/



























































































































































































































































































































































































































































































Текст
ИЗДАТЕЛЬСТВО
«М ИР»
Reagents for
Organic Synthesis
LOUIS F. FIESER
Sheldon Emery Professor of Organic Chemistry
Harvard University
MARY FIESER
Research Fellow in Chemistry
Harvard University
JOHN WILEY AND SONS, INC.
NEW YORK - LONDON SYDNEY
1968
Л. ФИЗЕР, М. ФИЗЕР
Реагенты
для органического
синтеза
ТОМ III
(О-Т)
ПЕРЕВОД С АНГЛИЙСКОГО
доктора хим. наук Н. С. ЗЕФИРОВА,
канд. хим, наук В. С. ПЕТРОСЯНА,
доктора хим. паук А. Ф. ПЛАТЭ
и канд. хим. наук С. С. ЧУРАНОВА
ПОД 'РЕДАКЦИЕЙ
академика И. Л. КНУНЯНЦА
и доктора хим. наук Р. Г. КОСТЯНОВСКОГО
МОСКВА 1970
ИЗДАТЕЛЬСТВО «МИР»
УД/' 661.7/54-41
Редакция литературы по химии
Инд. 2-5-3
~ 70
О ===---------------------- —-" 1 ' —
ОЗОН, Оз, газ голубоватого цвета, нестабильный; жидкий озон
окрашен в темно-синий цвет; т. пл. —192,5°, т. кип. —111,9°.
Оборудование. Лабораторные озонаторы создают концентрацию
О. в воздухе до 4 и в кислороде — до 8 вес. %. В озонаторе имеется
приспособление для непрерывного определения концентрации О,
в газе-носителе, принцип действия которого основан на измерении
теплопроводности. Полученные данные регистрируются самописцем
с движущейся лентой. Озонатор снабжен портативным осушителем
воздуха, представляющим собой автономную систему, состоящую
нз компрессора и осушителя; за ним не требуется наблюдения.
В бюллетене Уэлсбаха «Реакции озонирования органических
соединений и аппаратура» (4-й выпуск, январь 1962 г.) содержится
много полезных сведений о механизмах реакций и эксперименталь-
ных условиях, описываются лабораторные реакторы, приведена
библиография, включающая 91 ссылку. «Общий справочник по при-
менению и лабораторной технике озонирования» Уэлсбаха содержит
описание свойств и применения О. и рекомендации по технике экспе-
римента и методам анализа.
Конструкция генераторов озона. Смит и сотр. [1] дают подробное
описание и схемы устройства лабораторного генератора типа, пред-
ложенного Харрисом, с тремя трубками Бертло. Предельно допу-
стимая концентрация О. в воздухе составляет от 0,15 до 1 ч. на мил-
лион, поэтому выделяющийся газ пропускают через устройство для
каталитического разложения О., состоящее из двух колонок высотой
75 см, наполненных битым стеклом, смоченным 5%-ным раствором
едкого натра. Хенне и Хилл [2] использовали генератор с тремя
трубками Бертло и пропускали ток кислорода, содержащего 6%
О., в раствор 0,5 моля 6-метилгептена-1 в 200 мл хлористого мети-
лена при—78° со скоростью 20 л!час в течение 12 час. Полученный
раствор постепенно добавляли к смеси 32,5 а цинковой пыли н
300 мл 50 %-ной уксусной кислоты; озонид разлагается экзотермиче-
ски с отгонкой хлористого метилена. После кипячения и перемеши-
вания смесь экстрагировали эфиром, перекиси разлагали промыва-
нием раствором иодида калия и при разгонке получали 62% 5-метил-
гексанола, 5% 5-метилгексановой кислоты и 9% полимера. Вслед-
ствие перегрева электродов и хрупкости трубок Бертло Хенне и
Перилыптейн [31 сконструировали новый генератор из стекла
пирекс с охлаждаемыми водой электродами. Разложение озонида
осуществляют гидрированием в присутствии 1 % палладия на кар-
бонате кальция. Для уменьшения числа операций озонирование и
5
гидрирование озонида проводят в том же растворителе и в том же
сосуде. В качестве растворителя нельзя использовать хлористый
метилен, так как он затрудняет гидрирование; лучше применять
этил ацетат и этанол. Выходы простых альдегидов и кетонов из
олефинов составляют 30—60%.
Десен и Ныомен [4] использовали генератор Хенне и Пер ил ь-
штейна для озонирования технического пирена. Раствор 25 г угле-
водорода в 100 мл ДМФА обрабатывают О. в 50%-ном избытке в те-
чение приблизительно 6 час. Раствор озонида добавляют при пере-
мешивании к 500 мл 1%-ной водной уксусной кислоты; при этом об-
разуется тонкая суспензия, которая за 12 час коагулирует и дает
зернистый коричневый осадок. Этот осадок отделяют и несколько
раз экстрагируют 10%-ным водным едким кали, а темно-коричневый
фильтрат обрабатывают раствором гипохлорита калия и оставляют
на ночь. При нагревании на кипящей водяной бане в течение 6 час
цвет раствора изменяется на оранжевый. 5-Формилфенантрен-4-кар-
боновую кислоту осаждают в виде калиевой соли, выделяют из соли
и перекристаллизовывают.
о3; кося-кон
32-38%
Для озонирования фенантрена с образованием дифенил-2,2'-
диальдегида Бэйли и Эриксон 15] специальным образом высушивали
кислород и пропускали его через озонатор Уэлсбаха. При проверке
этой методики кислород высушивали пропусканием через колонку
(высотой 30 см) с силикагелем и использовали простой озонатор, да-
ющий 3,8 вес. % О. при скорости пропускания 20 л/час.
Раствор 10 г фенантрена (0,056 моля) в 200 мл горячего метанола
быстро охлаждают для образования мелкозернистой суспензии
и переносят в высокую цилиндрическую склянку для поглощения
газа с доходящей до дна газоподводящей трубкой, охлаждаемую до
— 30° в сосуде Дьюара смесью сухой лед — ацетон. Озонирование
продолжают до тех пор, пока не прореагирует весь суспендирован-
ный углеводород, для чего требуется 1,1—1,3 же О. Для восстапов-
6
ления перекисных продуктов озонолиза и метанолиза добавляют
раствор 0,17 моля иодида натрия в 30 мл уксусной кислоты и спустя
1 час выделившийся иод восстанавливают раствором тиосульфата
натрия. При удалении метанола током воздуха выделяется продукт.
Добавляют воду, продукт отделяют и кристаллизацией из смеси
эфир — лигроин получают бледно-желтые кристаллы с т, пл.
62—63°.
Шмидт и Графен [6] показали, что озонирование эфиров енолов
легкодоступных циклоалканонов является удобным методом полу-
чения и-альдегиде кис л от. Кетон (1) превращают через дпэти л аце-
таль (2) в эфир енола (3), его озонируют при 0° и озонид разлагают
гидрированием. Неустойчивый альдегидоэфир (4) выделяют в виде
диэтплацеталя (5). Сравнительный эксперимент, в котором смесь
I " 1 С=О I------С(ОС:НДг ।------С—ОСгНз
/ргг \ НССОСаНЩ i /ри1 \ А у fPW 'i I О’! Td v
।-------СН2 1------6Н2 !------
С1) (2) (з)
П = 3,4, 5,6, ю
р------СО2С2Н5 г-------СО2С2Н5
----> (СН2) п нщосщщ у (СН2)
I-------СНО I--——CH(OC2HS)S
(4) (5)
I же эфира енола (1-этоксициклогексен) и 1 же аналогичного оле-
фина (1-н-пропилциклогексен) озонировали при —80° 0,75 же
окислителя, показал, что 80% окисленного продукта образуются из
эфира енола.
Фремери и Филдс [7] разработали процесс озонирования цикло-
олефина в виде эмульсии в водно-щелочном растворе перекиси во-
дорода с образованием в одну стадию дикарбоновой кислоты обыч-
но с хорошим выходом. Эмульгирующий агент, полиоксиэтилиро-
ванный лауриловый спирт, растворяют в олефине, раствор добавля-
ют к водному раствору едкого натра и перекиси водорода в колбе,
снабженной скоростной механической мешалкой, затем пропускают
кислород, содержащий около 3% О, Ниже приведены типичные
примеры:
Циклооктеп —> Пробковая кислота
цис, цис-1,5-Циклооктаднен (большой избыток)—> А3-Гексен-1,6-дикарбоновая
кислота
Ипдел —•> Гомофталевая кислота
Аддукт бутадиена и малеинового ангидрида —> Бутан-1,2,3,4-тетракарбоповая
кислота
На одной из стадий расщепления биосинтетического холестерина,
полученного из меченого ацетата, Корнфорз, Хантер и Попжак [81
для установления распределения метки в кольце А превратили
Д5-холестен в озонид с 80%-ным выходом, пропуская О. в раствор
7
углеводорода в сухом н-гексане до тех пор, пока разбавленный раст-
вор брома в уксусной кислоте не стал быстро обесцвечиваться при
действии нескольких капель этого раствора в н-гексане. Центри-
фугирование, промывание и высушивание дали стеклоообразный
озонид, который восстанавливали с хорошим выходом в соответству-
ющий кетоальдегид встряхиванием раствора в уксусной кислоте с
цинковой пылью при 25° в течение нескольких часов, пока прибав-
ление кристаллика иодида калия к пробе раствора уже не будет со-
провождаться выделением иода.
Разрабатывая эффективный метод получения прогестерона (4)
из стигмасгерина, Хейл и Герр [9] показали, что стигмастадиенон
(1), полученный окислением стигмасгерина по Оппенау эр у, атакует-
ся О. преимущественно в боковую цепь с образованием бис-
норальдегида (2). Сломп и Джонсон [10] изучили озонолиз стиг-
м астад иен о н а, измеряя скорость уменьшения интенсивности
полосы при 10,26 мк, обусловленной двойной связью боковой цепи;
скорость взаимодействия двойной связи в кольце измерялась
изменением поглощения А4-енон-3-овой системы при 6,0 мк.
Они нашли, что добавление пиридина в виде 1%-ного (по объему)
раствора в хлористом метилене к раствору (1) существенно повы-
шает селективность реакции, и определили условия, в которых
8
альдегид (2) образуется с выходом 90%, Альдегид конденсировали
с пиперидином, используя в качестве катализатора гс-толуолсуль-
фокислоту, а енамин (3) окисляли в прогестерон бихроматом натрия
в смеси безводная уксусная кислота — бензол III],
Использование тетрацианэтилена в качестве буфера при озони-
ровании олефиновописано в разделе, посвященном этому реагенту.
Первые попытки Джонсона и сотр, [121 превратить фурфурили-
денкетон (5) в дикарбоновую кислоту (6) путем озонирования пока-
(6)
зали, что реакция, которая с насыщенными стероидами давала пре-
красные выходы, шла плохо и по-разному, возможно, из-за атаки
избыточного О, по бензильному атому С9. Этого удалось избежать
при насыщении соответствующего объема хлористого метилена О.
и
Рис. O-I, Прибор для озонирования.
при —78° в камере В специального прибора для озонирования [13]
(рис, 0-1) и выдавливании его азотом под давлением в камеру А
с перемешиваемым магнитной мешалкой раствором субстрата в хло-
9
ристом метилене (субстрат берут в таком количестве, чтобы не было
избытка О.). Голубая окраска О. сразу же исчезает и при разложе-
нии озонида иодной кислотой выделяется с высоким и воспроиз-
водимым выходом дикислота (6). Из диолефина аналогичным обра-
зом получен с удовлетворительным выходом диальдегид [14].
Стайл и Фостер [15] изучили озонирование аценафтилена (1)
в гидрат нафталин-1, 8-диальдегида (2) и показали, что с О. в токе
азота можно получить выход 73,5%, тогда как с обычной смесью
О.— кислород выход составляет лишь 16,5%.
73, 5%
НОНС^^СНОН
(1) (г)
1. S m i t h L. 1., Greenwoo d F. L., Hudrlik O,, Org. Syn., Coll.
Vol., 3, 673 (1955).
2. llenne A. L., H i 1 1 P., J. Am. Chem. Soc., 65, 752 (1943).
3. H e n n e A, L., Perilstein W. L., J. Am. Chem. Soc., 65, 2183 (1943).
4. D e s s у R. E., N e w in а п M. S., Org. Syn., Coll. Vol., 4, 484 (1963).
5. В a i 1 e v P. S., Erickson R. E., Org. Syn., Coll. Vol., 41, 41 (1961).
6. Sclimfdt U., Grafen P. Ann., 656, 97 (1962).
7. F r e m e г у M. I., Fields E. R., J. Org. Chem., 28, 2537 (1963).
8, Cor И 1 or I h J. W., Hunter G. D., P о p j a k G., Biochem. J., 54,
590 (1953).
9. H с у 1 F. W., Herr M. E., J. Am. Chem. Soc., 72, 2&I7 (1950).
10. S 1 о m p G., Jr., Johnson J. L., J. Am. Chem. Soc., 80, 915 (1958).
II. H e r r M. E., Hey I F. W., J. Am. Chem. Soc., 74, 3627 (1952); S h e p-
h e г d D. A. ct al., ibid., 77, 1212 (1955).
12. M e у e r W. L., Camcron D. D., Johnson W. S., J. Org. Chem.,
27, ИЗО (1962).
13. Rubin M. B., J. Chem. Ed., 41, 388 (1964).
14. J ohnson W. S., R ub in M. B. et al., J. Am. Chem. Soc., 85, 1409 (1963).
15. S t i I 1 c J. R., Foster R. T., J. Org. Chem., 28, 2703 (1963).
CH2 —CHa
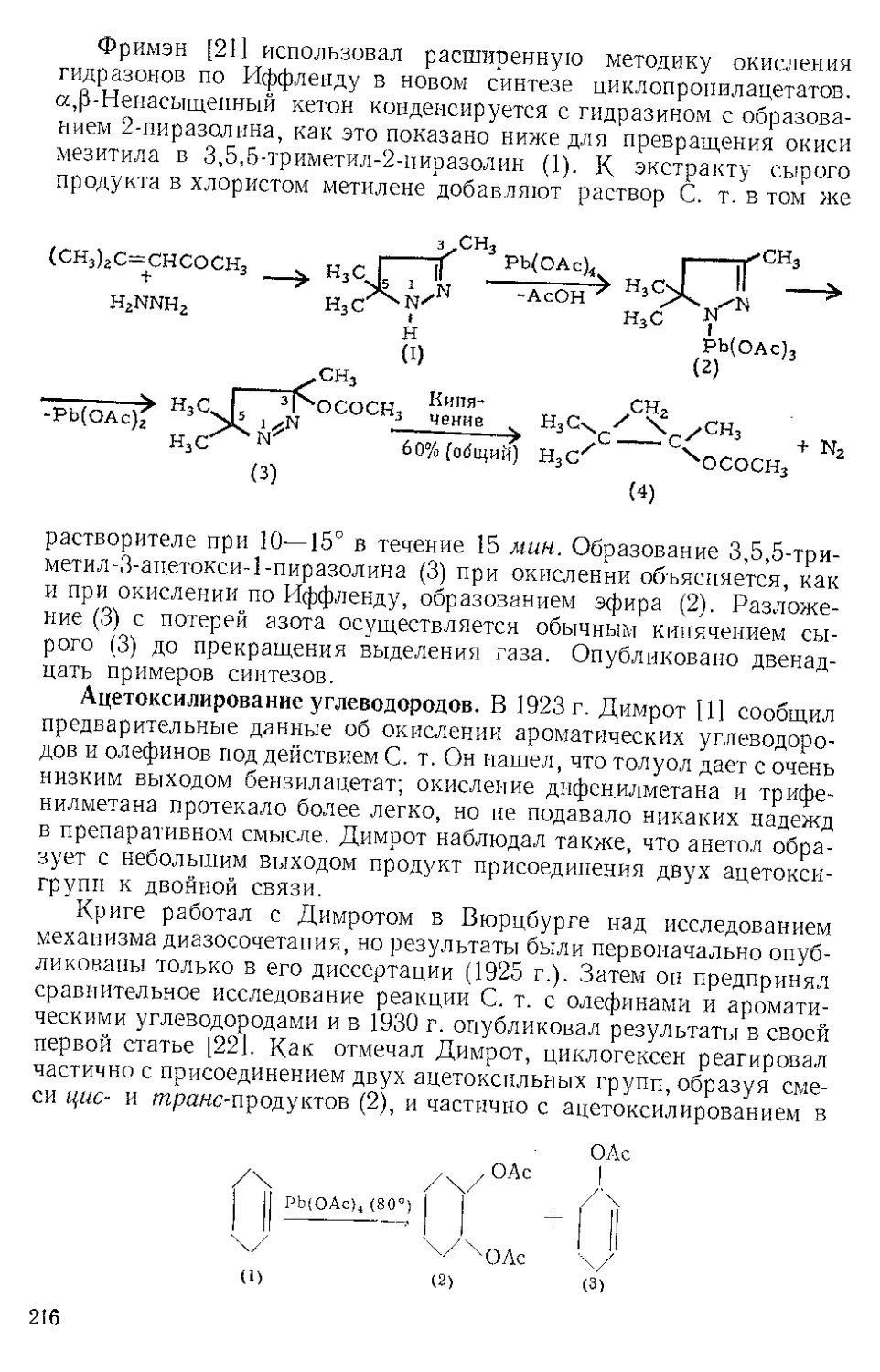
ОКИСИ МЕЗИТИЛА ЭТИЛЕНКЕТАЛ Ь, ,
(СН3)2С=СНССН3
Мол. вес 142,19, т. кип. 58725 мм. Получение ]1].
О. м. э. применяют для превращения А1-3-кетостероидов в 3-эти-
ленкетальные производные обменной диоксоланацией [2]. Например,
смесь 1 г ацетата кортизона (1) и 2 мл О. м. э. в 7,5 мл сухого ТГФ
обрабатывают 0,06 мл конц. серной кислоты, перемешивают 2 час
при комнатной температуре и оставляют на ночь. После обработки
получают 844 мг почти чистого (2).
10
1. S a 1 m i E. J., Rannikko V., Ber., 72, GOO (1939).
2. Cons t a n t i n J. AL, Haven A. C., S a r e 11 L. H., J. Am, Cliem. Soc.,
75, 1716 (1953).
1-ОКИСЬ ГФЕНИЛ-З-МЕТИЛ ФОСФОЛ EHA-3, (3). Мол. вес
192,19, т. пл. 60—65°, т. кип. 163—168°/0,65 мм.
Получение [1], В склянку для отсасывания на 1 л загружают
1 моль фенил дихлорфосфина, 300 мл технического изопрена (около
3 молей) и 2 г технического антиоксиданта ионола (2,6-ди-трет-
бутил-4-метилфенол). Склянку закрывают пробкой, боковой отвод
заглушают трубкой с зажимом и гомогенный раствор выдерживают
при комнатной температуре. Обычно через 2—4 час появляется бе-
лый осадок, и спустя 5—7 дней жидкая фаза заполняется белым кри-
сталлическим аддуктом (2). Зернистый аддукт промывают, суспен-
дируют в петролейном эфире и собирают на пористом стеклянном
c6h5fci2(1)
рг=1^3 н2о, NaOH
I I ----~------—>
57-63% (общий)
сИ^СбНз
(3)
фильтре. Дихлорид (2) гидролизуют ледяной водой в окись (3),
раствор нейтрализуют щелочью и продукт экстрагируют хлоро-
формом .
Применение. О. ф. катализирует превращение изоцианатов в кар-
бодиимиды [21. По сравнению с 1-окисью 1-этил-З-метилфосфолепа-З
реагент несколько менее реакционноспособен, однако он более до-
ступен .
1. McCormack W. В., Org. Syn., 43, 73 (1963).
2. С a m р b е 1 1 Т. W., Monagle J. J., Org. Syn., 43, 31 (1963).
ОКИСЬ ЭТИЛЕНА. Мол. вес 44,05, т. кип. 10,7°, уд. вес 0,89.
В синтезе н-гексанола из н-бутилмагнийбромида и О. э. эфирный
раствор реактива Гриньяра охлаждают льдом с солью и при
И
перемешивании прибавляют окись этилена (4—6 час) так, чтобы
температура не превышала 10° [11.
нао
«-C4H9MgBr-pCHa—СНг —> н-С4НэСНгСН,ОМ§Вг ———н-С4НэСН.,СН,ОН
\ / 60-62%
xoz
Очень удобно поместить баллончик с жидкой О. э. на весы, чтобы
контролировать расход по убыли веса. Конец трубки для ввода газа
должен находиться примерно на 2 см выше поверхности жидкости,
чтобы газообразная О. э. конденсировалась в холодной реакционной
смеси.
1. Др ед же р Е., «Синтезы органических препаратов», ИЛ, ЛЬ, 1949, сб. 1,
стр. 154.
F=iCH3
1-ОКИСЬ 1-ЭТИЛ-З-МЕТИЛФОСФОЛЕ- =
НА-3, (1). Мол. вес 144,15, т. кип. 115— . СзНб
11971,2—1,3 мм.
Новый метод получения карбодиимидов состоит в катализируе-
мой О. э. димеризации изоцианата с отщеплением двуокиси углеро-
да:
2ArN — С = О — ArN = С= NAr+COa
Согласно кинетическим данным, эта реакция обратима, имеет низ-
кую энергию активации и осуществляется в две стадии. Предпола-
гаемый механизм реакции приведен ниже:
О О
It б+ м II
q Q— Медленно q q
II + I |f"|!
Ar—N6 +PR3 Ar-N —PR3
О
II
c=o
Ar —X = PR3
R3P6 + 6 О R3P....O
IL_ II В В
Ar— N6 +6+C-N — ArzZAr—N—C=N—Ar
Быстро
( R3P?—O_
\ Ar—N—C=N—Ar
1 .Campbell T. W., Monagl e J.J., Fol di V. S., J. Am. Chem. Soc.,
84, 3673 (1962); Monaglc J. J., C a m p b e 1 1 T. AV., M eShane H. F.,
Jr., J. Am. Chem. See., 84, 4288 (1962).
ОКСАЛИЛБРОМИД, (COBr)3. Мол вес 215,85, т.кип. 102—103°.
Получение [1].
Трейбс [21 показал, что О. взаимодействует с алифатическими
ацилбромидами в четырех хлор истом углероде при повышении тем-
пературы бани от 100 до 135° с введением группы СОВ г в a-положе-
ние. Ход реакции можно контролировать, поглощая бромистый во-
дород и измеряя объем выделяющейся окиси углерода. Выходы сос-
12
тавляют38—68%. О. плохо реагирует с олефинами типа RCH=CH2
RCH.COBr (С0В12щ RCI-l/С0ВГ +СО + НВГ
ХСОВг
и R2C=CH2, однако бромкарбонилирование стирола и 1,1-ди-
фенилэтилена осуществляется с умеренным выходом [3]. О. реаги-
(СОВг),
С6Н5СН = СНа-----> С6Н5СН = СНСОВг
J 0 3 40% ”
(СОВг)«
(CfiHs)2C —СН2 -(С8Н3)2С = СНСОВг
рует с высокореакционноспособными ароматическими соединени-
ями: антрацен бромангидрид антрацен-9-карбоновой кислоты
(52%). Кетоны подвергаются а-бромкарбонилированию с выходами
порядка 10—40% [4].
l.Staudinger Н., А п t h е s Е., Ber., 46, 1431 (1913).
2. Treibs W., Orttmann H., Chem. Ber., 91, 297 (1958).
3. Treibs W., Orttmann H., Chem. Ber., 93, 545 (I960).
4. Treibs W., Riemer J., Orttmann H., Chem. Ber., 93, 551 (1960).
ОКСАЛИЛХЛОРИД, (COC1)2. Мол. вес 126,93, т. кип. 62°,
уд. вес 1,50.
О. получают из тонкоизмельченной безводной щавелевой кисло-
ты и пятихлористого фосфора [1, 2].
Получение ацилхлоридов. Адамс и Ул их [31, описавшие приме-
нение реагента для превращения карбоновых кислот в ацилхлори-
ды, предложили две основные методики: а) реакция кислоты с О.
в бензоле; б) превращение кислоты в натриевую соль и последую-
щая обработка О. с (или без) добавлением пиридина в качестве ка-
тализатора. Уилдс и Шунк [4] исследовали постадийно синтез дез-
оксикортикостеронацетата (5) из кислоты (1) и показали, что реакция
(1) с т ион ил хлоридом с образованием ацил хлорида (3) осуществляет-
ся неудовлетворительно вследствие атаки реагента по кольцу А.
Значительно лучшие результаты получают растворением кислоты
в 1 же водной щелочи, замораживанием раствора и отгонкой досуха
при пониженном давлении (лиофилизация). Затем высушенную соль
(2) суспендируют в бензоле, содержащем следы пиридина, и обра-
батывают О. при 0°. Реакцией ацилхлорида с диазометаном полу-
чают (4) с выходом 81% [в расчете па (1)] и взаимодействием (4)
с уксусной кислотой завершают синтез дезоксикортикостеронацетата
(5). Рейх штейн и сотр. [5], применяя ту же последовательность
реакций для синтеза 11-эпикортикостерона, повторили работу Уилд-
са и Шунка с соединением (1) в качестве модельного соединения, од-
нако получили иные результаты; вместе с метиловым эфиром кисло-
ты (1) обязательно получался диазокетон (4). Более хорошие резуль-
таты дала обработка свободной кислоты (1) О. в бензоле при 20°
без добавления пиридина. В течение 1 час раствор не обесцвечивает-
13
ся, и с выходом 75% получают чистый диазокетон (4) без примеси
сложного эфира. Этот метод дает хорошие результаты при синтезе
11 -эпикортпкостерона.
Энгел и Джаст Гб] успешно использовали метод с применением
О.— бензол для превращения (6) в ацплхлорид; в этом случае при
обработке натриевой соли в присутствии пиридина выходы оказа-
лись неудовлетворительными.
(6)
Кейзон [71 показал, что реакция полуэфира несимметричной ди-
кислоты типа (7) с хлористым тионилом осуществляется с перегруп-
пировкой, возможно, через ангидрид, с образованием смеси двух
возможных хлор ангидридов эфир окис л от. Штельберг-Штенхаген
[81 изучил реакцию тионил хлорида с двумя энантиомерами эфироки-
сл оты (8), где перегруппировка приводит к рацемизации. С хорошо
ОД
НО2ССЩСНЙССО.2СН;
СэН5
(7)
сн3
НОйССНаССН2СО2СН3
н
(8)
очищенным тионилхлоридом рацемизации не происходит, если ре-
акцию проводят при 30°, а избыток реагента удаляют при темпера-
туре ниже 50°. При более высокой температуре или с менее чистым
14
реагентом происходит рацемизация. О. дает лучшие результаты,
так как с этим реагентом в бензоле при 50° не было отмечено рацеми-
зации. Линстед [91 показал, что эта методика эффективно предотвра-
щает перегруппировку при превращении полуэфнров янтарной,
(9)
1) (СОС1)г
2) сн3он
---------->
7 6%
глутаровой и фталевой кислот в хлорангидриды эфирокислот. Шмущ-
ковпч [101 при этерификации кислоты (9) диазометаном получил
сложный эфир (10) лишь с 55%-ным выходом. Одиако путем обработ-
ки кислоты О. в бензоле, последующей отгонки досуха и кипячения
остатка с метанолом соединение (10) было получено с высоким вы-
ходом.
На одной из стадий полного синтеза ротенона [И] было необхо-
димо превратить тубовую кислоту в хлорангидрид, однако обычные
методы были непригодны из-за исключительной лабильности не-
насыщенной боковой цепи. Проблема была решена превращением
кислоты в сухую калиевую соль (методика не приведена) и ее взаи-
модействием с О. в бензоле.
Тубовая кислота
Введение COCI (хлоркарбонилирование). Караш и Браун 112]
показали, что некоторые насыщенные углеводороды взаимодейст-
вуют с О. под действием света или в присутствии перекиси с заме-
щением водорода на СОС1. Так, смесь 0,3 моля циклогексана, 0,2 мо-
ля О. и 1, 2 г перекиси бензоила при кипячении в течение 24 час
дает с умеренным выходом хлорангидрид циклогексанкарбоновой
НХ/СОС1
|/Х1 (сосщ, (сен5сод2 |/Х'| (
I [ ---*~' ) I Г I llx^rl
I. J 50% I J
кислоты. В некоторых случаях можно заменить на СОС1 атом водо-
рода в молекуле олефина 112]. Так, 1,1-дифен ил этилен (6,5 а) при
]5
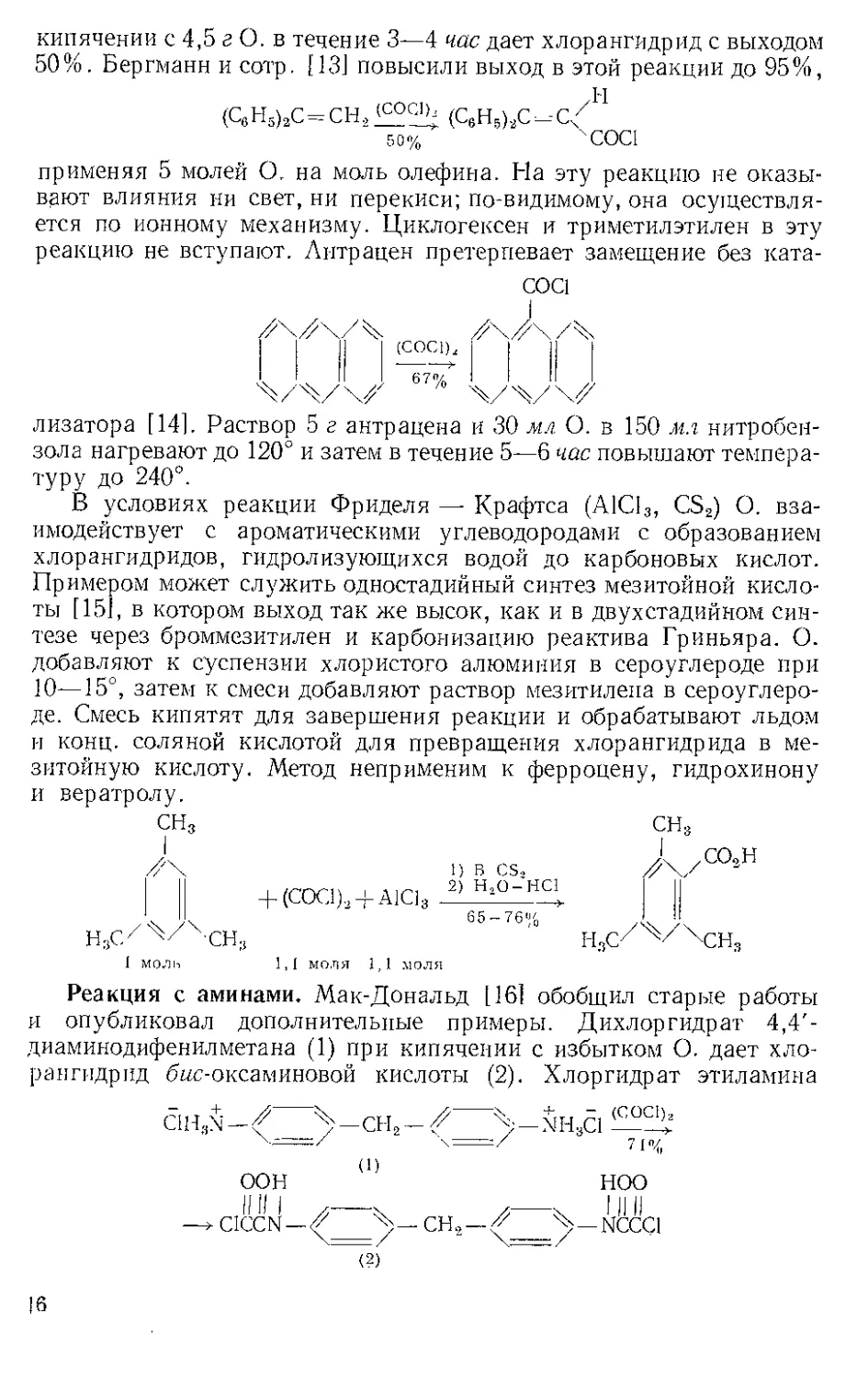
кипячении с 4,5 г О. в течение 3—4 час дает хлорангидрид с выходом
50%. Бергманн и сотр. [13] повысили выход в этой реакции до 95%,
(ceH5)2c-cH2^2E!i (с6н5),с_-с</11
50% ХСОС1
применяя 5 молей О, на моль олефина. На эту реакцию не оказы-
вают влияния ни свет, ни перекиси; по-видимому, она осуществля-
ется по ионному механизму. Циклогексен и триметил эти лен в эту
реакцию не вступают. Антрацен претерпевает замещение без ката-
СОС1
лизатора [14]. Раствор 5 г антрацена и 30 мл О. в 150 мл нитробен-
зола нагревают до 120° и затем в течение 5—6 час повышают темпера-
туру до 240°.
В условиях реакции Фриделя — Крафтса (A1CI3, CS2) О. вза-
имодействует с ароматическими углеводородами с образованием
хлорангидридов, гидролизующихся водой до карбоновых кислот.
Примером может служить одностадийный синтез мезитойной кисло-
ты [151, в котором выход так же высок, как и в двух стадийном син-
тезе через броммезитилен и карбонизацию реактива Гриньяра. О.
добавляют к суспензии хлористого алюминия в сероуглероде при
10—15°, затем к смеси добавляют раствор мезитилепа в сероуглеро-
де. Смесь кипятят для завершения реакции и обрабатывают льдом
и конц. соляной кислотой для превращения хлорангидрида в ме-
зитойную кислоту. Метод неприменим к ферроцену, гидрохинону
и вератролу.
СН3
I
1) в CS,
III + (СОС1У А А1С]3 ,2) н--0~н^
А 1 ' 65"76%
CH;J
I моли 1,1 моля 1,1 моля
Реакция с аминами. Мак-Дональд [16] обобщил старые работы
и опубликовал дополнительные примеры. Дихлоргидрат 4,4'-
диаминодифенилметана (1) при кипячении с избытком О. дает хло-
рангидрид бяс-оксаминовой кислоты (2). Хлоргидрат этиламина
СИГА —сн2_ / % — \НаС1 —
(1)
ООН нею
О I Z-----к z---X III II
—CICCN — СН2 — ____J—NCCC1
(2)
[6
СН3
/СОСОС1
xCOCOCi
(0,06 моля) при кипячении с 1,25 моля О. в течение 50 час превраща-
ется в N-этил оксимидохлор ид.
СН3СН2ЙН3С1 — СН3СН,
77%
С N,N-диметил ан и л ином О. образует а-ди кетон [17]. К суспен-
зии 1 моля хлористого алюминия в сероуглероде при 0° добавляют
при перемешивании 1,5 моля амина и образующуюся однородную
пасту обрабатывают сероуглеродным раствором 0,25 моля О.
О О
2 (CH3).,NT~^""^ C1C0C?2L (CH3)aN —С—С—N(CH3)2
38-42% \=/ \=/
Продолжительная обработка и кристаллизация из ацетона дают
4,4'-бмс-(диметиламино)-бензил с т. пл. 200—202°. Несмотря на низ-
кие выходы, эта методика исключительно важна, так как 4-диметил-
аминобеизальдегид не удается превратить в бензоин.
ROH->RC1. О. взаимодействует со спиртами при комнатной
или несколько более высокой температуре с образованием алкил-
хлороксалата, который в присутствии пиридина разлагается при
100—125° с образованием алкилхлорида [18].
ОО
II I! 100- 1 25°
ROH-|-{COC1)2 —ROCCCI——— RC1+CO4-CO3
— НС1 Ру
Амид—>изоцианат. Общий метод превращения амидов в ацил-
изоцианаты [19] является более удобным источником этих соединений,
чем реакция ацилхлоридов с цианатом серебра. В нижеприведенном
примере смесь а-хлорацетамида и 100 мл дихлорэтана охлаждают до
2° и при перемешивании обрабатывают сразу 0,6 моля О. Затем уда-
ляют охлаждающую баню, смесь перемешивают 1 час и кипятят при
83° с перемешиванием в течение 5 час. Раствор охлаждают до 0—10°,
холодильник заменяют колонкой высотой 120 мм со стеклянной на-
садкой и при перемешивании в вакууме отгоняют растворитель.
О О
II II
C1CH.,CNH,H-(СОС1)а___» С1СН2С— N^C = O + CO + 2HC1
65%
При 68—70°/70 мм собирают продукт. Дальнейшее изучение [20]
показало, что ароматические амиды дают отличные результаты, а
с алифатическими амидами выходы неудовлетворительны. Исклю-
чение составляют алифатические амиды, не имеющие а-водородных
атомов, а также содержаитмр эл е кто оотпнца тельный заместитель в
а-положении.
Реакция с ненасыщенные,! нс гонами. Дегенги и Гаудри [21]
показали, что О. взаимодействует с А4-3-кетостероидами с образо-
ванием 3-хлор-А3,5-стероидов, как это показано на примере про-
17
гестерона. Реакцию проводят в бензоле при комнатной температуре
с щавелевой кислотой в качестве катализатора. Можно использо-
вать также пяти хлор истый фосфор, хлорокись фосфора и ацет и л-
хлорид, однако они менее эффективны [22].
Перемешивание
при 25° 2час-
50-70%
Реагент взаимодействует с А1 2 3 4 5 6 7 8 9 10 11 12 ,+-3-кетостероидом (1) в бензоле при
комнатной температуре с образованием 3-хлор-А1,ч,5-триена (2),
который легко выделяется, однако очень неустойчив и под действием
кислоты перегруппировывается в ароматический 1-хлор-4-метил-
1,3,5 (Ю)-триен (3) [23]. Продукт перегруппировки (3) можно по-
лучить непосредственно из соединения (1), увеличивая реакционный
период, добавляя щавелевую кислоту и используя в качестве раст-
ворителя хлороформ.
1. Staudinger Н., Вег., 41, 3563 (1903).
2. К h а г a s с h М. S., В г о w п Н. С., J. Am. Chem. Soc., 64, 329 (1942).
3. A d a m s R., U 1 i c h L. H., J. Am. Chem. Soc., 42, 599 (1920).
4. W i I d s A. L., Shunk С. H., J. Am. Chem. Soc., 70, 2427 (1948).
5. Reber F., Lardon A., Reichstein T., Helv. Chim., Acta, 37, 45
(1954).
6. Engel Ch. R., Just G., Can. J. Cbem., 33, 1515 (1955).
7. C a s о я J., J. Am. Chem. Soc., 69, 1548 (1947).
8. Stallberg-Stenhagen S., J. Am. Cbem. Soc., 69, 2568 (1947).
9. Hancock J. E. H., L i n s t*ena\d J. Chem. Soc., 1953, 3490.
10. Szmuszkovicz J., J*Org» ChtitmjXS, k843 (1964).
11. Miyano M., J. Am. Chem. Soc., 87, 3958 (1965).
12. К h a r a s c h M. S., К a n e S. S., Brown H. C., J. Am. Chem. Soc., 64,
333 (1942).
18
13. Bergmann F., Weizmann M-, Dimant E.,P at ai J., S z m u-
szkovicz J., J. Am. Chem. Soc., 70, 1612 (1948).
14. La t ham H. G., Jr., May E. L., Moset t i g E., Am. Chem. Soc., 70,
1079 (1948).
15. S о к о 1 P. E., Org. Syn., 44, 69 (1964).
16. McDonald R. N., J. Org. Chem., 24, 1580 (1959).
17. Tiiziin C.,Ogl i aruso M., Becker E. I., Org. Syn., 41, 3 (1961).
18. Rhoads S. J., Michel R. E., J. Am. Chem. Soc., 85, 585 (1963).
19. Speziale A. J., Smith L. R-, J. Org. Chem., 27, 3742 (1962); 28, 1805
(1963); Org. Syn., 46, 16 (1966).
20. S p e z i a 1 e A. J., Smith L. R., F e d d e r J. E., J. Org. Chem., 30,
4306 (1965).
21. Deg h eng hi R., Ga u dry R., Can. J. Chem., 40, 818 (1962).
22. M о e г s c h G. W-, Neuklis W. A., Can. J. Chem., 41, 1627 (1963).
23. Moersch G. W., Neuklis XV. A., Culbertson T. P., Mor-
r о w D. F., Butler M. E., J. Org. Chem., 29, 2495 (1964).
о-бмс-(<ЖСИМЕТИЛ)-БЕНЗОЛ, HOCH,CeH4CH,OH-o. Мол. вес
138,16, т. пл. 64°.
Реагент получают восстановлением фталевого ангидрида алюмо-
гидридом лития в эфире (выход 87%) [1]. В новом эффективном син-
тезе циклопропанонгидрата Греве и Струве [2] использовали о-кси-
лиленовую ацетальную защитную группу (1), которую после отщеп-
ления бромистого водорода (2) и реакции Симмонса — Смита (3) уда-
ляли гидрогенолизом (4). Бензильные группы в этом случае непри-
годны ввиду облегчения клайзеновской перегруппировки.
1. N у s t г о m R. F., Brown W. G., J. Am. Chem. Soc., 69, 1197 (1947).
2. G r e w e R., Struve A., Chem. Ber., 96, 2819 (1963).
N-ОКСИМЕТИЛФТАЛИМИД. Мол. вес 177,16, т. пл. 138—141°.
О. получают с выходом 90% реакцией фталимида с формалином.
При взаимодействии О. с ароматическими аминами при кипячении
с обратным холодильником (0,5—2 час) образуются кристалличе-
ские И-(ариламинометил)-фталимиды, плавящиеся в интервале
112—242° [1].
/\/С°\ . - Ч
| || NCH2OH — | * || NCH3NAr
^/ХСО^ V\co/
1. W i n s t e a d M. B., Heine H. W., J. Am. Chem. Soc., 77, 1913 (1955).
19
2-ОКСИ-1,4-НАФТОХИНОН. Мол. вес 174,15, т. пл. 195—196°
(с разл.).
Получение. В одном из способов получения О. используют
аммонийную соль 1,2-нафтохинон-4-сульфокислоты [1L К 1 л ме-
танола при охлаждении льдом с солью при 0 ° медленно добавляют
80 мл конц. серной кислоты. Затем убирают охлаждающую баню,
добавляют 1 моль аммонийной соли, перемешивают до образования
однородной пасты и постепенно повышают температуру до кипения,
с отделением эфира (2) добавляют еще порцию метанола и смесь ох-
лаждают. Полученный эфир достаточно чист, и его гидролиз водной
щелочью дает оксинафтохинон в виде ярко-желтого зернистого твер-
дого вещества высокой чистоты.
Лучший метод [21 начинается с присоединения по Тиле уксус-
ного ангидрида к 1,4-нафтохинону (можно также использовать
1,2-нафтохинон). При добавлении 2 мл эфирата трех фтор истого бо-
ра к суспензии 15,8 г хинона в 40 мл уксусного ангидрида хинон
медленно растворяется со слабым разогреванием смеси. Получают
чистый бесцветный 1,2,4-триацетоксинафталин, который гидроли-
зуют, перемешивая с метилатом натрия в метаноле при 0°. Выделяю-
щийся гидрохинон окисляется кислородом воздуха с образованием
ярко-красной натриевой соли оке и нафтохинон а. Подкисление вод-
ного раствора дает ярко-желтые кристаллы оксинафтохинона с т.
пл. 195—196°.
1. F 1 с s е г L. F., Martin Е. L., Org. Syn., Coll. Vol., 3, 465 (1955).
2. F i e s e r L. F., J. Am. Chem. Soc., 70, 3165 (1948).
OH
I ,CH2Br
2-ОКСИ-5-НИТРОБЕНЗИЛБРОМИД, fY
Мол. вес 232,05, т. пл. 145°. 1 |
no2
20
О. получают бромметилированием п-питрофенола [11. Реагент
исключительно селективно реагирует с триптофановыми остатками
белков. Единственная другая реагирующая с О. аминокислота —
цистеин; однако эта реакция более медленная [11.
1. К о s h 1 a n d D. Е., Jr., К а г k h a n i s Y, D., Latham Н. G., J. Ат.
Chem. Soc., 86, 1448 (1964); Horton Н. R., К о s h 1 a n d D. E., Jr., ibid.,
87, 1126 (1965).
N-ОКСИПИПЕРИДИН, (2)^(3). Мол. вес 101,15, т. пл. 39s,
т. кип. 110°/55 мм.
Получение. О. получают окислением пиперидина 3%-ной пере-
кисью водорода [1]. Более удовлетворительный метод состоит в оки-
слении N-этилпиперидина до N-окиси с последующим элиминирова-
нием этилена по Коупу [21.
Строение. С фен ил изоцианатом О. взаимодействует как гидрок-
силамин (2) с образованием соединения (1) и как окись амнна (3),
образуя N-бензильное производное (4) [1].
(Б
(4)
(г) (3)
Пептидный синтез. В дициклогексилкарбодиимидном методе
синтеза реагент используют для превращения N-защищенных ами-
нокислот (5) в активированные эфиры (6) [2,3], конденсирующиеся
с эфиром аминокислоты с образованием дипептида.
R R о
! / \ дцк I II /—к
CbNHCHCOiH + l-ION рКЕЕК CbNHCHCON/
{5) (2) (6)
Вейганд и сотр. [41 при тщательном сравнении 16 различных ме-
тодов образования пептидной связи нашли, что в данном методе
полностью отсутствует рацемизация.
l.Wolffenstein R., Вег., 25, 2777 (1892); Haase F., W о 1 f f е n s-
t е i л R., Вег., 37, 3228 (1904).
2. Handford В. О., Jones J.H., Young G. Т., Johnson F. N.,
J. Chem. Soc., 1965, 6814.
3. В e a u m о n t S. M., H a n d f о r d В. O., J о n e s J. H., Y о u n g G. T.,
Chem. Comm., 53 (1965).
4. Weygand F., Pros Л., К 6 n i g \V., Chem. Ber., 99, 1451 (1966).
3-ОКСИПИРИДИН. Мол. вес 95,10, т. пл. 126s.
Пептидный синтез. Преимущество эфиров О. по сравнению
с обычно используемыми n-нитрофениловыми эфирами состоит в том,
21
что непрореагировавший эфир легко удаляется благодаря раство-
римости в разбавленной кислоте 111. Эфиры легко получают из СЬ-
пептида и О. с дициклогексил кар боднимидом в этилацетате в при-
сутствии триэтиламина.
1. Т aschner Е., Rzeszortarska В., Angew. Chem., Internal. Ed., 4,
594 (1965).
CH2—CO
N-ОКСИСУКЦИНИМИД, I NOH. Мол. вес 115,09, разла-
CH3—CO
гается при 175°.
Получение. См. [1, 2].
О. используют в пептидном синтезе, подобно N-оксифтал имиду,
с тем преимуществом, что побочным продуктом конденсации акти-
вированного эфира с эфиром аминокислоты является растворимый
в воде О.
1. W е g 1 е г R., G г е w е F., Meli 1 ose К., пат. США 2816111 (1957).
2. Anderson G. W., Zimmerman J.Е,, Callahan F. M.,J. Am.
Chem. Soc., 86, 1839 (1964).
N-ОКСИФТАЛИМИД, (4). Мол. вес 163,13, т. пл. 237—240°.
Получение О. с выходом 70% реакцией N-кар бэтоксифтал имида
(1) с гидроксиламином и триэтил амином в кипящем абсолютном эта-
ноле включает, по-видимому, раскрытие гетероцикла (2) и последу-
ющее его образование (3) с отщеплением уретана; с образованием
триэтиламмонийной соли О. (4) наблюдается красное окрашивание
раствора, и после подкисления н разбавления водой О. выделяется
в виде почти бесцветных игл [1].
О II, | 1 N—COoEt | II E‘sN Ч/ II О (1) ( ОН н 7С— NCO2Et ОН -h2nco2eT II О (3) О II ZC—NHCO2Et V—МНОЙ II О 2) О II /\/\ I |l N—ОН—> -Ч/Хс/ II О (4)
22
о
II
R О С /ч R'
I II / \/У\ !
ськн-сн-со2н ч/ | II h2nchco,ch,
---------—— —CbNCrlC— О — N ] -----------
C<H11N = C = NCeH11 |
Н
(5)
О н
—> CbNCHC — NCHC02CH3+(4)
I I I
HR R'
(6)
О. используют в пептидном синтезе следующим образом. Конден-
сацией (4) с N-защищенной аминокислотой в присутствии ДЦК
получают активированный эфир (5), который легко реагирует с ами-
иоэфиром с образованием защищенного дипептида (6); освобождаю-
щийся О. удаляют встряхиванием с водным раствором бикарбоната.
В отличие от синтеза О. по Нефкеису [(!)-> (4) [ разработанный
позднее Мазуром и Плюмом [2] прямой метод синтеза короче и да-
ет более высокий выход (91%).
К раствору 1,1 моля хлоргидрата гидроксиламина в 1,5 л пи-
ридина в одиогорлой круглодонной колбе, охлажденному до 30°,
добавляют сразу 1 моль фталевого ангидрида и смесь перемешивают
до получения прозрачного раствора (42°). В течение 15 мин раствор
выдерживают при 90° и пиридин упаривают на роторном испари-
теле в вакууме водоструйного насоса. Горячий вязкий остаток бы-
стро добавляют к 1 л 1 н. уксусной кислоты и выделяющийся оса-
док отделяют и промывают 0,01 н. СН3СООН.
1. N е 1 k е n s G. Н. L_, Tesser G. I., J. Агп. Chem. Soc., 83, 1263 (1961);
N с f к е n s G. Н. L., Т е s s е г G. I., Ni var d R. J. F., Rec. trav., 81,
683 (1962).
2. Mazur R. H., Plume G., procedure submitted to Org. Syn.
ОКТАНОЛ-2, CH3CH2CH2CH2CHaCHaCH(OH)CH3. Мол. вес
130,22, т. кип. 179°, уд. вес 0,819.
О. используют для устранения вспенивания [1].
1. Хартман В,, Дикки Дж., Стемпфли Дж., «Синтезы органических
препаратов», ИЛ, М., 1949, сб. 2, стр. 187.
ОЛЕИНОВАЯ КИСЛОТА, СН3(СН,)7СН-СН(СН.,)7СО3Н. Мол.
вес 282,47, т. пл. 13,0—13,2°, 16,0—16,3° (полиморфные формы),
т. кип. 200—201° /1,2 л-ш.
Очистка. Браун и corp. [1] приводят четыре известных из лите-
ратуры метода очистки О. к. Сверн и сотр. [21 описали методику,
включающую низкотемпературную кристаллизацию, перегонку и
затем перегонку в виде эфира. Однако в методике получения трео-§,
23
10-диоксистеариновой кислоты гидроксилированием О. к. надму-
равьиной кислотой Сверн и сотр. [3] указывают, что их метод очист-
ки О. к. более трудоемкий и менее удобный, чем очистка диоксики-
слоты.
Акцептор водорода. Силвервуд и Орчин [4] показали, что при
дегидрировании 2,5 лшолей гвайена 7,5 кмолями селена при 290°
в течение 1 час выход повышается с 14 до 28,6% , если в качестве ак-
цептора водорода добавить 15 зшолей О. к.
1. Khan N. A., Deatherage F. Е,, Brown J. В., Org. Syn. Coll.
Vol., 4, 851 (1963).
2. Knight H. B., J or dan E. F., Jr., S w e r n D., Biochem. Preparations,
2, 100 (1952).
3. Swern D., Scanlan J. T., Dickel G. B., Org. Syn., Coll. Vol. 4,
317 (1963).
4. Sil verwood H. А., О r c h i n M., J. Org. Chem., 27, 3401 (1962).
ОЛОВО, Sn. Ат. вес 118,70.
В классической методике получения анилина 11, 2] лучше ис-
пользовать гранулированное О. и соляную кислоту, чем дорогое
хлористое олово, так как продукт после подщелачивания выделяют
перегонкой с паром. Однако при восстановлении антрахинона в ан-
трон система SnCl3— НС1 — АсОН (см. Олово хлористое) более
пригодна по сравнению с Sn — НС1 — АсОН 13], так как при этом
исключается трудоемкое фильтрование горячего кислого раствора
и выход выше на 10%. В тех случаях, когда применялось только О.,
не ясно, использовали ли хлорид и нашли его непригодным или
просто не использовали. При восстановлении анизоина до дезок-
сианизоина используют порошок О. (200 меш) в 40%-ном избытке
от теории, причем при уменьшении количества металла выход сни-
жается 141.
60 л.1 95%-ного этанола
СН3О~^~VcH-C-^У-ОСНз A sn + 2НС1 Кипячение 24
| || 86-92%
он о
0,19 моля 0,33 моля 52 -u.i
-+ СН3О—%>-СНчС —%—ОСН3-I. S11C1. -I- н2
х=/ - || ' =/
о
В методиках восстановления 2,6-дибром-4-питрофенола до ами-
нофенола [5], нитробарбитуровой кислоты до амина (урамила)
0,87 моля 7,0 г-атом [9,6 моля
24
IG] и при получении флороглюцина [7} рекомендуется использовать
металл, но, по-видимому, реакцию можно провести и с хлористым
оловом.
В 1886 г. Гвидо Гольдшмидт при восстановлении папаверина
действием О. в соляной кислоте выделил 1,2,3,4-тетрагидропапа-
верин и кристаллическое основание, известное сейчас как павин,
строение которого было установлено Бэттерсби и Бвнксом [8[.
Павин
Амальгамированное олово. Шефер [91 получил этот реагент,
встряхивая смесь 100 г порошка О. (30 меш), 15 г сулемы и 100 мл
воды в закрытой колбе в точение нескольких минут, пока все олово
не покроется ртутью. Амальгамированное олово и соляная кислота
легко и без побочных реакций восстанавливают сопряженные ен-
дионы до насыщенных дикетонов. В противоположность этому
восстановление цинком и уксусной кислотой часто сопровождается
значительными побочными реакциями.
О о
[1 !l
CJE.CCH-CHCQFE+ Sn(Hg)--95%-ный С2Н-,ОН-^kohii. НО —>
5 ? 10 ; 150 ,vj 20 л/.i 90%
О о
!.| !.|
СГ,Н5ССН2СИ2ССД15
I. Fischer Е., «Anleilung ztir Darstellring orsranisclier Praparatc», 8th ed,,
Friedr, View eg and Sohn, 1908.
2. Org. Expts., 177,
3. M e й e p, «Синтезы органических препаратов», ИЛ, AE, 1949, сб. 1, стр. 45.
4. Картер П. X,, К р э г Дж. Ц., Л а к Р. Э., А1 о й л ь ЛЕ, «Синтезы ор-
ганических препаратов», изд-во «Мир», ЛЕ, 1964, сб. 12, стр. 22,
5, X а рты а и В., Д н к к н Дж., Стсм лфл п Дж., «Синтезы органических
препаратов», ИЛ, ЛЕ, 1949, сб, 2, стр. 187.
6. Хартман В., 141 с п п а р д О., «Синтезы органических препаратов», ИЛ,
М., 1949, сб. 2, стр. 491.
7. К л а р к, Хартма и, «Синтезы органических препаратов», ИЛ, ЛЕ, 1949,
сб. 1, стр. 446.
8. BattcrsbyA.R., В inks R.,J, Chem. Soc., 1955, 2888.
9. Schaefer .1. P., J, Org. Chem., 25, 2027 (I960).
25
ОЛОВО БРОМИСТОЕ, SnBr2. Мол. вес 278,53.
ж-Хлорбензальдегид получают из м~нитробензальдегида восста-
новлением хлористым оловом в соляной кислоте, последующим ди-
азотированием путем добавления нитрита натрия к охлажденной ки-
слой смеси и обработкой хлористой медью и соляной кислотой [1].
ии-Бромбензальдегид, полученный этим способом с помощью CuBr —
НВг, содержит до 20% м-хлорбензальдегида, но этот недостаток
можно устранить, используя при восстановлении на первой стадии
О. б. и НВг. Раствор реагента готовят из гранулированного олова
и 46 %-ной бромистоводородной кислоты [21.
1. Бук И., A ft д н В., «Синтезы органических препаратов», ИЛ, М., 1949, сб. 2,
стр. 551.
2, «Синтезы органических препаратов», ИЛ, М., 1949, сб. 2, стр. 553, см. прим. 6.
ОЛОВО ХЛОРИСТОЕ, SnCL, -2НХ). Мол. вес 225,65.
ArNO.,->ArNH,. Эффективное восстановление ароматических
нитросоединений до соответствующих аминов часто осуществляют
добавлением иитросоединений к раствору дигидрата О. х. в конц.
соляной кислоте (1 мл на 1г); примеры см. [1—31. По данным Оль-
шлегера [4], при восстановлении нитросоединений в больших коли-
чествах для смягчения реакции часть соляной кислоты лучше за-
менить хлористым натрием. Умеренно растворимый динитродурол
растворяют в уксусной кислоте и восстанавливают, добавляя раст-
вор О. х. в конц. соляной кислоте 151. 1,5-Динитро-2,6-диоксинафта-
лин восстанавливают действием О. х. в уксусной кислоте (выход
80%) с образованием неустойчивого на воздухе дихлоргидрата ди-
амина [61.
Ходгсон и Смит [71 нашли, что при использовании 3 экв дигидра-
та О. х. можно селективно восстановить 2,4-динитронафтол-1 до
2-нитро-4-аминонафтола-1. К суспензии дииитросоединения в сме-
си конц. соляной кислоты и этанола при перемешивании добавляют
в течение 1 час раствор О. х. в этаноле. Продукты восстановления
ОН ОН
/^WN°2 /Л- A/n°2
\ |Г Т Ниже 30° f if Y
| || | +3SnCI2.2H,O—Г | || |
Ч/Ч/ Д/ЧЧ
I I
no. nh.2.hci
Суспензия 5 г 15 г n 20 мл EtOH
вещества в смеси
20 мл HCI п 10 мл EtOH
выделяются в виде светло-желтых игл хлоргидрата.
Антиоксидант. О. х. при добавлении в небольших количествах
(2 г на 0,5 моля) является эффективным антиоксидантом в процессе
превращения 1-аминонафтола-2 или 4-аминонафтола-1 в хлоргидра-
ты и при перекристаллизации этих неустойчивых на воздухе продук-
тов [8].
26
—СН2Х>—СН3. Риикес [91 предложил способ получения 5-ме-
тилфурфурола (4), включающий кислотный гидролиз сахарозы (1),
превращение фруктозы (2) в производное хлорыетилфурана (3)
и восстановление под действием О. х. до (4).
SnCH-HCI
----1---->
20-22%
сахароза носн-снон JTjI
НОЩС-С СНСН^ОН СШаС^0 <ЗНО
(1) о он
(2) (3)
Сандин и Физер [10] разработали эффективный способ получе-
ния сильнодействующего канцерогенного вещества 9,10-диме-
тил-1,2-бенз антрацен а из 1,2-бензантр ах икона (1), включающий
присоединение реактива Гриньяра, реакцию аддукта (2) с иодисто-
водородной кислотой с образованием 9-метил-Ю-иодметнльного
производного (3) и восстановление последнего О. х. в смеси ди-
оксана (Di) с соляной кислотой.
(3) (4)
Синтезы этилениминов. Клосс и Бройс [И] предложили новый
метод синтеза, в котором тетр аал к ил этилен превращают в голубое
хлор нитрозопроизводное; это соединение восстанавливают О. х.
в соляной кислоте, и образующийся хлорамин без выделения цикли-
зуют под действием основания с образованием, например, 2,2,3,3-
тетраметилэтиленимина. Другие восстановители, такие, как LiAlH4,
NaBHj, Al(Hg), Zn — АсОН, при этом неэффективны. Синтез при-
меним только к тетразамещенным этиленам.
27
NOCI SnCk—HCl
(CH3;2C = C(CH3)2-----> (CH3)3C----C(CH3)2 ----—
-70° I |-
(CH3)2C----C(CH3)3
I I
nh2 Cl
NaOH
79%
(общип)
(CH3)2C-C(CH3)3
N
H
Расщепление N-нитрозаминов. Бек и Ферри [12] очищали
N-этил-л-толуидин превращением последнего в N-нитрозо!цзоизвод-
ное и разложением его путем восстановления О. х.
Расщепление 2,4-динитрофенилгидразонов. Демекер и Мартин
[13] нашли, что 2,4-динитрофен ил гидразоны насыщенных н нена-
сыщенных 3-кетостероидов расщепляются с высоким выходом
(84—98%) при кипячении с большим избытком ацетона для частич-
ного обмена остатков гидразина и последующем добавлении О. х. н
соляной кислоты, приводящем к восстановлению одной или
обеих нитрогрупп. Куллинейн и Эдвардс [14] предложили метод
для характеристики и очистки маслообразных о-оксикетонов,
получаемых реакцией Фриса. 2,4-Динитрофенилгидразон 2-окси-
З-метилацетофенона кипятят с ацетоном до получения прозрачного
раствора, затем добавляют раствор О. х. в соляной кислоте и кипя-
тят еще в течение 30 мин. Если вместо 6,5 а дигидрата О. х. взять
6,0 а или 7,0 г, то выход снижается от 81 до 70%.
сн3 н
I I Л—X ,
с = NN—//___NO2
Н0\/\ o3n/
I II +6,5 г SnCI2-2H2O в
. L / 80 л/ л НС! + 120 лм Н2О
н3с/
4 г в 500 «ил гцетоиа
сн3
со
по I
Кипячение ч /7\
30 мин у н
81% । Н
HgC/ 'V
28
Хиноны. Загер 115] разработал получение тетраокси-1,4-бен-
зохинона путем добавления 30% -ного раствора глиоксаля к раствору
сульфита натрия в бикарбонаты ом буфере и обработкой раствора то-
ком воздуха. Отделяются зеленовато-черные кристаллы динатрие-
/СНО ОН О
сно сно N..SO, Н0\1/0Н <. Н0АА0Н NaHCOj А У А А, А
сно сно - 1 II — 1! II
чсно НО/^/^ОН Si,CIj НО/ху' ЧОН он о
вой соли тетраоксихинона; подкисление дает блестящие черные кри-
сталлы тетраоксихинона с выходом 8%. Восстановлением послед-
него О. х. в соляной кислоте получают гексаоксибензол с выходом
70—77%.
Восстановление хинонов, минуя стадию гидрохинонов, из-
вестно лишь в антраценовом ряду. В лучшем лабораторном методе
получения антрона [16] смесь антрахинона, О. х. в конц. соляной
кислоте и уксусной кислоты осторожно кипятят до полного исчез-
новения кристаллов антрахинона (8—10 мин) и затем еще 15 мин.
Раствор нагревают на водяной бане и добавляют воду порциями по
1 мл ло получения насыщенного раствора (примерно 12 мл). Антрон
получается в виде бледно-желтых игл высокой чистоты.
О
I!
УА/А/А
| |i || |-'г-SnCH 2Н.,0 НС1
А/ у АА
О
40 м.1 ЛсОН
Кипячение
25 мин
85%
5 г (0,026 моля) 13 е (0,058 моля) 13
Восстановление (C(jHr,)3COH*. Восстановление трифенил карби-
нола О. х. в уксусной кислоте при нагревании на кипящей водяной
бане дает трифенилметильный радикал, который димеризуется в
л-бензгидр ил тетрафен ил метан (т. пл. 224°) (см. схему на стр. 30).
Теоретически для восстановления требуется 0,5 моля О. х., но
для эффективности процесса берут пятикратный избыток реагента,
по-видимому, для того, чтобы поддерживать необходимый восста-
новительный потенциал.
Восстановление иодгидринов. В стереоспецифическом синтезе
цис- или транс-олефинов по Корнфорсу [17] в качестве промежуточ-
ного продукта получают хлоргидрин предсказанной конфигурации,
который нельзя непосредственно восстановить в олефин. Превраще-
* Эта реакция была предложена как задача в книге Org. Expts., 93.
Ответ дан в книге <П. Физера и М. Физер «Органическая химия», т. I,
изд-во ,,Химия“, М., 1966.
29
(с6н5)3сон + snci/ z нго + на J_vat:np 87_>
86%
1 г в 20лл АсОН 2 г 5 мл
иие осуществляется в три строго стереоспецифические стадии: об-
разование эпоксидного цикла, расщепление нодистоводородной ки-
слотой (Nal — АсОН — EtCOoH) и восстановление полученного
иодгидрина под действием смеси О. х. — РОС13— пиридин.
>с---С7
7 [ I7
ОН С1
кон
HI V / SnCl..—POC1,-CrHsN v /
—ус—с/ —:---------------Ас=с<
7! Iх 77
НО L
Промотор гидрирования. Райландер и Каплан [18] обнаружили,
что О. х. является самым эффективным из ряда промоторов в про-
цессе гидрирования гептальдегида в присутствии платины и рутения
на угле. 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18
1. Бек И., Ай ди В., «Синтезы органических препаратов», ИЛ, М., 1949,
сб. 2, стр. 551.
2. Ф е р р и R., Б е к Дж., Б алтцл иР., «Синтезы органических препара-
тов», ИЛ, М-, 1952, сб. 3, стр. 160.
3. Вудвард Р., «Синтезы органических препаратов», ИЛ, М,, 1952, сб. 3,
стр. 363.
4. Oelschlager Н., Ann., 641, 81 (1961).
5. Смит Л., «Синтезы органических препаратов», ИЛ, М., 1949, сб. 2, стр. 262.
6. Paul Н., Zimmer G., J. prakt. Chem., 18, 219 (1962).
7. Hodgson Н. Н., S m i t h E. W., J. Chem. Soc., 1935, 671.
8. Ф и з e p Л., «Синтезы органических препаратов», ИЛ, М., 1949, сб. 2, стр, 44.
9, Р и и к е с И., «Синтезы органических препаратов», ИЛ, М., 1949, сб. 2,
стр. 338.
10. S a n d i n R. В., Fi eser L. F., J. Am, Chem. Soc., 62, 3098 (1940).
11. С 1 о s s G. L., В г о i s S, J., J. Am. Chem. Soc., 82, 6068 (1960).
12. Бек Дж., Ферри К., «Синтезы органических препаратов», ИЛ, М., 1949,
сб. 2, стр. 605.
13. Demaecker J., Martin R. 14., Nature, 173, 266 (1954); Bull. soc.
chim. Belg., 68, 365 (1959).
14. Cullinane N. M., Edwards B. F. R., J. Chem. Soc., 1958, 1311.
15. F a t i a d i A. J., Sager W. F., Org. Syn., 42, 66, 90 (1962).
16, Org. Expts., 199.
17. С о r n f о r t h J. W., Cornfort h R. H., M athc w R. R., J. Chem.
Soc., 1959, 112.
18. R у 1 a n d er P. N., К a p 1 a n J., Engelhard Ind. Tech. Bull., 2, 48 (1961).
30
ОЛОВО ХЛОРИСТОЕ БЕЗВОДНОЕ, SnCla. Мол. вес 189,61,
т. пл. 247°.
О. х. б. получают из дигидрата под действием уксусного
ангидрида [1].
Растворимый в эфире безводный реагент используют для получе-
ния Р-нафтальдегида реакцией Стефена 11] и о-толуилового альде-
гида реакцией Сонна — Мюллера [21. Раствор реагента в эфире на-
сыщают хлористым водородом и добавляют р-нафтонитрил в эфире.
Смесь снова насыщают хлористым водородом, перемешивают 1 час
и оставляют на ночь для полного осаждения желтого комплекса
альдимина с хлорным оловом. Эфирный раствор декантируют и
твердый продукт перегоняют с перегретым паром (8—10 час, 8—10 л
дистиллята). При получении о-толуилового альдегида восстановле-
ние ицидохлорида проводят аналогичным образом. Для перегонки
продукта с паром из кислой среды в этом случае требуется не более
1 час.
С1
0,14 моля
и
0,20 моля SnCl;, НС1
Эфир
Перегонка с паром
1, Уильямс Дж., «Синтезы органических препаратов», ИЛ, М., 1952, сб. 3,
Стр 331.
2. Уильямс Дж., В иттен Ч., Кринкцки й Дж., «Синтезы органи-
ческих препаратов», ИЛ, М., 1953, сб. 4, стр. 484.
ОЛОВО ХЛОРНОЕ, SnCl4. Мол. вес 260,53, т. пл. - 30 , т. кип.
114°, уд. вес 2,226.
О. х.— более мягкий катализатор, чем А1С13, в реакции Фриде-
ля—Крафтса. Тиофен достаточно активен для ацилирования в при-
31
сутствии SnCLj в бензоле [11. Под действием А1С13 происходит по-
лимеризация тиофена. О. х. и бензол в качестве растворителя
[|^ jj + CH3COCI
SnCl.
0-25°
79-83%
0, 2моля 0, 2 моля
сосн3
О, 2 моля
В 2ОС мл С6Н6
используют также для внутримолекулярной циклизации у-фенил-
бутироилхлорида [2].
СО.Н
СОС1
0,6 моля PCJ
1,28 моля SnCJ4
С6ГЦ,от— I Одо—15°
О
Кипячение
85—9 1%
0,6 моля
в 200 лл С6Н,
О. х. является умеренно эффективным катализатором для кон-
денсации мезоксалевого эфира с и-ксилолом [3]. Смесь эфира
СО2С2Ы5
С-0
СО,С2Н5
1 моль
-I- SnCl4 °-s5° :
51,5—57%
1,25 моля
ОН
I /СО2С2Н5
с<
ХСО2С2Н&
КОН; НС1
сз—7 о%
и углеводорода обрабатывают, добавляя по каплям О.х., при ох-
лаждении на ледяной бане и затем перемешивают при комнатной тем-
пературе 3 час. После обработки реакционной смеси, омыления, де-
карбоксилирования и перекристаллизации продукта нз бензола
получают чистую 2,5-диметилминдальную кислоту.
Реагент применяют в синтезе тетраэтил станнана [4].
1. Джонсон Дж., М э п Дж., «Синтезы органических препаратов», ИЛ, М.,
1949, сб. 2, стр. 76.
2, Джонсон Г., «Синтезы органических препаратов», ИЛ, М., 1954, сб. 5,
стр. 67.
3. Р и б з о м е р Дж., Ирвин Дж., «Синтезы органических препаратов», ИЛ,
М., 1952, сб. 3, стр. 195.
4. В а и дер Керк Дж., Люптен Дж., «Синтезы органических препара-
тов», ИЛ, М., 1958, сб. 8, стр. 57.
3'2
ОЛОВО ХЛОРНОЕ, ПЕНТАГИДРАТ, SnCl4 -5Н,О. Мол. вес
350,61.
Луц и Бэйли [1] в кратком сообщении указывают, что этот ре-
агент не только катализирует реакции метилв ин ил кетона или акро-
леина с изопреном, но и заметно влияет на соотношение образую-
щихся изомеров, как это показано в таблице для первого диенофила
Катализатор Растворитель Температура, °C Количество изомера, %
Ш IV
Без катализатора Толуол 120 71 29
SnCl4.5HaO Бензол 25 93 7
1. L u t z Е, F,, Bailey G. ЛЕ, J, Am. Chem. Soc,, 86, 3899 (1964).
ОРТОМУРАВЬИ ной кислоты ТРИЭТИЛОВЫЙ ЭФИР,
НС(ОС2Н5)3. Мол. вес 148,20, т. кип. 146°, уд. вес 0,94.
О. к. т. э. получают [1] добавлением в течение 2 час металличе-
ского натрия к раствору хлороформа в абсолютном этаноле:
2СНС1з + 6СгН5ОН + 6Na — 2НС(ОС2Н5)36NaCl + ЗН2
45%
Получение альдегидов. Реактив Гриньяра реагирует с О. к. т. э.
с заменой одной этоксигруппы и образованием ацеталя, который
гидролизуется до альдегида разб. кислотами. При получении гекса-
наля-1 [2] сырой ацеталь гидролизуют, нагревая с разб. серной кис-
НС(ОС.,Нг)3 на
H-C5H„MgBr ----—-----> н-С5Н1гСН(ОС2Н5)2 ----------->h-C6Hl1CHO
Выход 45—50%
(общий)
лотой, и отгоняют альдегид в раствор бисульфита натрия. Раствор
бисульфитного производного перегоняют с паром для удаления
н-амилового спирта, обрабатывают бикарбонатом натрия и перегон-
кой с паром выделяют свободный альдегид.
Исходным соединением для получения фенантреи-9-альдегида
[31 является сырой 9-бромфенантрен, который получают бромиро-
ванием технического фенантрена (90%) и очищают только перегон-
кой (т. пл. 54—56°; кристаллизация [41 дает чистый продукт с т. пл.
65—66°). В колбу на 5 л, снабженную мешалкой, делительной
2 Заказ А® 1166 33
воронкой на 500 мл, трубкой для подачи азота и эффективным обрат-
ным холодильником, на конце которого закреплена делительная
воронка на 1 л, помещают магний (50,3 г). Сырой бромфенантрен
(514 а) расплавляют и помещают в меньшую делительную воронку,
yMgBr СН(ОСгН5);
//---€____'/----\ НС(ОСЛ-1,),; ф_________Н
\=:--/ \----- - /' 40—42%
(общий)
щно
по,одерживая температуру около 70" осторожным нагреванием с по-
мощью микрогорелки. В большую делительную воронку помещают
1 л эфира. Прибор продувают азотом и, вытеснив весь воздух, до-
бавляют эфир и расплавленный бромфенантрен с такой скоростью,
чтобы поддерживалось слабое кипение и содержимое обеих воро-
нок было добавлено одновременно. Через некоторое время реактив
Гриньяра начинает осаждаться на стенках колбы, и для его раство-
рения добавляют 1 л бензола. После окончания реакции из нижней
делительной воронки постепенно добавляют О. к. т. э. (296,4 а) и
кипятят смесь в течение 6 час. Реакционную смесь охлаждают в ба-
не со льдом и прибавляют 1 л 10%-ной соляной кислоты, органиче-
ский слой отделяют и растворитель удаляют в вакууме. К остав-
шемуся ацеталю добавляют 1 л 25%-ной серной кислоты и смесь
кипятят 12 час для полного окончания гидролиза. Сырой альдегид
очищают через бисульфитное производное, так как аптрацен-9-аль-
дсгид, образующийся из антрацена, присутствующего в исходном
соединении, не образует продукта присоединения и таким образом
отделяется; кристаллизация дает чистый альдегид, т. пл. 101°.
Синтез а, [3-ацетиленовых альдегидов основан на том, что О.к.т.э.
в присутствии йодистого цинка как катализатора реагирует с тер-
минальными ацетиленами с выделением этанола и образованием
ацеталя, как это иллюстрируется синтезом фенилпропаргилового
альдегида [5J. Реагенты осторожно нагревают с катализатором при-
близительно до 135° с одновременной отгонкой этанола (около 1 час).
Zril,
СнН5С СИ з- НС(ОЕ 1 )3 —-1 СвН5С ССН(ОСгН5), ф С2Н,ОН
_ | НЮ(Н + )
СеН5С = ССН - О
О.к.т.э. используют для формилирования фенолов в хлористом
метилене в присутствии хлористого алюминия [61.
34
Диэтилацетали и кетали. Клайзен [7] показал, что О.к.т.э.
реагирует с альдегидами и кетонами, образуя диэтилацеталп и ди-
этилкетали. Реакцию обычно проводят в абсолютном этаноле в при-
сутствии каталитических количеств хлорного железа, хлористого
R3C О + НС(ОС2Н5)з —> R2C(OC2H5),НСО2С2Н5
или азотнокислого аммония, либо при комнатной температуре
(6,8 час), либо при кипячении (30 мин,). Примером может служить
превращение неустойчивого акролеина в более устойчивый диэтил-
ацеталь [81. Теплый раствор 3 г нитрата аммония в 50 мл абсолют-
ного этанола добавляют к смеси акролеина и О.к.т.э.; после выдер-
С113 = СНСНО -J- HC(OC2HS)3 СН2 = СНСН(ОС2Н-)2 4- НСОгС2Н5
0,79 моля 0,97 моля 72—80%
живания в течение 7 час при комнатной температуре светло-красный
раствор отфильтровывают и перегоняют над карбонатом натрия. Ди-
этилацеталь акролеина был получен с 620/0-ным выходом из аль-
дегида и этанола в присутствии n-тол у ол сульфо кислоты как
катализатора [91.
Синтез фенилпропаргилового альдегида, менее эффективный,
чем описанный выше, интересен тем, что он иллюстрирует образо-
вание ацеталя с целью защиты альдегидной группы в процессе
дегидрогалогенирования сильным основанием [10].
Вг, К »СО,
СД15СН = СНСНО -4 [QH^HBrCHBrCHO] у—СйН5СН = СВгСНО—>
НС(ОСД-Ц), кон
—-— > С6Н5СН = СВгСН(ОСаН5Ц--------С(;1-1Г1С^ССН(ОС3НВ)2 —>•
82-86% V 80-86%
Об удобном синтетическом приеме, включающем реакцию диэ-
тилацеталя альдегида с ал кил виниловым эфиром, см. Этилвиинло-
вый эфир.
Реакции с соединениями, содержащими активные метиленовые
группы. КлЭйзен [11] разработал метод получения диэтилэтокси-
метиленмалоната нагреванием смеси О. к. т. э., малонового эфира
и уксусного ангидрида с каталитическим количеством хлористого
2*
35
цинка. Указанный на схеме выход получен по стандартной методи-
ке [121.
ХпСЦ
С2НВОСН(ОС2НГ>)2 + СН,(СО2С2НЙ)3Н 2(СН3СО)гО -
50—6 0%
С3Н5ОСН С(СО2С2Н0)2 + 2СН3СО2С2Н5 - j - 2СН3СО.,Н
Реакция N-н-гептилмочевины (1) с О.к.т.э. (2) и малононитри*
лом (3), при которой образуется 3-«-гептилуреидометиленма-
лононитрил (4), вероятно, включает первоначальную конденсацию
(2) и (3) с образованием этоксиметил енмалононитр ил а (5) [131.
h-C7H15NHCONH2-PHC(OC2H5)3 + CH2(CN)2
(1) (2) (3)
н-С7 H13N HCON НСН = C(CN )а
(4)
(1)
(2Н-(3) СН=--С(С1\Т)2 (4)-J-C2H-OH
I
ОС,И5
(5)
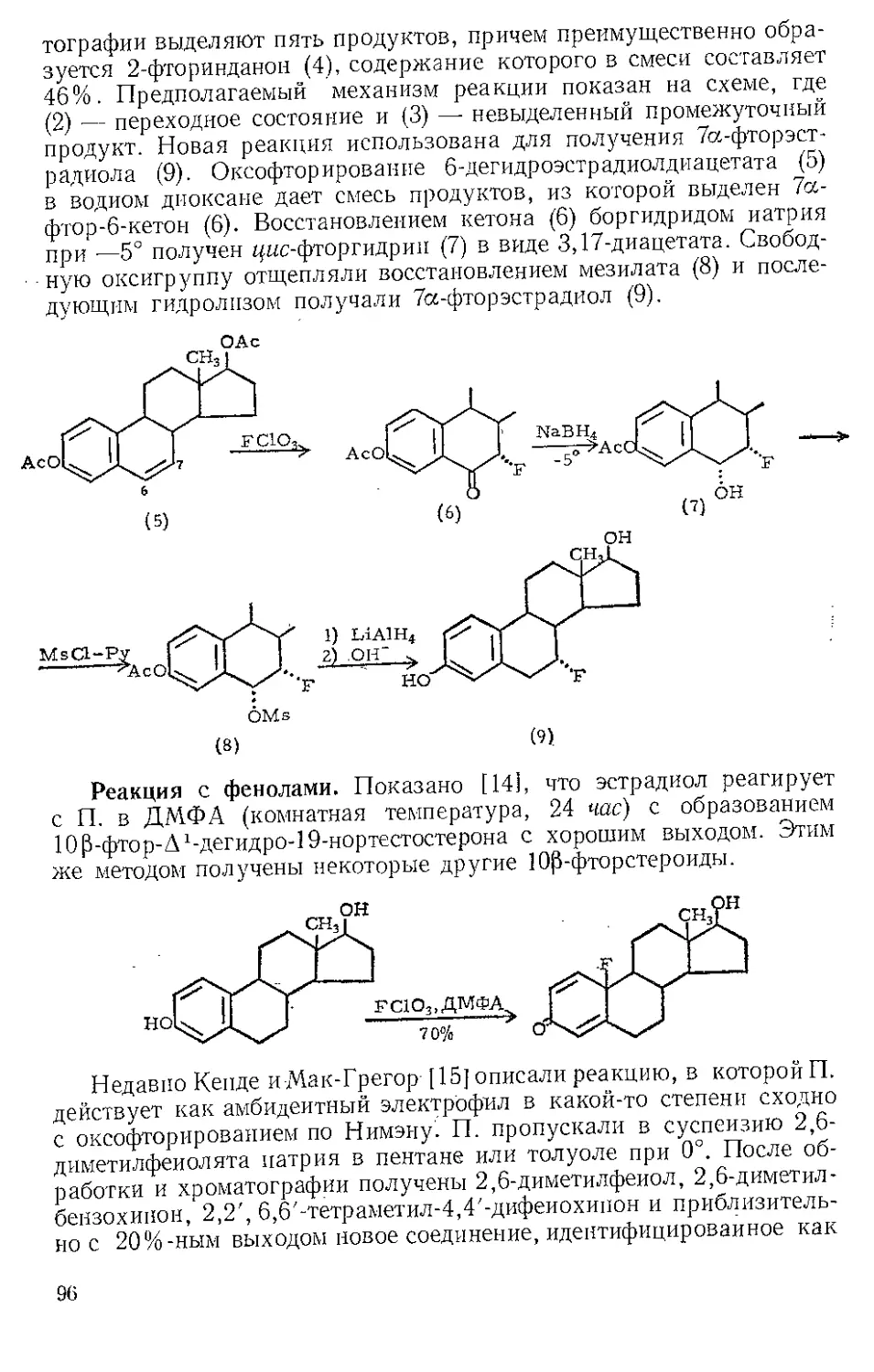
Получение эфиров енолов. Конденсация 3-кето-5з-стероида (1)
с О. к. т. э. в этаноле, содержащем следы хлористого водорода,
дает диэтилкеталь (2) с хорошим выходом; при кипячении в ксилоле
кеталь отщепляет молекулу этанола и образует эфир Д--енола (3)
[14]. Д4-3-Кетостероид (4) точно так же конденсируется с О. к. т. э.,
образуя непосредственно этиловый эфир диенола (5), а не диэтилке-
таль. В обычных условиях селективное гидрирование эфира диенола
(5) сопровождается перемещением связи и приводит к эфиру Да-енола
(3). Однако при гидрировании (5) в присутствии основания (NaOH,
пиридин) восстанавливается 5,6-двойная связь и образуется эти-
ловый эфир Д3-егюла(6) [15]. Превращение циклогекеандиона-1,3
в эфиры енолов также достигается при использовании О. к. т. э,
[16].
36
Реакция с первичными ариламинами. Исследования Уичелхауса
[17] и Клайзена [18] реакции анилина с О. к. т. э. были развиты
позднее Робертсом и Фогтом [19], которые показали, что удачный
выбор катализатора и экспериментальных условий позволяет полу-
чить с хорошим выходом любой из трех продуктов. Первоначаль-
ным продуктом в каждом случае является этил-М-фенилформимидат
(1). В отсутствие кислого катализатора соединение (1) реагирует
C6H5NH2-1(CoH50)2C—ОС2Н5 —> CBH5iV=C—ОС2Н5-рС.2Н5ОН
н н
(П II
CcH-,NH, J СДЦОН 1 (HsSO4), 140'
CcHe,N = C--NC6I-T5 C2H5N— сн=о
II И С2НВ
(2} (3)
с анилином, давая N, N'-дифенилформамидин (2), который также об-
разуется при взаимодействии 2 молей анилина с 1 молем О. к. т. э.
в присутствии хлористого водорода как катализатора. Серная ки-
слота является специфическим катализатором для проведения пере-
группировки соединения (1) в N-этилфор манил ид (3). Так, (3) мож-
но получить из 0,5 моля анилина, 0,75 моля О. к. т. э. и 0,02 моля
конц. серной кислоты, удаляя отгонкой образующийся на первой
стадии этанол и повышая затем температуру до 140° для осуществле-
ния перегруппировки. Этим методом н-хлораннлин был превращен
в N-этил-н-хлорформанилпд с выходом 87—92% [201. Хлористый
водород катализирует образование (2), но не перегруппировку в (3).
Для получения этил-П-фенилформимидата [21] 1,01 моля анилина
нагревают с 1 мл конц. соляной кислоты, для удаления воды, при-
сутствующей в кислоте, добавляют 1,5 моля О. к. т. э., этанол от-
гоняют до тех пор, пока его количество не будет близко к теоретиче-
скому (2 час), затем смесь фракционируют в вакууме. Выход соеди-
нения (1) 78—84%.
Синтез гетероциклов. Смесь 2-аминотиофенол а (0,17 моля),
О. к. т. э. (0,25 моля) и конц. серной кислоты (0,007 моля) постепенно
(СгН5О)гСНОСгН5
н+
37
нагревают до 180° с отгонкой выделяющегося этанола; последую-
щая перегонка в вакууме дает бензтиазол [221. Использование
ортоэфиров типа RC(OC2H5)3 приводит к 2-алкилбензтиазолам. Из
о-аминофенола этим путем получают бензоксазолы. Выходы дости-
гают 75—85%.
Ортоэфиры стероидов. В условиях кислого катализа О. к. т. э.
реагирует со стероидами, имеющими диокснацетоновую боковую
цепь (I), образуя защищенное производное (2) [231. Д4-3-Кетогруппа
в этих условиях в эфир енола не превращается. Возможны два пра-
вовращающих стереоизомера. С хлор гидр атом пиридина в качестве
(2)
катализатора образуется исключительно изомер с большим удель-
ным вращением, а с /г-толуолсульфокислотой — с меньшим. Ор-
тоэфиры устойчивы к действию оснований, но гидролизуются с вы-
соким выходом при непродолжительной обработке 1 н. раствором со-
ляной кислоты в метаноле. Гидролиз щавелевой кислотой дает смесь
17а-моноформиата (3) и 21-моноформиата (4), которые можно разде-
лить хроматографическп. Этот путь по существу является единствен-
ным для получения 21-окси-17-моноэфиров.
Этерификация. При нагревании с избытком О. к. т. э. карбо-
новые кислоты превращаются в соответствующие этиловые эфиры.
Этерифицируются даже пространственно затрудненные кислоты,
такие, как 2,4,6-триметилбензойная кислота [241. Кислый катали-
затор обычно не требуется и даже может оказаться вредным; однако
этерификация никотиновой и гиппуровой кислот проводится в при-
сутствии и-толуолсульфокислоты в ДМФА.
Циклические амидины [25]. Циклизация (1) в (2) достигается
кипячением основания с О. к. т. э. Реагент вводит в (2) атом угле-
рода в положение 6.
38
Оксаадамантаиы. Фогль, Андерсон и Симонс [26] установили,
что сциллит (1) реагирует с О. к. т. э. в ДМСО при 200°, образуя
гексаоксаднамантан Г(2), т. пл. 303—305°]. Продукт интересен как
аналог углеводорода диамантана (конгрессан), второго представи-
теля ряда адамантана, Штеттер и сотр. 127] синтезировали триок-
саадамантан (4) реакцией цнс-флороглюцита (3) с О. к. т. э. в
метаноле в присутствии хлористого водорода.
(3) (4)
1. Кауфман, Д р еджс р, «Синтезы органических препаратов», ИЛ, iM.,
1949, сб, 1, стр. 554,
2. Бахман Дж., «Синтезы органических препаратов», ИЛ, М,, 1949, сб. 2,
стр. 295.
3. Д орнфельд К., Колеман Дж., «Синтезы органических препара-
тов», ИЛ, Москва, 1953, сб. 4, стр. 22.
4. Дорпфельд К-, Каллен Дж,, Кол ьмэ н Дж., «Синтезы органи-
ческих препаратов», ИЛ, Москва, 1953, сб. 4, стр. 96.
5. И о \v к В. W,, Sauer J. С., J. Am, Chem. Soc,, 80, 4607 (1958); А л л ен
Ч., Эдене К., мл., «Синтезы органических препаратов», ИЛ, М., 1952, сб. 3,
стр. 444.
6. Gross Н., R i е с h е А., М a t t е у G., Cliem. Вег., 96, 308 (1963).
39
7. С 1 a i s e n I., Ber., 26, 2729 (1893); Ann., 297, 76 (1897).
8, Ван Аллан Дж., «Синтезы органических препаратов», ИЛ, М., 1953, сб. 4,
стр. 70.
9. Weisblat D. I, et al,, J. Am. Chem. Soc., 75, 5893 (1953).
10. Аллен Ч„Эденс !<., мл., «Синтезы органических препаратов», ИЛ, М.,
1952, сб. 3, стр. 444.
11. С 1 а 1 s е п I., Вег., 29, 1005 (1896); 31, 1019 (1898); 40, 3903 (1907).
12. Пархам В., Р и д Л., «Синтезы органических препаратов», ИЛ, М., 1953,
сб. 4, стр. 596.
13. Дем Б,, Уайтхед К., «Синтезы органических препаратов», ИЛ, М.,
1959, сб. 9, стр. 14.
14. S е г i n i A., Koster Н., Вег., 71, 1766 (1938); I п h о f f е п И. И. et
ah, Chem. Вег., 84, 361 (1951).
15. G а г d i R., Castelli P. P., E г с о 1 i A., Tetrahedron Letters, 497 (1962).
16. M e e k E. G., T u r n b u 1 1 J. H., Wilson W., J. Chem. Soc., 1953, 811.
17. Wichelhaus H., Ber., 2, 115 (1869).
18. Claisen L., Ann., 287, 360 (1895).
19. Roberts R.M.,Vogt P, J., J. Am. Chem. Soc., 78, 4778 (1956).
20. P о б с p т с P., Фогт П., «Синтезы органических препаратов», ИЛ, М.,
1960, сб. 10, стр. 80.
21. Р о б е р т с Р., Фогт П., «Синтезы органических препаратов», ИЛ, М.,
1957, сб. 7, стр. 88.
22. J е п k i n s G. L., К и е v е 1 А. М., Davis С. S., J. Org. Chem., 26, 274
(1961).
23. G а г d i R., Vitali R., E г с о 1 i A., Tetrahedron Letters, 448 (1961).
24. Cohen H.,Mier J. D., Chem. Ind., 1965, 349.
25. Butler K., Partridge At. W., Waite J. A., J. Chem. Soc., I960,
4970‘ Partridge M. W., Slorach S. A., V i p о n d H. J., J. Chem.
Soc., 1964, 3670.
26. V о g 1 O., Anderson B.C., Simons D. M., Tetrahedron Letters, 415
(1966).
27. S t e t t e r H., Steinach er К- H., Chem. Ber., 86, 790 (1953).
ОРТОУГОЛЬНОЙ КИСЛОТЫ ТЕТРАЭТИЛОВЫЙ ЭФИР,
С(ОС2Но)4. Мол. вес 192,25, т. кип. 158°, По' 1,3907, уд. вес 0,92.
К раствору этилата натрия в абсолютном этаноле при 58—60е
в течение 2 час при перемешивании добавляют хлорпикрин. Раство-
CCI3NO2-!-4C2Hr,ONa A C(OC2H&)4H-3NaCl + NaNO2
ритель удаляют отгонкой, смесь обрабатывают водой и экстрагиру-
ют эфиром Ill.
Гофман [2] установил, что ортоэфир реагирует с водным аммиа-
ком с образованием гуанидина. Реакция с анилином приводит к об-
разованию какого-либо из нескольких продуктов [3].
1. Робертс Дж., Мак-Магон Р., «Синтезы органических препаратов»,
ИЛ, М., 1953, сб. 4, стр. 584.
2. Hofmann A, W. von, Ann., 139, 114 (1866).
3. Tieckelmann Н., Post Н. W., J. Org. Chem., 13, 268 (1948).
ОСМИЯ ЧЕТЫРЕХОКИСЬ (осмиевый ангидрид), OsO4. Мол. вес
254,20, т. пл. 40°, т. кип. 135°. О. ч. растворяется в эфире и бензоле.
Растворимость в воде при 15° 5,88 а/100 г.
40
Этот дорогой реагент продается в запаянных стеклянных ампу-
лах, содержащих по 1 а и упакованных в деревянный ящичек с при-
крепленным к нему стеклорезом. Ампулу надрезают в середине,
переламывают, извлекают из нее кристаллы О. ч. и растворяют их
в эфире. При добавлении олефиновой компоненты медленно выпада-
ет осмат (обычно черный). Приблизительно через 24 час эфир отго-
няют, а осмат подвергают восстановительному расщеплению одним
из нижеописанных методов с образованием (обычно с высоким вы-
ходом) вицинального гликоля. В тех случаях, когда возможна изо-
мерия, продукт представляет собой чистый цис- или эритро-изомер,
так как он образуется из цис-осмата без затрагивания связей С—О.
I I 1
-с Oso “С-0 о и -С-ОН
Г- I X “ I
— С - С —СИ ЫЭ —С —он
I I I
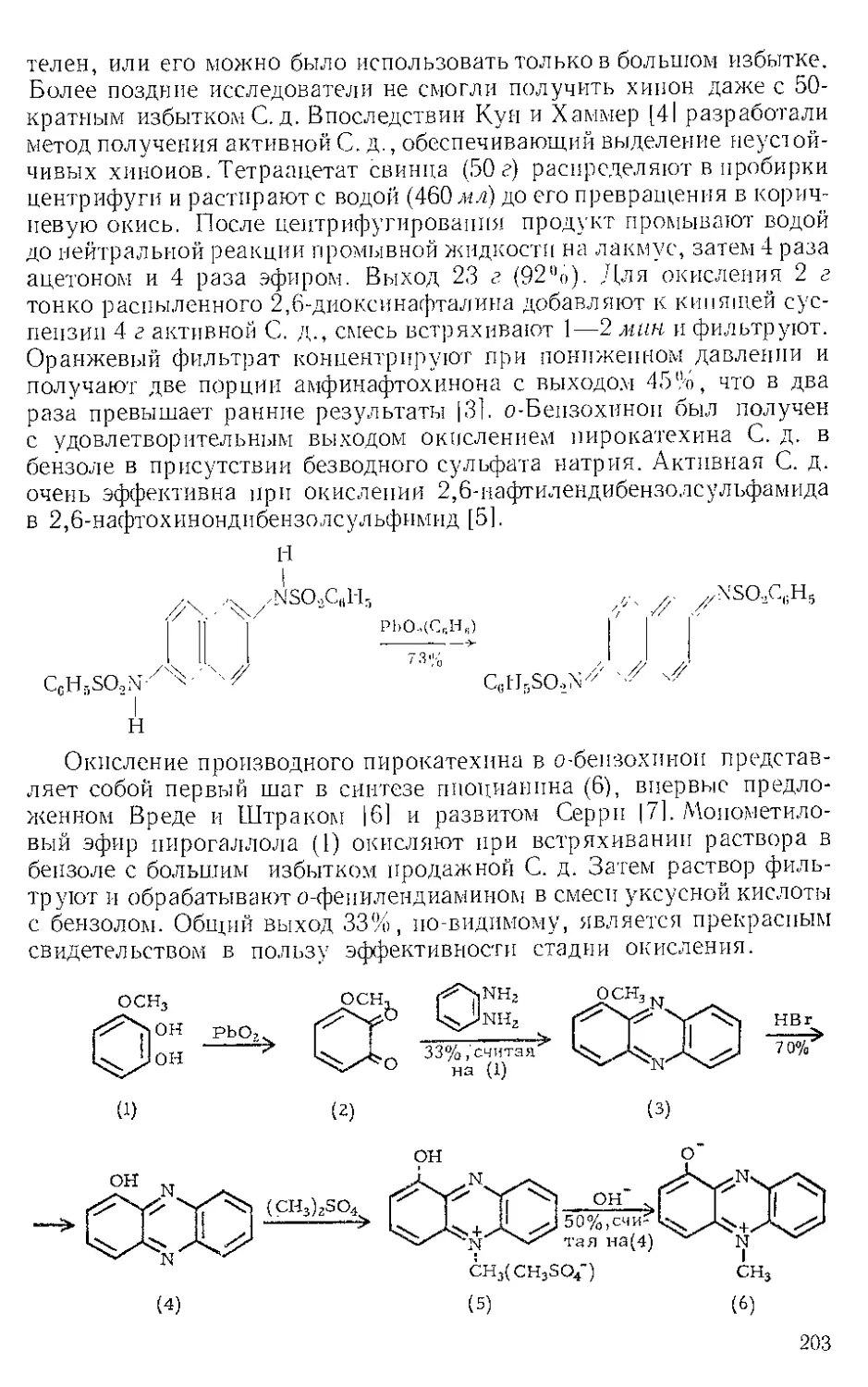
Описывая открытие этого удобного метода получения цас-гли-
колей, Криге III сообщил о том, что стимулировало его исследова-
ния. Гофманн [2] показал, что органические н неорганические сое-
динения, абсолютно устойчивые к действию хлората натрия или
калия в нейтральном водном растворе, легко окисляются при добав-
лении каталитических количеств О. ч. Например, раствор 2 г гид-
рохинона и 2 г хлората натрия в 100 мл воды не претерпевает при
20° никаких изменений, но при добавлении раствора 7,5 мг О. ч.
в 1,5 мл воды сразу же становится темно-коричневым и вскоре из
него выпадают зеленые иглы хингидрона. Система NaC103— OsO4
является также специфическим окислителем ненасыщенных соеди-
нений до вшргликолей. Окисление фумаровой кислоты в од-вин-
ную кислоту и малеиновой кислоты в мезовинпую кислоту подтвер-
ждает, что реакция осуществляется как стереоспецифическое цис-
присоединение, подобное окислению перманганатом калия. Бозекеи
13] предполагает, что образование ^uc-диола в реакции с перманга-
натом калия обусловлено промежуточным циклическим соединени-
ем, которое, естественно, может иметь только цис-конфигурацию.
Циклический манганат
Предполагаемый промежуточный продукт можно представить в виде
циклического манганата, который, по-видимому, невозможно выде-
лить, так как он растворим в воде и очень легко гидролизуется.
Бёзекен предполагает также, что при окислении водным раствором
41
NaC10;i— OsOi образуется аналогичное промежуточное соединение,
которое подвергается окислительному расщеплению хлоратом с ре-
генерацией О. ч.
СО.Н СО.2Н
Н — С—СО3Г1 OsOt Н-С-0 /Ч’аСЮз н-с-он
II Л 1 /°\ ~н7Г I + °S°4
Н-С—СО2Н Н — С—О %О 2 И— с—он
I !
со.2н со2н
Криге [1] подтвердил гипотезу Бозекена при изучении реакции О. ч.
с олефинами в неводных растворителях (эфир, бензол, циклогексан).
Из аценафтилена, индена, Л^-дитралина и А!,-окталина он получил
мелкокристаллические ярко окрашенные осматы с 89—99%-ным
выходом; для перекристаллизации веществ не удалось подобрать
растворитель, но анализы полученных образцов дали приемлемые
значения содержания осмия. Криге осуществил восстановительное
расщепление осматов кипячением с водно-спиртовым раствором суль-
фита натрия; осмий выделяется в виде иеидентпфицироваиного ко-
ричнево-черного комплекса, а из фильтрата получается диол. Выхо-
ды четырех цис-диолов составили 84, 66, 78 и 81 % . За исключением
четырех приведенных примеров, осматы обычно представляют со-
бой микрокристаллические или аморфные темно-коричневые или
черные твердые вещества.
В следующей работе {4] Криге сообщил, что пиридин заметно ка-
тализирует реакцию О. ч. с олефином и что осмат дает с 2 молеку-
лами пиридина комплекс, который можно перекристаллизовать из
смеси хлористый метилен — петролейный эфир. При проведении ре-
акции в бензоле, содержащем пиридин, О. ч. присоединяется даже
к тетрафенилэтилену (который, как известно, не присоединяет бром)
с образованием комплекса ди пиридин — осмат. При добавлении 0,005
моля фенантрена к раствору 0,005 моля О. ч. в 15 мл бензола сразу
появляется оранжевое окрашивание (образуется молекулярный
комплекс). При добавлении 0,01 моля пиридина цвет изменяется
до желтого и затем постепенно до коричневого. Через 7 дней обра-
зуется 2,8 г (95%) красно-коричневых кристаллов комплекса (2).
Для расщепления раствор 2,4 г комплекса (2) в 25 мл хлористого ме-
тилена встряхивают с раствором 10 г маннита и 1 г едкого кали
в 100 мл воды до обесцвечивания органического слоя (около 1 час).
Отгонка хлористого метилена дает остаток (0,55 г), который после
кристаллизации из разб. этанола (с норитом) и затем из толуола
42
дает бесцветные иглы чистого диола (3) с т. пл. 178 — 179'. Едкое
кали расщепляет осмат до положения равновесия, а маннит смещает
равновесие за счет переэтерификации.
Криге установил, что диены образуют с О. ч. 1,2-, а не 1,4-аддукты.
Метод с использованием щелочи и маннита не имеет широкого
применения, как и другие предложенные методы: с сульфитом на-
трия п цинковой пылью 151, со щелочным формальдегидом [61 и
аскорбиновой кислотой [6]. Бэран [71 эффективно усовершенствовал
методику Криге с применением пиридина, что иллюстрируется на
примере апдростенолона. Раствор 1,14 г (3,9жмоля) андростенолона
и 1,0 г (0,394 л/моля) О. ч. в 15 мл пиридина перемешивают 2 час
при комнатной температуре и при непрерывном перемешивании об-
рабатывают раствором 1,8 г бисульфита натрия в 30 мл воды и 20 мл
пиридина. Через 5 мин комплекс расщепляется с образованием оран-
жевого раствора, который экстрагируют хлористым метиленом пор-
циями 100, 50 и 50 мл. Отгонкой высушенного экстракта и растира-
нием остатка с этилацетатом получают 1,05 г (86%) продукта с т. пл.
240—243°.
Бартон и Элад 181 при исследовании природных соединений вве-
ли новый метод расщепления осмата. Раствор олефина и О. ч. в ди-
оксане выдерживают 48 час и затем обрабатывают сероводородом
для осаждения черной двуокиси осмия, которую удаляют фильтро-
ванием, а из фильтрата выделяют диол. Очень показательно приме-
нение этой методики при работе с большими количествами дорого-
стоящих реагентов (Хиршманн и сотр. 191) в превращении предни-
зона (1) в А-норкортизол (9), который, как было показано, не обла-
дает физиологической активностью. Раствор 100 г преднизона BMD
(2) в 720 мл пиридина охлаждают до 5° и обрабатывают раствором
43
69,9 г О. ч. (избыток 10%!) в 408 мл пиридина. Через 5 мин начинает-
ся выделение черного вещества, а через 5 дней (при комнатной тем-
пературе) реакционную смесь перемешивают с 13,4 л пстролейного
эфира, осадок отделяют, промывают и растворяют в 8 л диоксана.
Раствор охлаждают в бане со льдом и насыщают сероводородом.
Осаждающуюся двуокись осмия отделяют и большую часть раство-
рителя отгоняют. Дальнейшей обработкой получают смесь стерео-
изомерных виц-диолов (3) с выходом около 50%.
Новый метод осмилироваиия был использован Сареттом 110] в
синтезе кортизона для введения 17а-оксигруппы (см. схему иа
стр. 45). Циангидрин (II) дегидратируют, первичную 21-оксигруппу
в (Ш) селективно ацетилируют с образованием (IV). Присоедине-
нием О. ч. получают осмат (V), который, как показал Саретт,
является удобным производным для окисления у С3. Расщепление
осмата сульфитом натрия дает свободную 17сс-оксигруппу и сопро-
вождается ретроциангидринной реакцией с освобождением 20-кето-
группы и образованием (VI), имеющего желаемую диоксиацето-
новую боковую цепь.
Осмилирование а, p-ненасыщенного кетона — ключевая стадия в
синтезе триамцинолона, осуществленном Бернштейном и corp. [Ц].
Исходное вещество, 3,20-бмс-этиленкеталь кортизол-21-ацетата (1),
44
взаимодействует при —-5° с тионилхлоридом в пиридине с отщепле-
нием оксигрупп при Сы и С17 и образованием (после омыления,
декетализации серной кислотой в метаноле и ацетилирования)
21-ацетокситриендиона-3,20 (2). Затем селективно гидроксили-
руют 16,17-двойную связь реакцией с О. ч. в бензоле, содержащем
пиридин; расщепление осмата осуществляют в мягких условиях
(NaaSO;i — К2СОл в смеси вода — метанол — бензол) для пред-
отвращения перегруппировки с расширением кольца D.
При идентификации несопряжен кого кетона (2), полученного
из Д*-холестенопа-3 (1) взаимодействием с диазометаном в присут-
ствии трехфтористого бора, Джонсон и сотр. [12] открыли новую
реакцию. Обработка (2) О. ч. дает полукеталь (3), который легко
образует метиловый эфир.
ОН
(1) (2) (3)
1. С г i е g е е R., Ann., 522, 75 (1936).
2. Hofmann К. А., Вег., 45, 3329 (1912); Hofmann 1\. A., Е h г h а г t
О., S с h п е i d е г О., Вег., 46, 1657 (1913).
3. В б е s е к е п J., Rec. Irav., 41, 199 (1922).
4. С г i е g е е R., М а г с h а п d В., Wannowius Н., Ann., 550, 99 (1942).
5. S е г j n i A. et а|., Вег., 71, 1362 (1938); 72, 391 (1939).
6. Reich Н,, Sutter М., R е i с h s t е 1 п Т., Helv. Chilli- Acta, 23, 170
(1940).
7. В а г а п J. S., J. Org, Chem,, 25, 257 (i960).
8. Barton D. И. R.,Elad D.,J. Chem. Soc., 1956, 2085; see also U s koko-
v 1 с M. et al., J. Am. Chem, Soc., 82, 4965 (1960).
9. Hirschmann R., Bailey G. A., Walker R., Cliemerda
J. M., J. Am. Chem. Soc., 81, 2822 (1959).
10. S a r e t t L. H., J. Am. Chem. Soc., 70, 1454 (1948); 71, 2443 (1949).
11, Bernstein S., Lenh ar d R. H,, Allen W. S., Heller M., L i t-
t e 1 1 R., S t о 1 a г S. M., Feldman L. I., В 1 a n к R. HL, J. Am. Soc.
Chem., 78, 5693 (1956).
12, J ohnson W. S., N e c m a n M., Birkela n d S. P., Fedor uk
N. A., J. Am. Chem. Soc., 84, 989 (1962).
ОСМИЯ ЧЕТЫРЕХОКИСЬ — БАРИЯ ХЛОРАТ.
Показано [11, что промежуточный продукт (1) в синтезе алкалои-
дов типа резерпина гидроксилируется под действием реагента с
более высоким выходом диола (2), если четырехокись осмия взята
не в эквивалентном [2], а в каталитическом количестве. Исходное
вещество (1) встряхивают с водным хлоратом бария и небольшим ко-
личеством четырехокиси осмия до исчезновения сине-фиолетовой ок-
раски (10—12 час). Диол (2) легко выделяется и после перекристал-
лизации получается в чистом виде.
46
он
Н j II
ущ :/ОАс НО. /.^у^/ОАс
| I I H-Ba(ClO3), + OsO.1 I I I
/А /\ А 5.77 е I г ' /А \ Д
О" ч/: А ''ОСНЯ '-----------------' г/ Х/: Y ''ОСН3
н со2сн3 200J!'? що и согсн3
(I) 10 г (2)
I. Р 1 a h a L., W е i с h е t J., Z v а с с к J., S m о И к S., Хакас В.,
Coll. Czech., 25, 237 (1960); Ernest I., ibid., 29, 266 (1964).
2. Woodward R. B., Bader F. E., Bickel H,, Frey A. J., Kier-
stead R. W-, Tetrahedron, 2, 1 (1958).
ОСМИЯ ЧЕТЫРЕХОКИСЬ — ПЕРЕКИСЬ ВОДОРОДА, см. Пе-
рекись водорода — осмия четырехокись.
ОСУШИТЕЛИ. Пирсон и Оллереншо [ 1] указывают, что наиболее
просто определять содержание воды в растворителях спектрофото-
метрическим измерением поглощения обертона полосы валентных
колебаний Н — О-связи воды в области 5300 слГ1. Метод определе-
ния по Карлу Фишеру довольно трудоемок и требует расхода боль-
шого количества образца. Сравнение скорости и эффективности вы-
сушивания бензола, диэтилового эфира и этилацетата при комнат-
ной температуре показывает, что хлористый кальций, безводный
сульфат кальция и молекулярные сита 4А являются быстродей-
ствующими осушителями, поглощающими значительное количе-
ство воды. Сульфаты магния и натрия поглощают воду значительно
медленнее и лишь в небольших количествах. Диэтиловый эфир
обезвоживается значительно быстрее, чем бензол или этилацетат.
1. Pearson В. D., Ollerenshaw J. Е., Chem, Ind., 1966, 370.
ПАЛЛАДИЕВЫЕ КАТАЛИЗАТОРЫ, см. также Линдлара ката-
лизатор.
Мозииго [1] описал приготовление четырех катализаторов
А 5%-ный Pd на BaSO4
Б 5%-пый Pd на С
В PdCl3 на С (5% Pd)
Г 10%-ный Pd на С
Катализаторы А и Б получают восстановлением щелочным
раствором формальдегида. Они не содержат адсорбированного во-
Na2PdCl4 + СН2О + 3NaOH Pd + HCO.Na Д- 4NaCl + 2НгО
дорода и менее пирофорны по сравнению с катализатором Г, получае-
мым восстановлением водородом (PdCla+H2—>Pd+HCl). Для по-
лучения катализатора В соль палладия восстанавливают по мере
необходимости водородом, причем катализатор не теряет активности
при хранении.
Согласно Краму и Аллинжеру [2], при неполном гидрирова-
нии тройной связи катализатор Pd — BaSO4, частично отравлен-
ный хинолином, дает даже более воспроизводимые результаты, чем
катализатор Линдлара.
Гидрогенолиз. Розенмунд [3] показал, что активность катали-
затора Pd — BaSO4 при гидрогенолизе спиртов типа АгСН(ОН)—
может быть заметно увеличена добавлением небольшого количества
хлорной кислоты. Так, гидрогенолиз (1) во (2) в уксусной кислоте
значительно ускоряется при добавлении небольшого количества
СН3О — Ч—СН—снсн,сн3
\=/ f I
(1) ОН + NH3C1
Pd—BaSO4.H2(HClO,)
—> СН3О—Ч____
(2)
СНХНСНХНз
+ NH3CH
раствора 70 %-ной хлорной кислоты в уксусной. Киндлер [4] уста-
новил, что серная кислота является эффективным катализатором
превращения ацетофенона в этилбензол. Палладиевую чернь про-
С6Н-,СОСН3 Нз’ С6Н5СНОНСН3 С6Н5СН2СНз
4§
мывают разб. серной кислотой, а затем водой и метанолом до пол-
ного удаления кислоты. Полученный таким образом катализатор
обладает высокой активностью.
Дегидрирование. Линстед [5] показал, что дегидрирование
гидр ©ароматических углеводородов в присутствии палладия на угле
в жидкой фазе ускоряется как при пропускании через раствор тока
двуокиси углерода, способствующего удалению выделяющегося во-
дорода из равновесной смеси, так и при интенсивном кипячении, ус-
коряющем удаление водорода с поверхности катализатора. Кон-
троль реакции можно осуществить, поглощая двуокись углерода и
измеряя объем выделившегося водорода. Тетралин (т. кип. 208°)
дегидрируется в жидкой фазе только при кипячении. Дегидрирова-
ние можно осуществить и при 185°, если обеспечить кипение пони-
жением давления или добавлением разбавителя с более низкой тем-
пературой кипения. Однако в отсутствие кипения реакция при
200° не осуществляется, цис- и траке-Декад ины дегидрируются
крайне медленно при температуре кипения (около 190°), но удовлет-
ворительно в паровой фазе при 300°.
Фрайд и сотр. [6] в процессе расщепления эбурикоевой кислоты
(1) до 14-метилпрегнана ацетат этой кислоты превращали в норке-
токислоту (2), циклизацией которой получали смесь двух енольных
лактонов (3) и (4) с преобладанием (3); затем пытались дегидри-
49
ровать эти вещества до а-пирона (5). При исследовании таких
дегидрирующих агентов, как двуокись селена, ацетат ртути (II)
и бром, показано, что реакция осуществляется гладко с хорошим
выходом на 10%-ном палладии иа угле в кипящем и-цимоле
(т. кип. 177°). Из эндоцикл и чес ко г о лактона (3) получается чи-
стый (х-пирон (5) с 75%-ным выходом. Экзоцикл ический лактон (4)
также дегидрируется до (5), однако в этом случае выход состав-
ляет только 60% и продолжительность реакции возрастает от 2 до
6 час. По-видимому, медленная стадия процесса состоит в миграции
двойной связи с образованием лактона (3). В интер штейнер и Моор
[7] применили этот метод для дегидрирования соединения (6), про-
дукта расщепления изоиервина, и получили ароматическое произ-
водное вератрамнна (7).
Фенантрол-1 получают с хорошим выходом кипячением гидро-
ароматического кетона с палладиевым катализатором в нафталине
в течение суток [7а1.
Фенантрол-1
(г. ПЛ. 157°)
Катализатор палладий — платина на угле, по-видимому, наибо-
лее эффективен для дегидро конденсации двух бензольных циклов
с образованием третьего, например для превращения о-терфенила
(8) в трифенилен (9) [81 и 1,Г-динафтила (10) в перилен (11) [91.
50
Катализатор получают 1101 добавлением 400 г гранулированного
древесного угля (для абсорбции газов) к раствору 7,3 а платино-
хлористоводородной кислоты и 4 а хлористого палладия в 400 мл
воды с последующим добавлением 635 мл 35%-иого водного раст-
вора формальдегида при механическом перемешивании смеси и
охлаждении до —10°. Затем добавляют раствор 580 г едкого кали
в 580 мл воды с такой скоростью, чтобы температура смеси не под-
нималась выше 5°. Перемешивание продолжают при 60° в течение
15 мин. Катализатор тщательно промывают декантацией водой, за-
тем 1 %-ным раствором уксусной кислоты до отрицательной реак-
ции на ионы хлора и сушат хлористым кальцием.
Дегидрирование проводят в вертикальной стеклянной трубке,
нагреваемой в электрической печи до 490°. В нижней части этой
трубки находится катализатор; верхнюю часть заполняют фарфо-
ровыми шариками, и она служит в качестве подогревателя и испа-
рителя. Раствор 4,5 го-терфеиила в 15 мл очищенного декалина про-
пускают вместе с водородом над горячим катализатором в течение
3 час. Реакционную смесь хроматографируют.
Крауфорд с сотр. [10а] нашли интересную систему реагентов
для эффективного дегидрирования: палладий на древесном угле и
сера. Аддукт Дильса — Альдера (3) получали восстановлением те-
тралона (1) в пинакон, дегидратацией в (2) кипячением в уксусной
кислоте и далее реакцией с малеиновым ангидридом. Соединение (3)
не удалось дегидрировать в присутствии палладия на древесном угле
или серы в отдельности. Однако если тонкоизмельчениую смесь 0,5 г
(3), 0,05 г 30 %-кого Pd — С и 0,15 г серы нагревают на масляной
51
бане при 300° в течение 5 мин, то наблюдается выделение сероводоро-
да, а после экстракции хлороформом и кристаллизации из уксус-
ного ангидрида получается 0,22 г темно-красных призм (4), т. пл.
323,5°.
Сера или Pd — С в отдельности дегидрируют (5) до (6) при 300 —
310°, однако их сочетание дает при этой температуре равные коли-
чества (6) и (7). Поскольку дегидрирование 1,1'-динафтила (6) в (7)
в этих условиях не происходит, дегидроциклизация, приводящая
к образованию (7), должна предшествовать полной ароматизации.
Действие катализатора можно объяснить следующим образом: пал-
ладий отщепляет атомы водорода с образованием бирадикала, кото-
рый далее циклизуется. В отсутствие серы должно происходить бы-
строе насыщение палладия водородом, но сера способствует удале-
нию водорода с металла в виде сероводорода. Гидрирование в при-
сутствии металлического катализатора может осуществляться пу-
тем переноса водорода от богатого водородом донора. Описанная
реакция представляет собой обратный процесс: дегидрирование за
счет вещества, не содержащего водорода, т. е. серы.
Декарбонилирование альдегидов. Некоторые ароматические аль-
дегиды декарбон ил ируются при нагревании в присутствии 5%-ного
палладия на угле, например [111:
Pd — С, 1 час, 17 9°
с6н6сно —-----—--------. С6Н6+СО
Pd—С, 1 час, 243°
o-CH3OCsH4CHO -------------—> С6Н&ОСН3 - со
94%
Pd —С, 0,5 час, 205°
zbO2NQH4CHO-------- СВН5\Ю3ШСО
' ' %
Алифатические и некоторые ароматические альдегиды реагируют
неудовлетворительно. Так, 1-нафтальдегид превращается в нафта-
лин при 210°, тогда как из 2-нафта л ьдегида даже при 250—255°
образуются только следы газа.
Восстановление гидразином и Pd — С, см. Гидразин, раздел
Гидразин и металлический катализатор.
Гидроокись палладия на угле [121. Этот непирофорный катали-
затор сухого гидрирования, описанный Перлманом, получают сле-
дующим образом: смесь 100 г PdCl2, 240 г угля марки Darco G-60
и 2 л деионизированной воды перемешивают при 80° и обрабатывают
52
раствором 50 г 1ЛОН-Н2О в 200 мл воды; затем нагревание прекра-
щают и перемешивание продолжают в течение ночи. Твердый оса-
док отфильтровывают, промывают разб. уксусной кислотой и сушат
в вакууме при 60°. Перед употреблением катализатор смачивают
растворителем; даже в случае органического растворителя специаль-
ных мер предосторожности не требуется. Катализатор, полученный
восстановлением гидроокиси металла, высокоактивен.
1. Mozingo R., Org. Syn., Coll. Vol3, 685 (1955).
2. Cram D. J., Al linger N.L..J. Am. Chem. Soc., 78, 2518 (1956).
3. Rosenmund K. W., Karg E., Ber., 75, 1850 (1942).
4'. Kindler K-, S c h a r f e E., Henrich P., Ann., 565, 51 (1949).
5. Linstead R. P., M i 1 1 i d g e A, F., Thomas S. L. S., W a 1-
p о 1 e A. L., J. Chem. Soc., 1937, 1146; Linstead R. P., Thomas S. L. S.,
ibid., 1940, 1127.
6. Rosenthal D., Gr a bowi ch P., Sub о E.F-, Fried J.,J, Am.
Chem. Soc., 85, 3971 (1963).
7. Winters! einer O., Moore M., Tetrahedron Letters, 795 (1962).
7a. M о s e t t i g E., Duvall H. M., J. Ащ. Chem. Soc., 59, 387 (1937).
8. Copeland P. G., Dean R. E., M c N e i 1 D., J. Chem. Soc., 1960, 1687.
9. Copeland P. G., Dean R. E., M c N e i 1 D., J. Chem. Soc., 1960, 1689.
10. Baker W., Warburton W. К., Breddy L. J., J. Chem. Soc.,
1953, 4149.
10a. Blair H.S.,Crawfor d M., Spence J. M., Supanekar V. R.,
J. Chem, Soc., 1960, 3313; Crawford M., Supanekar V. R., ibid.,
1964, 2380.
11. HawthorneJ.O., W i 1 t M- H., J. Org. Chem., 25, 2215 (1960).
12. Pearlman W. AL, procedure submitted to Org. Syn.
ПАЛЛАДИЙ ХЛОРИСТЫЙ, PdCl2. Мол. вес 177,32.
Новый метод получения изоцианата состоит в реакции амина
с окисью углерода в присутствии П. х. [11.
G5° 1 атм, 48 час
H-C4H9NH2-FCO-pPdCl2 .H-C4H9N = C = O+Pd+2HC1
1. S t е г и Е. W*, Spector М. L,3 J. Org. Chem+, 31, 596 (1966).
ПАЛЛАДИЯ (II) АЦЕТАТ, Pd(OCOCH3)2, коричневые кри-
сталлы, получаемые из нитрата палладия и уксусной кислоты и.и
растворением губчатого Pd в горячей уксусной кислоте, содержащей
незначительное количество HNO3 111.
Под действием П. а. А1-олефины окисляются главным образом
до енолацетатов и А2-олефины — преимущественно до аллилаце-
ОАс
HPdOAc I
Pd(OAc)2-yCH2-CHCH.2R —> Г СН2—CHCFLR -----------> CH2 = CCH2R
AcOPd OAc
Pd(OAc)2 + CH3CFI = CHCH2R Г CH3C
AcOPd OAc
CH3 = CHCHCH2R
OAc
татов [2]. Считают, что реакция включает стадию оксипалладиро-
вания с последующим отщеплением элементов HPdOCOCH3.
1. Morehouse S. М., Powell А. R., Н е f f е г J. Р., Stephenson
Т. A., Wilkinson G., Chem, Ind., 1964, 544.
2. К i t c h i n g W,, Rappoport Z., Winst ein S., Young W- G.,
J. Am. Chem, Soc., 88, 2054 (19G6).
ПЕНТАДИЕН-1,4, CH2-CHCH.CH = CH.. Мол. вес 68,11, т. кип.
26—27,5°.
Получение (а) [1]. Ацетилированием пентандиола-1,5 получают
диацетат, который добавляют по каплям в стеклянную реакционную
Капилляр
(о,б лш х 8 слг)
Рис. П-1. Капельная воронка
Гершберга [2].
Р и с. П-2. Капельная воронка
Гершберга с отводом для выравни-
вания давления [\].
трубку, установленную вертикально в электрической печи. Нагре-
ваемую часть трубки заполняют стеклянными кольцами, поддержи-
СНоСН3СНпСНйСНа —СН.2 = СНСН2СН=СНо + 2СН3СООН
| i 67 — 71%
ОАс ОАс
54
ваемыми тампоном из стекловолокна, который зафиксирован вы-
ступами в трубке. Трубка в верхней части снабжена вводом для азо-
та и капельной воронкой Гершберга [2] (рис. П-1) с отводом для вы-
равнивания давления (рис. П-2 или рис. П-3). Выводная трубка про-
ходит в круглодонную колбу емкостью 1 л, охлаждаемую баней со
льдом и имеющую отвод, через который пары последовательно по-
падают в соединенные ловушки, охлаждаемые льдом и смесью сухо-
го льда с ацетоном. Среднюю часть трубки нагревают до 575°, в
Рис. П-3. Капельная воронка* (мо-
дель «Асе Glass 7340»),
* В этой новой модели дозировка обе-
спечивается при помощи точной опоры
и стержня. Грубая дозировка производится
перемещением стержня вверх пин вниз,
точная — вращением нарезного найлонового
держателя. Четыре оборота вблизи откры-
того положения изменяют скорость при-
капывания в 2 раза, вблизи закрытого
положения — ji 0, 1 раза. Использование
платиновой проволоки внутри капилляра
позволяет работать с сильнокорродирующи-
Mji веществами. Легко снимаемый тефло-
новый конус с более эластичным витоновым
или силиконовым кольцом позволяет со-
здать избыточное давление. Минимальная
скорость пр и ка пы п ан и я 10 капель/мин,
максимальная — 6 мл/мин.
трубку через реометр пропускают азот, затем в течение 3,5 час до-
бавляют 645 мл диацетата пентандиол а-1,5. Содержимое трех прием-
ников объединяют, разгоняют и перегоняют повторно.
Получение (б) [3]. Последовательность реакций показана на
приведенной ниже схеме. Обработкой паральдегида и абсолютного
этанола газообразным хлористым водородом при —5° получают
а-хлорэтилэтиловый эфир (I), который бронируется до а, fJ-дибром-
этилэтилового эфира (2). Взаимодействие с алл ил магнийбр омидом
55
приводит к (3), из которого реакцией с цинковой пылью в н-бутило-
вом спирте получается П. Таким образом, одна двойная связь обра-
зуется за счет бромистого аллила, другая — при Отщеплении
ВгОС2Н5 от этилового эфира бромгидрина (3).
СН3СНО + С2Н5ОНЗ-НС1 .. СН3СНОС,Н5
87 — 92% | 66-73%
С1
(1)
—> ВгСНХНОС,Н5 СНг = СНСН2СНОСоН5~*
| “ 77-82% I
Вг СНаВг
(S) (3)
--!1—+ СН2 = СНСН,СН —СНа
72-76"% (4)
I. Benson R. Е., М с К u s i с к В. С., Org. Syn., Coll. Vol., 4, 476 (1963).
2. Ф изер Л„ Хершберг Е., «Синтезы органических препаратов», ИЛ,
М-, 1949, сб. 2, стр. 349.
3. G г u m m i 11 О,, Budewitz E. P., Chu d d C.C., Org. Syn., Coll.
Vol., 4, 748 (1963).
ПЕНТАХЛОРФЕНОЛ, C6C15OH. Мол. вес 266,34, т. пл. 189—191°.
Синтез полипептидов. Конденсация дициклогексил кар бод и имид-
ным методом аминокислоты с П. приводит к активированному эфи-
ру, который в ДМФА в присутствии третичного амина легко полиме-
ризуется с образованием полиаминокислоты [1]. Полученные таким
путем полимеры имеют большие молекулярные веса по сравнению
с полимерами, синтезированными другими методами. Этот метод
активированного эфира применяют для синтеза полипептидов, со-
держащих повторяющуюся последовательность аминокислот. На-
пример, дипептид превращают в активированный эфир, который по-
лимеризуется.
1. Kovacs J.,Kapoor A., J. Аш. Chcm. Soc., 87, 118 (1965); Kovacs J.,
Ballina R., Rodin R.L., В alasubramani an D., Apple-
q u i st J., ibid., 87, 119 (1965); Kovacs J.t Ceprini M. Q., Chem. Ind.,
1965, 2100; Kovacs J., Johnson B.J., J. Chem. Soc., 1965, 6777; К o-
v a c s J.,Gi annot 1 i R., Kapoor A., J. Am. Chem. Soc., 88, 2282 (1966),
ПЕРЕКИСЬ ВОДОРОДА, HaO3. Мол. вес64,02. 30%-ный раствор
соответствует 11,6М; 86жл--=1 молю. Обсуждается возможная опас-
ность при работе с конц. растворами П. в. [Ц. При работе с боль-
шими количествами даже 30%-ного раствора необходимо использо-
вать предохранительный защитный экран.
Для удобства рассмотрения многочисленные реакции П. в. клас-
сифицированы далее в зависимости от условий их проведения: в
кислой, основной или нейтральной среде, с различными добавками.
1. S h а п 1 е у E.S., Greens pan Е. Р., Ind. Eng. Chem., 39, 1536 (1947);
Schumb W. C., ibid., 41, 992 (1949),
56
ПЕРЕКИСЬ ВОДОРОДА В КИСЛОЙ СРЕДЕ.
Надмуравьииая кислота. Надмуравьиная кислота получается
при слабом нагревании П. в. (30%-ной или более высокой концен-
трации) с избытком муравьиной кислоты. Надкислота реагирует
с олефинами с образованием эпоксидов, которые далее расщепляют-
ся муравьиной кислотой с раскрытием цикла в одном или двух воз-
можных направлениях с образованием моноформиата или смеси
двух возможных моноформиатов. Обычно продукт реакции выса-
живают водой и гидролизуют щелочью до диола. Ациклические
h2o2IL5£™ НСО3Н
НСООН
> С—С< + >с—с<
II II
он осмо осно он
ок-
он он
цис-олефины образуют mpco-диол; циклогексен дает трдас-диак-
сиальный диол. В обоих случаях реакцию можно объяснить как
транс-гидроксилирование.
Например [1J, смесь 0,5 моля олеиновой кислоты, 425 мл муравь-
иной кислоты (90—98%-ной) и 0,5 моля 30%-ной П. в. нагревают
при 40° в течение 3 час. Избыток муравьиной кислоты удаляют от-
гонкой при 50°/125 мм в токе азота или двуокиси углерода для пре-
дотвращения толчков при кипении. Остаток нагревают с избытком
н н нсо3н, н он
| I NaOH
CH3(CHn)7C^C(CH2)7CO.,H—---— СН
v - г»0—55% I |
онн
3(сн2)7(1-~с(сн2)7со2н
3 и. раствора едкого натра и выливают в соляную кислоту. Вод-
ный раствор декантируют, а остаток расплавляют под водой для уда-
ления неорганических солей. После трех кристаллизаций из 95%-
ного этанола получают чистый mpco-диол с т. пл. 94—95°.
транс-Гидроксилирован не холестерина [2] осуществляют на-
греванием суспензии 20 г холестерина в 200 мл 88%-ной муравьиной
кислоты при 70—80° при перемешивании в течение 5 мин, после
57
чего формиат холестерина отделяется в виде верхнего маслянистого
слоя. Смесь охлаждают до 25° и добавляют 20 мл 30%-ной П. в.
Реакционную смесь постепенно нагревают до 35—40° и затем очень
медленно охлаждают. Спустя 6—15 час добавляют 300 м.л кипящей
воды, смесь перемешивают, охлаждают и отделяют продукт реакции
(на этой стадии можно выделить 3,6-диформиат, по с низким выхо-
дом). Влажное вещество растворяют в 600 мл метанола и добавляют
20 мл 25%-ного едкого натра. Смесь нагревают на кипящей водяной
бане в течение 10 мин, фильтруют, разбавляют и подкисляют соля-
ной кислотой. При этом осаждается холестаитриол-3р,5а,6|3, ко-
торый плавится (полностью высушенный) при 236—238°. Перекри-
сталлизация из метанола дает иглы с т. пл. 237—239°. Реакцией ци-
клогексена с надмуравьиной кислотой по аналогичной методике
после гидролиза получается /прй«с-циклогександиол-1, 2 с выходом
65—73% 13].
Наличие фенилы-юй группы у одного или двух углеродных ато-
мов олефина изменяет стереохимию реакции — эпоксидный цикл
раскрывается с сохранением конфигурации у атома углерода, свя-
занного с фенильным радикалом, и образуется цис-диол. Недавно
НСОгН илиО зО4
7^ 'он%=-
и Ън
Ривер [4] установил, что при гидроксилировании 1-фенилциклогеп-
тена (1) надмуравьиной кислотой получается цис-диол (2), идентич-
ный продукту гидроксилирования под действием тетраокиси осмия.
Аналогичные данные получены и другими авторами, например Вас-
серманом [5] и Кертином 16]. Полагают, что ц/гс-эпоксиды образу-
ются так же, как из обычных олефинов, но фенильная группа обес-
печивает раскрытие цикла с сохранением конфигурации.
Получение надбензойной кислоты. Согласно улучшенной мето-
дике [7J, 0,45 моля 70 %-ной П. в. прибавляют по каплям (защитный
экран!) к смеси 0,3 моля бензойной и 0,9 моля метансульфо кислоты
при перемешивании и охлаждении льдом так, чтобы температура не
CH3SOaH
С6Н5СО2Н + НаО2----1—3-^ СеН6СО3Н
6 " 2 ‘ 2 85-90%
превышала 25—30° (30 мин). Бензойная кислота быстро растворяет-
ся, и после перемешивания в течение 2 час раствор охлаждают до
15°, добавляют лед и холодный раствор сульфата аммония и смесь
трехкратно экстрагируют бензолом. Бензольный раствор дважды
промывают раствором сульфата аммония для удаления метансуль-
фокислоты, сушат, фильтруют, надбензойную кислоту используют
непосредственно в растворе, определяя концентрацию иодометр иче-
58
34-4 0%
(общий)
ским титрованием. В случае необходимости из бензольного раствора
можно выделить чистую надбензойную кислоту.
Гидроперекиси как промежуточные соединения или продукты
реакции. Неопентиловый спирт (3) можно получить кислотно-
СН3 СН3
I н + I
(СН3)3ССН.2С-СН2ЩН3Оа —> (СН3)3ССН,С- сн3
(1) (2) |
о2н
—> (СН3)3ССНйОН + О-С(СН3)а
(3)
катализируемым присоединением П. в. к диизобутилену (1) [8]
через промежуточное образование гидроперекиси (2), которая в бо-
лее кислой среде перегруппировывается в неопентиловый спирт и
ацетон. Так, 800 г 30 %-ной П. в. перемешивают при охлаждении
льдом, прибавляют разб. серную кислоту, диизобутилен и переме-
шивают при 25° в течение 24 час. Верхний слой гидроперекиси от-
деляют и прибавляют при перемешивании к 70 %-ной серной кислоте
при 15—25° для осуществления перегруппировки. Неопентиловый
спирт отделяют перегонкой и затем фракционируют.
В условиях кислотного катализа П. в. присоединяется к окиси
мезитила (I) в положение 1,4 с образованием гидроперекиси (2), ко-
торая циклизуется в перекись (3), а при последующей реакции с П. в.
