшое
вспенивание), охлаждают, добавляют 20 г бикарбоната натрия и
через раствор пропускают азот. Смесь нагревают до 150 , выдержи-
вают при этой температуре 3 мин и быстро охлаждают; альдегид
выделяют в виде 2,4-д1П1Птрофенилг11Дразопа. Бензплтозплаты
гладко окисляются при 100".
Было показано, что обычные галогениды не удается окислить
этим способом и их приходится перед окислением переводить в тозп-
латы, однако ДМСО быстро окисляет w-октплнодид до альдегида при
ДМСО, 4 мин 150=
СН3(СН,)„СН21------_41|. ' СН3(СН2)вСНО
150° (Джонсон и Пельтер 1371). Другие первичные подпды также
достаточно хорошо окисляются. Однако для первичных хлоридов п
вторичных йодидов выходы плохие.
Эффективная методика получения этилового эфира глиоксиловой
кислоты заключается в окислении этилового эфира бромуксусной
кислоты 3 же ДМСО при 70 в присутствии нещелочного 1,2-эпоксп-
3-феноксипропана для связывания бромистого водорода и добавле-
- 7 0-
BrCH2CO2C2H3 -L(CH3),S — о—O^CHCO.,C2H5-I HBr ; (CH3)2S
1 *-* %
327
нии бромистого метила для превращения диметилсульфида в триме-
тилсульфонийбромид и подавления тем самым побочных реакций
[381. Эпоксиды гладко окисляются ДМСО в присутствии каталити-
ческих количеств эфирата трехфторпстого бора на кипящей водя-
22 час при 97°
(CH3)2S-O(BF,),
76%
ной бане; например, так получают 2-оксициклогексанон [391. 2|3,
ЗР-Эпоксихолестан и 2а, За-эпоксихолестан (последний при добав-
лении диоксана) при окислении этим способом дают холестанол-ЗР-
он-2 (с экваториальной ОН-группой) и небольшие количества 2,3-
диона и 2Р,За-диола.
Пфитцнер и Моффатт [40] нашли, что З'-О-ацетилтимидин (I) в
ДМСО при комнатной температуре в присутствии безводной ортофос-
форной кислоты и дициклогексилкарбодиимида (ДЦК) окисляется в
альдегид (II) с высоким выходом. По даннььм электрофореза карбок-
I
п
синуклеозида при этом не образуется. Очевидно, ДМСО
является окислителем, а ДЦК в присутствии кислоты реагирует с
образующейся водой.
Этот метод применили для окисления первичных спиртов в аль-
дегиды и вторичных спиртов в кетоны 1411. Например, окисление
ДМСО (избыток), ДЦК
O.2NСН2ОН <з^,),1щодо.5.имолм 0Д1_^ у_сно
'=/ 92% ‘ \=/
328
я-нитробензилового спирта в растворе сухого ДМСО в присутствии
ДЦК и безводной ортофосфорной кислоты дает количественный вы-
ход (тонкослойная хроматография) л-нитробензальдегида, В при-
сутствии таких кислот, как фосфорная, фосфористая, циануксусная
или фосфат пиридиния, для окисления при комнатной температуре
требуется несколько часов. Более сильная кислота, трифторуксус-
ная, дает плохие результаты, а соляная и серная кислоты полностью
ингибируют окисление. Однако пиридиниевые соли всех трех кислот
являются хорошими катализаторами. Приведем результаты окис-
ления в ДМСО в присутствии ДЦК и ортофосфорной кислоты или
трифторацетата пиридиния:
Октапол-1 --------> Октаналь-1 (70%)
Тестостерон ------> Д4-Андростендион-3,17 (92%)
Холестанол-3 [5---> Холестанон-3 (68%)
Холанол-24--------> Холаналь-24 (85%)
Для окисления тестостерона в дион лучше всего придерживаться
условий, указанных па схеме. Окисление холестерина по этой мето-
дике дает А5-холестенон-3 с выходом 66%.
Экваториальный 1 la-оксистероид легко окисляется смесью
ДМСО — ДЦК, тогда как 110-эпимер в этих условиях инертен [41].
Таким образом наблюдается ситуация,обратная той, которая встре-
чается при окислении хромовой кислотой (Эшенмозер). Предложена
гипотеза, объясняющая механизм этой реакции.
Дпэр и др. 1421 окисляли этим методом (ДМСО — ДЦК — пи-
ридин и йфосфат) производные фураноз; Олбрайт и Голдман [431
успешно применили методику Пфитцнера — Моффатта для окисле-
ДМС0-ДЦК-Н3Р04
80% 3
329
ния иохимбина и других индольных алкалоидов. Чувствительность
индольного цикла к окислению препятствует применению больши-
нства окислителей, и только окисление по Оппенауэру дает умерен-
ные выходы кетонов.
Бэкер и Басс [441 нашли, что методика Пфитцнера — Моффатта
пригодна для окисления мезилатов углеводов, имеющих свободный
гидроксил, например, мезилата гексита (1). Для сравнения окисление
по Сарретту дает кристаллический продукт с выходом лишь 0—5?о.
МвО—
Н3СК хо —
с
Н3С О—1
н3с. о—
с
н3с о—
MsO —
(1)
дмсо,дцк,н3ро4
74%
(2)
Опыты с фуранозидами и пиранозидами показали в то же время, что
окисление чувствительно к стёрическим затруднениям. В соединени-
ях (3), (4) и (5) свободный гидроксил экранирован с двух сторон эфир-
ной пли ацетальной группами, в силу чего эти соединения при ком-
(3) (4) (5)
натной температуре не вступают в реакцию. Бэкер и Басс [45]
использовали методику Пфитцнера—Моффатта для окисления метил-
330
3-бензамидо-4,6-О-бензилиден-3-дезокси-а-в-альтропиранозида (6),
имеющего 2Р-окси-За-бензамидную конфигурацию, и его 2а-оксп-3|3-
бензамидоизомера (7). В обоих случаях был получен один и тот же
кетон-2 с очень хорошим выходом. Окисление (6) в кетон (8) сопро-
вождается изомеризацией соседнего центра с образованием более
стабильного экваториального ЗР-оксипроизводного (4).
Метод Пфитцнера — Моффатта в применении к фенолам дает в
основном о-тиометоксиметильные производные [461. Фенолы с заме-
стителями в орто-положен и и дают /шра-алкнлированные продукты.
О-Алкилироваиие наблюдается для более кислых фенолов.
он он он
J J CH»SCH, CH,SHoC. X ,CH2SCH3
I ||^ЦМ 9 + HI
'V 'Ч/ 'И/
27% 17%
Реактив Пфитцнера — Моффатта является лучшим окислителем
треониновых звеньев пептида в звенья 0-кетокислоты [46аI. Обра-
СН3 СН3
Н—С—ОН R' дмсо-ДЦК. СГО R'
I I П3Р04 I I
...NH—СН—С—NCHC- • •-------------> —NH-CH-C-N—СНС------►
ii I !1 I! I I!
ОНО оно
H3Q ,N
сД NC0H5 R'
c„h;nhnh2 I | I
---------> — \H— CH-C = O 4-H„\CHC—
il
О
ботка окисленного пептида фенилгидразином позволяет осущест-
вить показанную на схеме избирательную фрагментацию.
Трайнелис и Хергенротер [471 нашли, что бензиловый спирт мо-
жно окислить в бензальдегид с выходом 80% кипячением раство-
ра 0,1 моля этогоспирта в 0,7 молях ДМСО при 190J и пропус-
кании через реакционную смесь воздуха. Хотя известно, что беизаль-
ДМСО (воздух)
свн5сн2он ———- С0Н5СНО
О V /о
дегид легко окисляется, однако в присутствии ДМСО реакция оста-
навливается на стадии альдегида. При кипячении раствора бензаль-
дегида в ДА1СО в течение 24 час и пропускании через раствор воздуха
получено только 1,6% бензойной кислоты, а 87% бензальдегида выде-
лено обратно. Хотя воздух облегчает окисление, тем не менее кис-
лород не поглощается. Выделение диметилсульфида как такового
или в виде комплекса с сулемой (т пл. 150°) показывает, что окис-
331
лителем является ДМСО. Несколько замещенных бензиловых спир-
тов были окислены таким же методом в альдегиды ароматического
ряда. В частности, коричный спирт дает альдегид с прекрасным вы-
ДМСО (воздух)
С6Н5СН = СНСНоОН-------------с„н-,сн =--- снсно
° - 60";, (1ЖХ) ’ ’’
ДМСО (воздух)
С0Н5СН.,СН2СН„ОН-------------('Н-,СН.,СН.,СНО
ходом; однако с гидрокоричным спиртом выход был низким.
ДМСО окисляет некоторые тиоэфиры в сульфоксиды с умеренным
выходом (481. Тиоэфир нагревают с 50"6-пым избытком Д1ЧСО при
160—175' в течение нескольких часов и отгоняют диметилсульфид
по мере образования. Сульфоксиды имеют высокую степень чистоты
и не содержат примеси сульфона. Дп-н-бутилсульфоксид и тетра-
этилепсульфоксид были получены с аналогичным выходом, но неко-
торые сульфиды практически не вступают в реакцию. ДМСО —
превосходный окислитель для превращения меркаптанов в дисуль-
фиды (491. Раствор тиола в ДМСО перемешивают при 80—90' в те-
IM Н2 NH= NH2
) /SH J ,S-S I
// ' / /Z / ' ^4
i if + — 8 час, 80 - 9(>' । и Li
I || + (chj.s-o ——-| || Il | .HCH.,),S + H,0
%' 'Ч/ ,z
чеиие не более 8 час и с очень хорошим выходом получают чистый ди-
сульфид. Выходы как алифатических, так и ароматических дисуль-
фидов находятся в пределах 80—10096. Проведение реакции в атмо-
сфере азота дало такие же результаты, как и па воздухе.
В присутствии 5 мол. % брома ДМСО окисляет изонитрилы в изо-
цианаты, возможно, по механизму цепной реакции 1501. Реакцию
Вг. (Cll.).,s-O
(CH3)2CHN = С —* (CH3)2CHN = CBr,--:----►
—> (СН з)2СН N — С — О + (СН з ).,S -у В г.,
проводят в кипящем хлороформе в течение 24 час.
Рассел и др. 1511 исследовали автоокисление ди- и триарилмета-
нов в системе ДМСО—mpcm-BuOH — mpcm-BuOK при 25'. Раст-
о,
(C6H5)3CJ I -г ДМСО — трет - В uO Н (80:20) znpcm-BuOK —* (СсН5)3СОН
3,14 .нмоля 25 лг I 6 ммолей 3 .нмоля
вор 3,14 лгмоля трифенилметапа при встряхивании в атмосфере кис-
лорода поглощает 2,6 лшоля газа за 2 мин и 3,2 лгмоля за 20 мин. Три-
фенилкарбииол получается с высоким выходом. Дифенилметан оки-
сляется в бензгидрол, бензофенон и продукт присоединения ДМСО
к кетону
ОН о-
I I
(C0H5)2CCH2S < СНз
33:
Кетены и кетимины подвергаются окислительному гидролизу
при обработке в ДМСО несколькими каплями конц. соляной кислоты
[521.
он
води. IICI - ДМСО [
(С0Н5).,С = С = О------—---------> (СвН5),С-СО.2Н
/----к води. НС1 - ДМСО
(С0Н5),С = С=Х
он о н
I II I z-------х
—- (СЛН5).,С-С— N-C V- сн3
Методика окисления спиртов. Бартон и др. 153) предложили
метод превращения первичных и вторичных спиртов в альдегиды или
кетоны в нейтральных условиях при комнатной температуре. Спирт
обрабатывают в эфире эфирным раствором фосгена с образованием
хлорформиата (2), растворитель отгоняют в вакууме и неочищенный
остаток перемешивают с ДМСО, получая нестабильный аддукт (3).
Y ।
COCI, |‘ CH,S + CH,
(СН3)2СН2СН2ОН------> (СН3),СНСН2ОСС1-------->-
(1) (2)
О
II + - со.
—> (СН3)2СНСН3О —С—OS(CH3),C1--------»-
(3)
н
I + (C2H,),n
—> (CH,),CHCH-O-S(CH3), -------->-
'у(. _ 70%(об1ции)
CI
— (СН3)2СНСН = О + (CH3),S + (C2H5)3NHC1"
(5)
Последний разлагается с бурным выделением углекислого газа
и образованием диметилсульфоксониевой соли (4). При добавлении
триэтиламина и охлаждении соединение^) разлагается на карбониль-
ное соединение (5) и диметилсульфид. Окисление тетраметиленгли-
коля этим методом дает янтарный диальдегид с выходом 80%, это
соединение трудно получить другими способами. Однако выходы
(—)-ментона (35%) и холестанона. (20%) невелики.
Метиленирование кетонов. При нагревании Д'-5а-андростенона-3
с ДМСО в присутствии эфирата трехфтористого бора вместо ожидае-
333
мого 1,3-диона был получен с выходом ,30"Ь -1-метилеикетон (2) 1511.
Добавление к реакционной смеси нараформа повышает выход до
75%. ДМСО легко взаимодействует и с насыщенным 3-кетоном, но
дает смесь четырех продуктов с низким выходом.
1. Aga in i С., Bull. soc. chim. France, 1965, 1021.
2. 1 s s i <1 <> r i cl e s С. H., К i t a g a w a I., .'1 о s e t t i g E., J. Org. Chem.,
27, 4693 (1962).
3. Wiikop B., Experivntia, 12, 372 (1965).
4. Smile у R. A., Arnold C., .1. Org. Chem., 25, 257 (I960).
5. Friedman L., S Ii e c 11 t e r H., J. Org. Chem., 25, 877 (1960) see also
A r g a b r i g h t P. A., Hail D. H., Chem. Ind., 1964, 1365.
6. Miller J., P a r k e r A. .1., J. Ain. Chem. Soc., 83, 117 (1961).
7. C a s о n J., Cor re i a .1. S., .1. Org. Chem., 26, 11645 (1961).
8. Cr a m D. J., R ickbor п В., К i n g s b u r v C. A., .1. Am. Chem. Soc.,
83, 3678 (1961).
9. C r a m D. J., R i c k b о r n B., KnoxG. R., J. Am. Chem. Soc., 82, 6412
(1960).
10. KornblumX., В 1 a c k xv о о d R. K., Org. Syn., Coll. Vol., 4 , 454 (1963).
II. CopeA.C., M e h t a A. S., J. Am. Chem. Soc., 86, 5626 (1964).
12. Eades E. D. M., Ball 1). H., Longe L., Jr., J. Org. Chem., 31, 1159
(1966).
13. Eaton P. E., J. Am. Chem. Soc., 84, 2344 (1962).
14. X ace H. R., .1. Am. Chem. Soc., 81, 5428 (1959).
15. lacuna R. X., R о xv lang A. T., X а с e H. R., J. Am. Chem. Soc., 29,
3495 (1964); X а с e H. R„ I acona R. N„ ibid., 29, 3498 (1964).
16. W a 1 1 a с e T. .1., H о f m a n n .1. E.. Schr iesliei in A., J. Am. Chem.
Soc., 85, 2739 (1963); H о 1 m a n n .1. E„ W a 1 I a с e T. J., ibid., 86, 1561
(1964).
17. Cram D. J., R i c k b о г n В., Knox G. R., J. Am. Chem. Soc., 82, 6412
(1960).
18. S a h у u n M. R. V., Cram D. J., Org. Syn., 45, 89 (1965).
19. P r i с e С. C., Snyder W. H., J. Am. Chem. Soc., 83, 1773 (1961).
20. C u n n i n g h a in J., G i g g R., W a г г e n C. D., Tetrahedron Letters,
1191 (1964).
21. S c h r i e s h e i m A.. H о f m a n n J. E., R о xv e C. A., J. Am. Chem. Soc.,
83, 3731 (1961).
22. В irch A. J., Gra ves J. M. H., S i d d a 1 1 J. B., J. Chem. Soc., 1963,
4234.
23. Dev a pr abb a kar a D., Car den as C. G., Gardner P. D., J.
Am. Chem. Soc., 85, 1553 (1963).
24. Zaugg H. E., J. Am. Chem. Soc., 83, 837 (1961).
25. Bloomfield J. J., J. Org. Chem., 27, 2742 (1962).
26. В 1 о о in f i e I (1 .1. J., .1. Org. Chem., 26, 4112 (1961).
27. В I о о m 1 i e I d J. J., F e n n e s e у P. V., Tetrahedron Letters, 2273 (1964),
28. C r a in .1., S a h у u n M. R. V., К n о x G. R., J. Am. Chem. Soc., 84, 1734
(1962).
29. К u h n R., T r i s c h mann H., Chem. Ber., 96, 284 (1963).
30. S r i v a s t a v a H. C., Sing h P. P., HarshcS. N., Virk K., Tetra-
hedron Letters, 1964, 493.
31. Tommila E., M u r t о M.-L., Chem. Scand., 17, 1947 (1963).
32. T г а у n e 1 i s V. J., Hergenrother \V. L., Livingston.!. R., V a-
I i c e n t i J. A.. .1. Org. Chem., 27, 2377 (1962); T г а у n e 1 i s V. J., Il e r-
g e n г о t h e r W. L., H a n s о n H. T., V a I i c e n t i, ibid., 29, 123 (1964);
T г а у n e I i s V. J., Hergenrother W. L., ibid., 29, 221 (1964).
33. G i I 1 i s В. T., В e c k P. E„ J. Org. Chem., 28, 1388 (1963).
34. LeGoff E., J. Am. Chem. Soc., 84, 3975 (1962).
35. К о r n b 1 u m X., et al., .1. Am. Chem. Soc., 79, 6562 (1952).
334
36. Kornblum Jones W. J., A ndersonG. J., J. Am. Chem. Soc.,
81, 4113 (1959).
37. J о h n s о n Л. P., P с 1 t e r A., J. Chem. Soc., 1964, 520.
.38. 11 u n s b e г g e г I. M., T i e n J. M., Chem. Ind., 1959, 88.
39. Cohen T„ Tsuj i T„ J. Org. Chem., 26, 1681 (1961).
40. P f i t z n e г К. E., Moffatt J. G., J. Am. Chem. Soc., 85, 3027 (1963).
41. P f i t z n e г К. E., M о f f a t t J. G., J. Am. Chem. Soc., 85, 3027 (1963);
87, 5661, 5670 (1965).
42. D у er J. R., M c G о n i g a 1 W. E., R i с e К. C., J. Am. Chem. Soc. 87,
654 (1965).
43. A 1 b r i g h t J. D., G о 1 d ni a n L., J. Org. Chem., 30, 1107 (1965); J. Am.
Chem. Soc., 87, 4214 (1965).
44. В aker В. R., Bus s D. H., J. Org. Chem., 30, 2304 (1965).
45. Baker B. R„ В u s s D. H., J. Org. Chem., 30 , 2308 (1965).
46. Burdon M. G., Moffatt J. G., J. Am. Chem. Soc., 87, 4656 (1965);
P f i t 7, п e г К. E., Mar inoJ.P., О 1 о f s о n R. A., ibid., 87, 4658 (1965).
46a. D’A n g e 1 i F. , Scaffone E., F i 1 i r a F., Giormani V., Tetra-
hedron Letters, 1966, 2745.
47. T г a v n e 1 i s V. J., Her g e и г о t h e r W. L., J. Am. Chem. Soc., 86,
298 (1964).
48. Searle s S. Jr., H a у s H. R., .1. Org. Chem., 23, 2028 (1958).
49. Y i a ii n i о s С. X., 1< a r a b i n о s J. V., J. Org. Chem., 28, 3246 (1963);
W a 1 1 a с e T. J., J. Am. Chem. Soc., 86, 2018 (1964).
50. Johnson H. W., Jr., D a u g h h e t с e P. H., J r., J. Org. Chem., 29,
246 (1964).
51. R u s s e 11 G. A., J a nzen E. G., Becker H.-D., S m e n t о w s к i F. J.,
J. Am. Chem. Soc., 84, 2652 (1962).
52. L 1 1 1 i e n J., J. Org. Chem., 29, 1631 (1964).
53. В a r t о n D. H. R., Garner B, J., W i g h t m a n R. H., J. Chem. Soc.,
1964, 1855.
54. Lunn W. H. W„ J. Org. Chem., 30 , 2925 (1965).
ДИМЕТИЛСУЛЬФОКСИДА ПРОИЗВОДНЫЕ, а) МЕТИЛСУЛЬ-
ФИНИЛМЕТИЛИДНАТРИЙ (димсилиатрий) (1). Кори и Чайков-
ский f 1 ] получили это чрезвычайно реакционноспособное соединение в
виде раствора в ДМСО путем добавления тонкоизмельченного гид-
рида натрия к избытку Д.МСО в атмосфере азота при перемешивании
при 65—70; до прекращения выделения водорода. Димсилнатрпн (1)
реагирует при комнатной температуре с этилтрифенилфосфонпй-
бромидом (5) с образованием илида (2), который вступает в реакцию
CH3SCH3 + ХаН
1 (С.НлЛ’ + СНдВг-)
(С(!Н-,);[Р СНСН»
(-’)
—(С,||Д,РО (С,На).С=О
(СвН5);>Р---СН..
(1)
86% ; Циклогексанон
(СвН5).,С-СНСН3
335
Виттига с бензофеноном с образованием 1,1-дифенилпропена-1
(3) 12). Добавлением к раствору димсилнатрия (1) метилтрифенил-
фосфонийбромида получают метиленилид (4), под действием
которого циклогексанон, камфора и холестанон-3 превращаются
в соответствующие метиленовые соединения с выходом 86, 73 и
69%. Таким образом, реагент Кори (1) позволяет легко получать
илиды Виттига.
Кори и Чайковский [31, а также Бекер и сотр. [41 независимо
нашли, что конденсация сложных эфиров с димсилнатрием приводит
к Р-кетосульфоксидам (6) с высоким выходом. В работе Кори и Чай-
ковского |3] сложный эфир (без растворителя, если жидкий, и в ТГФ,
если твердый) добавляют при 0J в атмосфере азота к раствору 2 экв
димсилнатрия (I), полученному из ДМСО и NaH. Выходы 70—9О?о.
О О
II II -
RCOCH3-I-CH3S - CH,(Na +)
(1)
о о-
дмсо II |
----« RCCH,S+CH3 + CH3OH
(6)
AI(Hg)
О
RCCH3
(7)
Бекер и сотр. 141 получили из сложных эфиров под действием дим-
силкалия (из mpe/n-бутилата калия и ДМСО) Р-кетосульфоксиды
с высоким выходом. Кори и Чайковский показали далее, что Р-ке-
тосульфоксиды(б), легко получаемые этим методом, гладко восстанав-
ливаются амальгамой алюминия до метилкетонов (7), и особо под-
черкнули эту характерную особенность нового метода. Бекер и Рас-
сел [5] получили димсилнатрий под действием метилата натрия и
конденсацией с диэтилфталатом осуществили новый удобный син-
тез нингидрина.
Диэтилфталат добавляют по каплям к суспензии основания в
ДМСО и перемешивают в токе азота. Первоначально образующийся
Р-кетосульфоксид (II) претерпевает внутримолекулярную сложно-
эфирную конденсацию с образованием индандиона-1,3 (III), который
в присутствии соляной кислоты подвергается перегруппировке Пум-
мерера с образованием 2-хлор-2-метилтиоиндапдпона-1,3 (V). Это
кристаллическое вещество получают с выходом 80% и при гидро-
336
лизе кипящей водой почти количественно превращают в нингидрин
(VI).
Димсилкалий, получаемый из ДМСО и трет-бутилата калия,
является, по-видимому, эффективным реагентом в трехуглеродной
конденсации п-метоксибензальдегида с дифенилметаном и ДМСО в
присутствии mpem-бутилата калия. В качестве первичного продукта
этой реакции получается соединение (1); увеличение времени реак-
ции или применение более высокой концентрации основания при-
водит к его расщеплению с образованием соединения (2) [6].
| ' (С.Н.ЦСОК I |
/г-СН3ОС,Н4С , CH3S-CH3----------< n-CH3OCeH4C-CH3S <Н3
(С„Н6).2СН2
(С6Н5)2СН
I (СНЩСОК
н
л-СН3ОС0Н,С
II
(СвН5)2С
Шемякин и сотр [7] при изучении синтеза тетрациклиновых анти-
биотиков нашли, что димсилнатрий является более удовлетворитель-
ным агентом для циклизации соединения (3) в (4), чем гидрид нат-
рия, амид натрия или диспергированный натрий.
\аН-1-ДМСО
1,5—3 час при 20°
СО.ХНС(СН3)3
4 моля Избыток
80%
СсН5СН2О
ОС2Н5
он
С6Н5СН.,О
Кори [8] применил димсилнатрий на трех стадиях полного син-
теза (/,/-кариофиллена. Первая стадия —замещение карбонильного
кислорода метиленовой группой по реакции Виттига. Вторая —
циклизация по Дикману соединения VII в VIII, возможно, через
337
промежуточно образующийся мостиковый лактон, который подвер-
гается атаке и расщеплению метилат-ионом. Третья стадия, где
применялся этот реагент,— реакция внутримолекулярного расщеп-
ления тозилата IX с образованием кетона X, ключевая стадия в син-
тезе кариофиллена. Тозилат (IX) обрабатывают димсилнатрием(Зэкв)
VII
в ДМСО при 25э в течение 30 мин, затем добавляют избыток трет-
бутанола и смесь выдерживают в течение 2 час. При этом происходит
изомеризация сочленения циклов и образуется кетон (X) «с очень
хорошим выходом».
Гарднер и сотр. 191 нашли, что димсилиатрий — эффективный
агент для дегалогенирования дибромпдов. Например, основной про-
дукт превращения вицинального дибромида 30-.хлор-5а,60-дпбром-
холестаиа—зр-хлор-А5-холестеи (2) (выход после перекристалли-
зации 77?<>), тогда как под действием ш/7Г/и-бутплата калия в ДМСО
(2) превращается в А:'"’-холестадпен (3) и смесь пеидеитифицировап-
иых диенов. Таким образом, первый более эффективен как дегало-
генирующий, а второй — как дегидрогалогенпрующий реагент. Ге-
O'
CH,S=CHz(Na*)
ДМСО *
72%
Н
н
(5)
мпиальный дибромид —7,7-дибромиоркараи (4) с дпмсплнатрпсм в
ДМСО при комнатной температуре дает 7-бромбицикло-14,1,01-геп-
тан (5) с преимущественным образованием /иряяс-изомера. Дальней-
шее дегидрогалогенирование (5) не наблюдается даже при длитель-
ной обработке реагентом. Геминальный дпбро.мпд (6), имеющий би-
338
циклическую [6,2,0]-спстему, па первой стадии также дает моно-
бромид (7), который, однако, при дальнейшем взаимодействии легко
образует аллен (8). Превращение гсм-дибромидов в бромциклопро-
паны (5) и (7) почти несомненно включает нуклеофильное замеще-
ние атома брома с промежуточным образованием бромциклопропил-
карбаниона, который затем протонируется растворителем. Наиболее
вероятный механизм перехода от монобромида (7) к аллену (8) вклю-
чает а-элиминированпе НВг под действием ДМСО", играющего роль
основания, с промежуточным образованием циклопропилкарбена.
Робертс и Уайтинг 1101 разработали метод эффективного безвод-
ного гидролиза сложных эфиров и нитрилов в ДМСО. Раствор дпм-
силпатрпя, полученный из ДМСО и гидрида натрия, титруют раст-
вором соответствующего количества воды в ДМСО с применением в
качестве индикатора трифенилметана. При этом образуется тонкая
суспензия едкого натра, под действием которой этилбензоат легко
гидролизуется при комнатной температуре. По сравнению с реак-
циями в гидроксилсодержащих растворителях в данном случае ско-
рость возрастает в 104—10" раз. Бензонитрил гидролизуется до
бензамида. Метиловый и этиловый эфиры мезитойной кислоты
легко гидролизуются при 25”.
Смесь ДМСО и /ирс/н-бутилата калия (образуется димсил-
калпй) метилирует диолефины и реакционноспособные ароматиче-
ские углеводороды, например [111:
I
СН3
Ь2 1»
б) ДИМЕТИЛСУЛЬФОНИЙМЕТИЛИД, (CH:1),S-СН.,. Этот реа-
гент, неустойчивый при нагревании, получают в растворе дегидро-
галогенированием триметилсульфонийиодида, -бромида или -пер-
хлората. Этот исключительно избирательный метиленирующий агент
превращает карбонильные соединения в эпоксиды (оксираны).
Методика Кори 1121. Димсилнатрий (2) получают прибавлением
в атмосфере азота измельченного гидрида натрия (0,1 моля) к из-
339
бытку ДМСО при нагревании до 75; и перемешивании до прекра-
щения выделения водорода (около 30 мин). После этого раствор дим-
силнатрия (2) охлаждают до комнатной температуры, разбавляют
Сг
+ - I
(СН3), S—СН3 (I)4-CH3S-. СН., (Ха •) — (СН,)., S-C.H, ]-(СН3), S-0
(1) (2) (3)
Ы%'(С.Д1Д,С = О
С\
(СОН5)2С-—-сн,
(4)
равным объемом сухого тетрагидрофурапа (чтобы предотвратить
замерзание смеси) и охлаждают смесью льда с солью. Затем без
перемешивания в течение 3 мин прибавляют раствор 0,1 моля три-
метилсульфопийиодида (1) в 80 мл ДМСО, перемешивают 1 мин и
медленно добавляют карбонильное соединение (либо в чистом виде,
либо в ДМСО или ТГФ). Перемешивают еще 15 мин при охлаждении
смесью льда с солью и затем еще 30—60 мин без охлаждения; смесь
разбавляют тремя объемами воды и продукт реакции экстрагируют
подходящим растворителем.
Простые карбонильные соединения и аф-непредельные кетоны
взаимодействуют с этим реагентом исключительно с образованием
эпоксидов (оксиранов), а не циклопропанов. Выходы следующие:
бензальдегид 75%, циклогептанон 97%, бензальацетофенон 87%,
карвон 89%, эйкарвон 93%, пулегон 90%, Д 4-холестенон-3 90%.
Этот реагент при взаимодействии с бутадиеном дает винилцикло-
пропан, возможно, путем 1,2-присоединения и последующего эли-
минирования диметилсульфида [12а].
СН,
(CH,)3S = CH, | \_
СН, = СНСН = СН,---------г СН2 СНСН = сн, —►
I
+S (СН3)2
сн,ч
| ;снси-сн,-!-(СНз), s
СН/
17-Кетостероиды под действием реагента превращаются в произ-
водные 17р, 20-эпокси-21-норпрегнана, которые можно восстановить
до 17Р-окси-17а-метплстероидов [131.
Методика Францена [14]. Раствор mpem-бутилата калия в ДМСО
добавляют по каплям к раствору триметилсульфонийбромида или
перхлората в ДМСО при комнатной температуре. В качестве осно-
вания можно применять метилат или этилат натрия, а в качестве
340
растворителя — ДМФА. Реагент, полученный этим способом, прев-
ращает монофункциональные альдегиды и кетоны в эпоксиды с хо-
рошим выходом. Франнен первоначально принял продукт реакции
с бензальаиетофеноном за соответствующий циклопропан; ошибоч-
ность этого была позднее доказана [151.
О
и
в) ДИМЕТИЛСУЛЬФОКСОНИЙМЕТИЛИД, (С.ГЦД= СН2. Этот
более стабильный реагент, предложенный Кори [16], при взаимодей-
ствии с монофункциональными карбонильными соединениями дает
эпоксиды (оксираны); однако с а, Р-непредельными кетонами обра-
зует циклопропилкетоны. Реагент получают [17,18] в трехгорлой
круглодонной колбе с мешалкой, одно горло закрывают медицинс-
кой резиновой пробкой, а в другое помещают обратный холодиль-
ник, закрытый сверху пробкой с трехходовым краном для подачи
сухого азота и для соединения с насосом для откачивания. В колбу
загружают по 0,1 моля измельченных гидрида натрия и трпметил-
сульфоксонийиодида (1) [17,18] и систему многократно промывают
О О
II дмсо II
(СН3)., S+ —СН3 (I-)ф NaH-> (CH3),S = СН., Д Nal -СН,
(2)
(С„Н„)гС = ОрО%
/°'
(СвН5)2Сл——-сн, -Ь (СНз), S = о
(3)
азотом (последовательно заполняя и откачивая), затем отключают
насос, на выходе помещают ртутный затвор и ток азота уменьшают
до едва ощутимого. Колбу охлаждают на водяной бане до 10—15°
п при перемешивании через резиновую пробку вводят с умеренной
скоростью 100 мл ДМСО, перегнанного в вакууме над гидридом каль-
ция, с помощью шприца для подкожной инъекции. Сразу начина-
ется бурное выделение водорода. Через 5 мин убирают охлаждаю-
щую баню и через15—30 мин,когда выделение водорода прекратится,
получают молочно-белый раствор илида (2). В реакции с бензофено-
ном раствор 0,08—0,09 моля кетона в ДМСО или ТГФ довольно быс-
тро вводят с помощью шприца в реакционную смесь (может потребо-
ваться небольшое охлаждение), перемешивают 10—15 мин при ком-
натной температуре, затем 1 час при 50°, охлаждают, разбавляют
тремя объемами воды, продукт реакции эпоксид (3) экстрагируют под-
ходящим растворителем и очищают кристаллизацией или перегон-
кой. Аналогично получены эпоксиды из 4-фенилциклогексано-
на (72%) циклогептанона (71/6) и бензальдегида (56%).
В случае получения и хранения больших количеств реагента
лучше исходить из триметилсульфоксонийхлорида, который полу-
чают, пропуская газообразный хлор в концентрированный раствор
соответствующего иодида до прекращения осаждения иода, после чего
341
раствор декантируют и освобождают от пода экстракцией эфиром.
Выпариванием водного раствора получают кристаллический хло-
рид. Смесь эквимолярных количеств триметилсульфоксонийхло-
рида и гидрида натрия в ТГФ кипятят при перемешивании до прек-
ращения выделения водорода (2 —3 час). Смесь охлаждают и выпав-
шую соль удаляют быстрым фильтрованием с отсасыванием в токе
азота па воронке Бюхнера через слой осушителя (целит). Филь-
трат собирают в трехгорлую круглодонную колбу, которую затем
заполняют азотом. Два горла колбы закрывают стеклянными проб-
ками и одно резиновой пробкой, через которую шприцем отбирают
пробу, добавляют к ней воду и титруют стандартным раствором кис-
лоты по фенолфталеину. Бесцветные растворы илнда можно хра-
нить при О' без заметного разложения в течение месяца.
