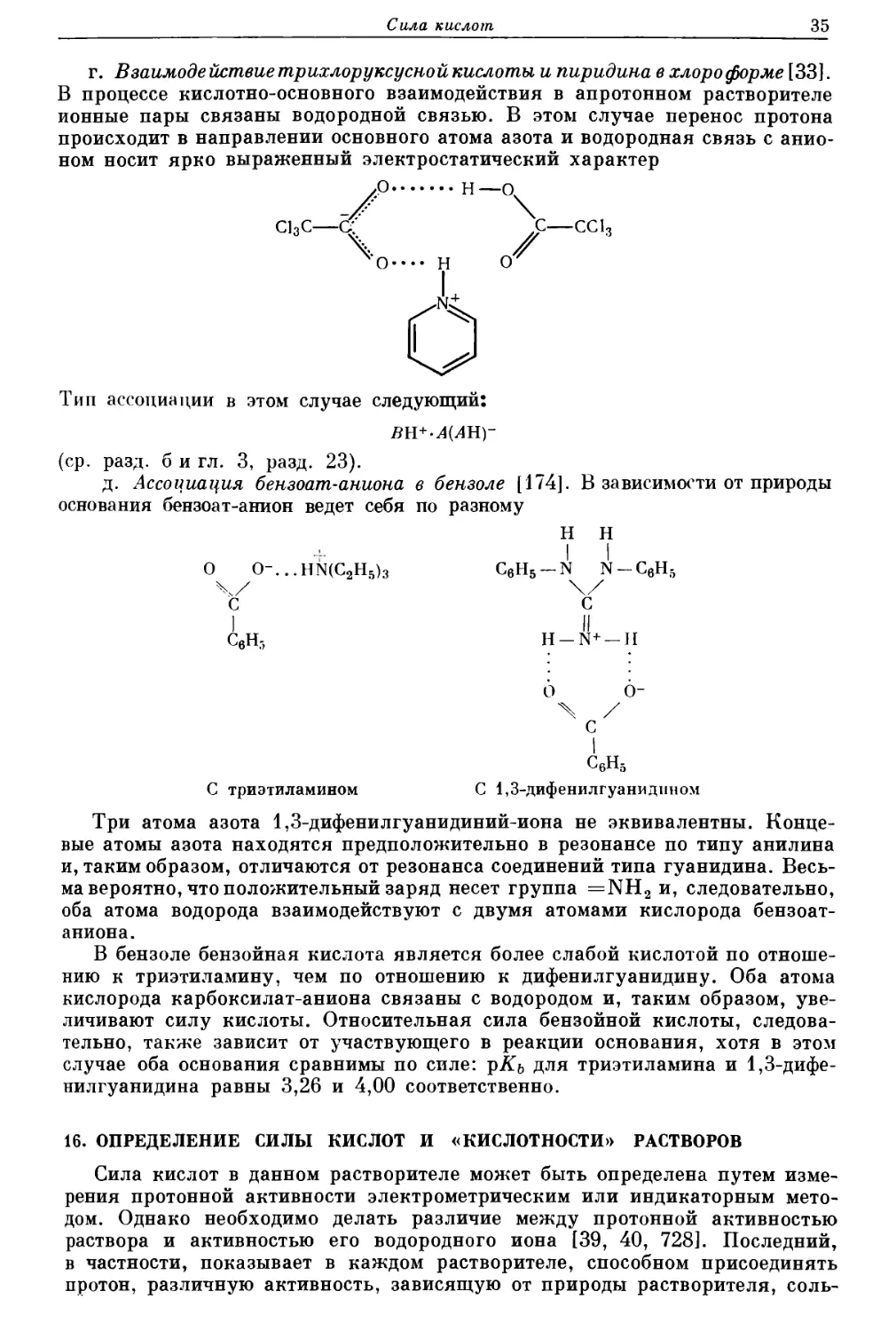
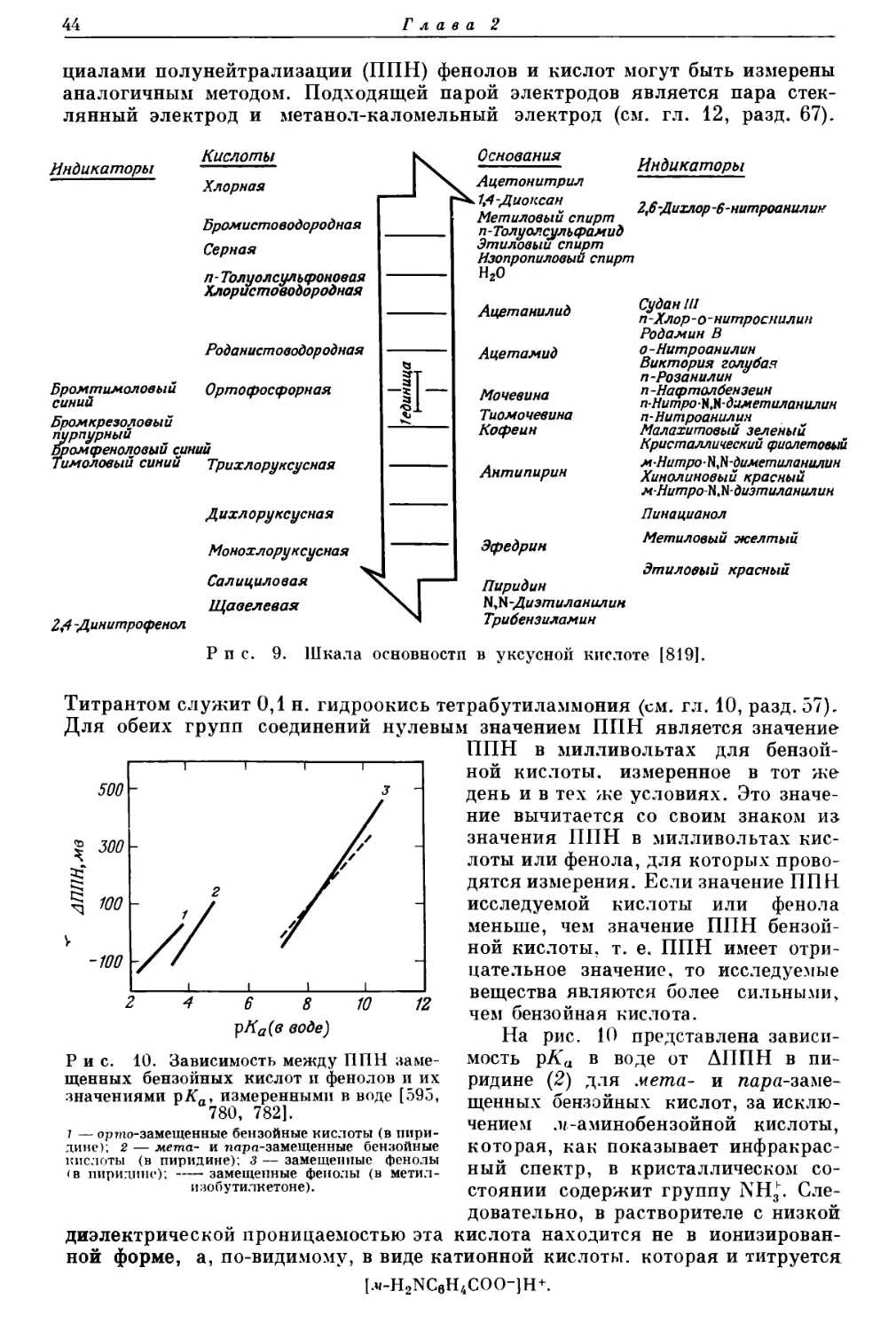
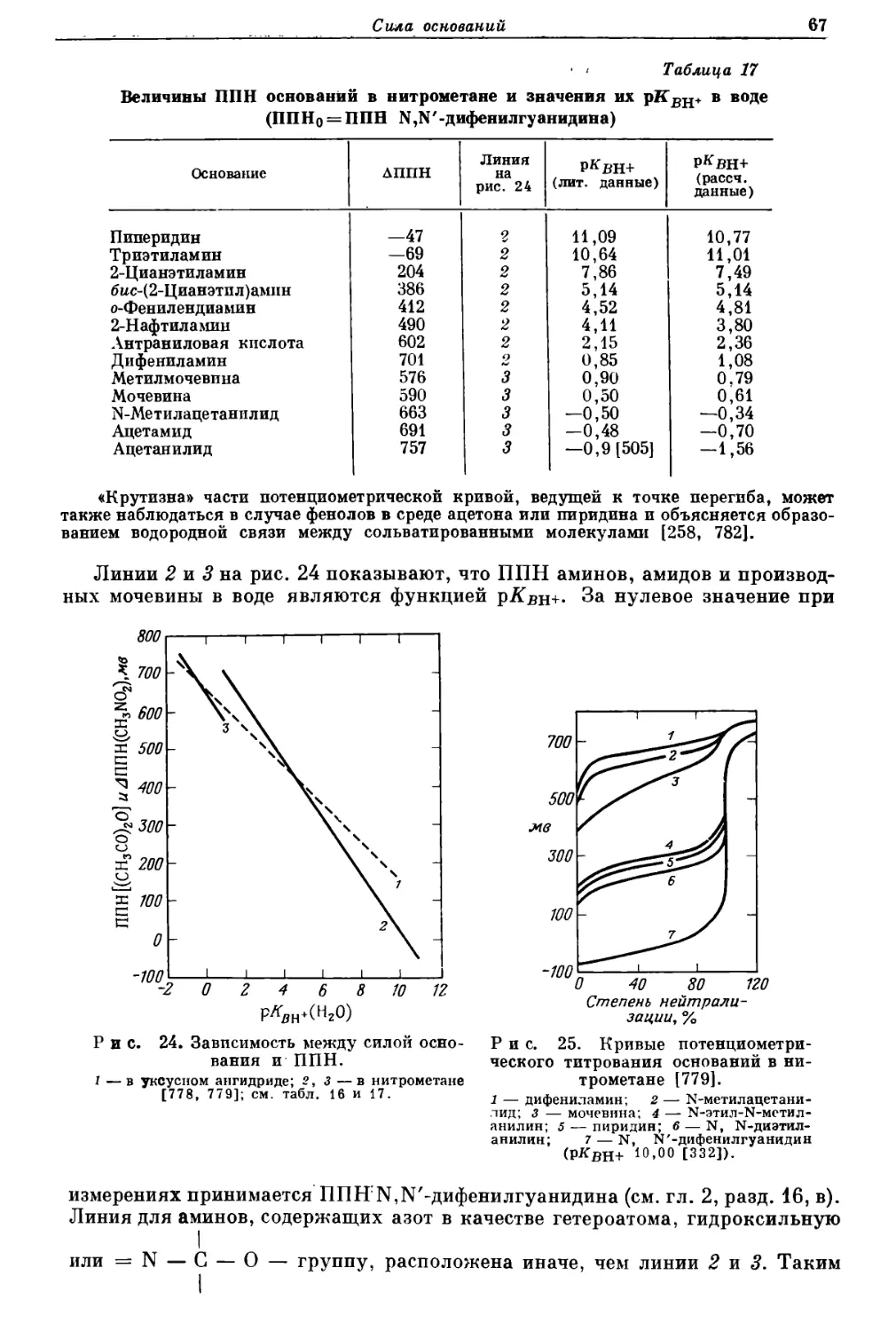
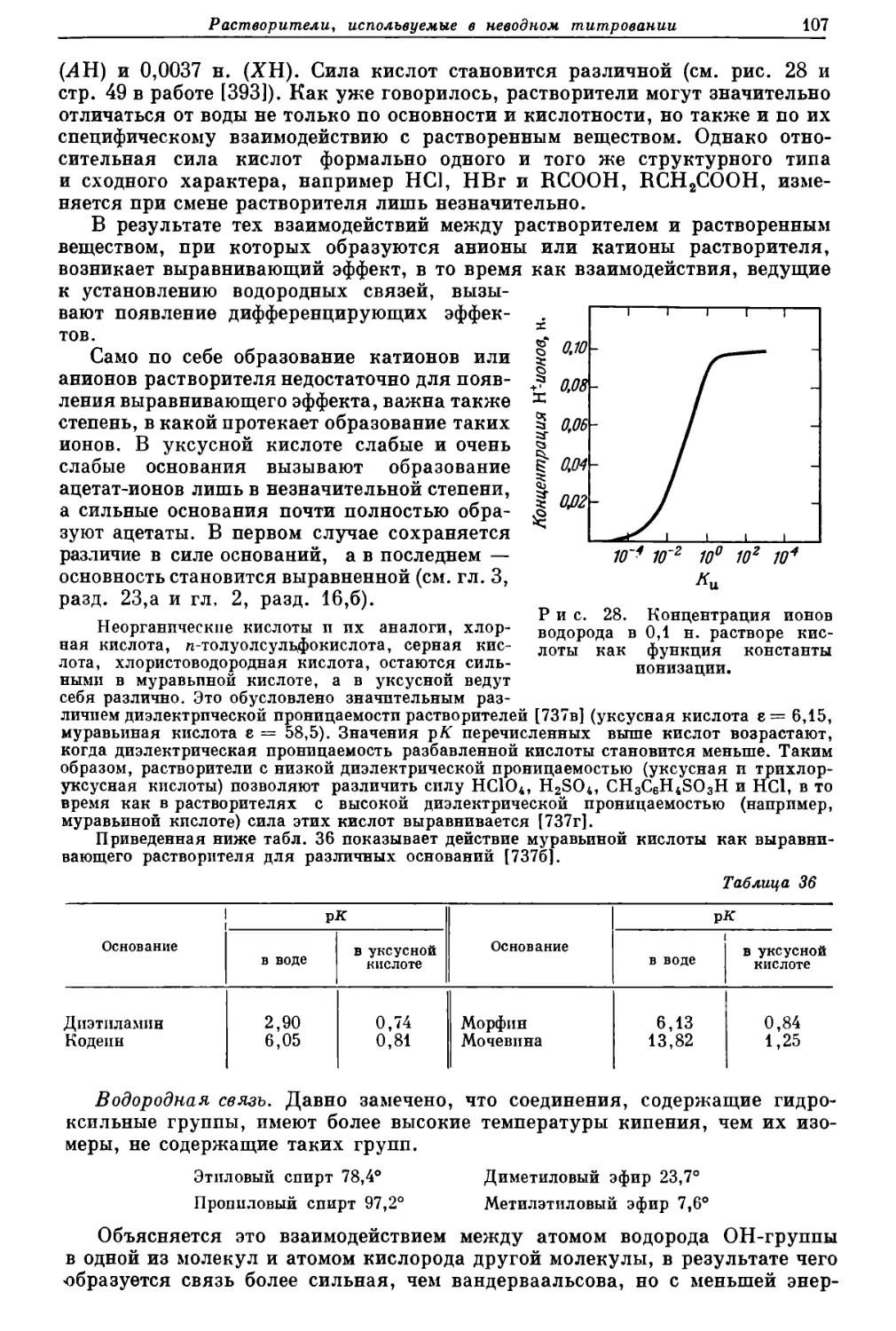
/


























































































































































































































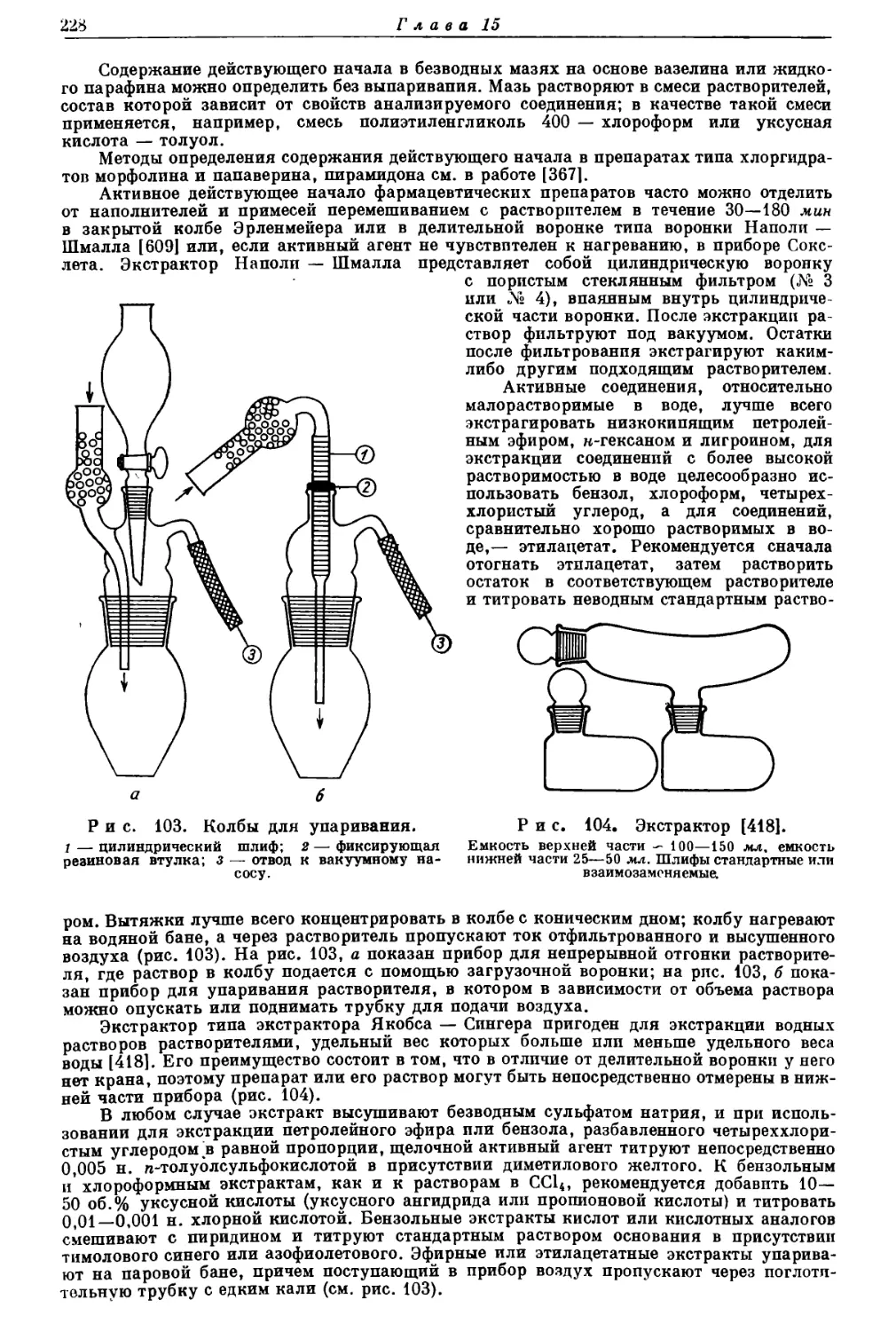


























































































































































































Текст
И.Денеш
"*п****яЯЕ№ВВвЯЯЯцЁВнВЯВЗН9ЯмфВ£п9Ямм908р*Яяб%
ТИТРОВАНИЕ
В НЕВОДНЫХ
СРЕДАХ
TITRATION IN NON-AQUEOUS MEDIA
I. GYENES C. Sc. (chim.)
Head of Department, Physico-chemical Research Laboratory Chemical
Works, Gedeon Richter Ltd., Budapest Reader in Chemistry,
University of Veszprem, Hungary
ENGLISH TRANSLATION EDITED
BY D. COHEN. M. A., Ph. D., A.R.I.C.
LECTURER IN CHEMISTRY
UNIVERSITY OF KEELE, STAFFORDSHIRE
AND I. T. MILLAR, B. Sc., Ph. D., F.R.I.C.
PROFESSOR OF ORGANIC CHEMISTRY UNIVERSITY OF KEELE,
STAFFORDSHIRE
AKADEMIAI KIADO, BUDAPEST, 1967
И. Денеш
ТИТРОВАНИЕ В НЕВОДНЫХ
СРЕДАХ
Перевод с английского
канд хим. наук И. Ф. ДОЛМАНОВОЙ
п канд. хим наук С. С ЧУРАНОВА
Под редакцией
доктора хим наук И П. БЕЛЕЦКОЙ
ИЗДАТЕЛЬСТВО «МИР» МОСКВА 1971
УДК 543
Английское издание монографии венгерского химика
И. Денеша посвящено вопросам теории и практического
применения сравнительно новой области объемного анализа —
титрования в неводных средах. Изложенные в книге вопросы
весьма актуальны, поскольку метод начинает широко приме-
няться как для исследования кинетики реакций в растворе, так
и в практике анализа; в частности, детально описаны методы
определения ряда фармацевтических препаратов, что особенно
важно для химико-фармацевтической промышленности. В кни-
ге подробно рассматривается определение важнейших классов
соединений и разнообразных функциональных групп.
Книга рассчитана на самый широкий круг химиков-ана-
литиков, органиков, фпзико-химиков, фармакологов — работ-
ников научно-исследовательских институтов, учебных заведе-
ний, заводских лабораторий.
„ 2-5-5
ЯНД* 83-70
Редакция литературы по химии
ПРЕДИСЛОВИЕ
В предлагаемой вниманию читателей книге рассматриваются вопросы
теории и практического использования сравнительно новой области объемно-
го анализа — титрования в неводных средах. Применение методов неводного
титрования оказалось очень перспективным и чрезвычайно расширило воз-
можности аналитического определения самых разнообразных веществ
и их смесей. В настоящее время эти методы уже достаточно широко исполь-
зуются как для аналитического определения большого числа соединений,
так и для исследования кинетики реакций в растворах.
Многие из рассмотренных в книге вопросов, например проблема относи-
тельной силы кислот и оснований в различных растворителях, дифференци-
рующее влияние растворителей и т. д., интересны и с теоретической и с прак-
тической точек зрения в связи с широким внедрением в химическую практику
различных растворителей и прежде всего полярных растворителей апротон-
ного типа.
Ценность данной книги состоит в том, что она содержит многочислен-
ные практические рекомендации по использованию метода неводного титро-
вания для определения органических соединений различных классов. Все
известные методы титрования: индикаторный (визуальный и фотометриче-
ский), спектрофотометрический без индикатора, потенциометрический и их
различные модификации изложены очень подробно и легко могут быть освое-
ны. В книге даны характеристики наиболее часто используемых раствори-
телей, титрантов (кислого и основного характера) и индикаторов.
Основная часть книги посвящена изложению методик неводного титрова-
ния кислот (неорганических и карбоновых) и кислотных аналогов (ангидри-
дов, спиртов, фенолов и т. д.), азотсодержащих органических оснований и их
солей, соединений, содержащих карбонильную группу, металлоорганиче-
ских соединений и т. д. Во многих случаях рассмотрены методики как полу-
микро-, так и микротитрования. В конце книги приведен большой список
оригинальной литературы.
Таким образом, книга Денеша систематизирует и обобщает огромный
материал, накопленный к настоящему времени по неводному титрованию;
это делает ее ценным справочным пособием, которое будет полезно химикам
самых разнообразных специальностей, работающим в области аналитической,
физической, органической химии и химии природных соединений, а также
работникам фармацевтической промышленности.
И. Белецкая
ПРЕДИСЛОВИЕ АВТОРА
К ВЕНГЕРСКОМУ ИЗДАНИЮ
Объемный анализ является одним из старейших и, пожалуй, наиболее
распространенных методов аналитической химии. Хорошо известно, что
определения в неводных средах можно рассматривать как разновидность
классической титриметрии. Однако этот современный метод берет свое начало
с исследований, проведенных еще в 1912 г. Фолином и Фландерсом, которые
титровали нерастворимые в воде карбоновые кислоты метилатом натрия
в хлороформе, бензоле или этиловом спирте и показали, что определение
проведено с достаточной точностью и наблюдается резкое изменение цвета
индикатора.
С другой стороны, последующие исследования в области использования
методов неводного титрования заставляли вновь и вновь возвращаться к рас-
смотрению полученных ранее теоретических выводов. Цель этой небольшой
книги — обратить внимание на широкие возможности, которые открывает
перед исследователями и химиками-аналитиками неводное титрование. В то
время как свойства единственного растворителя — воды ограничивают
область применения титрования лишь водными растворами, тот набор раство-
рителей, который может быть использован при неводном титровании,— бен-
зол, ацетон, уксусная кислота, пиридин и т. д.— делает доступным многое
из того, что раньше было практически невозможным.
Я не стремлюсь к исчерпывающей полноте ни в изложении теории, ни
в цитируемой литературе, но надеюсь, что и на основе тщательного отбора
данных из все растущего объема литературы по неводному титрованию может
быть получена полезная информация. Надеюсь также, что книга поможет
химикам, работающим в заводских лабораториях и занимающимся рядовыми
анализами.
И. Денеш
ПРЕДИСЛОВИЕ АВТОРА
К АНГЛИЙСКОМУ ИЗДАНИЮ
Шесть лет, прошедшие со времени выхода в свет первого венгерского
издания, вновь показали, что определения в неводных средах оказывают
в настоящее время существенную помощь в повседневной работе исследова-
тельских и контрольных лабораторий органической химии и фармацевтиче-
ской промышленности. Английское издание существенно расширено по срав-
нению с венгерским изданием как в теоретической части, так и в описании
методик определения. Оно состоит из 31 главы вместо 20 и в соответствии
с пожеланиями и замечаниями коллег, работающих в области практического
применения описанных методов, значительно полнее иллюстрировано по
сравнению с первоначальным изданием. Я надеюсь, что все это окажет суще-
ственную практическую помощью тем, кто интересуется этой областью
химии.
При изложении теоретической части, особенно кислотно-основных тео-
рий, я придерживался первоначально принятого построения, основанного
на историческом развитии вопроса. Однако я должен подчеркнуть, что эволю-
ция кислотно-основных теорий не может рассматриваться как завершенная
и дальнейшее развитие может приподнести еще ряд неожиданностей. Это,
однако, лишь укрепит и обогатит как теорию, так и практику неводного
титрования.
И. Денеш
Будапешт, сентябрь 1966 г.
Глава 1
РАЗВИТИЕ ПРЕДСТАВЛЕНИЙ О КИСЛОТАХ
И ОСНОВАНИЯХ
Хотя понятия «кислота» и «основание» являются основополагающими
и давно известными в химии, они еще никогда не были четко определены.
Аналогично не существует единого мнения относительно определения поня-
тия «силы» кислот и оснований. Прежде чем рассматривать вопросы титро-
вания в неводных средах, необходимо хотя бы кратко остановиться на разви-
тии теоретических представлений о кислотах и основаниях [325].
Важность проблемы определения понятий «кислота» и «основание» оче-
видна давно. Еще в 1786 г. Морво писал, что дать определение кислот —
вот ключ к химии. Но по мере развития химии теоретические представления
о кислотах и основаниях неоднократно становились непригодными для
объяснения реакций, происходящих при титровании в неводных средах,
особенно когда последние теоретические исследования открыли новые пер-
спективы применения методов неводного титрования. В данной книге не ста-
вилась цель дать подробный анализ различных кислотно-основных теорий;
основное внимание в ней обращено на различия между наиболее распростра-
ненными теориями.
1. РАННИЕ ТЕОРИИ О ПРИРОДЕ КИСЛОТ
Первая теория относится к XVII столетию. Согласно этой теории, соли
образуются при взаимодействии кислот и оснований, которые сами при этом
«исчезают». Бойль (1627—1691) отмечал, что кислоты и основания обладают
противоположными химическими свойствами, экспериментальным подтверж-
дением чего служило обратимое изменение окраски фиалкового сока при дей-
ствии кислоты или основания. В то же время существовало представление
о кислотах как о частицах, обладающих остриями, позволявшими им прони-
кать в поры металлов. По существу это было материалистическое представ-
ление, примитивное выражение более поздней донорно-акцепторной теории.
Кислородная теория Лавуазье (1789) пользовалась широким признанием
до начала XIX века. Согласно этой теории неметаллические элементы при
сгорании превращались в кислоты, т. е. в окислы: СО2, SiO2. Большое зна-
чение для выяснения свойств кислот имели эксперименты Дэви, проведен-
ные в 1816 г. Он доказал, что хлор представляет собой элемент, а не соеди-
нение кислорода, и что, поскольку хлористый водород состоит из хлора
и водорода, он не может содержать кислорода. Отсюда следует, что концеп-
ция Дэви противоположна кислородной теории Лавуазье, согласно которой
кислород является единственным кислотообразующим элементом. Дэви
утверждал, что «кислотность зависит не от присутствия какого-либо особен-
ного элемента, а от особого соотношения различных элементов». Открытые
Дэви безкислородные кислоты, содержащие водород, например НС1 и HCN,
были названы Гей-Люссаком «водородными кислотами». Термин «водород-
ная кислота» (Н-кислота) и в настоящее время все еще применяется в теории
неводного титрования.
В основу «дуалистической» теории Берцелиуса положено представление
о том, что атомы связаны друг с другом электрическими силами. И хотя все
10
Глава 1
атомы несут как положительный, так и отрицательный заряды, один из этих
двух типов заряда может преобладать, в результате чего и образуются хими-
ческие соединения. Таким образом, свойства кислот не связаны со специфи-
ческой кислотонесущей частью вещества, а зависят от строения вещества,
и почти каждое вещество может обладать как кислотными, так и основными
свойствами. Согласно Берцелиусу, хотя он и не говорил об этом прямо,
амфотерность характерна для любого вещества.
Более поздняя точка зрения, согласно которой кислотный характер
соединения определяется присутствием особой частицы иона Н+, является,
строго говоря, шагом назад. Это произошло вследствие того, что стали все
больше внимания уделять процессам в водных средах [827], а не изучению
минералов, как во времена Берцелиуса.
В настоящее время к кислотам относят ангидриды (окислы), подобные
СО2 и SiO2, Н-кислоты и L-кислоты (кислоты Льюиса: А1Вг3, BF3 и т. п.).
2. ТЕОРИЯ! АРРЕНИУСА — ОСТВАЛЬДА
Согласно теории электролитической диссоциации Аррениуса — Остваль-
да (1887), химическое соединение может рассматриваться как кислота или
основание только тогда, когда при диссоциации в водной среде оно образует
•соответственно ионы водорода или гидроксил-ионы. При этом нейтрализация
представляет собой взаимодействие «кислоты» и «основания», приводящее
к образованию воды и соли. Из теории Аррениуса следует, что основным
признаком взаимодействия Н-кислоты с ОН-основанием служит образование
дополнительной молекулы воды и твердой или растворенной в воде соли.
Следовательно, при взаимодействии кислоты и основания образуется вода:
Н+ + ОН- = Н2О. Это уравнение лежит в основе понятия нейтральности
и служит для построения шкалы значений pH, определяющих кислотность
или щелочность среды.
Недостатки гипотезы Аррениуса можно проиллюстрировать на следующем
примере: раствор аммиака в ацетоне является основанием, хотя ион гидро-
ксила в растворе отсутствует. Изучение реакций в неводных средах обнару-
жило недостатки гипотезы Аррениуса и привело к разработке большого
числа новых кислотно-основных теорий.
3. СОЛЬВЕНТНАЯ ТЕОРИЯ
Уолден и сотр. [855] в Европе, Франклин [239], Краус [472] и позднее
Одриет [21, 22] в США обнаружили, что не только вода, но и другие раство-
рители способны к автопротолитическим реакциям. Формально аммиак
является в такой же мере родоначальным веществом для некоторых азот-
содержащих соединений, как вода для соединений, содержащих кислород.
При изучении влияния растворителей это, данное в самом общем виде поло-
жение может оказаться в определенных пределах полезным при решении
проблемы. Когда вода в незначительной степени ионизирована, образуются
ион гидроксония (сольватированный протон) и гидроксил-ион. Жидкий
аммиак ионизируется с образованием иона аммония и амид-иона
2Н2О Н3О+4-НО-; 2NH3 NH+4-NH7.
|Таким образом, если рассматривать жидкий аммиак с позиций изложен-
ных выше, соединения, увеличивающие в растворе концентрацию ионов
NH+, можно считать кислотами, в то время как основаниями будут веще-
ства, увеличивающие концентрацию иона NH2. В безводной уксусной кисло-
те соединение, повышающее концентрацию иона ацилония, можно рассматри-
вать как кислоту, а соединение, увеличивающее концентрацию ацетат-иона,—
Развитие представлений о кислотах и основаниях
И
как основание
2СН3СООН СН3СООН|Ч-СН3СОО-.
Однако во многих случаях это формальное сходство становится более
глубоким. Например, соли аммония (NH4+C1_) в жидком аммиаке и ионы
гидроксония (Н3О+С1_) в воде ведут себя как кислоты. Отсюда следует, что
кислотный или основной характер некоторых соединений в одних раствори-
телях остается скрытым, тогда как в других он четко проявляется. Харак-
терным примером может служить мочевина, которая в воде является очень
слабым основанием, в уксусной кислоте — более сильным основанием,
а в жидком аммиаке приобретает кислотные свойства
NH2CONH2 + H2O nh2conh2...h+ + oh-,
(ОН- — анион растворителя)
NH2CONH2 + CH3COOH NH2CONH£4-CH3COO-,
(СН3СОО_ — анион растворителя)
nh2conh2+nh3 nh2conh-+nhj.
(NH4—катион растворителя)
Эта теория допускает, что любой содержащий протоны растворитель
способен в принципе к автопротолизу с образованием сольватированных
протонов — носителей кислотных свойств — и анионов растворителя (табл. 1).
Таблица 1
Исходное соединение Сольватированный протон плюс анион растворителя Исходное соединение Сольватированный протон плюс анион растворителя
Вода Метиловый спирт Н3О+4-ОН- СН3ОН+4-СН3О- Аммиак Уксусная кислота NHJ + NHj сн3соощ+сн3соо-
Изложенные выше кислотно-основные теории могут быть разделены на
две группы: одни характеризуют кислоты и основания с точки зрения строе-
ния вещества, другие — с точки зрения наличия функциональных групп.
Согласно первым теориям, кислотой является соединение, содержащее водо-
род, согласно вторым — в кислоте по крайней мере один Н+ должен быть спо-
собен отщепляться. Те же критерии могут быть применены и к основаниям:
основание или содержит ОН-группу или способно диссоциировать с образо-
ванием гидроксил-иона.
Со временем эти два критерия оказались недостаточными. По мере разви-
тия теории растворителей стало очевидным, что кислотно-основные взаимодей-
ствия возможны также в жидкой двуокиси серы и в жидком фосгене. В этом
случае соединение, соответствующее кислоте, не имеет протонов, а соедине-
ние, аналогичное основанию, не содержит ионов гидроксила
2S02 SO2+ + SO1-,
2СОС1а СОС1+4-СОС1з.
4. ПРОТОННАЯ ТЕОРИЯ БРЕНСТЕДА — ЛОУРИ
Протон Н+ — единственный катион, не имеющий ни одного электрона
и обладающий радиусом действия 10-13 см, в то время как радиус действия
других ионов составляет в среднем 10-8 см. Подвижность иона Н+ весьма
велика по сравнению с подвижностью других ионов, поэтому концентрация
водородных ионов в растворе в первом приближении пропорциональна про-
12
Глава 1
водимости раствора и в определенных случаях концентрация ионов Н+,
каталитическое действие и сила кислоты изменяются параллельно
(Оствальд, 1884). Данные, приведенные в табл. 2, хорошо иллюстрируют
сказанное выше. Проводимость и каталитическое действие хлористоводород-
ной кислоты приняты за 100.
Таблица 2
Кислота Ка Относитель- ная проводи- мость Каталитическое действие
гидролиз метилацетата инверсия сахарозы
Хлористоводородная 100 100 100
Азотная 99,6 92 100
Серная 65,1 73,9 73,2
Трихлоруксусная 1,3-10-1 62,3 68,2 75,4
Дихлоруксусная 5-10-2 25,3 23,0 27,1
Монохлоруксусная 1,4-Ю-з 4,9 4,3 4,8
Уксусная 1,75-10-5 0,42 0,35 0,4
Напряженность электрического поля вблизи протона довольно велика,
поэтому водородные ионы обладают большим сродством к частицам со свобод-
ными электронными парами. Таким образом, в воде частицей, определяющей
кислотность раствора, является сольватированный протон. В то же время
доля несольватированных протонов в воде ничтожно мала. Поскольку спо-
собность связывать протоны у частиц со свободными электронными парами,
будь то молекулы растворителя или растворенного основания, изменяется,
весьма существенно изменяются в зависимости от природы растворителя
и свойства кислот. Этот факт долгое время игнорировался, потому что
в кислотно-основном титровании вода рассматривалась лишь как «раствори-
тель», хотя среди большого числа растворителей именно вода обладает весь-
ма примечательными свойствами: она имеет высокую диэлектрическую про-
ницаемость, молекулы воды в значительной степени ассоциированы и т. д.
Прошло более трех десятилетий с тех пор, как Бренстед и Лоури неза-
висимо друг от друга преодолели существовавшую до тех пор ограниченность
кислотно-основной теории [99, 522, 523]. Согласно Бренстеду и Лоури, осно-
ваниями являются не только соединения, содержащие гидроксил-ион, но
и все те соединения, которые способны присоединять протон.
Основные положения протонной теории изложены ниже:
1. Эта теория устанавливает вполне определенную связь между характер-
ными признаками кислот и оснований: кислота является донором протонов,
а основание — их акцептором. Эти определения сохраняют силу и для случая
неводного титрования.
S В- Н+
(Кислота) (Основание) + (Протон)
HG1 С1- Н+
NHj- NH3 + Н+
НСОз СО| -i- На-
следует, однако, подчеркнуть, что протон существует не per se (сам по себе),
а лишь в виде сольвата.
2. Бренстед разграничивает реакции образования солей и реакции ней-
трализации, поскольку по его теории взаимодействие кислоты и основания
никогда не приводит к нейтрализации, а во всех случаях образуются новые
Развитие представлений о кислотах и основаниях
13
кислоты и новые основания.
51 + 52 +
(Кислота) (Основание) (Кислота) 4- (Основание)
н2о + н2о н3о+ 4- НО-,
НС1 + н2о н3о+ + ci-,
сн3соон + сн3соон СН3СООЩ + сн3соо-,
нсю4 + сн3соон ^4 СН3СООЩ 4- C1OJ,
(СвН5)3СН4- nh2 тг NH3 4- (С6Н5)3С-*,
НВг +H2N — NHj ^2 [H3N — MI3]2+ ; Br-.
Сопряженными основаниями кислот, приведенных выше в первой или
третьей колонках, являются соединения (ионы), представленные в четвертой
и второй колонках. Образование соли не является характерным для кислот-
но-основных взаимодействий, поскольку соль может образоваться в любой
другой реакции
(CH3)3N + CH3I = (CH3)4NI.
3. Соединение с кислотными свойствами может потерять свой протон
только в присутствии соединения, обладающего большей способностью
присоединить протон (т. е. с большим сродством к протону), чем основание,
с которым протон связан. Бренстед не учитывает влияния растворителя,
хотя в зависимости от специфических свойств последнего образование соли
следует рассматривать с учетом следующих равновесий:
ЛН л- + н+,
s + H+ «Н+,
В + Н+ ВН+,
где ЛН — протонодонорная кислота, например хлорная; s — молекула
растворителя, например уксусной кислоты; В — растворенное основание,
например анилин; $Н+ — ониевый ион, образованный растворителем («ион
лиония» [68], т. е. СНэСООЩ); 2?Н+ — катион основания (катионная
кислота).
4. «Кислотой» может быть нейтральная молекула, катион или анион.
Все они могут отдавать протон растворителю или протон-координирующему
основанию, при этом образуется ион гидроксония или в общем случае оние-
вый ион
Н2О + Н2О Н3О+ + НО-
Увеличение
кислотных
свойств
НСОз+Н2О Н3О+ + СО23-
NHt + H2O H3O+ + NH3
Увеличение
основных
свойств
НС1 + Н2О Н3О+ +Cl-
Молекула растворителя, превращающаяся при присоединении протона
в катион, называется «катионом растворителя» независимо от того, является
ли она молекулой воды или иного растворителя.
Недостаток теории Бренстеда состоит в том, что она в своей первоначаль-
ной форме действительна лишь для протонсодержащих растворителей
и исключает возможность того, что кислотами могут быть не содержащие
* В жидком аммиаке с амидом натрия: NaNH2 Na+ + NH2-
14
Глава 1
водорода соединения. Эта теория не может дать объяснения для всех реакций
солеобразования, поскольку известно большое число солеобразующих про-
цессов, происходящих в отсутствие протонсодержащего растворителя или
соединения, диссоциирующего с образованием водородного иона, например:
СО24-СаО СаСО3,
SiO2-j-Na2O ' Na2SiO3,
A1C13 + C5H5N (C5H5N-A1C12)C1.
5. ЭЛЕКТРОННАЯ ТЕОРИЯ ЛЬЮИСА
Согласно электронной теории Льюиса, кислота и основание также обла-
дают противоположными свойствами, хотя и в ином смысле, чем это следует
из протонной теории [509, 528]. Льюис провел различие между конститу-
циональным критерием, в основе которого лежало строение молекул,
и функциональным критерием, базировавшимся на поведении молекул
в химических реакциях. Последний описан Льюисом как феноменологиче-
ский критерий.
1. Реакция нейтрализации заключается в мгновенном образовании коор-
динационной ковалентной связи. Образование этой связи является первич-
ным процессом, за которым в подходящем растворителе может следовать
ионизация и, возможно, диссоциация.
Основание Кислота = Продукт нейтрализации.
R3N-lh+=-R3NH+,
R3N + HC1 = R3N-HC1 —» R3NH+4-Cl-,
R3N + SO2 = R3N-SO2 —> R3NSO2++O2-,
R3N-J-EC]3= R3N-BC13,
COC12 + A1C13==COC12.A1C]3 —> СОС1+ + А1СЦ-,
2HC1 + SnCl4= SnCl4*2HCl,
2NH3-{-Ag+ = Ag(NH3)£,
C5H5N + AsC13 = C5H5N- AsC13 —» [C5H5NAsC12] + ^C1-,
(C2H5)2O + SO3 = (C2H5)2O. so3,
ROH+A1(OR)3 = ROH-A1(OR)3 —» H++[A1(OR)4]-,
(C6H5)2CO + SOC12 = [(C6H5)2CO. S0C1)] +C1-,
CH3C< -- AICI3 - [C6H5O. A1C13]_[CH3CO]+,
XO-C6H5
2CH3CN 4- 2AICI3 = [CH3CN • A1C12. NCCH3]+ + AlClj.
2. Кислоты и основания вызывают обратимые изменения окраски инди-
каторов.
3. Любая кислота может быть оттитрована соответствующим основанием
в присутствии подходящего индикатора. Присутствие ионов Н+ не является
абсолютно необходимым для этой реакции. Так, например, индикатор кри-
сталлический фиолетовый, добавленный в раствор пиридина в воде, уксусной
кислоте или бензоле, дает «основное» (фиолетовое) окрашивание; он дает
также «кислотное» (желто-зеленое) окрашивание во всех этих трех раствори-
телях в присутствии хлорида бора
C13B4-NC5H5 = C13E -- NC5H5
(Кислота) 4-(Основание) —> (Продукт нейтрализации)
Развитие представлений о кислотах и основаниях
15
4. Более слабая кислота (основание) вытесняется более сильной кисло-
той (основанием), например:
NH3 + H3O+ —> NHt+H2O,
н3сч
)СО ВС13
н3с/
(СН3)2СО + ВС13
Образование соли
[G5H5N ВС13]+(СН3)2СО.
ГИзс\
C5H5N ; )СО ~- ВС13
1н3с/
Согласно Льюису, каждый кислотно-основной процесс можно объяснить
исходя из простого и универсального утверждения: кислота представляет
собой координационно ненасыщенное соединение и, следовательно, способна
присоединять электронную пару с образованием координационной ковалент-
ной связи. Для основания характерно наличие свободной электронной пары.
Таким образом кислота, так же как и основание, характеризуется опреде-
ленной электронной конфигурацией. Тенденция образовывать ониевые соли
(аммониевые, оксониевые, сульфониевые, карбониевые) является признаком
основности даже в том случае, если в реакции образования солей не участву-
ют ионы водорода
(CH3)3N + CH3I = (GH3)4nT.
Теория Льюиса объясняет поведение кислот и оснований независимо от
природы растворителя, а также в системах, не содержащих протонов. В свете
теории Льюиса становятся более понятными взаимодействия «кислых»
и «основных» окислов, а также многочисленные процессы органической
химии. Эта теория может, несомненно, рассматриваться как фундаменталь-
ная. Однако, как это случилось со многими более ранними теориями, она
тоже может оказаться неприменимой к химическим явлениям, которые Льюис
не мог предвидеть.
По Бренстеду кислота является донором, а основание — акцептором про-
тонов. С другой стороны, согласно Льюису, кислота является акцептором
электронов, а основание — их донором. Однако эти два утверждения только
кажутся противоречивыми.
Усанович в своей обобщенной кислотно-основной теории попытался све-
сти воедино эти две теории.
При титровании в неводных средах кислоты, имеющие дефицит электро-
нов и не содержащие протона, называются кислотами Льюиса (L-кислоты),
например:
А1С13, FeCl3, ВС13, (GH3)3B, AsCl3.
Фторид или хлорид бора выделяет двуокись углерода из суспензии кар-
боната кальция в смеси ацетона и четыреххлористого углерода.
По этой классификации катион со структурой инертного газа, например
NH+, нельзя рассматривать как кислоту. Хлористый водород не может так-
же считаться кислотой, так как он может быть получен при взаимодействии
двух частиц — акцептора и донора электронов
ci-+h3o+ НС1 + Н2О
Хлористый водород не только не обладает дефицитом электронов,
а наоборот, имеет три свободные электронные пары
Н:С1:
В этом смысле его следует рассматривать как основание.
L-Кислоты подобным же образом отличаются от Н-кислот, поскольку
химические реакции, основанные на передаче электронов, в отличие от про-
тотропных реакций часто требуют значительной энергии активации [52, 5101.
Каталитическое действие кислот — одно из их характерных свойств.
Изучение этих свойств способствует развитию наших представлений о кисло-
тах и основаниях. Кислотно-основная концепция Льюиса оказалась непри-
16
Глава 1
менимой при рассмотрении каталитического действия кислот Льюиса —
BF3, А1С13, TiCl4. Поляньи с сотрудниками показал (1947), что если реаги-
рующие вещества тщательно очищены, то кислоты Льюиса не способны ката-
лизировать полимеризацию изобутилена и что полимеризация инициируется
следами Н-кислоты (ср. стр. 213 в работе [231]).
BF3 + H2O —> [BF3OH]-H+.
Опытным путем получен следующий ряд кислот Льюиса по силе их
действия на реакцию полимеризации:
BF3 > А1Вг3 > А1С13 > TiCl4 > BCI3 > SnCl4 > H2SO4.
Однако сам Льюис подчеркивает, что сила кислот и оснований зависит не
только от природы растворителя, но также и от выбранных в качестве этало-
на кислоты или основания. Согласно классическим представлениям, NH3 —
более слабое основание, чем ОН-; однако при их сравнении с ионом Ag+,
типичной льюисовской кислотой, это соотношение изменяется, поскольку
AgOH диссоциирует, в то время как [Ag(NH3)2]+ остается стабильным ком-
плексом.
Таким образом, практически существует различие между протонодонор-
ными Н-кислотами (бренстедовские кислоты) и электроноакцепторными
L-кислотами (льюисовские кислоты). Для сравнения кислотно-основных
теорий Бренстеда и Льюиса см. также работу [833].
Приведенные выше примеры показывают неприменимость электронной
концепции Льюиса в отдельных случаях, и это побудило к созданию новых
более совершенных теорий.
Как было показано, по гипотезе Аррениуса химические соединения могут
быть разделены на кислоты и основания на основе структурного и функцио-
нального критериев. В теории Бренстеда они классифицируются частично
по их структурным, частично по функциональным признакам. Согласно тео-
рии Льюиса, соединения относят к кислотам или основаниям исключительно
по структурному признаку. Функциональная теория кислот и оснований,
предложенная Эбертом и Конопиком [201], не содержит вообще никаких
структурных критериев.
6. ПРЕДСТАВЛЕНИЯ О КИСЛОТАХ И ОСНОВАНИЯХ ЭБЕРТА — КОНОПИКА
Среди растворителей, способных к автопротолизу (самоионизации), разли-
чают два типа кислот и оснований:
1. Донор-кислота (*$о) отщепляет катион растворителя или какую-либо
другую кислоту или образует с растворителем катион растворителя.
2. Акцептор-кислота (SА) связывает анион растворителя или любое дру-
гое основание.
3. Донор-основание (В D) отщепляет анион растворителя или какое-либо
другое основание или образует с молекулой растворителя анион раство-
рителя.
4. Акцептор-основание (ВА) способно связывать катион растворителя
или любую другую кислоту.
Соединения, относящиеся к группам $ n vlBd, хорошо известны: Н-кисло-
ты, ОН-основания. NH3 принадлежит к группе ВА. К группе SA относится,
например. РО-, который, присоединяя гидроксил-ион, превращается в НРО2“.
Пять типов кислотно-основных реакций могут протекать в растворите-
лях, способных к самоионизации
5ц-|-Во = Соль4-Растворитель; например: НС1-|-КОН = КС14-Н2О, (6.1)
*^d + ^a — Соль; например: НС14- NH3 = NH^Cl-, (6.2)
*^А-г^1) = Соль; например: Zn(OH)24-2NaOH = 2Na+[Zn(OH)4]2-, (6.3)
SА +А = РаствоРитель— Соль; например: SO2 + 2NH3 + Н2О = (NH4)|+SO|_, (6.4)
ВА + А = Соль; например: ВС13-|-Х(СНз)з = С1зВ-Х(СНз)з. (6.5)
Развитие представлений о кислотах и основаниях
17
В инертных растворителях — гексане, четыреххлористом углероде,
бензоле — возможны лишь реакции типа (6.2), (6.3) и (6.5). Такие термины,
как, например, акцептор-кислота, донор-основание и т. п., обычно исполь-
зуются в области неводного титрования.
7. ИОНОТРОПНАЯ ТЕОРИЯ ГУТМАНА — ЛИНДКВИСТА
Широко известны следующие четыре кислотно-основные концепции [545]:
1. Сольвентная теория.
2. Протонная теория.
3. Катион-анионная теория.
4. Электронная теория (ср. [528]).
Современная кислотно-основная концепция Гутмана и Линдквиста позво-
ляет привести первые три из этих теорий в соответствие с теорией переноса
протонов и в более широком смысле с теорией переноса ионов (ионотропная
теория) [295, 296]. Гарни в той части своей работы, которая посвящена меха-
низму переноса протонов, поддерживает ионотропную теорию Гутмана
и Линдквиста [249].
Кислотно-основные свойства молекулы (или иона) проявляются в процес-
се взаимодействия с растворителем. Молекула обладает кислотными свой-
' ствами, если она отдает молекуле растворителя положительно заряженную
частицу. С другой стороны,[если молекула отдает отрицательно заряженную
частицу, то она обладает основными свойствами. Справедливо также и обрат-
ное: молекула растворителя отдает отрицательно заряженную частицу
растворенной молекуле с кислотными свойствами и т. д. В более упрощенном
и обобщенном виде вышеизложенное может быть записано следующим обра-
зом:
а) Перенос катиона внутри молекулы, растворителя'.
2sP sP£4-s“.
1« Кислотное взаимодействие
AP + sP sP+ + A~.
2. Основное взаимодействие
B + sP BP+H-S-.
б) Перенос аниона внутри молекулы, растворителя'.
2sN sN^A-s*.
1. Кислотное взаимодействие
А sN ( '' AN~ s+.
2. Основное взаимодействие
B.V-^sN ~> B+ + sNi,
где Р — положительно заряженная частица (протон, катион); iV — отрица-
тельно заряженная частица (анион); АР — донор-кислота; В — акцептор-
основание; А — акцептор-кислота; BN — донор-основание; s — раствори-
тель. принимающий участие в ионотропной реакции.
Примером реакций «а» является взаимодействие хлорной кислоты и азо-
тистого основания в уксусной кислоте в качестве растворителя. Примером
реакций «б» может служить
2AsCI3 Г AsClJ + AsClj.
Хлорид железа (III) при растворении в хлориде мышьяка (III) является
акцептор-кислотой (1 в группе «б»), а хлорид тетраметиламмония—донор-
18
Глава 1
основанием (2 в группе «б»)
FeCl3 + (GH3)4N+Gl- — (FeGl4)-N(CH3)4.
Экспериментальными данными по растворителям, где мигрирующим
катионом является не протон, автор не располагает.
Процесс переноса в кислотно-основных реакциях представляет собой
фактически миграцию ионов (ионотропию) от одного соединения к другому.
В зависимости от того, что переносится — катионы или анионы — говорят
о катионотропных или анионотропных сольвосистемах [295, 296].
Наиболее известный пример катионотропии — миграция протонов (пере-
нос протонов). В катионотропных сольвосистемах кислота является донором
катионов, а основание — их акцептором. И наоборот, в анионотропной соль-
восистеме кислота служит акцептором анионов, а основание — их донором:
Катионотропные сольвосистемы
Донор протонов -)- Акцептор протонов Акцептор протонов-(-Донор протонов
NH3 + NH3 NHt + NHJ
сн3соон СН3СООН СН3СОО- + CH3COOHt
нсю4 + сн3соон 7Z СЮ4 -L CH3COOHf
Анионот ропные сольвосистемы
Донор анионов-)- Акцептор анионов < > Акцептор анионов-)-Донор анионов
so2 + so2 SQ2+ + S02-
СаО + со2 ;± Са2+ + GOf-
FeCl3 + AsGl3 AsGlJ + FeGl4
Таким образом, прототропная система Бренстеда представляет собой
частный случай катионотропной сольвосистемы. Теория Гутмана — Линд-
квиста не противоречит концепции Льюиса. Она отличается от кислотно-
основных представлений Усановича, поскольку сама идея ионотропии исклю-
чает все сложные ионы как мигрирующие единицы, а также все окислительно-
восстановительные процессы. Вопрос об энергии, необходимой для переноса
протона, будет рассмотрен в гл. 2.
Кислотно-основные теории, рассмотренные выше, не содержат сколько-
нибудь цельного представления о способах образования солей; более того,
в некоторых случаях они рассматривают последние как не зависящие от
кислотно-основных взаимодействий.
8. КИСЛОТНО-ОСНОВНАЯ ТЕОРИЯ УСАНОВИЧА
Характерная черта кислотно-основной теории Усановича состоит в том,
что образование солей рассматривается как обычный результат кислотно-
основных реакций. Следовательно, согласно Усановичу, эти две реакции не
могут быть разделены [269, 325, 825—827]. Так, реакция
(GH3)3N + GH3I = [ (CH3)4N] +1-
представляет типичную кислотно-основную реакцию. Йодистый метил отдает
катион CH* основанию — триметиламину — так же, как в реакции
(CH3)3N + HI = [(GH3)3NH]+I-
иодистый водород отдает протон (катионотропия). Алкилоксониевые соеди-
ения, например, являются донорами алкилкатиона.
Развитие представлений, о кислотах и основаниях
19
Из теории Усановича можно сделать необычный и весьма спорный вывод,
что окислительно-восстановительные процессы тоже должны рассматривать-
ся как кислотно-основные реакции. Например, натрий («основание») отдает
электрон хлору («кислоте») так, что образуется Na^-катион-кислота и С1“-
анион-основание; их соединение приводит к образованию соли.
Согласно Усановичу, кислоты — это вещества, способные отдавать либо
ион водорода, либо другой катион или присоединять какой-либо анион.
В более широком смысле кислоты способны отдавать электроположительные
частицы и присоединять электроотрицательные. Основания — это соедине-
ния, способные отдавать электроны или анионы или присоединять протон
или другой катион. Таким образом, Усанович объединяет протонную
и электронную теории.
С точки зрения валентности характер насыщенного соединения может
быть изменен путем координации ионов. Координационное число централь-
ного атома определяет предел координирующей способности. Вследствие
этого соединение способно выполнять функции кислоты или основания до
тех пор, пока оно не будет координационно насыщено. Координационно
ненасыщенные электроположительные атомы, молекулы и ионы ведут себя
как кислоты, а координационно ненасыщенные электроотрицательные части-
цы — как основания. Так, например, молекула двуокиси углерода является
кислотой, поскольку ее электроположительный атом обладает более высокой
валентностью, чем электроотрицательный атом кислорода, хотя оба атома
координационно ненасыщены; присоединяя кислород, двуокись углерода
превращается в карбонат-анион. Углерод, однако, остается ненасыщенным
и все еще способен присоединить ион кислорода, превращаясь, таким обра-
зом, в анион ортоугольной кислоты. Этот ион уже не проявляет каких-либо
кислотных свойств, так как углерод стал координационно насыщенным.
В то же время основные свойства этого иона становятся ярко выраженными
из-за его отрицательного заряда
СО2 —> CQ2- СО$-
Это весьма верное представление Усановича логически основывается
на разделении элементов на кислотные и основные. Сила оснований и кислот
обратно пропорциональна силе сопряженных кислот и оснований. Это может
быть очень просто проиллюстрировано в случае простейших катионных
кислот и анионных оснований. Чем ниже ионизационный потенциал металла,
тем сильнее основание, например: К—е=К+; другими словами, металлы
проявляют основные свойства, отдавая электроны. Чем выше ионизацион-
ный потенциал, тем сильнее катионная кислота. Таким образом, сильная
кислота соответствует слабому основанию’, обратное также справедливо.
С14-е = С1-; К+-Н = К
Сильная Слабое Слабая Сильное
кислота основание кислота основание
Хорошо известно, например, что при титровании в неводных средах
уксусная кислота является «слабой кислотой», в то время как сопряженное
основание — ацетат-ион представляет собой «сильное основание». Анилин —
слабое основание, но катионная кислота (ион анилиния), полученная из
него в результате присоединения протона, представляет собой сильную
кислоту.
Согласно Усановичу, органические основания не способны отдавать
анионы, потому что они содержат координационно ненасыщенные электро-
отрицательные атомы (акцептор-основание Эберта — Конопика), подвер-
гающиеся взаимной поляризации, предшествующей образованию соли,
например:
б+ в- б+ б-
C5H6N + AsC13 -> C5H5N-AsC13 —> [C5H5N.AsC12]+ + C1-.
20
Глава 1
Таким образом возникает связь между противоположными зарядами
индуцированных диполей пиридина и хлорида мышьяка(Ш), и после уста-
новления координационной связи в продукте присоединения ион хлора
диссоциирует под действием сил отталкивания со стороны отрицательно
заряженного электронного облака азота.
Ниже перечислены критерии, используемые в кислотно-основной кон-
цепции Усановича:
1. Кислотно-основное взаимодействие не всегда является взаимодействи-
ем между протонсодержащими веществами. При отсутствии протонов тоже
существует кислотно-основное равновесие.
2. Соединения, способные отдавать катионы или присоединять анионы,
можно рассматривать как кислоты. Соединения, способные связывать катио-
ны или отдавать анионы, являются основаниями.
3. Кислотно-основные свойства зависят от координационной ненасыщен-
ности атомов или ионов, входящих в состав данного соединения.
4. Кислоты — это соединения, в которых имеются координационно нена-
сыщенные электроположительные атомы. Отсутствие координационно нена-
сыщенных электроположительных атомов придает соединению основной
характер.
5. Поскольку в большинстве соединений присутствуют координационно
ненасыщенные атомы, несущие заряды различные по знаку, амфотерность
может рассматриваться как общее свойство полярных соединений, хотя
амфотерный характер не исключает преобладания кислотных или основных
свойств. Кислотные или основные свойства зависят от природы как реаги-
рующего вещества, так и растворителя.
Из вышесказанного можно сделать вывод, что вопрос о природе кислот
и оснований нельзя рассматривать как окончательно решенный. Между тем,
для титрования в неводных средах развитие кислотно-основных представле-
ний имеет первостепенное значение. Для того чтобы объяснить явления,
происходящие со многими соединениями в процессе титрования в неводных
средах, необходимы современные кислотно-основные представления, даже
если теоретические взгляды на кислоты и основания не разделяются всеми
исследователями.
Глава 2
СИЛА КИСЛОТ
«Сила кислот» подробно обсуждается в специальной литературе, но удов-
летворительного определения’этого понятия пока^не дано. Хорошо известны
физико-химические свойства кислот, однако происхождение этих свойств
до конца не изучено. Объяснить эти свойства становится все труднее, посколь-
ку число используемых в аналитической и в органической химии раствори-
телей все возрастает. Например, для титрования в неводных средах в настоя-
щее время применяются более сорока различных растворителей.
Прежде чем обсуждать титрование в неводных средах, необходимо под-
робно познакомиться с относительной силой кислот и оснований, ее изме-
нением в зависимости от природы растворителя и титранта и главным образом
с влиянием на силу кислот основной молекулярной структуры. Поэтому
опыты по установлению относительной силы кислот, например в процессе
потенциометрического титрования на основе потенциала полунейтрализа-
ции, имеют неоценимое значение.
9. ИОНИЗАЦИЯ ОРГАНИЧЕСКИХ СОЕДИНЕНИИ
В настоящее время хорошо известно, что соединения с ковалентной
связью в неводных средах образуют ионы. Лэпуорт высказал предположение,
что большинству превращений в органической химии предшествует слабая
электролитическая диссоциация, т. е. возникают ионные промежуточные
соединения. На основе большого числа наблюдений можно сделать заклю-
чение, что некоторые типичные реакции присоединения по двойной связи
происходят в две стадии с образованием ионных промежуточных соединений
(см. стр. 206 в работе [653]).
Эти ионные промежуточные соединения могут рассматриваться как л-ком-
плексы, в которых катионная часть диполя брома в процессе бромирования
связана с л-электронной системой двойной связи [184]
(ср. гл. 27, разд. 155).
Льюис и Ленгмюр показали, что при разрыве ковалентной связи могут
образоваться либо ионы противоположного знака (9.1), либо два нейтраль-
ных радикала (9.2). Атом углерода является амфотерным: он может обладать
как электрофильными, так и нуклеофильными свойствами. В жидкой двуоки-
си серы гексафенилэтан распадается на ионы, и раствор, следовательно,
обладает электропроводностью.
(СбНй)зС С(СвН5)3 <——(С6Н5)зС+ + (С6Н5)3С-
Гексафенилэтан Трифенил- Трифенил-
метил-катион метил-анион
(9.1)
22
Глава 2
В то же время в неионизирующем растворителе
(С6Н5)3С — С(С6Н5)з (С6Н5)3С. 4- -С(СвН5)з (9.2)
Трифенилметил-радикалы
С точки зрения кислотно-основных взаимодействий производных три-
фенилметана основания типа трифенилкарбинола обладают ярко выраженны-
ми основными свойствами. С сухим хлористым водородом в бензоле трифенил-
карбинол дает хлорид
(С6Н5)3С - он + НС1 = (С6Н5)3СС1 + Н2О.
Таким образом, эта реакция подобна реакции нейтрализации. Анионный
характер трифенилметильной группы иллюстрируется поведением трифенил-
метилтетраметиламмония, который как электролит ионизируется во всех
растворителях.
(CeH5)3C-+N(CH3)4.
Известны также органические соединения с сопряженными связями, которые про-
водят ток, будучи растворенными в бензоле, содержащем трихлоруксусную кислоту,
например циклопента диен, 0-каротин, витамин А [861, 862].
Разделение соединений, имеющих структуру ионных решеток, на кине-
тически независимые ионы требует большой энергии, которая при комнатной
температуре не может быть получена за счет теплового движения. Диссо-
циация происходит потому, что ионы взаимодействуют с растворителем
и при этом освобождается значительное количество энергии.
Соли, образующиеся из органических катионов или анионов, как прави-
ло, растворяются в органических растворителях. (Так, п-толуолсульфонат
Х-о-бромбензил-Х-этил-Х,]У-диметиламмония растворяется в диоксане, в то
время как его тетрафенилборат не растворяется [316]).
В растворителе с малой диэлектрической проницаемостью эффективный
размер растворенных частиц обычно больше размера простого сольватиро-
ванного иона: агрегаты состоят из нескольких ионов или иона и одной или
более неионизированных молекул.
В гидроксилсодержащих растворителях образование сольватированных
ионов сопровождается возникновением оксониевых ионов (протолитическая
реакция)
HCI4-CH3OH СН3ОН£ + С1-.
Сравни: HC1 + CH3NH2 CH3NH£ + C1-.
Различия между органическими и неорганическими ионными реакциями
не являются резкими, хотя и существуют [263]. Образование промежуточных
продуктов при разрыве ковалентных связей иногда протекает медленно.
«Кислый водород» (водород, способный ионизироваться), характерный для
протонодонорных кислот, полностью соответствует «активному водороду»
в органической химии с тем лишь ограничением, что кислотность отражает
подвижность водорода. Значение ^Ка может достигать значения 30 единиц,
но атом водорода в этих соединениях обладает лишь незначительной актив-
ностью
Р*а
Трихлоруксусная кислота 0,9
Фенол 9,9
а-Глюкоза [582] 12,44
Фенплацетилеи 21
Трифенилметан 33
Эмпирически принято считать, что граница между кислым и активным
водородами лежит в интервале рХа 10—12.
Сила кислот
23
Для экспериментального определения активного водорода важно, чтобы
используемая реакция была необратимой. С другой стороны, по ряду при-
чин определение активного водорода является задачей, требующей соблюде-
ния ряда предосторожностей (см. гл. 19).
10. ВОДОРОДСОДЕРЖАЩИЕ КИСЛОТЫ (Н-КИСЛОТЫ)
Сила водородсодержащих кислот определяется их протонодонорной спо-
собностью, а сила оснований — их протоноакцепторной способностью. Таким
образом, для определения степени протонного переноса необходимо прини-
мать во внимание природу взаимодействующих частиц (под «частицей» под-
разумевается в данном случае кинетически независимая молекула, атом или
ион). По данным Фаянса [221], теплота сольватации свободного прото-
на составляет 232 ккал/г-атом. Однако ионизация кислоты не может
рассматриваться как «эмиссия» протона, потому что эта ионизация про-
исходит только в присутствии протоноакцепторного основания. Сродство же
последнего к протону уменьшается с увеличением числа свободных электрон-
ных пар; стабильность заполненного октета приводит к резкому исчезнове-
нию сродства к протону
|сНз > iNHj > |он" > |?Г
В то же время сродство к протону (сила основания) также уменьшается
в следующей последовательности:
02-> ОН-> он2> он+> ощ+.
Силу кислот и оснований нельзя измерить непосредственно; она может
быть определена лишь сравнительно: 1) при введении растворителя, способ-
ного к автопротолизу, 2) в нейтральном апротонном растворителе, при этом
необходимо добавление эталонной кислоты или основания (например, для
определения относительной силы ароматических кислот в бензольном раство-
ре эталонным основанием может быть дифенилгуанидин [173]) и 3) в газо-
образном состоянии; например, взаимодействие триметилбора и триметил-
амина (льюисовские кислота и основание).
Отношения, характеризующие относительную силу кислот и оснований:
рК, Но, Н+, Н_, РА*, основаны на экспериментальных данных, полученных
с растворителями, большинство из которых способно к самоионизации.
И. ПРОТОННЫЙ ПЕРЕНОС И ЕГО ЭНЕРГИЯ
По кислотно-основной теории Гутмана — Линдквиста в растворителях,
способных к автопротолизу, кислотные свойства являются результатом
взаимодействия молекулы растворителя и растворенной молекулы кислоты,
когда при частичной диссоциации кислоты происходит ионный перенос (пере-
нос протона) между взаимодействующими частицами [295, 296].
а. Энергия протонного переноса. Энергия, необходимая для переноса
протона (J), может определять кислотный эффект и его силу (см. стр. 34, 64
и 105 в работе [294]; [544, 614]). Когда кислота ЛН растворяется в раство-
* Р^а и Р*Ь представляют собой показатели степени константы диссоциации кисло-
ты и основания, причем рАа = — logAa.
Функция кислотности Гаммета (Но, Н+, НА определяет степень протонодонорной
способности растворителя по отношению к электрически нейтральным, положительно
и отрицательно заряженным индикаторам. Согласно универсальной шкале кислотности
Измайлова РА — — logMyyo, где М — концентрация раствора, у — коэффициент актив-
ности ионов в бесконечно разбавленном неводном растворе, у0 — коэффициент активно-
сти тех же ионов в бесконечно разбавленном водном растворе [76, 179, 342, 413, 625].
24
Глава 2
рителе, способном к автопротолизу, имеются две возможности для про-
тонного переноса: между молекулами растворителя и между кислотой
и растворителем
2$Н + или ЛН-f-sH Л- + $Н|
Вероятность протонного переноса от молекулы кислоты 4Н к молекуле
растворителя sH пропорциональна
ехр (— J/kT),
где J — энергия протонного переноса, к — постоянная Больцмана, Т — абсо-
лютная температура.
Энергия протонного переноса может быть выражена следующим урав-
нением:
J = (— кТ log Ка) 4- кТ log М,
где Ка — константа кислотности Бренстеда, М — число молей растворенного
вещества в 1000 г растворителя.
Если энергия, требуемая для протонного переноса между растворенной
кислотой и растворителем, больше, чем энергия, необходимая для такого
перехода между молекулами растворителя (J > Jo), то концентрация катио-
нов растворителя $Н+ в растворителе не увеличивается и, таким образом,
кислотный характер вещества ЛН остается скрытым. С другой стороны, если
J<0, то в данном растворителе молекула ЛН будет вести себя как сильная
кислота.
Чем ближе величина функции кислотности Гурни X = 1 — J/Jo к едини-
це, тем сильнее кислота; когда кислота очень слабая, значение этой функции
стремится к нулю.
Энергия протонного переноса складывается из двух частей: 1) внутрен-
ней специфической части энергии и 2) части энергии, проявляющейся после
диссоциации в процессе различных взаимодействий с растворителем (диполь-
ное взаимодействие, образование водородной связи, влияние диэлектриче-
ской проницаемости и т. д.). Первая часть зависит только от разности между
значениями наиболее низких уровней квантовой энергии иона (или молеку-
лы) до и после протонного перехода («внутренний» протонный переход),
в то время как та часть энергии, которая основывается на взаимодействии,
в значительной мере зависит от сольватации. Константа равновесия при
этом зависит от того, имеют ли эти две части энергии одинаковые или про-
тивоположные знаки.
Деление энергии протонного перехода на две составляющие связано
с разделением водородсодержащих кислот на ионогенный и изоионный типы
[218]. После протонного переноса число заряженных частиц в ионогенных
кислотах увеличивается, в то время как в изоионных кислотах оно остается
неизмененным. В последнем случае энергия протонного переноса, отражаю-
щая взаимодействие растворителя и растворенного вещества, меньше
J = J14-/2»
где Ji — энергия «внутреннего» протонного переноса, не зависящая от тем-
пературы, J2 — энергия частиц после протонного переноса, обусловленная
различными электростатическими взаимодействиями («энергия взаимодей-
ствия») [545]. Эта величина зависит от температуры и является сложной
функцией диэлектрической проницаемости растворителя.
Небольшая величина части энергии (J2), связанной с электростатически-
ми взаимодействиями, характерна для изоионных кислот, так как с точки
зрения взаимодействия растворенного вещества и растворителя состояние
после протонного переноса не претерпело существенного изменения по сравне-
нию с состоянием до протонного перехода. Следовательно, значения Ка
Сила кислот
25
для изоионных и ионогенных кислот изменяются различно с изменением
температуры.
б. Механизм протонного переноса приблизительно состоит в следующем
[546]: сначала между молекулой кислоты и молекулой растворителя обра-
зуется водородная связь ЛН . . . $Н. Миграция протона происходит
в (ЛН ... $Н)Раств-комплексе до тех пор, пока Н-связь не перестанет суще-
ствовать. В результате дипольного взаимодействия (ЛН ... sH) образуются
ассоциаты (Л"*«Н£) с растворителем, представляющие собой комплекс.
Диссоциация комплекса (Л “ ««Н^раств начинается с постепенного разделения
частиц и возрастающей сольватации, при этом образуются (Л“)раств
и («Щ)раств, где ЛН — протонсодержащая кислота, sH — протонсодержа-
щий растворитель, Л ~ — анион, вЩ — катион растворителя.
в. Типы протонных переносов [294, 887].
1. ЛН+В А-+(ВН)+
Пример:
НСЮ4+СН3СООН ClO7 + CH3COOHt
2. (ЛН)+4-В Л+(ВН)+
Пример:
CH3COOH£ + R3N < HsCOOH+R-jNH4-
3. (ЛН)"+В (ВН)++42-
Пример:
CH2(COOH)COO-+H2NCH2CH2NH2 СН2(СОО-)£ + [Н2НСН2СН2ТШз]
4. 4Н + В- 4-4-ЯН
Пример:
СвН6СООН + СН3О- СвН5СОО-4-СН3ОН
12. КОНСТАНТЫ ДИССОЦИАЦИИ КИСЛОТ И ОСНОВАНИЙ
Согласно протонной теории Бренстеда и Лоури, кислоты являются доно-
рами протонов, а основания — их акцепторами
Кислота Основание + Протон
АН А- 4- Н+
Для осуществления протонного переноса присутствие протонакцепторно-
го основания совершенно необходимо. Молекула растворителя, способная
как присоединять, так и отдавать протоны, может выступать в качестве-
протонакцепторного основания. Такими амфотерными растворителями,
обозначаемыми символом sH, могут быть вода, спирты, карбоновые кислоты
(муравьиная, уксусная, пропионовая)
ЛН + «Н Л-+«Щ (12.1)
где $Нз — ион лиония, образующийся в процессе присоединения протона,
т. е. сольватированный протон. Состояние равновесия определяется относи-
тельной основностью sH и Л“, молекулы или иона. Закон действия масс для
состояния равновесия может быть записан следующим образом:
у [А-][Ш+]
[АН] [sH] *
Поскольку в разбавленных растворах количество растворителя может
рассматриваться как практически постоянное, константа диссоциации кис-
26
Глава 2
М-] [sHt]
[АН]
лоты АН может быть записана как
(12.2)
Кислоты, для которых равновесие, изображенное уравнением (12.1),
существенно сдвинуто вправо, являются сильными и в первом приближении
константа диссоциации кислоты пропорциональна основности растворителя.
Однако на равновесие влияет большое число других факторов и не существу-
ет растворителя, который отличался бы от воды только своей основностью.
Константа равновесия, подобная (12.2), может быть записана также и для
других сопряженных кислотно-основных систем. Например, в случае заря-
женного акцептора — основания А~ (например, бензоат-аниона), которое
координируется с протоном, с протонодонорной молекулой устанавливается
следующее равновесие:
Л-+$Н ЛН-Н",
где s —ионы ОН , СН3О , СН3СОО“ и т. д. (лиат-ион). В этом случае кон-
станта основности к _ [ЛН] [*-] Къ [Л-] ’ (12-3)
откуда [4Н]= (12-4)
После подстановки в уравнение (12.2) значения [АН] ния (12.4) становится очевидным следующее соотношение между из уравне- константами
кислотности и основности для сопряженных кислотно-основных систем:
(12.5)
If
В случае воды произведение
кЩ [s-]
является ионным произведением. В общем виде для молекул растворителя,
способных к прототропным взаимодействиям, это выражение приобретает вид
[sHJ] [$"] = ЛГавто = КаКъ (12.6)
и
v- ^авто
Вместо Ка и Кь чаще употребляются по аналогии с pH значения рХа
и рЛ&, т. е. отрицательный десятичный логарифм Ка и Кь (рК = — logff).
Для случая воды
Р^н2о = рКа -|- рКъ
или
рКа=1А — рКь.
Константа автопротолиза для уксусной кислоты равна
^авто=[СНзСООЩ][СН3СОО-] =2,5-10-13 [677], см. также [102].
Уравнение (12.6) иллюстрирует зависимость контант кислотности и основ-
ности от способности растворителя к автопротолизу. В 0,1 М растворе уксус-
ной кислоты теоретически самая высокая концентрация иона лиония, создан-
ная сильной кислотой, составляет 0,1, а наиболее низкая его концентрация
при растворении сильного основания может быть 2,5 -10"12. Аналогично
концентрация иона лиония в муравьиной кислоте находится в пределах от
Сила кислот
27
Таблица 3
Растворитель Ион ЛИОНИЯ Лиат-ион Р^авто Литература
Ацетонитрил (CH3CNH)+ ch2cn- 19,5 689
Серная кислота H3SOt HSO4 3,0 342
Муравьиная кислота HCOOHt нсоо- 6,2 343
Уксусная кислота СН3СООЩ сн3соо- 12,6 677
Метиловый спирт СН3ОН| сн3о- 16,7 342
Этиловый спирт С2Н5ОНг С2Н5О- 19,1 677
Аммиак а NHt NH7 22 677
а При -33,4°.
0,1 до 6 «10-6 и в этиловом спирте от 0,1 до 8 -IO-19. Константы автопротолиза
некоторых растворителей приведены в табл. 3.
Протонный перенос обычно происходит в направлении свободной электрон-
ной пары растворенного основания, когда протонакцепторные основания
(например, анилин) растворены в амфотерных растворителях (например,
уксусная кислота).
Равновесие диссоциации может быть представлено следующей реакцией:
B-f-sH BH+H-s-
и константа диссоциации основания
|ВН[Д]1И (12.7)
где s- — например, ацетат-ион.
Таким образом, в процессе присоединения протона возникает новая
кислота — ониевый ион, соответствующий основанию. Чем сильнее донор-
ная кислота, тем слабее соответствующее акцепторное основание. Приведен-
ное ниже соотношение показывает протонодонорную роль ониевого иона
(катионная кислота)
BH+4-sH
К __ iz [В]
А а = Л вн+ = [ВН+] ' (14.0)
где sHj—ацилониевый ион СНзСООЩ и sH — уксусная кислота.
В табл. 14 (стр. 61) приведены данные о связи между различными обозна-
чениями, такими, как К, рК, Квн+, р^вн+- Корреляции, подобные имеющим-
ся между уравнениями (12.2), (12.3), (12.7) и (12.8), действительны и для
любого другого амфотерного растворителя, однако значения К&то неодина-
ковы для различных растворителей.
В случае инертных апротонных растворителей приведенные выше соот-
ношения не выполняются (см. разд. 16).
В неводных средах необходимо учитывать еще один фактор, который
для водных растворов имеет второстепенное значение: это тенденция соль-
ватированных ионов к ассоциации с образованием ионных пар и больших
агрегатов, особенно в растворителях с низкой диэлектрической прони-
цаемостью. В растворителях этого типа ионная диссоциация незначительна
по сравнению с образованием ионных пар.
В общем значение Ка кислот зависит как от протонодонорной способности
кислоты, так и от протонакцепторной способности растворителя. Например,
для уксусной кислоты величина Ка не может рассматриваться как абсолют-
ная мера силы кислоты.
Помимо силы кислоты необходимо также принимать во внимание «кислот-
ность» раствора, т. е. степень ионизации данной кислоты не обязательно
28
Глава 2
соответствует кислотности ее раствора [350]. В первом из вышеупомянутых
примеров кислотность раствора зависит от протонодонорной способности
иона лиония, в данном случае — от протонодонорной способности ацило-
ниевого иона. В случае замены растворителя, например уксусной кислоты
на изопропиловый спирт, изменяется не только значение К.а, но также
и «кислотность» раствора. Протонодонорная способность ацилониевого иона
значительно превосходит протонодонорную способность иона (СНз^СНОЩ.
Изопропиловый спирт обладает большей основностью, чем уксусная
кислота, и его диэлектрическая проницаемость также вдвое больше. Поэтому
хлорная кислота лучше диссоциирует в изопропиловом спирте, хотя «кислот-
ность» раствора уксусная кислота — хлорная кислота выше вследствие более
высокой протонодонорной способности ацилониевого иона.
Абсолютное значение силы кислот и оснований не может быть определено
в растворителях, способных к автопротолизу. Действительно, даже отно-
сительная сила кислот может быть лишь приблизительно установлена
с помощью уравнений. Сила кислот и оснований зависит и от диэлектриче-
ской проницаемости среды, константы автопротолиза и от трудно определяе-
мых специфических взаимодействий.
13. ВЛИЯНИЕ ПРИРОДЫ РАСТВОРИТЕЛЕЙ НА СИЛУ КИСЛОТ
В протонсодержащем растворителе относительная сила кислот зависит
от природы растворителя и определяется энергией, необходимой для пере-
носа протона от молекулы или иона на бесконечно большое расстояние [546].
Однако роль растворителя не является
Сильные кислоты Сильные основания
Рис. 1.
тельно, относительная сила кислот (и
телях не всегда одинакова [88, 103,
пассивной. Кислотные и основные
свойства тесно связаны со’взаимо-
действием растворенного вещества
и растворителя [295, 296]. Влияние
растворителя определяется его
диэлектрической проницаемостью
и прототропными свойствами.
Вследствие этого относительная
сила кислот (и оснований) может
изменяться в зависимости от при-
роды растворителя.
Свободная энергия кислот ;(и
оснований) изменяется от раство-
рителя к растворителю. Следова-
оснований) в различных раствори-
172, 763].
Например, в хлороформе и хлорбензоле более сильными основаниями
являются третичные амины, в то время как в w-гептане, бензоле и 1,4-диокса-
не более сильными основаниями оказываются вторичные амины [849].
В прототропном растворителе при реакции нейтрализации наблюдается
перенос протона (миграция протонов). Протонодонорная способность молекул
и ионов кислотного типа и тенденция молекул основного типа присоединять
протоны связаны с их молекулярной структурой. В данном растворителе
протонодонорная или протонакцепторная способность поляризованных
молекул уменьшается постепенно от наиболее сильной кислоты к наиболее
сильному основанию. Таким образом, шкала кислотности, будучи функцией
потенциала протонного переноса, сравнима со шкалой окислительно-вос-
становительных потенциалов. На рис. 1 вода условно принимается за ней-
тральное соединение. Расположенные в левой части рисунка соединения
проявляют себя как доноры протонов по отношению к веществам, располо-
женным в правой части, тем в большей степени, чем дальше друг от друга
они расположены, т. е. чем больше их потенциал протонного переноса
Сила кислот
29
(ср. [847]). Хлорная кислота является сильной кислотой в уксусной кислоте,
но она еще сильнее в воде, а по отношению к хлорной кислоте вода ведет
себя как основание. Этилендиамин, напротив, образует в воде ониевый ион,
т. е. является акцептором протонов.
Способность молекул и ионов проявлять свойства кислот или оснований
зависит не только от их структуры, но и от их «собственной внутренней
полярности».
Каждый растворитель выявляет «собственную полярность» растворенных
в нем молекул в различной степени. Монохлоруксусная кислота является
Соединение в растворе
ведет себя как кислота
Соединение в растворе
ведет себя как основание
Серная кислота
▼ НША
©EOV
Уксусная кислота Ш А ® Е О V
Метилизобутилкетон а • ▼ [] й ® В О V
А • ▼ О Н Пиридин
Этилендиамин
Хлорид аммония Ш Мочевина
А Трихлоруксусная кислота А м-Нитроанилин
• Монохлоруксусная кислота ® Анилин
▼ Бензойная кислота И N, N -Диэтиланилин
К п-Нитросренол О Бензиламин
И Фенол V Пиперидин
Рис. 2. Характеристика кислотных и основных свойств соединений (идеализирован-
ная схема)
сильной кислотой в пиридине, но в уксусной кислоте она уже не обладает
кислотными свойствами. Метилмочевина и гуанидин ведут себя как основа-
ния в серной кислоте, но в жидком аммиаке они проявляют свойства кислот
(рис. 2).
Относительная кислотность некоторых соединений и ионов в жидком
аммиаке [864] изменяется в ряду
ион аммония > ион гуанидиния > тиомочевина > мочевина > гуанидин < анион
тиомочевины > карбампд-анпон > анион гуанидина > амид-анион.
В аммиаке амид-ион является анионом растворителя.
Помимо фактора, зависящего от молекулярной структуры растворителя,
на силу кислот и оснований влияет также ряд факторов, зависящих от при-
роды растворителя: это эффект сольватации, диэлектрическая проницаемость
и прототропный эффект (например, кислотность или основность растворите-
ля) (рис. 3).
В растворителе, кислотность которого выше кислотности воды, сила
кислот уменьшается или совсем не проявляется, в то время как сила основа-
ний возрастает, основность сильных оснований выравнивается, а слабых или
очень слабых — дифференцируется. В растворителе с более высокой по
сравнению с водой основностью сила кислот возрастает, в то время как сила
оснований уменьшается или основные свойства совсем пропадают.
30
Глава 2
В дифференцирующих растворителях с низкой диэлектрической прони-
цаемостью типа метилизобутилкетона могут быть в определенных пределах
оттитрованы как кислоты, так и основания.
Большим преимуществом титрований, проводимых в неводных средах,
является то, что относительная сила кислот и оснований может меняться
от растворителя к растворителю. Это показано на рис. 4.
Для растворов, кислотность которых не может быть уже выражена через
pH, область существования кислых и основных растворов в воде обозначает-
ся границами рисунка справа и слева. В пропионовой кислоте число и сила
полностью ионизированных кислот уменьшаются в противоположность осно-
ваниям, число и сила которых возрастают.
В этилендиамине число и сила оснований уменьшаются, а для кислот
и их аналогов — возрастают. В промежуточной области, отделяющей очень
Прототропный эффект,
сольволиз
Диэлектрический эффект
Рис. 3. Влияние смеси растворителей на
кислотность пикратов диалкиламмония (идеа-
лизированная схема).
Рис. 4. Влияние растворителя на
силу кислоты или основания.
ДФГ— дифенилгуанидин; ТБАГ — гидроокись
тетрабутиламмония.
сильные основания и кислоты друг от друга, эти показатели изменяются
в зависимости от автопротолиза растворителя. Пределы этой области для
воды pH 0—14, для уксусной кислоты pH 0—12,6 (ионное произведение
2,5 »10-13), для муравьиной кислоты pH 0—6,2 (ионное произведение 6,3 «10-7).
В инертных растворителях эта промежуточная область может рассматри-
ваться как бесконечная, так как катионы и анионы растворителя отсутству-
ют. В растворителях такого типа сила кислот и оснований не зависит от
природы растворителя. В растворителях с низкой диэлектрической прони-
цаемостью и пониженной сольватирующей способностью образование агре-
гатов, ионных пар, ионных диполей и продуктов присоединения существенно
влияет на кислотно-основные взаимодействия.
14. ВЛИЯНИЕ СОЛЬВАТИРУЮЩИХ СВОЙСТВ РАСТВОРИТЕЛЕЙ НА СИЛУ КИСЛОТ
Сольватирующая способность растворителя является важным фактором.
По мере ее увеличения увеличивается и ускоряется образование ионов
и, наоборот, уменьшение сольватирующей способности способствует исчезно-
вению ионов [402]. Это находит свое подтверждение при сравнении констант
диссоциации кислот с различными типами зарядов в растворителях различ-
ной основности.
Константы диссоциации алифатических карбоновых кислот в воде в 105—
106 раз больше, чем в этиловом спирте, в то время как для замещенных ионов
аммония они только в 10 раз больше (см. стр. 54 в работе [393]).
В воде равновесие реакции
R3NH++H2O R3N + H3O+
Сила кислот
31
смещено вправо. Вода является примерно в 15 — 20 раз более сильным осно-
ванием, чем этиловый спирт [84]
R3NH+ + C2H5OH R3N + C2H5OHt
В случае карбоновых кислот в левой части уравнения будут находиться
две нейтральные молекулы, в то время как справа — два заряженных реаген-
та. Образование заряженных частиц промотируется растворителем с большей
сольватирующей способностью
RCOOH + H2O rcoo- + h3o+,
RCOOH + CH3COCH3 RCOO--HCH3COCH3]H+.
Сольватирующее действие растворителя, способствующее ионизации, зави-
сит также от возможности резонанса (гл. 4, разд. 27). Константа ионизации
пикриновой кислоты в воде примерно лишь в 1050 раз больше, чем в этиловом
спирте, потому что заряд пикрат-аниона вследствие резонанса распределен
по всей молекуле и поэтому она сольватирована не сильно; стабильность
молекулы почти не изменяется и тогда, когда меняются сольватирующие
свойства растворителя. В противоположность этому заряд карбоксилат-
аниона распределяется лишь между двумя атомами кислорода, поэтому,
например, монохлоруксусная кислота в воде в 96 000 раз сильнее, чем
в этиловом спирте.
Электрически нейтральные кислоты в этиловом спирте слабее, чем в воде;
заряженные положительно кислоты отличаются друг от друга по силе
в меньшей степени (табл. 4) [185].
Таблица 4
*Ка
Кислота
в воде
в этиловом
спирте
Азотная
Пикриновая
Трихлоруксусная
Щавелевая
Монохлоруксусная
2,4-Динитрофенол
0,8
0,7
1,3
2,9
3,9
3,57
4,0
5,46
6,58
7,74
7,74
₽квн+
Ион
в воде
в этиловом
спирте
Анилиния
N, N - Диметилани линия
Пиридиния
4,6
5,0
5,3
3,73
4,13
4,33
Растворитель с более сильными сольватирующими свойствами способ-
ствует возникновению ионов, в то время как электрически нейтральные
молекулы приводят к образованию ^ионных пар
ЛН-J-s 4~ + sH+ (образуются два иона, АН—ионогенная кислота [218])
ВН+4-s В + «Н+ (число ионов остается неизменным, ВН+ —изоионная кислота [218])
В реакциях, где заряженные частицы образуются из нейтральных,
диэлектрическая среда способствует образованию и разделению ионов.
Растворители с низкой диэлектрической проницаемостью препятствуют обра-
32
Глава 2
Таблица 5
Реакция В этиловом спирте (е = 25) В воде (е = 80) Отношение
1 0,000015 2,7 180000
2 0,015 0,0095 0,6
зованию ионов и способствуют образованию ионных пар и ионных агрегатов:
например, уксусная кислота, ацетонитрил, нитрометан, бензол и т. д.
Если в прямой и обратной реакциях участвует одно и то же число одина-
ково заряженных ионов, влияние
диэлектрической проницаемости среды
меньше. Это можно проиллюстрировать
на примерах взаимодействия (1) бен-
зойной кислоты и анилина и (2) бензой-
ной кислоты и соли о-нитробензойной
кислоты в воде и этиловом спирте (см.
стр. 205 в работе [380]).
1. CeH5COOH + CeH5NHa
CeH6COO- + CeH5NH+f
2. CeH6COOH4-OaNCeH4COO-
CeH5COO-+O2NCeH4COOH.
Рис. 5. Влияние диэлектрической
проницаемости растворителей на потен-
циал полунейтрализации (ППН) [168].
Значения констант равновесия при-
ведены в табл. 5.
При потенциометрическом титрова-
нии кислот потенциал полунейтрали-
зации (ППН) примерно пропорциона-
лен константе ионизации кислот. Соот-
ношение между ППН, сольватирующей
способностью растворителя и диэлект-
рической проницаемостью (е) не явля-
ется однозначным. На рис. 5 графически
изображено изменение потенциала по-
лунейтрализации иона пиперидиния,
уксусной кислоты и бисульфат-аниона
как функции величины 1/е: влияние диэлектрической проницаемости раст-
ворителя является наименьшим в случае катионных кислот.
По утверждению Вольфа [882], при уменьшении диэлектрической проницаемости
растворителя относительная сила незаряженных кислот остается постоянной, для поло-
жительно заряженных — возрастает и, наконец, для отрицательно заряженных кислот
уменьшается.
15, ДЕЙСТВИЕ ДИФФЕРЕНЦИРУЮЩИХ И НЕ АНАЛОГИЧНЫХ ВОДЕ!
РАСТВОРИТЕЛЕЙ НА ИОНИЗАЦИЮ И СОЛЬВАТАЦИЮ КИСЛОТ
Влияние растворителей с дифференцирующими свойствами часто не похо-
же на сольватирующее действие растворителей, аналогичных воде. Это под-
тверждается, например, существенными различиями между силами кислот,
обладающих близкими константами диссоциации, и, кроме того, тем,
что анионы, образующиеся в процессе прототропных реакций, не соль-
ватируются, а образуют ионные пары и большие агрегаты ионов; могут
наблюдаться также и другие специфические взаимодействия, даже не под-
дающиеся детальному определению.
Сила кислот
33
а. Ионизация кислот в ацетонитриле [424]. Диэлектрическая прони-
цаемость ацетонитрила GH3GN довольно высока, даже выше диэлектрической
проницаемости метилового спирта (табл. 34). Как амфотерный растворитель
он обладает, подобно ацетону, очень слабыми кислотными и основными свой-
ствами. Практически, однако, он может быть отнесен к инертным раствори-
телям. Полярографическое изучение инертных растворителей обнаруживает
их дифференцирующие свойства [138, 467].
Галогеноводородные кислоты ионизируются в ацетонитриле двумя путя-
ми: с образованием соответственно ионов типа нитрилия или имино. Хло-
ристый водород диссоциирует по первому типу
СН3С -= N + HC1 СН3С= N-HC1 Xt СН3С = NHC1 СН3С = N11++ С+ольв (15.1а)
С1сольв + НС1 > С1(НС1)сольв (15.16)
Благодаря повышенной поляризуемости бромистого и йодистого водорода
эти кислоты ионизируются в ацетонитриле с образованием иминоионов
_4- — ___________> ц- —
СН3С = N4-HBr СН3С = NHBr <— CH3C = NH.Br, (15.2а)
Ч- —> + —> + _
CH3C = NHBr-4-HBr CH3C = NH2Br- СН3С = МН24-ВгСольв (15.26)
Вг Вт
О гетеросопряжении и гомосопряжении в ацетонитриле см. [446]. Гетеросопряжение
определяется как сопряжение аниона А~ с донором водородной связи HR
Л-4-nHR Л(НИ)п,
а гомосопряжение как A~-i-nHA х > Л(НЛ)л.
Для ацетонитрила характерно, что его протонакцепторное действие
по отношению к кислотам и их аналогам проявляется постепенно и является
функцией времени. Это не мгновенный процесс, как в случае метилового
спирта (относительно кетонов, пропионитрила и бензонитрила см. [598]).
Вследствие слабой полярности группы СН3 для достижения ионизационного
равновесия пикриновой кислоты требуются недели в отличие от растворов
в ацетофеноне и бензонитриле, где благодаря л-электронной системе арома-
тического ядра этот процесс происходит значительно быстрее.
На сольватирующую способность растворителей и их способность сти-
мулировать ионизацию оказывает влияние электрофильный характер ради-
калов, связанных с полярной группой молекулы растворителя. G увеличе-
нием электроноакцепторного характера этих радикалов (R) электронодо-
норные свойства полярной группы молекулы растворителя по отношению
к растворенной кислоте уменьшаются, т. е. стабильность ассоциата протон —
растворитель ослабевает, что приводит к уменьшению ионизации (табл. 6)
[281, 397, 494, 598].
В табл. 6 показано изменение силы пикриновой кислоты в зависимости
от дифференцирующей способности растворителя.
Таблица 6
Растворитель е (20°) Электроноакцепторная способность радикалов, связанных соответственно с группами СОСНз и CN [281, 397, 494] Константы ионизации пикриновой кислоты К- 10“»
Ацетон 21,5 -1,4 1,38
Метил этплкетон 17,8 -1,45 1,98
Ацетофенон 18,1 +4,2 0,15
Проппонптрпл 27,0 -1,45 4,25
Бензонитрил 26,5 +4,2 1,02
34
Глава 2
Дулова с сотрудниками исследовала силу различных кислот в ацетофеноне, цикло-
гексаноне и циклогексаноле. Эти растворители обладают в определенной степени диф-
ференцирующими свойствами; так, разность в значениях рА? для дихлоруксусной и три-
хлоруксусной кислот в воде составляет всего 0,4 единицы, однако в перечисленных выше
растворителях она в 3 раза больше (табл. 7) [194, 195, 196].
Таблица 7
Кислота рК (Н2О) д рК (ацето- фенон) д рК (цик- логек- санон) д
Дихлоруксусная 1,30 0,4 7,41 1,15 6,86 1,19
Трихлоруксусная 0,9 6,26 5,66
б. Ионизация серной кислоты в нитрометане. Специфическое действие
дифференцирующих растворителей можно хорошо показать также на при-
мере нитрометана. Хотя диэлектрическая проницаемость нитрометана не
отличается существенно от диэлектрической проницаемости метилового спир-
та, он не сольватирует кислоты. В нитрометане серная кислота (ЛН) с инди-
каторными основаниями типа нитроанилина (В) вступает в следующие
реакции:
1. При низкой концентрации
ЛЦ-р? ~ ВН+.Л-.
2. При концентрациях выше 0,1 М
ЗЛН + В В11+-Л(ЛН)7.
Свободный ион ВН+ и ионные пары ВН+-Л" и ВН+-Д(ЛН)2 обнаружи-
вают одинаковую незначительную способность к абсорбции [520]. Анион,
образующийся в процессе протонного переноса, должен быть «сольватиро-
ван» молекулой кислоты, а не молекулой растворителя, так как нитрометан
не обладает сольватирующей способностью.
В прототропных реакциях с участием растворителей, подобных воде,
преобладает сольватация аниона, например за счет образования водородной
связи между подвижным водородом растворителя и свободной электронной
парой аниона. Таким образом, отсутствие стремления образовывать водо-
родную связь является одной из причин, не говоря о других, обусловли-
вающих это свойство нитрометана, которое отличает его от воды.
в. Сольватация аминокислот в хлороформе и четыреххлористом углероде
[34]. Аминокислоты сольватируются в воде как цвиттер-ионы (амфотерные
ионы). В растворителе с низкой диэлектрической проницаемостью сольвата-
ция амино- и карбоксильной групп может происходить с протонным перено-
сом или без него. В концентрированном хлороформном растворе Х,Х-диэтил-
глицин присутствует в виде димерного цвиттер-иона (I), а в более разбавлен-
ном растворе СНС13 или в четыреххлористом углероде — в виде мономера,
имеющего внутреннюю водородную связь (II).
О.
С —О
н2с
(c2h5)2n—н
н —N (С2Н5)2
сн2
О —с
н5с2Х хс2н5
/N''
Н2С '*н
<U__I
I
II
Сила кислот
35
г. Взаимодействиетрихлоруксуснойкислоты и пиридина в хлороформе [33].
В процессе кислотно-основного взаимодействия в апротонном растворителе
ионные пары связаны водородной связью. В этом случае перенос протона
происходит в направлении основного атома азота и водородная связь с анио-
ном носит ярко выраженный электростатический характер
Тип ассоциации в этом случае следующий:
ВН+-Л(ЛН)-
(ср. разд, б и гл. 3, разд. 23).
д. Ассоциация бензоат-аниона в бензоле [174]. В зависимости от природы
основания бензоат-анион ведет себя по разному
о о-...нМс2н5)3
С
I
СвН5
н н
I I
С6Н5- N N-C6H5
С
H-N+-II
С триэтиламином
О о-
X Z
С
I
С6Н3
С 1,3-дифенилгуанидином
Три атома азота 1,3-дифенилгуанидиний-иона не эквивалентны. Конце-
вые атомы азота находятся предположительно в резонансе по типу анилина
и, таким образом, отличаются от резонанса соединений типа гуанидина. Весь-
ма вероятно, что положительный заряд несет группа = NH2 и, следовательно,
оба атома водорода взаимодействуют с двумя атомами кислорода бензоат-
аниона.
В бензоле бензойная кислота является более слабой кислотой по отноше-
нию к триэти ламину, чем по отношению к дифенил гуанидину. Оба атома
кислорода карбоксилат-аниона связаны с водородом и, таким образом, уве-
личивают силу кислоты. Относительная сила бензойной кислоты, следова-
тельно, также зависит от участвующего в реакции основания, хотя в этом
случае оба основания сравнимы по силе: рКъ для триэтиламина и 1,3-дифе-
нилгуанидина равны 3,26 и 4,00 соответственно.
16. ОПРЕДЕЛЕНИЕ СИЛЫ КИСЛОТ И «КИСЛОТНОСТИ» РАСТВОРОВ
Сила кислот в данном растворителе может быть определена путем изме-
рения протонной активности электрометрическим или индикаторным мето-
дом. Однако необходимо делать различие между протонной активностью
раствора и активностью его водородного иона [39, 40, 728]. Последний,
в частности, показывает в каждом растворителе, способном присоединять
протон, различную активность, зависящую от природы растворителя, соль-
36
Глава 2
ватирующего протон. В воде образуются ионы гидроксония, в уксусной
кислоте — ацилониевые ионы, в пиридине — ионы пиридиния. По протон-
ной активности может быть оценена способность растворенной кислоты пере-
давать свой протон молекуле растворителя. Однако протонная активность
сольватированного протона может отличаться от протонной активности
растворенной кислоты. Это тесно связано с основностью растворителя.
Ганч [351] считал, что константа диссоциации, измеренная в воде, не может
быть точной мерой силы кислот; сила кислоты и электролитическая диссо-
циация не всегда изменяются параллельно. Спектры поглощения в ультра-
фиолетовой области для ряда кислот в диэтиловом эфире (в этом случае кисло-
ты проявляют себя как слабые кислоты) отличаются от спектров в раствори-
телях, в которых они ведут себя как сильные кислоты. В первом случае
спектр поглощения подобен спектру эфира кислоты (псевдокислотная форма),
в то время как во втором — спектру соли кислоты (кислотная форма). Соглас-
но Брендстеду, в этом случае могут наблюдаться спектры сопряженного
кислоте основания или сопряженной кислотной формы
{RCOO}H
a^u-Форма
Псевдокислотная
форма
Сопряженное кислоте
основание
а. Индикаторный метод. Константа равновесия ассоциации замещенных
бензойных кислот с 1.3-дифенилгуанидином. Индикаторы можно рассматри-
вать как кислотно-основные системы; спектры поглощения в видимой обла-
сти обоих компонентов этих систем различны. В водной среде, так же как
и в неводной (способной к прототропной реакции), активность протона может
быть определена, если известно, например в результате спектрофотометри-
ческих измерений, отношение кислотной и основной форм индикатора.
J + H+ ZH+*
Константа кислотности индикатора определяется уравнением
[Н+] ГЩ1 ____ К* КН+]
ЛКИСЛ — JZH + ] И П J “ Лкисл jyj
Значение Х^кисл должно быть определено другим методом, после чего актив-
ность протона [Н+] может быть вычислена как результат спектрофотометри-
ческого измерения отношения /Н+/1. В неводной среде, однако, значение
Ккнся индикатора зависит от природы растворителя и, кроме того, требуется,
чтобы отношение /Н+/1 примерно равнялось единице.
Последовательность, в которой изменяется сила различных кислот, была изучена
Ганчем. Он добавлял определенные индикаторы в растворы кислот в хлороформе и опре-
делял максимальное разбавление, при котором происходило резкое изменение окраски
индикатора и его превращение в кислотную форму. Таким образом может быть уста-
новлена приведенная ниже последовательность:
НСЮ4 > С10Ни8О3Н > НС1 > СС13СООН > СН2С1СООН >
> НСООН > СН3СООН > С2Н5СООН > С8Н7СООН.
Даже сильные кислоты ионизируются в хлороформе и бензоле только в незначитель-
ной степени. Однако опыт органической химии показывает, что сильные кислоты в таких
растворах могут проявлять сильные кислотные свойства. Кислоты Льюиса в органиче-
ских растворителях могут быть почти так же эффективны, как Н-кислоты. Таким обра-
зом, хотя протонная активность отсутствует, взаимодействие веществ может рассматри-
ваться как следствие сродства кислоты к свободной электронной паре.
Тремийон и сотр. [499, 819] измерили относительную силу различных кислот и осно-
ваний в уксусной кислоте. Их метод является в основном индикаторно-спектрофото-
метрическим. Последовательность относительной силы кислот и оснований в уксусной
кислоте см. разд. .6, стр. 41.
Сила кислот
37
Дэвис и Хетцер [173] определили относительную силу моно-, ди- и три-
замещенных производных бензойной кислоты в бензоле. В неполярной среде
относительная сила кислот может быть определена только при добавлении
выбранного для сравнения основания. Дэвис и Хетцер в этих целях исполь-
зовали 1,3-дифенилгуанидин, кислотным индикатором служил этиловый
эфир тетрабромфенолфталеина. Исследовавшиеся кислоты и индикаторная
кислота реагировали с основанием сравнения по следующей схеме:
В /Н 7 У ВШ (16.1)
или
2? + ЛН Т* ВИА, (16.2)
где В — дифенилгуанидин, /Н — этиловый эфир тетрабромфенолфталеина,
ЛН — замещенная бензойная кислота.
Таким образом, этим методом определяется константа равновесия кислот-
но-основной ассоциации
Л'асс = |Д| |/Н) (16.3а)
<16-3б>
ВШ или ВНА могут рассматриваться как соли с водородной связью, обла-
дающие свойствами ионной пары [172].
При исследовании кислотно-основных равновесий в неполярных растворителях
необходимо принимать во внимание способность веществ к образованию ассоциатов
различного характера, особенно при высоких концентрациях веществ. Измерения пони-
жения температуры замерзания указывают, например, на возникновение межмолеку-
лярной ассоциации дифенилгуанидина за счет образования Н-связи; при концентрации
5-10-5 моль/л этот тип межмолекулярной ассоциации практически отсутствует. Наряду
с этим хорошо известно, что карбоновые кислоты ассоциируют с образованием димеров,
которые находятся в равновесии с мономерной формой. Эксперименты Дэвиса и Хетцера
кратко описаны ниже.
Концентрации реагентов в бензольном растворе следующие: этиловый эфир тетра-
бромфенолфталеина Са, = 5-10-5 молъ/л, дифенилгуанидин Съ — пСа, (где п = 0,5—4),
замещенная бензойная кислота Са„ = п"Са, (где п " = 0,5—0,6). Для того чтобы свести
к минимуму попадание влаги из воздуха, приготовление растворов и т. д. осуществля-
лось в сушильных камерах.
Максимум поглощения комплекса дифенилгуанидина с индикатором наблюдался при
540 нм. При добавлении различных замещенных бензойных кислот светопоглощенпе
уменьшается вследствие конкурентного взаимодействия кислот с дифенилгуанпдиновой
частью (В) комплекса IH.B. Использовали кварцевый спектрофотометр типа Beckman DU,
кюветы с толщиной слоя 2,5, 5, 10 и 20 мм, подобранные таким образом, чтобы светопро-
пускание было в пределах 20—70%. Температуру поддерживали равной 25,0 ±0.1°.
Для расчета констант ассоциации можно использовать следующую формулу:
К" - Х~У
асе- у -[Са„ — (х — у)] ’
причем
х — (nL — Z)
п
где Z — поглощение раствора при 540 нм в кювете с толщиной поглощающего слоя 5 мм,
L — экспериментально установленное предельное поглощение комплекса IHB, равное
1,017 при использовании кюветы с толщиной поглощающего слоя 5 мм и при концентра-
ции 5-Ю-5 молъ/л', значение Касс (ВШ) при 25° составляет 2,5-Ю6.
В табл. 8 приведены некоторые экспериментальные данные, полученные для .и-питро-
бензойной кислоты.
Этими экспериментами было показано, что реакционная способность
кислот в бензоле, выраженная через константу равновесия их реакции ассо-
38
Глава 2
Таблица 8
С „ а п Z Касс
5,015-10-5 1,0 0,1581 6,65-Юв
10,140-10-5 2,0 0,2124 6,64-106
20,047-10-5 4,0 0,3072 6,25-10е
циации с соответствующим основанием сравнения, может быть связана с их
реакционной способностью в водных средах. Это значит, что если константа
ионной диссоциации в воде выражается значением рК (—log ЛдИСс), то
константа ассоциации (/Гасс) показывает обратную зависимость.
Рис. 6. Зависимость между рЛГа замещенных бензойных кислот и их константами
равновесия ассоциации (logK"cc), измеренными в бензоле [173].
1 — 3t-NH2; 2 — СНЭ; 3 — бензойная кислота; 4 — jh-OCH3; 5 — ОН; 6 — F; 7 — J; 8 — С1; 9 — Вг
10 — CN; 11 — NO,; 12 — n-NH,; 13 — ОН; 14 — ОСН,; 15 — СНЭ; 16 — F; 17 — С1; 18 — I; 19 —
Вг; 20 — CN; 21 — NO,; 22 — o-NH,; 23 — СН3; 24 — ОН.
Так, значение ЛдИСС бензойной кислоты равно 6,30-10~5 (рК = 4,20), а значение
ее Касс с дифенилгуанидином составляет 182 000 (log КЛСС — 5,26). Чем сильнее кислота
в воде, тем меньше ее значение рК и чем сильнее та же кислота в бензоле, тем больше
для нее значение рЛГасс (см. гл. 4, разд. 25).
В водной среде кислотно-основная реакция сопровождается вторичными
процессами: ионизацией, сольватацией и т. д. В инертных растворителях
проявляются различные типы водородной связи. По этой же причине обна-
руживаются отклонения от линейной зависимости между двумя константами
равновесия. Для объяснения этих отклонений необходимо экспериментально
исследовать влияние положения и действия заместителей в бензойной
кислоте.
Последовательность изменения относительной силы .м-замещенных бен-
зойных кислот (за исключением .м-аминобензойной) одинакова в воде, мети-
ловом, этиловом, н-пропиловом, н-бутиловом спиртах, этиленгликоле и бен-
золе. В случае орто- и пара-замещенных бензойных кислот индуктивное
действие заместителей сильно зависит от резонансных эффектов, стерических
факторов и возможного образования водородных связей. На рис. 6 пред-
ставлена зависимость силы .м-замещенных бензойных кислот от рК в воде;
Сила кислот
39
жирная линия дает ясное представление о «стандартном» поведении этих
кислот. Кислоты, находящиеся в области над этой линией, оказываются
более сильными в воде и более слабыми в бензоле.
.и-Ампнобензойная кислота попадает в область ниже этой линии, поскольку кон-
станта ее ионизации в воде не отражает точно ее силы (рис. 6,7). Аминобензойные кисло-
ты имеют по две константы ионизации (табл. 9) [456].
Таблица 9
Положение заместителя Потенциометрический метоп. Спектрофотометрический метод
РК1 рК2 рК1 рК2
орто 2,05 4,95 2,14 4,80
мета 3,07 4,75
пара 2,38 4,89 2,29 4,86
Индуктивное действие аминогруппы и ее резонанс с бензольным кольцом зависят
от взаимодействия аминогруппы с молекулами воды. В воде л-аминобензойная кислота
присутствует в впде цвиттер-иона [142].
Благодаря образованию цвиттер-иона ле-аминобензойная кислота не растворяется
в некоторых инертных растворителях, например в o-нитротолуоле, хлор- и бромбензо-
лах [595]. Однако, будучи растворенной, например в пиридине, ацетонитриле, пзобутил-
метплкетоне, 2-нитропропане, нитробезоле или диметил формамиде, она ведет себя в этих
же растворителях как кислота более сильная, чем другие кислоты. Ионизация л-амино-
бензойной кислоты может быть представлена следующей реакцией:
4' —х-
•M-H3NCeH4COO- + s _.4-H2NCeH4COO--^sH+,
а ионизация n-аминобензойной кислоты
n-H2NCeH4COOH + $ n-H2NC6H4COO--L sH+.
Таким образом, ионизации лста-замещенной кислоты предшествует разделение
зарядов в цвиттер-ионе. Влияние диэлектрической проницаемости растворителя на силу
л-аминобензойной кислоты меньше, чем в случае n-аминобензойной кислоты (см. гл. 2,
разд. 2 и рис. 5). Более того, с уменьшением диэлектрической проницаемости растворителя
относительная сила .и-аминобензойной кислоты увеличивается по сравнению с другими
кислотами, поскольку внутримолекулярная «ионизация» уже произошла, в то время
как в других кислотах ионизация является результатом межмолекулярного взаимо-
действия между растворенными кислотами и молекулами растворителя. Увеличение
диэлектрической проницаемости растворителя может привести к увеличению межмоле-
кулярного взаимодействия, а ее понижение — к уменьшению этого взаимодействия.
Однако п-ампнобензойная кислота может вступать и в другие весьма характерные
для нее взаимодействия, например с нитробензолом (см. гл. 7, разд. 44).
В отличие от ле/па-замещенных бензойных кислот зависимость между
значениями рК и log Kacc пара- и opmo-замещенных кислот отличается от
«стандартного» вида этой зависимости.
В случае n-галогенбензойных кислот это отличие невелико. Производные
бензойной кислоты с электронодонорными заместителями (ОН, ОСН3, NH2)
являются относительно более сильными в бензоле, чем в воде, в то время
как пара-замещенные бензойные кислоты, содержащие группы GN и NO2,
слабее в бензоле, чем в воде.
Согласно Дэвису и Хетцеру, способность групп CN и NO2 координиро-
ваться водой увеличивает их влияние на силу кислот вследствие усиления
резонанса с кольцом. Однако гидратация групп —ОН, —ОСН3 и —NH2
уменьшает электронодонорное действие, т. е. уменьшает их способность
понижать силу кислот из-за уменьшения резонанса с кольцом (см. гл. 4,
разд. 27). Значения рК для пара-замещенных бензойных кислот в воде вполне
соответствуют существующей в настоящее время неопределенности в отно-
шении оценки силы этих кислот.
40
Г л а в а 2
Хорошо известно, что влияние заместителей, подобных группе СООН,
в общем не может быть предсказано. На рис. 6 пунктирная линия показы-
вает, что opmo-замещенные бензойные кислоты (за исключением о-амино-
бензойной кислоты) попадают в область выше «стандартной» линии, т. е.
они являются сильными кислотами в воде и слабыми в бензоле.
Пунктирная линия на рис. 6, иллюстрируя связь между значениями рА?
в воде и Касс в бензоле, может дать лишь приближенное представление
о последовательности, в которой происходит изменение силы орто-замещен-
ных бензойных кислот.
о-Метоксибензойная кислота — наиболее слабая в бензоле (она даже
не представлена на рис. 6): рК (в воде) = 4,09 и log Касс (в бензоле) = 3,7.
Ее ассоциации с дифенилгуанидином в бензоле мешает стабильность хелат-
ной структуры, которая образуется, когда протон карбоксильной группы
удерживается силовым полем атома кислорода метоксигруппы (Н-связь).
В воде, однако, образование водородной связи с растворителем мешает обра-
зованию хелатной структуры и видоизменяет индуктивное влияние метокси-
группы на карбоксильную группу, так же как и ее резонанс с бензольным
кольцом. Гидратация могла бы усилить стерическое требование метоксигруп-
пы до такой степени, что это привело бы к пространственному торможению,
связанному с нарушением преимущественно копланарного расположения
карбоксильной группы и ароматического ядра.
о-Оксибензойная кислота (салициловая кислота) так близка к «стандартно-
му» соотношению pAVlog Касс, соответствующему жирной линии на рис. 6,
что можно сделать заключение, что салицилат-анион присутствует в основном
в той хелатной форме, которой объясняется повышенная кислотность в вод-
ном растворе.
Поведение о-метилбензойной кислоты (о-толуиловой кислоты) в бензоле
также представляет интерес. В бензоле метильная группа стерически не
препятствует резонансу карбоксильной группы с кольцом. Однако в водном
растворе концентрация диполей воды вокруг карбоксильной группы стери-
чески затрудняет резонанс карбоксильной группы о-толуиловой кислоты
с кольцом. Гидратация в водной среде карбоксильной группы в орто-заме-
щенных бензойных кислотах с электронодонорными заместителями приводит
обычно к стерическому ингибированию резонанса, который в противном слу-
чае проявился бы в уменьшении силы кислоты.
Таким образом, на основании вышеизложенного можно сказать, что поря-
док изменения относительной силы бензойных кислот, содержащих замести-
тели в лгета-положении, в бензоле такой же, как и в растворителях, способ-
ных к автопротолизу.
Что же касается бензойных кислот, содержащих заместители в napa-
положении, то сольватация уменьшает или увеличивает их кислотность,
поэтому последовательность изменения их относительной силы в бензоле
отражает присущую им кислотность более точно, чем значение рК в воде.
При замене растворителя, даже при замене воды на растворитель, «подоб-
ный воде», последовательность изменения относительной силы бензойных
кислот, содержащих заместители в орто-положении, может измениться на
обратную. Карбоксильная группа в орто-положении увеличивает силу
кислот в водной среде, так как она сольватируется. Фталевая кислота,
будучи довольно сильной в воде, обладает «стандартной» кислотностью
в бензоле.
Образование внутримолекулярной водородной связи уменьшает силу
opmo-замещенных бензойных кислот в бензоле (например, о-метоксибензой-
ной кислоты). Возможность возникновения хелатной структуры уменьшается
при образовании Н-связи между растворенным веществом и растворителем.
Большая сила о-нитробензойных кислот в воде может быть частично
объяснена гидратацией.
Сила кислот
41
Из вышесказанного становится очевидным, что сила кислот, измеренная
в воде, не дает точного представления о силе этих кислот. В прототропных
растворителях протон существует только в сольватированной форме. Однако
вода способна сольватировать не только протон, но также и карбоксилат-
анион бензойной кислоты, а также, возможно, и другие ее заместители,
и, таким образом, электростатический, резонансный и стерический эффекты
тоже претерпевают изменения. Эти исследования, а также тщательные иссле-
дования Измайлова по изменению силы кислоты в зависимости от природы
растворителя [412] показывают, что не может существовать универсальной
шкалы силы кислот независимо от растворителя. В инертном растворителе
структура основания сравнения тоже оказывает влияние на силу ислот
[174]. Однако на практике с учетом структурных и сольватирующих факторов
могут быть построены пригодные для применения шкалы кислотности.
б. Порядок изменения относительной силы кислот и оснований в уксусной
кислоте. Согласно данным Кольтгофа и сотрудников, в растворителе с низкой
диэлектрической проницаемостью, например в уксусной кислоте, разделение
ионизирующихся соединений на ионы не имеет большого значения. Степень
кислотно-основного взаимодействия может быть выражена величинами
кислотности и основности в соответствующих шкалах. Протонакцепторные
основания могут быть разделены на классы по их взаимодействию с одной
и той же кислотой, например хлорной. По аналогии с рК может быть построе-
на шкала рнсю4- Те же основания с хлористоводородной кислотой дают дру-
гую шкалу — шкалу рнш [819].
Сказанное выше относится также к прототропным реакциям кислот
с основаниями. Для того чтобы показать степень протонного переноса между
водородсодержащими кислотами и, например, дифенилгуанидином (ДФГ)
может быть построена шкала кислотной силы Рдфг- Число шкал основности
р.иь которые можно построить, будет соответствовать числу выбранных
кислот сравнения. Верно также и обратное: каждое основание сравнения
позволяет построить новую шкалу кислотности. Следовательно, одна шкала
(шкала pH) не является универсальной.
В этом смысле кислотно-основная теория Бренстеда
Кислота ' У Основание—Н+
может быть распространена на прототропные реакции в растворителях с низ-
кой диэлектрической проницаемостью. Обычный процесс протонного пере-
носа изменяется, однако, под влиянием следующего важного фактора:
в растворителях с низкой диэлектрической проницаемостью ионы могут
существовать только в небольших количествах, так как в растворе присут-
ствуют в основном незаряженные частицы, ассоциаты и ионные пары.
В уксусной кислоте константа диссоциации солей составляет приблизи-
тельно 10-6 или даже несколько меньше [102, 412]. Константа диссоциации
хлорной кислоты равна 10-4-85 [102]. За некоторым исключением процессы
ионизации могут сравниваться только для растворителей, диэлектрическая
проницаемость которых превышает 40.
Среди различных типов переноса протона наиболее важным является
случай, когда молекула кислоты ЛН (хлорная, п-толуолсульфокислота,
хлористоводородная и т. д.) одновременно с протонным переносом ассоцииру-
ет с некоторым основанием В (амины, гетероциклические основания, амиды
кислот) (см. гл. 2, разд. 11, в и работы [294, 887])
ЛН+В-МЫ; *=М,
и А~ (анионное основание) и /?Н+ (ониевая кислота) образуют ионную пару,
поскольку растворитель с низкой диэлектрической проницаемостью не спо-
1,4 -Диоксан —————г
Метиловый спирт______
Этиловый спирт--------
Н20---------------
Салициламид 1
Бензамид —
Ацетанилид------{
Нитроанилин------г
Ацетамид---------г
2,4-Динитро-К,И-диэтил^~
анилин
Р НС104
---Ацетонитрил
---2,6 -Дихлор -6- нитроанилин
---п - Толуолсульфамид
' —Изопропиловый спирт
.4- Хлор-2-Humpo-N-метиланилин
2,6-Диметил-ос-пирон'
Тиомочевина
Кофеин
Антипирин
Ацетат серебра
Ацетат натрия
Эфедрин ———
Ацетат тетраэтил-
аммония, Н,И-диэтил-
анилин (ацетат),три-
бензиламин (ацетат)
п-Хлор-о-нитроанилин
— Родамин В
— Виктория голубая
2,6 -Дихлор -4- нитро -N.N- диметиланилин
у^п-Розанилин
Мочевина
\\ п-Нафтолбензеин
Малахитовый зеленый
^Кристаллический фиолетовый
M-Humpo-N,N- диметиланилин
^''-Хинальдин красный
---Пи нациа нол
---Метиловый желтый
---Этиловый красный
—10 —
— 1
—Z —
—3—
— 4—
---6 —
— 7 —
— 8 —
— 9 —
Р и с. 7. Шкала основности Рнсю< [819] (см. также [101, 102, 388—390, 463, 499, 505
763].
—10 —
— 9 —
---Хлорная кислота
— 8—
---Хлористоводородная
кислота
---7 —
— 6 —
Бромтимоловый
синий —______
Бромкрезоловый__
пурпурный
___Роданистоводородная
кислота
---Ортофосфорная
кислота
___Бромфеноловый
синий
4—
--3—
Тимоловый
синий
---Трихлоруксусная
кислота
—2 —
----1 —
___Дихлоруксусная
кислота
___Монохлоруксусная
кислота
Салициловая кислота
-Диэтил анилин
Рис. 8. Шкала кислотности Рм>к-дИэТИЛанилин I819! (см- также [102, 499]).
Сила кислот
43
собен полностью разделить заряженные ионы. Константа ионизации пер-
хлората пиридина равна лишь 10-6-1.
Основания в зависимости от их силы растворяются в уксусной кислоте
либо давая ацетаты, либо в молекулярной форме. Пиридин, являющийся
в уксусной кислоте сильным основанием, на 80% присутствует в виде
(С5Н5]>Ш+)СН3СОО_ и на 20% в молекулярной форме. Метиловый желтый
растворяется на 10% в виде ацетата и на 90% в молекулярной форме. Соот-
ветствующим образом мочевина, как слабое основание, не образует ацетата
в уксусной кислоте.
Сказанное выше применимо и к кислотам. Хлорная кислота в уксусной
кислоте находится в виде перхлората ацилония (СН3СООН2)ЧС1О7, в то вре-
мя как трихлоруксусная кислота присутствует в растворе в молекулярной
форме.
При сравнении взаимодействия различных оснований с одной и той же
кислотой, например хлорной, можно видеть, что константа ассоциации К
и рХ характеризуют степень этого взаимодействия, в результате которого
образуется комплекс ВНС1О4
к = [ВНС1О4] 1НС1°41
Значения рХ различных оснований по отношению к хлорной кислоте
могут быть представлены общей шкалой.
Это рнс1о4-шкала основности, где [НС1О4] = 1 и рнсю4 = 0- Такая
шкала основности может быть также построена для хлористоводородной
и других кислот [819] (рис. 7).
Сравнивая значение рК различных кислот по отношению к Х,Х-диэтил-
анилину можно построить подобную, хотя и обратную шкалу; это
Рм,м-диэтиланилин_шкала [102, 499, 819] (рис. 8).
При сопоставлении различных рдн-шкал основности можно видеть, что
положение или последовательность сильно отличающихя друг от друга
оснований, которые сравниваются, хорошо совпадают с относительным
положением Н-кислот в рв-шкалах кислотности [819]. Из этого следует,
что различные рдн- и рв-шкалы могут быть объединены в одну общую «шка-
лу кислотности», действительную для среды уксусной кислоты, причем
сила кислот возрастает при движении вверх, а оснований — при движении
вниз по- этой шкале. Интервал изменения рК составляет примерно 9,6 еди-
ницы (рис. 9).
Практическая ценность этой шкалы заключается в том, что она наглядно
показывает, что любая кислота способна вступать в реакцию с основанием,
расположенным на более низком уровне шкалы, и любое основание спо-
собно вступать в прототропную реакцию с кислотой, расположенной на более
высоком уровне шкалы.
Построение кислотно-основных шкал и их применение описано в работах [499, 819].
Измерение проводилось индикаторно-спектрофотометрическим методом Гаммета — Дей-
рапа [342, 343].
в. Связь между потенциалом полунейтрализации и значением рКа.
Стреули [780, 782] использовал для определения значений р7Га замещенной
бензойной кислоты и производных фенола электрометрический метод. Кис-
лотные свойства замещенных бензойных кислот и фенолов в пиридине могут
быть определены по значениям p7fa в воде, причем во внимание принимается
молекулярная структура: соединения с одинаковым молекулярным строе-
нием должны вести себя при потенциометрическом титровании одинаково.
Это справедливо, в частности, в отношении связи между потенциалом полу-
нейтрализации и р7Га.
Кислотность фенолов в пиридине возрастает до относительно более высо-
кого уровня, чем кислотность карбоновых кислот. Различия между потен-
44
Глава 2
циалами полунейтрализации (ППН) фенолов и кислот могут быть измерены
аналогичным методом. Подходящей парой электродов является пара стек-
лянный электрод и метанол-каломельный электрод (см. гл. 12, разд. 67).
Кислоты
Индикаторы --------------
Хлорная
Бромистоводородная
Серная
п-Толуолсулыроновая
Хлористоводородная
Основания
Ацетонитрил
1,4-Диоксан
Метиловый спирт
п-Толуолсулъфамид
Этиловыи спирт
Изопропиловый спирт
Н20
Индикаторы
2,6 -Дихлор -6-нитроанилин
Роданистоводородная
Бромтимоловый Ортофосфорная
синий
Бромкрезоловый
пурпурный
Бромфеноловый синий
Тимоловый синий Трихлоруксусная
Дихлоруксусная
2,4 -Динитрофенол
Монохлоруксусная
Салициловая
Щавелевая
Ацетанилид
Ацетамид
Мочевина
Тиомочевина
Кофеин
Антипирин
Эфедрин
Судан III
п -Хлор -о~ нитроснилин
Родамин В
о-Нитроанилин
Виктория голубая
п-Розанилин
п -Нафтолбен зеин
п-Нитро-Н,Н-диметиланилин
п-Нитроанилин
Малахитовый зеленый
Кристаллический фиолетовый
м-Нитро№,Бдиметиланилин
Хинолиновый красный
м-Нитро-^Мдиэтиланилин
Пинацианол
Метиловый желтый
Этиловый красный
Пиридин
Uji-Диэтиланилин
Трибензиламин
Рис. 9.
Шкала основности в уксусной кислоте [819].
Рис. 10. Зависимость между ППН заме-
щенных бензойных кислот и фенолов и их
значениями p/G, измеренными в воде [595,
780, 782].
7 — ортпо-замещенные бензойные кислоты (в пири-
дине); 2 — мета- и пара-замещенные бензойные
кислоты (в пиридине); 3 — замещенные фенолы
(в пиридине);---замещенные фенолы (в метил-
изобутилкетоне).
Титрантом служит 0,1 н. гидроокись тетрабутиламмония (см. гл. 10, разд. 57).
Для обеих групп соединений нулевым значением ППН является значение
ППН в милливольтах для бензой-
ной кислоты, измеренное в тот же
день и в тех же условиях. Это значе-
ние вычитается со своим знаком из
значения ППН в милливольтах кис-
лоты или фенола, для которых прово-
дятся измерения. Если значение ППН
исследуемой кислоты или фенола
меньше, чем значение ППН бензой-
ной кислоты, т. е. ППН имеет отри-
цательное значение, то исследуемые
вещества являются более сильными,
чем бензойная кислота.
На рис. 10 представлена зависи-
мость рАГ1 в воде от ДППН в пи-
ридине (2) для мета- и пара-заме-
щенных бензойных кислот, за исклю-
чением .u-аминобензойной кислоты,
которая, как показывает инфракрас-
ный спектр, в кристаллическом со-
стоянии содержит группу NH3k. Сле-
довательно, в растворителе с низкой
кислота находится не в ионизирован-
диэлектрической проницаемостью эта
ной форме, а, по-видимому, в виде катионной кислоты, которая и титруется
[.w-H2NCeH4COO-]H+.
Сила кислот
45
Линия 1 на рис. 10 соответствует ортпо-замещенным бензойным кислотам.
Кислоты, расположенные на рис. 10 слева от линии, соответствующей
отношению кислотностей, измеренных в пиридине и воде, в пиридине являют-
ся более слабыми. Кислоты, расположенные справа от этой линии, будут
более сильными в пиридине, чем в воде. Большинство ортпо-замещенных
бензойных кислот оказываются в пиридине слабее, чем в воде, т. е. проис-
ходит ослабление ортпо-эффекта. Однако о-окси- и о-аминобензойная кислоты
Степень нейтрали-
зации, %
Гис. 11. Кривые потенциометри-
ческого титрования о-толуиловой (7),
бензойной (2) и п-нитробензойной
(5) кислот в пиридиновом растворе.
Титрант: 0,1 н. раствор гидроокиси
тетра бутиламмония.
Р и с. 12. Кривые потенциометриче-
ского титрования замещенных фено-
лов [780].
Растворитель: пиридин, титрант: 0,1 н.
раствор гидроокиси тетрабутиламмония.
1 — крезолы; 2 — фенол; 3 — хлорфено-
лы; 4 — нитрофенолы; 5 — бензойная
кислота.
в пиридине сильнее, чем в воде, возможно, потому, что они находятся в ста-
билизованной анионной или катионной форме.
Линия 3 на рис. 10 относится к замещенным фенолам.
Различие между фенолами и бензойными кислотами заключается в том,
что в пиридине на силу кислот определяющее влияние оказывают замести-
тели, в то время как для фенолов этого не наблюдается.
На рис. 11 изображены кривые потенциометрического титрования неко-
торых производных бензойных кислот, а на рис. 12 показаны те же кривые
для некоторых замещенных фенолов.
В табл. 10—12 обобщены данные о зависимости между рКа и АППН
некоторых хорошо изученных производных бензойной кислоты и производ-
ных фенола [780, 782].
Таблица 10
Значения рКа в воде и АППН в пиридине
для орто -замещенных бензойных кислот (ППНо = ППН
бензойной кислоты) [780, 782]
Кислота А ППН, мв рКа [189] Рассчитанное значение »Ка
Толуиловая +44 3,91 4,05
Метокспбензойная т-33 4,09 3,95
Хлорбензойная -65 2,94 2,96
Бромбензойная -77 2,86 2,84
Нптробензойная -142 2,17 2,19
46
Глава 2
Таблица 11
Значения рКа в воде и ЛППН в пиридине для мета- и пара-замещенных
бензойных кислот (ППН0 -ППН бензойной кислоты) |780, 782]
Кислота А ППН, мв РКа [189] Рассчи- танное значение ?Ка Кислота А ППН, мв Р^а [189] Рассчи- танное значение РКа
п-Аминобензой- ная + 105 4,92 4,82 п-Хлорбен- зойная -25 3,98 3,99
п-Оксибензой- ная +80 4,54 4,66 п-Бромбен- зойная -30 3,97 3,96
п-Метоксибен- зойная +45 4,47 4,44 .и-Хлорбен- зойная -49 3,83 3,84
ле-Оксибензой- ная +23 4,08 4,30 ле-Бромбен- зойная -63 3,81 3,75
.и-Толуиловая п-Толуиловая +16 +3 4,27 4,37 4,25 4,17 ле-Нитробен- зойная -97 3,49 3,53
ле-Метоксибен- зойная -5 4,09 4,20 п-Нитробен- зойная -НО 3,42 3,44
Таблица 12
Значения в воде и АППН в пиридине для замещенных фенолов
(ППН0 = ППН бензойной кислоты) [780, 782]
Фенол А ППН, мв РКа Рассчи- танное значение РКа Фенол А ППН, мв РКа Рассчи- танное значение РКа
п- Аминофенол +463 10,68 10,58 п-Хлорфенол -4-248 9,38 9,16
п-Метоксифенол +403 10,16 10,18 ле-Хлорфенол +220 9,02 8,97
о-Аминофенол +389 9,95 10,09 о-Хлорфенол +190 8,48 8,77
.м-Крезол +386 10,01 10,07 ле-Нитрофенол + 139 8,35 8,44
Фенол +340 9,95 9,77 2,4-Дихлорфенол +78 7,85 8,03
п-Крезол +387 10,17 10,07 о-Нитрофенол -25 7,23 7,35
о-Крезол +386 10,20 10,07 п- Нитрофенол -50 7,14 7,19
м - Метоксифенол +310 9,33 9,57
На основании вышеизложенного можно сделать заключение: наличие
линейной зависимости между ППН и рКа в пиридине позволяет приблизи-
тельно оценить значения рКа для производных бензойной кислоты и фенола.
Точность определения для случая фенолов составляет 0,18 единицы рК,
а для мета- и пара-замещенных бензойных кислот — 0,1 единицы рК
[780, 782].
г. Нq-функция кислотности Гаммета. В соответствии с классическими
представлениями степень кислотности в водных растворах определяется
концентрацией гидроксониевых ионов. Законы ионного равновесия дей-
ствительны только для очень разбавленных растворов и неприменимы для
растворов с более высокими концентрациями. Концентрация гидроксоние-
вого иона может быть измерена электрометрическим или спектрофотомет-
рическим методом, однако для некоторых кислотно-катализируемых реак-
ций скорость реакции не пропорциональна измеряемой концентрации гидро-
ксониевого иона. В концентрированной серной кислоте (фактически
не содержащей воды) концентрация гидроксониевого иона мала, и поэтому
ее сила должна быть малой. Однако практически этого не наблюдается.
Функция кислотности Гаммета и Дейрапа (Но) оказывается во многих
случаях более удобной для выражения силы концентрированных растворов
Сила кислот
47
кислот, чем значение pH, с которым сила кислоты идентична в сильно раз-
бавленных, но не в концентрированных растворах, где она имеет другие
значения [339, 340, 342, 625]. Их метод основывается на ионизационных
равновесиях особого класса индикаторов, ведущих себя по Б ренете ду как
незаряженные основания. При добавлении индикатора В к водному раствору
кислоты низкой концентрации равновесие может быть выражено следующим
образом:
В л Н3О+ ЯН++Н2О.
Постоянная индикатора, т. е. кислотная постоянная сопряженной кис-
лоты 7?Н+, выражается как
Р#вн+ = log + pH.
Подставляя Но вместо pH, получаем для постоянной индикатора выра-
жение
pA'BH* = log + и H0 = pABHt-logl^! ,
где В — незаряженное индикаторное основание, величина р/Свн+ кото-
рого связана с величиной, определенной в разбавленном водном растворе
косвенным и последовательным ступенчатым методом.
Рис. 13. Логарифм отношения Cbh+Z^b
ионизации индикаторов в H2SO4 — Н2О
(вес.%) [342, 625].
1 — о-нитроани.тин (рКВН+ 2 — о-нитроани-
.1ин (—0,25); 3 — 4-хлор-2-нптроанилин (—0,97);
4 — n-нитродифени ламин (—2,50); 5 — 2,4-дихлор-
6-нитроанилин (—3,34); 6 — 2,4-динитроанилин
(—4,50); 7—2-бензоилнафталин (—6,04); 8 — 6-бром-
2,4-динитроанилин (—6,71); 9 — антрахинон
(—8,26); 10—2,4,6-тринитроанилин (—9,41).
-?\------1------1-----1____-1______
0 20 40 60 80 100
H2S04,%
Значение рХвн+ Для индикаторов с не слишком большим и не слишком
малым отношением [5Н+]/[5] (т. е. подходящим для спектрофотометриче-
ских измерений) может быть рассчитано в результате сочетания спектрофото-
метрических и электрометрических измерений. Зависимость между отно-
шением кислотной и основной форм индикатора и светопоглощением (напри-
мер. в сернокислой среде) выражается [230, 625] как
[ДН+] _ ев —е
l^l Е Евн+
где ев — коэффициент молярного поглощения основной формы при данной
длине волны; £вн+ — коэффициент молярного поглощения раствора,
содержащего только кислотную форму; е — коэффициент молярного погло-
щения растворов, содержащих частично ионизированный индикатор.
Для растворов с возрастающей концентрацией Гаммет использовал серии
индикаторов с постепенно понижающейся основностью. Поэтому отношение
[БН+]/[Б] могло быть измерено с большой точностью (см. [230]). Величины
этих отношений оказались адекватными для определений, проводившихся
дробно в кислых растворах различной концентрации, а также для измерений
в сильно разбавленных растворах. Этот процесс схематически изображен
на рис. 13.
Для измерений с двумя незаряженными индикаторными основаниями
В и С при одинаковых концентрациях кислот может быть использовано
следующее соотношение:
„ . [СН+] , [ЯН+]
РА СН+- РАвн+ - lpg [<7] ~1 og ''
48
Глава 2
Если значение р/Свн+ индикатора В известно, то можно также рассчитать
р!Ссн+- По мнению Гаммета, приведенное выше уравнение применимо для
сред с высокой концентрацией кислоты и большой диэлектрической прони-
цаемостью; для иных растворов следует учитывать другие факторы.
Из рис. 14 видно, что значение HQ возрастает быстрее в концентрирован-
ных водных растворах сильной кислоты, чем в концентрированных водных
растворах слабой кислоты. Таким образом, можно допустить, что в сильно
разбавленных растворах значение HQ идентично значению pH. Протонодо-
норная способность растворов с высокой кислотностью по отношению к неза-
ряженным индикаторам выражается через —log HG. Чем больше эта вели-
чина, тем «более кислым» является раствор.
Бейтс и Шварценбах подвергли критике функцию кислотности Гаммета
и пришли к заключению, что значение Нь существенно зависит от индиви-
дуальных свойств используемого индикатора. Таким образом, «функция
Рис. 14. Функция кислотности уме-
ренно концентрированных водных раст-
воров кислот [340].
Концентрация H2S04,
вес. %
Рис. 15. Изменение значений II0
в смесях серная кислота—вода и сер-
ная кислота — трехокись серы [625]
(см. также [82, 342]).
кислотности» не применима при измерениях в растворах с низкой диэлек-
трической проницаемостью. Это необходимо принимать во внимание при
определении кислотности или основности индикаторов в неводной среде [40]
(ср. [84, 505]).
На рис. 15 показана зависимость Но от концентрации серной кислоты
для водных растворов H2SO4 с концентрацией от 0 до 100% и для смеси
H2SO4 — SO3, содержащей 31 вес. % SO3 [82, 342, 625].
17. СРАВНЕНИЕ СИЛЫ КИСЛОТ В РАЗЛИЧНЫХ РАСТВОРИТЕЛЯХ
Для выяснения степени протонного переноса в качестве кислоты или
основания сравнения обычно используется сам амфотерный растворитель.
Значения Ка и Кь, приведенные в литературе, основываются на использова-
нии воды в качестве кислоты или основания сравнения. Вода — очень слабые
кислота и основание, однако существует ряд других растворителей с подоб-
ными амфотерными свойствами: метиловый спирт, изопропиловый спирт,
уксусная кислота и т. д.
В протонакцепторном растворителе растворенная кислота вступает
в протолитическое взаимодействие с молекулами растворителя, например:
НС1О4^СН3СООН сн3соощсю4,
(C6H5)2NH£ + CH3COOH zz сн3соон2+ ... HN(C6H5)2
Сила кислоты определяется смещением равновесия вправо.
Сила кислот
49
а. При сравнении значений Ка кислот весьма важным является выбор
подходящего растворителя. В растворителе с высокой основностью различие
между константами диссоциации сильных и средних кислот будет почти
незаметно из-за нивелирующего действия растворителя. Сильно кислая
среда также не подходит: n-толуолсульфокислота вряд ли будет диссоции-
ровать в серной кислоте, а трихлоруксусная кислота является в уксусной
кислоте слабой кислотой. Растворители со слабой способностью к автопро-
толизу были бы подходящими для сравнения силы кислот, однако вследствие
слабых диэлектрических свойств этих растворителей электролиты сольва-
тируются ими лишь в незначительной степени. Подходящими растворите-
лями для сравнения кислот электрохимическим методом являются пиридин
(ср. разд. 16, в), л1-крезол и н-бутиловый спирт. Их диэлектрические про-
ницаемости соответственно равны 12,5, 13 и 15,8. Из неполярных раствори-
телей применим бензол; в качестве основания сравнения используется дифе-
нилгуанидин и как индикатор для измерения конкурентного равновесия —
этиловый эфир тетрабромфенолфталеина (ср. разд. 16, а).
В очень сильно разбавленных растворах в бутиловом спирте или л«-кре-
золе значения констант диссоциации ряда кислот весьма близки значе-
ниям констант диссоциации этих же кислот, получаемым в воде. В уксусной
кислоте кислоты по силе располагаются в следующий ряд: хлорная >
> бромистоводородная > серная > хлористоводородная > азотная (ср.
с рис. 9). Вследствие низкой диэлектрической проницаемости уксусной кисло-
ты полученные для нее значения констант ионизации вызывают сомнение.
Кондуктометрические измерения для кислот (при сравнении с азотной кис-
лотой, константа ионизации которой принята за единицу) дают следующие
значения: хлорная 400, бромистоводородная 160, серная 30, хлористоводо-
родная 9, азотная 1. По методу Гаммета и Дейрапа (ср. с разд. 16, г) получен
следующий ряд изменения силы кислот при растворении их в безводной
уксусной кислоте: метандисульфокислота >► хлорметандисульфокислота >
>• хлорная > хлоркарбоксиметансульфокислота > хлорметансульфокисло-
та > серная > метансульфокислота > хлористоводородная [625, 763].
Вследствие нивелирующего действия воды (см. гл. 6, разд. 38) хлорная,
n-толуолсульфокислота, серная и хлористоводородная кислоты довольно
близки по силе. Азотная кислота, однако, оказывается слабее в этиловом
спирте, а хлорная кислота сильнее в уксусной кислоте, если сравнивать
их с хлористоводородной кислотой.
Таким образом, даже этиловый спирт и уксусная кислота проявляют
дифференцирующее влияние, хотя оно и невелико. Вышеупомянутые кис-
лоты, за исключением хлорной, являются слабыми кислотами в ацетоне,
ацетонитриле, диоксане и нитрометане. По этой причине основная составляю-
щая часть сульфатов алкалоидов, а также n-толуолсульфонаты и этансуль-
фонаты некоторых органических оснований в ряде случаев могут быть отти-
трованы в диоксане хлорной кислотой [123, 321].
При титровании в неводных средах относительное изменение силы кислот
в результате замены растворителя является весьма важным фактором. Пара
кислота — основание приводится в равновесие со стандартной парой кисло-
та — основание АН0 — Во и определяется состояние равновесия. Пара
АН0 — Во может быть парой лионий — лиат, образовавшейся из раство-
рителя в процессе автопротолиза: sH+ — s_. В этом случае сравниваются
константы диссоциации двух кислот. Пара АН0 — Во может также быть
индикаторной системой и в этом случае измеряется отношение Ш+/1. Опыт
показывает, что относительная сила кислот, идентичных по типу заряда
и подобных по структуре, грубо говоря, не зависит от природы растворителя
(см. разд. 16, а и [173].)
Рис. 16 показывает величину относительной силы некоторых кислот
в воде и н-бутиловом спирте по сравнению с бензойной кислотой. Если бы
50
Глава 2
относительная сила кислот была одинаковой в обоих растворителях, то точки
должны были оказаться на одной прямой [340].
б. Связь Кота и АрК с диэлектрической проницаемостью растворителя. На силу
кислоты, т. е. на величину рХя влияет основность растворителя (гл. 2, разд. 13 и 14).
Однако сравнение может быть проведено
2-
0-
-2
-4
С>1
19q /
18 о/
я-&6
40
7ЙХ К
6<^Ъ9
г
/05
30 /
20/
-ffk--1----1----1---i----
-6-4-2 0 2 4
АрК(в и-бутиловом спирте')
таким образом, что вместо зависимости Ка от
диэлектрической проницаемости берется зави-
симость log Хотн от 1/е, где Хотн = KIKQ
и log Кот = log (К/К0), т. е. А%тн представ-
ляет собой отношение констант диссоциации
исследуемой и стандартной (например, бен-
зойной) кислот [593, 727, 873, 886].
Рис. 17 иллюстрирует эту зависимость.
Значение Хотн пяти различных кислот изме-
Р и с. 16. Относительная сила кислот в воде
и н-бутиловом спирте при сравнении с бен-
зойной кислотой [340].
1 — 2,4,6-тринитрофенол; 2 — трихлоруксусная ки-
слота; 3— малеиновая кислота (К,); 4— 2,4-
динитрофенол; 5 — о-нитробензойная кислота; 6 —
о-оксибензойная кислота; 7 — л-нитробензойная
кислота; 8 — n-нитробензойная кислота; 9 — о-хлор-
бензойная кислота; 10 — л-хлорбензойная кислота;
11— n-хлорбензойная кислота; 12— о-толуиловая
кислота; 13 — уксусная кислота; 14 — л(-толуиловая
кислота; 15 — n-толуиловая кислота; 16 — масляная
кислота; 17—триметилуксусная кислота; 18—ма-
леиновая кислота (Кг); 19 — п-нитрофенол.
4
няется лимиейно с изменением 1/е [593]. Однако весьма существенно отметить, что танген-
сы угла наклона прямых различны, так как диэлектрический эффект меняется от соеди-
нения к соединению.
При сравнении кислот различного заряда и структуры становится очевидным, что
в отдельных случаях диэлектрическая проницаемость растворителей и их основность
оказывают на силу кислот противоположное,
а в других случаях — одинаковое действие. Таким
образом, не следует соединять эти два представ-
ления в одно представление об «основности раст-
ворителей» ([677], стр. 259). Линейная зависи-
мость между log Хотн и ДрХ и 1/е соответственно
существует только для определенных пар кислот.
Рис. 18. Значение ДрХ различных пар
кислот в растворителях с разной диэлек-
трической проницаемостью [412, 545].
А — формамид; Б — вода; В — метиловый
спирт; Г — этиловый спирт; Д — н-бутиловый
спирт. Пары кислот: 1 — уксусная — бензой-
ная; 2 — монохлоруксусная — бензойная;
А уксусная — пикриновая; • монохлорук-
сусная — пикриновая.
Рис. 17. Относительная сила кис-
лот в растворителях с различной
диэлектрической проницаемостью
[593].
А — вода; Б — диоксан + вода (е =?45);
В — метиловый спирт; Г — этиловый
спирт, диоксан + вода (е = 25). Кис-
лоты: 1 — циануксусная; 2 — монохлор-
уксусная; 3 — гликолевая; 4 — п-хлорбен-
зойная; 5 — уксусная.
например уксусная — бензойная (рис 18, прямая 1), монохлоруксусная — бензойная
(рис. 18, прямая 2); однако в случае пар уксусная кислота — пикриновая кислота (Д)
или монохлоруксусная кислота — пикриновая кислота (ф) приведенная выше зависи-
мость уже не соблюдается [412, 545].
Сила кислот
51
в. Связь между возможным интервалом значений рК растворителей и значениями
рК кислот (оснований) (см. гл. 2, разд. 12). Значения рК для пары кислота — основание
sH+ — характерное для данного растворителя, определяет возможный интервал
значений рК растворенной пары кислота —
основание. Если нижнее значение рК раст-
ворителя обозначать рК', а верхнее — рК"
и если рК < рК", то исследуемая кислота
«нивелирована», так как она образует
катион растворителя. Когда для иссле-
дуемого основания рК > рК’, оно «ниве-
лировано» вследствие образования аниона
растворителя. На рис. 19 изображены
приблизительные интервалы изменения
значений рК некоторых растворителей [53].
Аммиак
Вода
Этиловый
спирт'
Уксусная
кислота
Муравьиная
кислота ’
Серная
кислота
20
30
Диэтиловый
эфир ‘
Бензол —
-20 -10 0 10
рК в воде
Рис. 19. рХ-интервалы растворителей
[53].
18. ПРОТОНОДОНОРНЫЕ СВОЙСТВА
ОЧЕНЬ СЛАБЫХ КИСЛОТ
Химикам-органикам известно, что
между «кислым» и «активным» водо-
родами имеется определенная связь
(гл. 2, разд. 9). Слабой кислотой
считается такая, для которой значе-
ние Ка составляет лишь 10-21 (фенилацетилен). В общем случае для установ-
ления силы кислот требуется определить равновесие между двумя кислотами
АН + В А + ВН
Кислота + Основание Основание-(-Кислота
В равновесии такого типа кислота или основание может быть растворите-
лем или анионом растворителя. В случае более сильных кислот А может
быть удовлетворительно определено, если основание В является одновре-
менно и молекулой растворителя
НС1О4 + СН3ОН СЮд+сн3он+-.
Однако в случае очень слабых кислот вместо В необходимо использовать
сопряженное растворителю основание, чтобы из ЛН образовалось измери-
мое количество А. Сдвиг равновесия зависит от основности В (которое
должно быть пчень сильным), т. е. кислота ВН, соответствующая основа-
нию В, должна быть очень слабой. Для изучения очень слабых кислот
может быть использован жидкий аммиак [22], а для их титрования — этилен-
диамин [176, 359, 601]. В последнее время применяется также тетраметилен-
сульфон (сульфолан) [111, 498].
Даже такая слабая кислота, как п,п'-диоксидифенилметан, обнаруживает
два скачка при потенциометрическом титровании в этилендиамине гидро-
окисью тетрабутиламмония (гл. 18, разд. 108, б).
Пример взаимодействия очень слабой кислоты
HOCeH4CH2CeH4OH + H2N(CH2)2NH2 6C6H4CH2C6H4O + H3N(CH2)2NH3.
В жидком аммиаке даже углеводороды ведут себя по отношению к амиду
натрия как доноры’протонов. Амид натрия в жидком аммиаке диссоциирует,
причем образующийся амид-ион NH“ является по отношению к аммиаку
сопряженным основанием
2NH3 NH+ + NH2.
Пример взаимодействия чрезвычайно слабой кислоты:
(CeH5)2CH2 +NH2 (С6Н5)2СН- +NH3.
Дифенилметан Дифенилметил-анион
52
Глава 2
При растворении в жидком аммиаке соли аммония обнаруживают свой-
ства кислот
[NH4]+Cl- + NaNH2 —> NaCl + 2NH3,
что аналогично реакции соляной кислоты с едким натром
[Н3О]+С1- + NaOH —> NaCl + 2OH2.
Раствор хлористого аммония в жидком аммиаке растворяет металлы
с выделением водорода и как кислота катализирует многие реакции. При
добавлении соответствующих индикаторов наблюдается изменение окраски.
Мочевина в аммиаке ведет себя как кислота, хотя в уксусной кислоте она
является основанием.
В аммиаке:
;CO(NH2)2 + (NHpNHt) CO(NH2)NH- + NH3-|-NH1-.
При этом концентрация катиона растворителя увеличивается.
В уксусной кислоте:
CO(NH2)2 + (CH3COOH^CH3COO-) CO(NH2)NHJ + CH3COOH + CH3COO-,
т. е. увеличивается концентрация аниона растворителя.
Константа автопротолиза в аммиаке
2NH3 NHI + NHJ; Яавт0= ~ 10"22.
Амид-анионы проявляют очень большое сродство к протону; по этой
причине даже дифенилметан способен к протолитическим реакциям.
Другой метод изучения чрезвычайно слабых кислот основан на изучении состояния
равновесия между исследуемой кислотой и сопряженным кислоте сравнения основанием
в диэтиловом эфире, бензоле или жидком аммиаке, т. е. в растворителях с исключительно
низкой кислотностью. Сопряженным основанием кислоты сравнения может быть соль
щелочного металла этой кислоты, например дифенилметилнатрий [147] (ср. стр. 765
в работе [231])
(С6Н5)2СН2 (CeH3)2CHNa [С6Н5СНС6Н5]-
Дифенилметан Дпфенилметил- Сопряженное дифенилметану
натрий основание
Согласно этой классификации сила кислот изменяется в следующем порядке: «-бути-
ловый спирт — ацетофенон > фенилацетилен — инден > дифениламин > ацетилен >
анилин > трифенилметан > дифенилметан > аммиак > толуол > бензол [147, 884]
(см. также гл. 4, разд. 27, табл. 28).
Ацетилен — очень слабая кислота, но с нитратом или перхлоратом серебра в соответ-
ствующей среде он образует карбид серебра и при этом выделяется кислота (см. гл. 27,
разд. 157).
Спиртовая гидроксильная группа по аналогии с кислотой может быть оттитрована
в диэтиловом эфире в присутствии индикатора этиопорфирина трифенплметилнатрпем [567]
(C6H5)3C-Na++ ROH (C6H5)3CH + RONa.
Борнеол и трифенилкарбинол могут быть оттитрованы подобным же образом кумилкалп-
ем [896], а активный водород производных пиррола — трпфенилметилнатрием [150].
19. ОНИЕВЫЕ ИОНЫ, КАТИОННЫЕ КИСЛОТЫ
Ониевые ионы играют важную роль в неводном титровании: молекулы
амфотерных растворителей образуют ониевые ионы при протонировании.
Примером может служить ацилониевый ион СН3СООН£. Азотсодержащие
основания также превращаются при присоединении протона в ониевые ионы,
например в ионанилиния С6Н5]МНз. По Бренстедувсе эти соединения являют-
ся катионными кислотами. Кислотность катионных кислот находится
в обратной зависимости от основности сопряженных оснований. Уксусная
кислота является очень слабым основанием и значение К
снзсоон^ ониевой
Сила кислот
53
кислоты равно 106*2 (Р^СНзСООН+ = —6,2). С другой стороны, М,РТ-дифе-
нилгуанидин — настолько сильное основание, что кислотность иона дифе-
нилгуанидиния составляет только рЛвн+ = 10,00 (К = 10-10) [779].
Под ониевыми соединениями здесь подразумеваются все соединения, в которых атом,
обладающий неподеленной электронной парой, образует координационную связь и ста-
новится насыщенным (см. стр. 40 в работе [231]).
а. Карбениевые ионы*. Трифенилметановые красители являются произ-
водными трифенилметильного катиона (С6Н5)3С+, важного карбениевого
иона. Из числа трифенилметановых красителей в качестве индикатора часто
применяется кристаллический фиолетовый. В карбениевых ионах координа-
ционное число атома углерода на единицу меньше его валентности. В лите-
ратуре термин ион карбонил чаще всего применяют для обозначения [R 3С]+.
Обозначения, предложенные в школе в Бонне, нам кажутся более правиль-
ными: согласно им координационное число ониевых комплексов на единицу
больше, а ениевых комплексов на единицу меньше, чем валентность цен-
трального атома [17, 186].
[R3C]+ [R3NR]+ [R3C]- [R5C]+
Ион карбения Ион аммония Карбанион Истинный ион карбония
(теоретически невозможен)
Известен также дикарбениевый ион; он образуется из трпхлорметилпентаметил-
бензола в концентрированной серной кислоте [363]
Карбениевые ионы являются акцепторными кислотами, и ацетилиевый ион (ацетил-кати-
он) также можно рассматривать как карбениевый ион СН3С+О. Он играет важную роль
при ацилировании. Известна также соль этого иона — перхлорат ацетилпя
Н3С — СО — С1 -|-НС1О4 [Н3С —С = О]+С1О4 +НС1.
Ацетилхлорид Перхлорат ацетплия
Собирательное название для карбениевых ионов типа [RCO]+ — оксакарбениевый
ион. Ион этого типа образуется из 2,4,6-триметилбензойной кислоты в растворе серной
кислоты (гл. 3, разд. 22, б).
б. Сольватация ионов аммония. На основность аминов существенно
влияет сольватация аммониевых ионов [55, 629, 820] (см. гл. 3, разд. 24).
Энергия сольватации ионов аммония уменьшается с увеличением числа
алкильных заместителей. Замещение каждого атома водорода в ионе NH*
на одну метильную группу понижает теплоту сольватации приблизительно
на 8 ккал [631]. В противоположность аммониевым ионам энергия сольвата-
ции свободных аминов остается примерно той же. Это обстоятельство сильно
влияет на равновесие
R3NH+4-4- ?1H + R3N.
Сольватация ионов аммония происходит двумя путями: 1) кислый водород
ониевого иона сольватируется теми частями молекул растворителя, которые
богаты электронами, по аналогии с кислотно-основными реакциями, зави-
* Термин «карбениевые ионы» иногда употребляется вместо термина «карбониевые
ионы» (в немецкой литературе) для указания на возможность превращения СН2: +Н+ -►
-► CHJ.— Прим. ред.
54
Глава 2
сящими от электродонорных свойств молекул растворителя
[rh2nh]++:o—r (rh2n...h... :o-r)+
I I
R R
2) сольватирующее действие обусловлено поляризацией молекул раство-
рителя. Теплота сольватации для первого процесса 10 ккал/молъ, для вто-
рого — 69 ккал/молъ [8].
Равновесие кислотно-основного взаимодействия этил-, диэтил- и триэтил-
аминов (В) с 2,4-динитрофенолом (ЛН) зависит от сольватирующей способ-
ности растворителя [631]
ВЧ-ЛН вн+л-.
Константы равновесия образования ионных пар приведены в табл. 13. Рас-
творители, содержащие основные атомы кислорода, обладают повышенной
сольватирующей способностью, например диоксан, этилацетат. Богатые
Таблица 13
Основание Бензол е = 2,3 Диоксан е = 2,2 Хлороформ е = 4,8 Хлорбензол е = 5,6 Этила цетат е = 6,3
Этиламин Дпэтпламин Триэтиламин 1200 2940 4900 6230 1460 1 730 15 800 2500 9680 65 500 47 500 11900
электронами части молекул растворителя стабилизуют ионную пару, вероят-
но, вследствие образования водородной связи. В случае ониевых ионов чет-
вертичных аминов этого не происходит, так как Н+ образует водородную
связь с атомом кислорода иона нитрозамещенного фенолята
R3N ... Н+ ... "О -CeH3(NO2)2.
в. Оксониевые и сульфониевые ионы. Атомы кислорода и серы способны координиро-
вать протон своими свободными электронными парами, образуя при этом ионы оксония
и сульф они я. Моногидрат хлорной кислоты можно представить как перхлорат гидроксония,
который является изоморфным перхлорату аммония
[Н3О]+С1О4 [NHJ+ClOf.
Перхлорат гидроксония образует стабильную соль, имеющую кристаллическую
решетку, в которой чередуются ионы Н3О+ и ClOj [63, 851]. (Об электронной структуре
перхлората ацилония см. гл. 7, разд. 44.)
Эфиры и в значительно большей степени тиоэфиры также способны к образованию
онпевых соединений.
R —S —R + HC1 —» [R — S — R]+C1~
Хлорид диэтил-
сульфония
Тенденция к образованию ониевых ионов у атомов кислорода и серы меньше, чем
у азота. Существуют также следующие соли оксония:
[(СН3)3О]+ООССН3 [(CH3)3O]+OCeH3(NO2)3.
Ацетат триметилоксония Пикрат триметилоксония
Эти ионы можно рассматривать как результат замещения в ионе Н3О+ [572]. Ион
триалкилоксония является акцепторной кислотой по отношению к иону [BFJ_; с другой
стороны, он выступает в качестве донора алкил-катионов [R]+ (ср. [231] и гл. 1, разд. 8).
Известны также гетероциклические оксониевые соединения с кислородом в качестве
гетероатома, например:
С1СН2-СН
С1СН2 — СН
|/° —R
сн
+ r20
[241]
Сила кислот
55
2,6- Диметил-у-пирон — слабое основание и может быть оттитровано хлорной кисло-
той [148, 332]
5-Окси-2-(оксиметил)-у-пирон (койевая кислота) — слабая кислота и может быть оттит-
рована метилатом натрия
О
Койевая кислота, см. гл. 18, табл. 83.
г. Известны отрицательно заряженные аналоги ониевых ионов, например
[ВН4]~. В ионе аммония протон координируется с атомом азота, в то время
как в боргидрид-ионе гидрид-ион координируется с бором
NH3+H+ = [NH4]+; ВН3+Н-=[ВН4]-.
Такой тип ионов существует в алюмогидриде лития. Алюмогидрид лития
в тетрагидрофуране является подходящим титрантом для очень слабых кис-
лот, например спиртов и углеводородов ([391] и гл. 19). Когда титруют осно-
вания хлорной кислотой в уксусной кислоте, ацилоний-ион служит донором
протонов, когда же титруются очень слабые кислоты, ион тетрагидроалю-
мината [А1Н4]_ является акцептором протонов, немедленно выделяющим
водород
н+ + н-=н2
и
R— ОН +1/4 LiAlH4 = 1/4 LiAl(OR)4+H2.
Эта реакция необратима и, следовательно, отлична от обычных кислотно-
основных реакций. Недавно для титрования спиртов был применен алюмо-
тетрамид лития [387]. Его основность меньше основности гидрида, и поэтому
он пригоден для титрования спиртов в присутствии карбонильных, эфирных
и амидных групп. Восстановление карбонильной группы гидридом можно
рассматривать как необратимую реакцию между кислотой Льюиса и гид-
ридным основанием [383]
\б+ в- \rZH
>с=о + н- —> /С\
/ z хо-
Кислота Льюиса-j-Основание = Продукт
нейтрализации.
Структура иона алюмоамида лития не ясна (стр. 123 в работе [383]).
Тетралкилбораты легко образуются из триалкилбората и алкоголята
натрия
B(OR)3 + RONa —> [B(OR)J-Na+.
Аналогичным соединением является тетрафенилборат натрия
[B(C6H5)4]-Na+-
Такие комплексы большей частью нерастворимы в воде; комплексы, полу-
ченные из алкалоидов и четвертичных аммониевых оснований, могут быть
56
Глава 2
оттитрованы хлорной кислотой в диоксане или в смеси уксусного ангидрида
и метилэтилкетона [267, 316] (гл. 24, разд. 134 и 138)
[R4N]+[B(CeH5)4]- + CH3C00H£C104 —> R4N+C1O4 + [В(С6Н5)4]Н + СН3СООН.