дает конечный продукт — кристаллическую перекись [(4), т. пл. 122°]
19].
(СН3)2С=СНСОСН3 . Н2°2—> (СН3)2С—СН2СОСН3
5h.H2SO4 £он
(2)
Нзсх /£^СН
0—0
(3)
(1)
ИО, 3:о с 'с с
---Н3С^| |\ /| рсн3
49% (ойщий) о—о ° ° 0—0
(4)
Насыщенные 5fJ- и А4-ненасыщенные стероидные 3-кетоны при
взаимодействии с П. в. в mpem-бутаноле, содержащем соляную ки-
слоту, образуют с хорошим выходом [101 бис-гидроперекиси (5), (6).
59
Реакция циклогексанона с П. в. в эфире приводит к образованию
перекиси (7), а также моно- или бис-гидроперекиси в зависимости от
ноон /-----^-0----/...... \
\---/ \----/\он но/4— /
(Р
соотношения реагентов [11]. Возможно, перекись (7) получается в
результате первоначального присоединения П. в. к карбонильной
группе с образованием гидроперекиси. По-видимому, аналогичное
присоединение наблюдается при реакции 2-ацетилциклогексанона
(8) с П. в. в mpem-бутаноле, содержащем следы серной кислоты
[12]. Первоначально образующаяся перекись (9) подвергается кис-
лотно-катализируемой перегруппировке с сужением цикла и обра-
(8) (9) (10)
зованием циклопентанкарбоновой кислоты (11), которую получают
с выходом 87%.
Перекись водорода — уксусная кислота, см. также Надуксусная
кислота. Если водный раствор П. в. (обычно 30%-ный) прибавить
к уксусной кислоте при комнатной температуре, то начинается мед-
ленная реакция и постепенно наступает равновесие.
О О
II II
СН3СОН-|-НООН СН3СОО1-1-|-НОН
Реагент, полученный таким способом, содержит некоторое коли-
чество надуксусной кислоты, зависящее от концентрации П. в.,
количества взятой уксусной кислоты, температуры, времени реак-
ции, присутствия или отсутствия катализаторов. Реагент следует
отличать от продажной 40 %-ной надуксусной кислоты и безводной
надуксусной кислоты, которые рассмотрены отдельно.
Смит [13] изучил равновесие между уксусной кислотой и водным
раствором П. в. с помощью титрования последней перманганатом,
который не реагирует с надуксусной кислотой. В разбавленном
растворе при 0° гидролиз надуксусной кислоты происходит настоль-
ко медленно, что надкислоту можно титровать иодометрически. При
комнатной температуре равновесие достигается только через не-
сколько дней. Однако эта реакция очень сильно ускоряется 1%-ной
серной кислотой или нагреванием до 70°. Так, Фернхольц [14] на-
гревал смесь 30 мл 30 %-ной П. в. и 300 мл уксусной кислоты в те-
чение 5 час при 70° и выдерживал 2 дня при комнатной температуре.
В полученном растворе содержалось 2—2,5% надуксусной кислоты.
60
Хотя ароматические надкислоты в общем лучше эпоксидируют,
олефины, тем не менее реагент, получающийся просто при смешении
30%-ной П. в. с уксусной кислотой, также может быть использован
для этой цели, поскольку в таких мягких условиях (25°) эпоксиды
не расщепляются уксусной кислотой в отличие от более сильной
муравьиной кислоты. В таблице приведены данные Сверна [15J по
скоростям окисления некоторых олефинов.
Скорость окисления олефинов надуксусной (25,8°) или надбензойной
(20 — 30°) кислотой в уксусной кислоте
Соединение Формула 1г- 103
сн.,со;,н С6Н;СО;,Н
Этилен сн2 - сн.. 0,19
Пропилен сн3сн=сн2 4,2
Децен-1 СН3(СН3)7СН^СНг 4,7
Бутен-2 СН3СН = СНСН3 93
Нонен-4 СН3(СНо)2СН = СН(СНа)3СН3 105
н н
Олеиновая кислота С113(СН,)7С = С(СН.,)7СО,11 67
и
Элаидиновая кислота СН3(СН2);С^С(СН2);СО.2Н 59
н
2-Метил бутен-2 (СН3),С = СНСН3 1240
Циклобутеп 20,4
Циклопентен 195
Циклогексен 129
Циклогептен 175
Аллнлбензол CRH5CH3CH=CH3 1,9 G-15
Стирол С6Н5СН^СН2. 11,2 35
1, СДифеинлэтплен (С6И5)2С = СН2 48
Коричная кислота С6Н5СН^СНСООН 0,13
Реакции другого типа включают расщепление углерод-углеродной
связи. Фенантренхинон гладко окисляется П. в. в теплой уксусной
кислоте в дифеновую кислоту [161. Эту реакцию использовали для
решения структурных вопросов [17]. Так, одну из пяти дисульфоки-
слот, образующихся при сульфировании фенантрена, превращали
в диметоксифенантрен, который при окислении дает диметоксифенан-
тренхинон-9,10. Дальнейшее окисление этого хинона П. в.—
СН3СООН приводит к кислотному продукту, идентифицированному
как 3,3'-диметоксидифеновая кислота (2). Таким образом, продукт
61
сульфирования является 2,7-дисульфокислотой. Окисление надки-
слотой дает также значительное количество нейтрального вещества,
которое оказалось лактоном (3), Это соединение получается при раз-
рыве связи между карбонильной группой и бензольным циклом.
(2)
Дифеновая кислота получается при окислении фенантрена П. в.
в кипящей уксусной кислоте без выделения промежуточно образую-
щегося фенантренх инона 118],
П. в, в уксусной кислоте находит также применение для окис-
ления ароматических углеводородов и фенолов до п-х анонов. По Ар-
нольду и Лаусону [19] смесь 10 г нафталина, 25 мл 30%-ыой П. в.
и 50 мл уксусной кислоты нагревают при температуре немного выше
80° в течение 45 мин, отгоняют половину растворителя, для осажде-
ния продукта реакции добавляют воду и после кристаллизации по-
лучают достаточно чистый нафтохинон-1,4 с выходом 20%, Дурол
(5 г) нагревают с П, в,— СН3СООН 15 час на кипящей водяной бане
и дурохинон (2,1 г) выделяют перегонкой с паром. Неочищенные
2-метнл- и 2,3-диметилнафтохпионы-1,4 получают с выходами 30
и 78%. В обоих случаях в реакционной смеси присутствовал
неокисленный исходный углеводород,
Кэйв и Шир лей [201 нагревали раствор 2 г углеводорода (1) в
30 мл уксусной кислоты и 6 мл 30 %-ной П. в. при 80—85° в течение
5 час и выделили ярко-желтый хинон (2) с небольшим выходом.
Хинон обладает очень интересными свойствами. Он реагирует с бу-
тадиеном при 90—100° в течение 90 жни, давая бесцветный аддукт (3)
62
с выходом 93%, В этих условиях 2, З-д имет ил нафтохинон-1,4 не
изменяется даже через 5 дней. Повышенную реакционную способ-
ность хинона (2) как диенофила объясняют уменьшением напряже-
ния цикла при переходе циклобутена (2) в циклобутан в аддукте
(3). При повышенной температуре хинон (2) реагирует как диен по
Дильсу — Альдеру, вероятно, за счет разрыва циклобутенового
кольца с промежуточным образованием диена (4). Так, пиролиз в
присутствии N-фенилмалеинимида приводит к производному антра-
хинона (5). Низкий выход обусловлен, по-видимому, дегидрирова-
нием первоначально образующегося аддукта за счет хинона (2),
Брайс-Смит [21] установил, чтоП. в. эффективно окисляет 2,5- и
2,6-диметилфенолы в ц-бензохиноны. Например, раствор 15 а 2,5-
диметилфенола в 100 мл уксусной кислоты обрабатывают 1 мл конц.
серной кислоты и при охлаждении до 20° добавляют по каплям 12 мл
ОН О
/СНз А/СНз
H3czY
о
80%-ной П. в. в течение 45 мин. Реакционную смесь выдержи-
вают при 20° более 1 час, затем 10 мин при 60э до окончания
экзотермической реакции. При разбавлении и охлаждении хинон
выпадает в виде желтых кристаллов, т. пл. 125° (12,5 а).
Окисление ариламииов, нитрозосоединеиий и азобензолов. Не-
которые 2,6-дигалогенанилины окисляются до нитрозосоединений
30%-ной П. в. в уксусной кислоте при комнатной температуре [22].
Из раствора реагентов через некоторое время начинает кристалли-
63
зоваться димер нитрозосоединения, и продукт реакции легко выде-
ляют, отфильтровывая кристаллический осадок. Выходы 20—90%.
NO
Вг
Если проводить окисление в течение большего времени при нагре-
вании на кипящей водяной бане, то получается нитросоединение.
Окисление до нитрозосоединения, очевидно, требует наличия
двух объемистых ор/гсо-заместителей; так, гс-нитроанилин окисляет-
ся в мягких условиях в гс,п'-динитроазобензол и гс,гс'-динитроазок-
сибензол [231.
Для окисления 1,2-диметил-4-нитро-5~нитрозобензола в соответ-
ствующий о-динитробензол (Куи и ван Клаверен [24]) 10 а нитрозо-
соединения растворяют в 300 мл уксусной кислоты, добавляют смесь
150 мл уксусной кислоты и 150 мл 33%-ной П. в., а затем 10 мл
H"CW\/N0 HSC NO,
| || НгО2 —HNO3 । ||
азотной кислоты (уд. вес 1,40). При нагревании в течение несколь-
ких минут окраска раствора изменяется от темно-зеленой до оран-
жевой; разбавляя смесь водой, осаждают динитроксилол. Три дру-
гих алкилированных о-диннтросоединения получены с выходом
62—88%. Другие окислители дают низкие выходы ввиду окисления
алкильных групп.
П. в. с серной кислотой — хороший окислитель для превращения
аминопиридинов в нитропиридины [25]. Например, раствор 20 г
4-амино-2-метилпиридина в 100 мл конц. серной кислоты добавляют
по каплям к смеси 350 мл 15%-ного олеума и 175 мл 30%-ной П. в.
при 10—20°. Смесь перемешивают при 20°в течение I час, оставляют
на 2 дня и выливают в лед. После нейтрализация нитросоединение эк-
страгируют бензолом.
H,02-H2SO.,
NO3
64
Тэйлор и Мак-Киллоп [26] нашли, что смесь П. в. с трифторуксус-
ной кислотой является очень хорошим окислителем легкодоступ-
ных 5-нитрозопиримндннов в 5-нитропирнмидины.
НгОг~СР3СО2Н
85%
Азобензолы окисляются до азоксибензолов при нагревании с
30%-ной П. в. в уксусной кислоте при 65° или при температуре ки-
пения [27]. В некоторых случаях для окисления требуется 24 час.
Выходы в пределах 50—90%.
Получение галогенов. 2,6-Дихлоранилин получают хлориро-
ванием сульфаниламида и гидролитическим отщеплением сульфа-
HoN—SO2NH3
2НС1, H2OS
65—71%
CIx
H2N—SO2NHa —>
С1/
70%-ная H2SO4
75-80%
C1\
H2N—
C\/
мидной группы [28]. Необходимый для этой реакции галоген полу-
чают in situ при окислении соляной кислоты П. в. К перемешивае-
мому раствору сульфаниламида (0,29 моля) в разб. соляной кислоте
при 60° добавляют по каплям П. в. (0,58 моля) и перемешивают при
этой температуре до начала осаждения продукта реакции. Дихлор-
сульфаниламид отфильтровывают и кипятят 2 час с 70 %-ной серной
кислотой. Раствор выливают в воду и 2,6-дихлоранилин выделяют
перегонкой с паром. 2,6-Диброманилин получают аналогичным об-
разом.
П. в.—BE, в реакции Байера — Виллигера. Расщепление про-
стых алифатических кетонов по Байеру — Виллигеру с образова-
нием сложных эфиров можно1 осуществить под действием комплекса
П. в. с эфиратом трехфтористого бора [29]. Раствор октанона-2 в
эфире при охлаждении льдом (защитный экран!) обрабатывают
смесью 90 %-ной П. в. н эфнрата трехфтористого бора. После пере-
о о
15 h2o3-bf3 II
СН3СН2СН2СН2СН2СН2ССН3 --------—> сн3сн2сн2сн2сн2сн2оссн3
мешивания эфирный слой промывают раствором бикарбоната
натрия, сушат и отгоняют растворитель. Фракционирование дает в
основном гексилацетат, а также 8% гексанола и 32% исходного
соединения.
3 Заказ № 1166
65
RSCOCH RSO H. Сверн и сотр. [30] разработали удобный
метод синтеза алкилсульфокислот путем фотокаталитического при-
соединения тиоуксусной кислоты к терминальному олефину и окис-
лением полученного аддукта 90 %-ной П. в. в уксусной кислоте.
н,о,
RCH = CH, + CH3COSH .___RCH2CH.,SCOCH3 -----------
5 0-~9 0% Количеств.
RCH,CH,,SO3I-I
Окисление проводят в течение 12 час при 65—70°, применение П. в.
без уксусной кислоты дает плохие результаты. Этот метод позво-
ляет получать «-ал кил сульфокислоты, не содержащие изомеров.
Окислительная перегруппировка. Кори и сотр. [31] обнаружили,
что Зр,11а-диокси-Л1~-пентациклическин тритерпеноид (1) в хло-
ристом метилене при обработке раствором 30%-ной П. в. и «-то-
луол сульфо кис лоты в mpcm-бутаноле дает после ацетилирования
11а,12а-эпоксид, образующийся в результате скелетной перегруп-
пировки (миграция метила С14—-С13 и сдвиг двойной связи). Свобод-
ный эпоксиспирт интересен как продукт фотоокисления |3-амприна.
N-Окиси пиридинов. N-Окиси производных пиридина получают
нагреванием основания с П. в, и уксусной кислотой пли с над-
уксусной кислотой. Так, 2,76 моля 30%-ной П. в. добавляют при
встряхивании к раствору 2,15 моля 3-метил пиридина в 600 мл ук-
сусной кислоты, смесь нагревают при 70° в течение 24 час и 500 мл
растворителя отгоняют в вакууме [32]. Затем добавляют 200 мл
воды и вновь отгоняют 200 мл дистиллата. Смесь подщелачивают
и жидкую N-окись выделяют экстракцией хлороформом и перегон-
кой. N-Окись никотинамида получают аналогичным образом и пере-
кристаллизовывают из воды (т. пл. 291—293°, выход 73—83%) [33].
66
Для получения N-окиси пиридина [34] 1,50 моля 40%-ной надуксус-
ной кислоты прибавляют при перемешивании к 1,39 моля пиридина
с такой скоростью, чтобы температура не превышала 40°, отгоняют
Ряс. П-4. Прибор для перегонки
твердых веществ.
растворитель и продукт реакции (т. пл. 65—66°) перегоняют в при-
боре для перегонки твердых веществ (рис, П-4).
I. С в е р н Д., С к а и л а н Дж. Т., Дикел Г. Б., «Синтезы органических
препаратов», ИЛ, М., 1961, сб. И, стр. 19.
2. F i е s с г L. F., R a j a g о р а I а п S., J. Am. Chem. Soc., 71, 3938 (1949).
3. Р о бэ к А., Ад к и н с Г., «Синтезы органических препаратов», ИЛ, М.,
1953, сб. 4, стр. 559.
4. Riviere Н., Bull. soc. chim. France, 1964, 97.
5. Wasserman H. H., Aubrey N. E., J. Am. Chem. Soc., 78, 1726
(1956).
6. Cur t i n D. Y., Bradley A,, H en dr i ck s о n Y. G., J. Am. Chem.
Soc., 78, 4064 (1956).
7. S i I b e г t L. S., S i e g e I E., Swern D., Org. Syn., 43, 93 (1963).
8. Hoffman J., Org. Syn., 40, 76 (1960).
9. R i e c h e A., Schmitz E., Grundemann E., Chem. Bcr., 93, 2443
(I960).
10. Warnant J., Joly R., M a t h i e u J., Velluz L., Bull. soc. chim.
France, 1957, 331; V e 1 1 u z L., A m i a r d G., M a г t e 1 J., W a r n a n t J.,
ibid., 1957, 879, 1484.
11. M i I a s N. А., пат. США 2223807 (1939); Cr i eg e e R., Sch n orren-
b e r g W., Becke J., Ann., 565, 7 (1949).
12. P a у n e G. B., J. Org. Chem., 26, 4793 (1961); Виноградова Л. П.,
Завьялов С. И-, Изв. АН СССР, ОХН, 1961, 2050.
13. Smit W. С., Rec. trav., 49, 675 (1930).
14. Fernholz Н., Chem. Вег., 84, ПО (1951).
15. Swern D., J. Am. Chem. Soc., 69, 1692 (1947).
16. Hollcma n A. F., Rec. trav., 23, 169 (1904).
17. F i c s e r L. F., J. Am. Chem. Soc,, 51, 2471 (1929).
18. O’C о п n о r W. F., M or i с о n i E. J., J . Am. Chem. Soc., 73, 4044 (1951);
Ind. Eng. Chem., 45, 277 (1953).
19. Arnold R. T., L a w s о n R., J. Org. Chem., 5, 250 (1940).
20. Cava iM. P_, Shirley R. V-, J. Org. Chem., 26, 2212 (1961).
21. В г у c e-S m i t h D., Gilbert A., J. Chem. Soc., 1964, 873.
22. H о 1 m e s R. R., Bayer R, P„, J. Am. Chem. Soc., 82, 3454 (1964).
3* 67
23. Gutmann H, R., Experientia, 20, 128 (1964).
24. Kuhn R., van Klaveren W., Ber., 71, 779 (1938).
25. Kir pal A,,B6hm W., Ber., 64, 767 (1931); 65, 680 (1932); W I 1 e у R.H.„
Hartman J.L., J. Am. Chem. Soc., 73, 494 (1951); В г о w n E. V., ibid.,
76, 3167 (1954).
26. Taylor E. С., M с К i I 1 о p A., J, Org. Chem., 30, 3153 (1965).
27. Newbold В. T., J. Org. Chem., 27, 3919 (1962).
28. Сей к ел M., «Синтезы органических препаратов», ИЛ, М., 1952, сб. 3,
стр. 236.
29. М с С 1 и г е J. D., Williams Р. Н., J. Org. Chem., 27, 24 (1962).
30. Show ell J. S,, Russell J.R., Swern D.,J. Org. Chem., 27, 2853
(1962).
31. A g a t a I.,Corey E.J., Hortmann A. G., KleinJ., Proskow
S., U r s p r u n g J. J., J. Org. Chem., 30, 1698 (1965).
32. T э п л о p Э., мл., Kp оветт и А., «Синтезы органических препаратов»,
ИЛ, М., 1958, сб. 9, стр. 28.
33. Taylor Е. C.,Crovetti A. J., Org. Syn., Coll. Vol., 4, 704 (1963).
34. Мошер Г., T e p н e p Л., Карлсмит А., «Синтезы органических
препаратов», ИЛ, М., 1954, сб. 5, стр. 56.
ПЕРЕКИСЬ ВОДОРОДА В НЕЙТРАЛЬНОЙ СРЕДЕ.
Окиси аминов. Метод [1] получения алифатического амина (4),
превращение его в N-окись (5) и пиролиз последней до метиленцик-
логексана (6) (реакция Коупа) показаны на следующей схеме:
2HN(CH3),
86—89% из (1)
О~
и i
7 ch2n+(Ch3)2
Н2Ог
16 0°
79—88%
из (4)
+ (CH3).2NOH
(6)
Гомогенный раствор 0,35 моля N,N-диметилциклогексилметилами-
на (4) и 0,35 моля 30 %-ной П. в. в 45 мл метанола выдерживают при
комнатной температуре в течение 36 час. При этом через 2 и 5 час
прибавляют по 0,35 моля П. в. (с другими аминами может потребо-
ваться охлаждение). Окисление можно считать законченным, если
капля раствора не вызывает изменения окраски фенолфталеина.
Для разложения избытка П. в. добавляют небольшое количество
платиновой черни и раствор перемешивают до прекращения выде-
ления кислорода, после чего раствор фильтруют и концентрируют
при 60° в вакууме до затвердевайия остатка гидрата N-окиси амина.
68
При последующем нагревании в вакууме (100° при 10 мм) вещество
плавится и вновь затвердевает. Затем при повышении температуры
до 160° осуществляют реакцию Коупа приблизительно за 2 час.
Отгон промывают водой, углеводородный слой отделяют, сушат и
перегоняют. Метиленциклогексан получается очень чистым и не со-
держит 1-метилциклогексана. Из водного раствора можно выде-
лить И,Кг-диметилгидроксиламин в виде чистого хлоргндрата с вы-
ходом 78—90%.
Окисление серу- и мышьяксодержащих соединений. Под дейст-
вием П. в. в ацетоне тноэфиры окисляются до сульфоксидов; П. в,
в 50 %-ной уксусной кислоте дает соответствующие сульфоны [21.
Так, 16,1 мл 30%-ной П. в. при охлаждении добавляют к раствору
о-
cei-iaCH2sci-i3 cbh5ch2s-ch3
77%
°- о-
CH3SCH.,CN Нг°- CH3S!'CH2CN ch3s+ch2cn
54% 69% ‘ li
о
21 г метил бензил сульфид а в 60 мл ацетона и раствор оставляют на
ночь. Получающийся сульфоксид очищают кристаллизацией
(т. пл. 54°). П. в.— лучший окислитель для последней стадии получе-
ния бис-(п- аминофен ил)-дисульфида [3]. Первые стадии конденсации
O3N—С1 O3N—SNa —-Щ.
SNa °2—> H2N— SS—/ NH2
\= / 58 — 64% X. - /
(общий)
и восстановления проводят в водном растворе, который затем филь-
труют для удаления нерастворимых соединений, главным образом
/г-хлоранилина. Фильтрат концентрируют и при 65—70° в течение
2 час обрабатывают 30 %-ной П. в. При охлаждении раствора выде-
ляется твердый дисульфид, который очищают кристаллизацией
(т. пл. 75—76°).
Для окисления трифенил арсина до трифенил а рсиноксид а к пере-
мешиваемому раствору 0,33 моля трифениларсина в 200 мл ацетона
добавляют 0,41 моля 30%-ной П. в. в течение 20—30 мин при ох-
лаждении до 25—30° [4]. Ацетон удаляют отгонкой в вакууме и оста-
ющееся желтое масло сушат азеотропной отгонкой с бензолом. При
(CeI-I5)3As —lx—(C6H&)3AsO
84-87%
растирании с бензолом получают желтые кристаллы с т. пл. 189°.
1-Алкокси гидроперекиси. Рихе и Бишоф [51 получили безводнуто
П. в. обработкой раствора 60 а 84%-ной П. в, в эфире безводными
69
сульфатом натрия и карбонатом магния. Смесь выдерживают
2 дня, фильтруют и определяют содержание П. в. титрованием алик-
вотной пробы. К 1 же эфирного раствора П. в. добавляют 1 же
ацеталя и смесь осторожно нагревают при 70°. Эфир при этом отго-
няется, и после нагревания при 70° в течение 15 час выделяется
1-алкоксигидроперекись с выходом 40—70%.
yOCoHf,
RCHZ -h НООН
ЧСО5
0,1 моля 0,1 моля
/OCJ-C
RCHZ +С2Н5ОН
хоон
1. Коуп А. Ч., Ц и г а н е к Э., «Синтезы органических препаратов», ИЛ, М.,
1961, сб. 11, стр. 36.
2. Gazdar М., Smiles S., J. Chem. Soc., 93, 1833 (1908); Н ii п i g S.,
Boes O., Ann., 579, 23 (1953).
3, П p a n с 4., Стэси Г., «Синтезы органических препаратов», ИЛ, М., 1953,
сб. 4, стр. 150.
4. Shriner R. L., W о 1 f С. N., Org. Syn., Coll. Vol., 4, 910 (1963).
5. Rieche A., Bischoff C., Chem. Bcr., 94, 2722 (1961).
ПЕРЕКИСЬ ВОДОРОДА В ЩЕЛОЧНОЙ СРЕДЕ.
Реакция с еионами и хинонами. Для реакции а, ^-непредельных
кетонов с П. в. необходима щелочная среда, вероятно, для образо-
вания нуклеофильного гидропероксильного аниона 11, 2]. 1,4-
б+ б- о.н | - ,/ х
—С-С—С-0 ——С—С = С—О —> —С-------------с—с_о+он-
III III III
(I) (2)
Атака этой частицы приводит к аниону (1), отщепление гидроксиль-
ного иона в котором ведет к замыканию эпоксидного цикла (2).
Эпоксидная группа расщепляется кислотами, но устойчива по от-
ношению к основаниям.
Для превращения 2-метилнафтохиноиа-1,4 в 2,3-эпокснд [21 1 мл
30%-ной П. в. прибавляют к раствору 0,2 г безводного карбоната
натрия в 5 мл воды и эту смесь приливают к теплому раствору 1 г
хинона в 10 мл 95%-ного этанола. Желтая окраска сразу исчезает,
и при охлаждении выпадает эпоксид в виде бесцветных кристаллов
(0,97а) ст. пл. 93,5—94,5° (т, пл. чистого эпоксида 95,5—96,5°).
Этот метод применим для нафтохинона-1,4 и его 2,3-диалкилпроиз-
водных, включая витамин К, [3]. Для первой стадии окисления
2-алкил-3-оксинафтохннона-1,4(3) по Хукеру [410,01 моля хинона (3)
70
с молекулярным весом от 244 до 384 растворяют в 25 мл диоксана
(допустимо небольшое нагревание) и добавляют раствор 1,2 г кар-
боната натрия в 25 мл воды. Полученный темно-красный раствор
или суспензию натриевой соли нагревают на водяной бане при 70°,
вытесняют воздух слабым током азота и прибавляют 2 мл 30”/о-ной
П. в. Через 20—40 мин раствор обесцвечивается, и после охлажде-
ния и подкисления с высоким выходом выделяется диокспиндапон-
карбоновая кислота (6). Вероятно, реакция включает стадии при-
соединения П.в. к аниону (4) с образованием аниона (5) и его бензиль-
ную перегруппировку с образованием аниона кислоты (6). О второй
стадии окисления по Хукеру см. Меди сульфат — щелочь.
Изофорон (7) Г5| и 2,3-дифенилиндепон (8) 161 превращают в
соответствующие эпоксиды в метаноле пли этаноле с применением
едкого натра в качестве основания.
з № н2о2
СН3ОН + О, ъэкв ВОДЯ. NaOH
^Час ПРИ15 -25°
70-72%
HZO2
СгН5ОН-НгО -NaOH
84%
Первые попытки эпоксидирования простых а, |3-непредельных
альдегидов П. в. в присутствии щелочи приводили только к кислым
продуктам реакции. Однако Пайн [7] показал, что чувствительные
к щелочам альдегиды, такие, как акролеин и метакролеин, можно
эпоксидировать, поддерживая pH около 8—8,5. Так, акролеин при-
бавляют к разб. П. в., причем pH среды поддерживают около 8—8,5
71
(по pH-метру) постоянным добавлением разб. едкого натра. Выход
глицидного альдегида, определенный титрованием, составляет 75—
сн,=снсн^о
НД, pH 8—8,5
75-85%
/О\
СН2 — снсн = о
85%. Этиловый эфир этнлиденмалоиовой кислоты также эпоксиди-
руется этим способом в метаноле [81. В последующей работе Пайна
ШгС,Н5
СН3СН = С
ХСО2С2Н5
Н.Ог, pH 7,5-8
/О^/СОгСоН,,
сн,сн—с
'•со2с.,н
[9] было показано, что эту методику можно упростить, применяя
в качестве буфера бикарбонат калия.
2,5-Диоксибензохинон-1,4 получают, перемешивая раствор гид-
рохинона со смесью 37%-ной П. в. и 50%-ного раствора едкого
натра [10]. Эту реакцию можно рассматривать как окисление до
хинона, присоединение воды (или ОН”) с образованием окснгидро-
хинона (или аниона), окисление в оксихннон и дальнейшее присое-
динение и окисление. Продукт реакции выделяется в виде оран-
жево-красной динатриевой соли, которую растворяют в воде и под-
кисл яюг.
Реакция Дэкина. Эта реакция также основана на применении
П. в. в щелочной среде. Она применима к фенолам, имеющим в орто-
положении альдегидную или кетонную группу. Так, салициловый
альдегид под действием щелочного раствора П. в. превращается
с хорошим выходом в пирокатехин [111. Механизм реакции приве-
ден на схеме [12]. Раствор 1 а моля салицилового альдегида в 1 л
1 н. едкого натра обрабатывают при комнатной температуре 1,2
моля 3%-ной П. в. Раствор темнеет, и температура повышается до
45—50°. Через 15—20 час смесь нейтрализуют уксусной кислотой
и выпаривают досуха при комнатной температуре, а остаток экстра-
72
гнруют толуолом. Эта реакция применима к производным о-вани-
лина; в этом случае ее проводят в токе азота [13].
С но
^^ОСНз
ОН
I он
АГ11
H2O2-NaOH II |
68-80% II I
\"хосн3
Реакция Дэкииа для о-оксиацетофенона затруднена тем, что ис-
ходное соединение образует плохо растворимую хелатную натрие-
вую соль. В этом случае вместо едкого натра или кали лучше при-
менять гидроокись тетраметил аммония, несколько хуже тритон Б
[14].
/ОН ,он
П NaOH |/
----> | -|--CH3CO2Na
/\оссн3 /Хюн
Расщепление с/.-кетокислот и а-дикетонов. Расщепление а-ке-
токнслоты является заключительной стадией синтеза гомовератро-
вой кислоты (4) из вератрового альдегида (1) через аз лактон (2)
[15]. Под действием щелочи азлакгонный цикл разрушается с отщеп-
/СО2Н
СН3О—____^-СНО + сн2 °
CH3q/ ССбНэ
(1) й
Ac3O, NaOAc
69—73%
КаОН
О
сн3о—сн.ссо.н
снуо/
(3)
—°2’ сн3о—снасоан
CH3OZ
(4)
лением бензойной кислоты и аммиака и образованием а-кетокислоты
(3). 30%-ную П. в. добавляют при перемешивании и охлаждении
до 15° и смесь оставляют на ночь. Подкисление и экстракция дают
смесь гомовератровой и бензойной кислот, которую разделяют эте-
рификацией и фракционированием. При гидролизе образуется кис-
лота (4) с выходом 51 % в расчете на азлактон.
73
Ягер и сотр. [161 применяли щелочной раствор П. в, для расщеп-
ления 11,12-связи ненасыщенного трикетона (1), полученного при
окислении А8-л аностена.
Окисление аллодунниона (1). Прайс и Робинсон [17] окисляли
аллодуннион щелочным раствором П. в. и выделили кристалли-
ческий продукт, которому приписали строение лактоиокислоты.
Перегруп-
пировка
-НгО
(7) (8)
Однако впоследствии было показано [181, что это соединение не
является лактонокислотой, а представляет собой уже описанную
а-изопропилиденгомофталевую кислоту (8). Возможный механизм
этой реакции включает показанную на схеме бензильную пере-
группировку на стадии (6) —> (7).
Реакция с нитрилами. Реакция нитрилов с П. в. в присутствии
оснований, приводящая к образованию амида и выделению моле-
кулярного кислорода, была впервые описана Радзишевским [ 19.1.
Смесь 0,75 моля о-толунитрила, 2,6 г моля (300 мл) 30%-ной П. в.,
400 мл 95%-ного этанола и 300 мл 6 н. едкого натра образует гомо-
74
генный раствор, который вскоре самопроизвольно разогревается с
выделением кислорода [201. Смесь в течение 1 час охлаждают до 40—
50“ и затем нагревают’ 3 час при 50°. Смесь нейтрализуют, перего-
няют с паром до получения 1 л дистиллята, который оставляют для
N
2НД. NaOHT С2Нг,ОН
9 0—92%
сн3
CONH,
що3щн,о
кристаллизации продукта реакции. По аналогичной методике полу-
чен амид вератровой кислоты с выходом 87—92% [211. Ароматичес-
кие нитрилы обычно превращаются в амиды с выходом 80—90%;
выходы для алифатических соединений составляют толь ко 50—60%.
Виберг [22] установил, что реакция бензонитрила с П. в. имеет
первый порядок по нитрилу, П. в. и ионам гидроксила; окись бен-
зонитрила не является промежуточным соединением в этой реакции.
Кислород, входящий в состав амида, берется из П. в., а не из воды
или гидроксильных ионов. Влияние заместителей на скорость реак-
ции такое же, как и в известных реакциях с нуклеофильной атакой
нитрильной группы. Эти результаты показывают, что первой ста-
дией реакции является атака нитрила гидропероксильным анио-
ном с образованием и мин опад кислоты (3). Эго соединение не было
выделено, но полагают, что оно является окислителем. В отсутствие
субстрата это соединение реагирует с П. в., являющейся в данном
случае восстановителем, с образованием амида (4) и кислорода.
оон о
z— тт —- кт оон тт 1. ,н?о тт ХК ... —о о ьт 4 ктт_г 1 1 тт
CjH5C=N --C6H5C=N ~-2—> СЙН5С—NH -----------> с6н5с—nh2 + о2 + нго
-он'
9) (2) (3) (4)
Пайн и сотр. [231 установили, что если имиионадкислота
(3) образуется в присутствии олефина, то последний как более силь-
ный восстановитель, чем П. в., превращается в соответствующий
эпоксид, а (3) переходит в амид (4). Эпоксидирование лучше прово-
дить в присутствии бензонитрила или ацетонитрила. Например,
раствор 1,5 моля циклогексена и 2 молей ацетонитрила в 300 мл.
метанола перемешивают при 60° и одновременно обрабатывают
1 молем 50%-ной П. в. и 1 н. раствором едкого натра. Последний
добавляют с такой скоростью, чтобы поддерживать pH раствора в
интервале 9,5—-10 (по pH-метру). Окись циклогексена получают
с выходом 85% в расчете на прореагировавшую П. в. При окислении
этим способом пиридин дает N-окись пиридина (79%), а анилин —
азобензол (62%). В упрощенной методике в качестве основания
используют бикарбонат калия (pH 8), что делает ненужным приме-
нение рН-метра и контролируемое добавление щелочи [91. Например,
75
к перемешиваемой смеси 0,6 моля 2-аллилциклогексанона [II, 0,5
моля бензонитрила, 300 мл метанола и 10 г бикарбоната калия до-
бавляются моля 50 %-ной П. в. и перемешивают при комнатной тем-
пературе 40 час. Выход эпоксида приведен в расчете на прореагиро-
вавшее исходное вещество. При обработке кетона (1) надуксусной
(3)
кислотой происходит расщепление цикла по реакции Байера —
Виллигера с образованием лактона (3). Окисление метилеииикло-
гексаыа по вышеуказанной методике с применением бикарбонат-
ного буфера приводит к соответствующему эпоксиду с выходом 73%.
I. К о h 1 е г Е. Р., Richtmyer N. К., Hester W. F., J. Am. Chem.
Soc., 53, 205 (1931); Р 1 a 11 п е г Pl. et al., Helv. Chim. Acta, 31, 1822 (1948).
2. F i e s e г L. F., J. Biol. Chem., 133, 391 (1940).
3. Fi eser L. F., T i s h 1 e г M., Sampson W. L., J. Am. Chem. Soc.,
62, 1628 (1940).
4. Fieser L. F., F i e s e г M., J. Am. Chem. Soc., 70, 3215 (1948).
5. Wasson R. L., House H. O., Org. Syn., Coll. Vol., 4, 552 (1963).
6. Ullman E. F., Milks J. E., J. Am. Chem. Soc., 84, 1315 (1962);
Weitz E., Scheffer A., Ber., 54, 2341 (1921).
7. Payne G. B., J. Am. Chem. Soc., 81, 4901 (1959).
8. Payne G. B., J. Org. Chem., 24, 2048 (1959).
9. P a у n e G. B., Tetrahedron, 18, 763 (1962).
10. J ones R. G., Shonle IT. A., J. Am. Chem. Soc., 67, 1034 (1945).
11. Д э к и и Г., «Синтезы органических препаратов», ИЛ., М., 1949, сб. I,
стр. 346.
12. В u t о n С. A., in Edwards J. О., «Peroxide Reaction Mechanisms»,
Interscience, 1962, p. 14—15.
13. C io p p e й Ф., «Синтезы органических препаратов», ИЛ, М., 1953, сб. 4,
стр. 340.
14. Baker W. et al., J. Chem. Soc., 1952, 1825; 1953, 1615.
15. Снайдер X., Бек Дж., Айди В., «Синтезы органических препара-
тов», ИЛ, М., 1949, сб. 2, стр. 164.
16. Kyburz Е., Riniker В., Schenk H.R., Heusser Н.,
J eg er О., Helv. Chim. Acta, 36, 1891 (1953).
[7. P г i c e J. R., Robinson R., J. Chem. Soc., 1939, 1522; 1940, 1493.
18. Oxman M. A., E t t 1 i n g e г M. G., Bader A. R., J. Org. Chem., 30,
2051 (1965).
19. Radziszewski Br., Ber., 18, 355 (1885).
20. Нолл ер Ч., «Синтезы органических препаратов», ИЛ, М., 1949, сб. 2,
стр. 29.
21. Бек Дж., Айди В., «Синтезы органических препаратов», ИЛ, М., 1949,
сб. 2, стр. 32.
22. Wiberg К. В., J. Am. Chem. Soc., 75, 3961 (1953).
76
23. P a у n e G. В., Wi ] li ams Р. H., J. Org. Chem., 26, 651, 659 (1961);
Payne G. P., Deming P. H., Williams P. H., ibid., 26, 659 (1966).
ПЕРЕКИСЬ ВОДОРОДА — СЕЛЕНА ДВУОКИСЬ. Штолль [1]
изучил гидроксилирование олефинов П. в. в присутствии стехио-
метрического количества двуокиси селена в трет-бутаноле при 0°
Н2О2—SeO2
цис. и' транс
цис и транс
Н2О2—SeO2^
407s
(65 час). Циклопентадиен реагирует по схеме как 1,2-, так и 1,4-
присоединения с образованием смеси цис- и траяс-циклопентендио-
лов-1,2 и -1,3. Циклопентен дает тряяс-циклопентандиол-1,2.
Смит [2] изучил окисление П. в. в присутствии каталитических
количеств двуокиси селена. Акролеин гладко окисляется до акри-
ловой кислоты 15%-ным раствором П. в. в mpem-бутаноле в при-
сутствии 5 г двуокиси селена на 1 моль П. в. Интересно, что под
H2OHSeO2)
СН2 = СНСНО --:СН2^СНСО2Н
d ОПП/ £
о
ГЦОДЗеОД
(СН2%
(С1-Ь)п
СО.,Н
п = 2, 34%
п = 1, 32%
0, 23%
действием этого реагента циклогептанон, циклогексанон и цикло-
пентанон окисляются с сужением цикла и образованием цикло-
гексан-, циклопентан- и циклобутанкарбоновых кислот, выходы
которых приведены на схеме [31. Сужение пятичленного цикла —
о 3 час
+ 30% Н2о2 + SeO2 ттрелг-ВиОН ПРИ >
350 3 .52 ХЛ
546 г Е 500 Л7Л трет -ВиОН
77
необычная реакция. Не известно ничего, относящегося к механизму
этой реакции, за исключением того, что циклогександион-1,2 не
окисляется этим реагентом до кислоты с сужением цикла. Цикло-
додекапон окисляется под действием реагента до циклоупдеканкар-
боновой кислоты с выходом 32% [4J. В качестве побочного продукта
с выходом 17% выделена 1,10-декапдикар боновая кислота.
Окисление стероидных кетонов под действием Н2О2 — SeO2
изучено Касии [51. Окисление холестанона-3 дает смесь кислот с
циклом меньшего размера и продукт расщепления по Байеру —
Виллигеру. А4-3-Кетостероид (1) дает е-лактон (2), который легко
перегруппировывается в у-лактон (3).
со2н
(!) (О (3)
1, S I о 1 I A., Lindenmann A., J ucker Е., Hclv. Chiin. Acta, 36', 268
(1953).
2. Smith C. W., Holm R. T., J. Org. Chem., 22, 746 (1957).
3. Payne G.B., Smi th C. W., J. Org. Chem., 22, 1680 (1957).
4. Dittmann W. D., Kirchhoff W-, Stumpf W., Ann., 681, 30 (1965).
5. C a s p i E., Balasubrahmanvam S. N., Tetrahedron Letters, 745
(1963); Expericntia, 19, 396 (1963); 3. Org. Chem., 28, 3383 (1963); C a s p i E.,
Shimizu Y., В a ] a$u b r a h m а п у a m S. N-, Tetrahedron, 20, 1271
(1964); H e 1 1 m а п H. M., J er us si R. A., ibid., 20, 741 (1964).
ПЕРЕКИСЬ ВОДОРОДА — СОЛИ И ОКИСЛЫ МЕТАЛЛОВ
в качестве катализаторов.
Перекись водорода — соли железа. Смесь П. в. и соли двухва-
лентного железа используется для получения гидроксильных ради-
калов и известна как реактив Фентона [I]. Гидроксильные радикалы,
образующиеся при разложении П. в. сульфатом железа (II), реаги-
Fe-+H3O.> рез+-1-ОН“-!- -ОН
р уют с трет-бутанол ом с образованием углеродного радикала, кото-
рый димеризуется с образованием 2,5-диметилгександиола-2,5 [2].
78
сн3 сн3 сн3 сн3
! -он I I
СНз—СОН — -СН, —С-ОН ________ Н0-ССН.2СН,С—он
I I 4()__4(3о- I I
СНз СН;) '° СН,, сн3
В колбу с боковыми отводами, снабженную эффективной мешалкой, за-
гружают 900 мл трет-бутанола (9,5 моля) и раствор 0,5 моля серной
кислоты в 1,5 л воды. В одну из бюреток помещают 1 моль 35%-ной
П. в., а в другую — раствор 1 моля сульфата железа(П) в 1 моле
разб. серной кислоты. В реакционную смесь пропускают ток азота
и одновременно в течение 20 мин добавляют оба раствора из бюреток,
поддерживая при этом температуру 10° (охлаждение льдом). Затем
смесь нейтрализуют 1 молем едкого натра и прибавляют 450 г суль-
фата натрия (растворяется не полностью). Органический слой отде-
ляют, водный экстрагируют тремя порциями по 400 мл трет-бута-
нола, из объединенных экстрактов отгонкой удаляют основное
количество растворителя и остаток экстрагируют эфиром. После
испарения эфира получают твердый остаток, который при расти-
рании с эфиром и циклогексаном дает диол удовлетворительной сте-
пени чистоты ст. пл. 87—88°.
Гидроксильные радикалы, образующиеся при взаимодействии
П. в. с сульфатом железа(П), при комнатной температуре атакуют
нитробензол с образованием небольших количеств о-, м- и /г-нитро-
фенолов [31; бензол дает небольшие количества фенола и дифени-
ла [4].
Дегидрирование 2-амнно-3,4-дигидрохиноксалина до 2-аминохи-
ноксалина гладко осуществляется при взаимодействии с П. в. в при-
сутствии следов хлористого железа, сульфата железа(Ш) или дву-
окиси свинца в качестве «переносчика» [51. Подробности методики
н
\ н2 Н2ОДВЫ+ пли Fc^) \
не приведены, и природа истинного окислителя не ясна. Неясной
остается также природа истинного окислителя при взаимодействии
а-оксикислот с П. в. в присутствии солей трехвалентного железа.
-СНСО.Д-1 —> —сн^о-усо3
ОН
Так, при расщеплении углеводов по Руффу [6], например при пере-
ходе от d-глюкозы к о-арабинозе, альдозу электролитически окис-
ляют до альдоновой кислоты, которая при обработке П. в. и Fe(OAc)3
дает 2-кетоальдоновую кислоту, с потерей СО2 превращающуюся
в D-арабинозу. Выход альдопентозы из альдоновой кислоты возрас-
тает до 44% при добавлении ионообменных смол [7]. Однако окис-
79
СНО СО2Са1/3 СО2Н
неон неон 1 с=о
НОСИ Электролиз (Са-+) 1 поен Н,О2 Fc(OAc)3 НО(!н
неон 77% неон неон 25%
неон неон неон
CIHOH С1ДОН с!наон
D-Глюкоза сн=о
носи
> неон
неон
снйон
D-Арабиноза
ление альдозы до альдоновой кислоты и окисление последней до
альдопентозы можно провести гипохлоритом, и этот двухстадийный
процесс [8], выполняемый без выделения промежуточных веществ,
имеет явное преимущество по сравнению с расщеплением по Руффу.
Первую стадию проводят с 3 же гипохлорита при pH 11 (выход
80%) и вторую стадию — с 1,4 же этого реагента при pH 4,5—5
(выход 65—70%).
Уденфринд и сотр. [91 предложили систему для гидроксилиро-
вания ароматических соединений, включающую сульфат железа(Н),
кислород, аскорбиновую кислоту и этилендиаминтетрауксус-
ную кислоту (ЭДТК) в фосфатном буфере. Авторы указывают, что
во всех изученных примерах кислород можно заменить П. в. в атмос-
фере азота. Взаимодействие осуществляется и без добавления ЭДТК;
однако в присутствии ЭДТК значительно повышается скорость реак-
ции. Диоксималеиновая кислота и диэтиловый эфир дикетоянтар-
ной кислоты имеют приблизительно такую же активность, как и
аскорбиновая кислота, тогда как аллоксан и нингидрин менее ак-
тивны. Примеры приведены на следующей схеме;
СО
нос
il
Fe2+ -|-ЭДТК [-О., ЩНОС
I
нс-
НОСН
CHSOH
nh2
80
ch2ch,nh3
'OH
OH
В этих и пяти других случаях продукты окисления идентичны обра-
зующимся при метаболизме этих соединений in vivo.
Катализ соединениями ванадия. Трейбс и сотр. [10] получают
надванадиевую кислоту, добавляя 1 г пятиокиси ванадия при охла-
ждении к 10 мл 30%-ной П. вд образующийся желто-красный раст-
вор встряхивают, разбавляют 100 мл ацетона и фильтруют. Раствор
катализатора добавляют к 500 г циклогексена в 5 л ацетона и
при перемешивании, поддерживая температуру 30°, к смеси добав-
ляют 100 г 30 %-ной П. в. и кипятят 1 час. Получают 32 е транс-
циклогександиол а-1,2 и 41 г адипиновой кислоты, образующихся
в результате некатализируемой реакции циклогексена с П. в. и
40 г соединения, описанного как А2-циклогексенон-1 (2). Однако
позднее методом ГЖХ было найдено [11], что эта фракция является
О ОН
И I
смесью кетона (2) и спирта (3) в соотношении 1 : 2,3. При окисле-
нии таким способом тетралин дает а-тетралон с выходом 65% [10].
После сообщения о взрыве при проведении окисления с катали-
затором, полученным указанным способом, Трейбс [12] опубликовал
более подробную методику и особо подчеркивал, что необходимо
исключить даже следы минеральных кислот.
Катализ соединениями вольфрама. Первичные амины, содер-
жащие группу CH2NH.>, окисляются 35%-ной П. в. в присутствии
каталитических количеств вольфрамата натрия (Na2WOd-2H2O)
через промежуточное образование гидроксиламина до альдоксимов
[13]. Реакцию проводят в воде или водном этаноле при комнатной
температуре.
«-С.,Н7СП,№На h-C3H?CH,NHOH -----> н-С3Н7СН = NOH
57%
81
Пайн и Уильямс [14] нашли, что малеиновая, фумаровая и кро-
тоновая кислоты, очень стойкие по отношению к надуксусной и над-
бензойной кислотам, легко превращаются в эпоксиды с выходом
77, 50 и 50% при действии 50%-ной П. в. и вольфамата натрия при
pH 4—5,5. Прибавление П. в. вызывает экзотермическую реакцию,
II—с—СО2Н
|i +NaOH+30%H.,O2-7Na2WO4-2H2O__>
Н—С —СО2Н
1 моль 1,5 моля 1,2 моля 0,02 моля
360 мл Н,О, 63-65° НОИСЧ /СО., 11
---------------► нХ-с<н
о
которую регулируют, поддерживая температуру 63—65°. Для под-
держания необходимого значения pH (pH-метр) надо добавлять
щелочь. Когда титрование покажет, что прореагировало 1,02 же
П. в., раствор концентрируют в вакууме и обрабатывают ацетоном
для осаждения натриевой соли эпоксида.
Шульц и сотр. [15] нашли, что окисление сульфидов до сульфо-
нов П. в. в уксусной кислоте требует продолжительного кипячения
и дает плохие выходы, но добавление вольфрамового (или вана-
диевого) катализатора улучшает результаты. Для получения ката- i
лизатора вольфрамовый ангидрид (WO3) перемешивают в дистилли-
рованной воде, добавляют едкий натр до полного растворения и
доводят pH до 5—6 добавлением уксусной кислоты. Для окисления
2-фенилтиоэтанола его добавляют к водному раствору катализатора
и затем 30%-ную П. в. с такой скоростью, чтобы поддерживать тем-
пературу 63—75"; сульфон получают с высоким выходом. При не-
C6H5SCH3CH2OH + H2O2(H.>WO4) —CBHsSO;CH2CH2OH
катализируемом окислении главным продуктом является соответст-
вующий сульфоксид—2-фенилсульфинилэтанол (65%). Если суль-
фид плохо растворим в воде, то можно применять этанол или
диоксан.
Н2О2 — OsO4. Олефины гидроксилируют до /.(«с-диолов с выхо-
/\ /'-• Х'0И
I II НзОДОЮ.,} |
L '/ 58% I z х
дом 30—60% обработкой безводной П. в. в трет-бутаноле в присутст-
вии каталитических количеств четырехокиси осмия [161. Олефины
с четырехокисью осмия дают осматы, которые под действием П. в.
расщепляются с образованием ^ис-гликоля и регенерацией четырех-
окиси осмия. Этот метод использован для окисления стероида —
82
17 ,20-ена в 17а-ол-20-он 1171. Для этой реакции безводные условия
не являются необходимыми; можно применять 30%-ную П. в.,
используя в качестве растворителя ацетон или смесь ацетон —эфир
[18]. Так, к раствору 1 г аддукта фуран — малеиновый ангидрид
(1) в 8 мд ацетона и 2 мл эфира добавляют 1 мл раствора 1 г OsO4
нго2 (OsO4)
50% *
в 200 мл mpem-бутанола и затем 3 капли 30%-ной П. в. [19]. В этих
условиях ангидрид гидролизуется до ди кислоты (2), которая начи-
нает осаждаться. Смесь выдерживают 24 час при 30°, добавляют
эфир, диоксидикислоту отделяют и кристаллизуют; т. пл. 200°,
(1)
н
HzO;(OsO4) но
со2н
со2н
выход 50%. В то время как легко выделяемый аддукт малеинового
ангидрида и циклопентадиена имеет эндо-конфигурацию, присое-
динение малеинового ангидрида к фурану дает экзо-аддукт (1).
Указанная конфигурация диола строго не доказана *.
Окисление А4-холестенона-3 30%-ной П. в. в эфире в присут-
ствии каталитических количеств OsO4 дает оба возможных qwc-диола,
выходы которых приведены на следующей схеме [20];
I, Fenton Н. S. И., J. Chem. Soc., 65, 899 (1894).
2. J е n п с г Е. L., Org. Syn., 40, 90 (1960).
3, L о e b I IL, Stein G., Weiss W-, J. Chem. Soc., 1949, 2074.
4. Smith J. R. L., Norman R. О. C., J. Chem. Soc., 1963, 2897.
5. P f i s t e r K., 3rd., Sullivan A. P., Jr., Weijlard J., Tishler
M., J. Am. Cbem. Soc., 73, 4955 (1951).
6. Ruff O., Ber., 31, 1573 (1898); 32, 550, 3677 (1899); 34, 1362 (1901); R u f f O.,
Ollendorff G., ibid., 33, 1798 (1900).
* Доказательство конфигурации см.: ЖОХ, 34, 1074 (1964). — Прим, перев.
83
7. Fletcher H. G., Jr,, Diehl H. W., Hudson C. S., J. Am. Chem.
Soc., 72, 4546 (1950).
8. W h i s t 1 e r R. L., Schweiger R., J. Am. Chem. Soc., 81, 5190 (1959);
Whistler R. L., Yagi K., J. Org. Chem., 26, 1050 (1961).
9. U d e n f r i e n d S., Clark С. T., Axelrod J., Brodie В. B.,
J. Biol. Chem., 208, 731 (1954); В г о d i e В. B., A x el г о d J., S.h o-
r e P. A., Udenf riend S., ibid., 208, 741 (1954).
10. T r e i b s W-, Franke G., LeichscnringG., Ruder H., Chem.
Ber., 86, 616 (1953).
И. E isenbraun E.J.,Bader A.R., Polacheck J.W., Reif E.,
J. Org. Chem., 28, 2057 (1963).
12. T r e i b s W., Angew. Chem., Internat. Ed., 3, 812 (1964).
13. К a hr !<., В er th er C., Chem. Ber., 93, 132 (1960).
14. P a у n e G. B., Williams P. H., J. Org. Chem., 24, 54 (1959); see also
Liwschitz Y., Rabinsohn Y., Perera D., J. Chem. Soc., 1962,
1116.
15. Schultz H.S., Freyermuth H.B., Buc S.R., J. Org. Chem.,
28, 1140 (1963).
16. M i 1 a s N. A., Sussman S., J. Am. Chem. Soc., 58, 1302 (1936); 59,
2345 (1937).
17. Mi escher K.,Schm i d 1 in J., Helv. Chim. Acta, 33, 1840 (1950).
18. M u g d a n M., Young D. P., J. Chem. Soc., 1949, 2988.
19. Daniels R., Fischer J. L., J. Org. Chem., 28, 320 (1963).
20. E as th am J. F., Miles G. B., Krauth C. A., J. Am. Chem. Soc.,
81, 3114 (1959).
ПЕРЙОДАТЫ. Перйодат натрия (метапериодат), NaIO4 (мол. вес
213,90); парапериодат натрия, Na3HJO6 (мол. вес 293,90); пер-
йодат калия, КЮ4 (мол. вес 230,01).
Циклические 1,3-дикетоиы. Уолфром и Боббит [I] показали, что
некоторые циклические 1,3-дикетоны восстанавливают перйодат
натрия в водном растворе и при этом окисляются до двухосновных
кислот. Например, циклогександион-1,3, для которого практически
отсутствует дикетоформа (1), окисляется избытком перйодата (5,7 же)
с образованием чистой глутаровой кислоты с 86,5%-ным выходом.
Считают, что при этом в качестве промежуточных продуктов обра-
зуются соединения (3)—(5). Соединения (3) и (4) были синтезированы
О О г- О о О п
(1) (2) (3) (4) (5)
zCOaH
1^/СОаН
(6)
и действительно поглощали 3 и 2 же перйодата, давая с количест-
венными выходами глутаровую кислоту и двуокись углерода. Ендио-
ловая, или редуктоновая, структура (3) способствует дальнейшему
84
окислению без непосредственного расщепления углерод-углерод-
ной связи. Окисление подобной редуктоновой системы перйодатом
наблюдалось в случае 1,2-диоксиантрахинон-З-сульфоната натрия
12]. Из 5-метил- и 5,5-диметилциклогександиона-1,3 получают
3-метил- и 3,3-диметил глутаровые кислоты с выходом 90 и 92%. 2-За-
мещенные циклогександионы-1,3 окисляются до глутаровой и моно-
карбоновой кислот. Аналогично окисляются пятичленные цикли-
I II +rco.2h
^/^он
R = CH3, сан5, сен5сн2
ческие 1,3-дионы. Скорость реакции максимальна при pH 5—6;
показано, что при этих значениях pH наблюдается наибольшая кон-
центрация периодат-ионов Юу.
Расщепление кетонов по Корифорзу. Корнфорз, Корнфорз и
Попжак [3] разработали этот новый метод расщепления на одной
из стадий изящного синтеза R(—)-мевалонолактона (6) из S-(-]-)-
линалоола (1). Гидратацией обеих двойных связей против правила
Марковникова путем гидроборирования — окисления получают
триол (2). Для защиты первичной спиртовой группы проводят ката-
лизируемую кислотой конденсацию с ацетальдегидом, которая дает
1,3-диоксан. При окислении в кетон (4) раствор 37,5 г (3) в 100 мл
пиридина добавляют к системе реагентов СгО3—Н2О—Ру, получен-
ной из 60 г СгО3, смесь оставляют при комнатной температуре на
3 дня, затем разбавляют водой и экстрагируют эфиром. Продукт
окисления, пригодный для использования на следующей стадии,
получают с 88%-ным выходом. Необходимый далее метод окисли-
тельного расщепления —СОСН2— —СО2Н+НО2С — был пред-
ложен в работе Хюбнера и сотрудников [4], которые показали, что
некоторые 1,3-ди карбон ильные соединения расщепляются перйо-
датом в водном растворе. Поскольку а-оксиметиленкетон экви-
валентен 1,3-дикетону, вернее, кетоальдегиду, продукт (4) конден-
сировали с метилформиатом с образованием соединения (5), которое,
не выделяя, сразу окисляли водно-метанольным раствором перйо-
дата натрия. Ацетальная группа гидролизуется в процессе обра-
ботки и получается R-(—)-мевалонолактон. Аналогично получен
85
S-(-[-)- мевалонолактон из (-)-линалоола. Кроме того, в этой работе
установлена абсолютная конфигурация двух природных линалоолов
(ctDn-16,9 и —16,9). Мевалонаты натрия сравнивались в качестве
(4) (5)
субстрата фермента мевалонаткиназы, выделенного из печени крыс.
Расход R-мевалоната был практически полным, тогда как для S-
мевалоната он почти не обнаруживался.
Сульфиды ^сульфоксиды. Леонард и Джонсон [51 показали, что
при добавлении сульфида к небольшому избытку 0,5 М водного
раствора NaIO4 образуются с высоким выходом сульфоксиды, не
содержащие примесей как сульфида, так и сульфона, В двух ра-
R2S4-NalO4R2S+— O~+NalO3
ботах [6, 7] этот метод использован для окисления 4-замещенных
тианов до сульфоксидов. Окисление (1) [7] дает смесь цис-(2) и
транс-(3) сульфоксидов, наиболее устойчивых в конформации с
аксиальным положением сульфоксидного кислорода. При хромато-
графическом разделении смеси вначале элюируется транс-изомер.
Окисление смесью перйодат — перманганат [8—111. В этом
каталитическом методе (Лемье и Рудлоф) под действием водного
раствора 0,019 Л1 по перйодату натрия и 0,0034 М по перманганату
калия (pH 7—8) происходит легкое расщепление двойной связи
олефина при 25°. Перманганат восстанавливается только до стадии
86
манганата, из которого регенерируется под действием перйодата.
В отдельности сам перйодат с олефином не реагирует. Симметрично
дпзамещенные олефины (1) или тризамещенные олефины (2) превра-
щаются в а-кетолы, которые в некоторых случаях можно выделить;
1) RCH
R'CH
кмпо4-RCHOH RCHOH RCO,H
R'CHOH R'C = O R'CO^H
21 RaC = CHR'
KMnO4- NaIO4
R,C—CHR'
I I
OH OH
3) RCH=CH,
KMn04-NalO4
4) RCH = C(CH3)2
KMiiO4-NaIOrI
R,C —CR'
, | 11 , R,C = O+ HO,CR'
ОН О
RCH = O-]-CH,= O
RCH = O + O = C(CH3)2
при дальнейшем окислении образуются кислоты или кетоны. Тер-
минальные олефины (3) дают с высоким выходом формальдегид.
Олефины, содержащие изопропилиденовую группу (4), расщепля-
ются с количественным выходом до ацетона. Мейнвальд и Гэссмен
[12] для окисления 2-метил ей-5,5-диметилбицикл о-[2,1,1]-гексана
(I) в кетон (II) встряхивали смесь 2,2 a (I) (жидкость), 14 а перйодата
калия, 0,5 г перманганата калия и 20 г карбоната калия в 200 мл
воды в течение 16чйс. Нерастворимые в воде твердые вещества можно
окислять в водном трет-бутаноле [11, 13], пиридине [11] или диок-
сане [13].
Реагент нашел применение при установлении строения ненасы-
щенных природных соединений. Например, Ганстоун и Моррис
16 15 12 9
СН3СН2СНСНСН2СН = СНСНоСН - СН(СН,)7СО2Н
II
ОНОН (1)
К104- КМпО4-КЮ0я
----------- Л... '----> НО,С(СН.2)7СО,Н
Води. /нр<?/77-бутанол
(2)
[14] осуществили окисление 15,16-диоксилинолевой кислоты (1)
в азелаиновую (2) в смеси вода (300 мл) — mpem-бутанол (90 мл)
при добавлении карбоната калия для поддержания pH 8—9. Якоб-
сон н corp. [15] установили строение (3) полового аттрактанта
непарного шелкопряда, окисляя подобным образом 4 мг этого ве-
щества. Реагент атакует только двойную связь без затрагивания
87
ацетокси- и оксигрупп. При этом с 92%-ным выходом получена
3-ацетоксинонановая кислота (4) и со-оксикислота (5), которая при
окислении перманганатом в щелочной среде дает пимелиновую кис-
лоту с выходом 71%.
Чеше и сотр. [16] применили метод Лемье и Рудлофа для окис-
Ы Н
| | К1О4-КМпО4-К,СО,
CH3(CI I ЩСНСН.С = С(СН Л 5СН,ОН ”---------—:—*
। х “ Води, трет-бутанол
ОСОСНз
(3)
СН3(СН2)дСНСН.,СО3Н -ь НО2С(СН2)5СН2ОН
ОСОСНз
(4) (5)
ления карбинолфосфата (6) в 5-фосфат d.l-мевалоновой кислоты
(7). По окончании реакции избыток перйодата разлагают этилен-
гликолем, иодат-ион осаждают в виде йодата бария и катионы уда-
К1О4-КМпО4(рН 7-8)
8 0%
СН3
Т^СН.СОД!
"ОР+(ОН)2
О’
(?)
ляют на ионообменнике дауэкс-50. Фосфат мевалоновой кислоты
(7) выделяют в виде кристаллической /ирцс-циклогексиламмоние-
вой соли. При высушивании в вакууме эта соль теряет 1 моль
циклогексиламина и превращается в бпс-циклогекси л аммониевую
соль.
Окисление смесью перйодат — четырехокись осмия (Лемье и
Джонсон [17, 18]). Действиё’периодата в присутствии четырехокиси
осмия состоит в следующем. Четырехокись осмия присоединяется
к двойной связи с образованием осмата, который окисляется перйо-
датом с расщеплением до карбонильных соединений и регенерацией
четырехокиси осмия. Открытию этого метода способствовал успех
RCH-CR. RCH — CR2 RCII —O-|-O = CR^4-OsO4-|-2NaIO3
I I
о 0
метода окисления смесью перйодат — перманганат, по которому
олефин указанного типа превращается в кислоту и кетон. Новый
метод дает те же продукты, которые получаются при озонировании
88
и восстановительном расщеплении. Следует отметить, что этот метод
в некоторой степени сходен с каталитическим (NaC103 — OsO4)
методом Гофмана, с помощью которого Криге открыл осматы, хотя
при этом олефин окисляется до щщ-гликоля.
В работе [18] описаны две методики:
а) Смесь 5 мл воды, 15 мл диоксана, 0,77 г додецепа-1 и 11,3 мг
OsO4 перемешивают 5 мин до тех пор, пока смесь не станет темно-
коричневой. Затем добавляют 2,06 г тонкоизмельченного перйодата
натрия (NalOi) порциями в течение 30 мин, поддерживая при этом
температуру 24—26°. Перемешивание продолжают при этой темпера-
туре 1,5 час до тех пор, пока темная густая жидкость не станет
бледно-желтой. Смесь экстрагируют эфиром, экстракт сушат и об-,
рабатывают раствором 1 г 2,4-дииитрофенилгидразина в смеси 5 мл
конц. серной кислоты, 7,5 мл воды и 35 мл 95%-ного этанола. Смесь,
разделяющуюся на два слоя, перемешивают 70 мин, упаривают до
объема 50 мл и затем два раза отделяют желтые кристаллы 2,4-ди-
нитрофен ил гидразона, выход 1,09 г (68%).
б) Смесь 15 мл эфира, 15 мл воды, 0, 405 а циклогексена и 65,4 мг
OsO4 перемешивают при 24—26°, добавляя 2,32 г тонкоизмельчен-
ного перйодата натрия, в течение 40 мин. Перемешивание при 24—
26° продолжают 80 мин до появления желтого окрашивания смеси
и выделения значительного количества йодата натрия. Превращение
в бис-2,4-динитрофенилгидразон адипинового альдегида проводят по
методике «а»; выход 77%.
Дворник н Эдвардс [19] описали успешное применение реагента
в 80%-ной уксусной кислоте для окисления производного алкалоида
(1) в а-кетолацетат (2). Форбргогген и Джерасси [20] окисляли
(4)
F-дигидрогаррофолиндиацетат (3) в (4) также в 80%-ной уксусной
кислоте (при 4°). Следует отметить, что в этих двух реакциях окис-
ление происходит без затрагивания атома азота. При использовании
в качестве растворителя 80 %-ной уксусной кислоты каталитическое
количество четырехокиси осмия удаляют отгонкой при 40°/20 мм,
причем осмий можно регенерировать из дистиллята, добав-
ляя бисульфат натрия. При использовании в качестве растворителя
смеси диоксан — вода неочищенный продукт реакции, получаемый
после отгонки растворителя при пониженном давлении, растворяют
в. смеси бензол — эфир и раствор фильтруют через дезактивирован-
ную окись алюминия (степень активности II или III) [20].
89
Представляет интерес применение метода [21] в случае 8а,20-
эпоксиманоола (5). При окислении в водном диоксане первоначально
образующийся а-оксиальдегид (6) расщепляется с образованием
метилкетона (7) и формальдегида.
Тарбелл н сотр. [22], исследуя возможность синтеза трополон-
4-карбоновой кислоты (10), пытались расщепить стирольную двойную
связь 4-замещенного трополона (8) путем озонирования, окислением
надкислотами, азотной или хромовой кислотой, смесью перйодат —
перманганат и окислением по Прево. Во всех случаях наблюдалось
преимущественное окисление трополонового цикла. Однако окисле-
ние по Лемье и Джонсону в водном диоксане с последующей возгон-
кой продукта дает желтый 4-формилтрополон, т. пл. 154—157°,
выход 84%. Окисление альдегида окисью серебра в основной среде
дает трополон-4-карбоповую кислоту с 53%-ным выходом.
Особенность каталитического метода состоит в том, что по ско-
рости образования темноокрашенного осмата можно оценить отно-
сительную реакционную способность олефина [20].
Для соединений с пространственно не затрудненной метиленовой
группой изменение окраски наблюдается через 10—30 мин, тогда
как для пространственно затрудненных олефинов лишь через 4—
12 час; в таких случаях перйодат добавляют в течение 1—4 дней
при 4°.
Окисление смесью перйодата н четырехокиси рутения. Эту
реакцию, описанную Паппо и Бекером [23], можно продемонстри-
ровать на приведенном Сторком и сотр. [24] окислении при расщеп-
лении по Барбье — Виланду. Окисление кетодифеннлэтилена (3)
до кетокислоты (4) можно осуществить озонолизом, но наилучшие
выходы получаются при обработке четырехокисью рутения и перйо-
датом натрия в водном ацетоне. Перйодат является окислителем, а
четырехокись рутения действует как катализатор. К перемешивае-
90
мому раствору 2,22 г (3) в 100мл ацетона добавляют раствор 500 мг
четырехокиси рутения в четыреххлористом углероде и затем 2 г
перйодата натрия в 40 мл воды. По окончании отделения черного
осадка RuO3 для его растворения добавляют 3 г перйодата. Добав-
ление повторяют еще дважды в течение 10 час. Избыток реагента раз-
лагают пропанолом, черный осадок RuO2 отделяют фильтрованием
и растворитель выпаривают. Поскольку эта стадия объединялась
с другими, выход отдельно не определен.
Зондхеймер и сотр. [251 успешно применяли каталитический ме-
тод в другом окислительном расщеплении дифеиилэтилеиа по Бар-
бье — Виланду, где озонолиз и окисление хромовой кислотой дают
плохие результаты. Сарел и Янука [26] показали успешное приме-
нение метода в случае дифеиилэтилеиа (2), который не окисляется
перйодатом и четырехокисью осмия. К раствору олефина (2) в вод-
ном ацетоне (80—85%) добавляли 5 мол. % RuO4 и 140 мол. % NaIO4
при 15—25°.
1. W о 1 I г о m М. L., Bobbitt J. М., J. Am. Chem. Soc., 78, 2489 (1956).
2. Ramachandran L.R., Sarma P. S., J. Sei. Ind. Res. (India) 10B,
147 (1951) (С. A., 47, 2160 (1953)].
3. Cor nf or th R.H., C or n f or t h J.W Ponjak G., Tetrahedron
18, 1351 (1962).
4. Huebner C.F., Ames S.R., Bubl E.C., J. Am. Chem. Soc., 68
1621 (1946).
5. Leonard N. J., J olinson C. R., J. Org. Chem., 27, 282 (1962); J. Am.
Chem. Soc., 84, 3701 (1962).
6. J ohnson C. R., McCants D., Jr., J. Am. Chem. Soc., 86, 2935 (1964).
7. M artin J. C., Uebel J. J., J. Am. Chem. Soc., 86, 2936 (1964).
8. Lemieux R. LL, von R u d I о f f E., Can. J. Chem., 33, 1701 (1955).
9. Lemieux R.U., vonRudolff E., Can. J. Chem., 33, 1710 (1955).
10. R u d 1 о I I E., von, Can. J. Chem., 33, 1714 (1955).
11. R u d 1 о I f E., von, Can. J. Chem., 34, 1413 (1956).
12. Mei nwal d J., Gassman P. G., J. Am. Chem. Soc., 82, 2857 (1960).
91
13. W a 1 1 M. E,, Ser of a S., J. Org. Chem., 24, 741 (1959).
14. G unstone F. D., Morris L. J., J. Chem. Soc., 1959, 2127.
15. Jacobsen M., Beroza M., Jones W. A., J. Am. Chem. Soc., 83
4819 (1961).
16. Machlei d t H., Cohnen E., Tschesch e R., Ann. 655, 70 (1962);
672, 215 (1964).
17. Contributed in part by Vorbrueggen H., Stanford University.
18. P a p p о R., A 1 I e n D. S., Jr., Lemieux R. U., Johnson W. S.
J. Org. Chem., 21, 478 (1956).
19. D v о r n i к D., Edwards О. E., Can. J. Chem., 35, 860 (1957).
20. Vorbrueggen FL, Djcrassi C., Tetrahedron Letters, 119 (1961);
J. Am. Chem. Soc., 84, 2990 (1962).
21. Schei degger U., Schaffner K., J e g e r O., Helv. Chim. Acta,
45, 400 (1962).
22. T a r b e 1 1 D. S., Williams К. I. FL, S e h m E. J., J. Am. Chem. Soc.,
81, 3443 (1959).
23. P a p p о R., Becker A., Bull. Res. Council Israel, 5A, 300 (1956).
24. Stork G., M e i s e 1 s A., Davies J.E., J. Am. Chem. Soc., 85, 3419
(1963).
25. Sondhei mer F., Meclioulam R., Sprecher M., Tetrahedron,
20, 2473 (1964).
26. S a r e 1 S., Y anuka Y., J. Org. Chem., 24, 2018 (1959).
ПЕРХЛОРИЛФТОРИД, FC1O3. Мол. вес 102,46, газ, т. кип.
—46,8°.
П. устойчив, однако его смеси с окисляющимися веществами пред-
ставляют опасность.
Перхлорилирование. При пропускании П. в суспензию хлори-
стого алюминия в бензоле наблюдается взаимодействие типа реакции
Фриделя — Крафтса и образуется перхлорилбензол — бесцветная
жидкость с т. кип. 232° [1]. При щелочном гидролизе этого продукта
С6Н6-!-FCIOH-1-А1С13 C6H5C1O3-PHC1 + A1CI2F
образуется фенол, а при нитровании 3-нитроперхлорилбеизол. Реак-
ция перхлорилирования не осуществляется в случае нитробензола
и подобных дезактивированных ароматических соединений.
Фторирование соединений с активной метиленовой группой.
Сообщалось [2], что обработка малонового эфира 2 же этилата нат-
рия в этаноле и пропускание П. при охлаждении приводит к дифтор-
малоиовому эфиру, получаемому с высоким выходом. Одиако пов-
торное исследование [31 показало, что эта реакция намного сложнее,
СН2(СОгС.,Н5)г 2NaO^HL^ Na2C(CO,C2H5)2 —F3C(CO4C2FL,).,
JO- 15°
причем выделены следующие пять продуктов: дифтормалоновый
эфир, этилмалоновый эфир, малоновый эфир, этилфтормалоновый
эфир и фтормалоновый эфир.
Синтез а-фторкетонов и винилогов. Габбард и Еисен [4]
превращали холестанон в пирролидиленамин и в бензольный раст-
вор этого соединения пропускали П. до исчезновения оранжевой
окраски (30 сек). Обработкой смеси и кристаллизацией выделен 2ои
92
фторхолестанон-3 с высоким выходом. Жоли и Варнан [5] изучали
реакцию П. с енаминами семи /V-3-кетостероидов (1). Раствор ке-
тона в 8 об. метанола кипятили с обратным холодильником в атмос-
фере азота и затем обрабатывали 1 об. пирролидина. Енамин, отде-
ляющийся после охлаждения смеси, собирали и растворяли в 10
об. метанола, содержащего 10% воды. В раствор при охлаждении
до —20° пропускали П. в течение 20 мин. Избыток П. удаляли током
азота и 4-фтор-А5-3-кетон (3) осаждали ледяной водой. Изомериза-
цию в сопряженный кетон (5) проводили следующим образом: нео-
чищенный продукт (3) растворяли в 10 об. ДМФА, добавляли 1 об.
конц. соляной кислоты и раствор оставляли на 20 час при комнат-
ной температуре. Показано [6], что реакция Д3,5-диенамигюв типа
(2) с П. в эфире при 0° дает смесь продуктов, из которой 4,4-ди-
фтор-Анкетой (4) выделен с выходом 25—50%. Если в качестве
растворителя применяют эфир, содержащий 4% пиридина, то по-
лучают смесь (3 : 1) дифторкетона (4) и сопряженного 4-фторке-
тона (5).
Енаминовый метод неприменим к 20-кетостероидам, поскольку
эти кетоны очень редко удается превратить в енамины. Однако ена-
мид (Ш), полученный из А((}-20-кетона (I) через оксим (II) с после-
дующей перегруппировкой Бекмана, легко реагирует с П. в пири-
дине при комнатной температуре. Удалением одной трети пиридина,
подкислением до pH 2, экстракцией эфиром, хроматографией и
93
кристаллизацией получают чистый 16 ct-фтор-17-кетон (IV) с выхо-
дом 70% в расчете па ем амид (III).
Показано [8], что в качестве промежуточных продуктов можно
использовать также эфиры енолов. Например, эфиры енолов трех
3-кето-5а-стероидов при взаимодействии с П. в пиридине при ком-
натной температуре в течение 2 мин и последующем подкислении
дают 2 а-фтор-3-кетоны с выходом 75—90%. Эфир енола Л4-холес-
тенона-3—3-этокси-Д3,5-холестадиен реагирует с П. в пиридине при
—20^ с образованием 6р-фтор-Дг1-холестенона-3. Эдвардс и Риигольд
[91 получили 2а-фтортестостерон реакцией натриевой соли 2-окси-
метилентестостероиа (1) с П. в бензоле с последующим расщепле-
нием получающегося 2-альдегидо-2-фторпроизводного (2) ацетатом
калия в метаноле. 16-Формил-(оксиметил)-17-кетостероиды (4)
реагируют с П. в mpem-бутаноле в присутствии трет-бутилата калия
с образованием 16,16-дифтор-17-кетостероидов (7) с 25—30%-ными
выходами [10]. Предполагаемый механизм реакции указан на схеме:
94
Разработан [111 метод получения 6-фторстероидов из диенол-
апетатов А4-3-кетонов. В условиях ацетилирования енола ацетили-
руется 17а-оксигруппа моноацетата прегнадиендиондиола (1) с об-
разованием соединения II.
Реакция соединения II с П. в 65% -ном водном диоксане дает
смесь 6-эпимеров с преобладанием трудно отделимого 6[3-фторпроиз-
водного (Ш). Однако при обработке раствора этой смеси хлори-
стым водородом в хлороформе происходит эпимеризация в экватори-
альное ба-фторпроизводное (IV), которое, таким образом, получается
из соединения II с общим выходом 45%.
3,11-Дикетостероиды превращают [12] в 3-енол ацетаты, 3-енол-
эфиры и 3-енамины, из которых реакцией с П. получают 4- и 6-фтор-
производные.
Оксофторирование. Этот термин был предложен Нимэном
[13] для новой электрофильной амбидентной реакции П. с олефи-
(4)
нами типа индена (1). Раствор индена в водном диоксане насыщают
П. при комнатной температуре в течение 4 час. С помощью хромато-
графии на колонке, фракционной кристаллизации и газовой хрома-
95
тографии выделяют пять продуктов, причем преимущественно обра-
зуется 2-фторинданон (4), содержание которого в смеси составляет
46%. Предполагаемый механизм реакции показан на схеме, где
(2) — переходное состояние и (3) — невыделенный промежуточный
продукт. Новая реакция использована для получения 7а-фторэст-
радиола (9). Оксофторирование 6-дегидроэстрадиолдиацетата (5)
в водном диоксане дает смесь продуктов, из которой выделен 7сс-
фтор-6-кетон (6). Восстановлением кетона (6) боргидридом натрия
при —5° получен ^ис-фторгидрин (7) в виде 3,17-диацетата. Свобод-
ную оксигруппу отщепляли восстановлением мезилата (8) и после-
дующим гидролизом получали 7а-фторэстрадиол (9).
ОАс
Реакция с фенолами. Показано [14], что эстрадиол реагирует
с П. в ДМФА (комнатная температура, 24 час) с образованием
10Р-фтор-Д ^дегидро-! 9-нортестостерона с хорошим выходом. Этим
же методом получены некоторые другие 10(3-фторстероиды.
Недавно Кепде и Мак-Грегор [15] описали реакцию, в которой П.
действует как амбидентный электрофил в какой-то степени сходно
с оксофторированием по Нимэну. П. пропускали в суспензию 2,6-
диметилфеиолята натрия в пентане или толуоле при 0°. После об-
работки и хроматографии получены 2,6-диметилфеиол, 2,6-диметил-
бензохшюн, 2,2', 6,6'-тетраметил-4,4'-дифеиохипон и приблизитель-
но с 20%-ным выходом новое соединение, идентифицированное как
96
димер (3) 6-фтор-2,6-диметилциклогексадиеп-2,4-она (2). Димери-
зация осуществляется по 4,5-двойной связи одной молекулы и
2,5-диеновой системе другой.
Тауб [16] описал другой метод синтеза дпенонов. 3,5-Диметок-
сифепол (4) реагирует сП, в пиридине при 20°, давая с хорошим вы-
ходом смесь (1 : 1) диенонов (5) и (6). 2- и 4-Фторпроизводиые фенола
(4) идентифицированы в качестве промежуточных продуктов.
R ,F
но\хх/ 0СНз %/\z0СИз
I II I г
X/ 'Ч/
I I
осн3 осн3
(4) (5)
о.
ОСН3
(6)
Реакции с нитроалканами и оксимами. Шехтер и Роберсон [17]
показали, что натриевые соли вторичных нитроалкаиов при реакции
анионов с П. дают удовлетворительные выходы фторнитросоедине-
ний (36—42%), Реакция сопровождается также образованием соот-
ветствующих кетонов (32—55%) и небольших количеств вициналь-
ных динитросоединений. Первичные нитроалканы дают главным
образом альдегиды. Среди других примеров этой и подобных реак-
о- О-
R..CH-N+ =0 RX-N+~O-(Na+) _ FCI°3 (°°-U
сн3он CH3OH
RSCFNO24-R2C=O+R3C—CR2
NO3 NO,
ций Фримэн [18] привел превращение аниона 2-нитро-З-фенилнор-
бориена (1) в 2-фтор-2-иитро-3-фенилнорборнен (2) с хорошим вы-
ходом с одновременным образованием небольшого количества
кетона (3).
СйН5 1) ЦаН
2) FC1O:
no, ~
(1)
(2) 70% (3) 3. 5%
4 Заказ № 11 6G
97
Простые кето кс и мы превращаются в соответствующие кетоны
при обработке основанием и затем П.
(СПН5)2С-\О11 —Д. (C(!Il5)2C-NO-(Na-t) (СВН5)2С=О
Фторирование гетероциклических соединений. Шуец и сотр.
[191 показали, что 2-фтортиофен и 2-фтор тио нафтен получаются с
выходами 49 и 70% экзотермической реакцией газообразного П.
с соответствующим лптийорганическим соединением в эфире. Из
фениллития образуется незначительное количество фторбензола.
ксю3ч
-----
Реакция с [3-кетоэфирами 1201. Карбанион ₽-кстоэфира (1) реа-
гирует с П. с образованием а-фторф-кстоэфира (2), который под дей-
ствием метанольного раствора едкого натра претерпевает кетонное
расщепление и превращается в З-фтор-2-кетон (3). При обработке
каталитическим количеством этилата натрия продукт (2) подвер-
гается ретросложноэфирной конденсации с образованием сложного
эфира а-фторкарбоновой кислоты (4).
1) Na. С..1Т,0Г1
2) ВСЮ.,-
(СН3)2С- СНСНХНСОСНз -———------------
I
СО3СаН5
{И
NaOH, к
СИ ..ОН I
(сн3)2с=снсн.,снсосн3
/ 2%
(3)
F
[
(СНа)2С = СНС1-1.,ССОСН3
СОаС2Н5
(2)
F
1
(СН3);;С^СНСН..СНСО.,С2Н5
ЙВ
Непрямое фторирование. Дин и Паттисон [211 получили диэти-
ловый эфир фторянтарной кислоты (4) следующим путем: конден-
сацией диэтилсукцината с днэтилоксалатом в присутствии едкого
кали получили соединение (2), этоксалильная группа которого спо-
СО2С.,Нг, СО2С,Н5
L ' [
СН„ с.н.ок СН2
I ' -Н(СО2С21 -Г5)_, -[ FC1O,
СН2 КССОСО.2С2Н0 "
io2c.3H6 со2с2н5
(1) (2)
98
CO2C.,H5 COaCaH5
Y tl-L,
[ КНСОз I
fccocoah. * CHF
1 CO2C2H- CO.,C,H5
(3) (4)
собствуст фторированию под действием П. до (3). Атом фтора в (3)
делает, возможным щелочное расщепление этого [J-кетоэфира в мяг-
ких условиях с образованием (4).
I. Inman С. Е., Oesterling R. Е., Tyczkowski Е. A., J. Am.
Chem. Soc., 80, 5286 (1958),
2. Inman C.E., Oesterling R. E., Tyczkowski E. A., J. Am.
Chem. Soc., 80, 6533 (1958).
3. Gershon II., Renwick J. A. A., Wynn W. K., D’A s с о 1 i R.,
J. Org. Chem., 31, 916 (1966).
4. Gabbard R. B., Jensen E. V., J. Org, Chem., 23, 1406 (1958).
5. J о 1 у R., Warnant J., Bull. soc. chim. France, 1961, 569.
6. Nakanishi S,, M о r g a n R. L., Jensen E. V., Chem. Inci., 1960,
1136.
7. Nakanish i S., Jensen E. V., J. Org. Chem., 27, 702 (1962); N a k a n-
i s h i S., J. Med. Chem., 7, 108 (1964).
8. Nakanishi S., Ken-ichi Mor i I a, J cnsen E. V., J. Am. Chem.
Soc., 81, 5259 (1959).
9. E dwar ds J., R i n g о 1 d H. J., J. Am, Chem. Soc., 81, 5262 (1959).
10. R ob inson С. H., Bruce N. F., 01 i vet о E. P., J. Org. Chem., 28,
975 (1963).
II. В 1 oom В. M., В о g e r t V. V., Pinson R., Jr., Chem. Ind., 1959,
1317.
12. Magerl ein B. J., P i k c J. E., Jackson R. W., Vandenberg
G. E., Kagan F., J. Org. Chem., 29, 2982 (1964).
13. Necinan M., Osawa Y., J, Am. Chem. Soc., 85, 232 (1963); Tetrahedron
Letters, 1963, 1987.
14. Mills J.S., Barrera J., Olivares E., Garcia H., J. Am.
Chem. Soc., 82, 5882 (1960),
15. К e n d e A. S., MacGregor P., J. Am. Chem. Soc., 83, 4197 (1961).
16. T au b D., Chem. Ind., 1962, 558.
17. S h с c h t e r H., Roberson E. B., J. Org. Chem., 25, 175 (I960).
18. Freeman J,, J. Am. Chem. Soc., 82, 3869 (1960).
19. Schuetz R. D., Taft D, D., O’B ricn J. P,, J. L. She a, M or k
H. M., J. Org. Chem., 28, 1420 (1963).
20. M a c h 1 e i d t H., Ann., 667, 24 (1963); 676, 67 (1964).
21. Dean F. H., Pattison F. L. M., Can. J. Chcrn., 41, 1833 (1963).
ПИВАЛИНОВАЯ КИСЛОТА, см. Тримстилуксусная кислота.
N = N
O.->N. । ZNO2
ПИКРИЛАЗИД, " ) || М°Л' BeC 254,13> T' ПЛ' 93°‘
Y
NO3
4*
99
Получение. П. получают реакцией пикрилхлорида с азидом
натрия [1] или нитрованием о-или/г-Питр оазидобензо л а [2]. Подобно
циаиазиду, П. реагирует с олефинами с образованием либо основа-
ний Шиффа (2), превращаемых в соответствующие кетоны (3), либо
этилеиимина (4). Простые олефины дают основания Шиффа. П.
намного безопаснее в обращении, чем цианазид.
I
(l)
(4)
1. Schra der E., Ber., 50, 777 (1917).
2. Bailey A. S., Merer J. J., W kite J. E., Chem. Comm., 1, 4 (1965).
ПИКРИЛХЛОРИД, 2,4,6-(NO2);iC6H2Cl. Мол. вес 247,50, т. пл.81°.
П. при взаимодействии с азуленом в кипящем этаноле дает 1-
пикрилазулен с высоким выходом [1]. 2,4-Динитрохлорбензол
не реагирует с азуленом ни в аналогичных условиях в этаноле, ни
в условиях реакции Фриделя — Крафтса.
П., по-видимому, перспективный реагент для образования меж-
пуклеотидных связей [2].
100
I. T r e i b s W., Jost к. -H., Kurpjun C., Grundk e-S c h г о t h Q.t
Chem., Ber., 94, 1728 (1961).
2. Cramer F. et al., Angew. Chem., Internat. ed., 2, 43 (1963).
ПИКРИНОВАЯ КИСЛОТА, 2,4,6-(NO2)3C0H2OH. Мол. вес 229,10
бледно-желтые кристаллы, т. пл. 122,5°, рКа 0,80.
П. к. образует ионные соли с аминами — пикраты, твердые ве-
щества с характеристическими температурами плавления; напри-
мер, пикраты л-анизидина, N,N-диэтил ан ил и на и N-метил анилина
плавятся при 169, 142 и 144° соответственно.
П. к. образует л-комплексы (1 : 1) с нафталином и высшими угле-
водородами, обладающие более высокой температурой плавления,
меньшей растворимостью и более интенсивной окраской, чем исход-
ные соединения, например:
Л-Осионание л-Комплекс
окраска т. пл., СС
Нафталин Желтый 150
Аценафтен Оранжевый 162
Антрацен Красный 142
Фенантрен Желтый 145
3,4-Бензпиреп Пурпурно-коричне- 198
вый
Метилхолантрец Пурпурно-черный 182,5
Комплексы П. к, используются для выделения и характеристи-
ки веществ. Комплекс расщепляют экстракцией бензольного раст-
вора водным аммиаком или хроматографией на окиси алюминия,
которая удерживает П. к.
Азулены, имеющие ярко-синюю окраску, можно экстрагировать
из петролейного эфира 62%-ной серной кислотой в виде бесцветных
сопряженных кислых сульфатов. Соответствующие соли с П. к.,
Гвайен Гвайазулен (т.ПЛ.31°) Пикрат (т.пл.123°)
которые интенсивно окрашены, используют для выделения углево-
дородов. Так, гвайазулен, присутствующий в маслообразной смеси,
получаемой дегидрированием технического гвайена серой, выде-
ляют обработкой масла горячим насыщенным раствором П. к. в
101
95 %-ном этаноле с последующим охлаждением раствора [11. Пик-
рат, выделяющийся в виде темно-синих игл, собирают и расще-
пляют хроматографией на окиси алюминия; углеводород, имеющий
синюю окраску, быстро элюируют петролейным эфиром.
Пикраты аминов и углеводородов можно использовать для микро-
определения молекулярного веса [2]. Для этого измеряют вели-
чину поглощения при 380 ммк и молекулярный вес рассчитывают
по уравнению
13,440-С-п
Мол. Bec = -j—.. ... ,
log(/0/7 ’
где С — концентрация, г/л, а п — молярное отношение П. к. к
амину или углеводороду. Точность определения порядка +2%,
1. Org. Expts., 298—301.
2, Cunningham К. G., Dawso n W., Spring F. S., J. Chem. Soc.,
1951, 2305.
ПИПЕРИДИН, / NH. Мол. вес 85,15, т. кип. 105, уд. вес
0,86, рКЬ 2,9; смешивается с водой.
Расщепление по Брауну. Получение пентамстилендибромида [1].
N-Бензоилпиперидин обрабатывают при охлаждении трехбромис-
тым фосфором, затем добавляют бром, смесь нагревают до прекра-
щения выделения газа и перегоняют. Возможные промежуточные
продукты реакции указаны на следующей схеме:
Ynz
COC6ii5
I
ь СВг2СбН5
Вг(СН2)?,Вг-Р CGH-aCN -у РОВг3
65-72%
Этилирование. Ниже описан общий метод этилирования аминов
[2]. В промытую азотом колбу загружают 4 моля П., 4,4 г натрия
и 5 г пиридина и быстро нагревают при интенсивном перемешивании
в атмосфере азота. Полученную суспензию (Н2 не выделяется) пере-
носят в атмосфере азота в автоклав и под давлением этилена пе-
ремешивают при 100".
Получение 6-алкил-Д ’-циклогексенонов. Метод получения этих
соединений разработан Сторком и Уайтом 13]. 6-Ацетильное произ-
водное получают восстановлением о-толуидина по Берчу в смесь
102
дигидридов, которые при кислотном гидролизе в мягких условиях
дают смесь ненасыщенных кетонов (3) и (4) и небольшого количе-
ства (10—30%) насыщенного кетона (5). При непродолжительном
кипячении П. гладко присоединяется к кетонам (3) и (4) с обра-
зованием пиперидии кетона (6), тогда как насыщенный кетон (5)
Ы. гжпдк, NH3,
77?Щ?Л7-В[ГОН
остается неизменным. Разделение осуществляют экстракцией про-
изводных пиперидина разб. кислотой. Затем выделяют основание,
превращают его в кристаллический иодметилат (7), который на-
гревают с пиридином. Продукт представляет собой чистый
6-метил-А2-циклогексенон (8).
Катализатор реакции Клайзеиа — Шмидта. Изящный метод
Робинсона для построения кольца А стероидов иллюстрируется син-
тезом бициклического дикетона (2) [4]. Присоединение по Михаэлю
0,5 моля 2-метилциклогександиона-1,3 к метилвин ил кетону в присут-
ствии в качестве катализатора метанольного раствора едкого кали
дает соединение (1), которое циклизуется в (2) при кипячении в
бензоле, содержащем 3 щл П. Общий выход (2) составляет 63 -65%.
Ббнзол!
(пиперидин).
।
(2)
Катализатор реакции Кневенагеля. Реакцией Кневенагеля (мо-
дификация Кневенагеля — Дебнера) обычно называют конденсацию
альдегида или кетона с дважды активированной метиленовой груп-
пой, например малонового, ацетоуксусного или циануксусного
103
эфира, катализируемую П. или пиперидинацетатом. Одиако Кун
[5] в сообщении об интересном наблюдении, относящемся к этой
реакции, описал самоконденсацию кротонового альдегида под влия-
нием катализатора реакции Кневенагеля. Кун и Хоффер получили
полиепальдегцды действием П. на ацетальдегид или смесь ацетальде-
CHSCH = СНСНО -рсн3сн = СНСНО СН3(СН = СН)3С1-Ю+ Н2О
гида и кротонового альдегида, причем сообщалось, что реакция не
имеет места при действии П. на чистый кротоновый альдегид. Одиако
Бернхауэр и Валдан считали это сообщение ошибочным и утверждали,
что при этом осуществляется взаимодействие с образованием окта-
триеналя. Кун подтвердил свое сообщение об отсутствии реакции при
действии П. на чистый кротоновый альдегид. Однако он установил,
что П. промотирует самоконденсацию альдегида, который вначале
подвергался непродолжительному облучению ярким светом. Вывод
о том, что свет катализирует автоокислеиие в кротоновую кислоту,
был полностью подтвержден. Экстракция «активного кротонового
альдегида» основанием подавляет реакцию, а добавление крото-
новой или уксусной кислоты к чистому альдегиду способствует реак-
ции при добавлении П. Таким образом, действительным катализа-
тором оказалась соль П.
Для конденсации ароматических альдегидов с чистой малоновой
кислотой не требуется добавления другой кислоты, реакцию удобно
проводить в пиридине [6]. Смесь нагревают при 80—85° в течение
1 час и затем кипятят. Малоновую кислоту берут в 100%-ном избытке,
поскольку при меньшем избытке выход составляет лишь 50%; оче-
видно, декарбоксилирование малоновой кислоты конкурирует с
конденсацией по Кневенагелю.
^._сно +сн3(СО2Н)2
z~ 'С 2 моля 87—98%
СН3СИ хосн3
1 моль
/ У~сн=снс°гн
СН3о/ хОСНз
Методика проведения конденсации бензальдегида с малоновым
эфиром [7] была предложена в то время, когда бензальдегид, содер-
жащий 2—8% бензойной кислоты, считался вполне пригодным.
Если бензальдегид содержит 0,2% бензойной кислоты, выход со-
ставляет только 71%; в этом случае рекомендуется добавлять 2%
бензойной кислоты.
С6Н5СНО + СН2(СО2С,Н5)2 Г1иперидин ~ бе1132Ц С6Н-,СН = С(СОПС2Н5),
89—9 [%
0,66 моля 0,63 моля
Конденсацию салицилового альдегида с малоновым эфиром с
образованием 2-карбэтоксикумарина проводят в абсолютном эта-
ноле при добавлении 4 мл П. и 0,5 мл уксусной кислоты (кипяче-
ние 3 час) [8].
104
,сно
'COaCjHg
+ СН3(СО,С2НГ))3 ГС; ,~А^
0,55 молл 80—84%
0,5 моля
Для конденсации ацетальдегида с 2 экв ацетоуксусного эфира
19] к 1,61 моля сложного эфира при охлаждении до 0° добавляют
СН3СОСНСО2С8Н5
и. I
СН3СНО6-2СН3СОСН2СО2СгН5-----> СН3СН
о
CH3COCHCO3C2H5
о
AcOH-H2SO4
NaOH. H,SO.
(общий) J,J
3
H CO2C2HS
0,78 моля ацетальдегида и затем при охлаждении от —5 до 0° до-
бавляют раствор 2 мл П. в 5 мл абсолютного этанола. Раствор, кото-
рый вскоре мутнеет вследствие выделения воды, выдерживают при
0° в течение 30 час и обрабатывают еще двумя небольшими порциями
П. в этаноле с интервалом в один день. Продукт кристаллизуется
в виде желто-белых кристаллов. Кислоту не добавляют в расчете на
то, что прн окислении кислородом воздуха образуется достаточ-
ное количество уксусной кислоты, чтобы вызвать медленное про-
мотирование реакции. Выход для этой стадии синтеза 3,5-диме-
тил-А2-циклогексенона не приводится.
Пнперидииацетат также катализирует внутримолекулярную цик-
лизацию диальдегидов, например циклизацию (1) в (2) по Вудварду
и сотр. [10], превращение (3) в (4) по Сторку и сотр. [11]. Реакции
проводят в бензоле или уксусной кислоте при 60—70° в атмосфере
азота.
105
Синтезы гетероциклических соединений. 3-Циан-6~метпл-2(1Н)-
пирпдон (3) удобно синтезировать, исходя из 1-димотил ацеталя
3-кетобутиральдегида (1) [12]. Смесь 5 молей (1) и 5,5 моля цианацет-
амида (2) в 1 л воды обрабатывают раствором, полученным доба-
влением около 25 мл П. к 100 мл 20%-ной уксусной кислоты до
/CHfOCH^
СН3
I +
СО
н3с/
(3)
H3N
(О (2)
pH 9—10. При нагревании до 80° образуется прозрачный раствор,
который затем кипятят 24 час; продукт начинает выделяться при-
мерно через 1 час.
Енамины. Герр и Хейл [13] предложили метод защиты карбо-
нильной группы в стероидах прн помощи катализируемой кислотами
реакции с П. или морфолином с образованием енаминного производ-
ного; карбонильная группа освобождается при кислотном гидролизе.
С другой стороны, пипер ид и л енамин был использован для расщеп-
ления стигмастерина (А&,“--диепола-Зр растительного происхожде-
ния) в прогестерон (4) с высоким выходом. Окисление по Опленауэру
дает стигмастадиенон (1), селективным озонированием которого в
хлористом метилене, содержащем 1% пиридина, получают альдегид
(2) [14]. Последний превращают в енамин (3) кипячением с П. в
бензоле, содержащем следы n-толуолсульфокислоты, при непрерыв-
106
ном удалении воды [3,15]. Енамин окисляют в прогестерон бихро-
матом натрия в смеси безводная уксусная кислота — бензол [13, 151.
1. фоп-Б р а у н 10., «Синтезы органических препаратов», ИЛ, М., 1949, сб. I,
стр. 124.
2. W о 1 1 с n s a k J., С I о s s о п R. D., Org. Syn., 43, 45 (1963).
3, S t о г к Ст., White W. N., J. Am. Chem. Soc., 78, 4604 (1956).
4. Рамачандрап С., Ньюмен М. С., «Синтезы органических препа-
ратов», изд-во «Мир», М., 1964, сб. 12, стр. 52.
5. К и h п R., В a d s t u b n e г W-, Grundmann C., Ber., 69, 98 (1936).
6. К о о J., Fish M. S., Walker G. N., Blake J., Org. Syn., Coll. Vol.,
4, 327 (1963).
7. Allen C.F.H., Spangler F. W„ Org. Syn., Coll. Vol., 3, 377 (1955).
8, Horning E. C., Horning M. G., Dimmi g D. A., Org. Syn.,
Coll. Vol., 3, 165 (1955).
9. Horning E. C., D e n e к a s M. O., Field R. E., Org, Syn., Coll.
Vol., 3, 317 (1955).
10. Woodward R. B. et al., J. Am. Chem. Soc,, 74, 4223 (1952).
11. S t о г к G. et al., J. Am. Chem. Soc., 75, 384 (1953).
12. В i n о v i L. J., A r 1 t H. G., Jr., J. Org. Chem., 26, 1656 (1961).
13. Herr M. E., H e у 1 F. W., J. Am. Chem. Soc., 74, 3627 (1952).
14. S 1 о in p G., Jr., Johnso n J. L., J. Am. Chem, Soc., 80, 915 (1958),
15. Shepherd D. A. et al., J. Am. Chem. Soc., 77, 1212 (1955).
ПИПЕРИЛЕН, CH3CH = CHCH = CH3. Мол. вес 68,11, т. кип. 44°.
П. легко получается реакцией кротонового альдегида с метил-
магнийхлоридом с образованием пентен-З-ола-2 [1] и последующей
его дегидратацией над окисью алюминия при 450°.
СН3СН = СНСНО -СНзМ^ СН3СН =-. СНСНСНз > сн3сн = снсн = сн2
ю-86% I «0%
он
1. Coburn E.R., Org. Syn., Coll. Vol., 3, 696 (1955).
ПИРАЗОЛ, н Мол. вес 68,08, т. пл. 70°, т. кип,
188°, pKb 11,5. О применении П. в качестве бифункционального
катализатора в синтезе пептидов см. 1,2,4-Триазол,
ПИРИДИН, C5H5N. Мол. вес 79,10, т. кип. 115°, уд. вес 0,98,
рКЬ 8,8.
Чистый пиридин остается абсолютно сухим, если его хранить
над молекулярными ситами (4А), окисью бария или гидридом
кальция. Гранулированное едкое кали (80°/п) удаляет воду только
из очень влажного пиридина.
Дегидратация (см. также Фосфора хлорокись — пиридин; Тио-
нил хлористый — пиридин). При дегидратации спиртов терпенового
ряда под действием кислотных реагентов образуется несколько изо-
мерных олефинов. Рудлоф [1] установил, что при дегидратации
терпеновых и сесквитерпеновых спиртов под действием нейтраль-
107
ной окиси алюминия (вёльм 1) с добавкой 1—2% П. или хинолина
образуется либо один, либо небольшое число продуктов реакции.
Спирт нагревают с обратным холодильником при200—230° в течение
1—6 час с двойным избытком (по весу) предварительно обработан-
ной окиси алюминия и продукт реакции выделяют перегонкой с
водяным паром.
Ацетилирование. Чаще используется методика, согласно ко-
торой первичный или вторичный спирт растворяют в П., добавляют
избыток уксусного ангидрида и смесь оставляют иа ночь при ком-
натной температуре. Согласно другой методике [2], смесь 150 г
холестерина, 300 лл П. и 150 мл уксусного ангидрида нагревают на
кипящей водяной бане в течение 30 мин, и разбавляют значительным
количеством воды. Выпадающий в виде гранул продукт реакции
отфильтровывают и хорошо промывают; выход безводного холесте-
рилацетата составляет 166 г (100%).
Избирательное превращение метилового эфира холевой кислоты
в 3,7-диацетат осуществляют следующим образом [3]: 50 г исходного
эфира растворяют в смеси 100 л/л диоксана и 100 мл П. Смесь охлаж-
дают до комнатной температуры, обрабатывают 150 мл уксусного
r = -сн(сн3)снгснгсорн3
ангидрида и выдерживают при 26—28° в течение 20 час. После доба-
вления 200 мл воды смесь нагревают до полного растворения и оста-
вляют для кристаллизации продукта. После охлаждения смеси до
Зэ бесцветный диацетат отфильтровывают; выход 30,8—33,6 а
(51—56%). Выход снижается как при уменьшении, так и прн уве-
личении времени реакции. Обработкой маточного раствора полу-
чают 20,3 г чистой холевой кислоты; таким образом, выход продукта
в расчете на израсходованную холевую кислоту составляет 88,6%.
Другие сложные эфиры. Марвел н Секера [4] для превращения
додеканола в тозилат обрабатывали п-толуолсульфохлоридом смесь
додека пол а и П. прн перемешивании и охлаждении. После переме-
СН3(СН2)10СН2ОН -|- C5H5N -|- n-CH3C6H4SO2Cl —“
0,5 моля 2 моля 0,55 моля
—> CH3(CH2)10CH2OSO2CeH4CH3-«
108
шивания при 20° в течение 3 час смесь обрабатывали соляной кисло-
той и льдом и выпавший осадок отфильтровывали, промывали и
хорошо отжимали. Затем его переносили в стакан с 300 мл метанола,
смесь нагревали до плавления сложного эфира и интенсивно пере-
мешивали при охлаждении для получения тонкой суспензии слож-
ного эфира. Препарат сушили на воздухе при температуре ниже
20е (т. пл. 20—25°) и перекристаллизовывали из петролейного эфира;
т. пл. 28—30°.
При бензоилировании о-оксиацетофенона по Уиллеру [5] готовят
смесь фенола, бензоилхлорида и П., оставляю'!1 ее до начала слабо-
экзотермической реакции (15 мин), а затем выливают в смесь льда и
соляной кислоты. Продукт перекристаллизовывают из метанола.
yOCOCGH5
| И +C0I-IsCOCl-f-C5IIsN 5^-/ I ||
^/ХСОСН3 ^/XCOCII3
0,1 моля 0, [ моля 20 мл
Катализатор конденсации. Условия реакций конденсации ма-
лой овой кислоты с фурфуролом [6] ис м-нитробензальдегидом [7]
приведены вместе с уравнениями реакции. При конденсации с фурфу-
ролом берут больше П., чем в реакции с ж-нитробензальдегидом,
100°, 2 час
65-70%
2 /иоля
Л1-КТО2С6Н5СНО +
2 моля 1; 2 моля
Кипячение
в EtOH
сн2(со2н)2 + CgHjN —6-8 час > yH-NO2C6H4CH = CHCOzH
1м6ль 1; 1 моля 0; 3 моля
но другого растворителя не используют, причем продолжительность
реакции меньше, чем в том случае, когда в качестве растворителя
применяют этанол. При конденсации фурфурола с циануксуснон
кислотой Паттерсон [8] добавлял небольшое количество ацетата ам-
О'^'спо
СН2СО2И +
CN
c5h5n + ещео'щ и4
200 мл толуола
Кипячение
74-78%
1,1 моля
1, 0 моля 1, 4 моля 3 г
моння (никаких объяснений этого не дано), использовал толуол
в качестве растворителя и кипятил смесь с водоотделителем Дина —
Старка в течение 20 час, хотя теоретическое количество воды было
собрано за 1 час.
Выделение а-амино кислот из хлоргидратов. Для превращения
дихлор гидрата лизина в монохлор гидр ат Экк и Д^арвел [91 добав-
ляют раствор П. в 25 мл горячего 95%-ного этанола к горячему
109
раствору дихлоргпдрата в 1 л 9511 /п-иого этанола. Выпадающий
кристаллический монохлор гидр ат отфильтровывают после тща-
тельного охлаждения. Этот метод использован также для получе-
СН„СНпС1-1.>СН,СНСО,Нф Ру “'° —>
I “ "| “ 91—П4%
+ \Н:5С1- +NH3C1~
0,23 моля 0,32 моля
—- GH,CHXH0CH,CHCCU-I -KPv+HCl-
I ' “ ’ ’I
+ NH3C1~ NH2
ния свободной а-амииоизомасляной [101 и а-амино-а-фенилпропионо-
вой [111 кислот из их солей.
Получение ангидридов карбоновых кислот. Адкинс и Томпсон
[121 установили, что при добавлении фуроилхлорида к раствору
П. в петролейном эфире при —20° выделяется аналитически чис-
тый комплекс 1:1 ст. пл.~60° (с разл.). Комплекс реагирует с
0,5 моля воды в бензоле или диоксане с образованием ангидрида.
Таким путем был получен бензойный ангидрид с выходом 97%.
Ангидрид также образуется при взаимодействии комплекса с 1 молем
свободной кислоты, Этот метод был использован для получения энан-
тового ангидрида 113]. При добавлении хлор ан гидрида к раствору
П. в 25 мл бензола наблюдается небольшое разогревание и отделе-
ние комплекса. После добавления свободной кислоты температура
смеси повышается до 60—65° и выпадает хлоргидрат П. Гигроско-
пичную соль отфильтровывают и промывают бензолом; после отгонки
растворителя и перегонки получают энантовый ангидрид, т. кип.
155—162°/2 мм.
н-Сп1113СОС1 + Ру —> юС(ф1]3СОС1-Ру-
сл моля п-СсН,,СО3Н
78—83%
0J моля
0,2 моля
(я-СбН13С0),0 Щ Ру+I-IC1 -
Попытка применить этот метод к алкансульфокислотам оказа-
лась неудачной; эти ангидриды удалось получить взаимодействием
кислот с фосфорным ангидридом [14].
Дегидрогалогенирование. В синтезе 2-винилтиофена смесь тио-
фена и паральдегида при перемешивании обрабатывали хлористым
водородом и полученный неочищенный 2-(а-хлорэтил)-тиофен дей-
ствием П. превращали в четвертичную соль, которую пиролизо-
вали [151.
(сн3сно)3 + нс1 >
0 Ру (4 моля)
2) Нагревание
4 МОЛЯ 1, 33 МОЛЯ
СНСН3
С1
50-55%
(спящий )
СН=СНг
110
Бромирование. Согласно Вильсону [16], к раствору 1-метилами-
ноаптрахинона в П. при кипячении и перемешивании прибавляют
бром в течение 9—10 мин, после чего смесь нагревают на кипящей
водяной бане при перемешивании в течение 6 час. По охлажде-
нии хлоргидрат П. и 4-бромпроизводное выпадают в осадок, который
отфильтровывают и тщательно промывают водой. Темно-красный
хинон сушат и перекристаллизовывают из П., т. пл. 195—196°.
О \HGIIg
8 7"1, (> час
I- Ру ———
600 .11.'! 7
0,5 моля
I. I
О Вг
При увеличении количества взятого П. выход уменьшается.
Ароматические нитрилы. Ньюмен [17] установил, что при до-
бавлении П. к смеси а-бромнафталина и цианида меди (I) происходит
экзотермическая реакция [образуется комплекс П.— цианид меди
(I)]. Полученную смесь кипятят с обратным холодильником.
Перегруппировки. о-Бензоил окси ацетофенон претерпевает пе-
регруппировку с миграцией бепзоплы-юй группы и образованием
о-оксидибензоилметана при добавлении к его раствору в П. 7 г горя-
чего растертого в порошок гранулированного КОН. "Едкое кали из-
О
II
/ч ,-ОССбН5 /х. /01-1
|Г 6°° А
+ Pv -ИКОН —----------->
I II 7_-; 1 8°-^% । I!
^/'^сосщ " " ч:с1-1.,ссйн3
и и
о о
мельчают в ступке, предварительно «нагретой до 100°» (по-видимому,
ступку нагревают на кипящей водяной бане примерно до 87° и выти-
рают досуха) [18]. При перемешивании в течение 15 мин выпадает
объемистый желтый осадок калиевой соли. После охлаждения при
подкислении 10%-ной уксусной кислотой образуется светло-жел-
тый осадок фенольного дикетона.
Ill
1,4-Дифенил-5-амиио-1,2,3-триазол при кипячении в П. в тече-
ние 24 час перегруппировывается в изомер кислотного характера —
4-феяил-5-фениламино-1,2,3-триазол [19]. Раствор выливают в воду
H,N 5 4 Кипячение
-С=СС6Н5 + Ру
N М3 92-93%
2
О, 025 толя .0,25 моля
Н
6н5
со льдом, осадок отфильтровывают и перекристаллизовывают из
этанола (т. пл. 167—169°).
C-Ацилирование. Эта реакция, открытая Дэкином и Уэстом
[20], имеет общий характер. Примером может служить взаимодей-
ствие аланина с уксусным ангидридом в П. с образованием 3-ацетил-
аминобутанона-2 и двуокиси углерода [21]. Смесь перемешивают при
нагревании па водяной бане до полного растворения и нагревание
продолжают еще в течение 6 час. После отгонки летучих соедине-
CHyCI-ICCUl —>
I
ХЦСОСНз
СН3СНСО2Н + (СН3СО)2О Ру
| 2,35 моля 1,98 моля
nh2
0.39 моля
сосн3 -
^СН3ССО2Н ~СОз_-^ СН3СНСОС113
I 8 1 — 88% |
NHCOCH3_i NHCOCHs
ний ь вакууме продукт перегоняют при 102—10672 мм.
KOH RX. см. Фосфор трехбромистый. Тионил хлористый.
Окисление, см. Меди сульфат.
Более полное использование анилина. При реакции имидохло-
рида с анилином П. добавляют для того, чтобы взятый анилин пол-
ностью использовать в реакции 1221.
ci nhc8h5
I I
СаНГ)С = NCeH5-[--ChH5NH2 у Ру —-сйп5с^хсан5-ррутнс1-
0,46 моля 0,46 моля 0,46 73—80%
моля
Специальные синтезы. Миддлтон [23] получил 2,5-диамшю-3,4-
дицпантиофен обработкой сероводородом раствора тетрацианэти-
лепа в смеси ацетона и сероуглерода при охлаждении до 0—5°
и перемешивании с последующим быстрым добавлением П. Раствор
становился прозрачным, после чего начинало выпадать желтое про-
изводное тиофена. Сероводород пропускали еще около 30 мин,
112
а затем перемешивание при 0—5° продолжали еще 30 мин, после
чего продукт отфильтровывали.
NC-C=C-CN
i I
NC CN
0, 2 McwiiF
+ 2 H2S + Py + CS2 __________> NC[j T1CN
92-95% H2N^ J>NH2
Избыток 300 МЛ 300 МЛ S
По реакции Крёнке бензилгалогенид или а-галогеикетон пре-
вращают в соль пиридиния, которая реагирует с м-нитрозодиметил-
анилином с образованием нитрона; последний в свою очередь при
кислотном гидролизе дает альдегид или кетон. Ружичка и
QII5CH2C1 C6H5CH2NC5H5(C1~) fI.'2^-CaH^(CHs2L>
о-
— С6ЩСН- N f-C6H3N(CH3)2 с0н5сн—о
сотр. [241 использовали эту реакцию для превращения холеста-
нона через 2а-бр ом производное в смесь енольных форм холестан-
диона-2,3. Выход был на 10% выше, чем при окислении холестанона
двуокисью селена.
Интересная реакция осуществляется при добавлении порошко-
образного тетрацианэтилепа при перемешивании к смеси малоно-
нитрила, П. и воды [25]. Смесь нагревают до полного растворения,
и горячий темный раствор выливают в водный раствор хлористого
NC.
CN
NCX CN
0,1 толя
0,1 моля
0,1 м ол я(СН3)4Ы+ С1~
81.-85%
CH2(CN)Z
0; Ц ГИОЛЯ
.CN
NC
NC
C(CN)z
NC. CN
—~с
NCX ^C(CN)2
(СН3)Л +
тетраметиламмония, после чего смесь нагревают до полного раство-
рения и оставляют для кристаллизации продукта. После перекрис-
113
таллизацни (норит) получают тетраметил аммоний-1, 1, 2, 3, 3-пента-
циаиироиенид в виде ярких оранжево-желтых игл.
I, R u d I о f f Е,, von, Сап. J. Chem., 39, 1860 (1961).
2. F i е s е г L. F., J. Am. Chem. Soc., 75, 4400 (1953).
3. Fieser L. F., R a j ago p al an S., Wilson E., Tishler M.,
J. Am. Chem. Soc., 73, 4133 (1951),
4. Марвел K-, Секера В., «Синтезы органических препаратов», ИЛ, М,,
1952, сб. 3, стр. 249.
5. У и л л е р Т., «Синтезы органических препаратов», ИЛ, М., 1953, сб. 4,
стр. 516.
6. Р а д ж а г о п а л а и С., Раман П., «Синтезы органических препаратов»
ИЛ, М., 1952, сб. 3, стр. 456.
7. У а й л и Р.т С м и т Н., «Синтезы органических препаратов», ИЛ, М., 1954,
сб. 5, стр. 51,
8, Паттерсон Дж. М.т «Синтезы органических препаратов», изд-во «Мир»,
М.., 1964, сб. 12, стр. 167.
9. Экк Дж., Марвел Д., «Синтезы органических препаратов, ИЛ, М.,
1949, сб. 2, стр. 308,
10. Кларк X., Бин X., «Синтезы органических препаратов», ИЛ, iW, 1949,
сб. 2, стр. 34.
11. Штейгер Р., «Синтезы органических препаратов», ИЛ, М.т 1952, сб. 3,
стр. 58.
12. Adkins Н., Thompson Q, Е., J, Am. Chem. Soc., 71, 2242 (1949).
13. Аллен 4., Киблер Г., M а к-Л а х л и и Д., Вильсон К-, «Син-
тезы органических препаратов», ИЛ, М.т 1953, сб. 4, стр. 60.
14. F i с 1 d L., S е 1 t 1 a g с Р. FL, J, Am. Chem. Soc., 76, 1222 (1954).
15, Эмерсон У., П а т р и к Т., мл., «Синтезы органических препаратов»,
ИЛ, М., 1960, сб. 10, стр, 16.
16. Вильсон К., «Синтезы органических препаратов», ИД, М., 1953, сб. 4,
стр. 300.
17- Ньюмэн М., «Синтезы органических препаратов», ИЛ, М., 1932, сб. 3,
стр, 344.
18. Уилер Т., «Синтезы органических препаратов», ИЛ, М., 1953, сб. 4,
стр. 517.
19. Л и б е р Ю. и др,, «Синтезы органических препаратов», ИЛ, М-, [959, сб. 9,
стр. 23.
20. Dakin Н. D., West R., J, Biol. Chem., 78, 91 (1928).
21. У илей Р., Бор ум О., «Синтезы органических препаратов», ИЛ, М.,
1954, сб, 5, стр. II,
22. Хонтц А., В а г н е р Э., «Синтезы органических препаратов», ИЛ, М.,
1953, сб. 4, стр. 224.
23. Миддлтон У, Дж., «Синтезы органических препаратов», И.П, М., 1961,
сб. 11, стр. 11.
24, R ц z i с k a L., Р 1 a t t п с г PL A. Furrer М., FIclv. Chim, Acta, 27,
524 (1944).
25. М и д д л топ У. Дж., У и л с ii Д. У., «Синтезы органических препаратов»,
изд-во «Мир», М., 1964, сб. 12, стр. 132.
ПИРИДИНА N-ОКИСЬ, С5НЖ — О". Мол. вес95,10, т. пл. 67°.
Некоторые карбоновые кислоты или их ангидриды при кипяче-
нии с П. о. в бензоле, толуоле или ксилоле превращаются в альде-
гиды [1, 21.
C6H5CHXO2H + 2C-HrjN+-O--yAc2O — CSHBCHO+2CBH5N Д- 2АсОН + СО,
1, Riichardt С., Eichler S., К г ii t z О., Tetrahedron Letters, 233 (1965).
2, С о h е n Т., Song I. FL, Fa ger J. H., Tetrahedron Letters, 237 (1965).
114
ПИРИДИНА ПЕРБРОМИД C5H5NBr2. Мол. вес 238,93, т. пл.
62—63° [11.
Мак-Эльвен и Моррис [2] безуспешно пытались превратить
тетраэтилацеталь броммалоиового альдегида (1) в дибромпроизвод-
ное (2), используя ряд методов бромирования. Казалось, что весьма
важно связать образующийся бромистый водород и тем самым пре-
дотвратить его разрушающее действие на ацетальные группы, но
этого нельзя было достигнуть путем обработки (1) бромом в присутст-
вии пиридина, поскольку температура бромирования выше, чем
температура, необходимая для окисления амина бромом. Однако
при использовании заранее приготовленного П. п. были получены
удовлетворительные и воспроизводимые результаты. Для приго-
товления П. п. смешивали растворы эквимолярных количеств пири-
дина и брома в петролейном эфире; красный осадок отфильтровы-
вали, промывали и высушивали. Смесь П. п. и (1) перемешивали и
90°, 2,5 час
(С2Н5О)2СНС1-1ВгСН(ОС2Н&)2 4- C5H5NBr? -1—1-
(1) 0,36 моля 0,38 моля
—> (С2Н5О)2СНСВг2СН(ОС2Н5)2
(2)
нагревали при 90°, причем окрашенный в красный цвет реагент по-
степенно превращался в бурый бромгидрат.
Имеется одно упоминание о безуспешном применении этого реа-
гента. Джерасси и Шольц [3] использовали пербромид-бромид
пиридиния в качестве реагента для бромирования кетонов, но все
«попытки использовать П. п. в качестве реагента, который должен
был связать выделяющийся бромистый водород, не привели к жела-
емым результатам».
[.Williams D. М., J. Chem. Soc,, 1931, 2783.
2. М с Е 1 v a i n S. М., Morris L. R., J. Am. Chem. Soc,, 73, 206 (1951).
3. D jerassi C., Scholz C. R,, J. Am. Chem. Soc., 70, 417 (1948).
ПИРИДИНА ХЛОРГИДРАТ, C-,H5NHC1. Мол. вес 115,56, т. пл.
144°, т. кип. 218°.
Получение. По Прею [1]: во взвешенную колбу с пиридином,
снабженную трубкой, не доходящей до уровня жидкости, пропус-
кают хлористый водород до поглощения! же. Осадок перекристал-
лизовывают из смеси хлороформ — этилацетат и промывают эфи-
ром.
По Тейлору и Гранту [2]: для получения хлористого водорода
к 75 мл конц. соляной кислоты и 50 г хлористого натрия по каплям
добавляют 24 мл конц. серной кислоты. Полученный газ пропускают
в раствор 69 г пиридина в 200 мл абсолютного эфира до прекращения
выпадения осадка. Избыток эфира отгоняют, остаток его удаляют
в вакуум-эксикаторе, Выход 98%; степень чистоты 99,8%. Соль
115
весьма гигроскопична и ее необходимо предохранять от влаги воз-
духа.
Расщепление метиловых эфиров фенолов. Прей [3] установил,
что П. х. можно использовать для расщепления метиловых эфиров
фенолов. Например, при нагревании анизола с 3 ч. П. х. при 200—
220° в течение 5—6 час выход фенола достигает 83%. Простые эфиры
ди- и триоксибензолов при этом также легко расщепляются,
С6Н5ОСН3-рРу+НС1- 100~22Л СйН5ОН4-Ру+СН3С1-
о.днако дифениловый эфир в реакцию не вступает. Прей [4] предло-
жил использовать эту реакцию для определения метоксильных
групп, находя количество израсходованного реагента титрованием.
Согласно данным Шихана и сотр. [5], 3-метиловые эфиры 16а,
17р-эстриола, а также 16(3,17(3-эстриола при сплавлении с П. х.
при 200—220° дают с хорошим выходом эстрон. Под действием реа-
гента не только расщепляется метоксильная группа, но и дегидра-
тируется цис- или транс-16,17-гликольная группировка.
Филлер и сотр. (61 установили, что при попытке расщепления
метилового эфира (2) сильными кислотами или основаниями затра-
гивается группа CF3 с образованием 5-питросалициловой кислоты
(1). Расщепление с образованием желаемого фенола (3) удалось осу-
ществить реакцией с П. х. при 210° в течение 20 мин. При более
ОН ОСН3
J. ZCO2H A zCF3
|| НВг—АсОН ]| Ру. НС]
| .-------------- ]
"Ч/ "Ч/
I I
NO, NO2
(1) (2)
ОН
I
NO3
(3)
высокой температуре или увеличении времени реакции образуется
во все возрастающем количестве побочный продукт (1).
Эртман и сотр. [7] пытались деметилировать замещенный дифе-
нил (4) действием П. х., однако обнаружили циклизацию с образо-
ванием 2-оксикарбазола (5). Деметилирование в (6) осуществляется
при кипячении бромгидрата (4) с бромистоводородной кислотой в
течение 24 час.
116
Другие реакции. Бликкенстафф и Чанг [8] обнаружили, что
тозилаты первичных спиртов легко превращаются в соответствующие
хлориды реакцией с П. х. в ДМ ФА при комнатной температуре.
N-Ал кил амиды и анилиды карбоновых кислот дезалкилируются
с хорошими выходами при нагревании с избытком П. х. при 190—
200° [9].
О О
II 4- II /Н +
КС-N< +Cr.Hf,NHCl-RC — _pC5H5N(Cl“)
CSH6 Хн5 |
C2H5
Кетализация. П. х. является прекрасным катализатором кета-
лизации За, 17а-диокси-16Р-метилпрегнандиона-11,20 [10]. Смесь
реагирующих веществ и катализатора в бензоле кипятят при пере-
мешивании в течение 24 час с водоотделителем Дина — Старка, со-
держащим около 20 г безводного сульфата натрия для связывания
117
отделяющейся воды. Смесь упаривают до объема 3 л, добавляют
14 мл 50%-ного едкого натра, продолжают упаривать до появления
кристаллов и выливают в смесь воды и льда (10 л). Твердый продукт
отфильтровывают; после промывания и высушивания он пригоден
для использования на следующей стадии; выход 95%. В более ран-
них исследованиях по обычной методике с использованием в ка-
честве катализатора n-толуолсульфокислоты кеталь получили «с
незначительным выходом».
1. Prey V., Вег., 75, 445 (1942).
2. Taylor М. D., Grant L. R., J. Chem. Ed., 32, 39 (1955).
3. Prey V., Ber., 74, 1219 (1941).
4. Prey V., Ber., 75, 350 (1942).
5. Sheehan J.C., Erman W. F., С г u i c k s h a n k P, A., J. Am. Chem.
Soc., 79, 147 (1957).
6. F i 1 I e r R., Khan В. T., McMullen C, W., J. Ore, Chem., 27, 4660
(1962).
7. Er dtman FI., H a g 1 i d F,, S t j e г n s t r 6 in N. E., Chem, Scand.,
15, 1761 (1961).
8. В I i c k e n s t a f f R. T.,Cliang F. C,, J. Am, Chem. Soc., 80, 2726 (1958).
9, К 1 a m a n n D., Schaffer E., Chem. Ber., 87, 1294 (1954).
10, R ausser R., Ly neheski A. iM., H arris H., G г a с e 1 a R.,
Murrill N., Bellamy E,, Perching er D.,Gebert W,, Her-
z о g FI. L., Hershberg E, B.,OI i vet о E. P., J. Org. Chem., 31, 26
(1966).
ПИРИДИНБОРАН, C5H5NBH3. Мол. вес 92,94. П.—светло-
желтая жидкость, затвердевающая в белое твердое вещество, т. пл.
10—11°. П. устойчив в отсутствие влаги в воздухе, почти нераство-
рим в воде и лишь слегка гидролизуется водой, хорошо растворим
в спирте и эфире. Тейлор и сотр. [1] описали удобный метод полу-
чения П. из боргидрида натрия и хлоргидрата пиридина в пиридине.
Барнес, Грэхэм и Тейлор [2] установили, что П. восстанавливает
альдегиды и кетоны при кипячении в эфире, бензоле или толуоле
с выходами 24—94% [21. Кислоты и хлорангидриды восстанавли-
ваются этим способом с выходами лишь 21—40%, а сложные эфиры
в реакцию не вступают.
Джонсон и сотр. [31 восстановили метиловый эфир эстрона (1):
раствор 0,38 мг (1) в 0,030 мл уксусной кислоты обрабатывали раст-
вором 0,23 мг. П. в 0,005 мл. уксусной кислоты и смесь оставляли
в атмосфере азота при комнатной температуре па 3 час. После при-
бавления 1 капли конц. соляной кислоты смесь экстрагировали эфи-
ром, экстракт промывали, высушивали и выпаривали. На тонко-
118
слойной хроматограмме обнаружено только одно пятно, соответст-
вующее по RI метиловому эфиру эстрадиола (2). Джонсон и сотр.
показали, что П. восстанавливает также 17-кето- 18-иорстероид (3),
имеющий, как и (1), транс-сочленение колец С/D, тогда как
C/D-цнс-изомер (4) при этом не восстанавливается [31.
X ауто рн [4] разработал новый путь получения триал кил боранов
реакцией олефина с П. в диглиме в склянке для реакций под давле-
нием при нагревании на кипящей водяной бане. Продукт, получен-
ный из терминального олефина, при окислении перекисью водо-
рода дает первичный спирт с высоким общим выходом.
ЗСН3(СН,),СН = СН, с‘н^-н* в Д1,ГЛ,1МС (87Л
82%
[CH3(CH2)5C14,CI-I213BNC5H5 —ЗСНдС1-1а)йС1ЩСН.,ОН
ио%
1. Т а у 1 о г М, D., G г a n t L, R., Sands С. Л,, J. Am. Chem, Soc., 77, 1506
(1955),
2. Barnes R. P., Graham J. H., Taylor M. D., J. Org. Chem., 23,
1561 (1958).
3, Johnson W. S.,Y orka R. V., Tetrahedron Letters, 8, 11 (1960); Meyer
W. L., Cameron D.D.,Johnson W. S., J. Org. Chem., 27, 1130 (1962);
Yorka R. V., T г u e t t M. L., Johnson W, S., ibid., 27, 4581 (1962).
4. Hawthorne M. F., J. Org. Chem., 23, 1788 (1958).
ПИРИДИНИЯ БРОМИД-ПЕРБРОМИД, C5H5NHBr;j. Мол. вес
319,86, т. пл. 132—-134° (с разл.).
Это стабильное кристаллическое соединение более удобно, чем
бром; небольшие количества П. б. п. легко отвешивать с большой
точностью при работе с микро- или полумикро количествами. Этот
реагент дает такие же удовлетворительные результаты, как свобод-
ный бром; ниже приведены примеры, когда П. б. п. дает заметно
лучшие результаты.
Получение (Физер [1]). 15 мл пиридина смешивают с 30 мл
4896-ной бромистоводородной кислоты и смесь охлаждают; при пере-
мешивании постепенно добавляют 25 г брома, смесь охлаждают
и продукт отфильтровывают; для ополаскивания колбы и промы-
вания осадка используют уксусную кислоту. Не высушивая, про-
дукт кристаллизуют из 100)щл уксусной кислоты и получают в виде
красных игл; выход 33 г (69%).
119
Бромирование кетонов. Джерасси и Шольц [2] успешно исполь-
зовали П. б. п. для а-бромир овация стероидных кетонов. На-
пример, к раствору 38 мг холестанона (1)в 1 мл уксусной кислоты
при 40—60° добавляют 31 мг (1 же) П. б. п. Выделяется бромистый
водород, раствор обесцвечивается, и через 1 мин начинает кристал-
лизоваться 2а-бромхолестаноп (2). По охлаждении получают чис-
тый продукт (2) с выходом 81%. Из 3-кетона 5р-ряда (3) получают
4р-бром-3-кетон (4) с несколько меньшим выходом.
Дибромиды из олефинов. Исследование бромирования цис- и
транс-стильбенов бромом и П. б. и. показало, что последний обла-
дает значительно большей селективностью, чем бром. При реакции
цис-стильбен а со свободным бромом в сероуглероде Штраус 131
выделил 2 ч. d.Z-стильбендибр омида (транс-присоединение) и 1 ч.
л/езо-дибромида (^ас-присоединение). Физер [41 установил, что при
бромировании этого углеводорода П. б. п. в уксусной кислоте об-
разуется исключительно d,Z-дибромид, причем не обнаруживается
даже следов плохо растворимого л/сзо-дибромпда. По данным Брой-
ера и Цинке [5], при взаимодействии транс-стильбена с бромом в
хлороформе или сероуглероде при тщательном охлаждении, чтобы
избежать замещения в фенильных группах, максимальный выход
Л4езо-дибромидасоставляет79%. Вислиценус и Зеелер [6], используя
ту же методику, нашли, что значительно лучше растворимый
сЦ-дибромид можно легко выделить из маточного раствора с выходом
13%. В резком противоречии с этими результатами находится сле-
дующая методика [41: суспензию 20 г транс-стильбена и 40 а П. б. п.
в 400 мл уксусной кислоты перемешивают механической мешалкой
при комнатной температуре. Через 30 мин весь реагент расходуется,
сць
Н-С-Вг
С6н5-С-Н сщщнвгу [
° II -------—Н-С-Вг
Н —С—CfiH5 93-5% I
СЙН,
120
выпавший бесцветный кристаллический чистый мезо -дибромид от-
фильтровывают и высушивают; выход составляет 35,3 г (93,5%).
Обработкой маточного раствора получают еще 0,3 г лезо-дибромида;
в небольшом остатке маслянистой жидкости d, /-дибромид обнару-
жить не удается.
Перельман и сотр. [71 установили, что 19-пор-А5(10) - стероид
(19-нор-А511и)-андростенол-17рюн-3) (1) легко дегидрируется в
19-нор-А4,9-3-кетостероид (2) при взаимодействии с бромом в пири-
дине или, еще лучше, с П. б. п. При действии брома на соединение
(1) в уксусной кислоте, хлороформе или в твердом состоянии про-
межуточный 5,10-дпбромид разлагается с образованием преиму-
щественно фенольных продуктов.
Аркус и Штраусс [8] пытались превратить 1-фенил алл иловый
спирт (3) в дибромид (5) реакцией с бромом в хлороформе, четырех-
хлористом углероде или сероуглероде, однако поглощение брома фак-
тически прекращалось после добавления 0,6—0,7 экв брома и удава-
лось выделить только 1,2,3-трибром-1-феиилпропан с выходом 5—
CHa = CHCHCGH5-J-Br2 ВгСНаСНСНС6Н5
I | |
ОН Вг Вг
(3) (4)
C5H6NHBr7
I- ' - ВгСН2СНСНСеН5 % ВгСН2СНСНС6Н5
Вг ОН Вг Вг
(5) 47% (4) (5%
10%. При обработке П. б. п. дибромид (5) был основным продуктом
реакции, а трибромид (4) получался в незначительном количестве.
Фридман и Дуракян [91 описали удивительное действие П. б. п.
как мягкого селективного бронирующего агента, в частности для
соединений, склонных к разложению. При обработке комплекса
тетрафеиилциклобутадиен — бромистый никель (6) бромом в хло-
ристом метилене наблюдается значительное разложение с образо-
ванием лишь следов желаемого дибром циклобутен а (7). Бромиро-
вание П. б. п. дает значительно лучшие результаты, однако на
ранних стадиях исследования получались невоспроизводимые резуль-
таты и выход (7) колебался от 40 до 80%. Дальнейшие исследования
121
показали, что выход (7) изменяется обратно пропорционально коли-
честву побочного продукта 2,5-ди-/г-бромфенил-3,4-днфенилфураиа
(8), который образуется под действием воды. При этом было установ-
ОД/сйн5
I
сьн/ С6Н3
РуТНвг3~ В СНгС1г
-/Ру NiBr^c, -НВГ
ОД ___ход
+ Хх
, . .К /г-ВгСДЩ ° СДЦВг-тг
С6Н3 ВгВгСбН5- 4
ОДЧ
(6)
(8)
лево, что источником воды является бронирующий агент. С приме-
нением свежеприготовленного реагента по методу Физера, описан-
ному выше, выход (7) составил 60%, а (8)—20%. Если же исполь-
зовался реагент, который хранили без защиты от влаги воздуха,
то выход (7) снижался до 10%, а выход (8) увеличивался до 65%.