<х,р-Непредельные кетоны реагируют с дпметилсульфоксонийме-
тплпдом ио углерод-углеродной двойной связи с образованием
производных циклопропана, как показано на схеме:
л _ хсн2
С6Н5СН=СНСОС6Н5 ,кс»з)гЬ-СН2> СбН5СН — СНСОС6Н5 + (CH,),S=O
9 5J/'o
Подобно этому карвон и эйкарвон дают циклопропаны за счет
двойной связи, сопряженной с карбонильной группой. Реак-
о
(сн3)2Н=сн2ч
43% *
(2)
цпей илнда с М-метплхииолоном-2 получен циклопропан (2) [19].
Необходимо подчеркнуть, что по сравнению с днметплсульфоний-
метилидом диметилсульфоксонийметилид более стабилен и не под-
вергается самопроизвольному термическому разложению в инертной
среде. Вместе с тем последний — менее активный нуклеофил, как
это видно на примере 1,1-дифенилэтилена, который реагирует с
(CH;t)2S-CH2, но не взаимодействует с (CH;J)2S(==O)-СН2.
При обработке 2 же диметилсульфоксонийметилида 1 же хлор-
ангидрида кислоты наблюдается ацилирование илида с образова-
нием стабильных0-кетосульфоксоиийилидовтипа (1) (т. пл.120 ). Это
соединение интересно тем, что при УФ-облучении (А. 253 ммк) пре-
терпевает разрыв диполярной связи S — Си гладко превращается
342
в карбоновую кислоту (2), по-видимому, через промежуточное обра-
О О
ТГФ. ! <<-1С
С„Н.-,СС1 2C1L S (С.Н;!)2-----——
’ - о
о о о
11 н
—> CgH-,C—CHS ’ (СН3), СН3 S (СН.;С(С1-)
(1)
/,vpi=O
о
и
С0Н-,ССН2СО„Н
(2)
зование кетокарбена и кетена. Этот двухстадийный процесс подобен
удлинению цепи по Арндту — Эйстсрту. Этиловые эфиры <х, |3-не-
предельных кислот вступают в реакцию сопряженного присоеди-
нения с последующей циклизацией, как показано на схеме для этило-
вого эфира А’-циклогексенкарбоновой кислоты. Продукт конденса-
ции (3) при УФ-облучении дает сульфоксид (4), который после оки-
сления надкислотой и циклизации в присутствии основания дает ке-
той (5). Восстановление его амальгамой алюминия приводит к
;п/?анс-гидринданону-2 (6). Эту последовательность реакции можно
рассматривать как новый метод синтеза карбоциклических
соединений [201.
С амидом а,0-непредельной кислоты реагент дает пирролидон
пли амид циклопропанкарбоновой кислоты, либо смесь этих про-
I I I
О —С-СН -С-------------------СН., —С —CHCOXHR
!' I I I '
(ch3),s--ch, с-о-^н.,с с^о-р сн„
'X''
НХ I
дуктов в зависимости от характера заместителей! [211.
343
Диметилсульфоксонийметилид с хорошим выходом метилирует
гидразоны, оксимы, кислоты, фенолы, а также некоторые аромати-
ческие углеводороды [22|. Примеры приведены на следующей схеме:
о
и
(CHa),S = CH.
С6Н5СН = NNHC6H5---------* CeH5CII = NN -С„Н5 -г (СН3), S = 0
00,0 |
СН3
О
II
(CH,1.S = CH.,
n-NO2CeH4CO2H —;—* n-NO2C0H4CO,CH3
no2 °no2 no2
I
CH3
CH3
С дигидротестостероном диметплсульфонийметнлид и диметил-
сульфоксонийметилид реагируют стереоспецифично, но в обратном
порядке [231.
Взаимодействие диметилсульфоксонийме.тилида с третичными а-
ацетоксикетонами сопровождается циклоприсоединением и дегид-
ратацией с образованием бутенолидов [241.
1. С о г е у Е. J., С h а у к о v s k v М , J. Am. Chem. Soc., 84, 866 (1962); 87,
1345 (1965).
2. Greenwald R., Chaykovsky AL, С о r e v E. J., J. Org. Chem., 28,
1128 (1963).
3. Corey E. J., Chaykovsky AL, J. Am. Chem. Soc., 86, 1639 (1964).
4. Becker H.-D., M i к о I G. J., Russell G. A., J. Am. Chem., Soc., 85,
3410 (1963).
5. Becker H.-D., R u s s e 1 1 G. A., J. Org. Chem., 28, 1896 (1963).
6. R u s s e 1 I G. A., Becker H.-D., .1. Am. Chem. Soc., 85, 3406 (1963).
7. Гуревич А. И., Шем яки н M. AL и сотр., Tetrahedron Letters, 877 (1964).
8. С о г е у Е. J., А1 1 t г a R. В., U d а Н., J. Am. Chem. Soc., 86, 485 (1964).
9. Cardenas С. G., Khafaj i A. N., Osborn C. L., Gar d пег P. D.,
Chem. Ind., 1965, 345; Osborn C. L., SliieldsT.C., ShouldersB. A.,
Cardenas C. G., Gardner P. D., ibid., 1965, 766.
344
10. Roberts W., W h i t i n g M. C., J. Chem. Soc., 1965, 1290.
11. A r g a b r i g h t P. A., 11 о f m a nn J. E., Schri e shei m A., J. Org.
Chem.. 30, 3233 (1965); Russell G. A., Weiner S. A., ibid., 31, 248 (1966).
12. Cor e v E. .1., C h a v к о v s к v At., .1. Am. Chem. Soc., 84, 3782 (1962);
Tetrahedron Letters, 169 (1963); J. Am. Chem. Soc., 87, 1353 (1965).
12a. К i j i J., Iwamoto At., Tetrahedron Letters, 2749 (1966).
13. D r e i a h 1 G., P о n s о 1 d K... S c li i с к IL, Chem. Ber., 97, 3529 (1964).
14. Franzen V., Driesen H.-E., Tetrahedron Letters, 1962, 661; Chem.
Ber., 96, 1881 (1963).
15. Corey E. J., C h а у к о v s к у At., Tetrahedron Letters, 169 (1963).
16. Corey E. J., Chav к о v s к у M., J. Am. Chem. Soc., 84, 867 (1962); 87,
1353 (1965).
17. Kuhn R., T r i s c h m a n n H., Ann., 611, 117 (1958).
18. Maj or R. T., Hess H.-J., J. Org. Chem., 23, 1563 (1958).
19. L о e v В., К о г m e n d у Al. F., Snader К. M., Chem. Ind., 1964, 1710.
20. Corey E. J., C h а у к о v s к у At., J. Am. Chem. Soc., 86, 1640 (1964).
21. M e t z g e r H., S e e 1 e г t K., Angew. Chem., Internat. Ed., 2, 624 (1963).
22. Al e t z g e r H., К 6 n i g H., S e e I e r t K-, Tetrahedron Letters, 867 (1964);
Tr ay nel i s V. J., McSweeney J. V., О. P., J. Org. Chem., 31, 243
(1966).
23. С о о к С. Е., С о г 1 е у R. С., Wall М. Е., Tetrahedron Letters, 891 (1965).
21. L е h m a n n H.-G., Angew. Chem., Internat. Ed., 4, 782 (1966).
ДИМЕТИЛСУЛЬФОН, (CH:!)2SO2. Мол. вес 94,14, т. пл. 102—
109А Д. очищают кристаллизацией из хлороформа.
Этплбензоат прибавляют по каплям в атмосфере азота к суспен-
зии //фе/п-бутилата калия в растворе Д. в ДМСО, перемешивают при
50—60J в течение 9 мин и после подкисления и обычной обработки
(СИ,), со к
C6H5CO»C,H-,-lCH,SO.,CH3------—----> С6Н3СОСН.,5О„СНЭ
6 5 - ’ 3 - (CH.ms--o- - - з
получают (о-(метансульфоиил)-ацетофенон с выходом 91 % (сырой
продукт) [11.
Описано применение Д. как очень хорошего растворителя в реак-
{ П КГ в (CH:,),SO, (1НГ). 21 сутки С
I II 50% | ||
V \ 'Д
N Cl N F
циях обмена галогена, например для превращения 2-хлорпиридина
в 2-фторпиридин [2|.
1. Becker H.-D., R u s s е 1 1 G. A., J. Org. Chem., 28, 1896 (1963).
2. S t а г г L. D., Finger G. C., Chem. Ind., 1962, 1328; F i n g e r G. С. et
al., J. Org. Chem., 28, 1666 (1963).
N, N-ДИМЕТИЛ-п-ФЕНИЛЕНДИАМИН, n-(CH:1)2NCGH4NH,.
Мол. вес 136, 20, т. пл. 38. Д. получают восстановлением N.N-ди-
метил-н-нитрозоанилина цинковой пылью в этаноле в присутствии
хлористого аммония [1]. Д. реагирует с ароматическими альдеги-
дами с образованием кристаллических окрашенных анилов, удоб-
ных для идентификации. С кетонами Д. не реагирует; так, например,
345
ацетон можно применять как растворитель для получения анилов
АгСНО--------------АгСН - NCeH,X (СН..,),-/!
альдегидов. Алифатические альдегиды также образуют анплы, по,
за несколькими исключениями, они не являются кристаллическими
веществами. Однако их можно идентифицировать по растворимости
в 30%-ной уксусной кислоте.
1. I; t z i п ц е г G. Ed., Regenass F. A., Helv. Chim. Acta, 37, 1901 (1954).
ДИМЕТИЛФОРМАМИД (ДМФА), HCON(CH3),. Мол. вес 73,10,
т. кип. 153 .
Эффекты растворителя. ДМФА применяется как растворитель
для углеводов, поскольку в нем мутаротацпя происходит очень мед-
ленно [11. ДМФА ускоряет алкилирование фталимида калпя^|2|, нат-
риймалоновых эфиров [31, натрпйацетоуксусных эфиров |4| и щелоч-
ных солей других соединений с активной метиленовой группой |4|.
По общей методике 1 моль соединения, содержащего активную мети-
Nall - (СНЩСНВг
СН3СОС11,СО,С,Н-, --> CIL,COCIIC(),C,IIr>(\;i : )
2 чче, 9 7
1МФА " - - -
- CH3(1()CI --ХаВг
I
CH(CI
Выход 63%
неновую группу (если оно твердое, то растворяют в минимальном ко-
личестве ДМФА), прибавляют по каплям при перемешивании в атмос-
фере азота к суспензии 1 моля гидрида натрия в ДМФА, поддержи-
вая охлаждением температуру 40—50 А После прекращения выделе-
ния водорода, когда гидрид натрия полностью прореагирует, при-
бавляют 1 моль алкилирующего агента, изменяя скорость добавле-
ния в зависимости от экзотермического эффекта реакции. В случае,
изображенном на схеме, смесь затем нагревали на кипящей водяной
бане в течение 2 час. Сравнение с другими системами металл — раст-
воритель показывает определенные преимущества системы NaH —
ДМФА.
В случае превращения арилгалогенидов в нитрилы при кипяче-
нии галогенида и цианистой меди в ДМФА сокращается время реак-
ции и выходы несколько улучшаются[5]. Этим методом 1-нафтонптрил
Вг CN
ДМФА: 4 час
был получен из 1-бромнафталина с выходом 94% кипячением в те-
чение 4 час-, при использовании в качестве растворителя пиридина
реакционную смесь нагревают 15ч«сна масляной бане при 215—225J
и выход составляет 82—90% 161. Удобная методика для выделения
346
полученного нитрила из комплекса с солью меди состоит в вылива-
нии коричневой реакционной смеси в водный! раствор хлорного же-
леза. При этом прел.сходит окисление Си : в Си2:, который! уже не
дает комплекса с нитрилом. Можно также выливать реакционную
смесь в водный раствор этилендиамина (дает комплекс и с Си'
и с Си2') или цианистого натрия (образуется растворимый купро-
цнанид натрия). Более высоко кипящий N-метилпирролидои-2 (т.
кип. 202 ) также дает хорошие результаты [7]; однако он трудно
доступен.
При электролизе солей карбоновых кислот ио Кольбе выход ди-
мера R — R в обычных растворителях, воде и метаноле, лимити-
руется протеканием побочных реакций, приводящих к простым эфи-
рам, спиртам, сложным эфирам, углеводородам RH н олефинам. При
использовании в качестве растворителя нереакционноспособного
высокополярного ДМФА в смеси с триэтилампном, применяемым в
качестве основания, выходы существенно улучшаются 181. Так, гид-
Злсктродпв Д \
С,Н-С11СО.,Н- (С.,И-)., ,\------------- С|(Н1-СНС„Н-,
I ' 4|"" i I
С.Н;! СН„ CIL,
ратроповая кислота дает 2,3-дифенплбутан (мезо- и <7, /) с выходом
41"<>, в параллельном опыте с трпэтиламшюм в метаноле выход этого
димера был только 21 °о ;выделено также несколько продуктов, пре-
вращения а-метилбензильного радикала.
Согласно методике окисления спиртов, хромовый ангидрид пере-
мешивают с раствором спирта (например, холестанола) в ДМФА до
растворения и добавляют каталитическое количество серной кислоты.
Реакция протекает при комнатной температуре 191.
Пространственная направленность реакции пропионового аль-
дегида с реактивом Виттига (2) с образованием 0-этилстпрола (3)
очень чувствительна к условиям проведения этой реакции 1101.
Если ее проводить в ДМФА с использованием в качестве основания
этилата натрия и с добавлением подида лития, то соотношение
цис : транс равно 96 : 4, тогда как в эфире без каких-либо добавок
соотношение 31 : 69.
NaOC.H-, сп.с.п.сно
С„Н-СН.,Р (С„1 ----------- -* СД1-СН-- Р (С,,Н-,).,-:-->
ДМФА ' ДМФА, Li I
(I) (2)
-^С„1Ь,СП- СНСН.,сн.,
(3)
При детальном подборе реагентов и условий проведения син-
теза пси гидов через активированные эфиры Плесе и Буассон 1111
нашли, что в ДМФА реакция карбобензоксиэфпра (4) с бензилами-
CI
/.---. с. и си ,\н.
Cb.XHCHCOO — 7 ---CI------------- CbNlICHCOXlICII.C,,!!-,
I = ДМФЛ I
С„П- С1 С,,11-,
(4) (3)
.347
ном протекает быстрее, чем в других, обычно используемых раст-
ворителях. Это тем более благоприятно, что ДМФА обладает вы-
сокой растворяющей способностью по отношению к высокомолеку-
лярным пептидам.
ДМФА рекомендован как разбавитель в синтезе диарилов из
арилгалогенидов по Ульману [121 и как растворитель для бекма-
новской перегруппировки стероидных еноиокснмов 112а1.
Реакция Вильгеродта — Киндлера (реакция карбонильных сое-
динений со вторичным амином и серой с образованием тиоамида) в
ДМФА протекает при 50—60~ с хорошим выходом; пиридин дает
худшие результаты [131. Майер и Ведь [14] проводили реакцию в
RCOCH3 -LS + R'R'NH —> RCH2CSNR'R' -L Н.2О
ДМФА с добавкой /z-толуолсульфокислоты в качестве катализатора
для ускорения образования промежуточного енамина. В этих усло-
виях реакция протекает при комнатной температуре.
S
TsOH S
RCOCH3T(CH3)2XH— — RC —СН2 — -- RCI1.,C\ (СН3),
N (CH3)2
Дегидрогалогенирование. Кун и corp. 1151 растворяли 2,4-ди-
бром- 1,1,5,5-тетрафепилпентадпен-1,4 в ДМФА, продували при-
бор азотом пк смеси медленно добавляли метанольный раствор едкого
С.Н5 С6н5 С6Н5 CGH5
ССН.,С^С' Н-Конц. КОН —-О- СС^С-СС
| ‘I в СН3ОН "°" /
С6Н5 Вг Вг сбн5 с6н5 с6н5
3 г в 50 мл ДМФА 5 мл 1,6 г
кали. Через 10 мин, когда сине-зеленая окраска изменится до ко-
ричнево-желтой, прибавляют метанол (20мл) и затем 50%-ный вод-
ный метанол (20 мл). Тетрафенилпентатетраен выделяется в виде
красивых желтых призм.
Бернштейн и сотр. 1161 провели дегидрогалогенирование самим
ДМФА. Если стероидный 9а, 11Р-дпхлор-А1,4-диеион-3 (1) или 9а-
бром(хлор)-! 1§-окси-А1,4-диенон-3(2) кипятить 30 мин в ДМФА,то по-
лучаются два продукта (3)и (4)с выходом 20—25%. При ароматизации
19-метпльная группа отщепляется в виде хлористого (бромистого)
348
метила и получается соединение (4). Если вместо ДМФА применить
пиридин, то необходимо кипячение по крайней мере в течение 6 час.
Реакции. (См. также ДМФА — РОС1;!, ДМФА — SOCI2). Для
получения N.N-диметпламидов кислот из хлорангидридов вместо
диметиламина, который довольно неудобен в работе (т. кип. 7, 4 ),
можно использовать ДМФА 117]. При этом реакцию можно прово-
дить в каком-либо ином растворителе, нежели ДМФА. Если вместо
хлорангпдрида используют ангидрид кислоты, то необходимо доба-
()
C(ilJ.-,COCl --IICX (Clip., CeH5 СОХ (СН3)2 ; НС1 г со
СН,СО °
I о-r 11с\ (сн3)2 к"";|ЧД-1 цо,ссн,сн,сох(сн:)), со
C1I..CO У0"”
вить каталитическое количество серной кислоты.
ДМФА присоединяется к октепу-1 в присутствии трст-б\ \и.'\-
перекиси при 132' с образованием смеси амидов (1) и (2) в соотиоше-
СН3 (СН,)5СН = СН2 л- НСОХ (СН3), СН3 (СН,)-, СН2СН,СОХ (CI 13), +
О)
сн.(
I
+ СН.( (СН,)-, CHCON (CI13),
(2)
пии 60 : 40 [181.
ДМФА реагирует с алкпллптпем с образованием алифатических
альдегидов с выходом 50—80% 1191. Алкениллитий реагирует анало-
О OLi
I н. о
w-CHI^-Ai ; 11СХ (C.11,з)2. - ,//-C;II13CN(CII3), н-С7Н15СНО
| ^2%
Н
гичным образом, но с меньшим выходом 1201.
ДМФА конденсируется с ароматическими аминами, такими, как
(3), с образованием соответствующих форманилидов (4) [211. В этом
случае ДМФА берется в избытке и используется не только как реа-
гент, но и как растворитель. Реакцию проводят в присутствии мети-
лата натрия при кипячении. Однако в этих условиях хорошие резуль-
таты получаются не со всеми замещенными анилинами. Так, 2,5-ди-
метокспаиилпн в присутствии метилата натрия дает выход всего 48?о.
О
N;iOCH,
n-FC„H4NH,-~ HCN (СН3),----— /i-FCgHl|XHCHO-2 (СН3), Nil
1 * I)
(3) (4>
В присутствии гидрида натрия выход возрастает до 62%.
349
Применение ДМФА для синтезам, Р-непредельных ацетиленовых
альдегидов показано на следующей схеме 1221:
О OMgBr
' I HOSO.OH
СНз (СН2)з с ; CAlgBr НСХ (СН.(), —л СНз (СН2)з С =_ ССНХ (СНз),-*
- II.о
—► СНз (СН,)з с == сен - X (СН..,)., (X -) —СНз(СН,):,С..zCCHO 4- н\ (СН;,)2
ДМФА реагирует с диалкилхлорфосфатом (5) с образованием жел-
того кристаллического аддукта (6), который достаточно стабилен в
Cl II
(СН3)2Х О (CH;i)2 X 4 О R OPH
НС = О-ГС1Р (OR), —-НС —OP (OR)., н
(о) (b) | ,н
Основание| R ОН j (СН.,)Л'С —
О О О
:1
R’OP(C)R), R'C)P-O-P(OR)2
(О
отсутствие влаги 123]. Этот аддукт (6) реагирует со спиртами в при-
сутствии оснований с образованием эфиров фосфорной кислоты (7),
а также с эфирами фосфорной кислоты (8) с образованием пирофос-
фатов (9). Аналогичные результаты получены при реакции ДМФА
с алкилдихлорфосфатами.
1. Kuhn R., Haber F., Chem. Вег., 86, 722 (1953).
2. S h e e Ii an.l. С., В о 1 li о f e r W. A., J. Am. Chem. Soc., 72, 2786 (1950).
3. Z a u g g H. E., H о г г о m В. W.. В о r g w a r d t S., J. Am. Chem. Soc., 82,
2895 (1960).
4. Z a u g g H. E., Dunnigan D. A., At icliacls R. ,1., Swett L. R..
W a n g T. S., Sommers A. H., D e X e t R. \V., J. Org. Chem., 26, 641
(1961).
5. 1- г i e (I in a n L., S ii e c h 1 e r H., .1. Org. Chem., 26, 2522 (1961).
6. H ь io м э и Al., „Синтезы органических препаратов", ИЛ, М., 1952, сб. 3,
с гр. 344.
7. X е w m a n М. S., В о d е n Н., J. Org. Chem., 26, 2525 (1961).
8. F i и k e I s t e i n AL, Petersen R. C., J. Org. Chem., 25, 136 (1960).
9. S n a 1 z 1: e G., Chem. Ber., 94, 729 (1961).
10. В e p r e .i ь с о и Л. Д., II! смяк и и AL AL, Tetrahedron, 19. 149 (1963).
11. P I e s s .1., Boissonnas R. A., Helv. Chim. Acta, 46, 1609 (1963).
12. К о r n b I ii m X., К e n d a 1 1 D. 1.., J. Am. Chem. Soc., 74, 5782 (1952).
12a. К о h e n F., Chem. Ind., 1966, 1378.
13. С a г а у о n-G e ri t i I A., At i n о t AL, C h arbier P., Bull. soc. chim.
France, 1964, 1420.
14. At aver R., Wehl .1., Angew. Chem., Internat. Ed. 3, 705 (1964).
15. К u h ii R., Fischer II., F i s c h e r H., Chem. Ber., 97, 1760 (1964).
16. Heller AL, L e n h a r d R. H., Berns! e i n S., ,1. Am. Chem. Soc., 86,
2309 (1964).
17. С о p p i n g e r G. Al., J. Am. Chem. Soc., 76, 1372 (1954).
350
IS. F r i e <1 in и и I... S 11 v c ii t < г H.. Tetrahedron Letters, 238 (1961).
19. E v a ns E. A., .1. Chem. Soc., 1956, 4691.
20. В r a u d e E. A., E v a n s E. A., J. Chem. Soc., 1955, 3331.
21. P e t t i t G. R., T h <> in ;i s E. G., .1. One Chem., 24, 895 (1959); Pet-
ti t G. R., К a 1 n i n s M. V., LiuT. M., T li о in a s E. G., P a rent K.,
ibid., 26, 2563 (1961).
22. .1 о n e s E. R. H., S к a t t e b 0 1 L., W h i t i и Д M. C., J. Chem. Soc.,
1958, 1054.
23. C r a in e r F„ Winter M„ Chem. Ber., 94, 989 (1961).
ДИМЕТИЛФОРМАМИДА ДИНЕОПЕНТИЛАЦЕТАЛЬ (3). Мол.
вес 231,37. (См. Диметилформамида диэтилацеталь.)
Ацеталь (3) применяют для этерификации карбоновых кислот
спиртами бензильного типа (2) |1|. Преимущество этой методики в
использовании только 1 жн Д. д. и образовании летучих побочных
/ Cll:i у,
RCO.,H ArCll.i’l! (СП..)., \С11| (Х'.Н.,ССН., !---->.
(1> ('*) (3) ' i > —ч'1".,
\ СП., / '
—- rc( ).,ос! i.,Ar ; (сн:1).;.\'сн<) ; 2Н<х:п2С(СП:1)3
С)
продуктов. Д. д. был использован для получения и-метоксибеизп-
лового и п-додецилбензилового эфиров различных Ы-защшцеиных
аминокислот и пептидов.
1. В ii с h i If., S t e e n K., Esch e n in <> s e r A., Ansjew. Chem., Internat.
Ed., 3, 62 (1964); В r e e 11 b ii 11 1 e r 11., В ii c 11 i 11., H li t z E., S c h r e i-
b e r .1., E s e h e in user A., Helv. Chim. Acta, 48, 1746 (1965).
ДИМЕТИЛФОРМАМИДА ДИЭТИЛАЦЕТАЛЬ,
(СН:,)Л'СН(ОС2НAlo.i. вес 131,22, т. кип. 134—136 .
Получение 111. 1 моль борфторида триэтилоксоиия (1) при
охлаждении льдом с солью заливают ДА1ФА, после чего нагревают
до кипения при перемешивании. Через некоторое время начинается
довольно энергичная реакция и борфторид диметиламиноэтоксикар-
бония (2) отделяется в нижнем слое. Его отделяют и прибавляют по
каплям к раствору этилата натрия в этаноле. Выделяющийся борф-
торид натрия отделяют и после фракционирования полмчают ацеталь
(3).
О
(СИ3)2К1СН + (C2H5)3O#BF4* >(CH3)2NCH=OC2H5(BF4“) + (С2Н5)2О
(1) (2)
С*\ |NaOC2Hs
fVcHNlCH,), (CH,)2N-C>I(OC2lI5)2 + NaBF,
(А (2)
Конденсация с соединениями, имеющими активную метиленовую
группу. Д. д. конденсируется с циклопептадиеном при кипячении
без растворителя с образованием б-диметнламииофульвена 1(4) ,
351
т. пл. 67 1. Подобная конденсация описана с ацетофеноном, динитри-
лом малоновой кислоты, нитрометаном и ацетоуксусным эфиром.
Этерификация. Д. д. можно использовать для этерификации
карбоновых кислотв мягких условиях [2,31. Так, например, бензой-
ная кислота (0,4 моля) при реакции с этим ацеталем (2 же) даетэтпл-
бензоат в следующих условиях: в хлористом метилене за 5 час при
40°, в бензоле за 1 час при 80е и в ацетонитриле за 36 час при компат-
С6Н-,СО,Н -НСН3)2 ХСН (ОС2Н5)2 С0Н5СО2С2Н5-|-С2Н3ОН + (СН3), хено
ной температуре [31. Д. д. реагирует также с фенолом в дихлорэтане
в течение 46 час. при 84° [2].
Гетероциклы. Д. д. конденсируется с гуанидином с образо-
ванием 2,4-диамино-с//лпи-триазина [4].
ХН
NH
(C.HtO),CHN(CH3).
nh2
(1)
il
НА’- С -}-2С.,НвОН
I
N = CHN(CH3)3
(2)
ХН.,
I
А С-Х'Н:
ХН
-NH.
-<СН3)2 к и
(общий)
N
Н.,Х — h—NH,
J L
У1
(3)
1. Meer we i n H., Florian W., Schon N., Stopp G, Ann., 641, 1
(1961).
2. Verbruggen H., Angew. Chem., Internal. Ed., 2, 211 (1963).
3. Breclibuhler H., В ii c h i H., HatzE., SchreiberJ., Eschen-
m о s e r A., Angew. Chem., Internet. Ed., 2, 212 (1963); Helv. Chim. Acta,
48, 1746(1965).
4. Bredereck H., Effenberger F., Hofmann A., Chem. Ber.,
97, 61 (1964).
ДИМЕТИЛФОРМАМИДА ЭТИЛ EHKET АЛЬ,
.0—CH2
/ I
(CH3), X’CH |
XO-CH2
Мол. вес 117,15, т. кип. 142—144°.
Д. э. получают обменной реакцией диэтплкеталя ДМФА с эти-
ленгликолем [1]. В присутствии каталитических количеств уксусной
н
352
кислоты Д. э. избирательно взаимодействует с насыщенными 3-кето-
стероидами с образованием З-этиленкеталя в присутствии других ке-
тонных групп (как в обычных предельных кетонах, так и в непредель-
ных) [2|. В этих очень мягких условиях Д4-3-кетостероиды и 12- и
20-кетостероиды не реагируют.
1. М е е г w е i n Н., Florian W., Schon G., Stopp G., Ann., 641, 1
(1961).
2. Vorbrueggen H., Steroids, 1, 45 (1903).
ДИМЕТИЛФОРМАМИД — АЦЕТАТ НАТРИЯ.
Дегидратация. Рейхнгтейн 111 нашел, что эта смесь является
наиболее удобным дегидратирующим агентом для избирательного
превращения гптоксигенина (1) в 10-ангидрогитоксигенин (2). Смесь
3 мг гптоксигенина (1), 0,3 мл ДМФА и 1 мг безводного ацетата нат-
рия нагревают в запаянной трубке при 95J в течение 4 час. Хромато-
графия и кристаллизация позволяют получить 1,8 мг чистого 16-
ангидрогитоксигенина (2).
1. Russel J. Н., Schindler О., ReichsteinT., Helv. Chim. Acta,
43, 167 (1960).
ДИМЕТИЛФОРМАМИД — ДИМЕТИЛСУЛЬФАТ. ДМФА и ди-
метилсульфат реагируют в отсутствие растворителя с образованием
И(СН3)2
н-с=о +
сн’°^°
сн3ох
СН,О^ s-sO
о'Д^о__________
N(CH3)2 +n(ch3)2
н-с-осн3 *—> н-с-осн3
Комплекс
комплекса состава 1:1 (2 дня при 25\ 2 час при 60—80°) [ 1 ]. При по-
пытке перегонки этот комплекс разлагается на компоненты. Он
реагирует с циклопентадиенилнатрием с образованием 6-диметила-
минофульвена [2].
мснэ)г
нс+-осн3 снз5О<
> [Г\\ + CH3(M’a)SO4 + СН3ОН
сн
N(CH3),
12 Заказ № 965
353
1. Bredereck H., Effenberger F., Simchen G., Angew, Chem.,
73, 493 (1961).
2. H a f п e г K. et al., Angew. Chem., Internal. Ed., 2, 123 (1963).
ДИМЕТИЛФОРМАМИД — ТИОНИЛХЛОРИД (реактив Вильс-
мейера [1]) *.
Получение хлорангидридов кислот. Метод получения хлорангид-
ридов кислот с помощью хлористого тиснила непригоден для синтеза
сульфокислот и даже некоторых карбоновых кислот, например п-
NO2CdH4 СО2Н. Боссхард и сотр. [2| нашли, что ДМФА выступает в
роли катализатора этих реакций, когда применяется как раствори-
тель или когда присутствует в небольших количествах в инертном ра-
створителе. При реакции эквивалентных количеств ДМФА nSOCl2
в качестве промежуточного продукта был выделен хлорметплен-
диметилпминийхлорид — гигроскопичное и реакционноспособное
твердое вещество с т. пл. 140°, которое также получают при взаимо-
О
II soci.. ZC1 + .Cl
(сн3).,\с—н —* (CH3).,N+ = с; (CH3)2n -с '
-so-’ а- Н | чн
JRCO..H СГ
(CH.t).,XCH l-RCOCI -1-НС!
действии ДМФА с фосгеном, оксалилхлоридом или пятихлористым
фосфором. При реакции этого хлорида с кислотами регенерируется
катализатор ДМФА. В одном из опытов 0,3 моля /z-нитробепзойной
кислоты быстро нагревают при 90—95' с 0,315 моля хлористого тис-
нила и 0,03 моля ДМФА в 160 мл хлороформа и получают хлоран-
гпдрид с выходом 88?о. В другом опыте суспензию 0-нафталинсуль-
фоната натрия в ДМФА обрабатывают при 10—15° хлористым
тиенилом, через пять минут отделяют выпавший хлористый натрий и,
добавляя ледяную воду, высаживают 0-нафталннсульфохлорид; вы-
ход количественный (т. пл. 76°).
Использование этого метода в ряду гетероциклов открыло путь
к таким хлораигидридам, получение которых требовало специаль-
ных приемов. Так, хлорапгпдрнд 5-метилизоксазол-З-
<Г°гН карбоновой кислоты ранее получали только при взаимо-
с = Г| действии ее натриевой соли с пятихлористым фосфором
сДсх° [31 или хлористым тионилом [41, так как свободная
(Lj* кислота с хлористым тионилом не реагирует. По улучшен-
ной методике [51 50 г ДМФА добавляют по каплям в тече-
ние Юмин при перемешивании к суспензии 508 г 5-метилизоксазол-
3-карбоновой кислоты в 700 г хлористого тионила и кипятят 1 час
(кислота растворяется через 15 мин). После перемешивания в тече-
* Авторы работы [1] использовали для синтеза /г-алкиламинобеизальдегидов
примерно эквивалентную смесь хлорокиси фосфора и А'-алкилформанплпда. Фос-
ген и пятихлористый фосфор также применяются вместо хлористого тионила.
В этой книге описана еще одна смесь типа реактива Вильсмейера: диметилформа-
мид — хлорокись фосфора.
354
ние 12 час при комнатной температуре отгоняют избыток хлористого
тионила в вакууме водоструйного насоса (20 .пп), используя 15-с.и
колонку Вигро. Отгонку продолжают до тех пор, пока температура
верхней части колонки не достигнет 80 , после чего выдерживают ре-
акционную смесь при 4 в течение нескольких часов. Когда большая
час। ь одержимого колбы закристаллизуется, колбу переворачивают
над стаканом, чтобы стекал остаток ДМФА. Выход сырого хлорап-
гпдрпда 95—100|,о. Перегонка, которая часто сопровождается бур-
ным разложением [41, нс является необходимой. Полученный таким
образом препарат загрязнен лишь небольшим количеством диметнл-
формамида и может успешно применяться для получения замещен-
ных амидов, сложных эфиров и кетонов (метод с использованием
CilCI.). Эта методика \ спешно применялась для синтеза 3-метплизок-
сазол-5-карбоновой кислоты, 1шрпдш1-2,6-дпкарбоновой кислоты,
никотиновой, изопикотиповой н фуран-3-карбоновой кислот. Опти-
мальное молярное соотношение SOU.: кислота 1,47 : 1,0; при соот-
ношении 1 : 1 смесь сильно загустевает и ее трудно перемешивать.