20. КИСЛОТЫ ЛЬЮИСА
Кислоты Льюиса и кислотно-основная теория Льюиса описаны в гл. 1.
Определение кислот Льюиса в неводных средах с нуклеофильными основа-
ниями и взаимодействие азотсодержащих оснований с кислотами Льюиса
имеют практически меньшее значение, чем аналогичные реакции водород-
содержащих кислот и N-содержащих оснований. Однако исследование кислот
Льюиса имеет большое значение для органической химии (реакции Фриделя—
Крафтса, ацилирование, сульфирование и т. д.).
а. Примеры реакций кислот Льюиса. Хорошо известным взаимодейст-
вием является координация аммиака и фторида бора
Н F Н F
II II
Н —N: 4-В —F -» Н— N В —F
II II
Н F Н F
Эта реакция протекает с большой скоростью в диоксане и может использо-
ваться как индикаторная. Уже в 1938 г. Льюис оттитровал пиридин и подоб-
ные ему основания в органических растворителях фторидом бора и хлоридом
олова(1У) [509].
В органической химии имеются характерные типы гетеролитических
(полярных) реакций, в которых в качестве реагентов участвуют кислоты
Льюиса (электрофильные реагенты) и основания Льюиса (нуклеофильные
реагенты). Движущей силой процесса является сродство электрофильного
реагента к свободной электронной паре и сродство нуклеофильного реагента
к электронодефицитной частице.
Реакция присоединения:
СН3
[
снз-n: +
I
СНз
СНз
I
В —СНз
I
СНз
СНзх /СНз
СНз—N В-СНз
СН3Х ХСН3
Реакция замещения:
(CH3)3N -> B(CH3)3 + NH3 (СНз)зВ NH3 + N(CH3)3,
(CH3)3N -> B(CH3)34-BF3 (CH3)3N -> BF3 + B(CH3)3.
Кислоты Льюиса, например BF3 и ВС13, вытесняют двуокись углерода
из суспензии карбоната натрия в смеси ацетона и четыреххлористого угле-
рода.
Подобно иону гидроксония, BF3 реагирует с сульфат-анионом
OHJ + SQ2- OH2 + HSO^,
BF3 + SO2- _„ [SO4BF3]2-
Таким образом, существует аналогия между реакциями нейтрализации
протонсодержащих кислот и кислот Льюиса.
Кислоты Льюиса, такие, как BF3, А1Вг3, TiCl4, взаимодействуют с бензо-
феноном, ацетофеноном, дипропилкетоном и ароматическими нитросоеди-
нениями [122, 261, 786]. В случае кетонов «основным» атомом, донором
электронов, является атом кислорода карбонильной группы. В нитросоедине-
Сила кислот
57
ниях в нитрогруппе имеются два теоретически неразличимых электроно-
донорных атома кислорода, хотя на основании ИК-спектра можно предполо-
жить, что только один из этих атомов образует координационную связь
с А1Вг3.
Монография Ашуорса [20] содержит превосходное обобщение данных по титрованию
льюисовских кислот и оснований галогенидами металлов II—VIII групп периодиче-
ской системы.
б. Реакции Фриделя — Крафтса также происходят благодаря каталитическому
действию кислот Льюиса, например А1С13, А1Вг3 и т. д. При взаимодействии бензола,
н-бутилхлорида и А1С13 ион карбения реагирует с кислотой Льюиса по его ковалентной
связи: ион карбения образует л-комплекс с бензолом, после чего происходит замещение
с выделением протона. Затем протон взаимодействует с комплексным ионом AICI7.
Отсюда порядок изменения силы кислот следующий: кислота Льюиса >> ион карбения >
бензол.
(СН3)3СС1 + А1С13 -> (СН3)3С+ + A1C1J,
(СН3)3С+ + С6Нв -> (СН3)3С—СвН5+Н+,
A1C1J + H+ А1С134-НС1.
Вышеописанные реакции изображены упрощенно, без учета образования л-комплек-
сов [231]. Об изменении относительной силы кислот Льюса см. в работах [181, 365, 734].
в. Ионы карбения как кислоты Льюиса. С теоретической точки зрения при
неводном титровании электронодефицитные ионы карбения и оксакарбения
также являются кислотами Льюиса (см. разд. 19, а). Ион ацетилия СН3СО +
играет важную роль при ацетилировании (см. гл. 25, разд. 150).
Ацетилперхлорат образуется при взаимодействии ацетилхлорида и пер-
хлората серебра. Этот процесс протекает в уксусном ангидриде, растворитель
диссоциирует на ацетил-катион и ацетат-ион [531] (см. гл. 7, разд. 44).
CH3CO+CI- + Ag+ClOj —CH3CO+C1Oj + AgCl
Эта реакция подтверждает существование иона ацетилия (ацетил-катио-
на). Для случая ацетил-иона справедливо следующее представление: льюи-
совская кислота (электронодефицитная частица), ион карбения и акцептор-
ная кислота эквивалентны.
Гетероциклические четвертичные аммониевые соли могут быть представлены мезо-
мерными формами. Четвертичный аммониевый ион в его карбениевой форме может рас-
сматриваться как кислота Льюиса и, следовательно, может вступать в реакции с основа-
ниями Льюиса. Титрование хлорида котарнина стандартным раствором цианистого
калия в смеси ацетона и изопропилового спирта подтверждает этот факт. Высоконукле-
офильный цианид-ион атакует ион карбения и при этом образуется ковалентная связь.
Конец титрования может быть определен потенциометрически или визуально [215]. Четвер-
тичное аммониевое основание не может быть оттитровано цианидом. Хлорид 2,4-динптро-
фенил-6,7-диметоксиизохинолиния также может быть определен подобным образом [215].
Спектрофотометрические исследования показали, что доноры электронов, такие, как
бутиламин, в среде хлороформа легко вступают в обратимую реакцию, давая аддукты
с соединениями типа катиона Г^-2,6-дихлорбензилхинолиния. При более высокой^концен-
трации бутиламина образуется аддукт следующего типа:
\/Ч Щ
Z N ХН
I
R
(Определение ацетилхлорида см. гл. 16, разд. 101.)
58
Г л а в а 2
г. Реакции кислот Льюиса в ацетилхлориде [626]. Высокий дипольный момент
ацетилхлорида и большая реакционная способность делают возможным его распад
на пон ацетилия и хлорид-ион [см. выше разд, (в)]
СН3СОС1 (СН3СО)+ +Cl-
Измерения электропроводности показали, что в некоторых случаях ацетилхлорид реаги-
рует как ионная пара
C5H5N.CH3COC1 C5H5N.(CH3CO)+4-Ci-,
Комплекс пиридина
с ацетилхлоридом
2СН3СОС1+ SnCl4 SnClj[”4-2(CH3CO)+.
Кислота
Льюиса
В растворе ацетилхлорида соединения, повышающие концентрацию иона СН3СО+
или С1“, являются соответственно или кислотами или основаниями. Аналогично в уксус-
ном ангидриде ионы СН3СО + дают кислую реакцию, а ионы СН3СОО_ — основную
(см. гл. 7, разд. 44).
Кислоты Льюиса, образующие комплексы с более высоким координационным числом,
косвенно способствуют повышению концентрации иона ацетилия, понижая соответ-
ственно концентрацию основания С1"
SnCl4 + 2Cl- SnClg",
поскольку концентрация продукта остается неизменной [СН3СО+][С1_]. По этой при-
чине кислоты Льюиса в ацетилхлориде называют сольвокислотами.
Третичные азотсодержащие основания, например пиридин или хинолин, косвенно
способствуют повышению концентрации хлорид-иона в ацетилхлориде, так как реагируют
с ионом ацетилия, образуя (C5H5N)-CH3CO+. Благодаря своему сольволитическому дей-
ствию вышеуказанные основания называют сольвооснованиями. Четвертичные хлориды
аммония также являются сольвооснованиями, так как повышают концентрацию С1"-иона.
Четвертичные соединения, например бензилтриметиламмонийхлорид, и льюисовские
кислоты в отношении 2 : 1 образуют нейтральный комплекс
2(R4N)Cl + SnCl4 (R4N)2.SnCl6,T. е. (R4N)2+SnCl§-
Льюисовские кислоты SnCl4, TiCl4 не являются электролитами, хотя их растворы
в ацетилхлориде лучше проводят электрический ток, чем сам растворитель. Максимум
проводимости в случае SnCl4 наблюдается при добавлении избытка SnCl4, когда обра-
зуются комплексы состава 1 : 1
(R4N)2-SnCl6 + (CH3CO)2SnCl6 —> 2R4N-SnCl6(CH3CO) 2(CH3CO)+4-2[R4N.SnCle]".
Относительно изменения электропроводности как функции комплексообразования
см. гл. 5, разд. 33.
Вышеприведенные сольволитические реакции объясняют, почему титрование может
быть выполнено в ацетилхлориде даже в присутствии индикатора кристаллического
фиолетового. Окраска индикатора в этом случае совпадает с окраской в уксусном ангид-
риде: в ацетилхлориде она голубая, при прибавлении TiCl4 или SnCl4 становится темно-
желтой или желтой, а при добавлении хинолина переходит в фиолетовую.
д. Реакции льюисовских кислот и оснований в нитробензоле [379]. Пиридин и хино-
лин могут быть оттитрованы кондуктометрически в нитробензоле, содержащем измеренное
количество ВВг3 (т. е. обратным титрованием, когда титрант ВВг3 оттитровывают раст-
вором определяемого соединения). В результате применения этого теоретически интерес-
ного метода, который, однако, на практике требует большой осторожности, было найдено,
что первоначально ВВг3 взаимодействует с нитробензолом и образуется комплекс состава
1:1, который затем диссоциирует по схеме
2C6H5NO2.BBr3 [(C6H5NO2)2BBr2]+ + BBri.
Пиридин и хинолин, как более сильные основания, вытесняют более слабое основа-
ние — нитробензол из его комплексов
C6H5NO2-BBr3-|-: NC5H5 —> CeH5NO2 + C5H5N -> ВВг3.
Диссоциация комплекса пиридин — ВВг3 сильнее чем комплекса растворитель — ВВг3.
Приведенная выше реакция замещения указывает на сходство с, Н-кислотами: титрование
Сила кислот
59
хлорной кислоты пиридином в уксусной кислоте может быть изображено следующим
образом (без указания зарядов):
CH3COOH-HC1O4 + NC5H5 —> CH3COOH + C5H5NHC1O4.
Пиридин, как более сильное основание, вытесняет более слабое основание — уксусную
кислоту — из перхлората ацилонпя (см. гл. 1, разд. 5, д).
е. Реакции триэтилалюминия с хинолином [222, 833]. Льюисовская
кислота триэтилалюминий может быть оттитрована в инертном раствори-
теле 0,5 н. раствором изохинолина. Добавление индикатора излишне, так
как при соотношении 1 : 1 образуется комплекс желтого цвета, а при соот-
ношении 1:2 — комплекс красного цвета.
(С2Н5)3А14- :NC9H9 ->
Кислота Основание
Льюиса
+:NC9He
(С2Н5)3А1 <- NCgHg --------- (C2H5)3A1(NC9H9)2
Желтый комплекс Красный комплекс
Комплекс красного цвета образуется только после того, как весь изохино-
лин израсходуется на образование желтого комплекса.
Так как окраска раствора изменяется постепенно, следует проводить титрование
методом обратного тирования Ганча [331] (см. гл. 30, разд. 166 [222]).
ж. Кислотность смеси льюисовских кислот и Н-кислот в уксусной кислоте [711]. При
растворении SnCl4 в уксусной кислоте реакция идет с выделением тепла; образующийся
комплекс SnCl4(CH3COOH)2 проявляет характерные свойства кислоты Бренстеда [830].
Этот комплекс в растворе сольватирован тремя молекулами уксусной кислоты; уксусная
кислота протонируется этим комплексом SnCl4(CH3COOH)3, вследствие чего проводимость
'раствора повышается.
SnCl4(CH3COOH)3 [HSnCl4(CH3COO)2]- + CH3COOH£.
Прп добавлении хлорида олова(1У) к раствору индикатора типа нитроанилпна в уксус-
ной кпслоте УФ-спектр индикатора постепенно изменяется, что свидетельствует об обра-
зовании кислотной формы индикатора. В этом смысле кислота SnCl4(CH3COOH)2 в уксус-
ной кислоте по отношению к 4-хлор-2-нптроанилину равна по силе серной кислоте или
даже сильнее.
21. ВТОРИЧНЫЕ КИСЛОТЫ
При дальнейшем развитии своей электронной теории [510] Льюис рас-
сматривал как кислоту «любое вещество, один из атомов которого способен
принимать в свою валентную или координационную оболочку пару элек-
тронов другого атома». На этом основании каждое соединение является
гипотетической кислотой или основанием, но с очень различной реакционной
способностью, особенно если принимать во внимание фактор времени. Боль-
шое число кислот для взаимодействия с основанием не требует энергии акти-
вации — это первичные кислоты. Для вторичных кислот, наоборот, для
взаимодействия требуется энергия активации. Представление о вторичных
кислотах связано с представлениями Ганча о «псевдокислотах» (см. гл. 2,
разд. 16, а также разд. 15, а).
Нитроэтан реагирует со щелочами очень медленно; при нейтрализации
образующегося продукта хлористоводородной кислотой электропроводность
раствора медленно возрастает, приближаясь к проводимости NaCl. Моно-
нитроалканы являются кислыми соединениями, которые существуют в тау-
томерном равновесии с их нитроновыми кислотами
R — СН2— N
Нитро-изомер
(слабокислый)
+ /°-
“ R — CH=N<
ХОН
Енольный изомер
(гораздо более кислый)
60
Глава 2
Равновесие нитроалканов, нитроновых кислот и их ионов изображено на
следующей схеме:
Медленно
Нитро-форма > Кислотная форма
Медленно fl If Быстро
Н+ + Нитронат-анион
(см. гл., разд. 44).
Теория «псевдокислот» Ганча касалась только таутомерных перегруппи-
ровок водородных кислот. Своей теорией о вторичных кислотах Льюис
сделал шаг вперед. Это показано на примере двуокиси углерода, которая
из-за наличия двух двойных связей не может рассматриваться как акцептор
электронов, пока эти две двойные связи не затронуты. Нейтрализация дву-
окиси углерода происходит медленно, так что двуокись углерода является
вторичной кислотой [510]. Двуокись углерода в растворе этилового спирта
при температуре —60° очень медленно реагирует с этилатом натрия в раз-
бавленном этиловом спирте, и этот процесс может быть зафиксирован
с помощью индикатора. Когда, однако, эти вещества предварительно сме-
шивают и раствору дают постоять, сразу же происходит изменение окраски
индикатора. Это означает, что СО2 с его очень низким уровнем энергии
не обладает кислотным характером. Двуокись углерода может быть оттитро-
вана как кислота в среде ацетона или пиридина метилатом натрия [71]
и гидроокисью тетрабутиламмония в пиридине [168, 286]. Резонансные формы
двуокиси углерода и сероуглерода можно представить следующим обра-
зом [628]:
Двуокись углерода как льюисовская кислота вступает в реакцию с ОН "-ионом
гидроокиси тетрабутиламмония как льюисовским основанием и образуется
бикарбонат-ион
+Z’ - Z5!
с + он’ —► с —о]
\>Г Хон
В этом смысле хлорангидриды и ангидриды органических кислот, а также
трифенилметил-катионы являются вторичными кислотами (стр. 242 в работе
[677]; см. гл. 16, разд. 96, 101 и работу [828]).
Глава 3
СИЛА ОСНОВАНИЙ
22. СИЛА СЛАБЫХ ОСНОВАНИЙ
Основность слабых оснований может быть выражена или через силу
сопряженной кислоты или через ее обратное значение Квн+, определяемое
из следующего общего уравнения (см. гл. 2, разд. 12):
^вн+
[*Щ] [5]
[ЯН+]
Чем больше константа кислотности сопряженной кислоты, тем меньше
ее обратное значение и тем слабее основание. Константа кислотности иона
анилиния равна 10-4-58, и, следовательно, он является более сильной кисло-
той, чем уксусная кислота, константа кислотности которой равна 10-4,76.
Зависимость между константой диссоциации оснований Кь и их отрицательными
логарифмами (—logAj, = рКь) иллюстрируются данными, приведенными в табл. 14.
Таблица 14
У величе- ние основ- ности Основание кь РКЬ квн+ рКвн+ 1 КВН+ Увеличе ние протоно- донорной способности ониевого иона
2 - Пропплпи ридпн 1,2,3-Трпфенил- гуанпдин N-Аллплморфо- лин ]\,]М-Диметил- анилпн Нарцеин Дифениламин Ю-з 1,26-10-5 1,15-10-’ 1,15-10-9 2-10-ы [497] 0,71-10-13 3,00 4,90 6,94 8,94 10,7 13,15 ю-n 7,95-10-ю 8,71-10-8 8,71-10-6 5-10-4 1,4-10-1 11,00 9,10 [333] 7,06 [333] 5,06[333] 3,3 0,85 [332] юн 1,26-109 1,15-107 1,15-105 2-103 7,09 1 1
а. В сильнокислотных растворителях вода является акцептором прото-
нов. В муравьиной, уксусной и пропионовой кислотах вода действует как
основание, и поэтому она является нежелательной примесью. Реальная
основность воды различна в зависимости от природы растворителя
н2о+$н ДГ н3о++«-,
где $Н — например, этиловый спирт или муравьиная кислота.
По аналогии с формулой из разд. 22, где Z?H+ — ониевый ион (NHJ,
Н3О+), константа кислотности сопряженной воде кислоты
к _ [ШИ [Н2О]
Л-НзСН- [Нз0+]
Поскольку р/Са = р/^авто — Редисе значение рАн3О+ сопряженной воде
кислоты может быть определено экспериментально, например индикаторным
62
Глава 3
методом. Показатель степени константы диссоциации воды в этиловом спирте
равен 18,3, а в муравьиной кислоте — 4,8. Поскольку в этиловом спирте
Р^н3о+ = 19,1 — 18,3 = 0,8 и в муравьиной кислоте рА^н3о+ = 6,2 —
4,8 = 1,2, то ион гидроксония является более сильной катионной кисло-
той в этиловом спирте, чем в муравьиной кислоте, и, следовательно, сама
вода является более слабым основанием в этиловом спирте, чем в муравьиной
кислоте (IO-0-8 > 10-1*2). Таким образом, становится понятным, почему
кислотные растворители необходимо дегидратировать перед использова-
нием их в титровании очень слабых оснований (гл. 8, разд. 47).
б. Ионизация очень слабых оснований в серной кислоте (серная кислота
как растворитель для измерения основности очень слабых основании). Очень
слабые основания могут быть изучены только на основе их взаимодействия
с очень сильными кислотами, и, наоборот, сила очень слабых кислот может
быть определена по их взаимодействию с очень сильными основаниями
(см. гл. 2, разд. 18). Сам растворитель может быть такой сильной кислотой,
как, например, серная кислота, в которой ионизация слабых оснований
может быть определена или методом понижения температуры замерзания
или спектрофотометрическим методом [344, 349, 352, 818]. За исключением
уксусной кислоты, серная кислота исследована наиболее полно как раство-
ритель. Первая работа в этой области была выполнена Ганчем (1907—1930).
Более точные измерения были проведены Гамметом (1933—1937), Гилеспи
и Ингольдом (1946—1950) [271, 272]. Серная кислота как растворитель имеет
большое преимущество, состоящее в следующем: когда ионные или неионные
соединения растворены в ней, то понижение температуры замерзания в очень
широких пределах пропорционально концентрации растворенных соедине-
ний, поскольку коэффициент активности участвующих в реакции ионов
остается почти постоянным вплоть до необычайно высоких концентраций.
Способность серной кислоты сольватировать ионы, возможно, наивысшая
среди всех растворителей.
Многие молекулы, содержащие атомы кислорода и азота (амины, простые
эфиры, кетоны, альдегиды, спирты и сложные эфиры) способны координиро-
вать протоны в серной кислоте. Для большинства из них характерно пони-
жение температуры замерзания вдвое большее по сравнению с рассчитанным
на моль; это свидетельствует о том, что реакция
>O + H2SO4 >OH+ + HSOj
протекает следующим образом:
С2Н5ОН + 2H2SO4 —C2H5SO4H-f-H3O+-|-HSOj,
(C2H5)2O + H2SO4 [(C2H5)2OH]+ + HSO7.
Большое число алифатических и ароматических карбоновых кислот
присоединяет протон, образуя при этом протонированный ион кислоты,
т. е. они ведут себя как основания. Образование протонированных молекул
кислоты в случае дихлорукусусной кислоты, алифатических и ароматиче-
ских нитросоединений и сульфокислот происходит лишь частично. В процес-
се протонного переноса некоторые соединения претерпевают даже более
сложные изменения, например:
(СН3СО)2О + 2H2SO4 —> (СН3СО)+ + СН3СООН£ + 2Н8О4
Уксусный Ацетил- Протониро-
ангидрид катион ванная ук-
сусная
кислота
Во всех этих реакциях очевиден основной характер атома кислорода, обусловленный
наличием у него свободной электронной пары. В ароматических карбоновых кислотах
Сила оснований
63
(при возможном наличии стабилизованной вследствие резонанса формы оксокарбения)
образуются четыре иона [611]
2,4,6-(СН3)3С6Н2СООН + 2H2SO4 -* Н3О++ 2HSO4 +
+ Н3С
В серной кислоте трифенилметилкарбинол образует темно-желтый раствор, в котором,
как показано методом понижения температуры замерзания, содержится четыре иона
(CeH5)3COH4-2H2SO4 — (СвН5)3С+ + Н3О+ 4- 2HSO4.
Эта реакция аналогична реакции трифенилметилхлорида с хлоридом алюминия
(СвН5)3СС1 + А1С13 -> (CeH5)3C+ + A1C1J.
Существование иона карбения установлено путем выделения перхлората трифенпл-
метилкарбения
' [(С6Н5)3С]+СЮ4- .
1,1- Дифенил эти лен в серной кислоте дает подобный же сильноокрашенный раствор;
углеводород ведет себя как основание и его сопряженной кислотой является ион карбения
(CeH5)2C = CH2 + H2SO4 (СвН5)2С+—CH3 + HSOj.
Многие азотсодержащие соединения ведут себя в серной кислоте как электролиты:
нитроанилин, азобензол, ацетонитрил, бензамид и т. д. Мочевина проявляет свойства
основания.
CO(NH2)2 + 2H2SO4 CO(NH3)i+ + 2HSO4.
Флексер, Гаммет и Динкуэлл [230, 625] разработали специфический метод спектро-
фотометрического определения значения рАвн+ очень слабых оснований в смесях серная
кислота — вода. Если известно значение Но среды, то можно вычислить величину рАвн+
(см. гл. 2, разд. 16,г)
pXBH+ = K„ + log-5^-
е—евн+
В табл. 15 приведены значения рАвн+ некоторых очень слабых оснований: мочевина
является слабым основанием, а бензойная кислота — исключительно слабым основанием.
Таблица 15
Основание рКвН+ Литература Основание ' рКвН+ Литература
п-Нитроанилин 4-0,99 625 Уксусная кислота -6,2 279
Мочевина Ацетамид 4-0,4 4-0,25 505 279 Бензойная кис- лота -7,38 230
Ацетонитрил 1,4-Диоксан -4,3 -4,4 505 505 2,4,6-Тринитро- анилин -9,41 625
Из данных табл. 15 следует, что уксусная кислота — очень слабое осно-
вание и, таким образом, ее сопряженная кислота (протонированная уксус-
ная кислота) должна быть очень сильной кислотой.
23. ИЗМЕРЕНИЕ ОСНОВНОСТИ АЗОТСОДЕРЖАЩИХ ОРГАНИЧЕСКИХ ОСНОВАНИЙ:
ЗАВИСИМОСТЬ МЕЖДУ ПОТЕНЦИАЛОМ ПОЛУНЕЙТРАЛИЗАЦИИ
И ВЕЛИЧИНОЙ рК
Потенциометрическим титрованием азотсодержащих оснований в уксус-
ной кислоте, уксусном ангидриде, нитрометане, ацетонитриле и т. д. может
быть определен порядок изменения относительной основности этих соеди-
64
Глава 3
нений. Во многих случаях удалось установить количественную зависимость
между потенциалом полунейтрализации (ППН), измеренным в органиче-
ском растворителе, и величиной р2С в водной среде [146, 247, 333, 337, 778,
779, 838].
Бейлис п Четвпн, а также Пирсон и Фогельсон провели спектрофотометрические
исследования по определению равновесия азотсодержащих оснований с 2,4-динитрофено-
лом как кислотой сравнения в растворителях с различными диэлектрическими проница-
емостями и различной сольватирующей способностью [46, 631]. Сила оснований изме-
няется сложным путем как функция диэлект-
рической проницаемости и сольватирующего
действия растворителя. Исследования измене-
ния силы оснований, вызванного сольватирую-
щим действием растворителя и стереохимией
молекул, часто приводили к противоречивым
результатам [8, 96, 629, 631, 820].
Рис. 20. Кривые потенциометриче-
ского титрования оснований в уксус-
ной кислоте [332, 337].
1 — очень слабые основания; 2 — слабые
основания: * 3 — основания средней [силы:
4 — сильные основания.
а. Относительная сила оснований в уксус-
ной кислоте проиллюстрирована на рис. 20
и 21 [332, 337, 838]. Подавляющее боль-
шинство потенциометрических кривых имеет
Рис. 21. Относительная сила оснований
в воде и уксусной кислоте [332]. рНсц3соон
является pH, взятым в средней точке
потенциометрического титрования.
В уксусной кислоте: ...не титруемые очень слабые
основания; —неточно титруемые слабые основа-
ния; ... основания средней силы;---сильные
основания. В интервале рКвц+ от 4,5 до 14 осно-
вания в уксусной кислоте нивелированы.
характерную S-образную форму. Для слабых оснований начальная точка потенциометри-
ческой кривой находится на оси ординат, крутизна кривой в точке скачка потенциала
уменьшается постепенно, как, например, в случае ацетоксима, мочевины и ацетамида
(рис. 20). Прп титровании 0,05 М растворов очень слабых оснований (Къ < Ю-14) 1 М
хлорной кислотой протонная активность резко увеличивается даже при добавлении
очень небольших количеств титранта, но точки перегиба не наблюдается совсем или
в случае некоторых слабых оснований она слабо выражена (для в интервале 10-14—
10-10). В случае оснований средней силы или сильных оснований потенциометрическая
кривая плавно поднимается от начала кривой до точки перегиба, так как при сольвата-
ции образуется ацетат-ион, который взаимодействует с хлорной кислотой. Слабые и очень
слабые основания в уксусной кислоте образуют в различных пропорциях сольват уксус-
ной кислоты и ониевый ион.
Очень слабые основания:
R2NH...HOOCCH3 R2NHJ.CH3COO-
Слабые основания:
R2NH...HOOCCH3 R2NH£.CH3COO-
Порядок изменения силы оснований, оттитрованных потенциометрически хлорной
кислотой в уксусной кислоте, следующий ([332], рис. 20 и 21):
1. В уксусной кислоте не могут быть оттитрованы следующие очень слабые основания:
пропионитрил, 4-нитро-2,6-дихлоранилин, диацетилмоноксим, форманилид, диэтил-
дифенилмочевина, ацетанилид, триброманилин, N-пропилацетанилид, N-метилацетани-
лид, о-нитроанилин, фенилмочевина, ацетамид.
Сила оснований
65
2. В уксусной кислоте не точно титруются следующие слабые основания: п-нитро-
димети л анилин, мочевина, метилмочевина, дифениламин, 2,5-дихлоранилин, п-нитроани-
лин (в интервале рАвн+ от 0 до 2).
3. Основания средней силы: ацетоксим, 2,4-дихлоранилин, 2-нитро-4-хлоранилин,
.м-нитродиметиланилпн, o-броманилин, о-хлоранилин, антипирин (в интервале рЛрН+
от 2 до 3,5).
4. Сильные основания: .и-хлоранилин, .и-броманилин, n-нитрозодифениламин, семи-
карбазид, n-хлоранилин, n-броманилин, а-нафтиламин, м -то луп дин, м-анизи дин, п-ани-
зидин, бензиланилин, o-толуидин, n-толуидин, метиланилин, о-анизидин, метил-о-толу-
идин, хинолин, метил-п-толуидин, этиланилин, диметиланилин, пиридин, диметиламин,
2-пиколин, дифенилгуанидин, пиперидин, диэтиламин, трифенилгуанидин, метил-к-пропил-
анилин, n-аминодиметиланилин, три-н-бутиламин, ди-н-бутиламин, гуанидин, ди-и-про-
пиланилин, диэтиланилин, триэтаноламин, дпме-
тил-о-толуидин, диэтил-п-толуидин. триэтил амин,
диэтил-о-толуидин (выше рАвщ 3,5). О связи
между силой основанпя и молекулярной струк-
турой см. [334].
б. Зависимость между силой основания
и ППН послужила предметом многих
исследований. Из рис. 21 очевидно, что
основание, значение рА\вн+ которого равно
1—2, может быть оттитровано (при благо-
приятных обстоятельствах) только потен-
циометрически в уксусной кислоте. Рис. 22
показывает, что между ППН и значением
рА^ для оснований, у которых рК изме-
няется от 9 до 13, существует линейная
зависимость [332, 838].
в. Зависимость между силой основания
и ППН в уксусной кислоте. Стреули уста-
новил существование линейной зависимо-
сти между ППН азотсодержащих основа-
ний и величинами р&вн+ сопряженных
этим основаниям кислот в уксусном ангид-
риде [778]. Нейтральные незаряженные
основания при сравнении с отрицательно
заряженными основаниями-анионами ока-
зываются слабее в уксусном ангидриде,
Р п с. 22. Зависимость между ППН
и рК [838].
.4 — скачок потенциала при добавлении
0,1 н. хлорной кислоты между 9,5— 11 .пл;
Б — потенциал полунейтрализации: 1 —
пиперидин; 2 — пиридин; 3 — п-хлорани-
лии; 4 — At-хлоранил ин; 5 — ли-нитроани-
лин; 6 — о-хлоранилин; 7 — п-нитроани-
лин; 8 — о-нитроанилин.
чем в воде.
Ряд исследователей изучали использование уксусного ангидрида в каче-
стве растворителя [249, 288, 831]. Азотсодержащие основания, которые
нельзя оттитровать как основания в ацетонитриле или уксусной кислоте,
могут быть оттитрованы потенциометрически в уксусном ангидриде. Недо-
статок этого растворителя заключается в том, что он реагирует с аминами,
способными к ацилированию, в результате чего образуются амиды кислот
значительно более слабой основности.
Для исследований, проводимых в уксусном ангидриде, подходящим
титрантом оказался 0,05 н. раствор хлорной кислоты в смеси уксусной кисло-
ты и уксусного ангидрида (1 : 1) [778]. Для потенциометрического титрова-
ния были использованы стеклянный электрод и электрод сравнения серебро —
хлорид серебра. Последний был погружен в уксусный ангидрид, предвари-
тельно насыщенной хлоридами серебра и лития (гл. 12, разд. 67). На рис. 23
изображены кривые потенциометрического титрования 100 мл 0,25 мМ
растворов оснований. Очевидно, сила оснований не нивелируется уксусным
ангидридом и, следовательно, он может быть использован для дифферен-
цирующего титрования. Краткий перечень некоторых величин ППН и рК
оснований приведен в табл. 16 [778].
На рис. 24 (прямая 1) изображена зависимость ППН в среде уксусного
ангидрида от значения рАвнъ измеренного в воде. Основания, содержащие
66
Глава 3
Таблица 16
Величины ППН оснований в уксусном ангидриде и значения их рКвн+ в воде
Основание ППН рКвн+ (лит. данные) рКВН+ (рассч. данные) Основание ППН рКВН+ (лит. данные) рКВН+ (рассч. данные)
N, N-Диметил- бензиламин 196 9,02 9,01 Диметилсульф- оксид 605 1,0
N, N - Диэти л ани - ЛИН М-Этил-М-метил- анилин 322 358 6,52 5,99 6,55 5,85 1-Метил-2-пир- ролидинон Трифенилфосфин Кофеин 676 620 633 0,61 -о,з 0,49
М,М-Диметиланн- лин 389 5,21 5,25 Фенилмочеви на 660 -0,30 -0,04
гетероатомы азота, находятся дальше от линии 1. Примечательно, что три-
фенилфосфин (табл. 16) может быть оттитрован в уксусном ангидриде, в то
время как его азотсодержащий аналог трифениламин не удается оттитровать,
так как он является очень слабым основанием, рК = 16,83 [47]. Замещенные
фосфины могут быть оттитрованы в нитрометане
хлорной кислотой (см. гл. 31 и работу [781]).
Диметилсульфоксид (CH3)2SO титруется как
основание в уксусном ангидриде [878].
Р и с. 23. Кривые потенциометрического титрования
оснований в уксусном ангидриде [778].
Номер соедине- ния Соединение рАГВН+ Литера- тура
1 Ацетанилид -2,9 505
2 Ацетамид -0,48 382
3 Кофеин 0,61 497
4 Мочевина 0,50 505
5 Метилмочевина 0,90 332
6 М,М-Диметиланилин 5,21 778
7 Хинолин 5,06 778
8 Пиридин 5,30 332
9 N.N-Диэтилапилин 6,52 332
10 М.М-Диметилбензиламин 9,02 497
И Три-н-бутиламип 9,85 332
г. Зависимость между силой основания и ППН в нитрометане [333, 765,
779]. Нитрометан является хорошим растворителем для титрования слабых
или средней силы органических оснований. Он обладает очень слабыми кис-
лотными свойствами, поэтому никакого нивелирующего действия не наблю-
дается. Определение оснований с величиной р7Гвн+ больше 14 оказалось
невозможно провести из-за следов воды. Стреули титровал 100 мл 0,25 мТИ
раствора оснований в нитрометане 0,05 н. раствором хлорной кислоты в нит-
рометане. Некоторые кривые потенциометрического титрования изображены
на рис. 25, а результаты приведены в табл. 17. Рис. 25 показывает, что «кру-
тизна» потенциометрических кривых в интервале степени нейтрализации
от 20 до 80% одинакова для дифениламина, N-мети л ацетанилида, N-этил-
N-метиланилина, пиридина, N,N-диэтил анилина и N,N'-дифенил гуанидина.
В случае оснований, величины р7Гвн+ которых меньше 8, 10%-ная нейтра-
лизация вызывает скачок потенциала 11 + 1 мв. Однако в случае мочевины
наблюдался скачок потенциала 23 мв, т. е. кривая идет «круче». Такая
кривая типична также для замещенных производных мочевины, амидов
кислот и их монозамещенных.
Сила оснований
67
< Таблица 17
Величины ППН оснований в нитрометане и значения их рКвн+ в воде
(ППНо = ППН И,М'-дифенилгуанидина)
Основание ДППН Линия на рис. 24 РКВН+ (лит. данные) РКВН+ (рассч. данные)
Пиперидин —47 2 11,09 10,77
Триэтиламин —69 2 10,64 11,01
2-Цианэтиламин 204 2 7,86 7,49
бис-(2-Цианэтпл)ампн 386 2 5,14 5,14
о-Фенилендиамин 412 2 4,52 4,81
2-Нафтиламин 490 2 4,11 3,80
Антраниловая кислота 602 2 2,15 2,36
Дифениламин 701 2 0,85 1,08
Метилмочевина 576 3 0,90 0.79
Мочевина 590 3 0,50 0,61
N-Метилацетанилид 663 3 —0,50 —0,34
Ацетамид 691 3 -0,48 —0,70
Ацетанилид 757 3 —0,9 [505] —1,56
«Крутизна» части потенциометрической кривой, ведущей к точке перегиба, может
также наблюдаться в случае фенолов в среде ацетона или пиридина и объясняется образо-
ванием водородной связи между сольватированными молекулами [258, 782].
Линии 2 и 3 на рис. 24 показывают, что ППН аминов, амидов и производ-
ных мочевины в воде являются функцией рЛ\вн+- За нулевое значение при
Рис. 24. Зависимость между силой осно-
вания и ППН.
I —в уксусном ангидриде; 2, з —в нитрометане
[778, 779]; см. табл. 16 и 17.
Рис. 25. Кривые потенциометри-
ческого титрования оснований в ни-
трометане [779].
1 — дифениламин; 2 — N-метилацетани-
лид; 3 — мочевина; 4 — М-этил-М-мстил-
анилин; 5 — пиридин; 6 — N, N-диэтил-
анилин; 7 — N, N'-дифенилгуанидин
(РКВН+ Ю.ОО [332]).
измерениях принимается linH N.N'-дифенилгуанидина (см. гл. 2, разд. 16, в).
Линия для аминов, содержащих азот в качестве гетероатома, гидроксильную
I
или = N — С — О — группу, расположена иначе, чем линии 2 и 3. Таким
68
Глава 3
образом, хинолин, изохинолин, пиридин, ди- и три-(2-оксиэтил)амин при
сравнении с другими аминами оказываются более сильными основаниями
в нитрометане, чем в воде.
д. Зависимость между силой основания и ППН в ацетонитриле. Большое
число растворителей, слабо или совсем не способных к автопротолизу, при-
менялось для измерения силы оснований с использованием главным образом
индикаторного метода. К этим растворителям относятся бензол [172, 5596],
хлорбензол [54], ацетонитрил [247, 333, 455], нитробензол [469], нитрометан
(см. разд, г и работы [333, 765]), хлороформ и четыреххлористый углерод [35].
Согласно Фрицу, в ацетонитриле между ППН и силой основания суще-
ствует линейная зависимость. Однако другие растворители также пригодны
для потенциометрических измерений потенциала полунейтрализации: это
высокополярные растворители — нитрометан и нитробензол (ацетонитрил
также полярный растворитель), умеренно полярные растворители — этилаце-
тат и неполярные растворители — дихлорэтан [333]. Холл мл. установил,
что среди ароматических аминов п- и особенно о-нитроанилин являются
слишком слабыми основаниями, чтобы давать резкие пики. Нитроанилин,
однако, может быть легко оттитрован. Пиролидин был сильнейшим из иссле-
дованных оснований. В дихлорэтане осаждаются перхлораты метил- и этил-
аминов.
Концентрация аминов, использовавшихся с пятью вышеприведенными
растворителями, была такой, что на 50—75 мл растворителя требовалось
для титрования 0,3—1,1 мл 0,5 М хлорной кислоты в диоксане. Так как
добавление такого малого количества титранта требует большой осторожно-
сти, Холл мл. использовал микробюретку — пипетку Джильмонта
емкостью 1 мл. Титрант добавляли порциями по 0,03 мл. Электроды типа
Бекмана стандартизировали в диизобутиламинперхлоратном буферном
растворе.
Таким образом, относительная сила оснований не зависит от титранта,
например хлорной кислоты или n-толуолсульфокислоты, и в первом при-
ближении не зависит также от природы растворителя. Следовательно, сила
исследованного основания в первую очередь является функцией структуры:
электроноакцепторные группы, например винильная или фенильная, вызы-
вают заметное уменьшение силы оснований. Порядок изменения силы алкил-
аминов в пяти вышеприведенных типах растворителей (и в воде) следующий:
NH3<RNH2, r2nh>r3n.
Однако «идеальный» ряд оснований по силе должен был быть следующим:
NH3 < RNH2 < R2NH < R3N.
Отклонения, наблюдаемые, например, в метиловом и этиловом спиртах,
могут быть обусловлены образованием сольватов за счет водородной связи
между ионами аммония и гидроксильной группой растворителя. Идеальный
порядок изменения основности не всегда наблюдается в инертных раствори-
телях, поскольку водородные связи могут образоваться и с другими молеку-
лами, например молекулами аминов, кислот и солей (ср. [35, 172, 469, 5596]).
Сила основания определяется изменением энтропии, а также стерическим
и сольватирующим эффектами, и их суммарное действие на каждый из аминов
может быть различным. Результаты исследований представлены на рис. 26.
Величины рХвн+ для 16 монофункциональных оснований из 18 соответ-
ствуют величинам, измеренным в воде с точностью до ±0,2 единицы рК.
Полученные зависимости показывают, что порядок изменения относитель-
ной силы оснований в воде является полезной проверкой для титрования
в неводных средах. Хотя степень сольватации может быть различной, это
не может вызвать очень больших расхождений.
Сила девяти различных оснований, содержащих кислород или азот
по-соседству с аминогруппой, увеличивается в органических растворителях
Сила оснований
69
по сравнению с силой этих оснований в воде. Функция, представленная более
короткой линией на рис. 26, показывает в среднем, что рЛвн+ исследован'
ных оснований с точностью до ±0,18 единиц рХ согласуется со значениями,
измеренными в воде. Так, пиперазин сильнее в ацетонитриле, чем в воде
(см. гл. 23, разд. 123). Можно сделать заключение, что богатые электронами
полярные группы молекул сольватируют ониевые ионы в результате образо-
вания промежуточной Н-связи. Это стабилизует такие ионы по сравнению
с ионами, не образующими водородной связи (см. гл. 2, разд. 19).
Эксперименты Холла мл., Стреули, Фрица и Вейбела показали, что
измерение потенциала полунейтрализации и относительной силы оснований
в воде является полезным при титровании оснований. Сольватация может
привести к незначительным или даже большим отклонениям. Однако вели-
чина рЛвн+, определенная на основе измерения, отклоняется лишь иногда
Рис. 26. Зависимость между силой
основания и ППН в ацетонитриле
[333].
1 — ди-н-бутиламин; 2 — пиперидин; з —
триэтиламин; 4— триметиламин; 5— ме-
тиламин; 6 — диметиламин; 7 — н-бутил-
амин; 8 — иэобутиламин; 9 — N-этилпи-
перидин; 10 — 1,3-ди-о-толилгуанидин;
11 — аммиак; 12 — 1,2,3-трифенилгуа-
нидин; 13 — 2,6-лутидин; 14 — диэтил-
анилин; 15 — п-метоксианилин; 16— п-то-
луидин; 17—анилин; 18— п-бромани-
лин; 19—- этилендиамин; 20— пиперазин;
21 — этанол амин; 2 2— морфолин; 23 —
1-карбэтоксипиперазин; 24 — бенэоилпи-
перазин; 25 — N-зтилморфолин; 26 —
2-тозилпиперазин: 27 — N-аллилморфо-
лин.
на величину более ±0,5 единицы рК. Отклонениями, вызванными колеба-
ниями температуры в процессе титрования, можно пренебречь. Невоспроиз-
водимость показаний, полученных со стеклянным электродом, может при-
вести к большим ошибкам, хотя эти ошибки могут быть устранены тщатель-
ной стандартизацией. Непостоянство ионной силы может быть источником
серьезных ошибок, особенно в растворителях с низкой диэлектрической
проницаемостью. В таких растворителях степень ионизации изменяется
в большей степени с изменением концентрации раствора.
Построение графика зависимости потенциала полунейтрализации от зна-
чения рЛвн+, измеренного в воде, показывает, что последняя величина
зависит в первую очередь от действия заместителей и во вторую — от при-
роды растворителя. Порядок изменения основности алкиламинов в воде
следующий:
NH3<RNH2 и r2nh>r3n.
В ацетонитриле, этилацетате, нитробензоле и нитрометане этот порядок
не меняется. В хлороформе и хлорбензоле третичные амины сильнее,
но в н-гептане, бензоле и 1,4-диоксане сильнее вторичные амины [849].
Вследствие способности алкильных групп отдавать электроны можно было
бы ожидать следующий ряд изменения основности:
NH3 < RNH2 < R2NH < R3N.
Однако результаты экспериментов, проведенных в метиловом и этиловом
спиртах, не подтверждают этой последовательности. Это может быть объясне-
но стабилизацией образовавшихся в процессе сольватации ионов аммония
путем образования Н-связи. Вышеприведенный «идеальный» порядок изме-
70
Глава 3
нения основности не будет соблюдаться в случае инертных растворителей,
потому что ионы алкиламмония образуют водородную связь или с молекулой
амина или с молекулой кислоты соответственно [35, 172, 479].
О взаимосвязи между рКъ (Н2О) фентиазинов и симпатомиметических
аминов и величинами ППН в ацетоне, ацетонитриле, изопропиловом спирте
и нитрометане см. работу [125].
24. ВЛИЯНИЕ СОЛЬВАТИРУЮЩИХ ЭФФЕКТОВ НА СИЛУ АЗОТСОДЕРЖАЩИХ
ОСНОВАНИЙ; ЭФФЕКТ АССОЦИАЦИИ
В гл. 2 обсуждалось изменение основности ал.киламинов в зависимости
от природы применяемого растворителя. Браун и сотрудники объяснили
это изменение стерическими эффектами, другие исследователи — эффектами
сольватации.
а. Предметом спектрофотометрических исследования Пирсона и Фогель-
сона было определение порядка изменения основности алкиламинов по срав-
нению с 2,4-динитрофенолом [631]. На основании констант равновесия обра-
зования ионных пар было установлено, что наиболее сильными основаниями
в хлороформе, хлорбензоле и дихлорэтане являются третичные амины,
в н-гептане, бензоле и диоксане — вторичные амины и, наконец, в этилацета-
те — первичные амины (ср. гл. 2, разд. 19, б).
В теории «обратного напряжения» Брауна отмечено, что валентные углы в третичном
амине несколько больше, чем это должно было бы соответствовать нормальной тетраэдри-
ческой конфигурации [96]. В результате координации протона молекула «теснится»
в тетраэдрическую форму. Чем компактнее молекула в пространстве и чем больше
электростатическое отталкивание заместителей у атома азота, тем больше они про-
тиводействуют этому напряжению. Таким образом, несмотря на индукционный эффект
электронодонорных заместителей, триметиламин является более слабым основанием, чем
метиламин. Это, конечно, аргумент, противоположный аргументу, основанному на
эффектах растворения (см. выше).
Более того, экспериментальные данные доказывают, что сила оснований
зависит от эффекта сольватации растворителем [219, 629, 631, 820]. Распре-
деление аминов между водой и органическим растворителем также показы-
вает, что тенденции к сольватации и образованию водородной связи у алкил-
аминов могут значительно меняться. Скорости распределения для метил-
аминов, т. е. концентрация амина в водной фазе к концентрации амина
в органической фазе, приведены в табл. 18 (см. также гл. 5, разд. 30).
Таблица 18
Алкиламин В четырех- хлористом углероде В бен- золе В хлорбен- золе В диэтило- вом эфире В хлоро- форме
Аммиак 246 210 179 180 26
Метиламин 31,1 27.8 57,6 43,5 11,4
Дпметиламин 13,5 11,2 13,6 18,2 2,75
Триметпламин 1,3 2,1 2,0 2,2 0,45
В хлорбензоле порядок изменения основности «-бутиламинов следую-
щий: моно < ди < три [46]. Таким образом, в противоположность теории,
основанной только на пространственных затруднениях, порядок изменения
основности зависит и от влияния возрастающего индуктивного эффекта
заместителей (см. гл. 4, разд. 27). Согласно теории «обратного напряжения»
стерический эффект должен быть особенно сильным в инертных раствори-
телях, особенно в случае 2,4-динитрофенола. В хлорбензоле имеются ассо-
циаты основание — кислота, тогда как в водной среде — отдельные ионы.
Сила оснований
71
Поэтому порядок изменения основности бутиламинов в хлорбензоле обрат-
ный порядку изменения основности, определенной в воде. Все это показы-
вает, что пространственные затруднения нельзя рассматривать как един-
ственный фактор, определяющий силу основания, особенно в неводных средах.
СН2СН2СН2СН3
“ * 4-и
СН2СН2СН2СН3
Ассоциат в растворах три-н-бутиламина и 2,4-динптрофенола
б. Ассоциация в инертных растворителях. При взаимодействии уксус-
ной кислоты и триэтиламина в хлороформе и четыреххлористом углероде
соответственно реагирующие вещества образуют ассоциаты различного
типа в зависимости от концентрации раствора ([35 — 37] и гл. 2, разд. 15, г).
В четыреххлористом углероде'.
......и —
н3с —с С—сн3
-1^0 • • • • н СГ
"N(C2H5)3
Тип ассоциации: ^Н-Д(ЛН)-
При дальнейшем добавлении триэтиламина:
...О
н3с-(\
хО-.... HN(C21I5)3
Тип ассоциации:
В хлороформе:
_,^о .... НСС13 о .... НСС13 N(C2H5)3
н3с—с; ----► н3с—с; + н+
\ •..• HN(C2H5)3 _?\) .••• НСС13 N(C2H5)3
/2 /2
Тип ассоциации: ВН+-Л"; растворитель5(5Н+)-Л~ • 2 молекулы раствори-
теля.
Резонансная форма аниона стабилизована образованием водородной
связи с молекулой растворителя хлороформа таким образом, что отрицатель-
ный заряд распределился равномерно по атомам кислорода.
Приведенные выше примеры показывают, что при взаимодействии Н-кис-
лоты и акцепторного основания в инертных растворителях в результате
образования водородной связи образуются ионные пары [33, 54, 171]. Однако
степень переноса протона может меняться в широких пределах; кроме того,
при взаимодействии кислот и оснований перенос протона к основанию
не является необходимым. Если основание превращается в ониевые ионы,
водородная связь, образованная с анионом, по существу носит электроста-
тический характер. Продукты реакции слабых кислот и слабых акцепторных
оснований в неполярных растворителях представляют собой простые ком-
плексы с Н-связью: протон остается присоединенным за счет ковалентной
связи и благодаря электростатическому притяжению амин связан с «кислым
водородом». Помимо этих двух предельных случаев существуют также
72
Глава 3
другие переносы. Так, когда пиридин титруют трифторуксусной кислотой
в хлороформе, сначала образуется комплекс типа В «НА в отношении
1:1. Последующее добавление кислоты приводит к комплексу ВН+ -А (НА)~
в отношении 1 : 2
Аналогично взаимодействие карбоновой кислоты с более слабыми прото-
нодонорными свойствами и пиридином может быть представлено следующим
образом:
R—С( = О) —О —Н .... NG5H5 т> R —С(=О) —о- .... HNG5H5
Таким образом, порядок изменения силы оснований зависит в основном
от структурных характеристик. Этот порядок может изменяться в зависимо-
сти от природы растворителя. Однако эти изменения не столь значительны,
когда, учитывая структурные характеристики, значение потенциала полу-
нейтрализации сравнивают с величиной рАвн+, измеренной в воде. Теории,
объясняющие причины различия в силе оснований, часто противоречивы.
Это может быть отнесено, помимо других причин, за счет неодинаковых
экспериментальных условий, применяемых, например, при определении
кислотно-основного равновесия спектрофотометрически с использованием
потенциометрического или термометрического титрования [236].
Глава 4
СВЯЗЬ МЕЖДУ КИСЛОТНОСТЬЮ,
основностью и молекулярной структурой
Связь между кислотностью, основностью и молекулярной структурой
очень важна при исследовании химических связей и электронных смещений
в молекуле.
В пределах одного периода периодической системы кислотность гидридон
RH возрастает с увеличением электроотрицательности R.
НСН3 < HNH2 < НОН < HF.
Такое изменение можно объяснить увеличением заряда ядра и уменьше-
нием числа электроположительных атомов водорода, связанных с централь-
ным атомом. Это справедливо также для катионных кислот с зарядом одина-
кового типа
HNH£ < НОН2 < HFH+.
На основании кислотности порядок изменения силы соответствующих
оснований следующий:
СНз > NH2 >НО-> F- и NH3>H2O>HF.
(ср. гл. 1, разд. 8).
Состояние равновесия определяется тремя факторами: 1) потенциальной энергией,.
2) кинетической энергией и 3) энтропийным (вероятностным) фактором. Различия в потен-
циальной энергии могут быть обусловлены изменениями энергии связи, вызванными
резонансом, электрофильными или нуклеофильными эффектами функциональных групп
и взаимодействием ионов и диполей. Кинетическая энергия может изменяться вследствие-
вращательных и поступательных движений молекул пли колебательных и вращательных
движений внутри молекулы.
Во многих случаях факторы, вызывающие изменения в положении равновесия реак-
ции, не могут быть определены экспериментально. Однако при изучении структурных
изменений в связи с кислотно-основным равновесием удалось установить, что смещение-
электронного облака и образование диполя также может вызвать изменение в потен-
циальной энергии. Трудно ожидать, что теория, принимающая во внимание только
молекулярную структуру кислот и оснований, позволит всегда решить в любом и каждом
случае проблему силы кислот или оснований без учета природы растворителя. Весьма
трудно установить единую закономерность для органических соединений, поэтому
связь между молекулярной структурой и кислотностью и основностью будет про-
демонстрирована на некоторых характерных примерах.
25. ВЛИЯНИЕ! ЗАМЕСТИТЕЛЕЙ, ИНДУКЦИОННЫЙ И ДРУГИЕ ЭФФЕКТЫ
В случае связи углерод — галоген вследствие изменения электронного
сродства элементов валентные электроны смещены по направлению к гало-
гену («неэквивалентность электронного участия» [628]). Электроноакцептор-
ная (электрофильная) группа участвует в большей степени в смещении элек-
тронной пары, индуцируя положительный заряд на атоме, с которым она
связана. Неодинаковое распределение заряда приводит к частичной поля-
ризации молекулы, например Н3С -> С1. Индуцированный положительный
заряд сам по себе также оказывает электроноакцепторное действие на сосед-
ние атомы, которые в свою очередь вызывают последовательно убывающее
индукционное смещение электронной плотности с других связей в том же
направлении (/-эффект).
74
Глава 4
Индукционный эффект атома хлора передается вдоль цепи углеродных
атомов без уменьшения только по сопряженной системе двойных связей
(стр. 201 в работе [206] и стр. 101 в работе [605]). Направление смещения
электронов показано стрелками
н С1 он
I I I
Cl—СН, С1— С*—С-"—он
I I 1
Н CI он
Гидрат трихлоруксусной
кислоты
Электроноакцепторный эффект увеличивается по мере увеличения электро-
положительности электроноакцепторного атома или группы
— SRJ > — SR> — S-; —NR2H+>-NR2>NR-.
Он увеличивается также с увеличением заряда ядра у элементов с одинако-
вым числом внутренних электронных оболочек
_ F > — OR > — NR2 < — CR3
и уменьшается по мере увеличения числа внутренних электронных оболочек
— F > — С1 > — Вг > —I
Нитрогруппа, атом азота в которой обладает положительным зарядом
в обеих основных резонансных структурах, является относительно сильной
электрофильной группой
Двойные или тройные связи также могут обладать электроноакцепторным
действием — С = CR > — CR = CR2 > — CR3.
а. Влияние заместителей на силу алифатических карбоновых кислот.
Льюис и Лукас дали следующую интерпретацию электроноакцепторного
действия заместителей на кислотность карбоновых кислот. В связи CI <- С
хлоруксусной кислоты центр электронной плотности валентных электронов
смещен по направлению к атому хлора, и это выражается в наличии у моле-
кулы дипольного момента. Это внутримолекулярное дипольное взаимодей-
ствие распространяется также на соседние атомы
Н
I //°
С1 с -- с/
Индукционный эффект, обусловленный атомом хлора
Вследствие этого электронная плотность на карбоксильной группе умень-
шается, координация с протоном становится слабее и, таким образом, прото-
нодонорная способность, т. е. сила кислоты, увеличивается. Термодинами-
ческое обсуждение проблемы, основанное на изменениях энтропии, приводит
к подобным же выводам.
, < В галогензамещенных алифатических кислотах анион стабилизован
в результате взаимодействия диполя G1 <- С с отрицательным зарядом.
Н
3- 1л4. ур/2"
О I и + *
CI—с—cf
Н
‘О’/г-
Связь между кислотностью, основностью и молекулярной структурой
75
Трихлоруксусная кислота в 93 раза сильнее, чем монохлоруксусная,
I
хотя дипольные моменты групп —СС13 и —CGI очень близки. Эффект двух
I
групп одинаков на большем расстоянии, но в области, соизмеримой с вели-
чиной диполя, более важным фактором является распределение заряда
на атоме углерода. Группа I обладает большим положительным зарядом
на углеродЬ, чем группа II
б-? б+
-С^_С1 -сг_н
^С1 \ н
I II
При кислотно-основном равновесии атом, несущий лабильный протон,
обладает относительным дефицитом электронов, находясь в кислотной фор-
ме, и избытком электронов в сопряженном основании. Поскольку электроно-
акцепторная группа усиливает фактически существующий дефицит элек-
тронов, она увеличивает силу кислоты и уменьшает способность анионного
основания координировать протон.
Бранч и Калвин также обсудили влияние индукционных эффектов
на силу кислот. На основе индукционных констант можно видеть, что сила
кислот типа X — СН2СООН увеличивается (—Z-эффект) или уменьшается
(+/-эффект) вследствие индукционного эффекта электрофильных или нуклео-
фильных групп в следующем порядке (стр. 67 в работе [79]).
Х=— SO2R> — NH3+> — NO2> — CN> 'J
> — F>—Cl> — COOH> — Br> l —/-Эффект
> — OCH3>-I > —OR>— SR> — C6H5> — NR,>J
> H>
> - C2H5 > - CH3 > - C(CH3)3 > - coo-
4- /-Эффект
Этот статический постоянный индукционный эффект обозначается Ингольдом Is', если
валентные электроны притягиваются ключевым атомом сильнее, чем атомом водорода,
расположенным на том же атоме углерода, т. е. если ключевой атом более электроотрица-
телен, используют обозначение — Is, в то время как символ Ч“/я применяют в том случае,
если ключевой атом более электроположителен. Влияние ключевого атома быстро умень-
шается с увеличением длины углеродной цепи. По логике вещей влияние заместителей
на силу оснований противоположно (см. этот разд. е).
Приведенные ниже значения рКа различных галогензамещенных алифа-
тических карбоновых кислот показывают, что —Z-эффект является функ-
цией либо числа электроноакцепторных заместителей (Л), либо расстояния
от группы СООН (Б)
А
СНзСООН 4,76*
FCH2COOH 2,7** CF3COOH 0,30
С1СН2СООН 2,86* СЬСНСООН 1,30* СС13СООН 0,89*
ВгСН2СООН 2,87*
1СН2СООН 3,0*
Б
СН3СН2СНС1СООН 2,9**
СН3СНС1СН2СООН 4,1**
СН2С1СН2СН2СООН 4,5**
В табл, 19 приведены данные относительно влияния заместителей Xit
Х2, Х3 в карбоновых кислотах типа .Х\Х2ХзССООН на их силу (ср. [83,
206, 693]). а- и 0-Ацетиленкарбоновые кислоты сильнее, чем насыщенные
* См. работу [497].
** Стр. 68 в работе [393].
76
Глава 4
Влияние заместителей на силу карбоновых кислот
Таблица 19
Кислота Заместители Аа.1О5 1-эффект Литература
*1 х2 Хз
Анион малоновой кислоты соо- Н н 0,20 + 206
Триметилуксусная СНз СНз СНз 0,89 497
Пропионовая СНз Н н 1,35 — 497
Масляная с2н5 Н н 1,51 — 497
Уксусная Н н н 1,75 ± 497
Фенилуксусная с6н5 н н 4,88 — 497
Дифенилуксусная С6Н5 СбНб н 11,5 — 497
Гликолевая он н н 15,1 — 497
Моноиодуксусная I н н 100 — 497
Монобромуксусная Вг н н 135 — 497
Монохлоруксусна я С1 н н 138 — 497
Малоновая СООН н н 141 — 497
Монофторуксусная F н н 200 — 393, стр. 68
Циануксусная CN н н 356 — 206
+NH3CH2COOH NHJ н н 502 — 393, стр. 68
Нитроуксусная no2 н н 2 100 —
Дихлоруксусная Cl С1 н 5 000 —• 497
Трихлоруксусная Cl С1 С1 13 000 — 497
Примечание: Приведенная выше последовательность справедлива и для индукционного эффекта
электрофильных заместителей. Значения Ка определяли в водной среде, и поэтому они могут изме-
няться в каждом случае в зависимости от природы растворителя. Сила кислот зависит также и от
других эффектов.
или олефиновые кислоты, вследствие сильного электроноакцепторного дей-
ствия ацетиленовой связи [189, 547].
рА'а
НС = ССООН 2,23-2,66
RC = ССН2СООН 3,60 - 3,44
RC = С(СН2)ПСООН 4,20-4,58
где R = алкил или фенил и п = 2 — 4.
Существует сопряжение между л-электронной системой и свободной
электронной парой атома кислорода, т. е. электронная система оттягивает
электронное облако с группы СООН и, таким образом, электронная плот-
ность на ней уменьшается.
б. Ароматические кольца, такие, как фенильная группа, обладают
электроноакцепторными свойствами, а электронный секстет ароматического
кольца сопряжен с атомом, имеющим свободную электронную пару. Это
по существу мезомерный эффект (см. ниже), который, как показывает следую-
щий пример, может быть направлен противоположно индукционному эффекту
СН3—СН2-*С1[
2,03 D
1,66 D
1,55 D
Введение фенильной группы в молекулу в общем увеличивает силу кислоты
и уменьшает силу основания. Фенил- и дифенилуксусные кислоты сильнее,
чем уксусная кислота, но анилин и дифениламин являются более слабыми
основаниями, чем метиламин (табл. 20).
в. Влияние заместителей на кислотность фенолов и енолов. Кислотность
замещенных фенолов увеличивается при действии заместителей в неводных
средах в следующем порядке [258]:
NO2 > СНО > COR > CI > Вг > С6Н5
(см. также гл. 17).
Связь между кислотностью, основностью и молекулярной структурой
77
Таблица 20
Кислота Ка.1°5 [497] Основание кь.юв [332, 497]
Уксусная 1,75 Метиламин 43 700
Фенилуксусная 4.88 Анилин 0,0383
Дифенилуксусная 11,5 Дифениламин 0,0000071
В случае салициловой кислоты кислотность фенольного гидроксила
уменьшается СООН-группой. орлго-Заместители, содержащие неподеленные
пары электронов (NO2, СООН, СНО), образуют с фенольным гидроксилом
водородную связь, приводящую к изменениям свойств обеих функциональ-
ных групп, т. е. кислотность карбоксильной группы увеличивается, в то вре-
мя как кислотность фенольного гидроксила уменьшается, и молекула легче
растворяется в полярных растворителях.
Енолы и амиды можно титровать как кислотные аналоги метилатом щелоч-
ного металла или гидроокисью алкиламмония (RCH2R' или RNHR', где
R и R' — электрофильные группы COR, CONH2, СНО, COOR, CONHAr).
Однако, когда электрофильной группой является амидо- или иминогруппы,
титрование не всегда удается осуществить [246]. Так, например, гидантоин
может быть оттитрован в диметилформамиде как кислотный аналог, а ацетил-
мочевина таким образом не титруется
HN —СН2 —СО —NH—СО СНз —СО —NH —CO-NH2
|| Ацетилмочевпна
Гидантоин
г. Нитрогруппа — один из наиболее эффективных электрофильных
заместителей (стр. 248 в работе [863]): нитрометан является «кислотным»
растворителем, рЛ"а = 10,6 [780]. Фенольные соединения превращаются
в сильные кислоты (о- и n-нитрофенолы выделяют двуокись углерода из кар-
боната натрия); с другой стороны, нитроанилин растворяется только в кон-
центрированных минеральных кислотах.
Большая способность ароматической нитрогруппы в орто- и пара-поло-
жениях увеличивать силу кислоты обусловлена ее дипольным моментом,
а иногда стерическими эффектами или ее участием в анионной стабилизации
благодаря резонансу (делокализация заряда).
о -Н итробензоат- аннон
78
Глава 4
Нитрогруппа в ор/ио-положении также стабилизует анион в результате
смещения электронов карбоксилат-аниона в силовое поле атома азота;
о-нитробензойная кислота примерно в 18 раз сильнее, чем п-нитробензой-
ная (ср. [529]).
Таблица 21
Константы диссоциации замещенных бензойных кислот в воде
( X Ю6) и константы ассоциации в бензоле (Х10-5) [173]
Заместитель орто мета пара
IS ДИСС в воде яасс в бензоле IS ДИСС в воде Аасс в бензоле IS ДИСС в воде ^асс в бензоле
сн3 12,0 0,87 5,37 1,35 4,26 1,07
С1 115,0 12,0 14,8 11,5 10,5 6,60
он 106,0 282,0 8,32 2,24 2,63 0,88
no2 676,0 274,0 32,8 66,0 37,6 62,8
ОСНз 8,13 0,05 8,13 2,39 3,39 0,83
nh2 1,12 0,88 1,78 0,85 1,29 0,28
CN 25,0 36,3 28,2 33,8
В табл. 21 приведены константы диссоциации замещенных бензойных
кислот в воде и их константы ассоциации в бензоле (см. также гл. 2^
разд. 16, а и работу [173]).
д. Аминогруппа проявляет основные свойства, т. е. обладает способ-
ностью координировать протоны (нуклеофильный заместитель). Однако
ее индукционное влияние на силу кислоты является электрофильным, так
как заряд ядра атома азота больше, чем заряд ядра атома углерода (стр. 248-
в работе [863]). В аминокислотах вследствие образования цвиттер-иона
+H3N — СН2СОО“ —Z-эффект остается скрытым, однако он становится
очевидным при сравнении, например, силы анилиндиуксусной кислоты
с силой 2-фенилглутаровой кислоты. Первая из них приблизительно
в 3,5 раза более сильная кислота (стр. 248 в работе [863]):
CeH5N(GH2GOOH)2 СвН5СН(СН2СООН)2
Анилиндиуксусная кислота 2-Фенплглутаровая кислота
(Я-105 = 270) (/С-105 = 77)
Другой пример: р/Га нитрогуанидина равен 12,20, а 1-амино-З-нитро-
гуанидина 10,60 (см. гл. 20, разд. 115).
е. Влияние заместителей на силу азотсодержащих оснований. Индук-
ционное влияние заместителей на силу оснований аналогично наблюдаемому
в вышерассмотренных примерах для кислот. Заместители в основаниях
типа X’jX^X^N связаны непосредственно с протоноакцепторным атомом,
обладающим свободной электронной парой. Таким образом, они оказывают
непосредственное влияние и вместе с другими эффектами существенна
влияют на силу основания.
В кислотах типа Х^гХзССООН, наоборот, имеются три (I) или четыре (II) атома
между электронной парой, поделенной водородом и кислородом, и атомом или группой (Х)„
оказывающим индукционное влияние
Н О
I II
х —с — с — о —н
I
н
I П
Относительно индукционного эффекта алкильных заместителей в алифати-
ческих аминах см. работу [348].
Связь между кислотностью, основностью и молекулярной структурой 79
Таблица 22
Влияние заместителей на силу органических оснований
Основание Заместители къ (X 108) Г-эффект Литера- тура
Х2
Мочевина nh2co н н 0,0000032 + 779
Дифениламин с6н5 С6Н5 н 0,0000071 + 332
1-Нафтиламин С10Н7 н н 0,00836 + 332
2-Нафтиламин С10Н7 н н 0,0129 + 332
Анилин СвЩ н н 0,0383 + 497
Фенилгидразин c6h5nh н н 0,16 + 497
Гидроксиламин он н н 1,07 + 497
Гидразин nh2 н н 302 + 497
Аммиак н н н 1 790 -+-
Бензиламин СбНбСН2 н н 2 000 — 497
Триметиламин СНз СНз СНз 5 270 — 497
Этилендиамин nh2ch2ch2 н н 8 510 — 497
Метиламин СНз н н 43 700 — 497
Диметиламин СНз СНз н 51200 — 497
Этиламин С2Н5 н н 56 000 — 497
Триэтиламин с2н5 С2Н5 С2н5 56 500 — 497
Диэтиламин С2Н5 С2н5 н 126 000 — 497
Примечание: Приведенный выше порядок справедлив и для последовательности индукционных
эффектов отдельных заместителей. Значения определены в водной среде и могут изменяться
в каждом случае в зависимости от природы растворителя. На силу оснований влияют также и другие
эффекты.
Аналогично табл. 19 данные, приведенные в табл. 22, иллюстрируют
влияние заместителей на силу оснований типа -X\X2X3N.
26. ВЛИЯНИЕ ЭЛЕКТРОСТАТИЧЕСКОГО ПОЛЯ
Наряду с индукционным эффектом, передаваемым вдоль углеродной цепи,
существует также взаимодействие через пространство, например между
диполями, ассоциированными с карбоксильной группой или анионом карбо-
ксилата и электроотрицательной группой [458, 869]. В галогензамещенных
алифатических карбоновых кислотах положительный конец диполя С -+
С1 ближе к аниону карбоксилата, чем отрицательный конец, в резуль-
тате чего происходит стабилизация аниона. Степень стабилизации зависит
от величины диполя, расстояния между карбоксилат-анионом и диполем
и, наконец, от диэлектрической проницаемости среды, в которой происходит
взаимодействие. Позднее это явление было названо электростатическим
взаимодействием и в некоторых случаях оно должно приниматься во вни-
мание вместе с индукционным эффектом.
Карбоксильная, а также группы СООС2Н5 и CONH2 обладают электроно-
акцепторными свойствами. В случае дикарбоновых кислот, однако, ион
СОО" уменьшает кислотность второй СООН-группы, поскольку протон
труднее отделить от отрицательно заряженного иона, чем от нейтральной
молекулы.
В симметричных дикарбоновых кислотах две карбоксильные группы обладают одина-
ковыми возможностями отдать один протон. Но анион карбоновой кислоты, например
НООС(СН2)ЛСОО_, может координировать только один протон. При диссоциации второго
протона возникает обратная ситуация: имеется одна возможность для потери и две —
для координации протона. На основе этих простых рассуждений можно было бы ожидать,
что константа диссоциации первого порядка будет в 4 раза больше, чем константа диссо-
циации второго порядка. В действительности отношение К': К" больше 4 вследствие вли-
яния электростатического поля, особенно в случае двух соседних карбоксильных групп
(табл. 23) (ср. стр. 384 в работе [380]).
80
Глава 4
Таблица 23
Первая (К') и вторая (К") константы диссоциации
карбоновых кислот (Ка-105)
Кислота К’ К" К' : К" Литература
Щавелевая 6500 6,1 1065 497
Малоновая 139,7 0,08 1750 497
Янтарная 6,9 0,247 28
Глутаровая 4,5 0,38 12 380, стр. 384
27. ВЛИЯНИЕ МЕЗОМЕРИИ НА СИЛУ КИСЛОТ И ОСНОВАНИЙ
Различное сродство к электрону ди- или полиатомных заместителей
с двойной или тройной связью углерод — кислород или углерод — азот
вызывает несимметричное распределение л-электронной плотности в двой-
ной связи. Это состояние может быть описано полярной мезомерной струк-
турой [16]
\с=Оч->\с — о
В противоположность смещению, обусловленному /-эффектами, в о-свя-
зях, M-эффекты вызывают смещение л-связей. Это постоянное смещение
л-электронной системы может привести к общей поляризации молекулы.
Направление смещения определяется структурой соединения. Мезомерный
эффект может быть вызван или одной из двух следующих причин, или, воз-
можно, обеими:
1. Присутствием гетероядерных л-электронных орбиталей
С=£о
2. Распространением л-электронной системы, например перекрывание
электронной пары р-орбитали с л-системой
СН2=СН— сЦ
В первом случае М-эффект действует в том же направлении, что и /-эф-
фект, а во втором — в противоположном направлении (см. разд. 25, б).
Существует различие между электронодонорными и электроноакцептор-
ными группами; эти группы по их действию можно расположить в определен-
ной последовательности. К электронодонорным группам (+-Л/) относятся
Карбоксил ат-ион > амино > гидроксил > метил.
К электроноакцепторным группам (—М) относятся
гл
СГ< —S=O| < —С=О| < —С=О| < — N = O| < — N = O|
I _ I - - I -
|OR 1ОГ
«Мезомерной структуре» не соответствует какое-либо реальное состояние молекулы,
но такие структуры помогают в понимании молекулярной структуры (стр. 200 в работе
[26]). Электромерный эффект (Е) является важным в ненасыщенных соединениях: он обу-
Связь между кислотностью, основностью и молекулярной структурой
81
словливает поляризуемость молекул в момент реакции
С = С
«-Лс+^с-ЛЛс-с/
Относительно индукционного (Zs), мезомерного (М), индуктомерного (7^) и электро-
мерного (Е) смещений электронов см. стр. 182—185 в работе [231].
Теоретически сила каждой кислоты или основания, представляющих
собой ненасыщенные или ароматические соединения, зависит от распределе-
ния электронов, которое может быть изображено при помощи мезомерных
структур. Рассмотрим, например, карбоксилат-анион, у которого отрица-
тельный заряд рассеян и сродство к протону мало (стр. 258 в работе [605])
или
Приведенные выше формулы показывают возможность смещения двух л-электронов
к одному из атомов, в результате которого возникает «семиполярная» связь (Лоури
и Сидвик). Эти резонансные структуры, возможно, существуют только как промежуточ-
ные переходные состояния в ходе реакции (стр. 376 в работе [380]).
- Мезомерный эффект описывает распределение заряда в неактивирован-
ном «основном состоянии» молекулы и изображается при помощи канониче-
ских резонансных форм, например:
О = С — СН = СН— NH2*-*-O — С = СН — CH = NH£
I I
СН3 СН3
Локализацию электронов и направление перемещения электронного облака
можно также изобразить изогнутыми стрелками
Cd=L С-О СРьСк’Н,
I
сн3
Ионизация кислот обратима, поэтому константа диссоциации зависит
от электронной конфигурации как кислоты, так и аниона (стр. 246 и 258
в работе [863]).
Константа равновесия диссоциации представляет собой отношение ско-
рости образования Н+-иона из молекулы R — ОН к скорости присоедине-
ния Н+-иона к иону (R — О)~. Отсюда на силу кислоты влияют два не зави-
сящих друг от друга фактора: 1) освобождение протона и 2) стабилизация
аниона вследствие резонанса.
Фенолы обычно более сильные кислоты, чем спирты, а анилин более
слабое основание, чем метиламин. Для фенолов, так же как и для анилина,
сила кислоты или основания определяется резонансными эффектами, кото-
рые могут быть изображены различными структурными формулами. Эти
формулы не соответствуют реальному состоянию соединения, а являются
предельными структурами, в которых электронное перемещение завершено.
Истинное состояние не может быть выражено ни формулой Кекуле, ни хино-
идными мезомерными структурами, приведенными ниже. Например, в фено-
лят-анионе отрицательный заряд увеличивает электронную плотность в бен-
зольном кольце и электронная пара смещена по направлению к л-системе
82
Глава 4
К хиноидным мезомерным структурам относятся следующие *
Фенод
В структурах б — г положительная часть силового поля диполя спо-
собствует удалению протона, в то время как в структурах е — з отрица-
тельный заряд распределен по всей молекуле; следовательно, заряд, лока-
лизованный на атоме кислорода в ионе СбН5О_, меньше, чем в алкоголят-
анионе, например СН3О~. Таким образом, фенол может быть оттитрован
метилатом калия
С6Н5ОН -Ь СН3О-К+ = С6Н5ОК + СН3ОН.
Основной характер атома кислорода уменьшается до такой степени, что
фенол становится слабой кислотой. В результате удаления протона в феноле
две неподеленные электронные пары атома кислорода стабилизуются.
Мезомерные эффекты влияют также на силу ароматических оснований.
Так, например, неподеленная электронная пара, определяющая протонко-
ординирующую способность анилина, на атоме азота сопрягается с л-элек-
тронным секстетом ароматического кольца (+М-эффект). Результирующая
электронная конфигурация может быть приближенно представлена следую-
щими резонансными структурами:
Н —N-1I
В хиноидных резонансных структурах анилина (б — г) атом азота оказы-
вается в аммониевой (или иммониевой) форме и, таким образом, его протон-
* Wheland, Pauling, J. Am. Chem. Soc., 57, 2086 (1935).
Связь между кислотностью, основностью и молекулярной структурой
83
связывающая способность уменьшается. Ион анилиния не может стабили-
зоваться путем образования хиноидной формы, потому что октет атома
азота заполнен, и хиноидная форма может быть достигнута только в резуль-
тате выделения протона. По этой причине анилин — более слабое основа-
ние, чем метиламин. Основные свойства ослабляются при введении либо
двух или трех фенильных групп (Л), либо нитрогруппы в ароматическое
ядро анилина (Б) (табл. 24).
А
Таблица 24
Основание рК Литература Основание рК Литература
Анилин 9,42 334 и-Толуидин 8,21 337
Дифениламин 13,15 334 Ди-и-толуидин 11,77 47
Трифенплампн 16,83 47 Три-и-толуидин 15,33 47
Основание РКВН+ Литера- тура Основание РАВН+ Литера- тура
Анилин 4,58 2,4-Динитроанилин —4,53 625
л-Нитроанилпн 2,62 333 2,4,6-Тринитроанилин —9,41 625
2,50 625 (ипкрамид)
и-Нитроанилин 0,99 625
о-Нитроанплин —0,29 625
а. Влияние дополнительного ионного резонанса на силу гетероцикличе-
ских оснований [2—5]. Азотистые гетероциклические соединения играют
важную роль в синтезе лекарственных веществ и в химиотерапии. Очевидно,
соединения этого типа можно титровать хлорной кислотой, поэтому зависи-
мость силы основания от его структуры будет рассмотрена более детально.
Пиррол является очень слабым основанием, так как неподеленная элек-
тронная пара атома азота участвует в образовании ароматического секстета
и в координировании протона. Пиррол при растворении в концентрирован-
ных водных растворах минеральных кислот полимеризуется, поскольку при
протонировании эта электронная пара удаляется из электронного секстета
ядра и молекула приобретает диеновую структуру.
Н
Н+%"
Другая ситуация возникает в случае N-гетероциклических соединений
с шестичленным циклом. В пиридиновом кольце неподеленная электронная
пара азота не участвует в образовании ароматического секстета. Таким обра-
зом, азот пиридина обладает явно выраженными нуклеофильными свойст-
вами, и пиридин ведет себя как циклический третичный амин очень высокой
84
Глава 4
ОСНОВНОСТИ.
Альберт и сотрудники детально исследовали изменение основности гете-
роциклических оснований в зависимости от числа атомов в кольце и влия-
ние на основность второго атома азота, вводившегося в кольцо. Эффект
дополнительного резонанса ионов в гетероциклических основаниях под-
тверждают результаты многих экспериментов. Введение второго атома азота
в пиридиновый цикл вызывает уменьшение силы основания. Феназин
и хиноксалин — более слабые основания, чем акридин и хинолин соответ-
ственно.
Z\zN4/4
II I I I
Феназин, р^вн+ 1»2
Хиноксалин, р&\дН+ — 0.8
Соединения, содержащие пиррольное кольцо, приобретают основные свой-
ства при введении второго атома азота в цикл, хотя сама молекула, по суще-
ству, проявляет слабые основные свойства. Индазол (бензпиразол) и бенз-
имидазол — более сильные основания, чем пиррол и индол соответственно.
I rS I II CH
4Z4Z
Бензпиразол, р£лн+1,3
Бензимидазол, рХвн+5,53
Основность насыщенных гетероциклических оснований подобна основ-
ности соответствующих алифатических аминов: рЛ\вн+ ~ 10. Потребова-
лись многочисленные исследования, чтобы определить силу ненасыщенных
гетероциклических оснований и установить связь между силой этих соеди-
нений и их молекулярной структурой [2, 3].
По своей природе основность ненасыщенных гетероциклических основа-
ний отличается от основности насыщенных оснований. В процессе иониза-
ции оснований, содержащих ароматическое кольцо, резонирующие системы
электронов играют значительную роль в изменении запаса энергии. Кон-
станта ионизации — мера изменения свободной энергии ионизации — содер-
жит также фактор, связанный с изменением резонансной энергии.
На примере акридина показан результат действия дополнительного
ионного резонансного эффекта, способствующего изменению силы основа-
ния. Сам акридин является слабым основанием и его сила увеличивается
только в небольшой степени при введении аминогруппы в положение 3 или 4.
Основность уменьшается при введении НН2-группы в положение 1, в то
время как аминогруппы в положении 2 или 5 увеличивают основность моле-
кулы вследствие дополнительного ионного резонансного эффекта.
NH2
1~Аминоакридин, 4,40
Связь между кислотностью, основностью и молекулярной структурой
85
Акридин, р/СдНТ 5,60
NH2
4-Аминоакридин, рКдц+ 6,04
Резонансные структуры иона 5 - амииоакридиния
Дополнительный ионный резонансный эффект ясно виден на примере очень сильного
основания гуанидина (рХвн+ 13,71). Иону гуанидиния отвечают три возможные и экви-
валентные структуры, каждая из которых содержит катионную группу == NHJ и двойную
связь: следовательно, это катион с симметричной электронной структурой.
+nh2 nh2 nh2
II I + +1
h2n—c—nh2 H2N-C=NH2 h2n = c—nh2
nh2
h2n-c—nh2.
В случае ацетамидина ситуация подобна, хотя это соединение — более слабое осно-
вание, потому что для него возможны только две резонансные эквивалентные структуры,
Р^ВН+ = 12,52.
СНз СН3
I + + .1
h2n-c=nh2 h2n = c—nh2
5-Амидоакридин сравним с ацетамидином, а
2,5-диаминоакридин — с гуанидином
РКВН+
5-Аминоакридин 9,92
2,5-Диаминоакридин 11,49
Ацетамидин 12,52
Гуанидин 13,71
Удаление одного или двух бензольных колец ведет лишь к незначитель-
ным изменениям
рквн+
Акридин 5,60
Хинолин 4,94
Пиридин 5,23
86
Глава 4
Таблица 25
Основание Р#ВН+ Основание РКВН+
3-Аминопиридин 5,98 3-Аминохинолин 4,95
4-Аминопиридин 9,17 4-Аминохинолин 9,17
2-Аминопиридин 6,86 8-Аминохинолин 3,99
В случае пиридина и хинолина при введении аминогруппы в положе-
ние 3 основность также увеличивается лишь незначительно.
Аналогично 5-аминоакридин, 4-аминопиридин и 4-аминохинолин являют-
ся сильными основаниями вследствие дополнительного резонансного эффек-
та (табл. 25)
2-Аминопиридин
2-Пиридонимид-
ная структура
3-Аминопиридин
(ведет себя как
ароматический
амин)
4-А минопиридин
4-Пиридон-
имидная
структура
Ион 2-аминопиридиния
Ион 3-аминопиридиния
Ион 4-аминопиридиния
(см. также гл. 23, разд. 121 и работы [262, 732]).
В шестичленных гетероциклических основаниях, содержащих только
один атом азота в цикле, введение второго атома азота в кольцо уменьшает
силу основания. Это относится также к расплавам гетероциклических
Связь между кислотностью, основностью и молекулярной структурой
87
соединений (хинолин, акридин):
N
/ ч
Пиридин, Пиридазин Пиримидин
рЛ*лн+5,23 (1,2-диазин), (1,3-диазин),
Р^вн+^’ЗЗ Р^вн+2>30
\ Z
N
Пиразин
(1,4-диазин),
Р-^вн+
/\/Ч
1
N
Хинолин,
Р^вн+
Фталазин,
Р^вн+ ^>47
Хиназолин,
Р-&ВН+ 3>2
Хиноксалин,
Р^вн+ ~ 0’8
В пятичленных гетероциклических основаниях, содержащих один атом
азота, введение второго атома азота в кольцо приводит к увеличению
основности.
\Z
I
Н
Пиррол,
Р^вн+ 0,4
Индазол (бенз-
пиразол),
Р^вн+ 1’3
|—1“
I NH
Пиразол,
рХ вн"^ 2,53
Бензимидазол
Р-^вн+ 6,53
Имидазол
(глиоксалин),
Р^вн+ ^’ОЗ
Таким образом, становится очевидным отличие шести- и пятичленных
систем. Введение атома азота в пятичленное кольцо, вызывающее допол-
нительные ионные резонансные эффекты, приводит к увеличению силы
•основания
।--NH
I
I сн
\ Z
+N
I
Н
Ион имидазо линия
Н —NH
Н
H-NH
I
нч +
ЧСН3
I
н
Ион ацетамидия
88
Глава 4
б. Комбинация М- и /-эффектов. Необходимо очень осторожно подходить
к решению вопроса об основном или кислотном характере соединения на осно-
ве его структурной формулы. Так, я-оксибензойная кислота слабее, чем
jt-оксибензойная, так как в первой нуклеофильная ОН-группа сопряжена
с СООН-группой, находящейся в пара-положении (4-Af-эффект). По этой
причине электронная плотность и, следовательно, сродство к протону кар-
боксилат-аниона увеличиваются.
Как известно, гидроксильная группа обладает —/-эффектом; однако
в этом случае индукционный эффект гасится вследствие смещения электронов
к ароматическому кольцу за счет +Л/-эффекта. В л-оксибензойной кислоте
4-М-эффект в значительной степени ослаблен и доминирующим оказывается
—/-эффект ОН-группы (см. также разд. в).
В случае ароматических аминов, например анилина, сродство к протону
группы NH2 меньше, чем в метиламине, из-за акцепторного влияния бензоль-
ного кольца. В бензиламине вследствие того, что группа СН2 находится
между Кольцом и 1\тН2-группой, сопряжение между ароматическим кольцом
и азотом аминогруппы нарушается, поэтому бензиламин является более
сильным основанием, чем анилин. В насыщенных алифатических аминах
диполь ориентирован R NH2, в ароматических аминах направление
ориентации обратное Аг NH2 [26]. В этих системах существует противо-
положно направленный мезомерный электрический момент, который в неко-
торых случаях превышает электростатический момент
(J+ ) Алкил---*• NR2 (д—)
(i—) Арил
NR2(J+)
в. Стабилизация, обусловленная резонансом. Стабилизация вследствие
резонанса в кислотах и сопряженных йм основаниях является очень важным
фактором. Сравнение кислотности спиртов и алифатических карбоновых
кислот показывает, что более сильный кислотный характер этих кислот
обусловлен стабилизацией карбоксилат-аниона в результате резонанса.
Два атома кислорода аниона обладают одинаковой, хотя и меньшей, про-
тонсвязывающей способностью, поскольку отрицательный заряд более
рассредоточен
Енолят-анион также может стабилизоваться вследствие резонанса, кото-
рый усиливает силу кислот (стр. 442 в работе [346]). Диэтилмалонат может
титроваться как кислота в диметилформамиде или в смеси бензол — пиридин
(гл. 17 и работа [893]).
ос2н5
ОС2Н5
ОС2Н5
Rx ^С —О-
/С=О
R--Cz
о^С\ос2н5
^ОС2Н5
“Oz ХОС2Н5
г. Влияние резонанса и заместителей на силу замещенных бензойных
кислот [79, 173, 189, 863]. Заместитель влияет на силу бензойной кислоты
в соответствии с его электрофильной или нуклеофильной природой и в зави-
симости от положения группы СООН. Замещение атома водорода бензоль-
ного кольца более сильным, чем водород, электрофильным заместителем
Связь между кислотностью, основностью и молекулярной структурой 89
является причиной того, что электроны О — Н-связи карбоксильной
группы становятся более локализованными на атоме кислорода, облегчая
таким образом диссоциацию протона карбоксильной группы (увеличивая его
подвижность)
°-
-CZ Н+
^0
Влияние заместителя передается как через молекулу, так и через про-
странство («эффект поля»). Оба эффекта действуют в одном и том же направ-
лении и уменьшаются с увеличением расстояния от СООН-группы (табл. 26).
Таблица 26
Кислота А- 105 а
С6Н5СООН 6,30
С6Н5СН2СООН 4,88
С6Н5СН2СН2СООН 2,19
Таблица 27
Кислота К • 105 а
п-Оксибензойная 2,63 [173]
га-Нитробензойная 37,6[173]
Отношение 1: 14,3
Так как наряду с указанными имеют место и другие эффекты, то вопрос
о силе бензойной кислоты требует дальнейшего рассмотрения. Имеются раз-
личные причины, по которым величина Ка бензойной кислоты не может
превышать 6,3 «10-5. Например, бензойная кислота стабилизуется в недис-
социированной диполярной ионной форме (б); в то же время стабильность
бензоат-аниона уменьшается вследствие наличия резонансных структур
в и г. В резонансной структуре г происходит увеличение плотности отрица-
тельного заряда на атомах кислорода. Диссоциация протона, т. е. проявле-
ние кислотных свойств, до некоторой степени затруднена.
а б
Резонансные структуры бензойной кислоты
в г
Резонансные структуры бензоат-аниона
Такое объяснение подтверждается тем фактом, что ортпо-замещенные
бензойные кислоты (в водной среде) являются независимо от природы заме-
стителя более сильными, чем сама бензойная кислота. Заместители, по-види-
мому, препятствуют образованию плоских конформаций и, таким образом,
уменьшают возможность образования структур биг (см. табл. 21, кроме
о-аминобензойной кислоты).
Кислотность, обусловленная эффектом сопряжения в резонансных струк-
турах бензойной кислоты или бензоат-аниона, может уменьшаться или уве-
90
Глава 4
личиваться при введении заместителей в орто- или пара-положение
ОН )
ОСН3 ?
NH2 J
Электронодонорные
группы
NO2'I Электроноакцепторные
ON J группы
д
Резонансная структура
п-оксибензойной
кислоты [173]
е
Резонансная структура
п-нитробензойной
кислоты
Вышеприведенная формула показывает, что ОН-группа в пара-положе-
нии увеличивает, a NO2-rpynna в пара-положении уменьшает резонансный
эффект б и г. При рассмотрении структуры д видно, что резонансный эффект
противоположен индукционному эффекту ОН-группы, увеличивающему
силу кислоты (—1- и +М-эффекты). Структура е указывает, что индук-
ционный эффект ЫО2-группы, увеличивающий силу кислоты, усиливается
вследствие резонанса (—I- и —М-эффекты) (табл. 27).
Заместитель в лета-положении по существу не принимает участия в резо-
нансном взаимодействии. Его влияние, следовательно, почти полностью
обусловлено индукционными эффектами «второго порядка».
Образование водородных связей также влияет на силу орто-замещенных
бензойных кислот. В некоторых случаях в зависимости от природы раство-
рителя оно увеличивает или уменьшает кислотный характер соединения
(см. гл. 2, разд. 16,а и работу [173]). Кислотность салициловой кислоты
увеличивается за счет образования хелатного цикла между фенольным
водородом и кислородом карбоксила. Анион стабилизуется благодаря обра-
зованию хелатного цикла; такая стабилизация происходит даже в большей
степени в случае 2,6-диоксибензойной кислоты (см. также разд. 28,в этой
главы и работы [29, 81, 227]).
Эти примеры показывают возможность титриметрического определения
данного соединения, кислоты или основания в соответствии с его струк-
турой.
д. Резонансные структуры и кислотность кетонов, 1,3-дикетонов, пир-
ролов и циклопентадиена. В кетонах и в гораздо большей степени в 1,3-дике-
тонах атом водорода в a-положении имеет кислый характер (табл. 28). Резо-
нансный гибрид аниона, возникающий при отщеплении протона, можно
изобразить следующим образом:
О 1О|
II I
СН3—с—£н—снз ------------- снз—с=сн—СНз
Анион метилэтилкетона (очень сильное основание)
сн3—с —сн2—с —сн3
Ацетилацетон (может быть оттитрован в
бутиламине Метилатом натрия в присутствии
тимолового синего 12461)
Связь между кислотностью, основностью и молекулярной структурой
91
|0| z0
I II
сн3— с = сн — с —сн3
СН3— С — СН — С — СН3—► ит.д.
Анион ацетилацетона (резонансные .структуры)
Кето-енольная таутомерия может рассматриваться как результат миграции протона
внутри молекулы; тогда понятно, что енолизация происходит под действием оснований-
катализаторов (В) и начинается с образования енолят-аниона (стр. 368 в работе [380])
О
R—С — СН3
О
(R—С — СН2)" + ВН+
Сопряженное ацетону основание также может быть представлено гибридной резо-
нансной структурой
JO-н +/2~н
СН3 —Cz СНз-Ск
хсн3 сн3
Эта структура является промежуточной между структурами ионов оксония и карбония
(см. гл. 2, разд. 19а и в). Относительно сопряженного ацетону основания см. табл. 28,
гл. 6, разд. 36 и работы [380, 729].
Таблица 28
Чрезвычайно слабые кислоты, обладающие подвижным водородом (псевдокислоты)
Соединение Р*а Структура сопряженного основания Замечания
Аммиак Дифенилметан Трифенилметан Анилин Ацетилен Дифениламин Инден Фенилацетилен Ацетон Ацетофенон /претп-Бутиловый спирт Бензиловый спирт Пиррол Метиловый спирт Этилмалонат Ацетилацетон ~ 36 ~ 35 - 33 27 26 23 21 21 19 19 19 18 16,5 16—17 12,9 9,03 а (С6Н5)2С = / и т. д. /\/\ и 1 \/ с6н5с = с- (CH3COCH2)- (СвН5СОСН2)- (СНз)зС- о- Отрицательный заряд может быть на ор- то-углеродном ато- ме Отрицательный заряд может быть на Cj или С3 Содержание енола в ацетоне составляет 2,5-10-4% [380, 729] Гл. 4, разд. 27,д Гл. 4, разд. 27,в Для солей натрия возможно образо- вание аниона Гл. 4, разд.27,д
Maron i, Calmon, Bull. soc. chim. France, 1964, 519.
92
Глава 4
В дибензоилметане и со-нитроацетофеноне оди-форма стабилизуется
за счет внутримолекулярной водородной связи. В растворе толуола содержа-
ние а^и-формы со-нитроацетофенона по литературным данным составляет
приблизительно 10% (стр. 233 в работе [745] и [744]), однако результаты
последних ПМР-исследований показали, что он существует исключительно
в форме простого нитрокетона [Simmons Т. et al., J. Org. Chem., 31,
2400 (1966)]. atyu-Форма дибензоилметана может быть оттитрована метилатом
натрия в диметилформамиде в присутствии тимолового синего [246].
Дибензоилметан (аци- форма)
Суммарный эффект ароматического кольца и возможность проявления
резонанса позволяют объяснить очень слабые основные свойства пиррола
и наличие кислотных свойств у циклопентадиена, который не содержит
енольного гидроксила (стр. 65 в работе [393] и стр. 340 в работе [870]).
Пиррол
(очень слабые кислота и основание)
р*а 16,5
Катион пиррола
(очень сильная кислота),
Р-^вн+ 0»^
Анион пиррола
(очень сильное
основание)
НС —СН
II II
НС СН
чсх
Н2
-Н+ НС —СН НС-СН
----> II I! I II
НС СН НС СН
\ / "Ч /
-с с
н н
НС-СН
II I
НС СН
\ 7
с
н
Ци клонен тадиен
Анион циклопентадиена
Связь между кислотностью, основностью и молекулярной структурой 93
Таким образом, ароматические соединения, содержащие группы ОН,
= NH, GH, являются кислотными аналогами, хотя это нельзя считать
общим правилом. Кислотность более сложных молекул увеличивается, когда
отрицательный заряд аниона не сконцентрирован только на одном атоме,
а в результате перераспределения электронов рассредоточен по всем атомам
внутри молекулы [53, 263, 567, 884] (табл. 28).
28. ВЛИЯНИЕ СТЕРИЧЕСКИХ ПРЕПЯТСТВИЙ НА СИЛУ КИСЛОТ
И ОСНОВАНИЙ
Диссоциация кислот, сила оснований и образование солей зависят
от стерических требований, предъявляемых заместителями, и их простран-
ственного расположения внутри молекулы.
а. Влияние стерических затруднений на резонанс. Рассмотрение стериче-
ских затруднений важно с точки зрения резонанса. Характерным примером
являются бензохинуклидин [867] и Х,Х-диметилпикрамид и кислотные ана-
логи, например 3,5-диметилфенол, 3,5-диметил-4-цианфенол и 3,5-диметил-4-
нитрофенол.
Бензохинуклидин, p^ 6,21
Бензохинуклидин — более сильное основание, чем производные диал-
киланилина и фенилпиперидин.
рК
Диметиланилин 8,94
Дпэтиланилпн 7,44
N-Фенилппперидин 8,80
Основные свойства этих соединений не могут быть отнесены за счет бици-
клической системы (Б), так как сам хинуклидин — более слабое основание,
чем пиперидин (рК 2,80). Можно предположить, что в случае бензохинукли-
дина образование резонансных структур, уменьшающих силу основания,
зависит от стерических факторов: атом углерода, непосредственно связан-
ный с атомом азота, не может быть копланарным с ароматическим кольцом,
так что вероятность существования резонансных структур, приводящих
к ослаблению основных свойств, уменьшается. Таким образом, хинукли-
дин — самое сильное основание, за ним следует бензохинуклидин, в то время
как производные диалкиланилина благодаря возможности резонанса явля-
ются более слабыми основаниями. Например
94
Глава 4
Г^,1^-Диметилпикрамид почти в 40 тысяч раз более сильное основаниег
чем пикрамид (2,4,6-тринитроанилин). Такое большое различие в основ-
ностях вряд ли можно объяснить введением двух метильных групп, посколь-
ку диметиланилин лишь в 3,3 раза более сильное основание, чем анилин.
Нитрогруппы в ор/по-положении препятствуют копланарности диметилами-
ногруппы с кольцом [88].
Введение метильных групп в положения 3 и 5 уменьшает кислотность
фенола на 0,19, n-оксифенилцианида на 0,26 и n-нитрофенола на 1,11 единиц
рК [871] (табл. 29).
Таблица 29
Кислотные аналоги рХ Кислотные аналоги рК
Фенол 9,99 3,5-Диметил-4-циан фенол 8,21
3,5-Диметилф енол 10,18 и-Нитрофенол 7,14
п-Оксифени лцианид 7,95 3,5-Диметил-4-нитрофенол 8,25
n-Нитрофенолят-анион стабилизован вследствие образования хиноидной
мезомерной формы, и нитро-группа копланарна с хиноидным кольцом.
Рис. 27. 3,5-Диметил-4-нитрофенокси-анион.
Образованию копланарной формы препятствует введение метильной группы
в ор/по-положение, и резонанс нитрофенолят-аниона с кольцом затруднен
(рис. 27)
Электронная плотность на атоме кислорода в 3,5-диметил-4-нитрофено-
ляте увеличивается, так что его сродство к протону уменьшается и умень-
шается сила кислоты [871].
Иначе обстоит дело при введении двух метильных заместителей в положе-
ния 3 и 5 в кольцо, содержащее группу CN. Введение метильной группы
лишь незначительно уменьшает кислотность благодаря линейной структуре
группы С = N; анион стабилизуется вследствие резонанса.
б. Теория обратного напряжения. Браун и сотрудники детально обсудили
стерический эффект несколько другого характера, чем тот, который обсуж-
дался выше, но также влияющий на силу органических оснований [94—98].
Алкильные группы обладают электронодонорными свойствами, вслед-
ствие чего атом азота в метиламине более нуклеофилен, чем атом азота
в аммиаке. Тем не менее диметиланилин лишь немного более сильное осно-
Связь между кислотностью, основностью и молекулярной структурой
95
вание, чем метиламин, а триметиламин менее сильное основание, чем метил-
амин. В случае ароматических заместителей бензольное кольцо притяги-
вает электронную пару азота, и его способность связывать протон и его
основность уменьшаются. Сила основания уменьшается в ряду:
Аммиак > анилин > дифениламин > трифениламин.
Согласно Брауну, сила основания в ряду алифатических аминов опре-
деляется не только числом алкильных заместителей и характером передачи
электронов, но и их стерическими требованиями и электростатическим
эффектом отталкивания. Данные термических измерений для комплексов
триметилбора с аминами показывают, что комплексы, образованные (СН3)3В
и третичными аминами, легко диссоциируют. По отношению к кислотам
Льюиса с большими стерическими препятствиями основность аминов умень-
шается в следующем порядке:
Аммиак < метиламин < диметиламин триметиламин,
(СН3)3В Н- NH3 = (СНз)зВ — NH3.
Стерическое препятствие необходимо учитывать при рассмотрении взаимо-
действия три-трет-бутилбора как кислоты Льюиса и этиламинов как осно-
ваний
Аммиак < этил амин > диэтиламин > триэтиламин.
По гипотезе Брауна принимается, что если стерические препятствия,
обусловленные заместителями, значительны и, кроме того, если они имеют
несомненно полярный характер, то заместители отталкивают друг друга,
следствием чего является так называемое внутреннее напряжение в моле-
куле амина. Близкое расположение заместителей затрудняет переход аминов
в ониевые ионы. У алифатических аминов стерический эффект противостоит
электростатическому эффекту. У ароматических аминов основная сила
уменьшается за счет как стерического эффекта заместителя, так и электрон-
притягивающего мезомерного эффекта кольца. Следовательно, в последнем
случае действие этих двух эффектов суммируется.
Белл и сотрудники полагают, что ослабление основных свойств третичных аминов
в водной среде может быть отнесено за счет пониженной растворимости сопряженной
кислоты (ониевый ион), так как в третичных аминах меньшее число атомов водорода
проявляет способность к образованию водородной связп, чем, например, в первичных
аминах [45, 54, 55].
Н\/Н
О
R Н
И\ +1 Н\ I /Н
О...Н — N— R Хо ... H-+N —Н .. . (У
н/ I HZ I хн
R R
Валентный угол в воде п спиртах составляет 109° и он не меньше в сопряженных им
кислотах, в тригональных ионах оксония [394]. Валентный угол у кислорода в диэтпло-
вом эфире больше. По этой причине эфиру гораздо труднее присоединять протон, оп
более слабое основание, чем вода или спирт. Валентный угол С — О — Св 1,4-диоксане
фиксирован (около 109е), поэтому диоксан более спльное основание, чем дп-н-бутило-
вый эфир [505].
в. Орто-эффект. Термин «opmo-эффект» охватывает факторы, имеющие
различную природу и иногда направленные в противоположную сторону.
Многообразие этих факторов объясняет затруднения, часто встречающиеся
при истолковании opmo-эффекта. Если атомы водорода в феноле в орто-
положении заместить изопентильными группами, то его растворимость
в щелочи очень мала. Он также не дает цветной реакции с хлоридом желе-
96
Глава 4
за(Ш). Этот пример иллюстрирует стерический характер орто-эффекта.
Однако объяснение opmo-эффекта будет иным в том случае, когда орто-
заместитель способен образовывать водородную связь с гидроксилом фенола.
ОН
Салициловая кислота,рКа2,97
Салициловый альдегид
Приведенные выше соединения обладают свойствами, отличными
от свойств их мета- и пара-изомеров. Например, они могут возгоняться
и довольно легко растворяться в неполярных растворителях. Сила кислот
и оснований сильно зависит от введения заместителей в орто-положение.
Почти все opmo-замещенные производные бензойной кислоты более сильные
кислоты, чем сама бензойная кислота или ее мета- или пара-изомеры
(табл. 21).
Внутримолекулярная водородная связь в салициловой кислоте как акцеп-
тор электронов понижает электронную плотность на карбонильной группе,
поэтому салициловая кислота является сравнительно сильной кислотой.
Глава 5
ОБЩИЕ СВОЙСТВА РАСТВОРИТЕЛЕЙ
Так как растворители могут оказывать влияние на положение равновесия,
успешность определений, проводимых в неводных средах, зависит от выбора
растворителя. Особенно большое значение выбор растворителя имеет при
титровании очень слабых оснований или кислот, так как с помощью некото-
рых нейтральных растворителей, например ацетона, метилэтилкетона и метил-
изобутилкетона, можно дифференцировать слабый основной или кислотный
характер соединений. Растворители сильно кислотного характера, например
уксусная кислота, или сильно основного характера, например этилендиамин,
не только повышают силу слабых или умеренно сильных оснований и кислот,
но и выравнивают различия. Это называют выравнивающим эффектом.
Именно поэтому при титровании органических соединений все чаще исполь-
зуют смеси растворителей.
29. СОЛЬВАТИРУЮЩАЯ СПОСОБНОСТЬ
На растворимость органических соединений часто вследствие их сложной
структуры оказывают влияние внутримолекулярные и межмолекулярные
силы.
Растворимость органических веществ зависит от следующих факторов:
1) наличия функциональных групп в молекулах растворенного вещества
и растворителя, их числа, полярности, кислотности, основности и относи-
тельного расположения в молекулах; 2) диэлектрической проницаемости
растворителя; 3) внутримолекулярных и межмолекулярных водородных
связей; 4) числа и положения атомов углерода (в пределах гомологического
ряда); 5) наличия двойных связей и (или) ароматических ядер; 6) наличия
пространственных затруднений. Выбор растворителя определит, какой
из этих факторов будет оказывать решающее влияние на растворимость
вещества.
При растворении ионных соединений важную роль играет диэлектриче-
ская проницаемость растворителя, т. е. степень, с которой ослабляется
электростатическое взаимодействие между ионами противоположного заря-
да. Сила взаимодействия двух противоположно заряженных ионов состав-
ляет в уксусной кислоте только г/9, в н-бутиловом спирте г/16 и в циклогекса-
не г/2 силы их взаимодействия в вакууме. Поэтому вероятность образования
ионных пар в бутиловом спирте мала, в уксусной кислоте она увеличивается
и в еще большей степени возрастает в циклогексане. Трудно установить
различие между отдельными видами ассоциации, и в растворителях с малой
сольватирующей способностью возможно образование многих промежуточ-
ных дискретных образований между диссоциированными ионами, ковалент-
ными молекулами и ионными агрегатами.
Между диэлектрической проницаемостью растворителя, полярностью
растворенного вещества и его растворимостью существуют хорошо установ-
ленные соотношения. В действительности, хотя высокая диэлектрическая
проницаемость и необходима, все же она не является условием, достаточным
для предсказания растворимости ионных соединений. Так, диэлектриче-
98
Глава 5
ская проницаемость е жидкого HGN равна 116, тем не менее он плохой рас-
творитель для хлорида натрия. В случае органических соединений диэлек-
трическая проницаемость растворителя — фактор второстепенный; так,
диэлектрическая проницаемость пропионовой кислоты равна только 3,4,
однако эта кислота растворяет полярные органические вещества намного
легче, чем, например, хлорбензол (е = 5,6). Благодаря наличию неподелен-
ной пары электронов пропионовая кислота склонна акцептировать ионы
водорода и способна образовывать координационные связи.
Большинство органических соединений содержит как полярные, так
и неполярные группировки, и взаимодействие их с молекулами раст-
ворителя влияет на растворимость. Если большее значение имеет угле-
водородная часть молекулы, возрастает растворимость соединения в непо-
лярных растворителях, однако, если молекула содержит полярные функцио-
нальные группы, то растворимость в «водоподобных» растворителях увели-
чивается, а в неполярных падает. Типичным примером служит алкалоид
томатин, который почти нерастворим в четыреххлористом углероде, но рас-
творяется в нем при добавлении к растворителю полярного соединения —
фенола (гл. 24, разд. 136).
Вообще увеличение молекулярного веса приводит к возрастанию роли внутримоле-
кулярных сил в твердой фазе, что влечет за собой уменьшение растворимости (молекуляр-
ный вес томатина 1034). Так, например, метакрилат и глюкоза растворимы в воде, в то вре-
мя как их полимеры нерастворимы.
Еще более важным, чем диэлектрическая проницаемость, является спе-
цифическое взаимодействие между растворенным веществом и растворителем.
Примером взаимодействия такого типа служит ион-дипольное взаимодействие.
При растворении хлорида натрия атомы кислорода диполей воды настолько
тесно окружают ион Na+, насколько это позволяет соотношение между
притяжением и пространственными затруднениями для ион-дипольного
взаимодействия. Первичная сольватная оболочка, окружающая хлорид-ион,
удерживается за счет взаимодействия типа образования водородных связей
(мнения о точном числе диполей воды в первичной оболочке расходятся [73]).
нх /Н
н °+ /н
Н-0 Na °-Н
хх
w н
(Данные о сольватации солей карбоновых кислот и щелочных металлов
в гликоле см. в гл. 22, разд. 118).
Специфическое взаимодействие между растворяемым веществом и раство-
рителем становится заметным, когда, например, экстрагируют водный рас-
твор гидрохлорида органического основания, содержащего функциональные
группы аналогичного характера, причем раствор не подщелачивают,
а экстракцию проводят растворителем, не смешивающимся с водой, и тит-
руют экстракт свободного основания, образующегося в результате гидролиза.
Можно привести следующий пример: водный 0,1 М раствор гидрохлоридов
пиперидинмети лто ли лпропанона (ПМТП), пиперидинметилциклогексанона
(ПМЦГ) и пиперидинметилтетралона (ПМТ) экстрагировали различными
растворителями и после добавления уксусной кислоты в неводную фазу
определяли содержание оснований (0,01 н. хлорной кислотой). Приведенная
ниже табл. 30 показывает (в процентном отношении) количество оснований,
растворенных в органическом растворителе [313].
Общие свойства растворителей
99
Таблица 30
Растворитель пмтп пмцг пмт Растворитель ПМТП пмцг пмт
н-Гексан 2,6 1,0 2,3 Хлорбензол 5,2 1,2 1,2
Бензол 3,8 1,2 3,3 Хлороформ 11,6 1,0 6,7
Эфир 4,0 1,0 0,8
Степень перехода указанных органических оснований в органический
слой зависит от коэффициента распределения между водой и не смешиваю-
щимся с ней растворителем или, другими словами, от очень различного
взаимодействия между органическими молекулами и растворителями двух
типов. Из-за специфического взаимодействия можно ожидать довольно
разных результатов в случае растворителей, отличающихся по своей склон-
ности к образованию водородных связей (например, хлороформа), по диполь-
ному моменту (для хлорбензола ц = 1,5, бензола ц = 0, н-гексана ц =
= 0,08) или имеющих свободную пару электронов (эфир).
Взаимодействие между растворителем и растворенным веществом прояв-
ляется не только при растворении. Известны также и другие типы взаимо-
действия: сольватация, сольволиз, ионизация и ассоциация молекул
и ионов.
30. СОЛЬВАТАЦИЯ
Сольватация тесно связана с процессом растворения. Вообще говоря,
сольватация включает все типы взаимодействия между растворителем
и ионами или молекулами растворенного вещества, поскольку нельзя про-
вести никакого различия между свободными молекулами растворителя
и молекулами растворителя, связанными с ионами или молекулами раство-
ренного вещества (см. стр. 26 в работе [294]). Ионы или полярные молекулы
в полярном растворителе ориентируются под действием электростатических
сил, их энергия уменьшается и система становится более устойчивой. Вели-
чины энергии сольватации часто имеют тот же порядок, что и энергия кова-
лентных связей. Когда катионы или льюисовы кислоты сольватируются нук-
леофильным растворителем, молекулы размещаются таким образом, что
сольватируемые частицы окружаются оболочкой, вплоть до образования
ковалентной связи; электронодефицитные молекулы растворителя, не содер-
жащие подвижного водорода (например, жидкая двуокись серы), взаимодей-
ствуют с электронодонорными анионами. В случае растворителей, содержа-
щих подвижные, или «кислые», атомы водорода, сольватация аниона может
быть связана с «кислотностью» растворителя или его способностью образо-
вывать водородную связь (ср. гл. 6, разд. 38,а и стр. 47 в работе [393]).
Устойчивость образующихся таким образом аддуктов может быть самой
различной. Вследствие энергетических затрат на образование водородных
связей этот процесс понижает свободную энергию, например, аминов или
амидов кислот; отсутствие образования Н-связей увеличивает основность.
Таким образом, становится понятным, что сила кислот и оснований в водных
растворителях не всегда сравнима с этими же характеристиками, определен-
ными в неводных растворителях.
31. сольволиз
Сольватация может быть непосредственно связана с сольволизом в тех
случаях, когда благодаря электростатическому взаимодействию аниона
или катиона с диполями растворителя возникает ион-дипольный аддукт
100
Глава 5
вплоть до образования растворителем аниона или катиона:
ВН+С1О7 + 2(СН3СООН)2 (ВН-2СН3СООН)++(СЮ4.2СН3СООН)-,
(ВН-2СН3СООН)+ В-СН3СООН+СН3СООН|
и
(СЮ4-2СН3СООН)- С1О4Н-СН3СООН + СН3СОО-,
где 2?Н+ — катионная кислота, образованная за счет протонирования
азотсодержащего основания; (СН3СООН)2 — димер уксусной кислоты;
СН3СООНз — протонированная молекула растворителя и СН3СОО_ —
ацетат-ион.
В «водоподобных» растворителях [240], т. е. в растворителях, где был
обнаружен автопротолиз, процесс нейтрализации сопряжен с сольволизом
ВН+СН3СОО- + СН3СООН+С1О4 ВН+С1О; + (СН3СООН)2.
Если сильные или умеренно сильные азотистые основания растворять
в уксусной кислоте, образуется ацетат основания и, следовательно, появ-
ляется ацетат-ион, который титруют хлорной кислотой. Вследствие вырав-
нивающего эффекта сила оснований кажется одинаковой (см. гл. 3, разд. 23).
Карбоновые кислоты также сольватируются с образованием протониро-
ванного иона с участием молекул растворителя, например в этилендиамине:
RCOOH4-C2H4(NH2)2 rcoo- + h2nc2h4nhj.
В зависимости от относительной электрофильности используемых раство-
рителей, имеющих основной характер, в этих растворителях изменяется
также и относительная сила кислот и кислотных аналогов (см. табл. 31) [176].
Таблица 31
Кислота В воде В диметилформ- амиде В этилендиамине
Хлористоводо- родная Бензойная Фенол Сильная Слабая Не титруется Сильная Более сильная Очень слабая Сильная Сильная Слабая
В инертных растворителях, например гексане, бензоле и четыреххлори-
стом углероде, растворенное вещество взаимодействует с растворителем
совершенно отлично от рассмотренного выше, поскольку сольволиза при
этом не происходит и сила кислот и оснований лишь в малой мере зависит
от растворителя. Тем не менее найденные значения «кажущейся» силы кислот
и оснований могут отличаться от приведенных в литературе данных по кис-
лотности и основности (в воде) и в результате различной ассоциации
(см. гл. 2, разд. 13, 14 и гл. 3, разд. 24).
Типичные кислотно-основные реакции, подобные, например, нейтрализации или
изменению окраски индикаторов в апротонных растворителях, весьма способствовали
развитию теорий кислот и оснований, в большей или меньшей мере не учитывавших свой-
ства растворителя. В инертных растворителях возможно образование ионных пар, но не
наблюдаются диссоциация и сольволиз и не могут наблюдаться выравнивающие эффекты.
Перенос протона происходит в растворе непосредственно от кислоты к основанию. По тео-
рии инертные растворители должны были бы быть простыми для рассмотрения, но на
практике ряд экспериментальных фактов усложняет титрование в таких растворителях:
их диэлектрическая проницаемость низка (в среднем е = 2—6); ионные пары, образую-
щиеся из растворенного вещества в ходе взаимодействия, в значительной степени ассоции-
рованы, и карбоновые кислоты существуют в виде димеров. Индикаторы также ассоции-
Общие свойства растворителей
101
рованы и в связи с неприменимостью закона Ламберта — Бера не могут больше исполь-
зоваться. Из этих соображений данные, относящиеся к кислотно-основным реакциям
в апротонных или инертных растворителях, применимы только к определенным
растворителям и служат только для грубой ориентации при переходе к другим
растворителям.
32. ВЛИЯНИЕ ИОНИЗАЦИИ И ДИССОЦИАЦИИ
В теории титрования в неводных средах проведено четкое различие между
понятиями диссоциации и ионизации. Константы ионизации кислот и осно-
ваний, измеренные в растворителях с низкой диэлектрической проницае-
мостью, выражают силу кислот и оснований более точно, чем константы дис-
социации [101]. Если константу ионизации сильного основания, например
n-диметиламиноазобензола, в уксусной кислоте обозначить Кн, константу
диссоциации ионной пары обозначить 7ГДИСС и «общую» (суммарную) кон-
станту диссоциации — К, то в этом случае каждый из процессов будет харак-
теризоваться следующими величинами:
/4-СН3СООН 7Н+СНзСОО- Аи = 0,1
Ионизация
7Н+СН3СОО" 7Н+-НСН3СОО- Адпсс = 5,0-Ю'6
Диссоциация
при этом суммарная константа диссоциации равна
к = *Г*Усхс=4,б-1о-7,
(1-Г^и)
где I — индикатор, а 7Н + — кислотная форма индикатора.
В тех растворителях, где возможно образование водородных связей
(например, в уксусной кислоте), первая стадия процесса заключается в обра-
зовании непрочного аддукта, после этого возникает водородная связь
и только затем достигается стадия ионизационно-диссоциационного равнове-
сия. Таким образом, например, в азотистых основаниях может осуществ-
ляться перенос протона к атому азота (см. также гл. 2, разд. 11,6):
СН3СООН + В СН3СООН-В СН3СОО ...Н...В7*
Образование аддукта Образование водо-
родной связи
:z сн3соо-нв+ СН3СОО-4-ВН+
Ионизация и диссоциация
В органических растворителях с диэлектрической проницаемостью
е < 40, склонных к самоионизации, даже перхлораты оснований диссоции-
руют лишь частично; по большей части они образуют ионные пары (см. также
гл. 2, разд. 16,а и б).
33. АССОЦИАЦИЯ
В большинстве органических растворителей между молекулами раство-
ренного вещества, а также между молекулами растворенного вещества
и молекулами растворителя протекают процессы ассоциации, причем степень
ассоциации зависит не только от выбора растворителя, но также и от концен-
трации. Как следствие этого отношения равновесия в органическом раство-
рителе становятся более сложными и закон разбавления Оствальда оказы-
вается неприменимым даже к разбавленным растворам. В растворителях
с малой диэлектрической проницаемостью активность ионов заметно откло-
няется от единицы, и это отклонение также можно объяснить образованием
ионных пар и других ассоциатов [106, 260, 292, 389, 413, 447, 464, 5596].
Полярность ионных пар, константы их диссоциации и степень основности
102
Глава 5
зависят от растворителя и, в определенных случаях, от концентрации. Так,
например, из молекул веществ, имеющих аминогруппу и растворенных
в уксусной кислоте [561], образуются заряженные комплексные ионы.
(М3СН3СО)2+ 4- 2СН3СО- |
(М(СН3СО)3)2- + 2М+ / <—
(М2СН3СО)++(М(СН3СО)2)- (МСН3СО)3 ЗМСН3СО ЗМ+ + ЗСН3СО-
где М — катион.
Вследствие вторичной диссоциации комплексных ионов комплексы,
образующиеся из больших катионов, диссоциируют в уксусной кислоте
в большей мере, чем простые соли. В 0,01 н. растворах (в правой
части приведенной выше схемы) происходит обычная диссоциация солей.
Однако с повышением концентрации электропроводность проходит через
минимум, поскольку появляются ассоциаты типа (МСН3СОО)3. Тем не менее
при дальнейшем повышении концентрации (примерно до 0,1 н.) этот три-
мерный комплекс распадается на два комплексных иона так, что электро-
проводность снова возрастает. Наконец, в левой части схемы показан про-
цесс, при котором возрастающая ассоциация сопровождается увеличиваю-
щейся диссоциацией. Кислотность или основность комплексных ионов может
меняться при изменении концентрации.
34. АМФОТЕРНОСТЬ
Амфотерные соединения могут в зависимости от растворителя проявлять
как кислотный, так и основной характер. Можно различить два варианта:
1) амфотерность, обусловленную единственной функциональной группой
(например, в пирроле); 2) амфотерность, обусловленную двумя различными
функциональными группами (например, в сульфаминовой кислоте). Хоро-
шим примером служит кислый фталат калия (стандартное вещество для
установки титра), который можно титровать в воде как кислоту, а в уксус-
ной кислоте как основание. В первом случае образуется катион растворителя
Н3О+, а во втором — анион растворителя СН3СОО~:
[С6Н4(СООК)СОО]Н + Н2О СвН4(СООК)СОО- + Н3О+,
[С6Н4(СООН)СОО]К + СН3СООН С6Н4(СООН)2 + СН3СОО-К+.
Другие примеры амфотерных соединений приведены в табл. 32.
Таблица 32
Соединение Можно титровать
как кислоту (растворитель, титрант) как основание (растворитель, титрант)
Бензимидазол Гидразид изоникоти- новой кислоты Нптрогуанидин и его производные Пиррол Сульфаминовая кис- лота (стандарт для установки титров) Теобромин Сульфамиды N - Бензоилбенз ами- дин ДМФ, СН3ОК Диэтиламин, СН3ОК ДМФ, СН3ОК Чрезвычайно слабое основание и кислота, не титруется ДМФ, c4h9nh2, СН3ОК c5h5n, СН3ОК ДМФ, C5H5N, C4H9NH2, аце- тон, сн3ок c5h5n, тбаг С2Н5СО2Н, НСЮ4 СН3СООН — СНС13, нсю4 Трифторуксусная кислота, НСЮ4 СН3СООН, НСЮ4 СН3СООН — (СН3СО)2О — С6Н6, нсю4 СН3СООН —С6Н6, НС1О4 Этиленгликоль — изопропиловый спирт (1 :1), НС1О4
Общие свойства растворителей
103
Большинство амидов кислот «нейтральны» в уксусной кислоте, однако
в уксусном ангидриде их можно титровать потенциометрически; в жидком
аммиаке они ведут себя, как кислоты.
35. ВЫВОДЫ
В водоподобных растворителях взаимодействие между растворенным
веществом и растворителем происходит аналогично процессам, протекающим
в воде, но наблюдаются качественные и количественные отличия. Эти отличия
связаны с меньшими диэлектрическими проницаемостями, образованием
разнообразных ионных пар и их ассоциацией, а также с изменением электро-
статического поля в зависимости от концентрации (табл. 33).
Таблица 33
Влияние растворителя на растворенное вещество
_ Растворители кислотного или основного
Инертные растворители * характера
Соединения, способные к ионизации,
совсем не диссоциируют в инертных рас-
творителях; сохраняется собственная кис-
лотность или основность, но не наблюдает-
ся какого-либо выравнивающего эффекта;
сила слабых кислот или оснований не уве-
личивается
Катионы или анионы растворителя не
образуются; растворитель совсем не участ-
вует в процессе нейтрализации. Образую-
щееся вещество носит характер аддукта
или ассоциата
Нуклеофильность титрующего пли тпт-
руемого основания должна быть выше
нуклеофильности основания, соответствую-
щего титрующей или титруемой кислоте
В некоторых случаях необходимо ис-
пользовать специальные индикаторы
Потенциометрическое [титрование, осу-
ществляемое в неполярных растворителях,
требует добавления электролитов. Для про-
ведения потенциометрического титрования
необходимо специальное оборудование
В растворителях кислотного или основ-
ного характера вещество ионизируется,
а также (в меньшей степени) диссоциирует.
Оно образует с растворителем комплекс,
в результате чего собственная кислотность
или основность вещества изменяется (чаще
всего увеличивается или выравнивается)
Растворитель способствует миграции про-
тона, что приводит к образованию анионов
или катионов растворителя, т. е. раство-
ритель участвует в реакции нейтрализа-
ции. Продуктами реакции являются соль
и молекула растворителя
Растворитель должен быть более слабой
кислотой или основанием, чем титруемая
кислота или основание
Для определения конечной точки при-
годны многие обычные индикаторы и элек-
троды
Глава 6
РАСТВОРИТЕЛИ,
ИСПОЛЬЗУЕМЫЕ В НЕВОДНОМ ТИТРОВАНИИ
Растворитель участвует в прототропных процессах только в тех случаях,
когда его молекулы имеют атомы водорода, способные диссоциировать, или
если сам растворитель может образовывать водородные связи, например
уксусная кислота, пиридин, метиловый спирт. Это относится к простым слу-
чаям. Тем не менее существуют и некоторые другие, хотя и не особенно много-
численные растворители, в которых в реакции нейтрализации участвуют
ионы ацетилия или хлорид-ионы (гл. 1, разд. 5 и гл. 2, разд. 20,д). Однако
не способные к прототропному обмену растворители, обладающие низким
или нулевым дипольным моментом и не способные образовывать комплексы,
едва ли оказывают непосредственное воздействие на процесс нейтрализации,
тем более что он всецело зависит от реагентов, принимающих участие в реак-
ции. К таким апротонным инертным растворителям принадлежат н-гексан,
циклогексан, бензол и четыреххлористый углерод. В бензоле бензойная кис-
лота по отношению к дифенилгуанидину является более сильной, чем
по отношению к триэтиламину (гл. 2, разд. 15,д). Некоторые растворители
относятся к промежуточной группе; они в определенной степени влияют
на реакцию нейтрализации вследствие их способности образовывать водо-
родные связи, наличия дипольных моментов, комплексообразующих и нук-
леофильных или электрофильных свойств. Примерами таких растворителей
являются хлороформ, хлорбензол, ацетон, ацетонитрил, нитрометан, метил-
изобутилкетон и т. д.
Диэлектрическая проницаемость этих растворителей ниже, чем у воды,
поэтому поле силового действия ионов растворенного вещества и электро-
статическое притяжение возрастают:
p = (l/e)-(7ig2/r2),
где е — диэлектрическая проницаемость (табл. 34). В результате умень-
шается диссоциация, а сила взаимодействия между ионами возрастает, что
вызывает тенденцию к ассоциации. Все это влияет на цветовые переходы
окраски индикаторов и вызывает несистематические отклонения при потен-
циометрическом определении конечной точки.
Таблица 34
Диэлектрическая проницаемость растворителей е (20°) [560, 866]
(Следует иметь в виду, что литературные данные часто различаются
в последнем десятичном знаке, а иногда даже в предпоследнем)
Растворитель X арактеристика е
н-Гексан, н-гептан Инертный (неполярный) 1.9
Циклогексан » » 2,0
1,4-Диоксан » » 2,2
Четыреххлористый углерод » » 2,2
Бензол » » 2,3
Пропионовая кислота Кислотный 3,4 (40°)
Растворители, используемые в неводном титровании
105
Продолжение табл. 34
Растворитель Характеристика е
Эфир Инертный 4,3
Хлороформ » 4,8
н-Бутиламин Основной 5,3
Хлорбензол Инертный 5,6
Уксусная кислота Кислотный 6,2
1,1,1,- Трихлорэтан Инертный 7,5
трет-Бутиловый спирт Амфипротонный 10,9 (30°)
Пиридин Основной 12,5
Метилизобутилкетон Дифференцирующий (инерт- 13,0
ный)
Этилендиамин Основной 14,2
2-Метоксиэтанол (целлозольв) Амфипротонный 16,0
к-Бутиловый спирт » 17,1 (25°)
Изопропиловый спирт » 18,3 (25°)
Пропионовый ангидрид Кислотный 18,3 (16°)
Метилэтилкетон Дифференцирующий (инерт- 18,5
ный)
н- Пропиловый спирт Амфипротонный 20,1 (25°)
Уксусный ангидрид Кислотный (дифференцирую- 20,7
щий)
Ацетон Дифференцирующий (инерт- 21,0
ный)
Этиловый спирт Амфипротонный 24,3 (25°)
Пропиленгликоль-1,2 » 32,0
Метиловый спирт » 32,6 (25°)
Нитробензол Инертный 34,8 (25°)
Нитрометан Дифференцирующий (инерт- 36,0 (30°)
ный)
Ацетонитрил То же 37,5
Диметилформамид [503] Основной (дифференцирующий) 36,7а
Этиленгликоль Амфипротонный 37,7 (25°)
Гидразин Основной 53,0
Муравьиная кислота Кислотный 58,0
Вода Амфипротонный 80,4
Формамид Основной 109,0
Цианистый водород Кпслотный (инертный] 115,0
N-Метилформамид [503] Основной 190,5
е = 26,6 [595].
Мы рассмотрим пять групп растворителей, используемых при неводном
титровании: 1) апротонные инертные растворители, 2) дифференцирую-
щие растворители, 3) протогенные растворители, 4) протофильные раство-
рители, 5) смеси растворителей.
Растворители, способные высвобождать или связывать протоны, в равной
степени способны к переносу или акцептированию ионов водорода, т. е. к про-
тонному обмену. За исключением тех случаев, когда эти растворители обла-
дают явно выраженными кислотными или основными свойствами, их назы-
вают «амфипротонными растворителями».
36. АПРОТОННЫЕ ИНЕРТНЫЕ РАСТВОРИТЕЛИ
В инертных растворителях, подобных гексану, бензолу, четыреххлори-
стому углероду, хлорбензолу, проявляется собственная кислотность или
основность соединений (ср. табл. 32).
Некоторые инертные растворители оказывают дифференцирующее дейст-
вие на кислоты и основания, и при титровании в этих растворителях смеси
106
Глава 6
кислот или оснований не происходит выравнивания силы кислот или основа-
ний (см. гл. 15, разд. 95 и гл. 25, разд. 141). Тем не менее соединения с очень
низкой основностью или кислотностью нельзя титровать в них, поскольку
у апротонных растворителей отсутствует повышающее кислотно-основные
характеристики действие сильнокислых или сильноосновных раствори-
телей.
Согласно последним теориям взаимодействие растворенного вещества с растворителем
никогда не ведет к полному переносу протона в отсутствие эффекта сольватации. Можно
считать, что в неполярных растворителях система водородсодержащая кислота—акцепти-
рующее основание представляет собой устойчивую форму, стабильность которой опреде-
ляется водородной связью, возникающей между донором и акцептором протона [766].
37. ДИФФЕРЕНЦИРУЮЩИЕ РАСТВОРИТЕЛИ
Вальден и сотр. [855—857], а позднее Фриц, Брюс и Уайлд [105, 248]
рассмотрели дифференцирующее влияние ацетона, ацетонитрила, метил-
этилкетона, метилизобутилкетона, нитрометана и некоторых других раство-
рителей на электролиты. Измайлов и сотр. [411, 414, 415], как и Шкодин
с сотр. [735—739], занимались изучением дифференцирующего действия
некоторых растворителей (муравьиной, уксусной кислот и пр.) на кислоты,
фенолы и основания. Крешков и сотр. [474,475,479,485], подробно изучив
превосходные дифференцирующие свойства метилэтилкетона, особенно реко-
мендуют использовать в качестве титранта растворы хлорной кислоты
в метилэтилкетоне.
Показатели степени констант диссоциации кислот и оснований в подходя-
щих дифференцирующих растворителях могут заметно различаться, даже
если при измерении в воде они оказываются близкими (табл. 35) [411].
Таблица 35
Кислота РК
в воде в ацетоне
Пикриновая кислота 0,8 3,17
Салициловая кислота 2,97 9,53
Разность 2,17 6,36
Протонодонорное и протонакцепторное сродство дифференцирующих
растворителей является вообще гипотетическим и неизмеримым. Соединение
образует водородную связь или взаимодействует с растворителем тем или
иным маловыясненным путем и реагирует с титрантом в виде сольватиро-
ванной молекулы или иона.
38. ПРИРОДА ДИФФЕРЕНЦИРУЮЩЕГО И ВЫРАВНИВАЮЩЕГО ЭФФЕКТОВ
Дифференцирующий эффект можно частично приписать специфическому
взаимодействию между растворенным веществом и растворителем и частично
объяснить тем, что основность или кислотность растворителя отличается
от соответствующей характеристики воды. Так, например, если константы
диссоциации двух кислот ЛН и ХН равны 104 и 102, концентрация ионов
водорода в их 0,1 н. водных растворах составляет 0,099999 н. и 0,0999 н.,
т. е. сила кислот остается той же самой в пределах экспериментальной точ-
ности. Однако в растворителе, основность которого в 106 раз меньше основ-
ности воды, константы ионизации этих кислот равны 10-2 и 10-4 соответ-
ственно и концентрация ионов водорода в их 0,1 н. растворе равна 0,027 н.
Растворители, исполъвуемые в неводном титровании
107
(ЛН) и 0,0037 в. (ХН). Сила кислот становится различной (см. рис. 28 и
стр. 49 в работе [393]). Как уже говорилось, растворители могут значительно
отличаться от воды не только по основности и кислотности, но также и по их
специфическому взаимодействию с растворенным веществом. Однако отно-
сительная сила кислот формально одного и того же структурного типа
и сходного характера, например НС1, НВг и RCOOH, RCH2COOH, изме-
няется при смене растворителя лишь незначительно.
В результате тех взаимодействий между растворителем и растворенным
веществом, при которых образуются анионы или катионы растворителя,
возникает выравнивающий эффект, в то время как взаимодействия, ведущие
к установлению водородных связей, вызы-
вают появление дифференцирующих эффек-
тов.
Само по себе образование катионов или
анионов растворителя недостаточно для появ-
ления выравнивающего эффекта, важна также
степень, в какой протекает образование таких
ионов. В уксусной кислоте слабые и очень
слабые основания вызывают образование
ацетат-ионов лишь в незначительной степени,
а сильные основания почти полностью обра-
зуют ацетаты. В первом случае сохраняется
различие в силе оснований, а в последнем —
основность становится выравненной (см. гл. 3,
разд. 23,а и гл, 2, разд. 16,6).
Неорганические кислоты и их аналоги, хлор-
ная кислота, n-толуолсульфокислота, серная кис-
лота, хлористоводородная кислота, остаются силь-
ными в муравьиной кислоте, а в уксусной ведут
Рис. 28. Концентрация ионов
водорода в 0,1 н. растворе кис-
лоты как функция константы
ионизации.
себя различно. Это обусловлено значительным раз-
личием диэлектрической проницаемости растворителей [737в] (уксусная кислота е= 6,15,
муравьиная кислота е = 58,5). Значения рК перечисленных выше кислот возрастают,
когда диэлектрическая проницаемость разбавленной кислоты становится меньше. Таким
образом, растворители с низкой диэлектрической проницаемостью (уксусная и трихлор-
уксусная кислоты) позволяют различить силу НС1О4, H2SO4, CH3C6H4SO3H и НС1, в то
время как в растворителях с высокой диэлектрической проницаемостью (например,
муравьиной кислоте) сила этих кислот выравнивается [737г].
Приведенная ниже табл. 36 показывает действие муравьиной кислоты как выравни-
вающего растворителя для различных оснований [7376].
Таблица 36
Основание рК Основание рК
в воде в уксусной кислоте в воде в уксусной кислоте
Диэтил амин 2,90 0,74 Морфин 6,13 0,84
Кодеин 6,05 0,81 Мочевина 13,82 1,25
Водородная связь. Давно замечено, что соединения, содержащие гидро-
ксильные группы, имеют более высокие температуры кипения, чем их изо-
меры, не содержащие таких групп.
Этиловый спирт 78,4° Диметиловый эфир 23,7°
Пропиловый спирт 97,2° Метилэтиловый эфир 7,6°
Объясняется это взаимодействием между атомом водорода ОН-группы
в одной из молекул и атомом кислорода другой молекулы, в результате чего
образуется связь более сильная, чем вандерваальсова, но с меньшей энер-
108
Глава 6
гией, чем у обычных ковалентных связей. Этот тип химической связи, харак-
терный для взаимодействия между атомом водорода и атомом, обладающим
свободной парой электронов, был назван Хаггинсом, а также Латимером
и Родбушем [87, 500] водородной связью или водородными мостиками.
Образование водородной связи и ее прочность зависят от кислотности
водорода в молекуле, отдающей свой водород (ее протонодонорной способ-
ности), и от основности акцепторного атома, несущего неподеленную пару
электронов. Для того чтобы могла образоваться водородная связь, акцептор-
ный атом должен обычно быть по меньшей мере столь основного характера,
как азот, кислород или фтор в молекулах, не имеющих заряда, а связываю-
щий атом водорода должен обладать более кислым характером, чем атомы
водорода в насыщенных углеводородах, так как в противном случае возни-
кающая очень слабая водородная связь имеет весьма малое значение. Однако
если Н-атом слишком подвижен (лабилен), т. е он действительно является
«кислым водородом», а атом-акцептор слишком основен, то Н-атом переходит
в поле действия сил акцепторного атома, и в результате переноса протона
возникает ковалентная связь.
X ... Н ... :У Х-Н:У+.
Известно, что карбоновые кислоты существуют в виде димеров, и димери-
зация происходит за счет образования водородных связей:
_1,66А
о....Н---о
н—< с—н
/4
/ о----н....о
1.36А 1.07А 1,25А
В приведенной схеме атомы водорода расположены не на одинаковом рас-
стоянии от атомов кислорода (выраженном в ангстремах) (ср. [399, 440, 327]).
Мезомерная стабилизация димера не играет роли, поскольку она требует
существования резонансного гибрида, в котором атомы водорода должны
находиться на равном расстоянии от атомов кислорода; может играть роль
также и дипольное взаимодействие. Коулсон [151] считает, что при образо-
вании водородных связей определенное значение имеют также и электроста-
тические взаимодействия.
Подводя итоги, можно сказать, что на образование водородных связей
одновременно оказывают влияние а) относительная нуклеофильность ато-
мов, участвующих в связывании атома водорода, б) электростатиче-
ское взаимодействие диполей и в) в определенной степени резонансные
эффекты [605].
Образование водородной связи и образование катионов или анионов рас-
творителей — процессы взаимосвязанные:
(ЛН ... Ш) (Л-«Н|); (В ... sH) (BH+s-),
где ЛН — Н-кислота; sH — амфипротонный растворитель; А~ — анионное
основание; sH* иг — катион и анион растворителя; В — основание; 2?Н+ —
«ониевый» ион, образованный из основания при акцептировании протона
(см. также гл. 2, разд. 11,6 и гл. 5, разд. 31). Кислотность или основность
Растворители, используемые в неводном титровании
109
растворенных веществ становится различимой в тех случаях, когда они
образуют водородные связи с растворителем, в то время как значительное
образование «ониевого» соединения ведет к выравниванию. Тем не менее
исходный кислотный или основной характер веществ в обоих случаях изме-
няется.
Способность растворителей к образованию водородных связей можно определить
с помощью анализа инфракрасных спектров. Смещение (Ар.) поглощения в ИК-области,
характерного для —О—D-колебаний, при растворении CH3OD в исследуемом растворите-
ле по сравнению со значениями, найденными в бензольном растворе, пропорционально
склонности растворителя к образованию водородных связей (табл. 37) [282]. Прочность
Таблица 37
Растворитель Др Растворитель Др Растворитель Др Растворитель Др
Бензол 0,00 Метилэтилке- 0,11 Ацетон 0,14 Пиридин 0,27
Хлорбензол 0,02 тон 1,4-Диоксан 0,14 Этиленгли- 0,31
Нитробензол 0.04 Диэтилкетон 0,11 Эфир 0,19 коль
Ацетонитрил 0,09 Этилацетат 0,12 Анилин 0,27 Пиперидин 0,37
D-связи можно коррелировать с основностью растворителя и растворимостью протоно-
донорного соединения. Однако никакого соответствия между прочностью D-связей
и дипольным моментом растворителя установить не удалось.
Теория водородных связей и их экспериментальное определение детально рассматри-
ваются в работах [18, 19, 87, 90, 167, 282, 327, 341, 571, 771, 870].
39. КИСЛОТНЫЕ РАСТВОРИТЕЛИ
Для определения слабых оснований, нерастворимых в воде, используют
протонодонорные кислотные растворители, такие, как уксусная и пропионо-
вая кислоты. Так, азотистые органические основания взаимодействуют
с этими растворителями, и поэтому их можно титровать как относительно
сильные основания.
Я-j-sH 5Н+ + «-;
например,
CeH5NH2 + СН3СООН CeH5NHJ + СН3СОО-.
Основание Растворитель «Ониевый» Анион
ион растворителя
Сила аминов как оснований в результате взаимодействия с растворителем
возрастает, но и выравнивается; при этом образуется ацетат-ион, который
и определяют
СН3СОО- + C6H5NHJ + CH3COOHJCIO; —> CeH5NH+C10; 4- 2СН3СООН.
Ацетат-ион «Ониевый» Перхлорат Перхлорат Растворитель
ион ацилония анилиния
Реакция протекает до конца, если основание, которое надо оттитровать,—
более сильный акцептор протонов, чем растворитель, и если для титрования
используют сопряженное основание той же кислоты. В приведенном выше
случае это означает:
CeH5NH2 > СН3СООН > сю;.
Следовательно, протон хлорной кислоты мигрирует к анилину, взаимодей-
ствуя на промежуточной стадии с уксусной кислотой. Для титрования
оснований пригодны следующие растворители.
Инертные растворители (перечислены в порядке возрастания диэлек-
трической проницаемости): н-гексан, циклогексан, диоксан, четыреххлори-
стый углерод, бензол, хлороформ, хлорбензол, метилизобутилкетон, метил-
этилкетон, ацетон, ацетонитрил.
но
Глава 6
Кислотные или амфипротонные растворители (перечислены в порядке
уменьшения кислотности): муравьиная кислота, уксусная кислота, про-
пионовая кислота, уксусный ангидрид, нитрометан, нитробензол, этилен-
гликоль, пропиленгликоль, целлозольв, изопропиловый спирт. При титро-
вании оснований 1,4-диоксан считают инертным растворителем.
40. ОСНОВНЫЕ РАСТВОРИТЕЛИ
Протонакцепторные основные растворители, такие, как этилендиамин,
пиридин, н-бутиламин и диметилформамид, используют главным образом
для растворения кислот и кислотных аналогов (фенолов, енолов, имидов,
сульфамидов и т. д.), причем в этом случае образуется сопряженное основа-
ние растворенной кислоты и сольватированный протон:
АН + » sH++X-
например:
СвН5СООН + C5H5N C5H5NH+ + свн5соо-
Кислота Раство- «Ониевый» Сопряженное
рптель нон с кислотой
основание
Следовательно, кислотность при этом возрастает и становится выравненной
и появляется «ониевый» ион, который и титруют.
C5H5NH+ + CeH5COO- + CH3ONa —> CeH5COONa + C5H5N + CH3OH
Ион пири- Бензоат-анион Метилат Бензоат Растворители
диния натрия натрия
Определение возможно при условии, что, во-первых, молекула раство-
рителя способна образовывать «ониевый» ион и, во-вторых, титрант имеет
большую основность, чем сопряженное основание, образованное раство-
рителем, при этом могут быть определены кислоты (или кислотные аналоги)
сн3о-» C5H5N > свн5соо-.
Миграция протона к анионному основанию — метилат-иону — включает
промежуточную стадию взаимодействия с растворителем. Получить надеж-
ные результаты при титровании можно, если растворитель 1) имеет низкую
степень самоионизации, при этом ошибки, вызываемые сольволизом, стано-
вятся несущественными, и 2) обладает не слишком малой диэлектрической
проницаемостью, так как в противном случае может усложниться потенцио-
метрическое определение конечной точки.
Для определения кислот (и кислотных аналогов) пригодны следующие
растворители.
Инертные растворители (перечислены в порядке возрастания диэлектри-
ческой проницаемости): бензол, толуол, хлорбензол, метилизобутилкетон.
метилэтилкетон, ацетон, ацетонитрил.
Основные или амфипротонные растворители (перечислены в порядке
уменьшения основности): этилендиамин, н-бутиламин, пиридин, диметил-
формамид, 1,4-диоксан, тпретп-бутиловый спирт, изопропиловый спирт,
н-пропиловый спирт, н-бутиловый спирт, этиловый спирт, метиловый спирт,
эфир, метилцеллозольв, пропиленгликоль.
41. СМЕСИ РАСТВОРИТЕЛЕЙ
Преимущества смесей растворителей, приготовленных из двух или трех
компонентов, заключаются в следующем.
1. Не все соединения можно титровать в чистых растворителях отчасти
вследствие их малой растворимости, а отчасти из-за выпадения продуктов
Таблица 38
Смеси растворителей, пригодные для определения кислот (и кислотных аналогов)
Смесь растворителей Литература Смесь растворителей Литература
Ацетон — бутиловый спирт 692 Хлороформ — диметилформамид 153
Ацетон—пиридин Бензол — изопропиловый спирт 307 159, 530 Хлороформ — полиэтиленгли- коль 400 788
Бензол — метиловый спирт 25, 153, 252, 665 1,4-Диоксан — бутиловый спирт 692
Бензол — пиридин 307 Этиленгликоль — ацетон 170
Хлорбензол — эфир Хлорбензол — метиловый спирт (вместо смеси бензол — мети- ловый спирт) 753 Легкокипящий петролейный эфир — изопропиловый спирт Толуол — изопропиловый спирт 530 273
Таблица 39
Смеси растворителей, пригодные для определения оснований
Смесь растворителей
Уксусная кислота — уксусный ангидрид
Уксусная кислота — ацетон
Уксусная кислота — бензол
Уксусная кислота — четыреххлористый углерод
Уксусная кислота — хлорбензол
Уксусная кислота — хлороформ
Уксусная кислота — циклогексан
Уксусная кислота — дихлорэтан
Уксусная кислота—диоксан
Уксусная кислота — диоксан — уксусный ангидрид
Уксусная кислота—диоксан — нитрометан
Уксусная кислота — этиленгликоль
Уксусная кислота — нитрометан
Уксусный ангидрид—муравьиная кислота
Уксусный ангидрид — нитробензол
Уксусный ангидрид—нитрометан
Ацетон — фенол
Ацетонитрил — хлороформ — фенол
Бензол—уксусная кислота
Бензол — уксусный ангидрид
Бензол — хлороформ
Бензол — хлороформ — уксусный ангидрид
Бензол —нитробензол — уксусная кислота и бензол—нитробензол—
трихлоруксусная кислота
Бензол — нитрометан
Четыреххлористый углерод—фенол
Хлороформ—уксусная кислота
Хлороформ — изопропиловый спирт
Хлороформ — изопропиловый спирт — фенол
м - Крезол — ацетонитрил
Ацетоуксусный эфир—уксусная кислота
Этиленгликоль — фенол
Гексан — ацетон
Нитробензол — метиловый спирт
Нитробензол — ксилол
Нитробензол — бензол—муравьиная кислота
Пропионовая кислота—хлорбензол—пропионовый ангидрид
Литература
158, 288, 306, 310,
311, 314, 642,
707, 732, 854
453
575
314, 658
175, 317, 638
12, 28
860
130, 453
644
130
401
635
132
676
865
58, 249
700
123, 639
496, 575
703
158
158
590
586
302, 323
28, 297, 587, 790
702
701, 702
787
453
622
586, 587
579
579
890
311, 314
112
Глава 6
нейтрализации в виде желеобразных осадков, как, например, при титрова-
нии бензойной кислоты метилатом калия в бензоле. Этого можно избежать,
если добавить к бензолу метиловый или изопропиловый спирт.
2. Сольватирующую способность высококислотных или высокоосновных
растворителей можно сочетать с подходящими свойствами инертных или
дифференцирующих растворителей. В таких растворителях изменение
окраски индикаторов происходит четко, а скачок потенциала часто более
крутой, чем в кислотных или основных растворителях. Растворители, обла-
дающие выравнивающим действием, смешивают с инертными растворителями
чаще всего в соотношении от 1 : 1 до 1 : 20. Так, уксусную кислоту смеши-
вают с четыреххлористым углеродом, пиридин — с бензолом и т. д. Приме-
ры таких смесей даны в табл. 38 и 39.
Соотношение кислотного и инертного растворителей следует определять
экспериментально. В зависимости от растворимости можно улучшить резуль-
таты анализа, если увеличить количество инертного растворителя примерно
в 9 раз по отношению к кислотному растворителю, например уксусной кисло-
те. В таких случаях четкость определения конечной точки оказывается
достаточной даже при титровании 0,001 н. хлорной кислоты (гл. 13 и разд. 72
в работе [319]).
Так называемые G — Н-смеси растворителей (т. е. смеси гликоль — угле-
водород [170, 489, 621]) можно также отнести к этой группе смесей раство-
рителей. В качестве G-компонента (гликольного) такой смеси могут высту-
пать этилен-, пропилен- или диэтиленгликоль, а также смеси этилового
и метилового эфиров этиленгликоля (целлозольв, метилцеллозольв), а в каче-
стве Н-компонента (углеводородного) * могут служить хлороформ, диоксан,
изопропиловый, бутиловый, амиловый и бензиловый спирты и т. д. G-ком-
понент способствует растворению соединений, содержащих высокополярные
группы, в то время как Н-компонент понижает вязкость гликолей и облег-
чает растворение алифатических и ароматических соединений.
3. Инертные растворители способствуют выделению продукта нейтра-
лизации в кристаллической форме. Это часто помогает при титровании, так
как усиливает четкость цветового перехода индикатора [658], например при
определении сульфамидов (см. гл. 23, разд. 131) [575].
42. ВЫБОР РАСТВОРИТЕЛЕЙ
Растворители должны удовлетворять следующим требованиям: 1) они
не должны вступать в нежелательные побочные реакции с анализируемыми
веществами или с рабочими (стандартными) растворами (это ограничение
не включает процессы сольватации и образование катионов и анионов рас-
творителя); 2) они должны растворять вещество по меньшей мере настолько,
чтобы можно было получить 0,01 н. раствор; 3) растворители должны позво-
лить определять точку эквивалентности либо с помощью индикаторов, либо
потенциометрически; 4) наконец, их очистка должна быть простой и легкой.
При выборе растворителя желательно, хотя и не необходимо, распола-
гать полными сведениями о растворимости анализируемого соединения,
его чувствительности к свету и примерными константами кислотности или
основности, измеренными в водной среде (см. табл. 10—12, 16, 17, 19, 21,
22, 73 и 74). Наиболее подходящий индикатор обычно подбирают эмпириче-
ски. Точное изменение окраски индикатора контролируют потенциометриче-
ским титрованием (гл. 12).
В случае сильных или умеренно сильных кислот и оснований используют
растворители кислотного или основного характера. При титровании слабых
кислот или оснований кислые или основные растворители желательно раз-
бавлять инертными.
* Точнее, водородсодержащего компонента.— Прим, перев.
Растворители, используемые в неводном титровании
113
Таблица 40
Кислота рК Основание рКа рК6
Сильная <2 Сильное <8 < Ю,5
Умеренно сильная 2-8 Умеренно 8—12 10,5-12
Слабая 8—10 сильное
Очень слабая > 10 Слабое > 12 >12
а Индикаторное титрование.
® Потенциометрическое титрование (ср. рис. 20 и 21).
Приведенные ниже (табл. 40) сведения систематизируют данные по «силе»
кислот и оснований в неводных средах, основанные на измерении величин
рК в воде.
Из-за специфического взаимодействия растворенного вещества с раство-
рителем нельзя предложить какие-то единые правила выбора растворителя.
Но, например, при титровании органических оснований целесообразно посту-
пить следующим образом. Вначале приготовляют примерно 0,02 н. раствор
органического основания в 20 мл уксусной кислоты, если используют хлор-
гидрат основания, то на 20 мл уксусной кислоты берут еще 70—75 мг
(0,011—0,012 молъ/л) ацетата ртути(П). В колбы для титрования емкостью
25—50 мл или же в центрифужные пробирки помещают по 1 мл этого запас-
ного раствора и разбавляют пробы указанными в табл. 41 очищенными рас-
творителями. Полученный раствор титруют из микробюретки 0,01 н. рас-
твором хлорной кислоты в смеси уксусной кислоты с четыреххлористым угле-
родом (1 : 1) в присутствии индикатора, причем на титрование монофункцио-
нальных оснований расходуется примерно 2 мл стандартного раствора. Этот
способ дает возможность подобрать наиболее подходящие для определения
растворитель и индикатор.
Таблица 41
Растворитель
Индикатор
10 мл четыреххлорпстого углерода
или СНС13
10 мл пропионовой кислоты
10 мл хлорбензола
10 мл диоксана
20 мл диоксана
20 мл ацетонитрила
10 мл ацетонитрила
10 мл смеси уксусной кислоты
с уксусным ангидридом (5:1)
10 мл нитрометана
10 мл смеси нитрометана с бензолом
(1:1)
10 мл метил целлозольва
Кристаллический фиолетовый, 1-нафтолбензепн
Кристаллический фиолетовый, метанпловый жел-
тый
Кристаллический фиолетовый, метиловый фиоле-
товый, бромфеноловый синий, 1-нафтолбензеин,
тропеолин ОО
Метиловый фиолетовый, кристаллический фиоле-
товый
Метиловый фиолетовый, метиловый красный
Метиловый красный
Метиловый фиолетовый, 1-нафтолбензепн, тро-
пеолин ОО
Кристаллический фиолетовый, крезоловый крас-
ный
Кристаллический фиолетовый, 1-нафтолбензеин
Хинальдиновый красный
Тимоловый синий, конго красный
Если для титрования берут навески больше указанных, то необходимо
потенциометрически проверить переход окраски индикатора в выбранном
наиболее подходящем растворителе отдельно в присутствии исследуемого
вещества и в присутствии стандартного вещества, например дифенилгуа-
нидина, по которому устанавливается титр рабочего раствора. Эта проверка
в высшей степени необходима при разработке методов определения новых
соединений.
Глава 7
ХИМИЧЕСКИЕ И ФИЗИКО-ХИМИЧЕСКИЕ
ХАРАКТЕРИСТИКИ РАСТВОРИТЕЛЕЙ
43. ИНЕРТНЫЕ РАСТВОРИТЕЛИ
н-Гексан СН3(СН2)4СН3, н-гептан СН3(СН2)5СН3 и циклогексан (гекса-
гидробензол) С6Н12 представляют собой инертные растворители и исполь-
зуются в смеси с уксусной кислотой, пропионовой кислотой и ацетоном
[587, 860]. Содержание воды в последних, так же как и в стандартном раство-
ре хлорной кислоты, не должно превышать 0,05%; в противном случае пер-
воначально гомогенный раствор в процессе титрования расслаивается,
причем водная уксусная кислота с индикатором составляют нижнюю фазу.
О приготовлении совершенно не содержащего следов воды 0,03 н. стандарт-
ного раствора хлорной кислоты в диоксане см. гл. 9, разд. 52, б.
Перечисленные выше растворители легко воспламеняются (табл. 42).
Таблица 42
Инертные растворители
Растворитель Т. тип., °C tf20 "D
м-Гексан 68,7 0,659 1,3749
н-Гептан 98,4 0,684 1,3877
Циклогексан 80,7 0,779 1,4262
Хлороформ Четыреххлорпстый угле- 61,2 1,489 1,4455
род 76,8 1,594 1,4604
Бензол 80,1 1,879 1,5011
Хлорбензол 131,7 1,106 1,5248
Ацетон 56 ,2 0,791 1,3588
Метплэтпл кетон 79,5 0,804 1.3785
Метилпзобутилкетон 115,7 0,800 1,3958
Ацетонитрил 81,6 0,783 1,3441
Сольватирующая способность хлороформа СНС13 выше, чем у гексана.
Этот растворитель пригоден для экстракции миллиграммовых количеств
органических оснований в тех случаях, когда не требуется концентрирова-
ние экстракта (гл. 8, разд. 49,к). Благодаря склонности к образованию
водородных связей сольватирующая способность хлороформа выше, чем
у четыреххлористого углерода (гл. 3, разд. 24, б).
Сольватирующая способность четыреххлористого углерода СС14 выше,
чем у гексана, но ниже, чем у хлороформа. Растворитель применяют для
экстракции органических оснований, которые затем оттитровывают 0,01 н.
хлорной кислотой, причем к экстракту перед титрованием добавляют уксус-
ную кислоту, пропионовую кислоту или уксусный ангидрид. Вместо этих
растворителей для титрования можно использовать 0,005 н. раствор
п-толуолсульфокислоты [299, 305]. Четыреххлористый углерод применяют
также в смеси с фенолом [302, 323].
При приготовлении 0,02—0,01 н. стандартных растворов хлорной кислоты
с целью усиления резкости перехода окраски индикатора рекомендуется
использовать смесь уксусной (или пропионовой) кислоты с равным коли-
чеством четыреххлористого углерода.
Химические и физико-химические характеристики растворителей
115
Бензол в смеси с метиловым спиртом, изопропиловым спиртом или пири-
дином наиболее широко применяется в качестве растворителя для неводного
титрования кислот. Для определения оснований бензол смешивают с уксус-
ной кислотой или уксусным ангидридом в соотношении, определяемом эмпи-
рически. Бензол менее удобен для экстрагирования органических оснований,
чем четыреххлористый углерод или хлороформ, так как он часто образует
эмульсии. Толуол и ксилол можно применять в тех же целях, что и бензол.
Бензол дает аддукты с электрофильными кислотами Льюиса [243].
Хлорбензол CfiH5CI в смеси с уксусной кислотой, пропионовой кислотой
или уксусным ангидридом (1 : 1 —10 : 1) наиболее широко применим из хло-
рированных ароматических углеводородов; в этих смесях повышается рез-
кость перехода окраски индикатора и становится легче обнаружить конеч-
ную точку потенциометрическим методом. Сольватирующая способность
хлорбензола выше, чем у бензола, хотя и ниже, чем у уксусной кислоты.
Более высокая сольватирующая способность хлорбензола по сравнению
с бензолом частично может быть объяснена его дипольным моментом.
Наблюдаемый дипольный момент обусловлен противоположно направленными
индукционным и мезомерным эффектами атома хлора (см. стр. 429 в работе [870]).
Дипольные моменты всех хлорпропзводных насыщенных алифатических углеводо-
родов близки к 2.0 D. Отрицательный конец диполя направлен к атому хлора. В случае
хлорбензола дипольный момент (ц) из-за возможности резонанса уменьшается до 1,56D.
В качестве индикатора в среде хлорбензола наиболее пригоден бром-
феноловый синий, а в присутствии уксусной кислоты — трифенилметановые
красители. При увеличении силы растворенного основания окраска бром-
фенолового синего изменяется от пурпурной к розовой и, наконец, к желтой
(например, рК атропина 4,35, стрихнина 6,0, амидопирина 9,30). В точке
эквивалентности желтая окраска исчезает, раствор обесцвечивается [638]
(гл. 23, разд. 124, б).
Хлорбензол непригоден для определения оснований со значением рА7
в воде >> 10. Соли алкалоидов растворимы в хлорбензоле лишь в присут-
ствии уксусной кислоты. Смеси хлорбензола с уксусной кислотой и уксус-
ным ангидридом или с пропионовой кислотой и пропионовым ангидридом
(9:9:2) пригодны для определения эквивалентного веса пикратов (гл. 24,
разд. 135,в) [311, 314].
При титровании слабых кислот или аналогов кислот (например, фенолов)
в качестве растворителя широко применяют ацетон СН3СОСН3 или его
смесь с пиридином в отношении 4 : 1 [307]. По мнению Фритца и Ямамуры
[258], титрование кислот и их аналогов стандартным раствором гидроокиси
тетрабутиламмония (ТБАГ) удобно проводить в ацетоне потому, что кислот-
ные и основные свойства этого растворителя не препятствуют определению.
В некоторых условиях ацетон может давать анион (СН3СОСН2)_, способный
за счет свободной электронной пары образовывать координационную связь
с кислотами Льюиса. Диэлектрическая проницаемость ацетона достаточно
велика для того, чтобы проводить в нем потенциометрическое титрование.
Ацетон во всех отношениях смешивается с водой, метиловым спиртом,
уксусной кислотой, бензолом, четыреххлористым углеродом и пиридином;
он является огнеопасным растворителем.
116
Глава 7
Ацетон — исключительно слабая кислота с константой диссоциации порядка 10-19
(см. табл. 28), хотя константа образования карбаниона (аниона ацетона) равна 2,8-10-8.
Чистый ацетон содержит лишь 0,00025% енольной формы [729]. В ацетилацетоне равно-
весная смесь кетонной и енольной форм содержит 80% енола [145]. Это различие может
быть приписано стабилизующему влиянию внутримолекулярной водородной связи.
Поскольку кетонная форма в водном растворе тоже образует водородные связи с моле-
кулами воды, ацетилацетон в воде содержит лишь 15% енола, но в растворе гексана
(а также в парах) содержание енольной формы составляет 92% (см. также табл. 28):
.О.
3 H
снз
сн3
Метилэтилкетон СН3СОС2Н5 и метилизобутилкетон (4-метилпента-
нон-2) — растворители с хорошими дифференцирующими свойствами.
Последнее вещество с успехом используется при дифференцирующем титро-
вании как аналогов кислот, так и алифатических и ароматических аминов
[105, 479]. Значение потенциала полунейтрализации и рК для большого
числа замещенных фенолов в среде метилизобутилкетона примерно то же,
что и в пиридине [595, 780].
Ацетонитрил CH3CN представляет собой практически инертный раство-
ритель, хотя и ведет себя как кислота по отношению к пиколину [829] и как
основание по отношению к сильным льюисовским кислотам [396, 501].
Вследствие своей «основности» он служит хорошим растворителем для льюи-
совских кислот, например А1С13. По-видимому, хлористый алюминий в аце-
тонитриле образует комплекс с молекулами растворителя [396]. Константа
самоионизации ацетонитрила по схеме
2CH3GN CH3GNH + CH2GN-
равна 3,16 «10-20 [689]. По данным Ван-дер-Хейде и Дамена [371], различие
в кислотности определяемых в ацетонитриле кислот еще больше, что под-
тверждается более новыми данными для константы самоионизации: 3-10~26
[140]. Степень самоионизации ацетонитрила очень трудно определить вслед-
ствие того, что нестабилен CH3CNH+, являющийся очень сильной кислотой,
а также потому, что очень сильное основание CH2CN~, вступая в реакцию
конденсации с ацетонитрилом, образует слабое основание СН3—C = N"[66].
СН2 — C N
Вследствие этого ацетонитрил, хотя и является полярным растворителем,
может быть отнесен к инертным растворителям и широко применяется
для дифференцирующего титрования кислот, их аналогов и оснований
[155, 247].
Основность ацетонитрила связана также с полярным характером его
молекулы [828].
Ацетонитрил как основание [501]:
CH3C = N: + BF3 CH3CN:BF3
Ацетонитрил как кислота [829]:
GH3CN + 5 СН3 — C = N~ —(5CH3)+J СХ-
В+
Относительно высокое значение диэлектрической проницаемости ацето-
нитрила должно способствовать диссоциации на ионы комплекса ацетонитрила
Химические и физико-химические характеристики растворителей
117
с хлористым алюминием [396]
2CH3CN.AICI3 [сн3см.а1С13-мссн2]++а1С1;
Хлорная кислота является сильной кислотой в среде ацетонитрила [138]
HCIO4 + CH3CN CH3CNH4 С1О7
Эта реакция заканчивается не сразу, поскольку сольватация протона проис-
ходит медленно. Так как ион CH3CNH неустойчив, сила кислот в растворе
ацетонитрила постепенно уменьшается [468], весьма возможно, либо за счет
химического взаимодействия кислоты с молекулами растворителя, либо за
счет разложения ацетонитрила [404, 424, 502].
Кислотно-основное равновесие в ацетонитриле изучали Кольтгоф, Котце
и сотр. [140, 465, 466, 603]. Они установили, что хлорная кислота в ацетони-
триле полностью диссоциирует. Серная, азотная, соляная и бромистоводо-
родная кислоты диссоциируют
2НЛ Н+ + Н^2.
Основной обратимой реакцией дифенилгуанидина (В) с ацетонитрилом
при концентрациях дифенилгуанидина > 5 • 10-2 моль!л является
22? CH3CN HB£4-CH2CN-
Последовательность изменения силы некоторых кислот в растворе ацето-
нитрила следующая: уксусная >• хлористый ацетил >• пикриновая > моно-
хлоруксусная >• трихлоруксусная [828] и хлор-
ная >» бромистоводородная >• ион 2,5-ди-
хлоранилиния >• соляная >• п-толуолсуль-
фоновая >• серная >• ион диэтиланилиния >•
>• фторвалериановая >• щавелевая > фосфор-
ная >• бензойная >• уксусная.