При использовании безводного реагента, полученного in situ, вы-
ход (7) составил 3,3 г (85%). Для этого к раствору 4,3 а (7,5 л/моля)
(6) в 125 мл хлористого метилена добавляют 2,4 г (15 .ммолей) без-
водного пиридинийбромида и затем 2,4 а (15 л/моля) брома. Смесь
перемешивают в течение ночи, затем выпаривают растворитель и
остаток растирают в ацетоне.
Глицидная кислотанепредельный альдегид. Корнфельд
и сотр. [ 101 па одной из стадий полного синтеза лизергиновой кислоты
обрабатывали натриевую соль глицидной кислоты (1) в ацетонит-
риле П. б. п. и затем семикарбазидом. Свободный альдегид (4) выде-
ляли обменной реакцией с переносо*м сем и карбазидного остатка
на пировиноградную кислоту.
Бромирование этиленкеталей. Итон [111 установил, что наи-
более подходящий способ получения дважды ненасыщенного дике-
тона из соответствующего предельного ди кетона состоит в обработке
бмс-этиленкеталя (1) реагентом с образованием дибромида (2) и в
122
последующем дегидрогалогенировании под действием трет-бути-
лата калия в ДМСО 111].
Другие реакции. Имеются указания, что П. б. и.— наиболее
подходящий реагент для превращения индола в 3-броминдол [121.
П. б. п. использовали для селективного бромирования циклических
кеталей в тетрагидрофуране 1131, однако сейчас для этого более пред-
почтителен пербромид фен илтр и метил аммония.
1. Org. Expts., 68.
2, Djerassi С., S с b о I z C, R., J. Am. Chem. Soc., 70, 417 (1948).
3. St rails F., Ann., 342, 262 (1905).
4. Fieser L. F., J. Chem. Ed., 31, 291 (1954).
5. Breuer A., Zi ncke Th., Ann., 198, 154 (1879).
6. W i s 1 i c e n u s J.,Seel er F_, Ber., 28, 2694 (1895).
7. Perelman M., Farkas E., Fornelel d E. J., К г a a у R. J.
R a p a 1 a R. T., J. Am. Chem. Soc., 82, 2402 (I960).
8. Arcus C. L., S t г a u s s H. E., J. Chem. Soc., 1952, 2669.
9. Freedman H. FI., Dooraki an G. A., Tetrahedron, 20, 2181 (1964).
10. Rorn I eld E. C., Fornefeld E. J. Kline G. B., Mann M. J.,
M orr i sonD. E-, J ones R. G., Woodward R. B., J. Am. Chem.
Soc., 78, 3087 (1956).
11. Eaton P.E.,J. Am. Chem. Soc., 84, 2344 (1962).
12. Piers 1<., Meimarogl C., Jardine R. V., Brown R. R., Can.
J. Chem., 41, 2399 (1963).
13. M arquel A. et al., Compt. rend., 248, 984 (1959).
ПИРОВИНОГРАДНАЯ КИСЛОТА, CH3COCO3H. Мол. вес 88,06,
т. пл. 14°, т. кил. 165°, уд. вес 1,27. Получение пировиноградной
кислоты дегидратацией винион кислоты см.Калия бисульфат.
Гершберг [1] разработал эффективный метод выделения кетонов
из их семикарбазонов, феиилгидразоиов или оксимов путем обменной
реакции с П. к. В случае семикарбазона а ндр осте нол он ацетата эту
реакцию проводят в присутствии ацетата натрия для того, чтобы из-
бежать деацетилирования и чтобы образовавшийся в результате
обменной реакции сем и кар ба зон оставался в растворе после разбав-
ления смеси водой. 10 г чистого семикарбазона андростенолоиацетата
123
в 30 мл уксусной кислоты обрабатывают при 110° раствором 3,2 г
ацетата натрия и 7 г 50%-ной П. к. в 20 мл воды. Через 10 мин.
медленным добавлением воды (всего 100 мл) при кипячении осаж-
дают кристаллический продукт. Выход чистого андростенолонаце-
тата, т. пл. 170,2—170,9°, составляет 8,28 г (97%). Другие при-
меры см. в работе [2].
1. Н е г s h b е г g Е. В., J. Org. Chem., 13, 542 (1948).
2. Engel С. R., J. Am. Chem. Soc., 78, 4727 (1956); Taub D., Hollsom-
m e r R. D., Wendler N- E., ibid., 79, 452 (1957); Slates H. L., W end-
ler N. L., J. Org. Chem., 22, 498 (1957).
ПИРОКАТЕХИНА ДИХЛОРМЕТИЛЕНОВЫЙ ЭФИР, (3). Мол.
вес 191,01, т. кип. 82—89712 мм.
Карбонат пирокатехина [(2), т. пл. 1194 получается при встря-
хивании охлажденного раствора пирокатехина в раз б. едком натре
с 20%-ным раствором фосгена в толуоле [1]. Реакцией карбоната
(2) с РС15 получают П. д. э. [2,3]. В условиях реакции Фриделя —
Крафтса П. д. э. ацилирует ароматические углеводороды с образо-
ванием продукта (4), гидролизующегося водой в присутствии слабого
основания с образованием ароматических кислот (6) [31; выход
70—100%.
1. Einhorn A., Lindenberg Е., Ann., 300, 141 (1898).
2. Gross Н., R i eche А., Н о f t Е., Chem. Ber., 94, 544 (1961).
3. Gross EL, R usche J., Mirscb M-, Chem. Ber., 96, 1382 (1963).
124
а-ПИРОН (2). Мол. вес 140,09, т. пл. 5°, т. кип. 110726 мм.
Получение [11. П. получают декарбоксилированием кумалиновой
кислоты (1), которая образуется при действии конц. серной кислоты
на яблочную кислоту.
Реакция Дильса — Альдера. П. легко взаимодействует с диено-
филами при комнатной температуре, образуя нормальные аддукты;
при более высокой температуре аддукты могут отщеплять СО3 [21.
Циммерман [31 синтезировал интересный дикето!i (3) по остро-
умной схеме двойного диенового синтеза, нагревая П. с избытком
метилвин ил кетона.
о сн,
II II
СН3С“СН
(2)
СНСОСНз
(3)
1. Zimmerman Н. Е., Gruncwald G. L., Pauller R. М., Org.
Syn., 46, 101 (1966).
2. F i c s e r L. F., fi a dda di n M. J., Can, J. Chem., 43, 1599 (1965).
3, Zimmerman H. E., Paufler R. M., J. Am. Chem. Soc,, 82, 1514
(1960).
О О
ПИРОФОСФОРИЛТЕТРАХЛОРИД (ПФТХ), II II
v С12Р —О —РС13
Мол. вес 251,78, т. кип. 66—6870,01 мм, 90712 мм.
П. был получен с выходом около 30% при нагревании хлорокиси
фосфора с пятиокнсью фосфора в запаянной трубке с последующим
фракционированием полученной смеси [1]. Второй метод состоит
в реакции хлорокиси фосфора с рассчитанным количеством воды:
2РОС1Н-Н3О^Р2О3С14+2НС1 [2].
При низкой температуре без растворителя П. реагирует с пер-
вичной спиртовой группой 2\ З'-изопропилиденнуклеозидов с об-
разованием 5'-дихлорфосфата, который при обработке водой дает
О О
Il I1,
RC1-I2OH+ С12Р — О -РС1,
II II
RC112ОР — С1 -У С1 — Р-ОН
CI С1
1 0
НЮ
I—-^RCH.OP—ОН
I
ОН
о
о
125
5'-монофосфат [31; выходы порядка 80%. Этот метод позволяет избе-
жать применения щелочного катализа и удаления органических
остатков.
Грубер и Лайнен [4] обнаружили, что П. в присутствии три-
этиламина реагирует со свободной спиртовой группой 2',3'-изопро-
пил идеи аденозина (1) с образованием трихлорпирофосфата (2).
В результате гидролиза и удаления изопропилидеиовой группы под
действием муравьиной кислоты получают продукт реакции, содер-
жащий 22% аденозиндифосфата (АДФ). Показано также [4], что с
помощью П. можно получить несимметричные пирофосфаты, и удает-
ся, хотя и с небольшим выходом, осуществить синтез кофермента А.
1. Grunze И., Z. anorg. a 11g. Chem., 296, 63 (1958).
2. Besson A., Compt. rend., 124т 1099 (1897); Beck e-G oehring AL,
Sambcth J., Angew. Chem., 69, 640 (1957).
3. Koransky W., Grunze H., M й n c h G., Z. Naturforsch., 17b, 191
(1962).
4. Gruber W., Lynen F., Ann., 659, 139 (1962).
ПИРРОЛИДИН, Мол. вес 71,12, т. кип. 88°, уд.
Н J
вес 0,87, рКЬ 2,9, смешивается с водой.
Енамины. Обзор методов получения и использования енаминов
см. [1|. Из трех аминов, чаще всего применяемых для получения
енаминов (морфолин, пиперидин, П.), наиболее реакционноспособ-
ным является П. [1].
Хейл и Герр [2] разработали эффективный метод превращения
А4-андростендиона-3,17 в тестостерон, первой стадией которого
является селективное образование енамина при С3. Дикетоп кипятят
с 4 же П. в присутствии следов п-толуолсульфокислоты до выделе-
ния 1 моля воды в водоотделителе. При защите карбонильной груп-
пы при С3 [2] гладко восстанавливается кетогруппа приС17. Гидро-
126
лизом енаминной группы в слабокислом буферном растворе полу-
чают тестостерон.
В 1954 г. Сторк впервые предложил методы алкилирования
и ацилирования енаминов, а в 1963 г. опубликовал обзор
исследований в этой области [3]. Пирролидиновый енамин цикло-
гексанона (1а) в форме (16) подвергается электрофильной атаке
предельным алкилгалогенидом с образованием продукта С-алкили-
(6)
(?)
127
рования (2), который при гидролизе дает а-ал кил кетон (5). Однако
выходы снижаются из-за конкурентной реакции N-алкилирования
(1а) в (3). С высокоэлектрофильными галогенидами эта конкурент-
ная реакция подавляется, и в случае аллил-, бензил- и пропаргил-
галогенидов, а также а-галогенкетонов, сложных эфиров и нитри-
лов выходы обычно составляют 50—70%. Несколько необычно нап-
равление енолизации: стабильной формой енамина несимметрич-
ного кетона, например 2-метилциклогексанона [5], является та, в
которой у двойной связи имеется наименьшее число заместителей
(7). Поэтому при алкилировании (7) получают 2,6-диалкилпроиз-
водное (6).
Енамины альдегидов и кетонов алкилируются электрофильными
олефинами, такими, как а,р-нелредельные нитрилы, сложные
эфиры и кетоны. В этом общем методе (Сторк [4]) N-алкилиро-
вание — процесс обратимый и выходы продуктов С-алкилироваиия
часто достигают 60—85%. В приведенном примере енамин цик-
лопентанона кипятят с 1,3 экв акрилонитрила в диоксане в тече-
ние 12 час и продукт реакции гидролизуют водой. В качестве раство-
рителей применяют также бензол, ацетонитрил и абсолютный эта-
нол.
СН2=СНСЫ; Н2О
67%
ch2ch2cn
Конденсация с нингидрином. Пролин конденсируется с 1 же
нингидрина с образованием пролина желтого (3), а с 2 же нингид-
рина получается пролип красный (4), который образует анионтемно-
синего цвета [51. В первом случае реакционная смесь имеет сильный
запах П. Аналогичный красный пигмент получают конденсацией
нингидрина с П.
128
Катализатор конденсации. П. в каталитических количествах
эффективен в качестве инициатора циклизации (3) в (4) [6].
(з)
Оказалось, однако, что для циклизации ди кетон а (5) в (6) тре-
буется более 1 же П.; в этом случае возможно образование промежу-
точного енамина [7,].
СОгС2Н5
(5)
(6)
1, Шмушкович Дж., Успехи органической химии, изд-ва «Мир», М., 1966,
т. 4, стр. 5.
2. Н е у 1 F. W., Herr М. Е., J. Am. Chem. Soc., 75, 1918 (1953).
3, Stork G., Brizzolara A., Lan desman H. K., Szmuszko-
vicz J., Terrell R.,J. Am. Chem. Soc., 85, 207 (1963).
4. Stork G,, Landesman Ii., J. Am. Chem. Soc., 78, 5128 (1956).
5. Grassmann W., Arnim K., v., Ann., 509, 288 (1934).
6. P а м а а а н д p а и С. А., Ньюмен M. С., «Синтезы органических препа-
ратов», изд-во «Мир», М., 1964, сб. 12, стр. 52.
7. Meyer W. L., Levinson A. S., ,1. Org. Chem., 28, 2184 (1963).
ПЛАТИНОВЫЕ КАТАЛИЗАТОРЫ. Мол. вес: Pt 195,23, пла-
тинохлористоводородная кислота (хлорид платины) PtCU-2HCl-
•6Н2О 518, 08, хлор платинат аммония (NH4)3 PtCl6 443, 91.
Адамс и сотр. [1] приводят следующий метод регенерации ката-
лизатора: остатки нагревают с царской водкой, фильтруют и упари-
вают досуха. Для очистки осаждают хлорилатинат аммония, соль
прокаливают с образованием пористой платины и растворяют ее в
царской водке [21. Простейший метод получения катализатора
Адамса состоит в сплавлении хлорплатината аммония с нитратом
натрия 13].
См. также Хейна катализатор.
Катализатор Адамса, РЮ2 -Н2О, мол. вес 245,25. Сплавление
платинохлористоводородной кислоты с нитратом натрия при 500—
550° и отмывание охлажденного расплава водой дает коричневую
окись [1]. Она стабильна при хранении и активируется встряхива-
нием в присутствии водорода и растворителя как до, так и после
добавления субстрата. Адамс и Фоорхиз [4] описали аппаратуру
5 Заказ № 1166
129
для гидрирования. Имеются прописи для гидрирования бензальаце-
тофенона в бенз ил ацетофено и [5] и этил-и-иитробензоата в этил-и-
аминобепзоат [6].
Хенце и сотр. [7] установили, что активность различных образ-
цов катализатора Адамса значительно колеблется; разработан метод
для получения окиси, обладающей более воспроизводимой актив-
ностью, которая зависит по существу от скорости нагревания плати-
нохлористоводородной кислоты до 520° в присутствии нитрата нат-
рия.
Платиновая чернь. Усовершенствование ранних методов, описан-
ное Фейлгеном [8], заключается в восстановлении раствора плати-
нохлористоводородной кислоты щелочным раствором формальдегида
при энергичном встряхивании для коагуляции коллоидального
продукта. Этот катализатор используется для разложения избытка
перекиси водорода при перемешивании (в смеси спирт— вода) до
тех пор, пока не начнется выделение кислорода [9].
Катализатор Брауна и Брауна. При добавлении боргидрида
натрия к 10%-ному раствору платинохлористоводородной кислоты
образуется мелкодисперсный черный осадок по существу чистой
платины, который является активным катализатором гидрирова-
ния олефинов и ацетиленов [10]. Восстановление платиновой соли
в присутствии активированного угля дает'катализатор на носителе,
обладающий большей активностью [11]. Методика: раствор борги-
дрида натрия в этаноле, стабилизированный едким натром, добав-
ляют к суспензии активированного угля в растворе платинохлорис-
товодородной кислоты в этаноле для получения катализатора на
носителе. Добавляют избыток соляной кислоты, после чего вещество
гидрируют; твердое вещество добавляют в растворе, а жидкий оле-
фин можно ввести шприцем через резиновую пробку. Затем из капель-
ной воронки добавляют стабилизированный раствор боргидрида
натрия с такой скоростью, чтобы в системе поддерживалось атмос-
ферное давление. Разработана автоматическая аппаратура, удоб-
ная для проведения медленных реакций и гидрирования в больших
масштабах [12].
В видоизмененном методе Брауна и Брауна [13] в колбу Эрлеи-
мейера с боковым отводом, к которому присоединяют резиновую
камеру, загружают воду, раствор хлористой платины, норит, добав-
ляют стабилизированный раствор боргидрида и затем раствор субст-
рата (эн5о-норборненщнс-5,6-дикарбоновая кислота) н конц. соляную
кислоту. Колбу закрывают медицинской резиновой пробкой, закре-
пленной проволокой, через которую раствор вводят шприцем до
тех пор, пока резиновая камера достаточно раздуется. Тогда шприц
вынимают и колбу вращают, пока уменьшение объема камеры не
укажет на то, что необходимо дальнейшее добавление боргидрида.
При этом легко наблюдать за ходом реакции, а избыточное давле-
ние в камере около 0,7 кг/см2 обеспечивает достаточно быстрое
взаимодействие.
130
Активный платиновый катализатор. Катализатор более актив-
ный, чем катализатор без носителя Брауна и Брауна, получают вос-
становлением плати но хлор и стоводородной кислоты трибензилсила-
ном или другими гидридами кремния [14].
Катализатор платина — хлористое олово [15], Раствор ката-
лизатора получают в отсутствие кислорода растворением 10 лгмолей
SnCk-H3O и 1 .ммоля платинохлористоводородной кислоты в 120 мл
метанола. При введении в перемешиваемый раствор катализатора
смеси (1 : 1) этилена и водорода при атмосферном давлении проис-
ходит быстрое и почти количественное гидрирование. Смесь ацети-
лена и водорода (1 : 1) дает этан и этилен в молярном отношении
3:1. Поскольку высшие олефины труднее гидрируются на ком-
плексных катализаторах, можно предполагать, что легкость гид-
рирования соответствует комплексообразующей способности олефина
по отношению к платине. Действие хлористого олова сводится к
предохранению платины от восстановления до металла. Исполь-
зуемый раствор катализатора не содержит коллоидных частиц.
Селективное гидрирование. Восстановление ненасыщенных аль-
дегидов в ненасыщенные спирты обычно затруднительно, однако
Тулей и Адамс [16] осуществили восстановление коричного альдегида
С(;Н5СН - СНСНО+Н2 + Pt — РЮ, ZC-C-12~Ztl(0A!lA С0Н5СН = снсн2он
в коричный спирт с высоким выходом, используя смесь окиси пла-
тины и платиновой черни в этаноле с добавкой хлористого железа
(II) и ацетата цинка. Этот эксперимент подтвердили Райландер и
сотр. [17], которые установили, что достаточно использовать одну
окись платины, но что платина является высокоспецифичным метал-
лом в этой реакции. Восстановление кротонового альдегида в кроти-
ловый спирт оказалось более затруднительным и требовало реакти-
вации катализатора.
Окись платины на кремневой кислоте. Этот модифицированный
катализатор Адамса высокоактивен при комнатной температуре и
применяется для определения степени ненасыщенности вследствие
малой продолжительности и четкого окончания реакции [18]. Гид-
рирование над катализатором сложных метиловых эфиров в этаноле
осуществляется без переэтерификации [19].
I. А д а м с Р., Ф о о р х и з В., Шрайнер Р., «Синтезы органических
препаратов», ИЛ, М., 1949, сб. 1, стр. 357.
2. W i с h е г s Е., J. Am, Chem. Soc., 43, 1268 (1921).
3. В г u с е W. F., J. Am. Chem. Soc., 58, 687 (1936).
4, Адамс Р,, Фоорхиз В., «Синтезы органических препаратов», ИЛ,
М., 1949, сб. 1, стр. 46.
5. Адамс Р., Керн И., Шр айнер Р., «Синтезы органических препара-
тов», ИЛ, М., 1949, сб. 1, стр. 87.
6. Адамс Р., Коэн Ф,, «Синтезы органических препаратов», ИЛ, М., 1949,
сб. 1, стр. 533.
7. Frampton V. L., Edwards J. D., Jr., Henze H. R.,J. Am. Chem.
Soc., 73, 4432 (1951).
131
5*
8. F e u I g e n R., Ber., 54, 360 (1921).
9. Co p e A.C.,Cigane к E., Org. Syn., Coll. Vol., 4, 612 (1963).
10. Brown H. C., Brown C. A., J. Am. Chem. Soc., 84, 1493, 1494 (1962).
11. Brown H. C., Brown C. A.,J. Am. Chem. Soc., 84, 2827, 2829 (1962).
12. Brown 11. C., Sivasankaran K., Brown C. A., J. Org. Chem.,
28, 214 (1963}.
13. Org. Expts., 86.
14. Bott R. W., Eab orn C., Peeling E.R.A., Webster D.E.,
Proc. Chem. Soc., 337 (1962).
15. Cramer R.D., Jenner E.L., Lindsey R.V., Jr., Stolberg
U. G., J. Am. Chem. Soc,, 85, 1691 (1963).
16. Tuley W. F., Adams R., J. Am. Chem. Soc., 47, 3061 (1925).
17. R у 1 a n d e r P. N., Himelstein N., Kilroy M-, Engelhard Ind.
Techn. Bull., 4, 49 (1963).
18. Van denheu vel F. A., Anal. Chem., 28, 362 (1956).
19. Ackman R.G., Burgher R. D., J. Lipid Res., 5, 130 (1964).
ПЛАТИНЫ СУЛЬФИД НА УГЛЕ. Этот катализатор гетероген-
ного гидрирования менее эффективен, чем палладиевый катализа-
тор, для восстановления ароматических нитросоединений. Одиако
он обладает тем преимуществом, что является менее чувствительным
к ядам и не восстанавливает галогенные заместители [1]. Так, 2,5-
дихлорнитробензол восстанавливается количественно в 2,5-дихло-
ранилин без дегалогенирования.
1. DovclIF. S., Greenfield И., J. Am. Chem. Sac., 87, 2767 (1965).
ПОЛИ ФОСФОРНАЯ КИСЛОТА (ПФК),
о~ о- о-
НО—Р+О( — Р+— О)„Р-'ОН
он он он
П. к. представляет собой вязкую, почти стеклообразную жид-
кость, которая после нагревания на паровой бане становится под-
вижной, легко текучей и остается такой в течение нескольких дней.
Обзоры см. [1, 2].
Как реагент для циклодегидратации П. к. иногда используется
в тех случаях, когда нельзя применить серную кислоту, фтористый
водород или натрийалюминийхлорид. Она обладает хорошей раство-
ряющей способностью и содержит ангидридные группы, за счет
которых связывает выделяющуюся воду, вследствие чего ее эффек-
тивная кислотность не изменяется. В отличие от сериой кислоты П.к.
не является окислителем, не вступает в реакции ароматического за-
мещения и менее склонна к инициированию перегруппировок. Недос-
татком ее является то, что при температуре ниже 90° затруднительно
перемешивание вязкого продукта от руки. Гидролиз реагента в кон-
це реакции также часто затруднителен.
Смеси фосфорной кислоты и фосфорного ангидрида, использо-
вавшиеся в pai-ших работах, очевидно, сильно варьируют по составу
и степени полимеризации в зависимости от времени и температуры
132
после смешения, так как равновесие при комнатной температуре
устанавливается очень медленно.
Циклодегидратация. Впервые о применении П. к. сообщается
в работе, в которой сравнивалась реакционная способность П. к.
и трех других реагентов. В синтезе дибензкоронена и коронена Шолл
и Мейер }3] для циклизации гексакарбоновой кислоты (1) с образова-
нием первых четырех колец использовали конц. серную кислоту при
комнатной температуре. Красно-фиолетовый продукт (2) был получен
в аналитически чистом виде осаждением из аммиачного раствора.
Циклизацию в (3) осуществляли под действием 20%-ного олеума при
100°; продукт был выделен в виде фиолетово-черной натриевой соли
добавлением хлористого натрия к раствору (3) в водном аммиаке.
Наконец, для осуществления двойной циклизации в (4) 1,5 а (3)
перемешивали с 10 г расплавленной безводной фосфорной кислоты,
затем добавляли при перемешивании 25 а пятиокиси фосфора и
смесь нагревали до 340— 350°. Продукт осаждали горячей водой,
сушили и перемешивали 8 час с конц. серной кислотой при 100°,
причем большая часть продукта растворялась с образованием
темно-коричневого раствора. После фильтрования через воронку
Шотта раствор разбавляли водой и получали аналитически чистый
(4). Попытки осуществить эту циклизацию с применением плавле-
ного натрийалюминийхлорида дали галогенированные продукты.
133
В другом сообщении о циклизации с образованием П. к. Робин-
сон и сотр, [4, 5], очевидно не знавшие о работе [3], перемешивали
За кетокислоты (5) в смеси Юг Р3О5 и 10 мл сиропообразной фосфор-
ной кислоты (уд, вес 1,75) и нагревали смесь до 100". Выход (6) низ-
кий, однако попытки применить другие реагенты, включая фторис-
тый водород, не дали продукта циклизации. Берч, Ягер и Робинсон
(Я
[5] отмечают между прочим, что подобным путем у-фенилмасляная
кислота дает а-тетралон с выходом 86% и |3-л4-метоксифенилпропио-
новая кислота—5-метоксиинданон-1 с61 %-ным выходом. В первом
случае выход приближается к максимальному выходу а-тетралона по
прописи в сб, «Синтезы органических препаратов» с использованием
продажной П. к. [6], 22 г у-фен ил масляной кислоты, нагретой до
65—70°, добавляют в стакан, содержащий 80 г горячей (90°) П, к.,
стакан снимают с паровой бани и перемешивают от руки 3 мин, пока
температура не поднимется примерно до 90°, Затем к смеси добавляют
еще 70 г П, к., нагревают на паровой бане и перемешивают 4 мин,
дают охладиться до 60° и обрабатывают льдом н водой. Смесь пере-
мешивают и растирают до исчезновения оранжевого вязкого про-
дукта, экстрагируют эфиром и перегоняют; выход 75—86%,
В другом примере циклизации с высоким выходом [7] кислоту
(7) растворяют в 50 ч. горячей П. к. и желтый раствор перемешивают
при нагревании на паровой бане в течение 2 час.
П.к., 2 vac при 90°^
93% (крист-)
(7) (8)
Ньюмен и Сешадри [8] с успехом использовали П. к. для цикли-
зации альдегида (10) в бензантрацен (11). Обычный метод синтеза из
134
кислоты (9) через антрон оказался неудачным, поэтому карбоксиль-
ную группу восстанавливали (1лЛ1Н4), спирт окисляли в альдегид
(CrO;j— Ру) и последний нагревали сП. к. на паровой бане 15 мин.
Промежуточные продукты не очищали, однако общий выход указан
для чистого продукта.
Один из нескольких примеров циклизации с П. к., изученных
Ку [9, 10], представляет собой получение этилового эфира 6,7-
диметокси-3-метилинден-2-карбоновой кислоты (13). Кетоэфир (12)
добавляют к П. к., предварительно охлажденной до 5° в стакане, и
смесь тщательно перемешивают 15 мин прочным шпателем, поддер-
живая температуру 20—25° периодическим охлаждением льдом.
Полученную темно-желтую пасту выливают в воду и смесь переме-
шивают и растирают до полного превращения частиц желтой пасты
в бесцветный продукт. Экстракция хлороформом дает бледно-жел-
тое затвердевающее масло. После кристаллизации из этанола полу-
чают бесцветные иглы чистого соединения (13).
Хотя выход при циклизации (14) в (15) очень низкий, этот метод
лучше, чем два других способа получения 9-кето-9Н-пирроло-[1,
2а]-индола (15), который образуется в виде ярко-желтых пластинок,
100 2 И.к.
70° 1,25 час
28-32%
(14) 10 е
(15)
т. пл. 121—122° [II, 12]. Особенность обработки состоит в том, что
реакционную смесь выливают в хорошо перемешиваемую смесь
200 мл этилацетата и 300 мл воды. Через 1 час органический слой
отделяют и водный слой экстрагируют этил ацетатом. Хромато-
графией отделяют соединение (15) от других продуктов.
Другие реакции циклизации. Летсингер и сотр. [13] установили,
что в горячей П. к. кетон (1) конденсируется с 2 молями уксусной
кислоты с образованием 4Н-пиранона-4 (2). В аналогичном синтезе
дибензилкетон и уксусная кислота дают 2,6-диметил-3,5-днфенил-
4Н-пиранон-4 (3) [14, 15], а дезоксибензоин конденсируется с 2
135
молями уксусной кислоты с образованием бензофлуоренона (5), хотя
и с меньшим выходом. При нагревании в П. к. дсзоксибензоии под-
о
II
с6н5сн/с^сн2сйн^
о о
II II
н3с—с с СНз
I 1
он он
вергается конденсации в 1,2,3-трифенилнафталин [14]. Родственная
реакция — конденсация ацегофенона в 11. к. с образованием
1,3,5-трифенилбензола [15].
Ацилирование и алкилирование ароматических соединений.
Бензофеноны получают с хорошими выходами следующим путем:
смесь фенилового эфира и алкоксибензойной кислоты обрабатывают
П. к., полученной перемешиванием смеси 8 вес. ч. РЙО5 и 5 об. ч.
136
90 %-ной ортофосфорной кислоты (уд. вес 1,75) при 85° в течение
30 мин (Айерс и Денни [16]). Так, вератрол и ванилиновая кислота
дают 4-окси-3,3',4/-триметоксибеизофенон.
СН3О^ zoch3
си,о-У Ч+ногс—У У-он п“"80’’ 3±х
------------------/ 96% (нсочищ.)
0,03 4 моля 0,03 моля
СН3ОХ ° /ОСНз
> СН3О—As—С Д—ОН
Симметричные диарил кетоны можно получить новым методом
мереацилирования (Фыозон ]I7j). При нагревании 2,3,5,6-тетра-
мет и л бен зонной кислоты с ж-ксилолом в П. к. при 78° в тече-
н3сх хсн3 н3сх
СО2Н -I- А~~А>_сн3
Н3с/ %СН3
Н3СХ /СН3 о Н3С
с // А _ СН3 -«-Ксилол ?
Н3с/ хсн3
Н3С ZCH3 О Н3СХ
А -у н3С— А_______________А—СН3
НзС/ Асн3 Асн3
ние 4,5 час дурил-2,4-диметилфенилкетон был получен с выходом
62%. Если после первоначального нагревания до 78° в течение 4,5
час смесь нагревать еще 5 час до 150°, то 2,2',4,4'-тетраметилбен-
зофенон получается с 27%-ным выходом.
Гарднер [18] показал, что П. к. служит растворителем и катали-
затором ацилирования и алкилирования (потипу реакции Фриделя—
Крафтса) фенолов и фениловых эфиров, например:
200 г П.к..
5 0°, 3,75 час
83,5%
(СН3СО)аО или СН3СООН4-^~^>—осн3
0,09 моля
О
0,5 моля 0,5 моля
137
Попытки провести эти реакции с бензолом, толуолом и циклогексе-
ном окончились полной неудачей вследствие ничтожной раствори-
мости этих углеводородов в П. к.
П. к. используется для формилирования ароматических соеди-
нений гексаметилентетрамином; выходы при этом невысоки, однако
метод достаточно прост [191,
Ацетилирование 2,3-диметокситолуол а уксусным ангидридом и
П. к, дает 6-ацетильное производное как единственный выделяемый
СН3
(СНзСОЦО— П.к.
СНд
сн3соч^х/осн3
хосн3
продукт; ацетилирование хлористым ацетилом и хлористым алюми-
нием в CS3, CfiHr> илиС6Н5МО2 дает 5-ацетилпроизводное (52—59%)
и 6-ацетилпроизводное (41—45%) [20].
Фениловые эфиры получают нагреванием кислоты и фенола
в П. к. (Бадер [211). В приведенном примере реакционную смесь
охлаждают, обрабатывают толуолом и толуольный слой экстраги-
руют раствором бикарбоната натрия, возвращая 14 г салициловой
кислоты.
Синтезы гетероциклов. Соединение (2) можно получить с высоким
выходом смешением о-фенилендиамина и бензойной кислоты с соот-
ветствующим количеством П. к. с образованием пасты, которую мед-
ленно нагревают до 250° и перемешивают при этой температуре
(1)
nh2
+ С6Н5СО2Н -—У’250^
NH2 9 5% (сырой)
н
(2)
4 час [22], Метод является довольно общим. 2-Алкилбензимидазолы
получают при использовании алифатических кислот. Замена о-фени-
лендиамина на о-аминофенол или о-аминотиофенол дает бензокса-
золы и бензтиазолы. Метод использован также для синтеза бис-
бензимидазолов [231 и бис-бензтиазолов [24]. (См., однако, о ПФЭ
на стр. 146.)
138
П. к. является эффективным реагентом в синтезе индолов по Фи-
шеру [25], Так, 2-фенилиндол (4) получают нагреванием с П. к.
либо предварительно полученного фенилгидразона ацетофенона (3),
(3)
П.к.
120°
76°/»
либо смеси фенилгндразина и ацетофенона. Другие примеры см. в
работах [26, 27].
N-Ацилированные циклоалкиланилины, например (5), при наг-
ревании с П. к. подвергаются циклизации и дегидрированию с обра-
зованием 3,4-алкиленхпиолинов (6) с очень низким выходом [281.
(5) (6)
Херд и Хаяо [29] установили, что П. к. особенно эффективна
для циклодегидратации 3-арилоксипропионовых кислот в хрома-
ионы-4, 3-арилмеркаптопропионовых кислот в тиохроманоны-4 и N,
N-диарил-р-аланинов в 2,3-дигидро-1-арил-4(11)-хинолины.
По Ку [30], в присутствии П. к. циклизация (1) в 4-кетотетра-
гидрохинолян (2) проводится непосредственно в одну стадию, тогда
как в других методах циклизации такого типа необходима то-
зильная защита вторичной аминогруппы.
О
н н
(I) (2)
Реагент используется для циклодегидратации 4-амино-5-ароил-
аминопиримидинов в пурины [311.
П.к.
----->
100%
139
N-Фенил-а, непредельные амиды также циклизуются с вы-
сокими выходами [32].
Перегруппировки. Хорнинг и Штромберг показали, что П. к.
является прекрасным растворителем я реагентом для осуществления
перегруппировки кетоксимов [33] и альдоксимов [341 по Бекману.
Например, оксим бензофенона (2 а) при перемешивании от руки
с П. к. (60 а) при 130° растворяется примерно через 10 мин, и после
разбавления прозрачного раствора водой выделяется кристаллпче-
NOH
Л П.к., 130 , 10 мин
С6Н6СС6Нб 99% СсЩ1УНСОСвН5
ский бензанилид, судя по температуре плавления и ИК-спектру,
чистый. Другие примеры цитируются в работе [35].
Хилл и Чортик [361 установили, что оксим 9-ацетил-^мс-дека-
лина (2) под действием n-толуол сульфохлорида в пиридине при ком-
натной температуре претерпевает бекмановскую перегруппировку
с сохранением конфигурации (1),а при действии конц. серной кис-
лоты или П. к. при комнатной температуре в течение 12 час образует
NHCOCH3 CH3x^NOH NHCOCH3
(1) (2} (3)
транс-ацетамид (3). Таким образом, было экспериментально под-
тверждено, что инверсия в кислых условиях включает фрагментацию
протонированного оксима (4) с образованием нона 9-декал ил карбо-
нил и ацетонитрила (5) с последующей рекомбинацией этих фраг-
ментов .
140
Оксим флуоренона не изменяется при обработке П. к. при 120%
но при 180" претерпевает перегруппировку с почти количественным
выходом (Конли [37]). Конли установил также, что П. к. является
лучшим растворителем и катализатором для перегруппировки Шмид-
та. Он добавлял азид натрия по частям к смеси симметричных или
несимметричных диарил- и арилалкилкетояов с 15—20 ч. П. к.
при нагревании в интервале 25—75°до прекращения выделения азота
и получил амиды с выходами 80—90% , Дурен бос и By [38] изучили
превращение холестанона и копростанона в З-аза-А-гомооны-4 бек-
мановской перегруппировкой оксимов в присутствии П. к. при 120—
130° (10—20 мин) и реакцией Шмидта в П. к. при 50—60° (10 час).
Во всех случаях выходы были около 90%. Для сравнения они
провели реакцию Шмидта в бензоле с раствором азотистоводород-
ной и конц. серной кислот при комнатной температуре и полу-
чили 5а- и 5[3-аза стероиды с выходами 81 и 71% .
Пирсон и Стоун [39] изучили кинетику бекмановской перегруп-
пировки ацетофенопоксимов и установили, что в П.к. скорость пере-
группировки в 12—35 раз выше, чем в серной кислоте, а скорости
перегруппировки замещенных ацетофенон оксимов в П.к. во всех
случаях примерно одинаковы. Они пришли к выводу, что механизмы
перегруппировки в П. к. ив серной кислоте различны.
Обычно при использовании П.к. в реакции Шмидта процесс осу-
ществляют при 60—100° или даже при еще более высокой темпера-
туре. Согласно данным Стокеля и Холла [40], лучшие выходы можно
СО.2Н NHa
HI +NaN3 + n.K. -- | J
OCH3 OCH3
141
получить при комнатной температуре (азид натрия в избытке).Однако
выход зависит от природы заместителей. Так, из л-нитробензойной
кислоты /г-нитроанилии получают с выходом лишь 50%, тогда как
л-метоксианилин из я-метоксибензойной кислоты— с выходом 80%.
Фирма «Universal Oil Products» выпускает П. к. па кизельгуре
(твердый катализатор полимеризации № 2). При изучении действия
П. к. па цис, транс, транс-циклододекатриен-1,5,9 Уеллмеи и сотр.
[41] показали, что такой твердый катализатор удобнее сиропооб-
разного. Твердый катализатор можно отфильтровать и тем самым
исключить экстракцию продукта из водной П. к. В результате реак-
ции получают смесь, содержащую аценафтен и декагидроаценафтен.
4- Бициклыч-Смолы
Изомеризация у-ненасыщенных спиртов. Согласно данным Ко-
лонжа и Брюнн [42], смесь 60 мл 85 %-ной фосфорной кислоты и
51 г Р2О5 нагревают в течение 1,5 час па кипящей водяной бане, после
чего добавляют 20 г спирта (1), смесь перемешивают в течение 1,5
час на кипящей водяной бане, причем окраска смеси изменяется от
красной до бурой; кетон (3) выделяют с выходом 80%. По-видимому,
сн3ч ;с^ снсн,сн2снсн3 П.к. сн3 ,ч | | П.к.
СН3' ' 1 9 0°Г 9 9®
он OHg^ Q СН3
(I) (2)
О
СИ3х I!
>CHCH.,CHnCH,CCH3
си/
(3)
реакция осуществляется через стадию 2,2,6-триметилтетрагидропи-
рана (2), поскольку из этого соединения в тех же условиях кетон
(3) образуется с выходом 90%.
Расщепление эпоксидов. Томоеда и сотр. (431 приводят инте-
ресные данные по расщеплению 4р, 5р-эпоксикопростанона-3 (2) при
комнатной температуре. В результате ацетолиза при каталитическом
действии П. к. образуется аномальный продукт (1), тогда как взаи-
модействие с эти лмер каптан ом в диоксане при каталитическом
действии П. к. осуществляется нормально с образованием (3), пос-
ледующим превращением которого получен (5). Подобно этому, ди-
меркаптоэтан при каталитическом действии П, к. реагирует с (2) с
образованием (4).
142
Нитрование [44]. Смесь 50 г 100%-ной азотной кислоты и 80 г
П. к. перемешивают при 60° до полной гомогенизации, после чего
добавляют по каплям в течение 15—30 мин диэтиловый эфир алкил-
малоновой кислоты, охлаждением поддерживая температуру смеси
около 60°. Затем смесь нагревают при 60° еще в течение 1 час, охлаж-
дают н выливают на лед, Ранее для этой цели применяли смесь азот-
н-С4НэС1-1(СО.,С.,Ы5)2 н-С1НэС(СО.,С2Н5)а
75% |
NO.,
ной кислоты и уксусногоангидрида, которая представляет опасность.
Гидролиз. При действии горячей П. к. енаминонитрил (1) пре-
вращается в кетоамид (2) с хорошим выходом (1 г—>1,09 г почти чис-
того вещества) [45]. Серная кислота и смесь серной и уксусной кис-
лот дают продукт, растворимый в воде; соляная п уксусная кислоты
дают смеси; при щелочном гидролизе не образуется желаемого про-
дукта.
Кислоты амины или амиды. Снайдер и сотр. [461 показали,
что некоторые ароматические кислоты превращаются в соответствую-
143
щие амины при механическом перемешивании смеси кислоты, соляно-
кислого гидроксиламина и П. к. [46]; смесь постепенно нагревают
Хх/Х/С°*Н
| || | -j-I-IONHo-IlCi
0,23 моля
до 160° и поддерживают при этой температуре до прекращения выде-
ления двуокиси углерода. В случае о-, м- и /i-нитробензойпых кислот
и каприловой кислоты взаимодействия не наблюдается. Уксусная
кислота при взаимодействии со слабоосновпыми ароматическими
аминами дает амиды; с бензойной кислотой реакции не происходит.
O3NX О HO,N
ci-i3co.2h-[-i-ln~/ no2 ch3c-n—S-nto2
67% \=/
В качестве источника гидр окси л амина можно использовать
нитрометан [47]:
CH3NO2 NH2OH-PCO
Так, при нагревании смеси и-хлорбензойной кислоты, нитроме-
тана и П. к. при 115° в течение 10 мин образуется n-хлоранилип с
выходом 60 %.
Нитрилы амиды. П. к.— прекрасный реагент для гидролиза
пространственно не затрудненных ароматических нитрилов в амиды
(Снайдер, Элстон [48]). Цианомезитилен при 120° в реакцию не всту-
C^N CONHS
[ 1ч
П.к , 1 10°, 1 час
пает, а при 155° превращается в мезитллеп.
Фосфорилирование. Шербюльс и Рабинович [491 применили П. к.
для получения монофосфатов амипоспиртов и гидроксилсодержащих
сложных эфиров. Холл и Корана [50] использовали смесь 85%-ной
фосфорной кислоты и Р2О5 для синтеза уридин-5'-фосфата.
Расщепление простых эфиров. Согласно Стоуну и Шехтеру [51],
простые эфиры легко расщепляются с образованием нодидов под
дейстием 95%-ной фосфорной кислоты и йодистого калия. При при-
ROR+aKl + SHsPO.i 2RI-I-2КН2РО4-| НгО
менении этого метода к эфиру (1) Коуп и сотр. [52] использовали
П. к., полученную из фосфорной кислоты и Р2О5; выход иодида (2)
практически количественный..
144
сн3
I 1 35—140°,
СН3О(СН2)10СНСН2СН3+85%-ная Н3РО4-J-PaOr, + KI 5*/4
(1) 0,90 г 2 ян 0,8 з 3,32 г "%
сн3
I
1(СНа)10СНСНаСН3
(2)
I. Р о р р F.D,,McEwen W. Е,, Chem. Revs., 58, 321 (1958).
2. Uhlig F.,Sny der H. R., Adv. Org. Chem,, 1, 35 (1960).
3. S c h о 1 1 R., Meyer K., Ber., 65, 902 (1932).
4. К о e b n e r A., Robinson R., J. Chem. Soc., 1938, 1994.
5. В i r c h A. J., J a eg e r R., Robinson R., J. Chem. Soc., 1945, 582..
6. S n у d e r H. R., Werber F. X., J. Am. Chem. Soc., 72, 2965 (1950);
Org. Syn., Coll. Vol,, 3, 798 (1955).
7. Cope A.C,, Smith R. D., J. Am. Chem. Soc., 77, 4596 (1955).
8, Newman M.S., Sesha dri S., J. Org. Chem., 27, 76 (1962),
9. Roo J., J. Am. Chem. Soc., 75, 1891 (1953).
10. Ку Дж., «Синтезы органических препаратов», изд-во «Мир», М,, 1964, сб. 12,
стр. 190.
И. Josey A. D., Jenner Е. L.,J. Org. Chem., 27, 2466 (1962).
12. J os е у A. D., procedure submitted to Org. Syn.
13, Letsinger R. L., Jamison J. D., Hussey A. S., J. Org. Chem,,
26, 97 (1960).
14. Le t s i n g e г R. L., J a m i s о n J. D., J. Am. Chem, Soc., 83, 193 (1961).
15. E m m i c k T. L., Letsinger R. L,, procedure submitted to Org. Syn.
16.. Ayres D.C.,Denney R.C.,J. Chem. Soc., 1961, 4506.
17. Fuson R.C., Bakker G. R., V i 11 i m b erga B., J. Am. Chem.
Soc., 81, 4858 (1959).
18. G a г d л e r P. D., J. Am. Chem. Soc., 76, 4550 (1951).
19, Denton D. A,,Suscliitzky H., J. Chem, Soc., 1963, 4741.
20. Edwards J. D., Jr., McGuire S. E,, II i g n i t e C., J. Org. Chem.,
29, 3028 (1964).
21. Bader A. R-, К о n t о w i c z A. D., J. Am. Chem. Soc,, 75, 5416 (1953).
22. H e i n D. W-, Aiheim R. J., L e a v i t t J. J., J. Am. Chem, Soc.,
79, 427 (1957);
23. Wang L. L., J о u 1 1 i ё M. M., J. Am. Chem. Soc,, 79, 5706 (1957).
24. Rai C., Braunworth J. B., J. Org. Chem., 26, 3474 (1961).
25. Kissman H. M., Farnsworth D. W., Witkop B., J. Am. Chem.
Soc., 74, 3948 (1952).
26. G г а у A. P., Archer \V. L., J. Am. Chem. Soc,, 79, 3554 (1957). '
27. M a с P h i 1 1 a m у H. В., D z i e m i a n R. L,, L u c a s R, А,, К u e h-
п e M. E., J. Am. Chem. Soc., 80, 2172 (1958).
28. D e n t о n D. A., S m a 1 1 e у R. К., Suschitzky H,, J. Chem. Soc.,
1964, 2421.
29. Hurd C. D., II а у a о S., J. Am. Chem. Soc., 76, 5065 (1954).
30. Koo J., J. Org. Chem., 26, 2440 (1961); 28, 1134 (1963).
31. F u S.-C. J., C h i n о p о г о s E., Terzeau H., J. Org. Chem., 30,
1916 (1965).
32. С о n 1 e у R.T., Knopka W. N-, J. Org, Chem., 29, 496 (1964).
33. H о r n i n g E. C., Stromberg V. L,, J. Am. Chem. Soc., 74, 2680
(1952).
34. Horning E.C., S t г о m b e r g V- L., J. Am. Chem. Soc., 74, 5151 (1952).
35. Barnes R. A,, В e a ch em M- T., J. Am. Chem. Soc., 77, 5388 (1955).
36. Hill R.K-, Chortyk О. T-, J. Am. Chem. Soc., 84, 1064 (1962).
37. С о n 1 с у R. T., J. Org. Chem., 23, 1330 (1958).
£8. p о о r e n b о s N, J., W u M. T., J. Org. Chem,, 26, 2548 (1961),
145
39. Pearson D. E., Stone R. M., J. Am, Chem. Soc., 83, 1715 (1961).
40, S t о с к e I R. F,, Hall D. M., Nature, 197, 787 (1963),
41. Wellman W. F., В г en n erm а л M. C., RoneckyM. P,,J, Org.
Chem., 30, 1482 (1965).
42. Colonge J., Brunie J.-C., Bull, Soc. chim. France, 1963, 1799.
43. T о m о e d a M. et al., Chem. Pharm. Bull., 12, 383 (1964).
44. Kispersky J. P., К 1 a g e г R., J. Am. Chem. Soc,, 77, 5433 (1955).
45. Baldwin S., J. Org. Chem., 26, 3280 (1961).
46. S п у d e r H. R., Elston С, T., R e 1 1 о m D, B., J. Am. Chem, Soc.,
75, 2014 (1953).
47. Bachman G, B., Goldmacher J. E., J. Org. Chem., 29, 2576 (1964).
48. S n у d e г H. R., Elston С. T., J. Am, Chem. Soc., 76, 3039 (1954).
49. C h e r b u 1 i e z E,, Rabinowitz J., Helv, Chim. Acta, 39, 1455, 1461
(1956); 41, 1168 (1958).
50. Hall R. H., R h о r a n a H. G., J. Am. Chem. Soc., 77, 1871 (1955).
51. S t on e H., S h e c h t e r H., J. Org. Chem., 15, 49_1 (1950).
52. С о p e A, C. et al,, J. Am, Chem. Soc., 87, 5452 (1965).
ПОЛИФОСФОРНОЙ КИСЛОТЫ ЭФИРЫ (ПФЭ), CsH20O12P4.
Мол. вес 432,14.
Для получения П. к. э. [1] смесь 150 г фосфорного ангидрида
(Р4О10), 150 мл эфира и 300 мл хлороформа кипятят до получения
прозрачного раствора (15—30 час), фильтруют через стеклянную
вату и растворитель упаривают на роторном испарителе.
Продукт, первоначально полученный Лангхельдом [21 и обычно
называемый эти л метафосфатом, Ретц и Тило [3] рассматривают как
смесь циклических эфиров (2) и (3). Последующее исследование Кел-
вина и сотр. [41 методом ЯМР-спектроскопни показало возможное
присутствие, кроме циклического, еще и эфира с открытой цепью.
PiQio (1)
О OEt
' R°
I OEt
p
EtO Р
ЧР
Etc/ "'О
(2) (3)
Реакции, особенно представляющие биохимический интерес, рас-
смотрены в обзоре [51. Под действием реагента аминогруппы прев-
ращаются в реакционноспособные фосфамидогруппы, конденсирую-
щиеся с карбоксильной группой. Аминокислоты и пептиды в присут-
ствии П.к.э. образуют высокомолекулярные пептиды в мягких усло-
виях и без рацемизации, П. к. э. активируют также гидроксильные
группы. Например, при обработке П. к. э. раствора о-глюкозы и
метилф-о-глюкопиранозида (2) в ДМФА глюкоза превращается в
«активированную глюкозу» (1), которая конденсируется с (2) с от-
щеплением метанола и дает с хорошим выходом дисахарид, идентифи-
цированный хроматографически как чистая целлобиоза (3). «Актн-
146
вированная глюкоза» не выделена, но известно, что это не глюкозо-1-
фосфат,так как этот эфир в аналогичных условиях не образует глюко-
зида. Под действием П. к. э. глюкоза полимеризуется в линейный
полиглюкозид со средним молекулярным весом 50 000. Аденозин
(з)
(6) получают с хорошим выходом конденсацией о-рибозы (4) с адени-
ном (5), Полинуклеотиды высокого молекулярного веса получают с
хорошим выходом из различных нуклеотидов в присутствии П.к.э.
Однако Хейес и Хансбури [6] установили, что, несмотря на поли-
меризацию 5'-тимидиловой кислоты с П.к.э., полимер с истинными
фосфодиэфирными связями (полинуклеотид) не образуется.
Реагент для циклодегидратации. Японские исследователи [7]
показали, что полученные вышеописанным способом П. к. э. в хлоро-
формном растворе являются прекрасным реагентом для реакции Биш-
лера— Напиральского. Так, фин ил этил амиды (1) гладко цикли-
зуются в дигидроизохинолины (2) прн кипячении с П. к. э. в хлоро-
форме. Фенилпропиламиды (3) в аналогичных условиях дают 3,4-ди-
гидро-5Н-2-бензопины (4). Сообщалось [81, что П. к. э. лучше,
(1) (2)
147
П.к.-СНСЦ
63-76%
В = II, осн3, сн3
чем ПФ К, в синтезах 2-замещенпых бензимидазолов, конденсацией
о-фен ил енд и амин а с карбоновыми кислотами. Алифатические кисло-
ты нагревают с амином и избытком П. к. э. при 100° в течение 10 мин
(выходы около 70%). В случае ароматических кислот необходимо
нагревание до 120° в течение 20—40 мин (выходы 50—60%). П. к. э.
с успехом используют в синтезе индолов из фен ил гидразонов по
Фишеру [9].
1. Pollmann W., Schramm G., Biochem.- Biophys. Acta, 80, 1 (1964).
2. Langheld K-, Ber., 43, 1857 (1910); 44, 2076 (1911).
3. R a t z R,, T h i 1 о E., Ann., 572, 173 (1951).
4. Burckhardt G., Klein M.P., Calvin M., J. Am. Chem. Soc., 87,
591 (1965).
5. Schramm G., Grotsch H., P о 1 1 m a n n W., Angew. Chem., Inter-
nal. Ed., 1, 1 (1962).
6. Hayes F. N., Hansbury E.,J. Am. Chem. Soc., 86, 4172 (1964).
7. Kanaoka Y., Sato E., Yonemitsu O., Ban Y., Tetrahedron
Letters, No. 35, 2419 (1964).
8. Kanaoka Y,, Sato E,, Y о n e m i t s u О., В a n Y., Chem. Pharm.
Bull., 12, 793 (1964).
9. Kanaoka Y., Ban Y., Yonem i tsu О., I r i e K.., AV i у a s h i t a
IG, Ciiem. Ind., 1965, 473.
ПРЕВО РЕАКТИВ, см. Иододибензоат серебра.
ПРОПАНДИТИОЛ-1,3, HSCH2CH2CH2SH. Мол. вес 108,23,
т. кип. 152°. Получение [11.
Сравнительные исследования Хауптмана [2] показали, что П.
образует тиокетали с более высоким выходом, чем этандитиол, од-
нако этот реагент нашел лишь ограниченное применение 13,4].
Например, [3].
В этом случае реакцию проводят в атмосфере азота, иначе образует-
ся очень цизкоплавкий продукт.
148
I. A u t e n г i e t h W,, Wolff R., Ber., 32, 1368 (1899),
2, Hauptmann H., Ca m p os M. M., J. Am. Chem. Soc., 72, 1405 (1950),
3. Archer S,, Lewis T. R., Martini С. M., Jackman M., J. Am.
Chem. Soc.,. 76, 4915 (1954).. ..
4. Sheehan J, C,, Coderre R. A., Cruickshan к P. A., J. Am.
Chem. Soc., 75, 6231 (1953).
ПРОПАНДИТИОЛА-1,3 ДИ-я-ТОЛУОЛСУЛЬФОНАТ (3).
Получение. По Вудварду и Патчетту, не опубликовано.
Применение [11. П. д. использовали для защиты положения 2
в холестеноне (1) при алкилировании в положение С4 в синтезе ла-
ностерина. Для удаления защитной группировки продукт алкили-
рования гидрируют над никелем Ренея.
1. Woodward R.B., Patchett A. A., Barton D.H.R., Ives
D. A. J., Rel 1 у R. В., J. Chem. Soc., 1957, 1131.
CHXHXO
Р-ПРОПИОЛАКТОН, ] 0J Мол. вес 72,06, т. кип. 51°/10 мм,
уд. вес 1,15.
Это реакционноспособное соединение было получено взаимодей-
ствием кетена с формальдегидом. Грешам, Джансен и сотр. [Г] опи-
сали применение и многочисленные реакции П. Баркли и сотр.
[21 использовали П. на одной из стадий полного синтеза стероида —
присоединение П. по Михаэлю к непредельной системе (1). После
гидролиза (2) выход (3) из. (1) составлял только 47%. Это превраще-
ние было осуществлено также с применением акрилонитрила, но
выход повысился лишь незначительно (22%).
149
Согласно новому методу карбоксиэтилирования [31, кетон (3)
превращают в енамин морфолина (4), при нагревании которого
с П. осуществляется конденсация и перегруппировка в амид (5),
В результате щелочного гидролиза морфолида получают 2-кето-
циклогексилпропионовую кислоту (6).
1. Gresham Т. L., Jansen J. Е., et al. (Papers I and XIII), J, Am. Chem.
Soc., 70, 999 (1948); 74, 1323 (1952).
2. В a г k 1 e у L. В., Far г а г M. W., Knowles W. S., R a f f e 1 s о n
H., Thompson Q. E., J. Am. Chem. Soc., 76, 5014 (1954).
3. S c h г о 1 1 G., К lemmensen P., La wesson S.-О., Chem. Scand.,
18, 2201 (1964).
ПРОПИОНОВОЙ кислоты ЭТИЛОВЫЙ ЭФИР,
СН3СН3СО3С2Н5. Мол. вес 102,14, т. кип. 99°, уд. вес 0,896.
При катализируемой алкоголятами конденсации ароматических
альдегидов с фталидом получаются 2-арилиндандионы-1,3 с выходом
всего около 34%. Однако при добавлении П. к. э. э. для связывания
образующейся воды выходы достигают 100% [1]. Сложный эфир бе-
рется в избытке и используется в качестве растворителя.
+ О=СНС6Н5 ♦ СН3СО2С2Н6 ♦ C2HsONa -lygc П.Р.^ЛА.,.»
100%(сырой)
I ЭКв 3 ЭКв 3 Жв
I же
150
Хотя в соответствии со стехиометрией этой реакции необходим
1 это этилата натрия для получения натриевого енолята продукта
и 1 же — для образования пропионата натрия, практически лучше
применять 3 жв. Для связывания воды можно применять и другие
пространственно не затрудненные сложные эфиры.
1. S h а р i г о S. L., G е i g е г К-, Freedman L., J. Org. Chem., 25, 1860
(1960); Shapiro S. L., Geiger Youlus J., Freedman L.,
ibid., 26, 3580 (1961).
ПФИЦНЕРА — МОФФАТТА РЕАГЕНТ, см. Диметил сульфок-
сид.
РАЗДЕЛЕНИЕ НА ОПТИЧЕСКИЕ ИЗОМЕРЫ, РЕАГЕНТЫ,
см. также 3|3-Ацетокси-А5-этиеновая кислота; ь-(-)-)-Бутандитиол-
2,3; Дегидроабиетиламин. Вельюз fl] и Илиел [2] опубликовали
обзоры методов и привели список реагентов и методики для их полу-
чения. Дополнительные методы и реагенты перечислены ниже.
В новом синтезе г-лизипа исходя из капролактама Бреннер и
Рикенбахер [3] разделили па оптические изомеры промежуточно
ьгнг
СНгСЫгСНСО2Н
НС1 I
—CH2CHzNHz- НС1
L
образующийся D.L-a-аминокапролактам с помощью ь-пирролидон-
карбоновой кислоты, полученной из l-глутаминовой кислоты. В ре-
зультате остроумной схемы кристаллизации, расщепления соляной
кислотой, рацемизации и вновь циклизации из г>,ь-а-аминокапро-
лактама получили примерно равные количества хлоргидрата ь-
лизина и о.ь-аминокапролактама и выделили обратно неисполь-
зованный реагент.
Ньюмен и Ледницер [4] разделили на оптические изомеры гек-
сагелицен (1) с помощью оптически активной комплексообразующей
л-кислоты (2), полученной из 2,4,5,7-тетранитрофлуоренопа, и оп-
тически активного гидроксиламина. й,/-Гексагелицен (1) и л-кис-
лота (2) (оба окрашены в светло-желтый цвет) образуют в бензоль-
ном растворе темно-красный комплекс. При обработке горячего
концентрированного бензольного раствора (1) и 0,5 же (—)-(2)
|52
контролируемым количеством эта иол а с последующим промыва-
нием смеси оказалось, что в осадок выпадают желтые пластинки
частично разделенного на оптические изомеры (—)-гексагелицена.
Повторное проведение этого процесса и перекристаллизация Дают
чистый (—)-гексагелицсн, [alD=:—3640°. При использовании
(-;')-(2) получают (Д)-гексагелицен, (а1в=+3707°.
Коуп и сотр. [5] разделили на оптические изомеры (+)-транс-
циклооктен (1) взаимодействием его с комплексом (2), полученным
нз этилена, хлорида платины(П) и (+)-1 -фенил-2-аминопропана.
Вытеснение этилена в этом комплексе циклоалкеиом приводит к сме-
си диастереомерных комплексов, которые можно разделить кристал-
лизацией при —20°. В результате выделения (—)-углеводорода из
комплекса действием цианистого калия получают препарат с [cd о--
=—411°.
I. Vclluz L.t Substances naturelle de synthese, 9, 119—174 (1954).
2. И л не л Э,, Стереохимия соединений углерода, изд-во «Мир», М., гл. 4, 1965.
3. Brenner M.,R i ekenb acher H. R_, Helv. Chini. Acta, 41, 181 (1958).
4. Newman M.S., L c d n i c e r D., J. Ain. Chem. Soc., 78, 4765 (1956).
5. С о p e A.C., Ganell in C, R., Johnson H. W., Jr,, Van Auken
T. V., Winkler H, J. S., J. Am. Chem. Soc., 85, 3276 (1963).
РАСТВОРИТЕЛИ, см. также Дильса — Альдера реакция, раст-
ворители. В таблице приведен 51 чистый Р. Они разделены на че-
тыре группы, в каждой из которых Р. расположены в порядке воз-
растания температур кипения.
Один из поставщиков растворителей в США отмечает, что их Р.
перегоняются в стеклянных достаточно эффективных колонках по-
сле необходимой физической и (или) химической обработки. Интер-
вал кипения не превышает 1—2°.-ДМФА, ДМСО и N-метилпирро-
лидон перегоняют в вакууме (т. кип. в вакууме не приведены
в таблице).. Неустойчивые Р. хранят в коричневых бутылях и, если
необходимо, в атмосфере азота. Большинство Р. достаточно чистые и
сухие и не содержат никакого нелетучего остатка. Исключения со-
ставляют диоксан и 2-метоксиэтанол, которые содержат 1-Ю-'1 %
антиоксиданта /г-бенз ил аминофенол а; хлороформ и диэтиловый эфир
стабилизируются этанолом. Для большинства Р. даются ИК- и
УФ-спектры и данные хроматографического анализа.
153
В современных методах анализа применяются Р. высокой сте-
пени чистоты для спектрофотометрии. Они не содержат примесей,
поглощающих в УФ-, ИК- и видимой области спектра. Высокая
степень чистоты и низкое содержание нелетучего остатка позволяют
Чистые продажные Р.
Растворители Т. кип., °C
Апротонные Р,, нерастворимые в воде
Изопентан (CH3)2CHCH2CHS 28
Пентан 35—36
Хлористый метилен 40—41
Петролейный эфир 30—60
Сероуглерод 46,3
Циклопентап 48,5—49,5
Неогексан (СН3)3ССН2СНз 49—50
Изогексан (СН3)2СНСН2СН2СН3 59—63
Хлороформ 60—61
Гексан 68—69
Четыреххлористый углерод 76—77
Бензол 80,1
Циклогексан 80,8
Дихлорэтан С1СН2СН3С1 83,8
1-Хлор-2,2-диметилпропан 84—85
Гептан 95—99
Изооктан (СНЭ)3ССН2СН(СН8)3 99—100
Мети лци к логексан 100,5—101,5
Толуол 109—111
Т етра хлорэтилен 120,8
м-Ксилол 138—139
Бромоформ 149,6
Апротонные Р., растворимые в воде а
Метилформиат (30 г при 20°) 32
Дпэтиловый эфир (7,5 г при 20°) 34,6
Этилформпат (11 е при 18°) 54
Этилацетат (8,5 г при 15°) 77—78
Метилэтилкетон (37 г при 25°) 79—80
Этилпропионат (2,4 г при 20е) 99,1
Нитрометан (9,5 г при 20°) 100—102
Изобутанол (10 г при 15°) 108—109
Изобутилацетат (0,6 г при 25°) 115—117
н- Б ути л ацетат (0,7 е при 25°) 126,5
154
Растворители
продолжение
Т. кип.. °C
Апротонные Р., смешивающиеся с водой
Ацетон
Тетрагпдрофуран
Ацетонитрил
Глим СН3ОСНаСНаОСН3
Диоксан
Пиридин
Диметилформамид
Диглим
Диметилсульфоксид
Триглим
1-Метилпирролидон-2
56—57
65,4
81—83
83
100—102
115,2
153
161
189
222
222
Протонные Р., частично растворимые в водеа
Бутанол-2 (12,5 г при 20°) Бутанол-1 (9 г при 15°) Изоамиловьш спирт (2,8 г при 15°) 99—100 116—118 131,5
Протонные Р., смешивающиеся с водой
Метанол
Изопропанол
2-Метоксиэтанол
Глицерин
64,7
82,3
123—124
290
а В скобках приведено количество вещества, растворяющееся в 100 г
воды.
использовать их в тех случаях, когда для получения малых коли-
честв продукта необходим большой объем Р.
Некоторые фирмы дают спецификации, включающие типичные
кривые поглощения каждого растворителя; другие выпускают спект-
рально чистые реагенты, для которых прилагаются данные спектро-
графического контроля.
Промывалки. В качестве промывалок для обычных Р. можно
рекомендовать полиэтиленовые фляги емкостью 500 мл. Название
и формулу Р. можно написать восковым карандашом. Объем до
25 мл можно отобрать из иих, выдавливая Р. через носик в мен-
зурку; большие количества наливают из центральной горловины.
155
Рис. Р-I. Полиэтиленовая про-
мыва л ка.
РЕЙНЕКЕ СОЛЬ, NH4[Cr(NH,)2(SCN)J -Н2О. Мол. все 354,47.
Получение 11].
Это ценный реагент для осаждения первичных и вторичных ами-
нов, пролина, оксипролина и некоторых других аминокислот.
1. Дэкнн X., «Синтезы органических препаратов», ИЛ, М., 1949, сб. 2, стр. 432.
РЕНЕЯ КАТАЛИЗАТОРЫ КОБАЛЬТОВЫЕ. Эти катализаторы
получают действием едкого натра на сплав кобальта с алюминием
(40 : 60) по той же методике, которую Биллика и Адкинс [ 1] исполь-
зовали для приготовления никеля Ренея W-7.
При гидрировании простых оксимов Р. к. к. менее эффективны,
чем никель Ренея (21. Оксим эфира 2-хииолилпировиноградной
кислоты (1) поглощает 2 моля водорода в присутствии никеля Ренея
I 1!ч j 2нг
ХХХСН2ССО2С2Н5 —
il н,
О) NOH
20% ХСН2СНСО2С2Н5
46% (2) NHz
Со’Ренся, 80°, 13 0 атя | || |
Х/ХСНаССО.СоН5
О NH
156
и дает эфир хинолилаланина (2), а в присутствии Р. к. к. реакция
завершается на стадии образования сс-иминопропиопового эфира
(3) [3]. Р. к. к. также менее активны, чем никель Ренея, в реакциях
десульфуризации и при гидрировании нитрилов [5].
1. Б и л л и к а Г., Адкинс Г., «Синтезы органических препаратов», ИЛ,
М., 1953, сб. 4, стр. 349.
2. R е с v е W., Christian J., J. Am. Chem. Soc., 78, 860 (19o6).
3. Ried W., Schiller H., Chem. Ber., 86, 730 (1953).
4. Badger G. M., Kowanko N., S a s s e W. H. F., J. Chem. Soc., 1959,
440.
5. Gould F. E., Johnson G. S., Ferris A. F., J. Org. Chem., 25, 1658
(1960).
РЕНЕЯ КАТАЛИЗАТОРЫ НИКЕЛЕВЫЕ. Эти катализаторы
поступают в продажу в виде 50 %-ного губчатого никеля, получае-
мого по методу Ренея смешиванием никель-алюминиевого сплава
(1 : 1) с щелочью, а также в виде кизельгуровых гранул, обраба-
тываемых водородом и измельчаемых перед употреблением
[см. Org. Syn., Coll. Vol., 3, 278 (1955)].
Получение. Способы получения катализатора из никель-алю-
миниевого сплава Ренея различаются между собой по методу при-
бавления сплава, концентрации едкого натра, температуре, продол-
жительности перемешивания и, наконец, по методу промывания ката-
лизатора для удаления алюмината натрия и щелочи. Адкинс и сотр.
получили несколько катализаторов: W-ПП, W-2 12], W-3 13, 4],
W-4 13, 41, W-5 [5], W-6 [5, 6] и W-7 [5, 6] (номера отражают глав-
ным образом хронологию их открытия). Адкинс и Крзек [7] срав-
нили активность этих катализаторов па примере гидрирования
(3-нафтол а. Чаще других употребляются катализаторы W-2 и W-6;
W-5, хотя и менее активный, считается отличным катализатором.
Утверждается [81, что активность увеличивается при добавлении
небольшого количества хлор платината триэтиламина.
Обычно гидрирование проводят при умеренно высоких давле-
ниях водорода. Нагреванием 50%-ного губчатого никеля с 10%-ным
раствором едкого натра Домингес [91 получил очень активный ката-
лизатор Т-1, который может быть использован при низких темпе-
ратурах (40—60°) и низких давлениях (3,5—4,2/щ/с.и2), например: фе-
нол ц и кл о ге кса н о л (92 %); к у ма р и н 3,4-диги дроку мар и и (83 %).
Восстановление С =С и С=С. Ньюмен и др. [10] показали, что
резорцин можно прогидрировать в щелочном растворе в присут-
ствии катализатора W-2; после этого катализатор тщательно промы-
вают для удаления алюминия 5%-ным раствором щелочи и затем
дистиллированной водой.
ОН О
I 11
//\. Води. NaOH, Н» (133 атм) /\
. I II 9 II
157
Фили и Бокелхайд [11] использовали 5 г катализатора W-2
для гидрирования 108 г этоксиметиленмалонового эфира.
N1, Н^ (70—105 атм)
САОСН-С(СОгС,НЛ С^ОСНгСНтед),
0,5 моля Нагревание]. 91 — 94%
СНа=С(СО2С2И5)3
Р. к. н., дезактивированный пиперидином и ацетатом цинка,
был использован для частичного гидрирования тройной связи [12].
Эрман и Флаутт [13] пытались прогидрировать и-анизилборни-
лен (1) в присутствии Pt и Pd в качестве катализаторов в различных
растворителях, однако неизменно получали смеси эндо-и экзо- про-
дуктов. Использование катализатора W-5 привело к восстановле-
нию как ароматического кольца, так и двойной связи. Однако при
применении 50%-ного губчатого никеля (см. выше) с хорошим вы-
ходом был получен однородный экзо-днгидрид.
24
72%
Альдегиды. Реакционную смесь, содержащую диальдегид, нейт-
рализовали твердым бикарбонатом натрия, помещали в автоклав
и гидрировали в присутствии 39 г Р. к. н. W-2 [14].
СН3
2,6 моля
СН3
NaHCOy N i- Н2 (114 J2S^ pj0CHХНСН.2СН2ОН
RNO2-^RNH2. Дау бен и др. [15] гидрировали 1-нитрометил -
циклогексанол в 450 мл уксусной кислоты в присутствии трех пол-
158
ных чайных ложек катализатора W-4 при очень низком давлении,
поддерживая температуру смеси при 35° внутренним охлаждением.
НО CH2NO2 но ch2nh2
NaOCH3 AcOH.Ni,
Ас ОН Н2(2,8 -ЗДЛ7Л^
35°
2 МОЛЯ
hno2
40-42% (общий)
Для получения промежуточного соединения, необходимого
при синтезе 5,5-диметилпирролидона, Моффетт (161 использовал
катализатор W-5 и абсолютный этанол в качестве растворителя.
Ni, Нг (/Ойготлт)
(СН3)гССН2СН2СОгСН3 -----------------*(сн3)гссн2сн2со2сн3 - -Д-»
I I ac’vDw
no2 nh2
(сщ),
н
Амины. Первичные амины RNH2, в которых R—алкил, аралкил
или циклоалкил, при кипячении с никелем Реиея (без водорода)
превращаются во вторичные (80%) и отчасти в третичные амины
(10%) [171. н-Гексиламин в тех же условиях дает ди-н-гексиламин
(70%) и трн-я-гексиламин (9%). Считают, что реакция протекает
через дегидрирование в альдимин с последующим присоединением
исходного амина, отщеплением аммиака и гидрированием.
RCH.NH, -СЛд. RCH —NH .. RCHNH2------>
| -NH,
HNCHoR
RCH 2Н RCH2
11 |
nch2r hnch„r
Алкилирование первичных аминов можно осуществить Р. к. и.
(W-2) в присутствии этанола, как при получении N,N'-диэтилбен-
зидина [ 18J. Смесь в процессе кипячения энергично перемешивают.
___ Ni (125 г),
H.N-У Ч--------^~y_NH.-|-2C.H,OH
\=/ \=./ ' “ ‘ 47—52%
1 моль 500 ,ил
н н
C2H6N ^>-NCaH6
159
Бэлком и Нолер [19] получили вторичный амин конденсацией
метиламина с альдегидом в условиях гидрирования. Катализаторы
W-2 и W-6 были эффективны в этой реакции. Метиламин брали
в трехкратном избытке; при эквимолярных соотношениях реаген-
тов выход уменьшался до 75%.
, СНО
Гн
^/хосн3
осн3
0,25 моля
+ CH3NHS
0,75 ноля
Кипячение, этанол-
вода, Ni-H2 (3 атм)
80—93%
ch2nhch3
''У^ОСНз
осн3
Нитрилы. Ароматические [201 и алифатические [211 нитрилы
при гидрировании с никелем Ренея в этаноле, содержащем фенил-
гидразин, восстанавливаются в альдегиды, выделяемые в виде
фен ил гидразонов (8 час при атмосферном давлении или 5 час
под давлением). Выходы составляют 30% при использовании 1. же
фенилгидразина, но увеличиваются до 90% при применении 4 же
этого реагента.
Гидрирование нитрила при обычных условиях осложняется
тем, что образующийся первичный амин конденсируется с проме-
жуточно образующимся альдимином, давая вторичный амин. Гоулду,
Джонсону и Феррису [22] удалось исключить эту побочную реакцию
проведением гидрирования в уксусном ангидриде, так что первич-
RC = N RCH-NH 2Л. RCH2NH2
--------—> (RCHa)2NH
ный амин ацетилировался по мере образования. 2—3 г сырого
Р. к. н. или никель-хромового катализатора Ренея, необходимого
для восстановления 0,1 моля нитрила, непосредственно-перед ис-
пользованием отфильтровывают от воды и промывают двумя пор-
циями абсолютного этанола по 20 мл каждая и двумя порциями
уксусного ангидрида по 20 мл. Оба катализатора обладают приб-
лизительно одинаковой эффективностью. В качестве основного со-
катализатора обычно используют ацетат натрия; с едким натром
Ас2О, 5 0°
CH^CHA^CN + NffH^ I-NaOAc -----------> CH3(CH2)nCH3NHAc
1 оо%
0,1 моля 2-3 г 12г
как сокатализатором реакция протекает очень быстро, и металличе-
ский катализатор может быть вновь использован. С некоторыми
нитрилами типа бензонитрила сокатализатор не требуется. Выше-
приведенное восстановление (тридеканнитрил) проводили при на-
чальном давлении водорода 3,5 атм, поглощение заканчивалось
приблизительно через 1 час. Амин выделяли в виде ацетата или гид-
ролизовали его соляной кислотой. При гидрировании нитрилов
160
с Р. к. н. в водной уксусной кислоте или в смеси водной уксусной
кислоты с пиридином в присутствии гипофосфита натрия (NaHaPO2),
окисляющегося в фосфат, они превращаются в соответствующие
альдегиды [23]. Реакция протекает при комнатной температуре и
атмосферном давлении. Выходы составляют 50—90%. Например,
раствор 1 г бензонитрила и 2 а водного гипофосфита натрия в 29 мл
смеси вода — уксусная кислота — пиридин (1:1:2) перемешивают
с 0,3—0,4 г Р. к. и. при 40—-45° в течение 1 час; бензальдегид выде-
ляют в виде 2,4-динитрофенилгидразона.
C6H5CN + NaH2PO3+Ni(II2) —СаН5СНО
U 7у
Превращение галогенидов и тозилатов в углеводороды. Р. к. н.
в щелочном растворе осуществляет восстановительное замещение
фтора, хлора, брома и иода в ароматических и алифатических кис-
лотах и фенолах (60—70%) [241. При проведении реакции в D2O
получают дейтерированные производные. В этих условиях трифтор-
метильная группа, не подвергающаяся гидрированию обычными
методами, восстанавливается в метильную группу [25].
СО2Н СО3Н
II + Ы.{Щ) Вод». NaOH, 3^ | ||
FA/ 30 г 70% U
10 г
Кеннер и Мэррей [26] показали, что гидрированием Р. к. н.
при комнатной температуре и атмосферном давлении можно восста-
новить фенилтозилаты в соответствующие углеводороды. Сущест-
вуют и другие примеры [27]. Тозилаты алифатических спиртов гид-
ролизуются главным образом в соответствующие спирты.
OTs
’ ,СО2СН3 / /ХЦСНз
|| I Ni, Нд, СД-ЦОН | ||
Десульфуризация. В обзоре по десульфуризации Р. к. и. Петтит
и Ван-Тамелен [28] отдают должное Бу го [29], впервые наблюдав-
шему в 1940 г. реакцию, которая приобрела большое значение как
для синтеза, так и для расщепления. Проводя подготовительную
работу для исследования сер у содержащего биотина, Мозинго и др.
[30] показали, что сульфиды, дисульфиды, сульфоксиды и суль-
фоны при реакции с Р. к. и. подвергаются гидрогенолизу с замеще-
нием атома серы на два атома водорода. Добавления водорода не
требуется, так как значительное количество его поглощается ката-
лизатором или растворяется в нем. Показано, что на 4 з катализатора
6 Заказ № 11 G6
161
W-2 приходится от 170 до 460 мл водорода, в зависимости от темпе-
ратуры в конце получения катализатора. Пирофорный Р. к. н. из-
вестен как эффективный агент для удаления из субстратов примесей,
вероятно содержащих серу и отравляющих другие катализаторы
гидрирования [311. Десульфуризация биотина (1) в дезтиобиотин
1321 сыграла главную роль в установлении структуры витамина.
(и
Бэдгер [331 и Бу-Хой [341 широко использовали десульфуриза-
цию никелем в синтезе. В одной общей схеме используются произ-
водные тиофена [331. Например, у-2-тиепплмасляную кислоту
[(1), сукциноилирование тиофена и восстановление! ацетилируют (в
виде сложного эфира) в положение 5 (2) и восстановлением по методу
Хуанга — Минлопа и десульфуризацией W-6 превращают в капри-
новую кислоту (5). Высшие гомологи можно получить ацилирова-
нием (1) соответствующими ацил хлор идами. Ацетокислоту (2) можно
окислить в дикислоту (3), которая при десульфуризации дает азелаи-
новую кислоту (4). Десульфуризацию этих кислот удобно проводить
----смоа* lj—j|
[^ _Д(СНЩСО2Н SnCU СН3СО[^ ^(CHJjCOsH
s s
В) (2)
NaOBr
по Вольфу-Кижнеру:Ni
1
но:с
(СН2)3СО2Н
(3)
—> HOjCXCH^tCOjH
СНдСН^ьСО^Н
(4)
(5)
в водном растворе карбоната натрия; при этом обычно получают
очень высокие выходы. Имеются и другие примеры [351. Бэдгер и
сотр. [36] применили этот метод для синтеза 2,3,10,11-дибенз-
перилена (3). Тетрафенилтиофен (1), легко получаемый нагреванием
тетрафенилциклопентадиенона с серой, при нагревании с расплав-
ленным хлор алюминатом натрия претерпевает дегидрирование
с циклизацией в флавофен (2). Десульфуризация, осуществляемая
кипячением флавофена с дезактивированным Р. к. н. в мезитилене,
дает гептациклическпй углеводород (3).
162
Превращение кетона в этилентио кета ль и десульфуризация ни-
келем иногда полезны для селективного удаления карбонильной
вг3
HSCH2CIi2SH
группы после того, как она сыграла специфическую роль, как,
например, в синтезе Л9(1,)-холестенола из А7-холестенилацетата (1)
1371. Мопоэтилентиокеталь (8) десульфуризуется кипячением в те-
чение 22 час 147 мг вещества в 60 мл метанола с 3 г Р. к. н., получен-
ного из 6 г сплава. Мозинго, Спенсер и Фолкерс 138] показали, что
при десульфуризации часто протекают побочные реакции. Так,
они установили, что при обычных условиях десульфуризации про-
6*
163
исходит восстановление двойных связей, карбонильных и нитро-
групп, азоксибеизолы и гидразобензолы претерпевают восстано-
вительное расщепление связи 14—N, а бензиловый спирт дает
толуол. Для того чтобы избежать этих побочных реакций, в после-
дующих работах [39] дезактивировали катализатор кипячением его
с ацетоном. Однако этот прием не всегда эффективен и побочные
реакции по-прежнему вызывают осложнения. Например, Джерасси
и Уильямс [40] получили меченый 5 а-андростаион-11 (2) десульфу-
ризацией этилентиокеталя (I) Р. к. н., содержащим активный дей-
терий, однако образующийся в качестве основного продукта АI * 3-
олефин приводит к дезактивации катализатора. Аналогичным
образом при взаимодействии с умеренно активным катализатором
W-2 17-этилентиокетали дают в основном А10-стероиды, выделяе-
мые с выходами 65—75% [41].
В синтезе о-ди-трет-бутилбензола Бергсталер и Абдель-Рахман
[42] провели последнюю стадию — десульфуризацию сначала со
свежеприготовленным Р. к. н. W-6 в этаноле с перемешиванием
при 45'—50°, однако выход был низок из-за восстановления арома-
тического кольца. В этом случае дезактивация катализатора кипя-
чением с ацетоном сделала его неэффективным для десульфуризации.
В появившейся позднее работе [43] был предложен следующий
упрощенный метод приготовления Р.к.н., в присутствии которого
десульфуризация в кипящем этаноле осуществляется за 10 мин,
при этом легко восстанавливаемое ароматическое кольцо подвер-
гается лишь незначительной атаке [43]. 150 г порошка сплава добав-
н3с. ,сн3 Н3С ,СН3
//\/CCrwn LiAlH.,. TSOCI, /с<снз)з
p с7 8 СНО н-BuSK p II * * 4CH.2SBu-h ж p и *
I II ZCHO I II /CI-I2SBu-H * L jl
^/^C(CH3)3
H3(/ XCH3 H3CZ ZCH3
ляют в течение 30 мин при перемешивании к раствору 195 г едкого
натра в 750 мл воды при 75°. После этого смесь перемешивают еще
в течение 30 мин при постепенном понижении температуры. Отстояв-
шуюся жидкость декантируют и дистиллированной водой перено-
сят катализатор в одно- или двухлитровый цилиндр с делениями,
который затем помещают в раковину. Через трубку длиной 7 или
8 мм, достигающую дна цилиндра, пропускают дистиллированную
164
воду с такой скоростью, чтобы всплывший металл находился в 5—
7 см от верхнего края цилиндра. Через 10—15 мин промывные во-
ды дают нейтральную реакцию по индикаторной бумажке. После
этого трубку вынимают, воду декантируют и заменяют этанолом.
Десульфуризация применима при синтезе гетероциклов, в ко-
торых желаемая циклическая система наиболее легко может быть
построена в виде сульфгидрильного производного. Примером может
служить синтез 4-метил-6-оксипир имид ина через 2-тио-6-метилу-
рацил [44]. Сырое тиопроизводное было десульфуризовано 45 г
Р. к. п. (влажная паста).
СН3
CH3CZ ХСОСЧН5
II II
о + о
Н3\\ /NH,
с/
NaOEL
69-84%'
Ni(Ha)-H2O-NH+OH,
кипячение 1,5 час
90-93%
0,07 ноля
Диспропорционирование. Клейдерер и Корнфельд [451 показали,
что никель Ренея W-2 в присутствии акцептора водорода (циклогек-
санон) катализирует дегидрирование вторичных спиртов. Холесте-
рин при кипячении с 2 ч. катализатора и циклогексанона в толуоле
Кипячение в
С6Н5СНЭ 24тас
Никель Ренея
80%
дает холестеион; флуоренол превращается в флуоренон с 76%-ным
выходом. В присутствии донора водорода (циклогексанол, диэтил-
карбинол) металл катализирует восстановление карбонильных
групп и активированных двойных или тройных связей, например:
транс-стильбен-*-дибензил; холестанон->холестапол (выход 50%).
СнН5С—Н Н. /ОН
И “Г /X
Н—С — СГН- ( % Кипячение с СвН6СН3) 22 час
03 Р.к.ц.
60%
О
Jl^
С6Н5СН3СН3СаН5 + | |
165
Дегалогенирование. Ароматическую трифтор метильную группу
можно восстановить в метильную группу либо Р. к. и., либо ко-
бальтом Ренея [461.
Дегидрирование. Сассе [471 описал дегазированный Р. к. н.
и его применение для превращения пиридина в 2,2'-дипиридил.
Неренеевские никелевые катализаторы. Тиман [481 при обра-
ботке никель-алюминиевого сплава (1 : 1) водой при 70° получил
катализатор приблизительного состава Ni/Al2O3. Для этого ката-
лизатора не требуется интенсивного промывания, и он не подвер-
жен влиянию следов щелочи.
Ляппорт и Шуэтт [49] получили исключительно эффективный
для гидрирования ароматических соединений катализатор, добав-
ляя триэтилалюминий к раствору двухвалентной соли никеля
2-этилкапроновой кислоты в бензоле или гептане. При этом проте-
кает экзотермическая реакция с выделением газа, на 95% состоя-
щего из этана, и образованием черного непирофорного раствора
катал итичес к и а ктивного компл екса.
1. С о v с г t L. W., Adkins IL, J. Am. Chem. Soc., 54, 4116 (1932).
2. Mozingo R., Org. Syn., Coll. Vo]., 3, 181 (1955).
3. P a v 1 i c A. A., Adkins IL, J. Am. Chem. Soc., 68, 1471 (1946).
4. Adkins IL, P av 1 ic A. A., J. Am. Chem. Soc., 69, 3039 (1947).
5. Adkins FL, В i 1 ] i с a FL R., J. Am. Chem. Soc., 70, 695 (1948).
6. В i 1 1 i с a H. R., A <1 k i n s 14., Org. Syn., Coll. Vo]., 3, 176 (1955).
7. Adkins H., К rsek G., J. Am. Chem. Soc., 70, 412 (1948).
8. Levering D.R., Lieber 13., J. Am. Chem. Soc., 71, 1515 (1949),
9. Dominguez X. A., Lopez I.C., Franco R., J, Org. Chem., 26,
1625 (1961).
10. Mekler A. B., Ramachandran S., Swaminathan S., N e w-
man M.S., Org. Syn., 41, 56 (1961).
IL Feely W., В ocke 1 h e i de V-, Org. Syn., Coll. Vol., 4, 298 (1963).
12, Or oshn 1 k W., Karmas G-, Mebane A. D., J. Am. Chem. Soc,,
74 295 (1952).
13. E r m a n W. F,, F 1 a u t t T. J., J. Org. Chem., 27, 1526 (1962).
14. L о n g ] e у R. L, Jr., Emerson W. S., Org. Svn., Coll. Vol., 4, 660
(1963).
15. D a u b e n H. J., Jr., R i n g о ] d H. J., Wade R. H., Pearson
D. L., Anderson A. G., Jr., Org. Syn., Coll. Vol., 4, 221 (1963).
16. Moffett R. B., Org. Syn., Coll. Vol., 4, 357 (1963).
17. KJ n d 1 e r K., Melamed G., M a t t h i e s D., Ann., 644, 23 (1961).
18. R i c e R. G., Kohn E. J., Org. Syn., Coll. Vo]., 4, 283 (1963).
19. В a 1 с о m D. M., Nollcr C. R., Org. Svn., Coll. Vol., 4 603 (1963).
20. G a i f f e A., P о 1 ] a u d R., Compt. rend.“ 252, 1339 (1961).
21. G a i f f e A., P о 1 1 a u d R., Compt. rend., 254, 496 (1962).
22. Gould F. E., Johnson G. S., Ferris A. F., J. Org. Chem., 25,
1658 (1960); Ferris A. F., G о u ] d F. E., J ohnsonG.S., Stange
H., J. Org. Chem., 26, 2602 (1961).
23. В a c k e b e r g O. G., S t as k u л B., J. Chem. Soc., 1962, 3961.
24. В u u-I-J о i N. P., X uong N. D., van В a c N., Bull. soc. Chim. France,
1963, 2442.
25. В u u-l-l о i N-Р., Xuon g N. D., van Вас N-, Compt. rend., 257, 3182
(1963).
26. Kenner G. W., Murray M. A., J. Chem. Soc., 1949, S178.
27. S a r i п P. S., Scshadri T. R., Tetrahedron, 8, 64 (1960); C a s p i E.,
Cullen E., Grover P. K., J. Chem. Soc., 1963, 212.
166
28. Р е t t i ( G. R., van TamelenE. E., Org. Reactions, 12, 356 (1962).
29. Bougault J., Cattelain E., Chabrier P., Bull. soc. chim.
France (5), 7, 781 ([940).
30. Mozingo R., Wolf D. E., Harris S. A., Folkers !<., J. Am.
Chem. Soc., 65, 1013 (1943).
31. Adkins H., «Reactions of Hydrogen», The University of Wisconsin Press,
Madison, Wisconsin, 1937, p. 28,
32. Du Vignea u d V., Melville D. B.; F oik ers K-, Wolf D. E.,
Mozingo R.f Keresztesy J. C., Harris S. A., J. Biol. Chem., 146,
475 (1942).
33. Badaer G. M., Rodda H. J., S a s s e W. H. F,, J. Chem. Soc., 1954,
4162.
34. В u u-H о i N. P. et al., J. Chem. Soc., 1954, 1975; Compt. rend., 240, 442,
785 (1955); Bull. Soc. chim. France, 1955, 1175, 1583.
35. M i 1 1 e r I<. E., Haymaker C. R., Gilman H,, J. Org. Chem., 26,
5217 (1961); McGhi e J. F., R о s s W.A., Evans D., T о m 1 i n J. E.,
J. Chem. Soc., 1962, 350.
36. Badger G. M., Christie B.J., Pryke J. AU, Sasse W. H. F,,
J. Chem. Soc., 1957, 4417.
37. F i e s c r L. F., Huang W.-Y., J. Am. Chem. Soc., 75, 5356 (1953).
38. M о z i n g о R., Spencer C., Folkers I<., .1. Am. Chem. Soc., 66,
1859 (1944).
39. S p e г о G. B. et a]., J, Am, Chem. Soc., 70, 1907 (1948); Rosenkranz
G. et al., ibid., 71, 3689 (1949); Barkley L. B. et al., ibid,, 76, 5017 (1954).
40. D j e r a s s i C., W i 1 1 i a m s D. FL, J Chem. Soc., 1963, 4046.
41. Fishman J.,Torigoe M-, G u z i к H., J. Org. Chem., 28, 1443 ([963).
42. В u r g s t a h 1 e г A- W., Abdel-Rahman M. O., J. Am. Chem. Soc.,
85, 173 (1963).
43. Private communication from Burgstahler A. W.
44. Foster H. M-, Snyder H. R., Org. Syn-, Coll. Vol,, 4, 638 (1963),
45. I< 1 e i d e r e r E. С., К о r n f e 1 d E, C., J. Org. Chem., 13, 455 (1948).
46. В u u-H о i N. P., X uong N. D., Вас N. V., Compt. rend., 257, 3182
(1963).
47. Sasse W. H. F., Org. Syn., 46, 5 (1966).
48. T у m a n J. H, B., Chem. Ind., 1964, 404.
49. Lapporte S. J.,Schu ett W. R,, J. Org. Chem., 28, 1947 (1963).
РЕНЕЯ НИКЕЛЬ, см. Никель-алюминиевый сплав.
РЕНИЕВЫЕ КАТАЛИЗАТОРЫ. Бродбент и сотр. 11] опубли-
ковали методы получения ряда окислов рения (Re.2O7, ReCU, ReO3,
ReO), которые используются в качестве катализаторов гидриро-
вания, особенно при восстановлении карбоновых кислот в первич-
ные спирты. Для восстановления ароматического ядра, двойной
связи, карбонильных и нитрогрупп Р. к. менее эффективны, чем
никелевые или платиновые; следовательно, можно проводить се-
лективное гидрирование. Бензильные оксигруппы устойчивы к гид-
рогенолизу.
I. Broadbent FI. S, et al., J. Org. Chem., 24, 1847 (1959); 27, 4400 (1962);
28, 2343, 2345, 2347 (1963).
РЕНИЯ ГЕПТАСЕЛЕНИД, Re2Se7. Бродбент и Уайттл 11]
описали приготовление этого катализатора гидрирования и указали,
что он более активно восстанавливает группу С=О, чем связи С=С
167
(если только последние не сопряжены). Катализатор не вызывает
Гидрогенолиза связи С—S.
1. Broadbent Н. S., W h i t t 1 e C. W., J. Am. Chem. Soc., 81, 3587 (195?).
РОДИЙ (5%) НА окиси АЛЮМИНИЯ, Rh — AlaO:i.
Гидрирование фенолов. Катализатор обладает тем преимущест-
вом, что промотирует гидрирование фенолов при минимальном гид-
рогенолизе. Бергсталер и Битое [1] гидрировали галловую кислоту
в условиях, указанных ниже, и выделили полностью цис-гексагид-
о, 266 моля в
этаноле
+ Rh—А12О3
8 г
Н2 ((Ьбатг/лт)
90-100°, 8-12 час
38-43%
ОН
СО2Н
рогалловую кислоту в качестве единственного кристаллического
продукта реакции. Выход более чем в 2 раза превышал суммарный
выход (13—19%), полученный ранее по двухстадийной методике:
сначала галловую кислоту гидрировали с никелем Ренея в щелоч-
ном растворе до дигидрогалловой кислоты, которую затем подвер-
гали гидрированию над платиновым катализатором. Смит и Стамп [21
и Кей и Мэтьюс [31 также добились лучших результатов при гидри-
ровании одно- и многоатомных фенолов действием Rh—А1ЙО3, чем
Юг в этаноле
Н2 (0,4 amjvi}
25° 12 час
62%
при использовании других катализаторов. Мейерс и сотр. [4] ус-
тановили, что этот катализатор безусловно лучше никеля Ренея или
платины при превращении а-нафтола в цме-цме-декалол-1. В ре-
зультате гидрирования в этаноле в очень мягких условиях и двух
кристаллизаций был получен чистый ис-спирт. ГЖХ показа-
ла, что гидрогенолиз прошел лишь примерно на 3%.
Низкотемпературное гидрирование резорцина в присутствии
Rh—А12О3 в щелочном растворе является удобным методом полу-
чения циклогексаидиона-1,3 [5].
110\/\/он
| || + NaOH-j-H,
85%
Rh-А1гОз>
ЗД атм. 25°, 16 — 18 час
168
Гидрирование бензиловых спиртов. Стокер [6] показал, что гид-
рогенолиза удается полностью избежать при применении Rh—А12О3.
В очень мягких условиях из (+)-миндальной кислоты получили
СО2Н
I
снон
112 (3 — 4 атм),
25°, 1,5 час
I || -у Rh —А].2О3
0,05 моля
н 40 мл СН3ОН + 0,5 мл
с н,со ОН
(±)-гексагидроминдальную кислоту, а из D-миндальиой кислоты —
чистую d-гексагид ром инд ал ьну го кислоту. Отмечают, что при ра-
боте по этой методике требуется небольшое количество уксусной
кислоты. Повторив работу Стокера с добавкой уксусной кислоты,
Райландер [7] нашел, что восстановление протекает именно так,
как это было описано; при удалении уксусной кислоты восстанов-
ление полностью прекращается.
Гидрирование связей ('=(', C==NOH, C=N. Хэм и Кокер 18]
показали, что Rh—А12О3 является лучшим катализатором гидри-
рования винил- и аллилгалогенидов при минимальном гидрогено-
лизе, например:
ОСН = СНСН,С1 + Rh — Д12О3 . О(СН2)3С1 Щ СН3СН2СН2С1
0,334 моля 2,4 г " 48% 34%
Оксим циклогептанона можно превратить с хорошим выхо-
дом в амин гидрированием оксима при высоком давлении в при-
сутствии никеля Ренея и аммиака 19], однако, согласно данным
Фрейфелдера ПО], эта реакция настолько экзотермична, что при
проведении опытов в большом масштабе реакцию трудно контро-
лировать. Кроме того, он установил, что гидрирование протекает
NOH
+ НИ—A.I-.O3
450 г
30,6 моля
В метаноле
Н2 (1 <?тж),6о°
80% >
гладко при низком давлении в присутствии Rh—А12ОЯ. Темпера-
туре давали повыситься самопроизвольно до 60° и эту температуру
поддерживали до завершения восстановления. Фрейфелдер III],
восстановил 16 алифатических нитрилов в первичные амины
с хорошими выходами гидрированием в аппарате Парра над
169
Rh—А120я при комнатной температуре и низком давлении. Особый
интерес представляет восстановление 3-индол ацетонитрила в трип-
тамин в мягких условиях, поскольку гидрирование этого соедине-
0,15 моля
в смеси 10%-ного
этанола с аммиаком
ch2c=n Н2 (0,2атм)
, nV. л 1 л 25°,2 час .
+ R h — Al 2О,----------->
78%
4 г
СНг CH2NHz
нпя в присутствии других катализаторов при более высоких темпе-
ратурах сопровождается интенсивно протекающими побочными
реакциями.
Гидрогенолиз кеталей [121. В присутствии Rh — А12ОИ и следов
соляной кислоты кеталь претерпевает восстановительное расщепле-
ние и образуются простой эфир и спирт. Платина и рутений в этих
случаях почти неактивны, а палладий примерно в 2 раза менее ак-
тивен, чем родий.
СН3 /ОС6Ни
;Cy -j-Rh — АЦО3 -j- НС1 (следы) —>
СН3' ЮС6НП
Щ (2,8 шм), 25°, 3 чк СВ;!. /Н
Гидрирование гетероциклов. Для синтеза одного алкалоида
Рапале и сотр. 1131 требовалась цпе-декагидроизохинолинкарбоно-
вая кислота (2), и им удалось ее получить гидрированием 1,2,3,4-
тетрагидроизохинолин-З-карбоновой кислоты (1) в присутствии
Rh—А12О3.
Rh-Al,O3
5 г
Н, (1 5 атя},
50—100°
20 с в 5%-ном СН3ОН
(1) (2)
Фрейфелдер [141 установил, что пиридинкарбоновые кислоты
можно гладко гидрировать над Rh—А12О3 в аммиачном растворе.
Наиболее наглядным примером является гидрирование никотино-
вой кислоты в нипекотиновую, поскольку ранее эту реакцию не уда-
валось осуществить так, чтобы избежать декарбоксилирования.
I I + Rh —А1,О3
0,05 моля
в 50 мл Н2О+5 мл водн. NH3
170
Осуществляя синтезы алкалоидов крестовника, Адамс н сотр.
[ 15] получили 1-оксипирролизидин определенного стереохимического
строения (2) путем гидрирования (1) с Rh—А12ОЯ; при этой реакции
Rh-A12Oj
(1) 14 г в АсОН
Н7 ( 3 атм)
2 5°, 2 vac
64%
(2)
происходит полное насыщение диеноновой системы. Среди про-
дуктов реакции был обнаружен только один стереоизомер. Другие
исследователи безуспешно пытались восстановить (1), проводя гид-
рирование с окисью платины или с палладием на угле.
Катализатор Rh—ALO3 был успешно использован и для полного
гидрирования хиноксалина [16], а также для восстановления двой-
ной связи в положении 4,5 в пиримидиновом ядре нуклеозидов и
нуклеотидов [171.
Дегидрирование. Ньюмен и Ледницер [18] нашли, что Rh—А12ОЯ
является эффективным катализатором для реакции переноса водо-
рода от гексагидрогексагелицена (1) к бензолу с целью получения
гексагелицена (2). Аналогичные реакции обмена в присутствии
палладиевого катализатора протекают с плохими выходами. При
Rh—A12Oj
СДЩЗОС^
73%
(2)
кипячении гексагидрогексагелицена с 2,3-дихлор-5,6-дициан-1,4-
бензохиноном в ксилоле в течение 7 час образуется дигидропроиз-
водное, которое далее не дегидрируется. Обмен водорода с бензолом
в присутствии Rh—А12О3 представляет собой также наилучший ме-
тод дегидрирования (3) в пироцен (4). Андерсон и Андерсон [191
нг
171
помещали запаянную трубку, содержащую 0,24 г (3), 0,1 г катализа-
тора и 15 мл бензола, во вращающийся автоклав, наполненный стек-
лянной ватой и загруженный бензолом, и смесь перемешивали
при 290Г
Гидрирование арилами нов. Rh — А12О3 является, по-видимому,
лучшим катализатором для низкотемпературного гидрирования
анилинов, особенно их алкоксипроизводных, в циклогексил амины
[201, Гидрогенолиз протекает в незначительной степени, и вторич-
ные амины обычно образуются только в небольших количествах.
1. Burgstahler A. W., В i t h о s Z. J., J. Am, Chem. Soc., 82, 5466
(1960); Org. Syn., 42, 62 (1962),
2. Smith H. A., Stump B. L., J, Am. Chem. Soc., 83, 2739 (1961).
3. К а у e I. A., Matthews R. S., J. Org. Chem., 28, 325 (1963).
4. Meyers A. I., В evening W. N., Gar ci a-M unoz G., J. Org.
Chem., 29, 3427 (1964); Meyers A. I., В everung W. N., procedure
submitted to Org. Syn.
5. Sircar J. C., Meyers A. I., J. Org. Chem., 30, 3206 (1965).
6. Stocker J. H., J. Org, Chem., 27, 2288 (1962),
7. R ylan der P. N. (Engelhard Industries), private communication,
8. Ham G. E., Coker W. P., J. Org. Chem., 29, 194 (1964),
9. С о p e A. С., P i k e R. A,, Spencer C. F., J. Am. Chem. Soc., 75, 3212
(1953).
10. Freilel der M., Smart W. D., Stone G. R., J. Org. Chem., 27,
2209 (1962).
11. Freifeldcr M., J. Am. Chem. Soc., 82, 2386 (I960).
12. Howard W. L., В r own J. H., Jr., J. Org. Chem., 26, 1026 (1961).
13. R a p a 1 a R. T., L avagnino E. R., Shepard E. R., Farkas E,,
J. Am. Chem. Soc., 79, 3770 (1957).
14. Freifelder M., J. Org. Chem., 27, 4046 (1962); 28, 602, 1135 (1963).
15. Ada m s R., M i у a n о S., F 1 e § D-, J. Am. Chem. Soc., 82, 1466 (1960).
16. Broadbent H. S., et al., J. Am. Chem. Soc., 82, 189 (1960).
17. С о h n W- E., Doherty D. G., J. Am. Chem. Soc., 78, 2863 (1956).
18. N ewm ап M. S., L e dn i cer D., J. Am. Chem. Soc., 78, 4765 (1956),
19. Anderson A. G.,Jr.,An der.son R.G.,J. Org. Chem., 22, 1197 (19,57).
20. Freifelder M., N g Y. II., Helgren P. F., J. Org. Chem., 30, 2485
(1965).
РОДИЙ (5%) НА УГЛЕ (норит), Rh —С.
Брейтнер и др. [11 провели сравнение активностей продажных
препаратов Rh—С, Ru—С, Pt—С и Pd—С (все па норите). По-ви-
димому, Rh—С и Ru—С являются лучшими катализаторами для
гидрирования кетонов в нейтральной или щелочной среде. Эти ка-
тализаторы можно использовать для восстановления а, р-непредель-
ных кетонов (через стадию предельных кетонов) в предельные
спирты,
Фрейфелдер и сотр. [21 показали, что Rh—С — более подходя-
щий катализатор, чем родий на окиси алюминия, для восстановле-
ния пиридинового кольца. Отравляющее действие пиперидиновых
оснований можно преодолеть, если брать достаточное количество
катализатора.
172
/£Н2СНгСН.,ОН
11 + Rh—С
О 4-7 ‘
N
0,1 моля в абе. EtOH
Н2 (2,7 атм).
5—60°,
СН2СНЙСН2ОН
час
I
н
Швейцер и Лайт [3] установили, что Rh—С пригоден для поста-
дийного гидрирования ЗН-пирролизина (1).
(2)
Rh, 3 Нг, EtOH
68%'
(3)
-1
При кипячении норборнадиена с Rh—С углеводород превра-
щается с почти количественным выходом в продукт, состоящий из
70—80% смеси димеров и 20—30% три мер а [4].
1. В г е i t п е г Е., R о g i n s k i E., R у lander P. N., J, Org. Chem., 24,
1855 (1959).
2. Freifelder M., Robinson R. M., Stone G, R., J, Org. Chem.,
27, 284 (1962),
3. Schweizer E. E., L i g h t К. K., J. Am. Chem. Soc., 86, 2963 (1964).
4. Mrowca J. J., К a t z T. J., J, Am. Chem. Soc., 88, 4012 (1966).
РТУТИ АЦЕТАТ, Hg(OCOCH3)2. Мол. вес 318,70.
Растворимость в 100 а НХ): 25 г при 10° и 100 г при 100°.
Дегидрирование. В классических исследованиях эргостерина
и витамина D Виндаус предложил использовать. Р. а. в качестве
агента для дегидрирования некоторых ненасыщенных соединений.
Фотохимическим окислением [1] он превратил эргостерин (I) в
трансаннулярную перекись II и восстановил ее до триола III цин-
ковой пылью в кипящем спиртовом растворе щелочи [21. Этот триол
имеет две аллильные третичные оксигруппы и при перегонке в ва-
кууме в результате отщепления 2 молей воды дает дегидроэргосте-
рин (IV). Этот триен представляет интерес для исследования фото-
химической и кислотной изомеризаций и других реакций,однако
метод получения его через перекись в больших масштабах неудобен.
Поэтому была предпринята попытка его получения действием Р. а.
непосредственно на эргостерин. Раствор Р. а. в этаноле, содержащем
немного уксусной кислоты, добавляли к кипящему раствору 25 а
эргостерина в 1250 мл этанола и смесь кипятили в течение 40 мин.
Очистка без применения хроматографии дала 40%-ный выход про-
дукта, чистота которого была подтверждена по оптическому вра-
щению (см. значение [а]о под формулой). Позже [3] аналогичная
173
IV(q-d+ 149)
реакция была проведена с Р, а. в кипящей смеси диоксан — уксус-
ная кислота, и чистота продукта, полученного хроматографической
очисткой с выходом 37%, проверялась по экстинкции УФ-погло-
щения.
Внндаус применил реагент для дегидрирования 5-дигидроэрго-
стерина в эргостернн-D; соответствующие ацетаты—(1) и (4) [4|.
В более поздней работе, посвященной кортизону, эргостерин-D и
Д7,!*ип~диены холестерина, желчной кислоты и сапогенина приоб-
рели большое значение как ключевые промежуточные соединения
174
при получении 11-оксистероидов, поэтому была проделана большая
работа для улучшения методов превращения А7-енов в Д7гли).
диены. Получены [51 доказательства образования в реакции мерку-
рированного стероида типа (2), одиако часть диена (4) образуется
из термически неустойчивого промежуточного соединения, не со-
держащего ртути, выделенного в определенных условиях реакции
и охарактеризованного как (5). Согласно этой методике, раствор
200 а (0,44 моля) ацетата Д7,2г-эргостадиепола-ЗР (1) в 1,1 л хлоро-
форма обрабатывают раствором 0,91 моля Р. а. в 1670 мл уксусной
кислоты и суспензию перемешивают при 25—30° в течение 23 час.
Раствор отфильтровывают от ацетата одновалентной ртути (выход
98%) и концентрируют до объема 600 мл при 70—80° в вакууме;
в процессе медленной перегонки происходит полное превращение
(5) в (4). Кристаллизация при 25у дает диен (4) с чистотой 76,5%
и выходом 78%. Этот продукт пригоден в большинстве случаев;
дальнейшая очистка исключительно трудоемка.
Швейцарские химики 161 провели ту же реакцию кипячением
(1) с Р. а. в смеси диоксан — уксусная кислота в атмосфере азота.
Реакция завершается через 10 мин и дает с 70%-ным выходом сырой
продукт с чистотой 84%, пригодный для непосредственного исполь-
зования.
Третья, изученная Виндаусом [71 реакция — это дегидрирова-
ние н-нитробензоата пзодегидрохолестерина (Ав,,ч-холестадиенола-
ЗР) (I) — побочного продукта в промышленном синтезе А3’7-диена,
необходимого для получения витамина D3. Позднее реакция была
повторно изучена Бартоном и Розенфельдером [81 с образцом сте-
роида, полученного от Виндауса. Меркурпрование пространственно
незатрудненного конца диеновой системы с образованием II и
карбониевого иона III объясняет образование 6-эпимерных спиртов
IV и V, дающих при дегидратации трнены VI и VII.
175
Ацетоксилирование, Трейбс [9] показал, что олефины и кетоны
ацетоксилируются при взаимодействии с Р. а.
22%
Н£(ОАс)г, 150°
н
OAc + Hg
Первоначально реакцию проводили без растворителя при 150°.
Смесь 0,2 моля циклогексена и 0,2 моля Р. а. запаивают в стеклян-
ную ампулу и нагревают в бомбе в течение 2 час; при этом выделяют
металлическую ртуть с выходом 100%, а из сильно осмелившегося
органического продукта перегонкой выделяют небольшое количество
бензола н Д^циклогексенил-З-ацетат с 22%-ным выходом. В этой
реакции обычно получается низкий выход продукта; с несимметрич-
ными олефинами результаты сильно варьируют. (Н-)-Лимонен (1)
дает (+)-ацетоксипроизводное [9], но (+)-ментен-3 (3) окисляется
в (±)-эфир [10].
Окисление (3) протекает в две стадии,
необходимо лишь для осуществления
причем нагревание до 150°
второй стадии.
В C10H18 + 2Hg(OCOCH3)2 C10H17OCOCH3 + 2HgOCOCH3 + CH3COOH
2) C10H184-2HgOCOCH3 —> Cl0H17OCOCH3-h2Hg -|-СН3СООН
Например, при окислении 1-фенилциклогексена смесь 0,1 моля
углеводорода и 0,2 моля Р. а. в уксусной кислоте перемешивают
при 95° и получают ацетоксипроизводное с 70%-ным выходом [111.
Алькони [12] окислял по этой методике а-циклогераниол (5), од-
Л/0Ас
I |ХН + 2HgOAc
нако не получил столь высокого выхода; на основании того, что он
охарактеризовал полученный продукт как ацетат (6), можно счи-
тать, что все приведенные в этом разделе реакции осуществляются
не через аллильное ацетоксилирование, а включают атаку двойной
связи с ее миграцией.
176
2Hg(OAc)3, 75°
r 2HgOAc
(6)
Трейбс [9] исследовал действие Р. а, на кетоны при высокой тем-
пературе. При ацетоксилироваиии циклогексанона наблюдается
сильное осмоление и образуются небольшие количества а-ацето-
ксипроизводного и фенилацетата. В аналогичных условиях тетралин
Hg(OAc)3, 150°
+ Hg
и индан не реагируют.
Дегидрирование третичных аминов. Леонард [13—15] показал,
что циклические третичные амины окисляются в енамины под дей-
ствием 4 экв Р. а. в 5%-ной водной уксусной кислоте при 100°.
При обработке хинолизидина этим способом в течение 1,5 час ко-
личество выделяющегося ацетата одновалентной ртути соответ-
ствует 92%-ному отщеплению двух атомов водорода, а выход Д1(10)-
дегидрохинолизидина, выделенного в виде перхлората иммония,
Хинолизидин
Перхлорат иммония
(1696 гл?’1)
составляет 59% [13]. Следует отметить, что енамин и ион иммония
легко отличить по их ПК-спектрам. Леонард предполагает, что
первоначально образуется меркурированиый комплекс (2) за счет
р-электронов азота. Отрыв протона от третичного атома углерода
осуществляется одновременно с разрывом связи азот — ртуть и
выделением HgOAc, который очень быстро реагирует с Р. а. и
образует нерастворимый ацетат одновалентной ртути. Соль амина
остается в растворе в виде резонансно стабилизированного иммо-
ний-карбониевого иона (3); при подщелачивании от атома Сх отщеп-
ляется протон с образованием Д1(1 °’-енамина (4).
В реакции трансЛ-метилдекагидрохинолина (5) с 4 молями Р, а.
в 5%-ной уксусной кислоте (30 мин при 90°) дегидрирование сопро-
вождается гидроксилированием и после подщелачивания образуется
177
10-окси-1-метил-Др-окта гидрохинол ин (6) [15]. /{ис-Изомер (5) так-
же даст (6).
Бенкерт [16] предложил остроумную схему биогенеза винка- и
ибога-алкалоидов. Ее подтверждением является интересный синтез,
осуществленный Кутнеем, Брауном и Пирсом [17], который состоит
в трансаннулярной циклизации карбметоксидигидрокливамипа (I)
под действием Р. а. в уксусной кислоте при комнатной температуре.
Хроматографией в качестве основного продукта получают винка-
алкалоид винкадифформин (II). В форме 1а циклизация включает
атаку связанной с азотом метиленовой группы на индольное кольцо
и миграцию двойной связи. Два побочных продукта реакции иден-
тифицированы как эпимерные ибога-алкалоиды: коронаридин (Ш)
п ш, r - сгн5, R- - н
IV, К - н, R ' СгН5
178
и дигидрокатарантин (IV). Образование III и IV означает, что I реа-
гирует в форме 16. Изомеризация у атома С4 объясняется образова-
нием из 16 промежуточного иммониевого иона с 5,6-двойной связью,
находящегося в равновесии с енамином, содержащим 4,5-двойную
связь.
Для подавления побочных реакций при дегидрировании тре-
тичных аминов Р. а. Кнабе [18] проводил окисление алкалоидов
типа папаверина (1) в присутствии эквивалентного количества свя-
зывающего агента — этилеидиаминтетрауксусиой кислоты (в виде
ее динатриевой соли). Продукты реакции — соль папаверииия
(2) и дигидросоль (3); выходы указаны под формулами. В отсутствие
комплексообразующего агента происходит расщепление метилено-
вого мостика папаверина.
Обменные реакции виниловых сложных эфиров. Виниллаурат
получают реакцией 0,4 моля лауриновой кислоты с 2,4 моля винил-
ацетата в атмосфере азота с добавлением в качестве катализатора
1,6 г Р. а. [19]. Смесь встряхивают в течение 30 мин и после добав-
ления 0,15 мл 100%-ной H3SO4 кипятят в течение 3 час. Обменная
реакция подробно изучена Ватанабе и Конлоном [20], которые пока-
ОСОСНз
сн3(сн3)10со„н+он=сн2 -g(0Acb’
53—59%
о
II
СН3(СН,)1ПСОСН = СН2 + СН3СООН
зал и, что только ртутные соли слабых кислот (уксусной и бензой-
ной) являются специфическими катализаторами реакции. Берг-
сталер [21] считает, что большое значение имеет чистота катализа-
тора, и предлагает кристаллизовать реагент нз абсолютного этанола
и затем тщательно сушить. Бергсталер использовал обменную реак-
цию как одну из стадий при введении в циклическую систему ан-
гулярного заместителя. Так, виниловый эфир (3) при перегруппи-
ровке Клайзена дает ненасыщенный альдегид (4). Эта реакция
стереоспецифичиа; она была применена к А4-холестеиолу-3(3 [21]
и для синтеза продукта расщепления атизииа [22]. Бюхи и Уайт
179
ососн3
СН = СН31 Hg(OAc)s
[23] использовали реакцию обмена для получения р-циклогеранил-
винилового эфира (6), промежуточного соединения в полном син-
тезе (±)-туйопсена.
ососн.,
CH = CHS1 Hg(OAc)3
51%
сн=сн2
<6>
Меркурированные ароматические соединения. о-Хлормеркур-
фенол получают нагреванием фенола с Р. а. в кипящей воде и до-
бавлением хлорида натрия для расщепления первоначально обра-
зующегося о-ацетоксимер кур производного (Уитмор и сотр. [24]).
ОН
Ч/
Hg(OCOCHB)2
-CH3COOH
ОН
Д zHgOCOCH3
/ Д NaCl
II 44%
он
/1 /Hgcl
Ангидро-2-оксимеркур-З-нитробензойную кислоту получают при
добавлении раствора 1,1 моля Р. а. в разбавленной уксусной кис-
лоте к водному раствору динатриевой соли 3-нитрофталевой кислоты;
температуру постепенно повышают и смесь нагревают при 165—
175° в течение 70 час 125].
COzNa
Hg(QAc)2, 170°
СО2Ма 82 "90%
no2
180
Реакция с а, ^-ненасыщенными кислотами. Реакция, предло-
женная Лбдерхальденом и Хейнсом (26) и усовершенствованная
Картером [27], имеет особое значение для синтеза серина [28] и
треонина [29]. Кротоновую кислоту (1), обрабатывают Р. а. в мета-
ноле. Образующийся аддукт, возможно (2), дает при обработке
бромидом калия соединение (3), при бромировании которого полу-
чается а-бром-[3-метоксимасляная кислота (4), превращаемая в
Нй О А с)- С Н, О Н К В г
СН3СН - CHCO,I I :---'--- СН3СН------ СНСО2Н1 —“
О) I I
ОСН3 HgOAc
СН3СН CHCCMI1
I I
ОСН, HgBr
(3)
(2)
Вг.,
— —> СН3СН-----------СНСОаН
-85% f
ОСН3 Вг
(4)
D, L-треоиин. Метиловый эфир акриловой кислоты служит исходным
веществом в аналогичном синтезе предшественника о, ь-серина.
Гидратация ацетиленов. В некоторых случаях гидратацию аце-
тиленов с образованием соответствующих карбонильных соединений
можно осуществить непосредственно обработкой горячей кислотой.
Однако предпочтительнее использовать либо каталитическое коли-
чество сульфата ртути в разбавленной серной кислоте, либо 2 моля
Р. а. В последнем случае промежуточно образующийся комплекс
pj НОН(Н'Р 7 „ |
С ' СИ ' О-12
I I I
ртути расщепляют соляной кислотой или сероводородом с образо-
ванием карбонильного соединения. гЧиддлтоп [30] выделил некото-
рые комплексы и на основании их свойств предложил следующий
механизм реакции:
OHgOCOCH3
1) PC срф2Hg(OCCCH3)2ф 1Ь0 —> R — С~ С—Кф2СН3СООН
HgOCOCII3
OHgOCOCH,
2)
HgOCOCHs
' он
RC —CHR
о
II
RCCH.R
ф2Нф8ф2СН3СООН
Коулкес [31] разработал метод аналитического определения тройной
связи, основанный на образовании в реакции (1) 2 молей уксусной
кислоты на каждую тройную связь.
Жак [32] предложил метод гидратации тройной связи в некис-
лотных условиях применительно к ряду стероидных этинилкарби-
181
нолов, которые чувствительны к кислотам. Суспензию 2 г Р. а.
в растворе 1 a (I) в 100 мл этилацетата перемешивают при 20° в тече-
ние 24 час, обрабатывают током сероводорода для осаждения сое-
динений ртути и обработкой фильтрата получают З-ацетил-З-ацето-
ксипроизводное (II) с 84%-ным выходом. Метод применим в случае
чувствительного к кислотам 17-этиленкеталя 17-кетоиа, соответ-
ствующего I. Следует отметить, что третичная оксигруппа при С3
ацетилируется, тогда как вторичная 17₽-оксигруппа в I остается
неизмененной. Это, по-видимом у, объясняется промежуточным об-
разованием ртутного комплекса (1); миграция ацетильной группы
может происходить через циклизацию в (2) с последующим обра-
зованием гем- а цетил ацето кс и соединен и я (4).
Катализатор бромирования. Гризеофульвин (1) не реагирует
с бромом в уксусной кислоте в обычных условиях. Однако при об-
работке бромом в присутствии 0,5 моля Р. а. (1) дает 5-бромпроиз-
водное (2) [331. По-видимому, происходит мер курирование (1) с об-
разованием 5-ацетоксимер кур производного с последующим заме-
щением на бром.
Замещение галогена в производных сахаров. Уолфром и др. [34]
считают Р. а. отличным реагентом для замещения галогена в аце-
тилированных гл и ко пираноз ил галогенидах. Поскольку как tz-,
182
так и P-D-гликозилгалогениды дают р-п-ацетаты, пространственное
направление реакции, по-видимому, определяется эффектом орто-
эфирыой группы [35|.
(3)
Присоединение к олефинам. Р. а. реагирует с олефинами в спир-
товом растворе по механизму электрофильного присоединения [36]
ОС2Н5
Hg(OAc)2 + ")С-с/^С2Н5ОН '^С-С^+АсОН
HgOAc
В случае простых олефинов, например циклогексена (1), реакция
осуществляется как транс-присоединение. Однако Трейлор |37]
сн3он
осн3
HgOAc
Асон
показал, что в случае норбориена (2) и других напряженных сис-
тем наблюдается цис-присоединение.
(2)
НгО;
> + Hg(ClO4)j -£££-
-он
HgCl
1. , W indans А., В г u п k е п JAnn., 460, 225 (1928).
2. W indaus A., L i n s e г t О., Ann., 465, 148 (1928).
3, Z й r c h e r A., Heusser H,, J e g с г О., G e i s t 1 i c h P., Helv.
Chim. Acta, 37, 1562 (1954).
4, W indaus A,, D i t hmar K-, M ii г к e H-, S и с к f ii 1 1 F., Ann,,
488, 91 (1931),
5. Ryule W. V., Jacob T. A., Chemecda J. M,, Chamberlin
E. M., Roscnburg D. W., S i t a G. E., Erickson R, L., A 1 i-
minosa L. M., T i s h 1 e r M., J. Am. Chem. Soc., 75, 2604 (1953),
6. S а и с у G., G c i s t 1 i c h P,, H e I b 1 i n g R., Heusser H., Helv.
Chim. Acta, 37, 250 (1954).
7. W i n d а и s A,, Riemann U,, Z ii h I s d о r I f G., Ann., 552, 135,
142 (1942).
8. Barton D. H. R., Rosen folder W, JJ, Chem. Soc., 1951, 238f.
183
9, Т г e i b s W., Bast H„ Ann., 561, 165 (1949).
10. T r e i b s W., Luci us G., Kogi er H., В r e s 1 a u с r FI., Ann., 581,
59 (1953).
11. Treibs W., W eissenfels M., Chem., Ber., 93, 1374 (1960).
12. Alkonyi 1., Chem. Ber., 95, 279 (1962).
13. Leonard N. J., H a у A. S., Fulmer R. W., Gash V. W., J. Am.
Chem, Soc., 77, 439 (1955).
14. Leonard N. J., Fulmer R, W-, Hay A. S., J. Am. Chem, Soc., 78,
3457 (1956).
15. Leonard N.J. Miller L. A., Thomas P, D,, J. Am. Chem. Soc.,
78, 3463 (1956).
16. W'cnkert E., J. Am. Chem. Soc., 84, 98 (1962).
17. Kutney J.P., Bro w л R. T., Piers E., J. Am. Chem. Soc., 86, 2286,
2287 (1964).
18. Knabe J., Arch. Pharm., 292, 416 (1959); 293, 121 (1959); Knabe J.,
Grund G., ibid., 296 , 854 (1963); Knabe J., R о! о f f H., Chem. Ber,,
97, 3452 (1964).
19, Swern D., J ordan E. F., Jr., Org. Syn., Coll. Vol., 4, 977 (1963).
20. W atanabe W. H,, Conlon L. E., J. Am, Chem. Soc., 79, 2828 (1957).
21, Burgstahler A. W-, Nordin I, C., J. Am. Chem. Soc,, 83, 198 (1961).
22. О g i s о A., [wai E, Chem. Pharm. Bull., 12, 820 (1964).
23. В u ch i G., Wh i te J. D., J, Am, Chem. Soc., 86, 2884 (1964).
24. \V h i t m or e Г. C,, West H. D., Hanson E. R., Org, Syn., Coll.
Vol., 1, 161 (1941).
2э. У и т м о р Ф. R., Кульген П., Herep X., «Синтезы органических
препаратов», ИЛ, М., 1949, сб. 1, стр. 41.
26. Abdcrhalden Е., Heyns К. Вег., 67, 530 (1934).
27, Schiltz Е. R., Carter Н. Е., J. Biol. Chem., 116, 7.93 (1936).
28. С а г t е г Н. Е., West Н. D., Org. Syn,, Coll. Vol., 3, 774 (1955).
29. С а г t е г Н. Е., W es t Н. D., Org. Syn., Coll. Vol., 3, 813 (1955).
30. M у d d 1 e t о n W. W., Barrett A, W-, S e a g e r J, H., J. Am- Chem.
Soc., 52, 4405 (1930).
31. Koulkcs M., Bull. soc. chim. France, 1953, 402.
32. Kagan H. B., Mar q u et A,, Jacques J., Bull. soc. chim. France,
1Я60, 1079.
33. Walker T., Warburton W. K., Webb G. B., J. Chem. Soc., 1962,
1277,
34. Thompso n A., Wolfro m M. L., Inatome M-, J. Am. Chem.
Soc., 77, 3160 (1955).
35. Wolirom M. L., G г о e b k e W., J. Org- Chem., 28, 2986 (1963).
36. c h a t t J., Chem, Revs., 48, 7 (1951).
37. Traylor T. G.,B aker A. W., J, Am. Chem. Soc., 85 , 2746 (1963); Tra-
ylor T. G., ibid., 86, 245 (1964).
РТУТИ ОКИСЬ (ЖЕЛТАЯ, КРАСНАЯ), HgO. Мол. вес 216,61;
т. пл. 630°.
Окисление гидразонов. Метод Штаудингера [1] получения ди-
фенилдиазометана приведен с небольшими изменениями в сб. «Син-
тезы органических препаратов» [2]. В толстостенную бутыль поме-
щают 0,1 моля гидразона бензофенона, 0,1 моля желтой Р. о. и
100 мл петролейного эфира (т. кип. 30—60°), бутыль закрывают,
обертывают влажным полотенцем и встряхивают при комнатной
температуре в течение 6 час. Смесь фильтруют для удаления образо-
вавшейся ртути и небольшого количества азина бензофенона и вы-
паривают досуха в вакууме при комнатной температуре. Получен-
ный таким образом продукт плавится прн комнатной температуре,
184
однако он удобен для использования в качестве реагента. Чистое
I-I,NNH2 HgO +
'C6H5h_C = O (CfiH5),C —N = N
соединение имеет т. пл, 29—30°, ио очистка его кристаллизацией
затруднительна.
Дифен ил кетен получают [31 термическим разложением фен ил-
бензоилдиазометана, приготовленного перемешиванием смеси 0,25
моля бензилмоногидразона, 0,38 моля окиси ртути, 35 а безводного
сульфата кальция и 200 мл бензола при 25—35° в течение 4 час.
Бензольный раствор добавляют по каплям в колбу для перегонки,
нагретую на бане со сплавом Вуда до 100—110°; бензол мгновенно
отгоняется, а диазосоединение превращается в дифенил кетен. В том
случае, когда хотят получить фенилбензоилдиазометап, раствори-
тель должен быть более летучим, чем бензол, Штаудингер [1] от-
С6Н5С-С = О С6Н5С----------- С--0
Ii 1 II I
N Сбн5 N- CGH5
I Я
nh2 n-
H;i г рсванис
64%
(общи ji )
)C = C--O-N3
метил, что окисление гидразона в эфире протекает гораздо медлен-
нее, чем в бензоле или петролейном эфире, однако, как показали
Неницеску и Соломоника [4], реакция существенно катализируется
щелочью. Согласно предложенной имн методике, в склянку с проб-
кой помещают 0,134 моля бензилмоногидразона, 0,28 моля Р. о.,
15 г безводного сульфата натрия, 200 мл абсолютного эфира и 4 мл
холодного насыщенного раствора едкого кали в этаноле. Смесь
встряхивают 10—15 мин, фильтруют и выпаривают в вакууме
водоструйного насоса при температуре не выше 40°. Желтый крис-
таллический продукт высушивают на пористой пластинке и крис-
таллизуют из абсолютного эфира; выход продукта с т. пл. 79°
(с разл.) составляет 87-—94%.
При получении дифен и л ацетилен а из бензилди гидр а зон а послед-
ний суспендируют в бензоле .в колбе, снабженной мешалкой и об-
ратным холодильником, и добавляют порциями Р. о. с такой ско-
ростью, чтобы раствор равномерно кипел (Коуп [5]). Бензил-
С6Н5С-СС6Н5
2HgO
78-84%
моногидразон, необходимый для получения фенилбензоилдиазоме-
тана, приготавливают медленным добавлением гидразипгидрата
к горячему раствору бензила в этаноле; реакция заканчивается по-
сле 5-минутн.ого кипячения. Для получения дигидразоиа Коуп ки-
пятил бензил с 2 же гидразипгидрата в н-пропаполе (т. кип. 97°)
в течение 60 час.
185
Катализатор присоединения к ацетиленам. Превращение 1-эти-
нилциклогексанола в 1-ацетилцнклогексапол гидратацией тройной
связи осуществляется следующим образом: 5 г красной Р. о. Мал-
линкродта растворяют в смеси 8 мл конц. серной кислоты в 190 мл
воды, нагревают до 60° и 0,4 моля 1-этинил циклогексанол а добав-
ляют в течение 1,5 час [6|.
/----\/С~ Hg0. разб H2sq4 у---------Х/СОСН3
\----/\он 65-67% \----/\эн
Винилхлорацетат получают катализируемым присоединением
хлоруксусной кислоты к ацетилену 17]. Смесь 2,12 моля хлоруксус-
ной кислоты (т. пл, 63°), 0,2 г гидрохинона (ингибитор полимери-
зации) и 20 г желтой Р. о. перемешивают, нагревают до тех пор,
пока хлоруксусная кислота не начнет плавиться, и через стеклянную
трубку, доходящую до дна сосуда, пропускают ацетилен. Через
30 мин температуру можно понизить до 40—50° без затвердевания
смеси. Поглощение ацетилена, очень быстрое вначале, через 3 час
становится очень медленным, и реакция прекращается.
ГЫ О
С1СН,СО,Н-!-НС -=СН ~> С1СН„СО,СН = сн.,
I 42—49%
Реакция Хунсдикера. В оригинальной реакции Хунсдикера
сначала получают серебряную соль кислоты, а затем ее обрабатывают
бромом. Кристол 181 показал, что соль можно не выделять; добав-
ление брома к кипящей суспензии красной Р. о. в растворе кислоты
в четыреххлористом углероде дает с хорошим выходом соответст-
вующий ал кил галогенид. Вместо брома можно использовать иод [91.
2СН3{СН,)1ССО,Н Щ 2Вгг Щ HgO > 2CH3(CH2)IfiBr+ 2СО, -) - HgBr2-|- Н2О
J о
Методика получения бромциклопропана следующая ПО]: раствор
0,2 моля циклопропанкарбоновой кислоты и 0,2 моля брома в 50 мл
1, 1,2, 2-тетрахлорэтана добавляют по каплям при перемешивании
сн2. о-щ
2| >CHC(XH + HgO + 2Br, ~' "CHBri--HgBr3-j-HoO+2CO,
СН/ ’ ' 41~46% сн/
к суспензии 0,11 моля красной Р. о. в 60 мл тетрахлорэтана при наг-
ревании на водяной бане при 30—35° в течение 45 мин, и затем пере-
... мешивание продолжают до прекращения
ГЙГ[ выделения двуокиси углерода. При провер-
ке эт°й методики Фосетт и Мак-Казик
? показали, что выделение СО2 соответст-
р вует 52-—60% от теоретического. Они
: фи л ьтр ов а л и смесь и а но р исто м сте к л я н ном
uTSy фильтре под давлением, чтобы уменьшить
\ f потерн (рис. Р-2).
Рис, Р-2.
186
Применение методики Кристола к бицикло-[2,2,2]-октан-1-кар-
боновой кислоте (1) с бромом и Р. о. в четыреххлористом углероде
дает в качестве продукта смесь ожидаемого бромида (2) и большого
количества неожидаемого хлорида (3) [II?. Хлорид безусловно об-
разуется за счет отрыва хлора от растворителя промежуточно об-
(1) (2) 32% (3) 68%
разующимся бициклическим радикалом. Чистый бромид был полу-
чен при использовании в качестве растворителя бромтрихлор мета-
на или 1,2-дибромэтана.
Дэвис и сотр. [12] показали, что метод Кристола — Фирса яв-
ляется наилучшим для проведения реакции Хунсдикера, и предло-
жили ряд усовершенствований.
Синтез Кенигса — Кнорра. Реакцию поли-О-ацилгликозилга-
логеиида с гидроксильной компонентой, приводящую к глюкопи-
ранозиду 1131, обычно проводят в присутствии окиси или карбоната
серебра. Гораздо более дешевым реагентом является желтая Р. о.,
которая вместе с небольшим количеством бромида ртути действует
так же хорошо и не чувствительна к свету [14]. Будучи акцептором
кислоты, Р. о. взаимодействует с образующимся бромистым водоро-
дом и дает дополнительное количество катализатора.
1. Staudinger fl., Antlies E., Pf enni nger F., Ber., 49, 1928
(1916). О невозможности улучшить методику окисления см. В а 1 t z 1 у R.
et al., J. Org. Chem., 26 , 3672 (1961).
2. S m i 111 L. T., Н о w a r d K. L., Org. Svn., Coll. Vol., 3, 351 (1955).
3. Smit h L. I., Hoehn H. H., Org. Syn., Coll. Vol., 3, 356 (1955).
4. H еницее к у K-, Сол омо» ика Е., «Синтезы органических препара-
тов», ИЛ, М., 1949, сб. 2, стр- 506.
5. С о р е А. С., S m i t h D. S,, Cotter R. J., Org. Syn., Coll, Vol,, 4, 377
(1963).
6. S t а с у G. B-, Mikulec R. A., Org. Svn., Coll, Vol., 4, 13 (1963).
7. W i 1 e у R. H-, Org. Syn., Coll. Vol., 3, 853 (1955).
8. С c i s t о 1 S. J., F i г t h \V- C,, Jr., J. Org. Chem., 26, 280 (1961).
9, C r i s t о 1 S. J., Gaston L. !<., Tiedeman T,, J. Org. Clicm., 29,
1279 (1964),
10. /М e e k J. S., Osuga D. T., Org. Syn., 43, 9 (1963).