Японские исследователи 161 нашли, что превращение кислоты
(1) в хлорангидрпд «значительно улучшается» при испольювании
II СН.2СО.2Н II СН/.ОС1
I I + 5ОС12+ДМФА —д—у—* j I
//'. 42 "л Следы °''° //
О N О ОХО
I I
Н II
(1) 10; (2)
ДМФА в качестве катализатора. Смесь кислоты и SOC1., нагревают
15 мин при 100", охлаждают до комнатной шмпературы, добавляют
несколько капель ДМФ.Х и кипятят еще 15 мин; выход хлорапгид-
рпда 91%.
Формулирование. Группа Боссхарда [21 нашла, что ДМФА —
SOCl.j удобны для синтеза /z-.тпметплампнобензальдегида [7|:
/ _ —'(Ji,), X — (.IK.!; (J) /-к
(СН3)А—Ф '----------* /СН;),\— ' Хх—СНО
• • \=/
Комплекс ДМФА — SOCI, легко реагирует со стероидными спир-
тами, образе я с почти количественным выходом эфиры муравьиной
кислоты е сохранением конфигурации 181. Даже третичные спирты
форматируются под действием ДМФА—SOC1.> |9|. С эпоксидами
ДМФА—SOC1., образует Гформилокси-2-хлор- пли 1,2-дихлорпро-
12' 355
изводные [101.
Пептидные синтезы с использованием этого комплекса описаны
в работе 1111.
Дегидратация. Лоутон и Мак-Ричи [121 безуспешно пытались
провести дегидратацию амида пиромеллитовой кислоты (1) до соот-
ветствующего нитрила (2) обычными способами. В конце концов такое
превращение им удалось осуществить при действии хлористого тио-
нила в ДМФА (60, 7 час, выход не указан). Бэйли и др. [131 повто-
рили эту реакцию и получили нитрил (2) с выходом 35по. Термэн
[141 нашел, что эга реакция зависит от температуры; ему удалось
Н2\ОС CONH3 NC CN
\/ 0—2.5’ \/
I II I II
/Д. 60 ” /д/х
H3NOC CONHj КС CN
II) (2)
повысить выход до 60%, начиная дегидратацию при 0? и постепенно
повышая температуру до комнатной. Такой усовершенствованный
способ дегидратации стал общепринятым, в частности для циклоде-
гидратации N.N'-дибензоилгидразина [14].
C6H5CNHNHCC4H5 ЗОСЦ-ДМСРА С6Н,-Д V-c6h,
N-N
Хлорирование. Комплекс ДМФА — SOC12 можно использовать
в качестве хлорирующего агента. Так, пикриновая кислота превра-
щается в пикрилхлорид, а 1,3,5-триазин (1) в соответствующее ди-
хлорпроизводное (2) [151.
(1) (2)
При обработке нуклеозида (1) хлористым тнонилом в присутст-
вии только каталитических количеств ДМФА получается хлорид
356
(2) с почти количественным выходом [161. Однако если применять
хлористый тионил в избытке ДААФА, то с высоким выходом получается
диметиламинопроизводное (3).
. .о~~
CjHjCOO OCOCjH,
О у^^^”НгОСОСбН5
CiHjCOO OCOC6HS
CbHjCOO ОСОС6Н5
O'
Конденсация с дегидратацией. Икехара и Уно [17] показали,
что комплекс ДМФА — SOCU можно использовать для образова-
ния 3',5'-межнуклеотидной связи.
1. VilsmeierA., Н о а с k А., Вег., 60В, 119 (1927).
2. Bossh a rd Н. Н., Могу R., Schmid М., Zollinger Hch., Helv.
Chim. Acta, 42, 165.3 (1959).
3. Q u i I i с о A., P a n i z z i L., Gazz., 68, 625 (1938).
4. G ar d ne r T. S., Wen is E., LeeJ., J. Org. Chem., 26, 1514 (1961).
5. Private communication from Gar dner T. S., and W e n i s E., Hoffmann—
LaRoche Inc.
6. E g a w a Y., Su z uk i M., Ok u d a T., Chem. Ph arm. Bull., 11, 589 (1963).
7. В о s s h a r d H. H., Zollinger Hch., Helv. Chim. Acta, 42, 1659 (1959).
8. Morita K., Noguchi S., N i s h i к a w a M., Chem. Pharm. Bull., 7,
896 (1959).
9. A r n о 1 d Z„ Coll. Czech., 26, 1723 (1961).
10. Ziegenbein W., Franke W., Chem. Ber., 93, 1681 (1960); Z i e g e rl-
b e i n W., Hornung К.-H., ibid., 95, 2976 (1962).
11. Z a о r a 1 M., Arnold Z., Tetrahedron Letters, No 14, 9 (I960).
12. LawtonE. .A., M c R i t c h i e D. D.. .1. Org. Chem., 24, 26 (1959).
13. Bailey A, S., Henn B. R., Langdon J. M., Tetrahedron, 19, 161
(1963).
14. Thurman J. C., Chem. Ind., 1964, 752.
15. E i 1 i n g s f с I d H_, S e e f e I d e r M., Weidingen H., Angew. Chem.,
72, 836 (1960).
16. Z e in 1 11 i с к a J.,.Smrt J., Sorm F., Tetrahedron Letters, 397 (1962);
Z e m I i c.k a ,1., S о r m F., Coll. Czech.. 30, 2052 (1965).
17. I к e h а г a M., L n о H., Chem. Pharm. Buli12, 742 (1964).
357
ДИМЕТИЛФОРМАМИД — ХЛОРОКИСЬ ФОСФОРА (ИЛИ ФОС-
ГЕН), ДМФА — РОС1;>.
Формилирование. ДМФА и хлорокись фосфора образуют комплекс
состава 1:1, который начинает диссоциировать при температуре
около 100 и давлении 10 мм. Этот комплекс можно представить
резонансными структурами (1). Для форматирования комплекс
° °+ сг О' сг о-
(СН3)2Л —СН (СН3)2К’=СНОН — ,lP Cl;3 (CH3)2N = CHOP+Cl2«t—>(CHj)2N-CHOP + C12
-НС1 s_________T- 2
(1)
применяют в избытке ДМФА, взятого в качестве растворителя. Так,
например, хлорокись фосфора (0,055 моля) добавляют по каплям к
ДМФА (0,22 люля) при 10—20' для получения раствора комплекса
(1) и после этого прибавляют раствор индола (0,50 моля) в ДМФА.
Смесь выдерживают 45 мин при 35\ выливают в воду со льдом и
получают прозрачный светло-красный раствор соли (3) 111. После
подщелачивания раствора и нагревания до кипения с высоким выхо-
дом выделяется З-формилипдол (5) в виде белых кристаллов. При
нейтрализации до слабокпслой реакции раствора соли (3) при —5J
после экстракции холодным хлороформом получается 3-диметилами-
нометплениндоленин (4), который можно восстановить алюмогид-
рпдом лития до грамина (6). Применение этой реакции к аромати-
ческим углеводородам описал Снайдер |2|. Эта реакция в приме-
нении к эфиру стероидного диенола (7) позволяет удобно получить
А4-3-кетостероид (11) |3|. В этом случае хлорид диметилформиммо-
ния был получен из диметплформамида и фосгена. После проведе-
ния реакции замещения соль (8) восстанавливают до амина (9), гид-
рируют до диена (10) и кислотным гидролизом получают кетостероид
сн»:
Синтезы органических препаратов включают методики форми-
лнрования смесью ДМФА — РОС1:< с образованием п-дпметиламп-
нобензальдегида (выход 80—84?о) |4|, 2-ппрролальдегпда (выход
358
78—79%) (51, а также формилирования этим реагентом производных
тиофена [6].
Комплекс ДМФА—POCL, превращает даже очень слабооснов-
ные 4,5-диаминоп11рпмидпны в пурины в достаточно мягких усло-
виях [71. Так, например, смесь 0,2 г этилового эфира 4,5-диампно-
2-хлорпирпмидинкарбоновой-6 кислоты [(12), рКа 2,61, Юлы ДМФА
со2с2н5
(12)
(13)
и 1 мл хлорокиси фосфора выдерживают 1 чос, после чего нагревают
15 мин на кипящей водяной бане; выход этилового эфира 2-хлор-
п ур ин карбоновой-6 кислоты (13) составляет 0,16 г. Углеродный
атом в положении 8 пуринового соединения (13) берется из фор-
мильной группы ДМФА.
Комплекс ДМФА — фосген реагирует с эпоксидом (1) в трихлор-
этилене с образованием 1-хлор-2-формилокси-А5,1'-цнс, tpvc-цикло-
додекадиена (2) [81. Комплекс ДМФА — СОС12 реагирует с 3,3-
(CH3);N = CHC1(C1~)
84%
(О (2)
диалкокси-5а-стероидом с образованием соответствующей иммони-
евой соли (не выделена), которая при гидролизе водным раствором
ацетата натрия дает 2-формпл-3-алкокси-Д'2-стероид по следующей
359
схеме |9|:
1. S m i t h G. F., J. Chem, Soc., 1954, 3842; ДжеймсФ., Сна й дер X. «Син-
тезы органических препаратов», ИЛ, М., 1941, сб. II, стр. 30.
2. М о о г е Н. W., Snyder Н. R., J. Org. Chem., 29, 97 (1964).
3. В u г и D., Е I I i s В., F е a t h е г Р., К i г k D. X'., Р е t г о w V., Chem.
Ind., 1962, 1907; В u г п D. et al., Tetrahedron, 20, 597 (1964).
4. Каыпень Э., Арчер У., «Синтезы органических препаратов», ИЛ, М.
1954, сб. 5, с гр. 22.
5. Сил ьверштейн Р., Р ы ш к е в и ч Э., Уиллард К., «Синтезы
органических препаратов», ИЛ, М., 1958, сб. 8, стр. 44.
6. С a m р а I § n е Е., А г с h е г W. L., J. Am. Chem. Soc., 75, 989 (1953).
7. ClarkJ., ListerJ. H., J. Chem. Soc., 1961, 5048.
8. Ziegenbein W., Franke W., Chem. Ber., 93, 1681 (I960).
9.. В u r n D. et al., Tetrahedron Letters, 73, 733 (1964).
ДИМЕТИЛФОСФИТ, (CH3O)2POH. Мол. вес 110,05, т. кип.
64—66‘/15 мм.
При добавлении Д. к перемешиваемой суспензии 2,4,6-трибром-
4-метилииклогексадиенона (1) в бензоле при комнатной темпера-
туре взаимодействия не наблюдается. Однако после индукционного
периода (10 мин) желтый цвет изменяется на оранжевый и смесь
разогревается [ 11. Еще через 3 мин твердое вещество растворяется
и окрашивание исчезает. Обработкой смеси получают 2,6-дибром-
О
Вг ^|| Вг 0
|| || -ИСН3О) ,^Н (СН3О)2РОН
\/ (2а) (26)
Вг" ЧСН3
||>
II
+ (СН3О)2РВг
(4)
4-метилфенол (3) с высоким выходом. Автокатализ в этой реакции
связывают с медленным таутомерным превращением фосфонатной
формы (2а) в более реакционноспособную фосфитную форму (26),
в которой осуществляется реакция с кетоном (1). Такое таутомерное
превращение катализируется следами бромистого водорода, образую-
щегося при гидролизе бромфосфата (4). Если в качестве раствори-
теля использовать метанол, то образование бромистоводородной
кислоты при метанолизе бромида (4) ускоряется и время индукцион-
ного периода сокращается до 30 сек.
1. Miller В., J. Org. Chem., 28, 345 (1963).
360
3,5-ДИМЕТИЛ-4-ХЛОРМЕТИЛИЗОКСАЗОЛ, Н3С
Мол. вес 145,59, т. кип. 67,571 мм.
СН2С1
СН3
Получение. 3,5-Диметилизоксазол хлорметилируют действием
параформа и хлористого водорода в присутствии хлористого цинка.
111.
Аннелирование. Сторк описал синтез, изображенный на схеме,
без указания деталей [21. Алкилирование енолята 9-метилокталинди-
она-1,6 (1) этим реагентом даете хорошим выходом соединение (2)
Двойную связь енона восстанавливают с образованием соединения (3)
и затем осуществляют превращение изоксазолилметильного замес-
тителя с образованием аннелированной системы обработкой бор-
фторидом триэтилоксония с последующим действием основания. Воз-
можные стадии приведены на схеме. Промежуточный симметричный
р-дикетон (5) может расщепляться только в одном направлении.
Циклизация кетона (6) дает трициклический кетон (7).
1. Кочетков Н. К., Хомутова Е. Д., Б а з и л е в с к и й А1. В., ЖОХ,
28, 2736 (1958).
2. Stork G., «The Chemistry of Natural Products», 3, 131, Butterworths, 1964;
Pure and Appl. Chem., 9, 131 (1964).
2,2- ДИМЕТОКСИПРОПАН (диметилкеталь ацетона),
(2Нз)гС(ОСНз)г. Мол. вес 104,15, т. кип. 79—8Г. Получение [1].
В качестве примера использования этого реагента для получения
кеталей обменом с первичными или вторичными спиртами см. 2,2-
Дибутоксипропан.
361
Кетали. Применение Д. для превращения кетонов в кетали
можно иллюстрировать на примере получения диаллплкеталя цикло-
гексанона 121. Смесь циклогексанона, Д., аллилового спирта и ката-
о
I > т<он
Г СН, - СНСН,ОН - (CI 13),С(ОСН3), —
II 63 /
СН, == СНСН,0 ОСН2СН - сн,
+ СН3СОСН3-;-2СН3ОН
литического количества /ьтолуолсульфокислоты в бензоле нагре-
вают с одновременной отгонкой на колонке образующегося ацетона и
азеотропной смеси бензол — метанол. По окончании отгонки реак-
ционную смесь охлаждают, нейтрализуют катализатор метилатом
натрия и перегоняют.
Ацетониды (изопропилиденовые производные). Д. реагирует
с диоксиацетоновой боковой цепью кортикостероидов с образова-
нием 17,21-ацетонида, который можно рассматривать как защитную
2011 Pl2°\ /сн’
ОН <СНЛС(ОСН3)2 (TsOH) CHJCO
в ДМФА______ S^S° СН’
Г J + 2 СН3ОН
группировку для боковой цепи [3, 4], которая устойчива к основа-
ниям, но может быть удалена нагреванием на кипящей водяной бане
с водной уксусной или муравьиной кислотой.
Как цис-, так и трйнс-флавондиолы-3,4 при реакции с ацетоном
в обычных условиях дают ацетонид qwc-диола; с Д. реагирует только
цис-изомер (TsOH — катализатор) |5|.
Это соединение применяется для связывания воды при получе-
нии ацетонидных производных нуклеозидов [6|. (См. Ди-/г-нитрофе-
нил фосфат.)
Метиловые эфиры. Д. был использован для получения хлор-
гидратов метиловых эфиров аминокислот [7|. Этот реагент выпол-
няет роль растворителя, донора метоксильных групп и поглотителя
воды. Аминокислоту суспендируют в Д., прибавляют немного кон-
362
центрированной соляной кислоты и смесь выдерживают при ком-
(СН,).С(ОС.Н,), -
H,NCHCO„H ----------—- С1Н.,ХСНСО.,СН,
- | * Разб. НС.1 । -
R R
натной температуре в течение 18 час. Жирные кислоты количествен-
но этерифицируются при обработке метанолом и большим избыт-
ком Д. в присутствии следов разб. соляной кислоты; реакция закан-
чивается за 1 час при комнатной температуре |8|.
Эфиры енолов. Реакция Д4-3-кетостероида с Д. в ДМФА при
кислотном катализе приводит к эфир)’ енола [9].
Реакция с нуклеозидами [101. Д. селективно реагирует с пер-
вичной 5'-гидроксильной группой нуклеозида (1) с образованием
(1) (2)
5'-О-метоксиизопроппльного производного, которое можно при-
менять вместо обычно используемого тритильного производного 1101.
1. LoretteN.B., HowardW.L., В г о w n J. Н., Jr., J. Org. Chem., 24,
1731 (1959).
2. Lore t t e.X. B., Howard \V. L., J. Org. Chem., 25, 521 (1960); Org. Syn.,
42, 34 (1962).
3. Tanabe M., В i g 1 e у B. J., .1. Am. Chem. Soc., 83, 756 (1961).
4. Robinson С. H., Fi nckc nor L. E.,T i ber i R., Ol i vetoE. P.,
J. Org. Chem., 26, 2863 (1961).
5. Brown B. R., Mac Br i de J. A. H., J. Chem. Soc., 1964, 3822.
6. Hampton A., J. Am. Chem. Soc., 83 , 3640 (1961); Mof f a t t J. G., ibid.,
85, 1118 (1963).
7. R a c h e 1 e J. R., J. Org. Chem., 28 , 2898 (1963).
8. RadinN.S., HajraA. K., A k a h о r i Y., J. Lipid. Res., 1, 250 (1960).
9. N u s s b a u m A. L., Y nan E., D i n c e r D., О 1 i v e t о E. P., J. Org.
Chem., 26, 3925 (1961).
10. Hampton A., J. Am. Chem. Soc., 87,
5,5-ДИМЕТОКСИ-1,2,3,4-TETPA-
ХЛОРЦИКЛОПЕНТАДИЕН,
Мол. вес 263,95, т. кип. 108—110 ,11
4654 (1965).
Cl
Cl J
\ ОСН3
I X
/X/ ОСНз
С1 I
С1
мм.
363
Д. получают действием метанольного раствора едкого, кали на
гексахлорциклонентадиен 111. Это соединение более реакционноспо-
собно в реакции Дильса — Альдера., чем гексахлорциклопентадиен,
и реагирует с различными диенофилами в относительно мягких ус-
ловиях Г1,2]. Получающиеся аддукты интересны тем, что они легко
ароматизуются. Необходимые для этого стадии показаны на при-
мере ароматизации аддукта с акриловой кислотой, полученного ки-
пячением компонентов в течение 48 час. На заключительной стадии
о
С1
окисление перманганатом в ацетоне дает в качестве единственного
продукта 2, 3, 4, 5-тетрахлорбензойную кислоту; окисление хромо-
вым ангидридом в кислоте приводит к 25% ароматической кислоты
и 39% 1, 2, 3, 4-тетрахлорбензола.
В то время как гексахлорциклопентадиен образует аддукт с
нафтохиноном-1,4 с выходом всего 53%, Д. реагирует с этим хино-
ном в трихлорбензоле при постепенном повышении температуры до
180° с образованием аддукта (1) с выходом 88%. Этот аддукт можно
превратить различными способами в тетразамещенный антрахинон
131.
(1) н = осн.
1. Newcomer J. S., М с В е е Е. Т., J. Am. Chem. Soc., 71, 946 (1949).
2. McBee Е. Т., Diveley W. R., BurchJ. E., J. Am. Chem. Soc., 77,
385 (1955); L'ngnade H. E., M с В e e E. T., Chem. Revs., 58, 249 (1958).
3. К n i e 1 P., Helv. Chim. Acta, 46, 492 (1963).
4,4 -ДИМЕТОКСИТРИФЕНИЛХЛОРМЕТАН (2). Мол. вес 326,81,
т. пл. 114°.
364
Получение |1]
ОН
I
2 n-CH3OCeH4MgBr 4-СвН5СО2СН3 —п-СН3ОС3Н4 С—С6Н4ОСН3-л
(П |
свн5
С1
л-СН3ОСеН4— С-С0Н4ОСН3-л
Защитная группа. Тритильную группу нельзя считать удов-
летворительной в качестве защитной группировки для нуклеозидов,
поскольку гликозидные связи довольно лабильны в кислых усло-
виях, необходимых для ее удаления. Например (Корана и сотр.
[1,2|) в уридиновом производном (За) невозможно осуществить уда-
ление 5'-О-тритильной группы без затрагивания 2'-О-тетрагидропи-
ранильной группы, лабильной в кислой среде.
Однако при введении в защитную группировку одной, двух или
трех метоксильных групп скорость гидролитического отщепления
в 80%-ной уксусной кислоте повышается в 10 раз на каждую введен-
ную группу. Введение /z-метоксильных групп увеличивает скорость
реакции тритилгалогенида со вторичной гидроксильной группой и
таким образом ограничивает избирательность реакции с первичным
гидроксилом в положении 5'. Наилучшее соотношение этих противо-
положных факторов достигается в случае Д. и его производного (Зв).
1. Smith М., R a m гп 1 е г D. Н., Goldbergl.H., К h or а па Н. G.,
.1. Am. Chem. Soc., 84, 430 (1962).
2. Schaller Н., Weimann G., Lerch В., К h or a n a H. G., J. Am.
Chem. Soc., 85, 3821 (1963).
1,2-ДИМЕТОКСИЭТАН («глим»), CH3OCH2CH2OCH3. Мол. вес
90,12, т. кип. 83°, уд. вес 0,863; смешивается с водой.
365
Показано, что реакция Фаворского стереоспецифична в неполяр-
ных растворителях— эфире 111 и Д. |2| и нестереоспецпфична в по-
лярном растворителе — метаноле |2|.
Виттиг 131 нашел, что в эфире о-дибромбеизол реагирует с амаль-
гамой натрия с образованием гексамерной фенпленртути с выходом
52%, однако в Д. выход этого соединения снижается до G%, а глав-
ным продуктом (41%) является о-терфениленртуть.
Д.— предпочтительный растворитель при генерировании дегпд-
робензола безкислотным диазотированием антраниловой кислоты
изоамплнитритом 141.
Д.— гораздо лучше эфира для проведения реакции Симмонса —
Смита с ацетиленами; образование полимерных продуктов в этом
случае наб'людается в значительно меньшей степени [5].
По сравнению с другими растворителями Д. предпочтителен для
реакции щелочных металлов с нафталином или дифенилом [61, с
эфирами фенолов |7], а также для реакции натрия с трс/п-нитробу-
таном |8|.
Д. лучше, чем ДМСО, в качестве растворителя для формнлиро-
ваипя кетонов окисью углерода в присутствии алкоголятов щелоч-
ных металлов 191. Это обусловлено тем, что ДМСО является
донором протонов в присутствии сильных оснований (щрс//г-бутилат
калия) и поэтому не может рассматриваться как инертный раство-
ритель.
I. Stork G., В о г о w i t z I. J., J. Am. Chem. Soc., 82, 4307 (1960).
2. House H. O., G i 1 m о r e W. F., J. Am. Chem. Soc., 83, 3980 (1961).
3. W i t t i g G. et al., Chem. Ber., 95, 431, 1962 (1962).
4. Org. Expts., 316.
5. E m p t ozG., V o-Q u a n g L., Vo-Q u a ng Y., Bull. soc. chim. France, 1965, 2653.
6, Scott N. D., Walker J. F., H a n s 1 e v V. L., J. Am. Chem. Soc., 58,
2442 (1936).
7. W i 1 d s A. L., Nelson N. A., J. Am. Chem. Soc., 75, 5360 (1953).
8. HoffniannA. K., HendersonA. T.,J. Am. Chem. Soc., 83, 4671 (1961).
9. van der Zeeuw A. J.. Gersmann H.R., Rec. trav., 84, 1535 (1965).
ДИНАТРИЙФЕНАНТРЕН. Д. дебромирует с хорошим выходом
«циклооктатетраендибромид» (1) до бпцикло-|4, 2, 0|-октатриеиа-
2, 4, 7 (2) (Фогель и др. |1|). Самый чистый образец, содержащий
95% вещества (2) и примесь циклооктатетраена (3), получается при
низкотемпературной (—20е) перегонке.
(1) (2) (3)
1. Vogel Е., Kiefer Н., Roth W. R., Angew. Chem., Internat. Ed., 3,
442 (1964).
2,4-ДИНИТРОБЕНЗАЛЬДЕГИД. Мол. вес 196,12, т. пл. 70°.
Получают из 2,4-динитротолуола и л-нитрозодиметиланилина [1].
366
Д. применяется для идентификации первичных аминов, с которыми
СН — \С6Н4\(СП3)2
XN0-
/!-ONC«H4N(CH;,)2 I II
СНО
Разб. НС1
24—32%
(общий)
он дает основания Шиффа. Реакция очень легко протекает в горячем
этаноле, содержащем несколько капель уксусной кислоты [2|. Д.
также реагирует (в кипящем уксусном ангидриде) с а-пиколином,
2,6-диметилхинолином и другими соединениями, содержащими акти-
вированную метильную группу, с образованием производных 2,4-
динитростирола 131.
1. Беннет Дж., Белл Е., «Синтезы органических препаратов», ИЛ, М.,
1949, сб. 2, стр. 224.
2. Wild F., «Characterization of Organic Compounds», Cambridge, 1947, p. 229.
3. В e n n e t t G. M., P r a t t W. L. C., J. Chem. Soc., 1929, 1465.
ДИ-«-НИТРОБЕНЗИЛХЛОРФОСФАТ, (n-NO2C6H4CH2O)2POCl.
Мол. вес 386,70, т. пл. 107°.
Д. получают из п-нитробензилфосфата и пятихлористого фосфора.
Это стабильное, кристаллическое соединение применяется для фос-
форилирования окси- и аминосоединений. Нитробензильные группы
можно удалить каталитическим гидрированием |1].
1. ZervasL., Dilarisl., J. Am. Chem. Soc., 77, 5354 (1955).
3,5-ДИНИТРОБЕНЗОИЛ ХЛОРИСТЫЙ. Мол. вес 230,57, т. пл.
68°. Соответствующая кислота получается нитрованием бензойной
кислоты [1]. Описана [21 методика ее очистки для использования при
определении креатинина.
Д. применяется для выделения низкоплавких спиртов; реакция
в пиридине дает сложные эфиры с пониженной растворимостью и
повышенной температурой плавления. Так, витамин D (т. пл. 82—
84°) был выделен в виде 3,5-динитробензоата (т. пл. 132°) из концент-
рата (полученного из жира печени рыб) после этерификации Д. и
очистки хроматографией и кристаллизацией [31.
1. Б р е с т е р Р., Уильямс Б., Филлипс Р., «Синтезы органических
препаратов», ИЛ, Л1., 1952, сб. 3, стр. 214.
2. Jansen А. Р., So m brock W., N о у о n s E. C., Chem. Weekblad, 43,
731 (1947); |C. A., 42, 2307 (1948)].
3. BrockmannH.,Z. physiol., 245, 96 (1937); В r ockm a n nH.,B u sseA.,
ibid., 249, 176 (1937).
367
3,4-ДИНИТРОБЕНЗОЙНАЯ КИСЛОТА. Мол. вес 212,12, т. пл
166\ Получают из п-толуидина 111:
Д. к. при восстановлении сахарами и аскорбиновой кислотой
образует краситель, который можно использовать для микроопре-
деления сахара после разделения гидролизатов полисахаридов с
помощью хроматографии на бумаге |1|.
1. Borel Е., Deuel Н., Helv. Chim. Acta, 36, 801 (1953).
2,4-ДИНИТРОБЕНЗОЛСУЛЬФЕНХЛОРИД. Мол. вес 234,62,
т. пл. 96 . Д. получают из 2,4-диннтрохлорбензола [11. Обзор по
применению [21.
Д. вступает во многие ионные реакции, при которых нук-
леофильный реагент атакует атом серы и замещает атом хлора, как,
например, в реакции с анилином [31. Д. лег ко реагирует со спиртами,
SNHC„H5 SCI SOCR3
I сн,сн=снсн,
SCH(CH3)CHaCH3
I
I г
no2
в том числе и третичными [21. Д. присоединяется стереоспецифично
(транс-присоединение к пространственно незатрудненным олефинам,
например, к бутену-2. Тетразамещенные олефины с Д. не реагируют
[41. Д. можно использовать для замещения по Фриделю—Крафтсу
различных ароматических соединений [5], включая азулен [61.
368
Образующиеся производные, содержащие 2,4-динитрофенилсульфе-
нильный радикал, являются окрашенными твердыми веществами и
часто применяются для идентификации, так как хорошо очищаются
кристаллизацией и хроматографией.
Защитная группа. 2,4-Динитробензолсульфеиильную группу
можно использовать для защиты гидроксильных групп в нуклеози-
дах [7]. Она устойчива к действию фосфорилирующих агентов,
дициклогексилкарбодпимида, кислот и оснований, но ее можно се-
лективно отщепить тиофенолом или тиосульфатом. Так, из тимидина
(1) можно получить 5'-ацетильное производное (5) тритилированием
первичной 5'-оксигруппы, обработкой (2) Д. в ДМФА с образованием
соединения (3), отщеплением тритильной группы с образованием
соединения (4), ацетилированием его и отщеплением защитной груп-
пировки.
Было показано (Бартон и др. [8|), что 2,4-динитрофенилсульфеио-
выеэфирытипа (1) расщепляются при фотолизе в мягких, нейтраль-
ных условиях. Так, облучение эфира (1) в бензоле в атмосфере азота
дает уксусную кислоту с выходом 90%. Образование в качестве
О
SOCCH3 S
N0* ^'ып
I II +| |Дсн"с“н+| II '
\/ °
NO2 1\о2
(1) (2) 73%
главного побочного продукта соединения (2) показывает, что необ-
ходимый атом водорода берется из растворителя.
1. KharasehN. et al., J. Am. Chem. Soc., 69, 1612 (1947); 71, 2724 (1949);
Kharasch N., Lanford R. B., Org. Syn., 44, 47 (1964).
369
2. Kharasch X., PotempsS. J., W e h r m e i s t e r H. L., Chem. Rev.,
39, 269 (1946).
3. В i 1 1 m a n J. H., Garrison J., AndersonR., Wolnak B., J. Am.
Chem. Soc., 63, 1920 (1941).
4. Kharasch N. et al., J. Am. Chem. Soc., 71, 2724 (1949); C r a m D. J., J.
Am. Chem. Soc., 71, 3887 (1949).
5. KharaschN. ct al., J. Am. Chem. Soc., 72, 3529 (1950); 75, 1081, 6035 (1953).
6. Anderson A. G., Jr., McDonald R. N., J . Am. Chem. Soc., 81, 5669
(1959).
7. L e t s i n g e r R. L., Fontaine F., Mahadevan V., S c h c x n a y-
der D. A., Leone R. E., J. Org. Chem., 29 , 2615 (1964).
8. В ar ton D. H. R., Cho w Y. L., Сох A., К i r b у G. W., J. Chem. Soc.,
1965, 3571.
4,4 -ДИНИТРО-3,3'-ДИКАРБОКСИДИФЕНИЛДИСУЛЬФИД,
Мол. вес 368,33, т. пл. 238°. Получение [11.
В водном растворе при pH 8 этот реагент взаимодействует
с сульфгидрильными группами с образованием глубоко окрашен-
ного аниона (Х,П.|Ч 412 мяк, е 13 600), поэтому его используют для
количественного определения сульфгидрильных групп [11, а также
__со; ___________соон
S—//_S--___________^"КО,
RSH- j “ ~~
S-^___\\О2 RSS-^_______^.\О2
4 СООН соон
для изучения микробиологического переноса серы [21 и влияния
веществ, обладающих радиозащитной активностью, на содержание
сульфгидрильных групп в тканях [3|.
1. Е 1 1 m a n G. L., Arch. Biochem. Biophys., 82, 70 (1959).
2. Flavin M., J. Biol. Chem., 237, 768 (1962).
3. S 6 r b о B., Arch. Biochem. Biophys., 98 , 342 (1962).
2,4-ДИНИТРОИОДБЕНЗОЛ, 2.4-(\О2).,С6Н31. Мол. вес 294,01,
т. пл. 90°.
Реакцию 2,4-дпнптрохлорбензэла с подистым натрием проводят
в кипящем этиленгликоле [II, но в качестве растворителя лучше
использовать ДМФА [2|. Баннет и Коннер [21 определили константы,
С! 1
\о
I II Mil в HCON’ (СИ-
1^ '1 ДАДГ I,
NO, NO,
скорости и равновесия для обменных реакций атомов хлора, брома
и пода в I-галоген-2,4-динитробензолах.
370
1. Bennett G. M., Vernon J. H,, .1. Chem. Soc., 1938, 1783.
2. Bunnett J. F., Conner R. M., Org. Syn., 40, .'34 (1960).
2, 4-ДИНИТРОФЕНИЛ ГИДРАЗИН. Мол. вес 198,14, т. пл. 197°.
Получение |11. Растворяют 100 г 2,4-динитрохлорбензола при
перемешивании в 200 мл триэтиленгликоля, охлаждают до 15'
(некоторое количество вещества может выкристаллизоваться) и при-
бавляют по каплям 28 мл 64%-ного раствора гидразина, поддержи-
вая температуру смеси в интервале 20 дЗ' (~25 мин). После окон-
чания сильно экзотермической реакции кипятят полученную пасто-
образную массу на кипящей водяной бане, прибавляют 100 мл мета-
нола, снова кипятят, затем охлаждают, фильтруют и промывают
метанолом. Выход 98 г (100%), т. пл. 190—192°.
Д. реагирует с альдегидами и кетонами в условиях кислотного
катализа с образованием плохо растворимых и высокоплавких 2,4-
динитрофенилгидразонов, которые можно с успехом использовать
для идентификации.
Д.— высокоплавкое вещество красного цвета, плохо раствори-
мое в этаноле. Для получения растворов этого соединения можно
рекомендовать две методики.
Идентификация карбонильных соединений, а. [1, 2|. Растворяют
при нагревании 2,0 г Д. в 50 мл 85%-ной фосфорной кислоты, охлаж-
дают,прибавляют 50 мл 95 /o-ного этанола,снова охлаждают и отфиль-
тровывают с отсасыванием от небольшого количества нерастворив-
шегося препарата. Получившийся 0,1 М раствор очень стабилен.
Производные динитрофенилгидра-зонов получают прибавлением
0,001 моля альдегида или кетона к 10 мл 0,1 /Й раствора Д. с после-
дующим кратковременным нагреванием до начала кристаллизации.
Хотя раствор Д. в фосфорной кислоте разрушает неустойчивые в
кислой среде вещества гораздо в меньшей степени, чем растворы Д.,
содержащий минеральные кислоты, тем не менее при действии выше-
приведенного раствора иногда наблюдается элиминирование тре-
тичной гидроксильной группы 13J.
б. [4]. Растворяют при нагревании 4гД. в 120 мл диметилового
эфира диэтиленгликоля (диглима) и дают раствору охладиться до
комнатной температуры (этот раствор может храниться при комнат-
ной температуре в течение нескольких дней). Прибавляют 5 мл это-
го раствора к раствору 0,1 г карбонильного соединения в 1 мл этано-
ла (илидиглима), нагревают на кипящей водяной бане и прибавля-
ют 3 капли концентрированной соляной кислоты. Красный цвет
раствора переходит в желтый и вскоре выпадает гидразон.