При определениях сульфатов алкалоидов,
большинство которых плохо растворимо в уксус-
ной кислоте, в качестве растворителя приме-
няют смесь ацетонитрила с хлороформом и фе-
нолом (10 : 4 : 1) [123]. Ацетонитрил обладает
хорошими дифференцирующими свойствами, по-
Таблица 43
Растворитель Число пере- гибов кривой Число свя- зан- ных прото- нов
Уксусная кис- 1 2
лота
Ацетонитрил 2 2
Ацетон 1 1
этому он очень успешно применяется для по-
тенциометрического титрования, например, диалкилпроизводных п-фени-
лендиамина (табл. 43) [521]. Аналогичная картина наблюдается и при
определении производных пиперазина (гл. 23, разд. 123) [132, 811]. Подроб-
нее об ионизации кислот в ацетонитриле см. в гл. 2, разд. 15, а. Ацетони-
трил смешивается во всех отношениях с водой, этиловым спиртом, диэтило-
вым эфиром, бензолом и уксусной кислотой.
44. КИСЛОТНЫЕ РАСТВОРИТЕЛИ (ТАБЛ. 44)
Муравьиная кислота НСООН имеет высокую диэлектрическую прони-
цаемость е = 58,5 [812]. Штройли [777] титровал амины и азотсодержащие
гетероциклы в смеси нитрометана с муравьиной кислотой.
В продажной муравьиной кислоте часто содержится лишь 90% вещества.
В тех случаях когда основание или его хлоргидрат нерастворимы в уксусной
или пропионовой кислоте, но растворяются в 90 %-ной муравьиной кислоте,
их растворяют в муравьиной кислоте известных концентрации и качества,
а затем разбавляют равным объемом уксусной кислоты. К этому раствору
118
Глава 7
Таблица 44
Растворитель т. кип., °C С/20 „20 71D
Муравьиная кислота 100,7 1,220 1,3714
Уксусная кислота 117,7—118,1 1,049 1,3716
Пропионовая кислота 140,8—141,4 0,993 1,3865
Уксусный ангидрид 140,0 1,049 1,3904
Пропионовый ангидрид 167,0 1,011 1,4046
Нитрометан 101,3 1,13 1,3819
Нитробензол 210,8 1,203 1,5524
Метиловый спирт 64,7 0,791 1,3286
Этиловый спирт 78,4 0,789 1,3614
н-Пропиловый спирт 97,2 0,803 1,3855
Изопропиловый спирт 82,4 0,785 1,3747
(25°)
Этиленгликоль 197,9 1,113 1,4318
1,2-Пропиленглпколь 188—189 1,136 1,4331
Монометиловып эфир этиленгликоля (метил- 124,5 0,965 1.402
целлозольв)
Моноэтпловый эфир этиленгликоля (целло- 134,5 0,93 1,4076
зольв)
mpem-Бутпловый спирт 82,4—82,6 0,779
(т. пл. 25,6) (26°)
добавляют избыток стандартного раствора хлорной кислоты и такое коли-
чество уксусного ангидрида, которого достаточно для реакции с водой,
содержащейся в муравьиной кислоте. При добавлении уксусного ангидрида
необходимо применять охлаждение, так как хлорная кислота катализирует
реакцию уксусного ангидрида с водой. Избыток хлорной кислоты определяют
обратным титрованием при помощи раствора ацетата натрия в уксусной
кислоте.
Основания, нерастворимые в уксусном ангидриде, вначале необходимо
растворить в 5 мл 98 %-ной муравьиной кислоты, а затем смешать с 50 мл
уксусного ангидрида. Так поступают, например, при определении кофеина
и теобромина (гл. 23, разд. 126) [676]. Солянокислый хинин можно титровать
в уксусном ангидриде непосредственно, без добавления ацетата ртути(П)
(гл. 24, разд. 136,н) [676].
При определении оснований в качестве растворителя чаще всего приме-
няют уксусную кислоту СН3СООН, поэтому ее свойства будут рассмотрены
более подробно *.
Уксусная кислота — хороший растворитель для многих органических
и неорганических соединений. При растворении кислот в уксусной кислоте
образуются оксониевые ионы в самых различных концентрациях. В случае
метандисульфокислоты, хлорной кислоты и хлорметансульфокислоты кон-
центрация оксониевых ионов высока, однако монохлоруксусная кислота
в основном растворяется в молекулярной форме и концентрация оксоние-
вых ионов в этом случае незначительна. Ацилониевый ион является более
сильной кислотой, чем ион гидроксония
НС1О4+СН3СООН СН3СООЩ + С1О7,
НСЮ4 + Н2О Н3О+4-СЮ7.
* См. также Heymann and Klaus: «Химия в безводной уксусной кислоте, химия
низших жирных кислот и их производных» в книге «Chemistry in Non-aqueous Ionizing
Solvents», vol. IV, Ed. Jander, Spandau and Addison, Interscience, New York, 1963.
Химические и физико-химические характеристики растворителей
119
Ион ацилония СНдСООЩ представляет собой сопряженную кислоту
слабого «основания» — уксусной кислоты. Это ион стабилизован за счет
резонанса
Уксусная кислота как растворитель «сходна с водой» [240]; она способна
к самоионизации с образованием сольватированного протона и ацетат-иона —
аналога основания (гл. 1, разд. 3). Ионное произведение уксусной кислоты
К = Ссн3соон.^сн3соо- = 3,5-10"15 [102].
Как правило, карбоновые кислоты (и уксусная кислота в том числе)
в жидкой фазе присутствуют в димерной форме (стр. 261 в работе [605]).
Димер образуется за счет возникновения водородных связей: функциональ-
ные группы, содержащие подвижный водород, вступают в донорно-акцеп-
торное взаимодействие с неподеленной парой электронов атома кислорода
другой молекулы [87]
X—Н ... У (X— донор протона, 1’ — акцептор протона)
,0-Н ... О.
СН3 —С Х'С — СН3 обозначается как (СН3СООН)2
... н-о/
(см. также гл. 6, разд. 38).
Уксусная кислота сама плохо проводит электрический ток, однако рас-
творы способных к ионизации в уксусной кислоте соединений являются
хорошими проводниками. Это свидетельствует в пользу того, что, по крайней
мере частично, эти вещества находятся в диссоциированном состоянии
несмотря на низкую диэлектрическую проницаемость уксусной кислоты.
Это кажущееся противоречие можно объяснить, если принять во внимание
ассоциацию аминов, а также большую способность образовавшихся ком-
плексов к диссоциации (гл. 5, разд. 33) [561].
Уксусная кислота координационно ненасыщена и образует комплексы
с некоторыми насыщенными соединениями [561]. Одним из наиболее важных
комплексов является перхлорат ацилония, впервые полученный Беером
[354]. Перхлорат ацилония — стабильное соединение, образующее ионную
кристаллическую решетку; за счет электромерного эффекта компенсируется
электронная недостаточность, возникающая при поляризации карбоксиль-
ной группы [205].
В случае уксусной кислоты нейтрализация и сольволиз также являются
конкурирующими процессами (гл. 5, разд. 31); в некоторой степени они
происходят между димерными или ассоциированными молекулами и ионами.
В процессе нейтрализации сольватированный протон координируется с ана-
логом основания — анионом кислоты и образуются две молекулы раство-
рителя
СН3СОО-4-СН3СООЩ —> (СН3СООН)2.
Слабые основания в среде уксусной кислоты образуют «ониевые» ацетаты
лишь в небольшой степени. Поэтому слабые основания непосредственно пре-
вращаются в перхлораты оснований при титровании стандартным раствором [85].
c6h5nh2 ... сн3соон+сн3соощсю; c6h5nh3cio;+(ch3cooh)2.
Сильные основания и основания средней силы в уксусной кислоте из-за
выравнивающего эффекта последней присутствуют в основном в виде ацетатов.
120
Глава 7
Существует ограниченное число данных, на основании которых можно
судить, превратились ли основания средней силы в «ониевые» соли (пол-
ностью или частично) (гл. 2, разд. 16,6). Однако вследствие существования
равновесия
В ... СН3СООН 7Z ЯН+СНзСОО-
уксусная кислота обладает выравнивающим эффектом по отношению к силь-
ным основаниям и основаниям средней силы и дифференцирующим дей-
ствием по отношению к слабым и очень слабым основаниям (гл. 6, разд. 38).
На рис. 20—22 в гл. 3 показано выравнивание силы сильных оснований
и оснований средней силы и дифференцирование слабых оснований (табл. 45).
Таблица 45
Основание рК Действие уксусной кислоты Основание рК Действие уксусной кислоты
Гуанидин 0,5 Выравнивающее о-Хлоранплин 11,4 Дифференцирую-
Пиперидин 2,8 » щее
Пиридин 8,8 » п-Нитроанилнн 13,0 То же
п-Хлоранплпн 10,1 Переходное Мочевина 13,5 » »
.и-Хлоранплпн 10,5 Дифференцирую- о-Нптроанилпн 14,2 » »
щее Ацетамид 14,5 » »
-к-Нптроанилпн 11,4 То же
Пропионовую кислоту СН3СН2СООН можно использовать так же, как
и уксусную, для выравнивания силы оснований, p^вн- протонирования
пропионовой кислоты составляет —6,8 [202]. (р/*Свн+ уксусной кислоты
равен —6,2 [279]). Переход окраски индикатора (кристаллического фио-
летового, метанилового желтого) в пропионовой кислоте часто четче, чем
в уксусной. При потенциометрическом титровании отклонение в милли-
вольтах в конечной точке титрования также больше, чем в уксусной кислоте
[314, 374].
Пикрат ппперидпнометплтолилпропанона (20 мг) можно титровать 0,01 н. хлорной
кислотой в смеси хлорбензол — уксусная кислота — уксусный ангидрид с точностью
±0,7%, в то время как при применении смеси хлорбензол — пропионовая кислота —
пропионовый ангидрид точность составляет ±0,1%.
Пропионовая кислота использовалась в титриметрии с 1938 г. [801].
Уксусный ангидрид (СН3СО)2О — жидкость, обладающая раздражаю-
щим действием на кожу,— используется в качестве растворителя при невод-
ном титровании, а также как дегидратирующий и ацетилирующий агент.
Его сольватирующая способность меньше, чем у уксусной кислоты, посколь-
ку он менее склонен к образованию водородных связей; доказательства ассо-
циации молекул уксусного ангидрида отсутствуют [531]. За исключением
анализа соединений, содержащих аминогруппы, способные к ацетилирова-
нию, реакции нейтрализации в уксусном ангидриде можно проводить так же,
как и в уксусной кислоте.
В чистом состоянии уксусный ангидрид диссоциирует в соответствии
со следующей схемой [531, 561, 722]:
(СН3СО)2О (СН3СО)+ 4- СНзСОО-
Ацетил-катион Ацетат-ион
Константа самодиссоциации при 20° К = 3 «10-15 [423]. Ацетил-катион
(ион ацетилия) ведет себя в среде уксусного ангидрида так же, как протон
в воде, однако он не сольватирован [531].
Химические и физико-химические характеристики растворителей
121
В соответствии с вышеприведенной схемой диссоциации вещества, спо-
собные генерировать ацетил-катион, являются аналогами кислот (например,
хлористый ацетил), а вещества, дающие при диссоциации ацетат-ион,—
аналогами оснований. В смеси уксусной кислоты с уксусным ангидридом
могут существовать три частицы с кислотными свойствами [110, 531]; чет-
вертая, ион кислотного характера, может существовать лишь в чистом
уксусном ангидриде [831].
1. Ион ацилония СН3СООН2 Донорная кислота
2. Ион ацетангидридия (СН3СО)2ОН+ Донорная кислота
3. Ион ацетилия (СН3СО)+ Акцепторная кислота
4. Ион ацетилоксония [(СН3СО)3О]+ Акцепторная кислота
По силе они располагаются в следующий ряд [878]:
СН3СООН£ < (СН3СО)2ОН+ < (СН3СО)+.
Катион (СН3СО)2ОН+ реагирует, например, с ароматическими N-окисями
в среде уксусного ангидрида
О ОН
II II
-СН3-С —О —С-СН3
/ч
СЮ; -- | | СЮ;ч-СН3СООН
N +
ОСОСН3
О титровании ароматических N-окисей хлорной кислотой см. работы
[604, 880].
Подробные сведения о кислотно-основных взаимодействиях в среде
уксусного ангидрида можно найти в работе Усановича [831]. Хлористый
ацетил, хлористый бензоил и трихлоруксусная кислота в этом амфотерном
растворителе ведут себя как кислоты и способны титроваться ацетатом
натрия [831]. В среде уксусного ангидрида большое количество солей подвер-
гается сольволизу: например, хлористый алюминий образует хлористый
ацетил и А1С1(СНзСОО)2.
То, что уксусный ангидрид является более «основным» растворителем, чем уксусная
кислота, по-видимому, можно объяснить следующим. Хотя электронная плотность,
локализованная на атоме кислорода (отмеченном звездочкой) в молекуле уксусной кисло-
ты, и уменьшается при введении ацетокспгруппы и вследствие этого «основность» моле-
кулы уменьшается, уксусный ангидрид способен к координации с протонами, вероятно,
за счет образования хелата, в то время как уксусная кислота представляет собой
дпмер [531].
Теоретическое уменьшение «основности»
О* О* О
II II II
сн3—с—он —> СН3—С—О—с—сн3
Практическое увеличение «основности»
Н
О
II II
с с
crif 'V7 ^снз.
Ион ацетангидридия
(протонированный уксусный
ангидрид)
На основании большого числа экспериментальных данных можно сделать вывод, что
уксусный ангидрид может быть как донором протонов, так и донором карбаниона, как,
122
Глава 7
например, в реакции Перкина [429]. В этом случае самоионизация уксусного ангидрида
протекает следующим образом [676]:
2 (СН3СО)2О
(см. гл. 24, разд. 136,н).
При потенциометрическом титровании оснований добавление уксусного
ангидрида увеличивает скачок потенциала в точке эквивалентности. Это
может быть скорее приписано увеличению ионной активности хлорной кис-
лоты, чем дегидратирующему действию ангидрида, т. е. в этом случае можно
предположить образование более сильной комплексной кислоты [77]
[С1О4(СН3СООСОСНз)п]Н.
Подробнее об «ацетилперхлоратах», обладающих свойствами льюисовских кислот,
см. гл. 23, разд. 130,а.
На основании вышесказанного можно сделать вывод, что уксусный
ангидрид является «водоподобным» растворителем, однако в некоторых отно-
шениях более простым, чем вода: он не ассоциирован и ион ацетилия не соль-
ватирован. Постулирование существования стабильного иона ацетилия
облегчает понимание многих реакций [531]. Например:
Реакция алифатических кислот с ангидридами:
2R'COOH + (R"CO)2O = 2R"COOH + (R'C0)2O.
Диспропорционирование смешанных ангидридов:
2R'COOOCR" (R'CO)2O4-(R"CO)2O.
Доказательством существования стабильных ацил-катионов служит ионизация
2,6-дизамещенных бензойных кислот в серной кислоте, которую можно обнаружить
криоскопически [818]
RCOOH + 2H2SO4 = RCO+ 4- ОН£ < 2HSO;.
Пропионовый ангидрид (СН3СН2СО)2О более подходит для обезвоживания
пропионовой кислоты, чем уксусный ангидрид; его удобно применять при
анализе пикратов оснований [311] и определении числа гидроксильных
групп [640].
Нитрометан CH3NO2 — растворитель слабокислого характера: \)Ка =
= 10,6. Его можно титровать метилатом натрия в н-бутиламине и стандарт-
ным раствором гидроокиси тетрабутиламмония в пиридине [252, 780].
Он образует кристаллические аддукты со стрихнином, эфедрином и кодеи-
ном [126]. Его часто используют для проведения дифференцирующего
титрования [132, 249].
В серной кислоте нитрометан ведет себя как основание [270].
Среди немногих растворителей, содержащих нптрогруппу, редко используемый
нитроэтан также проявляет слабокислые свойства, однако его способность отдавать
протон невысока. В соответствии с представлениями Пирсона и Диллона [630] следует
отличать константу ионизации от константы образования карбаниона (см. гл. 2, разд. 21).
До сих пор не установлено, какая пз двух точек зрения на образование карбанио-
нов — более ранние представления Ганча пли высказанные позднее соображения —
является более обоснованной [369, 370, 371]. В случае алифатических нитросоединений
имеет место таутомерное равновесие между обычной и а^и-формой нитросоединений.
yrO ZO
СН3 — N CH2 = N
^он
Химические и физико-химические характеристики растворителей
123
Такое превращение можно обнаружить кондуктометрически при солеобразовании,
которое является медленным процессом, т. е. поддается измерению; возможно, устанав-
ливается следующее равновесие:
/° +/°
ch3-no2 ZZ ch2 = n ZT ch2 = n дн+
^он \о-
I II
ац п-Форма Л-4~Н_,
где Л- —анион.
Содержание а^и-формы нитрометана в водном растворе, как указывает Ганч, мало
К = 1Щ-= 1,1.10-7 и А'а = 1 =5,6-10-*
Нитробензол в качестве растворителя используется редко, хотя во многих
случаях он более удобен, чем нитрометан. Иногда применяют смеси нитро-
бензола с уксусным ангидридом, бензолом и гептаном [579, 590, 865].
Дипольный момент нитробензола составляет 3,97 D, а у насыщенных алифатических
нитросоедпнений никогда не превышает 3,7 D. Больший дипольный момент нитробензола
объясняется мезомерией
Нитробензол является слабой льюисовской кислотой, так как способен
образовывать л-комплексы, например с ароматическими аминами [585, 595]
Смесь нитробензола с анилином приобретает желтую окраску из-за образо-
вания комплекса с переносом заряда.
В среде нитробензола (а также и о-нитротолуола) n-аминобензойная кис-
лота является более сильной, чем в воде, так как образование л-комплекса
понижает электронную плотность бензольного кольца
В резонансной структуре, изображенной слева, кислотность понижена
за счет увеличения электронной плотности на карбоксильной группе; кис-
лотность л-комплекса, изображенного на схеме справа, повышена, поскольку
нитробензол, оттягивая электроны, уменьшает электронную плотность
на СООН-группе.
Аналогично в случае n-толуиловой кислоты гиперконъюгационному
эффекту противодействует электронное смещение при образовании л-ком-
плекса, т. е. образованию резонансных структур, уменьшающих кислот-
124
Глава 7
ность вещества, противодействует образование комплекса с переносом
заряда.
Н+ Н+
СН2 СН,
А А"
|| ' 'I | и т. д.
V А
С С
\)Н ох/ ^он
Спирты являются амфотерными растворителями. Кислотность спиртов
изменяется в следующем порядке [394]: изопропиловый < н-пропиловый
и н-бутиловый < этиловый <С (вода) <С метоксипропиловый <С бензиловый
< метиловый <С 2-аминоэтиловый <С метоксиэтиловый (метилцеллозольв) <
< этоксиэтиловый (целлозольв) <; пропи-
LdgN2
Р и с. 29. Зависимость активности
соляной кислоты от ее мольной доли
в различных растворителях [382].
1 — бензол, 2 — нитробензол, 3 — уксус-
ная кислота, 4 — этиловый спирт, 5 — ме-
тиловый спирт, 6 — вода.
ленгликоль <С этиленгликоль.
Для титрования кислот п их аналогов
спирты разбавляют бензолом (1 : 6—1 : 10)
или смесью пиридина с ацетоном (2: 70:30).
При определении альдегидов и кетонов
оксимным методом используется смесь изо-
пропилового и метилового спиртов (65 : 35)
[314, 574]. Это один из компонентов
G — Н-системы растворителей [621, 750].
Гликоли, например этиленгликоль,
пропиленгликоль и т. д., в смеси с изо-
пропиловым спиртом, 1,4-дпоксаном или
хлороформом в отношении 1 : 1 также мо-
гут быть компонентами G — Н-смесей
растворителей. Для приготовления 0,1 н.
хлористоводородной кислоты очень часто
используют смесь пропиленгликоля с хло-
роформом (1 : 1). Смесь монометилового
эфира гликоля с ксилолом (1 : 1) служит
хорошим растворителем при определении
числа омыления жиров и восков [330].
Смесь полиэтиленгликоля «400» с хлоро-
формом применяется в качестве раствори-
теля при анализе производных барбитуровой кислоты [788]. Для приготов-
ления стандартного раствора литийалюминийамида вместо тетрагидрофу-
рана используют диметиловый эфир этиленгликоля [761].
Амфотерность — основное свойство большого числа растворителей — во
многих случаях не обнаруживается. Амфотерные растворители представляют
собой переходное звено между кислотными и основными растворителями.
Основные свойства воды вследствие небольшой силы воды как основания были обна-
ружены сравнительно поздно. При концентрировании раствора бромистого водорода
в жидком аммиаке остается твердый бромистый аммоний, так как равновесие
NH34-HBr NHjBr-
при комнатной температуре смещено вправо. Напротив, константа равновесия реакции
Н2О + НВг Н3О+^-Вг-
значительно меньше, т. е. в равновесной смеси присутствуют большие количества воды
и бромистого водорода. При выпаривании раствора образуются все новые и новые коли-
чества летучих реагентов, так как равновесие сдвигается влево.
Химические и физико-химические характеристики растворителей 125
Для большинства растворителей, применяемых в неводном титровании, нельзя
определить отношение основной формы растворителя к сопряженной кислоте на основа-
нии поглощения в УФ-области, так как максимум поглощения лежит в области очень
коротких длин волн. Хестон и Холл [382], определив электрометрически активность
протонов протонных кислот, установили ряд относительной основности некоторых раство-
рителей (рис. 29). На этом рисунке изображена зависимость логарифма активности соля-
ной кислоты (а2) от логарифма концентрации соляной кислоты (TV2 — мольная доля
соляной кислоты). Из рисунка следует, что вода — более основной растворитель, чем
метиловый спирт, уксусная кислота—очень слабое основание и бензол обладает самой
низкой основностью.
45. ОСНОВНЫЕ РАСТВОРИТЕЛИ (ТАБЛ. 46)
Этилендиамин NH2GH2GH2NH2 применяется в качестве растворителя
при титровании аналогов кислот (фенолов, имидов), поскольку их кислот-
ность возрастает в этом растворе за счет образования катионов из раство-
рителя (гл. 5, разд. 31). Это вязкая едкая жидкость, дымящая на воздухе
Таблица 46
Основные растворители
Растворитель Т. кип., °C d24° ,20 nD
Этилендпамин 116—117 0,898 1,4568
Пиперидин 106,4 0,860 1,4530
н-Бгтплампн 76,2—77,8 0,741 1,401
Пиридин 115,5 0,983 1,5100
Этаноламин 171,1 1,022 1,4539
Диметилформамид 153,0 0,945 1,4269
1.4-Дпоксан 101,3 1,033 1,4224
Диэтпловый эфир 34,6 0,708 1,3527
Тетрагпдрофуран 64—65 0,888 1,4076
и поглощающая атмосферную влагу и двуокись углерода. Этилендиамин
в чистом состоянии бесцветен, но при стоянии желтеет и из него выпадает
кристаллический осадок состава C9H20N6, поэтому рекомендуется применять
растворитель, свежеперегнанный над натрием в атмосфере азота.
н-Бутиламин GH3(GH2)3NH2 — более слабое основание, чем этилендиа-
амин, но все же он чувствителен к действию двуокиси углерода, поэтому
титрование с использованием этих двух растворителей необходимо проводить
в изолированной системе или в атмосфере азота. Перед титрованием раство-
ритель следует нейтрализовать титрантом (гл. 10, разд. 56). н-Бутиламин
применяют в качестве растворителя для титрования фенолов и сульфамидных
производных. Так как он более сильное основание, чем диметилформамид,
его используют также при титровании тех аналогов кислот, которые обла-
дают недостаточной кислотностью в растворах диметилформамида.
Пиридин G5H5N, несмотря на свой неприятный запах,— один из наиболее
часто употребляемых растворителей (обычно в смеси с бензолом или ацето-
ном) при определении кислот и их аналогов. Пиридин можно применять
вместо эти лен диамина, н-бутиламина и в некоторых случаях вместо диметил-
формамида. Несмотря на низкую диэлектрическую проницаемость он приго-
ден в качестве растворителя при титровании ионных соединений. Более
эффективным и дешевым растворителем, чем чистый пиридин, является
смесь изомерных монометилпиридинов (пиколинов) с т. кип. 114—155°.
Одно из преимуществ пиридина — его ограниченная дифференцирующая
способность, позволяющая, например при потенциометрическом титрова-
нии, определить относительную кислотность большого числа производных
бензойной кислоты и соединений, содержащих фенольный гидроксил. При
126
Глава 7
титровании фенолов с р7£а, равными 10,6 и 7,2, получают разность потенциа-
лов полунейтрализации, превышающую 500 мв (см. табл. 12).
Этаноламин NH2CH2CH2OH, обычно используемый для приготовления
стандартного раствора аминоэтилата натрия, является высокоосновным
растворителем.
N,N-Диметилформамид HCON(CH3)2 (ДМФ) — один из наиболее удоб-
ных растворителей для титрования кислот и их аналогов. Он бесцветен,
не имеет запаха и менее гигроскопичен, чем этилендиамин; чувствительность
его к двуокиси углерода столь невысока, что ею можно пренебречь, и поэтому
при титрованиях не нужно соблюдать мер предосторожности. Диапазон pH
дпметилформамида охватывает более 18 единиц, что позволяет применять
его для дифференцирующего титрования [803].
Молекулы диметилформамида благодаря своей высокой полярности,
вероятно, образуют «полимерные» цепи, вследствие чего положительный
заряд на атоме азота уменьшается и соответственно возрастает его способ-
ность связывать протоны [768]
N(CII3), N(CH3), N(CH3),
- I - I I
... O — C+ O —C+ O —c+
I I I
H H H
ДМФ является акцептором протонов по отношению к сульфаминовой
кислоте, которая имеет амфотерный характер п применяется для определе-
ния титра стандартных растворов [118]
О О
II /° - И /0Н+
НО — S — NH,--(CII3),N-CZ — О —S —NH2 —(CH3)oN —С
И " ~ чн II ‘ хн
о о
Анионы в растворах дпметилформамида, по-видимому, не сольватированы, а катионы
окружены сольватной оболочкой [662].
В диметилформамиде сильные основания превращаются в соединения
с основностью, близкой к основности формиата натрпя. Примеси, содержа-
щиеся в диметилформамиде (формамид или метилформамид), под действием
воды и сильных оснований, вероятно, гидролизуются с образованием аммиака,
метиламина и муравьиной кислоты. В соединениях типа R3NH+«HCOO~
формиат-анион является анионным основанием [176].
1,4-Диоксан применяется довольно часто в качестве растворителя бла-
годаря своей устойчивости и заметной сольватирующей способности. Он сме-
шивается как с водой, так и с бензолом. В неводном титровании диоксан
используется в качестве инертного растворителя со слабоосновными или
дифференцирующими свойствами. Он также употребляется для приготовле-
ния стандартных растворов хлорной кислоты. Дифференцирующие свой-
ства диоксана основаны на его способности к образованию водородных
связей. Его сольватирующая способность по отношению к соединениям,
содержащим фенольную или спиртовую гидроксильную группу, возможно,
объясняется образованием оксониевых соединений за счет водородных
связей [242]
сн2—сн2 _Cfb
0.^ /О —- '^О ... Н — OR
ХСН2 — СН2 -СН-Х
Как показывают кондуктометрические измерения, диоксан ведет себя как
основание по отношению к муравьиной кислоте [565] {DO-диоксан)
DO + HCOOH 7^ DO HCOOH оон+нсоо-.
Химические и физико-химические характеристики растворителей
127
В то же время основность диоксана так низка, что он может служить рас-
творителем при титровании алифатических и гетероциклических аминов.
Диэтиловый эфир С2Н5ОС2Н5 редко применяется как растворитель.
В тех случаях, когда органическое основание, подлежащее определению,
находится в эфирном растворе, рекомендуется испарить растворитель
на водяной бане в слабой струе воздуха (см. рис. 103), а затем провести
титрование хлорной кислотой в смеси уксусной кислоты с четыреххлори-
стым углеродом.
Диэтиловый эфир является «водоподобным» растворителем
С2Н5ОС2Н5 С2Щж-0С2Н5.
В процессе реакции бромистого этила с литием в эфирном растворе вначале образуется
этилат лития, а затем регенерируется растворитель
С2Н5Вг ж ЫОС2Н5 = С2Н5ОС2Н5 4- ПВг.
Для большого числа как органических, так п неорганических соединений при раство-
рении в эфире благодаря самоионизации растворителя повышается электропроводность
(см. выше).
Процесс диссоциации алкилгалогенидов в эфире такой же, как и у галогеноводород-
ных кислот в воде. По отношению к льюисовским кислотам эфир является основанием [422]
А1Вгз4-С2Н5ОС2Н5 > C2Hj 4" [ВгзА1ОС2Н5]_.
Образующееся соединение может быть оттитровано алкоголятами щелочных метал-
лов [422]
CH3ONa
--------- [Br3A10C2H5]Na . С2Н5ОСН3
I Сольволиз
ч
NaBr-4 С2Н5[Вг2А1(ОС2П5)2]
Фотометрическое титрование диэтплового эфира хлорной кислотой в 0,25 М уксус-
ной кислоте пли уксусном ангидриде в присутствии Судана III указывает на очень сла-
бую основность эфира [385].
«Основность» растворителей — понятие относительное. Известно, что при образова-
нии донорно-акцепторных комплексов ароматические соединения в таких апротонных
растворителях, как четыреххлористый углерод, ведут себя как основания по отношению
к донорам протонов. Анализ ИК-спектров позволяет написать следующий ряд «основно-
сти» растворителей по отношению к пирролу (донору водорода):
Хлорбензол < бромбензол < бензол < толуол < ксилол
(см. гл. 7, разд. 44. О пирроле см. гл. 4, разд. 27, д, е. О комплексометрическом опре-
делении ароматических углеводородов см. гл. 30, разд. 167).
Глава 8
ОЧИСТКА РАСТВОРИТЕЛЕЙ
Применяемые растворители должны быть бесцветными, безводными
и не должны содержать примесей кислот и оснований. Кислые примеси
могут образовываться при хранении растворителей: например, в хлороформе
образуется соляная кислота. Этилендиамин и «-бутиламин жадно погло-
щают углекислоту из воздуха. Если в уксусной кислоте присутствуют даже
следы воды, то это следует считать загрязнением растворителя, так как вода
нейтрализует хлорную кислоту (см. гл. 3, разд. 22,а). Случайно образовав-
шийся ацетат щелочного металла (например, при хранении в стеклянной
посуде) также относится к числу нежелательных примесей, так как ацетат-
анион в среде уксусной кислоты является основанием.
Очистка растворителей включает три операции: 1) удаление примесей,
2) высушивание, 3) фракционная перегонка. Удаление примесей можно
осуществить экстракцией либо водой, либо концентрированной серной кис-
лотой, либо концентрированным раствором едкого натра. В зависимости
от количества и химических свойств примесей способы очистки и их последо-
вательность могут быть различными. Иногда применяют специальные методы
обработки растворителей; например, для удаления примесей альдегидов
из спиртов используют 2,4-динитрофенилгидразин, для удаления уксусного
ангидрида из уксусной кислоты — бензиламин; разработаны методы осво-
бождения эфира от перекисей и т. д.
46. ИСПОЛЬЗОВАНИЕ КОНЦЕНТРИРОВАННОЙ СЕРНОЙ КИСЛОТЫ
И КОНЦЕНТРИРОВАННОГО РАСТВОРА ЕДКОГО НАТРА ДЛЯ ОЧИСТКИ
РАСТВОРИТЕЛЕЙ
Обработка концентрированной серной кислотой или концентрирован-
ным раствором едкого натра проводится перед высушиванием и перегонкой
растворителей и может применяться лишь в тех случаях, когда растворитель
не смешивается с водой и не реагирует в обычных условиях ни с серной
кислотой, ни с едким натром, как, например, «-гексан, хлороформ, четырех-
хлористый углерод, бензол, толуол, ксилол и хлорбензол.
Использованные растворители необходимо собирать и подвергать повтор-
ной очистке. Это имеет смысл делать и в тех случаях, когда используется
смесь растворителей, один из которых можно экстрагировать водой. Однако
не рекомендуется собирать и подвергать очистке такие двойные и тройные
смеси растворителей, которые нельзя разделить при помощи экстракции
водой, например, смеси бензола с хлороформом или ацетона с пиридином.
Если загрязненные растворители содержат эмульгаторы или вещества, реа-
гирующие с серной кислотой экзотермически и с заметным осмолением,
перед экстракцией и фракционированием растворители следует профильтро-
вать через слой инфузорной земли.
К числу эмульгаторов относятся производные полиоксиэтилена, высокомолекуляр-
ные алифатические кислоты и их соли, алкилсульфонаты, холестерин, глюкоза, алкалоиды
и т. д. Концентрированной серной кислотой осмоляются также фенолы, гликоли и их
производные.
Очистка растворителей
129
Чтобы отделить уксусную кислоту и пиридин от таких не смешивающихся
с водой растворителей, как бензол и четыреххлористый углерод, проводят
предварительную обработку — экстрагируют смесь большим объемом воды.
В делительную воронку емкостью 2—3 л помещают 1 л растворителя
и прибавляют 100—200 мл концентрированной серной кислоты (необходимо
пользоваться защитными очками). Вначале каждый раз после осторожного
встряхивания открывают кран, чтобы выравнять давление. После прекра-
щения разогревания органический растворитель энергично встряхивают
с серной кислотой в течение 2—3 мин и оставляют стоять.
После разделения фаз серную кислоту отделяют и органический слой
встряхивают с 500—1000 мл воды до нейтральной реакции последних про-
мывных вод по конго красному (обычно достаточно 3—5 экстракций). Затем
растворитель встряхивают с 100—200 мл 20—30%-ного водного раствора
едкого натра и промывают от следов щелочи в делительной воронке до тех
пор, пока промывные воды не будут нейтральными по фенолфталеину.
Если под действием серной кислоты растворитель темнеет, то встряхива-
ние с кислотой повторяют до получения бесцветного слоя.
При тщательной работе потери растворителя составляют 5—10%.
47. ВЫСУШИВАНИЕ РАСТВОРИТЕЛЕЙ
В принципе всегда целесообразно применять осушители, практически
не реагирующие с растворителем и обладающие в выбранных условиях
наибольшей эффективностью. Если для этой цели применяют металлический
натрий или Р2О5, необходимо предварительное высушивание растворителя.
Относительная эффективность некоторых осушителей показана ниже:
Р2О5 > А12О3 > CaSO4 (драйерит) > СаО > КОН > SiO2 (силикагель) >
NaOH > СаС12 > Na2SO4, К2СО3.
В зависимости от экспериментальных условий порядок эффективности осушителей
может изменяться. Приведенные ниже сведения (табл. 47), относящиеся к высушиванию
газов (сокращенное изложение из [821]), могут быть использованы и для оценки эффектив-
ности высушивания растворителей (применять перхлорат магния не рекомендуется из-за
опасности взрыва).
Обезвоживание (предварительное высушивание) сульфатом натрия или
поташом проводится следующим образом. К 1 л растворителя добавляют
50 г осушителя, предварительно высушенного при 100—110° и охлаждавшего-
ся в эксикаторе. После 5—10-минутного встряхивания растворитель с осу-
шителем оставляют стоять в темноте и затем фильтруют через ватный тампон
в перегонную колбу. Такие растворители, как диоксан и бензол, перед
обработкой металлическим натрием предварительно высушивают.
Таблица 47
Осушители Остается воды, мкг/л Осушители Остается воды, мкг/л
Mg(C104)2-0,12H20 0,2 CaS04-0,02H20 (драйерит) 67
Mg(C104)2-1,48Н2О (ангидрон) 1,5 Силикагель 70
ВаО 96,2% 2,8 CaSO4-0,21H2O (ангидроцел) 206
д|,о3.о,ион2о 2,9 NaOH-0,03H2O 513
р2(\ 3,5-3,6 СаО-0,(ЮН2О 656
•СаС12- 0,18Н2О 67 КОН-0,52Н2О 939
Если применить металлический натрий или фосфорный ангидрид нельзя,
как, например, при высушивании хлороформа, четыреххлористого углерода
130
Глава 8
или ацетона, используют сульфат натрия или поташ как для предваритель-
ного, так и для окончательного высушивания.
Поташ не только высушивает растворители, но и нейтрализует также
кислотные примеси. В первую очередь он используется для высушивания
ацетона, метилэтилкетона и т. д. Об использовании поташа для высушива-
ния ацетона см. в работе [434].
Гранулированные хлористый кальций и окись кальция широко приме-
няются в качестве осушителей; они снижают содержание воды примерно
до 0,05%. Неудобство применения хлористого кальция заключается в том,
что он образует молекулярные соединения с некоторыми растворителями.
В то же время окись кальция активна лишь при условии, что она не содер-
жит карбоната кальция.
Окись алюминия, двуокись кремния (силикагель) и безводный сульфат
кальция (драйерит) применимы в тех случаях, когда содержание воды
в растворителе не превышает 0,05%. Сульфат кальция не реагирует с рас-
творителями и не катализирует их разложение (как это происходит, напри-
мер, при применении окиси алюминия).
Перхлорат магния, хотя и является великолепным осушителем, часто
остается в растворе, что при перегонке может вызвать взрыв.
Высушивание едким кали проводят, добавляя твердый КОН к пиридину,
этилендиамину и бутиламину. После энергичного встряхивания оставляют
смесь на 2—3 дня для осаждения мути. Окрасившееся в желтый или корич-
невый цвет при высушивании пиридина и бутиламина едкое кали можно
в случае необходимости заменить свежим. После отделения растворителей
от КОН их фракционируют. Сильные щелочи вызывают гидролиз диметил-
формамида.
Высушивание металлическим натрием производится лишь в тех случаях,
когда растворитель (например, эфир, диоксан, бензол) с ним не реагирует.
Чтобы избежать бурного протекания реакции, можно применять охлаж-
дение и затем отогнать обезвоженный растворитель от продуктов реакции,
например спирты от алкоголятов.
Никогда нельзя обрабатывать металлическим натрием хлорпроизводные
углеводородов, это может привести к взрыву\
Очищенный растворитель, предварительно обезвоженный обработкой
сульфатом натрия или поташом, фильтруют и прибавляют к нему металли-
ческий натрий (на 1 л инертного растворителя добавляют 0.5—1,0 г натрия,
на 1 4 метилового или изопропилового спиртов — 2—3 г). Бензол или гексан
можно непосредственно перегонять над остатками металлического натрия,
но при перегонке над натрием растворителей, склонных к образованию пере-
кисей, например эфира, перекиси должны быть предварительно удалены.
После охлаждения прибора непрореагировавший металлический натрий
разлагают в перегонной колбе небольшим количеством спирта.
Остаток при перегонке составляет от 5 до 20% в зависимости от темпера-
туры кипения растворителя.
Для высушивания углеводородов и ацетонитрила применяют фосфорный
ангидрид (см. при ацетонитриле). В этом случае так же, как и при примене-
нии металлического натрия, высушивание заканчивают перегонкой.
Алюмогидрид лития применяют для обезвоживания тетрагидрофурана
и диметилового эфира этиленгликоля (моноглима) [391, 761].
а. Обезвоживание уксусной кислоты.
1. Содержание воды в уксусной кислоте можно определить несколькими
способами. Температура плавления безводной уксусной кислоты равна
16,65° [381]. В табл. 48 показано, как изменяется температура плавления
уксусной кислоты при увеличении содержания воды от 0 до 1% [23].
На температуру плавления уксусной кислоты почти не оказывает влияния
присутствие хлорной кислоты, поэтому содержание воды в стандартном
Очистка растворителей
131
растворе хлорной кислоты можно определить по его температуре плавления,
используя точный термометр с ценой деления 0,01°.
Еще проще метод, основанный на определении количества 0,1 н. хлорной
кислоты, необходимого для изменения окраски кристаллического фиолето-
вого, и на оценке относительной резкости перехода окраски. К 10 мл уксус-
ной кислоты прибавляют 1 каплю 0,1%-ного кристаллического фиолето-
BOPcrfB зависимости от содержания воды в уксусной кислоте переход окраски
происходит как указано в табл. 49 [23].
Таблица 48 Таблица 49
Содержание воды, % Т. пл., °C Содержание воды, % Переход окраски
0.0 0 В 9 16,65 16 28 0,1 Четкий переход: фиолетовая —> зеленая
0 4 15 84 0,2—0,3 Переход: фиолетовая —> синяя —синевато-
О к зеленая —> зеленая
0 8 15 12 Выше 0,4 Нечеткий переход, но окраска изменяется,
1,0 14*80 как описано выше
Р и с. 30. Прибор для титрова-
ния по Карлу Фишеру.
1 — макробюретка; 2—микробюретка;
3 — корковая пробка; 4 — отверстие;
5 — платиновые электроды.
то количество воды, которое было
Очень небольшие количества воды в уксусной кислоте (0,009—1,7%) можно опреде-
лить спектрофотометрическим титрованием (используя длину волны 256 ммк [100]) уксус-
ного ангидрида в присутствии серной кислоты
в качестве катализатора. Этот метод, однако, не-
удобен для проведения серийных анализов содержа-
ния воды в уксусной кислоте.
2. Титрование по Карлу Фишеру более
удобно проводить, используя так называе-
мый метод биамперометрического титрования
[200, 610, 868].
В закрытую пли полузакрытую колбу для
титрования на 30—40 мл, снабженную двумя эле-
ктродами из гладкой платины, отмеряют 15 мл реак-
тива Карла Фишера (реактив КФ) и поляризуют
электроды, подводя к ним напряжение 30—40 мв
(рис. 30). Под действием реактива КФ электроды
деполяризуются и стрелка чувствительного гальвано-
метра (10-6 а) смещается. Постоянный объем реак-
тива КФ титруют растворителем из бюретки с ценой
делений 0,01 мл (обратное титрование). В конечной
точке стрелка гальванометра устанавливается на
нуле. Титр раствора КФ устанавливают по мета-
нольному раствору дпгпдрата щавелевой кислоты.
[Взвешивают 1,750 (0,175) г дпгидрата щавелевой
кислоты, эквивалентные 500 (50) мг воды и раство-
ряют в 50 мл метилового спирта, перегнанного над
металлическим натрием.] 1 мл этого раствора содер-
жит 10,0 (1,0) мг воды, введенной с дигидратом
щавелевой кислоты. Находят водный эквивалент
раствора КФ (глухой опыт). Поскольку объем реак-
тива КФ постоянен (скажем 15 мл}, объем метило-
вого спирта (42), требуемый для глухого опыта,
содержит такое же количество воды, что и объем
раствора дигпдрата щавелевой кислоты в метиловом
спирте (Hj). Последний раствор содержит не только
введено в виде дигпдрата щавелевой кислоты (Внго)» н0 и небольшие количества воды,
первоначально присутствовавшие в метаноле.
Расчеты
Разность А 2 — At так относится к количеству воды Л\ -ВигО, как -^2 относится к х мг
воды, где х — количество воды, эквивалентное раствору КФ, мг\ Вц2О — вес воды, вве-
денной в виде дигидрата, мг/мл; Ai •ВнгО — количество гидратной воды, израсходованной
при титровании, мг.
Пример. 15 мл реагента КФ титруют раствором 1,749 г дпгпдрата щавелевой кисло-
ты в 50 мл метилового спирта, т. е. 10 мг Н2О/.ил. Среднее значение трех определений
132
Глава 8
показывает, что на титрование раствора КФ потребовалось 9,33 мл метилового спирта,
перегнанного над натрием и 0,895 мл метанольного раствора дигидрата щавелевой кисло-
ты. Следовательно, водный эквивалент раствора КФ составит
9,33.(0,895-10)
9,33 — 0,895 ~9’9 Мг Нг°
п содержание воды в метиловом спирте равно 1,06 мг/л. Достоинство метода обратного
титрования заключается в том, что чем меньше содержание воды в исследуемом растворе,
тем больше раствора требуется израсходовать на постоянный объем реактива КФ.
После установления водного эквивалента мпкробюретку споласкивают уксусной
кислотой и титруют 15 мл реактива исследуемой уксусной кислотой.
Кроме метода биамперометрического титрования, титрование реактивом КФ можно
также проводить в присутствии индикаторов, таких, как метиленовый синий, в среде
пиридина или метилового спирта *. При прямом титровании смесь индикаторов хинали-
зарин — нафтоловый зеленый — метиленовый синий меняет окраску от цвета зеленого
плюща до коричневого [581]. Титрование раствором КФ можно осуществить и фотометри-
чески (см. стр. 55 в работе [200]).
3. Уксусный ангидрид очень удобно применять для обезвоживания
уксусной кислоты, аналогично для обезвоживания пропионовой кислоты
удобно применять пропионовый ангидрид. Реакция проходит при 5—6-часо-
вом кипячении в присутствии хромового ангидрида в качестве катализатора.
Если в качестве катализатора применяют хлорную кислоту, смесь уксусной
кислоты с уксусным ангидридом следует оставить на 48 час [657]. Для полу-
чения хороших результатов необходимо точно определить содержание воды
(титрование раствором КФ).
В 3-литровую колбу на шлифах помещают 2 л уксусной кислоты и добав-
ляют уксусный ангидрид в количестве, рассчитанном на содержание воды
в уксусной кислоте. Поскольку уксусный ангидрид не всегда бывает 100%-ной
чистоты, его может потребоваться пропорционально большее количество.
Добавляют 1—2 г хромового ангидрида, кипятят смесь с обратным холодиль-
ником 5—6 час, защищая от влаги воздуха трубкой, наполненной силикаге-
лем и затем фракционируют. Поскольку присутствие даже небольших коли-
честв уксусного ангидрида будет мешать определению первичных и вторич-
ных аминов, следует провести качественные пробы на присутствие уксусного
ангидрида (см. разд. в).
4. Уксусную кислоту можно обезводить также, пропустив ее через колон-
ку длиной 122 см и диаметром 2,5 см, содержащую молекулярные сита
Линде 5А, после чего подвергнуть перегонке. При таком способе очистки
можно снизить содержание воды в уксусной кислоте до 0,007% [109].
б. Влияние воды на титрование хлорной кислотой. Небольшие количе-
ства воды в уксусной кислоте акцептируют протоны и присутствуют в виде
ацетата гидроксония [644]
Н2О + СН3СООН Н3О+СН3СОО-
(см. также гл. 3, разд. 22,а).
В совершенно безводной уксусной кислоте количество хлорной кислоты, связываемое
водой, можно определить электрометрически. Однако на практике при использовании
хлорной кислоты концентрации не ниже 0,1 н. присутствие воды в количестве до ~1%
не мешает переходу окраски кристаллического фиолетового или же потенциометрическо-
му определению конечной точки (см. рис. 31, 32 и работы [644], [809]). Стандартный рас-
твор хлорной кислоты при разбавлении водой реагирует следующим образом [470, 763]:
СН3СООН+С1О74-Н2О H3O+CIO74-CH3COOH.
Образовавшиеся при этом вещества обладают слишком низкой кислотностью, чтобы
вступать в реакцию с индикатором в среде уксусной кислоты.
в. Содержание уксусного ангидрида в уксусной кислоте определяют
с помощью ряда полукачественных — полуколичественных методов.
♦ Fischer Е., Angew. Chemie, 64, 592 (1952).
Очистка растворителей
133
1. Качественная проба [57]. Приготавливают 5%-ный (об.%) раствор
свежеперегнанного анилина в бесцветном пиридине (реактив I) и 1%-ный
(об.%) раствор свежеперегнанного фурфурола в
тив II). Смешивают 10 мл исследуемого образца
постоять 1 мин и добавляют 1 мл реактива II. При
содержании в исследуемом образце примерно
0,5% уксусного ангидрида раствор приобретает
соломенно-желтый цвет, при более низком содер-
жании — ярко-красный.
2. Качественная проба [226]. Приготовление
реактива: 0,5%-ный раствор хлорида железа(Ш)
в этиловом спирте подкисляют каплей соляной
кислоты и насыщают солянокислым гидроксил-
амином.
Две капли этого реактива и 1 мл исследуемой
уксусной кислоты выпаривают досуха в неболь-
шом фарфоровом тигле. В присутствии уксусного
ангидрида появляется фиолетовое окрашивание.
3. Колориметрический контроль [23]. Смеши-
вают 1 мл исследуемой уксусной кислоты с 5,00 мл
0,50%-ного раствора 2,4-дихлоранилина в уксус-
ной кислоте (= 25 мг) и оставляют в закрытом со-
суде на 3 час при комнатной температуре. Затем
добавляют 30 мл разбавленной соляной кислоты
и доводят объем раствора водой до 100 мл.
1 мл этого раствора смешивают с 1 мл 0,25 %-ного
уксусной кислоте (реак-
с 1 мл реактива Z, дают
Рис. 31. Влияние содер-
жания воды на четкость
потенциометрического опре-
деления конечной точки при
титровании хлорной кисло-
той [644]. Цифры рядом
с кривыми показывают про-
центное содержание воды.
раствора нитрита натрия и 1 мл разбавленной соляной кислоты и остав-
ляют на 5 мин. По окончании выдержки добавляют 1 мл водного
СН3С00Н,л<л
1 %-ного раствора натриевой соли 8-это-
кси-1-нафтол-3,6-дисульфокислоты, под-
щелачивают 3 мл 15 %-ного раствора
едкого натра и разбавляют водой до 100 мл.
Через 20 мин измеряют поглощение раст-
вора, применяя цветной фильтр (типа
Пульфрих S53), и, пользуясь калибровоч-
ной кривой, подсчитывают количество не-
продиазотированного 2,4-дихлоранилина.
Р и с. 32. Влияние содержания воды на коли-
чество хлорной кислоты, необходимое для титро-
вания уксусной кислоты в присутствии кристал-
лического фиолетового [809].
Титрование проводят до появления следующей окраски:
1 — до синей окраски, вода отсутствует; 2 — до зеленой
окраски, вода отсутствует; 3 — до зеленой окраски,
0,2% воды; 4 — до зеленой окраски, 0,4% воды; 5 — до
зеленой окраски, 0,6% воды; 6 — до зеленой окраски,
1% воды; 7 — до желтой окраски, вода отсутствует;
8 •— до желтой окраски, 0,2% воды; 9 — до желтой
окраски, 0,4% воды.
Процентное содержание уксусного ангидрида = (25 — х) X 0,063, где
х — количество дихлоранилина (мг), определенное при помощи кривой
поглощения.
4. Титриметрический контроль может быть осуществлен реакцией
с морфолином [431] (гл. 16, разд. 96, а).
г. Удаление уксусного ангидрида из уксусной кислоты. При титровании
легко ацилируемых аминов даже минимальные примеси уксусного ангидрида
могут привести к ошибкам в определении, так как амин подвергается пол-
134
Глава 8
ному или частичному ацетилированию, а образующийся амид не связывает,
или почти не связывает, хлорную кислоту. Примеры легко ацетилируемых
аминов: 2-нафтиламин, n-фенилендиамин, анилин, /п/?етп-бутиламин [612],
гидразид изоникотиновой кислоты [732], гидразид изолизергиновой кис-
лоты [306], п-анизидин [767]. При определении легко ацилируемых аминов
уксусная кислота должна быть освобождена от примеси уксусного ангид-
рида, который может образоваться при высоких
температурах даже в кислоте, первоначально
свободной от ангидрида.
1 л уксусной кислоты предварительно обез-
воживают, добавляя уксусный ангидрид, сме-
шивают с 3 г бензиламина и после этого фрак-
ционируют. Приготовленная таким способом
уксусная кислота не содержит даже следов
уксусного ангидрида [767].
48. ФРАКЦИОННАЯ ПЕРЕГОНКА
Все безводные растворители подвергают пере-
гонке и хранят по 1—5 л. Перегонку проводят
в приборе на шлифах, снабженном специальной
головкой (см. рис. 33 и работу [200]). Эта голов-
ка колонки эффективна в тех случаях, когда
температура кипения растворителя не превышает
120°, причем для нагрева употребляют масляную
баню. Перегонку растворителей с температурой
кипения выше 120° проводят в колонке, окру-
женной рубашкой с подогревом. При перегонке
Р и с. 33. Колонка для очи- Дистиллят защищают от влаги воздуха.
стки растворителей фракци- Если перегонке подвергают растворители
онной перегонкой [200]. с достаточно высокой температурой плавления
(табл. 50). необходимо внимательно следить,
чтобы температура охлаждающей воды не была слишком низкой, иначе
растворитель может затвердеть в трубке холодильника. При этом рекомен-
дуется использовать длинный змеевиковый холодильник и давать слабый ток
воды; иногда приемную колбу, кроме того, охлаждают льдом. Если не при-
нять этих мер предосторожности, может произойти взрыв.
Таблица 50
Растворитель T. пл., °C Растворитель Т. пл., °C
mpem-Бутилоный спирт 25,6 Этилендпамин 8,6
Уксусная кислота 16,6 Бензол 5,5
1,4-Диоксан 11,8
49. СПЕЦИАЛЬНЫЕ МЕТОДЫ ОЧИСТКИ
а. Если метиловый и изопропиловый спирты применяют при определении
карбонильных групп оксимным методом, то предварительно для удаления
альдегидов и кетонов их обрабатывают 2,4-динитрофенилгидразином (1)
или нитратом серебра (2).
1. Помещают 2 л спирта в 3-литровую колбу, добавляют 1—2 мл концен-
трированной соляной кислоты, 1—2 г динитрофенилгидразина и кипятят
с обратным холодильником 1—2 час, после чего подвергают перегонке.
Дистиллят обрабатывают 50 г смеси, составленной из равных частей пред-
Очистка растворителей
135
варительно высушенных при 100—110° поташа и активированного угля,
фильтруют и, добавив 2—3 г металлического натрия, фракционируют.
2. Растворяют 5 г нитрата серебра в 1 л изопропилового спирта и остав-
ляют на 2 час на прямом солнечном свету. Кипятят с обратным холодиль-
ником 30 лик, пропуская слабую струю азота, затем фракционируют.
б. Испытание на присутствие перекисей и очистка диэтилового эфира.
1. Проба на содержание перекисей в диэтиловом эфире *. Перегонка
диэтилового эфира и тетрагидро фурана, содержащих перекиси, может при-
вести к взрыву с тяжелыми последствиями. Поэтому перед перегонкой убе-
дитесь в отсутствии перекисей и не перегоняйте эфир и тетрагидрофуран
в случае положительной пробы на содержание перекисей!
В колбу емкостью 30 мл, снабженную пришлифованной пробкой, поме-
щают 10 мл раствора роданида железа(П) (получение см. ниже); воздух
в колбе предварительно вытесняют СО2. К раствору приливают исследуемый
эфир до самого горла колбы и закрывают пробкой так, чтобы между срезом
пробки и поверхностью жидкости не оставалось свободного пространства.
Энергично встряхивают и оставляют в темноте на 5 мин. Если слой эфира
окрашивается в розовый цвет, содержание перекисей выше 0,00015%. При-
готовление реактива: смешивают 3 мл 2,5 М серной кислоты с 35 мл дистил-
лированной воды и кипятят, чтобы удалить кислород. В кипящем растворе
растворяют 1 г сульфата железа(П) и после охлаждения добавляют 0,5 г
роданида калия. Красную окраску, появляющуюся в присутствии роданида
железа(Ш), устраняют, прибавляя небольшие кусочки проволоки из чистого
железа. Хранят раствор в атмосфере, свободной от кислорода. Примеси
роданида железа(Ш) можно удалить из раствора также экстракцией эфиром.
Следы перекиси в диэтиловом эфире, диоксане и тетрагидрофуране можно
определить при помощи реактива, содержащего Х,]\т-диметил-п-фениленди-
амин, который дает синевато-красное окрашивание с максимумом поглоще-
ния при 570 ммк уже при содержании перекиси 10 мг/мл. Примеси алифати-
ческих альдегидов, начиная с концентрации 1000 мкг/мл, мешают определе-
нию. Приготовление реактива: 300 мг сульфата Х,Х-диметил-п-фенилен-
дпамина растворяют в 10 мл воды и разбавляют 90 мл метилового спирта.
Чтобы установить наличие перекиси, при комнатной температуре, защищая
раствор от света, смешивают 2 мл исследуемого вещества с 2 мл реактива
и через 5 мин сравнивают окраску с окраской контрольного раствора, при-
готовленного с использованием растворителя, перегнанного над металличе-
ским натрием. Если для стабилизации эфира применялся диэтилдитиокарба-
мат, цветную пробу провести не удается [193].
2. Для очистки от перекисей встряхивают 1 л диэтилового эфира с неболь-
шими порциями раствора, полученного из 100 г сульфата железа(П) и 200—
250 мл 10 %-ной серной кислоты. При саморазогревании раствора следующую
порцию сульфата железа добавляют после охлаждения раствора. Раствор
встряхивают 15 мин, отделяют эфир, тщательно промывают его водой и при-
бавляют к смеси грубоизмельченного перманганата калия (20 г) и едкого
кали (30 г), некоторое время встряхивают и оставляют на ночь. Очищенный
таким способом эфир можно перегонять над металлическим натрием.
3. О высушивании диэтилового эфира безводным сульфатом магния
см. [108].
Об освобождении метилцеллозольва от перекисей см. [417].
в. Вместо того чтобы производить очистку хлорбензола серной кислотой
и концентрированным раствором едкого натра, можно оставить 1 л хлор-
бензола на 24 час с 20 г гранулированного хлористого кальция и перегнать,
а затем провести повторное высушивание и фракционирование.
* Analyst, 87, 401 (1962).
136
Глава 8
г. Для очистки выпускаемого в продажу ацетонитрила его предвари-
тельно сушат сульфатом натрия, кипятят 1—2 час с фосфорным ангидридом
(25—30 г на 1 л) и фракционируют. Недостатком очистки ацетонитрила
повторными перегонками над фосфорным ангидридом является полимериза-
ция растворителя. Рекомендуемые методы очистки см. в работе [139].
д. Наиболее трудно удалить из ацетона воду. Чтобы связать ее, удобнее
всего пользоваться поташом [434], так как металлический натрий, фосфор-
ный ангидрид и хлористый кальций катализируют самоконденсацию ацетона
с образованием веществ, загрязняющих растворитель. Двуокись кремния
(силикагель) и окись алюминия повышают содержание воды в ацетоне,
возможно, за счет альдольно-кротоновой конденсации (стр. 382 в [866]).
Содержание воды в кетонах, а также в сложных и простых эфирах можно
определить фотометрически при помощи реактива, представляющего собой
ацетоновый раствор хлорида лития и перхлората меди [416].
Примеси метилового спирта в ацетоне можно удалить обработкой пер-
манганатом калия. Для этого к 1 л ацетона прибавляют 1—2 г тонкоизмель-
ченного перманганата калия и кипятят 1—2 час. Затем раствор охлаждают,
дают ему отстояться и, если он сохранит розовую окраску после перегонки,
сушат поташом и фракционируют повторно. После такой обработки в ацетоне
остается 0,1% воды. При использовании в качестве осушителя безводного
сульфата кальция содержание воды можно уменьшить до 0,001%.
Очистка метилэтилкетона и метилизобутилкетона проводится анало-
гично. Об очистке метилэтилкетона и ацетонитрила пропусканием через
колонку с активированной окисью алюминия см. в работе [105].
е. Определение примесей в 1,4-диоксане и его очистка. Выпускаемый
в продажу диоксан содержит различные примеси, часть которых попадает
в реактив в процессе производства, а часть образуется при хранении.
К такого рода примесям относятся этиленацеталь уксусного альдегида,
уксусный альдегид, кротоновый альдегид, этиленгликоль, окись этилена,
вода и перекись диоксана [784].
О
/О — СН2 п/7 \н — О — О —II
спз-cir I ।
\0-сп2 н2с сн2
о
Этиленацеталь Перекись диоксапа
уксусного альдегида
Поскольку диоксан — простой эфир, он окисляется кислородом воздуха
и при стоянии дает перекиси, из которых в свою очередь образуются альде-
гиды, уксусная кислота и некоторые другие вещества. Перегонка над метал-
лическим натрием позволяет лишь частично удалить этиленацеталь уксус-
ного альдегида (т. кип. 82,5°). Выбор метода очистки диоксана зависит
от того, для какой цели предназначается растворитель. При использовании
его для визуального титрования оснований достаточны самые быстрые и самые
простые методы очистки, в то время как для потенциометрического микро-
и полумикротитрования (особенно дифференцирующего титрования) реко-
мендуется использовать тщательно очищенный диоксан.
1. Качественная проба на присутствие перекисей в диоксане [224]. Рас-
творяют 9 г семиводного сульфата железа(П) в 50 мл соляной кислоты (1 : 1)
и добавляют 5 г роданида натрия и немного гранулированного цинка. После
обесцвечивания раствора в нем растворяют еще 12 г роданида натрия и декан-
тируют с непрореагировавшего цинка. При смешивании одной капли этого
реактива с двумя каплями диоксана, содержащего перекиси, появляется
красная окраска: бесцветный роданид железа(П) окисляется до окрашен-
ного в красный цвет роданида железа(Ш).
Очистка растворителей
137
2. Определение содержания перекисей в диоксане. Смешивают 20 мл дио-
ксана с 5 мл насыщенного водного раствора иодида калия, 2 мл 2 н. серной
кислоты и 2 мл раствора крахмала, после чего разбавляют примерно 50 мл
воды и титруют 0,1 н. раствором тиосульфата натрия до исчезновения синей
окраски. 1 мл стандартного раствора эквивалентен 0,05 мг-экв активного
кислорода.
3. Обнаружение воды в диоксане [115, 225]. Раствор дипикриламина (гек-
санитродифениламина) в безводном диоксане окрашен в бледно-желтый цвет.
При прибавлении незначительного количества воды раствор становится
оранжево-желтым. Вода диссоциирует на ионы Н + и ОН-, и диоксан, будучи
слабым акцептором протонов, образует некоторое количество катионов,
а дипикриламин под действием иона ОН- дает анион с хиноидной структу-
рой, который с протонированным диоксаном образует растворимую соль,,
окрашенную в желтый цвет
4. Очистка диоксана на ионообменных смолах. В течение 24 час встряхи-
вают среднюю фракцию перегнанного диоксана с сухой катионнообменной
смолой карбоксильного типа Амберлит IRC-50. После фильтрования кипя-
тят 1—2 час над твердым едким натром и затем фракционируют. Очищенный
таким способом диоксан пригоден для проведения потенциометрического
титрования [507].
5. Очистка диоксана едким натром. Диоксан оставляют на несколько
дней над твердым едким натром, ежедневно сменяя осушитель до тех пор,
пока он не перестает коричневеть; после этого растворитель фракционируют,
обрабатывают его металлическим натрием и фракционируют повторно [747].
6. Об очистке при помощи асбестового волокна см. [741].
7. Очистка диоксана для специальных целей [748]. В колбе с обратным
холодильником кипятят по крайней мере 10 час 1 л технического диоксана
и 100 мл 1 н. соляной кислоты (в этих условиях этиленацеталь уксусного
альдегида гидролизуется). Во время кипячения из трубки, опущенной
через холодильник до расстояния в 1 см от поверхности жидкости, пропу-
скают слабую струю азота. По окончании кипячения диоксан охлаждают
в струе азота, переносят в делительную воронку на 2 л, прибавляют 50 г
едкого натра и оставляют на 1 день при частом встряхивании, после чего,,
отделив нижний слой, повторяют обработку едким натром. После заверше-
ния операции диоксан оставляют на 2 дня в склянках из темного стекла,
добавив к нему металлический натрий. Затем диоксан декантируют, добав-
ляют еще 30 г натрия и кипятят в токе азота в течение одного дня. Если
поверхность натрия потеряет блеск, металл необходимо заменить.
Последней стадией очистки диоксана является фракционная перегонка.
Диоксан и новую порцию натрия (30 а) переносят в другую колбу, снабжен-
ную высокоэффективной колонкой высотой не менее 50 см.
К приемнику для защиты от влаги присоединяют трубку, наполненную
безводным хлористым кальцием. Перегонку проводят в токе азота, который
подается по двум трубкам. Одна из трубок, как уже говорилось, не достает
на 1 см уровня жидкости, другая оканчивается на 2 см выше ртутного шарика
термометра, и проходящая по второй трубке струя азота подается в направле-
138
Глава 8
нии движения паров испаряющегося диоксана. Перед перегонкой система
заполняется азотом через нижнюю трубку, но во время перегонки газ посту-
пает только через верхнюю трубку, что исключает возможность случайных
изменений в показаниях термометра. Перегонку проводят очень медленно,
предгон (пятую часть дистиллята) отбрасывают, в колбе остается до х/10
исходного количества растворителя.
Диоксан нужно хранить в склянках темного стекла, снабженных при-
шлифованной пробкой; склянка должна быть наполнена доверху. Так же
как и для диэтилового эфира, хранение над металлическим натрием уско-
ряет образование перекисей [784].
ж. Очистка диметилформамида. В присутствии следов воды диметил-
формамид гидролизуется. Выпускаемый в продажу продукт часто содер-
жит примеси воды и муравьиной кислоты. При перегонке диметилформамида
при обычном давлении идет незначительное разложение вещества, приводя-
щее к образованию диметиламина и окиси углерода; такое разложение, ката-
лизируемое кислыми примесями, может идти и при более низких температу-
рах [8051. При хранении над едким кали также образуется диметиламин.
С водой диметилформамид образует соединение ДМФ-2Н2О. Если смешать
ДМФ с 20% воды, температура смеси повышается на 20°. Все это свидетель-
ствует. что высушить диметилформамид довольно трудно.
1. Очистка с помощью обработки ионообменной смолой [600]. К 500 мл
диметилформамида прибавляют 50 г сухой ионообменной смолы дауэкс
1X10 в гидроксильной форме, перемешивают несколько часов, деканти-
руют и затем фильтруют.
2. Очистка обработкой окисью бария [660]. После фракционирования
на колонке, заполненной кольцами Рашига, дистиллят оставляют на
48 час над окисью бария. Когда взвесь осядет, диметилформамид деканти-
руют и перегоняют в вакууме в присутствии 1 % борной кислоты. Об
очистке ДМФ, применяемого для кондуктометрического титрования, см.
в работе [805].
з. Очистка нитрометана [135].
1. Смешивают 75 г ионообменной смолы амберлит IR-120 (в Н ^фор-
ме) с безводным метиловым спиртом, дают взвеси осесть и декантируют рас-
творитель. Эту процедуру повторяют несколько раз. Обработанной таким
образом смолой заполняют хроматографическую колонку размером 2 х
X 25 см, пропускают через нее 1 л метилового спирта со скоростью 1 —
2 мл!мин и вымывают растворитель 300 мл нитрометана. Приготовленная
таким образом смола пригодна для очистки 15—25 л нитрометана при скоро-
сти пропускания 2—5 мл/мин.
2. Энергично встряхивают 1 л нитрометана с тремя порциями по 300 мл
водного раствора бикарбоната (2,5%) и бисульфита (2,5%) натрия и после-
довательно промывают водой (3 х 200 мл), 5%-ной серной кислотой и вновь
водой (3 х 200 мл). После сушки сульфатом натрия фракционируют.
3. Об очистке нитрометана для специальных целей см. в работе [520].
и. Очистка пиридина. 1. Очистка с использованием окиси алюминия [30].
В бутыли с притертой пробкой встряхивают в течение 5 мин 500 мл пиридина
и 20 г активированной окиси алюминия (Мерк, 71707), фильтруют через
стеклянный фильтр диаметром 9 см, покрытый слоем 5 г диатомитовой земли.
На 100 мл неочищенного пиридина при титровании расходуется 0,2 —
0,4 мл 0,1 н. раствора гидроокиси тетрабутиламмония, а после вышеуказан-
ной очистки — только 0,05 мл.
Обычно pH суспензии, приготовленной из 10 г активной окиси алюминия и 90 мл
дистиллированной воды, составляет 10,0—10,2.
2. Очистка с использованием окиси бария [687]. В течение 3 час пиридин
кипятят над порошкообразной окисью бария в колбе с обратным холо-
Очистка растворителей
139
дильником, не допуская попадания в колбу влаги и СО2 из воздуха, а затем
подвергают фракционной перегонке. Приготовленный таким образом пири-
дин совсем не содержит двуокиси углерода, которая могла бы повлиять
на точность титрования гидроокисью тетрабутиламмония, а содержание
воды составляет 0,02—0,04%.
3. Высушивание пиридина азеотропной перегонкой с 10 об. % бензола
{см. рис. 33). При 67° отгоняется смесь воды, пиридина и бензола, затем отго-
няется бензол и, наконец', при 116° пиридин, содержащий 0,05—0,08% воды.
Для высушивания пиридина можно также применять едкий натр; методика
обработки такая же, как для диоксана (см. метод 5).
Для визуальных методов определения барбитуровых кислот в качестве
растворителей можно также применять гомологи пиридина (фракцию, кипя-
щую при 114—155е). Для потенциометрического определения предпочтитель-
но использовать фракцию с т. кип. 134—140° [370—372].
к. Примеси реакционноспособных алкилгалогенидов в хлороформе', опре-
деление этилового спирта в хлороформе', хранение хлороформа. Выпускаемый
в продажу хлороформ может содержать примеси таких галогенопроизвод-
ных, как хлористый метилен и хлорбромметан [874, 875J. Последнее соеди-
нение реагирует с триметиламином с образованием бромида хлорметил-
триметиламмония [С1СН2Х(СН3)3]+Вг_. В некоторых случаях содержание
хлорбромметана в хлороформе достигает 0,25%.
Сильные основания, например пирролидин, пиперидин и эфедрин, взаи-
модействуют с реакционноспособными галогенопроизводными и, вероятно,
даже с хлороформом (если какое-то время реакция проходит при 60°). Из-за
возможности такого взаимодействия не рекомендуется концентрировать
хлороформные экстракты алкалоидов, так как при этом, например, стрих-
нин образует бромид хлорметилстрихнина.
Очистка хлороформа от примесей реакционноспособных алкилгалогени-
дов. В колбе с обратным холодильником около 70 час кипятят 250 мл хлоро-
форма п примерно 7 г пирролидина, затем фракционируют, проводят очи-
стку концентрированной серной кислотой и едким натром и снова фракцио-
нируют. Предохранить от разложения хлороформ и четыреххлористый
углерод можно, добавив после обычной очистки 1 об. % низкокипящего
петролейного эфира. В закупоренных бутылях защищенные от света хлоро-
форм и четыреххлористый углерод могут храниться 5—6 месяцев без обра-
зования фосгена. Выпускаемый в продажу хлороформ часто предохраняют
от разложения, добавляя этиловый спирт, присутствие которого так же неже-
лательно, как и присутствие воды, если в смеси хлороформа с уксусной
кислотой проводят микротитрование раствором хлорной кислоты в уксус-
ной кислоте. Хлороформ можно освободить от примеси спирта обработкой
концентрированной серной кислотой с последующей промывкой концентри-
рованным раствором едкого натра (см. раздел 46). Для качественного опре-
деления примеси этилового спирта в хлороформе применяется раствор ком-
плекса ванадия с 8-оксихинолином (оксинат ванадия) либо в бензоле, либо
в о-дихлорбензоле (см. стр. 172 в работе [224], а также работы [69, 112]).
В микропробирке нагревают на водяной бане при 60° в течение 2—8 мин
1—2 капли хлороформа с 4—8 каплями реактива, одновременно нагревают
4—8 капель реактива в другой пробирке (глухой опыт). В присутствии
140
Глава 8
этилового спирта серовато-зеленая окраска реактива переходит в розовую
или красную. Приготовление реактива'. 1 мл водного раствора, содержащего
1 мг ванадия в виде ванадата аммония, смешивают с 1 мл 2,5 %-ного водного
раствора 8-оксихинолина, содержащего 6% уксусной кислоты, после чего
водный раствор экстрагируют 30 мл бензола и отделяют органический слой.
Бензольный раствор, содержащий комплекс оксината ванадия, может хра-
ниться не больше 1 дня. Этим методом можно определить 20 мкг этилового^
спирта.
л. О методах очистки редко употребляемых растворителей см. следую-
щие работы: анилин — [866], стр. 442, нитробензол — [866], стр. 432 и [469],
бензиловый спирт — [555], трихлорэтан — [38], тетрагидрофуран — [391],
целлозольв — [692]. Об очистке растворителей см. также подробную моно-
графию В. Бунге *.
50. ФИЗИОЛОГИЧЕСКОЕ ДЕЙСТВИЕ РАСТВОРИТЕЛЕЙ
Любой органический растворитель, растворяющий жиры и масла и обла-
дающий высоким давлением пара при комнатной температуре, является
Р и с. 34. Приспособления для заполнения пипетки токсич-
ными растворителями.
1 — резиновый конус; 2 — резиновая пробка; 3 — зажим: 4 — мерная
колба; 5 — сосуд для вакуумирования: 6 — резиновая втулка;
7 — широкая стеклянная трубка; 8 — широкогорлая склянка объе-
мом 250 дал; 10 — резиновая груша.
физиологически активным. Отравление может происходить либо через дыха-
тельные пути, либо, в меньшей степени, за счет абсорбции растворителя непо-
врежденной кожей; иногда вещество разрушает и эпителиальные ткани. При
тщательной работе отравление через желудочно-кишечный тракт мало-
вероятно.
Наиболее типичное токсическое действие некоторых растворителей: насы-
щенные углеводороды (гексан) обладают наркотическими свойствами, вызы-
вают конвульсии и раздражают кожу; бензол и толуол вызывают раздраже-
* Houben-Weil, Methoden der Organischen Chemie, Band 1,2; Allgemeine Labo-
ratoriums praktis, Band II, p. 765. Stuttgart, Thieme Verlag, 1959.
Очистка растворителей
141
ние кожи, конвульсии и при продолжительном действии малокровие; хло-
рированные углеводороды обладают значительной токсичностью, в первую
очередь они поражают печень. Отравляющее действие спиртов хорошо
известно, однако следует отметить, что более опасны вторичные спирты.
Метиловый спирт поражает зрительные нервы, гликоли действуют на почки.
Кетоны и простые эфиры обладают наркотическим действием, диоксан
может вызвать тяжелое поражение печени и почек. Уксусная и пропионовая
кислоты разъедают кожу, а их ангидриды оказывают раздражающее дейст-
вие на глаза. Пиридин ядовит, вдыхание его паров вызывает головную боль.
Многие растворители вызывают устойчивые дерматиты (экзему, раздра-
жение кожи), причем сила поражающего действия в большой степени зависит
от индивидуальной восприимчивости.
Все это следует знать, чтобы обеспечить безопасную работу в лаборато-
рии. На рис. 34 показано приспособление, которым следует пользоваться
при заполнении растворителями пипеток (рабочих титрованных растворов);
для этой же цели может служить также, например, пипетка с резиновой
грушей, снабженной трехходовым краном.
Для отмеривания едких растворителей, обладающих неприятным запа-
хом (уксусная кислота, уксусный ангидрид, раствор ацетата ртути(П)
в уксусной кислоте, пиридина и т. д.), в количествах 5—50 мл в тех случаях,
когда ошибкой в 0,5—1 мл можно пренебречь, удобно применять прибор,
показанный на рис. 35 [367]. Если колбу наклонить, калиброванная бюретка
с краном заполняется раствором и необходимое количество этого раствора
можно отмерить в другой сосуд.
Глава 9
КИСЛОТНЫЕ ТИТРАНТЫ
Органические азотистые основания можно точно оттитровать раство-
рами неорганических кислот в органических растворителях [144, 146]_
Кондуктометрические исследования показали, что самая сильная из этих
кислот хлорная [470]; она растворяется
в большинстве органических растворителей
и стабильность ее растворов, например
в уксусной кислоте или диоксане, удовле-
творительна. Хлорметандисульфоновая,
метантрисульфоновая [763] и фторсульфо-
новая кислоты [627] в среде уксусной
кислоты даже более сильные кислоты,
чем хлорная, однако широкого примене-
ния они пока не нашли. На рис. 36 даны
значения функции кислотности Гаммета
Но для сильных кислот в среде уксусной
кислоты [625, 763] и графически показана
зависимость этой функции от отрицатель-
ного логарифма концентрации кислоты
Сл. Как можно видеть, в растворе уксус-
ной кислоты хлорная кислота сильнее
сульфокислот и соляной кислоты (схг.
гл. 2, разд. 16).
Более того, используя хлорную кис-
Р п с. 36. Значения Но некоторых
кпслот в уксусной кислоте, содержа-
щей 0,12% воды [763], см. также
[625].
1 — метантрисульфокислота; 2 — хлормс-
тантрисульфокислота; 3 — хлорная кис-
лота; 4 — хлоркарбоксиметансульфоки-
слота; 5 — хлорметансульфокислота;
6 — серная кислота; 7 — метапсульфо-
кислота; 8 — соляная кислота.
лоту, можно приготовить сильно разбав-
ленные титранты (0,001 н.), и при этом
резкость перехода окраски останется удо-
влетворительной. Для получения 0,005—
0,001 н. растворов пригодны также
/г-толуолсульфокислота и п-нитротолу-
олсульфокислота [299, 318].
Хорошо зарекомендовали себя также
стандартные растворы соляной кислоты
в G — Н-смесях растворителей; например, 0,1 н. раствор соляной
кислоты в смеси пропиленгликоля с хлороформом.
Все титранты, о которых ниже будет идти речь, приготовлены с исполь-
зованием очищенных растворителей.
51. СОЛЯНАЯ КИСЛОТА
а. Приготовление 0,1 н. раствора соляной кислоты в смеси пропилен-
гликоля с хлороформом [169, 621]. В широкогорлой конической колбе смеши-
вают 10 мл соляной кислоты (уд. вес 1,18) с 500 мл пропиленгликоля (уд. вес
1,43), затем добавляют 500 мл хлороформа и переносят в бюретку с резервуа-
ром (автоматическая бюретка, рис. 37). Титр полученного раствора устанав-
ливают по раствору сплл-дифенилгуанидина в смеси метилового и изопро-
пилового спиртов (35 : 65).
Кислотные титранты
143
спльм-Дифенилгуанидин СвН5 — NH — С — NH — СвН5 (мол. вес
II
NH
211,3; рКь = 4,00) удобно использовать как стандартное вещество для
установки титра вследствие высокого молекулярного веса. Это основание
связывает лишь один протон С6Н5 — NH — С — NH — С6Н5. Подходящим
.1
+ NH2
индикатором служит метанольный раствор диметилового желтого — мети-
ленового голубого (см. табл. 63). На 20 мл титранта нужны три капли инди-
катора, при этом зеленая окраска раствора переходит в янтарную. 1 мл
0,1 н. соляной кислоты эквивалентен 21,13 мг дифенилгуанидина.
Для очистки дифенилгуанидин перекристаллизовывают последовательно
из 95%-ного этилового спирта и толуола и сушат при 110° [117]. Препарат
можно также сушить, не опасаясь разложения, 24 час при 80° или 5 час при
117° [173]. Температура плавления очищенного таким способом дифенил-
гуанидина 149,4—149,9°, он не гигроскопичен и растворяется в уксусной
кислоте, диоксане, этиловом спирте, хлороформе, бензоле, хлорбензоле,
ацетонитриле и т. д.
Титр раствора соляной кислоты можно также установить по окиси
ртути(П). С этой целью берут навеску примерно 200 мг окиси ртути(П)
с точностью до 0,1 мг, растворяют в 5 мл уксусной кислоты при слабом
нагревании и выпаривают почти досуха. Остаток растворяют в 25 мл смеси
пропиленгликоля с хлороформом (1:1) и нейтрализуют в присутствии
тимолового синего. Полученный в результате раствор ацетата ртути тит-
руют 0,1 н. соляной кислотой, 1 мл которой эквивалентен 10,83 мг окиси
ртути(П). Децинормальный раствор соляной кислоты в смеси пропиленгли-
коля с хлороформом, титрованный таким способом, пригоден для определе-
ния, например, нормальности раствора ацетата ртути(П) в метиловом спирте
(см. гл. 27, разд. 154) [804].
б. Приготовление раствора соляной кислоты в метиловом спирте или
монометиловом эфире гликоля. Чтобы получить 0,5 н. раствор, смешивают
50 мл соляной кислоты (уд. вес 1,18) с 950 мл метилового спирта, охлажден-
ного до 5—10°, или с монометиловым эфиром гликоля при комнатной тем-
пературе. Титр устанавливают или вышеописанным способом, или по 0,1 н.
раствору едкого кали в изопропиловом спирте в присутствии тимолфталеина.
52. ХЛОРНАЯ КИСЛОТА
При растворении хлорной кислоты в уксусной образуется ион СНзСООЩ.
По сравнению с водой уксусная кислота — значительно более слабый акцеп-
тор протонов, поэтому при растворении сильной протонной кислоты равно-
весие не смещено в сторону образования продуктов диссоциации; следова-
тельно, сильная протонная кислота — более слабый электролит в среде
уксусной кислоты, чем в воде [420]. Тем не менее раствор хлорной кислоты
в уксусной более «кислый», чем водный раствор, так как ацилониевый ион
более сильный донор протонов, чем И3О+. На «сверхкислый» характер хлорной
кислоты в уксусной кислоте впервые обратили внимание Конант и Холл [144].
Вследствие низкой диэлектрической проницаемости уксусной кислоты
лишь небольшая часть растворенной соли находится в виде свободных
ионов, большая часть соли образует ионные ассоциаты. У протонных кислот
способность к протонизации не обязательно приводит к образованию свобод-
ных ионов [728]. Скорее, следует постулировать равновесие между иониза-
цией и диссоциацией [102, 464]
сн3соон+нсю4 сн3соон£сю; ch3coohj -- сю;.
Ионизация Диссоциация
144
Глава 9
Кондуктометрические измерения позволяют судить о диссоциации [4701,
тогда как кислотность раствора более важна для определения наличия иони-
зации. Криоскопические измерения доказывают, что в растворе уксусной
кислоты хлорная кислота в основном присутствует в мономолекулярной
форме, а не в диссоциированном состоянии [421].
а. Приготовление 0,1 н. раствора хлорной кислоты в уксусной кислоте.
К 975 мл безводной уксусной кислоты, свободной от уксусного ангидрида,
добавляют 9 мл 65—70%-ного водного раствора хлорной кислоты и 15—
16 мл уксусного ангидрида. Раствор приготавливают непосредственно
в резервуаре со стеклянным шлифом от автоматической бюретки (рис. 37).
Так как хлорная кислота катализирует
Рис. 37. Бюретка с резервуаром
для неводного титрования, (см.
также Kirsten W. J., Burette
arrangement for some special titra-
tions, Analyt. Chem., 29, 461
(1957).)
1 — шприц объемом 50 или 100 мл.
гидролиз уксусного ангидрида, стандартный
раствор можно использовать уже после одно-
дневного стояния. При приготовлении раст-
вора может наблюдаться слабое разогрева-
ние.
Определение титра по дифенилгуанидину
[244, 450]. Взвешивают 150—250 мг дифе-
нилгуанидина с точностью до 0,1 мг и раст-
воряют при слабом нагревании в 20—30 мл
предварительно нейтрализованного раство-
рителя. При использовании уксусной кисло-
ты добавляют 2—3 капли 0,1%-ного кри-
сталлического фиолетового и титруют из бю-
ретки с ценой делений 0,05—0,01 мл до появ-
ления синей окраски (см. гл. 13, разд. 72).
21,13 мг дифенилгуанидина эквивалентны
1 мл 0,1 н. хлорной кислоты.
Определение титра по би фталату калия
[175, 491, 730]. В среде уксусной кислоты
бифталат калия ведет себя как основание
(гл. 5, разд. 34). Преимущество этого веще-
ства заключается в том, что оно менее гигро-
скопично, чем дифенилгуанидин, однако оно
нерастворимо в апротонных растворителях,
например в четыреххлористом углероде, хлор-
бензоле и т. д. 20,41 мг бифталата калия
эквивалентны 1 мл 0,1 н. хлорной кислоты.
Другие вещества для определения титра. Так как сульфаминовая кис-
лота NH2SO3H — соединение амфотерное, она пригодна для установки титра
как хлорной кислоты, так и растворов метилатов щелочных металлов.
9,71 мг сульфаминовой кислоты соответствуют 1 мл 0,1 н. хлорной кислоты
[118].
Содержание уксусного ангидрида в стандартном растворе хлорной кис-
лоты можно определить титрованием по пиперазинадипату. В присутствии
небольших количеств уксусного ангидрида пиперазин быстро ацилируется,
причем основность атомов азота настолько понижается, что они уже не спо-
собны титроваться хлорной кислотой. Сравнивая результаты такого титрова-
ния с результатами титрования по другим стандартам, например дифенил-
гуанидину, можно выяснить, загрязнен ли раствор хлорной кислоты уксус-
ным ангидридом [575]. Об использовании трифенилгуанидина для определе-
ния титра см. [390].
4-Аминопиридин (Къ = 1,3 «10-8) также можно применять для опреде-
ления титра [335]; в уксусной кислоте он также связывает один протон.
.9,41 мг аминопиридина эквивалентны 1 мл 0,1 н. хлорной кислоты.
Кислотные титранты
145
В последнее время стал шире применяться 7п/шс-(оксиметил)аминометан
(238], Къ = 1,2.10-%
NH2
I
НОСН2 —С —СН2ОН
I
СН2ОН
1 мл 0,1 н. хлорной кислоты эквивалентен 12,11 мг ?п/шс-(оксиметил)амино-
метана. Это вещество легче растворимо в уксусной кислоте (1,7 молъ/л), чем
бифталат калия или дифенилгуанидин [877].
Другие вещества для определения титра, например карбонат натрия
и метилникотинат, теперь употребляются редко.
Рекомендуется для установления титра применять примерно тот же
объем, что и для титрования. Суммарный расход титранта на раствор и на
индикатор можно определить также при помощи глухих опытов. В этом
случае измерения следует проводить 4—5 раз, а предварительная нейтра-
лизация раствора не нужна. Если для определения титра используются
различные вещества, то применение одинакового количества рабочего титро-
ванного раствора в присутствии одного и того же индикатора может привести
к различной «правильной» окраске в зависимости от стандарта, применяе-
мого для установления титра. Так, чтобы получить одинаковые результаты
в присутствии кристаллического фиолетового, бифталат калия нужно тит-
ровать до синей окраски, метилникотинат — до синевато-зеленой и дифенйл-
гуанидин — до изумрудно-зеленой [491, 730]. При установлении титра
хлорной кислоты получают различные значения, например, при использо-
вании оксината алюминия и бифталата калия.
Определение титра 0,25 н. хлорной кислоты в уксусной кислоте фотомет-
рическим титрованием [390] (см. также гл. 14). Взвешивают примерно
0,97—1,0 г трифенилгуанидина (мол. вес 287,35) в колбе объемом 100 мл
и растворяют его в 50 мл уксусной кислоты. Нейтрализуют примерно 90%
трифенилгуанидина хлорной кислотой, 100 мл которой содержат 8 мл насы-
щенного раствора хинальдинового красного в уксусной кислоте, и доводят
объем до метки. Пробы по 25 мл титруют фотометрически при 533 нм, добав-
ляя стандартный раствор порциями по 0,01 мл и отмечая результаты до тех
пор, пока минимум поглощения не приблизится к нулевому поглощению.
Фотометрическое титрование удобно проводить с циркуляцией раствора
в колбе Эрленмейера, соединенной с колориметрической кюветой так, как
это сделано в приборе «Спектроник 20» Бауша и Ломба (см. гл. 14). Содер-
жимое колбы можно полностью перемешать за 20—25 сек. Поглощение
индикатора в его основной форме измеряется для той же концентрации,
в которой он присутствует в растворе, подлежащем анализу. Значение вели-
чин ///II+ рассчитывается по разности А/А в — А, где А в — поглощение
ацетата индикатора; А — поглощение, измеренное на различных этапах
титрования. По полученным результатам строится график зависимости
отношения ///Н+ от израсходованного объема (в миллилитрах). Точка пере-
сечения кривой с осью определяет точку эквивалентности (гл. 14, разд. 79
и 80). 71,84 мг трифенилгуанидина эквивалентны 1 мл 0,25 н. хлорной кис-
лоты.
В качестве смазки для стеклянных шлифов рекомендуется следующий
состав. 8 г растворимого крахмала тщательно смешивают с 20—25 г глице-
рина, помещают на асбестовую подставку и нагревают до 140—160° при
постоянном перемешивании. Вязкая жидкость становится прозрачной
п светло-коричневой. Еще горячей ее выливают в широкогорлую склянку
и добавляют для предупреждения заплесневения 1—2 кристаллика фенола.
Приготовленная таким способом смазка нерастворима или почти нераство-
рима в большинстве органических растворителей.
146
Глава 9
б. При употреблении раствора хлорной кислоты в диоксане для титрова-
ния диоксанового раствора основания часто образуется нерастворимый:
в диоксане перхлорат, что усиливает резкость перехода окраски.
Приготовление раствора хлорной кислоты в диоксане, содержащего 0,2—
0,3% воды. 4,5 мл 65—70%-ной хлорной кислоты смешивают с 1000 мл диок-
сана, перегнанного над натрием. 1 мл 0,05 н. раствора, полученного таким
способом, эквивалентен 10,56 мг дифенилгуанидина, растворенного в том же
растворителе (или смеси растворителей), в котором растворен титрант.
В качестве индикатора предпочтительно применять метиловый красный.
Приготовление раствора безводной 0,03 н. хлорной кислоты в диоксане-
[557]. В мерную колбу объемом 1 л помещают 8,5 мл уксусного ангидрида,
охлаждают его колотым льдом и осторожно по каплям добавляют 2,85 мл
65 %-ной хлорной кислоты так, чтобы избежать повышения температуры.
Разбавляют до 1 л диоксаном, свежеперегнанным над едким кали и пропу-
щенным через колонку с ионообменной смолой пермутит C-50-D.
3. Использовать 0,01 н. хлорную кислоту в смеси уксусной кислоты
с четыреххлористым углеродом целесообразно в тех случаях, когда эта смесь,
растворителей является не только средой для титрования, но употребляется
и для приготовления стандартного раствора. Растворяющая способность
уксусной кислоты и инертный характер четыреххлористого углерода допол-
няют друг друга и дают возможность получить большую резкость перехода
окраски индикатора и большую крутизну скачка потенциала. Раствор хлор-
ной кислоты в смеси уксусной кислоты и четыреххлористого углерода гото-
вят так. Смешивают 0,9 мл 65—70%-ной хлорной кислоты с 500 мл уксусной
кислоты (содержание воды в ней должно быть ниже 0,05%) и 5 мл уксусногп
ангидрида, оставляют в хорошо закупоренной склянке на ночь, затем доли-
вают 500 мл четыреххлористого углерода и переносят в бюретку с резер-
вуаром. Чтобы определить титр раствора, растворяют точную навеску
(90—110 мг) дифенилгуанидина в предварительно нейтрализованном раство-
рителе, доводят в мерной колбе объем раствора до 100 мл и титруют пор-
циями этого раствора по 10 мл. 2,113 мг дифенилгуанидина эквивалентны
1 мл 0,01 н. хлорной кислоты. Анализируемое вещество также должно быть
растворено в нейтрализованном растворе.
Анализируемый раствор должен отбираться той же пипеткой (сполосну-
той растворителем), которая применялась при установке титра, так как
пипетки калиброваны по водным растворам, а толщина пленки растворителя,
остающейся на внутренней стенке пипетки, отличается от толщины пленки
воды. При таком способе анализа может быть достигнута точность до 0,2—
0,4%, если работать с 0,01 н. растворами.
г. Стандартный раствор 0,001 н. хлорной кислоты в смеси уксусной кис-
лоты с четыреххлористым углеродом получают из 0,01 н. раствора десяти-
кратным разбавлением соответствующей смесью растворителей (1:1, 1:5,
1 : 10) (см. также гл. 13, разд. 72).
53. п-ТОЛУОЛСУЛЬФОКИСЛОТА
Ферлендер [852], а позднее Дитце ль и Пауль предложили использовать
протонные кислоты в неполярных растворителях. Применение сульфокислот
для титрования в неводных средах представляет интерес, так как сульфо-
группа обладает сильными кислотными свойствами.
Очень хорошая точность достигается при потенциометрическом титровании метан-
и этансульфокислотами в среде уксусной кислоты (средняя ошибка 0,2%). В отличие от
хлорной кислоты эти кислоты не образуют осадков прп титровании бифталата калия [119].
Моногидрат n-толуолсульфокислоты легко растворим в смеси четырех-
хлористый углерод — фенол или хлороформ — фенол (10 : 1). 0,001 или
0,005 н. стандартные растворы удобно использовать в серийных анализах,
Кислотные титранты
147
если нужно проводить микротитрование некоторых органических оснований
в растворе хлороформа (как это бывает, например, при проведении экстрак-
ций) [299, 301, 302, 305, 318]. Вместо раствора n-толуолсульфокислоты в хло-
роформе для микроопределения алкалоидов можно применять 0,002—
0,005 н. раствор n-толуолсульфокислоты в этиленгликоле или в смеси этилен-
гликоля с диоксаном [700—702].
Приготовление раствора. В стакан объемом 100 мл помещают 50 г средней
фракции предварительно перегнанного кристаллического фенола и 90 г
моногидрата n-толуолсульфокислоты и нагревают при перемешивании стек-
лянной палочкой до растворения сульфокислоты. Полученный раствор
разбавляют 200—300 мл хлороформа, содержащего в качестве консерви-
рующего вещества 1 об. % низкокипящего петролейного эфира, переносят
в мерную колбу на 500 мл, доводят объем раствора до метки, после чего
оставляют на 24 час и затем фильтруют в микробюретку с резервуаром [299].
Стандартный (0,005 н.) раствор получают, исходя из 450 мг моногидрата
п-толуолсульфокислоты.
Нейтральность четыреххлористого углерода или хлороформа может быть
установлена следующим способом. К 2—3 мл растворителя прибавляют
2—3 мл свежедистиллированной воды, энергично встряхивают 30 сек, затем
после разделения фаз добавляют две капли бромтимолового синего. Водный
слой при этом не должен окраситься ни в желтый (кислотный), ни сине-зеле-
ный (основной) цвет.
Установление титра по атропину (основанию). В 3 порциях хлороформа
по 100 мл растворяют 3 навески атропина по 20 мг, взятые с точностью
до 0,1 мг. Для каждых 5—10 мл этого раствора используют одну каплю рас-
твора диметилового желтого в хлороформе, для 15—20 мл — две капли.
При титровании наблюдается переход окраски из желтой в розовую.
289,4 мкг атропина соответствуют 1 мл 0,001 н. раствора п-толуолсульфо-
кислоты. Для установления титра используется также дифенилгуани-
дин.
54. ПРИМЕНЕНИЕ ПОПРАВОК
При неводном титровании следует иметь в виду два вида поправок:
поправки на температуру и поправки, учитывающие растворители и индика-
торы. Для определения последних известны три метода.
1. Анализируемое и стандартное вещество растворяют в одинаковых
предварительно нейтрализованных растворителях в присутствии одного
и того же индикатора.
2. Суммарный расход стандартного раствора на растворитель и на инди-
катор определяют путем раздельных титрований и вычитают из объема,
израсходованного на титрование анализируемого и стандартного вещества
соответственно.
3. Почти эквивалентные количества анализируемого и стандартного
вещества растворяют в равных объемах растворителей (или их смеси) одина-
кового состава и качества. При этом фактор пересчета для раствора будет
включать и поправку глухого опыта. Этот метод удобен при потенциометри-
ческом титровании.
Первый метод наиболее прост, он может применяться и при потенцио-
метрическом титровании. Второй дает результаты, заслуживающие доверия
только в том случае, если для введения поправки вычислено среднее значе-
ние по крайней мере из трех определений. Так как точность визуального
определения зависит и от толщины слоя раствора и от преломления им света,
не рекомендуется отбирать пробы растворителя с индикатором в 10 раз
большие необходимых и затем делить объем раствора, пошедшего на титрова-
ние, на десять.
148
Глава 9
Безводная уксусная кислота, а также стандартный раствор хлорной кис-
лоты имеют относительно высокий коэффициент расширения. Поэтому при
определении титра температура титранта измеряется термометром, при-
Р и с. 38. Номограмма теплового расширения 0,005 н. раствора п-толуолсульфокислоты.
Отклонению от стандартной температуры на 1° соответствует увеличение пли уменьшение
объема 1000 мл стандартного раствора на 1,25 мл. Если температура, измеренная в про-
цессе титрования, ниже стандартной, то должна быть введена поправка со знаком (-[-),
если выше — то со знаком (—).
соединенным к бюретке, и вводится поправка ±0,0011 мл на каждый
миллилитр и каждый градус [386, 730].
^ = ^(1 ±0,0011 А),
где N! — нормальность при температуре, наблюдаемой во время титрования;
N0 — нормальность при температуре установки титра; Д — измеренная
разность температур (ЭС) установки титра и титрования. Для титрованных
растворов n-толуолсульфокислоты в хлороформе это значение составляет
0,00125 (рис. 38). Во время титрования изменение температуры не должно
превышать ±0,5°.
Глава 10
ОСНОВНЫЕ ТИТРАНТЫ
55. ГИДРООКИСИ ЩЕЛОЧНЫХ МЕТАЛЛОВ
а. При использовании стандартных растворов едкого кали (или едкого
натра) в метиловом (или этиловом) спирте необходимо учитывать, что
в чистом этиловом спирте или спирте, содержащем лишь очень незначитель-
ное количество воды, равновесие
с2н5он+-он и2о-^с2н5о-
сдвинуто в сторону образования этилат-ионов. Из этого следует, что стан-
дартный раствор алкоголятов щелочных металлов часто можно заменить
раствором едкого кали в спирте, получить который проще [153] (см. табл. 51).
Таблица 51
Содержание воды В С2Н5ОН, % Содержание ОН- (С2Н5О-4-НО-), %
0,1 н. 0,5 н. 1,0 и.
0 0,8 3,7 6,9
0,2 1,5 4,2 7,5
1,0 4,1 6,8 9,6
Приведенные выше данные подтверждаются уменьшением скорости гидролиза слож-
ных эфиров, что соответствует уменьшению содержания воды в этанольном растворе
едкого кали. Данные инфракрасных спектров также свидетельствуют о 75—100%-ном
связывании НО "-ионов [116].
Приготовление 0,1 н. раствора. 6 г твердого едкого кали (4 г едкого натра)
растворяют в 1000 мл абсолютного метилового (этилового) спирта, тщательно
защищая от СО2 и влаги воздуха. Титрование проводят 0,1 н. соляной кис-
лотой или раствором чистой бензойной кислоты в метиловом спирте или
пальмитиновой кислотой или бифталатом калия в присутствии тимолфта-
леина. 12,21 мг бензойной кислоты, 25,64 мг пальмитиновой кислоты или
20,42 мг бифталата калия эквивалентны 1 мл 0,1 н. раствора.
Твердые гидроокиси можно также растворять непосредственно в резервуаре бюретки,
пропуская струю азота (предварительно очищенного барботированием через концентри-
рованной спиртовый раствор щелочи и высушенного силикагелем). Струя азота одновре-
менно перемешивает раствор.
б. Приготовление 0,1 н. раствора едкого кали в изопропиловом спирте.
В 1000 мл изопропилового спирта, содержащего около 0,5—1% воды, рас-
творяют 6 г твердого едкого кали и кипятят 10—15 мин, предварительно
защитив его от попадания СО2 из воздуха. Для этой цели можно применять
либо надетую на обратный холодильник трубку «двойного поглощения»
с силикагелем и твердым едким кали, либо пропускать в кипящий раствор
струю промытого и высушенного азота. Охлажденный раствор оставляют
на несколько часов, за это время из него выпадает нерастворимый карбонат
калия. Титр раствора устанавливают по бифталату калия в присутствии
150
Глава 10
1-нафтолбензеина или фенолфталеина [176, 530]. Раствор едкого кали в изо-
пропиловом спирте или монометиловом эфире этиленгликоля используется
для определения содержания кислот в нефтепродуктах и битуминозных
веществах [530, 692].
в. Раствор едкого кали в смеси моноэтилового эфира гликоля с ксилолом
благодаря большой растворяющей способности и высокой температуре
кипения (около 130°) более удобен для определения числа омыления (слож-
ных эфиров), чем, например, раствор едкого кали в «-пропиловом спирте.
Для приготовления 0,5 г раствора 30 г едкого кали растворяют в 20 мл воды
и доводят объем раствора до 1000 мл, добавляя смесь моноэтилового эфира
гликоля (целлозольва) с ксилолом (1 : 1 или 1 : 2). Через 2 дня раствор
декантируют с кристаллов карбоната калия, которые иногда осаждаются
на стенках сосуда [450]. Титр раствора устанавливают по бензойной кислоте,
пальмитиновой кислоте или бифталату калия в присутствии тимолфталеина.
г. Приготовление раствора едкого натра в монометиловом эфире гли-
коля см. в гл. 11, разд. 60, к.
56. АЛКОГОЛЯТЫ ЩЕЛОЧНЫХ МЕТАЛЛОВ
К числу самых удобных растворителей для этой группы титрантов отно-
сятся метиловый, этиловый и изопропиловый спирты в смеси с бензолом
в отношении 1 : 12—1 : 6 [252, 259, 372, 502, 694]. Реже употребляются
моноэтаноламин, этилендиамин и пиридин [190, 423, 446, 601].
а. Раствор метилата натрия в смеси метилового спирта с бензолом
получают, используя безводный бензол и свободный от примесей альдегида
и воды метиловый спирт. Для приготовления 0,1 н. раствора кусок метал-
лического натрия с чистой поверхностью весом 2,2—2,5 г помещают в смесь
150 мл метилового спирта со 150 мл бензола, охлажденную до 5—10° (метал-
лический натрий предварительно погружают при помощи пинцета на 8—
10 сек в охлажденный льдом метиловый спирт). Соединяют колбу с шарико-
вым холодильником, снабженным трубкой, наполненной едким кали. Чтобы
выделение газа не было слишком бурным, применяют охлаждение. После
растворения натрия раствор разбавляют бензолом до 1 л и сразу же пере-
носят в бюретку с резервуаром (см. рис. 37). Если после добавления бензола
раствор мутнеет, приливают 2—10 мл метилового спирта, пока раствор
не станет совсем прозрачным. Чем меньше содержание метилового спирта,
тем четче переход окраски индикатора. Для полного растворения алкоголя-
тов в бензоле в случае метилата натрия необходимо не менее 12—13 об. %
метилового спирта, а в случае метилата калия — 8 об.%. Метилаты калия
и лития получают таким же способом. Преимущество метилата лития заклю-
чается в том, что при титровании, например, производных барбитуровой
кислоты не образуется осадка, мешающего определению конечной точки.
Титр раствора определяют по бензойной или 2-фенилхинолин-4-карбоно-
вой кислоте.
Определение титра по бензойной кислоте. 60—100 мг бензойной кислоты,
взвешенные с точностью до 0,1 мг, растворяют в 20—40 мл растворителя
(например, смеси бензола с метиловым спиртом или пиридином, смеси пири-
дина с ацетоном, диметилформамиде), выбор которого зависит от вещества,
подлежащего определению. Растворитель должен быть предварительно ней-
трализован по тимоловому синему, азофиолетовому или другому подходя-
щему индикатору. Для титрования используется бюретка с ценой деления
0,05 мл. Нейтрализацию, растворение и титрование лучше проводить в отсут-
ствие двуокиси углерода (рис. 39—42). Большинство применяемых индикато-
ров чувствительны к действию двуокиси углерода, но при работе с раство-
рами «-бутиламина и этилендиамина можно примириться с ее присутствием.
Однако в растворах пиридина и диметилформамида при достаточных навы-
Основные титранты
151
как титрование можно провести быстро, и тогда все указанные меры пред-
осторожности становятся излишними. При проведении серийных анализов
имеет смысл устанавливать титр каждые два дня, но если анализы проводят-
ся нерегулярно, титр нужно устанавливать перед каждым определением. При
установке титра и проведении анализа концентрация индикатора должна быть
одной и той же. 1 мл 0,1 н. раствора эквивалентен 12,21 мг бензойной кислоты.
Определение титра по 2-фенилхинолин-4-карбоновой кислоте. Неудоб-
ство применения бензойной кислоты заключается в том, что образующаяся
Р и с. 39. Колбы для титрования и при-
способление, применяемое при работе с ин-
дикаторами и растворителями, чувствитель-
ными к СО2.
а — фиксированный сосуд; б — переносная кол-
ба, снабженная тонкой перфорированной резино-
вой пленкой; 1 — корковая пробка с двумя от-
верстиями; 2, а — магнитная мешалка; 2,6 — ре-
зиновая пленка.
Р и с.< 40. Прибор для титрования
[453].
1 — эластичное сцепление; 2 — шприц;
3 — сосуд для титрования; 4 — фикси-
рующий стержень; 5 — эластичное рези-
новое поддерживающее устройство; 6 —
резиновая пробка.
Рис. 41. Ячейка для потенциомет-
рического титрования.
1 — стеклянный электрод; к 2 — электрод
Ag/AgCl или каломельный электрод; 3 —
пористое стекло или фарфор.
Рис. 42. Простейшее устройство для
титрования [в атмосфере азота — титруе-
мый раствор перемешивается током инерт-
ного газа, допускается также встряхивание
раствора вручную.
152
Глава 10
по мере титрования соль выпадает в виде желеобразного осадка. Этого можно
избежать, добавив в титрант 10% метилового или изопропилового спирта,
однако четкость перехода окраски индикатора при этом уменьшается. Калие-
вая соль 2-фенилхинолин-4-карбоновой кислоты более растворима, и эту
кислоту удобно применять для установки титра [307]. 24,925 мг 2-фенилхино-
лин-4-карбоновой кислоты эквивалентны 1 мл 0,1 н. раствора.
Для определения титра можно применять и другие вещества: стеариновую
кислоту (т. пл. 69,6—69,8°), перекристаллизованную из ацетона и высушен-
ную при 100° в вакууме над серной кислотой; пальмитиновую (т. пл. 92,4—
92,7°) и миристиновую (т. пл. 54,4—54,8°) кислоты, в частности для установ-
ки титра 0,05 н. раствора этилата натрия в этиловом спирте, применяемого
для определения кислотного числа.
б. 0,02 н. раствор в смеси бензола с метиловым спиртом приготовляют,
разбавляя 200 мл 0,1 н. раствора метилата натрия смесью бензол — метило-
вый спирт (9 : 1) до 1000 мл. При этом в смесь барботируют азот, пропущен-
ный через раствор едкого кали и высушенный силикагелем. Если 0,02 н.
раствор необходимо хранить длительное время, его следует держать в пласт-
массовой склянке или склянке из боросиликатного стекла в атмосфере азота
(см. также гл. 21).
57. ГИДРООКИСИ АЛКИЛАММОНИЯ
Среди первых ученых, применивших гидроокись тетрабутиламмония
для титрования слабых кислот, были Дил и Уайльд [176]. Однако они приме-
нили раствор гидроокиси тетрабутиламмония в изопропиловом спирте, содер-
жащем 10% воды, что могло неблагоприятно сказываться на результатах
потенциометрического титрования слабых кислот. Позднее эти ученые приме-
нили гидроокись тетрабутиламмония (ТБАГ) в безводной среде. ТБАГ обыч-
но получают из йодистого тетрабутиламмония (мол. вес (G4H9)4NI = 369,4).
Этот реактив имеется в продаже, а также может быть получен кипячением
с обратным холодильником йодистого бутила с три-н-бутиламином; получен-
ное вещество перекристаллизовывают из бензола [493].
Многочисленные методы приготовления стандартных растворов ТБАГ
можно разделить на три группы [159, 160, 164, 359, 361, 540].
Получение гидроокиси алкиламмония из галогенидов алкиламмония 1) при
помощи ионообменных смол основного характера [359], 2) взаимодейст-
вием с окисью серебра [159, 540] или же комбинацией этих двух методов
[160, 164, 550] и, наконец, 3) под действием едкого кали [361].
О методиках титрования и условиях, способствующих сохранению стабильности тит-
рантов из четвертичных аммониевых оснований, см. работы [356, 357].
При действии окиси серебра на иодистый тетрабутиламмоний в метиловом
спирте образуется смесь ТБАГ с метилатом тетрабутиламмония (ТБА) [136]:
2(C4H9)4N+I- + Ag2O + СНзОН PacTBOptlTU1t; 2AgI + (c4H9)4N+OH- + (C4H9)4N+OCH3-.
Получить чистое алкоксипроизводное трудно [359], даже следы воды могут
привести к образованию гидроокиси. ТБАГ в среде изопропилового спирта
на самом деле представляет собой смесь ТБАГ с изопропилатом ТБА [358].
Приготовить раствор ТБАГ для визуального титрования проще, чем для
потенциометрического титрования, во время которого случайные загрязне-
ния могут влиять на определение конечной точки титрования. Например,
в тех случаях, когда проводится дифференцирующее титрование сильных
кислот, стандартный раствор, полученный с использованием окиси серебра,
необходимо пропустить через ионообменную колонку, заполненную смолой
амберлит IRA-400 [160].
Основные титранты
153
В растворах ТБАГ могут присутствовать в виде примесей трибутиламин,
карбонат трибутиламмония и небольшое количество растворенного серебра
в виде комплексного аниона [164, 550]. Ионы калия, начиная с некоторой
предельной концентрации, также оказывают нежелательное влияние на
результаты потенциометрического титрования [355]. Карбонаты могут
быть внесены с выпускаемым в продажу препаратом окиси серебра, хотя,
поскольку ТБАГ сильно поглощает СО2, они могут появиться и при приго-
товлении раствора. Поэтому при приготовлении стандартного раствора сле-
дует избегать загрязнения его атмосферной углекислотой. Выпускаемый
в продажу препарат окиси серебра может содержать 0,08—0,2% СО2. Как
правило, содержание СО2 в стандартном растворе, полученном с использова-
нием окиси серебра, составляет 0,5 —1,0%, в то время как при применении
анионообменного метода загрязнение СО2 составляет 0,3% [550].
а. Окись серебра может быть приготовлена с использованием окиси
бария (1) или едкого натра, не содержащего карбоната (2).
1. Получение окиси серебра с использованием окиси бария [754]. В 200 мл
дистиллированной воды растворяют 34 г нитрата серебра, пропуская через
раствор ток азота. К раствору добавляют суспензию, полученную из 20 г
тонкоизмельченной окиси бария и небольшого количества дистиллированной
воды, не содержащей СО2. Выпавшую в осадок окись серебра через 30 мин
десять раз промывают декантацией дистиллированной водой, свободной
от СО2; все это время через раствор должен барботировать азот. Рекомен-
дуется также насытить азотом промывные воды. Отмытую окись серебра
высушивают метиловым спиртом.
2. Получение окиси серебра с использованием едкого натра, свободного
от карбоната [164]. В 50 мл дистиллированной воды, свободной от СО2,
растворяют 30 г нитрата серебра. Двуокись углерода можно удалить либо
кипячением, либо пропуская в воду ток азота; можно применить оба метода
одновременно. После растворения добавляют 55 мл 4 н. раствора едкого
натра, свободного от СО2, энергично встряхивают суспензию и фильтруют
через стеклянную воронку с пористым дном (фильтр № 3) в атмосфере
азота. Слой окиси серебра на фильтре промывают сначала 750 мл дистилли-
рованной воды, свободной от СО2, а затем 300 мл метилового спирта. Окись
серебра, смоченную спиртом, сразу же употребляют для приготовления
стандартного раствора. Опыт показывает, что окись серебра, смоченная
метиловым спиртом, реагирует с иодистым тетрабутиламмонием более энер-
гично, чем сухая [540].
б. Приготовление стандартных растворов, содержащих гидроокись тет-
рабутиламмония. 1. О приготовлении 0,2 н. раствора ТБАГ в изопропи-
ловом спирте при помощи ионообменной смолы см. [359].
2. Приготовление 0,1 н. раствора ТБАГ в смеси бензола и метилового-
спирта с использованием Ag2O и ионообменной смолы амберлит IRA-400
[160]. В склянке с хорошо притертой стеклянной пробкой растворяют 80 г
химически чистого йодистого тетрабутиламмония в 180 мл абсолютного
метилового спирта. Помещают склянку в колотый лед и, когда температура
снизится до 1—2°, добавляют 40 г свежеприготовленной окиси серебра, гер-
метически закрывают склянку и держат ее во льду в течение часа, часто-
и энергично встряхивая. Полученный раствор фильтруют через плотный
стеклянный фильтр прямо в сосуд, в котором будет храниться раствор, или
же в бюретку с резервуаром. Склянку и фильтр промывают (3 X 50 мл}
холодным бензолом и разбавляют профильтрованный раствор до 2 л бензо-
лом, перегнанным над металлическим натрием. На сосуде заранее должен
быть отмечен уровень, соответствующий объему 2 л.
Вместо йодистого тетрабутиламмония можно использовать эквивалент-
ное количество бромистой соли, причем в этом случае достаточно 15-минут-
ного встряхивания.
154
Глава 10
Иногда раствор ТБАГ необходимо очистить. Для этого хроматографи-
ческую колонку размером 2,5 х 40 см наполовину заполняют амберлитом
IRA-400 в основной форме и промывают 2 н. раствором едкого натра до отри-
цательной реакции на галоген-ионы в промывных водах. Затем промывают
дистиллированной водой, свободной от СО2, до тех пор, пока элюат не станет
нейтральным. Обезвоживают колонку 500 мл абсолютного метилового спир-
та и, наконец, пропускают через нее 500 мл смеси бензола с метиловым спир-
том. Через подготовленную таким образом колонку пропускают раствор
ТБАГ со скоростью 15—20 мл/мин. Раствор собирают непосредственно
в резервуар бюретки, защищенный от влаги и СО2 воздуха.
Приготовленный стандартный раствор практически пригоден для диф-
ференцирующего титрования смеси сильных, средних и слабых кислот
в пиридине, так как он не содержит примесей, которые могли появиться
за счет побочных реакций йодистого тетрабутиламмония с Ag2O.
Для визуального титрования удобнее применять растворы ТБАГ, приго-
товленные более простыми способами.
3. 0,1 н. раствор ТБАГ приготавливают как описано выше, но вместо
очистки на ионообменной колонке в раствор в течение 5—10 мин пропу-
скают струю сухого и свободного от СО2 азота [159].
4. Приготовление раствора гидроокиси три-я-бутилметиламмония в сме-
•си бензол — изопропиловый спирт (8 : 2) [540]. Окись серебра, полученную
из 40 г нитрата серебра действием едкого натра и пропитанную изопропиловым
спиртом, суспендируют в растворе 32,8 г CH3(C4H9)3N+I_ в 90 мл изопропи-
лового спирта. Суспензию встряхивают 15 мин в закупоренном сосуде,
после чего фильтруют суспензию через стеклянный фильтр непосредственно
в темный сосуд, где будет храниться раствор, или в резервуар полумикро-
бюретки. Промывают фильтр (3 х 25 мл) изопропиловым спиртом и раз-
бавляют фильтрат бензолом до 1 л. Рабочий раствор может храниться около
месяца без значительной потери активности.
О приготовлении для потенциометрического титрования раствора гидро-
окиси тетрабутиламмония в смеси бензол — изопропиловый спирт (8 : 2)
из раствора моногидрата ТБАГ в пгретп-бутиловом спирте см. в работе [550].
5. О приготовлении растворов гидроокисей тетраметиламмония и три-w-
бутилметиламмония в пиридине см. в работах [369—371].
6. Приготовление 0,02 н. раствора ТБАГ в смеси бензол—метиловый
спирт [164]. Необходимую для работы окись серебра получают при помощи
NaOH, свободного от карбоната (см. выше, метод а). Для приготовления
1 л 0,1 н. раствора ТБАГ используется смоченная метиловым спиртом свеже-
осажденная окись серебра, полученная из 30 г нитрата серебра. В 95 мл
абсолютного метилового спирта, помещенного в склянку с пробкой, раство-
ряют 40 г чистого ИТБА. Охлаждают раствор в смеси снега с солью до
(—5) (—10°), добавляют окись серебра и насыщают свободным от СО2
азотом. Закрывают склянку, снова помещают ее в охладительную смесь
и оставляют там на 1 час, часто и энергично встряхивая. После добавления
900 мл бензола раствор снова насыщают азотом и помещают в охладительную
смесь еще на 15 мин, затем фильтруют через воронку с мелкопористым дном
в атмосфере азота прямо в резервуар бюретки, защищая от влаги и СО2
воздуха. Когда раствор примет комнатную температуру, его разбавляют
до 1 л бензолом, перегнанным над натрием. 0,02 н. раствор готовится
из этого запасного раствора следующим образом. К 200 мл 0,1 н. раствора
добавляют 30 мл абсолютного метанола и разбавляют до 1 л бензолом. Таким
способом может быть приготовлен стандартный раствор, свободный от кар-
боната.
На холоду растворимость окиси серебра значительно уменьшается,
но низкая температура препятствует образованию аминов за счет реакции
между галогенидами алкиламмония и окисью серебра.
Основные титранты
155
При потенциометрическом титровании воспроизводимость результатов
лучше, если в качестве электролита в обычном каломельном электроде
употребить 0,01 М раствор бромистого тетрабутиламмония. Электролити-
ческий раствор необходимо менять ежедневно. При титровании 0,02 н. рас-
творами рекомендуется анализируемый раствор освобождать от СО2, про-
пуская в него перед титрованием в течение 5 —10 мин струю промытого
азота. Таким способом можно уменьшить поправку на глухое титрование
раствора.
7. Приготовление раствора гидроокиси гексадецилтриметиламмония
в изопропиловом спирте с использованием едкого кали [361]. В изопропиловом
спирте реакция между едким кали и галоге-
нидом четвертичного основания идет следую-
щим образом:
Изопропиловый I
R4NC1 + KOH------------’R.NOH-1KC1' .
спирт 4
Степень превращения составляет примерно
0,92. Отделение хлористого калия способст-
вует протеканию реакции. Применение бро-
мистых и иодистых солей менее удобно, так
как КВг и KI растворимы в изопропиловом
спирте лучше, чем КС1.
Сухой хлористый гексадецилтриметилам-
моний (70 г) добавляют к 1000 мл 0,22 н.
раствора едкого кали в изопропиловом спирте,
энергично встряхивают в течение 15 мин
и оставляют стоять до полного осаждения
хлорида калия. Прозрачную жидкость пере-
носят в сосуд для хранения.
Этим методом можно за 1 час пригото-
Р и с. 43. Влияние ионов калия
на показания стеклянного элект-
рода. Потенциометрическое титро-
вание фенола в пиридине 0,25 н.
гидроокисью тетрабутиламмония
[355].
Концентрация ионов калия: 1 — ионы
калия отсутствуют; 2— 0,0028 н.;
3—0,0046 н.; 4—0,0056 н.; 5—0,018 н.;
6 — 0,18 н.
вить раствор гидроокиси алкиламмония; для
метода с использованием окиси серебра необходимо 2—3 час, а для метода
с применением ионообменных смол — 4—8 час. Полученные таким обра-
зом растворы дают хорошие результаты при визуальном титровании, но
при потенциометрическом титровании могут возникать помехи из-за высокой
концентрации иона калия (0,1—0,2% К+).
Присутствие ионов калия в стандартном растворе ТБАГ приводит к значительным
помехам при потенциометрическом титровании очень слабых кислот в пиридине [355];
невоспроизводимые скачки потенциала наблюдаются также в тех случаях, когда ионы
калия из каломельного электрода переходят в анализируемый раствор [358]. Поэтому,
вероятно, имеет смысл заполнять каломельный электрод 1 н. водным раствором хлористого
тетрабутиламмония вместо насыщенного раствора хлористого калия. На рис. 43 показано
влияние ионов калия, искажающее форму потенциометрических кривых.
8. Для определения ангидридов используется 0,1 н. раствор гидроокиси бензилтри-
метиламмония (тритона Б) в пиридине [623]. О приготовлении раствора гидроокиси три-
этил-н-бутиламмония в смесп бензола с метиловым спиртом см. [258].
58. ТИТРАНТЫ, СОДЕРЖАЩИЕ АЦЕТАТ НАТРИЯ
Соединения основного характера, лишь слабо растворимые в уксусной
кислоте, можно перевести в раствор, добавляя избыток стандартного раство-
ра хлорной кислоты, причем иногда необходимо нагревание. Так, например,
при титровании динатриевой соли винной кислоты применяют обратное
титрование избытка хлорной кислоты 0,1 н. раствором ацетата натрия
в уксусной кислоте [70] (см. также гл. 22, разд. 117, б).
Стандартный раствор ацетата натрия пригоден для определения серной
кислоты [288] или хлористого ацетила [831]; оба анализа проводятся в среде
уксусного ангидрида.
156
Глава 10
Приготовление 0,1 н. раствора ацетата натрия в уксусной кислоте.
Навеску 8,2 г безводного ацетата натрия, взятую с точностью до 0,01 г,
растирают в тонкий порошок, сушат над пятиокисью фосфора и после повтор-
ного растирания растворяют в 1000 мл уксусной кислоты. К 970 мл уксус-
ной кислоты и 30 мл уксусного ангидрида прибавляют 13,6 ацетата натрия,
содержащего три молекулы кристаллизационной воды. Этот раствор можно
применять через два или три дня.
Стандартный раствор ацетата натрия можно также приготовить, исходя
из карбоната натрия, поскольку он подвергается сольволизу.
Na2CO3 r2CII3COOII 2Na(CH3COO) - H2CO3.
Угольная кислота в этой среде так же неустойчива, как и в воде, и разла-
гается на двуокись углерода и воду. С точностью до 0,01 г берут навеску
5,30 г тонкоизмельченного карбоната натрия, предварительно высушенного
при 270 ± 10°, и растворяют его в 500 мл уксусной кислоты при слабом
нагревании, добавляют 10—11 мл уксусного ангидрида и разбавляют pacTBopt
уксусной кислотой до 1000 мл (ср. [208, 769]).
Глава 11
РЕДКО ПРИМЕНЯЕМЫЕ ТИТРАНТЫ
Стандартные растворы, параметры которых (нормальность, фактор пере-
счета, активность) определяются при помощи глухих опытов во время титро-
вания, будут обсуждаться детально при описании определений такого рода.
59. СТАНДАРТНЫЕ РАСТВОРЫ, ПРИГОТОВЛЕННЫЕ ИЗ ВЕЩЕСТВ, СОДЕРЖАЩИХ
АМИНОГРУППУ ИЛИ КАРБОКСИЛЬНУЮ ГРУППУ
Известны различные стандартные растворы, полученные из соединений,
содержащих амино- или карбоксильную группы. Для приготовления этих
растворов твердые химически чистые вещества (которые часто одновременно
используются для установки титра) или свежеперегнанные органические
основания в количествах, указанных в табл. 52, растворяют в органических
растворителях и доводят объем до 1000 мл.
Таблица 52
Стандартные растворы, приготовляемые из соединений,
содержащих аминогруппу или карбоксильную группу
Вещество Нормаль- ность Эквива- лентный вес Навеска Растворитель
Бензойная кислота а 0,1 122,1 12,21 г Бензол
Циклогекспламнн 6 1,0 99,17 115 мл Метиловый спирт
Цпклогексиламин в 0,5 99,17 57 мл ТГФ
Дпфенплгуанидпн г 0,01 211,3 2,113 г Бензол, уксусная кислота, хлорбензол, хлороформ
Изохпнолин Д 0,5 129,15 59 мл Бензол
N-Этплппперпдпне 0,1 113,2 14 мл Ацетон
Пиперидин ж 0,1 85,15 9,9 мл Изопропиловый спирт
Пиридин 3 0,1 79,1 8,1 .мл Уксусная кислота
Салициловая кислота 11 1,0 138,1 138,1 г Изопропиловый спирт — этиленгликоль (1:1)
Трпэтиламин к 2,0 101,2 — Бензол
трис-( Окспметил)аминоме- тан л 0,1 121,5 12,15 г Метиловый спирт
Трп-н-бутпла.мин м 0,1 185,3 24 мл Дпоксан
Три-н-иропилампн 11 0,1 143,3 19 мл Хлорбензол Ацетон
аДля обратного титрования избытка стандартных растворов щелочей [121].
бДля дифференцирующего титрования нитрующих или сульфирующих кислотных смесей [783].
пДля определения хлорангидридов [519].
‘Для обратного титрования избытка [519] 0,01 н. стандартного раствора хлорной кислоты.
ДДля определения алкильных соединений алюминия [222].
еДля определения малеиновой и фталевой кислот в присутствии их ангидридов [746].
жДля дифференцирующего титрования смеси серной кислоты с другими кислотами [170].
3Для дифференцирующего титрования соляной и хлорной кислот [736].
пДля обратного титрования избытка лауриламина, применяемого при титровании альдегидов
с образованием шиффова основания [7 51].
кДля определения уксусной кислоты в присутствии уксусного ангидрида [564].
лДля титрования хлорной кислоты, выделяющейся из перхлората серебра при определении
ацетиленового водорода [32].
мДля определения кислот в присутствии их хлорангидридов [623].
11 Для титрования соляной кислоты в присутствии хлорангидридов кислот [753].
158
Глава 11
60. ДРУГИЕ ТИТРАНТЫ
а. Для получения раствора хлорной кислоты в смеси уксусной кислоты
с уксусным ангидридом прибавляют по каплям 4,2—4,5 мл 70—72 % -пых
хлорной кислоты к 500 мл охлажденной льдом уксусной кислоты, после чего
разбавляют уксусным ангидридом до объема 1 л при непрерывном перемеши-
вании и охлаждении. Полученный таким способом 0,05 н. стандартный
раствор пригоден для титрования азотистых оснований в уксусном ангидри-
де [778] (см. гл. 3, разд. 23, в).
б. Для приготовления стандартного раствора хлорной кислоты в нитро-
метане 4,2—4,5 мл 70—72 %-ной хлорной кислоты разбавляют до 1 л очи-
щенным нитрометаном. Этот стандартный раствор можно хранить в течение
месяца. Его титр устанавливают по бифталату калия. Стандартный раствор
хлорной кислоты в нитрометане пригоден для потенциометрического титро-
вания органических оснований, содержащих азот или фосфор (фосфинов)-
[779, 781] (см. гл. 3, разд. 23, г и гл. 31, разд. 170).
в. Чтобы приготовить стандартный раствор хлорной кислоты в про-
пионовой кислоте, 35 г пропионового ангидрида добавляют к 1 л чистой про-
пионовой кислоты и при перемешивании приливают 10 мл хлорной кислоты
(уд. вес 1,690). После 24-часовой выдержки раствор можно применять. Титр-
раствора устанавливают по дифенилгуанидину; в среде пропионовой кислоты
особенно резок переход окраски метанилового желтого от бледно-желтой
к пурпурной [374—377]. 0,01 и 0,001 н. стандартные растворы хлорной
кислоты в смеси пропионовой кислоты с четыреххлористым углеродом при-
готовляют так же, как и аналогичные растворы в среде уксусной кислоты
(см. гл. 9, разд. 52, в, г).
г. Для приготовления 0,1 н. раствора хлорной кислоты в метилэтил-
кетоне медленно при перемешивании смешивают 8—9 мл 72 %-ной хлорной
кислоты с 1000 мл очищенного метилэтилкетона и насыщают раствор сухим
азотом. Титр устанавливают по бифталату калия [479].
д. Приготовление 0,5 н. раствора хлорной кислоты в монометиловом
эфире этиленгликоля. К 500 мл монометилового эфира этиленгликоля (метил-
целлозольв), помещенным в колбу объемом 1 л, осторожно по каплям при-
бавляют при перемешивании 40 мл 70—72 %-ной хлорной кислоты, послн
чего разбавляют монометиловым эфиром этиленгликоля до 1000 мл. Этот
стандартный раствор употребляется при определении спиртовых гидроксиль-
ных групп по изоцианатному методу для титрования дпбутиламина, добавляе-
мого с целью связать избыточный фенилизоцианат [669]. Титр раствора уста-
навливают по тприс(оксиметил)аминометану в присутствии 0,1 %-ного мета-
нольного раствора бромкрезолового зеленого. Наблюдаемое изменение окра-
ски индикатора: синяя —> зеленая -> желтая. Более разбавленный (0,2 н.)
стандартный раствор используется при определении карбонильных групп
методом образования оксимов для обратного титрования избытка гидроксил
амина [259].
При помощи 0,01 н. раствора хлорной кислоты в метилцеллозольве можно-
определять небольшие количества третичных аминов в присутствии большого-
избытка амидов кислот [690] (см. гл. 25, разд. 146, б).
е. При использовании формиата гидроксиламина для анализа соединений,
содержащих карбонильные группы, чтобы оттитровать избыток реактива,
употребляют 0,5 н. раствор азотной кислоты в монометиловом эфире гликоля'-
[691] (см. гл. 26, разд. 152, г). Для приготовления стандартного раствора
в 500 мл метилцеллозольва растворяют 35 мл концентрированной бесцветной
азотной кислоты. Затем для стабилизации раствора добавляют 1 г моче-
вины и 0,1 г 1,4-диэтоксибензола и разбавляют метилцеллозольвом до-
1000 мл. Титр устанавливают по тпрш?-(оксиметил)аминометану (см. гл. 26г
разд. 152, г).
Редко применяемые титранты
159
ж. Приготовление стандартного 0,1 н. раствора бромистого водорода
в уксусной кислоте. Под тягой сухой бромистый водород пропускают в 1 л
уксусной кислоты, время от времени проверяя титр раствора. Взвешивают
около 100 мг безводного карбоната натрия, высушенного над фосфорным
ангидридом, растворяют его в 5 мл уксусной кислоты и раствор титруют
в присутствии кристаллического фиолетового до сине-зеленой окраски. Этот
стандартный раствор употребляют для определения оксиранового кислорода
(а-эпоксигруппы) [197, 198] (см. гл. 27, разд. 156).
3. Для приготовления стандартного 0,2 н. раствора аминоэтилата нат-
рия в смеси этилендиамина с аминоэтанолом при охлаждении растворяют
4,5 г металлического натрия в 200 мл аминоэтанола и разбавляют этилен-
диамином до 1000 мл. Титр раствора устанавливают потенциометрически
по бензойной кислоте и фенольным соединениям (n-оксибензойной кислоте,
3,4-ксиленолу). Раствор используют для определения надкислот [554] (см.
гл. 15, разд. 93).
и. Стандартный 0,5 н. раствор метилата натрия в пиридине употребляют
для обратного титрования избытка ацетилацетона или салицилового альде-
гида при определении первичных аминов в присутствии вторичных и тре-
тичных аминов [157] (см. гл. 25, разд. 144).
В мерную колбу помещают 167 мл приблизительно 3 н. метанольного
раствора метилата натрия и 40 мл метилового спирта и разбавляют до метки
пиридином. Поскольку раствор жадно поглощает углекислоту из воздуха,
его необходимо хранить в соответствующим образом приспособленной бюрет-
ке (см. рис. 37). Титр раствора устанавливают по бензойной кислоте, раство-
ренной в пиридине, в присутствии 1%-ного пиридинового раствора тимол-
фталеина в качестве индикатора.
О приготовлении стандартных растворов гидроокисей тетраметил- или
метилтрибутиламмония в пиридине см. [369—371].
к. Для приготовления стандартных 0,15 и 0,55 н. растворов метилата
натрия в монометиловом эфире этиленгликоля поступают следующим обра-
зом. 185 мл насыщенного водного раствора едкого натра, свободного от карбо-
ната, смешивают с 430 мл дистиллированной воды и затем с 5400 мл метил-
целлозольва. Не следует забывать, что метилцелозольв на воздухе желтеет.
Таким способом получают примерно 0,55 н. стандартный раствор. Так же
получают и 0,15 н. раствор: смешивают 55 мл насыщенного водного раствора
едкого натра, 55 мл воды и 5400 мл метилцеллозольва. О применении этих
стандартных растворов см. [257, 715, 716] (см. гл. 19, разд. 112).
л. Для непосредственного титрования соединений, содержащих спирто-
вую гидроксильную группу, применяют стандартный раствор алюмо-ди-н-
бутиламида лития в диметиловом эфире этиленгликоля (что удобнее, чем
раствор в тетрагидрофуране [761]). Из-за чувствительности 0,25 н. стандарт-
ного раствора к влаге воздуха при его приготовлении необходима особен-
ная тщательность; хранить раствор следует в атмосфере азота. Методика
заключает в себе возможные источники ошибок, поэтому необходимо озна-
комиться с оригинальной литературой [383, 387, 391, 513, 514].
м. Для приготовления стандартного раствора нитрата серебра в изо-
пропиловом спирте взвешивают с точностью до 1 мг 1,699 г нитрата серебра
и растворяют его в 1000 мл изопропилового спирта, очищенного нитратом
серебра (см. гл. 8, разд. 49, а). Полученный 0,01 н. раствор применяется для
определения тиолов [490] (гл. 28, разд. 160, а).
н. Стандартный раствор цианида калия в изопропиловом спирте. В 200 мл
дистиллированной воды растворяют 3,25 г цианистого калия и разбавляют
в мерной колбе до 1 л изопропиловым спиртом. Приготовленный таким спо-
собом 0,05 н. стандартный раствор пригоден для определения элементарной
серы, например в веществах, применяемых для защиты растений [214] (см.
гл. 23, разд. 158).
160
Глава 11
о. Стандартный раствор роданида {тиоцианата) аммония в ацетоне
употребляется для количественного титрования метил- и фенилхлорсиланов
[796, 797]. Получить 0,3 н. раствор можно следующим образом. В 100 мл
ацетона при нагревании растворяют 22,8 г роданида аммония (перекристал-
лизованного из метилового спирта и высушенного в вакуум-эксикаторе)
и доводят объем до 1000 мл. Титр устанавливают по стандартному раствору
нитрата серебра в присутствии соли трехвалентного железа (см. гл. 31).
п. Стандартный 0,1 н. раствор 2-этилгексаналя в диоксане используют для
фотометрического титрования первичных аминов [518]. Примерно 130 г
2-этилгексаналя (мол. вес 128,4), свежеперегнанного и хранившегося в атмо-
сфере азота, растворяют в диоксане, перегнанном над металлическим натрием
и пропущенном через колонку с активной окисью алюминия. Раствор разбав-
ляют до 1 л. Титр устанавливают по раствору н-бутиламина (см. гл. 25,
разд. 147).
р. Стандартный раствор хлордиизопропоксиалюминия в хлороформе исполь-
зуют для определения алкалоидов. Формула хлордиизопропоксиалюминия
[(RO)2A1C1] *НС1, где R —изопропильная группа.
О приготовлении и применении этого раствора см. в работах [755,
756, 810].
с. Раствор брома в уксусной кислоте. Для получения 0,1 и 0,05 н. раство-
ров берут соответственно 2,5 и 1,25 мл брома и растворяют в 1000 мл уксус-
ной кислоты. Эта операция должна проводиться под хорошо действующей
тягой. Раствор следует хранить на холоду в темных склянках с притертой
стеклянной пробкой. Титр раствора устанавливают либо по водному раство-
ру арсенита натрия, либо тиосульфатным методом (см. гл. 29, разд. 162, а)
[216]. Раствор брома в уксусной кислоте используется также для потенцио-
метрического определения фенольных соединений или для определения сое-
динений, содержащих двойную связь. В первом случае идет реакция заме-
щения бромом, во втором — присоединение брома, как, например, к холесте-
рину [813, 199] (см. гл. 27, разд. 155).
т. Стандартный 0,05 н. раствор ацетата меди(П) в смеси этилового
спирта с пиридином применяется для определения соединений, содержащих
карбонильные группы [107]. В 1000 мл смеси пиридина с этиловым спиртом
(1:1) растворяют 11 г моногидрата ацетата меди(П) и через 3 дня отфильтро-
вывают раствор. Об установлении титра и применении раствора см. гл. 29,
разд. 163.
у. При помощи стандартного раствора церий{1У)аммонийнитрата
в ацетонитриле можно титровать алкилксантогенаты (ксантаты, дитиокарба-
маты). При этом церий(1У) восстанавливается до церий(Ш) [666 а] (см.
гл. 29, разд. 164).
ф. 0,05 М раствор циклопентадиена с этиловом спирте и 0,1 М раствор
тетрацианэтилена в хлористом метилене используются для комплексометри-
ческого титрования диенов и ароматических углеводородов [618, 717].
Для получения стандартного раствора циклопентадиена (уд. вес 0,805
при 19°) деполимеризуют димер циклопентадиена крекингом при 160° и полу-
ченный таким способом мономер перегоняют еще раз. Фракцию, кипящую
при 40J, охлаждают смесью сухого льда с ацетоном; растворяют 0,8 г веще-
ства в 250 мл абсолютного этилового спирта. Титр раствора устанав-
ливают по тетрацианэтилену.
Приготовление стандартного раствора тетрацианэтилена (ТЦЭ). При
слабом нагревании растворяют 640 мг тетрацианэтилена в 40 мл хлористого
метилена. Чтобы довести растворение до конца, добавляют 0,5 мл диметокси-
этана и разбавляют хлористым метиленом до 50 мл. Из-за образования
комплекса ТЦЭ с диметоксиэтаном раствор окрашен в бледно-желтый цвет.
Ежедневно необходимо готовить свежий раствор. Чистоту ТЦЭ можно про-
верить, применяя реакцию Дильса — Альдера с антраценом [618].
Глава 12
ПОТЕНЦИОМЕТРИЧЕСКОЕ ОПРЕДЕЛЕНИЕ
КОНЕЧНОЙ ТОЧКИ
Точка эквивалентности (конечная точка титрования) может быть опреде-
лена либо при помощи индикаторов, либо физико-химическими методами.
К числу последних относятся фотометрическое определение конечной точки
[86, 275—277, 675, 774, а, б, 776], измерение электропроводности [104, 470,
565, 577—580], определение изменения электроемкости раствора и высоко-
частотное титрование [74, 177, 291, 395, 441, 495, 515, 664, 774, а, б, 776,
892], термометрическое титрование * [433, 451, 895], диэлектрический метод,
основанный на различии в диэлектрических проницаемостях кислот, основа-
ний и солей [410], и потенциометрическое титрование.
При построении графика зависимости э. д. с., определенной на подходя-
щем потенциометре, от объема израсходованного стандартного раствора обыч-
но, хотя и не всегда, получают S-образную кривую титрования, причем точка
ее перегиба соответствует точке эквивалентности. Потенциометрические
измерения как в водной, так и в неводных средах производятся одинаковым
способом при помощи одних и тех же приборов. Отличие состоит только
в составе солевого мостика, устанавливаемого между электродом и исследуе-
мым раствором, а при титровании в инертных растворителях имеются разли-
чия в способах экранирования и заземления сосуда, в котором производится
титрование. При проведении кислотно-основного титрования потенциал
индикаторного электрода, погруженного в исследуемый раствор, зависит
от активности ионов водорода в растворе. Электродвижущая сила опреде-
ляется трубчатым потенциометром с большим внутренним сопротивлением.
В качестве используемой аппаратуры можно назвать титри-рН-метр типа
ОР 401/1 (фирма «Radelkis», Будапешт); «Титрископ» типа Е 166 или Е 150А
(фирма «Metrohm», A. G.); потенциограф Е 366 (фирма «Metrohm»); «Титра-
тор» ТТТ1 (фирма «Radiometer», Копенгаген); двойной титратор с прецизион-
ной ячейкой АС (фирма «Precision Scientific Со»); регистрирующий спектро-
электротитратор (фирма «Е. Н. Sargent and Со.»); регистрирующий титратор
(фирма «Е. Н. Sargent and Со»); титратор («Н. Tinsley and Со. Ltd); преци-
зионный автоматический записывающий титратор Доу. Для титрования
в инертных растворителях: модель 31 Кери с нуль-гальванометром (фирма
«Applied Physics Corp.).
61. ОБЛАСТЬ ПРИМЕНЕНИЯ
Кроме применения для серийных макро- и полумикроанализов потенцио-
метрическое титрование используется также (табл. 53) для 1) дифференци-
рующего титрования смесей кислот или оснований, 2) титрования окрашен-
ных растворов, 3) определения исключительно слабых кислот или оснований
в разбавленных растворах (ниже 0,01 н.), 4) определения относительной силы
кислот или оснований, 5) определения эквивалентного веса, 6) оценки пере-
хода окраски индикаторов (рис. 44). Важность решения этой последней зада-
* Термометрический метод, в частности, используется для определения слабых
и очень слабых органических основании, например дифениламина, мочевины, ацет-
амида и ацетанилида, титрованием хлорной кислотой [451].
Таблица 53
Области применения потенциометрического титрования
Растворитель Стандартный раствор Литература
1. Дифференцирующее ти- Ацетон (C2H5)3C4H9NOH 258
трование смесей кислот Пиридин (C4h9)4noh 160
или оснований Метилизобутилкетон (C4h9)4noh 105
Этиленгликоль — ацетон Пиперидин 170
Метилизобутилкетон НС1О4 105
Ацетонитрил НС1О4 247
Уксусная кислота—1,4-ди- НС1О4 401
оксан—нитрометан
2. Титрование окрашенных Бензол — изопропиловый кон 530, 665
растворов спирт
Петролейный эфир (низко- кон 530, 665
кипящий) — изопропило-
вый спирт
3. Микротитрованпе очень ЭДА, ДМФА (C4H9)4NOH 176, 359
слабых кислот Бензол, СН3ОН, ДМФА, CH3ONa 562
пиридин
4. Измерение относительной Ацетонитрил, метплэтплке- (C4H9)4NOH 595, 780
силы кислот или осно- тон, пиридин
ваний Ацетонитрил НС1О4 333
Нитрометан НС1О4 779
Уксусный ангидрид нсю4 778
Рис. 44. Потенциометрическое титрование и изменение окраски индикаторов.
а— изменение окраски метилового фиолетового в (А) уксусной и (Р) пропионовой кислотах [375]: 1 —
желтая; 2—зелено-желтая; 3—желто-зеленая; 4—зеленая; 5—зелено-синяя; 6 — синяя; 7-—фио-
летово-синяя. б — изменение окраски метиленового синего — хинальдинового красного в смеси нитро-
метан — бензол — муравьиная кислота (100 : 10 : 2) [890]: 8 — зеленая; 9 — зеленовато-синяя; ю —
синяя; 11 — пурпурно-синяя; 12—пурпурно-красная, в—изменение окраски метилового красного
в смеси ацетонитрил — хлороформ—фенол (100:20:10): 13— красно-фиолетовая; 14— розовая;
15 — ярко-розовая; 16— розовато-оранжевая [123]. г — изменение окраски метилового красного
в смеси гексан — ацетон (2 : 1): 17 — фиолетово-розовая; 18 — оранжево-розовая; 19 — оранжевая.
Потенциометрическое определение конечной точки
163
чи ясна хотя бы из следующего примера. При титровании ацетата натрия
хлорной кислотой в среде уксусной кислоты точке эквивалентности соответ-
ствует зелено-синяя окраска метилового фиолетового, в то время как при
титровании в среде пропионовой кислоты точке эквивалентности отвечает
зеленая окраска (рис. 44, а). При применении индикаторов, изменение
окраски которых проходит через различные оттенки, для установления
правильного оттенка должна быть проконтролирована область, лежащая
вблизи (на 5% до и после) точки эквивалентности. Это особенно важно в тех
случаях, когда титруют соединения, ранее не подвергавшиеся анализу.
Большое количество таких индикаторов применяется и для неводного титро-
вания; это метиловый фиолетовый, кристаллический фиолетовый, метиловый
красный и т. д. (рис. 44, б). На оттенки окраски индикатора влияет также
состав растворителя (рис. 44, виг).
62. УСЛОВИЯ, ОБЕСПЕЧИВАЮЩИЕ ТОЧНОСТЬ ИЗМЕРЕНИЙ
Для точных измерений необходимо, чтобы разность между потенциалом
точки эквивалентности и потенциалом полунейтрализации (потенциал в точке,
в которой израсходована половина раствора) составляла 200—300 мв!мл.
Таблица 54
Аналоги кислот Изменение потен- циала, мв Аналоги кислот Изменение потен- циала, мв
титрова- ние CH3OLi титрова- ние ТБАГ титрова- ние СН3ОЫ титрова- ние ТБАГ
Резорцин 10 75 Пропил-п-оксибензоат 50 300
Эвгенол 15 100 Пикриновая кислота 1200 1500
Фенол 25 125
Эта разность потенциалов меняется в зависимости от силы анализируемой
кислоты или основания, от природы растворителя и стандартного раствора.
Так, например, при титровании аналогов кислот 0,1 н. раствором метилата
лития или гидроокиси тетрабутиламмо-
ния (ТБАГ) в среде диметилформамида
вблизи точки эквивалентности был опре-
делен очень широкий ряд скачков по-
тенциала (табл. 54) [7].
Приведенные в таблице результаты
показывают значительное различие
в силе аналогов кислот и свидетельст-
вуют, что даже для такой слабой кисло-
ты, как резорцин, можно провести из-
мерение, подобрав подходящий титрант.
Рис. 45. Прибор для потенциометрического
титрования в инертных растворителях [358].
1 — бакелитовая крышка; 2 — латунный экранирую-
щий стакан; 3 — стеклянный электрод; 4 — цилинд-
рический стакан объемом 250 мл\ 5 — магнитная
мешалка; в — соединение стеклянного электрода
с нуль-гальванометром; 7 — стенка вытяжного шка-
фа; 8 — каломельный электрод.
Самый низкий приемлемый предел при измерении скачка потенциала состав-
ляет 75 мв!мл. При титровании оснований средней силы 0,01 н. раствором
хлорной кислоты в смеси пропионовой кислоты с четыреххлористым углеро-
164
Глава 12
дом можно отметить вблизи точки эквивалентности изменение потенциала
порядка 500—1000 мв!мл [311, 314].
При проведении потенциометрического титрования необходимо знать
характеристический диапазон потенциалов растворителя, ограничивающий
изменение напряжения от начала до конца титрования, а также необходимо
обеспечить постоянство температуры вовремя титрования (±0,5°), подыскать
соответствующую систему электродов, что в большинстве случаев делается
эмпирически, необходимо избегать влияния флуктуаций потенциала жидко-
стного соединения, а также попадания в систему влаги и углекислоты из
воздуха. При работе с инертными растворителями необходимо тщательно
экранировать и заземлить сосуд, в котором производится титрование, и при-
менять специальный потенциометр, чтобы избежать ошибок из-за возможной
перезарядки и блуждающих токов [358] (рис. 45). Однако даже при выпол-
нении этих всех требований точность потенциометрического титрования не
всегда превосходит точность визуального титрования.
63. ВЛИЯНИЕ РАСТВОРИТЕЛЯ
Рассмотрим факторы, влияющие на потенциал полунейтрализации (ППН)
кислот и оснований в различных растворителях [333, 369—371, 778,
780, 782].
а. Кислотность или основность растворителя. Измерения можно про-
изводить лишь при условии, что способность катиона растворителя (sH*)
отдавать протон меньше, чем у анализируемой кислоты, и что основность
аниона растворителя ($“) остается ниже основности анализируемого основа-
ния. При значениях кислотности или основности выше некоторой величины
ППН зависит лишь соответственно от силы и (эффект выравнивания)
и от концентрации растворенного соединения. Чем сильнее растворитель
связывает протоны растворенной кислоты, тем труднее будет происходить
отдача этих протонов, скажем, электроду. Поэтому, хотя в растворе слабой
кислоты в сильноосновном растворителе количество сольватированных про-
тонов возрастает, этому противодействует уменьшение протонодонорной
способности слабой катионной кислоты, образованной молекулами сильно-
основного растворителя. Например:
[C9HeNO]H + H2N(CH2)2NH2 H2N(CII2)2NII+ + CgHeNO-
Оксихинолин, Этилендиамин,
слабая кислота сильное основание
Протонизированный
этилендиамин,
слабая кислота
Анион оксихи-
нолина, силь-
ное основание
H2N(CII2)2NII+ H2N(CH2)2NH2 + H+,
а также
[CH3C6H4SO3]H + СН3СОСН3
п-Толуолсульфо-
кислота, сильная
кислота
Ацетон,
слабое
основание
[СН3СОСН3]Н+ -{-
Протонизирован-
ный ацетон,
спльная кислота
ch3c6h4so-
Анпон толуол-
сульфокислоты,
слабое основание
[СН3СОСН3]Н+ СН3СОСН3 + Н+
б. В тех случаях, когда используется растворитель дифференцирующего
действия, ППН в первую очередь определяется собственной кислотностью
или основностью кислоты или основания. Примером могут служить ацето-
нитрил, ацетон, метилизобутилкетон, пиридин, уксусный ангидрид и т. д.
Однако, если в результате нейтрализации образуется очень устойчивая соль
(нерастворимая или слабо диссоциирующая), ППН зависит либо от раствори-
мости вещества, либо от его константы ионизации.
в. На силу кислоты и основания, как и на ППН, влияет диэлектрическая
проницаемость растворителя, однако механизм этого влияния еще не
выяснен.
Потенциометрическое определение конечной точки
165
В общем случае ППН катионных кислот не зависит от диэлектрической
проницаемости растворителя, тогда как ППН других кислот вследствие их
взаимодействия с растворителем не подчиняется никакой закономерности
[186] (см. рис. 5).
г. Нерегулярность потенциометрических кривых, вызванная ассоциацией.
В инертных растворителях, например толуоле, кислоты и фенолы ассоцииро-
ваны за счет образования водородных связей.
В слабополярных или основных растворителях эта ассоциация либо
совсем не имеет места, либо проявляется незначительно. Во время титрова-
ния, однако, образуется сопряженное основание кислоты, т. е. анион, срод-
ство которого к протону значительно выше
RCOO->RCOOH.
Таким образом образуются комплексы кислота—анион кислоты, более
стабильные, чем димеры кислот [35, 447, 559, б]
о ... н------о
Образование таких комплексов в инертной среде часто подтверждается дан-
ными кондуктометрических [104, 559, а], криоскопических [447], а также
ИК-спектроскопических исследований [33].
Аналогично ведут себя фенолы в пиридине [369] и толуоле [358]. В про-
цессе потенциометрического титрования монофункциональных кислот и фено-
лов 1,5 н. раствором гидроокиси тетрабутиламмония в изопропиловом спирте
иногда удается заметить два перегиба на кривой; это показывает, что тит-
руются две кислоты разной силы. Когда первый перегиб появляется в сред-
ней точке титрования, это говорит об образовании комплекса 1:1.
(С6Н5О)- ... НОС6Н5.
Образование комплекса фенолят-анион — фенол за счет водородной связи
видоизменяет нормальную реакцию нейтрализации фенола (например, мети-
лат- или изопропилат-анионами) следующим образом [358]:
1. Титрование фенолов изопропилат-анионами [стандартный раствор
(G4H9)4NOH в изопропиловом спирте в действительности состоит из смеси
((G4H9)4N)+OH- и ((G4H9)4N)+(GH3)2GHO-]
СвН5ОН + (СНз)2СН —О- СвН5О- + (СНз)2СНОН.
2. Образование комплекса фенол — фенолят
С6Н5О-4-С6Н5ОН (С6Н5О- ... Н-О-С6Н5).
3. Титрование комплекса фенол—фенолят
(С6Н5О-... Н - О - СвН5) + (СН3)2СН - О- 2СвН5О- + (СН3)2СНОН.
Комплекс фенол — фенолят-анион значительно стабильнее, чем димер фенола,
и увеличивает кажущуюся кислотность фенола. В средней точке титрования,
когда количество свободного фенола невелико и концентрация Н+ значитель-
но уменьшается, начинается титрование водорода комплекса. К этому вре-
мени кислая форма уже стабилизована за счет образования комплекса, при-
чем кислотность комплекса ниже, чем у свободного фенола. Хотя катион
тетрабутиламмония и не обозначен в приведенной схеме, очень вероятно,
что в инертных растворителях он в основном присутствует в виде ионных
пар [104], т. е.
(С6Н5ОН ... OCeH5)-((C4H9)4N)+ п (CeH5O)-((C4H9)4N)+.
166
Глава 12
Комплекс кислоты с ее анионом за счет водородной связи не получается
в тех случаях, когда растворитель сам образует водородные связи с раство-
ренным веществом. Поэтому при титровании фенола в толуоле 0,1 н. раство-
ром (а не 1,5 н.) гидроокиси тетрабутиламмония в изопропиловом спирте
избыточное количество изопропилового спирта (~1%), вносимое с более
разбавленным раствором, препятствует образованию комплекса фенол —
фено лят-аниона.
Титрование о- и ле-нитрофенолов подтверждает присутствие комплексов
с водородной связью [358]. Так, в случае о-нитрофенола образуются внутри-
молекулярные, а в случае ле-нитрофенола — межмолекулярные водородные
связи [628]
О-Нитрофенол л-Нитрофенол
Это также влияет на образование комплекса кислота — анион кислоты.
На кривой титрования о-нитрофенола наблюдается один перегиб, а на кривой
титрования лг-нитрофенола — два перегиба (первый — в средней точке
титрования).
Для фенолов комплексы кислота — анион кислоты менее прочны, если
в ортпо-положении находятся алкильные заместители, поэтому перегиб
в средней точке титрования становится тем менее заметным, чем больше
возрастают пространственные препятствия. Ряд стабильности комплексов
фенол — фенолят-анион имеет такой вид [358]:
фенол > 4-метил-2-7претп-бутилфенол > б-метил-2-тпреш-бутилфенол >
2,4,6-три(тпрепг-бутил)фенол
Аналогично ведут себя в толуольном растворе и двухатомные фенолы
[770]; при благоприятной ориентации одна из гидроксильных групп может
приобрести более кислый характер, в то время как другая может стать
настолько слабой, что ее нельзя оттитровать.
Форма нерегулярных потенциометрических кривых может быть самой
различной. На четырех кривых, представленных [369] на рис. 46, показано:
1) дополнительное повышение потенциала в точке полунейтрализации
(рис. 46, а);
2) асимметричное плато («горб» [369], рис. 46, б);
3) невоспроизводимость потенциометрических кривых (рис. 46, в);
4) внезапное падение потенциала при 25, 50 или 75 %-ной нейтрализации
(рис. 46, г). Сильнополярные анионы недостаточно экранируются инертным
или слабоосновным растворителем в процессе титрования, и поэтому при
их титровании образуются комплексы кислота — анион кислоты или фенол —
фенолят-анион, а также димерные молекулы, что приводит к нарушениям
формы кривых типа 1 и 2 [369].
Потенциометрическое определение конечной точки
167
Полярные молекулы в инертных растворителях ориентированы под дей-
ствием поверхности электрода и частично ею адсорбированы из-за малой
сольватирующей способности растворителя; так возникают аномалии типа 3
и 4, хотя они могут появиться и за счет влияния ионов калия [355]. Чем
слабее титруемая кислота, тем более основным должен быть растворитель,
чтобы молекулы кислоты ассоциировались или сольватировались не аниона-
ми кислоты, а молекулами растворителя. Карбоновые кислоты средней силы
дают нерегулярные кривые титрования в растворителях с основностью ниже,
чем у пиридина. Благодаря хорошей сольватирующей способности амфи-
протонных растворителей достаточно добавить 1% метилового или изопро-
пилового спиртов, чтобы предотвратить образование комплекса растворенной
кислоты с ее собственным анионом. 4-Метил-2,6-дибутилфенол не ассоции-
руется с собственным анионом из-за пространственных препятствий. Поэтому
его кривая титрования имеет обычный вид, в то время как кривая титрова-
ния 4-метил-2-бутилфенола имеет неожиданный подъем в точке полунейтра-
лизации. Тиофенол также дает нормальную S-образную кривую, поскольку
сера, как это хорошо известно, обладает меньшей тенденцией к образованию
водородных связей, чем кислород.
•Л7Л
Рис. 46. Нерегулярные потенциометрические кривые, полученные Хейде [369].
а — титрование уксусной кислоты в ацетоне стандартным раствором СНЯ(С4Н,)ЯЫОН в пиридине,
система электродов: стекло — каломель; б — титрование уксусной килоты в ацетонитриле (проводится
так же, как в случае а); в — титрование муравьиной кислоты в ацетоне стандартным раствором
(CH,)4NOH в пиридине, система электродов: стекло — каломель) г — титрование (А) фенола в пири-
дине, (Б) цетилфенола в бутиламине, система электродов: стекло — каломель, и (В) бензойной
кислоты в ацетонитриле, система электродов: Sb — каломель.
168
Глава 12
Опыт показывает, что при потенциометрическом титровании в раствори-
телях, которые сами дают водородные связи, не образуется ассоциатов,
искажающих форму потенциометрических кривых. В связи с отсутствием
таких межмолекулярных ассоциатов можно было бы проводить дифферен-
цирующее титрование слабых кислот в метиловом или изопропиловом спирте,
однако собственная кислотность этих растворителей не способствует успеш-
ному проведению анализа. тпретп-Бутиловый спирт (триметилкарбинол,
2-метилпропанол-2) характеризуется меньшей кислотностью, чем названные
выше простые спирты (см. табл. 10), и поэтому может применяться как для
потенциометрического титрования кислот и фенолов, так и для дифферен-
цирующего анализа смеси кислот и фенолов [253]. Из сильных кислот —
хлорная кислота (но не соляная или серная) дает очень стабильные растворы
в пгретп-бутиловом спирте, не обна-
Степень нейтрализации, %
Рис. 47. Потенциометрическое титро-
вание п-нитрофенола [258, 253].
А — в ацетоне 0,1 н. гидроокисью триэтил-
бутиламмония и Б — в mpem-бутиловом
спирте 0,116 н. гидроокисью тетрабутиламмо-
ния в смеси бензол — изопропиловый спирт.
руживающие признаков разложения
в течение неограниченного времени.
Гидроокись тетрабутиламмония устой-
чива в течение короткого времени,
поэтому избыток основания, введенный
в процессе титрования со стандартным
раствором, дает стабильный потенциал.
Благодаря этому тпрепг-бутиловый спирт
имеет преимущество по сравнению с пи-
ридином или ацетоном, так как в этих
растворителях избыток основания часто
приводит к флуктуациям потенциала.
К тому же одно характерное свойство
тпретп-бутилового спирта делает его
удобным для дифференцированного тит-
рования фенолов и нитрофенолов: при
потенциометрическом титровании боль-
шинства карбоновых кислот и фенолов
потенциометрическая кривая в области
от 0 до 9596 нейтрализации должна пройти очень большой диапазон зна-
чений потенциала. Это связано с образованием комплексов кислота—
анион кислоты [369], и в связи с их неустойчивостью во многих раст-
ворителях часто не удается провести дифференцирующего титрования.
При применении пгретп-бутилового спирта этот диапазон изменения потен-
циала меньше, поэтому в пределах значений потенциала, зависящих от при-
роды растворителя, может оказаться большее число скачков потенциала.
На рис. 47 представлены кривые титрования n-нитрофенола в ацетоне
и в тпретп-бутиловом спирте. В тпрепг-бутиловом спирте можно проанализи-
ровать, например, смесь пикриновой кислоты, 2,4-динитрофенола, о-нитро-
фенола и фенола [253].
Из-за разложения гидроокиси тетрабутиламмония в пиридине при титро-
вании этим раствором более слабых кислот, чем фенол (например, о-этилфе-
нола), результаты измерения составляют 102—104% от теоретического
значения, в то время как титрование в третп-бутиловом спирте дает 99,7%
[253]. На упомянутые выше отклонения при потенциометрическом титрова-
нии обычно не обращают внимания, если не строят графика от начала до
конца измерений, а делают лишь ограниченное число измерений до и после
ожидаемой точки эквивалентности. Необычный вид потенциометрических
кривых не свидетельствует о том, что титриметрические измерения нельзя
провести или что количественные результаты невоспроизводимы.
д. Требования к растворителям и титрантам. Стандартный раствор
должен обладать высокой основностью или кислотностью. В данном раство-
Потенциометрическое определение конечной точки
169
рителе он не должен образовывать с определенным веществом нерастворимых
солей или стабильных комплексов и не должен отрицательно влиять на рабо-
ту электродов. Итак, для титрования оснований удобнее всего использовать
хлорную кислоту. Алкоголяты щелочных металлов менее пригодны для
титрования кислот в растворителях с низкой диэлектрической прони-
цаемостью, так как здесь велика опасность образования осадков; кроме
того, высокоподвижные ионы К+, Na+ или Li+ влияют на режим работы чув-
ствительного стеклянного электрода (см. рис. 43).
При проведении особо точных анализов рекомендуется растворять
в одном и том же растворителе как титрант, так и определяемое вещество,
иначе во время титрования могут измениться диэлектрические, кислотные
или основные свойства растворителя.
На основании всего сказанного можно сделать вывод, что потенциометри-
ческие определения кислот, в частности микроопределения, представляют
значительно большие трудности, чем титрование оснований.
64. ДИАПАЗОН ПОТЕНЦИАЛОВ РАСТВОРИТЕЛЕЙ
Термин «кислотность» или «основность» в применении к растворителям,
употребляемым для потенциометрических определений, имеет особый смысл.
Значения, полученные при взаимном титровании 0,01 н. сильных кислот и
оснований, дают эмпирический ряд,
Р и с. 48. Диапазон потенциалов раст-
ворителя [369—371].
7 — этилендиамин; 2 — н-бутиламин; 3 — пири-
дин; 4 — диметилформамид; 5 — вода; 6 — изо-
пропиловый спирт; 7 — метиловый спирт; 8 —
ацетонитрил; 9 — ацетон; ю — хлорбензол;
77 — уксусная кислота.
Р п с. 49. Выбор растворителей
[369—371].
7 — участок кривой потенциометрического
титрования вблизи точки полунейтрализа-
ции; 2 — диапазон потенциалов раствори-
теля основного характера; з — диапазон
потенциалов кислотного растворителя; 4 —
диапазон потенциалов апротонного раст-
ворителя.
обладают ни большой «кислотностью», ни большой «основностью» по срав-
нению с инертными или дифференцирующими растворителями. Потенциалы
полунейтрализации различных соединений можно сравнивать лишь при
строго определенной концентрации. Например, ППН сильной кислоты
в пиридине меняется от —1100 до —200 мв при изменении концентрации 'от
0,005 до 0,5 н. .
170
Глава 12
65. ВЫБОР РАСТВОРИТЕЛЕЙ
Для кислот и оснований ППН отражают в определенных пределах их
собственную кислотность или основность (см. гл. 2, разд. 16, в, и гл. 3,
разд. 23, б—д). Измерение ППН, однако, ограничено диапазоном потенциа-
лов, достижимых в растворителе (рис. 49). На рис. 49 приведены три возмож-
ных случая: 1) наинизшее значение потенциала растворителя выше ППН
анализируемых кислоты или основания, в этом случае сила кислот увеличе-
на и выравнена, в то время как основания не поддаются титрованию
(рис. 49, В); 2) наивысшее значение потенциала растворителя ниже потен-
циала полунейтрализации анализируемых кислоты или основания, в этом
случае сила оснований увеличена и выравнена, а кислоты титровать невоз-
можно (рис. 49, в); 3) ППН определяемой кислоты или основания лежит
в пределах диапазона потенциала растворителя, в этом случае проявляется
собственная сила кислоты или основания, и их титрование становится выпол-
нимым (рис. 49, г).