11. Baker F. W., Holtz H. D,, Stock L. M., J. Org. Chem., 28, 514 (1963);
Holtz H. D,, Stock L. M-, J. Am. Chem, Soc,, 86, 5183 (1964).
12, Davis J.A., Herynk J.,Car roll S.,Bunds J,, Johnson D.,
J. Org. Chem., 30, 415 (1965).
13. W о I f г о m M. L., L i n cb a c k D.-RMethods in Carbohydrate Chemist-
ry, II, 342 (1963).
14. Schroeder L, R., Green J. W., J. Chem. Soc., 1966, 530,
РТУТИ окись — иод. Эта смесь реагентов была использо-
вана в надежде на то, что она будет более эффективной при окисле-
187
нии ангулярных метильных групп, чем тетраацетат свинца и иод.
Так, 6(3-о кс истер о ид (1) реагирует cP. о. и иодом на свету с образо-
ванием 6,19-окиси (2) через промежуточно образующийся гипоио-
ДИТ [11.
1. A h t t а г М., Barton D. Н. R., J. Am. Chem. Soc., 83, 1528 (1964).
РТУТИ(П) СУЛЬФАТ, HgSO4. Мол. вес 296,68.
Реагент обычно используется в комбинации с сильной кислотой
в качестве катализатора присоединения к ацетиленам. Гидратация
ацетиленового гликоля (1) происходит при добавлении 25 г тонкоиз-
мельченного вещества в течение 20 мин к энергично перемешивае-
мому охлаждаемому льдом раствору 1,25 г Р. с. в 125 мл 85%-ной
муравьиной кислоты [1]. При добавлении каждой порции темпера-
тура повышается на 2—3°. По окончании добавления перемешивают
\/он СН3 \ /он СНа
/чсн2с^с-с—он 2 х—/хсн2сн2с-Loh
I «2% Il [
сн3 о сн3
(1) (2)
еще 30 мин, после чего появляются красивые кристаллы. Добав-
ляют насыщенный раствор сульфата аммония, продукт экстраги-
руют бензолом и кристаллизуют его из пентана. Гидратация ацети-
ленового производного является главной стадией в интересном и
высокоэффективном синтезе гистамина (7), предложенном Фразером
и Рафаэлем [2]. Продажный бутин-2-диол-1,4 (1) превращают через
дихлорид (2) в дифталоил аминопроизводное (3). Гидратацию в ке-
тон (4) осуществляют следующим образом: раствор 3 а (3) в 140 мл
90%-ной уксусной кислоты обрабатывают 0,75 г Р. с. и 0,5 мл
конц. серной кислоты и смесь кипятят 4 час. При добавлении воды
высокоплавящийся кетон (4) отделяется, его собирают и промывают.
Кислотный гидролиз дает кетодиамин (5), который при обработке
тиоцианатом калия циклизуется в 2-меркаптогистамин (6). Послед-
няя стадия — замещение SH на Н—осуществляется с высоким вы-
ходом окислением хлоридом железа (III). Эта удивительная реакция
аналогична десульфуризации меркаптопиримидинов окислением
перекисью водорода или азотной кислотой и обусловлена образо-
ванием сульфокислоты и гидролизом [3[,
188
носн2с == ссн2он 22*3. асн2с ссн2а JLCC™
нгг kcns НС —С—СН2СН2ХН
_CH,-C-CHXH8NH2-2HC1 — I I
83% ] j| " 90% HN N
ННг О (о)
SH
(6)
FcCb hc-c-ch2ch2nh3
8 одГ HN N
(в виде дипикрата)
сн
(7)
Ньюмен [4] предложил новый катализатор гидратации, полу-
ченный пропиткой сульфированной полистирольной смолы дауэкс 50
Р. с. Смесь 39 a I-этинилциклогексанола (1), 100 мл уксусной кис-
лоты, 10 мл воды и 20 г пропитанной Р. с. смолы нагревали при пе-
ремешивании до кипения в течение 45 мин; смолу отфильтровывают и
обработкой получают 1-ацетилциклогексен (4) с 86,7%-ным выходом.
В присутствии Р. с. и серной кислоты к тройной связи ацетилена
можно присоединить 2 моля толуола [5]. Ацетилен пропускают в
IlgSOd CI‘I3Cf;H,ly
2СвН5СНз + СН = СН >снсн3
60-64% СН3С6Н/
смесь 606 г толуола, 70 мл конц. серной кислоты и 7 г Р. с., поддер-
живая при помощи льда температуру 10—15°.
I. Mondon А., Апл., 585, 43 (1954); for a further example of the reactions see
H ojasZ. G., D oeb el I(. J., Goldberg M. W., J. Org. Chem., 29, 2527
(1964).
2. Fraser M. M., Raphael R.A.,J, Chem. Soc., 1952, 227.
3. E 1 d e r f i e 1 d R. C., «Heterocyclic Compounds», 6, 284 (1957).
189
4. N e w in а п M. S., J. Am, Chem. Soc,, 75, 4740 (1953); for a fiither examples of
the reaction see H о j о s Z. G., D о e b e 1 K- J , G о 1 d b e r g M. W., J . Org,
Chem., 29, 2527 (1964).
о. Рейхерт Дж,, Ньюленд Дж., «Синтезы органических препаратов»,
ИД, М., 1949, сб. 1, стр, 202,
РТУТИ ТРИФТОРАЦЕТAT, Hg(OCOCF:j)2. Ньюмен и Аркелл
И1 получили реагент растворением окиси ртути в безводной трифтор-
уксусной кислоте и использовали его для окисления сс-кетогидра-
зона (1), дипивалоилмоногидразона, в дназокетон (2).
NNH3 it'—N
(CHS)3CCCC(CH3)3 -Hg<OS.°CF±~C£3^ (СН3)3СССС(СН8)3
Il II
о о
(I) (2)
1. Newman M.S., Ark efl A., J. Org. Chem., 24, 385 (1959).
РТУТИ(П) ЦИАНИД, Hg(CN)2. Мол. вес 252,65.
Гельферих [11 сообщил, что использование Р. ц. в качестве осно-
вания иногда является более предпочтительным, чем применение
карбоната серебра, в синтезе гликозидов и дисахаридов. Например,
конденсация тетра-О-бензоилбромглюкозы (1 же) с бензиловым спир-
том (1 же) в нитрометане в присутствии Р. ц. (1 же) дает тетра-О-бен-
зоилбензилф-о-глюкозу с 90%-ным выходом. Зорбах и др. [2],
используя в качестве растворителя дихлорэтан, применили эту
общую методику для синтеза сердечноактивного гликозида эво-
монозида (3) из бромида (1) и дигитоксигенина (2). После омыления
с 44%-ным выходом выделяют эвомоиозид (3). Попытка провести
190
синтез с карбонатом серебра была безуспешной, так как при этом
происходило отщепление третичной оксигруппы у С14.
1. Helferich В., W е i s К., Chem. Вег., 89, 314 (1956); Helferlch В.,
S t e i п p r e i s R., Chem. Ber., 91, 1794 (1958).
2. Z or b ach W. W., Vai i a vee dan G. D., Kash el i k ar D. V., J.
Org. Chem., 27, 1766 (1962).
РТУТЬ ХЛОРНАЯ (сулема), HgCl2. Мол. вес 271,52; т. пл. 277°;
т. кип. 304°; растворимость в 100 г Н2Ь; 3,6 г при 0й, 61,3 е при 100°;
растворима в ацетоне и 95%-т-юм этаноле.
Амальгамы. По Клемменсену — Мартину при восстановлении
Р-бензоилпропионовой кислоты в у-фенилмасляную кислоту Ш смесь
120 г цинковой пыли, 12 г Р. х., 200 мл воды и 5—6 мл конц. соляной
кислоты встряхивают в течение 5 мин, декантируют жидкость
с амальгамированного цинка и добавляют 75 .ил воды, 175 мл конц.
соляной кислоты, 100 мл толуола и 0,28 моля кетокислоты. Смесь
свн5сосн,сы.,со.,н свн3сн.2сн2сн2со2н
- 82—8П%
интенсивно кипятят 25—30 час, добавляя трижды по 50 мл конц.
соляной кислоты, и толуольный слой обрабатывают для выделения
продукта. При восстановлении по Клемменсену ванилина в креозол
[2] и бензоина в транс-стильбен [3] для получения амальгамы не
используют соляную кислоту и процесс более длителен. В одной
из методик гранулированный цинк оставляют с 5%-ным водным
раствором Р. х. на 2 час, периодически встряхивая смесь; согласно
другой методике, смесь цинковой пыли, воды и Р. х. механически
перемешивают втечение 20—30мин. Более низкие выходы продуктов
восстановления (60—67 и 53—57%) по сравнению с выходами, по-
лученными Мартином, по-видимому, можно объяснить тем, что
Мартин использовал толуол.
При восстановлении ацетона в пинакон [4] амальгаму магния по-
лучали in situ прибавлением по каплям раствора 90 г Р. х. в 505 мл
ацетона к суспензии 80 г магниевых стружек в 800 мл бензола.
Амальгаму алюминия получают in situ и используют ее для полу-
чения этилата или mpem-бутилата алюминия взаимодействием
с абсолютным этанолом 151 или /лрст-бутанолом 16]. В первом слу-
чае 27 г алюминиевых опилок обрабатывают 276 а абсолютного эта-
нола, 0,2 г Р. х. и «несколькими кристаллами иода» и смесь кипятят
в течение нескольких часов. С более высококипящим спиртом ка-
тализ иодом не требуется. Смесь 64 г алюминиевых стружек, 254 мл
mpem-бутанола и 5—10 г mpem-бутилата алюминия нагревают до
кипения и добавляют 0,4 г Р. х. По мере нагревания раствора смесь
постепенно чернеет, н нагревание прерывают на 1 час. Затем добав-
ляют 309 мл mpem-бутанола и 200 мл бензола; в заключение смесь
кипятят еще 10 час.
Ртутьорганические соединения. Несмеянов [7] получил р-на-
фтилмеркурхлорид диазотированием хлоргидрата [3-нафтиламина и
191
добавлением раствора 1 моля Р. х. в конц.соляной кислоте для
осаждения желтой комплексной соли. Комплекс собирают, промы-
вают водой и ацетоном, высушивают па воздухе (взрывоопасен) и
суспензию комплекса и порошка меди в ацетоне перемешивают при
ArNtCl". HgCl3
-ф!Ч2-ф2СиС1
Си
4 0 — 4 9%'
(общий)
70° для осуществления гладкого разложения с выделением азота.
Образующийся твердый осадок собирают и продукт (т. пл. 267°)
экстрагируют кипящим ксилолом.
Уитмор и сотр. [8] получили л-толилмеркурхлорид нагреванием
эквимолярной смеси /г-толуол сульфината натрия и Р. х. в кипящей
воде до прекращения выделения двуокиси серы (2 час)
n-CH3C6H4SO2Na • 2Н.0 -| - HgCl2 o-CH3C6H4HgCl -ф SO2 -ф NaCl -ф 2Н2О
о 1 о 7 %
Выделение гистидина. Гистидин образует плохо растворимый
комплекс с Р. х. (другие природные аминокислоты не образуют таких
комплексов) и может быть выделен в виде этого производного [9].
Например, 1,4 кг продажной высушенной пасты гемоглобина кипя-
тят с 4,5 л конц. соляной кислоты в течение 18—20 час и большую
часть избытка кислоты удаляют отгонкой. Остаток растворяют в 8 л
I ..1 сн2снсо2н
HN.^N NH2
воды, смесь доводят до pH 4,5 и оставляют до выпадения пиг-
мента. После фильтрования и осветления фильтрата норитом блед-
но-желтый раствор разбавляют до 25 л и обрабатывают раствором
600 г Р. х. в 2 л горячего 95%-ного этанола. Раствором карбоната
натрия доводят pH до 7,0—7,5 и смесь оставляют стоять до выпаде-
ния комплекса. Жидкость удаляют сифон и ров ан нем и сосуд запол-
няют до первоначального объема водой. После повторного промыва-
ния водой комплекс собирают, приготавливают суспензию в воде и
насыщают ее сероводородом при перемешивании. Сульфид ртути
удаляют, раствор концентрируют до объема 1 л, осветляют нори-
том и обрабатывают 3 об. этанола. После выдерживания в ящике
со льдом в течение 3—4 дней собирают кристаллизат монохлоргид-
192
рата г-гистидина (85—90 г). Перекристаллизация дает 75—80а
оптически чистого продукта.
Поправка. В методике, приведенной в сб. «Синтезы органиче-
ских препаратов» [101, для превращения 1,5 моля циклогексена
в транс-2-хлорциклогексапо л рекомендуется проводить реакцию
с хлорноватистой кислотой, полученной пропусканием хлора в ле-
дяной раствор едкого натра, после добавления 0,1 моля Р. х. для
образования осадка окиси ртути. Роль окиси ртути не ясна ни из
этой методики, ни из ранее опубликованной статьи Форти 1.111,
на которой основана эта методика. Форти утверждает, что он полу-
чил желтоватое масло, разлагающееся при перегонке при нормаль-
ном давлении. Остерберг и Кендалл [12] получили 2%-ный раствор
гипохлорита, пропуская СО3 в суспензию хлорной извести и отфильт-
ровывая карбонат кальция. Циклогексен, взятый в избытке, при
встряхивании с этим раствором дает транс-2-хлорциклогексанол
в виде эмульсии белого масла в воде, перегоняющейся при 760 мм
лишь со слабым разложением. Мы считаем, что окись ртути не ока-
зывает никакого полезного действия, и даже вредна.
Расщепление диэтилмеркапталей. Фишер [131 предложил Р. х.
для расщепления диэтилмеркапталя пента-О-ацетил-о, ь-глюкозы;
теперь используется [151 усовершенствованная методика Уол-
фрома [14]. Например [ 161, диэтилмеркапталь растворяют в водном
ацетоне, добавляют при энергичном перемешивании избыток вод-
ного раствора карбоната кадмия и затем постепенно добавляют из-
быток Р. х. в ацетоне. Смесь перемешивают 24 час при 25°, периоди-
чески добавляя свежий карбонат кадмия, и затем быстро нагре-
вают раствор до кипения.
RCH(SC2H5)2-|-2HgCU+CdCO3 RCHO4-2ClI-[gSC2H5-yCdCl2 + CO3
1. М а р т и и Е., «Синтезы органических препаратов», ИЛ, М., 1949, сб. 2,
стр. 511.
2. Schwarz R., Hering Н., Org. Syn., Coll. Vol., 4, 203 (1963).
3. S h r i п c r R. L., Berger A., Org. Syn., Coll. Vol., 3, 786 (1955).
4. Адамс P,, Адамс E., «Синтезы органических препаратов», ИЛ, М.,
1949, сб. 1, стр. 342.
5. Чалмерс У., «Синтезы органических препаратов», ИЛ, М-, 1949, сб. 2,
стр. 485.
6. Wayne W., Adkins Н., Org. Syn., Coll. Vol., 3, 48 (1955).
7. Несмеянов A. H., «Синтезы органических препаратов», ИЛ, М., 1949,
сб. 2, стр. 557.
8. Уитмор Ф., Гамильтон Ф., Турман Н., «Синтезы органичес-
ких препаратов», ИЛ, М., 1949, сб. 1, стр. 478.
9. Фостер Дж,, Шем и и Д., «Синтезы органических препаратов», ИЛ,
М., 1949, сб. 2, стр. 160.
10. Колс м а и Дж., Джонстон X., «Синтезы органических препаратов»,
ИЛ, М., 1949, сб. 2, стр. 496,
11. F о г t е у Е. С., J. Chem. Soc., 73, 932 (1898).
12. О s t е г b с г g А. Е., Kendall Е. С., J. Am. Chem. Soc., 42, 2621 (1920).
13. Fischer Е., Вег., 27, 673 (1894).
14. W о 1 f г о т М. L., J. Am. Chem, Soc., 51, 2188 (1929).
15. Arnold Н. W., Evans W. L., J. Am. Chem, Soc., 58, 1950 (1936),
7 Заказ № Il66
193
16. English J., Jr., Griswold P, H., Jr., J. Am. Chem. Soc., 67, 2040
(1945).
РУБИДИЯ ФТОРИД, RbF. Катализатор для реакции Кневе-
нагеля, см. Цезия фторид.
РУТЕНИЕВЫЕ КАТАЛИЗАТОРЫ ГИДРИРОВАНИЯ . 5%-иые
рутениевые катализаторы на угле (норит) и АЦО3. Некоторые ис-
следователи применяют двуокись рутения, которую восстанавли-
вают до металла in situ.
Карбонильные группы, Брейтнер и сотр. [11 показали, что
Ru—С и Rh—С заметно превосходят Pt—С и Pd—С при гидрирова-
нии кетонов в нейтральной или щелочной среде. Хасек и др. [2]
попытались восстановить тетр аметил цикл обута ндион-1,3 па пла-
Н2 (70'106 атт )
______125°,1 vac
9 8%
Т.ПЛ. 163° Т.ПЛ 148°
4оо г
в боолм метанола
типовом, палладиевом и родиевом катализаторах, однако результаты
оказались плохими. В случае медно-хромового и никелевого ката-
лизаторов выходы диолов были умеренными, и продукт реакции
содержал примеси низкоплавких веществ. При использовании про-
дажного 5%-ного рутения на угле гидрирование в метанольном
растворе протекало быстро, и с почти количественным выходом
была получена смесь, состоящая из почти равных частей цнс- и
транс-диола.
Ароматические циклы. Айрланд и Шисс [3] успешно осущест-
вили восстановление фенольного кольца в |(1), из подокарповой
(3) (4)
194
кислоты] гидрированием с добавлением двуокиси рутения в этаноле
в мягких условиях. Продукт реакции представляет собой смесь
стереоизомеров, образовавшихся, по-видимому, в основном в ре-
зультате атаки из a-области 8,9-двойной связи; при окислении
СгО^ по Джойсу образуется лабильный кетон (3), который при
эпимеризации по Сй на окиси алюминия дает стабильный ке-
тон (4). При проведении всех стадий без выделения промежуточных
продуктов кетон (4) получают с высоким общим выходом. Другие
примеры восстановления фенольного кольца в полициклических
соединениях приведены Уолтоном и сотр. [4], которые устано-
вили, что 10%-ный рутениевый катализатор на активированном
угле (дарко) превосходит ряд других катализаторов, поскольку он
не вызывает гидрогенолиза, а также Джонсоном с сотр. [5], которые
применяли двуокись рутения.
Рапала и Фаркаш 161 нашли, что при гидрировании в присут-
ствии рутения эстрона, эстрадиола и родственных стероидов (1)
в качестве главных продуктов реакции образуются 3|3-окси-5а,
10а-эстраны (2). Поскольку, по-видимому, реакция протекает через
стадию 3-кетопроизводного, стереохимия продукта по всем трем но-
вым асимметрическим центрам может быть объяснена с помощью
того же механизма атаки из и-области. В подтверждение такой
точки зрения, Коуиселл [7| позднее гидрировал эстрадиол в ще-
лочном растворе в присутствии RuO2 при 65'’ и давлении водо-
рода 103 атм и выделил 19-норстероид типа (2) с выходом 78%.
Рапала и Фаркаш [8] синтезировали другой ряд 19-норстероидов
путем гидрирования в присутствии рутения 19-нортестостерона (3),
с последующим окислением N-бромацетамидом; в качестве главного
продукта реакции образуется 5р,10р-эстрандион-3,17 (4).
Н I Н j
(3) (4)
Колобельски [91 исследовал гидрирование эиЗо-аддукта (5)
малеинового ангидрида и антрацена при высоком давлении н уме-
ренной температуре и установил, что поглощение водорода пре-
кращалось после насыщения одного из двух бензольных колец.
Второе бензольное кольцо не гидрируется, по-видимому, потому,
т
195
что молекула аддукта изогнута, вследствие чего только одно из
двух колец может расположиться на поверхности катализатора.
(5)
Ru-A12O3
Н2 (95 атм)
147° 16 час
84%
Джонсон и сотр. ПО] синтезировали транс-декалиндион-1,4
гидрированием 1,4-нафтохипона над RuOa с последующим окисле-
нием полученного диола хромовой кислотой и изомеризацией
цис-днопа в транс-дион.
Нг, 1ЫОг
СДЦОН (70°).
Фрейфелдер и Стоун [111 показали, что фен ил ал кил амины мож-
но восстанавливать в циклогексилалкиламипы с прекрасными выхо-
дами гидрированием на рутении. В аналогичных условиях пириди-
ны эффективно восстанавливаются в соответствующие пиперидины
[12].
СНаСНСН3
NHCH3
I I "|_ RuOa
X/ 2%
ЫД70 an,v),
9 0°, 1 час
90%
H. .CHaCHCH3
>< I
( ) NHCH3
Восстановление связен C=C и C=C. Беркович и Райландер [131
указывают, что при гидрировании в присутствии 5%-ного рутения
на порите избирательно восстанавливаются однозамещеиные, но
не двух- и трехзамещенные олефины. Например, в случае смеси
(1), (2) и (3) происходит преимущественное восстановление (1).
СН, СН3 СН,
I 1 !
сн3снсн2сн---сн2 СН3СН2СН2С = СН2 СН,СН2СН-ССН3
U) (2) (3)
Селективность, свойственная только рутению, подтверждается так-
же при восстановлении октеиа-1 в присутствии октена-2. Гидри-
рование проводили с дисперсией олефина в воде. Оказалось, что
ацетилены легко гидрируются до алканов, ио их нельзя превратить
в результате избирательного частичного гидрирования в олефины.
В отличие от реакций в присутствии обычных катализаторов реак-
ция гидрирования в присутствии рутения очень чувствительна к
196
природе растворителя и восстановление часто не происходит в от-
сутствие воды [14]. Это справедливо для гидрирования как при
низком, так и при высоком давлении и даже в тех случаях, когда
гидрируемое вещество в воде нерастворимо.
Хотя рутений — наиболее активный из числа известных катали-
заторов для гидрирования карбонильной группы, однако в его при-
сутствии можно количественно восстановить а, 6-непредельные аль-
дегиды до предельных альдегидов [151. Однако сотрудники фирмы
«Энгельгард» считают палладий наилучшим катализатором для та-
кого превращения. Для гидрирования до предельного спирта они
предпочитают двухстадийный процесс: восстановление двойной свя-
зи под действием палладия с последующим восстановлением альде-
гидной группы в присутствии рутения.
ArNO - ArNHNHAr. Ароматические питросоединения удобно
восстанавливать до гидразосоединений с помощью гидразина и
5%-ного Ru—С в 5%-ном спиртовом растворе едкого кали [16].
Можно также использовать и 5%-ную Pt — С, однако при этом не-
обходимы меры предосторожности, чтобы предотвратить дальнейшее
восстановление до аминов.
н н
NO PbNNHa’ СГ1Ирт- К0Н /Z~4-N
\=/ 2 80% ’ 4:=/
1. В г е i t п е г Е., R о g i п s k i E.: Rylander P. N., J. Org. Chem., 24,
1855 (1959).
?. Hasek R. H., Elam E. U., Martin J. C., Nations R. G., J.
Org. Chem., 26, 700 (1961).
3. Ireland R.E., S ch i ess P. W., J. Org. Chem., 28, 6 (1963).
4- Walton E. et al., J. Am. Chem, Soc., 78, 4760 (1956).
5. J oh nson W. S., R ogier E. R., Ackerman J., J. Am. Chem. Soc.,
78, 6322 (1956).
6. Rapala R. T., Farkas E., J. Org. Chem., 23, 1404 (1958).
7. С о и n s e 1 1 R. E., Tetrahedron, 15, 202 (1961).
8. Rapala R. T., Farkas E.,J. Am. Chem. Soc., 80, 1008 (1958).
9. Kolobielski M., J. Org. Chem., 28, 1883 (1963).
10. Johns о n W. S., A 1 1 e n D. S., Jr., liindersiun R.R., Sausen
G. N., P a p p о R., J. Am. Chem. Soc., 84, 2181 (1962).
11. Frei [el der M., Stone G. R., J. Am. Chem. Soc., 80, 5270 (1958); J.
Org. Chem., 27, 3568 (1962).
12. Freifeider M., Stone G. R.,J. Org. Chem., 26, 3805 (1961).
13. В e r k о w i t z L. M., R у 1 a n d e г P. N., J. Org. Chem., 24, 708 (1959).
14. Ryland er P.N., Rakoneza N., Steele D., Bollinger M.,
Engelhard Ind. Techn. Bull., 4, 95 (1963).
15. Rylander P. N., Himelstein N., Kilroy M., Engelhard Ind.
Techn. Bull., 4, 49 (1963).
16. P i e t г a S,, R es M., Ann. chim (Rome), 48, 299 (1958).
РУТЕНИЯ ЧЕТЫРЕХОКИСЬ, RuO4. Мол. вес 165,10, желтые
иглы, т. пл. 25,5°, желто-красный раствор реагента в четыреххло-
ристом углероде является стабильным.
Получение. Беркович и Райландер [1] получали этот реагент
окислением хлорида рутения(Ш) в разб. соляной кислоте водным
197
раствором бромата натрия, пользуясь сравнительно сложной ап-
паратурой. Позднее было найдено [21, что для отщепления брома
от того же исходного соединения более удобна экстракция его
раствора в четыреххлористом углероде водным раствором бикар-
боната. Согласно простой препаративной методике, описанной На-
ката 131, исходят из черной двуокиси рутения. Суспензию 0,4 г
RuO3 в 50 мл четыреххлористого углерода перемешивают при 0° и
обрабатывают раствором 3,2 г метапериодата натрия в 50 мл воды.
Примерно через 1 час черная окись растворяется, прозрачный слой
четыреххлористого углерода отделяют и фильтруют через стек-
лянную вату. Можно сделать приблизительный анализ этого ра-
створа, для чего аликвотную пробу обрабатывают этанолом для
восстановления Р. ч. до черной двуокиси, которую отфильтровы-
вают и взвешивают [4].
Растворители. Джерасси и Энгл [4], сравнив этот реагент с че-
тырехокисью осмия, установили, что эфир, бензол и пиридин, обыч-
но используемые в качестве растворителей для OsO4, в данном слу-
чае неприменимы, поскольку они мгновенно и бурно реагируют
с RuO4. Они нашли, что из числа обычных растворителей приме-
нимы только четырех хлор истый углерод и хлороформ; в большин-
стве работ использовали четыреххлористый углерод. Кори и др.
[51 применяли фреон И (CC1:JF, т, кип. 22°) при окислении в микро-
масштабах норборнеола-D в поркамфору-D с помощью RuO4.
Олефииы, см. также каталитический метод (стр. 201). Дже-
расси и Энгл [41 описали окисление фенантрена Р. ч. в четыреххло-
ристом углероде; реакционная смесь содержала значительное ко-
личество исходного вещества и небольшое количество фснаитрен-
хиноиа. Беркович и РаЙлапдер [41 окисляли аналогичным образом
циклогексен и получили адипиновый альдегид с низким выходом
в качестве единственного продукта реакции, который удалось выде-
лить; по их мнению, этот метод неприменим для получения альдеги-
дов и кислот, поскольку эти вещества сильно адсорбируются на об-
разующейся двуокиси рутения.
Дин и Найт [21 показали, что таким путем 3-алкилпден-2'-гри-
зены (1) можно окислить в гризеноны-3 (2). Выходы составляют
только 5—30%, однако озонирование и окисление хромовой кисло-
той вообще педали никаких результатов.
(2)
Кастелло и сотр. [6] разработали метод определения положения
изолированной двойной связи в стероидах, заключающийся в сле-
198
дующем: вещество кипятят в эфире с четырехокисью осмия, осмат
восстанавливают алюмогпдридом лития в ТГФ, расщепляют диол
тетраацетатом свинца и дикарбонильиые соединения характери-
зуют по ИК-спектрам. Дизамещенная двойная связь (Д^холестен)
дает две альдегидные группы с полосами при 2704 см~1 (слабая)
и 1730 см~' (интенсивная). Трехз вмещенная связь, как, например,
в (1) или (2), дает альдегидную группу и кетогруипу в шестичленном
(О
2700 сл.
1725 инт.
(2)
2704 сл
1728 инт.
(3) (4)
1712 ИНТ. 1705 инт.
1735 ИНК
1705инт 1705 инт
кольце с полосой при 1705 а'1. Этот метод позволяет хорошо раз-
личать АНС14) и Д8-стероиды, хотя оба являются четырехзамещен-
ными. В продукте реакции из Л8(14)-стероида (3) содержится одна
карбонильная группа в шестнчленном кольце (1712 см~1) и одна
в пятичленном кольце, поглощение которой имеет более высокую
частоту 1735 см'1; продукт реакции из Д8-ена (4) имеет две кетогруп-
пы в десятичленном кольце и поглощает только при 1705 см'1.
Снатцке и Фе л хабер [7( упростили эту методику и применили Р. ч.
для получения дикарбонильного соединения в одну стадию. На-
веску стероида (5—10 мг) окисляли с помощью RuO4 в СС14 в те-
чение 1 час, избыток реагента разрушали метанолом, раствор разбав-
ляли и отфильтровывали RuO2.
R.2CHOH-R,C = O. В предварительном эксперименте Беркович
и Райландер [1 ] окисляли транс-циклогександиол-1,2, используя
воду в качестве растворителя, и выделили продукт реакции, кото-
рый они идентифицировали, по-видимому, как циклогександион-
1,2. Наката [31 окислил несколько вторичных спиртов ряда стерои-
дов до кетонов с очень высокими выходами. Например, приготавли-
вали раствор 0,295 г 5и-андростанол-За-она-17 в 30 мл четыреххло-
ристого углерода, сверху приливали 2 мл воды и при перемеши-
вании прибавляли раствор RuO4 в СС14 до появления устойчи-
вой желтой окраски (большая часть черной двуокиси рутения
199
удерживалась в водной фазе, и роль воды сводилась, по-видимому,
к тому, чтобы желтая окраска от избытка реагента была более за-
метна). Избыток реагента разлагали 1 мл изопропанола, смесь
фильтровали, органический слой отделяли, сушили и выпаривали.
После кристаллизации получали две пори и и (всего 0,272 г) диона
у дов л етвор пте л ыюй чистоты.
Окисление простых эфиров. Беркович и Райландер [1] устано-
вили, что после краткого индукционного периода ди-н-бутиловый
эфир быстро окисляется RuO4 в СС14 при комнатной температуре.
По данным ИК-снсктроскопии продукт реакции представляет со-
бой н-бутиловый эфир н-масляной кислоты; выход практически
количественный.
При синтезе альдостерона исходя из алкалоидного предшест-
венника по Вольфу и др. [8] одной из решающих стадий этого
синтеза является окисление простого эфира (1) в лактон (2). При
окислении хромовой кислотой были получены лишь следы лактона.
Окисление Р. ч. протекало медленно и давало малые выходы, од-
нако все же удалось получить необходимое промежуточное соеди-
нение.
Серусодержащие соединения. Сульфиды инертны по отношению
к четырехокиси осмия, по быстро окисляются Р. ч. при 0° в сульфок-
сиды, которые в свою очередь окисляются тем же реагентом в суль-
фоны [4].
Окисление углеводов. Бейнон и сотр. [9] показали, что Р. ч.
лучше, чем СгО3 — пиридин (обычный окислитель) при окислении
соответствующим образом защищенных метилглюкозидов в глюку-
л о пираноз иды. Например, (1) окисляется RuO4 в четыреххлористом
углероде при 20° с образованием (2) с хорошим выходом. Если окис-
КчО.,
Г70%'
(2) (3)
200
ление проводить СгО3 в пиридине при 80°, то отщепляется метанол
и образуется в качестве главного продукта (3). Ди-О-изопропилиден-
D-глюкофураноза (4) гладко окислялась в соответствующий кетон (5).
Согласно Бейиону, окислитель следует получать из двуокиси ру-
тения и для окисления спирта брать не меиее 1 же Р. ч. Парик и
Джонс [10] усовершенствовали методику путем объединения обеих
стадий, т. е. они действовали на углевод в хлороформе или четырех-
хлористом углероде очень малым количеством (следы) Р. ч. (20 мг
на 1 г сахара), затем добавляли 5%-ный водный раствор метаперио-
дата натрия (1,3 же) при энергичном перемешивании, поддерживая
pH 6—7 (бикарбонат). Реакция заканчивается при изменении чер-
ной окраски на желтую. По этой методике кетосахара получаются
с высокими выходами, расходуется малое количество ценной двуо-
киси рутения и экономится время.
[.Berkowitz L. М., R у 1 a n d е г Р. N., J. Am. Chem. Sac., 80, 6682
(1958).
2. D е a n F. М., Knight J. C., J. Chem. Soc., 1962, 4745.
3. Nakata H., Tetrahedron, 19, 1959 (1963).
4. Djerassi C., Engle R. R., J. Am. Chem. Soc., 75, 3838 (1953).
5. С о г e у E. J., Casanova J., Jr., Vatakencherry P. A., Win-
ter R., J. Am. Chem. Soc.., 85, 169 (1963).
6. Castells L, Meakins G. D., Swindells R.,J. Chem. Soc., 1962,
2917.
7. Sna tzke G., Fell! haber H.-W., Ann., 663, 123 (1963).
8. W о 1 f f M. E., Kerwin J. E., Owings F. F., Lewis В. B.,
Blank B., J. Org. Chem., 28, 2729 (1963).
9. Bey non P. J., С о 1 1 i n s P. M., Overend W. G., Proc. Chem. Sow,
1964, 342; В e у n о n P. J., Col 1 j ns P. M., D о g a n g e s P. T., Ove-
rend \V. G., J. Chem. Soc. (C), 1966, 1131.
10. P a r i k h V.M., J ones J. K. N., Can. J. Chem., 43, 3452 (1965).
СВИНЦА ДВУОКИСЬ. Мол. вес 289,21, разл. при 290°.
HOAtCHv-> НОАгСО2Н. Гребе и Крафт [1J показали, что метиль-
ная группа метилированного фенола превращается в карбоксильную
методом, который можно охарактеризовать как окислительное ще-
лочное плавление, причем окислителем служит С. д. Этот метод был
успешно использован в случае трех крезолов, трех толуиловых
кислот, 2,4-диметилфенола и о-крезотиновой кислоты (1). Препара-
тивная методика [2] для превращения (1) в 2-оксиизофталевую кис-
лоту (2) заключается в следующем.
СН3 СОоН
/А./он А/он
I II КОН + ЗРЬО., (240-250°), HCI I II
I i.L 46-gi% * I и
'CO2H <O.,H
(1) <2)
Горячей смеси 240 г гранулированного едкого кали и 50 мл воды
в стакане из нержавеющей стали дают1 остыть и при перемешивании
добавляют к ней 40 г (0,263 моля) о-крезотиновой кислоты. Стакан
помещают в холодную масляную баню и прибавляют при перемеши-
вании 240 г (1 моль) С. д. Непрерывно перемешивая реакционную
массу, температуру бани медленно повышают до 240°; при этом сос-
тоящая из кусков коричневая масса быстро превращается в ярко-
оранжевый расплав, содержащий тяжелые кристаллы окиси свинца.
Через 15 мин температуру быстро поднимают до 250е и расплаву
дают остыть; при этом застывшая масса распределяется по стенкам
стакана. После добавления воды и сульфида натрия в количестве,
достаточном для осаждения всех ионов свинца, раствор фильтруют
и подкисляют. Рекомендуется брать 25%-ный избыток С. д.; при тео-
ретическом количестве С. д. выходы уменьшаются на 20%.
Гидрохиноны •>хиноны. Вильштеттср и Парнас 13] предложили
использовать суспензию С. д. в бензоле для окисления 2,6-диоксн-
нафталина в амфинафтохинон — крайне неустойчивый дикетон с
высоким окислительным потенциалом, разлагающийся даже водой.
Авторы обратили внимание на значительное влияние качества про-
дажного окислителя, который или был абсолютно неудовлетвори-
202
телен, или его можно было использовать только в большом избытке.
Более поздние исследователи не смогли получить хинон даже с 50-
кратным избытком С. д. Впоследствии Кун и Хаммер [41 разработали
метод получения активной С. д., обеспечивающий выделение неустой-
чивых хиноиов. Тетраацетат свинца (50 г) распределяют в пробирки
центрифуги и растирают с водой (460 мл) до его превращения в корич-
невую окись. После центрифугирования продукт промывают водой
до нейтральной реакции промывной жидкости на лакмус, затем 4 раза
ацетоном и 4 раза эфиром. Выход 23 г (92%). Для окисления 2 г
тонко распыленного 2,6-диоксинафталина добавляют к кипящей сус-
пензии 4 г активной С. д., смесь встряхивают 1—2 мин и фильтруют.
Оранжевый фильтрат концентрируют при пониженном давлении и
получают две порции амфинафтохинона с выходом 45%, что в два
раза превышает ранние результаты |3]. о-Бензохинон был получен
с удовлетворительным выходом окислением пирокатехина С. д. в
бензоле в присутствии безводного сульфата натрия. Активная С. д.
очень эффективна прн окислении 2,6-нафти леи ди бензолсульфа ми да
в 2,6-нафтохинондибензолсульфимнд [5].
И
,^zNSO.,C(tH5 Z/\SO.AH5
I II I I I ;
CeH5SO,M-CJCSCbN"
"I
H
Окисление производного пирокатехина в о-бешзохинои представ-
ляет собой первый шаг в синтезе ппоцианпна (6), впервые предло-
женном Вреде и Штраком |61 и развитом Серри 171. Моиометило-
вый эфир пирогаллола (1) окисляют при встряхивании раствора в
бензоле с большим избытком продажной! С. д. Затем раствор филь-
труют и обрабатывают о-фенилендиамином в смеси уксусной кислоты
с бензолом. Общий выход 33%, по-видимому, является прекрасным
свидетельством в пользу эффективности стадии окисления.
203
Окислительное декарбоксилирование. Деринг с сотр. [8] нашли,
что С. д., не подвергнутая специальной обработке, окисляета,Р-дикар-
боновую кислоту, например (1), или ангидрид до двуокиси углерода
и соответствующего олефина. Для немногих изученных примеров вы-
ходы составляют 20—35%. Кислоту или ангидрид измельчают вместе
с окислителем в шаровой мельнице и порошок осторожно нагревают
О) (2)
иногда вместе с тонкоизмельченным стеклом. Позднее Деринг и
Финкельштейн [9] сообщили о трудностях в воспроизведении выхо-
дов при использовании различных образцов С. д., включая и образец,
полученный по методу Куиа и Хаммера [4]. Затем они нашли, что
С. д., полученная взаимодействием тригидрата ацетата свинца со
щелочью и окислением гипохлоритом кальция, дает выходы около
19%. Тетраацетат свинца, по-видимому, является наилучшим реа-
гентом для окислительного декарбоксилирования такого типа.
Мак-Элвен и Эйзенбраун [ 10] нашли, что при окислении непето-
новой кислоты (1) по методу Деринга выделяются элементы муравьи-
ной кислоты и образуется а, ^-ненасыщенный кетон с выходом 34%.
(О
(2)
Окислительная циклизация. Активная С. д., полученная по
методу Куна и Хаммера, была использована в трех реакциях (схе-
мы которых приведены на стр. 205).
О первых двух реакциях сообщили Хассалл и Льюис [11]. При
использовании в качестве окислителя феррицианида выходы очень
низки. Третья реакция, описанная Таубом, Вендлером и др. [12],
представляет собой протекающую с высоким выходом окислительную
циклизацию п-оксибензофснона в дегидрогризеофульвеи. В этом слу-
чае окисление путем встряхивания эфирно-ацетонового раствора
с С. д. Куна — Хаммера эффективнее, чем при обработке активной
двуокисью марганца. С окисью серебра выход составлял лишь 5—
10%.
Уилмарт и Шварц [13] описали форму С. д., которая, по их
мнению, активнее формы Куна и Хаммера. Ее получают (ссылка 9 в
цитируемой работе) добавлением воды к раствору тетраацетата свинца
в уксусной кислоте и хлороформе и промыванием выпавшей С. д.
204
согсн3 СО2Н
еодой и затем растворителем, в котором она будет применяться,
Реагент использовали для окисления 1,1,4,4-тетрафенил-2,3-дибсн-
зоилтетразина (1) в гидразильный радикал (2) и для получения очень
нестабильных бногидразильных радикалов (4) [14].
(C6H5)2N-N — N—-N(C6H5)2 2(CeH5)2N — N •
L । 1
O=c C = O C-0
I I L
C6H5 CGH5 C.gHg
(1) (2)
O2Nx у NO, O.,Nx /ч 'NO,
v/'h — "ПГ
(C6H5)2NN (C6H5)2NN"z V 4'NX(C6H5)2
(3) (4)
I. G г a eb e C., Kraft II., Ber., 39, 799 (1906).
2. Тодд Д., Мартелл А., «Синтезы органических препаратов», изд-во
«Мир», М., 1964, сб. 12, стр. 116.
3. W i 1 1 s t a t t с г R., Par п as ,1., Вег., 40, 1406 (1907).
4. Kuh п R., Hammer I., Chem. Вег., 83, 413 (1950).
5. Adams R., Wankel R. A., J. Am. Chem. Soc., 73, 2219 (1951).
6. W г e d e F., S t г a c k E., Ber., 62, 2051 (1929); Z. physiol., 181, 74 (1929).
7. Surrey A. R., Org, Syn., Coll. Vol., 3, 753 (1955).
8. D о с r i n g W. E., von, Farber AA,, S a у i g h A., J. Am. Chem. Soc,,
74, 4370 (1952).
9. Doering W. E,, von, Finkelstein M., J. Org. Chem., 23, 141 (1958).
10. McE Iva i n S. M., Eisenbraun E.J., J. Am. Chem. Soc 77, 1599
(1955).
11. H a s s a 1 1 С. H,, L e w i s J. R., J. Chem. Soc., 1961, 2312.
12. T a u b D., Kuo C. Th, S 1 a i e s II. L., Wendler N- L., Tetrahedron
19, I (1963).
13. Wilmarth W. K., Schwartz N., J. Am. Chem. Soc., 77, 4543 (1955).
14. H e i d b c r g J., W e i 1 J. A., J. Am. Chem. Soc., 86, 5173 (1964).
205
СВИНЦА ДИАЦЕТАТА ТРИГИДРАТ, РЬ(ОСОСН,)2 -ЗН2О. Мол.
вес 379, 35, растворимость (г иа 100 мл Н.,О): 45,6 (15е), 200 (100е).
Нейтрализация НС1, Гидроокись свинца, полученная гидро-
лизом ацетата, является лучшим, чем окись свинца, реагентом для
выделения «-аминокислоты из ее хлоргидрата. В приведенном ниже
синтезе а-аминодиэтил уксусной кислоты )11 сырой хлоргидрат, полу-
ченный из 1 моля диэтил кетон а, нагревают в 2л воды с влажной пастой
гидроокиси свинца, образующейся при добавлении 1,5 л 2 и. едкого
натра к раствору 1,5 моля ацетата свинца в 1,3 л воды, при переме-
шивании и последующем фильтровании смеси. По неуказанной при-
чине суспензию разбавляют водой до объема около 3,5 л и, прежде
чем отфильтровать от солей свинца, концентрируют при пониженном
давлении до объема около 2 л. Фильтрат насыщают сероводородом
для осаждения сульфида свинца, который отделяют фильтрованием с
отсасыванием. Фильтрат концентрируют при пониженном давлении
почти досуха и аминокислоту перекристаллизовывают из этанола.
(сгн5)2со NaCN+NH^ (c,,h5)2ccn 2^ (c,h5),ccn ——
] | (Холодная)
OH NH2
(C2hs)2cconh2 ——(сан.)2ссо2н —(с2н6)2ссо2н
(Горячая) | 39—43 % |
+\H3C1- -NH;)C1- (°бщий) KJH2
Удаление селена и селенистой кислоты. Использование в ка-
честве окислителя двуокиси селена или селенистой кислоты ослож-
нено тем, что реакционную смесь трудно освободить от коллоидного
селена и избытка окислителя. Метод |2] окисления паральдегида в
глиоксаль и выделения его в виде бнс-бисульфитиого производного
с использованием ацетата свинца для удаления селенистой кислоты
удобнее способа с применением двуокиси серы только в том случае,
если нет большого избытка реагента, и при охлаждении раствора.
Смесь 1,72 моля селенистой кислоты, большого избытка параль-
(СЩСНОЬ ——* О = СНСН^О NaH5°U NaOgS—CH—CHSO3Na
7 2- ”4% I I
(об.’Ци.) 0H QH
дегпда, 540 мл диоксана и 40 ли 50%-ной уксусной кислоты кипятят
с обратным холодильником в течение 6 час и декантируют с осадка
селена. Затем раствор перегоняют для удаления паральдегида и
диоксана, декантируют с оставшегося небольшого количества
селена и титруют 25%-ным раствором ацетата свинца до прек-
ращения выпадения селенита свинца. Отфильтрованный раствор
насыщают сероводородом для удаления ионов свинца, обра-
батывают норитом и фильтруют. Бесцветный фильтрат концентри-
руют при пониженном давлении и перемешивают в течение 3 час
с отфильтрованным раствором бисульфита натрия в 40%-ном эта-
ноле. бис-Бисульфит глиоксаля отделяют и промывают этанолом, а
затем эфиром.
206
Отщепление H2S. Реагент использовался для превращения
тиомочевины (1) в цианамид (2) путем отщепления H2S |4]. Суспензию
0,2 моля о-хлорфенилтиомочевины (1) в кипящей воде обрабаты-
вают кипящим раствором едкого кали и затем горячим насыщенным
раствором С. д. т. После охлаждения до 0е сульфид свинца отфильт-
ровывают и бесцветный фильтрат подкисляют уксусной кислотой.
Выпавший о-хлорфенилцианамид (2) перекристаллизовывают из смеси
бензола с петролейным эфиром.
S
HN — С — NH.
Л /С1
Н\т—C^N
J zCl
РЬ(ОСОСН3)2 + КОН I
+ PbS-|-2CH3CO2K
(2)
1. Steiger R.E., Org. Syn., Coll. Vol., 3, 66 (1955).
2. R onzi о A. R., W a u g h T. D., Org. Syn., Coll. Vol., 3, 438 (1955).
3. Kurzer F,, Org. Syn., Coll. Vol., 4, 172 (1963).
СВИНЦА ОКИСЬ (ЖЕЛТАЯ), PbO. Мол. вес 223,21.
Тиле и Шлеусснер [11 получили транс, транс- 1,4-дифен ил бута-
диен реакцией коричного альдегида с фенилацетатом натрия и уксус-
ным ангидридом при температуре, необходимой для декарбоксили-
рования первоначально образующегося по конденсации Перкина
продукта. Кун и Винтерштейн [2] использовали С. о. в качестве осно-
вания; при этом выход сырого продукта повысился с 20 до 34%.
СО2Н
I Лс-Ю РЬО
С6Н5СН = СНСНО + СН.,С6Н5 ' --- ' —> с6нйсн-снсн = снс6н5
23—25%
Для чистого углеводорода в работе [3] приведен более низкий
выход, составлявший 23—25%. По этой методике смесь 1,1 моля
фенил уксусной кислоты, 1,1 моля коричного альдегида, 0,55 моля
С. о. (свинцового глета) и 155 мл уксусного ангидрида кипятят с об-
ратным холодильником в течение 5 час.
С. о. использовалась ранее |4] для превращения хлоргидрата
аминокислоты в свободную аминокислоту. Раствор хлоргидрата ала-
нина в воде кипятят в течение 1 час с С. о., смесь охлаждают,
отфильтровывают хлорид свинца, кипятят со свежей порцией С. о. и
СН3СНО CH3CHCN —2L сн8снсо,н
Nl-I3 +NH3C1-
--сн3снсо.,н
52-f>0% I
(общий) VU
207
снова охлаждают и фильтруют. Затем уменьшают содержание хло-
рид-иоиов гидроокисью свинца, осажденной из раствора ацетата
свинца. Остаточное содержание хлор ид-ионов определяют титрова-
нием и осаждают, добавляя эквивалентное количество нитрата
серебра. О других методах выделения нейтральных аминокислот
из их хлор гидратов см. Свинца тетр а ацетат, Ионообменные смолы,
Пиридин, Серебра карбонат.
1. Thiele J., Schleussner К., Ann., 306, 198 (1899).
2. Kuhn R., Winterstein A., Helv. Chim. Acta, 11, 103 (1928).
3. Корсо и Б., «Синтезы органических препаратов», ИЛ, Д'!., 1949, сб. 2,
стр. 231.
4. Кендалл Е., Мак-Кснзи Б., «Синтезы органических препаратов»,
ИЛ, М., 1949, сб. I, стр. 20.
СВИНЦА ТЕТРААЦЕТАТ, РЬ(ОСОСНЙ)4. Мол. вес 443,39.
Продажный продукт на 96—97% состоит из небольших кристал-
лов тетраацетата свинца, увлажненных уксусной кислотой и следами
уксусного ангидрида для предотвращения гидролиза. Пасту лучше
всего хранить при 5°. Для получения сухого вещества пасту пе-
реносят на вороику Бюхнера, промывают ее уксусной кислотой для
образования ровного осадка; осадок отжимают и сушат при ком-
натной температуре в темном месте под тягой. Обычно присутствие
уксусной кислоты в реакционной смеси вполне допустимо, и в этом
случае насту можно использовать сразу же после получения и иодо-
метрического анализа. Навеску вещества (около 0,5 г) растворяют
в 5 мл уксусной кислоты при слабом нагревании и добавляют
раствор 12 г безводного ацетата натрия и 1 г иодида калия в 100 мл
воды. После перемешивания в течение нескольких минут стенки
колбы обмывают водой и выделяющийся иод титруют 0,1 н. тио-
сульфатом натрия с крахмалом.
%Pb(OAc)..jz^ 2,217х(лы Ма252О3/цавеска).
После реакции избыток С. т. перед добавлением воды можно раз-
ложить этиленгликолем, в противном случае выпадает окрашенная
в коричневый цвет двуокись свинца.
Получение [1, 21. К смеси 600 жл уксусной кислоты и 400 мл
уксусного ангидрида, нагретой до 55°, в трехгорлой колбе с широ-
РЬ3О4-| 8СН3СОаН РЬ(ОСОСП3)4^2РЬ(ОСОСН3)2 + 4Н.,О
кими шлифами при механическом перемешивании добавляют 700 г
(1,03 моля) красной окиси свинца (РЬЙО4) порциями по 15—20 г.
Каждую последующую порцию прибавляют лишь после того, как
исчезнет окраска, обусловленная добавлением предыдущей пор-
ции; температуру поддерживают в пределах от 55 до 80°. В конце
реакции густой темный раствор охлаждают, собирают выделяющийся
С. т. и промывают его уксусной кислотой. Сырой продукт, не высу-
шивая, растворяют в горячей уксусной кислоте, осветляют раствор
норитом, фильтруют и охлаждают. Бесцветный кристаллический
208
продукт высушивают в вакуумном эксикаторе иад едким кали в тем-
ноте; продукт хранят в эксикаторе; выход 320—350 г (70—77%).
Обзоры [31.
Гидрохиноны—> хиноны. Использование С. т. в качестве реагента
для окисления органических соединений началось после классиче-
ских исследованийДимротомкрасителей,полученных из кошенили —
карминовой, кермезиновой и лаккаиновой кислот. Было показано,
что эти красители можно превратить мягким окислением в нестойкие
продукты, которые вначале не удавалось выделить, но которые легко
превращались мягким восстановлением в исходные вещества.
Охарактеризовав карминовую кислоту как антрахинон, содержащий
восемь гидроксильных групп, Димрот предположил, что нестойкий
продукт окисления является антрадихиноном, и с этой точки зрения
изучал окисление полиоксиантрахинонов, таких, как хинизарин и
ализарин [4]. Окисление хинизарина двуокисью свинца в бензоле
(метод Вилыитеттера и Кальба [5]) дает чистый хииизаринхинон.
Одиако последующее исследование этой реакции привело Димрота
к использованию для окисления С. т. в уксусной кислоте, и вскоре
этот реагент был признан паилучшим [6, 71. Для получения хини-
заринхипона [7] растирают в ступке смесь 20 а хинизарина, 40 г
РЬ(0Ас)4 - АсОН
65%
С. т. и 50лмуксусной кислоты и через 5 мин получают иглы дихино-
на. После растирания еще в течение 10 мин или до исчезновения
покраснения при разбавлении пробной порции водой продукт соби-
рают, промывают водой и высушивают. Для перекристаллизации
продукт растворяют в 200 мл нитробензола, раствор фильтруют на
предварительно нагретой воронке Бюхнера и разбавляют 2 объе-
мами сероуглерода; 1,4,9,10-дихинон выделяется в виде желтых игл.
Димрот [61 нашел, что карминовая кислота при реакции с уксус-
ным ангидридом, содержащим следы серной кислоты, дает полностью
ацетилированный октаацезат, однако при нагревании на кипящей
209
водяной бане смеси красителя с уксусным ангидридом без катализа-
тора до исчезновения покраснения пробной порции при разбавлении
ее водой образуется гексаацетат (см. вышеприведенную формулу).
Образование антрадихинона при окислении этого гексаацетата С. т.
явилось ключевой стадией при установлении строения карминовой
кислоты. Изучение модельных полиоксиантрахинонов показало, что
«-гидроксильные группы ацилируются значительно труднее, чем
p-гидроксильные. Современная интерпретация наблюдений Димрота
следующая. Карминовая кислота при взаимодействии только с уксус-
ным ангидридом дает гексаацетат, так как две «-гидроксильные
группы связаны водородной связью с карбонильными атомами кисло-
рода хинона. В присутствии каталитических количеств серной кис-
слоты протонируются карбонильные атомы кислорода с освобожде-
нием «-гидроксильных групп для ацетилирования и образованием
октаацетата. В дальнейших исследованиях (81 Димрот указал на
образование 1,4,5,8,9,10-антратрнхипона при окислении С. т., од-
нако продукт не был выделен в чистом виде.
Кениг и сотр. |91 нашли, что получение очень неустойчивого
дифенохипона окислением 4,4'-диоксидифенила двуокисью свинца
не удовлетворительно [5] даже при использовании активной двуокиси
свинца, полученной гидролизом тетраацетата свинца по методу Куна
и Хаммера. Однако было показано, что окисление можно успешно
осуществить действием С. т. Раствор 7,14 г С. т. в 140 г безводной
уксусной кислоты добавляют в течение 1—2 мин приЗСЙ к раствору
2 г гидрохинона в 80 мл дпоксапа. Еще через 3—5 мин слегка ох-
лаждают переливающуюся всеми цветами радуги кристаллическую
массу, продукт быстро отфильтровывают и перекристаллизовывают
из ацетона. Хинон образует коричнево-фиолетовые кристаллы с
т. пл. около 165° (с разл.).
РЬ(ОЛс)ь
/ -X /--------х. лиоксаи —ЛсОН / - - ——
но—К Д—ф" Д—он — - —о=< z^o
= / 45% %==/
Диацил ди имиды представляют собой интенсивно окрашенные
соединения, имеющие в растворе красный цвет. Обычно это кристал-
лические, устойчивые при хранении вещества; например, дибеизоил-
диимнд C(tH5CON-- NCOC,;Hr, плавится при 119,5—121,5°. Клемент
J10] нашел, что эти соединения удобнее получать окислением соответ-
ствующего диацилгидразина действием С. т. При попытке окисления
О Н Н О О О
rl I I 'I РЬ(ОЛс), । 'I
RC-N-N-CR —rC-N = N-CR
фталгидразида (1) С. т. в ацетонитриле при 0е был получен зеленый
раствор, который быстро обесцвечивался с выделением не поддаю-
щегося обработке белого полимера. Окислением в присутствии бута-
диена в качестве улавливающего агента и идентификацией продукта
210
реакции в виде аддукта (3) было доказано, что зеленый цвет раствора
обусловлен образованием диазахинона (2). Для сравнения аналогич-
ным образом (в присутствии бутадиена) С. т, были окислены цикли-
ческие гидразиды (4) и (6), и промежуточное существование цикличе-
ских диацилдиимидов (5) и (7) было доказано идентификацией аддук-
тов. Три циклических диацилдиимида проявляют необычно высокую
реакционную способность, как диенофилы в реакции Дильса — Аль-
дера. Бутадиеновые аддукты соединений (2), (5) и (7) были получены
с выходами 90, 76 и 54% соответственно. Наилучший способ окисле-
ния состоит в следующем. К смеси циклического гидразида (10 кмо-
лей) и диена (не менее 10 шмелей) в ~50 мл хлористого метилена,
содержащего 1 мл уксусной кислоты (для предотвращения гидролиза
окислителя), при комнатной температуре и перемешивании добавляют
1 же С. т. маленькими порциями по мере расходования. Непрореаги-
ровавший С. т. можно обнаружить по появлению коричневой ок-
раски (двуокись свинца) раствора, нанесенного на фильтров алии у го
бумажку с последующим испарением растворителя и увлажнением
водой (можно также реакционный раствор наносить на увлажненную
иодкрахмальную бумажку). Антрацен — относительно нереакциоп-
носпособный диен, однако все три циклических диацилдиимида дают
аддукты антрацена с выходами 71% (2), 23% (5), 3% (7). Окисле-
ние во всех случаях осуществляется довольно быстро, а уменьшение
выходов обусловлено уменьшением скорости присоединения в ряду
(2) у>(5) Д>(7). При использовании около 10 ммолей С. т. реакционную
смесь обрабатывают 7 г нейтральной окиси алюминия (первой степени
активности) и весь раствор досуха упаривают при пониженном дав-
лении. Образующееся твердое вещество хроматографируют на ко-
211
лонке с окисью алюминия первой степени активности (50 г, де-
зактивированная 10% воды, CCld, СН2С12).
а-Ацетоксилирование карбонильных соединений* Ранее в своих
исследованиях по использованию С. т. Димрот [1] отмечал, что в
кипящем растворе С. т. в уксусном ангидриде окислитель быстро вос-
станавливается. Исследования показали, что этот реагент осуществ-
ляет ацетоксилирование двух метильных групп с образованиемО-аце-
тилгликолевого ангидрида. Тетраацетат свинца выделяется по мере
О о
О
II
сна
>0
СН3С
II
о
Кипячение,
РЬЮАсЦ
40%
II II
СН3СОСНА
)О + РЬ(ОАс)3
СНзСОСНДУ
протекания процесса окисления. Ацетон реагирует при кипячении в
уксусной кислоте с образованием смеси сс-ацетокси- и а, а'-диацето-
ксипроизводных. Реакция особенно удобна для препаративных целей
в применении к соединениям с реакционноспособной метиленовой
группой и для окисления при пониженных температурах, для кото-
рого исключены побочные процессы. Ацетоуксусный эфир реагирует
особенно хорошо; в этом случае в качестве растворителя лучше ис-
пользовать бензол. С. т. (О, 84 же) добавляют четырьмя порциями к
раствору ацетоуксусного эфира в очищенном от тиофена бензоле,
поддерживая температуру 35° при охлаждении раствора. С. т. нераст-
ворим в бензоле и выпадает в виде хлопьев, а затем в кристалли-
ческой форме.
ососн3
РЬ(0АсЦ |
СН3СОСН2СОйСоН5-----------> СН3СОСНСО,СгН5
Ай-Холестенон-3 (1) имеет высокореакционноспособную метиле-
новую группу и окисляется С. т. в смеси бензол — уксусная кислота,
образуя 4а-ацетоксипроизводное (2) с умеренным выходом [11].
Для ацетоксилирования насыщенных 3-кетостероидов и стероид-
ных А4-енонов-3 необходимы высокие температуры; выходы низкие,
как, например, в реакции Л4-холестенона-3 (5) [12, 13].
212
Приведенные результаты подтверждают предположение, что
енолизация является стадией, определяющей скорость реакции аце-
токсилирования карбонильных соединений, так как склонные к еио-
лизации соединения легче вступают в эту реакцию. Кэвилл и Соло-
мон [14] предложили следующий радикальный механизм:
О ОН 0-0
|| I РЬ(ОАс)4 | II ОАс
RCH2CR'7^RCH = CR'-----> RCH = CR'^RCH-CR'—>
OAc О
I II
RCH —CR'
Хенбест и сотр. [15], допуская, что образование побочных про-
дуктов при дегидрогенизационном сочетании при высоких темпера-
турах свидетельствует о конкурентной радикальной реакции, пред-
полагают, что ацетоксилирование по обычному механизму заклю-
чается в превращении енола (7) в сложный эфир (8) и внутримоле-
кулярной передаче ацетоксигруппы к соседнему атому углерода
но- с=с- рь(°ас)4> £(АсО)3РЪО--С=С- J —>
t7)i I (8>
Q ! -> (AcO)zPb + О=С—С —
О-СОСН3 ОАс
АсО ОАс
(9)
с образованием (10). Авторы предполагают участие переходного
состояния (9). Основываясь на том, что кислота Льюиса должна
ускорять енолизацию кетона и потому способствовать его ацето-
ксилированию, Хенбест пытался провести реакцию холестанона-3
(I) с С. т. в уксусной кислоте, содержащей эфират трехфтористого
бора, в атмосфере азота. Он показал, что реакция при 25° заканчи-
вается за 145 мин. Наилучшие выходы при 21-ацетоксилировании
20-кетостероидов были получены при использовании в качестве раст-
213
верителя 5%-ного раствора метанола в бензоле. 11-Кетопрегнан
ацетоксилируется медленнее, но при 50° 9а-ацетокси-11-кетон полу-
чается с 15%-ным выходом.
Моноэфир малоновой кислоты, например III, полученный с 76 %-
ным выходом частичным омылением диэфира, окисляется С. т. в
бензоле при 50° в а-ацетоксипроизводное V, которое при 200° те-
ряет двуокись углерода, образуя cz-ацетоксиэфир VI [161. Выходы
составляют 35—80%. То, что моноэфиры окисляют гораздо легче,
чем нейтральные малонаты, объясняется образованием цикличес-
кого промежуточного соединения IV, ускоряющего енолизацию.
г о
СО2Н il
н-С4НчСНСО,СоН5 2^5^ н-С,Н9Сч< Ч;РЬ(ОАс)г
I
L ОС2Н5
III IV
СО2Н
! 200°
—>«-С4Н9ССО.,С.,Н5-------я-С4Н9СНСО.,С2Н5
I " ' 60% ' |
ОСОСНз (Общлго ОСОСНз
V VI
Окисление р, у-ненасыщенной
аллильные ацетоксипроизводпые
кислоты типа VII С. т. дает либо
VIII или IX, либо их смесь [171.
I
RCH — С—С —СО.,Н
I I
Аг СН3
VII
РЬ(ОАс1.
сн3
RCH = С —С — ОСОСН3
1 1
Аг СН3
VIII
сн3
I
RCHC—С
I ? I
СН3СОО Аг СН3
Хотя ацетоксилироваиие кетонов С. т. и ацетатом ртути обычно
дает одни и те же продукты, Залков и др. [181 сообщили, что пу-
легон с первым реагентом образует эпимерную смесь 4-ацетокси-
производных, и со вторым —2-апетоксиэпимерную смесь.
214
Родственные реакции ацетоксилирования. Иффленд [19] нашел,
что С. т. гладко окисляет кетоксимы Г в нитрозоацетаты Ш. Выше-
приведенный механизм ацетоксилирования кетона (7-МО) по Хен-
бесту предполагает образование промежуточного соединения II и пе-
R1R2C=NOH .РЬ-[,9Лс.Ь-> R1R2C=N-OPb(OAc)3 --->
I П
R. Л.
1чС-‘ О _д
р/: : ДЭАс
2 О...РЬХ
СОСН3 ОАс
Па
________ У r1r2c-n = o
~РЬ(ОАс)2 ОСОСНз
III
реходного состояния Па. Аналогичный механизм был предложен
для окисления гидразонов IV в азоацетаты VI. Реакцию проводят
при 0—10= в хлористом метилене. Можно использовать также
Н РЬ(ОАс)3
I РЬ(ОАс)* |
R2R3C=N —NR3-----------> R1R2C^=N—NR3 RiR2C —N = NR3
CH2Ci, при I
°~10° OCOCH3
IV
бензол и уксусную кислоту, но хлористый метилен предпочти-
тельнее, так как он обладает большей растворяющей способностью
по отношению к обоим реагентам.
Третья реакция, описанная Иффлеидом [20], представляет собой
окисление семикарбазона (1) в карбамат (4) с выделением элемен-
тарного азота. Можно считать, что реакция включает первоначальное
образование (2) через сложный эфир четырехвалентного свинца и
передачу ацетоксильной группы, как и при образовании соедине-
ния VI. При отщеплении азота образуется ионная частица (3),
которая путем 1,2-сдвига перегруппировывается в карбамат (4).
<с6н5),с^ Ч’Н
I
СХ(С2Н5)2
о
(Сйн5)2с~
СН3С-О+-С\(СгН5)2
il II
о о
(3)
о
II
(С6Н5)3С-ССН3
OCN(C.2H5)3
(4)
215
Фримэн [21] использовал расширенную методику окисления
гидразонов по Иффленду в новом синтезе циклопропил ацетатов.
а,Р-Ненасыщенный кетон конденсируется с гидразином с образова-
нием 2-пиразол ина, как это показано ниже для превращения окиси
мезитила в 3,5,5-триметил-2-пиразолин (1). К экстракту сырого
продукта в хлористом метилене добавляют раствор С. т. в том же
(СН3)2С=СНСОСН3
3 СН3
+
h2nnh2
Н3С
Н3С
х*СН3
-Pb(OAc)f
Н3С
(3)
сн3
н
(I)
Кипя-
> чение
60% (общий)
ът'-'К
н3с у
РЪ(ОАс)3
(2)
СН2
ВзС''с12>с/сн> + w
Н3с чососн3
(4)
растворителе при 10—15° в течение 15 мин. Образование 3,5,5-три-
метил-З-ацетокси-1-пиразол ина (3) при окислении объясняется, как
и при окислении по Иффленду, образованием эфира (2). Разложе-
ние (3) с потерей азота осуществляется обычным кипячением сы-
рого (3) до прекращения выделения газа. Опубликовано двенад-
цать примеров синтезов.
Ацетоксилирование углеводородов. В 1923 г. Димрот II] сообщил
предварительные данные об окислении ароматических углеводоро-
дов и олефинов под действием С. т. Он нашел, что толуол дает с очень
низким выходом бензилацетат; окисление дифенилметана и трифе-
нилметана протекало более легко, но не подавало никаких надежд
в препаративном смысле. Димрот наблюдал также, что анетол обра-
зует с небольшим выходом продукт присоединения двух ацетокси-
групп к двойной связи.
Криге работал с Димротом в Вюрцбурге над исследованием
механизма диазосочетапия, но результаты были первоначально опуб-
ликованы только в его диссертации (1925 г.). Затем он предпринял
сравнительное исследование реакции С. т. с олефинами и аромати-
ческими углеводородами и в 1930 г. опубликовал результаты в своей
первой статье [22]. Как отмечал Димрот, циклогексен реагировал
частично с присоединением двух ацетокспльиых групп, образуя сме-
си цис- и транс-иродуктов (2), и частично с ацетоксилированием в
216
аллильное положение (3), однако выходы были очень низки и цен-
ный высокой ипящий продукт остался неидентифицированным.
Инден реагировал аналогичным образом. С циклопентад неком
и 2,3-диметилбутадиеном были получены главным образом неболь-
шие количества 1,2-диацетоксгшроизводных.
В 1911 г., за десять лет до того, как Димрот ввел использование
С. т. в органическом синтезе, Мейер [23] показал, что добав-
ление двуокиси свинца к раствору антрацена в уксусной кис-
лоте приводит к образованию 9-ацетоксиантрацена с 40—50 %-ным
выходом и небольшого количества оксаитронапетата, Это наблюде-
ние не привлекало внимания до 1938 г., пока Гершберг [241 не нашел,
что некоторые полиядерные углеводороды при окислении С. т. в
уксусной кислоте дают только ацетоксильиые производные, причем
в некоторых случаях с исключительной легкостью. Окисление 1,2-
бензантрацена в уксусной кислоте при нагревании на кипящей водя-
ной бане дает чистое 10-ацетоксипроизводное (1) с умеренным выхо-
дом, тогда как 10-метил-1,2-бензантрацен реагирует более сложным
образом, давая 10-ацетоксиметильное производное (2) с низким выхо-
дом. 1,2,5,6-Дибензантр ацен (3). не реагирует вовсе, и окислением
С. т. можно эффективно отделять желтую примесь от углеводорода,
полученного по реакции Элбса. Метилхолантрен окисляли при мед-
ленном добавлении по каплям раствора С. т. в уксусной кислоте к
охлаждаемому льдом раствору углеводорода в бензоле при перемеши-
вании. Главным продуктом был 15-ацетокси-20-метилхолантрен (4),
продукт бензильного окисления аналогичный (2). Небольшое коли-
чество 15-кетона (5) образуется, возможно, через 15,15-диацетоксисое-
динение. 3,4-Бензпирен окисляли при смешении растворов окисли-
теля в уксусной кислоте и углеводорода в бензоле при комнатной
температуре; реакция заканчивалась за 0,5 час, а выход почти чи-
217
стого 5-ацетокси-З, 4-бензпирена (6) составлял 94%. Метилхолант-
рен и 3,4-бензпирен — наиболее сильные из известных канцерогенных
веществ; 1,2-бензантрацен неканцерогенен, а 1,2,5,6-дибензантрацен
обладает значительным канцерогенным действием.
Бензильное ацетоксилирование под действием С. т. имеет препа-
ративное значение в случае аценафтена [251, причем выход с заранее
полученным реагентом не лучше, чем с реагентом, генерируемым в
момент реакции из РЬ3О4. 7-Ацетоксипронзводное, очищенное пере-
30—82%
РЬаО4 — АсОН (60—7 0 s)
70-Н %
(общий)
гонкой, содержит следы аценафтена и аценафтенона, но после омы-
ления и кристаллизации получают чистый аценафтенол-7.
Путнам |261 повторно исследовал окисление антрацена С. т.
и получил интересные результаты. Окисление в уксусной кислоте
при 35—40е или в смеси бензол — уксусная кислота при 50—55°
дает небольшое количество нового соединения, идентифицированного
как 9,10-диацетокси-9,10-дигидроантрацен (2) с т. пл. 173е. При
окислении в кипящем бензоле в течение 45 час образуются значи-
РЪ(ОАс)4
в с6н&
(2) цис и -транс
Горячая
АсОН
- АсОН
ОАс
РЬ(ОАс)4 в С6Нй
Pb(OAc)4 I В. АсОН
АнтрЯ'
хинон
218
тельные количества этого соединения и его геометрического изоме-
ра с т. пл. 127°. При нагревании раствора любого из этих изомеров
в уксусной кислоте с хорошим выходом получается 9-ацетоксиант-
рацен (3). Таким образом, ацетоксилирование антрацена в уксус-
ной кислоте в (3) состоит в 1,4-присоединении ацетоксильных групп
к диеновой системе и 1,4-элиминировании уксусной кислоты. Изо-
мерные продукты присоединения (2) достаточно устойчивы к дей-
ствию горячего бензола, но нестойки в уксусной кислоте. Аце-
токсилирование 3,4-бензпирена — это, безусловно, прямое замеще-
ние, так как углеводород не обладает способностью к присоедине-
нию, например к малеиновому ангидриду.
Второй продукт, идентифицированный Мейером,— «оксантрон-
ацетат» [(5), точнее 10-ацетокси-9-антрон] — был получен с умерен-
ным выходом окислением 9-ацетоксиантрацена (3) С. т. в уксусной
кислоте. Окисление (3) в кипящем бензоле привело к 1,4-присоеди-
нению с образованием триацетоксипроизводного (4). Это соедине-
ние при нагревании в уксусной кислоте превращается главным обра-
зом в Ю-ацетокси-9-антрон (5) при потере уксусного ангидрида и в
меньшей степени в 9, 10-ди ацетокси антрацен (7) при потере уксусной
кислоты. Если (4) является промежуточным соединением при окис-
лении (3) в (5) в уксусной кислоте, то ацетоксигруппа в продукте
(5) должна быть присоединена к иным м.езо-углеродным атомам,
чем в (3). Правильность этого заключения была подтверждена окис-
лением 2-метил-9-ацетоксиаптрацена и идентификацией продукта
встречным синтезом 2-метил-10-ацстокси-9-аитрона. При дальней-
тем окислении С. т. в уксусной кислоте (5) и (7) дают антрахи-
нон, по-видимому, через продукты ацетоксидирования (5) и 1,4-
присоединения к (7).
Расщепление виц-гликолей. Вторая статья Криге [271 (1931)
также основана на его экспериментальной работе в Вюрцбурге и
посвящена описанию ставшего теперь классическим расщепления
ешргликолей С. т. в уксусной кислоте при комнатной температуре
до альдегидов, кетонов или их смесей в зависимости от строения
гликоля. Криге выделил карбонильные компоненты в виде п-нитро-
фенилгидразонов с выходом 80—90% и показал, что в/тщ-гликоль
можно количественно титровать 0,1 н. раствором С. т. в уксус-
ной кислоте.
—С—ОН рЬ(ОАс)< —с^О
—С—ОН ~С = О
1 1
Типичное препаративное использование реакции заключается
в следующем [281. К смеси 1,24 моля ди-и-бутил-о-тартрата и 1,25 л
бензола при энергичном перемешивании в течение 25 мин добав-
ляют 1,3 моля С. т., периодическим охлаждением поддерживая тем-
пературу 30\ После перемешивания в течение 1 час до превращения
219
С. т. из вязкого в кристаллическое неорганическое вещество его
отфильтровывают и промывают бензолом. Бензол и уксусную кис-
лоту отгоняют при 65740 мм и перегоняют н-бутил гл иоксал ат при
65—79720 мм.
СОаС4Нэ-а
неон
носн
I
С02С4Н,-н
РЬ(ОАсЦ В СеН,
77—8 7%
СО.,С4Н9-н
2 I ' + РЬ(ОСОСНз)афСН3СООН
сно
Для реакции расщепления гликолей характерна специфичность,
высокая скорость и общность применения. Расщепление легко осу-
ществляется для соединений со следующими группами:
—С—ОН
—С-ОН
I
-С—nh2
—С—NH?/
-С—ОН
—i—NH<
а также для а-оксикислот, а-оксиальдегидов, а-оксикетонов и щаве-
левой кислоты.
Таблица 1
Скорости расщепления гликолей
Соединение цис транс
Индандиол-1,2 27 800 0,47
Тетралииднол-1,2 40,2 1,86
Циклогекса идиол-1,2 5,0 0,22
у-Лактон D-манненовой кислоты у-Лактон D-арабоновой кислоты 39 0,01
а-Метил- D-маппофурацозид Этил- D-глюкофураиозид-5,6-карбонат 900 0,01
В первом из трех сравнительных исследований относительных
скоростей расщепления гликолей под действием С. т. в уксусной
кислоте Криге [29—311 отметил, что в шести парах цис-транс-изо-
меров или близких изомеров, приведеиых в табл. 1, цис-изомер ре-
агирует гораздо быстрее транс-изомера. Допустив, что это соотно-
шение носит общий характер и что гидроксильные группы в цис-изо-
1 I i
-С—ОН РЬ(0Асц -С—О. -С^О
| ------| 5РЬ(ОАс), Н-РЬ(ОАс)2
—С—ОН —2АсОН —С—О7 ' —С = О
I I !
(о
220
мере более сближены, чем в транс-гликоле, и отметив бимоле-
кулярпость реакции, Криге предположил, что расщепление про-
текает через промежуточный циклический сложный эфир I.
Кун [32] измерил расщепление полос ОН-групп в ИК-спект-
рах нескольких пар диолов. Полученные им результаты приве-
дены в табл. 2. Измеренные значения величин расщепления Av
обратно пропорциональны расстоянию между гидроксильными
группами. Константы скоростей расщепления гликолей совпадают
Таблица 2
Расстояние между ИК-полосами и скорости
расщепления тетраацетатом свинца
Циклический 1,2-диол ~1 k 00
С5-цнс С5-транс цис 61 транс 0 цис 40 000 транс 12,8
С^-цис Се,-транс 38 33 - 0,2
С7-цис С7-транс 44 37
С^ци.с С^транс 51 43
С9-цис CS)- транс 49 45 2,9 20,7
44 45 2,6 100
Сп-транс 38 51 1 А 73,6
Сггцис Cv,-транс — 50 7,8 91,2
со значениями, полученными Куном из литературных данных. Из
рассмотрения значений Av следует, что в циклах, содержащих
менее 10 углеродных атомов, гщс-гидроксильные группы более
сближены, чем транс, однако в циклах, содержащих 10 и более
углеродных атомов, более сближены транс ОН-группы. Обращение
221
наблюдается также и для относительной реакцион неспособности, но
уже начиная сС(), а не с С1(). Из сравнения полученных данных сле-
дует, что между скоростями реакции и Av нет простой корреляции,
и, следовательно, расстояние между ОН-группами не является
основным фактором, определяющим скорость реакции, В более
поздней работе Криге [311 опубликованы константы скоростей
^г°° 0,04 1.1,8
лЮ'лг’ щ
46
2 39 0
60
3000
65
для ряда бициклических пинаконов, а также определенные Куном
значения Av. Наблюдается примерный параллелизм этих значений.
Однако при изучении изомерных 9,10-диметилдигидрофенантрен-
диолов-9,10 выявилось сильное несоответствие, В 1940 г. единст-
венно известному в то время производному фенантренхинона —
изомеру с т, пл. 164° —- на основании высокой скорости расщепле-
ния (/г20°^= 192) приписывалась цис-конфигурация [301. Однако
быстро расщепляющийся диол не реагировал с дикалийтетра-
мет и л осматом — очень чувствительным реактивом на цис-диолы,
что стимулировало исследование осмилирования 9,10-диметилфе-
нантрена [311. Это привело к получению второго изомера ст. пл.
104°, который реагировал более медленно (/г20°-^7,5), хотя, судя по
методу его синтеза, он должен был бы быть ^мс-диолом.
Т. пл .164° Т.пл. 104°
192, д„0 7,5, д^27с^-1
Таким образом, Криге установил, что быстро расщепляющийся
диметилдигидрофенантрендиол является транс-изомером, а мед-
ленно расщепляющийся — цис-изомером. Аналогичное соотноше-
ние скоростей наблюдается для двух 9,10-дигидрофенантрендио-
лов-9,10; Бут и Бойланд [341 с помощью расщепления доказали, что
изомер, полученный восстановлением фенантренхинона, является
трдас-изомером, и нашли, что только другой изомер образует аце-
тонид.
222
Приведенные результаты опровергают предположение, что рас-
щепление гликоля протекает через циклический сложный эфир,
так как такой эфир вряд ли может образовываться из транс-диги-
дрофенантрендиол а, в котором гидроксильные группы ориентиро-
ваны диаксиально и антипараллельно. Кроме того, этот механизм
не объясняет сильный основной катализ водой, метанолом или аце-
татом калия [35]. Например, константа скорости для ^ас-диметил-
дигидрофенантрендиола увеличивается с 7,5 в уксусной кислоте
до 202 в смеси CH;iCOOH с СМ3ОН (75 : 25) и до 2430 в смеси
СНяСООН с СНдОН (25 : 75). Вода еще более эффективна, чем мета-
нол, но увеличение содержания воды выше 15°6 вызывает гидролиз
реагента. Для объяснения каталитического эффекта и быстрого
расщепления транс-дигидрофенаптрендиолов предложен 1321 со-
гласованный механизм с участием в образовании переходного сос-
тояния II основания Льюиса (НО-, С113О~), так как диакспальные
антипараллельныс гидроксилы копланариы и поэтому идеально
ориентированы для образования и распада переходного состояния.
Гораздо более медленное расщепление изомерных цис-диолов, а
также С5- и С6-диолов может быть обусловлено тем, что гидроксиль-
ные группы находятся в неблагоприятной скошенной ориентации;
катализ основаниями указывает на ациклический механизм II
реакций.
/ jO~^Pb(OAc)3 __ >с=о + РЬ-(ОАс)3
НО—*Н-0^-е^ НОН 4- о=с<^
II
Аналогичный процесс, по-видимому, имеет место при взаимо-
действии С. т. с антраценом в уксусной кислоте; реакция приводит
к 9,10-диацетоксипроизводному и сильно катализируется водой,
метанолом или ацетатом калия [36]. При добавлении лишь 0,6% воды
время расхода 0,5 моля реагента при 25° сокращается с 40 до
14 час.
Белл [37] нашел, что расщепление гликоля С. т. заметно катали-
зируется трихлоруксусной кислотой; кислотный катализ объяс-
няется переходным состоянием III. О практическом применении
I
СН3
>С=О 4- РЬ(ОАс)а
И4" 4- О-С< (К Д)Н
I
СНз
ш
катализируемого кислотами расщепления гликоля сообщил Гроб
[38]. Диаксиальное соединение IV при обычных условиях не расщеп-
ляется в Д3'8-циклодекадиендион-1,6 (V), однако в метаноле в при-
223
сутствии трихлоруксусной кислоты оно образует высокоплавкий
тетраметилкеталь VI с 75%-ным выходом.
он
он
IV,Т пл.83°
V, г.пл.185°
СИЮН
CChCOiH
СН3О ОСН3
VI,т пл 202°
Интересный случай получения из холестерина четырех изомер-
ных В-кольцевых енц-диолов изучен Анжиалом и Янгом [391. Коль-
цо В, расположенное между двумя другими кольцами, является
жестким и недоступно для атаки.
Константы скоростей расщепления под действием С. т. в уксусной
кислоте приведены под формулами. Большую реакционную способ-
ность 6{3,7р-диола (8) по сравнению с 6а,7а-диолом (9) можно
объяснить отталкиванием 6р-гидроксильной группы аксиальной ме-
тильной группой, что способствует связыванию с экваториальной
ОН-группой, а также тем, что при расщеплении (8) снимается прост-
ранственное напряжение, обусловленное взаимодействием СН3: ОН.
т/аднс-Диэкваториальный диол (10) менее реакционноспособен, чем
любой из двух цис-диолов. Диаксиальный диол (11) не расщепля-
ется в экспериментальных условиях; кислотный катализ не исследо-
вался. Холестантриол~3р,5а,6р (13) с диаксиалытой еиц-гликоль-
ной группой легко окисляется по методике Гроба, образуя 6,6-
диметоксипроизводное 5,6-секо-5-кето-6-альдегида (14) [401.
224
Перлин 14П нашел, что пространственно затрудненные виц-
гликоли ряда сахаров, устойчивые к расщеплению под действием
С. т. (в уксусной кислоте) или иодной кислоты, часто быстро взаимо-
действуют с С. т. в пиридине. На холестантриол-3р,6(3,7о. Анжиала
расходуется 0,8 моля реагента в пиридине при 0° за 6 час.
В поисковом эксперименте 1932 г. Криге [421 показал, что
D-глюкоза быстро расходует С. т. без конкурентного образования
формальдегида или муравьиной кислоты. При тщательном изуче-
нии реакции Перлин и Брайс [43] нашли, что D-глюкоза реагирует
с С. т. гораздо быстрее, чем цис-циклогександиол-1,2; в 98%-ной
уксусной кислоте при 25° 2 моля реагента расходуется за 3 мин.
d-Глюкопираноза (I) первоначально претерпевает расщепление
сс-оксиполуацетальной группы по Сх—-С2, давая II—4-формил-о-ара-
бинозу, стабилизирующуюся циклизацией в пиранозу III. Обра-
зование затем циклического эфира IV за счет миграции формиль-
ной группы приводит к З-формил-о-арабинозе V.
1 - Ci HOCH о=сн 1 с2 нс—он
С2 неон нс = о носн
НОСН ( j носи с 5 —> неон с )
неон ИСОН нсосно 1 НС-Оч /ОН
1 1 1 1 >с \н нс—о н
НС НС- СН2— i
I I
сн,,он СН.2ОН
I “ II III
I
Н— С —он
носн
I
нсосно о
I
неон
1
сн,__-
V
РЩОАсЦ
1
нс=о 1 неон
о=сн 1 1 нсосно п
НСОСНО О I 1
[ НС —О. юн нсосно
неон 1 J
1 СНо- пс-о/чн 1 сн,
VI VII
8 Заказ ХЬ 1 1 6G
225
а-Оксипол у апетальная группа V расщепляется вторым молем С, т.,
образуя 2,4-дпформил-о-эритрозу (VI), которая после мигра-
ции ацильной группы и циклизации дает конечный продукт 2,3-
диформпл-о-эрптрозу (VII). Приписанные формильным группам
положения возможны, но не однозначны; все эти сложные эфиры
очень лабильны и гидролизуются даже водой. Расщепление гликолей
С. т. нашло применение для определения структуры и конфигура-
ции природных октулозы [44] и нонулозы [45].
Окисление фенолов. Бамбергер [46] получил диенон н-толухи-
нола (2) катализируемой кислотой перегруппировкой /ытолилгид-
роксиламипа (1), В современной методике (Гудвин [471) гидрокси-
ламин добавляют к частично замороженной разбавленной серной
кислоте, смесь встряхивают и доводят до комнатной температуры; из
полученной сложной смеси хроматографически выделяют (2) с вы-
ходом 33%. Менее удачным способом получения (2) является окисле-
ние n-крезола надуксусной кислотой в нейтральной среде (Бамбер-
гер [48]). Кеннер [49] нашел, что 2,6-диалкил-4-нитрофенолы окис-
ляются С. т. в 2,6-диалкил-1,4-бензохиноиы. Затем Вессели [501
провел ряд экспериментов по окислению замещенных фенолов С. т.
в /?- и о-арепоны типа (3) и (8) [501. Например, n-крезол при окисле-
нии дает смесь n-толухинолацетата (3) и диацетокси-о-аренона (8).
Гудвин и Виткоп [47J использовали этот метод для получения орто-
продукта (8) с 27%-ным выходом, но не сообщили о выходе пара-
продукта (3), выделенного из смеси. Очевидно, получение (3) через
(7) (8) т.пл. 142®
(1) и (2) — паилучшпй препаративный метод, хотя выход на пер-
вой стадии очень низкий. Отметим сходство этих продуктов с про-
226
дуктами, образующимися при окислении антрацена С. т. о-Толу-
хинол, по-видимому, получается следующим образом:
РЬ(ОАс)
РЪ(ОАс)4^
-АсОН'*’
Окисление эстрона С. т. дает диенон с ~ 20%-ным выходом [511.
Замещение стероидных ангулярных метильных групп. С. т. ис-
пользовался для окисления стероидных спиртов, содержащих гид-
роксильную группу в положении, удобном для атаки на аннуляр-
ную метильную группу, как в Зр-ацетокси-20р-окси-5а-прегнане
(1). Аригони, Ягер и сотр. [521 кипятили (1) в бензоле в течение
ночи с 2 г свежевысушенного С. т,, хроматографировали реакцион-
ную смесь и выделили 200 мг исходного вещества и 250 мг 18,
20-окиси (2).
Б оу ер с и сотр. [531 сообщили об окислении с хорошим выходом
а ндростан-Зр, 6р, 17Р-триол-3,17-диацетата (3) С. т. в кипящем бен-
золе (18 час) в 6р,19-окись (4).
8*
227
Превосходные результаты при осуществлении окислительной
циклизации указанного типа были получены группой химиков
[54], применивших сочетание иод — С. т. Известный как «гипоио-
дитная реакция» вследствие генерирования реагентами HOI, этот
метод по простоте и эффективности превосходит метод с исполь-
зованием одного С. т. [55], а также другие три метода без примене-
ния С. т. По улучшенной методике [541 ацетат прегненолона (5)
сначала восстанавливают трис-(трет-бутокси)-алюмогидридом ли-
тия в ТГФ в 3[3-ацетокси-20[3-окси-АГ)-прегнен (6); 20а-изомер обра-
зуется в малых количествах. В конце восстановления по каплям
при перемешивании добавляют водный раствор сульфата аммония
для разрушения избытка реагента и осаждения металлических ком-
понентов. Отфильтрованный раствор упаривают досуха и продукт
восстановления (6) кристаллизуют из ацетона. На следующей ста-
дии смесь 0,0835 моля (6), 0,37 моля продажного С. т., содержащего
10% уксусной кислоты, и 0,095 моля иода в 3 л циклогексана при
перемешивании нагревают до кипения при помощи 1000-ваттной
лампы. После исчезновения окраски иода (60—90 лшн) смесь охлаж-
дают до комнатной температуры, фильтруют для удаления ацетата
свинца и соответствующей обработкой выделяют смесь 18-иод- и
18-окси-18,20-окисей (7). На заключительной стадии смесь окис-
ляют по методике Джонса в 18 -+ 20-лактон 3§-ацетокси-20|3-оксн-
А5шрегнен-18-овой кислоты (8). О других примерах «гипоиодитной
реакции» см. работу [56].
При изучении окислительной циклизации 20-оксистероидов
в 18,20-окиси Ягер и сотр. [571 наблюдали фрагментацию с образо-
ванием 17-ацетоксипроизводных. Ими исследовано окисление раз-
личных монооксистероидов под действием С. т. в кипящем бензоле.
19-Оксикетостероид (9) превращался с высоким выходом в 10-ацето-
228
ксистероид (Ю). 19-Окси-Л5-стероид (11) давал 6р-ацетокси-19-нор-
стероид (12). Другие наблюдавшиеся реакции фрагментации приве-
дены ниже. Применять для фрагментации иод не следует.
Бартон указал на два интересных случая применения смеси
иод — С. т. Это, во-первых, гладкое декарбоксилирование первич-
ных и вторичных карбоновых кислот в соответствующие иодиды,
как в следующих реакциях [58]:
РЩОАсЦ -1,
СН3(СН.,)5СН(СН2)10СО2Н --—-—СН3(СН2)5СН(СН2)ВСН21
j °* /о |
ОАс ОЛс
Pb(OAc)4- is
Циклогексанкарбоновая кислота -- 1 ->• Циклогексил иодид
9 * %
РЬ(ОАсЦ- 12
НО2С(СН2)4СО2Н ----------- 1СН2СН2СН2СН31ЦСО2
33%
К 5%-ной суспензии С. т. в четырех хлор истом углероде при пере-
мешивании и освещении электрической лампой добавляют карбоно-
229
вую кислоту, а затем иод до появления устойчивой окраски. Если
кислоту не добавлять, протекает следующая реакция:
РЬ(ОСОСЫ3)4 -I-13 2СН31 + 2СО. + РЬ(ОАс),
Во-вторых, превращение стероидного амида (17) через N-иода-
мид (18) в лактон (19) [591. Смесь амида (17) с 3 молями
С. т. и 4 молями пода в хлороформе облучают при 15° в течение
5 час.
Окислительное декарбоксилирование. Пытаясь получить 1-кар-
бэтокси-А2-бицикло-[2, 2, 2]-октен (8), Гроб и сотр. [601 синтезиро-
РЬ(ОАс)4-Ру-С6Н^
7)%
230
вали ангидрид (6) и исследовали окислительное декарбоксилиро-
вание двуокисью свинца по Дерингу (см. Свинца двуокись). Даже
при использовании активной двуокиси свинца Куна и Хаммера
реакция протекает плохо и выход (8) составляет лишь 30—37%.
Швейцарские исследователи показали, что выход можно удвоить
окислительным декарбоксилированием дикислоты (7) С. т. и пири-
дином (1 же) в кипящем бензоле. Позднее было показано, что этот
реагент еще более эффективен в ДМСО, содержащем 1 же пиридина
[611.
Бёчи 1621 осуществил окислительное декарбоксилирование моно-
карбоновой кислоты (9) кипячением ее с С. т. в бензоле в атмосфере
азота в течение 14 час. Олефиновые изомеры (10) и (11) разделяли
газохроматографически и выделили в небольших количествах. Груп-
па Бёчп столкнулась со случаями, когда в результате окисления
монокарбоновой кислоты получался не олефин, а сложный эфир,
в котором карбоксильная группа была замещена на ацетоксигруппу
[631. Такой же случай встретился Коупу [641 при исследовании оки-
сления 4-циклогептен-1-карбоновой кислоты (6), полученной по
новому методу, опубликованному Сторком [65]. При взаимодейст-
вии пирролидипенамина циклопентанона (1) с 1 же акролеина в диок-
сане первоначально при 0° образуется бициклический аминокетон
(4) путем присоединения по Михаэлю к (2). По-видимому, происхо-
дит обмен енаминной группы между кетокарбонильной группой и
(1) (2) (3)
55%, считая
на (1)
(3) (4)
231
более реакционноспособной альдегидокарбонильной группой с обра-
зованием (3) и последующая циклизация в (4). Соответствующий иод-
метилат (5) расщепляется основанием до 4-циклогептен-1-кар бо-
новой кислоты (6). Енамин циклогексанона тем же путем дает
с 75%-ным выходом бициклический аминокетон, аналогичный (4),
образующий далее 4-циклооктен-1-карбоновую кислоту. Коуп и
сотрудники показали, что циклогептен кар бонов а я кислота (6) при
окислении С. т. в уксусной кислоте дает с хорошим выходом
1 -ацетокси-А4-циклогептен (7).
РЬ(ОАс)4 в АсОТЦ
70%
^^^-ососн3
Таким образом, окисление кислоты с карбонильной группой при
насыщенном атоме углерода может дать либо олефин, либо ацето-
ксипроизводное; возможны следующие превращения:
—С—Н
I Z
^с-с/<—он
1
РЬ(ОЛс)4
- АсОН
— С— н —с—н
I/O -^ | о+'РЬ(°Ас>=
— С—с—ОРЬ(ОАс)3 — С—С^О
РЬ(ОАс)3 РЬ(ОАс)2+ -ОАс
— С
I! ДАсОН
— С
•ОАс / |
I
— С —н
I
— С—ОАс
При синтезе бензола Дьюара (10) Ван-Тамелен и Паппас [66]
осуществили окислительное декарбоксилирование ангидрида (9)
действием С. т. в пиридине при 43—45°,
Эфир
РЪ(ОАс)4 -Руч
-2 СОг '
<9) (10)
232
В модельном опыте Мейнвальд и сотр. [67] нашли, что малоно-
вую кислоту (11) можно удовлетворительно превратить в 5-ионанон
окислением С. т. в ацетонитриле (детали не опубликованы). Однако
применение этой реакции к циклической малоновой кислоте (13)
дает лишь следы желаемого кетона (14), как это было показано газо-
вой хроматографией и спектральным анализом.
сн3 сн2 снгснг . ✓ со2н
С.
CH3CH2CH2CH2Z со2н
РЪ(ОАс)4
CHjCN Л
СН3СН2СП2СН,
;с=о
СН3СН2СН2СН2
(Н) (12)
РЬ(ОАс)4
сн3ск *
(13) (14)
Кори и Казанова [68] нашли, что окисление мезо- или d,1-1,2-
дифенилянтарной кислоты С. т. в пиридине дает транс-стильбен
(выход 40—45%). Было показано, что г(пс-стильбеи в качестве про-
межуточного соединения не образуется. Аналогичное окисление
РЬ(ОАс)4
ОСОСНз -------Л.
py
эндо- или экзо-норбориан-2-карбоновой кислоты привело к экзо-
норборнилацетату (24—67%). Имеются доказательства промежу-
точного образования карбоииевого иона. Применение этой реакции
к кислоте (1) привело к образованию лактона (2) в качестве единст-
венного продукта [69],
РЬ(ОАс)4—Ру
35%
(2)
Окислительное декарбоксилирование — ключевая стадия при
объяснении строения фотохимического аддукта бензола с 2 молями
малеинового ангидрида (3) [701. Продукт окисления (4) является
аддуктом малеинового ангидрида и циклооктатетраена.
233
Pb(OAc).-Ру
—-———-----
56%
(4)
Уинстейн и сотр, [70а] действием С. т. и пиридина в бензоле
окислили кислоту I в ацетат II с почти 60%-ным выходом.
РЬ(оАс)4Ру-С6Н6>
Окислительное деметилирование. 1 моль С. т. в бензоле окисляет
21-дезоксиаймалин-17-ацетат (1) в 2-оксипроизводное (2) [711; при
избытке окислителя образуется 1-деметилированное А1-соединение —
индолении (3).
(2)
Окислительная перегруппировка. При окислении ароматического
амида С. т. протекает реакция, родственная перегруппировке N-
бромамидов по Гофману, с образованием изоцианата или его произ-
водного с выходами от умеренного до очень хорошего [721. Простые
ал иф ат ичес кие а м иды л ег ко р еа г нр уют в ДМФ А, содержащем тр и -
эти ламин.
О О
II РЬ(ОЛс)* I!
RC—NH2------------ RC—N: RN=C=O
Гало де карбоксилирование. Коши [731 нашел, что обработка
карбоновой кислоты С. т. и ионным галогенидом, обычно хлорис-
тым литием, приводит к реакции, в результате которой карбоксиль-
ная группа замещается на атом галогена. Соль добавляют к раство-
RCO2H-|-Pb(OAc)4-[-LiCl —> RCl + COa-pPb(OAc)3 + LiOAc-pAcOH
234
ру кислоты и С. т. в бензоле и смесь нагревают до 80° при перемеши-
вании в атмосфере азота. Выходы алкилхлоридов в этой реакции
высокие, однако бензойная кислота превращается в хлорбензол лишь
с 8%-иым выходом.
Расщепление циклопропанов. Согласно предварительному сооб-
щению, С. т. в уксусной кислоте расщепляет алкил- и ар и л цикло-
пропаны в 1,3-диацетоксиалканы и моноацетоксиолефины [74]. Реак-
цию рассматривают как электрофильную атаку С. т. на цикл опро-
СН2- РЬ(ОАс)з
-> С6Н5-СИ ---->
Х’СН2ОАс
^СЩОАс
С6Н6—СН
''"'‘СЩОАс
6 5%
шена
с6н£-сн |
^сн2
ОАс рЬ(ОАс)3
"^С6Н5-СН Jni, ----------->
"Х’Н2
Оде ОАс
I I 5%
с6н5—сн ^сн2
"сн2
РЬ(ОАс)3
-> С6Н5-СН СЩОАс
хсн2
пановое кольцо с образованием разлагающегося далее у-ацетоксисви-
пецорганического ацетата. Результаты, приведенные для расщеп-
ления фенилциклопропана при 75°, включают и аналитические дан-
ные, полученные газовой хроматографией. п-Бромфенилциклопро-
пан реагирует примерно в два раза медленнее фенилциклопропана
и образует больше продуктов симметричного расщепления. Моноал-
килциклопропапы также реагируют медленнее фенилцйклопропана.
Метилирование. Открытие этой реакции было результатом слу-
чайного наблюдения, сделанного одним из авторов при попытке
усовершенствовать известную методику превращения аддукта бута-
диен— толухинон (I) в 2-метил-1,4-нафтохинои (IV) 175]. Окисле-
ние изомеризованного продукта II окнсыо серебра останавливается
на стадии образования весьма чувствительного хинона III. Хромо-
вая кислота доводит окисление до желаемого продукта IV, однако с
выходом около 50%. С. т. может либо ацетоксилировать одну из
активированных метиленовых групп соединения III, либо присо-
единяться к двойной связи с образованием диацетата гликоля, либо
промежуточное соединение может легко терять уксусную кис-
235
о
IV
лоту, давая IV. При нагревании гидрохинона II на кипящей водя-
ной бане с 2 молями С. т. в уксусной кислоте образовывался кристал-
лический желтый продукт, сходный с IV, но плавящийся несколько
выше чистого IV (106—107°). При повторении эксперимента с 3
молями окислителя продукт четко плавился при 126,5—127,5°
и был идентифицирован как 2,3-диметил-1,4-нафтохинон. Физер
и Чанг [75] затем показали, что метилирование хинонов С. т. явля-
ется общей реакцией и распространяется на введение высших
алкильных групп путем постепенного добавления Pb30i к горя-
чему раствору хинона в данной карбоновой кислоте. Использова-
ние вместо перекиси ацила сложного эфира четырехвалентного
свинца как источника алкильных радикалов дает исключительно
удобный метод алкилирования хинонов [761. Хотя реакция не имеет
препаративного значения, ароматические нитросоединеиия можно
о-, ц-метилировать радикалами, генерирующимися при разложении
С. т. [77].
T. пл. S0,6°
РЬ(ОСОСН3Ц
32%
Т. пл. 182°
Различные реакции окисления. С. т. в уксусной кислоте при 35°
окисляет 1-бензолсульфамидонафталин (1) в N-бензолсульфонил-
1,4-нафтохинон-4-имин (2) и 2-бензолсульфамидонафталин (3) в
2-бензолсульфамидо-1,4-нафтохинон (4) [78].
(О
Pb(OAc)t
42% (Сырой)
NSO2CeH5
Ii
(2)
236
о
NHSO2C6Hr, !| NHSOaCeH5
Pb(OAe)* . 74 /x 7
I I) | 73% (Сырой)
X/X37
(3)
М,М-Диметиланилин при обработке реагентом в уксусном ангид-
риде и хлороформе при комнатной температуре дезалкилируется
и дает N-метилацетанилид и формальдегид [79]. Реакция является
общей для третичных аминов с одной ароматической группой.
СОСН3
РЪ(ОЛс% I
CcHr>N(CH3)2 —--—> C6H5NCH3-!-CH2O
83% 61%
Филд [80] показал, что С. т. эффективен для окисления тиолов
(алкильных, арильных, бензильных или гетероциклических) в ди-
сульфиды:
2RSH + Pb(OAc)4 RSSR + РЬ(ОАс)2 -] - 2АсОН
Впоследствии он нашел, что дисульфид далее можно окислить
С. т. в смеси метанола с хлороформом до метилсульфината [81],
как, например, в методике получения метилбензолсульфината 182].
о-
(Сеад3фЗРЬ(ОАс)4 -НСН3ОН 2C6H6S-’-OCH3+3Pb(OAc)2 +
+ 4АсОН Щ 2АсОСН3
К кипящему раствору 0,25 моля дифенилдисульфида в смеси хлоро-
форм — метанол при перемешивании добавляют с часовыми интер-
валами восемь порций С. т. по 0,125 моля; каждую порцию перед
добавлением растворяютв 250мл хлороформа (хлороформные раство-
ры при стоянии разлагаются).
Фьюзон и сотр. [83] окислили реагентомдиарилвиннловые спирты
(1) в ацетаты соответствующих диарилгликольальдегидов (3). Аддукт
(2), по-видимому, промежуточное соединение. С. т. окисляет пер-
РЫОАсц В Леон Г(Мезитил)2С-----СНОН
(Мезитил)аС = СНОН -— ---------—-—> [ [
40 [ ОАс ОАс
(2)
- АсОН
—----—> (Мезитил)оС— СНО
'I
ОАс
(3)
вичиые амины, содержащие а-метиленовую группу, в нитрилы,
вероятно, через альдимнны [84]. Выходы почти удваиваются при
237
отношении амина к окислителю 1 : 2, но тем не менее наибольший
выход составляет 61%.
РЬ(ОАс),
RCHoNH..---------> [RCH=NH] —> RCN
По-видимому, родственной реакцией является превращение аль-
дегида в нитрил под действием С. т. (2—3-кратный избыток) в бен-
золе при пропускании в раствор тока аммиака 184 al. В этом случае
для ароматических альдегидов получают высокие выходы; для али-
фатических альдегидов выходы составляют меньше 50%. Считают,
что реакция протекает через образование альдимина.
С6Н5 Н
Чсно
NH;t, РЬ(ОАс)4
80%
с,.нл и
с-с
XCN
Кэмпбелл и Рис [85] сообщили, что при окислении 1-аминобензо-
триазола С. т. в бензоле в мягких условиях быстро и почти коли-
чественно можно генерировать дегидробензол. В качестве промежу-
точного соединения, по-видимому, образуется нитрен (2). При окис-
лении в присутствии тетрафенилциклопентадиенона образуется 1,2,
3,4-тетрафен ил нафталин с выходом 95 %.
С, т. окисляет 2-аминобензотриазол (4) в цис,цис-ыукодинитрил
(7) с 64%-ным выходом, возможно, через нитрены (5) и (6) [86].
Тот же продукт получают с 50 %)-ным выходом при окислении о-
фенилендиамина (8) С. т. [87].
(4) (5) (6) (7)
| РЩОЛсЦ
"4/\NH2
(8)
238
Партч [88] изучил окисление первичных и вторичных спиртов
С. т. в пиридине.
Циганек [89] нашел, что гидразон динитрила мезоксалевой кис-
лоты (1) можно легко получить реакцией диброммалонитрила с гид-
разином в ТГФ, однако все попытки окислить гидразон в диазоме-
тан (2) обычными реагентами были безрезультатными. Затем он
нашел, что дициандиазометан 2) можно получить с почти количест-
венным выходом окислением (1) С. т. в ацетонитриле. Дициандиа-
NC.
СВг.. -j-H3NNH*
NCZ
ТГФ при - 70° NC-
--------> %С —NNIR
35—40%
(1)
NC - -
рС —N—N
NfCx
(2)
Pb(OAc)*-CB3CN
<56%
зометан — высокоэлектрофильный диазоалкан — представляет боль-
шой интерес в качестве источника дицианкарбена. 'Методика
окисления, ограниченная диазосоединениями, устойчивыми к дей-
ствию уксусной кислоты (продукт реакции), иашла также примене-
ние для получения бис-(трифторметил)-диазометана 190].
С. т. реагирует с амидом (1) в кипящем бензоле или смеси бен-
зол — уксусная кислота, образуя ациламин (5) и следы диалкил-
мочевины (6) [91]. Спектроскопическое обнаружение алкилизоциа-
ната (3) позволяет предположить, что реакция включает образо-
вание и перегруппировку ацилнитрена (2). Выходы ариламинов
составляют 40—80%.
О
РЬ(ОДс)4 || .
RCONH,---------> RCN- -рРЬ(ОАс).,-рАсОН
(1) (2)
I НО н нон
4. Асон п I I ;! I
RN^C^O---------* RNCOAc RNAc-RRNCN'R
(3) (4) (5) (6)
Реакция может также применяться для расщепления.
1. D i тп г о t h О., Schweizer RBer., 56, 1375 (1923).
2. Эта методика представляет собой модифицированный метод Димрота и Швей-
цера; Н е 1 1 m u t Ii R. Dissertation. Wurzburg (1930).
3. С г 1 eg с е R., Angew. Chem., 70, 173 (1958); Newer Methods of Preparative
Organic Chemistry, 2. 368 (1963).
4. Dimrotli O., Schultze E., Ann., 411, 345 (1916).
5. W i 1 1 s t a t t er R., К a I b L., Ber., 38, 1235 (1905).
6. Dimroth O., Kammerer H., Ber., 53, 471 (1920).
7. Dimrotli O., Fri edemann O-, Kammer er H., Ber., 53, 481
(1920).
8. Dimroth О., H i 1 e k с n V., Ber., 54, 3050 (1921).
9. К о n i g К. H., Schulze Moller G., Chem. Ber., 93, 554 (I960).
10. Cle m e n t R. A., J. Org. Chem., 25, 1724 (1960); 27, 1115 (1962).
239
II. Fieser L. F., Stevenson R., J. Am. Chem. Soc., 76, 1728 (1954).
12. S e e b e с к S., R ei ch s t e i n T., Flelv. Chim. Acta, 27, 948 (1944).
13. F 1 e s e г L. F., R om er о Al A,, J. Am. Chem. Soc., 75, 4716 (1953).
14. C a v i I I G. W. K., S ol о m on D. H., J. Chem. Soc., 1955, 4426.
15. Henbest H. B., Jones D.N., Slater G. P., J, Chem. Soc., 1961,
4472; Cocker J.D., Henbest H. B., Phillipps G. H., Slater
G. P., Thomas D. A., ibid., 1965, 6,
16. V i 1 к a s M., R ouh i-L ari djaui At., Compt. rend., 251, 2544 (i960);
R ouhi-Lari dj an i M., V i 1 к a s M., ibid., 254, Ю90 (1962).
17. Jacques J,, Wei dm апл C., Horeau A., Bull. soc. chim, France,
1959, 424.
18. Z a I к о w L. H., Ellis J. W., Brennan M. R., J. Org. Chem., 28,
1705 (1963); Z a 1 к о w L.H,Ellis J. W., ibid., 29, 2626 (1964).
19. I f f 1 a n d D.C.,Criner G, X., Chem. Ind., 1956, 176; I f fl a n d D. S.,
Salisbury L., Schafer W. R., J. Arm Chem. Soc., 83, 747 (1961).
20. I f f I a n d D. C., Davies T. M., J. Am. Chem. Soc., 85, 2182 (1963).
21, Freeman J . P., J. Org. Chem., 29, 1379 (1964).
22. C r i e g e e R., Ann., 481, 263 (1930).
23. Meyer К. H., Ann., 379, 37 (1911).
24. F i e s e r L. F., Hershberg E. B., J. Am. Chem. Soc,, 60, 1893, 2542
(1938).
25. Fieser L, F-, C a s о n J., J. Am. Chem. Soc., 62, 432 (1940); Cason J.,
Org. Syn., Coll. Vol., 3, 3 (1955).
26. Fieser L. F., P u t n a m S. T., J. Am. Chem. Soc., 69, 1038 (1947).
27. С r i e g e e R., Ber., 64, 260 (1931).
28. Wolf F. J., W e i j 1 a r d J., Org. Syn., Coll. Vol., 4, 124 (1963).
29. C r i e g e e R., К r a f t L., R a n к В., Ann., 507, 159 (1933).
30. С r i e g e e R.,B ii chner E., W a 1 t h e r W., Ber., 73, 571 (1940).
31. C r i e g e e R., HogerE., Huber G., Kruck P., M a rkt s ch of-
f e 1 F., Scliellenberger H., Ann., 599, 81 (1956).
32. Kuhn L. P., J. Am. Chem. Soc., 76, 4323 (1954).
33. Criegee R,, Marchand B., W annowi us H., Ann., 550, 99
(1942).
34. Booth J., Boyland E., Biochem. J., 44, 361 (1949); Booth J.,
Boyland E., Turner E.E,,J. Chem. Soc., 1950, 1188, 2808.
35. Criegee R., В iich л er E., Ber., 73, 563 (1940).
36. F i e s e r L. F., P u t n a m S. T., J. Am. Chem. Soc., 69, 1041 (1947).
37. В el 1 R. P., R i v 1 i n V.G., Waters W. A., J. Chem. Soc., 1958, 1696.
38. G г о b C. A., S c Ii i e s s P. W., Helv. Chim. Acta, 43, 1546 (I960).
39. Angyal S. J.,Young R.J.,J. Arn. Chem. Soc., 81, 5251 (1959).
40. F i e s e г L. F., Mukawa F., unpublished.
41. G о I d s c h m i d H. R., P e r 1 i n A. S., Can. J. Chem., 38, 2280 (1960).
42. Criegee R., Ann., 495, 211 (1932).
43. P e г 1 i n A. S., В г i с e C., Can. J. Chem., 34, 541 (1956).
44. Charlson A. J., R i c h t m у e г N. K-, J. Am. Chem. Soc., 81 1512
(1959); 82, 3428 (1960).
45. S e p h t о п H. H., R i ch tmy er N. K-, J. Org. Chem., 28, 2388 (1963).
46. В a m b e г g e r E., Ber., 34, 61 (1901).
47. Goodwin S.,Witkop B., J. Am. Chem. Soc., 79, 179 (1957).
48. Bamberger E., Ber., 36, 2028 (1903).
49. Jones E., Kenner J., J. Chem. Soc., 1931, 1851; Kenner J. Mor-
ton F., ibid., 1934, 679.
50. Paper I: Wessely F_, L a u t e r b a c h-K e i 1 G., Si n w el F., Mo-
natsh., 81, 811 (1950); Paper ]X: Wessely F., Z b i r a I E., Jorg J.,
ibid., 94, 227 (1963).
5]. G о 1 d A. M., Schwenk E.,J. Am. Chem. Soc 80, 5683 (1958); герм. пат.
1015802.
52. C a i n e 1 I i G,, M i h a ii ovic M. L., A r i g о n i D., J e g e r O..
Helv. Chim. Acta, 42, 1124 (1959).
240
53. Bowers A., D еп о t E., Cuellar Ibanez L., C a b ez a s Ma.
Elena, Ringold H. J., J. Org. Chem., 27, 1862 (1962).
54. Meystre Ch., Heusler K-, К a 1 v о d a J., Wiel and P., A n-
ner G., Wettstein A., Experientia, 17, 475 (1961); Helv. Chim. Acta,
45, 1317 (1962); Heusler K-, Wieland P., Meystre Ch., Org. Syn.,
' 45, 57 (1965).
55. В a g 1 i J.F., Moran d P. F., Gaudry R., J, Org. Chem., 28, 1207
(1963).
56. Meystre Ch., К a 1 v о d a J., Anner G., Wettstein A., Helv.
Chim. Acta, 46, 2844 (1963).
57. Amorosa M., A г i g о n i D., J eger O. et al., Helv. Chim. Acta, 45,
2674 (1962).
58. Barton D. H. R., Serebryakov E. P., Proc. Chem. Soc., 1962, 309;
Barton D. H. R., Faro H.P., Serebryakov E. P., Woolsey
N. F. , J. Chem. Soc., 1965, 2438.
59. В a r t о n D. H. R., Beckwith A. L. J., Proc. Chem. Soc., 1963,
335.
GO. G г о b С. A., О h t a M., R enk E., W e i s s A., Helv. Chim. Acta, 41,
1191 (1958); further examples in this series arc reported by C h a p m a n N. B.,
Sotheeswaran S., Тоупе K. J., Chem. Comm., 1965, 214.
61. Private communication from Grob C. A., cited by Criegee R. [see ref. 3[.
62. В u c h i G., Erickson R. E., W a к a b a у a s h i N., J. Am. Chem.
Soc., 83, 927 (1961).
63. Private communication from В ii chi G.
64. Cope A.C., Park С. IL, Scheiner P., J. Am. Chem. Soc., 84, 4862
(1962).
65. S t о г к G., L a n d e s m a n H. K., J. Am. Chem. Soc., 78, 5129 (1956).
66. T a m e I e n van E. E., Pappas S. P., J. Am. Chern. Soc., 85, 3297 (1963).
67- Mein w aid J., Tufariello J.J., Hurst J.J., J. Org. Chem., 29,
2914 (1964).
68. С о г e у E. J., Casanova J., Jr., J. Am. Chem. Soc., 85, 165 (1963).
69. M e i n w a 1 d J., Wheeler J. W-, N i m e t z A. A., L i u J. S., J. Org.
Chem., 30, 1038 (1965).
70. Grovenstein E., Jr.,Rao D.V.,Taylor J. W., J. Am. Chem. Soc.,
83, 1705 (1961).
70a. Birladeanu L., H a n a f u s a T., Winstein S., J. Am. Chem.
Soc., 88, 2315 (1966).
71. Bartlett M. F., Lambert B. F., Taylor W. L, J. Am. Chem.
Soc., 86, 729 (1964).
72. Baumgarten H. E., S t а к 1 i s A., J. Am. Chem. Soc., 87, 1141 (1965).
73. Rochi J. K., J. Am. Chem. Soc., 87, 2500 (1965).
74. Ouellette R. J., S h a w D. L., J. Am. Chem. Soc., 86, 1651 (1964).
75. Fieser L. F., C h a n g F. C., J. Am. Chem. Soc., 64, 2043 (1942).
76. F j e s e r L. F., Oxford A. E., J. Am. Chem. Soc., 64, 2060 (1942).
77. Fieser L. F., С 1 a p p R.C., Daudt W. H., J. Am. Chem. Soc., 64,
2052 (1942).
78. R i c n t e r H. J., Dressier R. L., J. Org. Chem., 27, 4066 (1962).
79. H о r n e r L., W i n к e 1 m a n n E., К n a p p K- FL, Ludwig W.,
Chem. Ber., 92, 288 (1959).
80. Field L., Lawson J. E., J. Am. Chem. Soc., 80, 838 (1958).
81. F i e 1 d L., H о e 1 z e 1 С. B., L о с к e J. M., Lawson J. E., J. Am.
Chem. Soc., 83, 1256 (1961); 84, 847 (1962).
82. F i с 1 d L., L о с к e J. M., J. Org. Syn., 46, 62 (1966).
83. F u s о n R. C. et al., J. Am. Chem. Soc., 79, 1938 (1957).
84. M i h a i 1 о v i с M. Lj., S t о j i 1 j к о v i t A., Andrejevic V.,
Tetrahedron Letters, 461 (1965). For other papers by this Yuogoslavian group
on «Reactions with Lead Tetraacetate», see Mihailov! t M. Lj. et al.,
Tetrahedron, 21, 955 (1966). See also ref. [52].
84a. Parameswaran K- N-, Friedman О. M., Chem. Ind., 1965, 988.
241
85. C a m p b е 1 1 C. D., Rees C. W-, Proc. Chem. Soc., 1964, 296.
86. C a m p bell C. D., Rees C. W., Chem. Comm., 1965, 192.
87. N akagawa К., Onou e H., Chem. Conirn., 1965, 396.
88. Parte h R. E., J. Org. Chem., 30, 2498 (1965).
89. Ciganek E., J. Org. Chem., 30, 4198 (1965).
90. G a 1 e D. M., Mi d d 1 e t on W- J., К г c s p a п C. G., J. Am. Chem.
Soc., 87, 657 (1965).
91. А с к о t t B., Beckwith A. L. J., Chem. Comm., 1965, 161.
СВИНЦА ТЕТРАБЕНЗОАТ, Pb(OCOCflH5)4- Мол. вес 691, 65.
Получение [1,2]. Реагент является окислителем, аналогичным
тетраацетату свинца [1J.
l.Criegee R., Ann., 481, 263 (1930).
2. Hurd С. D., А и s t i n P. R., J. Am. Chem. Soc., 53, 1543 (1931).
СВИНЦА ТЕТРАФТОРИД, PbF4. В классическом исследовании
по тетраацетату свинца Димрот [1] изучал реакцию тетраацетата
свинца в хлороформе с жидким фтористым водородом с целью полу-
чения С. т.:
Pb(OAc)4-4HF PbF,-i-4AcOH
Показав, что эта реакция обратима и равновесна, Димрот ис-
следовал реакции С. т. лишь с темп олефинами, которые не
взаимодействуют с тетраацетатом свинца. Одним из таких оле-
финов является 1,1-дифенилэтилен (1); он образует с С. т. в момент
его выделения с умеренным выходом кристаллическое вещество, кото-
рому приписывалось строение 1,2-аддукта (2). Позже было показано,
что аддукт Димрота является продуктом перегруппировки—1,2-
дифен ил -1,1 -дифторэтаном (3) [2J.
ит, (ю; Г(СД1-,).,С—СН.,1
(Свн5)гс=.сн2 ~— I | -> CGH6CFaCH2C6H5
(I) '° I F F J (3)
(2)
Открытие влияния фторных заместителей на биологическую
активность адренокортикостероидных гормонов побудило группу ав-
торов [3] исследовать реакцию ацетата прегненолона (4) в хлористом
метилене с С. т., генерируемым из тетраацетата свинца и фтористого
водорода при низких температурах. Лучший результат с 27%-ным
выходом 5а,ба-дифторпроизводного (3) получен при продолжитель-
ности реакции 15тшн при —75°, по-видимому, через промежуточное
соединение (2). Омыление в Зр-ол (4), окисление в 3-кетон (5) и
дегидрогалогенирование ацетатом натрия в метаноле привело к об-
разованию уже известного 6а-фторпрогестерона (6). С. т., генерируе-
мый из двуокиси свинца и тетрафторида серы, был использован in
siiu для присоединения фтора к двойным связям некоторых галоге-
нированных олефинов [41.
242
(4) (5) (6)
1. Di mrolh О., Bockemiiller W,. Ber., 64, 516 (1931).
2. Bernstein J., Borden M. R., Chem. Ind., 1958, 441.
3. Bowers A., Holton P. J., Denot E., Loza M. C., Urquiza
R , J. Am. Chem. Soc,, 84, 1050 (1962).
4. В i s s e 1 1 E. R., Fields D. B., J. Org. Chem., 29, 1591 (1964).
СЕЛЕН, Se. Ат. вес 78,96.
Дегидрирование. Ранние исследования строения бициклических
сесквитерпенов и трициклических дитерпенов путем дегидрирования
серой с отщеплением ангулярных метильных или карбоксильных
групп (если они присутствуют) и образованием ал кил замещенных
нафталинов или фенантренов дали богатую информацию о строении
природных соединений. Этот метод имеет тот недостаток, что сера
довольно реакционноспособна и может реагировать с непредель-
ными промежуточными соединениями, поэтому выходы обычно
очень низки. В 1925 г. Дильс и Гедке [1] пытались внести ясность
в проблему строения холестерина, которому приписывали приве-
денную ниже частичную формулу (воспроизводимую из “Berichte,,).
Они подвергли холестерин дегидрированию в ароматический угле-
водород. Попытки с применением серы и платиновой черни не дали
ожидаемых результатов, одиако при нагревании 30 а холестерина
Ci 8H3S —
СН,
НС[><С^СН2
hcLjI^Jch,
I СИОН
н
243
с палладироваиным углем (температура точно не указана) отщепля-
лась вода, отгонялся летучий фрагмент нз боковой цени и после
экстрагирования остатка было получено 1,2—1,5 а продукта с т. пл.
180—200°; позднее [21 было найдено, что он содержит хризен.
В надежде получить в более мягких условиях фрагмент боль-
шего размера Дильс и сотр. в 1927 г. исследовали дегидрирование
в присутствии С. [3,41. Несмотря на то что дегидрирование С. тре-
бует более высокой температуры, чем дегидрирование серой, дест-
рукция с С. меньше, чем с серой. Результаты, полученные с более
простыми соединениями, оказались обнадеживающими [4]. При де-
гидрировании 30 г абиетиновой кислоты было получено 22гретена,
тогда как реакция 50 а смоляных кислот с серой дала только
3,2 г этого углеводорода (Фестерберг). Из додекагидрида в присут-
ствии С. был получен трифенилен с выходом 32%.
Дегидрирование холестерина [4] — значительно более трудная
проблема, поскольку при этом образуется очень сложная смесь
и выделение кристаллического продукта требует больших количеств
исходного вещества и значительного времени. Пять порций по 40 а
холестерина нагревали иа бане при 330° и в течение 30 час к каждой
порции прибавляли по 50 г С. 3—4 порциями. Выделяющийся газ
отводили в пламя горелки Бунзена для разрушения H2Se, который
сгорает голубым пламенем, что позволяет следить за ходом реакции.
По охлаждении все порции соединяли вместе, экстрагировали эфи-
ром и разделяли иа пять фракций с температурами кипения от
200° до немного более 285° при 12 мм; суммарное количество сос-
тавляло около 100 г. В результате обработки было получено не-
сколько граммов углеводорода ст. пл. 127—128° и гораздо меньшее
количество углеводорода с т. пл. 219—220° (C2sH34). Дильс при-
писал продукту расщепления, полученному в большем количестве,
правильную формулу C1SHJ6 и показал, что его можно наиболее
надежно идентифицировать в виде желтого нитрозопроизводного
CjSHj3O2N с т. пл. 239°, однако в течение последующих пяти лет
не опубликовал каких-либо данных о его строении. К 1933 г. откры-
тие витамина D и половых гормонов С18 и С1Э послужило стимулом
для развития исследований в области стероидов, и нз ряда лабора-
торий стали появляться публикации, посвященные образованию
244
и строению вещества, известного под названием «углеводород
Дильса» [51. Этот углеводород был получен дегидрированием эрго-
стерина и холевой кислоты. Изучение спектра поглощения позво-
ляло предполагать, что вещество имеет строение 1,2-циклопептено-
фенантреиа (С17) или его Г,2'- или 3'-метильного гомолога (С18),
и некоторое время обсуждалась его возможная формула С^Н^.
Синтез всех четырех упомянутых выше углеводородов все еще
оставлял открытым вопрос о выборе между 1,2-цнклопентенофенан-
треиом и его З'-метильным производным, поскольку смешанная
проба с синтетическими углеводородами ие давала депрессии темпе-
ратуры плавления, а метод идентификации, предложенный Диль-
сом, не использовали. Наконец, в 1934 г. Дильс и Кларе убеди-
тельно подтвердили мнение, что вещество это является З'-метнл-
1,2-циклопентенофенантреиом, показав, что 3 а синтетического
1,2-циклопеитенофепантрена, полученные от Дж. В. Кука, при
обработке окислами азота в эфире не дали и следов иитрозопронз-
водного с т. пл. 239°, которое давал углеводород Дильса, имеющий
третичный атом водорода.
Для решения проблемы строения холестерина потребовалось
9 лет, ио установлено оно было на основе других доказательств
(в 1932 г.). Интерпретировать дегидрирование следует таким об-
разом, что ангуляриая метильная группа у CJ3 мигрирует к С17
с отщеплением боковой цепн. Однако дегидрирование С. оказалось
все же ценным приемом при исследовании эстрогенов [6] н стероид-
ных алкалоидов [7], а также при установлении стероидной при-
роды сапогенипов 18].
Результаты количественного исследования дегидрирования С.
гвайеиа до гвайазулена и дегидрирования трех гндроарематических
соединений опубликовали Силвервуд и Орчин [9], которые пред-
ложили также и механизм реакции. Практический интерес пред-
ставляет их наблюдение, что при дегидрировании 2,5 .нмоля гвайена
с 7,5 лшоля С. при 290° в течение 1 час выход повышается с 14 до
29,6% при добавлении 15 лшолей олеиновой кислоты в качестве
акцептора водорода.
Селеиофенолы. Фостер [10] описал методику получения селено-
фенола взаимодействием фенилмагнийбромида с С. в кипящем эфире
в атмосфере водорода с последующим гидролизом.
245
Кипящий эфир НС1
C6HsMgBrH-Se -........CGH5SeMgBr CcH5SeH
0,5 моля 0,5 моля
Катализатор изомеризации. Олеиновая кислота изомеризуется
в элаидиновую в присутствии ряда катализаторов, однако лучшим
является селен. Реакцию проводят при 220—225° в течение 1 час
в атмосфере углекислого газа нли азота [И], В тех же условиях
ю-фторолеиновая кислота превращается в замещенную элаидиновую
кислоту'' с выходом 75% 112].
СНДСН2)-С—Н sc СН3(СН,),С — Н
' II > 1|
НОгС(СН2)7С—Н 02-64% Н-С(СНа)7СОгН
Олеиновая кислота Элаидиновая кислота
1. D i е 1 s О., Gadke W-, Вег., 58, 1231 (1925).
2, D i е 1 s О., G a d k е W-, Вег,, 60, 140 (1927).
3. Diels О,, Karstens Л., Вег., 60, 2323 (1927).
4. Diels О., G a d к е W., К б г d i я g Р., Ann., 459, I (1927).
5. Fieser L. F., Fieser AL, «Steroids», Reinhold, 1959, p, 83—87.
6. F i e s e r L. F., Fieser AL, «Steroids», Reinhold, 1959, p. 455—461.
7. F i e s e г L, F., Fieser M., «Steroids», Reinhold, 1959, p. 870—871, 883—
884.
8. F i c s e г L. F., Fieser AL, «Steroids», Reinhold, 1959, p. 812—813.
9. Si 1 verwoo d H. А., О r c h i n AL, J. Org. Chan., 27, 3401 (1962).
10. Фостер Д., «Синтезы органических препаратов», ИЛ, At., 1952, сб. 3,
стр. 386.
11. S w er n D., Scanlan J.T., Biochem. Prepns., 3,118 (1953).
12. Dear R. E. A., Pattison F. L. AL, J. Am. Chern. Soc., 85, 622 (1963).
СЕЛЕНА ДВУОКИСЬ, СЕЛЕНИСТАЯ КИСЛОТА, SeO2, H2SeO3.
Мол, веса 110,96; 128,98. Обзор и описание способов превращения
выделяющегося в реакциях Se в SeO2 11].
При реакциях окисления и дегидрирования как с предвари-
тельно полученной С. к., так и с С. д. и водой (обычно в уксусной
кислоте, этаноле или диоксане) раствор сначала становится желтым,
затем красным, а вслед за этим часть селена выделяется в виде
формы красного цвета, которую трудно отфильтровать, часть се-
лена остается в виде коллоидного раствора и некоторое количество
связано с органическим веществом. Часто представляется трудным
удалить весь селен из реакционной смеси. Иногда путем кипячения
(если это допустимо) удается превратить коллоидное красное ве-
щество в черный металлический селей. В некоторых случаях можно
использовать хроматографию на колонке с окисью алюминия и слоем
продажного осажденного серебра [2]. Другие способы предложены
в приведенных ниже методиках, одиако общего метода (если не
считать перегонки) пока еще нет.
Окисление. Использование С. д. для окисления реакционно-
способных метиленовых групп до карбонильных групп предложили
Рилей, Морли и Френд [3]. Согласно методике, которую разрабо-
тали Рилей и Грей [4] для получения фен илгл иоксал я, раствор
24G
ацетофенона в диоксане нагревают с С. к. или с эквивалентными
количествами С. д. и воды и кипятят смесь при перемешивании.
Препарат выделяют перегонкой при 25 мм. Аналогичным образом
CeH5C0CH3--pH3SeO3
1 моль н I моль
600 .ил диоксана
Псремсшин.чннс при кипячении
4 час
60-7 2%
с1;н5сосно
получают бисульфитное производное глиоксаля [5], но при этом
паральдегид берут с большим избытком, а растворителем служит
смесь 540 мл диоксана и 40 мл уксусной кислоты; последняя однов-
Кипячепие 6 час
(CH3CHO)3-|-3H,SeO3-------------
00
р
неси
2 70 мл
NaHSO3
7 2-74%
1,72 моля
OHCCHO-2NaHSO3HzO
ременно ускоряет реакцию и является ингибитором перегруппировки
в гликолевую кислоту. Для окисления циклогексанона в циклогек-
сандион-1,2 [6] раствор С. к. в 500 мл диоксана и 100 мл воды добав-
ляют в течение 3 час при перемешивании и охлаждении к взятому
в большом избытке циклогексанону и затем нагревают смесь в те-
чение 5 час на кипящей водяной бане. Продукт перегоняют дважды
при 16 мм.
/' '% /°
/ А" 3 «<7С + 5 час при 8 7° / j/z
I -J-H.^SeOy считая па H„ScO3 | |
17,4 моля 3 моля
Кори и Шефер [7] исследовали кинетику окисления дезоксибеи-
зоина в бензил и предложили механизм этой реакции. Они разра-
ботали также эффективный синтез а-кетоэфиров, заключающийся
в окислении а-бромкетона в безводном растворителе; в качестве
примера можно привести методику получения этилового эфира
О г ОО I ОО
II II 1,1 СЛТ-ОН !' II
C6H5CCHsBr^SeO2 —> [сеН5ССВг] _> С6Н5ССОС,Н5
70%
15 г 9 а
фенил глиоксалевой кислоты [81. Раствор реагирующих веществ
в 75 мл горячего этанола кипятили в течение 12 час и затем разбав-
ляли водой. Продукт реакции экстрагировали эфиром и перегоняли
при 97-—98°/2 мм.
Радлик 19] описал методику, которая требует мало времени,
хотя и дает низкий выход, для получения тропона из циклогепта-
247
триена-1,3,5 путем окисления С. д. в водном диоксане с буфером;
функция буфера не ясна, но ои повышает выход примерно вдвое.
о,46моля в ззо лиг
диоксана
SeOj +
0) 48 моля
КНгРО4
0,1 МОЛЯ в
33 Л7Л нао
89° 15 час
25%
Мелнвальд и сотр. [101 нашли, что при окислении хлоркетона
(1) можно получить более чистый продукт, если реакцию проводить
в бромбензоле (а не в толуоле или ксилоле) с последующей обработ-
кой водой.
+ SeO2 + С6Н5Вг
(1) 200 г 164 г 1,5 л
150-155°
12 час
58%
(2)
Гидроксилирование в аллильном положении. На основании собст-
венных исследований и анализа литературных данных Гнльмона [11]
предложил следующие правила гидроксилирования олефинов в ал-
лильном положении С. д/.
I) Гидроксилирование происходит в а-гюложении к более заме-
щенному концу двойной связи;
СН3 СН2ОН
сн3-с=снсн3 222 сн3-с=снсн3
2) Легкость окисления убывает в ряду СН2>СН3>СН:
сн3 сн3 сн.он
I С | |
СН3СНа-С=СНСН3 — 2 СН3СН-С = СНСН3-1-СН3СН2С^СЫСН3
он
34% 1 %
Из следующего примера видно, что первое правило имеет преиму-
щество перед вторым:
СН3 СН3ОН
сн3—с=снсн2сн3 222 сн3-(Ь=снсн2сн3
3) Если двойная связь находится в кольце, окисление, если оно
возможно, происходит в кольце и опять-таки в сс-положеиии к заме-
щенному концу двойной связи:
248
4) При окислении концевой двойной связи образуется первичный
спирт в результате аллильного перемещения двойной связи:
СдП
СН3(СН2)2СНгСН--=СН2 СН3(СН,)аСН = СНСНгОН
Гильыона отметил также, что окисление СН до третичного спирта
часто сопровождается дегидрированием с образованием сопряжен-
ного диена. При рассмотрении экспериментальных результатов,
полученных в течение более чем 20 лет, прошедших после появления
работы Гильыона, интересно отметить, насколько хорошо эти ре-
зультаты совпадают со сделанными им предсказаниями.
Обсуждение возможных механизмов см. В ибер г и Нильсен [12].
Зоидхеймер и сотр. [13] окислили а,p-непредельный эфир (1)
в растворе уксусной кислоты и получили с хорошим выходом лактон
(2). Наказаки и Наемура [14] синтезировали кислоту (4) восстано-
вительным расщеплением бр-лактона (3) цинковой пылью в уксусной
кислоте и стереоспецифически окислили ее по методике Аби и сотр.
[15] в (транс) ба-лактон (5), (—)-р-сантонин. Окисление как (1),
(4)
так и (4) возможно только в одном направлении, однако следует
отметить, что обе реакции подчиняются правилу атаки с тыльной
стороны.