в. Паррик н Рэсберн [5] рекомендуют использовать в качестве
растворителя ДМФА или ДМСО. Д. хорошо растворим в этих
растворителях; можно приготовить настолько концентрированный
раствор Д., что из него будут самопроизвольно выкристаллизовы-
ваться даже хорошо растворимые производные. Канадские исследо-
ватели смогли получить гидразон (2) только этим способом.
371
(2)
Дегидрогалогенирование. Д. используют на последних стадиях
синтеза кортизона из желчных кислот в качестве дегидрогалогени-
рующего агента. Дегидрогалогенирование соединения (1) обычными
методами протекает довольно плохо вследствие цос-ориентации 50-
атома водорода и 60-атома брома. Однако Д. нестереоспецифично
конденсируется с (1) и производит дегидрогалогенирование с обра-
зованием (2) (выход высокий) [6].
1. Org. Expts., Chapt. 16.
2. J о h n s о и С. D., J. Am. Chem. Soc., 73, 5888 (1951).
3. Reich H., Cr a neK.F.,S a n f i 1 i p p oS. J., J.Org. Chem., 18, 822 (1953).
4. Shine H. .1., J. Org. Chem., 24, 252 (1959).
5. P a r r i c k J., Rasburn J. W., Can. J. Chem., 43, 3453 (1965).
6. Mattox V. R., Kendall E. C., J. Am. Chem. Soc., 72, 2290 (1950); M c-
GuckinW. F., KendallE. C., J. Am. Chem. Soc., 74 , 3951 (1952).
^«?-(2,4-ДИНИТРОФЕНИЛ)-КАРБОНАТ (1). Мол. вес 394,25,
т. пл. 148’. Получение 111.
При взаимодействии Д. (1) с эфирами аминокислот (2)
получают N-активированные производные (3), которые конденсиру-
ются с N-защищенными аминокислотами (4), с образованием
372
эфиров дипептидов (5).
___no2 __no2 °
о Ч_о CO-i-H,XCHCO„Et — O2N^ ^'-OC\CHCO2Et—>
'=z ь I - =/ II
R HR
(1) (2) (3)
R'
I
(4> CbNHCHCO2H
----------> CbNHCHCONHCHCO,Et
I I
R' R
10)
1. Glathard R., Matter M., Helv. Chim. Acta, 46, 795 (1963).
ДИ-п-НИТРОФЕНИЛФОСФАТ (2). Мол. вес 340,19, т. пл.
175—175,5', рКа 1,7 в 50%-ном этаноле.
Получение 11].
О
2O2N ^-ONa + РОС13 —> O2N—ОРО ^'-NO2 —>
\=/ .=/ , =
С1
(1)
о
LiOH z---х II Z----------X
--------—/ Чк_о-Р -о—f T-N’O2
Ь6% (общий» --- । -- z
он
(2)
Применение в качестве кислотного катализатора 121. Этот реа-
гент Д. имеет преимущества перед zi-толуолсульфокислотой как ка-
тализатор при получении 2',3'-О-пзопропилиденовых производных
ынг оО ЭН2С N °снз \.1 + (СН3)2СО + СН3ССН3 У У °СН3 ОН ОН 10 мл 8 лнолей (3) 1 лгмоль N ’ НОН2С 87% \ 7 о хо J^C.. HjC^ сн3 ( о и + n-O2NC6H4OPOC6H4NO2-n—> ОН 1,2 лжоля гшг ©
373
(ацетонидов), а также других кеталей рибонуклеозидов, например
аденозина (3). Смесь нуклеозида, ацетона, катализатора и 2,2-дпме-
токсипропана (который служит для связывания выделяющейся воды)
перемешивают до получения прозрачного раствора (1—3 час). По-
сле этого прибавляют метанольный раствор аммиака, насыщенный
углекислым газом, для переведения катализатора в аммонийную соль
и разделяют на ионообменной смоле. Выходы в пределах 85—95%.
Преимущество Д. как катализатора этой реакции частично обус-
ловлено тем. что лиипдофильность этого соединения примерно
вдвое больше, чем у н-толуолсульфокислоты, поэтому его соли с нук-
леозидами лучше растворимы в ацетоне. Другим преимуществом этой
методики является то обстоятельство, что образование ацетонида
нуклеозида можно сочетать с фосфорилированием, которое дает со-
ответствующий нуклеотид. Так, после получения раствора 2', 3'-
ацетонида аденозина (4) ацетон и 2,3-диметоксипропан можно отог-
нать, а остаток обработать тетра-ц-нитрофенилпирофосфатом для
превращения в ацетонид нуклеотида.
1. Moffatt J. G., К h or а п а Н. G.. J. Am. Chem. Soc., 79, 3741 (1957).
2 Hampton A., J. Ат. Chem. Soc., 83, 3640 (1961); Biochem. Prepn., 10, 91
(1963); Hampton A., F г a t a n t о n i J. С., С a г г о 1 1 P. M., Wang S.,
J. Am. Chem. Soc.. 87, 5481 (1965).
F
A?*
I II
2,4-ДИНИТРОФТОРБЕНЗОЛ (ДНФ), Мол. вес V
186,10, т. кип. 13772 мм. Получение II, 2). \'о2
Анализ пептидов. ДНФ был использован [31 для идентификации
концевых аминогрупп в белках и пептидах. Конденсация с образо-
ванием 2,4-динитрофеннлированиого белка проводится в мягких ус-
ловиях. При кислотном гидролизе концевые аминокислоты выделя-
ются в виде ярко-желтых 2,4-динитрофенильных производных, ко-
торые легко отделяются от смеси аминокислот и могут быть иденти-
R R
I (КО.) ,С«Н,Г | Гидролиз
Н2\СНСО—белок--------> (NO2)2CeH3NHCHCO— белок-►
zNO2 R
—> О2\’ —Ч"—NHCHCOoH -у Аминокислота
фпцированы бумажной хроматографией.
Характеристика оксисоединений. ДНФ в присутствии триэтила-
мина в качестве катализатора реагирует с фенолами в ацетоне с обра-
374
зованием 2,4-дннптрофениловых эфиров, которые могут быть испо-
льзованы для идентификации [4|. В сходных условиях ДНФ дает
также твердые производные спиртов {51.
Расщепление альдоз. ДНФ применяется для расщепления по
Волю оксимов альдоз; при этом образуется ариловый эфир оксима,
который разлагается на низшую альдозу, 2,4-диннтрофенол и циа-
О.Х
СН = ХОН СН = N — О—/ — хо2 o2n
СНОН —*СНОН —-сноу НО—/ Ч— NO,4-HCN
I I I х=/
нистый водород 161.
АгОН -АгН. Ппркл и Забришке 171 описали метод замещения
гидроксильной группы фенола на водород. Этот метод Ппркл и
Гэйтс 181 применили для исследований потенциальных аналгети-
ков, например для превращения цис-тетрагидродезоксикодеина (1)
в соединение (2). Арплпрованне соединения (1) ДНФ в смеси толуол—
Д/МФА в присутствии гидрида натрия как катализатора дает с по-
чти количественным выходом 2', 4'-динитрофениловый эфир (2). Ги-
дрирование эфира (2) приводит к диаминопроизводному (3), которое
не удается выделить из-за его легкого автоокисления и которое сразу
расщепляют с высоким выходом натрием и жидким аммиаком в эфире
до дезоксипроизводного (4). Для модельных соединений на стадии
образования и восстановления динитрофениловых эфиров выходы
очень хорошие, однако на стадии расщепления выходы колеблются
от 0 до 100% [71. Расщепление протекает достаточно эффективно
только для соединений, имеющих рядом с фенольным гидроксилом
метокси- или фенильную группу, как в соединении (1). Аналогичный
375
метод использовали [9] для отщепления 4-гидроксильной группы
в алкалоидах, родственных морфину, однако при этом образовывался
и расщеплялся 4-фениловый эфир. Получение фениловых эфиров по
реакции Ульмана является более трудной операцией, чем гладкое
арилирование динитрофторбензолом. К тому же динитрофениловые
эфиры лучше кристаллизуются, а диаминофенильные эфиры легче
растворяются в тех растворителях, которые применяются на стадии
расщепления; наличие аминогрупп способствует также расщепле-
нию эфира в требуемом направлении.
1. Cook Н. G., Saunders В. С., Biochem. J., 41, 558 (1947).
2. Finger G. С., FinnertyJ. L., Biochem. Preps., 3, 120 (1958).
3. Sanger F., Biochem. J., 39, 507 (1945); 40. 261 (1946); 45. 563 (1949); Por-
ter-R. R., Sanger F., Biochem. .1., 42, 287 (1948).
4. Reinheimer .1. D., Doug lass J.P., Le i st erH., VoelkeIM.B.,
J. Org. Chem., 22, 1743 (1957).
5. Whalley W. B„ J. Chem. Soc., 1950 , 2241.
6. Weygand F., Lowenfeld R., Chem. Ber., 83, 559 (1950).
7 P i r k 1 e W. H., Z a b r i s k i e J. L., J. Org. Chem., 29, 3124 (1964).
8. Pirkle W. FL. Gates M., J. Org. Chem., 30, 1769 (1965).
9. S a w a Y. К,., T s u j i XL, Maeda S., Tetrahedron, 15, 144, 154 (1961).
ДИОКСАН, O(CH2CH„)2O. Мол. вес 88,10, т. пл. 11,8°, т. кип.
101,3°.
Очистка. Бенсон, Мак-Би и Рэнд| 1), согласно методике получения
N-иодсукцинимида, очищали Д. «только перегонкой над натрием»
[2]. Для очистки Д. для кинетических измерений Берштейн п Рин-
гольд 131 применили метод Физера 121 и затем хранили растворитель
над молекулярным ситом не менее недели. Физер |2| приводит две
методики очистки Д., причем первый метод позволяет удалить воду
и гликольацеталь гидролизом до ацетальдегида.
а. [41. Смесь 2л продажного Д., 27 мл конц. соляной кислоты и
200 мл воды кипятят 12 час, пропуская через смесь ток азота для уда-
ления ацетальдегида. После этого раствор охлаждают, медленно до-
бавляют при встряхивании гранулированный едкий кали до прек-
ращения растворения и расслоения жидкости. Д. декантируют,
снова прибавляют гранулированный едкий кали для удаления
механически захваченных капель водного слоя, декантируют в чис-
тую колбу п кипятят с натрием 10—12 час; при этом поверхность ме-
талла должна оставаться чистой. Затем Д. перегоняют над натрием
и хранят без доступа воздуха.
б. 151. Удаляют перекиси и уменьшают содержание альдегида,
пропуская Д. через колонку с окисью алюминия (80 г на 100—
200 мл растворителя). Это метод применим для растворов в эфире
и других безводных растворителях.
Недавно был предложен другой способ [61, заключающийся в
перегонке продажного Д. над алюмогндридом лития.
376
Эффект растворителя |7|. В качестве примера рассмотрим реак-
цию превращения амидов в тиоамиды:
О S
ii , p=s5
RCNR2 RCNR.
(R-алкил. арил; R'—H. алкил, арил)
В литературе описаны самые различные растворители для этой ре-
акции, в том числе пиридин, ДМФА, толуол, Д., хлороформ и т. д.
В большинстве случаев пиридин или ДМФА дают очень хоро-
шие результаты, но иногда образуются трудно отделяемые
смолы. Д. оказался наилучшим растворителем; он неизменно давал
высокие выходы (80—90%) легко очищаемых продуктов даже в тех
реакциях, в которых с другими растворителями вообще не удалось
получить никаких продуктов.
1. BensonW. R., МсВееЕ. Т., Rand L„ Org. Syn., 42, 73 (1962).
2. F i е s е г L. F., „Experiments in Organic Chemistry", 3rd ed., D. C. Heath,
Boston, 1955, p. 284.
3. BursteinS. H., R ingold H. J., J. Am. Chem. Soc., 86, 4952 (1964).
4. Hess K., Frahm H., Ber., 71, 2627 (1938); Eigenberger E., J. prakt.
Chem., 130, 75 (1931).
5. D a s 1 e r W., В a u er C. D., Ind. Eng. Chem., Anal. Ed., 18, 52 (1946).
6. Augustine R. L., C a p u t о J. A., Org. Syn., 45, 80 (1965).
7. Contributed by B. Loev, S. Kline and French Laboratories.
ДИОКСАНДИ БРОМИД, OC4HgOBr2. Мол. вес 247,93, т. пл.
64°, желто-оранжевый.
К диоксану при перемешивании и охлаждении прибавляют бром,
горячий раствор выливают в ледяную воду и выпавший осадок оран-
жевого цвета отфильтровывают и сушат 111. Полученный комплекс
очень летуч, и его следует хранить в сосуде с притертой пробкой.
Безводный реагент .может быть получен смешиванием раствора в дио-
ксане с петролейным эфиром при —20’ 121.
Д. применяется для контролируемого бромирования фенолов.
Например, 25 г Д. прибавляют при охлаждении к 9,4 г фенола и смесь
выливают в воду; экстракция эфиром дает n-бромфенол |1|. Ацетил-
ОН ОН
I I
// //\
I ||ОС4Н,ОВг.г| |1
НО /СОСНз
у \
I I 0С4Н,0Вг
7
I
Вг
НО. /СОСНоВг
Н3С Z H Н3С/ХН
377
карбинолы можно бромировать Д. в эфирном растворе при силь-
ном облучении красного раствора видимым светом [2|.
Раствор или суспензию Д., полученную добавлением брома к
избытку диоксана, используют |31 для контролируемого бромиро-
вания реакционноспособных соединений, например аннлин-'П-бром-
анилпн (выход 68%).
1. Яновская Л. А., Терентьев А. П., Беленькп й Л. И., ЖОХ,
22, 1594 (1952).
2. В i 1 1 i in о г i a J . D.. М а с 1 a g a n X. F., .1. Chem. Soc., 1954, 3257.
3. К о s о 1 а р о f I G. М., J. Am. Chem. Soc., 75, 3596 (1953).
H . OPO (OH)2
°;
ДИОКСАНДИФОСФАТ, | |
4O z
H XOPO(OH)2
Мол. вес 222,15, т. пл. 83—87', очень гигроскопичен, разлага-
ется водой; растворим в диоксапе, эфире, этаноле, этплацетате;
нерастворим в углеводородах.
Получение 111. По простейшей методике смешивают 1 моль
диоксаиа и 2 моля 88"»-ной водной ортофосфорной кислоты (темпера-
тура повышается до 60 ) и перемешивают до начала кристаллизации.
Маточный раствор отсасывают, используя резиновый колпак (см.
стр. 220), а кристаллы сушат в вакууме над едким натром. Перекри-
сталлизация 10 г комплекса из 20 мл эфира при —603 не изменяет
температуру плавления. Полагают 111, что Д. можно использовать
для очистки диоксана или вместо безводной фосфорной кислоты.
Фосфорилирование. По данным Чамберса и сотр. [2] Д. можно
использовать для получения нуклеозидпнрофосфорной кислоты [(3),
1? = аденин, урацил нт. д.]. Нуклеотид (1) при реакции с аммиаком и
дициклогекснлкарбодиимидом (ДЦК) превращается в 5'-фосфамид(2),
Дифосфат диоксаиа
(3)
378
который затем обрабатывают Д. в о-хлорфеноле (3 час, О3). Пнро-
фосфорную кислоту (3) осаждают петролепным эфиром и очищают
в виде солиаммонпя.Вы.ходыоколо 90%,тогда какпрн использовании
85%-ной фосфорной кислоты выходы составляют 50—60%. Однако
реакцию следует проводить в безводных условиях, а в случае сырой
погоды — в сухой камере.
1. Baer Е., J. Am. Chem. Soc., 66, 303 (1944).
2. Chambers R. W., Shapiro P., Kurkov V., J. Am. Chem. Soc.,
82, 970 (1960).
ДИТИОЦИАН (родан), (SCN)2. Мол. вес 140,19.
Д. получают добавлением 10%-ного раствора брома в четырех-
хлорпстом углероде к суспензии тиоцианата свинца (взятого в 10% -
ном избытке) в том же растворителе до обесцвечивания; раствор
фильтруют II]. Тиоцианат свинца готовят из нитрата свинца и
тиоцианата натрия.
По реакциям Д. до некоторой степени напоминает бром, за исклю-
чением каталитического действия. Прямое замещение атомов водо-
рода на тиоцнаногрунну, тноцианпрованне пли роданированпе, про-
текает только для ароматических аминов и фенолов, а также для
высокореакцнонноспособных углеводородов. Анилин превращается
в 4-тиоцианоанилин с 97%-ным выходом, о-толуидин в 4-тиоцнано-
2-метнланилин с выходом 80% и антраниловая кислота в 5-тпоцпа-
ноантраниловую кислоту с 80%-ным выходом. Фенолы тиоцпанп-
руются аналогично в /щра-положение, а если оно занято, то в орто-
положение.
NH2 NH3
J СООН J соон
//'у, // /
I
SCN
Углеводороды ряда бензола и нафталина не подвергаются тиоциа-
нированию. 3,4-Бензпнрен и метилхолантрен реагируют с Д. в че-
тыреххлористом углероде при 25; с образованием соответственно
5- и 15-тиоцианопроизводных (выходы приведены на схеме) 111.
Антрацен дает 9, 10-дитноциноантрацен с более низким выходом.
87%
379
Вторая реакция Д. заключается в присоединении. Циклогексен
присоединяет Д. с образованием 1, 2-днтиоцианоциклогексана с
73?'о-ным выходом (стереохнмическн реакция не изучена). Бутадиен
и изопрен дают 1,4-аддукты с выходом 80% и 19% соответственно.
СН, СН - СН = СН, т (SCN )2-СН, - СН — СН — СН,
SO% I I -
SCN SCN
Роданирование осуществлено для М.М-диметпланилина [2|, азу-
лена |3|, индола [4|. Первичные и вторичные амины дают с Д.
N-роданиды [5].
R2NH 4- (SCN),-----> R,NSCN
50- 70%
1. Органические реакции, ИЛ, М., 1950, сб. 2, стр. 431; Wood J. L., F ieser L. F.,
J. Am. Chem. Soc., 63, 2323 (1941).
2. Синтезы органических препаратов, ИЛ, М., 1949, сб. 2, стр. 431.
3. Anderson A. G, Jr., McDonald R. N., J. Am. Chem. Soc., 81, 5669
(1959).
4. Gr an t M. S, S n у d e r H. R., J. Am. Chem. Soc., 82, 2742 (I960).
5. Kuhn M., Mecke R., Chem. Ber.. 93, 618 (I960).
ДИ-п-ТОЛИЛКАРБОДИИМИД, n-CH3CeH4N-C-NCeH4CH3-n.
Мол. вес 222, 28, т. пл. 56’.
Получают окислением ди-п-толилтиомочевины желтой окисью
ртути [1, 21.
Присоединение спиртов. Д. количественно присоединяет спирты
в диоксане в присутствии оснований с образованием эфиров N.N’-дя-
п-толил-О-алкилнзомочевины (2) |21. Этот эфир (2) реагирует с диал-
килфосфатом (3), давая нейтральный третичный фосфат (4) и диарил-
мочевину (5). Дициклогексилкарбодипмид не присоединяет спирты.
or-
I
R'O-P-OH (3)
II
RONa О
ArN—C = NAr~bROH------* ArN = C - NHAr------------
DI !
OR
(1) (2)
OR' H О H
I I II I
—> RO - P-OR' + ArN —C—NAr
II
О
(4) (5)
Дегидратация сульфокислот. Под действием Д. ароматические
и алифатические сульфокислоты превращаются в ангидриды с высо-
ким выходом 131. Преимущество Д. перед дициклогексплкарбодипми-
дом состоит в том, что образующаяся в этой реакции мочевина
хуже растворяется в бензоле п легко отделяется от ангидрида.
380
Получение эфиров пирофосфорной кислоты. Д. реагирует с моно-
и диэфирами фосфорной кислоты при комнатной температуре, об-
разуя соответствующие симметричные ди- и тетраэфиры пирофэсфор-
ной кислоты 14). Выходы почти количественные. Дициклогексил-
карбодиимид реагирует аналогично, но выделение тетра-и-нитрофе-
нплпирофосфата представляет некоторую трудность, так как раст-
воримость этого эфира такая же, как у замещенной мочевины. При
использовании ди-/г-толилкарбодиимида в диоксане эти трудности
устраняются.
Нуклеозидфосфиты. Реакция нуклеозида с фосфористой кис-
лотой и Д. в пиридине приводит к образованию нуклеозидфосфита
с хорошим выходом |5). Так, смесь дезоксиаденозина и фосфористой
кислоты тщательно высушивают трехкратным выпариванием с пири-
дином при 10мм и затем растворяют остаток в сухом пиридине,
прибавляя Д. Выдерживают в течение 3 дней при комнатной темпе-
ратуре и выделяют в основном в виде аммонийной соли дезоксиаде-
нозин-5'-фосфит (42%) и в меньших количествах З'-фэсфит (29%).
1. Zetzsche F., Meyer Н. Е., Ov er beck Н., N er jcr W., Вег., 71,
1512 (1938); Zetzsche F., Nerger W., Ber , 73, 467 (1940).
2. К h о r a n a H. G., Can. J. Chem., 32, 227 (1954).
3. К h о r a n a H. G., Can. J. Chem., 31, 585 (1953).
4 К hor a na H. G., Tod d A. R., J. Chem. Soc., 1953, 2257.
5. Schofield J. A„ Todd A. R„ J. Chem. Soc., 1961, 2316.
ДИ-п-ТОЛУИЛ-Э-ВИННАЯ КИСЛОТА,
со„н
нсо - с — S— сн3
Z---X I || /
сн3 - " Д-- сосн о
li I
о СО2Н
Мол. вес 386, 34, т. пл. 169-171°, aD —141° (EtOH).
Д. получают 11, 21 перемешиванием смеси d-винной кислоты
с п-толуилхлоридом в течение 3 час в ксилоле, последующим разбав-
лением бензолом, фильтрованием образовавшегося кристаллическо-
го ангидрида и гидролизом его в дикислоту.
Эта оптически активная дикислота применяется для расщепления
dl -гидразида изолнзергиповой кислоты |1] и синтетических полупро-
381
дуктов [31. Она применяется также для идентификации аминов, так
как ее соли (главным образом кислые) являются стабильными твер-
дыми веществами с высокими температурами плавления [2, 4|.
Соли можно получать смешиванием эфирных растворов кислоты
и амина |2|. Амин можно извлечь встряхиванием раствора соли в сме-
си .хлороформ—метанол с водной щелочью 121.
1. Stoll A., Hofmann Л., Helv. Chim. Acta, 26, 922 (1943).
2. Kidd D. A. A., J. Cheni. Soc., 1961, 4675.
3. Woodward R. B. ct al., Tetrahedron, 2, 50 (1958).
4. Co 1 1 i n s R. I7., J. Client. Soc., 1960, 2053.
ДИФЕНИЛАЦЕТИЛЕН, C.l 1 С CC..H:,. Мол. вес 178,22, т. пл.
61". Д. получают из бензила превращением в бпе-гидразон и окисле-
нием последнего желтой окисью ртути в бензоле (67—7.3% в расчете
на бензил) [ 11.
Получение из /пранс-стильбена [21. шракс-Стильбен (20 г)
нагревают на кипящей водяной бане с 400 мл уксусной кислоты до
растворения, прибавляют 40 г иербромида бромида пиридиния,
нагревают смесь на кипящей водяной бане и сильно перемешивают
(до образования воронки) в течение 5 мин. /Иезо-дибромстпльбен
сразу начинает выпадать в виде жемчужных пластинок. После ох-
। ’ кон
о u ru 7---- и А и НО(СНЮН.СЛ,,Н
С6Н5СН Н—С—Вт 160°
НССсН5 87% ^Н-С—Вг 84%
СсН5
С„Н3
I
с
I!
С
I
СВН-,
лаждения осадок отделяют и промывают метанолом. Смесь этого
дибромида (32,4 г, т. пл. 236—237°), 65 г едкого кали и 130 мл три-
этиленгликоля энергично перемешивают при нагревании на откры-
том пламени, доводя температуру до 160', при которой начинает
осаждаться бромистый калий. Через 5 мин (при 160—170') реакция
заканчивается, и горячую смесь выливают в стакан, а колбу промы-
вают попеременно водой и этанолом. После охлаждения продукт
реакции собирают, промывают водой и сушат (16,5 г). Кристаллиза-
ция из 50 мл 95% -ного этанола дает Д. в двух формах: 11,8 г, т. пл.
61,5—62,5°, 2,4 г т. пл. 58—59=.
Применение Д. как диенофила можно иллюстрировать на примере
его реакции с тетрафенилциклопеитадиеноиом (см. Дильса —
Альдера реакция, растворители).
1. Соре А. С., SmithD.S., CottcrR.J., Org. Syn., Coll. Vol., 4, 377 (1963).
2. F i e s e r L. F„ Org. Syn., 46, 46 (1966).
2-ДИФЕНИЛАЦЕТИЛ-1.3-ИНДАНДИ0Н-1-ГИДРА30Н. Дол,
вес 354,39, т. пл. 305° с разложением.
Исходный ацилиндандион-1,3 (1) получают конденсацией диме-
тилфталата с метилбензгидрилкетоном в присутствии метилата нат-
382
рия [II. Гидразон (2) реагируете альдегидами и кетонами в условиях
кислотного катализа с образованием желтых или оранжевых сильно
(И
флуоресцирующих высокоплавких азинов, которые можно применять
для идентификации.
1. Braun R. Л., Mosher W. A., J. Am. Chem. Soc., 80, 2749, 3048 (1958).
2,3-ДИФЕНИЛБУТАДИЕН-1,3(2). Мол.вес206,27, т. пл. 46-47°.
Получение. Лучший метод [11 состоит в приготовлении раствора
ацетофенона в изопропиловом спирте в присутствии следов уксус-
ной кислоты, после чего колбу закрывают, переворачивают и выстав-
ляют на солнечный свет на несколько месяцев. Образовавшийся
2,3-дифенплбутандпол-2,3 (1,35 г) обрабатывают 105 .и.? ацетилхло-
рида и 20 мг N-фенпл-р-нафтиламина (ингибитор полимеризации).
С3Н5 свн5 свн5
| СН3ч ,ОН hv | | СНЭСОС1
Сх J-АсОН--------->-СН3С----ССН3---------*
so.. СН/ 4 И Ь6°“ | |
ОН он
После первоначально бурной реакции избыток ацетилхлорпда уда-
ляют в вакууме, реакционную смесь охлаждают и нейтрализуют вод-
ным раствором карбоната натрия. Неочищенный диен (2) экстраги-
руют хлороформом, сушат, выпаривают растворитель и получают
1,4-дибромид (3), т. пл. 144—147°. Это соединение устойчиво при хра-
нении. Д. по мере надобности можно получать из дибромида (3) (вы-
ход 92%) кипячением с цинковой пылью в ацетоне в течение 2 час.
Д. можно хранить в темноте в атмосфере азота в течение нескольких
недель; в иных условиях он быстро густеет и приобретает окраску.
Более быстрый метод получения Д. (2) (21 заключается в прибав-
лении к диметилсульфоксиду в атмосфере азота при перемешивании
2CH3SOCH3 + 2\aH (50% в масле) —2Д 2CH3SOCH,\a+4-2Н2
30 .4.1 2,4 г
CeH5C CC6H3 + CH3SOCH2Na— С,Н5С —СС6Н5
I! II
СН, СН,
383
гидрида натрия в масле; при этом образуется темно-серый раствор
метилсульфенилметплида натрия, к которому прибавляют по кап-
лям раствор дифепилацетилепа в ДМСО п нагревают смесь при 65°
в течение 2 час. Обработка реакционной смеси и кристаллизация из
метанола дает чистый Д. с т. пл. 47—48е (стабилен при хранении
в холодильнике в темноте около года).
1. А 1 1 е n С. F. Н., Е 1 i о t С. G., В е 1 1 A., Can. J. Research, 17В, 75 (1939).
2. Iwai I., I <1 е J., procedure submitted to Org. Syn.
траяс-т/>аяс-1,4-ДИФЕНИЛБУТАДИЕН. Мол. вес 206,27,
т. пл. 153'. Этот углеводород легко и быстро получается по схеме (1 ]:
ОС2Н5
(C2HS,O)3 Р | Кипячение 1 час
СвН6С.Н2С1------:---> СвН6СН.,Р<- (ОС2Н5), (СГ)------———>
н н
О-
—► CSH5CH2U (ОС2Н6),
свн6сн=снсно
NaOCHj, ДМФА
Реакция Дильса — Альдера. Синтез и-терфенила [1|. Исполь-
зование Д. в качестве диена в реакции Дильса — Альдера показано
на примере получения /г-терфенила:
С6Н5
I
//
I
//
I
QII5
С6Н5
\
X
I
с6н5
СО2СН3
I
с
с
I
со.,сн3
220-230’
транс-трин :• 1 4-Дифе-
нил бутадиен
(1, мол. вес 206,27)
Н„ zCeH5
X со,сн3
Ilf -
' " 'СО.,сн3
Н свн5‘
III, 98°, мол. вес
348,38
Н СО2СН3
! 1
: 'Н
z) СО2СН3
11. мол вес 1 4 2,11
V л-Терфенил
(VI, мол вес
230,29; 21 И
+ 2СО2
IV 170°
X
1. Org. Expts., Chapt. 24; see also McDonald R. Campbell?. W.,
Org. Syn., 40, 36 (1960).
384
1,1-ДИФЕНИЛГИДРАЗИН, (CeH5)2NNH2. Мол. вес 184,24, т. пл.
34, 5“. Д. получают из дифениламина превращением его в N-нитро-
замин и восстановлением цинковой пылью и уксусной кислотой в эта-
ноле [1, 21.
Д. реагирует с альдогексозами, давая ограниченно растворимые
и хорошо кристаллизующиеся дифенилгидразоны [2[. Д. приме-
няется для выделения дополнительного количества D-арабинозы
из маточного раствора при синтезе ее путем расщепления глюкозы
СНО CH = NN(CeH5)2 СНО
I I снго I
(СНОН)3 — (СНОН)з --------------> (CHOH)s + CH2 = NN(CeH5)2
I I I
СН2ОН сн„он СН2ОН
по Волю [31.
В то время как фенилгидразин реагирует с озонами (глюкозонами)
с образованием фенилозазонов, Д. избирательно взаимодействует с
сн=о CH = NN (СвН5)2
1 С = О H2NN(CeH6)2 1 С-0
(СНОН)3 (СНОН)3
1 СН2ОН 1 СН2ОН
альдегидной группой, давая кристаллический дифенилгидразон
озазона [41.
1. Fischer Е., Ann., 190, 174 (1877).
2. S t a h е 1 R„ Ann., 258, 242 (1890).
3. Б р а у н Г., «Синтезы органических препаратов», ИЛ, М., 1952, сб. 3, стр. 77.
4. HansekeG., L i eb е now W., Chem. Ber., 87, 1068 (1954).
ДИФЕНИЛДИАЗОМЕТАН, (CeH5)2CN2. Мол. вес 194,23, т.
пл. 32°.
Получение. Д. в жидком виде получают окислением гидразона
бензофенона желтой окисью ртути в петролейном эфире в течение
6 час [11. Улучшенная методика [2] основана на катализе этой реак-
ции основаниями; возможен следующий механизм реакции:
HgO +
(C„H5)2C = NNH2-> (CeH5)2C = NNH2HgO—»
он-
(C6H5)2 C=NNHHgOH —(CeH6)2 C = NNHgOH
— Н 2«Д
-»(CeH5)2C = N = N + Hg + OH~
Смесь 13 г гидразона бензофенона, 15 г безводного сульфата
натрия, 200 мл эфира, 5 мл насыщенного этанольного раствора ед-
13 Заказ Л» 965 385
кого кали и 35 г желтой окиси ртути встряхивают в течение 75 мин
в толстостенной склянке, завернутой в мокрое полотенце. Раствор
фильтруют, выпаривают и остаток экстрагируют иетролейным эфи-
ром. После удаления растворителя получают масло, кристаллизую-
щееся в темно-красные кристаллы с т. ил. 29—32 ; выход 89%.
Этерификация. Одно из применений Д.— этерификация карбо-
новых кислот для защиты карбоксильной группы или для получения
легко идентифицируемых производных. Даже простраиственноза-
трудиениые карбоновые кислоты с карбоксильной группой при
третичном атоме углерода реагируют с Д., образуя сложные эфиры
бензгидрола. Защитная группа при необходимости удаляется гидро-
генолизом |3|. Ацетилсалициловая кислота реагируете 1,5.адД. в
СО..СН (('.„11.-,)..
ОСОСНз
СО.,11
ОСОСНз
кипящем бензоле в течение 20мин (окраска изменяется от красно-фио-
летовой до желтой); при этом получается кристаллический беизгпд-
рпловый эфир (т. ил. 107 ) с количественным выходом. Для иденти-
фикации карбоновых кислот и анализа их смесей можно использовать
спектрофотометрическое определение скоростей реакций этих ве-
ществ с Д. в спиртовом или бензольном растворе (41.
Присоединение. Направление реакции Д. с с/., Р-непасыщсииымп
карбонильными соединениями было выяснено после тщательного
изучения реакции с метилакрилатом 151. Если реакционную колбу
промыть кислотой, прополоскать три раза дистиллированной водой
Основание
СН —ССО2СН,
N
I
Н
(3)
/СН2
(С4Н5)гС—CHCOjCH, + N;
(4)
38G
п высушить, то добавление раствора Д. в пентане или гексане при
постоянном перемешивании к раствору метилакрилата в том же
растворителе при 0—5J приводит к почти количественному выделе-
нию азота и образованию метилового эфира 2,2-дифенилциклопропан-
карбоновой кислоты (4) (выход до 89%). Очевидно, производное цик-
лопропана (4) получается при разложении первоначально образовав-
шегося пиразолина-1 (2). Однако если реакционную колбу промыть
раствором тринатрийфосфата, затем три раза дистиллированной во-
дой и высушить, то в результате реакции образуется с высоким вы-
ходом пиразолни-2 (3). При добавлении к реакционной смеси еще
и трпэтплампна перед введением Д.азот практически не выделяется,
а получается чистый 5,5-дифенил-3-карбометоксиппразолин-2 с вы-
(C6H5)zN2
С6н6 >
ходом 70% (3, т. пл. 140°, разл.). При добавлении Д. к нафтохинону-
1,4 первоначально образующийся аддукт еиолизуется в гидрохинон,
прежде чем успевают пройти какие-либо другие реакции [61.