Для незаряженных протонных кислот и катионных кислот типа R3NH+,
величина рК для которых в воде меньше 2, в качестве растворителей пригод-
ны изопропиловый спирт, кетоны и ацетонитрил, а в качестве стандартных
растворов — едкий натр или гидроокись тетраалкиламмония в изопропило-
вом спирте. Для растворения незаряженных протонных кислот и кислот,
имеющих положительный или отрицательный заряд, а также кислот Льюиса
со значением рК в водных растворах между 2 и 8, могут употребляться диме-
тилформамид, пиридин, инертные растворители или смеси этих веществ,
а в качестве стандартных растворов, кроме едкого натра и гидроокиси тетра-
алкиламмония в изопропиловом спирте, растворы гидроокиси тетрабутил-
аммония в смеси бензола с метиловым спиртом (гл. 6, разд. 40, 41 и табл. 38).
Для кислот со значением рК в воде больше 8 в качестве растворителя могут
быть использованы н-бутиламин, этилендиамин или смеси растворителей,
а в качестве стандартного раствора — гидроокись тетраалкиламмония
в смеси бензол — метиловый спирт или в пиридине (см. гл. 10, разд. 57,6).
Для ориентировки в случае льюисовских кислот используют константу
диссоциации соответствующей протонной кислоты, например А^о2 ~ ^h2so3«
При потенциометрическом титровании сильных оснований и оснований
средней силы (гл. 6, разд. 42) применяются уксусная или пропионовая кисло-
та в качестве растворителя и 0,1—0,001 н. раствор хлорной кислоты в уксус-
ной кислоте в качестве стандартного раствора. В случае слабых оснований
уксусную или пропионовую кислоту смешивают с хлорбензолом или четы-
реххлористым углеродом, а в случае третичных аминов — с уксусным анги-
дридом; в качестве стандартного раствора используют 0,01 н. раствор хлор-
ной кислоты в смеси уксусная кислота — четыреххлористый углерод (гл. 6,
разд. 39, 41 и табл. 14).
66. ИСПОЛЬЗОВАНИЕ ФОНОВЫХ ЭЛЕКТРОЛИТОВ
Чтобы увеличить электропроводность инертного растворителя с низкой
диэлектрической проницаемостью, добавляют фоновый электролит (или
раствор соли). Для этой цели пригодны, например, GH3(G4H9)3NI в концен-
трации 1,5 «10-3 молъ!л, а также хлорид лития, 200 мг которого растворяют
в 50 мл смеси бензола с метиловым спиртом (10 : 1), используя магнитную
мешалку [153, 252, 665]; можно использовать также раствор ацетата меди(П)
в уксусной кислоте. При титровании солянокислых солей оснований употре-
бляют ацетат ртути(П), так как он, во-первых, повышает электропровод-
ность, а во-вторых, выделяет свободное основание из хлоргидрата [369—371,
207, 462, 644]. Когда величина рК определяемой кислоты больше 2, в каче-
стве фонового электролита можно использовать иодид тетраалкиламмония.
Потенциометрическое определение конечной точки
171
Однако если рК кислоты меньше 2, в нейтральных растворителях в процессе
реакции образуются HI и затем иод. В таких случаях необходимо использо-
вать перхлораты или n-толуолсульфонаты оснований (например, перхлорат
диэтиланилина [192]). Если потенциометрические измерения производятся
лишь вблизи точки эквивалентности, образовавшийся продукт нейтрализа-
ции сам может выполнять роль фонового электролита, как, например, пер-
хлорат амина, но при условии, что соль не выпадает в виде нерастворимого
осадка.
67. ЭЛЕКТРОДЫ
а. Индикаторные электроды. При проведении неводного титрования
в качестве индикаторного электрода обычно применяется стеклянный элек-
трод. Его можно применять для титрования оснований и их солей с галоге-
новодородными кислотами в среде уксусной кислоты [11, 60, 208, 244, 247—
249, 450, 548, 459, 643—645, 732, 742, 769, 854] и в смесях уксусной
кислоты с четыреххлористым углеродом или уксусной кислоты с хлорофор-
мом, а также в смесях пропионовой кислоты с пропионовым ангидридом
и хлорбензолом [314], уксусной кислоты с хлорбензолом [175], уксусной
кислоты с толуолом [599] и т. д. Стеклянные электроды могут использоваться
не только в кислых растворителях, но и в некоторых дифференцирующих
и основных растворителях: ацетоне [124, 258, 746], ацетонитриле [247, 248,
155, 539], метилизобутилкетоне [105, 359], пиридине [159, 161, 162, 780,
782], нитрометане [779], диметилформамиде [58, 176], смеси бензола с мети-
ловым спиртом [58, 783], а также в G — Н-системах [621, 788] и др.
В неводных средах и особенно в инертных растворителях стеклянные
электроды даже одного и того же образца могут проявлять разные свойства.
Эффективность выбранных электродов не всегда удается регулировать или
улучшить, вводя вспомогательные электролиты. Свойства электрода сильно
зависят от его предварительной обработки. Но, конечно, невозможно знать
предысторию каждого стеклянного электрода. В большинстве случаев, одна-
ко, электрод перед употреблением оставляют «набухать» на 12—48 час
в растворителе, в котором предполагается его использовать [894]. Эта про-
цедура также необходима для проведения относительных потенциометриче-
ских измерений. Все это затрудняет выбор подходящего растворителя;
трудности особенно возрастают, если, следуя методике, необходимо заменить
растворитель в процессе выполнения титрования. Так, может случиться, что
стеклянный электрод, предварительно находившийся в уксусном ангидриде,
не будет работать безошибочно в среде уксусной кислоты. При титровании
хлорной кислотой в уксусной кислоте часто достаточно предварительно
погрузить стеклянный электрод на 1—2 час в смесь уксусного ангидрида
с уксусной кислотой (1 : 10). После употребления стеклянный электрод спо-
ласкивают вначале чистым растворителем, затем метиловым спиртом и водой
и хранят его в дистиллированной воде.
Как указывает Цейдлер [894], даже небольшой избыток хлорной кислоты
оказывает настолько вредное действие на стеклянный электрод, что при
последующих определениях скачок потенциала оказывается либо сглажен-
ным, либо вообще не проявляется. В то же время замечено, что чувствитель-
ность стеклянного электрода возрастает, если перед титрованием хлорной
кислотой электрод выдерживают в смеси уксусной кислоты с уксусным анги-
дридом (10 : 1), к которой добавлены 1—2 капли хлорной кислоты [314].
Такое противоречие в данных привело к необходимости применения других
типов индикаторных электродов и новых приемов работы. Тем не менее
и сейчас для потенциометрического титрования обычно используются
стеклянные электроды, потому что те образцы стеклянных электродов, кото-
рые обладают большой чувствительностью, работают без ошибок и иногда
172
Глава 12
могут служить в течение ряда лет. На практике невозможно получить стек-
лянный электрод, поверхностный слой которого не содержал бы некоторого
количества воды в геле (даже в процессе изготовления стеклянный электрод
находится в контакте с парами воды, как, например, в пламени газовой
горелки), поэтому возможно, что даже в безводной среде определение потен-
циала возможно именно из-за наличия содержащего воду гелеобразного
поверхностного слоя (см. стр. 79 в работе [725]).
Бадоз-Ламблинг и сотр. [27] для кислотно-основного титрования в неводной среде
применяли стеклянные электроды, наполненные органическим растворителем, например
ацетонитрилом. Как следует из проведенных ими сравнительных опытов, при применении
стеклянных электродов, заполненных ацетонитрилом, постоянный потенциал получается
скорее и воспроизводимость составляет ±10 мв, в то время как при применении «водо-
содержащего» стеклянного электрода воспроизводимость составляет лишь ±30 мв.
Стеклянные электроды замедленного действия. При неводном титровании
бывают затруднения из-за того, что в жидкостном соединении электрода
сравнения (например, каломельного) между насыщенным водным раствором
Рис. 50. Ка-
пиллярный эле-
ктрод.
Р и с. 51. Прибор для титрования с задержи-
вающим стеклянным электродом.
1 — простой стеклянный электрод; 2 — задерживающий
стеклянный электрод.
хлорида калия и органическим растворителем вследствие диффузии возника-
ют неустойчивые потенциалы. Чтобы обойти это затруднение, применяют
большое количество систем электродов, среди которых необходимо упомянуть
о стеклянном электроде замедленного действия (электроде капиллярного
типа) (рис. 50 [420, 569]), о биметаллической паре электродов, о каломель-
ном электроде, составленном с использованием хлоридов калия, лития, тетра-
бутиламмония и органического растворителя.
Стеклянные электроды замедленного действия могут использоваться для
так называемого непосредственного дифференцирующего титрования [444,
457] (не путать с титрованием в растворителях, обладающих дифференци-
рующими свойствами). Один из двух идентичных стеклянных электродов
помещается в трубку с узким отверстием. Когда электрод вставлен в трубку,
она может вместить примерно 1 мл жидкости. Если использовать в качестве
электрода сравнения стеклянный электрод замедленного действия, можно
Потенциометрическое определение конечной точки 173
избежать флуктуации диффузионного потенциала. Однако измерения при
этом проводятся медленнее, чем при обычном потенциометрическом титрова-
нии, и сопротивление электродной системы составляет около 500 Мом, так
что электроды и сосуд для титрования после чистки и высушивания должны
обрабатываться силиконом (например, силиконом марки «Desicote R» фирмы
«Beckman Instruments») с последующим электростатическим экранированием
и заземлением (рис. 51).
Разность э. д. с. (АЕ) измеряется после добавления каждых 0,1—0,25 мл
(ДУ) стандартного раствора. После каждого измерения потенциала жидкость
вытесняется при помощи резинового баллончика и титрование продолжается
до тех пор, пока не будет достигнуто наивысшее значение АЕ. Точка эквива-
лентности может быть определена затем либо графически, либо расчетом.
В обоих случаях конечная точка будет соответствовать максимуму значения
&E/&V или же моменту, когда величина №EI&V2 превратится в нуль
(табл. 55).
Таблица 55
AV, мл ДЕ, мв AE/AV A2E/AV2 Расчет
5,00 250 5,20 + 0,1 / 72 \ „„„
13 ( 72 + 45 )-5’262-“Л
5,10 263 28 +15 или
5,20 291 +72 5,20 + 0,1 / 100—28 \ _
100 \ 100—28+100—55 /
5,30 391 55 —45 = 5,262 мл
5,40 446 —33
22
5,50 468 10 —12
5,60 478
Приведенный метод расчета можно применять к обычному потенциометри-
ческому титрованию и для любой системы электродов. Присутствие раствора
около стеклянного электрода замедленного действия вызывает лишь незна-
чительную ошибку в измерениях (примерно 0,03%) [444].
Различные графические методы и способы расчета критически рассматриваются
в работах [237, 517, 588].
При потенциометрическом титровании нет необходимости прибавлять
стандартный раствор порциями по 0,1 мл. После предварительных проб
будет достаточно 3—4 отсчетов до и после точки эквивалентности. Другой
способ заключается в том, что добавляют такие количества стандартного
раствора, которые изменяют потенциал на 30 мв. При этом объем порций
стандартного растворителя уменьшается постепенно вблизи точки эквива-
лентности [258].
Потенциал индикаторного электрода ртуть — ацетат ртути(1) в уксус-
ной кислоте определяется концентрацией ионов СН3СООН2 и СН3СОО“
так же, как потенциал стеклянного электрода определяется Н+-ионами. Та-
ким образом, оба электрода, хотя и отличаются по устройству, служат одной
и той же цели [713]. Электрод Hg/Hg2(CH3COO)2 представляет собой золотую
проволоку, на одном конце которой электролитическим способом создан
внутренний слой металлической ртути в форме капельки и внешний слой
ацетата ртути(1). Конструкция этого электрода дана в работе [713].
К индикаторным электродам также могут быть причислены некоторые
металлические электроды.
174
Глава 12
б. Металлические электроды во многих случаях пригодны для измерения
относительного изменения потенциала. При этом обычно можно обойтись
без добавления хингидрона или хлоранила в качестве окислительно-восста-
новительной системы. В качестве индикаторных электродов пригодны плати-
на, золото, сурьма, висмут [894], молибден [56], платина с 10% родия [540]
и т. д. Согласно Цейдлеру [894], для потенциометрического определения
Рис. 52. Прибор для титрования с систе-
мой электродов платина — платина [359].
1 — отвод для соединения с сосудом с раствори-
телем; 2 — трубка для подачи растворителя;
3 — трубка для подачи титранта; 4 — внутрен-
няя поверхность камеры; 5 — отвод к подающему
устройству титратора; 6 — проволочный платино-
вый электрод сравнения; 7 — стенка камеры;
8 — внешняя поверхность камеры; 9 — магнит-
ная мешалка; 10 — платиновый индикаторный
электрод; 11 — держатель электродов (резина).
Рис. 53. Прибор для титрования
с системой электродов сурьма — сурьма
[601].
1 — электрод сравнения; 2 — индикаторный
электрод; 3 — магнитная мешалка.
оснований в метиловом, этиловом, пропиловом и бутиловом спиртах, ацето-
нитриле, уксусной кислоте и уксусном ангидриде наиболее удобен золотой
электрод с поверхностью 1 см2.
Можно также использовать платиновый индикаторный электрод в среде
этилендиамина, как, например, при титровании фенолов [285], применяя
в качестве стандартного раствора аминоэтилат натрия в смеси аминоэтанола
с этилендиамином [446]. Гарлоу и сотр. [359] рекомендуют для титрования
фенолов в этилендиамине использовать платиновый электрод с предвари-
тельной анодной поляризацией (рис. 52). Нормальная работа платинового
индикаторного электрода зависит от величины его поверхности и ее предва-
рительной обработки. Предпочтительнее заранее обрабатывать поверхность
электрода, и эта обработка должна повторяться перед каждым определением.
Даже при такой обработке нельзя избежать старения электрода; когда это
происходит, электрод следует оставить на 3—4 час в водной соляной кисло-
те (1 : 1). Анодную поляризацию производят в 1%-ной серной кислоте при
потенциале Зев течение 1 мин. Хотя роль этой поляризации еще не совсем
ясна, возможно, что поляризованный электрод функционирует как кисло-
родный электрод. Применяя предварительно аноднополяризованный плати-
новый индикаторный электрод, можно получить скачок потенциала в среде
этилендиамина в точке эквивалентности втрое больший, чем со стеклянным
электродом. При выполнении потенциометрического титрования в этилен-
диамине предварительно поляризованный платиновый электрод следует
оставить в растворе на 2—3 мин, прежде чем добавлять стандартный раствор.
Потенциометрическое определение конечной точки
175
Описание способов употребления различных биметаллических электрод-
ных систем в среде метилового спирта и уксусной кислоты можно найти
в литературе (см., например, [615]). Сурьмяные электроды можно исполь-
зовать в качестве индикаторных электродов либо в сильноосновных раство-
рителях, либо в смеси бензол — метиловый спирт [252, 444, 601, 665].
О методах приготовления и сохранения сурьмяных электродов см. в работе
[329, 860].
На результаты титрования, полученные при использовании сурьмяных электродов,
не всегда можно положиться [7, 285, 770] даже в тех случаях, когда и индикаторный элек-
трод, и электрод сравнения оба сурьмяные (см. [601]).
В качестве электрода сравнения можно использовать любой электрод,
если постоянна активность ионов раствора, находящегося в контакте
Р п с. 54. Ртутный
«J-образный» элект-
род [см. Schmidt,
Chemist-Analyst, 51,
56 (1962)].
1 — ртуть в капилляре;
2 — чашечка со ртутью;
3 — платиновая прово-
лока.
Рис. 55. Прибор для титро-
вания с системой электродов
алюминий — алюминий [222].
1 — индикаторный алюминиевый
электрод; 2 — алюминиевый элект-
род сравнения; 3 — пористая стек-
лянная пластинка; 4 — магнитная
мешалка.
Р п с. 56. Электродный сосуд
Уоркера — Хаскелла [860].
1 — каломельный электрод; 2 —
стеклянный электрод; з — пори-
стая стеклянная пластинка; 4 —
магнитная мешалка.
с электродом. Этого можно достичь, применяя остроумное приспособление,
благодаря которому металлический электрод (из сурьмы, платины), закреп-
ленный у выхода из бюретки, находится в стекающем титранте (рис. 52 и 53)
[92, 105, 183, 359, 446, 601, 633, 770] (см. также гл. 21).
в. J-образный ртутный электрод можно причислить к специальным метал-
лическим электродам, он применяется при титровании тиолов в ацетоне
(рис. 54) [255, 674]. Электрод этого типа применяется для стандартных
растворов, содержащих перхлорат ртути(П) (см. гл. 28, разд. 160,г). Систе-
ма электродов алюминий — алюминий может применяться при анализе
алкильных производных алюминия. Полуэлементом сравнения биметалли-
ческой системы электродов является алюминий, погруженный в бензольный
раствор триэтилалюминия (рис. 55) [222] (см. также гл. 30, разд. 166).
г. В специальной литературе широко описаны электроды сравнения,
причем чаще всего это тот или другой вариант каломельного электрода.
В новом варианте «заполненного каломельного электрода» имеется солевой
мостик, который сообщается с анализируемым раствором через мелкопори-
стый стеклянный фильтр. Так устроены электроды типа Уорнера — Хаскел-
ла (рис. 56) [860] и типа Иена № 9401 и 9422 (рис. 57—60). Солевым мостиком
Рис. 57. Каломельный эле-
ктрод (Иена № 9401).
1 — отверстие для введения жид-
кости солевого мостика; 2 — пори-
стая стеклянная пластинка; 3 —
винтовая нарезка; 4 — платиновая
проволока; 5 — асбестовая нить.
Рис. 58. Прибор для потенциометрического полу-
микротитрования.
1 — стеклянный электрод; 2 — боковой отвод с каломельным
электродом (Иена № 9422); 3 — сосуд для титрования с двой-
ными стенками (Иена № 9457); 4 — асбестовая нить; 5 — под-
ставка и пята (Иена № 9454); 6 — приспособление для прекра-
щения подачи тока азота, используемого для перемешивания.
Рис. 59. Закрытый прибор для фото-
метрического полумикротитрования.
Титруемый раствор перемешивается
струей азота.
1 — стеклянный электрод; 2 — каломельный
электрод или электрод Ag/AgCl; 3 — боковой
отвод для подачи анализируемого образца или
инертного газа.
Рис. 60. Закрытый прибор для потен-
циометрического полумикротитрования.
Титруемый раствор перемешивают с помо-
щью магнитной мешалки.
1 — стеклянный электрод; 2 — каломельный эле-
ктрод или электрод Ag/AgCl; з — магнитная
мешалка; 4 — боковой отвод для введения анали-
зируемого образца или инертного газа.
Рис. 62. Элект-
род серебро—хло-
рид серебра с муф-
той.
1—насыщенный раст-
вор КС1 в СН,СООН
или 10%-пый раст-
вор LiCl в метиловом
спирте.
Рис. 64. Система эле-
ктродов Ag/AgCl и стек-
лянный электрод [368].
1—электролиты КС1—AgCl
или KNOj—AgCl; 2— се-
ребряная ^проволока, конец
которой покрыт AgCl; з—
стеклянный электрод; 4 —
пробка; 5 — льняная пить.
Рис. 61. Каломельные электроды с нитью (а)
и с муфтой (б).
1 — насыщенный раствор LiCl в уксусной кислоте;
2 — ртуть; 3 — каломельная паста; 4 — кристаллы LiCl;
5 — кристаллы КС1; 6 — тонкие капилляры, удержи-
ваемые винтовой нарезкой; 7 — резиновая прокладка;
8 — насыщенный раствор КС1 в метиловом спирте;
9 — пришлифованная стеклянная муфта.
Р и с. 63. Электрод серебро — хло-
рид серебра [273].
1 — спай; 2 — пробка из неопрена; 3 —
зажим из тайгона; 4 — 0,2 М раствор
тетраметиламмонийхлорида в изопропи-
ловом спирте; 5 — серебряная проволока
(№ 14) с покрытием из тайгопа, предо-
храняющим от коррозии (коррозия приво-
дит к появлению искаженного потенциала);
6 — трубка из боросиликатного стекла;
7 — пористая фарфоровая пластинка.
178
Глава 12
в последнем может быть насыщенный раствор хлорида лития в уксусной
кислоте; анализируемый раствор перемешивается струей азота, как это
показано на рисунке. В более простом варианте каломельный электрод
соединяется с исследуемым раствором при помощи тонкой асбестовой нити,
запаянной в капилляр (рис. 61, а, электрод с нитью). Американские иссле-
дователи предпочитают каломельный электрод с пришлифованной муфтой,
с помощью которой анализируемый раствор можно отделить от солевого
мостика (рис. 61, б).
Водные каломельные электроды имеют много недостатков; так, при при-
менении их в среде, содержащей уксусный ангидрид, на поверхности электро-
да наблюдается выделение тепла, что вызывает аномальное протекание
диффузии (см. разд. 68).
Электрод типа Ag/AgCl также может быть использован в качестве элек-
трода сравнения, он представляет собой серебряную проволоку, покрытую
слоем AgCl. В простейшем случае этот электрод непосредственно погружает-
ся в раствор [244], однако предпочтительнее отделять электрод сравнения
от исследуемого раствора. В этом случае способ применения электрода
Ag/AgCl идентичен показанному на рис. 57; на рис. 62—64 можно видеть
другие три типа электрода [273, 368, 592]. Для того чтобы стабилизовать
диффузионный потенциал, надо использовать для электролита солевого
мостика органические растворители.
Стеклянный электрод пригоден также и в качестве электрода сравнения,
например в этилендиамине, так как стеклянный электрод не реагирует на
изменение pH в присутствии ионов Na+. В этом случае в качестве индикатор-
ных электродов применяют сурьмяные или платиновые электроды [10, 190,
252, 372, 446].
68. УСТРАНЕНИЕ ФЛУКТУАЦИЙ ПОТЕНЦИАЛА ЖИДКОСТНОГО
СОЕДИНЕНИЯ.
РАЗЛИЧНЫЕ СИСТЕМЫ ЭЛЕКТРОДОВ
Для устранения флуктуаций диффузионного потенциала используются
различные типы электродов. В тех случаях, когда анализы проводят в среде
уксусного ангидрида, водный раствор хлорида калия из каломельного
электрода реагирует с этим растворителем, и из-за выделения тепла меняется
потенциал. Ацетат ртути(П), применяемый при титровании хлоргидратов
аминов, загрязняет электроды, что проявляется во флуктуациях напряже-
ния [643, 644]. Если в ртутных электродах в качестве соединительной жидко-
сти применять не водный, а насыщенный метанольный ^раствор хлорида
калия, можно в значительной степени устранить источник ошибок, вызван-
ных потенциалом жидкостного соединения. В подавляющем большинстве
работ по потенциометрическому титрованию применяется именно такой тип
каломельного электрода и когда употребляется термин «каломельный элек-
трод», то подразумевается элшстрод,'наполненный насыщенным метанольным
раствором хлористого калия. Этот тип электрода пригоден для проведения
титрования в среде ацетонаП258], ацетонитрила, смеси бензола с изопропи-
ловым спиртом, пиридина [159], уксусной кислоты и смеси бензол — хлоро-
форм — уксусная кислота [158] (рис. 61).
Если определение проводят в уксусной кислоте или в смеси уксусной
кислоты с уксусным ангидридом, для солевого мостика каломельного электро-
да вместо раствора хлорида калия можно применять насыщенный раствор
хлорида лития в уксусной кислоте [144, 146, 314, 337, 730] (рис. 58—60).
При проведении титрования в уксусном ангидриде (например, при определе-
нии амидов кислот) каломельный электрод заполняют 0,1 н. раствором пер-
хлората лития в уксусном ангидриде, так как хлорид лития малорастворим
в уксусной кислоте [878]. Употребляя «каломельный электрод», заполненный
Потенциометрическое определение конечной точки
179
насыщенным раствором ацетата ртути(1), хлорида натрия и перхлората
натрия в уксусной кислоте, можно проводить измерения потенциала с вос-
производимостью ±0,25 мв [102]. При титровании в среде бензола, толуола
и низкокипящего петролейного эфира эффективен каломельный электрод,
содержащий вместо насыщенного раствора хлорида калия 1 н. раствор хло-
рида тетрабутиламмония [358]. Из-за разложения каломели нельзя пригото-
вить каломельный электрод с насыщенными растворами солей (C4H9)4N +
или (CH3)4N+ [550]. При титровании в этилендиамине в качестве солевого
мостика можно использовать насыщенный раствор хлорида лития и каломе-
ли в этилендиамине [285] (рис. 65).
При проведении определений в уксусной кислоте и диоксане широкое
распространение получила система из стеклянного индикаторного электрода
Рис. 66. Прибор для титрования с си-
стемой электродов графит—платина [894].
1 — боковой отвод для засасывания раствора аце-
тата меди(П) под вакуумом (внутреннее простран-
ство должно быть полностью заполнено); 2— гра-
фитовый электрод; 3 — пористая стеклянная
пластинка; 4 — мешалка; 5 — платиновый (или
золотой) электрод.
Z~2,5j4m
Рис. 65. Каломельный электрод с эти-
лендиамином [285].
1 — отвод для заполнения электродного прост-
ранства под вакуумом; 2 — платиновая прово-
лока, впаянная в тонкую стеклянную трубку;
3 — этилендиамин, насыщенный каломелью
и L1C1; 4 — каломель—ртутная паста; 5— ртуть .
и серебряного электрода сравнения [244, 368]. В современной практике
электрод Ag/AgCl не погружают непосредственно в органический раствори-
тель, а соединяют с исследуемой жидкостью при помощи солевого мостика,
содержащего фоновый электролит. При титровании в уксусной кислоте
с этой целью применяют насыщенный раствор хлорида калия в уксусной
кислоте [592] (рис. 62), при титровании метилатами щелочных металлов —
10%-ный раствор хлорида лития в метиловом спирте [592] (рис. 62), при
титровании в уксусном ангидриде — уксусный ангидрид, насыщенный Lid
и AgCl [778], при титровании гидроокисью тетрабутиламмония в среде толу-
ол — изопропиловый спирт подходящим электролитом фона является 0,2 н.
раствор (C4H9)4NC1 в изопропиловом спирте [273] (рис. 63). О комбинирован-
ных электродах стекло — Ag/AgCl см. рис. 64 и работу [368].
Вместо каломельного и серебряного электродов применяют также графи-
товый электрод (рис. 66). В принципе он сходен с капиллярным электродом
и может быть сделан из углеродного стержня сухой батарейки для лампы-
180
Глава 12
вспышки. Этот стержень погружают в раствор ацетата меди(П) в уксусной
кислоте, который отделен от анализируемого раствора пористым стеклянным
фильтром (графитовый электрон Цейдлера [894]). Иногда вместо ацетата
Рис. 67. Система электродов стекло — каломель, используемая для потенциометри-
ческого микротитрования [754].
а — чашечный стеклянный электрод, который должен быть заполнен (заштрихованная часть) бу-
ферным раствором с pH ~ 6,8; б — сосуд для титрования; в — каломельный электрод сравнения; 1 —
стеклянная электродная мембрана из стекла корнинг 015; 2 — стеклянный шлиф 15 (чтобы предот-
вратить взаимное проникновение образца и буферного раствора через поверхность стеклянного шлифа,
шлиф смазывают силиконовой смазкой); 3 — водяная рубашка для поддержания в электроде постоян-
ной температуры и для защиты его от изменений электрической емкости; 4 — электродное соединение;
5 — керамическая диафрагма для соприкосновения между образцом и насыщенным раствором КСГ,
6 — отвод электрода сравнения (в), разделенный (7) на два отдельных канала; 8 — ввод для подачи
азота (для перемешивания образца и удаления СОг), до пуска азота ввод должен быть заполнен раст-
ворителем.
меди(П) используют сульфат меди(П) [698]. Индикаторный электрод изго-
тавливают из платины или золота. Для дифференцирующего титрования
Таблица 56
Система электродов Максимум ДЕ/AV, мв/мл
система а система б
Pt — поляризованная Ptа 12 500 6 500
Pt — поляризованная Pt 6 4 900 3100
Pt — PtB 6 750 4 750
Поляризованная Pt — поляризован- 6 000 3 500
ная Pt в
Pt — каломельг 4350 5 000
Pt — каломель л 5 750 4 250
Стекло — каломель г 5 600 750
Стекло — каломель 5150Д 700 е
Ag — стекло 3 650 1 250
Комбинированный электрод: стек- 3 250 300
ло — Ag/AgCl
а Неполяризованный электрод омывается титрантом.
б Поляризованный электрод омывается титрантом.
в Один из электродов омывается титрантом.
г Электролит — насыщенный водный раствор хлорида калия.
д Электролит — насыщенный.’раствор хлорида лития в уксусной кислоте.
е Электролит — насыщенный раствор хлорида калия в метиловом
•спирте.
Потенциометрическое определение конечной точки
181
Рис. 68. «Двухфазный» каломельный электрод [550].
1 — насыщенный водный раствор НС1; ‘2—раствор трет-
бутилового спирта, содержащий около 10% воды и примерно
0,056 молъ/л КС1; 3 — ультрамелкопористый стеклянный
фильтр; 4 — насыщенный раствор бутилового спирта, содержа-
щий около 4,3 молъ/л КСГ, 5 — тиретп-бутиловый спирт, насы-
щенный бромистым тетрабутиламмонием.
в ацетоне Мальмстадт и Вассало применяли
в качестве индикаторного платиновый электрод,
содержащий 10% родия, а в качестве электрода
сравнения — графитовый стержень каранда-
ша 9Н.
Для микротитрования оснований, кислот
и их аналогов пригоден модифицированный
стеклянный электрод типа «Ингольд», исполь-
зуемый в термостатируемой ячейке [407, 754]
(электрод марки «Napf — Glasselektrode», тип
232-1, фирма «W. Ingold» Франкфурт-на-Майне).
Набор приспособлений для микротитрования
показан на рис. 67 [754].
Иногда могут возникать затруднения и в том
случае, если, следуя литературным данным,
выбирают для анализа «наиболее подходящий
электрод». Пеллерин и Демай [633] провели
детальные исследования десяти различных пар
электродов в среде уксусной кислоты и диме-
тилформамида. В первом случае бифталат калия
титровали 0,1 н. раствором хлорной кислоты в уксусной кислоте (а), а во
втором — бензойную кислоту титровали 0,1 н. раствором метилата натрия
в смеси бензола с метиловым спиртом (б). В приведенной ниже табл. 56 пред-
ставлены результаты этих определений; чтобы эти результаты было удобнее
сравнивать, они представлены в виде значений \Е1 ДУ. Приведенные данные
показывают, что наиболее резкие скачки потенциала наблюдаются при при-
менении сочетания электродов Pt — Pt, особенно в том случае, когда неполя-
ризованный электрод помещен в титрант с постоянной ионной силой (у вы-
ходного отверстия бюретки).
В своих экспериментах Пеллерин и Демай пользовались впаянным платиновым
электродом длиной 1—3 см и диаметром 0,5—1,0 мм, предварительно поляризованном
в 1%-ной серной кислоте при 3—5 в в течение 1 мин.
Свобода и Шайн при изучении неводного титрования слабых кислот
и оснований использовали обычные методы работы, применяемые для потен-
Р и с. 69. Потенциометрическое определение бензойной кислоты в пиридине метилатом
натрия с использованием системы электродов стекло — каломель (7), серебро — кало-
мель (2), серебро — стекло (3} [888].
182
Глава 12
циометрических измерений постоянного тока * [733, 787]. Для этого изме-
рялась разность потенциалов двух платиновых электродов, поляризованных
постоянным током силой 1 мка. Эти электроды были сделаны из платиновой
проволоки (диаметром 0,016), впаянной в трубку из мягкого стекла таким
образом, чтобы выступающие концы проволоки имели длину около 1 см.
Таблица 57
Перечень различных каломельных электродов и электродов Ag/AgCl
Раствор электролита Номер рисунка Литература
Каломельные электроды
Метиловый спирт, насыщенный КС1 61,6 156, 158—160,
0,02 н. раствор LiCl в уксусной кислоте 258, 540, 595, 780 389
Уксусная кислота, насыщенная LiCl 1 н. раствор (C4H9)4N+C1- в воде 58—60 314
61,6 355
0,01 н. раствор (C4H9)4N+Br- в метиловом спирте 61 ,а 164
Уксусная кислота, насыщенная Hg2Cl2, NaCl, NaC104 102
Изопропиловый спирт, насыщенный КС1 579
0,1 н. раствор LiC104 в уксусном ангидриде 61,а 878
Этилендиамин, насыщенный LiCl и Hg2Cl2 65 285
КС1 в двухфазной системе Н2О — znpezn-бутиловый спирт 68 550
Электроды Ag/AgCl
Уксусная кислота, насыщенная КС1 10%-ный раствор LiCl в метиловом спирте 62 592, 767
62 592
Уксусный ангидрид, насыщенный AgCl-|-KCl 778
AgNO3 в ацетонитриле 648
0,2 н. (C4H9)4N+C1~ в изопропиловом спирте 63 273
Таблица 58
Системы электродов, используемые в анализе фенолов
Растворитель Система электродов Стандартный раствор Литерат ура Номер рисунка
ЭДА Сурьма — сурьма Аминоэтилат нат- рия 601 53
ЭДА Платина — каломель 285 54
ЭДА Сурьма — сурьма То же 446 53
ЭДА, бензол, метило- Платина — платина » » 92, 601
вый спирт
ЭДА, ДМФ Стекло — каломель (C4H9)4NOH 176 61 ,б
ЭДА, пиридин, ме- тилэтилкетон Предварительно поляри- зованная платина (C4H9)4NOH 359 52
Пиридин, ДМФ
Ацетонитрил Стекло — каломель (C4H9)4NOH 159 61,6
ДМФ Сурьма — каломель СН3ОК ИЗ
Пиридин Сурьма — стекло CH3ONa 10
ДМФ Стекло — каломель (C4H9)4NOH 7 61 ,б
Пиридин Стекло — каломель (C9H9)4NOH и CH3OK 25, 355, 780
Метилизобутилкетон Стекло — платина (C4H9)4NOH 105
Ацетон Стекло — каломель (C2H5)3C4H9NOH 258 61
Бензол, толуол Стекло — каломель (C4h9)4noh 358
/иретп-Бутиловый спирт Стекло — «двухфазный» каломельный электрод (C4H9)4NOH 253, 550 68
* См. также Reilley, Cooke, Furman, Anal. Chem., 23, 1223 (1951).
Потенциометрическое определение конечной точки
183
Типичные пикообразные кривые титрования были получены при анализе
различных органических кислот и оснований. Основания титровались в сме-
си л-крезола с ацетонитрилом (1 : 1) при помощи хлорной кислоты в л-кре-
золе или ацетонитриле. Различные типы электродов сравнения и систем
электродов, применявшихся для анализа слабых кислот (фенолов), пред-
ставлены в табл. 57 и 58 (см. также гл. 18 и 21).
69. ФОРМА КРИВЫХ ПОТЕНЦИОМЕТРИЧЕСКОГО ТИТРОВАНИЯ
Форма кривых потенциометрического титрования в неводной среде зави-
сит от используемого электрода, растворителя, стандартного раствора и силы
анализируемых кислоты или основания. На форму кривых влияют также
загрязнения ионами К+ или Na+, ассоциации между растворенным веществом
и растворителем, а также возможное образование комплексов кислота —
анион кислоты. На рис. 43 и 46 показаны неустойчивые (неравновесные)
кривые, а на рис. 44 можно видеть корелляцию между потенциометрической
кривой и переходом окраски индикатора.
При проведении анализа с использованием пары электродов — стеклянно-
го в качестве индикаторного и серебряного как электрода сравнения —
наблюдается V-образный максимум или минимум, но при титровании фено-
лов сохраняется обычная S-образная кривая [888]. Внезапное изменение
потенциала в точке эквивалентности можно приписать изменению роли
электродов в процессе титрования (рис. 69, в). Из этого явления можно сде-
лать вывод, что в какой-то момент титрования оба электрода играют роль
индикаторных электродов. Изменение потенциала можно наблюдать при
применении обоих электродов. Вероятно, стеклянный электрод вплоть до
точки эквивалентности является индикаторным электродом, а серебряный —
переменным электродом сравнения. При достижении точки эквивалентности,
которая совпадает с переходом окраски тимолового синего, если использо-
вать для титрования метилат натрия, потенциалы электродов меняют знак
и направление э. д. с. становится обратным. Таким образом, режим работы
электродной пары стекло — серебро в среде пиридина при титровании мети-
латом натрия зависит как от химических, так и от поляризационных факто-
ров [888].
Все это можно наблюдать при применении электродной пары стекло —
серебро в ацетоне, ацетонитриле, смеси бензола с метиловым спиртом, изо-
пропиловом спирте, пиридине и диметилформамиде. При работе в этих
растворителях кривая с максимумом получается в ацетонитриле, а в осталь-
ных растворителях — кривая с минимумом в точке эквивалентности. Та же
пара электродов дает S-образные кривые в воде и н-бутиламине. Изгиб на
кривой не появляется при работе в бензиламине, этилендиамине и пипери-
дине.
Серебряный электрод представляет собой проволоку с поверхностью 0,11 см2; время
от времени ее необходимо полировать или погружать в концентрированный раствор NH4OH.
Можно использовать также серебряный электрод Бекмана № 4949-V6B [888] (см. так-
же гл. 15, разд. 95).
Глава 13
ИНДИКАТОРЫ
Индикаторы оказались пригодными при определении конечной точки
титрования, особенно при резких подъемах потенциальных кривых (крутых
кривых) в случае потенциометрического титрования (см. рис. 44). В принци-
пе, индикаторы, применяемые в водной среде, пригодны для определений
в неводных протонсодержащих или инертных растворителях. Практически
разница лишь в том, что оттенок перехода окраски часто отличается от того
оттенка, который наблюдается при титровании в водной среде, поскольку
в зависимости от природы растворителя активность протонов в растворе
часто меняется и в точке эквивалентности окраска индикатора определяется
природой и концентрацией иона [48, 207, 338].
70. ИОНИЗИРОВАННЫЕ КРАСИТЕЛИ, ИНДИКАТОРЫ
Окраска вещества обусловлена поглощением молекулами этого вещества
квантов света в видимой области спектра. Взаимодействие света с веществом
переводит электронную систему молекулы с более низкого на более высокий
энергетический уровень, т. е. эта система приходит в состояние возбуждения.
У соединений, которые можно представить в виде гибридов нескольких
резонансных структур, поглощение смещается в сторону больших длин волн
(более низких энергий). Этот эффект усиливается с увеличением степени
сопряжения, т. е. по мере уменьшения разницы между основным и возбуж-
денным состоянием.
Трифенилметан бесцветен, но радикал трифенилметила, трифенилметил-
катион и трифенилметил-анион окрашены
Н5Свх Н5С6х _ (С6Н5)зС-
(С6Н5)3С:Н ЩС6-С+ С=/______>+ и т. д. и
Н5Св/ Н5Св/ (С6Н5)зС-
Резонансная стабилизация увеличивается, если в орто- и пара-положе-
ниях ароматического кольца находятся заместители ОН, NH2 и N(GH3)2.
Такими соединениями являются кристаллический фиолетовый (хлоргидрат
гексаметил-м-розанилина), метиловый фиолетовый (хлоргидрат пентаметил-
n-розанилина), малахитовый зеленый (хлоргидрат тетраметилди-м-амино-
фуксина) и т. д.
Можно привести много примеров, подтверждающих, что ионизированная форма
веществ окрашена либо иначе, либо более глубоко по сравнению с неионизированной фор-
мой (галохромизм). Так, например, оксониевая форма бензальацетофенона, имеющая
сопряженную систему, окрашена сильнее, чем исходное основание (бензальацетофенон
применим в качестве индикатора для очень сильных кислот). При растворении бензаль-
ацетофенона в серной кислоте понижение температуры замерзания последней указывает
на образование ионов
С6Н5 - СН = СН — СО — С6Н5
Бензальацетофенон
С6Н5—СН = СН--С —С6Н5 С6Н5 —СН —СН = С —С6Н5 С6Н5 —СН = СН —С-С6Н5
Оксониевая форма
Индикаторы
185-
Подобно протону, кислоты Льюиса обладают также большим сродством к неподелен-
ной паре электронов. Хлористый алюминий образует координационный комплекс с одной
молекулой бензальацетофенона, и в этом случае продукт также более сильно окрашен,
чем исходное основание.
Индикаторы представляют собой слабые основания или кислоты, меняю-
щие свою окраску при образовании соли благодаря изменению их электрон-
ной конфигурации и вследствие этого специфической способности поглощать-
свет. Сила оснований или кислот относительна и лишь в первом приближе-
нии не зависит от природы растворителя. Так, в неводных растворах величи-
на pZT индикаторов различного типа заряда и различной структуры может
отличаться от их величины pZT в водной среде. Благодаря образованию ассо-
циатов константа диссоциации и степень основности зависят от природы
растворителя и в некоторых случаях также от концентрации вещества.
Образование солей по-разному влияет на изменение цвета индикатора [292,
389, 464]. Из вышеизложенного следует, что для точности определений
вещество, применяемое для установки титра, и анализируемое вещество-
должны иметь примерно одинаковую ионную силу.
В водных средах существует известная зависимость между соотношением
кислотной и основной форм индикатора и концентрацией водородных ионов.
Однако Кольтгоф и Бруккенштейн показали, что при титрованиях хлорной
кислотой в уксусной кислоте отсутствует простая корреляция между соотно-
шением кислотной и основной форм индикатора и величиной pH раствора,
которая фактически зависит от общей концентрации хлорной кислоты и от
константы образования перхлората индикатора [101, 102, 464]. Это явилось
поворотным пунктом в области исследований системы кислота — основание
в среде уксусной кислоты. Метод, применявшийся этими исследователями
(в основном спектрофотометрическое титрование в присутствии индика-
тора), пригоден для измерения константы диссоциации кислоты и константы
образования перхлората индикатора при взаимодействии умеренно слабой
кислоты с основной формой индикатора *.
В уксусной кислоте из-за ее низкой диэлектрической проницаемости даже
самые сильные электролиты диссоциируют слабо и в основном присутствуют
в виде ионных пар. Окраска основной формы индикатора в чистой уксусной
кислоте определяется равновесием
Z4-CH3COOH ZH+CH3COO- ZH+4-СНзСОО-,
Ионизация Диссоциация
где кислотными формами индикатора являются ZH+CH3COO_ и ZH+; их спектры
идентичны. Если пренебречь диссоциацией, то константа ионизации индикатора
определяется уравнением
Ki [ZH+СНзСОО-] [Za]
[/] [/ь] ’
где 1а — кислотная форма индикатора; 1Ъ — основная форма индикатора.
Если для индикатора = 1 в уксусной кислоте, т. е. если ZH+GH3GOO~ =
= 50% и Z = 50%, то соотношение ~ не будет уменьшаться при добавле-
ъ
нии основания. В этом состоит основное различие между водной средой и сре-
дой уксусной кислоты (так, например, в воде количество ионных пар ZH+OH_
ничтожно мало и, следовательно, не определяет цвета «кислого» окрашивания
индикатора. В уксусной кислоте преобладают ионные пары ZH+CH3COO_.
В уксусной кислоте может быть применен индикатор очень слабой основности
с величиной = 0,01 или ниже. В этом случае в чистой уксусной кислоте-
концентрация кислотной формы индикатора ZH+CH3COO_ ничтожна и ей
можно пренебречь).
* Подробно этот вопрос обсужден в книге: Kolthoff, Elving, Sandelin
Treatise on Analytical Chemistry, part I, Vol. 1, p. 475, Interscience, New York, 1959.
-186
Глава 13
Диссоциацией 7Н+С1О~ в процессе взаимодействия между основной фор-
мой индикатора и кислотой можно пренебречь, поскольку при титровании
оснований концентрация перхлорат-иона так велика, что в равновесии
/4-нсю4 zh+cio; zh++cio7
Ионизация Диссоциация
диссоциация на
ния
[ZH+C1O7]
1Л
ионы подавлена, и окраска индикатора зависит от соотноше-
При титровании хлорной кислотой константа образования
перхлората индикатора равна
Ьг1НС1О4 _ [ZHC1O4]
ло6р ~ ТТТс
Снсю4
и отношение кислотной формы индикатора к его основной форме, а следова-
тельно, и окраска индикатора зависят от общей концентрации хлорной кисло-
ты и константы образования перхлората индикатора [101, 102, 464]
[Za] [ZHCIOJ KrIHC104 /7П п
щу =—jjj— = Л-обр сНсю4
где /НСЮ4 — кислотная форма индикатора; I — основная форма индика-
тора; Я^РС1°4 — константа образования перхлората индикатора (табл. 59).
Таблица 59
Некоторые константы образования перхлората индикатора
в уксусной кислоте (см. [388—390])
Индикатор Константа образования Индикатор Константа образования
Этиловый красный 4-109 Малахитовый зеленый 2-105
Хинальдиновый красный 1-Ю7 1-Нафтолбензеин 1-105
Перхлорат кристаллического Нильский голубой А 4-10*
фиолетового Судан III 7-102
Ki 6-105
2-105
В процессе титрования величина снсю4 определяется константой образо-
егВНСЮ4
вания АОбр
54-НСЮ4 ВНСЮ4, (70.2)
егВНС1О4 Свнсю4
где Кобр — — — •
снсю4св
•Объединяя уравнения (70.1) и (70.2), получим
[Za] _ ^о?рИО4свнсю4
[/&] ^ВНрС1О4св •
Другие спектрофотометрические исследования [389] свидетельствуют, что
изменение окраски индикатора в уксусной кислоте зависит от соотношения
концентраций присутствующего в растворе основания и сопряженной с ним
кислоты. В водном растворе, содержащем слабую (например, бензойную)
кислоту и ее соль в присутствии индикатора, реакция может быть выражена
уравнением
AH4-Z /н+ + л-,
где I — основная форма индикатора; ZH+ — протонированная форма инди-
катора. Константа равновесия этого процесса равна
к = [/Н+][Л-] АИНД
[АН] [Z] Акисл *
И ндикаторы
187
Константа равновесия определяется константами диссоциации как инди-
катора, так и кислоты и может быть вычислена из констант ионизации кисло-
ты и индикатора.
Ионы очень склонны образовывать в уксусной кислоте ассоциаты,
и индикаторы не являются исключением из этого правила. Поэтому пове-
дение индикаторов, например в уксусной кислоте, можно схематически
выразить следующим уравнением (см. [389]):
ЖЛ-ь/НЛс /НЛ-рНЛс,
где s — растворитель или основание, способное присоединять протон;
$НЛ — продукт ассоциации кислоты ЛН и молекулы s; 7НЛс — продукт
ассоциации индикатора с уксусной кислотой, образующийся в уксусной
кислоте (а именно основной формы индикатора); /НА — продукт ассоциации
индикатора и сильной кислоты.
Очень слабые основания в уксусной кислоте не образуют «ониевых» аце-
татов. Поскольку индикаторы обычно являются очень слабыми основаниями,
в уксусной кислоте «основное» окрашивание раствора зависит от относитель-
ной концентрации ассоциата индикатор — уксусная кислота. В этом случае
константа равновесия определяется уравнением [389]
к _ (/НЛ) (ЖЛс)
Л “ (ЖЛ) (/НЛс) •
। Между /НАс и /НА имеется значительное различие, так как степень
переноса протона различна у обоих ассоциатов. В первом случае перенос
протона сводится, самое большее, к образованию водородной связи, тогда
как во втором, особенно если титрование проводится хлорной кислотой,
характер переноса протона скорее приближается к образованию «ониевого»
иона, вследствие чего спектральные свойства /НАс и /НА различны. Степень
переноса протона также меняется в зависимости от того, является ли s основ-
ной молекулой, способной к значительной координации с протоном, или это
только молекула растворителя, обладающего низкой основностью (напри-
мер, уксусной кислоты). В последнем случае суммарная реакция может быть
выражена упрощенно следующим образом:
5Н+Л--^/НЛе 7НЛ-]-ВН+Лс-
и
к _ [7НЛ] [ЯН+Лс-]
Л ~ [ЯН+Л-] [/НЛс] ’
где БН+Л~ — ионная пара, образованная сильной кислотой (например,
НС1О4) и основанием (например, дифенилгуанидином); ВН+Ас~ — соответ-
ствующий ацетат.
Константа равновесия в среде уксусной кислоты включает в себя кон-
станты четырех процессов
, _КТ„.
ВН+Дс 1НЛ
Л = ~К-----К-----•
1. Константа диссоциации ацетата основания
к _ [5Н+] [Л<г]
ВН+4с [ЯН+Лс-]
2. Константа диссоциации перхлората основания
к [ВН+] [Л-1
А. RH+д- = ..—.
188
Глава 13
3. Константа диссоциации ацетата индикатора
к _ [ZH+] [Лс-]
лшлс— [/НЛс]
4. Константа диссоциации перхлората индикатора
На величину каждой из этих констант диссоциации влияют диэлектри-
ческая проницаемость растворителя и присутствие в растворе других ионных
пар или посторонних ионов; следовательно, величина К тоже может в изве-
стной степени изменяться. Если ацетат индикатора будет более сильным
основанием, чем анализируемый ацетат основания (как, например, в случае
титрования очень слабых оснований), то индикатор будет давать «кислое»
окрашивание до наступления точки эквивалентности, и по этой причине
не может быть использован для анализа. В качестве примера можно привести
определение эфедрина {Eph) при помощи хлорной кислоты в присутствии
индикатора этилового красного {Ег)
к-ю
EPhH+ ... СЮг+Яг-НЛс -----£г.НС1О44-Ер/гН+Лс-
Если же ацетат основания является более сильным основанием, чем ацетат
индикатора, то образование перхлората индикатора происходит после обра-
зования перхлората основания и точка эквивалентности может быть обнару-
жена при помощи индикатора
К-10-2
£рЛН+ ... С1О4 +Qr НЛс ' —» (?г.НСЮ4 + ^рЛН+Лс-
где Qr — хинальдиновый красный (переход окраски при титровании хлорной
кислотой: красная -+ бесцветная).
Таким образом, хинальдиновый красный пригоден для титрования алка-
лоидов и других азотсодержащих оснований с величиной р/Г = 4,7 или более
низкой.
71. ВЫБОР ИНДИКАТОРА
Исследований по вопросу о теоретическом методе выбора индикаторов
было проведено немного. Наиболее важными в этой области являются работы
Хигучи с сотр. [389], Кольтгофа и Бруккенштейна [464] и Мальмштадта [540].
Их идеи оказались плодотворными в особенности для развития фотометриче-
ского метода определения конечной точки (см. гл. 24). Подробная статья
Стока и Парди [775] посвящена применению индикаторов.
В табл. 60 и 61 приведены данные о переходе окраски индикаторов при
титровании кислот. Из этих данных следует, что, например, в ацетоне азо-
фиолетовый применим для титрования фенолов, а тимоловый синий более
пригоден для титрования замещенных бензойных кислот [540]. Табл. 62—65
могут помочь в выборе некоторых индикаторов в зависимости от природы
растворителя. Выбор же самого растворителя определяется силой исследуе-
мых кислот или оснований.
Наиболее часто при титровании в неводной среде применяют такие инди-
каторы, как азофиолетовый [4-(п-нитрофенилазо)резорцин], тимоловый синий
(тимолсульфофталеин), относящийся к группе сульфофталеинов, и кристал-
лический фиолетовый (хлоргидрат гексаметил-п-розанилина), принадлежа-
щий к классу трифенилметановых красителей (обычно в смеси с некоторыми
соответствующими пента- и тетраметильными соединениями). Кроме пере-
численных, довольно часто применяются индикаторы, приведенные в табл. 66.
Таблица 60
Переход окраски индикатора в псевдобуферных растворах (пб),
содержащих кислоту и СНз^Нд^КГОН в ацетоне [540j
Соединение (растворитель) (0,01 н.) Азофиолето- вый: кислот- ное окраши- вание -> ще- лочное окра- шивание Тимолфтале- ин: кислот- ное окраши- вание -> ще- лочное окра- шивание Тимоловый синий: кис- лотное окра- шивание -> -► щелочное окрашивание Нейтральный красный: кис- лотное окра- шивание -> -► щелочное окрашивание Отклонение, измеряемое системой электродов стекло — ка- ломель, мв
НС1О4 Желтое Бесцветное Красное Красное +685
НС1О4 (пб) » » Оранжевое » +580
HNO3 (пб) » » Желтое » +200
Ацетон » » » Желтое —150
Бензойная кислота (пб) » » » » —250
Антраниловая кисло- Розовое » » » —350
та (пб)
о-Нитрофенол (пб) Красное » » » —450
Фенол (пб) » Синее Синее » —580
€Нз(С4Н9)3МОН (избы- Фиолетовое » » » —750
ток)
Таблица 61
Выбор индикаторов для кислот разной силы [540]
Кислота или ее аналог 1%-ный раствор ин- дикатора в метило- вом спирте Переход окраски Фильтр, нм Увеличение (+) или уменьше- ние (—) погло- щения света в точке эквива- лентности
•Очень слабые кисло- Азофиолетовый Красная — ►синяя 600 +
ты
Замещенные уксус- ные кислоты Тимолфталеин Бесцветная няя —> си- 575 +
Замещенные бензой- Тимоловый синий Желтая —> синяя 575 +
ные кислоты
Неорганические кис- лоты Нейтральный красный Красная —5 тая жел- 550 —
Примечание: Вышеуказанные кислоты могут титроваться фотометрически в ацетоне раствором
гидроокиси метилтри-н-бутиламмония в смеси бензола с изопропиловым спиртом (8 : 2) [540].
Таблица 62
Выбор индикаторов для применения в инертных растворителях
П Инертный раство- ритель Индикатор Переход окраски в среде: (а) основной -> кислой, (б) кислой -► основной
Ацетон 1%-ный раствор азофиолетового в метиловом спирте 0,3%-ный раствор тимолового синего в метиловом спирте 0,2%-ный раствор фенолфта- леина в метиловом спирте 0,2%-ный раствор тимолфта- леина в метиловом спирте 1%-ный раствор нейтрального красного в метиловом спирте Красная —> синяя (в случае очень слабых кислот) (б) Желтая —> красная (а) Желтая —> синяя (в случае за- мещенных бензойных кислот) (б) Бесцветная —> красная (б) Бесцветная —> синяя (б) Красная —> желтая (в случае неорганических кислот) (б)
Продолжение табл. 62
Инертный раство-
ритель
Индикатор
Переход окраски в среде:
(а) основной -+ кислой,
(б) кислой -> основной
Ацетон, ацето- нитрил Ацетон, ацето- нитрил, метил- этилкетон, метил- изобутилкетон 0,2 %-ный раствор п-оксиазо- бензола в бензоле Насыщенный раствор метилово- го красного в ацетонитриле 0,1 %-ный раствор метилового фиолетового в уксусной кислоте 1?о-ный раствор 1-нафтолбен- зеина в изопропиловом спирте 0,5%-ный раствор тропеолина ОО в уксусной кислоте
Ацетонитрил Раствор 1% диметилового жел- того и 0,1% метилового синего в метиловом спирте 0,3%-ный раствор тимолового синего в метиловом спирте
Оранжевая —> желтая (б)
Желтая —> оранжево-красная —>
—> розовая —> фиолетово-красная
(а)
Фиолетовая —> темно-синяя —>
—> светло-голубая (а)
Желтая —> светло-зеленая —>
—* травянисто-зеленая (а)
Желтая —> цикламеново-крас-
ная —> розово-красная (в случае-
сильных и средней силы основа-
ний) (а)
Красно-коричневая —> зеле-
ная (б)
Бензол, хлорбен- 0,1 %-ный раствор диметилово-
зол го желтого в хлороформе Насыщенный раствор фенолфта- леина в бензоле
Насыщенный раствор бромкре- золового пурпурного в бензоле Насыщенный раствор бромкре- золового зеленого в бензоле
Хлорбензол Насыщенный раствор бромфе- нолового синего в хлорбензоле
В случаях слабых кислот: жел-
тая —> зеленая —> синяя (б)
В сильных кислотах: красная —>
—> желтая —> синяя (б)
Желтая —> розовая (а)
Бесцветная —> красная (б)
Пурпурная —> желтая (а)
Синяя —> желтая (а)
В зависимости от силы раство-
ренного основания:
пурпурная
розовая
ярко-желтая
светло-желтая
—> бесцветная
Хлороформ, че-
тыреххлористый
углерод
Дихлорметан
(метиленх лори д,
хлористый мети-
лен)
Метилэтилкетон
0,1%-ный раствор метанилово-
го желтого в метиловом спирте
0,1 %-ный раствор диметилово-
го желтого в хлороформе
1%-ный раствор крезолфталеи-
на в смеси метилового спирта и
хлороформа (1 :1)
0,1%-ный раствор диметилами-
ноазобензола в хлороформе
Раствор 0,6% тимолфталеина
и 0,4% тропеолина О в диметил-
формамиде
Желтая —> фиолетовая (а)
Желтая —> розовая (а)
Бесцветная —> фиолетово-розо-
вая (б) (барбитураты)
Желтая —> розовая (а)
Желтая —> зеленая (б) (барби-
тураты)
Таблица 63
Кислотный раство- ритель Индикатор Переход окраски в среде: (а) щелочной-+кислой, (б) кислой-+щелочной
Уксусная кисло- та, пропионовая кислота 0,1—1,0%-ный раствор кристал- лического фиолетового в уксусной кислоте 0,2 %-ный раствор метилового фиолетового в хлорбензоле 0,5 %-ный раствор малахитового зеленого в уксусной кислоте Фиолетовая —> темно-синяя —> —> сине-зеленая —> зелено-жел- тая (а) Фиолетовая —> синяя —> сине- зеленая —> желтая (а) Сине-зеленая —> зеленая —* —> желтая (а)
Продолжение табл. 63
Кислотный раство- ритель Индикатор Переход окраски в среде: (а) щелочной -> кислой, (б) кислой -> щелочной
Уксусная кисло- та, пропионовая кислота Уксусный ангид- рид Этиловый спирт Изопропиловый спирт Метиловый спирт Метилцеллозол ьв Пропионовая кислота 0,5%-ный раствор крезолового красного в смеси уксусной кисло- ты с хлорбензолом (1:1) 0,02%-ный раствор этилового красного в уксусной кислоте 0,5%-ный раствор тропеолина 00 в уксусной кислоте 0,02%-ный раствор нильского голубого А в уксусной кислоте 0,02%-ный раствор Судана III в уксусной кислоте 0,02 %-ный раствор хинальдино- вого красного в уксусной кислоте Насыщенный раствор хиналь- динового красного в уксусной кислоте 0,02 %-ный раствор 1-нафтолбен- зеина в уксусной кислоте 1 % -ный раствор целитонового прочного синего FR в уксусной кислоте 0,1 %-ный раствор кристалли- ческого фиолетового в уксусной кислоте 0,5%-ный раствор тимолфталеи- на в метиловом спирте -{-0,02 %- ный водный раствор метилового оранжевого 0,1—1,0 %-ный раствор 1-нафто л- бензеина в метиловом спирте или изопропиловом спирте 0,3 % -ный раствор тимолового синего в метиловом спирте Раствор 0,1% тимолового сине- го-{-0,025% патент — голубого А в метиловом спирте 0,3%-ный раствор тимолового синего в диметилформамиде 0,2%-ный раствор метанилового желтого в смеси пропионовой кислоты и диоксана (1:1) Желтая —» розовая —крас- ная (а) Красная —> бесцветная (а) В случае сильных и умеренно сильных оснований: желто-оранжевая —> пурпурная—> —розово-красная (а) Голубая —> бесцветная (а) Желтая —> красная —> синяя (а} Темно-красная —> бесцветная (а} Темно-красная —> желтая (а) Желтая—> зеленая (а) Синяя —> красновато-фиолето- вая (а) Синяя —> зелено-желтая —> жел- тая (а) Желтая —> зеленая (б) Оранжевая —> коричневато-зе- леная —> зеленая (б) Желтая—> красная (а) В случае сильных кислот: пурпурная —г зеленая (б) Желтая —> оранжевая -> крас- ная (а) Светло-желтая —> пурпурная (а)
Таблица 64
Щелочной растворитель Индикатор Переход окраски в среде: (а) щелочной-* кислой, (б) кислой-* щелочной
н-Бутиламин, пири- дин, диметилформ- амид Этилендиамин Этилен диамин, н- бутиламин, пиридин, диметилформамид 0,3 %-ный раствор тимолового синего в метиловом спирте или диметилформамиде 0,15%-ный раствор нитроанили- на в бензоле 0,05 Уо -ный раствор п-нитро-п'- аминоазобензола [798] в бензоле Насыщенный раствор азофиоле- тового в бензоле Красная —> желтая —> синяя (в зависимости от силы кисло- ты) (б) Желтая —> оранжевая (б) Красная —> (синяя) —> бес- цветная Кислоты: желтая —> (оран- жевая) —> синяя —> фиолето- вая (б); фенолы: оранжевая—> —> (красная) —> синяя —> фио- летовая (б)
П родолжение табл. 64
Щелочной растворитель Индикатор Переход окраски в среде: (а) щелочной-»• кислой, (б) кислой -»• щелочной
Диметилформамид 4,4-Диоксан 0,2%-ный раствор ализариново- го желтого в диметилформамиде 0,3%-ный раствор тимолового синего в диоксане 0,1%-ный раствор метилового красного в диоксане Желтая —> фиолетово-си- няя (б) Красная —> желтая —> си- няя (б) Желтая —> оранжевая —> ро- зовая —> темнеет (а)
Таблица 65
Выбор индикатора для применения в смесях растворителей различного состава
Смесь растворителей Индикатор Переход окраски в среде: (а) щелочной -»• кислой, (б) кислой-+щелочной
Ацетон — уксусная кислота 0,25%-ный раствор метилового оранжевого в ацетоне Желтая —> оранжевая (а)
Ацетон—пиридин 0,3%-ный раствор тимолового синего в метиловом спирте Насыщенный раствор азофиоле- тового в бензоле Желтая —> синяя (б) Желтая —> оранжевая —* си- няя (б)
Ацетон — уксусная Насыщенный раствор метилово- Оранжевая —> розовая цвета
кислота го красного в ацетонитриле семги (а)
Ацетон — уксусная кислота — уксусный ангидрид Насыщенный раствор метилово- го оранжевого в ацетоне Желтая —> оранжевая (а)
Ацетонитрил — хло- Насыщенный раствор метилово- Оранжевая —> розовая —
роформ—фенол го красного в ацетонитриле фиолетово-красная (а)
Бензол — уксусная 0,5%-ный раствор 1-нафтолбен- При обратном титровании
кислота зеина в уксусной кислоте ацетатом натрия: темно-зеле- ная —=” травянисто-зеленая — —> желтая (б)
Бензол — изопропи- 0,1—1,0%-ный раствор 1-наф- Оранжевая —> коричневато-
ловый спирт толбензеина в изопропиловом спир- те или смеси бензол — метиловый спирт зеленая (б)
Бензол—метило- 0,3%-ный раствор тимолового Красная —> желтая —си-
вый спирт синего в метиловом спирте няя (б)
Бензол — нитроме- тан Бензол—пиридин 1%-ный раствор тропеолина 00 в метиловом спирте Аналогично смеси ацетона и пи- ридина Желтая —розово-красная (а)
Дихлорэтан—ук- сусная кислота—ук- сусный ангидрид 0,5%-ный раствор тропеолина 00 в уксусной кислоте^ Желтая —* розово-красная (а)
Диоксан — уксусная 0,2%-ный раствор кристалличе- Фиолетовая —> голубая —>
кислота—уксусный ангидрид ского фиолетового в уксусной кислоте зелено-желтая (а)
Диоксан — хлоро- форм 0,1 %-ный раствор конго красно- го в метиловом спирте Красная —синяя (а)
Уксусная кислота— 0,5 %-ный раствор тропеолина Желтая —* оранжевая —* пур
бензол в уксусной кислоте пурная —* розово-красная (а)
Уксусная кислота— дихлорэтан и ук- сусная кислота—ди- оксан 0,1—1,0%-ный раствор метило- вого фиолетового в уксусной кис- лоте Синяя —=” зелено-желтая (а)
Уксусная кислота— 0,2 %-ный раствор метилового Фиолетовая —> синяя —зе-
нитрометан фиолетового в хлорбензоле 0,5 %-ный раствор малахитового леная (а)
Уксусная кислота— Оттитровывается ацетатом
уксусный ангидрид зеленого в уксусной кислоте 0,5%-ный раствор крезолового красного в смеси уксусная кисло- та — хлорбензол натрия: зелено-желтая —=” жел- то-зеленая (б) Желтая —> розовая —> крас- ная (а)
Индикаторы
193
Продолжение табл. 65
Смесь растворителей
Индикатор
Переход окраски в среде:
(а) щелочной -+ кислой,
(б) кислой -> щелочной
Уксусная кислота—
хлорбензол — ук-
сусный ангидрид
Уксусный ангид-
рид — бензол
Уксусный ангид-
рид — нитрометан
G — Н-смесь раство-
рителей
Гексан — ацетон
Метилэтилкетон —
уксусный ангидрид
Нитрометан — бен-
зол
Нитрометан — му-
равьиная кислота
Низкокипящий пет-
ролейный эфир — изо-
пропиловый спирт
Пропиленгликоль —
хлороформ
0,2 %-ный раствор метилового
фиолетового в хлорбензоле
Аналогично смеси уксусная
кислота — бензол
0,5 %-ный раствор 1-нафтолбен-
зеина в уксусной кислоте
0,1%-ный раствор метилового
оранжевого в метиловом спирте,
диоксане
Водный раствор 0,15% метило-
вого оранжевого —J-0,08% ксилол-
цианола FF
0,05—0,1 %-ный раствор мети-
лового красного в метиловом
спирте
1%-ный раствор диметилового
желтого в хлороформе
0,1%-ный раствор метилового
красного в уксусной кислоте
0,1 %-ный раствор метилового
фиолетового в уксусной кислоте
Раствор 0,1% метиленового си-
него + 0,2% хинальдинового
красного в метиловом спирте
Аналогично смеси нитрометан —
бензол
0,1—1,0%-ный раствор 1-наф-
толбензеина в изопропиловом
спирте
0,2 %-ный раствор тимолового
синего в метиловом спирте
Фиолетовая —> голубая —>
—> желто-зеленая (а)
Желтая —> травянисто-зеле-
ная —> темно-зеленая (а)
Желтая —> оранжевая (а)
Зеленая —> оранжевая (а)
Желтая —> ярко-розовая (а)
Желтая —> оранжевая (а)
Оранжевая —> ярко-розо-
вая (а)
Фиолетовая —> зелено-си-
няя (а)
Пурпурная —> синяя —> зеле-
ная (а)
Оранжевая —> коричневато-
зеленая (б)
Желтая —> розовая (а)
Таблица 66
Класс соединений
Индикатор
Азосоединения
Сульфофталеины
Фталеины
Нитросоединения
Производные трпфенил-
метана
Ализариновый желтый R, метаниловый желтый, диметило-
вый желтый, метиловый оранжевый, метиловый красный,
тропеолпн ОО, конго красный
Крезоловый красный, бромфеноловый синий, бромтимоловый
синий, бромкрезоловый зеленый
Фенолфталеин, тимолфталеин
о-Нптроанилин
Метиловый фиолетовый, малахитовый зеленый
Приведенный в табл. 61—65 список индикаторов не является полным,
так как в практике неводного титрования число применяемых индикаторов
быстро увеличивается. Большое число индикаторов, приводимых в опубли-
кованных работах, не означает, что все они обычно употребляются для
серийных анализов. Следует отметить, что в настоящее время, кроме боль-
шого числа смесей растворителей, применяются уже более 40 чистых раство-
рителей и в практике титрования насчитывается около сорока индикаторов
[775]. Из сказанного ясно, что здесь не представляется возможным ознако-
мить читателя со всеми комбинациями этих веществ.
194
Глава 13
В отношении концентраций растворов индикаторов нельзя дать каких-
либо общих указаний. На практике достаточно бывает добавить от 1 до
5 капель относительно разбавленного раствора (0,1—1%) к раствору 10—
30 мл. При полумикро- и микротитрованиях, особенно при серийных анали-
зах, проводимых с использованием растворов сравнения, рекомендуется
пользоваться более разбавленными (0,01%) растворами индикатора, добавляя
при этом точно 1,0—5,0 мл при помощи, например, автоматической пипетки,
а не вручную. При микротитрованиях важно, чтобы растворитель индикато-
ра был идентичен или по меньшей мере аналогичен растворителю титруемого
вещества. Например: 10 мл 0,002 М основания алкалоида в растворе хло-
роформ — уксусная кислота (10 : 1) титруют 0,01 н. хлорной кислотой
в растворе уксусная кислота — четыреххлористый углерод (1:1). В этом
случае разбавленный раствор индикатора должен содержать кислоту и инерт-
ный растворитель в таком соотношении, чтобы не снижалось усиливающее
влияние инертного растворителя. Однако может случиться, что в смеси
растворителей, идентичной по составу со смесью, примененной для раство-
рения титруемого вещества, индикатор будет неустойчив или будет лишь
незначительно растворяться. В таком случае непосредственно перед титро-
ванием готовят очень сильно разбавленный раствор индикатора в соответ-
ствующем объеме, нейтрализуют его и из полученного раствора приготовля-
ют запасной раствор, содержащий вещество, подлежащее анализу.
По этой причине трудно точно оценить влияние смесей растворителей,
обсуждаемых в специальной литературе, так как в конце титрования полу-
чается смесь растворителей, отличающаяся от исходной. Поэтому при фото-
метрическом титровании из микробюретки добавляют стандартный раствор
такой концентрации (например, 1,0—2,0 н.), чтобы концентрация индика-
тора в стандартном и в исследуемом растворах была идентична.
Фотометрическое титрование дает значительно более точные результаты
по сравнению с обычным визуальным титрованием. Однако недостатком фото-
метрических титрований является необходимость применения спектрофото-
метра. В тех случаях, когда четкость конечной точки невысока, проще
и удобнее пользоваться методом контроля поглощения раствора при помо-
щи, например, ступенчатого фотометра Пульфриха с применением разбавлен-
ного раствора индикатора; после добавления избыточного количества стан-
дартного раствора применяют обратное титрование, добавляя по каплям
запасной раствор исследуемого вещества до тех пор, пока не будет достигну-
то ранее определенное поглощение. Большинство индикаторов (кристалли-
ческий фиолетовый, метиловый красный, метаниловый желтый, ализари-
новый желтый Бит. д.), употребляемых при неводных титрованиях, дает
значительное смещение максимума поглощения вблизи точки эквивалентно-
сти, что позволяет легко контролировать перетитрование. Таким образом,
объем израсходованного стандартного раствора эквивалентен стандартному
объему запасного раствора, например, 20 мл плюс количество запасного
раствора, израсходованного для обратного титрования, например 0,15 мл,
т. е. всего 20,15 мл.
72. ПЕРЕХОД ОКРАСКИ КРИСТАЛЛИЧЕСКОГО ФИОЛЕТОВОГО
Кристаллический фиолетовый содержит ион три- (п-диметил аминофе-
нил) карбония, центральный атом углерода которого координационно нена-
сыщен и имеет ионный характер ([187] и стр. 285 в работе [597])
R3C+C1- (или ОН-)
где
R = n-(CH3)2NC6Hj
Центральный атом углерода благодаря своей электронной ненасыщенно-
сти обладает электроноакцепторными свойствами (антиауксохром); аромати-
Индикаторы
195
ческие кольца образуют сопряженную систему между этим атомом углерода
и электронодонорной диметиламиногруппой (ауксохром). Ауксохром и анти-
ауксохром находятся в сопряжении, и, следовательно, для них могут быть
написаны резонансные структуры. Резонанс обусловливает поглощение света
этими соединениями в видимой области [80, 726, 881].
Атомы азота кристаллического фиолетового также являются координа-
ционно ненасыщенными. Поэтому ионизированный краситель может реаги-
ровать как основание, способное связывать несколько протонов. По мере
присоединения протонов взаимодействие нуклеофильных аминогрупп
с электрофильным центральным атомом углерода постепенно уменьшается
и вследствие этого изменяется окраска индикатора
в + Н+
Желтый
Для равновесия в системе кристаллический фиолетовый —'хлорная кислота
Кольтгоф и Бруккенштейн предложили следующие уравнения:
/+С1О7 + НС1О4 ^2 /Н2+(С1О4)2
7Н2+(С10г)24-НСЮ4 JH3+(C10j)3
/+С1О7 и 7Н2+(СЮ7)2 имеют максимумы поглощения при 590 и 630 нм соот-
ветственно, а 7Н3+(С1О7)з — ниже 450 нм.
Таким образом, переход окраски кристаллического фиолетового является
постепенным и резкость каждого перехода понижается по мере присоедине-
ния протонов, так как при присоединении второго и третьего протонов обра-
зуется соединение, близкое к положительно заряженному ионизированному
красителю. Однако в уксусной кислоте первый переход очень резок, посколь-
ку сродство аналогов оснований к протону значительно больше в уксусной
кислоте, чем в воде, и сила катионных кислот значительно понижается.
(Кристаллический фиолетовый является аналогом основания, пока он не
присоединит протона, но в то же время он является катионной кислотой вви-
ду своего положительного заряда.) Во время титрования максимум погло-
щения кристаллического фиолетового составляет 591 нм за 1% до точки
эквивалентности (фиолетовая окраска), 595 нм в точке эквивалентности
196
Глава 13
(синяя окраска) и 627 нм при перетитровании (зелено-синяя окраска). Опас-
ность перетитрования таким образом практически устраняется, так как+1 %
титранта сопровождается смещением максимума поглощения на 36 нм [312].
Следует отметить, что чувствительность глаза очень велика. Так, разли-
чие между зеленой и светло-зеленой окрасками ясно замечается даже тогда,
когда разделение максимумов, измеряемое спектрофотометром, составляет
всего 2 нм. При титровании основных п-толуолсульфонатов и этансульфона-
тов хлорной кислотой в диоксане при концентрации 10~2 М ошибка визуаль-
ного наблюдения, например при установлении различия между сине-зеленой
и зеленой — светло-зеленой окрасками соответственно может вызвать погреш-
ность 1—2% в случае оснований средней силы и 2,5% для слабых оснований.
Переход окраски кристаллического фиолетового в диоксане происходит
постепенно (табл. 67).
Таблица 67
Окраска Максимум поглощения, нм А, нм Окраска Максимум поглощения, нм А, нм
Фиолетовая 594—598 4 Зеленая (изумруд- но-зеленая) 632—636
Синяя 598—602 2
24 Светло-зеленая 634—638
Зелено-синяя —сине- 622—626 3
зеленая 10 Желто-зеленая 636—642
Титрование слабых оснований, например анилина, в уксусной кислоте
не вызывает резкого перехода окраски кристаллического фиолетового.
В этом и аналогичных случаях правильный оттенок окончательно определяет-
ся потенциометрически [15, 208], а степень поглощения света определяется
при данной длине волны для данных концентраций индикатора и перхлората
основания. На основании этих данных можно затем определить конечную
точку титрования.
Для титрования аминов в ледяной уксусной кислоте был предложен в качестве индика-
тора 2-пиридилазо-п-диметиланилин. Точность результатов такого титрования превосхо-
дит точность результатов, получаемых с кристаллическим фиолетовым [120].
Переход окраски кристаллического фиолетового при определении слабых
оснований с помощью 0,001 н. хлорной кислоты [319]. Как уже упоминалось
выше, согласно общепринятому методу, правильный переход окраски про-
веряется электрометрически в присутствии данного вещества и индикатора
в идентичном растворителе. В случае необходимости титрование проводят
до идентичного оттенка окраски, наблюдаемой при стандартизации титранта
по дифенилгуанидину или бифталату калия и т. д. Однако этот способ может
привести к ошибочным результатам. Если, например, 1т-фенил-2,3-диметил-
4-диметиламинопиразолон (пирамидон) титруют в присутствии кристалли-
ческого фиолетового 0,001 н. хлорной кислотой в смесях уксусной кислоты
с четыреххлористым углеродом (10 : 1, 1:1, 1 : 10) и титр стандартного
раствора устанавливают в аналогичной смеси растворителей по дифенил-
гуанидину примерно такой же молярной концентрации, то величина 100 ±
± 0,25% может быть получена только в смеси растворителей 1:1, где отте-
нок окраски обоих оснований весьма различной силы идентичен в конечной
точке. Дифенилгуанидин — сильное (К = 6,1 ЛО-5), а пирамидон — слабое
основание (К = 6,0-IO-10).
В дифференцирующей смеси растворителей (1 : 10) результат для пира-
мидона составлял 99,4—100,2%, но только в том случае, когда оттенки окра-
ски при титровании стандартного вещества и исследуемого основания не
были одинаковыми. В смеси растворителей с выравнивающим действием
Индикаторы.
197
(10 : 1) возможная ошибка определения была больше: ±0,8%. В последнем
случае результаты, полученные в интервале изменения оттенка окраски от
красно-фиолетового и бирюзово-голубого, колебались в пределах от 80 до
120% в зависимости от коэффициента, получаемого для дифенилгуанидина.
Поэтому при микротитровании слабых оснований рекомендуется уста-
навливать титр 0,001 н. раствора по идентичному соединению стандартной
чистоты или по основанию, содержащему идентичные функциональные груп-
пы, и почти идентичной силы с исследуемым основанием. При неводном
Количество 0,ДО/н.НС1О4, мл
Рис. 70. Различные переходы окраски
кристаллического фиолетового при изме-
нении состава смешанного растворителя
(СН3СООН — СС14). Титрование амидопи-
рина [319].
Рис. 71. Различные переходы окраски
кристаллического фиолетового при изме-
нении состава смешанного растворителя
(СН3СООН — СС14). Титрование дифенил-
гуанидина [319].
титровании, особенно при применении концентраций 0,001 н., по-видимому,
для установки титра необходимо пользоваться серией стандартных веществ,
обладающих основностью в пределах Къ 10-6—10-12.
Усиливающее влияние инертных растворителей на переход окраски при
микротитрованиях показано на рис. 70 и 71.
73. ПЕРЕХОД ОКРАСКИ ТИМОЛОВОГО СИНЕГО
В водных средах с повышением основности окраска тимолового синего
переходит от красной к желтой (pH 1,5) и от желтой к синей (pH 8,9). В невод-
ных средах этот индикатор ведет себя аналогично и, следовательно, пригоден
для определения смесей кислот и аналогов кислот в метиловом спирте, диок-
сане, ацетоне, ацетонитриле и т. д.
Так, например, в ацетоне в зависимости от кислотности можно наблюдать
следующие окраски:
С соляной кислотой
С хлорангидридами
С азотной кислотой, карбоновыми кислотами,
их ангидридами, фенолами, солянокислыми
аминами
С алкоголятами щелочных металлов или
(C4H9)4NOH
Красная
Розовая
Желтая
Синяя
198
Глава 13
Если в качестве стандартного раствора пользоваться более слабым осно-
ванием, например три-к-бутиламином, то в присутствии тимолового синего
можно титровать только сильные неорганические кислоты [623] (см. также
табл. 60).
74. ПЕРЕХОД ОКРАСКИ АЗОФИОЛЕТОВОГО
Поведение азофиолетового [4-(п-нитрофенилазо)резорцин] аналогично
поведению тимолового синего [307, 540, 798]. Таким образом, например,
в смеси ацетона и пиридина (4:1) наблюдаются следующие окраски в зави-
симости от силы кислоты:
Фенилхинолинкарбоновая кислота
Бензойная кислота
п-Оксипроппофенон
Фенол
8-Оксихинолпн
Лимонная
Темно-желтая
Оранжево-желтая
Красная
Фиолетовая
При титровании кислот метилатом калия окраска переходит от желтой
к красной и по добавлении нескольких избыточных капель стандартного
раствора — от красной до синей. С фенолами раствор вначале окрашен
в оранжевый цвет или в желто-красный, а для конечной точки характерна
явно синяя окраска, которая, однако, в зависимости от природы применяемо-
го растворителя может иметь различные оттенки (фиолетово-синий, василь-
ковый, серо-синий). В ацетоне азофиолетовый дает желтую окраску с хлор-
ной, азотной или бензойной кислотами, розовую — с антраниловой кислотой
и синюю — с фенолом или избытком (C4H9)4NOH. Такиура и Такино [798]
наблюдали следующие окраски, возникающие в конечной точке титрования
в пиридине (сокращено):
о- и л-Нитрофенол
n-Нитрофенол и п-оксиацетофенон
Эфиры n-оксибензойной кислоты
2,4-Динитрофенол
2,4,6-Тринитрофенол
2,4,6-Триоксибензальдегпд
Темно-красная
Зеленая
Синяя
Темно-коричневая
Темно-красная
Светло-фиолетовая
Из этих результатов следует, что при разработке нового метода — с при-
менением азофиолетового — практически необходим потенциометрический
контроль перехода окраски индикатора, и в отдельных случаях для установ-
ки титра должен применяться реагент, идентичный с исследуемым соеди-
нением, но обладающий стандартной чистотой.
75. ПЕРЕХОД ОКРАСКИ ДИМЕТИЛОВОГО ЖЕЛТОГО
Диметиловый желтый (4-диметиламиноазобензол) впервые был использован
при титрованиях в среде бензола [852]. Он применяется преимущественно
для микротитрований оснований в хлороформе и четыреххлористом угле-
роде. 0,005 н. раствор n-толуолсульфокислоты в хлороформе можно употреб-
лять для точного титрования ряда оснований алкалоидов в присутствии диме-
тилового желтого, который в конечной точке дает переход окраски от желтой
к розовой. Существуют две гипотезы относительно изменения структуры
4-диметиламиноазобензола при присоединении протона. Более ранняя, пред-
ложенная Ганчем и Хилыпером [353] на основании их спектроскопических
исследований, предполагает хиноидную перегруппировку (г, см. ниже).
С другой стороны, в присутствии сильных кислот данные УФ-спектра ука-
зывают на бензоидную структуру (в) [460, 568]. Большая основность диме-
тиламиногруппы по сравнению с азогруппой свидетельствует в пользу
последней теории. Однако после присоединения протона хиноидная система
Индикаторы
199
двойных связей не может включать атома азота диметил аминогруппы, так
как октет атома азота заполнен
(CH3)2N-C6H4-N = N-C6H5 <—> (CH3)2N ^>= N —n-c6h5
(CH3)2N-C0H4 — N = N-CeH5 ИЛИ (CH3)2N =/____\=N-N-C6H5
H г Н
в
76. ТОЧНОСТЬ ИЗМЕРЕНИЙ. РАСЧЕТЫ
При применении 0,1 н. — 0,01 н. хлорной кислоты для неводного титро-
вания вероятные пределы ошибки составляют от ±0,2 до ±0,4% в случае
оснований, константа диссоциации которых, измеренная в воде, не ниже
10-10. При использовании пропионовой кислоты вместо уксусной точность
может быть повышена до ±0,2% [313]
(рис. 72). В инертном растворителе точ-
ность определения 0,005 н. п-толуолсуль-
фокислотой даже при применении микро-
метода (например, 0,5—5 мг алкалоидов)
составляет ±1 % . В соответствующих усло-
виях опыта слабые основания (рК >• 12)
можно титровать с точностью ±1%. Точ-
ность определений сильно повышается при
определении конечной точки фотометри-
ческим методом даже в случае таких осно-
ваний, которые можно считать чрезвы-
чайно слабыми (например, мочевина, тио-
мочевина и т. д.).
На точность измерений отрицательно
влияет нечеткий переход окраски в конеч-
ной точке титрования. Ошибка постепенно
Рис. 72. Точность определения
оснований в зависимости от их силы
в воде [313].
1 — в уксусной кислоте; 2 — в пропионо-
вой кислоте; 3 — в хлорбензоле.
увеличивается из-за ступенчатого перехода окраски кристаллического
фиолетового или малахитового зеленого [463] (табл. 68).
Таблица 68
Переход окраски малахитового зеленого Ошибка определения
Сине-зеленая —> зеленая Зеленая —> желто-зеленая Желто-зеленая —* желтая ±0,18 ±0,52 ±1,6
Таблица 69
Среднее количество израсходо- ванного стандартного раствора, мл Число измерений Вероятные пределы ошибки одного измерения, %
2,06 10 -4-0,5
10,25 5 -4-0,3
51,40 8 ±0,7
Для получения более точных результатов при определении кислот и их
аналогов необходимо тщательно изолировать реакционную смесь от попада-
ния двуокиси углерода из воздуха. Чем меньше кислотность определяемого
вещества, тем большей основностью должен обладать растворитель, однако
вместе с увеличением основности возрастает и способность растворителя
поглощать двуокись углерода. Точность титрования сильных карбоновых
200
Глава 13
кислот и кислот средней силы или фенолов составляет от ±0,3 до ±0,8%,
тогда как для аналогов кислот с очень низкой кислотностью точность 1—2% .
При обсуждении данных анализа будут отмечены случаи, когда точность
титрования отличается от приведенных выше значений.
При неводных титрованиях переход окраски индикатора и промежуточ-
ные оттенки окраски зависят не только от количества используемого индика-
тора, силы кислоты или основания и концентрации ионов в растворе, но
также и от изменений в составе растворителя в процессе титрования. В смеси
пиридина с ацетоном наблюдается резкий переход окраски азофиолетового,
но добавление метилового спирта уменьшает резкость перехода. Нельзя
ожидать одинаковой точности при проведении анализа, если, например, при
титровании п-оксипропиофенона 0,1 н. раствором метилата натрия в смеси
бензол — метиловый спирт с содержанием метилового спирта 10 об. % на
20 мл раствора n-оксипропиофенона в смеси ацетона с пиридином (4 : 1)
израсходованы различные количества стандартного раствора: в одном случае
2 мл, во втором 10 мл и в третьем 50 мл. В первом случае содержание мети-
лового спирта в оттитрованном растворе составляет 1, во втором — 3,3
и в третьем — 7 об.%. В том случае, когда на титрование израсходовано
только 2 мл стандартного раствора, ошибка при измерении объема не соответ-
ствует точности анализа, в то время как при расходе стандартного раствора
50 мл уменьшается четкость перехода окраски индикатора за счет увеличе-
ния концентрации метилового спирта (табл. 69).
Хотя глаз человека и очень чувствителен к изменениям в оттенках цвета, все же
могут встречаться индивидуальные различия в восприятии, и, кроме того, чувствитель-
ность глаза меняется со временем (табл. 70).
Таблица 70
Аналитик Определение пикрата осно- вания полумикрометодом
«А» (с большим опытом) «Б» (малоопытный) Год спустя: «А» «Б» 99,7±0,2 (5 определений) 99,9-j-0,55(7 определений) 99,7±0,6 (4 определения) 99,8 ± 0,25 (6 определений)
Сходство этих результатов не обязательно свидетельствует о точности анализа.
Так, два аналитика в двух соседних лабораториях в одинаковых условиях анализировали
алкалоиды стандартной чистоты мпкрометодом, применяя один и тот же стандартный
раствор. Первый аналитик получил значение 100,0 ± 0,8%, а второй — 99,6 ± 0,3%.
Следует отметить, что точность метода, приводимая в литературе, зависит и от индивиду-
альных факторов, например чувствительности глаза аналитика к восприятию цвета.
Расчеты. Ниже кратко приведены расчеты, часто применяемые в тптрпметрпи.
Фактор стандартного раствора f
Е.
г G“&rifj
1= b ’
где амг — количество стандартного вещества, мг; Ег_экд — грамм-эквпвалентный вес
стандартного вещества; N — нормальность стандартного раствора; b — израсходован-
ный объем, мл.
Нормальность стандартного раствора N
а-100
'v=-—
где а — вес вещества, использованного для установки титра, г; Е — 1/10 эквивалентного
веса вещества для установки титра; с — израсходованный объем стандартного раство-
ра, мл.
Индикаторы
201
Эквивалентный вес анализируемого вещества Ег_экв
_а-1000
Е'г-экв ’
где а—вес исследуемого вещества, г; с — израсходованный объем, мл.
Процентное содержание определяемого соединения
а
где Е — 1/10 эквивалентного веса известного соединения.
Процентное содержание функциональной группы, например ОН
где 1,7—1/10 эквивалентного веса гидроксильной группы.
Если необходимо учитывать расход титранта на глухой опыт, то его вычитают
из объема, пошедшего на титрование в целом; когда объем титранта, израсходованного
в глухом опыте, больше объема, пошедшего на титрование, поступают наоборот — объем
раствора, израсходованного на титрование, вычитают из объема титранта, пошедшего
на глухой опыт.
Содержание определяемого соединения (%) •
(c — b)NE ,
а ’
где с — объем, израсходованный на титрование; b — поправка глухого опыта на раст-
воритель + индикатор
(b-c)NE
% =----------- ,
где b — объем стандартного раствора, пошедший на глухой опыт.
Подобный случай встречается, например, когда реагент представляет собой раствор
хлоргидрата гидроксиламина, содержащий трибутиламин. Раствор титруют соляной
кислотой, чтобы найти объем титранта, пошедший на глухой опыт; после образования
оксима выделившаяся соляная кислота реагирует с трибутиламином, и, таким образом,
объем стандартного раствора при последующем титровании будет меньше объема, израс-
ходованного в глухом опыте.
Глава 14
ФОТОМЕТРИЧЕСКОЕ ОПРЕДЕЛЕНИЕ КОНЕЧНОЙ
ТОЧКИ
Среди успешно применяемых физико-химических методов определения
конечной точки при неводном титровании использование поглощения моно-
хроматического (или почти монохроматического) света относится к числу
наиболее новых методов, хотя предварительные исследования такого рода
были проведены еще в начале этого столетия [806]. Начало этим исследова-
ниям положили Мюллер и Патридж [606], а основные принципы метода были
изложены Годдом и Хьюмом [275—277], Мика (см. стр. 52—60 в работе [584])
и Осборном с сотр. [617]. Позднее Хигучи и сотрудники детально разработа-
ли методику титрования в среде уксусной кислоты и опубликовали различ-
ные типы графиков и расчетов [148, 385, 390, 670, 671]. Большое количе-
ство исследователей занималось фотометрическим титрованием, изучая
поглощение в ультрафиолетовой области [86, 675, 679]. Эллерт и сотр. [209,
211] разработали фотометрический анализ некоторых лекарственных препа-
ратов. Несколько более трудной задачей является фотометрическое опреде-
ление фенолов, хотя в специальной литературе можно найти много примеров
практического применения этого метода [405]. Хидридж обсуждает метод
фотометрического титрования в своей краткой и поучительной монографии *.
77. ОСНОВНОЙ ЗАКОН ФОТОМЕТРИИ
При фотометрическом титровании стандартный раствор добавляется
к исследуемому образцу порциями по 0,01—0,2 мл и одновременно измеряет-
ся поглощение света, проходящего через раствор. Основной закон фотомет-
рии — закон Ламберта — Бера
Поглощение А (или оптическая плотность Е) = log -у- = &cd,
где 10 — интенсивность падающего света; I — интенсивность пропущенного
света; е — коэффициент поглощения; с — концентрация поглощающего
раствора; d — толщина слоя, т. е. длина пути света, проходящего через
поглощающую среду, см\ Т — (пропускание) = ~ и —log ~ = log Т =
*о, 'о
= log .
78. ПРЕИМУЩЕСТВА И ОГРАНИЧЕНИЯ МЕТОДА ФОТОМЕТРИЧЕСКОГО
ТИТРОВАНИЯ
Фотометрический метод определения конечной точки титрования часто
позволяет получать более точные результаты, чем потенциометрический
метод, особенно если используются логарифмические функции (ср. [774а]
и [7746]). Так, например, при фотометрическом титровании оксината алю-
миния точность достигает ±0,8%, тогда как потенциометрическое определе-
* Headridge I. В., Photometric Titrations, Pergamon Press, Oxford, 1961.
Фотометрическое определение конечной точки
203
ние дает ошибку ±2,3% [659]. Преимущество фотометрического титрования
по сравнению с прямыми колориметрическими определениями состоит в том,
что соединения, не участвующие в реакции, но поглощающие при той же
длине волны, что и реагирующие вещества, не мешают определению, так как
при фотометрическом титровании существенно лишь изменение поглощения
в точке эквивалентности. Фотометрическое титрование чрезвычайно полезно
при дифференцирующем титровании многокомпонентных систем. Этим мето-
дом можно определить концентрацию каждого компонента в смеси, состоящей
из ди-н-бутиламина (рК 2,6), ?<,?<-диэтиланилина (рК 7,5), анилина (рХ 9,4)
и о-хлоранилина (рК 11,4) [404]. Другое преимущество фотометрического
титрования заключается в том, что определение конечной точки не зависит
от измерений, проводимых в непосредственной близости от точки стехиомет-
рической эквивалентности. Две прямые части кривой титрования могут быть
экстраполированы, и проекция точки их пересечения на ось х даст точку
эквивалентности.
В то же время при фотометрическом титровании необходимо применять
спектрофотометры, которые часто нуждаются в некоторых усовершенство-
ваниях, а также специальные кюветы или сосуды для титрования. Из-за
изменения объема анализируемого образца приходится либо вводить поправ-
ки, либо вести титрование стандартными растворами высокой концентрации
(в последнем случае необходимо пользоваться микробюретками с ценой деле-
ния 0,01 мл).
79. СПОСОБЫ ФОТОМЕТРИЧЕСКОГО ТИТРОВАНИЯ
Необходимо различать так называемое прямое фотометрическое титро-
вание (самоопределяющиеся системы) и непрямое титрование (в систему
вводится индикатор). В первом случае измеряется изменение поглощения
(например, в ультрафиолетовой области) в точке эквивалентности либо
для стандартного раствора, либо исследуемого соединения, либо продукта
реакции; во втором — измеряется поглощение света добавленным индика-
тором. Поглощение света индикатором, добавленным в небольших количе-
ствах, как правило, изменяется линейно лишь вблизи точки эквивалентно-
сти [390].
80. ГРАФИЧЕСКОЕ ПОСТРОЕНИЕ КРИВЫХ ФОТОМЕТРИЧЕСКОГО
ТИТРОВАНИЯ.
ФОРМЫ И ТИПЫ КРИВЫХ ТИТРОВАНИЯ. ПРИМЕРЫ
Основной вывод из закона Ламберта — Бера состоит в том, что величи-
на А, измеренная спектрометрически, прямо пропорциональна концентрации
молекул поглощающего вещества. Если при данной длине волны при прове-
дении титрования свет поглощается либо стандартным раствором, либо
исследуемым веществом, либо продуктами реакции, строят кривую зависи-
мости измеренных значений А от объема израсходованного стандартного
раствора V. Если реакция закончилась, точка перегиба полученной кривой
указывает на конечную точку титрования. В идеальных условиях зависи-
мость А от V графически представляет собой две прямые линии, которые
можно экстраполировать до пересечения в точке эквивалентности.
Форма кривых титрования зависит от стандартного раствора, свето-
поглощающей способности анализируемого вещества или добавленного
индикатора, от длины волны света, от константы диссоциации окрашенного
соединения и от растворителя.
Для фотометрического титрования в неводных средах можно использо-
вать те же приборы, что и при анализах в водной среде. Однако следует
иметь в виду, что даже сильные электролиты не полностью диссоциируют
204
Глава 14
Рис. 73. Кривые фотометрического тит-
рования цинка дитизоном в бензоле при
640 нм [552].
1 — глухой опыт: 2 — стандартный раствор
цинка; 3 — масло, содержащее цинк.
на ионы в неводных растворителях. По этой причине индикаторы ведут
себя в неводных растворителях иначе, и не существует прямой корреляции
между отношением 1а11ь индикатора и значением pH, как это имеет место
в водной среде (см. гл. 13). Точность измерений, как и форма кривых титро-
вания, сильно меняется в зависимости от того, применяется ли прямое фото-
метрическое титрование или же становится необходимым добавление инди-
катора. В последнем случае результат фотометрического титрования
не всегда более точен, чем при применении визуального или потенциометриче-
ского способа определения конечной
точки [583]; однако в некоторых слу-
чаях точность этого метода все же пре-
восходит точность обычных методов
титрования [148, 385, 390, 670, 671].
а. Изменением поглощения света
в точке эквивалентности из-за избыт-
ка стандартного раствора пользу-
ются при определении содержания
цинка в бензольном растворе смазоч-
ного масла, содержащего диалкилди-
тиофосфат цинка, причем в качестве
стандартного раствора используется
0,05 %-ный раствор дитизона в бен-
золе (рис. 73) [552].
анализируемым веществом положено
б. Изменение поглощения света
в основу методики определения оксината алюминия и 8-хлорхинолина
титрованием раствором хлорной кислоты в уксусной кислоте (рис. 74 и 75)
[404, 659, 675].
в. Для фотометрического титрования с использованием индикаторов
Хигучи и сотрудники, а также Эллерт и сотрудники разработали детальные
Рис. 74. Кривая фотометрического
титрования оксината алюминия
(226 мг) при 450 нм [659].
Рис. 75. Кривая фотометрического тит-
рования 8-хлорхинолина (130 мг) при
380 нм [404].
методики. Форма кривой зависимости А от V и ЦТ от V во многих случаях
имеет сходство с некоторыми кривыми потенциометрического титрования.
Точка перелома кривых зависимости А от V и ЦТ от V является точкой,
в которой отношение первых производных достигает максимума (рис. 77
и 78). Чтобы точнее определить конечную точку титрования, т. е. точку,
в которой индикатор внезапно меняет цвет, можно также построить график
зависимости АЛ/АУ от V и A (1/T)/AV от V.
Для индикаторного метода можно применять, например, фотоколориметр
с кюветой 15 мм. В 20 мл уксусной кислоты растворяют навеску основания
порядка 0,5—1 мг-экв и титруют 0,1 н. раствором хлорной кислоты в уксус-
ной кислоте, добавив 2 капли 0,5%-ного раствора метиленового синего
в уксусной кислоте. Стандартный раствор вводят порциями по 0,05—0,2 мл
Фотометрическое определение конечной точки
205
и после добавления каждой порции измеряют поглощение света. Применяя
цветной фильтр (550—580 нм), можно анализировать большое число органи-
ческих оснований (рис. 76—78), например 1- и 2-нафтиламины, нарцеин,
адреналин, сульфатиазол, пиридин, солянокислый хинин (последнее веще-
ство в присутствии ацетата ртути(П) в растворе нитрометана, остальные —
в уксусной кислоте) [209]. При анализе кофеина форма кривых титрования
зависит от примененного индикатора. Так, например, примечательная кри-
вая получается в присутствии сафранина (смесь диаминопроизводных соли
фенилфеназония) в среде уксусного ангидрида: при добавлении последних
Рис. 76. Кривые фотомет-
рического титрования нар-
цеина в присутствии мети-
ленового синего с фильтром
при 570—600 нм (7) и при
550—580 нм (2).
Рис. 77. Фотометрическое
титрование!' 1-нафтиламина
(7) и 2-цафтил амина (2)
в присутствии индикатора
метиленового синего [209].
Рис. 78. Фотометрическое
титрование 1-нафтиламина
(7) и 2-нафтил амина (2)
в присутствии индикатора
метиленового синего [209].
двух капель 0,1 н. раствора хлорной кислоты поглощение света раствором
внезапно возрастает, а затем заметно уменьшается (рис. 79, кривая 4). Конеч-
ная точка титрования определяется из этой кривой непосредственно [211].
При фотометрическом титровании, кроме уже упомянутых зависимостей,
можно получить кривые зависимостей ZH+/Z от V, 1/1Н+ от V, 1Н+/1
от HV и т. д.
Фотометрическая кривая типа I [390]. При обычном визуальном опре-
делении конечной точки титрования, когда индикатор присутствует лишь
в очень небольших количествах, интенсивность окраски изменяется про-
порционально количеству стандартного раствора вплоть до точки эквива-
лентности. При титровании слабой кислоты сильным основанием в водных
растворах имеет место следующее равновесие:
Л- + 7Н+ ЛН + 7,
где ЯН и А~ —слабая кислота и сопряженное с ней основание соответ-
ственно; ZH + и Z — протонированная (кислота) и основная форма индика-
тора. Следовательно, константа равновесия
к_ МН] [7] _ К,
[Л-] [ZH+] КА '
Примем, что количество анализируемой кислоты в титруемой системе экви-
валентно объему S стандартного раствора щелочи, а X — объем стандарт-
ного раствора, прилитого в данный момент титрования. Тогда
S — X
X
К
(80.1)
206
Глава 14
В системах, для которых значение К меньше 0,05 и где X приближается
к величине S, фотометрическая кривая вблизи точки эквивалентности пред-
ставляет собой почти прямую, т. е.
ХК (80.2)
Если построить зависимость значения Ш+/1, найденного из величины
поглощения, от X, получается прямая линия, пересекающая ось X в точке
эквивалентности (X = S).
Когда слабые кислоты титруют сильными основаниями, отношение 1Н+/1
уменьшается в процессе титрования. Зависимость такого типа можно исполь-
зовать для расчетов лишь в том случае, если значение К невелико, а значе-
ние 7Н+/7 становится меньше 1 лишь к концу титрования (>99%). Такой
Р п с. 79. Кривые фотометрического
титрования кофеина в уксусном ангид-
риде [211].
Индикаторы: 1— малахитовый зеленый; 2—ниль-
ский голубой; 3—нейтральный красный; 4—Су-
дан III; <5 —сафранин.
Рис. 80. Фотометрические кривые
типа I [390].
Идеализированные кривые: К = 0,1, 0,01
и 0,001. А—идеализированные кривые тит-
рования кислот; Б — фотометрическое титро-
вание 242,6 мг трифенилгуанидина 0,25 н.
хлорной кислотой в присутствии хинальдино-
вого красного (533 н.м).
случай представлен на рис. 80, где показана конечная стадия трех идеальных
фотометрических кривых для К = 0,1, К = 0,01 и К = 0,001. Полученная
при фотометрическом титровании кривая зависимости /Н+/7 от V (а для
оснований кривая зависимости 77/Н + от V) при экстраполяции пересекает
ось х, она известна под названием кривой типа I [390] (рис. 80, кривые А и £>).
Фотометрическая кривая типа II [390]. В некоторых случаях не удается
применить индикаторы с величиной К порядка 0,01 и меньше, а иногда
бывает необходимо использовать индикатор с относительно высоким значе-
нием К, например 0,05, так как надо определить более сильную кислоту
в присутствии более слабой. В этих случаях можно использовать другую
модификацию уравнения (80.1)
т- (зо-з)
Если выразить графически зависимость экспериментально найденного
значения /Н+// от величины, обратной значению объема израсходованного
стандартного раствора, то пересечение прямой с осью х наступит при
ИХ = 1/5, т. е. когда титрование достигнет точки эквивалентности. Кри-
Фотометрическое определение конечной точки
207
выми типа II называются те фотометрические кривые, которые выражают
зависимость 7Н+/7 от 1/V (для кислот) и зависимость Z/ZH + от 1/V (для осно-
ваний) (рис. 81).
Применение модифицированной кривой титрования типа II при анализе
небольших количеств очень слабых оснований [148]. Построение кривых типа II
для чрезвычайно слабых оснований иногда приводит к неточностям в ана-
лизах. Поскольку образующийся перхлорат основания претерпевает сольво-
лиз даже в уксусной кислоте, можно рекомендовать применение другого
графика, хотя в этом случае придется провести
титрование дважды с количествами вещества
20—30 и 5—7 мг. Реакцию можно представить
следующим образом:
^обм
ВНЛ + 7 -' /НЛ + B.J
(80.4)
где I — например, нильский голубой А, а В — моче-
вина. Обозначение, использованное для константы
равновесия вышеприведенной схемы, выражает кон-
станту обмена
1,2
0.8
0,4
°0,05 0,15 0,25 0,35
мл~1
Рис. 81. Кривые фото-
метрического . титрования
. типа II.
Титрование мочевины хлорной
кислотой в присутствии индика-
тора нильского голубого А (I;
Кэксп = 0;47) или малахитового
зеленого (2; Кэксп = 2,5) [390].
izJHA
г ____ сгна'св ___ обр
Л»6-- С,-СВНА - КВНЛ •
(80.5)
где КобрА — константа образования В НА,
—’константа образования кислой формы
индикатора, которая относится
равновесия В + АН В НА.
к положению
Диссоциацией образовавшегося вещества можно пренебречь ввиду низкой
диэлектрической проницаемости уксусной кислоты [101, 102, 464].
Уравнение (80.6), полученное из (80.4), представляет условие равновесия
при титровании [148]:
(80.6)
где S — объем стандартного раствора кислоты, эквивалентный навеске
основания; X — объем добавленного стандартного раствора в любой момент
титрования; 1вИа — экспериментально найденное соотношение основной
и кислой форм индикатора.
Если графически выразить зависимость величины соотношения основной
и кислой формы индикатора от 1/Х, то получится кривая типа II. Если
принять во внимание сольволиз в растворе уксусной кислоты, но пренебречь
диссоциацией, то общая концентрация хлорной кислоты в процессе титро-
вания основания перед конечной точкой составит X', а общая концентрация
основания будет равна S, т. е.
х' = Снсю4 + Свнскц (80.7)
8' = с в + Свнсю4 (80.8)
Между концентрациями X' и S' и упомянутыми выше объемами X и S
существует следующая связь:
v, XN q, S-N
Х =— и 5 = —
где N — нормальность стандартного
ренного образца.
Примем, что объем стандартного
момент титрования первого раствора
раствора; V — общий объем раство-
раствора кислоты в определенный
(меньшая навеска) равен Xit объем
208
Глава 14
второго раствора (большая навеска) равен Х2, а концентрация кислоты,
эквивалентной основанию, соответственно равна и S2. При заданном
постоянном отношении 1вИд титрование можно представить уравнением
При данном значении 1ВИА величина Н(Х2 — Х^ может быть рассчитана
из кривых зависимости IBUA от 1/Х4 и 1вИд от 1/Х2 (см. рис. 82).
Пересечение прямой на графике «модифицированной кривой титрования
типа II» с осью X дает значение величины S2 — Si, т. е. точка пересечения
представляет собой исправленную точку эквивалентности для количества
вещества, представляющего собой разность между второй и первой навесками.
Пользуясь этим методом, удалось получить очень точные результаты при
титровании в присутствии нильского голубого А в качестве индикатора сла-
бых и чрезвычайно слабых оснований (табл. 71).
Таблица 71
Основания а Вероятная ошибка единич- ного определе- ния, % ^обм Основания а Вероятная ошибка единич- ного определе- ния, % ^сбм
Ацетамид 1,7 3,66 Кофеин 0,9 0,16
Мочевина 1,6 0,46 Антипирин 0,8 0,00
2,6-Д иметил-у-пи-' 0,3 0,28 Трифенилгуани- 0,7 0,00
рон Т иомочевина 0,5 0,25 дин
а Первая навеска 5 — 7 мг, вторая — 25 — 35 мг; стандартный раствор—0,1 н. хлорная кислота
в уксусной кислоте.
При построении модифицированных кривых титрования типа II конечную точку
можно определить с высокой точностью, если величина ЛСОбм очень мала. Этот метод под-
счета, однако, в основном применяется для определения основности, причем в этом слу-
чае рекомендуется применять дифференцирующий индикатор, значение константы обмена
которого больше нуля.
Для проведения анализов с использованием «модифицированных кривых титрования
типа II», как уже упоминалось, пригодны колориметры фирмы «Bausch and Lomb Spectro-
nic 20» и циркуляционный колориметр Эрленмейера (см. рис. 99). Раствор индикатора
в уксусной кислоте смешивают с раствором хлорной кислоты в уксусной кислоте в таком
соотношении, чтобы он содержал достаточно высокую концентрацию каждого вещества.
Нормальность раствора индикатора и хлорной кислоты устанавливают по бифталату
калия в присутствии 1-нафтолбензеина. Небольшие количества индикатора, например
нильского голубого А, не влияют на четкость перехода окраски 1-нафтолбензеина.
На рис. 82 представлены две кривые и «модифицированная кривая», полученные при
таком титровании.
Расчеты с использованием «модифицированной кривой типа II» можно
применять для определения (или оценки) основности таких слабых основа-
ний, как хлорацетамид или диэтиловый эфир [385]. Сольволиз очень слабых
оснований весьма значителен даже в уксусной кислоте. Концентрации кис-
лоты и основания выражаются уравнениями (80.7) и (80.8). Видоизмене-
нием этих уравнений является уравнение (80.9)
(80.9)
снсю4 нужно определять независимо от результатов глухого опыта с уксус-
ной кислотой, содержащей индикатор, например Судан III, в той же ко-
центрации, что и анализируемый раствор (см. рис. 85, кривая Б). Для рас-
четов находят зависимость величины (1/Х) —снсю4 от I в/1 л. Поскольку
Фотометрическое определение конечной точки
209
в глухом опыте индикатор является единственным основанием, величина
£нсю4 пропорциональна индикаторному отношению данного индикатора
и может быть рассчитана по реакции образования перхлората индикатора
/+НСЮ4 ZHC1O4.
с1НС104
с1’сНС104
где
к-1НС104
Л-обр =
СНСЮ*
Зависимость IАЦ в от снсю4 имеет вид прямой, проходящей через начало
координат (рис. 85, кривая Б). Очевидно, количество кислоты, идущее
Рис. 82. Кривые фотометрического тит-
рования «модифицированного типа II».
Фотометрическое титрование мочевины
0,098 н. хлорной кислотой в присутствии
нильского голубого А при 632 нм [148].
1 — кривая титрования 24,2 мг мочевины; 2 —
кривая титрования 4,84 мг мочевины; 3 — раз-
ностная кривая титрования (19,36 мг).
Рис. 83. Фотометрическое титрование
хлорацетамида 0,5 н. хлорной кислотой
в присутствии индикатора судан III [385].
1 — кривая «модифицированного типа II»; 2 —
кривая типа II.
на образование перхлората, например, Судана III, очень невелико. Кон-
станта образования Судана III равна примерно 700 [389].
В уксусной кислоте в качестве основания выступает вода, и чтобы исклю-
чить ее конкурирующее влияние, уксусная кислота, употребляемая в каче-
стве растворителя, должна содержать 0,25 молъ/л уксусного ангидрида
(табл. 72).
Таблица 72
Основность некоторых соединений в растворе уксусной кислоты по сравнению с
Суданом III
Соединение ^обм Соединение ^обм
1-Ацетилпиперидин N,N-Диметилформамид Хлорацетамид 2,4,6-Т рибромацетанилид 0,014 0,14 2,7 16 Диэтиловый эфир 5-Этил-5-фенилбарбитуровая кислота (фенобарбитал) 1400 Не прояв- ляет основ- ных свойств
На рис. 83 показано, что для такого слабого основания, как хлорацета
амид, кривые титрования различного типа имеют различный тангенс угл-
наклона. Точное значение Хобм находят, используя «модифицированную
кривую типа II» (кривая Л).
210
Глава 14
Величина Лобм с данным индикатором может быть рассчитана следующим образом:
^обм- !b/Ia S
или
1/(Х —сНС1О1) —1/5
----------------s-
На рис. 84 представлены идеализированные кривые, тангенсы углов
наклона которых соответствуют величинам Лобм Для оснований различной
силы (взяты эквимолекулярные количества оснований; S = 3 мл стандарт-
ного раствора).
«Фотометрические кривые типа III» [671]. Смысл этого метода в построе-
нии зависимости изменения поглощения от объема израсходованного стан-
дартного раствора после установления точки эквивалентности. Избыток стан-
Р и с. 84. Идеализированные тан-
генсы дляЛГэксп 0»1; 1 »0; 2,5; 5,0и 10.
Рис. 85. Кривые фотометрического титрования
типа III [671].
А — титрование 65,6 мг мочевины в присутствии индика-
тора Судан III (615 юн); Б — глухое титрование 25 мл
уксусной кислоты, содержащей 0,2% воды и судан III.
дартного раствора равен (X — S), где X — общее количество добавленного
стандартного раствора, S — объем стандартного раствора, в действитель-
ности эквивалентный определяемому соединению
X-S = K^Vt
где V — общий объем титруемой системы.
При анализе оснований находится зависимость ГН+Ц от X и пересечение
экстраполированной прямой с осью X дает точку эквивалентности. Для
этого типа титрования применимы те индикаторы, переход окраски которых
происходит только в большом избытке хлорной кислоты. Переход окраски
соответствует равновесию
Z4-HCIO4 ZHCIO4,
для которого
к_ ИНСЮ4]
[ZJ [НС1О4] ’
Если преобразовать предыдущее равенство
[НСЮ4] = К
Фотометрическое определение конечной точки
211
и построить график зависимости [ZHC1O4]/[Z] от объема хлорной кислоты,
то получится прямая линия, пересечение которой с осью х соответствует
точке эквивалентности (рис. 85, кривая А: титрование мочевины хлорной
кислотой в присутствии Судана III и кривая Б: холостой опыт титрования
уксусной кислоты, содержащей Судан III).
Судан III С6Н5 — N = N — С6Н4 — N = N — С10Н6ОН в основной
форме окрашен в желтый цвет, в кислой — в синий. Его кажущаяся основ-
ность настолько невелика, что в 0,05 н. растворе перхлората мочевины
в уксусной кислоте лишь 10% индикатора переходит в кислую форму с мак-
симумом поглощения при 615 нм. Зависимость типа III имеет вид прямой,
Рис. 86. Фотометрическое тит-
рование о-хлоранилина (312 нм)
[675].
если приведенное ниже равновесие значи-
тельно смещено влево
74-BH+ClOj /H+ClOj + B,
где В —титруемое основание, аВН+С1О7—
ионная пара перхлората основания. Отно-
нм
Рис. 87. Изменение пропускания света [675].
1 — ион о-хлоранилиния; 2 — о-хлоранилин (5-Ю-4 н.);
3—хинолин (5-10-4 н.); 4—ион хинолиния.
шение кислой формы индикатора к основной определяется по поглощению
IR+/I = (A- AB)/(AS — А),
где As — оптическая плотность перхлората индикатора, А в — оптическая
плотность основной формы индикатора, А — оптическая плотность, най-
денная из измерений.
Оптическую плотность кислой формы индикатора определяют в конце
титрования после того, как прибавят большой избыток хлорной кислоты’,
величину оптической плотности для основной формы индикатора измеряют
перед началом титрования.
Об использовании бриллиантового зеленого, родамина Б и] виктории синей
см в работе [463].
г. Фотометрическое титрование оснований с использованием поглощения
в ультрафиолетовой области. Этот метод титрования, впервые примененный
Брикером и Суитсером [86], имеет много преимуществ, так как большое
количество органических соединений обладает значительным поглощением
не в видимой, а в ультрафиолетовой области. Этот метод дает результаты,
очень хорошо согласующиеся с законом Ламберта — Бера, поскольку
благодаря высокой чувствительности метода можно использовать сильно
разбавленные растворы [675], как, например, при титровании о-хлорани-
лина (рис. 86).
Наиболее удобная длина волны для измерения о-хлоранилина и иона
о-хлоранилиния определяется эмпирически, и при этой длине волны (312 нм)
затем ведут титрование (рис. 87). По мере титрования поглощение падает
(рис. 86). Для гетероциклических оснований, например хинолина и 8-хлор-
хинолина, максимум поглощения кислой формы (ониевого иона) смещен
в сторону больших длин волн по отношению к поглощению основания,
212
Глава 14
и поэтому при титровании значение А увеличивается, хотя положение мак-
симума поглощения почти не меняется (см. рис. 75).
Фотометрическое титрование ароматических аминов, способных к ацетилированию,
может быть проведено 0,001 М раствором уксусного ангидрида в пиридине [679].
В качестве растворителей для фотометрического титрования слабых
оснований применяются уксусная кислота и ацетонитрил (табл. 73) [404].
Таблица 73
Длины волн, рекомендуемые для фотометрического
определения 10 '2 М растворов оснований в ацетонитриле
или уксусной кислоте; толщина поглощающего слоя 5 см;
стандартный раствор: 0,5 н. раствор хлорной кислоты [404]
Соединение X, нм
рК в ацето- нитриле в уксусной кислоте
N.N-Диэтил анилин 7,5 337
N.N-Диметил анилин 8,9 334
Анилин 9,4 319
п-Х лор-N, N-диэтил анилин 9,5 342
га-Хлоранилин 10,0 333 327
ле-Хлоранилин 10,5 325
N, N-димети л-.н-нитроани л ин 11,0 518
8-Хлорхинолин а 11,2 380
5-Нитро-1-аминонафталин 11,2 555 555
о-Хлоранилин 11,4 324 320
ле-Нитроанилин 11,5 464
2-Метил-5-нитроанилин 12,0 470
2,4-Дихлоранил ин 12,2 335
N, N-Диметил-п-нитроанилин 12,5 470
2,5-Дихлоранилин 12,8 328
га-Нитроанилин 13,0 448
2-Хлорпиридин а 13,3 294
4г-Метил-2-нитроанилин 13,5 520
а Поглощение увеличивается в процессе измерения.
Объем титруемого раствора, содержащего 0,5—2,5 мг-экв монофункциональ-
ного основания, составляет 100—НО мл. Поскольку в качестве стандартного
раствора используется 0,5 н. раствор хлорной кислоты, можно пренебречь
разбавлением раствора в процессе титрования и не вводить поправок. Если
при титровании поглощение увеличивается, то’перед добавлением стандарт-
ного раствора спектрофотометр устанавливают на нуль. Если же поглощение
во время титрования уменьшается, вначале помещают на пути светового
луча кювету с чистым растворителем, устанавливают прибор на нуль и затем
заменяют растворитель исследуемым веществом. Это рекомендуется делать
потому, что при этом меньше ошибки, связанные с разбавлением и действием
постороннего света [277]. Большинство ароматических аминов дает уменьше-
ние поглощения при титровании, если выбрана длина волны, смещенная
от максимума поглощения свободного основания в сторону более длинных
волн. При протонировании поглощение уменьшается и максимум смещается
в сторону более коротких длин волн.
Основания со значениями рК меньше 10 (в воде) нельзя анализировать
фотометрическим титрованием в уксусной кислоте, в которой они полностью
или частично протонированы. Эти основания растворяют в ацетонитриле,
протонодонорные свойства которого невелики, хотя такие чрезвычайно
слабые основания, например n-нитроанилин (p/f 13,0) и 4-метил-2-нитро-
анилин (рК 13,5) лучше титровать в уксусной кислоте. О титровании о-нитро-
анилина (р/С 14,3) см. в работе [484].
Фотометрическое определение конечной точки 213
д. Дифференцирующее титрование оснований. Фотометрическое титрова-
ние используется для анализа смесей соединений с очень близкой основ-
ностью, например смеси 2-метил-5-нитроанилина и 4-метил-2-нитроанилина,
в которой более слабое основание поглощает при 522 нм (рис. 88) [404].
В то же время в ацетонитрильном растворе анилина (рА" 9,42; 290 нм)
и 1Ч,1Ч-диэтиланилина (рК 7,48; 310 нм) более сильное основание поглощает
в более длинноволновой области, но после протонирования поглощение
смещается в область более коротких длин волн. После получения первой
точки перехода (при 377 нм) прибор настраивают на режим работы в более
коротковолновой области и продолжают титрование (рис. 89). Если система
состоит более чем из трех компонентов и рК оснований отличаются меньше,
чем на 1,5, дифференцирующее титрование не удается провести в уксусной
Рис. 88. Фотометрическое титро-
вание 0,5 мг-экв 2-метил-5-нитро-
анилина и 1,0 мг-экв 4-метил-2-нитро-
анилина в уксусной кислоте при
522 нм [404].
Рис. 89. Фотометрическое титрова-
ние 0,512, мг-экв N .N-диэтил анилина
и 1,104 мг-экв анилина в ацетонитриле
[404].
кислоте. Поскольку ацетонитрил не обладает выравнивающим действием,
используя его как растворитель, можно анализировать системы из четырех
компонентов.
е. Фотометрическое титрование хлоргидратов аминов. На титрование
солянокислого хинина в среде уксусной кислоты без добавления ацетата
ртути(П) расходуется 1 экв хлорной кислоты, но в растворе нитрометана
в присутствии ацетата ртути(П) солянокислый хинин ведет себя как первич-
ный амин (см. гл. 24, разд. 136). Перхлорат хинина не растворяется в уксус-
ной кислоте, его титруют в нитрометане [209]. В качестве индикатора исполь-
зуется 0,5%-ный раствор метиленового синего в уксусной кислоте, титро-
вание ведут в колориметре Ланга с фильтром с полосой пропускания 550—
580 нм. Для этого соединения получены кривые необычного вида (рис. 90).
ж. Фотометрическое титрование фенолов — задача более трудная, чем
титрование оснований, и работ, посвященных этому методу, опубликовано
меньше. Титрование обычно проводят в бутиламине [570]. Растворители,
содержащие кетогруппы, сильно поглощают в ультрафиолетовой области
и могут поэтому применяться лишь при титровании в видимой области.
Из растворителей со спиртовыми гидроксильными группами пригодным
оказался изопропиловый спирт. Для титрования в ультрафиолетовой области
используют спектрофотометр Бекмана модели DU [405]. Над анализируе-
мым раствором пропускают ток азота, чтобы в раствор не попадала из воздуха
двуокись углерода. В качестве стандартного раствора применяют 0,4 н.
раствор гидроокиси тетрабутиламмония в изопропиловом спирте, содержа-
щем 10—15% метилового спирта. На результаты анализа оказывают влия-
ние два фактора — состав стандартного раствора и образование фенол-
фенолятного комплекса. Стандартный раствор не должен содержать слабо-
основных примесей типа карбонатов. В среде изопропилового спирта фенол
обладает кислотностью, достаточной чтобы превратить СО|“ в НСО-, однако
214
Глава 14
2,4,6-трихлорфенол реагирует уже и с НСО9, а реакция между пентахлор-
фенолом и НСО3 протекает количественно. Таким образом, на нормальность
стандартного раствора оказывают влияние и примеси карбонатов, и кислот-
ность вещества, примененного для установки титра. Если титр стандартного
раствора (G4H9)4NOH был установлен по слабой кислоте, а затем этот раствор
использовался для титрования смесей, содержащих сильную и слабую кис-
лоту, то для более сильной кислоты получают заниженные результаты,
а для более слабой — завышенные, хотя общая кислотность смеси может
быть определена правильно. Поэтому стандартный раствор (G4H9)4NOH
следует пропустить через ионообменную колонку, прежде чем приме-
нять для титрования фенолов (см. гл. 10,
разд. 57).
Фенолят-ион поглощает в более длин-
новолновой области, чем фенол. Выбран-
ная для измерений длина волны должна
находиться в длинноволновой области
вблизи пика поглощения фенолят-аниона,
Рис. 91. Кривая фотометрического
титрования 1,012 мг-экв фенола
(329 нм) [405].
Рис. 90. Фотометрическое титрование
солянокислого хинина в присутствии мети-
ленового синего (550—580 нм).
1 — кривая титрования 'солянокислого хинина
как монофункционального основания в среде
уксусной кислоты; 2 — кривая титрования соля-
нокислого хинина как бифункционального основа-
ния после добавления ацетата ртути.
тогда теоретически, фотометрическая кривая поглощения должна получаться
линейной. Однако из рис. 91 можно видеть, что кривая титрования не линей-
на и что она поднимается после точки эквивалентности; это можно объяснить
поглощением титрантом. Нелинейные кривые титрования характерны для
большинства фенольных соединений. Известно, что фенол образует с фено-
лят-анионом комплекс за счет водородной связи (см. гл. 12, разд. 63, г).
В инертных растворителях устойчивость этого комплекса увеличивается
и кривая титрования становится менее линейной (рис. 92). При титровании
о-нитрофенола фенол-фенолятного комплекса не образуется из-за наличия
внутримолекулярной водородной связи (рис. 93).
Интересно, что образование фенол-фенолятного комплекса значительно
резче обнаруживается при фотометрическом, а не при электрометрическом
титровании. При потенциометрическом титровании перегиб, наблюдаю-
щийся при достижении потенциала полунейтрализации, объясняется незна-
чительным различием в кислотности фенола и фенол-фенолятного комплекса,
при кондуктометрическом титровании — различием в константах диссо-
циации двух ионных пар; при фотометрическом титровании степень погло-
щения непосредственно коррелируется с образованием комплекса. Интен-
сивность УФ-поглощения зависит от образования водородных связей, длина
волны при этом не изменяется [233—235].
Спектральные характеристики замещенных фенолов не благоприят-
ствуют проведению дифференцирующего фотометрического титрования; веще-
Фотометрическое определение конечной точки
215
ства, имеющие более кислый характер, как правило, поглощают в более
длинноволновой области, чем вещества с меньшей кислотностью. На погло-
Рис. 93. Кривые фотометри-
ческого титрования о- и п-нит-
рофенолов [405].
1 — о-нитрофенол в смеси изопро-
пилового спирта с бензолом (5 : 105)
при 545 нм', 2 — n-нитрофенол при
490 нж.
ZHh.(C4H9)4N0H
Рис. 92. Кривые фотометрического титрования
ж-нитрофенола [405].
1 — 0,503 мг-экв в смеси изопропилового и этилового спиртов
(5 : 105) при 353 нм', 2 — 0,503 мг-акв в смеси изопропилового
спирта с бензолом (5 : 105) при 560 нм', 3 — в бензоле при
560 нж»
щение вещества с меньшей кислотностью оказывает влияние поглощение
продуктами титрования фенола с более высокой кислотностью. Этого можно
Рис. 94. Кривые фотометрического тит-
рования смеси фенолов [405].
1 — 0,291 мг-зкв 2,4,6-трихлорфенола (358 нж);
2— 0,484 мг-зкв 2,4-дихлорфенола (345,5 нм).
Рис. 95. Кривая: фотометрического
титрования смеси фенолов [405].
1 — 0,509 мг-экв п-нитрофенола; 2 — 0,503 мг-зкв
ж-нитрофенола (555 нм).
избежать, если сменить рабочую длину волны спектрофотометра после пер-
вого перегиба на кривой и увеличить щель прибора. Примером может слу-
"Таблица
Длины волн, рекомендуемые для фотометрического титрования 10-2 М
растворов фенолов в изопропиловом спирте; стандартный раствор:
раствор (C4H9)4NOH |405]
Соединение рК X, нм (кювета с поперечным сечением 5 см) Соединение рК X, нм (кювета с поперечным сечением 5 см)
Пентахлорфенол 4,0 387 о-Хлорфенол 8,4 337
2,4-Динитрофенол 4,0 507 о-Бромфенол 8,4 342
2,4,6-Трихлорфенол 6,0 360 ж-Хлорфенол 9,0 333
п-Нитрофенол 7,1 495 п-Бромфенол 9,3 342
«-Нитрофенол 7,2 541 п-Хлорфенол 9,4 341
2,4-Дихлорфенол 7,7 351 Нафтол-1 9,5 408
2,4-Дибромфенол 7,7 359 Нафтол-2 9,7 408
ж-Нитрофенол 8,3 570 Фенол 9,9 329
216
Глава 14
жить титрование смеси 2,4,6-трихлорфенола (рЛГ 6,0; кривая 7) и 2,4-дихлор-
фенола (рЛГ 7,7; кривая 2) (рис. 94). Дифференцирующее титрование
м- и n-нитрофенолов проходит более удовлетворительно; анион более кис-
лого n-нитрофенола (рХ 7,1) поглощает в более коротковолновой области,
чем л4-нитрофенолят-анион (рХ 8,3), поэтому нет необходимости в смене
длины волны в процессе титрования (рис. 95). Обычно титруют 0,3—
0,55 мг-экв фенола в 100 мл растворителя (табл. 74).
81. ПРИБОРЫ ДЛЯ ФОТОМЕТРИЧЕСКОГО ТИТРОВАНИЯ
Для фотометрического титрования можно использовать такие спектро-
фотометры, как спектрофотометры Бекмана модель В [277] или Колемана
модель 14 [275], а для измерений в ультрафиолетовой части спектра — квар-
цевый спектрофотометр Бекмана модели DU. Фото-
метры с цветными фильтрами, например колори-
метр Ланге модель IV и колориметр Эвелина, рас-
сматриваются в работах [209] и [141].
У колориметра Бекмана модели В (320—
1000 нм) удаляют держатель ячейки, верхнюю
и нижнюю стенки и вместо них вставляют сосуд
для титрования емкостью 150 мл с магнитной
мешалкой [277]. Внутренняя поверхность ящичка,
вставленного вместо удаленных перегородок ячей-
ки, должна быть окрашена в черный цвет. Для
титрования применяется микробюретка на 5 мл,
ее кончик должен быть погружен в раствор. Ниж-
няя часть бюретки (выше крышки и вплоть до
начала шкалы) должна быть также покрашена
в черный цвет, чтобы воспрепятствовать, насколько
это возможно, прониканию света. Сосуд для тит-
рования необходимо очень прочно закрепить, так
как даже самое небольшое смещение может выз-
вать значительные изменения оптических харак-
теристик.
Стандартную металлическую крышку (10 см)
держателя кюветы в спектрофотометре Бекмана
модели DU нужно заменить крышкой с двумя
отверстиями, через одно из них вводят кончик
бюретки, через другое подают ток азота. Для
измерений в видимой области (до 240 нм) можно
использовать сосуды из боросиликатного стекла
и сосуды из стекла викор (марки 7910, Corning
Gloss Works) емкостью 150 мл, помещаемые вместо
держателя кюветы. Для точной установки сосуда для титрования служат
просверленные прокладки из пористой резины, перемешивают раствор магнит-
ной мешалкой[404].
Очень удобным*прибором для проведения фотометрического титрования
является автоматический спектроэлектротитратор модели SE Сарджента
и Малькольма (Е. Н. Sargent and Со.), позволяющий использовать автома-
тический контроль при применении индикатора, а также определять конеч-
ную точку потенциометрически [537, 538, 540]. Прибор имеет шесть
интерференционных фильтров, охватывающих диапазон 370—650 нм.
В качестве ячеек для титрования используются сосуды емкостью 30—
600 мл.
Если в конечной точке титрования происходит резкое изменение окраски,
великолепные результаты дает автоматический титратор Стона (Е. Leitz,
Рис. 96. Ячейка для фото-
метрического титрования
[275].
1 — кончик микробюретки; 2 —
мешалка; 3 — слой черной
краски; 4—бакелитовая крыш-
ка; 5 — сосуд для титрования
емкостью 150—200 мл; 6 — дере-
вянные подпорки; 7 — верхняя
крышка прибора’ 8 — трубка
диаметром 15—24 мм или под-
ставка размером 18 X 30 мм;
9 — путь луча света; 10 — на-
правляющая планка.
Фотометрическое определение конечной точки
217
Inc., New York). Титратор Кере (Н. Tinsley and Со. Ltd., London) можно
снабдить приспособлениями для записи кривых фотометрического титрова-
ния и хроматограмм.
Для автоматического фотометрического титрования можно использовать
титратор CENCO (Central Scientific Со., Chicago), в котором подача титранта
прекращается при достижении заданного объема раствора.
В фотометрическом комплекте для титрования ТТ А4 (Radiometer A/S,
Copenhagen) используется полихроматический свет и в качестве детектора
фоторезистор на основе CdS, включенный в простую мостиковую систему,
соединенную с радиометрическим титратором типа ТТТ1 или ТТТ11,
а также титрографом типа SBR 1 или SBR 2.
Прибор для одновременного потенциометрического и фотометрического
титрования [137]. Упрощенный прибор для фотометрического титрования
описан Агацци и Бондом [1]. Комплект для титрования
состоит из фототрубки и источника света, заключенных
в стеклянные трубки, так что они могут быть погру-
жены в стакан емкостью 250 мл. В приборе могут при-
меняться различные типы кювет, показанные на рис. 96
[86, 141, 275, 552, 553, 659, 824], рис. 97 [789], рис. 98
Рис. 97. Ячейка для
фотометрического титрова-
ния [789].
Рис. 98. Ячейка для
фотометрического титро-
вания [364].
Рис. 99. Циркуляционная
колба Эрленмейера для фото-
метрического титрования [670].
[364]; колба Эрленмейера с циркуляцией жидкости и колориметр Бауша
и Ломба Спектроник 20 показаны на рис. 99 [670].
Чувствительность различных методов повышается, если использовать
для измерения поглощения как кислой, так и основной форм индикатора
прибор с двумя фотоячейками [232].
Из факторов, влияющих на точность измерений, следует отметить разба-
вление титруемого раствора при прибавлении стандартного раствора. Можно
ввести поправку на изменение концентрации индикатора с изменением объема,
умножая найденное значение поглощения на (V + v)/V, где V — начальный
объем раствора, a v — объем прибавленного стандартного раствора. Однако
часто не имеет смысла вводить эту поправку, как, например, в тех случаях,
когда к 100—150 мл раствора добавляют 2—3 мл стандартного раствора.
Само собой разумеется, что при нахождении точки эквивалентности графи-
ческим методом объем стандартного раствора, откладываемый на оси х,
необходимо умножить на фактор титранта / (F).
В процессе титрования необходимо обеспечить гомогенность титранта
механическим перемешиванием (при помощи магнитной мешалки или про-
пуская через раствор инертный газ), однако перемешивание не должно
влиять на прохождение света через раствор. Цилиндрический сосуд для
титрования нужно поместить в кювету большего размера с параллельными
стенками, содержащую растворитель с тем же показателем преломления,
что и показатель преломления стекла сосуда для титрования [200]. В про-
218 Глава 14
цессе титрования необходимо поддерживать постоянную температуру. При
выборе длины волны для измерений нужно обратить внимание на следующие
факторы. Необходимо устранить мешающее поглощение соединений, кото-
рые не определяются в процессе титрования; максимальное светопоглощение
должно оставаться в пределах измерения прибора, поскольку высокая сте-
пень поглощения может быть серьезным источником ошибок. Если у кривых
фотометрического титрования около точки пересечения происходит значи-
тельное искривление прямолинейного участка, при экстраполяции могут
возникнуть отрицательные ошибки.
Объективный метод расчета конечной точки описан Грюнвальдом [293].
Глава 15
ОПРЕДЕЛЕНИЕ
ВЕЩЕСТВ КИСЛОГО ХАРАКТЕРА
В этой главе рассматриваются методы определения донорных кислот,
содержащих способные диссоциировать атомы водорода. Константы диссо-
циации (в воде) ароматических кислот, содержащих карбоксильную группу,
меньше констант диссоциации неорганических кислот, и большинство аро-
матических кислот лишь слабо растворимы в воде. Сила карбоновых кислот,
содержащих нитрогруппу в пара-положении, как и сила сульфокислот,
приближается к силе неорганических кислот.
Карбоновые кислоты характеризуются наличием функциональной группы
СООН; между ионизированной и неионизированной формами этих кислот
существует равновесие:
RCOOH [RCOO]- + H+
(Н — ионизирующийся атом водорода).
Методы определения карбоновых кислот основаны на нейтрализации их
алкоголятами щелочных металлов, четвертичными гидроокисями аммония
(алкоксидами) и соединениями, содержащими аминогруппу, проводимой
в инертных или основных растворителях, а также в смесях растворителей.
Конечная точка нейтрализации определяется либо с помощью индикаторов
(визуально или фотометрически), либо электрометрическими методами.
Титрование, в котором участвует активный атом водорода енола, имида
двухосновной кислоты, фенола или некоторых других протонодонорных
групп, также можно рассматривать как реакцию нейтрализации. Соедине-
ния, содержащие одну из таких групп, представляют собой донорные кис-
лоты, в то время как соединения, содержащие ацетильный катион или частич-
но поляризованное ароматическое ядро, являются акцепторными кислотами.
Примером последних может служить хлористый ацетил и .м-динитробензол
(см. гл. 16 и 20). Все эти соединения называют аналогами кислот и их опре-
деление обсуждается в гл. 16—21.
Выбор растворителя зависит от растворимости соединения и его относи-
тельной кислотности. Когда титруют карбоновые кислоты, желательно пред-
варительно знать вероятную силу кислоты (гл. 2 и 4); в выбранном раствори-
теле должно быть лучше всего потенциометрически определено правиль-
ное изменение окраски индикатора по отношению к чистому веществу
(гл. 12).
Для определения органических кислот в первую очередь используют
метиловый, этиловый и изопропиловый спирты с тем, чтобы раствор всегда
оставался однородным, даже если в качестве титранта применяют водный
раствор щелочи. Этиловый спирт использовали как растворитель для опре-
деления высших жирных кислот уже 40 лет назад [67].
Наиболее типичное изменение окраски некоторых индикаторов при титровании
в этиловом спирте показано в табл. 75 (стандартный раствор: этилат натрия в этило-
вом спирте).
Резкость изменения окраски можно усилить, используя индикаторную смесь тимол-
фталеин — метиловый оранжевый [709] (ср. табл. 64).
220
Глава 15
Таблица 75
Индикатор
«Кислая» форма -+ «Основная»
форма
Бромфеноловый голубой,
0,1%
Метиловый фиолетовый,
0,1%
Фенолфталеин, 1%
Тимолфталеин, 1%
Желтая —> зеленая —> голубая
Фиолетовая
Бесцветная
Бесцветная
-> бесцветная
-> красная
-> голубая
Позднее спирт заменили инертными растворителями и смесями раствори-
телей, такими, как ацетон, хлороформ, бензол, бензол — изопропиловый
спирт, и вместо стандартных водных растворов начали употреблять спирто-
вой раствор щелочи или алкоголята щелочного металла. Соли слабых кислот
подвергаются алкоголизу в значительно меньшей степени, чем гидролизу,
так что изменение окраски индикатора выражено обычно четче.
Затем выяснилось, что вместо спиртового раствора щелочи или алкого-
лята щелочного металла целесообразно использовать четвертичные алкилам-
мониевые основания, поскольку алкил-
аммониевые соли карбоновых кислот
в отличие от солей щелочных металлов
растворяются в инертных раствори-
телях.
Для определения слабых кислот и
кислотных аналогов оказались пригод-
ными растворители основного харак-
тера, например диметилформамид, пири-
дин, н-бутиламини этилендиамин, а для
определения смесей кислот — такие
растворители, как ацетон, метилизобу-
тилкетон, пгреттг-бутиловый спирт и др.
Пиридин относится к числу тех
растворителей основного характера,
которые допускают потенциометриче-
ское или визуальное определение отно-
сительной силы слабых кислот [780,
782] и дифференцирующее титрование
кислотных смесей [159]. При потенцио-
метрическом титровании в пиридине
м- и n-оксибензойные кислоты обна-
Р и с. 100. Кривые потенциометриче-
ского титрования оксибензойных кислот
[159].
руживают два перегиба на кривой, в то
время как орпго-производное дает только один перегиб. Это показывает,
что в орпго-производном титруется только группа —СООН (рис. 100). Ана-
лизы большого количества образцов с применением обычных методов потен-
циометрического титрования требуют много времени, и поэтому все чаще
конечную точку титрования определяют с помощью индикаторов. Алифати-
ческие карбоновые кислоты, бензойная кислота, салициловая кислота, нико-
тиновая кислота, о- и n-нитрофенолы, 2,4-динитрофенол, 2-меркаптобенз-
тиазол и многие аминокислоты можно титровать в пиридине стандартным
раствором (C4H9)4NOH в присутствии тимолового синего. В присутствии
азофиолетового в пиридине n-бензилфенол, нафтол-1 и нафтол-2, 2,5-диметил-
фенол, n-бромфенол, пирогаллол, фталимид, салицилальдоксим, сукцин-
имид, ацетилацетон, n-хлортиофенол, бензилмеркаптан и 5-аминобензимид-
азолтиол-2 могут быть оттитрованы как монофункциональные кислотные
аналоги. В то же время м- и n-оксибензойные, щавелевая, яблочная, мало-
Определение веществ кислого характера
221
новая, малеиновая, фумаровая, янтарная, фталевая и лимонная кислоты
в присутствии азо фиолетового требуют расхода 2 экв титранта.
Относительно использования стандартного раствора, содержащего пиперидин или
диэтаноламин в пиридине, см. [815].
Среди растворителей основного характера наибольшее распространение
получил диметилформамид. Многие авторы рассмотрели титрование кислот
и кислотных аналогов в диметилформамиде [7, 113, 159, 176, 246, 448, 457,
562]. В диметилформамиде можно титровать большинство соединений, содер-
жащих фенольный гидроксил, особенно если, кроме него, имеется и электро-
ноакцепторный заместитель, усиливающий кислотный характер, например
СНО, COR, COOR, CONH2 или NO2 [176, 251]. Группа СООН не дает того
эффекта увеличения силы кислоты, который типичен для вышеупомянутых
заместителей; так, фенольный гидроксил салициловой кислоты содержит
менее активный водород. В определенных случаях диметилформамид приго-
ден также для дифференцирующего титрования. Например, при использо-
вании платино-каломельных электродов может быть выполнено титрование
двойных смесей, таких, как фталевый ангидрид — монометилфталат, олеи-
новая кислота — монометилфталат, олеиновая кислота — фталевый ангид-
рид [457]. Относительно дифференцирующего титрования смесей кислот
см. разд. 95 данной главы. Высокочастотное титрование кислот и кислотных
аналогов в диметилформамиде рассматривается в работах [177, 441, 664].
Из инертных растворителей наиболее пригодным для обычного анализа
оказался ацетон: он доступен, дешев и его легко очистить [258]. Так как
ацетон практически совершенно не обладает кислотными свойствами, в нем
можно титровать даже очень слабые кислоты, а вследствие его низкой основ-
ности незначителен и выравнивающий эффект. Ацетон широко исполь-
зуется при дифференцирующем титровании [258], хотя в этом отношении
более пригодны метилэтилкетон [359] и метилизобутилкетон [105]. Электроды,
используемые обычно при потенциометрическом титровании, могут быть
применены также и в ацетоновых растворах, так как достаточно большая
диэлектрическая проницаемость ацетона позволяет наблюдать стабильные
перегибы на потенциометрических кривых в процессе титрования. Сильные
кислоты и основания, однако, реагируют с ацетоном.
Карбоновые кислоты, соединения, содержащие енольный и фенольный
гидроксилы, сульфамидные производные, кислотные аналоги с имидной
группой и т. д., можно титровать в ацетоне [258]. В качестве стандартных
растворов целесообразно применять гидроокись тетрабутиламмония или
гидроокись три-н-бутилметиламмония. Раствор последнего в смеси бензол —
изопропиловый спирт (4 : 1) очень удобен для потенциометрического титро-
вания, если для индикаторного электрода используется платина (содержа-
щая 10% родия), а в качестве электрода сравнения — графит [540]. Крайне
слабые фенолы можно титровать либо используя индикаторы, либо фото-
метрически в среде ацетона в присутствии азофиолетового. Если вблизи
конечной точки титрования раствор содержит по меньшей мере 90% аце-
тона, можно применять и другие растворители (см. табл. 23).
Дифференцирующие свойства ацетонитрила сходны с дифференцирующими свой-
ствами ацетона. Некоторые смеси, например смесь миндальной кислоты и 2-ацетил-
нафтола-1, можно титровать метилатом калия.
Изменение окраски индикатора n-оксиазобензола от бесцветной до желтой позволя-
ет определить карбоксильные группы, а последующее титрование в присутствии азо-
фиолетового дает возможность определить и фенольные гидроксилы. При этом окраска
индикатора меняется от оранжевой до желтой, затем до голубой и зеленой. Такие смеси
можно также дифференцировать потенциометрическим титрованием [251].
Монометиловый и моноэтиловый эфиры гликоля (метилцеллозольв, цел-
лозольв) часто используют как растворители либо самостоятельно, либо
в смеси с другими растворителями, такими, как изопропиловый спирт, кси-
222
Глава 15
лол, анизол, ацетон, диоксан, н-бутиловый спирт и т. д. Составы смесей
должны определяться экспериментально. Это, однако, зависит также от того,
титруют ли кислоту потенциометрически или с использованием индикатора.
Гораздо реже в качестве растворителей применяют бензиловый и а-метил-
бензиловый спирты; их используют для определения кислотных групп
в полимерах, таких, как полиамиды, полиэтилентерефталаты, полиамино-
капроновые кислоты и др. [650, 714, 723, 858]. Титрование обычно производят
в горячих растворителях.
Основной принцип, который следует соблюдать при определении веществ
кислотного характера, заключается в том, что растворитель не должен
содержать никаких кислотных или основных примесей; кроме того, перед
титрованием СО2, попавший из воздуха, должен быть нейтрализован стан-
дартным раствором. Вещество, содержание которого исследуют, растворяют
в подходящем растворителе, чтобы израсходовать на титрование 5—8 мл
0,05—0,1 н. стандартного раствора. Раствор вещества для визуального
титрования должен иметь объем 20—30 мл, в то время как при потенцио-
метрическом определении конечной точки объем составляет 25—50 мл. Сле-
довательно, состав растворителя в ходе титрования изменяется очень незна-
чительно. Если изменение диэлектрических или других свойств во время
титрования нежелательно, используют меньшую навеску и более концен-
трированный стандартный раствор в микробюретке (ср. гл. 14). Навеска
анализируемого вещества должна быть такой, чтобы количество использован-
ного стандартного раствора не превысило 10% объема растворителя.
Достаточно трудно найти подходящие смеси растворителей для потен-
циометрического определения содержания кислот в сильно окрашенных
промышленных и природных продуктах. Ликкен и сотрудники из примерно
40 растворителей и их смесей выбрали только две смеси, пригодные для
определения кислотности нефтяных смазочных масел, растительных и живот-
ных масел, восков, жиров, производных асфальтов и битумов, погонов дегтя,
а также природных и синтетических смол. Оказалось, что наиболее при-
годны смеси бензол — изопропиловый спирт или петролейный эфир —
изопропиловый спирт (1:1) с содержанием примерно 0,5% воды [530].
Выбор растворителя может оказывать решающее влияние на воспроиз-
водимость результатов определений. Так, если кислотное число раститель-
ного масла или китовой ворвани определяется титрованием раствором едкого
кали в нейтрализованном изопропиловом спирте в присутствии фенолфта-
леина, точность титрования составляет ±0,025 мг-экв, в то время как точ-
ность потенциометрического контроля составляет ±0,043 мг-экв [9].
82. АЛИФАТИЧЕСКИЕ КИСЛОТЫ
При определении алифатических кислот берут такую навеску кислоты,
чтобы при титровании было израсходовано максимум 9—10 мл 0,05 н.
стандартного раствора; кислоту растворяют в 10—20 мл абсолютного этило-
вого спирта. В качестве индикатора (табл. 63) добавляют 5 капель тимол-
фталеина и 3 капли метилового оранжевого, нагревают на водяной бане
до 65° и титруют из бюретки с ценой делений 0,05 мл 0,05 н. раствором эти-
лата натрия в этиловом спирте до появления зеленой окраски (раствор срав-
нения: 20 мл этилового спирта и 3 капли метилового оранжевого как инди-
катора). Следует учитывать количество стандартного раствора, расходуемое
в аналогичных условиях на нейтрализацию растворителя. Титр стандарт-
ного раствора устанавливают по чистой стеариновой кислоте [709]
(см. гл. 10, разд. 55).
Алифатические кислоты с большим числом атомов углерода можно также
оттитровать 0,1 н. раствором метилата натрия в смеси бензол — метиловый
спирт, к-бутиламине или ацетоне в присутствии тимолового синего [252].
Определение веществ кислого характера
223
Олеиновая кислота может быть оттитрована в этиловом спирте раствором
едкого кали в этиловом спирте.
Определение основности технического стеарата цинка [527]. В 50 мл эти-
лового спирта, нейтрализованного в присутствии фенолфталеина, раство-
ряют 1 г технического стеарата цинка. Добавляют 25 мл 0,1 н. раствора
олеиновой кислоты в этиловом спирте и кипятят 30 мин с обратным холо-
дильником. Охлаждают раствор льдом и быстро фильтруют, осадок промы-
вают нейтрализованным этиловым спиртом. Олеиновую кислоту, не погло-
щенную основным стеаратом цинка, титруют 0,1 н. раствором едкого кали
в этиловом спирте в присутствии фенолфталеина. Глухой опыт выполняется
подобным образом.
Эквивалент нейтрализации алифатических кислот высокого молекуляр-
ного веса рекомендуется определять в полумикромасштабе после растворе-
ния в растворе, содержащем избыток метилата натрия, соответствующий
нормальности, например, 0,01 н., обратным титрованием избытка щелочи
стандартным раствором, содержащим щавелевую кислоту [807]. Описание
микрометода см. в работе [808].
В маленькой мерной колбе из стекла пирекс взвешивают примерно 10 мг
анализируемой кислоты. Образец переносят вместе с мерной колбой в кони-
ческую колбу для титрования из жаростойкого стекла объемом 150 мл, снаб-
женную устройством для ввода азота, и растворяют кислоту в смеси 5 мл
метилового спирта с 15 мл бензола. Чтобы удалить воздух, систему проду-
вают в течение 2 мин током очищенного азота; добавляют 4—5 капель инди-
каторной смеси (см. ниже) и 10 мл 0,01 н. раствора метилата натрия в смеси
бензол — метиловый спирт. Раствор метилата натрия приготовляют так же,
как раствор 0,02 н. концентрации; подробно это описано в гл. 10, разд. 56, б.
Избыток алкоголята щелочного металла оттитровывают 0,01 н. раство-
ром щавелевой кислоты в смеси метиловый спирт — бензол до перехода
окраски от зеленовато-синей к желтой. Глухой опыт выполняется подоб-
ным же образом. Во время титрования система должна быть защищена
от попадания влаги и углекислого газа из воздуха (см., например, приборы
на рис. 39—42).
Нормальность стандартного раствора метилата калия устанавливают
по щавелевой кислоте. Индикаторную смесь готовят непосредственно перед
титрованием, смешивая в соотношении 3:1:1 0,4%-ные растворы феноло-
вого красного, крезолового красного и бромтимолового синего в метиловом
спирте. Чтобы приготовить 0,01 н. раствор щавелевой кислоты в смеси бен-
зол — метиловый спирт, 126 мг дигидрата щавелевой кислоты растворяют
в 10 мл метилового спирта, а затем разбавляют до 100 мл бензолом.
Этот метод пригоден для определения эквивалентов нейтрализации
лауриновой, миристиновой, пальмитиновой, стеариновой, олеиновой и лино-
леновой кислот.
83. ЗАМЕЩЕННЫЕ АЛИФАТИЧЕСКИЕ КИСЛОТЫ
Потенциометрическое титрование алифатических кислот проводится в тех
случаях, когда окраска раствора меняется и раствор темнеет в ходе титро-
вания и, особенно, если переход окраски в конечной точке становится мед-
ленным или незаметным. Это может наблюдаться при титровании сырого
ланолина, а также замещенных жирных кислот, таких, как 12-кетостеари-
новая, 9,10-дегидростеариновая, 9,10-эпоксистеариновая, аминостеарино-
вая, 1-бром- и меркаптопальмитиновые кислоты [665].
Анализируемое вещество, взятое в таком количестве, чтобы на его титро-
вание расходовалось 6—8 мл стандартного раствора, растворяют в 50 мл
смеси бензол — метиловый спирт (3:1). Сосуд для титрования должен быть
защищен от попадания углекислого газа из воздуха, поэтому определение
224
Глава 15
замещенных алифатических кислот удобно проводить в титровальной колбе,
предложенной Оггом и сотрудниками для определения карбоксильного числа
жирных кислот. Прибор представляет 250-миллилитровую плоскодонную ко-
ническую колбу с притертой пробкой и боковыми отводами, в которые введены
сурьмяный (индикаторный) и каломельный (электрод сравнения) электроды
[252, 616]. Электроды предварительно выдерживают в течение 30 мин в смеси
бензол — метиловый спирт, куда в качестве электролита добавляют 0,2 г
хлорида лития. После растворения образца и хлорида лития (примерно
через 10—15 зеин) анализируемый препарат титруют 0,1 н. метилатом натрия,
перемешивая раствор с помощью магнитной мешалки. Нормальность стан-
дартного раствора устанавливают в аналогичных условиях по чистой паль-
митиновой, бензойной или фенилхинолиновой кислоте (гл. 10, разд. 56
и гл. 15, разд. 86 и 88).
84. ЧИСЛО ОМЫЛЕНИЯ
При проведении анализа жиров и восков, например хлопкового, кунжут-
ного, касторового и кокосового масел, животного и рыбьего жиров [330],
для определения числа омыления очень удобно использовать 0,5 н. раствор
едкого кали в смеси этиленгликоль—ксилол. Для омыления достаточно 10-ми-
нутного^ нагревания в открытой колбе на водяной бане. Избыток едкого кали
оттитровывают 0,5 н. соляной кислотой в метиловом спирте или этиленгли-
коле с тимолфталеином в качестве индикатора.
85. КИСЛОТНОЕ ЧИСЛО НЕФТЯНЫХ ПРОДУКТОВ
i При определении кислотного числа нефтяных продуктов в качестве раство-
рителя используют смесь 500 мл 'изопропилового спирта, 3—5 мл воды
и 500 мл бензола; воду и бензол добавляют последовательно. Титрование
проводят 0,1 н. раствором едкого кали в изопропиловом спирте в присут-
ствии индикатора 1-нафтолбензеина (ср. табл. 27).
В колбе емкостью 250 мл взвешивают нефтепродукты:
20 г, если ожидаемое кислотное число 0,0 — 3,0;
2 г, если ожидаемое кислотное число 3,0 — 25,0;
0,2 г, если ожидаемое кислотное число 25,0 — 250.
Навеску образца растворяют в 100 мл растворяющей смеси (это может
быть также смесь низкокипящего петролейного эфира и изопропилового
спирта), добавляют 3 мл индикатора и титруют при температуре выше 30°
до перехода оранжево-желтой окраски в коричневато-зеленую.
Сильно окрашенные растворы можно титровать потенциометрически,
применяя стеклянный (индикаторный) и каломельный электроды (электрод
сравнения) [530].
При потенциометрическом титровании минеральных масел с кислотным
числом меньше 0,05 не удается получить отчетливых точек перегиба; в этих
случаях проводят визуальное определение конечной точки, сравнивая
титруемый раствор с эталонным. Титрование удобно проводить в колбе с боко-
вым тубусом, показанной на рис. 101; такая колба может быть полезна
и в других случаях [486, 487].
Когда исследуют темные или окрашенные растворы, наблюдать переход
окраски помогает устройство, изображенное на рис. 102 [14]: луч света,
проходя через раствор, падает на экран в виде резко очерченных ярких
цветных пятен. Измерения должны производиться в темноте. Если необхо-
димо, то между источником света и колбой можно установить цветной фильтр.
Об определении кислотных чисел темных, окрашенных масел с использова-
нием люцигенина (нитрата диметилдиакридиния) — хемилюминесцентного
Определение веществ кислого характера
225
индикатора см. в работе [889]. Титрование проводят 0,05 н. раствором мети-
лата, этилата, бутилата или изоамилата натрия в среде метиловый спирт —
бензол (2:1).
Относительно бюретки с краном для тит-
рования в темноте см. на стр. 151 в ра-
боте [31].
Чтобы в различных сортах дегтя и смолы
или в их фракциях, полученных быстрой
перегонкой, оттитровать соединения, содер-
жащие фенольный гидроксил, в качестве
растворителя используют этилендиамин,
бутиламин или пиридин; титрование про-
Рис. 102. Прибор для титрования в темноте
[14].
1 — белый экран; 2 — магнитная мешалка; з — свето-
фильтр; 4 — источник света.
Р и с. 101. Колба для титрования
с боковым отводом для анализа окра-
шенных растворов [486].
I — микробюретка Коха на 5 мл\ 2 — от-
метка на колбе, соответствующая объему
120 мл\ з — воронкообразное углубление,
образующееся при перемешивании; 4 —
траектория движения капель титрующего
раствора; 5 — удлиненное горло колбы;
6 — боковой отвод; 7 — магнитная мешал-
ка и подставка.
водят 0,05-0,1 н. раствором метилата или аминоэтилата натрия в смеси
бензол — метиловый спирт, применяя следующие пары электродов: платина
(содержащая 10% родия) — сурьма, сурьма —Ag/AgCl, платина — кало-
мель или сурьма — каломель [287, 486, 492, 530, 661]. Для удаления осадка
в растворитель можно добавить 2,6-ксиленол [287].
86. БЕНЗОЙНАЯ КИСЛОТА И ЗАМЕЩЕННЫЕ БЕНЗОЙНЫЕ КИСЛОТЫ
Титрование ароматических кислот обычно проводят в смеси бензол —
метиловый спирт (10 : 1), пиридине, ацетоне или диметилформамиде. Кон-
дуктометрические измерения показывают, что растворенные в бензоле кар-
боновые кислоты находятся почти полностью в недиссоциированном состоя-
нии. Однако четкость визуального контроля и четкость скачка потенциала,
наблюдаемые в точке эквивалентности, в водной или спиртовой среде часто
возрастают. В чистом бензоле соли щелочного металла выпадают в виде
осадка или образуют студенистую массу, а это влияет на определение конеч-
ной точки. В некоторых случаях такая же картина наблюдается и при исполь-
зовании в качестве растворителя ацетона или бутиламина. В смеси бензол —
метиловый спирт хорошим индикатором служит тимоловый синий, тогда
как в растворителях основного характера титрование можно вести в при-
226
Глава 15
сутствии тимолового синего, тимолфталеина, ализаринового желтого или
азофиолетового (см. табл. 60—66).
Для потенциометрического титрования в смеси бензол — метиловый
спирт употребляют стеклянный и каломельный электроды, а в растворите-
лях основного характера — сурьмяные и каломельные (ср. табл. 58). Во вре-
мя титрования раствор должен быть защищен от попадания углекислого
газа из воздуха, так как чувствительность индикаторов к СО2 зависит от при-
роды растворителя. При быстром титровании ошибка, вызванная присут-
ствием примеси СО2, незначительна, но в случае потенциометрического
определения, проводимого в течение продолжительного времени, влияние
СО2 необходимо учитывать. Поэтому потенциометрическое титрование лучше
проводить в закрытых приборах, показанных на рис. 39—42, 45, 52. 53
и в гл. 21.
Если углекислый газ удален из растворов и прибор промывается очи-
щенным азотом, то при титровании 0,02 н. раствором гидроокиси тетрабутил-
аммония (приготовленного из 0,1 н. раствора разбавлением бензолом) можно-
проводить измерения с 1—2 мг кислоты с точностью ±2%. Примерно равные
количества анализируемого вещества и стандартного вещества растворяют
в одинаковых объемах одного и того же растворителя. Существенно, что при
любом титровании концентрация индикатора должна оставаться постоянной.
Этого можно добиться, используя 1,0—3,0 мл сильно разбавленного раствора
индикатора. Рекомендуется, чтобы растворитель для индикатора и для
исследуемого соединения был одним и тем же.
Бензойную, о-, м- и n-нитробензойные, оксибензойную, ароматические
дикарбоновые, аминокарбоновые, пиридинкарбоновые кислоты и т. д. можно
титровать в смеси бензол — метиловый спирт.
СООН
СООН
Ароматическая
дикарбоповая
кислота
Пиридинкарбоновая
кислота
Бензойная кислота пригодна также для определения титра стандартных
растворов оснований (см. гл. 10).
Выпадения образованных алкоголятом щелочного металла студенистых
осадков можно избежать, если использовать раствор ТБАГ в смеси бензол —
метиловый спирт или в изопропиловом спирте (гл. 10, разд. 57). Применяя
при потенциометрическом титровании вместо растворов алкоголятов щелоч-
ного металла свободный от карбонатов стандартный раствор ТБАГ, можно
добиться более воспроизводимого изменения потенциала.
При потенциометрическом титровании в пиридине с использованием
стандартных растворов ТБАГ м- или n-оксибензойной кислоты на кривой
обнаруживается два перегиба, в то время как при титровании о-оксибензой-
ной кислоты (салициловой кислоты) на кривой наблюдается только один
перегиб, указывающий точку эквивалентности для карбоксильной группы
[159] (см. рис. 100 и гл. 2, разд. 16, а также гл. 4). В пиридине в присутствии
азофиолетового п- и л-оксибензойные кислоты требуют расхода 2 экв стан-
дартного раствора ТБАГ, в то время как о-оксибензойная кислота в присут-
ствии тимолового синего поглощает только 1 экв.
Кислотности производных бензойной кислоты см. в табл. 10, 11 и 21.
Определение веществ кислого характера
227
Никотиновую и изоникотиновую (пиридин-4-карбоновая кислота) кисло-
ты можно оттитровать метилатом натрия соответственно в бензоле и пири-
дине [832].
87. ОПРЕДЕЛЕНИЕ ДЕЙСТВУЮЩЕГО НАЧАЛА В ФАРМАЦЕВТИЧЕСКИХ
ПРЕПАРАТАХ
Активные терапевтические агенты кислотного или основного характера в таблетках
и порошках могут быть оттитрованы непосредственно в неводной среде, если определе-
нию не мешают другие кислотные или основные связующие вещества (см. табл. 76).
Из тонкоизмелъченной таблетки приготовляют взвесь в соответствующем растворителе
и проводят титрование; кислоты или кислотные аналоги титруют алкоголятом щелочного
металла или гидроокисью алкиламмония, а основания — хлорной кислотой. Мешающие
примеси, например стеариновую кислоту, можно удалить, если тонко измельченный
порошок предварительно проэкстрагировать петролейным эфиром или н-гексаном. Боль-
шинство соединений, содержащих полярные функциональные группы, а также соли
карбоновых кислот, основные гидрохлориды (сульфаты и т. д.) не растворяются в этих
растворителях. Стеарат магния растворяется в уксусной кислоте и переходит в ацетат.
В таком случае порошкообразный образец суспендируют в очищенной уксусной кислоте,
иногда при слабом нагревании, и титруют хлорной кислотой. К этому раствору добавля-
ют нейтрализованный 3%-ный раствор ацетата ртути(П) в уксусной кислоте, при этом
образуются недиссоциирующий галогенид ртути(П) и свободное основание (ацетат)
из основного хлоргидрата. Это основание титруют хлорной кислотой. На титрование
фармацевтических препаратов хлорной кислотой в различной степени влияют связующие
вещества таблеток. Степень мешающего воздействия зависит от использованного раство-
рителя; так, в хлороформе она меньше, чем в уксусной кислоте [133] (табл. 76). (В табл. 76
Таблица 76
Расход хлорной кислоты на титрование инертных наполнителей таблеток [133]
Инертный наполнитель Расход 0.05 н. хлор- ной кислоты, мл Инертный наполнитель Расход 0,0 5 н. хлор- ной кислоты, мл
в хлоро- форме в уксус- ной кис- лоте в хлоро- форме в уксус- ной кис- лоте
Пчелиный воск 0 0 Стеарат магния 0,14 >2,00
Карбонат кальция 0 0,13 Сульфат магния 0 0,8
Фосфат кальция, двух- 0,04 >2,00 Метилцеллюлоза 0 0,34
основный Полиэтиленгликоль 400 >2,00 0,06
Фосфат кальция, трех- 0 0,58 Поливинилпирролидон >2,00 >2,0
основный Альгинат натрия 0 0,98
Стеарат кальция 0,06 >2,00 Натрийкарбоксиметил- 0,06 >2,00
Сульфат кальция 0 0,70 целлюлоза
Карнаубский воск 0 >2,00 Силикат натрия 0 >2,00
Цетиловый спирт 0,02 0 Сульфат натрия 0 1,18
Желатин 0 >2,00 Картофельный крахмал 0 0,06
Лактоза 0 0,02 Сахароза 0 0
Карбонат магния 0 >2,00 Тальк 0,14 0,5
Окись магния 1,06 >2,00 Трагакант 0 0,38
приведен расход хлорной кислоты для титрования насыщенного раствора наполнителя
(связующего) в 50 мл хлороформа или уксусной кислоты. Если расход титранта более
2 мл, то он не указывается.) Отношение числа молей основного хлоргидрата, присутствую-
щего в таблетке в качестве действующего начала, к числу молей стеарата магния должно
быть по возможности не менее 40 : 1—50 : 1, иначе можно получить невоспроизводимые
и ошибочные результаты, особенно при титровании в смеси хлорбензол — уксусная
кислота и при использовании визуального индикатора, например тропеолина ОО.