Зоидхеймер и сотр. [16] окисляли а,р-иепредельный эфир (6)
С. д.; эта реакция — ключевая стадия синтеза ацетата дигитокси-
генина (7). В соответствии с правилами Гильмона гидроксилиро-
вание происходит в «-положении к более замещенному атому угле-
249
рода двойной связи и предпочтительно по группе СН3, а не СН. В дру-
гой работе Зондхеймера и сотр. [17] описано гидроксилирование
в 17а-положен не агликонов (8), влияющих на сердечную деятель-
ность. В этом случае правила Гильмона неприменимы, поскольку
у одного из атомов углерода в a-положении к более замещенному
углероду двойной связи имеется кислородсодержащий заместитель.
5еО2'диоксан
Кипячение \bvac
ьо%
Розенгейм и Старлинг [181 осуществили окисление холестерина
(10) в АБ-холестендиол-3]3,4р (11); при этом атаке подвергается
а-положен не к наиболее замещенному непредельному атому угле-
рода. Дальнейшая обработка состоит в прибавлении 100 г ацетата
натрия и быстром нагревании смеси для превращения коллоидаль-
ного селена в черный селен. Последний отфильтровывают, пр ил и-
бензола АсОН
(10) (11)
вают по л у насыщенный раствор хлористого натрия, выпавший про-
дукт собирают, кипятят с петролейным эфиром и дважды пере-
кристаллизовывают. Получают бесцветные иглы длиной около
2,5 см, т. пл. 176—177°. Одним из авторов этой книги эксперимент
был повторен; оказалось, что удалить селей трудно и результаты
250
плохо воспроизводятся. В двух случаях выход был порядка 60—70%,
однако при точном соблюдении методики воспроизвести эти резуль-
таты не удалось.
Дегидрирование карбонильных соединений. Способ введения
9,11-двойной связи в 12-кетожелчную кислоту путем дегидри-
рования селеном открыли Швенк и Сталь [19]; эта реакция стала
решающей стадией в промышленном производстве кортизона. Кен-
далл н сотр. [20] тщательно изучили эту реакцию применительно
к метил-За-бензоилокси-12-кетохоланату, используя в качестве
растворителя смесь хлорбензола и уксусной кислоты (4 : 1). Они
нашли, что следы соляной кислоты замедляют реакцию, но повы-
шают выход. Так, без добавления минеральной кислоты максималь-
ный выход составлял 67% при длительности кипячения 24 час,
I моль в 3560 мл
ЩЩО-АсОН (4:1)
тогда как при добавлении соляной кислоты (концентрация в смеси
0,0006 н.) и кипячении в течение 72 час выход достигал 84%. Про-
дукт отделяют после гидролиза; специальным требованием яв-
ляется очистка его от селена, чтобы можно было восстановить кар-
бонильную группу гидрированием над платиновым катализато-
ром. Из многих изученных методик единственно эффективной ока-
залась обработка хромовой кислотой. Смеси, полученные окис-
лением трех порций (всего 508 г) кетоэфирбензоата, объединяли,
фильтровали, обрабатывали 150 г хромового ангидрида в 150 мл
воды и перемешивали 2 час. Хлорбензольный слой декантировали
н перемешивали его со второй порцией раствора хромовой кислоты.
Органический слой отделяли, промывали, сушили и растворитель
отгоняли в вакууме. После омыления и кристаллизации из смеси
ацетон — вода (4 : 1) получали три порции За-окси-12-кето-Д9(11)-
холсновой кислоты, пригодной для использования в следующей
стадии.
Диацетат ]2-кетоэфира (1) был превращен в Ду(| 1’-производное
кипячением его (82 а) в 800 мл уксусной кислоты с 48 а С. д, в те-
чение 18 час [21]. Соляную кислоту не добавляли, поскольку про-
дукт реакции неустойчив и отщепляет 7а-ацетоксигруппу при
действии кислых катализаторов. Смесь фильтровали, разбавляли
водой и выпавший осадок для удаления селена обрабатывали в раст-
воре уксусной кислоты водной хромовой кислотой при 20° в течение
4 час. После повторного осаждения был получен почти чистый
251
препарат с выходом 75,5%. Поскольку спирокетальная группи-
ровка сапогенина чувствительна к действию кислот, дегидрирова-
ние 12-кетонов этого ряда проводят в трет-бутаноле, содержащем
небольшое количество пиридина [22].
Эта реакция применима не ко всем стероидным кетонам, в том
числе и имеющим третичный атом водорода в ^-положении к карбо-
нильной группе; это очевидно из того факта, что в 6-кето-3р,5а-
диацетоксихолестан (2) не удается ввести двойную связь в положе-
ние 7,8 даже при кипячении с С. д. в уксусной кислоте или в
нитробензоле [23].
Учитывая приведенные выше отрицательные результаты, ка-
жется удивительным, что 3-кето стер о иды как 5а-, так н 5[3-рядов,
а также А1- и А4-3-кетостероиды претерпевают дегидрирование под
действием С. д. в mpem-бутаноле до А1,4-диенонов (4) с хорошим
выходом. Это открытие сделали одновременно и независимо друг
от друга в 1956 г. группа исследователей фирмы «Циба — Базель»
[24] и группа фирмы «Органон» 125]*. Группа Гершберга (фирма
«Шеринг») [26] за год до этого обнаружила, что в результате мнкро-
* Примерно в это же время было отмечено [Rin go Id Н. J., Rosen-
kranzG., Soudheimer F., J. Org. Chem., 21, 239 (1956)], что при кипя-
чении 1 г тестостерона с двуокисью селена в бензоле, содержащем немного
воды, в течение 64 час образуется 0,35 9 ДМ-аидростадиенол-Пр-она-З.
252
биологического дегидрирования Л4-3-кетостероидов кортизона и кор-
тизола (6) до /\1;1-дпенонов — преднизона и преднизолона (4) —
заметно повышается биологическая активность. Дегидрирование
через бромкетон или с помощью тетраацетата свинца осуществляется
с очень низкими выходами. Поэтому новый одностадийный процесс,
применимый к разнообразным исходным кетонам, имел огромное
значение для производства аналогов гормонов. Группа исследова-
телей фирмы «Шеринг» была очень близка к этому открытию.
В 1954 г., в поисках нового метода введения двойной связи в поло-
жение 4,5 предельногоЗ-кстопа (8), которое сократило бы послечипе
(8) (9)
стадии синтеза кортизона из желчной кислоты, эта группа исследо-
вала действиеС. д. на кетон (8), но в качестве растворителя был выб-
ран метанол [271, Селен в осадок не выпал, а полученный с хорошим
выходом продукт реакции оказался диметил кеталем (9). Позднее
реакция 3-кетостероидов с этиленгликолем и С. д. в хлористом
метилене была использована для получения 3-этиленксталсй [28!.
По-видимому, С. д. играет роль дегидратирующего агента, а об-
разующаяся С. к. катализирует образование кеталя. Кетогруппы при
Си и С20, как и А4-3-кетостероиды, инертны по отношению к С. д.
в метаноле [29]. Бернштейн и Литтелл 130] сообщали о следующих
примерах использования реакции дегидрирования. Смесь 2 а сое-
динения (10), 21-ацетата 16а-ацетокси кортизол а, 2,2 г С. д. и 200 мл
шрт-бутанола кипятили в атмосфере азота в течение 24 час, после
чего фильтровали. Фильтрат выпаривали в вакууме досуха и для
удаления селена остаток перемешивали в течение 2 час с 50 мл
метанола и 3 г дезактивированного никеля Ренея (см. [30], приме-
чание 22). Хеллер и Бернштейн [31] использовали гу же методику
253
в другом случае. Другие примеры этой реакции приводят Фрид
и corp. 132] и Аллеи и Лустин 1331.
Барнес и Бартон (34] нашли, что под действием С. д. тритерпе-
ноидпые 1,4-дпкетоны дегидрируются до ендионов в том случае,
когда оба подлежащие удалению атома водорода находятся в /{не-
положен ни. Реакцию осуществляли при кипячении в уксусной
кислоте.
О i I н О о о
Хилл [351 показал, что феи и л малеиновый ангидрид, удобный
диенофил, можно легко получить при взаимодействии фепилянтар-
иой кислоты с С. д. в уксусном ангидриде. По-видимомм, здесь
НС -С-0
СН.,СО2Н || \0
QH.CHCO.il | SeO. + .W) К"""чс""с 3 Ц С,Н,С-с4о
4,9 г 3,3 е 2 0 m.i
существенно наличие фенильной группы, так как па янтарную
и этцлянтарную кислоты этот реагент не действует.
При дегидрировании Д’-З-кетостеропдов до Д',4-3-кетостеропдов
выходы обычно ниже 50% вследствие образования селеностеропд-
нон структуры, природа которой еще не выяснена. Кокор и Туши-
Макзка [361 отмечали сильное изменение окраски при взбалты-
вании бензолытых экстрактов реакционной смеси с водным сер-
нистым аммонием и нашли, что этот реагент можно использовать
для полного удаления селена. Неочищенный продукт окисления
растворяют в этаноле, обрабатывают сернистым аммонием, остав-
ляют на ночь, а затем кипятят. Смесь разбавляют водой, экстра-
гируют бензолом и экстракты промывают водным раствором сер-
нистого аммония до обесцвечивания водного слоя. Выходы в трех
случаях составили 75—90%.
Установление типа ненасыщенности. А!-Холестсиилапстат
окисляют С. д. в смеси уксусной кислоты п бензола и получают
7а-анетоксн-ДИ!4)-холестен (2), по-видимому, в результате гидро-
ксилирования в аллильное положение у С)4, аллильной перетруп-
254
пировкп и ацетилирования [21. Поскольку окисление легко проте-
кает при комнатной температуре, тогда как холестерин взаимо-
действует лишь при 55- 60', реакцию можно использовать для
обнаружения небольших количеств Ат-холестепола, который обычно
сопутствует холестерину в большинстве тканей [37]. С помощью
проб, проводимых с навесками по 1 мг в капиллярах для определе-
ния температуры плавления 1381, легко отличить холестерин желч-
ного камня, содержащий около З'’о А7-изомера, от чистого холесте-
рина, Проба с С. д. характерна для стероидов типов (1) и (3) — (6),
т. е. для А7- и Ая-степолов 5а-ряда, для А;,!1(|1)- и Д|!’4-диеновтого же
ряда и для Дг”т-диенов. Так, положительную пробу дают только
5а- или А71-стероиды с двойной связью, соседней с 14а-водородным
атомом. 5р-Изомеры соединений типов (1), (3) и (4) дают отрица-
тельную пробу, возможно, потому, что отклонение кольца Л с об-
разованием «клетки» делает 14а-водородный атом недоступным для
5а ИЛИ j3
(?)
атаки из а-областп. /Другие структурные типы, с которыми проба
отрицательна, изображены формулами (7) — (И). Поведение (7)
и (8) показывает, что активированный 80-водород неуязвим, а по-
ведение (9), (ТО) и (И) показывает, что активированный а-водород-
ный атом у С9 не обладает той чувствительностью, которую имеет
водород у Си.
Природа продукта реакции, образующегося в результате окис-
ления С, д. елового пли диенового соединения, зависит от осо-
бенностей его строения и от условий реакции. При окислении
Ажхолестсн ил ацетата (1) в уксусной кислоте образуется Ая(||)-7а-
ацетоксисоедипение (2), тогда как при окислении в смеси этанола
и бензола образуется Ач( 14’-7а-этиловый эфир [2]. В других случаях
ей дает диен, а диен — триеи.
255
1. R a b j о h n N., Org. Reactions. 5, 331 (1949).
2. Fieser L. F., Ourisson G., J. Am. Chem. Soc., 75, 4404 (1953).
3. Riley H. L,, Morlcv J. F., Friend N. A. C., J. Chem. Soc., 1932
1875.
4. Рилей X,, Грей А., «Синтезы органических препаратов», ИЛ, ДЕ, 1949,
сб. 2, стр. 507.
5. Р о н з и о А,. В о Т,, «Синтезы органических препаратов», ИЛ, М., 1952,
сб. 3, стр. 106.
6. Н а с h С. С. Banks С. V., D i с 11 1 II., Org. Syn., Coll. Vol., 4, 229
(1963).
7, С о г e у Е. ,L, Schaefer J, Р., J. Am. Chem. Soc., 82, 918 (1960); sec
also S c h a с I e г J. P., H orva IhB., Tetrahedron Letters, 2023 (1964).
8. Schaefer J.P., Corcv E.J., J. Org. Chem,, 24, 1827 (1959).
9. R a d 1 i c k P., J. Org. Syn.,'29, 960(1964).
10. M e i n w a 1 d ,1., Jensen С, B., Lewis A., S w i t h e n b a n k C.,
J. Org. Chem., 29, 3469 (1964).
II. Gu i I 1 emonat A., Ann. chim., 11, 143 (1939).
12. Wiberg К. B., Nielsen S. D., J. Org. Chem., 29, 3353 (1964).
13. D ani ei i N., Mazur Y., Son dheimer F,, Tetrahedron Letters,
310 (1961).
14. X a k a z a k i M., N a c m u r a К , Chem. Ind., 1964, 1708.
15. Abe Y., et al. J. Am. Chem. Soc., 78, 1422 (1956).
16. Daniell N., Mazur Y_, S о n d h c i m e r F., J. Am. Chem. Soc., 84,
875 (1962).
17. Da ni el i M., Mazur Y., Son dheimer F., Tetrahedron Letters,
1281 (1962).
18. Rosenheim O., Starling W, W-, 9- Chem. Soc., 1937, 377.
19. Schwenk E., Stahl E., Arch. Biochem., 14, 125 (1947).
20. McKenzie B. F,, Mattox V. R., Engel L. L., К e n d a I 1 E. C.,
J. Biol. Chem., 173, 271 (1948).
21. F i e s c r L. F., Rajagopalan S., Wilson E., T is 11 1 er, J.
Am. Chem. Soc., 73, 4133 (1951).
22. Bowers A., Denot E., Sanchez M. B., Neumann F., D j e-
r a s s i C., J. Chem. Soc., 1961, 1859.
23, Fieser L. F_, Rajagopalan S., J. Am. Chem. Soc., 71, 3938 (1949).
24. Meystre Ch., Frey H., V о s e r \V., W e t t s t e i n A., Helv. Chim.
Acta, 39, 734 (1956).
25, S z p i 1 f о g e 1 S., P о s t h u m u s T., D e Winter M., van Dorp
D. A., Rec. trav., 75, 475 (1956).
26. iierzog H.L., Nobile A., Tolksdorf S,, Charney W.,
Hersh berg E. B., Perlman P. L., Science, 121, 176 (1955).
27. О 1 i v e t о E.P., G e г о 1 d С., II e r s h b e r g E. B., J. Am. Chem. Soc.,
76, 6113 (1954).
28. Nussbaum A. L., Popper T.L., Oil veto E.P., Friedman
S., W e n d с r I., J. Am. Chem. Soc., 81, 1228 (1959),
29. Mager lein B. J,, J. Org. Chem., 24, 1564 (1959).
30. Bernstein S., L i t t c 1 [ R., J. Am. Chem. Soc., 82. 1235 (i960).
31. H e 1 1 e r M., Bernstein S., J. Org. Chem., 26, 3876 (1961).
32. Fried J. H., A r t h G. E., S a r e t t L. H., J. Am. Chem. Soc., 81, 1235
(1959).
33. Allen G.R., Austin N. A., J. Org. Chem., 26, 4574 (1961),
34. Barnes C. S., Barton D. H, R., J. Chem. Soc., 1953, 1419.
35. Hill R. K.,J. Org. Chem., 26, 4745 (1961).
36. К о c 6 r M., T u s z y-M a c z k a M., Bull. acad. Polon., Sci. Chem.. 9, 405
(1961) (in English).
37. Fieser L. F., J. Am. Chem. Soc., 75, 4395 (1953).
33. Org. Expts., 74—75.
256
СЕЛЕНА ХЛОРОКИСЬ, SeOCl2. /Мол. вес 165,87, т. кип. 177,6°,
уд. вес 2,44.
Шефер и Соинепбург [1] добавляли С. х. при охлаждении
к раствору ацетофенона в бензоле и выдерживали раствор при ком-
натной температуре. Кристаллический продукт реакции (1) выде-
О
2СЙ1-Ь,СОСН3-; SeOCL, [С6Н.,СОСИ.ф5еС12 2С6Н:ДСН,С1Se
5411,',
{/.2 моля 0,1 моля (1) (2)
лился полностью спустя 1 час\ в результате его пиролиза был полу-
чен «-хлорацетофенон (2) с умеренным выходом.
1. S с 11 л с Г е г J. Р., Sonnen burg Г\, J. Org. Chem., 28, 1128 (I9G3).
СЕМИКАРБАЗИДА ХЛОРГИДРАТ, НС1 H.NNHCONH,. Мол.
вес 111,54, т. пл. 173" (свободное основание неустойчиво, т. пл. 96°).
Получение семикарбазона 111. 0,5 мл исходного раствора, при-
готовленного из 1,11 г С. х. и 5 мл воды, содержит 1 лшоль реагента.
К 0,5 мл такого раствора прибавляют 1 жмоль ацетофенона и доста-
точное количество метанола (1 мл), чтобы раствор стал прозрачным,
затем добавляют 10 микрокапель пиридина и смесь осторожно на-
гревают па кипящей водяной бане до выпадения кристаллов
(т. пл. 198°).
1. Org. Expts., 97.
СЕРА, S. Ат. вес 32,07, светло-желтые кристаллы, т. пл. 120°,
т. кип. 444,6, незначительно растворима в CS.,.
Дегидрирование [1]. Дегидрирование гидроароматических сое-
динений проще проводить с С., чем с селеном, иногда дегидриро-
вание с С. предпочитают каталитическому дегидрированию. 3,4-
Днгпдро-1,2-иафтойный ангидрид (синтез см. Этилат натрия)
дегидрируют перегонкой с 1 экв серы в колбе Клайзена на 50 мл,
од ?-атом
о, I моля
к которой припаяна перегонная колба в качестве приемника [2].
Колбу погружают в сплав Вуда или в смесь 10 ч. KNOa и 7,5 ч.
NaNCB (т. пл. около 150), предварительно нагретую до 230—235°.
Колбу встряхивают до растворения серы и повышения температуры
в течение 30 мин до 2503 1,2-Нафтойный ангидрид перегоняют при
12—13 лыг и кристаллизуют из смеси бензол — лигроин в виде
257
9 Заказ № 1166
светло-желтых игл с т. пл. 166—167°. Аналогичным образом был
дегидрирован Ьфеиил-Д'-дналпн [3].
Кокер и сотр. [4] обнаружили, что производные тетралина (1)
можно дегидрировать серой при 230—240°, одиако при дегидриро-
вании селеном при 330—350° они отщепляют н-бутнльную группу
с образованием соединения (3). Дегидрирование на палладирован-
ном угле при 260—280 ’ дает смесь (2) и (3).
сн3сн2сн..сн,сн3 CH3CILCILCH,CH;J сн3
/1^ нас HF 1 \ 1 11 \ Н3С ) \ А V у 1 i 1 1
1 И 1 ! 1
1 11 1 1 11 г А/ // 1 Д/ X г/
сн3 сн3 сн3
in Ы) (3)
С другой стороны, для дегидрирования с отщеплением боковых
метильных групп селен обычно эффективнее серы, как это видно
из результатов, полученных с холестерином и ретеном (см. Селен).
Селен, вероятно, эффективнее серы и при дегидрировании гидро-
азуленов, хотя нагревание с серой при 220° применяется чаще.
Удобна методика, состоящая в кипячении гвайена с серой в
триглиме (т. кип. 222') в течение 1 час. Полученный гвайазулен
перегоняют с паром и переводят в кристаллический пикрат [5].
Выход азулена при дегидрировании серой, вероятно, лимити-
руется реакцией серы с промежуточным насыщенным соединением.
Одни из авторов [61 обнаружил сер у со держащие продукты реакции
при попытке дегидрировать 1,6-диметил-7,12-дигидроплейаден (1) до
антраценоподобиого плейадена (2). Продукт (3) имеет состав аддукта
Дильса — Альдера соединения (2) с серой. Выход приведен для
продукта, перегнанного и перекристаллизованного до постоянной
точки плавления.
258
Реакция Гриньяра. Реактив Гриньяра, полученный из 2-иод-
тиофена, образует с серой производное, удобное для метилирования
О, 3 3 МОЛЯ
мё ;
Эфир
0,36 моля СНД
Кипячение 10 vey
Ь 3 - 6 0%
ОДЗмоля S
Кипячение
4 5 мин
Реакция Фриделя — Крафтса. Дифениловый эфир, взятый в
в большом избытке в качестве растворителя, реагирует с серой
в присутствии хлористого алюминия и дает феиоксатпн [8]. Смесь
реагентов после энергичного встряхивания становится пурпурной,
ее нагревают в течение 4 час на водяной бане и выливают в лед с со-
ляной кислотой. Слои разделяют, органический слой сушат и пере-
1 [ Ml),'!; it S i[
гоняют при 5 мм для выделения исходного дифенилового эфира
и затем продукта реакции.
Катализатор бромирования. При бромировании паральдегида
для получения бромаля использование С. в качестве катализатора
увеличивает выход на 5—10%, нс затрудняя при этом обработку
[9]. Паральдегид прибавляют к смеси брома и серы в течение около
4 час, причем необходим внешний обогрев, чтобы в течение еще 2 час
поддерживать температуру 60—80?.
(CICCHO)- 9Вг., -Г S ЗСВгзСИО : Ш1Вг
52-57%
11,52 м >/1 ',t 4,5 мо/ы 1,6
Реакция Вильгеродта [10]. Вначале эта реакция состояла в на-
гревании арилалкилкетона в запаянной трубке при 210—230°
с водным раствором желтого полисульфнда аммония, полученным
9*
259
при растворении серы в растворе сернистого аммония. Продукт
реакции — арилзамещенный амид алифатической кислоты, содер-
жащий некоторое количество соответствующей карбоновой кислоты
и (часто) углеводорода. Примером служит реакция ацетофенона,
дающая фепилацетамид и аммонийную соль феннлуксусиой кис-
лоты [11]. Первоначально реакция нашла лишь ограниченное при-
C6H5COCH3 + (NH4)2Sx C6H5CI I2GONII2 •C6HaCHaCO..NIil
5{)%
менение вследствие малых выходов и необходимости использовать
запаянную трубку. Физер и Кильмер [12] улучшили методику,
предложив использовать в качестве растворителя диоксан, что
позволило работать при температуре 160°. По этому методу 1-ацсто-
аценафтен превращают в 1-аценафтилуксусную кислоту с 57%-иым
I [I I д(юксл|, 11) 0° I АМПД |
I >1 I ----------v I' -
Xx /- /7\ I 5 г ле водород J
"'7 чсосн3
CH3—CH2
4" I II !
CH,CO2H
CH2—CH2
выходом. Другой возможный путь синтеза (окисление гипогалоге-
нитами, превращение через хлораигидрид кислоты в диазокетон
и реакция Арндта — Эйстерта) приводит к 1-аценафтилуксусной
кислоте с общим выходом 50%.
Важной модификацией реакции, предложенной Киндлером [13],
является нагревание кетона с эквивалентным количеством серы
и амином, который препятствует окислению; образуется тиоамид,
дающий при гидролизе карбоновую кислоту. Особенно удобен для
этой реакции морфолин; об этом варианте реакции см. Морфолин.
1. Платтпер 11. Л. А., Армстронг Е. С., Дегидрирование серой,
селеном н платиновыми металлами. В сб. «Новые методы препаративной орга-
нической химии», ИЛ, М., 1950.
2. Г е р ш б е р г Е., Физер Л., «Синтезы органических препаратов», ИЛ,
М., 1949, сб. 2, стр. 53.
3. Вейс Р., «Синтезы органических препаратов», ИЛ, М., 1952, сб. 3, стр. 440.
4. С о с к е г W, Cross В. Е., Edward j. Т., Jenkinson D. S.,
McCormick J., J, Chem. Soc., 1953, 2355.
5. Org. Expts., 298.
6. Fieser L. F., J. Am. Chem. Soc,, 55, 4977 (1933).
7. Ц н м с p м а и - К p э г Дж., Лодер Дж,, «Синтезы органических препа-
ратов», ИЛ, М., 1956, сб. 7, стр. 39.
8. Сутер К., М а к с в е л л Ч-, «Синтезы органических препаратов», ИЛ,
М., 1949, сб. 2, стр. 529.
260
9. Лонг Ф., Хоуард Дж,, «Синтезы органических препаратов», ИЛ, М.,
1949, сб. 2, стр. 102.
10. Обзоры: К а р м а к М., III п и л ьма н М. А., «Органические реакции»,
ИЛ, М., сб. 3, 1951, стр. 88; W е g 1 е г R., К u h 1 с Е., Schafer W.,
Newer Methods of Preparative Organic Chemistry, 3, 1 (1964).
11. Willficrodl C., Merk F. H., J. prakt. Chem., [2], 80, 192 (1909).
12, F i e s с г L, F., Kilmer G. \V., J. Am. Chem. Soc., 62, 1354 (1940).
13. К i n d 1 e r K. Ann., 431, 187 (1923).
СЕРА ДВУХЛОРИСТАЯ, SCI,. Мол. вес 102,98. Темно-красная
жидкость, т. кип. 59°, уд. вес 1,621.
Три группы химиков независимо исследовали присоединение
С. д. к циклическим диенам [2—41. При соответствующих условиях
разбавления С. д. присоединяется к песопряженным циклическим
диенам, образуя мостиковые серусодержащис системы. Так, Кори
(3) тлит. 97,9-99,9° (4)
и Блок [21 при перемешивании раствора 1,5-циклооктадиеиа в хло-
ристом метилене при — 50’ в атмосфере азота и добавлении при ох-
лаждении С. д. получили с высоким выходом кристаллический
аддукт, охарактеризованный как 2,б-дихлор-9-тиабшшкло-13,3,11-
нонан (3), Предполагают, что промежуточно образуется 1,2-эпи-
сульфонпй-ион (2). Агомы хлора можно удалить восстановлением;
при этом с высоким выходом получают 9-тпабицикло-13,3,11-ноиан (4).
1. Брауэр Г. «Руководство но препаративной неорганической химии», ИЛ,
М., 1956.
2. Со г е v Е. J., В 1 о с k Е., J. Org. Chem., 31,1663 (1966),
3. W с 1 ГЕ. D., Smith K.E,Grubcr R. J., J. Org. Chem., 31, 1669 (1966).
3. L a u I e n sc h I a eg er F., J, Org. Chem., 31, 1679 (1966).
СЕРА ОДНОХЛОРИСТАЯ, S 2C12. А\ол. вес 135,03, красновато-
желтая жидкость, т. кип. 137°, уд. вес 1,687.
Очистка. Шмушкович 11| рекомендует перегонку 500 г С. о. в при-
сутствии 20 г серы и 5 с угля; фракцию, кипящую при 135 —137°,
хранят в темной склянке в холодильнике.
Дифенилсульфид легче получать но реакции бензола с С. о,
в присутствии хлористого алюминия [2],
261
2С6Н3 4- S.CI, + А1С1;; Cr,H5SCJ{„-:-S + 2MCl
b I - Q 3 n,,
16 MO.'IL'j'i 3 миля 3,4S
Для обычного электрофильного замещения в пиридиновом кольце
требуются очень жесткие условия, однако Хансбергер и сотр, [3|
показали, что нагреванием пиридина с бромом и С. о. при умерен-
ной температуре можно получить с небольшим выходом 3,5-дибром-
пиридин. Для реакции характерно введение брома в |3-положенне
к азоту пиридинового кольца независимо от уже имеющегося за-
местителя. Хансбергер предположил, что реакция идет через про-
межуточные состояния типа (2) — (4).
I. Private communication.
2. Хартман В.,Смит .1, Дпкк
ратов», ИЛ, М., 1949, сб. 2, стр. 241.
3- G а г с i а Е. Е., Greco С. V., И и
82, 4430 (I960).
(3) (4) (5)
п Дж., ^Синтезы органических прспа-
nsberger I. M., J. Дт. Clicin. Soc.,
СЕРА ЧЕТЫРЕХФТОРИСТАЯ, SF4. Мол. вес 108,07.
Внимание! Газообразный реагент почти в два раза токсичнее
фосгена. Он легко гидролизуется до фтористого водорода, поэтому
применяемая аппаратура должна быть тщательно высушена.
Получение. Наиболее удобно получать С. ч. кипячением дву-
хлористой серы с фтористым натрием в ацетонитриле [2|.
3SCL + 4NaF SF4- S,,C1.2 -R 4\аС1
Реакции 13|. Для С. ч. наиболее характерны реакции типа
4 С--О^ /CR И — СООН -CF;i
Эти реакции требуют специальной техники эксперимента; их
проводят в реакторе для работы под давлением, покрытом изнутри
нержавеющей сталью; необходима емкость для хранения из поли-
этилена или другого материала, устойчивого к HF. В лаборатор-
ных углов и их предпочитают использовать трехфтористую феи пл-
ееру.
Реакция с карбоновой кислотой осуществляется в две стадии;
RCO,H-;-SFt -> RCOF-i-UF l-SOF..
RCOF + SF4 RCF3-RSOF3
262
Первая стадия протекает при комнатной температуре, для вто-
рой требуется нагревание до 100—200 \ При превращении энан-
ювой кислоты в 1,1,1-трифторгептан |4| реагенты нагревают в реак-
торе под давлением при перемешивании в течение 4 час при 100”'
и 6 час при 130'. Ангидриды и хлораигидриды карбоновых кислот
дают те же продукты, что и соответствующие кислоты, по требуют
[ f) л _ 1 3 П*
CH;!(CH.J-CO.II -:-2SF11 ---СНЙ(СН.,)5СР3
7 О - 8 ид,
0,2 моля 0,.'7 моля
нагревания до 300 -350'. Сложные эфиры реагируют при более
низкой температуре в присутствии трехфтористого бора в качестве
катализатора.
Реакцию можно использовать и для аминокислот, если прово-
дить ее в присутствии фтористого водорода для защиты амино-
группы [51. Оптическая активность аминокислот сохраняется.
Так, ь-лейцин даст левовращающий 3-метил-1-(трифторметил)-
бутиламин.
8 час при 1 20п СН3
О.ЧЯ моля ST, L 2 5 моля IIF
-------------------СНЙСНСН.,СНСР,
22%----------------.1
С. ч. способна частично обменивать галоген с галогенмета нам и,
например CCU-l-SF^CFCn (выход 66%) Гб].
Применение реакции с С. ч. в области стероидов описано Мар-
тином [7].
Эппльквист и Сирл 18], пытаясь синтезировать 9-фторантрацен,
нагревали антрон с С. ч. и фтористым водородом в хлористом ме-
тилене в течение 16,3 час при 69', но неожиданно получили 10,10-
дифтораитрон с высоким выходом.
О О
cii/hch,chco.,h
I
NTI,
Ь-Леицпн (0.4 моля)
Н' ЧН F/XF
1. Technical Information Bulletin, 1959.
2. T u 1 1 о с k С. W., F a \v с с t t F. S., Smit h \V. C., Coffman D. D.,
J. Am. Chem. Soc., 82, 539 (i960).
3. Smith \V. С., Angew. Chem,, Internal, Ed., 1. 467 (1962).
4. H asek W. R., Smith \V. C.t Engelhardt V- A., J. Am. Chem.
Soc. 82, 543 (1960).
5. R a a s c h M. S., J. Org. Chem., 27, 1406 (1962).
6. Tull о c k C. С a г h о n i R. .A., Harder R. J., Smit h \V. C.,
Coffman D. D., .1. Am. Chem. Soc., 82, 5107 (I960).
7. Martin D. Ci., Каца n IT, J. Org. Chem., 27, 3164 (1962); M a r-
tinD.G, Pike J. E., ibid., 27, 4086 (1962).
8. Applcquist D.E., Searle R.,J. Org. Chem., 29, 987 (1964).
263
СЕРЕБРА БОРФТОРИД, AgBE4. Мол. вес 194,70, г. разл. 200°.
Получение. Согласно методике Меервейпа и сотр. |11, С. б.
получают реакцией между эфиратом трехфторпстого бора и суспен-
зией окиси серебра в нитрометане, однако по другим данным прп
этом происходит «сильный взрыв» [2]. Простой и надежный метод
получения чистого С. б. состоит во взаимодействии трехфтористого
бора с монофторидом серебра в нитрометане [31 или в бензоле [4].
AgF I BF3 AgBF4
900-
Методика Ола и Квинна. Безводный С. б. лучше всего получать
из фтористого серебра и трехфторпстого бора в нитрометане [3].
Суспензию 1 моля фтористого серебра в 170 мл нитрометана пере-
мешивают магнитной мешалкой и одновременно, защищая смесь
от влаги воздуха, пропускают газообразный трехфтористый бор.
Температура повышается до 60?, и ее поддерживают иа этом уровне
охлаждением смеси. Пропускание трехфтористого бора прекращают
после почти полного растворения фтористого серебра и образова-
ния бесцветного, но мутного раствора. В течение 1 час в раствор
пропускают ток сухого азота для удаления избытка трехфтористого
бора; раствор фильтруют в атмосфере азота через стеклянный по-
ристый фильтр и нитрометан отгоняют в вакууме при 70и. Белова-
тый твердый остаток помещают в ступку в сухой камере, заливают
н-нентаиом и растирают в порошок. С. б. высушивают в вакууме
при нагревании до 70°; выход 86—94%. Хранят С. б. в «-пен-
тане.
Комплексы. С. б. нерастворим в циклогексане, но хорошо
растворяется в воде, эфире, толуоле и нитрометане и умеренно —
в бензоле и циклогексене [1,5]. Растворимость в непредельных угле-
водородах объясняют образованием л-связи между непредельным
соединением и ионом серебра. Наблюдалось образование комплексов
как с донорами л-электронов, так и с донорами неподеленнон элект-
ронной пары; так, Меервейн 111 получил стабильные комплексы
с бензолом, мезитиленом, циклогептатрнеком, диэтиловым эфиром,
ДМСО и с нитрилами ряда кислот. Одиако данные о составе ком-
плекса С. б. с бензолом противоречивы: 1:1 [1], 1 : 2 |4[ и 2 : 3 16|.
Алкилирование. Меервейн fl| указывает, что простые эфиры
и кетоны можно эффективно алкилировать бромистым этилом
и С. б. в дихлорэтане при комнатной температуре:
CaH5OC2Hsi-C,H3Br-|-AgBF4----> <С4Н5)3О ; (BF“) - AgBr
R2C — О (камфора) —+ R2C~O“(BF7)
59% |
с. Hr,
(С.,Н50).,С=--0 --> (C2115O),C^6c,H5(BF7)
72%
264
В случае у-бромэфнра
алкилирование:
можно осуществить внутримолекулярное
сн2— сн,
I I AgBF4
сн2
I О'" ос,н5 88%
Вг г 5
сн—сн2
сн2 хс <—>
ХОХ ОС2Н5
сн—сн2
СН2 с,.
хр ^ОС2Н5
вг4“
Эшенмозер и corp. [7] использовали этот реагент в новом синтезе
N-метил аминокислот. Хлоргидрат метилового эфира l-трипто-
фана (1) конденсировали с хлор ан гидридом у-хлормасляной кислоты
в присутствии пиридина и полученное производное (2) циклизовали
в имннолактон (3) при действии С. б., взятого с небольшим избыт-
ком, в смеси хлористый метилен — беизол при температуре от —20
(4)
до 4 20 . Затем иминолактон алкилировали подпетым метилом
в ацетонитриле (41 час при 30") и получили четвертичное мономе-
тил ыюе производное, от которого отщепляли защитную группу
бикарбонатом калия (2 час при 20е). Продукт выделяли в виде
хлор гидрата (4).
R,CHBr - >R2C-=O. Ломал и Фрей [2| использовали С. б. в одном
из вариантов методики окисления под действием ДМСО по Корн-
блюму. 3-Нортрициклилбромид (1) добавляли к раствору эквива-
лентного количества С. б. в ДМСО, в результате чего в осадок вы-
падало бромистое серебро. Через 1 час, когда образование диметил-
3-нортрнцпклплсульфоилевой соли, по-видимому, заканчивалось,
добавляли избыток триэтилампна и темпо-бурую смесь выдержи-
265
вали прн комнатной температуре, а затем быстро нагревали на
кипящей водяной бане. Полученную смесь фильтровали и кетон (2)
экстрагировали эфиром.
ArCHCl-xArCHN НСОСН. Циммерман и Пасковпч f81 исполь-
зовали С. б. для превращения димезитил хлор метана (1) в эти л кар-
бамат (2). Смесь 0,00385 моля С. б. и 5 г уретана обрабатывали
при 60J раствором 0,0035 моля хлорида (1) в 20 мл диоксана.
В осадок сразу же выпадало хлористое серебро, после нагревания
в течение 5 мин на кипящей водяной бане добавляли воду и пре-
парат экстрагировали хлороформом.
Катализатор электрофильного замещения в ароматическом ядре.
В 1957 г. Ола и сотр. 191 сообщили, что С. б, активен в качестве
катионобразующего агента в реакциях электрофильного замещения
в ароматическом ядре. Он может служить катализатором для нит-
рования с помощью NO..C1 [101, галогенирования элементарным
галогеном 111 I и форматирования формилфторидом [12|. Реакции
протекают бурно и с почти количественными выходами.
I. Meerwcin II., II е fl е г i с Ii V., Wunderlich К., Arch. Pharm.,
291, 541 (1958).
2. Letnal D. М., Fray Л. J., Tetrahedron Letters, 775 (1961).
3. О I a li G. A., Quinn II. W., J. Inorg. Nuel. Chem., 14, 295 (I960); private
communication from Drs Olah and Quinn of Dow Chemical.
4. H e у n s K-, Paulsen FL, Angew. Chem., 72, 349 (1960).
5. Sharpe A. G., .1. Chem. Soc., 1952, 4538.
6. Sharp D. W. A., Sharpe A. G., J. Cliem. Soc., 1956, 1855.
7. Peter H., Bruggcr M., Schreiber J., Eschenmosor A.,
Helv. Chim. Acta, 46, 577 (1963).
8. Zimmerman И. E., Paskovich D. FL, J. Am. Chem. Soc., 86 2149
(1964).
9. Olah G. A., Pavla th A., Kuhn S., Chem. Ind., 1957, 50.
10. Kuhn S. J., Olah G. A., ,L Arn. Chem. Soc., 83, 4564 (1961).
11. Olah G. A., Kuhn S. J., 1; 1 о о d S. H., Hardie B. A., J. Am. Chem.
Soc.. 86, 1039 (1964); Olah G. A.. Kuhn S, J., Hardie B. A., ibid., 86,
1055 (1964).
12. Ola li G. A., Kuhn S. J., J. Am. Chem. Soc., 82, 2380 (1960).
СЕРЕБРА ДИФТОРИД, AgF2. Мол. вес 145,85.
Шеппард [1] описал реакцию С. д. с дифенил дисульфидом с целью
получения трпфторфенилсеры. В колбу, предварительно продутую
266
сухим азотом, помещают 435—470 г черного порошкообразного С. Д.,
добавляют 500 мл фреона ИЗ (т. кип. 47 ) и перемешивают смесь
(СДЬДМ 6AgF., J:2^1rX2C0H5SF3-6AgF
0.438 моля 3,1 моля
в атмосфере азота, постепенно прибавляя дифенилдпсульфид при
температуре 35—40' (45—60 мин'). Реакционную смесь фильтруют
в атмосфере азота для удаления желтого фторида серебра и продукт
отделяют перегонкой.
1. S Ii е р р а г d W. A., Org. Syn., 44, 82 (1964).
СЕРЕБРА КАРБОНАТ, Ag2CO3. Мол. вес 275,77. Чувствите-
лен к действию света. Получение [1].
Для получения 2,3,4,6-тетра-О-ацетил-р-п-глюкопирапозы [11
раствор 2,3,4,6-тетра-О-ацетил-Р-0-глюкопирапозилбромида в 125 мл
ацетона охлаждают в бане со льдом и прибавляют в течение 15 мин
ОАс
0, Z моля
небольшое количество воды и С. к., свежеприготовленного на рас-
сеянном свету. Взбалтывают еще 30 мин, раствор фильтруют
и фильтрат упаривают в вакууме при самоохлаждении. После одной
кристаллизации из смеси эфир — лигроин получают почти чистый
препарат с. т. пл. 132—134“.
1. Ма к-К л <т с к п Ч., К о л ьмэн Дж,, ^Синтезы органических препаратов»,
ИД, М., 1952, сб. 3, стр. 401.
СЕРЕБРА НИТРАТ, AgNO,. Мол. вес 169,89.
Окисление, см. также Серебра окись. В одной из стадий синтеза
липолевой кислоты Вальборскпй и сотр. [1] окисляли Д9',’-стеарди-
иналь до соответствующей кислоты путем перемешивания раствора
2,34 г альдегида п 2,32 г С. и. в абсолютном этаноле в атмосфере
азота и добавления за 10 мин 4,5 мл 5 и. едкого натра, разбавленного
до 40 мл абсолютным этанолом. После выдерживания в течение
,VXO, - К'гЮН
СН3(СН..)4С -3 ССНХ — (ДСНЩСПО
л СН,(СН ,)4С — CCII./2 С(СНДТСО.2Н
ночи темно-бурый осадок почернел.
л-Кислотиые комплексы. Уинстейн и Лукас 121 исследо-
вали комплексы С. и. с непредельными углеводородами и обна-
ружили, что в некоторых случаях эти комплексы твердые и их
267
можно выделить и очистить. Стилле и Фрей [3] исследовали при-
соединение по Дильсу — Альдеру норборнадиепа (1) и циклопен-
тадиена (2) и в качестве главного продукта реакции получили
аддукт 1:1 (3) наряду с небольшими количествами аддукта 1 : 2,
а также димера, тримера и тетрамера циклопентадиена. После
повторной перегонки аддукт (3) имел степень чистоты около 97%;
53%
(9 (2) (3) (4)
в совершенно чистом виде его удалось выделить через комплекс
с С. н. (4), который получали медленным добавлением при пере-
мешивании 10 мл насыщенного водного раствора С. н. к 7,9 г
(0,05 моля) углеводорода. Смесь застывала в твердый гель, который
отделяли фильтрованием и кристаллизовали из горячего абсолют-
ного этанола. Полученный таким путем комплекс плавился при
157—158° (с разд.) и был аналитически чистым. Для выделения
углеводорода комплекс (14,8 а) растворяли в 150 мл теплой воды
и раствор подвергали непрерывной экстракции эфиром. Углево-
дород (5), являющийся дигидропроизводным углеводорода (3), но
полученный путем взаимодействия цпклопентадиена с норборне-
ном (4) [41, Стилле и Уитерелл [5] подвергали очистке с целью изу-
чения скорости реакций через комплекс с С. и. 1:1. Чистый ком-
плекс был получен с выходом 77% и разложен путем кипячения
(100 а) с 400 мл водного аммиака в течение 3 час с последующими
охлаждением и экстракцией эфиром (выход 71%).
Комплексообразование — простой метод очистки а, со-диенов
с прямой цепью, содержащих 12 и более углеродных атомов [6|.
Очистка Д1,,,-додекадиеиа, полученного по методике Др ахов цал я [7|,
представляет специфические трудности, поскольку он перегоняется
вместе с 1,6-дихлоргексаном. При добавлении 1 моля неочищенного
маслянистого диена к насыщенному раствору 2,37 моля С. н.
в 243 мл дистиллированной воды при энергичном перемешивании
выпал твердый комплекс, который отделяли, промывали холодным
С1(СН.,)(!С1 - ClMg(Cl I-JuMgCl ---CH.2 CH (CH ,)hCJ 1 - CH.,
абсолютным этанолом, сушили на воздухе и перекристаллизовы-
вали из горячего этанола [81. Очищенный комплекс разлагали
268
водой, углеводород экстрагировали эфиром и перегоняли. Газовая
хроматография дала только один ник. Выход, считая па взятый
в реакцию 1,6-дихлоргексаи, составлял 18,9%.
Силикагель, пропитанный водным раствором С. н. и высушен-
ный при 120°, обладает высокоизбирательными адсорбционными
свойствами относительно геометрии, степени замещения и числа
двойных связей непредельных липидов [91. Так, оказалось, что
при элюировании смесями петролейного эфира с бензолом или
эфиром можно достигнуть четкого разделения смеси метиловых
эфиров стеариновой, элаидиновой и олеиновой кислот и смеси ме-
тиловых эфиров олеиновой, линолевой и линоленовой кислот.
При применении этого адсорбента, образующего л-комплексы с кис-
лотами, де Врис 19] добился количественного разделения холесте-
рина и холестапола; такое разделение ранее было осуществлено
с применением хроматографии иа окиси алюминия, по при этом
пришлось преодолеть значительно большие трудности [10]. Сили-
кагель, пропитанный С, и., оказался также пригодным для разде-
ления 1-олеодистсарина и 2-олеоднстеарнна ]11Г
Песнель п Оуриссоп [12] успешно использовали силикагель,
пропитанный AgNO3, для разделения углеводородов (2) и (3),
полученных дегидратацией сесквитерпенового спирта (1). Главный
продукт реакции 1(2), транс-отщепление] имеет трехзамещенную
двойную связь; продукт, образующийся в меньшем количестве [(3),
а-гурыонен, цне-отщепление], имеет тетразамещенную двойную
связь, и этот изомер удерживается сильнее селективным адсорбен-
том в соответствии с правилом, предложенным для моноэтилено-
вых сесквитерпенов, согласно которому величины R1 лежат между
0,90 и 0,95 для тетразамещенных типов, между 0,60 ц 0,75 для трех-
замещенных и между 0,25 и 0,40 для углеводородов с метиленовой
группой.
Гэпстон п сотр. [13] нашли, что разделение глицеридов предель-
ных и непредельных жирных кислот путем кристаллизации при
— 10' можно улучшить, если добавить С. н. в метаноле, так чтобы
образовался комплекс с непредельными компонентами. Моррис 1141
описывал разделение цис- и транс-жирных кислот и окисленных
кислот с помощью тонкослойной хроматографии на силикагеле,
пропитапом С. п. С необработанным силикагелем разделения не
наблюдалось.
269
Воловский [ 151 описал окислительную конденсацию гексадиина-
1,5 с последующей прототропной перегруппировкой при нагрева-
нии с /77/?с/72-бутилатом калия в смеси mpem-бутаиол — бензол при
100 . Полученная смесь непредельных углеводородов легко разде-
ляется с помощью вытеснительной хроматографии на колонке
с окисью алюминия, пропитанной 20%-ным раствором азотнокис-
лого серебра; таким путем были получены тетрадегидро-[ 18]-анну-
лсн (111) и два изомерных тридегидро-[181-аннулена (1) и (II).
Комплексы с азотнокислым серебром удобны для разделения
смеси 1,3-, 1,4- и 1,5-никлооктадиеиов, полученных восстановле-
нием циклооктатетраена натрием и спиртом [16]. Три аддукта
различаюшя по стабильности в водном растворе С.н. Все они ста-
бильны при 0—5 ; аддукт, полученный из 1,3-диена, диссоциирует
при 30 ; аддукт из 1,4-диена — выше 60°; аддукт из 1,5-изомера
устойчив при 90—ЮО' . Чистые диены регенерируют перегонкой
с водяным паром.
Гумулсп (I) можно легко очистить в виде аддмкта с С.н.,
СПС,• 2Ag\O:;. Регенерируют его перегонкой с водяным паром
или обработкой водным аммиаком [17].
Дегидробромирование. С. н. в этаноле оказался очень эффек-
тивным реагентом для дегидробромированпя кетона (Г) 1181.
О
(2)
270
1. Wa I h or s к v H. M., Davis R. 11., H о w t о n D. R , J. Am. Chem.
Soc., 73, 2593 (1951).
2. \V i ns t ei n S., L ucas H. J., J. Am. Clicin. Soc.. £0, 839 (1938).
3, S t i ) 1 e ,1. K., Frey D. A., J. Am. Chem. Soc., 81, 4273 (1959).
4. S n 1 o\v a v S. B., J . Am. Chem. Soc., 74, 1027 (1932).
5. S t i I 1 e J. K., Withei-ell D.R.,J. Am. Chem. Soc., 86, 2188 (1964).
6. Contributed bv Klimstra P. D., Searle G. D. and Co.
7. Draho w 7. a 1 F,, Monatsh., 82, 793 (1931).
8, \V a wzonck S., Klimstra P. D., К a 1 1 i о R. E., S t ew a г t J. E.,
.1, Am. Chem. Soc., 82, 1421 (1960).
9. Vries B., de, Chem. Ind., 1962, 1049.
10. F i e s e r L. F., J. Am. Chem. Soc., 75. 4395 (1953).
11. В а г r c t t С. B., Dallas M. S. J., P a d 1 e v I-’. B., Chem. Ind., 1962,
1050.
12. Pesnelle P-, Ourisson G., J. 0гц. Chem., 30, 1744 (1963).
13. G ii n s t о n e F. D., Hamilton R. J., Q u r es li i At I., J . Chem.
Soc., 1965, 319.
14. M or r i s L. J., Chem. Ind., 1962, 1238.
15. W о 1 о v s к у R., J. Am. Chem. Soc., 87, 3638 (1965).
16. J о n c s W. ,1., J. Chem. Soc., 1954, 312.
17. Hildebrand R. P., Sutherland M. D.. Australian .1, Chem., 14,
272 (1961); see also D a m о d а г а п X. P., Dev S., Tetrahedron Letters,
1977 (1965).
18. С г о rn well N. II., А у c r R. P., Foster P. \V., J. Arm Chem. Soc.,
82, 130 (1960).
СЕРЕБРА НИТРИТ, AgNCt. Мол. вес 153,89, разлагается при
1407 в 100 г Н2О растворяется при 60: 1,06 г, чувствителен к дейст-
вию света, Получение 11—3].
Лкйер |4] при кипячении йодистого амила с С. н, получил смесь
соответствующего алкилнитрпта и шггроалкана. Пользуясь усо-
вершенствованной методикой 11, 2], .можно с высокими выходами
получать чистые шггропарафины из первичных ал кил галогенидов
с нормальной цепью, Примером может служить получение 1-нитро-
октана 121, 1-Бромоктан добавляют в течение 2 час при перемешп-
CHjCH-hBr-Ugxo. А—’Щ’ТМЛсн3(СИ..)7хо^-а1!вг
73-80’.,,
0,5 моля 0,7 5 моля
вании и охлаждении к суспензии С. н. в эфире. Смесь перемешивают
24 час при охлаждении в бане со льдом и еще 40 час при комнатной
температуре. Смесь фильтруют, растворитель отгоняют и в резуль-
тате фракционирования получают 14% 1-октилнитрита (т. кип,
37 /3 мм) и 1-питрооктаи (т. кип, 66 /2 -юн).
1. Kornhlu m N., Taub В., U ngnade H. E.. J. Am. Chem. Som, 76,
3209 (1954); К op н блюм H., «Органические реакции», изд-во «Мир», 1965,
сб. 12, сгр. 117.
2. Корн блюм Н., Унгнаде X., «Синтезы органических лрепараюв»,
ИЛ, М., I960, сб. 10, стр. 59.
3. Plummer С. W., Drake N. L., J. Am. Chem. Soc., 76, 2720 (1954).
4. M e у e г V., Stuber O-, Ber., 203 (1872).
271
СЕРЕБРА ОКИСЬ, Ag»O. Мол. вес 231,76.
Окисление гидрохинонов, о-Бензохннон — высокореакционно-
способное соединение красного цвета, чувствительное даже к дейст-
вию воды,— удавалось получить окислением пирокатехина только
после того, как Вильштсттер и Пфанненштиль 111 обнаружили,
что эту реакцию можно осуществить, применяя С. о., выделенную
осаждением из водного раствора и тщательно промытую несколь-
кими порциями дистиллированной воды, затем ацетоном и, нако-
нец, абсолютным эфиром. К раствору пирокатехина в абсолютном
эфире прибавляют безводный сульфат натрия для поглощения
образовавшейся воды и при взбалтывании добавляют сухую С. о.
до тех пор, пока не перестает углубляться окраска. Раствор фильт-
ОН О
А /ОН Д ,о
| || V/) ХаЗО, || |
'Ч'
2г в 15 0 ял 1 0,5 г 8 г
абсолютного эфира
руют и упаривают, причем о-бензохиноп выпадает в виде блестящих
ярко-красных пластинок. Выход небольшой. Этот метод пригоден
и для получения других малостабпльпых хинонов с высоким окис-
но - X.-a I _снД —К _oi1
-- - •= 40- 50%
ZZZ2—' - Д/
лительным потенциалом, например стильбенхинона [2]. Гидрохи-
нон кипятят в ацетоне с большим избытком реагента.
С. о.— лучший реагент для окисления гидрохинона витамина
К, 131, поскольку это соединение хорошо растворимо в эфире и прев-
ращение его протекает количественно; после фильтрования и упа-
ривания раствора получают аналитически чистый витамин в
виде желтого масла. В этом случае в качестве исходного можно
использовать продажный реагент.
АгСНО-> ArCO.H. J 1ля окисления З-тиофсиальдегида в тиофсн-3-
карбоновую кислоту Кампень и Ле Сюер [4] смешивали растворы
нитрата серебра и избытка щелочи для осаждения бурой С. о., при-
ll ПСНО + AgNO; +
iS
0, 88 моля
0,425 моля В 300 мл Н2О
NaOH —НЯ-, [0“°“
95-97%
1,75 моля
В 300 мл Н2О
бавляли при перемешивании альдегид, отфильтровывали черный
осадок серебра и фильтрат подкисляли. Черное серебро можно
растворить в конц. азотной кислоте н использовать вновь.
272
Для окисления ванилина в ванилиновую кислоту Пирл [5]
применил весьма экономичную методику, которая специфична
для получения и-оксибепзальдегидов, В случае альдегидов других
типов, например 3-тиофенальдегида, при окислении в щелочной среде
необходимо 2 моля нитрата серебра, в то время как для и-окси-
бензальдегида требуется только 1 моль реагента *. По Пирлу,
СНО СО2Н
I -LAgNO3 4-NaOH
। 'к 82-95%
у хосн3 !
он он
1 моль 1 моль 5,85 моля
смешивают водные растворы, содержащие по 1 молю нитрата се-
ребра и едкого натра, перемешивают смесь 5 мин, отфильтровывают
С, о. и отмывают ее от нитратов водой. Влажную С. о, заливают
2 л воды и обрабатывают 4,85 моля гранулированного едкого натра
при энергичном перемешивании, поддерживая температуру 55—
60 , после чего добавляют 1 моль ванилина. Реакция, начинающаяся
через несколько минут, сильно экзотермична и включает две ста-
дии, Первая стадия — окисление С. о. в присутствии щелочи, при
котором образуются натриевая соль ванилиновой кислоты и губ-
чатое металлическое серебро. Вторая стадия состоит в превращении
еще некоторого количества ванилина в кислоту в результате взаимо-
действия с губчатым серебром и щелочью (огромный избыток ед-
кого натра). После перемешивания в течение 10 мин смесь фильт-
руют, а фильтрат и промывную жидкость обрабатывают сернистым
газом, чтобы препарат не приобретал рыжевато-коричневой окраски.
После подкисления получают белые иглы с т. пл. 209—210° (чистое
вещество имеет т. пл. 210—21 Г). Пирл [61 обсуждает возможность
протекания в данном случае специфической реакции Канниццаро,
катализируемой серебром. Однако если бы это действительно было
так, то максимальный выход составил бы 0,5 моля в результате
окисления С. о. и 0,25 моля по реакции Канниццаро; фактический же
выход значительно превышает 75%, Пирл [7] получал ванилино-
вую кислоту с выходом 89—95% сплавлением со смесью едкого
натра и едкого кали при 180 — 195°; в этом случае реакция сопро-
вождается выделением водорода:
АгСНО%2КОН - > АгСО4К + Н, + Н2О
Интересно, происходит ли выделение водорода при реакции с губ-
чатым серебром и водной щелочью при 55—60°.
* Механизм згой реакции объяснил Р. А. Шрайнер,
273
Окисление сахаров. Эванс и сотр. [8] изучали окисление глю-
козы, фруктозы, маннозы и галактозы в виде 0,025 Л1 раствора
как свежеосажденной С. о., так и С. о. с добавлением щелочи. Все
продукты окисления (двуокись углерода, щавелевая, муравьиная
и гликолевая кислоты) можно определить количественно. Смесь
серебра и С. о. отделяют фильтрованием, экстрагируют С. о. вод-
ным аммиаком и по весу оставшегося серебра вычисляют количество
израсходованного кислорода.
Окисление гидразонов. Шрёдер и Кац [9] показали, что при
окислении гидразона (1) обычным методом с использованием окиси
ртути результаты плохо воспроизводятся и что гораздо лучшим
окислителем является С. о. При взбалтывании гидразона (1) с С. о.
(1)
U)
и сульфатом магния в эфире был получен дпарплдиазометап с вы-
ходом 99%. Окисью ртути не удалось также окислить гидразон (2),
однако соответствующее диазососдинение было получено с выходом
89(% при кипячении (2) в течение 5 час с С. о. в ТГФ 110]. Хют-
тель и сотр. [11] показали, что С. о. в качестве окислителя лучше
окиси ртути.
Дегидрогалогенирование. Для дегидрогалогенирования (1), имею-
щего атом брома в аллильном положении, Штейнер и Шинц [121
осаждали С. о. из 5 г AgNO3, промывали ее водой, затем этанолом
и сушили в высоком вакууме. С. о. при охлаждении добавляли
к раствору 4 г галогенида (1) в бензоле и смесь взбалтывали в те-
чение 15 час.
н3с сн3 н3с сн;.
7 ClLOAc СИ, ОАс
II | Ag.O. СДЦ., || [
II Н —г II I н
н.,с у/ Н3С А
Н' Вг
(1) (2)
Шмидлпп и Веттштейи [13] превратили ш/дще-диэкваториаль-
пый бромгидрии (3) в эпоксид (4), действуя на раствор 1,132 г этого
вещества в 21,5 мл пиридина 4,143 г свежеосажденной и высушен-
ной С. о. в темноте в течение 48 час, после чего добавили еще 2,072 г
С. о. и взбалтывание продолжали еще 72 час.
274
АсО
'CH,
С. о. превращает гомоаллилгалогениды в производные цикло-
пропана, например [14]:
СН=СНСНгСНВг
сн.
- -АёгО>
Количеств.
•СН’
!
ОН
сн,
калия также
и иже.
МОЖНО осуществит!.! эту
В присутствии карбоната
реакцию, однако выходы
Аминокислоты из их хлоргидратов. р-Алании можно выделить
из водного раствора его неочищенного хлоргидрата путем переме-
шивания с влажной осажденной С. о. с последующим фильтрова-
нием и обработкой фильтрата сероводородом [15]. Для выделения
е-аминокапроновой кислоты из ее хлоргидрата Экк [16] исполь-
зовал 2 ч. измельченного сурика, 1 ч. осажденной гидроокиси
свинца и 1 ч. С. о. с последующей обработкой сероводородом.
Синтез полиоксн-ь-пролина (6). По усовершенствованной мето-
дике, описанной Канальским и сотр. [ 17], О-ацетил-ь-оксипролии (2)
был превращен в карбон ил хлор ид (3), а последний подвергнут цик-
лизации в ангидрид Лейхса (4) действием С. о. в ацетоне. Кристал-
лический ангидрид в результате полимеризации в пиридине дал
пол и-О-ацетил оксипрол ни (5), при деацетилировании которого вод-
ным аммиаком
был получен полимер (6).
СНОН
СЩ^СНг
HN---СНСОгН
(НСЮ4)'
СНОАс
снг хсн2
HN---СНСОгН
СОСЦ
СНОАс
снгхснг
N---СНСОгН
СОС1
АёгО
(б
(2)
СНОЛс
СН, \сн,
I
N----СН
I I
ОС .СО
-сог
СНОАс
СН,^СН, О
1 г I II
N---СН —С-
NH4OH
-CHjCONH,
^СНОН
сн, хсн, о
I I II
-N--СН —С-
(5)
(Ь)
J
(4)
275
Разложение диазокетонов. Диазокетоны общей формулы (1)
при действии на них свежеприготовленной С, о. в диоксане раз-
лагаются при комнатной температуре до а,0-непредельных кето-
нов 1181. Эту реакцию можно осуществить и путем фотолиза, но
с более низкими выходами, В качестве промежуточного продукта
в реакции образуется кетокарбен.
О N = N О
I! II Ag,0 II
RC —С—CH2R' -------> RCCH=CHR'-l\2
2 50-90% 2
(1) (2)
I. W i I 1 s t a t t е г R., Pf annenst iel A,, Ber., 37, 4744 (1904).
2. К 5 n i g К.-H., Schulze W., Moller G., Chem. Ber., 93, 555 (1960).
3. F i e s e r L. F., J, Am. Chem. Soc., 61, 3467 (1939).
4. Кам пень Э., Л е-Сюср У., «Синтезы органических препаратов», ИЛ,
М., 1954, сб, 5, стр. 69.
5. П и р л И., «Синтезы органических препаратов», ИЛ, ДУ, 195.3, сб. 4, стр. 119.
6. Р е а г 1 1. A,, J. Am. Chem. Soc., 68, 429 (1946); J. Org. Chem., 12, 79 (1947).
7. Пир л И., «Синтезы органических препаратов», ИЛ, ДЕ, 1953, сб. 4,стр. 120.
8. Evans W. L., et al. J. Org. Chem., 1, 1 (1936).
9. Schroeder W-, Katz L., J. Org. Chem., 19, 718 (1954).
10. С о И er A. K„ W ang S. S., J. Org. Chem., 27, 1517 (1962).
11. H ii t t e 1 R., R i e d I J., Mart in H., Fra n ke K., Chem. Ber., 93,
1425 (I960),
12. Steiner U., Schinz II,, Helv, Chim. Acta, 34, 1176 (1951).
13. S c h ni i d 1 i n J., Wet tst ein A., Helv. Chim. Acta, 36, 1241 (1953).
14. Hanack M., Eggensperger H., Chem. Ber., 96, 1259 (1963); H a-
nack M,, Gorier K., ibid., 96, 2121 (1963).
15. Кларк X., Бэр Л., «Синтезы органических препаратов», ИЛ, ДЕ, 1949,
сб. 2, стр. 20.
16. Э к к Дж., «Синтезы органических препаратов», ИЛ, ДЕ, 1949, сб. 2, стр. 36.
17. Kurtz J., Fasman G.D., Berger A., Katchalski Е., J.
Am. Chem. Soc., 80, 393 (1958).
18. Franzen V., Ann,, 602, 199 (1957),
СЕРЕБРА СУЛЬФАТ, Ag„SO4. Мол. вес 311,83. Дербишир
и Уотерс [1] нашли, что при добавлении С. с. к раствору брома
в серной кислоте образуется высокореакционноспособный броми-
рующий электрофильный агент, по-видимому AgBr.t. Аромати-
ческие соединения, дезактивированные заместителями, ориенти-
рующими в лета-положение, например нитробензол и 2,4-динитро-
толуол, можно бромировать с хорошими выходами. Одновременно
образуется бромистое серебро, которое отфильтровывают.
NO2 no2
Br2 — H2SC\-Ад2504
1-Фенилпиразол (1) в обычных условиях бронируется в поло-
жение 4 пиразолыюго кольца, однако Линч и сотр. [2] показали,
что в условиях, описанных Уотерсом, продуктом реакции является
1-л-бромфенилпиразол (выход 62%). В сильнокислой среде в ре-
276
зультате протопирования образуется сопряженная кислота (2),
в которой пиразолыюе кольцо дезактивировано в отношении электро-
фильных реагентов, тогда как -| М-эффект 1-азота облегчает заме-
(1)
(3)
щение в ядра-иоложение. Этот реагент с высоким выходом броми-
рует нереакционноспособный 2-фенил-1,2,3,2Н-триазол (3) до п-
бромфенильного производного [3].
1. Derbyshire D. I I., Walers W. A., J. Chem. Soc., 1950, 573.
2. Khan ALA. Lynch В, M., flung Y.Y., Can. J. Chem., 41, 1540 (1963).
3. Lynch В. M., Can. J. Chem., 41, 2380 (1963).
СЕРЕБРА ХЛОРАТ, AgClO3. Мол. вес 191,34, т. пл. 230°, в 100 г
Н.;О при 153 растворяется 10 г С. х.
С. х. нашел ограниченное применение в сочетании с каталити-
ческим количеством четырехокиси осмия для гидроксилирования
растворимых в воде непредельных кислот [11 и спиртов 12] в цис-
вицинальные диолы:
СН3
I
н—с—он
СН3 —С —Н AgCiO3(OsO<) I
lj---------------> НО—С—Н
н-с-со2н |
со.,н
В осадок выпадает хлористое серебро. Другие хлораты дают смеси
гликоля и хлоргидрина.
1. Braun G., J. Am. Chem, Soc., 51, 228 (1929),
2. Posternak Th., E г i e d I i H., Helv. Chim. Acta, 36, 251 (1953).
СЕРЕБРА ХРОМАТ, Ag2CrO4. Мол. вес 331,77.
Ключевой стадией в синтезе кортизона из желчной кислоты по
Кендаллу и сотр. [1] является окислительный гидролиз 1ф,12а-
дибромида, формула которого приведена ниже, в 12сс-бром-11-кетон.
Реакция определяется активацией 110-брома окисным кислородом
0,16 МОЛЯ , 2, ! Л
ацетона
277
у Ся, поскольку соответствующий стероид без За,9а-ок ясного
мостика не реагирует. Суспензию дибромида в ацетоне переме-
шивают, добавляют С.х. и раствор хромового ангидрида в 200
воды и путем охлаждения поддерживают температуру 25—285 еще
в течение 2 час до полного растворения твердого вещества. При-
ливают 77 мл 5 н. серной кислоты, раствор фильтруют и упарива-
ют в вакууме.
(.Turner R. В., Mattox V. R., Enyel L. L., McKenzie В. I7.,
Kendall E. C., J. Biol. Chem., 166, 345 (1946).
СЕРЕБРА ЦИАНИД, AgCN. Мол. вес 133,90.
Для получения этил изоцианида [1] С. ц. добавляют к иодистому
этилу, и смесь перемешивают и нагревают на кипящей водяной
бане до превращения в бурую вязкую жидкость, представляющую
собой комплекс, который иногда закристаллизовывается. Мешалку
поднимают так, чтобы она была несколько выше уровня жидкости,
и через обратный холодильник приливают сначала воду, а зате.м
2KCN(H.O)
CJKN-AgCN —> С2НЭ\Т —C-Agl — ~ C.,H5N-C^KAg(CN)2TKI
/ — о о %
Зт4 моля 3,4 моля
раствор цианистого калия. Этил изоцианид выделяется в виде бу-
рого слоя.
1. Джеке а и Ч., М а к-К у з и к Б., «Синтезы органических препаратов»,
ИЛ, М., 1956, сб. 7, стр. 83.
СЕРНИСТОЙ КИСЛОТЫ N, N-ДИЭТИЛАМИДА МЕТИЛОВЫЙ
О
ЭФИР, CH;)OSN(C.H5)2. Мол. вес 151,23, т. кип. 73—74 °/10 мм.
Реагент получают из диэтиламина и метилового эфира хлорсуль-
фоновой кислоты [11. В пиридине при комнатной температуре он
реагирует с карбоновыми кислотами (с карбонильной группой при
первичном и вторичном углероде) в течение нескольких дней, об-
разуя соответствующие \Д!-дпэтплампды. Кислоты с карбоксиль-
ной группой при третичном углероде и а-аминокислоты в реакцию
не вступают [2].
О о
I
RCO.2H-L-CH3()SXV:2Hr>)., RCX(C2Ha)2
1. Zinner G., Chem. Ber., 91, 966(1958).
2. В e c k er ft. G. O., Fun k K. F., J. prakt. Chem., 14, 55 (1961).
СЕРНИСТЫЙ АНГИДРИД, S02. Мол. вес 64,07, т. кип. —10°.
Изомеризация полиенов. Группой авторов [1] установлено, что
С. а. в пиридине изомеризует эр гостер ил ацетат (1) в Д6,8(и)- нли
ЕК-пзомер (4); реакция, вероятно, осуществляется через образо-
вание аддукта Дильса — Альдера (2). Изомеризация дегидроэрго-
стер пл ацетата, А&,7,аИ1)-триена, в ДВ’8(]4)’Э(] шТрПен действием SO.
278
в пиридине лежит в основе синтеза кортизона.
1. L a u b а с h G. D.,Schreiber Е.С., AgnelloE.J., L
E. N., Brunings K. J-. J- Am. Chem. Soc., 75, 1514 (1953};
G. D., Schreiber E.C.,AgnelIo E. J., Brunings
78, 4743 (1956); sec also Agnello E. J., Pinson R., Jr.,
G. D., ibid., 78, 4756 (1956).
i g h t f о о t
Laub ac h
K. J ., ibid.,
L a u b a c h
СЕРНЫЙ АНГИДРИД, SOa. Мол. вес 80,07, т. пл. 17", т. кип.
около 45". В органической химии С. а. используют главным обра-
зом в виде комплексов с диоксаном, ДМФЛ и пиридином.
3—5 Л4 раствор С. а. в нитрометане реагирует с а рил галогени-
дами при О', образуя ангидрид сульфокислоты, который осаждается
из раствора и при гидролизе дает сульфокислоту [1],
CH.rNO,
3SO3-r2C6H5I (lC6H4)2S2O.4-HaSO4
[.Christensen \т. IL, Chem. Scand., 15, [307 (1961).
СЕРНЫЙ АНГИДРИД — ДИМЕТИЛФОРМАМИД, SO, x
xHCO\r(CH;i).,. Мол. вес 153,17.
Этот реагент получали (Гарбрехт [1]) постепенным добавлением
в течение 4—5 час 903 г технического SO:i к 10—11 л ДМФА при
охлаждении (0—5) и перемешиванием с обратным холодильником.
При этом наблюдается осаждение кристаллического комплекса,
но перемешивание продолжают до полного растворения.
Синтез амидов. Гарбрехт [1] использовал реагент в улучшенном
синтезе амидов (-(-)-лизергиповой кислоты. К раствору моногпд-
279
рата кислоты и моногидрата гидроокиси лития в метаноле добав-
ляют ДМФЛ и отгонкой в вакууме удаляют метанол и затем воду.
При добавлении к смеси 2 да реагента образуется смешанный
ангидрид, который при взаимодействии с 1 экв вторичного амина
дает амид кислоты.
Ци клодегидратация. Реагент с успехом используют для цикли-
зации в синтезах азлактонов [2] и бутенолидов [3—61.
N,\'-Диацилгидразины циклизуются реагентом с образованием
1,3,4-оксадназолов с выходом около 90% 151.
RCNHNHCR* —Д|ДФ>> N N
О
Синтез пептидов
CbNHCHCO.Li ---— ----— > CbXHCHCO2SO3Li —>-
К'
1
н„хт:нс.о.хм
———CbNHCHCOXHCFICOA’a -1- IJHSO4
i I
К R' j
I. Garhrerht \V. I., J. Org. Chem., 24, 368 (1959). t
2. Ballazz i E., Davis E. A., Chem. Ind., 1962, 929.
3. В a I f a 7, 7. i E., Davi s E. A., Chem. Ind., 1962, 1653.
4. Rao Y. S,, Killer R., Chem. ind., 1964, 280.
5. В a 1 1 a z z i E., \V ysorki A. J., Chem. Ind., 1963, 1080.
6. Kenner G. \V., Stedman R. JJ. Chem. Soc., 1952, 2069; Clayton
D. \V., P a r r i n g t о n J. A., К e n n e r G. W., T u r n er J. At, J . Chem.
Soc., 1957, 1398; Albertson N. E’., Org. Reactions, 12, 255—260 (1962),
280
СЕРНЫЙ АНГИДРИД —ДИОКСАН, SO3 -ОС4НЙО. Мол. вес
168,17.
Получение. По Рондестведту и Бордуэллу, 800 а охлажденного
дихлорэтана насыщают серным ангидридом,полученным из 60 ° о-кого
олеума, до привеса 300 г. К раствору серного ангидрида upir ох-
лаждении до — 5:J и перемешивании добавляют по каплям эквива-
лентное (в расчете на SO3) количество диоксана; прн этом выпадает
комплекс в виде мелких гранул. Этот кодшлекс намного реакционно-
способнее, чем пиридин — серный ангидрид; он гидролизуется
холодной водой.
Сульфирование олефинов. Раствор 1 моля стирола в 2 об, ди-
хлорэтана добавляют при охлаждении к суспензии реагента в ди-
хлорэтане (Рондестведт и Бордуэлл), смесь кипятят с обратным хо-
лодильником и стирол-Р-сульфокнслоту выделяют в виде натриевой
соли Ш.
ОСД-[8О SO3 NaOH
СвН5СН^СН2 -ггт7—7Г CeH5CH=CHSO3H C6H5CH^CHSO3Na
C-j 11—> Н Э—' Гт [ I.) о и |Д jj
Сульфирование олефинов под действием реагента г сследовано
в работах 12] — [4].
Сульфирование фенантрена. 88 г (1,1 моля) SO3 добавляют по
каплям при перемешивании и охлаждении льдом к 100 л/л диоксана
в 350 мл дихлорэтана. Затем добавляют 1 моль фенантрена и смесь
нагревают в течение 5 час при 60° [5]. На схеме показаны выходы
моносульфокислот, полученных сульфированием реагентом и конц.
серной кислотой (Соломон и Хеннесси) 16].
Сульфирование при 60°
Сульфирование альдегидов и кетонов. Трюс и Олфьери [7]
использовали реагент для превращения простых альдегидов и ке-
тонов в а -сульфонаты с выходом около 70%.
[.Рондестведт К., мл., Б орду э л л Ф., '.Синтезы органических пре-
паратов», ИЛ, АС, 1956, сб. 6, стр. 57.
2. Bordwell F. G. et al., J. Arn. Chem. Soc., St, 2002 (1959), and preceding
papers of the series.
3. Su t er C. At, Truce \V. E., J, Am. Chem. Soc., 66, 1105 (1944).
4. Sperling R., J. Chem. Soc., 1949, 1925.
5. Solomon At G., ff enness у D. J-, J. Org. Chem., 22, 1649 (1957).
6. ф и з e p Л., 'Синтезы органических препаратов», ИЛ, М., сб. 2, 1949, стр. 493.
7. Truce W. Е., A I f i е г г С. С., J. Am. Chem. Soc., 72, 2740 (1950).
281
СЕРНЫЙ АНГИДРИД — ПИРИДИН, SO. C.H N. Мол. вес
159,16, т. ил. 175 .
Получение [11. а) Пиридин (1 моль) медленно при перемешивания
н охлаждении прибавляют по каплям к смеси измельченного сер-
ного ангидрида (1 моль) и четырсххлорпетого углерода (4 ч.),
кристаллический продукт отделяют и промывают небольшим коли-
чеством ледяной воды для удаления следов пиридина и серной
кислоты.
б) Хлорсульфоновую кислоту (0,5 моля) при охлаждении и пере-
мешивании прибавляют по каплям к раствору пиридина (I моль)
в четырех хлор истом углероде; продукт промывают ледяной водой
для удаления хлоргидрата пиридина:
2СГ1Н-Х CISO.H C-H5\ SO,5 i СГ1Н5ХНС|
Сульфаты. Баумгартен |21 показал, что реагент сульфирует
такие соединения, как гидразин, диэтпламин, фенол и нафталин.
Его можно использовать для получения сульфатов углеводов 13],
стероидов [4| и фенолов [51,
2-Оксп-З-алкнл-1,4-нафтохинон, противомалярийный препарат,
можно отделить от продуктов обмена, содержащих гидроксильные
группы в боковой цепи, превращением гидроксилированных про-
дуктов обмена в растворимые в воде сульфоэфиры с последующей
экстракцией водой [6]. Продукт обмена гидролапахола, иденти-
фицированный как оксигидролапахол (1), имеет третичную спир-
товую группу, за счет которой легко превращается в сульфат (2)
под действием хлорсульфоновой кислоты в пиридине. Эфир (2)
СН..СН.,С ' (Г
| ”CJis i ClSO;fH4-Py—>
ОН 0,1 .11.2 1 ,11.2
ОН
° .СН3
ХНХНХУ
II I di'3
II OSO.OPyH
Y хон
о
( 1 ) 100 .rh' П 1 .11.2 Ру
стабилен в пиридине, но неустойчив в присутствии воды. Несмотря
на это, найдены эффективные методы расщепления, гидролиза или
расщепления кислотами НХ до олефинов. Реакции можно выпол-
нить в микромасштабах и количественно проанализировать колори-
метрически.
282
Баддили и сотр. [7] использовали реагент для превращения
аденозин- 3',5'-дифосфата в аденоз и и-3'-фосфат-5'-сульфофосфат.
В присутствии соответствующего фермента «активный сульфат»
передает сульфатную группу различным субстратам.
Класс 18] обнаружил, что алифатические альдегиды реагируют
с этим комплексом в кипящем дихлорэтане, образуя с .хорошим
выходом соли бетаина. Ацетон и бензальдегид в тех же условиях
не реагируют.
RCHO + C5HsN-SO3
> RCHOSO3
1. В a u ш й а г t е п Р., Вег., 59, 1166 (1926).
2. В и и in sjarten Р., Вег., 59. 1976 (1926).
3. Duff R. В., .1. Clicm. See., 1949. 1597.
4. S о b с 1 А. Е., D г с k ter I. 4., Na t elsn n S., .J. Biol. Chem.. 115,
38 1 (1936); Sobel Л. E., Spocrri P. E., J. Am. Chem. Sue., 63, 1259
(1941); 64, 361 (1942).
5. Bur к h а г d t G. X., Lap yx orth A., ,1. Chem. Soc., 1926, 684,
6. F i c s e r L. I'., J. Am. Chem. Soc., 70, 3232 (1948).
7. В a d d i 1 с у .1., Buchanan J . G.. Letters R., Sanderson
A. R., J. Chem. Soc., 1959, 1731; Ba d d i le у J., Sander son A. R., Bio-
cliern. Prcpns., 10, 3 (1963).
8. К 1 a s s D. L., J. Org. Chem., 29, 2666 (1964).
СЕРНЫЙ АНГИДРИД — ТРИМЕТИЛАМИН, (CHa);{N4'— SOy.
Мол. вес 139,18, т. пл. 236—240’’ (с разл.), медленно гидролизуется
водой, по может быть перекристаллизован из воды.
Вещество является мягким сульфирующим агентом. Со спир-
тами и фенолами образует аммонийные соли мопосульфата 11].
Смесь о- и п-фен ил фенолов (ио 0,05 моля каждого) можно разделить
при обработке 0,05 моля реагента в слабощелочном растворе с по-
следующей экстракцией эфиром; n-фенилфенол реагирует селек-
тивно, образуя чистый сульфат ]2].
/i-CfiH5CriHjOl 1 (CH3):iX-SO.; — mCJI.CjIjOSOTfCIIJ.jN
I. Traub c W., Z a n d e r IL, G a f f г о и H.. Clicm. Ber., 57, 1045 (1924).
2. liar d у W. B., Scalera M., J. Am. Cbem. Soc., 74, 5212 (1952).
СЕРОУГЛЕРОД, CS.;. Мол. вес 76,14, т. кип. 46,3”, уд. вес 1,263.
Серебряные соли карбоновых кислот при нагревании с С. в за-
паянной ампуле при 100 ' в течение 8 час превращаются в соответ-
ствующие ангидриды 11].
4RCO,Ag -; CS., 2(RCO).O СО., 2Ag,S
f. В г у с. e-S ru i t h D., Proc. Chem. Soc., 1957, 20.
2S3
СЕРЫ ШЕСТИОКИСЬ, S_.Of;. Получение С, ш. и се использо-
вание для превращения 1,5-динитроиафтал1ша в нафтазарин опи-
сано в патентной литературе [11 и в статьях Шарье [21 и одного из
авторов книги [31. По методике Физсра 25 г серы растворяют
в 375 ли 18%-ной дымящей серной кислоты и раствор добавляют
к суспензии 50 г 1,5-дпнитронафталнна в 230 мл кони,, серной кис-
лоты при 60°. После окончания реакции смесь охлаждают и вы-
ливают в лсд.
1. Герм. пат. 71386.
2. С 11 а г г i с г G., Т о с с о G., Gazz. chim. Ital., 53, 431 (1923).
3. Г i с s е г L. F., J. Am. Chem. Sue., 50, 439 (1928).
СЕФАДЕКС (Sephadex).
С. представляет собой химически модифицированный декстран,
полученный в результате брожения тростникового сахара. Благо-
даря своей сшитой структуре он может действовать как молеку-
лярное сито для отделения низкомолекулярных соединений от
сопутствующих нм высокомолекулярных веществ с помощью про-
цесса, известного под названием гель-фильтрации. С. выпу-
скается пяти тнпов; G-25, G-50, G-75, G-100, G-200; G-25 имеет
наиболее сшитую структуру, a G-200 — наименее. Так, с помощью
С. G-100 можно отделять вещества с молекулярным весом около
100 000 и менее от более крупных молекул; G-200 позволяет отде-
лять вещества с молекулярным весом менее 200 000; G-50 — ве-
щества с молекулярным весом 7000—10 000.
Эти материалы были введены в практику в 1959 г. [1, 2] для
фракционирования водорастворимых веществ. Мелкие зерна С.
набухают в водной среде, и колонка с набухшими зернами образует
стационарную фазу геля, состоящую из сшитых цепей декстрозы.
Большие молекулы не могут внедриться в гелевую фазу и движутся
в водной фазе, которой окружены зерна. Меньшие молекулы про-
никают в поры геля и проходят через колонку в водной фазе как
через сами зерна, так и вне их и таким образом продвигаются
дольше и элюируются позднее, чем большие молекулы.
В первом сообщении [1] было описано использование С. для
обессоливания белков сыворотки крови; этот процесс подобен диа-
лизу. Вероятно, наиболее важное применение С.— разделение
протеинов, пептидов н аминокислот, которое подробно исследовал
284
Порат [3]. С., набухший в фосфатном буфере (pH 6,8), переносят
в хроматографическую колонку и медленно пропускают через нее
раствор смеси, которую надо разделить. При этом легко достигается
групповое разделение; для фракционирования в пределах одной
и той же группы может потребоваться декстрановый гель .меньшей
степени сшивания. Преимуществом является то, что гель можно
легко регенерировать в самой колонке и снова использовать в те-
чение нескольких месяцев.
Гросс и Виткоп [4| пришли к выводу, что фильтрование через С.
G-25 — совершенно незаменимый способ разделения фрагментов,
получаемых в результате расщепления метионин-пептидных свя-
зей в рибонуклеазе под действием бромциана. В этом случае в ка-
честве растворителя используют 0,2 н. уксусную кислоту.
В брошюре «А. В. Pharmacia» о С. и его использовании в ка-
честве фильтрующего геля приведено около 100 ссылок за время
с 1959 по 1962 г. (о применении С. для выделения, разделения
и очистки протеинов, пептидов и аминокислот). В брошюре № 1
(1965 г.) приведено уже 638 ссылок и даны также ссылки на рефе-
раты.
В мен[,шей степени С. был использован в области нуклеиновых
кислот [5, 61. С. G-50 пригоден для отделения высокомолекулярной
РНК от нуклеозидов или оснований. Однако с помощью С. нельзя
разделить сложную смесь нуклеотидов (от моно до гекса), получае-
мых ферментативным гидролизом РНК. С. G-25 был использован
в качестве носителя при разделительной хроматографии дрож-
жевой Т-РНК [7].
Применение сефадекса в области углеводов показало, что можно
разделять смеси олигосахаридов, отличающихся по своей величине
лишь на одну глюкозидную единицу [81. Низкомолекулярные декст-
раны удалось разделить гель-фильтрацией [9|. Описано 110) частич-
ное разделение ряда декстринов крахмала па колонках с С. Так,
амилодскстршг может быть полностью отделен от амилозы. Этот
метод особенно удобен в случае полисахаридов, которые по свой-
ствам мало отличаются друг от друга и обычно различаются лишь
ио молекулярному весу.
1. Рог ,i I h J-, F 1 о d i п Р., Nature, 183, 1657 (1959).
2. G с 1 о t t е В.. Krantz А.-В., Chem. Scanit., 13, 2127 (1959).
3. Р о г a t li J ., Biochem. Biophvs. Acta, 39, 193 (1960).
1. Gross E., A' i t k о p B., j. Biol. Chem., 237, 1856 (1962).
5. Gclotte B., Naturv. Es., 48, 554 (1961).
6. Z a d r a z i I S., S о r m ova Z., Sor m F., Coll. Czech., 26 2643
(1961).
7. Tanaka K., Richards 11.-fl., C a n t о n i G. I.., Biochem. Biophvs.
Acta, 61, 816 (1962).
8. ( I о d i n P., A s p b ii г у I\., IUB/IUBS Symposium on «Biological Struc-
ture and Function», Stockholm, I960.
9. G r a n n t h I\. A.. F 1 о d i n P._ Makromol. Chem., 48, 160 (1961).
10. N or d i п P., Arch. Biochem. Biophys., 99, 101 (1962).
285
СИММОНСА — СМИТА РЕАКТИВ, ICH2ZnI или его димер.
Природа реагента. Химики фирмы «Дюпон» |1|, предложившие
этот реагент, позднее точно описали методику его получения и при-
менения в реакции с циклогексеном, приводящей к норкараиу 12L
Реагент получают при взаимодействии йодистого метилена в эфире
с активной цинк-медной парой, полученной но Шенку и Шехтеру [3]
из промытой кислотой цинковой пыли ii раствора сернокислой меди.
Затем цинк-медную пару промывают водой, абсолютными спиртом
и эфиром. К суспензии такой пары в абсолютном эфире прибавляют
+ Zn пыль(Си)
О, 75 моля
О, 65 гчоля
СН21г
О, 71 моля
Эфир(кип.ячение 15 час
56-58%
кристаллик пода, а затем олефин и подпетый метилен. После ки-
пячен ия в течение определенного срока эфирный раствор деканти-
руют от мелко раздробленной медн, промывают насыщенным раст-
вором хлористого аммония, с^шат в выпаривают. Выходы па ос-
новании 30 примеров обычно в пределах 30—60%. Реакция при-
менима к винилацетату (выход 30°о), днгидропнрапу (65%), сти-
ролу (32%), 1-(о-, л- и п-метокс и)-фен ил пропен у (60—70%>). Реа-
гент характеризуется значительной стереоспецифичностью, так
как даже с избытком реагента о-лимонен образует моноаддукт.
СНа
О-Лимснен(-|- 106°)
СН,
+ 51°
Вначале реагент рассматривали [II как подпетый подмети л цинк,
ICFLZnl,— сравнительно прочно связанный комплекс карбена
и подпетого цинка (а), в котором атом углерода электрофилен.
.. Znl
сн2 :
I
С. .'Znl
—> Г: \'сн2 :
С’* •'!
, > I .СН2 К Znlj
(а)
Внттнг [41 получал реагент Симмонса — Смита из подпетого цинка
и дпазометапа и приписал ему формулу подпетого подметил цинка.
Хоберг [51 высказал мнение, что реакция Симмонса — Смита не
ZuL -: СН2\Ц-^ ICH.Znl -Г-Х2
286
включает промежуточного образования карбена, а происходит
через простое присоединение высокореакциоииоспособного йодис-
того иодметилципка:
- с 7н1 — C-Zni —С
|| --1 — | -> | сп._.: ztli3
- с сн.д —с-снл -с
I
Однако несколько позднее химики фирмы Дюпон |6, 7] выска-
зали точку зрения, что, по-видимому, реагент представляет собой
не простой мономер, а комплекс бпс-(подметил)-цинк — подпетый
нипк (ICH,) >Zn-Znl.,.
Если олефин или образующееся цпклопропановое производное
чувствительно к иодистому цинку, который является кислотой
Льюиса, то обычную методику можно изменить, применив в качестве
растворителя диэтиловый эфир, содержащий 1 же тлима (1,2-диме-
токсиэтана) 171. Йодистый цинк удаляют из раствора количественно
в виде кристаллического нерастворимого комплекса с тлимом (1 : 1).
Jle-Гофф [8| получил очень активный цинк-медный комплекс,
при использовании которого нет необходимости в активации иодом
и можно применять нс только подпетый, но и бромистый метилен.
Для его получения на гранулированный цинк пли цинковую пыль
действуют горячим раствором моногидрата ацетата меди (II) в го-
рячей уксусной кислоте с последующей промывкой уксусной кис-
лотой и эфиром.
В процессе синтеза спирановых углеводородов типа, приведен-
ного ниже, Винеман и сотр. [9] усовершенствовали методику: они
отделяли подпетый метилен до обработки смеси, для чего добавляли
2-метилбутен-2 и дополнительное количество цинк-медной пары
и смесь кипятили еще 24 час.
Стерическая направленность реакции. Образование цпклопро-
панового кольца при использовании реагента протекает стереосис-
цпфично и является /щс-пр исосдинеписм; например, из цис- и
/нранс-гексенов-3 образуются чистые цис- и транс- 1,2-диэтнл-
циклопронапы.
Кори и сотр. [101 нашли, что реакция Симмонса — Смита лучше
идет при наличии гидроксильного заместителя. Так, Д"-циклопенте-
нол реагирует быстрее и с лучшими выходами, чем соответствую-
щий ацетат или циклопентадиен. Поскольку циклопропановое
кольцо находится в цис-положении к гидроксильной группе, Кори
полагает, что атом кислорода в исходном соединении образует
287
координационную связь с цинком, Даубен и Березин 1111 показали,
что гидроксильная группа в спирте (3) аллилового типа также уско-
ряет реакцию Симмонса — Смита и что циклопропановос кольцо и
гидроксильная группа в (4) находятся в цпе-положении друг к другу.
Это наблюдение открыло путь для стереоспецифического синтеза
углеводорода (5) 1121, который оказался идентичным сесквитерпену
(+)-туйопсену; таким образом была выяснена стереохимия этого
углеводорода.
Удивительно, что обычно образуется только один изомер. Так,
сотрудники фирмы «Дюпон» [7] нашли, что порборнен (6) дает
экзо-аддукт (7); эндо-изомер не обнаружен. Уинстейн и сотр. [13]
показали, что цис,цис,цис-\ ,4,7-циклононатрнен (8) с реагентом
образует с хорошим выходом только цис-аддукт (9).
СНг1г Zn
(?)
СНг1г-Zn^
80-9 0%
(9)
Реакция с ацетиленами. Стеркуловая кислота была синтези-
рована путем кипячения стеародовой кислоты в эфире с подпетым
метиленом и цинк-медной парой [141. Дробной кристаллизацией
аддуктов, образовавшихся при действии мочевины, была выделена
288
в чистом виде цнклопропеновая кислота (т. пл. 19°) с выходом 4%
СН21,— Zn
Кипячение в эфире Я час
СН3(СН2)7С = С(СН..)7СО.,Н ------—--------->
4 о
сн2
СНГ,(СН,)7С=С(СП2)7СО;Н
после проведения реакции в течение 9 час; через 12 час выход сни-
зился до 2,5%.
Реагент взаимодействует с соединениями, имеющими «-ацетиле-
новую связь, образуя в качестве основного продукта метильное
производное [151. В качестве растворителя рекомендуется смесь
СИЛ., —Zn
СЙН6С-=СН ——• > с6н5с--.сси3 + с6н5сп=с = сн2
6%
1,2-диметоксиэтан — эфир; если применять только первый раство-
ритель, то реакция протекает слишком бурно и ее трудно контро-
лировать.
Вассерман н Клагетт [16] осуществили синтез циклопропанолов,
замещенных в положении 1, исходя нз 1-алкоксивипилового эфи-
ра (2), который можно получить из алкокснацетнлена (1). Например,
сгн5о-с=сн ЧЬд°?-н:
Hg(OAc)2
С2Н5О
^с=сн2
СНзСО^
I!
CHzI2-Zn
35%
С2Н5О
СН3СО
Ml
о
(3)
CH3MgI^
4 6%
(4)
при синтезе 1-этоксициклопропилацетата (3) добавляли глим для
осаждения йодистого цинка и защиты нестойкого соединения (3),
которое по реакции Гриньяра было превращено в 1-метилцикло-
пропанол (4).
1. Simmons Н. Е., Smith R. D,, J. Am. Chem. Soc., 81, 4256 (1959).
2. Смит P. Д., Симмонс X. Э., «Синтезы органических препаратов»,
изд-во «Мир», 1964, сб. 12, стр. 110.
3. Shank R. S., S h е с h t е г Н., J. Org. Chem., 24, 1825 (1959).
4. W i t t i g G_, Schwa rzenb ach K., Ann., 650, 1 (1961); \V i t t i g G.,
Wing] er F., Ann., 656, 18 (1962).
5. Hoberg H., Ann., 656, 1, 15 (1962).
6. В la n char d E. P., Simmons II. E., J. Am. Chem. Soc., 86, 1337
(1964).
7. Simmons H. E., Blanchard E. P., Smith R. D(, J. Am, Chem.
Soc., 86, 1347 (1964),
10 Никаз № [I6G
289
8. L c G о f f E., .). Огд Chem., 29, 2048 (1964).
9. К о c lj S. D., К 1 i s s R. M., L о p i с к cs D. V., W i n e m л n R. J.,
J. Огд. CZhcin., 26, 3122 (1961).
10. Core у E. J., Da w son R. L., .]. Am. Chem. Soc.. 85, 1782 (1963); C o-
rev E. J., U (I ;i 11J . Am. Chem. Soc., 85, 1788 (1963).
11. 1) и' if b e n W. G.. Berezi n G. 11., J. Ain. Chem. Soc., 85, 468 (1963).
12. I) ;i и b e n \V. G., A s h c r ;i i't Л. C., J. Am. Chem. Soc.. 85, 3673 (196.3).
13. Boi к ess R. S., i ns t ei n S.,J.Am. Chem. Soc., 85, 343 (1963); R ;i d-
I i с к P., Winst ei n S., J . Am. Chem. Soc., 85, 344 (1963).
f4. C, a s t c I I it c e i M. T., Griffin С. E., J. Am. Chem. Soc., 82, 4107
(I960).
15. Q и a n у L. V., C a diol P., Bull, soc, cliini. Trance, 1965, 1525.
16. \V a s s e r ni a n П-ll., fl 1 a Melt D. C., Tetrahedron Leiters, 341 (1964).
СТЕАРИНОВОЙ КИСЛОТЫ АНГИДРИД, (C, TH:i ,CO)?O. Мол. вес
550,92, т. пл. 7Г'.
С. к. а. получают кппячеииехМ стеариновой кислоты с уксусным
ангидридом. С. к. а. рекомендуют применить вместо уксусного
ангидрида для аналитического определения первичных и вторичных
спиртов и фенолов 11]. Анализируемое соединение кипятят с на-
веской С. к. а. в ксилоле, избыток ангидрида гидролизуют водой
и стеариновую кислоту оттитровывают.
1. Sull у В. 1)., Analyst, 87, 640 (1962).
О О
СУКЦИНОИЛА ПЕРЕКИСЬ, НОХСН.СН-хСОСХГН-.СНХОФ!.
Мол. вес 243,16.
С. и. получают из янтарного ангидрида и водной перекиси во-
дорода и используют для эпоксидирования олефинов 11]. В качестве
растворителя пригоден ДМФА, содержащий небольшое количество
воды, необходимой для гидролиза перекиси.
1. Bln m J., Compt. rend., 248, 2883 (1959).
СУЛЬФАМИНОВАЯ КИСЛОТА, HASO.OH. Мол. вес 97,10.
С. к. используют для разрушения избытка азотистой кислоты
при диазотировании.
СУЛЬФОЛАН, см. Тетраметплснсульфон,
СОЛ
/ он
HO3SZ^
СУЛЬФОСАЛИЦИЛОВАЯ КИСЛОТА,
Мол. вес 254,22, растворяется в воде,
спирте, эфире.
Кендалл н Мак-Кензи 11] этернфидпровали сырую (З-бромпропно-
новую кислоту (около 4 молей) нагреванием с водоотделителем
в смеси четыреххлористый углерод —этанол, содержащей 10 аС. к,,
и получили этпл-р-бромпропионат с общим выходом 85—87%.
290
Разработана [21 методика определения относительных скоростей
енольного ацетилирования кетостероидов уксуспььм ангидридом
в присутствии кислотного катализатора и установлено, что С. к.
но каталитической активности превосходит п-толуол сульфо кислоту.
С. к. используют как катализатор для получения циклических
1,2-ацсталей глицерина обменной ацетализанисй [3]. Напри-
мер, смесь 30 г гексадецснальдпметилацеталя, 18 г глицерина
.осн, сн2он
н-С15Н:иСН -| СИОН
ХОСН3 CPI..O1I
и 50 мг С. к. нагревают при перемешивании при 130 — 170° в колбе
с насадкой для перегонки и собирают выделяющийся метанол до
тех пор, пока не оттопится примерно теоретическое количество
спирта. Продукт перекристаллизовывают из метанола, выход 93%,
т. пл. 47—48°.
1. Кендалл Е. К., Ala к-Ке из и Б., «Синтезы органических н реи ара гов»,
ИЛ, ЛЕ, 1949, сб. I, стр. 543.
2. Anderson И., Garret Е. R., Lincoln Е. II., Jr., \г a t li а п
Л. Н., И ogg J. А., .1. Am. Chem. Soc., 76, 743 (1954).
3. Р i а и t a d о s i G., Anderson С. E., В г e c h t E. А., A’ a r 1) г о
C. L., J. Am. Clicri). Son., 80, 6613 (1958); P i a n t a d о s i С., И i г s c h A. F.,
Yarbr о C. L., Anderson С. E., J. Org. Chem., 28, 2125 (19b3).
СУЛЬФОУКСУСНАЯ КИСЛОТА, CH:{CO2S(WH.
Раствор С. к. получают добавлением ио каплям охлажденной
льдом конц. серной кислоты к уксусному ангидриду II]. Кристал-
лический продукт был выделен Штиллихом 12].
Хаузер и сотр. [3] превращали алифатические кетоны в их ено-
ла цетаты реакцией с кетеном в присутствии 0,5% С. к. Шнейдер
и Зак 141 получили перхлорат 2,4,6-тримегплпприлия добавлением
при охлаждении 8 мл копи, серной кислоты к 33 мл уксусного
ангидрида, последующим добавлением 8 мл окиси мезитила и
(спустя 24 час) нагреванием до 40—45°. Добавление льда и затем
хлорной кислоты дает 3,5 г перхлората пиридия. В усовершенст-
вованной методике [5] применяют хлорную кислоту в качестве
кислотного катализатора.
озо2он
10'
291
Проба Либермана — Бурхарда на ненасыщенные стерины сос-
тоит в добавлении С. к. к раствору стернна в хлороформе; при по-
ложительной пробе появляется яркое окрашивание [61.
1. Franchimont М., Compt. rend., 92, 1054 (1881).
2. S t i 1 I i с h O., J. prakt. Chem., 73, 541 (1906).
3. Y oung F. G., F г о s t i c k F. C., Jr., Sanderson J.J., Hauser
C. R., J. Am. Chem. Soc., 72, 3635 (1950).
4. Schnei der W., Sack A., Ber., 56, 1786 (1923).
5. H a f n с г К., К a i s e г H., Oig. Syn., 44, 101 (1964).
6. Физер Л., Физер M., «Стероиды», изд-во «Мир», М., 1964.
СУЛЬФУРИЛ ХЛОРИСТЫЙ, CLSO.,. Мол. вес 131,98, т. кип.
69,2°, уд. вес 1,667. С. х. получают взаимодействием двуокиси
серы с хлором в присутствии активированного угля в качестве
катализатора [1].
Об использовании С. х. в синтезе 2-амино-б-метилбензтиазола
см. Натрия тиоцианат.
а-Хлорирование карбонильных соединений. При хлорировании
кетонов С. х. атакует наиболее замещенное положение, соседнее
с карбонильной группой [2]; реакционная способность убывает
в ряду: метин >метилен>метил 13]. В качестве примера можно
привести получение 2-хлор-2-метилциклогексанона Ц]. Реагенты
растворяют при охлаждении в четыреххлористом углероде н смею-
выдерживают в течение 3 час при комнатной температуре.
О
’L /снз
О
/СН;,
| |ХС1 +SO2-pH(l
Cl/Z Чэ 83—85%
2 моля 2,2 моля
Аналогичным способом превращают пировиноградную кислоту
в хлорпировиноградную кислоту (выход 98%) 15] и ацетоуксус-
ный эфир в а-хлорацетоуксусный эфир 16].
А1-3-Кетостероиды реагируют с большим избытком С. х. (2 эке)
в пиридине, образуя 4-хлор-ДЧЗ-кстостеропды с выходом 70—90%
17]. Согласно датскому патенту [8], при использовании бензола
в качестве растворителя получают 2-хлор-АЧЗ-кетостероиды. Ясуда
[9] хлорировал ацетат тестостерона в бензоле и показал, что при этом
образуется смесь ацетата 2а-хлортестостерона и ацетата 2а,4-ди-
х л ортестостер он а.
Уайман и Кауфман 131 сообщили, что хлорирование ацетона
и мети л эти л кетона приводит главным образом к а,а-дпхлорпроиз-
292
водным, даже если кетон и С. х. взяты в эквивалентных количест-
вах. Имеются данные н об а-хлорировании альдегидов [10], одиако
при этом были получены главным образом циклические тримеры
эфиров и линейные полиэфиры fill.
С. х. имеет преимущества перед хлором при хлорировании
N-бензоил-е-каиролактама [12]. Раскрытие кольца, аммонолиз
и гидролиз приводят к о>ь-лизину с общим выходом 7326, считая
на е-капролактам.
о
^NCOC6H5
) C12SO2
___/ 89%
Хлорирование ароматических соединений. Бензол довольно
инертен по отношению С. х. и используется как растворитель (см.
выше). С. х. в мягких условиях хлорирует этнл-4-окспбензоат 1131.
Эта реакция является первой стадиен синтеза 2,6-дпхлорфенола.
ОН
А
I || 2C12SOo
) 3,9 моля
Г
СО,С2Н5
1,5 моля
сдцкцснщ,
130—15Ю
8 0-91%
ОН
Тиофен н алкилтиофены легко хлорируются С. х. в а-положе-
нпе [14]. Так, тиофен дает 2-хлортиофен (43%) и 2,5-дихлортио-
фен; 3-метилтиофен образует 2-хлор-З-метилтиофен с выходом 79%.
С. х.— прекрасный реагент для избирательного хлорирования
2-тениламинов в положение 5 115]
R
ch2nz
CbSO,
—-—
50-80%
Алифатические углеводороды. Карат и Браун [16] разработали
метод низкотемпературного хлорирования алканов С. х. с перекисью
293
бензоила в качестве инициатора. Распределение изомеров при хло-
(ЩЩСОЩ
RH+Ci2SO2 --------> RCI-;-HCl -|-SO.,
40—80% '
рировании «-гептана и 1-хлорбутана показано на следующей
схеме:
—15%-
СНзСН2СН2СН2СНкСН2СНз
85%
24%. 22%
1 г I
F t
СН3СН2СН2СН2С1
4 4
। 1
47% 7%
Хлорирование по Карашу
Расщепление S-бензильной связи. Такое расщепление исполь-
зовано Карашем и Лэнгфордом [17] при получении 2,4-динитро-
бензолсульфенхлорида.
а
I моль в 400 мА
СН3ОН
Кипячение 16 час,
1 моль IISCH.CjHs
1,1 моля Ру
Реакция с бензоином. С. х. реагирует с бензоином, образуя
циклический эфир диенола, который самопроизвольно распадается
на бензил и двуокись серы [18].
ci о
Xs/?
ci о
СвН6СОСНОНС6Н5----------
ГСеЫаС —О 01
I:
[С6Н-С — СИ %oj
с6н5с=о
I
СЧ1-15С = О
+ SO3
95% 94%
Ди-«-бутилсульфат. Этот эфир получается с выходом 74—83%
при нагревании 1,6 моля С. х. с 3,2 моля ци-«-бутилсульфпта и уда-
лении образующегося «-бутилхлорида перегонкой 119]. Реакция,
по-впдимому, протекает но следующей схеме:
н-ВиО СЦ /О //-ВиОч /О
4-H.BHC1-1-SO.2
h-BuO-7 СР X) СИ X) '
н-ВиОч «ВиОч /О w-BuCl zO
)S = O+ )S< — w-BuCl-j-SO3
«-BuO/ Ciz 4) w-BuCK ZO
294
Катализатор этерификации. С. х.— эффективный катализатор
этерификации, особенно удобный в химии пептидов, поскольку он
не вызывает рацемизации [20]. N-Ацилпрованную аминокислоту
пли пептид растворяют в метаноле, содержащем следы катализа-
тора, и смесь выдерживают 48 час при 20 . Выход, как правило,
составляет 85—95%. Аналогичным образом получают бензиловые
эфиры, только в качестве растворителя используют сши.н-тетра-
хлорэтан.
Хлорирование метилсульфидов и метиловых эфиров. Метпл-
сульфиды [21] и метиловые эфиры [22] под действием С. х. превра-
щаются в хлорметплпроизводные.
RSCH3 + SO2C12 -> RSCHXl -I-so., 4- HC1
Ц.11-OCH.J SO.,CL, --------СД1-ОС1 ECI SO., Щ HCI
93-95%
Сульфанилиды [23]. Наиболее удобный путь получения суль-
фанилилов— реакция анилина с С. х. в пиридине при 0? (в от-
сутствие пиридина образуются продукты окисления и хлориро-
вания).
II Н
Ру 0° |
2//-С1СД1Л1 -I..•; C|SO,C1 —> n-CICfiH4NSO2NCGH4CFn
' ' " GM,)
Реагент ВМС. Этот мощный перхлориругощий агент назван по
инициалам открывших его испанских исследователей 1241. Для
пер хлорирования органических соединений реагент получают из
монохлор истой серы (S.,C12, 5 а), С. х. (850 мл) и хлористого алю-
миния (2,5 а) в 750 мл С. х. Баллестер [251 описал получение этим
методом некоторых высоконапряженных ароматических хлоругле-
водородов, например перхлорфенантрена и перхлортолуола.
I. «Неорганические синтезы», т. I, ИЛ, ДУ, 1949.
2, Т с h о u h а г В., S а с 1< н г О., Compt. rend., 208, 1020 (1939); D е 1 b а е-
г с Р., Bull. soc. chim. Belg., 51, I (1942).
3. \V у m a n D. P., К a u I m я n P, R-, J. Org. Chem., 29, 1956 (1964); W y-
ni а л D. P., Kaulma n P. R., F г e e m a n W. R., J. Ore. Chem,, 29,
2706 (1964).
4. Уорнх о ф Э., Мартин Д., Джоцсо и У., «Синтезы органических
препаратов», ИЛ, М., 1959, сб. 9, стр. 77.
5. К Р э г о Э. Дж., мл., Робб Ч. М., «Синтезы органических препаратов»,
изд-во «Мнр», 1964, сб. 12, стр. 123.
6. IS ё м е В., «Синтезы органических препаратов», ИЛ, М., 1954, сб. 5, стр. 35,
37.
7. М о г I IL, Chem. Pharm. Bull., 10, 429 (1962).
8. Датск. пат. 8363] (1957) [С. Л., 53, 11452 (1959)].
9. Yasu da К., Chem. Pharm. Bull., 12, 1217 (1964).
10. Stevens C. L., Farkas E., Gillis B,, J. Am. Chem. Soc., 76, 2695
(1954).
11. W v ni a n D, P., Kaufman P. R., Freeman W. R., J. Org. Chem.,
29, 2706 (1964).
12. T u 1 I R., O'X e i 1 1 R. С, O., McCarthy E. P., Pappas J. J.,
C h e m er (1 a J. M., J. Org. Chem., 29, 2425 (1961).
295
13. Та рбе л л Д., В пл ьсо и Дж., Ф а п г а П., «Синтезы органических
препаратов», ИЛ, М., 1953, сб. 4, стр. 236.
14. Campaifine Е.. L е S u е г W. М., J. Am, Chem. Soc., 70, 415 (1948).
15. Ci о d t И. C., Jr., Wann R. E., J. Org. Chem., 27, 1459 (1962).
16. Karasch M.S., Brown H. C., J. Am. Chem. Soc., 61, 2142 (1939);
Brow n II. C., A s h A. B., ibid., 77, 4019 (1955); Russell G. A.,
В г о w n II. C., ibid., 77, 4031 (1955); Ford M. C., W at ers \V. A., J .
Chem. Soc., 1951, 1851; 1952, 2240; M oo r a d i a n A., С 1 о k c J . B., .J. Am,
Chem. Soc., 68, 785 (1946).
17. Kharasch N., Langford R. B., Org. Syn., 44, 47 (1964).
18. F i es e г L. F., Okumura Y., J. Org. Chem., 27, 2247 (1962).
19, Сутер К., Герхарт К., «Синтезы органических препаратов», ИЛ, М.,
1949, сб. 2, стр. 135.
20. Т a s с Ii n е г Е., Wasie I ew s k i C., Ann., 640, 136, 139 (1961).
21. Truce \V. E., В i г n in G. IL, McBee E. T., J. Am. Chem. Soc., 74,
3594 (1952); BordwellF. G., Pitt В. M., J. Am. Chem. Soc., 77, 572
(1955).
22. Davis C. S., Long heed G. S,, procedure submitted to Org. Syn.
23. P а г n e I t E. W., .1. Chem. Soc., 1960, 4367.
24. В a 1 I es t er M о I i n el C., Casta n er J.,J. Am. Chem. Soc., 82,
4254 (I960).
25. В a 1 1 e s t e r M., Bull. soc. chim. France, 1966, 7.
СУРЬМА ПЯТИХЛОРИСТАЯ, SbCI5. Мол. вес 299,04, т. пл.
2,3°, т. кип. 92730 льи; уд. вес 2,346.
При комнатной температуре в растворе (CS2) С. п. взаимодейст-
вует с некоторыми углеводородами, например с трифен ил мет а ном,
образуя гексахлорантимонаты карбон иевых ионов II]. Эта соль
(СеН5)3СН2SbCl5 (CeH5)3CrSbCI6---|-SbCl3+HCl
кристаллизуется из нитрометана в виде желтых призм с т. пл. 218°.
Реакция С. п. с циклогептатрнеком в сероуглероде аналогичным
образом приводит к гексахлорантимонату тропилня (т. пл. 190е).
[.Holmes J., Pettit R., J. Org. Chem., 28, 1695 (1963).
ТАЛЛИЯ ТРИАЦЕТАТ, Т1(ОСОСН3)3. А1ол. вес 381,53, т * п л. 1боL *
Получение по Уинстейну. и Андерсону [1]. Смесь окиси таллия
и уксусной кислоты перемешивали при 65° до тех пор, пока вся
Т1,О3-‘-СН3СООН —>2Т1 (ОСОСН3)3 + зн2о
50 г ’ 300 мл 99,5%
черно-коричневая масса не перешла в раствор (приблизительно
1 день). Раствор фильтровали и при охлаждении выделяли 73 г
(87,5%) белых кристаллов. Концентрирование маточного раствора
дает еще Юг (12%) препарата. Твердое вещество собирают деканта-
цией и сушат в вакууме, так как прн стоянии па воздухе оно ста-
новится коричневым. Высушенный в вакууме над РгО3 и сохраняю-
щийся в закрытой колбе реагент устойчив по крайней мере I год.
Он ядовит.
Окисление олефинов. Кабби 12] кратко описал окисление цикло-
гексена, стирола и о-аллилфенола и пришел к выводу, что Т. т.
по своей активности является промежуточным между тетр а ацета-
том свинца и ацетатом ртути. Андерсон и Уинстейн [31 изучили
ТЦОАс)
АсОН
(5)
2-3%
окисление циклогексена в уксусной кислоте при комнатной тем-
пературе (несколько дней) и с помощью ГЖХ количественно
определили образование всех 5 продуктов: цис- и транс-диацетатов
(I) и (2), диацетата (3) и альдегида (4), получившихся в результате
сужения цикла, и продукта аллильного окисления (5). В сухом
растворителе образуется преимущественно /npawc-дпаиетат (2),
а во влажном растворителе преобладает цне-диацетат (I). Те же
пять продуктов образуются при окислении тетраацетатом свинца,
по количество аллильного ацетата (5) возрастает до 37%. Окисле-
ние ацетатом ртути дает исключительно аллильный ацетат (5).
Расщепление циклопропанов. Оуеллетте ,4] установил, что Т. т.
образуя фактически единственный
фенилциклопропан,
расщепляет
297
продукт — I-фенил-1,3-дпацетоксипропап (6). Анализ методом ГЖХ
показал присутствие только следов транс-имт[амилацетата (7). Ис-
пользования чувствительного к влаге 'Г. т. можно избежать, вместо
пего можно взять окись таллия, так как в условиях реакции она
превращается в Т. т.
С611-СН ^НОЛсЦ-ЛсОН
1 \ с| ] 1 - О 'tac 11 гн ~ж
: СН2ОЛс
С(;ЫГ)СН I
си
/II
у-_.с
Н 'СИ,ОАс
Л)
В параллельных опытах по окислению фенплниклоироиана
тетраацетатом свинца [51 в уксусной кислоте при 75 реагент посте-
пенно растворялся и реакция заканчивалась за 10 час (ср. 120 час).
Продукты (6) и (7) получались в результате разрыва связи Q- Са,
как и при окислении Т. т., но реакционная смесь на одну треть
состояла из диацетата (8), образовавшегося в результате разрыва
С->—Сз-связп.
с6н5сн
^СНгОАс
(8) 32% (разрыв СЖСз)
I. \V i n s t е i п S., private communication.
2. Kabbe II. J., Ann., 656, 204 (1962).
3. Andcrso n С. B., Winstcin S., J. 0гц. Client., 28, 605 (1963).
4. Ouel 1 e 11 eR. J., Sh awD. L.,Sou th A., Jr., J. Am. Chem. Soc., 86,
_ 2744 (1964).
5. О u c e 1 1 c t t e R. J., Shaw D. L., J. Am. Clitmi. Soc., 86, 1651 (1964).
TETPA-w- БУТИ ЛАММОНИ Й ИОДТЕТРАХЛ ОРИД,
(h-CiHJjNICI.j. Мол. вес 511,20, т. ил. 137—139 ' (с разл.). Получе-
ние [II.
При взаимодействии Т. с алкенами и алкинами происходит
стереоспецифическое транс-и рисоед и пение хлора. Так, ipic- и транс-
стильбены дают соответственно мезо- и d, /-дихлориды 12[. Раствор
0,005—0,06 моля олефина в 25 мл дихлорэтана обрабатывают 1 же
реагента и оставляют при комнатной температуре на 3 дня; выход
298
каждого дихлорпда стильбена составляет 50%. Реакция с ацетофено-
ном приводит к хлорацетофенону с выходом 54%.
I. Р о р о v Л. J., Buckles R. Е., ]пощ. Syn., 5, 176 (1957).
2. Buckles R. E., I< n a a c k D. Г., .1. Or”. Chem., 25, 20 (1960).
ТЕТРАГИДРОФУРАН (ТГФ). Мол. вес 72,10, т. кпп. 65,4°,
уд. вес 0,88; смешивается с водой.
Очистка [I]. I л Т. помещают в колбу, снабженную обратным
холодильником с хлоркальциевой трубкой, п при перемешивании
магнитной мешалкой добавляют небольшими порциями 2—4 г
алтомогпдрида лития. Пробу отстоявшейся жидкости добавляют
к нескольким миллилитрам воды; если не происходит интенсивного
выделения газообразного водорода, то прибавляют еще 2—3 г
LiAlH1. Затем Т. перегоняют при атмосферном давлении. Так как
LiAlHi разлагается при температуре выше 150д перегонку не сле-
дует проводить досуха. Очищенный Т. следует хранить под азотом.
Гидрид металла рекомендуется прибавлять из колбы Эрлеп-
мейера, присоединенной к колбе при помощи трубки Гуча (рис.
А-I). Эфир и дпоксан также можно эффективно высушить алюмо-
гпдрпдом лития; во избежание взрыва растворитель не следует
отгонять досуха.
Эффект растворителя, см. также Гриньяра реактивы (виннл-
галогениды, сорастворители).
Т. используют в синтезах с малоновым эфиром благодаря его спо-
собности растворять различные натриевые производные 12|. При-
менение водного Т. облегчает диазотирование солей аминогюлифс-
нпльпых соединений, лишь незначительно растворимых в воде 131;
кроме того, он рекомендуется также для реакции Шимана 141.
Т. используют вместо эфира как растворитель при реакции ацети-
ленового реактива Гриньяра с пропаргилгалогенпдами [5Г Кремлпн
и Чпзхолм |6| установили, что Т. — более подходящий раство-
ритель по сравнению с диоксаном пли целлозольвом для гидриро-
вания холестерина при обычных температуре и давлении в присут-
ствии хлорной кислоты в качестве катализатора.
Реактивы Гриньяра можно легко приготовить в углеводородной
среде (толуол, ксилол), если на каждый моль галогенида взять
I моль Т. [7]. Процесс экономичен и не представляет опасности, так
как 3'. связан с реактивом Гриньяра.
Реагент оказывает значительное стабилизирующее действие па
cz-хлорал кил литиевые соединения, что позволяет получать и хра-
нить их без разложения при температуре —100J 181. Например,
/С1Г,
' ' с—с
едцы сн* сн;[ /• / '
С6Н5СС13— -->ceH-ca2Li———C|V /С ч <н;1
’ “ Cl
И) (2) (3)
299
а,а-дихлорбеизиллптпп (2) получают взаимодействием бензотрихло-
рнда с фсниллптием; при разложении при —65° в присутствии оле-
фина соединение (2) превращается в производное циклопропа-
на (3).
Реакционная способность мстпллитня в Т. выше, чем в эфире.
Так, мстпллптнй и тетрафепилфосфонийбромпд в эфире реагируют
очень медленно (4 месяца, N..), давая смесь различных фосфорсо-
держащих соединений [9), но если эту реакцию проводить в смеси
Т.— эфир (4 : 1), то легко осуществляется следующий процесс [10]:
Ч- т. Ч- —
(CGH5)4PBr--| -CII3Li — (CGIi5)3P - Cl Г QIV-LiBr
Бензол образуется почти с количественным выходом, а ил ид — с вы-
ходом не меньше 70%, как это следует из реакции с циклогексаноном.
6-Метнл-2-бромнафтал1н1 ие дает реактива Гриньяра в эфире
или смесн эфир — бензол, но при использовании Т. реактив обра-
зуется легко и с высоким выходом [11].
Реакции. Т.—исходный материал для получения следующих
соединений:
Тетраметилен хлор гидр ин [12]
Кипяче-
U + НС1 ниеД05 > С1СН2СН2СН2СН2ОН
54-57% ‘
- (Газообразный)
1, 58 (ПОЛЯ
4-Хлорбутилбепзоат [13]
__________________________________ о
С6Н5СОС1 + ZnCIj. + I I —87° > С6Н5СО(СН2)4С1
78-83%
О, 27 МОЛЯ 5 2
1,4-Днподбутан 1141
Кипячение
Г Л + 2 KI + 85%-ная Н3Р04+Р205 --------—> 1СН2СН2СН2СН21
С» 2 моля 135 Л7Л 65 г
О, 5 МОЛЯ
4,4'-Дихлордибутиловый эфир [15]
U 4- POCI3 + H2SO4 °'1QQP > С1(СН2)4О(СН2)4С1
52-54%
1, 67 моля 50 мл
5 молей
300
1. Submitted by Zweifcl G., University of California, Davis, California,
2. Law ess о ii S. -(J., Busc h T., Acta Chem. Scand., 13, 1717 (1959).
3. C a d e J. NP i 1 b e a m A,, Chem. {nd., 1959, 1578.
4. Fletcher T, L., Namkung AL J., Chem. fnd., 1961, 179.
5. E у о S. N., W о 1 о v s к v R., G c n s 1 e r \V. .1., J. Am. Chem- Soc., 83,
3080 (1961).
G. С г c m 1 у n R. J. W., Chisholm AL, .1. Chem. Soc., 1965, 5117.
7. Leigh T., Chem. Jnd., 1965, 426.
8. 11 о e g D.F., Lusk D. L, С r n m b I i s s A. L.,J. Ain. Chem. Soc., 87,
4147 (1965).
9. \V i t L i g G., G e i s s I e r G., Ann., 580, 44 (1953).
10. S e у f e r t h D., Hughes W. B., Hecren J. K., J- Am. Chem. Soc.,
87. 3467 (1965).
11. Lapin 11., ( Inmia, 18, J 41 (1964).
12. Старр Д., X и i; <, о н P., «Синтезы органических препаратов», ИЛ,
AL, 1949, сб. 2, стр. '150.
13. Сайнер хол м AL, «Синтезы органических препаратов», ИЛ, AL, 1953,
сб. 4, стр. 528.
14. С т о у н Г., Шехтер Г., «Синтезы органических препаратов», ИЛ,
М.,1953, сб. 4, стр. 164.
15. Александер К., Тоулз X., «Синтезы органических препаратов»,
ИЛ, М., 1953, сб. 4, стр. 232.
ТЕТРАЗОЛИ ЕВ ЫЙ СИНИЙ. Мол. вес 658,74, т. пл. 244°,
получение III, очищают кристаллизацией из пиридина [2].
Тетрдзочиг;:г,гй синий
Диформазанозый краситель
Т. с. представляет собой светло-желтое растворимое в воде
соединение, которое в присутствии следов щелочи окисляет аль-
дозы, к егозы и другие а-кетолы, восстанавливаясь до интенсивно
синего диформазана, нерастворимого в воде. Это чувствительная
проба на восстанавливающие сахара, позволяющая отличить а-
кетолы от простых альдегидов. Порядок реакционной способности
следующий: фруктоза>-глюкоза>лактозад>мальтозад>«-маслянын
альдегид.
Реагент используется для определения дегидрогеназ в тканях,
клетках н бактериях II], а также определения всхожести семян
и обнаружения кортикостероидов [3].
301
1. R n t e n b u r g Л. M., G о f s t e i n R., S e 1 i g in a n A. iM., Cancer Res., 10,
113 (1950).
2. Ried W., G i с к H.. Ann., 581, 16 (1953).
3. M a d e г W. J.. В u <' к R. R., Anal. Chem., 24, 666 (1952).
ТЕТРАЛИН, Мол. вес 132,20, т. кип. 207—208'', уд. вес
0,97.3, 1,5461.
Продажный Т. содержи!’ в качестве примеси нафталин, что ме-
шает получению 1-гидроперекиси Т. |1] или использованию угле-
водорода как донора водорода в реакциях с гидрпдпым переносом [2].
Первоначально процесс очистки Т. [II включал следующие стадии:
фракционирование, обработку ртутью (для удаления сер у содержа-
щих примесей), раствором ацетата ртути (для удаления олефинов),
серной кислотой и повторное фракционирование. Для очистки по
Бассу [2] сырой углеводород сульфируют конц. серной кислотой
и добавляют хлористый аммоний для осаждения тетр ал ин-6-е ул ьфо-
ната ам.мония. Соль очищают кристаллизацией и гидролизуют пере-
гонкой с паром из сернокислого раствора. Перегонка над натрием
дает продукт, который в ультрафиолетовой области не имеет пог-
лощения, характерного для нафталина.
1. 11 я ii т X., С в е р и Д., «Синтезы органических препаратов», ИЛ, .\1., 1956
<6. 6, стр. 19.
2. В a s s К. С., J. Chem. Soc., 1964, 3498.
ТЕТРАМЕТИЛАММОНИЯ АЦЕТАТ, (СН,)Лю..СС1Д. Мол. вес
133,19.
Получение Ill. Водный раствор гидроокиси тетраметпламмоппя
нейтрализуют уксусной кислотой и выпаривают досуха при по-
ниженном давлении. Твердый остаток кристаллизуют из ацетона
и сушат в вакуум-экс и каторс.
Замещение галогена. Штейгман п Гаммет [2] обнаружили, что
реакция а-фен ил этил хлорида с реагентом имеет второй порядок и
осуществляется с преобладающим обращением конфигурации. На-
против, сольволиз уксусной кислотой является реакцией первого
порядка, причем происходит значительная рацемизация. Аллплхло-
риды взаимодействуют с реагентом в сухом ацетоне, давая соответ-
ствующие ацетаты без перегруппировок и с преимущественным
обращением конфигурации 11, 3].
Физер и Ромеро [41 показали, что при реакции 2а-бромхолестан-
опа-3 с ацетатом калия в кипящей уксусной кислоте образуется
комплекс 2а- и 4а-апетокспхолестанона-3 1(2) и (3)1, неразделимый
хроматографпчески. Уильямсон и Джонсон 15] подтвердили это
и, кроме того, приготовили идентичный комплекс из эквимолярных
количеств компонентов. Затем они обрабатывали а-бром-3-кетой (1)
Т. а. в кипящем ацетоне и получили смесь ацетоксикетонов, из
которой в чистом виде удалось выделить только За-ацетоксихолестан-
он-2 (4). 2Р-Ацетоксихолестанон-3 (5), ожидаемый продукт реакции,
при обработке Т. а. изомеризуется в (4). Реакция 4а-бромхолестано-
302
на-3 с Т. а. в ацетоне при 25" дает Д’-холестенон-З (20%) и 4% комп-
лекса (3) и (4),
1. Gi>er ing 11. L., N с v i t t T. D., S i I v ersm i 1 h E. F., ,1. Am-
Chem. Soc., 77, 4042 (1955),
2. S 1 e i ц in a n J., 11 a m m e t t L. P., J. Am. Chem. Soc., 59, 2530 (1937): see also
S t г c i t w i c s о г A., Jr., \V о 1 f e J. R., Jr.. J. Ain. Chem. See., 79, 903 (1937).
3. R о b e г t s J. D., 5 о u n ц W. G., \V i n s t c i п S., J. Ain. Chem. Soc.,
64, 2157 (1942).
4. Fi esc r L. F., R о m с г о M. A., J. Am. Chem. See.. 75, 4710 (1953).
5. Williamson 1\. L., Johnson W. S., .1, 0гц. Chem., 26, 4503 (1901).
ТЕТРАМЕТИЛАММОНИЯ БОРГИДРИД, (СНДЛ’ВЩ. Мол. вес
89,00.
T. б.— стабильное белое вещество, обладающее такими же вос-
становительными свойствами, как п боргидрид натрия, ио более
удобное, поскольку лучше растворимо в неполярных раствори-
телях и даже в бензоле UJ. По сравнению с боргидридом натрия
Т. б. менее чувствителен к гидроксилсодержащим растворителям;
например, реагент устойчив в этаноле и этиленгликоле, с которыми
боргидрид натрия реагирует. Аналогично ведут себя боргидрпды
тетраэтил аммония (C2H5)4NBH4, мол. вес 145,10, цетилтриметилам-
мония мол. вес 299,39 и трикапрнлметнлам-
мония (w-CKH]7);iCll:ДВН4, мол. вес 383,54.
1. S u 1 I i v а п Е. А., 11 i n с k 1 с у A. A., J. Огц. Chem., 27, 3731 (1952).
ТЕТРАМЕТИЛАММОНИЯ БРОМИД, (CHJ.NBr. Мол. вес 153,67.
Т. б. активнее, чем ацетат натрия, в качестве катализатора
восстановительного ацетилирования хинонов под действием цинко-
вой пыли и уксусного ангидрида |1[.
1. F i е s е г L. In, «Experiments in Organic Chemistry», 2nd ed., p. 399.
303
ТЕТРАМЕТИЛАММОНИЯ ТРИБРОМИД, (CH4)4NBr3. Мол. вес
313,90, т. пл. 118,5°.
Т. т. получают из бромистого тетраметиламмония и брома [1].
Т. т.— слабый бромирующий агент [21. В бензольном растворе
в присутствии перекиси бензоила углеводороды, такие, как толуол,
подвергаются бензильному бромированию. В уксусной кислоте
осуществляется замещение в ароматических соединениях. При реак-
ции с циклогексеном получается только 1,2-дибромциклогексан
(80%) независимо от природы растворителя и наличия или отсут-
ствия перекиси бензоила.
l.Chattaway F. О., 11 о у 1 е G., J, Chem. Soc., 123, 654 (1923).
2. A v г a in о П М., Weiss J., S с li а с h t е г О., J. Org. Chem., 28, 3256
(1963).
ТЕТРАМЕТИЛГУАНИДИН, (CH3)2NC( NH)N(CH3)2. Мол. все
115,18, т. кип. 165°.
Реагент используют в качестве катализатора конденсации по
Михаэлю Зр-оксл!47|3-иитро-5р-андростана с метилакрилатом [I].
При использовании тритона Б или этилата натрия выход продукта
только 10%.
1. N у s t с d L. N., Burtner R. R., J. Org. Chem., 27, 3173 (1962).
ТЕТРАМЕТИЛЕНСУЛЬФОКСИД. Мол. вес
104,17, т- кип. 45°/3 мм. Реагент очищают пере- сн2—сн2
гонкой при пониженном давлении иад молекул я- снг сн2
рными ситами (Linde 13 X). Продукт хранят на
холоду, так как при комнатной температуре его
можно хранить примерно 3 месяца [1].
Подобно ди мсти л сульфо кс ид у, этот циклический сульфоксид —
прекрасный реагент для окисления тиолов в дисульфиды [2].
СП,— СН, СН,— СН,
2RSH + | )>S-0 — > RSSR -| • | /S -L Н,0
СП,—СН, СН2— СН,
1. Private communication from Wallace T. J., Esso Research and Enginee-
ring Co.
2. Wallace T.J., J. Am. Chem. Soc., 86, 2018 (1964); Wallace T, J.,
Mahon J . J., J. Am. Chem. Soc., 86, 4099 (1964); J. Org. Chem., 30, 1502 (1965).
304
ТЕТРАМЕТИЛЕНСУЛЬФОН (сульфолан). Мол.
вес 120,17, т. пл. 28°, т. кип. 130/6,5 мм, диполь-
ный момент 4,69, диэлектрическая проницаемость 42,
смешивается с водой.
Очистка. Продажный растворитель, полученный присоедине-
нием SO-j к бутадиену и гидрированием, содержит различные при-
меси. Один из методов очистки [1] состоит в пропускании через ко-
лонку с молекулярными ситами Линде и фракционировании. Сог-
ласно другому методу [2], проводят многократную вакуумную
перегонку над гранулированным едким натром до тех пор, пока
при смешении 1 мл растворителя с 1 мл 100%-ной серной кислоты
не будет появляться окраска в течение 5 мин, а затем перегоняют
еще раз над гидридом кальция. Остальные методы [3] заключаются
в обработке Т. перманганатом и серной кислотой для удаления
окисляющихся примесей; затем добавляют достаточное количество
пиросульфита натрия для осветления раствора, экстрагируют
хлористым метиленом, обрабатывают Р2О5, декантируют и пере-
гоняют.
Применение. Т.— хороший растворитель для ароматических
соединений; его с успехом применял Ола с сотрудниками для нит-
рования [4], проводимого по реакции типа Фриделя — Крафтса,
и для изучения механизма нитрования ал к ил бензолов [5] и галоген-
бензолов [6] тетрафторборатом нитрония в гомогенных растворах.
Он является наилучшим растворителем для четвертизации третич-
ных аминов алкилгалогепидамп, так как имеет высокую диэлект-
С6Н5СН2Вг
(CH2)4SO
9 3%
ричсскую проницаемость и не вступает в побочные реакции, наблю-
даемые в случае нитробензола и ДМФА 17]. Например, в синтезе
акридизиниевой соли (3) Брэдшер и Пархам [81 проводили с хо-
рошим выходом четвертизацию (1) бензилбромидом в Т. при ком-
натной температуре. Аналогично различные соли, подобные (2),
получены с хорошим выходом в кристаллическом виде, в то время
как при использовании других растворителей получаются окрашен-
ные масла. Арнетт и Доути [2] характеризуют растворитель как очень
слабое основание со слабо выраженной тенденцией сольватировать
катионы; в этом отношении он подобен нитрометану, но более удобен.
Фуллер [9] обнаружил, что Т. — особенно эффективная реак-
ционная среда для галогенного обмена в реакциях перхлор- и пер-
бромароматических соединений с фтористым калием. Его преиму-
305
щества перед другими некислотными растворителями заключаются
в высокой температуре кипения (230—240 ), термической и хими-
ческой стабильности и низкой сольватации анионов. Т. также ис-
пользуют при кинетическом исследовании чегвертизации произ-
водных пептидов и иоли-З-впнилппрндпна |7, 10|.
1. С г a m I). J., R i с k Ь о г п В.. Kingsbury С. A., IJ а Ь с г f i е J cl
Р., J. Am. Clicrn. Зое.. 83, 3678 (1961).
2. А г п е t 1 Е. AL, D о u t v С. I7., ,1. Am. Chem. Soc., 86, 409 (1964).
3. A Ider R. W., W h i t i*n £ M. C., J. Chem. Soc., 1964, 4707.
4. К и h n S. .1., 0 1 a h G. A., J. Am. Chem. Soc., 83, 4564 (1961).
3. 01a li G. A., К li h и S. J., F 1 о о d S. H., J. Am. Chem. Soc., 83, 4571
(1961).
6. Olah G. А., К и h н S. J., F I о о d S. FI., J. Am. Chem. Sue., 83, 4o8I
(1961).
7. Co loin а и B. IF, Fuoss R. AL, J. Am. Chem. Soc., 77, 5472 (1955).
8. В r a dsh er С. К.., P a rh a in J. C., J. 0гц Chem., 28, 83 (1963).
9. F и 1 1 e г G., J. Chem. Soc., 1965. 6264.
10. Arcus C. L., 11 a 1 1 \V. A., ,1. Chem. Soc., 1964, 5995.
О
II
ТЕТРАМЕТИЛМОЧЕВИНА, (СН:,)ЛХЛЧСНЯ)2. Мол. вес 116,17,
т. кип. 176,5 , смешивается с водой.