Пептидный синтез. Сложные эфиры бензгидрола, получаемые
при взаимодействии Д. с N-защищенными аминокислотами, приме-
няются в пептидном синтезе, особенно для получения цистеиновых
пептидов [71. Эфиры бензгидрола по низкой устойчивости в кислой
среде можно сравнить с /ире/и-бутиловыми эфирами. Эти эфиры нель-
зя синтезировать прямым путем, однако их можно получить этери-
фикацией и-толуолсульфоната аминокислоты 181.
1. Спит Л., X о у а р Д К., «Синтезы органических препаратов», ИЛ, ЛЕ, 1952,
сб. 3, стр. 232.
2. М i 1 1 с г J. В., J. Org. Cliein., 24, 560 (1959).
3. Hardegger Е., Е 1 Н е w с i h i Z., Robinet F. G., Helv. Chim.
Acta, 31, 439 (1948).
4. Roberts J. D., R eg a n С. M., Anal. Chem., 24, 360 (1952).
5. Jone s W. M., Glenn T. H., Baarda D. G„ J. Org. Chem., 28, 2887
(1963).
6. Fie s e r L. F., Peters At. A., J. Am. Chem. Soc., 53, 4080 (1931).
7. H i s k e у R. G., A d a m s J. B., Jr., J. Am. Chem. Soc., 87, 3969 (1965).
8. A b о d e r i n A. A., Delpierrc G. R., Fruton J. S., J. Am. Chem.
Soc., 87, 5469 (1965).
а, у-^«с-(ДИФЕНИЛЕН)-р-ФЕНИЛАЛЛИЛЬНЫЙ РАДИКАЛ
(БДФА). Мол. вес 417,50, т. пл. 188—191 .
БДФА — стабильный радикал — получен Кельшем 111 реакцией
соответствующего хлорида со ртутью и Куном и Нойгебауэром [21
13* 387
по схеме:
БДФА
Радикал кристаллизуется из бензола в виде зеленого комплекса 1 : 1
(т. пл. 231—233°) или из уксусной кислоты (без включения раст-
ворителя) в виде коричневых игл (т. пл. 188—19Г). БДФА в высокой
степени диссоциирован, но тем не менее очень устойчив к действию
кислорода. Интересно, что первоначальная статья с описанием этого
вещества, направленная Кельшем в печать в 1932 г., была отклонена
из-за его несоответствия, по мнению рецензента, свойствам свобод-
ного радикала. Из всех доступных радикалов БДФА оказался наи-
более подходящим для улавливания радикалов при разложении цик-
логексилформил- и изобутироилперекисей (31.
1. К о е 1 s с h С. F., J. Am. Chem. Soc., 79, 4439 (1957).
2. Kuhn R., N eugeb a uer F. A., Monatsh., 95, 3 (1964).
3. Lamb R. С., P a c i f i c i J. G., А у e r s P. W., J. Am. Chem. Soc., 87,
3928 (1965); see also Lamb R. C., S p a d a f i n о L. P., Webb R. G.,
S m i t h E. В., P a c i f i c i J. G., J. Org. Chem., 31, 147 (1966).
1,3 - ДИФЕНИЛИЗОБЕНЗОФУРАН (2,5 - дифенил - 3,4 - бензофу-
ран). Мол. вес 270,31, т. пл. 13Г.
Получение. Один из двух возможных путей синтеза этого сое-
динения включает восстановление о-бензоилбензойной кислоты до
фенилфталида (2), реакцию с фенилмагнийбромидом и осторожную
кислотную дегидратацию (3) с образованием ярко-желтого Д. (4)
[11. По методике, разработанной Ньюменом [2], превращение (1)
в (2) проводят с выходом 87%, а реакцией Гриньяра и дегидратацией
получают Д. с выходом 87%. По второму способу [3] бутадиен при-
соединяют к транс-Х,2-дибензоилэтилену. Полученный аддукт бро-
мированием и дегидробромированием превращают в о-дибензоилбен-
зол (7), из которого конечный продукт (4) получают частичным вос-
становлением либо активированным цинком в этанольном растворе
388
гидроокиси натрия [31, либо боргидридом калия [4] и последующим
подкислением.
СОС6Н5
сн
II
НС
СОС6Н5
(6) (7)
Улавливающий агент. Д.— высоко реакционноспособный агент
по отношению к диенофилам, так как при этом взаимодействии аро-
матизуется о-хиноидный цикл. Д. реагирует с большим числом раз-
личных этиленовых диенофилов 151. С ацетиленди карбоновой кис-
лотой Д. дает биг-аддукт (8) даже в присутствии большого избытка
кислоты 161.
С6н5
СО2СН3
СО2СН3
С диметиловым эфиром ацетилендикарбоновой кислоты образует-
ся исключительно аддукт состава 1 :1 (9). Оба аддукта (8) и (9) были
получены с высоким выходом.
Эффективность Д. как улавливающего агента можно показать на
примере его реакции с дегидробензолом, которая приводит к аддукту
(10) с выходом 85% 17]. Как улавливающий агент Д. применялся для
доказательства промежуточного образования сильно напряженных
389
циклоалкинов [8] и бензоциклобутадиенов 191.
НцО
С6н5
1. Guyot Л., Cat el J., Bull. soc. chim. France. |3], 35, 1124 (1906).
2. X e w in a n M. S., J. Огд. Cliein., 26, 2630 (1961).
3. A cl a in s R., Gold M. II.. J. Am. Chem. Soc., 62, 56 (1940).
4. C a v aM. P„ Mitchell M. J„ Dean a A. 11., J . Ore. Chem., 25, 1481 (1960).
5. X or t о n J. A., Cliein. Rees., 31, .319 (1942).
6. В er son J. A., J. Ain. Chem. Soc., 75, 1240 (1933).
7. W i t t i д G., К n a u s s E., X i e t li a in m e r K.. Ann., 630, 10 (1960).
8. \V i t t i g G„ Krebs A., Chem. Ber., 94, 3260 (1961); W i t t i ц G., Pohl-
ke R., Chem. Ber., 94, 3276 (1961),
9. C a v a M. P., Pohl k e R., J. On*. Chem., 27, 1564 (1962); X e n i t z e s-
c и C. D. et al., Ann., 653, 79 (1962).
ДИФЕНИЛИОДОНИЙБРОМИД, (С„Н5)Д+Вг~. Мол. вес 361,03,
т. пл. 208°, разлагается. Получают из иодозобепзола (11.
Д. широко используется как фенилирующий агент; например,
он реагирует с фенолятом калия с образованием дифенилового эфи-
ра (76%) и нодбеизола и фенилирует карбанионы по атому углеро-
да 121.
1. В е г i n е е г F. М. et al., .1. Am. Chem. Soc., 75, 2705 (1953).
2. В e r i n у e r F. M. et al., .1. Am. Chem. Soc., 75 . 7708 (1953); 84, 2819 (1962);
Mayer W. et al., Ber., 93, 2761 (1960).
ДИФЕНИЛ ИОДОНИЙ-2-КАРБОКСИЛАТ (МОНОГИДРАТ).
Мол. вес 341,13, т. пл. 220 (с разд.).
Моногидрат Д.— стабильный и безопасный реагент, применяю-
щийся для получения дегидробензола в апротонных растворителях.
Реакцию проводят в присутствии реакционноспособного диена, свя-
зывающего образовавшийся дегндробепзол 111. Ранее предложенная
методика |2| была улучшена 131, и 2 г о-иодбензойной кислоты мож-
OSO2OK
с6н5
\ J1 НН4ОН^
со2н
390
но превратить в 2,1 г моногидрата Д. за 1,5 — 2 час. Синтез 1,2,3,
4-тетрафенпл11афталпна осуществляют нагреванием смеси моногид-
рата Д. (небольшой избыток) и тетрафенилцнклоиентадпеиона
в дпметпловом эфире трпэтиленгликоля при 200—205 до исчезнове-
ния фиолетовой окраски (3—4 мин) или кипячением реагентов в сме-
си м- и /(-диэтилбензолов в течение 45 мин. Выход углеводорода 92%,
моногидрат Д. используется на 69%.
1. L е G о 1 f Е., J. Am. Chem. Soc., 84, 3786 (1962); sec also BeringerF. M.,
И u а и g S. J„ ,1. Org. Chem., 29, 445 (1964).
2. Beringer F. At, L i 1 1 i e n I., .1. Ant. Chem. Soc.. 82, 725 (1960).
3. Org. Expts., Chapt. 61; F i e s e г L. F., Haddadin M. L., Org. Svn., 46,
107 (1966).
ДИФЕНИЛИОДОНИЙХЛОРИД, (С„Н ,)Л СГ. Мол. вес 316,57,
т. пл. 228—229'.
Получение [11. Смесь бензола, йодата калия, серной кислоты
и уксусного ангидрида перемешивают до завершения реакции и за-
тем разбавляют водой. Смесь экстрагируют эфиром; при добавлении
водного раствора хлористого аммония Д. выпадает в осадок (выход
63%).
Фенилирование анионов. Д. фенилирует кислые ди- и трикетоны
с высоким выходом [21. В качестве растворителя применяется трет-
бутанол; металлический натрий используют для получения анионов.
Для этой реакции предложен следующий механизм:
СНС6Н,
(СН3)зСОН
(С6Н5)Д С1
8 5%
+ C6HsI
Хаузер [31 нашел,чтоД. можно применять дляфенплпрованпяметиль-
ной группы ацетилацетона, который при обработке амидом натрия
в жидком аммиаке дает промежуточную соль (2). При соотношении
соли (2) и Д. 2 : 1 выход составляет 92%, но при соотношении 1 :1
падает до 47?о. Очевидно, соль (2) превращается в фенплпрованную
соль (3), которая в результате обменной реакции с (2) и образует
391
динатриевую соль (4):
О О О О
|| II 2NaNH2 II- II.. (CeH5)J + CI-
СН3ССН,ССН3-----—-СНХСНССН., ------— ------<-
NH3 \a+W -c'Hs‘
(1) (-)
О О О О
II - II (2) II _ II _ Н.О
—+ сн3сснссн,с6н5----> сн3сснсснс6н5 ——
\’я+ \:я+ Мя + 92°°
Ла iNd (общий)
(3) (4)
О о
II II
—+ СН3ССН2ССН2С6Н5
(5)
1. BeringerF. М., G е е г i n g Е. J., Kuntz!., Mausner Л!., J. Phvs.
Chem., 60, 141 (1956).
2. Beringer F. M., F о r g i о n e P. S., Y u d is M. D., Tetrahedron, 8, 49
(1960); BeringerF. M., G al tonS. A., H u a ngS. J., J. Am. Chem. Soc.,
84, 2819 (1962).
3. Hampton K. G., Harris T. Л4., Hauser C. R., J. Org. Chem., 29,
3511 (1964).
ДИФЕНИЛКАРБАМИНОИЛХЛОРИД, (C6H5) NCOCl. Мол. вес
231,68, т. пл. 86J.
Д. может быть использован для введения карбоксильной группы
в ароматические соединения, например в полистирольные смолы [1].
Ацилирование по Фриделю — Крафтсу дает амиды, которые при
О О
II Aid II Гндполиз
(C6H5)2NCC1 + HAt —LA (CGH5)2 NC—Аг-------->
о’
II
--> (С6Н6), NH + НОС—Аг
гидролизе образуют ароматические кислоты.
1. L е t s i n g e r R. L., К о r n e t M. J., J. Am. Chem. Soc., 85, 3045 (1963).
ДИФЕНИЛКАРБОДИИМИД, C6H5N=C=NCcH5. Мол. вес
194,23, т. кип. ПО—11270,2 мм.
Эффективный метод синтеза ароматических и алифатических кар-
бодиимидов основан на реакции изоцианата с 1-окисью 1-фенил-З-ме-
тилфосфолена-3, являющимся наиболее доступным соединением этого
типа [1].
г c6h5n=c=o
О +/-/СНз
C6hZ\J
--72.93%--» C6H5N=c=NC6H5 < СО;
1. Campbell Т. W., М о n a g 1 е J. J., Org. Syn., 43, 31 (1963).
392
ДИФЕНИЛКЕТЕН, (СвН6)2С=С=О. Мол. вес 194,22, т. кип.
12073,5 мм.
Получение. Впервые Д. был получен Штаудингером [1] обра-
боткой а-хлордифенилацетилхлорида цинковой стружкой в эфире
с очень низким выходом. В настоящее время этот хлорангидрид лег-
ко получают действием пятихлористого фосфора на бензиловую кис-
лоту(1) [21. Штаудипгер пыталсядегидрогалогенироватьдифенила-
цетилхлорид (2) трипропиламином или хинолином [3], но позднее [41
PCI-, Zn
1) (CeH5)2CCO2H (С0Н5)2С-С = О (CGH5)2C = C = O
ОН С1 С1
Основание
2) (СеН5)2С-С = О----------► (С6Н5)2С = С = О
I I
Н С1
отдал предпочтение методу, разработанному Шретером [5]. Этот ме-
тод описан в сб. «Синтезы органических препаратов» [61. Моногидра-
3) С6Н5С- СОС6Н5
II
NNH.,
HgO, с.н,
CaSO4, 35°
Г С6н5
I
CGH5C—С = О
(С6Н5)2С = С=О
зон бензила окисляют желтой окисью ртути в бензоле в присутст-
вии дегидратирующего агента и отфильтрованный раствор прибав-
ляют по каплям в колбу, нагретую до 100—110°, для разложения
диазокетона и удаления бензола. Полимеризация Д. ингибируется
в присутствии следов гидрохинона; Д. хранят в атмосфере азота.
Была предложена модификация второго метода Штаудингера
(Тэйлор и сотр. [7]). Дифенилацетилхлорид получали (выход 82—
94%) реакцией кислоты с хлористым тионилом в эфире с последую-
щим добавлением по каплям (охлаждение льдом, атмосфера азота)
раствора триэтиламина в эфире. Д. получен перегонкой при 118—
119°/1 мм с выходом 78—84% . В этом методе используются дешевые,
нетоксичные исходные вещества, он состоит из двух простых опера-
ций. Кроме того, за исключением последней перегонки, Д. ни разу
не нагревается выше 30—35°, что сводит полимеризацию к минимуму.
Пептидный синтез. Для получения Д. для пептидного синтеза
(Лосс и Демут [8]) был использован метод Штаудингера [II. Карбо-
бензоксиаминокислоту обрабатывали кетеном и эфиром аминокисло-
ты в ТГФ в присутствии триэтиламина. Выход составлял около 50%;
393
оптическая активность сохранялась. Смешанный ангидрид, полу-
R R О О
I (C.HS)3N I 'I "
СЬХНСНСО2Н-гО = С = С(СаН5)2 —----* СЬХНСНС-О—ССН (СвН5), —►
R' R R'
' 1 I
h2nchcox;H^ Cb\HCHCO\HCHCO.,C2H3 + HO.,CCH (С6Н5).,
ченнып из N-ацнламинокислоты (1) п Д., например (2), при реакции
с фенолом дает оптически чистый ариловый эфир (3) 19 J.
О
" ТГФ
Но\ССН.,СН.,СНСО.,Н-гО = С = С (СвН5),->
НХСОСН2С8Н5
(1)
о о о ____
II н 'I но - '_у - \ Oj
—* Н,ХССНХН,СНС—О—ССН (СвН-,)п----:---—<
I 60
Н\СОСН»С6Н5 (общи»)
II
о
(2)
ОС) о
II !' z------X II
—> Н2ХССН„СН,СНС—О-/ ХО24-НОССН (С„Н6)а
НХСОСН,СвН5
II
о
(3)
1. Staudinger Н., Вег., 38, 1735 (1905).
2. Bill in а п J. Н., Н i d у Р. Н., J. Am. Chem. Soc., 65, 760 (1943).
3. Staudinger Н., Ber., 40, 1148 (1907); 44, 1619 (1911).
4. Staudinger H., Вег., 44, 1623 (1911).
5. Schroeter G., Ber., 42 , 2346 (1909).
6. Смит Л., X о e u X., «Синтезы органических препаратов», ИЛ, Л1., 1952,
сб. 3, стр. 234.
7. Т а у 1 о г Е. С., М с К i 1 1 о р А., И awks G. Н., procedure submitted to
Org. Syn.
8. Losse G., Demutli E., Chem. Ber., 94, 1762 (1961).
9. E 1 m о г e D. T., S m у t h J., Proc. Chem. Soc., 1963, 18.
ДИФЕНИЛ КЕТЕНА/z-ТОЛИЛ ИМИН, (CH,)/. C --NCeH4CH3-n.
Мол. вес 283,36, т. пл. 83,5 ’, желтый.
394
Получение 111:
О
PCI, H.NC0H.CH3-n
(СеН5)2ССО2Н—(C,H5)2CCC1 ---------->
I 8,% I
ОН Cl
он
I’ I PCI, Nal. ацетон
-> (C6H5)2CCXC6H4CH3-n —(C6H5).,C—C = _\'C6H4CH3-n
I O/°(1 I I 83.0%
Cl CI CI
—> (C6H 5)2C = C X - { - CH3
Реакции. Д. т. реагирует с карбоновыми кислотами, давая
имиды [21. Д. т. превращает янтарную и фталевую кислоты в ангид-
O О
Н I!
(CeHs).,C=-C- ХАгТ RCOH —- (С„Н5),СНС—ХАг
RCO
риды с незначительным выходом. Так же как и дициклогексилкарбо-
дпимпд, этот кетимин можно использовать для синтеза пептидов
131.
1. S t е v е n s С. L., F г е n с h J. С., J. Am. Chem. Soc., 75, 657 (1953).
2. StevensC. L., M u n k M. E., .1. Am. Chem. Soc., 80, 4065 (1958).
3. StevensC.L,, M u n k M. E., J. Am. Chem. Soc., 80, 4069 (1958).
N, М-ДИФЕНИЛ-М-ПИКРИЛГИДРАЗИЛ (радикал, см. 4).
Мол. вес 394,32, т. пл. 137—138°, фиолетовый.
Получение. Этот стабильный, темно-фиолетовый радикал полу-
чают конденсацией 1,1-дифенилгидразина с пикрилхлоридом [11
и окислением гидразина активной двуокисью свинца по Куну и
Хаммеру [21. Очистка [31.
О2Х\
2 (СвЩ),ХХН„ + С1 — ХО2 —
\ - /
O2NZ
(1) (2)
Н О2хх
— (СеН5).,ХХ--X (CeH5)2XXHsC1
(3) СйХ-7
%ьо2
О2ХХ
(С8Н5)2Х'Х V хо2
O.,\z
(4)
395
Дегидрогенизация. Д. может быть использован для переноса
водорода (Линстед [4]). Реакция протекает очень быстро в кипящем
СНС13 или СС14 и может сопровождаться изменением окраски от
Н
I
ХН2 + 2 (С6Н5),\\Аг —> X + 2 (CcH6).,NXrAr
темно-фиолетовой до красной или бледно-оранжевой. Гидразин осаж-
дается почти количественно. Выходы хорошие и иногда достигают
90—95%.
Полагают (Бартлетт и Куарт [51), что этот радикал является иде-
альным ингибитором цепных радикальных реакций, однако показа-
но (Хэммонд [61), что реакция Д. с азодиизобутиронитрилом несте-
хиометрична и чувствительна к кислороду.
Дегидрирование соединения (5) до (6) проведено при кипячении
с избытком Д. в бензоле в течение 72 час (Дауд |7|).
1. Goldschmidt S., Renn К., Вег., 55, 628 (1922).
2. Kuhn R., Н a m m е г I., Chem. Вег., 83, 413 (1950).
3. L у о n s J. A., W a t s о n W. F., J. Polymer. Sci., 18, 141 (1955).
4. В г a u d е Е. А., В г о о k A. G., L i n s t е a d R. Р., J. Chem. Soc., 1954,
3574.
5. В а г t 1 е t t Р. D., Kwart Н., J. Am. Chem. Soc., 72, 1051 (1950).
6. Hammond G. S., Sen J.N., BoozerC. E., J. Am. Chem. Soc., 77, 3244
(1955).
7. Dowd P., Chem. Comm., 1965 , 568.
ДИФЕНИЛСИЛАН, (C6H6)2SiH2. Мол. вес 184,31, т. кип. 75—
76°/0,5 мм.
Д., полученный восстановлением дифенилдихлорсилана алюмо-
гидридом лития [1| (лучше всего в ТГФ [2]), восстанавливает неко-
торые диарилкетоны до соответствующих углеводородов при кипя-
чении (температура ~260°). Бензофенон дифенилметан (37?6);
антрахинонантрацен (45%) [2].
1. BenkeserR.A., Landesman Н., F о s t е г D. J., J. Am. Chem. Soc.,
74 . 648 (1952).
2. Gilman Н., D i е h 1 J., J. Org. Chem., 26, 4817 (1961).
ДИФЕНИЛСТАННАН, (C6H5),SnH2. Мол. вес 298,94, т. пл.
—20°.
Д. получают восстановлением дифенилдихлоролова алюмогидри-
дом лития в эфире [1]. Д. можно выделить кристаллизацией из сме-
си петролейный эфир — хлористый метилен. Его раствор в бензоле
умеренно стабилен при комнатной температуре.
396
Д. восстанавливает альдегиды и простые кетоны до спиртов при
уС = О + (CeH5)2SnH2 —* ^СНОН + (CeHe),Sn
комнатной температуре с выходом около 80%. В отличие от восста-
новления гидридами металлов в данном случае не требуется стадии
гидролиза [2]. Образующееся дифенилолово нерастворимо в эфире
и легко отделяется.
1. К u i v i ) а Н. G., Sawyer А. К., Armour A. Q., J. Org. Chem., 26,
1426 (1961).
2. К u i v i 1 а Н. G., В е u m е 1 О. F., J. Am. Chem. Soc., 83, 1246 (1961).
ДИФЕНИЛСУЛЬФОКСИД, C6H5SC6H5. Мол.вес 202,27, т.пл. 71°.
о-
Хотя Д. при комнатной температуре твердое вещество, он был
предложен в качестве растворителя для изучения автоокисления
слабокислых углеводородов (толуол, ксилол) в присутствии трет-
бутилата калия при 100° [1]. В этих условиях ДМСО быстро окисля-
ется и непригоден в качестве растворителя.
1. Wallace Т. J., Schriesheim A., Jacobson N., J. Org. Chem.,
29, 2907 (1964).
ДИФЕНИЛТЕЛЛУРИД, (С6Н5)2Те. Мол. вес 281,81, т. кип.
312—320°, разл., 174710 мм.
Д. (1) получают [1] по реакции теллурдибромида с фенилмагний-
бромидом в эфире и очищают, превращая в кристаллический, жел-
тый дифенилтеллурдибромид (2, т. пл. 199—202°). Д. регенерируют
2C6H5MgBr-|-TeBr2 —> (C6H5)2Te + 2MgBr2
О)
(CeH6)->Te + Вг2 >• (С6Н5)2ТеВг2
(2)
реакцией дибромида (2) с бисульфитом натрия.
Вследствие сродства к брому Д. применяется для дебромирова-
ния вицинальных дибромидов [2], например:
С6Н5СНВгСНВгСО2Н +(С0Н3)2Те —>• CSH6CH = СНСО2Н + (С6Н5),ТеВг2
Однако для дибромида холестерина реакция протекает довольно
плохо.
1. Lederer К-, Вег., 46, 1345 (1915).
2. de MouraCampos М., PetragnaniN., Thome С., Tetrahedron
Letters, 15, 5 (1960).
N,N'-ДИФЕНИЛФОРМАМИДИН, C6H5NHCH=NC6H5. Мол.
вес 196, 25, т. пл. 141°. Получают из анилина и муравьиной
397
кислоты 111:
С0Н,\Н2 1IOCH =--= о + Н2\С„Н5 —7^2 С„Н5Хнсн = \сен5
С соединениями, имеющими активные метиленовые группы, на-
пример с ацетплаиетоном, Д. конденсируется с выделением анилина
121:
z сосн,
-соси, ZCOCH3
CjH-XHCH - --------C.(1Hr,XHCH —C- НДСД
140' ' 'COCH.,
Синтез 2,4-дпоксппзофталевого днальдегпда (4) по усовершенст-
вованному (Куп, Штааб 111) методу Шесмита и Холдейна 131 вклю-
чает конденсацию Д. с резорцином, который, вероятно, реагирует
в дпкетонной форме (2). Реагенты нагревают в вакууме для удале-
ния образующегося анилина и уменьшения окисления воздухом.
Основание Шиффа (3), имеющее красный цвет, гидролизуют разб.
щелочью до диальдегида (4). Общий выход 2,4-диоксипзофталевого
СН =-з хс„н5
। ОН
i' | +2Сг,Н5\Н2
11 /
СН=-\СЙН5
ОН
(3) \
ХаОн\ сно
2 г»
сно
он
днальдегпда составляет 21—24%.
(4)
1. К и 11 и I?., St a :i b 11. A., Chem. Bit., 87, 272 (19.54).
2. D a i и s 1;. В., Вег., 35, 2504 (1902).
3. S 11 о е s нт i t h J. В., H a I <1 ane J., J. Chem. Soc., 1923, 2704.
О
ДИФЕНИЛФОСФАТ СЕРЕБРА, (свНг,О)2Р OAg. Мол. вес 357,05.
Получение [11.
Постернак |2| использовал Д. с. для синтеза 1-фосфата а-о-глю-
козы (4). Тетра-О-ацетпл-а-п-глюкопирапозилбромпд (1) обычно
реагирует с обращением конфигурации у С,, но при взаимодейст-
вии с Д. с. конфигурация сохраняется. Эфир фосфорной кислоты
398
(4) выделен в виде кристаллической калиевой соли.
1. Posternak Т., J. Biol. Cliein., 180, 1269 (1949).
2. Posternak .1., J. Лпт. Chem. Soc., 72, 4824 (1950).
ДИФЕНИЛФОСФИНЛИТИЙ (2). Эго соединение получают из
дпфеннлфосфппа (1) и бутиллития в ТГФ 111. При кипячении в те-
чение 4 час с Д. в ТГФ анизол дезалкилируется с хорошим выходом
ТГФ -
(С6Н-).,РН -1 «-BuLi-г (C(iH-,).,PLi -j- w-BuH
(I) (2)
[21. В этих условиях выход фенола из фенетола составляет всего 911 о.
осн3 он
I I
I ||--}-(C6H5).2PLi+ .. .'ГГФ; HaO_iZ || + (С6Н5)2РСН3
I II кипячение -1 час \ Н
'Ч/ 'Ч/
7 Д о' 7 О '
• ° 0 ‘ О , о
Поэтому в случае смешанного эфира можно осуществить селектив-
ное деметилирование без затрагивания этильной эфирной группы.
Бензиловые и аллиловые эфиры расщепляются этим реагентом.
1. Witten b е г " D., G i 1 m а n Н., J. Or". Chem., 23, 1063 (1958'.
2. MannF.G., Р г a g n е 1 1 М. J., Chem. Ind., 1964, 1386.
ДИФЕНИЛФ0СФ0РИЛИ30ТИ0ЦИАНАТ (2). Мол. вес 291,26,
л. кии. 210'0,1 мм, уд. вес 1,29.
21. получают реакцией дифенилхлорфосфата (1) с тиоцианатом
калия в ацетонитриле; при этом сразу осаждается хлористый калий.
О О
" il
(CeH5O)2PCl -I К XCS —- (С„Н3О).,Р \ S KC1
(1) (-’)
Д. реагирует с трпэтиламмоипевымп солями N-ацплпрованных
пептидов с образованием смешанных ангидридов, разлагающихся
до замещенных ацилтпогидантоинов, по которым можно идентифи-
цировать аминокислоту, находящуюся па карбоксильном конце
пептидной цени 111.
399
1. К e n n е г G. W., К h о г a n а Н. G., S t е d m a n R. J., J. Chem. Soc., 1953,
673.
О
ДИФЕНИЛХЛОРФОСФАТ, (C6H5O)2PC1. Мол. вес 268,63, т. кип.
147—14871,3 мм.
Получают нагреванием смеси 1,1 моля хлорокиси фосфора и
2 молей фенола до 180° с последующей перегонкой продукта реак-
ции [1]. Д. применяют для получения 3-фосфата оь-глицеринового
альдегида [1], фосфата диоксиацетона [21, а также дифенплфосфорп-
лизотиоцианата (см. стр. 399). Д. используется для синтеза моноал-
килфосфатов; он (1) реагирует со спиртами в пиридине с образованием
ООО
II HOR II H..Pt II
(С6Н5О),РС1--<- (СвН5О)2Р —OR-> (НО)2Р—OR
(1) (2) (3)
триэфира (2), фенильные группы которого отщепляются гидрогено-
лизом. Бригль и Мюллер [3] впервые применили это свойство Д.
для синтеза фосфатов глицерина и фруктозы; другие примеры ис-
пользования Д. рассмотрены в обзоре Брауна [4].
Пептидный синтез. В новом методе пептидного синтеза (Зервас
[5]) эфир аминокислоты защищают, превращая его в дибензилфосфо-
роильные производные (1), после чего эфирную группу удаляют ще-
лочным гидролизом. Реакция кислоты (2) с Д. дает смешанный ангид-
рид (3), который конденсируется с бензиловым эфиром аминокисло-
О ОН
II h2nchrco2ch3 II | он-
(ЛгСН20).,Р-С1------------> (ArCH2O)2P-NCHRCO2CH3-------
[Ar = n-N02C,H,] (I)
0 Н °
II | (СЙН5О)2РС1
— (АгСН2О)2Р - NCHRCO2H —— -+
(С «п 5)3 IN
(2)
ОН 0 0
II | II II H2NCHR'CO..CH.C«H,
(ArCH2O)2P —NCHRCOP (OCGH5)2------11------
(3)
ОН он
II I II I Н.. Pd
—>• (ArCH2O),P —NCHRC—NCHR'CO2CH2CeH5 —--у
(4)
ОН он
II I II I
—»(НО)2Р—NCHRC—NCHR'CO.,H + 2CH3C6H4\H., + CcH.-,CH3
(5)
-НзРО- Н20
► H»NCHRCONHCHR'CO,H
(Н + )
(6)
400
ты в производное дипептида (4). Гидрированием удаляют все бен-
зильные группы и получают N-фосфорилированный пептид (5), ко-
торый в кислой среде дефосфор ил ируется до пептида (6). Связь Р —N
в О-защищенных фосфамидах (2), (3) и (4) при комнатной температу-
ре устойчива к действию кислот и щелочей, тогда как эта же связь
фосфамида (5) с незащищенной карбоксильной группой стабильна
в щелочах, но расщепляется кислотами даже при pH 4.
1. Baer Е., Biochem. Preps., 1, 50 (1951).
2. В а 1 1 о u С. Е., Biochem. Preps., 7, 45 (1960).
3. Rrigl Р„ М й 1 1 е г Н„ Вег., 72, 2121 (1939).
4. Бра у п Д. М., «Успехи органической химии», изд-во «Мир», М., 1966, т. 3,
стр. 79.
5. С о s in a t о s Л., Р h о t a k i I., Z е г v a s L., Chem. Ber., 94, 2644 (1961).
ДИФЕНИЛЦИНК, (C0H5)2Zn. Мол. вес 219,58, г. пл. 107°.
Получение, а. Из дифенилртути. ДифенилртуТь, очень токсич-
ное вещество ст. пл. 121 —123°, получают реакцией бромбензола
с 3%-ной амальгамой натрия в ксилоле [1]. Смесь дифенилртути
2С8Н5Вг J \a.,Hg (C0H5)2Hg-j-2NaBr
(C0H5)2Hg-|-Zn---> (CeH5).,Zn 4-ZnHg
и цинковых опилок кипятят в ксилоле в атмосфере углекислого газа,
не содержащего кислорода. Этот же газ применяют при фильтро-
вании и промывании кристаллического вещества [2].
б. Более удобный метод основан на реакции фениллития и возог-
нанного бромистого цинка в эфире [31.
2CcH5Li 4-ZnBr2—> (C0H5)2Zn + 2LiBr
Реакция с солями диазония. Кертин и Твитин [31 нашли, что Д.
(независимо от способа получения) с борфторидом фенилдиазония
в ДМФА дает /ирпйс-азосоедппения с высоким выходом. Однако
V \ \ (BF4-) + (CeH6)2Zn
' ' 97%
ч=/ N-N\y—ч,
если Д., полученный методом (а), взят в большом избытке, то обра-
зуется преимущественно цис-изомер.
1. Калвер и, «Синтезы органических препаратов», ИЛ, М., 1949, сб. 1, стр. 204.
2. К о ч е ш к о в К. А., Несмеянов А. Н., Потросов В. И., Вег.,
67, 1138 (1934).
3. Curtin D. Y., Т v е t е п J. L., J. Org. Chem., 26, 1764 (1961).
N, N-ДИФЕНИЛЭТИЛЕНДИАМИН. Мол. вес 212,29, т. пл.
67,5°.
401
О получении и применении Л- в качестве специфического реагента
для альдегидов см. |1|; кетоны с Д. не взаимодействуют. Раствор
Д. и альдегида в метаноле в присутствии следов уксусной кислоты
CH.,NHC6H6
| ’ + RCHO
СН.ЛНС0Н5
сн,он
АсОН
СИ, —Nz
С6Н5
| CHR
СН_хч
Xhs
дает кристаллический осадок имидазолидина через 1—2 мин. Про-
изводное формальдегида плавится при 1263. Для регенерации аль-
дегида это производное встряхивают с 10?о-ной соляной кислотой
и альдегид экстрагируют эфиром.
1. W a n z 1 i с k II.-W., L б с h е 1 W., Chem. Вег., 86, 1463 (1953).
ДИФТОРАМИН, HNF,. Мол. вес 53,02, т. кип. — 233.
Получение. Исходным веществом для синтеза Д. является тет-
рафторгидразин, который получают реакцией трехфторпстого азо-
та с различными металлами при З753 (1) [1 ]. Д. получают нагрева-
нием тетрафторгидразпна с тиофенолом в вакууме; выход 74% 121.