Содержание действующего начала в препаратах для инъекиий можно определить
титрованием раствора сухого вещества в неводных растворителях. Этот метод, однако,
приемлем только при условии, что активный агент не разрушается под действием воды
и нагревания. Некоторые буферные вещества мешают титрованию; так, цитрат натрия
в уксусной кислоте титруется как основание, но в растворителе основного характера
он ведет себя как кислота.
228
Глава 15
Содержание действующего начала в безводных мазях на основе вазелина или жидко-
го парафина можно определить без выпаривания. Мазь растворяют в смеси растворителей,
состав которой зависит от свойств анализируемого соединения; в качестве такой смеси
применяется, например, смесь полиэтиленгликоль 400 — хлороформ или уксусная
кислота — толуол.
Методы определения содержания действующего начала в препаратах типа хлоргидра-
тов морфолина и папаверина, пирамидона см. в работе [367].
Активное действующее начало фармацевтических препаратов часто можно отделить
от наполнителей и примесей перемешиванием с растворителем в течение 30—180 мин
в закрытой колбе Эрленмейера или в делительной воронке типа воронки Наполи —
Шмалла [609] или, если активный агент не чувствителен к нагреванию, в приборе Сокс-
лета. Экстрактор Наполи — Шмалла представляет собой цилиндрическую воронку
с пористым стеклянным фильтром (№ 3
или № 4), впаянным внутрь цилиндриче-
ской части воронки. После экстракции ра-
створ фильтруют под вакуумом. Остатки
после фильтрования экстрагируют каким-
либо другим подходящим растворителем.
Активные соединения, относительно
малорастворимые в воде, лучше всего
экстрагировать низкокипящим петролей-
ным эфиром, к-гексаном и лигроином, для
экстракции соединений с более высокой
растворимостью в воде целесообразно ис-
пользовать бензол, хлороформ, четырех-
хлористый углерод, а для соединений,
сравнительно хорошо растворимых в во-
де,— этилацетат. Рекомендуется сначала
отогнать этилацетат, затем растворить
остаток в соответствующем растворителе
и титровать неводным стандартным раство-
Рис. 104. Экстрактор [418].
Емкость верхней части — 100—150 мл, емкость
нижней части 25—50 мл. Шлифы стандартные или
взаимозаменяемые.
Р и с. 103. Колбы для упаривания.
1 — цилиндрический шлиф; 2 — фиксирующая
резиновая втулка; 3 — отвод к вакуумному на-
сосу.
ром. Вытяжки лучше всего концентрировать в колбе с коническим дном; колбу нагревают
на водяной бане, а через растворитель пропускают ток отфильтрованного и высушенного
воздуха (рис. 103). На рис. 103, а показан прибор для непрерывной отгонки растворите-
ля, где раствор в колбу подается с помощью загрузочной воронки; на рис. 103, б пока-
зан прибор для упаривания растворителя, в котором в зависимости от объема раствора
можно опускать или поднимать трубку для подачи воздуха.
Экстрактор типа экстрактора Якобса — Сингера пригоден для экстракции водных
растворов растворителями, удельный вес которых больше или меньше удельного веса
воды [418]. Его преимущество состоит в том, что в отличие от делительной воронки у него
нет крана, поэтому препарат или его раствор могут быть непосредственно отмерены в ниж-
ней части прибора (рис. 104).
В любом случае экстракт высушивают безводным сульфатом натрия, и при исполь-
зовании для экстракции петролейного эфира пли бензола, разбавленного четыреххлори-
стым углеродом'в равной пропорции, щелочной активный агент титруют непосредственно
0,005 н. n-толублсульфокислотой в присутствии диметилового желтого. К бензольным
и* хлороформным экстрактам, как и к растворам в СС14, рекомендуется добавить 10—
50 об.% уксусной кислоты (уксусного ангидрида или пропионовой кислоты) и титровать
0,01—0,001 н. хлорной кислотой. Бензольные экстракты кислот или кислотных аналогов
смешивают с пиридином и титруют стандартным раствором основания в присутствии
тимолового синего или азофиолотового. Эфирные или этилацетатные экстракты упарива-
ют на паровой бане, причем поступающий в прибор воздух пропускают через поглоти-
тельную трубку с едким кали (см. рис. 103).
Определение веществ кислого характера
229
Водные растворы хлоргидратов (сульфатов, этансульфонатов, фосфатов и нитратов)
оснований экстрагируют в делительной воронке после подщелачивания не смешивающи-
мися с водой растворителями и экстракты титруют как описано выше 0,005 н. п-толуол-
сульфокислотой или 0,01 н. хлорной кислотой. В ходе экстракции в присутствии эмуль-
гирующих или растворяющих реагентов, таких, как поливиниловый спирт, поливинил-
пироллидон, поверхностное натяжение увеличивают добавлением силиконового масла
или октилового спирта. Подщелачивание водных растворов не должно приводить к разло-
жению оснований (алкалоидов). Проше всего предупредить разложение, заменив раствор
карбоната калия или едкого натра насыщенным раствором бикарбоната калия. Полезно
также применять буферные растворы, так как они фиксируют значение pH и обладают
солевым эффектом. Используя буферные растворы, можно также проводить селективную
экстракцию. Так, активный реагент можпо экстрагировать при низких значениях pH,
в то время как продукт разложения, особенно если он легко растворяется в воде, экстра-
гируется только прп высоких pH или за счет высаливающего действия КНСО3 4- КС1.
Экстрактор Шмалла [721, 883] пригоден для длительных экстракций и применим для
растворителей, которые тяжелее или легче воды. Однако вместо него можно пользовать-
ся более простой делительной воронкой. При образовании эмульсий (недостаток делитель-
ной воронки) применяется экстракция с центрифугированием. Нерастворимые в воде
соединения отделяются от водного раствора, содержащего действующее начало, в виде
соли под действием щелочи или серной кислоты. После экстракции подходящим раство-
рителем экстракт центрифугируют 5—15 мин со скоростью 3000—4000 об/мин. Органи-
ческий растворитель отсасывают шприцем и фильтруют через воронку с сульфатом натрия
в колбу для титрования. Эту операцию повторяют несколько раз. Экстракт титруют
стандартным раствором хлорной кислоты или основания.
О способах быстрого титрования фармацевтических препаратов, обладающих свой-
ствами кислот и кислотных аналогов, см. работу [24].
88 2-ФЕНИЛХИНОЛИН-4-КАРБОНОВАЯ КИСЛОТА
2-Фенилхинолин-4-карбоновая кислота может быть использована вместо
бензойной кислоты для установки титра щелочных титрантов [307]. В каче-
стве растворителей пригодны смеси ацетон — пиридин (4 : 1), бензол —
метиловый спирт (9:1), бензол — пиридин (2:1), ацетон — пиридин —
метиловый спирт (70 : 30 : 2) и пиридин [832] в присутствии тимолового
синего или азофиолетового. При титровании стандартным раствором метилата
натрия не наблюдается появления осадка и переход окраски индикатора
четче, чем в случае бензойной кислоты: желтая — голубая (тимоловый
синий), желтая — красная — голубая (азофиолетовый).
В мерной колбе в 100 мл нейтрализованного растворителя растворяют
600—700 мг 2-фенилхинолин-4-карбоновой кислоты. 20 мл запасного (основ-
ного) раствора титруют 0,1 н. раствором алкоголята щелочного металла или
гидроокиси алкиламмония до появления голубой окраски. Можно также
непосредственно в колбу для титрования взвесить 120—130 мг препарата;
на титрование этого количества кислоты расходуется примерно 5 мл стан-
дартного раствора. 24,93 мг фенилхинолин-4-карбоновой кислоты эквива-
лентны 1 мл 0,1 н. раствора основания.
89. п-АМИНОСАЛИЦИЛОВАЯ КИСЛОТА |124|
50 мл ацетона нейтрализуют в присутствии 4-х капель 0,3%-ного тимоло-
вого синего до появления голубой окраски, добавляют 80—100 мг тонко
измельченной n-аминосалициловой кислоты и после растворения титруют
0,1 н. раствором едкого кали в спирте или метилатом калия в смеси бензол
и метиловый спирт, пока раствор не станет ярко-голубым. В качестве раство-
рителя можно использовать и пиридин [832]. 1 мл 0,1 н. стандартного раство-
ра эквивалентен 15,31 мг n-аминосалициловой кислоты.
Определение п-аминосалициловой кислоты в таблетках. В колбу Эрлен-
мейера с притертой стеклянной пробкой взвешивают 15—20 тонко измель-
ченных таблеток (~80—100 мг кислоты), добавляют 40 мл нейтрализован-
ного ацетона и экстрагируют при перемешивании вручную или магнитной
мешалкой. Затем раствор оставляют стоять на несколько минут, осторожно
230
Глава 15
сливают прозрачную жидкость на фильтр и промывают колбу и фильтр
(3x5 мл) нейтрализованным ацетоном. Объединенные растворы титруют
как указано выше. О мешающем действии стеариновой кислоты см. в разд. 87.
90. ОПРЕДЕЛЕНИЕ АЦЕТИЛСАЛИЦИЛОВОЙ КИСЛОТЫ В ПРИСУТСТВИИ
ФЕНАЦЕТИНА И КОФЕИНА |883]
Тонко измельченные таблетки, в которых содержится примерно 500—
600 мг ацетилсалициловой кислоты, освобождают от стеарина в колбе Эрлен-
мейера с притертой стеклянной пробкой экстракцией (3 х 30 мл) низко-
кипящим петролейным эфиром (т. кип. 30—60е) и фильтруют через бумажный
фильтр диаметром 9 см. Сухой порошок вместе с фильтровальной бумагой
снова помещают в колбу и после добавления 20 мл 5%-ной серной кислоты
экстрагируют (5 х 40 мл) смесью хлороформ — эфир (1 : 3). Объединенные
вытяжки высушивают сульфатом натрия и выпаривают почти досуха на водя-
ной бане в колбе для упаривания, пропуская струю сухого воздуха над
раствором, как, например, показано на рис. 103. Остаток растворяют в 80 мл
диметилформамида (нейтрализованного в присутствии тимолового синего),
вливают в мерную колбу емкостью 100 мл и разбавляют до метки. Получен-
ный раствор титруют порциями по 20—25 мл 0,1 н. метилатом лития [646]
до появления голубой окраски. Метилат лития в меньшей степени, чем мети-
лат калия, склонен к образованию студенистого осадка солей кислот в орга-
нических растворителях. На титрование 18,02 мг ацетилсалициловой кис-
лоты расходуется 1 мл 0,1 н. раствора.
91. ХОЛЕВАЯ, ДЕЗОКСИ- И ДЕГИДРОХОЛЕВЫЕ КИСЛОТЫ (153J
50—100 мг препарата либо растворяют в 50 мл смеси бензол — метило-
вый спирт (10 : 1), либо суспендируют в 2 мл диметилформамида при поме-
шивании стеклянной палочкой, затем добавляют 50 мл хлороформа. После
этого соединение титруют 0,1 н. раствором метилата калия в смеси бензол —
метиловый спирт в присутствии двух капель тимолового синего до появления
голубой окраски или в смеси хлороформ — диметилформамид до появления
пурпурно-голубой окраски. На титрование 40,85 мг холевой кислоты,
39,25 мг дезоксихолевой кислоты и 40,25 мг дегидрохолевой кислоты расхо-
дуется 1 мл 0,1 н. раствора.
Холевую кислоту можно также титровать 0,1 н. метилатом натрия в при-
сутствии гексаметилентетрамина в среде пиридина [832].
92. МНОГООСНОВНЫЕ КИСЛОТЫ [159, 160, 176, 252, 309, 324, 360, 480
Анион кислоты, образующийся в процессе титрования, несет отрицатель-
ный заряд, поэтому диссоциация второго и третьего протонов многоосновных
кислот затруднена (см. гл. 4, разд. 26). Во многих случаях, однако, в соот-
ветствующих растворителях могут быть оттитрованы обе карбоксильные
группы двухосновных кислот. Так, в смеси бензол — метиловый спирт
малоновая кислота только грубо титруется метилатом натрия в присутствии
тимолового синего, в то время как в н-бутиламине ее можно оттитровать
с высокой точностью как двухосновную кислоту.
При потенциометрическом титровании используются закрытые приборы,
имеющие магнитную мешалку и пару электродов стекло — каломель. При-
боры подобного типа показаны на рис. 112—114 в гл. 21. Кривые титрования
для различных кислот могут иметь различную форму. Со стандартным
раствором ТБАГ и в диметилформамиде три изомера фталевой кислоты дают
две точки перегиба; первый перегиб значительно четче выражен длл фталевой
Определение веществ кислого характера
231
кислоты, в то время как второй перегиб сильнее для м- и n-фталевых кислот
(т. е. для изо- и терефталевой кислот). В качестве растворителей лучше всего
использовать пиридин или изопропиловый спирт, так как двухосновные
кислоты незначительно взаимодействуют с этими растворителями. Реак-
ционная способность растворителей возрастает в следующем ряду: пири-
дин < изопропиловый спирт < ацетон < метилизобутилкетон или ацетони-
трил < диметилформамид.
Растворители основного характера, имеющие большой диапазон потен-
циала, более удобны для потенциометрического титрования (гл. 12).
Характерно, что выравнивающее и повышающее величину констант действие пири-
дина ведет к тому, что при титровании в пиридине различие между первой и второй точка-
ми перегиба для серной или янтарной кислоты мало (350 и 250 мв соответственно), хотя
константы диссоциации серной кислоты в воде (Л\ = 10-1 и К2 = 1,2-10-2) значительно
олыпе, чем у янтарной кислоты (Ki = 6,9-10-4 и К2= 2,5*10-в) [159].
Для приготовления раствора взвешивают такое количество двух- или
трехосновной кислоты, на титрование которого потребовалось бы не менее
3—5 мл стандартного раствора при потенциометрическом титровании
и 5—8 мл при визуальном определении конечной точки. В первом случае
кислоту растворяют в 50 мл пиридина, во втором — в 25 мл пиридина. В каче-
стве индикатора используют 0,3 %-ный раствор тимолового синего в изо-
пропиловом спирте, а для слабых кислот — насыщенный раствор азофиоле-
тового в бензоле. Концентрация стандартного раствора метилата калия
или ТБАГ должна быть по возможности меньше 0,1 н. Для каждой анали-
зируемой двухосновной кислоты рекомендуется потенциометрически
контролировать, соответствует ли точка эквивалентности фиолетово-голубой
окраске азофиолетового.
При титровании трех- и четырехосновных кислот (лимонной, фосфорной
и этилендиаминтетрауксусной) в пиридине или диметилформамиде стандарт-
ным раствором ТБАГ в пиридине или диметилформамиде на потенциометри-
ческой кривой можно наблюдать три точки перегиба [159]. Если фосфорную
кислоту титруют метилатом калия в смеси ацетон — пиридин (4 : 1), при
первом изменении окраски азофиолетового (красная окраска) образуется
дигидрофосфат калия, так что 30—80 лег фосфорной кислоты могут быть опре-
делены с точностью ±0,8% (так же как фосфорнокислые соли алкалоидов
[309]). (См. также гл. 24, разд. 140.) Винная, о-фталевая и малеиновая кислоты
могут быть оттитрованы как двухосновные кислоты 0,1 н. метилатом калия
в среде пиридина с тимоловым синим. Этот метод пригоден также для опре-
деления кислотных компонентов алкалоидов спорыньи [324]. Щавелевая,
малоновая, малеиновая, фумаровая, яблочная, янтарная, о-фталевая и лимон-
ная кислоты титруются как двухосновные кислоты 0,1 н. раствором ТБАГ
в пиридине в присутствии азофиолетового [159].
Важность точного определения щавелевой кислоты в неводной среде очевидна преж-
де всего для «косвенных» методов. Приведем два примера. Во-первых, это определение
эквивалентов нейтрализации высших жирных кислот с помощью полумпкро- и микро-
методов [807, 808], когда производится обратное титрование избытка метилата калия
стандартным раствором щавелевой кислоты в смеси метиловый спирт — бензол (гл. 15.
разд. 82). Второй пример связан с определением производных карбодиимида; в этом
случае избыток щавелевой кислоты, не вступивший в реакцию с карбодиимидом, определя-
ется обратным титрованием стандартным раствором метилата натрия в смеси бензол —
метиловый спирт в присутствии тимолового синего [891].
Карбодиимпды общей формулы RNCNR' (где R и R' - алкил, арил или циклогек-
сил) взаимодействуют в диоксане с избытком щавелевой кислоты с образованием дизаме-
щенной мочевины, СО2 и СО
RNCNR' + (COOH)2 -> RNHCONHR/4-CO2±CO.
232
Глава 15
93. НАДКИСЛОТЫ И ГИДРОПЕРЕКИСИ
Из органических перекисей надкислоты, гидроперекиси и перекиси аль-
дегидов (кетонов) можно титровать как кислоты
У0
К — Се Надкислота
ХЮН
Величина рК надкислот в среднем на 3,5 единицы больше, чем величина рК
соответствующей органической кислоты; гидроперекиси и перекиси альдеги-
дов представляют собой очень слабые кислоты со значением рА 11,6—12,8.
Надкислоты, как и перекись водорода, можно титровать потенциометри-
чески 0,2 н. раствором аминоэтилата натрия в этилендиамине в относительно
простых смесях, используя пару электродов сурьма — сурьма [554, 601].
В колбу герметического прибора для титрования, изображенного на рис. 53,
взвешивают 2—5 мг-экв надкислоты или гидроперекиси, растворяют в 50 мл
этилендиамина и титруют 0,2 н. раствором аминоэтилата натрия в смеси
этилендиамин — этаноламин. (О приготовлении стандартного раствора
см. в гл. 11.) 1 мл 0,2 н. стандартного раствора эквивалентен 15,21 мг над-
уксусной кислоты. Точность измерения равна ±2%.
94. АМИНОКИСЛОТЫ
Растворители основного характера значительно уменьшают основность
аминогрупп и в то же время увеличивают кислотность карбоксильной груп-
пы. Большинство аминокислот растворяют в пиридине, содержащем 1—3%
воды, после чего их можно титровать потенциометрически или 0,1 н. раство-
ром ТБАГ в смеси бензол — метиловый спирт в присутствии тимолового
синего. Навеску сначала растворяют в 0,5—1,5 мл воды, затем добавляют
50 мл нейтрализованного пиридина. Так титруют аланин, лейцин, треонин,
аспарагин, метионин, оксипролин, глутамин, глутаминовую и аспарагино-
вую кислоты. Когда аминокислоты титруют в пиридине, содержащем
1—3% воды, титр стандартного раствора можно устанавливать по бензойной
кислоте или любой аминокислоте известной и достаточной чистоты. Пири-
дин должен содержать одно и то же количество воды и в 50 мл растворителя
должны находиться примерно эквимолярные количества как стандартного,
так и анализируемого вещества. Концентрации индикаторов должны быть
также одинаковы как при установке титра, так и при анализе.
95. ОПРЕДЕЛЕНИЕ СМЕСЕЙ КИСЛОТ В ДИФФЕРЕНЦИРУЮЩИХ
РАСТВОРИТЕЛЯХ
Дифференцирующему титрованию минеральных и органических кислот
посвящено много работ, тем не менее общепринятых методов анализа не суще-
ствует [105, 159-161, 170, 212, 258, 360, 414, 415, 480, 539, 550, 736, 783, 888].
Различные смеси кислот, в состав которых входит серная кислота, исполь-
зуются в органических реакциях, таких, как сульфирование (серная кис-
лота — сульфокислота), нитрование (серная кислота — азотная кислота),
этерификация и т. д. Во многих случаях анализ этих смесей может быть
выполнен в неводной среде.
Задача дифференцирующего титрования двойных смесей моно-, ди- или
трихлоруксусной кислоты с соляной кислотой в растворителе кетонного
характера была решена Измайловым и сотр. [414, 415]. Уксусная кислота,
позволяющая дифференцировать сильные кислоты, была использована Шко-
диным и Измайловым [736] для анализа смесей кислот: хлорная — соляная,
хлорная — серная и и-толуолсульфокислота — азотная. В качестве основ-
Определение веществ кислого характера
233-
НЫХ агентов для титрования применяли пиридин или диметил анилин, раство-
ренные в уксусной кислоте. Лавин и Тоеннис [502] использовали ацетонитрил
при титровании смесей хлорная кислота — уксусная кислота; в течение
некоторого времени этот растворитель очень широко применялся для диф-
ференцирующего титрования кислот и оснований. Критчфельд и Джонсон
[155] анализировали смеси кислот: серная — хлорная и серная — азотная
титрованием в ацетонитриле стандартным раствором, содержащим морфолин.
Тем не менее некоторые исследователи используют для дифференцирующего
титрования кислот растворители основного характера, например диметил-
формамид или пиридин [159, 176, 359]; Фриц и Ямамура [258] применяют
в этом случае ацетон. Наиболее широкое применение на практике нашли
ацетон, ацетонитрил, метилэтилкетон, метилизобутилкетон, смесь этилен-
гликоль — изопропиловый спирт, метиловый спирт и пиридин. Многообе-
щающим растворителем является пгрепг-бутиловый спирт [253, 550].
При дифференцирующем титровании смесей кислот различной силы
желательно, чтобы растворитель как можно меньше взаимодействовал с рас-
творенным соединением; только при этом условии обнаруживается собственно
кислотность, зависящая от молекулярной структуры. С теоретической
точки зрения наиболее пригодны инертные растворители (четыреххло-
ристый углерод, бензол), но вследствие их низкой электропроводности
потенциометрические измерения могут вызвать большие трудности и, более
того, низкая диэлектрическая проницаемость растворителя способствует
образованию неупорядоченных ассоциатов (ср. гл. 12, разд. 63, г). Для
анализа двойных смесей минеральных кислот рекомендуется применять
растворители с возможно меньшей основностью: их выравнивающее дей-
ствие, противоположное дифференцирующему эффекту, в некоторой степени
согласуется с основностью растворителя. (Пиридин составляет удачное
исключение, так как, несмотря на его основные свойства, он пригоден для
дифференцирующего титрования.)
В растворителях, обладающих дифференцирующими свойствами, можно
проводить титрование сильных (минеральных) кислот в относительно простой
смеси; для этой цели используют стандартные растворы ТБАГ. Раствори-
тели сильноосновного характера (этилендиамин, пиперидин) могут быть
использованы для титрования чрезвычайно слабых кислот; минеральная
и карбоновая кислоты в этих растворителях не дифференцируются. В диме-
тилформамиде, пиридине и ацетонитриле четкая точка перегиба обнаружи-
вается даже в случае очень слабых кислот; при этом весьма сильные и уме-
ренно сильные карбоновые кислоты могут быть проанализированы диффе-
ренцирующим титрованием, в то время как для анализа сильных мине-
ральных кислот эти растворители непригодны. Для анализа смесей мине-
ральных кислот можно использовать метилэтилкетон, но перегибы на кри-
вых титрования очень слабых кислот (или кислотных аналогов) в этом слу-
чае выражены несколько менее четко.
а. Титрование в метилизобутилкетоне. Метилизобутилкетон как раство-
ритель сочетает полезные свойства ацетонитрила и метилэтилкетона
[105], что благоприятствует потенциометрическому титрованию оснований
и кислот. Сильные и слабые кислоты могут быть оттитрованы в простых
смесях 0,2 н. раствором ТБАГ в изопропиловом спирте даже по полумикро-
методу; например, возможен анализ смеси хлорной, соляной, салициловой,
уксусной кислот и фенола (рис. 105) [105]. Благодаря широкому диапазону
изменения потенциала (около 1400 мв) этот раствор ТБАГ пригоден как для
измерения относительной кислотности, так и для титрования смесей кислот
различной силы, если разность между точками перегиба для отдельных ком-
понентов составляет 200—300 мв.
Так как выпускаемый в продажу метилизобутилкетон может содержать
кислотные загрязняющие примеси, его следует перед употреблением очистить.
234
Глава 15
(гл. 8, разд. 49, д). Так как метилэтилкетон слабо поглощает СО2, нет необ-
ходимости проводить титрование в атмосфере азота.
Для титрования в метилизобутилкетоне вполне пригодны 0,2 н. растворы
ТБАГ в изопропиловом спирте. В 20 мл метилизобутилкетона растворяют
примерно 0,2 мг-экв смеси кислот и титруют 0,2 или 0,1 н. раствором ТБАГ
в изопропиловом спирте из бюретки, градуированной по 0,01 или 0,02 мл.
Для титрования используют небольшой сосуд, предложенный Дилом
и Уайльдом (см. рис. 114). Электродом сравнения может служить платино-
вый наконечник на конце бюретки (см. рис. 52) или насыщенный каломель-
ный электрод (гл. 12 и 21).
Для титрования кислот в простых смесях лучше использовать метилизо-
бутилкетон, а не метилэтилкетон, ацетонитрил, пиридин, диметилформамид
Рис. 105. Титрование пятикомпонент-
ной смеси в метилизобутилкетоне. Система
электродов стекло — платина [105].
---Избыток основания
Z, 6 -Ди-трет-бутил-4 .метилфенол
500------Фенол
300 /Серная кислота (как двухосновная)
-----------Уксусная кислота
1qq __бензойная кислота
--Салициловая кислота
-100 —
у Азотная кислота
-300 — X
--Хлористоводородная кислота
~500 Серная кислота (как одноосновная)
-JQQ — ^"Хлорная кислота
Рис. 106. Относительное положение
потенциалов кислот [105].
Растворитель: метилизобутилкетон; титрант: 0,2 н.
раствор гидроокиси тетрабутиламмония в изопро-
пиловом спирте; система электродов: стекло —
каломель.
или изопропиловый спирт (рис. 105 и 106). тпрет-Бутиловый спирт, как
и метилэтилкетон, очень подходит для дифференцирующего титпования
кислотных аналогов (ср. рис. 108).
В неводных растворителях порядок относительно кислотности обычно обозначается
точкой полунейтрализации исследуемого соединения относительно некоторой кислоты,
выбранной в качестве эталона. При потенциометрическом титровании сильных кислот
в метилизобутилкетоне характеристика перегиба (скачок, измеренный в милливольтах)
примерно в пять раз четче, чем в среде этилендиампна (в обоих случаях титрование про-
водят стандартным раствором ТБАГ). Относительное положение точек полунейтрализации
различных кислот показано на рис. 106.
При титровании ТБАГ салициловой кислоты в метилизобутилкетоне можно видеть
одну точку перегиба, соответствующую нейтрализации карбоксильной группы. Однако
при титровании в метилизобутилкетоне раствором едкого кали на кривой появляются две
точки перегиба, что указывает на нейтрализацию как группы СООН, так и фенольного
гидроксила. Титрование в этиленгликоле, например, п, п'-диоксидифенилметана стан-
дартным раствором ТБАГ в изопропиловом спирте дает два перегиба на кривой, а при
использовании стандартного раствора едкого кали в изопропиловом спирте [359] наблю-
дается только один перегиб.
Относительно титрования кислотных смесей в метилэтилкетоне см. работы [114, 359,
474, 480, 485]. Так, например, о-, м-, n-нитробензойные кислоты и нитрофенолы можно
оттитровать в присутствии азотной кислоты в среде метилэтплкетона 0,1 н. раствором
гидроокиси тетраэтиламмония [478]. 30 мл растворителя нейтрализуют до голубого цвета
в присутствии тимолового синего; 50—100 мг пробы растворяют в нейтрализованном раст-
ворителе и титруют потенциометрически стандартным раствором (С2Н5)^ОН в смеси
бензол — метиловый спирт в атмосфере азота с использованием электродной пары стек-
ло — каломель. В этом случае анализ смеси кислот можно провести с точностью +1%.
Определение веществ кислого характера
235
б. Определения в ацетоне. Безводный ацетон мало пригоден для диффе-
ренцирующего титрования сильных минеральных и карбоновых кислот, так
как в процессе потенциометрического титрования, продолжающегося доволь-
но долго, он взаимодействует с этими кислотами [160, 258]. Тем не менее
ацетон пригоден для титрования смесей карбоновых кислот. Так, в ацетоне
дихлоруксусная кислота может быть определена в присутствии уксусной
кислоты, а салициловая кислота — в присутствии метилсалицилата. Кроме
того, в ацетоне, содержащем немного воды, можно привести анализ смесей
кислот: соляная — серная или азотная — серная, если использовать стан-
дартный раствор гидроокиси три-н-бутилметиламмония. Конечную точку
титрования можно определить потенциометрически или визуально с помощью
индикаторной смеси из нейтрального красного и тимолового синего [539].
Навеску 0,4—0,8 мг-экв смеси кислот растворяют в 40 мл нейтрализованного
ацетона и титруют потенциометрически 0,1 н. раствором ТБАГ, используя
стеклянный и каломельный электроды.
Грибова и Левин [289] исследовали потенциометрическое титрование двойной смеси
серной кислоты с антрахинонмоно- или антрахинондисульфокислотой, нафталинмоносуль-
фокислотой или бензолсульфокислотой в ацетоне или ацетонитриле. В качестве стандарт-
ного раствора они использовали раствор морфолина или дифенилгуанидина в этиловом
спирте, содержащем 10—15% глицерина. Точность определения составляла ±2%.
Смесь малеиновой и фталевой кислот анализируют титрованием в среде ацетона [543].
Навеску безводного образца < 0,04 мг-экв растворяют в 100 мл тщательно обезвоженного
ацетона. Разбавляют 10 мл запасного раствора 40 мл ацетона и титруют 0,1 н. раствором
едкого кали в этиловом спирте, используя стеклянный и каломельный электроды.
На потенциометрической кривой дифференцирующего титрования наблюдаются три
точки перегиба. Первый перегиб указывает на первые атомы водорода любой из двух
кислот, второй перегиб — на второй атом водорода фталевой кислоты, а третий пере-
гиб показывает второй атом водорода малеиновой кислоты.
Критчфельд и Джонсон [155] титровали серную кислоту в присутствии других кислот
в среде ацетонитрила, используя стандартный раствор, содержащий морфолин.
Пиридин широко используется в качестве растворителя для дифферен-
цирующего титрования минеральных и других кислот [159—162].
в. Определение в пиридине. Взвешивают 0,7—0,8 мг-экв смеси кислот и при
необходимости растворяют ее в 1,5 мл воды, после чего разбавляют 50 мл
пиридина и титруют в атмосфере азота стандартным раствором ТБАГ в смеси
бензол — метиловый спирт, используя стеклянный и каломельный элек-
троды.
При титровании смеси серной и азотной кислот первая точка перегиба отвечает
первому эквиваленту серной и азотной кислоты, в то время как второй перегиб соответ-
ствует нейтрализации бисульфат-иона. При титровании смеси серной и фосфорной кислот
могут наблюдаться три перегиба на кривой: 1) первая ступень нейтрализации серной
кислоты, 2) вторая ступень нейтрализации серной кислоты и первая ступень нейтрализа-
ции фосфорной кислоты и 3) вторая ступень нейтрализации фосфорной кислоты. Такие
смеси, как соляная — серная кислоты, серная — хлорная кислоты, азотная — фосфор-
ная кислоты, серная кислота — бензолсульфохлорид, серная кислота — бензолсуль-
фокислота, серная кислота — n-толуолсульфокислота, серная кислота — метансульфо-
кпслота можно оттитровать обычным путем [161, 162]. Точность определения сильных
кислот ±0,3%, слабых кислот ±0,5% и очень слабых кислот ±1%.
Определение алкоксилъной группы служит примером остроумного приема
для выполнения дифференцирующего титрования в пиридине, ибо в этом
случае, используя двухступенчатое изменение окраски индикатора азофио-
летового, можно титровать в одной смеси сначала HI (оранжевая —> красная),
затем (C5H5NR+)I~ (красная —> фиолетовая), применяя 0,02 н. раствор ТБАГ
(см. гл. 31, разд. 168) [163].
Титрование смесей соляной и бензойной кислот или бензойной кислоты
и фенола осуществляется потенциометрически в среде пиридина метилатом
натрия в смеси бензол — метиловый спирт при использовании стеклянного
и серебряного электродов. Если нанести на график зависимость изменения
потенциала от израсходованного объема, то соляная кислота и фенол обна-
236
Глава 15
руживаются по обычной S-образной кривой, втовремя как бензойную кислоту
можно выявить по минимальному пику на перегибе (рис. 107) [888] (ср. гл. 12.
разд. 69).
г. Титрование в G — Н-смеся.; [170]. Серную кислоту можно титро-
вать потенциометрически в среде этиленгликоля, для этого удобно исполь-
зовать растворы едкого натра в смеси этиленгликоль — изопропиловый
спирт или 0,1 н. раствор пиперидина в изопропиловом спирте. Однако раствор
едкого натра, приготовленный ранее чем за неделю, непригоден для диффе-
ренцирующего титрования. При использовании для титрования раствора,
содержащего пиперидин, это влияние примесей и побочных продуктов устра-
няется, но слабые кислоты (например, уксусную) нельзя титровать в смеси
Рис. 107. Кривые потенциометрического
титрования смеси кислот [888]. Титрова-
ние ведут 0,1 н. раствором метилата натрия
в смеси бензол — метиловый спирт со стек-
лянным и серебряным электродами.
1 — соляная кислота; 2 — бензойная кислота;
3 — фенол.
Рис. 108. Титрование четырехкомпонент-
ной смеси в тгарегга-бутиловом спирте [243].
Титрование ведут раствором гидроокиси
тетрабутиламмония в смеси бензол — изо-
пропиловый спирт (электроды см. на
рис. 68).
с минеральной кислотой. Преимущество такого стандартного раствора
состоит в том, что при титровании серной кислоты не появляется осадка
сульфата. Смесь серной и других кислот можно титровать потенциометри-
чески, используя стандартный раствор, содержащий пиперидин, и применяя
электродную пару стекло — насыщенная каломель. Удобным растворителем
для этого служит смесь этиленгликоль — изопропиловый спирт (1 : 1).
Точность измерения ±1%. Определение этим методом тройных смесей дает
худшие результаты.
Содержание серной или соляной кислоты в ацетилирующих смесях в среде диэтилен-
гликоля можно определить с помощью 0,05 или 0,1 н. раствора триэтаноламина в про-
пиловом спирте [213].
д. Метиловый спирт применяют при серийных анализах состава суль-
фирующих или нитрующих кислотных смесей [783]. В качестве титрую-
щего агента для потенциометрического титрования используют циклогексил-
амин в метиловом спирте, причем титр этого раствора устанавливают по бен-
зойной кислоте, применяя пару электродов стекло — каломель. Навеску
образца, содержащую около 8 мг-экв кислоты (S — вес пробы, мг), раство-
ряют в 50 мл метилового спирта и титруют при перемешивании до первого
скачка потенциала (а), а затем продолжают титрование до второго скачка (&).
В случае бинарных смесей многоосновных минеральных кислот с серной
кислотой расчет ведут по формулам
Р, „ (6 — а) -98,08 -100 0/
Содержание серной кислоты = ---------------%,
„ „ (2а — Ь) л. 100 0,
Содержание второго компонента кислотной смеси = -------£-----"о,
4J
Определение веществ кислого характера
237
где А — молекулярный вес второго компонента (например, бензолсульфо-
кислоты).
Титрование 100—300 мг смеси Na2SO4—CeH5SO3Na-H2O можно провести с исполь-
зованием ионообменной смолы, например вофатит KS, следующим путем. Смесь солей
растворяют в 3 мл воды, пропускают ее через колонку с ионообменной смолой (30 X 2 см),
затем вымывают метиловым спиртом (150 мл) и титруют, как указано выше [783].
е. Относительно дифференцирующего титрования в тпретп-бутиловом
спирте см. работы [253, 550, 551], а также рис. 68 и 108 и гл. 12, разд. 63, а.
ж. Потенциометрическое дифференцирующее титрование большого числа
кислот и их смесей, кислотность которых меняется в широких пределах
(от минеральных кислот до фенолов), может быть осуществлено при исполь-
зовании в качестве растворителя смеси М,]Х-диметиламидов жирных кислот,
содержащих примерно 95% М,М-диметиллаурамида, 3% М,]Х-диметилми-
ристамида и 2% М,М-диметилкапрамида [678].
Навеску пробы растворяют в 25 мл растворителя и титруют в атмосфере
азота стандартным раствором гидроокиси тетра-«-бутиламмония в смеси
бензол — изопропиловый спирт — вода. Для титрования можно использо-
вать микроячейку (рис. 106). Поскольку хлористый калий очень слабо
растворяется в указанном растворителе (Hallcomid М-12, С. Р. Hall Со.,
Illinois), к электроду сравнения из насыщенной каломели добавляют водный
хлористый литий.
При использовании в этом растворителе пары электродов стекло —
каломель воспроизводимость отсчетов потенциала находится в пределах 2 мв.
Глава 16
ОПРЕДЕЛЕНИЕ АНГИДРИДОВ КИСЛОТ,
ГАЛОГЕНАНГИДРИДОВ, РЕАКЦИОННОСПОСОБНЫХ
АЛКИЛ- И АРАЛКИЛГАЛОГЕНИДОВ
Хлорангидриды и ангидриды кислот относятся к числу акцепторных
кислот, причем сам ацетил-катион СН3СО+ в определенном смысле также-
представляет собой кислоту.
96. АНГИДРИДЫ КИСЛОТ
В водной среде и особенно в присутствии пиридина как катализатора
уксусный ангидрид достаточно быстро растворяется, при этом образуются
две молекулы уксусной кислоты; в дальнейшем при нейтрализации уксусный
ангидрид поглощает 2 экв гидроокиси щелочного металла. При реакции
между уксусным ангидридом и метилатом щелочного металла, растворен-
ным в метиловом спирте, смеси бензол — метиловый спирт или ацетоне,
образуются ацетат щелочного металла и метилацетат, т. е. расходуется
только 1 экв титранта [762].
(СН3СО)2О + СН3ОК = СН3СООК + СН3СООСН3.
Ангидриды кислот таким же образом реагируют с едким кали в этиловом
спирте и ацетоне [613]. Смеси уксусного ангидрида и карбоновой кислоты
можно титровать как в водной, так и в неводной среде, например, в при-
сутствии тимолового синего в качестве индикатора [762].
Фталевый ангидрид титруют в смеси бензол — метиловый спирт (3 : 1)
метилатом натрия в присутствии тдмолевого синего [252] или проводят
потенциометрическое титрование раствором едкого кали в диметилформамиде
с использованием пары электродов платина — каломель [457].
а. Морфолиновый метод [431]. На практике очень большое значение
имеет определение небольших количеств ангидрида в присутствии больших
количеств карбоновых кислот и определение небольших количеств карбоно-
вых кислот в присутствии больших количеств анигидрида. В первом случае
можно использовать морфолиновый метод, так как с ангидридами морфолин
O(CH2CH2)2NH образует эквимолярные количества амида кислоты и карбо-
новую кислоту
HN(CH2CH2)2O + (CH3CO)2O —> CH3CON(CH2CH2)2O + CH3COOH.
Взаимодействие с уксусным ангидридом завершается, например, в течение
30 сек. В метиловом спирте избыток морфолина можно определить обратным
титрованием 0,1 н. соляной кислотой.
В колбу Эрленмейера с притертой пробкой емкостью 250 мл помещают
10 мл уксусной кислоты, содержащей 0,01—0,5% уксусного ангидрида,
добавляют 50 мл 0,02 н. раствора морфолина в метиловом спирте (1,74 мл
морфолина -|- 1000 мл спирта), перемешивают и оставляют на 5 мин, плотно
закрыв пробку. Затем титруют в присутствии 4—5 капель индикаторной
смеси диметиловый желтый — метиленовый голубой (см. табл. 63 и гл. 9.
разд. 51, а) 0,1 н. соляной кислотой в метиловом спирте или смеси пропи-
ленгликоль — хлороформ до появления желтой окраски. Расход стандарт-
ного раствора морфолина определяется следующим образом. Разность в коли-
честве истраченного титранта пропорциональна содержанию ангидрида;
10,21 мг уксусного ангидрида требуют 1 мл 0,1 н. соляной кислоты (~ 5 мл
Ангидриды кислот, галогенангидриды и алкил- и аралкилгалогениды
239
0,02 н. раствора морфолина). Этот метод применяется для определения содер-
жания ангидрида в безводных уксусной и пропионовой кислотах (см. гл. 8,
разд. 47, в). Для определения больших количеств ангидрида используются
0,5 н. растворы. Раствор морфолина должен быть защищен от попадания С02
из воздуха. Содержание в смесях пропионового, масляного, глутарового
и янтарного ангидридов рассчитывают исходя из того, что 1мл 0,1 н. соля-
ной кислоты эквивалентен 13,01; 15,82; 17,02; 10,01 и 14,81 мг перечислен-
ных ангидридов соответственно.
Морфолиновый метод неприменим для определения ангидридов кислот
с константами ионизации больше 2-10-2 (например, малеиновый и цитрако-
новый ангидриды), так как карбоновая кислота, образующаяся в метиловом
спирте, не очень сильно отличается от рекомендованного индикатора по своей
кислотности. Присутствие других соединений — кетенов, дикетенов, ацил-
хлоридов и минеральных кислот, которые реагируют с морфолином, вызывая
образование солей с ним, оказывает нежелательное влияние на титрование.
б. Анилиновый метод [208]. Уксусный ангидрид легко реагирует с ани-
лином
(CH3CO)2O + H2NCeH5 —» CH3CONHCeH5 + CH3COOH,
а избыток анилина можно определить обратным титрованием хлорной кис-
лотой.
В мерной колбе емкостью 100 мл с точностью до 1 мг взвешивают 1 г
уксусного ангидрида и разбавляют до отметки уксусной кислотой, не содер-
жащей уксусного ангидрида (гл. 8, разд. 47, в и г). Переносят 50 мл 0,1 н.
раствора анилина в уксусной кислоте (9,1 мл анилина +1000 мл кислоты)
в колбу Эрленмейера на 200 мл, отмеряют в нее пипеткой 20 мл исследуемого
раствора и оставляют смесь на 40 мин при комнатной температуре, затем
после добавления 0,1 мл 0,1%-ного раствора кристаллического фиоле-
тового в уксусной кислоте титруют 0,1 н. хлорной кислотой в уксус-
ной кислоте до появления светло-зеленой окраски. При титровании реко-
мендуется использовать растворы сравнения, так как образующийся амид
кислоты в уксусной кислоте обладает до известной степени также и свой-
ствами основания. Расход хлорной кислоты на 50 мл раствора анилина
определяется аналогичным образом. Разность между двумя определениями
(кислоты и ее смеси с ангидридом) пропорциональна содержанию уксусного
ангидрида.
Сиггпа и Ханна [748] одними из первых предложили использовать анилиновый метод
для протенцпометрпческого титрования. Согласно их методу, растворителем для анализи-
руемого вещества и 0,2 н. раствора соляной кислоты служит смесь этиленгликоль —
изопропиловый спирт в соотношении 1 : 1 (ср. [621]). Аддукт, образующийся из анилина
и избытка карбоновой кислоты, в этом случае также можно оттитровать как основание.
97. ОПРЕДЕЛЕНИЕ МАЛЕИНОВОЙ И ФТАЛЕВОЙ КИСЛОТ В ПРИСУТСТВИИ
ИХ АНГИДРИДОВ
Три-к-пропиламин и N-этилпиперидин не реагируют с ангидридами
кислот, поэтому карбоновые кислоты с константой диссоциации не ниже
10-3 можно определять в присутствии ангидридов карбоновых кислот [56,
746] с помощью стандартного раствора три-к-пропиламина и N-этилпипери-
дина в ацетоне или в метилэтилкетоне (табл. 18, е).
В 30—50 мл ацетона растворяют навеску образца, содержащую примерно
0,2—2 ммоля анализируемой кислоты, и титруют потенциометрически 0,1 и.
раствором три-к-пропиламина в ацетоне, используя электродную пару
стекло — каломель. Этот метод пригоден для определения 0,2—75 %-ной
малеиновой или фталевой кислоты в присутствии их ангидридов. Для опре-
деления фталевой кислоты, присутствующей как загрязняющая примесь,
хорошо использовать электродную пару молибден — каломель [56].
240
Глава 16
Определение титра стандартного раствора проводят по чистой малеино-
вой кислоте, исходя из предположения, что реагирует только одна группа
GOOH. На титрование 11,61 мг малеиновой и 16,61 мг фталевой кислоты
расходуется 1 мл 0,1 н. раствора.
98. ОПРЕДЕЛЕНИЕ ПРИМЕСЕЙ УКСУСНОЙ КИСЛОТЫ В УКСУСНОМ АНГИДРИДЕ
Уксусная кислота, содержащая уксусный ангидрид, определяется более
точно, если ее титровать непосредственно [564], напимер, 2 М раствором
триэтиламина в бензоле (табл. 52).
При содержании уксусной кислоты 0,6—3,5% точность этого метода
составляет ±0,07% (абс.); недостатком является сложность приготовления
стандартного раствора основания.
Любая примесь амина, способного к ацилированию, удаляется кипячением триэтил-
амина (500 мл), предварительно высушенного твердым едким кали, с ангидридом (25 мл)
в течение 30 мин. Избыток ангидрида осторожно при постоянном перемешивании разла-
гают 125 мл 10 н. едкого натра и триэтиламин экстрагируют 500 мл бензола. Бензольную
вытяжку промывают еще раз 125 мл щелочи и фракционируют; около 200 мл головного
погона отбрасывают, заменяют приемник и продолжают перегонку, пока остаток не соста-
вит примерно 100 мл. В ходе перегонки растворитель защищают от попадания влаги
из воздуха. Вторую фракцию разбавляют равным количеством бензола (содержание
воды < 0,05%). Приготовленный таким образом раствор основания титруют водной
0,5 н. соляной кислотой.
В колбу для титрования приливают 100 мл уксусного ангидрида, содер-
жащего 5—50 мг-экв уксусной кислоты, и сразу же титруют в присутствии
5 капель насыщенного метилового красного в ацетонитриле. Для сравнения
окраски индикатора готовят раствор: пять капель индикатора ±100 мл бен-
зола; на 3,13 моля триэтиламина расходуется 1 моль уксусной кислоты.
99. АЦИЛХЛОРИДЫ
а. Ацетилхлорид можно титровать как кислоту в среде уксусного ангид-
рида ацетатом натрия в уксусной кислоте [831]
СН3СОС1 + CH3COO-Na+ NaCl + (CH3CO)2O
или
СН3СОС1 + (СН3СО)2О —> (СН3СО)3О+±С1-
и
(СН3СО)3О+ + СН3СОО- —> 2(СН3СО)2О.
Предлагается следующая методика анализа. В мерную колбу емкостью
25 мл, содержащую 5 мл 100%-ного уксусного ангидрида, взвешивают
90—110 мг ацетилхлорида. Раствор переносят в мерную колбу на 100 мл.
промытую уксусным ангидридом, и заполняют до метки. 20 мл приготовлен-
ного раствора отмеряют пипеткой в колбу Эрленмейера емкостью 200 мл.
с притертой пробкой (соблюдая правила безопасности, рис. 34), добавляют
0,05—0,1 мг индикатора твердого диметиламиноазобензола и очень медленно
титруют полученный красный раствор 0,1 н. раствором ацетата натрия
в уксусной кислоте до появления желто-розового окрашивания (~30лшн).
(Приготовление стандартного раствора ацетата натрия см. в гл. 10, разд. 58.)
Колбу с закрытой пробкой оставляют на 30 мин, затем титрование продол-
жают, пока желто-розовая окраска не перейдет в желтую.
В начале титрования после добавления 3—5 капель ацетата натрия раствор становит-
ся желтым, однако он вновь приобретает исходную окраску и уже вскоре наблюдается
медленный переход желтой окраски в розовую. После стояния в течение 30 мин пере-
ход оранжевой окраски в желтую становится более ярко выраженным.
Ангидриды кислот, галогенангидриды и алкил- и аралкилгалогениды
241
Титр раствора ацетата натрия определяют по хлорной кислоте в присут-
ствии диметилового желтого как индикатора; на 7,85 мг ацетилхлорида
расходуется 1 мл 0,1 н. ацетата натрия. Точность определения ±2%.
б. Хлорангидриды кислот могут быть также определены титрованием
(в ацетоне или диоксане) соляной кислоты, образующейся при взаимодей-
ствии ацилхлорпда со спиртом [623, 753]
НСОС1 + С2Н5ОН —> ВСООС2Н5 + НС1.
В этом случае титруют алкоголятом щелочного металла или трибутилами-
ном. В бензоле, смеси бензол — метиловый спирт или диоксане ацилхлориды
(а также ангидриды кислот) титруются как одноосновные кислоты алкоголя-
том щелочного металла в присутствии тимолового синего до появления голу-
бой окраски [252, 623]
RCOC1 + CH3ONa —- НСОССН3 + NaCl.
Например, 3,5-динитробензоилхлорид в бензоле и хлористый бензоил
в смеси бензол — метиловый спирт (3 : 1) можно оттитровать в присутствии
тимолового синего [252]. Однако в пиридине они титруются раствором гидро-
окиси триметилбензиламмония в пиридине (тритон В) как двуосновные
кислоты.
R'COCl -L 2R4NOH —[R'COO] NR4+R4NC1 + H2O.
в. Гидролиз хлорангидридов кислот проводят в диоксане, содержащем
небольшое количество воды и пиридина [378]. Избыток воды связывают уксус-
ным ангидридом и образующийся хлорид пиридиния определяют титрованием
раствором хлорной кислоты в присутствии ацетата ртути(П). Это определе-
ние может быть также выполнено в присутствии карбоновых кислот и их
ангидридов.
Навеску образца помещают в колбу с притертой пробкой и нагревают
на водяной бане с обратным холодильником в течение 1 час с 10 мл реагента,
содержащего пиридин. После охлаждения смеси добавляют еще 10 мл реа-
гента, содержащего ангидрид, и нагревают еще 1 час. После охлаждения
добавляют 25 мл уксусной или пропионовой кислоты и титруют 0,1 н. хлор-
ной кислотой в присутствии нескольких капель 0,2%-ного раствора мета-
нилового желтого как индикатора (в диоксане), пока не появится розовая
окраска. Добавляют 25 мл реагента ацетата ртути(П) и вновь продолжают
титрование до появления розовой окраски. Количество раствора, расходуе-
мое на второе титрование, пропорционально количеству хлорангидрида
кислоты.
Реагент, содержащий пиридин. Смесь 20 г пиридина и 1,8 г дистиллированной воды
разбавляют до объема 500 мл диоксаном. Реагент, содержащий ангидрид. 11 г 98%-ного
уксусного ангидрида разбавляют до 500 мл дпоксаном. Реагент, содержащий ацетат
ртути(П). 22 г окиси ртути(П) растворяют в смеси 11 мл уксусного ангидрида и 1000 мл
уксусной кислоты.
г. Определение ацилгалогенидов карбоновых кислот и определение
хлорангидридов сульфокислот может быть также выполнено в присутствии
органических кислот и алкилгалогенидов. Хлорангидриды вводят в реакцию
с гексаметиленимином (ГМИ) и избыток последнего оттитровывают 0,1 н. со-
ляной кислотой в метиловом спирте [799]
RCOHal + 2(CH2)6NH —> RCON(CH2)6+ (CH2)6NH-HHal,
RSO2C1 + 2(CH2)6NH RSO2N(CH2)6 + (CH2%NH-HCl.
30—70 мг образца оставляют с 10 мл раствора ГМИ на 5—10 мин при
комнатной температуре. Избыток гексаметиленимина определяют обратным
титрованием 0,1 н. соляной кислотой в присутствии 5 капель индикаторной
смеси в метиловом спирте. Глухой опыт проводят так же и разность объемов
242
Глава 16
рассчитывают по формуле
{Vi-V^.d
6-200 ’
где а — содержание гексаметиленимина, %; Vi — расход стандартного
раствора в глухом опыте, мл; V2 — расход стандартного раствора при титро-
вании образца, мл; d — молекулярный вес анализируемого соединения;
b — вес образца, г. Таким образом могут быть определены ацетилхлорид,
ацетилбромид, ацетилиодид, хлорангидриды монохлоруксусной, про-
пионовой, олеиновой, изовалериановой, капроновой и фенилуксусной кис-
лот, а также бензолсульфохлорид, n-толуолсульфохлорид и 1,4-циклогек-
сандисульфохлорид.
Приготовление раствора ГМИ. Водный технический раствор ГМИ насыщают едким
натром, верхний слой отделяют и высушивают над твердым КОН. 20 г продукта
с т. кип. 137—138°, собранного при дробной перегонке высушенного ГМИ, растворяют
в 1000 мл метилового спирта.
И ндикаторная смесь. 10 мг метиленового голубого и 40 мг метилового красного рас-
творяют в 100 мл этилового сппрта и разбавляют 100 мл дистиллированной воды. Наблю-
даемое изменение окраски индикатора: зеленая фиолетовая. (Соляная кислота в мети-
ловом спирте см. гл. 9.)
д. Относительно титрования в хлористом ацетиле см. работу [758].
100. АНАЛИЗ СМЕСИ АРОИЛХЛОРИДА, СОЛЯНОЙ КИСЛОТЫ И КАРБОНОВОЙ
КИСЛОТЫ
а. Анализ смеси хлористого бензоила, соляной кислоты и бензойной кислоты
проводят следующим образом [623]. К 2—5 мл * исследуемого раствора
в диоксане, содержащем 0,2—1 ммоля ароилхлорида, помещенного в колбу
Эрленмейера емкостью 25 мл, добавляют 2 капли тимолового синего
(ср. гл. 13, разд. 13 и табл. 65) и титруют свободную соляную кислоту 0,1 н.
раствором трибутиламина (табл. 52) в диоксане до появления желтой
окраски (Ti). Титрование рекомендуется вести при перемешивании с помощью
магнитной мешалки, кончик бюретки должен быть погружен в жидкость.
После добавления 5 мл этилового спирта смесь нагревают в течение 5—10 мин
до 80°, чтобы полностью выделить в свободном состоянии соляную кислоту
из ацилхлорида. После повторного нагревания титруют соляную кислоту
0,1 н. раствором метилата натрия в смеси бензол — метиловый спирт
до устойчивой желтой окраски (Т2). Карбоновую кислоту титруют в остав-
шейся части анализируемого раствора метилатом натрия до появления
голубой окраски тимолового синего (Т3).
Расчет’.
a = Ti-Ni
b = T2-N2
c^T3-N2-(TiNi-YT2N2),
где а, Ъ и с — количества соляной кислоты, хлорангидрида бензойной кис-
лоты и бензойной кислоты соответственно, мг-экв; 7\ — объем израсходо-
ванного стандартного раствора трибутиламина, мл; Т2 и Т3 — объем израс-
ходованного раствора метилата натрия, мл; и N2 — нормальность стан-
дартных растворов трибутиламина и метилата натрия соответственно, г-экв!л.
Изменение окраски тимолового синего в диоксане под действием реагентов можно
описать следующим образом.
1. НС1 + тимоловый синий’, слабая красная окраска, усиливающаяся при добавле-
нии спирта. НС1 можно титровать достаточно точно либо трпбутпламином (наблюдае-
мое изменение окраски индикатора: бледно-красная ярко-желтая), либо метилатом
натрия в смеси бензол—метиловый спирт (бледно-красная -> ярко-красная -> желтая)
и с незначительным избытком CH3ONa до голубой. Объем стандартного раствора, израс-
ходованного до изменения окраскп от желтой к голубой, показывает степень загрязнения
слабой кислотой.
Ангидриды кислот, галогенангидриды и алкил- и аралкилгалогениды
243
2. Бензойная кислота + тимоловый синий', желтая окраска, сохраняемая при добав-
лении метилата или гидроокиси алкиламмония, голубая в точке эквивалентности.
3. Хлористый бензоил + тимоловый синий', бледно-розовая окраска от 1—2 капель
трибутил амина переходит в ярко-желтую, при добавлении эквивалентного количества
метилата натрия становится голубой.
б. Определение соляной кислоты в присутствии хлорангидрида кислоты
[753]. Навеску образца, содержащую не больше 1 ммоля соляной кислоты,
растворяют в 50лел смеси эфир — хлорбензол (1 : 1) и титруют свободную НС1
потенциометрически 0,1 н. раствором трипропиламина (см. табл. 52). Титр
последнего устанавливают по малеиновой кислоте в ацетоне. 1 мл 0,1 н. три-
пропиламина эквивалентен 3,6465 мг соляной кислоты или 11,61 мг малеино-
вой кислоты (титруется только одна группа СООН).
В диоксане НС1 можно также титровать 0,1 н. раствором трибутиламина
в диоксане [623] в присутствии тимолового синего до перехода окраски
от красной к желтой. Титр раствора трибутиламина устанавливают по 0,1 н.
соляной кислоте в диоксане.
101. ОПРЕДЕЛЕНИЕ АЦИЛХЛОРИДОВ В ПРИСУТСТВИИ ЛЕГКО
ГИДРОЛИЗУЮЩИХСЯ ГРУПП
Ацилхлориды алифатических и ароматических карбоновых кислот
можно титровать в тетрагидрофуране непосредственно 0,5 н. стандартным
раствором циклогексиламина (ЦГА) в тетрагидрофуране (ТГФ) (табл. 52),
нормальность которого определена потенциометрически по соляной кислоте
с использованием электродов стекло — каломель. Реакция протекает по сле-
дующей схеме [519]:
RCOC1+ 2C6H10NH2 —» ВСО^С6Н10Н-С6Н10Н2 НС1.
Примеси карбоновых кислот не влияют на титрование, но присутствие
соляной кислоты сразу сказывается на результатах, так как циклогексил-
амин реагирует с сильной кислотой. Преимущество ТГФ заключается в том,
что определение ацилхлоридов может быть выполнено даже в присутствии
легко гидролизующихся групп, например СН2С1, СНС12, СС13.
В сухую мерную колбу с притертой пробкой взвешивают ацилхлорид
в количестве, соответствующем расходу 35—40 мл раствора основания.
В химический стакан на 150—200 мл наливают 10—15 мл ТГФ и растворяют
в нем взвешенную пробу. Мерную колбу ополаскивают 30—40 мл ТГФ.
После установки стеклянного и каломельного электродов очень медленно,
Терефталоил- Терефталевая
дихлорид кислота
д-Толуиловая Фталевый
кислота ангидрид
Бензилхлорид
244
Глава 16
со скоростью не более 3 мл/мин, титруют (данные о ходе титрования и описа-
ние кривой см. в работе [519]). Таким методом, например, могут быть опре-
делены следующие вещества (в скобках указано количество вещества, мг,
эквивалентное 1 мл 0,5 н. стандартного раствора): пропионилхлорид (23,13)
в присутствии масляной или уксусной кислоты; лаурилхлорид (54,69) в при-
сутствии лауриновой кислоты; изофталоилдихлорид (25,38) в присутствии
изофталевой кислоты, п-трихлорметилбензоилхлорид (67,49) в присутствии
n-трихлорбензойной кислоты и терефталоилдихлорид (25,38) в присутствии
семи загрязняющих примесей: терефталевой кислоты, n-толуиловой кислоты,
фталевого ангидрида, трихлорбензола, бензотрихлорида, бензилхлорида
и бензальхлорида.
102. РЕАКЦИОННОСПОСОБНЫЕ АЛКИЛГАЛОГЕНИДЫ
Реакционноспособные алкил- и аралкилгалогениды реагируют с анили-
ном с образованием фенилалкиламинов и соли анилина с галогеноводород-
ными кислотами [623]
RC1 • 2C6H5NH2 —> c6h5nhjci- + c6h5nhr.
Образующуюся катионную кислоту можно титровать в анилине мети-
латом натрия в присутствии тимолового синего
C6H5NHJ ± СН3О- C6H5NH2-bCH3OH.
Взвешивают такое количество реакционноспособного алкилгалогенида,
чтобы для реакции с ним потребовалось 2—5 мл 0,1 н. раствора основания.
Навеску растворяют в 5 мл свежеперегнанного анилина и кипятят 20—
30 мин (продолжительность кипячения зависит от реакционной способности
галогенида). После охлаждения раствор разбавляют 10 мл анилина, доба-
вляют 3 капли тимолового синего и титруют 0,1 н. раствором метилата
натрия в смеси бензол — метиловый спирт до появления зеленой окраски.
Таблица 77
Соединение Продолжи- тельность кипячения, .VUH Эквива- лент, At 2 Соединение Продолжи- тельность кипячения, лиги Эквива- лент, мг
Бензилхлорид 30 12,66 Цетилбромид 30 30,53
Дибромэтилен 30 18,59 1-Бромпентан 30 15,11
н-Бутилиодид 20 18,40 га-Нитробензилхлорид 25 17,16
га-Бромацетофенон 25 19,90
1-Бромдиэтилацетил- 20 23,71
мочевина
В табл. 77 приведены соединения, которые могут быть определены этим
методом со средней точностью ±0,7%.
Глава 17
ОПРЕДЕЛЕНИЕ СОЕДИНЕНИИ,
СОДЕРЖАЩИХ ЕНОЛЬНЫЙ ГИДРОКСИЛ
ИЛИ ИМИДНУЮ ГРУППУ
Соединения, содержащие енольную гидроксильную группу и способные
к кето-енольной таутомерии, являются слабыми кислотами (кислотными
аналогами)
R —С = О R —С —ОН
I II
CHR' Т ' CR'
I I
R-С () R-C = o
Электроноакцепторные группы увеличивают кислотность (гл. 4, разд. 25 в;
см. также стр. 230—236 в работе [605]). Соединения типа RCH2R', где R
и R' — электроноакцепторные группы (например, COR, СНО, СООН,
CONH2, CONHAr или CN), можно титровать как кислоты в диметилформамиде
в присутствии азофиолетового [246]. Электрофильность амидной группы
невелика. Так, изменение окраски индикатора в случае малонитрила резкое,
с цианацетамидом окраска изменяется лишь слегка, а малонамид
не проявляет никаких кислотных свойств. Соединения типа R — СО —
— СО — СН, — R' (например, диацетил) существуют только в кето-форме
и их не удается титровать как кислоты. При наличии групп CONH2, CONHR
и COONa результаты титрования предсказать нельзя, особенно в случае
имидов.
Кето-енольная таутомерия зависит не только от молекулярной структуры, но также
и от природы растворителя: содержание енольной формы ацетилацетона в воде составляет
19%, в хлороформе — 79%, в бензоле — 85% и в гексане — 92% (стр. 226 в работе [605]).
103. СОЕДИНЕНИЯ, СОДЕРЖАЩИЕ ЕНОЛЬНЫЙ ГИДРОКСИЛ
Для титрования соединений, содержащих енольный гидроксил, удобно
использовать следующие растворители: смесь бензол — метиловый спирт
(3 : 1), ацетон, пиридин, бутиламин, этилендиамин и диметилформамид.
Последний рекомендуется для енолов, у которых величина рК не превы-
шает 10 (в присутствии тимолового синего или азофиолетового) или 10,5—11
(для потенциометрического титрования). В качестве титранта используют
0,1 н. метилат щелочного металла или гидроокись алкиламмония [159, 246,
252, 258].
Образец монофункционального соединения растворяют в нейтрализо-
ванном растворителе так, чтобы на титрование пошло примерно 5—8 мл
основания. При визуальном контроле конечной точки берут 20—30 мл раство-
рителя, а при потенциометрическом титровании — 40—50 мл. При всех
потенциометрических измерениях используют электродную пару стекло —
каломель, за исключением этилендиамина, в случае которого применяют
пару сурьма — сурьма (см. рис. 53). В качестве индикаторов используют
2—4 капли тимолового синего или азофиолетового и 3—5 капли о-нитро-
анилина (табл. 61, 62, 64 и 65). По мере уменьшения кислотности в качестве
индикаторов применяют тимоловый синий, азофиолетовый и о-нитроани-
лин, а как растворители используют диметилформамид, пиридин, бутиламин
и этилендиамин (табл. 78).
246
Глава 17
Определение енольных соединений
Таблица 78
Соединение Растворитель Стандартный раствор Определение конеч- ной точки (метод или индикатор) Литера- тура
Ацетилацетон Ацетон (C4H9)4NOH Потенц. а 258
Бутиламин CH3ONa Тимоловый синий 252
Ацетплацетанилид Ацетон (C4H9)4NOH Потенц. 258
ДМФ CH3ONa Азофиолетовый 246
Цианацетамид ЭДА CH3ONa о-Нитроанилпн 246
Ацетон (C4H9)4NOH Потенц. 258
Этплцианацетат ДМФ CH3ONa Азофиолетовый 246
Ацетон (C4H9)4NOH Потенц. 258
Дибензоилметан ДМФ CH3ONa Тимоловый синий 246
Ацетон (C4H9)4NOH
CH3(C4H9)3NOH Потенц. 258, 540
3,5-Дикето-1,2-дифенил- Бензол CH3ONa Тимоловый синий 303
4-к-бутилниразолидин
5,5-Диметилциклогек- ДМФ CH3ONa Азофиолетовый 246
сандион-1,3 (димедон)
Ацетон (C4H9)4NOH Потенц. 258
Этилмалонат ДМФ CH3OK Азофиолетовый 893
Малонитрил Ацетон (C4H9)4NOH Потенц. 258
ДМФ CH3ONa Азофиолетовый 246
а Потенц. —потенциометрическое титрование.
а. Определение эфиров малоновой кислоты. Диэтилмалонат можно также
титровать в смеси с диэтил-(1-метилбутил)малонатом в диметилформамиде
в присутствии азофиолетового (7) 0,1 н. метилатом калия в смеси бензол —
метиловый спирт (метилат натрия в этом случае дает заниженные значения,
а метилат лития не пригоден).
[(C2H5OOC)2CR] H + KOCH3=[(C2H5OOC)2CR] К + СН3ОН.
Диэтилмалонат
Однако диэтил-(1-метилбутил)малонат может титроваться с достаточной
точностью в смеси с диэтилэтил-(1-метилбутил)малонатом в этилендиамине
в присутствии о-нитроанилина {2) [893].
Замещенные диэтилмалонаты, будучи слабыми кислотными аналогами,
требуют при определении по методу (7) небольшого количества стандартного
раствора (в процентах к весу образца): диэтил-(1-метилбутил)малонат 2;
в/пор-бутилмалонат 2,3; изопропилмалонат 4 и н-бутилмалонат 13. При
увеличении размера заместителя кислотность эфира замещенной малоновой
кислоты уменьшается.
б. Определение диэтилмалоната (диэтилового эфира малоновой кислоты)
в присутствии диэтилциклогексилмалоната (диэтилового эфира циклогек-
силмалоновой кислоты) [406]. 250 мг образца растворяют в мерной колбе
на 100 мл в смеси пиридин — бензол (5 : 1). Пипеткой берут 20 мл этого
раствора, помещают его в колбу на 50 мл (ср. рис. 39—42), добавляют
3 капли насыщенного азофиолетового индикатора в бензоле и насыщают
азотом в течение 1—2 мин. Титруют 0,02 н. метилатом калия в смеси бензол —
метиловый спирт до появления интенсивной голубой окраски (гл. 10,
разд. 56). Таким же образом определяют расход стандартного раствора
на титрование растворителя и вносят поправку. Для титрования 3,20 мг
диэтилмалоната требуется 1 мл 0,02 н. метилата калия.
в. Определение смеси диэтилмалоната, диэтилциклогексилмалоната
и диэтилдициклогексилмалоната [406]. Общее количество диэтилмалоната
с моно замещенными компонентами можно определить титрованием 50—100 лег
Соединения, содержащие енольный гидроксил или имидную группу 247
образца 0,1 и. метилатом калия в этилендиамине в присутствии о-нитроани-
лина в качестве индикатора. Однако, если титрование проводить 0,02 н.
метилатом калия в смеси пиридин — бензол (5 : 1) в присутствии азофиоле-
тового, то можно определить только диэтилмалонат. Дизамещенный эфир
малоновой кислоты не содержит активного водорода и его нельзя титровать
как кислотный аналог.
104. СОЕДИНЕНИЯ, СОДЕРЖАЩИЕ ИМИДНУЮ ИЛИ АМИДНУЮ ГРУППУ
Хотя соединения, содержащие группы — GONHGO — или — NHGONH —,
являются очень слабыми кислотами, некоторые из них можно точно оттитро-
вать в соответствующих растворителях (табл. 79). Соединения типа RNHR'
представляют собой более слабые кислоты, чем соединения типа RCH2R'
Таблица 79
Определение соединений, содержащих имидную группу
Соединение Растворитель Стандартный раствор Определение конеч- ной точки (метод или индикатор) а Литера- тура
1 - Ацетил-2-тиогпдантоин Ацетон (c4h9)4noh Потенц. 258
Барбитуровая кислота Пиридин (C4H9)4NOH » 780
Д пциклогексил ка рбоди- пмид Диоксан CH3ONa Тимоловый синий (косвенный ме- тод) Азофполетовый 891
си.м.ч-Дифенилтпомоче- ДМФ CH3ONa 246
вина
5,5'-Дифенилгидантоин Ацетон (C4H9)4NOH Потенц. 258
2,4-Дитиобиурет ДМФ CH3ONa Азофиолетовый 246
Дитпооксамид ДМФ CH3ONa » 246
1-Фенил-З-циклогексил- ЭДА CH3ONa о-Нитроанилин 246
тиомочевина
1-Фенил-З-нафтил- и 1- ДМФ CH3ONa Азофиолетовый 246
фенил-3-(2-пирпмп- дил)тиомочевпна
Фталимид Ацетон, пи- ридин (C4H9)4NOH Потенц. 258, 540, 780
ДМФ CH3ONa Азофполетовый 246
Гидантоин ДМФ CH3ONa » 246
Ацетон (C4H9)4NOH Потенц. 258
п-Нптробензолсульфамид Ацетон CH3(C4H9)3NOH » 540
Сукцинимид Пиридин, ацетон (C4H9)4NOH » 159, 258, 540, 780
Теобромин Пиридин, СНС13 CH3ONa Азофиолетовый 246
Теофиллин ДМФ CH3ONa Тимоловый синий 566
8-Хлортеофиллин Пиридин, СНС13 CH3ONa То же 576
Тиазолидиндион-2,4 Ацетон (C4H9)4NOH Потенц. 258
Т иобарбитуровая кис- ДМФ CH3ONa Тимоловый синий 246
лота
Замещенные фенплмо- чевины Бутилампн (C4H9)4NOH Потенц., кондук- тометрический, спектрофотомет- рический 137
а Потенц. — потенциометрическое титрование.
[246]; однако если R или R' — GOR, GHO, GOOR или GONHAr, то эти соеди-
нения можно титровать как в присутствии индикатора, так и потенцио-
метрически. В некоторых случаях кислотность имидов увеличивается
при наличии в молекуле ароматического ядра: так, ацетилацетанилид
248
Глава 17
GH3GOGH2GONHG6H5 можно оттитровать метилатом натрия в диметилформ-
амиде в присутствии азофиолетового, в то время как ацетилмочевину
GH3GONHGONH2 титровать не удается. Кислотность повышается при замене
карбонильной группы на тиокарбонильную группу (>GS): силл-дифенил-
мочевина не может быть оттитрована, в то время как симм-дифенилтиомоче-
вина титруется в диметилформамиде в присутствии азофиолетового [246].
Многие типы соединений, в том числе и широко используемые в химио-
терапии, можно отнести к этой группе, например: барбитуровую кислоту,
сульфамиды и производные пурина. Два последних типа включают также
соединения, которые можно титровать как основания (гл. 5, разд. 34). Прин-
ципы, изложенные при обсуждении титрования енольных соединений, сохра-
няют также силу и в этом случае. Из пуриновых
оснований теофиллин и аминофиллин можно
определять в диметилформамиде в присутствии
тимолового синего, а теобромин — в пиридине
или этилендиамине с азофиолетовым в качестве
индикатора [49, 246, 566, 646]. Другие соеди-
нения, обладающие подвижным атомом водо-
рода, связанным с атомом углерода или азота
(если в молекуле отсутствует фенольная, гидро-
ксильная или карбоксильная группа), можно
титровать подобным образом в пиридине. Для
потенциометрического титрования в качестве
стандартного раствора применяют раствор ги-
дроокиси алкиламмония в смеси бензол — мети-
Степень нейтрализа-
ции,%
Р п с. 109. Кривые потенциометрического титрования
кислотных аналогов в пиридине [780].
1 — дициандиамид: 2 — нитрометан; з — цианамид: 4 — сук-
цинимид; 5 — фталимид; 6 — циануровая кислота; 7 — барби-
туровая кислота.
ловый спирт (см. [780] и рис. 109). Этот метод применим к следующим соеди-
нениям: барбитуровая кислота (рК 4,0), фталимид (pTf 9,9), сукцинимид
(рК 9,6), цианамид (рК 9,4), дициандиамид (pTf 14,2), нитрометан (рК 10,6)
(см. также гл. 4, разд. 25, г). Цианамид и дициандиамид можно титровать
также и в смеси [780].
105. БАРБИТУРОВАЯ КИСЛОТА И ПРОИЗВОДНЫЕ ГИДАНТОИНА
Производные барбитуровой кислоты и гидантоина можно титровать
в инертных или основных растворителях (или в смесях растворителей) стан-
дартными растворами алкоголята щелочного металла или гидроокиси алкил-
аммония в присутствии тимолового синего, фенолфталеина, тимолфталеина,
о-крезолфталеина, азофиолетового, ализаринового желтого R (натриевая
соль 4'-нитро-4-оксиазобензол-3-карбоновой кислоты) как индикаторов или
потенциометрически, используя электродную пару сурьма — стекло
Н
Основной вид таутомерии в 5,5-дизамещенных барбитуровых кислотах
Соединения, содержащие енольный гидроксил или имидную группу
249
Заместители R и R' в барбитуровой кислоте или гидантоине влияют
на кислотность. Барбитуровая кислота почти в 6 раз сильнее, чем уксусная
(рЯ 3 ,98 и 4,76 соответственно); 5-этилбарбитуровую кислоту можно титро-
вать водной щелочью в водной среде в присутствии фенолфталеина (рЯ 4,42),
но диэтилбарбитуровая кислота является слабой кислотой (7,91) (табл. 80).
Таблица 80
Кислотность некоторых производных 5,5-дизамещенной барбитуровой кислоты3
Производное барбитуровой кислоты рК
колориметрически потенциометрически
Этилфенплбарбптуровая кислота 7,30 7,21
Этил циклогексен-1 '-плбарбитуровая кислота 7,35 7,51
Изопропплбромаллилбарбитуровая кислота 7,55 7,63
Бромаллил-втор-бутплбарбитуровая кислота 7,62 7,69
Х-Метил-этилфенил барбитуровая кислота 7,65 7,76
Дпэтплбарбитуровая кислота 7,81 7,91
Х-Метил-метплцпклогексен-1'-плбарбитуровая 8,04 8,08
кислота
aPoethke, Horn, Arch. d. Pharm., 287, 487 (1954).
а. Титрование в пиридине. Для титрования производных барбитуровой
кислоты как растворители пригодны пиридин или смесь гомологов пиридина
[372]. В качестве основания можно использовать 0,1 н. раствор метилата
натрия в смеси бензол — метиловый спирт, а в качестве индикатора —
тимоловой синий, фенолфталеин или тимолфталеин. Окраска индикатора
изменяется по-разному для различных соединений. Диэтил-, диаллил-
и этилбутилбарбитуровые кислоты следует титровать до появления голубого
оттенка тимолового синего, в то время как этилаллилбарбитуровую кислоту
титруют до тех пор, пока не появится зеленая окраска. Для титрования
аллилизопропилпроизводных можно использовать тимолфталеин, а для
этилфенилбарбитуровой кислоты — фенолфталеин. Этилциклогексенилбар-
битуровая кислота не дает хороших результатов при титровании как с инди-
каторами, так и потенциометрически, однако N-метилпроизводные могут
быть определены потенциометрически.
Совместное действие заместителя, индикатора и растворителя заметно проявляется
в точности измерений: N-метил-этилфенилбарбитуровая кислота дает со всеми тремя
индикаторами сильно заниженные значения (—2,5%), точность анализа N-метпл-
этилциклогексенильного производного в среднем на 5% выше, в то время как значения
для этилфенилбарбитуровой кислоты постепенно увеличиваются в ряду: тимоловый
синий < фенолфталеин < тимолфталеин [372].
Этилфенплбарбптуровая кислота, пзопропплбромаллилбарбптуровая кислота и неко-
торые другие барбитураты можно титровать с точностью ±1,2% в пиридине и смеси
пиридин — бензол (5:1) при фотометрическом определении конечной точки [212]. Наве-
ску (0,5 мг-экв) производного барбитуровой кислоты растворяют в 30 мл пиридина, содер-
жащего 0,003% тимолового синего. Титруют 0,1 н. метилатом натрия в смеси бензол —
метиловый спирт, содержащей такое же количество тимолового синего, как и анализиру-
емое соединение. После добавления каждой порции стандартного раствора измеряют
поглощение и результаты наносят на график (см. гл. 14).
В пиридине этилфенилбарбитуровую кислоту можно определять в присутствии
кофеина стандартным раствором метилата натрия с тимоловым синим в качестве инди-
катора [832].
б. Титрование в диметилформамиде. Этилфенилбарбитуровую кислоту
можно титровать метилатом натрия в диметилформамиде в присутствии
тимолового синего в качестве индикатора, а этилбутилбарбитуровую кислоту,
более слабую кислоту, в присутствии азофиолетового [130]. В качестве
индикатора можно применять 0,2 %-ный раствор ализаринового желтого R
250
Глава 17
в диметилформамиде (табл. 64) [453]. 150—200 мг барбитурата растворяют
в 50 мл дпметилформамида, предварительно нейтрализованного в присут-
ствии одной капли ализаринового желтого R, и титруют, защищая от попа-
дания СО2 из воздуха, 0,1 н. метилатом натрия в смеси бензол — метиловый
спирт до появления голубовато-фиолетовой окраски (рис. 39—42). Таким же
образом можно титровать диэтил-, этилфенил, этилбутил-, этилпентил-
и N-метилэтилфенплбарбитуровые кислоты.
Размельченные таблетки, содержащие производные барбитуровой кислоты и их нат-
риевые соли, можно анализировать в безводной среде [130]. Одну часть мелко размель-
ченных таблеток суспендируют в диметилформамиде при комнатной температуре, а дру-
гую — в уксусной кислоте при слабом нагревании. После охлаждения добавляют дпок-
сан, так чтобы отношение уксусной кислоты к диоксану было 2:1. Вместо диоксана
можно использовать дихлорэтан. Свободную барбитуровую кислоту титруют стандартным
раствором основания в присутствии ализаринового желтого R, а ацетат натрия, образо-
вавшийся в среде уксусной кислоты,— 0,1 н. хлорной кислотой в присутствии метило-
вого фиолетового до перехода голубой окраски в желто-зеленую.
Если в качестве растворителя используют диметилформамид (10—20 мл)
и титруют стандартным раствором метилата лития, то желатинообразный
осадок не выпадает, и большинство производных барбитуровой кислоты
можно титровать до получения густой голубой окраски с тимоловым синим
в качестве индикатора [280, 452, 694, 846].
в. Титрование в смеси бензол — изопропиловый спирт. Для потенцио-
метрических измерений очень хорошо использовать смесь бензол — изопро-
пиловый спирт (10 : 1). Конечная точка не затемняется, даже если присут-
ствует 40 об.% хлороформа (чистый хлороформ легко поляризует электроды).
Это имеет очень важное значение в том случае, когда соли барбитуровых
кислот экстрагируют после подкисления хлороформом. В этой смеси раство-
рителей путем титрования стандартным раствором 0,1 н. ТБАГ в присутствии
тимолового синего можно определять диэтил-, этилфенил-, этилизоамил-,
аллилизопропил-, диаллил- и аллилизобутилбарбитуровые кислоты с точ-
ностью от ±0,1 до ±0,2% [504]. Навеску (0,5—0,8 мг-экв) производного
барбитуровой кислоты, предварительно высушенного в течение 4 час при 80°,
растворяют в 20 мл диметилформамида или в 50 мл смеси бензол — изопро-
пиловый спирт, нейтрализованного в присутствии 5 капель тимолового
синего, и титруют из бюретки, градуированной по 0,02—0,05 мл, стандарт-
ными растворами метилата лития или ТБАГ до появления интенсивной
голубой окраски. 1 мл 0,1 н. стандартного раствора эквивалентен 0,1 мг-экв
производного барбитуровой кислоты.
г. Титрование в хлороформе [794]. 40—60 мг производного барбитуровой
кислоты растворяют в 20 мл хлороформа и после добавления 3—4 капель
1%-ного о-крезолфталеина (в СН3ОН — СНС13, 1 : 1) титруют 0,1 н. мети-
латом калия в метиловом спирте или 0,1 н. метилатом натрия в смеси бен-
зол — метиловый спирт. Окраска индикатора изменяется от бесцветной
до фиолетово-розовой. Если титрование проводится метилатом натрия,
то в раствор рекомендуется предварительно добавить 1—2 мл метилового
спирта.
д. Титрование в метилэтилкетоне [684]. 200 мг производного барбитуро-
вой кислоты растворяют в 20 мл метилэтилкетона, добавляют 2 капли смеси
индикаторов (0,6 г тимолфталеина и 0,4 г тропеолина О в 100 мл диметил-
формамида) и титруют 0,1 н. раствором гидроокиси тетрабутиламмония,
пока цвет не изменится от желтого до зеленого.
е. Определение солей производных барбитуровой кислоты с помощью
экстракции. Навеску 1,8—2,2 мг-экв натриевой соли растворяют в 10 мл
0,1 н. водного раствора едкого натра, подкисляют 30—35 мл 0,1 н. серной
кислоты и экстрагируют (6 X 20 мл) хлороформом (7) или (5 X 20 мл) эфи-
ром (2). Хлороформный раствор сушат безводным сульфатом натрия, упа-
ривают до малого объема (см. колбу для упаривания на рис. 103) и разба-
Соединения, содержащие енольный гидроксил или имидную группу
251
вляют до 50 мл в мерной колбе полиэтиленгликолем 400 или смесью бен-
зол — изопропиловый спирт: 20 мл этого раствора титруют потенциометри-
чески стандартными растворами метилата натрия [788] или ТБАГ [504].
Эфирный раствор (2) выпаривают досуха, остаток растворяют в диметил-
формамиде и титруют 0,1 н. метилатом лития в присутствии тимолового
синего.
ж. Определение солей производных барбитуровой кислоты с помощью
катионообменных смол [848]. 90—100 мг барбитурата растворяют в 20—
25 мл диметилформамида, раствор переносят в ионообменную колонку, запол-
ненную смолой амберлит IRG-50 (Н+-форма), и промывают диметилформ-
амидом, пока объем элюата не составит примерно 50 мл. Титруют метилатом
натрия в присутствии азофиолетового до устойчивой голубой окраски.
В случае анализа растворов разбавляют количество раствора, эквивалент-
ное 100 мг производного барбитуровой кислоты, 25 мл диметилформамида.
а затем анализируют, как указано выше.
В случае анализа таблеток суспендируют мелкоразмельченную таблетку
в диметилформамиде и после энергичного взбалтывания фильтруют в мерную
колбу; повторно промывают на фильтре связующее вещество таблетки диме-
тилформамидом. Титруют 50 мл элюата (~100 мг барбитурата). Точность
в случае относительно чистых образцов составляет ±1,1%, а в случае фар-
мацевтических препаратов ±1,5%.
з. Производные барбитуровой кислоты реагируют с нитратом серебра
в пиридине и образуют соли серебра, при этом протон освобождающейся
азотной кислоты связывается пиридином. Образующийся ион пиридиния
можно титровать стандартным раствором едкого натра в спирте в присут-
ствии тимолового синего [266] (гл. 28, разд. 160, в).
106. СУЛЬФАМИДНЫЕ ПРОИЗВОДНЫЕ
Поскольку группа —SO2 — является сильным электрофилом, водород,
связанный с атомом азота в сульфамидах, можно титровать как кислоту
в растворителях основного характера [220, 250, 252, 452, 846] и в ацето-
не [258]
R — SO2 — NH — R' + СН3ОК = R — SO2 — NK — R' + СН3ОН.
Кислотность сульфамидов в зависимости от природы R' увеличивается
в следующем порядке: фенил Z> пиридил > тиазоил > водород > нафтил >
> бензил Z> алкил.
Сульфагуанидины не имеют кислотного характера. Большинство суль-
фамидных производных можно титровать в диметилформамиде и пиридине
в присутствии тимолового синего, а тг-толуолсульфамид и аминобензол-
сульфамид титруют в бутиламине в присутствии азофиолетового.
Для определения вышеупомянутых производных навеску мелкоразмель-
ченного сухого образца (0,2—1,0 мг-экв} (количество зависит от растворимо-
сти) растворяют в 10—50 мл нейтрализованного растворителя, затем тит-
руют 0,1 н. метилатом щелочного металла из бюретки, проградуированной
по 0,02—0,05 мл.
В диметилформамиде можно титровать с помощью метилата натрия
в смеси бензол — метиловый спирт (до появления голубой окраски тимоло-
вого синего в конечной точке) следующие соединения: тг-аминобензолсуль-
фаметилпиримидин, тг-аминобензолсульфамдиметилпиримидин, тг-аминобен-
золсульфапиримидин, тг-аминобензолсульфапиридин, тг-аминобензолсуль-
фатиазол, тг-аминобензолсульфаметилтиазол. В качестве индикатора часто
используют ализариновый желтый R вместо тимолового синего (табл. 64)
[453]. В w-бутиламине в присутствии азофиолетового можно определять
аминобензолсульфамид, а также соединения, в которых заместителем R
или R' является нафтил, анизил, бензил, толил или тг-бромфенил. Сукци-
252
Глава 17
нилсульфатиазол, n-аминобензолсульфатиазол, п-аминобензолсульфапири-
мидин, п-аминобензолсульфадиметилпиримидин и п-аминобензосульфаме-
тилпиримидин можно титровать в пиридине метилатом натрия в присутствии
тимолового синего [220].
Титрование сульфамидов лучше всего проводить в приборах, надежно
защищенных от попадания двуокиси углерода из воздуха (рис. 39—42).
а. Определение 2-ацетиламино-1,3^4-тиадиазол-5-сулъфамида (ацетазол-
амида) *. Около 300 мг образца растворяют в 30 мл пиридина, нейтрализо-
ванного в присутствии азофиолетового, и титруют метилатом натрия, либо
100 мг образца растворяют в 50 мл диметилформамида и титруют метилатом
натрия в присутствии 0,3 мл ализаринового желтого R до появления серо-
голубой окраски **. В обоих случаях для титрования 11,11 мг ацетазол-
амида требуется 1 мл 0,1 н. раствора основания
N — N
II II
CH3CONH-C С —SO2 — nh2
б. ?\т-(4-Метилбензолсульфонил)-?Г-бутилмочевину (толбутамид) и Х-(4-
аминобензолсульфонил)-1\т'-бутилмочевину (карбутамид) (антидиабетики)
можно титровать метилатом натрия в ацетоне или пиридине в присутствии
фенолфталеина [471]. Поскольку молекула карбутамида содержит амино-
группу, его можно титровать хлорной кислотой в уксусной кислоте. 1 мл
0,1 н. метилата натрия эквивалентен 27,04 мг метилбензосульфонилбутил-
мочевины или 23,13 мг аминобензолсульфонилбутилмочевины.
107. КОНДЕНСИРОВАННЫЕ ГЕТЕРОЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ,
КИСЛОТНЫЕ АНАЛОГИ
Бензимидазол, бензтриазол, 2-меркаптобензтиазол и 5-амино-2-меркапто-
бензимидазол можно титровать алкоголятом щелочного металла или гидро-
окисью алкиламмония (табл. 81).
Таблица 81
Соединение Растворитель Стандартный раствор Определение конеч- ной точки (метод или индикатор) а Литера- тура
Бензимидазол ДМФ 0,025 н. CH3ONa Азофиолетовый 562
Бензтриазол ДМФ 0,025 н. CH3ONa Тимоловый синий 562
Пиридин (C4H9)4NOH Потенц. 780
2-Меркаптобензтпазол Пиридин (C4H9)4NOH Тимоловый синий 159
Бутиламин CH3ONa То же 252
Ацетон (C4H9)3CH3NOH Потенц. 540
5-Амино-2-меркаптобенз- Пиридин (C4H9)4NOH Азофиолетовый 159
имидазол
а Потенц. — потенциометрическое титрование.
В пропионовой кислоте бензимидазол можно титровать как основание 0,1 н. хлорной
кислотой в пропионовой кислоте [377]; ср. [398] и гл. 5, разд. 34.
Замещение группы —NH — СН = N — на группу — S — C(SH) = N —
увеличивает кислотность, и поэтому 2-меркаптобензтиазол является силь-
* Drug. Std., 26, 63 (1958).
** Ann. pharm. fran^., 20, 402 (1962).
Соединения, содержащие енольный гидроксил или имидную группу
253
ным кислотным аналогом. Пятичленное ядро бензтриазола стабилизуется
за счет резонанса с бензольным ядром. Эти соединения также способны
к таутомерным превращениям.
Н
Н
а. Определение
(хлортиазида R).
6-хлор-7-сульфамил-1,2,4-бензтиадиазин-1,1-диоксида
Хлортиазид,
мол. вес 295.7
Дигидрохлортиазид (Х=С1)и
дигид ротрифторметилти азид
(Х=СРз)
109 — 150 мг хлортиазида растворяют в 50 мл диметилформамида, предва-
рительно нейтрализованного в присутствии 2 капель ализаринового жел-
того R. и титруют 0,1 н. метилатом натрия в смеси бензол — метиловый
спирт до появления фиолетовой окраски. Во время титрования раствор
защищают от попадания двуокиси углерода из атмосферы *. Вместо ализа-
ринового желтого R можно использовать азофиолетовый [129]. Для титро-
вания 14,79 мг хлортиазида требуется 1 мл 0,1 н. раствора основания.
В диметилформамиде в присутствии тимолового синего хлортиазид реагирует
только с 1 экв метилата натрия [180]. Дигидрохлортиазид и дигидротрифтор-
метилтиазид можно титровать в этилендиамине в присутствии азофиолето-
вого как бифункциональные кислотные аналоги (табл. 82).
* Ann. pharm. franc., 20, 295 (1962).
254
Глава 17
Таблица 82
Соединение Растворитель Индикатор Количество, экви- валентное 1 мл 0,1 н. стандартного раствора, мг
Хлортиазид ДМФ Тимоловый синий 29,57
Дигидрохлортиазид ЭДА Азофиолетовый 14,89
Дигидротрифторметил- ЭДА » 16,55
тиазид
б. Среди производных триазола изомер I дифенил-5-амино-1,2,3-триазола
можно титровать в уксусной кислоте как основание хлорной кислотой,
а изомер II в диметилформамиде в присутствии азофиолетового титруется
как кислота. Таким образом, положение заместителя определяет поведение
изомеров [511] и, следовательно, два изомера могут быть также определены
в смеси.
I
Основной изомер: 1,4-дифенил-5-амино-
1,2,3 - триазол
III
С6Нг---С С-------NH----С6Н5
14з 151
n<2 >—н
II
Кислотный изомер: 4-фенил-5-фенил-
амино-1,2 ;3-триазол
IV
в. 5-А цетиламинотетр азол (III) можно титровать аминоэтилатом натрия
как бифункциональный кислотный аналог, а 5-фенилтетразол(1У) — как
монофункциональный кислотный аналог в этилендиамине потенциометриче-
ски, используя пару электродов сурьма — сурьма [534].
Глава 18
ОПРЕДЕЛЕНИЕ СОЕДИНЕНИИ,
СОДЕРЖАЩИХ ФЕНОЛЬНЫЙ ГИДРОКСИЛ
108. ФЕНОЛЫ
Кислотный характер соединений, содержащих фенольный гидроксил,
существенно зависит от природы заместителя. По данным Фитца [258],
кислотность в зависимости от природы заместителя R изменяется в следую-
щем порядке:
R = NO2 > СНО > COR > С1, Вг > С6Н5.
Алкильные заместители уменьшают силу кислот в следующем порядке:
метил > этил > изопропил.
На силу кислотных аналогов большее влияние оказывает замещение
в орто- или пара-положении, чем в леета-положении. Если электроноакцеп-
торная группа находится в орто-положении, то в результате образования
водородной связи кислотность может уменьшаться. Например, все фенолы,
имеющие в пара-положении группы NO2, СНО и СОСН3, являются более
кислыми, чем соответствующие орто-соединения (см. также гл. 2, табл. 12
и гл. 4, разд. 25, в).
Для определения фенолов предпочтительны основные растворители
(табл. 83), в то время как метилизобутилкетон, ацетон, пиридин и трет-
бутиловый спирт более удобны для потенциометрического дифференцирую-
щего титрования простых смесей фенолов с более сильными кислотами
(кислотными аналогами) [105, 159, 253, 258], см. рис. 105—108 и гл. 12,
разд. 63, г. Систему электродов лучше всего подбирать эмпирически. В пири-
дине обычно предпочтительны электродные пары: Pt — каломель и Sb —
каломель.
Из основных растворителей многими исследователями использовался
этилендиамин [176, 251, 285, 446, 601], но из-за его неприятного запаха
он не нашел широкого применения. В известной степени это справедливо
и в отношении пиперидина. Подходящим растворителем является диметил-
формамид. Он не имеет запаха, очень слабо гигроскопичен, не реагирует
с СО2, хорошо растворяет феноляты и во многих случаях может заменить
этилендиамин [7]. В n-бутилдиамине и тем более в этилендиамине может
наблюдаться выпадение осадка даже с титрантом типа ТБАГ.
При потенциометрическом титровании фенолов с использованием стеклянного
и каломельного электродов следует предотвратить попадание в раствор ионов натрия
и калия. Эти ионы могут привести к получению нерегулярных потенциометрических
кривых [355], рис. 43 и гл. 12, разд. 63, г. Ионы калия могут попадать в раствор из образ-
ца, каломельного электрода или из стандартного раствора ТБАГ. Загрязнений ионами
калия из насыщенного хлоридом калия раствора каломельного электрода можно избе-
жать, если солевой мостик электрода заполнить хлоридом тетра-н-бутпламмония.
Использование стандартного раствора ТБАГ для титрования фенолов в сильнооснов-
ных растворителях позволило усовершенствовать потенциометрическое титрование
с использованием стеклянного (индикаторного) электрода. В этом случае нельзя исполь-
зовать стандартные растворы, содержащие ионы калия или натрия. К искажению резуль-
татов титрования ведут не только значительные загрязнения щелочным металлом стан-
дартного раствора; даже малейшие их примесп могут влиять на измерения, так как потен-
циометрическая кривая смещается в область более отрицательных потенциалов и пере-
гибы становятся меньше [355]. Влияние ионов калия не зависит линейно от их концентра-
ции. Влияние стандартного раствора, содержащего примесь ионов калия, проявляется
в виде некоторой нерегулярности кривой, как это можно видеть на рис. 43. 0,25 н. стан-
256
Глава 18
дартный раствор ТБАГ также содержит ионы калия в переменной концентрации; при
0,0055 н. концентрация ионов К+ непосредственно перед нормальным перегибом появляет-
ся обратный перегиб. Также неблагоприятно влияют и ионы натрия. Очевидно, что при
титровании (например, в пиридине) стандартным раствором, содержащим одни и те же
количества примеси иона натрия, искажение потенциометрической кривой становится
тем большим, чем больше навеска фенольного соединения, так как оно «связывает» боль-
шее количество стандартного раствора и, таким образом, концентрация ионов натрия
к концу титрования возрастает.
Малые количества ионов калия или натрия не слишком искажают результаты титро-
вания, если требуется только количественное определение, но если, например, определяют
кислотность слабых кислот на основании потенциала полунейтрализацип, загрязнения
ионами щелочного металла следует тщательно избегать в ходе приготовления стандарт-
ного раствора ТБАГ (гл. 10, разд. 57): с большой тщательностью должны быть промыты
окись серебра и анионит. Титрование фенолов лучше выполнять в приборах, защищен-
ных от попадания углекислого газа из воздуха (рис. 39—42).
а. Титрование в этилендиамине 1251]. Используя градуированный
цилиндр, 25 мл этилендиамина переносят в колбу Эрленмейера емкостью
100—150 мл с притертой пробкой и нейтрализуют до появления красной
окраски в присутствии двух капель 0,15%-ного о-нитроанилина в бензоле.
Колбу немедленно закрывают. Взвешивают такое количество анализируе-
мого соединения, чтобы на его титрование потребовалось 3—6 мл 0,1 н.
стандартного раствора. В процессе титрования пропускают ток азота над
раствором и для перемешивания используют магнитную мешалку. Тем самым
удается избежать искажения результатов за счет попадания углекислого
газа из воздуха. В конечной точке окраска изменяется от желтой к оранже-
вой. В соответствии с работой [392] бензопурпурин 4В или тропеолин 00
(оранжевый IV) в качестве индикаторов имеют преимущество перед о-нитро-
анилином в этилендиамине; окраска первого изменяется от красного к голу-
бому, а второго — от оранжево-желтого к голубому. Такиура и Такино [798]
рекомендуют использовать для титрований в среде этилендиамина тг-нитро-
n'-аминоазобензол, для которого окраска изменяется следующим образом:
красная —(голубая) -> бесцветная.
б. Титрование п,п'-диоксидифенилметана [359]. На примере этого
бифункционального фенола можно проиллюстрировать преимущества при
титровании раствора ТБАГ в изопропиловом спирте по сравнению с КОН
в изопропиловом спирте. В случае ТБАГ на потенциометрической кривой
заметны два перегиба, в то время как с КОН в изопропиловом спирте —
только один перегиб, что указывает на поглощение 2 экв стандартного раство-
ра. При нейтрализации первой фенольной ОН-группы раствором ТБАГ
образуется слаборастворимая соль, которая, однако, растворяется в конце
нейтрализации. Следует отметить, что это один из редких случаев, когда
со стандартным раствором ТБАГ образуется осадок.
в. Титрование в диметилформамиде [251]. К 20 мл диметилформамида
добавляют две капли насыщенного азофиолетового в бензоле и нейтрализуют
0,1 н. метилата натрия или калия в смеси бензол — метиловый спирт
до появления голубого цвета. В этом растворе растворяют количество
анализируемого соединения, эквивалентное 3—8 мл стандартного раствора,
и продолжают титрование до появления голубой окраски.
г. Потенциометрическое титрование в ДМФ [7]. Для серийных анализов
этилендиамин менее пригоден, чем, например, н-бутиламин или диметилфор-
мамид.
Последний является даже более основным, чем «-бутиламин, но он осо-
бенно подходит для потенциометрического титрования большинства феноль-
ных соединений. Его преимущество состоит в том, что он меньше поглощает
углекислый газ из воздуха и легче растворяет феноляты, чем н-бутиламин
или этилендиамин. В последнем осадок может появляться даже со стандарт-
ным раствором ТБАГ (б).
Таблица 83
Определение соединений, содержащих фенольный гидроксил
Соединение Растворитель Стандартный раствор Определение конечной точки (метод или индикатор) Литера- тура
м-Ацетаминофенол ЭДА CH3ONa п- Нитро - п' -аминоазобензол 798
2-Ацетил -1 - нафтол ДМФ CH3ONa Азофиолетовый 251
Бензилмеркаптан Пиридин (C4H9)4NOH Потенц. или азофио- летовый 159
л-Бензилфенол Пиридин (C4H0)4NOH Потенц. 159
о-, л-, п-Бромфенол п - Бромсалици л альде- гид Ацетон (C4H9)4NOH » 258
ЭДА CH3ONa п-Нитро - п' -аминоазобензол 798
о-, ж-, п-Хлорфенол Крезолы Пиридин (C4H9)4NOH Потенц. 780
Пиридин (C4H9)4NOH » 780, 159
Диенэстрол ДМФ (C4h9)4noh » 7
Диоксидиметилрезор- цин Пиридин (C4H9)4NOH Потенц. или - азофиолетовый 159
Этилванилат ДМФ CH3OK Потенц. 113
Эвгенол ДМФ CH3OK » 113
о -0 ксиацетофенон Ацетон (C4H9)4NOH » 258
п - 0 ксиацетофенон Пиридин CH3OK Азофиолетовый 798
0-, М-, п-0ксибензальдегид Ацетон (C4H9)4NOH Потенц. 258
Сложные эфиры Пиридин CH3OK Азофиолетовый 798
п-оксибензойной кислоты
н-Пропил-п-оксибен- ДМФ (C4H9)4NOH Потенц. 7
зоат
Гидрохинон ДМФ (C4H9)4NOH » 159
п-Оксипропиофенон ДМФ, ацетон Алкоголяты Азофиолетовый 251, 307
п-Оксипропиофенон Пиридин KOH в этаноле Потенц. или тимолфталеин 178
о-0ксибензиловый спирт Пиридин Аминоэтилат натрия Потенц. 287
8- Окси- 5,7 -дииодхи- ДМФ CH3ONa Тимоловый СИНИЙ 448
нолин
Производные 4 - оксикумарина Пиридин КОН в этаноле 4-(п-Нитрофенил а- зо)-пирокатехин 419
о-Оксибифенил ЭДА СН3ОК о - Нитроанилин 251
8-Оксихинолин Ацетон—пири- дин (4 : 1) сн3ок Азофиолетовый 307
Койевая кислота Пиридин CH3ONa » 506
[5-ОКСИ-2-(оксиме- тил)-1,4-пирон] (C4H9)4NOH
Метилсалицилат Ацетон Потенц. 258
Нафтол-1 и нафтол-2 Пиридин (C4H9)4NOH Азофиолетовый или потенц. 159
Нитрофенолы трет-Бутило- вый спирт, ацетон, пири- дин (C4H9)4NOH Тимоловый синий 159, 258, 780
п-трет- А ми лфено л ЭДА CH3OK о -Нитроанилин, 251
Фенол ЭДА, пиридин, ДМФ, ацетон КОСНэ, (C4H9)4NOH о - Нитроанилин, азофиолетовый или потенц. 7, 159, 251, 258, 780
н-Пропилгаллат ДМФ (C4H9)4NOH Потенц. 7
Флороглюцин ЭДА CH3OK о -Нитроанилин 258
о-Фенилфенол Пиридин (C4H9)4NOH Азофиолетовый 159
Пирокатехин » (C4h9)4noh Потенц. 159
Резорцин ДМФ (C4H9)4NOH » 7, 159
Салициламид ДМФ NaOCH3 Азофиолетовый 251
Салицилальдегид Ацетон (C4H9)4NOH Потенц. 258
Тиосемикарбазон салицилальдегида ЭДА CH3ONa п -Нитро -п'- аминоазобензол 798
Тимол ДМФ (C4H9)4NOH Потенц. 7
Трихлорфенол ДМФ CH3ONa Азофиолетовый 251
Ванилин 1 ДМФ CH3ONa 251
17-284
258
Глава 18
0,2мл 0,2 н. ТБАГ
Рис. НО. Потенциометрическое
титрование фенолов [105].
1 — в пиперидине; 2 — в метилизобу-
тилкетоне; 3 — в пиридине; 4 — в аце-
тонитриле; 5 — в метил этилкетоне;
6 — в этилендиамине.
Навеску примерно 0,5 мг-экв соединения, содержащего фенольный
тидроксил, растворяют в 25 мл ДМФ, нейтрализованного в присутствии
тимолового синего или азофиолетового, или иногда ализаринового жел-
того В.
Титруют в стакане емкостью 50—100 мл, закрытом корковой пробкой
с четырьмя отверстиями (резиновые пробки разрушаются ДМФ). Во время
титрования используют магнитную мешалку. Через отверстия в пробке
вводят стеклянный (индикаторный) и каломельный (сравнения) электроды,
кончик бюретки и трубку, через которую в стакан подается промытый
и осушенный азот.
Кончик бюретки вставляют свободно так, чтобы дать возможность току
азота выходить наружу (см. также рис. 39—42). Используют бюретку, гра-
дуированную по 0,05 мл; для титрования применяют 0,1 н. раствор ТБАГ
в смеси бензол — метиловый спирт. Скачок
потенциала в конечной точке в 6 раз больше,
чем скачок, наблюдаемый, например, при
использовании метилата лития [7]. Скачок
в милливольтах, наблюдаемый в конечной
точке, отражает в некоторой степени относи-
тельную кислотность фенолов: резорция
(75jte), эстрадиол и этинилэстрадиол (100 мв),
тимол (120 мв), диенэстрол (120 мв), гексэст-
рол и фенол (125 мв), стильбэстрол (140 мв),
эстрон (160 мв), м-пропилгаллат (180 мв),
ванилин (300 мв).
Батлер и Чепель проводили титрование
0,05 н. метилатом калия в смеси бензол —
метиловый спирт с использованием сурьмяно-
го и каломельного электродов (табл. 58 и 83).
д. Титрование соединений, содержащих карбоксильную группу, в присут-
ствии соединений, содержащих фенольный гидроксил, в ацетоне или ацето-
нитриле [251]. Образец растворяют в 30—40 мл растворителя и титруют
0,1 н. метилатом калия в присутствии двух капель 0,2%-ного п-оксиазобен-
зола в бензоле до появления светло-желтой окраски; таким путем определяют
возможное присутствие соединений, содержащих карбоксильную группу.
Теперь, если титровать другой образец в эти лен диамине, как описано выше
(а), то разность между титрованиями будет пропорциональна количеству
соединения, содержащего фенольный гидроксил.
е. Потенциометрическое титрование в ацетоне [258]. Навеску 0,4—
0,8 мг-экв фенольного соединения растворяют в 40 мл очищенного ацетона.
Титруют, применяя стеклянный электрод в качестве индикаторного и кало-
мельный в качестве электрода сравнения. В каломельном электроде вместо
водного KG1 используется насыщенный раствор KCI в метиловом спирте.
В качестве стандартного раствора применяют либо 0,1 н. (G4H9)4NOH, либо
С4Н9(С2Н5)з?ЮН, оба в смеси бензол — метиловый спирт, и добавляют его
из бюретки, градуированной по 0,02 или 0,05 мл. Вносят поправку, получен-
ную в глухом опыте титрования растворителя. Если наблюдаются два или
больше перегиба, то для расчета расхода стандартного раствора используют
разность между двумя последующими точками. Титрант можно добавить
в таком количестве, чтобы увеличение отклонения точки перегиба составило
около 30 мв. Таким образом, при приближении к конечной точке титрования
количество добавленного стандартного раствора постепенно уменьшается.
ж. Потенциометрическое титрование в метилизобутилкетоне [105].
На рис. НО изображены кривые потенциометрического титрования фенолов
в этом растворителе (см. также гл. 15, разд. 95). При потенциометрическом
титровании фенольных соединений значительные перегибы могут наблюдать-
Определение соединений, содержащих фенольный гидроксил
259
ся в тех случаях, когда используют 0,2 н. ТБАГ в качестве стандартного
раствора. Платиновые электроды, установленные в потоке титранта, или
каломельный электрод типа втулки пригодны в качестве электродов срав-
нения (ср. рис. 41, 52, 61 и 114).
з. Титрование в пиридине [159]. 25 мл пиридина, нейтрализованного
в присутствии 4 капель тимолового синего или азофиолетового в бензоле,
помещают в колбу Эрленмейера емкостью 100—150 мл и растворяют в нем
такую навеску, чтобы на титрование пошло 6—8 мл 0,1 н. стандартного
раствора. Быстро титруют 0,1 н. ТБАГ в смеси бензол — метиловый спирт
(10 : 1) до появления голубой окраски тимолового синего или пока окраска
азофиолетового не станет вновь фиолетовой, а в некоторых случаях голу-
бой. Тимоловый синий используют, например, для определения нитрофено-
лов, так как они являются более сильными кислотными аналогами.
и. Потенциометрическое титрование в пиридине [159, 780]. В стеклян-
ный сосуд емкостью 100—200 мл (стакан или соответствующая колба для
титрования, изображенная на рис. 41) помещают навеску соединения,
содержащего фенольный гидроксил, для титрования которой требуется
2—10 мл 0,1 н. раствора ТБАГ. Добавляют 50 мл пиридина и титруют из полу-
микробюретки, используя стеклянный и каломельный электроды. В этом
случае также желательно пропускать ток азота над поверхностью раствора
с тем, чтобы исключить действие углекислого газа из воздуха; используют
магнитную мешалку. Стандартный раствор добавляют порциями по 0,1 мл,
а вблизи эквивалентной точки — по 0,05 мл. Стандартный раствор добавляют
до тех пор, пока потенциал ячейки не достигнет максимума и при дальней-
шем добавлении стандартного раствора будет относительно стабильным.
Если наблюдаются две точки перегиба, расчет проводят, используя разность
в миллилитрах между двумя соответствующими точками перегиба.
При титровании полиоксисоединений в пиридине иногда может наблю-
даться интенсивное изменение окраски. Так, например, пирогаллол дает
темно-голубую, флороглюцин — ярко-пурпурную окраску. Это можно
объяснить окислением ионизированной молекулы, так как в атмосфере азота
подобного окрашивания не наблюдается [780].
к. п-Оксипропиофенон можно определять с точностью ±0,3% в смеси
ацетон — пиридин (4 : 1) [307]. Для установки титра можно пользоваться
также и стандартными растворами метилата калия, если последний исполь-
зуют для титрования фенольных соединений. Изменение окраски индикатора
азофиолетового от красного к голубому происходит резко. Фенолы, замещен-
ные в пара-положении, являются более сильными, чем сам фенол. Для
титрования 16,42 мг n-оксипропиофенона требуется 1 мл 0,1 н. метилата
калия в смеси бензол — метиловый спирт.
В пиридине м- и n-оксибензойные кислоты дают две точки перегиба,
в то время как о-оксибензойная кислота — только одну. В качестве стан-
дартного раствора используют ТБАГ и применяют стеклянный и каломель-
ный электроды (рис. 100).
л. Титрование в трет-бутиловом спирте [253]. В сосуд с боковыми отво-
дами помещают 50 мл раствора, содержащего такое количество фенольного
соединения, чтобы на титрование пошло 3—4 мл 0,1 н. раствора ТБАГ в смеси
бензол — изопропиловый спирт (8 : 2). Сосуд соединяют с полуячейкой,
как показано на рис. 68. Стеклянный электрод погружают в раствор.
По-видимому, потенциометрическое титрование, проводимое в тпретп-бутило-
вом спирте, удобно, в частности для определения нитрофенолов [253]
и в основном для дифференцирующего титрования фенольных соединений
[152] (рис. 47 и 108).
Относительно сильное основание 1,1,3,3-тетраметилгуанидин (содержащий 1—3% во-
ды) также можно применять как растворитель при титровании фенольных соедпненпй [876].
260
Глава 18
109. СОЕДИНЕНИЯ, ОБЛАДАЮЩИЕ ЭСТРОГЕННЫМ ДЕЙСТВИЕМ
а. Титрование в диметилформамиде [7]. Хотя синтетические соединения
с эстрогенным действием обычно представляют собой бифункциональные
соединения, их можно титровать как одноосновные кислоты в диметилформ-
амиде [7] (табл. 84). Навеску 0,4—0,5 мг-экв соединения растворяют в 25 мл
ДМФ, предварительно нейтрализованного в присутствии азофиолетового,
и титруют потенциометрически 0,1 н. раствором ТБАГ, используя стеклянный
и каломельный электроды. Предварительно стеклянный электрод помещают
на 24 час в ДМФ.
Таблица 84
Соединение Скачок потенциала в эквивалент- ной точке, мв Количество, эквивалент- ное 1 мл 0,1 н. стан- дартного раствора, мг Соединение Скачок потенциала в эквивалент- ной точке, мв Количество, эквивалент- ное 1 мл 0,1 н. стан- дартного раствора, мг
Диэтилстильбэст- 140 26,83 Эстрадиол 100 27,24
рол Эстрон 160 27,04
Гексэстрол 125 27,04 Этинилэстрадиол 100 31,64
Диенэстрол 120 26,63
б. Определение в пиридине [25]. Диенэстрол, диэтилстильбэстрол и гекс-
эстрол при титровании в пиридине требуют 1 экв 0,1 н. раствора ТБАГ или
2 экв метилата калия; в обоих случаях конец потенциометрического титрова-
ния определяется только одной точкой перегиба (ср. гл. 18, разд. 108).
бпс-Фенольные соединения, содержащие две opmo-OH-группы, требуют
только 1 экв щелочи вследствие существования водородной связи ([770]
и гл. 12, разд. 63, г). В бис-фенольных эстрогенах, как, например, в стильб-
эстроле, расстояние между фенольными функциональными группами состав-
ляет 8,55 А, так что внутримолекулярная водородная связь не может воз-
никнуть.
Навеску 0,5 мг-экв анализируемого соединения растворяют в 80 мл
пиридина, нейтрализованного в присутствии азофиолетового. Титруют
потенциометрически метилатом калия или ТБАГ. Используют магнитную
мешалку и пропускают во время титрования ток азота над раствором (прибор
изображен на рис. 38). Диенэстрол также можно титровать визуально в аце-
тоне в присутствии азофиолетового.
в. Определение в смеси ацетон—пиридин — метиловый спирт (70 : 30 : 2)
[309]. 20—60 мг диэтилстильбэстрола растворяют в 20 мл смеси раствори-
телей, нейтрализованной в присутствии 0,2 мл 0,1 %-ного азофиолетового
в хлорбензоле и титруют 0,1 н. метилатом калия в смеси бензол — метило-
вый спирт до появления серо-голубой окраски. Для титрования 13,42 мг
4,4'-диокси-а,а'-диэтилстильбена требуется 1 мл 0,1 н. стандартного раствора.
110. СЛОЖНЫЕ ЭФИРЫ ФЕНОЛОВ
Сложные эфиры, образованные карбоновыми кислотами с фенолами или
с замещенными фенолами, подвергаются аминолизу метиламином или дру-
гим сильным основанием, например этилендиамином:
CeH5OCOR + H2N(CH2)2NH2 —> CeH5OH +H2N(CH2)2NHCOR (110.1)
CeH5OCOAr + H2N(CH2)2NH2 —> C6H5OH + H2N(CH2)2COAr (110.2)
CeH5OCOArOH + H2N(CH2)2NH2 —> CeH5OH + H2N(CH2)2NHCOArOH (110.3)
Таким образом, после реакции образуются фенол и амид кислоты. Навеску
примерно 0,5 мг-экв вещества растворяют в 20 мл этилендиамина, нейтрали-
Определение соединений, содержащих фенольный гидроксил
261
зованного в присутствии двух капель о-нитроанилина, и титруют 0,1 и.
метилатом калия до появления оранжевой окраски. Фенилацетат (110.1),
фенилпропионат (110.1), фенилбензоат (110.2), о- и л-толилацетаты (110.1),
1-нафтилацетат (110.1), дифенилфталат (110.2), фенилсалицилат (110.3)
и т. д. могут быть определены с точностью от ±1 до ±2% [274].
Различные сложные эфиры фенолов в зависимости от природы заместителей ведут
себя по-разному при титровании в ацетоне ТБАГ в смеси бензол — метиловый спирт. Хотя
точность измерения и не превышает точности по методу Гленна и Пика [274], все же этот
метод пригоден для дифференцирующего титрования различных сложных эфиров фенолов
[764]. Так, при титровании фенилацетата переэтерификация происходит благодаря ката-
литическому действию основного стандартного раствора, содержащего метиловый спирт.
RCOOCeH5 + CH3OH БСООСНз + СвН5ОН.
Следовательно, в случае сложных эфиров фенолов и алифатических кислот (напри-
мер, фенилацетата) освобождается один фенольный гидроксил. Однако, если в орто- или
пара-положении ароматического ядра имеются ОН- или МН2-группы, они препятствуют
переэтерификации, как, например, в случае n-аминофенилацетата. В случае ацетил-
салициловой кислоты в противоположность м- и n-изомерам сложноэфирная связь
не расщепляется.
Титрование сложных эфиров п-нитрофенола (ПНФ) [872]. ПНФ-эфиры
ведут себя как одноосновные кислоты при титровании метилатом натрия
O2NCeH4OCOR4-CH3ONa —> CH3OCOR + O2NCeH4ONa
Найдено, что при растворении ПНФ-эфиров в спирте или смеси спирт —
диоксан, добавлении тимолового синего и титровании до появления голубой
окраски в конечной точке можно определять с точностью ±2% некоторые
ПНФ-эфиры карбобензилоксиаминокислоты. Сложные эфиры тиофенолов
реагируют значительно медленнее, чем ПНФ-эфиры.
Глава 19
ОПРЕДЕЛЕНИЕ СОЕДИНЕНИИ,
СОДЕРЖАЩИХ СПИРТОВЫЙ ГИДРОКСИЛ
В большинстве случаев аналоги кислот можно определять ацидиметри-
ческим титрованием, если только удается подобрать подходящий титрую-
щий раствор и индикатор или систему электродов. Так, трифторэтиловый
спирт титруют в среде бути л амина, применяя раствор аминоэтилата натрия
в смеси этаноламина и этилендиамина и используя стеклянный и сурьмяный
электроды [686].
Соединения, содержащие спиртовый гидроксил, обладают очень низкой
кислотностью. Однако сопряженная кислота основания, используемого для
определения, должна быть еще слабее. Таким образом, например, фенолы
можно титровать метилатами щелочных металлов, поскольку метиловый
спирт — более слабая кислота, чем фенол. Следовательно, можно ожидать,
что амиды металлов (например, литийалюминийамид) будут пригодны для
титрования спиртового гидроксила. Кислотность и сродство к протону умень-
шаются соответственно в таком порядке:
CeH5OH > СН3ОН > R2NH; R2N~ > СН3О- > СвН5О-
«Нейтрализация» спиртов амидами металлов происходит в соответствии
со следующим уравнением:
R2NM + HOR —> MOR + R2NH,
где М — металл [387]. Для определения активного водорода используют
раствор ди-н-бутиламида лития в тетрагидрофуране в присутствии, напри-
мер, n-фениламиноазобензола как индикатора. Таким способом можно
определять этиловый, н-бутиловый и н-пентиловый спирты, нафтол-1, моно-
этаноламин, N-этилэтаноламин и многие другие соединения. Точность
определений колеблется в пределах от ±2,5 до ±5%. Этот метод был исполь-
зован для исследования химического строения при определении числа
активных атомов водорода, относящихся к спиртовым функциональным
группам. Однако он может послужить источником многих ошибок, поэтому
обязательно следует ознакомиться с оригинальной литературой [383, 387,
391, 513].
111. ОПРЕДЕЛЕНИЕ СОЕДИНЕНИЙ, СОДЕРЖАЩИХ СПИРТОВУЮ
ГИДРОКСИЛЬНУЮ ГРУППУ, С ПОМОЩЬЮ РЕАКЦИИ ЭТЕРИФИКАЦИИ
Спиртовую гидроксильную группу можно определять не только ациди-
метрическим титрованием, но также и другими методами, например ацилиро-
ванием хорошо известными классическими реагентами, все представители
которых принадлежат к акцепторным кислотам. К их числу относятся
уксусный, пропионовый, фталевый ангидриды, хлористые ацетил, бензоил,
3,5-динитробензоил, стеарил и т. д. (ион ацетилия СН3СО+ представляет
собой акцепторную кислоту). В большинстве случаев при ацилировании
необходимо добавлять катализаторы — хлорную, серную кислоты, п-толуол-
сульфокислоту, треххлористый бор или пиридин. Гораздо труднее проходит
ацетилирование третичных спиртов.
Определение соединений, содержащих спиртовый гидроксил
263
Для определения спиртовых функциональных групп пригоден также
фенилизоцианатный метод, так как в ходе процесса образуется N-фенил-
карбамат в таутомерной форме.
Найдено, что триэтилендиамин, 1,4-диазабицикло-[2,2,2]-октан, обладает почти
а 100 раз большей каталитической активностью, чем пиридин, при реакциях изоцианатов
со спиртами *. Шенк, Вайнс и Мойцис [719] нашли, что при ацилировании органических
гидроксильных групп в присутствии оснований каталитическая активность триэтилен-
диамина выше, чем у пиридина.
а. Определение гидроксильного числа с помощью пропионового ангидрида.
Пропионовый ангидрид в присутствии n-толуолсульфокислоты как ката-
лизатора пригоден для определения содержания гидроксилов (пропиониль-
ное число), например в гликолях, производных полиоксиалкиленгликолей
или стероидных спиртах [640, 772, 850].
В ходе взаимодействия пропионового ангидрида с п-толуолсульфокисло-
той образуется протонированная форма пропионового ангидрида, которая,
подобно уксусному ангидриду, превращается в пропионовую кислоту
и пропионильный катион (ср. гл. 7, разд. 44).
(СН3СН2СО)2О + Н+ —> (СН3СН2СО)2ОН+ —> (СН3СН2СО)+4-СН3СН2СООН.
Пропионильный катион является ацилирующим ионом и в благоприятных
случаях им удается ацилировать даже третичные спирты.
Смесь для ацилирования готовят растворением 1 г очищенной п-толуол-
сульфокислоты (свободной от примесей серной кислоты) в 30 лед уксусной
кислоты и последующим добавлением 5 мл пропионового ангидрида. Реагент
следует использовать не позже чем через 15 мин после его приготовления.
В конической плоскодонной стеклянной колбе для ацилирования
емкостью 5 мл взвешивают 0,5—1,5 мг-экв монофункционального соедине-
ния (а), добавляют 2,0 мл ацилирующего реагента и после растворения
оставляют на 2 часа при комнатной температуре или на 30 мин при 100°.
Затем смесь охлаждают 15 мин, переносят содержимое конической колбы
в толстостенную колбу на 500 мл, в которую предварительно отмеряют пипет-
кой 25 мл 0,1 н. раствора анилина (0,9%-ного) в бензоле и 30 мл уксусной
кислоты. Анилин реагирует с избытком пропионового ангидрида, образуя
анилид и пропионат анилиния. Реакционную смесь выдерживают 5—15 мин,
затем титруют 0,1 н. хлорной кислотой в присутствии пяти капель раствора
кристаллического фиолетового до появления зеленой окраски (Ь мл). В иден-
тичных условиях проводят глухой опыт (с мл). Разность двух измерений
(& — с) пропорциональна расходу пропионового ангидрида.
(GHgCHaCOjjO 4-ROH —> CH3CH2COOR-|-CH3CH2COOH,
(GH3CH2CO)2O + 2CeH5NH2 —> CH3CH2CONHCeH5-|-CH3CH2COO-H3NCeH5.
Пропионат анилиния титруют хлорной кислотой. Анализ в присутствии
примесей аминов, способных к ацилированию, дает неточные результаты.
Гидроксильное число равно количеству КОН в миллиграммах, эквивалентному
количеству кислоты, израсходованному на ацилирование 1 г соединения, содержащего
спиртовую (фенольную) гидроксильную группу.
ю (Ь-с)х56
Гидроксильное число =------- ,
а
где а — вес, г; с — объем стандартного раствора, израсходованный на глухой опыт, мл‘,
b — объем стандартного раствора, пошедший на титрование образца, мл.
Приведенные в табл. 85 данные показывают точность измерений [640].
О методе микропропионилирования см. в работе [62].
б. Определение гидроксильных чисел в поверхностноактивных сорбитоно-
вых аддуктах с помощью ацетилирования в присутствии п-толуолсулъфо-
♦ В и г k е u s J., J. Org. Chem., 26, 779 (1961).
264
Глава 19
Таблица 85
Соединение Гидроксиль- ное число Соединение Гидроксиль- ное число
вы- чис- ленное изме- рен- ное вы- чис- ленное изме- рен- ное
Бензиловый спирт 518 512 Кортизон 311 319
Циклогексиловый спирт 560 560 н-Октиловый спирт 430 435
Дезоксихолевая кислота 285 280 Изопропиловый спирт 933 933
Дезоксикортикостерон 169 168 Пропиленгликоль (ацилиро- 1473 1473
Гидрокортизон 464 458 ванне 2 час при 18°)
Метил - п - оксибензоат 368 369 Трифенилметиловый спирт 215 210
кислоты [772]. Сорбитов (моноангидросорбит) СвН8О(ОН)4; спан (Span) —
поверхностноактивное вещество большого молекулярного веса представляет
собой сложный эфир жирных кислот и сорбитона; твин (Tween) —поли-
оксиэтиленовое производное сорбитонового ангидрида, частично этерифици-
рованное жирными кислотами,
О
HoG^ \hch2ocor
I I
хоне снох
'V'
нох
COR — олеоил или лаурил и др., X — полиоксиэтиленовая группа.
В колбе Эрленмейера емкостью 250 мл с притертой пробкой взвешивают
5—6 ммолей соединения, содержащего ОН-группу, добавляют точно 5,00 мл
2 М уксусного ангидрида в этилацетате, нагревают 5 мин при 50 ± 1° на бане;
перемешивают и после выравнивания давления нагревают раствор еще
10 мин. После ацетилирования гидролизуют избыток уксусного ангидрида,
добавляя последовательно 2 мл воды и 10 мл водного раствора пиридина
(3 : 1), энергично встряхивают, оставляют на 5 мин и затем титруют обра-
зовавшуюся уксусную кислоту 0,56 н. едким кали в метиловом спирте (Ь мл).
Аналогично проводят глухой опыт (с мл); вычисляют разность между двумя
измерениями. В качестве индикатора пользуются раствором смеси крезоло-
вого красного и тимолового синего.
100
Гидроксильное число соединения = ——*
где а — навеска образца, мг; с — объем раствора, израсходованный в глу-
хом опыте, мл; b — объем раствора, определенный после ацилирования
0,56 н. раствором основания, мл; f — фактор пересчета для титрованного
раствора.
Приготовление реагентов. Раствор ангидрида. 14,4 г моногидрата п-то-
луолсульфокислоты растворяют в 360 мл этилацетата. Перемешивают (лучше
магнитной мешалкой), защищая от света, и во время перемешивания
добавляют небольшими порциями 120 мл уксусного ангидрида. Раствор
окрашивается в бледно-желтый цвет, однако содержание ангидрида остается
в нем неизменным в течение нескольких дней.
0,56 н. росте р едкого кали. 334 г КОН растворяют в 2000 мл абсолют-
ного метилового спирта, дают ему охладиться, предварительно защитив
Определение соединений, содержащих спиртовый гидроксил
265
от попадания влаги и двуокиси углерода из воздуха, и, если необходимо,
отфильтровывают выпадающие карбонаты. В заключение разбавляют абсо-
лютным метиловым спиртом до объема 9000 мл.
Смесь индикаторов. Смешивают 1 ч нейтрализованного 0,1%-ного крезо-
лового красного с 3 ч нейтрализованного 0,1%-ного тимолового синего
(ср. [616]).
Вследствие того что хлорная кислота как катализатор атакует также
и сложноэфирные связи, полученные при титровании значения оказываются
на 20—30% выше теоретических, а при титровании с соляной кислотой
они примерно на 15% ниже.
в. Определение спиртовой гидроксильной группы по реакции с 3,5-дини-
тробензоилхлоридом [687]. 3,5-Динитробензоилхлорид — реагент, обычно
используемый для этерификации соединений, содержащих гидроксильную
группу [516]. Его недостатком является гигроскопичность, а преимущест-
вом по сравнению с уксусным и фталевым ангидридами служит то, что при
его применении могут быть этерифицированы даже третичные спирты,
а кетоны и альдегиды при содержании их в смеси ниже 40% совсем не влияют
на результаты определений.
Спирты реагируют в пиридине по следующему уравнению:
ROH + 3,5-(NO2)2CeH3GOGl + G5H5N 3,5-(NO2)2C0H3COOR + C5H5NH+C1-
3,5- Динитробензоилхлорид гидролизуется водой
HOH + 3,5-(NO2)2C6H3GOCl + C5H5N —> 3,5-(NO2)2GeH3COOH + C5H5NH+Gl-
Из продуктов реакции динитробензойную кислоту и хлорид пиридиния
можно титровать одновременно как сильные кислоты потенциометрически,
используя первую точку перегиба. Точка эквивалентности сложного эфира
динитробензойной кислоты — слабой кислоты — может быть обнаружена
по второму перегибу на кривой. Динитробензоилхлорид и динитробензой-
ную кислоту можно титровать как бифункциональные соединения стандарт-
ным раствором ТБАГ в смеси бензол — метиловый спирт (ср. гл. 20). Коли-
чество эфира динитробензойной кислоты пропорционально содержанию
ОН-групп в анализируемом соединении.
Определить конечную точку довольно легко, так как в точке эквивалент-
ности сильных кислот желтая окраска раствора переходит в красную;
раствор эфира динитробензойной кислоты становится красным от одной
капли стандартного раствора основания (причины появления окраски еще
не выяснены). Это изменение окраски отвечает первой точке эквивалентности,
т. е. позволяет определить количество динитробензоилхлорида, пошедшего
на этерификацию.
Визуальное титрование. В колбе Эрленмейера на 100—150 мл с притертой
пробкой взвешивают 4,0 мл динитробензоилхлорида и добавляют точно
1,0 мл раствора анализируемого соединения. (Раствор анализируемого соеди-
нения приготавливают следующим образом: в предварительно взвешенной
мерной колбе емкостью 10 мл, в которую отмерено пипеткой 3 мл пиридина,
взвешивают примерно 4 мг-экв жидкости. После повторного взвешивания
доливают пиридин до метки.) Раствор перемешивают и оставляют в плотно
закрытой колбе на 5—15 мин. Гидролизуют избыток хлорангидрида в кис-
лоту 7—10 каплями воды. К гидролизованной смеси прибавляют 40 мл
пиридина, нагревают почти до кипения, затем охлаждают и титруют 0,2 н.
раствором ТБАГ до первого четкого появления красной окраски. Полу-
микробюретку, в которой находится титрованный раствор, лучше защищать
от попадания влаги и двуокиси углерода. Кончик бюретки должен находить-
ся под поверхностью анализируемого раствора, причем раствор также необ-
266
Глава 19
ходимо защитить от влаги воздуха. Аналогично выполняют глухой опыт.
При вычислении концентрации анализируемого вещества исходят из раз-
ности между объемами в миллилитрах, израсходованными на глухой опыт
и титрование образца. 1 ммоль монофункционального соединения требует
1 мл 0,2 н. раствора ТБАГ. Методика расчетов рассматривается в гл. 13,
разд. 76.
Приготовление реагентов. 0,2 н. раствор ТБАГ в смеси бензол — мети-
ловый спирт готовят так, как описано в гл. 10, разд. 57, с той лишь разницей,
что расходуется двойное количество иодида алкиламмония и окиси серебра
и берут не 180, а 300 мл метилового спирта. Раствор пропускают через
анионообменную колонку со скоростью 7—10 мл!мин.
Пиридин очищают над окисью бария (см. гл. 8, разд. 49, и).
Раствор динитробензоилхлорида получают непосредственно перед тит-
рованием. 1,15 г тонкоизмельченного 3,5-динитробензоилхлорида осторожно
нагревают с 25 мл пиридина, тщательно защитив от попадания влаги из воз-
духа. Сухой реагент сохраняют в эксикаторе над пятиокисью фосфора.
Первичные алифатические спирты и такие спирты, как холестерин,
пропиленгликоль, циклогексиловый спирт, ментол, бензиловый спирт,
цетиловый спирт, триэтиленгликоль, глицерин, декстроза, сахароза, пен-
таэритрит, манноза, стигмастерин и т. д., можно определять этим методом
с точностью примерно от ±0,2 до ±0,6%.
Из пяти ОН-групп (—)-сорбозы и фруктозы четыре ацетилируются в тече-
ние 5 мин при комнатной температуре, после чего раствор сразу же разбав-
ляют пиридином и титруют. С третичными спиртами, например тпрепг-бути-
ловым или 2-метилбутанолом-2, реакцию следует проводить 24 или 48 час.
Трифенилметиловый спирт не реагирует даже через 100 час; 0,4 моля этило-
вого спирта можно определить с точностью ±2% в присутствии 0,1 —
0,4 ммолей трифенилкарбинола, трибензиламина, ацетона, циклогексанона
и бензальдегида.
г. Определение спиртовых гидроксильных групп с помощью фенилизоциа-
ната [669]. Метод заключается, по существу, в реакции соединения, содер-
жащего спиртовую ОН-группу, с фенилизоцианатом в присутствии октаноата
олова(П) как катализатора. После того как реакция закончится, непрореа-
гировавший фенилизоцианат связывают определенным количеством дибутил-
амина, взятого в избытке. В заключение избыток дибутиламина оттитро-
вывают хлорной кислотой.
1. CeH5N = С = О + ROH —> С6Н5 —N = C —OR Т± CeH5 —NH—GOOR
Ан
Эфир N-фенилкарбаминовой кислоты
2. G6H5N = C = О + (G4H9)2NH —» CeH5—NH —СО —N(C4H9)2
^^Дибутил-К'-фенплмочевина
3. (C4H9)2NH-[-HC104 = (C4He)2NHjC10<
Октаноат олова(Н) Sn(G8H15O2)2 — катализатор, обладающий в какой-то степени
селективным действием в реакции изоцианатов со спиртами. Он используется при про-
изводстве пенообразователей на основе сополимеров мочевины.
Взвешивают две порции анализируемого вещества, так чтобы ни в одной
из них не содержалось больше 11 мг-экв ОН-групп, и переносят в две колбы
Эрленмейера по 500 мл с притертыми пробками. Если реакцию следует
проводить при 98°, то используют сосуды, выдерживающие повышенное
давление и нагревание до высоких температур. Образцы растворяют в 25 мл
диметилформамида каждый и перемешивают до полного растворения. Как
ДМФ, так и толуол, используемые для реакции, не должны содержать
более 0,01 % воды, так как фенилизоцианат количественно разлагается водой.
После растворения прибавляют реагент, содержащий точно 20 мл фенилизо-
Определение соединений, содержащих спиртовый гидроксил
267
цианата и 1,0 мл раствора катализатора и оставляют в закрытой стеклянной
пробкой колбе (см. также табл. 86). Одновременно, используя идентичные
пипетки и реагенты, в двух колбах Эрленмейера емкостью 500 мл каждая
проводят глухой опыт.
После истечения указанного времени во все четыре колбы добавляют
точно по 20 мл раствора, содержащего дибутиламин, и после перемешива-
ния оставляют на 15 мин, затем содержимое каждой колбы разбавляют 100 мл
метилцеллозольва и титруют 0,5 н. хлорной кислотой в метилцеллозольве.
В качестве индикатора используют 0,1%-ный раствор бромкрезолового
зеленого в метиловом спирте; первое характерное изменение окраски на жел-
тую указывает на конечную точку. 1 мл 0,5 н. хлорной кислоты эквивален-
тен 8,5 мг ОН-групп. Следует принимать во внимание объем хлорной кис-
лоты, пошедшей на глухой опыт; расчеты ведут по разности (см. гл. 13,
разд. 76).
Растворители — тщательно высушенные диметилформамид и толуол (Н2О < 0,01%).
1 н. раствор фенилизоцианата готовят следующим образом: 109 мл (119 г) фенилизоциана-
та растворяют в 500 мл толуола и объем раствора доводят до 1000 мл. Раствор хранят
в склянке, закрытой пробкой, обернутой алюминиевой фольгой. Если из раствора выпал
осадок, следует приготовить свежий раствор. Чтобы получить 2 н. раствор дибутиламина,
337 мл (256 г) дибутиламина разбавляют толуолом до объема 1000 мл. При приготовлении
раствора катализатора 21 г октаноата олова(П) растворяют в 100 мл толуола. О приготов-
лении стандартного раствора хлорной кислоты в монометиловом эфире гликоля см.
в гл. 11, разд. 60, а, д.
С помощью описанного метода, согласно литературным данным [669],
можно определять приведенные в табл. 86 соединения, содержащие
ОН-группы.
Таблица 86
Соединение Продол- житель- ность реакции, мин Температура, °C Точность определения
«-Бутиловый спирт 15 Комнатная 99,9±0,34%
mpem-Бутпловый спирт 15 98
2,2- Диметплбутандио л -1.3 30 98
2,2- Диметплпропан дио л -1,3 60 Комнатная
Сорбит 60 »
Аддукт окиси пропилена с сорбитом 30 » 110±0,9 вес.экв.
Трипропиленгликоль (следует использовать 30 »
2 мл раствора катализатора)
Вода, первичные и вторичные амины мешают определению, так как количественно
реагируют с фенилизоцианатом. Третичные амины с ним не реагируют, однако они реаги-
руют с хлорной кислотой. Ароматические ОН-производные в количествах больше 1%
также мешают анализу.
112. ПРИМЕНЕНИЕ ХЛОРНОЙ КИСЛОТЫ КАК КАТАЛИЗАТОРА
ДЛЯ АЦЕТИЛИРОВАНИЯ СМЕСЬЮ УКСУСНЫЙ АНГИДРИД —
ЭТИЛАЦЕТАТ ИЛИ ПИРИДИН — УКСУСНЫЙ АНГИДРИД
Сильные кислоты, например хлорная или n-толуолсульфокислота, ката-
лизируют ацилирование соединений, содержащих ОН-группу: ацетилиро-
вание первичных и вторичных спиртов уксусным ангидридом в этилацетате
заканчивается уже за 5 мин [257].
Предложен следующий механизм ацетилирования в уксусной кислоте
[110] или этилацетате [257], катализируемого хлорной кислотой:
(СН3СО)2О + Н+ —> (СН3СО)2ОН+; (СН3СО)2ОН+ СН3СО+ + СН3СООН
СН3СО*4-ВОН —> R—ООССН3-[-Н+ (ацетилирование)
268
Глава 19
В пиридине реакция протекает по такому механизму [257]:
C5H5NH+-HCH3CO)2O (C5H5NCH3CO)+ + CH3COOH
(C5H5NCH3CO)+4-ROH ROOCCH3H-C5H5NH+ (ацетилирование)
В приведенных схемах (СН3СО)2ОН+ — ион ацетангидридия (протониро-
ванный уксусный ангидрид), СН3СО+ — ион ацетилия (ср. гл. 7, разд. 44);
о пиридин-ацетилиевом ионе см. в работе [278].
Уксусным ангидридом в этилацетате можно ацетилировать не только
спирты, но и многие другие соединения, включая фенолы, полифенолы, гек-
созы, меркаптаны, амины, кетоксимы и вицинальные диоксимы [715, 716].
Преимущество этого метода связано с тем, что можно анализировать также
и пространственно затрудненные фенолы. При анализе смесей соединений,
содержащих спиртовые и фенольные ОН-группы, общее содержание
ОН-групп определяют ацетилированием, а содержание фенольных гидро-
ксилов — ацидиметрическим титрованием. Производные нитроанилинов
обладают свойствами либо слабых оснований, либо чрезвычайно слабых
кислот. Определять их методом кислотно-основного титрования сложно,
но с помощью ацетилирования можно добиться их количественного опре-
деления. 4-Метоксифенол и N-метиланилин можно определять 0,25 М уксус-
ным ангидридом в пиридине с достаточной точностью даже в присутствии
пространственно затрудненных фенолов и менее реакционноспособных
производных анилина.
Для ацетилирования используют уксусный ангидрид в этилацетате или
пиридине в зависимости от того, что растворяют. Уксусную кислоту, обра-
зовавшуюся после разложения избытка уксусного ангидрида, титруют 0,55
или 0,15 н. раствором едкого кали в метилцеллозольве. Следовательно, послед-
няя стадия метода заключается в титровании уксусной кислоты сильным
основанием. Методики приготовления реагентов приводятся ниже.
Реагент А
Этилацетат (растворитель)', 2 М уксусный ангидрид', 0,15 М хлорная кислота.
4 г (2,35 мл) 72%-ной хлорной кислоты прибавляют к 150 мл этилацетата (максималь-
ное содержание воды 0,1%). При энергичном перемешивании прибавляют 8 мл уксусного
ангидрида, снова перемешивают и оставляют раствор в закрытой колбе на 30 мин при
комнатной температуре. Охлаждают до 5° и, продолжая перемешивание, прибавляют еще
42 мл уксусного ангидрида, после чего выдерживают 1 час при 5°. Приготовленный таким
способом реагент можно использовать в течение примерно двух недель.
Реагент Б
Этилацетат (растворитель)', 0,25 М уксусный ангидрид', 0,006 М хлорная кислота.
К 200 мл этилацетата прибавляют 0,10 мл 72%-ного хлорной кислоты и по каплям
6,5 мл уксусного ангидрида; затем перемешивают 5 мин магнитной мешалкой. Реагент Б
можно использовать примерно в течение двух недель.
Реагент В
Этилацетат (растворитель)', 0,25 М уксусный ангидрид', 0,003 М хлорная кислота.
Реагент В готовят так же, как реагент Б, используя как катализатор только 0,05 мл
хлорной кислоты. Применяют после однодневной выдержки.
Реагент Г
Пиридин (растворитель)', 2 М уксусный ангидрид', 0,15 М хлорная кислота.
Осторожно по каплям и при постоянном перемешивании прибавляют 0,8 г (0,47 мл)
72%-ной хлорной кислоты к 30 мл пиридина и затем, продолжая перемешивание, 10 мл
уксусного ангидрида. Реагент Г необходимо готовить ежедневно.
Реагент Д
Пиридин (растворитель)', 0,25 М уксусный ангидрид', 0,02 М хлорная кислота.
0,12 мл 72%-ной хлорной кислоты прибавляют к 80 мл пиридина, содержащего мень-
ше 0,1% воды, затем смешивают с 2,5 мл уксусного ангидрида и встряхивают 5 мин.
Реагент Д можно использовать в течение примерно одного дня.
Определение соединений, содержащих спиртовый гидроксил
269
Реагент Е
Пиридин (растворитель)’, 0,35 М уксусный ангидрид’, 0,04 М хлорная кислота.
Реагент Е готовят так же, как реагент Д, используя 0,24 мл хлорной кислоты
и 3 мл уксусного ангидрида.
Внимание! Следует тщательно выполнять правила техники лабораторной
безопасности при работе с реагентами, содержащими хлорную кислоту.
Надевают защитные очки и перчатки и работают в вытяжном шкафу. При-
готовленные реагенты нельзя нагревать. Оттитрованные растворы, как
и растворы, полученные при проведении глухих опытов, необходимо немед-
ленно разбавлять водопроводной водой.
Кроме описанных реагентов, необходимы 0,55 и 0,15 н. растворы едкого
натра (см. гл. И, разд. 60, и) и смесь индикаторов (см. разд. 111, б).
а. Определение первичных и вторичных спиртов [257]. В колбе Эрленмейе-
ра емкостью 100—150 мл с притертой стеклянной пробкой взвешивают
3—4 ммоля соединения, содержащего ОН-группу, добавляют точно 5,0 мл
реагента А, взбалтывают до растворения и оставляют на 5 мин. Гидроли-
зуют избыток уксусного ангидрида 1—2 мл воды и 10 мл водного пиридина
(1 : 3) и вновь оставляют на 5 мин. После добавления индикаторной смеси
титруют 0,55 н. раствором едкого натра до перехода желтой окраски в фиоле-
товую в конечной точке (с мл)\ точно так же выполняют глухой опыт (Ъ мл).
Используют для расчета разность между объемами, пошедшими на титрова-
ние (ср. гл. 13, разд. 76).
В течение 5 мин способны ацилироваться алифатические первичные
и вторичные спирты, бензгидрол, бензоин, циклогексанол, глицерин, лак-
тоза, манноза, мальтоза, за 35 мин ацилируется целлобиоза, за 45 мин —
глюкоза и т. д., которые можно определять этим способом со средней точ-
ностью ±0,4%.
Реагент Г — более мягкое ацетилирующее средство, чем реагент А,
используют для определения первичных и вторичных спиртов в присутствии
третичных спиртов. Так, его применяют для определения следующих спир-
тов (продолжительность реакции в минутах указана в скобках): метилового
(7—10), 2-этилгексилового (10—15), циклогексилового (18—20), изопропи-
лового (20—25).
б. Ацетилирование фенолов, тиолов и аминов [716]. 1. В колбе Эрлен-
мейера на 100—150 мл с притертой пробкой взвешивают 3—4 ммоля соеди-
нения, содержащего ОН-, SH- или NH2-rpynny, добавляют точно 5,0 мл
реагента А, перемешивают до растворения и оставляют на 5 мин. Избыток
уксусного ангидрида гидролизуют, как описано выше в разд, (а), и титруют
в присутствии смеси индикаторов 0,55 н. раствором едкого натра в метилцел-
лозольве до перехода желтой окраски в фиолетовую (с мл). Аналогично
выполняют глухой опыт (Ь мл)’, для расчета процентного содержания исполь-
зуют разность объемов (Ь — с) (см. гл. 13, разд. 76).
2. Если располагают только 0,4—0,6 ммоля вещества, то ацетилирование
проводят реагентом Б в колбе Эрленмейера на 50—100 мл. Остальные опера-
ции выполняют, как в методе (1), но добавляют для гидролиза уксусного
ангидрида только 5 мл водного пиридина (1:3). Титруют 0,15 н. раствором
едкого натра. Этим методом можно определить со средней точностью ±0,4%
следующие соединения (в скобках указана продолжительность реакции
в минутах и тип реагента): 2-тпретп-бутилфенол (5, А): 2,6-ди-77грет?г-бутил-4-
крезол (5, А); резорцин (5, А); 2,6-диизопропилфенол (5, Б); 2,6-диметилфенол
(5, Б); тпретп-бутилмеркаптан (5, А); тиокрезолы (5, А); «-бутиламин
(15, А); N-метиланилин (10, Б); 2-нитроанилин (60, А); 3-нитроанилин
(20, А); 4-нитроанилин (75, А); о-фенилендиамин (5, А); дифениламин (45, А).
Описанным методом, используя реагент Д, можно определить 4-метоксифенол
и N-метиланилин в присутствии пространственно затрудненных фенолов или анилинов
(см. табл. 87) [716].
270
Глава 19
Для определения 2-трет-бутилфенола в присутствии 2,6-ди-тргтп-бутилфенола
применяют реагент В и метод (6,2) (25—66 мол.% бутплфенола).
в. Определение оксима циклогексанона [715]. В колбе Эрленмейера на 50 мл с при-
тертой стеклянной пробкой взвешивают 0,3—0,7 ммоля образца, добавляют точно 5,0 мл.
реагента Е и после растворения оставляют на 5—10 мин. Избыток уксусного ангидрида
гидролизуют 0,10—0,15 мл воды и после 10-минутной выдержки разбавляют 30 мл
диметилформамида, нейтрализованного в присутствии тимолового синего. В заключение
титруют 0,15 н. метилатом натрия. Аналогично выполняют глухой опыт. Для расчета
используют разность между двумя измерениями. 16,97 мг оксима циклогексанона требуют
1 мл 0,15 н. раствора основания. В этом методе ацилируется гидроксил CNOH-rpynn.
Таблица 87 Таблица 88
Пространственно затруднен- ные фенолы Концен- трация, мол. % (через 20 мин)
2,6-Ди-тпрет-бутил-и-крезол 5—10
2,6-Дн-трет-бутилфенол 5—10
2-т рет-Бутилфенол 5—10
2,6-Диизопропилфенол 80
Метилсалицилат 80
Амины Концентра- ция, мол.% (через 15 мин)
Дифениламин 5-14
2- Нитроанилин 5—14
4-Нитроанилин 80
ИЗ. АНАЛИЗ СМЕСЕЙ СОЕДИНЕНИЙ, СОДЕРЖАЩИХ ГИДРОКСИЛЬНЫЕ
ГРУППЫ
Из-за различных размеров и различной структуры молекул скорости
реакций данного реагента с различными органическими соединениями, содер-
жащими одни и те функциональные группы, могут значительно отличаться.
Второй порядок реакции — это такой порядок, при котором скорость
пропорциональна произведению молярных концентраций обоих реагентов.
Обозначим реагенты символами А и Б, их концентрации — а и b и х —
эквивалентное количество А и Б, которые прореагируют за определенное
время t. Скорость реакции в этом случае равна
dxldt = k(b — х)-(а — х).
Проведем интегрирование (приняв ж = 0 при f = 0 и х=-х при t = i),
в результате получим для константы скорости реакции следующее выражение'
. 2,303 . а (Ь — х)
k:=7W=^loeb^T)
Если условия исследования и соотношение концентраций выбраны
удачно, то можно анализировать смеси пропилового и изопропилового,
бутилового и изобутилового спиртов [749] и т. д. Для реакции второго
порядка зависимость log [(& — х)/(а — z)] от t имеет вид прямой. Если в про-
стой смеси протекают две реакции второго порядка, то на графике получают-
ся два прямолинейных сегмента (рис. 111). Это хорошо выполняется при
условии, что скорости реакции различны, например для реакции бутило-
вого и изобутилового спиртов с уксусным ангидридом. Определение проводят
следующим образом. Известным методом, например ацетилированием, про-
пионилированием и т. д., устанавливают общее содержание бутилового
спирта в бинарной системе, например смеси бутилового и изобутилевого
спиртов. Для этого отбирают часть запасного раствора и определяют общее
содержание бутилового спирта (а мол.%):
/л r-\ N Е
a (D С) у ' м '
где D — объем стандартного раствора, потребовавшийся для титрования
смеси пиридин — уксусный ангидрид — вода (т. е. в глухом опыте), мл\
Определение соединений, содержащих спиртовый гидроксил
271
С — объем стандартного раствора, пошедший на титрование образца, мл;
N — нормальность стандартного раствора, г-экв/л; Е — эквивалентный вес
соединения, содержащего гидроксил; М — молекулярный вес; V — алик-
вотная доля запасного раствора, мл.
В мерной колбе емкостью 250 мл взвешивают такое количество анализи-
руемого соединения, чтобы в нем содержалось не больше 0,05 молъ/л
ОН-групп. Добавляют примерно 240 мл пиридина, 10 мл уксусного ангид-
рида и быстро разбавляют до метки пиридином. Немедленно замечают время.
Рис. 111.
В колбы Эрленмейера с притертыми пробками с интервалом 5—10 мин
пипеткой отбирают аликвотные части раствора по 10 мл и добавляют по 5 мл
воды, чтобы предотвратить дальнейшее ацилирование. Опять отмечают
время и через 10 мин образовавшуюся уксусную кислоту титруют 0,1 н.
раствором едкого кали в присутствии индикаторной смеси. Для приготовле-
ния индикаторной смеси берут две части 0,1%-ного раствора нильского
голубого в 50%-ном этиловом спирте и одну часть 10%-ного раствора фенол-
фталеина в 95 %-ном спирте. В глухом опыте аналогичным образом опреде-
ляют объем стандартного раствора, израсходованный на реагент, содержа-
щий уксусный ангидрид. Для этого в мерной колбе на 250 мл пиридином
разбавляют 10 мл уксусного ангидрида. 10 мл этого раствора смешивают
с 5 мл воды и через 10 мин титруют, как описано выше. Молярную концен-
трацию раствора уксусного ангидрида (6) определяют по формуле
, D-N Е
где Е — эквивалентный вес уксусного ангидрида.
Уменьшение молярной концентрации соединения, содержащего гидро-
ксил, наносят на график как функцию от времени (см. рис. 111). Наносят
на график также значение log [(Ь — х)/{а — .г)] в зависимости от времени.
Прямую линию, отвечающую менее реакционноспособному изобутило-
вому спирту, экстраполируют к нулевому моменту времени. Линия, парал-
272
Глава 19
лельная оси абсцисс, соединяет точку пересечения A (log 0,369) с прямой
линией, отвечающей более реакционноспособному бутиловому спирту.
Точка В, найденная при этом, соответствует определенному времени Т
(например, Т = 59,5 мин). По графику зависимости х от t можно найти
уменьшение концентрации в момент Т. Таким образом, количество более
реакционноспособного компонента бинарной смеси выражается в процентном
отношении как (z/a)«100.
Расчет можно провести и таким образом: подставить в приведенное ниже
уравнение значение концентрации, соответствующее точке А, и известные
концентрации спирта и растворов ангидрида.
где а — общая молярная концентрация бинарной смеси, содержащей
ОН-группу; b — молярная концентрация уксусного ангидрида в пиридине;
А — антилогарифм значения для точки А, найденной экстраполяцией
на графике.
Простой пример позволяет проиллюстрировать изложенное [749]. Если
принять, что концентрация бутилового спирта а = 0,2 молъ/л, концентрация
уксусного ангидрида Ъ = 0,415 моль/л, а А' — антилогарифм 0,3690 равен
2,339, то при подстановке значений в уравнение получим
2,339-0,2 — 0,415 А АОАС
х =----2ЖЙ--------= °’0395-
Следовательно, содержание бутилового спирта в бинарной системе равно
* = 2^2.100=19,8%.
Расчет функции х по отношению к t для уменьшения молярной концен-
трации: наносят молярную концентрацию общего содержания бутилового
спирта, меньшую молярной концентрации, найденной после ацетилирова-
ния через интервалы t.
Рейли и Папа предложили три более углубленных метода расчета, дозволяющие
определить и менее реакционноспособные компоненты [673].
При проведении анализа целесообразно использовать смеси, в которых
молярное соотношение уксусного ангидрида и гидроксилсодержащего
соединения равно 2:1.
Если содержание более реакционноспособного компонента превышает
70%, графический расчет и оценка становятся более трудными. Слишком
высокая скорость реакции может быть другой помехой при анализе бинар-
ной смеси. Молярные концентрации реагентов не должны быть равными,
так как при а = b log [(& — х)/(а — ж)] = 0.
Этот метод можно также применять и для анализа смесей альдегидов,
кетонов и первичных аминов [347, 749]. Его преимущество состоит в том,
что можно анализировать бинарные смеси (а в некоторых случаях и тройные)
изомерных или гомологичных соединений, содержащих одинаковые функ-
циональные группы. Однако метод, предусматривающий определение рас-
хода реагента во времени, требует значительного количества растворителей
(например, 500 мл пиридина), и это можно считать его недостатком.
Глава 20
ОПРЕДЕЛЕНИЕ НИТРОСОЕДИНЕНИЙ
114. АРОМАТИЧЕСКИЕ НИТРОСОЕДИНЕНИЯ
Нитропроизводные бензола или фенола можно титровать как кислоты
в пиридине или этилендиамине [91, 93, 254, 731]. Фенольный гидроксил
и карбоксильная группа в ароматических нитросоединениях являются
донорными кислотами, в то время как частично поляризованное ароматиче-
ское ядро служит акцепторной кислотой, и реагент, обладающий избытком
электронной плотности, координируется с электронодефицитными атомами
углерода или с атомами с пониженной электронной плотностью.
При потенциометрическом титровании пикриновой кислоты аминоэтила-
том натрия в этилендиамине наблюдается только один перегиб на кривой,
хотя расходуется 3 экв основания. Таким образом, помимо фенольного гидро-
ксила еще два атома углерода бензольного кольца выступают в роли акцеп-
торов. В случае л^-динитробензола расход стандартного раствора составляет
2 экв, что подтверждается двумя перегибами на потенциометрической кривой;
это также свидетельствует о координации с двумя атомами углерода в коль-
це. Нитробензол нельзя титровать в этилендиамине. С точки зрения взаимо-
действия донор-основание — акцептор-кислота реакционная способность
поляризованного ароматического кольца проявляется не только по отно-
шению к алкоголят-анионам, но в определенной степени также и по отноше-
нию к спиртам, диоксану и соединениям, содержащим аминогруппу.
В случае нитроанилинов и нитродифениламинов можно предположить,
что один из атомов водорода аминогруппы имеет кислотный характер [254].
274
Глава 20
В случае тринитробензола ОН-группа гидроокиси тетрабутиламмония
присоединяется к бензольному ядру, причем отрицательный заряд в свою
очередь оказывается распределенным по кислородным атомам нитрогрупп.
Многочисленные нитроароматические соединения можно титровать
потенциометрически в пиридине 0,1 н. раствором гидроокиси тетрабутилам-
мония в смеси бензол — метиловый спирт [254]. Навеску 0,4—0,8 мг-экв
вещества растворяют в 40 мл пиридина, нейтрализованного в присутст-
вии тимолового синего или азофиолетового, и, используя стеклянный
и каломельный электроды, титруют стандартным раствором, содержащим
(C2H5)3C4H9N+OH_. Соединения, приведенные в табл. 89, дают четкие
перегибы на потенциометрической кривой.
Таблица 89
Нитроанилиновые производные Нитродифениламиновые производные Полинитрованные ароматические соединения
2,6- Дпх л ор - 4 - нптроанпл пн 2,4-Динптроанилин 2,4,6-Тринптроанилин (пи- крамид) 1-Нитродпфениламин 2,4-Динптродпфенплампн Г ексанптродифенплампн 2,4,6-Тринитротолуол 1,3,5-Тринптробензол 2-Бром-4,6-дипптротолуол 2,4-Динптротолуол
Точность определений меньше, чем при анализе енолов или фенолов,,
и полученные значения иногда завышены: 2- и 4-нитроанилины, так же
как и 2-нитро дифенил амин, титровать не удается.
Определение 2-нитродифениламина в присутствии 2,4-динитродифенил-
амина рассмотрено в работе [558]. N-Нитрозодифениламин нельзя титровать
ни как кислоту, ни как основание. Его определяют косвенным методом
посредством реакции с избытком 1-нафтиламина (см. гл. 23, разд. 120, д).
Орчин и Вульфольк сообщили, что 2,4,7-тринитрофлуоренон образует комплексы
с многоядерными соединениями, например периленом, флуорантеном, фенантреном, хризе-
ном, пиреном, антраценом *. Этп комплексы относительно легко приготовить и они
могут оказаться полезными для характеристики этой группы ароматических соединений.
Кундифф и Маркунас [165] показали, что молекулярные комплексы трпнптрофлуорено-
на в пирпдпне можно титровать как кислоты: 2—20 мг трпнитрофлуоренонового комплек-
са растворяют в 50 мл пиридина и титруют потенциометрически в атмосфере азота 0,1 н.
раствором гидроокиси тетрабутиламмония; см. также [159, 164]. Использование индикато-
ров при этом анализе не дает удовлетворительных результатов, так как все растворы
в ходе титрования становятся интенсивно окрашенными.
Помимо вышеупомянутых соединений, можно титровать как кислоты
2,4-динитрофенилгидразин, а также 2,4-динитрофенилгидразоны, получен-
ные из альдегидов и кетонов с гидразином [731]. 2—20 мг гидразона раство-
ряют в 50 мл пиридина и титруют потенциометрически в токе азота 0,01 н.
или 0,02 н. раствором ТБАГ в бензоле, используя стеклянный и каломель-
ный электроды. Этот метод служит хорошим методом идентификации
(по определению эквивалентных весов) соединений, содержащих карбо-
нильную группу; точность метода 2% даже при навесках 1—2 мг. Относи-
тельно титрования n-нитрофенилгидразонов см. [688].
Тринитротолуол, динитротолуол и тетранитрат пентаэритрита можно титро-
вать в метилизобутилкетоне 0,1 н. раствором ТБАГ в смеси бензол — метиловый спирт.
Потенциометрическое титрование проводят с платиновым (содержащим 40% родия)
индикаторным электродом и каломельным электродом в качестве электрода сравне-
ния [710].
Триметплентринитрамин (гексоген, гексагидро-1,3,5-тринитро-си.мл«-триазпн, мол.
вес 222,1) и октагидро-1,3,5,7-тетранитро-сил«л«-тетразин (мол. вес 296,2) можно титро-
вать ТБАГ в качестве титранта с азофиолетовым как индикатором в пиридине, пипериди-
не, этилендиампне, диметилформамиде, диоксане, тетрагидрофуране, ацетоне, метил-
изобутилкетоне и ацетонитриле. Триметилентринитрамин можно также титровать мети-
латом натрия в диметилформамиде [223, 449, 710].
* J. Аш. Chem. Soc., 68, 1727 (1946).
Определение нитросоединенич
275
Определение гексагидро-1,3,5-тринитро-симм-триазина (RDX) в смеси с октагидро-
1,3,5,7-тетранитро-симм-тетразином (НМХ) [759]. Метод состоит в потенциометри-
ческом титровании в смеси метилизобутплкетон — изопропиловый спирт (4 : 1) 0,1 н.
метилатом натрия в смеси бензол — метиловый спирт. Система электродов представляет
собой каломельный электрод втулочного типа с хлоридом калия в метиловом спирте
и электрод сурьма — окись сурьмы.
При применении смеси метилизобутилкетона с изопропиловым спиртом в отношении
80 : 20 кривая потенциометрического титрования смеси RDX и НМХ имеет один пере-
гиб, который соответствует содержанию RDX. НМХ даже при концентрации около 80%
не влияет совершенно на титрование RDX. В метилизобутилкетоне RDX ведет себя как
более сильная кислота по сравнению с НМХ. При понижении основности растворителя
различие в кислотности RDX и НМХ может увеличиваться.
115. НИТРОГУАНИДИН И ЕГО ПРОИЗВОДНЫЕ
Кислотный характер нитрогуанидина и его производных немедленно
проявляется в диметилформамиде (ср. гл. 2, разд. 21). Можно предположить,
что кислотность водорода аминогруппы в нитрогуанидине вызвана электро-
нооттягивающим действием акцепторного заместителя, т. е. нитрогруппы,
приводящей к следующим предельным мезомерным структурам [182,
183, 488]:
+
RHNX + .О- RHNX + .О- RHN. + ,О~
>C = N —N< >С —N = N< +\С —N = N<
H2Nz X) h2nz хо- h2n^ хо-
На примере 1-амино-З-нитрогуанидина можно продемонстрировать способность амино-
группы увеличивать кислотность (ср. гл. 4, разд. 25, д)
1
H2N —NH з
>C = N — NO2
h2n
2
„ 10,60 [182]
рл“ 10,47 [488]
Навеску нитрогуанидина (рКа 12,20) или его производного в таком коли-
честве, чтобы на нитрование пошло 3—9 мл 0,1 н. раствора метилата натрия
в смеси бензол — метиловый спирт, растворяют в 50 мл диметилформамида,
нейтрализованного в присутствии пяти капель азофиолетового, и титруют
до появления голубой окраски, лучше в атмосфере азота. Этим способом
можно определять 1-амино-, 1-фенил-1-метил, 1-(2-пиридил)-, 1-бензил-,
1-бензоил-, 1-(1- или 2-фенилэтил)-, 1-(п-диметиламинофенил)- и 1-(п-циан-
фенил)нитрогуанидины, нитрогуанилморфолин и т. д. Из перечисленных
соединений 1-бензоил- и (или) 1-(п-цианфенил)нитрогуанидины [рКа 8,10
и (или) 8,60] имеют наиболее кислый характер.