Получение. Из диметил амин а и фосгена 111
о
2 (СН3).А;Н— С1СОС1-—---- (CHU)2XCOC1 ——1 (СН3)АС.\ (СН3)2
-~(С нд,х н д:1
Из триметиламина и фосгена [2.1
О
(СН:?)зX -у C1COCI [(СНд, \COC1 J С1 “ _т СН3С1 :-(СН;;)АС\ (С113)2
Растворитель. Т. как растворитель подобна пиридину
(т. кип. 115), но имеет более высокую температуру кипения.
Т. смешивается во всех отношениях не только с водой, но п со всеми
обычными органическими растворителями и даже с пстролейпым
эфиром. Она является прекрасным растворителем для аромати-
ческих и гетероциклических соединений.
Дезаминирование, Новый метод дезаминирования состоит в
осаждении гексафтор бората арилдназония, например, как описано
Резерфордом и Редмондом 131, и самопроизвольном экзотерми-
ческом разложении этой соли в Т., которая действует удовлетво-
рительно в тех условиях, когда другие неводные растворители
разрушаются |4|. Выход паплучшпй (75—85%), когда в аромати-
ческом кольце имеются электроотрицательные заместители (NO2,
CO.JI, Вг). В случае анилина, о- и п-толупдинов и о-анпзидииа
выходы порядка 25 -30%. Сухую соль прн комнатной температуре
306
порциями добавляют к 'Г., температуру поддерживают при 65"
и, когда выделение азота прекращается, раствор выливают в воде.
Реакция Ульмана. Пели для проведения реакции Ульмана тре-
буется высокая температуря, то вместо ДМФА (т. кип. 153 ) ре-
комендуется использовать Т. 121.
Алкилирование енолятов. Определение констант скорости ал-
килирования, например, натрий-н-бутилмалонового эфира н-бутил-
бромидом в бензоле в присутствии различных добавок, показало,
что при менее чем 5%-ной концентрации Т. скорость реакции уве-
личивается во много раз. ДМФА немного эффективнее, но при ис-
пользовании 'Г. выход выше 12|.
1. М i с li I е г W., Е s с 11 е г i с h С,, Вег., 12, 1162 (1879).
2. I. ii t t г i и н h a u s A., D i rksen В, \V., Annexe. Chern., Intermit. Ed., 3,
2G0 (1964).
3. Rutherford K, G., Redmond W., Ощ. Syn., 43, 12 (1963).
4. Rutherford K. G., Redmon d .1. 0гц. Chem., 28, 368 (1963).
ТЕТРАМЕТИЛТИОМОЧЕВИНА, (CH:i)2NC(^S)N(CH3)... Мол.
вес 132,24, т. пл. 78°.
При исследовании различных сер у со держащих соединении для
отравления катализатора в реакции восстановления бензоил хло-
рида по Розенмупду Т. оказалась наиболее эффективной, предотвра-
щая гидрирование па стадии бензальдегида 111.
1. AHrossman S., Thomson S. J., J. Chem. Sue., 1962, 2024.
ТЕТРАНИТРОМЕТАН, C(NO2)4. Мол. вес 196,04, т. пл. 13,8°,
т. кип. 126", уд. вес 1,639.
Получение. Метод, описанный в «Синтезах органических пре-
паратов» [1], заключается в реакции уксусного ангидриде! с безвод-
ной азотной кислотой
И if же ]()°, тттем
4 (CH;!CO)aO-'-HNO;i—-->6 (ХОД,-К 7СН:;СООН — СО2
О,:, моля 0,5 моля (1Г'
(безн.)
В настоящее время этот способ предпочитают более эффектив-
ному методу Дарзапа и .Певи [2] с использованием кетена.
4С11о-С --- О : 4НХО:(—» С(\О2)>1- СО. : ЗСП:!СООН
о 11 °
Проба на ненасыщенность. Т. образует окрашенные л-комплексы
со многими олефинами и другими ненасыщенными соединениями,
что используется для обнаружения ненасыщенности. Ввиду высо-
кой стоимости реагента определение рекомендуется выполнять
в капилляре для определения температуры плавления 13]; поскольку
Т. очень летуч, но замерзает в холодильнике, реагент смешивают
с эквивалентным объемом хлороформа и хранят на холоду. На-
пример, для пробы на ненасыщенность в капилляр вводят 0,5-мил-
лиметровый слой холестерина. Другой капилляр, конец которого
307
вытянут в форме пипетки, погружают в раствор СНСР -СРЮ,), и
набирают столбик жидкости высотой 5 мм, кончик помещают в
первый капилляр и раствор стряхивают резким движением. Желтая
окраска комплекса легко наблюдается, если поместить капилляр
на фойе белого листа бумаги.
По мере увеличения ненасыщенности окраска изменяется от
желтой до том но-крас пой. Проба положительна для соединений,
содержащих двойную связь, устойчивую к гидрированию. л-Ком-
плекс с бензолом — желтый, с нафталином —оранжевый. Проба по-
ложительна с циклопропаном, но отрицательна с циклобутаном.
а, fJ-Нспредельныс карбонильные соединения дают слабую окраску;
с алкинами и аллиловыми спиртами проба отрицательна.
Хсйлброииер [4] предложил метод определения типа двойной
связи путем измерения длины волны поглощения комплекса
олефина с Т. при стандартном коэффициенте экстинкции. Каждый
алкильный заместитель вызывает высокочастотный сдвиг примерно
на 56 ммк, как видно из приведенных данных для стероидных оле-
финов: дизамсщенная двойная связь, А1 2 3, 478 ммк; тризамещениая,
А4, 530 ммк; А5 (2 примера), 530, 531 ммк; тетразамещенная,
АК(,4) (3 примера), 570, 559, 580ммк.
Нитрование. Совершенствуя ранние методы, Андерсон н сотр.
[5] обрабатывали азулен в пиридине небольшим избытком?, в эта-
ноле. Окраска смеси за 18 мин изменяется до красной, экстракцией
и хроматографией получают 1-нитроазулен с высоким выходом.
Реагент, по-видимому, превращается в триннтрометан.
Риордан с сотр. [6] обнаружили, что Т. пригоден для нитро-
вания тирозина и тирозильных остатков в белках. Прн pH 8 нитро-
вание сопровождается вытеснением двух протонов и образованием
3-нптротирозина. ч
он с —
/к /X /NO,
ji Ч 20° V7
;-C(NO2)4->| +С(.\О.Д3-+21П
Н \ рН8 J I
I I
С11СОД1 снсо.,н
I I
NH3 NH2
1. Л ианг П., «Синтезы органических препаратов», ИЛ, М., 1952, сб. 3, стр. 411.
2. Pdfzens G., Levy G., Compt. rend., 229, 1081 (1949).
3. Org. Expts., 75.
4. Hei Ibronner E., Helv. Chim. Acta, 36, 1121 (1953).
308
5. Anderson A. G., Jr., Scot oni R., Jr., Cowles E. J,, F r i t z C. G.,
J. Org. Chem., 22, 1193 (1957).
r.Riordan J. F., Wacker \V. E, C., Vallee B. L., J. Am. Chem.
° Soc., 88, 4104 (1966).
ТЕТРА-л-НИТРОФЕНИЛПИРОФОСФАТ. Реагент (2) получают
in situ реакцией кислого ди-n-нитрофенилфосфата (1) с ди-п-толил-
карбодиимидом в безводном дпоксане 111. Т. реагирует со спиртом
в отсутствие основных катализаторов с образованием нейтрального
ди-п-нитрофеиилфосфата (3). Одну из п-нитрофенильных групп Т.
удаляют обработкой 1 и. щелочью в течение 30 мин при 25'’, дру-
о
г’ / ———“ х ”1 1 А г N —- С А1*
2 о,,\—Чч-О- — Р—ОН--------—---->
~ О)
О о
/ Ч~О—] -Р—О—Р—[о—NO.,
. „ . (2) “
О о
коп^о,\— —P-OR-I ___/—0—] ~р~он
(3)
гую — 1 и. щелочью в течение нескольких часов при 100°. Эти
группы можно также удалить гидрогенолизом: одну — в нейтраль-
ной среде, а другую — в присутствии кислотного катализатора.
С помощью Т. получен гуанозин-5'-фосфат с выходом 70% [2|.
Хлорокись фосфора в этом случае дает выход не больше 20%.
1. М о f f a t t J. G,, к h о r a n а II. G , J. Arn. Chem. Soc., 79, 3741 (1957).
2. C li a in b c rs R. W., M <j f f a t 1 J Д1., К h о r a n a 11. G., J. Ain, Chem.
Soc., 79, 3747 (1957),
(+)- и (—)-а-(2,4,5,7-ТЕТРАНИТРО-9-ФЛ УОРЕНИЛИДЕН-
АМИНООКСИ)-ПРОПИОНОВАЯ КИСЛОТА. Мол. вес 447,27, т. пл.
200°.
кю2 no2
Ньюмен и сотр. [1] получили эти оптически активные л-кислот-
ные комплексообразующие агенты и использовали их для разде-
ления на антиподы полициклических ароматических углеводородов,
например гексагелицена [2].
1. N с w nr a n М. S,, Lutz W. В., J. Am. Chem. Soc., 78, 2469 (1956); Block
Р., Jr., N c \v m a п M. S., Procedure subbiiied io Org. Syn.
2. Newman M. S.( Led nicer D., J, Am. Clicm. Soc., 78, 4765 (1956).
309
2,4,5,7-ТЕТРАНИТРОФЛУОРЕНОН, Jj>
Мол. вес 360,19, т, пл. 254,59 Полу- Г I]____________|1 \
чают нитрованием флуоренона с выходом
77"/„ (1|. ' NOi
Т. образует л-комплексы с сильными л-основаппямп (нафталин,
антрацен, гексаметилбензол), температура плавления которых на
50° выше температуры плавления соответствующих комплексов
2,4,7-тринитрофлуоре1Юна 111. Установлен следующий ряд изме-
нения силы л-кислот 12]: Т. >2 4,7-тринптрофлуоренои >4,3,5-
тринитробензол >ппкрплхлорид.
1. X с w ni а п А). 8., Lutz \У. В., J. Am. Chem. Sue., 78, 2469 (1956).
2. Colter А. К., С I е rn ens L. ,\L, J. Am. Chem. Sue., 87, 847 (1965).
ТЕТРАФЕНИЛЦИКЛОПЕНТАДИЕНОН. Мол. вес 384,45, т. пл.
220°, темно-красные пластинки.
Получение. Т. легко получают конденсацией бензила с дибен-
знлкетопом в присутствии основного катализатора. Оригинальный
метод Дилтея |1] приведен в «Синтезах органических препаратов»
[21; растворителем служит этанол, основанием — раствор едкого
кали в этаноле. Недостаток .метода — низкая температура кипения
спирта, ограниченная растворимость едкого калл п продукта реак-
ции в этаноле. В усовершенствованном методе 13, 4] в качестве
C6IISC=O
I +
сд-цс-о
с6н5
СН^с=0
сн/
С6н5
носн2сн2оснгдн2оснгснгОн
______СДЦСН/n (СЛЦфСГн
93%
растворителя используют триэтиленгликоль, что позволяет ра-
ботать при более высокой температуре, и легкорастворимую гидро-
окись бснзплтримстиламмоппя в качестве основания. Смесь 0,2 моля
бензила и 0,2 моля дибепзплкетона в 200 мл триэтиленглпколя
нагревают до 100 и обрабатывают 20 мл 40"» -кого раствора гидро-
окиси бенз плтрп метил аммония в метаноле, перемешивают до раст-
ворения и оставляют для кристаллизации, которая начинается
менее чем через минуту. После охлаждения реакционную смесь
разбавляют этанолом, темно-красные пластинки промывают эта-
нолом до тех пор, пока растворитель не станет чисто розовым.
Продукт растворяют в три этилен гл и коле (Юлл/с) при 220 : и раствор
оставляют для кристаллизации (большие пластинки) или переме-
шивают магнитной мешалкой при охлаждении (маленькие плас-
тинки); выход после кристаллизации составляет 92"о.
Реакция Дильса - - Альдера. Благодаря высокой реакционной
способности и окраске, которая позволяет наблюдать за реакцией,
взаимодействие Т. изучено с большим числом диенофилов [5].
310
Т. реагирует с .малоиловым ангидридом в кипящем бензоле (т. кии.
80?) с сохранением карбонильного мостика и образованием (3) 161,
в то время как в кипящем хлорбензоле (т. кии. 132') |61 пли бром-
бсизоле [7] (т. кип. 155,5 ) выделяется окись углерода и получается
тетрафен илдигпдрофталсвый ангидрид (4). Взаимодействие Т. с ди-
метиловым эфиром ацетцлсндпкарбоповой кислоты с целью полу-
чения диметилтстрафенилфталата, о чем впервые сообщил Дплтей
О'рнзоле
Кипяч (1)
3,5час и
СДЦС1 )
-со
161, изучалось одним из авторов при использовании широкого ряда
растворителей; полученные результаты приведены в разделе «Раство-
ри гели для реакции Дильса — Альдера». Примеры взаимодействия
реагента с дифенплацетиленом и дегидробензолом, см. Дифенил-
ацетилен и Дифенилиодоний-о-карбоксилат.
Аллеи и сотр. 171 получили аддукт диснона с этиленом реакцией
при высоком давлении и нагревании до 100 с хлористым алюми-
нием в качестве катализатора. Без катализатора при 100 взаимо-
действия не наблюдается, а при 180 реакция сопровождается
дскарбонилированием с образованием (6) и (7).
С6Н5
CtH5
I. D i 1 t Ii t> у W., Q unit F., J. prakt. Cheni. (2), 128, 146 (1930).
2. Д ж <> и с о и Дж., Г p у .я я итт О., «Синтезы органических препаратов»,
ИЛ, М., 1952, сб. 3, стр. 415.
3. Огц. Expts., 301; 1' iescr L. Р’., Огц. Syn., 40, 44 (1966).
4. F i е s с г L. F., 11 a d <1 a d i п М. J., Can. J. Chem., 43, 1399 (1965).
5. A I 1 с п С, F. 11., Chem. Revs., 37, 209 (1945); 62, 653 (1962).
6. D i I t h с у \V., Thcwal t I., T г 6 s k e n O., Ber., 67, 1959 (1934).
7. Allen C. F. IL, R v an R. W., Jr., V а и Allan J. A., .1. Ory. Chem.,
27,778 (1962).
311
ТЕТРАФТОРГИДРАЗИН, F2NNFa. Мол. вес 104,02, т. кип. —73°.
Реагент получают реакцией трифторида азота с различными
металлами. При этом следует соблюдать предосторожности, так
как при смешении с органическими соединениями возможен взрыв,
если не исключено присутствие кислорода [11- Т.— универсальный
реагент для введения дифтор аминогруппы. Так, фотолиза-дикетона
в присутствии N2F4 дает М,И-дифторамиды
ftv
RCOCOR+ F2NNF2 2RCONF2
которые образуются также при нагревании Т. с альдегидами [2].
150°
RCHOH-H,NNF2 —> RCONF2 + HNF2
Смесь алкана и Т. при облучении 2337 А в результате радикаль-
ной реакции дает алкилдифторамины [31. Например, из я-бутана
получены 1- и 2-дифтор а ми иобута и ы. Алкены образуют как продук-
ты замещения с введением группы NF2, так и присоединения NF3:
г, nn в»
ci 13сн _ снсн3 —* а 13сн=chch.nf2 щ сн3сн~сн сн3
I I
F i\F2
I, Col burn С. В., Kennedy A., J. Am. Chem. Soc., 80, 5004 (1958).
T Petry R. C., Freeman J. P., J. Am. Chem. Soc., 83, 3912 (I960).
3. Burner dner C. L., Tetrahedron Letters, 3683 (1964).
1^,2,4,6-ТЕТРАХЛОРАЦЕТАНИЛИД, Cl3CeHaNCICOCH3. Мол.
вес 272,96, т. пл. 75°.
Получение из 2,4,6-трихлорацетаиилнда и гипохлорита натрия [1].
Хлорирование. В присутствии перекиси бензоила как инициа-
тора реагент хлорирует алканы, циклоалканы и ал кил бензолы
(в боковую цепь) [2].
1. Avad К. N., Beard С., Garwood R. F., Hickinbottom
W. J., J. Chem. Soc., 1957, 2981.
2. В о о с о c k J. R. В., H i ok i nb ot t om W. J., J. Chem. Soc., 1963,
1234.
ТЕТРАХЛОР-1,2-ДИБРОМЭТАН, BrCChCCLBr. Мол. вес 325,68;
разлагается при нагревании выше 105 \ легко очищается возгонкой
в вйкууме.
Т.— эффективный агент для аллильного бромирования, кото-
рое заметно ускоряется при освещении 11]. Циклогексен, например,
бромируется при освещении лампой (275 <?т). Реакцию проводят
Вг
\ и 50 мин ирл ОО"1 d ।[
| || , ВгСС13СС12Вг-!-СС14—1г
0,2 моля 0,5 моля 30 лы
+ С1.,С-^-СС12л-НВг
312
в гомогенном растворе с избытком Т. или в СС14 как разбавителе.
Т. — более дешевый реагент, чем N-бромсукцинимид (NEC). Кроме
того, если при реакции получается смесь продуктов, то с Т. и NEC
их соотношение различно 11].
I, Н u yser Е. S., DeMott D. N., Chem. Ind., 1963, 19.54.
ТЕТРАХЛОРФУРАН. Мол. вес 205,87, т. кип. 6Г/10 мм.
Реагент (3) нолучаюТ хлорированием ТГФ до гексахлор дигидро-
фурана (2) и дехлорированием под действием магния [1]. Он ис-
пользуется как диен в реакции Дильса — Альдера; например,
при нагревании с малеиновым ангидридом в автоклаве при 180'
образуется аддукт (4). Для проведения реакции с менее реакцион-
носпособными диенофилами — акриловой кислотой и циклогексе-
ном — в качестве катализатора добавляют небольшие количества
гидрохинона или трихлоруксусной кислоты.
ТЕТРАХЛОРЦИКЛОПЕНТАДИЕНОН, (3). Т., реакциоиноспо*
собный диен в реакциях Дильса — Альдера, неизвестен в виде моно-
мера, ио может быть получен in situ из легкодоступного гексахлор-
циклопентадиена (1). Ныокамер и Мак-Би 11] установили, что это
соединение при обработке конц. серной кислотой при 80° превра-
щается с хорошим выходом в пентахлорциклопептенон (2) с т. пл.
80°
ЩЗО4
78%
AcONaj,
CHjCN
(3)
Cl Н
Cl н
О) (2)
82—83°. Дегидрогалогенирование (2) можно осуществить действием
ацетата натрия в ацетонитриле или иодида натрия в 1,2-диметокси-
этане 121. При получении дпенона (3) в присутствии норборпадиена
с хорошим выходом получается аддукт диенового синтеза 1(4),
т. пл. 108—1101.
1. Newcomer J. S., МсВе е Е. Т., J. Am. Chem. S( с., 71, 946 (1949).
2. D i е I s с h е W. И., Tetrahedron Letters, 201 (1966).
313
ТЕТРАЦИАНХИНОДИМЕТАН. Мол. вес 204,18, NCCCN
т. пл. 296 . Т. получают конденсацией циклогексан- ''
дпоиа-1,4 с малоноиптрилом, бромированием и после- ц г]
дующим дегидробромпрованнем иод действием пиридина ||
[11. Т. по свойствам подобен тетрациапэтилеиу и яв-
ляется л-кислотой, сравнимой с ним по силе. Константа \CCCX
равновесия для реакции образования л-комплекса с
пиреном равна 78,4, а для комплекса тетр а циан этил ей— пирен 29,5.
1. A i- к с г D, S., И с г I 1 е г W. R., J. Am. Chem. Soc., Я4, 3370 (1962).
ТЕТРАЦИАНЭТИЛЕН, (Х]=С)Д2- С(С-N)2. Мол. вес 128,10,
т. пл. 200° (в запаянном капилляре); светло-бежевые кристаллы;
кристаллизуется из девяти кратного по весу количества хлорбен-
зола.
Получение [1]. К смеси малононитрпла и бромида калия в 900 мл
воды добавляют бром в течение 2,5 час для получения твердого
комплекса дпброммалопонитрпла с бро.мпдом калия. Комплекс
перемешивают с бензолом, добавляют свсжеосажденную порошкооб-
разную медь и смесь кипятят при перемешивании для завершения
реакции. Продукт экстрагируют горячим бензолом и перекристал-
лизовывают из хлорбензола.
З-НН
4СН., (СХ)„ -i 8Вг., KBr— - KBr [Br.CtCX’Ph : ННВг
- ' - Г, (s < । 11 - -ii.
I, ”, миля 3,03 м(1 а',1 <), 3 3 мо.'гя
J IcpcxieiTi rt и п и ис
К Вг [ В г.,С (С X)., 14 I 4 О i н(. АЬ1,И" И"1’-- >( NC).,C С (С X).,
’ П>— ] Г. ‘tdi'
Ит25 моля 1.57 г-атом 1 л
л-Комплексы. Т.— сильная л-кислота, образующая окрашенные
л-комплексы с ароматическими соединениями, некоторые из них
приведены в таблице [21. Гексаметилбензол — значительно более
сильное л-оеиоваине, чем бензол; компоненты комплекса распола-
гаются одни над другим с максимальным перекрыванием л-орби-
.п-Комплексы Т. с ароматическими соединениями в CHoCL
\ Р t Л] ,9 т л 9 I2CB ОС' соединение К1 lil ст,[ нт лсеогuoi Hi г 1 f (К) 7 М.т КС .If-lffi'
Бензол 2,01) 3«-1 3570
Толуол 3,70 406 3.330
о-Ксплол 6,97 430 3S60
Гексаметилбензол 263 545 4390
Гексаэтплбепзол 3,11 550 56
Нафталин 11,7 550 1210
Пиреп 29,5 724 1137
314
талей. Так как все 12 атомов углерода гексаметилбензола лежат
в одной плоскости, метильные группы не препятствуют тесному
сближению с другим компонентом на расстояние около 5 А. Тетра-
этилбензол, напротив, ие копланарен. Т. образует также л-ком-
плекс с циклогексеном, однако К. в этом случае составляет только
около 0,1 от константы бензольного комплекса. Комплекс с бен-
золом — желтый, с нафталином — красный и с антраценом — ярко-
зеленый.
Реакции Дильса—Альдера [3]. Зеленый цвет л-комплекса Т.
с антраценом вскоре исчезает, так как комплекс быстро превра-
щается в неокрашенный аддукт диенового синтеза. При добавле-
нии к охлажденному льдом раствору Т. в ТГФ небольшого избытка
бутадиена раствор вследствие образования л-комплекса становится
ярко-желтым, но через несколько минут начинает выпадать бесц-
ветный аддукт. В тех же условиях для завершения реакции малеи-
нового ангидрида с бутадиеном требуется много часов.
. ''CN
C(CN)2 ю
। С\
CN
Т. оказался наиболее реакционноспособным из 24 диенофилов
при кинетическом исследовании реакции Дильса — Альдера с цпк-
лонентадпеном и 9,10-диметплаитраценом [41. Пакьстт [51 не смог
получить кристаллический аддукт при обработке азепиноиа (1)
малеиновым ангидридом или N-фенилмалепмпдом при нагревании
в интервале 35—120°. Реакция осуществляется при нагревании (1)
с диметиловым эфиром ацетиленднкарбоновой кислоты без раство-
рителя при 130', по выход аддукта составляет лишь 20%. В про-
тивоположность этому раствор эквивалентных количеств (1) и Т.
в ТГФ при комнатной температуре первоначально приобретает
фиолетово-коричневую окраску, которая в течение 30 лшн пол-
ностью переходит в светло-желтую; аддукт (2) получают с количест-
венным выходом.
Присоединение с образованием циклобутанов. Бломквист и Мейи-
вальд 161 показали, что диен, не образующий аддукты Дильса —
Альдера, может реагировать с Т. с образованием производного
циклобутана. Например, Уильямс [7] исследовал реакцию диена
315
(1) с Т. при комнатной температуре, приводящую в основном к про-
дукту (2), образующемуся в результате присоединения по наименее
>сн3
(СН3)2С
(I)
C(CN)2
C(CNh
ТГФ,
80%
(CN),
(CN)Z
пространственно затрудненной двойной связи. Однако если экзо-
циклическая двойная связь не экранирована (3), то получается
спироциклпческий аддукт (4).
(О
C(CN)Z
II
C(CN)2
ТГФ, 25°
7 5%
(NC)2I----l(CN)z
(4)
Озонирование. Т. устойчив к действию озона даже при ком-
натной температуре. Поэтому Криге и Гюнтер 18] озонировали
тетраметилэтилсн в этилацетате ври —70J в присутствии 1 же
Т. в надежде, что предполагаемая промежуточно образующаяся
«окись карбонила» —О—О<-> ^С-=О— О будет улавливаться
реагентом в результате 1,3-диполярного присоединения. Неожиданно
продуктами реакции оказались ацетон и кристаллическая окись Т.
Оз
(СН3)Х —С (СН3), 4~ (NC).,C —С
о
- ,2((Д1;)ьСО (ХС)2С--С(С.\>2
so %,
Механизм этого превращения неясен, но полученные результаты
дают новый метод расщепления озонидов с образованием непо-
средственно альдегидов и кетоиов с хорошим выходом. Так, Му-
навалли и Оуриссон 19] обнаружили, что озонирование лоигифо-
лсиа (1) в стандартных условиях дает необычный продукт — эпок-
сид, но в присутствии Т. в качестве основного продукта получается
316
ожидаемый кетой (2) и небольшое количество диона (3), образую-
щегося в результате расширения цикла.
Реакция со спиртами. В присутствии мочевины Т. реагирует со
спиртами, например с этиленгликолем, с выделением цианистого
водорода [101. Продукт, этиленксталь дицианкетена, в одну стадию
можно превратить в пиримидин, пиразол или изоксазол.
НОСН2
[ 70 -7 70
(\С).,С--С (CN)3HOCII2 + ILNCONH.,>
0,2 моля п 300 ли 50 лы 0,057 моля 77 — 85%
ациона и 300 m.i CS2
О-СН,
(NCpC-C; I
XJ-CH2
Замещение в ароматическом ряду. Т. реагирует с М.М-дпметил-
анилином в ДМФА в мягких условиях с образованием п-трицпан-
винильного производного [111. Это вещество образует темио-сиипе
иглы и является красителем. В тех же условиях 2,6-дпметилфенол
лае г 4-трпциа нв । шильное производное, пиррол образует 2-трициан-
впнн. ширрол, а фенантрен превращается в 9-триц.ианвипилфепаит-
fen [12].
(CH3)2N—+ (NC)2C = C (CN).,Д- ДМФА -
---х “ 52-58°
0,22 моля 0,20 моля 65 молен
(<. ,H3)SN— <fZZ^/~C = C (CNP
CN
Дегидрирование. T. - эффективный pcai сит для ароматизации
1,4 дигидробензолов [131
i'r Кипячение 4 idir fz и
|| ,|-HNC)!C = C(CN)?— - — — -| ||-1'.C!r.-C(CX)._
'о’>% 11 И
98%
Другие реакции. Подобно фенолам и третичным арилампнам,
малононитрпл легко реагирует с Т, [141. Реакцию проводят в при-
сутствии пиридина и продукт выделяют в виде ярких желто-оран-
жевых игл 1,1,2,3,3-пентаииаш1ро1В’нида тетраметиламмония.
25 м-i Н .О
(\СщС^С(С\).2 + СНЩСЫ)2Д-Ру ----
0.1 МОЛЛ 0,1 мол: II.II МОЛЯ
-(ХОХ-с-Е (Сэдр(н1
| 81—85% (общий)
CN
(NC),C-C-C(CN)2 l(CH3)4N[4- Pv NCI
I
CN
317
Интересная реакция получения 2,5-дпа1мино-3,4-д11Циант1[офе1ш
состоит в пропускании сероводорода в раствор Т., содержащий
пиридин 1151. В отсутствие пиридина или другого основания реак-
ция прекращается на стадии образования тетрацианэтана и серы.
NC CN
I I
С = С + Нг5
NC CN
0,2моля В 300мл
NC CN
I I
+ Ру —> СН—СН
100 МЛ NC CN
92-95%*
ацетона и ЗООлтт CS2
Дпамшюдпцпантиофеп изомеризуется в 2-а мино-3,4-дицпан-5-мер-
каптониррол при нагревании его с водной щелочью до полного
растворения п последующем подкислении раствора. Обычно тио-
фены и пирролы, содержащие амино- или меркаптогруппы, очень
неустойчивы, по присутствие двух электроотрицательных циан-
групп стабилизирует эти соединения.
1. К л р б о н п Р. А., «Синтезы органических препаратов», ИЛ, М., 1961,
сб. 11. стр. 54.
2. М с г г i f i е I (1 R. Е., Phillips \V. D.f J. Am. Chem. Soc,, 80, 2778
(1958).
3. Middleton W. J., Heckert R. E., Little E.L.Krcspan
C. G., J. Am. Chem. Soc., 80, 2783 (1958); Stewart C. A., ,1 r., ,J. Org. Clicm.,
28, 3320 (1963).
4. Saner ,1., \V i c s t IL, M i c 1 e г t A., Cbem. Ber., 97, 3183 (196 I).
5. P a q и c t t c L. A., J. Org. Chem., 29, 3447 (1964).
(j, В 1 о m q nisi A. T., Me i n w aid Y. C., J. Am. Chern. Soc., 79, 5316
(1957).
7. \V i 1 I i a m s J. K., J. Am. Chem. Soc., 81, 4013 (1959).
8. Cr i egee R., Gunther P., Chem. Ber., 96, 1564 (1963).
9- 31 u и a v a 1 1 i S., Ourisson G., Bull. soc. chim. France, 1964, 729.
10. Д и к и и с о li 4. Л., M ельб и Л. P., «Синтезы органических препара-
тов», ИЛ, М., 1961, сб. 11, стр. 72.
11. Ма к-К у з и к Б. И., М с.чьб п Л. Р., «Синтезы органических препара-
тов», ИЛ, М., 1961, сб. 11, стр. 67.
12, Sauscn G. N., Engelhardt V. А., М i d d 1 е t о n W. J., J. Am.
Chem. Soc., 80, 2815 (1958); Sola n d ,L R., M cKusic k В. C., J. Am. Chem.
Soc., 83, 1652 (1961).
13. L о n g a n e D. T., S rn i t h G. L., Tetrahedron Letters, 205 (1962).
14. M и д д л e т о к У. Дж., 5' и л e ii Д. У., «Синтезы органических препара-
тов», изд-во «Мир», М., 1964, сб. 12, стр. 132.
15. М i d d I e t о n W. J., Engelhardt V. A., Fisher B. S., J. Am,
Chem. Soc., 80, 2822 (1958).
ТЕТРАЦИКЛОН, см. Тстрафеннлцпклоиентадисшш.
ТЕТРАЭТИЛАММОНИЯ АЦЕТАТ, (CH.-^NfOCOCH,). Мол. вес
189,29, вязкое масло. Получение Ц, 21.
Т. а. взаимодействует с (—)-а-фенил этил хлоридом в ацетоне
с образованием ( )-а-фенплэтплацетата; рацемизация при этом осу-
ществляется в меньшей степени, чем при сольволизе уксусной
318
кислотой 11], Т. а. реагирует также с алл ил галогенидами без анио-
нотропной перегруппировки |2, 3|.
СН.{ СН,
QH-CHCI (С2Н-) ] \’ (C)COCH;t) - , CJ L.CHOCt)СН, (С,! L0J\С1
(-) (-1 )
Корана и сотр. 141 получили 2',5'-диацетат у р иди н-3'-фосфата
обработкой (1) в пиридине 10-кратным избытком уксусного ангид-
рида и I же Т. а. в качестве источника ацетат-ионов. Реагент полу-
о
о=р-он
он
чают нейтрализацией водного раствора гидроокиси тетраэтилам-
мония уксусной кислотой и обезвоживают азеотропной сушкой
с пиридином.
1. Steinman ,1Hammett L. Р,, J. Am. Chem. Soc., 59, 2536 (1937).
2. R i e in sc Ii n e i d c r R., N c ti г i п y RAmi., 66(1, 42 (1962).
3. Roberts J. D., У о u п и \V. G., \V i n s t e i n S., J. Am. Chem. Soc.,
64, 2157 (1942); О w c n L. X'., Smit |i P. X., J. Chem. Soc., 1952, 4035;
C n m p bell J, R. В., I s Hi m A. AL, R a p h a e 1 R. A., ibid., 1956,
4096.
4. R a ni in I c r 1). 11L a p i d о t 5',, 1\ h о г a n a 11. Q., .1. Am. Chem. Soc.,
85, 1989 (1963).
ТЕТРАЭТИЛАММОНИЯ ГИДРООКИСЬ, (C2H-,) ЛТОН. Полу-
чение и применение подобно гидроокиси трл-н-гексилэтиламмония
111.
1. \V е i ш а п п G., Schaller IL, Khor a n а II. G., J. Аш. Chem. Soc.,
85, 3835 (1963); Lohrmann R,, 1\ h о г a n a H. G., ,1. Am. Chem. Soc.,
86, 4188 (196-1).
О
ТЕТРАЭТИЛАММОНИЯ ФОРМИАТ, (C, HENOCH [1]. Это гиг-
роскопичное кристаллическое вещество применяют при эпимери-
зации вторичных спиртов. Для его получения смешивают эквива-
лентные количества 90%-ной муравьиной кислоты и 23%-ного
водного раствора гидроокиси тетраэтил аммония, 'Воду удаляют
319
отгонкой в вакууме и последующей азеотропной перегонкой с бензо-
лом. Кристаллический продукт тщательно предохраняют от влаги.
Примером использования служит следующая реакция:
о
о
(СДЦЧОСН
Ацетон,
кипячение
Преимущество этого эпимеризующего агента по сравнению,
например, с ацетатом калия состоит в том, что промежуточные фор-
миаты легко гидролизуются в таких- мягких условиях, когда эфиры
других кислот не изменяются.
1. Contributed by L е v i n e S, G., North Carolina State Univ.
ТЕТРАЭТИЛАММОНИЯ ХЛОРИД, (C2H5)4NC1. Мол. вес 165,71.
Получение [11.
Т. х. в ацетонитриле действует как катализатор дегидрогало-
генирования [1, 2]
Н3СХ/СН3 Н3С^/СН3
I II I (С-.ИЩХС1 f и' й
| || |/СН2СвН5-------^^| II И +НВг
"V Y'^Br у хсн„сли,
н II
О О
1. Coppens G., к е v i 1 1 D.N., Cromwell N. Н., J, Org. Chem., 27,
3299 (1962).
2. К e v i 1 1 D, NС о p p e n s G. A., Cromwell N. H., J. Org, Chem.,
28, 567 (1963).
ТЕТРАЭТИЛПИРОФОСФИТ, (QH,O)2POP(OC2HY Мол. вес
258,19, т. кип. 8871 мм.
Получение. Первоначальную методику [1] можно заменить
простым методом [2] с использованием более доступных реагентов,
320
К суспензии карбоната серебра в бензоле при перемешивании мед-
ленно добавляют дпэтилхлорфосфит. Экзотермическая реакция
сопровождается выделением газа; продукт перегоняют из реакцион-
ной смеси без отделения твердого осадка.
(С- н О-..2Р-С1 -4Z21) (СДКОЦР^Р/ОГДЦ), -12£ыс,н5о)2р-о-р(ос2н5)г
I 1. 00%
Применение в пептидном синтезе 11, 31,
CbNHCHCO2H -рНаХСНС0Х2Н5-!-(С,Иг>0)2 POP (ОС2Нф
_v Cb\rHCHCONHCHCO2C2H-, ’-‘2 (С.Н-,0).. РОН
I I
R R'
1. Anderson О. W,, В 1 о d i и g е г J., Welclier A. D., J. Am. Chem.
Soc., 74, 5309 (1952).
2. Samuel D., Silver В. L., J. Org. Chem., 28, 1155 (1963).
3. V igneau d V., du, Ressler C., Swan J. M., Roberts C. W.,
К a t s oy a n n i s P. G., J. Am. Chem. Soc., 76, 3115 (1954); Gish D. T.,
du Vigneaud V., ibid., 79, 3579 (1957); Ressler C., duVigneaud
V., ibid. 79, 4511 (1957); Ratsoyannis P. G., Gish D. T., du Vigne-
a u d V., ibid., 79, 4516 (1957).
ТЕТРА ЭТИЛ СУКЦИНИЛ ФОСФО HAT, (3). Мол. вес 309,28.
Т. получают обработкой полуэфира малеиновой кислоты три-
этилфосфитом, образующийся продукт превращают в фосфонатиый
карбанион при перемешивании с 50%-ной суспензией гидрида нат-
рия в 1,2-диметоксиэтане 11]. Карбанион реагирует с карбонильны-
ми соединениями подобно илидам Виттига, но более энергично.
О-
(СДКО^Р ф СНСО..Н (С.,Н5О).,!р + - СНСО.,СгНг,
J 1
СНСО.2С.Л 1Я СН,СО2С2НЙ
(1) (2) (3)
о-
(СЛ!ЙО).,Р L — ССО.,С,Н5(\а *) СсН-СН-ССО.С.Н-
। " ‘их; I
СНаСО.2С.,НГ1 СН.СОаС.Д15
I. Wadsworth \V. S. Jr., Е rn m о и s \V, D., J. Am. Chem. Soc., 83, 1733
(1961).
CH, ZCH3
ТИГЛИНОВОЙ КИСЛОТЫ ХЛОРАНГИДРИД, n c c cocl
Мол. вес 118,57, т. кип. 69'35 леи.
11 Заказ № UGG
321
Получение. Т. к. х. получают реакцией треххлористого фос-
фора с тиглшювой кислотой 111.
Защита ОН-групп 121. Превращение спирта (1) в эфир тцглипо-
вой кислоты (2) используют для защиты ОН-группы в процессе син-
теза. Защитную группу удаляют действием OsO4 — НЮ4, в резуль-
тате чего образуется эфир пировиноградной кислоты (3), который
легко расщепляется под действием слабого основания в течение ночи.
1. В а г у с г О., .М ;1 г t 1 и W. !е, М i t с h с 11 W.. J. Chem. Soc., 1937, 1822.
2. К u р е li а n S. .М., В а I о n A. D. I’ и j i 1 а Е., .1. Огц. ('hem., 27
3103 (1962).
ТИОГЛИКОЛЕВАЯ КИСЛОТА, HSCH2CO,H. Мол. вес 92.12,
т. пл. —16,5, т. кип. 96 /3 мм, рКа 3,83, уд. вес 1,325. Т. к. обла-
дает сильным неприятным запахом и легко окисляется на воздухе.
Натриевая и аммониевая соли Т. к. используются для холодной
завивки волос и в качестве модификаторов шерсти, так как они вызы-
вают восстановительное расщепление поперечных связей —S — S —
в кератине.
Пептидный синтез 111. Нежелательные побочные реакции, часто
наблюдающиеся при синтезе пептидов, содержащих метионин, можно
предотвратить путем превращения на соответствующей стадии син-
теза тиоэфпрноп группы метионина в сульфоксидную. Под дейст-
вием небольшого избытка перекиси водорода окисление до сульфок-
сида осуществляется без образования сульфона. Под действием Т. к.
о—
Ts CH.CH.SCH, Ts CH,CH..S юн,
I i I ШТ ! "
CbPro — T yr-Lys-N нецел, CH ; —v cb • Pro - Туг-l.v< - N HCJ JCO^CH ;
"%
(11 (2)
J-JCI j 70%
0“
Ts C’H .CIKSCH., Ts CH .CH.S-l-CIb
| HSCH..CQ,H I
HPro-Tyr-I-V'-NHCHCO,H -•---------—------- H Pro - Тут - Ly--X HCHCO, H
(41 Ы"„ (3)
322
сульфоксид легко восстанавливается до сульфида. В противополож-
ность карбобепзоксипроизводному (СЬ) мстиопина удаление карбо-
бензоксигруппы из соответствующих сульфоксидов гладко осуществ-
ляется осторожной обработкой конц. соляной кислотой в мягких
условиях. Например, попытки прямого декарбобепзоксилировапия
(1) оказались безуспешными, однако желаемый продукт (4) был
легко получен через сульфоксиды (2) и (3).
Изомеризация олефинов. Под действием Т. к. транс,транс,
рас-[ ,5,9-циклододскатриен (1) изомеризуется в транс, транс,
транс-изомер (2) при комнатной температуре (экзотермическая
реакция) 12]. Обычные методы изомеризации оказались безуспеш-
ными 13].
1. Iselin В., Helv. Chim. Acta, 44, 61 (1961).
2. Due k E. W., L о c k e J. AL, Chem. Ind., 1965, 507.
3. Wilke G., Angcw. Chem., Inlernat. Ed., 2, 105 (1963)
N—k S
D- 4 I
М-С-Й
piwi. isvu i. iui. jw-iuu . '--'
T. получают реакцией тпофосгена с 2 же имидазола 111. С пер-
вичными аминами Т. реагирует прн компа гной температуре с обра-
зованием тиокарбамоил имидазолов (2), при разложении которых по-
лучают изотиоцианаты (3) и исходный имидазол 12].
(п
ямнг ----
R-X=C=5
(3)
Об использовании реагента в стереоспецифическом синтезе оле-
финов см. Триметплфосфпт.
1. S t a a b Н. A., W а 1 t h е г G., Ann., 657, 96 (1962).
2. Staab Н. A., Walther G., Ann., 657, 104 (1962).
11’
323
ТИОМОЧЕВИНА, S=C(NH2)2. Мол. вес 76,12, т. пл. 178э,
растворимость: 9,2 а/100 г воды прн 13°.
Соединения включения с тиомочевиной (см. Мочевина, соеди-
нения включения). Очевидно, не зная о комплексах с мочевиной,
Англа 111 нашел, что Т. соединяется с различными веществами с
образованием, как он думал, нестабильных стехиометричных сое-
динений, содержащих 3 моля Т. В настоящее время установлено,
что Т. образует нестехиометричные комплексы, подобные соедине-
ниям с мочевиной, но способные к включению несколько больших
молекул, так как атом серы больше кислорода. Шисслер и Флиттер
|2| определили способность ряда углеводородов к образованию ад-
дуктов с мочевиной и с Т., но ие анализировали комплексы. Линейные
предельные углеводороды, образующие комплекс с мочевиной, слабо
удерживаются Т. Объемистый 2, 2, 4-три мети л пен тан образует ком-
плекс с Т., ио не с мочевиной. Шисслер и Флиттер анализом на мо-
делях Фишера — Хиршфельдера — Тейлора установили, что наи-
более подходящее поперечное сечение,, молекулы для образования
комплекса с Т. составляет 5,8 х6,8 А, что согласуется с рептге-
ноструктурными исследованиями Смита [31.
Для построения цилиндра, моделирующего канал Т., мы взяли
за основу адамантан — молекулу с хорошо известными парамет-
рами, образующую стабильный комплекс с Т., но нес мочевиной[4].
В комплексах с Т. на каждую гостевую молекулу углеводорода при-
ходится 3,4 молекулы хозяина-—Т. Цилиндр, легко охватывающий
модель Фншера, имеет длину окружности 83 см и диаметр 26,4 см
(или 5,28А), и, следовательно, зазор равен (6,80—5,28)/2- =0,64 А.
Такое тесное расположение атомов водорода и молекул Т. объяс-
няется, ио-видимому, жесткой структурой молекулы углево-
дорода. В случае 2,2,4-триметилпентапа расположение более
свободное.
Исследование на моделях с помощью подобного цилиндра дало
возможность одному из нас предсказать, что п-ди-(трет-бутил)-
бензол будет образовывать комплекс с Т., а его 2,5-диметокснпропз-
воднос — нет. Это наблюдается в действительности 1.51. Для полу-
чения комплекса 3 г/?-ди-(трет-бутил)-беизола и 5 г Т. растворяют в
50 мл горячего метанола и раствор оставляют на льду для кристал-
лизации; выход 5,8 г (4,4 моля Т. на 1 моль углеводорода).
Очистку сквалена проводят следующим образом [61: получают
насыщенный раствор Т. в кипящем метаноле, выдерживают этот
раствор в течение дня и фильтруют. К 10 мл этого раствора добав-
ляют сквален (100 л/а), сосуд закрывают пробкой н встряхивают в
течение 6—8 час. Углеводород постепенно растворяется и выделя-
ются кристаллы комплекса. Кристаллический продукт отделяют п
сквален выделяют после добавления воды экстракцией петролей-
ным эфиром.
В сквалепе двухзвенные метиленовые цепочки могут быть либо
в цие-, либо в транс- положении относительно каждой из четырех
324
внутренних двойных связей. Николаидес и Лейве [7] новым мето-
дом доказали полностью транс-конфигурацию, как показано в
формуле I:
Из данных рентгеноструктурного анализа монокристаллов соедине-
ний включения мочевины и Т. можно определить длину насыщенно-
го соединения и сравнить ее с соответствующим непредельным сое-
динением. Сравнение раз*меров олен новой (цис) и элаидиновой (транс)
кислот со стеариновой кислотой в комплексах с мочевиной показы-
вает, что одна транс-двойная связь укорачивает молекулу на 0,19 А,
тогда как /(нс-двойная связь — на 0,88 А. Сравнение комплексов Т.
с иергпдросквалеиом и скваленом показывает, что четыре двойные
связи укорачивают молекулу только на 0,73 А, или 4 х 0,182 А. По-
лагая, что эффект изолированной двойной связи одинаков для комп-
лексов с мочевиной и с Т., можно сделать вывод, что все четыре
двойные связи имеют транс-конфигурацню, как и показано в фор-
муле I. Молярное соотношение Т. к сквалену в комплексе по данным
рентгеноструктурного анализа равно 14,65 181.
Две изомерные формы 4-изопропил циклогекс ан карбоновой кис-
лоты удалось разделить [91 благодаря тому, что высокоплавкая
транс-форма (94—94, 5°) образует комплекс с Т., а низкоплав-
кая /^ас-форма (40--41°) не образует. При этом отмечается, что мети-
ловые эфиры обеих изомерных кислот образуют комплексы. Пола-
гают 13J, что эти различия обусловлены тем, что в соответствии с
данными Николаидеса и Лейвса 171 жирные кислоты включаются
в мочевину в виде димеров.
Получение меркаптанов. Ал кил галогениды, например лаурил-
бромид, реагируют с тиомочевиной с образованием бромгидрата
изотиомочевины, который при щелочном гидролизе дает мер кап-
S
н-СДЩДг Н2ХС\1134-95%-ный C.JI5OH Ки”Я
3 час
0,5 моля 0,5 моля 25 0 лы
/ДН Кипячение 2 час с поди. NaOII
-НВг ------------------—---------H-C12I-I.,6SH
' *' XNH2 ] 79—83%
тан. При получении лаурил меркаптана ПО], не выделяя промежуточ-
ные продукты, соль кипятят с водным раствором щелочи. При полу-
325
чешпг этапдитпола из дпбромэтапа промежуточный этилендиизотиу-
ронийбромид выделяют пз 95°о - ного этанола с выходом 90"о и гидро-
лизуют с выходом 55—62% 1111. Применение в качестве исходных
продуктов спиртов бензилового тина показано на примере полтче-
пня 2-фурфурилмеркантапа [12].
s
п
+ HHXCNIIz + НС1
снгон
5 молем -100 Л7Л
5 мол ей
NaOll,отгонкя с паром
Производные. Ниже приведены методы получения сульфата
S-метилпзотиомочевпиы [13] и гуанидиноуксусноп кислоты [14].
S SCII3
Н.,NCNИ., ж (СН:{).,SO4 (НX - С — NН>) I I.SO4
7 9-Ь 1 %
2 моля 1Д моля
S
С.,Н5Вг ---Н.>ХСМ1, - ,?бс- едзе/11 • НВг ^^^ОДГ-ХаОН
93-99% ХН-. 90%
2,29 моля 1,97 моля
/NH
HOXCH .NHC'/ %C.,ri5SH
XNH2
Эпоксиды -+ тиираны. Эпоксиды, по крайней мере получаемые
с хорошим выходом указанным методом, можно превратить в ти-
ираны с выходом 90—95% реакцией с Т. в присутствии разбавленной
серной кислоты при 5—103 П5[.
roh CH..-CUC1I.CI ДК_, roch.chch.ci
„ „ \ / I эфир При 0°
Изиьнок \,,/ Ат.
4 моля О ОН
S
JI
ROGH.,CH-CH,________HjNCN1Ij ROCHXII-CH.,
\ / [’гиб. IKSO, (5—100 /
Гетероциклические соединения, см. также Этилат натрия (ге-
тероциклы). 2-Лмиио-4-метилтиазол (1) получают кипячением смеси
хлора погона и Т. в 200 мл воды и обработкой после охлаждения 200 г
твердого едкого натра [161. Приведенные примеры показывают учас-
тие Т. в енольной форме как в этой реакции, так п в реакции (2), в
результате которой образуется пссвдотиогпдангоин 1171. Продук-
том реакции (3) является 4-метпл-6-оксиниримпдпн [181. В реакции
(4) из 1, 1,3, З-тетраэтокспиропана получается хлоргидрат 2-меркап-
топнримпдипа [19].
I)
CH3CO nh2
CHzC1HS-'C^NH
I) НгО,КИГТЯЧ.
г) Na ОН
70-75%
1 моль 1 моль
NH2
2)
O = COC2H5
cn ri + +95%-нЫЙ,СгН5ОН
ОН2О1 ндх^ XNH
CH3CO2Na
79-82% '
1 моль 1 моль
3)
со
СН/ хСНгСО2СгН5 (1моль)
H2N + NH2 (I моль)
%'С/
II
S
CH3ONa НзСГ^ 7|OH
СН3ОН.
69-84%
sh
H2-Ni
90-93%
4)
СН (ОС.,Щ)„ NIL. 600/M
/ ‘ ' 1 95'«„ C..H-OH
C1L + Г -Ч1С1 ——— -
I H,N'Z s 60-64 %,
CH (OCJF,),
0.8 моля 0,8 моля 200 ли
11 N • HCl
4 N SH
Дебромирование. Вицинальные дибромиды дебромируются Т.
с хорошим выходом. Реакцию проводят в этаноле при 50° в тече-
ние 8 час [201.
1. Angla В., Bull. soc. chim. France, 16, 12 (1949); Ann. chim. 112], 4, G39 (19-19),
2. Schiessler R. W., Flitter 1)., J. Am. Cliein. Soc.., 74, 1720 {1932).
3. Smit Ii A, E., J. Chem. Phys., 18, 150 (1050).
4. Landa S., M a ch а с с k V. Coll. Czech. 5 1 (1933); Landa S., И a 1 a
S., Coll. Czech., 24, 93 (1939).
5. Org. Expts., 184, 187.
6. В 1 о с h К., private communication.
7. N i с о 1 a i d es N., Laves F., J. Am. Chem. Soc., 76, 2596 (1954); 80,
5752 (1958).
8. Nicolaides N., private communical ion.
9. В e k k u m H., van, Kleis А. А. В., M e d e m a D., V e r k a de P. E.,
WcpsterB. M., Rec. trav. chim., 81, 833 (1962).
10. Уркварт Г., Гейтс Дж., мл., Копп о р I-’., «Синтезы органических
препаратов», ИЛ, М., 1952, сб. 3, стр. 246.
11. Специале А., «Синтезы органических препаратов», ИЛ, М., 1953, сб. 4,
стр. 569.
12. К ° ф о Д X., «Синтезы органических препаратов», ИЛ, М., 1957, сб. 7, стр. 85.
13. Ш и л ь д н с к II., У п и д у с У., «Синтезы органических препаратов»,
ИЛ, М., 1949, сб. 2, стр. 326.
14. Бра ид Э., Бранд Ф., «Синтезы органических препаратов», ИЛ, М.,
1952, сб. 3, стр. 148.
15. S с h u е t z R. D., Jacobs R. L., J. Org. Chem., 26, 346/ (1961).
16. Банере Дж., Д п к к и Дж., «Синтезы органических препаратов», ИЛ,
М., 1949, сб. 2, стр. 40.
17. Л л л е п Ч., Ва п Алл а п Дж., «Синтезы органических препаратов»,
ИЛ, М., 1953, сб. 4, стр. 434.
18. Фостер Ч., Снайдер X., «Синтезы органических препаратов», ИЛ,
М., 1957, сб. 7, стр. 37.
327
19. Crosby D. G., Berthold R. V., Johnson II. E., 0гц. S\ n., 43,
68 (1963).
20. I b ii e-R a s а К. M., Muhammad N_, Hasi bul lah, Chem. Ind.,
1966, 1418.
ТИОНИЛ БРОМИСТЫЙ, SOBr.. Мол. вес 207,89, т. кип.
68740jf.n, уд. вес 2,68.
Т. б. получают с хорошим выходом из хлористого тионила и
бромида калия Ш. Для более экономичного превращения спирта
в бромид [2] раствор спирта в эфире обрабатывают 0,5 моля хлорис-
того тиопила и 1 молем пиридина при —10°; раствор отфильтровы-
вают от солянокислого пиридина, упаривают и оставшийся сульфит
(RO)jSO нагревают с 0,5 моля Т. б.
По более ранней методике [3] спирт растворяют в бензоле при
нагревании около 100J и Т. б. добавляют по каплям.
1. Frazer М. J., Gerrard \V., Chem. Ind., 1954, 280; «Неорганические
синтезы», ИЛ, iM., 1951, сб. 1.
2. F г a z е г М. J., Gerrard \V. М а с h е I 1 G., S h е р h е г d В. I).,
Chem. Ind., 1954, 93i.
3. Е 1 d е г f i е I d R. С., К г e in e г С. В., К u р с h a n S. М., В i г s t е i n
О., Cortes G., J. Am. Chem. Soc., 69, 1258 (1947).
N, N'-ТИОНИЛДИИМИДАЗОЛ, (II). Мол. вес 182,21, т. пл. 78 -
79°.
Т. получают при взаимодействии хлористого тионила с 4 же
имидазола в ТГФ и используют раствор, отфильтрованный от соля-
нокислого имидазола III. Т, сходен с N, N'-карбонилдиимидазолом,
но более активен. Реагент используют в синтезе пептидов [2], пап-
I II
СНэ
I
CbNHCHCOaH
-T(-so2 >
CINHsCHiCOjCiHe,
(CjHsJjN
CHj
CbNHCHCONHC^COAHs 4- __
IV
ример для конденсации СЬ-d ,ь-алаиипа с этиловым эфиром гли-
цина. Реакция защищенной аминокислоты с Т. в ТГФ дает N-ацил-
имидазол (III), который эквивалентен активированному эфиру.
Хлор гидрат этилового эфира глицина присутствует в смеси с самого
начала; при добавлении триэтил амина освобождается свободный
аминоэфир, который конденсируется с промежуточным продуктом
III с образованием производного дипептида IV.
328
I. Staab H. A., Wcndel K-, Angew. Chem., 73, 26 (1961); Staab H. A.,
\V al thcr G., Ann., 657, 98 (1962).
2. Wieland T., V о g e 1 e г К., Angew. Chem., 73, 435 (1961).
ТИОНИЛ ХЛОРИСТЫЙ, SOC13. Мол. вес (18,98, т. кип. 75—76°,
уд. вес 1,67.
Обращение. Так как Т. х. разрушает резину, а в реакциях с ним
обычно выделяются НС1 и SO.2, все операции необходимо проводить
в полностью стеклянной аппаратуре с силиконовой смазкой на стек-
лянных шлифах н с обязательным отводом газов. Лучше всего про-
водить реакцию под тягой в круглодонной колбе с обратным холо-
дильником и хлор кальциевой трубкой; избыток Т. х. отгоняют на
водяной баие в вакууме водоструйного насоса и затем продукт пе-
регоняют в вакууме. Многие исследователи используют Т. х. в
большом избытке, хотя, очевидно, в этом нет необходимости, если
реакция идет при комнатной температуре или если смесь кипятят с
эффективным обратным холодильником, охлаждаемым водой до
температуры 20° и ниже.
Очистка HI. К 1 л технического Т. х. добавляют 160 мл трифснил-
фосфита и перемешивают 30 мин (перемешивание уменьшает мест-
ные реакции фосфита с Т. х.). Смесь перегоняют на колонке дли-
ной 31 см, заполненной стеклянными спиральками и снабженной при-
емником с хлоркальциевой трубкой. После отделения незначитель-
ного количества головного погона собирают Т. х. ст. кип. 75,5—
76°. Исходная температура куба 79°, и когда она поднимается до
85—86°, дистиллат (т. кнп. 76°) желтеет. Выход частично очищен-
ного хлористого тиоинла 60% (80% вместе с желтой фракцией).
Перегонка частично очищенного вещества с добавкой трифенил-
фосфита (15—20 мл/л) дает чистый Т. х. с т. кип. 76°; выход
более 90%. Перегонку прекращают, когда температура куба дос-
тигает 84°. В малых количествах Т. х. кажется бесцветным, в боль-
ших он имеет слабо-желтый цвет. Спектр частично очищенного
хлористого тионила в видимой области почти сравним со спектром
чистого продукта.
RCOOH -RCOC1. В большинстве методик, описанных в «Син-
тезах органических препаратов», рекомендуется использование реа-
гента в большом избытке. По методике, предложенной Кейзоном
[2] (СН3О2ССН.2СН.2СОС1), необходим 100%-иый избыток Т., но
при проверке этой методики Аллен и Вильсон получили тот же
выход (90—93%) с 20%-ным избытком реагента. При получении
аналогичного соединения —СН3О2С(СН.2)СОС1 Бишоп (31 исполь-
зовал 27%-ный избыток Т. х.; после кипячения реакционной
смеси в течение 5 час выход составил 83—86%. При получении
СН;(О..С(СН.г)4СОС1 реакционную смесь оставляют на ночь при
комнатной температуре; применение 100%-ного избытка Т. х. в
этом случае оказывается неоправданным 141.
Вомак и^Мак-Унртер 15] кипятили смесь I моля коричной кислоты
и 1 моля Т. х. в течение 1 час и затем сырой хлор ангидрид смеши-
329
вали с I молем фенола; при этом получался фенилцинпамат с выхо-
дом 83—89"о. Методика превращения кислоты (1) в оранжево-крас-
ный хлорангидрид (2) с г. пл. 94,5—95,5" предусматривает добав-
ление к реакционной смеси твердого карбоната натрия, который,
как утверждают, предотвращает разложение и осмоленис [61.
Cf!II-,N - \CfiH,СО..Н | SOCi„ -I- Xa.CO-i L5 C6115\ - \CfiH jCOCl
Ы)%
(!) 0,22 моля 3,5 моля 0,17 моля (2)
Следует упомянуть методику лаборатории авторов [7], чтобы об-
ратить внимание на эффективную аппаратуру (том I, рис. А-4) для
перетопки с паром tz-тетралоиа. Круглодонная колба слева служит
жидкостным затвором или ловушкой; в пей всегда находится немного
конденсата, за исключением тех случаев, когда для проверки или
при окончании перегонки она опорожняется струей холодной воды.
Нитробензол можно перегонять со скоростью 400 г/час.
Аллен и сотр. 181 разработали методику получения хлораигид-
рида олеиновой кислоты и других хлорангцдридов кислот, нс выдер-
живающих длительного нагревания в процессе реакции с противо-
точной перегонкой, по которой хлорангидрид нагревается лишь в
течение нескольких минут. Хлорангидрид олеиновой кислоты удов-
летворительной чистоты получается с выходом 97—98%.
Получение чистого, бесцветного феи ил ацетил хлорида и других
хлорангидридов кислот с активным а-водородом несколько затруд-
нительно, так как они образуют окрашенные продукты конденса-
ции. Баклес н Купер [91 разработали метод получения бесцвет-
ного феи ил ацетил хлорида, В смесь кислоты с 1 же Т. х. помещают
кипятильные камешки для самопроизвольного выделения газа и
периодически перемешивают ее при комнатной температуре до
прекращения выделения газов, а затем нагревают до 35—40° для
завершения реакции. Добавляют 1/4 объема бензола и тщательно
с6н-,сн,со.,н щ soci,. 2и с6н5снасос!
80—85%
4 моля 4 моля
удаляют все летучие продукты низкотемпературной перегонкой,
используя две ловушки, охлаждаемые ацетоном с сухим льдом,
вначале водоструйным насосом и затем при 1 мм. После удаления
всех летучих продуктов, которые придают красную окраску, хлор-
ангидрид перегоняют при 55—57'=/1 мм.
ззо
В синтезе эфиров «-бромкарбоновых кислот Т. х. служит не
только для получения хлораигпдрида кислоты, по и растворителем
для последующего бромирования 110]. Кислоту кипятят с Т. х,,
затем добавляют бром и снова нагревают. Этот метод пригоден для
моно- и дпкарбоповых кислот [11 [.
О получении хлорангпдридов карбоновых и сульфокислот,
которые не реагируют с одним Т. х., см. Диметилформамид—
тпонилхлорид.
Сульфохлориды. Приведенные ниже примеры показывают,
что Т. х. легко взаимодействует как со свободными сульфокислотами
[121, так и с их гидратированными натриевыми солями [131.
CH3SO.,H J- SOCK, -'5 ",,с. CH.{SO,C1
7 1-83%
1,5 моля 2 моля
/oCH3CrH4S0A’a.2IL,0 -|- SOCL — „-СН3С01-Г,SO..C1
0,2 моля 1,5 моля
Эфиры аминокислот. Бреннер 1141 рекомендует для этерифика-
ции аминокислот смесь метанола с Т. х. (метод Фишера обычно
более труден). К смеси аминокислоты и метанола при температуре
от —5 до —10" добавляют Т. х., поднимают температуру смеси до
40" н выдерживают при этой температуре в течение 2 час. Преимуще*
RCHCCKH -1- SOC1,2 -С СН.ОН RCIICO..CH3 , SO.,IIC1
’ 70 —80% [
NHa NH.rHCl
1 моль 1,1 моля 8 молей
ство метода в том, что не образуется вода. В частности, Уле и Харрис
[15] успешно получили этим методом диметиловый эфир метилами-
номалоновой кислоты (2). После нескольких превращении кислота
подвергается декарбоксилированию до метилового эфира саркозина
(3):
СН3ХНСН(СО.,1))., СИо\НСН(СО.,СН3).. CH;iNHCI 12СО.,СН3
(1) (2> (3)
Пател и Прайс 116] использовали Т. х. как кислотный катали-
затор и дегидратирующий агент при получении бензиловых эфиров
аминокислот, как показано, например, для и-фенилаланина:
СвН,СН.,СНСО.,Н CGI I - SOC1. _____СДЬ.СНХД [СО/'.НДД 1-
| ’ “ Д
NH2 NH3C1
3,3 г 125 мл 20 мл
ROH—RC1. Условия реакции аналогичны условиям получения
хлорангпдридов кислот, как показано, например, для Р-хлорэтнл-
метпл сульфида [17]- Раствор Т. х. добавляют по каплям к раствору
спирта в том же растворителе при перемешивании в течение 2 час.
331
CH3SCH,CH2OH + SOC!., 6 CH3SCH.,CH2C1
75-85%
1,63 моля 1,7 моля
в 200 мл СНСЬ в 135 мл СНС13
Аналогичная методика используется для получения этпл-а-хлор-
фен ил ацетата (181.
15 час при 25"
СJb,CHCO.,C.,H3-НS0C1, ки,,п,1е|1ис 30 С0Н-СНСО.,СЛh
I 81-85% I
он а
0,75 моля 0,82 моля
Дарзан 119] осуществил такую же реакцию в присутствии 1 моля
пиридина или другого третичного амина. В приведенном примере
промежуточный хлорсульфинат выделяется вначале в виде крис-
таллов, которые затем превращаются в жидкость без выделения
СН3СНСО2С2НЙ Н- SOCIj i- C5H5N CII3CHCO2C2H3 1 СД-15Х-НС1
I 1 . I
1,03 моли 1 моль Q]
I моль
газов. Реакция завершается при 110°с выделением SO2 и НС1. Дар-
заи отмечает, что третичный спирт дает «^-ненасыщенный эфир,
вероятно, через хлорид. Жеррар и сотр. 120] использовали модифи-
цированную методику, по которой 1 моль спирта смешивают в эфире
при —10° с 0,5 моля Т. х. и полученный раствор ди-н-бутилсульфита
«-BuOH+SOCl3-pC-H5N (h-BuO)2S-O-!-G,H6N.HCI
1 моль 0,5 моля 1 моль 0,5 моля
О
II
(h-BuO)2S = O 4-SOCI., -v 2h-BuOSCI ySO.,
0,5 моля
О
5|
2h-BuOSC1 —> 2rt-BuCl + SO2
отфильтровывают от солянокислого пиридина. Растворитель уда-
ляют и остаток нагревают с 0,5 моля Т, х.; быстро образующийся
хлорсульфит медленно разлагается на н-бутил хлор ид и SO2.
Брукс и Снайдер [21] использовали методику Дарзана при полу-
чении хлорида из тетрагидрофурфурилового спирта.
Г I + SOC12 + Ру ________________> 1 1
СН2ОН 73-75% ^О''"^,СН2С1
4. 2 МОЛЯ 4, 7 МОЛЯ
4 МОЛЯ
332
При получении траяс-стильбепа [221 бензоин нагревают с Т. х.
иа Кишинев водяной бане, избыток Т. х. удаляют на водоструйном
насосе, дезилхлорид (2) восстанавливают боргидридом натрия в
этаноле и раствор стереоизомерных хлоргидриноз (3) обрабатывают
нинковой пылью и уксусной кислотой и кипятят 1 час. При кристал-
лизации образуются алмазоподобные переливчатые пластинки чис-
того транс-стильбена с т. ил. 124— 125°. Эта мегодика быта исполь-
Zn,
С6Н5СН СС6Н5 SOC\ С6Н5СНССЙН5 C6H5CHCHC6H5_^£21i> с6н5с-н
I II Я7° I II II /Хй, гчи- И
ОНО CIO Cl ОН ЬЬ/0,с<и Н_С_С1^
тая на (1)
(1) (2) (3) (4)
SOCIz> С6Н5С==СС6Н5 ~5О-> С6Н5С—CC6HS
I । И II
СИ /О ------- О О
SX /44
b (6)
(5)
NaB1S CfeH5CH2CC6Hs
О
(П
зована в студенческом практикуме, и у большинства студентов
получился шрюяс-стильбеп, ио у некоторых образовался низко-
плавкий продукт, который идентифицировали как дезоксибензоин.
Исследования [23, 241 показали, что если смесь бензоина и Т. х.
не нагреть сразу, а оставить на некоторое время при комнатной
температуре (или ниже), то протекает другая реакция с образова-
нием ендиола бензоинсульфита (5). При дальнейшем стоянии
этот эфир разлагается до бензила, а при восстановлении боргид-
ридом патрия превращается в дезоксибензоин.
Дегидратация. Т. х. в пиридине является более сильным де-
гидратирующим агентом для стероидных спиртов, чем хлорокись
фосфора в пиридине (см. Фосфора хлорокись).
Метод получения 2-этплкапронитрила 1251 показывает, что ус-
ловия, необходимые для дегидратации амида, пичем не отличаются
от условий получения хлор ан гидридов кислот.
CII.i(CH.,).iCHCONH., [ SOCI., 1''’ час при СН3(СН.,),СНС\
| ‘ I
СН3СН3 СВД,
2 моля в 300 j'j С6Н3 3 моля
Реакция Брауна. Воган и Карлсон [261 нашли, что при расщеп-
лении амида иа алкплхлорпд и нитрил Т. х. имеет некоторые пре-
333
имущества перед пя гп хлор истым фосфором, первоначально исполь-
зованным Брауном. Т. х. берут в избытке (3—4 экв), а в качестве
RN'HCOR' SOCI, —> RCI р R'CX - SO., - MCI
растворителя применяют нитрометан, если он не мешает выделению
продуктов.
Ди-я-бутилсульфит. Этот эфир получают при добавлении
Т. х. к и-бута пол у в течение 2 час 1271.
35 |3'
2^-BuOH I SOCL. _ —» Щ-ВцО)25О-!-2НС1
Ч wMi 4.2
Пиридин-4-сульфокислота. Образование ароматических сульфо-
кислот при взаимодействии с Т. х. является особой реакцией произ-
водных пиридина; возможный се ход привете!! ниже 128]:
soci2
1) НОСС
2) С Л ЦОИ
4 0-45%'
(3)
N'a,SO-5 ,
Пиридин'
(4)
45-50%, считая
ИЛ (3)
Дегидрирование. Бенн н .Пукас 1291 сообщили о повой реакции,
в которой Т. х. осуществляет дегидрирование (I) до (4). Взаимодей-
ствие ппрролииона (1) с Т. х. при комнатной температуре дает жел-
334
тый продукт (4) с высоким выходом. Хлорсульфпт (2) II продукт
его разложения (3) являются, по-видимому, промежуточными.
N-Метил производное (1) устойчиво к действию Т. х.
1. Contributed by F г i с d in a n L., Wetter W. P., Case Institute of Techno-
logy.
2. К e ii з о н Дж., «Синтезы органических препаратов», ИЛ, AL, 1952, сб. 3,
стр. 459.
3. Бишоп В., «Синтезы органических препаратов», ИЛ, At, 1952, сб. 3,
стр. 312.
4. Durha m L. J., McLeod D. ,L, Cason J., Org. Syn., Coll. Vol.,
4, 556 (1963).
5. В ома к Э., Мак-У п р т о р Дж., «Синтезы органических препаратов»,
ИЛ, At, 1952, сб. 3, стр. 442.
6. К о я ь м э н Дж., II и к о л ь с Г., М а к-К л о с к и Ч., А и с п о и Г.,
«Синтезы органических препаратов». ИЛ, АТ, 1952, сб. 3, стр. 458.
7. Март и п В., Физе р Л., «Синтезы органических прела pa юв», ИЛ,
AL, 1949, сб. 2, стр. 448.
8. А л л с и Ч., Б a ft е р с Дж., мл., X у м ф л с т т У., «Синтезы органичес-
ких препаратов», ИЛ, At, 1959, сб. 9, с гр. 70.
9. Buckles R. Е., Cooper J. A., procedure submitted to Org. Syn.
10. S c li w с n k E., Papa D., J. Am. Сйсчп. Soc., 70, 3626 (|948),
11. Radin N. S., private communication.
12. X и p с т II., 11 о л л e p К., «Синтезы органических препаратов», ИЛ,
Al., 1953, сб. 4, стр. 296.
13. Курцер Ф., «Синтезы органических препаратов», ИЛ, At, 1956, сб. 6,
стр. 81.
14- Brenner AL, Huber W., Helv. Chim. Acta, 36, 1109 (1953).
15. U h 1 e F. G, Harris L. S., .1. Am. Clicm. Soc., 78, 381 (1956); U Ii I c
F. G, J. Org. Clicm., 27, 4081 (1962).
16. Patel R. P.. Price S., J. Org. Chem., 30, 3,575 (1965).
17. К и p h e p В., У и и д у с В., «Синтезы органических препаратов», ИЛ, At,
1949, сб. 2, стр. 562,
18. И л и э л в Э., Ф и с к At, Прос с е р Т-, «Синтезы органических препа-
ратов», ИЛ, At, 1958, сб. 8, стр. 75.
19. D а г z е ns (i., Compt- rend., 152, 1601 (1911).
20. Frazer At J Gerrard W., Al ache I 1 G., Shop h e r d B. D.,
Chem. Ind., 1954, 931.
21. Брукс Л., С и a it д e p X., «Синтезы органических препаратов», ИЛ, At,
1952, сб. 3, стр. 374.
22. Org. Expts., 219.
23. F i c s с r L. F., Okumura Y., J. Org. Chem., 27, 2247 (1962).
24. О kumura Y., J. Org. Chem., 28, 1075 (1963).
25. К P и и и u к и ii Дж., К a p x a p т Г., «Синтезы орыпшческих препара-
тов», ИД, At, 1949, сб. 2, сгр. 136.
26. Vaughan W. R., Carlson R. D-, J. Am. Chem. Soc., 84, 769 (1962).
27. Cvrcp К., Герхарт X., «Синтезы органических препаратов», ИЛ, At,
1949, сб. 2, стр. 136.
28. Evans R. Г., Brown Н. С., van d с г Р I a s И. С., Org. Syn.,
43, 97 (1963).
29. В ii с li i G., Lu k a s G., J. Ain. Chem. Soc., 86, 5654 (1964).
H s
I .
ТИОСЕМИКАРБАЗИД, FLNN—CNHa. Мол. вес 91,14, т. пл.
180—181° (с разл.).
Получение гетероциклов, см. 1,2,4-Триазол.
335
ТИОУКСУСНАЯ КИСЛОТА, CH3COSH. Мол. вес 76,12, т. кип.
87° (347'100 мм), уд. вес 1,07.
Получение. Реагент получают обработкой уксусного ангидрида
сероводородом (до небольшого привеса) в присутствии каталити-
ческих количеств порошкообразного едкого натра [11.
О
6 час прн ПО -53° Г
(CJI3CO)2O-|-HsS^NaOH ----------------> CH3CSH | CH:tCOOH
I моль 1
Радикальное присоединение к олефинам [21. Хольберг [31 и
Ипатьев [41, исследовавшие эту реакцию, нашли, что Т. к. присое-
диняется почти исключительно против правила Марковникова:
3 час при [00е
CH.,-C(CH3b J-CH3COSH ....CI-I:,COSCH..CH(CHs).,
ь о0 D
Выход увеличивается при добавлении нескольких капель аскари-
дола или небольшого количества перекиси бензоила [5]. Бордуэлл
и Хьюэтт [61 показали, что в основном происходит транс- при-
соединение. Например, присоединение Т. к. к 1-метил-
циклогексену и 1-метилциклопентену с последующим щелочным
гидролизом до тиола в каждом случае протекает с преобладаю-
щим образованием продуктов /щс-конфигурации.
15%
30%
Радикальное присоединение Т. к. к терминальным олефинам
является основной стадией в новом методе получения алкилсульфо-
кислот [7). Тиоацетат высокой чистоты получается с хорошим выхо-
ду, !)0%-Н,О .-(лцеоон. ми
RGii-CH,-rc.H3cosH —RCH3CH.,SCOCH3-------------------——--------------
5 j - !Л % 'ЛЩ
R(nk(:ii.,so;(n
дом н окисляется до сульфокислоты под действием надуксусной кис-
лоты, получаемой in situ из перекиси водорода и уксусной кислоты.
Радикальное присоединение к алкинам [21. Т. к. энергично
реагирует с алкинами, образуя моно- и диаддукты. В некоторых
случаях выход можно повысить облучением или добавлением пере-
336
киси, однако более важная роль инициирования — это улучшение
воспроизводимости эксперимента. Реакция позволяет превра-
щать терминальные ацетилены в альдегиды [81. Эта реакция явля-
ет 2 моля (л ,<:os11
h-CJLjO- СН ———------
О,2 моля
О
I'
— * я-С411аСН - С -SCCH, - н-С4Н.)СН - CH..SCOCH,
1 I
Н SCOCH:i
5J% i9%
ВТ1’,', | H.NM-ICOxNIIj
н-С41 БОСС—XNIICON’H,
H
ется ключевой стадией в общем синтезе линолевой кислоты по Валь-
борскому [9],
СН ,COSH II.AOH
IIC - С(СН6С - СН ----------ПС - С(С11 >)6С11 - CI ISCOCH.J ———’
° носнюнюн .O-GH./
_НС-_2C(CH2)7CIU-NOH ------НС=С(СН,)7СН |
Х О —СП,
С.НДЦВг zO —СН„
(.]],,(. = осн .Вг 1 " н % Анкет
— ---------— -> QHUC- -ССН..С -СДСНщСН ——”—
5з<';} а 11 “ “'щ I зз'.'о
' О —СП,
H N i
—- СДИпС- ССКС -- С(СН„)7СО,П ———*
н н н’н ’ ”
I ! I I
__ СН3(СН,)4С ССН..С -- С(С11,)7СО,Н
Бломквист и Волинский [101 запаивали в ампулу из стекла
пирекс смесь 20 г пропаргилового спирта, эквивалентного коли-
чества Т. к. и 0,1 г перекиси бензоила и облучали в течение ме-
сяца. При перегонке получили 30 г смеси дитиол ацетатов и, воз-
можно, тритиол ацетатов, кислотный гидролиз которой дает 2,3-ди-
меркаптопропапол (БАЛ) с т. кин. 86—9071 мм (в атмосфере
азота).
но
СН- -ССКОНД-СНуСОЗН Смесь-----------> СН.,СИСН.,О11
1 i
SH SH
Присоединение ка, ^-ненасыщенным карбонильным соединениям.
Эта реакция является важной стадией синтеза липоевой кислоты.
Кетон (2) нагревали [11) сТ. к. на кипящей водяной бане в течение
337
20мин и получили продукт 1,4-прпсосдпненпя (3) с высоким выходом.
В сип юзе по Мерку 112] реагент добавляют к аф-пенасыщенной
О
С2Г1-О.2С(СН2)4СОС1 J-CI-L-СН2 C2l-l,5O.JC{CH01CCH-~-CH2
(I)
о о
СоНьОдхсн.лсснхн.зссНз —i
91%
Ы)
о
—> C.,H-jO.,C(CH.I)iCl-ICH.>CH.,SCCH3
1
он
11 )
—4 ногс(снг)4снснгснг
ОН SH
(О
11 HI + 5=С(МНгЦ
2) ^аОН-------> ногс(снг)4снснгснг
SH SH
(61 (7)
кислою (8) и выдерживают 17 дней при комнатной гсмисратх ре.
Разделение (ч=)-лппоевой кислоты не удалось провести полностью,
и поэтому преимущество синтеза по Мерку состоит в том, что разде-
ление можно проводить на рапипх стадиях, например аддукт (9)
разделить с /-эфедрином.
С2Н7,О2С(СП2)4СН-СНСО2Н C2H7j02C(CHJ.jCHCH2c0..H—►
OS) !П% Ы) I
SCOCII3
—C2H5O,C(CH2)4CHCH2COCI iLkklk
HOoC(CH2)4CHCH.,CH.,OH — --
j XjOH
SH
(111
> СНгСНгСН(СНг)4СОгН
SH SH
(12)
(СН2)4СОгН
(13)
Присоединен не Т.к. к а, ^-ненасыщенным 3-кетостероидам исполь-
зуется для получения соединений с повьппенпойбиологической актив-
ностью (облучение при этом необязательно 1131). Например, Д1Ч-3-
кетостеронды дают 1а-ацетнлтио-Л]-3-кетостероиды; А-1 ,(;-3-кето-
стероиды образуют 7а-а цетил тио-А [-3-кетоны.
Замещение брома. Боннер 114! обрабатывал тетр а а цетил-а-о -
глюкозплбромпд в хлороформе действием Т. к. и едкого кали (оба
с 10%-ным избытком) и получил с хорошим выходом иеитаанетпл-!-
тпоф- D-глюкозу.
Вт
S-Ацетилированис. Т. к. пли водный раствор ее натриевой соли
был использован для Б-ацетилировання кофермента А 1151 п пан-
тетеина 1161. Группа исследователей 1161 растворяли 10 г пантетеина
в 60.ил воды, добавляли 20 .ил Т. к. (большой избыток) и оставляли
смесь при комнатной температуре на ночь; за это время смесь ста-
СН3 ОН О Н о
1 । I li ('ll CSH
носим:-----СНС - NCHaCH,C - ХСЩСН.ЭН —
| I - "
сн;) ОН
11 ;i нтетеин
CH.J он он о
I 'I .1
—.> HOCHX— СНС — NCILCHX — NCH.1CH2SCCH3
1 I
сн;! он
З-Лцетилпантстеии
повилась гомогенной. Концентрирование при пониженном давле-
нии дает S-ацет пл пантетеин — вязкое светло-желтое масло ана-
литической чистоты. Выход количественный.
1. Эллпигбо Э., лСпнтсзы органических препаратов», ИЛ, М., 1953, об. 4,
стр. 471.
2. У о л л и и г Ч., «Свободные радикалы в растворах», ИЛ, М., 1960.
3. II о 1 b е г у В., Arkiv. Kemi Alin. Gcol., 12В, № 17, 3 (1938).
4. Ipatieff V- N., Friedman B. S., J. Am. Clicm. Soc., 61, 71 (1939).
5. Brown R., J on es W. E., Pinder A. R., J. Chem. Soc., 1951, 2123.
6. В о r d we I I F. G., II c. w e t t W. A., J. Am. Clicm. Soc., 79, 3493 (1957),
7. Showe 11 ,f. S., Russell J. R., S w e г n D_, ,1. Or^. Chem., 27, 2853
(1962).
8. В a d с г II., Cross L. С., II ci 1 br on 1., J ones E. R. IE, J. Chem.
Soc., 1949, 619; Bchrini’er H., Ann., 564, 219 (1949).
9. \V a 1 b (> r s k у II. M., Davis R. H-, llowton D. R., J. Am. Chem.
Soc., 73, 2590 ('1951).
339
10. В I о iTi q и i s t А. T., \V о I i n s к у J., J. Org. Chem., 23, 551 (1958).
11. Bullock M. Brockman ,1. A., Jr., Patterson E. 1..,
Pierce J. V., von Sal iza M. H., Sandor s Г., SI oksta d
E. L. R., J. Am. Cheni. Soc., 76, 1828 (1954).
12. Walton E., \V а и n e г A. F., Bachelor F. \\r., Peterson
L. IE, 11 о 1 1 у F. \V., Folkers K., J. Am. Chem. Soc., 77, 5144 (1955).
13. Dodson R. At, Twci t R. C., J. Am. Chem. Soc., 81, 1224 (1959); Celia
J. A., I weit R. C., J. Org. Chem., 24, 1 Ю9 (1959).
14. Bonner W. A., J. Am. Chem. Soc., 73, 2659 (1951); see also Horton D.
Wolfrom At L., J. Org. Chem., 27, 1794 (1962).
15. Wilson J. B., J. Am. Chem. Soc., 74, 3205 (1952).
16. Walton E., Wilson A. N., Holly F. W., Folkers K. J. Am.
Chem. Soc., 76, 1146 (1954).
ТИОУКСУСНОЙ КИСЛОТЫ КАЛИЕВАЯ СОЛЬ, CH3COSK-
Мол. вес 114,21. Получение Ill.
Чепмеп и Оуэн [21 предложили реакцию первичных тозилатов
с Т. к. к. с. в качестве удобного метода получения тиолацетатов и
последующим гидролизОхМ — тиолов. При взаимодействии тетра-
ги дрофу рфурилтозплата с Т. к. к. с. в течение 1 час в кипящем эта-
ch3cosk
CHzOTs 69-71%
КОН
55%
ноле или ацетоне образуется тиол ацет ат с хорошим выходом. Со-
ответствующий мезилат при реакции в кипящем ацетоне в течение
1 час дает этот эфир с выходом только 20%; при кипячении в тече-
ние 6 час выход возрастает до 60%. Вторичные тозилаты реагируют
иначе.
1. Ulrich С., Ann., 109, 272 (1859).
2. С hapman J. Н., Owen L. N., J. Chem. Soc., 1950, 579.
ТИОФЕНОЛЯТ НАТРИЯ, C(iH5S’Na+. Мол. вес 142,17.
Реагент — активный нуклеофил, хотя и является более слабым
основанием, чем этилат или фенолят натрия Ш. Шихан и Дэйвс
[21 использовали Т. н. в ДМФА при комнатной температуре (пли
ниже) для расщепления активной сложноэфирпой группы ( в основ-
ном фенацильной) по С — О-связи с образованием натриевых со-
лей соответствующих карбоновых кислот с прекрасным выходом,
как это видно на примере фенацилбензоата. Реагент получают при
добавлении тиофеиола к эквимолярному количеству натрия, тонко
диспергированного в эфире, смесь перемешивают в течение 72 час,
О О О
С,Д5СаиО—ССГ,П5 I 2C„H5SNa CfitI5('.CH2SC6H,, | C(iH-CO.,Na
! моль 2 моля 3 .чин ь“%
соль отделяют фильтрованием и хранят в вакуумэксикаторе. Реа-
гент можно использовать, в частности, для удаления защитной сло-
жпоэфирной группировки в синтезах ряда таких чувствительных к
340
воздействиям соединений, как пенициллины и пептиды. Например,
под действием Т. и. избирательно удаляется фенацильная сложно-
эфирная группировка в молекулах, содержащих высоколабильную
фталимидную систему.
Т. п. является, по-впдпмому, лучшим реагентом для избиратель-
ного деметилирования четвертичных аммониевых солей [31. В ка-
честве растворителя при этом используют метил этил кетон. Реакция
осуществляется по механизму 5\2 с атакой тиофенолят-анионом
N-метилыюй группы, как, например, при деметилировании хлор-
метилата (+)-лауданозина в лауданозин. Реакция осложняется при
наличии сложноэфирной группы, которая превращается при этом
в тиофениловый эфир соответствующей карбоновой кислоты [21.
1, М а г е de la Р. В. D., Vernon С. A., J. Chem. Soc., 1956, 41.
2. S heehan J. C., Daves G. D., Jr,, J. Org. Chem., 29, 2006 (1964).
3. Shamma M., Deno N. C., Rcmar J. I'., Tetrahedron Letters, 137,5
(1966).
ТИОЦИАНАТ НАТРИЯ, NaSCN. Мол. вес 81,09.
Для получения изопропилтиоцианата [1] к суспензии Т.н. в
Кипячение 6 час,
(СН;})аСНВг [-NaSCN J2’50 50 90^1,иго Et?H (CH3)2CHSCN
7 В_7 по/
В молен мо;ы 1 и J /о
90%-ном этиловом спирте при перемешивании и кипячении прибав-
ляют в течение 1 час изопроиилбромид, затем смесь кипятят при
перемешивании еще в течение 6 час. Хлористый натрий удаляют
фильтрованием, большую часть этилового спирта отгоняют, добав-
ляют воду и продукт экстрагируют эфиром.
Применение реагента для получения гетероциклических соеди-
нений иллюстрируется синтезом 2-а ми ио-6-мет ил бензтиазол а [21.
Конц, серную кислоту добавляют по каплям при перемешивании к
раствору п-толуидина в хлорбензоле и к образующейся тонкой сус-
пензии п-толуидинсульфата прибавляют тиоцианат натрия. При наг-
ревании смеси в течение 3 час при 110° образуется /г-толилтпомоче-
вина и сульфат натрия. Смесь охлаждают до 30°, обрабатывают при
30—50J хлористым сульфурилом и нагревают при 503 еще в течение
2 час. Продукт отделяется в виде хлоргидрата; его собирают и раст-
341
н
1 м о л ь
вор я ют в воде. После перегонки с водяным паром для удаления остав-
шегося хлорбензола продукт осаждают гидроокисью аммония и
кристаллизуют из этанола.
I. Ill р a й н е р Р., «-Синтезы органических препаратов», ИЛ, ХУ, 1919, сб. 2,
стр. 2GS.
2. А л л с п И.. В а в-Л л л а и Дж., «Синтезы органических препаратов», ИЛ,
М.. 19,52, сб. 3, стр. 49.
ТИТАН ЧЕТЫРЕХХЛОРИСТЫЙ, TiClМол. вес 189,73, т. кип.
136 , уд. вес 1.726.
Реакция Фриделя — Крафтса. Реагент менее активен по срав-
нению с хлористым алюминием. Например, бензол плохо алкили-
руется первичными алкплгалогенндами 111. Однако Т. ч. является
эффективным катализатором алкилирования более активными тре-
тичными алкплгалогенндами и более удобен, так как растворим в
органических растворителях. При взаимодействии бензола с трет-
С(СН;1)3 C(CII3):J
С0НйрЩСН3)3 СС1 ———----
моли ПР"
С(СН3)3
бутплхлоридом в течение 6 час при 10° образуется в основном л-ди-
///ре/л-бутилбензол и немного ш/лдл-бутплбензола. В тех же усло-
виях толуол реагирует в течение 1—3 час, давая л-тр£/?г-бут ил то-
луол с выходом 77%.
Реакция Фриса. Т. ч. применяется как катализатор в перегруп-
пировке Фриса — превращении фениловых эфиров в орто- или
ларл-оксикетоны 12, 3]. Например, перегруппировка л-крезилаце-
тата в нитробензоле осуществляется с высоким выходом 13].
ОСОСН;
он
) ХОСП;
сн3
сн3
342
Под действием Т. ч. Тауб и сотр. (41 осуществили перегруппи-
ровку фенилового эфира (I) в бензофенон (2), Хлористый алюминий
в этом случае приводит к распаду (1) на компоненты, а наибольший
выход (2) составляет 5%.
/ н О ОСН;} _/~°Н
v '' . х __QH Ti(.[4____
CIi3O/ ° У------------------"ДЩДД СН3О OIlUki
Cl Н/7 Cl
fl) {-J
Реактивы Гриньяра. Финкбейнер и Купер [51 обнаружили, что
в присутствии небольших количеств Т. ч. пзопронплмагнийбромпд
в эфирном растворе изомеризуется в н-пропплмагнийбромид. Они
предположили, что реакция включает стадии элиминирования п
присоединения с образованием пропилена, который обменной реак-
цией с реактивом Гриньяра превращается в «-нрошглмагннйбромпд.
Это подтверждается новым методом образования реактива Гринь-
яра, реакцией катализируемого обмена терминального олефина с
н-прон ил магний хлоридом, который в присутствии Т.н. не изомери-
зуется. Действительно, при кипячении в течение 18 час эфирного ра-
створа 4-метилпентена-1 с эквивалентным количеством н-пропилмаг-
нпйхлорида в присутствии 0,03 же Т. ч, с последующей! обработкой
ацетальдегидом получают 6-метилгептанол-2 с выходом 37’4. В 10
опытах выход составлял 22—60%. Внутренняя двойная связь в
реакцию нс вступает.
СН:1 СН:!
I 11С.Ц I
CH3CJ]Cl-l2CH_Cii2H-«-PrAlgCl--------- CI iyCl 1CI L.ClCCl J._,MgCl
ch.,ciio
CIi3
I
CH-jCHCI J.,Cl I..CH.,CHC1I3
' ;,h '
Четыреххлористый титан — триэтилалюминий, TiClj— (СаН.-,);1А1>
так называемый катализатор Циглера S.6I, используется для поли-
меризации «-олефинов, Кроме того, он является катализатором пик-
лотримернзацшр ацетиленовых производных в производные бен-
зола 17]
3RC=CR'
T1CJ, ™ (СДЦЦА!
и Л'си । а и. 4 3°.
I ‘2 чеи
20—80%
R\/' /R'
hi
R/ 'R
R'
343
Восстановительная конденсация спиртов. Ван Тамелен и Шварц
18] использовали Т. ч. в реакции восстановительной конденсации
спиртов с образованием углеводородов: 2ROH^RR. Спирт под дей-
ствием гидрида натрия превращают в алкоголят (1) и затем в присут-
ствии Т. ч. в промежуточный продукт (2), который восстанавливается
ROH -A RONa 217% (RO)2TiCI2 A (RO).Ti 2А.2.А RR ( TiO2
(1) (2) (3} (4)
металлическим калием до диалкоксититана (3), При нагревании до
100—140 (3) превращается в углеводород и двуокись титана. При
этом выход дифенилэтана из бензилового спирта составляет 51%.
1. С u I 1 i n а п е N. М., Leys h on D. AL, J. Chem. Soc., 1954, 2944-
2. С и 1 1 i n а п е N. At, L I о у d Е. Т., Т u d b а 1 [ J. К., J. Chem. Soc.,
1954, 3894.
3. С и 1 1 i n а п е ГУ. At, Edwards В. F. R., J. Chem. Soc., 1957, 3016;
Cullinane N. At, Woolhous e R. A., Edwards B. F. R., ibid.,
1961, 3842.
4. Taub D., J и о С. H., Slates ILL., Wendler N. L., Tetrahedron,
19, 1 (1963).
5. Finkbciner H. L., Cooper G. D., J. Org. Chem. 26, 4779 (1961).
G. Natla G., Pasquori I., «Advances in Catalysis and Related Subjects»,
II, 1 (1959); Гейлорд H. Г., Марк Г. Ф., Линейные и стереорегулярцые
полимеры, ИЛ, М., 1962.
7. Lutz Е. F.. J. Am. Chem. Soc., 83, 2551 (1961); Н о р I I IL, G a t i A.,
Helv. Chim. Acta, 48, u09 (1965).
8. T a m e 1 e n E. E., van, Sch w a r t z AL A., J. Am. Chem. Soc., 87, 3277
(1965).
N -ТОЗИЛ ИМИ НОУ КС УСНОЙ КИСЛОТЫ н-БУТИЛОВЫЙ
ЭФИР, rt-CH3CeH4SO.,N -СНСО,С4Н9-«. Мол. вес 283,35, т. кип.
130 —135710”3 мм.
Получение. Нагревают до кипения смесь «-бутил гл иоксилата
(Пи N-сульфин ил-/г-толуол сульфамида в бензоле и медленно при-
бавляют раствор хлористого алюминия в нитробензоле [2]:
— SO;
n-CH3C((H,SO2N SO ! О-СНСОХШэ-н —->
«7%
n-CH3C6H4SO2N=-CIiCO.2C4HrH
Диенофил Дильса— Альдера (2]. Этот реагент образует аддукт
с 2,3-диметилбутадиепом (выход не указал) при кипячении смеси
компонентов в бензоле в течение 10 час. Охмыление аддукта дает
4,5-диметилпиридин-2-карбоновую кислоту с низким выходом.
Нзс, .SOoCeHjCIR-n
' Кипячение
I 1 [зН Сапа
Нр' х \
4,1 J CO.CJI^H
н.;Сх /ч .so^HjCiR-/!
- II г
НзС/ ^ХОДН^
344
С2Н0ОН - КОН
H3C/Z х^хсо.дн
ТОЗИЛ ХЛОРИСТЫЙ, см. п-Толуолсульфохлорид.
1. W о 1 I Г. J., W е i j 1 ti г d J., Org. Syn., Coll. Vol., 4, 124 (1963).
2. A 1 b r e c li t R.T К r e s z e G., chem Ber., 98, 1431 (1965).
ТОЛЛЕНСА РЕАКТИВ Hl.
Определение восстановительной активности f2], В пробирке
к 2 л/л 5%-ного раствора азотнокислого серебра добавляют 1 ль?
10%-ного раствора едкого натра для осаждения окиси серебра.
Готовят разбавленный раствор аммиака, смешивая 1 мл конц. раст-
вора аммиака с 10 мл воды. Добавляют 0,5 мл этого раствора к оса-
дку окиси серебра, закрывают пробирку и встряхивают. Повторяют
эту операцию до полного растворения осадка (3 мл, следует из-
бегать избытка) и раствор разбавляют до объема 10 мл.
Пять пробирок промывают 10%-ным раствором едкого натра
при нагревании па водяной бане и затем дистиллированной водой.
В пробирки помещают по микрокапле 0,1 М раствора сахара и н-
масляного альдегида и no 1 мл приготовленного раствора Т. р. Реак-
цию проводят сначала при комнатной температуре, оценивая актив-
ность но изменению цвета и по времени появления серебра. Через
несколько минут пробирки помещают на горячую баню. Наблюдает-
ся следующий порядок изменения активности: фруктозау>глюкозау>
>лактоза у> мальтоза>н-масляпый альдегид.
Т. р. используют для окисления ретинина в кислоту, соответст-
вующую витамину А (выход 50%) [31.
1. То! lens В., Вег., 15, 1635 (1882).
2, Org. Expts., 132.
3. Bar u a 1<. K-, Barna IL B., Biochem. J., 92, 21C (1964).
ТОЛУОЛДИ ИЗОЦИАНАТ, CH3CeH3(N-C=O)a [1]. Мат. вес
174,16. Продажный продукт представляет собой смесь (80 : 20)
2,4- и 2,6-изомеров с т. кип. около 240°.
Реагент применяют для получения алифатических изоцианатов
обменной реакцией с алифатическими изотиоцианатами. Оп, в част-
ности, очень удобен для получения аллилизопианата и металлил изо-
цианата, так как соответствующие изотиоцианаты легкодоступны.
Другие известные способы синтеза аллилизоцианатов менее удобны.
Получение аллилизоцианата. В колбе, снабженной колонкой со
стеклянной спиральной насадкой, I же алл ил изотиоцианата раство-
ряют в Заке Т. и смесь нагревают при 190—210°. Реакция осуществ-
ляется через промежуточное образование уретидинтионона; отгонка
летучего аллилизоциапата способствует сдвигу равновесия в желае-
мом направлении. Колонка работает при полном орошении, пока в
345
CH2^CHCH2
CH, - СНСНЛ = C o- SCN — "_____- CH3
T. кип. 87—8‘J°
головке колонки не образуется достаточное количество а длил изо-
цианата, который затем отбирают. Реакция становится экзотерми-
ческой при температуре выше 210 \ особенно на стадиях развития
реакции обмена, и ее прекращают, как только обнаруживают разог-
рев.В реакционной смеси образуется ограниченно растворимый поли-
мер, Выход аллилизоцнаната 23'\). Металл ил изоцианат (т. кин.
107—109 ) получают с выходом 31 %.
1. Cf tn! г i but ed by Erncr W. E., Air Products and Clicinicals, Inc.
бИс-(н-ТОЛУОЛСУЛЬФАМИД)-РТУТЬ, (л-Cl I;ICJ1 jSO,NH),Hg.
Мол. вес 241,03. Реагент получают нагреванием 2,1 .моля п-толуол-
сульфампда и 1 моля окиси ртути при 195—200’ в течение 2,5 час,
измельчением в порошок охлажденного продукта и экстракцией
избытка амида этанолом 111.
Реагент используют для гидратации стероидных 17-этииилкар-
бпнолов типа 1, для которых обычные методы дают плохие резуль-
таты 111, Например, смесь 1 с соединения I, I, 80 г реагента и 50 мл
95%-ного этанола кипятят в течение 72 час и для осаждения ртути
раствор обрабатывают сероводородом. После фильтрования из раст-
вора выделяют 1 г 11.
346
I. Goldberg M. W., Acschbachcr R., Her de ggcr E.( Helv-
Cliini. Acta, 26, G80 (1943).
/2-ТОЛУОЛСУЛЬФОКИСЛОТА, /z-CH3C6H4SO3H -H2O(TsOH •
• H,O). Мол. вес 190,22, т. пл. 105\
Т.— эффективный кислотный катализатор, как и серная кислота,
но более удобна, так как в меньшей степени разрушает реагенты н
является твердым веществом. Т. выделяют, разбавляя реакционную
смесь небольшим количеством воды и добавляя конц. соляную кис-
лоту для уменьшения растворимости; при охлаждении кристалли-
зуется в виде моногидрата.
Этерификация. у-Хлорпронплацетат Ш получают нагреванием
компонентов в бензоле с небольшим количеством Т. в колбе, снаб-
СН3СО3Н + НОСН2СН.,СН3С1 й- TsOH 11,0 4- С6Н6
3 моля 2 моля 2 г .300 мл
—> СН3СО,СН,СН,СН,С1
женной колонкой Вигре, соединенной с конденсатором и автомати-
ческим сепаратором для возвращения в реакционную колбу бо-
лее легкой фракции 121; применяют также колонку Clarke — Rahrs
]3|, Азеотропную перегонку проводят в течение 7—9 час. Колонку
Clarke — Rahrs используют при получении метилового эфира пиро-
[ 30-
СН;}СОСО,Н 4-CH3OH-|-TsOH H,O4-CBH(. —_ > СН3СОСО,СН3
I моль I моль 0,2 с 350 Л/.1 (Л—71%
виноградной кислоты (перегонка в течение полутора дней) 14]. Пре-
вращение цис- Д4-тетра гидрофтал евого ангидрида в соответствующий
диэтиловый эфир осуществляют кипяченном реакционной смеси при
95—105° в течение 12—16 часе последующими добавлением 270 мл
толуола,азеотроппой перегонкой,добавлением этанола и кипячением;
после этого добавляют толуол и проводят азеотропную перегонку [5].
н
сгн5он 4*
9 молей
1, 5 моля
Тбон- гн2о
2 г
согс2н5
\xSco2c2h5
н
Переэтерификация. При получении н-бутилакрилата [6] из
метилакрилата в качестве ингибитора полимеризации добавляют
гидрохинон. Смесь нагревают сначала при полном орошении, отго-
СН, = СНСОгС113^ч-ВиОН4- TsOH-2Н2О —► СН2^cнCO,Bu-«
10 молей 5 молсп К) г 0
няют азеотропную смесь метанола и метилакрилата по мере ее обра-
зования, а затем (через 6—10 час) метилакрилат и н-бутил акрил ат.
447
Этерификация еиола. 3-Этокси-А3-циклогексенон, енольный эфир
дигидрорезорцнна, получают аналогично при кипячении с последую-
щей азеотропной перегонкой в течение 6—8 час [7].
ONa О
I II
Z-x Н. — Ni, /\
\ II НС] I II
I ii || + Абе. СЛОН
Жн хОн
0,4 7 2 моля 23 0 .ил
О
II
4 TsOIi-HaO-| C6H6 —-----+ I II
2 3г 900 м.1 66—68,5% ч
ЧОС2Н5
Получение кеталей. Превращение глицерина в нзопрогшлиде-
повое производное (ацетонид) осуществляют кипячением с водоот-
делителем при перемешивании смеси компонентов и катализатора
в 300 мл петролейного эфира (т. кип, 35—55°) в течение 21—36 час
[8]. Смесь нейтрализуют ацетатом натрия, фильтруют, упаривают
и продукт перегоняют.
СП.,ОН
СН3ч I ' 35-3.7,- СПз П-СН.,
;С-0-!СНОН -pTsOHH.,O—С; |
СНз7 | за' СН/ Ч)-СН
СП,ОН |
4,09 моля 1,09 моля ClIaOH
Превращение А4-3-кетостероида в этилен кеталь сопровождается
миграцией двойной связи из 4,5- в 5,6-положение 19]. В реакции с
холестенономЦО] смесь реагентов в указанных количествах кипятят
при перемешивании в течение 4,5 час (с постоянным удалением во-
ды). После охлаждения смесь обрабатывают насыщенным раствором
31, 8 г
снгон +
СН2ОН
40 Л1Л
бикарбоната натрия, продукт выделяют из бензольного слоя и крис-
таллизуют. Выход приведен для продукта с т. пл. примерно на 2°
ниже необходимой п очень низок. Возможное объяснение этому можно
найти в последней работе Петерсена и Соуерса [11], которые изу-
чили влияние количеств взятого катализатора в реакциях с 1,02 г
холсстенона. Чистый, пе подвергшийся перегруппировке этилен-
кеталь А4-холестеиона-3 удалось получить с выходом 77% только в
присутствии 0,0052 с моногидрата Т. Для получения продукта пере-
348
группировки — этиленкеталя А5-холестенона-3 — достаточно увели-
чить количество катализатора до 0,05 г. Поскольку ранее упомя-
нутые исследователи использовали промежуточное количество ката-
лизатора, это, видимо, и привело к образованию смеси двух кеталей,
t Аналогичным образом Т. используют для получения этилентио-
кеталей. Реагент — более мягкий катализатор, чем эфират трехфто-
ристого бора в конденсации кетона с этилендитиолом, и исполь-
зуется в уксусной кислоте для селективного превращения, напри-
мер, А+-андростептриона-3,Н,17 (0,6 г) в 3-этиленкеталь (0,40 г)
[121.
Ацетилирование. Холестантриол-3|3,5а,6|3 при ацетилировании
уксусным ангидридом в пиридине дает 3,6-диацетат; триацетат мо-
жно получить непродолжительным нагреванием триода с уксусным
ангидридом п Т. при 100° [131. Многие стероиды, содержащие 5|3-
гпдроксильную группу, не поддаются ацетилированию всеми извест-
ными методами. Тернер [14] и группа других исследователей [151
независимо друг от друга установили, что третичный 17а-гидрок-
сил стероидов легко ацетилируется при комнатной температуре ук-
сусным ангидридом в присутствии Т. в качестве катализатора.
По этой реакции легко получить 17сс-ацетоксипрогестерон — ши-
роко применяемый прогестогенный препарат для приегла рсг os.
Енолацетилирование. При разработке общего метода синтеза
а-бромкетонов Бедокян [161 искал катализатор для превращения
кетонов в енолацетаты. Ацетат калия, фосфорная кислота и серная
кислота в различных концентрациях не дали положительных резу-
льтатов, однако Т, оказалась вполне приемлемой.
349
ОСОСНз
TsOH-H.O |
н4ХНяСНХОСН3-у (CH3CO)2O-----—н-С4Н9СН -ССНз —►
ОСОСНз ‘ о
Вг, I Сг 1лО11
---нЛ^НдСНССНз -------------> н-С4Н9СНССН3
cci, । ( ' j
Вг Вг Вг
Указанный метод енолапетплпровапня имеет большое значение
в ряду стероидов, например в синтезе 17а-оксикортш<оидов по Гал-
лахеру |17|, включая кортизон 1181, по схеме (1)-^(4). Этот метод,
подробно описанный только для мопокетонов, состоит в следующем
[191, Раствор 2 .нмолей кетона и 2 .нмолей Т. в 75 мл уксусного анги-
дрида медленно перегоняют па незаполненной колонке, пока не от-
топится основное количество уксусного ангидрида (4—5 час). После
охлаждения и добавления воды продукт экстрагируют эфиром и
хроматографируют; выход 60—70%.
Енамины. Получая енамин из .морфолина и циклогексанона,
Хюппг и сотр, [201 проводили азеотропную перегонку с водоотде-
лителем, описанным Нательсоном и Готтфрпдом [21Г Морфолин
0 0
I I - I | .rTSOH.H.O + Csll3CH3-My^^-. %
\ ч 1 1,о мо;1я 300 .кл (! 1
1,и моля 1,3 моля
350
берут в избытке, так как отделяющаяся вода содержит значитель-
ное количество этого амина.
Дегидратация. Джонсону с сотр. 1221 при определении разности
энергий конформаций «кресло» п «ванна» циклогексана требо-
вались изомерные лактоны (2) п (4), и поэтому было необходимо син-
тезировать у-о к с и кислоты (1) н (3). Лактон получали из (1) путем
кипячения раствора I г (1) и 0,4 г Т. в 600.ил бензола в атмосфере
азота в течение 10 мин при перемешивании и сборе дистнллата в во-
доотделителе. По завершении операций и кристаллизации получали
почти количественный выход лактона (2). Этот метод нельзя при-
менять к транс-диаксиальнон оксикислоте (3), которая дает лактон
лишь в том случае, когда одно из колец имеет неустойчивою конфор-
мацию «ванны» (4). Этот же катализатор неэффективен в кипящем
толуоле, однако при замене толуола на ксилол выделяется вода
и лактон (4) получается после хроматографии с выходом 73(|».
Цпклодегидратацшо бутаптрпола-1, 2, 4 до 3-оксптетрагидрофурана
проводят при нагревании триола с Т. в установке с колонкой для
вакуумной перегонки 1231, После перегонки (2—2,5 час) получают
300—306 г продукта с т, кип. 85—87 /22 мм. Вторичная перегонка
на том же аппарате дает фракцию весом 50—60 ?, кипящую при
42—44724 лкп н содержащую в основном воду, незначительную про-
межуточную фракцию и 215—231 г чистого продукта с т. кип. 93—
95726 мм.
Перегонка
НОСН2СН2СНСН2ОН + TsOH' Н2О Р ракУУМе»
<1Н 81-86%
3 моля 3 г
351
Т. в кипящем бензоле — наиболее подходящий реагент для
дегидратации эфиров кетоспиртов (1) [241. Бенкерт и Стивенс [251
сообщили в общих чертах, что при дегидратации кетоспирта (3) в
(4) ими использовалась смесь Т. и хлористого кальция; выход про-
дукта (4) 170 мг. Установлено, что смесь Т. и хлористого кальция
является наилучшей для дегидратации (5), так как в присутствии
только Т. происходит ароматизация кольца А [261.
(3) 200
В 25 мл
TsOH- НгО'
Кипячение
В
TsOH - Н2О
100 №
ТеОН-СаСЦ-СЦЩ
74% **
Изомеризация. Айс л ер и сотр. [271 сообщили, что при кипячении
100 г оксиизофорола (2) и 2 г Т. в 200 мл бензола происходит изо-
меризация (2) в дион (3). После встряхивания с твердым бикарбо-
Н3СЧ ХН3 Н3СХ/СН3 И3СХ/СН3
/А. TsOH H.O, С„Н„ /ч /Р
। .j СНаС03Н । । Кипячение 20 час । ।
I II —> | | 7()-К0% I |
о-А сн3 о-А" А ' сн;1 о-А < сн3
О) (2) (Ц
патом натрия раствор фильтруют через колонку с окисью алюминия,
концентрируют и разбавляют петролейным эфиром; дион (3) выде-
ляется в виде белых игл.
Фенилирование ароматических соединений. Кэзлоу и Сэммсрс
[281 получили 3-нитродифенил из л-нитроанилина диазотированием,
превращением в лг-ннтрофенил-К.У-диметплтрпазин и реакцией
последнего с бензолом в присутствии Т. в качестве катализатора.
К кипящему раствору триазина в бензоле при перемешивании
= NN(CH3)3
। 11 Кипячение
• + C6Hc-LTsOH-H.,O -------- —
1 “ L 1 - on__jft»'
L J 2 л 0,8 г в U 0
у 2,5 л С„Н.
NO.;
0,6 моля
+
— I || 4- TsO3"NH2(CH3)2-}-N2
NO.,
медленно добавляют раствор катализатора в бензоле. В конце ре-
акции добавляют воду, продукт выделяют из бензольного слоя,
перегоняют при 0,1 мм и кристаллизуют.
Ангидрид п-толуолсульфокислоты. Для получения этого про-
изводного [29] предлагается добавлять инертное вещество, чтобы
облегчить смешение твердых реагентов и экстракцию продукта:
кизельгур смешивают с РаО6, а асбест с Т. Общая смесь разогре-
9 час при 125°
/Z-CH3C6H4SO2OH.H2O-PP.2O5 ——*
I моль 1.5 моля
о- о-
•—> п-СН3С6Н4^+ — О — S+—С6Н4СН3-п
II II
о о
вается и спустя некоторое время ее нагревают на масляной бане.
Ангидрид выделяют многократной экстракцией дихлорэтаном и
кристаллизуют из смеси бензола с эфиром.
Алкоголиз нитрилов. Т.~ наилучший реагент для этой реакции
[30]. Смесь эквимолярных количеств нитрила, спирта и Т. кипятят
в течение нескольких часов, затем добавляют воду для растворения
солн аммония, а эфир выделяют обычным способом.
Кипячение
RCN + R'OH-!-TsOH‘H,O —.. " —> RCO,R'-rTsO-NH.t
1 1 “ 2;>—85%
12 Заказ 1 166
353
1. Алле и Ч., Сиэи г л с р Ф., «Синтезы органических препаратов», ИЛ,
М., 1953, сб. 4, стр. 538.
2. Схему аппаратуры см. «Синтезы органических препаратов», ИЛ, At, 1949,
сб, 1. стр. 327.
3, Syntli. Org. Chem., 9, № 3 (May 1936), Eastman Kodak- Co.
4. Be ii c Geprcp А., К п б л с p 4., «Синтезы органических препаратов»
ИЛ, At, 1952, сб. 3, стр. 314.
5. К о \ п А., X е р р н к Э., «Синтезы органических препаратов», ИЛ, М.,
1953, сб. 4, стр. 250.
6. Р с б с р г Ч., «Синтезы органических препаратов», ИЛ, At, 1953, сб. 4,
стр. 116.
7. Г а н п о п В. Ф., X а у з с Г, О., «Синтезы органических препаратов»,
изд-во «Мир», 1964, At, сб. 12, ciр. 192.
8. Р е и о ,т л М-, Ньюмен At, «Синтезы органических препаратов», ИЛ,
At, 1953, сб. 4, стр. 259.
9. F е г п h о И z Е., пат. США 2378918 |С. А., 39, 5051 (1954)|.
10. А п t о и п с с i R., Bernstein S., L i t t с 1 1 R., Sax K. J. W i 1-
liams ,1. 11., ,J. Org. Chem., 17, 1341 (1952).
11. P e t с г s e ii Q. R., Sowers E. E., ,J. Org. Chem., 29 1627 (1964).
12. Ralls ,1. \V., Riegel B., ,1. Ain. Chem. Soc.. 76, 4479 (1952).
13. 1) a v i s At. Pet г о w V., .). Chem. Soc., 1949, 2536.
14. T и г п e r R. B., J. Am. Chem. Soc., 74, 1220 (1952).
15. И и a n g-Al inion, Wilson E., Wendler X. L., Tishlor At,
J. Am. Cl)cm. Soc., 74, 5394 (1952).
16. В e d о и k i a n P. Z., J. Am. Chem. Soc., 67, 1430 (1945).
17. К г j t c li c v s k у T. 1-1., G a I Higher T. E., .1. Am. Chem. Soc., 73,
184 (1951); К о e c h 1 i n В. А., К r i t c h e у s k у T. И., Gallagher
T. I ., ibid., 73, 189 (1951).
18. К r i t c Ii e v s k у Т.Н, G a r in a i s e D. L., Gallagher T, F.,
J. Am. Chem. Soc., 74, 483 (1952).
19. M a г s c h л 1 1 C. W.. Kri tclievsky T. И., Tie b e г in а п S,,
G a I 1 a g h e г T. I’., J. Am. Cliem. Soc,, 70, 1837 (1948).
20. X io и и г (А, Л io к к e 3., Б p e и и и n rep У., «Синтезы органичес-
ких препаратов», изд-во «Мир», At, 1964, сб. 12, стр. 96.
21. На гельсоп С., Готтфрпд С., «Синтезы органических препаратов»,
ИЛ, At, 1952, сб. 3, етр. 504.
22. J о h n s о п \V. S., В а и с г V. .1., М а г g г a v e J. L., F г i s с h At A,,
Greger I,. ЕЕ, 11 п b b a r d W. X., .1. Ain. Chem. Soc., 83, 606 (1961).
23. У a ii и б с p г Г., B а и т ж с с А., «Синтезы органических препаратов»,
ИЛ, М., I960, сб. 10, стр. 66.
24. S о п d h с i in о г М е с h о и 1 a m R., S р г е с h е г AL, Tetrahedron,
20, 2473 (1964).
25. W enkert Е., Stevens Т. Е., J., Am. Chem. Soc., 78, 2318 (1956),
26. S р е п с с г Т. A., S с h in i с g с 1 К. К., Sc h in i с g e I W. \V., .1. Org.
Chem., 30, 1626 (1965).
27. Isler О.. L i n d 1 а г IE, Al о n t a v о n M., R ii e g g RS а и c v G.,
Zeller P., Helv. Chim. Ada, 39, 2041 (1956).
28. К э з л о у К., С э м м с р с Р., «Синтезы органических препаратов», ИЛ,
At, 1954, сб. 5, стр. 46.
29. Ф п л д Л., М а к-Ф а р л е ид Дж., «Синтезы органических препаратов»,
11.4. At, 1958, сб. 8, етр. 5.
39. J a m е s F. L., В г с g а п W. И., J. Org. Chem., 23, 1225 (1958).
ТОЛУОЛСУЛЬФОКИСЛОТЫ л-НИТРОБЕНЗИЛОВЫЙ ЭФИР,
/HCH;iCliH3SC)2OCH2C(lH3NO:,-n. Мол. вес 307,32; т. пл. 103'.
Реагент получают взаимодействием л-толуолсульфоната серебра
с н-нитробепзилбромидом (88°п сырого, 80% чистого) [1]. Он взап-
354
модействует с натриевой или триалкилам.моииевой солью карбобен-
зокс и аминокислоты или пептида в ацетоне пли диметилформамнде
с образованием с высоким выходом п-нитробеизилевого эфира. За-
щитная группа устойчива к действию кислоты и легко удаляется
каталитическим гидрированием,
1. Т 11 с о do г о р о и I os D., Т s а n а г i s JJ. Огц. Chem., 29, 2272 (1961).
л-ТОЛУОЛСУЛЬФОМЕТИЛНИТРОЗОАМИД (диазальд), см. Ди-
азометаи.
/г-ТОЛУОЛСУЛЬФОНАТ СЕРЕБРА, n-CH;iCJI4SO;!Ag- Мол. вес
279,08.
По Корнблюму и сотр. [11, реагент получают с количественным
выходом смешением эквивалентных количеств окиси серебра и и-
толуолсульфокислоты (моногидрата) в ацетонитриле (при защите
от света). Через 30 мин хлористое серебро отфильтровывают и
фильтрат упаривают досуха.
С алкплгалогенндами Т. с. образует тозплаты, которые при окис-
лении смесью ДМСО и бикарбоната натрия дают альдегиды 111.
Cll.yCl -r /еС1 l:!C((H.1SO2OAg —-—°'
I 1 г 1! i 00 -if.7 ch.,<:n
/U.i) ЦЮЩШЫ n 1Л1,Щ ДН'Г
—> CH;;(C112)5C1JO
ДГ1Ф — дчнлтрофеиплгндразол
Например, тозплат, полученный из 1-иодгептаиа в виде масла,
экстрагируют эфиром и добавляют в атмосфере азота к смеси ДМСО
и NallCO.p нагретой до 150°. Окисление завершается за 3 мин\
в случае бензилгалогенида окисление проводят при 100; в течение
5 мин.
Обычно при взаимодействии Т. с, с вторичным или третичным
галогенидом образуются лишь продукты реакции отщепления 121.
Однако проведение реакции при охлаждении до —25" и ниже в
минимальном количестве ацетонитрила позволяет выделить слож-
ные эфиры даже третичных спиртов [31.
I. К о г п b 1 li in Г\г., Jones XV. J., А п ii с г s о и G, JJ. Ann Chcui. Soc.,
81, 41 13 (1959).
2. Е in ru (ins \V. D.. 1-' e r r i s A. 1’.. J. Ain. Chem. Soc., 75, 22o7 (1953).
3. H о 1 f ru a n n H. AV R,. J. Chem, Sue., 1965, 67-18.
О
) _l _
w-TOJl УОЛСУЛ ЬФОН И Л АЗИД, X. Мол.
вес 197,22, т. пл. 22“. О"
Получают из тозилхлорпда и азида натрия с выходом 83% [I, 2[.
12*
355
Диазосоединения. Деринг и Де Пюи [2] получили из циклопен-
тадиеииллития и Т. диазоциклопентадиен — в высшей степени устой-
чивое темно-красное соединение (т. кип. 53'750 мм) с выходом 35%.
Первоначально образующийся триазин самопроизвольно разлага-
о
и
Ar—S=N—N=N
I
о-
Li+CsHs-
-ArSO»N,~HLi+
ется с выделением литиевой соли n-толуолсульфамида. Т. реаги-
рует с р-дикетонами в присутствии основных катализаторов (эгплат
калия, водио-этавольный раствор КОН) с образованием а-диазо-
р-дикетонов [3J. Например, димедон дает 2-диазодимедон в виде
N-
II
N+
ярко-желтых игл с т. пл. 108°. Антрон превращается в 9-диазоант-
рон (I) с высоким выходом. Реакцию с Т. проводят в этаноле при
комнатной температуре с добавкой пиперидина при перемешива-
нии магнитной мешалкой [41. Если За 9-диазоантрона (I, коричне-
вый) добавить при перемешивании к смеси, состоящей из 20 мл пипе-
ридина и 20 мл пиридина, и осторожно нагреть, то выделится азот
и образуется оранжево-красный антрахиноназин (II).
20 г в 300 мл EtOH
Пиперидин — пиридин
82%
TsN3
пиперидина
356
Теддер п Уэбстер [5] описали реакцию, аналогичную реакции Де-
ринга и Д,е Пюи 12|. Сухая натриевая соль р-нафтола с Т. в эфире
даст триазин (2), который самопроизвольно разлагается с выделе-
нием и-толуолсульфамида натрия и образованием диазо-оксида (3);
в щелочной среде (3) взаимодействует с Р-нафтолом, давая 2,2'-
диокси-1, Г-азонафталин (4). В эту реакцию вступают только актив-
ные фенолы: нафтолы, резорцин, флороглюцин и 3-метоксифенол.
Соли менее активных фенолов дают н-толуолсульфонат и азид
натрия.
]. С и г t i u s Т., К г а с m с г G., J. prakt. Cliein., 125, 323 (1930).
2. D о с г i п у \V. Е., von, D е Р u v С. 1!., J, Аш. Chem. Soc., 75, 5955 (1953).
3. R су j t z M., Ann., 676, 101 (1964).
4. R e у i t z M,, Chem. Ber., 97, 2742 (1964).
5. Tedder J. M-, Webster B., J. Chem. Soc., 1960, 4417.
л-ТОЛУОЛСУЛЬФОНИЛГИДРАЗИД, n-CH;jCttH4SO2NHNH2 (то-
зил гидразид, TsNHNH-,). Мол. вес 186,23, т. пл. 104—107°.
Получение [1]. Раствор тозил хлорида в ТГФ перемешивают
so2\:hnh3
10—15"
90%
V-
I
СН3
при 10 : и добавляют раствор гидразина с такой скоростью, чтобы
температура не поднималась выше 10—15°. Через 15 мин нижний
слой отделяют, а верхний промывают насыщенным солевым раство-
ром, фильтруют через осушитель, упаривают и добавляют петролей-
пый эфир для осаждения продукта.
357
Реакция элиминирования. Тозил гидразон, полученный конденса-
цией Т. с бе изпл метил кетоном, взаимодействуй! с раствором натрия
в этиленгликоле, давая диазосоединение, которое разлагается с
выделением азота и образованием олефина [2,31. Тозплгпдразон
2-мстилиропаналя взаимодействует аналогично с метилатом натрия
в диэтиленгликоле с образованием главным образом 2-мстнлпропена;
если реакцию проводить в апротонном растворителе, например в
СН;! СН;{
I ХаОСН,СН.,ОИ [ !-
CgII-CI-LC-XNIIT.s >СйН.СН.,С^\-\
' - — Т‘Ха
С(;Н-СН - СНСН а -1-
диэтиловом эфире диэтиленгликоля, то 2-мстплпропен и хметилцикло-
пропаи образуются в примерно равных количествах [41. Тозилгид-
разоиы циклоалканонов разлагаются в диэтиловом эфире диэтилен-
гликоля иод действием метилата натрия на диазоуглеводороды с вы-
ходом 60—70% [51.
Реакция применима для получения а-диазокетонов из соот-
ветствующих сс-ди кето нов [6|. 1,2-Инда и дион, полученный через
2-оксимипопронзводное, взаимодействует с Т, с образованием
монотознлгидразона. В присутствии основания отщепляется тозил-
окси-аипон и с хорошим выходом образуется 2-д[1азо-1-индаиои,
2,6-Дпметпл-1,4-бензохинон даст 2,6-дпметпл-1,4-бензохш[Он-4-ди-
азид, а 4,5-диметил-1,2-бензохпнон— 4,5-димстил-1,2-бснзохинон-
1-диазид [7|.
Литиевые соли тозилгидразонов разлагаются при пиролизе в
вакууме (0,2—0,3 льи) при 70—140 ’ с образованием дпазососди-
нсшш [8]. Тозплгпдразон можно получить в реакционной смеси из
эквивалентных количеств альдегида или кетона и Т. в ТГФ и пере-
TsNHNH2 выл
(C11;J}.,CHCH - О----— (Cl 1;;),СНСН — \ N HTs--
—> (СНД.СНСП \\Ts-(СН.р.,С11СН=х\ TsLi
Li-
вести, в соль при помощи бутлллптия. Этот метод, в частности, ис-
пользуют для получения дназосоединепий в том случае, если карбо-
нильное соединение более доступно, чем соответствующий амии.
Тозилгидразоиы -ненасыщенных альдегидов под действием
основных катализаторов разлагаются до циклопропенов [9|.
358
сн3
снэ /
?Нз
C=C-CH=NNHTs
72%
CH3ONa в диглиме
150°
В результате перегруппировок углеродного скелета прп разложе-
нии тозил гидразонов ни накол ина и камфоры образуются 2,3-днме-
тилбузен-2 и камфен ]2|. Прп обработке горячен щелочью тозил-
гидразон хеконгсшша подвергается расщеплению и перегруппи-
ровке с сокращением кольца С и расширением кольца D {10].
Идентификация сахаров. Глюкоза взаимодействует с Т. с обра-
зованием «прекрасного гидразона» (ein schones Hydrason), который
легко превращается снова в сахар при взаимодействии с бензальде-
гидом |11|, и-Рибоза в метаноле образует с количественным выхо-
дом высококрнсталличный тозплгндразон (г. нл. 164 ) |12|. Анало-
гичные производные получены из ь-арабинозы, о-ксилозы, о-лик-
созы и ь-фукозы.
Дехлорирование. 5-Хлоракрпдин взаимодействует с Т. в хлорофор-
ме с образованием соли, которая при нагревании со щелочью разда-
ст)
TsNHNH,
сись
NHNHTs
Cl'
Водн. NaOH
НОСНгСН2ОН
2 vac при 97°^
48% (общий)
гается до акридина! 131, Метод позволяет дехлорировать 5-хлор-
акридины, содержащие восстанавливающиеся группы (\О., CN).
Получение диимида, см. Диимнд,
Необычная реакция. Чанг [141 осуществил реакцию 12-кето-
холана с Т. прп кипячении в этаноле л, кроме тозилгидразона (2),
получил азин (3), который, очевидно, образуется в процессе мед-
ленного разложения Т. до гидразина. Присутствие соляной кислоты
^/С-0 —> Ч С-- NNHSO,,Ar-h‘>C =
(!)
(2)
(3)
в качестве катализатора приводит к достаточно быстрому образо-
ванию (2), так что азин вовсе не образуется.
359
1. Фридма и Л., Литл Р. Л., Рейхл В., «Синтезы органических пре-
паратов», изд-во «Мир», М., 1964, сб. 12, стр. 143.
2. Bamford W, R., Stevens Т. S., J. Chem. Soc., 1952, 4735.
3. Powell J. W., Whiting M. C., Tetrahedron, 7, 305 (1959); De Pu v
С. H., Froemsdorl D. H., J. Am. Chem. Soc., 82, 634(1960).
4. Friedman L., Shech ter H., .1. Am. Chem, Soc., 81, 5512 (1959).
5. fricdman L., S h e c h t e г ]]., J. Am. Chem. Soc., 83, 3159 (1961).
6. C a v a M. P., L i t 1 e R. L., Napier D. R., J. Am. Chem. Soc., 80, 2257
(1958); Blomquist A. T., S c h 1 a e f er F. W,, ibid., 83, 4547 (1961).
7. Ried W., Dietrich R., Chem. Ber., 94, 387 (1961).
8. Kaufman G. M., Smith J.A., Vander Stouw G.G., Shech-
t e r II., J. Am. Chem. Soc., 87, 935 (1965).
9. С 1 о s s G. L., С 1 о s s L. E., J. Arn. Chem. Soc., 83, 2015 (1961).
10. Hirschrnann R., Snoddy C. S., Jr,, II i s k с у C, F., W e n d-
1 c r N. L., J. Am. Chem. Soc,, 76, 4013 (1954),
11, Freudenberg K-, В 1 ii m m e I F., Ann., 440, 45 (1924).
12. Easterby D.G., Hough L., J ones J. K, N-, J - Chem. Soc., 1951,
3416.
13. A 1 b e г t A., Royer R., J. Chem. Soc., 1949, 1148.
14. Chang F. C,, J. Org. Chem., 30, 2053 (1965),
л-ТОЛУОЛСУЛЬФОНИЛПЕРХЛОРАТ, rt-CH,CeH4S0;C10;.
Мол. вес 254,60.
Реагент получают в нитрометане при 0° взаимодействием тозил-
бромида с перхлоратом серебра.Т. является сильным сульфонирую-
щим агентом, который реагирует даже с относительно инертными
галогенбензоламн, образуя сульфоны [1L
[n-CH3CeH4S02] + C10.r +АгН —>n-CH3C6H4SO2Ar-|- НС1О4
I. К 1 a g е s F,, М а 1 е с k i F. Е., Ann., 691, 15 (1966).
2-л-ТОЛУОЛСУЛЬФОНИЛЭТИЛХЛОРФОРМИАТ,
n-CH3CeH4SO2CHzCHaOCOCl [11. Мол. вес 262,72, т. пл. 48°.
Получение. га-Толуолсульфинат натрия нагревают с избытком
2-хлорэтанола в ДМФА; полученный сульфон перемешивают в
сухом бензоле с избытком фосгена.
HC0N(CHa)..
n-CH;)C(.H4SO.,Na А С1СН2СН..ОН-~ >
' / G J,,
[ моль 3 моля
COCL в СЙНВ при 0—25°
—*/i-CH:iCeI-I.jSO2CI-I2CH2OH--------—
—> n-CH3C6H4SO2CH2CH2OCOCl
N-Защитная группа. Т. взаимодействуете аминокислотами в вод-
ном диоксане в присутствии окиси магния, образуя N-защищенную
аминокислоту с выходом 85—92%. В отличие от других защитных
групп н-толуолсульфонилэтоксикарбонильная группа вполне устой-
чива как к действию кислоты, так и к каталитическому гидрирова-
нию. Защитную группу удаляют обработкой избытком 1 н. водно-
этаиольного раствора едкого натра при комнатной температуре или
с помощью сильно основной ионообменной смолы. Реагент исполь-
360
зуют в синтезе пептидов как по азидному методу, так и через
л-нитрофсн иловый эфир.
1. Kader Л, Т.„ Stirling С. J. М., J. Chem. Soc., 1964, 258.
л-ТОЛУОЛСУЛЬФОХЛОРИД (тозил хлористый). Мол. вес
190,65, т. пл. 67,5—68,5°, т. кии. 146715 мм.
Очистка. Высококачественный продажный продукт можно ис-
пользовать при условии, что он взят из недавно открытой склянки,
так как при длительном хранении в незащищенном состоянии он
может содержать значительное количество п-толуолсульфокислоты.
Пельтье [1] проверил два опубликованных метода очистки и, найдя
их неудовлетворительными, предложил следующий вариант; 10 г
неочищенного Т. ст. пл. 66—86° растворяют в минимальном коли-
честве хлороформа (около 25 мл) и раствор разбавляют 5 об. петро-
лейного эфира (т. кип. 30—60е) для осаждения примесей. Раствор
фильтруют, очищают норитом, упаривают до объема 40 мл на водя-
ной бане, а затем в вакууме до очень малого объема и получа-
ют 7,0 а мелких белых кристаллов аналитически чистого Т. ст.
пл. 67,5—68,5°. Нерастворимая часть (1,4 г, т. пл. 101 —104°)
содержит в основном /г-толуолсульфокислоту.
Тозилаты для исследования сольволиза 12]. Рекомендуется приме-
нять Т., свежеперекристаллизованный но методу Пельтье. Пиридин
из только что откупоренной бутыли сушат кипячением над окисью
бария с последующей перегонкой. Раствор 3—5г спирта в 50—75 мт
пиридина помещают в колбу Эр л ей мейер а на 125 м л, охлаждают
до 0° н смешивают с 1 молярным избытком Т. [3|. Затем раствор
помещают в холодильник иа 12—24 час (с некоторыми спиртами
реакция протекает дольше). Реакция может сопровождаться появ-
лением окраски (желтой, коричневой, а часто розовато-пурпурной)
и выделением солянокислого пиридина в виде длинных игл. Оса-
док не образуется, если пиридин влажный. По окончании реакции,
когда образования осадка уже не наблюдается, смесь выливают при
перемешивании в 300—400 г воды со льдом. Обычно тозилаты крис-
таллизуются сразу же или после перемешивания еще в течение
15 мин', в этом случае продукт отфильтровывают, промывают на
фильтре водой, сушат в вакууме при комнатной температуре и
кристаллизуют, как описано ниже. Если тозилат остается маслооб-
разным, его растворяют в эфире и водный слой дважды экстраги-
руют эфиром. Эфирный раствор дважды промывают холодным раст-
вором соляной кислоты (1 : 1) для удаления пиридина, а затем водой,
сушат над K2CO.{— Na..SO4 и упаривают при комнатной температуре
до получения бесцветного или светло-желтого масла. Не нагревать
тозилат выше комнатной температуры!
Для очистки маслообразного или твердого тозилата его раство-
ряют в минимальном количестве петролейиого эфира (т. кип. 30—
60е) при комнатной температуре. После перемешивания с норитом
361
смесь фильтруют через мелкозернистый фильтр и промывают. Чис-
тый бесцветный раствор медленно охлаждают до —75 в ацетоне с
сухим льдом, проводя палочкой по внутренним стенкам стакана,
чтобы вызвать кристаллизацию и предотвратить маслообразование.
Пели кристаллизация началась, то она протекает очень хорошо, но
охлаждение прекращают перед фильтрованием. После фильтрования
с отсасыванием влажный осадок сразу же сушат в вакууме при
комнатной или более низкой температуре.
Если вызвать кристаллизацию не удается, то в результате про-
цесса маслообразования при —75 образуется белая стеклообразная
масса, которая иногда медленно кристаллизуется при стоялпи при
этой температуре; в этом случае твердый продукт фильтруют, как
указано выше. Если вещество остается вязким и стеклообразным,
то верхнюю жидкую фазу сливают; тозплаты настолько слабо раст-
воримы в петролейпом эфире, что таким способом отделяются даже
небольшие количества примесей. Аналитически чистое вещество
в виде масла можно получить, повторив этот процесс один или два
раза с последующим высушиванием в вакууме при комнатной темпе-
ратуре.
Хотя многие тозплаты устойчивы при комнатной температуре,
их следует хранить на холоду. Некоторые тозплаты, особенно непре-
дельные, разлагаются при 25' в течение нескольких часов. Эти не-
стабильные эфиры следует получать непосредственно перед употреб-
лением ii образны, посланные па анализ, снабжать пометкой «срочно,
нестабилен».
Тозплаты дают узкий интенсивный дублет при 8,4 и 8,5 як в
ИК-спектрах.