1) 2XF3М — F..X — XF-PMF.,
2) F.,X-XF., J C0H5SH — 2HXF.,-;-C(;H-,SSC(iH5
Дезаминирование. Д. позволяет осуществлять прямое деза-
минирование первичных алифатических и ароматических аминов,
например:
Зн-С,Н9ХН., -i- НXF2 —* 2н-С4НяХH3F " + X., -l h.Qaн,„
Реакции конденсации. Под действием Д. дибензиламин, этиле-
нимнн и азетидин расщепляются с выделением азота и образованием
соответственно дибензила, этилена н циклопропана 131:
СДуСЩ HNF
N-H - - 5. С6Н5СНгСН2С6Н5 + Щ
СьН5СНгХ 53
Эгпиленимин
ЛзетиОин
Синтез диазирина. Грэхем 14I разработал простой метод полу-
чения чистого диазприна (бесцветный газ, т. кип —14 ). Конденса-
цией ///prw-бутплампна с формальдегидом (а) получают исходный
/преш-бутнлазометип (т. кип. 65 ); при его взаимодействии с Д. в
CCI, в вакуумной системе образуется диазирин, который собирают
402
в ловушку, охлаждаемую до —128 . Возможные стадии реакции
(а) (СН:,):)СХН., СН.,0 —> (СН;,):,СХ -СН., |- Н..О
(б) СН, X —С (CHJ., HXF, — - :\Fy-CH,=- ХНС (CH3).,(F)
X—С(СН3)3 ZN
(в) СН, = X —С (СН3)3+ :NF — Н,С ч | Н*С х i
приведены на схеме (б) и (в).
Реакция с С-иитрозосоединениями. Эта реакция дает соответст-
вующую N-окись N'-фтордиимпда [51:
о-
Пиридин |
RNO НХТ,--------->RN+-NF
1. С о I b и г п С. В., Kennedy Л., J. Ain. Chem. Soc., 80, 5004 (1958).
2. F r e e m an J. P., К e n n c d у A., Col burn С. B., J. Am. Chem. Soc.,
82, 5304 (1960).
3. В u in yard n e r C. L., M a r t i и К. -I., F r e e m a n J. P., J. Am. Chem.
Soc., 85, 97 (1963); Bumgardner C. L., Free m a n J. P., J. Am. Chem.
Soc., 86, 2233 (1964).
4. G r a h a m W. H., ,1. Am. Chem. Soc., 84, 1063 (1962).
5. S t e v e n s T. E., Free m and. P., J. Ory. Chem., 29, 2279 (1964).
1,1- ДИФТОР-2,2-ДИХЛОРЭТИЛ EH, CI,C=CF,. Мол. вес 132,93,
т. кип. 18,9 /758 мм [1]. Технический продукт с т. кип. 19—21J
перегоняют на колонке длиной 122 см со стеклянной насадкой и
получают индивидуальный продукт (по данным ГЖХ) с т. кип.
18,9“.
Бутадиен и цис- и /прднс-пиперилен реагируют с Д., взятым
в избытке, при 80J в запаянной трубке и в присутствии ингибитора
полимеризации, образуя продукты только 1,2-присоедпнения типа
1 111
I п
1. В а г t 1 е t t Р. D., М о n t ц о m е г у L. К., S е i d е I В., J. Am. Chem. Soc.,
86, 616 (1964).
симм- ДИ ФТОРТЕТР АХ Л ОР АЦЕТОН, CFC12COCFCI2. Мол. вес
231.86, т. кии. 124 .
Д. применяется для получения фторхлоркарбена [1]. К суспен-
О О
11 BuO- II _
CUFCCCFCl,-----> C1..FCCOBU-I- :CFCI л-CI
зпп 1 моля трет -бутилата калия в 500 мл циклогексена при 0° при
перемешивании добавляют 0,5 моля Д. и перемешивают смесь еще
403
3 час при 0—5°. После фильтрования и фракционирования получают
7-фтор-7-хлорноркаран (1) с выходом 36%. а-Метилстирол дает со-
зС-Д-F
С6Н5 С1
(2)
единение (2) с выходом 46?6.
1. Farah В., Н о г n е s к у S., J. Org. Chem., 28, 2494 (1963).
ДИФТОРХЛОРУКСУСНОЙ кислоты НАТРИЕВАЯ соль,
CClF2CO2Na. Мол. вес 152,48.
Получение из кислоты и едкого натра в метаноле [1].
Предшественник дифторкарбена. При кипячении в течение
30 час суспензии Д. к. н. с. в диглиме, содержащем циклогексен,
получают 7,7-дифторноркаран с выходом 11% 12}.
При кипячении смеси Д. к. н. с., альдегида и трифенилфосфина
в 50 мл 1,2-диметоксиэтана в атмосфере азота в течение 72 час (пока
не выделится 0,11 моля углекислого газа) дифторкарбен взаимодей-
ствует с трифенилфосфином с образованием плпда, который реаги-
рует с альдегидом, давая 1,1-дифторолефнн 131. Распространение
F—Ч—СНО | ClF2CO2\'a-i-(C6H5)3P—гт* F—/ CH=CF2
0,1 моля 0,11 моля 0, II моля
этой реакции на кетоны, в целом, не дало положительных результа-
тов [4]. Однако Бэртон и Херкес [5] нашли, что Д. к. н. с. дает
с а,а,а-трифторацетофеноном 2-фенилпентафторпропен с высоким
выходом.
Диглим, 105° 2
20 час (No) I!
C0H5COCF3 + 2 (С0Н5)3Р + CClF„CO.,\a-----С6Н5С—СК,
S и ° о
50 яял 100 яял 100 мял
I. FuquaS. Л., Duncan W. G., Sil v erst e i и R. M., J. Org. Chem.,
30, 1027 (1965); procedure submitted to Org. Syn.
2. В i r c h a 1 1 J. M., С г о s s G. W., Haszeldine R. \., Proc. Chem. Soc.,
1960, 81.
3. Fuq u a S. Л., D uncan W. G., S i 1 verst ei n R. M., Tetrahedron Let-
ters, 1461 (1964).
4. FuquaS.A., Duncan W.G., SilversteinR. M., Tetrahedron Letters,
521 (1965); J. Org. Chem., 30, 2543 (1965).
5. В u r t о n D. J., H e r k e s F. E., Tetrahedron Letters, 1883 (1965).
ДИХЛОРАМИН, HNCIo. Д. является очень реакционноспо-
собным соединением, его можно получить в водном растворе из хло-
ра и аммиака при pH 3—5; при pH выше 8 образуется хлорамин
H2NC1 [1]. Д. получают также из 5%-ного раствора гипохлорита
натрия и хлористого аммония в буферном растворе муравьиная кис-
404
лота —формиат натрия (Грэхем [21). Д. доступнее и безопаснее ди-
фторамина, который, также как и Д., используют для синтеза ди-
азирина (6). Другой реагент, необходимый для синтеза диазирина,—
трет-октилазометин (2) получают при добавлении раствора фор-
мальдегида при перемешивании к mpem-октиламину [(1), диметил-
неопентилметиламин] (Ром н Хаас [3]). Для получения диазирина
(6) 5%-ный раствор гипохлорита прибавляют по каплям при 5—12°
сн3 СН3
СН3-С~СН2-С—NH, + 37%-ньшводн.СН20 40°»
СН, СН, 82%
сн3 СН,
ch3-c-ch2-cn=ch2----->
ё:н3 сн,
(1) 2 моля
2 05 лл
/NHCgH,
СН2
xnci2
n-c8h17
сн2 р
Х.х-С1
<0 (5) (6)
к смеси формиатного буфера, хлористого аммония и трет-октилазо-
метина; летучие продукты удаляют в вакууме через ряд ловушек
(—35, —80, —142 и —196°).
При —142° собирают основное количество диазирина, который
обычно содержит до 15% СО.,; средний выход 25—33%.
1. С h а р i п R. М., J. Am. Chem. Soc., 81, 2112 (1929); CorbettR.E.,Met-
с а 1 f W. S„ S о р е г F. G., J. Chem. Soc., 1953, 1927.
2. Graham W. H., J. Org. Chem., 30, 2108 (1965).
3. Graham W. H., private communication.
Cl
1,3- ДИХЛОРБУТЕН-2, CH3C = CHCH2C1. Мол. вес 125,00,
т. кип. 127—128° (39°/30 мм), n2D° 1,4740.
Физические константы чистых транс- и цис-изомеров Д. описа-
ны Хатчем и Перри [1]. Продажный препарат представляет собой
темно-коричневую жидкость с лакримогенными свойствами; его мож-
но очистить перегонкой на эффективной колонке при атмосферном
давлении, тщательным высушиванием над К2СО3 или СаС12 и пов-
торной перегонкой при 30 мм [2]. ГЖХ показала, что препарат со-
держит транс- и цис-изомеры в соотношении 70 : 30. ИК-Спектро-
скопия указывает на присутствие следов загрязнений ароматическо-
го характера.
Получение. Первый метод заключается в гидрохлорировании
хлоропрена при катализе хлористой медью [3]:
С1 С1
I HCI (Cu.CI2) |
сн2 = ссн=сн2----------> СН3С = СНСН2С1
405
Второй путь синтеза — из ацетоуксусного эфира [II:
CI
PCl6 I LiAIH*
снясосн2со2с2нг>—> сн:|с=-снсо.,с.,н..,------►
С1 С1'
I Пиридин. PCI., |
—лСН:1С-СНСН..ОН--------------* С.НЯС^СНСН,С1
Д. успению использован Вельюзом и др. [41 для построения
кольца А в полном синтезе 19-норстероидов и эстрогенов.
В модельном синтезе с использованием насыщенного кетона гид-
ролиз хлорвинильной группировки приводит главным образом к
продуктам нежелательной циклизации |5|:
сщ
(Главный продукт)
1. Н a t с h L. F., Perry R. Н., J. Ат. Chem. Soc., 77, 1136 (1955).
2. Т о г о т а п о [ I Е. R., private communication.
3. Н a t с 11 L. F., В а 1 1 i п S. G., J. Ат. Chem. Soc., 71, 1039 (1949);
406
4. V е 1 1 u z L., Nomine G., Mathieu J., Angew. Chem., 72, 725 (1960);
Veil u z L., X о m i ti e G., M a t h i e u J.. T о r <> in и и о f 1 E.. В e r-
t i и I)., Tessier.!., P i e r d e t A., Conipt, rend., 250, 1084 (I960); V e I-
1 ti z L., X о ni i и ё G., В ueour t R., P i e r d e t A., T e s s i c r ,1., ibid
252, 3903 (1961); V e 1 1 u z L., X о m i n ё G., Bucourt R., Pierdet A.,
I) u f a у Ph., Tetrahedron Letters 1961, 127; Bucourt R., Tessier J.,
X о m i n 6 G., Bull. soc. chim. France, 1963, 192.3.
5. J u 1 i a S., Bull. soc. chim. France, 21, 780 (1954).
1,1- ДИХЛОРДИМЕТИЛОВЫЙ ЭФИР, CH:1OCHCI... Мол. вес
114,96, т. кип. 85—87’.
Д. э. получают обработкой метилформиата пятихлористым фос-
фором 111. Д. э. превращает карбоновые кислоты или их ангидриды
в хлорангидриды с высоким выходом 12]. Сульфокислоты или их
натриевые соли превращаются в сульфохлориды, карбонильные сое-
RCO.,H СЬСНОСН. —RCOC1 + НСО.,СН..(
(RCO),O I С12СНОСН3——> 2RCOC1-I- НСО.,СН;(
дипсиия — в гелг-дихлориды |2|. В условиях реакции Фриделя —
Крафтса Д. э. взаимодействует с ароматическими углеводородами,
образуя альдегиды 131:
1. R i е с h е A., Gros s И. G., Chem. Teclin., 10, 515, 659 (1958).
2. R i с с h с A., GrossH., H 6 f t E., Chem. Ber., 93, 88 (1960).
3. R i e c h e A., G г о s s H., Chem. Ber., 92, 83 (1959); G г о s s H., R i e c h e A.,
Mall he у G., ibid., 96 . 308 (1963).
2,3-ДИХЛОР-5,6-ДИЦИАН-1,4-БЕНЗОХИНОН (ДДХ). Мол. вес
227,01, т. пл. 213 К
Получение. ДДХ был синтезирован по схеме Тиле 111. По усо-
у См. обзор Д ж е к м а и а Л. в кн. <Успехи органической химии», изд-во
tMiip" М., 1964. том 2, ciр. 328.— Прим, перес.
407
вершенствованной методике [2] реакцию можно провести за два
дня; общий выход ДДХ 70%, считая на бензохинон. Совсем недавно
хлорированием 2,3-дициангидрохинона в кипящей уксусной кислоте
был получен с выходом 40—50% 5,6-дихлор-2,3-дициангидрохинон
(Митчелл |3|), окисление которого двуокисью свинца в водном эта-
ноле, содержащем 5% соляной кислоты, дает ДДХ. В отсутствие
кислоты окисления не наблюдается [3|. Однако по новой методике
14] перемешиванием 2,3-дициангидрохинона с разбавленной соляной
кислотой (1:1) при 35° и добавлением 70%-ной азотной кислоты
ДДХ получают с выходом 90%. В этой методике используется ста-
бильность ДДХ в разбавленной минеральной кислоте и способность
2,3-дииианхинона и 2-хлор-5,6-дицианхпнона быстро присоединять
хлористый водород в водном растворе.
Ароматизация. ДДХ, обладающий высоким окислительно-вос-
становительным потенциалом 12), используется для дегидрирования
гидроароматическпх соединений. Например, в кипящем бензоле он
превращает тетралин в нафталин, аценафтен в аценафтилен.
Гидроароматические соединения с геминальными блокирующими
группами (1) ароматизируются с 1,2-сдвигом метильной группы.
ДДХ можно использовать для получения солей стабильных аро-
матических катионов, в частности перхлоратов тропиллия и трифе-
нилциклопропенилия [5]:
+ ддх
+ нсю4
Н С$Нд СаН
X + ДДХ + НСЮ4 -----------> А 5 С1О4‘ + ддх-н2
с6н5£=Ас6н5 с6н5'^' с6н5
В стероиде (3) под действием ДДХ ароматизируется кольцо А с об-
разованием эквилина (4) [61:
408
Дегидрирование карбонильных соединений. ДДХ используется
для введения в стероидные кетоны двойных связей, наличие кото-
рых повышает их биологическую активность. Насыщенный 3-кето-
стероид (5) дегидрируется ДДХ в кипящем бензоле или диоксане,
давая Д1,4-кетон-3 (6) 17, 8]. А4-Кетостероид-3 (7) дегидрируется
с образованием двух продуктов. В апротонных условиях или в при-
сутствии слабых доноров протонов, например n-нитрофенола, проис-
ходит 1,2-дегидрирование до Д1,4-кетона-3 [9—12|. В присутствии
хлористого водорода осуществляется 6,7-дегидрирование доД4,ь-ке-
тона(8) [121. Д4,0-Кетон-3 (8) подвергается только 1,2-дегидрирова-
нию [9, 10).
(8)
(9)
Эти продукты получаются с высоким выходом. Особое преимущество
этого метода дегидрирования состоит в том, что ДДХ не действует
на функциональные группы, обычно встречающиеся в стероидах.
б-Лактоны дегидрируются в соответствующие «.^-непредельные
лактоны, но выходы колеблются от низких до удовлетворительных
[131. В сухом бензоле ДДХ превращает Д3,®-диенольный эфир (11)
в 1,4,6-триенон-З (10), а в присутствии воды в 4,6-диенон-З (12) [141.
409
Аналогично в водном ацетоне 17а-ацегокси-6-окспметил-3-метокси-
Д:‘’’’-прегпаднепоп-20 дегидрируется ДДХ до 17а-ацетоксн-6-оксн-
метил-А1’“-прегнадиендиона-3,2б 114аI.
Окисление кислородсодержащих функциональных групп. ДДХ
применяется для селективного окисления аллиловых |15—171 и
бензиловых [181 спиртов. Этот реагент имеет преимущество перед
двуокисью марганца, активность которой может варьировать.
(2)
ДДХ дегидрирует 2-окспметилен-З-кетостеропды с выходом 50 % 1191.
Для этого стероид обрабатывают при комнатной температуре в те-
чение 1—5 мин 1,1—1,5 же ДДХ в растворе дноксана, смесь раз-
бавляют хлористым метиленом и продукт реакции выделяют хрома-
тографически.
При изучении кинетики окисления аллиловых стероидных спир-
тов Берштейн и Рипгольд 1201 нашли, что 30-окси-Д 4-стероиды (эк-
ваториальные) окисляются почти в 6 раз быстрее, чем За-эппмеры
(аксиальные), а водородсодержащпй За-оксистероид (4) окисляется
в 5 раз быстрее, чем соответствующее соединение, содержащее дей-
терий (5). Последнее указывает, что стадией, определяющей скорость
реакции, является разрыв связи С — Н или С — D. Самое быстрое
окисление наблюдается в /нреш-бутаноле, самое медленное — в ук-
сусной кислоте.
Бензильное окисление. При кипячении с ДДХ в бензоле в те-
чение 5 дней 8-дифенплметилнафтойная кислота [3] подвергается
410
окислительной циклизации в 6-лактон (4) с высоким выходом 1211.
О
/\
(С6Н5)„СН СО2Н (С6Н5), с cz он
' J I ' 1 1
' ^ч fl Z/' ' Г'\Т
I II I Hi I +° I II “
ОН
(3) (4)
Окисление фенолов 1221. ДДХ—очень сильный окислитель
для фенолов; реакция протекает гладко в метаноле при комнатной
температуре. Как правило, главными продуктами являются димеры,
образованные за счет С — С- или С — О-связывания. 2,6-Димето-
ксифенол со свободным //-положением дает главным образом дпфено-
хинон (2) и следы 2,6-диметоксихпнона (3).
(И
Пространственно затрудненный фенол (4) дает димерный продукт
он о
(СНз1зС I .С(СН3)3 (СН,),С С(СН3)3
I II S II Г(1 с,
'V ч/ 1;________/
С(СН3)3 (СН313С у__________V-OH
XCZ "С\
(4)
(3)
(5) с С — О-связью. Из 2,6-ди-Аг/2е/7г-бутил-4-метоксифе1юла (6)
образуется желтое масло, которому поданным ИКС и ЯМР припи-
сана структура диметплкеталя (7); гидролиз кеталя (7) приводит к
411
хинону (8).
ОН
О
(СН3)3 С. 1 .С (СН3)3 (СН3)з (\ JI С (СН3)3
I II II II
\./
ОСН3 СНзО/^ОСНз
(6) (7)
О
(СН3)3СЧ (СН3)3
'll II
Гидролиз
(8)
2,4,6-Триметилфенол (9) окисляется до альдегида (14) с выходом
85%; предполагали, что промежуточными продуктами здесь являют-
ся производные хинонметана (10) и (12):
Окислительная димеризация. Енолы и енолизирующиеся кетоны,
ва,Р-положение которых нельзя ввести двойную связь, под действи-
ем ДДХ подвергаются окислительной димеризации [231. Например,
2-фенилиндандион-1,3 превращается в тетракетон.
1. Т h i е I е J., G ii n t h е г F., Ann., 349, 45 (1906).
2. Braude Е. А., В г о о k A. G., L i n s t е a d R. Р., J. Chem. Soc., 1954,
3569; L i n s t е a d R. Р., В г а и d е Е. A., J а с к m a n L. М., В е а-
412
m e s A. N., Chem. Ind., 1954, 1174; C r e i g h t о n A. M., Jackman L. M.,
J. Chem. Soc., I960, 3138.
3. M i t c h c 1 1 P. W. D„ Can. J. Chem., 41, 550 (1963).
4. W a 1 к e r D., Waugh T. D„ J. Org. Chem., 30, 3240 (1965).
5. R c i d D. H., Fraser M., M о 1 1 о у В. В., Payne H. A. S„ Sut her-
1 a n d B. G., Tetrahedron Letters, 530 (1961).
6. В a g 1 1 J. F., M о r a n d P. F., Wiesner K-, G a u d г у R., Tetrahedron
Letters, 387 (1964).
7. M u 1 1 e r G., Mar t el J.. H uy n h C., Bull. soc. chim. France, 1961, 2000.
8. Z d e r i c J. A., farpioH., LimonD.C., J. Org. Chem., 27, 1125 (1962).
9. Burn D., К i г к D. N., Petrow V., Proc. Chem. Soc., 1960, 14.
10. В urn D., Pet row V., Weston G., J. Chem. Soc., 1962, 29.
11. C a s p i E., Grover P. K., Graven N., Lynde E. J., Nussbau-
m er Th., J. Chem. Soc., 1962, 1710.
12. RingoldH.J., TurnerA., Chem. Ind., 1962 , 211.
13. В e г к о z В., Cuellar L., G r e 7, e m к о v s к у R., Avila N. V.,
Cross A. D., Proc. Chem. Soc.. 1964, 215.
14. P r a d h a n S. K., R i n g о 1 d H. J., J. Org. Chem., 29, 601 (1964).
14a. В u rnD.,Coom bsR. V., E 1 1 isB.,Mc В r i de J.A. H.,Pet r owV.,
Yardley Y. P., Chem. Ind., 1966, ’.97.
15. В r a u d e E. A., L i n s t e a d R. P., W о о 1 d r i d g с K. R., J. Chem. Soc.,
1956, 3070.
16. Burn D., Petrow V., Weston G. O., Tetrahedron Letters, № 9, 14
(1960).
17. Bowers A., H о 1 t о n P. G., Necoech ea E., К i n cl F. A., J. Chem.
Soc., 1961, 4057.
18. Becker H.-D., Adler E., Chem. Scand., 15, 218 (1961).
19. В о w e r s A. and co-worker, J. Org. Chem., 29, 3481 (1964); M о r a n d P.,
S t a v r i c S., G о d i n D., Tetrahedron Letters, 49 (1966).
20. BursteinS. H., R i n g о I d H. J., Am. Chem. Soc., 86, 4952 (1964).
21. С r e i g h t о n A. M., .1 ackman L. M., .1. Chem. Soc., 1960, 3138.
22. Becker H.-D., J. Org. Chem.. 30, 982 (1965).
23. Becker H.-D., J. Org. Chem., 30, 989 (1965).
1,1-Д ИХЛОРДИЭТИЛОВЫЙ ЭФИР, CH3CCI2OC2H5. Мол. вес
143,01, т. кип. 104,5—105,5=.
Д. э. получают присоединением 2 экв сухого хлористого водорода
к этокснацетилепу. Д. э. взаимодействует с карбоновыми кислота-
ми (при 40°), образуя с хорошим выходом соответствующие хлоран-
гидриды [1]:
О О
О С1 II II
II I -НС1 R—С—Cl R— С-С1
R-C+C1—С—ОС..Н-,_____, ! ! +
| | 6—С—ОС.,Hr, о=с—ос.,н5
ОН СНз | I
СН3 сн3
Пептидный синтез осуществляется при кипячении указанных на
схеме компонентов в этплацетате в течение 15—45 мин [2]. Амино-
группу защищают карбобепзоксн- или фталоильной группой:
R R'
I I
CbNHCHCO.H + НС1 • H.,N’CHCO.,CoH5 + СН3СС1.,ОС.,Н5
ROHR'
I II I I
—. CbXHCHC—N CHCO.,C.,H5 -F 3HC1 4-CH3COgC„H5
413
1. Heslinga L., KaterbergG. J., Arens J. F., Rev. trav., 76, 969
(1957).
2. Il e s 1 i ii g a L., A r e n s J. I7., Rec. trav., 76, 982 (1957).
ДИХЛОРКЕТЕН, CI..C C O (не выделен).
Д., полученный «в момент выделения», быстро реагирует с цикло-
пентадиеном, образуя аддукт Дильса — Альдера, который далее
превращается в трополон (5) 11). Раствор дихлорацетнлхлорида
и десятикратного избытка циклопентадиена в гексане обрабатывают
при 0—5 ' раствором триэтпламниа в гексане. После удаления твер-
дого хлоргидрата амина и перегонки выделяют аддукт (3) с выходом
70—75°о. Этот аддукт (3) гидролизуют водным ацетатом калия в
уксусной кислоте до трополона (5); возможный механизм приведен
на следующей схеме.
Независимо была разработана аналогичная методика получения Д.,
причем продукт присоединения его к пнклопептадпену был охаракте-
ризован как бициклический дихлоркетон (3); подобным образом Д.
реагирует с циклопентеном, образуя кетон |21:
с12снсос1 ДДДДД. с1.с=с=о
Тернер н Седеп|3]описали получение 4,5-беизтрополона из инде-
на и Д; однако выходы при этом низкие.
1. Stevens Н. С., Reich D. A., Brandt D. R., Fountain К. R.,
G a u g h а ii Е. J., J. Am. Chem. Soc., 87, 5257 (1965).
2. G 11 о s e z L., M о n t a i g n e R., M о 1 1 e t P., Tetrahedron Letters, 135 (1966).
3. T u г и er R. \V., S e d e n T., Chem. Comm., 1966, 399.
ДИХЛОРМЕТИЛЕНТРИФЕНИЛФОСФОРАН, (CeH5)3P=CCl2.
Мол. вес 345, 20.
414
Исходный трет-бутплат калия был получен по новой методи-
ке (Сиезил и corp. 111) из калия и/?;/7с/7(-бутанола в атмосфере азо-
та, очищенного пропусканием через две промывных склянки с раст-
вором Физера и промывные склянки с кони, серной кислотой и с без-
водным хлористым кальцием. Избыток шрегп-бутаиола отгоняют,
добавляют пентан н продолжают перегонку, трет-Бутилат калия
используют в виде суспензии в пентане. Суспензию охлаждают до
О—5', добавляют трнфенилфосфин и к хорошо перемешиваемой сме-
си по каплям прибавляют раствор хлороформа в гептане. Хлороформ
с /п/ячи-бутилатом калия дает дихлоркарбен, который присоединяет -
о—.т
<СКН:Х. Р + (СНОСОК +СНСЫ Г-С..Н,...
0,5 моля 0.5 моля 0.5 моля 2 л
/МСП()„ NC..H.CHO
—> (С..Н,)..Р --С(:---------------------- М-(СНЧ)..Х('.„Н4СН CCL
’ ’ J - (.X - ;т\ (сырч») - 4
(I) Ы)
ся к трифепилфосфипу и образует фосфоран (1), находящийся в виде
суспензии в гептане. При 0—10 и перемешивании прибавляют л-дп-
метнламипобензальдегнд (шесть порций), удаляют выпавший трп-
феннлфосфпноксид, фильтрат выпаривают на роторном испарителе
и после перекристаллизации из метанола твердого коричневого
остатка получают сырой Рф-днхлор-н-днметиламиностирол (2). Ос-
новная примесь — непрореагнровавший трнфенилфосфин, который
можно удалить добавлением хлорной ртути к сырому (2) в абсолют-
ном этаноле. Нерастворимый комплекс хлорной ртути и трпфенил-
фосфпна отфильтровывают, фильтрат упаривают и остаток кристал-
лизуют.
Д. получают также непосредственно из трифенилфосфпна и че-
тыреххлористого углерода |2, 3|. Это простой метод синтеза илидов
тина (4). из которых в одну стадию можно получить 1,1-дигалогено-
лефины. Например, нагревают при 60 в течение 2 час раствор трн-
сонг>),, р —(с6н5'3 р........- сс13 —+ (с0н5);) р-са32£212.дЛ
° S
(С6Н3)з Р — СС1., (С,;Н5):1Р=СС1., + С1.,Р(С6Н5)Я
—* (-»> (5)
(] Q ссН5е.гь-о j-(С„Н5).,РО -(С.,н5).,Р0 | О-СИС.Н,
X /, С(!Н-,СН = CCL CLCHCJ13
Р(( -с>Н3)з
(3) (G) (7)
феннлфосфнна и бензальдегида в четырех хлор истом углероде, осаж-
дают трифенилфосфннокснд петролепным эфиром и после разгонки
фильтрата получают Рф-днхлорстнрол (6) и бензальхлорид (7)
с выходами 72%. В реакции не происходит промежуточного образо-
вания свободных радикалов или карбенов; процесс включает пере-
415
ходные состояния (1) и (3) и илид (4). Взаимодействие четырехбро-
мистого углерода с трифенилфосфином и бензальдегидом заканчи-
вается за несколько минут и приводит к р,Р-дибромстиролу с выхо-
дом 84 %.
1. S р е z i а 1 е A. J., R a t t s К. W., J. Am. Chem. Soc., 84, 855 (1962); Spe-
zialeA. J., RattsK. W., BissingD, E., Ory. Syn., 45, 33 (1965).
2. R abi nowi tz R., Mar cus R., J. Am. Chem. Soc,, 84, 1312 (1962).
3. R am irez F., Desai N. B., Al с К e I v i c N., J. Am. Chem. Soc., 84,
1745 (1962).
ДИХЛОРМЕТИЛЛИТИЙ, ТРИХЛОРМЕТИЛЛИТИЙ, LiCHCL,
LiCCI.,.
Эти реагенты получают реакцией бутиллптия с хлористым
метиленом или хлороформом в смеси ТГФ — эфир — петролейный
эфир (4:1:1) при —110° [1]. Первый реагент более стабилен, чем
второй. Д., т. реагируют с кетонами, давая с хорошим выходом кар-
бинолы.
LiCHCI,
(С.Н8)2С = О—
LiCCl3
(С6Н5)2СОНСНС12
(С6Н5)2СОНСС13
При добавлении N-бензилиденанилина в эфире к избытку ди-
хлорметиллития при температуре ниже —70э получают с высоким
выходом неочищенный монохлоразиридин, первое известное соеди-
нение этого класса, которое слишком реакционноспособно, чтобы
его можно было очистить для анализа. Реакция протекает стереоспе-
цифично с образованием qwc-изомера (Дьеруп и Гринвальд [21).
-70» ¥_
C6HSCH=NC6HS + LiCHCIj -—|Г7ТК'С(,Н5 + ЫС1
н5су
С1
Трихлорметиллитий получали (Миллер и Уэйлен (31) прибавлением
СС1;)Вг к суспензии метиллития в эфире при —115 в присутствии
2 экв циклогексена; после перемешивания при—100 в течение часа
был выделен 7,7-дихлорноркаран с выходом 77%. Бреслоу и Альтман
отмечали [41, что Т. функционирует как донор дихлоркарбена и при
—100° легко реагирует с газообразными ацетиленами (так как при
этой температуре их можно получить в виде концентрированных
—95°
сн3сн.,сн.,с^ссн.,сн,а1, у- ca 3cl i->
н,о+
—> CH3CH2CH2C = ССН2СН.,СН3 ——* СН3СН.,СН2С = ССН2СН2СН3
с' V
/\ II
С1 С1 о
растворов. Так, трихлорметиллитий реагирует с октином-4 при
—95°, и после подкисления при низкой температуре и обработки
416
смеси водой получается ди-н-пропилциклопропенон (выход 19%).
Эта реакция осуществляется также с пентином-1, пропином, бути-
ном-2, но не наблюдается для ацетилена.
1. KobrichG., Flory К., D г i s с h е 1 W., Angew. Chem., 76, 563 (1964);
ibid., Internat. Ed., 3, 513 (1964).
2. Deyrup J. A., Greenwald R. B., Tetrahedron Letters, 321 (1965); J.
Am. Chem. Soc., 87, 4538 (1965).
3. Mil I e r W. T., Jr., Whalen D. M., J. Am. Chem. Soc., 86, 2089 (1964).
4. В r e s 1 о w R., A 1 t m a n L. .1., J. Am. Chem. Soc., 88, 504 (1966).
ДИХЛОРФОРМОКСИМ, CI2C=NOH. Мол. вес 113,94.
Д. сильно раздражает кожу, а его пары токсичны. Получают при
восстановлении хлорпикрина оловом и конц. соляной кислотой
[II:
Sn-HCI
СС1;|ХО., ---> C1.,C = NOH
Д. медленно разлагается при комнатной температуре, но при хра-
нении в холодильнике устойчив в течение нескольких месяцев.
Синтезы гетероциклов. Д. был использован для синтеза 3-хлор-
изоксазолов на основе реактивов Гриньяра, полученных из тер-
минальных ацетиленов (Квилико и сотр. [2]).
«cscMgx * cicci _hf<25<’)> |j ПС‘
II RlCzN
N °
но"
R = H. выход 36%
1. M i 1 1 er С. E., procedure submitted to Org. Syn.
2. Bravo P., Ga udiano G., Q u i 1 i с о A., Ricca A., Gazz., 91,
47 (1961); |C. A., 56, 12869 (1962)].
1,1-ДИХЛОРЭТИЛЕН, CH2=CCI2. Мол. вес 96,95, т. кип. 37°.
Промышленное получение 111 Д. основано на хлорировании ви-
нилхлорида и дегидрохлорировании продукта присоединения вод-
ной щелочью или гидроокисью кальция:
С1г Основание
СН,=СНС1 —> С1СНХНС1.,----------СН2 = СС1.,
Д. относительно стабилен при хранении только с добавкой 0,3% три-
этиламина или /прет-бутилпирокатехина в качестве ингибиторов
полимеризации.
Д. реагирует со спиртами, способными к образованию карбоние-
вых ионов, давая карбоновые кислоты:
— ОН- СН2=СС12 + +Вода
R3COH--------* R3C+------- R3CCH2CC12----< R3CCH2CO2H
Реакция протекает в 80—100%-ной серной кислоте и сильно уско-
ряется трехфтористым бором |2]. Норборнен превращается с хо-
14 Заказ № 965 417
рошим выходом вжзо-норборнилуксусную кислоту (2); адамаитанол-1
(3) дает адамантил-1-уксусную кислоту (4):
1. Bott К., private communication.
2. Во t t К., Angew. Chem., Internal. Ed., 4, 956 (1965).
ДИЦИАНАЦЕТИЛЕН (динитрил ацетплендикарбоновой кис-
лоты), NCC=CCN. Мол. вес 76,06, т. пл. 21', т. кип. 761, уд. вес
0,970.
Д. получают дегидратацией амида ацетилендикарбоновой кисло-
ты пятиокисью фосфора [II *.