Нитрогуанидин можно титровать также и как основание хлорной кисло-
той в трифторуксусной кислоте [183].
18*
Глава 21
ПОЛУМИКРО- И МИКРОТИТРОВАНИЕ КИСЛОТ
И КИСЛОТНЫХ АНАЛОГОВ
В результате действия двуокиси углерода из воздуха полумикро-
и микротитрование кислот и их аналогов значительно сложнее, чем микро-
титрование оснований в уксусной кислоте. То, что уменьшение нормально-
сти титранта приводит к понижению его эффективности, вызывает допол-
нительные трудности. Нормальность стандартной хлорной кислоты может
быть уменьшена даже до 0,001 н. без изменения точности измерения. При
титрованиях, проводимых в основных растворителях, этого делать нельзя,
так как расход растворителя в глухих опытах и действие двуокиси углерода
из воздуха искажают результаты. Этого можно частично избежать, если
взять для титрования по возможности меньший объем — 3 мл для 2—10 мг
образца.
При таком небольшом объеме адекватные соотношения между поверх-
ностью и объемом жидкости и между внутренней поверхностью сосуда для
титрования и титруемым объемом могут быть достигнуты, только если
применять по возможности сосуд для титрования меньшего размера и уда-
лить все усложняющие его части. По этой, а также по другим причинам
водородные электроды неприменимы [92, 176, 562, 601]; см. рис. 112—114.
В приборе, изображенном на рис. 112, индикаторный электрод, введен-
ный под кран бюретки, может быть использован как электрод сравнения,
так как в этом случае он погружен в жидкость с постоянной ионной силой.
Стандартный раствор в носике бюретки служит солевым мостиком, а пере-
вернутый крючкообразный конец препятствует конвекции. При введении
стандартного раствора капилляр постоянно заполняется свежим раствором,
следовательно, ошибка вследствие диффузии устраняется. Простой плати-
новый электрод можно также использовать как электрод [285]. Применяя
технические приемы, описанные ниже, и прибор, изображенный на рис. 112,
можно добиться хороших результатов, даже если оба электрода — индика-
торный и электрод сравнения — являются платиновыми. Раствор необхо-
димо перемешивать с помощью магнитной мешалки.
Вследствие поляризуемости электродов потенциометрическое измерение
следует проводить по возможности таким образом, чтобы ток, проходящий
через систему, не превышал 10-10 а. Поэтому компенсационные методы изме-
рения применять нельзя даже при использовании трубчатого гальванометра.
Вследствие большого сопротивления системы, особенно в таком раство-
рителе как бензол — метиловый спирт, возникают емкостные помехи, поэтому
желательно поместить прибор для титрования в ячейку Фарадея. При
применении этилендиамина пленка последнего может вызвать появление
тумана над бюреткой и прибором. Вследствие электропроводности конден-
сированных паров могут появиться блуждающие токи; поэтому все оборудо-
вание следует сохранять в чистоте и сухим.
Для микротитрования используют бюретку на 2 мл-, эту бюретку, а также
сосуд, содержащий стандартный раствор, защищают от влаги воздуха
с помощью пришлифованной поглотительной трубки. Колбой для титрова-
ния служит колба Эрленмейера емкостью 5—10 мл, причем ее боковой отвод
снабжен приспособлением для введения индикаторного электрода. Стер-
Полумикро- и микротитрование кислот и кислотных аналогов
277
жень магнитной мешалки делают из проволоки длиной 8—10 мм, впаянной
в капилляр.
Платиновые электроды представляют собой пластины длиной 6 мм и тол-
щиной 3 мм, вытянутые на конце в виде проволоки и впаянные в стеклянную
трубку длиной 7 см. Перед использованием электроды прокаливают в несве-
тящем пламени газовой горелки. Вводы должны быть заземлены.
Титрование в этилендиамине. Навеску 2—10 мг вещества помещают
в сухую колбу, вносят в нее сухую магнитную мешалку, заполняют бюретку
до метки и протирают кончик капилляра бюретки, вытянутый в конус,
смоченной в спирте ватой. Из бюретки, снабженной трубкой с аскаритом,
1 — кран для выравнивания давления; 2— тит-
рант; 3 — трубка для ввода азота; 4 — бюретка;
5 — двухходовый кран из тефлона; 6 — стеклян-
ный шлиф; 7 — отверстия для ввода электродов;
V—отверстие для вывода азота; 9— сосуд для
мнкротитрования емкостью 50 лл; 10 — магнит-
ная мешалка: 11 — стеря.’снь магнитной мешалки;
12 — реостат.
1 — индикаторный платиновый электрод; 2 —
платиновый электрод сравнения; 3 — магнитная
мешалка.
добавляют в колбу 3 мл этилендиамина и прибор немедленно соединяют
с бюреткой (рис. 112). Осторожно обтирают колбу ватой, смоченной в спирте,
особенно около мест ввода электродов, затем высушивают, чтобы избежать
появления блуждающих токов. Когда установится постоянный потенциал
(1—5 мин), титруют 0,05 н. аминоэтилатом натрия в этилендиамине.
120 мг металлического натрия ополаскивают этиловым спиртом и аминоэтанолом:
растворяют (при 35°) в 2 мл аминоэтанола в мерной колбе емкостью 100 лл, предваритель-
но продутой азотом, и затем разбавляют эти л енди амином до метки.
После добавления каждой порции стандартного раствора смесь переме-
шивают, затем выключают магнитную мешалку и оставляют стоять до уста-
новления постоянного значения потенциала. Длительность каждого потен-
циометрического измерения 20—40 мин. Точность измерения может быть
278
Глава 21
значительно увеличена, если соблюдать следующий порядок проведения
опытов: глухой опыт — проверка титра — титрование — проверка титра —
глухой опыт.
Прибор, изображенный на рис. 113, пригоден для полумикротитрования
кислот и их аналогов [562]. Одно из пяти горл колбы для титрования
емкостью 50 мл находится на осевой линии колбы и служит для введения
выводной трубки автоматической бюретки. Через два горла вводят электро-
ды, а еще два других горла служат для ввода и вывода азота. Используют
магнитную мешалку. В качестве стандартного раствора пригоден 0,02 н.
метилат натрия, стеклянный электрод применяют в качестве индикаторного,
а электродом сравнения служит каломельный электрод. Стандартный рас-
твор лучше сохранять в боросиликатной посуде в атмосфере азота. Во время
титрования электроды должны быть погружены в жидкость на 2 мм и нахо-
диться на расстоянии 5 мм друг от друга; в качестве индикаторов следует
применять 0,3%-ный тимоловый синий в метиловом спирте или 0,5%-ный азо-
фиолетовый в смеси бензол — метиловый спирт (1:2).
Загрязнение растворителя кислотными примесями может приводить
к значительным ошибкам при полумикротитровании кислот, поэтому их
необходимо нейтрализовать перед титрованием. В этом отношении пири-
дин и диметилформамид более пригодны, чем метилизобутилкетон, так как
в 5 мл этих растворителей может содержаться в среднем 0,002 мг-экв кислот-
ных примесей, в то время как в 5 мл метилизобутилкетона — 0,09 мг-экв.
Перед определением к 5 мл растворителя добавляют 0,03 мл тимолового
синего или азофиолетового и ток азота регулируют таким образом, чтобы
Таблица 90
Титрование кислот и аналогов кислот 0,02 н. метилатом натрияа [562]
Соединение Растворитель Количество, эквивалент- ное 1 МЛ стандартного раствора, мг
диметилформамид пиридин метил- изобутил- кетон
Ацетоацетанплпд Азофполетовый 3,544
п-Ампнобензолсульфацетанплпд п-Аминобензолсульфамид Азофполетовый (-{-) Азофиоле- товый ТС(-) 4,325 3,444
2-(п-Аминобензол сульфамидо)пи- римидпн ТС(-) ТС(+) 5,005
2-(п-Ампнобензолсульфамидо)-4- метплпиримидпн ТС(--) 5,286
2-(п-Ампнобензолсульфамидо)-4,6- диметплпиримидпн ТС(-) ТС(+) 5,566
2-(п-Аминобензолсульфампдо)тиа- зол тс 5,106
Бензимидазол Азофполетовый 2,363
Бензтрпазол тс 2,382
Диэтилбарбитуровая кислота тс 3,684
Этилбутилбарбитуровая кислота ТС 4,245
Этилизоамилбарбптуровая кислота тс 4,525
Этиламилбарбитуровая кислота ТС тс 4,525
Этилфенплбарбптуровая кислота ТС тс 4,645
п-Оксибензальдегид ТС 2,442
Фталимид Азофполетовый 2,943
Резорцин (с добавкой бензола) » 2,202
Сукцинимид » 1,982
Теофиллин (безводный) ТС 3,604
Тимол Азофполетовый 3,004
а Сокращения: ТС — тимоловый синий; (-}-) — незначительное завышение; (—) — незначительное
занижение.
Полумикро- и микротитрование кислот и кислотных аналогов
279
можно было считать пузырьки, затем нейтрализуют
Ю,02 н. раствором основания до появления голубой
окраски. Взвешивают приблизительно 0,1 мг-экв
образца в маленькой сухой мерной колбе и навеску
вносят в прибор через один из боковых отводов.
Снова взвешивают маленькую колбу. После раст-
ворения навески титруют в атмосфере азота до
появления ярко-голубого окрашивания. Анало-
гично определяют титр стандартного раствора по
отношению к бензойной кислоте.
При потенциометрическом титровании 15 мл
растворителя нейтрализуют, а затем титруют как
описано выше, но конечную точку определяют,
используя ранее упомянутые электроды.
Удобными растворителями для титрования
являются диметилформамид, пиридин и метилизо-
бутилкетон; в них можно точно анализировать
многочисленные соединения, содержащие феноль-
ный и енольный гидроксилы или иминогруппу
(табл. 90).
Рис. 114. Ячейка для титрования [176].
1 — отверстие для ввода азота; 2 — медная рукоятка; з — стек-
лянный и каломельный электроды; 4 — стержень магнитной
мешалки; 5 — магнитная мешалка; 6 — микробюретка: 7 — баке-
литовая крышка ячейки; S — винт для фиксации электродов.
А — крышка ячейки (вид сверху).
Как при потенциометрическом, так и при визуальном титровании кислот,
величина рК которых не превышает 10,5—11, подходящим растворителем
является диметилформамид. Пиридин можно применять в качестве раство-
рителя в том случае, если не удается установить конечную точку титрова-
ния в диметилформамиде потенциометрически или визуально (например,
сульфаниламид в присутствии азофиолетового). В некоторых случаях, одна-
ко, барбитураты в присутствии тимолового синего в пиридине дают быстрое
изменение окраски, а в присутствии азофиолетового — медленное. Конечная
точка в пиридине наблюдается отчетливее, чем в ДМФ, но потенциометриче-
ские кривые в последнем случае имеют более характерный вид: потенциал
достигает максимума, а затем резко понижается [562]. Для полумикротитро-
вания кислот с рК < 9 удобным растворителем служит метилизобутилкетон.
Прибор, изображенный на рис. 114, применяют для полумикротитрова-
ния стандартными растворами КОН и ТБАГ в изопропиловом спирте [176].
Хорошо известно, что стеклянный электрод нечувствителен в этиленди-
амине, если для титрования применяют стандартный раствор, содержащий
ионы Na+; поэтому в некоторых случаях стеклянные электроды можно
использовать в качестве электродов сравнения [446]. Однако со стандарт-
ными растворами, содержащими едкий натр или гидроокись тетраалкил-
аммония в изопропиловом спирте, стеклянные электроды могут применяться
и как индикаторные электроды. В качестве электрода сравнения можно
использовать каломельный электрод (рис. 61, б). Для титрования приме-
няют бюретку на 5 мл, калиброванную по 0,02 мл. Стандартный раствор
(0,1 н.) предохраняют от влаги и двуокиси углерода воздуха с помощью труб-
ки с аскаритом, вставленной в верхнюю часть бюретки. Применяют магнит-
ную мешалку диаметром около 4 мм и длиной 18 мм. На внешней части
стакана для титрования отмечают объем 20 мл.
Для потенциометрических титрований применяются прецизионный титрометр Preci-
sion Dual AC Titrometer co шкалой измерений от +1650 до —1650 мв или автоматический
регистрирующий прецизионный титратор Precision-Dow Recordomatic Titrator.
280
Глава 21
Навеску образца, на титрование которой потребуется 1—2 мл 0,1 н.
КОН или ТБАГ в изопропиловом спирте, растворяют в 20 мл растворителя
при перемешивании с помощью магнитной мешалки, пропуская при этом
через прибор слабый ток азота. Немедленно вводят в прибор электроды и гер-
метизируют его так, чтобы изолировать от действия влаги и СО2. Добавляют
стандартный раствор порциями по 0,02—0,05 мл и записывают показания
прибора после того, как потенциал примет постоянное значение. (Потенциал
считается постоянным, если отклонение pH-метра или милливольтметра
в течение 30 сек не превышает 2 мв.) Немедленно добавляют следующую пор-
цию титранта. Когда потенциал достигнет своего максимального значения
Рис. 115. Кривые потенциометрического титрования о,о'-дизамещенных фенолов
в диметилформамиде [176].
Стандартный раствор: 0,1 н. гидроокись тетрабутиламмония в изопропиловом спирте. Электроды: стек-
лянный и каломельный (см. рис. 114).
и затем начнет уменьшаться, записывают максимальное значение и немедлен-
но добавляют следующую порцию стандартного раствора. Титрование про-
должают до тех пор, пока потенциал не достигнет того же значения в милли-
вольтах, что и в глухом опыте с растворителем, и не будет оставаться постоян-
ным или не начнет непрерывно уменьшаться.
После титрования электроды, сосуд для титрования и остальные части
прибора необходимо тщательно очистить.
Аналогичным образом проводят глухой опыт по потенциометрическому
титрованию 20 мл растворителя (например, этилендиамина, диметилформ-
амида).
Если на потенциометрической кривой в этилендиамине обнаруживается
два перегиба, то при расчетах в качестве эквивалентного объема принимают
разность между первой и второй точками перегиба. В случае только одного
перегиба большое количество очень слабой кислоты может затемнять конеч-
ную точку титрования небольшого количества сильной кислоты. Перегиб
на кривой, полученный таким образом, соответствует кривой потенциомет-
рического титрования очень слабой кислоты только в том случае, когда две
кривые или конечные точки титрования сравнимы с данными, полученными
в модельном опыте с фенолом. Для вычисления результатов титрования
слабых кислот используют разность объемов титранта при титровании
образца и в глухом опыте.
Для титрования смеси кислот, например хлористоводородная кислота —
уксусная кислота — фенол, ДМФ является более удобным растворителем,
чем ЭДА (о дифференцирующем титровании смеси кислот см. гл. 15).
В случае соединений, содержащих две фенольные группы (например,
катехин) даже в ДМФ можно наблюдать два перегиба на кривой, хотя первый
перегиб едва заметен. Для гидрохинона обе конечные точки титрования
Полумикро- и микротитрование кислот и кислотных аналогов 281
нечетки; в этом случае вместо КОН в изопропиловом спирте следует исполь-
зовать ТБАГ, так как после нейтрализации первого гидроксила образуется
осадок.
Полифункциональные фенолы, содержащие две ОН-группы в орто-
положении, обнаруживают увеличение кислотных свойств по сравнению
с незамещенными фенолами, что, по-видимому, обусловлено взаимодейст-
вием гидроксильных групп, как, например, в случае о,о'-диоксидифенилме-
тана. Вследствие возникновения водородных связей удается титровать
только один фенольный гидроксил [176, 770]; ср. гл. 12, разд. 63, г. Кривые
потенциометрического титрования соединений, приведенные на рис. 115,
дают дополнительные сведения об их кислотности.
Титрование п,п'-диоксидифенилметана описано в гл. 18, разд. 108, б
и в работах [176, 359]. Приборы для микротитрования с визуальным и элек-
трометрическим определением конечной точки титрования см. работу [62].
Приборы пригодны для определения 10—25 мкг-экв монофункциональных
соединений. Объем кювет для титрования растворов равен 0,15—10 мл.
О применении электродов «Napf-Glaselectrode» типа Ингольда см. рабо-
ты [407, 754] и рис. 67. Методика микротитрования подробно описана в рабо-
те [589].
Глава 22
ОПРЕДЕЛЕНИЕ СОЛЕЙ
КАРБОНОВЫХ КИСЛОТ
Р и с. 116. Кривые потенциомет-
рического титрования солей кар-
боновых кислот в уксусной кис-
лоте [548].
1 — бензоат лития; 2 — ацетат натрия;
3 — дигидроцитрат 2-оксиэтилтриме-
-тиламмопия; 4 — ацетат аммония; 5 —
ацетат калия.
Соли кислот в подходящих растворителях и при выборе соответствую-
щего титранта можно титровать либо как кислоты, либо как основания.
Соли ароматических и алифатических аминов с сильными минеральными
или сульфоновыми кислотами можно титровать как кислоты алкоголятом
щелочного металла или гидроокисью алкиламмония в качестве титрантов
в смеси бензол — метиловый спирт или в основном растворителе. Соли орга-
нических оснований с галогеноводородными кислотами в уксусной кислоте
после добавления ацетата ртути(П) можно
титровать как основания хлорной кислотой.
Этот метод детально описан в гл. 27.
Соли карбоновых кислот с щелочными
и щелочноземельными металлами, аммиаком
и органическими основаниями можно титро-
вать в подходящем основном растворителе
как кислоты. Однако многое зависит от раст-
воримости в используемом растворителе. Так,
например, многие аммонийные соли моно-
функциональных карбоновых кислот с аммиа-
ком или аминами растворяются в ДМФ и их
можно титровать с помощью стандартного
раствора основания в присутствии тимоло-
вого синего; с другой стороны, соли поли-
функциональных карбоновых кислот в боль-
шинстве случаев нерастворимы в ДМФ. В этом
случае целесообразно растворить образец в не-
большом количестве воды (0,1—1,0 мл) и по-
сле добавления 15—20-кратного количества
этилендиамина титровать кислотные группы
соединения в присутствии азофиолетового.
Соли карбоновых кислот со щелочными, щелочноземельными металлами
и сильными или средней силы органическими основаниями подвергаются
сольволизу в уксусной кислоте с образованием эквивалентного количества
ацетат-ионов. Таким образом, эти соединения можно титровать хлорной
кислотой как основания [15, 48, 442, 470, 548, 732, 769, 682].
В уксусной кислоте в зависимости от природы катиона и аниона соли
проявляют кислотный или основной характер; кислотность или основность
раствора определяется солеобразующими катионами или анионами. Ацетаты
ртути(П), меди(П) и кадмия(П) в уксусной кислоте нейтральны. Ацетаты
щелочных и щелочноземельных металлов обладают основным характером.
Органические кислоты, константы диссоциации которых (в воде) 10"3 или
меньше, при растворении в уксусной кислоте обычно являются нейтраль-
ными. Соли таких кислот, как уксусная, винная, лимонная, глюконовая,
яблочная, малеиновая, фумаровая, бензойная, салициловая, а также произ-
водные барбитуровой кислоты и т. д. можно титровать хлорной кислотой
подобно ацетатам.
Определение солей карбоновых кислот
283
Перхлораты, соли галогеноводородных кислот, бисульфаты, персуль-
фаты, ферри- и ферроцианиды обладают кислотными свойствами, в то время
как фосфаты, нитраты и анионы большинства органических кислот в уксус-
ной кислоте являются основаниями [645] (ср. [470, 548]).
Кислотность катионов увеличивается с уменьшением объема иона или
с увеличением его заряда [470, 645].
Mg2+ > Са2+ > Ва2+ > Li+ > Na+ > NH+, К+.
Это влияет также на величину скачка (мв) при потенциометрическом титро-
вании (рис. 116). Кислотность калиевых солей изменяется в следующем
порядке:
ClOj > Г > Вг- > С1- > NOj.
116. ТИТРОВАНИЕ В ОСНОВНЫХ РАСТВОРИТЕЛЯХ
Навеску такого количества образца, на титрование которого требуется
5—6 мл 0,1 н. метилата натрия, растворяют в 15—20 мл нейтрализованного
диметилформамида, пиридина или этилендиамина. Если необходимо,
смесь осторожно нагревают. В качестве индикаторов в зависимости от кис-
лотности используют тцмшшвый синий или азофиолетовый [245]. Техниче-
ские приемы титрования (например, использование азота и др.), подробно
списанные в гл. 15—18, применимы также и в этом случае (см. также
рис. 39—42).
Если образуется осадок, используют колбу Эрленмейера со стеклянной
пробкой и вблизи конечной точки титрования после добавления стандарт-
ного раствора энергично встряхивают, оставляют раствор на 30—60 сек
и наблюдают за изменением окраски индикатора. Если необходимо, продол-
жают титрование до тех пор, пока окраска индикатора в течение этого вре-
мени не будет оставаться постоянной.
а. Определение лимонной кислоты в цитрате кофеина. Навеску 80—
100 мг цитрата кофеина растворяют в 15 мл ДМФ, нейтрализованного
в присутствии двух капель азофиолетового, и титруют 0,1 н. метилатом калия
в смеси бензол — метиловый спирт до появления голубой окраски. На тит-
рование 9,606 мг лимонной кислоты расходуется 1 мл стандартного раствора.
б. Определение бензойной кислоты, содержащейся в бензоате аммония.
Навеску 60—80 мг образца растворяют сначала в 1 мл дистиллированной
воды, затем добавляют 20 мл этилендиамина, нейтрализованного в присут-
ствии двух капель азофиолетового. Титруют, как описано в разд. «а». На тит-
рование 13,92 г бензоата аммония требуется 0,1 мл 0,1 н. метилата калия.
Титр стандартного раствора устанавливают по бензойной кислоте в той же
смеси растворителей.
в. Определение пикриновой кислоты, содержащейся в пикратах оснований.
Пикриновую кислоту, входящую в состав пикратов оснований, можно опреде-
лить непосредственно в диметилформамиде, в то время как основания, входя-
щие в состав этих пикратов, можно титровать хлорной кислотой в уксусной
кислоте [58] (гл. 20, разд. 114 и гл. 24, разд. 135). Следовательно, пикраты
оснований можно анализировать двумя способами.
Навеску пикрата, эквивалентную 0,1—0,2 мг-экв пикриновой кислоты
(последняя в ДМФ ведет себя как монофункциональная кислота), раство-
ряют в 15 мл ДМФ, предварительно нейтрализованного в присутствии тимо-
лового синего, и титруют (из бюретки, калиброванной по 0,01 мл) метилатом
натрия в смеси бензол — метиловый спирт до появления сине-зеленого
окрашивания. На титрование 22,91 мг пикриновой кислоты расходуется
1 мл 0,1 н. раствора.
г. Определение содержания винной кислоты в тартрате эрготамина
[324]. Навеску 200—250 мг тартрата эрготамина (высушенного над пяти-
284
Глава 22
окисью фосфора) помещают в колбу со стеклянной пробкой и растворяют,
защищая от света, в 20—30 мл пиридина, нейтрализованного в присутствии
0,2 мл 0,1%-ного тимолового синего. Титруют 0,1 н. метилатом калия в смеси
бензол — метиловый спирт до появления устойчивой синей окраски. Для
титрования 7,504 мг винной кислоты требуется 1 мл стандартного раствора.
Вместо тимолового синего можно взять одну каплю 1%-ного нафтолбензеина
в пиридине, который также является подходящим индикатором. В этом слу-
чае окраска изменяется от желтой до зеленой. Точность: ±1% [793].
Этим методом можно также определить кислоту, входящую в состав
малеата эргометрина, дифталата эрготамина и фосфата эрготоксина [308,
324].
Кальциевые соли ацетилсалициловой, салициловой и фенилхинолпнкарбоновой
кислот можно титровать метилатом натрия в пиридине [832]. Кислотные компоненты суль-
фата атропина, фосфата кодеина, хлоргидратов котарнина и скополамина, а также неко-
торые другие соли алкалоидов можно определить титрованием в пиридине метилатом
натрия в присутствии фенолфталеина [832].
д. Определение метансулъфоната п-аминобензоата N,N'-диэтиллейци-
нола. Кислотные компоненты солей азотистых оснований можно также опре-
делять при помощи стандартного раствора ТБАГ в смеси бензол — метиловый
спирт. Так, например, если растворить 100—200 мг образца в 20 мл диметил-
формамида, нейтрализованного в присутствии тимолового синего, метан-
сульфонат-ион можно титровать 0,05 н. ТБАГ в смеси бензол — метиловый
спирт. Для титрования 19,42 г «-аминобензоата диэтиллейцинолметансуль-
фоната требуется 1 мл 0,05 н. раствора [322].
117. ТИТРОВАНИЕ В КИСЛОТНЫХ РАСТВОРИТЕЛЯХ
Большинство солей органических кислот с щелочными металлами или
аминосоединениями можно титровать хлорной кислотой в уксусной кислоте
как ацетаты в присутствии кристаллического фиолетового или потенцио-
метрически [15, 48, 442, 548, 682, 732, 769].
Индикатор, содержащий производное дифениламина, целлитоновый
чисто синий FR (Cellitonechtblau FR) дает отчетливое изменение окраски
от голубой до краснофиолетовой; даже присутствие 4% воды совершенно
не влияет на отчетливость изменения окраски [473].
Около 3,5 мг-экв образца растворяют в 30 мл уксусной кислоты, если
необходимо, при осторожном нагревании и титруют 0,1 н. хлорной кислотой
в присутствии одной капли 1%-ного кристаллического фиолетового до появ-
ления сине-зеленой окраски. Этим способом, например, можно титровать
следующие соединения (в скобках приведено число миллиграмм-эквивален-
тов соли, соответствующее 1 мл раствора 0,1 н. концентрации): бензоат
аммония (13,92), салицилат аммония (15,52), ацетат натрия (8,204), салици-
лат натрия (16,01), бензоат натрия (14,41), ацетат калия (9,815), ацетат гуа-
нидина (11,91).
В некоторых случаях удобнее растворить образец в избытке стандарт-
ного раствора хлорной кислоты и затем провести обратное титрование ацетатом
натрия в уксусной кислоте [70]. В случае тартратов вследствие их плохой
растворимости лучшие результаты часто можно получить, используя раз-
бавленный раствор и 0,01 н. стандартный раствор [48]. Ацетаты цинка,
кобальта и марганца, которые обнаруживают слабый потенциометрический
скачок в конечной точке титрования в уксусной кислоте, дают удовлетво-
рительные результаты при определении конечных точек титрования в мети-
ловом спирте [667].
а. Определение п-аминосалицилата натрия. 40—50 мг «-аминосалицилата
натрия растворяют в 50 мл метилового спирта, нейтрализованного в при-
сутствии четырех капель тимолового синего, и титруют 0,05 н. хлорной
Определение солей карбоновых кислот
285
кислотой в диоксане до появления желтой окраски [124]. На титрование
10,56 мг n-аминосалицилата натрия требуется 1 мл 0,05 н. раствора. n-Амино-
салицилат натрия легко растворим в метиловом спирте, и аминогруппа
титруется хлорной кислотой.
В уксусной кислоте образуются ацетат-ионы и при потенциометрическом титровании
хлорной кислотой в уксусной кислоте можно наблюдать на кривой два перегиба, соот-
ветствующие ацетат-иону и аминогруппе (эквивалентный вес в этом случае соответствует
половине молекулярного веса). В уксусной кислоте, содержащей 10% уксусного анги-
дрида, расходуется 1 же хлорной кислоты, потому что первичные аминогруппы в этом
случае ацетилированы [732].
б. Определение тартрата натрия [70]. 90—160 мг образца растворяют
в 30 мг уксусной кислоты, добавляют точно 25,00 мл 0,1 н. хлорной кислоты
и перемешивают с помощью магнитной мешалки до растворения (5—10 мин).
Затем проводят обратное потенциометрическое титрование избытка хлорной
кислоты 0,1 н. раствором ацетата натрия в уксусной кислоте, используя
стеклянный электрод в качестве индикаторного и каломельный как элек-
трод сравнения. Точка эквивалентности точно определяется графическим
путем на основании зависимости AE7AV от V (см. гл. 12)
v 0/ [(25 мл N^ — {bNo)] х Е
а
где а — вес образца (г); Ъ — количество израсходованного стандартного
раствора ацетата натрия (А2 — его нормальность); Nx — нормальность
стандартной хлорной кислоты; Е — 0,1 г-экв титруемого соединения.
Тартрат натрия можно также определить визуальным титрованием в при-
сутствии двух капель 1 %-ного метилового фиолетового по изменению окра-
ски от зелено-синей до синей. Тартрат калия-натрия, гидротартрат калия,
ацетат калия, цитрат лития, бензоат натрия и салицилат натрия можно тит-
ровать таким же образом [70].
в. Дигидрат цитрата натрия в уксусной кислоте можно оттитровать
как потенциометрически, так и в присутствии индикатора орацетового синего
(Oracet blue) [49, 680].
г. Определение тартрата калия-натрия. 0,6 г KNaG4H6O6-4H2O раство-
ряют в 50 мл 0,1 н. хлорной кислоты в уксусной кислоте (тартрат калия-
натрия почти нерастворим в уксусной кислоте вследствие образования моно-
тартрата калия) при сильном пермешивании в течение 20 мин с помощью
магнитной мешалки. Затем проводят обратное титрование 0,1 н. раствором
ацетата калия в уксусной кислоте.
Приготовление стандартного раствора ацетата кцлия. 6,91 г карбоната
калия (предварительно высушенного при 120°) растворяют при осторожном
нагревании в 200—300 мл уксусной кислоты. После охлаждения разбав-
ляют до 1 л.
д. Определение натрийкарбоксиметилцеллюлозы [743]. Взвешивают около
500 мг натрийкарбоксиметилцеллюлозы и нагревают на водяной бане в тече-
ние 20 мин с 80 мл уксусной кислоты, затем охлаждают до комнатной тем-
пературы и титруют потенциометрически 0,1 н. хлорной кислотой в диокса-
не. Для титрования 2,299 мг соединения требуется 1 мл 0,1 н. раствора.
е. Определение бензоата натрия. Навеску приблизительно 150 мг бен-
зоата натрия помещают в колбу Эрленмейера емкостью 250 мл и растворяют
при нагревании в 150 мл уксусной кислоты, затем охлаждают, добавляют
10 мл нейтрализованного 3%-ного ацетата ртути(П) для того, чтобы стабили-
зовать индикатор. Индикатор: 0,5%-ный раствор жирового синего (Fett-
Ыан В, Ciba) в уксусной кислоте. Титруют 0,1 н. хлорной кислотой в уксус-
ной кислоте до перехода окраски индикатора сначала в фиолетовую, затем
в розовую. На титрование 14.41 мл бензоата натрия расходуется 1 мл стан-
дартного раствора. Результаты контрольных титрований без ацетата рту-
ти(П) дают 99,45%, а в присутствии ацетата ртути(П) — 100,01%.
286 Г лав a 22
Натриевую соль диэтил- или этилфенилбарбитуровой кислоты, п-аминосалицплат
натрия, салицилат натрия и растворимый сахарин (натриевая соль) можно титровать-
аналогичным способом, но без ацетата ртутп(П) [682].
ж. Определение растворимого сахарина [326]. Навеску около 200 мг
тонкоизмельченного образца растворяют в 30 мл уксусной кислоты и после-
довав ления одной капли 1%-ного кристаллического фиолетового титруют
0,1 н. хлорной кислотой до появления синей окраски. 1 мл стандартного
раствора эквивалентен 24,12 мг дигидрата натриевой соли или 18,32 мг
сахарина.
з. Т итриметрическое определение солей аци-формы нитросоединений.
например калиевой соли оди-формы нитрогексана (циклогексилнитроната
калия), см. работу [228].
118. ТИТРОВАНИЕ В СРЕДЕ ГЛИКОЛЯ (G — Н-СИСТЕМЫ)
Гликоли сольватируют соли карбоновых кислот со щелочными металлами
за счет образования водородных связей
R—С
'о •• • но —сн2
Na +
Изменение окраски индикатора происходит резче, если добавить к гликолю
изопропиловый спирт, хлороформ или бензиловый спирт.
Навеску приблизительно 0,5—0,8 мг-экв соли монофункциональной карбо-
новой кислоты со щелочным металлом растворяют в 5—10 мл этилен- или
пропиленгликоля при встряхивании и осторожном нагревании. Затем раз-
бавляют 5—10 мл изопропилового спирта, хлороформа или бензилового
спирта. Добавляют 3—5 капель 0,05%-ного метилового красного в качестве
индикатора и, продолжая перемешивание, медленно титруют 0,2 н. соляной
кислотой или хлорной кислотой в смеси пропиленгликоль — хлороформ
до тех пор, пока окраска раствора не изменится от желтой до розовой: изме-
нение очень четкое.
Этот метод можно использовать для титрования наряду с другими соеди-
нениями натриевых солей капроновой, лауриновой, олеиновой, стеариновой,
масляной, коричной, миндальной и бензойной кислот [621]. Определение
солей кислот более сильных, чем муравьиная (р/С 3,75), не дает точных
результатов.
119. ТИТРОВАНИЕ В ДИФФЕРЕНЦИРУЮЩИХ РАСТВОРИТЕЛЯХ
Ограниченная растворимость некоторых солей в уксусной1 кислоте, пиридине,
диметилформамиде и т. д. вызывает необходимость в других методах. Известно, что-
сильные, средней силы и слабые кислоты можно титровать в простых смесях в пириди-
не [161]. Если к концентрированному водному раствору соли карбоновой кислоты доба-
вить серную кислоту, то образуются сульфат металла и свободная карбоновая кислота.
Последнюю можно оттитровать в присутствии серной кислоты с помощью ТБАГ [162].
Cu(CH3COO)2 + H2SO4 (избыток) —> CuSO4 + 2CH3COOH.
0,5 мг-экв солп взвешивают в мерной колбе емкостью 100 мл и растворяют в смеси
1 мл воды и 2 мл 1 н. серной кислоты. Если необходимо, добавляют еще 3 мл воды, раз-
бавляют до метки пиридином (или ацетоном), энергпчно встряхивают и потенциометри-
чески титруют 50 мл прозрачного раствора 0,1 н. ТБАГ. В глухом опыте определяют
расход стандартного раствора на титрование растворителя с добавкой 2 мл серной кисло-
ты. Выделяющаяся карбоновая или слабая минеральная кислота вызывает появление-
дополнительного перегиба на кривой. Динатриевая соль фосфорной кислоты, фосфит
натрия, ацетаты меди(П) и никеля(1), гидрофталат калия и цитрат натрия можно титро-
вать в ацетоновом растворе с точностью 100 ± 0,4%. Точность определения бромида
Определение солей карбоновых кислот 287
натрия, бпхромата калия и кислого фосфата натрия в пиридине составляет 99 ± 0,3%.
Нитрат и нитрит натрия можно определить в смеси с точностью 99,7 ± 1,3% или 99 ± 2%
Крешков с сотр. [485] использовал как растворитель метилэтилкетон (обладающий
хорошими дифференцирующими свойствами) для титрования натриевых или калиевых
солей карбоновых кислот п для анализа смесей солей, таких, как салицилат калия —
бензоат калия, ацетат натрпя — салицилат калия. Смеси солей с соответствующими
кислотами, например олеат натрия — олеиновая кислота, антранилат натрия — антра-
ниловая кислота, бензоат калия — бензойная кислота и т. д., можно титровать анало-
гичным образом. Карбоновая кислота, однако, выделяется в свободном виде не только
при действии серной кислоты, но и 0,1 н. хлорной кислоты, причем полученную смесь
кислот титруют потенциометрически стандартным раствором гидроокиси тетраэтил-
аммония с использованием стеклянного и каломельного электродов.
Глава 23
ОПРЕДЕЛЕНИЕ АЗОТИСТЫХ
ОРГАНИЧЕСКИХ ОСНОВАНИЙ
Применение метода неводного титрования для определения сильных
и средней силы органических оснований, содержащих азот, связано с раз-
витием фармацевтической промышленности: стали необходимы анализ сме-
сей природных органических оснований (алкалоиды, гликоалкалоиды,
антибиотики) и количественный контроль за производственным процессом,
а также анализ синтетических терапевтических средств, содержащих раз-
личные типы основных групп. Сейчас можно определить, например, содер-
жание действующего начала в лекарственных препаратах, в которые входят
алкалоиды, проследить производственный процесс (экстракция) и проанали-
зировать конечный фармацевтический продукт методами объемного микро-
анализа. При этом точность результатов титрования в неводных средах
выше.
Те монофункциональные гетероциклические основания, перхлораты
которых плохо растворимы и константа диссоциации (в воде) не ниже 10-12,
можно точно оттитровать хлорной кислотой в уксусной кислоте. Перхлораты
алифатических и жирноароматических аминов более растворимы, и точно
можно определить только те из них, константа диссоциации которых
не меньше 10-11. Это, однако, уже предельное значение и в большинстве
случаев необходимо использовать методы потенциометрического титрова-
ния. В растворе 0,01 н. концентрации основания с константой диссоциации
10*10 можно титровать визуально в уксусной кислоте с точностью ±0,3%,
если выбраны благоприятные условия для эксперимента (ср. рис. 72).
Для более слабых оснований предпочтительно использовать смеси раствори-
телей, например смеси пропионовая кислота — хлорбензол, уксус-
ный ангидрид — нитрометан, уксусный ангидрид — бензол, уксусная кис-
лота —четыреххлористый углерод, нитробензол — уксусный ангидрид
(ср. табл. 14).
Микротитрование проводят 0,01—0,001 н. хлорной кислотой или 0,005 н.
тг-толуолсульфокислотой. Первую лучше всего готовить в смеси уксусной
или пропионовой кислот и четыреххлористого углерода (1 : 1). Добавляют
2 капли 0,02%-ного метилового фиолетового в хлорбензоле к 3—5 мл рас-
твора 0,03—0,05 мг-экв органического основания в пропионовой кислоте,
четыреххлористом углероде или хлорбензоле и титруют 0,01 н. хлорной кис-
лотой в центрифужной пробирке на 15—25 мл над белой подложкой, пока
фиолетовое окрашивание не перейдет в синее.
Все сказанное выше служит руководством при выборе возможного метода
определения азотистых органических оснований. Для упомянутых в этой
главе соединений характерно то, что их молекулы содержат один или больше
координационно ненасыщенных атомов азота. Основность зависит от присут-
ствия электроноакцепторных или электронодонорных заместителей, и сила
оснований может изменяться в различных растворителях (см. гл. 3 и 4).
Аминосоединения в виде солей можно титровать в уксусной кислоте как
основания в зависимости от их кислого компонента. Если соли образованы
органическими кислотами, то в отличие от солей галогеноводородных кислот
добавление ацетата ртути(П) для титрования основного компонента в уксус-
Определение азотистых органических оснований
289
ной кислоте не обязательно (как, например, при титровании малеата эрго-
метрина). В результате добавления ацетата ртути(П) образуется недиссоции-
рующий хлорид ртути(П) и свободное основание из галогеноводородных
солей (см. гл. 25).
Основания, растворимые в уксусной кислоте, растворяют в виде ацета-
тов или аддуктов оснований с уксусной кислотой. Успех титрования зависит
от того, остается ли основность соединения, содержащего остатки амино-
группы, неизмененной в растворе, а если уменьшается, то в значительной
ли степени (например, при ацетилировании) или же исчезает полностью
(R3NH+). В зависимоти от природы заместителя в этом случае можно варьи-
ровать растворитель и силу аниона кислоты.
Сила оснований, содержащих азот, увеличивается в уксусном ангидриде
[249, 288, 732]. Очевидно, его нельзя применять для аминов, способных
к ацетилированию, за исключением тех редких случаев, когда титрование
можно проводить при 0° (например, дифениламин [557]). Уксусный ангидрид
и смесь нитрометан — уксусный ангидрид относятся к числу растворителей,
пригодных для потенциометрического титрования тех слабых оснований,
константа диссоциации которых (в воде) меньше 10“12: дифениламин (р#
13,15), хиноксалин (~13,2), теобромин (13,32), кофеин (13,40), мочевина
(13,50), никотинамид (13,60), тиомочевина (14,96) и т. д.
Титрование хлорной кислотой неприменимо в случае большинства неорга-
нических катионов, за исключением Hg2+ и Си2+, так как в результате соль-
волиза они образуют ацетаты.
В заключение укажем некоторые соединения, которые нельзя титровать
хлорной кислотой в уксусной кислоте; это пиррол, N-алкилпирролы, индол,
карбазол, бензоксазол, пиразин, бензтриазол, ацетамид, фентиазин, изатин,
нитроанилины и др.
Потенциометрическое дифференцирующее титрование возможно для о-, м- и п-нитро-
анплинов как оснований, если в качестве растворителя использовать 2 М раствор нитро-
бензола в бензоле, к которому добавлена уксусная или трихлоруксусная кислота до
концентрации 2,5 молъ/л [590]. о-Нитроанилин можно титровать спектрофотометрически
при 505 нм хлорной кислотой в уксусной кислоте [484].
120. АЛИФАТИЧЕСКИЕ, ЖИРНО АРОМАТИЧЕСКИЕ, АРОМАТИЧЕСКИЕ
И ГИДРОАРОМАТИЧЕСКИЕ АЗОТИСТЫЕ ОСНОВАНИЯ
RNH2
Первичный
R2NH
Вторичный
R3N
Третичный
RNHNH2
Производные гидразина
•CH2NH2
рК 4.70
Анилин
рК 9,42
7 Н
Дифенилами
рК 13,15
Циклогексилами
рК 3,21
(N — основной атом азота, N — атом азота пониженной основности).
Уксусная кислота, содержащая 10—20% уксусного ангидрида, пригодна
как растворитель при титровании неацетилирующихся аминов. Если амины
способны ацетилироваться, уксусный ангидрид применять нельзя и в каче-
стве растворителей используют пропионовую кислоту, хлорбензол, диоксан,
ацетонитрил и смеси растворителей, но не содержащие уксусной кислоты
(ср. табл. 39, гл. 6, разд. 39).
Взвешивают 0,3—0,8 мг-экв монофункционального основания, раство-
ряют в 20—30 мл нейтрализованного растворителя и титруют 0,05—0,1 н.
хлорной кислотой в уксусной кислоте или диоксане; выбор индикатора опре-
290
Глава 23
деляется растворителем (относительно выбора индикатора см. гл. 13
и табл. 62, 63 и 65).
При потенциометрическом титровании используют пару электродов стек-
ло — каломель; для определения конечной точки рекомендуется нейтрали-
зовать растворитель после добавления подходящего индикатора. Относи-
тельно поправки на температурный эффект см. гл. 9.
Если анализируются летучие жидкие основания, лучше сначала пригото-
вить раствор в бензоле, толуоле или (что предпочтительно) в хлорбензоле
и затем смешать адекватную часть с уксусной или пропионовой кислотой
или уксусным ангидридом. Таким образом можно снизить увеличение тем-
пературы, вызываемое взаимодействием между аминами и кислотным рас-
творителем и сопровождающееся потерей летучего основания. Для раство-
ров 0,01 н. концентрации подобная предосторожность не нужна. Основания,
плохо растворимые в хлорбензоле, можно растворить в смеси, содержащей
уксусную кислоту.
В случае бифункциональных оснований после протонирования первой
аминогруппы способность акцептировать протоны у второй группы в некото-
рой степени уменьшается. Однако при потенциометрическом титровании,
например в ацетонитриле, могут наблюдаться два скачка и изменение окра-
ски кристаллического фиолетового происходит иногда после добавления
1 или 2 экв хлорной кислоты. На кривой потенциометрического титрования
о-фенилендиамина наблюдается два скачка, а на кривых м- и п-фениленди-
аминов — один скачок, соответствующий двум эквивалентам хлорной кис-
лоты [732].
При анализе N-аминопиперидина образующаяся при протонировании
группа NH' благодаря сильному индукционному эффекту (—/) уменьшает
электронную плотность на соседнем атоме азота, так что он уже не способен
акцептировать протон [732]. Поэтому потенциометрическое титрование
дает только один скачок, указывающий на связывание 1 экв хлорной кислоты.
В уксусной кислоте, содержащей уксусный ангидрид, титруется только азот
пиперидинового кольца, так как первичная аминогруппа ацетилируется.
Пиперидин — сильное основание (рК 2,80), однако замещение при атоме азота ведет
к образованию менее сильного основания [333]: N-этилпиперидин (р/С 3,60), N-аллил-
пиперидин (рА^ 4,32).
Хлорной кислотой в уксусной кислоте можно титровать многочисленные
алифатические, ароматические и другие основания [72, 175, 244, 345, 374,
548, 599, 732, 802]. Перечислять их все не имеет смысла, и мы приведем толь-
ко некоторые типичные примеры.
а. Определение анилина. Взвешивают тонкостенную широкогорлую ампу-
лу для инъекции на 1—2 мл, вносят в нее по каплям 0,4—0,5 мл анилина
и снова взвешивают. Наливают 80—85 мл хлорбензола в колбу из прочного
стекла емкостью 200 мл и нейтрализуют в присутствии 5 капель 1%-ного
раствора кристаллического фиолетового 0,1 н. хлорной кислотой до появ-
ления сине-зеленого окрашивания. Ампулу с анилином помещают в раствор
и разбивают стеклянной палочкой. Переносят раствор через воронку в мер-
ную колбу на 100 мл, обмывают колбу уксусной кислотой (5x2 мл) и напол-
няют мерную колбу до метки. Полученный раствор порциями по 20 мл
титруют 0,1 н. хлорной кислотой в уксусной кислоте до появления сине-
зеленого окрашивания. 9,312 мл анилина требуют 1 мл стандартного рас-
твора.
О высокочастотном титровании замещенных производных анилина (п-анизидина,
и-толуидина, хлоранилина и n-аминоацетофенона) см. в работе [515].
б. Фотометрическое титрование о-хлоранилина [675]. Приготавливают
100 мл примерно 0,1 н. раствора о-хлоранилина в уксусной кислоте, поме-
щают его в специальную кювету спектрофотометра типа Бекман DU и тит-
Определение азотистых органических оснований
291
руют при длине волны 312 нм 0,1 н. хлорной кислотой. Наносят на график
зависимость поглощения от расхода титранта с учетом поправки на измене-
ние объема для каждого значения. На титрование 12,76 мг о-хлоранилина
расходуется 1 мл 0,1 н. хлорной кислоты (рис. 86, гл. 14 и рис. 96).
в. Определение трибензиламина. 140—160 мг образца растворяют в мер-
ной колбе емкостью 100 мл в смеси уксусная кислота — уксусный ангидрид
(10 : 1), нейтрализованной в присутствии кристаллического фиолетового.
Порциями по 10 мл раствор титруют 0,01 н. хлорной кислотой до перехода
фиолетовой окраски в синюю. 2,874 мг трибензиламина требуют 1 мл 0,01 н.
хлорной кислоты.
г. Определение циклогексиламина. В 100 мл пропионовой кислоты, ней-
трализованной в присутствии 6—8 капель метанилового желтого, раство-
ряют 0,5—0,6 мл циклогексиламина, как описано выше (а), и порциями
по 20 мл титруют 0,1 н. хлорной кислотой. Конечную точку титрования
определяют либо потенциометрически, используя пару электродов стекло —
каломель, либо визуально по изменению окраски метанилового желтого
до пурпурного. На 9,917 мг циклогексиламина расходуется 1 мл 0,1 н. хлор-
ной кислоты.
д. Фотометрическое титрование 1-нафтиламина (рК 10,08) и 2-нафтил-
амина (рК 9,89) проводят так же, как и определение нарцеина [209], см.
рис. 77, гл. 14 и 23, разд. 121, 1.
Определение N-нитрозодифениламина может быть выполнено обратным
титрованием с помощью 1-нафтиламина [558]. Порцию раствора, содержащую
около 10 мг N-нитрозодифениламина, растворяют в 10 мл уксусной кислоты
и смешивают с 75 мг 1-нафтиламина в 15 мл уксусной кислоты. Далее добав-
ляют 75 мл уксусной кислоты и, защищая от света, нагревают 30 мин
на водяной бане при 60°, охлаждают в темноте и оттитровывают избыток
1-нафтиламина 0,03 н. хлорной кислотой в уксусной кислоте. Глухой опыт
проводят так же. Титрование проводят потенциометрически, применяя
систему электродов стекло — Ag/AgCl. Для вычислений используют
разность двух титрований. 1 мл 0,03 н. хлорной кислоты эквивалентен
5,946 мг нитрозодифениламина и 4,296 мг 1-нафтиламина.
е. Определение аминокислот. Чтобы приготовить 0,1 н. раствор амино-
кислоты в уксусной кислоте, компоненты смеси необходимо встряхивать,
иногда в течение нескольких часов. Скорость растворения можно увеличить
нагреванием. Для растворения аминокислот используют также смесь кислот
муравьиная — уксусная (иногда солюбилизирующуюся от избытка хлорной
кислоты); избыток стандартного раствора оттитровывают раствором глици-
на, ацетата натрия, дифенилгуанидина или гуанидинацетата в уксусной кисло-
те в присутствии кристаллического фиолетового или 1-нафтолбензеина.
Применимость метода в каждом отдельном случае определяется раствори-
мостью аминокислот. Потенциометрически можно оттитровать непосред-
ственно 0,1 н. хлорной кислотой в уксусной кислоте следующие аминокис-
лоты (в скобках указано количество вещества (в миллиграммах), эквива-
лентное 1мл 0,1 н. раствора): аспарагин (13,21); антраниловая кислота
(13,71); n-аминобензойная кислота (13,71); глицин (7,507); d-валин (11,72),
изолейцин (13,12); Z-пролин (11,51); /-триптофан (20,42). Некоторые амино-
кислоты сначала растворяют в 1—2 мл муравьиной кислоты, затем разба-
вляют 20 мл уксусной кислоты и титруют как описано выше. Так титруют
d-глутаминовую кислоту (14,71), Z-аспарагиновую кислоту (13,31), /-цистин
(24,03), d,/-валин (11,72) и d,/-фенилаланин (16,52). Аспарагиновую и глут-
аминовую кислоты (14,71), цистин и аланин (8,909), /-серин (10,51), /-лей-
цин (13,12) и /-тирозин (18,12) растворяют (приблизительно 0,8 мг-экв)
при небольшом нагревании в 12,00 мл 0,1 н. хлорной кислоты и обратным
титрованием 0,1 н. гуанидином в уксусной кислоте в присутствии индикатора
1-нафтолбензеина [607, 732, 809] определяют избыток стандартного раствора.
292
Глава 23
Осаждение нерастворимого перхлората часто благоприятно влияет
на визуальное определение с индикатором. Осадок выпадает при добавлении
бензола или четыреххлористого углерода в раствор аминокислот в уксусной
кислоте [575]. При этом n-аминобензойную и антраниловую кислоты можно
титровать 0,1 н. хлорной кислотой в уксусной кислоте в присутствии
1-нафтолбензеина. На 1 ммоль аминокислоты берут 10 мл уксусной кислоты
и после добавления индикатора разбавляют 25 мл бензола или четыреххло-
ристого углерода. На 1 ммоль антраниловой кислоты берут 5 мл уксусной
кислоты и разбавляют 40 мл бензола или четыреххлористого углерода.
Известен еще один метод: к раствору добавляют небольшой избыток хлорной
кислоты и через 30 мин оттитровывают избыток ее с помощью стандартного
раствора, содержащего ацетат натрия или дифенилгуанидин.
ж. Определение N,N'-дизамещенных производных п-фенилендиамина. N-3a-
мещенные производные n-фенилендиамина в зависимости от заместителя
можно титровать потенциометрически в уксусной кислоте, ацетонитриле или
ацетоне. Если 1У,]\т'-диалкил-п-фенилендиамин титруют в уксусной кислоте,
то наблюдается один скачок: протонируются одновременно два атома азота;
при титровании в ацетонитриле получают два скачка для различных атомов
азота. При титровании в ацетоне наблюдается один потенциометрический
скачок только для одного атома азота; следовательно, после присоединения
первого протона основность молекулы понижается в такой степени, что
дальнейшее протонирование невозможно [521].
В перечисленных растворителях 1М-алкил-]У'-арил-п-фенилендиаминовые
производные реагируют только с 1 экв хлорной кислоты.
Производные 1Ч,К'-диалкил-(а), ?»1-алкил-]У'-арил-(б), Х,К'-диарил-п-фе-
нилендиамина (в), используемые в резиновой промышленности в качестве
антиоксидантов, могут быть определены титрованием в неводной среде.
20—100 мг антиоксидантов типа а и б растворяют в 50 мл растворителя
и титруют потенциометрически 0,1 н. хлорной кислотой в уксусной кислоте,
используя стеклянный электрод в качестве индикаторного и Ag/AgCl в каче-
стве электрода сравнения (гл. 12, рис. 62—64). Титрование антиоксидантов
типа в проводят следующим образом. К раствору образца в 50 мл уксусной
кислоты добавляют примерно 2 экв хлоранила (тетрахлорхинона), при этом
раствор окрашивается в темно-синий цвет, и затем титруют. К,Ат'-Диарил-п-
фенилендиаминовые производные — очень слабые основания и при добавле-
нии хлоранила они окисляются в так называемые соли Вюрстера, при этом
образуется сильное основание ацетат-ион или анион тетрахлоргидрохинона.
.. .. СНзСООН
2R _ HN — С6Н4 — NH — R + С6С14( = О)2 + 2Н+--
—> 2R — HN — C6H4-NH— R + C6C14(OH)2
121. ГЕТЕРОЦИКЛИЧЕСКИЕ ОСНОВАНИЯ
Многие используемые в медицине природные или синтетические соеди-
нения содержат гетероциклический атом азота.
Во многих соединениях гетероатом азота не обладает основными свой-
ствами (как, например, в фентиазине), но если молекула также содержит
обычный первичный, вторичный или третичный атом азота, то последний
подвергается титрованию. В таких соединениях, содержащих гетероцикли-
ческий азот и аминогруппы при использовании визуального индикатора
в уксусной кислоте количество расходуемой хлорной кислоты иногда экви-
валентно только содержанию аминогрупп, поскольку основность гетеро-
атома азота очень низка (например, в амидопирине, 3-аминопиразолоне,
диметиламиноацетилфентиазине) или гетероатом азота совсем не обладает
основными свойствами (например, в циклосерине, d-4-аминоизоксазолидино-
Определение азотистых органических оснований
293
не-4). В уксусной кислоте, содержащей 10% уксусного ангидрида, амидо-
пирин дает два потенциометрических скачка и требует 2 экв хлорной кислоты
[732], в то время как феназон (антипирин, 2,3-диметил-1-фенил-5-пиразолон)
присоединяет только один протон.
СбН5
ОС 5 1 2 N-снз
I 4 3 |
RC=C-------СН3
Феназон (антипирин) R=H
Амидопирин R= (СНз)2Ы
Н,С NH
I I
НС-------СО
I
nh2
Циклосерин
Однако в других соединениях, например 4-аминопиридине и 2-амино-
пиридине, гетероатом азота обладает основными свойствами, а в 3-амино-
пиридине оба N-атома имеют основной характер [262] (см. также гл. 4,
разд. 27, а).
NH2
4--*Аминопиридин 2'Аминопиридин З-Амииопиридин
В N-аминопиперидине основными свойствами обладает N-атом амино-
группы, а в N-ацетиламинопиперидине — N-гетероатом [732]
N- Ацетиламинопиперидии
N- Аминопиперидин
Как правило, основность второго атома азота уменьшается или совсем
исчезает в соединениях, содержащих два гетероциклических N-атома,
например в пиримидине, имидазоле, пиразоле (а также гидразинах), вслед-
Г 1 1 +
ствие индукционного или мезомерного эффекта группы —NH , образую-
щейся при протонировании (см. также гл. 4)
Пиримидин
I
Н
Пиразол
Н
Имидазол
Пиразолон-5
294
Глава 23
а. Определение пиридина и родственных соединений. В зависимости
от природы соединения анализ его проводят методом, описанным для анили-
на и N-аминопиперидина, или же раствор готовят непосредственным взве-
шиванием без предварительного приготовления раствора. Хорошим раство-
рителем как для акридина, так и для пиридина, алкилпиридинов, хинолина,
изохинолина и их алкильных производных является нейтрализованная
смесь хлорбензол — уксусная кислота (1 : 1) [175]. Конечную точку опре-
деляют или потенциометрически или визуально с кристаллическим фиолето-
вым или 1-нафтолбензеином (см. табл. 91).
Таблица 91
Соединение Количество вещества, эквивалент- ное 1 мл 0,1 н. НС1О4, мг рК Литература
Пиридин 7,910 8,85 497
Р-Пиколин (3-метил- 9.312 7,96 175
пиридин) 2,6-Лутидин 10,72 6,58 333
у-Коллидин (2,4,6-трп- 12,12 6,68 175
метилпиридин) Хинолин 12,92 9,20 497
Изохинолин 12,92 8,70 779
Хинальдин (2-метилхи- 14,32
нолин) Акридин 17,92 8,40 2,3
б. Определение пиридинкарбоновой кислоты и (или) фенилхинолин-4-кар-
боновой кислоты. Потенциометрическое титрование пиридинкарбоновой
кислоты, никотинамида и фенилхинолинкарбоновой кислоты рассматри-
вается в ряде работ [249, 437, 442, 732]. Так, выяснено, что в уксусной кис-
лоте, содержащей 10% уксусного ангидрида, можно титровать следующие
соединения [732] (в скобках указано количество вещества в миллиграммах,
эквивалентное 1 мл 0,1 н. хлорной кислоты): пиридин-2-, пиридин-3- и пири-
дин-4-карбоновые кислоты (12,31), никотинамид (12,21), диизоникотинил-п-
фенилендиамин (15,91), изоникотиназид (14,82), N-изоникотиниламинопипе-
ридин (10,62), диизоникотинилгидразид (12,11) и изоникотинилгидразоны
пировиноградной кислоты, ацетиламиноацетофенона, циклогексанона и цикло-
гептанона. Никотинамид можно также титровать визуально в присутствии
индикатора трифенилметанового ряда в смеси нитрометан — уксусный
ангидрид (4 : 1) [249] (см. разд. 125).
В смеси уксусная кислота — хлорбензол (10 : 1) в качестве индикатора
можно использовать 5 капель 0,05%-ного раствора метилового фиолетового.
Во время титрования иногда выпадает осадок кристаллического перхлората;
это увеличивает резкость изменения окраски индикатора. Выпадение осадка
наблюдается при титровании никотинамида и фенилхинолинкарбоновой
кислоты; в последнем случае 1 мл 0,1 н. хлорной кислоты соответствует
~ 24,93 мг анализируемого вещества. Никотинамид можно также титровать
фотометрически 0,1 н. хлорной кислотой в уксусной кислоте с малахитовым
зеленым в качестве индикатора при длине волны 620 нм [65].
в. Определение N-аминопиперидина. 0,4—0,5 мл взвешивают так, как
это описано в методике определения анилина; поскольку это соединение
гигроскопично, его запаивают в ампулу (см. разд. 120, а) (отпаянную часть
взвешивают вместе с ампулой), чтобы во время измерения не абсорбирова-
лась влага. Ампулу помещают в колбу Эрленмейера с притертой стеклянной
пробкой, наливают туда смесь 10 мл уксусного ангидрида и 20 мл уксусной
кислоты и разбивают ампулу стеклянной палочкой. Смачивают пробку
колбы Эрленмейера уксусным ангидридом и оставляют в темном месте
Определение азотистых органических оснований
295
на 30 мин для ацетилирования первичных аминогрупп. После этого перено-
сят раствор в мерную колбу емкостью 100 мл, ополаскивают колбу Эрлен-
мейера уксусной кислотой и доводят объем раствора до 100 мл. Полученный
раствор порциями по 25 мл титруют 0,1 н. хлорной кислотой, используя стек-
лянный и каломельный электроды. При визуальном определении конечной точ-
ки титрование ведут в присутствии крезолового красного в качестве индика-
тора. 10,02 мг N-аминопиперидина эквивалентны 1 мл 0,1 н. хлорной кислоты.
г. Определение хлорхинолиновых производных’, фотометрическое титро-
вание 8-хлорхинолина [404]. 100—110 мл раствора уксусной кислоты, содер-
жащей около 0,8 мг-экв 8-хлорхинолина (рК 11,2), помещают ячейку для
титрования на 150 мл (Wycor grade 7910, Corning Glass, Works, Corning, N. Y.)
и закрепляют ее в держателе для кювет толщиной 10 см спектрофотометра
типа Бекман DU. Измерения проводят при длине волны 380 нм (гл. 14,
табл. 73). Поскольку в процессе титрования поглощение усиливается, прежде
чем добавлять стандартный раствор, устанавливают стрелку прибора на
нулевую отметку. В начале титрования добавляют 0,5 н. хлорную кислоту
порциями по 0,2 мл, а вблизи конечной точки — порциями по 0,05—0,1 мл.
Фотометрическую кривую строят на графике как функцию А от vlf, где / —
израсходованный объем стандартного раствора. Проекция точки пересече-
ния прямых линий на ось х указывает на израсходованное количество рас-
твора (в миллилитрах), т. е. на эквивалентную точку (ср. рис. 75). 81,8 мг
8-хлорхинолина требуют 1 мл 0,5 н. хлорной кислоты.
Определение 5,7-дихлор-8-оксихиналъдина (хлорхиналъдолъ) *. J3 широко-
горлой колбе объемом 200 мл взвешивают примерно 300 мг образца и раство-
ряют в смеси 25,00 мл 0,1 н. НС1О4, 25 мл уксусной кислоты и 2 мл уксусного
ангидрида, осторожно нагревая на водяной бане. Колбу накрывают неболь-
шим часовым стеклом и большим химическим стаканом. После охлаждения
оттитровывают избыток хлорной кислоты 0,1 н. раствором ацетата калия
в уксусной кислоте, добавив 1 каплю индикатора 0,5 %-ного раствора мала-
хитового зеленого. Первоначально зелено-желтая окраска раствора в точке
эквивалентности переходит в желто-зеленую. 22,81 мг хлорхинальдоля
эквивалентны 1 мл 0,1 н. стандартного раствора. 5,7-Дихлор-8-оксихинолин
можно титровать в уксусном ангидриде [620].
Определение производных пиразолона. Вайбель и сотр. [839—841] иссле-
довали влияние заместителей у атомов Ni, С3 и С4 пиразолонового кольца
и объяснили основные свойства пиразолонов типа феназона (структура анти-
пирина) вкладом фенолбетаиновой резонансной структуры
Структура
фенолбетаина
Drug. Std., 26, 106 (1958).
296
Глава 23
На титрование пиразолона идет 1 экв хлорной кислоты, но если у С3 углеводо-
родный радикал замещен на аминогруппу, то расход хлорной кислоты уве-
личивается, что, вероятно, связано с тем, что структура 3-аминопиразолона
подобна структуре гуанидина
I II
н2с—с—nh2
R
I
N
ос \н / nh2
II I
Н2С — G = NH \ср. Н2.\—C --NH
Гуанидин
Кривые потенциометрического титрования указывают на следующий
порядок основности производных фенилпиразолона (в уксусной кисло-
те) [445]:
4-амидоантипирин >> 1-фенил-З-метилпиразолон >> антипирин Э> амидопирин
В работе [126] рассматривается титрование амидопирина (рЛГ9,30) в невод-
ной среде. Титрование можно проводить потенциометрически в уксусной
кислоте, содержащей 10% уксусного ангидрида [732], а также в уксусной
кислоте в присутствии диэтилбарбитуровой кислоты [13]. Амидопирин
можно оттитровать в хлороформе в присутствии диэтилбарбитуровой кисло-
ты 0,05 н. n-толуолсульфокислотой в хлороформе [649]. В хлорбензоле его
можно определять как потенциометрически, так и визуально с бромфеноло-
вым синим в качестве индикатора [638], а в смеси бензол—уксусная кислота
(3:1) амидопирин титруют потенциометрически или визуально в присут-
ствии индикатора тропеолина 00 [496].
Ни аналоги кислот, ни хлоргидраты оснований, подобные, например,
этилфенилбарбитуровой кислоте, бромизовалерилмочевине, кофеину или
эфедрину, не мешают определению амидопирина в уксусной кислоте
[695].
Взвешивают 200—250 мг амидопирина и растворяют при небольшом
нагревании в 20 мл смеси бензол — уксусная кислота (3 : 1), нейтрализован-
ной в присутствии двух капель 1%-ного раствора тропеолина ОО в уксус-
ной кислоте. Титруют 0,1 н. хлорной кислотой в уксусной кислоте до розо-
вого окрашивания; 23,13 мг амидопирина эквивалентны 1 мл стандартного
раствора.
Определения амидопирина, хлоргидрата папаверина, фенил этил барбитуровой
кислоты и оксихинолинсульфокислоты в фармацевтических препаратах описано в ра-
ботах [835 и 837].
е. Определение 2,4-диамино-5-(4'-хлорфенил')-6-этилпиримидина (пиримет-
амин) *. 200мг образца нагревают со 100 мл ацетона на водяной бане, остав-
ляют раствор на 1—2 мин, затем охлаждают и после добавления 1 мл мети-
лового оранжевого в ацетоне титруют 0,1 н. хлорной кислотой в диоксане
до появления розовой окраски. На титрование 24,87 мг пириметамина рас-
ходуется 1 мл 0,1 н. раствора.
ж. Определение меламина в меламиновых смолах [461]. Содержащийся
в меламиновой смоле меламин (2,4,6-триамино-1,3,5-триазин) можно титро-
вать непосредственно хлорной кислотой. Меламин ведет себя как монофунк-
Апп. pharm. franc., 20, 86 (1962).
Определение азотистых органических оснований
297
циональное основание.
Н Н
I
С
/ ч
N N
:i ।
н с с н
I I
H H
Содержание азотистых оснований в различных промышленных смолах можно опре-
делить с хорошей точностью, если его вычислять по результатам титрования в неводной
среде, результаты определения сравнимы с результатами определения по методу Кьельдаля.
Взвешивают точно 0,5 г раствора смолы, содержащего 50—70% сухого
вещества, и растворяют в 20 мл смеси уксусная кислота — уксусный ангид-
рид (4 : 1). Титруют 0,1 н. хлорной кислотой в уксусной кислоте в присут-
ствии кристаллического фиолетового до желтого окрашивания индикатора.
„ „ с-N-12,6-100 а
Содержание меламина в твердой смоле =-------------%,
где а — вес раствора смолы, a; N — концентрация стандартного раствора,
г-экв/л’, с — израсходованный объем кислоты, мл: d — содержание в растворе
смолы сухого вещества, %.
з. Определение 4-амино-З-изоксазолидинона (циклосерина) [698]. 50—
60 мг образца растворяют в 25 мл уксусной кислоты, нейтрализованной
в присутствии 2 капель 0,2%-ного раствора кристаллического фиолетового,
и титруют потенциометрически 0,05 н. хлорной кислотой в уксусной кисло-
те, используя платиновый электрод в качестве индикаторного, а графитовый
электрод, погруженный в насыщенный раствор ацетата меди (II), в качестве
электрода сравнения (графитовый электрод Зайдлера см. гл. 12, разд. 67
и 68). 5,10 мг циклосерина эквивалентны 1 мл 0,05 н. раствора кислоты.
и. Бензимидазол и его производные можно титровать в пропионовой кис-
лоте 0,1 н. раствором хлорной кислоты в пропионовой кислоте в присутствии
индикатора фенацилового желтого [377], а бензтиазол и хиноксалин можно
титровать потенциометрически в смеси нитрометан — уксусный ангидрид
(4 : 1) стандартным раствором хлорной кислоты" в уксусной кислоте." 1 мл
0,1 н. раствора эквивалентен 11,81 мг бензимидазола, 13,52 мг бензтиазола
и 13,01 мг хиноксалина (см. также разд. 125 этой главы)
Бензимидазол Бензтиазол Хиноксалин
к. Акридин можно титровать потенциометрически в уксусной кислоте
(см. также разд. 121, а). 30—180 мг акридина растворяют в таком количе-
стве уксусной кислоты, чтобы получить приблизительно 0,1 М раствор,
добавляют 0,6 г хлоранила и 0,8 г тетрахлоргидрохинона и титруют 0,1 н.
хлорной кислотой, используя платиновый и каломельный электроды. Элек-
тролит каломельного электрода — уксусная кислота, насыщенная нитратом-
лития [608]. 17,92 мг акридина требуют 1 мл 0,1 и. стандартного раствора.
298
Глава 23
л. Титрование алкалоидов в присутствии малахитового зеленого как
индикатора [822]. Титрование хлорной кислотой кодеина (р/£ 6,05), крипто-
пина, морфина (рК 6,17), тебаина (рК 6,05), наркотина (рК 7,82) и папавери-
на (рК 8,10) лучше проводить в присутствии малахитового зеленого, а не
кристаллического фиолетового, так как при его использовании результаты
определения более близки к данным потенциометрического титрования.
250—350 мг алкалоида растворяют в 100 мл уксусной кислоты и после
добавления 1 мл 0,001 М раствора малахитового зеленого титруют 0,05 н.
хлорной кислотой в уксусной кислоте. Вблизи конечной точки титрования
титрант добавляют по каплям до тех пор, пока синяя окраска не перейдет
в зеленую. Чтобы определить суммарный расход стандартного раствора
растворителем и индикатором, растворяют 100 мг перхлората натрия в 100 мл
уксусной кислоты и титруют в присутствии 1 мл раствора индикатора до
зеленого окрашивания.
1. Определение нарцеина с фотометрическим определением конечной
точки [209]. Взвешивают 250—500 мг нарцеина (рК 10,7) в 20 мл уксусной
кислоты, добавляют 2 капли 0,5 %-ного раствора метиленового синего в уксус-
ной кислоте и помещают смесь в кювету фотометра толщиной 15 мм (напри-
мер, фотометр модель Ланге IV). Раствор титруют 0,1 н. хлорной кислотой
в уксусной кислоте при длине волны 550—580 нм, используя цветные филь-
тры типа OG-2. Наносят на график расход стандартного раствора как
функцию от 1/Т. Величина 1/Т в эквивалентной точке резко возрастает
(ср. рис. 76, кривая Б). 54,55 мг нарцеина эквивалентны 1 мл 0,1 н. стан-
дартного раствора кислоты.
2. Методом потенциометрического титрования в пропионовой кислоте
можно анализировать следующие алкалоиды, имеющие важное значение
в фармацевтической промышленности: бруцин, кодеин, нарцеин, наркотин,
хинин, вератрин [663].
122. ПРОИЗВОДНЫЕ ФЕНТИАЗИНА (ТАБЛ. 92)
При определении производных фентиазина следует избегать применения
растворителей, содержащих окислители, и в частности перекиси (например,
диоксан). Наряду с ацетоном, ацетонитрилом и хлорбензолом используют
такие смеси растворителей, как 1,2-дихлорэтан — уксусная кислота, бен-
зол—нитрометан или гексан — ацетон.
а. Титрование в смеси 1,2-дихлорэтан — уксусная кислота [130]. 150—
200 мг хлорпромазина, прохлорперазина или прометазина растворяют
в 30 мл уксусной кислоты, содержащей 10% уксусного ангидрида, добавляют
60 мл дихлорэтана и титруют 0,1 н. хлорной кислотой в уксусной кислоте
в присутствии индикатора тропеолина ОО.
б. Титрование в ацетонитриле или хлорбензоле [795]. 150—250 мг
N-диметиламиноацетилфентиазина (агистан) растворяют в 100 мл раство-
рителя, нейтрализованного в присутствии метилового фиолетового или
тропеолина ОО, и порциями по 10—20 мл титруют 0,01 н. хлорной кислотой.
2,844 мг диметиламиноацетилфентиазина требуют 1 мл 0,01 н. хлорной
кислоты.
в. Титрование бифункциональных производных фентиазина в смеси
бензол — нитрометан [586]. 50 мг прохлорперазина, тиопропазата или
перфеназина растворяют в 150 мл смеси бензол — нитрометан (2 : 1) и тит-
руют в присутствии 3 капель 1%-ного тропеолина ОО 0,05 н. хлорной кис-
лотой в диоксане до появления розово-фиолетовой окраски. Если анализи-
руемый образец представляет собой дималеат или дихлоргидрат основания,
чтобы воспрепятствовать образованию эмульсии, водный раствор насы-
щают сульфатом натрия; соль переводят в основание, добавляя минимально
необходимое количество едкого натра и экстрагируют (6 х 15 мл} бензолом.
Определение азотистых органических оснований
299
Таблица 92
Соединение Производные фентиазина а 1 Z Молеку- лярный вес
X Y Z
1. Хлорпромазин (хлоргидрат) н С1 — CH2CH2CH2N(CH3)2 318,9 355,3
2. Прометазин (хлоргидрат) н н -CH2CH(CH3)N(CH3)2 284,4 320,9
3. Прохлорперазин (дпхлоргидрат) н С1 CH2CHy-N^~H~^N—СН3 373,9 476,9
Прохлорперазин (дималеат) 606,1
4. Агистан и -COCH2N(CH3)2 284,4
5. Тиопропазат (дпхлоргидрат) н С1 ~(СН2)3—n^~h~^n—СН2СН2ООССН3 446,0 518,9
6. Перфеназин (дпхлоргидрат) н С1 —(CH2)3-N^H^N—сн2сн2он 403,9 476,9
7. Пеказин (хлоргидрат) н н — CH2C5H9NCH3 (N-метилпиперидил) 310,4 346,9
3. Трифторпромазпн (хлоргидрат) н CF3 - CH2CH2CH2N(CH3)2 352,4 388,9
9. Ацепромазпн (хлоргидрат) Ацепромазин (малеат) н ОССНз — CH2CH2CH2N(CH3)2 326,5 362,9 442,5
а Величины рК летучих производных фентиазина приведены в работе [125].
Добавляют 40—50 мл нитрометана для полноты экстракции и титруют как
указано выше. Относительно определения малеата прохлорперазина см.
также разд. 123 этой главы.
Кислый компонент вышеупомянутых транквилизаторов можно определять
в хлороформе титрованием едким кали в метиловом спирте или метилатом
калия в диметилформамиде; в обоих случаях можно применять как индикатор
тимоловый синий [586].
Все три указанных производных фентиазина содержат пиперазиновое
кольцо; следовательно, их можно титровать как бифункциональные основа-
ния. Хлоргидраты пеказина и трифторпромазина и малеат ацепромазина,
содержащие только один титруемый атом азота, ведут себя как монофункцио-
нальные основания.
г. Титрование монофункциональных производных фентиазина в ацетоне
или смеси гексан — ацетон [586]. 70 мг производного фентиазина растворяют
в 50 мл ацетона. Хлоргидрат переводят в свободные основания действием
300
Глава 23
0,5 мл 6%-ного раствора ацетата ртути(П) в уксусной кислоте и титруют
0,05 н. хлорной кислотой в диоксане в присутствии 2 капель 1%-ного диме-
тилового желтого в хлороформе до перехода желтой окраски в оранжевую.
Титрование малеата ацепромазина проводят в присутствии 2 капель 5%-ного
индикатора; оранжево-желтая окраска при этом переходит в оран-
жево-красную. Из водного раствора хлоргидрата пеказин экстрагируют
(4 х 20 мл) гексаном (см. разд, в); объединенные вытяжки разбавляют 40 мл
ацетона и титруют, как указано.
д. Определение хлоргидрата хлорпромазина экстракцией [587]. Примерно
50 мг хлоргидрата 10-(3-диметиламинопропил)-2-хлорфентиазина помещают
в делительную воронку, растворяют в 20 мл дистиллированной воды, пере-
водят в основание действием едкого кали и экстрагируют (4 х 20 мл) гек-
саном. Объединенные вытяжки смешивают с 40 мл ацетона и титруют 0,05 н.
хлорной кислотой в диоксане в присутствии 3 капель 0,1%-ного метилового
красного в уксусной кислоте, пока оранжево-желтая окраска индикатора
не перейдет в розовую. Из двух третичных аминогрупп можно оттитровать
только ту, которая находится в алкильной группе. Это определение можно
провести потенциометрически, используя систему электродов стекло —
каломель (рис. 44, а, гл. 12). См. также гл. 24, разд. 13 в, г.
123. ПРОИЗВОДНЫЕ ПИПЕРАЗИНА
а. Основность пиперазина и его производных. Рассмотрим влияние метиль-
ной группы на основность производных пиперазина и (или) пиразина
(1,4-диазина) [454]
Пиразин (1,4-диазин)
Пиперазин (гексагидро-1,4-диазин)
Метильная группа снижает основность пиперазина, особенно если заме-
ститель связан с атомом азота. Сила основания удваивается при введении
метильной группы на атом углерода пиразина в положение 2, увеличивается
в 10 раз при введении второй метильной группы в положение 5 или в 30 раз —
в положение 6. Метильные группы, находящиеся во всех четырех возможных
положениях, увеличивают основность соединения в 50 раз по сравнению
с самим пиразином. При титровании хлорной кислотой протонируется один
N-атом [454].
Пиперазин рК\ 3,97 и рТС2 8,34; 2-метилпиперазин 4,10 и 8,54; 2,5-диметилпипера-
зин: ^ис-изомер 4,02 и 8,77; mpawc-изомер 4,16 и 8,66; 2,3,5,6-тетраметилпиперазин
4,06 и 8,89; 1,2,4-триметилпиперазин 5,64 и 10,06.
Пиразин рК 12,9; 2-метилпиразпн 12,5; 2,5-диметилпиразин 11,9; 2,3,5,6-триметил-
пиразин 11,2.
1,4-Дизамещенные производные пиперазина при потенциометрическом
титровании дают в зависимости от растворителя и заместителя один или два
перегиба на кривой. Так, если 1-п-хлорбензгидрил-4-.м-метилбензилпипера-
зин (меклизин) титровать потенциометрически хлорной кислотой в уксус-
ной кислоте, то наблюдается один перегиб на кривой, соответствующий двум
эквивалентам, но в дифференцирующих растворителях, например в ацето-
Определение азотистых органических оснований
301
нитриле или нитрометане, наблюдается два перегиба, указывающие на про-
тонирование двух третичных атомов азота [132]. Конец титрования наи-
более отчетлив, если в окончательном объеме содержится 22% уксусной
кислоты.
В случае симметричного замещения, например для 1,4-дифенилпиперази-
на в ацетонитриле или нитрометане, можно наблюдать один перегиб на кри-
вой и расход 1 экв хлорной кислоты, так как в этих растворителях второй
N-атом слишком слабо координирует второй протон; в уксусной кислоте,
однако, расходуется 2 экв хлорной кислоты и наблюдается два перегиба
на кривой. При титровании пиперазина в ацетонитриле или уксусной кис-
лоте наблюдается один перегиб и расходуется 2 экв хлорной кислоты.
Влияние заместителей на основность уменьшается в следующем порядке: карбэток-
сп > фенил > п-хлорбензгидрил > бензил > 2-(2'-оксиэтокси)этил > Н.
Значение р^дц+ некоторых производных пиперазина [333]: 1-карбэтоксипиперазин
8,28: 1-бензоилппперазин 7,78; 1-тозилпиперазин 7,44.
Эти примеры показывают, что титрование оснований, способных связы-
вать 2 экв кислоты, следует проводить раздельно для каждого случая, когда
растворители способны облегчать или затруднять протонирование (а сле-
довательно, и титрование).
б. Определение дигидрохлорида 1-(п-хлорбензгидрил)-4-[2'~(2"-оксиэтокси)-
этил\пиперазина (гидроксизин) *. 50 мг гидроксизина помещают в дели-
тельную воронку емкостью 100 мл, растворяют в 10 мл воды, переводят соль
в свободное основание действием 20 мл 0,1 н. щелочи и экстрагируют основа-
ние (4 X 25 мл) хлороформом. Объединенные экстракты высушивают безвод-
ным сульфатом натрия, упаривают приблизительно до 15 мл (например,
в колбе, изображенной на рис. 103) и после добавления 10 мл уксусного
ангидрида титруют 0,02 н. хлорной кислотой в уксусной кислоте. 4,478 мг
гидроксизина требуют 1 мл 0,02 н. раствора хлорной кислоты
Интересным примером служит 3-хлор-10-[3'-(Г'-метил-4"-пиперазинил)-
пропил] фентиазин (прохлорперазин), который содержит три третичных атома
азота. Его малеат можно титровать непосредственно (без экстракции) 0,05 н.
хлорной кислотой в уксусной кислоте в присутствии кристаллического фиоле-
тового [699]. Молекула реагирует с 2 экв хлорной кислоты; так, на титрова-
ние 9,35 мг основания (мол. вес 373,9) необходим 1 мл 0,05 н. хлорной
кислоты.
Об определении диэтансульфоната Ы-(0-3,4,5-триметоксибензоилоксиэтил)-
К'-[у-(3'-хлор-10'-фентиазинил)пропил]пиперазина см. в работе [321]
(гл. 24, разд. 140).
в. Определение пиперазина и оксиэтилпиперазина в одной смеси [811].
При титровании N-оксиэтилпиперазина в ацетонитриле расходуется 2 экв
хлорной кислоты. Пиперазин, как и оксиэтилпиперазин, можно ацетилиро-
Drug. Std., 26, 108 (1956).
302
Глава 23
вать в смеси ацетонитрил — уксусный ангидрид или хлороформ — уксус-
ный ангидрид. N, N'-Диацетилпиперазин не связывает хлорную кислоту, но
Х-ацетил-№'-оксиэтилпиперазин реагирует с 1 экв хлорной кислоты. Так,
смесь пиперазина и оксиэтилпиперазина можно анализировать титрованием
хлорной кислотой в сочетании с ацетилированием.
Для ацетилирования берут смесь уксусной кислоты с уксусным ангид-
ридом (2:8).
50—70 мг образца растворяют в 25 мл ацетонитрила, отбирают 10 мл
этого раствора в колбу с притертой пробкой, добавляют 10 мл ацетилирую-
щей смеси и оставляют хорошо закрытой на 90—100 мин, а затем титруют
0,05 н. хлорной кислотой в уксусной кислоте (расход титранта а мл) в при-
сутствии кристаллического фиолетового до перехода фиолетовой окраски
в голубую. Другую порцию раствора, тоже 10 мл, титруют без ацетили-
рующей смеси (расход титранта — Ъ мл). Чтобы определить объем титранта,
расходуемый на реакцию с пиперазином, вычитают удвоенное количество,
израсходованное после ацетилирования (2а мл) из объема, пошедшего на
титрование без ацетилирования (Ь мл). 1 мл 0,05 н. хлорной кислоты экви-
валентен 2,153 мг пиперазина или 6,51 мг оксиэтилпиперазина (последний
в виде ацетата).
Этот метод можно применять только при условии, что продукт реакции
не содержит третьего компонента, например бис-(оксиэтил)пиперазина,
который не ацетилируется, хотя и обладает основными свойствами. Для опре-
деления такой тройной смеси описанный метод следует модифицировать.
Пиперазин образует с сероуглеродом соединение C4H10N2-CS2, которое
не растворимо в хлороформе *; в случае пиперазина превращение происхо-
дит на 98,3%, а оксиэтилпиперазина (в расчете на формулу C6H14N2O -CS2) —
на 90—92%. бис-(Оксиэтил)пиперазин не образует осадка и может быть
оттитрован хлорной кислотой в уксусной кислоте или смеси хлороформ —
сероуглерод.
Примерно 300 мг тройной смеси 0—10% пиперазина, 80—90% оксиэтил-
пиперазина, 0—10% бис-(оксиэтил)пиперазина растворяют в 50 мл хлоро-
форма. Отбирают 10 мл раствора в колбу емкостью 50 мл, добавляют 0,5 мл
сероуглерода и кипятят с обратным холодильником 40—80 мин на водяной
бане. После охлаждения фильтруют через стеклянный пористый фильтр
№ 4, промывают осадок на фильтре (5x3 мл) хлороформом и титруют бис-
(оксиэтил)пиперазин в объединенном фильтрате 0,05 н. хлорной кислотой.
4,536 мг бис-(оксиэтил)пиперазина требуют 1 мл 0,05 н. хлорной кис-
лоты.
Две следующие порции основного раствора по 10 мл титруют хлорной
кислотой: одну титруют после ацетилирования, другую — без ацетилирова-
ния. Вычитают объем, пошедший на титрование фильтрата, из объема, кото-
рый пошел на титрование после ацетилирования. Таким образом получают
количество стандартного раствора, израсходованное на реакцию с окси-
этилпиперазином, исправленное на количество стандартного раствора,
пошедшего на титрование бие-(оксиэтил)пиперазина.
124. ТИТРОВАНИЕ ОСНОВАНИЙ В ХЛОРБЕНЗОЛЕ
а. Потенциометрическое определение [638]. В 25 мл хлорбензола раство-
ряют 0,23—0,26 мг-экв основания и титруют 0,05 н. хлорной кислотой
в уксусной кислоте, используя стеклянный электрод как электрод сравне-
ния и каломельный как индикаторный. При отсчете показаний милливольт-
метра останавливают магнитную мешалку, применяемую для перемешива
ния во время титрования.
*Castiglioni, Z. anal. Chem., 119, 118 (1940).
Определение азотистых органических оснований
303
Если анализируемые основания нерастворимы в хлорбензоле, 0,5 мг-экв
образца растворяют в 50 мл хлорбензола, содержащего 20% уксусной кис-
лоты, и титруют, как описано выше. Подходящий пропорцией стандартного
раствора по отношению к пробе считается 2 : 10. Точность определения
0.1 мг-экв атропинового основания составляет 100,5 ± 1,1%, а 0,2 мг-экв —
99,7 ± 0,55%.
б. Визуальное определение конечной точки [638]. При визуальном опреде-
лении конечной точки в качестве индикатора рекомендуется использовать
бромфеноловый синий. Исходная окраска раствора зависит от силы раство-
ренного основания: атропин и эфедрин дают пурпурную окраску, стрихнин —
розовую, амидопирин — желтую. Титрование проводят также 0,05 н. хлор-
ной кислотой до обесцвечивания индикатора. Слабые основания образуют
нерастворимые перхлораты, что уменьшает точность анализа. 1 мл 0,05 н.
раствора эквивалентен 14,47 мг атропина (р/Г 4,35), 8,261 мг эфедрина
(рК 4,68), 16,72 мг стрихнина (рК 6,00) или 11,56 мг амидопирина (рК 9,30).
125. ТИТРОВАНИЕ ОСНОВАНИЙ В НИТРОМЕТАНЕ
Соединения низкой основности можно титровать потенциометрически
в нитрометане или смеси нитрометан — уксусный ангидрид 0,05 н. хлорной
кислотой в диоксане [126] или нитрометане [779].
а. Взаимодействие нитрометана с основа-
ниями [126]. Четтен и сотрудники изучили по-
ведение многочисленных алкалоидов в нитро-
метане. Перегибы на потенциометрической кри-
вой наводят на мысль, что основания, рК кото-
рых ниже 11, взаимодействуют с нитрометаном;
к таким основаниям относятся (в скобках дано
значение рК): атропин (4,35), эфедрин (4,68),
цинхонин (5,85), стрихнин (6,00) и кодеин (6,05)
(рис. 117), соланин (6,66), наркотин (7,82),
папаверин (8,07), амидопирин (9,30), и нарцеин
(10,7). Взаимодействие может быть отнесено
к реакции с aifu-формой нитрометана, образую-
щей с алкалоидами продукты присоединения
(гл. 7, разд. 44 и гл. 2, разд. 21).
б. Титрование 0,05 н. хлорной кислотой
в нитрометане [779]. Между значением рХвн+
оснований и потенциалом полунейтрализации
(ППН) существует линейная зависимость (гл. 3,
разд. 23, г). Титрование проводят потенциомет-
рически в нитрометане со стеклянным и кало-
мельным электродами. Примерно 0,25 мг-экв
основания титруют в 100 мл растворителя
0,05н. HCL04 в диоксане,мл
Рис. 117. Дифференциаль-
ная кривая титрования коде-
ина с использованием нитро-
метана в качестве раствори-
теля [126].
А — первый максимум, обозначает
нейтрализацию около 64% коде-
инового основания; В — второй
максимум 100%-ное титрование)
указывает на титрование соедине-
ния типа CH2NO2-BH, где В— ко-
деин — основание.
(см. табл. 17 и рис. 24 и 25).
Приготовление стандартного раствора. 4,2 мл 72%-ной хлорной кислоты раство-
ряют в 1000 мл нитрометана. Титр устанавливают по бифталату калия. Стандартный
раствор можно хранить приблизительно в течение месяца.
в. Потенциометрическое титрование в смеси нитрометан — уксусный
ангидрид (4 : 1) [249]. К 0,3—0,8 мг-экв образца приливают 20 мл смеси
растворителей. Если соединение плохо растворимо, его растворяют предва-
рительно в небольшом количестве уксусной кислоты, если необходимо при
легком нагревании. Титруют потенциометрически 0,1 н. хлорной кислотой
в уксусной кислоте со стеклянным и каломельным электродами. Этим мето-
дом, помимо третичных алифатических аминов, можно также титровать еле-
304
Глава 23
дующие гетероциклические основания (в скобках указано количество веще-
ства в миллиграммах, эквивалентное 1 мл стандартного раствора): бензтиазол
(13,52), безводный кофеин (19,42), 5,7-дихлор-8-оксихинолин (21,40) (см. так-
же разд. 121, г), никотинамид (12,21), 8-нитрохинолин (17,42), хиноксалин
(13,01), теобромин (18,02), безводный теофиллин (18,02).
Примечательно, что трифенилкарбинол можно титровать как основание и в то же
время он используется как индикатор для титрования бензтиазола, никотинамида
и хиноксалина; при титровании кофеина в качестве индикатора можно использовать
нейтральный красный.
126. ТИТРОВАНИЕ ОСНОВАНИЙ В УКСУСНОМ АНГИДРИДЕ
Превосходные свойства уксусного ангидрида как растворителя для опре-
деления слабых оснований получили всеобщее признание [58, 211, 249, 288,
676, 732, 778, 878].
Даже те слабые основания, которые нельзя определять визуально или
потенциометрически в уксусной или пропионовой кислоте, можно анали-
зировать в уксусном ангидриде. Примерами таких веществ служат кофеин,
амиды кислот (рА? 13,4) (см. в этих разделах). Ацетанилид по своему значе-
нию рК в уксусном ангидриде лежит точно на границе возможностей потен-
циометрического титрования (рК 13,39 [497]; рАвн+ — 0,9 [505]).
В некоторых случаях соли оснований с галогеноводородными кислотами
также можно титровать без добавления в раствор ацетата ртути (II) (гл. 24,
разд. 136, н).
Уксусный ангидрид действует на слизистую оболочку глаз, носа и рта,
поэтому титрование следует проводить в хорошем вытяжном шкафу.
В уксусном ангидриде можно титровать только не ацетилирующиеся осно-
вания или основания, содержащие два основных N-атома, один из которых
третичный; ацетилирование первичных и вторичных аминов способствует
успешному титрованию. Сольватирующая способность уксусного ангидрида
не так высока, как сольватирующая способность уксусной кислоты. Образец
растворяют в 1—2 мл муравьиной кислоты и затем разбавляют уксусным
ангидридом. Если в качестве индикатора используют кристаллический
фиолетовый, то окраска изменяется от синей или сине-зеленой до желто-
зеленой и желтой. Растворы, полученные после титрования в уксусном
ангидриде следует разложить водой.
а. Визуальное титрование [676]. Взвешивают образец, на титрование
которого требуется 5—10 мл 0,1 н. раствора хлорной кислоты в уксусной
кислоте. Добавляют под тягой 50 мл уксусного ангидрида, накрывают колбу
небольшим стаканом и перемешивают магнитной мешалкой до растворения.
Титруют в присутствии 5—6 капель 0,1 %-ного раствора кристаллического
фиолетового в уксусном ангидриде. При расчете концентрации учитывают
поправку на титрование растворителя (в большинстве случаев этим можно
пренебречь). Этим способом, например, можно определять антипирин (18,82)
и никотинамид (12,21) (в скобках указано количество вещества в миллиграм-
мах, эквивалентное 1 мл 0,1 н. раствора). Некоторые соединения, например,
теофиллин (18,02), 7-(2'-оксиэтил)теофиллин (22,42), никотиновая кислота
(12,31) растворяются только в процессе титрования. Образцы кофеина
и теобромина сначала растворяют в 5 мл 98 %-ной муравьиной кислоты,
затем добавляют 50 мл ангидрида и титруют, как указано выше. В последнем
случае расход растворителя на глухой опыт больше (см. также гл. 23.
разд. 129).
Относительно определения 8-оксихинолина, З-метил-8-оксихинолина
и 3-метил-5,7-дихлор-8-оксихинолина в уксусном ангидриде см. в работе [620].
Как указывается в литературе последних лет, органические основания,
содержащие азот, протонируются растворителем, в то время как уксусный
Определение азотистых органических оснований
305
ангидрид превращается в анион, который реагирует с ионом ацилония [676]:
(СН3СО)2О + В ВН++(СН3СОООССН2)- (126.1)
(СН3СОООССН2)"4- СН3СООН£ —* (СН3СО)2О -4- СН3СООН (126.2)
б. Потенциометрическое титрование. Потенциометрическое титрование
в среде уксусного ангидрида основывается на линейной зависимости между
значением рЛвн+ оснований и потенциалом полунейтрализации [778] (гл. 3,
разд. 23, в, табл. 16, рис. 23 и 24).
Титрование выполняют 0,05 н. хлорной кислотой в смеси уксусной кислоты
и уксусного ангидрида (1 : 1) (гл. 11, разд. 60,а) или хлорной кислотой
в уксусной кислоте.
1 .мг-экв образца растворяют в мерной колбе в 100 мл уксусного ангидрида
и с помощью приспособления, изображенного на рис. 34, отбирают 25 мл
раствора в колбу емкостью 150—200 мл. Добавляют 75 мл уксусного ангидри-
да и титруют с использованием пары электродов стекло — Ag/AgCl. Электрод
сравнения представляет собой спиральную серебряную проволоку, поверх-
ность которой электролитически покрыта AgCl и погружена в насыщенный
раствор AgCl и LiCl в уксусном ангидриде (ср. рис. 62—64). Другие авторы
для титрования амидов кислот используют пару электродов стекло — кало-
мель. В этом случае в качестве электролита используется 0,1 М раствор
LiC104 в уксусном ангидриде [878].
в. Определение ароматических N-окисей титрованием хлорной кислотой
в уксусном ангидриде рассматривается в работе [604, 880].
127. ПО Л У МИКРО-, МИКРО- и УЛЬТРАМИКРОТИТРОВАНИЕ АЛКАЛОИДОВ
И ДРУГИХ ОСНОВАНИЙ
Большинство алкалоидных оснований терапевтически эффективны уже
в незначительных количествах (например, протовератрин эффективно дей-
ствует уже в количестве 0,1 мг) и среднее содержание их в лекарственных
препаратах также очень мало (0,2—2%), хотя фармацевтические полу-
фабрикаты могут содержать значительное их количество. Следовательно,
для определения могут оказаться необходимыми микрометоды и в исключи-
тельных случаях ультрамикрометоды титрования.
Полумикроанализ представляет промежуточную ступень между макро-
и микроопределениями. Если принять средний эквивалентный вес равным
200, то для титрования в неводных средах используют следующие навески:
Макрометод 100—300 мг
Полумикрометод 10—50 мг
Микрометод 0,1—5 мг
Ультрамикрометод 5—50 мкг
(ср. данные работы [218] и [584])
Условия, применяемые для макро- или полумикроопределения (например,
количество индикатора, объем растворителя и т. д.), нельзя непосредственно
переносить на микро- или ультрамикроопределение. Для титрования осно-
ваний можно использовать 0,001 н. хлорную кислоту, но если сила исследу-
емого основания значительно отличается от силы основания, применяемого
для установки титра раствора титранта, то могут возникнуть трудности.
Об этом уже упоминалось в гл. 13, разд. 72. Иногда, особенно при потенцио-
метрическом титровании, желательно использовать 0,5 н. хлорную кислоту;
в этом случае применяют так называемую микробюретку Гильмонта (см. рабо-
ту [333] и стр. 168 в работе [584]).
а. Титрование 0,005 н. п-толуолсулъфокислотой [299, 304, 696, 700].
Преимущества титрования 0,005 н. п-толуолсульфокислотой кратко заклю-
чаются в следующем [299].
306
Глава 23
1. Для анализа достаточно 0,5—3,0 мг образца алкалоидного основания.
2. Самая выгодная особенность метода в том, что после экстракции алка-
лоида из его щелочного раствора хлороформом и высушивания экстракта
безводным сульфатом натрия хлороформный раствор можно титровать
непосредственно; нет необходимости выпаривать раствор, растворять осно-
вание в 0,01 н. стандартной соляной кислоте и оттитровывать избыток
кислоты 0,01 н. щелочью. Этот метод исключает три лишние операции^
а следовательно, и три источника ошибок.
3. Титрование n-толуолсульфокислотой дает гораздо более точные
результаты, чем титрование в водной среде. Это в первую очередь обуслов-
лено очень резким изменением окраски диметилового желтого, используемо-
го в качестве индикатора.
4. Одна капля стандартного хлороформного раствора титранта соот-
ветствует (при использовании микробюретки, см. рисунок в работе [299])
объему 0,007 мл. Фактически это означает, что, если вблизи конечной точки
титрование ведут по каплям и даже не прибегают к обычному техническому
приему, когда, касаясь стенки сосуда кончиком бюретки, добавляют часть
капли, вероятная ошибка на 1 мл титранта составляет только +0,7%.
Еще об одном достоинстве этого метода говорится в статье Чафарика *.
Используемый в качестве растворителя при титровании п-толуолсульфокис-
лотой хлороформ — один из лучших растворителей для органических осно-
ваний: он неполярен, не образует солей ни с кислотами, ни с основаниями
и, следовательно, не влияет на равновесное состояние реакции нейтрали-
зации.
Измерение вращения плоскости поляризации оптически активных алка-
лоидов следует проводить в тех же растворителях, что и неводное титрова-
ние. При определении основания гиосциамина примерно 50 мг образца раство-
ряют в 25 мл хлороформа. Измеряют вращение плоскости поляризации раст-
вора (2%-ный раствор в СНС13; [a]D = —25,2°). Из остатка раствора отби-
рают пробу 5 мл, разбавляют ее хлороформом до 100 мл и титруют после
добавления одной или двух капель 0,1%-ного диметилового желтого в хлоро-
форме. Титруют 5 мл этого раствора до перехода желтой окраски в розовую.
1,447 мг гиосциамина (атропина) эквивалентен 1 мл 0,005 н. п-толуолсуль-
фокислоты.
Вращение плоскости поляризации и процентное содержание, например
эрготамина, определяют аналогично.
Основания с константой диссоциации (в воде) не меньше 10"9 можно ана-
лизировать аналогичным образом, так как п-толуолсульфокислота — менее
сильный донор протона, чем перехлорат ацилония. В качестве примера
приведем следующие основания (в скобках указано количество вещества
в миллиграммах, эквивалентное 1 мл 0,005 н. раствора): гиосцин (1,517),
стрихнин (1,672), хелидонин (1,766), кодеин (1,497), эрвин C28H37O3N (2,057),
винцамин (1,772), протовератрины А + В (4,02), папаверин (1,695), томатин
(5,171), томатидин (2,078), резерпин (3,044) ** [41, 299, 301, 304, 305, 323, 701,
791, 792]. При титровании еще более слабых оснований (рАь ~ И) вместо
хлороформа более целесообразно использовать хлорбензол или дихлорме-
тан и 0,005 н. n-толуолсульфокислоту в смеси хлороформ — этиловый спирт
(99 : 1) [696].
б. Определение алкалоидов спорыньи [305]. Взвешивают 5—10 мг алкалои-
дов спорыньи на полумикровесах, растворяют в 5—10 мл очищенного хлоро-
форма, стабилизированного 1 об. % легкокипящего петролейного эфира, и тит-
руют 0,005 н. раствором n-толуолсульфокислоты в присутствии 1—3 капель
индикатора диметилового желтого. Желтая окраска индикатора переходит
* Safor i с, Cesk. Farm., 11, 387 (1962).
** p/f 7,7; см. Krebs, Fiitscher, Arzneimittel-Forschung, 10, 75 (1960).
Определение азотистых органических оснований
307
в светло-розовую. 3,048 мг эргокристина требуют 1 мл стандартного раствора.
Смеси эрготамин — ацетон, эрготамин — бензол, эрготоксин — бензол мож-
но анализировать таким же способом.
в. Определение хлоргидрата диэтиламиноэтилбензилатпа (хлоргидрата
бенактизина) [757]. Тонко измельчают таблетку и взвешивают такое количе-
ство образца, чтобы на титрование его расходовалось 1—3 мл 0,005 н. п-толу-
олсульфокислоты; навеску помещают в делительную воронку емкостью
100 мл. Суспендируют взвешенный образец в 10 мл воды, добавляют 0,2 г
тонко измельченного бикарбоната натрия и после растворения экстрагируют
основание (2 х 40 и 2 х 30 мл) хлороформом. Объединенные вытяжки
сушат безводным сульфатом натрия и упаривают до 5 мл. Эту операцию
можно выполнить в колбе для упаривания, изображенной на рис. 103.
К сконцентрированному экстракту добавляют 4 капли 0,1% -ного диметилового
желтого в хлороформе и титруют 0,005 н. п-толуолсульфокислотой до появ-
ления розовой окраски (1 мл стандартного раствора эквивалентен ~ 1,8194 мг
хлоргидрата бенактизина).
г. Определение резерпина в таблетках. Определить малые количества
(0,1—0,25 мг) алкалоидных оснований с высоким молекулярным весом
часто довольно трудно из-за большого количества наполнителя (связующего
вещества), адсорбирующего действующее начало таблетки. Если адсорбция
обратима или алкалоиды не разрушаются как в процессе приготовления таб-
летки, так и при хранении, то количественный анализ можно проводить
в неводной среде [302].
Разрушение алкалоидов часто сопровождается хемосорбцией на наполни-
теле. В этом случае неразложившийся алкалоид связан в более значитель-
ной степени, чем разложившийся продукт (селективная адсорбция). Это явле-
ние, которое можно объяснить адсорбцией, чаще всего наблюдается в таблет-
ках, приготовленных водным гранулированием [300].
Экстракцию действующего начала увеличивают добавлением 2—3% фено-
ла к экстрагирующему растворителю (СНС13 или СС14) [323], фенол не меша-
ет титрованию п-толуолсульфокислотой.
Из таблеток, каждая из которых содержит 0,1—0,25 мг резерпина, при-
готавливают тонко измельченный гомогенный порошок и в широкогорлой
плоскодонной колбе емкостью 100 мл взвешивают количество порошка,
на титрование которого расходуется 1—3 мл 0,005 н. п-толуолсульфокислоты.
Добавляют 15 мл смеси четыреххлористый углерод — фенол (100 : 2), ней-
трализованной в присутствии диметилового желтого. Оставляют смесь на
30 мин, вращая колбу, особенно вначале, чтобы увеличить полноту экстра-
гирования резерпина. Затем дают смеси отстояться и фильтруют ее на филь-
тровальной бумаге (0 9 см) типа Schleicher-SchiiU «Schwarzband». На дно
стакана, гораздо большего, чем воронка, помещают слой ваты и над ним
плотную фильтровальную бумагу, пропитанные СС14. Во время фильтрова-
ния прикрывают фильтр подготовленным таким образом стаканом. Оставшийся
на фильтре наполнитель промывают (3x3 мл) хлороформом и объединенные
вытяжки титруют в присутствии диметилового желтого. На 3,044 мг резерпи-
на расходуется 1 мл 0,005 н. стандартного раствора [41, 323]. Действующее
начало таблетки, содержащей протовератрин А + В, можно определить
таким же способом [323].
Резерпин можно также экстрагировать хлороформом, не добавляя в него
фенол, если технологические приемы, используемые в ходе приготовления
таблеток, препятствуют адсорбции алкалоидного основания на носителе [792].
д. Определение папаверина (основания) 0,005 н. п-тслуолсулъфокислстпой
в смеси этиленгликоль — изопропиловый спирт [700]. 10—15 мг папаверина
(основания) растворяют в ацетоне, метилизобутилкетоне или хлороформе
и титруют потенциометрически. Если к раствору в ацетоне добавить неболь-
шое количество фенола, то титрование можно проводить в присутствии мети-
308
Глава 23
лового оранжевого как индикатора. 1,695 мг папаверина требует 1 мл стан-
дартного раствора.
Морфин, кодеин и тебаин можно титровать в смеси хлороформ — изопропиловый
спирт — фенол (10 : 1 : 1) 0,002 н. хлорной кислотой или п-толуолсульфокислотой
в смеси этиленгликоль — изопропиловый спирт (1 : 1) или этиленгликоль — диоксав
(1 : 1) [702].
е. Титрование 0,001 н. хлорной кислотой. Для приготовления стандарт-
ного раствора используют смеси растворителей четыреххлористый углерод —
уксусная кислота или смеси четыреххлористый углерод — пропионовая
кислота (гл. 9, разд. 52, г). Готовят раствор алкалоида, каждый миллилитр
которого содержит около 0,005 мг-экв основания; для этого 15 мг алкалоида
со средним молекулярным весом 300 растворяют в 10 мл нейтрализованного
растворителя. Во время титрования раствор нельзя ставить вблизи источника
тепла. Растворитель или смесь растворителей, применяемую для растворе-
ния, оставляют стоять, чтобы они приняли комнатную температуру.
С помощью точной пипетки титруют 1 мл раствора в присутствии одной кап-
ли 0,1%-ного раствора кристаллического фиолетового или метилового
фиолетового в конической центрифужной пробирке на белом фоне, при
титровании на раствор глядят сверху. Если образец представляет собой не-
ацетилируемое основание, в качестве растворителя используют предвари-
тельно нейтрализованную уксусную или пропионовую кислоту или уксус-
ный ангидрид или смесь его с апротонным растворителем в соотношении
от 1 : 10 до 1:1. Титр раствора устанавливают в таком же растворителе
с индикатором такой же концентрации. Если титруемое основание имеет
рК << 10-9, то для определения титра используют, если это возможно, такое
же соединение или одно из оснований приблизительно такой же силы, что
и исследуемое основание, но заведомо известной чистоты. Для определения
титра сильных оснований можно использовать дифенилгуанидин. В обоих
случаях, однако, как анализируемое соединение, так и вещество для опреде-
ления титра должны иметь в растворе приблизительно одинаковую молярную
концентрацию. Для определяемого соединения и для раствора, применяе-
мого для определения титра, используют одну и ту же пипетку. Титро-
вание проводят с одинаковой скоростью и избегают нагревания пипетки
рукой.
В смеси уксусной кислоты и хлороформа (1 : 3) даже основания с р# 10-12,
а в уксусном ангидриде и с рК 10-13*5 можно титровать в присутствии
кристаллического фиолетового [297]. 0,5 мг анилина можно титровать потен-
циометрически в смеси циклогексан — уксусная кислота (1 : 1) 0,005 н. хлор-
ной кислотой в уксусной кислоте, используя для титрования сосуд Варне-
ра — Хаскеля [860] (см. рис. 56, а также работу [450]).
ж. Улътрамикротитрование оснований требует особенно большой точ-
ности [51, 584]. 50 мкг органического основания растворяют в 0,4 мл тща-
тельно обезвоженной уксусной кислоты. В качестве сосуда для титрования
используют небольшую стеклянную трубку (30 х 11 мм}, закрытую корко-
вой или резиновой пробкой, которая покрыта полиэтиленовой пленкой для
предотвращения адсорбции уксусной кислотой воды через пробку. Констан-
та диссоциации воды как основания равна 12,53 [464], причем ошибка титро-
вания, вызванная примесью воды, при потенциометрическом титровании
меньше, чем при визуальном определении. Вблизи конечной точки
титрования помехи увеличиваются, так как, будучи слабым основанием, вода
мешает образованию перхлората индикатора (индикатор также представ-
ляет собой очень слабое основание).
При потенциометрическом ультрамикротитровании расход титранта на
титрование растворителя только незначително увеличивается небольшим
количеством воды, содержащимся в 0,4 мл уксусной кислоты, но при посте-
пенном возрастании содержания воды потенциометрическая кривая выравни-
Определение азотистых органических оснований
309
вается. При визуальном определении расход стандартного раствора на титро-
вание растворителя в глухом опыте значительно увеличивается (табл. 93) [51].
Так как вода мешает титрованию, маленькую колбу для титрования
и пипетку следует ополоснуть уксусной кислотой, содержащей кристал-
лический фиолетовый и минимальное количество хлорной кислоты, пока
будет сохраняться устойчивая сине-зеленая окраска уксусной кислоты.
Таблица 93
Объем титранта, расходуемый на глухой опыт, мкл Содержание воды, %
0,02 0,2 0,5 0,75 1,0 1,5
При потенциометрическом 1,0 1,0 1,1 1,1 1,2 1,2
титровании При визуальном определении 1,2 1,4 1,9 2,3 3—3,5 4—5
Трубку для титрования ополаскивают уксусной кислотой, высушивают в токе
азота и закрывают пробкой, обернутой полиэтиленом. Самым подходящим
индикатором является кристаллический фиолетовый [51]; если титруют 50 мкг
основания в 0,4 мл уксусной кислоты, то для полного перехода фиолетовой
окраски в зеленую нужно 0,2—0,4 мкл 0,01 н. хлорной кислоты. Теорети-
чески для каждого основания переход окраски нужно подбирать индиви-
дуально; во многих случаях, однако, достаточно сравнивать два раствора:
«синий» (а) и «зелено-синий» (б).
а. В 78—80 мл уксусной кислоты растворяют 0,6 г мочевины, добавля-
ют 3,0 мл 0,01 н. хлорной кислоты, 3,3 мл 0,01 %-ного кристаллического фио-
летового и разбавляют уксусной кислотой до 100 мл.
б. Приготавливают как описано выше (а), но с 6,0 мл хлорной кислоты.
Может случиться, что нейтрализация 30 мкг основания даст окраску,
отличную от окраски, вызываемой 60 мкг [146]. Например, если титровать
50 мкг ацетата натрия 0,01 н. хлорной кислотой в присутствии кристал-
лического фиолетового, точный эквивалент может совпасть с появлением
синей окраски, в то время как для 300 мкг он совпадает с появлением зеле-
ной окраски, хотя значения pH для конечной точки титрования одинаковы.
Для ультрамикротитрования взвешивают такое количество образца, что-
бы расход 0,01 н. хлорной кислоты не превышал 30—50 мкл. Если используют
большие количества растворителя (1—2 мл), расход титранта на глухое
титрование растворителя может возрасти до 10 об. % стандартного раствора,
расходуемого на титрование, но для 0,4 мл растворителя эта величина
не превышает 1 мкл.
Результат анализа зависит также от продолжительности титрования.
При визуальном определении титрование должно заканчиваться за 2—3 мин,
в то время как при потенциометрическом определении титрование может
длиться 7—8 мин. Из ультрамикробюретки Бельтера [51] 4—5 мкл раствора
испаряются за 90 мин.
Вследствие летучести некоторых оснований их нельзя хранить в эксикаторе: так,
73 мкг и-аннзидина теряют 30% веса через 3 час. n-Толуидин настолько летуч, что его
нельзя взвесить даже на высокочувствительных аналитических весах в открытом сосуде.
Для потенциометрического ультрамикротитрования используют стеклян-
ный сферический электрод диаметром 3,5 мм и Ag/AgCI в качестве электрода
сравнения. Раствор электролита представляет собой 0,02 н. раствор моно-
гидрата перхлората натрия в уксусной кислоте.
Точность определения даже в ультрамикроколичествах ±1%, но приобре-
тение необходимых навыков требует изучения метода и практики [51].
Результат потенциометрического микротитрования можно вычислить
графически (см. стр. 17—25 в работе [584]).
310
Глава 23
5мм
А
6-7мм
Б
32мм
Ммм
Р и
экстракции твердого вещества
жидкостью и жидкого вещества
жидкостью, используемый при
определении алкалоидов, со-
держащихся в лекарственных
травах [525, 526].
128. ДЕЙСТВУЮЩЕЕ НАЧАЛО В ЛЕКАРСТВЕННЫХ РАСТЕНИЯХ,
СОДЕРЖАЩИХ АЛКАЛОИДЫ
Проиллюстрируем на 4-х примерах определение действующего начала
в высушенных и измельченных лекарственных растениях. Первые три мето-
дики с незначительными изменениями можно использовать для анализа дру-
гих растений, содержащих алкалоиды.
а. Определение суммарного содержания алкалоидов в литорине маленькой (Vinca
minor L.) [790]. С точностью до 1 мг взвешивают 3 г высушенных и тонко размолотых
листьев и стеблей растения и переводят в основание обработкой 3 мл 10%-ного раствора
аммиака, экстрагируют бензолом (1 X 20 и 2 X 10 мл)
и разбавляют объединенные вытяжки бензолом до
100 мл. Экстрагируют 20 мл раствора (3 X 10 мл)
2%-ной серной кислотой, фильтруют, подщелачивают
20 мл кислого раствора раствором едкого натра до
pH 8 и экстрагируют (4x5 мл) хлороформом. Хлоро-
формный экстракт сушат безводным сульфатом натрия
и выпаривают 10 лед досуха. Остаток растворяют в смеси
10 мл хлороформа и 5 мл уксусной кислоты и титруют
0,01 н. хлорной кислотой в смеси уксусной кислоты
и четыреххлористого углерода (1:1) в присутствии
одной капли кристаллического фиолетового. 3,544 мг
винкаминового основания C21H2FO3N2 требуют 1 мл
стандартного раствора. Из двух N-атомов в молекуле
титруется только один. Этим способом определяют
алкалоиды, содержащиеся в 0,20 г порошка.
б. Анализ корней беладонны (Atropa belladonna L.)
[525, 526]. Описанные ниже методика и аппаратура
годны для серийного анализа. Экстракцию измельчен-
ных в порошок корней растения органическим раство-
рителем и перевод алкалоида в кислую (жидкую) фазу
выполняют обычными методами.
Сухой корень беладонны растирают в тонкий поро-
шок и просеивают через сито 50 меш с отверстиями
0,30 мм. Оставшийся на сите порошок размалывают
и вновь просеивают. Гомогенизируют продукт, полу-
ченный при различных просеиваниях, и сушат над
негашеной известью.
2 г приготовленного таким образом порошка рас-
тирают в ступке с 2 мл 12%-ного аммиака и помещают
на стеклянной вате (2) в муфту экстрактора, изобра-
женного на рис. 118. Открывают кран (5) между экст-
ракторами для экстрагирования твердого вещества
жидкостью (А) и жидкого вещества жидкостью (Б)
и в цилиндр экстрактора (Б) через загрузочный отвод
(4) заливают 10 мл 2%-ной серной кислоты. Вещество
в цилиндре (5) экстрактора (А) заливают таким коли-
чеством бензола, чтобы бензол (6) заполнил циркуля-
ционную трубку (7) в нижней части и покрывал анали-
зируемое вещество, когда кран закрыт.
После сборки включают аппарат, осторожно пода-
вая ток газа через трубку с краном (8), так что бензол,
который находится выше слоя (9) серной кислоты, раз-
брызгивается на анализируемое вещество через цир-
куляционную трубку, проходит через него и через
поступает в серную кислоту, собирается на ее поверх-
д на анализируемое вещество через циркуляцион-
18~20мм
с.
118. Экстрактор для
перфорированную трубку (70)
ности п затем разбрызгивается на анализируемое вещество через циркуляцион-
ную трубку (7). Таким образом, бензольная фаза находится в постоянном движении.
В то время когда бензол проходит через серную кислоту, экстрагированные бензолом
алкалоиды растворяются п переходят в сернокислотную фазу. Большая часть инертного
вещества, растворимого в бензоле, но нерастворимого в серной кислоте, остается в орга-
нической фазе.
Одновременную экстракцию твердого вещества жидкостью и жидкости жидкостью
проводят соответственно 1 X 2 и 3 X 1 час. После каждого рабочего периода выключают
прибор, снимая давление, в это время кран 3 должен быть закрыт. Затем открывают
кран 11 и из нижней части экстрактора Б спускают кислую фазу в мерную колбу емко-
стью 50 мл, а в аппарат через загрузочную трубку заливают 10 мл свежей 2%-ной серной
Определение азотистых органических оснований 311
кислоты. Таким способом получают примерно 40 мл кислого раствора и разбавляют его
до 50 мл 2%-ной серной кислотой.
40 мл кислой водной фазы подщелачивают в мерной колбе 10%-ным едким натром
до pH 8—9 и экстрагируют (4x5 мл) хлороформом. Объединенные вытяжки сушат
нейтральным безводным сульфатом натрия и 10 мл выпаривают на водяной бане досуха.
После охлаждения растворяют остаток в 10 мл хлороформа, добавляют 5 мл уксусной
кислоты п титруют 0,01 н. хлорной кислотой в уксусной кислоте в присутствии 0,1%-ного
кристаллического фиолетового до синего окрашивания.
Изображенный на рис. 110 прибор можно соединить с другим прибором через трубку
с краном (8), через который подают ток газа (жидкости), так что одновременно могут
функционировать 6—12 приборов. В 15 мл конечного раствора (смеси хлороформ —
уксусная кислота) содержится сумма алкалоидов из 0,8 г растертого в порошок расте-
ния. 2,894 мг атропина требуют 1 мл 0,01 н. раствора кислоты.
в. Определение томатина, содержащегося в Licopersicum esculentum var. ribesiforme
и Licopersicum esculentum var. pruniforme [302]. 100 г растертого в порошок сухого расте-
ния (первоначально размолотого в шаровой мельнице) просеивают через сито 40 меш
Рис. 119. Определение томатина [302].
(размер отверстия 0,42 мм) и затем через сито 80 меш (размер отверстия 0,177 мм). Взвеши-
вают с точностью до 0,1 г порошок, оставшийся на сите 40 меш, грубый порошок, просе-
янный через сито 40 меш, и тонкий порошок, просеянный через сито 80 меш, и вычисля-
ют процент составных частей помола.
Па фильтровальной бумаге (9 см), сложенной в форме воронки, взвешивают 1,00 ±
+0,01 г растертого в порошок растения и экстрагируют 40 мин в маленьком аппарате
Сокслета 30 мл метилового спирта. Добавляют 1 мл 25%-ного аммиака через холодиль-
ник и продолжают экстракцию еще 20 мин. Полученный экстракт выпаривают в фарфо-
ровой чашке на водяной бане досуха и после охлаждения тщательно растирают в поро-
шок с 1 г мелкозернистого кварцевого песка, а затем экстрагируют основание последо-
вательно 1 X 5 и 5 X 2 .мл 0,1 н. соляной кислоты при 5—10°. Солянокислый раствор
фильтруют в охлаждаемый льдом мерный стеклянный стакан на 25 мл и промывают
песок 2—3 мл дистиллированной воды. Большая часть хлорофилла удерживается квар-
цевым песком.
Чтобы выделить томатиновое основание из охлажденной смеси, отфильтрованный
раствор подщелачивают 1,5 мл 10%-ного аммиака. Определяют объем раствора и через
полчаса фильтруют осадок на фильтровальной бумаге (0 9 см), которая предварительно
промыта 200 мл кппящей дистиллированной воды и высушена при 100°. Фильтровальную
бумагу с осадком кладут на часовое стекло и сушат 20 мин при 100°, затем разрезают на
кусочки размером 1 ел2 и помещают в колбу Эрленмейера емкостью 100 мл, закрытую
стеклянной пробкой. Смывают сухие гликоалкалоиды с фильтровальной бумаги 15—
20 мл горячей смеси фенола и четыреххлористого углерода (1 : 4), нейтрализованной
0,005 н. n-толуолсульфокислотой в присутствии 6 капель дпметилового желтого как
индикатора, и титруют n-толуолсульфокислотой до появления устойчивого розового
окрашивания. Во время титрования раствор нужно неоднократно подогревать и после
энергичного встряхивания охлаждать. 5,171 мг томатина С5пН8зО21П (мол. вес 1033)
требуют 1 мл 0,005 н. раствора. В полученный результат вносят поправку на количество
гликоалкалоидов, содержащееся в водном растворе аммиака при 0 °(из расчета
0,17 мг/мл). Вышеописанные процессы иллюстрируются рис. 119.
Содержание томатина, определенное для каждой из фракций порошка после просе-
ивания, пересчитывают для всего образца в целом, учитывая соотношение фракций помола.
Таким же образом можно определять соласонин [318].
г. Экстракция лекарственных веществ, содержащих алкалоиды, уксусной кислотой,
свободной от уксусного ангидрида [23]. Согласно общепринятому методу, лекарственное
312
Глава 23
сырье экстрагируют лиофильным растворителем, в то время как основания из их солей
переводят в свободное состояние действием водной щелочи или аммиака. Это приводит
к двухфазной системе, что не всегда желательно. Лекарственные вещества, содержащие
алкалоиды, можно непосредственно экстрагировать уксусной кислотой, но этот метод
пригоден для работы с большим количеством вещества и не может применяться для всех
образцов. Другим недостатком метода является то, что в случае ацетилируемых алка-
лоидов уксусная кислота не должна содержать даже следов уксусного ангидрида (гл. 8,
разд. 47, в).
Определение содержания эметина (рК 5,77). 2 г растертого в порошок лекарственно-
го сырья нагревают до кипения с обратным холодильником с 20, 15 и 10 мл уксусной
кислоты в течение 5 мин. Каждый экстракт фильтруют через стеклянный фильтр
и объединяют экстракты в делительной воронке емкостью 500 мл. Промывают остаток при-
мерно 50 мл воды и уксуснокислый экстракт осторожно переводят в основание действием
раствора аммиака (делительную воронку охлаждают током воды). Экстрагируют, избегая
сильного встряхивания, хлороформом (1 X 100 мл и 2 X 50 мл). Экстракт сушат без-
водным сульфатом натрия, выпаривают на водяной бане под уменьшенным давлением
досуха, остаток растворяют в 10 мл уксусной кислоты и титруют 0,1 н. хлорной кисло-
той в присутствии кристаллического фиолетового. 24,03 мг эметина требуют 1 мл стан-
дартного раствора (т. е. сумма алкалоидов выражена через эметин).
Определение никотина и норникотина в измельченном табаке описано в гл. 25,
разд. 146, а, об определении каучука в растительном сырье см. в гл. 29, разд. 165.
129. ПУРИНОВЫЕ ОСНОВАНИЯ
Благодаря «кислой» иминогруппе некоторые пуриновые соединения
можно титровать как кислоты [246, 566] (гл. 17, разд. 104, табл. 32). Однако
уксусный ангидрид в смеси с нитрометаном, четыреххлористым углеродом
или бензолом способен увеличить основность гетероатома азота настолько,
что некоторые из этих соединений можно титровать как основания (см. гл. 23,
разд. 125, в, работу [249], разд. 126, а и работу [676]).
Кофеин и теобромин можно также титровать в бензоле в присутствии
индикатора Судана III [681]. Для определения теобромина пригодна также
смесь уксусного ангидрида и четыреххлористого углерода [658], а для
определения кофеина — смесь уксусного ангидрида и хлороформа, в качестве
индикатора используется Судан III [439].
Из-за большой важности некоторых пуриновых соединений (кофеин,
теобромин и теофиллин) предложено много других методов анализа. Эти
данные были кратко суммированы Сальвезеном [703] (см. также [11, 15, 636,
644, 681, 443]).
При титровании пуриновых оснований можно применять потенциометри-
ческое, фотометрическое и визуальное определение конечной точки. Кофеин
определялся Сальвезеном в смеси с салицилатом натрия или бензоатом натрия
в присутствии тропеолина ОО и метилового фиолетового как индикаторов.
Несколько оснований в одной смеси можно определить в растворителе с дифферен-
цирующими свойствами, но два соединения с различной основной силой можно титро-
вать и в одной смеси, используя два вида индикаторов при условии, что окраска инди-
каторов меняется в широких пределах и в то же время совпадает с концом титрования
двух оснований.
а. Определения кофеина и салицилата (или бензоата) натрия [703].
Примерно 400 мг образца растворяют в 10 мл уксусного ангидрида. После
охлаждения добавляют 20 мл бензола и 4 капли 0,5%-ного тропеолина ОО
в уксусной кислоте и титруют салицилат натрия 0,1 н. хлорной кислотой
до перехода желтой окраски в оранжево-желтую. На титрование 16,01 мг
салицилата натрия расходуется 1 мл стандартного раствора. Затем к раство-
ру добавляют 4 капли 0,1%-ного метилового фиолетового в уксусной кислоте
и продолжают титрование до изменения окраски смеси индикаторов от зеле-
ной до золотисто-желтой. Изменение окраски совпадает с эквивалентной
точкой кофеина. 19,42 мг безводного кофеина требуют 1 мл 0,1 н. раствора.
б. Определение теобромина, диоксипропилтеофиллина или теобромина
и салицилата натрия [704]. 300—400 мг теобромина или диоксипропилтео-
Определение азотистых органических оснований
313
филлина растворяют при нагревании в 20 мл смеси уксусной кислоты
и уксусного ангидрида (1 : 3) или 15 мл уксусного ангидрида. Добавляют 20—
25 мл бензола и 5 капель 0,1 %-ного метилового фиолетового в уксусной
кислоте и титруют 0,1 н. хлорной кислотой до изменения окраски от зеленой
до желтой. 1 мл стандартного раствора эквивалентен 18,03 мг теобромина
или 25,42 мг диоксипропилтеофиллина.
При определении теобромина и салицилата натрия примерно 350 мг
образца растворяют при нагревании в 25 мл смеси уксусной кислоты
и уксусного ангидрида (1 : 5) и после добавления 20 мл бензола титруют, как
при определении кофеина и салицилата натрия, но вместо метилового фиоле-
тового добавляют 7 капель 0,1%-ного кристаллического фиолетового.
в. Титрование кофеина с фотометрическим определением конечной точки
[211]. 0,5—1 мг-экв кофеина растворяют в 20 мл уксусного ангидрида. Добав-
ляют 2 капли 0,5 %-ного сафранина или нильского голубого А в качестве
индикатора, помещают в кювету колориметра (15 мм), например кювета
модель Ланге IV, и титруют 0,1 н. хлорной кислотой в уксусной кислоте.
И в том и в другом случае используют фильтр с длиной волны 550—580 нм
(OG2). Конечную точку титрования определяют графически по зависимо-
сти 1/Т от V (см. рис. 79 и также гл. 14). 19,42 мг безводного кофеина экви-
валентны 1 мл 0,1 н. хлорной кислоты. Другие пуриновые производные,
например аденин, гуанин, гипоксантин и аденозин, можно растворять при
осторожном нагревании в смеси уксусная кислота — хлорная кислота при
избытке последней. Избыток стандартного раствора можно оттитровать 0,1 н.
ацетатом натрия в уксусной кислоте в присутствии кристаллического фиоле-
тового. Точность метода только 1—2% [188] (см. также работу [15]).
г. Определение теофиллина [45, 654]. 100 мг образца растворяют в 2 —
—3 мл 0,1 н. хлорной кислоты в уксусной кислоте, добавляют 30 мл смеси бен-
зол — уксусный ангидрид (2:1), предварительно нейтрализованной в при-
сутствии 6 капель Судана III (в 0,5%-ной уксусной кислоте), и продолжают
титровать 0,1 н. хлорной кислотой. Перед окончанием титрования к раство-
ру добавляют 3—4 капли индикатора. 1 мл 0,1 н. стандартного раствора экви-
валентен 18,03 мг теофиллина.
Добавление ацетата ртути(П) увеличивает основность теофиллина, вероятно, вслед-
ствие того, что кислый водород иминогруппы в имидазольном кольце пуринового скелета
подвергается замещению. В уксусном ангидриде и в присутствии ацетата ртути(II)
теофиллин можно титровать потенциометрически как основание.
д. Одновременное определение теофиллина и 7-оксиэтилтеофиллина см.
в работе [512].
130. АМИДЫ КИСЛОТ
Большинство амидов кислот даже более слабые основания, чем вода,
но в то же время они также и слабые кислоты, способные образовывать соли
с металлами, например, в жидком аммиаке. Эффекты таутомерии и резонанса,
а также влияние образования водородных связей — факторы, определяю
щие поведение амидов кислот (см. стр. 90 в работе [380])
R —С
+
NH2
О—И
//
R —С
Н
314
Глава 23
Протонирование амидов в кислых растворах происходит по кислороду,
а не по азоту [403, 427, 438]
/70 Z0H
R —C<f R — С<
XNH2 xnh2
Алкилирование N-атома увеличивает основность амида кислоты в меньшей степени,
чем в аммиаке.
Относительно влияния ацилирования и алкилирования на силу оснований см. в рабо- те [403, 556], а сравнительный порядок основности амидов кислот при фотометрическом титровании хлорной кислотой в присутствии
Таблица 94 Судана III рассматривается в работе [385]. См. также гл. 14, разд. 80, в.
Амид кислоты рК [403] If ЭКСП а. Определение амидов кислот в уксус-
[385] ном ангидриде [878]. При добавлении
хлорной кислоты к уксусному ангидриду
Формамид -0,48 0,53 возможно образование «ацетилперхло-
N - Метилформампд -0,04 0,13 рата», кислоты Льюиса (СН3СО)+СЮ~,
N,N-Диметилформ- -0,01 0,14 образующего соль только с амидом
амид Ацетамид N - Метилацетампд +0,11 4-0,80 0,10 0,023 кислоты. Взвешивают 6—8 ммолей образца и растворяют в 100 мл уксусного анги- дрида. Разбавляют 10 мл раствора в уксусном ангидриде до 100 мл и титру-
ют потенциометрически 0,1 н. хлорной кислотой в уксусной кислоте, исполь-
зуя систему электродов стекло — каломель. Каломельный электрод в качест-
ве электролита содержит 0,1 М LiC104, растворенного в уксусном ангидриде.
Стеклянный электрод помещают за 12 час до использования в уксусный анги-
дрид. Значение Де/Др, наблюдаемое в конечной точке титрования, отражает
в определенной степени основность амида кислоты (в скобках указано коли-
чество миллиграммов, эквивалентное 1 мл 0,1 н. раствора, и первое значение
скачка потенциала в милливольтах): N,N-flHMeTH л ацетамид (8,712 мг, 300 мв),
NjN-диэтилацетамид (11,52 мг, 280 мв), тиоацетамид (7,513 мг, 470 мв),
N-формилпирролидин (9,913 мг, 300 мв), N-ацетилпиперидин (12,72 мг,
470 мв).
В работе Уимера описано определение 36 других соединений, помимо перечислен-
ных выше [878].
б. Фотометрическое определение мочевины [390]. Взвешивают пример-
но 1,1—1,2 ммоля мочевины, помещают в мерную колбу емкостью 100 мл,
растворяют в 50 мл уксусной кислоты, добавляют 10 мл индикатора
0,01%-ного малахитового зеленого в уксусной кислотен доводят до метки
уксусной кислотой. 20 мл раствора титруют 0,1 н. хлорной кислотой в уксус-
ной кислоте. Образец и титрант содержат малахитовый зеленый в одинаковой
концентрации. Титруют при 622 нм и снимают значение после добавления
каждых 0,2 мл.
Значение 777Н+ вычисляют по уравнению
I А
7Н+ ~ АЪ-А ’
где Аъ — поглощение основной формы индикатора; А — отдельные значе-
ния, снятые в процессе измерения.
Значение 777Н+ наносят на график как функцию, обратную объему хлор-
ной кислоты (в миллилитрах), и точка пересечения прямой линии, получен-
ной экстраполированием, с осью х соответствует точке эквивалентности
(см. гл. 14, разд. 79 и 80; [390]; рис. 81).
Вместо малахитового зеленого можно использовать в качестве индикато-
ра нильский голубой А (при 632 нле). Если изобразить на графике зависи-
Определение азотистых органических оснований
315
мость 777 Н + от 1/р в одинаковом масштабе, то при титровании с нильским
голубым А получается более крутой градиент: значение Аэксп при фото-
метрическом определении мочевины с малахитовым зеленым равно 2,5, а при
определении с нильским голубым А — 0,47 (см. рис. 81 и 84, гл. 14).
Титрование можно также выполнять с Суданом III при длине волны
615 нм. В этом случае, однако, строится фотометрическая кривая зависимо-
сти 7Н+// от объема, так как менее основной индикатор изменяет окраску
уже после достижения эквивалентной точки (см. рис. 85, фотометрическая
кривая типа III) [671]. Относительно фотометрического титрования неболь-
ших количеств мочевины см. гл. 14 (модифицированная кривая типа II).
в. Определение первичных амидов кислот с помощью динитробензоилхло-
рида в диоксане [596] (ср. гл. 19, разд. 11, в)
Амиды кислот реагируют с 3,5-динитробензоилхлоридом:
RCONH2 + 3,5-NO2C6H3COCl —* RCN 4-3,5-NO2C6H3COOH ~ НС1,
т. е. в ходе реакции образуются хлористый водород и динитробензойная
кислота; в то же время избыток 3,5-динитробензоилхлорида дает с метиловым
спиртом хлористый водород и метилдинитробензоат.
10 мг-экв амида кислоты помещают в мерную колбу емкостью 250 мл,
в которую первоначально было внесено 15 мл динитробензоилхлорида и 5 мл
пиридина, 30—60 мин нагревают на водяной бане при 60°, затем охлаждают
в бане с измельченным льдом. Избыток динитробензоилхлорида переводят
в метилдинитробензоат, осторожно прибавляя сначала 2 мл и через 5 мин
25 мл метилового спирта. Полученный раствор титруют 0,5 н. метилатом
натрия в присутствии фенолфталеина в качестве индикатора. Глухой опыт
проводят так же. Разность между двумя измерениями дает содержание
амида кислоты.
Приготовление реагента. 461 г 3,5-динитробензоилхлорида растворяют в диоксане
п разбавляют до 1 л, встряхивают с 3,4 г активированного древесного угля и, предохра-
няя от попадания влаги из воздуха, фильтруют непосредственно в резервуар бюретки.
Раствор окрашен в бледно-желтый цвет.
Ацетамид (29,53 мг), пропиоамид (36,54 мг), салициламид (68,56 мг),
изобутирамид (43,56 мг) и др. можно определить с точностью ±0,3% (в скоб-
ках указано количество миллиграммов, эквивалентное 1 мл 0,5 н. раствора).
Формамид и его производные можно гидролизовать кипячением с едким кали в эти-
ловом спирте, избыток щелочи оттитровывают уксусной кислотой в этиловом спирте [408].
131. СУЛЬФАМИДНЫЕ ПРОИЗВОДНЫЕ
Некоторые производные сульфамидов можно титровать аналогично кисло-
там в основном растворителе (гл. 17, разд. 106), а также благодаря наличию
аминогруппы или гетероатома азота анализировать в кислом растворителе
как основания. Во многих случаях помочь титрованию может образование
нерастворимого перхлората [264, 575]. Мейленхоф предлагает добавлять
к растворителю бензол, в котором производные сульфамида не выделяют
осадка перхлората во время титрования. Чтобы способствовать разделению
моно- и диперхлората, рекомендуется после добавления избытка хлорной
кислоты разбавить раствор бензолом и через некоторое время титровать
избыток стандартного раствора 0,1 н. ацетатом натрия в уксусной кислоте.
0,8—0,9 мг-экв сукцинилсулъфатиазола суспендируют в 10 мл уксусной
кислоты, добавляют 10 мл 0,1 н. хлорной кислоты в уксусной кислоте и раст-
воряют образец, нагревая его при сильнОхМ встряхивании (перхлорат может
выделиться до того, как растворится весь образец). После охлаждения разбав-
ляют 50 мл бензола и через полчаса оттитровывают избыток хлорной кислоты,
продолжая сильно встряхивать, стандартным раствором ацетата натрия
316
Глава 23
в присутствии 1-нафтолбензеина. 35,55 мг сукцинилсульфатиазола требуют
1 мл 0,1 н. хлорной кислоты.
Незначительно видоизменив этот метод можно титровать сульфагуанидин,
сульфапиридин и сульфатиазол [575]. При титровании сульфатиазола вместо
бензола для содействия осаждению перхлората сульфатиазола используют
хлороформ. 1 мл 0,1 н. раствора эквивалентен 11,62 мг сульфагуанидина,
12,57 мг сульфапиридина и 25,53 мг сульфатиазола.
132. КЕТИМИНЫ. БЕНЗАМИДИН И РОДСТВЕННЫЕ СОЕДИНЕНИЯ.
СОЕДИНЕНИЯ, СОДЕРЖАЩИЕ ЭТИЛЕНИМИННУЮ ГРУППУ
R\
>C = NH
R'Z
Rv
>C = NR"
R'Z
Кетимин Основание Шиффа
а. Кетимины можно титровать в уксусной кислоте 0,01 н. хлорной
кислотой в присутствии кристаллического фиолетового. 0,05—0,10 мг-экв
производного кетимина растворяют в 10 мл уксусной кислоты и титруют
0,01 н. хлорной кислотой в присутствии кристаллического фиолетового [642].
1,811 мг дифенилкетимина требуют 1 мл стандартного раствора.
б. Определение бензамидина и родственных соединений [50]. Бензамидин
и некоторые родственные соединения можно титровать потенциометрически
как кислоты в пиридине или диметилформамиде стандартным раствором
ТБАГ в смеси бензол — метиловый спирт или как основания в смеси этилен-
гликоль — изопропиловый спирт (1 : 1) соляной или хлорной кислотой
(табл. 95).
NH
II
с6п8-с
I
nh2
Бензамидин
NH О О
II II II
С6Н5-С c6H5-C-N-C-C6H5
I I
n=c-c6h5 н
I
R Дибензамид
N-Бензоилбепзамидип
(R = OH);
N-тиобензоил бензамидин
(R-SH)
NH
II
с6н5-с
I
ОСН2СН2ОСН3
Бензимидат метилцеллозольва
Таблица 95
Соединение 0,2 и. НС1 Титрант Примечания
0,1 н. НС1О4 0,05 н. ТБАГ
Бензамидин — Титруется как основа- ние
Бензоат бензамидина -1- Амфотерен
N - Бензоилбензамидин + Д- — »
N - Тиобензоилбензамидин — — —к- Титруется как кислота
Дибензамид — — То же
Бензимидат метилцелло- зольва —I- — Титруется как основа- ние
Определение азотистых органических оснований 317
Перегибы на кривой потенциометрического титрования позволяют ана-
лизировать смеси, сила оснований в которых уменьшается в таком поряд-
ке: бензамидин >» бензоат бензамидина > бензимидат метилцеллозольва >»
> N-бензоилбензамидин, а порядок кислотности следующий: дибензамид >»
>> Х-тиобензоилбензамидин >» бензоат бензамидина >» N-бензоилбензамидин.
В колбе для титрования емкостью 30 мл взвешивают такое количество
вещества, на титрование которого расходуется 2—5 мл стандартного раствора.
Титрование в пиридине преимущественно проводят в атмосфере азота. Если
определение выполняют в смеси этиленгликоль — изопропиловый спирт
(1 : 1), то проводить титрование в закрытой системе не обязательно. В обоих
случаях используют систему электродов стекло — каломель, в качестве
щелочного титранта применяют 0,05 н. раствор ТБАГ в смеси бензол —
метиловый спирт, а в качестве кислого титранта — 0,2 н. соляную кислоту
в G — Н-растворителе (смесь гликолей) или 0,1 н. хлорную кислоту в уксус-
ной кислоте. При двух или более перегибах на потенциометрической кривой
вычисления ведут последовательно по каждой из точек перегиба.
Если ТБАГ содержит в качестве примеси карбонат тетрабутиламмония
и титр раствора установлен по бензойной кислоте, то при титровании бензо-
илбензамидина (очень слабая кислота) получают завышенное значение. По-
этому рекомендуется устанавливать титр ТБАГ по чистому бензоилбенз-
амидину.
в. Определение соединений, содержащих этилениминную группу [720].
Соединения, содержащие этилениминную группу, реагируют с роданисто-
водородной кислотой с разрывом кольца. Согласно методу Шлитта, роданисто-
водородная кислота образуется in situ из роданида калия и п-толуолсульфо-
кислоты. Не вступившую в реакцию n-толуолсульфокислоту титруют едким
кали в метиловом спирте в присутствии соответствующего индикатора.
/СН2
— N< | -j- KSCN + n-CH3C6H4SO3H —> —NHCH2CH2SCN + n-CH3C6H4SO3K
ХСН2
Приведенная реакция быстро проходит до конца.
В колбу Эрленмейера емкостью 250 мл помещают 2,5 мг-экв образца
и растворяют в 50 мл раствора роданида калия и точно 10 мл раствора п-то-
луолсульфокислоты, перемешивают раствор магнитной мешалкой. Титро-
вание ведут в двухгорлой колбе Эрленмейера: одно горло служит для ввода
азота, в другое помещают кончик бюретки на 50 мм. Перемешивают раствор
несколько минут в атмосфере азота, добавляют 6 капель индикатора и титру-
ют смесь 0,1 н. едким кали в метиловом спирте. Для определения индивиду-
альных соединений используют различные индикаторы, изменение окраски
которых первоначально контролируют потенциометрически.
Глухой опыт следует проводить в тех же условиях.
Рекомендуется использовать свежеприготовленный раствор п-толу-
олсульфокислоты в метиловом спирте, так как последний взаимодействует
с n-толуолсульфокислотой с образованием метилового эфира. Изменение
окраски индикатора в случае несвежеприготовленного индикатора менее
четко, и часто можно наблюдать два перегиба на потенциометрической
кривой.
Некоторые препараты n-толуолсульфокислоты содержат в качестве при-
меси следы железа(Ш). Вследствие этого при добавлении п-толуолсульфокис-
лоты к раствору роданида калия появляется красное окрашивание. Оно не
не мешает определению, так как постепенное исчезновение красной окраски
указывает на близость конца титрования. Но выбранный индикатор следует
добавлять к раствору, когда красное окрашивание начинает исчезать.
Необходимые реагенты: 0,1 н. раствор едкого кали в метиловом спирте, титр раствора
устанавливают по бифталату калия: роданид калия, полученный растворением 160 г
'K.SCN в 1000 мл метилового спирта; п-толуолсульфокпслота, 73,2 г моногидрата п-толу-
318
Глава 23
олсульфокислоты растворяют в 50 мл дистиллированной воды и разбавляют метиловым
спиртом до 1 л. Вода добавляется для замедления образования метилового эфира п-
толуолсульфокислоты. Шлитт использовал для определения азиридиновых соединений
три различных индикатора: 0,1 %-ный бромтимоловый синий в метиловом спирте; 0,1 % -
ный раствор калиевой соли фенолового красного в метиловом спирте и, наконец, инди-
каторную смесь, приготовленную смешением равных объемов двух указанных выше
индикаторов.
Описанным методом можно определить следующие соединения (используе-
мый индикатор приведен в скобках): например, окись пг/шс-1-(2-метил)-
азиридинилфосфина (бромтимоловый синий), окись фенил-бис-1-(2-метил)ази-
ридинилфосфина (индикаторная смесь), тримезоил-1-(2-этил)азиридин (фено-
ловый красный), пг/ше-(2-метил)азиридинилтриазин (бромтимоловый синий).
133. ГИДРАЗИНЫ, ЗАМЕЩЕННЫЕ ГИДРАЗИНЫ, ГИДРАЗИДЫ И ГИДРАЗОНЫ
ч н+
ii2n—nh2-------- ii2n-nhj
Гидразин Ион гпдразпния
р/С 5,52
(CH3)2N-NH2
flci/jt.if-Замещенный гидразин [541]
n-NH2 - С6Н4 - CONH — NH2
Гидразид n-аминобензойной кислоты
потенциометрически титруется как би-
функциональное основание [732]
с6п5- nh-nii2
Фепилгидразин
рА 8,80
2,4-Динитрофенилгидразин и 2,4-динитро-
фенилгидразоны мало основны и тит-
руются как кислоты (гл. 20, разд. 114)
R — CH = N — NH2; IVKC = N-NH2
Гидразоны [541]
RR'C = N — NH —COR"
Неосновной гидразон
RR'C=N— NH — CO —R"
Гидразид изонпкотиновой кислоты, потен-
циометрически титруется как бифункцио-
нальное основание [732]
CONHNHCOCH3
Ацетилированный гидразид изоникотино-
вой кислоты титруется как гетероцикли-
ческое монофункциональное основа-
ние [732]
Изоникотинилгидразон, титруется
по N-гетероатому заместителя R" как
основание [732] (см. гл. 23, разд. 121,6)
R — СН = N — N = CII — R
Неосновной альдазин [541]
(N — основной атом азота, N — неосновной или слабоесновной атом азота).
Гидразин, замещенные гидразины, гидразиды и некоторые гидразоны —
монофункциональные основания, если заместитель R не содержит второго
протонируемого атома азота. Сенси и Галло [732] детально обсудили титро-
вание этих соединений хлорной кислотой.
Если уксусная кислота, используемая для растворения, содержит сле-
ды уксусного ангидрида, гидразиды частично или полностью ацетилируются
и результат получается ошибочным. Вследствие этого в случае гидразида
изоникотиновой кислоты и подобных соединений, т. е. тех соединений, где,
кроме ацетилируемой первичной аминогруппы, присутствует третичный
N-атом, рекомендуется использовать для растворения образца уксусную
кислоту, содержащую 10—20% уксусного ангидрида, и через 10—30 мин тит-
ровать моноацетилированный продукт хлорной кислотой [732].
Гидразиды следующих кислот можно титровать в уксусной кислоте„
свободной от ангидрида, со стеклянным и каломельным электродами (в ckocv-
Определение азотистых органических оснований
319
ках указано количество вещества в миллиграммах, эквивалентное 1 мл
0,1 н. хлорной кислоты): бензойной (13,61), п-оксибензойной (14,81), и-ами-
нобензойной (7,557), п-нитробензойной (18,11), фенилуксусной (15,02), циан-
уксусной (9,909).
Если молекулы содержат также и третичный атом азота, то желательно
ацетилировать первичную аминогруппу.
а. Определение гидразида изоникотиновой кислоты (ИНГ) [442, 732].
100—120 мг образца растворяют в 20—30 мл уксусной кислоты, содержащей
10% уксусного ангидрида, и титруют потенциометрически 0,1 н. хлорной
кислотой в уксусной кислоте, используя стеклянный и каломельный электроды.
13,71 мг ацетилированного продукта требуют 1 мл 0,1 н. хлорной кислоты.
В смеси уксусная кислота — хлороформ (4 : 1) ИНГ можно титровать как основа-
ние, а в диэтиламине — как кислоту — едким натром [12].
б. Определение гидразида изолизергиновой кислоты (ИЛГ) [306]. 60—
200 мг образца растворяют, защищая от света, в 20 мл уксусной кислоты,
содержащей 15% уксусного ангидрида, и титруют через 10 мин 0,05 н. хлор-
ной кислотой в присутствии кристаллического фиолетового. 14,12 мг ИЛГ
требуют 1 мл стандартного раствора.
Гидразиды никотиновой и пиколиновой кислот можно титровать так же,
но Х-гетероатом гидразида 2-тиазолкарбоновой кислоты обладает такой
слабой основностью, что при этом будет титроваться первичная аминогруппа.
НС—N
II II
НС С — СО — NH — NH2 Мол. вес 143,16
Гидразоны, в которых первичная аминогруппа замещена на группу COR^
можно титровать, только если заместитель R содержит основной N-атом
(см. гл. 23, разд. 121, б).
в. Определение смеси гидразина и 1,1-диметилгидр азина * [541]. В уксус-
ной кислоте с салицилальдегидом гидразин образует нейтральный салицил-
альдазин, в то время как 1,1-диметилгидразин образует гидразон основного
характера
N2H4 +2С6Н4(СНО)ОН —> НО —С6Н4 —CH = N —N = CH —С6Н4 —ОН + 2Н2О
(CH3)2N — NH2 + C6H4(CHO)OH НО —С6Н4 —CH = N —N(CH3)2 —Н2О
Основность 1,1-диметилгидразона салицилальдегида такая же, как
и у 1,1-диметилгидразина, и его можно титровать хлорной кислотой в уксус-
ной кислоте.
Определяют вес мерной колбы емкостью 50 мл, содержащей 40 мл уксус-
ной кислоты, и добавляют при охлаждении примерно 2 г смеси гидразинов.
Когда раствор примет комнатную температуру, колбу снова взвешивают
и доводят объем раствора до 50 мл, добавляя уксусную кислоту. 1,0 мл
полученного раствора разбавляют 20 мл уксусной кислоты, титруют хлорной
кислотой в присутствии метилового фиолетового и определяют сумму основа-
ний (А). Другую порцию раствора тоже 1,0 мл смешивают с 20 мл уксусной
кислоты и 2 мл салицилальдегида и выдерживают 10 мин при 50°; при этом
осаждается желтый салицилальдазин. К раствору добавляют 6 капель
метилового фиолетового и титруют 0,01 н. хлорной кислотой в диоксане до пере-
хода коричнево-красной (янтарной) окраски в темно-зеленую (В).
(В — ь2). N-6,01 п,
Содержание диметилгидразина = -------- %
[(л-^-св-ЬАЬлг.зда
Содержание гидразина = ---—-————---------- %
С/
* Гидразин, монометилгидразин и 1,1-диметилгидразин важны как ракетное топливо.
320
Глава 23
где А и В — количество расходуемой 0,1 н. НС1О4 в диоксане, мл; С — вес
образца; Ь{ — объем, необходимый для титрования растворителя и индика-
тора, мл; Ь2 — объем, пошедший на глухой опыт с салицилальдегидом, мл;
N — нормальность стандартного раствора; 6,01 = мг-экв X 100 для диме-
тилгидразина, 3,205 = мг-экв X 100 для гидразина. Использование потен-
циометрического и фотометрического (тип 77) определения конца титрования
при анализе смеси гидразина и 1,1-диметилгидразина методом Малоуна
с салицилальдегидом [541] было изучено Бернсом и Лоулером [109]. Исполь-
зуя двухступенчатое равновесие с кристаллическим фиолетовым в качестве
индикатора, сумму оснований определяют при 590 нм; причем конечную
точку титрования устанавливают четче, чем по кривой титрования, наблюда-
емой при 630 нм. Поскольку кристаллический фиолетовый в качестве инди-
катора дает нечеткую конечную точку (данные различных авторов значи-
тельно отличаются), которая зависит от природы источника света, метод
Малоуна включает значительно больше источников ошибок, чем метод
Бернса и Лоулера.
Фотометрическое определение конечной точки титрования довольно
сложно, поэтому серийные анализы рекомендуется проводить методом потен-
циометрического титрования [109] с использованием муфты Бекмана типа
каломельного электрода, содержащего ледяную уксусную кислоту, насыщен-
ную хлоридом лития.
г. Кислотно-основной метод определения смесей монометилгидразина
и 1,1-диметилгидразина см. в работе [542].
Глава 24
ОПРЕДЕЛЕНИЕ СОЛЕИ
АЗОТИСТЫХ ОСНОВАНИИ
В фармацевтической промышленности многие соединения, содержащие
азотистые основания, получают в виде солей. Большинство солей, образо-
ванных органическими кислотами, можно непосредственно титровать хлор-
ной кислотой в уксусной кислоте или в кислой смеси растворителей. При
анализе солей, образованных галогеноводородными кислотами, к раствору
добавляют ацетат ртути(П); при этом образуется недиссоциирующий гало-
генид ртути(П) и ацетат азотистого основания [207, 462, 644].
2В • НС14- Hg(CH3COO)2 = 2В .СН3СООН + HgCl2.
95% сульфат-ионов из сульфатов азотистых оснований связывается
бензидином, причем освобождающееся основание можно титровать хлорной
кислотой [265]. Сульфаты мало растворимы в уксусной кислоте, однако в сме-
си фенола, хлороформа и ацетонитрила (1:4: 10) многие сульфаты, фосфаты,
а также некоторые этансульфонаты и n-толуолсульфонаты (в диоксане) мож-
но титровать хлорной кислотой в диоксане [123, 321]. В такой смеси раство-
рителей нельзя титровать галогениды оснований вследствие того, что под
действием ацетата ртути(П) идут побочные реакции. Ацетат ртути(П)
можно использовать для определения хлоргидратов оснований в ацетоне
или в смеси ацетонитрил — уксусный ангидрид — уксусная кислота
(12 : 0,8 : 0,2) [314, 587].
134. СОЛИ АЛКАЛОИДОВ И ДРУГИХ АЗОТИСТЫХ ОСНОВАНИЙ
С ОРГАНИЧЕСКИМИ КИСЛОТАМИ
Органические кислоты, константы диссоциации которых (в воде) имеют
порядок 10-3 и ниже, при растворении в уксусной кислоте обычно образуют
нейтральные растворы (гл. 22). В основу метода определения этих соединений
положено то, что их можно титровать и как основания и как соли уксусной
кислоты. Однако влиянием кислотного остатка на степень перегиба при по-
тенциометрическом титровании нельзя пренебрегать. Это лучше всего про-
демонстрировать на примере дифференцирующего титрования различных
дифенгидраминовых солей со стеклянным электродом замедленного дей-
ствия (рис. 120).
Навеску вещества, эквивалентную 0,3—0,5 мг-экв основания, растворяют
в 20 мл уксусной или пропионовой кислоты или в случае неацетилирующихся
оснований в смеси уксусная кислота — уксусный ангидрид (10 : 1) или уксус-
ная кислота — хлорбензол (1 : 1), нейтрализованной в присутствии кристал-
лического фиолетового, и титруют 0,05—0,1 н. хлорной кислотой до перехода
окраски из фиолетовой в синюю. Алкалоиды спорыньи следует растворять,
защищая от действия света. Тартрат эрготамина, битартрат, бифталат
и малеат эргометрина можно титровать с точностью от ±0,5 до ±1%
[324, 697]. 1 мл 0,1 н. хлорной кислоты эквивалентен 16,27 мг эргометрина
и 29,08 мг эрготамина.
Для титрования салицилата физостигмина, ацетата морфина и тар-
трата дигидрокодеинона [123] в смеси фенол — хлороформ — ацетонитрил
322
Глава 24
(1:4: 10) берут такое количество образца, чтобы на титрование пошло
2—Змл 0,05 н. хлорной кислоты в диоксане. Добавляют 5 г кристаллического
фенола, 10—20 мл хлороформа и после их растворения — 50 мл ацетонитри-
ла и 1—2 капли 0,25%-ного мети-
+600
+400
-400
-600-
-800 -
+200
Я °
? -200
1 мл
Р и с. 120. Вторая производная диффе-
ренциальных кривых титрования кислых
солей дифенгидрамина (0,02 М) при
23 ± 1° [444].
1 — сукцинат; 2 — фумарат; з — малеат; 4 —
оксалат; 5 — нитрат; 6 — фосфат. Растворитель:
уксусная кислота; стандартный раствор: 0,05 н.
хлорная кислота в диоксане; система электродов:
стекло — стекло (электроды замедленного дейст-
вия, ср. рис. 51).
лового красного в смеси фенол —
хлороформ (1 : 50). Титрование мож-
но проводить и потенциометрически
(см. рис. 44, в).
Тартрат 1-(4'-оксифенил)-2-метил-
аминоэтанола (симпатол) можно
титровать в уксусной кислоте 0,05 н.
хлорной кислотой в диоксане в при-
сутствии кристаллического фиолето-
вого [651].
Определение лактата 2-этокси-
6,9-диаминоакридина (лактата) (этак-
ридин, риванол) проводят таким
образом [834]. В 15 мл метилового
спирта растворяют 400—600 мг эта-
кридина, добавляют 2—3 капли
0,5%-ного тимолового синего в мети-
ловом спирте и сразу же титруют
0,1 н. хлорной кислотой в диоксане
до перехода окраски из желтой
в оранжевую. 1 мл 0,1 н. кислоты
эквивалентен 25,33 мг основания, 34,34 мг лактата и 36,14 мг моногидрата
лактата. В процессе титрования основания или вытеснения молочной кислоты
из лактата основания при титровании расходуется 1 экв хлорной кислоты.
135. ПИКРАТЫ ОСНОВАНИЙ
Хорошо известен метод идентификации оснований по температурам
плавления их пикратов. Удобным методом определения эквивалентных
весов оснований является определение содержания оснований в пикратах
путем непосредственного титрования хлорной кислотой в подходящем
растворителе или смеси растворителей [28. 58, 134, 311, 314, 366, 400, 425,
426] и определение пикриновой кислоты титрованием метилатом натрия
в пиридине или диметилформамиде [58, 245] (ср. гл. 20).
В качестве растворителя при определении содержания оснований в пик-
ратах целесообразно применять уксусную кислоту или смесь уксусного анги-
дрида и хлорбензола, уксусного ангидрида и нитрометана или уксусной
кислоты и хлороформа. В полумикроанализе удобно использовать тройные
смеси: уксусная кислота — уксусный ангидрид — хлорбензол или пропионо-
вая кислота — пропионовый ангидрид — хлорбензол (9:2:9). Титрование
рекомендуется проводить 0,05—0,1 н. хлорной кислотой в уксусной кислоте
или 0,01 н. хлорной кислотой в смеси уксусной кислоты и четыреххлористого
углерода (1 : 1) в присутствии кристаллического фиолетового или метилового
фиолетового
C5H5N-; HOC6H2(NO2)3 (C5H5NH)+[(NO2)3C6H2OJ-
Пикрат пиридиния
Пикрат-анион и основание в уксусной кислоте выступают как акцепторы
протонов, а катион пиридиния и пикриновая кислота ведут себя в диметил-
формамиде как кислоты.
Подобно пикриновой кислоте, пикролоновая кислота также образует
нерастворимые соли с основаниями, и пикролонаты можно титровать как
Определение солей азотистых оснований
323
основания в уксусной кислоте, уксусном ангидриде или диоксане 0,01 н. хлор-
ной кислотой в уксусной кислоте в присутствии метилового фиолетового.
Пикролонат-анион определяют в пиридине действием метилата натрия в при-
сутствии тимолового синего [210]
Пикролоновая кислота (мол.вес 264,2)
а. Получение пикратов оснований [311, 314]. 1. В 10 мл дистиллированной
воды растворяют 50 мг монофункционального хлоргидрата основания
и нагревают до 60—70°. Добавляют 10 мл 1,5%-ного раствора пикриновой
кислоты в метиловом спирте и нагревают до тех пор, пока не растворится
образующийся пикрат, затем охлаждают и выдерживают 2 дня при 0°.
Отфильтровывают выделившиеся кристаллы на стеклянном пористом фильтре
№ 3, промывают (5x2 мл) водным метиловым спиртом (1 : 1) при 0° и в за-
ключение сушат 12 час в вакууме (5 мм рт. ст.) над КОН.
2. В стакане на 100 мл взвешивают 100—200 мг монофункционального
хлоргидрата основания и растворяют в 2—4 мл воды при нагревании на водя-
ной бане. Добавляют 20—40 мл кипящего водного 1 %-ного раствора пикри-
новой кислоты, раствор перемешивают и продолжают нагревание на водя-
ной бане еще в течение нескольких минут. После охлаждения отфильтровы-
вают кристаллы на стеклянном пористом фильтре № 3, промывают (5x5 мл)
дистиллированной водой при 0° и сушат 12 час в вакууме над Р2О5.
б. Титрование пикрата основания в уксусной кислоте [134]. Взвешивают
1—4 мг-экв пикрата амина и растворяют, если необходимо при слабом нагре-
вании, в 50 мл уксусной кислоты. Охлаждают до комнатной температуры
и титруют 0,1 н. хлорной кислотой в уксусной кислоте, добавив 6 капель
0,2%-ного метилового фиолетового в хлорбензоле. Цвет индикатора постепен-
но изменяется от зеленого к фиолетовому и в конце внезапно переходит
в голубой. К числу аминов, пикраты которых можно определять этим спо-
собом, относятся диметиланилин, этилендиамин, диэтаноламин, анилин,
2-аминопиридин, 2,4,5-коллидин и пиперидин.
в. Титрование пикрата основания в смеси хлорбензол — уксусная кисло-
та — уксусный ангидрид или хлорбензол — пропионовая кислота — пропио-
новый ангидрид [311, 314]. Пикраты оснований, содержащих третичный атом
азота, можно точно титровать как потенциометрически, так и визуально
в присутствии метилового фиолетового 0,01 н. хлорной кислотой в одной
из вышеупомянутых смесей растворителей (компоненты смеси берутся
в соотношении 9:9:2). Наблюдаемая в конечной точке титрования вели-
чина скачка потенциала в смеси, содержащей пропионовую кислоту, вдвое
больше, чем в смеси, содержащей уксусную кислоту.
20—40 .иг пикрата основания растворяют в 10—20 мл смеси растворителей
и титруют 0,01 н. хлорной кислотой или со стеклянным и каломельным элек-
тродами или в присутствии метилового фиолетового до тех пор, пока окраска
индикатора не перейдет в голубую. Раствор электролита в каломельном
электроде — уксусная кислота, насыщенная хлоридом лития. Стеклянный
324
Глава 24
электрод перед использованием помещают на 1 час в смесь уксусного анги-
дрида и уксусной кислоты (1 : 9), содержащей несколько капель стандарт-
ного раствора хлорной кислоты. Этим способом можно определять пикраты,
например, пиперидинометилтолилпропанона или пиперидинометилцикло-
гексанона.
г. Идентификация антигистаминов титрованием пикрата основания
хлорной, кислотой [28]. Примерно 40 мг пикрата антигистамина растворяют
в 15 мл уксусной кислоты, добавляют 15 мл хлороформа и титруют 0,02 н.
хлорной кислотой в уксусной кислоте потенциометрически со стеклянным
и каломельным электродами или титрование ведут в присутствии кристал-
лического фиолетового и определяют конечную точку визуально. Этим спо-
собом с помощью определения эквивалентного веса можно идентифицировать
20 различных антигистаминов, например монопикрат дифенгидрамина, дипи-
крат пирибензамина, монопикрат прометазина, дипикрат циклизина, дипи-
крат хлорциклизина и т. д.
д. Титрование пикратов меркаптопиридиновых производных [58]. Пик-
раты диалкиламинопроизводных 4-меркаптопиридина (Л) можно титровать
как бифункциональные основания в уксусном ангидриде или в смеси нитро-
метан — уксусный ангидрид. Пикрат 4-меркаптопиридинодиалкилацетами-
да (Б) можно титровать как монофункциональное основание. Пикраты также
можно титровать метилатом натрия в диметилформамиде в присутствии
тимолового синего. В этом случае определяют пикриновую кислоту, содержа-
щуюся в пикрате [58].
z--ч /R z--ч /R
S-CH2 —СН2 —N< N<f >—S —СН2—CON<
\R Х=/ \R
е. Определение зефирола [400]. По международной номенклатуре соеди-
нения типа
R4 +/СН3
>N<
Н3(У хсн2с6н5
Cl-
где R — алкильная группа с неразветвленной цепью С8Н17, называют хло-
ридами «бензалкония».