Очень небольшое число тозилатов плавится без разложения, давая
желтые нлп темные расплавы. Иногда температура плавления зави-
сит от температуры в момент внесения образца или от скорости
нагрева. В том случае, когда тозилат нельзя получить аналитически
чистым пли нельзя проанализировать, результаты сольволиза могут
все же оказаться правильными, поскольку линейность для кривой
первого порядка и сравнение экспериментально определенного титра
для бесконечного разбавления с вычисленным являются критериями
чистоты. Тозплаты, полученные из с,меси изомеров, та Kate могут дать
ценную инфор.мацию; если скорости образования компонентов от-
личаются в 10 или более раз, смесь можно анализировать кинетиче-
ски |4|.
Селективное тозилирование. В одной из стадий синтеза альдо-
стерона ио Джонсону 151 требовалось селективное тозилирование
первичной спиртовой группы кетальдиола (1) для окисления вторич-
ной группы до кетотозилата (2), который необходим для получения
информации о кольце D. Мезптпленсульфохлорид, хотя и вполне
пригодный для этой пело, нс имеет никаких преимуществ по сравне-
нию с Т., так как желаемая селективность обеспечивается лишь в
том случае, если реакцию проводить в абсолютно безводных ус-
362
ловиях при низкой температуре (8 9 в весьма разбавленном раст-
воре в пиридине. В этих условиях кетотозплат (2) получается с
высоким выходом.
Сульфамиды. В первой стадии синтеза 2-аминобеизофенона
Шейфел и Де Тар 161 превращали антраниловую кислоту в «-толу-
ол сульфонил антран иловую кислоту в присутствии в качестве ос-
нования карбоната натрия; если использовать едкий натр, то основ-
ным продуктом будет соль /i-тол уолсульфокнслоты и антраниловой
кислоты, В реакции используют технический Т. в 20%-ном избытке.
/ со-.н /z со., и
Д I. <i() -So У ]] PCI,
I || -|-ха!со3 •н,0—5- | 1| ----►
NJ-Ь 'XIITs
I моль 1,2 моля 2.-1 моля 1,5 .?
О о
II II
I
Ts
Боер и Бэкер [7] получали п-толуолсульфонилметилнитроз-
ампд — предшественник дпазометала — реакцией Т. с метилами-
ном по методу Шоттена — Баумана, заключающемуся в поочеред-
ном добавлении хлораиглдрпда и раствора едкого натра.
СНл— SO.CI CILA'H.. У- NaOH >
[,<’>> моля
н
2.1 моля
(ЗЗ11,,-111,111
води.
J’.I.'l 1?Ор)
ILO
хо
СНл—SOA’CI-B
AcOJJ. JJNO.
КЗ - !) ГМ>„
СН.л —// ЗО-.ХСНз
Превращение формамидов в изонитрилы. В синтезе дпгидроко-
нессина из Зр-ацетоксп-V-биснорхолсновоп кислоты Кори и Херт-
лер [81 обрабатывали формамид (I) Т. в пиридине, ожидая, что в
основном тозилпроваипе коснется Зр-гидрокспльной группы. Однако
363
полученный продукт оказался не (3), а тозил атом изоиитрила (2).
В начальной стадии реагент в пиридине отщеплял воду от форм-
амидной группы с образованием изоиитрила, а избыток реагента
этерифицировал гидроксильную группу при С3, Реакция (1) с I же
реагента дает изонитрил, почти не содержащий тозилового эфира.
От изонитрила (2) легко избавиться в процессе синтеза: при обработ-
ке сырого продукта (2) уксусной кислотой в эфире осуществляется
гидратация изонитрила с образованием желаемого продукта (3).
Неожиданно легкая дегидратация формамида послужила поводом
для исследования, показавшего общность метода с применением Т.
в пиридине [9].
Перегруппировки. Херд и Бауэр [101 добавляли Т. в хлороформе
к суспензии натриевой соли бепзгндроксамовой кислоты (1) в
том же растворителе. Они наблюдали бурную реакцию и охаракте-
ризовали продукт как О-(фенилкарбамипоил)-бензгидроксамовую
кислоту (6). Они также предположили, что исходный продукт —
тозил ат (3) — является гораздо более сильной кислотой, чем (I),
и отрывает натрий от соли (2), образуя соль (4), которая подверга-
ется одновременному расщеплению и перегруппировке Лоссена в
фен ил изоцианат (5) и тозилат натрия. Фен ил изоцианат затем ре-
агирует с (I) и дает конечный продукт (6).
ОН ОН ОН
Il I II I - тю 1 I 4(2)
CGH5C — \ — ОН — СДР- С— \ — ОХа + — > C«IфС — N — OTs —
(1) (2) (3)
О Na+ ОН ОН
II I (1) II I II I
— с.д 15с-N- - OTs-Nо=--л: - хс6н5 — с(!нд: -N- O-C-NCJP,
Ш (3) (6)
Т. можно также применять для осуществления перегруппировки
Бекмана [II, 121, например, оксима 9-ацетил-/щс-де калина [121.
364
Метил-n-толилсульфон. По методу Фнльда и Кларка [13] Т.
восстанавливают сульфитом натрия в буферном растворе с бикар-
бонатом натрия до п-толуолсульфината натрия, который выделяется
в виде твердого осадка. Полученную соль отделяют и перемешивают
О
1| Перемешивание при
n-CH3C(iH4S+Cl Н- Na3SO3 + NaHCO3 -f- НаО 3
q_ 4f7O моля 5 молей 2,4 л
2,54 моля
О
„ ru г U (CH3O)2SO2 + NaHCO.,
/i-CPJjjCgH4SONa------------——— zi-CHyCgW^SO^CHg
7 8—82%
с диметилсульфатом, бикарбонатом натрия и водой; образующийся
при этом сульфон экстрагируют бензолом.
Дегидратация, Винтерштейнер и Моор 1141 дегидратацией 30-
ацетоксихолестаиола-7а в присутствии Т. в кипящем пиридине по-
лучали смесь стенилацетатов, обогащенную Д7-изомером и в процессе
каталитической перегруппировки превращающуюся в чистый А8(14)
хол естен и л ацетат.
( И ИЗОМЕРЫ I
При исследовании строения окситетрациклина и хлортетрацик-
лина было установлено [15], что Т., взаимодействуя с антибио-
тиками в холодном пиридине, дегидратирует карбоксамидную
группу с образованием нитрила. Было также найдено, что более
простые амиды с достаточно хорошим выходом дегидратируются в
нитрилы при медленном добавлении 1 же Т. к смеси амида с 2,25 же
пиридина так, чтобы температура не поднималась выше 70°. Предпо-
лагаемый механизм включает стадию О-тознлирования:
О OSO3Ar
RC- NH2 гС1, RC = N Н RC - N + C5H5NH(ArSO" )
-CbHBN-HCI
Шихан и сотр. [16] использовали Т. и триэтиламин для прев-
ращения производных мочевины в карбодиимиды.
НОН
RN^C-NR' _LSEL RN-—С-- NR'
(6HS),N
365
Этерификация. При взаимодействии кислоты (1 экв) со спиртом
('I ,?к«) в присутствии Т. (2ж) в пиридине получаются с высоким вы-
ходом эфиры П7|, Предполагают, что на промежуточной стадии
реакции образуются ангидриды кислот. Метод был использован для
КСООпА4'1 11 . (RCC)).,0 —И RCOOH ! RCOOR'
t < Д1Л 1
‘I
получения эфиров N-аиетилированпых аминокислот (выход 51 —
97%) 1181 и особенно успешно — для этерификации третичных аце-
тиленовых спиртов 1191.
N-защитная группа. Т. взаимодействует с аминокислотами
в щелочном растворе с образованием N-тозпламипокислот 1201.
Защитная группа удаляется восстановительным расщеплением нат-
рием в жидком аммиаке, Т., в частности, используют для защиты
ы-ампногрупп лизиновых и орнитиновых пептидов.
Декарбонилирование. Шихан и Франкенфельд 1211 осуществили
реакцию а-фсниламино-а, а-дифепилуксусной кислоты (1) с Т. в
пиридине, предполагая получить К’-тозпльпое производное. Однако
они получили продукт, идентифицированный как ан ил бензофенон (3),
моноокись углерода и тозилат пиридиния. Ими постулировано обра-
зование иа промежуточной стадии смешанного ангидрида (2).
(С6]Ц)2ССО2Н 4
едцкн
о
, , ли
(QH5)2(p£-c-OTs
С6Н5гЛ
4— Ру
---+----
-РуНЩзО )
(С6Н5)гС
C6HSN
со
(1)
(%
(3)
I. Pelletier S. W-, Chem. Ind., 1953, 1034.
2. (.ontribiited by S c h 1 e у er P. von R., Princeton University.
3. M n p в е л К., С с к е р а В., «Синтезы органических препаратов», ИЛ, М.,
1952, сб. 3, стр. 249; получек тозилат лаурилового спирта по реакции со
спиртом в пиридине при 10- 20J, избыто/TsCI около 10%, выхот 88—90%
4. Brown Н. С., Fletcher R. S., J. Am. Chem. Soc., 71, 1851 (1949).
о. ,1 о h п s о n W. S., Colli n s J. C. Jr., P a p p о R., R u h i п M B.
К г о p p P. J., J о b n s W. F., Pike J. E., Bari m a n n W., .1. Am’
Chem. Soc., 85, 1409 (I9G3).
6. Ill e it фея X., Д e T я p Д., «Синтезы органических препаратов», ИЛ.
М., 1953, сб. 4, стр. 32.
7. Ьиер Т.. до, Бэкер X., «Синтезы органических препаратов» ИЛ AV
1956, сб. G, стр. 67; 1958, сб. 8, стр. 8.
8. Corey Е. .1.. Heftier AM R., J. Am. Chem. Soc., 81 5209. 1939
9. Hertler W. [?., С о г е у Е. J., J. Огщ Chcin., 23, [221 (1958).
10. Hurd С. D., Bauer I.., .J. Am. Chem. Soc., 76, 2791 (1954).
11. О x I e у P., Sh о r t W. F.. .1. Chem. Soc., 1947, 382.
12. Hill R. K., C h о r t у k О. T., J. Am. Chem. Soc...84, 1065 (1962).
13. Фильд Л., Кларк 1’., «Сивгезы органических препаратов», ИЛ М
I960, сб. 10, стр. 45.
14. Winterstciner О., Moore At, J. Am. Chem. Soc. 65 1503 1507
(1943). ’ ’
366
15. S t e p h с n s C. R., В i а п с о Е. .1., Pilgrim Г. J., J. Аш. Chem. Soc.,
77, 1701 (1955).
16. S li е е Ii а п J. С., С г и i с к s h и и к Р. А., В os h а г t С. L., J. Org.
Chem., 26, 2525 (1961).
17. Brewster J. II., C i о t t i C. J., Jr., J. Am. Chem. Soc., 77, 6214 (1955).
18. В 1 о t n у G., В i erna 1 .1. I, T a s c h пег E., Ann., 663, 194 (1963),
19. II enn ion G. F., Barrett S. O., J. Am. Chcni. Soc., 79, 2146 (1957);
К 1 о s a J., Annew. Chem., 69, 135 (1957).
20. Byaci’on P. А., Успехи органической химии, т. 3, нлд-во «Мир», АЕ,
1966.
21. S I] с е li а п J . С., F г а п к е n I с I d J. W,, J. Or”. Chem., 27, 628 (1962),
/г-ТОЛУОЛСУЛЬФОХЛОРИД -—ДИМЕТИЛФОРМАМИД. Тозпл-
хлорпд образует комплекс с ДгМФА [1|
(СН;!).,Х - СНОЗОХ(;Н4СН;!-/1(С1~)
(1)
который является великолепным реагентом для формил и ров ан ия
спиртов. Например, тестостерон, взаимодействуя с тозилхлорпдом
в ДМФА, образует формиат тестостерона с выходом 79% [2|. Реа-
гент взаимодействует также с ариламинами, давая сульфанилпды
(2) п (пли) формамидины (3).
.м-кн.,
(1)----- /i-CH3Cr,I liSO.,N,HAr4- (CH;)),N _-CH\HAr(/bCH;:CeH4SO2O-)
(2) (3)
1. Н a I 1 Н. I\., Jr., J. Am. Chem. Soc., 78, 2717 (1956).
2. A 1 b г i g Ii t J. D., Benz E., Lanzilotti A. E_, Goldina n L.,
Clieni. Comm., 1965, 413.
ТОРИЯ ДВУОКИСЬ, ThO2. Мол. вес 264,05.
Дегидратация вторичных спиртов в паровой фазе на Т. д.
как катализаторе даст почти исключительно I-олефины ПР
TliO,
СНДСН.^С! ECIICH;( -г— ' СНдСН.щСШД I CEL, н- CH;j(CI I.,)4CH - chch3
All
Напротив, дегидратация вторичных спиртов на окиси алюминия
дает смеси, обогащенные 2-олефииамп. Наиболее активный катали-
затор получают при прокаливании оксалата тор ня при 350—450“ в
течение нескольких часов.
1. L а п rl о с и A. J., Van Iloozcr R., J. Am. Chem. Soc., 85, 2180 (1963).
ТРЕХХЛОРИСТЫЙ АЗОТ, NQ;. Токсичное и взрывоопасное
вещество (особенно в водной среде); реагент необходимо получать
в неводном растворителе, используя надежную защиту.
Получение [1|. К суспензии 1,2 моля гипохлорита кальция
в 600 мл воды и 900 мл о-дп хлорбензол а, охлажденной до О' в бане
с ацетоном и сухим льдом, при хорошем перемешивании по кап-
лям добавляют смесь 1,2 моля хлористого аммония, 150 мл конц.
соляной кислоты и 450 мл Н .О при температуре не ниже IOJ. Спус-
367
тя 15 мин отделяется ярко-желтый органический слой, который
промывают водой, сушат сульфатом натрия, титруют иодометричес-
ки и хранят при 0°.
Реакция Фриделя — Крафтса [2]. Реакция Т. а. с кумолом,
катализируемая хлористым алюминием, приблизительно соответст-
вует стехиометрии, указанной па схеме
сн(сн3)2
Ч -1 (1°
3 1 + NC13 В C6H4Ct2-O + А1С13 - >
40-50%
СН(СНЭ)2
2
СН(СН3)г
О нс я о. 4М0ЛЯ 800 мл 0, 8 моля
3, 7 5 моля 1
Кумол н хлористый алюминий добавляют к раствору Т. а. в о-дих-
лорбензоле. Подобным же образом метилциклогексан, взаимодейст-
вуя с Т. а. и хлористым алюминием в хлористом метилене при 0°
в атмосфере азота, превращается в 1-метил-1 -аминоциклогексан с
выходом 86—87%.
1. К о v а с i с Р., Levinsky J, A., Case Inst. Techn., procedure submitted
to Org, Syn.; К о v a c i с P, et al., J. Am, Chem. Soc., 86, 1650 (1964),
2. К о v a c i с P., Chau dhary S. S., procedure submitted to Org. Syn.;
Kovac i с P,, Hopper R.J., Chaudhary S. S„tcvinsky
J. A., Licpkalns V, A., Chem. Comm., 1966, 232; KovaciC P., G о r-
m 1 s b J, F., J. Am. Chem. Soc., 88, 3819 (1966),
тет^ак«с-(ТРЕХХЛОРИСТЫЙ ФОСФОР)-НИКЕЛЬ(О),
(PCI:j)4Ni. Получение 111.
Циклотетрамеризация ацетиленовых соединений (21. Хотя три-
меризацию ацетиленовых производных в производные бензола ката-
лизируют различные никелькарбонилфосфины, этот катализатор, не
содержащий СО, вызывает тетрамеризацию с образованием цикло-
октатетраенов. Область применения реакции, однако, крайне огра-
ниченна, Среди целого ряда исследованных ацетиленов реагируют
R = СН3ияиС2Н5
и дают как тримеры, так и тетрамеры только наиболее реакционно-
способные метиловый и этиловый эфиры пропиоловой кислоты. Реак-
цию проводят в атмосфере азота при комнатной температуре в цикло-
гексане и бензоле; реакция сопровождается разогреванием до 50—
60° и завершается за 20 мин. Из метилпропиолата получают 1,2,
4,6-тстракарбметоксициклооктатетраен (1) с выходом 83% н 1,2,4-
368
трикарбметоксибензол (2) с выходом 17%. Этилпропиолат дает 28%
(1) и 1% 1,3,5,7-тетракарбэтоксициклооктатетраена (3); основным
продуктом (71%) является смесь (2) и (4)—1,2,4- и 1,3,5-трикарб-
этоксильных производных бензола.
I. I г v i n е J, W., W i 1 k i п s о n G., Science, 113, 742 (1951),
2. Leto J. R., Leto M. F., J. Am. Chem. Soc., 83, 2944 (1961).
1,2,4-ТРИАЗОЛ, (3). Мол. вес 69,06, т. пл. 121°, т. кип. 260°.
Получение [11. При конденсации тиосемикарбазида с муравьи-
ной кислотой образуется 1-формил-З-тиосемикарбазид (I), который
циклизуется в присутствии щелочи, давая 1, 2, 4-триазол-3(5)-тиол
(2); тногруппу удаляют окислением (см. Азотная кислота, окисле-
ние).
н s
1 11 87°
H,NN— CNH2 +90%-чая НСО2Н--------->
71-81%
П КНг
СН ХС=5
HN_____k BoAK.NaOH; НС^
72-81%
2 моля 400 мл
HNO,
-----*-►
52-58%
(г) (3)
Катализатор образования пептидной связи. Бейерман [2] уста-
новил, что Т., который имеет как слабокислотную группу (NJ,
так и слабоосиовную группу (N2), заметно катализирует образова-
ние пептидной связи. В присутствии этого катализатора при конден-
сации цианметилового эфира N-бензоилглицина с бензиламином в
ацетонитриле выход N-бензоилглицилбензиламина увеличивается
с 20 до 80%. При добавлении Т. к растворам п-иитрофеиилового
эфира глицина, ь-лейцииа или ь-фенилалаиина в ацетонитриле в те-
чение нескольких минут осаждается полипептид. Каталитическая
активность Т. обусловлена тем, что кислотные и основные группы
занимают смежные положения и, таким образом, допускают заме-
щение через циклическое переходное состояние по согласованному
механизму. Пиразол и 2-оксипиридин также удовлетворяют этим
условиям и обладают каталитической активностью. Три функцио-
нальные группы имидазола не являются смежными, и поэтому это
соединение малоэффективно. Бейерман исотр. и Вейгаид и сотр. в
совместной работе [3] установили, что Т. не вызывает заметной ра-
цемизации. В присутствии Т. незначительная рацемизация наблю-
далась только в одном случае. С другой стороны, использование
имидазола в ряде случаев приводит к рацемизации.
369
1. Э ii и с в о р т Ч., «Синтезы органических препаратов», изд-во «Мир», At. 1964,
сб, 12, ci р. 1 13.
2. В с v е г ш а и 11. С., v а п d с п В г i л k W. At, Proc. Client. Soc., 1963,
266.
3. В с v <? г гл а п Н. С. el al,, W с у у а n d В. et al., Rev. trav, chim., 84, 213
(1965).
(о-ТРИБРОМАЦЕТОФЕНОН, C(;H5COCBr:i. Мол. вес 356,87,
т. пл. 66 , т. кип. 176 16 .н.п.
Кронке111 описал получение этого реагента л сто применение для
селективного бромирования метилкетонов и боковой цепи аромати-
ч ос к п х у гл сводородов.
1. Krohnkt F'., Вег. 69, 921 (1936): К г 6 h п k е F., Е 1 ! eg a s t К., Clietn.
Ber., 86, 1556 (1953).
ТРИ-н-БУТИЛАМИН, (CH;iCH..CH,,CH2),N. Мол. вес 185,35,
т. кип. 216,5е, уд. вес 0,78.
Дегидрогалогенирование. Р. Лайл и Ж. Лайл |]| установили,
что продукт, образующийся при действии серной кислоты па диол
(2), является не эпоксидом, как ранее полагали, а 2,2-дифен ил цикл о-
гептаномом (3). Этот напряженный кетон можно бромировать, но
попытки провести дегидрогалогенирование в присутствии пиридина
или пиколина даже в кипящем тетралине не дали положительных ре-
зультатов. Реакция с \[,Х!-димети л анилином приводит к отщепле-
нию брома и образованию (3) 121. Наконец, желаемый результат был
получен кипячением в течение 12 чне смеси 5 г (4), 8,6 мл Т. и 25мл
тетралина.
1. Lyle R. Е., Lyle G. G.,.]. Am. Chem. Soc., 74, 4059 (1952).
2. L у 1 е R. Е., С о v с у R. А.. .1. Am. Chem. Soc., 75, 4973 (1953).
ТРИ-я-БУТИЛ БОРАН И ТРИИЗОБУТИЛБОРАН, (С4Н9)3В.
Мол. вес 182,16, т. кип. 109 .20 .из/, 9Г'.9 мл/ и 68"'/7 яки.
Гомогенное гидрирование. .Легкое присоединение боргидридов к
олефинам, а также тот факт, чтотриалкилборап может подвергаться
гидрогенолизу с образованием дналкплбораиа н алкана, наво-
370
дятла мысль, чтотриалкилборан может служить гомогенным катали-
затором гидрирования [1|. Хотя любой из простых алкилборанов
достаточно эффективен, оба первичных трибутилборана более удоб-
ны в обращении, Например, циклогексен или каприлен, содержащий
3,8% три-н-бутилборана, количественно гидрируется в течение 3 час
при 200° и давлении водорода 70 атм. Гидрогенолиз связи металл —
углерод приводит к образованию дибутилборгндряда (2), который
присоединяется к олефину, давая новый триалкилборан (.3), Гидро-
генолиз дает алкан (4) с регенерацией дпбутилборгидрида (2). Г1о
израсходовании всего олефина бор превращается в некоторые формы
гидрида .металла.
вв НВВи ^222; ксн,сн.2вв1ь 22 rchcim - нввщ
'’ - Bull
(I) (2) (3) ( О (М
I. De Wi t t E. J., Ramp F. L., Trapasso L. E., J. Am. Chem. Soc.,
83, 4672 (1961): J, Org. Chem., 27, 4368 (1962).
ТРИ-н-БУТИЛБОРОКСИН, (н-С,НчВО)3. Мол. вес 198,16, т. кип.
133,5716 лы/.
Получение III. Смесь три-н-бутилборана и безводного борного
ангидрида кипятят в атмосфере азота в течение 40 час и продукт пере-
гоняют. В тех же условиях три-вмор-бутплборан перегруппиро-
вывается в Т,
/°х
, ~ rr ' П , Г, Л Кипячение 40 члг (Кц б о и
(н-С,Нч) зВ -г- ВлО3---------- я * С4 Н э — в В — с4нэ-«
69% 1 [
0,27 моля 0,27 моля О О
%Н9-н
Разделение цис- и транс-диолов. Браун п Цвейфель [21 уста-
новили, что цпс-циклогександиол-1,4 реагирует с избытком Т.,
образуя умеренно летучий циклический борат (т. кип. 20070,05 лы/),
тогда как троне-изомер образует нелетучий линейный иол и мер и ый
371
борат. Например, 5 г смеси изомеров обрабатывают избытком реа-
гента и воду, образующуюся при этерификации, удаляют азеотроп-
ной отгонкой с бензолом. После вакуумной перегонки получают
вязкий продукт, из которого tfwc-диол (1,42 г) извлекают переэтери-
фикацией этиленгликолем с последующей вакуумной отгонкой бората
этиленгликоля и кристаллизацией остатка. Обработка полимерного
бората тем же способом дает 2,25 а транс- изомер а. Так же разделяют
цис- и трйяс-циклогександиолы-1,3, но бораты цис- и транс-цик-
логександиолов-1,2 незначительно различаются по температуре
кипения и разделяются с трудом,
В приложении этого метода к углеводам [3] при выделении
1,3-диола из бората предпочитают переэтерификацию с пропан-
диолом-1,3, а не с этиленгликолем. Борат обрабатывали в ацетоне
пропандиолом-1,3 и раствор упаривали досуха. Полученный борат
пропандиола-1,3 экстрагировали петролейным эфиром и кристал-
лизацией остатка выделяли углевод.
1. 11 еп n i on G. F., McCusker P. A., Ashby E. C-, Rutkowski
A, J„ J, Am. Chem. Soc., 79, 5194 (1957).
2. В г о w n H. C., Z w e i f e 1 G., J, Org. Chem,, 27, 4708 (1962).
3. Ferrier R. j., Prasad D., R u dowski A., Sangster I., J,
Chem. Soc., 1964, 3330.
ТРИ-я-БУТИЛСТАННАН, (н-С4Нэ)35пН, Мол. вес 291,04, т. кип.
7670,7 мм.
Т. получают по методу Ван дер Керка и сотр. (1] восста-
новлением три-н-бутилхлорстаннана алюмогидридом лития.
Т. - - бесцветная жидкость, которую в сухой атмосфере можно
хранить некоторое время, под действием влаги превращается в
щ/ш-н-бутилоксистаннан.
Восстановление органических галогенидов. Эта реакция была
открыта Ван дер Керком и сотр., и позднее было опубликовано
еще несколько примеров [2, 3]. Кювила [3] убедительно доказал,
R3SnH + R'X R3SnX + R'H
что восстановление является радикальной реакцией, и осуществил
ступенчатое восстановление бензотрихлорида в толуол [41:
СвН6СС13-^СвН6СНС12->СвНБСНаС1—с6н5сн3.
Сейферт и сотр. [5] установили, что геминальные дибромцикло-
пропаны, легко образующиеся при присоединении днбромкарбена
к олефинам, можно восстановить Т. до монобромциклопропанов с
хорошим выходом при температуре не выше 40°. Например, 1,1-ди-
бром -2, 2, 3-триметилциклопропан восстанавливается 1 же реагента
Вт
0, 08 моля
0,08м о ля BuaSnH^
79%
JAX гЧ
4 ч.
Тах ~ з, 8 гц.
372
в атмосфере азота с образованием смеси изомеров, конфигурация
которых установлена с помощью ЯМР. /Можно довольно легко осу-
ществить полное восстановление до циклопропана, как это по-
казано, например, для аддукта тетраметилэтилена и днбромкарбе-
на [6]. 7,7-Дихлорноркарап восстанавливается при ~140°, и поэто-
Вг
ZBUjSnH
69% *
му одновременно легко осуществляется преимущественное вос-
становление атома брома в 7-бром-7-хлорноркаране. Описано
также селективное восстановление 1-хлор-1-фторциклопропанов до
мопофторци клоп ропаков [6].
Кювила [3] установил, что реагент при комнатной температуре
восстанавливает хлорангидриды кислот до альдегидов.
Другие реакции. Недостатком Т. при восстановлении является
его высокая реакционная способность по отношению к различным
функциональным группам, например карбонильной группе кетонов
и альдегидов [7], двойной и тройной связям [1, 8], что ограничи-
вает его применение для восстановления.
Хаус и corp. [91 установили, что реагент эффективнее никеля
Ренея и платины прн гидрогенолизе связи С—-I в иодлактоне (1).
Я)
/ . - +
(?/-Bu)3SnH, ОН , Н3О
(2)
1. Van Der Kerk G. J. М., Noltes J.G., Suijtcn J.G.H., J.
Appl, Chem., 7, 366 (1937).
2. R othman L. A., Becker E. I., J. Org. Chem., 25, 2203 (1960); Ku p-
c h i k E. J., Connolly R. E., ibid., 26, 4747 (1961); К u p c h i k E. J.,
Kiesci R. J., Chem. Ind., 1962, 1654.
3. К u i v i 1 a H. G., J. Org. Chem., 25, 284 (1961); К u i v i 1 a H. G.t Wais h
E. J., Jr., J. Am. Chem. Soc., 88, 571 (1966).
4. Kui vil a 11. G., Menapace L. W., Warner C. R., J. Am. Chem.
Soc., 84, 3584 (1962).
5. Seyferth D., Y a mazak i H.,Alleston D.L.,J. Org. Chem., 28,
703 (1963),
6. О 1 i v e г J. P., Rao U. V., Emerson M. T., Tetrahedron Letters, 3419
(1964).
7. К u i v 1 1 a H. G., Beumel O. F., Jr., J. Am. Chem. Soc., 80, 3798 (1958);
83, 1246 (1961).
8. Van Der Kerk G.J., Noltes J.G.,J. Appl, Chem., 9, 106 (1959).
9. House 11.0., Boots S. G., Jones V, K-, J- Org. Chem., 30, 2519
(1965).
373
ТРИ-//-БУТИЛФОСФИН, (н-С4Н0):1Р. Мол. ве? 202,32, т. кип.
240 (130 20 льи), рКа 8,4; бесцветная жндкс съ. с чесночным за-
пахом, не смешивающаяся с водой, но смешивающаяся с большин-
ством органических растворителей. Получают взаимодействием н-
бутилмагнийбромпда с треххлористым фосфором [1].
Зн-СД^МйВг РСЦ :')ф1'р’ Nn*.a>(H-C;iH.)).,P 3MgBrCl
•’Л1',,
Триалкил фосфины окисляются на воздухе; с увеличением ал-
кильной группы способность к окислению сначала уменьшается, а
затем снова увеличивается. Т.— наиболее устойчивое соединение
->фТ! р — ПОД!!. X.'lOI I
CCI/IOCI ' Н\(С,Н.,),-—------—
- fUi—42%
(I) (-’)
.Cl
(н-СД1„),Р +
CC13COX1(C.2H5)2 CIX^C -H'oC4H<,pP —о
4N(CTI-P
(3) (4) (Г,)
этого ряда, и поэтому чаще всего используется. Спецпале и
Фриман 12,3] применяли этот реагент для превращения N,N-;in-
этил-2,2,2-трихлорацетамида (3) в 51,\г-диэтил-1,2,2-трихлорЕН-
ниламин (4) в атмосфере азота. Эта интересная реакция включает
окисление фосфора(Ш) амидом и миграцию галогена.
Боскин и Денни 141 нашли, что при 100J Т. отщепляет кислород
от эпоксидов цис- и /пранс-бутенов-2. трянс-Эиоксид даст 72 %
щ/с-бутена-2 и 28% транс-бхтеигА-2, тогда как из que-эпоксида полу-
чается 81 % транс-бутен а-2 и 19% рас-бутеиа-2.
— С -у (н-С4Н9):1Р —* \С = С^ (и-С4Нн):!РО
о- 7 4
Галоген кетоны подвергаются под действием Т. восстановитель-
ному дегалогенированию [5|.
X О ОР1%(Х-) и о
; : I и.,о I а
с- — - —с—с--унхфИчРО
I I I
1. Kauffman G. В., Teter L. А., Inorg. Syn., 6, 87 (1960).
2. S р с z i а 1 е A. JFreeman R. С., J. Arm Chem. Soc., 82, 903 (1960).
3. Cntmia л е А. Дж., Ф р и м а п Р. Ч.. «Синтезы органических препара-
тов» иза-ио «Мир» М., 1964, сб. 12, сгр. 70.
4. В о s к । и М. J., Denney D. В., Chem. Ind., 1959, 330.
5. Hollmann IL, Di ehr 11. J., Angcv.. Chem., Internal. Ed., 3, 145 (1964).
ТРИ-Я-БУТИЛФОСФИНА ОКИСЬ, (н-С4Н0)3Р O“. Мол. вес
218,32, очень гигроскопичное твердое белое вещество, т. пл. 51 —
52 (СС14), т. кип. 300%
374
Реагент катализирует изомеризацию эпоксидов в карбонильные
производные 111. Например, /и/ттшс-эиокснитпльбсн образует дезок-
сль, ,о п
Ы-ЩНДД’О
С - С ----------- • CJ l,COCI I..CJ 15 (Си11-Д5НС1 Ю
н срр,
сибензоии и дифенил ацетальдегид в отношении 9:1. цпс-Эиоксп-
стильбен дает тс же продукты. /и/яшс-4-Эиокспоктсн превращается
в 4-октанон только в присутствии м-хлорпадбензойной кислоты.
1. В i s s i n ” D. Е., S р е z i а I с Л. .1., J . Am. Cliein. Soc., 87, 1105 (I0631.
ТРИ-н-ГЕКСИЛЭТИЛ АММОНИЯ ГИДРООКИСЬ,
|(м-С(;Н,:фД'СН,](ОН-). Мол. вес 315,57.
Получение 111. Реагент получают взаимодействием три-н-гек-
силамнна с йодистым этилом в этплацетате: образовавшийся не-
растворимый четвертичный иодид превращают в гидроокись про-
пусканием через ионообменную смолу дауэкс-1.
Содействие при ацетилировании. Корана [11 использовал реа-
гент прп ацетилировании iрипуклсотпда, который практически
нерастворим во всех растворителях. Сильно основный реагент обра-
зует соль с нуклеотидом, которая растворяется в пиридине и затем
легко ацетилируется уксусным ангидридом.
I. S г li а Н с г Н., К И о г а п я 11. С., .1. Аш. Chem. Sue.. 85. 3841 (1963).
ТРИГЛИМ, СЯ;;()СЛГСН2ОСНГ.Н.ЮСЯ/:Н.2()СЬ1,. Мол. все
178,22, т. кип. 222 . Смешивается с водой во всех отношениях.
ТРИИЗОБУТИЛАЛЮМИНИЙ, КСН:!)2СНСН2|:(А1. Мол. вес
198, 32, т. пл. 4,3, т. кин. 86 /10 мм (чтобы избежать разложения
Т. на ди изобут пл алюмин ini гидр ид и изобутилен, рекомендуется
вести перегонку при 0,5 льп), уд. вес 0,7859. Получение 111.
Восстановление кетонов. Восстановление карбонильной группы
триалкнлалю.минпем (трлэтплалюмпний) впервые было описано Меер-
вепном 12]. Циглер [31 для этих же целей) использовал Т. и показал,
что реакция протекает с образованием изобутилена:
К,С -О-; АЦСДЙгША- ДДЮ- Л1(С41-1щыщ> ' 6-Н;Щ,С--С112
! п.о
R,C1 ЮН
В восстановлении кеышов участвует только одна пзобутильная
гр\ппа.
Хаубспсток и Дэвидсон |4| установили, что восслаповлснпе 3,3,5-
1рпмстплцнклогекса[1она (дигидроизофоропа) под действием 1'.
весьма стерсосиецифичпо, по зависит от условий реакции. При до-
бавлен ип раствора кетона в бензоле к избытку Т. продукты реакции
содержат 96% аксиального ш/мяс-сиирта; в этом случае в кинетиче-
ски коп।ролирусмой стадии восстановления реагент атакует кетон
3/5
н3с
Л1 (гаи> - J3u) j
ц ас
трапе
из более доступной a-области, образуя (J-спирт. Однако при избытке
кетона возможно установление равновесия, и тогда единственным
продуктом является термодинамически более устойчивый цис-
спирт. Восстановление протекает через циклическое переходное
состояние, как и восстановление по Меервейну — Понндорфу.
Восстановление ненасыщенных лактонов. Минато и Нагасаки
15] установили, чтосс, р-непредельныеу-лактоны восстанавливаются
как Т., так и ди изобутил ал юминийгидридом вТГФ, образуя соот-
ветствующие производные фурана, например (2).
б) V)
1. Z i е ц 1 е г К., Gellert H.-G., L е h гп k u h 1 Н., Р Г о h 1 \V., Z о-
s е 1 К., Ann., 629, I (i960).
2. М е е г w е i п hl., Hinz G,, М a j е г t Н., Sonke 11., J. prakt, Chem.
(2), 147, 226 (1936).
3. Z i е g 1 е г К.., Schneider К., Schneider J., Ann., 623, 9 (1959);
Циглер К., в кн. «Химия металлоорганических соединений».
изд-во «Мир», М.. 1964, стр. 257—262.
4. Haubenstock И., Davidson Е. В., J. Org. Chem., 28, 2772 (1963).
5. М i п a t о Н., Nagasaki Т., Chem. Ind., 1965, 899; Chem. Comm., 1965
377.
ТРИИЗОПИНОКАМФИЛБОРАН. Этот реагент для гидро-
борирования получают in situ добавлением 3 молей cz-пинена к 1
молю диборапа в ТГФ при 0° [1]. В отличие от диизол инокамф ил-
+ вгщ
или димер
борана реагент взаимодействует с достаточной скоростью с транс- и
пространственно затрудненными олефинами. Поскольку d- и ь-фор-
мыа-пинена легко выделяются из природного сырья, реагент, по-
376
лученный из той или иной формы, может использоваться для опре-
деления конфигурации оптически активных спиртов, полученных
гидроборированием и окислением.
1. Brown Н.С., Ay у angar N. R., Zweifel G., J. Am. Chem. Soc.,
86, 1071 (1964).
2,4,6-ТРИ ИЗОПРОПИЛ БЕНЗОЛСУЛЬФОХЛОРИД,
2,4,6-f(CH3)aCHl3CeH2SOaCl. Мол. вес 302,86, т. пл. 96—97°.
Получают сульфохлорированием 1,3,5-триизопропилбензола [1,2].
Реагент предпочтительнее мезитиленсульфохлорида (см. соот-
ветствующий раздел) для образования С3. — С2- межнуклеотидной
связи [2]. Основным преимуществом реагента является гораздо
меньшая скорость сульфирования 5'-гндроксильной группы нуклео-
зидов.
1. Newton A., J. Am. Chem. Soc., 65, 2439 (1943).
2. Lohrmann R., Khorana H. G., J. Am. Chem. Soc., 88, 829 (1966).
ТРИ ИЗОПРОПИЛ ФОСФИТ, [(СН3)аСНО1зР. Мол. вес 208,03,
т. кип. 60—61710 мм, 43,571 мм. Получение [1].
Дехлорирование. Этот мягкий дехлорирующий агент исполь-
зуется, как и триэтилфосфит, для получения перхлорфульвалена
[21.
Диизопропилметилфосфонат. Этот эфир получают постепенным
добавлением Т. к кипящему иодистому метилу и продолжают наг-
ревание после того, как закончится первоначальная экзотермическая
реакция [3].
О
Кипячение (СН3)2СНОХ ||
КСН3)3СНО]3Р + СН31 —>Р-СН3 НСНзЬСН!
2 моля 2 моля ° '° (СНОСНО
1. Ф о р д-М ур А., Перри Б., «Синтезы органических препаратов», ИЛ, М.,
1953, сб. 4, стр. 492.
2. Mark V., Tetrahedron Letters, 333 (1961); Org. Syn., 46, 93 (1966).
3. Ф о p д-М ур А., ПеррпБ., «Синтезы органических препаратов», ИЛ,
М., 1953, сб. 4, стр. 162.
ТРИМЕТИЛАМИНА ОКИСЬ, (СН8)3Й—О. Мол. вес 75,11,
т. пл. 213—2147
Получение [1]. Воду не удается удалить полностью при азеот-
ропной сушке с бензолом, нагревании при 1057200 мм и возгонке
[2].
Синтез альдегидов. Реагент используют для превращения га-
логенидов или тозилатов в соответствующие альдегиды 131:
RCH.3Br(Ts)-|-(CH3)3N—б —> RCH,ON(CH3);s(Br)~ RCH-O !- (СН3)3МНВг
Бромид или тозилат и Т. о. кипятят при перемешивании в хлороформе
в течение 30 лшн; выходы составляют30—60%. Найдено, что альде-
гиды, полученные этим способом, можно использовать в реакции
377
Виттпга, тогда как альдегиды, полученные но реакции Розенмунда,
содержат нежелательные примеси, взаимодействующие с реактивом
Виттпга |41.
1, II i с k i п b о t t о m W. .1., «Reaction of Organic Compounrfs», Longmans
Green and Co.. Y., 1936, p. 277.
2. Al unable J. J., J. Огщ Chem.. 27. 3851 (1962).
3. Franzen V., Otto S., Chem. Ber., 94, 1360 (1961).
4. Б e p г e л ь г о н Л. Д., 11.1 е v я к н н АЕ М., Апцечс. Chem., Internal. Ed.,
3, 250 (1964).
ТРИМЕТИЛАМИНБОРАН, ’ ВНу. Мол. вес 72,96, т. ил.
94°, т. кип. 171’, белые кристаллы, устойчив в течение нескольких
часов при 125\ Растворимость (г/л при 30): в воде 14,8, в метаноле
10, в эфире хорошо растворим, в гексане 7,4; в бензоле хорошо раст-
ворим, в уксусной кислоте растворим.
Получение. Реагент получают взаимодействием триметиламииа
с диборапом плис карбонплбораиом (BII.jC.O) 111. В новом методе
121 активированный алюминиевый порошок и хлористый алюминий
добавляют к раствору трифен илбората в трпметилампне и смесь
перемешивают при 180° под давлением водорода 140 атм.
2(CH;j);iN : 2ЩОСД1-)., ! 2Л1-: ЗН2 8U-2(C.B:i)3\BH;t ; 2Al(OC(iHr,h
'1" I,
Восстановление кетонов. В обычных растворителях Т. реагирует
с кетонами очень медленно даже при нагревании па кипящей водя-
ной бапе, но в присутствии эфирата трехфторпетого бора он являет-
ся эффективным восстанавливающим агентом; 4-mpem-бутилцикло-
гексанон легко восстанавливается с образованием 46% цис- и 54%
/пранс-спирта 131.
Восстановление оснований Шиффа. Основание Шиффа, например
бензальапилин, быстро и эффективно восстанавливается реагентом
в виде раствора пли суспензии в уксусной кислоте 141. В отличие от
других боргпдридов Т. можно применять в кипящей уксусной кисло-
3C6H5CI1 — X’CfilI- : (CH;t):iNBl L. ; ЗСНзСООН/
- -3C«IE,CH.,XCftH-,
: •>- (CH3).,N -I- В(ОН).5
СОСНз
тес небольшой потерей активного водорода (рекомендуется 25%-иый
избыток реагента), ^Действующим реагентом является, по-видимому,
боран, который постепенно выделяется из комплекса и вступает
в реакцию; выходы часто высоки, причем следующие группы не
затрагиваются: NO.., С1, ОН, СО..Н, СО.СН:!, SO.NH,. Реакция
примечательна тем, что образующиеся вторичные амины ацетили-
руются уксусной кислотой.
Реакция с олефинами. Олефины превращаются в трпалкплбо-
раны при нагревании с реагентом в автоклаве при 100—200 ’ в от-
378
сутствие растворителя [5]. Предполагается, что реакция включает
диссоциацию аддукта с образованием диборапа, Олефины с впутреп-
(Cl-bp.jN: ВН3 (CH.;);,N !- РЩ ВН-Щ Н—
8(1—
пей двойной связью изомеризуются и дают первичные три ал к пл бо-
раны, например гекссн-2 образует три-н-гексплбораи.
ci i!tcii..cH2ci-i снсн;} ——v (сн3сн,сн2снхн2с:н.,)3в
1. В и г ц Л. В., S с li 1 е s ) и ц с г Н. 1., J. Am. Chem- Sac., 39, 780 (1939).
2. A s h I) v Е. С., Foster W. Е., J. Am. Chem. Soc., 84, 340? (1962).
3. Jones W. M. J. Am. Cliern. Soc., 82, 2528 (I960).
4. Billman J. H. Al cDowell J. AV., J. Огщ Chem., 27, 2640 (1962).
5. A s h b у E. C., J.'Am. Chem. Soc., 81, 4791 (1959).
О-(2,4,6-ТРИМЕТИЛБЕНЗОИЛ)-ГИДРОКСИЛАМИН,
о сн;
и ,
Н,.\ОС---с _"м—сн3
сн3
Мол. вес 179,21 , т. пл. 31—32°.
Получение. Реагент получают общим метолом, разработанным
Карпино [1, 21 для синтеза О-ароилгидроксиламппов, как это по-
казано для О-бензоилгидроксилампна (5). /?грещ-Бутилазпдофор-
миат (1) при обработке хлоргпдратом гидроксиламина и щелочью
дает /лреш-бутил-N-OKcnкарбамат (2). Бензоилированием в присут-
ствии триэтлламипа получают О-бснзонлиное производное (3),
которое при расщеплении хлористым водородом в нитрометане
дает хлоргид.рат (4), образующий при нейтрализации О-бевзоплгид-
роксилампи (5) 111 —довольно нестабильную жидкость. При аци-
лировании пространственно затрудненным мезитоил хлоридом по-
лучают сравнительно устойчивый твердый Т. 121.
О О
И.NOH ’ CJU.OCI
N3COC(CH3)3 - HONCOC(CH3)3 —~уд*
11
0 0 о о
HCT-CH...XO, 1 + - NaHLO. !
—+ Cr,H-CONCOC(CH3)3------------ Щ1Е,СОХН3С1 C(il l-CONH2
(3) m
Аминирование. При слабом нагревании смеси Т. с дпбензилами-
ном происходит перенос амидогруппы от реагента с образованием
1,1-дибензилгидразнна |21. С пространственно не затрудненным
379
О-бснзоилгндроксиламином аминирование наблюдается лишь как
побочная реакция.
О О
!! /CH.Cfilh II zCH2CeHs
(CHa)3C6H2CONH2!-HN\ ' (CH;p3C6H2COH + H2NN/
СН А н Г1 3 у % хСН2СвНг,
Под действием реагента осуществляется аминирование натриевой
соли ди-/прг/л-бутилового эфира иминодикарбоновой кислоты (6)
О О
Il II
(CH3)3CfiH2CONH, -|- NaNjCOX(CH3)3]3 —-~Г (CH3)3C6H2CONa +
do 1U '0
(6) (7)
-| H2X\[CO.,C(CH3)3h
w
с умеренным выходом [31. Реагент применяют также для аминиро-
вания сульфамидов, амидов, имидов и пирролов [4].
Н
CH.XSO, —/ —СН;,
{. С а г р i п о L. A,, G i z а С. A., Carpino В. A. J. Am. Chem.
Soc., 81, 955 (1959).
2. Carpino L. A., J. Am. Chc-m. Sue., 82, 3133 (1960).
3. Carpino L. A., J. Org. Chem., 29, 2820 (1964).
4. Carpino L. A., J. Org. Chem., 30, 321 (1965).
ТРИМЕТИЛ БОРАТ, (CH3O)3B. Мол. вес 103,92, т. кип. 68,7°.
Небольшие количества реагента можно легко получить из борного
ангидрида и метанола [1].
2 В2О3-|-ЗСН3ОН —► 3 НВО2-уВ(ОСН3)3
Одно из применений Т. состоит в превращении а рил галогенидов
в фенолы, если невозможен непосредственный гидролиз [2]. Т. мед-
ленно обрабатывают раствором фенилмагнийбромида при —80э
и гидролизом аддукта получают смесь фенилборной и дифенилбор-
380
ной кислот, расщепление которых перекисью водорода в эфире дает
с хорошим выходом фенол.
С.Н1М6ВГ + (СН.О)3В да} с.н3он
1. ДжерардВ., «Химия органических соединений бора», Химия, М., 1966,
стр. 13 и сл.
2. Hawthorne М. F., J. Org. Chem., 22, 1001 (1957).
ТРИМЕТИЛБОРОКСИН, (СН3ВО)3. Мол. вес 124,75, т. кип.
79—80°.
Получение. Это соединение впервые получено с 12%-ным вы-
ходом дегидратацией метилборной кислоты, синтезированной из
триметилбората и метилмагнийгалогенида [11. Впоследствии Ратке
и Браун [21 нашлн препаративный метод — реакцию окиси угле-
рода с днборапом в ТГФ. Перегонка реакционной смеси дает триме-
сн3
I
/в \
о о
ЗВН3 1-ЗСО---> I [
84% I I
тилбороксин с превосходным выходом.
Разделение цис- и mpawr-диолов. Три-я-бутнлборокспн
(см. соответствующий раздел) применяют для разделения и идентифи-
кации изомерных цис-транс-диолов. Применение Т. для этой цели
имеет явное преимущество, так как циклический эфир более летуч
и более устойчив к действию атмосферного кислорода.
1. Hillman М. Е. D., J. Am. Chem. Soc., 84, 4715 (1962); 85, 982, 1636 (1963).
2. R a I h k е M. W., В r own Н. С., J. Am, Chem. Soc., 88, 2606 (1966).
ТРИМЕТИЛЕНА ОКИСЬ, (СН2)3О. Мол. вес 58,08, т. кип. 48°.
Получают из 3-хлорпропилацетата [1],
Реакция Гриньяра. Взаимодействием реагента с бензилмагний-
хлоридом, фенилмагнийхлоридом, фениллитием и 1-нафтилмагний-
о I) Присоединение
C6HsCH2MgCl + Г" I _|2)НгО с6н5сн2сн2сн2сн2он
1 1 83%
бромидом получают первичные спирты с выходами 80—85% [21.
В применении к вторичным алкилмагнийгалогенидам реакция не
дает таких хороших результатов. Например, циклогексилмагний-
бромнд дает нормального продукта меньше, чем побочного 3-бром-
пропанола-1, который, как показано, образуется из Т. о. и бромида
магния.
381
ан
MgBr
нение
Z) Н?О
-------
1) Присоеди-
ан
СНгСН2СНгОН
28%
|MgBr;
ВгСНгС11гСНгОН
4 0%
J, И о л л с р 1\., «Синтезы органических препаратов», ИЛ, At, 1953, сб- 4,
стр. 398.
2. S с а г 1 е s S., J. Ain. Chcui. Soc., 73, 124 (1951).
ТРИМЕТИЛОКСОНИЯ БОРФТОРИД, (СН:,)Я(У BF;. Мон вес
147,92, т. пл. 141 —143‘. Получение 111.
Подобно борфгориду триэтилоксоння, эта соль является потен-
циальным алкилирующим агентом. Она получается легче, чем
2,4,6-трип пиробензол сульфон ат гриме гил оксон ня, ио -менее устой-
чива при хранении.
1. А! е с г \\ о i п 11., Ощ. Syn., 46, 120 (1966).
ТРИМЕТИЛОКСОНИЯ 2,4,6-ТРИНИТРОБЕНЗОЛСУЛЬФОНАТ.
Мол. вес 311,25. т. пл. 181 —183°. Получение III.
АО.,
О.,.\ - — SO;111 -- (СН3),.О СМ. \ ,
,--/
Х\о..
А’О..
/
—> ОА—— SQ-(CM.)3On- N.
4 хо;
Этот пощпппальпып алкилирующий агент более труднодоступен,
чем борфторпд трпметплоксоипя, однако он не гигроскопичен и бо-
лее устойчив при хранении.
1. И с 1 ги 1< и Ill р G. к., Р с 1 1 i t 1 D. J., Grip Syn., 46, 122 (1906).
ТРИМЕТИЛСИЛАН, (CI Мол. вес 74,20, т. кин. 6,7°,
т. ил. —136’. Получение [1|.
1,4- Дигидропиридин. Хотя некоторые Х-этпл- и Х-арнлдпгпд-
ронпридины были известны, первый из пяти возможных изомеров
незамещенных дпгпдроппрпдинов был получен Куком и Лапонсо.м
|3|. Катализируемое палладием присоединение Т. к пиридину дает
1-трнметилсплпл-1,4-дигпдропнрндии (т. кип. 57 ), из которого при
обработке метаноло.м, содержащим 0,1% едкого кали, получают
1,4-днгидронпридин и метокситриметплсплаи. Соединения (1) и (2)
382
легко реагируют с кислородом, поэтому их следует очень тщательно
защищать от действия воздуха,
и н н н
Ipj: Т!. |р |р--сидаснл
\ х‘ N
I I
Si(CH;I)3 И
(О *2)
1, Tannenbaum S., К л у е S,, L o\v е n z G, F.. J. Лги. Chem. Soc., 75
3753 (J953).
2. Private communication.
3, C no k X. C., Lyons J. E. Am. Chem. Soc., 87, 3283 (I9(>5).
ТРИМЕТИЛСИЛ ИЛАЗИД, (CH:!)3SiN 111. Мол. вес
11 5,21, т. пл. 95'. Этот устойчивый азид получают с 87%-ным вы-
ходом реакцией триметплхлорсилана с азидом натрия в ТГФ;
хлористый натрий отфильтровывают и продукт перегоняют. Реагент
используют вместо взрывоопасной азотистоводородной кислоты
в синтезе 4-фенил-1,2,3-трназола, триметплсилильная группа затем
легко удаляется при обработке водой.
сн
(CH3)3SiN=^=N + CC6HS -------> (CH,)3SiN—СН —HN----------СН
I If I II
n^n^cc6h5 n^nx.cc6h5
Подобное циклоприсоединение реагента к диметпланетнлену
с последующим гидролизом дает 4,5-диметпл-1,2,3-трназол 12[,
I. В ir holer L., Kilter Л., Aiiycw. Chem., Inicrnnt. IS d., 4, 417 (19G5).
2. В i г к и f с r I.., \V с с п e r P,, procedure submitted to Ощ. Syn.
6uc-(O, N-ТРИМЕТИЛСИЛ ИЛ)-АЦЕТАМИД, (1). Мол. вес 203,44
т. кип. 71—73/35 лип. Т. получают с 8О‘’п-нЫхМ выходом реакцией
ацетахмида с трнметилхлорсиланом в присутствии триэтилампна
как катализатора. Этот реагент является эффективным средством
для триметилсилилирования амидов, мочевин, аминокислот,
фенолов, карбоновых кислот, сполов |1|.
osi(CHtl);i н СН(СХ
СН3С- XSi((’.Н;Щ,СДI- — Х'СОСН, 1
(1) (2)
Si(CH.,). о
I II
—> СеН5-.ХСОСН3 CH3(.XSi(C]I3)3
I
н
383
J. Klcbe J.F., Fi nkbei пег H., White D.M.. J. Am. Chem, Soc.,
88, 3390 (1966).
N-ТРИМЕТИЛСИЛИЛАЦЕТАМИД, CH3CONHSi(CH3):J.
Мол. вес 131,25, т. кип. 84713 мм.
Получение реакцией триметнлхлорсилана с ацетамидом в при-
сутствии триэтиламина [11.
Реагент силилирует спирты в очень мягких условиях; единствен-
ным побочным продуктом является нейтральный ацетамид. Исчер-
пывающее силилирование углеводов достигается ие продол житель -
ROH-J-CH3CONHSi(CH3)3 —> ROSi(CH3)3^CH3CO\H2
иым сплавлением с реагентом. Методом сплавления глюкоза превра-
щается в 1,2,3,4,6-лентш<«с-(О-триметилсилил)-глюкозу. В пиридине
реакция осуществляется более мягко; удивительно, что в этом слу-
чае первичная гидроксильная группа остается незатронутой и в ре-
зультате реакции образуется 1,2,3,4-тетракис-(О-триметилсилил)-
глюкоза (2) [21. Легкая доступность этого соединения открывает
путь к синтезу производного генциобиозы (4), которое получают
превращением (2) в натриевое производное (3) конденсацией с ацето-
бромглюкозой и десилилированнем водной или спиртовой соляной
кислотой.
неон
I
неон
I
носн о
I
неон
нс -——
I
СН..ОН
(1)
СН ;,CONIHSi (СНщ
Ру
I
HCOSi(CH3)3
Hcosi(CH3),
I
(CH3)3SiOCH
!
HCOSi(CH3\3
I
HC — -
CH,OH
I
H(.OSi(CH3)3
I
HCOSi(CH3)3
(CH:;)3sioc:H
HCOS’(CH3)3
HC—-—-
Лцртобромглюкоза
Деси.милированне (НС!)
I
С,Н..О\а
384
I---
I-ICOH
I
HCOH
I О
HOCH
I
HCOH
HC^—
CH3—
Г~~]
---CH
HCOAc
I о
AcOCH
I
HCOAc
CHnOAc
(4)
I. Birk of er L., Ritter A,, Dickopp H., Chem, Ber., 96, 1473 (1963)
2. Birkofer L„ Ritter A„ Bentz F., Chem. Ber., 97, 2196 (1964)
&/с-(ТРИМЕТИЛСИЛИЛ)-НАТРИЙАМИД, (2). Мол. вес 183,39,
т. пл. 165—167°. Реагент получают из гексаметилдисилазана и
амида натрия в виде суспензии в бензоле [11:
Н Na
(CH3)3SiNSi(CH3)s-i-NaNH2 —> (CH3)3SiNSi(CH3)3 +NH3
(I) (2)
В синтезе используют полученный таким образом раствор или,
удаляя растворитель, выделяют основание в виде бесцветного
кристаллического вещества.
Под действием Т. еиолизирующиеся кетоны почти количественно
превращаются в соответствующие еноляты. Нееиолизирующиеся
кетоны реагируют так, как это показано на примере бензофенона
[21:
/SifCHgh /ONa
(CflH5)2C = О-pNaN ——* (CeH5)2C Si(CH3)3 >
XSi(CH3)3 xnZ /
Si(CH3)3
—> (C6H5)2C= NSi(CH3)3 + NaOSi(CH3)3
В случае 1,4-бензохинона с реагентом конденсируются обе карбо-
нильные группы.
Реагент применяют для промотирования конденсации простран-
ственно затрудненных соединений, например: этилизобутират
этил изобутироил изобутир ат (30%) [31. Под действием Т. осущест-
вляют циклизацию суберодинитрила в а-циаициклогептанонимнн.
ZTcH3)3Si72NNa
Эфир (25°)
96%
Qnh
CN
Реагент используют также как основание в реакции Виттига (бен-
зофенон -> 1,1-дифенилэтилен; выход 92%).
13 Заказ № 1166
385
1. \V anna^al U., N i e d с r p r ii rn If., Chem. Ber., 94, J.>40 (1961).
2. К г ij ger C., R о e. h о w E. Ci., \V а п п a у a t U., Chem. Ber., 96, 2131
(1963).
3. I< г ii gerC. R., R о c h о \v E. G., An^cw. Chern., Intcrnat. Ed., 2, 617 (1963),
ТРИМЕТИЛСУЛЬФОКСОНИЯ ИОДИД, (CII;i)^-О(Г). Мол. вес
220,08, разд. прп 200 ,
Реагент получают кипячением ДМСО с йодистым метилом в те-
чение нескольких диен III. Склонность к превращению в ДМСО
1+
CH3S сн3
о"
он
+ (снОз^ор-)
33 7 80%
NO;
осн3
+ CH3S+CH3 + Agl
NOa
обусловливает высокую метилирующую активность реагента.
Т. и.— исходное вещество для синтеза диметилсульфоксон и имети-
лпда.
1. К и li ii R., Tr jsHi m и пп IF., Ann., 611, 117 (1958).
ТРИМЕТИЛСУЛЬФОНИЯ ИОДИД, (СНДДЧэ Мол. вес 204,08.
Это исходное соединение для синтеза диметилсульфоипйметплида
получают с почти количественным выходом путем смещения
диметплсульфпда (6 г) с подпетым метилом (14 г) 111. Продукт выпа-
дает в виде твердого сгустка, который отделяют через 24 час, кри-
сталлизуют из спирта и промывают эфиром.
1. Emele us II. J., Ileal If. G., J. Chem. Soc,, 1946, 1126.
ТРИМЕТИЛ-(ТРИФТОРМЕТИЛ)-СТАННАН, (CH3)sSnCF3.
Мол. вес 232,81, т. кип. 100—ЮГ.
Получение 11]:
(CETbSii Sn(CH3):j -1 CE;iI —v (СН:ЩЗп I ! (CHn)3SnCF3
Получение днфторкарбена. При нагревании в течение 20 час
в запаянной ампуле в отсутствие воздуха реагент гладко разлагает-
ся с образованием триметплфторстапиана и перфторциклопропана
3(CH3)3SnCF;t 3(CI-I;t).TSnF -| 3 :С1д —„ F,C~ CF,
'\ /
CF.,
11]. Перфторциклопропап образуется также при разложении реа-
гента в присутствии избытка тетрафтор этилена.
386
Сейферт н сотр. [2] установили, что дпфторкарбен образуется
при взаимодействии реагента с иодидом натрия в 1,2-дпметоксиэтане
прн 80“. Газовым хроматографическом анализом отфильтрованной
12 vac при 80°
„т т СН3ОСН,СН,ОСГН I (Хх17
Г П + (CH3)3SnCF3 + Nal -----------2---I--z----I l/X,
п,8 жмоля 15 лгмолей
100 Л7М0ЛЕЙ
реакционной смеси после взаимодействия с циклогексеном пока-
зано, что в ней содержится триметил подставная (90%) и 7,7-дифтор-
норкаран (73%).
1. Clark II. С., Willis С. J., J. Am. Chem. Soc., 82, 1888 (I960).
2. S с у I е г t h D., М u i J. Y.-P., Gordon At Lt, В u r 1 i t c h J. AV,
J. Am. Chem. Soc., 87, 681 (1965).
ТРИМЕТИЛУКСУСНАЯ КИСЛОТА ( пивал ивовая кислота),
(СН;ф(ССО;Н. Мол. вес 102,13, т. пл. 35J, т. кип. 164'7
Получение карбонизацией /лреш-бутплмагпийхлорида (61 —70%)
[11 и окислением пинаколнпа гнпобромптом (71—74%) [2[.
1. [] v и т ам бе к с р, Целльпср, «Синтезы органических препаратов*,
ИЛ, At, 1949, сб. 1, стр. 410.
2. Сандборн, Б v с к е г, «Синтезы органических препаратов», ИЛ, Ш,
1949, сб, 1, стр. 412.
ТРИМЕТИЛУКСУСНОЙ КИСЛОТЫ ХЛОРАНГИДРИД (пива-
лоил хлорид), (CH;!):iCCOCI. Мол. вес 120,58, т. кип. 105 —1067
Синтез пептидов. В ранних исследованиях применения сме-
шанных ангидридов в синтезах пептидов Воган и Оса го [11 кратко
упомянули о пивалоил хлор и де, считая, однако, что он несколько
хмже хлорангидрида изовалерпаповой кислоты. Наорал 121 вновь
ввел реагент в частности для экранирования карбонильной группы
вспомогательного кислотного компонента и получил выходы лучше,
чем прн использовании emop-бутплхлорформиага. Кеннер и сотр.
131 установили, что этот метод применим для синтеза пептидов,
содержащих пространственно затрудненный а-метплалаиин. При
обработке триэтпламмониевой соли СЬ-сс-метилаланина иивалоил-
хлоридом количественно образуется кристаллический смешанный
ангидрид:
СН:{
I <-
СЬ\НССОД.\Н(С2Н.Да : (С113)3CCOC1 —
СПз
СН3О О СЛл
И hi,
CbXHC — С — О-С — С-61% !-(С>Н-,)3 X HCI-
I I
СН3 СП3
13*
387
Швайцер и Зибер [4] применили указанный метод для конденса-
ции пептидов со стороны пролинового остатка.
1. Vaughan J. R., Jr., Osa io R. L., J. Am. Chem. Soc., 73, 5553 (1951).
2. Z a or a I M., Coll. Czech., 27, 1273 (1962).
3. L e p I a w у M. T., J ones D.S., Kenner G. \V., Sheppard R.C.,
Tetrahedron, 11, 39 (1960).
4. Schwyzcr R., Sieber P., Nature, 199, 172 (1963).
ТРИМЕТИЛФЕНИЛАММОНИЯ ТРИБРОМИД, см. Фенилтри-
метил аммония пербромид.
ТРИМЕТИЛ ФОСФ ИТ, (СН3О)3Р. Мол. вес 124,08, т. кип. 111°.
Аддукты с хинонами, ос-днкетонамн и ос-кетоэфнрами. Рамирец
и сотр. [11 установили, что хлоранил и другие 1,4-бензохиноны легко
реагируют с Т. с образованием метиловых эфиров гидрохинон-
монофосфатов, например (3), и доказали, что первичным продуктом
является диполярный ион (2). Фенаптренхинон реагирует с Т. в
бензоле при 20° и образует бесцветный кристаллический цикличе-
(СН.ЮЦР
О
N ZOCH3
О —Р(
I ОСНз
ОСНз
(3)
ский ненасыщенный оксифосфоран (5) с количественным выходом.
Озонолиз аддукта дает перекись дифеноила (6), Бензил и диацетил
дают аддукты, плавящиеся при 50 и при 46° соответственно.
С метиловым эфиром пировиноградной кислоты Т. образует
жидкую смесь двух возможных диастереомерных циклических орто-
фосфатов; при обработке 1 же 2 и. едкого натра ортофосфаты дают
диастереомерные диметил-С.С-диметилтартраты [2]. Реакция пред-
ставляет новый метод образования углерод-углеродной связи,
388
со2сн3
СО2СН3 сн3 — с—о.
i | Р(ОСН3)3 NaOH
2СН3С^О--(СН3О)аР —СН3-С~О/ ____>
83% ] 87%
СО2СН3
со2сн3
I
СН3-С—он
* СНз-С-ОН
I
со2сн3
Синтез олефинов. Кори и Уинтер [31 разработали новый метод
синтеза олефинов из 1,2-диолов, используя на первой стадии реак-
цию с тиокарбоиилдиимидазолом (2); реагент получали из 2 молей
имидазола и 1 моля тнофосгеиа. Кори предположил, что при обра-
ботке циклического тионкарбоната (3) реагентом, который отщепля-
ет серу с образованием карбена (4), должно осуществляться цис-
элиминироваиие до олефина, и установил, что под действием Т.
эта реакция осуществляется гладко и с высоким выходом. d,l-
Гидробензоин при такой же обработке дает чистый транс-стильбен
с выходом 87%. Метод Кори и Уинтера используют для синтеза
ненасыщенных сахаров [4].
s
и
CICCI
------►
.мезо -Гидробензоин
О) (2)
О^Схо (СНЭО)3Р
----> I I --------------------►
С6Н5-С — C-C6HS
А А
(3)
р ?
с6н5-с-----С-С6Н5
I I
н н
(4)
92%
СДЦС-П f(CH3O)jPS
II + \
C6HSC-H со2
Дегидрогалогенирование. Хунздикер и Мюльнер [5] осуществили
аллильное бромирование холестерил бензоата в кипящем петролей-
с6н5сог
389
ном эфире, кристаллическую смесь 7а- и 7₽-бромхолестер ил бензоатов
отделяли ii обрабатывали Т. в кипящем ксилоле. Кристаллический
продукт реакции содержит 56% 7-дсгидрохолестер ил бензоата, что
соответствует выходу 52%. Предполагают, что в продукте броми-
рования содержится диеновый сложный эфир, образующийся из
7а-холестерил бензоата (транс-элиминирование).
Отщепление кислорода. Озонолиз олефина в метаноле при —40а
дает гидроперекись и карбонильный фрагмент. При добавлении
к смеси Т. при —40° происходит восстановление гидроперекиси
до карбонильного соединения [6].
^С = с/+О3--СН3ОН -^С—ООН ]- х = о
ОСНз
1—х>с=о%сн3он%(сн3о)3р=о
Окиси нитрилов легко отщепляют кислород с высоким выходом
под действием Т. и трнэтилфосфита [7]. Реакцию проводят при на-
гревании смеси реагентов в бензоле иа кипящей водяной баие в те-
чение 5—10 мин. Выделение нитрила облегчается тем, что обра-
+ —
RC— N-0 4 (СН3О)3Р RC-^N-I (СН3О)3РО
зующийся триалкилфосфат растворяется в воде. В случае раствори-
мых в воде нитрилов используют трифенилфосфпн.
Дебромнрованне. Т. дебромирует вицинальные дибромиды, в ко-
торых один или оба галогена находятся рядом с карбонильной груп-
пой 181. Применение подпетого натрия приводит к осмолению; цин-
ковая пыль восстанавливает двойную связь.
RCHCHCOR' RCH —CHCOR'
! !
Br Вг
1. Ramirez F., Desa i X. B., J. Ain. Chem. Sue., 85, 3252 (1963); and ear-
lier papers cited.
2. R a m i г e z F., Desai №. B., R a manat lian X., Tetrahedron Letters,
323 (1963).
3. Corey L. J., Winter R. A. L., J. Am. Chem. Soc., 85, 2677 (1963).
4. Horton D., Turner \V. N., Tetrahedron Letters, 2531 (1964).
5. Hunziker F., M ii 1 1 n e r F. X., Helv. Chim. Acta, 41, 70 (1958).
6, К n о w 1 e s W. S., T h о m p s о n Q. L., J. Org. Chem., 25, 1031 (I960).
7. Grund rn a n n C-, From tn с I d H.-D.. J. Orc. Chem., 30, 2077 (1965).
8. Der s h о witz S., P г о s k a u e г S., J, Org. Chem., 26, 3595 (1961).
ТРИМЕТИЛХЛОРСИЛАН, (CH3)aSiCI. Мол. вес 108,65 т. кип.
57,3°.
Под действием Т. в присутствии триэтил амина или пиперидина
силилируются гидроксильная, амино- п карбоксильная группы.
390
Однако N-триметил с ил пл ацетамид — более подходящий реагент
для этой цели [ 1).
КОП HCHnhSiCl--(СЛЬЛЛ ROSi(CH3)!t -(С.,Н-)ЛнС1
1. В i г к о f е г L., R i t t о г Л., Atu'cw. ('hem., Internal. К;1., 4, 417 (1965).
ТРИМЕТОКСИБОРГИДРИДНАТРИЙ, NaBH(OCH.);- Мол. вес
127,93, т. пл. 230°.
Реагент получают из гидрида натрия и трпметплбората [11.
Х'аН -В(ОСНл)з 2LX NaBH(OCH3)3
Т. используют для восстановления альдегидов, кетоиов, ангид-
ридов и хлор а и гидридов кислот. Эфиры и нитрилы восстанавливают-
ся медленно прп повышенных температурах [21.
1. В г о w п И. С., Schlesinger Н. J., Shell ]., Ritter D, ДЕ, J,
Am. Chem. Soc., 75, 192 (1953).
2. Bro w n H. C., M e a d E. J., J, Ain. Chem. Soc., 75, 6263 (1953).
1,3,5-ТРИНИТРОБЕНЗОЛ, С(1Н,(\Ю.,).,. Мол. вес 213,10, т. пл.
1220
TH Б получают окислением тринитротолуола (ТНТ) би хроматом
натрия в разб. серной кислоте при 45—55е [11. 2,4,6-Трииитробен-
зойную кислоту отделяют от непрореагпровавшего ТНТ экстракцией
разб. щелочью и нагреванием фильтрата до кипения осуществляют
декарбоксилирование; общий выход 43—46% [2].
л-Комплексы с реакционноспособными углеводородами. Эта
л-кислота образует с углеводородами комплексы, удобные для их
идентификации и выделения, температуры плавления которых про-
межуточные но сравнению с комплексами пикриновой кислоты
(ПК) и трниитрофлуорсиона (ТНФ). Например, температуры плав-
ления п цвет комплексов метилхолаитрена следующие: ПК 182,5°,
фиолетово-черный; ТНБ 204,5°, темно-красный; ТНФ 254°, зеленый.
1. К л а р к, Хартма и, «Синтезы органических препараты;», ИЛ, М., 1949,
сб. 1, стр. 416.
2. К л а р к, Хартма и, «Синтезы органических препаратов», ИЛ, ДЕ, 1949,
сб. 1, стр. 418.
ТРИ-я-НИТРОФЕНИЛТИОФОСФИТ, P(SCfiH4\TO.>-n)3. Мол. вес
397,28, т. пл. 166—167", желтый. Получение [11-
Сннтез пептидов. Т. применяется в синтезе циклических пеп-
тидов при циклизации, когда С-концевая ам инок пел ота — глицин
(в случае оптически активной аминокислоты наблюдается рацеми-
зация) [2].
I. Farrington J.A., Hext a It P.J., Kenner G, \V., Turner
J. M,, J. Chem. Sue., 1957, 1407.
2. Kenner G. W., T liomson P. J., Turner J. M., J. Chem. Soc,,
1958, 4148; К e n п e г G. \V., Laird A. H,, Chem. Comni., 1965, 305.
391
2,4,7-ТРИНИТРОФЛУОРЕНОН, о NOi
Мол. вес 315,19, т. пл. 175—176°.
Т. получают из флуоренона с вы- ----L^^Jnoz
ходом 75—78?6 111.
Т. образует л-комплексьг, удобные для выделения и идентифи-
кации ароматических углеводородов [21.
I. В у л ф о л к Э., О р ч и и М., «Синтезы органических препаратов», ИЛ, М.,
1953, сб. 4, стр. 490.
2. О г с h i n Al., Woolfolk Е. О., J. Am. Chem. Soc., 68, 1727 (1946); О г-
с h i п At, R е g g е I L., W о о 1 f о I k E. O., J. Am. Chem. Soc., 69, 1225
(1947).
ТРИТОН Б (гидроокись бензилтриметил аммония),
С(}Н5СН2(СН:фДтОН. Мол. вес 167,25. Растворяется как в гидроксил-
содержащих, так и в бе зги д рокси ль ных растворителях.
Катализатор конденсации. Благодаря высокой основности и раст-
воримости в различных растворителях тритон Б широко исполь-
зуется в качестве катализатора реакций конденсации. Примером
реакции Михаэля является конденсация 2-иитропропана с метил-
акрилатом, приводящая к образованию метил-у-метил-у-нитровале-
рата Ц].
(CH3)2CHNO2-}-CH2-CHCO2CH3 ! Тритон Б s5~10_°^
I моль 1 моль 60—86%
В 50 .ил диоксана
(СН3)2ССН,СН2СО2СН3
no2
Гарднер и сотр. ]2] установили, что тритон Б является превос-
ходным катализатором конденсации альдольного типа аромати-
ческих альдегидов с этилиденмалоновыми эфирами, которая являет-
ся одной из стадий в новом методе синтеза бензосуберонов.
Тритон В;
С6Н5СНО + СН3СН=С(СОгСгН5)г Г^Р°ЯИЛ С6Н5СН = СНСН=С(СО2Н)г---->
98%
--Hz-pd> с6Н5СН2СНгСНгСИ(СО2Н)г ~СОг> с4н5снгснгснгснгсогн-->
84% 84%
Полифосфорная кислота(95°)' у ]] )
67% '
(В форме этилрчкеталя);
Реагент эффективно катализирует оксиметилирование произ-
водных ацетонитрила, содержащих по крайней мере одну а-ариль-
ную группу [31. При взаимодействии нитрила и параформальдегида
392
в пиридине в присутствии тритона Б получают р-ок с и пропионитрил
(CJIJ2CHCN-; СН.,- Oi Тритон Б - 'iar "'11--^ (CfiH5).,CCH.OH
I
CN
часто с прекрасным выходом. Тритон Б эффективно катализирует
также альдолыгую конденсацию, описанную при получении тетра-
фен илциклопентадиенона.
Тритон Б — очень подходящий катализатор для реакция циан-
этилирования [41. С катализируемого тритоном Б цианэтилирования
начинается построение кольца А в полном синтезе стероида, осущест-
вленном группой Вудварда [51.
Образование карбанионов. Сиринзак [6] показал, что соединения
ряда циклопентадиена под действием тритона Б в пиридине легко
превращаются в карбанионы, очень чувствительные к кислороду
воздуха. При добавлении реагента к раствору флуорена в пиридине
образуется оранжево-желтый раствор, который быстро поглощает
кислород; после перемешивания в течение 40 мин кристаллизующий-
ся флуоренон выделяют с высоким выходом. При низкой температу-
ре (от —15 до —40°) 9-алкилфлуорены превращаются в гидропере-
киси 9-алкилфлуоренов; при температуре 40° продуктом является
si%
Ог, Ру,Тритон В (-40°),
соответствующий 9-алкил-9-флуорснол. Индеи дает аморфный про-
дукт сложного строения, по при окислении 2,3-дпфенилиндена при
—40° образуются приблизительно равные количества кетона и
о
гидроперекиси. Эфиры дпарилуксусиых кислот н 2,3-днарилпро-
пиоповых кислот в системе пиридин — тритон Б также подвергают-
ся автоокислению до гидроперекисей с выходами 30—45%; наряду
с этим образуются а-оксиэфиры и кетоны [7]. Область реакций авто-
окислеиия расширяется при использовании mpem-бутилата калия
в апротонном растворителе, как это иллюстрируется реакцией
изопропилметплкетопа [8].
393
СН3СОСН(СН3)2 в (СН3)3СОН+СН3ОСН2СН.,ОСН3
I .ИЛ 1 ,11.t
О О.,Н
.wi/i НЬП — К- I ' /j,
oL„ кощснщ ,,u r
-------------——> k j г! з Си — С s
4 0% \СН3
При взаимодействии альдегидов с инденом и флуореном в ат-
мосфере азота в пиридине, содержащем тритон Б, осуществляется
конденсация альдольного типа [9].
1. Мофф ci г Р., «Синтезы органических препаратов», ИЛ, М., 1953, сб. 4,
стр. 319; other examples: Walker G. N., J. Am. Chem. Soc., 77, 3664 (1955);
Julia S., Bonnet Y., Compt. rend., 243, 2079 (1956).
2. G a r (1 п с r P. D., Horton W. J., Thompson G., Twelves
R. RJ. Am. Chem. Soc., 74, 5527 (1952); Horton W. J., P i t c h I о r t h
L. L., J. Org. Chem., 25, 131 (I960).
3. Avramoff M., Sprinzak Y., J. Ore. Chcin., 26, 1284 (1961).
4. Bruson H. А., 0гц. Reactions, 5, 79 (1949); Sheehan J. С., M u m a w
С. E., J. Am. Chem. Soc., 72, 2127 (1950).
5. W nod w ar d R. B., Sondhei m er F., Taub D., Heusler K-,
M cL a m or e W. M., J. Am. Chem. Soc., 74, 4223 (1932).
6. Sprinzak Y., J. Am. Chem. Soc., 80, 5449 (1958).
7. A v г a m о f f M., Sprinzak Y., J. Am. Chem. Soc., 85, 1655 (1963).
8. Gcrsni a n п H. R., Nieuwenhuis H. J. W-, Bickel A. F.,
Proc. Chem. Soc., 1962, 279.
9. Ghera E., Sprinzak Y., J. Am. Chem, Soc., 82, 4945 (1960).
ТРИФЕНИЛБРОММЕТАН, (CGH-,).CBr. Мол. вес 323,23, т. пл.
153—154°.
Получение Ц|. 2Мстодика аналогична получению трифен ил хлор-
метана [2]. В типичном опыте из 220,8 г трнфеннлкарбпнола, 122,9 г
ацетилбромпда в 100 мл бензола получают Т. с выходом 90%;
т, пл. 153—154°.
Тритиловые эфиры. Т. лучше, чем трифен ил хлорметап, для
превращения солей кислот в тритиловые эфиры 111, поскольку он
более активен, менее гигроскопичен и реагирует с сухими серебря-
ными, натриевыми и калиевыми солями кислот. В качестве раство-
рителя чаще используют бензол.
RCO.,Afi(K, Naj-l-(CGH5)3CBr —2^^ RCO.,C(CGH5)3 + AgBr
S5—45%
R — С1'13, C2H5, (CH3)3C, C8H5, CrH^CII—CH
1. Ber I in K. D., Gower L. J., White J. W., Gi li hs D. E.,S lur m
G. P., J. Org. Chem., 27, 3595 (1962).
2. Бахма и В., «Синтезы органических препаратов», ИЛ, М., 1952, сб. 3,
стр. 426.
394
ТРИФЕНИЛДИБРОМФОСФОРАН, (СГ[Н,КРВг2, см. Трифенил
фосфиндибромид.
ТРИФЕНИЛМЕТАНСУЛЬФЕНХЛОРИД, (С,;НГ1);:С8С1. Мол. вес
.310,84, т. ил. 137\ желтый. Получение |1|.
N-Защнтная группа. Зервас [21 применил этот реагент для
N-защиты эфиров аминокислот, в том число и активировании?;
п-нитрофен иловых эфиров, используемых в пептидных синтезах.
Защитная группа удаляется обработкой теоретическим количеством
хлористого водорода в спирте,
(GJ-L^CSCl -Н ,\CHRCOT'cHjXO2hi —V
—V (GH-):fCS-XHCHRCO2CriH4XO.,-/i
(i)
, НО
—> (C(;II5)3CS — XHCHRCO — XHCHR'CO3R->
. XILCHRCO— XHCHR'COJT
(3)
I. V or 1 а nder D.. Mi t t а ц П-. Ber., 52, 413 (1914).
2. 7. e r v ;i s L., В о г о v а ч [)., G ;i z i s D., J. Am. Chem. See., 85, 3060
(1963).
ТРИФЕНИЛМЕТИЛКАЛИЙ, (C«H;,):,CK (темно-красный). В од-
ном из методов [ I ] реагент получают взаимодействием трифен пл мета на
с калием в жидком аммиаке; замена аммиака эфиром приводит
к образованию суспензий реагента, причем эфир служит раствори-
телем, пригодным для ацилирования, алкилирования и карбони-
зации. Тритплнатрпй также можно получить этим методом из
трнфенилметана, ио он разрушается при замене аммиака эфиром;
получение эфирного раствора из тритил хлорида и натрия гораздо
более продолжительно, чем получение Т. из трнфенил мета на. Кроме
того, трифепилметан выделяется после конденсации с хорошим вы-
ходом и пригоден для повторного использования. Типичные реак-
ции, иллюстрирующие применение Т.: сложноэфирная конденсация
(1) [I!, ацилирование (2) [1], алкилирование (3) L11 и карбонизация
(4) 121.
(щнщск
1) 2 (CH3).>CHCII,CH2C(.).1GIB, - -
‘ <Л"„
—> (С) 1.ЩС1 ICHXHCO.,GHS
(СНд.СТГСНЩнД)
ю,йи Л ,с К
2) СКН5С( )С1 --(С.Нд,СНСН,СО,С,Н71 —— —>
—-(СН;1),СНСНСС).,С2Н5
С6Н5СО
395
3) С.ЛЬ,1 + (ClI3)XlXHXOX.,H5 (S“H£iCK> (СН.0ХНСНСОХ2Н5
ТЦи I
C3II5
о о
I I
C(CH;1)3 C(CH3)3
Хауз и Крамар [3] получили Т. из трифепилметана и калия в
1,2-диметокснэтапе и использовали его для получения енолятов
калия.
1, Levine R., Baumgarten Е., Hauser С. R., J. Am. Chem. Soc.,
66, 1230 (1944).
2. Sicher J., S i p о s F., T i c h у At., Coll. Czech., 26, 847 (1961).
3. House H.O., Кг a m a г V., J. Org. Chem., 28, 3362 (1963); see also H о u-
s e H. O., Trost B. At., ibid., 30, 1341 (1965).
ТРИФЕНИЛМЕТИЛЛИТИЙ, (CeEI5)3CLi. Мол. вес 250,25, крас-
ные растворы.
Гильман и Хей [1] получали этот реагент добавлением эфирного
раствора н-бутиллитпя к раствору трифенил метана в ТГФ; карбо-
низация в течение 15 мин при комнатной температуре дает трифенпл-
уксусную кислоту с хорошим выходом.
(C6Ha)3CH + BuLi (C6H5)3CLi —— — (СвН5)3ССОгН
— В u Н 7 8 %
0,0204 моля 0,021 моля
в 50 мл ТГФ в 20 мл В ЦО
Реагент — сильное основание. Например, при взаимодействии
с ацетоном он образует окись мезитила [2]:
(CGHfi)3CLi „ (CH.J..CO
СН3СОСНз--------—>CHXOCHs(Li+) —>(СН3)Х = СНСОСН3
-(СоНДзСН “ -ион ‘
Хауз и Трост [3] получали приблизительно 1 М раствор Т.
в 1,2-диметоксиэтане и широко использовали его для изучения
состава енолят-анионов из кетонов [31 и енолацетатов [4]. При этом
использовали следующий метод: из 15 мл 1 М эфирного раствора
металл ити я растворитель удаляли в вакууме и твердый остаток
растворяли в 15 мл 1,2-диметоксиэтана. Добавляли трифенилметан
(18,3 лгмоля) и смесь перемешивали в атмосфере азота до отрицатель-
ной пробы на металл итий. Реакцию енол ацетата с Т. можно прово-
дить как обычное титрование, конец которого определяется по появ-
лению светло-розовой окраски.
1. Gilman Н., G a j В. J., J. Org. Chem., 28, 1725 (1963).
2. Tombouli an P., J. Org. Chem., 24, 229 (1959).
3. House H. O., Trost B. At., J. Org. Chem., 30, 1341 (1965).
4. H о u s e H. O., Trost B. At., J. Org. Chem,, 30, 2502 (1965).
396
ТРИФЕНИЛМЕТИЛНАТРИЙ, (CcH5)3CNa. Красный раствор
в эфире.
Получение [1]. Раствор тритилхлорида в эфире встряхивают
с 1%-ной амальгамой натрия на механической качалке при слабом
охлаждении в течение около 3 час, оставляют для осаждения хло-
(CeHr,)sCCi -! Эфир 1-1%-пая NaHg~*(C6H6)3CNa-;’ NaCl^Hg
0,226 моля 1,5 л 1,50 г
ристого натрия и красный раствор сифонируют под давлением азота.
Из 3%-ной амальгамы натрия можно получить почти в пять раз бо-
лее концентрированный раствор.
Ацилирование и алкилирование. При бензоилировании [2]
этил изобутират добавляют к эфирному раствору Т. из 1%-ной
0,187 моля (C6Hs)3CNa
в эфире —
(С113)ХНСО2С2Н5 (СН3)2ССОаС,Н5
0,187 моля Ю„НЬ)3СН Na +
сн3
0Д87 моля^С6НйСОСЦ
50—55% |
сн,
амальгамы и через 10 мин при комнатной температуре к смеси
добавляют эфирный раствор хлористого бензоила. Наблюдается
разогревание раствора и образование осадка хлористого натрия.
Через несколько часов большую часть эфира отгоняют, добавляют
разб. уксусную кислоту и смесь обрабатывают.
Подобная методика применяется для алкилирования сложных
эфиров [3] и для карбонизации и карбэтоксилирования эфиров [4]
и кетонов [5]. В синтезе сс-амирина из глицирретовой кислоты Кори
и Кэнтрал [6] осуществили гладкое метилирование 20-кето-З-
бензоата (1) в 19 р-метильное производное (2), обрабатывая раствор
(1) в диоксане эфирным раствором Т. до тех пор, пока еще сохра-
няется красная окраска, и затем большим избытком йодистого
метила, чтобы уменьшить диметилирование. Избыток Т. мгновенно
связывается йодистым метилом, по-видимому, с образованием трифе-
нилэтаиа.
397
Полимеризация диенов. Циглер [71 отметил, что под действием
Т. бутадиен превращается в смесь полимеров, по-видимому, путем
1,2- и 1,4-нрисоединения с последующим присоединением к бутадне-
(C(lHJ.CX'a (_^±НС1Г_„С1Л (С,.\-)3ССН.,СНСН - СН,
(1)
(С„п5).в
. (С,еП-);,ССН2СНСН - сн2
С~) 1
—- (Ci;IIJ:{CCH.>CH.,CI l - сн,
(3}
Г пдрп.ппз
ну новых натриевых производных. Затем Виттиг и Шлоедер [8]
показали, что трпфенилбор блокирует реакцию на первой стадии
в результате присоединения к 1,2-аддукту (1) и образования комплек-
са (2). Комплекс растворяется в воде без разложения, но при кипя-
чении с разб. минеральной кислотой гидролизуется до 5,5,5-трифе-
п пл пентена-1 (3). В этих же условиях изопрен превращается в угле-
водород (4).
сн3
I (Со I 1ЩС X а Гидролиз
СН„ — Cl 1С СН., -----------Комплекс------------*•
(ЩНЩВ
сна
—> (C(!H5);tCCl Ш I.,C - C.I I,
(4)
В отсутствие добавок Т. полимеризует аценафтилен (5) [91.
В присутствии трифепилалюмиипя реакция завершается на стадии
образования комплекса (6), образующегося путем 1,2-присоедиые-
398
ния. В присутствии трнфенилбора происходи!' 1 ^-присоединение
и образуется комплекс (7), Эют комплекс осторожным кислоты м
гидролизом превращаю!' в (8;, который легко изомеризуется в 7-
тритилаценафiси (9),
Т. используют для получения кислых фталатов третичных спир-
тов но описанной ниже методике ПОI. Эфирный раствор Т. добавляю!
/z ДХ)ч Z/ CCUXCIU;
|Г К „ , 13<i m.i -j(|)ub.i ( 'I
I о . ((Лл.Г.О!! •iCJI/UAn -------------1 i,
z co ’ x- %oji
мо.тя (),O6<5 мочи
к перемешиваемому раствору трет-бутипола в эфире до постоянного
красного окрашивания, свидетельствующего о небольшом избытке
основания. Затем добавляют фталевый ангидрид и перемешивание
продолжают в течение 2 час. Последующей обработкой и крпшаллп-
заипей получают чистый полуэфир (т. ил. 87—88 ), который разла-
гается с выделением изобутилена при 151 —155 .
1. Р с п ф р о v В. мл., X 3V3Cp Ч„ иСшпез!,! opi алпческих Jipciiii|kin4i.,
ИЛ, М._ 1949, сс5. 2, стр. 481.
2. X a v з с р Ч. Р е н ф р о v В., мл.. Ллпнсзы опта и лчегкпх и репа p.i г< чт>,
И21,’М., 1940, си. 2, cj р. 582?
3. Н и d s л и В. Е., Jr., JI ;i и s сг С. R., J. Am. (Jieui. S.jc., 02, 245/ (1910);
63, 3156 (1941).
4. В а и тп к <i г ( с п E., Ihi u s er C. R., .1. Am, Chem. Soc., 66. 1037 (19111.
5. В ;i u in ,к a г 1 c n E., Levine R., H a u s c r (7 R.. J. Am. (.’hem. S<>c.,
66, 862 (1944).
6. С и г с у E. J ., Cantrall E. XV -, J. Am. Chem. Soc.. 81, i ?4л (19n9).
7. Ziegler I\., D e r s e Ii 1'., \V о I t Ii и n IL. Ann., 511, 13 (1934).
8. \V i I t i ц G., S c !j J (j> c <1 <' г IL, Ann.. 592, 38 (1955); XX i ( t i м (i.,
Vi j ( t e в I) e г c 1)., iI)id.. 606, 1(19o7).
9. XX’ i ( ( i g G.. R e p p e IL G., E i e h e Г T., Ann., 643, 47 (1961).
10. R u I li e г I n r d K. G., P г k i p e a k J. M-, E а п g D. P. C., ,J. Org.
Chem., 28, 582 (1963).
ТРИФЕНИЛМЕТИЛПЕРХЛОРАТ, (CJI-):;C CIO, . .Нол вес
342,77, т. пл. 143". ТРИФЕНИЛМЕТИЛБОРФТОРИД, (CJ4 J,C' Bly .
.Мол. вес 330,13, т. пл. 200" (с разл.),
Получение |1|. Реагенты получают взаимодействием чистого
трнфенилкарбинола с 71 %-пой хлорной кислотой или 48"п-поп
борфтористоводородной кислотой в присутствии уксусного ангид-
рида для удаления воды (выходы 76 и 92%). Борфторид имеем преиму-
щества ввиду сто большей устойчивости по сравнению с перхлора-
том.
Отщепление гидридного иона. Даубсп и сот)). |2| получали тро-
пил и евые соли реакцией цпклогеитатриспа с трнфенил метил-
перхлоратом пли трифсн и л мети л борфтор и дом в ацетоин (риле или
399
жидкой двуокиси серы при —20°. Реакция была использована так-
же для синтеза гепталена (6) [31.
Дегидрирование. Перхлорат является эффективным реагентом
ароматизации гидроароматических соединений. Он превращает
9,10-дигидроаитрацеи в антрацен с количественным выходом [4[.
С его помощью обеспечиваются наилучшие условия превращения
перинафтанонов в перинафтеионы [41 и хроманонов в хромоны [5].
Синтез гетероциклов. 1,5-Дикетон (1) при взаимодействии с три-
фенил мети л пер хлор атом подвергается дегидроциклизации в пирил-
лиевую соль (2), которая далее при взаимодействии с ацетатом аммо-
ния превращается в пиридиновое производное (3) [6].
со,н
I
^сн +
сн2 Vh2 (c6i-i5)3c сю,
С6Н5С СС6Н5 Вб% *
о о
АсОН
CH3CO2NH4
94% Сьн5
(3)
(1)
400
Реакция с диазосоединениями [71. Трифен ил метил перхлорат —
эффективный катализатор разложения дифенилдиазометана, кото-
рое сопровождается димеризацией в тетрафенилэтилен.
+ - (С6Н.,)3С + С1О4
2(CeH6)2C=N—N-----------------> (С6И5)2СС(С6Н5)., =-2N3
’ /О
С диазоуксусным эфиром в ацетонитриле реагент образует этил-
трифенилакрилат. По-видимому, реакция включает присоединение,
перегруппировку и элимииированне.
(С6Н5)3С * СЮГ Ч- N =-- N — СНСО2С3Н5 —*
' 2 /о
Свн6
(CeH5)aC = ССО2С2Н5 + N2 + НС1О4
1. Dauben Н. J., Jr-, Honnen L. R., Hanfion К. M., J. Org. Chem.,
25, 1442 (1960).
2. Dauben H. J., Jr., Ga decki F. A., Harmon KJ M., Pearson
D. L., J. Am. Chem. Soc., 79, 4557 (1957).
3. D a u b e n H. J., Jr., В e r t e И i D. J., J. Am. Chem. Soc., 83, 4657, 4659
(1961).
4. Bonthrone W., Reid D. H., J. Chem. Soc., 1959, 2773.
5. Schonberg A., Schutz G., Chem. Ber., 93, 1466 (1960).
6. Siemiatycki M., Fugni tto R., Bull. soc. chim. France, 1961, 538.
7. Whitlock H. W., Jr., J. Am. Chem. Soc., 84, 2807 (1962).
ТРИФЕНИЛСТАННАН, (CeH5)3SnH. Мол. вес 351,01, т. пл. 26 -
28°, т. кип. 17476 мм.
Т. получают [11 восстановлением трифенилхлорстаннана LiAl Н4.
Восстановительное действие. Т. восстанавливает органические
галогениды и амины до соответствующих углеводородов [2, 3].
По легкости замещения галогены располагаются в следующем
порядке: 1>Вг>.С1 [41. 1-Бром-4-хлорбензол восстанавливается
в хлорбензол с 75%-пым выходом [5]. См. также [6—8].
Т., полученный in situ из LiAIH4 и (CtiH-,)3SnCl, более стереоспе-
цнфнчеп по сравнению с действием цинковой пыли и спиртового
раствора этилата натрия при восстановлении смеси эпимерных
7-хлор-7-фенилноркаранов (1), полученных по следующей схеме:
401
При этом образуется смесь 80% jwdo-7-фенил норка рана, J % экзо-
изомера и 19% пспдентифпцпрованного олефина [9].
1. F i п h о I t А. Е., Bond А. С., Jr., AV i 1 z Ь а с h К. В.. Schlesin-
ger Н. J., J. Am. Chem. Soc., 69, 2692 (1947); V an Der Ker k Ci. J.
X о 1 t e s J. G., Luijtcn,|. G. A.. J. Appl. Chem. (London^ 7, 366 (1957);
Ku i v i 1 a 11. G., В e u rn e I O. F., Jr., .1. Am. Chem. Soc,, 63, 1246 (1961).
2. 1пц h a m R. K„ Rose n b c r g S. D., G i Iman 11., Clicm. Revs., 60,
459 (I960).
3. К u p c h i k E. J., С о ii п о 1 1 у R. E., J. Org. Chem., 26, 4747 (1961); К u p-
r h i k E. J., К i e s c 1 R. J., Chem. Ind., 1962, 1654; J. Org. Chem., 29, 764
(1964).
4 К u i v i 1 a El. G., M e na p a ce L. W., Warner C. R., J. Am. Chem.
Soc., 84, 3584 (1962).
5. Rothman L. A.. Becker E. I , J. Org. Chem., 25, 2203 (1960).
6. Lorenz D. FI., Shapiro P., Stern A., Becker E. 1., J. Org.
Chem., 28, 2332 (1963).
7. Kupcliik E. J., К i c s c 1 R. J., J. Org. Chem., 29, 3690 (1964).
8. К ii i v i 1 a EL G., «Advances in Organometallic Chemistry», 1, 81 (1964).
9. Jensen F. R., Patterson D. B., Tetrahedron Letters, 3837 (1966).
ТРИФЕНИЛФОСФИН, (СД15)3P. Мол. вес 262,28, т. пл. 79—80С
Реакция Виттига. В методе Виттита и Шеллкопфа 111 для син-
теза метиленциклогексана раствор Т. в бензоле загружают в ампулу,
охлажденную льдом с солью, п добавляют рапсе сконденсированный
(ДПДД ClL.Br _., (C6l|.);jp-CHj(Bi'-)
0 21 моля н 4;> м.1 мо.’ы
; ~~ О z_______
— (CJLsLP-AGL^CCJl.hP- СМ? —==~—Z \-СН2-(С6Н5)3РО
О О \__ .
бромистый метил, ампулу запаивают и выдерживают при комнат-
ной температуре 2 дня. Трифеиилметплфосфонийбро.мид выделяется
в виде белого твердого вещества, которое отделяют с помощью при-
близительно 500 мз горячего бензола. Для получения реактива
Виттига или илпда часть этой соли (0,1 моля) добавляют порциями
при перемешивании в атмосфере азота к эфирному раствору 0,1 моля
феппллития и затем добавляют циклогексанон.
В синтезе щранс',/пргшс-1,4-дифснилбутадпена-1,3 |2] необходи-
мый фосфонпйхлорид получают кипячением смеси Т. и цинизмил-
Кипячение 12 час
С(;ЩСН--СНСИ..С1 (Ci;H-,)3P i Ксилил
0,2Н моля 0,33 Мм.чя Г>00 .ил
I.fOl.JI-,
CfH.CH _CI1C11.2Р(С(111-)3(С1) —СвП-СН-СНС1-1_Р(СйН5)3
С„Н ,(Л1 - О
---------- 'СЛ-1-,С11..- СНСН . ;СНС,.Н.
р,, . II .1 I,
хлорида при перемешивании в течение 12 час. Соль, выделяющуюся
в виде бесцветных кристаллов, отделяют и высушивают; 0,145 моля
402
соли растворяют вместе с 0,155 моля бензальдегида в 200 мл абсо-
лютного этанола. Затем в присутствии карбонильного компонента
получают ил ид под действием раствора этилата лития в абсолютном
спирте.
Первую стадию в синтезе п-к в и и кв i [фенил а [3] осуществляют
кипячением при перемешивании в течение 3 час смеси 1,4-бпс-(хлор-
метил)-бензола, Т. и 1 л ДМФА. Коричный альдегид с этилатом
лития в качестве основания дает по реакции Внттпга тетраен, кото-
рый затем присоединяет 2 моля ацетилендпкарбонового эфира, обра-
зуя бис-аддукт. Гидролиз этого аддукта включает перемещен ire
связей и дает, как показано ria схеме, транс,шранс-тстракислогу в
соответствии с данными, опубликованными позднее, о подобном син-
тезе п-терфенила [41.
hcon(ch3)2
2 77-С1СНгC^HjCHjCl + 2Р(ОД3 КипячениеЗ^ n-^Ji^C^C^CH^CeH^ (2 С1)
93-98т»
0,48 моля г моль 1
2 СьН5СН = СНСНО I
69-75% I 2 1ДОС2Н5
2 С2Н5О2ССЕССО2С2НЬ
собЛ |он’ /пгу/Л/г \=/ Н"1—Р'СОан\=/ но2с н Н(>1с | 52%,считая на тетраен|к3Ге(СМ)6 Чсл усо2н5г=-' + 4 С0?
403
Основываясь на нуклеофильном характере Т, и электрофильном
характере карбенов, две группы исследователей пришли к выводу,
что эти два реагента должны взаимодействовать, образуя илиды
нового типа, которые можно использовать для расширения приме-
нения реакции Впттпга. Специале н corp, [51 нашли, что Т. при
обработке хлороформом и трет-бутилатом калия в пептане при
0° дает желтый трифенилдихлорметилепфосфоран, который с бензо-
феноном образует 1,1-дифенил-2,2-днхлорэтилен (1). Сейферт и сотр,
:СС1, (С6Н5)гСО
1) (С6Н5)3Р —------>(С6Н,,)3Р-СС12 ----------> (С6НГ1)2С---СС13
2) (СвН5)3Р Сн-BuLi ] СН2С12->(С6Нб)3р.- СПС1—>
(ДЩСОСН.,
----—7-------С6НГ|С(СН3)-СНС1
'°----------цис- и гцрапс-
[G1 получали хлоркарбеи из н-бутиллития и хлористого метилена
в присутствии Т. для синтеза трифенил хлор метиленфосфо рана, кото-
рый с ацетофеноном образует смесь цис- и транс- 1-хлор-2-фенил-1 -
пропенов (2),
Фагерлунд и Идлер [71 получили трнфенилизопропилиденфос-
форан (2), нагревая Т. с изопропилбромидом при 150^ и обрабатывая
фосфо и и йб ром ид (1) н-бутиллитием в эфире (N2), и илид использо-
вали в синтезе 24-дегидрохолестерина, Виттит и Хааг [81 обнару-
жили, что прй добавлении дифенилкетена темно-красный эфирный
раствор (2) сразу же обесцвечивается; выделяющийся в виде бледно-
желтых игл продукт идентифицирован как бетаин (3).вПри быстром
пиролизе в высоком вакууме бетаин расщепляется до аллена (4) и
окиси трифенилфосфина (5). Углеводород выделяют возгонкой, а окись
(С6НБ)3РСН(СН3),(Вг) -^^(С6Н5)3Р = С(СН3)2 —
(1) (2)
(С(П,),С=С=О + 160= (0,2 m.v)
•...> (Сг,Н5)3Р - С(СН3)2----------------------->
е J ,0 |
О — С — С(С6НГД,
(3}
— (CH3)SC - С - C(CGH5)2 4- (CGH5);iPO
64% 96%
(4) (5)
фосфина — кристаллизацией остатка. Трифен и л этил идеи- и три-
фен илметиленфосфорапы реагируют с дифенил кетеном, образуя бетаи-
ны (6) и (7) с выходом 45 н 12% соответственно, которые при пироли-
зе дают димерные продукты неустановленного строения.
(С6Нй)3Р-СНСП3
б-С-С(С6Н5)3
(6)
(С6н5)3р-сн2
.. i
О-С = С(С6Н5)2
(?)
404
Бестманн и Шульц [91 описали метод синтеза к, ^-ненасыщенных
у-кетоэфиров, который можно иллюстрировать реакцией фена-
цилбромида с 2 молями реактива Внттига (2), трифенилкарбметок-
с и метиленфосфо рапа.
CfiH5COCH2Br-!- СН - Р(С6Н5)з —' ГСеН3СОСН2. - CHP(CGH3)y(Br~)l —>
(1) ( I
со2снл [ со2сн3
(2) (3)
СН = Р(С«Н6),
I
СО..СН, +
----;-----------СеН5СОСН = СНСО2СНвжр(С6Н5)3-рСН,Р(С6Н5)3(Вг-)
(4) (5) i
со.2сн3
(Ь)
По-видимом у, промежуточная фосфониевая соль (3) реагирует со
второй молекулой реагента (2) с выделением Т. и образованием
кетоэфира (4) и фосфониевой соли (6), из которой можно получить
реагент (2). Синтез альдегидов, предложенный Внттигом [101,
можно иллюстрировать следующей схемой:
ecu (С(н5)3р
CfiHaOCH2SO2\a - ----* Сс1 ЦОСН2С1 — ------->
(1) (2)
+ с.ндл
->СвН5ОСН2Р(СйН5)3(С1-)---------->С6Н5ОСН^Р(С6Н-)3
(3) (4)
(СеН6),СО НС104
-----(СвН5)2С — СНОСЬН5 — -------------> (С8Н5)2СНСНО
р
Бергельсон и Шемякин [11] открыли возможность стерического
контроля реакции Витти га путем соответствующего выбора условий
и строения реагентов. В приведенной реакции образования р-этил*
+ Основание
СвН5СН2Р(СвН5)3С1 - ----------> свн5сн = Р(С6н5)3 —
([) (2)
сн.сщсно
------------—> С6Н5СН - СНСН2СН3
(3)
стирола (3) соотношение цис-транс-изомеров широко изменяется в
зависимости от условий, как это показано в таблице.
Растворитель Добавка Основание Соотношение изо- меров цас/т ране
Бензол BuLi 26:74
Бензол Анилин BuLi 40:60
Бензол LiBr BuLi 91:9
Бензол Lit BuLi 93:7
Эфир EtONa 31:69
Днметилформамид (ДМФА) Lil EtONa 96:4
405
Бромид и йодид лития добавляют к бензольному раствору в вйде
высокодисперсных суспензий, полученных нейтрализацией бензоль-
ного раствора бутиллития сух наг бромистым или подцепям водоро-
дом; хлористый литий эффекта по дает.
Если в молекуле нлнда имеется электроноакцепторная группа,
связанная с илидпым углеродом (4), илнды практически дают только
трпне-продукт (5) независимо от присутствия ионов галогенов или
используемого растворителя:
Н
еддпо I
(С6НД:1Р : CHCCEEt----------> CJ-I5C- CCO.,F.t 4 (СеН5)3РО
(4) I
и
Советские исследователи показали, что карбэтоксильиая группа,
удаленная от илидного углерода, не влияет на стереохимию реак-
ции, п на этой основе разработали несколько высокостереоспецифпч-
пых синтезов природных qwc-этплеиовых жирных кислот; некото-
рые из них приведены на схеме.
11 11 Н Н н И
I I осшсн л„<:о.сн, | I I I
RC —ССНХН ~ P(CfiI ---------г • д- - RC- -CC1EC С(СН2)„СОХНЯ
н н' |
I ,1 „ Г1ЛГ. (СВН,),РГ=СН(СП.)„СО,(:П3 | дмфа, i-
КС. С^.С.1 loLriU
11 ы
RC Ahxi-LCH - Р(С.Н,Ъ
ДМФА, I-
11 II 11 И
II II
—> RC- CCII..CHX •- С(СН..)„СО,СН;1
и н
СН.ГСН.,),- С—с «:но _ " Т У " "ЛС'_.
| I ДАККА. 1 ~
н и
И Н II 1-1
I I II
-—> Cl I.JCH ,)ЯС — С — С - С — C^C(CH.,)XO.J-t
I I
Н II
Метод Виттига для синтеза олефинов был распространен на али-
фатические альдегиды, например газообразный формальдегид и
параформальдегид, ацетальдегид, пропионовый альдегид, н-гек-
саиаль, акролеин [121.
Корн и сотр. [131 показали, что если порошкообразный гидрид
натрия перемешивать с избытком ДМСО в атмосфере азота при
65—70Д то выделяется водород и образуется очень активный метил-
106
сульфин пл метил нд (1). Эта соль реагирует с этилтрпфенилфосфонип-
бромидом при комнатной температуре, образуя реактив Виттита
(2), что подтверждается его быстрой реакцией с бензофеноном и обра-
О о-о
; I г -
CH3S-~CJJ3 щ NaH —’ CH;,S=^CH., < —СН2
- Н '----------,--------
Ха +
| (С,Н#)3Р-СН2СН,(Вг-)
(С,я]-],),со
(С6Нв).,С = СИСН3 - ---(СеН5)3Р = Cl (CH;t
(3) (2)
зовапием 1,1-дифенилпропена-1 (3). Силыюосновный характер ме-
тилсульфинил карбаниона иллюстрируется его быстрой реакцией
с трнфенилметаном; при этом образуется темно-красный раствор,
содержащий трнфенилметильный анион. Получаемый аналогично
низший гомолог (2) реагирует с циклогексаноном, камфорой
н холсстаноном-З с образованием метиленовых соединений с выхо-
дом 86, 73 п 69% соответственно.
Отщепление кислорода и десульфуризация. Высокая полярность
Т. очевидна нз сравнения дипольных моментов трифеиильпых
производных элементов пятой группы:
(CeH5)3N 0,26 D (CeH-,)3Sb 0,57 D
(CfiH5)3P 1,45 D (C«H6)3Bi 0 D
(%Hs)3As 1,07 D
Способность к присоединению подпетого метила изменяется в зави-
симости от полярности, Трнфениламин, трифени.тетпбнп и трнфенпл-
висмут не образуют иодметилатов, а трифен ил арсин реагирует
только при высокой температуре, тогда как Т, взаимодействует
с йодистым метилом при комнатной температуре, причем реакция
сильно экзотермична 1141. Высокий нуклеофильный характер Т.
подтверждается также его реакцией с фтор а цеп щеном, сопровождаю-
щейся образованием соли (С|.Н5)3Р+С^СН (F-) [151, и его реакция-
ми с карбенами, упоминавшимися выше. Хотя Г. в основном устой-
чив на воздухе, он реагирует с различными кислородсодержащими
соединениями, образуя окись трифепилфосфииа. Таким образом,
реагент используется для удаления кислорода из субстратов не-
скольких типов,
Хорнер и Г'осрфмал 116] сообщили об использовании Т. для рас-
кисления азоксисоединепий в азосоединения, перекисей — в про-
стые эфиры, нитронов — в шиффовы основания и окисей алифати-
ческих аминов в амины. Последняя реакция проводится в кипящей
уксусной кислоте; в этих условиях N-окиси пиридина и хиноли-
на устойчивы. Метод раскисления, более общий, чем при исполь-
зовании трех хлористого фосфора, представляет ценность, так как
эти N-окиси удобны для получения 2- и 4-замещенных продуктов.
407
Хоуард и Олшевски [17] установили, что Т. при достаточно высокой
температуре удовлетворителен для этой цели. Оптимальный про-
цесс заключается в нагревании реагентов в отсутствие растворите-
ля до температуры, при которой амии отгоняется из реакционной
смеси. Окись трифепнлфосфина можно выделить перекристаллиза-
цией остатка из смеси метанол — вода. 2-Пн кол ин, 4-пиколин,
+ (с6н5)3р
230-233°
(С6Н5)3ро
90% 73%
4-метоксипиридии и хинолин были удовлетворительно получены
из N-окисей по этому методу, но эта реакция оказалась непри-
менимой для синтеза 4-нитропиридина. Наиболее подходящим раст-
ворителем для данной реакции является триэтиленгликоль.
Виттиг и Хааг 118] установили, что окись стирола при нагрева-
нии с Т. до 165° раскисляется до стирола с выходом около 50%.
Реакция приобретает препаративное значение, так как она приме-
нима к эпоксидам, недоступным исходя из олефинов, таким, как
Р-фепил глицидный эфир (1), который получают конденсацией
Дарзана. Прп нагревании с эквивалентным количеством Т. при 210е
0.0 1 моля
СГ1Н5СН — СНСО2С2Н5 [ (СП1Ь,)3Р ' С6Н5С11 = снсоасцн5
'Tq,/ 0,03 моля (2)
0.03 моля
(П
этот эфир дает этнлцнннамат (2) с выходом 61%. Однако при
добавлении гидрохинона в качестве кислого катализатора и ингиби-
тора полимеризации реакция начинается при 125° и завершается
при 170°; выход этил циннамата достигает 80%.
Бойер и Эллзи [19] получили бензофуразан кипячением о-ди-
питрозобензола с Т. в 95%-ном этаноле в течение 30 мин (красный
раствор); продукт выделяли перегонкой с паром. Замена Т. три-н-
0, 010 моля
Кипя-
+ (сйн5)3Р + С2Н5ОН
6 8%
0,011 моля 25 мд
бутилфосфшюм даег бензофуразан со сравнимым выходом.
Вейс 1201 успешно применил Т, для раскисления аддукта
3,4-дицианфурана с дициана цетил еном (1). Суспензия аддукта (1)
в ацетонитриле реагирует с Т. даже при комнатной температуре
с образованием темно-синего умеренно растворимого бетаина (3),
408
причем его предшественник бетаин (2) образуется, по-видимому,
при первоначальном присоединении к двойной связи. Пиролиз (3)
дает 1,2,4,5-тетрациаибензол с хорошим выходом, а также окнсь
трнфенилфосфина. Другой аддукт, подобный (1), расщепляется
аналогично, но три других не реагируют с Т
Дэвис [21 ] установил, что под действием Т. тиираны десульфу-
рпзуются за 3 дня при 25° с образованием олефинов с высоким выхо-
дом.
| \s 1 (C9H5)3P —> II +(CeH5)3PS
Cz с
Сульфоксиды при действии Т. в четырех хлор истом углероде
раскисляются в сульфиды 1221. Механизм этого превращения
о-
n-CH3OC6H4S-CeH4OCHrra + (CGH5)sP {-СС1,
0,0! моля 0,02 моля 100.и.г
Кипячение 2 час
—/i-CH3OCGH4SCGH4OCH3- п
неизвестен, однако возможно промежуточное образование плида:
2 (С6Н5)зР 4 С.С14 -^-Д-сТ* <СеН^Р = СС)'^
— (Се Н ь) С12
R-.SO (С.И6)ДРС12
R2S = CC12 ----------—> R2S + CCi4-y(CBHfi)3P
Определение озонидов [23]. Обработка озонида в спиртовом
растворе избытком Т. в течение 3 дней при комнатной температуре
в отсутствие кислорода (который окисляет реагент) дает соответст-
вующие карбонильные соединения и окись трнфенилфосфина:
/°3Х
/С —Y/ (CGHa)3P-> + (СВНБ’|3РО
409
Избыток реагента оттмтровывают иодом.
Дебромирование. Хорнер и сотр. [241 применили реагент для
восстановительного замещения атомов брома или иода в орто- и
/юра-положеппях фенолов. В первую очередь замещается галоген
в орпш-поло/кенип. Смесь фенола, 1 оке Т. и бензола нагревают в
он
/I Вг
б7 || н.о
ф (С6н5)3р —> Аддукт —
III • • * о
В г
ОН
j || т (CflHjlsPO-p НВг
I
Вт
запаянной ампуле при 100—150? в течение 1—5 час и образующийся
аддукт обрабатывают 2 и. раствором едкого натра.
Т. реагирует с бромацетофеноном, нуклеофильно замещая бром
и образуя фенацилтрпфешЕлфосфонийбромид [251. В отличие от
этого вторичные п третичныеа-бромкетоны реагируют с Т. в кипящей
смеси бензол — метанол с образованием дебромпровапиых кетонов
Д) .X А)
I сн,он-с,н, ] | 7
I I : (C(,Hr>):ip —I | (CeH5):iP0a-CBr
В г
1261. В пяти изученных примерах выходы достигали 60—70%.
Реагент дебромпрует замещенные дибромнды метил- и этил-
акрплатов, переводя их в ненасыщенные эфиры с выходом 50—
65% [271.
Вг
I
CH3C-C-CO,R'-I- (С6Н5)3Р—>CH3C = CCO2R'%(C6H5)3PBri
I I !
Вг R R
Р-Н. СП3
Удивительно, что при взаимодействии Т. с бромметилтрифенил-
фосфонийбромпдом в кипящем этаноле образуется метплтрифенил-
фосфопийбромид с высоким выходом 1281.
j- - с.щон <
(СвНГ));1РС1Г,Вг(Вг) -(<%НГ))3Р —-> (СпН5)лРСН3(Вг} +
i-(CeII-r5PO ЩДН-Вг
Образование нитрилов и ацетиленов. Необычная реакция 1-бро.м-
1-нитрооктана с 2 эка Т., описанная Триппеттом и Уолкером [291,
дает капрплонитрил и окись трифен ил фосфина в виде бромгидрата.
Предполагают, что прп этом происходит атака по атому кислорода
аци-формы (2), отщепление бромгидрата окиси трифенплфоефнна
410
с образованием окиси нитрила [(4), не выделено! и раскисление
последней в нитрил (5). N-Бромфснил ацетамид (Ct.H5CH2CONHBr)
Вг Вг
I I ОН
к-С7Н1йСНХ'Оя дд «'C-Hj-C —i\ '
ХО~
(I) (2)
Вг
(С.ПЛ.Р , ! . ,ОР(С611а),.Ц
-———* н-СгНг5С-:--X
'О-
-(С.Н5)3РО.НВг
(4)
(ЩНЩР
72% (общий)
(3)
»-с7н15с
X(С6Н5)3РО
(S)
аналогично дает фен ил ацетонитрил (CfiH5CH2CN, 60%), а а-фенил-
фен ацил хлор ид (6) в кипящем бензоле — дифен ил ацетилен с выхо-
дом 90%.
CI О С1 он
I II II (СЙН{)3Р
С6НГ1СН — СС6Н5 CfiH5C -ссйн5------------>
(6)
Ci ОР(С,.Н-)3Н
I I
СЙН-,С—-CCflH-, — * CeH.-C=CCfiH, :-НС!-;-(С6Н-).,РО
Синтезы кетоальдегидов и кетоэфиров. Штаудпнгер и Мейер
1301 отметили, что Т. присоединяется к алифатическим диазосоеди-
нешгям, образуя соединения нового типа, названные фосфазипами.
Позднее Бестманн и сотр. [311 разработали следующий метод синтеза
а-кетоальдегидов. Дназокетоны реагируют с Т. в эфире, при этом
R х - X - N < (C6I I5)3P RX _ X - N _ Р (Сй116)3 -
R2C-N^X-P(Ci;H5);,
быстро выделяется кристаллический а-кстотрпфенилфосфазин с вы-
соким выходом. Фосфазин расщепляется азотистой кислотой с об-
разованием а-кетоальдегида.
О О
RCCH _ \ — N Р(С6Н5)3 RCCH = - PlC6H5)3
О
HNO. ||
----+ RCCH-О -|-Хо + (С6Н5)3РО
В других синтезах [321 исходят из a-диазо-^-кетоэфира (3),
который легко образуется нз хлорангидрида кислоты и дназоук-
сусного эфира. Эфир (3) реагирует с Т. в уксусной кислоте, давая
О О
:1 - + II
RCCI 1-2N -хХ-СНСО2Сг115 -* RCCCOaC2H3 С1СН2СОХгН5
(1) (2) (4)
J
N-
(3)
4И
о о оо
РССвЩ), .1 hno2 I! i1
rccco2c„h5 -------* rccco2c2h5 —rccco,c2h5
'I I! (6)
N+ N—N —P(C6H6)3
II (5)
I H20—ZnC]2
k О о
II Основание li
RCCCOaCaH&——RCCHaCO,C2H5
II "N* (8)'
NNHa
(?)
фосфазпн (5), который расщепляется азотистой кислотой до а, р-ди-
кетоэфира (6). С другой стороны, фосфазин (5) можно гидролизовать
до гидразона р-кетоэфира (7). При нагревании (7) до 60—70° с N-ме-
тил пиперидином выделяется азот и образуется р-кетоэфнр (8).
1. В п т т и г Г., Шеллкопф 10., «Синтезы органических препаратов»,
пзд-во «Мир», М., 1964, сб. 12, стр. 89.
2. М а к-Д опал ьд Р, Н., Кэмпбелл Т. У., «Синтезы органических
препаратов», изд-во «Мир», М., 1964, сб, 12, стр. 54,
3. Кэмпбелл Т. У,, Мак-Дональд Р, И., «Синтезы органических
препаратов», изд-во «Мир», М., 1964, сб. 12, стр. 78.
4. F 1 е s е г L. F., Н a d d a d i n M. J., J. Am, Chem. Soc., 86, 2392 (1964),
5. S p e z i a 1 e A. J., M а г с о G, J., Rails K- W., J. Am. Chem. Soc.,
82, 1260 (1960).
6. Scyferth D., Grim S, O., Read T. O., J, Am. Chem, Soc., 82, 1510
(1960).
7. Fager j u nd U. II. M., Idler D. R., J. Am. Clicm. Soc., 79, 6473 (1957).
8. Wittig G., Haag A., Chem. Ber., 96, 1535 (1963).
9. Bestmann IL JSchulz H., Chem. Ber., 95, 2921 (1962); В e s t-
mann H.J., Seng F., Schulz H., Chem. Ber., 96, 465 (1963).
10. Wittig G., Boll W., Kriick K.--H-, Chem. Ber,, 95, 2514 (1962).
11, Бергельсон Л. Д., Шемякин M. M., Tetrahedron, 19, 149 (1963).
12. Hauser C. F., Brooks T. W., Miles M. L., Raymond M.A.,
Butler G. B.,J. Org. Chem., 28, 372 (1963).
13, Greenwald R., Chaykovsky M., Corey E. J,, J. Org. Chem.,
28, 1128 (1963).
14. Davies W. C., L e w i s W. P. G., J. Chem. Soc., 1934, 1599.
15. M i d d 1 e t о n W. J., Sharkey W, IE, J. Am. Chem. Soc., 81,803 (1959),
16. Horner L., Hoffmann H., Angew. Chem., 68, 480 (1956),
17, Il о w a r d E., Jr., Olszewski, J, Am. Chem. Soc., 81, 1483 (1959).
18. W i t t i g G., Haag W-, Chem. Ber., 88, 1654 (1955).
19. Boyer J. H., E 1 I z e у S. E., ,1. Org. Chem., 26, 4684 (1961).
20. W e i s C, D., J. Org, Chem., 27, 3520 (1962).
21. D a v i s R. E., J. Org. Chem., 23, 1767 (1958).
22. C a s t r i 1 1 6 n J. P. A., Szmant 11. H., .1. Org. Chem., 30, 1338 (1965).
23. Lorenz O,, Analyl. Chem., 37, 101 (1965); Lorenz O., Parks C. R.
J. Org. Chem., 30, 1976 (1965),
24. Hof fmann H., Horner L., Wi ppcl H. G., Michael D.,
Chem. Ber., 95, 523 (1962).
25. Ramirez F., Dershowitz S., J. Org. Chem., 22, 41 (1957).
26. В о г о w i t z I. J,, G г о s s m a n L. I., Tetrahedron Letters, 471 (1962).
27. Tung С. C., S p e z i a 1 e A. J., J. Org. Chem., 28, 1521 (1963),
28. G r i s 1 e у D. W-, Jr., Tetrahedron Letters, 435 (1963).
412-
29. Т г i p p e t t S., Walker D. ,M., J. Chem. Soc., 1960, 2976.
30- St audinger IL, Meyer J., Helv. Chim. Acta, 2, 619 (1919).
31. Best tn ann H.J., Buckscliewski H., Leu be IL, Chem. Ber.,
92, 1345 (1959); Bestmann H. J., Klein O., GothlichL., Buck-
schewski H., Chem. Ber., 96, 2259 (1963).
32. В e s t m a n n H. J., К о 1 m H., Chem. Ber., 96, 1948 (1963); Bestmann
H. J., Klein O., Ann., 676, 97 (1964).
ТРИФЕНИЛФОСФИНДИБРОМИД И ТРИФЕНИЛФОСФИНДИ-
ХЛОРИД, (СбН^цРХо. Мол. вес: дибромид 422,11, дихлорид 333,19.
Реакция со спиртами и фенолами. Хорнер и сотр. 111 предло-
жили эти реагенты для получения алкил- и ар ил галогенидов из
спиртов и фенолов, а Уили и сотр. 12] установили, что реагенты
обладают значительным преимуществом по сравнению с пентагало-
генидами фосфора в реакции С — ОН —> С — X и рассмотрели воз-
можный механизм превращения [3]. Для получения н-бутилбромида
по Уили к раствору н-бутанола и трнфеннлфосфина в ДМФА в ат-
мосфере азота добавляют бром до тех пор, пока при добавлении
следующих двух капель раствор не приобретает устойчивый оран-
жевый оттенок. Летучие вещества отгоняют, добавляют вбду и слон
н-С4Н9О11-! (СД15)3Р-|-ПСОХ(СН3С рВг2 ———
0,1 Mo.'tii 0,107 моля 100.11.1
— > а-ДНдВг р(СвН5)аРО НВг
разделяют. Для получения неопентил хлорида трифен ил фосф ин
сначала хлорируют в четыреххлористом углероде, затем полностью
удаляют растворитель. Реагент растворяют в ДМФА, добавляют
неопентиловый спирт и смесь кипятят в течение 1 час. Последующая
обработка дает неонентилхлорид (выход 92%), который не дает
осадка со спиртовым раствором нитрата серебра. Превращение
вторичного амилового и неопентилового спиртов в соответствую-
щие чистые галогениды показывает, что замещение происходит
без перегруппировки. Превращение цнклопентанола и циклогек-
санола в бромиды свидетельствует об отсутствии элиминирования.
Фенолы при повышенной температуре гладко превращаются в бро-
миды без изменения положения заместителя.
ОН Вг
Леви и Стивенсон 14] нагревали на кипящей водяной бане в те-
чение 20 час холестерин и холестанол с трпфенплфосфиндпбромпдом
в ДМФА в молярном соотношении 10 : 1 и получили 3|3-6poM-V-
холестен и 2а-бромхолестан с выходами около 80%.
413
4,4-Дпмстпл-ЗР-ольная система в тритерпсноидах и триметил-
стеропдах, которая подвергается перегруппировке при обработке
пентагалогеипдами фосфора, при взаимодействии с (С(!Н3);(РВг.2
дает главным образом Д2-еп и небольшое количество 2а,3[-)-дпбро-
мида. 4,4-Дпметилкетои-З образует А'-ои-З. По реакционной спо-
собности три гидроксильные группы метплхолата по отношению
к реагенту располагаются в следующем обычном порядке: За>7а>
> 12а.
Бензальдегид реагирует с трифевплфосфиндпхлоридом, образуя
бензальхлорид с 59%-ным выходом, а циклогексанон дает 1-хлор-
циклогексен-1 с выходом 45%[ 1]* Из бензойной кислоты образуется
хлористый бензоил с 63%>-ным выходом.
Трифепилфосфиндибромид реагирует с карбоновыми кислота-
ми и ангидридами, образуя брома и гидр иды [4а В
RCO2H-|- (С({Нг,)3РВг2 ~0—"A RCOBr-r(CfiH-)3PO-;-HBr
Шефер и Вайнберг 15] показали, что методику Учли можно
использовать для превращения ( -ф-.эмЪ-иорборнеола (1) в ( '-)-жзо-
норборпилбромид (3). При проведении реакции в мягких условиях
можно выделить промежуточную соль (2), которая термически раз-
лагается с образованием (3). Это говорит о наличии вальденовского
обращения. Та же реакция с (—)-жзо-норборнеолом оказывается
более сложной 161. В этом случае наблюдается зависимость от раст-
ворителя и доминирует промежуточное образование карбон левого
иона. По методу У или был получен цпннамилбромнд [71.
Расщепление эфиров ]8[. Реагент, полученный но описанной
методике в растворе, расщепляет алифатические эфиры при уме-
ренных температурах в бензонитриле, ацетонитриле (медленнее)
пли хлорбензоле с образованием бромидов. Например, бром медлен-
но добавляют к охлажденному раствору трнфенилфосфина в бензо-
12.ГТ
50 .i/л СПНД-.Х’ 0.5 мо.гл (r/-C-I I,, )-.О
(С0Пй)3РЩВг----------------------- (CJIВс. — ——--
' Ю и
0,(М2 0.052
моля моля
— 2н-С;,Н11Вг ; (СЙН3)ЯРО
нитриле, смесь нагревают до 12о и обрабатывают ди-н-амиловым
эфиром. Анализ методом ГЖХ показал, что расщепление эфира
414
завершается приблизительно через 4 час. ТГФ дает 1,4-дпбромбу-
тан с выходом 75%. Двойная связь в аллиловых эфирах при действии
этого реагента нс затрагивается.
1. Horner L. Oediticr Н. Hoftmann II., Anu., 626. 26 (19.59).
2. W i 1 e у G. А., I I с г s Ii к о w i I z R. I.., Rein B. W, ('. Ii ч n В. C.,
.1. Aid. Chem. Soc., 86. 964 (1961).
3. W i I e v G. A.. R e i п B. A\., llersliko w i 1 z R. L., Tetrahedron Let-
ters, 2609 (1964).
4. Lew D., S t e v о ns о n R ., Tetrahedron Letters, 341 (196.5); .1. 0гц. Chem.,
30, 3469 (1965).
4a. Bcstiliann Mott L., Ann., 693. 132 (1966).
5. S c h ;i eler .1. P., W e i n h с г Ц D. S.. J. Ощ. Chem., 30. 2635 (196.5).
G. S e h a e ter J. P., W einber e !> S., .1, 0гц. Chem., 30. 2639 (1965).
7. A procedure for the preparation of einnamyl bromide hy tin* Wiley method has
been submitted to Огц. Syn. by Schaefer ,1. P., 11 i ц .Ц i n s ,1. G., S 11 e-
n о у P. К.
8. Anderson A. G., Jr., F r e e n or E. J., .1. Am. Cliem. Son., 86, 5037 (1964).
^пс-(ТРИФЕНИЛФОСФИН)-НИКЕЛЬДИКАРБОНИЛ,
|(СвНД)Р|.Л,цСО.,). Этот комплекс катализирует тример из а пню
ацетиленов в ароматические соединения | II, а также димеризацию
бутадиена в цпклооктадпеи-! ,5 121 в присутствии ацетилена как
промотора.
I. R с р р е W., S е h w ос k е п d i с k XV. ,1., Arm., 560, 1’04 (1948).
2. R ее d II, W. В., .1. Chem. Soc.. 1954, 1931.
тряс-(ТРИФЕНИЛФОСФИН)-РОДИЙХЛОРИД, |(СД1 Д;Р|:lRhCI.
Мол. вес 425,21.
Т. получают в виде красно-фиолетовых кристаллов взаимодейст-
вием спиртового раствора RIiCR-THT) с шестикратным молярным
избытком трифенилфосфина, действующим как комплексообразую-
щий и восстанавливающий агент. Комплекс является активным
катализатором гидрирования алкенов и алкинов (спирт — бензол,
250 , атмосферное давление) 111. Т. используют для гидроформили-
рования алкинов |1|, альдегиды гладко декарбопилируются с об-
разованием парафинов [2|:
RCHO 1 |(C(;II-):iP].,RhCl — RH - RhCI(CO)[(C(,FI;,);iP]; - (СГН-ЩР
1. А' о и п ц J. Г’., Osborn J. A., J а г d i п ь Г. II., Wilkins о il G.,
Chem. Comm., 1965, 131.
2. J a r d i пе Г. IL, Os И о r n J. A., W i 1 k i п ч о п G., Y о ц п и J. F.,
Chem. Ind., 1965, 560.
3. Т .ч и j 1 .1., О h п о К., Tetrahedron Letters. 3969 (1965).
ТРИФЕНИЛФОСФИН- УГЛЕРОД ЧЕТЫРЕХХЛОРИСТЫЙ.
Эта смесь превращает первичные и вторичные спирты в соответст-
вующие хлориты в мягких, по существу нейтральных, условиях
I1L
Карбооовые кислоты превращаются в хлор а и гидр иды, вероятно,
следующим образом 121:
415
RCO;H
(C6H-)3PH CC14 — (Сонг.)3р^ CCI3(C[-) — --
—> RCO2P 1 (C6H5);p- RC0C1--[-(C6H5)3PO
L Downie 1, M., Holmes J. B., Lee J. B., Chem. Ind., 1966, 900.
2. Lee J. B., J. Am. Chem. Sue., 88, 3440 (1966).
ТРИФЕНИЛФОСФИТ, (CeHsO)3P. Мол. вес 310,27, т. пл. 25°,
т. кип. 21071 мм. Получение Ill.
Получение алкилгалогеиидов. Т. взаимодействует со спиртами
п галогеноводородом, образуя галогенид (1) 12|. Вместо галогено-
водорода можно применять ал к ил галогениды (2) 121 или свободные
галогены (3) [31.
о-
1) (C6H5O)3P-;-ROH-J-НХ —RX->-HP+(OC6H5)2-!-CbH5OH
о-
2) (C6H5O)3P + ROH-LR'X-^RXH-R'P+(OCeH5)2 ]-С6Н5ОН
о-
3) (C6H5O)3P + ROH-LX2-> RX4-XP+(OCeH5)2 + CeH5OH
В случае ненасыщенных спиртов реакцию можно провести в две
стадии (4).
хл снЕ=снсн2он
4) (СсН5О)3Р -> (СЙН5О)3РХ2 --—------►
—> СН3 —СНСН2Х + ХРО(ОС6Н5)2 -| -СвН5ОН
1. Gottlieb Н. В., J. Am. Chem. Soc., 54, 748(1932).
2. L а п d а и е г S. R., Ry don H. N., J. Chem. Soc., 1953, 2224.
3. С о e D. G., Landauer S. R., R у d о п H. N., J. Chem. Soc., 1954, 2281.
ТРИФЕНИЛФОСФИТА ДИБРОМИД, (СвН5О).,РВг(Вг). Мол. вес
470,11.
Т. д.— наилучший реагент для превращения первичных и вто-
ричных ацетиленовых или алленовых спиртов в бромиды 111. Во
избежание присоединения бромистого водорода по двойной или
тройной связи для его нейтрализации применяют пиридин.
1. В 1 а с К Г>. К-, L a n d о г S. R., Patel A. N., Whiter Р. F., Tetra-
hedron Letters, 483 (1963).
ТРИФЕНИЛ ФОСФИТА ИОДМЕТИЛАТ (метилтрифепоксифос-
фонин иодистый), (СвНг,О)зРСН3(1). Мол. вес 452,22, т. пл. 146°.
Ландауер н Райдон П] описали получение Т. и. и его примене-
ние для превращения спиртов в алкилиодиды. Корн блюм и Иффленд
12] показали, что неопентилиодид, полученный этим путем, содер-
жит 6% трв/л-амнлиодида, но с помощью модифицированной мето-
дики можно получить чистый продукт.
416
СН3 СН3
I + I
си3сси,он-; (ссн-,о)3рснэ(1-)———> сн3сснг1 д-
сн3 сн3
о-
I
+ СН3Р:(ОС6Н5), [ С6Н5ОН
Т. н. используют для синтеза иода л ленов и иодацетиленов [3],
а также для прямого иодирования углеводного фрагмента в нуклео-
зидах [41.
1. L ап da u er S. R., Rydon Н. N., J. Chem. Soc., 1953, 2224.
2. Kornb lum N., 1 f f 1 a n d D. C., J. Am. Chem. Soc., 77, 6553 (1955).
3. Baker C. S. L., L a n d о г P, D., Landor S. R., Patel A. N., J.
Chem. Soc., 1965, 4348.