Д.— очень реакционноспособный диенофил; в реакции Дильса —
Альдера с простыми диенами при комнатной температуре он образует
аддукты с высоким выходом [2, 3]. С антраценом и дуролом он дает
CN
(2)
аддукты (1) и (2) [31. Д. реагирует с барреленом |(1), бицикло-[2, 2, 2]-
-октатриен-2,5,7] при комнатной температуре с образованием ад-
дукта (II б) с выходом 95%; в случае диметилового эфира ацетилен-
дикарбоновой кислоты [41 реакция протекает при 100° и выход со-
ставляет только 29%. Если аддукт Пб нагревать 3,5 час при 100°,
то образуется 1,2-дицианнафталин с выходом 63%. Пиролиз ад-
дукта Па при 150° в течение 4 час дает диметиловый эфир нафталин-
дикарбоновой кислоты (V) с выходом 33%. Полагают, что реакция
Новые способы получения см. [1а].— Прим, перев.
418
сопровождается приведенным на схеме синхронным электронным
сдвигом.
(а) МеО,СС=ССО2Ме
100°
или(6) NC-CHC-CN
RT
Па, R=-COOMe
6,R=-CN
1. В 1 о m q u i s t А. Т., W i n s 1 о w E. C., J. Org. Chem., 10, 149 (1945).
la. C i g a n e k E., К r e s p a n C. G., J. Ogr. Chem., 33, 541 (1968).
2. Cookson R. C., Dance J., Tetrahedron Letters, 879 (1962).
3. Weis C. D., J. Org. Chem., 28, 74 (1963).
4. Z i m m e r m a n H. E., GrunewaldG. L., J. Am. Chem. Soc., 86, 1434
(1964).
«ДИЦИАНДИАМИД» = N-ЦИАНГУАНИДИН, (1а)±^(1б). Этот
реагент получают димеризацией цианамида (1) в присутствии осно-
вания. Мол. вес 84,08, т. пл. 209—21 Г. Тривиальное название «ди-
циандиамид» неверное; этот реагент следует рассматривать как смесь
NH NH2
он- II I
2H.,NC=N-------> H2NCNC=N H,NC = N -fcN
H
(1) (Ы) (16)
таутомерных форм N-циангуанидина (1а)дД(1б).
Синтезы гетероциклов. В 1834 г. Либих впервые осуществил
синтез меламина (2,4,6-трнамино-сцлмг-триазпна) (2 моля) при нагре-
14
419
вании Д. (3 моля) под давлением
КН н
II I
Н.,К— С— К'—СзХ •— 2H2N—C=N
N
N С
|l| \ __z
2H.jN—C+2 NH —► 2НгК —z/___4n
HN-<(
nh2
Для получения 2,4-диамино-6-фенил-сшч.и-трназина (бензогуана-
мин) бензонитрил и Д. добавляют к смеси едкого кали и метилцелло-
CsN
0,48 моля 0.6 моля
NH
HN = C//
\’Н2
сольва при перемешивании. Вначале наблюдается экзотермическая
реакция, затем смесь кипятят 5 час. Продукт реакции отделяется
в виде белого мелкокристаллического вещества.
Модест и сотр. [21 разработали новый одностадийный синтез
2,4-диаминопиримидинов конденсацией Д. с монофункциональны-
ми кетонами, как показано на примере циклогексанона:
В обзоре Модеста рассмотрено применение этого реагента для син-
тезов ароматических триазинов (2) [31 и 4,6-диамино-1-арил-1,2-
дигидро-сп.иуи-триазинов (4) [41.
н
гш2
(2)
О)
NH2 . C6H5N^N\c^NH2
ОН или нпгревоние |6 4ц
--------> Ni N
н"
Вг
Н)
420
1. SimonsJ. К., Saxton M.R., Org. Syn.. Coll. Vol.. 4, 78 (1963).
2. M о d e s t E. J., C h a t t с r j e e S., Protopa pa H. K.. .1. Org. Cliem.,
30, 1837 (1965).
3. M о d e s t E. J., E 1 d e r f i e 1 d R. C., in (-Heterocyclic Compounds-, 7, 650—
656 (1961).
4. Modest E. J., El d e r f i e 1 d R. C., in «Heterocyclic. Compounds», 7, 697—
701 (1961).
ДИЦИАНМЕТИЛЕН-2,4,7-ТРИ НИТРОФЛУОРЕН,
Мол. вес 351,23, т. пл. 266—268’.
Д. получают с высоким выходом при конденсации 2,4,7-тринитро-
флуоренона с дннитрилом малоновой кислоты в присутствии пипе-
ридина в качестве катализатора 11 |.Д. представляет собой л-кислоту,
среднюю по силе между тетрацианэтплеиом и 2,4,7-трииитрофлуоре-
ноном.
1. Mukherjee Т. К., Levasseur 1.. А.. .1. Org. Chem., 30, 644 (1965).
N, N'-ДИЦИКЛОГЕКСИЛАМИДИН N-МОРФОЛ ИЛ КАРБОНО-
ВОЙ КИСЛОТЫ (3). Мол. вес 293,44, т. пл. 105,5°.
Д. получают кипячением морфолина (1)и дициклогексилкарбо-
диимида [(2) ДЦК1 в треш-бутаноле 111.
, г~ и кт г~ vr и Кипячение в (СН.,).,СОП. 1 час
О NH + CeHnN =С= ХС6НП--------------------------->
\___/ 89%
(1) (2)
/----« -АС.Н,,
_^О N-CC
\. чхс„нп
(3| i
Н
Д.реагирует с нуклеозид-5'-фосфатом (4)с образованием моноами-
да нуклеозид-5'-фосфата (5), который при обработке ДЦК в кипящем
пиридине дает циклический нуклеозид-3',5'-фосфат (6) Ц|. Реакция
незамещенного 5'-фосфата с ДЦК в пиридине иногда дает удовлетво-
рительные результаты, но в основном фосфаты недостаточно раст-
воримы в пиридине, лучшем из возможных растворителей 121.
1. Moffatt J. G., К b с. г а п а Н. G , .1. Am. Chem. Soc., 83, 649 (1961).
2. S т i t h M., D r u m т о n d G. I., К hor an a H. G., J. Am. Chem. Soc.,
83, 698 (1961).
421
ДИЦИКЛОГЕКСИЛАМИН,
Мол. вес 181,31, т. пл. 20э, т. кип. 256°.
Д. образует исключительно хорошо кристаллизующиеся соли со
многими N-защищенными аминокислотами |1|. Обычно кристалли-
зация соли начинается сразу же при сливании эквимолекулярных
количеств реагентов в спирте или этилацетате. После разбавления
эфиром соль отделяют и промывают эфиром.
I. К I i е g е г Е., Schroder Е., G i b i a n Н., Анн., 640, 157 (1961).
ДИЦИ КЛОГЕКСИЛ КАРБОДИ ИМИД(ДЦК),(,1Е С = МС„Н1г
Мол. вес 206,33, т. пл. 34—35”, т. кип. 155”/11 мм, 95—97'30,2 мм.
Техника работы. К действию ДЦК часто наблюдается индиви-
дуальная кожная чувствительность. Реагент плавят в склянке 111,
нагревая ее в теплой воле или в токе теплого воздуха. Затем жид-
кость выливают в теплую взвешенную колбу и доводят до необходи-
мого веса, прибавляя реагент предварительно разогретой пипеткой.
Получение. ДЦК получают окислением Ц,М'-дициклогексил-
тиомочевины С„НПМНС( SJNHCbH,, окисью ртути [2|. Разрабо-
тан общий метод получения карбодиимидов из алифатических и аро-
матических изоцианатов |3|, однако его не применяли для синтеза
дициклогексильного производного.
Конденсация гидроксилсодержащих соединений с отщеплением
воды. Применение ДЦК для конденсации гидроксилсодержащих
соединений с отщеплением воды в мягких нейтральных условиях
рассмотрено в обзорах |4, 51. Новый пример такого превращения —
получение симметричных перекисей ацилов конденсацией 2 молей
кислоты с 1 молем перекиси водорода |61. Эквивалентные количества
кислоты и ДЦК ( I 10% избытка) взвешивают в жидком состоянии
0°
2СвН5ЕО2Н-j- H2O2 + C0HllN-C = NCeHu —
Уи /о
о о нон
II II I II I
СвН5С—OO-CCeH54-CeHnN-C— NC6Hn (ДЦК-Н2О)
в реакционной колбе, снабженной мешалкой и термометром, охлаж-
дают в бане со льдом, прибавляют эфир и охлаждают раствор до 0°.
Затем добавляют сухой эфирный раствор перекиси водорода в коли-
честве, обеспечивающем пятикратный избыток. Раствор достаточно
хорошо растворимой кислоты в хлористом метилене прибавляют к по-
лученной смеси с такой скоростью, чтобы температура не поднима-
лась выше 5\ Если кислота нерастворима в хлористом метилене,
то его прибавляют первым и затем добавляют кислоту в виде тонкоиз-
422
мельченного порошка и перемешивают; окончание реакции опреде-
ляют по НК-поглощению в карбонильной области. Выпавшую N,
N'-днциклогексилмочевину отфильтровывают при отсасывании и про-
мывают хлористым метиленом. Несимметричные перекиси ацилов
могут быть получены при реакции эквивалентных количеств двух
кислот с избытком перекиси водорода в присутствии ДЦК.
Алкилариловые эфиры получают с хорошим выходом нагрева-
нием при 100—110° в запаянной трубке смеси фенола (1 экв), спирта
(1,2 же) и ДЦК (1,1 же) в течение 24 час 17]. Использование инерт-
ного растворителя или проведение реакции при комнатной темпера-
туре ведет к снижению выхода.
ДЦК быстро и почти количественно реагирует с моно- и диэфи-
рами фосфорной кислоты в эфирном растворе при 25° с образованием
соответствующих ди- и тетраэфиров пирофосфорной кислоты и быст-
о- о- о-
ROP4OH)., — Ч-. ROP+-O-P+OR сДЦК —Н2О
I I
ОН он
о- о- о-
(RO).,P + OH RO-P + — О-Р+ -OR +ДЦК-Н2О
<J)R <J)R
рым осаждением дициклогексилмочевины [8]. По-видимому, ДЦК —
наилучший реагент для полимеризации мононуклеотидов [91: ком-
поненты обрабатывают пиридином и двухфазную систему сильно
встряхивают.
ДЦК использовали для синтеза симметричных ангидридов N-за-
щищенных аминокислот (Корана [10]). Так, если перемешивать при
комнатной температуре Ы-карбобензокси-о,ь -фенилаланин и ДЦК
в эфире, то наряду с дициклогексилмочевиной осаждается образую-
щийся ангидрид. Ангидрид очищают экстракцией холодным этилаце-
татом и кристаллизацией (т, пл. 125—126°). При использовании раст-
дцк
2СвН6СН2СНСО2Н----> (СвН5СН2СНСО)2О
I 90% I
NHCO2CH2CeH5 NHCO2CH2CaH5
вора в ацетонитриле также получают ангидриды, но с несколько
меньшим выходом (Шюсслер и Цан [111).
ДЦК способствует этерификации первичных и вторичных спир-
тов в очень мягких условиях; в случае третичных спиртов выход
очень низкий [12|.
Сешадри и сотр. [131 успешно применили ДЦК для синтеза деп-
сидов лишайников, например метилэверната [3]. Такие эфиры ранее
получали при реакции хлорангидрида кислоты с фенолом в присутст-
вии водной щелочи или пиридина. Однако типичные депсиды, такие,
423
как (3), замещены в орто-положении по отношению к карбоксилу,
СН3
А/СОзН Н0\^\/0Н
I II + I II
СНзС/^7 \эн со2сн3 ЭФир
СНз
(I) (2)
/) х/°Н
~ I II I II
CHSO Z ' ОН "V ' со.,сн3
СНз
(3)
что весьма затрудняет получение хлорангидрида. Кроме того, сво-
бодные гидроксильные группы эверниновой кислоты (1) необходимо
защищать перед превращением кислоты в хлорангпдрид. Использо-
вание ДЦК позволяет обойти эти трудности и дает возможность
получить соединение (3) с хорошим выходом. С этой целью успешно
применяли также ангидрид трифторуксусной кислоты.
Пептидный синтез. Этот дегидратирующий агент нашел ши-
рокое применение в синтезе пептидов [141, хотя в ряде случаев он
вызывает рацемизацию [151. Чтобы свести к минимуму образование
ацилмочевины, рекомендуется применять в качестве растворителя
ДЦК
RCO2H + H2NR----► RCONHR' + ДЦК —Н2О
смесь ацетонитрила и ТГФ [16]. Активированные эфиры для пептид-
ного синтеза можно получить этерификацией N-ациламинокислот
R R
СЬ1Х'НСНСОгН + НО—NO2 —CbNHCHCO, \О2
с n-нитрофенолом, проводя дегидратацию ДЦК [17].
N-Ациламины. Амины реагируют с карбоновыми кислотами
в присутствии ДЦК при 0° с образованием амидов с выходом 70—
АсОСН
I
HCNH.
I
АсОСН
I
НСОАс
I
НС______
СНгОАс
I------
АсОСН
HCNHCOR
дцк |
O+RCO2H —АсОСН
С. 11 j с_Д j |
НСОАс
I
нс-------
о+дцк-н2о
I
СН2ОАс
424
90% [181. Этот реагент использован для получения N-апильных
производных D-глюкозамина.
Лактонизация. Вудвард и сотр. |19| показали, что ДЦК в пи-
ридине является более эффективным реагентом для лактонизации
у-оксикислоты (1), чем уксусный ангидрид в пиридине.
у-Оксикислота (3) не лактонизуется обычными методами, так как
такое превращение требует конверсии циклогексанового кольца с пе-
реходом в конформацию ванны (4) (Джонсон и сотр. 1201). В некото-
рых опытах лактонизацию удалось провести с помощью ДЦК в пи-
ридине, но результаты не всегда воспроизводимы. Так, выход 55%,
полученный в одном из опытов, повторить не удалось. Этот лактон
удалось получить с удовлетворительным выходом при использова-
нии /7-толуолсульфокислоты в ксилоле. .
₽-Лактамы. Синтез пенициллинов, например пенициллина V,
требует циклизации соответствующей пеницпллоиновой кислоты
ОН п О Н S
II । \ ДЦК 'I 1
CeH5OCH,C ХСН-СН С(СН3Ь — -^С6Н,<>СН.,К.\СН-СН С(СИ,Р
III" 'III'
СОгН NH—СНСО2Н О -С----N—СНСО2Н
Пенициллин v
с образованием напряженного Р-лактама. ДЦК позволяет осущест-
вить эту основную стадию реакции, хотя и с низким выходом 1211.
Перегруппировка Лобри де Брюина — ван Экенштейна. При
попытках синтеза гликозидов Пассерон и Рекондо [221 обнаружили,
что обработка ДЦК сахаров, обладающих восстановительными свой-
ствами, в горячем метаноле приводит к перегруппировке Лобри де
Брюнна — ван Экенштейна. Так, фруктоза изомеризуется в глюко-
зу, маннозу и психозу. После разбавления водой раствор трижды
экстрагируют эфиром, который затем отгоняют, осветляют норитом
и упаривают до получения сиропа. Сироп растворяют в фосфатном
буфере и прибавляют пекарские дрожжи для разрушения фруктозы,
425
глюкозы и маннозы. После фильтрования и деионизации раствора СН,ОН СН2ОН С = О С-=О 1 1 носн неон 1 16 час при 85° | неон + ДЦК н- сн3он > неон | 16г 160 .ил 1 неон неон 1 1 сн2он сн2он D-Фрук оза D-Псикоза' 39 г
на амберлите МВ-3 прибавляют трихлоруксусную кислоту для осаж-
дения белков и избыток кислоты удаляют экстракцией эфиром.
После концентрации раствора до образования сиропа выделяют пси-
козу в виде фенилгидразона с выходом 25%.
Димерные ангидриды. Производные фенилпропиоловой кислоты
димеризуются под действием ДЦК при температуре ниже 0° в произ-
водные ангидрида 1-фенилнафталиндикарбоновой-2,3 кислоты (Бра-
ун и Стивенсон 123]). Ранее эта реакция была осуществлена с уксус-
ным ангидридом и хлорокисью фосфора.
Дегидратация амидов. ДЦК в пиридине дегидратирует карбо-
бензокси-ь-аспарагин (1) до соответствующего нитрила (2) [24].
сн.,со\н2 сн.,е\
СНСО.,Н СНСОЦ1
I I
Cb.XH CbNH
(I) (2)
Синтез гетероциклов. Очень интересно применение ДЦК для
синтеза барбитуровых кислот [25|.
О цни II 1 с—он X / ’1 СвНп о /CeHu 1
сн., -+ 2 С -7-577’ •х II С—ОН X II 1 о Н2С >С = О ре^О и ХСвНи | CeHu
426
Нуклеотиды. При детальном изучении различных агентов для
образования 3',5'-межнуклеотидной связи Якоб и Корана [261 при-
шли к выводу, что наиболее эффективна комбинация ДЦК с /z-толу-
оле ул ьфох лор идом или мезитиленсульфохлоридом.
Гетероциклические стероиды. 2,3-Секо-5а-холестандиовая-2,3 ки-
слота (1) под действием ДЦК в диоксане при комнатной температуре
превращается в ангидрид (2) с высоким выходом 127). Аналогично
5-оксим 3,5-секо-4-норхолестанон-5-овой-3 (3) кислоты при дегидра-
тации дает 4-оксн-4-аза-Д6-холестенон-3 (4).
1. В о d а п s z к у М. et al., Biochem. Prep., 10, 122 (1963).
2. Sell ni i cl t E., Hitzler F., L a h d e E., Ber., 71, 1933 (1938).
3. C a m p b e I I T. W., Mo n a g I e J. J., Fo I d i V. S., J. Am. Chem. Soc.,
84, 3673 (1962).
4. Khorana H. G., Chem. Revs., 53, 145 (1953).
5. Б p а у н Д. M., «Успехи органической химии», изд-во «Мир», М., 1966, сб. 3,
стр. 79.
6. G г е е n е F. D., К a z a n J., J. Org. Chem., 28, 2168 (1963).
7. V о w i n k е 1 Е., Chem. Ber., 95, 2997 (1962); Angew. Chem., Internat. Ed.,
2, 218 (1963).
8. К h о r a n a H. G., T о d d A. R., J. Chem. Soc., 1953, 2257; Kho-
rana H. G., J. Am. Chem. Soc., 76, 3517 (1954); D e c k e r C. A., Kho-
rana H. G., ibid., 76, 3522 (1954).
9. К h о r a n a H. G., V i z s о 1 у i J . P., J . Am. Chem. Soc., 83 , 675 (1961);
Khorana H. G., Turner A. F., Vizsol у i J. P., ibid., 83, 686 (1961);
Khorana H. G., VizsolyiJ.P., Ralph P. K., ibid., 84, 414 (1962).
10. R a rn m I e r D. H., К h о r a n a H. G., J. Am. Chem. Soc., 85, 1997 (1963).
11. S c h ii s s 1 e r H., Zahn H., Chem. Ber., 95, 1076 (1962).
12. В и z a s A., E g n e 1 1 C.. Freo п P., Compt. rend., 252, 896 (1961); 255,
945 (1962); S t e m p e 1 A., L a n d g r a f F. W., J. Org. Chem.,27, 4675 (1962).
13. NeelakantanS., PadmasaniR., SeshadriT. R., Tetrahedron,
21, 3531 (1965).
14. Sheehan J.C., H e s s G. P., J. Am. Chem. Soc., 77, 1067 (1955); Good-
in a n M.., К e n n e r G. W., Adv. in Protein Chem., 12, 488 (1957); Альберт-
co н H Ф., «Органические реакции», изд-во «Мир», М., 1965, сб. 12, стр. 174.
15. Schwarz Н., В и т р и s F. М., J. Am. Chem. Soc., 81, 890 (1949).
16. Z a h n H., Diehl J. F., Z. Naturforsch., 12B, 85 (1957); F 6 1 s c h G.,
Chem. Scand., 13, 1407 (1959).
17. Bodanszky M., d и V i g n e a и d V., J. Am. Chem. Soc., 81, 5688 (1959).
18. Bonner W. A., McNameeP. J., Org. Chem., 26, 2554 (1961).
19. Woodward R. B., Bader F. E., Bickel H., Frey A. J., Kier-
stead R. W., Tetrahedron, 2, 1 (1958).
20. J о h n s о n W. S., В а и e г V. J., M a r g r a v e J. L., F r i s c h M. A.,
D r e g e r L. H., Hubbard W. N., J. Am. Chem. Soc., 83, 606 (1961).
21. Sheehan J. С., Hener у Loga n K. R., J. Am. Chem. Soc., 81, 3089
(1959).
22. Passer on S., Recondo E., J. Chem. Soc., 1965, 813.
23. Brown D., Stevenson R., Tetrahedron Letters, 3213 (1964).
24. R ess 1 er C., Rotzkin H„ J. Org. Chem., 26, 3356 (1961).
427
25. В о s е А. К., G а г г a t t S., J. Am. Chem. Soc., 84, 1310 (1962).
26. .1 acob T. AL, I< horan a H. G., J. Am. Chem. Soc., 86, 1630 (1964).
27. D oorenbos N . L., W u M. T., Chem. Ind., 1965, 648.
ДИЭТИЛАЛЮМИНИЙИОДИД, (C.H5)2A1I, Мол. вес 212,01.
Д. (1) реагирует с дпазометаном в пентане при —80" с выделением
азота и образованием кристаллического иодметилдиэтилалюминия
(2) (Хоберг 111). Это соединение (2) стабильно в интервале темпера-
тур от —80 до —50°, но при температуре от —10 до 0° разлагается
с выделением этилена. Образование этилена интерпретируется как
результат взаимодействия двух молекул соединения (2) с образова-
(С2Н5)2А11 +СИ2\2—(C2H5uA1CH21
(I) 12)
(С2Н5)2А1СН2СН21 I I
(3) I —с — с—
1 I I
(С2Н5).2А11+СН2 = СН2 1СН2 ацс2н5)2
I ,4)
I I
-С-С- 4-(С2Н5).,А11
сн2
нием соединения (3) и дальнейшего элиминирования диэтилалюми-
нпйиодида. Карбен нельзя рассматривать как промежуточный про-
дукт этой реакции, поскольку при температуре, при которой метал-
лоорганическое соединение (2) стабильно, он быстро реагирует с цик-
логексеном, давая норкаран. Эту реакцию можно также объяснить
простым присоединением с образованием соединения (4) и элимини-
рованием.
1. Hoberg Н„ Ann., 656, 1, 15 (1962).
ДИЭТИЛАЛЮМИНИЙЦИАНИД, (C.;H5)2A1CN. Мол. вес 111,12,
очень вязкое масло с т. кип. 15(1' 0,07 мм.
Получение |1|. К раствору триэтилалюминия в бензоле при
перемешивании магнитной мешалкой и охлаждении льдом прибав-
ляют по каплям раствор цианистого водорода в бензоле. После пре-
кращения выделения этана, растворитель испаряют и остаток пере-
(С2Н6)зА1 + HCN (C2H5)2A1CN + С2Нв
93%
гоняют.
Гидроцианирование. Д. позволяет осуществить более эффек-
тивное гидроцианирование,чем смесьпнанпстого водорода стриэтпл-
алюминпем. Так, холестеион в толуоле реагирует с Д. в 10 000
раз быстрее, чем с вышеуказанной смесью; в отличие от этой смеси
428
применение Д. позволяет раскрывать пространственно сильно за-
трудненные эпоксиды ряда стероидов, такие, как 9а, 11а-эпоксиды.
I. X j ; a t a W., Y о s 11 i о k а М., Tetrahedron Letters, 1913 (1966).
ДИЭТИЛКАРБОНАТ, С2Н.-,ОСО,С2Н5. Мол. вес 118,13, т. кип.
127 , уд. вес 0,975.
Д. применяют для карбэтоксплирования соединений, содержа-
R С Н,С.( ),Со Н 5 4- С., Н 5ОСОС2 Н 5 + С2 Н -,ON a
(1) (2;
Na +
2C,H5OH + RC(CO,C2H5)2
(4)
щих активную метиленовую группу. Этиловый эфир типа (1) обра-
тимо реагирует с лиэгилкарбонатом в присутствии этилата натрия,
давая натриевое производное (3). Даже при неблагоприятном поло-
жении равновесия желаемое превращение достигается применением
4—8 молен диэтил карбоната и отгонкой образующегося этанола Н I.
Обработка соединения (3) кислотой дает замешенный малоновый
эфир (4).
Аналогичная реакция Д. с кетонами в присутствии основных
катализаторов приводит к Р-кетоэфирам 12]. Этиловый эфир фенпл-
циануксусной кислоты получают 13, 41 из сухого свежеприготовлен-
ного этилата натрия (0,52 моля), фенилацетонитрила (0,5 моля) и Д.
О О
CeH5CI 1., - С„115ОС()С.,Н, С„Н5ССОС.Л г,(\а + )
' ’ '-2НОСДЦ j
С.\ CN .
j н +
СсН5СНС( ),С.,Н5
(2,5 моля) в толуоле. По мере отгонки этанола добавляют дополни-
тельное количество толуола. После подкисления енолята получают
этиловый эфир фенилциануксусной кислоты с выходом 70—78%.
При получении три-н-дифенилкарбинола 151 натрий постепенно
добавляют к раствору н-хлордифенила и Д. в бензоле при кипячении
и интенсивном перемешивании. По окончании реакции смесь под-
ЗС„Н5СвН4С1-п + (C.,H,O),CO+6Na-->
35 — 40%
—► 3NaCl -у ЗХаОС2Н5 + (n-C6H5C0H4)3CONa
кисляют и продукт выделяют обычным способом.
1. WallingfordV. Н., HomeyerA. Н., JonesD. М., J. Am. Chem.
Soc., 63, 2056 (1941).
429
2. W a I 1 i n g f о r d V. H., H о in е у е г А. Н., J о n е s D. М., J. Am. Cliem .
Soc., 63, 2252 (1941).
3. W а 1 I i n g f о г d V. H., H о ni e у с г Л. H., Jones D. M., .1. Am. Chem.
Soc., 64, 576 (1942).
4. X о p п и н г Э., Фи нел.1 ii Л., «Синтезы органических препаратов», ИЛ,
М., 1953, сб. 4, стр. 594.
5. Мортон Э., Майлс Дж., Эмерсон В., «Синтезы органических пре-
паратов», ИЛ, М., 1952, сб. 3, стр. 422.
ДИЭТИЛ КАРБЭТОКСИМЕТИЛФОСФОНАТ,
о-
(С,Н5О),Р 1 СН2СО,С,Н5.
Мол. вес 224,20, т. кип. 10970,8 мм, 140710 мм.
Д. получают нагреванием трнэтплфосфита с этилбромацетатом
и используют в модифицированной реакции Виттига 111. Гидрид нат-
рия отрывает активированный атом водорода в а-положенип, обра-
зуя анион соли (2), который действует подобно илиду в реакции с аль-
дегидом или кетоном и даета.р-ненасыщенный эфир (3). Растворите-
лем служит 1,2-диметоксиэтан. Анион соли (2) дает с дифенилкете-
ном аллен (5), а с окисью стирола — циклопропан (6).
О- О-
| NaH | -
(С,НГ>О),Р+СН,СО,С2Н5 —--> (С,НГ,О),Р+ СНСО..С.ЛI-,(\а )—>
— Н2
(I) (2)
(СН,),СО
(СН3),С - СНСО,С2Н,-|- (С.2Нг,О)2Р+О\’а
70/0 (3) I <4)
о-
о-
I -
(С2Н5О)2Р+СНСО2С2Нг,(Ма+) + (СвН5),С=-С=--О
(2)
(СвН5)2С = С = СНСО2С2Н5 + (4)
(5)
о- о СНСО,С,Н5
I - /\ /\
(C2H5O)2P+CHCO2C2H5(Na+) + CGHsCH—СН, CJI-СН —СН,
(2) ” (6)
Уэдсворт и Эммонс [2] к смеси 50%-ной дисперсии гидрида нат-
рия в минеральном масле с бензолом при перемешивании медленно
добавляли Д. при 30—35° в атмосфере азота. При этом энергично
о-
I
(С„Н5О),Р + СН,СО,С,Н, + NaH -!-СвН0
0,33 моля
O(N.)
0,33 моля
0,33 моля 100 мл
= СНСО2С2Н5 + Н2
выделялся водород и образовывался почти бесцветный раствор. За-
тем медленно добавляли циклогексанон и реакционную смесь сна-
430
чала выдерживали при 20—30°, а затем при 60—65°. Продукт реак-
ции, этилииклогексилиденацетат, собирали при 48—49°/0,02 мм.
Д. селективен по отношению к кетостероидам |3|. 3-Кетостероиды
и А 4-3-кетостероиды реагируют легко, но кетонные группы в поло-
жении Се, С7 и С17 с Д. не реагирует. Японские химики исполь-
зовали в качестве основания амид натрия; растворителем слу-
жил ТГФ [4].
1. Wadsworth W. S., Jr., Emmons W. D., J. Am. Chem. Soc., 83, 1733
(1961); WolinskyJ., Erickson K. L., J. Org. Chem., 30, 2208 (1965),
2. Wadsworth W. S., Jr., Emmons W. D., Org. Syn., 45, 44 (1965).
3. В о s e A. K., D a h i 1 1 R. T., Jr., Tetrahedron Letters, 959 (1963);J. Org. Chem.,
30 , 505 (1965).
4. T a k a h a s h i H., F u j i w a г а К. О h t a At., Bull. Chem. Soc., Japan,
35, 1498 (1962).
О О
ДИЭТИЛ КАРБЭТОКСИФОСФОНАТ, С.,Н5ОС-Р(ОСЛ15).. Мол.
вес 130,17, т. кип. 122,5—123°.
Д. получают 111 нагреванием этилового эфира хлоругольной кис-
лоты с триэтилфосфитом (реакция Арбузова):
О О О
II II II
С.,Н5ОСС1 + С..Н5ОР(ОС2Н5)., —С,Н5ОС-Р(ОС2Н5)., + С2Н5С1
Д. имеет преимущества перед диэтилкарбонатом [2] или диэтил-
оксалатом [21 при карбэтоксилировании кетонов 131. Например,
21,2 г этого реагента добавляют к 5 г 5%-ной суспензии гидрида нат-
рия в парафиновом масле и 150 мл дибутилового эфира, после чего
небольшими порциями прибавляют 9,8 г циклогексанона. В течение
часа поддерживают температуру ниже 30°, а затем растворитель
О о
° ° II II °
Д о о Д' С — Р(ОС2Н5)., JI СО2С,Н5
i CjrsOC-P(OC,Hs)2 I |Д Na+ C,H6OH-H2SO4 I V
I ,ViH-B(hO ”11 71% ”l j
(-СгН,ОН) ° / X/
и этанол отгоняют в вакууме. Смесь выливают в безводный этанол,
содержащий серную кислоту, и получают 2-карбэтоксициклогекса-
нон (т. кип. 93—94 ), идентифицированный в виде медного хелата
(т. пл. 178°).
1, N у 1 ё n Р., Вег., 57, 1023 (1924); also see R с е t z Т., С h a d w i с k D. H,,
H a r d у E. E„ К a u f m :: n S., J. Am. Chem. Soc., 77. 3813 (1955).
2. S w a m e r F. \V., HauserC. R., J. Am. Chem. Soc., 72, 1352 (1950).
3. Shahak J., Tetrahedron Letters, 2201 (1966).
ДИЭТИЛСУЛЬФАТ, (C2H5O)2SO?. Мол. вес 154,19, т. кип.
210°, уд. вес 1,17. (См. также Диметилсульфат.)
431
С-Алкилирование. Для получения н-пропилбензола [1] свеже-
перегнанный диэтилсульфат добавляют по каплям к бензилмагний-
хлориду.
Эфир 4 моля(С>Н5О) .SO,
С6Н5СН.,С1 +Mg---->C6H5CH2MgCI-----n СвН5(СН)2СН3
/ U— / о /0
2 моля 2г-атом
I. Гильмап Г., Катлин, «Синтезы органических препаратов», ИЛ, М.,
1949, сб. 1, стр. 364.
ДИЭТИЛ-U, 1,2-ТРИФТОР-2-ХЛОРЭТИЛ)-АМИН,
(C2H5)2NCF2CHC1F. Мол. вес 189,62, т. кип. 32—ЗЗэ/5,5—6,0 мм.
Д. получают из трифторхлорэтилена и диэтиламина [1 ];
(C2H5),NH + CF., = CCIF —+ (C2H5),NCF2CHCIF
Д. реагирует с первичными и вторичными оксистероидами при 25°
в хлористом метилене с образованием соответствующих фторирован-
ных соединений. Эта реакция сопровождается почти полным обраще-
нием конфигурации; только в случае А5-ЗР-оксистероидов наблюдает-
ся сохранение конфигурации. Например, А5-ЗР-оксиандростенон-17
дает соответствующее 30-фторпроизводпое с выходом 78% [21. Было
найдено [31, что направление этой реакции зависит от температуры
и растворителя и что фторирование сопровождается элиминирова-
нием и перегруппировками.
Д. при взаимодействии с 19-оксн-А4-андростеидионом-3,17 (1)
в кипящем ацетонитриле дает 5,19-циклостероид (2) и (3) [4].
Смит и сотр. [51 при попытке получения 1 -фторстеропда дейст-
вием Д. на 1а-окси-5а-андростандион-3,17 выделили три продукта
ретропинаколиновой перегруппировки метильной группы при С19 :
А4-, Д5(Ю, и д9-1р.метилэстреноны-3:
432
Нокс и сотр. [6] нашли, что при взаимодействии Д. с 19-окси-Д5-
3-ацетоксистероидами образуются главным образом три продукта
скелетной перегруппировки.
1. Р г u е t t R. L. et al., J. Am. Chem. Soc., 72, 3646 (1950).
2. Ayer D. E., Tetrahedron Letters, 23, 1065 (1962).
3. К n о x L. H., Velarde E., Berger S., C u a d r i e I 1 о D.,
С г о s s A. D„ ibid., 26, 1249 (1962); J. Org. Chem., 29, 2187 (1964).
4. Knox L. H., V e 1 a r d e E., С г о s s A. D., J. Am. Chem. Soc., 87, 3727
(1965).
5. S m i t h L. L„ F о e 1 1 T. J., T e 1 1 e r D. M., J. Org. Chem., 30, 3781 (1965).
6. Kuo x L. H., Velarde S., Berger S., Delfin I., Grezemkov-
s k у R., Cross A. D., J. Org. Chem., 30, 4160 (1965).
N, N-ДИЭТИЛ-1,2,2-ТРИХЛ0РВИНИЛАМИН,
CI2C - C(C1)N(C2H5)2. Мол. вес 202,52, т. кип. 67°/6,2 мм.