80—100 мг соединения растворяют в маленькой колбе в 5 мл воды,
добавляют 20 мл насыщенного при комнатной температуре водного раствора
пикриновой кислоты и 1 мл 10 %-ной соляной кислоты. Нагревают 1 час
на кипящей водяной бане, затем оставляют стоять на час при комнатной тем-
пературе. Осадок отфильтровывают на бумажном фильтре. Ополаскивают
колбу (3 X 5 мл) раствором пикриновой кислоты и этой жидкостью (3x5 мл)
промывают осадок на фильтре. Помещают воронку с бумажным фильтром
в колбу и небольшим количеством ацетона смывают осадок в колбу. От-
гоняют ацетон (для осаждения и концентрирования раствора можно
использовать колбу для выпаривания, изображенную на рис. 103), раство-
ряют остаток в 20 мл уксусной кислоты и титруют 0,1 н. хлорной кислотой
в уксусной кислоте в присутствии 2—3 капель 0,1%-ного кристаллического
фиолетового до появления голубого окрашивания. Содержание хлорида бенз-
алкония в растворе зефирола можно определять аналогичным образом.
Взвешивают 1—1,5 г раствора и после добавления 20 мл воды обрабатывают
20 мл раствора пикриновой кислоты, как описано выше. Расчеты выполняют,
исходя из предположения, что средний молекулярный вес равен 390 [400].
См. также разд. 137
Определение солей азотистых оснований
325
136. ГАЛОГЕНГИДРАТЫ (ГАЛОГЕНИДЫ) АЗОТИСТЫХ ОСНОВАНИЙ
Основной компонент хлористоводородной соли основания можно титро-
вать в уксусной кислоте в системах уксусная кислота — уксусный ангидрид,
уксусная кислота — хлоробензол, уксусная кислота — ацетонитрил и др.,
превратив сначала галогеноводородную соль кислоты действием ацетата
ртути(П) в хлорид ртути(П) (см. введение к гл. 24). Для этой цели исполь-
зуют 3%-ный раствор ацетата ртути(П) (приблизительно 0,2 н.) в уксусной
кислоте; причем добавляют 10 мл последнего к 10 мл 0,1 н. раствора моно-
функциональной хлористоводородной соли основания. В результате около
50% ацетата ртути(П) переходит в хлорид ртути(П). Если хлоргидрат осно-
вания нерастворим в уксусной кислоте, ацетат ртути(П) повышает раство-
римость (например, некоторых ароматических или жирноароматических
аминоспиртов). Целесообразно растворитель, как и ацетат ртути, предвари-
тельно нейтрализовать хлорной кислотой в присутствии подходящего инди-
катора, даже если титрование выполняют потенциометрически (см. гл. 9,
разд. 54).
Для 0,05 н. и более разбавленных растворов используют меньшие коли-
чества ацетата ртути(П): достаточно 10—20%-ного избытка.
Хлоргидраты многих фармацевтических важных соединений можно тит-
ровать в присутствии ацетата ртути(П) 0,01—0,1 н. хлорной кислотой как
с потенциометрическим, так и фотометрическим или визуальным определе-
нием конечной точки. Ниже мы рассмотрим некоторые типичные методы.
а. Определение хлоргидрата папаверина в хлороформе [836]. 500 мг хлор-
гидрата папаверина растворяют в 50мл хлороформа, добавляют 5л«л 6%-ного
раствора ацетата ртути(П) в уксусной кислоте и 2 капли индикатора
0,1%-ного раствора метанилового желтого в метиловом спирте. Титруют
0,1 н. хлорной кислотой в диоксане до перехода желтой окраски в фиолето-
вую. На 37,58 мг хлоргидрата папаверина расходуется 1 мл 0,1 н. НС1О4.
б. Фотометрическое титрование хлоргидрата хинина [211]. Хлоргидрат
хинина можно титровать как монофункциональное основание в уксусной
кислоте. 160—170 мг вещества растворяют в 20 мл уксусной кислоты и добав-
ляют 2 капли 0,5 %-ного раствора метиленового синего в уксусной кислоте
в качестве индикатора. Титруют в 15-миллиметровой кювете колориметра,
например модели Ланге IV, 0,1 н. хлорной кислотой (фильтр 0G2; область
пропускания 550—580 нм). Для вычисления результатов пользуются фото-
метрической кривой, показывающей зависимость ИТ от Vlf (см. гл. 14,
рис. 90). Хлоргидрат хинина можно титровать как бифункциональное
основание, если образец растворить в смеси 10 мл нитрометана и 10 мл
3%-ного раствора ацетата ртути(П) в уксусной кислоте. 1 мл 0,1 н. хлорной
кислоты эквивалентен в зависимости от метода анализа 36,09 или 18,04 мг
безводного хлоргидрата хинина.
в. Определение антибиотиков. Для определения антибиотиков как
в водных, так и в неводных средах не существует каких бы то ни было спе-
цифических общих методов весового или объемного определения, так как
эти соединения принадлежат к различным структурным типам.
Хлортетрациклин, тетрациклин и окситетрациклин (основания или
хлоргидраты) можно титровать потенциометрически 0,05 н. хлорной кисло-
той в диоксане. Растворителем для антибиотика служит нитрометан, содержа-
щий муравьиную кислоту. Определение конечной точки потенциометрически
(система электродов: стекло — Ag/AgCI) или визуально (индикаторная
смесь: метиленовый синий — хинальдиновый красный) дает одни и те же
результаты [890].
В стакане емкостью 150 мл взвешивают примерно 50 мг антибиотика и при
перемешивании магнитной мешалкой растворяют в смеси муравьиная кисло-
та — нитрометан (1 : 50). После растворения добавляют 5 мл бензола и 1 мл
326
Глава 24
6%-ного раствора ацетата ртути(П) и титруют 0,05 н. хлорной кислотой
в диоксане в присутствии 0,1 мл индикаторной смеси до появления зеле-
ного оттенка. 1 мл титранта эквивалентен 25,77 мг хлоргидрата хлортетра-
циклина, 24,85 мг хлоргидрата окситетрациклина или 24,05 мг хлоргидрата
тетрациклина.
Об определении антибиотиков в мазях, свечах и препаратах для инъекций см. в рабо-
тах [207, 742, 890]. Анализ стрептомицина и дигпдрострептомицинсульфата рассматри-
вается в работе [635]. Определение окситетрациклина (основания и хлоргидрата) с помо-
щью титрования при высоких частотах см. в работе [524]. Способы определения эквива-
лентного веса нистатина, фунгицидного антибиотика, в неводной среде обсуждаются
в работе [563].
Определение хлормицетина [хлорамфеникол, D-(—)-л?г/>ео-1-(п-нптрофенпл)-2-дпхлор-
ацетамидпропандиол-1.3]
ОН
I
O,N - С6Н4 - СН — СН - СН2 - ОН
I
NH-СО —СНС12
При гидролизе амидной связи 25%-ной соляной кислотой образуются дихлоруксус-
ная кислота и й-трео-1-(п-нптрофенил)-2-аминопропандиол-1,3, причем последний можно
титровать хлорной кислотой [705].
г. Определение антигистаминов (см. табл. 96). К числу антигистаминов
относятся соединения с весьма различной структурой; некоторые из них
содержат 1 или 2 гетероатома азота, а в алифатических боковых цепях также
и третичный N-атом, причем часто удается титровать только последний (ср.
гл. 23, разд. 122, и табл. 92).
Титрование в ацетоне. 50 мг хлоргидрата промазина растворяют в 40 мл
ацетона, добавляют 1 мл 3%-ного раствора ацетата ртути(Н) в уксусной кис-
лоте и титруют 0,05 н. хлорной кислотой в диоксане в присутствии метилово-
го красного. На 16,04 мг хлоргидрата расходуется 1 мл стандартного
раствора [587].
280—300 мг хлоргидрата хлорпромазина или прометазина растворяют
в смеси 5 мл 5%-ного раствора ацетата ртути(П) и 100 мл ацетона. Добав-
ляют 1 мл 0,25%-ного раствора метилового оранжевого в ацетоне и титруют
0,1 н. хлорной кислотой. 1 мл стандартного раствора эквивалентен 35,53 мг
хлоргидрата хлорпромазина или 32,09 мг хлоргидрата прометазина [453].
По сравнению с уксусной кислотой преимущество ацетона заключается в том, что
при анализе таблеток, содержащих стеарат магния как наполнитель, на реакцию
с последним не расходуется хлорная кислота.
Титрование в смеси бензол — уксусная кислота — уксусный ангидрид
[683]. Систематическое исследование 25 антигистаминов было выполнено
Ринком и Римхофером с использованием потенциометрического и визуального
определения конечной точки. В качестве индикаторов они использовали
бриллиантовый зеленый, кристаллический фиолетовый и жировой синий Б
(Fettblau В).
Взвешивают такое количество вещества, на титрование которого потре-
буется примерно 10 мл 0,1 н. хлорной кислоты. Растворяют, если необходи-
мо при слабом нагревании, в 10 мл раствора ацетата ртути(П) в уксусной
кислоте, охлаждают до комнатной температуры и добавляют к раствору
25 мл бензола и 5 мл уксусного ангидрида. Титруют хлорной кислотой в при-
сутствии бриллиантового зеленого. 1 мл хлорной кислоты эквивалентен
14,59 мг хлоргидрата трипеленамида (неоантергана), 15,84 мг совентола
и 16,31 мг хлоргидрата хлорпирамина.
Титрование в системе бензол — уксусная кислота [683] проводят так же,
как описано выше, но без уксусного ангидрида. 1 мл 0,1 н. хлорной кислоты
эквивалентен 25,30 мг хлоргидрата буклизина, 29,18 мг хлоргидрата дифен-
гидрамина, 34,03 мг систрала и 27,58 мг прагмана.
Определение солей азотистых оснований
327
Титрование в смеси растворителей уксусная кислота — хлороформ (а)
или ацетонитрил — хлороформ (б) [133]. Клер и Уэттен исследовали
18 различных антигистаминов.
Взвешивают тонко измельченные таблетки, содержащие 0,15—0,25 мг-
экв чистого соединения, добавляют 15 мл хлороформа, после чего перемеши-
вают 15 мин магнитной мешалкой, фильтруют через стеклянный фильтр,
промывают фильтр и осадок (2x5 мл) хлороформом и объединенные фильт-
раты смешивают с 25 лм уксусной кислоты (а) или ацетонитрила (б). Титруют
0,05 н. хлорной кислотой в диоксане, добавив 2 капли 0,5%-ного кристал-
лического фиолетового (а) или 2 капли 0,1 %-ного метилового красного
в метиловом спирте (б).
При проведении анализа солянокислых солей оснований добавляют
1 мл 6 %-ного ацетата ртути(П) к 50 мл смеси укусная кислота — хлороформ
или к 50 мл смеси уксусная кислота — ацетонитрил.
При анализе таблеток следует учитывать также и наполнитель. Влияние
примесей наполнителя на результаты анализа рассматривается в табл. 76.
Этот метод можно использовать для анализа ряда соединений (табл. 96);
соединения под номерами 1, 2, 4, 5 содержат эфирную группировку и один
высокоосновный третичный N-атом в молекуле (расходуется 1 экв хлорной
кислоты). В антигистаминах, обозначенных номерами 7 и 8 (сукцинат
и малеат), бензольное кольцо заменено на пиридиновое и атом азота в кольце
обладает меньшей основностью, чем алкиламинный атом азота. В системе
уксусная кислота — ацетонитрил эти 2 атома азота можно оттитровать
потенциометрически. Для визуального определения больше подходит сис-
тема уксусная кислота — хлороформ, в этом случае расходуется 2 экв хлор-
ной кислоты.
Соединения под номерами 10—12 (малеаты) можно титровать аналогично
в смеси уксусная кислота — хлороформ. Соединение номер 13 (тартрат)
представляет молекулу, в которой на титрование N-атома 2-метилтетрагидро-
пиридиндена в системе уксусная кислота — ацетонитрил расходуется 1 экв
хлорной кислоты. Это соединение слабо растворимо в хлороформе, но рас-
творимость его увеличивается при добавлении фенола [123].
Соединения 15, 17 и 18 содержат третичный алифатический N-атом наряду
с фентиазиновым или тиофенилпиридиламиновым кольцом. На титрование
соединений 15 и 17 в системе уксусная кислота — хлороформ расходуется
один эквивалент хлорной кислоты. На титрование соединения 18 расходуется
также 1 экв, но для этого следует использовать систему растворителей
уксусная кислота — ацетонитрил (азот в фентиазиновом кольце не про-
тонируется).
Соединения 19 (хлоргидрат), 20 (малеат), 21 и 22 (хлоргидраты) можно
рассматривать как замещенные производные этилендиамина и в смеси уксус-
ная кислота — хлороформ для их титрования требуется 2 экв хлорной кислоты.
Тонзиламин (соединение 22) содержит пиримидиновое кольцо, но два гетеро-
атома азота титровать не удается (ср. гл. 4, разд. 27,а), и в смеси уксусная
кислота — ацетонитрил расходуется только 1 экв хлорной кислоты, но изме-
нение окраски происходит резко.
Соединение под номером 25 содержит пиперазиновое кольцо; в смеси
уксусная кислота — ацетонитрил на его титрование расходуется 1 экв,
а в смеси уксусная кислота — хлороформ 2 экв хлорной кислоты (см. также
работу [132] и гл. 23, разд. 123).
(Анализ дифенгидрамина рассматривается также в работах [407] и [576],
рис. 120).
д. Анализ анестезирующих средств. Определение после ацетилирования
[707]. Прокаин (новокаин, n-аминобензоат 2-диэтиламиноэтилового спирта)
и ларокаин (n-аминобензоат 2,2-диметил-З-диэтиламинопропилового спирта)
содержат первичную аминогруппу, в то время как тетракаин (п-бутиламино-
Таблица 96
’^б\, С—О—CH2CH2N (СН3) 2 X II н? N аз X ио к II II II II И XN чЧ СЧ СО Дифенгидрамин, 0-диметил- аминоэтилбензгидрило- вый эфир Бромдифенгидрамин, 0-ди- метиламиноэтил-л-бром- бензгидриловый эфир Систрал, Р-диметиламино- этил-п-хлор-1-метилбенз- гидриловый эфир Дименгидринат, 8-хлортео- филлинат Р-диметилами- ноэтилбензгидрилового эфира
ВДв у < СН—О—у Н СНз Н5(£ '—' 5. Пипрингидринат, 8-хлортео- филлинат N-метилпипе- ридил-(4)-бензг пдрилово- го эфира
н5се /—\ N—/ Н N—СН3 Н5Се—Н2С ' ' 6. Совентол, М-фенил-Х-бен- зил-4-амино-1- метилпипе- ридин
х \Ол CH2CH2N (СН3)2 7. Х = Н Z = СН3 8. Х = С1 Z = H 9. Доксиламин, 2-[1-(2-диме- тиламиноэтокси)-1-метил- бепзил]пирпдин Карбиноксамин, 2-[п-хлор- 1-(диметиламиноэтоксп)- бензил]пиридин Прагман, 1-фенил-1-п-то- лил-3-диметилпропил- амин
^ун—CH2CH2N(CHa)2 10. Х = С1 11. Х = Н 12. Х = Вг Хлорфенамин, й-3-(п-хлор- фенил)-3-(2'-пиридил )- N, N-диметилпропиламин Фенирамин. 3-фенил-3-(2'- пиридпл)-Х,Х-диметил- пропиламин Бромфенирамин, 3-(п-бром- фенил)-3-(2' -пирид пл )- Х,Х-диметилпропиламин
С6Н5 /\/\/\ НЦ^СНз 13. Фениндамин, 2-метил-9-фе- нил-1,2,3,4-тетрагидро- пиридинден
Z 14. Х = Н Y = C г = 3-Диме- тиламинопро- пил Промазин, Х-(3-диметил- аминопропил)фентиазин
Определение солей азотистых оснований
329*
Продолжение табл. 96
15.
16.
17. Х = Н
Y = G ___
z=ch2-ch2n/_______|
18. Х = Н
Y = N
Z = 2-Диметилами-
нопропил
Прометазин (см. табл. 92)
Хлорпромазин (см. табл. 92)-
Паратиазин, М-(0-пирроли-
диноэтил)фентиазин
Изотипендил, N-диметил-
аминоизопропилтиофе-
нилпиридиламин
GH2-R
I
N-GH2GH2N(CH3)2
R'
19. R = Фенил
R' = 2-Пиридил
20. R = п-Метоксифенил
R' = 2-Пиридил
Трипеленнамин(неоантер-
ган), N, М-диметил-N' -(2' -
пиридил)-Х' -бензил эти-
лендиамин
Мепирамин, М,1М-диметил-
N' -(2' -пирид ил)-Х' -(л-
метоксибензил)этилен-
диамин
R' = 2-Пиридил
22 R — л-Метоксифенил
R' =2-Пиримидил
23. R = n-Хлорфенил
R' = Пиридил
24. R = л-Бромфенил
R' = Пиридил
Метапирилин, N.N-диме-
тил-М'-(2'-пиридил)-М'-
[2'-тиофенил-(2)]-этилен-
диамин
Тонзиламин, Х'-(2-пири-
мидил)-Х' -(п-метоксибен-
зил)-Х,Х-диметил этилен-
диамин
Хлоропирамин, М,1М-диме-
тил-N' -(2' -пиридил)-Х' -
(л-хлорбензил)этилендиа-
мин
Гибернон, М,М-диметил-М'-
(2' -пиридил)-Х' -(п-бром-
бензпл)этилендиамин
25. R = Метил
26. R = ^H2
I
С6Н4
I
С(СН3)з
Хлорциклпзин, сМ-1-(4'-
хлорбензгидрил)-4-метил-
пиперазин
Буклизин, 1-(4'-хлорбенз-
гидрил)-4-(л-лпрелп-бутил-
бензил)пиперазин
бензоат 0-диметиламиноэтилового спирта) — вторичную аминогруппу непо-
средственно в ароматическом кольце. Эти соединения используют в виде
хлоргидратов. Первичный, вторичный или третичный атомы азота в этих
соединениях можно титровать потенциометрически; при этом можно наблю-
дать два перегиба. В случае визуального определения (например, с кристал-
лическим фиолетовым) резкого изменения окраски не наблюдается; поэтому
целесообразно сначала ацетилировать первичную или вторичную амино-
группу, а затем титровать только третичный N-атом (см. гл. 23, разд. 133).
В уксусной кислоте на титрование прокаина расходуется 1 экв хлорной кислоты
в присутствии хинальдинового красного и 2 экв с 1-нафтолбензеином [386].
330
Глава 24
Примерно 200 мг образца растворяют в 20 мл смеси уксусная кислота —
уксусный ангидрид (5:1), нагревают до кипения, охлаждают и смешивают
с 20 мл диоксана. Добавляя 10 мл ацетата ртути(П), выделяют свободное
основание из соли и титруют в присутствии кристаллического фиолетового.
1 мл 0,1 н. хлорной кислоты эквивалентен 27,27 мг хлоргидрата прокаина,
31,47 мг хлоргидрата ларокаина и 30,08 мг хлоргидрата тетракаина.
Ацетилирование в ацетоне [453]. Тетракаин быстро ацетилируется в сме-
си ацетон — уксусный ангидрид без нагревания.
250 мг хлоргидрата тетракаина растворяют в смеси 5 мл 5 %-ного ацетата
ртути(П) и 5 мл уксусного ангидрида, добавляют 50 мл ацетона и титруют
0,1 н. хлорной кислотой в присутствии 1 мл насыщенного раствора метило-
вого оранжевого в ацетоне. 400мг хлорпрокаина (хлоргидрат 2-хлор-4-амино-
бензоат 2-диэтиламиноэтилового спирта) ацетилируют в смеси 80 мл уксусной
кислоты и 2 мл уксусного ангидрида при нагревании на кипящей водяной
бане; после охлаждения, чтобы выделить основание, добавляют 10 мл 5%-ного
ацетата ртути(П) и титруют 0,1 н. хлорной кислотой.
На 30,72 мг хлорпрокаина расходуется 1 мл 0,1 н. стандартного раст-
вора кислоты*.
е. Определение хлоргидрата метил(1-фенил-1 -пиперидил-{2)-ацетата)
(сентедрина) [317]. Полумикротитрование. 60—70 мг образца растворяют
в 100 мл смеси уксусная кислота — хлороформ (1 : 10), нейтрализованной
в присутствии тропеолина ОО. Смешивают 10 мл полученного раствора
с 0,2 мл 3 %-ного раствора ацетата ртути(П) в уксусной кислоте и, добавив
3 капли 0,5%-ного раствора тропеолина ОО в уксусной кислоте, титруют
0,01 н. хлорной кислотой в смеси уксусная кислота —четыреххлористый угле-
род (1 : 1) до тех пор, пока индикатор не даст окраски фуксина. Для 2,698 мг
сентедрина необходим 1 мл 0,01 н. раствора.
Титрование в ацетоне. Хлоргидрат метил-1-фенил-1-пиперидил-(2)-ацета-
та нерастворим в ацетоне, но растворяется в уксуснокислом растворе ацета-
та ртути(П).
250—300 мг образца растворяют в 100 мл смеси ацетона и 6?^-ного раство-
ра ацетата ртути(П) в уксусной кислоте (95 : 5), предварительно нейтрализо-
ванной в присутствии метилового красного. 20 мл полученного раствора раз-
бавляют 20 мл ацетона и титруют 0,05 н. хлорной кислотой в диоксане до пере-
хода оранжево-желтой окраски в оранжево-розовую. В качестве индикатора
используют насыщенный раствор метилового красного в ацетонитриле.
На титрование 13,49 мг сентедрина расходуется 1 мл 0,05 н. стандартного
раствора.
Действующее начало образца нельзя определить непосредственным титро-
ванием в неводной среде, поэтому вещество следует экстрагировать в форме
основания. Для этого в делительной воронке на 25 мл взвешивают 140—
160 мг тонкоизмельченной таблетки и суспендируют в 1 мл дистиллирован-
ной воды, добавляют 5 мл насыщенного раствора бикарбоната калия и экстра-
гируют 5 мин (1 х 10 и 3x5 мл) хлороформом. Объединенные вытяжки
высушивают безводным сульфатом натрия, смешивают с 2—3 мл уксусной
кислоты и титруют 0,01 н. хлорной кислотой. 1 мл 0,01 н. раствора эквива-
лентен 2,698 мг метил-1-фенил-1-пиперидил-(2)-ацетата (свободного осно-
вания).
Об определении солянокислого 2-пиперидилэтилового эфира 1-фенил-1-
пиперидилуксусной кислоты в свечах (спазмонал) см. в работе [367].
ж. Титрование хлоргидрата этилового эфира 1-метил-4-фенилпиперидин-
4-карбоновой кислоты (петидин) [453]. Примерно 240 мг образца растворяют
в 5 мл 5%-ного раствора ацетата ртути(П) в уксусной кислоте, добавляют
50 мл ацетона и титруют в присутствии 1 мл 0,25 %-ного раствора метило-
* Drug. Std., 27, 30 (1959).
Определение солей азотистых оснований
331
вого оранжевого в ацетоне 0,1 н. хлорной кислотой в уксусной кислоте.
На 28,38 мг петидина расходуется 1 мл 0,1 н. раствора. Хлоргидрат петидина
можно также титровать в уксусной кислоте в присутствии кристаллического
фиолетового [42].
з. Определение хлоргидрата пиридоксина (витамин В6) [130. 453].
НО - НоС
сн2он
Л /ОН
N ХСН3
Пиридоксин (витамин В6), 2-метил-3-окси-4,5-бис-(окспметил) пиридин
Взвешивают хлоргидрат пиридоксина в количестве, эквивалентном 3—
8 мл 0,1 н. хлорной кислоты, и растворяют в 20 мл уксусной кислоты,
содержащей 10% уксусного ангидрида, добавляют 5 мл 5%-ного ацетата
ртути(П) и 10 мл 1,2-дихлорэтана и титруют в присутствии 4 капель 0,2 %-но-
го 1-нафтолбензеина в уксусной кислоте. На 20,56 мг хлоргидрата пиридок-
сина расходуется 1 мл 0,1 н. хлорной кислоты. Растворять образец можно
непосредственно в 5 мл 5%-ного ацетата ртути(П), а затем разбавить 25 мл
дихлорэтана и титровать, как указано выше.
и. Определение тиаминхлорида (витамин Bj) [130, 453, 643]. Полумикро-
титрование [643]. 50—60 мг тиаминхлорида взвешивают в колбе Эрленмей-
ера емкостью 200 мл и растворяют при слабом нагревании в 5—10 мл уксус-
ной кислоты. После охлаждения добавляют 1 мл 6%-ного ацетата ртути(П)
и 70—80 мл бензола и титруют 0,01 н. хлорной кислотой в диоксане в при-
сутствии 2—3 капель 0,1%-ного кристаллического фиолетового. Для титро-
вания 1,686 мг тиаминхлорида необходим 1 мл стандартного раствора.
Определение действующего начала в таблетках витамина ВР Анализируе-
мые таблетки измельчают в тонкий порошок и взвешивают такое количество
порошка, чтобы в нем содержался приблизительно 1 мг-экв тиаминхлорида.
Навеску порошка суспендируют в 10 мл 5 %-ного ацетата ртути(П) в уксус-
ной кислоте и энергично перемешивают несколько минут магнитной мешал-
кой, затем добавляют 70—75 мл ацетоуксусного эфира. Причиной ошибки
при анализе может служить даже слабое нагревание. Титруют 0,1 н. хлорной
кислотой в уксусной кислоте, добавив 6 капель 25%-ного тропеолина ОО
(оранжевый IV) в уксусной кислоте. Ацетоуксусный эфир растворяет тиа-
минперхлорат. Чтобы избежать побочных реакций, титрование проводят
по возможности быстро. Для титрования 16,86 мг тиаминхлорида необходим
1 мл 0,1 н. хлорной кислоты.
NH2
II
„ Г/\ / ^(/^СНгСНоОЦ
rlglu О “
Тиамин (основание)
к. Определение адренергических соединений [651]. Эти соединения исполь-
зуют обычно в виде хлоргидратов.
20—100 мг образца растворяют в 15 мл уксусной кислоты, нейтрализо-
ванной в присутствии кристаллического фиолетового. Смешивают с 5 мл
5%-ного раствора ацетата ртути(П) и титруют 0,05 н. хлорной кислотой
в диоксане до изменения фиолетовой окраски индикатора на голубую. Таким
способом можно определять эфедрин, норэфедрин, 1-фенил-2-метил-амино-
пропан (первитин), 1-(3, 4-диоксифенил)-2-аминопропан (корбазил) и соля-
нокислые соли многих других адренергических средств.
332
Глава 24
Солянокислый эфедрин можно определять в ацетоне в присутствии ацетата ртути(П)
[453]. Относительно определения содержания алкалоидов в препаратах для инъекций,
содержащих солянокислый эфедрин, так же как и об анализе смесей эфедрин — фена-
цетин и эфедрин — фенилэтилбарбитуровая кислота см. в работе [651]. Методы опреде-
ления эфедрина в маслах и препаратах для ингаляции обсуждаются в работе [637].
Величины рЛГ для симпатомиметических соединений приведены в работе [125].
л. Определение дикарбамата 2-метил-2-н-пропилпропандиола-1,3 (мепро-
бамат) [708]. При его гидролизе соляной кислотой образуются хлористый
аммоний, СО2 и 2-метил-2-н-пропилпропандиол-1,3
СН2 — OCONH2 СН2ОН
I 2НС1 I
СН3 —С—с3н7 ——> CH3-C-C3H7 + NH4C1 + 2CO2
сн2—oconh2 сн2он
Эквивалентное количество образующегося хлористого аммония титруют
в уксусной кислоте хлорной кислотой в присутствии ацетата ртути(П).
В широкогорлой колбе Эрленмейера емкостью 250 мл взвешивают 60—
110 мг вещества, добавляют 40 мл чистой концентрированной соляной кисло-
ты, закрывают колбу часовым стеклом и раствор 45—60 мин осторожно кипя-
тят в вытяжном шкафу. После завершения гидролиза выпаривают соляно-
кислый раствор почти досуха, добавляют 10 мл уксусной кислоты и 10—
15 мл 5%-ного раствора ацетата ртути(П) в уксусной кислоте (нейтрализован-
ного в присутствии кристаллического фиолетового) и после растворения
разбавляют 20 мл диоксана. Титруют 0,1 н. хлорной кислотой до появления
ярко-синего окрашивания. На титрование 10,91 мг дикарбамата 2-метил-
2-н-пропилпропандиола-1,3 требуется 1 мл стандартного раствора кислоты.
Чтобы определить возможную примесь NH4C1 в соляной кислоте, реко-
мендуется провести глухой опыт. Следует избегать полного выпаривания
гидролизата, так как NH4C1 улетучивается при 100°, а кристаллический
NH4C1 плохо растворяется в уксусной кислоте.
м. Определение органических ртутных производных с помощью ацетоли-
за [149]. Применяемый для титрования хлоргидратов аминов модифициро-
ванный метод Пифера — Воллиша [644] может оказаться полезным для
определения некоторых органических производных ртути. Ртутные произ-
водные, представленные общей формулой R — Hg — R' (в которой одна
из связей может быть С — Hg-связью), подвергаются ацетолизу в уксусной
кислоте в соответствии со следующими уравнениями:
RHgR'-J- СН3СООН 7± RHgOOCCH3 + R'H
и
RHgOOGGH3 + СНзСООН Hg(OOCCH3)2+ RH
Если к реакционной смеси добавить избыток солянокислого метиламина,
то образуется эквивалентное количество метиламина (в виде ацетата), кото-
рый можно оттитровать хлорной кислотой.
RHgOOGCH3 + CH3NH2-HCl —> RHgGl-Ь GH3NH2-CH3COOH,
Hg(OOCGH3)24- 2GH3NH2-HG1 —> HgGl2-|-2GH3NH2-CH3COOH.
В колбе взвешивают образец, содержащий 0,5—0,8 мг-экв ртутного про-
изводного, и растворяют в уксусной кислоте, добавляют несколько капель
индикатора (0,4%-ного раствора 1-нафтолбензеина в ледяной уксусной
кислоте) и титруют 0,1 н. хлорной кислотой до перехода желтой окраски
в зеленую. Добавляют небольшой избыток 0,1 н. раствора солянокислого
метиламина в ледяной уксусной кислоте и доводят титрование до конца.
Объем раствора хлорной кислоты, израсходованный между двумя конечными
точками титрования, эквивалентен содержанию ртутного производного
в навеске.
Определение солей азотистых оснований
333
Ртутное производное
Мерк у р( 11 )с укцинимид
Фенилмеркур(П)ацетат
Эквивалентный
198,4
336,8
Тип связи
2Hg —N
Hg-O, Hg-C
СН3СОНа1 +
+ СН3СОО~+М+
Методика анализа тимерозала (экв. вес 202,4, тип связи Hg — S, Hg — С).
Образец растворяют в уксусной кислоте, добавляют индикатор и неболь-
шой избыток (1—2 мл) раствора солянокислого метиламина и титруют до
конечной точки 0,1 н. раствором хлорной кислоты. Объем израсходованной
хлорной кислоты эквивалентен содержанию ртутного производного плюс
содержание какого-либо основного компонента в навеске. Ацетолиз этого
соединения дает тиосалицилат натрия и этилмеркур(П)ацетат.
н. Титрование галогенидов оснований в среде уксусного ангидрида.
В некоторых случаях соли галогеноводородных кислот и азотистых основа-
ний, а также четвертичных оснований можно титровать без добавления аце-
тата ртути(П) [619, 676, 778].
При реакции между уксусным ангидридом и галогенидом металла удает-
ся обнаружить образование ацетилгалогенида и ацетата металла [722]
(СН3СО)2О сн3со+ ^сн3соо-
СН3СО+ + СН3СОО- + М+ + На1- СН3СОНа1 + СН3СООМ,
где М+ — катион металла и Hal- — анион галогена.
При этом, по-видимому, происходит дополнительная (реакция между
ионом металла из галогенида и карбонильной группой уксусного ангидри-
да. сопровождаемая нуклеофильной атакой галогенид-иона [676].
Hal
СН3—С —6:' СН3—С — 6—М
I *’ + _ I ”
О + М+На1 < о
СН3—С = О СН3—С=О
Навеску образца, на титрование которой должно пойти примерно 5—
10 мл 0,1 н. хлорной кислоты, растворяют под тягой в 50 мл уксусного
ангидрида и титруют в присутствии кристаллического фиолетового до тех
пор, пока синяя или сине-зеленая окраска не перейдет в желтую (см. гл. 13,
разд. 72). Этим способом можно определять, например, хлоргидраты лидо-
каина, хинина, наркотина и хлорид цетилпиридиния. Бромгидраты гио-
сциамина и гиосцина растворяются в процессе титрования. Хлоргидраты
тиамина, эфедрина, иохимбина и папаверина следует сначала растворить
в 5 мл 98 %-ной муравьиной кислоты и только потом разбавить 50 мл уксус-
ного ангидрида. Расход титранта на титрование самой смеси уксусный
ангидрид — муравьиная кислота следует определить в глухом опыте и вне-
сти соответствующую поправку в результаты титрования образца (см. также
гл. 23, разд. 126).
Титрование хлоргидрата хинина в смеси хлороформ — уксусный ангидрид с исполь-
зованием в качестве индикатора Судана III обсуждается в работе [655].
О титровании солянокислого анилина см. в работе [778].
Как уже упоминалось, соединения, содержащие фентиазиновое кольцо,
чувствительны к окислению, поэтому их нельзя титровать в растворителе,
содержащем, например, следы перекисей (см. гл. 23, разд. 122). Также нель-
зя титровать в уксусной кислоте хлоргидраты фентиазиновых производных,
так как предполагается, что окислительное действие хлорной кислоты ката-
лизируется ацетатом ртути(П) [619]. Красный оттенок не дает возможности
наблюдать изменение окраски кристаллического фиолетового. В уксусном
334
Глава 24
ангидриде красный оттенок отсутствует и хлоргидраты хлорпромазина, про-
метазина и диэтазина можно титровать без добавления ацетата ртути(П)
[619].
Примерно 250 мг хлоргидрата фентиазинового производного растворяют
в 10—15 мл 95—100%-ного уксусного ангидрида или смеси уксусный ангид-
дрид — хлороформ (1 : 1), предварительно нейтрализованной в присутствии
3 капель кристаллического фиолетового до появления желтой окраски,
и титруют 0,1 н. хлорной кислотой в уксусной кислоте до желтого окраши-
вания. 1 мл 0,1 н. хлорной кислоты эквивалентен 35,53 хлоргидрата хлор-
промазина, 32,09 мг хлоргидрата прометазина и 33,48 мг хлоргидрата ди-
этазина [хлоргидрата Х-(0-диэтиламиноэтил)фентиазина].
о. Определение галогенидов оснований в присутствии продуктов разло-
жения основного характера, растворимых в соляной кислоте (определение
феназоцина) [204]. Иногда галогенгидраты оснований или соли растворимы
в хлороформе. Это наблюдается главным образом в случае солей алкалоидо-
подобных оснований. Бромгидрат феназоцина растворяется даже в смеси
хлороформа с 0,1 н. соляной кислотой. Разложение происходит следующим
образом:
НО
Продукты разложения
100 мг образца растворяют в делительной воронке в 50 мл 0,1 н. соляной
кислоты и экстрагируют бромгидрат феназоцина (4 X 30 мл) хлороформом.
Продукт разложения остается в солянокислом слое. Хлороформ через вату,
смоченную хлороформом (вата может быть покрыта некоторым количест-
вом безводного сульфата натрия), фильтруют в колбу для титрования.
Делительную воронку и вату промывают хлороформом. Хлороформный
раствор упаривают досуха на кипящей водяной бане (для этой цели, так же
как и для титрования, может быть использована колба для упаривания,
изображенная на рис. 103), остаток растворяют в 25 мл уксусной кислоты
и 10 мл 6 %-ного раствора ацетата ртути(П) в уксусной кислоте и титруют
хлорной кислотой в присутствии кристаллического фиолетового. Исчезнове-
ние красного окрашивания наблюдают через желтый фильтр. 1 мл 0,02 н.
хлорной кислоты эквивалентен 8,04 мг бромгидрата феназоцина; точность
определения ±1%.
137. ЧЕТВЕРТИЧНЫЕ ОСНОВАНИЯ И ИХ СОЛИ
Сольватирующая способность смеси растворителей, содержащей гли-
коли, часто выше, чем у уксусной кислоты. Катионные мыла также иногда
хорошо растворяются в смеси пропиленгликоля и изопропилового спирта.
Определение солей азотистых оснований
335
Галогениды четвертичных солей аммония можно титровать как основания
хлорной кислотой при добавлении ацетата ртути(11) (соляная кислота
оттитровывает и избыток ацетата ртути(П)).
Найдено также, что можно титровать соли гетероциклических четвертичных аммони-
евых оснований; например, котарнинхлорид можно титровать раствором цианида калия
в изопропиловом спирте [215] (см. также гл. 28).
а. Титрование в гликолъной смеси растворителей. 200—300 мг образца
растворяют в 20—30 мл гликольной (G — Н) смеси (гл. 6, разд. 41), добав-
ляют избыток раствора ацетата ртути(П) (примерно 20—50%-ный) и титру-
ют 0,1 н. хлорной кислотой в изопропиловом спирте в присутствии бром-
фенолового синего (при потенциометрическом титровании получают более
точные результаты). Этим методом можно определять, например, бромид
лаурилтриметиламмония и хлорид триметилэтиламмония [489].
б. Титрование в уксусной кислоте. Титрование бромида декаметилен-
бис-(триметиламмония) (декаметонийбромида) *. Примерно 400 мг образ-
ца растворяют в 50 мл уксусной кислоты и после добавления 2 мл уксусного
ангидрида нагревают на кипящей водяной бане до полного растворения.
После охлаждения добавляют 15 мл 5%-ного ацетата ртути(П) и титруют
0,1 н. хлорной кислотой в уксусной кислоте в присутствии 2 капель инди-
катора (1%-ный кристаллический фиолетовый). 1 мл 0,1 н. стандартного
+
раствора эквивалентен 20,92 мг бромида декаметония [(CH3)3N — (СН2)ю—
N(CH3)3]2Br-.
в. Титрование в пропионовой кислоте [376]. 300—400 лег бромида цетил-
пиридиния или бромида триметилцетиламмония растворяют в 25 мл 0,1 н.
раствора пропионата ртути(П) и титруют 0,1 н. хлорной кислотой в пропио-
новой кислоте в присутствии метанилового желтого до перехода желтой
окраски в пурпурную (см. табл. 63). 1 мл стандартного раствора эквивален-
тен 38,44 мг бромида цетилпиридиния или 36,44 мг бромида цетилтриметил-
аммония.
Приготовление пропионата ртути(П). 22 г окиси ртутп(П) растворяют при слабом
нагревании в 1000 мл пропионовой кислоты, содержащей 14 г пропионового ангидрида.
Приготовление 0,1 н. хлорной кислоты в пропионовой кислоте. При охлаждении смеши-
вают 1000 мл пропионовой кислоты, 35 г пропионового ангидрида и 10 мл хлорной кисло-
ты плотностью 1,690 (см. также гл. 24, разд. 135, е).
г. Титрование в смеси бензол — хлороформ [409]. Бромид цетилпириди-
ния можно также титровать в системе бензол — хлороформ.
Примерно 300 мг образца, предварительно высушенного до постоянного
веса при 40—50° в вакууме, растворяют в смеси бензол — хлороформ (30 : 2),
добавляют 3 мл 6 %-ного раствора ацетата ртути(П) в уксусной кислоте
и титруют 0,1 н. хлорной кислотой в диоксане в присутствии метанилового
желтого.
138. ТЕТРАФЕНИЛБОРАТЫ ОСНОВАНИЙ
Известно, что многие органические основания, содержащие азот (включая
четвертичные основания), дают с тетрафенилборатом натрия осадок тетра-
фенилбората, нерастворимый в воде ** (см. также гл. 2, разд. 19, г). Тетра-
фенилбораты растворимы в различных растворителях, но выбор наилучшего
из них следует вести эмпирически. Тетрафенилборат-анион титруют как
* Drug. Std., 26, 136 (1958).
** Тетрафенилбораты образуются либо при взаимодействии с солями (гидроокисями)
четвертичных оснований, либо в присутствии влаги, вероятно, с гидратами аминов.—
Прим, перев.
:336
Глава 24
.основание хлорной кислотой [229, 267, 268, 316, 632]:
О поведении тетрафенилборной кислоты в растворе в безводной уксусной
кислоте см. в работе [632].
Тетрафенилбораты некоторых анестезирующих средств можно титровать
в смеси уксусная кислота — ацетон [127]. С этой целью растворяют 100 мг
.образца в 5 мл ацетона, добавляют 45 мл уксусной кислоты и титруют
0,05—0,1 н. хлорной кислотой при определении пиперокаина до появления
синей окраски, а при определении прокаина или тетракаина — до зеленой
окраски.
Для растворения тетрафенилборатов обычно употребляют ацетон или
диоксан. Тетрафени лборат N-о-бромбензи л-N-этил-N, N-димети ламмония
нерастворим в диоксане, но растворим в ацетоне. При титровании в ацетоне
0,01 н. раствором кислоты с метиловым красным, метиловым фиолетовым
или тропеолином ОО в качестве индикатора переход окраски нечеткий.
Однако эти соединения можно легко оттитровать в смеси метилэтилкетон —
уксусный ангидрид (1 : 1) до зелено-синего окрашивания метилового фиоле-
тового [316]. Ошибки титрования становятся минимальными, если к образцу
добавить избыток хлорной кислоты до изменения окраски индикатора и про-
вести обратное титрование: оттитровать избыток кислоты 0,01 н. ацетатом
натрия в уксусной кислоте.
Навеску тетрафенилбората, эквивалентную 10—15 мг бромбензилэтил-
диметиламмоний-иона, растворяют в нейтрализованной смеси 10 мл метил-
этилкетона и уксусного ангидрида (1 : 1), добавляют 1 каплю 1%-ного мети-
лового фиолетового и медленно титруют при постоянном перемешивании до
появления зелено-синей окраски. Продолжают титрование 0,01 н. хлорной
кислотой в смеси уксусная кислота — четыреххлористый углерод (1 : 1),
пока не добавят 10,00—12,00 мл кислоты, и полученный желтый раствор
оттитровывают 0,01 н. ацетатом натрия до появления зелено-синей окраски.
Эталонный раствор можно приготовить из 0,1 н. раствора (см. гл. 10, разд. 58).
Титр хлорной кислоты устанавливают по свежеприготовленному раствору
дифенилгуанидина в смеси метилэтилкетон — уксусный ангидрид. Для
проверки нормальности раствора ацетата натрия смешивают 10 мл хлор-
ной кислоты известной нормальности с 5 мл метилэтилкетона и 5 мл уксус-
ного ангидрида и титруют стандартным раствором ацетата натрия в при-
сутствии метилового фиолетового до появления зелено-синей окраски. 1 мл
0,01 н. хлорной кислоты эквивалентен 5,623 мг тетрафенилбората бромбен-
зилэтилдимети ламмония.
139. ОПРЕДЕЛЕНИЕ ОСНОВАНИЙ В ПРИСУТСТВИИ «КИСЛЫХ» SH-
ИЛИ S-ГРУПП
Кислые SH- или S-группы маскируют основность аминосоединений и
и мешают титрованию хлорной кислотой. После добавления ацетата ртути(Н)
в уксусной кислоте можно оттитровать следующие соединения: 1-метил-2-
Определение солей азотистых оснований
337
меркаптоимидазол (метотирин), аллилтиомочевину, тиомочевину, тетраэтил-
тиурамдисульфид (антабус), производные тиобарбитуровой кислоты (напри-
мер, интранаркон — калиевую соль 5-аллил-5-циклогексенил-2-тиобарбиту-
ровой кислоты) и т. д.
ЛИ
2HS — С< 4- Hg(OCOCH3)2 —>
xnh2
Тиомочевина
HNX /Nil
>CSHgSCf + 2СН3СООН
h2nz xnh2
/ ’ * \
Меркурдимеркаптид
50—70 мг метилмеркаптоимидазола растворяют в 25 мл 3%-ного раствора
ацетата ртути(П), добавляют 5 мл уксусного ангидрида и титруют, добавив
3—5 капель 0,1%-ного кристаллического фиолетового. 1 мл 0,1 н. раствора
расходуется [42—441 на 11,42 мг метилмеркаптоимидазола
СНз
N
(¥SH
и---N
Если при добавлении ацетата ртути(П) раствор мутнеет, то в качестве
растворителя используют смесь уксусный ангидрид — хлороформ (1 : 1)
и после добавления 10 мл 3%-ного ацетата ртути(П) оттитровывают избыток
хлорной кислоты 0,1 н. ацетатом калия в уксусной кислоте. 1 мл 0,1 н. хлор-
ной кислоты эквивалентен 14,83 мг ((Z^Hs^NCSSSCSN^Hs^.
Микроопределение тиомочевины рассматривается в работе [6].
140. СУЛЬФАТЫ, ЭТАНСУЛЬФОНАТЫ, ет-ТОЛУОЛСУЛЬФОНАТЫ,
ФОСФАТЫ И НИТРАТЫ ОСНОВАНИЙ
Для определения этих соединений пригодны многочисленные методы.
После перевода солей в основания действием NaOH, КНСО3 или NaHCO3
водные растворы, например солей алкалоидов, экстрагируют хлороформом
или четыреххлористым углеродом и безводный экстракт, высушенный суль-
фатом натрия, титруют 0,005 н. тг-толуолсульфокислотой или после добавле-
ния уксусной кислоты 0,01 н. хлорной кислотой. Так определяют атропин-
сульфат, эрготоксинэтансульфонат, стрихнинсульфат, кодеинфосфат [299].
Винкалейкобластин C46H58O9N4 можно титровать подобным же образом
0,005 н. тг-толуолсульфокислотой * [436].
Определение сульфата винкалейкобластина (велъбан, винбластинсульфат}.
Винкалейкобластинсульфат содержит воду, которую можно удалить высу-
шиванием в вакууме при 50°. Безводное вещество очень гигроскопично
и, если оставить его на воздухе, за 2 мин адсорбирует около 2% влаги. Рав-
новесие устанавливается примерно через 4 час, поэтому для точного взвеши-
вания образец оставляют на 4 час на воздухе. В делительной воронке с точ-
ностью ±0,1 мг взвешивают примерно 25 мг образца, растворяют его
в небольшом количестве воды, переводят в основание действием бикарбоната
натрия и экстрагируют освободившееся основание хлороформом (1 X 10
и затем 3x5 мл}. Фильтруют хлороформный экстракт через вату, покры-
тую слоем сульфата натрия, в мерную колбу емкостью 25 мл и разбавляют,
если необходимо, до метки. 5 мл полученного раствора титруют 0,005 н.
* Neuss, Gorman, Svoboda, Maciak, Вег r, J. Am. Chem. Soc., 81,
4754 (1959).
338
Глава 24
п-толуолсульфокислотой в присутствии диметилового желтого. 1 мл стан-
дартного раствора эквивалентен 2,026 мг винкалейкобластина основания
или 2,317 мг моногидрата сульфата винкалейкобластина.
Одновременно описанным выше способом, высушивая препарат в вакууме
при 50° в течение 16 час, определяют влажность образца.
При использовании так называемого бензидинового метода предвари-
тельно следует установить приблизительное содержание сульфат-иона.
95% сульфат-иона связывается в бензидинсульфат, который нерастворим
в уксусной кислоте, а остающееся небольшое количество свободной серной
кислоты в большинстве случаев не оказывает существенного влияния
на изменение окраски индикатора [265].
Взвешивают 1 мг-экв сульфата основания, растворяют в 10 мл уксусной
кислоты, добавляют 0,05 н. раствор бензидинового основания в количестве,
эквивалентном 95% сульфат-иона, содержащегося в навеске, и титруют без
предварительного фильтрования 0,1 н. хлорной кислотой в присутствии
1 капли 1 %-ного раствора метилового фиолетового. На определение 19,58 мг
двухводного сульфата хинина расходуется 1 мл стандартного раствора.
Об определении сульфатов стрептомицина и дигидрострептомицина см.
в работе [635].
Большинство сульфатов алкалоидов мало растворимо в уксусной кислоте;
Четтен [123] считает, что в таком случае следует проводить титрование в смеси
ацетонитрил — хлороформ — фенол (10 : 4 :1). Фосфат кодеина можно также
определять в этой смеси в присутствии ацетилсалициловой кислоты, фена-
цетина и кофеина [639].
а. Титрование в смеси ацетонитрил — хлороформ — фенол [123]. В процессе усо-
вершенствования методов неводного титрования использование смесей растворителей
стайо наиболее общим приемом также и для определения солей оснований с минеральны-
ми кислотами, так как величина скачка потенциала при потенциометрическом титровании
в них часто выше, а изменение окраски индикатора при визуальном определении конеч-
ной точки выражено четче.
Используя полумикробюретку с ценой делений 0,02 мл взвешивают
образец, на титрование которого будет расходоваться 2—3 мл 0,05 н. хлор-
ной кислоты в диоксане. Растворяют навеску в 5 мл расплавленного свеже-
перегнанного фенола или смеси 5 г фенола и 10—20 мл хлороформа и после
добавления 50 мл ацетонитрила титруют потенциометрически либо прили-
вают 1—2 капли метилового красного и определяют конечную точку титро-
вания по появлению фиолетово-красной окраски. Вблизи конечной точки
титрования можно наблюдать переход через разные оттенки окраски,
но последнее цветовое изменение тем не менее очень четко выражено
(см. рис. 44, а). Этим методом при потенциометрическом или визуальном
определении конечных точек можно титровать следующие соединения:
d, /-1-фенил-2-пропиламиносульфат (амфетамин), сульфаты эфедрина, мор-
фина, кодеина, стрихнина и физостигмина, фосфаты кодеина и димоксилина
(последний представляет собой 6,7-диметокси-1-(4'-этокси-3'-метоксибен-
зил)-3-метилизохинолинфосфат). Сульфаты хинина, цинхонина и цинхони-
дина можно определять только потенциометрическим титрованием.
б. Титрование в диоксане. Определение этансулъфонатов и п-толуолсулъ-
фонатов оснований [321]. Если соль образована органическим азотистым
основанием и сильной кислотой, например этансульфокислотой или п-толуол-
сульфокислотой, то при титровании в неводной среде могут протекать сле-
дующие реакции.
1. При растворении в уксусной кислоте ацетат не образуется совсем или образуется
в незначительном количестве. Ацетат-ион — сильное основание; уксусная кислота отно-
сится к слабым кислотам, в то время как СН3С6Н4ЗОз- и C2H5SO7-hoh — слабые осно-
вания (соответствующие кислоты — сильные). При титровании хлорной кислотой в уксус-
ной кислоте, где СН3СООН2+-ион представляет протонированное промежуточное соеди-
нение, нельзя ожидать резкого изменения окраски. В большинстве случаев индикатор
Определение солей азотистых оснований
339
показывает «кпслое» окрашивание сразу после прибавления первых несколькпх капель
титранта. Слабые основания и п-толуолсульфокислота образуют аддукт B...HS03-
•СВН4СН3, к тому же n-толуолсульфокпслота обладает более кислым характером в уксус-
ной кислоте; в солях, образованных сильным основанием, ион в ионной паре
BH+(CH3CBH4SO3)_ представляет собой сильную катионную кислоту, в то время как
анион n-толуолсульфокислоты — слабое основанпе.
Катионно-кислотный характер ониевого иона четвертичных оснований общей фор-
мулы R4N+X~ в отличие от обычных солей аммония с протонными кислотами нельзя
объяснить координированием протона. Если в солях этого типа Х~ — галоген-ион, то
благодаря влиянию ацетата ртути(II) в уксуснокпслой среде происходит его обмен
с ацетат-ионом и последний можно оттитровать как основание. Еслп, однако, Х~ пред-
ставляет собой (CH3CbH4SO3)_-hoh пз метпл-п-толуолсульфоната или метплсульфат-анион
CH3SOj из дпметилсульфата, то такие четвертичные соли не образуют ацетатов даже
после добавления ацетата ртути(П) в уксусной кпслоте, и поэтому титровать их нельзя.
2. В апротонном или дифференцирующем растворителе хлорная кислота сама ней-
трализуется или основанием или этансульфонат-анионом или п-толуолсульфонат-анпо-
ном без вмешательства катиона растворителя (ацилоний-иона)
R3NH+X-+HC1O4 (R3NH+C10j R3N ... НС104) + НХ,
R4N+X-4-HC104 —» R4N+C1Oj4-HX.
Взвешивают примерно 0,5 мг-экв этансульфоната (или п-толуолсульфоната)
монофункционального основания или 0,25 мг-экв для бифункционального
основания и растворяют в 10—15 мл нейтрализованного диоксана. Титруют
0,05 н. хлорной кислотой в диоксане в присутствии кристаллического фиоле-
тового этансульфонаты или n-толуолсульфонаты пиперидина, циклогексил-
амина, морфолина, диметиламиноацетилфентиазина, винкамина до появления
тускло-зеленой окраски, n-толуолсульфонаты трибензиламино-, пипе-
ридинометилтолилпропанона и бромбензилдиметиламмония до светлой изум-
рудно-зеленой окраски, а этансульфонат и n-толуолсульфонат эрготоксина
до светло-зеленой окраски. В последнем случае точность определения не пре-
вышает ±2%, в то время как при титровании других указанных соединений
ошибка составляет не более ±0,5%. Диэтансульфонат 1Ч-[0-3,4,5-триме-
токсибензоксиэтил]-№-[у-(3'-хлор-10'-фентиазинил)-пропил]пиперазипа, если
его титровать до появления изумрудно-зеленой окраски, можно определить
с точностью ±1%.
Относительно изменения окраски кристаллического фиолетового см. гл. 13.
в. Определение эрготоксин фосфата 1308]. 100—150 мг тонко измельчен-
ного эрготоксинфосфата растворяют в защищенной от света колбе в 20 мл
уксусной кислоты и титруют из полумикробюретки 0,05 н. хлорной кисло-
той, добавив 1 или 2 капли 0,1%-ного раствора кристаллического фиолето-
вого в уксусной кислоте до появления синего окрашивания. Для определе-
ния 34,37 мг эрготоксинфосфата необходим 1 мл 0,05 н. хлорной кислоты
(эффективный молекулярный вес смеси, содержащей 50% фосфатов эрго-
кристина, 25% эргоэриптина и 25% эргокорнина, равен 687,3).
Об определении фосфорной кислоты, содержащейся в эрготоксинфосфате,
см. гл. 22, разд. 116,г.
г. Определение пирробутаминфосфата *. Примерно 0,4 г 1-[4-(п-хлорфе-
нил)-3-фенил-2-бутенил]пирролидиндифосфата растворяют в 80 мл уксусной
кислоты и титруют 0,1 н. хлорной кислотой в уксусной кислоте в присутствии
кристаллического фиолетового. На 50,79 мг пирробутамина требуется 1 мл
0,1 н. хлорной кислоты.
Drug. Std., 27, 58 (1959).
340
Глава 24
д. Титрование в уксусной кислоте 0,05 н. хлорной кислотой в диоксане
[651]. В уксусной кислоте некоторые адренергические средства можно непо-
средственно титровать хлорной кислотой в диоксане.
20—100 мг образца растворяют, если необходимо при незначительном
нагревании, в 15 мл уксусной кислоты, нейтрализованной в присутствии
кристаллического фиолетового, и титруют из полумикробюретки до синего
окрашивания. 1 мл 0,05 н. стандартного раствора эквивалентны 18,43 мг
бензедрина (1-фенил-2-амино-пропансульфат), 21,43 мг веритола [1-(4-окси-
фенил)-2-метиламинопропансульфат], 25,83 мг бутедрина [1-(4-оксифенил)-
2-н-бутиламиноэтанолсульфат] и 27,83 мг алюдрина [дигидрат 1-(3,4-диокси-
фенил)-2-изопропиламиноэтанолсульфата]. Бензедрин и бутедрин легко
растворимы, алюдрин растворяется при непродолжительном, а веритол —
при продолжительном нагревании.
е. Прямое титрование сульфатов оснований в уксусном ангидриде или
в смеси уксусный ангидрид — хлороформ обсуждается в работах [655.
706].
ж. Определение нитрата стрихнина и метанитрата атропина [656].
Для восстановления нитрата в нитрит в уксусной кислоте используют аскор-
биновую кислоту, нитрит можно затем кипячением и пропусканием воздуха
через систему разрушить и удалить из раствора. Таким образом нитрат
основания превращают в ацетат основания. (О восстановительных свойствах
аскорбиновой кислоты см. гл. 29, разд. 162. На этом явлении основан
отроумный метод Посгея и Байера, с помощью которого можно прямо титро-
вать нитраты алкалоидов в смеси уксусная кислота — уксусный ангидрид
с кристаллическим фиолетовым как индикатором.
Известно, что при подщелачивании солей четвертичных оснований,
используемых в фармакологии, например метанитрата атропина, основание
не выделяется и его, следовательно, нельзя проэкстрагировать органиче-
скими растворителями. Нитрат пилокарпина можно определять только
потенциометрически в смеси фенол — хлороформ — ацетонитрил [183].
60—70 мг нитрата стрихнина растворяют при нагревании в 10 мл уксус-
ной кислоты, в горячем растворе растворяют 200 мг тонко измельченной
твердой аскорбиновой кислоты, пропуская над раствором струю воздуха
(для этой цели может быть использован аппарат, изображенный на рис. 42
и 103), и осторожно кипятят 1 мин, постоянно пропуская ток воздуха.
К охлажденному раствору добавляют 20 мл уксусного ангидрида и титруют
0,02 н. хлорной кислотой в смеси уксусная кислота — четыреххлористый
углерод (1:1) в присутствии 1—2 капель 0,2%-ного кристаллического
фиолетового в хлорбензоле до появления зеленой окраски. В полученные
результаты вносят поправку из данных глухого опыта. На 7,948 мг нитрата
стрихнина требуется 1 мл 0,02 н. хлорной кислоты.
Подобным же образом определяют и метанитрат атропина. 150 мг веще-
ства растворяют в 15 мл уксусной кислоты и восстанавливают нитрат
до нитрита 1 г аскорбиновой кислоты, а затем анализируют, как описано
выше. На 36,64 мг метанитрата атропина требуется 1 мл 0,1 н. хлорной
рис .ноты.
Глава 25
ОПРЕДЕЛЕНИЕ СМЕСЕЙ
АЗОТИСТЫХ ОСНОВАНИЙ
ИЛИ СМЕСЕЙ ХЛОРГИДРАТОВ ОСНОВАНИЙ
Хотя известно много методов анализа смесей аминов, нельзя рекомендо-
вать какой-то один метод, приемлемый во всех случаях. Если в дифферен-
цирующих растворителях сила оснований изменяется (гл. 6), то бинарные
и тройные смеси аминов можно анализировать потенциометрическим титро-
ванием [247, 401, 404, 579]. Ацетилированием в уксусной кислоте первичные
и вторичные амины можно превратить в амиды кислот, которые не реагируют
с хлорной кислотой [158, 311, 312, 314, 612, 750, 811, 854], а первичные
амины переводят действием салицилового альдегида [156, 432, 750], ацетил-
ацетона [157] и 2-этилгексальдегида в имины (в последнем случае возможно
непосредственное титрование с 0-этилкапроновым альдегидом [518]). В пири-
дине избыток салицилового альдегида можно определить обратным титро-
ванием метилатом натрия; вторичные и третичные амины, не реагирующие
с салициловым альдегидом, можно оттитровать в хлороформе стандартным
раствором кислоты [156, 432]. Первичные алифатические и ароматические
амины можно перевести действием фталевого ангидрида в фталимиды, ней-
тральные в уксусной кислоте [262].
В заключение отметим метод перегонки [166, 315] и некоторые другие
способы, основанные на различных изменениях окраски индикаторов [156,
247, 703, 704], а также некоторые кинетические методы, в которых учитывается
различная скорость реакций оснований [347].
141. СМЕСИ АМИНОВ В ДИФФЕРЕНЦИРУЮЩИХ РАСТВОРИТЕЛЯХ
Для анализа смесей аминов в качестве растворителей пригодны ацето-
нитрил, ацетон, метилэтилкетон, метилизобутилкетон, диоксан, нитрометан,
нитробензол и различные смеси растворителей, такие, как уксусная кис-
лота — хлороформ (от 1 : 10 до 2 : 100), нитробензол — гептан, уксусная
кислота — диоксан — нитрометан и т. д. (см. гл. 2, разд. 15; гл. 6,
разд. 37—38). Чаще всего и целесообразнее растворители или смеси раство-
рителей подбирать в каждом случае отдельно.
Для дифференцирующего влияния растворителей характерно, например,
следующее. При потенциометрическом титровании смеси пиперидина и пири-
дина в хлорбензоле соляной кислотой в изопропиловом спирте можно опре-
делить только пиперидин (рА 2,80), при этом наблюдается только один
перегиб на потенциометрической кривой. Однако при титровании в смеси
уксусная кислота — хлорбензол хлорной кислотой в уксусной кислоте
удовлетворительно определяется сумма оснований, здесь также наблюдается
только один перегиб на кривой. В метилизобутилкетоне при титровании
хлорной кислотой в диоксане появляются два перегиба на кривой в конеч-
ных точках титрования двух компонентов [105].
Дифференцирующее потенциометрическое титрование можно также про-
водить 0,01 н. хлорной кислотой в диоксане, например в смеси уксусная
кислота — хлороформ (1 : 10). При титровании смеси ацетат натрия (ацетат-
ион — сильное основание), «-бутиламин (рК 3,39) и пиридин (рА 8,85) каждое
из соединений дает один перегиб на кривой [647]. В среде ацетонитрила можно
анализировать бинарные смеси, например смеси бутиламин — пиридин,
342
Глава 25
бое основание и может быть
Рис. 121. Потенциометриче-
ское титрование смеси пипери-
дин — этилендиамин — п-толуи-
дин в нитробензоле [579].
этаноламин — анилин (рК 4,56 и 9,42), дибутиламин — салицилальдимин
бутиламина, анилин — о-хлоранилин (р/С 9,42 и 11,4), Р-фенилэтиламин —
анилин [247]. Этот метод можно применять и для анализа некоторых промыш-
ленных полупродуктов, состав которых близок к составу указанной модель-
ной смеси.
Если анализируется соединение с первичной аминогруппой, то при при-
менении растворителя, содержащего карбонильную группу может образо-
ваться кетимин, что приведет к заниженным результатам (кетимин — сла-
оттитрован только в уксусной кислоте [642]).
В таких случаях в качестве растворителя
можно использовать нитробензол или смесь
нитробензола и гептана, а в качестве ггитран-
та — хлорную кислоту в диоксане [579]. Так,
при потенциометрическом титровании в нитро-
бензоле смеси пиперидина, этилендиамина
и n-толуидина наблюдаются три перегиба на
кривой (рис. 121).
а. Титрование в ацетонитриле [247] мо-
жно проводить потенциометрически. В пос-
леднем случае для сильных оснований подхо-
дящим индикатором служит эозин Y, для
слабых — метиловый фиолетовый.
Взвешивают 0,6—1,0 мг-экв смеси бутил-
амин — пиридин, растворяют в 20 мл очищен-
ного ацетонитрила, добавляют 6 капель инди-
катора насыщенного раствора эозина У в аце-
тонитриле и титруют бутиламин (В{) 0,1 н.
хлорной кислотой в диоксане до слабого жел-
того окрашивания индикатора (т), затем добав-
ляют в раствор 20 мл уксусной кислоты
и 2 капли 0,02 %-ного метилового фиолетового в хлорбензоле и продолжают
титровать до появления сине-зеленой окраски (я). Второе изменение окраски
индикатора указывает на нейтрализацию пиридина (Z?2).
Расчет’.
Bi = (m — тъ) N
и
В2^(п — пъ) N,
где Bi — количество сильного и В2 — слабого основания, мг-экв; т — стан-
дартный раствор, израсходованный для достижения первой эквивалентной
точки, мл; п — стандартный раствор, израсходованный от первой до второй
конечной точки титрования, мл; тъ — стандартный раствор, израсходованный
в глухом опыте для достижения первой конечной точки титрования, мл;
пь — стандартный раствор, израсходованный в глухом опыте от первой
до второй конечной точки титрования, мл; N — нормальность хлорной
кислоты.
В этом и аналогичных случаях желательно определять и применять при расчетах
нормальность титранта, определенную в том и другом растворителях при различных
индикаторах.
Для физостигмина в присутствии тропеолина ОО эквивалентный вес равен молеку-
лярному весу алкалоида, в то время как в присутствии кристаллического фиолетового
эквивалентный вес в два раза больше молекулярного веса, т. е. расходуется 2 экв .хлорг
ной кислоты [264].
б. Титрование в ацетонитриле с фотометрическим определением конеч-
ной точки. Определение анилина и N,N-диэтиланилина в смеси [404]. Взве-
шивают смесь аминов (1,5—1,6 мг-экв) и растворяют в кювете (150 мл) для
Смеси азотистых оснований или смеси хлоргидратов оснований
343
титрования в 100—110 мл ацетонитрила (рис. 96). Титрование, к примеру,
смеси анилин — диэтиланилин рекомендуется проводить при соотношении
компонентов 2 : 1 (рис. 89) на приборе типа Бекман, модель В при 337 нм
(длина волны до первой конечной точки титрования). В ходе протонирования
поглощение К,К-диэтиланилина (р/£ = 7,48) смещается в коротковолновую
область; таким образом, если длина волны изменяется до 320 нм, в ходе
титрования можно оттитровать также и анилин. К образцу порциями
по 0,05—0,1 мл добавляют 0,5 н. раствор хлорной кислоты в уксусной кис-
лоте. На график наносят фотометрическую кривую — кривую зависимости
поглощения от расхода титранта. Точка пересечения прямой, экстраполиро-
ванной к оси х, показывает объем титранта, израсходованный в конечной
точке титрования. 1 мл 0,5 н. хлорной кислоты эквивалентен 74,61 мг диэтил-
анилина или 46,56 мг анилина.
в. Титрование в уксусной кислоте с фотометрическим определением
конечной точки. Анализ смеси 2-метил-5-нитроанилина и 4-метил-2-нитро-
анилина [404]. Для анализа пользуются описанным выше методом (б), тодько
вместо ацетонитрила в качестве растворителя применяют уксусную кислоту.
Измерения ведут* при длине волны 552 нм; наилучшее соотношение основа-
ний при анализе 4-метил- и 2-метилпроизводных — 1 : 0,5 (рис. 88). 1 мл
0,5 н. хлорной кислоты расходуется на 76,07 мг метилнитроанилинов
(см. гл. 14).
г. Титрование в метилэтилкетоне [474, 475, 479]. При потенциометри-
ческом титровании часто имеет смысл использовать метилэтилкетон как
растворитель и для анализируемого вещества и для титранта (хлорной кис-
лоты). С хлорной кислотой в метилэтилкетоне в определенных случаях
наблюдается больший скачок потенциала, чем в диоксане. Крешков
и сотрудники изучили возможность анализа бинарных и тройных смесей
по следующей методике. Анализируемое вещество помещают в стакан для
титрования, растворяют в 20 мл метилэтилкетона и, используя стеклянный
и каломельный электроды, проводят титрование 0,1 н. хлорной кислотой
в метилэтилкетоне. Чтобы исключить влияние двуокиси углерода и влаги
воздуха, целесообразно проводить титрование в атмосфере азота (достаточно
пропускать ток азота над поверхностью раствора). Титрование одного осно-
вания можно провести с точностью от ±1 до ±1,5%, в то время как анализ
смеси оснований проводится с точностью от ±2,5 до ±3,0%. Этим методом
можно проводить анализ следующих смесей: пиперазин — метилбензимида-
зол, пиперазин — 2,2'-диметил-5,5'-дибензимидазол, пиперазин — 2,2'-диме-
тил-5,5'-дибензимидазол — дифенилметан, пиперидин — метилбензимида-
зол — 3,5-диметилпиразол, пиперазин — метилбензимидазол — 3,5-диметил-
пиразол.
д. Определение азотсодержащих глюкозидов в присутствии п-анизидина,
п-фенетидина и п-толуидина [767]. Определение N-глюкозидов рекомен-
дуется проводить потенциометрическим дифференцирующим титрованием
0,01 н. хлорной кислотой в диоксане. При титровании в качестве индикатор-
ного электрода используют стеклянный электрод и в качестве электрода
сравнения Ag/AgCl с жидкостным мостиком из уксусной кислоты, насыщен-
ной KG1. В зависимости от состава смеси можно применять различные раство-
рители (см. табл. 97). Точность определения азотсодержащих глюкозидов
не превышает ±1%. В зависимости от силы основания скачок потенциала
в конечной точке титрования составляет 25—220 мв. При титровании тг-хлор-
и п-броманилин-К-глюкозидов скачок потенциала не удается фиксировать
длительное время. Обычно растворяют 0,1 ммоль образца соединения
и титруют в 25 мл растворителя (метиловый спирт и глюкоза не мешают
анализу).
е. Титрование в уксусном ангидриде. Эффективный анализ смесей аминов,
сульфоксидов и амидов кислот [64]. Алифатические и ароматические (или
344
Глава 25
Таблица 97
Соединение или смесь (в скобках указаны величина рК и количество вещества), эквивалентное 1 мл 0,01 н стандартного раствора Растворитель, число перегибов на кривой (римская цифра), экви- валентное количество потребляемого титранта (арабская цифра)
уксусная кислота этил- ацетат нитро- метан ацето- нитрил
1. n-Анизидин (рА 8,71; 1,232 мг), п-фенетидин I 1 I 1 I 1 I 1
(рА 8,66; 1,372 мг), га-толуидин (рА 8,70; 1,072 мг)
2. N-Глюкозиды (см. в тексте) I 1
3. Комплекс Амадори (сильное основание) I 1 I 1 I 1 I 1
4. n-Анизидин -j- л-анизидин-]\-глюкозид I 2 II 2 I 1 I 1
5. п-Толуидин л-толуидин-1Ч-глюкозид I 2 I 1 I 1 I 1
6. п-Броманилин 4- п-броманилин-1Ч-глюкозид I 1 I 1 I 1 I 1
7. п-Анизидин 4- л-анизидин-]\-глюкозид -|-ком- I 3
плекс Амадори
8. га-Анизидин-!Ч-глюкозид-j- комплекс Амадори I 2 II 2
9. Амины 4-комплекс Амадори 12 12 I 2 I 2
гетероциклические) амины, сульфоксиды и амиды кислот можно с успехом
определять в одном и том же образце. Анализируемый образец должен содер-
жать примерно 0,1—1 ммоля каждого индивидуального компонента. Его
растворяют в 50—75 мл уксусного ангидрида и титруют потенциометрически
0,05—0,1 н. хлорной кислотой в диоксане, используя стеклянный и кало-
мельный электроды. В качестве раствора электролита в каломельном элек-
троде применяют уксусную кислоту, насыщенную хлористым литием. Если
определяемый образец лишь незначительно растворим или совсем нерас-
творим в уксусном ангидриде, его следует предварительно растворить
в хлорбензоле, диоксане или нитрометане и затем разбавить уксусным
ангидридом. Точность определения образцов весом 2—20 мг от 1 до 2%.
142. САЛИЦИЛОВЫЙ АЛЬДЕГИД, МЕТОД I
В потенциометрическом титровании смесей аминов Вагнер, Браун и Петерс
1854] использовали салициловый альдегид для связывания первичных ами-
нов и определяли непрореагировавшие вторичные и третичные амины. Джон-
сон и Функ [432] определяли первичные амины косвенно обратным титрова-
нием непрореагировавшего салицилового альдегида метилатом натрия
в пиридине (гл. 18). Салициловый альдегид должен быть свободен от примеси
салициловой кислоты.
В колбу Эрленмейера емкостью 250 мл пипеткой отбирают точно 10 мл
0,5 н. раствора салицилового альдегида в пиридине, добавляют образец
смеси аминов, в которой должно содержаться не более 3 мг-экв первичных
аминов, и оставляют на 15 мин. После добавления 1 мл индикатора (1%-ного
тимолфталеина или фенолфталеина в пиридине) титруют 0,1 н. метилатом
натрия в пиридине (гл. 11, разд. 60,з). Для определения нормальности реа-
гента, содержащего салициловый альдегид, проводят глухой опыт. Соли
аммония и аминосоединения, как и пиперидин, пиперазин и морфолин, иска-
жают результаты анализа. Этаноламин не удается определять этим методом.
Общее содержание оснований в уксусной кислоте можно определять титро-
ванием хлорной кислотой. На 0,1 мг-экв алифатического первичного моно-
функционального амина требуется 1 мл 0,1 н. метилата натрия.
Типичным примером использования этого метода служит анализ смеси, содержащей
2,4% этиламина, 22,6% диэтиламина, 2,3% триметиламина, 0,9% этилового спирта
и 71,8% воды: при анализе было найдено 2,3% этиламина [432].
Смеси азотистых оснований или смеси хлоргидратов оснований
345
143. САЛИЦИЛОВЫЙ АЛЬДЕГИД, МЕТОД II
Согласно другому равноценному методу определения алифатических
аминов в смеси, сумму вторичных и третичных аминов, а затем салицилальд-
имин титруют в среде с дифференцирующими свойствами, используя два
различных индикатора [156].
В колбе Эрленмейера с притертой стеклянной пробкой емкостью 250 мл
взвешивают смесь аминов, содержащую максимум 12,5 мг-экв первичных
и 12,5 мг-экв вторичных и третичных аминов, добавляют 25 мл хлороформа
и 5 мл чистого салицилового альдегида. Если раствор получается мутным,
то его разбавляют небольшим количеством диоксана, выдерживают в тече-
ние 15 мин и титруют 0,5 н. хлорной кислотой в диоксане в присутствии
4—6 капель 0,5%-ного бромкрезолового зеленого до исчезновения зеленого
окрашивания. Объем израсходованного стандартного раствора эквива-
лентен количеству вторичных и третичных аминов. Смесь разбавляют 75 мл
диоксана и после добавления 8—10 капель 0,1%-ного конго красного
в метиловом спирте продолжают титрование до появления чистой зеленой
окраски. Салицилальдимин, образующийся из первичного амина, титруют
этим же способом.
Этот метод нельзя использовать для соединений, молекулы которых
содержат одновременно первичные и вторичные или третичные аминогруппы.
Соли аммония, гетероциклические вторичные амины (например, морфолин)
и вторичные аминоспирты мешают определению. Этим методом можно опре-
делять следующие индивидуальные амины: бутил амин, дибутил амин, диэтил-
амин, ди-(2-этилгексил)амин, дигексиламин, гексиламин, изопропиламин,
метиламин, триэтиламин, трипропиламин (хлороформ 50 мл; салицилальде-
гид 10 мл; диоксан 50 мл). Следующие амины можно определять также
и в смеси [156]: бензиламин — этилциклогексиламин, бутиламин — дибутил-
амин, этиламин — диэтиламин, этиламин — триэтиламин, 2-этилгексил-
амин — ди-(2-этилгексил)амин, этилендиамин — трипропиламин, изопро-
пиламин — диизопропиламин и пропиламин —трипропиламин.
144. МЕТОД С ИСПОЛЬЗОВАНИЕМ АЦЕТИЛАЦЕТОНА
Ацетилацетон (пентандион-2,4) реагирует с первичными аминами и обра-
зует имины. По существу при этом аналогично салициловому альдегиду
реагирует енольная форма ацетилацетона.
CH3COCH = C(OH)CH3 + RNH2 —» CH3C( = NR)CH==C(OH)CH3 + H2O,
Ацетилацетон
(енольная форма)
Салицилальдегидиминная форма стабилизована вследствие резонанса
с бензольным кольцом, а ацетилацетонимины — вследствие резонанса
с иминкетаминной формой. Ацетилацетон — слабая кислота, и его можно
титровать в среде пиридина метилатом натрия в присутствии тимолфта-
леина (ср. гл. 17 и табл. 78). Если к соединению, содержащему первичную
346
Глава 25
аминогруппу, добавить избыток ацетил ацетона, то его можно определять
обратным титрованием после образования имина 1157].
В двух толстостенных склянках емкостью 250 мл взвешивают точно
по 10 мл 2,5 н. раствора ацетилацетона. В одну из склянок помещают обра-
зец, содержащий 10—15 мг-экв смеси первичных аминов, и оставляют стоять
на определенное время при данной температуре (см. табл. 98). Перед титро-
ванием охлаждают раствор до комнатной температуры, добавляют 1 мл
1 %-ного тимолфталеина в пиридине и титруют 0,5 н. метилатом натрия
в пиридине до восстановления первоначального синего цвета индикатора
(см. гл. 11, разд. 60,и). Вычисления проводят, исходя из разности объемов
стандартного раствора, пошедшего на титрование в глухом опыте и на титро-
вание образца.
Приготовление раствора ацетилацетона. 250 мл средней фракции свежеперегнан-
ного ацетилацетона разбавляют свежеперегнанным пиридином до объема 1 л.
Ацетилацетонный метод позволяет определять амины, которые не удается
оттитровать при использовании салицилового альдегида; к их числу отно-
сятся этиленамины, аминоспирты, аминокислоты и первичные амины, содер-
жащие также гетероциклический атом азота. С точностью от ±0,1 до ±0,3%
этим методом могут быть определены соединения, приведенные в табл. 98.
Таблица 98
Соединение Температура, °C Про юлжитель- ность, мин
Аминоэтанол амин, этилендиамин 25 30—60
JN-Аминоэтилпиперазин 0 30-60
N-Аминопропилморфолин, бутиламин, этаноламин, гексиламин, 1-амино-2-пропиловый спирт 25 15—60
Пропйлендиамин 25 60—120
Изопропиламин 98 15—60
Бензиламин 25 15—120
Этот метод пригоден для определения, например, этаноламина в при-
сутствии диэтаноламина или триэтаноламина так же, как и для определения
изопропиламина в присутствии диизопропиламина и этилендиамина в при-
сутствии пиперазина.
145. МЕТОД С ИСПОЛЬЗОВАНИЕМ ФТАЛЕВОГО АНГИДРИДА
Основание Шиффа, образующееся при реакции между первичным амином
и салициловым альдегидом в уксусной кислоте, титруется хлорной кислотой
подобно первичному амину. Тем не менее первичные амины можно перевести
действием фталевого ангидрида во фталимиды, которые в уксусной кислоте
нейтральны |262|.
Взвешивают такое количество смеси аминов, чтобы фталевый ангидрид
(мол. вес 148,1), который берется в количестве 1 г, был в 2-3-кратном
избытке по отношению к первичному амину. После добавления 1 г фталевого
ангидрида к 20—25 мл раствора, содержащего уксусную кислоту, нагревают
30—40 мин на водяной бане и после охлаждения титруют непрореагировав-
шие вторичные и третичные амины потенциометрически 0,1 н. хлорной кис-
лотой. Нагревают другую порцию анализируемого раствора с 10—15 мл
уксусного ангидрида, как описано выше, и после охлаждения титруют тре-
тичные амины хлорной кислотой. Как только ацетилируемые амины перейдут
в амиды кислот, они не реагируют с хлорной кислотой.
Смеси азотистых оснований или смеси хлоргидратов оснований
347
146. МЕТОД С ИСПОЛЬЗОВАНИЕМ УКСУСНОГО АНГИДРИДА
а. Определение никотина, норникотина и суммы алкалоидов, содержа-
щихся в порошке табака [158]. В колбе емкостью 250 мл с притертой стеклян-
ной пробкой взвешивают 2,5—3,5 г измельченного в тонкий порошок табака,
добавляют 1 г гранулированной восьмиводной гидроокиси бария и 15 мл
насыщенного водного раствора гидроокиси бария. Экстрагируют тщательно
смоченный табачный порошок 100 мл смеси бензол — хлороформ (9 : 1),
встряхивая его 15 мин. Для облегчения фильтрования добавляют около 2 г
кизельгура (или целита), перемешивают и затем фильтруют органический
слой через фильтровальную бумагу ватман № 2 декантацией в другую колбу.
Отбирают пипеткой (2 X 25 мл) фильтрат в две колбы Эрленмейера по 100—
150 мл. Пропускают над поверхностью жидкости в первой колбе в течение
5 мин воздух, чтобы удалить образующийся аммиак (для этой цели пригодна
склянка для упаривания, показанная на рис. 103), во вторую колбу вносят
0,5 мл уксусного ангидрида. Титруют оба раствора 0,025 н. хлорной кисло-
той в присутствии 1 капли кристаллического фиолетового.
Расчет:
r t \ Г,ЛГ-32,45 0/
Сумма алкалоидов (в виде никотина) = —----------%,
Содержание никотина = —ГО /Г'32,43 %,
„ (2Г1-Г2)ЛГ-29,64 0/
Содержание норникотина =-----1----------— %,
где Vi — объем стандартного раствора, необходимый для нейтрализации
неацетилированного раствора, мл; V2 — объем стандартного раствора, необ-
ходимый для нейтрализации ацетилированного раствора, мл; N — нормаль-
ность стандартного раствора; b — вес образца, г.
Реакционная способность ацетилирующихся аминов по отношению к уксусному
ангидриду различна в зависимости от заместителей, возможных пространственных за-
труднений и др. Способность некоторых аминов к ацетилированию 1 М уксусным анги-
дридом в 0,1 н. растворе в уксусной кислоте в расчете на 90%-ное протекание реакции
уменьшается в следующем ряду [612]: 2-нафтпламин, n-фенплендиамин, анилин, 2-трет-
бутиламин (1 мин): 2-метиланилин, 1-нафтиламин, N-метил анилин, 2,6-диметиланилин
(1,5 мин): этилендиамин, дибензил амин, бензиламин (1—2 час); этанолампн, пиперидин,
н-пропплампн, н-бутпламин, диэтаноламин (2—6 час); изопропилампн (8—30 час); рн-н-
бутпламин, диметиламин, ди-н-пропиламин (одна неделя и больше); дпизопропиламин.
Следовательно, легкость ацетилирования индивидуальных оснований изменяется
в широких пределах, и это позволяет осуществить на практике анализ их смесей.
б. Определение небольших количеств {0,01—0,02%) третичных аминов
в присутствии аминов, способных к ацетилированию [690]. Амиды кислот,
образующиеся при ацетилировании уксусным ангидридом большинства пер-
вичных и вторичных аминов, хотя и представляют собой слабые основания,
тем не менее они могут мешать определению небольших количеств третичных
аминов в уксусной кислоте (или смеси уксусная кислота — уксусный ангид-
рид): переход окраски индикатора нечеткий, а потенциометрическая кривая
выровнена.
Определять небольшие количества третичных аминов в присутствии боль-
ших количеств ацетилированных аминов можно достаточно точно, если
в качестве растворителя использовать метилцеллозольв, а не уксусную
кислоту (уксусный ангидрид) и проводить титрование 0,01 н. хлорной кисло-
той также в метилцеллозольве, так как уксусная кислота в известной степени
увеличивает основность амидов кислот.
Показано, что метилцеллозольв как растворитель более пригоден, чем метиловый
и изопропиловый спирты, ацетонитрил и гликоли. '
348
Глава 25
В зависимости от силы оснований в качестве индикатора может быть
использован 0,3 %-ный раствор тимолового синего в диметилформамиде
или 0,1%-ный раствор конго красного в метиловом спирте (К = 10-4—10-7
или 10-8—10-10). В присутствии тимолового синего можно титровать триэтил-
амин (К = 5,7-10-4), 1М,]\т-диметилэтаноламин (1,6 -10-5) и N-этилморфолин
(3,1 -10-7), а в присутствии конго красного — у-пиколин (1,1-10-8) и диметил-
анилин (1,6 ЛО-9). Титрование лучше проводить 0,01 и. раствором хлорной
кислоты в метилцеллозольве, а не раствором хлористого водорода в этом же
растворителе. Если для потенциометрического титрования использовать
хлористый водород в метилцеллозольве, то кривая титрования становится
более пологой, а окраска тимолового синего изменяется нечетко вследствие
наличия в смеси ациламида.
Однако в среде метилцеллозольва уменьшается основность некоторых
третичных аминов; так, у М-^енилпиперазина протонируется только вто-
ричный атом азота (К = 1 «10-6), а третичный N-атом (1 ЛО-12) не титруется.
Таким образом, после ацетилирования основной характер N-фенилпипе-
разина в метилцеллозольве нивелируется. Поэтому в метилцеллозольве
можно титровать только амины, константы основности которых >> 10-10.
Предложенный Рачом и Критчфильдом метод определения аминов пре-
дусматривает их ацетилирование. В смеси метилцеллозольв — уксусный
ангидрид (10 : 2) реакция заканчивается через 30 мин. В этилацетате, аце-
тонитриле и хлороформе ацетилирование заканчивается уже через 15 мин,
но для титрования смесь необходимо разбавить метилцеллозольвом в отно-
шении 1 : 3.
Точность титрования третичных аминов увеличивается при добавлении
уксусного ангидрида. Титрование пиридина, однако, проходит менее точно,
поскольку образующийся ацетат ацетилпиридиния — более слабое основа-
ние, чем сам пиридин. Гидроксильная группа триэтаноламина ацетили-
руется в течение 90 мин, так как вследствие электроноакцепторных свойств
ацетоксигруппы основность атома азота понижена. Это влияние слабее
проявляется при титровании диметилэтаноламина, так как при этом обра-
зуется только одна ацетоксигруппа. Метод пригоден также для определения
триэтаноламина, содержащего примесь моноэтаноламина, если ацетилирова-
ние проводить в течение 30 мин. Кроме того, этот метод применяется для
определения минимального содержания триэтиламина в этиламине или
диэтиламине, триизопропиламина в изопропиламине, N-этилморфолина
в морфолине и 1\,К-диметиланилина в анилине.
5—7 г испытуемого образца (который, однако, не должен содержать более
0,5 мг-экв третичного амина) растворяют в 100 мл метилцеллозольва, доба-
вляют 20 мл уксусного ангидрида и выдерживают в течение 30 мин. Титрова-
ние ведут 0,01 н. хлорной кислотой в присутствии тимолового синего или
конго красного.
Относительная точность определения 0,01—0,2% третичного амина
составляет 80—100 ±5%.
147. ФОТОМЕТРИЧЕСКОЕ ТИТРОВАНИЕ ПЕРВИЧНЫХ АМИНОВ
В ПРИСУТСТВИИ ВТОРИЧНЫХ И ТРЕТИЧНЫХ АМИНОВ
Первичные алифатические амины можно титровать 0,1 М 2-этилгексан-
альдегидом в диоксане с уксусной кислотой в качестве катализатора даже
в присутствии вторичных и третичных аминов [518]. Из первичных аминов
при этом образуются соответствующие имины. Оптимальная концентрация
катализатора должна быть примерно равна 0,5 молъ/л (в диоксановом рас-
творе). При меньшей концентрации реакция идет медленнее, а при концен-
трации больше 0,5 молъ!л происходит протонирование аминов и последующие
реакции затрудняются [518].
Смеси азотистых оснований или смеси хлоргидратов оснований
349
1. Присоединение протона к альдегиду:
RCHO + H+ [RCHOH]+.
2. Присоединение амина к положительно заряженному атому углерода
протонированного альдегида:
ОН
+ I +
RCHOH + R'NH2 —> R — с — N-R'
I /\
НН н
3. Образование имина при дегидратации:
ОН
I +
R — С — N— R' R — CH = NR' + H2O + H+
I /\
НН н
Первичные алифатические амины нельзя определять в присутствии пер-
вичных ароматических аминов; хотя ароматические амины в сколько-нибудь
значительной степени не реагируют с этилгексанальдегидом, анализ ведут
при длине волны 305 нм и собственное светопоглощение ароматических колец
мешает определению. Добавление этилгексаналя после конца титрования
вносит значительные изменения в поглощение при 305 нм. Титр стандарт-
ного раствора устанавливают по н-бутиламину, нормальность которого про-
веряют титрованием хлорной кислотой (о его приготовлении см. гл. 11,
разд. 60,п). Нормальность стандартного раствора этилгексанальдегида
сохраняется неизменной до тех пор, пока он защищен от попадания кисло-
рода воздуха. Поэтому желательно заполнять сосуд с титрантом азотом.
Титрование проводят в кварцевых кюветах на 50 мл [570].
Смесь, содержащую 0,15—0,45 молъ/л алифатического первичного амина,
растворяют в 100 мл 0,5 М раствора уксусной кислоты в диоксане. Пипеткой
отбирают точно 1,00 мл этого раствора в кварцевую кювету, уже содержа-
щую 35 мл 0,5 М уксусной кислоты в диоксане. Используя монохроматиче-
ский свет с длиной волны 305 нм, титруют 0,1 н. раствором 2-этилгексаналя
в диоксане; во время определения пропускают через раствор непрерывный
ток азота. Максимум поглощения 2-этилгексаналя лежит при 295 нм, однако
в этой области поглощение исходного вещества и образовавшегося имина
различаются. Скорость образования имина невелика, но она увеличивается
под действием уксусной кислоты. Ацетаты н-бутиламина, н-гексиламина,
Р-фенилэтиламина, бензиламина и циклогексиламина растворимы в диоксане.
Однако для ацетата этилендиамина уксусная кислота в диоксане должна
быть заменена уксусной кислотой в ацетонитриле.
Точность описанного метода ±0,6%.
148. ОПРЕДЕЛЕНИЕ СМЕСЕЙ АМИНОВ КОМБИНИРОВАННЫМ МЕТОДОМ
а. Определение смесей ароматических аминов [750]. Третичные амины
титруют после ацетилирования первичных и вторичных аминов (титрова-
ние 1); затем по реакции с салициловым альдегидом (титрование 2) опреде-
ляют суммарное количество вторичных и третичных аминов; титрование 3
показывает суммарное содержание всех основных компонентов. Суммарное
количество первичных аминов вычисляется по разности результатов титро-
ваний 2 и 3; количество вторичных аминов — по разности результатов
титрований 2 и 1.
Титрование 1. Взвешивают трубку размером 20 X 150 мм с образцом,
в котором содержится примерно 0,02 моля третичного амина, и помещают
в лед. При осторожном и постоянном перемешивании добавляют к образцу
350
Глава 25
10 мл уксусного ангидрида и оставляют на 15 зеин при комнатной температуре.
Переносят содержимое трубки в колбу, промывая трубку 50 мл смеси этилен-
гликоль — изопропиловый спирт (1:1) и титруют амины потенциометри-
чески 1 н. соляной кислотой, приготовленной в аналогичной смеси раство-
рителей.
V-N
Мтрет = g.looo ,
где Мтрет — содержание третичного амина, молъ/г; V — объем соляной
кислоты, мл; N — нормальность раствора кислоты; g — вес образца, г.
Титрование 2. Растворяют образец, содержащий в сумме примерно
0,02 моля вторичных и третичных аминов, в 50 мл смеси этиленгликоль —
изопропиловый спирт (1 : 1), добавляют 5 мл салицилового альдегида, выдер-
живают в течение 30 мин, затем титруют потенциометрически и вычисляют
(как описано выше) суммарное количество вторичных и третичных аминов
в 1 г образца.
Титрование 3. В 50 мл смеси растворителей потенциометрически титруют
образец, содержащий в сумме около 0,02 моля аминов. Суммарное содержа-
ние аминов в 1 г образца вычисляют так же, как описано в титровании 1.
Расчет’.
, , М ^втоп “Натрет
Мперв -—------------------ ,
Л/Втор -Дотрет -Дотрет
‘"втор' - - ,
где МПерв, МВТОр и Мтрет — содержание первичных, вторичных и третич-
ных аминов соответственно, молъ/г; М — суммарное содержание аминов,
молъ/г; g — вес образца, г. Определение содержания третичных аминов
см. выше (титрование 1).
Грибова и Левин [290] определяли состав смеси анилина, гексиламина и дициклогек-
си л амин а, с этой целью они проводили два титрования. Если титрование выполнялось
в ацетоновом растворе 0,1 н. хлорной кислотой в дпоксане с использованием стеклянного
и каломельного электродов, то наблюдалось два скачка потенциала. Первый указывает
на общий эквивалент нейтрализованных циклогекспламина и дицпклогексиламина,
а второй соответствует титрованию анилина. Второе титрование, которому предшествует
реакция с салициловым альдегидом, снова дает два перегиба на кривой, причем первый
характерпзует дициклогексиламин, а второй соответствует общей эквивалентной точке
циклогекспламина и салицплальдимина анплина.
Бурник и Нойзер [78] титровали тройную смесь бутиламин — N-метиланилин —
N, N-диметпланилин в смеси диэтиленгликоль — изопропиловый спирт. Первый пере-
гиб на потенциометрической кривой соответствует бутиламину, второй — сумме двух
ароматических аминов. После ацетилирования титруется только М.М-диметпланилпн.
б. Титрование в тройной смеси растворителей [401]. Анализ большинства смесей
аминов можно выполнить, целесообразно комбинируя титрование в дифференцирующих
растворителях, ацетилирование и реакцию с образованием оснований Шиффа. Разность
в значениях p/f между алифатическими и ароматическими аминами в среднем составляет
5 единиц. Потенциометрическое дифференцирующее титрование можно проводить в слабо-
кислой, но в то же время спльнополярной среде, например ацетонитриле, нитрометане
или нитробензоле. Подходящей смесью растворителей является смесь уксусная кислота —
диоксан — нитрометан (5 : 75 : 20), титрование проводят 0,1 н. хлорной кислотой
в дпоксане в качестве титранта. Так, например, смесь из 6 компонентов можно проанали-
зировать с помощью трех титрований. В первом титровании определяют сумму аминов
титрованием хлорной кислотой части смеси бутиламин — дибутилампн — триметил-
амин — анилин — метиланилин — пиридин в уксусной кислоте. Второе титрование
проводят после ацетилирования уксусным ангидридом, когда титруются только алифа-
тические и ароматические третичные амины. После реакции с салициловым альдегидом
проводят третье потенциометрическое титрованпе. Первый перегиб на кривой указы-
вает на сумму вторичных и третичных алифатических аминов, т. е. сумму сильных осно-
ваний. Второй перегиб на кривой соответствует сумме вторичных п третичных аромати-
ческих аминов и оснований Шиффа, образованных первичными алифатическими аминами.
Третий перегиб на кривой указывает эквивалентную точку слабых оснований, т. е. осно-
ваний Шиффа, образованных первичными ароматическими аминами.
Смеси азотистых оснований или смеси хлоргидратов оснований
351
149. МЕТОДЫ ПЕРЕГОНКИ
а. Разделение азотистых оснований перегонкой с перегретым паром [301.
315, 3201. В некоторых случаях летучие основания можно отделить от отно-
сительно менее летучих или нелетучих оснований перегонкой с перегретым
паром растворителя, например бензола или хлороформа.
Относительный эмпирический порядок летучести оснований при перегонке с пере-
гретым паром растворителя после испарения 5 мл хлороформа из раствора анализируе-
мого соединения (в скобках указаны количество паров бензола, необходимых для пере-
гонки 90% летучего основания, выраженное в миллилитрах сконденсированного бензо-
ла, и температура кипения основания, взятые в основном из работы [315]: пропиламин
(0,7; 49°), бутиламин (1,8; 77,8°), дибутиламин (15; 159°), трибутиламин (73: 216.5°),
Рис. 122. Прибор для перегонки с использованием перегретых паров растворителя
[315].
1 — колба для титрования, н холящаяся в водяной бане; 2 — масляная баня для нагревания при
185 ± 2°; 3 — широкогорлая колба с притертой пробкой емкостью 200—250 мл, в этой колбе образу-
ются пары растворителя: 4—в — регулируемые электрические нагреватели; 7—электрическая схема
выключения нагревателей; 8 — холодильник; 9 — сферическое соединение на шлифе; 10 — асбестовая
изоляция.
циклогексиламин (6; 134°), дициклогексиламин (19; 247°), пиперидин (4,4; 106,4°), N-
(2-пксиэтил)ппперидин (7; 200—202°), пиперазин (10; 145—146°), М-(2-пксиэтил)пипе-
разин (100; 242—244°), пиридин (3; 115,5°), 2-метилпиридин (4,4; 128,8°), 2-аминопирпдин
(7,8; 204°), анилин (6; 184.4°), N-метиланплин (7,4; 196,3°), п-толуидин (8,7; 200,6°).
бензиламин; (6,0; 184—185°), N-метилбензиламин (7,0; 180—181°), 4-метилбензиламин
(9,6; 194—196°), морфолин (4,2; 129°), ]Ч-(2-окспэтил)морфолин (12,4; 220—222°), этанол-
амин (2, 171°), 4-аминобутанол-1 (7,6; 206°), диэтаноламин (83; 272°). К числу малолетучих
оснований относятся, например, N-ппперидинометилтолилпропанон [314], триэтаноламин
и бис-(оксиэтил)пиперазин (количество перегнанного амина пропорционально количе
ству перегнанного бензола). Атропиновые основания, трибензиламин и 1Ч-(2-пиридил)-
-]Ч-(п-хлорбензил)-]Ч',]Ч'-диметилэтилендиамин практически нелетучи.
Многие основания с температурами кипения 100—280° можно перего-
нять с парами бензола при примерно 95°. Основания с более высокими тем-
пературами кипения относятся к менее летучим или нелетучим. Прибор для
разделения азотистых оснований методом перегонки изображен на рис. 122.
Этим методом можно определять, например, трибензиламин в присутствии
бензиламина; пиперидинометилтолилпропанон после отгонки примесей
N-метилпиперидина, пиперидина и N-оксиэтилпиперидина; 21-дезокси-21-1\-
(1\'-метилпиперазипил)преднизолоп в присутствии N-метилпиперазина
и, наконец, атропиновые основания в смеси с различными летучими алка-
лоидами.
352
Глава 25
В колбе для титрования (рис. 122) взвешивают такое количество анали-
зируемой смеси, чтобы в ней содержалось примерно 5«10-2 ммолей моно-
функционального основания и растворяют ее в 3—5 мл хлороформа. При
использовании экстрактов отбирают в колбу пипеткой 3—5 мл. Хлороформ
отгоняют на водяной бане, поддерживая температуру пароперегревателя
185 ± 2°. После концентрирования хлороформного раствора в колбу 3
наливают бензол, предварительно подогретый выделяющимися парами
растворителя до температуры кипения, и пропускают пары бензола через
спираль нагревателя. Количество растворителя, проходящего через аппарат,
контролируют градуированным цилиндром, его объем обычно равен 10—
30 мл. Рекомендуется вначале подобрать нужное количество растворителя
на модельных опытах. После перегонки летучих оснований с перегретым
Рис. 123. Прибор для определения триметиламина в солях холина [166].
паром растворителя колбу для титрования вынимают из водяной бани,
охлаждают, добавляют к остатку 5 мл нейтрализованной уксусной кислоты
и титруют нелетучие основания 0,01 н. хлорной кислотой. В качестве инди-
катора используют 0,1 %-ный раствор кристаллического фиолетового. Если
остаток растворить в хлороформе или четыреххлористом углероде, остав-
шиеся основания, например атропин, можно титровать также 0,005 н.
лг-толуолсульфокислотой в присутствии диметилового желтого до появления
розового окрашивания.
Иногда летучесть малолетучих оснований пропорциональна количеству
пропускаемых паров бензола (конденсат, мл). И если это учесть, то можно
проанализировать смесь летучих и малолетучих оснований.
б. Определение летучих оснований в солях холина [166]. В качестве другого
варианта метода перегонки приведем метод определения примеси триметил-
амина в хлориде, глюконате, дигидроцитрате и гидрокарбонате холина.
Здесь летучие основания удаляют из системы током воздуха, улавливают
уксусной кислотой и титруют хлорной кислотой. Прибор, используемый
для такого определения, показан на рис. 123.
Взвешивают 50 мл 40—70%-ного водного раствора соли холина и пере-
носят в сосуд (7), добавляют 50 мл приблизительно 30%-ного (вес/объем)
раствора едкого натра. 50 мл уксусной кислоты, нейтрализованной в при-
сутствии кристаллического фиолетового, помещают в колбу Эрленмейера (2).
а 10 мл — в контрольную склянку (5). В течение 75 мин через аппарат про-
пускают ток очищенного воздуха, так чтобы пузырьки непрерывно про-
булькивали через щелочной раствор соли холина. Затем объединяют уксус-
ную кислоту из склянок 2 и 3, добавляют 15 мл уксусного ангидрида, кипя-
тят 5 мин и после охлаждения титруют 0,1 н. хлорной кислотой в уксусной
Смеси азотистых оснований или смеси хлоргидратов оснований 353
кислоте в присутствии кристаллического фиолетового. Для титрования
5,9 мг триметиламина требуется 1 мл 0,1 н. хлорной кислоты.
Методики определения солей холина с карбоновыми кислотами рассматри-
ваются в работе [549].
Анализ смеси пиперазина, оксиэтилпиперазина и бис-(оксиэтил)пиперазина см.
в гл. 23, разд. 23, в.
Анализ смеси гидразина и несилл-диметилгидразина см. в гл. 23, разд. 133, в.
Методы анализа смеси соединений, содержащих аминогруппы, основанные на раз-
личии в скорости реакций, обсуждаются в работе [347] (ср. гл. 19, разд. 113).
150. ОПРЕДЕЛЕНИЕ ХЛОРГИДРАТОВ ТРЕТИЧНЫХ АМИНОВ
В ПРИСУТСТВИИ ХЛОРГИДРАТОВ АМИНОВ, СПОСОБНЫХ
К АЦЕТИЛИРОВАНИЮ [310, 312, 707J
Известно, что третичные амины можно титровать в присутствии аминов,
способных к ацетилированию, в смеси уксусная кислота — уксусный ангид-
рид. Это было установлено до того, как было найдено, что хлоргидраты
первичных, вторичных и третичных аминов можно титровать раздельно,
если добавить к их раствору в уксусной кислоте ацетат ртути(П). Объединив
эти два метода, можно титровать и хлоргидраты третичных аминов в смеси
с хлоргидратами аминов, способных к ацетилированию. В качестве ацетили-
рующего агента используется смесь уксусного ангидрида и раствора ацетата
ртути(П) в уксусной кислоте, т. е. превращение хлоргидрата амина в ацетат
основания предшествует ацетилированию в той же смеси реагентов.
Взвешивают такое количество анализируемого вещества или раствора,
на титрование которого потребуется 6—8 мл 0,05 или 0,01 н. хлорной кис-
лоты. В колбу Эрленмейера к 5—10 мл образца добавляют достаточное коли-
чество ацетилирующего агента так, чтобы общий объем раствора равнялся
20—30 мл, причем в нем содержалось 40—80% уксусного ангидрида и в зави-
симости от навески образца 200—300 или 40—60 мг ацетата ртути(П). Широ-
кую часть колбы Эрленмейера смачивают уксусным ангидридом. Ацетилиро-
вание проводят при комнатной температуре в течение 60—180 мин. Затем
добавляют 1 каплю 1%-ного (или 2 капли 0,2%-ного) кристаллического
фиолетового и титруют 0,05 или 0,01 н. хлорной кислотой, пока фиолетовая
окраска индикатора не перейдет в синюю. В каждом случае учитывают рас-
ход титранта на реакцию с ацетилирующей смесью.
Что касается содержания в смеси ангидрида и продолжительности аце-
тилирования, то достаточно знать константу диссоциации (в воде) амина,
способного к ацетилированию. Реакцию ведут в условиях, приведенных
в табл. 99.
Таблица 99
Константа диссоциации основания Содержание ангидрида в ацетили- рующей смеси, % Продолжи- тельность, мин Константа диссоциации основания Содержание ангидрида в ацетили- рующей смеси, % Продолжи- тельность, мин
10-3 40—80 180 10“5 40 80
1(Г4 60 100 10~6 40 60
Если в смеси аминов количество амина, способного к ацетилированию,
превышает 10%, то после ацетилирования смесь разбавляют уксусной кис-
лотой до 40 %-ной концентрации и титруют хлорной кислотой в присутствии
5 капель 0,5%-ного крезолового красного, а не кристаллического фиолето-
вого, до перехода желтой окраски индикатора в красную.
354
Глава 25
На точность определения влияют константа диссоциации способного к ацетилиро-
ванию амина (измеренная в воде), способность амида кислоты образовывать связь с хлор-
ной кислотой, молярная концентрация третичного амина и ацетилируемого амина, содер-
жание уксусного ангидрида в смеси и выбор индикатора.
В смеси уксусная кислота — уксусный ангидрид при комнатной температуре весьма
эффективно ацетилируются те амины, константа диссоциации которых (в воде) мала. Ко-
личественные результаты ацетилирования сильноосновных аминов оказываются хуже
(титрование в присутствии крезолового красного): бензиламин 100%, этиламин 97—
98%, пиперидин 97% и диэтиламин 95%. Это можно объяснить тем, что амины раство-
ряются в уксусной кислоте частично как «ониевые» соли, а частично как сольваты ами-
нов с уксусной кислотой. Более основные амины образуют больше «ониевых» солей
R3NH+, которые, будучи донорными кислотами, вовсе не вступают во взаимодействие-
с ацетил-катионом, присутствующим в ацетилирующей смеси (СН3СО+ — акцепторная
кислота), но легко реагируют с сольватами аминов с уксусной кислотой R3N...CH3COOHr
которые являются донорными основаниями.
Этим методом можно анализировать, например, смесь диэтиламиноэтил-
хлорида и его хлоргидрата в присутствии хлоргидрата этиламина или диэтил-
амина, хлоргидрат трибензиламина в присутствии хлоргидрата бензиламина
и хлоргидрат пиперидинометилциклогексанона в присутствии хлоргидрата
пиперидина и т. д.
Глава 26
ОПРЕДЕЛЕНИЕ КАРБОНИЛЬНЫХ ГРУПП
151. ИДЕНТИФИКАЦИЯ АЛЬДЕГИДОВ И КЕТОНОВ
арил)имидазолидиновые производ-
а. Идентификация альдегидов в виде имид азоли диновых производных [8421.
Соединения, содержащие альдегидную группу, образуют с 1,2-бис-(фенил-
амино)этаном 1,3-дифенил-2-алкил( или
ные [859].
R-c/°
С6Н5 —N —СН2
I !
н сн2
н С6Н5
—Н2О С5Н5 — N — СН2
----> 2 | |
R - НС СН2
'n7
3I
С6Н5
N
Производные имидазолидина можно титровать в уксусной кислоте
0,1 н. хлорной кислотой, определяя конечную точку либо потенциометри-
чески, либо визуально с индикатором. Следовательно, зная молекулярный
вес образовавшегося аддукта, можно идентифицировать соединения, содер-
жащие альдегидную группу. Молекулярный вес альдегида можно вычислить,
если из удвоенной величины эквивалентного веса, найденной титрованием
имидазолидина, вычесть 194,3, например:
2-Метилпроизводное, мол. вес 238,3 —194,3 = 44,0 (ацетальдегид),
2-Этилпроизводное, мол. вес 252,3 —194,2 = 58,1 (пропионовый альдегид),
2-Пропилпроизводное, мол. вес 266,3 — 194,2 = 72,1 (масляный альдегид) и т. д.
200—300 мг альдегида растворяют в 5 мл метилового спирта, смешивают
с 10 мл 5 %-ного раствора 1,2-бис-(фениламино)этана в метиловом спирте
и по каплям добавляют 50%-ную водную уксусную кислоту. Производное
имидазолидина выделяется обычно в кристаллической форме; если оно не
выпадает, то смесь кипятят с обратным холодильником 5 мин. Примерно
0,05 ммоля высушенного продукта растворяют в 20—25 мл уксусной кислоты
и титруют, определяя конечную точку потенциометрически или визуально;
в последнем случае титруют 0,1 н. хлорной кислотой в уксусной кислоте
в присутствии кристаллического фиолетового. На титрование расходуется
2 экв титранта. Согласно исследованиям Вайбеля и Кроха Андерсена, при
добавлении хлорной кислоты образуется диперхлорат 1,2-бис-(фениламин)эта-
на, поэтому имидазолидиновые производные некоторых альдегидов (фурфу-
рол, тиофенальдегид) титровать не удается вследствие того, что образуются
окрашенные продукты распада альдегидов. Этого не наблюдается при титро-
вании бис-(и-метоксибензил)имидазолидиновых производных
C6H5-N —СН2 н+ CeH5NH2-CH2
| | 4-2СН3СООН —> |
R —НС СН2 СН2
\ / Л-/
N H2N
I I
СбНБ С6Н5
+ RCH(OCOCH3)2
Диацетат альдегида
356
Глава 26
В случае имидазолидиновых производных пиридилальдегидов расхо-
дуется 3 экв хлорной кислоты. Точность определения тем меньше, чем меньше
молекулярный вес альдегида.
Альдегиды (и кетоны) могут быть также идентифицированы по реакции с 2,4-динп-
трофенилгидразином и последующим титрованием образующегося гидразона стандартным
раствором ТБАГ в ппридине (см. гл. 20).
б. Идентификация ^-дикетонов [844]. Р-Дикетоны при реакции с п-карбоксифенпл-
гидразином образуют вместо гидразонов производные пиразола, кристаллизующиеся
с 1 молем воды. Первоначальные продукты конденсации, образовавшиеся из бензо-
илацетона, дибензоилметана или ацетилацетона, при перекристаллизации из водного
этилового спирта дают карбоксифенилгидразоны, которые можно титровать как кислоты.
Но если пиразольное производное высушить в вакууме над Р2О5 при 56°, то вновь обра-
зуется пиразольное производное и, следовательно, можно сделать вывод, что во время
•высушивания происходит замыкание кольца, а во время перекристаллизации кольцо
снова раскрывается. Производное пиразола можно определять как основание титрова-
нием хлорной кислотой [838, 844]
П2С — С — R' НС — С — R' zNH2 -Н2о НС — CR'
II 7± II II + HN< ----> II |1
R—С О R —С О ХС6Н4СООН RC N
II I \ /
О НО N
I
С6Н4СООН
Ароматические а-дикетоны дают моно-п-карбоксифенилгидразоны,
а у-дикетоны — производные дигидропиридазина, причем и те и другие
производные можно титровать как кислоты [838, 845].
152. ОПРЕДЕЛЕНИЕ КАРБОНИЛЬНЫХ ГРУПП
В хорошо известном методе определения соединений, содержащих альде-
гидную или кетонную группу, используется реакция образования оксимов
с гидроксиламином или образование гидразонов с производными гидразина.
Изменение окраски индикатора в неводных средах более отчетливо, чем
в водной фазе. В ходе образования оксимов в водной среде выделяются вода
и хлористый водород
RCHO4-NH2OH-HC1 7 л RCH=NOH-!-H2O+HCl.
Реакция происходит в присутствии основания, например пиридина,
октадецениламина, морфолина, «-трибутиламина и др., которые связывают
хлористый водород.
Меткаф и Шмитц [574] применили октадецениламин в смеси метиловый —
изопропиловый спирты, Фриц и сотр. [259] — 2-диметиламиноэтиловый
спирт в изопропиловом спирте, а Денеш [314] применил три-«-бутиламин
в смеси метилового и изопропилового спиртов. При использовании третич-
ного амина исключается образование оснований Шиффа. Вследствие опас
ности образования ацеталей и кеталей нужно строго выполнять порядок
смешения компонентов. Реакция протекает по следующей схеме:
1. Образование свободного гидроксиламина
(NH3OH)+C1- -В 5H+Cl-4-NH2OH,
где В — например, трибутиламин.
2. Образование оксима
NH2OH + >C = O \c = NOH + H26’.
3. Титрование гидрокси ламина, оставшегося в избытке,
NH2OH + H+ —(NH3OH)+.
Определение карбонильных групп
357
а. Определение альдегидов с использованием несимм-диметилгидразина
[752]. Альдегиды образуют гидразоны с несимметричным диметилгидразином
(CH3)2N — NH2. Избыток реагента определяют обратным потенциометри-
ческим титрованием соляной кислотой в метиловом спирте.
Для определения алифатических альдегидов 0,002 моля соединения
(0,01 моля ароматических альдегидов) растворяют в 25,0 мл диметилгидра-
зина и выдерживают при комнатной температуре; продолжительность выдерж-
ки зависит от активности альдегида. Затем добавляют 50 мл метилового
спирта и, используя стеклянный и каломельный электроды, титруют потен-
циометрически в метиловом спирте 0,1 или 0,5 н. соляной кислотой в зави-
симости от концентрации (0,2 или 1,0 моль/л) используемого диметилгидра-
зина (мол. вес 60,10; = 0,791).
Расчет
(b-a)-N-E 0/
Содержание альдегида —----------%,
где Ъ — объем стандартного раствора, пошедшего на титрование в глухом
опыте, мл; а — объем стандартного расхода, израсходованный на титрова-
ние образца, мл; N — нормальность стандартного раствора, г-экв/л;
Е — 1/ю эквивалентного веса соединения; g — вес образца, г.
Таблица 100
Альдегид Растворитель Концентра- ция, моль/л
Алифатический Этиленгликоль 0,2
Ароматический » 1,0
Дизамещенный Метиловый спирт 1,0
бензальдегид
Этим методом можно с точностью от ±0,3 до ±0,8% титровать следую-
щие альдегиды (в скобках указана продолжительность реакции в минутах):
формальдегид, ацетальдегид, пропионовый, масляный (15), коричный, кро-
тоновый, салициловый (30), анисовый, бензальдегид, лг-нитробензальдегид,
2,6-дихлорбензальдегид, 2,4-диметоксибензальдегид (120).
Ароматические альдегиды можно определить в присутствии кетонов,
а алифатические определить не удается.
б. Определение альдегидов и кетонов реакцией с гидроксиламином [259]
(табл. 101). В колбе Эрленмейера с притертой стеклянной пробкой взвеши-
вают такое количество вещества, чтобы в нем содержалось 1,5—2,5 ммоля
карбонильных групп, добавляют точно 20,0 мл 0,25 М раствора 2-диметил-
аминоэтанола и точно 25,0 мл 0,4 М раствора солянокислого гидроксил-
амина. Перемешивают содержимое и оставляют на некоторое время
(см. табл. 101). После этого добавляют 5 капель индикаторной смеси и тит-
руют хлорной кислотой до обесцвечивания желтой окраски индикатора или
до перехода ее в сине-серую. Расчет выполняют так же, как описано выше.
Реагенты.
Раствор диметиламиноэтанола получают, растворяя 22,5 г свежеперег-
нанного основания в 1 л изопропилового спирта.
Чтобы получить раствор солянокислого гидроксиламина, 27,8 г NH2OH -НС1
растворяют в 300 мл очищенного метилового спирта и разбавляют до 1 л
очищенным изопропиловым спиртом.
Для приготовления индикаторной смеси 67 мг марциуса желтого
(2,4-динитронафтол-1) и 4 мг метилового фиолетового растворяют в 50 мл
этилового спирта. Приготовление стандартного раствора см. в гл. 11,
разд. 60, д.
358
Глава 26
Таблица 101
Определение альдегидов и кетонов с помощью
гидроксиламина {259]
Карбонильное соединение а Продолжи- тельность реакции, мин Температура реакции, °C Молекуляр- ный вес
Ацетофенон 45—60 70 120,14
Бензальдегид 20 Комнатная 106,1
Бензоин 180 70 212,2
Масляный альдегид 20 Комнатная 216,3
Циклогексанон 30 » 98,1
Дпбензилкетон 30 » 210,3
Фурфурол 20 » 96,08
Метилизобутилкетон 30 » 100,2
п-Нитробензальдегид 20 » 151,1
п-Оксибензальдегид 5—15 » 122,1
Салициловый альдегид 5—15 » 122,1
Ванилин 5—15 » 152,1
а На титрование 2 ммолей альдегида расходуется 1 мл 0,2 н. хлорной
кислоты.
Соединения, которые можно определять с точностью от +0,1 до +0,3%,
приведены в табл. 101.
в. Определение солянокислого пиперидинометилтолилпропанона [314].
Если соединение содержит третичную аминогруппу в виде соли, то гидро-
ксиламинный метод определения карбонильных групп можно использовать
для анализа аминокарбонильных производных. |
В колбе Эрленмейера с притертой стеклянной пробкой емкостью 200 мл
взвешивают 130—150 или 260—300 мг образца, растворяют его в 10 или
20 мл примерно 0,1 М н-трибутиламина и добавляют 10 или 20 мл приблизи-
тельно 0,1 М раствора солянокислого гидроксиламина. Эти реагенты гото-
вят в смеси метилового и изопропилового спиртов (36 : 65). Между горлом
колбы и пробкой закладывают полоску фильтровальной бумаги, затем
закрывают колбу и ставят ее на 5—10 мин на водяную баню с температурой
71—73°. Снижают температуру до 69—70°, удаляют полоску фильтроваль-
ной бумаги и закрывают колбу герметически. Эту температуру поддержи-
вают 50—55 мин, а затем дают раствору охладиться до комнатной темпера-
туры и титруют 0,1 н. хлористоводородной кислотой в смеси пропиленгли-
коль — хлороформ (гл. 9, разд. 51, а). Индикатор —раствор 0,5 г диметил-
аминоазобензола и 0,05 г метиленового синего в 60 мл метилового спирта.
К 20 мл реакционной смеси добавляют 3 капли и к 40 мл смеси 5 капель
индикатора. Для глухого опыта выполняют одновременно три титрования
с 10 или 20 мл раствора гидроксиламина в трибутиламине. Расчет проводят
так же, как описано выше. На титрование 28,18 мг солянокислого 1-пипери-
дин-2-метил-З-п-толилпропанона-З расходуется 1 мл 0,1 н. соляной кислоты.
г. Макро- и полу микрометоды, определения альдегидов и кетонов с исполь-
зованием формиата гидроксиламина [691]. Преимущество этого метода
в сравнении с другими ему подобными заключается в том, что 0,003—0,3%
альдегида или кетона можно определить с точностью до +5%. Метод дает
надежные результаты, даже если в смеси присутствуют органические кис-
лоты, ацетаты, кетали и виниловые эфиры, так как все эти соединения легко
гидролизуются в кислой среде. Муравьиную кислоту в метиловом спирте
нейтрализуют по тимоловому синему, а избыток гидроксиламина можно
титровать хлорной кислотой в диоксане [641]. Возможно, что при титроваг
Определение карбонильных групп
359
нии гидроксиламина образуются перекиси и соединения, содержащие кар-
бонильную группу, что влияет на результаты титрования. Рач, Джонсон
и Критчфильд [691] использовали в качестве растворителя метилцеллозольв,
так как метиловый спирт имеет меньший дифференцирующий эффект. Эти
авторы предлагают проводить титрование не хлорной кислотой, а раствором
азотной кислоты в метилцеллозольве, так как раствор стабилен и не гидро-
лизует ацетали и кетали (приготовление см. гл. 11, разд. 60,е). Чтобы устано-
вить титр раствора, растворяют 1,5 г дприс-(оксиметил)аминометана в 50 мл
кипящего абсолютного метилового спирта, разбавляют 100 мл пропилен-
гликоля и титруют в присутствии 0,3 %-ного тимолового синего в диметил-
формамиде до перехода желтой окраски в отчетливую оранжевую. На титро-
вание 60,51 мг дприс-(оксиметил)аминометана расходуется 1 мл 0,5 н. азот-
ной кислоты в метилцеллозольве.
При проведении полумикроанализа реагенты разбавляют метиловым
спиртом, очищенным от альдегидов (см. гл. 8, разд. 49,а).
Определение альдегидов и кетонов с помощью ацетата гидроксиламина
обсуждается в работе [384].
Приготовление 0,5 н. раствора формиата гидроксиламина в метилцелло-
зольве. В 350 мл свежеперегнанного метилцеллозольва помещают 32 г КОН
и, чтобы ускорить растворение, добавляют 20 мл 90 %-ной муравьиной кис-
лоты. Затем раствор нейтрализуют в присутствии фенолфталеина, вновь
подщелачивают 2—3 горошинами гранулированного КОН и охлаждают
до 15°. В 650 мл метилцеллозольва растворяют 34 г солянокислого гидро-
ксиламина, охлаждают до 15° и смешивают с полученным раствором. Выпав-
ший в осадок KG1 отфильтровывают. Раствор стабилен в течение двух недель.
В колбу Эрленмейера на 100—200 мл с притертой пробкой отбирают
точно 5,0 мл 0,1 н. формиата гидроксиламина и растворяют в нем образец,
содержащий максимально 0,4 мг-экв СО-группы. Глухой опыт проводят
так же. Раствор, содержащий формальдегид, оставляют в плотно закрытой
колбе на 4 час при комнатной температуре; раствор, содержащий масляный
альдегид и глиоксаль, оставляют на 30 мин-, раствор а-этилмасляного альде-
гида оставляют на 90 мин, в то время как для ацетона достаточно 15 мин.
После этого раствор титруют 0,02 н. азотной кислотой в присутствии 3 капель
тимолового синего до перехода желтой окраски в оранжевую. Желательно
вначале провести глухой опыт, а затем титровать. При расчете учитывают
разность объемов стандартного раствора, израсходованного при титровании
анализируемого соединения и глухом титровании. Стандартный 0,02 н.
раствор азотной кислоты, как и 0,1 н. формиат гидроксиламина, готовят
из раствора с более высокой концентрацией, разбавляя его абсолютным
свободным от альдегидов метиловым спиртом. Перед определением колбу
споласкивают чистым растворителем и высушивают.
Точность макрометода (например, титрование индивидуального соеди-
нения) составляет приблизительно ±0,2%. При полумикротитровании точ-
ность определения меньше, но все же полученные результаты отвечают
требованиям; так, 0,09% примеси альдегида в виниловом эфире можно отти-
тровать с точностью 98,5%. Так как виниловые эфиры и ацетальдегид летучи,
реакцию проводят при —10°.
Кислоты, константа ионизации которых выше 10"2, как и основания,
соли кислот и ангидриды, мешают определению. Продолжительность реакции
и концентрации реагентов рекомендуется подбирать на модельном опыте.
Если применять 0,02 н. формиат гидроксиламина, то более подходящим
растворителем может оказаться изопропиловый спирт или бензол, так как
они увеличивают скорость реакции.
Для макроопределения берут 50 мл 0,5 н. формиата гидроксиламина
и растворяют в нем образец, содержащий не более 15 мг-экв СО-группы.
После завершения реакции разбавляют реакционную смесь 125 мл смеси
360
Глава 26
метиловый спирт — метилцеллозольв (50 : 75) и титруют 0,5 н. азотной
кислотой, добавив 5—6 капель тимолового синего.
При титровании летучих соединений навеску отбирают из раствора
соединения в метилцеллозольве (см. табл. 102).
Таблица 102
Определение альдегидов и кетонов с использованием гидроксиламина [6911
Карбонильное соединение Количество вещества, эквивалент- ное 1 мл стандартного раствора HNO3, мг
0,02 н. 0,5 н.
Ацетон 1,16 29,04
Формальдегид 0,60 15,01
Глиоксаль 1.16 29,04
Масляный альдегид 1,44 36,05
а-Этилмасляный альдегид (2-этилбутираль- 2,00 50,08
дегид)
Этилбутилкетон 57,09
Метилэтилкетон 36,05
Метилизобутилкетоп 50,08
д. Определение кетостероидов с использованием салицилата гидроксилам-
мония [284]. Кетостероиды можно отнести к малоактивным кетонам, образо-
вание их оксимов требует более длительного времени и более высокой тем-
пературы. Поэтому при их определении рекомендуется применять салицилат
гидроксиламмония, который более стабилен, чем ацетат или формиат гидро-
ксиламмония. 0,1 н. раствор салицилата гидроксиламмония в смеси хлоро-
форм — метиловый спирт снижает активность только на 0,7% после 3-часо-
вого кипячения. Раствор можно хранить при комнатной температуре в тече-
ние месяца.
В широкогорлой плоскодонной стеклянной колбе емкостью 50 мл взве-
шивают примерно 0,3 мг-экв образца; растворяют его в 0,1 н. растворе сали-
цилата гидроксиламмония, который готовят из точно 10 мл смеси хлоро-
форма и метилового спирта (2 : 1), и кипятят 2 час на водяной бане. Дают
смеси охладиться, затем после добавления одной капли смеси индикаторов
диметиловый желтый и метиленовый синий титруют 0,05 н. соляной кислотой
в смеси пропиленгликоль — хлороформ. Зеленая окраска раствора пере-
ходит в красную, хотя в конечной точке титрования он кажется коричневым.
Глухой опыт проводят аналогично. Разность между результатами двух
титрований служит мерой содержания кетостероида.
Этим методом можно определять не слишком сильно пространственно
затрудненные стероидные соединения, содержащие изолированную карбо-
нильную группу. Так, ^ис-еноны, например эстрон, дегидро-этш-андросте-
рон, прегненол-Зр-диол-5,20, трикетохолановую кислоту, определяют с точ-
ностью ±1,5%.
Приготовление салицилата гидроксиламмония. 40 г салицилата натрия растворяют
в 75 мл воды при небольшом нагревании, добавляют 17,4 г солянокислого гидроксилампна
в 75 .мл воды и оставляют смесь на ночь при 0°. Осадок отфильтровывают, промывают
20 мл дистиллированной ледяной воды и сушат 3 час в вакууме при 80°.
Глава 27
ОПРЕДЕЛЕНИЕ КРАТНЫХ СВЯЗЕЙ.
ПРИМЕНЕНИЕ АЦЕТАТА РТУТИ (И),
БРОМА В УКСУСНОЙ КИСЛОТЕ
ИЛИ 0,1 и. РАСТВОРА БРОМИСТОГО ВОДОРОДА
В УКСУСНОЙ КИСЛОТЕ
153. СВОЙСТВА КОНЦЕВОЙ ДВОЙНОЙ СВЯЗИ
И ДВОЙНОЙ СВЯЗИ В ^КС-ПОЛОЖЕНИИ
Опишем три метода определения кратных связей. В основе первого мето-
да (а) лежит реакция с ацетатом ртути(П) в метиловом спирте [169, 885];
второй метод (б) основан на реакции морфолина с а,0-ненасыщенными соеди-
нениями, в результате которой образуются третичные амины [154]. Третий
метод (в) похож на второй, однако вместо морфолина в реакцию с соедине-
нием, содержащим двойную связь, например метакрилатом, вводят гекса-
метиленимин [800].
СН3О HgOCOCH3
\ / СН3ОН \ I | /
Hg(CH3coo)2+2c = c\-----у" —с\ + сн3соон
Реакция идет быстро в метиловом спирте и медленно * в воде. Ацетат
ртути можно точно оттитровать по методу Палита [621].
Н2С-СН2 Н2С-СН2
Q ^NH + R — СН = СН — X Q7 ^N — CH(R) — СН2 — X,
ЩС — СН2 ЩС — сн2
где X — сильная электронооттягивающая группа.
СН2 = СН —COOCH3 + HN(CH2)6 —» (CH2)6NCH2CH2COOCH3.
а. Известны два варианта метода с применением ацетата ртути(1Г).
Согласно первому, избыток ацетата ртути(П) определяют обратным титро-
ванием, а во втором оттитровывают образовавшуюся уксусную кислоту.
1. В колбе Эрленмейера с притертой стеклянной пробкой взвешивают около
2 ммолей ненасыщенного соединения, добавляют 20—25 мл приблизитель-
но 0,25 н. раствора ацетата ртути(П) в метиловом спирте и, закрыв проб-
кой колбу, выдерживают 10—30 мин при 30°. Раствор ацетата ртути(П)
готовят растворением 20 г Hg(CH3COO)2 в смеси 500 мл метилового спирта
и 1 мл уксусной кислоты, перед использованием раствор фильтруют.
По окончании реакции продукты реакции разбавляют 25 мл смеси про-
пиленгликоля с хлороформом (1:1) и титруют в присутствии тимолового
синего 0,1 н. раствором хлористого водорода в такой же смеси пропиленгли-
коля и хлороформа (1 : 1) до перехода желтой окраски в розовую. Глухой
опыт выполняют аналогично.
Во время титрования избытка ацетата ртути(П) расходуется 2 экв хло-
ристого водорода; кроме того, продукт присоединения ртути также потреб-
ляет 1 экв кислоты [169, 536].
Hg(CH3COO)2H 2НС1 —> HgCl2-i-2CH3COOH,
хс-сх -J- нс1 хс-с/ 4 СН3СООН
7\ I4 ' Х1 1Х
Н3СО HgOGOCH3 Ы3СО HgCl
♦Wright G. F., J. Am. Chem. Soc., 62, 1993 (1935); см. также работу [885]..
362
Глава 27
Таким образом, разность между взятым количеством (мг-экв) ацетата
ртути(П) и израсходованным выражает количество ненасыщенных двойных
связей в миллимолях.
__(6— a)NM
ё~ 1000 ’
где g — навеска ненасыщенного соединения, г; Ъ — объем 0,1 н. НС1, израс-
ходованный в глухом опыте, мл; N — нормальность стандартного раствора;
М — молекулярный вес образца, г; а — объем стандартного раствора,
израсходованного при титровании, мл.
Этим методом можно определять стирол, циклогексен, аллиловый спирт,
аллилацетат, винилацетат и т. д. [169]. Для определения метил- и этилмет-
акрилатов пригоден 0,1 М раствор ацетата ртути(П) в метиловом спирте,
содержащий 0,01 М раствор НС1О4 в качестве катализатора [536]. Методика
определения 3-хлоропропена обсуждается в работе [535].
2. По другому варианту метода определение проводят следующим обра-
зом [430]. В колбу емкостью 250 мл с притертой пробкой отмеривают пипет-
кой 50 мл 0,12 М раствора ацетата ртути(П) в метиловом спирте и раство-
ряют в нем образец соединения, содержащий 3—4 мг-экв двойных связей.
Закрывают колбу пробкой и выдерживают при комнатной температуре;
глухой опыт проводят также. По окончании выдержки в колбу добавляют
2—4 г NaBr и перемешивают до растворения; ацетат ртути(П) при этом
превращается в бромид ртути(П). После добавления в качестве индикатора
1 мл 1%-ного фенолфталеина в метиловом спирте выделившуюся уксусную
кислоту титруют 0,1 н. раствором едкого натра в метиловом спирте до появле-
ния красной окраски. Разность объемов стандартного раствора, пошедшего
на глухой опыт и на титрование образца, пропорциональна содержанию
двойных связей во взвешенном образце. При определении виниловых эфиров
реакционную смесь охлаждают до —10°. В процессе титрования температура
не должна превышать 15°.
По этому варианту метода можно определять (в скобках указано время
выдерживания в минутах) аллиловый спирт (1), аллилацетат (60), циклогек-
сен (1), дихлорстирол (120), N-винилпирролидон (10) и т. д.
б. Метод с применением морфолина [154]. В колбе Эрленмейера емко-
стью 250 мл с притертой стеклянной пробкой взвешивают максимально
.2 мг-экв ненасыщенного соединения, добавляют 10 мл морфолина и 7 мл вод-
ной 50 %-ной уксусной кислоты из градуированной пипетки. Смесь в закры-
той колбе оставляют при комнатной температуре (см. табл. 103), затем при
постоянном помешивании добавляют 50 мл ацетонитрила и 20 мл уксусного
ангидрида. Когда установится постоянная температура, титруют 0,5 н. раст-
вором соляной кислоты в метиловом спирте в присутствии 5—6 капель смеси
метилового оранжевого и ксилолового цианолового ФФ до исчезновения
зеленого окрашивания. Глухой опыт проводят аналогичным способом.
Приготовление индикатора. В 100 мл воды растворяют 150 г
метилового оранжевого и 80 мг ксилолового цианолового ФФ.
Во время определения титруют третичный амин, образующийся из мор-
фолина и соединения, содержащего двойную связь, а избыток морфолина
действием уксусного ангидрида превращают в амид кислоты. Глухой опыт
требует очень малого количества стандартного раствора титранта, потому
что морфолин, который в этом случае не дает третичного амина, превра-
щается в N-ацетилморфолин. Основность последнего в смеси уксусного
ангидрида и ацетонитрила крайне низка.
Этим методом можно определять акриловые производные (см. табл. 103).
в. Метод с использованием гексаметиленимина [800]. Готовят раствор
метакрилата в метиловом спирте, 5 мл раствора переносят пипеткой в колбу,
закрытую стеклянной пробкой. Этот образец не должен содержать больше
Определение кратных связей
363
Таблица 103
Соединение Продолжи- тельность реакций, мин Температура, °C Мол. вес Дополнительный растворитель
Акриламид 5—120 25 71,08
-Акриловая кислота 15—120 98 72,06 4-10 мл СН3ОН
Акрилонитрил 5-60 25 53,06
Метилакрилат 5-60 25 86,09
Этилакрилат 5—60 25 100,1
.Диэтпловый эфир мало- новой кислоты 30—90 25 160,2 40 мл СН3ОН4~2 мл 50%-ной уксусной кис- лоты
0,5 мг-экв метилакрилата. Добавляют 5 мл 1%-ного раствора гексаметилен-
имина в диоксане и 0,2 мл уксусной кислоты (1 : 1) как катализатор и оста-
вляют стоять, хорошо закрыв пробкой, на четверть часа. После этого доба-
вляют к раствору по каплям 5 мл уксусного ангидрида, разбавляют 5 мл мети-
лового спирта и титруют образовавшийся 0(Г^-гексаметиленимин)пропио-
нат 0,1 н. НС1 в метиловом спирте в присутствии 5 капель индикаторной
смеси. На титрование 8,6 мг метакрилата расходуется 1 мл 0,1 н. раствора
хлористого водорода. Очистка гексаметиленимина и приготовление индика-
торной смеси рассматриваются в гл. 16, разд. 99 [799].
154. ОПРЕДЕЛЕНИЕ АЦИЛЦИКЛОПРОПИЛАМИНОВ РЕАКЦИЕЙ
С АЦЕТАТОМ РТУТИ (11) В МЕТИЛОВОМ СПИРТЕ [804]
Циклопропановое кольцо ацилированного циклопропиламина реагирует
•с ацетатом ртути(П), растворенным в метиловом спирте. Механизм реакции
сходен с тем, который предложен для других циклопропановых соединений
{508]. Циклопропановое кольцо раскрывается между замещенным и незаме-
щенным С-атомами, а метоксигруппа присоединяется к замещенному С-атому
/СП2
Hg(CH3COO)2 + СН3ОН+ RCONHNC< | —>
ХСН2
/ОСН3
—> RCONHHC< 4- СН3СООН
xCH2CH2HgOCOCH3
Предполагается, что при растворении ацетата ртути(П) в метиловом спирте
образуется метоксимеркур(П)ацетат [169]
IIg(ClI3COO)2-^CH3OH Hg(OCII3) —О —COCH3 + CH3COOII.
N-Ацилциклопропиламины (иногда проявляющие слабые кислотные свой-
отва) являются столь слабыми основаниями, что их нельзя оттитровать
в уксусном ангидриде (ср. [878]). Поэтому предварительно проводят реак-
цию с 0,12 М ацетатом ртути(П) в метиловом спирте. 40 г ацетата ртути(П)
растворяют в 1 л метилового спирта, добавляют 3—8 капель уксусной кис-
лоты и перед использованием фильтруют.
В колбе Эрленмейера с притертой стеклянной пробкой взвешивают
1—4 мг-экв образца. Растворяют навеску в 50 мл раствора ацетата ртути(И)
и выдерживают 30 мин. Добавляют 2—4 г тонкоизмельченного порошка
NaBr, перемешивают до растворения, титруют выделившуюся уксусную
кислоту 0,1 н. раствором едкого натра в метиловом спирте в присутствии
1 мл 1%-ного раствора фенолфталеина в метиловом спирте. Глухой опыт
проводят также. При добавлении NaBr ацетат ртути превращается в бромид
ртути(П). Бромид ртути(П) и ацетат натрия не реагируют с едким натром
364
Глава 27
в метиловом спирте, а производные циклопропана при комнатной темпера-
туре реагируют очень медленно, поэтому их кипятят с реагентом 30 мин.
Колбу с реакционной смесью тщательно защищают от попадания из воз-
духа влаги и СО2, используя поглотительную трубку, наполненную едким
кали.
Титр раствора ацетата ртути можно установить по 0,1 н. раствору хлористого водо-
рода в смеси пропиленгликоль — хлороформ (1:1) в присутствии тимолового синего..
Об определении эфиров метакриловой кислоты и 3-хлоропропена с применением ацетата
ртутп(П) см. в работе [535, 536].
155. ОПРЕДЕЛЕНИЕ ДВОЙНЫХ СВЯЗЕЙ РЕАКЦИЕЙ С 0,1 н.
РАСТВОРОМ БРОМА В УКСУСНОЙ КИСЛОТЕ
Раствор брома в уксусной кислоте (реагент У риджа — Левина) дли-
тельное время использовался для определения олефиновых связей [823].
В ледяной уксусной кислоте бром реагирует как окислитель по отношению,
например, к мышьяку(III), в то же время с насыщенными органическими
соединениями идет реакция замещения, а с ненасыщенными органическими
соединениями — реакция присоединения. На реакцию влияет присутствие
ацетата натрия, который, будучи акцептором протонов, уменьшает кислот-
ность среды [813].
а. Определение холестерина [813]. 150—300 мг холестерина растворяют
в смеси 20 мл уксусной кислоты и 3 мл уксусной кислоты, насыщенной ацета-
том натрия. Нагревают до 60° и титруют потенциометрически 0,1 н. раствором
брома в уксусной кислоте (см. гл. 11, разд. 60,2). В качестве электродов
используют блестящую платиновую спираль и насыщенный каломельный
электрод. На 19,33 мг холестерина расходуется 1 мл стандартного раствора.
б. Определение 4-оксикумарина и его производных [199]. Навеску 4-окси-
кумарина (0,1—0,5 ммоля) растворяют в 20 лы уксусной кислоты (в кипящей
кислоте растворение идет быстрее) и после охлаждения титруют потенцио-
метрически 0,1 н. раствором брома в уксусной кислоте с использованием
платинового и каломельного электродов. 1 моль 4-оксикумарина реагирует
с 2 экв брома.
Дикумарол, 3,3'-метилен-бие-(4-оксикумарин) можно оттитровать до.
появления желтой окраски, вызванной избытком брома.
20—100 мг дикумарола растворяют при нагревании в 20 мл уксусной
кислоты. После охлаждения смешивают с 3 мл уксусной кислоты, насы-
щенной ацетатом натрия, и титруют до появления неисчезающего слабого
желтого окрашивания. На 16,81 мг дикумарола расходуется 1 мл раствора
брома в уксусной кислоте.
в. Бромометрическое определение стероидов, содержащих двойные связи
в ^-положении с применением хлорида pmymuflI) как катализатора и ацета-
та натрия [283]. При определении соединений, содержащих двойные связи,
присоединение брома иногда представляет самую медленную стадию, даже
если титрование проводят непосредственно 0,1 н. раствором брома в уксусной
кислоте при 60° (как, например, при определении холестерина). Однако
бромометрическое титрование некоторых стероидов можно ускорить, если
в дополнение к ацетату натрия использовать в качестве катализатора хлорид
ртути(П). При этом рекомендуют пользоваться в качестве растворителя
безводной уксусной кислотой, перегнанной над хромовым ангидридом
и содержащей 0,5 молъ/л ацетата натрия и 0,08 молъ/л хлорида ртути(П).
С растворителем, содержащим такой катализатор, реакция присоединения
брома заканчивается за несколько секунд, причем нагревание нельзя счи-
тать необходимым, а побочные реакции замещения вызывают лишь незна-
чительные погрешности даже при проведении реакции на рассеянном сол-
нечном свету.
Определение кратных связей
365
При титровании бесцветных растворов конечную точку титрования опре-
деляют по появлению желтоватой окраски, вызванной присутствием избытка
брома. Это служит причиной перетитровывания приблизительно на 0,5—1%,
однако, когда титр 0,1 н. раствора брома в уксусной кислоте устанавливают
при аналогичных условиях по окиси мышьяка(Ш), этой ошибки можно
избежать. Окрашенные растворы титруют потенциометрически с использо-
ванием платинового и каломельного электродов (солевой мостик заполняют
уксусной кислотой, насыщенной хлоридом лития). При анализе методом
биамперометрического титрования можно употреблять два гладких плати-
новых электрода.
Взвешивают примерно 0,5 ммоля стероида и растворяют в 10 мл уксус-
ной кислоты, содержащей катализатор. Если необходимо, то раствор нагре-
вают. Титруют при комнатной температуре 0,1 н. раствором брома в уксус-
ной кислоте. Во время титрования колбу энергично встряхивают. Вблизи
конечной точки титрант добавляют небольшими каплями с такой скоростью,
чтобы их можно было сосчитать. Желтая окраска, вызванная присутствием
избытка брома, должна оставаться неизменной в последующие 15—20 сек.
Установка титра раствора брома. Растворяют 50 мг As2O3 и после добавления
7 мл 20%-ного едкого натра титруют бромом в уксусной кислоте до появления бледно-
желтой окраски.
Приготовление катализатора в уксусной кислоте. В уксусной кислоте, перегнанной
над хромовым ангидридом, растворяют 68 г тригидрата ацетата натрия и 21,7 г хлорида
ртути(П) и разбавляют до 1000 мл. Раствор можно хранить в темноте в течение несколь-
ких месяцев.
Этим методом можно определять, например, диосгенин, соласодин,
O.N-диацетилсоласодин, 3|3-ацетокси-Д5>16-прегнадиенон-20, Д5-прегненол-
ЗР-он-20, 16а,17а-эпокси-Д5-прегненол-ЗР-он-20, Д5-андростенол-3|3-он-17,
30-ацетокси-Д5-андростенон-17, 30-формилокси-17а,21-диацетокси-Д5-прег-
ненон-20, 17 а-мети л- Д5-андростендио л-30,170.
В случае стероидов, содержащих изолированные двойные связи, экви-
валентный вес равен половине молекулярного. Воспроизводимость этого
метода в зависимости от соединения составляет 98,2—101,0 + 0,3%.
156. КИСЛОРОД ОКСИРАНА (а-ЭПОКСИГРУППА) J197, 198, 373]
Бромистый водород в уксусной кислоте реагирует с а-эпоксигруппой
следующим образом:
II I I
— С —С—4-НВг—- — С —С —
\ / II
О Вг ОН
Эпоксигруппу можно непосредственно титровать с определением конеч-
ной точки по индикатору.
300—600 мг образца, содержащего эпоксигруппу, растворяют в неболь-
шом количестве бензола или хлорбензола и титруют 0,1 н. раствором бро-
мистого водорода в уксусной кислоте в присутствии 5 капель 0,1%-ного
кристаллического фиолетового в уксусной кислоте до появления сине-зеле-
ной окраски индикатора. Раствор перемешивают магнитной мешалкой
и проводят реакцию в герметизированном приборе. Кончик бюретки должен
быть расположен непосредственно над поверхностью анализируемой
жидкости. Относительно приготовления титранта и установления его титра
см. гл. 11. На титрование 1,6 мг оксиранового кислорода расходуется 1 мл
раствора бромистого водорода. Точность определения + 1%.
Присутствие карбоновой кислоты, альдегида, простого и сложного эфи-
ров или перекисей не мешает титрованию. Гидроперекиси в определенной
степени влияют на результаты, а присутствие аминов может вызвать боль-
366
Глава 27
шие ошибки. По более новому методу Дебетаки [198] кислород оксирана
можно непосредственно определять даже в солях эпоксикислот и в присут-
ствии аминов.
Метод Дебетаки дает быстрые и хорошие результаты, но реагент дымит
на воздухе, и поэтому его надо хранить в специальных условиях и часто
проверять концентрацию раствора. В методе, предложенном Джейем [428],
бромистый (или иодистый) водород генерируют непосредственно в реакцион-
ной смеси добавлением 0,1 н. хлорной кислоты к (C2H5)4NBr или (C4H9)4NL
Бромистый или иодистый водород быстро раскрывает оксирановое или азири-
диновое кольцо
/СН2
— N< | -f-HI —> ICH2CH2NH—
ХСН2
причем HI — более энергично действующий реагент, чем НВг.
В колбе Эрленмейера емкостью 50 мл взвешивают образец, содержащий
0,8 мг-экв оксиранового или азиридинового соединения, растворяют его-
в 10 мл хлороформа, ацетона, бензола или хлорбензола. Добавляют 10 мл
раствора бромида или иодида и четвертичного аммониевого основания
и 2—3 капли раствора кристаллического фиолетового и титруют 0,1 н. раство-
ром хлорной кислоты в уксусной кислоте. Особенно отчетливо конечная
точка наблюдается для эпокисей. Глухой опыт не проводят.
Приготовление раствора бромида тетраэтиламмония. 100 г (С2Н5)^Вг растворяют
в 400 мл безводной уксусной кислоты, добавляют несколько капель индикатора и нейтра-
лизуют 0,1 н. хлорной кислотой до нейтральной реакции.
Приготовление раствора иодида тетрабутиламмония. 50 г (C^g^NI растворяют
в 500 мл очищенного хлороформа. Этот реагент можно хранить только в том случае, если
он предварительно не был нейтрализован хлорной кислотой, и только в темноте.
157. ВОДОРОД В АЦЕТИЛЕНОВЫХ ПРОИЗВОДНЫХ
Монозамещенные производные ацетилена образуют соединения типа
R — С =; С — Ag при взаимодействии с нитратом серебра в водноаммиач-
ном растворе, и в то же время в спиртах они дают соединения типа
R—С = С — Ag.AgNO3,
если не слишком велик избыток нитрата серебра. Однако, если его избыток
значителен, то последнее соединение растворяется в виде комплекса
R — С = С — Ag-6AgNO3
а. Определение реакцией с нитратом серебра в пиридине [594]. При исполь-
зовании раствора нитрата серебра в пиридине ацетиленовое соединение
высвобождает 1 экв азотной кислоты, которую можно определить как потен-
циометрически, так и визуально.
Взвешивают 0,5 мг-экв ацетиленового прозводного и растворяют
в 5,0 мл 1 н. раствора нитрата серебра в пиридине, предварительно нейтра-
лизованного в присутствии 5—10 капель 1%-ного раствора тимолового-
синего до появления светло-голубой окраски. Окраска индикатора в про-
цессе растворения исчезает или становится бледно-желтой. Освобождаю-
щуюся азотную кислоту титруют из микробюретки 0,1 н. раствором едкого
натра в метиловом спирте до появления голубого окрашивания. Этим мето-
дом можно определять З-метилпентин-1-ол-З, этинилциклогексанол, карбамат
З-метилпентин-1-ола-З (см. также гл. 31) и многие другие ацетиленовые
производные.
б. Определение по реакции с перхлоратом серебра в метиловом спирте
[32]. Под действием перхлората серебра кислый водород монозамещенных
ацетиленовых производных (ср. табл. 28) можно заменить в метиловом спирте-
Определение кратных связей
367
на серебро
R —С = с—H + 2AgC104 -> R—С = С —Ag-AgC104-j-HC104.
Образующуюся хлорную кислоту можно титровать стандартным 0,1 н.
раствором тприс-(оксиметил)аминометана в присутствии индикаторной смеси
тимоловый синий — патент голубой А до изменения окраски от пурпурной
до зеленой. О приготовлении стандартных растворов см. табл. 52 примеча-
ние «л».
В 5—10 мл нейтрализованного метилового спирта растворяют 1—3 мг-экв
ацетиленового производного. Нейтрализуют 10 мл 1 М раствора перхлората
серебра в метиловом спирте в присутствии 3 капель индикатора до зеленой
окраски. Смешивают оба раствора и после добавления 5—10 капель индика-
тора титруют до зеленого окрашивания.
Приготовление раствора перхлората серебра. 104 г AgC104 растворяют в 500 мл
безводного метилового спирта и хранят в полиэтиленовом сосуде, чтобы избежать взрыва
перхлората [32]. Приготовление смеси индикаторов. 100 мг тпмолового синего и 25 мг
патента голубого А растворяют в 100 мл метилового спирта. Этот раствор неустойчив,
поэтому его следует приготовлять заново еженедельно. Однако смесь 1 г тимолового-
синего и 250 мг патента голубого А, растворенная в 200 мл диметилформамида, дает
более стабильный раствор, причем для титрования требуется 2—3 капли этой смеси
(Барнес, частное сообщение).
Если образец содержит сильную кислоту, которая окрашивает индикатор
в пурпурный цвет, ее нейтрализуют 0,1 н. раствором ?прис-(оксиметил)амино-
метана до зеленой окраски индикатора перед смешением растворов образца
и перхлората серебра.
В присутствии слабых кислот (К = 10“5 и ниже) нейтрализации не тре-
буется, однако, если выпадает нерастворимая соль серебра, то количество
метилового спирта следует увеличивать до 50 мл и использовать 15—20 капель
раствора индикатора.
В присутствии сильных оснований (гидроокисей щелочных металлов)
раствор окрашивается индикатором в синий цвет. В этом случае раствор
следует нейтрализовать 0,1 н. хлорной кислотой в метиловом спирте до появ-
ления пурпурной окраски. Если в присутствии органических оснований
(А = 10-3—10-6) раствор приобретает зеленый оттенок, его следует ней-
трализовать в метиловом спирте до пурпурной окраски.
Изменение окраски индикаторной смеси. С сильными кислотами индикатор
дает пурпурную окраску, с органическими основаниями и кислотами —
зеленую, с сильными основаниями — синюю.
Этим способом можно титровать пропин-2-ол-1, 2-метилбутин-3-ол-2,
З-метилпентин-1-ол-З, З-метилгексин-1-ол-З, 2-этилгептин-1-ол-3, 3-метил-
нонин-1-ол-З, а также ацетоновые растворы ацетилена, пентина-1 и гек-
сина-1. Методы микроопределения ацетиленового водорода рассматриваются
в работе [298].
Глава 28
-ОПРЕДЕЛЕНИЕ ЭЛЕМЕНТАРНОЙ СЕРЫ
И ОРГАНИЧЕСКИХ СЕРУСОДЕРЖАЩИХ СОЕДИНЕНИЙ.
ПРИМЕНЕНИЕ НИТРАТА СЕРЕБРА В ПИРИДИНЕ
158. ЭЛЕМЕНТАРНАЯ СЕРА В НЕВОДНЫХ СРЕДАХ
Скудж и Бартлетт [760] разработали метод объемного определения эле-
ментарной серы титрованием ее ацетоновых растворов цианидом калия
в изопропиловом спирте. Эрдей и сотр. [214, 215] нашли, что растворимость
серы в смеси бензол : ацетон (4:1) в десять раз превышает растворимость
серы в ацетоне, в 1 мл которого способно раствориться только 0,32 мг серы.
Кроме того, в последнем методе может быть также повышена точность опре-
деления.
Определение элементарной серы в химических средствах борьбы с вреди-
телями растений. Около 300 мг пробы, предварительно высушенной в вакуу-
ме, экстрагируют в вибрационном аппарате 80 мл бензола приблизительно
в течение 1 час. Раствор фильтруют в мерную колбу емкостью 100 мл
и добавляют к нему ацетон до метки. 10—20 мл раствора смешивают с 1 мл
воды, содержащей 2—3 капли раствора бромтимолового синего, и титруют
0,05 М раствором KCN в изопропиловом спирте, например в сосуде для
титрования при постоянной температуре около 50—60° до появления устой-
чивой голубой окраски. Вблизи конечной точки титруют медленно. Бензол
должен содержать не более 0,1% воды, так как более высокое содержание
воды мешает экстракции серы.
Методика приготовления стандартного раствора дана в гл. 11, разд. 60, н. Для
определения титра стандартного раствора разбавляют 20 мл 0,05 н. раствора KCN 50 мл
воды, добавляют 2 мл 6 н. раствора NH4OH и 0,6 г Nal и титруют 0,05 н. раствором
AgNO3 до помутнения
t a*^AgNO3*2
/KCN— 1 '
где а — количество израсходованного раствора AgNO3, мл\ b — количество раствора
KCN, мл.
Содержание серы в фармацевтических препаратах, имеющих в своем
составе элементарную серу и активированный уголь, также можно опре-
делять по описанному выше методу. В этом случае предпочтительно потен-
циометрическое титрование со стеклянным и каломельным электродами.
159. ОРГАНИЧЕСКИЕ СУЛЬФОКСИДЫ
Органические сульфоксиды (например, диметилсульфоксид) можно титро-
вать потенциометрически стандартным раствором хлорной кислоты в уксус-
ном ангидриде [778, 879].
Сульфоксидную группу можно представить в поляризованной резонансной гибрид-
ной форме, если допустить, что на внешней электронной оболочке атома серы могут
находиться более 8 электронов (см. работу [863], стр. 27). Асимметрия молекулы воз-
= ?. = 2 *—* = S— О:
никает вследствие того, что сера и три атома, связанные с ней, не лежат в одной пло-
скости (см. стр. 594 в работе [231]).
Определение серы и органических серусодержащих соединений
369
Взвешивают приблизительно 0,001 моля сульфоксидного соединения,
растворяют в 75 мл уксусного ангидрида и титруют потенциометрически
0,1 н. хлорной кислотой в диоксане, используя систему электродов стекло —
каломель или стекло — Ag/AgCl (см. разд. 130,а). Этим способом можно опре-
делять диметил-, дифенил-, дибензилсульфоксиды и т. д. Сульфиды и суль-
фоны не мешают определению.
Особо отметим, что этим же методом можно титровать фентиазин-5-оксид.
160. АЛКАНТИОЛЫ (МЕРКАПТАНЫ). ПРИМЕНЕНИЕ НИТРАТА СЕРЕБРА
В ПИРИДИНЕ
Для тиолов, представляющих собой очень слабые кислоты, характерно
то, что с ионом серебра Ag+ они образуют меркаптиды серебра, а с ионом
Hg2+ — меркаптиды ртути.
а. Титрование нитратом серебра в изопропиловом спирте [490]. 20 мл
смеси изопропилового спирта и 30%-ного раствора аммиака в соотношении
10 : 1 помещают в колбу, в пробке которой просверлено два отверстия
(рис. 39—42). Прибавляют 1—2 мл 0,01%-ного раствора дитизона в смеси
раствора аммиака с изопропиловым спиртом. Взвешивают 0,05—0,1 мг-экв
меркаптанового производного, растворяют его в приготовленном растворе,
пропуская над его поверхностью ток азота. Титруют в атмосфере азота 0,01 н.
нитратом серебра в изопропиловом спирте до изменения окраски от оран-
жево-желтой до глубоко красной (о приготовлении стандартного раствора
см. гл. 11, разд. 60,1). Наряду с другими меркаптанами таким путем можно
титровать от 5 до 20 мг гексантиола, бутантиола, додекантиола и др. Обра-
зующаяся в ходе реакции из азотнокислого серебра азотная кислота связы-
вается аммиаком, а яркая окраска координационного комплекса дитизона
с серебром, образующегося при избытке стандартного раствора, указывает
конечную точку.
Тиолы также реагируют с солями ртути, например ацетатом ртути(П), образуя
димеркаптид или ацетоксимеркур(П)меркаптид. Избыток ацетата ртути(П) в растворе
метилового спирта прибавляют к меркаптану и после окончания реакции оттитровывают
0,1 н. соляной кислотой в бутиловом спирте в присутствии тимолового синего [489].
б. Применение нитрата серебра в пиридине. Множество органических
соединений реагирует с ионами серебра с выделением протонов.
R —С = CH + Ag+ R —С = CAg + H+,
BH24-2Ag+ BAg24-2H+,
где ВН2 — например, молекула барбитуровой кислоты.
В пиридине подобный процесс приводит к образованию катиона пири-
диния, который можно титровать как сильную кислоту. Этот метод приме-
няют для определения ацетиленового водорода [594] и барбитуратов [266].
В колбе Эрленмейера с притертой пробкой взвешивают примерно
1 мг-экв монофункционального соединения, содержащего SH-группу, раство-
ряют в 15 мл пиридина, приливают 5 мл 0,5 М раствора AgNO3 в пиридине
и оставляют в плотно закрытой колбе на 15 мин. Полученный раствор тит-
руют 0,1 н. раствором едкого кали в этиловом спирте, добавив 5 капель
тимолфталеина или тимолового синего (1%- или 0,1%-ный раствор в этило-
вом спирте) (см. гл. 10, разд. 55,а).
Соединения, нерастворимые в пиридине, например цистеин, сначала
растворяют в 5 мл воды, этилового спирта или ацетона. В дальнейшем титро-
вание ведут так же, как описано.
Каждая отдельная функциональная группа SH образует эквивалентное
количество катиона пиридиния. К монофункциональным соединениям отно-
сятся, например, тиоглицерин, этил- или бутилмеркаптан; димеркапрол
(BAL, дитиоглицерин) — соединение бифункциональное. По другому вариан-
370
Глава 28
ту метода определения соединений, содержащих другую кислотную функ-
циональную группу, помимо SH-группы, карбоксильную группу оттитро-
вывают в пиридине и затем после прибавления нитрата серебра отдельно
определяют SH-группы (тиосалициловая кислота, цистеин). Этот способ
применим также и для определения тиогликолевой кислоты, тиомалоновой
кислоты, тиофенола, 1,2-димеркаптобензотиазола (см. также гл. 17, где
приведены кислотные аналоги конденсированных гетероциклических соеди-
нений), н-пропилтиоурацила и т. д.
НО N SH
SH NH2 \ / \ /
1 I С С
HS —СН2 —СН —СН2ОН HS —СН2 —СН —СООН I II
НС N
Димеркапрол Цистеин \ /
С
I
С3Н7
Пропилтиоурацил
Этот метод применим также для определения содержания действующего
начала в таблетках. Точность определения ±1%.
в. Определение производных барбитуровой кислоты нитратом серебра
в пиридине [266]. Соединения, содержащие иминогруппу, одновременно
с SH-группой освобождают также протоны при реакции с нитратом серебра
в пиридине
BH2 + 2Ag+4-2C5H5N BAg2-|-2C5H5NH+,
где -5Н2 — производное барбитуровой кислоты.
В конической колбе для титрования взвешивают 0,25—1 мг-экв барбиту-
рата (для этой цели применяют колбу для выпаривания, показанную
на рис. 103), растворяют в 5 мл пиридина, приливают 10 мл 0,5 М пиридино-
вого раствора нитрата серебра и титруют 0,1 н. раствором едкого натра
в этиловом спирте в присутствии 4—5 капель 1 %-ного раствора тимолового
синего в этиловом спирте. Диэтил-, этилбутил-, этилизоамил-, этилфенил-
и N-метилэтилфенилбарбитуровые кислоты могут быть определены с точ-
ностью ±1 %.
Содержание действующего начала в фармацевтических продуктах, кото-
рые имеют в своем составе свободное производное барбитуровой кислоты
и в то же время его натриевую соль, следует определять комбинированным
методом. Сначала ацетат-ион, образовавшийся в ходе реакции с раствори-
телем, титруют раствором хлорной кислоты в уксусной, вторую порцию
образца титруют согласно описанному выше методу [264]. При расчетах
необходимо учитывать, что на реакцию с натриевой солью барбитуровой
кислоты расходуется 1 экв хлорной кислоты, в то время как на реакцию
с азотнокислым серебром расходуется 2 экв едкого натра. Точность комби-
нированного метода составляет ±2%.
г. Прямое титрование меркаптанов (тиолов) перхлоратом ртути(11).
Тиолы реагируют с Hg2+-noHOM по реакции
Hg2+-r2RSH —> (RS)2Hg + 2H+.
Конечную точку титрования можно наблюдать визуально или фотометри-
чески в присутствии свежеприготовленного 0,01 %-ного ацетонового раствора
кетона Михлера [4,4'-бис-(диметиламино)бензофенона]. Для потенцио-
метрического титрования в качестве электрода сравнения используют кало-
мельный электрод, а в качестве индикаторного — ртутный, причем чаще
всего ртутный электрод J-типа * (см. также рис. 54) [674]. j
* Chemist-Analyst, 51, 56 (1962).
Определение серы и органических серусодержащих соединений
371
Этот метод применим также и для определения следов тиола; в этом слу-
чае фотометрическое определение конечной точки рекомендуется проводить
при длине волны около 580 нм (можно использовать модифицированную
кювету типа Beckman Model Band) [256].
Титрование можно проводить в ацетоне, смесях ацетон — бензол или
ацетон — легкокипящий петролейный эфир, содержащих до 40 об. % бен-
зола или легкокипящего петролейного эфира.
0,3—1,0 ммоль производного тиола растворяют в 100 мл растворителя,
добавляют 1 мл пиридина и индикатор в количестве, достаточном для того,
чтобы раствор приобрел яркую желто-зеленую окраску. Титруют водным
0,05 М раствором перхлората ртути до перехода желто-зеленой окраски
в голубую. Таким образом, в конечном объеме раствора содержится 3—10%
воды.
Приготовление стандартного' раствора. 26 г Hg(C104)2-3H20 растворяют в 1 л вод-
ного 0,1 М раствора хлорной кислоты. Титр полученного раствора устанавливают по
0,05 М этилендиаминтетрауксусной кислоте в присутствии пиридина в качестве буфера
(pH 6) и кетона Михлера как индикатора.
При фотометрическом титровании 0,006—0,5 ммоля тиола растворяют
в 100 мл растворителя и титруют, как указано выше, но используют соот-
ветственно более разбавленный 0,0005—0,01 М стандартный раствор.
д. Потенциометрическое титрование меркаптосоединений тетраацета-
том свинца рассматривается в работе [785].
161. ТИТРОВАНИЕ ХЛОРНОЙ КИСЛОТОЙ АЛКИЛКСАНТОГЕНАТОВ,
S-АЛКИЛ- И S-БЕНЗИЛТИУРОНИЕВЫХ ПРОИЗВОДНЫХ
И ДИАЛКИЛ-п-БРОМФЕНАЦИЛСУЛЬФОНИЕВЫХ СОЕДИНЕНИЙ
[59, 60, 724, 843 J
а. Реакция образования ксантогенатов (ксантатов, дитиокарбонатов)
используется для идентификации спиртов. Титрование алкилксантогенатов
калия хлорной кислотой делает возможным определение эквивалентного
или молекулярного веса спирта
ROCSS- + СН3СООЩ = ROCSSH + СН3СООН.
В уксусной кислоте ксантогенаты разрушаются
ROCSSK + CH3COOH — > ROCSS1I ^СН3СООК
и
ROCSSH —> ROH + CS2.
Образующийся ацетат калия можно титровать как основание. Таким
образом, ксантогенаты независимо от того, разрушаются они или нет, при
титровании требуют 1 экв хлорной кислоты.
200—300 мг алкилксантогената растворяют в 15—20 мл уксусной кислоты
и титруют 0,1 н. раствором хлорной кислоты в присутствии кристалличе-
ского фиолетового до перехода окраски в голубую.
б. Соли, образованные ионом S-алкил- или S-бензилтиурония с анионом
пикриновой или карбоновой кислоты, удобны для идентификации спиртов
и карбоновых кислот; их можно титровать как основания 0,1 н. хлорной
кислотой в присутствии кристаллического фиолетового.
-R — SC = NH,1+ [R — SC Xlf.,1 +
I " A'- i-CH3COOHJ -> | ‘ -;-XH-^CH3COOH,
NH2 J NII2 J
где R — алкильная или бензильная группа; Х~ — пикрат- или карбокси-
лат-анион.
О титровании пикратов см. гл. 24, разд. 135.
372
Глава 28
в. Определение диалкилсулъфидов (тиоэфиров) в виде п-бромфенацилсулъ-
фониевых солей. Диалкилсульфиды образуют п-бромфенацилсульфониевые
соли с п-бромфенацилбромидом
R
R2S + ВгС6Н4СОСН2Вг —> ВгС6Н4СОСН2 —S+Вг-
R
В ходе такой реакции Вг- заменяется на пикрат-анион и образующийся
пикрат диалкилбромфенацилсульфония можно титровать раствором хлорной
кислоты в уксусной кислоте в присутствии кристаллического фиолетового
до появления зеленой окраски.