4. Verhcyden J. P. H., Moffatt J. G., J. Am. Chem. Soc., 86, 2093
(1964).
ТРИФЕНИЛХЛОРМЕТАН, (CeH5)3CCl. Мол. вес 278,78, т. пл.
111—112°.
Получение. В одном из методов [11 трифенил карбинол нагре-
вают в бензоле с большим избытком хлористого ацетила, который
добавляют прн кипячении смеси одиннадцатью порциями. При ох-
лаждении добавляют петролейный эфир, кристаллический продукт
отделяют и промывают (т. пл. 111 —112°). Во втором методе [2|
Охлаждение, затем
(G(jHs)3COH-|-СНзСОС14-С6Н6 7g_g3<y * (С6П3)3СС1
используют реакцию Фриделя — Крафтса, которую начинают при
охлаждении льдом и заканчивают при кипячении.
Охлаждение, затем
кипя пение
С„П, -I- CS, .|. А1С13 -----------► (С,Н5)3СС1
5,2 моля 470 мл 4,51 моля /0
О-Защитиая группа. Гельферих и сотр. [3] показали, что пер-
вичные и вторичные спирты и фенолы превращаются в соответствую-
щие тритиловые эфиры при взаимодействии с Т. в пиридине при ком-
натной температуре, и установили, что альдогексозы селективно
трптнлпруются по первичной гидроксильной группе [41. Примене-
ние этого селективного процесса в химии углеводов иллюстрируется
получением 1,2,3,4-тетра-О-ацетилф-D -глюкопиранозида (4) [51.
Смесь углевода, Т. и пиридина нагревают на кипящей водяной бане
до полного растворения и к горячему раствору 6-тритилпроизводного
(2) добавляют уксусный ангидрид для ацетилирования четырех
вторичных гидроксильных групп. Раствор выливают тонкой струей
в смесь ледяной воды и уксусной кислоты при перемешивании ме-
ханической мешалкой н получают твердую смесь ф-изомера (3)
и а-изомера в виде гранул. Кипячением сухой соли в эфире отделяют
14 Заказ М Г [ 5G
417
большую часть а-изомера и повторной кристаллизацией из 95%-ного
этанола получают чистый (3). Для удаления тритильной группы раст-
вор тритил тетр а ацетата (3) в уксусной кислоте охлаждают до 10J
(1) 0, 67 моля
(С6Н5)зСС1 + Пиридин
0, 7 МОЛЯ 500 МЛ
(2)
(3)35%,считая на (1) (4)
(С6Н5)3СВг
и обрабатывают раствором сухого бромистого водорода в уксусной
кислого. Смесь встряхивают в течение 45 сек: выделившийся трптил-
бромпд быстро отделяют фильтрованием и фильтрат сразу выливают
в воду. Экстракцией хлороформом и унарпваниехМ получают сироп,
который сразу же кристаллизуется при растирании в эфире; обра-
зовавшиеся кристаллы затем перекристаллизовывают нз смеси
хлороформ — эфир.
В синтезе кофермента урпдпнглюкозодифосфата (УГДФ) Тодд
и сотр. 161 защищали первичную 5-гидроксильпую группу уридина
(5) тритплироваиием, бензилировали 2- и З-гидроксильпые группы
(6) и тритильную группу удаляли 80%-пой уксусной кислотой.
S-Защитная группа [7]. Цистеин легко превращается вБ-защпщен-
ное тритильное производное взаимодействием Т. с хлоргпдрагом
цистеина в ДМФА; в этих условиях аминогруппа не затрагивается.
Защитная группа отщепляется под действием Na — NH3, НВг
в АсОН и CF3CO;H. Недостаток первого метода — в расщеплении
имеющихся в пептиде S — S-связей; два последних метода кислот-
ного расщепления сопровождаются отщеплением N-карбобензокси-
групп. Для удаления S-тритилыюй группы пептид сначала обраба-
тывают прн 0‘ метанольным раствором нитрата серебра и пиридина.
418
Образующийся меркаптид серебра затем превращают в SH-соеди-
нение под действием конц. соляной кислоты в ДМФА:
НО НО
I 'I AgXO,-I>v | I!
-NCHC- -NCHC- -й(СД1Д,СОСН3-:-РуН+КО;
CrljUri i
CH2SC(C0H5)3 CH,SAg н о
| hci | |]
1----NCHC — -p AgCL
I
CH2SH
Для S-защиты используют также дифеиплметильную (бепзгид-
рпльпую) группу; она устойчива к солям металлов, но может быть
удалена другими методами: НВг — ЛсОН или CF;iCO2H. Таким
образом можно синтезировать пептид, содержащий одно цистеино-
вое звено, защищенное тритильной, и другое, защищенное дифенил-
метильной (бензгидрильной) группой. S-Трнтильную группу затем
можно удалить действием AgNO;j; при этом S-дифенилметильная
группа не затрагивается и может быть удалена действием CF3CO2H.
N-Защитная группа. Тритильная группа привлекала некоторое
внимание в качестве N-защиты аминокислот 181, оДпако серьезным
ограничением является то, что, за исключением производного гли-
цина, /1-нитрофеннловые эфиры N-тритиламинокислот не реагируют
с эфирами аминокислот [9].
1. Бахман В., «Синтезы органических препаратов», ИЛ, At, 1952, сб. 3,
стр. 426.
2. X а у з е р Ч_, X удзон Б., мл., «Синтезы органических препаратов»,
ИЛ, М., 1952, сб. З; стр. 427.
3. Flclferich В., S р с i d е 1 Р. Е., Т о с 1 d 1 е W., Вег., 56, 766 (1923).
4. Н с 1 f е г I с h В., *М о о g L., Junker Л., Вег., .58, 872 (1925).
5. Рейнольде Д., Эванс У., «Синтезы органических препаратов», ИЛ,
М., 19x52, сб. 3, стр. 399.
6. Kenner G. \V., Todd A. R., W с b b R. Г., J. Chem. Soc., 1954, 2843;
.Michelson A. \V., Todd .A. R., J. Chem. Soc., 1956, 3459.
7. Zervas L., Photaki 1., J. Am. Chem. Soc., 84, 3887 (1962).
8. Z cr v a s L., T h e о d or о p a u I os D. At, .1. Am. Chem. Soc., 78, 1359
(1956); Буассома P. А., в кн. «Успехи органической химии», том 3, пзд-во
«Мир», М., 1966, стр. 171; A in i а г d G., 11 е у rn csR., V е 1 1 u z L_, Bull,
soc. chim. France, 22, 191 (1955).
9. Zervas L., Borovas D., Gazis E., .1. Am. Chem. Soc., 85, 3660
(1963).
ТРИФЕНИЛХЛОРСТАННАН, ((AH^SnCl. Мол. вес 385,46,
т. пл. 104—106°, растворяется в бензоле и метаноле, нерастворим
в воде.
Получение аллиллития [1F Аллилтрпфенплстаинаи получают
следующим образом: к суспензии магния в эфире прп кипячении
и перемешивании добавляют раствор бромистого аллила и Т. в
ТГФ в течение 7 час, добавляют еще бензол и смесь кипятят в тече-
ние ночи. После гидролиза насыщенным раствором хлористого
аммония алл илтрифец ил станнан выделяют из органического слоя
141=
419
н кристаллизуют из лигроина (т. пл. 73—74°). На следующей ста-
дии к раствору 0,127 моля аллилтрифепилстапнана при перемеши-
вании в атмосфере азота добавляют эфирный раствор 0,127 моля
фениллитня. Тетрафен ил станнан быстро осаждается и через 30 мин
раствор аллиллития обрабатывают эфнрным раствором 0,12 моля
1) Кипячение, 7 час
2) NHjCI
(CeH5)3SnCl Сн3 — CHCH3Br -| - Mg у________*•
0,65 моля 1 моль 2,1 г-атом. ' ,г>
CcH,Li
—* (C8H5)3SnCH2CH---СН3 дД СН3 -- CHCH3Li —>
— (Си Н д) 4ОП
он
СН-^ОСИЮНССН^ I
—:------------->СН...---СНСН.- -С- СН,СН(СН3)а
70—75% * . '
(две стадии) I
СПз
4-метилпентанона-2, который добавляют с такой скоростью, чтобы
поддерживать легкое кипение. После гидролиза смеси водой твер-
дый тетрафен ил станнан отфильтровывают и из органического слоя
выделяют продукт 4,6-диметил-Л1 2-гептенол-4.
Этот единственный метод получения чистого аллиллития гножно
также использовать для синтеза внниллнтия.
1. Сейферт Д., Вейнер М., «Синтезы органических препаратов», изд-во
«Мир», 1964, сб. 12, стр. 3-1.
О, Н-бис-(ТРИФТОРАЦЕТИЛ)-ГИДРОКСИЛАМИН,
COCF3
HN —ОСОСЬ'з
Мол. вес 225,06, т. пл. 62°.
Реагент получают из ангидрида трифторуксуспой кислоты и
гндроксиламина 111. С альдегидами в смесн пиридин — бензол Т.
образует соответствующие нитрилы (выходы 70—90% [I]).
COCF3 г ОН COCF3
I II’
СеНйСН = СНСНОД HN—ОСОСРД -. —С -N
I I
L Н OCOCF3
“>C6H6CH^CHCN
Под действием Т. оксиметиленстероиды превращаются в циан-
стероиды [2].
HNOCOCF3
COCF3
1. Pomeroy J. IL, Craig С. A., J. Am. Chem. Soc., 81, 6340 (1959).
2. Kissman H. M-, Hoffman A. S., Weiss M. J. J. Org. Chem., 27,
3168 (1962).
420
ТРИФТОРАЦЕТИЛГИПОГАЛОГЕНИТЫ, CF3CO2X. Т. полу-
чают взаимодействием брома или иодас трифторацетатом серебра [1].
Т. применяют для галогенирования ароматических соединений;
примеры [2];
CeH6I + CF3CO2I ~ п-1С6Н41
C6H5CH3^CF3CO2Br —л-ВгС6Н4СН,
c6h5nh2 -i-cf3co2] — ^-n-IC6H4NH2
1 /л
1. Н с п n е A. L., Z i m m er W. F., J, Am. Chem. Soc., 73, 1362 (1951).
2. Haszeldinc R. N., S li а г p e A. G., J. Chem. Soc., 1952, 993.
N ТРИФТОРАЦЕТИЛИМИДАЗОЛ, (1). Мол. вес 176,10, т. пл.
136—137°.
Получение 11]. Т. получают из имидазола и трифторуксусного
ангидрида в ТГФ или из \т,Гч'-карбонилдиимидазола и трифторук-
сусной кислоты в ТГФ.
Ангидриды карбоновых кислот [2]. При обработке 1 моля Т.
2 молями карбоновой кислоты в ТГФ в результате равновесных реак-
ций образуется смешанный ангидрид (2), а затем чистый ангидрид
(3). Реакция идет до конца благодаря выделению ограниченно раство-
римой соли имидазола и трифтор уксусной кислоты (4), и прн вы-
держивании при комнатной температуре в течение примерно 5 час
с высоким выходом получается чистый ангидрид.
о
II
леон +
о о
II и
rc—о—ссг3
(2)
о О о о
X 11 Й у II
2) RC-O-CCF3 + RCO2H RC—O—CR + CF3CO2H
(3)
3)
cf3co2h + HN
> CF3COO
(4)
Для получения п-нитрофен иловых эфиров аминокислот Т. более
эффективен, чем N,N'-карбонилдиимидазол, одиако этот метод менее
удовлетворителен, чем стандартный метод с применением дицикло-
гексил карбодиимида [31.
[.Staab Н. A., Walther G.., Chem. Ber., 95, 2070 ([962).
2. S t a a b H. A., Walther G., Rohr W., Chem. Ber., 95, 2073 (1962).
3. La w H. I)., J. Chem. Soc., 1965, 3897.
421
а, а, а-ТРИ ФТОР АЦЕТОФЕНОН, C«HSCOCF3. Мол. вес 174,12,
т. кнп. 1517740 льи.
Применение Т. для синтезов р-замещенных перфторолефннов
можно продемонстрировать на примере получения 2-фенилперфтор-
пропена [2]. Смесь эквимолярных количеств a,а,a-трпфтор ацетофе-
нона, трифенплфосфина и хлордифторацетата натрия (взятого
в 100%-ном избытке для получения CF._.) нагревают в диглиме в ат-
мосфере азота.
Дим им,
C6H5COCF3-Н (СЙН5)3Р + СС1Р2СОЛ% С6Н-,С - CF, 4- (СвН5)3РО
30—Ь0% I
0r2j моля 0 25 моля 0,5 моля v |
CF3
1. Dishart К. Т., Levine R., J. Am. Chem. Soc., 78, 2268 (1956).
2. Herkes F. E., Burton D. J., procedure submitted to Ощ. Syn.
ТРИФТОРИОДМЕТАН, CFJ. Мол. вес 195,92, газ.
Трифторметилирование. При облучении в пиридине Т. взаимо-
действует со стероидным 3-этиловым эфиром енола (1), образуя
6-трифторметильное производное (2) [1]. Катализируемый кислотой
гидролиз дает экваториальный 6а-трифторметил-Д4-3-кетостероид
(3) с хорошим выходом.
Радикальное присоединение к олефинам детально изучено Хас-
цельднном; ссылки приведены в работе 12L
l.Gotfredsen XV. О., Va nged а 1 S., Chem. Scand., 15, 1786 (1961).
2. У о л л н и г Ч., Свободные радикалы в растворах, ИЛ, М., 1960.
ТРИФТОРНАДУКСУСНАЯ КИСЛОТА, CF3CO3H.
Т. к., легко синтезируемая и обладающая замечательной окис-
лительной способностью, была открыта Эммонсом и corp. [ 11 в 1953 г.
—NH>—*—NO2. В первом сообщении [II отмечено, что очень
сильная трифтор уксусная кислота (р[<а 0,3) реагирует с водной пе-
рекисью водорода намного быстрее, чем муравьиная или уксусная
кислота, и равновесие смещено в сторону образования Т. к. Для
окисления 0,05 моля п-ампнобензонптрнла в л-нптробензошггрпл
раствор Т. к. получают добавлением 5,1 мл (0,2 моля) 90%-ной пе-
ХС-<T”Vnh2 2££°Д
рекпеи водорода к 40 мл трпфторуксусной кислоты при 207 Затем
при добавлении амина температура повышается до 507 Эгу темпера-
422
туру поддерживают периодическим охлаждением в течение 1 час.
Продукт осаждают водой; выход высокий. Из анилина получен
нитробензол с выходом 79%; окисление надуксусной кислотой
дает 11% нитробензола и 71 % азоксибензола. Высокая кислотность
трифтор уксусной кислоты препятствует такой вторичной кон-
денсации. Этим методом из дпэтилнптрозамина было получено76%
диэтнлнитрамина.
СЩСО,П
(С,Н Г1) 2 У NO —- - •> (С, НД. N NO.,
Однако в последующих исследованиях Эммонс [2] усовершенство-
вал этот метод, применив безводную Т. к., полученную смешива-
нием 90"о-пой перекиси водорода с соответствующим количеством
трнфторуксусного ангидрида, причем хлористый метилен оказался
наиболее пригодным растворителем (т. кип. 41°). Раствор безвод-
ного реагента в хлористом метилене не терял активного кислорода
при кипячении в течение суток; стабильность этого реагента выше,
чем надмуравьиной кислоты. Реакцию при температуре кипения
легко контролировать.
В типичном окислении 4,1 мл (0,15 моля) 90 %-ной Н2О2 и 100 ли
хлористого метилена перемешивают и охлаждают льдом, затем до-
бавляют в один прием 26 мл (0,18 моля) трнфторуксусного ангидри-
да. Через 5 мин (экзотермическая реакция) смесь нагревается до
комнатной температуры, и в течение 30 мин добавляют 0,1 моля
диэтилпитрозамнна в 10 мл хлористого метилена при интенсивном
кипении. После кипячения в течение 1 час органический слон отде-
ляют, промывают водой, сушат н упаривают. Выход остатка (ди-
этилнитрамин) составляет 91%, выход после перегонки — 76%.
Раствор реагента (безводного) в хлористом метилене или хлоро-
форме применяли для окисления анилинов (особенно содержащих
электроотрицательные заместители) в нитробензолы 131. В рассмат-
риваемом примере растворителем служит хлороформ; выход при-
веден для чистого продукта, перекристаллизованного из этанола.
NH, no2
ГУ ,NO2 I /NOa CF3CO.,H C ijz 88 % ’ | ||
Y
NO., no2
о- и п-Динптробепзолы, а также 2,4,6-тригалонитробензолы получа-
ются с хорошими выходами. Ппкрампд не окисляется; окисление
и-аиизидина и р-нафтилампна проходит слишком бурно.
Метод окисления анилинов и питрозаминов применим для окис-
ления оксимов до резонансно-стабилизированных ацинитроалканов
[4].
сг3со,н. СНЙСЦ
HON — С(СО2С2Нй)а-------------O.2NCH(CO2CsH6)3
ь о %
423
Окисление монофункционального н-октанальдоксима в хло-
ристом метилене приводит к смеси продуктов, одним нз которых
оказался нитрил, образовавшийся, возможно, при отщеплении три-
фторуксуспой кислоты от трифтор а нети л оксима:
С[',СО(Н
н-С71115СН -- ХОН — > w-CTHl5CII NOCOCI'u
__^н.С7ц15С — х ;-CF/:0Jl
Добавление твердой динатриевой соли фосфорной кислоты в ка-
честве буфера, удаляющего трифторуксусную кислоту из системы,
предотвращало побочную реакцию образования нитрила, однако
при этом образовалось незначительное количество нитроалкаиа.
Дальнейшее изучение условий реакции показало, что нитроалканы
можно получить с высокими выходами окислением Т. к, в ацетонит-
риле в присутствии буфера (Na2HPOd, NaHCO3, NaaCO3). Например,
бензальдоксим окислялся безводным реагентом в ацетонитриле в
CF3C0,H, Na2HPO4; CO(NH.),
в CH3CN
C6H5CII - хон--------———--------* C6H5CII3NO2
%
присутствии динатриевой соли фосфорной кислоты и небольшого
количества мочевины для удаления окислов азота.
Олефины. Эммонс и сотр. [5] показали, что в хлористом мети-
лене безводный реагент мгновенно взаимодействует прн 0° со всеми
испытанными олефинами; он оказался намного активнее надмуравь-
иной кислоты. Высшие терминальные олефины реагируют быстро,
тогда как при 40° реакция с надмуравьиной кислотой протекает
в течение 8—24 час. Первоначально образовавшийся эпоксид рас-
щепляется Т. к. до моноэфнра диола, который при метанолизе
превращается в диол. В первых опытах моноэфир в некоторой сте-
пени взаимодействовал с эпоксидом, но эта побочная реакция за-
медлялась при добавлении трифторацетата триэтиламмония, повы-
шавшего эффективную концентрацию иона трнфторацетата. Напри-
н ОН он
|/Ч|1 сгщо.,н Ах СЦСОД \ "|-н СП..0Н \ ЧГ”Н
I ч j!---> ф,!----------U-OCOCF, -ST (-он
Н ZH II
мер, раствор 50 мл хлористого метилена, 37,2 ль? (0,264 моля) трп-
фюруксусного ангидрида н бл/л 90%-ноп перекиси водорода (0,264
моля) добавляли при кипячении в течение 20 мин к раствору 0,2 моля
циклогексена и 0,1 моля трифтор ацетата триэтиламмония в 50 мл хло-
ристого метилена и перемешивали 15 мин. После удаления летучих
растворителей при пониженном давлении перегонкой было выделе-
но 46,7 г сырого моноэфнра, нз которого при кипячении с 300 мл
хметанола в течение 20 час с последующим выпариванием получали
424
19,0 г (82%) бесцветного кристаллического транс-циклогександио-
ла-1,2. Продукт очищали однократной перекристаллизацией из
ацетона, т. пл. 103—104\ Мстанолпз заметно ускоряется хлористым
водородом. В синтезе пентандиола-2,3 кипячение в метанольном
растворе хлористого водорода (3%) заканчивали через 2 час
(выход 74%).
Эммонс н Пагано [6] предложили оригинальную схему выделе-
ния первоначально образующегося эпоксида, основанную на до-
пущении, что Т. намного слабее трифторуксусной кислоты. Кон-
станту кислотности Т. к. определить невозможно, однако на основа-
нии констант для двух других пар кислота — надкислота можно
предположить значение рКа около 3,7 (см. таблицу). В присутствии
твердого буфера (Na2CO3, NaHCO3, Na2HPO4) трнфторуксусная
кислота нейтрализуется по мере добавления, однако надкислота
намного быстрее реагирует с олефином. По израсходовании олефина
Кттслот J рКл Надкислота рКа
СН3СООН 1,8 СН3СО3П 8,2
НСО2Н 3,7 НСОдН 7,1
CF3CO2H 0,3 С1%СО31-[ (3,7, при- близительная оценка)
буфер разлагает любой избыток надкислоты, и единственным ве-
ществом, присутствующим в растворе, является эпоксид. Для
эпоксидирования 0,2 моля пентена-1 к смеси 8,2 мл 90%-ной пере-
киси водорода (0,3 моля) и 50 мл хлористого метилена при переме-
шивании и охлаждении льдом добавляли 50,8 мл (0,76 моля) трифтор-
уксуспого ангидрида и затем перемешивали еще 15 мин. Полу-
ченный раствор добавляли в течение 30 мин к перемешиваемой смеси
0,9 моля твердого карбоната натрия с раствором 0,2 моля пентена-1
в 200 мл хлористого метилена (интенсивное кипение). После кипяче-
ния в течение 30 мин соли отделяли центрифугированием. Пентен-
1,2-эпоксид выделяли перегонкой; т. кип. 89—90°, выход 81%.
Этим методом были получены эпоксиды из метилметакрилата (84%)
и этил к ротон ата (73%).
CF/..0 ПЛ .СО СМЫСЛ.
СН,С1! ,CILC11 -СН., ------------- -CH.iCH.,CiI.,ClI--ClH
" -‘л1;,, - - / -
Ч>
Мишели 171 применял эту методику к а цетил метил у рсол ату,
имеющему сильно защищенную двойную связь, и выделял обра-
зовавшийся кетон с высоким выходом,
Реакция Байера — Виллигера. Эммопс п Лукас 181 показали,
что Т. к. в отличие от других надкислот мягко и быстро окисляет
с высокими выходами большинство ациклических кетонов до слож-
ных эфиров. Вначале 5—10% продукта теряли при переэтерифика-
О С)
1| СГ,СО;|Н 'I
RCR' -----RC-OR'
ции эфира с Т. к. Но эту побочную реакцию можно подавить добав-
лением динатриевой соли фосфорной кислоты в качестве буфера
для удаления большей части трифтор уксус пой кислоты в момент
се образования. Стедует отметить применение этой реакции для
П
Г>-ССНз
СГ3СО3Н^
53%
окисления мсти л циклопропил кетона до ппклопропи л ацетата [81.
Некоторое количество непрореагировавшего кетона, присутствую-
щего в реакционной смеси, нужно удалять реактивом Жирара Р,
кроме того, выход был невысоким. Однако метилциклопропилкетон
совершенно инертен по отношению к надбеизойиой кислоте. Благо-
даря высокой кислотности трифторуксусная кислота, возможно,
играет роль катализатора:
о
11С)с?сс1-з ОН
R-C-0-------------R-Cr
| | ' ооссг.,
R' R' г
О
426
Высокие выходы также достигались при окислении циклических
кетонов в лактоны с расширением цикла |9|.
сьдсо3п^
81-88%
Окисление ароматических соединений. Чамберс и сотр. [10]
исследовали окисление ароматических углеводородов безводной
Т. к. в хлористом метилене и, кроме случая бензола п толуола, полу-
чили смесь продуктов, которая содержала незначительное коли-
чество фенолов п хинонов, как показано на примере л/-ксилола:
Из мезитнлена был получен мезитол и 2Д5-трпметпл-1,4-беизо-
хинон — продукт перегруппировки. Мак-Клур [111 окислял анизол
и дифениловый эфир и выделил небольшие количества (7—35%)
орто- и /шрп-оксипропзводпых.
Мак-Клур показал, что медленное добавление перекиси водорода к
раствору 2,6-дпметилфснола и трифторуксусной кислоты в хлористом
427
метилене приводит сначала к продукту орто-окисления — димеру
III 2,6-диметил-о-хинола (II, ср. окисление хлорилфторидом).
Однако при смешивании реагентов в одни прием хинон V получался
с 72%-ным выходом. Это объясняется тем, что в кинетическом урав-
нении для V имеется по крайней мере один член более высокого
порядка по окисляющему агенту, чем в соответствующем уравнении
для соединения III. При высокой концентрации окислителя наблю-
дается преимущественное образование V, однако при низкой кон-
центрации окислителя, добавляемого за 20 час, скорость образо-
вания V уменьшается и главным продуктом реакции становится
соединение III. Образование о-хинола II объясняется через проме-
жуточный комплекс VII, в котором Т. к. связана водородной
связью в положении, благоприятном для атаки орто-углеродного
атома электрофильным кислородом Т. к. в переходном состоя-
нии VIII.
Баклер и Харт [121 предположили, что координация кислоты
Льюиса с органической надкислотой может способствовать отщеп-
лению ионного электрофильного гидроксила и повышению активно-
сти окислителя в мягких условиях. Действительно, применение
трехфтористого бора значительно улучшило метод синтеза мезитола.
Для уменьшения окисления первичного продукта углеводород брали
в избытке. Раствор готовили при 0° из 35 г (0,167 моля) трифторук-
сусного ангидрида, 50 мл хлористого метилена и 4 мл (0,148 моля)
90%-ной перекиси водорода, давали ему нагреться до комнатной
температуры, затем охлаждали льдом (температура ниже 7':) и в те-
чение 2,5 час добавляли 56,1 г (0,468 моля) мезитилена в 100 мл
хлористого метилена при пропускании трехфтористого бора в реак-
ционную смесь. После соответствующей обработки было получено
32 г мезитилена п 17,7 а (88%) мезитола.
428
При окислении пренитола (1) было получено только 9% ожида-
емого продукта (2), а также меньшие количества соединений (3) —
(7), очевидно образовавшихся вследствие перегруппировок. Обра-
зование диенона (4) объясняется атакой ОН+ и миграцией метиль-
ной группы:
Предположив, что гексаалкилбепзолы не способны к образова-
нию фенола, и, таким образом, миграция алкила должна иметь
большое значение, Уоринг и Харт [131 применяли реакцию, ката-
лизируемую BF3, к гексаметил- и гсксаэтплбензолам при 0° и полу*
Сн3 О
H3Q J ZCH3 Н3С Д /СНз
t Д CF,CO,H. вг3. СН2С1, и к
- -СИ
I II 03% II I
н,с/^/хсн3 H3cz 'Y' \сн3
си3 сн3
Желтое масло, А 330 ммк (log е 3,62)
429
чили диепоиы с высоким выходом. 2,3,4,5,6,6-Гексаметилцнклогек-
садцеп-2,4-он не димеризуется, а при кипячении в эфире реагирует
с образованием продукта присоединения к малеиновому ангидриду
(т. пл. 140').
N-Окись 2,6-дибромпиридина. Вследствие индуктивного влия-
ния двух атомов брома в 2,6-дибромпиридине неподеленная пара
электронов азота практически неспособна к координации, и N-окпсь
CF3CO3H
75%
О'
нельзя получать окислением иадбепзойноп или надуксусной кисло-
той, однако удовлетворительные результаты даст применение более
электрофильной Т. к. 114|. Смесь 3 г амина, 37 мл трпфторуксусной
кислоты и 4 мл 30"о-ной перекиси водорода нагревали иа кипящей
водяной бане в течение 3 час и полученный продукт однократно
перекристаллизовывали из метанола.
Синтез бензимидазола. Нейр и Адамс [151 показали, что Т. к.
окисляет 2-замещениые анилины типа (1) до бензимидазолов
(2) в очень мягких условиях с выходом от 60 до 90% в зависимости
от заместителей в ароматическом кольце. Удовлетворительные ре-
зультаты получены с иадмуравьиной кислотой 1161. Оба реагента
циклизуют N-ацилпроизводные типа (3) с отщеплением ацильной
группы с высоким выходом [13].
CF3CO3H
или НС03Н
----•--
(з) (4)
R = СНО, СОСН3, СОС6Н5-
~ (СН2)г, (сн2)3, СН2ОСН2, (СН2)4
Окисление /У-З-кетостероидов. Т. к. окисляет Д-*-холестенон-3
до альдегпдолактона (3), возможно, через образование и перегруп-
пировку продукта (2) — эпоксида енола лактона [171.
430
W. D., Ferris A. F., J. Am. Chem. Soc., 75, 4623 (1953).
W. D., J. Am. Chem. Soc., 76, 3468 (1954).
W. D., J. Aim Chem. Soc., 76, 3470 (1954).
W. D., Pagano A. S., J. Ann Chem. Soc., 77, 4557 (1955).
W. D., Pagano A. S., Freeman J. P., J. Ain. Chem.
1. Emmons
2. Emmons
3. E m ш о ns
4. Emmons
5. Emmons
Soc., 76, 3472 (1954).
6. Emmons W. D., Pagano A. S., J. Am. Chem. Soc., 77, 89 (1955).
7. M i c h с I i R. A., J. Org. Chem., 27, 666 (1962).
8. E m m о n s W. D., Lucas G. B., J. Am. Chem. Soc., 77, 2287 (1955).
9. S a g e г W. F., Duckworth A., J. Am. Clicrn. Soc., 77, 188 (1955);
Huisgen R., Ott H., Tetrahedron, 6, 253 (1959); Smissman E. E.,
M uren J. F., Dahlc N. A., J. Org. Chem., 29, 3517 (1964).
10. Cham bers R. D., Goggin P., Musgrave W. R. R., J. Chem.
Soc., 1959, 1804.
11. McClure J. D., W i I 1 i a m s P. H., J. Org. Chem., 27, 627 (1962);
McClure J. D., ibid., 28, 69 (1963).
12- В u с к 1 e г S. A., Hart H-, J. Am. Chem. Soc., 85, 21/7 (1963); Hart
H. et al., J. Org. Chem., 29, 2397 (1964); 30, 331 (1965).
13. Waring A. J., Hart FL, J. Am. Chem. Soc., 86, 1454 (1964).
14. E v a n s R. F., Van Am jnefs AL, Den Her tog H- J., Rec. trav.
chim., 78, 408 (1959).
15. Nair M. D., Adams R., J. Am. Chem. Soc., 83, 3518 (1961).
16- M c t h-C о h n O., Siischitzky FL, J. Chem. Soc., 1963, 4666.
17. P i n h e у J. T., Schaffner R., Tetrahedron Letters, 601 (1965).
ТРИФТОРТИОБЕНЗОЛ, C«H5SF3. Мол. вес 166,17, т. пл.
около 0°, т. кип. 6075 мм (4872,6 мм).
Т. получают взаимодействием дифенилдисульфида с дифто-
ридом серебра в фреоне-113 (1,1,2-три хлор-1,2,2-трифтор этан)
CeH5SSC(il-I3 + 6 AgF3 2 CgH-SF3-F6 AgF
(т. кип. 47°) (I J.T.медленно реагирует co стеклом,но может хранить-
ся без изменения в сосудах из алюминия или политетрафторэтилена
(тефлон). Подобно тетрафториду серы, он реагирует с карбониль-
ными соединениями с образованием дифторметиленовых соединений
(R2C=ORaCF3) и с карбоновыми кислотами с образованием три-
фторметильных производных (RCO>H^ RCF3). Он не столь токсичен,
как тетрафторид серы, и применение его не требует оборудования
для проведения реакций под давлением, выполненного из стойких
к действию фтора сплавов. Так, реакцию с бензальдегидом проводят
в стеклянной колбе, соединенной с сухой дистилляционной колонкой
при 100°; затем понижают давление до тех пор, пока не отгонится
c6h5cho+c6h5sf3 >C6FI5CHF3+C61-I5SOF
431
бензальфторид (т. кип. 45о/15льи). Второй продукт, бензосульфин ил -
фторид (т. кип. 60°/2,5 мм), образуется с выходом 82—89%.
1. S h с р р а г d W. A., J. Am. Chem. Soc., 84, 3058 (1962); Org. Syn., 44, 39, 82
(1964).
ТРИФТОРУКСУСНАЯ КИСЛОТА, CF3CO.2H. Мол. вес 114,03;
т. кип. 72—72,5°, рКа 0,3 (кислота более сильная, чем трихлорук-
сусная).
Получение карбаматов. Карбаматы спиртов, даже третичных
(которые легко дегидратируются), можно получить с высоким вы-
ходом при перемешивании спирта с 2 же цианата натрия и Т. к.
в бензоле или хлористом метилене при комнатной температуре в те-
чение нескольких часов (Лоев и Кормепди fl]). Наблюдается слабо
экзотсрмичная реакция, и выделяется некоторое количество газо-
СН3 СН3 О
СН3 — С — ОН 4- О = С — NH . н •*f i ic , СН3 - С - OCNHo
| 69% (чистый) |
сн3 сн3
Т. пл. 107—108°
образной синильной кислоты, трет-Бутил карбамат выделяют,
добавляя воду и отделяя органический слой, удаляют растворитель
и кристаллизуют сырой продукт (т. пл. 98—101°, выход 92%).
При использовании трихлор уксусной кислоты выход едва достигает
50%. То обстоятельство, что другие очень сильные кислоты (соля-
ная, метаисульфокислота) дают только следы продукта, привело
авторов [1] к предположению о том, что в реакции Т. к. обра-
зуется циклическое переходное состояние или смешанный ангид-
рид. Метод является весьма общим. Даже сравнительно неустой-
чивый пропаргиловый спирт образует карбамат.
Конденсация с последующей циклизацией. Вудс [2] разработал
простой метод синтеза кумаринов конденсацией фенола с (3-кетоэфи-
ром в присутствии Т. к. Однако в приведенном примере замыкание
цикла происходит в том положении, которое активировано орто-
и пара-гидроксильными группами. Фенол, пирокатехин, 4,6-дихлор-
ОН
|| +СВН5СОСН2СО.2СН5
CF;,CO,H
-СДНОН*
он с6н5
432
резорцин, гидрохинон и крезолы не вступают в реакцию. Реакция
успешно применена к 1-нафтолу, но 2-иафтол не реагирует [3].
Растворитель для гидрирования. По-видимому, Т. к. является
наилучшим растворителем для катализируемого платиной гидри-
рования кетоиов [41. Скорость реакции в этом растворителе прибли-
зительно втрое выше по сравнению со скоростью о уксусной кислоте.
Необходимо использовать сравнительно высокую концентрацию
кетона, так как в разбавленном растворе реакция протекает медлен-
но. Спирты часто получаются в виде эфиров.
Трифторацетат серебра. Используется вместе с иодом для иоди-
рования вератрола в положение 4 [51. Соль получают добавлением
Т. к. к суспензии осажденной окиси серебра в воде, фильтруют
и упаривают фильтрат досуха. Экстракция сухой соли эфиром в ап-
парате Сокслета и удаление растворителя дают бесцветное кристал-
лическое вещество с 88%-ным выходом.
Пептидные синтезы. Защитные N-карбобензоксигруппы ами-
нокислот и пептидов могут отщепляться с высоким выходом при
кипячении с Т. к. [6]. Кислота также рекомендуется в качестве раст-
ворителя для снятия защитных групп с помощью НВг вместо ук-
сусной кислоты, в которой происходит частичное ацетилирование
гидроксильной группы серина 171. В случае образования неболь-
ших количеств брома его удаляют, добавляя диэтилфосфат.
Т. к. удаляет защитные mpem-бутоксигруппы оксиаминокислот
при комнатной или более низкой температуре без рацемизации
и без расщепления пептидных связей [8]. трет-Бутильная и mpem-
бутокси карбон ильная защитные группы отщепляются 90 %-ной
Т. к. с прекрасным выходом [9].
Расщепление бензиловых эфиров. Т. к. расщепляет аромати-
ческие бензиловые эфиры при комнатной температуре [10].
Растворитель для изучения кинетики. Браун и Вирккала [111
установили, что Т. к. является наилучшим растворителем для изу-
чения кинетики ароматического электрофильного замещения (бро-
мирования, нитрования, мер курирования). В отличие от сложной
кинетики, наблюдающейся в уксусной кислоте, в данном раствори-
теле наблюдаются реакции второго порядка. Кроме того, реакции
в Т. к. протекают быстрее.
Hz/Pt (Rh)^
cf3co2h
Н /Г-Л. О н СО,Н
I' // II J I
R = Н ИЛИ ch2n—е у— C-N - сн
снгсн2со2н
433
Растворитель для гидрирования. Т. к. является панлучшим
растворителем для гидрирования (над Pt пли Rh) птеринов, так
как пиразиновое кольцо птерина, его метилированного производ-
ного и фолиевой кислоты гидрируется полностью и селективно за
короткое время (см. схему на стр. 433) [12!.
1. L о е v В., к о г П1 с и d v At 1'.. J. Org. Chem. 28, 3421 (1963).
2. W о о d s L. L., Sapp J. 0гц. Chem., 27, 3703 (1962).
3. W о о d s L. L., Steel i n g J.. J., 0гц. Chem., 29, 502 (1964).
4. P c t e r s о n P. E., Casey C., J. 0гц. Chem., 29, 2325 (1964).
5. Д ж а и с с с u Д., У и ,i ь с о и Ч., «Синтезы органических препаратов»,
ИД, М., 1958, сб. 8, стр. 22.
6. \\ с у g а п d Г7., Steel i с И W., Z. УatindorseJi.. 14В, 472 (1959),
7. G u t t rn annSt.,Boi ss о n n as 1?. A., Helv. Chiim Acta, 42, 1257 (1939);
X icolaides E. D., 1) e \V a Id 11. A., J. Org. Chem., 28, 1926 (1963).
8. В c v e г in а п H. 0., Bout с k о c J. S., Proc, Chem. Soc., 1961, 249;
Rec.' Trav., 81, 691 (1962).
9. Sell w v z cr R., Cos I op a na»i ot is A., S i ch er P., Helv. Chim.
Acta, 46, 870 (1963).
10. At arsh J. P., Jr.. G о о d man L., J. Org. Chem., 30, 2491 (1965).
И. В г a w n 11. C., W i r k k a 1 a R. A., J. Am. Cheni. Soc., 88, 1447, 1453,
1456 (1966).
12. В о b s t A., Viscont ini At., Helv. Ch hit. Acta, 49, 875 (1966).
ТРИФТОРУКСУСНОЙ КИСЛОТЫ n- НИТРОФЕН иловый
ЭФИР, п-NTH:нНЛХЮСКр Мол. вес 235,12, т. ил. 37—393
Реагент полу чают с количественным выходом кипячением
//-нитрофенола с трпфюруксуспым ангидридом. При взаимодействии
с карбобсизоксч[аминокислотой при комнатной температуре реагент
легко образует л-иптрофсииловый эфир 111. Реакция с карбобензок-
сипеитидом осуществляется медленнее и требует нагревания пли
длительного реакционного периода. //-Нитрофен илтр и хлорацетат
реагирует только в кииящехм пиридине и дает //-нитрофен иловые
эфиры с плохим выходом.
1. S я к а к i h а г a S., I и и к a i У., Bull. Chem. See, Japan, 37, 1231 (1964).
ТРИФТОРУКСУСНОЙ КИСЛОТЫ СЕРЕБРЯНАЯ СОЛЬ,
CF;{CO2Ag. Мол. вес 220,90.
Получение |1|. Трифторуксусную кислоту добавляют к свеже-
осаждениой окиси серебра в воде, раствор фильтрхют л упаривают
в вакууме досуха. Белое твердое вещество очищают экстракцией
эфиром в аппарате Сокслета; выход 88%.
Йодирование. Джанссен и Уильсои [II использовали реагент
в смеси с иодом для иодирования вератрола. К смеси вератрола
ocn;i oci ь
/ OClh; I /ОСН,
I
43-t
п Т. к. с. с. при перемешивании добавляют в течение 2 час раствор
подав 1,6.? хлороформа. Еще через 1 час подпетое серебро отфильтро-
вывают, фильтрат промывают, упаривают и продукт перегоняют,
Окисление. Ньюмен и Реид [2] нашли, что реагент лучше окиси
ртути при окислении по Курциусу бензилдпгпдразонов до дпарил-
ацетпленов. Смесь беизилдпгпдразоиа и Т. к. с. с. в 250 ма ацетонит-
рила перемешивают при комнатной температуре и в течение 2,5 час
СЙН;-С — ССДЩ 4CF;,CO.2Ag |- 4E1:iN __-
I .Ml J Tli.ll.I
N M
ME NIC
J.r, г
—> CfiH5C - CCOHS4- 2\г 4Ag 1 4СЕ3СОГ\;НРЛЯ
обрабатывают тр идти ламином. Контроль реакции осуществляют,
определяя объем азота, который выделяется в течение 6 час. Затем
смесь выливают в 200 ли копи, водного раствора аммиака.
>СВг.•<’ С О. Кэйв |3| нашел, что Т. к. с. с. является пре-
красным реагентом для гидролиза 1,1,2,2-тетрабромбензоцпклобу-
тена (1) до а-дпкетона (2).
(1) з,4з г
+ CF3COzAg + Н2О +
17, 67 2 4 Л/Л
(2)
Перечтет и вэние,
_тт т кипячение 12 час.в темноте
G Н3 U ГЧ и »| । ।
87
70 Л/Л
Присоединение к олефинам. При добавлении иода и V-холестена
к раствору реагента в хлористом метилене образуется трифтора це-
тил под ид, который присоединяется к олефину с образованием За-
вод-2р-трифтор ацетокси ход еста на с выходом 72%[4]. Под действием
алюмогидрпда лития осуществляется восстановление и деацетилиро-
вание до холестанола-2[Е
1. Дж а песен Д., 5' л л j, с о и Ч., ('Синтезы органических препаратов»,
ИЛ, At, 1958, сб. 8, стр. 22.
2. X’ewman At S., Reid D. Е., .1. Огр. Clieni., 2,3, 66о (1958).
3. С a v a At Р., Napier D. R., Pohl R. J., .1. Am. Chem. Soc., 85. 2076
(1963) .
4. Hey D. G., J. Chem. Soc., (C)r 1966, 1331.
ТРИФТОРУКСУСНОЙ кислоты ФЕНИЛОВЫЙ ЭФИР,
CF£O,C(iH-,. Мол. вес 190,12, т. кип. 148—1494
435
Получение. Один из методов синтеза состоит в нагревании
трифтор уксус ной кислоты и трифенилфосфита при 100 до прекра-
щения кипения кислоты н еще в течение 1 час с последующей пере-
CF3CO2H•! (CGH5O);,P .!.00°’ 1,5 'ш—>CF;jCO.,CfiH5-'-(Cf,H5O)2POH
0,2 моля 0.2 2 мол?! у
(перон г д к л мл)
гонкой [1]. Другой метод [2] включает реакцию фенолята натрия
с фосгеном и обработку полученного хлорформиата (1) 1 мдтрифтор-
уксусной кислоты и триэтиламина в ТГФ.
о
Г II
СЙН-ОХЛ - С1СС1 -——- >С,Н-ОСС1
(1)
ООП о
CGHaOC —О —CCFa 21?евани^ С,Н5ОССР3
ел
Синтез пептидов. Т. к. ф. э. используют для получения N-три-
фторацетильных производных аминокислот и пептидов [31. Субстрат
нагревают с 1,2—2 зкв реагента в расплавленном феноле при 120—
150“ в течение 2—20 мин. Избыток реагента и фенол удаляют в ва-
кууме либо отделяют, растворяя в петролейном эфире или четырех-
хлористом углероде, из которых производные аминокислот кри-
сталлизуются. Основное преимущество N-трифтор а цетильной за-
щитной группы состоит в том, что она удаляется в очень мягких
условиях.
1. Beno it on L., Ry don H. N., Willett J. E., Chem, Ind., 1960,
1060.
2. Green M., Chem. Ind., 1961, 435.
3. Weygand Ropsch A., Chem. Ber., 92, 2095 (1959).
ТРИФТОРУКСУСНЫЙ АНГИДРИД, (CF;iCO),O. Мол. вес 210,04,
т. кип. 39,5“. Обзор [1]. Получение: СсН,СЕ{—> ji-NO.»C6H4CFs
->3?-NHaC(iH4CF3->HO3CCF.^ Ангидрид (РгО5) 12].
Трифторацетилирование. Первичные и вторичные спирты очень
легко ацетилируются Т. а., но эфиры настолько чувствительны
к гидролизу, что иногда в процессе всего синтеза необходимо избе-
гать контакта с водой. Так, Татлоу и сотр. 13! в течение короткого
времени кипятили смесь н-нптробеизилового спирта, Т. а. и трифтор-
ацетата натрия и перегоняли смесь с четырьмя порциями четырех-
хлористого углерода при пониженном давлении, чтобы удалить
избыток ангидрида и трифтор уксусной кислоты. Эфир отделяли от
п-\О,Св114СН„ОН I (CF;iCO).,O-n CF3CO.2Na ...к"^|''ен.п2> п.\о2СеН4СН,ОСОСР3
0,407 г 2 мл 0J61 г Т. пл. 47°
трифтор ацетата натрия экстракцией петролейным эфиром и раствор
кон центр провали для кристаллизации. Эфир быстро гидролизовал-
436
ся водой. Сообщалось о применении Т. а. в синтезах саха-
ров [4].
Лардон и Рейхштейн 15] установили, что трифтор ацетилирова-
ние удобно для защиты стероидных На- и 110-гидроксильных групп
в процессе превращения 170-карбоксильной группы в —COCTLOH.
С этими объемистыми и нерастворимыми в воде веществами нежела-
тельный гидролиз в процессе работы ие представляет опасности, и
защитную группу можно легко удалить с помощью слабого основа-
ния. Ацетилирование выполняют без растворителя или в диоксане,
или в ппридппс. Аксиальная 110-гидроксильная группа устойчива
А
СН3ОН- водн.К2СО3
(з)
к ацетилированию в пиридине, но легко реагирует с Т. а. при комнат-
ной температуре. Так, раствор 30,110-диола (1) в ангидриде выдер-
живают в течение 16 час и экстрагируют эфиром. Раствор промы-
вают до нейтрализации раствором соды и высушивают; удаление
растворителя дает сырое бис-(трифтор ацетил иное) производное (2)
с хорошим выходом. После одной кристаллизации получают чистое
вещество. Для селективного удаления группы у С3 раствор 51 мг
(2) в 8 мл метанола обрабатывают раствором 200 мг бикарбоната
калия в 6 мл воды и выдерживают в течение двух дней при 20°.
Упаривание, экстракция эфиром и кристаллизация дают 21 мг
чистого 11-монотрифторацетата (3). При обработке этого соединения
водным метанольным раствором карбоната калия удаляется вы-
соко пространственно затрудненная группа у Сп и образуется (1).
Смешанные ангидриды. Эммонс и сотр. [6] нашли, что 1'. а.
легко реагирует с карбоновыми кислотами, образуя смешанные
ангидриды, которые можно выделить с выходом 50—65%. Так,
смесь бензойной кислоты и ангидрида перемешивают при кипяче-
437
нии в течение 30 мин, Т. а. и кислоту отгоняют в вакууме, а бензо-
Кипячение
30 МНН I; '
CJI-COoH + (С[’ЯСО)2О-----7^—- —> C(iH.-,COCCF3-CF3CO,H
2,5 моля 2 моля ' '* ”
плтрифторацетат перегоняют прн 10070,5 мм. Изучение этой реак-
ции в и-бутиловом эфире и в ацетонитриле методом инфракрасной
спектроскопии показало, что равновесие устанавливается немед-
ленно и беизоилтрнфторацетат образуется с количественным вы-
ходом,
Дюкуорт [71 использовал Т, а. для получения ангидридов мало-
новых кислот, которые интересны тем, что при пиролизе образуют
кетены.
R,C(CO,H).
(CF.COi ,0 z со,н Гу 4 „ А
1 — RaCy 1_R,C О
С—О—CCF3 — 95% % / 6U —S0%
li il СО
о о
—> R.,C-C^CH со..
Этерификация. В присутствии Т. а. спирт реагирует с кислотой
по следующей схеме [11:
ROH I R'CO.H--(CF.tC0),,0 — R'CO,R । 2CF3CO.H
Из работы Эммонса, цитированной выше, следует, что этерифи-
кация протекает через промежуточное образование смешанного ан-
гидрида. Эта исключительно мягкая реакция имеет значение при
этерификации пространственно затрудненных кислот. Так, Парит
и Сток 181 перемешивали смесь мезитпленкарбоновон кислоты и
СИ, 1
няс со,н Д1 •• С1 f 7 । к'' ' ОН Н.С /1 сн, //' U 1 2(1 мнн iijiif 25- (CF .СО) .О - - 0 мл “ 1Г.С Д сн О
1 сн, I,U S 1 сня 0,83 г няс С-0 J /СН, 1 II
и
СН3
мезитола в Т. а. в течение короткого времени при комнатной темпе-
ратуре, смесь экстрагировали бензолом и выделяли мезнтилмези-
тоат с высоким выходом.
438
Антибиотик хлорамфеникол (1) содержит первичную и вторичную
спиртовые группы; частичное ацилирование по обычной методике
дает первичный моноэфир, который, будучи безвкусным, находит
применение в педиатрии, а также используется для парентерального
введения. Альмиранте и Тосол ин и |91 разработали метод гюлуче-
Н NHCOCHC1.,
O,N— / Ч — С — С — СН,ОН-,- CH3(CH..)lBCO,HT(CF3CO).,O —
\=/ 5 | -
он н
’ И NHCOCHCJ,
O2N-<f с—CHsOCCF3 —н^*
i н о
(Lo
I
(CHJ|6CH3
(2)
Н NHCOCHCla
х I I •
—Оп\ — с____________у—с—с—сн.,он
\=/ j ,
О II
I
с-о
(снг)[всн3
(3)
ния моноэфиров по вторичной спиртовой группе, основанный на
методе Шмидта и Штааба 121, ранее использованном в ряду саха-
ров. Реакция хлорамфеникола (1) со стеариновой кислотой и Т. а.
дает производное (2), в котором стеароильный остаток присоединен
к вторичной, а трифторацетильный остаток — к первичной спирто-
вой группе. Слабое основание удаляет трифторацетильную группу,
образуя (3); таким образом, осуществляется селективное стеароили-
рование вторичной спиртовой группы в присутствии первичной
спиртовой группы.
Ацилирование. Т. а. катализирует ацилирование активирован-
ных ароматических соединений карбоновыми кислотами 1101. у-Фе-
ОСН3 ОСН3
I II ' I II
\/ W
I
СОСНз
иилмасляная кислота с высоким выходом превращается в а-тетр а-
лон при нагревании с Т. а. при 60—70° в течение 3 час 1111. В тех же
439
условиях Р-фенилпропионовая кислота превращается в инданои-1
с очень низким выходом.
Тот факт, что в приведенной выше реакции анизол дает ацетиль-
ное, а не трифторацетильиое производное, означает, что смешан-
ный ангидрид является более активным ацилирующим агентом,
чем Т. а. Однако чистый Т. а. способен к эффективному ацилиро-
ванию. Он реагирует с азуленом (синий) в четыреххлористом угле-
роде без катализатора при комнатной температуре, образуя 1-три-
фтор а цетил азулен (красный) с высоким выходом [12].
Хоппе и Теддер [13] сообщили об интересном, хотя и протекаю-
щем с низким выходом ацилировании олефинов и ацетиленов. При
нагревании смеси циклогексена, уксусной кислоты и Т. а. прибли-
зительно до 37" образуется эфир (1), формально — продукт присое-
динения смешанного ангидрида, который можно считать результа-
том атаки Н3СО+ и CF3COy. Этот эфир не выделен в чистом виде,
| || + СН3СООН -И (СР3СО).О
18,5 мл 10,5 мл 27 мл
+ CF3CO,H
так как он самопроизвольно разлагается до циклогексеиилметил-
кетона (2) и трифторуксусной кислоты. Гексин-1 реагирует сходным
образом; продукт присоединения (3) можно выделить в чистом виде
или после метаиолиза в виде октандиопа-2,4 (4).
ч-С4119С СН + СНзСООН + (CF3C0).,0 «-с4н3с=снсосн3
11,4 мл 5,7 мл 14,5 мл I
OCCF3
;1
О
О)
II II
О о
(4)
440
Харфепист [14] установил, что Т. а. значительно эффективнее
фосфорного ангидрида в циклизации фенотиазивового производного
(5). Кислоту в течение непродолжительного времени кипятят
с эквивалентным количеством ангидрида в бензоле.
Перегруппировки. Эммонс [15] установил, что Т. а. удобен для
проведения перегруппировки Бекмана,причем он образует раство-
римые в воде амиды. Раствор метилциклопропилкетоксима в 1,2-дп-
метоксиэтане обрабатывают при кипячении и перемешивании
Т. а. в течение 1 час и кипятят еще 1 час. Летучие растворители отго-
няют при атмосферном давлении, к смеси при перемешивании и ох-
лаждении льдом добавляют раствор 300 г едкого кали в смеси ООО мл
НГОН 400 мл
С— CHj + (CF3CO)2O Т СН3ОСНгСНгОСИз
1,92 моля
2, 2 моля
Кипячение
]^>—NHCOCHj
КОН ,
77%
(oduinilj
этиленгликоля и 300 мл воды. В качестве побочного продукта выде-
ляется метиламин, а циклопропнламин (т. кип. 49—51") перегоня-
ют на колонке в течение 8 час, отбирая продукт, кипящий ниже 51°.
н-Алкилфенолы, важные в биохимии, окисляются в процессе
обмена до д-хинолов, которые под влиянием ферментов перегруппи-
ровываются в алкилгидрохиноны. Хекер и Мейер [16] установили,
что диеиои-фенольная перегруппировка может осуществляться под
действием Т. а. при комнатной температуре с количественным вы-
ходом. Толухинол (1) дает толугидрохинон (2) и 2,4-диокситолуол
(3). Диеиои встряхивают с Т. а. до полного растворения и раствор
оставляют при комнатной температуре на 72 час, трифтор ацетиль-
ный остаток удаляют гидролизом водным диоксаном. Метиловый и
СН3 ОН
q/' тА7 HQ/ "Ч/ но7
(I) (2> (3)
тетрагидропиранпловый эфиры (1) дают только гидрохинон п не
образуют резорцинового производного. Тетралин-и-хипол (4) и его
эфиры перегруппировываются исключительно в 5,8-дпокситетралин
441
(5) или его моноэфиры. Стероидный дпенон (6) дает продукты (7)
и (8). Хекер [17] показал также, что эфир дпенона (9) перегруппиро-
вывается под действием Т. а. в ароматический продукт (16).
CI13СОО
(СГ;,СО)Ю
НО' 4 "z
(10)
[18] осуществили реакцию Шмидта для
(9)
Резерфорд и Ныомен
фснантренШкарбоновой кислоты. 5 г кислоты растворяли в J00 м-1
раствора, содержащего равные объемы Т. а.
кислоты, охлаждали до 0—5' и
и трифторуксусной
при перемешивании постепенно
CO..II
дооавлялп
(CFJlObO
CF.COjl
N-C--0
кон
выделялся азот,
количественным
избыток азида натрия. При этом быстро
и изоцианат кристаллизовался из раствора с почти
выходом. Следует отметить, что для перегруппировки не требуется
присутствия минеральной кислоты и что реакция не происходит,
если ангидрид пли кислота используется в отдельности.
Получение нитраминов. Т. а. используют для иптролиза диал-
киламидов с образованием диалкилиптрамииов 1191.
2HN’O3^(CF3CO)2O NCC J-NO; з_2СР..С(ХН
R4 . R
NO;- ;N—Ac —> O2N —N/ J-Ac+
“ ' R XR
442
Синтез глицеридов. Т. а. использовали для прямого синтеза
глицерида из глицерина и жирной кислоты; моноглицериды ti-
ll 2’) можно получить из соответствующим образом защищенных
глицеридов [201.
О- и N-защита. Ныомен [211 использовал трифторацетпльпую
группу для защиты гидроксильной и аминогрупп в 1-галогеисахаре
(1); после конденсации (2) со спиртовым компонентом, например
холестерином, защитные группы удаляли обработкой водио-мета-
польным раствором карбоната калия прн комнатной температуре.
(О (2)
К2СОз-водн.МеОН^
(4)
Уолфром и Бат [221 использовали трифторацезильную группу в ка-
честве N-блокирующей группы в синтезе нуклеозида D-глюкоза-
мина.
1. Tedder J. AL, Chem. Revs., 55, 787 (1955).
2. S с h m i d t О. Th., Staab >V., Chem. Ber., 87, 388 (1934).
3. Bourne E. JT a t I о w С. E. AL, T a t I о w J, C., J. Chein. Soc.,
1950, 1367.
4. Bourne E. J,, Stacey AL, T a t I о w С. E. AL, T a t I о w ,1. C.,
J. Chem. Soc., 1951, 826.
5. Lardon A., R eichstci п T., Helv. Chim. Acta, 37. 388, 443 (1934).
6. Emm о n s W. D., McCallum K. S., [’ er r i s A. J. Am. Chem.
Soc., 75, 6047 (1953). See also Bourne E. J., S t а ее у AL, T a t I о w J. C.,
W о г r a 1 1 R., J. Chem. Soc., 1954, 2006.
7. D u c k w о r t h A. C., J. Org. Chem., 27, 3146 (19621.
8. Parish R. C., Stock L. AL, J . Org. Chem., 30, 927 (1965).
9. A I ni i r a n t e L., Tosolini G., J. Org. Chem., 26, 177 (1961).
10. Bourne E. J., Stacey AL, Tallow J. C., Tedder J. AL, J.
Chem. Soc., 1951, 718.
II. Ferrier R. J., Tedder J. AL, J. ('.hem. Soc., 1957, 1435.
12. Anderson A. G., Jr., Anderson R. G-, J. Org. Chem., 27, 3578 (1962).
13, H c n n e A. L., T e d d e r J. AL, .1. Chem. Soc.. 1953, 3628.
14. Harfenist AL, ,J. Org. Chem., 28, 1834 (1963).
15. Emmons \V. D., J. Am. Chem. Soc., 79, 6522 (1957).
16. Hecker E., Aleyer E., Angew. Chem. Internal. Ed., 3, 229 (1964);
Chem. Ber. 97, 1926 (1964).
17. Heeker'E., Chem. Ber., 97, 1940 (1964).
413
18. Rutherford К. G., Newman M. S., J. Am. Chem. Soc., 79, 213
(1957).
19. R obson J.H., Rei n har t J., J. Am. Chem. Soc., 77, 2453 (1955); Fran-
kel M. B., Tieman С. H., V a n n e m a n C. R., Gold M. H., J.
Org. Chem., 25, 744 (1960).
20. Bourne E.J., Stacey M., Tatlow J.C., Tedder J. AL, J.
Chem. Soc,, 1949, 2976; Cook P. F. E., S h о w 1 e r A. J., ibid., 1965, 4594.
21. Newman H., J. Org. Chem., 30, 1287 (1965).
22. W о 1 f г о m M. L., В h a t H. B., Chem. Comm., 1966, 146.
ТРИХЛОРАЦЕТОНИТРИЛ, CC13CN. Мол. вес 144,40, т. пл. 44°,
т. кии. 84,67741 лш, уд. вес 1,44.
Получение. Крамер f 11 использовал несколько измененный
метод Штейикопфа 121: 200 г трихлорацетампда и 250 г Р2О5 хорошо
перемешивают и нагревают примерло до 200° в течение нескольких
часов на масляной бане. За это время отгоняется Т., и его снова
перегоняют над Р2О5; выход 70— 80%.
Применение. Для замены гидроксильной группы на хлор спирт
обрабатывают Т. в растворе в эфире при —15и и пропускают сухой
хлористый водород 121. Реакция протекает с полным обращением
конфигурации. Реагент используют для селективной этерификации
фосфорной кислоты (1) 13]. Ои превращает фосфат в симметричный
ROH 4 CC|3CN-|- НС1 —> RCl 4-CCI3CONH2
пирофосфат (2), а в присутствии спирта — в диэфир. Т. до некото-
рой степени сходен с карбодиимидами, по нс реагирует с диэфирами
О
1) С6Н5СН.,ОН-|- Н3РО4 ESbSL'^Si^^Cel^CFEOP-OH + CCbCONIL
77°' I "
1' ’u I
он
о о о
СГ1,СХ;-Ру 'ii l!
2) 2n-CICBH4OP—ОН —n-CIQH.jOPO —POCeH4Cl-n + CCl3CONH.
I 9|- ||
ОН он он
фосфорной кислоты и с карбоновыми кислотами. Т. применяется
для получения пирофосфатов терпеновых спиртов [4].
1. Cramer F., private communication.
2. StcinkopI W., Ber., 41, 2541 (1908).
3. Cramer F., Baldauf 11.-J., Chem. Ber., 92, 370 (1959).
4. C r a m c r F., R i t ters d or f \V., Bohm \V., Ann., 654, 180 (1962).
ТРИХЛОРМЕТАНСУЛЬФОБРОМИД, CCJ3SO2Br. Мол. вес
262,37, т. пл. 1397
Т. получают нз соответствующего хлор и да следующим образом [1 ]:
CC13SO2CI + KCN CCI3SO2K CCl3SO2Br
Под действием света или в присутствии перекисей как инициаторов
Т. реагирует с циклогексаном, циклопентаиом или толуолом с об-
444
разованием соответственно циклогексилбромида, циклопеитилбро-
мида или беиз ил бромида.
I. Р i п п е 1J R. Р., Н и у s е г Е. S., К 1 е i п b е г g JJ. Org. Chem., 30,
38 (1965).
ТРИХЛОРМЕТАНСУЛЬФОХЛОРИД, C1hCSO2C1. Мол. вес 217,89,
т. пл. 140—141°.
Получение. Т. получают с 78%-ным выходом окислением три-
хлорметансульфснилхлорида перекисью водорода в уксусной кисло-
те [11.
Радикальное хлорирование. В условиях радикальной реакции
Т. действует как хлорирующий агент [21:
RH -У ClaCSO2Cl —RC1-1 CI3CH-PSO2
(ЩЩСОЩ
Типичные реакции Т., протекающие с высоким выходом:
С6НйСП3 СВН5СН,С1
п.-ВгСеН4С113 —••> л-ВгСЦЦСН.дС!
Циклогексан —>- Циклогексилхлоряд
Этот метод хлорирования обладает значительной селективностью.
Так, я-гексап и этилбензол дают только 2-хлоргексап и а-хлор этил-
бензол. При хлорировании н-гептаиа образуется 2-хлоргептан с вы-
ходом 50%, но наблюдается замещение и у других атомов в цепи.
Последовательность реакций следующая:
R- Ц CI3CSO.,C1 RCI + C13CSO2
Cl;iC8O2-;-RH R.+Cl3CSOsH
C13CSO211 C13CH + so.
1. S о s п о v s k у G., J. Org, Chem., 26, 3506 (1961).
2. Huyscr E. S., J. Am. Chem. Soc., 82, 5246 (1960); Huyser E.S., Gi ri-
dings B., J. Org. Chem., 27, 3391 (1962); Huyser E. S., S c h i m k e H.,
Burham R. L., ibid., 28, 2141 (1963).
ТРИХЛОРМЕТИЛЛИТИЙ, CUCLi (см. tom I, стр. 416).
T. получают добавлением трихлорбромметана к взвеси метил ли-
тия в эфире при—115° [Ц. При —80° Т. разлагается экзотермически
с образованием смеси, содержащей тетрахлорэтилен. При взаимо-
действии с циклогексеном при —100° Т. дает дихлориоркараи с вы-
соким выходом. По имеющимся данным реакция, по-видимому,
осуществляется не через дихлоркарбен. Кёбрих и сотр. [21 вместо
эфира использовали ТГФ, в котором Т. более устойчив.
77%
445
1. Miller \У. T., Jr,, \V hale n D. *M., J. Am. Chem. Soc., 8fi, 2089 (1961).
2. Kobri e h G., !•’ I о г у К., Merkle 11. R., Tetrahedron Letters, 973
(1965).
ТРИХЛОРУКСУСНАЯ КИСЛОТА, CC1;(COOH. Мол. nee 163,40,
т. пл. 58 т. кип. 196уд. вес 1,62, рКа 0,08. Т. к. получают окисле-
нием хлораля азотной кислотой, содержащей окисли азота 111.
Т. к. рекомендуют в качестве катализатора в процессе диазоти-
рования антраниловой кислоты для получения бензолдпазоний-2-
/ХН- Ц<> 7 . N=N
I || -|-СС13СО.,Н-г Ледuae-Ain-NO, '5'22^ 1 ''Z- I ||
L Н ьб-ю% । I,
'Ч/ СОаИ 4 со.7
20 .кмолей 0,003 л 25 г 5 м.1
н 30 мл ТГФ
карбоксилата, предшественника дегидробеизола [21. Охлажденную
реакционную смесь перемешивают и выдерживают при комнатной
температуре в течение около 1 час. Выделившаяся соль диазония
взрывоопасна; ее собирают на пластмассовой воронке Бюхнера
пластмассовым шпателем, промывают холодным ТГФ, затем раст-
ворителем, применяемым на следующей стадии реакции, и исполь-
зуют в виде суспензии в этом растворителе. Уксусная кислота и
сильные минеральные кислоты пе являются катализаторами.
1. Parkes G. Г)., Hollingshead R, G. W., Chem, Ind., 1954, 222.
2. L о g u 1 I о I'. M., Friedman L., Case lust. Teclin., procedure submitted
Io Org. Syn.
ТРИХЛОРУКСУСНОЙ КИСЛОТЫ НАТРИЕВАЯ СОЛЬ,
CCl3CO2Na. Мол. вес 185,39.
Получение, а) |1|. К 12,8 а трихлоруксусной кислоты в колбе
Эрленмейера (на 125 мл) с боковым отводом при охлаждении льдом
добавляют около 9/10 охлажденного до О3 раствора 3,2 г гранули-
рованного едкого натра в 12 мл воды. При добавлении капли 0,04%-
ного раствора бром крезолового зеленого смесь приобретает слабо-
желтую окраску, и, пользуясь капиллярной пипеткой, ее тигруют
оставшимся раствором щелочи до изменения окраски от желтой
до сипей. Колбу закрывают резиновой пробкой, помещают иа водя-
ную баню, обертывают полотенцем для сохранения тепла, присое-
диняют колбу к водоструйному насосу и включают насос на полную
мощность. Выпаривание не требует внимания и завершается за
15—20 мин. Вес сухого порошка близок к теоретическому (14,5 а).
б) |2|. Раствор трихлоруксусной кислоты в абсолютном метаноле
нейтрализуют свежеприготовленным раствором метилата натрия
(в присутствии фенолфталеина) при температуре ниже 20Избы-
ток метанола удаляют на роторном испарителе при 20 ; соль остается
в виде белой твердой массы, которую высушивают при 50° над
РЙО5 в течение 20 час.
416
Получение дихлоркарбена. При термическом разложении соли
в апротонном растворителе, содержащем реакционноспособный
олефин, получается дихлоркарбеи, присоединяющийся к олефину
|3--5]. Например П|, цис, цис-1,5-цпклооктадиен дает смесь двух
кристаллических бис-аддуктов; при этом в качестве растворителя
используют тетрахлорэтилен, подходящий по температуре кипения
(121 ') и низкой реакционной способности. Однако поскольку Т. к.
п. с. мало пли вообще не растворяется в этом растворителе и не
распадается даже при кипячении, то в качестве дополнительного
растворителя применяют небольшое количество диглима. Диглим
является настолько сильным растворителем для соли, что при
использовании только одного диглима его следует добавлять не-
большими порциями, поддерживая температуру ниже 120’, причем
раствор сильно темнеет. Соль добавляют к смеси растворителей
сразу, и смесь кипятят без присмотра, так как соль растворяется
медленно по мере взаимодействия. Слабое окрашивание исчезает
после перегонки с водяным паром и промывания сырого продукта
метанолом.
CCljCO.Na
14, 5 г
Ьмл (4,4 г )
( 20 МЛ
МЛ
СКС-СС1,
Д и гл и м
В основном иис,
немного транс
Филдс 161 установил, что Т. к. н. с. лучше других предшествен-
ников CCL. для внедрения его по С—Н-связи в кумоле. При кипя-
чении смеси кумола и Т. к. и. с. в глимс до прекращения выделения
СО2 образуется р,р-дихлор-тл/«’/п-бутплбензол с выходом 33%.
При получении дихлоркарбена из хлороформа и трет-бхчплата
калия или из этилтр и хлор ацетата и метилата натрия этот продукт
СН3
Н3С —С— И
I
Л , Кипячение r CHjOCB^CHjOCH
I р (т, кип. 12 ча(
Р--CCl,CO,Na ..................
I 1; 11 33:lu
Д/
CH,
11аС —С —СНС1,
д
I
образуется с выходом всего 0,5—5%.
При получении пента хлор циклоп рои ан а путем присоединения
дихлоркарбена к трихлорэтилену (Тоби и Уэст |4|) смесь 1,600 кг
продажной Т. к. н. с., содержащей 6% воды, и 2,5 д трихлорэтиле-
на нагревают при перемешивании до полной отгонки воды (2 час).
Выделение СО., начинается только после добавления 750 м.1 диметок-
сиэтапа (глим, т. кип. 83 ) и при дальнейшем нагревании продол-
447
ждется в течение 2 дней; реакционная смесь при этом темнеет. Вы-
сушивание натриевой соли в вакууме не повышает выхода продукта.
С1 С1
СНС1—СС12 + :СС1г , С,Нз°^.гСй2°СНз> ГХ. * NaC1 + со2
22,4% Н XjX С1
С1
7. кип. 58°/7 мм
Трихлорметилирование ангидридов [2]. При разложении Т. к. н.с.
в глиме в присутствии ненасыщенного ангидрида реакция осущест-
вляется исключительно по карбонильной группе ангидрида, по-ви-
димому, с участием трихлорметильпого аниона, являющегося пред-
шественником дихлоркарбена. Выход колеблется от 80 до 8%.
о
Cl3CCO~Na+ (84°)
СН3ОСНгСН2ОСН3
7 5%
I. Fieser L. F., S а с h s D. Н., J. Org. СИрш-, 29, 1 i 13 (1961); Org. Expts.,
205.
2. Winston A., Bederka J. P. M., 1 s п e г W. G., Juliano P. C.,
Sharp J. C., J. Org. Chem., 30, 2784 (1965).
3. Wagner W. M., Proc. Chem. Soc., 1959, 229; Wagner W. M., Kloos-
terziel Fl., Van def Ven S., Rec. trav. chim., 80, 740 (1961).
4. Tobey S, W., West R., Tetrahedron Letters, 1179 (1963); J. Am. Chem.
Soc., 88, 2481 (1966).
5. Moore W. R., К r i k о г i a n S. E., L aPra de J. E., J. Org. Chem.,
28, 1404 (1963).
6. Fields E. K., J. Am. Chem. Soc., 84, 1744 (1962).
ТРИХЛОРУКСУСНОЙ кислоты ЭТИЛОВЫЙ ЭФИР,
СС13СО2СаН6. Мол. вес 191,44, т. кип. 55—58712 мм. Реагент при-
меняют для получения дихлоркарбена. Например, реакция с дигид-
ропираном дает 2-окса-7,7-дпхлорнокаран 11]. В промытую азотом
СС13СО2С2Н5 + СНэОТ4а^
68-75%
колбу загружают 0,92 моля метилата натрия, 0,8 моля дигидропи-
рана и 600 мл пентаиа, не содержащего олефинов. Светло-желтый
раствор перемешивают 15 мин при охлаждении в бане со льдом и
водой и затем в течение 3—4 мин обрабатывают 0,86 моля реагента.
Смесь перемешивают 6 час в атмосфере азота при 6° и оставляют
на ночь при комнатной температуре; при этом окраска изменяется
от желто-оранжевой до коричневой. Реакционную смесь обрабаты-
418
вают водой, водный слой экстрагируют пентаном и после фракциони-
рования получают продукт с т. кип. 74—7678 мм.
1. Parham W. Е-, Schweizer Е. Е., Mierz. w a S. A. Jr., Org. Syn.,
41, 76 (1961).
2,4,5-ТРИХЛ0РФЕН0Л, ChCJLOH. Мол. вес 197,46, г. пл.
62—63°, рКа 9,45.
Сравнение скоростей реакций различных замещенных фениловых
эфиров N-защищенных «-аминокислот показало, что 2,4,5-трихлор-
фениловые эфиры более реакционноспособны, чем н-нитрофениловые
эфиры, и являются новыми активными производными для синтеза
пептидов ] 1]. Одно из преимуществ состоит в том, что эфирная груп-
па не изменяется при каталитическом гидрировании для удаления
карбобензоксигруппы. Эфиры получают конденсацией N-защищен-
ных аминокислот и Т. в присутствии дициклогексилкарбодиимида.
В частности, в синтезе гексапептида, где другие методы давали низ-
кие выходы, Бентли и corp. 121, используя трихлорфениловый
эфир, получили выход 97%.
I. Pless J., Boissonnas R. A., Helv. Chim. Acta, 46, 1609 (1963).
2. В en 11 ey P. II., Gregory H., Laird A. H-, XI о r 1 e у J. S., J.
Chem. Soc., 1964, 6130.
ТРИЭТАНОЛАМИН, (HOCH2CH2)3N. Мол. вес 149,19, т. пл. 21°,
т. кип. 2797150 мм, уд. вес 1,12.
Липстед и сотр. [1] установили, что это основание — наилучший
катализатор конденсации алифатических альдегидов с малоновой
кислотой, приводящей к образованию чистых Р,у-ненасыщенных
кислот, например А3-гексеновой кислоты. Смесь альдегида, кислоты
и катализатора (каждого по 1 молю) смешивают до полного раство-
СН3СН2СН2СНО + СН2(СО.,Н).2 Н (HOCH2CH2)3N ——
4 0 — 4 2 %
1 моль I моль [ моль
СНзСН2СН-СНСН2СО2Н
рения, выдерживают 2 дня и затем нагревают на водяной бане
в течение ночи до прекращения выделения СОа. Побочными продук-
тами являются продукты само конденсации альдегида, а также про-
дукты его конденсации с 2 молями малоновой кислоты.
Гарднер и сотр. [2J разработали эффективный метод синтеза
пимелииовой кислоты (4) из фурфурола (1), включающий стадии
f Я + СН2(сОгН)2 ОН у [[ 11 С2Н5ОН (Н_)
Хо'^СНО 86% \оХ^СН=СНСОгН 75%
(1) о (2)
с2н5о2с( сн2)2с( сн2)2согсгн5 но,с( сн2)5согн
99} 5%
(3) (4)
15 Зиказ XI 1166
449
получения фур ил акриловой кислоты (2) и этерификации, которая
происходит с расщеплением колыш и диспропорционированием
и приводит к дпэтил-у-кетопимелату (3). Восстановление по Хуанг-
Миллону по обычной методике в этиленгликоле или диэтиленгли-
коле значительно менее удобно, так как при выделении продукта
восстановления путем продолжительной экстракции эфиром экстра-
гируется значительное количество растворителя. Задача была ре-
шена при использовании Т. в качестве растворителя и подкислении
реакционной смеси перед экстракцией эфиром в автоматически дей-
ствующей аппаратуре.
1. L i и s t с a d R. Р., Noble Е. G., Boor in а п Е. J., J. Clicm. Soc.,
1933, 557.
2. Gardner Р. D. R a n d L., Haynes G. R., J. Am. Chem. Soc., 78,
3425 (1956).
ТРИЭТИЛ АЛЮМИНИЙ, A i(C2H Мол. вес II4,16.
Получение. Наилучшим является двухстадийнып процесс [11.
А1-1 1,511..-- 2(С2Н5).,Л1 -- ЗЛ1(С..Н5),Н
ЗА1(С2Нг,).,Н f-3CH.,-CH.> —> ЗАl(C,H-,h
Реакция с нитрилами и изоцианатами [2]. Т. взаимодействует
с нитрилами в молярном отношении 2:1, причем образуются этид-
кетоиы с высоким выходом. При молярном соотношении 1 : 1 ре-
акция не идет. Реакция Т. с бензонитрилом, вероятно, протекает
через циклическое переходное состояние; выход пропиофенона 77%.
С6Н5—C=N СДТг,-СШ N С6Н5—C=NAlEt2 + _ AlEta
Et AlEt2 ---------► СН3бН2 y*AlEt2 * CH3CH2
ЧА1 Et A’’’Et |
/ \ Et Et 2
Et Et ОДС=О
CH3CH2
Аналогичным образом протекает реакция изоцианатов с Т.,
при этом образуются N-замещенныс амиды пропионовой кислоты
с выходом 90—95%.
RN С - О ~ СН3СН..CON Н R
Присоединение HCN. При 1,4-присоединении HCN к холесте-
нону в обычных условиях образуются 5сс- и 5р-цпапокетоиы в соот-
ношении 1:1. Однако если реакцию проводить в эфире, ТГФ или
бензоле в присутствии 4'. при соотношении кетон : HCN : (C..Hft%A 1 —
сил KCN-КП. CI вДМФА^ < 82% (°бщ,ий) ' СН,1 | j + Г Т CN CN Отношение 1’1
450
= 1 : 2 : 3, то 5а- и 5р-нптрилы образуются в соотношении 2:1,
хотя выход остается примерно гем же (85%) 131. Для реакции неко-
торых кетонов стсреоселсктпвпость выше. В случае реакции модель-
ного кетона (1) соотношение транс- и цас-продуктов 24 : 1.
В присутствии Т. и I1CN в молярном соотношении 6 : 10 происхо-
дит расщепление эпокепгрупп стероидов с образованием с высоким
выходом диаксиальных продуктов [4|:
1. Ziegler К., Gellert II. G., Lch m к п h I JI., Р f о li I W., Zose I
К.. Ann., 629 (I960).
2. R с i nil с с к е I FL, Jahnke I)., Client. Ber., 97, 2661 (1964).
3. N a g a t ;i \V., Yoshioka .XL, 11 i r a i S., Tetrahedron Letters, 461
(1962); X a g a t a W., T e r a s a w a T.. А о к i T., ibid , 865 (1963); X a-
g ata \V., К i к к a w a I,, F u j i in о t <i XL, Bui I. Chem. Pharm. 11, 226
(1963).
4. i\ a g a t a \V., X’ oshi о к a M., Oku in u г a T., Tetrahedron Letters,
847 (1966).
ТРИЭТИЛАМИН, (Cj-I5):iN. Мол. вес 101,19, т. кип. 89,5°,
уд. вес 0,73, рКЬ 3,36.
Очистка. Реагент выдерживают над едким натром и перегоняют
в присутствии приблизительно 2% фенил изоцианата 111 или пафтнл-
изоцпаната 121.
Получение диазокетонов. Обычная методика синтеза дпазоке-
тона состоит в медленном прибавлении хлорангидрида кислоты
к эфирному раствору 2 молей диазометапа; хлористый водород за-
тем вступает в реакцию 2. Если порядок изменить п реакцию про-
водить только с 1 молем диазометапа, то полученный в реакции
1 дпазокстон будет расходоваться в реакции 3. Ньюмен и Вил |.3|
установили, что в присутствии 1 эка Т., взятого для связывания
15*
451
НС1, можно получить ароматические диазокетоны с высоким выхо-
дом и при использовании I же диазометана. Применение пиридина
О о
!| + _ + -
1) RC—Cl-I-CH., -N-N RC-CH-X-N-i-HCl
2) IIC1-1-СНи — X — N СН3С1-р\2
О о
!1 - '!
3) RCCH=N —N : HCl RCCII,C1 -- N.,
неэффективно в случае хлорангпдридов алифатических кислот;
например, хлорангидрид капроновой кислоты дает смесь продук-
тов.
Кетены и их производные. В присутствии Т. происходит дегид-
рогалогенирование хлора и гидридов кислот, i имеющих а-водородный
атом, с образованием кетена, выделяющегося в виде димера, кото-
рый можно превратить в (3-кетокислоту или симметричные кетоны
(Зауср П, 4]). Примером служит получение лаурона из хлорангид-
рида лауриновой кислоты. В эфирный раствор хлорангидрида при
перемешивании и охлаждении быстро добавляют эквивалентное
। юле-жк; г
2С1011.,1СН.ДД-О----"СС. С10Н21С11 -С -О —>
-(.(лнщьша
0,7 моля в 1 2G0 .п.г
эфира
О
н.о -со., р
—. C10H21CIU=C—О —> С^Н^СНоС-О др—
CwH21CH-C-0 C10II2lCH-CO,H
количество Т. и выдерживают при комнатной температуре. Соляно-
кислый Т. экстрагируют 2%-ной серной кислотой и раствор димера
децилкстсна нагревают с 2%-ной серной кислотой, чтобы отогнать
эфир, гидролизовать димер и декарбоксилировать р-кетокнелоту.
Полученный лаурон перегоняют при давлении 3 мм и кристаллизу-
ют из ацетона. Другим примером может служить синтез 6-кето-
уидекандиовой кислоты из хлорангидрида б-карбметоксивалериа-
новой кислоты [2J.
1) моля (C.H,)tN О
2) КОН
ch.!o.,c(ch.,)1('.oci iLL’i1-— ho.,C(Ch2)4c.(Ch.,)(c.o,h
00 — 64%
0td моля в 500 мл
бензола
Синтез смешанных ангидридов. Этот метод, широко применя-
емый в синтезе пептидов [51 и являющийся общим методом ацили-
рования, можно иллюстрировать на примере бензоилирования дц-
452
этилмалоната [61. К раствору соли бензойной кислоты и Т. в толуоле
при 0° добавляют этилхлорформиат; при этом образуется смешанный
ангидрид и выделяется солянокислый Т. Другой компонент — это-
ксимагниймалоновый эфир — получают из диэтилмалоната в раст-
воре в смеси этанол — эфир в присутствии каталитических коли-
честв СС14. После отгонки растворителей остаток растворяют в
эфире и при (У добавляют к смешанному ангидриду. Реакция про-
СвН5СО.,Н-г\(С2115)з
0,2 моля и,2 моли
I
20'» v.( ;
( „11-(.ДI|
СеНГ)СО7 гЛ
О'’ |с1СО_С;Н. {Пт1? моля)
О О
I г
СвН5(>-0-С.ОС2Н5
СН,,(СО.,СаН5), -I- Mg + СС14
0,2 моля 0,2 г-лтом 0,2 моля
21 ,ил абс. С2Н5ОН
GO .11/1 эфира
Оггопка растворителя
в вакууме
CJI5OMgCH(CO2C2H5).,
0° | 68-75%
о
I;
С81 f - С—CI I (СОгС,115)о СОг + Mg(OO.H0)2
тскаст быстро с выделением двуокиси углерода. Диэтил бензоил ма-
ловат перегоняют и фракционируют.
Катализатор конденсации. Т. является основным катализатором
конденсации по Перкину онитробензальдсгида с фенилуксусной
кислотой и уксусным ангидридом с образованием а-фенил-транс-о-
нитрокоричной кислоты 171. Методика конденсации бензальдегида
с теми же реагентами включает и простой способ разделения цис-
и транс-изомеров 181.
СО2Н
o-NOaCj 1 jCl 10 -уСН2С6Н‘ (С.,Н&)3\ -1 - Ас.,О --+
0,2 моля 0,2 0 моля 0,2 моля 100.ил
Кипячение И, мин
--------------0-NOAH.-C-H
С„Н6—С—СО2Н
Т.— катализатор реакции образования гидразонов [91 и реакции
аминокислот с фталевым ангидридом с образованием защищенных
фтал силам инокисл от, как это видно на примере и-фенилаланина
а СО Кипя-
S + Ь-С4Н5СНгСНСОгН + С4Н5СН} + (СгН5)3П -чение.»
/ 91,5-95%
rn NH*
0, 1 моля
0, I молл 150 МЛ I, 3 мл
С4П5СНгСНСОгН
453
1101. Смесь указанных веществ кипятят с водоотделителем 2 час,
отгоняют растворитель при пониженном давлении, остаток пере-
мешивают с водой в присутствии небольшого количества соляной
кислоты до образования однородной массы и собирают.
Т. является эффективным катализатором реакции цианэтилирова-
ния ]111. Так, при выдерживании раствора ацетилацетона, акрило-
нитрила и Т. в смеси /п/дтщбутанола (45 лш) и воды (15 ты) в течение
2 дней при 25" образуется очень чистый уцу-диацетилпнмелоцптрпл
с высоким выходом. Эффект растворителя виден из данных, при-
веденных в таблице. Повышение скорости реакции с увеличением
сосн;}
СНчСОСН.СОСНз + 2С11,-CHC.N Л’> NCCH.,CH.,CCH.,CI I.,CN
77"„ |
COCi I;,
сольватирующей способности растворителя означает, что реакция
протекает через промежуточное образование ионов. Цианэтили-
рование мет ил под] [до в 4- и 2-метил пиридина проводят следующим
образом: пиридиниевую соль растворяют в воде, добавляют акрило-
нитрил в 2—3-кратном избытке и 0,3 моля Т. на 1 моль нитрила и
кипятят. 4-Метил пиридин левая соль образует ш/7Ш?-(2-цпанэтил)-
нроизводное, а из 2-метплпиридиниевой соли получается только
бш>(2-цпанэтил)-пропзвоД11ое. Последнее соединен не образуется
в присутствии катализатора — метилата натрия; выход 34%.
СН(СНгСНгСЫ)г
Г
Цианэтилирование ацетилацетона при 25°
I’.ICTBOprrTC.il, AU'JTh IIЫ1 [ ЦЬ1- ХОД, % Время достижения ко л они in.! предель- ного выходя, час
93% -шли этане л—вида
||11 (обвемн.)| 50 2
95%-пып этанол 57 23
Изопропанол 67 89
шрглвБутанол 51 106
Т. использовали для различных целен па двух стадиях полного
синтеза стероидов 1121. Алкилирование 1,3-дикетопа (2) галогени-
дом (1) проводят в трнт-бутаноле в присутствии Т. Впутримоле-
454
кулярпая альдолизацпя продукта (3) с отщеплением воды и замы-
канном кольца С требует более жестких условий, а (3) крайне чув-
ствителен, особенно к основания.м. Циклизацию (3) проводят с хоро-
шим выходом в растворе в ксилоле с эквивалентными количествами
бензойной кислоты и Т., образующими бензоат Т. при к пи ячеи пи
раствора с удалением образующейся воды.
RCOCH .X - - RCOCH.OCOR'. Морланд [13] разработал превос-
ходный метод получения эфиров из а-галогенкетонов и других
соединений, содержащих активный галоген, которые реагируют
с трпэтнла ми новой солью кислоты в органическом растворителе.
Например, растворяют в ацетоне эквивалентные количества бен-
зойной кислоты и Т_, затем добавляют 1 экв бромацетофенона и гомо-
СЦ1-СОЛ1 I Х(С.,Н-);) —-1О-' C.J-L>CO."NH(C.,Hr>)ii
СДЬСОШКВг
---' с(,1 L.COCI POCOCJI-
гепный раствор выдерживают при комнатной температуре в тече-
ние 2 час. Прп использовании менее активного п-нитробензплхло-
рида необходимо добавлять каталитические количества Nal и ки-
пятить в течение 2 час (выход 50—70%). Реакцию можно использо-
вать для превращения 21-иодкетостероидов в 21-ацстокспсоеди-
пеиия [14].
ch3co2nh(c2h5)3
Дегидрогалогенирование. Т. вызывает циклизацию N-хлорфор-
мплампнокпелот до ангидридов в ацетоне плп дпоксане 115). Т.
455
удобнее, чем другие основания, так как образующийся хлоргидрат
легко отделяется от раствора.
С0С1г
СО2Н
О=СС1
(сгн5)д
Ацетон
88%
(общий)
4* *
+ (С2Т1Е)3МНС1
0=0
О=с---о
Дицианокарбен получали дегидробромированием броммалэпо-
нитрила Т. в присутствии тетр а метил этилен а в качестве улавливаю-
щего агента и растворителя 1161.
КС CN
H'/C(GN), ’ с ; :С1-Д /21 (СН.Д,
Brz ’ --1%
Бреслоу и сотр. 117] использовали Т. для проведения перегруп-
пировки Фаворского а,а'-дибромдибензилкетона (1) в (2) и дегидро-
галогенирования этого промежуточного продукта с образованием
дифепилциклопропенона (3).
° ?
О с с
I; {С.НдЩ / \ ЮН.Щ
СЧН5СН —С—СНС(!Н5 C6HSC -ССДЦ — С(;Н-С-СС(;Н5
। । '11 4п"'’ (o,),iuiZ
Вт Вг Н Вг
(I) (2) (3)
Пране и Джадж [18] осуществили дегидробромирование а-бром-
у-бутиролактона (2) в у-кротонолактон (3) под действием Т. в эфире.
Восстановительное ацетилирование [19]. Суспензию 0,5 г цин-
ковой пыли и 0,5 г 2-метил-1,4-нафтохипона в 3 мл уксусного ангид-
рида обрабатывают при 25° одной каплей Т. и перемешивают;
появляется красное окрашивание, температура возрастает и при-
мерно через 2 мин окрашивание исчезает. Смесь кипятят в течение
1 или 2 мин и экстрагируют несколькими порциями горячей уксусной
кислоты (10 мл). К отфильтрованному раствору добавляют корун-
довые кипятильники и при кипячении медленно добавляют воду
до насыщения (20—25 мл); из бесцветного раствора при охлаждении
выпадает 0,6 г чистого дяацетата 2-метилД ,4-нафтогидрохинона,
т. пл. 112— 113°.
45G
При получении продукта с низкой температурой плавления,
такого, как дпанетат гидрохинона витамина Кг 1201, реакционную
смесь экстрагируют эфиром, экстракт фильтруют, промывают
разб. соляной кислотой и сильно встряхивают с 3 пли 4 порциями
разб. щелочи до разрушения уксусного ангидрида; упаривание
сухого эфириого раствора дает продукт. В ранней методике 1211
рекомендуется применять пиридин в качестве основного катализа-
тора, но при этом возможны затруднения, так как реакционная смесь
может окрашиваться в желтый цвет в результате побочной реакции
между уксусным ангидридом, пиридином и цинком. Использование
Т. позволяет избежать этих затруднений.
Сложные эфиры можно обычно получать с хорошим выходом
при нагревании смеси кислоты с ал кил галоген идами и Т. в течение
нескольких часов [221. Полученный эфир отмывают от галогенгид-
рата амина и очищают обычным методом.
Реактивы Гриньяра. Реактивы Гриньяра можно получать в бен-
зольном или толуольном растворе, если в качестве комплексообра-
зующего агента использовать стехиометрическое количество Т. [23].
Реакция начинается с большим трудом, чем в диэтиловом эфире,
но этот метод позволяет избежать применения дорогих и опасных
в обращении растворителей.
1. S a u er J, С., Org. Syn., Coll. Vol., 4, 560 (1963).
2. Durham L. J_, McLeod D.J., Cason J., Org. Svn., Col I, Vol.
4, 555 (£963).
3. N e w m a п M. S., В e a 1 P., Ill, J. Am. Chem. Soc.,71, 1506 (1949); sec
also Franzen V., Ann., 602, 199 (1957).
4. S ;ni er J. C., J. Am. Clicm. Soc.. 69. 2444 (1947).
5. V a n g h a n J. R., Jr., J. Am. Chem. Soc., 73, 3547 (1951): В о i s s о n п a s
R. A., Helv. Chim. Acta, 34, 874 (1951); Wieland T., Bernhard H.,
Ann., 572, 190 (1951).
6. Прайс Дж., T a p б e л л Д., «Синтезы органических препаратов», ИЛ,
М., 1959, сб. 9, стр. 28.
7. Де Л ос Ф., Дс Тар Д., «Синтезы органических препаратов», ИЛ. М.,
1956, сб. 7, стр. 47,
8. Org. Expts., 224.
9. Barton D. Н. R., O'B rich R. E., S t e г n h e 1 1 S. J. Chem. Soc.,
1962, 470.
10. Бозе А., «Синтезы органических препаратов», изд-во «Мпр», AL, 1964, сб. 12,
стр. 165.
11. Adamcik J. A., Miklasicwicz E. J.. J. Org. Chem., 28, 336 (1963);
A d a m c i k J. A., Flores R. J., ibid., 29, 572 (1964).
12. Wieland P., Ueberwasser H., Anner G.( Mi csclier K-,
Helv. Chim. Acta, 36, 376 (1953).
13. Moreland W. T., Jr., J. Org, Chem., 21, 820 (1956).
14. Rotliman E, S., Per 1st ei n T., W л 1 1 M- E., J. Org. Chem. 25,
1966 (I960).
15. Randall A. A., J. Chem. Soc., 1962, 374; Dvonch W., A 1 b n r n
IL E., J. Org. Chem., 29, 3719 (1964).
16. Swenson J. S., Renaud D. JJ. Am. Chem. Soc., 87, 1391 (1965).
17. Breslow R., Eicher T., Krebs A., Peterson R.A., Pos-
ner J., J. Am. Clicm. Soc., 87, 1323 (1965).
18. Price C.C., Judge J. M., Org. Syn., 45, 22 (1965).
19. Fieser L. F., «Expts, in Org. Chem., 2 nd ed,, 1941, p. 399.
457
20. Fieser I,. Г., J. Am. Clicm. Soc., fil, 3407 (1939).
21. F i e s c r I.. F., Campbell W. P., F r v E. AL, Gates AL, J. Am.
Chem. Soc.. fil, 3216 (1939).
22. Mill R. II., a rrar AL W., W e i n к а и 1 I O. .1., Chem. Ind., 1962,
2144.
23. A li Ь \ E. C., R e c d R., .1. Огд. Chcrn., 31. 971 (1966).
ТРИЭТИЛ ЕНДИАМИН,
Мол. вес 112,18, у. пл. 159--I60 , t. кип. 174 . pKa, 2,95, pKa2
8,60, Растворимость: 46?/100a H..O при 26 ; около 50г/100г бензола
ирп 26 .
Скретгас и Истхэм III установили, что это основание образует
кристаллические комплексы с металлоорганическими соединения'
мп магния, лития и цинка. Так, например, при добавлении бензоль-
ного раствора Т. к растворе бутпллитня выделяется кристалли-
ческий комплекс Bu.J.i., — Т. - Bn.LiТриэтпламин (1) и хину-
клпдин (2) образуют такие же комплексы, но они значительно более
растворимы, чем соответствующие комплексы Т.
сн3 сн3 сн3
СНг СП2 снг
(2)
р)
Комплексы применяю!ся для идеи 1 пфпкацпп металлооргани-
ческих соединений; для соединений лития, большинство из которых
жидкости, аминные комплексы - первые кристаллические произ-
водные, получение которых пе сопровождается разложенцем. Эти
комплексы используют в некоторых реакциях металлоорганических
соединений. Кроме того, найдено, что эти три основания катализи-
руют реакции ал к пл литиевых соединении, в частности металли-
рование, Так, реакция толуола с ал кил литием, приводящая к обра-
зованию бепзпллнтпя, промотпрхется всеми тремя комплексообра-
зующими основаниями.
I. S с г с t t я s С. G.. F a s t li и m .1. Е., J. Ain. Chcrn. Soc., 87, 3276 (1965).
ТРИЭТИЛЕНТЕТРАМИН, 1 1,NCU.2CH2M ICH2CH..NHCH,CHAEL.
Мол. вес 146,24, т. кип. 155—161 .18 мм.
Металлсодержащие ферменты, такие, как лейшшамннопепти-
даза, катализируют гидролиз Х-коицевых пептидных связен
в ходе процесса, включающего хелатообразование между фермен-
том, субстратом п ионом металла. Колман |1| сообщил о селектив-
ном гидролизе N-концевых связей простых пептидов ионами цис-
оксиаквагрпэтплснтетрампнкобал ьта(111). Формула (1) представ-
ляет собой цис-а-форму, одну из двух возможных стереохимических
форм комплекса. В водном растворе при pH 7 Вп 65 под влиянием
комплекса быстро происходит гидролиз N-концевого аминокислот-
ного остатка, и комплекс превращается в неактивный металлический
комплекс (2). Этот процесс, скорее стехиометрический, чем катали-
тический, вероятно, является ла пл учтен моделью in vitro действия
экзо-металлпептидаз в живом организме.
1. (toll m a n J. Р., В и с k i п ,ц h a m 1). A., J. Am. Clicm. Soc., 85. .3039
ТРИ ЭТИ Л ОКСО НИ я БО РФ ТО РИД, (С2Н ,):;О В1ф Мол, вес
189,90, т. пл. 92 .
Получение [11. Эфирный раствор эпихлоргидрина добавляют
по каплям к кипящему эфирному раствору эфпрата трехфтористого
бора. Смесь перемешивают в течение 2 час, оставляют на ночь, кри-
сталлическую оксопиевую соль отделяют и промывают эфиром.
Образовавшийся эфир борной кислоты остается в маточном растворе
Т. б., не растворяется в эфпре, легко растворим в жидкой двуокиси
С1О-Ш1
3 | АО | 4(C.,H,).,O + BF; : 2(С.,11,).,0------.
СН.,-
~ А 3(С.,Н5);!О-; BF
С1СН.,СН
I
С., 11, осн..
в
459
серы и умеренно растворим в хлористом метилене. Об одном из при-
менений см. Диметилформамида диэтилацеталь.
Реакции, наблюдавшиеся Меервейном и сотр. 121. Т. б. этилирует
спирты, фенолы и карбоновые кислоты. Фенол и кислота взаимодей-
ствуют с Т. б. в виде натриевых солей в водном растворе, так как
он реагирует с солями легче, чем с водой. С аммиаком Т. б. образует
(С2н5)уо* bf;roh(С,н5).,сн roc3h5 । bf;! i hf
смесь ди- и триэтиламина, а с пиридином — N-этилпиридиниевую
соль. 1Мочевина и ацетамид при комнатной температуре превращают-
ся в соли. Нитрилы дают нитрилисвые соли:
СН3С — N < (С.Д !BF; (СН3С -г. NC.H-JBF; !-(С.,Н-).,()
Кетоны и эфиры также реагируют с образованием солей, например
R2c_o+(C.,Hr,)3CF bf; [rX-O ^c.mijbf;
Эфиры нитроновых кислот (2). Эти прежде малоизвестные сое-
динения образуются с количественным выходом при взаимодейст-
вии с натриевыми солями нитропарафинов при 0° 131. В качестве
+ /°"
RR'CХОрNa+ -у (С,,1Ь,)3СНВР Г RR'C-X\ t
(i) ' ’ (2) 'ОС2нЛ
-!-NaBF4 + (C2II5)2O
растворителя используют хлористый метилен, так как борфторид
натрия и Т. б., взятый в избытке, нерастворимы в нем и отделяются
фильтрованием.
Дебензоилирование. В синтезе трициклина Муксфельдт и Ро-
гальски 141 применили реагент Меервейна для снятия N-бензоиль-
ной группы. После обработки реагентом проводят гидролиз 3?'о-ным
I) (щнщо+вгр
X /14 2) 3% ЛсОН ч ZH
>С:/ ------------ - 'С •
/ \NHCOC6H5 / х NIL,
водным раствором уксусной кислоты в диоксане.
Иминоэфиры. Харли-Мэсон и Лини [51 установили, что при
действии Т. б. оксиндол (1) превращается в эфир енола 1(2), т. пл.
110'1, который при нагревании в вакууме изомеризуется в имино-
1 (C2Hs)3OBF4^ 1 и (1) КП .1 80°, 0,1 Л-7Л7 г ГТ~~~—г L N Н —2 > 1 11 1 С2Н5 ОС2Н5 н (2) (3)
460
эфир [(3), т. пл. бЗ°1.При нагревании выше температуры плавления
соединение (3) затвердевает и затем вновь плавится при НО3,
превращаясь, таким образом, в (2). Растворы (2) и (3) в четыреххло-
ристом углероде имеют идентичные спектры и, очевидно, содержат
как (2), так и (3). В синтезе системы коррипа, которая характерна
для структуры витамина В12, Эшснмозер и сотр. 161 использовали
реагент Меервейпа. Например:
сг3со2н
(CzHs)3QtB Г4“- СН2С1^
(5)
Пакьетт [7] показал, что обычные реагенты (диметплсульфат,
бензолсульфохлорид — пиридин) неэффективны для превращения
амидной группировки 1,3-дигидро-3,5,7-триметил-2Н-азеппнона-2
(6) в иминопроизводное, но при действии Т. б. это превращение легко
осуществляется и образуется ЗН-азеиин (7).
(с2н5)3о+вг4“
83, 5% *
[, Meerwein Н., Battenberg В., Gold Н., Р f е г 1 Е., Will-
fang G., J. prakt. Clicm., J2J, 154, 83 (1940); Meer w c i n It., Org. Syn.
46, 113 (1966).
2. Meerwein H. et al., J. prakt. Chem., 147, 257 (1937); Chem. Ber. 89 209,
2060 (1956).
3. К or n b 1 u m N., Brown R. A., J. Am. Chem. Soc., 86, 2681 (1964).
4. M u x f e I d t H., R о g a 1 s k j W., J. Am. Chem. Soc., 87, 933 (1965).
5. H a r 1 e y-M as on J., Leeney T. J., Proc. Clicm. Soc., 1964, 368.
6. Eschenmoser A. et al., Angew. Chem., Internal. Ed., 3, 490 (1964).
7. Paquette L. A., J. Am. Chem. Soc., 86, 4096 (1964).
ТРИЭТИЛСИЛАН,(CrH^SiH. Мол.вес 116,28,т. кип. 1077733 .ilw.
Т. получают взаимодействием трихлорсилана с этил маги вибро-
МИДОМ [11-
3C.,H-MgBr-;-ci.tsiH -—- -чсл-kSiH
12,6 моля 3 моля
461
Реакции с ацеталями и кеталями. Нециклические анетали и ке-
тали при взаимодействии с Т. в присутствии 5—10% хлористого
цинка превращаются в эфиры 12].
,ос,нч
' ~ ' ZnCI,
С«И5СН -I (С.Л~—-СвНлСН3ОС2Нлш (С,Н-,):.5ЮСгН$
(XJJL,
Расщепление N-карбобензоксипроизводных. В общем методе
селективного расщепления N-карбобензокспаминокислоту или пеп-
тид обрабатывают избытком Т. (который также служит раствори-
телем) в присутствии каталитических количеств хлористого палла-
дия и трнэтпламина |3|. Смесь кипятят в течение 3 час, фильтруют
и свободную аминокислоту или пептид осаждают добавлением ме-
танола. Этот метод позволяет получать свободную аминокислоту,
расщепление с НВг в уксусной кислоте дает бромгидраты. Бензи-
ловые эфиры также расщепляются иод действием Т., однако S-бен-
зильные группы не затрагиваются.
О Н R
I । । Кипячение
СЙН,,(Л 1..ОС - N — СНСО.Н - - (C..Hp,.Sil 1 + PdCl., (С.,Н-,),\--—>
—- Н 2
П. 01 моля (),(> 1 мпля оО .if.1 1 ипля
О Н R
i I 1 (С,И-Д sS I н
- - QH.-.CH-.OC —\ — CHCO.2Si{C.,Hг,-
HR R
I I СИ..ОН I
—> (C,I-N — CHCO.,Si(CJUh -----—* H..NCHCCU1 2(CJ 15);iSiOCH3
1. Whitmore ]•'. С., P i e t r u s z a E. W., Sommer L. IL, .1. Am.
Chem. Soc., 69, 2108 (19-17).
2. [•’ rainnet F,., E s e I a m a d о п Compt. rend., 254, 1814 (1962).
3. В irkoler L., Bierwirth E., Ritter A., Chem. Ber., 94, 821 (1961).
ТРИЭТИЛ-N-TPH КАРБОКСИ ЛАТ, XfCO.CjH,);!. Мол. вес 233,22,
т. кип. 146—147 /12 т.н.
Т. получают |1| кипячением эфирного раствора уретана над ку-
сочкам it натриевой проволоки до превращения болы пей части металла
в студенистый белый осадок натрийуретана. После этого продолжают
кипячение при перемешивании. Затем при охлаждении добавляют
этилхлорформпат и смесь перемешивают в течение ночи (реакция
1). Смесь фильтруют и продукт выделяют перегонкой. Этот триэфир
1) НАС0.,С.,Н- i-2ClCO.,C,Пз-i 2Na Х(СОД:Щ[Щ( ; 2NaClR-H.J
2) 2N(C0.,C.,H-):i-i-5IUNNH.,-l H.,0—-
—- HMCOXHNH.,), ЗНАЩНСоДН-СО./ ЗС.Н-ОН NIL;
реагирует с гидразином (реакция 2), образуя два продукта |21.
462
Первый, диаминобпурет, получается в виде нерастворимого в спирте
кристаллического вещества |т. пл. 205' (с разл.)] с 69—75%-ным
выходом. Второй, этплгидразннкарбокенлат, выделяют из фильтрата
перегонкой и кристаллизацией; т. пл. 52', выход 90—95%.
[. Diels О., Вег., 36, 740 (1903); Аллеи Ч., Б с л л Д., «Синтезы органиче-
ских препаратов», И.Ц, А)., 1952, сб. 3, стр. 515.
2. Л л л е н Ч.. Б е л л Д., «Сц1гге:лл органических препаратов», И/I, Л)., 1952,
сб. 3. стр. 510.
ТРИЭТИЛ ФОСФАТ, (С2НЛО);;Р-зО. Мол. вес 182,16, т. кип.
215—216', уд. вес 1,068; в 100 г воды при 25 растворяется 100 г.
Этот эфир является превосходным растворителем для реакций!
слабоосновных аминов с алкпл- пли арилбромида.ми или иодидами
111. Например, смесь эквивалентных количеств 2-аминофлуоренона,
9-бромфлуорена и бикарбоната натрия кипятят в Т. в течение 1 час,
охлаждают и разбавляют водой для осаждения продукта, который
кристаллизуется из смеси хлороформ — метанол в виде блестящих
красиовато-фиолстовых игл.
I. Pan Ii.-U, Fletcher Т. L., .1. Огщ Chem., 27, 3639 (1962),
ТРИЭТИЛФОСФИТ, (СаН;,О)зР. хМол. вес 166,16, т. кип. 155—
137 , (43—44°/10 мм), не растворяется в воде.
Получение [И:
РС1а + 3C;HftOH + 3CSI UN (С,, Нг,)2 - (С2П-,0);)Р ЗС;;Н51Х(С,Н6), -HCI
Т. очищают перетопкой над металлическим натрием 121.
463
Получение фосфонатов для модифицированной реакции Виттига
[3]. В типичном примере синтеза 141 хлористый бензил добавляют
к Т., образующийся бенз илтриэтоксифосфоний хлор ид при кипяче-
нии легко отщепляет хлористый этил и превращается в диэтилбен-
зилфосфопат. После охлаждения его добавляю!' к суспензии метилата
с,;[i-.cii,ci ; (CjisO);{p > едценд i (Осн5).,(сщ)
час
5 л<л
7,7 мл
о
2,4 г СЛЬОКа
5 4i.j С6Н,СН=СНС[1О
С6НГ|СН2Р(ОС2Н5)2
н
н
10 .IU псомспл
н
н
натрия в ДМФА и к реакционной смеси при охлаждении льдом и
перемешивании прибавляют коричный альдегид. Кристаллизация
продукта дает транс, транс-дифен и лбутадиен с выходом 64% (в рас-
чете на хлористый бензил).
Бициклические фосфиты. При нагревании триметилолэтана с Т.
в присутствии следов триэтиламина образуется кристаллический
фосфит, т. пл. 94:) [51.
CH3C(CH2OH)S + Р(ОС2Н5)3
100°
90%
Р
О^ О
I I I + з с,н8он
сн3
Алкилирование. Примером реакции является получение 1,2,3,4,5-
пснтахлор-5-этплциклопентадиена этилированием гексахлорцикло-
пентадиена (Марк [6]). К охлажденному льдом с солью раствору Т.
1 моль
100тл петрал.эфира
+ (С2Н5О)3Р -------->
1Д моля 92-96%
О"
+ (С2Н5О)2Р+С1
в пстролейпОхМ эфире при перемешивании добавляют в течение
4—6 час раствор гексахлорциклопентадиена в том же растворителе.
Продукт алкилирования можно отделить от диэтил хлорфосфата
гидролизом последнего или перегонкой.
464
Дехлорирование. Дехлорированием декахлор-б«с-(циклопентади-
енила) (1) иод действием Т. Марк [7| получил перхлорфульвален
(2) в виде темно-фиолетовых кристаллов.
а
а
P(OEt),
- EtCI,
(Et.OhPOCl
ci ci
CD (2) (3)
Восстановление гидроперекисей. T. энергично, но гладко вос-
станавливает гидроперекиси до соответствующих спиртов [8].
СИ, СН3
С6Н5С- О._,Н л- (С,Н5О)3Р С0Н5С-ОН I- (С,.Н5О)3РО
| 0,323 моля 25~30 I
сн3 сн,
0,323 моля
Отщепление кислорода. Реагент отщепляет кислород от диаро-
илперекисей с образованием ангидрида ароматической кислоты
и триэтилфосфата [9].
О
I! zO
ЛгС—О ЛгС^
[ -(СгН6О)-,Р— /О +(С,Ц.,О)ЛРО
ЛгС - О ЛгС/
li
о
Прп кипячении фталевого ангидрида с избытком Т. в качестве
растворителя почти в чистом виде выделяется дифтал иле н (Рамирец
ПО]). Предполагают, что первоначальным продуктом может быть
карбен. Три.метилфосфит менее эффективен из-за более низкой тем-
пературы кипения.
(С2Н5О)3Р
Перенос кислорода с окисей этилена и пропилена на триэтплфос-
фит происходит при 150—175t: [111, но реакция не является обшей.
СН3СН—СН2-|(С2Н5О)3Р
150—175°
------— СН,СН - СНп + (С2Н5О),РО
-165
Так, окись бутена-2 при 200° дает лишь незначительные количества
олефинов [ 12],
Образование изонитрплов из алкилизоцианатов [131—другой
пример отщепления кислорода под действием этого реагента (с
феннлизоцианатом реакция не осуществляется). Дифснилкетен при
RN — С = О-| (QH5O);;P —> RN ^С-Р (С21[&О)ДО
нагревании с Т. при 215'! в атмосфере азота превращается в дифенил-
ацетилен [13]. Реакция включает отщепление кислорода и миграцию
фенильной группы.
2 I 5,J
(СГ1Н5).,С-С-0(С.ЛГ>О);1Р СЯН5С CCflH5 + (С2Н5О)3РО
Десульфуризация. Десульфуризация эписульфидов (тинравов)
с образовапие.м олефинов осуществляется, гораздо легче, чем отщеп-
ление кислорода от эпоксидов [141. Реакция эпнеульфидов цис-
и транс-бутена-3 с реагентом приводит к удалению серы и образо-
ванию соответственно цис- и щранс-бутена-2 со стереоспецифич-
ностью свыше 97 % [121.
V - С• Z i (C2HdO).jP - ' ) С С < (GHsO),PS
' s 7
Гофман и corp. 1151 показали, что Т. при нагревании с алифати-
ческим спиртом подвергается некаталпзируемой переэтерификации,
включающей три отдельные стадии с приблизительно равными ско-
ростями, С другой стороны, при десульфуризации меркаптанов
образуется углеводород и триэтнлтноиофосфат с высокими выхода-
ми [161, Уоллинг и Рабинович [17! установили, что реакция осущест-
вляется по радикальному цепному механизму. Родственная реакция,
fiv 6,25 час
w-CbH1TSH-,-{C>H-OhP------------h-C8H1s HCJlyOLPS
6,6 моля 6,о моля нисколько luicoii ,,
протекающая при медленной перегонке смеси диэтплдисульфида
и трнэтилфосфита, дает диэт пл сульфид и трпэтплмопотпофосфат
[18|. В другом варианте дисульфид реагирует с Т. в присутствии
О
г
QH^SQHj-l-fCJbObP^ CJI5SC2H5+(CJ-ldO).2PSC2H5
окиси углерода, образуя тиоэфир по радикальной реакции [19].
О
«-CJ^SSCJlp-H 4-СО-р (С2Н5О)3Р ’ н-С4НэС—SC4H9-h t (CH&O)3PS
J J %
Щ6
Восстановление азотистых соединений, Нод действием Г. пнт-
розобепзол превращается в азоксибеизол, вероятно, через промежу-
точное образование азепа 1201. С избытком реагента азоксибензол
восстанавливается до азобензола П3|.
О-
(С,Н-.О),Р ЩН-.К'О | Д.
CGI ] - NO --— [ [ ЦХ ] — ~<йНдХ - N CJ-1-
о-
ckh,x -n4c6h5-; (с,нг,О):,р — c(;h5x--n6i;h5 : «ан-сщро
Реакция 2-нитродифенпла с Т. при [00° в атмосфере азота дает
карбазол и включает первоначальное восстановление нитросоеди-
неиия 121]. 2-Нитрозодифен вл под действием Т. аналогичным обра-
зом превращается в карбазол [221.
Под действием Т. осуществляется также восстановительная
циклизация аипла (1) в имидазол (2) |231.
^\^-CH = NCbH4©CH3- г;
(СДЦОДР*
8 3,5%
Родственная реакция — восстановление р-ал к ил-о-нитростирола
(3) до алкилпндолй (4) 1241.
с:н
|| ^CHR
(31
(C2H5O)3P>
Аналогично 2-нитрофспплфеиилсульфид (1) претерпевает вос-
становительную циклизацию до фенотпазина (2), а 2"-нитрохалкон
(3) дает З-стпрплантрапил (4) [25].
467
Груидманн [26] использовал Т. для восстановления фуроксанов
(5) до фуразанов (6). Трифепилфосфит также можно использовать
в этой реакции восстановления' но для препаративных целей лучше
Т., так как образующийся триэтилфосфат растворим в воде и легко
отделяется.
снзО --1"| сн3
Г\ /N
О
(CZH5O)3P^
64% *
+ (С2Н5О)зРО
(6)
Аддукты с а-дикетонами, Т. образует с а-дикетонами аддукты
1 : 1 [27]. При нагревании аддукта бензила и Т. при 215—225° в ат-
мосфере азота с избытком реагента образуются дифенил ацетилен
и триэтилфосфат [28].
с6н&с-а:вн5+(С2н5О)яр - +сйн6с - сс6н5 - -
i; 'I II
о о о о
ч;р/
(СгН;О),Р
cGH5c^cceH5 -у 2(С?нГ)О)3РО
1. Schuetz R. D., Jacobs R.L., J. Org. Chern., 26, 3467 (1961).
2, Ф о p д-М у p А,, Псрр и Б.т «Синтезы органических препаратон» ИЛ, М.,
1953, сб. 4, стр, 492.
3. Wadsworth W. S., Jr., Е m in ons W. D., J. .Am. Chern. Soc. 83, 1733
(1961); Sens E. J., Wilson С. V., J. Org.-Chem.', 26, 5243 (1961),
4. Org, Expts., 1 [9.
5. Wadsworth W. S., Jr., E m in ons W. D., J. Am. Chem. Soc. 84,
610 (1962).
6. Mark V., Wann R. E., G о d t H. C., Jr., Org. Syn., 43, 90 (1963).
7, Mark V., Tetrahedron Letters; 333 (1961).
468
8. Kharasch M.S., Mosher R. A., Bengelsdorf I, S., J. Org.
Chem., 25, 1000 (I960).
9. В u r n A. J., C a d о g a n J, I. G., Bunyan P. J., J. Chem. Soc., 1963,
1527.
10. Ramirez F., Yamanaka H., Basedow О. II., J. Am. Chem.
Soc., 83, 173 (1961).
11. S с о t t С. B., J. Org. Chem., 22, 1118 (1957).
12. X eu r e i t e г X'. P_, В о r d w e 1 1 F. G., J. Am. Chem. Soc., 81, 578 (1959).
13. M ukaiyama T., N a rn b u FI., Okamoto M., J. Org. Chem., 27,
3651 (1962).
14. Davis R. E., J. Org. Chem., 23, 1767 (1958); Schuetz R. D., J acobs
R. L., ibid., 23, 1799 (1958); see also ref [ 1].
15. Hoffmann F. \V., Ess R. J., Usinger R. P., Jr,, J. Am. Chem.
Soc., 78, 5817 (1956).
16. Hoffmann F. W-, Ess R.J., Simmons T. C., Hanzel R. S.,
J. Am. Chem. Soc., 78, 6414 (1956).
17. Walling C., Rabinowitz R., ,J. Am. Chem. Soc., 81, 1243 (1959).
18. Jacobson II. I., Harvey R.G., Jensen E.V., J. Am. Chem.
Soc., 77, 6064 (1955).
19. W a 1 I i n g C. Basedow О. H., S a v a s E. S., J. Am. Chem. Soc.,
82, 2181 (I960).
20. Bunyan P.J., Cadogan J. I. G., J. Chem, Soc., 1963, 42.
21. Cadogan J.I.G., Camero n-\V о о d M., Proc. Chem. Soc., 1962,
361.
22. Bunyan P. J., Cadogan J. I. G., Proc. Chem. Soc., 1962, 78; C a d o-
g a n J. 1. G., Cameron-Wood M., M а с к i e R. K-, Searle R. J .
G., J. Chem. Soc., 1965, 4831.
23. Cadogan J.I.G., Searle R.J. G., Chem. Ind., 1963, 1282,
24. Sundberg R.I.,J. Org. Chem., 30, 3604 (1965).
25. Cadogan J, I. G., Mackie R. K-, Todd M. J., Chem. Comm..
1966, 49’1.
26. Grundmann C., Chem. Ber., 97, 575 (1964).
27. Ramirez F., Desai N. B., J. Am. Chem. Soc., 85, 3056 (1963).
28. Makaiya rn a T., N a in b u H., Kumamoto T., J. Org. Chem., 29,
2243 (1964),
ТРИЭТИЛФОСФОЕНОЛПИРУВАТ. Мол, вес 252,21, т. кип.
95—10070,1 мм. Т. получают взаимодействием этилового эфира
бромпировиноградной кислоты и триэтилфосфата, В присутствии
сильных оснований Т. реагирует с соединениями, содержащими
активные метиленовые группы, с образованием циклопропаяовых
производных [11.
снг=ссо2сгн5
о
о=р(осгн5)г
(1)
ДМСО-NaH
80%
СОгСгН4
1. Schmidt U., Angew. Chem., Internal. Ed,, 4, 238 (1965).
ТРИЭТОКСИАЛЮМОГИДРИДНАТРИЙ, NaAlH(OGH5)3. Мол.
вес 37,95.
469
Т. получаю! взаимодействием этилата алюминия с гидридом
натрия; продукт --- бесцветный, мелкокристаллический порошок,
в отсутствие воздуха устойчив в течение нескольких недоль; приме-
няется в эфире или ТГФ [ 1 L Реагент - - более мягкий восстановитель
по сравнении» с алюмогидридом лития, используется, в частности,
для восстановления ароматических нитрилов в альдегиды:
CJI-CX^QPCIIO (95%)
1. Hesse G., S с li г о d е ] R., Ann., 607. 24 (1957).
ТРОПИЛ ИЯ БОРФТОР ИД, С7Н-!ВР/. Мол. вес 177,95. разла-
гается приблизительно при 200 . О получении см. Циклогептатрпеп.
Использование реагента для получения замещенных тропплиде-
нов описал Ковров 111 (см. литературу, цитированную в его статьях).
I.Conr о w К., J. Am. Chem. Soc.. 81, 5641 (1959); 83, 2313 (1961); С о п г о w
К., X a i k D. X., J. Med, Chem., 6, 69 (1963).
содержание
о
Озон.......................................................... 5
Окиси мезитила этиленксталь ............................... Ю
1-Окись 1-феи и л-3-мети л фосфол сна-3 . . ............ . . . И
Окись этилена............ . ............................. 11
1-Окись 1-этил-З-метилфосфолеиа-З...................... , • 12
Оксалилбромид . ........................................... 12
Оксалилхлорнд.........,...................................... 13
о-бнс-(Окснметил)-бсизол................................... 19
N-Оксиметилфталимид.......................... ......... 19
2-Окси-1,4-пафтохцнон........................................ 20
2-Окси-5-нитробеизилбромид .................................. 20
N-Оксипнперндин......................................- - 21
З-Оксипиридин . ,.......................................... 21
Х-Оксиеукцинимпд.............,....................., ... , 2'2
N-Оксифтал имид . . , ..................... 22
Октанол-2................................................... 23
Олеиновая кислота.......................................... 23
Олово . . .................................................. 24
Олово бромистое......................................... . . . 26
Олово хлористое.............................................. 26
Олово хлористое безводное................................... 31
Олово хлорное................................................ 31
Олово хлорное, пентагидрат................................... 33
Ортомуравьиной кислоты триэтнловый эфир...................... 33
Ортоугольной кислоты тетраэтнловый эфир...................... 40
Осмия четырехокись (осмиевый ангидрид) ...................... 40
Осмия четырехокись — бария хлорат............................ 46
Осмия четырехокись — перекись водорода....................... 47
Осушители.................................................... 47
П
Палладиевые катализаторы..................................... 48
Палладий хлористый .......................................... 53
Палладия(II) ацетат.......................................... 53
Пептадиен-1,4.............................................. 54
Пента хлорфснол.............................................. 56
471
Перекись водорода ............................................ 56
Перекись водорода в кислой среде ............................. 57
Перекись водорода в нейтральной среде ........................ 68
Перекись водорода в щелочной среде............................ 70
Перекись водорода — селена двуокись........................... 77
Перекись водорода — соли и окислы металлов в качестве катализа-
торов ........................................................ 78
Перйодаты................................................... 84
Перхлорилфторид............................................... 92
Пивалиновая кислота .......................................... 99
Пикрилазид.................................................... 99
Пнкрилхлорид ................................................ 100
Пикриновая кислота ......................................... 101
Пиперидин.................................................... 102
Пипсрилен.................................................... 107
Пиразол...................................................... 107
Пиридин...................................................... 107
Пиридина N-окись............................................. 114
Пиридина пербромид .......................................... 115
Пиридина хлоргидрат ......................................... 115
Пиридинборан ................................................ 118
Пиридипия бромид-пербромид................................... 119
Пировиноградная кислота...................................... 123
Пирокатехина дихлормстилеиовый эфир ......................... 124
а-Пирон...................................................... 125
Пирофосфорилтетрахлорид (ПФТХ)............................... 125
Пирролидин................................................... 126
Платиновые катализаторы...................................... 129
Платины сульфид иа угле...................................... 132
Полифосфорная кислота (ПФК).................................. 132
Полифосфор ной кислоты эфиры (ПФЭ)........................... 146
Прево реактив................................................ 148
Пропандитиол-1,3............................................ 148
Пропандитиола-1,3 ди-л-толуол сульфон ат.................... 149
Р-Пропиолактон............................................... 149
Пропионовой кислоты этиловый эфир............................ 150
Пфицпера — Моффатта реагент.................................. 151
Р
Разделение на оптические изомеры, реагенты ................. 152
Растворители................................................ 153
Репнекс соль................................................ 156
Ренея катализаторы кобальтовые............................. 156
Ренея катализаторы никелевые............................... 156
Ренея никель............................................... 167
Рениевые катализаторы....................................... 167
Рения гептаселенид......................................... 167
472
Родий (5%) на окиси алюминия.................................... 168
Родий (5%) на угле (норит)...................................... 172
Ртути ацетат.................................................... 173
Ртути окись (желтая, красная) .................................. 184
Ртути окись иод................................................. 187
Ртути (11) сульфат.............................................. 188
Ртути трифторацетат............................................. 190
Ртути (П) цианид................................................ 190
Ртуть хлорная (сулема).......................................... 191
Рубидия фторид.................................................. 194
Рутениевые катализаторы гидрирования............................ 194
Рутения четырехокись ........................................... 197
С
Свинца двуокись................................................ 202
Свинца диацетата тригндраг...................................... 206
Свинца окись (желтая)........................................... 207
Свинца тетр а ацетат............................................ 208
Свинца тетрабензоат............................................. 242
Свинца тетрафтор ид............................................. 242
Селен........................................................... 243
Селена двуокись, селенистая кислота ............................ 246
Селена хлорокись ............................................... 257
Семикарбазида хлоргидрат ....................................... 257
Сера............................................................ 257
Сера двухлористая.............................................. 261
Сера однохлористая.............................................. 261
Сера четырехфторие тая.......................................... 262
Серебра борфторид............................................... 264
Серебра дифторид................................................ 266
Серебра карбонат ............................................... 267
Серебра нитрат ................................................. 267
Серебра нитрит.................................................. 271
Серебра окись .................................................. 272
Серебра сульфат................................................. 276
Серебра хлорат.................................................. 277
Серебра хромат.................................................. 277
Серебра цианид.................................................. 278
Сернистой кислоты N,N-диэтил амида метиловый эфир............... 278
Сернистый ангидрид ............................................. 278
Серный ангидрид................................................. 279
Серный ангидрид — диметилформамггд.............................. 279
Сериый ангидрид — диокса)!...................................... 281
Серный ангидрид — пиридин....................................... 282
Серный ангидрид — триметпламин.................................. 283
Сероуглерод..................................................... 283
Серы jrrecTiioKHCb.............................................. 284
473
Сефадекс....................................................... 284
CiLMMOiica — Смита реагент..................................... 286
Стеариновой кислоты ангидрид .................................. 290
Сукциноила перекись........................................... ‘290
Сульфаминовая кислота.......................................... 290
Сульфолан...................................................... 290
Сульфисалициловая кислота ..................................... 290
Сульфоуксусиая кислота ........................................ 291
Сульфурил хлористый............................................ 292
Сурьма пятихлористая........................................... 296
Таллия триацетат............................................... 297
Тстра-н-бупиламмоннйиодтстрахлорид............................. 298
ТетрагпДрофураи (ТГФ).......................................... 299
Тетр азол певый синий.......................................... 301
Тетралип....................................................... 302
Тетраметиламмония ацетат....................................... 302
Тетраметиламмония боргидрпд.................................... 303
Тетраметиламмония бромид....................................... 303
Тетраметиламмония трибромнд.................................... 304
Тетраметилгуанидин ............................................ 304
Тетр а метил ей сульфоксид..................................... 304
Тстраметилепсульфоп (сульфолап)................................ 305
Тетраметилмочевипа............................................. 306
Тетраметилтиомочсвнна.......................................... 307
Тетр а нитрометан . . . . ..................................... 307
Тетра-я-питрофенпл пирофосфат.................................. 309
(-)-)- и (—)-а-(2,4,5,7-Тетранитро-9-флуорепплиденаминоокси)-про-
ппоионая кислота ............................................ 309
2,4.5,7-Тетрапнтрофлуореиои.................................... 310
Тетрафснилцпклолентадиенон..................................... 310
Тетр афтор гидр азин........................................... 312
Г\т,2,4,6-Тетрахлорацетапилид.................................. 312
Тетрахлор-1,2-дибромэтап....................................... 312
Тетрахлорфуран................................................. 313
Тетрахлорциклопентадиспон...................................... 313
Тетради а их и в оди метан..................................... 314
Тетради анэтилсв............................................... 314
Тетрациклин.................................................... 318
Тетраэтил аммония ацетат....................................... 318
Тетраэтил аммония гидроокись................................... 319
Тетраэтил аммония формиат...................................... 319
Тетраэтил аммония хлорид....................................... 320
Тетраэтил пирофосфит........................................... 320
Тетраэтплсукципилфосфонат...................................... 321
Тиглииовой кислоты хлораигидрпд ............................... 321
474
Тиоглпколевая кислота ........................................ 322
\,1\Г'-Тиокарбонилдиимндазол.................................. 323
Тиомочевина................................................... 324
Тионил бромистый.............................................. 328
N, N' -Тионил диимид азат..................................... 328
Тиопил хлористый ............................................. 329
Тиосемикарбазид............................................... 335
Тяоуксусная кислота .......................................... 336
Тиоуксусной кислоты калиевая соль ............................ 340
Тпофеиолят натрия............................................. 340
Тиоцианат натрия ............................................. 341
Титан четыреххлористый........................................ 342
N-Тозилимипоуксусной кислоты н-бутиловый эфир................. 344
Тозил хлористый.............................................. 345
Толленса реактив............................................. 345
Толуолдиизоцианат ........................................... 345
бис- (п- Толуол сульфамид) -ртуть............................ 346
п-Толуолсульфокислота........................................ 347
Толуолсульфокислоты rt-нитробензиловый эфир.................. 354
«-Толуолсульфомстилнитрозоамид (диазальд).................... 355
н-Толуолсульфонат серебра.................................... 355
/1-Толуолсульфонилазид....................................... 355
п-Толуолсульфонилгидразид.................................... 357
н-Толуалсульфонилперхлорат................................... 360
2-п-Толуолсульфопилэтилхлорформпат........................... 360
n-Т ол у ол сульфо хлор ид (тозил хлористый)................. 361
п-Толуолсульфохлорид — димстилформамид....................... 367
Тория двуокись............................................... 367
Треххлористый азот........................................... 367
тетра/а/г-(Треххлористый фосфор)-никель(О).................... 368
1,2,4-Триазол................................................ 369
со-Триб ром ацетофенон....................................... 370
Три-н-бутиламни.............................................. 370
Три-н-бутилборан и трнизобутплборап.......................... 370
Трн-н-бутилбороксин.......................................... 371
Трн-н-бутилстаннаи........................................... 372
Три-н-бутнлфосфнн............................................ 374
Три-н-бутнлфосфина окись..................................... 374
Три-я-гексилэтиламмония гидроокись........................... 375
Триглим...................................................... 375
Трнизобутил алюминий......................................... 375
Трнизопинокамфилборан........................................ 376
2,4,6-Триизопропилбепзолсульфохлорид......................... 377
Триизопропилфосфпт........................................... 377
Трнмстиламина окись.......................................... 377
Триметиламинборан............................................. 378
О-(2,4,6-Триметил бензоил)-гидроксилам и и................... 379
475
Триметилборат ................................................ 380
Триметилбороксии ............................................. 381
Триметилена окись............................................. 381
Триметилоксония борфторид .................................... 382
Триметилоксония 2,4,6-тр и нитробензол сульфонат.............. 382
Триметилсилан ................................................ 382
Триметилсилилазид ............................................ 383
бис (ОД’-Триметплсилил)-ацетампд.............................. 383
М-Три.метилсилилацетамид...................................... 384
бис-(Трпметилсилил)-натрийамид................................ 385
Трнмстилсульфоксония нодид ................................... 386
Три метил сульфония и од ид................................... 386
Т р им стп л-(трифтор мети л)-ста и н а и..................... 386
Триметилуксусная кислота (пивалпновая кислота)................ 387
Триметилуксусной кислоты хлорангпдрид (пивалоилхлорид) . . . 387
Триметилфениламмония трнбромид................................ 388
Триметилфосфит................................................ 388
Триметилхлорсилан............................................. 390
Триметоксибор гидр и дн атрий................................. 391
1,3,5-Трипитробензол.......................................... 391
Тр н-п-нцтрофенил тиофосфиТ................................... 391
2.4,7-Трниитрофлуоренои....................................... 392
Тритон Б (гидроокись1 бепзнлтриметилахммонпя)................. 392
Трнфенил бромметан............................................ 394
Трифенилдибромфосфоран........................................ 395
Трифепнлметансульфепхлорид.................................... 395
Трифенил метил кал ий........................................ 395
Трифенилметиллитий............................................ 396
Трифенилметилнатрий........................................... 397
Трифенилметилперхлорат и Трифсвилм стал бор фтор ид........... 399
Трифснилстаинан............................................. 401
Трифенилфосфин................................................ 402
ТрифепилфосфиндибрОхМпд и трифенилфосфиндпхлорид.............. 413
бно(ТрифС11илфосфин)-ннкельдикарбонил......................... 415
мр«с-(Трифен11лфосфи1г)-родий хлорид.......................... 415
Трифепилфосфин — углерод четыреххлористый..................... 415
Трифенилфосфит................................................ 416
Трифенилфосфита дибромид...................................... 416
Трнфенилфосфита иодметилат (метилтрифеноксифосфоний иодистый) 416
Трифснплхлорметан............................................ 417
Трифенилхлорстаппан........................................... 419
0,1\г-бис-(Трифтора11етил)-п1дроксилами11.................... 420'
Трифторацетилгипогалогениты.................................. 421
N-Трифторацетилимидазол....................................... 421
а,а,а-Трифторацетофенон....................................... 422
Трифториодметан............................................... 422
Трифторпадуксусиая кислота ................................... 422
476
Трифтор тиобензол............................................ 431
Трифтору ксуспая кислота..................................... 432
Трифгоруксусной кислоты п - ни тр офенилов ый эфир........... 434
Трифторуксусной кислоты серебряная соль..................... 434
Трифггор уксусной кислоты фениловый эфир.................... 435
Трифторуксусный ангидрид..................................... 436
Трихлорацстонитрил........................................... 444
Трихлорметансульфобромид..................................... 444
Трихлорметансульфохлорид..................................... 445
Трихлорметиллитий............................................ 445
Трихлоруксусная кислота...................................... 446
Трихлоруксусной кислоты натриевая соль....................... 446
Трихлоруксусной кислоты этиловый эфир........................ 448
2,4,5-Трихлорфепол........................................... 449
Триэтаноламин................................................ 449
Триэтил алюминий............................................. 450
Триэтиламин.................................................. 451
Триэтилендиамип.............................................. 458
Триэтилентстра.мип........................................... 458
Триэтилоксопия борфторид..................................... 459
Триэтилсилан................................................. 461
Триэтил-М-трикарбокснлат..................................... 462
Триэтилфосфат................................................ 463
Триэтилфосфит................................................ 463
Триэтилфосфоенол пируват..................................... 469
Триэтоксиалюмогидридпатрий................................... 469
Тропилия борфторид......................................... 469