Д. получают при взаимодействии М,М-диэтил-2,2,2-трихлорацета-
мида и триэтилфосфита, которое сопровождается окислением атома
фосфора под действием амида и миграцией атома хлора [1]. Получа-
О С)
’i I
Ci3CCN(C,H6)2 + (C;HsO)3P —► С1.,С = СХ’(С,Н5)., + (С2Н5О)3Р+ — о-
ющийся трихлоренамин легко реагирует с карбоновыми кислотами
или спиртами и дает соответствующие хлорангидриды или алкилхло-
риды с хорошим выходом. С первичными аминами Д. образует ами-
дины |2|.
C6H5COjH + C1.,C = CN(C,H5), CeH6COCl + CloCHCN(C,Hs)2
I - ’ '2"» II
Cl о
\С6Н5
C6HSNH, + С12С = С\(С,Н5), ——* С12СНС\(С.2Н5)2 + НС1
। 82%
С1
1. SpezialeA. J., FreemanR.C., J. Am. Chem. Soc., 82, 903 (1960).
2. SpezialeA. J., Freeman R.C., J. Am. Chem. Soc., 82, 909 (I960).
^ас-(2,6-ДИЭТИЛФЕНИЛ)-КАРБОДИИМИД,
CH2CH3 CH3CH,
— N = C = N — /
CH,CH3 CH3CH2
Мол. вес 306, 20, т. кип. 192—194/0,4 мм, уд. вес 1,007.
О
II
ДИЭТИЛФОСФИТ, (С2Н3О)2РН, или (С2Н5)2РОН. Мол. вес
138,11, т. кип. 187—188е (73—74'/14 мм). Д. получают с выходом
93% реакцией этанола с РС13 в СС14 или в эфире 111.
433
Восстановление фенолов. Д. позволяет осуществить удобное
восстановление фенолов до соответствующих углеводородов [2|.
Раствор Д. и фенола в СО4 обрабатывают триэтиламином и выдер-
живают 24 час’для полного осаждения хлоргидрата триэтиламина (1).
Образующийся фенилдиэтилфосфат растворяют в ТГФ и восстанав-
ливают натрием в жидком аммиаке (2). Выходы неочищенных угле-
О
II
1) ArOH + HP(OC2H6)2 + CCl4 + (C2H5)3N —
о-
—> АгОР + (ОС..Н5)„ 4- СНС13-|- (C„H-,)3N • НС1
о- " ' о-
I I
2) АгОР-* ((X.,H5)2 + 2Na + NH3 —> ArH + NaNH.,-;-N'aOP+(OC2H5)3
водородов 70—80%. Пельтье [31 нашел, что эту реакцию можно ис-
пользовать в полумикромасштабе (50—200 мг) для идентификации
фенольных соединений, получающихся при расщеплении природных
продуктов.
Применение в синтезе. Уэдсворт и Эммонс [41 получили диэти-
ловый эфир циклогексиламида фосфорной кислоты (3) конденсацией
Д. с четыреххлористым углеродом и циклогексиламином по Тодду
(1945 г.). Первоначально образующийся трихлорметилфосфонат (2)
О О ОН
II C,H,,NH, II C,H,,NH. И I Nall
(С2Н5О)2РН +СС14---(С2Н6О)2РСС13—(СНзО^РХСеНн— -
— ПО1 — С, г! О» i t — г 1 •>
О) (2) (3)
О о
II - C«HSN = C = O II
-> (C,H9O)2P-NC6H11(Na+)---—---> С6Н5\ =С== МС„НИ + (С2Н5О).,РО.\а
о и /о
(4) (5) (6)
при взаимодействии со второй молекулой амина (отщепление хлоро-
форма) дает соединение (3). Обработка фосфамида (3) гидридом нат-
рия в 1,2-диметоксиэтане дает очень реакционноспособное промежу-
точное соединение — натриевую соль фосфамида (4). Реакция этой
соли с карбонильными соединениями показана на примере взаимо-
действия с фенилизоцианатом с образованием N-фенил-М'-циклогек-
силкарбодиимида (5). В этой реакции можно применять и другие
первичные амины. Карбонильным компонентом может служить аль-
дегид, кетен, двуокись углерода или сероуглерод.
Специфический растворитель. Д. смешивается с водой и обыч-
ными органическими растворителями и обладает высокой растворя-
ющей способностью. Он рекомендован в качестве растворителя и ак-
цептора протонов в пептидных синтезах с применением тетраэтилпи-
рофосфита [51 или таких реагентов, как этилдихлорфосфит 161.
Д. применяют как растворитель для получения а рил гидразонов
и 2,4-динитрофенилгидразонов |7|. В этом случае не требуются кис-
434
лые катализаторы, так как следы воды гидролизуют этот реагент до
кислого этилфосфита НР(О) (ОС2Н5)ОН. Выходы повышаются при
добавлении небольшого количества растворителя. Обычно реакция
протекает достаточно быстро при комнатной температуре, но выходы
повышаются при нагревании смеси па кипящей водяной бане в те-
чение 1 чае.
1. М с С о m b 1 е Н., S a u л d ers В. С., StaccvG. J., J. Chem. Soc., 1945,
380.
2. К е n n е г G. W., W i I 1 i a m s N. R., J. Chem. Soc., 1955, 522.
3. Pelletiers. W„ Locke D. M„ J. Org. Chem., 23, 131 (1958).
4. Wadsworth W. S., Jr., E m mo л s W. D., J. Am. Chem. Soc., 84, 1316
(1962).
5. A nderson G. W., Biodinger J., Welcher A. D., J. Am. Chem.
Soc., 74, 5309 (1952).
6. Y oung R. \V., Wood К. H., JovceR. J., Anderson G. W., J. Am.
Chem. Soc., 78 , 2126 (1956).
7. M aynard J. A., Australian J. Chem., 15, 867 (1962).
ДИЭТИЛФОСФОНИОДУКСУСНОЙ кислоты этиловый
ЭФИР,
(QHsO^P+CHlCOAHs
Реагент получают in situ обработкой триэтилфосфонацетата
гидридом натрия в 1,2-диметоксиэтане, после чего добавляют иод
111. Он реагирует с бензальдегидом в присутствии 2 экв гидрида нат-
рия, образуяэтилпропиолат.
1) NaH
2) Г.
(С2Н,О)2Р+СН2СО,С2Н 5---—i--------> (С,Н 5О),Р+СН IСО2С,Н,
' 1 •' ' | 2 - 2 5СН,ОСН2СН2ОСН, ' - 5 72 > 2-5
о-
C,H,CHO+2NaH
CGH5C ССО.,С2Н5 - I- (C2H5O)2P + ONa ф Nal + Н2
1. W a d s w о г t h W. S., Jr., Emmons W. D., J. Am. Chem. Soc., 83, 1733
(1961).
ДИЭТИЛХЛОРФОСФАТ, (C2H6O)2POC1. Мол. вес 172,56, т. кип.
64: '6—7 мм.
Получение. Д. получают с выходом 81% реакцией диэтилфо-
сфита с четыреххлористым углеродом и триэтиламином [1].
АгОН —* АгН. Гольдкамп и сотр. [2] нашли, что для удале-
ния фенольного гидроксила эстрона по методу Кеннера и Виль-
435
ямса [31 лучше применять чистый Д., чем получать его из диэ-
тилфосфита in situ.
1. Steinberg G. M., J. Org. Chem., 15, 637 (1950).
2. К e n n e r G. W., W il 1 i a ms N. R., J. Chem. Soc., 1955 , 522.
3. Goldkamp A. H.et al., J. Med. Chem., 8, 409 (1965).
ДИЭТИЛХЛОРФОСФИТ, (C2H,O)2PC1. Мол. вес 156,55, т. кип.
57°/30 мм.
Д. получают кипячением 0,2 моля триэтилфосфита с 0,1 моля
РС1Э и перегонкой продуктов реакции [11:
2(C,H5O)3P-l РС13 —> 3(С2Н6О)2РС1
Применение Д. в пептидном синтезе можно иллюстрировать следую-
щей схемой [2j:
R R
I (С,Н6О),РС1 I
H2NCHCO2C2H5 ————> (С2Н5О),Р\,НСНСО2С2Н5—*
У' R' R
CbNHCHCO.H | |
--------CbXHCHCONHCHCO,C2H5 + (С„Н5О)2РОН
1. Со о k Н. G., Ilett J. D., Saunders В. С., Stacey G. J., Wat-
son Н. G., Wil ding 1. G. E., Woodcock S. J., J. Chem. Soc., 1949,
2921.
2. A n d e r s о n G. W., BlodingerJ., YoungR. W., W e 1 c h e r A. D.,
J. Am. Chem. Soc., 74 , 5304 (1952).
ДИЭТИЛЦИАНАМИД, (C2H6)2NCN. Мол. вес 98,15, т. кип.
68г/Ю мм. Д. получают реакцией бромциана (1 моль) с диэтилами-
ном (2 моля) [1].
Цианамид N^C — NH., в таутомерной форме является карбоди-
имидом HN~C=NH и может использоваться в пептидном синтезе.
Хотя Д. и не способен к таутомерии, тем не менее он действует
как дегидратирующий агент при конденсации N-защищенных амино-
R’
I
(C,H6).,NCN H.NCHCO2C2Hs
CbNHCHCO,H------;--> CbXHCHCOCX(C.,H5)2 —-------»-
I I i; ii
R RON
R OHR' О
I II I I II
—> CbNHC.HCNCHCO2C2H5 + H2XCX(C2H5),
кислот с эфирами аминокислот |2|. Наилучшие выходы получены
при нагревании трех компонентов без растворителя при 100е в тече-
ние 2 час.
1. Wallach О., Вег., 32, 1872 (1899).
2. Losse G., W е d d i g e H., Ann., 636, 144 (1960).
436
о-
ДИЭТИЛЦИАНМЕТИЛФОСФОНАТ, (C2H5O)2P+CH2CN. Мол.
вес 177,17.
Д. используют в модифицированной реакции Виттига; при взаи-
модействии Д. с альдегидами и кетонами в присутствии сильных
оснований в качестве катализаторов образуются а,0-ненасыщенные
нитрилы:
О- О-
| NaH I
(C.2H5O)2P+CH2CN +С.,Н5СОН СД15СН =СНС\’ 4- (С3Н5О),Р+ ONa
Реакцией Д. с эпоксидом (1) получают производное циклопропана (2):
о- сн, сн, о-
I \ NaH / \ |
(С,Н6О),Р + СН,С\ 4-CJUCH— О-' C6HSCH— CHCN + (C2H5O)2P+ONa
“ 51%’
(1) (?)
В первоначальной методике Уэдсворта и Эммонса 111 в качестве
основания применяли гидрид натрия и 1,2-диметоксиэтан как раст-
воритель. В методике японских авторов [21 используют амид натрия
и ТГФ. Д. реагирует со стероидами, имеющими кетогруппу при С3,
С17 или С,о, но менее селективно, чем диэтилкарбэтоксиметилфосфо-
нат (С,Н5О),Р+ СН2СО.,С2Н5, который взаимодействует только с
I
о-
3-кетостероидами [3].
1. Wadsuort Ii W.S., J, Е m mon s W. D., J. Am. Chem. Soc., 83, 1733 (1961).
2. Takahashi Н., Fujiwara К., О h t а М., Bull. Chem. Soc., Japan, 35,
1498 (1962).
3. В о s e A. К., D a h i 1 1 R. T., Jr., J. Org. Chem., 30, 505 (1965).
ДИЭТИЛЦИНК —МЕТИЛЕН ИОДИСТЫЙ. Этот реагент при
взаимодействии с циклогексеном дает норкаран с удовлетворитель-
1 9Кв
(C2Hs)2Zn + сн212
1 экв 1 экв
53%*
ным выходом [II. Сходное с реакцией Симмонса —Смита, это пре-
вращение значительно более быстрое; для предотвращения бурного
выделения тепла необходимо медленное добавление йодистого мети-
лена. Следует иметь в виду, что диэтилцинк возгорается на воздухе.
1. Furukawa J., Kawabata N, Nishimura J., Tetrahedron Let-
ters, 3353 (1966).
437
ДИЭТИЛЭТИЛЕНПИРОФОСФИТ (3). Мол. вес 228,12, г. кип.
68—69'/0,4 мм.
Д. получают из этпленхлорфосфита (1) и диэтилфосфита (2) II]
и подобно тетраэтил пирофосфиту применяют для образования пептид-
ных связей. Преимущество Д. в более легком получении.
О
сн„о
PCI т НР(ОС2Н
(C2H„).,N
СНЛ)
| ХР — ОР(ОС,Н5),
СН2С)
(3)
1. A ndf г s о н G. W., McGregorA. С., J. Am. Chem. Soc., 79, 6180 (1957).
сн.,о
ДИЭТОКСИМЕТИЛАЦЕТАТ, СН;1СО.СН(ОС ,НГ,)2. Мол. вес
162,18, т. кип. 79—80 ;24 мм, 85—87’/30 л/л/.
Д. получают равновесной реакцией уксусного ангидрида с орто-
муравьпным эфиром (41% превращения) 11]. Д. предпочитают сме-
(СН3СО)2О4 НС(ОС2Н6)3^СН3СО2С2Н,-|-СНзСО!!СН(ОСгН6)г
си уксусного ангидрида и ортомуравьиного эфира для превращения
2,6-д11ХЛор-4,5-диаминоппримидина (1) в 2,6-дихлорпурин (2) [2].
СН3СО2СН(ОС2Н5)2 (120°)^
С1
85-90%
С1
н
(2)
1. Post Н. \V„ Erickson Е. R., J. Org. Chem., 2, 260 (1937).
2. М о n t g о ш е г у J. A., Tempi еС., Jr., J. Am. Chem. Soc., 79, 5239 (1957);
Montgomery J. A., H о 1 u m L. B., ibid., 80, 404 (1958); Montgo-
mery j. A., TempleC., Jr., ibid., 80, 409 (1958).
2,2-ДИЭТОКСИПРОПАН(диэтилкеталь ацетона), (CH 3)2C(OC2H.-,).,
Мол. вес 132, 20, т. кип. 115с Д. получают из триэтилортоформиата,
ацетона, абсолютного этанола в присутствии следов //-толуолсульфо-
кислоты; выход 75% [1].
Д. кипит при более высокой температуре, чем 2,2-диметоксипро-
пан (т. кип. 80е), и поэтому более приемлем для превращения цикли-
тов в их полиацетонидные производные обменной реакцией |2].
он он
но!—I он
И N) + з (сн3)с(ос2н5)2
он он
438
Смесь измельченного циклита, 2,2-диэтоксипропана и каталитиче-
ских количеств /i-толуолсульфокислоты нагревают на кипящей водя-
ной бане 1—2 час. При использовании обычной методики (ацетонЧ-
хлористый цинк, 5 час) получается смесь ди- и триацетонидов.
1. Н ur d С. D., Р о 1 1 а с k М. A., J. Am. Chem. Soc., 60, 1905 (1938).
2. AngyalS. J., Hoskinson R. M., J. Chem. Soc., 1962, 2985.
содержание
Предисловие.......................................................... 5
Основные сокращения ................................................. 7
А
1-Адамантилхлорформиат............................................... 9
Адамса катализатор................................................... 9
Адипиновая кислота................................................... 9
Адкинса катализатор (смесь окисей меди и хрома)...................... 10
п-Азобензолкарбоновон кислоты гидразид............................... 11
Азодинзобутнронитрпл................................................. 11
Азоди карбоновой кислоты диамид..................................... 12
Азодпкарбоновон кислоты ди-трет-бутиловый эфир..................... 12
Азодикарбоновой кислоты дикалиевая соль............................ 12
Азодпкарбоновон кислоты дцэтиловын эфир............................ 13
Азот.............................................................. 15
Азота окислы (в основном NoOt и немного N,O3 и NO,)............... 16
Азота пятнокись (азотный ангидрид)................................ 17
Азота четырехокись................................................ 18
Азота четырехокись — бора трифторид............................... 24
Азотпстоводородная кислота.......................................... 25
Азотистый ангидрид — бора трифторил................................. 28
Азотная кислота................................................... 28
Акрилонитрил...................................................... 31
Р-Алаиии............................................................ 32
Алкиламинбораны..................................................... 32
Алкилсульфаты....................................................... 33
Алкилцианнды........................................................ 33
Алкилы иодистые..................................................... 33
Аллен............................................................... 34
Аллиллитий.......................................................... 35
Алюминия амальгама.................................................. 35
Алюминия бромид..................................................... 37
Алюминия гидрид..................................................... 38
Алюминия иодил.................................................... 39
Алюминия окись для дегидратации спиртов............................. 39
Алюминия окись для хроматографии.................................... 40
Алюминия окись для этерификации..................................... 41
Алюминия окись, окрашенная морином.................................. 41
Алюминия хлорид..................................................... 41
Амид калия.......................................................... 54
Амид лития.......................................................... 57
Амил натрия......................................................... 58
н-Амилнитрит........................................................ 66
п-Аминоазобензол.................................................... 66
1-Аминобензотриазол................................................. 67
2-Амино-2-метплпронанол-1........................................... 67
N-Аминофталнмид..................................................... 67
Аммония анегат и формиат............................................ 68
440
Аммония метаванадат.................................................. 69
Аммония нитрат....................................................... 70
Анизол............................................................... 70
Анилин............................................................... 70
Антрацен-9, Юдиимин.................................................. 70
Арилдназония гексафгорфосфаты........................................ 70
Арилдиазония тетрагалогенбораты...................................... 71
Ацетальдоксим........................................................ 73
Ацетамид.......................................................... 73
л-Ацетамидобензолсульфохлорид..................................... 74
Ацетилаиетон (2,4-пентандион)..................................... 74
Аиетилгексафторантимонат.......................................... 75
Аиетилгипобромит.................................................. 76
Ацетилен.......................................................... 77
Ацетиленди карбоновая кислота..................................... 78
Ацетиленди карбоновой кислоты диметиловый эфир.................... 78
Ацетплендикарбоновой кислоты диэтиловый эфир...................... 79
Ацетиленид лития.................................................. 79
Ацетиленид лития—этилендиамин..................................... 80
Ацетиленид натрия................................................. 81
N-Ацетилимпдазол.................................................. 81
S-Ацетилмеркантоятарной кислоты ангидрид.......................... 82
Ацетилнитрат...................................................... 82
а-АцетиЛ'Р-этокси-М-карбэтоксиакриламид . ........................ 83
а-Ацетоксиакрилонигрил............................................ 83
1-Ацетоксибутадиен................................................ 83
2- и З-Ацетоксипиридины........................................... 85
N-Ацетоксифталимид................................................ 85
зр-Ацетокси-Д6-этиеновая кислота.................................. 86
Ацетондикарбоновая кислота ....................................... 86
Ацетонциангидрин.................................................. 87
Ацетонциангидрина нитрат.......................................... 88
Ацетоуксусной кислоты mpem-бутиловый эфир. . .................. 89
Ацетоуксусной кислоты фторангидрид................................ 90
Ацилов перекиси .................................................. 90
Ацилы фтористые................................................... 91
Б
Бария перманганат.................................................... 93
Бензальдегид......................................................... 93
Бензиламин........................................................... 93
Бензилат натрия ..................................................... 94
Бензиллитий.......................................................... 94
Бензилтриметиламмонийиодид........................................... 95
Бензилтриметиламмониймезитоат........................................ 95
Бензилтриметиламмонийхлорид.......................................... 96
Бензилтриметиламмонийцианид.......................................... 96
О-Бензилфосфористый-О.О-дпфенилфосфорный ангидрид ................... 96
Бензилхлорметиловый эфир............................................. 97
Бензоила перекись ................................................... 98
Бензоилизотиоцианат ................................................. 100
Бензоил хлористый.................................................. 100
Бензойная кислота ............................................ 101
Бензойной кислоты натриевая соль ................................. 101
Бензойной кислоты серебряная соль ................................. 102
Бензойный ангидрид .................................................. 103
о-Бензолдиазонийкарбоксплат (предшественник дегидробензола) .... 104
Бензолсульфокислоты ангидрид......................................... 104
44)
Бензолсульфонат натрия............................................. 104
Бензолсульфопплизонпанат........................................... 105
Беизолсульфохлорнд................................................. 105
б«с-(Бепзонитрнл)-палладий(11)хлорил............................... 106
1,4-Бензохнпон (п-хпнон)........................................... 107
1,2,3-Бензтиадиазол-1,1-диокспд.................................... 108
Берча восстановление............................................... 108
Бора трибромид..................................................... 110
Бора трифторид..................................................... 111
Бора трифторид—метанол............................................. 112
Бора трифторида— уксусной кислоты комплексы........................ 112
Бора трифторида эфнрат ............................................ 113
Бора трифторида эфнрат — уксусный ангидрид......................... 116
Бора трихлорид..................................................... 116
Борная кислота..................................................... 118
Борфтористоводородиая кислота...................................... 122
N-Бромацетамид (NBA)............................................... 125
N-Бромацетамид — пиридин — S0.2.................................... 126
N-Бромацетамид — фтористый водород................................. 126
N-Бромацетамид — хлористый водород................................. 126
Бром — ацетат серебра.............................................. 127
о-Бромиодбеизол.................................................... 127
Бромистоводородная кислота, постоянно кипящая (48%-пая НВг) . . . 128
Бромистый водород безводный........................................ 131
Бромистый водород— уксусная кислота................................ 132
N-Бромкапролакгам.................................................. 132
Броммочевпна....................................................... 133
Бромноватистая кислота водная...................................... 133
Бром — окись серебра............................................... 133
Р-Бромпропионнлизоцианат........................................... 134
(5-Бромпропионитрил................................................ 134
N-Бромсукцинимид (NBC)....................................... .... 134
4-Бром-2,5,7-тринитрофлуорен....................................... 137
Бромтрифторметан.................................................. 138
Бромтрихлорметан................................................... 138
и-Бромфеиацилбромид................................................ 138
Бромцпан........................................................... 138
(J-Бромэтилметилкетона этнленкеталь............................... 141
Бутадиен.......................................................... 141
D-(—)-Бутандиол-2,3............................................... 141
ц-(-|-)-Бутандитиол-2,3............................................ 142
«рет-Бутила гидроперекись.......................................... 142
я-Бутилазид........................................................ 144
трет-Бутил амии.................................................... 145
mpe/n-Бутила перекись.............................................. 145
mpem-Бутилат алюминия.............................................. 147
/лре/л-Бутилат калия.............................................. 14
mpem-Бутнлгипобромит............................................... 168
/прещ-Бутнлгипоиодит............................................... 168
щрет-Бутнлгипохлорит............................................... 169
н-Бутиллитпй....................................................... 174
mpem-Бутиллитий.................................................... 175
я-Бутилнитрит...................................................... 176
щрет-Бутилнитрит................................................... 177
тре/л-Бутил-и-нитрофенилкарбоиаг................................... 177
mpewi-Бутил хлористый.............................................. 177
щре/л-Бутилхромат . ............................................... 178
2-я-Бутилциклогексилат натрия...................................... 178
у-Бутиролактои..................................................... 179
442
в
Вильсмсйера реагент.................................................. 180
Винилацетат.......................................................... 180
Впнил-(карбметоксиметил)-кетон (метиловый эфир акрилоилуксусной
кислоты)........................................................ 181
Винил-ф-карбметокснэтил)-кетои....................................... 182
Внпиллитий........................................................... 183
Випилтрифенилфосфонипбромид.......................................... 184
Винной кислоты натрий-калиевая соль, тетрагидрат (сегиетова соль) , . 185
Висмута трехокись ................................................... 186
Г
Газы инертные........................................................ 187
Галоформная проба.................................................... 187
Гальвиноксил ........................................................ 188
Гексаметилбензол .................................................... 188
Гексаметилдисилазан ................................................. 189
Гексаметилентетрамин (уротропин)..................................... 189
Гексаметилтриамид фосфористой кислоты................................ 193
Гексаметилтриамид фосфорной кислоты (гексаметапол)................... 195
Гексафторбутип-2..................................................... 196
Гексафторфосфорная кислота .......................................... 196
Гексахлорциклопентадиен.............................................. 196
Гексаэтилтриамид фосфористой кислоты................................. 196
Гиамин 1622 и гиамин 3500 ........................................... 197
Гидразин............................................................. 197
Гидразина ацетат..................................................... 211
Гидразина сульфат ................................................... 211
Гидроксиламин-О-сульфокислота........................................ 211
Гидроксиламина сульфат .............................................. 214
Гидроксиламина хлоргидрат ........................................... 215
Гидроперекиси алкилов ............................................... 219
Гипогалогеиитов растворы............................................. 219
Гиппуровая кислота .................................................. 222
Глиоксаль............................................................ 223
Глиоксиловой кислоты ,a-6yiилового эфира л-толуолсульфпмид .... 224
Глицин............................................................... 225
Глутаровая кислота .................................................. 225
Глутаровый диальдегид................................................ 227
Гриньяра реактивы.................................................... 227
д
Даутерм A (Dowtherm А)............................................... 239
Дегидроабиетиламип................................................... 240
1,5-Диазабицикло-(3,4,0|-ионен-5..................................... 240
Диазомеган........................................................... 242
Диазометандисульфонат калия ......................................... 248
Диазоуксусный эфир................................................... 249
9-Дпазофлуорен....................................................... 252
Дп-н-амила.мин....................................................... 252
Дп ацетилен.......................................................... 252
тр«нс-/пра«с-1,4-Диацстокс1|бутадисн................................. 254
Дибензилкетои........................................................ 256
Дибеизнлфосфат серебра............................................... 256
Дибензилфосфнт....................................................... 257
Дибензилхлорфосфат................................................... 257
транс-1,2-Дибеизоилэтилен............................................ 257
443
Дибор аи............................................................... 258
1,3-Дибром-5,5-диметилгидантони (< дибромантин»)....................... 269
Дибромдифтор,метан..................................................... 269
Диброммалоновой кислоты диамид......................................... 270
Дпбромэтан............................................................. 270
Ди-и-бутил амин........................................................ 271
Ди-тпрет-бутилиминоксил (радикал)...................................... 271
Ди-«-бутилоловодихлорид................................................ 272
2,6-Ди-тре/п-бутнлпиридин ............................................. 272
2,2-Ди-н-бутоксинропан (ди-«-бутил кеталь ацетона)..................... 272
Дивпинлртуть........................................................... 272
Дигпдропнран........................................................... 273
тстракнс-(\'-Дигидропиридил)-алюмииат.................................. 275
Диглим (днметиловый эфир диэтиленгликоля).............................. 275
Диизобутилалюминийгнлрид............................................... 275
Диизопинокамфилборан................................................... 279
Диизопропилпатрийамид.................................................. 279
Диизопропилпероксидикарбонат ........................................ 280
Ди им ид............................................................... 280
1,3-Дииод-5,5-ли.метилгидантоип........................................ 282
Ди кетен.............................................................. 283
Дикетен — ацетон, аддукт 1.1.5-триметил-2,6-диоксацпклогексен-4-он-3. . 286
Дикетен — ацетофенон, аддукт........................................... 286
Дикобальтоктакарбоиил.................................................. 286
Дильса — Альдера реакция, растворители.............................. 291
Днмедои (5,5-диметилццклогександион-1,3)............................... 299
/ир«с-(Д11метиламило)-фосфи11....................................... 301
л-Димстиламинобепзальдегид ......................................... 301
Диметиламиноборан...................................................... 302
З-Диметиламииопропиламнн............................................... 302
\-(3-Ди.метиламинопропнл)-л'-этнлкарбодиимнл, х.торгидрат.............. 303
Диметиламшгосульфонплметилидлнтпй...................................... 303
М.Х-Диметнлаиплин...................................................... 303
Диметилацетамид........................................................ 305
Диметилацетамида диэтнлаиеталь......................................... 306
2,3-Диметнлбута.диен................................................... 307
2,3-Днметилбутил-2-боран «гексилборан»................................. 307
М.М'-Диметнлгндразин .................................................. 308
N, М'-Диметилглициигидразил, хлоргидрат (реактив D) ................... 309
!,2-Диметил-4,5-ди-(меркаптометил)-бснзол.............................. 309
Димегилкетсн........................................................... 310
Ди.мстил магний ....................................................... 312
бис-( 1,2-Лиметиллропил-11-боран (< диизоамплборан>)................... 313
Диметилсульфат......................................................... 316
Днмегилсульфат — бария окись........................................... 318
Димстплсульфат—калия карбонат.......................................... 318
Диметилсульфит......................................................... 318
Диметилсульфоксид (ДМСО1............................................... 319
Диметилсульфоксида производные, а) Метилсульфинилметилиднатрий
(днмсилнатрий); б) Диметнлсульфоиипметилид; в) Диметилсульфоксо-
нийметилид........................................... 335, 339, 341
Диметилсульфои.................................................... 345
N, N-Диметил /г-фениленднамин..................................... 345
Диметилформамил (ДМФА)............................................ 346
Диметилформамнда динеопентилацеталь............................... 351
Диметилформамнда диэтилацеталь.................................... 351
Диметилформамнда эгиленкеталь................................... 352
Диметилформамид — ацетат натрия . ......................... 353
444
Диметилформамид — диметилсульфат....................................
Диметилформамид — тионилхлорид (реактив Вильсмейера)................
Диметилформамид — хлорокись фосфора (или фосген)....................
Диметилфосфит.......................................................
3,5-Диметил-4-хлорметилизоксазол....................................
2,2-Диметоксипропан (диметилкеталь ацетона).........................
5,5-Диметокси-1,2,3,4-тетрахлорциклопентадиен.......................
4,4'-Диметокситрифенилхлорметан...............................
1,2-Диметоксиэтан («тлим»)..........................................
Динатрийфенаитрен...................................................
2,4-Динитробензальдегид.............................................
Ди-га-нитробензилхлорфосфат.........................................
3,5-Динятробензоил хлористый........................................
3,4-Динитробензойная кислота........................................
2,4-Динитробензолсульфенхлорид......................................
4,4'-Дииитро-3,3'-дикарб(>ксидифенилди('ульфид......................
2,4-Динитроиодбеизол . .........................................
2,4-Динитрофенилгидразии............................................
бнс-(2,4-Динитрофенил)-карбонат.....................................
Ди-п-нитрофенилфосфат...............................................
2,4-Дииптрофторбензол (ДНФ).........................................
Диоксаи.............................................................
Диоксаидибромид.....................................................
Диоксандифосфат.....................................................
Дитиоциан (родан)...................................................
Ди-п-толилкарбодиимид...............................................
Ди-га-толуил-D-винная кислота.......................................
Дифенил ацетилен....................................................
2-Дифенилацетил-1,3-индандион-1 -гидразон...........................
2,3-Дифепилбутадиен-1,3.............................................
транс-транс-1,4-Дифенилбутадиен.....................................
1,1-Дифенилгилразин.................................................
Дифенилдиазометан..................................
а,у-бцс-(Днфенилен)-Р-фенилаллильный радикал (БДФА)
1,3-Днфенилизобензофуран (2,5-дифенил-3,4-бензофуран)
Дифенилиодопинбромид...............................
Дифенилнолоиий-2-карбокс11лат (моногидрат).........
Дифенил нодонийхлорнд..............................
Дифенилкарбаминонлхлорид...........................
Дифенилкарбодиимид.................................
Дифеиилкетеи.......................................
Дифенилкетена п-толилимин..........................
N, К:-Дифенил-\'-пикрнлгидразил (радикал)..........
Дифенилсилан ......................................
Дифенилстаннап............................... . . . .
Дифепилсульфоксид..................................
Дифенилтеллурид..............................
N, N'-Днфенилформамидии.........................
Дифенилфосфат серебра...........................
Дифенилфосфннлитий.................................
Дифенилфосфорилизотиоцианат........................
Дифенилхлорфосфат..................................
Дифенилцинк........................................
N, N'-Дифенилэтнлендиамин..........................
Дифторамин.........................................
1,1-Дифтор-2-2-дихлорэтилен........................
снлок-Дифтортетрахлорацетон........................
Дифгорхлоруксусной кислоты натриевая соль..........
353
351
358
360
361
361
363
364
365
366
366
367
367
368
368
370
т-0
373
374
376
377
378
379
380
381
382
382
383
384
385
385
387
388
390
390
391
392
392
393
394
395
395
395
397
397
397
368
399
399
400
401
401
402
403
403
404
445
Ди хлорамин......................................................... 404
1,3-Днхлорбутен-2................................................... 405
1,1-Дихлордиметиловый эфир.......................................... 407
2,3-Дихлор-5,6-дициан-1,4-бензох11ион (ДДХ) ........................ 407
1,1-Дихлордиэтиловый эфир........................................... 413
Ди хлор кетен....................................................... 414
Днхлорметилентрифеиилфосфоран....................................... 414
Дпхлорметиллитин, трихлорметиллитий ................................ 416
Дпхлорформоксим..................................................... 417
1,1-Дихлорэтилен.................................................... 417
Днипанацетилен (дипнтрил ацетилеиднкарбоновой кислоты) ............. 418
«Дициандиамид» = N-Циангуанидип .................................... 419
Дииианметилен-2,4,7,-триш1трофлуореп................................ 421
N, Х'-Дициклогексиламидин Ы-морфолплкарбоповой кислоты.............. 421
Дициклогексиламин................................................... 422
Днииклогексилкарбодиимид (ДНК)...................................... 422
Диэтплалюминийподид................................................. 428
Диэтилалюминийциаипд................................................ 428
Диэтил карбонат................................................... 429
Диэтилкарбэтоксиметилфосфонат....................................... 430
Диэтилкарбэтоксифосфоиат............................................ 431
Диэтнлсульфат....................................................... 131
Диэтил-(1,2,2-трифтор-2-хлорэтил)-амин ............................. 432
N,\-Диэтил-1,2,2-трихлорвиниламин................................... 133
бис-(2,6-Диэтилфенил)-карбодиимид................................... 433
Диэтилфосфит........................................................ 133
Диэтилфосфониодуксусной кислоты этиловый эфир....................... 435
Диэтилхлорфосфат.................................................... 435
Диэтил хлорфосфит................................................... 436
Диэтилнианампд...................................................... 436
Диэтилиианметилфосфоиат............................................. 437
Диэтилиинк — метилен йодистый....................................... 437
Диэтилэтиленпирсфосфит.............................................. 438
Диэтоксиметилаиетат................................................. 438
2,2-Диэтоксипропан (диэтилкеталь ацетона)........................... 438
Е—