Приготовление алкилксантогенатов [59]. 0,5—0,6 г (около 0,01 моля) едкого кали
растворяют при нагревании в 1 г исследуемого спирта. После охлаждения добавляют
по каплям 1 мл CS2 (около 0,017 моля). Если при этом смесь нагревается, ее необходимо
охлаждать. Перемешивают смесь в течение нескольких минут стеклянной палочкой,
затем приливают 20 мл эфира и энергично встряхивают. Отфильтровывают выпавшие
кристаллы ксантогената калия и промывают их несколькими миллилитрами эфира.
Растворяют в небольшом количестве кипящего ацетона. Для осаждения ксантогената
после фильтрования и охлаждения приливают 20 мл эфира.
Приготовление пикратов S-алкилтиурония из третичных спиртов *. Встряхива-
ют в течение нескольких минут один объем третичного спирта с 5—6 объемами концент-
рированной соляной кислоты или 48%-ной бромистоводородной кислоты. Первоначаль-
но гомогенная смесь разделяется на два слоя, причем верхний слой представляет собой
алкилгалогенид.
1 мл полученного алкилгалогенида кипятят в колбе с обратным холодильником
с раствором, приготовленным из 1 г тиомочевины, 3 мл воды и 2мл этилового спирта
до тех пор, пока не прореагирует весь галоидный алкил (2—3 час). Затем смесь выливают
в 200 мл 1%-ного водного раствора пикриновой кислоты; при этом выпадает пикрат
S-алкилтиурония. Через полчаса выпавший пикрат
Г R
I Лн2
R'_с — S — с<
I XNH2
-OC6H2(NO2)3
R
отфильтровывают и перекристаллизовывают из водного спирта.
Приготовление S-бензилтиурониевых солей карбоновых кислот *. 0,01 мг-экв карбо-
новой кислоты смешивают с 8 мл 1 н. раствора едкого натра, нейтрализуют и растворяют,
добавив 2 капли метилового красного как индикатора, в 1 н. щелочи так, чтобы раствор был
настолько слабощелочным, насколько это возможно. Нагревают раствор [до 90°, при-
бавляют 2 г хлорида S-бензилтиурония, растворенного в 5—10 мл воды, и немедленно ста-
вят колбу со смесью в лед. Раствор медленно кристаллизирующегося производного насы-
щают хлоридом натрия и для облегчения роста кристаллов оставляют при охлаждении
на ночь.
ZNH21
C6H5CH2-S-C<
\nh2J
-OCOR
Образовавшаяся соль легко гидролизуется в щелочной среде с образованием бензил-
меркаптана, обладающего характерным неприятным запахом.
Приготовление пикратов диалкил-п-бромфенацилсулъфония * В 10 мл ацетона,
содержащего 5% воды, растворяют 0,01 моля диалкилсульфида и 0,01 моля га-бром-
фенацилбромида. Раствор оставляют на сутки в закрытой колбе. После декантирования
кристаллы промывают эфиром, затем их растворяют в небольшом количестве воды п при-
ливают раствор к насыщенному водному раствору пикриновой кислоты. После фильтро-
вания и высушивания пикрат взвешивают и титруют хлорной кислотой.
Диарилсульфиды и тиоанизолы не реагируют с n-бромфенацилбромидом. Диаллил-
или дибензилсульфиды при кипячении с n-бромфенацплбромидом в колбе с обратным
холодильником дают бис-(га-бромфенацил)сульфиды [843].
* Байбель С., Идентификация органических соединений, ИЛ, М., 1959.
Глава 29
ОКИСЛИТЕЛЬНО-ВОССТАНОВИТЕЛЬНОЕ
ТИТРОВАНИЕ В НЕВОДНОЙ СРЕДЕ
Теория окислительно-восстановительного титрования еще не достигла
уровня развития теории кислотно-основного титрования. Исследование
этого процесса ведется во многих странах.
Систематическое изучение химических процессов, проходящих в невод-
ной среде, началось еще в начале этого столетия [855]. Ограниченные воз-
можности этого метода широко проиллюстрированы большим числом при-
меров кислотно-основного титрования, однако в настоящее время еще нельзя
предвидеть, найдет ли окислительно-восстановительное титрование в невод-
ной среде такое же широкое применение, как кислотно-основное титрование.
Однако можно предположить что при правильном выборе растворителей
и условий титрования методом окислительно-восстановительного титрования
можно будет определять соединения, не титрующиеся в водной среде.
Согласно Харнеду [362], зависимость нормальных потенциалов от вели-
чины, обратной диэлектрической проницаемости, имеет линейный характер.
В различных растворителях, однако, степень диссоциации и действие раство-
рителя в основном могут оказывать самое различное влияние. Ионы, напри-
мер, могут образовать большие агрегаты (см. гл. 5, разд. 33). Кроме того,
изменяется электродный потенциал. Растворители могут оказывать влияние
на реакции, проходящие между растворенными в них соединениями.
В основном электроды в водной и в неводной средах ведут себя одина-
ково. Конант и Чоу [143] считают, что окислительно-восстановительные
потенциалы некоторых хиноидных соединений изменяются в уксусной кис-
лоте в зависимости от кислотности раствора. Исследования Томичека [217]
доказывают, что окислительно-восстановительный потенциал системы
хром(У1) — хром(Ш) изменяется в соответствии с изменением водородного
потенциала в среде уксусной кислоты (табл. 104).
В окислительно-восстановительной системе аскорбиновая кислота —
дегидроаскорбиновая кислота в среде уксусной кислоты потенциал изме-
Таблица 105
Окислительно-восстановительная
система бром — бромид-ион
в уксусной кислоте
Таблица 104
Окислительно-восстановительная
система хром(Ш) — xpoM(VI)
в уксусной кислоте
Кислотность среды, н. (НСЮ4) Е, мв (система электродов: пла- тина — каломель)
1 1150
0,1 1107
0,01 1069
0,001 1026
0,0001 980
Уксусная кислота (880)[217]
Кислотность среды
Е, мв (система
электродов:
платина — кало-
мель)
1 н. CH3COONa
0,01 н. CH3COONa
0,001 н. CH3GOONa
Уксусная кислота
0,001 н. НСЮ4
0,01 н. НСЮ4
1 н. НС1О4
560
640
679
830
(790)[217]
930
1010
1050
374 Глава 29
няется под действием ацетата натрия, так как ацетат-ион как основание
способствует диссоциации аскорбиновой кислоты, связывая водородные
ионы [217]. Окислительно-восстановительный потенциал системы бромид-
ион — бром в уксусной кислоте (см. табл. 105) зависит от pH раствора*.
В присутствии ионов водорода некоторое количество бромид-ионов превра-
щается в бромистый водород, так как в безводной среде отсутствует акцептор
протонов. Таким образом, активность бромид-ионов уменьшается, а значе-
ние отношения окисленная форма — восстановленная форма увеличи-
вается. При добавлении ацетата уменьшается концентрация ионов ацилония,
увеличивается активность бромид-ионов и понижается окислительно-вос-
становительный потенциал.
Перечисленные данные ясно указывают, что в среде уксусной кислоты
потенциал окислительно-восстановительной системы зависит от соотношения
окисленной и восстановленной форм и во многих случаях от среды [217].
Число примеров окислительно-восстановительного титрования в невод-
ной среде относительно мало. Первые работы в этой области провели Томи-
чек и сотр. [816, 817]. Для титрования используют бром в уксусной кислоте
[216, 897], перманганат калия в уксусной кислоте [131], ацетат хрома(П)
в диоксане [589, 591], ацетат меди(П) в смеси пиридин — этиловый спирт
[107], церий(1У)аммонийнитрат в ацетонитриле [666], третп-бути л гипо-
хлорит в уксусной кислоте [336], бихромат калия в уксусной кислоте [685],
тетраацетат свинца в уксусной кислоте [785] и т. д.
162. ОПРЕДЕЛЕНИЕ АСКОРБИНОВОЙ КИСЛОТЫ В НЕВОДНОЙ СРЕДЕ
|216, 666в]
а. Аскорбиновую кислоту можно титровать 0,05 н. раствором брома
в уксусной кислоте в присутствии индикатора, обладающего восстанови-
тельными свойствами. К числу таких индикаторов относится, например,
производное вариамина синего [1\т,Х'-бис-(тг,тг'-диметоксидифениламин)тио-
мочевина (GH3OG6H4NHG6H4NHGSNHG6H4NHG6H4OGH3)], которое исполь-
зуется в виде 0,1%-ного раствора в пиридине. Хотя раствор при хранении
становится желтым или коричневым, его можно использовать в течение двух
недель.
Салициловую кислоту нельзя титровать в безводной уксусной кислоте, но ее можно
определить в водной уксусной кислоте (1 : 1) стандартным раствором брома в уксусной
кислоте [897]. Исследования показали, что в уксусной кислоте ацетат натрия играет
роль основания, и в такой среде может протекать большинство реакций.
30—60 мг аскорбиновой кислоты растворяют в 30 мл уксусной кислоты,
к раствору приливают 10 мл 1 н. ацетата натрия в уксусной кислоте и 4 капли
индикатора и титруют 0,05 М раствором брома в уксусной кислоте до появле-
ния фиолетовой окраски (которая должна сохраняться в течение 1 мин).
Вблизи конечной точки титруют медленно, так как скорость реакции умень-
шается. На титрование 4,4 мг аскорбиновой кислоты расходуется 1 мл
0,05 н. раствора брома. Точность измерения составляет 100,7 ± 0,9%.
О приготовлении брома в уксусной кислоте см. гл. 11, разд. 60,2. Установление тит-
ра. 10 мл титранта смешивают с 50 мл свежеприготовленного 2%-ного водного раствора
иодида калия и выделившийся иод сразу оттитровывают 0,05 н. раствором тиосуль-
фата натрия в присутствии 0,1 %-ного вариамина синего в пиридине как индикатора.
б. Аскорбиновую кислоту можно также титровать потенциометрически
в смеси уксусная кислота — ацетонитрил стандартным раствором иона
церия(1У) [666в]. 2,0-5,0 мл стандартного 0,05 н. раствора церий(1У)аммо-
* Tomice k. Ileyrovski, Coll. Czech. Communs., 15, 997 (1950).
Окислительно-восстановительное титрование в неводной среде
375
нийнитрата в ацетонитриле разбавляют 10 мл уксусной кислоты и затем
проводят титрование, используя стеклянный и платиновый электроды. При
этом стандартный раствор должен быть оттитрован (обратное титрование)
приблизительно 0,015 н. раствором аскорбиновой кислоты, приготовленным
из анализируемой аскорбиновой кислоты в уксусной кислоте. Целесообраз-
но использовать полумикробюретку и магнитную мешалку. Вблизи конеч-
ной точки может наблюдаться внезапное падение потенциала до 100—120 мв.
Окисление аскорбиновой кислоты проходит через стадию образования дегид-
роаскорбиновой кислоты; соотношение аскорбиновой кислоты и церияЦУ)
составляет 1 : 4,005 [666в]. Таким образом, в безводной среде окисление
стандартным раствором церияЦУ) является высоко эффективным.
Известно, что, например, щелочным раствором гипоиодита или кислым раствором
перманганата аскорбиновая кислота окисляется в конечном счете до триоксимасляной
п щавелевой кислот.
НО —С — С —О
II I
НО-С о
н1
НО —С —н
I
сн,он
О = с — с = о
I I
() = С о
н|
НО-С — н
I
СНоОН
СООН
II - С — ОН г (СООН»2
I
н-с-н
I
сн2он
163. ПОЛУМИКРООПРЕДЕЛЕНИЕ ФЕНИЛГИДРАЗИНА, ГИДРОКСИЛАМИНА
И СОЕДИНЕНИИ, СОДЕРЖАЩИХ КАРБОНИЛЬНУЮ ГРУППУ,
РЕАКЦИЕЙ С АЦЕТАТОМ МЕДИ(П) [107J
а. Ионы двухвалентной меди окисляют фенилгидразин или гидроксиламин,
восстанавливаясь до ионов одновалентной меди
C6H5NH — NH24 2Cu2+ 2ОН- CeHe± 2Cu+ -• 2Н,0 N2,
2NH,OH + 4Cu- + + 4ОН- N2O ± 4Cu+ Ч- 5H20.
Используя эту реакцию, потенциометрическим титрованием можно опре-
делить от 7 до 42 мг фенилгидразина и от 3,5 до 17,5 мг гидрокси ламина [107].
Анализируемые соединения растворяют в 5 мл пиридина и титруют 0,05 М
раствором ацетата меди(П) в смеси пиридин — этиловый спирт (1 : 1)
(см. гл. 11, разд. 60,г). Устанавливать титр стандартного раствора лучше
всего по чистому хлоргидрату фенилгидразина. При титровании платиновый
электрод используют как индикаторный и каломельный — как электрод
сравнения. Определение следует проводить в атмосфере азота в небольшом
специально сделанном приборе (рис. 124). Точность определения фенил-
гидразина ±0,1%, точность определения гидроксиламина ±0,5%. На титро-
вание 3,615 мг хлоргидрата фенилгидразина или 1,737 мг хлоргидрата гидро-
ксиламина расходуется 1 мл 0,05 М раствора ацетата меди(П).
б. Определение соединений, содержащих карбонильную группу. 0,15—
0,2 мг-экв образца (g мг) растворяют в 5 мл пиридина, добавляют точно
38—40 мг хлоргидрата фенилгидразина (А мг). Раствор насыщают в течение
5 мин азотом, чтобы исключить попадание воздуха, затем нагревают 5 мин
в реакционной колбе (7, рис. 124) на водяной бане. После охлаждения до ком-
натной температуры титруют потенциометрически в атмосфере азота
(а — объем израсходованного раствора). Электродную насадку (2) при
титровании помещают так, чтобы электроды доставали до дна реакционной
колбы.
376
Глава 29
Этим методом можно определять содержание карбонильных групп,
например в ацетоне, бензальдегиде, изатине, метилэтилкетоне и др.
Ва \
~Ь~) ’
g \
где х — содержание карбонильных групп, %; g— навеска образца, мг;
А—вес хлоргидрата фенилгидразина, мг; В—молекулярный вес хлоргидрата
Рис. 124. Прибор для окислительно-восста-
новительного потенциометрического титро-
вания стандартным раствором ацетата
меди (II) [107].
1 — реакционный сосуд; 2 — насадка для электро-
дов; 3 — платиновый электрод; 4 — каломельный
электрод.
Р и с. 125. Экстрактор’для опреде-
ления каучука в растительном сырье
[131]. Обозначения см. в тексте.
фенилгидразина (144,6), мг; а — объем стандартного раствора, израс-
ходованного на титрование образца, мл; Ъ — эквивалентный объем стандарт-
ного 0,05 М раствора (40 мл), мл.
164. ОПРЕДЕЛЕНИЕ АЛКИЛКСАНТОГЕНАТОВ (КСАНТАТОВ)
ЦЕРИЙ(1У)АММОНИЙНИТРАТОМ |666а]
Ксантат калия можно титровать в ацетонитриле 0,05—0,1 н. раствором церий(1¥)-
аммонийнитрата в ацетонитриле. На конечную точку титрования указывает желтая
окраска, появляющаяся при избытке стандартного раствора
zSK
2S = C< H-2Ce(NO3)4 —> S = C —S —S —C = S4-2KNO3 + 2Ce(NO3)3.
XOC2H5 | |
OC2H5 OC2H5
5—25 мг этилксантогената растворяют в 10—15 мл ацетонитрила и титруют. Стан-
дартный раствор после добавления обесцвечивается и выпадает белый осадок нитрата
церия(Ш).
Титр стандартного раствора устанавливают по подкисленному раствору иодида
калия, выделившийся иод титруют тиосульфатом.
165. ОПРЕДЕЛЕНИЕ КАУЧУКА В РАСТИТЕЛЬНОМ СЫРЬЕ [131]
Каучук, имеющий ненасыщенную (полиизопреновую) цепь, можно титро-
вать в смеси серная кислота — уксусная кислота — четыреххлористый
углерод 0,01 н. раствором перманганата в уксусной кислоте. При этом по двой-
Окислительно-восстановительное титрование в неводной среде
377
-J- 2MnSO4 K2SO4
ной связи присоединяются две ОН-группы.
5 / — СН2 — С = СН — СН2 — \ + 2КМпО4 + 3H2SO4 + 2Н2О —>
।
\ СН3 /
ОН он
I I
— СН2 —с — с—сн2—
I I
СНз Н
На 340 г каучука расходуется 316,06 г перманганата калия.
Каучук экстрагируют из растительного сырья в модифицированном аппа-
рате Сокслета (рис. 125). Растворитель вытекает из сосуда через распреде-
лительную трубку 1 и кран 2 в компенсационный сосуд 3, а оттуда через
кран 4 попадает в сосуд для экстракции 5. Экстракционная муфта 6 рас-
положена в верхней части сосуда. Растворитель в приборе можно заменить,
не прекращая экстракцию и не разбирая прибор, если открыть краны 2, 4 и 7.
При серийных определениях несколько экстракторов можно последова-
тельно соединить с распределительной трубкой 1. Экстракция проводится
при повышенных температурах, и поэтому муфта 6 наполовину погружена
в жидкость. Слив растворителя необходимо отрегулировать так, чтобы
экстрагирующий растворитель был теплым. Сосуд 5 нагревают на водяной
или масляной бане.
Взвешивают 1—5 г высушенных и измельченных в тонкий порошок расте-
ний и экстрагируют из него 50 мл кипящего ацетона (10 час) вещества, мешаю-
щие определению. Каучук экстрагируют трижды четыреххлористым углеро-
дом (5 час). Разбавляют экстракт до 100—200 мл и после добавления 1—5 ка-
пель смеси 96 %-ной серной кислоты и уксусной кислоты (1 : 1) титруют при
постоянном помешивании порциями по 1—5 мл 0,01 н. раствором КМпО4
в уксусной кислоте до появления красной окраски, устойчивой в течение
15 мин. Предварительно устанавливают титр стандартного раствора по щаве-
левой кислоте. Чтобы определить расход перманганата на титрование раство-
рителя, проводят глухой опыт.
Окисление иодной кислотой в неводной среде [668]. Вицинальные гликоли можно
окислять перйодатом трпэтпламмонпя в смеси этилового спирта и этил ацетата. Однако
обратное титрование подной кислотой следует проводить в разбавленной водной среде
и водным раствором титранта.
Глава 30
ТИТРОВАНИЕ В НЕВОДНОЙ СРЕДЕ,
ПРИВОДЯЩЕЕ К ОБРАЗОВАНИЮ КОМПЛЕКСОВ
166. АЛКИЛАЛЮМИНИЕВЫЕ СОЕДИНЕНИЯ
Молекулы алюминийорганических соединений являются электроноде-
фицитными и при образовании комплекса с электронодонорными реагентами,
такими, как ароматические амины (например, изохинолин,) эфиры, спирты,
их можно рассматривать как кислоты Льюиса. Окисление или гидролиз
одной из связей А1 — G приводит к понижению электрофильности молекулы.
Триалкилалюминий, диалкилалюминийгидрид и диалкилалюминийгало-
генид представляют собой «активные» молекулы и образуют стабильные
комплексы с некоторыми азотистыми основаниями. Дезактивированные
молекулы, такие, как алкоголяты диалкилалюминия, не образуют устой-
чивых комплексов с основаниями.
Алкилалюминиевые соединения A1R3 и их производные (например, моно-
и дигалогениды AlR2Hal и AlRHal2) можно титровать потенциометрически
стандартными растворами, содержащими изохинолин [222]. При этом алю-
минийалкилы реагируют с изохинолином в стехиометрическом соотношении
с образованием устойчивых комплексов [75]. С соединениями типа A1R3
образуются комплексы состава 1:1, окрашенные в желтый цвет, в то время
как соединения типа A1R2H образуют красные комплексы состава 2 : 1
(см. также гл. 2, разд. 20,е) [833]. Эти комплексы, растворенные в углеводо-
родных растворителях, обладают электропроводностью и поэтому реакцию
можно контролировать электрометрически. Как индикаторный электрод
используют алюминиевый стержень, а электродом сравнения может слу-
жить алюминиевый стержень, погруженный в раствор триэтилалюминия
в бензоле. Этот раствор сообщается через пористую поверхность (диаметр
пор 5—15 мк) с анализируемым раствором (ср. рис. 55). Для соединений
триалкилалюминия воспроизводимость измерений составляет 0,2—0,4%.
Вода, содержащаяся как в растворителе (бензоле или н-гептане), так
и в изохинолине, сильно влияет на титрование. Уменьшение расхода алю-
минийалкила находится в линейной зависимости от содержания воды.
Поэтому при определении чистоты алюминийалкила следует знать содержа-
ние влаги в реагентах и растворителях.
Моноалкоголяты алюминия в присутствии триалкилалюминиевых соеди-
нений не реагируют с изохинолином. Соединения типа AlR2Hal иАШНа!2
реагируют подобно соединениям A1R3, хотя окрашенных комплексов не обра-
зуется. При титровании определяют суммарное количество обоих типов
соединений. Если соотношение AlR3/AlRHal2< 1, то с изохинолином обя-
зательно будет наблюдаться характерное желтое окрашивание. Смесь триал-
килалюминия и его эфирата можно анализировать потенциометрически.
Первый скачок потенциала соответствует триалкильному соединению,
а второй — эфирату.
В сосуд для титрования (рис. 55) подают медленный ток азота. Электрод
сравнения содержит 1—2 мл 10%-ного раствора триэтилалюминия в бен-
золе. Электроды хранят в атмосфере азота в разбавленном растворе три-
этилалюминия. В ходе титрования расстояние между электродами должно
быть 5—15 мм. Используют магнитную мешалку. Для титрования 5—10 ммо-
лей образца растворяют в 20—40 мл абсолютного бензола. Стандартный раст-
Титрование в неводной среде, приводящее к образованию комплексов
379
вор, содержащий изохинолин (табл. 52,д), растворенный в бензоле или дибу-
тиловом эфире, может быть 0,2 или 0,5 М. В процессе титрования необхо-
димо полностью исключить попадание кислорода из воздуха и непрерывно
пропускать ток сухого азота. Чистые образцы алюминийалкилов крайне
реакционноспособны по отношению к влаге воздуха и, таким образом, точ-
ность и воспроизводимость результатов анализов в значительной степени
зависит от защиты пробы до и во время анализов. Показания измеритель-
ного прибора снимают с интервалами от 0,5 до 2 мин после добавления тит-
ранта. В случае эфиратов показания снимают через 10 мин\ см. также рабо-
ты [740, 853].
Для различных алкилалюминиевых соединений было предложено боль-
шое число отличающихся методов [328, 740, 853]. Хаген и Лесли установили,
что большинство алюминийалкилов с длиной цепи от С2 до С30 можно титро-
вать, используя индикаторы трифенилметанового ряда, например нейтраль-
ный фиолетовый, нейтральный красный, метиловый фиолетовый, основной
фуксин. Эти трифенилметановые соединения образуют обратимо глубоко
окрашенные комплексы с алюминийалкилами и могут быть использованы
в качестве индикаторов при объемном определении «активности». Интенсив-
ность окраски сильно увеличивается, когда в молекуле индикатора присут-
ствуют два или более азометиновых звена. Методику и прибор для титрова-
ния см. работу [328]. Приведенные ниже реакции иллюстрируют принципы
такого титрования и могут быть отнесены к реакциям дезактивации (1)
и замещения (2)
1. [I - A1R3]~ R'OII A1R2OR' -j-RH-t-I,
/\/СН-СН
2. |I-A1R3] + | I
- x сн= CH
Для замещения индикатора может быть использовано более сильное
основание: пиридин, изохинолин, гексиловый спирт (растворенные в кси-
лоле).
В присутствии A1R2H эти индикаторы теряют способность давать резкие
изменения окраски в конечной точке титрования, что обусловлено реакцией
с первичной аминогруппой или образованием ковалентной связи А1 — N.
Титрование с использованием визуальных индикаторов применимо к триал-
кильным соединениям алюминия, содержащим небольшое количество или
совсем не содержащим гидридов [328].
167. ДИЕНЫ, АКТИВНЫЕ В РЕАКЦИИ ДИЛЬСА — АЛЬДЕРА,
И АРОМАТИЧЕСКИЕ УГЛЕВОДОРОДЫ
а. Определение диенов [618]. 1,3-Диены с диенофилами образуют аддукты
в соответствии со следующей схемой (стр. 377 в работе [870]):
Дпен Диенофил Аддукт
Для определения 1,3-диенов, вступающих в реакцию Дильса — Альдера,
применяют тетрацианэтилен (ТЦНЭ) (NC)2C = C(CN)2. Навеску ТЦНЭ,
взятого в избытке, смешивают с анализируемыми соединениями в среде
380
Глава 30
хлористого метилена. После реакции Дильса — Альдера, протекающей при
комнатной температуре, определяют избыток ТЦНЭ медленным обратным
титрованием 0,05 М раствором циклопентадиена в этиловом спирте, в резуль-
тате которого образуется бесцветный аддукт
сн=сн
сн2
сн = сн
C(CN)2
+
C(CN)2
СН—СН—C(CN)
СН — СН — C(CN)
Для облегчения определения конечной точки используют два индика-
тора: 1) предупреждающий и 2) индикатор конечной точки.
1) Бензол, содержащий 20% фенантрена:
С5Н6 + ТЦНЭ[С14Н10] с5Н6- ТЦНЭ + С14Н10
Красно-фиоле- Бесцветный
товый
2) Хлористый метилен, содержащий 1% пентаметилбензола:
С5Н6 + ТЦНЭ[С6Н(СН3)5] С5Н6.ТЦНЭ + С6Н(СН3)5
Красный Бесцветный
Исчезновение красной окраски индикатора можно также проследить фотометри-
чески при длине волны около 525 нм.
Относительно приготовления стандартных растворов ТЦНЭ и циклопен-
тадиена см. гл. 11, разд. 60,ф.
320—390 мг ТЦНЭ взвешивают в колбе Эрленмейера, снабженной стек-
лянной пробкой. Добавляют 50 мл перегнанного хлористого метилена
и смесь нагревают до растворения. Летучесть хлористого метилена может
быть понижена добавлением хлороформа. Добавляют 1,25—1,5 ммоля
образца, колбу закрывают и оставляют на 10 мин. Затем титруют до исчез-
новения красно-фиолетовой окраски, добавляя по каплям 0,05 М раствор
циклопентадиена в присутствии 10 капель предупреждающего индикатора,
содержащего фенантрен. Прибавляют 5 капель индикатора, содержащего
пентаметилбензол, и титруют, добавляя стандартный раствор по каплям
до обесцвечивания. Таким методом можно определять антрацен, циклопен-
тадиен, циклогексадиен-1,3, 2,3-димети л бутадиен и др.
Константа ассоциации для л-комплекса пентаметилбензол — ТЦНЭ
высока (К = 123), а реакция комплекса с циклопентадиеном протекает
медленно, поэтому л-комплекс фенантрен — ТЦНЭ с меньшей стабиль-
ностью (К = 18) применяется как предупреждающий индикатор. Точность
определения составляет около 1 %.
б. Определение ароматических углеводоров [717]. л-Электронная система —
«функциональная группа», характерная для ароматического кольца,—
имеет относительно очень низкую основность [435]. Тем не менее некоторые
ароматические углеводороды образуют более или менее устойчивый окра-
шенный комплекс неионного типа с ТЦНЭ [573, 718].
Согласно Малликену [602], этот тип образования л-комплекса можно рассматривать
как льюисовское кислотно-основное взаимодействие, в котором комплексная связь
обусловлена переносом л-электронов на вакантную орбиталь кислоты Льюиса (ком-
плекс с переносом заряда).
Ароматические соединения определяют фотометрически и строят график
изменения поглощения в зависимости от количества израсходованного
ТЦНЭ (мл) (гл. 14). Молярное отношение при образовании комплекса обычно
равно 1. Так, например, светопоглощение комплекса 1,2,4,5-тетраметилбен-
зол — ТЦНЭ одинаково независимо от их соотношения — 5:1 или 1:5.
Титрование в неводной среде, приводящее к образованию комплексов
381
Согласно Меррифилду и Филлипсу [573], константа ассоциации (К) неко-
торых ароматических углеводородов может значительно изменяться
(табл. 106).
к =____________(£)__________
{(ТЦНЭ) — (С)}{[В1-[С]} ’
где (ТЦНЭ) — начальная концентрация тетрацианэтилена, молъ/л\ (С) —
концентрация л-комплекса в состоянии равновесия, молъ/л\ [5] и [С] —
мольные доли ароматического л-основания или комплекса при равновесии.
Таблица 106
л-Основание Устойчивость ТЦНЭ-ком- плекса (К) хмакс- нм [717] Точность [717]
Гексаметплбензол 263 [573] 545 99+1%
Пентаметилбензол 123 525 97+2%
Флуорантен - 90 [717] 540 100+1%
Дурол 54,2 [573] 480 99+2%
Бензо[а]пирен ~ 40 [717] 520 99+2%
Нафталин 11,7 [573] 550 90+8%
Бензол 2 [573]
Хризен 560 100+1 %
Перилеп 460 100+1 %
При фотометрических титрованиях в конечной точке, полученной экстра-
поляцией, не каждое ароматическое л-основание образует комплекс в соот-
ношении 1:1, а при добавлении дополнительного количества раствора
ТЦНЭ поглощение увеличивается, что часто делает расчет неточным, осо-
бенно для соединений с низким значением К.
Точность измерений, однако, можно повысить, если для определения
органических примесей использовать спектрофотометрический метод
(«метод анализа следов» [672]), например при анализе следов нафталина
во фталевом ангидриде.
В колбу Эрленмейера (рис. 99) емкостью 50—125 мл помещают навеску
около 0,3 ммолей (К ~ 120) или 0,6 ммолей (К ~ 12) ароматического угле-
водорода и растворяют в 60 мл хлороформа. Колбу соединяют с 2,5-сантимет-
ровой кюветой фотометра (Bausch and Lomb Spectronic 20), устанавливают
коэффициент пропускания на 100% и титруют раствором 0,1 н. ТЦНЭ при
выбранной длине волны (см. табл, выше), применяя магнитную мешалку.
Для построения графика титрант прибавляют порциями по 0,3 мл так, чтобы
до середины графика было нанесено по крайней мере четыре точки; титро-
вание продолжают до тех пор, пока молярное соотношение ароматического
л-основания и ТЦНЭ не станет приблизительно 1 : 2—1 : 3. На график
наносят не менее 4 точек после того, как соотношение компонентов ком-
плекса достигнет 1 : 1,5.
Глава 31
ОПРЕДЕЛЕНИЕ АЛКОКСИГРУППЫ,
ЭФИРОВ КАРБАМИНОВОЙ КИСЛОТЫ,
ЗАМЕЩЕННЫХ ФОСФИНОВ,
КРЕМНИЙОРГАНИЧЕСКИХ СОЕДИНЕНИЙ
168. АЛКОКСИГРУППА (163]
Описываемый в настоящей главе метод остроумно упрощает методику
определения алкоксильной группы, применявшуюся до настоящего времени.
Йодистый алкил, образовавшийся в результате реакции алкоксисоедине-
ния с иодистоводородной кислотой, а также отгоняющийся избыток иоди-
стоводородной кислоты поглощаются пиридином
r-I + C5H5N -> [C5H5NR]+I-.
Йодистый водород и иодид алкилпиридиния в смеси можно титровать
при помощи фотометрического дифференцирующего титрования в пиридине
(рис. 126). Титрование можно также проводить и в присутствии азофиолето-
вого как индикатора, так как двухстадийное изменение окраски индикатора
дает возможность визуального определения первой конечной точки для HI
по изменению оранжевой окраски в красную и второй эквивалентной точки
по изменению окраски индикатора из красной в голубую, соответствующей
иодиду алкилпиридиния (гл. 13, разд. 74). В качестве раствора для титрова-
ния используют 0,02 н. ТБАГ в смеси бензол — метиловый спирт (см. гл. 10,
разд. 57) и как индикатор 0,5%-ный азофполетовый в пиридине. Количество
стандартного раствора, израсходованное между двумя точками перегиба
или между переходами окраски прямо пропорционально содержанию алко-
ксильной группы. Алкилиодид и пиридин реагируют в стехиометрическом
отношении. Алкилиодиды нейтральны в ацетоне, ацетонитриле или метил-
изобутилкетоне.
Прибор для титрования изображен на рис. 127. Иод и сульфиды не мешают
титрованию. При определении пропокси- и бутоксигрупп смесь сначала
кипятят 2 час.
В реакционной колбе (7, рис. 127) взвешивают 10—15 мг образца. Лету-
чие соединения взвешивают в желатиновых капсулах. Добавляют 0,5 мл
ксилола и навеску растворяют, если необходимо при нагревании. Прибав-
ляют 5,0 мл 55—58%-ного раствора иодистоводородной кислоты и бросают
в колбу несколько кипятильников. В приемник помещают 50 мл перегнанно-
го над Ва(ОН)2 и нейтрализованного пиридина. Конец алонжа должен нахо-
диться ниже поверхности растворителя (рис. 127).
Через прибор пропускают ток азота со скоростью один пузырек в секунду.
Колбу нагревают при помощи нагревательной рубашки так, чтобы кон-
денсат находился только в шаре (2). Азот продолжают пропускать через
прибор в течение 20 мин и затем увеличивают скорость его тока до 2—3 пу-
зырьков в секунду. Нагревание продолжают 25 мин при определении мето-
ксигруппы и 40 мин для этоксигруппы. В случае пропокси- и бутоксигрупп
продолжительность нагревания увеличивается до 100 мин, а для определения
S-метильной группы — до 160 мин. Небольшое количество конденсата может
попасть в приемник.
Когда нагревание закончено, конец трубки обмывают небольшим коли-
чеством нейтрализованного пиридина. Пиридиновый раствор осторожно
кипятят в течение 2 мин в токе азота в вытяжном шкафу, затем охлаждают
и титруют в атмосфере азота 0,02 н. раствором ТБАГ. При расчетах прини-
Алкоксигруппы, карбаматы, фосфины, кремнийорганические соединения
383
мают во внимание расход титранта в глухом опыте. Определение нормаль-
ности титранта можно проводить по чистой триметоксибензойной кислоте.
Высокие температуры кипения алкилиодидов, содержащих более 4 атомов
углерода, мешают их количественному определению (изоамил-, гептил-
и гексадецилиодиды). Если расщепление проводят в запаянной ампуле,
то образовавшийся алкилиодид экстрагируют известным количеством бен-
зола * и аликвотную часть бензольного слоя обрабатывают анилином [624];
Рис. 127. Прибор для определения
алкоксигрупп [163].
7 — реакционная колба; 2 — шар; 3 — прием-
ник; 4 — нагревательная рубашка; 5 — тонкая
перфорированная резиновая мембрана.
Рис. 126. Титрование смеси HI и
н-С4Н91 [163].
Растворитель: пиридин. Стандартный раствор:
0,2 н. раствор гидроокиси тетрабутиламмония
в смеси бензол -— метиловый спирт.
образующуюся иодистоводородную кислоту можно оттитровать метилатом
натрия, используя в качестве индикатора тимоловый синий [203].
R — I + 2C6H5NH2 C6H5NH2.HI + C6H5NHR,
HRNC6H5Hl4-CH3ONa —► HRNC6H5 4- Nal + CH3OH.
169. ЭФИРЫ КАРБАМИНОВОЙ КИСЛОТЫ |121]
Многочисленные эфиры карбаминовой кислоты находят широкое приме-
нение в фармакологии и промышленном синтезе.
ОН
CO(NH2)2
Мочевина
CO(NH2)OH
Карбаминовая
кислота
CO(NH2)OR
Эфир карбами-
новой кислоты
C = NH
\r
Енольная
форма
Карбамоильная группа OGONH2 является общей функциональной груп-
пой для эфиров карбаминовой кислоты.
Эфир карбаминовой кислоты в своей енольной форме реагирует с мети-
латами щелочных металлов и при его расщеплении, легче протекающем
в основной среде, образуется цианат щелочного металла и спирт ROH [121]
ROC(OH) = NH4-CH3OK - - ROC(OK) = NH-j-СН3ОН,
ROC(OK) = NH —» ROH4-NCOK.
* Kirsten, Ehrlich-Rogozinski, Mikrochimica Acta, 4, 786 (1955).
384
Глава 31
Эта реакция не происходит с амидами кислот, мочевиной, семикарбазидом
и биуретом.
В колбе из стекла, устойчивого к щелочам, взвешивают 1—2 мг-экв эфира
карбаминовой кислоты, растворяют в 10 мл пиридина, добавляют точно
25 мл 0,1 н. раствора метилата щелочного металла в смеси бензол — метило-
вый спирт и кипятят с обратным холодильником в течение 1 час. Колбу
защищают от попадания влаги и двуокиси углерода из воздуха при по-
мощи трубки, заполненной едким кали.
Продолжительность кипя-
чения, мин
Тис. 128. Уменьшение эффектив-
ности 10 мл 0,1 н. КОН в смеси
ксилол — метилцеллозольв при ки-
пячении в колбе из стекла, устойчи-
вого к щелочам (2), и в колбе из
обычного стекла (2).
Аналогично проводят глухой опыт. После
охлаждения титруют 0,1 н. раствором бен-
зойной кислоты в бензоле (табл. 52, при-
мечание «а») в присутствии тимолового
синего до тех пор, пока голубая окраска
индикатора не превратится в желтую.
При расчете содержания карбаминовой
группы используют разность между двумя
измерениями. На титрование 0,1 мг-экв
эфира карбаминовой кислоты расходуется
1 мл 0,1 н. раствора (в скобках приведены
количества, мг, эквивалентные 1 мл 0,1 н.
раствора): этилуретан (8,908), изобутил-
карбамат (11,72), триметилендикарбамат
(8,11), бензилкарбамат (15,11), фенилкар-
бамат (13,71), 2-метил-2-н-пропилпропан-
диол-1,3-дикарбамат, мепробамат (10,91),
карбамат фенилэтинилкарбинола, нирво-
тин (17,52), хлорид карбамоил-0-метил-
холина, урехолинхлорид H2N — СО —О—
СН(СН3) — GH2N+(GH3)3G1_ (19,67), 3-метилпентин-1-ол-З-карбамат, обливон
{14,12; см. также гл. L27), 1-этинилциклогексилкарбамат, этинамат (16,72).
Вследствие вредного влияния примесей действующее начало таблеток
(Следует предварительно экстрагировать бензолом или метиловым спиртом
3 аппарате Сокслета.
На рис. 128 изображены кривые уменьшения эффективности щелочного титранта
при кипячении в колбе из стекла, устойчивого по отношению к щелочам, по сравнению
с кипячением в колбе из обычного стекла.
170. ЗАМЕЩЕННЫЕ ФОСФИНЫ [781]
Замещенные фосфины являются фосфорными аналогами аминов: R3P —
третичные, R2PH — вторичные, ИРН2 — первичные фосфины. Их относи-
тельная основность, определенная при помощи потенциометрически измерен-
ного потенциала полунейтрализации, находится в полном соответствии
со значениями, полученными другими методами. Фосфины можно титровать
в нитрометане 0,05 н. раствором хлорной кислоты в нитрометане со стек-
лянным и каломельным электродами (гл. 11, разд. 60,6).
Навеску 0,5—1 ммоля вещества растворяют в 100 мл нитрометана.
Используют магнитную мешалку. Таким методом можно титровать большое
число замещенных фосфинов (в скобках приведены значения рКа основа-
ний): например, трициклогексил-(9,70), триэтил-(8,69), три-н-пропил-(8,64),
лгрис-(2-фенилэтил)-(6,60), лгрис-(п-метоксифенил)-(4,46), трифенил-(2,73),
дициклогексил-(4,55), бис-(2-фенилэтил)фосфины (3,46). Наиболее силь-
ными основаниями являются третичные фосфины. Ароматические и электро-
отрицательные заместители уменьшают основность. Таким образом, порядок
изменения основности следующий: третичные > вторичные > первичные.
Иногда некоторые первичные и вторичные фосфины не удается титровать.
Алкоксигруппы,, карбаматы, фосфины, кремнийорганические соединения
385
171. КРЕМНИЙОРГАНИЧЕСКИЕ СОЕДИНЕНИЯ
Хлорсиланы общей формулы HnSiCl4_n, где R = метил, этил или фенил,
реагируют с роданидом калия или аммония в эфире с образованием хлорида
калия или аммония [796].
Навеску хлорсилана (около 1 мл), эквивалентную приблизительно 100 мг
хлора, растворяют в 10 мл эфира (содержание воды <0,15%), добавляют
две капли 0,5 %-ного раствора хлорида железа(Ш) и титруют 0,3 М раство-
ром роданида в ацетоне до появления красной окраски (гл. 11, разд. 60,е).
Точность определения в случае метилхлорсиланов ±1%, а в случае
фенилхлорсиланов +2%.
Визуальное или потенциометрическое определение индивидуальных алкил-
хлорсиланов в ацетонитриле органическими основаниями, например 0,05 н.
раствором антипирина в ацетонитриле или раствором амидопирина в бен-
золе, см. работу [482]. В качестве индикаторов можно использовать кри-
сталлический фиолетовый, диметиламиноазобензол, бромкрезоловый пур-
пурный, бромфеноловый синий и метиловый оранжевый. Амперометрическое
титрование в смеси метиловый спирт — бензол и в среде уксусной кислоты
описано в работе [476].
Определение бинарных и тройных смесей метилхлорсиланов основано
на количественном превращении (CH3)3SiCl, (CH3)2SiCl2 и CH3SiCl3 в алкил-
тиоцианатные производные при действии тиоцианата аммония. Дифферен-
цирующее кондуктометрическое титрование осуществляют в смеси ацето-
нитрил — диэтиловый эфир (2 : 3) 0,1 М раствором амидопирина в бензоле
[481].
Определение ацетоксигруппы в различных замещенных ацетоксисиланах
в метилэтилкетоне проводят 0,1 н. раствором метилата натрия в метиловом
спирте в присутствии тимолового синего как индикатора [191].
T?7lSi(OCOCH3)4_7l + (4 — n)CH3ONa —> RnSi(OCH3)4_n +(4 —n)CH3COONa.
Потенциометрическое определение азотсодержащих кремнийорганиче-
ских соединений описано в работе [483].
Определение следовых количеств соляной кислоты и хлорспланов в силок-
сановых материалах см. работу [533].
Определение алкоксигрупп в алкоксисиланах ацетилированием, ката-
лизируемым кислотами, описано в работе [532]. Этот метод был применен
главным образом Фритцем и Шенком [257] для определения гидроксильных
групп (см. гл. 19); его можно применять для анализа большого числа разно-
образных алкокси- и арилоксисиланов. Ацетилирование при комнатной тем-
пературе проходит количественно менее чем за 2 мин при использовании
0,06 М хлорной кислоты в качестве катализатора. Реакция протекает по сле-
дующему механизму [532]:
(Сн3со)2о-1-н+ (сн3со)2он+,
(СН3СО)2ОН+ СН3СО+ + СН3СООН,
СН3СО+4- —SiOR ^SiO+R,
/ /I
СОСН3
— SiO+R+(CH3CO)2O —- — SiOCOCH3-;CH3COOR CH3CO+,
Z СОСНз /
— SiC+R 4-CH3COOII —- — SiOCOCH3- CH3COOR —H+.
COCH3 7
386
Глава 31
Определение меркаптосиланов ацетилированием ацетатом ртути(П)
с использованием хлорной кислоты в качестве катализатора описано в рабо-
те [61]. Анализ связи Si — S — Св меркаптосиланах можно проводить мето-
дом ацетилирования по Фритцу и Шенку [257] в присутствии хлорной кис-
лоты как катализатора. Механизм реакции уксусного ангидрида с группой
Si — S — С, вероятно, такой же, как и предложенный для аналогичного
метода определения связей Si — О — С [532].
Гидролиз меркаптосиланов делает возможным косвенный анализ мер-
каптогруппы с помощью реакций с избытком ацетата ртути(П). Избыток
оттитровывают 0,2 н. раствором хлористого водорода в бутиловом спирте
(см. работу [489] и гл. 28).
В колбе Эрленмейера емкостью 250 мл , снабженной притертой пробкой,
взвешивают образец, содержащий около 4 мг-экв меркаптана. Навеску
растворяют в 50 мл толуола. Приливают точно 50 мл 0,2 н. раствора ацетата
ртути(П) в метиловом спирте и раствор перемешивают. Добавляют три
капли 0,2%-ного раствора тимолового синего (в этиловом спирте) и медлен-
но титруют 0,2 н. раствором хлористого водорода в бутиловом спирте
до появления красного окрашивания в конечной точке. Глухой опыт про-
водят таким же образом, но без анализируемого вещества. Содержание мер-
каптосилана рассчитывают по разности в объемах титранта, пошедшего
на титрование пробы и в глухом опыте.
ЛИТЕРАТУРА
1. Agazzi, Bond, Anal. Chem., 33, 972 (1961).
2. Albert, Goldacre, J. Chem. Soc., 1943, 454.
3. Albert, Goldacre, J. Chem. Soc., 1946, 706.
4. Albert, Goldacre, Phillips, J. Chem. Soc., 1948, 2240.
5. Albert, Phillips, J. Chem. Soc., 1956, 1294.
6. A 1 i c i n o, Microchem. J., 4, 551 (1960).
7. Allen, Geddes, J. Pharm. Pharmacol., 9, 990 (1957).
8. Altschuler, J. Am. Chem. Soc., 77, 3480 (1955).
9. A m e s, L i c a t a, J. Am. Oil. Chem. Soc., 25, 203 (1948).
10. Anastas i, Gallo, M eca re Hi, Mikrochim. Acta, 1956, 252.
11. Anastas i, Gallo, Novaci c, J. Pharm. Pharmacol., 7, 263 (1955).
12. Anastas i, Mecarelli, Novaci c, Mikrochim. Acta, 40, 53 (1952).
13. Anastas i, Mecarelli, Novaci c, Pharm. Acta Helv., 30, 55 (1955).
14. Арбузов, Кузнецов, Павлов, Завод, лаб., 27, 225 (1961).
15. van Arkel, К roonen berg, Pharm. Weekbl., 87, 137 (1952).
16. Arndt, Ber., 63, 2963 (1930).
17. A r n d t, L о r e n z, Ber., 63, 2124 (1930).
18. A r s h i d, G i 1 e s, J a i n, J. Chem. Soc., 1956, 559.
19. A r s h i d, G i 1 e s, M c L u г e, О g i 1 v i e, R о s e, J. Chem. Soc., 1955, 67.
20. Ashworth, Titrimetric Organic Analysis, Part I in Elving, Kolthoff, Chemical
Analysis XV, Interscience, New York, 1964.
21. Audrieth, Z. phys. Chem., 165A, 323 (1933).
22. Audrieth, К leinberg, Non-Aqueous Solvents, John Wiley and Sons, New
York, 1953.
23. Auterhoff, Becker, Arch. Pharm., 293, 1021 (1960).
24. В а с к e - H a n s e n, Norsk Farm. Selskap., 17, 1 (1955).
25. Backe-Hansen, Wickstrom, Norsk Farm. Selskap., 19, 193 (1957).
26. Badger, The Structures and Reactions of the Aromatic Compounds, University
Press, Cambridge, 1954.
27. Badoz-Lambling, Desbarres, Tacussel, Bull. soc. chim. France,
1962, 53.
28. Baggesgaard-Rasmussen, Berger, Folting, Dansk Tidsskr. Farm.,
32, 29 (1958).
29. Baker, Nature, 137, 236 (1936).
30. В a n i c k, Anal. Chem., 34, 296 (1962).
31. В a n у a i, Kemiai Indikatorok, Budapest, Miiszaki Konyvkiado, 1961.
32. Barnes, Anal. Chem., 31, 405 (1959).
33. В а г г о w, J. Am. Chem. Soc., 78, 5802 (19 6)
34. В а г г о w, J. Am. Chem. Soc., 80, 86 (1958).
35. В а г г о w, Y e r g e r, J. Am. Chem. Soc., 76, 5211, 5247, 5248 (1954).
36. В а г г о w, Y e r g e r, J. Am. Chem. Soc., 77, 4474 (1955).
37. В а г г о w, Y e r g e r, J. Am. Chem. Soc., 77, 6206 (1955).
38. В a r t о n, H о w 1 e t t, J. Chem. Soc., 1949,155.
39. Bates, Anal. Chem., 29, 15A (1957).
40. Bates, Schwarz enbach, Helv. Chem. Acta, 38, 699 (1955).
41. Bayer J., Magyar Kem. Folydirat, 62, 355 (1956).
42. В а у e г, P о s g a y, Pharm. Zhalle, 96, 561 (1957).
43. Bayer, P о s g a y, Naturwissenschaften, 45, 185 (1958).
44. В а у e г, P о s g a y, Pharm. Zhalle, 100, 65 (1961).
45. В а у e г, P о s g а у, M a j 1 a t, Pharm. Zhalle, 101, 476 (1962).
46. В а у 1 e s, C h e t w v n, J. Chem. Soc., 1958, 2328.
47. Becker, Ber., 86, 1150 (1953).
48. В e с к e t t, C a m p, M a r t i n, J. Pharm. Pharmacol., 4, 399 (1952).
49. В e с к e t t, T i n 1 e y, Titration in Non-aqueous Solvents, British Drug Houses Ltd,
50. В e g g s, S p e n c e r, Anal. Chem., 34, 1590 (1962).
51. В e 1 c h e г, В e r g e r, W e s t, J. Chem. Soc., 1959, 2877, 2882.
25*
388
Л итература
52. Bell A., Quart. Rev., London, 1, ИЗ (1947).
53. Bell R. P., Acids and Bases, J. Wiley and Sons, New York, 1956.
54. В e 1 1, В а у 1 e s, J. Chem. Soc., 1952, 1518.
55. Bell, Trotman-Dickenson, J. Chem. Soc., 1949, 1288.
56. В e 1 1 e n, Chem. Anal. (Warsaw), 6, 531 (1961).
57. В e n s о n, К i t s c h e n, Can. J. Research, Sect. F., 1949, 27.
58. Bergamini, Mattei, Sperimentale, 6, 13 (1955).
59. Berger, Acta Chem. Scand., 6, 1564 (1952).
60. Berger, Acta Chem. Scand., 8, 427 (1954).
61. Berger, Magnuson, Anal. Chem., 36, 1156 (1964).
62. В e г г e t t, Bull. soc. chim. France, 1960, 271.
63. Berthe 1, Sheppard, J. Chem. Phys., 21, 1421 (1953).
64. Безингер, Гальперин, Абдурахманов, Ж. анал. хим., 16, 91
(1961).
65. В i а п с h i, Il Farmaco, 16, 476 (196L.
66. Billon, Bull. soc. chim. France, 1962, 863.
67. Bishop, Kittredge, Hildebrand, J. Am. Chem. Soc.. 44, 135 (1922).
68. В i e r r u m, Chem. Rev., 16, 287 (1935).
69. В 1 a i r, P a n t о u y, Anal. Chim. Acta, 13, 1 (1955).
70. Blake, J. Am. Pharm. Assoc. Sci. Ed., 46, 163 (1957).
71. Blom, Edelhausen, Anal. Chim. Acta. 13, 120 (1955).
72. В lumrich, Bandel, Angew. Chem., 54, 374 (1941).
73. В о с к г i s, Quart. Rev., London, 3, 173 (1949).
74. Бонина, Заринский, Штифман, Ж. анал. хим., 19, 635 (1964).
75. В о n i t z, Вег., 88, 742 (1955).
76. Bonner, Lockhart, J. Chem. Soc., 1957, 364, 2840.
77. В о u b 1 i, Boll. chim. pharm., 94, 186 (1955).
78. Bournique, Neuser, Chemist-Analyst, 53, 41 (1964).
79. Branch, Calvin, The Theory of Organic Chemistry, Prentice-Hall, Inc., New
York, 1941.
80. Branch, Walba, J. Am. Chem. Soc., 76, 1564 (1954).
81. Branch, Y abroff, J. Am. Chem. Soc., 56, 2568 (1934).
82. Brand, Horning, Thornley, J. Chem. Soc., 1952, 1374.
83. Braude, Nachod, Determination of Organic Structures by Physical Methods,
Academic Press, New York, 1955.
84. Braude, Stern, J. Chem. Soc., 1948, 1971, 1976, 1982.
85. В e n n e c k e, Die Titration schwacher Basen in Eisessiglosung, F. Enke, Stuttgart,
1951.
86. Bricker, Sweetser, Anal. Chem., 24, 409 (1952).
87. Briegleb, Z. Elektrochem., 50, 35 (1944).
88. а) В r i e g 1 e b, Angew. Chem., 62, 262, 536 (1950); 6) Z. Elektrochem., 53, 350
(1949).
89. Briegleb, Ruttiger, Jung, Angew. Chem., 75, 671 (1963).
90. В г i 11, Z. Elektrochem., 50, 47 (1944).
91. Brockman n, Meyer, Naturwissenschaften, 40, 242 (1953).
92. Brockman n, Meyer, Ber., 86, 1514 (1953).
93. В г о c k m a n n, M e у e r, Ber., 87, 81 (1)54).
94. Brown, Recent Chem. Progr., 14, 83 (1953).
95. В г о w n, В а г b a r a s, J. Am. Chem. Soc., 75, 6 (1953).
96. Brown, Bartholomay, Taylor, J. Am. Chem. Soc., 66, 435 (1944).
97. Brown, Johannesen, J. Am. Chem. Soc., 75, 16 (1 953).
98. Brown, Kanner, J. Am. Chem. Soc., 75, 3865 (1953).
99. В ronst ed, Rec. trav. chim., 42, 718 (1923).
100. Bruckenstein, Anal. Chem., 31, 1757 (1959).
101. Bruckenstein, Kolthoff, J. Am. Chem. Soc., 78, 10 (1956).
102. Bruckenstein, Kolthoff, J. Am. Chem. Soc., 78, 2974 (1956).
103. Bruehlman, Verhoeck, J. Am. Chem. Soc., 70, 1401 (1948).
104. Bruss, Harlow, Anal. Chem., 30, 1836 (1958).
105. Bruss, W у 1 d, Anal. Chem., 29, 232 (1957).
106. Bryant, War drop, J. Chem. Soc., 1957, 895.
107. Budesinsky, Chem. listy, 52, 2292 (1958).
108. Burgstahler, Kulier, Worden, J. Chem. Educ., 39, 578 (1962).
109. Burns. Lawler, Anal. Chem., 35, 802 (1963).
110. Burton, Praill, J. Chem. Soc., 1950, 1203.
111. Burwell, Langford, J. Am. Chem. Soc., 81, 3799 (1959).
112. В uscarons, Marin, Clave r, Anal. Chim. Acta, 3, 310, 417 (1949).
ИЗ. В u t 1 e r, C z e p i e 1, Anal. Chem. 28, 1468 (1956).
114. Быкова, Казарьян, Труды комиссии по анал. химии АН СССР. 13, 309
(1963).
115. С а 1 d a s, Chemist-Analyst, 43, 100 (1954).
Л шпература
389
116. Caldin, Long, J. Chem. Soc., 1954, 3737.
117. Carlton, J. Am. Chem. Soc., 44, 1469 (1922).
118. Caso, Cefola, Anal. Chim. Acta, 21, 205 (1959).
119. Caso, Cefola, Anal. Chim. Acta, 21, 374 (1959).
120. Caso, Cefola, Anal. Chim. Acta, 29, 127 (1963).
121. Cerri, Spialtini, Gallo, Pharm. Acta Helv., 34, 13 (1959).
122. Chalandonand Susz, Helv. Chim. Acta, 41, 697 (1958).
123. C h a t t e n, J. Pharm. Pharmacol., 7, 586 (1955).
124. C h a t t e n, J. Amer. Pharm. Assoc., Sci. Ed., 45, 556 (1956).
125. Chatten, Harris, Anal. Chem., 34, 1495 (1962).
126. Chatten, P ernarowski, Levi, J. Am. Pharm. Assoc., Sci. Ed., 44, 332
(1955).
127. Chatten, Pernaro wski, Levi, J. Am. Pharm. Assoc., Sci. Ed., 48, 276
(1959).
128. Че ронис H., Микро- и полумикрометоды органической химии, ИЛ, М., 1960.
129. Chiang,!. Pharm. Sci., 50, 885 (1961).
130. С h о 1 v у, Ann. pharm. franc., 18, 138 (1960).
131. Chrastil, Chem. listy, 48, 781 (1954).
132. Ciaccio, Missan, McMullen, Grenfell, Anal. Chem., 29, 1670 (1957).
133. Clair, Chatten, J. Pharm. Sci., 50, 848 (1961).
134. C 1 a г k, W a n g, Anal. Chem., 26, 1230 (1954).
135. Clarke, Sandler, Chemist-Analyst, 50, 76 (1961).
136. Cluett, Anal. Chem., 31, 610 (1959)
137. Cluett, Anal. Chem., 34, 1491 (1962).
138. Coetzee, Diss. Abs., 16, 1071 (1956); Coetzee, Kolthoff, J. Am. Chem.
Soc., 79, 6110 (1957).
139. Coetzee, Cunningham, McGuire, Padmanabhan, Anal. Chem.,
34, 1139 (1962).
140. Coetzee, Padmanabhan, J. Phys. Chem., 66, 1708 (1962).
141. Cohen, Gordon, Anal. Chem., 28, 1445 (1956).
142. Cohn, McMeekin, Edsall, Blanchard, J. Am. Chem. Soc., 56, 784
(1934).
143. Conant, Chow, J. Am. Chem. Soc., 55, 3745, 3752 (1933).
144. Conant, Hall, J. Am. Chem. Soc., 49, 3047, 3062 (1927).
145. Conant, Thompson, J. Am. Chem. Soc., 54, 4039 (1932).
146. Conant, Werner, J. Am. Chem. Soc., 52, 4436 (1930).
147. Conant, Whelan d, J. Am. Chem. Soc., 54, 1212 (1932).
148. Connors, Higuchi, Anal. Chem., 32, 93 (1960).
149. Connors, Swanson, J. Pharm. Sci., 53, 432 (1964).
150. Corwin, Ellingson, J. Am. Chem. Soc., 64, 2098 (1942).
151. Коулсон Ч., Валентность, изд-во «Мир», М., 1965.
152. Crabb, Critchfield, Taianta, 10, 271 (1963).
153. Crisafio, Chatten, J. Am. Pharm. Assoc., Sci. Ed., 44, 529 (1955).
154. Critchfield, Funk, J ohnson, Anal. Chem., 28, 76 (1956).
155. Critchfield, Johnson, Anal. Chem., 26, 1803 (1954).
156. Critchfield, J ohnson, Anal. Chem., 29, 957 (1957).
157. Critchfield, J ohnson, Anal. Chem., 29, 1174 (1957).
158. Cundiff, Markunas, Anal. Chem., 27, 1650 (1955).
159. Cundiff, Markunas, Anal. Chem., 28, 792 (1956).
160. Cundiff, Markunas, Anal. Chem., 30, 1447, 1450 (1958).
161. Cundiff, Markunas, Anal. Chim. Acta, 20, 506 (1959).
162. Cundiff, Markunas, Anal. Chim. Acta, 21, 68 (1959).
163. Cundiff, Markunas, Anal. Chem., 33, 1028 (1961).
164. Cundiff, Markunas, Anal. Chem., 34, 584 (1962).
165. Cundiff, Markunas, Anal. Chem., 35, 1323 (1963).
166. Cundiff, Riddick, Anal. Chem., 24, 910 (1952).
167. Curran, J. Am. Chem. Soc., 67, 1835 (1945).
168. D a h m e n, Chim. anal., 40, 378 (1958).
169. Das, Anal. Chem., 26, 1086 (1954).
170. Das, Mukherjee, Anal. Chem., 31, 233 (1959).
171. D a v i s, H e t z e r, J. Res. Nat. Bur. Stand., 48, 381 (1952).
172. D a v i s, H e t z e r, J. Am. Chem. Soc., 76, 4247 (1954).
173. D a v i s, H e t z e r, J. Res. Nat. Bur. Stand., 60, 569 (1958).
174. Davis, Paabo, J. Am. Chem., Soc., 82, 5081 (1960).
175. Deal, Weiss, White, Anal. Chem., 25, 426 (1953).
176. Deal, W у 1 d, Anal. Chem., 27, 47 (1955).
177. Dean, Cain, Anal. Chem., 27, 212 (1955).
178. De Lorenzi, Cred al i, Boll. chim. farm., 95, 325 (1956).
179. D e n o, J. Am. Chem. Soc., 74, 2039 (1952).
180. De Paulis, Dipietromari a, Boll. chim. farm., 99, 15 (1960)
Литература
'ermer, Wilson, J о hn son, D e rm er, J. Am. Chem. Soc., 63, 2881 (1941).
• e Vries, Gantz, J. Am. Chem. Soc., 76, 1008 (1954).
i e Vries, Schiff, Gantz, Anal. Chem., 27, 1814 (1955).
• e w a r, The Electronic Theory of Organic Chemistry, Oxford, 1954.
• e у г u p, J. Am. Chem. Soc., 56, 60 (1934).
• ilthey, Dinklag e, Ber., 62, 1834 (1929).
• ilthey, Wizinger, J. Prakt. Chem., 2, 118, 321 (1928).
i imroth, Meyer-Bruno t, Biochem. Z., 323, 338 (1952).
ippy, Chem. Revs., 25, 151, 179 (1939).
• о ember g, Hubache r, Lysyj, J. Am. Phyrm. Assoc., Sci. Ed., 43, 418
1954).
, p о з д о в, Власова, Труды комиссии по анал. химии АН СССР, 13, 187 (1963).
lubois, Lacaze, С. В., 252, 748 (1961).
• u g a n, Anal. Chem., 33, 1930 (1961).
, у л о в а, К и м, Химическая наука и промышленность, 4, 134 (1959).
.улова, Леонтьев, Ким, Труды Среднеазиатского гос. университета
м. В. И. Ленина, Химия, 84, 69 (1958).
улова, Попова, Труды Среднеазиатского гос. университета им. В. И. Ле-
ина, Химия, 84, 63 (1958).
• urbetaki, Anal. Chem., 28, 2000 (1956).
। urbe taki, Anal. Chem., 30, 2024 (1958).
• usinsky G., Chem. listy, 48, 1868 (1954).
b e r i u s, Wasserbestimmung mit Karl Fischer-Losung, Verlag Chemie, Weinheim/
ergstr., 1958.
bert, Konopik, Osterr. Chem. Ztg., 50, 184 (1949).
dward, Wang, Can. J. Chem., 40, 966 (1962).
1 hr 1 i ch - R о g о z i n sk i, Patchornik, Bull. Res. Counc. Israel, HA,
90 (1962); Anal. Chem., 36, 840 (1964).
! i sdorf er, Rosen, Ellenbogen, J. Pharm. Sci., 50, 612 (1961).
! istert, Tautomerie und Mesomerie, F. Enke, Stuttgart, 1938.
I i stert, Chemismus und Konstitution, F. Enke, Stuttgart, 1948.
ikeblad P., J. Pharm. Pharmacol., 4, 636 (1952).
! lierington, Nicholls, Analyst, 82, 233 (1957).
lllert, Jasinski, Marcinko wska, Acta Polon. Pharm., 17, 29 (1960).
lllert, J asinski, Pavelczak, Acta Polon. Pharm., 19, 69 (1962).
Hlert, I asinski.Weclawska, Acta Polon. Pharm., 17, 145 (1960).
lllert, J asinski, Weclawska, Acta Polon. Pharm., 19, 75 (1962)
!мелин, Свистунова, Завод, лаб., 27, 1458 (1961).
! rdey, Gimesi, Rady, Acta Chim. Hung., 28, 179 (1961).
! r d e y, Gimesi, Rady, Taianta, 11, 461 (1964).
Irdey, Meisel, Rady, Acta Chim. Hung., 26, 71 (1961).
! r d e y, R a d y, Acta chim. acad. sci. Hung, 15, 81 (1958).
I verett, Ind. chim. Beige., 16, 647 (1951).
iverett, Wynne-J ones, Proc. Roy. Soc., A177, 499 (1941).
' a b e r, J. Pharm. Pharmacol., 6, 187 (1954).
a j a n s, Ber., 21, 709 (1919).
arina, Donat i, Ragazzini, Ann. chim. (Rome), 48, 501 (1958).
auth, Frandsen, Havlik, Anal, chem., 36, 380 (1964).
e i g 1, Spot Tests in Organic Analysis, Elsevier, Amsterdam (1956).
) айгль Ф., Капельный анализ органических веществ, Госхимиздат, М., 1962.
' е i g 1, Anger, Frehden, Mikrochemie, 15, 19 (1934).
erguson, J. Chem. Educ., 33, 276 (1956).
euer, Vincent, Anal. Chem., 35, 598 (1963).
laschka, Barnard, Jr., Tetraphenylboron as an Analytical Reagent in Re-
lley, Advances in Analytical Chemistry and Instrumentation, Vol. I, Interscience
nc., New York, 1960.
lexer, Hammett, Dingwall, J. Am. Chem. Soc., 57, 2103 (1935).
odor, Szerves Kemia I, II, Tankonyvkiado, Budapest, 1960.
1 о g g, Jellum, Analyst., 87, 302 (1962).
' о rbes, Knight, Can. J. Chem., 37, 334 (1959).
orbes, Templeton, Chem. and Ind. (London), 1957, 77, 600.
' orbes, Templeton, Can. J. Chem., 36, 180 (1958).
‘orman, Hume, J. Phys. Chem., 63, 1949 (1959).
' о r t u i n, Anal. Chim. Acta, 24, 175 (1961).
'ossum, Markunas, Riddick, Anal. Chem., 23, 491 (1951).
' г a n k 1 i n, J. Am. Chem. Soc., 46, 2137 (1924).
’ r a n k 1 i n, The Nitrogen System of Compounds, Reinhold Publishing Co., New
fork, 1935.
’reri, Gazetta Chim. I tai., 83, 61 (1953).
’ г e у m a n n, C. R., 204, 41 (1937).
Литература
391
243. Fripiat, Vankompernolle, Servais, Bull. soc. chim. France, 1960,
250.
244. Fritz, Anal. Chem., 22, 578, 1028 (1950).
245. Fritz, Anal. Chem., 24, 306 (1952).
246. Fritz, Anal. Chem., 24, 674 (1952).
247. Fritz, Anal. Chem., 25, 407 (1953).
248. Fritz, Anal. Chem., 26, 1701 (1954).
249. Fritz, Fulda, Anal. Chem., 25, 1837 (1953).
250. F r i t z, К e e n, Anal. Chem., 24, 308 (1952).
251. Fritz, Keen, Anal. Chem., 25, 179 (1953).
252. Fritz, Lisicki, Anal. Chem., 23, 589 (1951).
253. F r i t z, M a r p 1 e, Anal. Chem., 34, 921 (1962).
254. F r i t z, M о у e, Richard, Anal. Chem., 29, 1685 (1957).
255. Fritz, Palmer, Anal. Chem., 33, 98 (1961).
256. Fritz, Pietrzyk, Anal. Chem., 31, 1157 (1959).
257. Fritz, Schenk, Anal. Chem., 31, 1808 (1959).
258. Fritz, Y amamura, Anal. Chem., 29. 1079 (1957).
259. Fritz, Y amamura, Bradford, Anal. Chem., 31, 260 (1959).
260. Fuoss, Krauss, J. Am. Chem. Soc., 55, 2387 (1933).
261. Gagnaux, J anjic, Susz, Helv. Chim. Acta, 41, 1023, 1322 (1958).
262. Г а л ь и e p и н. Б e з и н г е р, Ж. анал. хим. 13, 603 (1958).
263. Gautier, Bull. soc. chim. France, 1960, 1263.
264. Gautier, Pellerin, Ann. pharm. franc., 10, 401 (1952).
265. Gautier, Pellerin, Ann. pharm. franc., 12, 505 (1954).
266. Gautier, Pellerin, Pineau, Ann. pharm. franc., 16, 625 (1958).
267. Gautier, Renault, Pellerin, Ann. pharm. franc., 13, 725 (1955).
268. Gautier, Renault, Pellerin, Ann. pharm. franc., 14, 337 (1956).
269. Gehlen, Z. phys. Chem., 203, 125 (1954).
270. Gillespie, J. Chem. Soc., 1950, 2542.
271. Gillespie, J. Chem. Soc., 1954. 1851.
272. Gillespie, Hughes, Ingold, J. Chem. Soc., 1950, 2473.
273. Glass, Moore, Anal. Chem., 33, 494 (1961).
274. Glenn, Peake, Anal. Chem., 27, 205 (1955).
275. Goddu, Hume, Anal. Chem., 22, 1314 (1950).
276. Goddu, Hume, Anal. Chem., 26, 1679 (1954).
277. Goddu, Hume, Anal. Chem., 26, 1740 (1954).
278. Gold, J efferson, J. Chem. Soc., 1953, 1409.
279. Goldfarb, Mele. Gutstein, J. Am. Chem. Soc., 77, 6194 (1955).
280. Goldstein, Dodge n, Drug. Std., 26, 113 (1958).
281. Goodhue, Hixon, J. Am. Chem. Soc., 56, 1329 (1934); 57, 1688 (1935).
282. Gordy, Stanford, J. Phys. Chem., 8, 170 (1940); 9, 204 (1941).
283. G о г о g, Acta Chim. Hung., 47, 1 (1966).
284. Goto g, To mcsany i, Acta Chim. Hung., 47, 121 (1966).
285. Gran, Althin, Acta Chem. Scand., 4, 967 (1950).
286. Grant, Hunter, Massie, Analyst, 88, 134 (1963).
287. Greenhow, Smith, Analyst, 84, 457 (1959).
288. Gremillion, Anal. Chem., 27, 133 (1955).
289. Грибова, Левин, Завод, лаб., 24, 1356 (1958).
290. Грибова, Левин, Завод, лаб., 25, 38 (1959).
291. Grove, Jeffery, Taianta, 7, 56 (1960).
292. Grunwald, Anal. Chem., 26, 1696 (1954).
293. Grunwald, Anal. Chem., 28, 1112 (1956).
294. Gurney, Ionic Processes in Solutions, McGraw-Hill, New York, 1953.
295. Gutmann, Lindqvist, Z. phys. Chem., 203, 251 (1954).
296. Gutmann, Lindqvist, Angew. Chem., 66, 330 (1954).
297. Gutter son, Ma, Mikrochim. Acta, 1960, 1.
298. Gutterson, Ma, Microchem. J., 5, 601 (1961).
299. G у e n e s, Magyar Kem. Folyoirat, 56, 383 (1950).
300. G у e n e s, Magyar Kem. Folyoirat, 57, 4 (1951).
301. G у e n e s, Magyar Kem. Folyoirat, 59, 12 (1953).
302. G у e n e s, Magyar Kem. Folyoirat, 59, 353 (1953).
303. G у e n e s, Acta. Pharm. Hung., 24, 11 (1954).
304. G у e n e s, Magyar Kem. Lapja, 10, 12 (1955).
305. G у e n e s, Magyar Kem. Folyoirat, 61, 89 (1955).
306. G у n e s, Magyar Kem. Folyoirat, 62, 26 (1956).
307. G у e n e s, Magyar Kem. Folyoirat, 62, 237 (1956).
308. G y’e n e s, Magyar Kem. Folyoirat, 62, 239 (1956).
309. G у e n e s, Magyar Kem. Folyoirat, 62, 242 (1956).
310. G у e n e s, Magyar Kem. Folyoirat, 63, 94 (1957).
311. G у e n e s, Magyar Kem. Folyoirat, 64, 10 (1958).
392
Литература
312. G у е n е s, Magyar Kem. Folyoirat, 65, 264 (1959).
313. G у e n e s, Acta Pharm. Hung., 30, 55 (1960).
314. G у e n e s, Magyar Kem. Folyoirat, 66, 55 (1960).
315. G у e n e s, Acta Chim. Hung., 26, 403 (1961).
316. G у e n e s, Magyar Kem. Folyoirat, 67. 162 (1961).
317. Gyenes, Magyar Kem. Folyoirat, 67, 371 (1961).
318. G у e n e s, Chemie u. Biochemie d. Solanum-Alkaloide, Tagungsberichte No. 27.
Dtsch. Akad. d. Landwirtschaftswissenschaften z. Berlin, 177 (1961).
319. G у e n e s, Acta Pharm. Hung., 32, 241 (1962).
320. G у e n e s, Acta Pharm. Hung., 34. 99 (1964).
321. Gyenes, Laszlo, Magyar Kem. Folyoirat, 67, 166 (1961).
322. Gyenes, Laszlo, Taianta, 10, 567 (1963).
323. Gyenes, Nemeth, Bayer, Acta Pharm. Hung., 27, 23 (1957).
324. Gyenes, Szasz, Magyar Kem. Folyoirat, 61, 356 (1955).
325. Gyenes, Szasz, Изв. АН Каз. ССР, 1, 34 (1961).
326. Gyenes, V a 1 i, Magyar Kem. Folyoirat, 61, 90 (1955).
327. Had zi, Thompson, Hydrogen Bonding, Pergamon Press, London, 1959.
328. Hagen, Leslie, Anal. Chem., 35, 814 (1963).
329. Haggard, Greenberg, Science, 93, 479 (1941).
330. Hahn, Anal. Chim. Acta, 4, 577 (1950).
331. Hahn, Anal. Chim. Acta, 9, 400 (1953).
332. Hall, J. Am. Chem. Soc., 52, 5115 (1930).
333. Hall, Jr., J. Phys. Chem., 60, 63 (1956).
334. Hall, Sprinkle, J. Am. Chem. Soc., 54, 3469 (1932).
335. van Hall Stone, Anal. Chem., 27, 1580 (1955).
336. van Hall Stone, Anal. Chem., 30, 1416 (1958).
337. Hall Werner, J. Am. Chem. Soc., 50, 2367 (1928).
338. Hammett, J. Am. Chem. Soc., 50, 2666 (1928).
339. Hammett, Chem. Revs., 16, 67 (1935).
340. Hammett, Physical Organic Chemistry, McGraw-Hill Book Comp., New York,
1940.
341. Hammett, J. Chem. Phys., 8, 644 (1940)
342. Hammett, Deyrup, J. Am. Chem. Soc.. 54, 2721 (1932).
343. Hammett, Deyrup, J. Am. Chem. Soc., 54, 4239 (1932).
344. Hammett, Deyrup, J. Am. Chem. Soc., 55, 1900 (1933).
345. Hammett, Paul, J. Am. Chem. Soc., 56, 827 (1934).
346. Хэммонд, в книге «Пространственные эффекты в органической химии», под ред.
Ньюмена, ИЛ, М., 1960.
347. Hanna, Siggia, Anal. Chem., 34. 547 (1962).
348. H a n s s о n, Svensk Kem. Tidskr., 67, 256 (1955).
349. Hantzsch, Z. phys. Chem., 61, 257 (1907).
350. Hantzsch, Z. Elektrochem., 29, 221 (1923).
351. Hantzsch, Die Theorie der ionogenen Bindung als Grundlage der lonentheorie,
Verlag Chemie, Leipzig, 1924.
352. Hantzsch, Z. phys. Chem., 63B, 1782, 1789 (1930).
353. Hantzsch, Hilscher, Ber., 41, 1171 (1908).
354. Hantzsch, Langbein, Z. anorg. Chem., 204, 193 (1932).
355. Harlow, Anal. Chem., 34, 148 (1962).
356. Harlow, Anal. Chem., 34, 1482 (1962).
357. Harlow, Anal. Chem., 34, 1487 (1962).
358. Harlow, Bruss, Anal. Chem., 30, 1833 (1958).
359. H а г 1 о w, N о b 1 e, W у 1 d, Anal. Chem., 28, 787 (1956).
360. H а г 1 о w, W у 1 d, Anal. Chem., 30, 69 (1958).
361. Harlow, W у 1 d, Anal. Chem., 34, 172 (1962).
362. H a г n e d, J. Am. Chem. Soc., 60, 336 (1938).
363. H a r t, F i s h, J. Am. Chem. Soc., 82, 5419 (1960).
364. Havemann, Biochem. Z., 310, 378 (1942).
365. Hawke, Steigman, Anal. Chem., 26, 1989 (1954).
366. H adi eke, Howorka, Pharm. Zhalle, 99, 312 (1960).
367. Hadicke, Howorka, Pharm. Zhalle, 100, 3 (1961).
368. Hefferren, Koehler, J. Pharm. Sci., 50, 582 (1961).
369. van der Heijde, Anal. Chim. Acta. 16, 392 (1957).
370. van der Heijde, Anal. Chim. Acta, 17, 512 (1957).
371. van der Heijde, Dahmen, Anal. Chim. Acta, 16, 378 (1957).
372. H e i z, Dansk Tiddskr. Farm., 20, 69 (1952).
373. Hennart, Merlin, Chimie analytique, 39, 269 (1957).
374. H e n n a г t, M e г 1 i n, Chimie analytique, 39, 385 (1957).
375. Hennart, Merlin, Chimie analytique, 40, 20 (1958).
376. Hennart, Merlin, Chimie analytique, 40, 167 (1958).
377. Hennart, Merlin, Chimie analytique, 40, 264 (1958).
Литература
393
378. Hennart, Vieillet, Chimie analytique, 44, 61 (1962).
379. Henry, Hazel, McNabb, Analyt. Chim. Acta, 15, 187 (1956).
380. Hermans, Theoretical Organic Chemistry, Elsevier, Amsterdam (1954).
381. Hess, Haber, Ber., 70, 2205 (1937).
382. H e s t о n, H a 1 1, J. Am. Chem. Soc., 56, 1462 (1934).
383. H i g u c h i, «Application of Lithium Aluminium Hydride to Organic Analyses» in
Mitchell, Kolthoff, Proskauer, Weissberger, Organic Analysis, II, 123 Interscience,
New York, 1954.
384. Higuchi, Barnstein, Anal. Chem., 28, 1022 (1956).
385. Higuchi, Barnstein, Ghassemi, Perez, Anal. Chem., 34, 400 (1962).
386. Higuchi, Concha, J. Am. Pharm. Assoc., Sci. Ed., 40, 173 (1951).
387. Higuchi, Concha, Kuramoto, Anal. Chem., 24, 685 (1952).
388. Higuchi, Connors, J. phys. Chem., 64, 179 (1960).
389. Higuchi, Feldman, Rehm, Anal. Chem., 28, 1120 (1956).
390. Higuchi, Rehm, Barnstein, Anal. Chem., 28, 1506 (1956).
391. H i g u c h i, Z u с к, J. Am. Chem. Soc., 73, 2676 (1951).
392. Hillenbrand, Pentz, «Determination of Amides and Related Compounds»,
in Mitchell, Kolthoff, Proskauer, Weissberger, Organic Analysis, III, Interscience,
New York, 1954.
393. Hine, Physical Organic Chemistry, McGraw-Hill, New York, 1956.
394. Hine, Hine, J. Am. Chem. Soc., 74, 5266 (1952).
395. H i n s w a r k. Stone, Anal. Chem., 28, 334 (1956).
396. Hitchcock, Elving, Anal. Chim. Acta, 27, 501 (1962).
397. Hixon, J о h n s, J. Am. Chem. Soc., 49, 1786 (1927).
398. Hofmann, Chemistry of Heterocyclic Compounds, Vol. IV, Interscience, New York,
1953.
399. Hof stadter, J. Chem. Phys., 6, 540 (1938).
400. Howorka. Hiidicke, Pharm. Zhalle, 98, 538 (1959).
401. Huber, Angew. Chem., 72, 865 (1960).
402. Hughes, Ingold, J. Chem. Soc., 1935, 244.
403. H u i s g e n, В r a d e, Ber., 90, 1432 (1957).
404. Hummelstedt, Hume, Anal. Chem., 32, 576 (1960).
405. Hummelstedt, Hume, Anal. Chem., 32, 1792 (i960).
406. Inczedy, Gimesi, Acta Chim. Hung., 31, 347 (1962).
407. Ingold, Helv. Chim. Acta, 29, 1929 (1946).
408. Иоффе, Сергеева, Ж. анал. хим., 12, 540 (1957).
409. 1 onescu, Enache, Constantinescu, Rev. chim. (Bucharest), 14, 543
(1963).
410. Ishidate, Nishizava, Sano, Horikoshi, J. Pharm. Soc. Japan,
81, 1303, 1307, 1310, 1313 (1961).
411. И з м а й л о в, Ж. анал. хим., 10, 275 (1949).
412. Измайлов, ЖФХ, 24, 321 (1950).
413. Измайлов, ЖФХ, 28, 2047 (1954).
414. Измайлов, Белгова, ЖОХ, 9, 453 (1939).
415. Измайлов, Шустова, Водорез, ЖОХ, 9, 598 (1939).
416. J ackwerth, Specker, Z. anal. Chem., 171, 270 (1959).
417. Jacobs, Analyst, 85, 257 (1960).
418. Jacobs, Scheflan, Chemical Analysis of Industrial Solvents, Interscience,
New York, 1953.
419. Jancik, Korbl, Ceskoslov. Farm., 5, 408 (1956).
420. Jander, Klaus, Z. Elektrochem., 58, 237 (1954).
421. Jander, Klaus, J. Inorg. Nuclear Chem., 1, 228 (1955).
422. Jander, Kraffczyk, Z. anorg. allg. Chem., 282, 121 (1955).
423. Jander, Surawski, Z. Elektrochem., 65, 527 (1961).
424. J anz, Danyluk, J. Am. Chem. Soc., 81, 3846, 3850 (1959).
425. J a s i n s к i, Acta Polon. Pharm., 14, 45 (1957).
426. J a s i n s к i, M i s i a k, Chem. Anal. (Warsaw), 9, 113 (1964).
427. Jaunin, Beretta-Piccoli, Charalam bous, Helv. Chim. Acta, 37,
216 (1954).
428. Jay, Anal, chem., 36, 667 (1964).
429. Джонсон, Органические реакции, т. I, ИЛ, М., 1948, стр. 263—344.
430. J ohnson, Fletcher, Anal. Chem., 31, 1563 (1959).
431. Johnson, Funk, Anal. Chem., 27, 1464 (1955).
432. Johnson, Funk, Anal. Chem., 28, 1977 (1956).
433. Jordan, Chimia, 17, 101 (1963).
434. Jordan, Fischer, Z. anal. Chem., 168, 182 (1959).
435. Josie n, Sourisseau, «Contribution a Г etude des molecules aromatique con-
si deres comme accepteurs de protons», in Hadii, Thompson, Hydrogen Bonding, Per-
gamon Press, London (1959).
436. Jovanovics, Mark, Horompo, Szasz, Acta Pharm. Hung, 34, 36 (1964).
394
Литература
437. К a h a n е, Bull. soc. chim. France, 1951, 92.
438. Kahovec, Knollmuller, Z. phys. Chem., B51, 49 (1941).
439. К a 1 e i s, Aptechnoe Delo, 7, 63 (1958).
440. Karie, Brockway, J. Am. Chem. Soc., 66, 574 (1944).
441. Karrman, Johansson, Mikrochim. Acta, 11, 1573 (1956).
442. К a s h i m a, Bull. Nat. Hyg. Lab. (Tokyo), 72, 145, 151 (1954).
443. Kashima, J. Pharm. Soc. Japan, 74, 1078 (1954).
444. Kashima, Chem. and Pharm. Bull., 6, 229 (1958).
445. Kashima, Syoji, Tsuchiy a, Eisei shikenjo Hokoku, 74, 15 (1956).
446. Katz, Glenn, Anal. Chem., 24, 1157 (1952).
447. Kaufman, Singleterry, J. Phys. Chem., 56, 604 (1952).
448. Kavarana, Am. J. Pharm., 131, 184 (1959).
449. Kaye, Anal. Chem., 27, 292 (1955).
450. К e e n, F r i t z, Anal. Chem., 24, 564 (1952).
451. К e i 1 y, Hume, Anal. Chem., 36, 543 (1964).
452. Keller, Weiss, Pharmazie, 12, 462 (1957).
453. Kern y, Billon, Bigear d, Ann. pharm. franc., 17, 284 (1959).
454. Key worth, J. Org. Chem., 24, 1355 (1959).
455. Kilpatrick, Kilpatrick, Chem. Revs., 13, 131 (1933).
456. Kilpi, Harjanne, Suomen Kemistilehti [B], 21, 14 (1948).
457. Kirrmann, Daune-Dubois, C. R., 236, 1361 (1953).
458. Kirwood, Westheimer, J. Chem. Phys., 6, 506, 513 (1938).
459. Kleckner, Osol, J. Am. Pharm. Ass., Sci., Ed., 41, 573 (1952).
460. Klotz, Fiess, Ho, Mellody, J. Am. Chem. Soc., 76, 5136 (1954).
461. Knapp e, Peter i, Z. anal. Chem., 194, 417 (1963).
462. Rolling, J. Am. Chem. Soc., 79, 2717 (1957).
463. Rolling, Smith, Anal. Chem., 31, 1876 (1959).
464. Kolthoff, Bruckenstein, J. Am. Chem. Soc., 79, 1 (1957); 78, 1 (1956).
465. Kolthoff, Bruckenstein, Chantooni, J. Am. Chem. Soc., 83, 3927
(1961).
466. Kolthoff, Chantooni, J. Am. Chem. Soc., 85, 2196 (1963).
467. Kolthoff, Coetzee, J. Am. Chem. Soc., 79, 870, 1852, 6110 (1957).
468. Kolthoff, Ikeda, J. Phys. Chem., 65, 1020 (1961).
469. Kolthoff, Stocesoca, Lee, J. Am. Chem. Soc., 75, 1834 (1953).
470. Kolthoff, Willman n, J. Am. Chem. Soc., 56, 1007, 1014 (1934).
471. Kraimarova, Kracmar, Ceskoslov. Farm., 7, 566 ((1958).
472. Kraus, The Properties of Electrically Conducting Systems, Chemical Catalog Co.,
1922.
473. Kraus z, Havas, Magyar Kem. Folyoirat, 70, 104 (1964).
474. Крешков, Алдарова, Труды комиссии по анал. химии АН СССР,13, 315
(1963).
475. Крешков, Алдарова, Ж. анал. хим., 19, 537 (1964).
476. Крешков, Борк, Апаршева, Ж. анал. хим. 18, 1149 (1963).
477. Крешков, Быкова, М хит а рьян, Ж. анал. хим., 14, 529 (1959).
478. Крешков, Быкова, Русакова, Казарьян, Завод, лаб., 28, 11 (1962).
479. Крешков, Быкова, Шемет, Ж. анал. хим. 16, 331 (1961).
480. Крешков, Быкова, Смолова, Ж. анал. хим., 19, 156 (1964).
481. Крешков, Дроздов, ДАН СССР, 131, 1345(1960).
482. Крешков, Дроздов, Изв. высш, учебн. заведений, Химия и химическая
технология, 3, 85 (I960).
483. Крешков, Дроздов, Власова, Завод, лаб., 26, 108 (1960).
484. Крешков, Васильев, Ж. анал. хим., 17, 908 (1962).
485. Крешков, Яровенко, Зельманова, Ж. анал. хим., 17, 780 (1962).
486. Kukin, Anal. Chem., 29, 461 (1957).
487. Kukin, Anal. Chem., 30, 1114 (1958).
488. К u m 1 e r, S a h, J. Org. Chem., 18, 669 (1953).
489. К u n d u, Das, Anal. Chem., 31, 1358 (1959).
490. Kunkel, Buckley, Gorin, Anal. Chem., 31, 1098 (1959).
491. Kiihni, Grossglauser, Richard, Stump, Pharm. Acta Helv., 29, 104.
(1954).
492. Lada, Rocznicki Chem., 29, 950 (1955).
493. Laitinen, Wawzonek, J. Am. Chem. Soc., 64, 1765 (1942).
494. Lande e, J ohns, J. Am. Chem., Soc., 63, 2895 (1941).
495. Lane, Analyst, 80, 675 (1955).
496. Lang, Tavaszy, Z. anal. Chem., 158, 339 (1957).
497. Lange, Handbook of Chemistry, McGraw-Hill Book Company Inc., New York, 1961.
498. Langford, Burwell, J. Am. Chem. Soc., 82, 1503 (I960).
499. Lapidus, Lucas, Tremillon, Bull. soc. chim. France, 1960, 1849.
500. Latimer, Rodebush, J. Am. Chem. Soc., 42, 1430 (1920).
501. Laubengayer, Sears, J. Am. Chem. Soc., 67, 164 (1945).
Литература
395
502. Lavine, Toennies, J. Biol. Chem., 101, 727, 732 (1933).
503. Leader, Gormley, J. Am. Chem. Soc., 73, 5731 (1951).
504. Leavitt, Autian, Drug. Std., 26, 33 (1958).
505. Lemaire, Lucas, J. Am. Chem. Soc., 73, 5198 (1951).
506. Leuts, Wagner, Chemist Analyst, 50, 43 (1961).
507. Levi, Chatten, Pemarowski, J. Am. Pharm. Ass.. Sci. Ed., 44, 61 (1955).
508. Левин, Костин, ЖОХ, 23, 1054 (1953).
509. Lewis, J. Franklin Inst., 226, 293 (1938).
510. Lewis, Seaborg, J. Am. Chem. Soc., 61, 1886 (1939).
511. Lieber, Ramachandra Rao, Tai Siang Chao, Anal. Chem., 29,
932 (1957).
512. Linek, Peciar, Chem. Zvesti, 16, 692 (1962).
513. Lintner, Schleif, Higuchi, Anal. Chem., 22, 534 (1950).
514. Lintner, Zuck, Higuchi, J. Am. Pharm. Ass., Sci. Ed., 39, 418 (1950).
515. Lippinkott, Timnick, Anal. Chem., 28, 1690 (1956).
516. Lipscomb, Baker, J. Am. Chem. Soc., 64, 179 (1942).
517. Liteanu, Cormos, Taianta, 7, 32 (1960).
518. Liu, Reynolds, Anal. Chem., 34, 542 (1962).
519. Lohr, Anal. Chem., 32, 1166 (1960).
520. van Loo y, Hammett, J. Am. Chem. Soc., 81, 3872 (1959).
521. Lorenz, Parks, Anal. Chem., 34, 394 (1962).
522. Lowry, Chem. and Ind., 42, 43 (1923).
523. Lowry, Chem. Revs., 5, 231 (1928).
524. Lobe r, Hesse, Pharmazie, 14, 214 (1959).
525. L о r i n c z, Magyar Kem. Folyoirat, 67, 414 (1961).
526. Lorincz, Szasz, Acta Pharm. Hung., 33, 84 (1963).
527. Lucchesi, H irn, Chemist Analyst, 50, 109 (1961).
528. Luder, Zuffanti, The Electronic Theory of Acids and Bases, Wiley and Sons,
New York, 1946.
529. L w о w s к y, Angew. Chem., 70, 483 (1958).
530. Lvkken, Porter, Ruliffson, Tuemler, Ind. Eng. Chem. Anal. Ed.,
16; 219 (1944).
531. Mackensie, Winter, Trans. Faraday Soc., 44, 159, 171, 243 (1948).
532. Magnuson, Ana], Chem., 35, 1487 (1963).
533. Magnuson, Chemist Analyst, 53, 15 (1964).
534. Maher, Y ohe, J. Org. Chem., 23, 1082 (1958).
535. M a 1 1 i k, Anal. Chem., 32, 1369 (1960).
536. M a 1 1 i k, Das, Chem. and Ind., 1959, 162.
537. Malmstadt, Roberts, Anal. Chem., 28, 1408 (1956).
538. Maltmstadt, Vassallo, Anal. Chim. Acta, 16, 455 (1957).
539. Malmstadt, Vassallo, Anal. Chem., 31, 206 (1959).
540. Malmstadt, Vassallo, Anal. Chem., 31, 862 (1959).
541. Malone, Anal. Chem., 33, 575 (1961).
542. Malone, Biggers, Anal. Chem., 36, 1037 (1964).
543. Малышев, Завод, лаб., 28, 927 (1962).
544. Mandel, Nature, 176, 792 (1955).
545. Mandel, Anal. Chim. Acta, 15, 301 (1956).
546. Mandel, Ind. chim. Beige, 23, 722 (1958).
547. Mansfield, Whiting, J. Chem. Soc., 1956, 4761.
548. Markunas, Riddick, Anal. Chem., 23, 337 (1951).
549. Markunas, Riddick, Anal. Chem., 24, 312 (1952).
550. Marple, Fritz, Anal. Chem., 34, 796 (1962).
551. Marple, Fritz, Anal. Chem., 35, 1223, 1305, 1431 (1963).
552. Marple, Matsuyama, Burdett, Anal. Chem., 30, 937 (1958).
553. Marple, Przybylowicz, Hume, Anal. Chem., 28, 1892 (1956).
554. Martin, Anal. Chem., 29, 79 (1957).
555. Martin, George, J. Chem. Soc., 1933, 1413.
556. Martin, Reece, J. Chem. Soc., 1960, 4697.
557. Marvillet, Trenchant, Chimie Analytique, 40, 293 (1958).
558. Marvillet, Tranchant, Chimie Analytique, 43, 169 (1961).
559a.M а г у о 11, J. Res. Nat. Bur. Stand., 38, 527 (1947).
559б. M а г у о 11, J. Res. Nat. Bur. Stand., 41, 1 (1948).
560. Maryott, Smith, Nat. Bur. Stand. Circ., 514 (1951).
561. Mass, J ander, Forschr. chem. Forsch., 2, 637 (1953).
562. M auermey er, Margosis, Ma, Mikrochim. Acta, 2, 177 (1959).
563. M a z о г, P a p a y, Z. anal. Chem., 184, 272 (1961).
564. McClure, Ro d er, Kinsey, Anal. Chem., 27, 1599 (1955).
565. McCurdy, Galt, Anal. Chem., 30, 940 (1958).
566. M с E n i r y, Ass. Agric. Chem., 40, 926 (1957).
567. M с E w e n, J. Am. Chem. Soc., 58, 1124 (1936).
396
Литература
568. McGuire, Izzo, Zuffanti, J. Org. Chem., 21, 632 (1956).
569. M с I n n e s, J о n e s, J. Am. Chem. Soc., 48, 2831 (1926).
570. McKinney, Reynolds, Taianta, 1, 46 (1958).
571. Mecke, Z. Elektrochem., 50, 57 (1944).
572. Meerwein, Hinz, Hofmann, Kroning, Pfeil, J. pract. Chem., 147,
257 (1937).
573. Merrifield, Phillips, J. Am. Chem. Soc., 80, 2778 (1958).
574. Metcalfe, Schmitz, Anal. Chem., 27, 138 (1955).
575. Meulenhoff, Pharm. Weekblad, 93, 262 (1958).
576. Meulenhoff, van Sonsbeek, Pharm. Weekblad, 91, 453 (1956).
577. van Meurs, Dahmen, Anal. Chim. Acta, 19, 64 (1958).
578. van Meurs, Dahmen, Anal. Chim. Acta, 21, 10 (1959).
579. van Meurs, Dahmen, Anal. Chim. Acta, 21, 193 (1959).
580. van Meurs, Dahmen, Anal. Chim. Acta, 21, 443 (1959).
581. Michaelis, Pharmazie, 13, 740 (1958).
582. Michaelis, Ron a, Biochem. Z., 49, 247 (1913).
583. Mika, Z. anal. Chem., 128, 159 (1948).
584. Mika, Die Methoden der Mikromassanalyse, F. Enke, Stuttgart, 1958.
585. Miller, Wynne-J ones, J. Chem. Soc., 1959, 2375.
586. M i 1 n e, J. Am. Pharm. Ass., Sci. Ed., 48, 117 (1959).
587. Milne, Chatten, J. Pharm. Pharmacol., 9, 686 (1957).
588. M inczewski, Chem. Anal. (Warsaw), 3, 453 (1958).
589. Minczewski, Zh. anal. Khim., 15, 151 (1960).
590. Minczewski, Kiciak, Chem. Anal. (Warsaw), 8, 239 (1963).
591. Minczewski, Kolyga, Chem. Anal. (Warsaw), 3, 463 (1958).
592. Minczewski, Lada, Roczniki Chem., 29, 919, 950 (1955).
593. Minnick, Kilpatrick, J. Phys. Chem., 43, 259 (1939).
594. Miocque, Gautier, Bull. soc. chim. France, 1958, 467.
595. Miron, Hercules, Anal. Chem., 33, 1770 (1961).
596. Mitchel, Ashby, J. Am. Chem. Soc., 67, 161 (1945).
597. Mohler, Chemische Optic, Sauerliinder, Aarau, 1951.
598. M о о r e, J о h n s, J. Am. Chem. Soc., 63, 3336 (1941).
599. Moore, McCutchan, Y oung, Anal. Chem., 23, 1633 (1951).
600. Moskalyk, Chatten, Pernarowski, J. pharm. Sci., 50, 179 (1961).
601. Moss, Elliott, Hall, Anal. Chem., 20, 784 (1948).
602. Mulliken, J. Am. Chem. Soc., 74, 811 (1952).
603. Muney, Coetzee, J. Phys. Chem., 66, 89 (1962).
604. Muth, Darla k, English, Hamner, Anal. Chem., 34, 1163 (1962).
605. Мюллер E., Новые воззрения в органической химии, ИЛ, М., 1960.
606. Miiller, Patridge, Ind. Eng. Chem., 20, 423 (1928).
607. Nadeau, Branchen, J. Am. Chem. Soc., 57, 1363 (1935).
608. N a i du, Krishnan, Anal. Chem., 33, 497 (1961).
609. Napoli, Schmall, Anal. Chem., 23, 1893 (1951).
610. N e m i t z, Chem. Z., 82, 222 (1958).
611. N e w m a n, J. Am. Chem. Soc., 63, 2431 (1941).
612. Nicksic, J udd, Anal. Chem., 32, 998 (1960).
613. Nicolas, Burel, Chimie analytique, 33, 341 (1951).
614. Nisonoff, Kuhn, Gurney, J. Chem. Phys., 20, 985 (1952).
615. Novak, Chem. listy, 49, 848, 934 (1955).
616. Ogg, Porter, Willits, Ind. Eng. Chem. Anal. Ed., 17, 394 (1945).
617. Osborn, Elliott, Martin, Ind. Eng. Chem. Anal. Ed., 15, 642 (1943).
618. Ozolins, Schenk, Anal. Chim., 33, 1035 (1961).
619. Omboly, D erz si, Z. Anal. Chem., 187, 29 (1962).
620. Omboly, Derzsi, Acta Pharm. Hung., 32, 246 (1962).
621. P a 1 i t, Ind. Eng. Chem. Anal. Ed., 18, 246 (1946).
622. Palit, Singh, J. Indian Chem. Soc., 33, 507 (1956).
623. P a tchornik, Rogozinski, Anal. Chem., 31, 985 (1959).
624. Patchornik, Rogozinski, Anal. Chem., 33, 803 (1961).
625. Paul, Long, Chem. Revs., 57, 1 (1957).
626. Paul, Singh, Sandhu, J. Chem. Soc., 1959, 315.
627. Paul, Vasisht, Malhotra, Pahil, Anal. Chem., 34, 820 (1962).
628. П а у л и н г Л., Природа химической связи, Госхимиздат, М.-Л., 1947.
629. Pearson, J. Am. Chem. Soc., 70, 204 (1948).
630. Pearson, Dillon, J. Am. Chem. Soc., 75, 2439 (1953).
631. Pearson, Vogelsong, J. Am. Chem. Soc., 80, 1038 (1958).
632. Pellerin, Ann. pharm. franc., 14, 193 (1956).
633. Pellerin, Demay, Ann. pharm. franc., 20, 661 (1962).
634. D ellerin. Gautier, Ann. pharm. franc., 19, 81 (1961).
635. Pena u, Saias, Ferdet, Ann. pharm. franc., 11, 740 (1953).
636. Pernarowski, Drug. St., 21, 189 (1953).
Литература
397
637. Pernaro wski, Drug St., 22, 1 (1954).
638. Pernaro wski, Blackburn, J. Am. Pharm. Ass., Sci. Ed., 47, 585 (1958).
639. Pernaro wski, Chatten, Levi, J. Am. Pharm. Ass., Sci. Ed., 43, 746 (1954).
640. P e s e z, Bull. soc. chim. France, 1954, 1237.
641. P e s e z, Bull. soc. chim. France, 1957, 417.
642. Pickard, Iddings, Anal. Chem., 31, 1229 (1959).
643. P i f e r, W о 1 1 i s h, J. Am. Pharm. Ass., Sci. Ed., 40, 609 (1951).
644. P i f e r, W о 1 1 i s h, Anal. Chem., 24, 300 (1952).
645. P i f e r, W о 1 1 i s h, Anal. Chem., 24, 519 (1952).
646. Pifer, Wollish, Schmall, J. Am. Pharm. Ass., Sci. Ed., 42, 502 (1953).
647. Pifer, Wollish, Schmall, Anal. Chem., 26, 215 (1954).
648. Плешков, ЖФХ, 22, 351 (1948).
649. Poethke, Horn, Pharm. Zhalle, 94, 41 (1955).
650. Pohl, Anal. Chem., 26, 1614 (1954).
651. Pohloudek-Fabini, Konig, Pharmazie, 13, 753 (1958).
652. P ohloud er-F a bin i, Konig, Pharm. Zhalle, 98, 176 (1959).
653. Polgar. Jungnickel, «Determination of Olefinic Unsaturation» in Mitchell,
Kolthoff, Praskauer, Weissberger. Organic Analysis, Vol. Ill, Interscience, New York,
1954.
654. P о s g a y, Pharm. Zhalle, 101, 471 (1962).
655. P о s g a y, Pharm. Zhalle, 101, 534 (1962).
656. P о s g a y, Bayer, Acta Pharm. Hung., 33, 9 (1963).
657. Posgay, Hoehmann, Acta Pharm. Hung., 27, 263 (1957).
658. Poulos, Anal. Chem., 24, 1858 (1952).
659. Powers, Day, Underwood, Anal. Chem., 30, 254 (1958).
660. Prey, Unger, Osterr. Chem. Ztg., 61, 10 (1960).
661. Pringle, Nature, 183, 815 (1959).
662. Prue, Sherrington, Trans. Faraday Soc., 57, 1795 (1961).
663. Przyszlakovski, Acta Polon. Pharm., 17, 395 (1960).
664. P u r d y, S t о c k, Drug. St., 26, 177 (1958).
665. R a dell, Donahue, Anal. Chem., 26, 590 (1954).
666a.R а о, M u r t h y, Z. analyt. Chem., 177, 86 (1960).
6666.R ao, Murth y, Z. analyt. Chem., 180, 169 (1961).
666b.R а о, M u r t h y, Z. analyt. Chem., 187, 97 (1962).
666r.R а о, M u r t h y, Z. analyt. Chem., 195, 406 (1963).
667. Rashbrook, Analyst, 87, 826 (1962).
668. R e d d a w a y, Analyst, 82, 506 (1957).
669. Rea d,Critchfield, Elder, Anal. Chem., 35, 571 (1963).
670. Rehm, Bodin, Connors, Higuchi, Anal. Chem., 31, 483 (1959).
671. Rehm, Higuchi, Anal. Chem., 29, 367 (1957).
672. Reilley. Crawford, Anal. Chem., 26, 716 (1955).
673. R eilley. Papa, Anal. Chem., 34, 801 (1962).
674. Reilley, Porterfield, Anal. Chem., 28, 443 (1956).
675. Reilley, Schweizer, Anal. Chem., 26, 1124 (1954).
676. R e i s s, Z. anal. Chem., 167, 16 (1959).
677. P e ми к А., Электронные представления в органической химии, ИЛ, М., 1950.
678. Reynolds, Little, Pattengill, Anal. Chem., 35, 973 (1963).
679. Reynolds, Walker, Cochran, Anal. Chem., 32, 983 (I960).
680. Richardson, Anal. Chim. Acta, 24. 46 (1961).
681. Rink, Lux, Deut. Apoth. Ztg., 99, 1051 (1959).
682. Rink, L u x, F r a n k e n, Deutsch. Apoth. Zt., 99, 157 (1959).
683. R i n k, R i e m h о f e r, in Arch. Pharm., 294 (1961), Mitteilungen d. deutsch. pharm.
Ges., 31 [11], 197.
684. Rink, Riemhofer, Deutsch. Apoth. Ztg., 102, 1567 (1962).
685. R i о 1 о, M а г с о n, Ann. Chim. (Rome), 46, 1121 (1956).
686. Roberts, McBee, Hathaway, J. Org. Chem., 21, 1369 (1956).
687. Robinson, Cundiff, Markunas, Anal. Chem., 33, 1030 (1961).
688. Robinson, Sensabaugh, Markunas, Anal. Chem., 35, 770 (1963).
689. Romberg. Cruze, Z. Elektrochem., 63, 404 (1959).
690. R u c h, С r i t c h f i e 1 d. Anal. Chem., 33, 1569 (1961).
691. Ruch, Johnson, Critchfield, Anal. Chem., 33, 1566 (1961).
692. R u e h 1 e, Ind. Eng. Chem. Anal. Ed.. 10, 130 (1938).
693. R u m p f. Chimie Analytique, 42. Ill (1960).
694. R у a n, Y a no wsk i, P i f er, J. Am. Pharm. Ass., Sci. Ed., 43, 656 (1954).
695. Safai i k, Ceskoslov. farm., 6, 515 (1957).
696. S a f а г i k, Mikrochim. et Ichnoanalyt. Acta, 1963 (1), 26.
697. Saf ar ik, В u m b a, Pharm. Zhalle, 96, 3 (1957).
698. S afarik, S p i n k о v a, J^eskoslov. farm., 7, 76 (1958).
699. § a f a i; i k, § pinkova, Ceskoslov. farm., 7. 144 (1958).
700. Sakurai, J. Pharm. Soc. Japan. 81, 164 (1961).
398
Литература
701. Sakurai, Awad a, J. Pharm. Soc. Japan, 76, 1026 (1956).
702. Sakurai, Y oneda, Nomura, J. Pharm. Soc. Japan, 78, 377 (1959).
703. Salvesen, Medd. Norsk Farm. Selskap., 19, 199 (1957).
704. Salvesen, Medd. Norsk Farm. Selskap., 20, 21 (1958).
705. Salvesen, Medd. Norsk Farm. Selskap., 20, 65 (1958).
706. Salvesen, Medd. Norsk Farm. Selskap., 23, 141 (1961).
707. Salvesen, Kristoffersen, Aasbo, Medd. Norsk Farm. Selskap., 18,
88 (1956).
708. Salvesen, Solli, Medd. Norsk Farm. Selskap., 21, 85 (1959).
709. Sandin, Kulk a, Woolley, Ind. Eng. Chem. Anal. Ed., 8, 355 (1936).
710. S a r s о n, Anal. Chem., 30, 932 (1958).
711. Satchel 1, J. Chem. Soc., 1958, 3910.
712. Saunders, Stacey, Wilding, Biochem. J., 36, 368 (1942).
713. Scarano, Ceglie, Anal. Chim. Acta, 12, 292 (1955).
714. Schaefgen, Flory, J. Am. Chem. Soc., 72, 689 (1950).
715. Schenk, Anal. Chem., 33, 299 (1961).
716. Schenk, Fritz, Anal. Chem., 32, 987 (1960).
717. Schenk, Ozolins, Anal. Chem., 33, 1562 (1961).
718. Schenk, Santiago, Wines, Anal. Chem., 35, 167 (1963).
719. Schenk, Wines, M о j z i s, Anal. Chem., 36, 914 (1964).
720. S c h 1 i 11, Anal. Chem., 35, 1063 (1963).
721. Schmall, Pifer, Wollish, Anal. Chem., 24, 1446 (1952).
722. Schmidt, Blohm, Jander, Angew. Chem., 59, 233 (1947).
723. Schnell, Makromol. Chem., 2, 172 (1948).
724. Schott e, Veibel, Acta Chem. Scand., 7, 1357 (1953).
725. Schwabe, Elektrometrische pH-Messung unter extremen Bedingungen, Verlag
Chemie, Weinheim/Bergstr., 1960.
726. Schwarzenbach, Brandenberger, Ott, Hagger, Helv. Chim.
Acta, 20, 490 (1937).
727. Schwarzenbach, Egli, Helv. Chim. Acta, 17, 1176, 1183 (1934).
728. Schwarzenbach, Stensby, Helv. Chim. Acta. 42, 2342 (1959).
729. Schwarzenbach, Wittwer, Helv. Chim. Acta, 30, 656, 659, 663, 669 (1947).
730. Seaman n, Allen, Anal. Chem., 23, 592 (1951).
731. Sensabaugh, Cundiff, Markunas, Anal. Chem., 30, 1445 (1958).
732. Sensi, Gallo, Ann. Chim. (Rome), 43, 453 (1953).
733. Shain, Svodoba, Anal. Chem., 31, 1857 (1959).
734. Sheldon, Tyree, J. Am. Chem. Soc., 81, 2290 (1959).
735. Ш ко дин, Ж0Х, 10, 1694 (1940).
736. Шкодин, Измайлов, ЖОХ, 20, 38 (1950).
737a.Ill кодин, Измайлов, Дзюба, ЖОХ, 20, 1999 (1950).
7376.Ш кодин, Измайлов, Дзюба, Ж. анал. хим., 6, 273 (1951).
737в.Ш кодин, Измайлов, Дзюба, ЖОХ, 23, 27 (1953).
737г.Ш кодин, Измайлов, Дзюба, Укр. хим. журнал, 20, 595 (1954).
738. Шкодин, Каркузаки, Клименко, ЖОХ, 27, 29 (1957).
739. Шкодин, Садовникова, Панченко, Ж. анал. хим., 17, 540 (1962).
740. Штпфман, Ларикова, Труды комиссии по анал. химии АН СССР, 13, 325
(1963).
741. S i d е г i, О s о 1, J. Am. Pharm. Ass., Sci. Ed., 42, 586 (1953).
742. S i d e r i, О s о 1, J. Am. Pharm. Ass., Sci. Ed., 42, 688 (1953).
743. S i d e r i, О s о 1, J. Am. Pharm. Ass., Sci. Ed., 44, 759 (1955).
744. S i d g w i c k, J. Chem. Soc., 127, 907 (1925).
745. Sidgwick, Taylor. Baker, The Organic Chemistry of Nitrogen, Second ed.,
Oxford University Press, 1937.
746. Siggia, Floramo, Anal. Chem., 25, 797 (1953).
747. Siggia, Hanna, Ind. Eng. Chem. Anal. Ed., 20, 1084 (1948).
748. Siggia, Hanna, Anal. Chem., 23, 1717 (1951).
749. Siggia, Hanna, Anal. Chem., 33, 896 (1961).
750. Siggia, Hanna, Ker venski, Anal. Chem., 22, 1295 (1950).
751. Siggia, Segal, Anal. Chem., 25, 830 (1953).
752. Siggia, Stahl, Anal. Chem., 27, 1975 (1955).
753. Siggia, Stahl, Anal. Chem., 28, 1971 (1956).
754. Simon, Kovats, Chopard-dit - J ean, Heilbronner, Helv. Chim.
Acta, 37, 1872 (1954).
755. Simony i, Tokar, Magyar Kem. Folyoirat, 64, 151 (1958).
756. Simony i, Tokar. Acta Chim. Hung., 25, 305 (1960).
757. Simony i, Toth, Szabo, Acta Pharm. Hung., 30, 129 (1960).
758. Singh, Paul, Sandhu, J. Chem. Soc., 1959, 845.
759. Sinha, Kulkarni, Rao, Anal. Chem., 36, 894 (1964).
760. Skoog, Bartlett, Anal. Chem., 27, 369 (1955).
761. Small, Analyst, 84. 117 (1959).
Литература
399
762. Smith, Bryant, J. Am. Chem. Soc.. 58, 2452 (1936).
763. Smith, Elliott, J. Am. Chem. Soc., 75, 3566 (1953).
764. Smith, Haglund, Acta Chim. Scand., 14, 1349 (1960).
765. Smith, Hammett, J. Am. Chem. Soc., 67, 23 (1945).
766. S о b c z у k, «Dielektrische Polarisation und die Wechselwirkung der Carbonsaure
und Amine» in Hadzi, Thompson, Hydrogen Bonding, Pergamon Press, p. 323, 1959.
767. Sokolowski, Kolk a, Chem. Anal. (Warsaw), 6, 429 (1961).
768. Soloway, Rosen, Anal. Chem., 29, 1820 (1957).
769. Spengler, Kaelin, Pharm. Acta Helv., 18, 542 (1943).
770. Sprengling, J. Am. Chem. Soc., 76, 1190 (1954).
771. Stanford, Gordy, J. Am. Chem. Soc., 63, 1094 (1941).
772. Stetzler, Smullin. Anal. Chem., 34, 194 (1962).
773. Stock, Purdy, Chem. Revs., 57, 1159 (1957).
774a.S tock, Purdy, Lab. Pract., 7, 523 (1958).
7746 .S t о о k, Purdy, Lab. Pract., 11, 116 (1962).
775. Stock, Purdy, Chemist-Analyst, 48, 22, 50, 55 (1959).
776. Stock, Purdy, Lab. Pract., 11, 21 (1962).
777. S t r e u 1 i, Anal. Chem., 27, 1827 (1955).
778. S t r e u 1 i, Anal. Chem., 30, 997 (1958).
779. S t r e u 1 i, Anal. Chem., 31, 1652 (1959).
780. S t r e u 1 i, Anal. Chem., 32, 407 (1960).
781. S t r e u 1 i, Anal. Chem., 32, 985 (I960).
782. S t r e u 1 i, M i г о n, Anal. Chem., 30, 1978 (1958).
783. S t u c k, Z. anal. Chem., 177, 338 (1960).
784. Stumpf, Chemie und Anwendung des 1,4-Dioxans, Verlag Chemie, Weinheim/Ber-
gstr., 1956.
785. Suchomelova, Z у к a, J. Electroanal. Chem., 5, 57 (1963).
786. Susz, Lachavanne, Helv. Chim. Acta, 41, 634 (1958).
787. Svoboda, Anal. Chem., 33, 1638 (1961).
788. Swartz, Foss, J. Am. Pharm. Ass., Sci. Ed., 44, 217 (1955).
789. Sweetser, Bicker, Anal. Chem., 25, 253 (1953).
790. Szasz, Kovats, Karacsony, Lorincz, Bayer, Planta Medica, 7,
234 (1959).
791. Szasz, Szporny, Bittner, Gyenes, Havel, Mag 6, Magyar Kem.
Folyoirat, 64, 296 (1958).
792. S z e n d e y, Arch. Pharm., 291, 215 (1958).
793. Szendey, J. Pharm. Sci., 51, 906 (1962).
794. Szendey, Arch. Pharm., 296, 126 (1963).
795. Takacsi-N agy, Luka ts, III. Gyogyszeresz Nagygyiiles, Budapest, 1959.
796. Takiguchi, Analyst, 83, 482 (1958).
797. Takiguchi, Hirata, J. Chem. Soc. Japan, Ind. Chem. Sect., 62, 527 (1959).
798. Takuira, Takino, J. Pharm. Soc. Japan, 74, 971 (1954).
799. Терентьев, Обтемперанская, Б у з л а н о в а, В л а с о в а, Ж. анал. хим.,
17, 900 (1962).
800. Терентьев, Обтемперанская, Володзко, Бузланова, Ж.
анал. хим., 19, 135 (1964).
801. Terjesen, Sandved, Kgl. Norske Videnskab. Selskabs Skrifter, 7, 20 (1938).
802. Terry, Eilar, Moe, Anal. Chem., 24, 313 (1952).
803. Те ze, Schaal, Bull. soc. chim. France, 1962, 1372.
804. T h e i w a g t, Anal. Chem., 33, 1391 (1961).
805. Thomas, Rochow, J. Am. Chem. Soc., 79, 1843 (1957).
806. T i n g 1 e, J. Am. Chem. Soc., 40, 873 (1918).
807. T i w a r i, S r i s t a v a, S h a r m a, Z. anal. Chem., 187, 161 (1962).
808. T i w a r i, Sharma, Z. anal. Chem., 195, 267 (1963).
809. Toennies, Callan, J. Biol. Chem., 125, 259 (1938).
810. Tokar, Simony i, Magyar Kem. Folyoirat, 64, 94, 379 (1958).
811. Toldy, Csillag, Bobak, Gyenes, Magyar Kem. Folyoirat, 67, 180 (I960).
812. Tomi cek, Sb. Celostatni Pracovni Konf. Anal. Chemiku, 1, 246 (1952).
813. Tomice k, Dolezal, Acta Pharm. Internet., 1, 31 (1950).
814. Tomi cek, Heyrovsky, Coll. Czech., 15, 997 (1950).
815. Tomice k, Krepelka, Chem. listy, 47, 526 (1953).
816. Tomice k, Stodolova, Herman, Chem. listy, 47, 516 (1953).
817. Tomice k, Valch a, Coll. Czech., 16, 113 (1951).
818. Treffers, Hammett, J. Am. Chem. Soc., 59, 1708 (1937).
819. Tremillon, Bull. soc. chim. France, 1960, 1940.
820. Trotman-Dickenson, J. Chem. Soc., 1949, 1293.
821. T r u s e 1 1, D i e h 1, Anal. Chem., 35, 674 (1963).
822. Tuthill, Rolling, Roberts, Anal. Chem., 32, 1678 (1960).
823. Uhrig, Levin, Ing. Eng. Chem. Anal. Ed., 13, 90, 194, (1941).
824. Underwood, Anal. Chem., 26, 1322 (1954).
400
Литература
825.
826.
827.
828.
829.
830.
831.
832.
833.
834.
835.
836.
837.
838.
839.
840.
841.
842.
843.
844.
845.
846.
847.
848.
849.
850.
851.
У санович, ЖОХ, 3, 443 (1932).
У санович, ЖОХ, 9, 182 (1939).
У санович, Что такое кислоты и основания, Алма-Ата (1953).
У санович, Дулова, ЖОХ, 16, 1978 (1946).
У санович, Дулова, ЖОХ, 17, 669 (1947).
У санович, Калабановская, ЖОХ, 17, 1235 (1947).
Усанович, Я ц и м iivp с к и й, ЖОХ, 11, 954, 957, 959 (1941).
Vacek, Kracmar, Ceskoslov. Farm., 5, 80 (1956).
Vaillant, Chemie Analytique, 39, 413 (1957).
Vasiliev, Mangu, Sisman, Rev. Chim. (Bucharest), 12, 231 (1961).
Vasiliev, Pazarina, Sisman, Rev. Chim. (Bucharest), 15, 163 (1964).
Vasiliev, Scintee-Pazarina, Sisman, Rev. Chim. (Bucharest),
14, 352 (1963).
Vasiliev, Sisman, Scintee-Pazarina, Rev. Chim. (Bucharest),
15, 46 (1964).
V e i b e 1, Chim. Anal. (Warsaw), 36, 145 (1954).
V e i b e 1, Chem. Anal., 3, 207 (1958).
Veibel, Eggersen, Linholt, Acta Chem. Scand., 6, 1066 (1952).
Veibel, Eggersen, Linholt. Acta Chem. Scand., 8, 768 (1954).
Veibel, Krogh-Andersen, Anal. Chem. Acta, 15, 15 (1956).
Veibel, Nielsen, Acta Chem. Scand., 10, 1488 (1956).
Veibel, Nielsen, Refn, Acta Chem. Scand., 12, 934 (1958).
Veibel, Vrang, Dansk Tiddskr. Farm., 17, 112 (1943).
Vespe, Fritz, J. Am. Pharm. Ass., Sci. Ed., 41, 197 (1952).
V i b e r g, Phys. Chem., A 171, 1 (1934).
Vincent, Blake, J. Am. Pharm. Ass., Sci. Ed., 48, 359 (1949).
Vogelsong, Univ. Microfilms, Ann. Arbor, Mich. Publ. No. 19 605; ref.: Rid-
dick, Anal. Chem., 30, 793 (1958).
Vogelzang, Stover, Pharm. Weekblad, 93, 550 (1958).
V о 1 m e r, Annalen, 440, 200 (1924).
852. Vorlander, Ber., 37, 1485 (1903).
853. W a d e 1 i n, Taianta, 10, 97 (1963).
854. W a g n e г, В г о w n, P e t e r s, J. Am. Chem. Soc., 69, 2609 (1947).
855. Walden, Elestrochemie nichtwassriger Losungen, Johann Ambrosius Barth, Lei-
pzig, 1924.
856. Walden. Audrieth, Birr, Z. Phys. Chem., 160, 337 (1932).
857. W a 1 d e n, В i r r, Z. Phys. Chem., 144, 269 (1929); 153, 1 (1931).
858. W a 1 t z, T а у 1 о r, Anal. Chem., 19, 448 (1947).
859. Wanzlick, Lochel, Ber., 86, 1463 (1953).
860. Warner, Haskell, Anal. Chem., 26, 770 (1954).
861. Wasserman n, J. Chem. Soc., 1959, 986.
862. Wasserman n, Chem. Eng. News, 38, H51, 46 (1960).
863. Waters, Physical Aspects of Organic Chemistry, Routledge and Kegan Paul, Lon-
don, 1953.
864. Watt, Sowards, McBridge, J. Am. Chem. Soc., 77, 5835 (1955).
865. Wei-hwa, Acta Pharm. Sin., 4, 217 (1956).
866. Вайсберге p, А., Проскауэр Э., Риддик Дж., Тупс Э., Орга-
нические растворители, ИЛ, М., 1958.
867. W е р s t е г. Rec. trav. chim., 71, 1171 (1952).
868. Werniment, Hopkinson, Ind. Eng. Chem., Anal. Ed., 15, 272 (1943).
869. Westheimer, Shookhoff, J. Am. Chem. Soc., 61, 555 (1939).
870. У э л а н д Дж. У., Теория резонанса и ее применение в органической химии, ИЛ,
М., 1948.
871. W h е 1 a n d, В г о w n е 1 1, М а у о, J. Am. Chem. Soc., 70, 2492 (1948).
872. Wilchek, Patchornik, Bull. Res. Council Israel, 11A (1962).
873. Willi, Helv. Chim. Acta, 40, 2032 (1957).
874. Williams, J. Pharm. Pharmacol., 11, 400 (1959).
875. Williams, Chem. and Ind., 1960, 900.
876. Williams, Custer, Taianta, 9, 175 (1962).
877. Williams. Harley, Chemist Analyst, 50, 114 (1961).
878. Wimer, Anal. Chem., 30, 77 (1958).
879. Wimer, Anal. Chem., 30, 2060 (1958).
880. Wimer, Anal. Chem., 34, 873 (1962).
881. W i z i n g e r, Organische Fasbstoffe, F. Dummler, Bonn., 1933.
882. Wolf, Anal. Chim. Acta, 1, 90 (1947).
883. Wollish, Colarusso, Pifer, Schmall, Anal. Chem., 26, 1753 (1954).
884. Wooding, Higginson, J. Chem. Soc., 1952, 774.
885. Wright, J. Am., Chem. Soc., 57, 1993 (1935).
886. W у n n e - J ones, Proc. Roy. Soc. A 140, 440 (1933).
887. W у n n e - J ones, Proc. Roy. Soc., A 140, 448 (1933).
Литература 401
888. Yakubik, Safranski, Mitchell, Anal. Chem., 30, 1741 (1958).
889. Я ровенко, Кошелева, Завод, лаб., 27, 407 (1961).
890. Y okoyama,Chatten, J. Am. Pharm. Ass., Sci. Ed., 47, 548 (1958).
891. Zarembo, Watts, Determination of the Carbodiimide Function, Microchemical
Techniques, ed. N. D. Cheronis, Interscience, New York, 1962.
892. Зарпцкий, Нуриев, Ж. анал. хим., 18, 1306 (1963).
893. Z a u g g, Garven, Anal. Chem., 30, 1444 (1948).
894. Z e i d 1 e r, Z. anal. Chem., 146, 251 (1955).
895. Zenchelski, Anal. Chem., 32, 289 (1960).
896. Ziegler, Dersch, Ber., 62, 1833 (1929).
897. Z у k a, Pharmazie, 9, 812 (1954).
898. Z у k а, В e r ka, Use of Lead Tetraacetate as a Volumetric and Oxidimetric Reagent
for Microanalytical Purposes, Microchemical Techniques, ed. N. D. Cheronic, Inter-
science, New York, 1962.
предметный указатель
Автопротолиз, константы 26, 27
Адренергические вещества, определение
331
Азофиолетовый как индикатор 198
Акридин, определение 297
Акцептор-кислоты 16
Акцептор-основанпя 16
Алкалоиды, определение 298, 305 и сл.
галогенгидраты 325
Алкилалюминивые соединения, определе-
ние 378
Алкилгалогениды, определение 244
Алкоксигруппы, определение 382 и сл.
Альдегиды, идентификация 355 и сл.
Амидные группы, определение 247 и сл.
Амидопирин, определение 296
Амиды кислот как основания, определение
313—315
с 2,4-динитробензоплхлоридом 315
основность 313
титрование в уксусном ангидриде 314
Аминокислоты, определение
как кислот 232
как оснований 291 и сл.
сольватация 34
N-Аминопиперидин, определение 294 и
сл.
n-Аминосалицпловая кислота, определение
229 и сл.
Амины, определение 117, 288—320; см.
также Основания азотистые органиче-
ские
первичных в присутствии вторичных
и третичных 348 и сл.
смеси 340—350
третичных (хлоргпдратов) в присут-
ствии ацетилирующих аминов (хлор-
гидратов) 347, 353 и сл.
Аммониевые ионы, сольватация 53 и сл.
Амфотерные
вещества 102
растворители 124
Ангидриды кислот, методы определения
238 и сл.
анилиновый 239
морфолиновый 238
Анестезирующие вещества, определение
327 и сл.
Анилин
определение 290
как растворитель 109
Анионы, перенос внутри молекулы рас-
творителя 17
Антигистамины, определение 326
Ароматические углеводороды, определение
380 и сл.
Аррениус —Оствальд, теория 10
Аскорбиновая кислота, титрование
бромом в уксусной кислоте 374
окислительно-восстановительное 373
и сл.
Ассоциация
бензоат-аниона 35
влияние на основность 70 и сл.
в инертных растворителях 71 и сл.
константы равновесия 36
эффекты в инертных растворителях
71 и сл.
Ацетилирование
аминов 269
спиртовых ОН-групп 263 и сл., 267
и сл., 270 и сл.
тиолов 269
фенолов 269, 270
Ацетилсалициловая кислота, определение
230
Ацетон как растворитель 104, 105, 106,
109, 114, 115 и сл.
высушивание 129, 130
Ацетонитрил как растворитель 104, 105,
106, 109, 114, 116 и сл.
высушивание 130
ионизация в 33 и сл., 116
очистка 136
Ацилониевый ион 118, 119, 121
Ацплхлориды, определение 240—242
Барбитуровая кислота, производные 248 —
кислотность 249
таутомерия 248
титрование в диметилформамиде 249
и сл.
— в метилэтилкетоне 250
— в пиридине 249, 370
— в смеси бензола и изопропилового
спирта 250
— в хлороформе 250
Бензамидин и родственные соединения,
определение 316 и сл.
S-Бензилтиуроний-производные, определе-
ние 371
Бензимидазол и его производные
амфотерность 102
определение 252, 297
Бензоат натрия, определение 312
N-Бензоилбензамидпн, амфотерность 102
Бензойная кислота
кислотность 100
определение 225—227
Бензойные кислоты, замещенные
константы равновесия 36
Предмепгный указатель
403
корреляция между рК и константами
равновесия ассоциации 38
эффект заместителей 36 и сл.
Бензол как растворитель 99, 104, 109,
114, 115
высушивание 129, 130
очистка 128, 129
Бензтиазол, определение 297
Бензтриазол, определение 252
Бренстед — Лоури, протонная теория
11 —14
n-Б ромфенацил су л ьфониевые соединения,
определение 372
н-Бутиламин как растворитель 105, НО,
125
высушивание 130
Бутиловые спирты как растворители 105
Вода
влияние на титрование 132
— как основания в кислотных раство-
рителях 61 и сл., 124 и сл.
определение в растворителях 62
как растворитель 105
Водород ацетиленовый, определение 366
и сл.
Водородная связь 107 и сл.
Выравнивающий эффект 106—109
Гаммет, функция кислотности 46—48
сульфоновых кислот 142
н-Гексан как растворитель 99, 104, 114
высушивание 130
очистка 128
н-Гептан как растворитель 104, 114
Гетеросопряжение 33
Гетероциклические соединения, определе-
ние
азотистых оснований 292—298
кислотных аналогов 252 и сл.
Гидразиды, определение 318—320
пзолизергиновой кислоты 318, 319
пзоникотиновоп кислоты 318, 319
никотиновой кислоты 319
пиколиновой кислоты 319
Гидразин, определение 319, 320
Гидразины и их производные 105, 318
Гидразоны, определение 318—320
Гидроксиламин, определение с ионами
Cu(II) 375
Гидроксильная группа, спиртовая, опре-
деление 262—272
с амидами металлов 262
с 2,4-динптробензоилхлорпдом 265
и сл.
с пропионовым ангидридом 263
в смеси 270—272
с уксусным ангидридом 264, 267—270
с фенилизоцианатом 266 и сл.
Гидроксильное число 263
Гликоли как растворители 118, 124
Гомосопряжение 33
Гутман — Линдквист, ионотропная
теория 17—18
Двойная связь, определение 361—366
бромом в уксусной кислоте 364 и сл.
с применением ацетата ртути(П)
361 и сл.
— гексаметиленамина 362 и сл.
— морфолина 362
стероидных кетонов 365
Двуокись
кремния как осушитель 130
углерода как кислота Льюиса 60
Диалкилсульфиды, идентификация 372
Диены, определение 379 и сл.
0-Дикетоны, идентификация 356
Диметиловый желтый как индикатор 198
и сл.
N,N-Диметилформамид как растворитель
100, 105, 110, 125, 126
очистка 138
1,4-Диоксан как растворитель 104, 109,
НО, 125, 126
обезвоживание 129
очистка 136—138
п, п'-Диоксидпфенилметан, определение
256
Диссоциация, константы 25—28, 101
Дифференцирующее титрование 172
Дифференцирующие растворители см. Рас-
творители дифференцирующие
Дифференцирующий эффект 106—109
Диэлектрические проницаемости 104, 105
эффект 31 и сл.
Диэтилкетон как растворитель 109
Диэтилмалонат 246
Диэтиловый эфир как растворитель 105,
109, 125, 127
очистка 135
Донор-кислота 16
Донор-основание 16
Едкое кали как осушитель 130
Изопропиловый спирт как растворитель
105
очистка 134 и сл.
Имидазолидин, производные 355
Имидогруппа, определение 247 и сл.
Индикаторы 184—201
выбор 113, 188—194
изменение окраски 100, 194—199
в инертных растворителях 189 и сл.
в кислотных растворителях 190 и сл.
в смесях растворителей 192—193
теория 185—188
трифенил метановые красители 184
в щелочных растворителях 191 и сл.
Индуктивный эффект 31, 80 и сл., 88
Ион-дипольное взаимодействие 98
Ионизация
в ацетонитриле 33 и сл.
константы 101
в нитрометане 34
органических соединений 21—23
— — и диссоциация 101
— — слабых оснований в H2SO4 62
и сл.
Карбаминовая кислота, эфиры 383 и сл.
Карбамоильная группа, определение 383
Карбениевый ион 52
как кислота Льюиса 57
Карбонильная группа, определение 354—
360
с гидроксиламином 357
с диметилгидразином 357
образование фенилгидразонов 375
полумпкрометод 358 и сл.
404
Предметный указатель
с салицилатом гидроксиламмония 360
стероидных кетонов 360
в формиатом гидроксиламина 358 и сл.
Карбоновые кислоты
димеризация 100, 108
соли, определение 282—287
— — в дифференцирующих раство-
рителях 286 и сл.
— — в кислотных растворителях
284—286
— — в основных растворителях 283
и сл.
— — в G — Н-системе растворителей
286
сольватация 100
титрование в дифференцирующих рас-
творителях 286 и сл.
— в кислотных растворителях 284—
286
— в основных растворителях 283 и сл.
— в G — Н-системе растворителей 286
Катион-анионная теория 17
Катионы, перенос 17
Каучук в природных материалах, опре-
деление 376 и сл.
Кетимины, определение 316, 317
Кетоны, определение 356—360, 355
Кетостероиды, определение 360
Кислота — основание, теории 9—20
Аррениуса — Оствальда 10
ионотропная Гутмана — Линдквиста
17-18
протонная Бренстеда — Лоури 11—14
сольвентная 10 и сл.
Усановича 18—20
Эберта — Конопика 16 и сл.
электронная Льюиса 14—16
Кислотность — основность, порядок из-
менения относительной силы в уксусной
кислоте 41—43
Кислотные вещества, определение 219—237
алифатические кислоты 222—224
аминокислоты 232
n-аминосалициловая кислота 229 и сл.
ацетилсалициловая кислота 230
бензойная кислота и ее производные
225—227
многоосновные кислоты, растворите-
ли 230 и сл.
надкислоты и гидроперекиси 232
полумикро- и микротитрование 276—
281
смеси в дифференцирующих раствори-
телях 232—237
2-фенилхинолин-4-карбоновая кисло-
та 229
холевые кислоты 230
Кислоты
вторичные 59 и сл.
катионные Бренстеда (Н-кислоты) 23
неорганические, сила 107
полумикро- и микротитрование 276—
281
растворители для титрования 110
сила, определение 35
— эффект мезомерии 80—93
слабые, протонодонорные свойства
51—52
смеси, определение 232—237
— — в ацетоне 235
— в mpem-бутиловом спирте 236,
— — в Ы,Ы-диметиламидах жирных
кислот 237
— — в метилизобутилкетоне 233 и сл.
— — в метиловом спирте 236 и сл.
— — в пиридине 235 и сл.
— — в G — Н-растворителях 236
— — в этпленглпколе 236
Кодеин, определение 308
Колориметры 216
Конечная точка титрования, определение
с индикаторами 184—201
потенциометрическое 161—183; см.
также Потенциометрическое опреде-
ление конечной точки
фотометрическое 202—218; см. также
Фотометрическое определение конеч-
ной точки
Константы
автопротолиза растворителей 26, 27
диссоциации кислота — основание 25
и сл.
Кофеин, определение 312, 313
Кремнийорганические соединения, опреде-
ление 385 и сл.
Кристаллический фиолетовый как индика-
тор 133, 194—197
Ксантогенаты, определение 371
Ксилол как растворитель 115
очистка 128
Лиат-ионы 27
Лионий-ионы 27
Льюис
кислоты 16, 55
— каталитический эффект 16
— реакции в ацетилхлориде 58
— — в нитробензоле 58 и сл.
электронная теория 14—16
Малеиновая кислота, определение в при-
сутствии ее ангидрида 239
Малоновая кислота, эфиры, определение 246
Мезомернып эффект 80—83, 88
Меламин, определение в меламиновых смо-
лах 296 и сл.
Меркаптаны, определение 369—371
с нитратом серебра в изопропиловом
спирте 369
--------в пиридине 369 и сл.
с перхлоратом ртути(П) 370 и сл.
2-Меркаптобензимидазол, определение (как
кислоты) 252, 253
2-Меркаптобензтиазол, определение (как
кислоты) 252, 253
Меркурпропзводные, определение посред-
ством ацетолиза 332
Метилизобутплкетон как растворитель 104,
105, 106, 114, 116
Метиловый спирт как растворитель 105
очистка 134
N-Метилформамид как растворитель 105
Метиэлтплкетон как растворитель 105,
109, 114, 116
высушивание 130
Микро- и полумикротитрование 276—281
кислот 276, 277—281
оборудование 277, 279
оснований 305—309
Предметный указатель
405
Многоосновные кислоты, определение
230—231
Морфин, определение 308
Мочевина, определение 314
Муравьиная кислота как растворитель
105, 107, 117 и сл.
выравнивающее действие 107
дифференцирующее действие 106
Надкислоты, определение 232
Нарцеин, определение 298
Натрий металлический как осушитель 130
Нафтиламины, определение 291
Ненасыщенные связи, определение
ацетиленовый водород 366 и сл.
двойная связь 361—363
Никотин, определение 347
Нитробензол как растворитель 105, 109,
118, 123
очистка 140
Нитрогуанидин, амфотерность 102
Нитрометан как растворитель 105, 106,
118, 122 и сл.
ионизация 34, 122 и сл.
очистка 138
а^и-форма 123
Нитросоединения, определение 273—275
ароматические 273—275
нитрогуанидин и его производные 275
n-Нитрофенол, сложные эфиры, опреде-
ление как кислот 261
Нитроэтан
как вторичная кислота 59
как растворитель 122
Окиси как осушители
алюминия 130
кальция 130
n-Оксипропиофенон, определение 259
Оксирановый кислород, определение 365
и сл.
Оксониевые ионы 54
Ониевые ионы 52 и сл., 108, 110
Основания азотистые органические, опре-
деление 288—354
алифатические 280
алкалоиды 298, 305 и сл.
амиды кислот 313—315
аминокислоты 291 и сл.
ароматические 289
бензамидин и родственные соединения
316-318
гетероциклические 292—298
гидразиды и гидразоны 318—320
гидразин и его замещенные 318—320
гидроароматические 289
кетимины 316 и сл.
в лекарственных растениях 310—312
микро-, полумикро- и ультрамикро-
методы 305—309
пиперазиновые производные 300—302
пуриновые 312—313
смеси в дифференцирующих растворите-
лях 341 и сл.
— — — с использованием ацетилаце-
тона 345 и сл.
— — — — салицилальдегида 344
и сл.
— — — — уксусного ангидрида 347
и сл.
— — — — фталевого ангидрида 346
— — — комбинированным методом
349—350
— — — методами перегонки 351—353
соли 321—340
— с азотной кислотой 340
— с галогеноводородными кислотами
321
— с органическими кислотами 321 и сл.
— с пикриновой кислотой 322—324
— с сульфокислотами 337—340
— с тетрафенилбором 335 и сл.
— с фосфорной кислотой 320
сульфамиды 315 и сл.
титрование в нитрометане 303
— в смеси нитрометан — уксусный
ангидрид 303
— 0,005 н. и-толуолсульфокислотой
305
— в хлорбензоле 115, 302 и сл.
— в уксусном ангидриде 120
фенотиазиновые производные 298—300
фотометрическая конечная точка 291,
298, 314
этилениминогруппа 317 и сл.
Осушители 129 и сл.
Перегонка фракционная растворителей
134—140
Перхлорат магния как осушитель 130
Пиперазин, производные, определение
301—303
Пиперидин
как растворитель 105, 109, НО, 125
и сл.
Пиразин, производные 300
Пиразолон, производные, определение 295
Пиридин
определение 294
как растворитель 105, 109, НО, 125
и сл.
— высушивание 130
— очистка 138 и сл.
Пиридинкарбоновая кислота, определение
294
Пиридоксин, хлоргидрат, определение 331
Полунейтрализации потенциалы 44 и сл.
Поташ как осушитель 129
Потенциометрическое определение конеч-
ной точки 161—183
изменение окраски индикатора 162,
163
нерегулярность потенциометрических
кривых 165, 167
область применения 161, 163, 164
оборудование 163, 172, 174, 175, 176,
177, 179, 180, 181
растворители 169, 170
— эффект 164—169
расчеты (конечная точка) 173
точность 163—164
фоновые электролиты 170 и сл.
форма кривой титрования 167, 183
электроды 171—183
Пропиленгликоль-1,2 как растворитель 105
Пропиловый спирт как растворитель 105
Пропионовая кислота как растворитель 98,
105, 118, 120
Пропионовый ангидрид как растворитель
105, 118, 122
406
Предметный указатель
Протон-акцептор 12
Протон-донор 12
Протон, перенос 23—25
механизм 25
определение 23
типы переносов протона 25
энергия 23 и сл.
Пуриновые основания, определение
как кислот 247 и сл.
как оснований 312 и сл.
Резерпин, определение в таблетках 307
Резонанс
влияние на силу замещенных бензой-
ных кислот 88—90
и кислотность кетонов 90—93
Растворители
амфипротонные 105
амфотерность 102, 110, 124
апротонные (инертные) 104, 105 и сл.,
114—117
— характеристики 114—127
ассоциация 101 и сл.
взаимодействие с растворенным веще-
ством 98, 103
влияние ионизации и диссоциации
101
— растворителя на растворенное ве-
щество 103
выбор 112 и сл., 170
выравнивающий эффект 106 и сл., 112
высушивание 129—134
димеризация 108
дифференцирующие 32, 105, 106 и сл.
диэлектрические проницаемости 97
и сл.
инертные 100, 109, НО
— в смесях 111, 112
кислотные 109 и сл., 117—125
— в смесях 111, 112
образование водородных связей 108,
109
общие свойства 97—99
определение воды 130 и сл.
— кислот 110
— оснований 109
основные НО, 125—127
очистка 128—141
— редко применяемых 140
перегонка фракционная 134—140
примеси, удаление 128 и сл.
смеси для определения кислот 111, 112
— — оснований 111, 112
сольватация 99
сольватирующая способность 97—99,
112
сольволиз 99 и сл.
физиологическое действие 140
Салицилат натрия, определение 312
Сера элементарная, определение 368
Сила кислот 21—60
влияние заместителей 74 и сл., 76 и сл.
— — в орто-положении см. орто-
Эффект
— индукционного эффекта 73 и сл.
— мезомерии 80—93
— пространственных затруднений
93—96
— электростатического поля 79 и сл.
и молекулярная структура 73—79
определение 35 и сл.
сравнение в различных растворителях
48-51
функция Гаммета 46—48
шкала 42, 43 и сл.
Сила оснований 61—72
влияние ассоциации 70 и сл.
— заместителей 78 и сл.
— мезомерии 80—93
— пространственных затруднений
93—96
— резонанса 83 и сл.
измерение 63—70
и молекулярная структура 71—96
полунейтрализации потенциалы в аце-
тонитриле 68—70
— — в нитрометане 66—68
— — в уксусном ангидриде 65
— — в уксусной кислоте 65—66
растворителей 127
слабых оснований в серной кислоте
62 и сл.
в уксусной кислоте 64 и сл.
шкала 42, 43
Силикагель как осушитель 130
Смазки для шлифов 145
G — H-Смесь растворителей 112
титрование кислот в 236
Сольватация 99
аминокислот 34
аммониевых ионов 53 и сл.
влияние на силу оснований 70 и сл.
Сольвентная теория 10 и сл.
Сольволиз 99 и сл.
Сольво-система
анионотропная 18
катионотропная 18
Спектрофотометры 216 и сл.
Спирты как растворители 118, 124
ряд кпслотности 124
Стероиды, определение
двойной связи 364
кетогруппы 360
Сукцинилсульфатиазол, определение 315
Сульфамидные производные 251 и сл., 315
и сл.
Сульфаминовая кислота, амфотерность 102
Сульфаты как осушители
кальция 130
натрия 129
Сульфоксиды, определение 368
Сульфониевые ионы 54
Тебаин, определение 308
Теобромин
амфотерность 103
определение 312 и сл.
Тетрагидрофуран как растворитель 135,
140
Тетразольные производные, определение
254
1,1,3,3-Тетраметилгуанидин как раство-
ритель 259
Тетрацианэтилен как реагент 280
Тиамин (витамин Bi), определение 331
Тимоловый синий как индикатор 197—
198
Тиолы, определение 369 и сл.
Тиоэфиры, определение 372
Предмепгный указатель
407
Титранты
кислотные 142—148
— определение титра 143, 144, 146, 147
— n-толуо л сульфокислота 146 и сл.
— соляная кислота 142 и сл.
— хлорная кислота 143—146
основные 149—156
— алкиламмония гидроокись 152—155
— алкоголяты щелочных металлов
150-152
— ацетат натрия 155 и сл.
— гидроокиси щелочных металлов
149 и сл.
— оборудование 151
— определение титра 149, 150, 152
определение титра кислот 142, 143,
144, 145, 147
— — — поправки 148
— — оснований 148, 150, 151, 152
редко применяемые 157—160
— — азотная кислота 158
— — алюмодп-н-бутиламид лития 159
— — аминоэтилат натрия 159
— — ацетат меди(П) 160
— — бром в уксусной кислоте 160
— — бромистый водород 159
— — изопропилат алюминия 160
— — метилат натрия в метилцелло-
зольве 159
— — — — в пиридине 159
— — нитрат серебра 159
— — содержащие аминогруппу 157
— — — карбоксигруппу 157
— — тиоцианат аммония 160
— — хлорная кислота в различных
растворителях 158
— — церий(1У)аммонийнитрат 160
— — цианид калия 159
— — 2-этилгексаналь 160
Титраторы 216 п сл.
Титрование
с образованпем комплексов 378
— — алкилалюминиевых соединений
378 и сл.
— — ароматических соединений 380
и сл.
— — диенов 379 и сл.
окислительно-восстановительное 373—
377
окрашенных растворов 224
в темноте 224
Томатин, определение 311
Толуол как растворитель 115
очистка 128
Точка эквивалентности см. Конечная точ-
ка титрования
Триазольные производные, определение
254
Трибензиламин, определение 291
Трихлоруксусная кислота, ассоциация
с пиридином 35
1,1,1-Трихлорэтан как растворитель 105
Триэтплалюминий, реакция с изохиноли-
ном 59]
Уксусная кислота 117, 118 и сл.
выравнивающий эффект 119, 120
димеризация 100, 119
дифференцирующее действие 106, 120
комплексы 119
обезвоживание 130 и сл.
как примесь в уксусном ангидриде 132
и сл.
как растворитель 105, 107, 118—120
самоионизация 119
содержание воды 130 п сл.
Уксусный ангидрид 120—122
ацетангидридий-ион 121
ацетилий-ион 121
ацетил-катион 120, 121
ацетилоксоний-ион 121
ацилоний-ион 118, 119, 121
как дегидратирующий агент 132
определение в уксусной кислоте 132
и сл.
как растворитель 105, 118, 120
— очистка 130 и сл.
самоионизация 120, 122
удаление из уксусной кислоты 133
и сл.
Фармацевтические препараты, определение
действующего начала 227—229
Фенилгидразин, определение с ионами
меди(П) 375
n-Фенилендиамин, М,Х-замещенные, оп-
ределение 292
Фенил мочевины, определение 247 и сл.
2-Фенилхинолин-4-карбоновая кислота,
определение 229, 294
Фенол, кислотность 100
Фенольная гидроксильная группа, опре-
деление 255—261
потенциометрическое титрование 256,
258, 259
титрование в ацетоне 258
— в mpem-бутиловом спирте 259
— в диметилформамиде 256, 260
— в метилизобутилкетоне 258
— в пиридине 259, 260
— в этилендиамине 256
Фенолы, эфиры, определение 260 и сл.
Фентиазиновые производные, определение
298-300
Формамид как растворитель 105
Фосфины, определенпе 384
Фосфорный ангидрид как осушитель 130
Фотометрическое определение конечной
точки 202—218
ограничения 202
основной закон 202
преимущества 202
приборы 216—218
титрование дифференцпрующее основа-
ний 213
— кривые 203—216
— — типы I 205 и сл.
------------II 206—210
------------III 210 п сл.
— в ультрафиолетовом свете 211—213
— фенолов 213—216
— хлоргидратов ампнов 213
ячейки 216, 217
Фталевая кислота, определение 239 и сл.
Хиноксалин, определение 297
Хлорбензол как растворитель 98, 99, 104,
105, 109, 114, 115
очистка 128, 135
408
Предметный указатель
Хлористый кальций как осушитель 130
Хлороформ как растворитель 104, 105, 114
высушивание 129
очистка 128, 139
Хлорхинолпн, производные, определение
295
Целлозольв (2-метокспэтанол) как раство-
ритель 105
Цианистый водород как растворитель 98,
105
Циклогексан как растворитель 104, 114
Циклогексанон, оксим, определение 270
Циклогексиламин, определение 291
Циклопропилампны, определение 363 и сл.
Циклосерин, определение 297
Четвертичные основания, определение 234
и сл.
Четыреххлористый углерод как раствори-
тель 104, 114
высушивание 129
очистка 128, 129
Число
гидроксильное 263 и сл.
кислотное 152, 224 и сл.
омыления 150, 224
Экстрактор
твердое вещество — жидкость и жид-
кость — жидкость 310
Шмалла 229
Якобса — Сингера 228
Экстракция
каучука 376 и сл.
твердое вещество — жидкость и жид-
кость — жидкость из лекарственных
растений 310—312
Электроды 171—183
алюминий — алюминий 175
графит — платина 179
двухфазные 181
задерживающие 172
индикаторные 171
каломельные 175, 176, 177, 178
капиллярные 172
комбинированные 175, 177
металлические 173 и сл.
для микротитрований 180, 181
J-образные ртутные 175
для определения фенолов 182
платина — платина 174, 180
ртуть — ацетат ртути(1) 173
серебро — хлорид серебра 177, 178
сравнения 175
стекло — каломель 180
стеклянные 171
сурьма — сурьма 174
Электростатическое поле, влияние 79
Эметин, определение 312
а-Эпоксигруппа, определение 365 и сл.
Эстрогенные вещества, титрование 260
в диметилформамиде 260
в пиридине 260
в смеси ацетон — пиридин — метило-
вый спирт 260
Этаноламин как растворитель 125, 126
Этилацетат как растворитель 109
Этиленгликоль как растворитель 105, 109
Этилендиамин как растворитель 100, 105,
ПО, 125
Этилениминогруппа, определение 317
Этиловый спирт как растворитель 118
ор/по-Эффект 95 и сл.
Эффект заместителей
в ароматическом ядре 76
на кислотность фенолов и енолов 76
и сл.
на силу азотистых оснований 78 и сл.
— кислот 37, 73 и сл.
Эффект растворителей
на ионизацию и сольватацию кислот
32—35
на силу кислот 28—30
смеси 30
сольватирующие свойства 30—32
СОДЕРЖАНИЕ
Предисловие.................................................................. 5
Предисловие автора к венгерскому изданию................................... 7
Предисловие автора к английскому изданию................................... 8
Глава 1. Развитие представлений о кислотах и основаниях...................... 9
1. Ранние теории о природе кислот .................................... 9
2. Теория Аррениуса — Оствальда...................................... 10
3. Сольвентная теория ............................................... 10
4. Протонная теория Бренстеда — Лоури................................. И
5. Электронная теория Льюиса......................................... 14
6. Представления о кислотах и основаниях Эберта — Конопика........... 16
7. Ионотропная теория Гутмана — Линдквиста........................... 17
8. Кислотно-основная теория Усановича................................ 18
Глава 2. Сила кислот ...................................................... 21
9. Ионизация органических соединений................................. 21
10. Водородсодержащие кислоты (Н-кислоты)............................. 23
И. Протонный перенос и его энергия................................... 23
12. Константы диссоциации кислот и оснований.......................... 25
13. Влияние природы растворителей на силу кислот...................... 28
14. Влияние сольватирующих свойств растворителей на силу кислот ... 30
15. Действие дифференцирующих и не аналогичных воде растворителей
на ионизацию и сольватацию кислот..................................... 32
16. Определение силы кислот и «кислотности» растворов................. 35
17. Сравнение силы кислот в различных растворителях................... 48
18. Протонодонорные свойства очень слабых кислот...................... 51
19. Ониевые ионы, катионные кислоты................................... 52
20. Кислоты Льюиса.................................................... 56
21. Вторичные кислоты ................................................ 59
Глава 3. Сила оснований..................................................... 61
22. Сила слабых оснований............................................. 61
23. Измерение основности азотсодержащих органических оснований: зави-
симость между потенциалом полунейтрализации и величиной рК . . . 63
24. Влияние сольватирующих эффектов на силу азотсодержащих оснований;
эффект ассоциации .................................................... 70
Глава 4. Связь между кислотностью, основностью и молекулярной структурой 73
25. Влияние заместителей, индукционный и другие эффекты............... 73
26. Влияние электростатического поля................................. 79
27. Влияние мезомерии на силу кислот и оснований...................... 80
28. Влияние стерических препятствий на силу кислот и оснований ... 93
Глава 5. Общие свойства растворителей....................................... 97
29. Сольватирующая способность........................................ 97
30. Сольватация ...................................................... 99
31. Сольволиз ........................................................ 99
32. Влияние ионизации и диссоциации................................ 101
33. Ассоциация....................................................... 101
34. Амфотерность..................................................... 102
35. Выводы............................................................ ЮЗ
410
Содержание
Глава 6. Растворители, используемые в неводном титровании.................. 104
36. Апротонные инертные растворители............................... 105
37. Дифференцирующие растворители.................................... 106
38. Природа дифференцирующего и выравнивающего эффектов.............. 106
39. Кислотные растворители........................................... 109
40. Основные растворители............................................. НО
41. Смеси растворителей .............................................. НО
42. Выбор растворителей............................................. 112
Г лава 7. Химические и физико-химические характеристики растворителей ... 114
43. Инертные растворители............................................ 114
44. Кислотные растворители........................................... 117
45. Основные растворители............................................ 125
Глава 8. Очистка растворителей............................................. 128
46. Использование концентрированной серной кислоты и концентрирован-
ного раствора едкого натра для очистки растворителей ............... 128
47. Высушивание растворителей ....................................... 129
48. Фракционная перегонка............................................ 134
49. Специальные методы очистки....................................... 134
50. Физиологическое действие растворителей........................... 140
Глава 9. Кислотные титранты ............................................... 142
51. Соляная кислота.................................................. 142
52. Хлорная кислота ................................................. 143
53. п-Толуолсульфокислота............................................ 146
54. Применение поправок.............................................. 147
Глава 10. Основные титранты................................................. 149
55. Гидроокиси щелочных металлов...................................... 149
56. Алкоголяты щелочных металлов...................................... 150
57. Гидроокиси алкиламмония .......................................... 152
58. Титранты, содержащие ацетат натрия................................ 155
Глава 11. Редко применяемые титранты......................................... 157
59. Стандартные растворы, приготовленные из веществ, содержащих ами-
ногруппу или карбоксильную группу.................................. 157
60. Другие титранты................................................ 158
Глава 12. Потенциометрическое определение конечной точки.................. 161
61. Область применения ............................................ 161
62. Условия, обеспечивающие точность измерений..................... 163
63. Влияние растворителя .......................................... 164
64. Диапазон потенциалов растворителей............................. 169
65. Выбор растворителей............................................ 170
66. Использование фоновых электролитов............................. 170
67. Электроды ..................................................... 171
68. Устранение флуктуаций потенциала жидкостного соединения. Различ-
ные системы электродов............................................. 178
69. Форма кривых потенциометрического титрования...................... 183
Глава 13. Индикаторы ........................................................ 184
70. Ионизированные красители, индикаторы............................ 184
71. Выбор индикатора.................................................. 188
72. Переход окраски кристаллического фиолетового...................... 194
73. Переход окраски тимолового синего................................. 197
74. Переход окраски азофиолетового................................... 198
75. Переход окраски диметилового желтого............................. 198
76. Точность измерений. Расчеты..................................... 199
Глава 14. Фотометрическое определение конечпой_точки........................ 202
77. Основной закон фотометрии........................................ 202
78. Преимущества и ограничения метода фотометрического титрования . . 202
79. Способы фотометрического титрования............................ 203
80. Графическое построение кривых фотометрического титрования. Формы
и типы кривых титрования. Примеры.................................. 203
81. Приборы для фотометрического титрования.......................... 218
Содержание 411
Глава 15. Определение веществ кислого характера........................... 219
82. Алифатические кислоты .......................................... 222
83. Замещенные алифатические кислоты................................ 223
84. Число омыления.................................................. 224
85. Кислотное число нефтяных продуктов.............................. 224
86. Бензойная кислота и замещенные бензойные кислоты................ 225
87. Определение действующего начала в фармацевтических препаратах 227
88. 2-Фенилхинолин-4-карбоновая кислота............................. 229
89. п-Аминосалициловая кислота ..................................... 229
90. Определение ацетилсалициловой кислоты в присутствии фенацетина
и кофеина............................................................ 230
91. Холевая, дезокси- и дегидрохолевые кислоты...................... 230
92. Многоосновные кислоты........................................... 230
93. Надкислоты и гидроперекиси...................................... 232
94. Аминокислоты ................................................... 232
95. Определение смесей кислот в дифференцирующих растворителях . . . 232
Глава 16. Определение ангидридов кислот, галогенангидридов, реакционноспособ-
ных алкил- и аралкилгалогенидов........................................... 238
96. Ангидриды кислот ............................................... 238
97. Определение малеиновой и фталевой кислот в присутствии их ангидри-
дов ................................................................. 239
98. Определение примесей уксусной кислоты в уксусном ангидриде . . . 240
99. Ацилхлориды..................................................... 240
100. Анализ смеси ароилхлорида, соляной кислоты и карбоновой кислоты 242
101. Определение ацилхлоридов в присутствии легко гидролизующихся
групп ............................................................... 243
102. Реакционноспособные алкилгалогениды ............................ 244
Глава 17. Определение соединений, содержащих енольный гидроксил или имидную
группу.................................................................... 245
103. Соединения, содержащие енольный гидроксил...................... 245
104. Соединения, содержащие имидную или амидную группу.............. 247
105. Барбитуровая кислота и производные гидантоина.................. 248
106. Сульфамидные производные....................................... 251
107. Конденсированные гетероциклические соединения, кислотные аналоги 252
Глава 18. Определение соединений, содержащих фенольный гидроксил.......... 255
108. Фенолы ........................................................ 255
109. Соединения, обладающие эстрогенным действием................... 260
110. Сложные эфиры фенолов.......................................... 260
Глава 19. Определение соединений, содержащих спиртовый гидроксил.......... 262
111. Определение соединений, содержащих спиртовую гидроксильную группу,
с помощью реакции этерификации....................................... 262
112. Применение хлорной кислоты как катализатора для ацетилирования
смесью уксусный ангидрид — этилацетат или пиридин — уксусный
ангидрид............................................................. 267
113. Анализ смесей соединений, содержащих гидроксильные группы . . . 270
Глава 20. Определение нитросоединений..................................... 273
114. Ароматические нитросоединения.................................. 273
115. Нитрогуанидин и его производные................................ 275
Глава 21. Полумикро- и микротитрование кислот и кислотных аналогов........ 276
Глава 22. Определение солей карбоновых кислот............................. 282
116. Титрование в основных растворителях............................ 283
117. Титрование в кислотных растворителях........................... 284
118. Титрование в среде гликоля (G — Н-системы)..................... 286
119. Титрование в дифференцирующих растворителях.................... 286
Глава 23. Определение азотистых органических оснований.................... 288
120. Алифатические, жирноароматические, ароматические и гидроаромати-
ческие азотистые основания........................................... 289
121. Гетероциклические основания.................................... 292
412
Содержание
122. Производные фентиазина ........................................... 298
123. Производные пиперазина............................................ 300
124. Титрование оснований в хлорбензоле................................ 302
125. Титрование оснований в нитрометане................................ 303
126. Титрование оснований в уксусном ангидриде......................... 304
127. Полумикро-, микро- и ультрамикротитрование алкалоидов и других
оснований ........................................................... 305
128. Действующее начало в лекарственных растениях, содержащих алка-
лоиды ............................................................... 310
129. Пуриновые основания............................................... 312
130. Амиды кислот...................................................... 313
131. Сульфамидные производные.......................................... 315
132. Кетимины. Бензамидин и родственные соединения. Соединения, содер-
жащие этилениминную группу........................................... 316
133. Гидразины, замещенные гидразины, гидразиды и гидразоны........... 318
Глава 24. Определение солей азотистых оснований............................. 321
134. Соли алкалоидов и других азотистых оснований с органическими кис-
лотами .............................................................. 321
135. Пикраты оснований................................................. 322
136. Галогенгидраты (галогениды) азотистых оснований................... 325
137. Четвертичные основания и их соли.................................. 334
138. Тетрафенилбораты оснований ....................................... 335
139. Определение оснований в присутствии «кислых» SH- или S-групп . . . 336
140. Сульфаты, этансульфонаты, n-толуолсульфонаты, фосфаты и нитраты
оснований ........................................................... 337
Глава 25. Определение смесей азотистых оснований или смесей хлоргидратов
оснований ................................................................ 341
141. Смеси аминов в дифференцирующих растворителях..................... 341
142. Салициловый альдегид, метод I.................................... 344
143. Салициловый альдегид, метод II................................... 345
144. Метод с использованием ацетилацетона............................. 345
145. Метод с использованием фталевого ангидрида....................... 346
146. Метод с использованием уксусного ангидрида....................... 347
147. Фотометрическое титрование первичных аминов в присутствии вторичных
и третичных аминов .................................................. 348
148. Определение смесей аминов комбинированным методом................ 349
149. Методы перегонки ................................................ 351
150. Определение хлоргидратов третичных аминов в присутствии хлор-
гидратов аминов, способных к ацетилированию.......................... 353
Глава 26. Определение карбонильных групп.................................... 355
151. Идентификация альдегидов и кетонов............................... 355
152. Определение карбонильных групп................................... 356
Глава 27. Определение кратных связей. Применение ацетата ртути (II), брома
в уксусной кислоте или 0,1 н. раствора бромистого водорода в уксусной,
кислоте .................................................................. 361
153. Свойства концевой двойной связи и двойной связи в ^ис-положенип 361
154. Определение ацилциклопропил аминов реакцией с ацетатом ртути(П)
в метиловом спирте .................................................. 363
155. Определение двойных связей реакцией с 0,1 н. раствором брома в уксус-
ной кислоте ......................................................... 364
156. Кислород оксирана (а-эпоксигруппа)............................... 365
157. Водород в ацетиленовых производных............................... 366
Глава 28. Определение элементарной серы и органических серусодержащих соеди-
нений. Применение нитрата серебра в пиридине.............................. 368
158. Элементарная сера в неводных средах.............................. 368
159. Органические сульфоксиды......................................... 368
160. Алкантиолы (меркаптаны). Применение нитрата серебра в пиридине 369
161. Титрование хлорной кислотой алкилксантогенатов, S-алкил- и S-бен-
зилтиурониевых производных и диалкил-п-бромфенацилсульфониевых
соединений........................................................... 371
Содержание
413
Глава 29. Окислительно-восстановительное титрование в неводной среде .... 373
162. Определение аскорбиновой кислоты в неводной среде............... 374
163. Полумикроопределение фенилгидразина, гидроксиламина и соедине-
ний, содержащих карбонильную группу, реакцией с ацетатом меди(И) 375
• 164. Определение алкилксантогенатов (ксантатов) церий(1У)аммонийнитра-
том . .................................................... 376
165. Определение каучука в растительном сырье........................ 376
Глава 30. Титрование в неводной среде, приводящее к образованию комплексов 378
166. Алкилалюминиевые соединения..................................... 378
167. Диены, активные в реакции Дильса — Альдера, и ароматические угле-
водороды ............................................................ 379
Глава 31. Определение алкоксигруппы, эфиров карбаминовой кислоты, замещен-
ных фосфинов, кремнийорганических соединений.............................. 382
168. Алкоксигруппа................................................... 382
169. Эфиры карбаминовой кислоты..................................... 383
170. Замещенные фосфины.............................................. 384
171. Кремнийорганические соединения ............................... 385
Литература ............................................................. 387
Предметный указатель...................................................... 402